/



















































































































































































































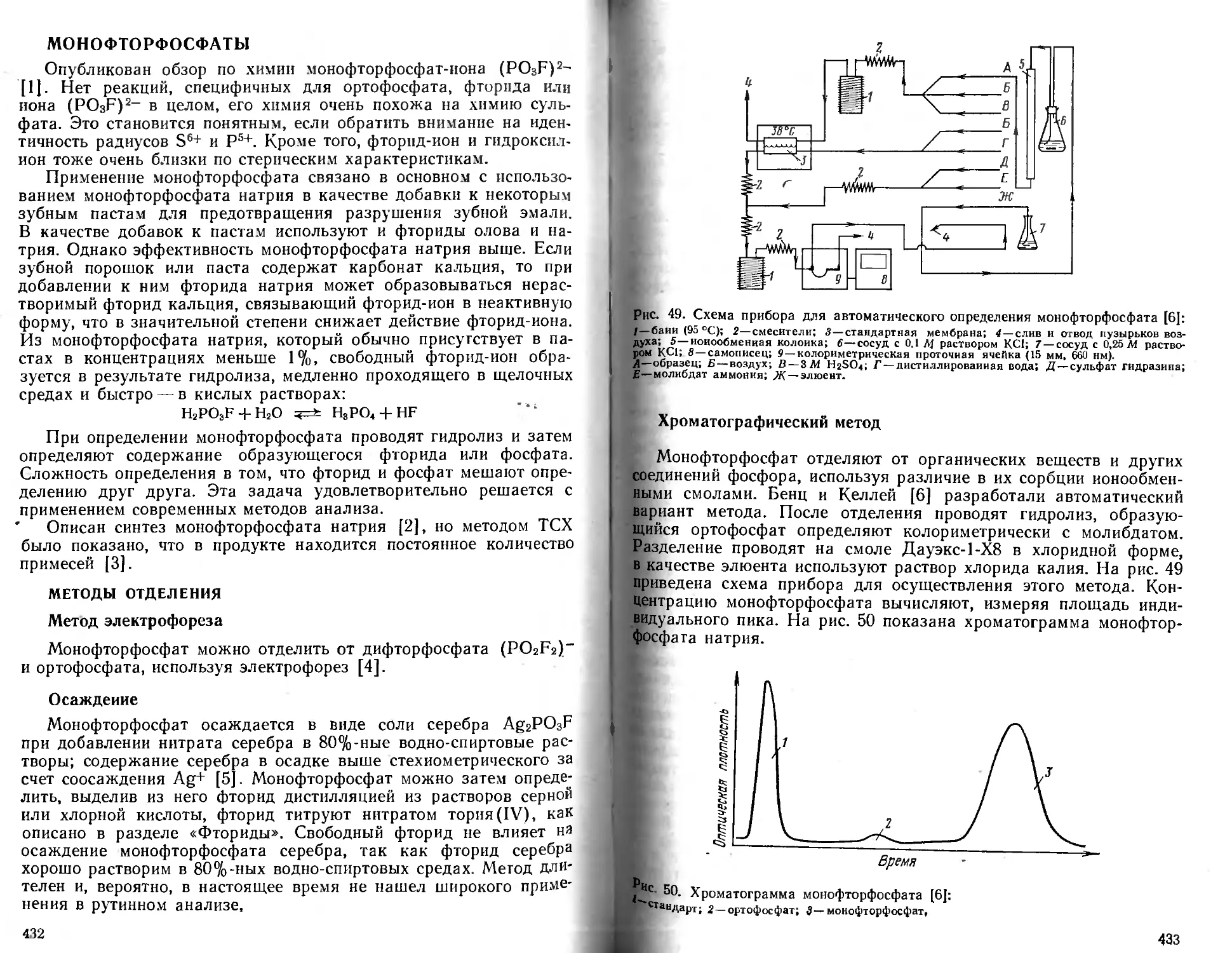



































































































Text
ОПРЕДЕЛЕНИЕ АНИОНОВ
У. Дж. Уильямс
Handbook of Anion Determination
W John Williams
School of Chemistry, University of Bath
Butterworths
London Boston Sydney Wellington Durban Toronto
ОПРЕДЕЛЕНИЕ
АНИОНОВ
У. Дж. Уильямс
Перевод с английского
канд.хим.наук С.У Крейнгольда, канд.хим.наук Л.А.Деминой и канд.хим. наук В.Н.Антонова
Москва «Химия» 1982
543
У368
УДК 543.217(031)
Уильямс У. Дж.
Определение анионов: Справочник. Пер. с англ. — М.: Химия, 1982 — 624 с., ил.
В данном справочнике для каждого аниона приведен материал о методах выделения, обнаружения и особенно определения в самых разнообразных материалах. Описаны наиболее эффективные классические (химические) и инструментальные методы определения (атомно-абсорбционный, газохроматографический и др.). Сведения о каждом анионе представлены таким образом, чтобы максимально облегчить выбор лучшего метода для конкретного случая анализа. Приведены наиболее важные методики и необходимые литературные источники.
Предназначен химикам-аналитикам, а также медикам и металлургам, связанным с химико-аналитической работой.
624 с., 42 табл., 69 рис., 3117 литературных ссылок
У
1804000000-014
050(01)-82 14,82
© Butterworth and Со (Publishers) Ltd 1979
© Перевод на русский язык, Издательство «Химия», 1982 г.
СОДЕРЖАНИЕ
ЧАСТЬ I. . 9
АНИОНЫ С, N, В, As, Si, Se И НЕКОТОРЫХ ПЕРЕХОДНЫХ ЭЛЕМЕНТОВ 9
Ацетаты.............................................................9
Арсенаты и арсениты................................................12
Бораты.............................................................29
Карбонаты и гидрокарбонаты ....................................... 43
Хроматы и бихроматы .............................................. 51
Цитраты............................................................63
Цианаты............................................................69
Цианиды ...........................................................72
Фторсиликаты.......................................................88
Формиаты...........................................................90
Гексацианоферраты(П) и (III).......................................93
Гипоиитриты.......................................................102
Молибдаты.........................................................103
Нитраты...........................................................118
Нитриты...........................................................142
Оксалаты..........................................................151
Перманганаты......................................................156
Перренаты и пертехнаты............................................164
Перрутеиаты.......................................................169
Селеиаты и селениты...............................................171
Селеноцианаты.....................................................187
Силикаты......................................................... 188
Тартраты..........................................................205
Теллураты и теллуриты.............................................208
Тетрафторобораты..................................................218
Тетрафенилбораты................................................. 220
Роданиды..........................................................222
Вольфраматы.......................................................233
Ванадаты..........................................................245
ЧАСТЬ II. 256
ГАЛОГЕНЫ........................................ . 256
Броматы..........................................................256
Бромиды..........................................................263
Бромигы..........................................................277
Хлораты..........................................................280
Хлориды..........................................................287
Хлориты..........................................................327
Фториды..........................................................331
Гипобромиты......................................................365
Гипохлориты......................................................370
Иодаты...........................................................376
Йодиды...........................................................383
Перброматы.......................................................399
Перхлораты . ....................................................402
Перйодаты........................................................410
5
ЧАСТЬ III, ОКСИАНИОНЫ ФОСФОРА..........................................., , 415
Дифосфаты (пирофосфаты) .........................................415
Гексафторфосфаты................................................420
Гипофосфаты......................................................422
Гипофосфиты.................................................... 425
Метафосфаты: трпметафосфат, тетраметафосфат, гексаметафосфат .... 429
Монофторфосфаты.................................................432
Ортофосфаты......................................................435
Фосфиты..........................................................476
Полифосфаты: трифосфаты, тетрафосфаты и высшие фосфаты...........479
ЧАСТЬ IV,
АНИОНЫ СЕРЫ.................................................. 489
Пиросульфиты...................................................489
Дитионаты......................................................490
Дитиониты......................................................492
Пероксодисульфаты..............................................498
Полисульфиды...................................................504
Политионаты....................................................509
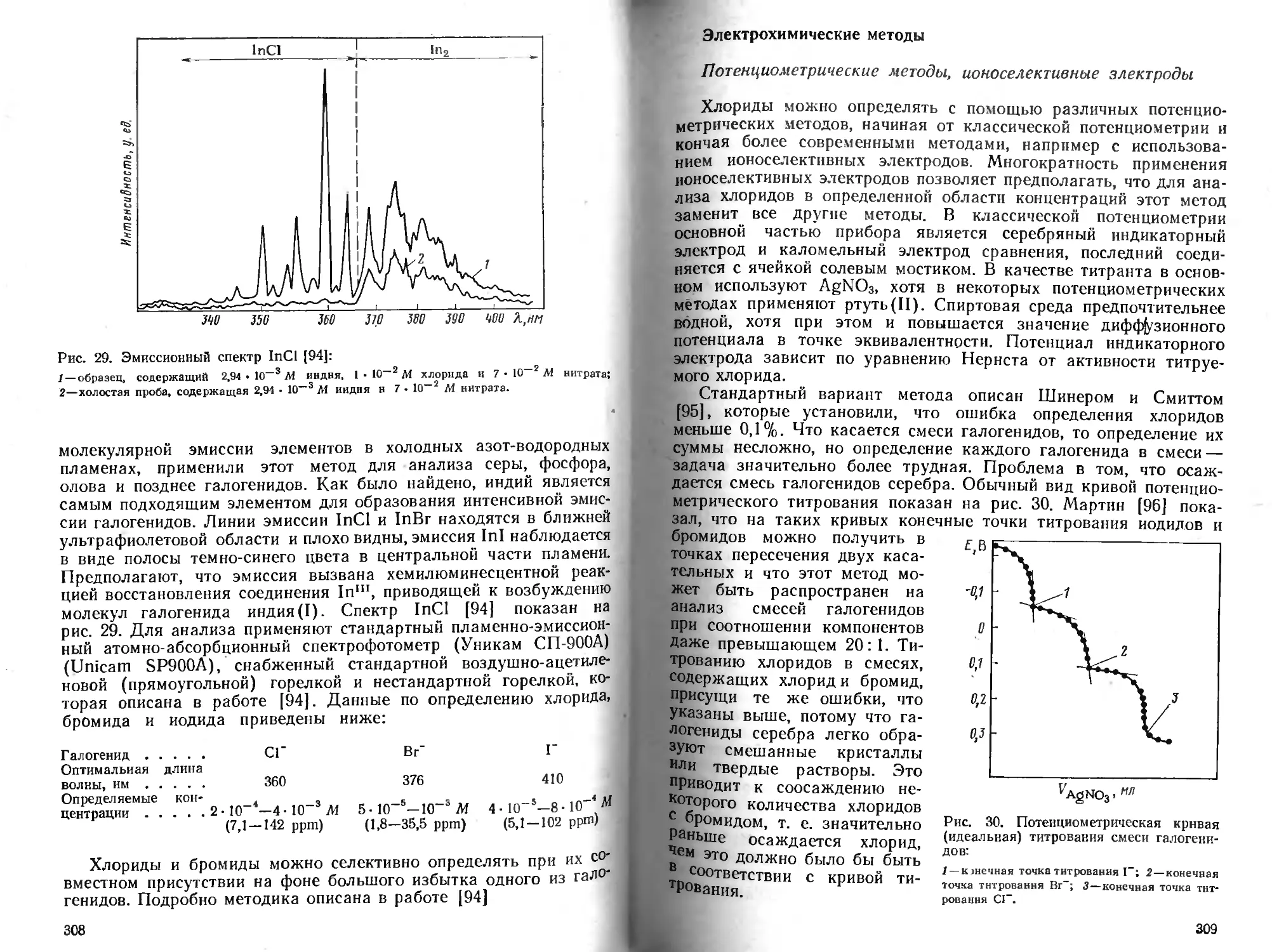
Сульфаты . ....................................................519
Сульфиды...................................................... 560
Сульфиты и диоксид серы........................................580
Тиосульфаты ...................................................595
Предметный указатель...........................................609
ПРЕДИСЛОВИЕ
Настоящая книга дает необходимую информацию об определении широкого круга анионов в различных объектах. Вряд ли найдутся возражения в необходимости такой книги. Анионы применяют в различных областях науки и технологии, и важно уметь их определять.
При написании этой книги учитывался ряд факторов. До настоящего времени нет книги (монографии), специально посвященной определению анионов. Есть, правда, монографии, посвященные определению некоторых анионов, но в них в центре внимания находятся теоретические вопросы или отдельные методы определения анионов. Определению некоторых анионов посвящены обзоры, но одни из иих ограничены в объеме, а другие являются лишь каталогами, указывающими иа метод определения, без необходимой критической оценки.
Наиболее важной особенностью книги является то, что она посвящена определению отдельных, (не объединенных в группы) анионов, причем каждому аниону посвящен самостоятельный раздел. Объем раздела определяется важностью аниона. Главным недостатком такого построения книги является неизбежность некоторых повторений при описании сходных анионов, однако он компенсируется практическими удобствами при поиске методов определения отдельных анионов.
Опыт помощи химикам и нехимикам в решении аналитических задач подсказал автору необходимость уделить больше внимания собственно аналитическим методикам, чтобы облегчить выбор эффективного метода анализа. В этом заключается основной принцип построения книги. Вероятно целесообразно отметить главные вопросы, на основании которых выбирают метод для решения конкретных задач, т. е. для какого интервала концентраций применим метод; какова его воспроизводимость; какова длительность анализа; требуются Ли дополнительные исследования; пригоден ли метод для анализа большого числа проб.
Даже для химиков-аналитиков выбрать лучший аналитический метод из большого числа возможных методов, затруднительно. Еще сложнее положение исследователей, не имеющих опыта химика-аналитика. Методы аналитической Химии широко применяют в различных областях науки: все возрастающее число биологов, физиков, металлургов и инженеров убеждаются в необходимости проведения химических анализов. Настоящая книга предназначена для них, а также для химиков, знакомых с методами анализа, характер изложения как раз отвечает запросам указанных исследователей.
Автор старался избежать изложения большого числа мало детализированных методов, избирая методы, содержащие аналитическую информацию об интервале определяемых содержаний, мешающих ионах и воспроизводимости определения. Выбор методов был непростой задачей; кроме того, одновременно отбиралась цитируемая литература. Так, многие работы, представляющие исторический интерес, не цитируются, при необходимости ранние работы можно найти в соответствующих источниках.
Структура разделов, посвященных различным анионам, идентична: методы отделения, гравиметрические, титриметрические, спектроскопические, электрохимические методы, затем каталитические, кинетические, радиохимические и термические. Этот порядок является условным и не связан со значимостью отдельных
7
методов. При квалификации гибридных методов определяющим признаком является методика, которой заканчивается определение. Включение гравиметрических методов требует пояснения, поскольку некоторые химики считают их устаревшими. Однако их продолжают использовать, когда нет других удовлетворительных методов, например для определения состава реактивов с высокой точностью или для определения вольфрамата, силиката.
Учитывая наличие превосходных монографий, посвященных методам разложения неорганических [1] и органических [2] проб, этот этап анализа анионов автором не описан, т. е. предполагается, что проба переведена в растворимое состояние. Однако в книге приведены методы отделения, как фундаментальный аспект анализа.
Всего приведено более НО методик анализа. Учитывая быстрый рост числа аналитических работ, выбор рекомендуемого метода является непростой задачей. За последние 10—15 лет предложено несколько новых методов определения анионов, например, с применением ионоселективных электродов, атом но-абсорбционной спектроскопии и газовой хроматографии. Это, разумеется изменило статус методов, считавшихся стандартными, при рекомендации тех или иных из них для практического применения мы руководствовались широтой их использования в аналитической практике.
В ряде методик приведены химические уравнения, поскольку они имеют большое значение для анализа. Концентрацию растворов чаще выражают молярностью (вместо нормальности), поэтому для расчета результатов возникла необходимость приводить полное уравнение реакции. В рекомендуемых методиках концентрации выражены в моль/л.
Автор нередко прибегает к помощи журнала Analytical Abstracts, в частности, при цитировании малодоступных работ.
Приятно высказать мою высокую оценку той поддержке, которую я получил при работе над книгой. Часто это были ценные советы, например, те, которые дали по разделам «Цитраты» и «Ортофосфаты» м-р Р. Витти и м-р С. Гринфилд. Косвенную поддержку я получал также будучи в течение 9 лет членом Совета по аналитическим методам (Аналитическое отделение Химического общества) под председательством д-ра Д. К. Гаррата. Благоприятное влияние оказали также мои коллеги по Западному региону Аналитического отделения, которые поделились своим опытом и сделали критические замечания по ряду аспектов определения анионов. Разумеется, мои коллеги ие ответственны за возможные ошибки, допущенные в книге.
Я благодарен д-ру Р. А. Чалмерсу, чей энтузиазм способствовал выходу книги. Наконец, я особенно благодарен моей жеие Анне, за непрерывную поддержку, проявленную ею при написании книги.
Автор
1. Dolezai G., Povondra Р., Sulcek Z. Decomposition Techniques in Inorganic Analysis. Iliffe, London, 1966.
2. Gorsuch T. T. The Destruction of Organic Matter. Pergamon, Oxford, 1970.
ЧАСТЬ I
АНИОНЫ С, N, В, As, Si, Se И НЕКОТОРЫХ ПЕРЕХОДНЫХ ЭЛЕМЕНТОВ
АЦЕТАТЫ
МЕТОДЫ ОТДЕЛЕНИЯ
Уксусная и другие летучие жирные кислоты могут быть отделены перегонкой с водяным паром [1]. Этот способ применяют как предварительную стадию, предшествующую определению уксусной кислоты методом газовой хроматографии [2]. Лестер [3] выделял уксусную кислоту из биологических материалов вакуумной дистилляцией и определял методом газовой хроматографии.
Метод газовой хроматографии широко используют как для отделения, так и для последующего определения уксусной кислоты. Условия эксперимента для некоторых определений приведены в табл. 1.
Таблица 1. Методики газохроматографического определения уксусной кислоты
Вещества, от которых отделяют уксусную кислоту Условия определения Литература
Пропионовая, масляная, «зо-масляиая, валериановая и «зо-валериановая кислоты 10% SP-1200 на хромосорбе, 150 °C (размер зерен 0,175—0,147 мм) 4
Муравьиная, пропионовая и масляная кислоты 10,5% этилеигликольадипината и 1,75% фосфорной кислоты на аиа-хроме (размер зерен 0,120—0,140 мм), 100 °C, газ-носитель — аргон 2
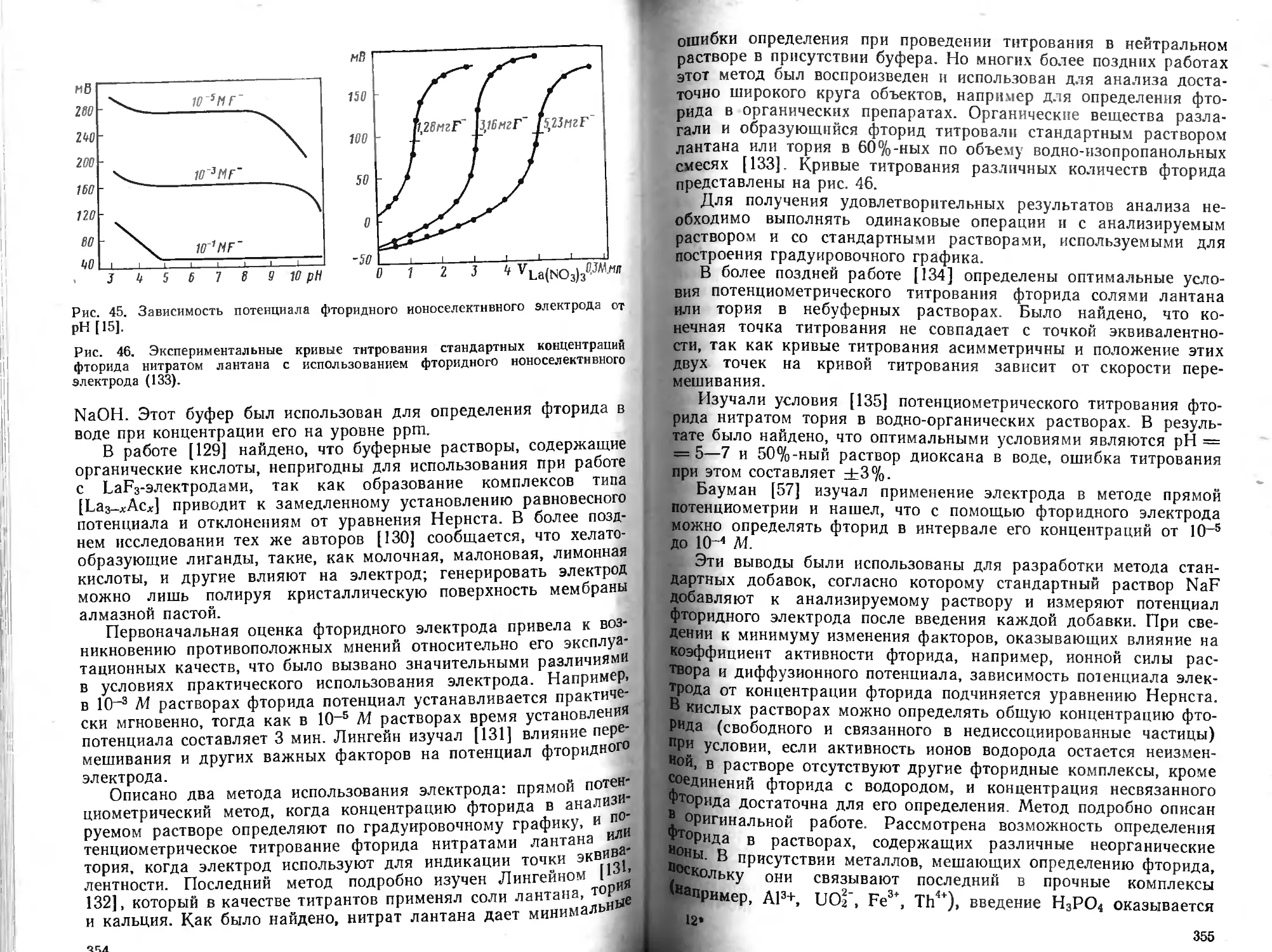
Ацетальдегид, ацетон, винилацетат, паральдегид, уксусный ангидрид и этилидеидиацетат Тетрафенилсилан, нанесенный на цеолит 545, 95 °C, газ-носитель — гелий 5
Биологические материалы 20% 2,2-диметилпропаи-1,3-диол-сукцииата и 1,7% фосфорной кислоты, 165 °C, газ-носитель — азот 6
Дитман показал, что муравьиную, уксусную, щавелевую, вин-йую и лимонную кислоты можно отделить от других карбоновых кислот методом тонкослойной хроматографии на целлюлозе [7]. Состав растворителей и значения Rf даны в разделе, посвященном цитратам. Бумажная хроматография может быть применена Для отделения ацетатов и формиатов [8].
Ацетат может быть количественно извлечен раствором три-фенилстаннония в бензоле при pH = 1 — 2,4.
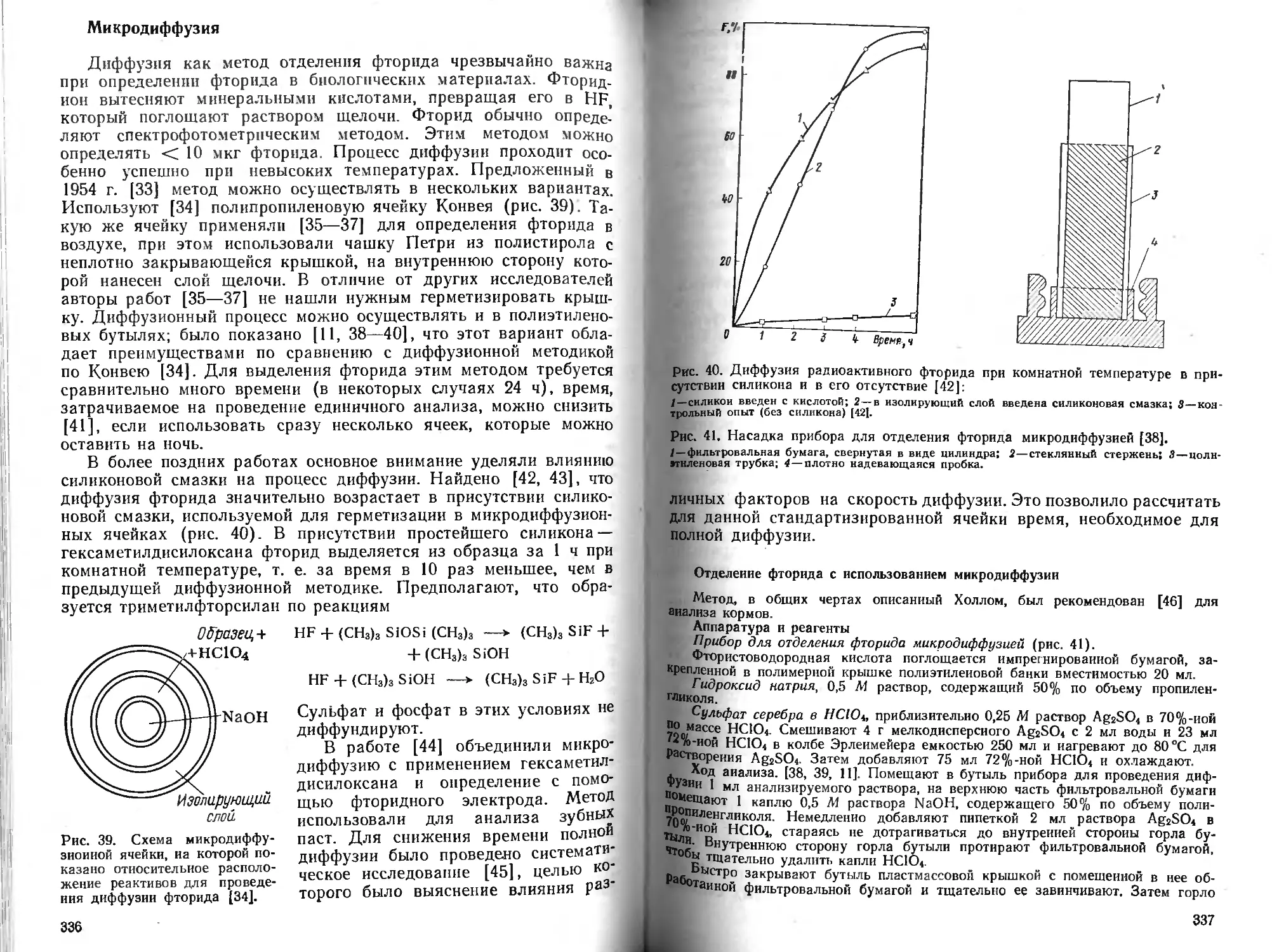
Для отделения ацетатов может быть использован метод диффузии. Лундквист, Фарман и Рассмусен изучили и применили Этот метод для анализа крови и тканей 110], отделение длительно.
9
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Данные о свойствах и методах анализа уксусной кислоты [11] опубликованы Британским институтом стандартов.
После отделения от мешающих компонентов для определения уксусной кислоты может быть использован ряд методов. Выбор метода зависит от определяемых количеств и требуемой точно-сти. Некоторые достаточно селективные методы позволяют определять ацетат в присутствии других карбоновых кислот.
Титриметрические методы
Кислотно-основные методы
Как и другие карбоновые кислоты, уксусную кислоту можно титровать в водном растворе щелочью, причем точка эквивалентности соответствует pH = 8,23. Фенолфталеин, тимоловый синий или тетрабромфенолфталеин могут быть применены в качестве индикаторов. Метод неселективен.
Эрдеи, Пикеринг и Вильсон предложили для титрования смешанные хемилюминесцентные индикаторы, например флуоресцеин с люцигенином (10,10'-диметил-9,9'-биакридиния динитрат) [12]. Действие индикатора обратимо, если титрование начинают при 60°С.
Иодиметрические методы
Прямое иодиметрическое определение по реакции
Ю; + БГ + 6Н+ —► 312 + ЗН2О
невозможно. Нельзя также усилить кислотные свойства уксусной кислоты добавлением солей Са, Ba, Mg или Zn, как это удается в случае лимонной и винной кислот содержащих гидроксильные группы. Можно, однако, ввести избыток тиосульфата в раствор уксусной кислоты, содержащей КЮз и KI и через некоторое время оттитровать этот избыток [13].
Иодиметрическое определение ацетата
Ход анализа. Приблизительно к 20 мл 0,1 М раствора уксусной кислоты добавляют 1 г KI, 5 мл 3%-иого раствора К1О3 и 25 мл 0,1 М раствора тиосульфата. Через 15—30 мин титруют избыток тиосульфата стандартным раствором иода.
Другие титриметрические методы
Для определения уксусной и муравьиной кислот при их совместном присутствии использована их способность окисляться ионами меди(Ш) при кипячении в течение 30 мин [14]. Избыток Си111 определяют с помощью мышьяка (III) и иода. Церий(IV) окисляет только муравьиную кислоту при кипячении с обратным холодильником в течение 1,5 ч в присутствии хрома (III). Оста-io
точное количество Ce!V титруют железом (II) с применением N-фенилантраниловой кислоты в качестве индикатора.
Для титрования миллиграммовых количеств ацетата предложен натрий-аммоний моногидрофосфат. Титрование ведут с применением водного раствора бромкрезолового пурпурного как индикатора [15].
Для определения анионов применен жидкостной мембранный электрод на основе растворов метилтрикаприлата аммония в 1-деканоле [16]. Для уксусной кислоты коэффициент Af/AlgC в уравнении Нернста равен 53,0 мВ. Время работы электрода составляет около месяца при условии отсутствия механических повреждений.
Для определения субмикрограммовых количеств ацетата может быть применен каталитический метод, основанный на обесцвечивании индигокармина уксусной кислотой в присутствии пероксида водорода [17].
После предварительного диффузионного отделения ацетат определяют ферментативным методом, который основан на взаимодействии ацетата с сульфаниламидом в присутствии фермента печени голубя. Не вступивший в реакцию сульфаниламид определяют фотоколориметрически при 540 нм с применением N-наф-тилэтилендиамина и сульфаминовой кислоты. Метод позволяет определять 1—15 мкг ацетата в объеме не более 3 мл и, вероятно, обладает специфичностью.
Описаны также фотоколориметрические методы определения ацетата [18, 19].
ЛИТЕРАТУРА
1. Official Methods of Analisis of the Association of Official Agricultural Chemists, Washington U. S. A., 10th edn., 1965, p. 274.
2. Shelley R. N., Salwin H., Horowitz W. — J. Ass. off. agric. Chem., 1963, v. 46, p. 486.
3. Lester D. — Analyt. Chem., 1964, v. 36, p. 1810.
4. Ottenstein D. M., Bartley D. A. — Analyt. Chem., 1971, v. 43, p. 952.
5. Smith B., Dahlen J. — Acta chem. scand., 1963, v. 17, p. 801; Analyt. Abstr 1964, v. 11, p. 1001.
6. Medzinradsky F., Lamprecht W. — Hoppe-Seyler’s Z. physiol. Chem., 1965, v. 343, p. 35; Analyt. Abstr., 1967, v. 14, p. 2702.
7. Dittman I. — J. Chromat., 1968, v. 34, p. 407.
8. Jong E. De, McCullough T. — Chmemist Analyst, 1967, v. 56, p. 80.
9. Bock R., Niederauer H.-Т., Behrends K. — Z. analyt. Chem., 1962, v. 190, p. 33.
10. Lundquist F., Fugmann U., Rasmussen H. — Biochem. J., 1961, v. 80, p. 393. II. British Standard 576: London, 1969.
12. Erdey L., Pickering W. F., Wilson C. L. — Taianta, 1962, v. 9, p. 371.
13. Kolthoff I. M. — Chem. Weekbl., 1926, v. 23, p. 260.
14. Jaiswal P. K, Chandra S. — Microchem. J., 1969, v. 14, p. 289.
15. Saxena A. K-— Microchem. J., 1970, v. 15, p. 171.
16. Coetzee C. J., Freiser H. — Analyt. Chem., 1968, v. 40, p. 2071.
17. Krauze A., Slawek Chemia analit., 1968, v. 13, p. 1329; Analyt Abstr 1970, v. 18, p. 2500.
18. Brada Z. — Chemike Listy, 1943, v. 37, p. 289; Chem. Abstr., 1950 v. 44 • p. 5762d. ’ ’
19. Kimura K-, Ikeda N., Nomura M. — Bull. chem. Soc. Japan, 1953, v. 26, p. 119;
Chem. Abstr., 1954, v. 48, p. 3201 c.
АРСЕНАТЫ И АРСЕНИТЫ
Мышьяк может находиться в растворе в виде молекул соответствующих кислот или в анионной форме (арсенаты, арсениты или комплексные анионы). После предварительной обработки проб мышьяк обычно присутствует в виде Asv. При необходимости мышьяк (V) можно восстановить путем пропускания SO2 через анализируемый раствор. Избыток SO2 удаляют пропусканием СО2. В присутствии соляной кислоты анализируемые растворы нельзя нагревать до кипения во избежание потерь летучего трихлорида мышьяка.
МЕТОДЫ ОТДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
Дистилляция
Дистилляция в виде трихлорида или трибромида остается одним из наиболее важных методов отделения мышьяка. Трихлорид мышьяка кипит при 130 °C, но обладает достаточной летучестью и при ПО °C. Для восстановления до As111 можно использовать различные реагенты. Например, сульфат гидразина имеет то преимущество, что позволяет одновременно удалять оксиды азота, которые могут мешать последующему определению мышьяка.
Отделеникг мышьяка путем дистилляции мешают нитраты и другие окислители.
Летучие хлориды образует ряд элементов, в том числе Sb, Ge, Sn, Те, Se, Hg, Re и Mo. Тетрахлорид германия кипит при 86 °C, SnCh при 115 °C, a SbCh, несмотря на более высокую температуру кипения (220°C), обладает достаточной летучестью уже при 130 °C. Если проводить дистилляцию при температуре не выше 108 °C, можно отделить мышьяк от большинства элементов, однако мешающее влияние могут оказывать Ge, Hg и Se. Селен и теллур можно восстановить до металлов путем кипячения с обратным холодильником с Н250з и хлоридом гидразина. Осадок отфильтровывают. Отгонку AsCla можно проводить в токе СОг.
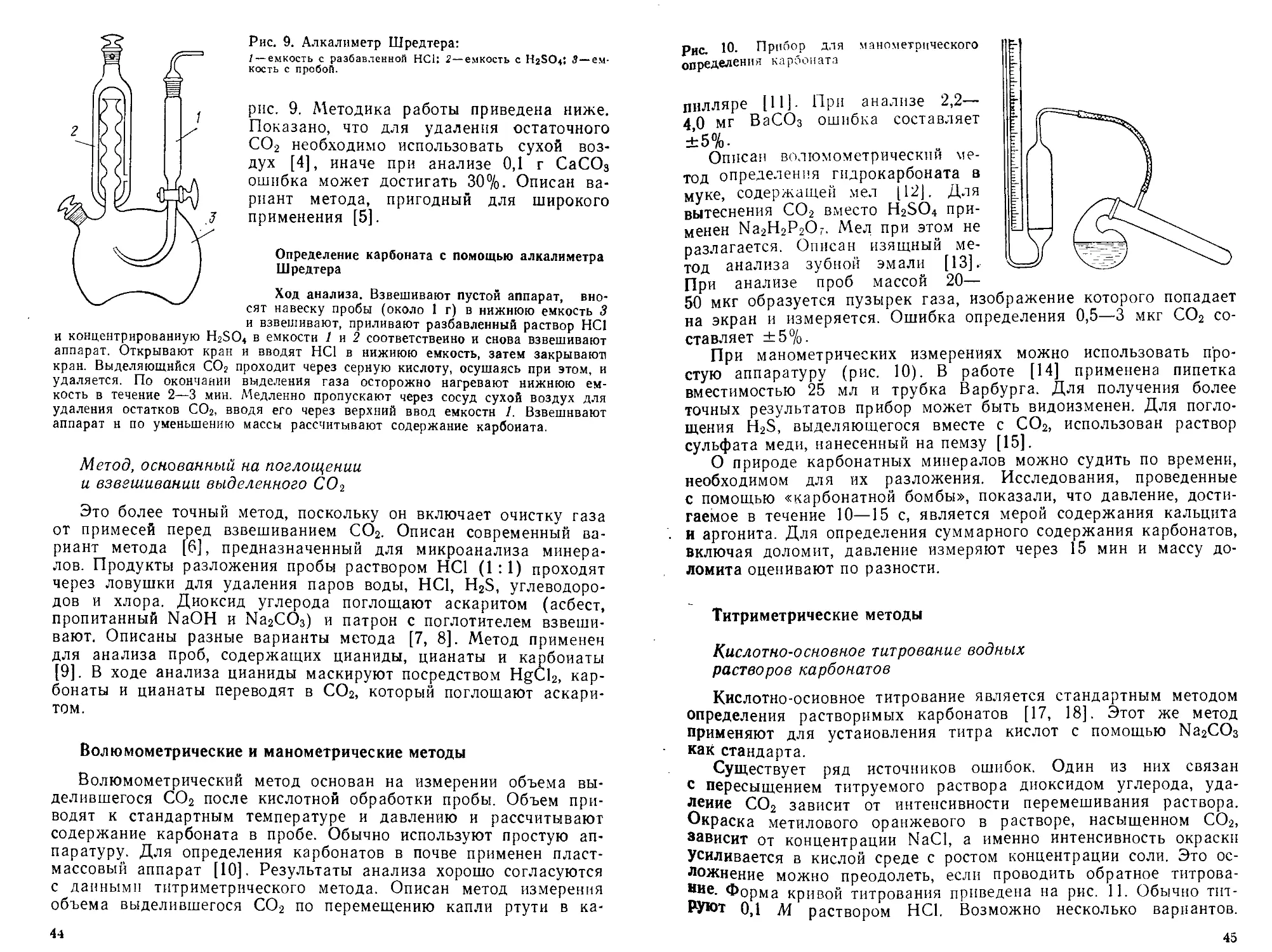
Описано большое число вариантов методики дистилляции применительно к анализу органических веществ [1], пищевых продуктов [2], пестицидов [3]. Основные детали аппаратуры для дистилляции показаны на рис. 1. Улавливать AsCl3 можно с помощью двух поглотительных склянок.
Отделение макроколичеств мышьяка дистилляцией
Ход дистилляции. Образец, содержащий ие более 0,4 г As, помещают в колбу вместимостью 500 мл, добавляют около 6 г NaBr и 1—2 г сульфата гидразина. Приливают концентрированный раствор НС1 до достижения соотношения НС1 : Н2О =1:1 и приливают раствор НС1 (1:1) до общего объема 200 мл.
Отгоняют 130—150 мл кислоты, приливают в колбу еще 80 мл НС1 (1 : 1) и продолжают отгонку до общего объема дистиллата 240 мл (в колбе остается
12
Рис. I. Прибор для дистилляции макроколичеств мышьяка (возможно последовательное подсоединение второго поглотительного сосуда).
Рис. 2. Прибор для дистилляции микроколичеств мышьяка с ловушкой.
*—общий вид; б—ловушка.
40 мл кислоты). Промывают холодильник, объединяя промывной раствор с ди-стиллатом, доводят раствор до определенного объема и анализируют аликвотную часть. В случае применения двух поглотительных склянок их содержимое объединяют и анализируют.
Для отделения малых количеств мышьяка в органических материалах, например в пищевых продуктах, рекомендована аппаратура [1], приведенная на рис. 2. Дистилляцией в виде АэВгз можно отделить 1—25 мкг As и определить его спектрофотометрическим методом по молибденовой сини.
Экстракция
' В разделе «Ортофосфаты» описаны методы отделения Р, As, Si и Ge путем экстракции гетерополикислот и приведена характеристика методов отделения. Следует отметить, что мышьяк можно восстановить в водном растворе до синего арсеномолибдата Восле удаления фосфора. Мышьяк на конечной стадии анализа Можно определять также методом атомно-абсорбционной спектроскопии.
Стара и Стары описали селективный метод экстракции мышьяка {4], основанный на работах Танаки. Мышьяк(III) экстрагируют Из сернокислого раствора, содержащего KI, четыреххлористым углеродом или другим неполярным растворителем. Можно .Экстрагировать также SnIV, GeIV и Sb111; экстракция SbHI подавляется в присутствии высоких концентраций KI. Мышьяк можно 1*8экстраг«ровать в водную фазу и затем определить соответст
13
вующим методом. Авторы работы [4] предложили метод определения мышьяка, основанный на образовании интенсивно окрашенного в желтый цвет комплекса при взаимодействии 8-меркаптохи-нолина с раствором Asl3 в четыреххлористом углероде. Спектрофотометрическим методом можно определять 0—30 мкг As. Определению мешает олово (11).
При определении мышьяка в стали Налл [5] экстрагировал АзОз хлороформом и реэкстрагировал мышьяк в водную фазу. Затем мышьяк определяли спектрофотометрическим методом по поглощению молибдомышьяковой кислоты. Фосфор определению не мешает. Фогг, Марриот и Барнс [6] модифицировали эту методику для включения ее в Британские стандарты [7], используя экстракцию Ash вместо AsCh. Показано также, что при использовании H2SO4 по способу [4] увеличивается скорость спонтанного окисления мышьяка в растворе.
В других экстракционных системах используют диэтилдитиокарбамат натрия и диэтил аммония. С помощью диэтилдитиокарбамата натрия можно отделить мышьяк путем экстракции хлороформом при pH = 5 — 6 от Ga, In и Т1 [8] или от Ge [9]. Чалмерс и Дик [10] экстрагировали мышьяк(Ш) или (V) чистым хлороформом или смесью его с ацетоном в отношении 5:2.
С помощью диэтиламмония можно экстрагировать As111 хлороформом из среды минеральных кислот (0,5—5 М H2SO4) [11]. Мешающие ионы металлов предварительно экстрагируют тем же реагентом, причем мышьяк(V) при экстракции остается в водной фазе. Боде и Нейман [12] использовали для экстракции СС14.
Отделение мышьяка экстракцией диэтилдитиокарбаматом диэтиламмония [11]
Ход экстракции. К 40 мл раствора, 2 М по HjSO4, содержащегоAsv,приливают 1 мл концентрированного раствора Н2О2 и экстрагируют мешающие иоиы последовательно двумя порциями по 10 мл 1%-ного раствора диэтилдитнокар-бамата диэтиламмония в хлороформе.
К отделенному от СНС1з водному слою приливают 1 мл 30%-ного раствора Н2О2 и осторожно кипятят в течение 15 мин. Приливают 1 мл бромной воды и кипячением удаляют бром. Охлаждают, приливают 1 мл раствора, содержащего иодид и аскорбиновую кислоту (15 кг KI и 2,5 г аскорбиновой кислоты в 100 мл раствора). Экстрагируют As111 последовательно двумя порциями по 10 мл 1%-ного раствора реагента в хлороформе. Промывают водную фазу, встряхивая ее с 5 мл хлороформа и присоединяют промывную жидкость к экстракту. Методика не позволяет отделить мышьяк от сурьмы(Ш).
Хроматографические методы
Метод тонкослойной хроматографии позволяет отделить As111 и Asv от большого числа анионов (табл. 2).
Для отделения As111 и Asv предложен [25] метод восходящей бумажной хроматографии с применением в качестве растворителя смеси метанол — 1 М раствор аммиака (4; 1). Ионообменную хро-
14
Таблица 2. Отделение As111 и Asv методом тонкослойной хроматографии *
Отделяемые ноны Адсорбент Растворитель Литература
F-, no;, s2o2-, sof, GrO2’, POf, AsOf, AsOp Силикагель + 5% крахмала МОН — н-бута-иол — Н2О (3:1: 1) 13
PO]’, NO2’; S20‘-, CrO2', N3’, CN', SCN’, BO|", S2’, Крахмал Ацетон — 3 М NH.OH (1:4) 14
AsO2’, AsO2’, no;, SO2’
AsO2’, AsO2’ Силикагель + 5% Ацетон — 15 М 15
CaSO, Н3РО4 (50 : 1) 16
CrO2’, cr, Br-, r, Bro;, C1O3’, Fe(CN)*’, Fe(CN)2’, SCN’, AsOj’, SO2’ (Циркулярная тонкослойная хроматография)
• Полуколнчественный метод тонкослойной хроматографии, описанный ранее [17, 18]. Полученные пятна нлн кольца сравнивают с эталонами.
матографию применяют для отделения мышьяка от катионов и других анионов. Сопутствующие катионы задерживаются сильнокислотными катионитами, в элюате можно определять мышьяк. Этот метод использован для отделения железа при анализе сплавов мышьяка с железом [19].
Основные аниониты используют для разделения других ионов. Например, мышьяк(III) и селен(IV) можно разделить на сильноосновном анионите Дауэкс-1 [20] с применением хлорида аммония в качестве элюента. Ионы мышьяка (111) и (V) можно разделить на слабоосновном анионите в гидрокси-форме (амберлит IRA-4B) при элюировании водой или разбавленным раствором NaOH [21].
Осаждение
Арсенаты можно осаждать, как и фосфаты, солями магния. Выделение в виде арсената магния-аммония MgNH4AsO4-6H2O позволяет отделить мышьяк от Sb, SnIV, Мо и Ge. Многие катионы мешают, осаждаясь в виде оксидов, гидроксидов и основных солей, поэтому их маскировка цитратами или тартратами повышает селективность отделения мышьяка.
Осаждение мышьяка в указанной форме можно использовать для его определения.
Друюй метод гравиметрического отделения основан на образовании гетерополикислот. Хеслоп и Пирсон при сравнительном
15
изучении молибдатов хинолиння и лутидиния показали, что эти реактивы осаждают фосфат, оставляя арсенат в растворе, причем молибдат хинолиння более эффективен, чем молибдат лутидиния.
Адсорбция на носителях
Следы мышьяка можно соосадить с гидроксидом железа. Кар и Сингх описали новый интересный метод, основанный на адсорбции [23]. Прокаленный осадок BaSO4 является адсорбентом мышьяка(У) и фосфора(У). Мышьяк(Ш) не адсорбируется, что можно использовать для разделения As111 и Asv. Для сорбции 15 мкг мышьяка необходимо 1,5 г BaSO4. Метод прост и удобен в исполнении.
Примечание. Если необходимо удаление мышьяка без последующего его определения, можно применить осаждение его сероводородом или отгонку в виде АзВгз. При необходимости определения других элементов, образующих гетерополикислоты, можно использовать прием селективного разрушения мышья-ково-молибденовой кислоты лимонной кислотой [24].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Эталонные растворы мышьяка, необходимые для анализа, готовят следующим образом.
Эталонный раствор мышьяка(Ш), 0,025 М. Растворяют 2,47 г чистого As2O3 в 20 мл 1 М раствора NaOH. Раствор слегка подкисляют по лакмусовой бумаге добавлением разбавленной H2SO4 или НС1, доводят объем раствора водой до 500 мл. Рассчитывают титр раствора (теоретически рассчитанная масса навески 2,4725 г).
Эталонный раствор мышьяка(У). Подкисляют титрованный раствор As111 или растворяют соответствующую навеску чистой соли динатрия-моногидроарсената (гептагидрата) в воде. Содержание воды в кристаллогидрате может меняться, поэтому титр раствора следует проверять гравиметрически.
Гравиметрические методы
Метод отделения мышьяка путем осаждения арсената магния-аммония описан выше. Осадок MgNH4AsO4-6H2O, полученный в присутствии ЭДТА для маскировки двухзарядных катионов, можно прокалить при 415—855 °C до пироарсената Mg2As2O7. Трех- и четырехзарядные катионы можно замаскировать Тайроном (1,2-дигидроксибензол-3,5-дисульфокислота) [26]; Возможно титриметрическое окончание анализа, при котором осадок растворяют в НО, вводят избыток ЭДТА и оттитровывают его раствором MgCl2 с применением эриохром черного Т в качестве индикатора.
Определение мышьяка осаждением в виде молибдоарсената хинолиння проводят методом, аналогичным методу определения фосфора. Описаны гравиметрический [27] и титриметрический [28] варианты определения, В первом случае, чтобы предотвратить осаждение молибденовой кислоты, в раствор вводят винную кислоту, а затем молибдат хинолиння В отличие от желтого синий молибдоарсенат не разрушается винной кислотой. Для рас
16
чета содержания мышьяка в пробе массу осадка нужно разделить на 30,127,
Для определения Asv и As'1' при совместном присутствии можно определить As111 в аликвотной части раствора путем окисления перманганатом, а содержание Asv — по разности, с учетом суммарного содержания мышьяка, найденного гравиметрически. Фосфаты, германаты, силикаты и нитраты мешают определению, ионы Fe11 и Fe111 мешают при их содержании более 2 г/л.
Арсенаты осаждаются в виде нерастворимой соли BiAsO4-H2O, что лежит в основе другого гравиметрического метода определения мышьяка. Осадок взвешивают после высушивания под вакуумом. Термогравнметрическое исследование осадка показало, что кристаллизационная вода удаляется не полностью, если температура ниже 645 °C [30].
Гравиметрическое определение арсената [29]
Ход анализа. Щелочной раствор, содержащий 0,1—0,2 г арсената, разбавляют до 100 мл и подкисляют, вводят 1 мл концентрированной HNO3. Нагревают раствор до кипения и добавляют по каплям, при перемешивании, 0,5%-ный раствор В1(\Оз)з до тех пор, пока в верхнем слое раствора ие перестанет появляться осадок. Следует избегать добавления большого избытка реагента, поскольку это приводит к ошибке, достаточно 1—2 капель избытка раствора соли висмута.
Осадок фильтруют через пористый фильтр № 4 и промывают 2%-ным раствором HNO3, этанолом и наконец диэтиловым эфиром. Осадок сушат под вакуумом и взвешивают в форме BiAsO<-H2O.
Примечание. Ошибка не превышает 0,22%.
Титриметрические методы
Редокс-методы
Литературные данные о редокс-методах определения мышьяка можно найти в работах [31—33].
Для титрования мышьяка(III) применен ряд окислителей, в том числе бромат калия, иод, иодат калия, гипохлорит натрия, хлорит натрия, тетраацетат свинца и соли церия(IV). Наиболее успешно применяют бромат калия и иод.
Если мышьяк отделяют дистилляцией, можно непосредственно титровать дистиллат броматом после подкисления раствора. Конечную точку определяют потенциометрически, а также визуально с применением обратимых или необратимых индикаторов:
ВгО’ + 3As3+ + 6Н+ —> Вг‘ + 3As5+ + ЗН2О
В конечной точке генерируется бром:
ВгО3‘ + 5Вг' + 6Н+ —> ЗВг2 + Н2О
В качестве необратимых индикаторов, окраска которых изменяется при взаимодействии с бромом, применяют метиловый оранжевый (0,1% -ный водный раствор) [34] или нафтоловый сине-чер-ный (BCI 246, 0,2%-ный раствор) [35]. Переход окраски от желтой до бесцветной и от зеленой до слабо-розовой соответственно.
17
Рис. 3. Зависимость ошибки титрования от кислотности среды при потенциометрическом титровании 25 мл 0,05 М раствора мышьяка (III) 0,01667 Л1 раствором КВгОз^
/ — среда НС1, начальная концентрация бромида 0,1 Л1; 2—среда HCI, бромид не введен; 3 — среда НС1, введено две капли 0,01 м раствора осмиевой кислоты; 4—среда H2SO4, введено две капли 0,01 М раствора осмиевой кислоты.
При использовании необратимых индикаторов следует избегать локального избытка реагента при титровании. В качестве обратимых индикаторов предложены а-нафтофлавон (0,5%-ный раствор в этаноле) [36, 37] и п-этоксихризоидин (0,1%-ный раствор в этаноле) [38]; переход окраски — от соломенно-желтой до оранжевой и от красной до желто-оранжевой соответственно. Лучшие результаты дает применение последнего индикатора.
Чтобы избежать добавления избытка титранта, рекомендуется в присутствии а-нафтофлавона титровать вблизи конечной точки медленно, при перемешивании. Показано, что недостаточная чистота а-нафтофлавона может приводить к снижению интенсивности окраски в конце титрования [39].
Важным условием получения правильных результатов при титровании броматом является выбор кислотности раствора. Отта-вей и Бишоп показали, что при концентрации НС1 менее 0,55 М в отсутствие Вг~ и менее 0,3 М в присутствии 0,1 М Вг_ результаты получаются завышенными [40]. При использовании в качестве индикатора розанилина солянокислого максимальная допустимая концентрация НС1 в обоих случаях составляет 2,5 М. Реакция протекает особенно медленно в отсутствие С1_ и Вг~, которые, вероятно, действуют как катализаторы.
С целью расширения области кислотности в качестве катализатора применен тетраоксид осмия [41]. Изменение области кислотности в присутствии катализатора видно из рис. 3. Следует отметить, что в этом исследовании применен потенциометрический метод, поскольку бром, взаимодействующий с индикатором, генерируется в конечной точке недостаточно быстро, если концентрация кислоты ниже 0,1 М. При потенциометрическом титровании применен гладкий платиновый проволочный электрод и насыщенный каломельный электрод сравнения. При более низких концентрациях кислоты постоянный потенциал устанавливается в конечной точке весьма медленно. При концентрации кислоты более 0,1 М максимальное время установления потенциала составляет 30 с.
В работе [41] показано, что осмиевая кислота действует как катализатор при титровании мышьяка (III) броматом и допускает' титрование в более широком интервале кислотности, чем в присутствии бромида и хлорида. Рекомендуется проводить титрова
18
ние броматом в среде 0,1 М НС1 пли H2SO4 с добавлением 1—2 капель осмиевой кислоты, конец титрования регистрировать потенциометрически.
Определение мышьяка(1П) титрованием броматом калия
Индикатор — л-этоксихрнзопдин, 0.1%-иып раствор в этаноле.
Ход анализа. К раствору мышьяка(Ш) приливают НС1 до концентрации 1— 1,5 Л4, добавляют 1 г КВг и 4 капли индикатора. Титруют раствором КВгО3 до появления желто-оранжевой окраски.
Один из лучших методов определения мышьяка(III) основан на титровании его иодом:
H3AsO3 + 12 + Н2О HAsOf+4Н+ + 2Г
Приведенная реакция обратима, положение равновесия зависит от концентрации ионов водорода в растворе. В сильнокислой среде равновесие смещено влево. Применение щелочей для смещения равновесия вправо невозможно, поскольку сильные основания взаимодействуют с иодом. В слабощелочной среде равновесие смещено вправо. В ходе реакции значение pH уменьшается вследствие образования HI, поэтому необходимо применение буфера. В разных исследованиях рекомендованы различные значения pH, но чаще всего реакцию проводят в присутствии избытка NaHCO3, Для установления титра растворов иода в качестве установочного вещества часто используют высокочистый триоксид мышьяка. Иод может быть получен также электрогенерированием, с применением установки для кулонометрии.
Иодиметрическое титрование мышьяка применяют широко. В Британских стандартах приведен метод определения мышьяка в стали, основанный на осаждении элементного мышьяка путем восстановления гипофосфитом. Затем мышьяк определяют иоди-метрическим титрованием [7].
Иодиметрическое определение мышьяка(Ш)
Ход анализа. Нейтрализуют при необходимости раствор арсенита, добавляют 1 г NaHCO3 и титруют стандартным раствором иода до устойчивой голубой окраски с применением крахмала как индикатора.
Хорошо известным титрантом арсенита, кроме КВгО3 и 12, является КЮ3:
2H3AsO3 + Ю3'+ 2Н++ СГ 2H3AsO4 + IC1 + Н2О
Титрование по Эндрюсу — Джеймисону протекает при высокой концентрации НС1; титрование мышьяка(III) идет удовлетворительно при концентрации НС1 3—6 М.
В этих условиях окисление йодатом проходит по стадиям
Ю3'+бН+ + 6е- —> 1' + ЗН2О
Ю3 + 5Г + 6Н+ —> 312 + ЗН2О
Ю3‘ + 21а + 6Н+ —> 51+ + ЗНгО
19
что отвечает суммарной реакции
ю; + 6Н+ + Ье' —> г + зн о
Для определения конечной точки применяют разные методы. Четыреххлористый углерод окрашивается в пурпурный цвет в присутствии свободного пода. Эта окраска внезапно исчезает при окислении последней порции иода до IC1, который не экстрагируется и окрашивает водную фазу в слабо-желтый цвет.
После первых работ Смита и Вилкокса несколько красителей использовали в качестве индикаторов [43]. Среди них нафтоловый сине-черный и амарант необратимо обесцвечиваются, а n-этоксихризоидин, предложенный Шулеком и Розса в качестве бромометрического индикатора, применен Белчером и Кларком [45] как обратимый индикатор при титровании йодатом калия.
Определение мышьяка(Ш) с помощью КЮ3
Ход анализа. Методика с применением четыреххлористого углерода. Устанавливают кислотность раствора арсенита равной 4—6 М по НС1, приливают 5 мл СС14 и титруют 0,025 М раствором КЮ3. Слой СС14 становится пурпурным. Продолжают титрование до тех пор, пока этот слой не обесцветится.
Методика с применением n-этоксихризоидина. Устанавливают кислотность раствора арсенита равной 4 М по НС!, титруют 0,025 М. раствором КЮ3 до почти полного исчезновения окраски иода и затем добавляют 12 капель 0,1%-ного раствора п-этоксихризоидина в этаноле. Продолжают титрование до перехода окраски от красной до оранжевой.
Примечания. 1. Обратимость этого индикатора ограничена вследствие его частичного разрушения. Допустимо всего 3 обратимых перехода.
2. Непосредственно перед конечной точкой индикатор приобретает интенсивную пурпурную окраску.
3. Необходимо проводить холостой опыт при смене раствора п-этоксихри-зоиднна, поскольку его значение может существенно изменяться.
Реакция между церием (IV) и мышьяком (III) протекает медленно, для ее ускорения необходимо вносить катализатор OsCh или IC1. Большинство методик основано на работах Глю [46], который предложил в качестве индикатора ферроин, а в качестве катализатора тетраоксид осмия. Окраска изменяется от оранжево-красной до слабо-голубой. Позднее было показано, что тетраоксид рутения также катализирует реакцию, но конечную точку при этом необходимо определять потенциометрическим методом [47].
Хотя суммарная реакция может быть представлена стехиометрическим уравнением
2Ce’v + As,n 2Ce'" + Asv
изучение кинетики показало, что механизм реакции сложен и большую роль играет среда [48, 49]. Важным аспектом является установление точного титра раствора церия(IV) по чистому триоксиду мышьяка [50—53]. Установлено, что существует значимое различие между результатами прямого и обратного титрования мышьяка(III) церием(IV). Несмотря на небольшое расхождение 20
(около 0,03%), оно может быть существенным для очень точных работ. Это различие связано, вероятно, с взаимодействием церия (IV) с водой.'Недавно установлено [53], что в качестве катализатора можно использовать иодид, но определению мешает нитрат, как и в случае применения OsO4 или IC1 в качестве катализаторов.
Цериметрическое определение мышьяка(Ш)
Ход анализа. К раствору арсенита, который должен быть подкислен до концентрации H2SO4, равной 1 М, приливают 0,5 мл 0,005 М раствора KI и 2 капли 0,001 М (0,25%-ного) раствора ферроина. Титруют 0,1 М раствором сульфата церия (IV) до появления бледно-голубой окраски в конечной точке.
Другие окислители также позволяют окислять мышьяк(III), в том числе гипохлорит, хлорит, тетраацетат свинца, хлорамин Т, N-бромсукцинимид и пероксомолибдат натрия. Большинство из них описано в качестве новых редокс-титрантов в книге Берка, Вултерина, Зыка [54]. V-Бромсукцинимид применен для определения арсенита в присутствии арсената [55]. Окисление проводят В присутствии NaHCO3:
СН2—СО
NBr + NaAsO2 + Н2О
СН2—СО
сн2—со
/NH + NaAsO3 + НВг
СН2—СО
Отмечено несколько преимуществ реагента по сравнению с иодом: нелетучесть, дешевизна, возможность определения малых количеств Аз20з (100 мкг). Однако растворы реагента не очень устойчивы [54].
Показана возможность титрования 0,6—6,0 мкг As”1 раствором N-бромсукцинимида с применением бордо красного как индикатора [50].
Недавно предложено применение пероксомолибдата [57]. Арсенит титруют потенциометрически с платиновым и насыщенным Каломельным электродами. Иодид калия является, вероятно, катализатором реакции.
Комплексометрические методы
' Предложен ряд комплексометрических методов определения Мышьяка (V), основанных на осаждении его нитратом висмута С Последующим определением избытка висмута путем титрования раствором ЭДТА [58, 59].
Другие титриметрические методы
Описан метод определения мышьяка, аналогичный известному ; Методу определения фосфора, путем осаждения молибдоарсената *йнолиния [60]. Осаждение ведут при температуре кипения 1 М
21
раствора НС1 в присутствии КС1О3, добавляемого для удержания мышьяка в степени окисления -J-5, и винной кислоты, предотвращающей выпадение молибденовой кислоты. Как и при определении фосфора, к промытому осадку добавляют отмеренный избыток NaOH, который оттитровывают раствором НС1 по фенолфталеину.
Воспроизводимость характеризуется вероятным отклонением ±10 мкг мышьяка, длительность определения около 20 мин.
Спектроскопические методы
Колориметрические методы
Старый метод Гудцайта все еще находит широкое применение для определения следовых количеств мышьяка в неорганических и органических материалах, в том числе в пищевых продуктах. Метод основан на взаимодействии растворов мышьяка с водородом в момент выделения с образованием арсина, который, реагируя с бромидом ртути, дает на импрегнированной бумаге желтокоричневое пятно вследствие образования Н (HgBr)2As, (HgBr)3As и Hg3As2. Несмотря на низкую воспроизводимость, метод позволяет оценивать очень малые количества мышьяка.
Метод может быть применен после дистилляции мышьяка. При анализе большинства пищевых продуктов необходимо проводить предварительное мокрое окисление органических веществ. На рис. 4 приведен прибор, рекомендованный официальной ассоциацией химиков-аналитиков (АОАС) [61]. Метод определения мышьяка (0,5—5,0 мкг) в органических материалах по Гудцайту детально описан в сообщении [62]. Этот метод официально принят также Британской фармакопеей [63].
Для получения пятен с воспроизводимой интенсивностью окраски необходимо точное соблюдение условий анализа. Определению мешают некоторые элементы, например фосфор образует РН3 (из гилофосфорной кислоты) и выделяется совместно с AsH3.
Образующийся при восстановлении H2S задерживают ацетатом свинца. Сульфат, фосфор(V) и небольшие количества фторида не мешают. Ионы Со, Ni и Си мешают.
Описан полуколичественный метод определения мышьяка с применением тонкослойной хроматографии [16]. Аппаратурное оформление метода описано ранее [17, 18]. Метод заключается в получении на хроматографической пластинке кольцевой хроматограммы. Б капле пробы можно определить 11 анионов (включая арсенит) с точностью ±5%.
Рис. 4. Прибор для определения мышьяка по Гудцайту.
22
Спектрофотометрические методы
Наиболее важный спектрофотометрический метод определения мышьяка основан на образовании гетерополпкпслоты. Арсенат, как и фосфат, взаимодействует с молибдатом в кислой среде с образованием гетерополикислоты, которую можно восстановить до молибденовой сини. Поглощение обычно измеряют при 840 нм. Из большого числа предложенных восстановителей наплучшим, вероятно, является сульфат гидразина.
Недостатком метода является необходимость строгого соблюдения условий анализа, в том числе значения pH для получения воспроизводимых результатов [64].
Ввиду мешающего влияния фосфора и кремния иногда проводят предварительное отделение мышьяка путем дистилляции. Показано, что часто в этом нет необходимости. Метод применен для анализа самых разных веществ от пищевых продуктов до сталей. Изучена возможность применения метода для анализа органических веществ (включая пищевые продукты), разработана методика анализа, которая подвергнута межлабораторным испытаниям [1]. Методика основана на работе Гофмана и Роу-сома [65]. Показано, что она более экспрессна, чем принятая ранее стандартная методика [66].
Для определения мышьяка в стали предложены две модификации метода с применением гетерополикислот [5, 6], в которых стадии образования молибденовой сини предшествует экстракционное выделение мышьяка.
Чалмерс и Синклер [67] использовали интересную особенность— существование гетерополикислот As, Ge и Si в а- и (3-форме. Добавлением ацетона можно стабилизировать (3-форму. При добавлении ацетона бесцветный молибдоарсенат приобретает желтую окраску. Поглощение сдвигается в сторону больших длин волн, но восстановления комплекса не происходит. Поглощение растворов измеряют при 440 нм. Определению мешает фосфор, не мешает кремний. Разработанная методика отличается большей воспроизводимостью, экспрессностью и чувствительностью. Закон Бера соблюдается до концентраций As 140 мкг/мл.
При определении As в присутствии Р, Si и Ge спектрофотометрическим методом проводят селективную экстракцию или маскировку мешающих ионов. Детальное обсуждение этих приемов описано в разделе, посвященном ортофосфатам. Существенны следующие факторы: 1) лимонная кислота разрушает фосфорномо-либденовую и мышьяковомолибденовую, но не кремнемолпбдено-вую кислоту; 2) добавление НСЮ4 до введения молибдата аммония предотвращает образование кремнемолибденовой кислоты, з соответствующие гетерополикислоты As и Р образуются; 3) гетерополикислоты Р, As, Si и Ge существуют в а- и (3-формах, причем устойчивость и спектры поглощения этих форм различны.;
В табл. 3 показано использование этих факторов для разработки избирательного метода определения мышьяка на основе образования гетерополикислот (ГПК).
23
Таблица 3. Методы определения мышьяка в присутствии Р, Si и Ge
Элементы Метод достижения избирательности Метод определения As Литература
As, Р Удаляют Р экстракцией изобутилацетатом в виде ГПК Спектрофотометрический 68
As, Р, Si Добавляют НС1О4 перед введением молибдата аммония для предотвращения образования Si-ГПК То же 24
As, Р, Ge Экстрагируют Р-ГПК изобутилацетатом при pH =1,0—0,8, экстрагируют Ge-ГПК изооктиловым спиртом при pH = 0,4, As остается в водной фазе 69
As, Si Экстрагируют Si-ГПК изооктиловым спиртом при pH = 4, As-ГПК остается в водной фазе 69
As, P, Si Экстрагируют Р-ГПК изобутилацетатом, экстрагируют As-ГПК смесью этилацетат — бутанол — изоамиловый спирт, экстрагируют Si-ГПК метил-изобутилкетоном в присутствии цитрата Атомно-абсорбционный спектроскопический (по Мо) 70
Исследования Пунгора и соавторов [71—73] показали, что свойства а- и р-гетерополикислот Р, As, Si и Ge, несмотря на близкие характеристики, существенно различаются, что позволяет определять их без разделения. Например, поглощение а-мышья-ковомолибденовой кислоты при 427 нм практически отсутствует, в то время как ГПК Р, Si и Ge поглощают в этой области спектра, и возможно определение примесей без операций восстановления и экстракции.
Весьма распространенным является спектрофотометрический метод определения мышьяка с применением диэтилдитиокарбамата серебра. Этот метод, связанный с образованием комплекса при пропускании арсина через раствор диэтилдитиокарбамата серебра в пиридине, проще в исполнении, чем метод образования ГПК. Однако метод имеет и ряд недостатков. В опубликованных результатах межлабораторных испытаний [1] отмечены большие расхождения в максимуме поглощения комплекса (от 520 до 550 нм), связанные со способом приготовления раствора реагента.
Мало известно и о природе самого комплекса. Исследовано применение метода для анализа питьевой воды; показано, что определению мешают следовые количества (2—500 ppb) Ст, Мо и Re, которые препятствуют образованию комплекса [74]. Ионы меди приводят к получению завышенных результатов. Тем не менее метод рекомендован АОАС [75] и Британским институтом стандартов для определения 1 —10 мкг As, он использован для анализа промышленных вод.
Атомно-абсорбционный метод
Методом атомно-абсорбционной спектроскопии можно определять непосредственно многие неметаллы, резонансные линии 24
которых лежат в вакуумной ультрафиолетовой области спектра. Мышьяк, как и Р и Si, можно определять косвенно, по молибдену, образующему с этими элементами стехиометрические соединения— гетерополикпслоты. Если определению предшествует экстракция гетерополикислот, метод отличается высокой селективностью и позволяет определять следовые количества As даже в присутствии Р, Si и Ge [70]. Основные характеристики метода даны в табл. 3.
Ямамото и соавторы описали аналогичный метод с тем отличием, что As111 окисляют подом до Asv и экстрагируют мышьяко-вомолибденовую кислоту метилизобутилкетоном [78]. Определению мешают силикаты и фосфаты.
Флуорометрические методы
В разбавленных сернокислых растворах церий (III) обладает характерной флуоресценцией в области 350 нм при возбуждении в области 260 нм. Это использовано для определения мышьяка по реакции
2Ceiv + As1” —► 2CenI-f-Asv
катализируемой тетраоксидом осмия [79]. Определяя образующийся Се!” по его флуоресценции, можно косвенно определять 7,5—37,5 мкг мышьяка.
Другой флуориметрический метод основан на образовании бромидных комплексов As”1 и Asv при —196 °C. Методика, требующая небольшой модификации ячейки для флуориметрирования, позволяет определять мышьяк в интервале концентраций 2-Ю-5 — 10~4 М [80].
Электрохимические методы
Известный полярографический метод определения мышьяка сложен и недостаточно точен. При восстановлении As”1 на электроде в кислой среде наблюдается несколько волн, причем продукты реакции не идентифицированы. Мышьяк(V) не восстанавливается. Обширная литература по данному вопросу приведена в обзоре [81].
Известен косвенный метод определения мышьяка, основанный на полярографировании пероксомолибдата, который образуется По схеме [82]
экстракция изобутанолом
As* —► арсеномолибдат-------------------► пероксомолибдат
г окисление посредством
нею, И Н2О2
• Некоторые варианты полярографии позволяют определять -Мышьяк в растворах с содержанием на уровне ppb (п-10-7%). Майерс и Остерюнг [83] с помощью дифференциальной пульс-по-Лярографии достигли предела обнаружения 4-10-9M.
. Описаны кулонометрические методы определения мышьяка, основанные на одновременном контроле тока и потенциала. Напри
25
мер, Визе и Вильямс [84] титровали мышьяк (III) электрогенери-рованным иодом при постоянном токе. Прекрасное описание этого метода определения мышьяка дано в монографии Милнера и Филлипса [85].
Успешно применяемый в анализе фторидный электрод использован для косвенного определения мышьяка (V). Арсенат при его содержании в растворе менее 1,5-10-3% осаждают солью лантана при pH = 8,65, а избыток лантана титруют фторидом с применением фторид-селективного электрода. Содержание арсената определяют графическим методом [86]. Определению не мешает 600-кратный мольный избыток хлоридов и нитратов, но сульфаты завышают результаты анализа. Влияние сульфатов можно устранить, осадив их ионами бария. Точность метода составляет ±5%. Для определения миллиграммовых количеств мышьяка (V) с более высокой точностью и воспроизводимостью в той же работе рекомендуется проводить потенциометрическое титрование раствором перхлората свинца и применять свинец-селективный электрод.
Кинетические методы
За последние годы появилось несколько работ по кинетическим методам определения мышьяка. Краузе и Славек предложили метод, основанный на окислении индигокармина пероксидом водорода. Скорость реакции определяли спектрофотометрически, методом фиксированной концентрации [87].
Богнар и Чеккел для определения более 1-10_6г/мл мышьяка предложили метод, основанный на образовании молибденовой сини. Определение проводят методом одновременного компарирова-ния [88].
В работе [89] предложен метод, основанный на реакции
As111 + Вг2 —► Asv + 2Br'
которая протекает очень быстро, но лимитируется скоростью генерирования брома:
ВгО3‘+ 5Вг'+ 6Н+ —> ЗВг2 + ЗН2О где
== к [Вго;] [вг-] [н+]2
причем k = 489 л3/(моль3-мин) при 25 °C и ионной силе, равной 1 М.
Завершение реакции окисления мышьяка(III) бромом индицируется визуально по обесцвечиванию метилового оранжевого. Описанный метод отличается простотой и экспрессностью и позволяет определять мышьяк при его содержании 0,005 мкг/мл в 20 мл раствора. Метод не селективен, определению мешают восстановители, взаимодействующие с бромом. Однако этот недостаток в некоторых случаях не препятствует анализу.
26
Радиохимические методы
Если осадить микрограммовые количества мышьяка в виде ам-моний-ураниларсената на фильтровальной бумаге, можно по результатам радиоактивационного анализа косвенно определить мышьяк, связанный с ураном, в количестве 1—8 мкг [90].
В последние годы для определения следов мышьяка применяют активационный анализ. Недавно предложен метод определения мышьяка в биологических материалах. В случае присутствия сурьмы проводят предварительную экстракцию иодидных комплексов толуолом из растворов, содержащих H2SO4 и KI [91]. Наиболее интересный пример использования активационного анализа — определение мышьяка в волосах Наполеона. Анализ выполнен, .чтобы установить, не погиб ли Наполеон вследствие отравления мышьяком. Это интересное исследование иллюстрирует значение анализа в судебной медицине [92, 93].
ЛИТЕРАТУРА
1. Analytical Methods Committee, Analyst, 1975, v. 100, p. 54.
2. British Standards 757: (1959).
3. Official Methods of Analysis of the Association of Official Agricultural Chemists, 10th edn., 1965, p. 35.
. 4. Stara V., Stary J. — Taianta, 1970, v. 17, p. 341.
5. Nall W. R. —Analyst, 1971, v. 96, p. 398.
6. Fogg A. G., Marriott D. R., Thorburn Burns D. — Analyst, 1972, v. 97, p. 657.
7., British Standards 1121: Part 38: 1967.
8. Назаренко В. А., Флянтикова Г. В., Лебедева Н. В. — Зав. лаб., 1957, т. 23, , с. 891; Analyt. Abstr., 1958, v. 5, р. 2954.
9. Saito К., Ikeda S., Saito M. — Bull. chem. Soc. Japan, 1960, v. 33, p. 884; Analyt. Abstr., 1961, v. 8, p. 1485.
10. Chalmers R. A., Dick D. M.— Analytica chim. Acta, 1964, v. 31, p. 520.
11. Wyatt P. F. — Analyst, 1953, v. 78, p 656; 1955, v. 80, p. 368.
12. Bode H., Neumann F. — Z. analyt. chem., 1960, v. 172, p. 1.
•13. Kawanabe K-, Takitani S., Miyazaki M., Tamura Z. — Japan Analyst, 1964, v. 13, p. 976; Analyt Abstr., 1966, v. 13, p. 5385.
14. Canit V. D., Turcic M. N., Bugarski-Vojinovic M. V. — Z. analyt. Chem., 1967, v. 229, p. 93; Analyt. Abstr., 1968, v. 15, p. 5211.
15. Oguma A. —Tai anta, 1967, v. 14, p. 685.
16. Hashmi M. H., Chughtai N. A. — Mikrochim. Acta, 1968, p. 1040.
17. Hashmi M. H.t Shahid M. A., Ayaz A. A. — Taianta, 1965, v. 12, p. 713.
•16. Hashmi M. H., Shahid M. A., Ayaz A. A., Chughtai F. R., Hassan N., Adil A. S. — Analyt. Chem., 1966, v. 38, p. 1554.
19. Duval G. R., Ironside R., Russel D. S. — Analytica chim. Acta, 1961, v. 25, - p. 51.
20. Krause K. A., Nelson F., A. S. T. M. Symposium No. 195, Philadelphia, 1958.
21. Blasius E. — Chromatographische methoden in der analytischen und praparati-‘ ven anorganishen chemie, Stuttgart, Enke Verlag, 1958.
22. Heslop R. B., Pearson E. F. — Analytica chim. Acta, 1967, v. 37, p. 516.
23. Kar K. R-, Singh G.— Mikrochim. Acta, 1968, p. 560.
24. Paul J. — Mikrochim. Acta, 1965, p. 836.
25. Miketukova V., Kohlicek J., Kacl K. — J. Chromat., 1968, v. 34, p. 284.
*6. Malinek M.. Rehak B. — Colin Czech chem. Commun. Engl. Edn., 1956 v 21, p. 777.
27. Filipov D. — Compt. Rend. Acad. Bulg. Sci., 1963, v. 16, p. 61; Chem. Abstr oo 1964, v. 60, p. 4797f.
48. Meyer $., Koch O. G. — Z. analyt. Chem., 1957, v. 158, p. 434.
27
29. Vancea M., Volusnivc M. — Stud. Cercet. Chim. Cluj., 1959, v. 10, p. 141; Analyt. Abstr., 1960, v. 7, p. 1713.
30. Duval C. — Inorganic Thermogravimetric Analysis, London, Elsevier, 1963.
31. Kolthoff I. M., Belcher R.— Volumetric Analysis, Vol. 3, New York, Interscience, 1957.
32. Wilson C. L., Wilson D. W. (Eds.), Comprehensive Analytical Chemistry, Vol. IB, Amsterdam. Elsevier, 1960.
33. Berka A., Vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Pergamon Press 196o
34. Gyory S. — Z. analyt Chem., 1893, v. 32, p. 415.
35. Smith G. F.. Bliss H. H. — J Am Chem. Soc., 1931, v. 53, p. 2091.
36. Uzel R. — Colin Czech chem. Commun. Engl. Edn., 1935, v. 7, p. 380; Chem. Abstr., 1936, v. 30, p. 1683.
37. Schulek E. — Z. analyt. Chem., 1935, v. 102, p. 111.
38. Schulek E., Rozsa R. — Z. analyt. Chem., 1939, v. 115, p. 185; Chem. Abstr., 1939, v. 33, p. 1621.
39. Belcher R., Nutten A. J. — Quantitative Inorganic Analysis, 3rd edn., London, Butterworths, 1970, p. 251.
40. Ottaway J. M., Bishop E. — Analytica chim. Acta, 1965, v. 33, p. 153.
41. Bhattarai D. R., Ottaway 1. M. — Taianta, 1972, v. 19, p. 793.
42. Bishop E. (Ed ), Indicators, Oxford, Pergamon Press, 1972.
43. Smith G. F., Wilcox C. S. — Ind. Engng Chem. analyt. Edn., 1942, v. 14, p. 49.
44. Schulek E., Rozsa P. — Z. analyt. Chem., 1939, p. 115, p. 185.
45. Belcher R., Clark S. 1. — Analytica chim. Acta, 1950, v. 4, p. 580.
46. Gleu K. — Z. analyt. Chem., 1933, v. 95, p. 305.
47. Keattch C. 1. — Taianta, 1961, v. 8, p. 620.
48. Rodrigues P. A., Pardue H. L. — Analyt. Chem., 1969, v. 41, p. 1369.
49. Habig R. L., Pardue H. L., Worthington J. B. — Analyt. Chem., 1967, v. 39, p. 600.
50. Zielen A. /. — Analyt. Chem., 1968, v. 40, p. 139.
51. Schlitt R. C., Simpson K.— Analyt Chem., 1969, v. 41, p. 1722.
52. Ziflen A. J. — Analyt. Chem., 1969, v. 41, p. 1905
53. Ohlweiler O. A., Meditsch J. O., Piatnicki С. M. S. — Analytica chim. Acta, 1973, v. 63, p. 341.
54. Berka A., Vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Pergamon Press, 1965.
55. Barakat M. Z., Abdalla A. —Analyst, 1960, v. 85, p. 288.
56. Sarwar M„ Randa A. K-, Hamdani S. P. — Microchem. J., 1971, v. 16, p. 184.
57. Kotkowski S., Lassocinska A. — Chemia analit. (Warsaw), 1966, v. 11, p. 789; Analyt. Abstr., 1967, v. 14, p 6774.
58. Yamamoto Y., Ban T., Ueda S. — J. chem. Soc. Japan, pure Chem. Sect., 1965, v. 86, p. 540; Analyt. Abstr., 1967, v. 14, p. 3935.
59. Vasiliev R., Anastasescu G. — Rev. Chim. Bucharest, 1960, v. 11, p. 298; Analyt. Abstr., 1961, v. 8, p. 89.
60. Meyer S., Koch O. G. — Z. analyt. Chem., 1957, v. 158, p. 434.
61. Official Methods of Analysis of the Association of Official Agricultural Chemists, 10th edn., 1965, p. 354.
62. Analyst, i960, v. 85, p. 629
63. British Pharmacopoeia, 1973, A74.
64. Chariot G. — Colorimetric Determination of Elements, London, Elsevier, 1964.
65. Hoffman I., Rowsome M. — Analyst, 1960, v. 85, p. 151.
66. Analytical Methods Committee of the Society for Analytical Chemistry, Analyst, 1960, v. 85, p. 629.
67. Chalmers R. A., Sinclair A. G. — Analytica chim. Acta, 1965, v. 33, p. 384.
68. Paul J. — Mikrochim. Acta, 1965, p. 830.
69. Paul J.— Analytica chim. Acta, 1966, v. 35, p. 200.
70. Ramakrishna T. V.. Robinson J. W. West P. W. — Analytica chim. Acta, 1969, v. 45, p. 43.
71. Halasz A., Pungor E. — Taianta, 1971, v. 18, p. 557.
72. Halasz A., Pungor E. — Taianta, 1971, v. 18, p. 569.
73. Halasz A., Pungor E., Polyak K-—Taianta 1971, v. 18, p. 577.
28
74. Martins H. M., Whitnack G. С. — Science 1971, v. P-383.
75. Official Methods of Analysis of the Association of Official Agricultural Chemists, 10th edn., 1965, p. 358.
76. British Standards 4404: (1968).
77. Rees T. D. — Proc. Soc. analyt. Chem., 1970, v. 7, p. 32.
78 Yamamoto Y., Kumamaru T., Hayashi У., Ranke M., Matsui A. — Taianta, 1972, v. 19, p. 1633.
79 Kirkbright G. F.. West T. S.. Woodward C. — Analytica chim. Acta, 1966, v. 36, p. 298.
80. Kirkbright G. F., Saw C. G.. West T. S. — Analyst, 1969, v. 94, p. 538.
81. Arnold J. P., Johnson R. M.— Taianta, 1969, v. 16, p. 1191.
82. Asaoka H. — Japan Analyst, 1968, v. 17, p. 736; Analyt. Abstr., 1970, v. 18, p. 1602.
83. Myers D. J., Osteryoung J. — Analyt. Chem., 1973, v. 45, p. 267.
84. Wise W. M,, Williams J. P.— Analyt. Chem., 1964, v. 36, p. 19.
85. Milner G. IP. C., Phillips G. — Coulometry in Analytical Chemistry, Oxford, Pergamon Press, 1967.
86. Selig U7.—Mikrochim. Acta, 1973, p. 349.
87. Krause A., Slawek J. — Z. analyt. Chem. 1969, v. 255, p. 44.
88. Bognar J., Czekkel J.—Mikrochim. Acta, 1970, p. 572.
89. Burgess A. E„ Ottaway J. M. — Analyst, 1972, v. 97, p. 357.
90. Wilson A. D., Lewis D. T. — Analyst, 1963, v. 88, p. 510.
91. Byrne A. R. — Analytica chim. Acta, 1972, v. 59, p. 91.
92. Forshufvud S., Smith H., Wassen A. — Nature, 1961, v. 192, p. 103.
93. Smith H.t Forshujvud S., Wassen A. — Nature, 1962, v. 194, p. 725.
БОРАТЫ
Бор в различных материалах находится чаще всего в виде растворимых или нерастворимых в воде боратов. Нерастворимые бораты полностью переходят в раствор при их обработке кислотами. После такой обработки бораты определяют различными методами. Содержание бората в железных и цветных сплавах, минеральных рудах, биологических материалах, пищевых продуктах, фармацевтических препаратах, почвах и удобрениях определяют рутинными методами анализа. Соединения бора применяют в производстве стекольных изделий, фарфора и глазури и в композициях для очистки металлов.
Бор — широко распространенный элемент. Установлено, что нет таких веществ, которые не содержали бы определенных количеств бора. Часто необходимо определять низкие концентрации бора, например, его содержание в чистой воде должно быть не выше 0,01 ppm (10-6%). В качестве стандартов используют в основном борную кислоту (Н3ВОз) и тетраборат натрия (Na2B4O7• ЮН20).
Аналитическая химия бора обобщена в ряде обзоров [1—3]. Методы определения бора в органическом микроанализе рассмотрены в работе [4].
Особое внимание необходимо уделять предотвращению улетучивания бора в ходе анализа или внесению его в анализируемый раствор из содержащих бор материалов, особенно стекла. При определении микроколичеств бора необходимо использовать стекла, не содержащие бор. Избежать внесения бора из посуды можно путем замены стекла на платину, фарфор или плавленный кварц.
29
Показано, что для определения около 20 мкг бора годится только платиновая и кварцевая посуда, а результаты, полученные при работе с так называемым безборным стеклом, являются неудовлетворительными [5].
Борная кислота отгоняется с водяным паром. Летучи также трифторид бора и HBF4. Метилборат имеет температуру кипения 68,5 3С, что используют в методах дистилляционного отделения бора. При кипячении, выпаривании и прокаливании подкисленных растворов, содержащих бор, необходимо учитывать возможность улетучивания бора. Для кипячения растворов необходимо использовать обратный холодильник. Потери бора при выпаривании кислых растворов рассмотрены в работе [6].
МЕТОДЫ ОТДЕЛЕНИЯ
Дистилляция в виде метилбората
Дистилляция является стандартным методом, который применяют в широком интервале концентраций от макро- до микроколичеств бора. Общий вид аппаратуры для дистилляции показан на рис. 5.
Ловушки в виде U-образных трубок содержат воду. Мешающий отделению фторид-ион можно улавливать в виде устойчивого комплекса с алюминием, если в сосуд 3 ввести А1С13. Следует, однако, избегать избытка А1С13, поскольку он занижает результаты определения бора. Вместе с бором отгоняется также ванадий(V).
В работе [5] предложена модификация аппарата для дистилляции менее 20 мкг бора (рис. 6). Аппарат изготавливают из кварца, за исключением рубашки холодильника, которая может быть из мягкого стекла. Дистиллат собирают в платиновой чашке.
Рис. 5. Прибор для дистилляции макро- и микроколичеств бора:
1, 6—ловушки с водой; 2—колба с метанолом;
3 — колба с пробой; 4—холодильник; 5 —колба для сбора дистиллята.
Рис. 6. Прибор для дистилляции микроколичеств бора.
30
Показано, что метанольный дистиллат можно упарить, не теряя бора, если предварительно добавить в него воду, NaOH и глицерин. Полученный раствор можно непосредственно анализировать спектрофотометрическим методом с применением куркумина [5]. Аналогичный аппарат для дистилляции предложен в работах [7,8] . В этом аппарате нагревание проводили инфракрасной лампой, а метилборат отгоняли в токе азота.
Отделение бората дистилляцией
Ход дистилляции. Применяют аппарат, показанный на рис. 5. Навеску пробы до 1 г растворяют в 5 мл 6 М раствора НС1 и вносят в колбу 3. Если проба не растворяется в НС1, необходимо сплавить ее с карбонатом. Для связывания воды в колбу 3 добавляют чистую безводную соль СаС12 (около 1 г на каждый миллилитр пробы). В колбу 2 вносят около 300 мл очищенного метанола, включают плитку с закрытой спиралью и отгоняют около 25 мл метанола в колбу 3. Затем нагревают колбу 3 на водяной бане, избегая улетучивания метанола. Собирают в колбе 5 около 180 мл дистиллата и приливают к нему содержимое ловушки. Для контроля полноты отгонки бора можно провести дополнительную дистилляцию. Дистиллат можно сконцентрировать упариванием после нейтрализации щелочью (NaOH). Щелочной раствор поглощает значительные количества СО2, которые можно удалить, если слегка подкислить раствор и прокипятить его в течение нескольких секунд. Затем раствор снова нейтрализуют по метиловому красному.
Ионообменный метод
Для определения боратов применяют катионный и анионный обмен. Мартин и Хайес при анализе ферробора применили катионный обмен для отделения бората от ионов металлов [9]. По методике определения бора в ферроборе, принятой в Британских стандартах [10], железо удаляют катионным обменом и затем титруют борную кислоту гидроксидом натрия в присутствии маннита.
Сузуки использовал сильноосновной анионит для разделения бората и силиката [И]. Карлсон и Пауль [12] применили избирательную к бору смолу Амберлит ХЕ-243 в полиэтиленовой колонке. Сконцентрированные бораты переводили в тетрафторборат Для определения с помощью ионоселективного электрода.
Экстракция
Для определения бората известно несколько экстракционных методов. Показано, что растворимые формы бора в удобрениях Экстрагируются 20%-ным раствором 2-этилгексан-1,3-диола в ме-ТИлизобутилкетоне. Органическую фазу анализируют методом атомно-абсорбционной спектроскопии. Позднее было изучено 40 Потенциальных экстрагентов борной кислоты [14]. Показано, что алифатические 1,3-диолы с шестью и более углеродными атомами являются более эффективными экстрагентами по сравнению с ди-Кетонами, гидроксикетонами и другими соединениями. Следует отметить, что ряд металлов, мешающих определению бора, можно отделить экстракцией в виде комплексов с 8-оксихинолином.
31
Метод тонкослойной хроматографии
Методом тонкослойной хроматографии на маисовом крахмале с использованием в качестве растворителя смеси ацетон — 3 М раствор NH4OH (1:4) удалось разделить 12 анионов: NO.', SjOs'-СгОГ, N3‘, CN', SCN', ВОз", S2', AsO?, NOj, SO? и РО? [15]. Борат и оксалат можно разделить на силикагеле, содержащем 5% растворимого крахмала, используя в качестве растворителя бутанол, насыщенный 2Л1 раствором HNOs [16].
Другие методы отделения
Ряд ионов металлов можно отделить от боратов путем электролиза на ртутном катоде. Бораты остаются в растворе. Можно также отделить некоторые ионы металлов путем осаждения в виде гидроксидов при pH = 5,0 — 5,5; при этом бораты остаются в фильтрате. Для отделения металлов можно экстрагировать их в виде оксинатов. Другие методы разделения будут приведены ниже.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Разработано несколько гравиметрических методов определения бора. Эти методы обычно рассматривали как второстепенные по сравнению с титрованием щелочью в присутствии маннита, и в.е-роятно они не нашли широкого применения. Информацию об этих методах можно найти в цитированных выше обзорах. Однако в последнее время появились новые работы. Например, Акимов, Бусев и Анджапаридзе сообщили об использовании производных антипирина (1,1-диантипирилбутан и а,а-диантипирилтолуол) для гравиметрического определения 0,5—37 мг бора в 0,5 М растворе HF. При использовании первого реагента относительная ошибка составляет всего 2% и определению не мешают Al, Zn, Cd, Ti, Zr, Cu, Fe, Co и Ni, однако Sbv, Nb и Та должны отсутствовать. Описан также титриметрический вариант метода [17]. Чедвик описал гравиметрический метод определения бората с применением 2,4,6-трифенил-пириллийхлорида. Подкисляют боратный раствор посредством НС1, добавляют реагент, а затем HF. Борат переходит в BF4, который затем участвует в реакции
(CeH6)3 СВН2О+ + BF" —> (СвН6)з C5H2OBF4 (осадок)
Осадок выдерживают 20 ч при комнатной температуре, 3 ч при 0°С, затем его высушивают при И0°С и взвешивают. Фактор пересчета равен 0,1561. Дополнительные данные о реагенте приведены в разделе, посвященном перхлорату.
Титриметрические методы
Наиболее важным для определения миллиграммовых количеств бора является метод титрования щелочью (NaOH) в присутствии маннита. При некоторых условиях титрование можно использовать
32
для определения .микроколичеств бора, но для определения малых количеств предпочтительны все же другие методы. Титриметриче-ский метод впервые был применен в 1899 г. (Гуч, Джонс [19], Джонс [20]), хотя взаимодействие полиспиртов с борной кислотой известно еще раньше.
Основы метода обсуждены в ряде аналитических работ. Борная кислота является слабой кислотой (ka = 6,4-10_|°), которую нельзя титровать в обычных условиях щелочью. Однако Н3ВО» взаимодействует с полигидроксисоединениями, например глицерином, сорбитом, маннитом, с образованием значительно более сильных кислот (/га»10-4):
I
—С (ОН)
в (ОН)з + 2 I
—С (ОН) I
+ н+ + зн2о
Обычно в анализе применяют маннит, однако было показано, что более эффективен сорбит [21]. При анализе чистой борной кислоты к ней добавляют маннит и титруют раствором NaOH, отмечая конечную точку по появлению окраски фенолфталеина. Если борная кислота присутствует в смеси с сильной кислотой, последнюю необходимо предварительно нейтрализовать с применением в качестве индикатора метилового красного или подобного ему индикатора. Затем после добавления маннита продолжают титрование до появления окраски фенолфталеина. Объем щелочи, пошедший на титрование между двумя переходами, эквивалентен содержанию борной кислоты. Возможно потенциометрическое титрование до pH = 8,5. В опубликованных методах потенциометрического титрования сильную кислоту нейтрализуют до' pH = 4 — 6. Диксон установил, что для получения правильных результатов необходимо при нейтрализации сильной кислоты и установлении титра щелочи по борной кислоте исходить из одного и того же значения pH раствора, особенно при микротитрованиях [22]. Вильсон и Пеллегрини использовали метод «идентичного значения pH» [23] при определении боратов в удобрениях. Раствор доводят до pH = 6,3 и после добавления маннита титруют 0,02 М раствором NaOH (без карбонатов) до того же значения pH. Метод рекомендован для анализа удобрений [24]. Назаренко и Ермак считают, что оптимальная кислотность при потенциометрическом титровании Н3ВО3 с маннитом отвечает pH = 7,75 — 7,9 [25].
Титрованию Н3ВО3 с маннитом мешают многие вещества, в том числе, разумеется, слабые кислоты и основания. Фосфат, который может быть захвачен при дистилляции, мешает определению. Фосфат можно осадить в виде магний-аммоний фосфата или фосфата железа(III). Можно использовать также метод осаждения нитратом висмута. Избыток висмута удаляют путем слабого подщелачивания [23]. Мешают определению борной кислоты ионы аммония, которые можно удалить кипячением раствора со щелочью. 2 Зак. 167
33
Тяжелые металлы могут маскировать конечную точку титрования или оказывать мешающее влияние вследствие гидролиза солей. В этом случае необходимо отделение бора дистилляцией метилбора та.
Титрование щелочью в присутствии маннита рекомендовано в качестве стандартного метода определения бора [26—28]. Определение бора в ферроборе [28] проводят после отделения ионов железа на катионите.
Спектроскопические методы
Спектрофотометрические методы
в видимой и ультрафиолетовой областях
Широкое распространение бора, встречающегося обычно в низких концентрациях, дало толчок развитию методов определения следовых количеств этого элемента. Среди них спектрофотометрические методы занимают значительное место. Последние исследования показали растущее значение метода атомно-абсорбционной спектроскопии для определения бора.
Большинство спектрофотометрических реагентов для определения бора, например, хинализарин, карминовая кислота, диан-тримид являются производными антрахинона. Окраска растворов обычно развивается в растворах концентрированной H2SO4. В этих случаях окраска зависит от температуры, кислотности раствора, времени нагревания и других условий анализа. Иногда в литературе утверждают, что один реагент имеет преимущество перед другими. Такие мнения, вероятно, базируются на сравнении по ограниченному числу критериев и не имеют достаточных оснований для обобщенных выводов. Опубликованы основные данные о 21 хромогенном реагенте для определения бората, в том числе о чувствительности, оптимальной длине волны и концентрации реагента [29]. Их характеристики приблизительно одинаковы.
Следует отметить другой аспект развития методов определения бора — их автоматизацию. До недавнего времени возможность автоматизированного определения бора с помощью некоторых реагентов, таких, как ализарин и карминовая кислота, принимали с оговорками, поскольку определение проводят в среде концентрированной H2SO4 [30]. Применение эсидфлекса и аналогичных материалов позволило преодолеть это затруднение. Описан упрощенный автоматический метод определения бора с куркумином [31]. В той же работе рассмотрена возможность применения еще И автоматизированных методов определения бора. Спектрофотометрические методы определения бората описаны в монографии Больца [53] и в последнем обзоре Брамана [3].
Рассмотрим некоторые реагенты. 1,1'-Диантримид (1,Г-бисан-трахиноиламин) в концентрированной серной кислоте окрашен в желтый цвет, который в присутствии бора переходит в синий. Окраска зависит от температуры, она пропадает при 90°C. Изме-34
рения обычно проводят при 620 нм, закон Бера соблюдается в интервале 0,5—6 мкг бора. Чувствительность определения выше, чем в случае применения хинализарина пли карминовой кислоты. Мешают определению бора сравнительно большие количества фторида. Определению мешают также XOJ, Х’Оз, CrOf, 10] и СЮ], но их влияние можно устранить. В работе [32] установлено влияние больших количеств катионов на результаты анализа. Установлены оптимальные условия и влияние мешающих ионов при определении 1 —10 мкг бора [33].
В присутствии бора растворы хинализарина (1,2,5,8-тетрагидр-оксиантрахинон) изменяют свой цвет от красно-фиолетового до синего. Поглощение измеряют при 615 нм. Определению мешают нитраты и фториды. Реагент более чувствителен к германию, чем к бору, поэтому германий должен отсутствовать. Большое влияние оказывают температура и концентрация H2SO4. По данным работы [34], чувствительность определения бора составляет 3,7-• 10-4 мкг/мл, оптимальный интервал содержаний бора 0,06— 0,25 мкг в пробе. Автоматический вариант метода не описан.
Кармин и карминовая кислота, глюкозид кармина дают идентичную цветную реакцию с боратом, поскольку глюкозидный радикал не сопряжен с хиноидной структурой реагента, которая определяет окраску раствора. Кармин в концентрированной H2SO4 изменяет окраску от ярко-красной до пурпурно-синей в присутствии бората. Поглощение измеряют в интервале длин волн 575 -610 нм. Окраска развивается медленно при комнатной температуре и быстро при 90°C. В обычном варианте осуществления метода определению мешают F\ NO], As111, Юз', Г, Vv и цитрат. Закон Бера соблюдается в интервале 0—20 мкг бора. Автоматический вариант метода имеет предел обнаружения 0,02 мг/л, и для содержаний бора 3,0 мг/л относительное стандартное отклонение составляет 0,03 мг/л [35]. Метод применен для анализа сточных и речных вод. Описание других автоматических методов дано в работе [31].
Метод определения бора с помощью карминовой кислоты нашел широкое применение и рекомендован для анализа пищевых продуктов [27] и воды [26]. Предложено два улучшенных
метода фотометрического определения бора в видимой и один но* вый метод в ультрафиолетовой области спектра [36]. В первых двух методах измерения проводят в среде 95%-ной H2SO4 при 615 нм и в смеси H2SO4— уксусная кислота при 548 нм. Чувствительность определения равна соответственно 1,5-10~3 и 3,8-10-4 мкг/мл. На рис. 7 приведены спектры поглощения комплексов бора с карминовой кислотой, из которых видно, что поглощение в УФ-области (300 нм) выше, чем в области 600—615 нм. Чувствительность определения В при 300 нм равна 7-10~4 мкг/мл. Влияние посторонних ионов описано в работе [36]. Определению спектрофотометрическим методом с помощью карминовой кислоты (в среде H2SO4 при 300 нм) не мешают следующие ионы (в скобках указаны допустимые содержания, мкг/25 мл): F’(2);GeVI, TilV (1); UOi+, NO3 (50), Fe2+(10), Ce‘"(100); АзОз'(150). Другие ионы мешают при более высоких содержаниях [36].
Изучено применение карминовой кислоты для определения бора в магнезитах [37], влияние магния, выщелачивание бора из стекла и потери его за счет летучести при химической подготовке^ Куркумин, 1,7-бис(4-гидрокси-3-метоксифенил) -1,6-гептадиен-3,5-дион) был первым реагентом, с которым не требуется применения растворов концентрированной H2SO4. Метод, основанный на образовании двух видов комплексов бора, подробно описан в работе [38].
Куркумин в среде уксусной кислоты образует с борной и щавелевой кислотой руброкуркумин. Руброкуркумин сравнительно легко подвергается гидролизу, поэтому воспроизводимость определения невысока. Необходимо контролировать содержание воды в растворе и воздухе. Соотношение борной кислоты, куркумина и щавелевой кислоты в комплексе равно (1:1:1). В сильнокислом безводном растворе борат образует с куркумином розоциа-нин, который очень устойчив. Молярный коэффициент поглощения розоцианина в два раза выше, кем руброкуркумина, поэтому этот метод определения бора более чувствителен. Соотношение борной кислоты и куркумина в комплексе равно 1 : 2 [39, 40]. При анализе водных растворов с помощью куркумина первой стадией является удаление воды, которая мешает определению. Ранее это достигалось упариванием раствора досуха на водяной бане. Затем растворяли красный комплекс в спирте или ацетоне и измеряли поглощение обычно при 560 нм. Можно также проводить отгонку метилбората или экстракцию бора. В последнее время эти стадии анализа упростились.
Куркумин взаимодействует с борной кислотой только в случае протонирования куркумина в сильнокислой среде. Однако эта форма куркумина неустойчива. Поэтому упаривание с целью удаления воды является тонкой процедурой, которая позволяет, с одной стороны, установить достаточно высокую кислотность для протонирования куркумина, а с другой — осуществить реакцию без разрушения реагента. Показано, что реакцию можно прово
зе
дить при комнатной температуре, если реагенты растворить в под* ходящем неполярном растворителе [41]. Отмечено также, что допустимо наличие небольших количеств воды, хотя чувствительность определения при этом снижается. Воду можно связать уксусным ангидридом в присутствии НС1 в качестве катализатора [42]. Важным моментом является удаление избытка протонированного куркумина, который мешает анализу. Это достигается разбавлением раствора этанолом.
Для связывания воды рекомендовано использовать пропионовый ангидрид в присутствии хлорангидрида щавелевой кислоты [38]. Реакция протекает по двум направлениям:
(СОС1)2 + Н2О —> 2НС1 + СО + СО2
(СОС1)2 + 2Н2О —> (СООН)2 + 2НС1
причем первая реакция преобладает в водном растворе. Затем протекает взаимодействие бора с кур кумином в гомогенной жидкой среде. Избыток протонированного куркумина разрушают, вводя ацетатно-аммиачный буфер. Методика с применением куркумина позволяет определять бор в интервале 0,0033—40 мг/л. В зависимости от уровня содержания бора используют одну из шести стандартных методик анализа. Для определения самых низких содержаний бора время анализа составляет 4,5 ч. Эта модификация метода с применением куркумина признана эффективной и использована в автоматическом анализе [43, 44].
Определению бора мешает германий, образующий комплекс, аналогичный розоцианину, а также фторид, который необходимо отделять, и нитрат. При определении 0,2—5 мкг бора влияние NOj. (до’30 мг) можно устранить обработкой раствора анилином сер* нокислым в платиновой чашке [41]. Некоторые ионы взаимодействуют с куркумином или замедляют реакцию образования окрашенного комплекса бора: Fe3+, Mo, Ti, Nb, Та, Zr, СгОГ. MnOi, NO2 и СЮз.
Метод применен для анализа различных веществ: воды [26], промышленных стоков [24], пищевых добавок [45] и биологических тканей [46].
1,Г-Диантримид, хинализарин, карминовая кислота относятся к одной группе спектрофотометрических реагентов для определения бора. Рассмотрим другие методы. Некоторые производные тионина, например метиленовый голубой, нильский голубой А, образуют с фторборат-ионом экстрагируемые ионные ассоциаты. В качестве экстрагентов используют 1,2-дихлорэтан, о-дихлорбен-э°л и аналогичные растворители. Метиленовый голубой впервые предложен для определения бора в работе [47]. Сведения об этом реагенте приведены в монографии Белчера и Вилсона [48]. Чувствительность определения высока, закон Бера соблюдается в интервале 0,1—6 мкг В в 50 мл. Влияние посторонних ионов ^определение 2,5-10-8 М Н3ВО3 в 0,1 М НС1 приведено в ра* OQTe [49]. Определению мешает ряд неорганических кислот и
37
силен, образующих с метиленовым голубым малорастворимые cot динения. Большинство из них растворимо в 1,2-дихлорэтане и экстрагируется совместно с комплексом метиленовый голубой — EFi- Авторы [49] показали, что бор присутствует во многих реагентах, даже в высокочистых.
Образование BF4 из бората протекает примерно за 18 ч. Реакцию катализируют ионы железа, снижая время анализа до 5— 10 мин. Описан метод определения бора в стали [50] с пределом обнаружения 2-10“5%. Определению не мешает до 20% Сг, 10% Ni и по 1% V, Мо пли W (даже при совместном присутствии), что позволяет анализировать легированные стали.
Для спектрофотометрического определения бората предложен реагент азометин Н [51, 52], получаемый конденсацией Н-кислоты (8-амино-1-нафтол-3,6-дисульфокислота) и салицилового альдегида. Получение реагента описано в работе [54], где приведен также метод определения 1—10 мкг бора в тканях растений. Изучено влияние ионов, которые обычно присутствуют в растениях, и показано, что определению мешают Си, Fe, Al, которые, однако, можно связать с помощью ЭДТА. Ионы Си и Fe можно замаскировать также тиоглпколевой кислотой, а А1 — цитратом или оксалатом.
Для предотвращения гидролиза реагента его следует хранить в холодильнике, где он устойчив в течение 14 дней. Показана возможность синтеза реагента в ходе анализа, что использовано для разработки автоматического метода [55]. Метод применен для определения бора в почвах и растениях [56]. Из 15 изученных ионов определению мешают только F~, Al3+, Fe3+, Cu2+ при их содержании в растворе, равном соответственно 0,5, 0,3, 0,02, 0,25%.
Для определения бора используют также реагенты: тетра-бромхризазии, фталеиновый фиолетовый, хромотроповую кислоту и виктория фиолетовая. Данные об этих реагентах приведены в обзорах Немодрука и Кораловой [1], Брамана [3] и в общем обзоре по анионам Больца [57].
Спектрофотометрическое определение бората с помощью карминовой кислоты [36]
Реагенты
Карминовая кислота. I. Растворяют 1,00 г карминовой кислоты в 1 л 95%• нон H2SO4. хранят в холодильнике. Для получения 0,018%-ного раствора разбавляют 18 мл исходного раствора до 100 мл.
II. Растворяют I г карминовой кислоты в I л ледяной уксусной кислоты, фильтруют и разбавляют 400 мл полученного раствора до 1 л ледяной уксусной кислотой.
Серная кислота. 98%-ный раствор с тлотиостью 1,84 г/см3. 95%-нын раствор (получают разбавлением 98%-ного раствора). Концентрацию контролируют титриметрнческн.
Ледяная уксусная кислота. 99,8%-ная.
Стандартный раствор бора I. Растворяют 1,427 г борной кислоты в 1 л 95%-ной H2SO4, получают 2,03-10”4 М раствор Н3ВО3 (2,20 мкг/мл бора).
Стандартный раствор бора /7. Растворяют 1,4302 г борной кислоты в 1 л воды. Разбавляют 100 мл полученного раствора ледяной уксусной кислотой до 38
I л. Помещают 40 мл этого раствора в мерную колбу вместимостью 1 л. приливают 21,8 мл уксусного ангидрида (97%-ною) н доводят объем раствора до метки ледяной уксусной кислотой Полученный раствор содержит 1 мкг В в 1 мл.
Ход анализа. 1. Переносят 1 —10 мл сернокислого раствора, содержащего 1 — 25 мкг В, в мерную колбу вместимостью 25 мл и приливают 95%-ный раствор H2SO4 до общего объема 10 мл. Добавляют 10 мл 0.1%-ного раствора карминовой кислоты(1) для измерения поглощения в области 615 нм или 10 мл 0,018%-ного раствора карминовой кислоты(1) для измерения ири 300 нм. Готовят контрольный раствор. Помещают сосуды с растворами в воду (температура 50 ± 2 °C) и через 15—20 мнн охлаждают их до комнатной температуры. Доводят объем растворов 95% -ной H2SO4 до метки и измеряют поглощение при 300 или 615 нм относительно контрольного раствора.
2. Помещают уксуснокислый раствор Н3ВО3 в мерную колбу вместимостью 25 мл, приливают 10 мл раствора карминовой кислоты(II), охлаждают во льду и вводят в 1 мл 98%-ного раствора H2SO4, встряхивают. Выдерживают сосуды иа водяной баие прн 50 ± 2 °C в течение 15—20 мин, затем охлаждают до комнатной температуры, доводят объем растворов до метки уксусной кислотой. Растворы перемешивают и через 1 ч измеряют поглощение при 548 нм, используя в качестве раствора сравнения контрольный раствор.
Примечания. 1 Хотя мерные колбы из боросиликатного стекла, в которых проводят определение, предварительно обрабатывают концентрированной H2SO4, при анализе -каждый раз проводят контрольный опыт.
2. Все растворы следует хранить в полиэтиленовой посуде.
3. Закон Бера соблюдается в интервале содержаний 2—40 мкг В в 25 мл.
При измерении поглощения в области 615 нм и интервале 0,5—8 мкг В в 25 мл раствора при 300 нм, чувствительность определения равна 1,5-10-3 и 7,ОНО-4 мкг/мл соответственно в ..реде 95%-ной H0SO4. При измерении поглощения в среде смеси серной и уксусной кислот (548 нм) чувствительность определения равна 3,8-10-4 мкг В в 1 мл.
Другие спектроскопические методы
Борная кислота образует флуоресцирующий комплекс с ди-бензоилметаном в стеклообразной среде H2SO4 — диэтиловый эфир (8—96% объемн.) при 77 К [58]. Эта реакция легла в основу метода определения бората при его содержании около нескольких нг/мл [59]. Фосфоресценция отнесена к Т -> So излучательному переходу хелата дибензоилметана с Н3ВО3 состава 2:1. Свечение измеряют при 508 нм и возбуждении 402 нм. Ана-
логичные светящиеся комплексы образуют W и Мо, в их присутствии получаются завышенные результаты. Люминесцирующие комплексы с Н3ВО3 образует также бензоилацетон.
Описан флуориметрический метод определения бора в среде концентрированной H2SO4 с применением 2-гидрокси-4-метокси-4-хлорбензофенона [60]. При возбуждении светом с длиной волны 365 нм и измерении при 490 нм можно определить 10— 100 про (10 6 10-5%) бора в анализируемом растворе.
Определению бора атомноабсорбционным методом препятствует образование ряда весьма устойчивых оксидов ВОХ. Степень атомизации низка даже при работе с высокотемпературным плавнем N2O ацетилен. По данным работы [62], возможно опре-ппйеНИе б°Ра в интервале 0—200 мкг/мл. Разработанный метод экстпеНеН ДЛЯ анализа водорастворимых удобрений [63] после ракции бора смесью 2-этнл-1,3-гександиола и изобутплкетона.
39
Поглощение измеряют при 249,7 нм. Определению не мешают элементы, обычно присутствующие в удобрениях. Метод рекомендован, в качестве стандартного при анализе удобрений [64]. Для анализа растений борат переводят в метилборат, атомизацию проводят в пламени N2O — ацетилен [65]. Из навески пробы 3 г можно определять бор при его содержании 25 мг/кг с приемлемой точностью.
Описано определение бора методом фотометрии пламени [66, 67]. Предложен полуавтоматический метод, включающий концентрирование бора дистилляцией с метанолом [68]. Производительность установки для дистилляции 60 проб в 1 ч, анализа — 180 проб в 1 ч. Чувствительность определения бора 0,004 мкг/мл. Методы определения бора с помощью атомной эмиссионной спектроскопии получили значительное развитие вследствие применения лампы с полым катодом [69]. Пробу, содержащую бор, помещают в полый катод и упаривают под инфракрасной лампой досуха. Затем систему вакуумируют, продувают аргоном и включают разряд. Получена чувствительность на уровне ниже 1 ppb (10~7%), не достигаемая флуоресцентным и другими методами.
Электрохимические методы
Потенциометрический метод
Описан потенциометрический вариант титрования Н3ВО3 щелочью в присутствии маннита, применяемый для определения борной и салициловой кислот в фармацевтических препаратах [70]. После концентрирования на избирательной к бору смоле Амберлит ХЕ-243 борат переводили в анион BF4, который определяли косвенно, потенциометрическим методом, основанным на применении электрода селективного к BF4 [12].
Кулонометрический метод
Кулонометрический метод отличается сравнительно высокой точностью. Он был применен для разработки весьма точного метода определения борной кислоты [71]. В качестве электролита использовали 1 М раствор КС1 и 0,75 М раствор маннита. При определении макроколичеств Н3ВО3 точность характеризуется относительным стандартным отклонением 0,0033%.
Термометрические методы
Кривые потенциометрического и термометрического титрования растворов Н3ВО3 и НС1 щелочью (NaOH) существенно различаются [72]. Четкий скачок на кривой потенциометрического титрования, характерный для НС1, при титровании Н3ВО3 едва заметен. С другой стороны, кривые термометрического титрования этих кислот весьма близки (рис. 8).
40
Рис. 8. Кривые термометрического титрования сильной и слабой кислот: ._титрованне 20 мл 0,00408 Л1 НС1 раствори NaOH (выделяется 56,0 кДж/моль); 2 —титрование 20 мл (0.00393 М Н3ВО3 раствором NaOH (выделяется 44,3 кДж/моль).
Объем титранта или время
Это связано с тем, что, хотя сила этих кислот (НС1 и Н3ВО3) весьма различна, теплота их нейтрализации отличается только на 30% (—56,0 и —44,3 кДж/моль). Описаны методики термометрического определения Н3ВО3 [73—75]. При определении 0,3 мг-экв борной кислоты относительное стандартное отклонение составляет около 1%. Удается анализировать смесь Н3ВО3 с H2SO4 (или НС1), причем сначала титруется сильная кислота [73, 74].
Другие методы определения
Бор можно определять методом газовой хроматографии в виде производных триметилсилана [76]. Масс-спектрометрический метод изотопного разбавления применен для определения бората в минеральных водах [77]. Для этого в пробу вводили сильно обогащенный изотоп 10В в виде Н3ВО3. После ионообменного отделения бората его переводили в NaBF4 и анализировали на спектрометре.
ЛИТЕРАТУРА
1. Nemodruk A. A. Karalova Z. К. — Analytical Chemistry of Boron, Trans, by R. Kondor, Distributed by Oldbourne Press, 1966, London.
2. nudswell F. — In Comprehensive Analytical Chemistry (Ed. C. L. and D. W. Wilson), Vol. 1C, Amsterdam, Elsevier, 1962, p. 89.
3. Braman R. S. — In An Encyclopedia of Industrial Analysis (Ed. F. D. Snell and C. L. Hilton), Vol. 7, New York and London, Interscience, 1968, p. 312.
4. Dixon J. P.— Modern Methods in Organic Microanalysis, Chapter 10, London, Van Nostrand, 1968
5. Spicer G. S., Strickland J. D. //. — Analytica chim. Acta, 1958, v. 18, p. 523.
6. Feldman C. — Analyt. Chem. 1961, v. 33, p 1916.
7. Luke C. L.— Analyt. Chem , 1958, v. 30, p. 1405.
8. Luke C. L., Flaschen S. S. — Analyt. Chem., 1958, v. 30, p. 1406.
.2- ™uiin J- Hayes J. R. — Analyt. Chem., 1952, v. 24, p. 182.
10. British Standards 1121: Part 49, London, 1966.
**• 7.— J- Chem. Soc. Japan, pure Chem. Sect., 1961, v. 82, p. 696; Analyt.
Abstr., 1962, v. 9, p. 4103.
Io £?Г£®ОЛ Paul J L ~ Analyt. Chem., 1968, v. 40, p. 1292.
lo. Melton J. R.t Hoover W. L., Howard P. A. — J. Ass. off. analyt. Chem., 1969, v. 52, p. 950.
4. Egneus B. Up psi г от L. — Analytica chim. Acta, 1973, v. 66, p. 211.
° сиП1С Turcic M. N., Bugarski-Vojinovic A4. B., Perisic N. U. — Z analyt.
16 ” 1967’ v' 229’ P- 93; Analyt. Abstr., 1968, v. 15, p. 5211.
n %' 1akitani S., Miyazaki J., Tamura Z. — Japan Analyst, 1964, v 13,
P- «76; Analyt. Abstr., 1966, v. 13, p. 5385,
41
17, Акимов В. К., Бусев .4. И., Анджапаридзе Д. И.—ЖАХ, 1971, т. 26, с. 2434, Analyt. Abstr., 1973, т. 24, с. 3373.
18. Chadwick Т. С.— Analyt. Chem., 1973, v. 45, р. 985.
19. Gooch F. A., Jones L. C.— Am. J Sci., 1899, v. 7, p. 34.
20, Jones L. C. — Am, J. Sci., 1899, v. 7, p. 147.
21. Belcher R., Tully G. W., Svehla G. — Analytica chim. Acta, 1970, v. 50, p. 261.
22, Ref. 4, p. 175.
23. Wilson H. N„ Pellegrini G. U. M.— Analyst, 1961, v. 86, p. 517.
24. Official Standardised and Recommended Methods of Analysis, 2nd edn,, Com-pilled and Edited by N. W. Hanson, London, The Society for Analytical Chemistry, 1973.
25. Назаренко В. А., Ермак Л. Д. — Зав, лаб., 1968, т, 34, с. 257; Analyt. Abstr., 1969, v. 16, р. 2908.
26. American Public Health Association, American Waterworks Association, and Water Pollution Control Federation, Standard Methods for the Examination of Water and Wastewater, 13th edn., New York, American Public Health Association, 1970.
27. Ref. 24, p. 264.
28. British Standards 1121: Part 49, London, 1966.
29. Goward G. W., Wiederkehr V. R.— Analyt. Chem., 1963, v. 35, p. 1542.
30. Basson W. D., Bohmer R. G., Stanton D. A. — Analyst, 1969, v. 94, p. 1135.
31. Ostling G. — Analyst, 1975, v. 78, p. 507.
32. Langmyhr F. J., Skaar О. B. — Analytica chim. Acta, 1961, v. 25, p. 262.
33. Gupta H. K. L., Boltz D. F. — Analyt Lett, 1971, v. 4, p. 161.
34. Gupta H. K. L., Boltz D. F. — Mikrochim. Acta, 1971, p. 577.
35. Lionnel L. J. — Analyst, 1970, v. 95, p. 194.
36. Gupta H. K. L., Boltz D. F. — Mikrochim. Acta, 1974, p. 415.
37. Shelton N. F. C., Reed R. A. — Analysti, 1976, v. 101, p. 396.
38. Uppstrom L. R. — Analytica chim. Acta, 1968, v. 43, p. 475.
39. Dyrssen D. W., Novikov U. P., Uppstrom L. R.—Analytica chim. Acta, 1972, v. 60, p. 139.
40. Spicer G. S., Strickland J. D. H. — Analytica chim. Acta, 1958, v. 18, p. 523.
41. Hayes M. R., Metcalf J. — Analyst, 1962, v. 87, p. 956.
42. Crawley R. H. A. — Analyst, 1964, v. 89, p. 749.
43. Hulthe P., Uppstrom L., Ostling G. — Analytica chim. Acta, 1970, v. 51, p. 31.
44. Ostling G. — Analytica chim. Acta, 1975, v. 78, p. 507.
45. Official Methods of Analysis of the Association of Official Analytical Chemists (Ed. W. Horowitz), 11th edn., Washington, D. С. AOAC, 1970.
46. Mair J. N., Day H. G. — Analyt. Chem., 1972, v. 44, p. 2015.
47. Ducret L. — Analytica chim. Acta, 1957, v. 17, p. 213.
48. Belcher R., Wilson S. L. — New Methods of Analytical Chemistry, 2nd edn., London, Chapman and Hall, 1964, p. 222.
49. Skaar О. B. — Analytica chim. Acta, 1963, v 28, p. 200.
50. Bhargava О. P., Hines W. G. — Taianta, 1970, v. 17, p. 61.
51. Capelle R. — Analytica chim. Acta, 1961, v. 24, p. 555.
52. Шанина T. M., Гельман H. E„ Михайловская В. С. — ЖАХ, 1967, т. 22, с. 782; Analyt. Abstr., 1969, v. 16, р. 204.
53. Porter G., Shubert R. C. — In: Colorimetric Determination of Nonmetals (Ed. D. F. Boltz), New York and London, Interscience, 1958, p. 339.
54. Basson W. D., Bohmer R. G., Stanton D. A. — Analyst, 1969, v. 94, p. 1135.
55. Basson W. D., Pille P. P., Du Preez A. L. — Analyst, 1974, v. 99, p. 168.
56. John M. K., Chuan H. H., Neufeld J. H. — Analyt. Lett., 1975, v. 8, p. 559.
57. Boltz D. F. — Critical Reviews in Analytical Chemistry, 1973, p. 147.
58. Marcantonatos M., Garnba G., Monnier D. — Helv. chim. Acta, 1969, v. 52, p. 538.
59. Marcantonatos M., Gamba G., Monnier D. — Analyt. chim. Acta, 1973, v. 67, p. 220.
60. Liebich B., Monnier D., Marcantonatos M. — Analyt. chim. Acta, 1970, v. 52, p. 305.
61. Mavrodineanu R.. Boiteux H. — Flame Spectroscopy, New York, Wiley, 1965.
62. Harris R. —Atom. Abs. News!., 1969, v. 8, p. 42.
42
63 Melton J. R-, Hoover 1. L., Howard P. A., Ayres J. L. — J. Ass, off, analyt, ’ Chem., 1972, v. 53, p. 682.
64. Ref. 24, p. 597.
65 Elton-Bott R. R. — Analytica chim. Acta. 19/6. v. 86. p. 281.
66 Agazzi E. J. — Analyt. Chem.. 1967, v. 39. p. 233.
67 Maeck W. J., Kussy M. E., Ginther В. E., Wheeler G. V',, Rein J. E. — Analyt. ' Chem., 1963, v. 35, p. 62.
68 Pierce F. D., Brown H. R. — Analyt, Chem., 1976. v. 48. p. 670.
69. Daughtrey E. H., Harrison ИЛ Ц7. — Analytica chim. Acta, 1973, v. 67, p. 253.
70. Mohay J-, Mohay-Farkas J. — Acta pharm. Hung., 1967, v. 37, p. 71; Analyt. Abstr., 1968, v. 15, p. 3567.
71 Marinenko G„ Champion C. E. — J. Res. nath Bur. Stand. A, 1971, v. 75, ' p. 421.
72. Jordan J. — J. chem. Edn., 1963, v. 40, p. A5.
73. Miller F. J., Thomason P. F. — Taianta, 1959, v, 2, p. 109.
74. Linde H. Wh, Rogers L. B., Hume D. N.— Analyt, Chem., 1953, v. 25, p. 404.
75. Pechar F. — Chemike Listy, 1965, v. 59, p. 1073; Analyt. Abstr,, 1966, v. 13, ' p. 6768.
76. Butts W. C., Rainey W. T.— Analyt Chem,, 1971, v. 43, p. 538.
77. Gorene B„ Marsel J., Tramsek G. — Mikrochim. Acta, 1970, p, 24.
КАРБОНАТЫ И ГИДРОКАРБОНАТЫ
Карбонаты определяют в разных материалах: макроколичества в почвах и минералах, микроколичества в крови и зубной эмали. Когда содержание карбонатов снижается до 20 мкг, используют субмикрометоды. Кроме стандартных титриметрических и гравиметрических применяют и другие методы, в том числе спектроскопические, термометрические, кондуктометрические и в последнее время — газохроматографические. Несмотря на относительную простоту, газометрические и манометрические методы не обладают достаточной точностью. Тем не менее эти методы часто используют для быстрого определения карбонатов. Ряд методов основан на измерении объема выделившегося СО2. Методы, применяющиеся для онределения углерода в стали и органических веществах, часто используют и для анализа других объектов.
Обзор методов определения СО2 и СО (кроме гравиметрического) приведен в работе [1].
Для получения эталонов карбоната обычно используют На2СОз. Сушить эту соль следует осторожно, поскольку потери СО2 наблюдаются уже при 400 °C [2], вероятно, вследствие гидролиза, однако возможно также взаимодействие Na2CO3 с кварцем и боросиликатным стеклом. Свойства и получение чистого Na2COs в качестве стандарта для кислотно-основного титрования описаны в работе [3].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Метод, основанный на потере массы при выделении СО2
Старый метод, основанный на использовании алкалиметра редтера, привлекает своей простотой. Схема прибора дана на
43
Рис. 9. Алкалиметр Шредтера:
/ — емкость с разбавленной НС1; 2—емкость с H2SO4: 3—емкость с пробой.
рис. 9. Методика работы приведена ниже. Показано, что для удаления остаточного СО2 необходимо использовать сухой воздух [4], иначе при анализе 0,1 г СаСОз ошибка может достигать 30%. Описан вариант метода, пригодный для широкого применения [5].
Определение карбоната с помощью алкалиметра Шредтера
Ход анализа. Взвешивают пустой аппарат, вносят навеску пробы (около 1 г) в нижнюю емкость 3
и взвешивают, приливают разбавленный раствор НС1 и концентрированную H2SO4 в емкости 1 и 2 соответственно и снова взвешивают аппарат. Открывают кран н вводят НС1 в нижнюю емкость, затем закрывают кран. Выделяющийся СО2 проходит через серную кислоту, осушаясь при этом, и удаляется. По окончании выделения газа осторожно нагревают нижнюю емкость в течение 2—3 мин. Медленно пропускают через сосуд сухой воздух для удаления остатков СО2, вводя его через верхний ввод емкости 1. Взвешивают
аппарат и по уменьшению массы рассчитывают содержание карбоната.
Метод, основанный на поглощении
и взвешивании выделенного СО2
Это более точный метод, поскольку он включает очистку газа от примесей перед взвешиванием СО2. Описан современный вариант метода [6], предназначенный для микроанализа минералов. Продукты разложения пробы раствором НС1 (1:1) проходят через ловушки для удаления паров воды, НС1, H2S, углеводородов и хлора. Диоксид углерода поглощают аскаритом (асбест, пропитанный NaOH и Na2CO3) и патрон с поглотителем взвешивают. Описаны разные варианты метода [7, 8]. Метод применен для анализа проб, содержащих цианиды, цианаты и карбонаты [9]. В ходе анализа цианиды маскируют посредством HgCl2, карбонаты и цианаты переводят в СО2, который поглощают аскаритом.
Волюмометрические и манометрические методы
Волюмометрический метод основан на измерении объема выделившегося СО2 после кислотной обработки пробы. Объем приводят к стандартным температуре и давлению и рассчитывают содержание карбоната в пробе. Обычно используют простую аппаратуру. Для определения карбонатов в почве применен пластмассовый аппарат [10]. Результаты анализа хорошо согласуются с данными титриметрнческого метода. Описан метод измерения объема выделившегося СО2 по перемещению капли ртути в ка-44
Рис. 10- Прибор для манометрического определения карбоната
пилляре [11]- При анализе 2,2— 40 мг ВаСОз ошибка составляет ±5%.
Описан волюмометрическпй метод определения гидрокарбоната в муке, содержащей мел [12]. Для вытеснения СО2 вместо H2SO4 применен Na2H2P2O7. Мел при этом не разлагается. Описан изящный метод анализа зубной эмали [13]. При анализе проб массой 20—
50 мкг образуется пузырек газа, изображение которого попадает на экран и измеряется. Ошибка определения 0,5—3 мкг СО2 составляет ±5%.
При манометрических измерениях можно использовать простую аппаратуру (рис. 10). В работе [14] применена пипетка вместимостью 25 мл и трубка Варбурга. Для получения более точных результатов прибор может быть видоизменен. Для поглощения H2S, выделяющегося вместе с СО2, использован раствор сульфата меди, нанесенный на пемзу [15].
О природе карбонатных минералов можно судить по времени, необходимом для их разложения. Исследования, проведенные с помощью «карбонатной бомбы», показали, что давление, достигаемое в течение 10—15 с, является мерой содержания кальцита и аргонита. Для определения суммарного содержания карбонатов, включая доломит, давление измеряют через 15 мин и массу доломита оценивают по разности.
Титриметрические методы
Кислотно-основное титрование водных растворов карбонатов
Кислотно-осиовное титрование является стандартным методом определения растворимых карбонатов [17, 18]. Этот же метод применяют для установления титра кислот с помощью Na2COs как стандарта.
Существует ряд источников ошибок. Один из них связан с пересыщением титруемого раствора диоксидом углерода, удаление СО2 зависит от интенсивности перемешивания раствора. Окраска метилового оранжевого в растворе, насыщенном СО2, зависит от концентрации NaCl, а именно интенсивность окраски Усиливается в кислой среде с ростом концентрации соли. Это осложнение можно преодолеть, если проводить обратное титрование. Форма кривой титрования приведена на рис. 11. Обычно тит-Р-УЮт 0,1 М раствором НС1. Возможно несколько вариантов.
45
о 20 оо 00 jo :оо НС1;.-со2- НСО3 Н2СО3
Рис. 11. Кривая титрования 0.5 .11 раствора МагСОз 0,1 .И раствором НС.1.
Например, добавляют кислоту до перехода окраски метилового оранжевого (pH х 3), затем индикатор окисляют добавлением капли бромной воды и проводят обратное титрование 0,01 ,11 раствором NaOH до перехода окраски соответствующего индикатора.
Методика прямого титрования карбонатов с помощью фенолфталеина и бромкрезолового зеленого по Кольтгофу [19] приведена ниже. Возможно титрование карбонатов в присутствии гидрокарбонатов, однако кривая титрования имеет весьма малый угол наклона в точке эквивалентности (pH = 8,3 — 8,4). Для раздельного определения карбонатов и гидрокарбонатов приме-
няют упомянутый выше метод'титрования с помощью двух индикаторов. При титровании смесей карбонатов и гидрокарбонатов ошибка определения компонента, присутствующего в значительно меньшем количестве, может достигать 60% вследствие неправильного выбора pH точки эквивалентности. В работе [20] приведены кривые зависимости правильного значения pH точки эквивалентности от соотношения карбонатов и гидрокарбонатов.
При определении гидрокарбонатов раствор титруют по метиловому оранжевому, а СО2 удаляют кипячением непосредственно вблизи точки эквивалентности. В ряде случаев растворы не кипятят [21, 22]. Значение pH точки эквивалентности зависит от концентрации Н2СО3, что может приводить к существенной ошибке, особенно при титровании разбавленных растворов. При титровании гидрокарбонатов (до pH = 4,4) другие слабые кислоты также титруются. Если для определения суммарного содержания СО2 (свободного растворенного СО2 и выделяющегося из гидрокарбоната) использовать гравиметрический метод [23], а затем оттитровать свободный растворенный СО2, то по разности можно найти содержание гидрокарбоната.
Карбонат в присутствии щелочи можно оттитровать раствором НС1. Объем кислоты, пошедший на титрование, в присутствии фенолфталеина эквивалентен сумме ОН' + '/гСОз". Затем в раствор вводят метиловый оранжевый и проводят титрование. Определяют содержание ‘/гСОг. Концентрацию щелочи находят по разности
двух результатов.
Описан метод титриметрического определения гидрокарбонатов в К2СО3 для фотографии, принятый в Британских стандартах [24].
Титриметрическое определение карбонатов
с применением фенолфталеина и бромкрезолового зеленого [19]
Ход анализа. К раствору, содержащему карбонаты, добавляют одну каплю фенолфтатеина и титруют 0,1 М раствором НС1 до обесцвечивания. Добавляют
46
несколько капель раствора бромкрезолового зеленого п продолжают титрование до перехода окраски в снне-зеленую. Кипятят раствор для удаления СО2 в те-ченне 1—2 мии, при этом цвет индикатора изменяется от голубого до фиолетового. Раствор охлаждают и продолжают титрование до перехода окраски в зеленую.
Титрование СО2, выделившегося из карбонатов
Диоксид углерода, выделившийся при обработке карбонатов кислотой, можно поглотить раствором гидроксида бария (очищенным от карбонатов) и оттитровать избыток щелочи кислотой. Метод применен для определения карбонатов в почве [25, 26]. Несмотря на высокую точность метода, длительность определения велика, особенно при анализе известняка. Для ускорения анализа рекомендуется вместо кислоты использовать растворы ЭДТА [27]. Прибор для описанного ниже метода показан на рис. 12.
Исследования последних лет позволили усовершенствовать титриметрический метод, основанный на поглощении СО2 пиридином и титрованием СО2 раствором метила.та натрия в среде метанол— пиридин [28]. Этот метод применен для определения углерода в стали и в микроанализе органических веществ. Разработан также быстрый метод определения карбонатов в угле [29]. ^Диоксид углерода поглощают раствором бензиламина, а образую-щуюся соль титруют раствором метилата калия в смеси метанол—бензол, используя в качестве индикатора тимоловый синий. Длительность определения, включая взвешивание, составляет 10 мин.
Модификацией описанного метода является микродиффузион-ный метод Конвея, пригодный для определения карбонатов в крови. Выделяющийся СО2 взаимодействует с Ва(ОН)2 с образованием осадка карбоната бария, который титруют ацидиметрически. Описана модифицированная ячейка Конвея, позволяющая титровать пробу объемом 0,1 мл, содержащую 126 ммоль СО2 с точностью ±0,8%.
' Для титрования выделившегося из карбонатов СО2 применяют ЭДТА, например, при анализе щелочных карбонатов в присутствии щелочей (при их отношении от 10:1 до 3 : 10 соответственно). Сначала титрованием аликвотной части устанавливают общую щелочность. К другой аликвотной части раствора добавляют отмеренный избыток раствора Sr(NO3)2 и
Рис. 12. Вакуумный дистилляционный прибор определения карбонатов в почвах [27]:
,|^*олба-реактор вместимостью 200 мл; 2 — трубка; Ц^~Рызгоуловитель: 4 — холодильник; 5 - колба-по-- ЮТЩЦфль вместимостью I л; 6—магнитная мешалка,
после отделения осадка SrSO3 в фильтрате титруют стронции ра створом ЭДТА с применением индикатора тимолового синего. По результатам двух титрований можно рассчитать содержание щелочей и карбонатов [31].
Поглощение карбонатов в ультрафиолетовой области (235 нм) использовано [32] для фотометрического титрования COj' в присутствии гидрокарбонатов и гидроксидов с высокой точностью. Фотометрическое титрование с применением бромтимолового синего как индикатора легло в основу автоматического определения суммарного содержания карбонатов и общей щелочности морской воды [33].
Определение карбонатов в почвах методом
вакуумной дистилляции [27]
Аппаратура и реактивы
Вакуумный дистилляционный прибор (см. рис. 12). При поглощении раствор Ва(ОН)2 непрерывно перемешивается магнитной мешалкой. Вакуум в системе поддерживается вследствие конденсации в холодильнике пара с температурой 90—95 °C. Ловушка для брызг и трубка, соединяющая ее с реактором, предотвращают попадание почвы при разбрызгивании в поглотительный сосуд. Трубки для ввода раствора ЭДТА и отсоса воздуха водоструйным насосом с той же целью впаяны в боковой отвод трубки, соединяющей реакционный сосуд с ловушкой.
ЭДТА, 0,5%-ный раствор.
Гидроксид бария, 0,02 М раствор.
Хлористоводородная кислота 0,02 М раствор.
Тимолфталеин (индикатор), 0,1%-ный раствор в 96%-ном этаноле.
Ход анализа. Тонко измельченную пробу почвы с содержанием СО2, достаточным для нейтрализации примерно половины вводимого гидроксида бария, помещают в колбу 1 вместимостью 200 мл. Включают водоструйный насос и приливают в поглотительную колбу 25 мл 0,02 М раствора Ва(ОН)2 и в реакционную колбу—150 мл 0,5%-ного раствора ЭДТА. Прикрывают кран и быстро нагревают суспензию почвы газовой горелкой до 90 °C. Поддерживают температуру 90—95 °C и регулируют положение крана так, чтобы температура в поглотителе была не выше 40 °C. Через 5 мин (или ранее в зависимости от природы известняка) убирают пламя и постепенно полностью открывают кран. Еще через 5 мин (для полного поглощения СО2) восстанавливают в сосуде атмосферное давление путем введения воздуха, не содержащего СО2. Затем избыток Ва(ОН)2 титруют 0,02 М раствором НС1 с применением тимолфталеина до слабо-голубой окраски индикатора.
Спектрофотометрические методы
Спектрофотометрический метод определения гидрокарбоната в плазме крови основан на увеличении pH водно-этанольного раствора КН2РО4 (12,5-10-3 М) при добавлении этанольного раствора плазмы или сыворотки, из которой удалены протеины. Увеличение pH измеряют по поглощению метилового красного при 520 нм. Результаты анализа хорошо согласуются с данными независимых методов [34].
Для определения выделенного СО2 и карбонатов в твердых пробах применен метод ИК-спектрометрии. Карбонаты в твердых образцах определяют по полосе 1429 см 4 [35]. Определение COj 48
в газовых кюветах (давление 67—1013 Па) проводят по поглощению в области 3676 и 2315 см-1 [36]. Метод отличается экспрессивностью и специфичностью. Чувствительность определения 100 ppm (по КзСОз), воспроизводимость характеризуется ошибкой ±0,04% при анализе 0,4%-ного раствора К2СО3. Описан метод определения СО2 в плазме крови из аликвотной части 50 мкл с прямым отсчетом результата на цифровом вольтметре [37].
Электрохимические методы
Конечную точку ацидиметрического титрования карбонатов кислотой можно определять потенциометрическим методом. •В последнее время применяют ионоселективные электроды. Для определения в крови газов при содержании СО2 более 20 мкг/мл применен электрод, селективный к СО2 [38]. Электрод содержит полимерную мембрану, которая отделяет анализируемый раствор от пленки раствора NaHCO3, заключенной между собственно электродом и чувствительной поверхностью стеклянного электрода. СО2 диффундирует через мембрану.
- Показано, что электрод этого типа (Радиометр Е-5036) для определения СО2 непосредственно в питательной воде котлов применить не удается [39]. Жидкостной перхлоратный ионоселективный электрод с применением перхлората бриллиантового 'зеленого реагирует на содержание НСО~ в интервале 10-1 — 10-3Л4 [40].
Предложенный электрод для определения карбонатов [41, , 42] имеет, по-видимому, ряд существенных преимуществ. Он включает жидкостную мембрану, которая представляет собой раствор четвертичной соли аммония в органическом растворителе. Зависимость сигнала от логарифма концентрации карбоната линейна в интервале 10-7—10-2 М. Рабочий интервал соответствует pH = 5,5 — 8,5. Электрод достаточно селективен по отношению к хлоридам, сульфатам и фосфатам. В интервале pH, важном Для физиологических исследований, гидроксид-ион не мешает определению. В цитированных работах подробно рассмотрены состав и свойства жидкой фазы, образующей мембрану.
Электрод для анализа СО2 в газовой фазе описан в работе [43].
Газохроматографический метод
В последнее время в неорганическом анализе все шире применяют метод газовой хроматографии. В обзоре [44] описаны газохроматографические методы определения анионов. Определению анионов посвящен ряд исследований. Показано, что при взаимодействии (ЬЦ-ЦгСОз с бистриметилсилилфторацетамидом в среде Аиметилформамида образуются производные триметилсилила, что позволяет разделить карбонат, борат, оксалат, фосфит, сульфат, арсенит, фосфат, ванадат, арсенат и сульфамат (см. раздел
49
«Сульфаты»), Карбонат и гидрокарбонат дают общий пик. Особенность метода состоит в том, что соли натрия и калия с рассматриваемыми анионами не образуют производных триметилси-лила, поэтому их предварительно переводят ионным обменом в аммонийную форму.
Другой метод анализа H2S, SO2 и СО2 основан на их выделении путем обработки солей кислотой, разделении методом газовой хроматографии и определении с помощью детектора теплопроводности [46].
Другие методы определения
Описано определение карбонатов методом термометрического титрования [47]. Показано, что точность дифференциального Термического анализа карбонатных минералов выше, чем рентгенофазового. При содержании карбонатов менее 50% относительная ошибка определения составляет 2% [48].
Описан быстрый и точный анализ минералов, почв и глин, основанный на методе динамической сорбции карбонатов [49]. В качестве газа-носителя применен гелий. При точном соблюдении условий анализа относительная ошибка определения составляет 0,1 %.
Для определения 10—100 мкг карбонатов применен метод изотопного разбавления [50].
ЛИТЕРАТУРА
1. Davies D. Н. — Taianta, 1969, v. 16, р. 1055.
2. Otterson D. А.—Analyt. Chem., 1966, v. 38, p. 506.
3. Analytical Methods Committee of the Society for Analytical Chemistry — Analyst, 1965, v. 90, p. 251.
4. Pauschmann H.— Z. analyt. Chem., 1965, v. 207, p. 14; Analyt. Abstr., 1966, v. 13, p. 2281.
5. Pieters H. A. J. — Analytica chim Acta, 1948, v. 2, p. 263.
6. Meyrowitz R.— Prof. Pap. U. S. geol. Surv., No. 700-S, 1970, s. 183; Analyt. Abstr., 1972, v. 22, p. 704.
7. Hillebrand IF. F., Lundell G. E. F., Bringht H. A., Hoffman }. I. — Applied Inorganic Analysis, New York, Wiley, 1953.
8. Yoyng R. S.— Industrial Inorganic Analysis, London, Chapman and Hall, 1953.
9. Harzdor C. — Z. analyt. Chem., 1968, v. 237, p. 161; Analyt Abstr., 1969, v. 17, p. 791.
10. ' Leo M. W. M.— J. agric. Fd Chem., 1963, v. 11, p. 452.
11. Горбенко-Германов Д. С., Зенкова P. A-, 1965, t. 20, c. 749; Analyt. Abstr., 1967, v. 14, p. 637.
12. Stephenson W. H., Hartley A. IF.— Analyst, 1955, v. 80, p. 461.
13. Weatherell J. A., Robinson C. — Analyst, 1968, v. 93, p. 244.
14. Pittwell L. R. — Mikrochim. Acta, 1968, p. 903.
15. Jordanov N.— Taianta, 1966, v. 13, p. 563.
16. Mueller G., Gastner M.—Neues. Jb. Miner. Mh., 1971, p. 466‘ Analyt. Abstr., 1972, v. 23, p. 1983.
17. Laitinen H. A. — Chemical Analysis. New York, McGraw-Hill, I960, p. 86.
18. Fischer R. B., Peters D. G. — Quantitative Chemical Analysis, Philadelphia, W. B. Saunders Co., 1968. p. 302.
19. Kolthoff I. M., Sandell E. B., Meehan E. J., Bruckenstein S. — Quantitative Chemical Analysis, 4th edn., Toronto, The Macmillan Co., 1969, p. 778.
50
80. Zangen M. — J. appl. Chem., 1962. v. 12, p 92.
21. British Pharmacopoeia. Pharmaceutical Press. London, 1968. p. 898.
«2. The U. S. Pharmacopoeia. 18th Revision. 1970, p. 610.
23' Rosopulo .4.. Quentin K.—E — Z. analyt. Chem.. 1971, v. 253. p. 27; Analyt. Abstr., 1971, v. 21. p. 4449.
94 British Standard 3751: (1964); British Standards Institute, London.
25 Schollenberger C. J. — Canad. J. Soil ScL, 1930, v. 30. p. 307.
2tL Schollenberger C. 7.— Canad. Sci.. 1945. v. 59, p. 57.
27. Vatkinson J. H. — Analyst, 1959. v. 84. p. 6o9.
28. Blom L, Edelhausen L. — Analytica chim. Aeta. 1955, v. 13, p. 120.
29. Pringle W. J. S . Fuel, Lond.. 1963, v. 42, p. 63.
30 Holm-Jensen L, Scand. J. Clin. Lab. Invest., 1960, v. 12, p. 269; Analyt Abstr., ' 1961, v. 8, p. 5123. , ,
31. Szekeres L., Bakacs-Polgar E. — Z. analyt. Chem., 1960, v. 177, p. 89; Analyt. Abstr., 1961, v. 8, p. 189!
32. Underwood A. L., Howe L. H. — Analyt. Chem., 1962, v. 34, p. 692.
ЙЗ.’ Graneli A., Anoalt T. — Analytica chim. Acta, 1977, v. 91, p. 175.
"34. Sinnema Y. A.— Pharm. Wekebl., 1968, v. 103, p. 837; Analyt. Abstr., 1969, v. 17, p. 3614.
35. Шербов Д. П; Гуляев A. M. — Труды Казах, научно-исслед. ин-та минерального сырья, 1962, с. 204; Analyt. Abstr., 1964, v. Il, p. 3584.
36. Ppbinger H.— Analyt. Chem., 1962, v. 34, p. 878.
37. Peterson J. /. — Clin. Chem., 1970, v. 16, p. 144; Analyt. Abstr., 1971, v. 20, p. 1820.
38. Severinghaus }. IF.— Ann. N. Y. Acad. Sci., 1968, v. 148, p. 115.
39 Midgley D. — International Symposium on Selective Ion-Sensitive Eectrodes, Cardiff, Wales, 1973.
40. Fogg A. G., Pathan S., Burns D. T. — Analytica chim. Acta, 1974, v. 73, p. 220.
41. Herman H. B., Rechnitz G. A. — Science, 1974, v. 184, p. 1074.
42. Herman H. B., Rechnitz G. A.— Analytica chim. Acta, 1975, v. 76, p. 155.
43. Ross J. IF., Riesman J. H„ Kruger J. 4.—Pure appl. Chem., 1973, v. 36, p. 473.
44. Rodriguez-Vazquez J. A.’—Analytica chim. Acta, 1974, v. 73, p. I.
45. Butts IF. S., Rainey W. TQ—Analyt. Chem., 1917, v. 43, p. 538.
46. Birk J. R., Larsen С. M„ Wilbourn R. G. — Analyt. Chem., 1970, v. 42, p. 273.
47. Priestley P. 7.— Analyst, 1963, v. 88, p. 194.
48. Gita G., Gita E. — Rev. Chim. Bucharest, 1964, v. 15; p. 214; Analyst. Abstr., 1965, v. 12, p. 3914.
49, Thomas J., Hieftze G. M.—Analyt. Chem., 1966, v. 38, p. 500.
50. Johannesson J. K. — Analyst, 1967, v. 92, p. 766.
ХРОМАТЫ И БИХРОМАТЫ
Соединения хрома находят широкое применение в гальванотехнике, для получения сплавов, особенно нержавеющих сталей, ДЛЯ получения ингибиторов коррозии. Поскольку хром (VI) присутствует в ингибиторах коррозии, он попадает и в окружающую среду, где проявляет токсические свойства. При определении хрома его обычно переводят в хроматы.
Хроматы и бихроматы щелочных и некоторых других металлов хорошо растворимы. Хроматы тяжелых металлов, как правило, малорастворимы, что используют для осаждения и определения хрома.
В кислой среде бихромат является сильным окислителем:
Cr20j- + 14Н+ + 6е 2Сг3++ 7Н2О Е° = +1,33В
5!
Редокс-снстема СггО?7Сг3+ необратима. В щелочной среде окислительная способность Crvi уменьшается:
CrO*' +4H2O + 3e Сг (ОН)3 • aq + 5ОН' Е° = — 1,13В
В умеренно кислой среде превалируют ионы СщОГ п НСгО). При pH > 8 преобладает ион СгОГ- Теоретические основы химического определения хрома даны в монографии {!]. Хром(VI) часто определяют в неорганических материалах, определение его в органических веществах весьма затруднительно. Обзор методов определения хрома дан в работах [2, 3]. В качестве стандарта при определении хрома(VI), как правило, применяют бихромат калия. При необходимости содержание хрома(VI) в бихромате можно определить с высокой точностью методом кулонометрии при постоянном токе, как описано ниже.
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение
Хромат количественно осаждается солями Ва2+, Ag+, Hg+ н Pb2+. Мешают анионы, осаждаемые этими катионами, и катионы, образующие нерастворимые хроматы. Мешающие катионы, например Fe3+, Ni2+ и А13+, можно предварительно отделить осаждением их в виде гидроксидов.
Экстракция
Бихромат экстрагируется метилизобутилкетоном из кислой среды [4]. Этот метод использован для определения хрома в биологических материалах. Органический экстракт анализируют атомно-абсорбционным методом [5].
В работе [6] хром(VI) экстрагировали раствором три-н-бу-тилфосфата в бензоле и определяли спектрофотометрическим методом. Определению не мешают Ni, ThiV, UO”, Cu2+, Co2+, Fe3+ н Vv.
Хром (VI) экстрагируют алифатическими аминами [7]. Показано, что CrVI почти количественно экстрагируется тридецилами-ном, дидециламином, дигексиламином и трибензиламином из 0,08—1,5 М растворов H2SO4. Полностью экстрагируют Crv’ 0,1 М растворы трибензиламина в хлороформе, дихлорэтане, циклогексаноле, трибутилфосфате. Для экстракции Crvl из перхлоратных сред использованы растворы трибензиламина в дихлорэтане [8]. Этот метод, позволяющий отделить Crvi от кальция и алюминия (III), применен для анализа силикатов.
Показано, что хромат селективно экстрагируется из 0,1-— 0,2 М растворов H2SO4 растворами триоктиламина или триоктил-метиламмонийхлорида в хлороформе [9], при этом не экстрагируются Fe, Ni, Со, Си, Al, Zn и другие ионы. Соэкстракцию вана
дия(У) и урана (VI) предотвращают добавлением Nad. Описан метод экстракции .хрома(VI) оксидом мезитила из растворов, содержащих НС1 и КС1, с последующей реэкстракцией Crvi аммиачным раствором [10]. Определению мешают Ptvl, Au", Fe3+, SCN', S2O)’, CN , F' и винная кислота.
Описаны также другие экстракционные системы [11].
Ионообменный метод
Ионы Mg, Ni и Al отделяют от хромата на катионите. Трудность метода состоит в том, что при малой скорости пропускания раствора через колонку хроматы восстанавливаются до Ст’11, который не элюируется. При скорости выше 8 мл/мин потери хрома не превышают 1 % [12].
Описаны методы ионообменного отделения хрома. Показано, что хром (VI) нельзя количественно отделить от других металлов на сильноосновном анионите в среде H2SO4, однако Crvi можно концентрировать в щелочных растворах [13], что позволяет отделить хром от таких элементов, как алюминий, который обладает амфотерными свойствами, и поэтому элюируется. Затем элюируют хром (VI) 8—10%-ным раствором Na2CO3 или смесью NaHCO3 и Na2CO3.
Хромат и сульфат можно отделить адсорбцией их на сильно-основном анионите [14]. Затем сульфат элюируют раствором NaCl, а хромат — раствором NaOH.
Методы бумажной и тонкослойной хроматографии
Описано разделение Crvi и Сг1” методом тонкослойной хроматографии (ТСХ) на оксиде алюминия с применением насыщенного водного раствора сульфата натрия в качестве растворителя [15]. Пятна при этом слегка размываются. Метод ТСХ [16] на А1аО3 с разными растворителями применен также для разделения Ю4, Юз, ВгО3 и СгО4. Описаны также другие варианты метода ТСХ для отделения СгО4’ от большого числа анионов [17, 18].
Метод высоковольтного электрофореза на бумаге
Методом высоковольтного электрофореза на бумаге при напряжении 1—1,5 кВ в среде 0,1 М KCI можно разделить Сг"1 и СгО4‘ за 5—10 мин [19].
Соосаждение
Метод соосаждения применен для отделения 0,05—0,5 мг хро-а(1Ц) от 005—юо мг xpoMa(VI) при pH = 8,0—9,5 с применением Ti(OH)4 [20].
53
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
В разделе, посвященном отделению хрома(VI), указано, что Crvi осаждается рядом металлов, например ионами Ag~, Ва2+, Hg;' п РЬ2+, в виде нерастворимых хроматов. Можно применять также органические осадители.
При осаждении хрома(\’1) ртутью(1) весовой формой может быть хромат ртути(1) пли Сг2О3 (после прокаливания осадка). Хромат серебра можно осадить из растворов галогенидов с помощью карбамида [21]. Более эффективен, вероятно, метод осаждения путем удаления аммиака из аммиачных комплексов серебра [22]. Определению не мешают Mg2+, Mn2+, Fe3+, Cu2+, Tilv, ZrIV, Ni2+, Co2+ и большие количества сульфатов.
Общим недостатком методов гравиметрического определения СгОГ является мешающее влияние анионов, образующих нерастворимые соединения с осадителями, и катионов, осаждающих хроматы.
Для осаждения СгОГ часто используют хлорид или ацетат бария, причем сульфаты необходимо предварительно отделить. Гравиметрические методы определения хромата сопоставлены в обзоре [2].
Тнтрнметрнческне методы
Важным моментом титриметрического определения хрома(VI) является установление титра раствора бихромата калия, применяемого в анализе. Это соединение можно получить высокочистым. После высушивания К2СГ2О7 при 150—180 °C его можно использовать в качестве стандарта. Для этого необходима точная навеска. Однако бихромат калия не отвечает требованиям, предъявляемым к стандартам, и для высокоточных анализов необходимо установить титр раствора. Наиболее подходящим методом установления титра раствора бихромата калия является метод кулонометрии при постоянном токе, а также приведенные ниже титриметрические методы.
Редокс-метод с применением железа(П)
Метод основан на добавлении избытка стандартного раствора железа(II) к подкисленному раствору бихромата и обратном титровании железа(II) стандартным раствором бихромата с применением дифениламинсульфоната натрия или ферроина как индикатора (для обратного титрования можно применять также раствор перманганата калия):
Сг2О2'+ 6Fe2++ 14Н+ —► 6Fe3++ 2Сг3++ 7Н2О
Определение ведут в среде НС1 или H2SO^. Фосфорную кислоту вводят для образования устойчивых фосфатных комплексов 54
железа(Ш), что снижает потенциал системы Fe2'—Fe’F Титрование применяют для определения хрома в некоторых сплавах. После растворения пробы хром(III) предварительно окисляют до хроматов. Метод имеет некоторые особенности, в том числе аномальное поведение некоторых индикаторов, нашедшее удовлетворительное объяснение в работах [22—24]. Механизм взаимодействия Fe2- с хроматом сложен, поэтому результат анализа зависит от того, приливают ли раствор Crvi к раствору Fe2+ или поступают наоборот.
Изменение потенциала вблизи конца титрования также зависит от порядка приливания реагентов. Причины такого поведения системы неясны. Механизм реакции описан в работах [25, 26, 27, 28]. В работе [28] обсуждено поведение ферроина как индикатора. Несмотря на аномальное поведение системы Fe11 — CrVI, эту реакцию широко применяют для определения хроматов.
t 1 Определение бихромата с применением
в качестве стандартов растворов Fe2+ и бихромата
Реагенты
Соль Мора, 0,1 М раствор, содержащий 20 мг концентрированной H2SO4 - иа 1 л.
Бихромат калия, 1/60 М раствор.
Дифениламин сульфонат натрия, 0,2 %-ный раствор.
Ход анализа. К слабокислому раствору бихромата объемом около 25 мл до-' бавляют раствор соли Мора до перехода окраски в зеленую и дополнительно - 5 мл избытка, точно замечая объем добавленного раствора. Приливают раствор HjSO4 (1 : 1) для создания концентрации H2SO4, равной примерно 2 М, вводят ♦—5 капель индикатора (дифениламина) и 5 мл концентрированной фосфорной • кислоты. Титруют стандартным раствором 1/60 М КзСггО? до перехода окраски индикатора в пурпурную. Затем продолжают титрование раствором соли Мора и стандартным раствором бихромата, как описано выше. Объем раствора Fe”, -.пошедшего на титрование, соответствует содержанию бихромата в смеси. Дру-. гой вариант определения описан в разделе «Спектрофотометрические методы».
Примечания. 1. Вместо раствора бихромата калия можно использовать стандартный раствор КМпО4. Этот метод рекомендован [29] для определения ipOMa в сталях.
2. Конец титрования можно устанавливать амперометрическим методом [30] (см. ниже). Метод применен для анализа смесей МпО'— Сг2О2' [31], при этом — раствор в качестве катализатора добавляют MnSO4. Титруют стандартным раствором оксалата натрия или щавелевой кислоты до перехода оранжево-красной окраски в желтую.
Йодометрический метод
Хром(VI) в кислом растворе взаимодействует с избытком KI с образованием иода, который можно оттитровать стандартным раствором тиосульфата:
Сг2О2' + 6Г+14Н+ —► 2Сг3++ 312 + 7Н2О
Как и в предыдущем методе, простые стехиометрические отношения не отражают сложного механизма реакции. Показана ВОЗМОЖНОСТЬ окисления подпстоводородной кислоты кислородом
55
воздуха, что приводит к завышенным результатам. Другая возможная ошибка связана с образованием при некоторых условиях комплексов хрома(III) с тиосульфатом [32]. Эти источники ошибок обсуждены в монографии [33].
Кислотность существенно влияет на протекание реакции. При избытке кислоты в'растворе, несмотря на быстрое протекание реакции, возможно окисление иодида кислородом. Для получения правильных результатов определение ведут в слабокислой среде, когда реакция завершается в течение 5 мин.
Иодиметрический метод дает возможность определять СгО4" (или МпО[) в присутствии ионов железа(III) [36], которые маскируют с помощью (ЫаРОз)б. При титровании концентраций СгО4‘, отвечающих содержанию 17—88 мг Сг, ошибка составляет ±0,3%.
Иодиметрическое титрование бихромата *
Ход анализа. Анализируемый раствор бихромата в колбе, закрывающейся пробкой, разбавляют водой до 100 мл и добавляют 2 г KI. Вводят 2 М раствор НС1 для создания концентрации кислоты 0,4 М. Закрывают сосуд пробкой и оставляют в темном месте на 5 мин, разбавляют раствор, приливают еще 50 мл воды и тнтруют выделившийся иод стандартным раствором тиосульфата с применением крахмала как индикатора.
Примечания. 1. Методика пригодна для титрования хромата с концентрацией ’/во Л4. Для более разбавленных растворов (примерно в 10 раз) концентрацию НС1 устанавливают равной 0,6 М.
2. Ионы железа (III), меди(II), мышьяка (V), молибдена (VI), ванадия (V) мешают определению, так как они окисляют иодид до иода. Влияние Fe111 и Си” можно устранить введением ЭДТА при pH = 1—2 [34].
Комплексометрические методы
Описан метод определения хромата, основанный на осаждении хромата бария введением избытка стандартного раствора Ва2+ и комплексометрическом титровании избытка бария [35,36].
Другие методы основаны на восстановлении Crvl до Сг1п и титровании хрома(III) раствором ЭДТА. Недостаток метода связан с малой скоростью взаимодействия Сг1” с ЭДТА. Показано, что если Crvl восстанавливать бисульфитом натрия при pH = 6,6 в присутствии ЭДТА, то комплекс Сг’” с ЭДТА образуется мгновенно [37], в то время как обычно равновесие достигается при длительном кипячении растворов.
Предложен метод, основанный на осаждении хромата серебра [38] в нейтральной или слабокислой среде. Осадок отфильтровывают, промывают и растворяют в аммиачном растворе K2(Ni(CN)4] [39].
Ag2CrO4±K2[Ni(CN)4] —К2 [Ag2 (CN)J + Ni2++ CrOf
Образующиеся ионы Ni2+ титруют стандартным раствором ЭДТА с ошибкой не более 1 %.
5
Другие титриметрические методы определения
Растворы хрома(VI), молибдена (VI) и вольфрама можно титровать барием(II) в водно-органической среде с применением нитхромазо как индикатора [40]. Реакция протекает количественно в среде 80—90%-ного ацетона при pH = 4,0— 5,5 в исходном водном растворе.
Описаны другие редокс-методы для титрования бихромата. При окислении хромом (VI) Fe(CN)s переходит в Fe(CN)e’, который титруют стандартным раствором аскорбиновой кислоты [41]. Можно использовать и другие окислители, например IOJ и ВгОз.
Спектроскопические методы
Колориметрический и спектрофотометрический методы в видимой и ультрафиолетовой областях
При взаимодействии с Crvi дифенилкарбазида в кислой среде образуется интенсивно окрашенный красно-фиолетовый комплексный катион [42, 43]. В максимуме поглощения при 540 нм молярный коэффициент поглощения равен 3,14-104. Интервал определяемых содержаний хрома 0,005—0,1 ppm. При кислотности ниже 0,05 н. реакция протекает медленно, при концентрации Нг5О4 более 0,2 н. комплекс неустойчив. Мешающее влияние молибдена (VI), который также образует красно-фиолетовый комплекс, устраняют оксалатами. Мешает медь(П) и в меньшей мере—железо (III). Возможно искажение результатов за счет остаточных количеств окислителей, применяемых для спбгсления хрома(Ш) до хроматов, и сорбции хроматов, используемых при мытье посуды. Окислители разрушают окрашенный комплекс. Этот комплекс разрушается и в отсутствие окислителей [44]. Метод нашел широкое применение и рекомендован для анализа железа и сталей [52].
Хромат и бихромат можно определять по их собственной окраске. Максимум поглощения хроматов наблюдается при 366 нм, е = 4416. Измерения можно проводить в видимой области, при 440 нм. В щелочной среде, при высоких значениях pH, когда доминирует СгОГ. закон Бера строго соблюдается. Возможно определение Crvi и Mnvn при их совместном присутствии. Мешают определению в щелочной среде уран(VI), церий(IV) и другие элементы, осаждающиеся в этих условиях. Определению не мешают Movl и WVI, малое влияние оказывает ванадий(V).
В кислой среде следует учитывать равновесия
Сг2О’Ч-Н2О 2НСгО4
НСгО4- Н'4-CrOf
Ионы CrOz', СггО?’, НСгО; характеризуются разными значениями е. Даже при постоянной кислотности закона Бера
57
нарушается вследствие влияния концентрации CrVl на соотношение ионов Сг-О)" и HCrOi. Тем не менее нелинейный градуировочный график можно использовать для определения в ограниченном интервале концентраций хрома(VI). При колориметрическом определении хрома следует учитывать присутствие НСгО( [45]. В разбавленных растворах НСгО] преобладает, при концентрации бихромата выше 10~4Л1 закон Бера не соблюдается независимо от концентрации кислоты в растворе.
Известны и другие спектрофотометрические методы определения хрома (VI). Некоторые окислители, в том числе Сг^О; , окисляют трифенилметановые красители [46]. Чувствительность определения СггО)" с применением бриллиантового зеленого и кристаллического фиолетового составляет 0,01 мкг/мл.
Хромат можно определять также с помощью 3,3-диаминобен-зидина [47].
Атомно-абсорбционный метод
Метод ААС часто применяют для определения микропримесей хрома (VI). Для определения используют воздушно-ацетиленовое пламя и аналитическую линию хрома 357,9 нм, обычно предел обнаружения составляет 0,1 ppm. Метод применен для анализа низколегированных сталей. Мешающее влияние ионов железа, которые подавляют поглощение хрома, можно устранить введением NH4C1 или алюминия [48].
Преимущество метода состоит в том, что он позволяет, используя экстракцию, определить содержание хрома в различных степенях окисления. Хроматы можно экстрагировать с помощью диэтилдитиокарбамата натрия [хром(III) не мешает], а затем хром (III) извлечь с помощью 8-оксихинолина и теноилтрифтор-ацетона [49]. Растворителем в обоих случаях является изобутил-метилкетон. Органический экстракт анализируют методом ААС. Метод является экспрессным и простым.
При анализе почв хром (III) и хром (VI) разделяли с помощью ионного обмена и определяли методом ААС [50].
Метод непламенной ААС, который предложил Львов в 1961 г. [51], позволяет определять хром с абсолютной чувствительностью 1-10~!0 г Атомизацию проводят в графитовой печи, используя аналитическую линию 425,4 нм. При использовании анализатора в виде углеродной нити и линии хрома 357,9 нм предел обнаружения составляет 1 • 10_ 11 г. Определению не мешают НС1 и HNO3> но серная кислота снижает чувствительность анализа; определению мешают Sr, Са, Ва и в меньшей мере — Ti, V, Мп и другие катионы.
Метод инфракрасной спектроскопии
Более 20 мкг хромата можно определять в тонких дисках из КВг одновременно с ферроцианидом (II и III), кобальтициани-дом, сульфатом и метаванадатом [54].
58
Электрохимические методы
Потенциометрический метод
Конечную точку титрования хромата железом(II) можно определять потенциометрически с помощью платинового и насыщенного каломельного электродов. Несмотря на сложный механизм реакции, метод дает точные результаты. Показаны некоторые преимущества титрования в среде 8,5 Л1 Н3РО4 [55]. Метод позволяет определять Сг, Мо и V. При титровании смеси CrV1 и Vv наблюдается два скачка потенциала; первый соответствует восстановлению смеси до Сг111 и Vlv, второй — переходу от V'v до V111. Реакция взаимодействия феррицианида с аскорбиновой кислотой применена для косвенного потенциометрпчесокго определения бихромата [56]. Описан метод автоматического титрования 0,2—2 мг СГ2О7', а также микрометод [57].
Для потенциометрического титрования хромата в щелочной среде применен раствор TiCI3 в глицерине, содержащем муравьиную кислоту [58]. Раствор устойчив на воздухе в течение четырех дней. Конец титрования можно устанавливать также визуально по переходу окраски резаруфина.
. Амперометрический метод
При титровании избытка железа(II) в кислой среде стандартным раствором хромата конечную точку можно определять амперометрически [30] с точностью 0,03—0,05%.
При последовательном амперометрическом титровании марганца (VII), хрома(VI) и ванадия в качестве титранта применяют раствор KI [59]. Хроматы титруют в среде 3 М H2SO4 с применением графитового дискового индикаторного электрода при потенциале 4-0,6 В относительно насыщенного каломельного электрода. Предел обнаружения хрома составляет 40 мкг в 20 мл раствора.
Можно использовать также вращающийся платиновый электрод с потенциалом 4-1,2 В относительно насыщенного каломельного электрода [60]. Определению Сг, Мп мешает 600-кратный избыток Al, Mg, Zn, Со, 100-кратный избыток NO3 и 10-кратный Избыток Си, Cd, Н3РО4, лимонной, винной и уксусной кислот. Не мешают равные количества Bi и Fe и большие количества U, Мо и Th, мешают Hg, V, Ag и 10-кратные количества Bi, Fe и ЭДТА.
Кулонометрический метод
Важной особенностью кулонометрического титрования хро-Ma(VI) является достижение высокой точности. Показано, что Ошибка титрования CrVI электрогенерированным железом (II) достигает л-10-3% [61]. Относительно высокая точность достигается даже при микротитровании. Например, ошибка определения < нг хрома(VI) составляет 3,7% [62].
59
Рис. 13. Схема установки для автоматического определения бихромата с применением каталитического пористого кислородного электрода [65]:
I — датчик с пористым кислородным электродом; 2—сепараторы; 3—смеситель; 4—устройство для подачи пробы; Д—проба (2,0—1,2 мл/мин); Б — Н2О2 (0.8—0,6 мл/мнн); S—N2 (7.8 мл/мин); Г, Д— сброс фаз (2,9, 2,0 и 0,8 мл/мин); Е— сброс остатка (5,4 мл/мин).
Кулонометрию при постоянном токе рассматривают как один из наиболее точных аналитических методов. Точность метода настолько высока, что в качестве стандарта предложена единица количеств электричества — кулон [63]. Кулонометрическим методом исследован бихромат калия в качестве вторичного стандарта [64]. Разработанная методика является одной из наиболее точных. Результаты анализа бихромата приведены ниже;
Найдено основ-
ного вещества, % 99,972 99,977 99,975 99,974 99,977 99,974
Среднее, % ... 99,975
Стандартное от-
клонение, % . . . 0,02
Паспортные дан-
ные NBS, % . . 99,98
Данные [61], % 99,977
При оценке столь точных результатов следует принимать во внимание правильность измерения атомных масс, вклад которых в ошибку измерения становится значимым.
Разработан новый метод кулонометрического автоматического определения СггО?" и МпСХ- Метод основан на применении пористого каталитического кислородного электрода, функция которого почти не зависит от температуры и давления. Кислород генерируется при взаимодействии бихромата (или перманганата) с НгО2 в кислой среде. Схема процесса следующая:
2СгОз -р Н2О2 —-> Сг2Оу -р Н2О
Сг2О7 -р 4Н2О2 —► Сг2О$ -р 4Н2О -р 4О2
В действительности механизм реакции сложнее. Схема установки для автоматического определения бихромата приведена на рис. 13. Существенно, что метод можно применять для определения показателя «химическое потребление кислорода».
60
Полярографический метод
Хром(VI) восстанавливается в 0,1—1 М растворе NaOH, образуя хорошо выраженную волну со значением =— 0,85 В относительно НКЭ [66]. В сильнощелочном растворе восстановление протекает по уравнению
CrOf + 2HkO + Зе' —> CrO; + 4ОН'
При полярографировании растворов Crvi в нейтральной среде или в небуферном растворе КС1 в качестве фонового электролита наблюдается четыре волны при значениях потенциалов полуволн, равных: —0,3, —1,0, —1,5, и —1,7 В (НКЭ). Последние три волны соответствуют восстановлению до Сгш, Сг11 и металлического хрома.
Хорошо выраженная волна восстановления CrOi в щелочном растворе использована для определения хрома в стали [67].
Другие методы определения
Для определения хрома (VI) применяют каталитические методы, например реакции окисления индигокармина [68] и метилового оранжевого [69] пероксидом водорода в присутствии хрома. Определение проводят спектрофотометрическим методом. Чувствительность определения хрома (VI) составляет от 6 до 21 нг/мл.
Описан автоматический метод, основанный на каталитическом действии хрома (VI) на взаимодействие Н2О2 с иодидом в сернокислой среде, позволяющий определить 0,6—3,0 мкг хром а ( VI) с ошибкой 1—2% в течение 10—50 с [70].
Реакция окисления метурина (1-гидрокси-3-метил-1-фенилкар-бамид) броматом в кислой среде, катализируемая хромом(VI), позволяет определять хром с чувствительностью около 5 нг/мл
Для определения СгО42 предложена реакция умножения, отличающаяся тем, что цикл умножения, приводящий к образованию AgCl, может быть многократно повторен [72].
Описан радиохимический метод определения хрома(VI), основанный на взаимодействии СГ2О7' с металлическим серебром* 110Ag [73].
ЛИТЕРАТУРА
1- Lingane J. J. — Analytical Chemistry of Selected Metallic Elements, New York and London, Reinhold, 1966, p. 39.
2. Chalmers R. A.— In: Comprehensive Analytical Chemistry (Ed. C. L. and „ 2; W. Wilson), Vol. 1C, Amsterdam and London, Elsevier, 1962, p. 581.
* ulsen E. D. and Foreback С. C. — Encyclopedia oi Industrial Chemical Analysis (Ed. F. D. Snell and L. S. Ettre), Vol. 9, New York and London, Inter-science, 1970, P- 632.
К ^elnhardt A. E, Hixson A. E. — Ind Engng Chem., 1951, v. 43, p. 1676.
ыап F' Knoblock E. C., Purdy W. C. — Analytica chim. Acta, 1967, *• aa, p. 489.
61
6. Tuck D. G. — Analytica chim. Acta, 1962, v. 27, p. 296.
7. Шевчук И. А., Симонова T. H.— Укр. хим. ж.. 1964. т. 30. с. 983.
8. Церковницкая И. .4.. Ильинская Г. И.. Беляев В. П., 1969. т. 24. с. 1357, Analyt. Abstr., 1971, v. 20. р. 1605.
9. Adam J., Pribil R.— Taianta. 1971, v. 18, p. 91.
10. Shinde Г. 41., Khopkar S. Al. — Z. analvt. Chem, 1970, v. 249, p. 239: Analyt. Abstr., 1971, v. 20. p. 2428
II. De A. K.. Khopkar S. AL, Chalmers R. A. — Solvent Extraction of Metals. London and New York, Van Nostrand Reinhold, 1970.
12. De A. K-, Sen A. K-— Taianta, 1966. v. 13, p. 1313.
13. Mulokozi A. M.— Analyst, 1972. v. 97, p. 820.
14. Субботина А. И.. Архангельская E. А.. Петров A. AL — Труды по химии и хим. технологии (Горький), 1963, с. 118.
15. Galateanu I. — J. Chromat., 1965, v. 19, p. 208.
16. Lederer M., Polcaro C. — J. Chromat., 1973, v. 84, p. 379.
17. Kawanabe K-, Takitani S., Miyzaki M., Tamura Z.—Japan Analyst, 1964, V. 13, p. 976; Analyt. Abstr., 1966, v. 13, p. 5385.
18 Canic V D„ Turcic M. N., Bugarski-Vo/inovic M. B., Perisic N. U. — Z. analyt. Chem, 1967, v. 229, p. 93; Analyt. Abstr, 1968, v. 15, p. 5211.
19. Баташова В. В., Бабешкин A. AL, Несмеянов А. Н. — Вестник Моск. гос. унта, Сер. хим, 1970, т. 11, с. 361; 1971, 20, р. 2429.
20. Плотников В. И., Кочетков В. Л. — ЖАХ, 1968, т. 23, с. 377; Analvt. Abstr, 1969, v. 17, р. 2564.
21. Gordon L., Firsching F. H. — Analyt. Chem, 1954, v. 26, p. 759.
22. Belcher R., Rees D. /, Stephen W. I. — Chim. Analit, 1959, p. 397.
23. Belcher R., Brazier J. V, Stephen W. I. — Taianta, 1965, v. 12, p. 778.
24. Belcher R., Brazier J. N., Stephen W. I. — Taianta, 1965, v. 12, p. 963.
25. Latinen H. A. — Chemical Analysis, New York and London, McGraw-Hill, 1960, p. 441.
26. Westheimer F. H. — Chem. Rev, 1949, v. 45, p. 419.
27. Belcher R., Chakrabarti C. L., Stephen W. I. — Analyst, 1969, v. 94, p. 20.
28. Spiramam K.— Taianta, 1972, v. 19, p. 1085.
29. British Standards Handbook No. 19, Chromium Method 2, London, British Standards Institute, 1970.
30. Kelly H. J., Edridge A, Hibbits I. O. — Analytica chim. Acta, 1959, v. 21, p. 135.
31. Rao G. G., Muralikrishna U. — Analytica chim. Acta, 1955, v. 13, p. 8.
32. Hahn F. L. — J. Am. chem. Soc, 1935, v. 57, p. 614.
33. Kolthoff I. M., Belher R. — Volumetric Analysis, New York, Interscience, 1957, p. 237, 332.
34. Pribil R., Sykora J. — Chemike Listy, 1951, v. 45, p. 105.
35. Isagai K-, Tateshita H. — Japan Analyst, 1955, v. 4, p. 222.
36. De Sousa A. — Chemist Analyst, 1961, v. 50, p. 9.
37. Aikens D. A, Reilley C. N.—Analyt. Chem, 1962, v. 34, p. 1707.
38. De Sousa A. — Taianta, 1973, v. 20, p. 1039.
39. Flaschka H,, Huditz F. — Z. analyt. Chem, 1952, v. 137, p. 104.
40. Крешков А. П., Кузнецов В. В., Мешлюмян П. Г.—ЖАХ, 1974, т. 29, с. 1349. Analyt. Abstr, 1975, v. 29, ЗВ, р. 138.
41. Erdey L., Kasa I. — Taianta, 1963, v. 10, p. 1273.
42. Saltzman В. E.— Analyt. Chem, 1952, v. 24, p. 1016.
43. Allen T. L. — Analyt. Chem, 1958, v. 30, p. 447.
44. Sandell E. B. — Colorimetric Determination of Trances of Metals, 3rd edn, New York, Interscience, 1959.
45. Kolthoff I. AL, Sandell E. B., Meehan E. J., Bruckensteiii S. — Quantitative Chemical Analysis, 4th edn, London, Macmillan, 1969, p. 1053.
46. Раманаускас E. И., Буникене Л. В., Сапрачонене AL С, Шулюене А. А, ленайте AL В. — ЖАХ, 1969, т. 24, с. 244; Analyt. Abstr, 1970, v. 19, р. 1022.
47. Cheng К- L. — Chemist Analyst, 1963, v. 52, p. 73.
48. Barnes L.— Analyt. Chem, 1966, v. 38, p. 1083.
49. Yanagisawa M„ Suzuki AL, Takeuchi T.— Mikrochim. Acta, 1973, p. 475.
50. Cresser Al. S, Maigritt R. — Analytica chi. Acta, 1976, v. 81, p. 196.
62
i L. — Analvt'ca chim.
1962. v. 9. n. 841.
23. c. 384. Analyt.
'2. London. Br’tRh
64.
Abstr., 1969. v. 17.
analyt. Chem., 1968, v. 240: p. 91; Analvt.
ci I'vov В V.— Spectrochim. Acta. 1961, v. 17, p. 761.
g- British Stand”.’- HenAhooC \\ '
Standard bmr.- t
53 Jackson К- II 4 est T. S.. В<.:.т.
' n 363.
м Haba F. R. Wilson C. L —lalanta.
55’ Вегр^го Л. И. -ЖАХ. 1068. т.
п. 2565.
56 Erdeu L Sve'do G Г> eber O. Z. Abstr., 1970, v. 18. p. 12.
57 KissEross K.. Bi:zas I.. Erdeu L. — Z. analvt. Chem.. 1971, v. 255, p. 273; Analvt. Abstr, 1972, v. 22. r>. 3117.
58 Ruzicks E.— Chemicke Zvesti, 1972, v. 26, p. 516; Analyt. Abstr, 1973, v. 24, n. 2789.
59 Сонгина О. А., Захаров В. .4, Бектурова Г Б.. Смирнова Л. И, —ЖАХ. 1974. т. 29. с. 1594: Analvt. Abstr, 1975. v. 29. р. 3814
60 Жданов А К.. Кондратова В. Ф.. Акентьева Н. .4 —Узбек, хим. ж, 1969, ' с. 13; Analyt. Abstr, 1970, v. 18, р. 2380.
64 G, Taylor J, K.—J. Res. nath Bur. Stand, A, 1963, v. 67A,
p. 453.
62. Christian G. D . Feldman F. J. — Analytica chim Acta, 1966, v. 34, p. 115.
63'- Cooper F. A.. Quale J- C. — Analvtica chim Acta, 1963, v. 91, p. 363.
64. Knoeck Diehl H. — Taianta, 1969, v. 16, p. 181.
65. Fleet B.. Ho A. Y. IF.— Analvt. Chem, 1972. v. 44, p. 2156.
66. Lingane J. J., Kolthoff I. M.— J. Am. chem. Soc, 1940, v. 62, p. 852.
67. Von Stackelberg №.. Klinger P., Koch W.. Krath E. — Tech Mitt. Krupp Forsch , 1939, v. 2. p. 59.
68. Ясинскене Э. И . Билидене E. Б. — ЖАХ, 1968, т. 23, с. 143; Analyt. Abstr,
1969, v. 17. n. 2084.
69. Ясинскене 9. И.. Билидене Е. Б. — ЖАХ, 1967, т. 22, с. 741; Analyt. Abstr, 1969. v. р. 124.
70. Hadjiioannou Т. Р. — Taianta, 1968, v. 15, р. 535.
71, Крейнгольд С. У.. Божевольнов Е. А.. Супин Г. С. и др.— ЖАХ, 1969, т. 24. с. 853; Analvt. Abstr, 1970. v. 19, р. 4848.
72. Weisz Н., Goenner №. — Analytica chim. Acta, 1968, v. 43, p. 235.
73. Richter H. G., Gillespie A. S. — Analyt. Chem, 1965, v. 37, p. 1146.
ЦИТРАТЫ
МЕТОДЫ ОТДЕЛЕНИЯ
Метод тонкослойной хроматографии
Лимонную кислоту можно отделить от других карбоновых кислот методом тонкослойной хроматографии. Разделение проводят методом восходящей хроматографии на целлюлозе трехкомпонентными растворителями [1]. Условия разделения приведены в табл. 4,
Ионообменный метод
Сильноосновные аниониты в карбонатной пли гидроксильной Форме позволяют выделить лимонную кислоту совместно с малоновой, янтарной, аскорбиновой и другими кислотами из расти-ельных соков [2]. Описано разделение лимонной, малоновой и КИСЛ0Т на Амберлите CG-120 (размер зерен 0,045— ’ 4 мм, Н+-форма) с помощью растворителя, содержащего аце-н, Дихлорметан, воду в соотношении 160: 100:9 [3].
Таблица 4. Значения Rj для карбоновых кислот при разделении их с помощью трехкомпонеитных растворителей
Кислота Содержание кислоты, мкг Значения при использовании растворителей типа
I i: Ill
Муравьиная 1.3 0,21+0,00
Уксусная 1,0 0,21+0,00
Щавелевая 1.0 0,58+0,02 0,62+0,04 0,04+0,01
Винная 1,0 0,35+0.02 0,45+0,03 0,03+0,01
Лимонная 0,5 0,42+0,08 0,57+0,04 0,01+0,01
Растворитель 1: амиловый спирт — муравьиная кислота — вода (40:40:2).
Растворитель II: 1-бутанол — муравьиная кислота — вода (60:10:20).
Растиоритель III: 2-бутанол—2М NH4OH (80:20).
Экстракция
Цитрат можно извлечь на 97—99% из водных растворов с pH = 1,6— 3,1 с помощью иона трифенилстаннония Ph3Sn+. Оксалат и ацетат извлекаются почти количественно при pH = = 1,0 — 2,3 и 1,0 — 2,4 соответственно, но тартрат и формиат не экстрагируются [4]. Нелетучие карбоновые кислоты, растворенные в воде, можно экстрагировать бутанолом [5].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Обзор распространенных методов определения лимонной кислоты дан в работе [6]. Ряд методов основан на превращении лимонной кислоты в пентабромацетон по реакциям
н2с—соон 1 нос—соон — НС—соон II -> нос — СНз 1 + со - СНВг2 -+ io
1 Н2С—соон 1 н2с—соон 1 СНз 1 СВг3
Определение заканчивают гравиметрическим, титриметриче-ским или спектрофотометрическим методом в зависимости от содержания лимонной кислоты в пробе.
Гравиметрические методы
Гравиметрическое определение в виде пентабромацетона
Гравиметрический метод дает наиболее точные результаты при содержании лимонной кислоты более 50 мг. Этот метод официально принят [7] для анализа фруктов и фруктовых продуктов. Сокращенное описание метода приведено ниже.
64
Гравиметрическое определение лимонной кислоты в виде пентабромацетона
Реагенты
Перманганат калия. 5с.'-иый рдствор.
Сульфат железа. 40%-нып раствор FeSO4-7H2O, содержащий 5 мл концентрированной H2SOj в 500 мл.
F Ход анализа. К 40—45 мл раствора лимонной кислоты добавляют 2 г КВг и 5 мл концентрированной H2SO4. Нагревают до 50 °C и оставляют на 5 мим. Медленно, порциями по 1—2 мл, перемешивая раствор каждый раз в течение нескольких секунд, приливают 20 мл раствора 1\МпО4 и оставляют раствор на 5 мин. Охлаждают раствор до 15 °C и медленно, при постоянном перемешивании, добавляют раствор FeSO4 до начала просветления раствора. Встряхивают 1 мин, продолжают добавлять раствор FeSO4 до растворения МпО2 и добавляют еще несколько миллилитров раствора.
Добавляют 20 г безводного Na2SO4 и перемешивают до растворения соли (если значительная масса Na2SO4 не растворится, определение повторяют). Охлаждают раствор до 15 °C и энергично встряхивают : . .дк-нче 5 мин. Затем фильтруют раствор через тигель с фильтром Гуча, содержащий асбест, промывая осадок на фильтре фильтратом и затем 50 мг холодной воды в течение нескольких минут. Сушат тигель с осадком ночь в эксикаторе, содержащем H2SO4, или помещают тигель в воздушную сушилку и доводят до постоянной массы (потери массы не более 0,2 мг). Первое взвешивание проводят через 20 мин.
Растворяют пентабромацетон в спирте, а затем в эфире, наполняя тигель по три раза каждым растворителем. Сушат тигель в течение 10 мнн при 100 °C, охлаждают в эксикаторе и взвешивают. Разность соответствует массе пентабромацетона в двух взвешиваниях. Для пересчета на массу безводной лимонной кислоты умножают полученное значение на 0,424 (теоретический коэффициент пересчета). Можно пользоваться эмпирическим коэффициентом, учитывающим потери за счет растворимости и летучести пентабромацетона. Относительное стандартное отклонение метода равно 0,33%.
Титриметрические методы
Метод кислотно-основного титрования
Лимонную кислоту можно определять (неизбирательно) титрованием водного раствора стандартным раствором NaOH с применением фенолфталеина [8]. Константы диссоциации кислоты следующие:
. Д’, = 7,4 • 10~4; 1,74-IO’5; Д3 = 4,0-1ГГ7
При потенциометрическом определении наблюдаются три перегиба на кривой титрования. В случае применения азофиолетового объем щелочи, пошедшей на титрование, соответствует двум ступеням нейтрализации.
Иодиметрический метод
Некоторые карбоновые кислоты можно определять прямым иодиметрическим методом, основанным на выделении иода из иодат-иодидной смеси:
Ю3- + 5Г + 6Н+ —> 312 + ЗН2О
иопим°бЫЧНЫХ Условиях лимонную кислоту не удается определять етрически, однако по методу Кольтгофа [9] получаются
8 Зак. 167
65
хорошие результаты. Метод основан на добавлении избытка стандартного раствора тиосульфата к раствору, содержащему лимонную кислоту, 1\Ю3 п KI. Затем через определенный интервал времени проводят титрование.
При взаимодействии лимонной кислоты с Са, Ba, Mg и Zn образуются комплексы, которые ведут себя как сильные кислоты, что дает возможность проводить прямое иодиметрическое титрование лимонной кислоты [10]. Подиметрпческий метод неселек-тпвен, поэтому, вероятно, он не нашел широкого применения.
Тпериметрический метод, основанный
на образовании пентабромацетона
Метод определения цитратов, основанный на образовании пентабромацетона, более селективен и может быть использован для определения субмикрограммовых количеств при анализе хроматографических фракций.
Принцип одного из вариантов метода следующий [11]. Окисление и бромирование лимонной кислоты до пентабромацетона проводят, как описано ранее. Затем продукт реакции экстрагируют и обрабатывают сульфидом натрия, при этом выделяется бромид. При взаимодействии бромида с йодатом серебра образуется иодат, который, окисляя иодид, образует иод. Иод титруют тисульфатом. Принцип метода представлен реакциями
ВГ + AgIO3 = AgEr + IO3"
Ю3' +5Г + 6H+= 3I2 + 3H2O
I2+2S20f = S40f+21-
Контроль условий получения пентабромацетона из лимонной кислоты (время реакции, кислотность раствора) становится менее строгим, если определение проводить в присутствии метафос-форной кислоты [11]. Улучшенный метод находит широкое применение.
Титриметрическое определение лимонной кислоты после превращения ее в пентабромацетои
Реагенты
Бромид калия, 1 М раствор.
Перманганат калия, 0,3 М раствор.
Сульфид натрия, 4%-ный раствор NajS-HjO, хранят в холодильнике. Раствор устойчив в течение нескольких недель.
Метафосфорная кислота, 20%-ный раствор. Готовят при комнатной температуре, хранят в холодильнике.
Петролейный эфир с температурой кипения 30—60 °C.
Хлорид калия, 0,0125 М раствор, получают растворением 0,9319 г высушенной солн в 1 л 0,085 М фосфористой кислоты.
Пероксид водорода, 3%-ный раствор.
Тиосульфат натрия, 0,01 М раствор (получают разбавлением 0,1 М раствора) .
Ход анализа. К раствору лимонной кислоты приливают 18 мл 1 М раствора H2SO4, 1 мл 20%-ного раствора метафосфорнон кислоты н разбавляют водой Д°
66
- [Щи температуре не выше 22 °C добавляют 2 мл раствора КВг и 5 мл рас-50 ' КМпО4. Примерно через 10 мин раствор, не переметчивая, охлаждают до
Ю’С и добавляют охлажденный до 0 °C раствор Н;О2 при перемешивании до обесцвечивания реакционной смети.
° Переносят раствор в грушевидную делительную воронку вместимостью 125 мл и ополаскивают сосуд небольшими порциями петролейного эфира (общий объем 25 мл). Растворы в делительной воронке энергично встряхивают, сливают водную фазу н промывают эфирный слой четырьмя порциями по 3 мл воды для удаления следов галогенидов.
1 Разрушают пентабромацетон, встряхивая эфирный слой последовательно с двумя порциями по 3 мл раствора Na2S. сливая окрашенный раствор в мерную колбу вместимостью 25 мл. Промывают эфирный слой порциями по 2 мл воды до тех пор, пока очередной водный слой не обесцветится; обычно достаточно двух промывок. Промывную воду сливают в мерную колбу, приливают 2 мл 2Q М раствора фосфористой кислоты, нагревают до кипения и осторожно кипятят 56 мин. Рекомендуется предварительно ввести в раствор несколько кусочков кварца, промытого кислотой и водой.
Раствор охлаждают, добавляют точно 5,00 мл раствора KCI, доводят объем раствора до метки водой н перемешивают. Затем раствор переливают, не ополаскивая колбы, в коническую колбу со стеклянной пробкой вместимостью 50 мл, содержащую 0,25 г сухой солн AgIO3. Колбу закрывают пробкой, встряхивают - в течение 5 мни и фильтруют раствор через сухой бумажный фильтр, не содержащий хлоридов, в мерную колбу.
Титруют аликвотную часть раствора (от 2 до 5 мл) 0,01 М раствором тиосульфата иатрня после добавления двух капель 0,085 М раствора фосфористой кислоты и 1 мл 10%-ного Nal нлн КД- Оставляют раствор на 5 мин. Проводят контрольный опыт.
Для расчета результатов анализа используют эмпирический коэффициент 0,980, учитывающий степень превращения лимонной кислоты в пентабромацетон, полученный по данным 42-х определений [И].
Комплексометрический метод
Предложен метод определения лимонной, щавелевой и винной кислот путем осаждения их ионами свинца (II) с последующим титрованием избытка соли раствором ЭДТА [12]. Метод позволяет быстро определить 8—25 мг лимонной кислоты.
Редокс-метод
Лимонная кислота (4—5 мг) может быть определена титрованием раствором КМПО4 [13]. Обратное титрование осуществляют через 1,5 ч раствором гидрохинона.
Колориметрические методы
Метод, основанный на реакции пентабромацетона с пиридином
„„^бромацетон при взаимодействии с щелочным раствором ннридина образует продукт красного цвета, что может быть по-сителк° В ОСНОВУ метода определения цитрата [14]. Метод отно-2 мкг° прост> селективен и позволяет определять примерно тоя лимонн°й кислоты. Описан ряд модификаций [15, 16]; ме-стаипяп™еНД0Ван в качестве стандартного [17]. Относительное Д ртное отклонение составляет 3,7% [6].
3*
67
Метод с применением пиридина и уксусного ангидрида
Простой колориметрический метод, основанный на взаимодействии лимонной кислоты с пиридином и уксусным ангидридом в присутствии трихлоруксусной кислоты, предложен в 1948 г [18]. Поглощение измеряли, используя светофильтры с максимумом пропускания при 400 нм. В последнее время показано, что метод недостаточно специфичен. Получены воспроизводимые результаты, однако следует учитывать влияние концентрации трихлоруксусной кислоты [19]. Описан вариант метода, который может быть использован в автоанализаторе для определения 25 ррш лимонной кислоты в растворе [20]. Критический обзор'методов определения лимонной кислоты в молоке приведен в работе [21] и рекомендована методика с применением трихлоруксусной кислоты [22].
При анализе образцов, содержащих протеины, может быть использована методика с применением пиридина и уксусного ангидрида [23].
Другие колориметрические методы
Лимонная кислота (а также щавелевая, малоновая, малеиновая, аскорбиновая и винная) снижает поглощение комплекса FeHi с 5-ннтросалиииловой кислотой в ацетатном растворе, что использовано для ее колориметрического определения [24] при 492 нм. Мешают определению ионы меди и нитриты.
Другой метод основан на изменении окраски растворов CeIV в присутствии ферроина при окислении разных органических веществ [25]. Метод не специфичен, аналогично лимонной кислоте определяются аскорбиновая, молочная, щавельная и винная кислоты. Поглощение Се в присутствии ферроина измеряют при 426 нм.
Более специфичным является, вероятно, метод определения цитрата, основанный на расщеплении цитрата в присутствии фермента цитрат оксалоапетат лиазы и цинка [26], который может быть применен для анализа тканей животных и фруктовых соков. При определении 0,02 мкмоль (0,004 мг) лимонной кислоты ошибка составляет 5%.
Другие методы определения
Для разработки высокочувствительного метода определении цитрата использован эффект гашения флуоресценции при 450 нм комплекса вольфрамата с флаванолом (1 : 1) [27]. Снижение флуоресценции, вероятно, связано с образованием более устойчивою комплекса вольфрамата с цитратом. Метод позволяет определить 0,2 мкг цитрата. Интервал определяемых солержапии нитрата 0—200 ppm, при некотором видоизменении можно определять 0—5 ррш.
68
Определению не мешают йодаты, хлораты, броматы, сульфаты, ульфиты, бромиды, хлориды и нитраты, небольшие содержания по Данилов и фторидов. Мешают даже небольшие концентрации тартратов, оксалатов и иодидов.
^Описан радиохимический вариант метода с использованием “Вт основанного на образовании пентабромацетона [28]. Результаты анализа плазмы крови согласуются с данными фотометрического метода.
ЛИТЕРАТУРА
1 Dittman —J. Chromat., 1968, v. 34, р. 407.
2* Goodbam А. Е., Stark J. В.— Analyt. Chem., 1957, v. 29, p. 283.
3 Seki T. —J. Chromat., 1966, v. 22, p. 498.
4. Bock R-, Niederauer H.— T., Behrends K.— Z. analyt. Chem., 1962, v. 190, p. 33.
5. Хоменко A. H., Гончарова И. A. — Гидрохимия, матер., 1968, т. 48, с. 77, ’ Analyt. Abstr., 1970, v. 18, p. 2830.
8. Witty A. R.— Proc. Soc. analyt. Chem., 1966, v. 3, p. 183.
7t Official Methods of Analysis of the Association of Official Analytical Chemists, Washington, U. S. A., Association of Official Analytical Chemists, 10th edn., 1965, p. 317.
8. Pharmacopeia of the U. S., 17th edn., Mack Printing Co., 1965, p. 135.
9. Koithoff I. M. — Chem Weekbl., 1926, v. 23, p. 260.
10. Koithoff I. M. — Pharm. Weekbl., 1920, v. 57, p. 63.
11. Hargreaves C. A., Abrahams M. D., Vickery H. B. — Analyt. Chem., 195f, v. 23, p. 467,
12. Crisan I. A., Krausz R. — Studia Univ. Babes-Bulyai, Ser. Chem., 1967, v. 12, p. 19; Analyt. Abstr., 1968, v. 15, p. 6720.
13. Berka A., Hilgard S. — Mikrochim. Acta, 1966, p. 174.
14. Ettinger R. H., Gadbaum L. R., Smith L. N. — J. biol. Chem., 1952, v. 199, p. 531.
15. Safronov A. P. — Biokhimiya, 1959, v. 24, p. 123; Analyt. Abstr., 1959, v. 6, p. 4514.
18. Jones G. B. — Analyt. Biochem., 1967, v. 21, p. 286.
17. Pro M. J., Nelson R. A. — J. Ass. off. analyt. Chem., 1956, v. 39, p. 952.
18. Saffan M„ Denstedt O. F. — J. biol. Chem., 1948, v. 175, p. 849.
19. Lowenstein J. M.— Meth. Enzym., 1969, v. 13, p. 513; Analyt. Abstr., 1970, в. 19, p. 5044.
20. Marler I. R„ Boulet M. — J. Dairy Sci., 1958, v. 41, p 1683
21. White J. C. D„ Davies D. T. — J. Dairy Res., 1963, v. 30, p. 171.
22. Marier J. R., Boulet M., Rose D. — J. Dairy Res., 1961, v. 44, p. 359.
23. Choy T. K, Quattrone J. J., Elefant M. — Analytica chim. Acta, 1963, v. 29, p. 1,14.
^ee b. W., Hwang J. V. — Analyt. Chem., 1968, v. 40, p. 2049.
no м .’ Kazimowitz W. L. — Mikrochim. Acta, 1969, p. 345.
28. Moellering H., Gruber W. — Analyt. Biochem., 1966, v. 17, p. 369.
27. Guyon J. C., Marks J. У. — Mikrochim. Acta, 1969, p. 731.
& L. — J. nucl. Med., 1964, v. 5, p. 297; Analyt. Abstr., 1965, v 12, p. 4069.
ЦИАНАТЫ
При анализе промышленных объектов, в том числе отходов, не-ОсЙ-ИМ° опРеделение цианатов OCN-. В нейтральных растворах медленно разрушается, в кислых растворах наблюдается
69
быстрый гидролиз цианатов. Даже в щелочных растворах OCN не очень устойчив. Поэтому эталонные растворы цианата не следует хранить более недели.
В течение многих лет стандартный метод определения OCN-был основан на гидролизе цианата до аммония и определении последнего. Поэтому важным моментом является отделение OCN- от N’H* при их совместном присутствии. Ввиду неустойчивости OCN-удаление NH3 кипячением раствора неприемлемо, единственно эффективным является метод ионного обмена. Сильнокислотные катиониты в Н+-форме непригодны, поскольку их применение приводит к разрушению цианата, поэтому обычно рекомендуется применять катиониты в \та+-форме [1, 2].
В качестве стандарта применяют KOCN. Синтез и очистка соли описаны в работе [10].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Цианат осаждается раствором AgNOs из нейтральных или разбавленных азотнокислых растворов [3]:
KOCN + AgNO3 —► AgOCN + KNO3
Осадок можно высушить при 100°C и взвесить. Показано, что AgOCN не разлагается до температуры 138 °C, однако при 240 °C выделение циана становится заметным [4].
Спектрофотометрические методы
Один из методов основан на отделении цианата от аммония ионным обменом, переведении цианата в аммоний путем гидролиза в серной кислоте и определении NH4 по Несслеру [2]:
OCN' + 2Н+ + 2Н2О —► nh; + h2co3
Определению не мешают карбамид и наиболее распространенные катионы и анионы. Гидролиз цианата в среде 0,05 М H2SO4 завершается в течение 10 мин. Интервал определяемых концентраций цианата составляет 40—200 мкМ.
В работе [1] после ионообменного отделения цианата реакцию гидролиза проводили в присутствии катализатора хлорамина Т. Последующее определение аммония основано на образовании в присутствии пиридина и пиразолона соединения пурпурного цвета, которое экстрагируется четыреххлористым углеродом и фо-тометрируется. Этот метод более чувствителен, чем с применением реагента Несслера. Интервал определяемых содержаний цианата 0,1—10 ppm. Мешают только цианиды и роданиды. Для анализа готовят реагент, приготовленный из бис-(3-метил-1-)-фенилпира-зол-5-она, пиридина и 3-метил-1-фенил-5-пиразолона.
70
При добавлении цианата к разбавленному раствору соли меди, содержащему пиридин, образуется сине-лпловый осадок ICufCsHsNhfOCNh], который растворяется в хлороформе, обра-зая растворы сапфирово-синего цвета. Указанный медь(П)-пиридиновый метод, менее чувствительный, чем метод, основанный на гидролизе цианата до аммония, долгое время был единственным спектрофотометрическим методом определения цианата [5]. В работе [6] показано, что чувствительность метода увеличивается в 30 раз, если измерения проводить не при 680—700 нм [7, 8], а при 315 нм.
Определение цианата медь( II)-пиридиновым методом [7]
Реагенты
Цианат калия, исходный раствор, содержащий 0,0193 г/мл в 0,04 М растворе NaOH.
Пиридинат меди(П), водный раствор, содержащий 1,38-10~4 моль/мл Cu(NO3)2-3H2O и 2,1 -10-3 моль/мл пиридина.
Ход анализа. К 10 мл раствора пиридината меди (II) при 25 °C приливают 5 мл раствора цианата калия. Раствор перемешивают и, приливая раствор NaOH, устанавливают с помощью рН-метра pH = 8,00. Раствор выдерживают при 25 “С в течение 10 мин, переносят в делительную воронку вместимостью 50 мл и экстрагируют комплекс четырьмя порциями хлороформа по 5 мл. Экстракт фильтруют через бумажный фильтр, доводят объем до 25 мл и измеряют поглощение при 680 нм.
Примечание. Интервал определяемых содержаний OCN~ равен 0,5— 5,0 мг. Цианиды не мешают даже в двукратном избытке.
Амперометрический метод
Описан амперометрический метод последовательного титрования CN~, G1- и OCN- раствором Ag+ [9]. Метод основан на различной растворимости AgCN, AgCl и AgOCN, он позволяет определить 0,5—2 мкмоль OCN~. Применяют платиновый и насыщенный каломельный электроды.
Если присутствуют карбонаты, их предварительно осаждают раствором нитрата бария. В присутствии равных количеств посторонних ионов ошибка определения цианата составляет ±2%.
5.
6.
7.
8.
9.
10.
ЛИТЕРАТУРА
1' MellonM. G.— Analyt. Chem., 1953, v. 25, p. 1188.
3 c’ H\R-’.B°rdea“x /-/--Analyt Chem., 1955, v 27, p. 136.
4 Analytica chim. Acta, 1951, v. 5, p. 506.
’ 1963 ,norSanic Thermogravimetric Analysis, 2nd edn, London, Elsevier,
VrtX r Aiir“J TC^T Soc ’ 1923’ v- 123‘ P- 2577.
19721 v- 66- P 189-
Marler f'₽’^lcCZ?1Za,w//-— Analyt. Chem., 1951, v. 23, p. 1519.
Mushn 4 °;; ~ Analyt. Biochem., 1964, v. 7, p. 304.
v. 14 n S'“JaPan Analyst, 1965, v. 14, p. 795; Analyt. Abstr., 1967,
^^McGraw-Hil/faie01lnorga':ic Synthesis- New York and Lon-
71
ЦИАНИДЫ
Цианистый водород применяют в производстве акрилового волокна, найлоне 66 и метилакрилата. Цианид натрия используют для промышленного извлечения серебра и золота, в гальванотехнике, при закалке стали и в качестве сырья для химической промышленности.
Высокая токсичность цианидов общеизвестна. Например, максимально допустимое содержание HCN в воздухе при 8-часовом рабочем дне составляет 10 ppm, поэтому необходимы методы чувствительного определения цианидов в воздухе и стоках. В сточных водах цианиды могут находиться в виде HCN, CN~ и ряда ионов типа M(CN)X, где цианиды связаны в комплексы с ионами металлов. Содержание всех форм цианидов оценивают как сумму цианидов или общее содержание ионов CN~.
Установлено, что токсичность цианидных растворов зависит от содержания HCN-. Концентрация CN- также важна, поскольку существует равновесие [1]
CN' + H2O HCN + ОН'
Обзор методов определения цианидов дан в работе [2], методы обнаружения и определения малых содержаний рассмотрены в работе [3]. В обзоре, посвященном анализу анионов, рассмотрены современные методы определения цианидов [4].
Цианиды маскируют формальдегидом:
C1V + нсно + Н2О —> CN—сн2—он + ОН'
Эту реакцию применяют при анализе цианидов, например при определении цианидов в присутствии галогенид-ионов по методу Фольгарда. Обычно стандартные растворы нельзя получить по точной навеске NaCN и KCN, необходимо установить точное содержание CN~ титрованием раствором AgNO3.
Ввиду токсичности цианидов необходима особая осторожность при работе с ними. При титровании растворов, в которых ожидается наличие цианидов, нужно пользоваться безопасными пипетками и бюретками. Все операции, при которых возможно выделение HCN, необходимо проводить в вытяжном шкафу. Цианистый водород можно отделить от некоторых газов барботированием через раствор NaOH.
МЕТОДЫ ОТДЕЛЕНИЯ
Дистилляция
Дистилляцию применяют как основной метод отделения макро-и микроколичеств. Этот метод обстоятельно рассмотрен в работе [5]. В качестве поглотителя используют растворы NaOH. Цианистый водород может выделяться из растворов при pH <Z 9 При подкислении раствора винной кислотой HCN можно отогна . ь
72
дистилляцией или вытеснить потоком воздуха. В присутствии комплексообразователей раствор необходимо подкислять более сильными кислотами, например фосфорной. Необходимая для дистилляции кислотность зависит от фо?мы. в которой на едятся цианиды.
Цианиды щелочных металлов легко перевести в HCN. Достаточно легко разрушаются при подкислении цианиды Cd, Си, Ni и Zn. Комплексные цианиды железа в тех же условиях разрушаются с трудом. Очень медленно разлагаются цианиды кобальта (Ш), Выделение HCN из комплексных цианидов облегчается при введении солей магния и ртути. Предложен эффективный метод отгонки цианидов, в котором для разрушения комплексных цианидов используют растворы хлоридов магния(Н) и ртути(II) в серной кислоте [6]. При этом FefCN)!- и Fe(CN)6- переходят в цианиды магния и ртути.
Дистилляцию цианидов можно проводить из растворов, содержащих Си’ и НС1. Этот метод рекомендован для выделения цианидов из растворов [7].
Показано, что при дистилляции HCN при пониженном давлении в присутствии ацетата цинка разложения ферроцианида не происходит [8]. Дистилляция при пониженном давлении из буферных растворов с pH = 5,2 — 5,8, содержащих соль цинка, рекомендована для отделения цианидов в присутствии ферри- и ферроцианида [9]. Соли свинца вводят для подавления отгонки сульфидов и других летучих соединений серы. Возможна отгонка с паром при различных условиях свободных и комплексных цианидов из промышленных образцов [10]. Методы дистилляции применяют при анализе разных образцов, в том числе биологических материалов [11].
Отделение суммы цианидов дистилляцией [7]
Реагенты и аппаратура
Кислый раствор хлорида меди(1), получают растворением 2 г хлорида меди(1) в 100 мл 5 М раствора НС1, готовят ежедневно.-
Гидроксид натрия, 0,5 М раствор.
Колба вместимостью 500 мл с ловушкой для брызг, обратным холодильником и поглотительной склянкой.
Ход отделения. В колбу для дистилляции вводят анализируемый раствор, Содержащий ие более 50 мг цианидов, и доводят водой до 200 мл. Приливают о мл кислого раствора хлорида меди(1) и несколько кусочков пористой керамики. Немедленно закрывают колбу, чтобы избежать потерь HCN. Помещают ли мг 0,5 А1 раствора NaOH в поглотитель. Проводят дистилляцию, собирая примерно 90 мл жидкости в поглотителе.
Микродиффузия
Метод отделения цианистого водорода с помощью микродиффу-ВаткН°Й ячеяки Конвея очень прост. Он позволяет сконцентриро-ид Чианиды, выделить их из образца в раствор NaOH, а затем ' ДОм МИТЬ титРи':стРическнм или спектрофотометрическим мето-метод дает возможность отделить свободные цианиды от
73
Рис. 14. Модернизированный прибор для мнкродиффузнон-ного отделения цианидов:
1 — чашка с раствором N’aOH; 2—чашка с H2SO4 п пробой.
ферри- и ферроцианидов [12]. Для мпкродпф-фузнонного отделения цианидов предложен модифицированный прибор (рис. 14) [13]. Прибор применен для определения цианидов в крови при содержании 0,2 мкг CN- в 2 мл. В чашке I находится поглотитель — раствор NaOH, в чашке 2 содержится раствор H2SO4 и проба. Реакция начинается при контакте пробы с кислотой, для полного отделения цианида необходимо около 2 ч.
Газохроматографический метод определения цианидов в биологических материалах включает их предварительное микродиффу-зионное отделение [14].
Метод жидкостной экстракции
Цианистый водород при содержаниях 0—2 мг/л можно экстрагировать из водных растворов с помощью 1,1,1-трихлорэтана, что позволяет отделить его от свободных и комплексных цианидов. Определение заканчивают спектрофотометрически.
Для экстракции менее 200 мкг цианидов применен комплекс Fe" с 1,10-фенантролином. Экстракцию проводят хлороформом из растворов с pH = 8,5 — 10. Мешают Г, ЮД S2', SCN', Со11, Си 1 Fe11 и Ni11 [15].
Хроматографический метод
Для отделения цианидов применяют тонкослойную хроматографию. На крахмале с помощью растворителя, содержащего ацетон и 3 М NH.)OH (1 : 4), удается разделить 12 анионов, в том числе SCN", AsOt', AsO®’ и POt [16].
На силикагеле с помощью разных растворителей можно отделить цианид от F’, СГ, Вг’, Г, N3, SCN', [Fe(CN)6]3‘ и Fe (GN)8]4‘ [17]. Метод тонкослойной хроматографии на алюминиевой фольге, покрытой силикагелем, с применением раствора FeCla позволяет отделить CN-от SCN", [Fe(CN)e]3_ и [Fe(CN)e]4-[18].
Газо-хроматографическпе методы описаны ниже.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Цианид можно осадить на .холоду избытком Ag+ в виде AgCN и высушить осадок при 100 °C. Однако этот метод редко используют.
74
Титриметрические методы
Титрование раствором AgNO3
Метод Либиха — Дениже основан на титровании цианида стандартным раствором AgNO3 в аммиачном растворе в присутствии KI. При добавлении AgNO3 к избытку цианида в нейтральном растворе образуется растворимый комплекс K[Ag(CN)2]:
Ag+ + 2CN’ — > Ag (CN)2-
После связывания всего цианида при добавлении Ag-ь образуется слаборастворимый комплекс дицианоаргентата серебра:
Ag+ + Ag (CN)~ —> Ag[Ag(CN)J
Связанное с этим помутнение раствора отвечает конечной точке титрования по Либиху [19]. Образование осадка наблюдается несколько раньше, чем предсказывает стехиометрия реакции, поскольку образующийся локально осадок AgCN растворяется очень медленно. Дениже [20] модифицировал метод, использовав аммиачный раствор, содержащий KI. В этом случае конечная точка индицируется по образованию осадка Agl:
Ag [Ag (CN)2] + 2NH3 —> Ag (NH3)2 + Ag (CN)'
Ag(NH3)2+ + r —> AgI| + 2NH3
Теоретическое обоснование метода обсуждено в работах'[21, 22]. Метод рекомендован для определения цианидов в KCN и NaCN [23].
Титриметрическое определение цианидов с помощью AgNOs
Ход анализа. К раствору, содержащему цианиды, приливают 15—20 мл 2 М раствора аммиака н разбавляют водой до 100 мл. Добавляют 0,2 г КТ, перемешивают для растворения соли. Титруют 0,1 М раствором AgNO3 до появления мути, которую лучше наблюдать иа черном фоне. Как видно из приведенного выше уравнения, 1 моль Ag+ соответствует 2 моль CN~.
Для определения цианидов применяют метод Фольгарда. К анализируемому раствору приливают избыток AgNO3 и раствор слегка подкисляют. Раствор отфильтровывают и аликвотную часть титруют стандартным раствором роданида аммония, используя в качестве индикатора железо(III)-аммонийные квасцы. Для анализа смеси CN~ и SCN~ применяют комбинацию метода Либиха и Фольгарда. Методом Либиха определяют только цианид, а методом Фольгарда — сумму анионов.
Для аргентометрического титрования цианидов предложено несколько индикаторов, например п-диметиламинобензилиденрода-НИН [24]. Метод с применением этого индикатора рекомендован для пределения цианида в промышленных растворах [7]. В конечной тооя6 окРаска изменяется от желтой до розовой. В качестве индика-Р предложен также родизонат натрия [25], а в водно-спиртовой
75
среде — дитизон [26]. В конечной точке в присутствии избытки серебра наблюдается резкое изменение окраски от желто-оранже-вой до глубокой красно-пурпурной. Чувствительность индикации примерно в 10 раз выше, чем методом Либиха.
Титриметрическое определение цианидов с помощью AgNOs в присутствии дитизона как индикатора [26]
Реагенты
Нитрат серебра 0,01 М раствор, готовят точным разбавлением 0,1 М рас* твора.
Дитизон, 0,01%-ный раствор в ацетоне.
Ход анализа. К раствору, содержащему меиее 5,0 мг CN~, добавляют 50 мл 95%-иого этанола, 1 мл 1 М раствора NaOH и 2,0 мл дитизона. Титруют 0,01 М раствором AgNO3 до интенсивной красно-пурпурной окраски. Проводят контрольный опыт и вносят соответствующую поправку.
Примечания. 1. В конце титрования объем воды, включая раствор титранта, не должен превышать 20 мл. Избыток воды снижает четкость перехода окраски.
2. Показано [26], что интенсивность окраски и четкость перехода в присутствии п-диметиламииобеизилиденродаиина меньше, чем в присутствии дитизона, который можно использовать даже для титроваийя окрашенных растворов.
Другой индикатор, 1,10-фенантролин (Phen) + бромпирогалло-ловый красный (BPR) также дает четкую точку перехода при титровании цианидов раствором AgNOs 27]. В основе метода лежит образование комплекса [(AgPhen2)+]2BPR2-, используемого для чувствительного (е = 5,1-104) спектрофотометрического метода определения серебра. Титриметрическое определение пригодно для макро- и микроколичеств. Определению мешают в эквимолярных количествах БгОГ, SCN’, Г, Fe3+, Со2+, Си2+ и Hg"+. Ионы меди (II) мешают при молярном отношении более 1/10, а Г-'е3+, Со2+ и Hg2+ — при отношении более 1/100.
Меркуриметрический метод
Цианид ртути диссоциирован значительно меньше, чем роданид ртути:
Hg2+ + 2CN' —> Hg (CN)2
Это позволяет титровать роданидом избыток стандартного раствора ртути в присутствии цианида ртути. Предложены различные индикаторы конца титрования, например нитропруссид натрия и дифе-нилкарбазон, а также вариаминовый голубой [28], в присутствии которого конечную точку устанавливают по устойчивой голубой окраске раствора.
Иодиметрический метод
Широкое распространение получил также метод, основанный на образовании бромциана [29]. Бром количественно взаимодействует со слабощелочным раствором цианида (и роданида) с образованием бромциана:
HCNВг2 —> BrCN + Н+ + Вг‘
76
который устойчив в слабокислых растворах. Избыток брома ч гз-створе можно связать сренолом, который не взаимодействует .• ВгС'-. Затем при взаимодейс:вин бромциана с йодидом образуется иод, который можно оттитровать тиосульфатом:
ВгСХ + 2Г —> Ь + Вт’ + CN*
Метод позволяет определять цианиды в присутствии СГ, Br'. S"' и ЗгОз’- Описан метод определения микроколичеств цианида в присутствии NH4, NO? и NOj [39].
Комплексометрический метод
Предложен ряд методов комплексометрическою определения цианидов. При взаимодействии цианида с никель-аммоиийсульфа-том образуется комплексный анион тетрацианоникелат (II) [Ni(CN)4]2-. Можно оттитровать избыток иона никеля и рассчитать содержание цианида [31—33]. Этот метод применен для анализа смесей цианида с хлоридами или роданидами с целью определения цианидов.
Для титрования цианидов использовано образование сметано-лигандных комплексов [34]. При титровании 0,025—2 мг CN" заствором комплекса ртути(II) с ЭДТА (H4Y) при pH — 4 —8 об-)азуется комплекс [HgY(CN)]3-, который взаимодействует с CN“. 1ри этом образуются Hg(CN)2 и ЭДТА. Этилепдиаминтетрааце-тат титруют раствором CuSO4. Определению мешают S2-, Vv, Cu2+, Со2+, Ni2+, Hg2+ и Ag1. Предложен метод последовательного титрования цианида и роданида, основанный на том, что комплекс Hg11 с CN~ и ЭДТА не поглощает при 240 нм, в то время как соответствующий роданидный комплекс поглощает в этой области спектра. Кроме того, образование цианидного комплекса протекает быстрее, чем роданидного. Титрование 5—100 мкг CN- и 10—100 мкг SCN~ проводят стандартным раствором Hg(NO3)s при pH = 7 в буферном растворе, в присутствии ЭДТА. Конец титрования цианидов устанавливают по росту поглощения при 240 нм, а роданидов — по достижению постоянного поглощения при 240 нм [35]. Разработан метод анализа смеси CN~, SCN~ и I- [36].
Спектроскопические методы
Колориметрические и спектрофотометрические методы
В обзоре [3] критически сопоставлены спектрофотометрические методы определения цианидов (до 1962 г.). Методы разделены на три группы: основанные на реакции Кёнига, включающие образование комплексов металлов и разные методы.
Первая группа методов основана на взаимодействии бромциана с соответствующим ароматическим амином и пиридином. Бромцнан «кисляет пиридин до тлутаконового альдегида, который ющденси-гуется с ароматическим амином, образуя интенсивно окрашенное
77
Рис. 16. Схема установки для автоматического определения цианидов 144]:
/—ыас^яный нагреватель. 98 СС; 2—5—смесители; 6—самописец; 7—колориметр; А — воздух (0,090 мл/мин); Б — НС1 (0,100 мл/мнн); В — проба (0,073 мл/мин); Г — бромат-бромидная смесь (0,056 мл'мн); Д — проба (0,110 мл/мин); Е—НС1 (0,056 мл/мнн); Ж’* арсен нт натрия (0,056 мл/мнн); 3-воздух (0,973 мл/мнн); Я—пиридин (0,081 мл/мий); К—л-фениленднамин (0,06э мл/мнн); Л— линчи протока через ячейку (0,081 мл/мин).
соединение Аг—N = CH—СН = СН—СН = СН—СН—Аг. Чувствительность зависит от природы ариламина. Разработан метод [37, 38], основанный на взаимодействии CN- с избытком брома (образуется бромциан), восстановлении избытка брома мышьяком(III) и определении бромциана по реакции с пиридином и бензидином. Описано несколько модификаций метода. При экстракции окрашенного соединения 1-гексанолом чувствительность достигает 0,02— 0,5 ppm [39]. Канцерогенный бензидин можно заменить барбитуровой кислотой [99], однако при этом окраска раствора по достижении максимума быстро исчезает. Из других соединений наиболее эффективным оказался п-фенилендиамин [40]. Метод позволяет определять 0,1—2 мкг CN~. Определение 0,005—0,1 ppm CN~ проводят после отделения CN- от SCN-, Fe24-, Ni2+, Cu2+ и цианидных комплексов Zn и Си методом дистилляции [41]. Метод рекомендован для анализа растворов [42]. Раствор реагента рекомендуется готовить непосредственно перед применением [43].
Разработан автоматический метод дистилляции и определения цианида [44]. При этом исключается фильтрование растворов проб, производительность анализатора 40 проб в 1 ч. Схема работы прибора дана на рис. 15.
Предложен автоматизированный спектрофотометрический метод, основанный на применении пиридина и бензидина [45].
Для определения цианидов широко применяют метод с использованием пиридина и пиразолона [46]. Цианиды окисляют хлорамином Т, а образующийся хлорциан взаимодействует с раствором пиридина, содержащим 3-метил-1-фенил-5-пиразолон и небольшую концентрацию бис(3-метил-1-фенил-5-пиразолона). Образующийся синий краситель фотометрирует при 630 нм. Чувствительность метода можно повысить экстракцией красителя 1-бутанолом. Недостатком является малая устойчивость растворов реагента, которые необходимо готовить ежедневно. Метод рекомендован для анализа воды [47]. Предварительная дистилляция позволяет устранить или уменьшить влияние посторонних ионов. Метод применен для опре
78
деления в крови свободного цианида в присутствии феррп-, ферроцианида и цианида. Отделение цианидов проводили известными методами.
Предложен метод определения цианидов, основанный на образовании относительно устойчивого соединения сине-фиолетового цвета при взаимодействии цианидов, обработанных хлорамином Т, с у-пиколином и барбитуровой кислотой [43]. Метод применен для определения 0—0,5 мкг/мл цианидов в воде. Определению 2 мкг CN~ в 10 мл раствора не мешают до 1 мг СГ, NO2, NO3, SO2"< СН3СОО" и CNO' (в виде солей Na или К), а также 100 мкг Fe111, [FefCNJe]3-, [Fe(CN)6]4-. В присутствии роданидов получаются завышенные результаты. Раствор реагента (у-пиколин + + барбитуровая кислота) устойчив в течение 3 недель при 10 °C.
Способность цианида образовывать устойчивые комплексы с ионами металлов использована для разработки прямых и косвенных спектрофотометрических методов определения CN-. Комплекс ртути(II) с ЭДТА поглощает при 250 нм, а при его взаимодействии с CN- образуется смешанный комплекс [HgY(CN)]3-, который не поглощает в этой области [48]. Снижение поглощения HgY2- при 250 нм пропорционально концентрации цианидов в интервале 0,01—1,2 мг в 50 мл раствора. Определению мешают^СЫ', S2’,Br", Г, Cu2+, Ni2+, Со2+, 32ОГ, Hg2+, СГ, NH1 и др. Аналогичный метод, основанный на взаимодействии цианидов с комплексом ртути(II) с метилфталеином (1:1) [49] позволяет определять 1—20 мкг CN- в 50 мл раствора. Влияние некоторых мешающих катионов можно устранить. Мешают сульфиды и иодиды.
Предложен косвенный метод определения цианидов, основанный на их взаимодействии с избытком Hg(NO3)2, при этом образуется Hg(CN)2. Раствор диэтилдитиокарбамата меди(П) в хлороформе взаимодействует с избытком Hg2+, после чего раствор фотометри-руют при 440 нм [49]. Определению-50 мкг CN~ мешают S2’, S2O2‘> Г (>1 мг), Ag и Со (>60мкг); Ni (>100мкг) и Си (>190мкг).
Образование тройного комплекса цианидов со ртутью(II) и метилтимоловым синим использовано для определения 5—60 мкг CN-. Градуировочный график линеен. Растворы фотометрируют при 615 нм. Некоторые мешающие катионы и анионы можно замаскировать [50].
При взаимодействии меди(II) с цианидом и вариаминовым голубым образуется окрашенный продукт [51]. Определению CN- не мешают NH4, NO3, SO2' и СГ.
Применяют методики спектрофотометрического определения цианидов, основанные на применении комплекса Fe11 с 1,10-фенан-тролином. Метод, предложенный в работе [52], основан на экстракции хлороформом и фотометрировании комплекса дицианобис-(1,10-фенантролнната) железа (II). Модифицированный вариант метода обладает большей селективностью и чувствительностью [53]. Определению не мешает до 1 г/л солей Fe и Си, S2-, Ca(CN)2,
79
акрилонитрил, фенол, метанол, формальдегид, карбамид, тиомоч-вина, капролактам и гексамин.
При взаимодействии цианида и труднорастворимого соединения трииодида с ферроином в раствор переходит эквивалентное содержанию цианида количество ферроина, который определяют спектрофотометрически [54]. После переведения цианида в роданид и образования ионного ассоциата ферроина с SCN~, п >следний можно экстрагировать нитробензолом и фотометрировать при 516 нм [55]. Метод позволяет определять менее 40 мкМ цианида.
Известно, что хлоранилат применяют для спектрофотометрического определения СГ, SOV и F. Этот метод применен также для определения цианидов (а также сульфидов и сульфитов) [56]. С применением хлоранилата ртути, в зависимости от выбранной длины волны, удается определять 0,4—4 и 20—200 мкг/мл цианидов.
Для определения цианидов применяют комплекс дифенилкарба-зона со ртутью(П) [57, 58]. Предложен косвенный метод, основанный на образовании тройного комплекса, использованный ранее для определения следов серебра [59]. Комплекс (AgPhen+)2BPR2~ образуется при взаимодействии серебра(I), 1,10-фенантролина и бромпирогаллолового красного и характеризуется молярным коэффициентом поглощения е= 5,1 -104 при 635 нм. Цианид ингибирует образование комплекса. Предел обнаружения цианида равен 0,2 ppm. Из 17 изученных катионов мешают только ртуть(II), не мешают определению 100-кратные количества 14 анионов. Мешают бромиды, иодиды и роданиды, однако их можно осадить, добавив растворы Pb(NO3)2, (NH4)2SO4 и Ba(NO3)2 и отцентрифугировав осадок.
При взаимодействии цианидов с аммиачным раствором ни-келя(П) образуется устойчивый анионный комплекс [Ni(CN)4J2-— тетрацианоникелат, поглощающий при 267 нм. Закон Бера соблюдается в интервале содержаний 5—200 мкг CN- в 10 мл раствора. При 267 нм значение е равно 1,1-104. Преимущество метода заключается в том, что аммиачный раствор N1C12 может служить для поглощения HCN при дистилляции. Летучий цианистый водород мгновенно образует цианидный комплексный анион, который можно непосредственно фотометрировать. Мешают сульфиды и сульфиты, однако их можно легко отделить. Метод может быть применен также для определения никеля.
Рассмотрим другую группу спектрофотометрических методов определения цианидов. Показана возможность осуществления реакции нуклеофильного замещения при атаке цианида на органический дисульфид с образованием аниона тиола и замещенного тиоцианата [60]:
RSSR-J-CN' —> RS’ + RSCN
Изучен ряд дисульфидов, в том числе 2,2/-дитиопиридин, 4,4'-ди-тиопиридин и 5,5'-дитнобис(2-питробензойная кислота). Относительно простой метод позволяет определять 0,2 —5,0 мкг/мл циа
80
нида. При выборе оптимального соотношения реагентов можно определять до 50 мкг/мл цианидов. Определению мешают меркаптаны, сульфиды, сульфиты, а также многие ноны металлов, которые, однако, можно замаскировать ЭДТА (кроме Си2+ и Hg2+). Недостатком является медленное протекание реакции, особенно при малых концентрациях цианидов. При использовании диметил-формамида продолжительность реакции снижается от 2 ч до 30 мин [61].
Описан очень чувствительный и, вероятно, специфичный спектрофотометрический метод определения цианидов, основанный на их взаимодействии с n-нитробензальдегидом в щелочном растворе с образованием циангидрина, способного при взаимодействии с разными реагентами давать окрашенные соединения [62]. Например, с динитробензолом циангидрин взаимодействует по схеме:
причем цианид регенерируется. Вследствие каталитического характера реакция обладает высокой чувствительностью. Скорость реакции измеряют по увеличению оптической плотности растворов. Содержание цианидов в интервале 45—450 нг/мл определяют по градуировочному графику, построенному в координатах &А/&1 — концентрация CN-, где А — поглощение растворов, t — время. Определению не мешает ни один из 30 изученных анионов. С применением 1-бензоил-1-гидроксиимино-ацетона можно определять минимально 1,3 нг/мл цианида. В присутствии ферри- и ферроцианида методика несколько видоизменяется [63].
Спектрофлуориметрические методы
Для спектрофлуориметрического определения цианида применены некоторые производные хинона [64, 65], в том числе п-бензо-инон, П-хлор-л-бензохинонимин и о-(«-нитробензол—сульфонил) инон монооксим. Метод с применением n-бензохинона имеет
81
чувствительность 0,2 мкг/мл. Предполагается, что реакция протекает по схеме
Определению не мешает ни один из большого числа изученных ионов при концентрации 0,1 Л1. Интервал определяемых содержаний цианида составляет 0,2—50 мкг.
Разработан метод определения 1,3 мкг цианида с помощью пиридоксаля [66]. Чувствительность метода в 10 раз выше, чем метода с применением реакции взаимодействия бромциана с бензидином.
При взаимодействии цианида с нефлуоресцирующим 2/,7/-бис-(ацетоксимеркури) флуоресцеином образуется флуоресцирующий продукт. Интенсивность флуоресценции пропорциональна содержанию цианида в интервале 3—9 мкг [67].
Атомно-абсорбционный метод
Описано косвенное определение цианида методом ААС. В работе [68] предложен метод, основанный на образовании комплекса дициано-трис(1,10-фенантролината) железа (II). Градуировочный график линеен в интервале 0,1—5 ppm CN-. При анализе сточных вод с чувствительностью 2,0-10-5 М предложен метод, основанный на образовании цианидного комплекса меди состава 3:1 [69].
Электрохимические методы
Потенциометрические методы
Аргентометрическое титрование цианидов, описанное выше, можно проводить потенциометрически с применением серебряного электрода.
Описан потенциометрический анализатор для непрерывного измерения HCN в атмосфере [70]. Анализируемый газ пропускают через щелочной поглотитель, после чего измеряется потенциал с применением серебряного и каломельного электродов.
Изучена возможность применения некоторых биметаллических электродов для титрования цианидов раствором AgNO3 [71], в том числе Pt — Ni, Pt — Ag, Pt — W, Pt — Pd.
Цианиды можно определять с помощью ионоселективных электродов, этот метод будет введен в качестве стандартного для анализа промышленных вод. Селективные электроды выпускают серийно, например Орион 94-06 и OP-CN-711 (Раделкис, Венгрия). Для определения цианидов можно применять галоге-нидные мембранные электроды, например иодидные (Орион 94-53, Corning Eel 47612700), отличающиеся высокой селективностью.
82
рис. 16. Кривая потенциометрического титрования смеси CN" и С1 0.1 Л/ раствором AgX'Oj с помощью сульфид-ссрееряниго электрода Орион 94-16:
у—скачок, соответствующий концу титрования Ag+ т 2CN —» Ag (CN),' ;
j—скачок, соответствующий концу титрования Ag (CN), +
ц. Ag+ —> 2 AgCN;
J—скачок, соответствующий концу титрования Ац + + С‘ —»
-> AgCl.
Можно применять также сульфид-серебряный электрод (например, Орион 94-16). При использовании электрода Орион 94-06 интервал определяемых концентраций равен 10-2 — 10s /И цианида. Определению не мешают ионы F’, NOJ и СОД, но мешают
S2-. Константы селективности по отношению к Cl-, I- и Вт- равны 10-6, 10 и 2-10~3 соответственно. При высоких концентрациях цианида время работы электрода ограничено.
Ионоселективные электроды можно использовать также для аргентометрического титрования цианидов. Иодид-серебряный электрод позволяет определять 10~6—- 10-1 М CN-. Он фиксирует
концентрацию цианидов и цианидных комплексов, которые менее устойчивы, чем Ag(CN)2‘[72]. Изучено влияние pH на показания иодид-серебряного электрода, связанные с взаимодействием цианида с поверхностью мембраны [73]. С применением сульфидсеребряного электрода методом «добавок» можно определять минимально 0,03 ppm цианидов [74]. При содержании CN~ равном 1 ppm ошибка составляет 2%. При анализе сточных вод пробы, содержащие Zn2+, Cd2+, Cr3+, Cu2+ и Ni2+, обрабатывали раствором ЭДТА для разрушения цианидных комплексов и последующего определения цианидов.
Сульфид-серебряный электрод использован в качестве индикаторного при потенциометрическом титровании растворов, содержащих CN~ и С1~ [75]. Кривая титрования показана на рис. 16.
Значительное внимание уделено автоматическому непрерывному анализу. Описано непрерывное измерение концентрации цианидов с применением компьютера [76]. Цианидный электрод применен Для анализа в потоке раствора [77]. Регистрируемый сигнал зависит от скорости потока и температуры. В той же работе предложен метод осаждения сульфидов обработкой раствора солью кадмия (II).
Цнанид-селективные электроды использованы для анализа разных материалов, в том числе растений при биохимических исследованиях. Гликозид, содержащий связанный цианид, не ток-J^h. Однако при его гидролизе образуется токсичный цианн-водород. Исследования превращений такого рода были сде-Ны с применением цианид-селектпвпого электрода [78]. Другие
83
области применения описаны в работе [79]. Опубликован обзоп применения галогенид- и циапид-еелективных электродов для анализа химикатов, пищевых продуктов и фармацев!нческих препаратов [80].
Амперометрические методы
Амперометрическое титрование цианида раствором серебра(I) с применением вращающегося платинового электрода описано в работе [81]. При использовании платинового микроэлектрода мешающего влияния иодида и сульфата можно избежать ир установлении рабочего потенциала —0,85 В [82]. Определению не мешают 100-кратные количества бромида и хлорида. Последовательное титрование мнкрограммовых количеств CN~, С1~ и OCN- описано в работе [83]. Примерно равные содержания анионов можно определять с ошибкой около 2%.
Полярографические методы
Цианиды образуют со ртутью устойчивые комплексы. Анодная волна цианидов на капающем ртутном электроде наблюдается при потенциале —0,4 В в среде 0,1 Л1 раствора NaOH. На этом основан полярографический метод, пригодный для рутинного анализа. Описан метод определения Ю-6—10-10 г цианида в растворе LiOH с применением быстро вращающегося золотого электрода [84].
Цианиды можно определять по каталитической волне, возникающей при полярографировании комплекса никеля(II) с этилендиамином в присутствии цианидов [85]. Чувствительность определения цианидов составляет 0,5 мкг/л. Высота волны пропорциональна концентрации цианидов в интервале 0,1—1 мкг/л. Изучена система никель(П) — гилроксиламин — цианид [86].
Методом катодно-лучевой полярографии определено содержание цианидов в воде (>0,05 мкг/мл). Определению не мешают большие содержания хлорида, мешают свободные хлор, бром и иод [87]. Описан косвенный полярографический метод [88], основанный на выделении хлоранилат-иона цианидом из хлорани-лата ртути, как и известные спектрофотометрические методы определения F', СГ, CN и SO)’. Концентрацию хлоранилат-иона определяют по волне двухэлектронного восстановления на капающем ртутном электроде. Ток восстановления пропорционален концентрации цианида.
Каталитические методы
Цианид ингибирует хемилюминесцентную реакцию окисления люминола пероксидом водорода в присутствии Си2+, причем индукционный период пропорционален содержанию цианидов. Пред* ложен метод фиксированного свойства для определения ппаннД8 по этой реакции [89]. Определению мешают Fe3+ (> 5 ppm), NO2 (>0,4 ppm), Г(>20 ррш) и SO3'(>0,5 ppm).
84
Гидролиз сахарозы, катализируемый ферментом р-фруктофу-ранозидаза (инвертаза), замедляется в присутствии следов металлов, являющихся ши пои юрам и. Микроколичества анионов, образующих устойчивые комплексы с этими металлами, например ионов цианидов, сульфидов и иодидов, понижают ингибиторное действие металлов. Этот принцип положен в основу метода определения ю-5—10-7 М цианидов [90]. Скорость гидролиза определяли по углу оптического вращения раствора.
Газохроматографические методы
Газовая хроматография была применена для выделения цианида из смеси, содержащей О2, N2, СН4, СО2 и СО [91]. Разделение проводили на двух колонках. Одна из них содержала хро-мосорб Р, пропитанный 25%-ным триацетатом глицерина, а другая— молекулярные сита. В качестве газа-носителя применен гелий.
Описан метод определения цианида и роданида, основанный на переводе их в бромцпан с помощью брома [92]. Анализ проводят газохроматографическим методом с детектором электронного захвата. Предел обнаружения равен 10~8 г, в интервале содержаний CN~ 0,0—0,5 ppm высота пика примеси пропорциональна содержанию CN-. Возможно повышение чувствительности анализа путем экстракции BrCN диизопропиловым эфиром с последующим введением экстракта в колонку. Метод позволяет обнаруживать 1 ppb CNBr. Для определения комплексных цианидов их можно предварительно разложить. Газохроматографический метод определения цианидов в биологических материалах описан в работе [14], причем предварительное отделение CN“ Проводят методом микродиффузии, в качестве абсорбента применяют раствор NaOH. Затем CN- переводят хлорамином Т в CNBr и хроматографируют на колонке, заполненной 7% хал-Комида М-18 на носителе Анахроме ABS (размер зерен 0,149— 0,160 мм) при 55 °C. В качестве газа-носителя применяют смесь метан — аргон (1:19), используя детектор электронного захвата. Градуировочный график линеен до содержания CN- 100 мкг/мл.
Радиохимические методы
Если суспензию Hg(103)2, содержащую изотоп 203Hg, ввести в раствор, содержащий CN~, образуется раствор Hg(CN)2. Для снижения растворимости Hg(IO3)2 в раствор вводят также КЮ3. Содержание цианидов определяют по активности раствора [93]. .♦ Аналогичный метод с поименением Agl предложен в работе [94]:
2CN' + "j'Ag! = 11 Ag (CN)' + Г Анализируемый раствор пропускают через колонку, содержащую •^81. Активность элюата измеряю-: с помощью кристалла Nal, 'д?егРоенного на у-излучение изотопа "°Ag с энергией 0,66 МэВ. '.“етод эксвреесен, чувствителен и весьма избирателен.
85
Другие методы определения
Мпкрограммовые количества цианида можно определять косвенно, методом рентгенофлуоресценгного анализа после осаждения цианида раствором серебра(I) [95].
Ряд неорганических веществ, в том числе цианиды, можно определять с помощью серийного CHN-анализатора [96]. Методом бумажной хроматографии, по площади белых пятен, образующихся на бумаге Ватман № 1, пропитанной сульфидом меди, можно определить 1—6 мкг CN~ [97]. Границы пятен становятся более четкими, если опрыскать хроматограмму 1%-ным раствором диэтилдитиокарбамата натрия в метаноле. Не мешает 100-кратный молярный избыток CN-, СП, Вг~, 1“ и OCN~ и 24-кратный избыток S2~,
Для определения микросодержаний цианидов или Ag (CN)> применена фильтровальная бумага, пропитанная Ag2CrO.i (или AgCl, AgBr) [98]. На узкую полоску бумаги наносят 0,01 мл раствора и элюируют водой в специальном аппарате. Бумагу сушат, обрабатывают H2S, белое пятно вырезают и взвешивают. Метод позволяет определять 8—80 мкг CN- при анализе смесей CN~ и Ag(CN)2-.
ЛИТЕРАТУРА
1. Montgomery Н. А. С., Gardiner D. К-, Gregory J. G. G. — Analyst, 1969, v. 94, р. 284.
2. Roberts G. L. — In: Encyclopedia of Industrial Chemical Analysis (Ed. F. D. Snell and L. S. Ettre), Vol. 14, New York, Interscience, 1971, p. 403.
3. Bark L. S., Higson H. G. — Analyst, 1963, v. 88, p. 751.
4. Boltz D. F. — Crit. Rev. analyt. Chem., 1973, v. 3, p. 147.
5. Ludzack F. J., Moore W. A., Ruchhoft С. C. — Analyt. Chem., 1954, v. 26, p. 1784.
6. Serfass E. J., Freeman R. B., Dodge B. F., Zabban W. — Plating, 1952, v. 39, p. 267.
7. Official Standardised and Recommended Methods of Analysis, Compiled and Edited by N. W. Hanson, 2nd edn., London. The Society for Analytical Chemistry, 1973, p. 455.
8. Roberts R. F., Jackson B. — Analyst, 1971, v. 96, p. 209.
9. Ref. 7, p. 457.
10. Hilbert F., Darwish N. A. — Z. analyt. Chem., 1971, v. 255, p. 357; Analyt. Abstr., 1972, v. 22, r. 3068.
11. Tompsett S. L. — Clin. Chem., 1959, v 5, p. 587.
12. Kruse J. M., Thibault L. E. — Analyt. Chem., 1973, v. 45, p. 2260.
13. Baar S. — Analyst, 1966, v. 91, p. 268.
14. Valentour J. C., Aggarwal V., Sunshine 7.— Analyt. Chem., 1974 v 46 p. 924.
15. Schilt A. A. — Analyt. Chem., 1958, v. 30, p 1409.
16. Canic V. D., Turcic M. N., Bugarski-Vojinovic M. B., Perisic N. U. — Z. an: lyt. Chem., 1967, v. 229, p. 93; Analyt. Abstr.. 1968, v. 15, p. 5211.
17. Gagliardi E., Pokorny G.—Mikrochim. A?ta, 1965, p. 699.
18. Thielmann H. — Z. analyt. Chem., 1974, v. 270, p. 128; Analyt Abstr, 1975 v. 28, p. 2B86.
19. Liebig J. von — Ann. Chem. Liebigs, 1851, v. 77, p. 102.
20. Deniges G. — Compt. rend., 1893, v. 117, p 1078.
21. Laitinen H. A. — Chemical Analysis, New York, McGraw-Hill, 1960, p. 225.
22. Butler J. N. — Ionic Equilibria, Mass, Addison-Wesley, 1964, p. 280.
86
23. Britisch Standards 622: 1967. Appendix B. London.
24. Ryan J. .4,, Culshaw G. W.— Analyst, 1944, \. 69, p. 370.
25. Mehlig 1. P. — Chemist Analyst, 1955, v. 44, p. 87.
26. Archer E. E. — Analyst, 1958, v. 83. p. 571.
27. Dagnail R. Al., El-Ghamry AL T., West T. S. — Taianta, 1966, v. 13, p. 1667.
28. Gregorowicz Z.. Buhl F. — Z. analyt. Chem., 1960, v. 173, p. 115; Analyt. Abstr., 1960, v. 7, p. 4150.
29. Schulek E. — Z. analyt. Chem., 1923, v. 62, p. 337.
30 Schulek E., Burger K.. Feher M. — Z. analyt. Chem., 1959, v. 167, p. 429; Analyt. Abstr., 1960, v. 7, p. 451.
31 De Sousa A. — Inf. Quim. Anal., 1961, v. 15, p. 61; Analyt. Abstr, 1962, v. 9, p. 127.
32 De Sousa A. — Inf. Quim. Anal., 1962, y. 16, p. 126; Analyt. Abstr., 1963, v. 10, p. 2243.
33. De Sousa A. — Taianta, 1961, v. 8, p. 782.
34. Komatsu S., Nomura T. — J. Chem. Soc. Japan, pure Chem. Sect., 1966, v. 87, p. 1060; Analyt. Abstr., 1968, v. 15, p. 720.
35 Nomura T. — J. chem. Soc. Japan, pure Chem. Sect., 1968, v. 89, p. 580; Analyt. Abstr., 1970, v. 18, p. 114.
36 Nomura T. — Bull. chem. Soc. Japan, 1970, v. 43, p. 104; Analyt. Abstr., 1971, v. 20, p. 934.
37. Aldridge W. N. — Analyst, 1944, v. 69, p. 262.
38. Aldridge W. N. — Analyst, 1945, v. 70, p. 474.
39. Nusbaum I., Skupeko P.— Analyt. Chem., 1951, v. 23, p. 875.
40. Bark L. S., Higson H. G. — Taianta, 1964, v. 11, p. 471.
41. Bark L. S., Higson H. G. — Taianta, 1964, v. 11, p. 621.
42. Ref. 7, p. 456.
43. Nagashima S. — Analytica chim. Acta, 1977, v. 91, p. 303.
44. Casapieri P., Scott R., Simpson E. A. — Analytica chim. Acta, 1970, v. 49, p. 188.
45. Royer J. L., Twichell I. E., Muir S. Al. — Analyt. Lett., 1973, v. 6, p. 619.
46. Epstein J. — Analyt. Chem., 1947, v. 19, p. 272.
47. American Public Health Association, Standard Methods for the Examination < of Water and Wastewater, 13th edn., New York, American Public Health Association, 1970.
48. Komatsu S., Nomura T. — J. chem. Soc. Japan, pure Chem. Sect., 1966, v. 87, p. 845; Analyt. Abstr., 1967, v. 14, p. 7419.
49. Komatsu S., Nomura T., Mochizuki F. — J. chem. Soc. Japan, pure Chem. Sect., 1969, v. 90, p. 944;; Analyt. Abstr., 1970, v. 19, p. 3801.
60. Nomura T. — Bull. chem. Soc. Japan, 1968, v. 41, p. 1619; Analyt. Abstr., 1969, v. 17, p. 3378.
51. Gregorowicz 7 Buhl F.-—Z. analyt. Chem., 1962. v. 187, g. 1; Analvt. Abstr..
1962, v 9, p. 4575. ". ’ " ’ ’
52. Schllt /1. ,1, — Analyt. Chem., 1958, v. 30, p. 1409.
53. Kodura I. Lada Z. — Chemia analit., 1972, v. 17, p. 871; Analyt. Abstr., 1973, r v. 24, p. 3842.
54. Lambert J. L., Manzo D. J. — Analyt. Chem., 1968, v. 40, p. 1354.
55. Hiiro K., Yamamoto Y.— Analyt. Lett., 1973, v. 6, p. 761; Analyt. Abstr., 1974, v. 26, p. 2596.
56. Humphrey R. E., Hinze W.— Analyt. Chem., 1971, v. 43, p. 1100.
57. Okutani T., Utsumi S. — J. chem. Soc. Japan, pure Chem. Sect., 1966, v. 87, P- 444; Analyt. Abstr., 1967, v. 14, p. 5341.
«8. Ohlweiler O. A., Oliveira J. — Zh. analit. Khim., 1959, v. 14, p. 303; Analyt. Abstr., I960, v. 7, p. 921.
Dagnail R. M., El-Ghamry AL T., West T. S. — Taianta, 1968, v. 15, p. 107.
Humphrey R. E., Hinze W. — Taianta, 1971, v. 18, p. 491.
g- Humphrey R. E., Alvares J. J.—Microchem. J., 1971, v. 16, p. 652.
Guilbault G. G., Kramer D. AL —Analyt. Chem., 1966, v. 38, p. 834.
H°Pescu G., Dura M. — Revta. Chim., 1970, v. 21, p. 7; Analyt. Abstr., 1971, W«P.3O4.
» Guilbault G. G„ Kramer D. N.- Analyt. Chem., 1965, v. 37, p. 918.
87
65. Guilbault G. G., Kramer D. N. — Analyt. Chem., 1965, v. 37, p. 1395.
66. Takanashi S, Tamura Z. — Chem. Pharm. Bull., Tokyo, 1970, v. 18, p. 1633;
Analyt. Abstr.. 1971, v. 21, p 118.
67. Colovos G., Haro AL, Freiser H.— Taianta, 1970, v. 17. p. 273.
68. Danchik R. S., Boltz D. F. — Analyt Chem., 1970, v. 49. p. 567.
69. .Manahan S. E., Kunkel R. — Anaijt. Lett., 1973, v. 6, p. 547.
70. Strange J. P.— Analyt. Chem., 1957, v 29. p. 1878.
71. Ueno K., Tachikawa T. — Muroran Kogyo Diagaku Hokaku, 1959, v. 2, p. 377; Chem. Abstr., 1960, v. 54, p. 1154a.
72. Toth K-, Pungor E. — Analytica chim. Acta, 1970, v. 51, p. 221.
73. Mascini Al.— Analyt. Chem., 1973, v. 45, p. 614.
74. Front At. S., Ross J. W., Riseman J. H. — Analyt. Chem., 1972, v. 44, p. 2227. 75. Conrad F. J. — Taianta, 1971, v. 18, p. 952.
76. Fleet В., Но А. У. W.— Taianta, 1973, v. 20, p. 793.
77. Fleet B., Von Sforp H.— Analyt. Chem., 1971, v. 43, p. 1575.
78. Gyorgy B., Andre L.. Stehli L., Pungor E. — Analytica chim. Acta, 1969, v. 46, p. 318.
79. Orion analytical methods guide, 7th edn., Crawley, East Sussex, UK, Orion Research. Inc., Cambridge, Massachusetts, 1975. Available from MSE Scientific Instruments.
80. Pungor E. — Zh. analit. Khim., 1970, v. 25, p. 1182.
81. Laitinen H. A., Jennings W. P., Parks T. D. — Ind. Engng Chem. analyt. Edn., 1946, v. 18, p. 574.
82. Shinozuka F., Stock J. T. — Analyt. Chem., 1962, v. 34, p. 926.
83. Musha S., Ikeda S. — Japan Analyst, 1965, v. 14, p. 795; Analyt. Abstr., 1967 v. 14, p. 3028.
84. Miller G. W„ Long L. E., George G. M., Sikes W. L. — Analyt. Chem., 1964, v. 36, p. 980.
85. Торопова В. Ф., Аверко-Антонович А. А. — ЖАХ, 1971, т. 26, с. 2437. Analyt. Abstr., 1973, v. 24, р. 3412.
86. Торопова В. Ф., Аверко-Антонович А. А. — ЖАХ, 1972, т. 27, с. 116.
87. Hetman J. — J. appl. Chem., 1960, v. 10, p. 16.
88. Humphrey R. E., Laird С. E. — Analyt. Chem., 1971, v. 43, p. 1895.
89. Musha S., Ho M., Yamamoto Y., Inamori У.— J. chem. Soc. Japan, pure Chem. Sect., 1959, v. 80, p. 1285; Analyt. Abstr., 1960, v. 7, p. 4186.
90. Mealor D., Townshend A. — Taianta, 1968, v. 15, p. 1477.
91. Isbell R. E. — Analyt. Chem., 1963, v. 35, p. 255.
92. Nota G., Palombari R. — J. Chromat., 1973, v. 84, p. 37.
93. Banyal E., Szabadvary F., Erdey L. — Taianta, 1963, v. 10.
94. Bowen H. J. M. — Analyst, 1972, v. 97, p. 728.
95. Stork G., Jung H. — Z. analyt. Chem., 1970, v. 249, p. 161; Analyt. Abstr., 1971, v. 20, p. 1523.
96. Noshiro M., Matsumoto K. — Japan Analyst, 1971, v. 20, p. 1069; Analyt. Abstr., 1973, v. 25, p. 20.
97. Kielczewski W., Schneider M., Uchman W. — Chemia analit., 1967, v. 12, p. 143; Analyt. Abstr., 1968, v. 15, p. 1932.
98. Ganchev N., Koev K. — Mikrochim. Acta, 1964, p. 92.
99. Murty G. V. L. N., Viswanathan T. S.—Anab'tica chim. Acta. 1961, v. 25, p. 293.
ФТОРСИЛИКАТЫ
Фторсиликат (гексафторсиликат) SiFT применяют для фторирования воды, при производстве синтетического криолита и обработке металлов. При отделении фтора традиционным методом дистилляции (по Виларду и Винтеру) часть фтора отгоняется в виде H2SiF6. Литература по методам определения SiFe' не систематизирована. Обзор ранних исследований приведен в работе [1].
88
Простой метод обнаружения кремнефтористой кислоты основан на добавлении KNO? и этанола к охлажденному льдом раствору. При этом осаждается гексафторсиликат калия и выделяются протоны, которые титруют стандартным раствором щелочи по фенолфталеину. Этот метод использован для определения фторсиликата в электролите хромирования [2]. Осажденный K2S1F6 промывают 20%-ным раствором КХ'О3 или КС1, растворяют в воде и охлажденный раствор нейтрализуют по фенолфталеину. Затем раствор нагревают до 80 °C и титруют стандартным раствором щелочи до розового окрашивания фенолфталеина.
Спектрофотометрический метод, основанный на образовании а-кремиемолибденовой кислоты, позволяет определять фторсиликат в присутствии фтористоводородной кислоты, которую связывают А1С13 [3]. Кремнемолибденовая кислота образуется при добавлении к сильнокислому раствору молибдата аммония при экспозиции 3 ч. Поглощение измеряют при 370 нм, закон Бера соблюдается до содержания SiO2 0,8 мг в 100 мл раствора.
Разработан метод экспрессного фотометрического титрования фторсиликата в ваннах хромирования металлов [4]. Раствор титруют нитратом тория в присутствии салицилата железа (III) как индикатора. При введении индикатора протекает реакция:
SiF|- + FeR2+ + 2Н2О 7=± FeF|" + R' + 4Н+ + SiO2
(HR — салициловая кислота). Титрование фторсиликата проходит по уравнению:
2SiF|_ + 3Th4+ + 4Н2О —> 3ThF4 + 8Н+ + 2SiO2
/В конце титрования FeFl’ взаимодействует с Th4+, вытесняя ион Fe3+, который образует с салицилатом окрашенный комплекс. В конечной точке поглощение салицилата железа (III) при 510 нм достигает максимума и при дальнейшем добавлении Th4+ не меняется. Для удержания SiO2 и ThF4 в коллоидном состоянии в раствор добавляют поверхностно-активное вещество.
Определение фторсиликата методом фотометрического титрования
Реагенты
Хлоруксусная кислота. 1,5 М раствор, нейтрализованный NaOH до pH = •» 2,8.
Салицилат железа(Ш), 5-10~3 М раствор.
Полиоксиэтилен сорбитан лаурат, 2%-ный раствор.
Нитрат тория, 0,25 М раствор.
Ход анализа. Отфильтрованный анализируемый раствор (10 мл), содержаний 2—12 г/л S1F2’, нейтрализуют раствором NaOH до pH = 3,0, доводят объ-«И раствора до 100 мл в мерной колбе. К аликвотной части раствора 10 мл при-"Ивают 5 мл раствора хлоруксусной кислоты, 2 мл раствора салицилата же-™*8(Н1) и 2 мл раствора полиоксиэтилен сорбитан лаурата. Полученный рас-титруют 0,25 М раствором нитрата тория до достижения максимума по-г«ОЩеиня при 510 нм.
ф-Примечания. 1. Определению не мешают Сг2О2', а также до 10 г/л ио-t- Ст3+ и Fe3+. 2. С .гибка определения не превышает 2%. 3. При наличии в
89
растворе Fe3+ вводят 1.2 мл 0,03 Л1 раствора салицилата натрия вместо салицилата железа (III). 4. В присутствии избытка Fe1" в растворе вводят 0,1 М раствор ЭДТА.
ЛИТЕРАТУРА
1. Kolthoff !. М., Stenger V. А. — Volumetric Analysis, Vol. II, Second revised edition. New York, Interscience, 1947.
2. Vysotskai/а V. A'.— Zav. Lab., 1971, v. 37, p. 415; Analyt. Abstr., 1972, v. 22, p. 763.
3. Graff P. R., Langmyhr F. J. — Analytica chim. Acta, 1959, v. 21, p. 429.
4. Musha S., Higashino T. — Japan Analyst, 1958, v. 7, p. 156; Chem. Abstr., 1960, v. 54, p. 1122h.
ФОРМИАТЫ
МЕТОДЫ ОТДЕЛЕНИЯ
Отделение формиата обычно проводят дистилляцией или хроматографическим методом. Тонкослойной хроматографией на целлюлозе можно отделить муравьиную кислоту от щавелевой, винной и лимонной с помощью растворителя 2-бутанол — 2 М раствор аммиака или смеси других растворителей [1]. Условия разделения описаны в разделах «Цитраты» и «Оксалаты». Отделение муравьиной кислоты от уксусной и молочной можно проводить на силикагеле G с помощью растворителя пиридин — петролейный эфир (1 : 2) [2].
При разделении формиата и ацетата методом бумажной хроматографии большинство растворителей дают близкие значения Rf, однако насыщенный водой фенол дает четкое разделение [3].
Отгонку с паром часто применяют для предварительного отделения муравьиной кислоты, например при анализе биологических материалов [4]. Последующее спектрофотометрическое определение проводят после восстановления формиата до формальдегида.
При анализе пищевых продуктов формиат отделяют дистилляцией с паром. Затем газохроматографическим методом определяют муравьиную, уксусную, пропионовую и масляную кислоты [5].
Изучена возможность экстракции органических кислот ионом трифенилстаннония Р1тз5п+ с помощью бензола как растворителя [6]. При pH = 2 формиат экстрагируется примерно на 90%.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Метод кислотно-основного титрования
Возможно прямое титрование муравьиной кислоты щелочью (NaOH) в присутствии фенолфталеина как индикатора. Метод неселективен, однако находит применение для обнаружения муравьиной кислоты [7]. Точка эквивалентности отвечает pH = 7,88.
90
Редокс-методы
Для редокс-тптрования муравьиной кислоты предложен ряд окислителей. Кроме всесторонне изученной реакции окисления формпата перманганатом [8], в ходе которой избыток щелочного раствора КМпО4 в присутствии ионов бария (связывающего перманганат) титруют восстановителем, предложен потенциометрический вариант титрования [9]. В связи с неустойчивостью перманганата предложено отделять гидратированный диоксид марганца (IV), образующийся при окислении формиата, восстанавливать его аскорбиновой кислотой до Мп2+, а затем Мп2+ титровать раствором ЭДТА [20]. Изучено влияние pH и времени окисления на полноту протекания реакции [10]. Предложен метод, основанный на растворении образующейся МпО2 в водном растворе пирофосфата натрия, подкислении раствора H2SO4 и титровании комплекса Мп3+ раствором гидрохинона.
Описана методика титрования формиата перманганатом калия [II]. При выбранных условиях удается титровать муравьиную, щавелевую и уксусную кислоту при их совместном присутствии.
Селективное окисление муравьиной кислоты в присутствии уксусной можно проводить растворами Celv. Сумму муравьиной и уксусной кислот определяют редокс-титрованием раствором меди(Ш), по разнице рассчитывают содержание ацетата [12]. При определении муравьиной и щавелевой кислот формиат окисляют избытком раствора Ag11. Оксалат определяют окислением раствором церия(IV) [13].
Метод окисления муравьиной кислоты иодом имеет ряд прей, муществ перед другими методами [14]. Реакция протекает по уравнению
HCOONa + 12 —► Nal + HI + СО2
в присутствии гидротартрата калия. Определению не мешают следующие органические вещества: кальциевые соли масляной, молочной, глюконовой кислот, натриевые соли уксусной, бензойной, борной, винной, фталевой, малеиновой, фумаровой, фенилуксусной, аминоуксусной и адипиновой кислот, не мешают также бензальдегид и глюкоза. Определению мешают натриевые соли щавелевой, салициловой, малоновой кислот, формальдегид, ацетальдегид и салициловый альдегид.
Определение муравьиной кислоты путем окисления иодом [14]
Ход анализа. В колбу вместимостью 250 мл вводят 5—20 мл нейтрального раствора, содержащего 25—90 мг формиата, добавляют 1 г сухого гидротар-грата калня н отмеренный избыток стандартного раствора иода (на х мл 0,1 М Р*-Твора формиата приливают 2х мл 0,1 Л1 раствора иода). Присоединяют об-холодильник высотой 100 см, закрыв его верхний конец перевернутым Г^ааном. Помещают колбу в кипящую воду на 25 мин, дают ей остыть и затем З^гельно обмывают холодильник 5°/о-ным раствором KI. Избыточный нод тн-стандартным 0,1 М раствором тиосульфата Проводят контрольный опыт. ^„Примечание. При определении 23—92 мг формиата ошибка определения ” выше 0,5%.
91
Формиат можно окислять хлоридом ртути, при этом не мешают молочная, масляная, уксусная, пропионовая, винная, лимонная и бензойная кислоты [15]. Рекомендованы и другие окислители: хлорамин Т [16], тетраацетат свинца [17], щелочной раствор феррицианида [18].
Газохроматографический метод
Формиат можно определять газохроматографическим методом [5]. Муравьиную кислоту и формиаты можно определять в присутствии минеральных и карбоновых кислот. Анализируемый раствор подкисляют соляной кислотой, вводят ароматический амин (анилин, о-, или n-толуидин и др.) и кипятят с обратным холодильником. Продукты конденсации определяют газохроматографическим методом [19].
Спектрофотометрические методы
После восстановления формиата магниевой стружкой образующийся формальдегид определяют путем конденсации с хромотроповой кислотой [4, 21]. Для определения микроколичеств формиата применяют также 6-амино-1-нафтол-3-сульфокислоту и 6-анилино-1 -нафтол-3-сульфокислоту [22].
Разработан метод определения муравьиной кислоты в природных водах [21]. Закон Бера соблюдается до содержаний формиата 20 мкг, чувствительность определения 1 мкг, не мешают фенол при содержании более 500 мкг/л н 10-кратный избыток органических кислот. В работе [23] отмечено, что восстановление формиата протекает всего на 30%. Воспроизводимость метода невысока, ошибка может достигать 30% [24]. Определению муравьиной кислоты мешает формальдегид, его следует отделять или связывать, например, фенилгидразином [23]. Разработан метод, основанный на восстановлении нитрата серебра до металлического серебра в слабокислом буферном растворе [24];
HCOOH + AgNO3 —* HCOOAg + HNO3 2HCOOAg —> НСООН + CO2 + 2Ag|
В присутствии защитного коллоида, например крахмала, раствор окрашивается в коричневый цвет, причем поглощение пропорциО' нально содержанию формиата. Относительное стандартное отклонение при доверительной вероятности 0,95 составляет ±4%.
При разложении муравьиной кислоты или формиатов в сред? концентрированной H2SO4 при 100 °C выделяется СО, которЫ" определяют спектрофотометрическим методом после взаимодейси. вия СО с п-сульфамоилбензоатом. Оксалат не мешает опреДе‘[ лению. 1
92
Другие методы определения
Возможно неселективное определение муравьиной кислоты по реакции:
1О3' + 51 + 6Н+ —> 312 + ЗН>0
однако для завершения реакции необходимо 30 мин [25].
Методом ядерного магнитного резонанса можно определять более 0,05% муравьиной кислоты [26].
Опубликованы стандартные методы определения муравьиной кислоты [27].
ЛИТЕРАТУРА
1. Dittman J.— J. Chromast., 1968, v. 34, p. 407.
2. Prey I7., Berbalk H., Rausz M. — Mikrochim. Acta, 1962, p. 449.
3. De Jong E., McCullough T. — Chemist Analyst, 1967, v. 56, p. 80.
4. Rietbrock N., Hinrichs W.-D. — Klin. Wschr., 1964, v. 42, p. 981; Analyt. Abstr., 1966, v. 11, p 831.
5 Shelley R. N., Salwin H Horowitz W.— J. Ass off. agric. Chem., 1963, v. 46, . ‘ p. 486.
6. Bock R., Niederauer H.-Т., Behrends K. — Z. analyt. Chem., 1962. v. 190, p 33.
7. British Pharmacopoeia. H. M. S. O. London 1973, A25.
8. Stamm H. — Angew. Chem., 1934, v 47, p. 791.
9. Tatwawadi S. V.— Z. analyt. Chem., 1959, v. 168, p. 15.
10. Berka A., Konopasek /.-—Mikrochim. Acta, 1968, p. 405.
11. Polak H. L. — Z. analyt. Chem., 1960, v. 176, p. 34.
12. Jaiswal P. K., Chandra S, — Microchem. J., 1969, v. 14, p. 289.
13. Jaiswal P. l(., Singh V. N. — Chim. analyt., 1971, v. 53, p. 253.
14. Verma R. Al., Bose S. — Analytica chim. Acta, 1962, v. 27, p. 176.
15. Verma R. M., Bose S. — J. Ind. Chem. Soc., 1960, v. 37, p. 47.
16. Singh B., Sood I\. C. — Analytica chim. Acta, 1955, v. 13, p. 305.
17. Perlin A. S. — Analyt. Chem., 1954, v. 26, p. 1053.
18. Deshmukh G. S., Rao A. L. J.— Z. analyt. Chem., 1963, v. 194, p. 110
19. Omeh E. O. — J. Chromat., 1970, v. 51, p. 139.
20. Flaschka H., Garrett J. — Chemist Analyst, 1963 v. 52, p. 101.
21. Страдомская А. Г., Гончарова И. A. — Гидрохимия, матер., 1967, т. 43, с. 57; Analyt. Abstr., 1968, v. 15. p. 6338.
22. Sawicki E., Hauser T. R., McPherson S. — Analyt. Chem., 1962, v. 34, p. 1460.
23. Grant W. H. — Analyt. Chem., 1948, v. 20, p. 267.
24. Higgs D. G., Charles A. F. — Analyst, 1971, v. 96, p. 502.
25. Koithoff 1. M. — Pharm. Weekbl., 1920, v. 57, p. 63.
«6. Burakevich J. V., O’Nell J. — Analytica chim. Acta, 1971, v. 54, p. 528.
«. Britisch Standards 4341: 1968. London. Also addendum No. 1 (1971) to this specification.
ГЕКСАЦИАНОФЕРРАТЫ(П) И (III)
Гексацианоферрат(П) (ферроцианид) натрия или калия наводит применение в фотографии, при обработке металлов, в галь-. *®Нотехнике, при гравировании, дублении, крашении и окраске Ц®ией- ГексацИаноФеРРат(Ш) (феррицианид) применяют для .Х?В°ДСТва синих пигментов. Соли щелочных металлов приме*
Ют в различных областях техники, в том числе в качестве ре* ,*jr ятора размера кристаллов хлорида натрия. Гексацианофеп-
93
рат(Ш) калия хорошо поддается очистке, поэтому его применяют в качестве вторичного стандарта при редокс-тптрованиях [1]. Например, потенциометрическое титрование кобальта стандартным раствором KsFe(CN)6 в аммиачной среде, вероятно, является наиболее точным методом определения кобальта.
Применение в анализе гексацианоферрата(III) как окислителя дано в обзоре [3]. Однако обзоры методов определения ферро-и феррицианидов в последнее время не публиковались.
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение гексацианоферратов
Гексацианоферрат(П) можно количественно отделить осаждением солями Ag1, Pb", Zn!l. Гексацианоферрат(II) можно отделить от гексацианоферрата(Ш), цианида, роданида и определить гравиметрически в виде (NH4)4Fe(CN)6-2Mo03.
Хроматографические методы
Условия отделения гексацианоферратов(II) и (III) методом тонкослойной хроматографии приведены в табл. 5.
Таблица 5. Отделение гексациаиоферратов (II) и (III) методом тэикослойиоб хроматографии
Носитель Растворитель Разделяемые ионы Литература
Силикагель Ацетон — вода (Ю:1) SCN", Г, ВГ, СГ, Fe(CN)’’, Fe(CN)J-, сю;, Bro;, ю;, no; 4
Силикагель Бутанол — пропанол — дибутнламин (9 : 9 : 2) F-, СГ, Вг”, Г, CN’, SCN', Fe(CN)j', Fe(CN)’- 5
Силикагель на алюминиевой фольге Бутанол — этанол — вода (20 : 60 : 20) CN', Fe(CN)-’, Fe(CN)g' 6
Методом распределительной хроматографии с обращенной фазой можно разделить Fe(CN)e' и Ре(СЫ)б"[7] при использовании в качестве стационарной фазы порошка Kel-F и растворителя три-бутилфосфата.
Другие методы отделения
В определенных условиях можно провести дистилляцию цианида без разложения Fe(CN)e~ и Fe(CN)e' (см. раздел «Цианиды»). Для разложения комплексных цианидов применены рас
94
творы MgClo, HgCl, в среде H2SO4 [8]. При этом образуются Mg(CN)2 и Hg(CN)2.
Гексацианоферрат(Ш) можно отделить термическим методом. Возможно селективное осаждение Fe(CN)e’солью тория [9]. Последующее количественное определение основано на колориметрировании роданидных комплексов железа. Описание печи для термического отделения Fe (CN)^ приведено в работе [10].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Гексацианоферрат (II) можно определять гравиметрически в присутствии Fe(CN)r> цианида и роданида [11]. Осаждение ведут молибдатом аммония из ацетатного буферного раствора с pH = 3,7. Раствор с осадком выдерживают в течение нескольких часов, фильтруют. Осадок сушат при 120 °C и взвешивают в виде (NH4)4Fe (CN) 6-2МоО3. Метод позволяет определять 300 мг Fe(CN)g’ с ошибкой около 1%.
Титриметрические методы
Редокс-методы
Для определения гексацианоферрата(III) предложено большое число реагентов. В монографии [3] описано 17 реагентов, в том числе аскорбиновая кислота, сульфат гидразина, нитрит натрия, хлорид олова(II) и нитрат ртути(I), однако некоторые из них не представляют практического интереса. Ниже рассмотрены методы титрования с участием аскорбиновой кислоты, церия(IV), тита-иа(Ш) и иодида.
Гексацианоферрат(III) взаимодействует с KJ в кислой среде, при этом выделяется иод, который можно оттитровать стандартным раствором тиосульфата:
2Ее(СМ)Г + 2Г —> 2Fe (CN)^ + I2
Реакция протекает медленно, однако в присутствии сульфата Цинка ее скорость увеличивается. Ионы цинка осаждают Fe(CN)e' и Fe(CN)|', однако растворимость гексацианоферра-та(П) значительно ниже, что сдвигает равновесие вправо.
Иодиметрическое определение гексацианоферрата(Ш)
Ход анализа. К раствору Fe (CN)g*,разбавленному до объема 40 мл в колбе со стеклянным шлифом, добавляют 2 г КД, 2 мл 4 Л1 раствора НС1 и 10 мл тЬ-иого раствора ZnSO4. Закрывают колбу, перемешивают и оставляют на конечной^ "ЮТ стандаРтным раствором тиосульфата, добавляя крахмал вблизи зована для стандартиза-прнготовлен по точной
примечания. 1. Методика может быть исполь иавескеС1^реа(С™0С^ЛЬ^аТа’ если гексацнан0ФеРРат
95
2. Иоднметрический метод рекомендован для испытания гексацианоферрата, применяемого в фотографии [12].
Для прямого титрования гексацианоферрата (II) применяют раствор Ce'v в 1 М НС1 или H2SO4. Цианиды не мешают:
2Се (SO4), + 2K4Fe (CN)e —> 2K3Fe (CN)6 + Ce (SO4)3 + K2SO4
В качестве индикатора применяют перхлорат ферроина, при использовании сернокислой соли конец титрования может быть нечетким [13]. Этот метод применен для титрования Fe(CN)j" в смеси с СГ [14]. Сначала определяют гексацнаноферрат(П), а затем в аликвотной части раствора — сумму Fe(CN)e и СГ путем титрования раствором AgNOs в присутствии хромата как индикатора.
Гексацианоферрат(Ш) можно определять титрованием аскорбиновой кислотой, определяя конечную точку визуально или потенциометрически [15,3]:
CeHeO6 + 2Fe(CN)r —> СсН6Об + 2Fe (СХ1)Г + 2Н+
Определение проводят в буферном растворе ацетата натрия или бикарбоната калия. При визуальном титровании применяют в качестве индикатора 2,6-дихлорфенолнндофенол, конечную точку определяют по исчезновению синей окраски раствора.
Определение гексациаиоферрата(Ш) титрованием
стандартным раствором аскорбиновой кислоты
Реагенты
Аскорбиновая кислота, 0,05 М раствор. Готовят растворением 8,90 г кристаллической аскорбиновой кислоты в дистиллированной воде с добавлением 0,5 г ЭДТА иа 1 л раствора. Раствор хранят в темноте.
2,6-Дихлорфенолиндофенол. Готовят смесь индикатора с NaCl (1:500), которая весьма устойчива. Раствор индикатора в воде (0,1%-ный) устойчив в течение 3 недель.
Ход анализа. К нейтральному раствору, содержащему 50—600 мг Fe (CN);]", добавляют 1—2 г твердой соли КНСО3 (или NaHCO3) и около 0,5 г смесн индикатора с NaCi. Титруют 0,05 М раствором аскорбиновой кислоты до исчезновения голубой окраски раствора.
Примечания. 1. Разложение аскорбиновой кислоты ускоряется ионами тяжелых металлов и облучением раствора. ЭДТА маскирует иоиы металлов и замедляет разложение аскорбиновой кислоты.
2. Раствор аскорбиновой кислоты можно стандартизировать по указанной методике по KaFe(CN)6 как вторичному стандарту.
3. Относительное стандартное отклонение метода ±0,2%.
Титрование гексацианоферрата аскорбиновой кислотой применяют при косвенном определении ряда веществ. Некоторые окислители, например хроматы, взаимодействуют с избытком гекса-цианоферрата(П), в результате чего образуется эквивалентное количество Fe (CN)e’, которое можно оттитровать аскорбиновой кислотой [17, 18]. Пероксид водорода количественно восстанавливает Fe(CN)e~ в щелочной среде. После нейтрализации раствора остаток гексацианоферрата (III) титруют аскорбиновой кис-
96
лотом. Приведено много примеров косвенного опредетенпя ряда веществ п>тем титрования Fe(CN)^' аскорбиновой кислотой.
Для титрования Fe(CN)6* предложено применять растворы титана (III) в глицерине в присутствии муравьиной кислоты [20]. Растворы устойчивы на воздухе в течение 4 суток. Титрование ведут в щелочной среде, конец титрования определяют визуально или потенциометрически.
Метод осадительного титрования
Гексацпаноферрат(Ш) можно титровать раствором AgNO3 при pH — 6,5 — 8,0 в присутствии хромата калия как индикатора [21]. Интервал определяемых содержаний Fe(CN)]' составляет 15—300 мг с точностью +0,5 мг. Метод применен для анализа смеси СГ и Fe (CN),- .
Гексацианоферрат(II) можно титровать раствором тория(IV) с применением ализаринового красного С, ксиленолового оранжевого или пирокатехинового фиолетового в качестве адсорбционных индикаторов [22J. Определению не мешают СГ, Вт”, Г, NO3 и ацетаты, мешают Ag”, Pb‘+, Zn2+, Cd2+, Cu2+, Со2+, Ni2*» Fe(CN)r, POt, СгОГ, IO3 и F".
Комплексометрический метод
Описан метод определения гексацианоферрата(III), основанный на восстановлении его иодидом, осаждении гексацианоферрата (II) в виде Zn2Fe(CN)6 и определении остаточного цинка(II) с помощью ЭДТА [23]. Описан метод определения нерастворимых в воде гексацианоферратов цинка, кобальта, никеля и меди. Соли растворяют в избытке ЭДТА, который титруют стандартным раствором свинца(II) [24].
Спектроскопические методы
Спектрофотомет рические методы в видимой и ультрафиолетовой областях
Ряд спектрофотометрических методик основан на образовании окрашенных комплексов при взаимодействии гексацианоферрата (II) и железа(III). Разработанные ранее методы [25] позволяют определять более 50 мкг Fe(CN)e'. Закон Бера соблюдается, если поглощение измерять через I мин после смешения реагентов. Модифицированный метод [26] основан на измерении поглощения при 690 нм и позволяет определять 5—80 мкг Fe(CN)e'. Описан метод анализа гексацианоферрата после предварительного концентрирования на кизельгуре [27]. Метод пригоден для анализа воды и растворов с чувствительностью 0,10 ppm, а с некоторыми изменениями даже на уровне 0,013 ppm. Определению
4 Зак. 167
97
Fe(CN)s' не мешают при сравнимых содержаниях ионы металлов, цианиды, роданиды и нитропруссид.
Показано, что гексацпаноферрат(П) взаимодействует с Fe11 в присутствии 4,7-дпфенил-1,10-фенантролина сернокислого. Образующийся комплекс характеризуется значением г = 20 500 при 535 нм, что позволяет определять 1 • 10~9 моль Fe (CN)e’ [28].
Гексацианоферрат(П) .можно определять по его собственному поглощению в УФ-области, при 237 нм [29]. Этот метод менее чувствителен, чем основанный на поглощении берлинской лазури, тем не менее он позволяет определять 1 • 10~5 моль Fe(CN)j' в присутствии 50-кратных количеств Fe(CN)ti . Определению мешают вещества, поглощающие в УФ-области.
Другой метод спектрофотометрического определения гексацианоферрата (II) основан на его разрушении и определении Fe11 с помощью 1,10-фенантролина. Разработан метод определения 10~5—10 ’4 моль Fe (CN)l’ [30]- Вероятно, чувствительность определения можно повысить, применив 4,7-дифенил-1,10-фенантролин.
Чувствительный спектрофотометрический метод определения окислителей использован для определения гексацианоферрата (III). Метод основан на окислении 1~ или Вг~ до 1+ или Вг+, которые, взаимодействуя с трифенилметановыми красителями (бриллиантовым зеленым, пирокатехиновым фиолетовым) при pH = 3 — 5, снижают поглощение растворов при 628 и 594 нм соответственно [31]. Интервалы определяемых содержаний Fe(CN)i" равны соответственно 0,021—0,64 и 0,02—0,428 мкг/мл.
Турбидиметрический метод
Описан турбидиметрический метод определения гексацианоферрата (III), основанный на его взаимодействии с триоксалато-кобальтатом (III) калия [32].
Спектроскопический метод в инфракрасной области
Ионы Fe(CN)e' и Fe(CN)g' имеют характерные полосы поглощения в ИК-областИ, в интервале 2100—2000 см“! [33]. Для К4ре(СЫ)б-ЗН2О характерны очень интенсивные полосы 2015 и 2040 см-1, а для КзРе(С1\Г)6 — интенсивные полосы 2100 и 2115 см-1. Предложен метод идентификации и определения микроколичеств гексацианоферрата (И) и (III), кобальтицианида, сульфата, хромата и метаванадата в смеси с использованием КВг [34]. Содержание Fe(CN)e’ и Fe(CN)e’ не должно превышать 20 и 5 мкг соответственно.
Трудность определения гексацианоферрата(II) и (III) в водных растворах связана с отсутствием водостойких материалов, прозрачных в ИК-области, пригодных для изготовления кювет. Хорошие результаты получены при изготовлении окон кюветы из Иртрана [35]. Спектры поглощения гексацианоферрата (II) и
98
Рис. 17 Инфракрасный спектр 8,0 10-’ М ра-Х™ппа гексацианоферрата (III) калия (]) и вк in-» М раствора гексацианоферрата(П) на-?рия (2) [35].
(III), полученные при использовании таких кювет, показаны на рис. 17.
Для растворов Na4Fe(CN)6 градуировочный график линеен до содержаний 0 04 М, значение е = 4,24-103 л-моль-1 • • см-1. Интервал определяемых содержаний 3-Ю-4—4 • 10-2 М. Интервал
0,0012—0,1 М, значе-
определяемых содержаний Fe(CN)iT равен ние 8 1Д8- 103 Л-МОЛЬ-1-СМ-1.
Электрохимические методы
Потенциометрический метод
Гексацианоферрат(Ш) можно титровать потенциометрически в 25—60%-ном растворе К2СО3 в инертной атмосфере [36]. При титровании используют платиновый и каломельный или два платиновых электрода (дифференциальный метод). Конечную точку можно устанавливать также амперометрически.
При потенциометрическом титровании гексацианоферрата(II) раствором КМпО4 или Ce(SO4)2 используют платиновый анод и катод из Au, W, Pd, Ag или Ni [37]. В качестве титранта применяют тетраацетат свинца [38]. Титрование ведут в среде НС! или НС1О4, реакция протекает количественно в присутствии сравнительно больших количеств хлорида. Определению не мешают Цианиды и Fe (CN)e’, но мешают фториды.
Гексацианоферрат(III) можно титровать перхлоратом ртути (I) [39] в присутствии KSCN в буферном растворе:
Метод позволяет определять или (II) после окисления до
2Fe (CN)|- + Hg22+ + 8SCN' —> 2Fe (CN)|" + 2Hg (SCN)2-
Метод титрования Fe(CN)e’ аскорбиновой кислотой описан выше. Титрование проводят потенциометрически с помощью пла-1 чинового и каломельного электродов [16]. Описан автоматический вариант микротитрования [40].
1 Ю мг гексацианоферрата (III) Fe(CN)g нитритом натрия.
Кулонометрический метод ^^Гексацианоферрат (II) можно Kdi"V"“CM ег
U-? 141]. При потенциале ниже 0,7 В определению не мешают Тпп ИДЫ’ ХЛ°РИДЫ’ ЦианиДЬ1- н0 мешают иодиды и роданиды. Ме-, Позволяет определять 50—150 мг Fe(CN)e B 100 мл раствора.
сксацианоферрат(11) можно определять кулонометрическим »1?СЛением его на платиновом электроде в среде 0,5 М раствора
99
Другой кулонометрический метод основан на электрогенерировании октацианомолибдата [42]:
Мо (СХ)Г + Fe (CN)]' —» Мо (CN).*' + Fe (С\)Г
Реакцию можно проводить в широком интервале pH.
Другие электрохимические методы
Показано, что амперометрическое титрование Fe(CN)a' раствором перхлората ртутп(1) с применением вращающегося платинового электрода дает точные результаты [43]. Изучены источники ошибок при титровании малых количеств [44]. В результате дальнейших исследований предложен простой биамперометрический метод с применением двух платиновых электродов [45].
Раствор тетраацетата свинца использован для титрования Fe(CN)e’ в среде НС1 или H2SO4. В работе применяют платиновые электроды [46]. Смесь ионов Fe(CN)g’ и Fe(CN)s' можно определять методом вольтамперометрии [47]. Пик анодного или катодного тока на пастовом графитовом электроде пропорционален концентрациям Fe(CN)l" и Fe(CN)e в интервале 10~5 — 10~3 М при отношениях ионов от 1 : 5 до 5 : 1.
Другие методы определения
Известны хроматографические методы определения гексацианоферрата (II) и (III) [9].
Описан метод определения 3—60 MKrFe(CN)e' с применением бумаги, пропитанной хроматом серебра [48]. Содержание Fe(CN)e' определяют с точностью ±1% субмикрогравиметриче-ским методом, по массе пятна Ag3Fe(CN)6 путем сравнения с пятнами, полученными на стандартной бумаге.
Для полумикроопределения гексацианоферрата(III) применен метод кольцевой печи [49]. Для количественного определения в качестве стандарта применен сульфид серебра. Новый вариант метода описан в работе [50]. Определение заканчивают титрпмет-рически. Приведена схема автоматизации метода.
ЛИТЕРАТУРА
1. Kolthoff I. М., Belcher R.— Volumetric Analysis, Vol. Ill, New York, Inter-science, 1957.
2. Lingane J. J. — Analytica chim. Acta, 1964, v. 30, p. 319.
3. Berka A., Vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Perganion Press, 1965.
4. Kawanabe R., Takitani S., Mirazaki AL, Tamara Z.—Japan Analyst, 1964, v. 13, p. 976; Analyt. Abstr., 1966, v. 13, p. 5385.
5. Gagliardi E., Pokorny G. — Mikrochim. Acta, 1965, p. 699.
6. Thielemann H. — Z. analyt. Chem., 1974, v. 270. p. 128- Analyt. Abstr., 1975, v. 28, p. 2B86.
7. Akaza I., Kiba T., Taba M. — Bull. chem. Soc. Japan, 1969, v. 42, p. 1291: Analyt. Abstr., 1970, v. 19, p. 1289.
8. Serfass E. J., Freeman R. B., Dodge B. F., Zabban IF. — Plating, 1952 v 39, p. 267; Chem. Abstr., 1952, v. 46, p. 6514a.
100
9 Leonard Л1- A., Shahine S. .4. A. R. Wilson C. L. — Mikrochim. Acta, 1965,
10 ieonard AL <4-, Shahine S. A. E. F.. WiRoa C. L.— Mikrochim. Acta, 1965,
11 Jimeno S. A.— Inf. Quim. Anal.. 1959, v. 13. p. 167, Analyt. Abstr., 1960, v. 7,
' p 3253.
12 British Standards 3/o2; London. 1964.
n Belcher R., batten A. /. — Quantitative Inorganic Analysis, 3rd edn., London, Butterworths, 1970, p. 239.
14 Rao К В., Rrishnamurthy С. I'.— Ind. J. appl. Chem., 1968, v. 31, p. 108; Analyt. Abstr., 1970, v. 19. p. 210.
15 Erdey L., Svehla G. — Chemist Analyst, 1963. v. 52. p. 24.
[6 Erdey L., Svehla G.— Z. analyt. Chem., 1956, v. 150, p. 407; Analyt. Abstr., ’ 1956, v. 3, p. 3055.
17 Erdey L., Svehla G.~Z. analyt. Chem., 1958, v. 163, p. 6; Analyt. Abstr., 1959, v. 6, p. 1620.
18 Erdey L., Svehla G. — Z. analyt. Chem., 1959, v. 167, p. 164; Analyt. Abstr., 1960, v. 7, p. 122.
jo Erdey L., Svehla G., Koltai L.— Analytica chim. Acta, 1962, v. 27, p. 164.
20. " Ruzicka E. — Chemicke Zvesti, 1972, v. 26, p. 516; Analyt. Abstr., 1973, v. 24,
p. 2789.
21 Bashkara R.— Reel. Trav. chim. Pavs-Bas., 1965, v. 84, p. 69; Analyt. Abstr., ' 1966, v. 13, p. 2283.
22. Dubey P. S., Tandon R. R. —Tai anta, 1966, v. 13, p. 765.
23. Вильборг С. С., Дроздов В. A. — Научи, докл. высш, школы, Хим. и хим. технол. 1958, с. 721; Analyt. Abstr., 1960, v. 7, р. 865.
24. Krtil J.— Cooln Czech, chem. Commun. Engl. Edn., 1967, v. 39, p. 4496; Analyt. Abstr., 1969, v. 16, p. 1870.
25. Lea С. H.— Analyst, 1947, v. 72, p. 336.
26. Marler !. R., Clark D. S. — Analyst, 1960, v. 85, p. 574.
27. Roberts R. F., Wilson R. H.— Analyst, 1968, v. 93, p. 237.
28. Avron M., Shavit N. — Analyt. Biochem., 1963, v. 6, p. 549.
29. Kidby D. R.— Analyt. Biochem., 1969, v. 28, p. 230.
30. Karas-Gasparec V., Pinter T. — Croat. Chem. Acta, 1962, v. 34, p. 131; Analyt.
Abstr., 1963, v. 10, p. 1804.
31. Раманаускас E. И., Буникене JI. В., Сапрогонене M. С. и dp. — ЖАХ, 1969,
I. 24, c. 244; Analyt. Abstr., 1970, v. 19, p. 1022.
32. Drew H. D., Fitzgerald J. M.— Analyt. Chem., 1966, v. 38, p. 778.
33. Miller F. A., Wilkins С. H. — Analyt. Chem., 1952. v. 24, p. 1253.
n‘ Haba F. R., Wilson C. L. — Taianta, 1962, y. 9, p. 841.
Drew D. M. — Analyt. Chem., 1973, v. 45, p. 2423.
Furman N. H., Fenton A. J. — Analyt. Chem., 1960,.v. 32, p. 745.
Ueno K., Tachikawa T. — J. chem. Soc. Japan, pure Chem. Sect., 1961, v. 82, P- 577; Analyt. Abstr., 1962, v. 9, p. 4211.
Толар В., Сантручек И. Немец И., Зыка И.—ЖАХ, 1967, т. 22, с. 664;
Analyt. Abstr., 1969, v. 16, р. 145.
Lucena-Conde F., Sanchez-Bellido I. — Taianta, 1958, v. 1, p. 305.
Erdey L., Svehla G., Weber O. — Z. analyt. Chem., 1968, v. 240, p. 91; Analyt.
Abstr., 1970, v. 18, p. 12.
ErishnanV. R. — Z. analyt. Chem., 1967, v. 231, p. 192, Ana-
Lucena-Conde F. —Taianta. 1969, v. 16, p. 1114.
Microchem. J., 1971, v. 16. p. 77.
Analytica chim. Acta, 1971, v. 53, p. 233.
Analyt. Abstr.,
p. 778.
34.
35.
36.
37.
38.
39.
40.
41.
49 J?*- A"bsir ’ 1968, v. 15, рЛг'бк ernandez-Mendez J.. Lucena-ьопае r. — laianta.
MerrerR J., stock J. T. — Analyst, 1967, v. 92, p. 98.
**• Stock J. T„ Merrer R. J.
tc д^ГГег I-' Stock J. T. — Analytica chim. Acta, 19/1, V. oJ, p. io™* ik K-’ Сираканян AL A. — ЖАХ, 1970, t. 25, c. 1677;
47 Fa/’ V' 22’ P- 4004-
G Tomcsanyi L. — J. electroanalyt. Chem., 1967, v. 13, p. 73.
u° n "' Getcheva T., Mandova B.—Mikrochim. Acta, 1974, p. 503.
ЯЖК W Weisz H- - Mikrochim Acta, 1962, p. 24.
’’ Goenner M. — Analytica chim. Acta, 1968, v. 343, p. 235.
ГИПОНИТРИТЫ
МЕТОДЫ ОТДЕЛЕНИЯ
Анионообменный метод
Гипонитрит NjOj’, нитрат, нитрит и Х-нитрогпдроксплампнат-анионы можно разделить на анионите [1]. Смолу перед анализом переводят в гидроксо-форму. Ионы элюируют раствором 1 А! Na2SO4 —0,1 Л1 ХаОН.
Методы бумажной хроматографии
и электрофореза на бумаге
Гипонитрит можно отделить от нитрата, нитрита и гидроксиламина методом бумажной хроматографии [2]. Разделение проводят на бумаге Ватман № 1 с помощью растворителя этанол — Н2О— NaOH (70 мл : 30 мл : 2 г). Методом электрофореза на бумаге можно разделить гипонитрит, гипонитрат, нитрат, нитрит, НС1, NH4C1, хлорид гидразина и гидроксиламин солянокислый [3].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Умеренные концентрации H2N2O2 в кислой среде можно определять путем отделения осадка гипонитрита серебра [4]. Избыток серебра в растворе титруют обычным методом.
Определение гипонитрита аргентометрическим методом
Ход анализа. К аликвотной части раствора H2N2O2 объемом 25 мл приливают 25 мл 0,1 М раствора AgNO3. Сразу же нейтрализуют 0,1 AI раствором NaOH до начала осаждения Ag2O, который растворяют несколькими каплями 0,1 М раствора уксусной кислоты. Приливают 5—10 мл ацетатного буферного раствора (0,1 М СН3СООН и 0,1 М CH3COONa) и доводят водой объем раствора в мерной колбе до 100 мл. В полученном растворе с pH = 4,5—5,0 гипонитрит серебра практически нерастворим, a Ag2O и Ag2CO3 растворимы.
Фильтруют раствор, отбрасывая первые порции фильтрата. В аликвотной части фильтрата определяют Ag+ (методом Фольгарда) и рассчитывают содержание гипонитрита в исходном растворе.
Спектроскопические методы
Молярный коэффициент поглощения N>O2" в кислой среде при 232 нм равен (6,5 ± 0,2) • 103 [5]. Это значение выше, чем приведенное в работе [6] и равное 4,0-103. При измерении поглощения в области 232 нм закон Бера соблюдается до концентраций N2O2’ 1,5 • 10 4 М.
Гипонитрит можно определять после переведения его в нитрит спектрофотометрическим методом Грисса [7]. Гипонптри'1 102
окисляют в щелочном растворе КМ11О4 в присутствии ионов Ag? иЧц Си2+. Избыток AlnOi и МпО? удаляют восстановлением формиатом натрия, а ХО2 определяют спектрофотометрически с помощью 1-нафтплампна. Метод позволяет определять 005 мкмоль N2Oj'co средней погрешностью 1,5%.
Разработан метод определения ХАОС в присутствии других азотсодержащих ионов [8, 9]. Гпдроксилампн, аммонии и другие азотсодержащие основания удерживаются сильнокислотным катионитом при 5 °C, а гипонитрит проходит, не задерживаясь. Описан вариант определения гипонитрита в присутствии нитрата, нитрита и нитрогпдрокспламината.
ЛИТЕРАТУРА
1 Guertler О., Holzapfel И. — Angew. makromol. Chem., 1969, v. 7, p. 194; Ana-’ lyt. Abstr., 1971, v. 20, p 960.
2. Stevens H. M. — Analytica chim. Acta, 1959, v. 21, p. 456.
3. Veprek-Siska I., Smirous F., Pliska V., Vesely F. — Chemike Listy, 1958, v. 52, p. 410; Analyt. Abstr., 1959, v. 6, p. 115.
4 Polydoropoulos C. N., Pipinis Л1. — Hem. Hron., 1963, v. 28, p. 107; Chem. ' Abstr., 1964, v. 60, p. 4800c.
5. Polydoropoulos C. N., Voliotis S. D. — Analytica chim. Acta, 1968, v. 40, p. 170.
6. Addison С. C., Gamlen G. A., Thompson R.— J. chem. Soc., 1952, p. 338.
7. Holzapfel H., Gurtler O. — J. prakt. Chem., 1967, v. 35, p. 59; Analyt. Abstr., 1968, v. 15, p. 3878.
8. Holzapfel n., Gurtler O. — J. prakt. Chem., 1967, v. 35, p. 68; Analyt. Abstr., 1968, v. 15, p. 3878.
9. Holzapfel H., Gurtler O. — J. prakt. Chem., 1967, v. 35, p. 70; Analyt. Abstr., 1968, v. 15, p. 3878.
применение в черной и цветной
МОЛИБДАТЫ
Молибден находит широкое
металлургии, поскольку добавление его в сплавы увеличивает их износостойкость, коррозионную стойкость и прочность. Молибден применяют в электронике и для изготовления высокотемпературных сплавов.
Роль молибдена как существенного микрокомпонента в высших растениях установлена в 1939 г. Содержание следов молиб-Дена определяют в растениях, животных организмах, воде и гео-огических пробах, в том числе почвах.
уШ,Ля определения молибдена используют косвенные методы д2п!аСТИем микРоколичеств ряда других анионов. Молибден опре-провоТ В виде гетерополикислот фосфора, мышьяка и кремния, оппеде*1 ИХ экстРакпию (при необходимости). Затем молибден методомЯЮТ спектР°ФотометРИческим или атомно-абсорбционным сЪед^°ЛИбДаТ'ИОН склонен к полимеризации. В сильнощелочной ЦМц М0ПлРиеб°бЛаДаеТ МоОГ. в слабокислой среде при конденса-И0Дата образуются полианионы, например димолибдат ’ гептамолибдат МО7О24 [известная соль гептамолибдата
(NH4)6Mo;O24-4H2O]. Обзор химических свойств молибдена в связи с методами его определения дан в работе [1].
Наиболее распространенной формой является Movi. Вторпч ные стандарты Movi готовят из молибдата натрия дигпдрата, геп тамолибдата аммония тетрагидрата пли молибденового ангид ряда.
Аналитическая химия молибдена рассмотрена в монографиях Элвелла и Вуда [2] и Бусева [3], опубликованы также обзорь К 5].
МЕТОДЫ ОТДЕЛЕНИЯ
Большинство методов определения молибдена неселективно поэтому проводят предварительное отделение молибдена. Обзор методов отделения приведен в монографии Коркиша [6]. Обсуж дены методы экстракции, соосаждения, ионного обмена и другие хроматографические методы. В последних работах большое внимание уделено экстракции растворителями.
для полного отделения необходимо
Осаждение
Растворимость молибдена (VI) в щелочной среде может быть использована для отделения его от ионов, которые в этих условиях осаждаются. Обычно повторное осаждение.
В присутствии ЭДТА и 8-гидроксихинолина в ацетатном буферном растворе молибдат осаждается в виде комплекса с оксином [7, 8]. Вследствие низкой устойчивости комплексов MoVI с ЭДТА молибден можно отделять от Fe2+, Al111, Be111, Zn2+, Ni"+, Co2+, Mn2+, Pb2+, Cd2+, Bi1", Cu2+, Hg2+ и V,v. Мешают ионы Ti, W, V и U.
Бензоин-а-оксим осаждает Movi, WVI, CrVl, Vv, Nbv, Pd11 и Tav из сильнокислых растворов. Это можно использовать для отделения Movi от элементов, которые не осаждаются бензоин-а-оксимом [9].
В ряде случаев можно осадить мешающий ион, например цинхонином осаждают вольфрам^!), а молибден^!) при этом остается в растворе.
Для отделения и определения молибдена в качестве осадителя предложен сульфид [10]. Показано, что осаждение Mov происходит полнее, чем Movi, и посторонние ионы соосаждаются в меньшей степени. Поэтому Movl предварительно восстанавливают кипячением с сульфатом гидразина в течение 3—4 мин.
Ионообменный метод
Молибден(VI) в слабокислых нейтральных и щелочных растворах существует в анионных формах и не поглощается на сильнокислотном катионите, поэтому можно отделить его от большого
104
числа металлов. Чтобы избежать осаждения малорастворимых молибдатов тяжелых металлов в слабо кислой среде, в раствор необходимо вводить комплексообразователи.
Поглощение молибдена(VI) на анионитах можно использовать для его отделения и предварительного концентрирования. Используя в качестве анионита диэтиламиноэтилцеллюлозу, можно разделить Movi, Revl1 и WVI. Разделение проводят в роданидных растворах. Порядок сорбции комплексов на анионите следующий:
> Movi > Rev”. Разделение металлов достигается выбором концентрации роданида и pH раствора [11, 12].
В работе [13] описано отделение и предварительное концентрирование следов молибдена в воде на анионитах. Затем молибден определяют методом атомной абсорбции. ч
Методы бумажной хроматографии и электрофореза
Для отделения молибдена используют также методы бумажной, тонкослойной хроматографии и электрофореза. Методом бумажной хроматографии можно отделить Movi от WVI [14], а методом электрофореза на бумаге MoVI от Crvl [15]. Условия разделения некоторых ионов методом тонкослойной хроматографии даны в табл. 6.
Таблица 6. Условия отделения MoVI методом тонкослойной хроматографии
Адсорбент Растворитель Отделяемые ионы Литература
Силикагель и поливиниловый спирт (связующий компонент) 1 М HSCN или 0,5—0,9 М малоновая кислота, 1,0—1,8 М уксусная кислота ReO; или WO2' 16
Оксид алюминия 1 — 1,16 М Н3РО4 ReO2', VO' WO2" 17
Силикагель Метанол — 3 М НС1 (7:3) или метанол — 1 М NH4NO3 - 3 М NH3 aq ReO2', WO2’ 18
Листы с нанесенным силикагелем («Chroma gram», Eastman) (14:5: 1) Метанол, содержащий 0,6 М НС1 — вода (9:1) ReO2’, Se,v 19
Экстракция
Экстракцию чаще всего применяют для отделения следов мо-иодена от мешающих ионов. Определение молибдена проводят ^посредственно в органической фазе, полученной при экстрак-нос’ ЧТ° позволяет сократить время анализа, повысить селектив-цеСсЬ и Устранить потери, которые неизбежно сопровождают про-экстп реэкстРакН”и молибдена в водную фазу. Обзор методов Чаях эКЦИИ молибдена дан в работах [20, 211. В некоторых слу-кстрагируют Mov и Мо111, но большинство методов основано
105
на экстракции Mov. Рассмотрим наиболее важные методы экстракции молибдена.
Молибден (V) экстрагируют в виде интенсивно окрашенного красного роданидного комплекса. Экстракцию проводят после восстановления молибдена до Mov. В качестве экстрагентов используют диэтиловый эфир, амилацетат, изобутанол, гексон. Палладий, родий, платина, вольфрам и рений соэкстрагпруются с Mov. Не мешают Fe, Al, Ti, Мп, Ni, Со, U и Та. Экстракцию вольфрама подавляют добавлением тартрата или цитрата. Молибден часто определяют по поглощению выделенного роданидного комплекса Mov. Молибден можно определить в экстракте методом атомной абсорбции. Показано, что в этом случае наиболее подходящим экстрагентом является метилизобутилкетон [22].
Образование комплекса Movi с толуол-3,4-дитиолом впервые описано в работе [23]. При экстракции комплекса эфирами, например амилацетатом, молибден отделяют от Со, V, Sn, Pb, Ni, Мп и А1. Малые количества железа соэкстрагируются. Вольфрам можно замаскировать лимонной кислотой и фотометрировать экстракт. Метод применен для анализа биологических, геологических объектов и сталей [24].
Бензоин-а-моноокспм является селективным реагентом для экстракции молибдена (VI).
Молибден(VI) количественно экстрагируется хлороформом из I М растворов НС1 [25]. Хром (VI) необходимо предварительно восстанавливать, поскольку он окисляет реагент. Ванадий (V) необходимо восстановить до VIV. Вольфрам^!) можно замаскировать фосфатом. В методике анализа стали предусмотрено отделение молибдена от Та, Nb и W [26].
Купферон образует комплекс с молибденом (V), который полностью экстрагируется хлороформом при pH < 2 [27]. Изоамиловый спирт количественно экстрагирует комплекс из 3 М растворов H2SO4 [28]. При pH = 1,6 экстрагируются также Fe111, TiIV, Vv, Sn’v, Си11 и WVI. С помощью купферона можно отделить Movi от WVI экстракцией хлороформом при pH — 1 из тартратных или цитратных растворов, однако интервал значений pH узок [29].
Молибден экстрагируется мезитилоксидом из 1 М раствора НС1 и 8 М раствора LiCL После реэкстракцни 0,5 М раствором аммиака молибден определяют в виде роданидного комплекса [30].
Для отделения молибдена от Ti, Zr, Сг, Мп, Fe, Со, Ni, U и Al в различных образцах предложен метод экстракции молибденовой сини [31]. При однократной экстракции метилизобутил-кетоном фосфорномолибденовая синь экстрагируется на 99,5%, затем ее реэкстрагируют в водную фазу и определяют.
Соосаждение и аналогичные методы
Соосаждение с гидроксидом железа(III), гидратированным диоксидом марганца и гидроксидом алюминия, было использовано для отделения и предварительного концентрирования молибдена, JOf?
Рис. 18- Флотационная установка: f — манометр; 2—игольчатый клапан; 3—рота-'тр- 4—увлажнитель газа; 5 —входной клд-„ян-6—клапан сброса давления; 7—фильтр из поойстого стекла; 8—раствор каучука; J—copot пены; /О—сброс; // — вывод воды; /2—резервуар с водой.
особенно из морской воды [32]. Использовано соосаждение комплекса бензоин-а-оксима с Movl с избытком реагента при pH = = 1,8 — 5,5 [33]. Использование гидроксида железа(Ш) в качестве носителя позволяет отделить молибден(VI) от рутения(VII), который остается в растворе [34].
Для выделения молибдена из морской воды применен метод адсорбционной коллоидной флотации [35]. Этот метод относится к группе методов разделения, называемой адсорбционным отделением на пузырьках [36]. Классификация методов адсорбционного отделения на пузырьках, в том числе адсорбционной коллоидной флотации, приведена на схеме 1. При отделении молибдена в раствор вводят носитель — свежеполученный гидроксид железа(III) — и поверхностно-активное вещество, затем через раствор барботируют воздух.
Схема I
ном(\ч\ИЦЫ гидРоксиДа вместе с адсорбированным молибде-хорош* вын°сятся на поверхность раствора, где они образуют боте 136метный слой, который отделяют и анализируют. В ра-троЛотп ДЛЯ опРеделения молибдена применен роданидный спек-Делить »ический метод. Из 500 мл морской воды можно вы-Схема ж ' ° содеР^ащегося в ней молибдена в течение 5 мин.
флотационной установки приведена на рис. 18.
107
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Молибден(VI) можно осадить в виде сульфида или в виде молибдатов Са, Ba, Pb, Ag и Hg1. Можно также использовать орга нические осадители, например 8-гидроксихинолин или бензоин-а оксим. Часто рекомендуют прокаливание до МоО3 как весовой формы, а при осаждении 8-гидроксихинолином осадок сушат при низкой температуре и взвешивают в виде комплекса. Для осаждения Movl в виде сульфида применяют тиоацетамид [37]. Хорошим методом обычно считают осаждение молибдена (VI) в виде молибдата свинца. Осадок прокаливают при 600—650 °C [38]. При осаждении используют медленное замещение ионов свинца в комплексе с ЭДТА ионом хрома(III).
Изучены органические осадители молибдена(VI). Как указано выше, бензоин-а-оксим применяют для отделения Mo(VI) осаждением и экстракцией. Реагент образует осадки с многими ионами, в том числе с Movi, Wvl, Vv и UO2+. При определении Mo(VI) после фильтрования осадок прокаливают при 600 °C и взвешивают в виде МоО3. Мешающее влияние CrVI и Vv устраняют восстановлением ионов до Сг'11 и V,v обычно с применением SO2. Вольфрам (VI) можно замаскировать фосфатами.
8-Гидроксихинолин осаждает молибден в виде комплекса MoO2(C9H6ON)2, который можно взвесить после высушивания при 200 °C. Определению мешают ионы некоторых металлов, однако при осаждении в присутствии ЭДТА определению не мешают Fe”1, Al, Be, Zn, Ni, Co, Mn, Pt, Cd, Bi, Си и Hg". Мешают Wvl, U, Vvn Ti [7]. Гравиметрический метод используют для точного определения больших количеств молибдена. Изучение гравиметрического метода определения MoVI с помощью 8-гидроксихинолина показало, что он характеризуется высокой воспроизводимостью и точностью [39]. Метод применен для внутри- и межлабораторных анализов при выпуске стандартных образцов ферромолибдена. Оценка результатов анализа показала, что метод характеризуется достаточно высокой точностью. Метод прост и экспрессен, включает одну операцию осаждения, он менее трудоемок чем рекомендованный ранее метод осаждения молибдата свинца.
Другие гравиметрические методы основаны на применении са-лицилгидроксамовой кислоты [40] и тритиокарбамата [41].
Гравиметрическое определение молибдена(У1)
с помощью 8-гидроксихииолина
Реагенты
Ацетатный буферный раствор, готовят, смешивая 3 объема 50%-ного раствора ацетата аммония и 4 объема 50%-ной уксусной кислоты.
8-гидроксихинолин. Растворяют 3 г 8-гидроксихииолина в 8,5 мл уксусной кислоты и разбавляют водой до 100 мл.
Ход анализа. К нейтральному раствору, содержащему около 0,07 г молибдена в 100 мл раствора, приливают 10 мл буферного раствора и нагревают до кипения. Приливают 10 мл 3%-ного раствора 8-гидроксихинолнна и оставляют на 2—3 мин. Фильтруют горячий раствор через пористый фильтр № 4 и промы
108
вают шестью порциями по 15 мл горячей воды, сушат до постоянной массы при 200сС. Взвешивают в виде Mo02(C9H6ON)2. Коэффициент пересчета для определения молибдена равен 0,2304.
Титриметрические методы
Редокс-методы
Редокс-методы определения молибдена (VI) недостаточно селективны. Большинство методов основано на восстановлении Movi до iMov или Мо111 с последующим окислением восстановленных форм стандартным раствором окислителя, обычно перманганатом или сульфатом церия. В качестве восстановителей применяют в основном амальгамы Hg, Ag, Zn и Cd, а также SnCl2, другие восстановители даны в работе [4]. При восстановлении Movl ртутью или серебром образуется Mov, а при взаимодействии с амальгамой цинка или кадмия образуется Мо111, в последнем случае восстановление можно проводить в редукторе Джонса. Ионы Ag, Fe, Ti, Сг, V, U и W, находящиеся в высшей степени окисления, а также азотная кислота восстанавливаются.
При восстановлении молибдена до Мо111 следует избегать окисления Мо111 кислородом воздуха. Для этого вытекающий из редуктора Джонса раствор должен попадать в сосуд, содержащий избыток стандартного раствора окислителя, который затем оттитровы-вают. Благодаря маскированию можно повысить избирательность определения молибдена. Например, при восстановлении Мо'1 амальгамой кадмия в среде HgSOi или НС1 вольфрам можно замаскировать фторидом, а образующийся Мо111 оттитровать раствором КМпО4[42].
При использовании серебра, ртути и Sn11 степень восстановления зависит от концентрации НС1. Изучен метод определения молибдена, основанный на его восстановлении и последующем окислении [43]. Показано, что восстановление до Mov в серебряном редукторе происходит только при концентрации НО, равной 2,0— 2,25 М. В том же исследовании подтверждены результаты ранее проведенной работы, показавшей, что при титровании Mov стандартным раствором бихромата наблюдается систематическая отрицательная ошибка около 1 % [44, 45]. Предполагается, что это связано с каталитическим действием ионов меди, катализирующих окисление Mov кислородом воздуха. Серебряный редуктор часто приготавливают путем обработки растворов серебра медью [46]. Описан метод приготовления серебряного редуктора без применения меди [47]. В работе [43] показано, что количественное восстановление Movl ртутью протекает в 2—4 М НС1, а раствором SnCl2 — в среде с концентрацией НО выше 2 М. При использовании редуктора Джонса влияние кислотности несущественно.
Метод осадительного титрования
Макросодержание молибдена (VI) можно определять титрованием растворами свинца или ртути (II). Предпочтительно титрование раствором Pb(NO3)2 при pH = 4,8 — 5,5 с применением дити
109
зона как индикатора [48]. Определению мешает ряд ионов. В работе [49] титрование проводили в гексаминовом буферном растворе в присутствии 4-(2-пиридилазо)-резорцина (ПАР). Метод позволяет определять сумму молибдена и вольфрама, затем молибден можно оттитровать отдельно раствором ЭДТА. Определению мешает более 2 мг сульфата и 3 мг фторида [50]. При титровании вольфрама допустимы большие количества этих ионов. Осадительное титрование раствором РЬ(ХО3)2 использовано для анализа смеси молибдата, перрената и фторида [51].
Комплексометрические методы
Существуют методы определения молибдена с помощью ЭДТА. В ранних работах MoVI осаждали солью кальция или свинца, а избыток соли титровали раствором ЭДТА. В некоторых методах использовано образование более устойчивого комплекса Mov (МоО2) 2V2- по сравнению с соответствующим комплексом MoVI. Комплекс получают восстановлением MoVI сульфатом гидразина в сильнокислой среде в присутствии ЭДТА. Избыток ЭДТА титруют обычным методом [52].
Предложен новый метод, основанный на образовании тройного комплекса состава 1:1:1, получаемого при кипячении растворов MoVI с солянокислым гидроксиламином и ЭДТА. Этот комплекс устойчивее, чем комплекс Movl с ЭДТА, однако избирательность определения невысока. В работе [53] предложено использовать тройной комплекс с ДЦТА (1,2-диаминоциклогексан-М, N, N', N'-тетрауксусная кислота). После образования комплекса избыток ДЦТА титруют при pH = 5—5,5 стандартным раствором цинка в присутствии ксиленолового оранжевого в качестве индикатора.
Метод позволяет определять 5—40 мг молибдена в присутствии вольфрама, если в растворе содержится достаточно солянокислого гидроксиламина (10 г).
Титриметрическое определение молибдата с помощью ДЦТА [53]
Реагенты
Гексамин, 10%-ный раствор.
Ксиленоловый оранжевый, 0,5%-ный раствор.
Ход анализа. К нейтральному раствору, содержащему 5—40 мг Movl, добавляют 1—2 г гидроксиламина солянокислого, разбавляют до 150—200 мл водой и нагревают до кипения. Раствор становится желтым или сине-зеленым в зависимости от концентрации молибдена. Добавляют избыток стандартного раствора ДЦТА и кипятят раствор в течение 15 мин. Раствор становится желтым или желто-зеленым. Раствор охлаждают, добавляют раствор гексамина для установления pH = 5—5,5, затем несколько капель ксиленолового оранжевого п титруют 0,05 М раствором ZnCl2 до красно-фиолетовой окраски, устойчивой в течение 3 мнн.
Примечание. В присутствии вольфрама титрование проводят, добавляя 10 г гидроксиламина солянокислого.
НО
Спектроскопические методы
Спектрофо то.'.; етри чес кий я е тод
Определение молибдена чаще всего проводят роданидным методом. Метод заключается в восстановлении молибдена (VI) в кислом растворе в присутствии роданида с образованием оранжево-красного оксипентароданида молибдена (V), который можно экстрагировать простыми и сложными эфирами, спиртами и т. д. и затем определять спектрофотометрически. Метод имеет недостатки, связанные с малой стабильностью и воспроизводимостью окраски. Для количественного восстановления до Mov необходимо строгое соблюдение условий анализа, поскольку возможно восстановление до Мо111, а в слабокислой среде — образование молибденовой сини. При восстановлении молибдена SnCl2 небольшие содержания меди и железа стабилизируют окраску раствора. Аналогичное влияние оказывают некоторые органические растворители и окислители Механизм реакции сложен и до конца не изучен [54]. Первые работы, посвященные роданидному методу определения молибдена, обсуждены Сейделом [55].
В последних работах комплекс рекомендуется экстрагировать «-бутилацетатом вместо диэтилового эфира, применяемого ранее. Поглощение растворов измеряют в интервале от 400 до 550 нм. Значение е комплекса зависит от выбранного метода и при экстракции изоамиловым спиртом составляет 1,5-104 при 475 нм [56].
Данные о влиянии мешающих ионов противоречивы, поскольку некоторые ионы мешают при сравнительно высоких содержаниях. Ниже приведены ориентировочные данные, которые следует уточнять по оригиналам работ. Определению мешают WVI, Vv, TinI, CrVI, UVI, Co11, Cu11, Pb", Fe111, Pt, Rh, ReVI1, Ag, Hg, A, Se, Те, F' н NOj. При анализе сталей вольфрам можно замаскировать лимонной, винной, щавелевой кислотой; или проводить восстановление в мягких условиях, не затрагивая вольфрама [54]. Методика, основанная на восстановлении хлоридом олова (II) и экстракции комплекса изоамиловым спиртом или изоамилацетатом, получила широкое распространение. Она рекомендована для определения молибдена в растениях [57] и стали [58]. Описан автоматический метбд анализа стали [59].
В последнее время используют образование смешаннолигандных комплексов, причем устойчивость роданидного комплекса молибдена увеличивается в присутствии другого лиганда. Сме-шанолигандный комплекс можно экстрагировать органическим растворителем. В качестве лигандов применяют тетрафениларсо-Г^„ХЛОРИД [54, 60], 2-меркаптобензо-у-тиопирон [61], этилксанто-ат калия [62] и а-анилинобензилфосфонат [63], при этом повы-а oafT чУВСТВительность и селективность определения. Например, оте [63] получено значение е = 5,9-103; определению не чрпц^°Т значительные концентрации Fe, W и V. В методике с - Нением тетрафениларсонийхлорида [54] молибден восста
111
навливают аскорбиновой кислотой и титаном (III), затем добавляют роданид и экстрагируют смешанный комплекс. При 470 нм е = 1,74-104; определению не мешает 40-кратный избыток W.
Описано применение ионного ассоциата роданидного комплекса Мо'' с катионом кристаллического фиолетового [64] пли родамина В [65]. При этом образуется комплекс состава 1:5:2
MoO(SCX)r+ 2RhB+ ч=Ь МоО (SCX)5 (RhB)2 Розовый Голубой нефлуогесцн»
флуоресцирую- рующий комплекс
щий катион
Комплекс характеризуется интенсивным поглощением в области 600 нм. Этот чувствительный и селективный метод применен для определения молибдена в почвах и растениях. При использовании спектрофотометрического и флуоресцентного методов предел обнаружения молибдена составляет 0,1 и 0,05 мкг соответственно.
Толуол 3,3-дитиол (дитиол) образует с MoVI в среде минеральных кислот комплекс темно-зеленого цвета. Взаимодействуют с реагентом также вольфрам(У1) и олово(П). Комплекс экстрагируется полярными и неполярными растворителями. Этот реагент, предложенный в работах [66, 67], нашел широкое применение. В бензоле значение е равно 2,86-104 при 665 нм. Мешают ионы Fe, As, Sb, W, Se, Pt, Re, Sn, Bi, Ti, V, Zr, Th, U, Cu, Pb, Cd, Co и Ni. Ряд работ посвящен устранению влияния этих ионов [68, 69]. Известно, что растворы дитиола неустойчивы. Описан метод получения раствора реактива, устойчивого в течение нескольких недель при хранении в холодильнике [70]. Показано, что специфичность и чувствительность метода повышаются при использовании 0,33%-ного раствора дитиола в 1%-ном растворе NaOH, стабилизированного добавлением 1% (по объему) меркаптоуксусной кислоты [71 ].
Ряд методик осуществляют в особых условиях. В работе [70] молибден (VI) экстрагируют три-н-бутилфосфатом из раствора НС1. Комплекс с дитиолом образуется затем в среде уксусная кислота— три-н-бутилфосфат — фосфорная кислота. В этих условиях подавляется взаимодействие дитиола с вольфрамом (VI), железом (III) и следами других элементов, которые соэкстрагируются с молибденом. Значение е равно 1,84-104.
В работе [72] экстракцию проводят изоамиловым спиртом. Чувствительность определения в биологических, геохимических образцах и сталях равна соответственно 0,05, 0,5 и 10 ppm. Влияние железа и вольфрама устраняют аскорбиновой и лимонной кислотами; введение K.I позволяет маскировать медь.
Предложены другие методы определения молибдена. В работе [73] обсуждена природа и механизм образования тройных комплексов с участием молибдена. Разработан спектрофотометрический метод, основанный на образовании тройного комплекса молибдена с пирокатехиновым фиолетовым и цетплтриметиламмо-нийбромидом. Молярный коэффициент поглощения комплекса в воде при 675 нм равен 4,6-104. Этот метод является одним из наиболее чувствительных методов определения молибдена. Соот
112
ношение реагентов в комплексе равно 1:1:2, закон Бера соблюдается в интервале содержаний молибдена 9,6—96 ppm. Определению мешают ионы, образующие комплексы с ппрокатехиновым фиолетовым, в работе [73] указаны мешающие ионы.
Вместо ппрокатехпнового фиолетового можно применять галлеин [74]. Чувствительность этого метода, основанного на образовании водорастворимого тройного комплекса синего цвета, вдвое выше, чем методов с использованием роданидного и дитполового методов. Однако определению мешают многие ноны, поэтому рекомендуется предварительное отделение молибдена.
Метод, основанный на восстановлении фосфорномолибденовой кислоты с образованием молибденовой сини, является чувствительным, но недостаточно воспроизводимым. В работе [75] показано, что положение максимума поглощения молибденовой сини меняется в интервале 680—810 нм в зависимости от природы восстановителя.
Предложен чувствительный метод, основанный на применении 2-амино-4-хлорбензол-тиола (е = 3,6-104 в хлороформе) [76]. Метод использован для определения фосфата, образующего фосфор-номолибденовую кислоту.
Исследовано 25 гидрокси- и азосоединений в качестве потенциальных спектрофотометрических реагентов для определения молибдена (VI) в неводной среде [77]. Наиболее чувствительным реагентом является n-диметиламинофенилфлуорон (е = 4300). Определению не мешает вольфрам при отношении Мо: W равном 2: 1.
Флуориметрический метод
Выше был рассмотрен флуориметрический метод, основанный на образовании тройного комплекса между молибденом, роданидом и родамином В.
Карминовая кислота в водном растворе взаимодействует с молибденом н вольфрамом, образуя флуоресцирующие комплексы [78]. Комплекс с MoVI при возбуждении 560 нм флуоресцирует при 590 нм. Указаны методы снижения мешающего влияния примесей.
Атомно-абсорбционный метод
Чувствительность и предел обнаружения элементов, образующих тугоплавкие оксиды, методом атомно-абсорбционной спектроскопии невысоки вследствие относительно высокой энергии диссоциации оксидов. Установлено, что чувствительность метода недостаточна для определения молибдена при содержании менее 100 ppm [79].
Чувствительность определения повышается при использовании ыЛ^сНт?ксид азота (И)— ацетилен. При использовании пламени жд Ч2Н.2 достигнута чувствительность определения молибдена hohJ^h1 ‘ВЭД- В этих условиях мешающее влияние посторонних выражено сильнее. При измерениях используют апалитиче-линию молибдена 313,3 нм,
ИЗ
Применяют метод жидкостной экстракции и введение органической фазы в пламя. Например, в работе [22] молибден экстрагируют в виде роданидного комплекса изобутилметилкетоном и экстракт распыляют непосредственно в пламя. Разработан метод анализа геологических образцов в интервале содержаний молибдена 1—500 ppm из навески пробы 1 г. Предел обнаружения равен 0,1 ppm. Метод анализа геологических образцов описан также в работе [81]. Экстракция алкиламина с длинной цепью применена для предварительного концентрирования Мо, W и Ре в виде роданидных комплексов с последующим определением методом атомной абсорбции [82]. Предел обнаружения в конечном растворе метил-изобутилкетона составляет 0,02 ppm.
Описан метод непламенного атомно-абсорбционного определения молибдена [83—85]. В работе [83] применена графитовая ячейка, нагреваемая до 2600°C, интервал определяемых содержаний молибдена 0,1 — 1 ppm.
Электрохимические методы
Потенциометрический метод
Потенциометрическое определение молибдена с помощью титана (III) и хрома (II) описано в работах [86, 87]. При титровании хромом реакция протекает количественно в среде НС1 или F^SOt при 80—100°C. Наблюдается два скачка потенциала, соответствующих переходам Movl—Mov и Mov—Мо111. Описано потенциометрическое титрование Movl раствором TiCl3 в присутствии лимонной кислоты [88].
Титрованию молибдена(VI) перхлоратом ртути(1) не мешает вольфрам [89]. В работе [90] молибден(VI) впервые восстановлен до Мо111 с помощью амальгамы цинка, затем Мо111 титровали потенциометрически железом (Ш). Показано, что для количественного восстановления необходимо высокое содержание хлоридов.
Описан метод потенциометрического титрования в среде концентрированной фосфорной кислоты [91].
Вольтамперометрический метод
В работе [92] показано, что молибден (VI) восстанавливается на капающем ртутном электроде в разбавленной HNO3, содержащей молочную и щавелевую кислоты. Описан высокочувствительный метод определения молибдена по каталитической волне восстановления водорода в азотной или хлорной кислоте. Высота волны, наблюдаемой при потенциале —0,3 В (н.к. э.) в нитратно-сульфат-ном электроде, линейно возрастает в интервале концентраций MoVI 10~7—10_5М [93]. В работе [94] в нитратной среде достигнута чувствительность определения молибдена 0,005 мкг/мл. Метод весьма селективен.
Изучено полярографическое восстановление MoVI в среде НС1 [95]. Наблюдаемая в этой среде волна отвечает восстановлению
114
MoVI до Mo111 п не связано с восстановлением ртути, как полагали ранее. В среде 4—6 A! НС1 и 0,24—1,08 .11 XH4F высота волны пропорциональна содержанию молибдена в интервале (1—6)-10~3Л1. Вольфрам не мешает определению.
Другие электрохимические методы
Описано амперометрическое титрование MoVI раствором 8-мер-каптохинолпна (тиооксин) с помощью платинового и иодпдртутного электродов в растворах разного состава [96]. Определению не мешает двухкратный избыток Bi, W и Те, мешают V и Си. Метод применен для анализа ферромолибдена, стали и других сплавов.
Описаны кулонометрические методы, основанные на электрогенерировании ТР" [97] и Fe" [98]. Метод позволяет определять до 7-10~5 М Movi. Не мешает 30-кратный избыток W.
При высокочастотном титровании молибдена (VI) применен купферон [99]. Излом кривых титрования соответствует отношению Movl и купферона, равному 1 : 1 и 1 :2. Метод может быть применен для анализа смесей MoVI — UVI.
Каталитические методы
Каталитические методы определения Мо" в основном связаны с реакцией окисления иодида Н2О2. Метод предложен Яцимирским и Афанасьевой в 1956 году [100] и впоследствии изучен другими авторами. В работе [101] описан метод определения 10—100 мкг/мл молибдена. Предложен метод измерения скорости иоднд-пероксид-ной реакции, основанный на непрерывном введении раствора иодида со скоростью, равной скорости окисления иодида, так что концентрация иодида в растворе остается неизменной [102]. Скорость введения I- пропорциональна содержанию молибдена. Реакцию окисления иодида Н2О2 катализируют также ионы W, Zr, Та и Fe. В работе [103] изучены оптимальные условия определения молибдена и методы устранения влияния этих ионов.
Описан метод определения молибдена с помощью автоматического анализатора [104—106]. Влияние Fe и W устраняют введением NH4F [106]. Для улучшения точности и правильности определения в работах [103] и [106] рекомендуется предварительное
Рис. 19. Схема установки Аля автоматического определения молибдата [104]: змеевик; 2—колориметр;
самописец; 4— устройство для сдачи пробы; 5—8—смесители, л—сброс (3,9 мл/мин); Б — 0.125 М
HCI (2,9 мл/мин); В-ра-ст»лР ^А2^2 (°*8 мл/мин); Г—ра-<0-8 мл/мин); Д-раба (°’8 мл/мин); Е —про-
(1Д м^инГ/М""); Ж-воздух
115
отделение Movl бензоин-а-оксимом. Предел обнаружения модно дена 0,003 мкг-мл. Метод применен для анализа растительных материалов. Схема потока растворов в автоматическом анализаторе дана на рис. 19.
Другие методы определения
Описан метод определения молибдена в морской воде методе • ЭПР [107]. Предварительно молибден выделяют в виде парамагнитного комплекса Mo(SCN)5 изоамиловым спиртом. Предел обит ружепия молибдена равен 0,46 мкг/л.
ЛИТЕРАТУРА
1. Lingane J. J. — Analytical Chemistry of Selected Metallic Elements, New-York, Reinhold, 1966, p. 85.
2. Elwell W. T„ Wood D. F. — Analytical Chemistry of Mo and W, Oxford, Per-gamon Press, 1971.
3. Бусев А. И. Аналитическая химия молибдена. Наука. 1979.
4. Chalmers R. A.— in: Comprehensive Analytical Chemistry (Ed. C. L, and D. W Wilson), Vol. IC, Amsterdam, Elsevier, 1962, p. 589.
5. Mann !. W. — In: Encyclopedia of Industrial Chemical Analysis (Ed F. D. Snell and L. S. Ettre), Vol. 16, New York, Interscience, 1972, p. 150,
6. Korkisch !. — Modern Methods for the Separation of Rarer Metal Ions, Chapter 17, Oxford, Pergamon Press, 1969.
7. Pribil R., Malat AL —Cooln. Czech, chem, Commun., 1950, v. 15, p. 120.
8. Malinek M. - Chemike Listy, 1954, v. 48, p. 30.
9 Yagoda H , Fales Fl. A. — J. Am. chem. Soc., 1938, v. 60, p. 640.
10. Yatirajan V., Shuja U., Kakkar L. R. — Taianta, 1976, v. 23, p. 819.
11. Ishida K., Kuroda R. — Analyt. Chem., 1967, v. 39, p. 212.
12. Kuroda R., Kawabuchi K.--7.. analyt. Chem., 1972, v. 261, p. 394; Analvt. Abstr., 1972, v. 24. p. 2809.
13. Korkisch J., Godl L.. Gross H.— Taianta, 1975, v. 22, p. 669.
14. Blasius E., Czekay A. — Z. analyt. Chem., 1957, v. 156, p. 81; Analyt. Abstr., 1957, v. 4, p. 3312.
15. Blum L. — Rev. Chim. Bucharest, 1958, v. 9, p. 28; Analyt. Abstr., 1958, v. 5, p. 3712.
16. Phipps A. M. — Analyt. Chem., 1971, v. 43, p. 467.
17. Shiobara Y.— Japan Analyst, 1968, v. 17, p. 1396; Analyt. Abstr., 1970 v. 19, p. 202.
18. Гайбакян Д. С. — Учен. записки Ереванск. гос. ун-та. Естеств. науки, 1971, с. 71.
19. Kuroda R., Kawabuchi К., Ito Т. — Taianta. 1968, v. 15, р. I486.
20. Бусез А. И., Родионова Т. В. — ЖАХ, 1971. т. 26, с. 578.
21. De А. К-, Khopkar S. М., Chalmers R. А. — Solvent Extraction of Metals, London, Van Nostrand Reinhold Co., 1970.
22. Kim С. H., Owens С. M., Smythe L. E. --Taianta, 1974, v. 21, p. 445.
23. Hamence J. Н, — Analyst, 1940, v. 65, p. 152.
24. Quin B. F., Brooks R. R. — Analytica chim. Acta, 1975, v. 74, p. 75.
25. Hoenes H. J., Stone K. G.— Taianta, 1960, v. 4, p. 250.
26. Luke C. L. — Analytica chim. Acta, 1966, v. 34, p. 302.
27. Stary J., Smizanska J. — Analytica chim. Acta. 1963, v. 29, p. 545.
28. Allen S. H„ Hamilton M. B. — Analytica chim. Acta, 1952, v. 7, p. 483.
29. Пятницкий И. Б., Кравцова Л. Ф. — Укр. хим. ж., 1969, т. 35. с. 77: Abstr., 1970, v. 18, р. 3098.
30. Shinde V. М., Khopkar S. М. — Ind. J. Chem , 1969, v. 7, р. 504; Analyt-Abstr., 1970, v. 19, p. 201.
31. Yatirajam V., Ram J.— Taianta, 1973, v. 20, p. 885.
116
32. Chan К. Al., Riley J. P. — Analytica chim. Acta, 1966. v. 36, p 220.
33. Weiss H. I'.. Lai Al. G.— Talanta, 1961. v 8. p. 72
34 Новиков А. //. — ЖАХ. 1961, т. 16, с. 388; Analvt. Abstr., 1962, v. 9
' p. 1903.
35 Kim У. S, Zeitlin H. — Separation Sci, 1971, v. 6. p 503: Analvt. Chem. ’ 1971, v. 43, p. 1390.
36. Karger B. L.. Grieves R. B., Lemlich R.. Rubin .4. J., Sebba F. — Separation Sci., 1967, v. 2. p. 401.
37. Burriel-Madi F., Madera I idan .4. — Analyt’ca chim. Acta. 1962, v. 26 p. 163.
38. Newcomb G.. Markham J. J. — Analytica chim. Acta. 1966 v. 35, p. 261.
39. Methods of Analysis Panel, Glasgow. MetaUurgia, 1971, v. 83. p. 129.
40. Chaudhuri N. K.. Sarkar A. K„ Das J. — Z. analyt. Chem., 1971, v. 254, p. 365 Analyt. Abstr., 1972, v. 22, p. 1561.
41. Johri K. N., Kaushik N. K., Singh K. — Talanta, 1969, v. 16, p. 432.
42. Сперанская E. Ф., Мерцалова В. E. — Сб. Хим. и хп.м. технол., Алма-Ата
1964, т. 2, с. 89; Chem. Abstr., 1966, v. 64, р. 1344.
43. Becker J., Coetzee C. J. — Analyst, 1967, v. 92. p. 166.
44. Wenier R., Boriss P. — Z. analyt. Chem., 1958, v. 160, p. 343; Analyt. Abstr.
1958, v. 5, p. 4086.
45. Grubitsch H., Halvorsen K-, Schindler G.— Z. analyt. Chem., 1960, v. 173 p. 414; Analyt. Abstr., 1960, v. 7, p. 4756.
46. Koithoff 1. AL, Belcher R. — Volumetric Analysis III, New York, Interscience
1957, p. 15,
47. Salam Khan M. A., Stephen W. I. — Analyst, 1968, v. 93, p. 26.
48, Veda S., Yamamoto Y., Takenuchi H. — J. chem. Soc. Jaoan, pure Chem Sect., 1967, v. 88, p. 1299; Analyt. Abstr., 1969, v. 16, p. 1857.
49. Lassner E., Scharf R. — Chemist Analyst, 1960, v. 49, p. 68.
50. Lassner E., Schedle H. — Talanta, 1966, v. 13, p. 326.
51. Cheng K. L., Goydish B. L. — Microchem. J., 1968, v. 13, p. 35.
52. Schwarzenbach G., Flaschka H. — Complexometric Determinations, 2nd English edn., London, Methuen, 1969, p. 223.
53 Pribil R., Vesely V. — Talanta, 1970, v. 17, p. 170.
54- F°gg A. G., Kumar J. L., Burns D. T. — Analyst, 1975, v. 100, p. 311.
55. Sandell E. B. — Colorimetric Determination of Traces of Metals, 3rd edn, New
York, Interscience, 1959.
56. Chariot G. — Colorimetric Determination of Elements, Amsterdam, Elsevier, 1964, p. 301.
57. Official Methods of Analysis of the Association of Official Analytical Chemists (Ed. W. Horowitz), 11th edn, Washington D. C., The Association of Official Analytical Chemists, 1970, p. 42.
58. British Standards Handbook No. 19, British Standards Institute, London 1970.
Braithwaite K-, Hobson J. D. — Analyst, 1968, v. 93, p. 633.
Tamhlna B.t Herak M. 1. — Mikrochim. Acta, 1976, p. 553.
Savariar С. P., Arunachalam M, K., Hariharan T. R. — Analytica chim. Acta, 1974, v. 69, p. 305.
Arunachalam AL К., Kumaran M. K. — Talanta, 1974, v. 21, p. 355.
Tamhlna B., Herak M. J., Jagodic V. — Analytica chim. Acta, 1975, v. 76,
P-417.
{«наго Л. И., Иванова И. Ф. — ЖАХ, 1972, т. 37, с. 713; Analvt. Abstr., ’973, v. 25, р. 1555.
Haddad Р. R,t Alexander Р. IV., Smythe L E. — Talanta, 1975, v. 22, Hamence J. //. — Analyst, 1940, v. 65, p, 152.
miller C. C.t Low A. J. — J. chem. Soc., 1940, p. 1258.
'effrey P. G. —Analyst, 1956, v. 81, p. 104.
59.
60.
61.
63.
64.
65.
66.
67.
68.
® -<//-=!/ r. u. —Analyst, 1956, v. 81, p. 104.
n }- B-~ J- agric. Fd Chem., 1959, v. 7, p. 269.
71 Knha M- E- M- S. — Analyst, 1975, v. 100, p. 517.
^ P 778Va Л1' ~Chemik§ Listy, 1972, v. 66, p. 768, Analyt Abstr., 1973,
B. F., Brooks R. R.— Analytica chim. Acta, 1975, v. 74, p. 75.
p. 61.
v. 24,
117
73 Bailey В. If., Chester J. E„ Dagnall R. .If.. ITcsf T. S. — Talar,ta. 1968. v. 15, p. 1359.
7-1. Leong C. L.— Analyst, 1970. v. 95. p. 1018.
75. Крисс Ё. E.. Руденко В. К.. Яцимирский К. Б.— Ж- неорг. химии. 1971, т. 16. с. 2147.
76. Kirkbright G. F., Yoe J. Н.— Taianta. 1964, v. И. р. 415.
77. Пономарев .4. .4.. Агринская Н. .1.—Труды Новочеркасск, политехи, ин-та, 1972, т. 266. с. 99: Analyt. Abstr, 1973, v. 24, р. 2797.
78. Kirkbright G. F.. West T. S. Woodward C.— Taianta, 1966, v. 13, p. 1637.
79. Dadd D. /. — Analyst, 1961, v. 86. n. 730.
80. Ramakrishna T. I'., West P. U7., Robinson J. U7. — Analvtica chim. Aeta. 1969 v. 44. p. 437.
81. Sutcliffe P. — Analyst, 1976, v. 101, p. 949.
82. Kim С. H., Alexander P. W,, Smythe L. E. — Taianta, 1975, v. 22, p. 739.
83. Бодров Af. В., Николаев Г. И. — ЖАХ, 1969, т. 24, с. 1314; Analyt. Abstr., 1971, v. 20, р. 1644.
84. Muzzarelli R. А. А. — Analytica chim. Acta, 1973, v. 64, p. 371.
85. Johnson D. J., West T. S., Dagnall R. M. — Analytica chim. Acta, 1974, v. 65, p. 171.
86. Willard H. H., Fenwick F. — J. Am. chem. Soc., 1923, v. 45, p. 928, 933.
87. Brintzinger G., Oschatz F. — Z. analyt. Chem., 1927, v. 165, p. 221.
88. Hahn H., Moosmueller A. F. — Z. analyt. Chem., 1966, v. 221, p. 261; Chem. Abstr., 1967, v. 66, p. 16236e.
89. Тараян В. M.. Овсепян Е. Н. — Зав. лаб., 1951, т. 17, с. 529.
90. Dolezal J., Moldan В., Zyka J.—Coin. Czech, chem. Commun., 1959, v. 24, p. 3769; Analyt. Abstr., 1960, v. 7, p. 3223.
91. Muralikrishna U., Rao G. G. — Taianta, 1968, v. 15, p. 143.
92. Uhi F. A. — Z. analyt. Chem., 1937, v. 110, p. 102.
93. Johnson M. G., Robinson R. J. — Analyt. Chem., 1952, v. 24, p. 366.
94. Violanda A. T., Cooke W. D. — Analyt. Chem., 1964, v. 36, p. 2287.
95. Сперанская E. Ф. Козловский M. T. — Зав. лаб., 1964, т. 30, с. 403; Analyt. Abstr., 1965, v. 12, p. 3857.
96. Павлова И. ЛЕ, Сангина О. А. — Сб. статей аспирантов н соискателей, Мин. высш, и среди, спец, образов. Каз. ССР. Химия и хнм. технол., 1965, с. 218; Analyt. Abstr., 1967, v. 14, р. 3160.
97. Binder Е., Goldstein G., Lagrange P., Schwing J. P. — Bull. Soc. chim. Fr., 1965, p. 2807; Analyt. Abstr., 1967, v. 14, p. 688.
98. Агасян П. K-. Теремова К- К., Николаева Е. Р., Катина Р. М. — ЖАХ, 1967, т. 33, с. 547; Analyt. Abstr., 1968, v. 15, р. 4670.
99. Riolo С. В., Soldi Т. F., Spini G. — Analytica chim. Acta, 1968, v. 41, p. 388.
100. Яцимирский К. Б., Афанасьева Л. П. — ЖАХ, 1956, т. 11, с. 319; Analyt. Abstr., 1957, v. 4, р. 119.
101. Svehla G.f Erdey L. — Microchem. J., 1963, v. 7, p. 206.
102. Weisz H., Klockow D., Ludwig H. — Taianta, 1969, v. 16, p. 921.
103. Бабко A. K-, Лисецкая E. С., Царенко Г. Ф. — ЖАХ, 1968, т. 23, с. 1342; Analyt. Abstr., 1970, v. 18, р. 238.
104. Hadjiioannou Т. Р. — Analytica chim. Acta, 1966, v. 36, r. 360.
105. Fudge R.—Analyst, 1970, v. 95, p. 171.
106. Bradfield E. G., Strickland J. F. — Analyst, 1975, v. 100, p. 1.
107. Hanson G., Szabo A., Chasteen N. D. — Analyt. Chem., 1977, v. 49, p. 461.
НИТРАТЫ
Нитрат — один из самых распространенных анионов. Избыточное содержание нитратов в питьевой воде связано с источниками загрязнения промышленного и сельскохозяйственного происхожД6' ния. Загрязнение атмосферы оксидами азота связано с выхлопными газами автомобилей; их определение часто проводят после переве-
118
пения в азотную кислоту. Вредное влияние избыточного содержания нитратов в воде хорошо известно. Нитраты и фосфаты вызывают сильный рост, а затем распад водорослей, что приводит к обеднению воды кислородом и как следствие — к гибели рыб и животного мира вод. Высокое содержание нитратов в питьевой воде вызывает цианоз у детей. В последнее время уделяют большое внимание взаимосвязи между содержанием нитратов и нитритоз и образованию нитрозаминов, этому посвящены обзоры [1,2].
Опубликованы обзоры .методов определения нитрата [3, 4]. Часто аналитическую химию нитратов и нитритов рассматривают совместно. Один из распространенных методов определения нитрата основан на восстановлении его до нитрита и последующем определении нитрита. Для некоторых объектов, например почв, важно знать содержание аммонийного, нитратного и нитритного азота. Описаны методы определения суммы нитратов и нитритов. Для определения только нитратов предварительно разрушают нитрит, например, обработкой сульфаминовой кислотой.
МЕТОДЫ ОТДЕЛЕНИЯ И ПРЕДВАРИТЕЛЬНОГО КОНЦЕНТРИРОВАНИЯ
Гравиметрические методы
Известный метод отделения нитратов основан на том, что нитраты бария и серебра растворимы. Это позволяет отделить осаждением анионы, образующие с барием и серебром нерастворимые соли. С помощью сульфата серебра можно отделить хлориды от нитратов. Этот метод применен для удаления мешающих примесей при определении нитратов в растворах гальванических ванн [5]. В этой работе в качестве осадителя использовали смесь хлорной кислоты и перхлоратов бария и серебра, компоненты которой не мешают последующему спектрофотометрическому определению нитратов. Раствор реагента приливают по каплям до тех пор, пока не перестанет выделяться осадок. Через 1 ч пли более раствор фильтруют, промывают три раза водой и объединенный раствор, содержащий фильтрат и промывные воды, анализируют.
Ионообменные методы
Методом ионного обмена можно отделять и концентрировать нитраты. Описано концентрирование нитратов из воды пропусканием ее через анионит Амберлит 1 R-4 В в хлоридной форме [6]. При элюировании 1%-ным раствором хлорида достигается 10-крат-вое концентрирование нитратов. Ионный обмен применен для отделения нитратов от хлоридов и гидрокарбонатов [7] с при.мене-
Двух катионитов. Первый, в Ag^-форме, удаляет хлориды, а g рон, в Н+-форме, сорбирует серебро, вытесненное из первого. ш-КИслом растворе после выхода из катионита в Н+-форме разрушается гидрокарбонат.
5МолЛЯ 0Тделення нитрата от сульфата применена ионообменная а в РЬ2+-форме [8]. При определении нитрата возникает необ
Пй
ходимость отделения нитрита. Разработан метод разделения ионов на тонкой пленке полимерного метакрилата, содержащего группы тетраалкиламмония на носителе цппакс (zipax) [9].
Отгонка в виде аммония
Восстановление до аммиака и отгонка последнего являются стандартным методом отделения нитрата. Выделенный аммиак можно определять спектрофотометрическим или тптриметрическпм методом и рассчитать содержание нитрата. Опубликовано описание реагентов и методов для восстановления нитратов [10]. Используемые ранее методы были длительными и трудоемкими. В последнее время созданы быстрые и автоматизированные методы [11]. Наплучшим является известный старый метод с применением сплава Деварда, состоящего из AI, Си и Zn (45, 50 и 5% соответственно) [12]. Основное восстановительное действие проявляет алюминии, взаимодействующий с нитратом по схеме
3NO] + 8А1 + 5ОН’ + 2Н2О —> 3NH3 + 8A10J
Реакция протекает в щелочной среде. Прибор для восстановления макроколичеств нитратов и отгонки аммиака показан на рис. 20. Для определения миллиграммовых содержаний нитрата используют модифицированную аппаратуру [14].
Для восстановления нитрата предложены другие восстановители, в том числе титан(III) и хром(II). Титан(III) взаимодействует с NO3 по схеме:
NO; + 8T*i3+ + 10Н+ —NHt + 8Ti4+ + ЗН2О
Метод дистилляции не только пригоден для определения NHt, NO] и NO] при совместном присутствии, но и позволяет определять эти ионы без помех со стороны глутамина и других лабильных в щелочной среде органических азотсодержащих соединений, что важно при анализе экстрактов из почв. Эти проблемы анализа обсуждены в обширном обзоре [13]. Отгонка с паром в присутствии оксида магния позволяет выделить аммиак в количествах, эквивалентных содержанию аммония в присутствии нитрата и нитрита. При добавлении сплава Деварда и продолжении дистилляции выделяется аммиак в количествах, эквивалент-
Рис. 20. Прибор для восстановления нитрата с помощью сплава Деварда и от" гонки аммиака.
120
них сумме NO] и NOj. Для удаления нитрита используют его способность быстро разлагался сульфаминовой кислотой с выделением азота, причем аммоний и нитрат не разрушаются. При использовании в качестве восстановителя раствора сульфата тита-на(Ш) (вместо сплава Деварда) восстанавливается только нитрат, а нитрит не восстанавливается [11].
Метод дистилляции может быть применен для анализа мутных и окрашенных растворов. Другое преимущество метода состоит в том, что при исследовании образцов, обогащенных изотопом ,5N, с помощью масс-спектрометрии легко проследить за превращением образующегося NH3 в N2.
Ранее метод Деварда, как правило, применяли для определения общего азота, но в настоящее время для этого применяют метод Кьельдаля. Однако определению азота по Кьельдалю должна предшествовать химическая подготовка пробы. Нитрат взаимодействует с салициловой кислотой в среде H2SO4 с образованием нитросалициловой кислоты, которая при восстановлении тиосульфатом образует аминосалициловую кислоту, после чего применяют метод Кьельдаля [15].
Микродиффузия
При анализе экстрактов почв используют метод микродиффузии. Нитрат восстанавливают до аммиака в микродиффузионной ячейке Конвея с помощью раствора сульфата железа (II) в 0,5 М H2SO4 в присутствии Ag2SO4 как катализатора [16]. Метод позволяет определять до 200 мкг нитрата в 5 мл экстракта почвы.
Экстракция
Нитрат можно экстрагировать с помощью тетрафениларсоний-хлорида [17]. При pH = 1 —2 экстракция хлороформом составляет 95—99%. Метод экстракции применен в спектрофотометрических методиках определения нитрата.
Метод тонкослойной хроматографии
Нитрат можно отделить от 12 других ионов с помощью тонкослойной хроматографии на маисовом крахмале, применяя в качестве растворителя смесь ацетон — 3 М раствор аммиака (1 : 4) [18]. Разделяемые ионы: NOJ, S2O3', СгОГ, N3, CN', SCN', ВОГ, S2', АзОз', AsOt, SOt и POt.
При анализе органических веществ пищевых продуктов нитрат выделен методом тонкослойной хроматографии на слое А12О3 толщиной 0,375 мм с помощью растворителя — 0,05 М раствора NaOH ‘4wrleT0Ha (15; 88) [19]. После выделения нитратной зоны спектро-,^^ОМетрическим методом можно определять не менее 10 ppm
121
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наиболее существенным достижением последних лет является разработка ионоселективного метода определения нитрата. Этот метод может конкурировать с трудоемкими методами, которые до сих пор приняты как стандартные. Метод получит в дальнейшем более широкое распространение, в связи с чем роль некоторых других методов уменьшится.
Гравиметрические методы
Обычно избегают применения гравиметрических методов при наличии других, более удобных и быстрых, но все же иногда их используют.
Метод количественного осаждения нитрата сильным основанием нитроном (1,4-дифенил-3,5-энданило-4,5-дигидро-1,2,4-триа-зол) предложен в 1905 г. Бушем [20]. Вероятно, реагент имеет структуру цвиттер-иона. Белый кристаллический осадок, содержащий 16,52% нитрата, сушат при 105 °C и взвешивают. Обсуждение гравиметрических методов определения нитрата приведено в обзоре [21]. Наиболее существенными недостатками метода являются следующие.
1. Относительно высокая растворимость осадка. Растворимость снижается в присутствии избытка реагента.
2. Недостаточная чувствительность метода.
3. Изменение состава осадка.
4. Мешающее влияние большого числа анионов: Вг', Г, С10з> СЮ4‘, СгОГ, Fe(CN)|', Fe(CN)t, SCN’, NO2, С2ОГ и Юз. Хлорид не мешает при сравнительно малых содержаниях.
Метод можно использовать только в том случае, когда в пробе отсутствуют указанные анионы. При добавлении ацетата свинца ряд примесей осаждается, другие анионы можно замаскировать или удалить принятыми методами. Например, нитрит можно разложить введением сульфаминовой кислоты; бромид — окислить и удалить в виде брома; хроматы удается восстановить сульфатом гидразина. Труднее удалить перхлорат.
Гравиметрическое определение нитрата с помощью нитрона
Реагенты
Нитрон, растворяют 10 г реагента в 100 мл 5%-ного раствора уксусной кислоты.
Ход анализа. К 80—100 мл нейтрального раствора, содержащего не более 0,1 г нитрата, добавляют 0,5 мл 1 М раствора HsSO4 или 1 мл ледяной уксусной кислоты. Нагревают раствор на водяной бане до достижения температуры кипения, затем вводят 10—12 мл раствора реагента. Перемешивают раствор, охлаждают до 0 °C и выдерживают при этой температуре 2 ч. Фильтруют раствор через стеклянный фильтр и промывают осадок 20—30 мл раствора, насыщенного нитратом нитрона, небольшими порциями. Б конце осадок промывают тремя порциями по 3 мл ледяной воды. Сушат осадок при 105 °C в течение I ч. Взвешивают в виде CzoHieNrHNOj. Фактор пересчета иа NO3 равен 0,1652.
Примечания. 1. Раствор реагента следует хранить в темной склянке.
2. Метод неприменим для микроколичеств (открывается 81 % от общего содержания нитратов) [22].
Известно, что ряд соединений образует малорастворимые нитраты. До 1960 г. такие соединения систематически не изучали. В работе [23] показано, что 8 известных реагентов для осаждения нитратов имеют разное строение. Все эти реагенты—органические основания с умеренно высокой молекулярной массой и молекулярным объемом. Исследователи пришли к выводу, что свойство образовывать малорастворпмые нитраты зависит скорее от молекулы в целом, чем от специфических групп атомов.
Исследование ряда реагентов показало, что ди-(1-нафтилметил) амин, предложенный впервые в 1923 г., более чувствителен к нитрату, чем нитрон. N-замещенные 1-нафтилметиламинов [24], в частности Ы-(4-хлорбензил)-1-нафтилметиламин, является особенно . эффективным реагентом для гравиметрического и титриметриче-ского определения NO3‘. Этот реагент аналогичен нитрону, но имеет ряд преимуществ, может быть применен для определения 1—50 мг NO3 [24].
В работе [24] предложены также Ы-бензил-1-нафтилметиламин и N-(4-метилбензил)-1-нафтиламин. Эти реагенты не образуют осадков при содержании 5 мг/млВгОз, Вг~, СГ, Юз, SO]’, Н2РО4, нс4н4О6 (гидротартрат) и S2Oi'; 4-хлорбензил-производное образует небольшие осадки с С1 и Вг~; Большинство реагентов образует . осадки с СЮз, СЮД Fe(CN)f*, Fe(CN)e', Г, NO2> MnO4', SCN’, WO4", C2O4’ и MoO4\ Представляет интерес применение М-(4-хлорбензил)-1-нафтилметиламина для последовательного определения перхлората и нитрата. Сначала осаждают перхлорат ионом тетрафенилфосфония и определяют гравиметрически: 1 мг (CeHs)4 Р+С1О] соответствует 0,2266 мг С1О4. В фильтрате определяют нитрат гравиметрически или титриметрически, с помощью Ы-(4-хлорбензил)-1-нафтилметиламина [25], причем 1 мг (Ci8Hi7ClNH2)+ NO3 соответствует 0,1800 мг NO3. Метод длителен, раствор с осадком выдерживают в течение 5—24 ч в зависимости от содержания нитрата (при содержании NO3 более 10 мг раствор фильтруют через 5 ч, при меньших концентрациях длительность экспозиции увеличивается).
В работе [26] для гравиметрического определения нитрата при-- Менен дициклогексил-таллий(Ш) сульфат как осадитель. Применение реагента и его синтез описаны в монографии [27]. Реагент был применен для титриметрпческого определения, при котором - конечную точку устанавливали потенциометрически или с помощью Ионоеелективного электрода.
Описан новый микрометод определения нитрата и нитрита, основанный на гравиметрическом определении СО2, образующегося ®и окислении муравьиной кислоты нитратом или нитритом [28]. поскольку N2O не поглощается щелочью, газообразные продукты
123
реакции пропускают через щелочной раствор в манометр и определяют объем N2O. В другом варианте предполагается гравиметрическое определение СО2 с применением обычных поглотительных трубок. Абсолютная ошибка определения нитрата и нитрита описанным методом составляет ±0,2%.
Титриметрические методы
Титриметрические методы определения нитрата основаны на его восстановлении. Образующуюся восстановленную форму, например аммиак, титруют непосредственно. В других вариантах применяют обратное титрование избытка восстановителя.
Восстановление нитрата до аммиака описано в разделе методов отделения нитрата. Применяют в качестве восстановителей Fe11, Tiin, сплав Деварда, Сг11 и UIV. В некоторых случаях определение проводят в отсутствие кислорода воздуха. Нитрат может быть восстановлен до NH3, NO, NO2 или гидроксиламина.
Метод, основанный на восстановлении
до NO методом Лейте
Метод восстановления нитрата избытком железа(П) в кислом растворе предложен в работе [29]. Избыток железа(II) оттитро-вывают чаще всего раствором бихромата:
3Fe2+ ± NOJ + 4Н+ —► 3Fe3+± NO + 2Н2О
При использовании в качестве катализатора молибдена [29] реакция завершается при кипении раствора в течение 10 мин. Чтобы предотвратить окисление NO и Fe2+ кислородом, используют атмосферу СО2. Лейте [31] предложил проводить реакцию в среде 6—8 М H2SO4, при этом отпадает необходимость в катализаторе и кислород воздуха не мешает определению. Время кипения раствора сокращается до 3 мин.
Применение свежеприготовленного раствора Лейте дает правильные результаты, однако через 1—2 ч появляется осадок и снижается результат контрольного опыта, что приводит к ошибочным результатам. В работе [30] предложен раствор реагента, устойчивый в течение 5—6 суток.
Титриметрическое определение нитрата модифицированным
методом Лейте
Реагенты
Раствор FeSO4—HCI. Растворяют 92 г FeSO4-7H2O в 200 мл воды, содержащей 20 мл концентрированной НС1, и доводят водой до 1 л.
Ферроин (индикатор), фенантролинат железа(П) сернокислый, 0,0025 М раствор.
Раствор К2Сг2О7, '/во А1.
Ход анализа. Отбирают пипеткой 25 мл раствора, содержащего 25—80 мг нитрата, и помещают в коническую колбу вместимостью 250 мл. Добавляют 5 г NaCl и точно 15 мл раствора FeSO4—НС1, затем 20 мл концентрированно!!
124
HjSOi- Нагревают раствор до кипения и осторожно кипятят 3 мин. Раствор охлаждают в проточной воде, приливают 50 мл холодной воды и 1 мл раствора индикатора. Титруют раствором бихромата калия до перехода коричневой окраски в сине-зеленую. Проводят контрольное титрование и рассчитывают количество железа(П). израсходованного на восстановление нитрата. Учитывают, что 1 мл */бс М раствора КзСггО? соответствует 2,067 мг NO"
Примечания. 1. Авторы работы [32] показали, что хлориды мешают анализу, и рекомендовали вводить в контрольный раствор столько хлорида, сколько его содержится в анализируемом растворе. Влияние хлорида в модифицированном методе Лейте аналогичное.
2. Определению мешают нитриты, ио с помощью карбамида можно устранить влияние 46 мг нитрита.
'Метод, основанный на восстановлении до NH3
Основной титриметрический метод определения нитратов основан на восстановлении их до аммиака с помощью сплава Деварда в щелочном растворе, отгонке аммиака, поглощении его стандартным раствором кислоты. Можно применять борную кислоту, которая удерживает аммиак и, как слабая кислота, не мешает последующему титрованию его сильной кислотой.
Метод подробно рассмотрен в разделе, посвященном отделению нитрата (см. рис. 20). Определению мешают ионы аммония и нитрита. Методы устранения их влияния обсуждены выше. При дистилляции аммиака следует отгонять около 2/з исходного раствора, контрольный опыт проводить в идентичных условиях. В некоторых условиях метод может дать ошибочные результаты. В работе [33] отмечено систематическое завышение результатов вследствие механического уноса щелочи с газом. Это происходит при использовании несовершенной аппаратуры. Рекомендован другой метод выделения и улавливания аммиака. Нитраты восстанавливают до аммиака металлическим железом в присутствии NiSO4 как катализатора. Аммиак осаждают в кислой среде в виде тетрафенилбората аммония, который фильтруют, промывают, растворяют в ацетоне и титруют стандартным раствором AgNO3 с применением ацетата вариаминового голубого как индикатора. Получены правильные результаты даже для микроколичеств нитрата.
Другие титриметрические методы
До сих пор используют старый метод, основанный на титровании нитрата раствором сульфата железа(II)-аммония в среде H2SO4 [34]. Полное восстановление нитрата до NO устанавливают по коричневой или розовой окраске комплекса FeSO4 — NO. При титровании используют H2SO4 с концентрацией выше 75%.. Вблизи 1°чки эквивалентности температура должна составлять от 40 до ° м ^етали метода описаны в работах [35, 36].
го МодиФичиР°ваннь>й, как описано ранее, метод Кьельдаля при-м ден Аля определения нитрата. Разработан ряд титриметрических титрА°В С пРименением органических реагентов как осадительных
125
Описан титриметрический метод, основанный на восстановлении нитрата железом(П) и (или) титаном(Ш) [37]. Титан(Ш) восстанавливает нитрат в сильнокислой, цитратной и ацетатной средах, при этом затрачивается соответственно 3, 6 и 8 эквивалентов титана(III). Глубина протекания реакции зависит не только от среды, но и от состава пробы. Восстановление завершается за несколько минут. При восстановлении раствором железа(П) время реакции зависит от содержания НС1. В присутствии молибдата аммония как катализатора реакция завершается за 2 мин.
Спектроскопические методы
Спектроскопический метод в видимой
и ультрафиолетовой областях
Спектроскопические методы широко применяют для определения нитратов. Методы можно разделить на 4 группы.
1. Методы, основанные на нитровании органических соединений, особенно фенолов. Применяют хромотроповую кислоту, 2,4-ксиленол, 2,6-ксиленол, фенолдисульфоновую кислоту и 1-аминопирен.
2. Методы, основанные на окислении органических соединений, например, бруцина.
3. Методы, основанные на восстановлении нитрата до нитрита или аммиака, которые затем определяют. Лучшим методом этой группы является восстановление до нитрита и определение последнего реактивом Грисса.
4. Метод, основанный на поглощении нитрата в УФ-области.
Другие методы, не относящиеся к рассмотренным группам, будут обсуждены ниже.
Метод нитрования является наиболее распространенным, несмотря на некоторые экспериментальные трудности, особенно при определении следов в присутствии хлоридов и нитритов. Эти ионы, как правило, мешают проведению реакций нитрования и окисления. Нитрит мешает даже в реакциях восстановления. Часто удается легко устранить мешающее действие этих ионов. Недостатки многих спектрофотометрических методов связаны с нелинейностью градуировочных графиков и длительностью анализа. Обзор спектрофотометрических методов определения нитратов дан в работе [4].
Особенности отдельных методов рассмотрены ниже.
Предложен специфичный и чувствительный капельный метод обнаружения нитратов, основанный на нитровании хромотроповой кислоты’ (4,5-дигидрокси-2,7-нафталиндисульфокислота) в концентрированной H2SO4 [38]. Эта реакция образования желтого продукта положена в основу спектрофотометрического метода [39]. Максимум поглощения наблюдается при 410 нм. Определению мешают хлориды, железо(III), хлор и некоторые друг,,е окислители. Мешающее влияние хлорида и железа (III) устраняют
126
введением сурьмы [40]. Модифицированный метод специфичен и позволяет определять 0,5—50 мкг нитрата. Эта реакция изучена в работе [41], она использована для анализа ряда объектов, в том числе почвы [42, 43].
Широко применяют метод, основанный на восстановлении нитрата до нитрита и определении нитрита реактивом Грисса. Спектрофотометрический метод определения нитрита обсужден ниже. Рассмотрим методы восстановления нитрата до нитрита. Из многих предложенных восстановителей наилучшпмп являются цинк и кадмий. При анализе пресной и соленой воды в качестве восстановителя предложен гидразнн [44]. Метод высокочувствителен и достаточно селективен, однако восстановление следует вести при постоянной температуре в узком интервале pH, в течение 20 ч. При увеличении температуры до 70 °C время реакции сокращается до 15 мин [45], что позволяет автоматизировать анализ.
В качестве восстановителя используют цинковую пыль [46— 48]. Показано, что цинк в присутствии хлорида аммония является более эффективным восстановителем, чем кадмий [49]. Нитраты можно восстанавливать амальгамой кадмия [50, 51] или металлическим кадмием [52—54]. Метод определения нитритов и нитратов, разработанный в работе [54], рекомендован для анализа пищевых продуктов для животных [55]. Восстановление нитрата кадмием с последующим определением нитрита методом Грисса рекомендовано при анализе металлов и экстрактов [56].
Улучшенный метод восстановления кадмием предложен в работе [57] при определении нитрата в беконе и лечебных рассолах. Растворы нитрата встряхивают с губкой кадмия в течение 5 мин, что эквивалентно обработке этого раствора в колонке в течение 40—60 мин. Применение омедненного кадмия изучено в работе [58]. Описан автоматизированный вариант этого метода [59]. Однако получение однородно-омедненного кадмия встречает затруднения [60]. Хорошие результаты получены при использовании в качестве катализатора смеси CuSO4, NH4C1 и двузамещенного фосфата натрия, а в качестве восстановителя — кадмиевой пыли.
Нитрат взаимодействует с алкалоидом бруцином C23H26N2O4 в среде H2SO4 с образованием красных, а затем быстро окрашивающихся в желтый цвет продуктов. Градуировочный график линеен при измерении в максимуме поглощения. Для получения точных результатов строят градуировочный график во всем интервале определяемых содержаний нитрата. Для анализа смеси нитратов и нитритов можно применить разные методы. Сумму ионов Жно определить при высокой кислотности, а при низкой кис-гимН°СТИ Т0ЛЬК0 НИТРИТ- Нитрит можно определить также дру-хлоп не3а т'симым методом. Кроме нитрита определению мешаюг Поль ДЫ ^ем не менее метод с применением бруцина широко ис-нитрптЮТ ДЛЯ опРеделения нитратов в воде. При этом влияние
Р а устраняют введением карбамида. При определении 0,05—
127
2,5 ppm нитрата отмечено мешающее влияние 1 мкг железа, 100 ppm меди и кадмия (занижают результаты). Влияние 100 ppm К, Na н Мп несущественно [61]. В той же работе описано определение суммы нитратов и нитритов, основанное на окислении нитритов раствором КМпО4, избыток которого разрушают щавелевой кислотой. Метод применен для анализа почв и экстрактов из растений [62].
Максимум поглощения нитрат-иона в УФ-области наблюдается при 193,6 нм [63]. Метод определения нитратов по собственному поглощению впервые предложен в работе [64]. Поглощение можно измерять также при 210 нм [65]. Хотя этот метод прост и экспрес-сен, определению мешает ряд ионов [66], в том числе Fe3+, Сгг+, Pb2+, Мо6+, Сгб+, V5+, Ti4+, Г, SCN', NO7, S2O33'и Вг‘. а также органические соединения. Влияние поглощения органических веществ можно учесть, измеряя поглощение растворов при 275 нм. Если к раствору, содержащему нитраты и хлориды, добавить H2SO4, то максимум поглощения сдвигается до 230 нм. В этой области мешающее влияние посторонних ионов выражено слабее. Применение сернокислых растворов предложено в работе [67]. Метод использован для анализа воды [68] и других объектов [69]. Измерение поглощения в УФ-области позволяет определять нитрит и нитрат при совместном присутствии [70], поскольку оба иона поглощают в области 302 нм, а нитрит — в области 355 нм. При использовании кюветы с толщиной слоя 1 см предел обнаружения нитрита равен 0,02 мг/мл, а нитрата 0,09 мг/мл. Определению мешает ряд ионов [70]. Описан косвенный метод определения нитратов, основанный на их восстановлении титаном (III) до аммиака и измерении поглощения аммиака в газовой фазе при 201 нм. Ионы кобальта, меди, железа и цинка подавляют сигнал, хотя не мешают определению аммония в аналогичном методе. Предполагается, что этот эффект связан с частичным окислением титана (III) или образованием неустойчивых промежуточных комплексов этих ионов, которые разлагаются с выделением не аммиака, а других соединений азота.
При взаимодействии фенолдисульфокислоты с нитратом образуются интенсивно окрашенные желтые продукты. Метод, несмотря на широкое применение, имеет недостатки; он длителен, так как предполагается выпаривание растворов нитрата досуха. Раствор выпаривают, поскольку нитрование проводят в малом объеме серной кислоты. Затем кислоту нейтрализуют раствором щелочи. Метод недостаточно надежен [71, 72]. Мешают хлориды, нитриты и органические соединения, а также аммоний, в присутствии которого наблюдаются потери до 50% нитрата [72]. Для удаления аммония рекомендуется выпаривание в присутствии КОП. Влияние нитрита устраняют введением сульфаминовой кислоты, карбамида или тиокарбамида. Хлориды осаждают ионами серебра.
При нитровании 2,4- и 2,6-ксиленолов образуются окрашенные продукты. При нитровании 2,4-ксиленола образуется 6-нитро-2,4‘
128
ксиленол, который можно отогнать с паром и поглотить NaOH или проэкстрагпровать толуолом п затем обработать щелочью [74], Нитрит также образует окрашенные продукты, но может быть разрушен карбамидом. Предложен улучшенный метод определения нитрата, основанный на нагревании 2,4-ксиленола с H2SO4 и нитратом. Продукты нитрования отгоняют и улавливают водно-аммиач-ным раствором, содержащим изопропанол. Поглощение раствора аммонийной соли 6-нптро-2,4-кспленола измеряют при 455 нм. Нитрит оказывает небольшое влияние. Метод характеризуется хорошей воспроизводимостью.
Для анализа промышленных вод предложена экстракция продуктов нитрования толуолом [76]. Метод позволяет определять до 30 мкг нитрата. Определению не мешают хлориды, сульфаты, же-лезо(Ш) и медь при их содержании не менее 1500, 5000, 500 и 250 мкг соответственно. Разработан автоматизированный метод определения нитратов в удобрениях [77].
Метод определения нитратов с помощью 2,6-ксиленола [78] примерно в 6 раз более чувствителен, чем с применением 2,4-ксиленола [79].
Один из самых старых спектрофотометрических методов определения нитрата основан на восстановлении его до аммиака и определении последнего. Метод восстановления нитрата описан в разделе методов отделения. Рассмотрим спектрофотометрическое окончание определения. Метод определения аммиака по Несслеру опубликован в 1956 г. Он основан на реакциях
2HgU' + 2NHs —+ 2NH3HgI2 + 4r
2NH3Hgi2 —> NH2Hg2i3; + г + nh;
Коагуляцию осадка предотвращают добавлением гуммиарабика. Поглощение измеряют при 410 нм, интервал определяемых содержаний NH3 равен 0,2—2,0 ppm. Метод применен в работе [80] и описан в монографии Шарло [81]. В качестве восстановителя предложен раствор СгС12 в присутствии NaOH [82].
Метод Несслера имеет недостатки. Реагент неустойчив, окраска раствора зависит от продолжительности реакции, температуры, концентрации реагента и посторонних катионов.
Описан метод определения нитратов, основанный на восстановлении их раствором FeSO4 в щелочной среде [83]; затем аммиак определяют пиридин-пиразолоновым методом [84]. Метод позволяет определять 0,01—0,1 мг нитратного азота. Определению мешают нитрит и аммиак, ион аммония можно отделить ионообменным методом. Цианиды, роданиды и цианаты мешают, поскольку образуют окрашенные продукты с реагентом, для отделения этих анионов можно использовать ионный обмен.
Аммиак можно определять по образованию индофенолового ^®Нег.° и индантриона. Индофеноловый синий образуется при взаи-f85e«RiBHH аМмиака со щелочным раствором фенола и гипохлорита Мия °” Метод применен для автоматического определения аммо-и нитрата в экстрактах почв [11]. Вначале отгоняют с паром
аммиак в присутствии MgO и рассчитывают содержание аммония. Если в раствор дополнительно вводят титан (III), то отогнанный аммиак соответствует сумме аммония и нитрата, причем нитрит не восстанавливается. Метод применен для анализа вод и удобрений. Индофенольный метод изучен в работе [87].
Метод, основанный на образовании индантрионгидрата [88,89], позволяет определять аммиак с чувствительностью 0,56 мкг; применен для анализа протеинов, пищевых продуктов и азотсодержащих гетероциклических соединений, однако может быть использован для определения нитратов [90].
Помимо методов, относящихся к четырем указанным выше группам, известны и другие. Предложен интересный спектрофотометрический метод, основанный на образовании ионных ассоциатов комплекса меди(1) с купроином (или неокупроином) с некоторыми анионами: нитратом, перхлоратом, фталатом, тетрафенилборатом. Ионные ассоциаты экстрагируются из водных растворов. Оптическая плотность экстрактов пропорциональна содержанию указанных анионов в водной фазе.. Метод позволяет определять 10~б — 10-5Л1 нитратов [91]. Мешают нитриты и хлориды в 10-кратном избытке, но хлориды можно осадить серебром (I), а нитриты разрушить сульфаминовой кислотой. Роданиды, цианиды, бромиды и иодиды мешают даже в эквимолярных количествах. Содержание нитратов можно определять косвенно, по концентрации меди, методом атомной абсорбции.
Другой экстракционно-фотометрический метод основан на применении кристаллического фиолетового [92]. Экстракцию проводят хлорбензолом при pH = 6, поглощение измеряют при 595 нм. Фосфат и сульфат не мешают в 100-кратном избытке. Хлорид в 10-кратном избытке дает небольшую положительную ошибку. Закон Бугера — Ламберта — Бера соблюдается в интервале (4—20)•IO-6 Л1 нитрата.
В работе [93] предложен метод, основанный на нитровании толуола. Продукт реакции экстрагируется толуолом в виде молекулярного комплекса, поглощение которого измеряют при 284 им. Мешающее влияние ЫОг, Вг", Г, SCN', SO§' и Hg2+ легко устранить. Интервал определяемых содержаний нитрата 1 —18 ppm.
Предложен новый спектрофотометрический метод определения нитрата, основанный на применении аминопирена [94]; он прост и весьма чувствителен (е = 94 000). Хотя градуировочный график нелинеен, метод может быть применен для анализа загрязнений воздуха. Нитрит и нитрат определяются совместно, определению не мешает 50-кратнын избыток гидросульфита.
Для анализа водопроводной воды предложен метод, основанный на нитровании тетрафениларсонийхлорида с последующей экстракцией продукта хлороформом [95]. Определению не мешает хлорид при содержании 200 ppm.
Для изучения загрязнений воды и воздуха, для санитарных анализов необходимы более чувствительные методы, чем описанные выше. Описан спектрофотометрический метод определения 130
Таблица 7. Характеристика некоторых спектрофотометрических методов определения нитратов
Приведенные характеристики следует рассматривать как ориентировочные, поскольку они зависят ог особенностей методики.
Для кюветы с толщиной слоя 1 см.
Поглощение завысит от длины волны.
нитратов на уровне концентраций мкг'.т, основанный на нитровании 2,6-диметнлфенола [96]. После экстракции продукта толуолом намеряют спектр поглощения раствора в области 400—600 нм. Измеряют разницу поглощения в максимуме 428 нм и на плато в области 520—560 нм. .Метающее влияние нитритов и хлоридов устраняют введением сульфампновой кислоты и ртути(II) соответственно. Линейная зависимость поглощения от концентрации нитрата наблюдается в интервале 0—900 мкг,л. Предел обнаружения 50 мкг/л нитрата. Ошибка определения ±5%.
Характеристика некоторых спектрофотометрических методов определения нитрата дана в табл. 7.
Спектрофотометрическое определение NO3
с помощью хромотроповой кислоты [40]
Реагенты
Хромотроповая кислота. Насыщенный раствор двунатрпевой соли 1,8-дпгндр-оксииафталпн-З.б-дисульфокпслоты обрабатывают дважды древесным углем. Раствор фильтруют и кристаллизуют при добавлении H2SO4. Кристаллы промывают несколько раз этанолом и сушат при температуре ниже 80 °C.
Готовят 0,1%-нЫ1Т раствор хромотроповой кислоты в концентрированной H2SO4. Раствор бесцветен в отсутствие нитратов. Раствор устойчив в течение 2 недель.
Раствор сульфита и карбамида. Растворяют 5 г карбамида и 4 г безводного Na2SO3 в 100 мл дистиллированной воды.
Раствор, содержащий сурьму. Растворяют 0,6 г металлической сурьмы в 80 мл концентрированной H2SO4 при нагревании. Раствор охлаждают и добавляют 20 мл ледяной воды. Если при стоянии выпадает осадок соли, его растворяют нагреванием.
Ход анализа. Отбирают 2,5 мл раствора, содержащего 0,2—20 мг/л нитрата, в сухую мерную колбу вместимостью 10 мл. В колбу добавляют 1 каплю раствора сульфита и карбамида, помещают в емкость с холодной водой (10— 20 °C) и добавляют 2 мл раствора сурьмы при перемешивании. Оставляют раствор в холодной воде на 4 мин, приливают 1 мл раствора хромотроповой кислоты, перемешивают и оставляют на холоду еще на 3 мин. Затем добавляют концентрированную H2SO4 до метки, закрывают колбу пробкой п перемешивают раствор, переворачивая колбу 4 раза. Затем оставляют раствор па 45 мин при комнатной температуре и снова доводят объем раствора до метки серной кислотой. Перемешивают осторожно раствор. Контрольный опыт проводят с бидистиллятом.
Через 15 мин после доведения раствора до метки измеряют поглощение при 410 нм. Ополаскивают кювету раствором и наполняют ее так. чтобы не было пузырьков воздуха (наклоняют кювету под углом 30° к горизонтали и осторожно приливают раствор по стенкам кюветы). В качестве раствора сравнения используют воду.
Примечания. 1. Метод может быть применен непосредственно для анализа воды без выпаривания пли осаждения. Однако раствор следует отфильтро* вать (или отцентрифугировать) от взвешенных частиц.
2. Раствор H2SO4 не должен содержать нитраты.
3. Определению не мешают 2000 мг хлоридов. При увеличении содержания сурьмы допустимое количество хлоридов составляет 4000 мг.
Флуориметрические методы
Описан чувствительный флуориметрический метод определения нитрата, основанный на одностадийной реакции [97]. При взаимо-действии нитрата с флуоресцеином в среде H2SO4 образуется Дй' 132
нитрофлуоресцеин. Флуоресценцию измеряют при 485 нм и возбуждении 435 нм. Предел обнаружения нитрата в растворе составляет 0 01 мкг/мл. Определению мешают хлориды, бромиды и йодиды. Влияние С1_ можно устранить введением SbL!! или 100-кратного избытка Fe11. Поны Fe11, однако, могут окисляться при наличии в растворе окислителен. Определению не мешает 10-кратный избыток нитрита, большие содержания мешают.
Для флуорпметрпческого определения _\Oj применен 2,3-дн-аминонафталин [98]. Сначала нитрат восстанавливают до нитрита, который взаимодействует с реагентом. Интервал определяемых содержаний нитрата в растворе — 0,05—5 мкг, мл. Предел обнаружения можно снизить в 5 раз.
Для определения нитрата применен 2,2/-дигпдрокси-4,4/-дпмет-оксибензофенон [99].
Разработан автоматический анализатор нитрата с производительностью 20 проб в 1 ч, использованный для анализа воды и осадков с предельным содержанием нитратного азота 5 мкг/л. Определению не мешают высокие содержания хлорида, сульфида и гуминовых кислот.
Метод инфракрасной спектроскопии
Для идентификации анионов методом ПК-спектроскопии применен нитрон. В спектрах поглощения нитрата нитрона наблюдаются характеристические полосы при 1370 и 1337 с.м-1. Разработан количественный метод определения нитратов, основанный на поглощении в области 1370 см-1 [100]. При определении миллиграммовых содержаний нитрата относительное стандартное отклонение составляет около 5%. Определению не мешают двукратные содержания NOa, CIO4, ВгОз, СЮз, СгО4', Ю4 и Г. Изучена возможность определения нитрита методом ИК-отражательной спектроскопии [101].
Атомно-абсорбционный метод
При определении анионов методом ААС обычно используют образование комплексов анионов с металлами. Содержание аниона определяют косвенно, по металлу. Например, нитрат определяют, используя образование ионной пары NO3 с бис(2,9-диметил-1,10-фенантролинатом) меди(1). Ионную пару экстрагируют метилизо-и Распь,ляют экстракт в пламя воздух — ацетилен .1 03, 104], используя поглощение при 325 им. Градуировочный график линеен в интервале (10 — 70)- 10~s М NO3~. Определению Не рпЗ-еТ ^'кРатны!”1 избыток сульфида, 100-кратный—СО?, SO? Sjq2_ 4 * Мешают Г, SCN", СЮ' и равные содержания СГ и ^^Аналогичный метод применен в работе [105] для определения .ством гЛс1ДтРата- Предварительно NO1 окисляют до NO3 носред- Ve(SO4)2 или КМпО4.
133
Электрохимические методы
Потенциометрические методы, ионоселективные электроды
Наибольшее распространение получил потенциометрический метод определения микросодержаний нитрата, основанный! на применении ионоселективных электродов. Этот метод во многих случаях позволяет получить результаты, достигаемые стандартными методами.
В настоящее время серийно выпускается более трех типов нитратных электродов, основанных на применении жидких анионитов. В электроде Орион 92-07 использован в качестве ионообменника раствор трис-замещенных о-фенантролпнатов никеля(II) в органическом растворителе, в электроде Корнинг 476134 — раствор тридодецилгекс а деци л аммоний нитрата в «-октил-о-нитрофениловом эфире. Выпускают также электроды Бекман 39618 с применением ионообменников. Описана конструкция двух нитратных электродов, в которых чувствительным элементом является мембрана из поливинилхлорида, содержащая серийно выпускаемый фирмами «Корнинг» и «Орион» жидкий ионообменник.
Существует два метода применения нитрат-селективных электродов. Один метод основан на построении эмпирического градуировочного графика зависимости электродного потенциала от концентрации нитрата или на построении этой же зависимости в полулогарифмических координатах. В последнем случае для определения NOj используют линейную зависимость потенциала от логарифма концентрации NO3. Электродом сравнения обычно служит каломельный электрод.
Другой метод, нашедший применение в анализе, основан на применении нитратного электрода для потенциометрического титрования, а именно для установления точки эквивалентности. Сравнение характеристик серийных нитратных электродов приведено в работе [106].
Интервал определяемых концентраций NO3 составляет 10~6 — 10-1 М. Определению не мешают умеренные содержания Cl-, SO< , РОГ и СОз', мешает нитрит, но его влияние можно устранить описанными ранее приемами. Электрод не чувствителен к катионам. Наибольшие помехи оказывают ионы Г, СЮз и СЮ'. В литературе имеются противоречивые сведения об избирательности ионоселективных электродов. Часто не учитывают ионные равновесия. Например, указана неверная селективность относительно ионов НСОз', СОз’ и РОГ, поскольку не учитывают их гидролиз В водном растворе, который приводит к образованию ОН~-ионов. Избирательность ионоселективных электродов обсуждена в работе [107].
Нитратный электрод может быть применен в широком интервале кислотности от pH = 2 до pH = 12. Описан электрод для ра-134
боты в сильнокпслой среде, в котором активный компонент тетра-деци.таммонппнптрат. находится в полимерной матрице из ноли-винилхлорида, пластифицированной дибутилфталатом пли диоктилфталатом [108]. Электрод позволяет определять Н\О3 в интервале концентраций 10~4 — 6.2.V.
Важное преимущество ионоселективного электрода заключается в возможности применения его для автоматизации анализа [109]. Пример автоматического анализатора нитратов показан на рис. 21.
Анионы, образующие устойчивые комплексы или малорастворимые осадки (например, хлориды) можно определять методом потенциометрического титрования, причем конечную точку устанавливают с помощью ионоселектпвного электрода. Для определения нитрата этот метод применить труднее. Тем не менее описан метод титрования нитрата сульфатом дпфенилталлпя(Ш) [НО] и нитроном [Н1]. Установлена константа кислотной диссоциации нитрона р/(а = 10,34 и произведение растворимости нитрата нитрона, равное (1,78 ± 0,03) • IO-6 при 20°С в 0,05 М растворе K2SO-. Оптимальная концентрация титруемого нитрата равна 0,01 Л1, метод применен для анализа удобрений. Нитрат осаждается сульфатом дифенилталлия (III) в виде (СбН5)2 ТГЫОз- Метод позволяет определять 0,01 — 0,1 М NO3. Характеристика нитратного электрода ухудшается при pH < 2, при pH > 4 начинает осаждаться гидроксид (С6Н5) гТНОНщ что приводит к завышенным результатам определения NO3. Нитрит не осаждается раствором (С6Н5)2Т1ф однако помехи могут возникать вследствие диспропорционирования NOj в кислом растворе с образованием NO.] и NO. Поэтому NO2' предварительно разрушают, устанавливая pH = 4 и вводя гидразин сернокислый. Основные недостатки метода связаны с мешающим влиянием галогенидов и сравнительно высоким уровнем определяемых содержаний нитрата.
Приведенные примеры являются лишь иллюстрацией широкого применения нитратных электродов [112].
Результаты анализа воды с применением нитратного электрода сравнимы с данными спектрофотометрического метода с примене пнем бруцина [113] и ксиленолового оранжевого [114]. Сопоставления результатов, полученных разными методами, даны в работе [115]. Результаты анализа почв с помощью нитратного
Рис. 21. Проточная ячейка с нитрат-селективным электродом [109]:
внеиив^ » в°Дяному насосу; 2 —электрод сра-омиого’птлВЫХОД В03ДУ*а; 4 — pH-метр высоко-трат-срлякт»Э с. Расширенной шкалой; 5 —ни-тРУбок Ыи1тпН1,1Й электрод; 6-входной па-»еиь " Диаметр 0.254 см); 7-vpo-
П~сливна'отг, 1\орпУс; 9— магнитная мешалка; » труба (внутренний диаме- р I‘,33 см).
д
135
электрода хорошо согласуются с результатами метода, основанного на дистилляции нитрата с паром; метод с применением фенолди. сульфокислоты дает слегка заниженные результаты. При анализе почв перемешивание раствора успешно заменено применением ультразвука [117]. Это ускоряет анализ, поскольку большая часть времени (несколько часов) затрачивается на предварительную подготовку проб, включая встряхивание и фильтрование раствора. Описан автоматический анализатор, рассчитанный на анализ 60 проб почвы [118].
Интересно использование нитратного электрода для анализа смеси нитратов и нитритов. Потенциал определяют дважды: до и после окисления NO2" раствором КМпО4 [120]. Метод применен для определения оксидов азота в сигаретном дыме. Нитратный электрод широко применяют для определения оксидов азота в воздухе, загрязненном выхлопными газами [121].
Определению оксидов азота с помощью нитратного электрода [122] предшествует окисление оксидов озоном до HNO3:
NO О3 < Л NO2 -р О2
2\тОз "р Оз < 3 N2O5 -р О2
Пентаоксид азота поглощается водой и гидролизуется:
N.Os-PH.O —> 2HNO3
Метод позволяет определять NO2 и NO на уровне концентраций ррш.
Описаны методы, не связанные с применением ионоселективных электродов. Пропуская раствор нитрата через катионит, можно выделить HNO3 [123]. Кислоту титруют потенциометрически в среде метанола или ацетона. Нитраты металлов третьей группы титруют раствором КОН, а нитраты I и II групп — гидроксидом тетраэтил-аммония. Нитриты можно титровать аналогично, раствором НС1О4 в метаноле или этилметилкетоне. Метод может быть применен для анализа нитритов и нитратов некоторых металлов, а также для анализа смеси нитритов и нитратов.
Вольтамперометрические и амперометрические методы
Поведение нитратов и броматов на ртутном капающем электроде изучено давно. Установлено, что потенциал восстановления сдвигается в положительную область с увеличением заряда катиона в ряду К+, Ва2+, La3+. Известно, что La3+ катализирует восстановление нитрата. Показано, что каталитический эффект связан свлия* нием продуктов гидролиза катионов [124]. Обзор полярографических методов определения нитратов дан в работе [4]. Кроме La3 катализаторами восстановления NO3~ являются другие катионы» в том числе ZrIV и UVI. Градуировочные графики для определения нитрата линейны в узком интервале концентраций [125, 126]. Д;|Я достижения линейной зависимости и снижения силы остаточного тока необходимо предварительное восстановление NO3 желе< зом (II) [125].
136
В разд. «.Спектрофотометрические методы» описано нитрование 2 6-кспленола в среде H2SO4. В этом случае происходит образование иона нитрония ХО>:
HXO3 + H.-SO4 NOT 4-HSO7+Н;О
Затем
протекает реакция: ОН
Описан метод, основанный на полярографическом восстановлении 4-нитро-2,6-ксплепола [127J, которое происходит обратимо, быстро и почти количественно. Контролируемый диффузией предельный ток пропорционален концентрации нитрата в интервале 2-Ю-5 — 10-3М. Мешающее влияние хлорида можно устранить. Влияние
кислорода существенно только при низких концентрациях нитрата.
Разработан полярографический метод определения HNO3 в H2SO4 йа фоне 0,5 М раствора СаС12 [128]. Потенциал восстановления NO.) равен Е- -, — —1,83 В относительно НКЭ. Высота волны увеличивается с ростом концентрации HNO3 от 5-10~5 до 10~3 А1. Показано, что потенциал восстановления NO3 на ртутной капле
относительно ртутного анода в среде уранила уксуснокислого равен Е[/2 = —1,0 В [129]. Метод несколько более чувствителен к нитратам, чем к нитритам. Определению мешают многие ионы, в том числе Al3*, Fe3+, Pb2+, Ni2+ и Н2РО4. При анализе смеси NO) и NO3' сначала определяют их сумму, а затем после удаления \'Oj с помощью метанола—только нитрат. Метод применен для анализа воды и удобрений.
Описан портативный полярограф для определения нитратов в воде [130]. Волны NO3‘ возникают на фойе солей ZrIV и UVI. Опи-
сан новый вольтамперометрический метод определения NOs на Уровне ррЬ [131] с применением электрода из пиролитического графита. Каталитическая волна NOj наблюдается при потенциале ' 1,0 В относительно НКЭ после предварительного осаждения на электроде металлических кадмия и меди. Определению не мешают ионы железа, хлориды, сульфаты и органические соединения; ме-ает нитрит (восстанавливается в тех же условиях). Градуировоч-ыи график линеен в интервале 10~6—10!Л1 (62 ррЬ — 62 ppm), вол рР°гРаФический метод эффективно использован для анализа
ДЬ1 [132], однако широкого применения в анализе он не получил.
_ фонометрические методы
Ййтпат РастВ0Рении в охлажденной ы и нитриты находятся в
концентрированной H2SO4 виде ионов NO) и NO+
137
соответственно. Пон NO* можно титровать электрогенерирован-ным железом (II) с применением платинового и Hg Hg2SO4 электрода [133]. Определению 12 мкмоль ХОз не мешает 70-кратнып избыток нитрита.
Предложен кулонометрический метод нодиметрического титрования нитрита, полученного восстановлением NO3 в редукторе, заполненном омедненным кадмием [134]. Восстановление NO3 при pH = 5 — 10 протекает по уравнению
Cd + NOJ + 2\Н3 + 2ХН4 —> Cd (NH3)24+ + NO; + Н2О
Нитрит определяют кулонометрическим методом [135]. Метод основан на восстановлении нитрита иодидом с последующим кулонометрическим восстановлением выделившегося иода. Интервал определяемых концентраций нитрата 0,01 — 100 мкг/мл. Длительность определения, включая восстановление NO3“, составляет 10 мин. Особое преимущество метода состоит в том, что он применим для анализа окрашенных растворов, исследование которых спектрофотометрическим методом часто невозможно. Метод применен для анализа окрашенных фруктовых соков, мяса и сточных вод. Вероятно, возможен анализ смеси нитратов и нитритов.
Другие методы определения
Опубликованы газометрпческие методы определения нитрата. Описан микрометод, основанный на восстановлении NO3 до NO в кислой среде [136]. В качестве восстановителей можно использовать железо(II), титан(III) и гидрохинон. При восстановлении NO3 гидрохиноном Movl является катализатором реакции, протекающей по схеме
С6Н4 (ОН)2 + 2MoVI —> С6Н4О2 + 2Mov + 2Н+
NO3 + 4Н+ + 3Mov —> NO + 2Н2О + 3Movl
Определение проводят в стеклянном аппарате специальной конструкции. В работе [137] в качестве восстановителей использованы иодид и иод:
2.NO3 +ЗГ + 6С1' + 8Н+ —► 2NO + 4H2O+3IC12
2NO3 -|-312 + 8Н++ 12СГ —> 2NO 4- 4Н2О + 6IC1J
Оксид азота NO собирают в нитрометре и измеряют объем газа. Содержание NO3‘ (х, в %) находят по формуле
(V - v) (Р - р) 273- 14,01 х (273 + t) W • 224 • 760
где V — объем NO, мл; v — объем NO в контрольном опыте, мл; Р— атмосферное давление, мм рт. ст.; р — давление паров воды при комнатной температуре /, мм рт. ст.; W — масса пробы, мг.
138
Оптимальная концентрация раствора SnCl2 для поглощения иода составляет 20%. Метод позволяет определять неорганические и органические нитраты.
Аналогичный метод, основанный на выделении NO при взаимодействии NO] со ртутью н H2SO4, описан в работе [138]. Метод применен для анализа смеси нитрита и нитрата, причем нитрит при реакции с сульфаминовой кислотой выделяет азот.
ферментативный метод определения нитрата основан на специфическом восстановлении NO] до NO], катализируемом разла-лагающей нитрат редуктазой, выделенной из Escherichia coli (штамм В) [139]. После инкубации раствора в течение 4 ч при 45°С проводят диазотирование и сочетание по реакции Грисса. Метод специфичен, предел обнаружения составляет 0,1 мкг азота (в виде NO]) в конечном объеме 10 мл.
Описано определение нанограммовых содержаний NO] и NO] (отдельно и при совместном присутствии) методом кольцевой печи [140]. При использовании метода сегментов отпадает необходимость в устойчивой шкале стандартов. Нитрат восстанавливают до NO] цинком, окраску проявляют с помощью реакции Грисса. Определению не мешает 10-кратный избыток сурьмы, мышьяка и свинца.
ЛИТЕРАТУРА
1. Wolff I. A., Wasserman А. Е,-—Science, 1972, v. 177, р. 15.
2. Egan Н. — Fd Cosmet. Toxic, 1971, v. 9, p. 81.
3. Clear A. J., Roth M. — In: Treatise on Analytical Chemistry (Ed. I. M. Kolt-hoff, P. J. Elving), Part II, Vol. 5, New York, Interscience, 1961, p. 268.
4. Streuli C. A., Averell P. A. — Analytical Chemistry of Nitrogen and its Compounds, Chapter 4, New York, Wiley-Interscience, 1970.
6. Dolance A., Healy P. W. — Ind. Engng Chem. analyt. Edn., 1945, v. 17, p. 718.
6. West land A. D., Langford R. R. — Analyt. Chem., 1956, v. 28, p. 1996.
7. Paul J. L., Carlson R. M. — J. agric. Fd Chem., 1968, v. 16. p. 766.
8. Рябинин А. И., Богатырев В. Л.—ЖАХ, 1968, т. 23, с. 894; Analyt. Abstr., 1970, у. 18, р. 879.
9. Schmit J. A., Henry R. А. — Chromatographia, 1970, v. 3, р. 497; Analyt. Abstr., 1971, v. 21, р. 2242.
10. Niedermaier Т. — Z. analyt. Chem., 1966, v. 223, p. 336.
11. Keay J., Menagi P. M. A. — Analyst, 1970, v. 95, p. 379.
*“• Devarda A. — Chemiker-zeitung, 1892, v. 16, p. 1952.
•* Bremner J. M. — In: Methods of Soil Analysis (Ed. C. A. Black), Part II: Chemical and Microbiological Properties, Agronomy No. 9, Madison, Wiscon-14 r>n’ American Society of Agronomy, 1965.
is ELemner E Keeney D. R. — Analytica chim. Acta, 1965, v. 32, p. 485.
JO. Dickinson W. E.— Analyt. Chem., 1954, v. 26, p. 777.
17 п-е?‘ P- Cornfield A. H. — Analyst, 1967, v. 92, p. 196.
!« n°. Beilstein G. M. — Z. analyt. Chem., 1963, v. 192, p. 44.
conic у D., Turcic M. N., Bugarski-Vojinovic M. B., Perisic N. U. — Z ana-.« Ip. Chem., 1967, v. 229, p. 93; Analyt. Abstr., 1968, V. 15, p. 5211.
M "«cfter D. W., Schall E. D. — J. Ass. off. agric. Chem., 1965, v. 48, p. 648
•g) flwscft Af._ Ber„ 1905, v. 38, p. 861.
ftjW'ns M — Ind. Chemist, 1954, p. 594.
W. I., Hassan S. S. M„ Zaki M. T. Mr-Taianta, 1971, v. 18, p. 219.
139
23. Stephen U" I. — Feigl Anniversary Symposium, Birmingham 1962 (Ed P. W. West, A. M. G. Macdonald and T. S. West), London, Elsevier, 1963. p. 156.
24. Hutton R. C., Salam S. A., Stephen W. I.— J chem. Soc., 1966, p. 1573(A).
25. Hutton R. C„ Stephen W. I. — Analyst. 1967, v. 92, p. 501.
26. Hartmann H., Bathge G.— Angew. Chem., 1953, v. 65, p. 107.
27. Belcher R., Wilson C. L. — New Methods in Analytical Chemistry, London, Chapman and Hall, 1955, p. 69.
28. Awad W. I., Hassan S. S. AL, Zaki Л!. T. AL —Taianta, 1971, v. 18, p. 219.
29. Kolthoff I. M., Sandell E. B., Muskovitz B. — J. Am. chem. Soc., 19.33, v. 55, p. 1454.
30. Latimer G. W., Chambers О. K. — Taianta, 1965, v. 12, p. 417.
31. Leithe W.— Mikrochemie, 1947, v. 33; p. 48; Analyt. Chem., 1948, v. 20, p. 1082.
32. Heitner-Wirguin C., Friedman D. — Taianta, 1960, v. 9, p. 121.
33. Erdey L., Polos L., Gregorowicz Z.— Taianta, 1959, v. 3, p. 6.
34. Bowman F. C„ Scott W. W.— J. Ing. Engng Chem., 1915, v. 7, p. 66.
35. Kolthoff I. AL, Belcher R. — Volumetric Analysis, Vol. Ill, New York, In-
terscience, 1957, p. 609.
36. Williams A. F. — In: Comprehensive Analytical Chemistry (Ed. C. L. and D. W. Wilson), Vol. 1C, Amsterdam, Elsevier, 1962, p. 207.
37. Awad IF. /., Hassan S. S. AL — Taianta, 1969, v. 16, p. 1383.
38. West P. W., Sarma P. — Mikrochim. Acta, 1957, p. 506.
39. West P. W., Lyles G. L. — Analytica chim. Acta, 1960, v. 23, p. 227.
40. West P. W., Ramachandran T. P. — Analytica chim. Acta, 1966, v. 35, p. 317.
41. Robinson J. W„ Hsu C. J. — Analytica chim. Acta, 1969, v. 44, p. 51.
42. Clarke A. L., Jennings A. C. — J. agric. Fd Chem., 1965, v. 13, p. 174.
43. Басаргин H. H., Чернова E. A. — ЖАХ, 1968, t. 23, c. 102; Analyt. Abstr.,
1969, v. 17, p. 4717.
44. Mullin J. B., Riley J. P. — Analytica chim. Acta, 1955, v. 12, p. 464.
45. Henriksen A. — Analyst, 1965, v. 90, p. 83.
46. Akisada T.—Japan Analyst, 1963, v. 12, p. 614; Analyt. Abstr., 1965, v. 12, p. 621.
47. Komatsu S., Hagino K- — J. chem. Soc. Japan, pure Chem. Sect., 1967, v. 88, p. 1157; Analyt. Abstr., 1969, v. 16, p. 1202.
48. Morimoto M., Hirakoba A., Ishibashi R. — Japan Analyst, 1967, v. 16, p. 1335; Analyt. Abstr., 1969, v. 16, p. 1203.
49. Matsunaga K., Hlshimura M. — Analytica chim Acta, 1969, v. 45, p. 350.
50. Morris A. W., Riley J. P. — Analytica chim. Acta, 1963, v. 29, p. 272.
51. Исаева А. Б., Богоявленский A. H.— Океанология, 1968, т. 8, с. 539; Analyt. Abstr., 1969, v. 17, p. 1210.
52. Kamm L., McKeown G. G., Smith D. M. — J. Ass. off. argic. Chem., 1965, v. 48, p. 892.
53. Kamm L., Bray D. F., Coffin D. E. — J. Ass. off. analyt. Chem., 1968, v. 51, p. 140.
54. Schall E. D., Hatcher D. IF.— J. Ass. off. analyt. Chem., 1968, v. 51, p. 763
55. Official Methods of Analysis of the Association of Official Analytical Chemistry (Ed. W. Horowitz), 11th edn, Washington, D. С., AOAC, 1970, p. 126.
56. Official, Standardised and Recommended Methods of Analysis (Compiled and Edited by N. W. Hanson), 2nd edn, London, The Chemical Society, 1973.
57. Elliott R. J., Porter A. G. — Analyst, 1971, v. 96, p. 523.
58. Wood E. D., Armstrong F. J. A., Richard F. A.— J. mar. Biol. Ass. UK, 1967, v. 47, p. 23.
59. Henriksen A., Selmer-Olsen A. R. — Analyst, 1970, v. 95, p. 514.
60. Lambert R. S., Dubois R. J- — Analyt. Chem., 1971, v. 43, p. 955.
61. Saito G., Sugimoto K-, Hagino K. — Japan Analyst, 1971, v. 20, p. 542; Analyt. Abstr., 1972, v. 23, p. 2415.
62. Baker A. S. — J. agric. Fd Chem., 1967, v. 15, p. 802.
63. Scheibe G. — Ber., 1926, v. 59, p. 1321.
64. Hoather R. C. — Proc. Soc. Wat. Treat. Exam., 1953, v. 2, p. 9.
65. Hoather R. C,, Rackham R. F. — Analyst, 1959, v. 84, p. 548.
140
56. Bastian R.. Wcberling R-. Palilla F. — Analyi. Chem.. 1957. v. 29, p. 1795.
57. Armstrong F. A. /. — Analxt. Chem. 1963. y. 35. p. 1292.
58. Kavone R. —J. Am. Wat. Wks Ass.. 1964. v. 56, p. 781.
59. American Public Health Assoc-'ation. Standard Methods for the Examination of Water and Wastemeter, 13th edn, Washington. D. C-. American Public Health Association. 1971.
70 Welters J. H.. L'gpim K. L.— Anoint Chem.. 19,0. v. 42. p. 335
71 Goldman E. J.^bs R. — J. Am. W.-,t. Wks Ass.. 1961, v. 53, p. 187.
72 Flora F. B.. Wehber P. J.— -\nalyst. 1960. v. 85. p. 567.
73. Cresser .11. S. — Analyst. 1977, y. 102. p 99.
74. Barnes H.— Analyst. 1950, v. 75. p 3SS.
75. Norwitz G._ Gordon FL — Analytica chim. Acta, 1977, v. 89. p. 177.
76. British Standard 2690: Part 7, 1968. London.
77. Docherttj .1. C. — Lab. Pract., 1969, y. 18, p. 47.
78. Hartley А. .11.. ,l«j> R. F— Analyt. Client., 1963, v. 35, p. 1207.
79. Andrews D. IV'. VC—Analyst, ib64. y. 89, p. 730.
80. Allerton F. IV'. — Analyst, 1947, v. 72, p. 349.
81 Chariot G. — Colorimetric Determination of Elements, Amsterdam, Elsevier, 1964, p. 320.
82. Harrison G. A. F. — Taianta, 1962, v. 9, p. 533.
83. Pappenhagen J. M.— Analyt. Chem., 1958, v. 30, p. 282.
84. Kruse J. AL, .Mellon 51. G. — Analyt. Chem., 1953, v. 25, p. 1188.
85. Rusell J. A. — J. biol. Chem., 1914, v. 156, p. 457.
. 86. Ref. 81, p. 321.
87. Weatherbum 51. II7. — Analyt. Chem., 1967, v. 39, p. 971.
88. Jacobs S. — Nature, bond., 1959, v. 183, p. 262.
89. Jacobs S. — Analyst, 1962, v. 87, p. 53.
90. Jacobs S. — Feigl Anniversary Symposium, Birmingham 1962 (Ed. P. W. West, A. M. G. Macdonald and T. S. West), London, Elsevier, 1963, p. 200.
91. Yamamoto Y., Okamoto N., Tao E.— Analytica chim. Acta, 1969, v. 47, p. 127.
92. Yamamoto Y„ Uchikawa S., Akabori K. — Bull. chem. Soc. Japan, 1964, v. 37, p. 1718; Analyt. .Abstr.. 1966, v. 13. p. 1233.
93. Bhatty 54. /<., Townshend A. — Analytica chim. Acta, 1971, v. 56, p. 55.
94. Sawicki E._ Johnson FL. Stanley T. II7. — Analyt. Chem., 1963, v. 35, p. 1934.
95. Burns D. T._ Fogg A. G., Willcox ,4. — Mikrochim. Acta, 1971, p. 205.
96. Tan Y. L. — Analytica chim. Acta, 1977, v. 91. p. 373.
97. Axelrod H. D., Bonelli J. E., Lodge J. P. — Analytica chim. Acta, 1970, v. 51,
p. 21.
98. Sawicki C. Afghan B. Magee R.
R.— Analyt. Lett., 1971, v. 4, p. 761.
K-, Ryan J. F. — Analyt. Chem., 1975, v. 47, p. 2347.
J., Shahine S. A. E. F., Wilson C. L. — Mikrochim. Acta,
1964,
'iihite r. see 1 ..... .. :
Vang R. T., Low M. J. D. — Analyt. Chem., 1973, v. 45, p. 2014.
N., Ellis R. F. — Appl. Spectroscopy, 1963, v. 17, p. 168.
V- 18, p. 359; Analyt. Abstr., 1972, v. 22, p. 105.
99.
100.
p. 1019.
101. Wilhite
102. T ,.e _ _________ _________....... ____, ,. t„_________
103. Kumamaru T., Tao E. Okamoto N., Yamamoto Y.— Bull. chem. Soc. Japan, ' 1965, v. 38, p. 2204: Analyt. Abstr., 1967, v. 14, p. 1934.
104. Yamamoto r., Kumamaru T.. Hayashi Y., Otani Y. — Japan Analyst, 1969, . .«e Y; 18> P- 359i Analyt. Abstr., 1972, v. 22, p. 105.
«Б. Houser M. E., Fauth M. /. — Microchem. J„ 1970, v. 15, p. 399.
P?vies J- E- w- Moody G. J., Thomas J. D. R. — Analyst, 1972, v. 97, p. 87.
J’’- G- J-’ Thomas J. D. R. — Taianta, 197Й, v. 19, n. 623.
' л E' А.,'Грекович А. Л., Гарбузова H. В.—ЖАХ, 1974, t. 29,
109.
110.
111.
112.
c. 1900. ' ,
Milham P. J.— Analyst, 1970, v. 95, p. 758.
«gregorio J, S., Morris M. D. — Analyt. Chem., 1970, v. 42, p.
nulantcki A„ Maj M. — Taianta, 1975, v. 22, p. 767.
МсрПс , search Analytical Methods Guide, 7th edn, Crawley, tLanrr ^CI,er,tific Instruments. Manor Royal.
StnHtPn1rP’’ Jacobson R. L., — Environ. Sci. Technol., 1970, v. 4, p. G\F- Г Analyt. Lett., 1975, v. 8, p. 503.
“non N. A., Crosby N. T. -Water Treat. Exam., 1969, v. 18, p. 338.
94.
Sussex, UK,
4, p. 834.
141
116. Mahendrappa M. К- — Soil Set., 1969, p. 132.
117. Sinieonov I'., Asenor.' /.. Diadov V. — Talanta, 1976. v 24, p. 199.
118. Goodman D. — Analyst, 1976, v. 101, p. 943.
119. Paul J. L., Carlson R. M. — J. agric. Fd Chem., 1968, v. 16, p. 766.
120. Morie G. P., Ledeord C. J., Glover C. A. — Analytica chim. Acta, 1972, v. 60, p. 397.
121. McFarland J. H„ Benton C. S. — J. Chem. Educ., 1972, v. 49, p. 21.
122. Di Martini R.— Analyt. Chem., 1970, v. 42, p. 1102.
123. Бирум А. Л1., Комарова К. А., Крешкова E. К., Яворенко А. И. — Известия высш, учебн. заведении. Химия и хим техно.!., 1966, т 9, с. 546; Analyt. Abstr., 1967, v. 14, р. 6065.
124. Progress in Polarography (Ed. P. Zuman), New York, Interscience, 1962, p. 225.
125. Rand M. C., Heukelekian //. — Analyt. Chem., 1953, v. 25, p. 878.
126. Hamm R. E., Withrow C. D. — Analyt. Chem., 1955, v. 27, p. 1913.
127. Hartley А. Л1., Curran D. J. — Analyt. Chem., 1963, v. 35, p. 686.
128. Гальперин Г. M., Ильина В. А.—Зав. лаб., 1970, т. 36, с. 1046; Analyt.
Abstr., 1971, v. 21, р. 165.
129. Montenegro М. I., Cruz М. J., Matias М. L. — Revta. port Quim., 1971, v. 13, p. 217; Analyt. Abstr., 1973, v. 24, p. 103.
130. Young R. L., Everett Spell J., Siu H. M„ Philip R. H., Jones E. R. — Environ. Sci. Technol., 1975, v. 9, p. 1075.
131. Bodini M. E., Sawyer D. T. — Analyt. Chem., 1977, v. 49, p. 485.
132. American Public Health Association, Standard Methods for the Examination of Water and Wastewater, 12th edn, Washington D. C., American Public Health Association, 1965.
133. Giuffre L., Losio E., Fontani Lamina F. — Annali Chim., 1967, v. 57, p. 1450; Analyt. Abstr., 1969, v. 16, p. 1831.
134. Karlsson R., Torstensson L.-G.— Talanta, 1975, v. 22, p. 27.
135. Karlsson R., Torstensson L.-G. — Talanta, 1974, v. 21, p. 945.
136. Awad W. I., Hassan S. S. M. — Talanta, 1969, v. 16, p. 1393.
137. Hassan S. S. M.— Analyst, 1971, v. 96, p. 59.
138. Van Slyke D. D., Lo Monte A. F. — Microchem. J., 1969, v. 14, p. 608.
139. NcNamara A. L., Meeker G. B., Shaw P. D. — J. agric. Fd Chem., 1971, v. 19, p. 229.
140. Weisz H., Hanif M.— Analytica chim. Acta, 1976, v. 81, p. 179.
НИТРИТЫ
Нитриты натрия и калия находят широкое применение в качестве консервантов и при фиксировании окраски пищевых продуктов. Нитрит входит в одно из звеньев цикла кругооборота азота в природе. Нитрит содержится в почвах, воде, стоках. Нитрит вводят в технологические растворы в качестве ингибитора коррозии.
Токсическое действие нитрита осуществляется по нескольким механизмам. Ион NOJ связывается с пигментом крови, образуя метагемоглобин, который не является переносчиком кислорода. Считают, что нитрит может связываться в желудке с аминами и амидами, образуя высококанцерогенные нитрозамины [1, 2]. Допустимые содержания NO- в некоторых пищевых продуктах строго ограничены. По санитарным нормам (Food Regulations, 1962) допустимое содержание NO2 (в виде NaNO2) составляет 500 ppm.
Содержание нитрита определяют также в воздухе. Оксиды азота, поступающие в воздух из выхлопных газов и промышленных выбросов, являются серьезным источником загрязнения атмосферы.
142
Известный метод Зальцмана для определения NO2 в воздухе, модифицированный в работе [4], основан на пропускании анализируемого воздуха через щелочной раствор и анализе этого раствора.
Методы определения NO; и NO; имеют много общего. Стандартный метод определения нитрата основан на восстановлении до нитрита и определении последнего. В свою очередь нитрит можно окислять до нитрата такими окислителями, как MnV11 и Celv, и определять косвенно содержание NO;. Определение NO; и NO3 при совместном присутствии является важной аналитической задачей, особенно при анализе почв. Один из способов заключается в определении суммы ионов. Затем определяют только нитрат после разрушения нитрита сульфаниламидом, карбамидом, сульфаминовой кислотой или гидразином сернокислым. Реакции NO; с сульфаминовой кислотой и карбамидом приведены ниже:
hoso2-nh2 + hno2 —> n2 + h2so4 + h2o
СО (NH2)2 + 2HNO2 —> 2N2 + CO2 + 3H2O
Аналитическая химия нитритов рассмотрена в обзорах [5—8].
МЕТОДЫ ОТДЕЛЕНИЯ
Метод тонкослойной хроматографии
Нитрит отделяют от некоторых других анионов методом ТСХ на крахмале с помощью растворителя ацетон — 3 М аммиак (1:4) [9]. Методом ТСХ на силикагеле, используя в качестве растворителя смесь метанол — бутанол — вода (3:1:1), можно отделить NO; от F’, S2O3’, SOt, СгОГ, РОГ, AsO]’ и AsO33‘[10],
Экстракция
Нитрит можно экстрагировать раствором трифенилстаннония в бензоле в интервале pH = 1,0 — 2,2 на 97% [11].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Метод кислотно-основного титрования
Метод основан на титровании аммиака, образующегося при восстановлении нитрита (и нитрата) сплавом Деварда. Современный вариант метода [12] использован для определения NO; и NO2 в почве и экстрактах растений и применим для проб, содержащих до 2 мг неорганического азота. В одной аликвотной части раствора оба аниона восстанавливают до аммония. Аммиак отгоняют ^иаром, поглощают раствором борной кислоты и титруют сильной *~слот°й. В другой аликвотной части раствора разрушают нитрит Д'У'Яьфаминовой кислотой и определяют нитрат.
143
Редокс-методы
Почти все титриметрические методы определения нитрита основаны на редокс-реакцпях. Для титрования применяют гппогалоге-наты, Ceiv, хлораты, хлорамин Т, тетраацетат свинца, КМпО-, Мп1’1, Со111, гексацианоферрат(III) и другие окислители. Эти методы описаны в работах [13, 14]. В большинстве случаев прямое титрование невозможно, поскольку реакция протекает медленно. Поэтому обычно приливают избыток окислителя, который оттитро-вывают. Важной проблемой является разложение или окисление HNO2 в ходе титрования, например, при использовании в качестве титранта КМпО4. Другая трудность связана с отсутствием подходящих индикаторов, например, при использовании гипохлорита в качестве окислителя. В этом случае рекомендуется конец титрования устанавливать потенциометрически или амперометрически. Можно применить также иодиметрический метод, восстанавливая избыток окислителя введением KI-
Прямое иодиметрическое определение возможно в строго контролируемых условиях:
2HNO2 + 2Г + 2Н+ —► 2NO + 12 + 2Н2О
Прямому титрованию иода препятствует выделяющийся NO, поскольку оксид азота окисляется кислородом с образованием HNO2. Титрование можно проводить только в отсутствие кислорода, что усложняет анализ.
Для определения ряда восстановителей применен щелочной раствор гексацианоферрата(III) в присутствии осмиевой кислоты в качестве катализатора. В работе [15] показано, что, хотя Fe(CN)e~ не окисляет нитрит в растворе, содержащем OsvnI, в присутствии сульфата цин,ка реакция протекает количественно;
2Fe (CN)t' + NO2 + 2ОН' —> 2Fe (CN)46‘ + NO3' + H2O 2K<Fe (CN)6 + 3ZnSO4 —> K2Zn3 [Fe (CN)6]2 + 2K2SO4
Метод прост. К известному объему раствора гексацианоферрата (III) добавляют 2—3 г ZnSO4 и раствор нагревают до кипения. Приливают раствор нитрита из бюретки до полного обесцвечивания титруемого раствора. Определению не мешают большие содержания нитрата. Метод рассчитан на определение 2—45 мг NOL
Возможность применения хлорамина Т в качестве окислителя изучена в работе [16]; предложен метод, являющийся модификацией известного п дающий удовлетворительные результаты [17]:
CH3C6H4SO2NC!Na + ,\'а\О2 + Н2О —► NaNO3 + CH3C6H4SO2NH2 + NaCl
CH3C6H4SO2NClNa + 2KI + 2HC1 —> CH3C6H4SO2NH2 + h + NaCl + KC1
Приведенная ниже методика обратного титрования основана на иодиметрическом окончании определения, 144
Титриметрическое определение нитрита с помощью гексацианоферрата(Ш) [16]
Реагенты
Хлорамин Т, примерно 0,05 Л1 раствор. Растворяют около 25 г натриевой соли п-толуолсульфохлорамида СНзСбРЦЗОгХОХа-ЗНгО в воде. Фильтруют в мерную колбу вместимостью 2 л и доводят объем раствора водой до метки. Хранят в темной склянке.
Ход анализа. Отбирают пипеткой 25 мл раствора хлорамина Т (или 50 мл при содержании нитрита более 30 мг) в коническую колбу вместимостью 500 мл с пробкой. Приливают раствор нитрита и разбавляют водой до 1.50 мл. Добавляют 10 мл 9 М раствора уксусной кислоты и через 2 мин—10 мл 10%-ного раствора KI. Помещают раствор на 5 мин в темное место и титруют стандартным раствором тиосульфата, применяя крахмал для определения конца титрования.
Аналогичным образом проводят контрольный опыт. Содержание нитрита рассчитывают по объему раствора тиосульфата, пошедшему на титрование, причем 1 мл 0,1 М раствора 52О2'соответствует 2,30 мг NO'.
Примечания. 1. Раствор хлорамина Т не очень устойчив, поэтому следует регулярно проводить контрольное титрование.
2. Максимально допустимые содержания посторонних ионов (дающих ошибку 0,3% при определении 25 мг NO') следующие: 1 мг S2', NH*, SCN'; 1 мг Fe(CN)g'; 5 мг Си"; 50 мг Г; 125 мг Вг', Fe (CN),', Hg11, Pbn, СЮ'.
3. Мешающее влияние Cu2+ и NH* можно устранить путем их ионообменного отделения.
Спектроскопические методы
Спектроскопические методы в видимой и ультрафиолетовой областях
Разработано большое число спектрофотометрических методов определения нитрита, некоторые из них являются разновидностью метода Грисса — Илосвая. Опубликован обзор [52] спектрофотометрических методов определения нитрита [18].
Большая группа методов основана на реакциях диазотирования и сочетания с образованием азокрасителей. В методе Грисса — Илосвая используют сульфаниловую кислоту и 1-нафтиламин [19, 20]. Показано, что 1-нафтиламин канцерогенен [21], поэтому его заменяют некоторыми другими соединениями, например 1-нафтиламин-7-сульфокислотой (кислота Клеве), N-1-нафтилэтилендиамином и Диметил-1-нафтиламином. Вместо сульфаниловой кислоты часто применяют сульфаниламид. Реакцию Грисса — Илосвая изучали .в течение нескольких десятилетий. Главные условия успешного применения метода описаны в работе [22]. Диазотирование следует проводить в сильнокислой среде при возможно более низкой температуре. Сочетание следует проводить после завершения диазотирования при минимально возможной кислотности.
Установлены оптимальные условия проведения реакции Грисса £ применением кислоты Клеве [23]. Метод менее чувствителен, ем метод с применением N-1-нафтилэтилендиамина, но следует ^метить большую устойчивость растворов кислоты Клеве [24].
Работе [25] применен один реагент, /г-фенилазоанилин, который
14Л
Рис. 22. Схема установки для автоматического определения нитрата и и нитрита в воде:
/ — змеевик; 2, 3, 5 —смесители; 4 — редуктор; 6 — устройство для подачи пробы; 7 — расширитель шкалы; S —самописец; 9 — к ?л метр.
Д-водт (0,*’65 мл мин); Б —проба (0,'J45 мл/мин); В—воздух (0,056 мл/мин); Г— о'о-ный раствор NH4CI + ХН4ОН (pH “ = 8,6, 0,665 мл, мин): Д-раствор (0,081 мл/мии); Е — 1,0%-ный раствор сульфаниламида в 1,2 Л1 растворе НС1 (0,040 мл/мни); Ж — 0,1%-ный раствор N-I-нафт и л эти леи-диамина дигидрохлорида (0,040 мл/мии); 3-линия протока через ячейку (0,073 мл/мии).
одновременно является диазотирующим и сочетаемым реа-тентом. Молярный коэффициент поглощения образующегося красителя равен 67000 при 959 нм, т. е. выше, чем красителя, образующегося в кислоты и N-1-нафтилэтилендиамина
присутствии сульфаниловой
(50 000 при 550 нм). При использовании кислоты Шеффера (6-гидр-оксинафталин-2-сульфокислота) [26] и сульфаниловой кислоты чувствительность определения низка (е = 12 000).
Описаны автоматические анализаторы для определения нитрата и нитрита в воде и экстрактах почв [27—29]. Схема потоков жидкости в анализаторе приведена на рис. 22.
В автоматическом анализаторе «Техникой» восстановление нитрата до нитрита осуществляют в колонке, заполненной омедненным кадмием; при этом определяется сумма нитратов и нитритов. Для
определения нитритов редуктор не используют.
Для повышения чувствительности метода Грисса образующийся краситель экстрагируют [30] или концентрируют на ионообменной смоле. Для диазотирования и сочетания используют один реагент — 8-аминохинолин, краситель экстрагируют н-гептанолом. Чувствительность определения составляет 0,0038 рргп при объеме раствора 40 мл, что отвечает оптической плотности 0,010, измеренной при 465 нм. Интервал определяемых содержаний ИО^равен 0,5—16 мкг. В работе [31] катион красителя экстрагируют четыреххлористым углеродом в виде ионного ассоциата с анионом к-додецилбензол-сульфатом, затем реэкстрагируют его соляной кислотой и измеряют поглощение при 543 нм. Реэкстракция необходима ввиду мутности органической фазы. Чувствительность метода (7-10~10 моль/л нитритного азота) значительно больше, чем без применения экстракции (2-Ю-8 моль/л нитритного азота).
Другой метод с применением экстракции описан в работе [62]. Диазотируют лг-аминоацетофенон, а сочетание проводят с л«-фени-лендиамином. При экстракции толуолом (pH = 9) и измерении поглощения в области 450 нм молярный коэффициент поглощения равен 2,3-104, что позволяет определять 1—30 ppm нитрита.
14R
шают S2O3’, Cu2+ и Fe3+, но влияние Feii! можно устранить введением фосфатов; применять ЭДТА для .маскировки нельзя, поскольку этот лиганд мешает определению.
Описано концентрирование красителя на ионообменной колонке [32], заполненной смолой Дауэкс Х-8, для элюирования применяют 60%-ную уксусную кислоту. Чувствительность определения азота 1 10_9 моль/л, интервал определяемых содержаний 10*9— Ю-7 моль/л нитритного азота при объеме анализируемой пробы 1—5 л.
Из приведенных выше данных следует, что невозможно указать чувствительность метода Грисса, поскольку она зависит от применяемых реагентов и условий определения. С применением N-1-наф-тнлэтилендиамина и сульфаниловой кислоты получено значение е = 50 000. Избирательность метода зависит от реагентов и условий определения. В ряде работ избирательность указана для 1-наф-тиламина, однако в настоящее время применять этот реагент не рекомендуется. Иногда авторы просто указывают мешающие ионы или приводят максимально допустимые их содержания. Например, в стандартном методе, где для определения 1 —15 мкг нитритного азота [33] в промышленных водах используют сульфаниловую кислоту и 1-нафтиламин-7-сульфокислоту (кислота Клеве), указаны следующие допустимые содержания ионов (в мкг): СГ, SO4' (5000), Са2+ (2 500), Cu2+, Mg2+, Fe2+ (1000), Fe3+, метафосфат (в пересчете на Р2О5) (500), NH4 (2000). Определению с помощью 1-нафтиламина мешают Cl2, NC13, Fe3+, Hg1, Hg”, Ag1, Bi”1, Sb1”, Pb2+, Au”1, Cu2+, хлорплатинат, метаванадат, карбамид, алифатические амины, сильные окислители и восстановители.
Для спектрофотометрического определения нитрита чаще всего используют разные модификации метода Грисса. Описаны методы анализа NH(NO3 [34], мяса [35], воздуха [4], крови [37], пищевых продуктов [38], в том числе для детского питания [39].
Нашли применение методы, основанные не на реакции Грисса. Предложен чувствительный метод, основанный на образовании интенсивно окрашенных свободных радикалов [36]. В качестве реагента применен 1-метил-2-хинолин азин, для которого е = *= 1270000 [26] значительно превышает предельное значение, ’рассчитанное теоретически (е = 200 00); закон Бера не соблюдается. Определению не мешает сульфит, который препятствует 'Определению нитрита по реакции Грисса.
В ряде работ изучен метод определения нитрита с помощью бруцина [40—42]. Линейность градуировочного графика наблюдается в смеси H2SO4 — НСЮ4, но не в среде H2SO4 [42]. Поглощение измеряют при 430 нм, определению мешают в основном ионы Железа (III).
В работе [43] показано, что методом с применением 2,6-ксиле-.£К>ла в отличие от метода Грисса можно раздельно определять ЙИтраты и нитриты при их совместном присутствии, причем, кроме ./Галогенидов, восстанавливающих нитрит, определению не мешаки
147
другие наиболее распространенные ионы. Другой недостаток метода Грисса заключается в том, что он позволяет определять только небольшие содержания нитрита ввиду малой растворимости азокрасителя.
Некоторые ароматические о-диамины взаимодействуют с нитритом [44], продукты реакции можно экстрагировать «-бутанолом или изоамиловым спиртом. Закон Бера соблюдается при «.одержании от 2 до 10 мкмоль NOj в 50 мл раствора. Например. З.З'-дна-минобензпдин образует с NO7 продукт, имеющий значение е — 3 500 при 350 нм. Определению не мешают многие ионы, мешают окислители. Влияние MoVI, Сг1’1, Си, Ni'n Fe3+ устраняют введением ЭДТА или тартрата; мешают серебро и селен.
Описан метод, основанный на восстановлении нитрита резорцином в кислой среде. Образующееся нитрозосоединение при взаимодействии с цирконил-ионом дает желтый хелат, характеризующийся значением s = 2,67-104 при 347 нм [63]. Окраска раствора устойчива в течение 24 ч. Метод достаточно селективен. Интервал определяемых содержаний нитрита от п-10 ppm до 1 ppm. Из присутствующих в воде ионов мешают только Fe3+ и фосфаты, но их влияние можно устранить.
Спектрофотометрическое определение нитрита модифицированным методом Грисса — Илосвая [23]
Метод позволяет определять 1—10 мкг нитритного азота.
Реагенты
Сульфаниловая кислота. Растворяют 0,5 г препарата в растворе, содержащем 30 мл ледяной уксусной кислоты и 120 мл воды. Хранят в темной склянке.
Стандартный раствор нитрита. Растворяют 0,493 г нитрита натрия (для анализа) в воде в мерной колбе вместимостью 1 л. Непосредственно перед применением разбавляют 10 мл раствора до 1 л, получают раствор, содержащий 1 мкг/мл нитритного азота.
Ход анализа. Аликвотную часть раствора, содержащего нитрит, помещают в мерную колбу вместимостью 50 мл и приливают воду до 40 мл, 2 мл раствора сульфаниловой кислоты и оставляют на 10 мин для диазотирования. Затем добавляют 2 мл раствора кислоты Клеве и оставляют иа 20 мин для завершения реакции сочетания. Измеряют поглощение при 525 нм в кювете с толщиной слоя 1 см. В качестве раствора сравнения используют воду.
Нитрит-ион сравнительно слабо поглощает в УФ-области (е » « 23 при 355 нм), поэтому его обычно переводят в соединения, которые можно определять с большей чувствительностью. Разработан метод раздельного определения NO3 и NO2 по поглощению в УФ-области [45], основанный на поглощении нитрита при 355 нм и обоих ионов при 302 нм. Метод позволяет определять 0,02 мг/мл нитрита и 0,09 мг/мл нитрата.
В работе [46] приведен обзор методов определения нитрита по поглощению в УФ-области. Для определения нитрита предложен антипирин, позволяющий определять 0,1 —12 ppm нитрита. Молярный коэффициент поглощения продукта реакции равен 7,61 • 103 при 343 нм. Определению не мешают Си2+, Мп2+, Hg", Mg, Ni, Са» Вг’, СГ, F’, NO3, РОГ, СЮ4 и оксалат. Полный перечень ионов приведен в работе [46].
148
Флуориметрический метод
Предложен флуориметрический метэд определения нитрита с применением диазотированного 2,3-дпампнонафталпна [47]. Определению 0—850 нг нитрита не мешают NH4 и NO/, мешают Си11, Al, Bi, Ni, Сг’’’, Sn", SeIV и Fe11’.
Очень чувствительный флуориметрический метод основан на диазотировании /г-хлоранилпна и сочетании с 2,6-диампноппридп-ном. Образующееся при реакции азосоединение взаимодействует с CuSO4 и образует интенсивно флуоресцирующий триазол. Максимум флуоресценции наблюдается при 430 нм при возбуждении 360 нм. Предел обнаружения нитрита составляет 2 нг/мл при объеме пробы 10 мл [48].
Атомно-абсорбционный метод
В работах [49, 50] отделяли путем экстракции метилизобутил-кетоном ионную пару нитрата с 2,9-диметил-1,10-фенантролинатом меди(1). Содержание нитрата определяли косвенно, методом ААС, по поглощению меди. В работе [51] этот метод применен для определения нитрита после его окисления церием (IV).
Электрохимические методы
Потенциометрический метод
Показано [52], что нитрит можно титровать тетраацетатом свинца в хлорцдном растворе (для связывания образующегося РЬ2+), используя платиновый и насыщенный каломельный электроды. В конечной точке скачок потенциала составляет 250 мВ на 0,01 мл титранта, а относительная ошибка определения 2—35 мг нитрита составляет 0,17%. В качестве титранта применяют также раствор Со111 [53].
Для определения нитрита применяют ионоселективные электроды. Нитратный электрод дает сигнал также в присутствии нитрита [54]. Нитратный электрод применен для анализа смеси нитратов и нитритов [55]. Сначала определяют потенциал смеси, а Мтем, после окисления нитрита раствором КМпО4, снова измеряют Потенциал раствора. Содержание компонентов определяют по уравнению. Метод применен для анализа сигаретного дыма после его поглощения щелочным раствором.
Другие электрохимические методы
Предложен метод кулонометрии при постоянном потенциале, основанный на прямом окислении нитрита до нитрата на платиновом электроде [56]. Метод обладает хорошей точностью, однако время электролиза превышает 20 мин.
В работе [57] показано, что при анализе мяса метод Грисса — "Лосвая дает недостаточно точные результаты. Разработан куло-
149
неметрический метод определения при постоянном потенциале, основанный на восстановлении нитрита йодидом с образованием иода
2HNO2 + 2Г + 2Н+ —► 12 + 2X0 + 2Н2О
Метод позволяет определять от 0,005 до 5 мг нитрита в течение 5 мин с ошибкой ±0,1%. Описан также метод анодного амперометрического титрования малых содержаний нитрита раствором сульфаминовой кислоты или церия (IV) [58].
Другие методы определения
Описан новый метод определения нитрита, основанный на определении газов СО2 и N2O, выделяющихся при добавлении к нитриту безводной муравьиной кислоты [59]. Содержание N2O определяют газометрически, а СО2 — гравиметрически. Аналогичный метод предложен для анализа смеси нитратов и нитритов [60].
Метод кольцевой печи использован для определения нанограм-мовых содержаний нитрата и нитрита порознь и при их совместном присутствии [61].
ЛИТЕРАТУРА
1. Wolff I. A., Wasserman А. Е.— Science, 1972, v. 177, р. 15.
2. Egan Н. — Fd Cosmet. Toxic., 1971, v. 9, p. 81.
3. Anon., Nature, bond., 1972, v. 239, p. 63.
4. Jacobs M. B.t Hochheiser S. — Analyt. Chem., 1958, v. 30, p. 426.
5. Clear A. J. — In: Treatise on Analytical Chemistry (Ed. I. M. Kolthoff and P. J. Elving), Part II, Vol. 5, New York, Interscience, 1961, p. 275.
6. Streuli C. A., Averell P. A. — Analytical Chemistry of Nitrogen and its Compounds, Chapter 4, New York, Wiley-Interscience, 1970.
7. Sawicki E., Pfaff J., Stanley T. W. — Rev. Univ. Ind. Santander, Columbia, 1963, v. 5, p. 337.
8. Bark L. S. — Ind. Chemist, 1961, p. 608.
9. Canic V. D., Turcis M. N., Bugarski-Vojinovic M. B., Perisic N. U. — Z. analyt. Chem., 1967, v. 229, p. 93; Analyt. Abstr., 1968, v. 15, p. 5211.
10. Kawanabe K., Takitani S., Miyazaki M., Tamura Z.— Japan Analyst, 1964, v. 13, p. 976; Analyt. Abstr., 1966, v. 13, p. 5385.
11. Bock R., Niederauer H.-T. — Z analyt. Chem., 1962, v. 190, p. 33.
12. Bremner J. M.. Keeney D. R. — Analytica chim. Acta, 1965, v. 32, p. 485.
13. Kolthoff I. M., Belcher R. — Volumetric Analysis, Vol. Ill, New York, Interscience, 1957.
14. Berka A., Vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Pergamon Press 1965
15. Sant B. R. — Analytica chim. Acta, 1958, v. 19, p. 523.
16. Agderdenbos J. — Taianta, 1970, v. 17, p. 238.
17. Mitchell A. D., Ward A. M. — Modern Methods in Quantitative Chemical Analysis, London, Longmans Green, 1932, p. 150.
18. Sawicki E., Sanley T. W., Pfaff J., D’Amico A. — Taianta, 1963, v. 10, p. 641.
19. Griess P. — Ber., 1879, v. 12, p. 427.
20. Ilosvay M. L. — Bull. Soc. chim., 1889, v. 2, p. 317.
21. Precautions for Laboratory Workers Who Handle Aromatic Amines, London. Chester Beatty Research Institute, 1965.
22. Rider B. F„ Mellon M. G.— Ind. Engng Chem. analyt. Edn, 1946, v. 18, p. 96. 23. Bunton N. G.. Crosby N. T., Patterson S. J.—Analyst, 1969, v. 94, p. 585. 24. Personal communication, N. T. Crosby.
25. Sawicki E., Stanley T. W., Elbert W. C. — Analyt. Chem., 1962, v. 34, p. 297. 26. Kieruczenkowa A. — Chemia analit., 1967, v. 12, p. 1031; Analyt. Abstr., 1968, v. 15, p. 7231.
150
27. Kamyhake L. J., Hannah S- A., Cohen 1. AL— Wat. Res., 1967, V. 1, p. 205; Analyt. Abstr.. 1968. v. 15. p. 3627.
28. Henriksen .1. — Analyst. 1965, v. 90, p. 83.
29. Henriksen A., Selmer-Olsen A. R.— Analyst, 1970, v. 95. p. 514.
30. Foris A., Sweet T. R. — Analyt. Chem., 1965, v. 37, p. 701.
31. Matsur.aga К., Oyatna T., Nishimura Al. — Analvtica chim. Acta, 1972, v. 58, p. 228.
32. Wada E., Hattori A.— Analytica chim. Acta, 1971, v. 56, p. 233.
33. British Standard 2690: Part 7 (1968) London.
34. British Standard 4267: (1968) London.
35. Official. Standardised and Recommended Methods of Analysis, 2nd edn (Edited and compiled by N. W. Hanson), London, The Chemical Society, 1973.
36. Sawicki E., Stanley T. IE., Pfaff J., Johnson H. — Analyt. Chem., 1963, v. 35, p. 2183.
37. Schechter H., Gruener N., Shuval H. I. — Analytica chim. Acta, 1972, v. 60, p. 93.
38. Schall E. D., Hatcher D. IE. — J. Ass. off. analyt. Chem., 1968, v. 51, p. 763.
39. Kamm L., Bray D. F., Coffin D. F. — J. Ass off. analyt. Chem., 1968, v. 51,
p. 40.
40. Fisher F. L., Ibert E. R., Beckman H. F. — Analyt. Chem., 1958, v. 30, p. 1972.
41. Fabrus H., Maly J. — Cesk Hug., 1968, v. 13, p. 300; Chem. Abstr., 1968, v. 69,
p. 61437.
42. Saito G., Sugimoto K-, Hagino K-— Japan Analyst, 1971, v. 20, p. 542; Analyt. Abstr., 1972, v. 23, p. 2415.
43. Hartley A. M., Asai R. I. — Analyt. Chem., 1963, v. 35, p. 1214.
44. Lin E., Cheng K. L.— Mikrochim. Acta, 1970, p. 652.
45. Welters J. H., Uglum K- L. — Analyt. Chem., 1970, v. 42, p. 335.
46. Weiss K. G., Boltz D. F. — Analytica chim. Acta, 1971, v. 55, p. 77.
47. Wiersma J. H. — Analyt. Lett., 1970, v. 3, p. 123.
48. Dombrowski L. J., Pratt E. J. — Analyt. Chem., 1972, v. 44, p. 2268.
49. Kumamaru T., Tao E., Yamamoto У. — Bull. chem. Soc. Japan, 1965, v. 38, p. 2204; Analyt. Abstr., 1967, v. 14, p. 1934.
50. Yamamoto Y., Kumamaru T., Hayashi Y., Otani Y.— Japan Analyst, 1969, v. 18, 1 p. 359; Analyt. Abstr., 1972, v. 22, p. 105.
,51. Houser AL £., Fauth AL L—Microchem. J., 1970, v. 15, p. 399.
52. Morales A., Zyka J. — Colin Czech, chem. Comtnun, 1962, v. 27, p. 1029; Analyt. Abstr., 1962, v. 9, p. 4637.
,53. Hanif M., Dolezal J., Zyka J. — Microchem. J., 1971, v. 16, p. 291.
'54. Digregorio J. S., Morris M. D. — Analyt. Chem., 1970, v. 42, p. 94.
55. Marie G. P., Ledford C. J., Glover C. A. — Analytica chim. Acta, 1972, v. 60, p. 397.
56. Harrar J. E. — Analyt. Chem., 1971, v. 43, p. 143.
57. Karlsson R., Torstensson L.-G. — Taianta, 1974, v. 21, p. 945.
§8. Stock J. T., Bjork R. G. — Taianta, 1964, v. 11, p. 315.
89. Awad W. L, Hassan S. S. AL, Zaki AL T. M. — Taianta, 1971, v. 18, p. 219.
60. Van Slyke D. D., Lo Monte A. F. — Microchem. J., 1969, v. 14, p. 608.
«1. Weisz H., Hanif M. — Analytica chim. Acta, 1976, v. 81, p. 179.
62. Toei K-, Kiyose T. — Analytica chim. Acta, 1977, v. 88, p. 125.
63. Gabbay J., Almog Y., Davidson M., Donagi A. E. — Analyst, 1977, v 102 p. 371.
ОКСАЛАТЫ
МЕТОДЫ ОТДЕЛЕНИЯ
. Для отделения оксалата предложено несколько методов.
Метод тонкослойной хроматографии
Методом ТСХ на целлюлозе можно отделять оксалат от солей ВРУгих карбоновых кислот [1] (см. табл. 6).
151
Газохроматографический метод
Метод газовой хроматографии может быть применен для отделения оксалата, а также бората, карбоната, фосфата, сульфата, арсената, фосфита и ванадата в виде их триметилсилил-производ-ных [2], которые образуются при взаимодействии с раствором аммонийной соли трифтор-бис (триметилсилил) ацетамида в диме-тилформамиде. Каждому аниону отвечает один пик на хроматограмме.
Экстракция
Оксалат можно экстрагировать на 99% раствором трифенил -станнония РЬз5п+ из раствора с pH = 1 —2,3.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Приведен обзор методов определения оксалата в биологических материалах [4].
Гравиметрические методы
Описаны методы гравиметрического определения оксалата в виде оксалата тория или кальция [5—7].
Соль тория взвешивают в виде дигидрата после высушивания при 105—110 °C или в виде моногидрата после сушки при 200— 220 °C. Определению не мешают малеиновая, лимонная, винная и янтарная кислоты.
Путем переосаждения оксалата кальция также можно отделить щавелевую кислоту от других карбоновых кислот. Мешающее влияние кислот убывает в ряду: винная, лимонная, янтарная кислоты. Хлорид натрия оказывает небольшое влияние, но в присутствии солей магния ошибка может достигать 5%.
Титриметрические методы
В литературе описаны методы кислотно-основного, окислительно-восстановительного и комплексометрического титрования оксалата. Известный ранее иодиметрический метод в настоящее время менее распространен.
Методы кислотно-основного титрования
Щавелевая кислота является достаточно сильной (pKai = 1,25, рК а2= 4,28) для прямого титрования гидроксидом натрия.
Хорошие результаты получены при титровании в присутствии хемилюминесцентных индикаторов. В щелочной среде люцигенин (диметилакридиния динитрат) флуоресцирует в присутствии Н2О2 зеленым светом. Флуоресценция усиливается при введении флуоресцеина. Смесь указанных индикаторов рекомендована для титрования оксалата гидроксидом натрия. Результаты улучшаются, если титрование начинать при 60 °C. Определению мешают уксусная, винная, лимонная кислоты [8].
152
Редокс-методы
Описаны методы титрования оксалата растворами KMnO4, CeIV, Со"1, КВгОз и КЮз.
Реак-ция взаимодействия оксалата с перманганатом характеризуется сложным механизмом [15]. Точные результаты можно получить в строго контролируемых условиях [16]. Сначала необходимо достаточно быстро добавить около 90% от общего объема раствора КМпО4, дождаться его обесцвечивания, нагреть раствор до 55— 60 °C и завершить титрование медленным добавлением КМпО4 до неисчезающей в течение 30 с слабой розовой окраски раствора. Высокая точность достигается, вероятно, вследствие компенсации ошибок разного знака.
При титровании оксалата церием (IV) возможен ряд осложнений. Титрант неустойчив в перхлоратной среде. В результате детального исследования [9] установлены источники ошибок, которые можно устранить. Важными обстоятельствами являются следующие.
1. При низких концентрациях реагентов скорость реакции велика в широком интервале концентраций НС1 от 0,2 до 6,0 М.
2. При высоких концентрациях реагентов скорость реакции сначала высока, но со временем снижается.
3. Сульфат уменьшает, a IC1 увеличивает скорость реакции.
Показано, что для проведения реакции в отсутствие катализатора и без нагревания раствора необходимо предварительно удалить сульфат путем осаждения его солью бария.
В работе [10] рекомендован метод потенциометрического титрования оксалата гексанитроцерратом(1У) аммония в среде НС1 или HNO3 в присутствии небольших содержаний KI и КЮ3, вводимых для установления постоянного потенциала. В этих условиях реакция протекает быстро даже при комнатной температуре. Относительная ошибка при титровании 0,05 и 0,01 М растворами CeIV равна соответственно ±0,1 и 0,4%.
Взаимодействие CeIV с оксалатом изучено недавно в связи с разработкой высокоточного метода стандартизации раствором Церия (IV) [11]. Для получения точных результатов рекомендуется потенциометрический метод. При визуальном титровании к раствору оксалата приливают избыток CeIV, который титруют раствором же-леза(П).
Титриметрическое определение оксалата
Ход анализа. Раствор 1 М H2SO4, содержащий оксалат, нагревают до 60 °C И добавляют достаточно быстро избыток 0,1 М раствора CeIv. Через несколько Минут раствор охлаждают и титруют 0,1 М раствором Fe11 с ферроином в качестве индикатора.
Комплексометрический метод
Если определяемый ион может количественно взаимодействовать с окислителем с образованием иона металла, дающего устойчивые Комплексы с ЭДТА, эта реакция может быть положена в основу Комплексометрического метода.
153
Гак, давно известен метод, основанный на окислении оксалата раствором 1\МпО4 в спльнощелочной среде, отделении MnCh, восстановлении диоксида марганца аскорбиновой кислотой с последующим титрованием марганца(II) раствором ЭДТА. В работе [12] восстановление КМпО4 проводят в кислой среде и после добавления аскорбиновой кислоты титруют марганец(П) раствором ЭДТА в щелочной среде.
Описан другой метод, основанный на осаждении оксалата избытком свинца(П) и титровании свинца(П) раствором ЭДТА в присутствии ппрокатехпиового фиолетового как индикатора [13]. Метод экспрессен и позволяет определять 3—10 мг щавелевой кислоты.
Комплексометрическое определение оксалата [12]
Реагенты
ЭДТА, 0,05 М раствор.
Метилтимоловый синий, смесь с KNO3 (1 -. 100).
Ход анализа. К 5—25 мл слабощелочного раствора, содержащего 40—220 мг оксалата, приливают 10 мл 20%-ного раствора H2SO4 и воду до объема 30— 40 мл. Нагревают раствор до 80 °C и добавляют по каплям 0,02 М раствор КМпО4, свежеотфильтрованный через стеклянный фильтр, до появления слабофиолетовой окраски.
Раствор охлаждают, добавляют 1 каплю 5%-ного водного раствора сульфита натрия для обесцвечивания раствора и 0,5 г аскорбиновой кислоты. Затем добавляют 15 мл 0,880 М раствора аммиака, несколько капель индикатора метилтимолового синего и титруют марганец(П) точно 0,05 М раствором ЭДТА.
Иодиметрический метод
Щавелевая кислота является одной из карбоновых кислот, которую можно определять прямым иодиметрическим титрованием. В результате взаимодействия оксалата со смесью иодида и йодата в присутствии солей кальция или магния выделяется иод:
Ю3" + 51' + 6Н+ —> 312 + ЗН2О
Спектроскопические методы
Предложены косвенные спектрофотометрические методы определения оксалата. Один из методов основан на восстановлении оксалата до гликолевой кислоты и последующем определении ее с помощью хромотроповой кислоты [14].
Опубликован очень чувствительный косвенный метод, основанный на уменьшении поглощения красного комплекса урана(Щ) с 4-(2-пиридилазо) резорцином (ПАР) в присутствии оксалата [17]. Это один из методов, позволяющий определять оксалат на уровне ppm. Уменьшение поглощения растворов при 515 нм пропорционально содержанию оксалата в интервале 0—3 ppm. Метод относительно прост в исполнении.
Ниже приведены ионы, мешающие определению оксалата с применением комплекса урана (IV) с 4-(2-пиридилазо) резорцином
154
[17]. В скобках указано содержание иона (в ppm), в присутствии которого относительная ошибка определения 4,00 ppm оксалата превышает 2%: Fe3+ (9), NH] (107), F~ (8), Br~ (114), I" (200), NO3, SO? Cl", SO? (200), ЭДТА (21), NOT(45), ацетат (138), тартрат (47), цитрат (32) PO? (18).
Один из прямых методов определения оксалата основан на взаимодействии его с индолом в определенных условиях с образованием красного или розового соединения [18]. Метод не очень чувствителен (0,05—1,00 мг/мл оксалата), но определению не мешают уксусная, пропионовая, винная, лимонная, бензойная и мочевая кислоты. Мешают большие содержания хлорида и фосфата.
Недавно предложен метод, основанный на уменьшении поглощения комплекса железа (III) с 5-нитросалицилатом при 492 нм при введении оксалата [19].
Микросодержания оксалата определяют также спектрофлуори-метрическим методом, основанным на том, что оксалат восстанавливает Ce'v до Се1'1, который флуоресцирует. Этот метод значительно чувствительнее, чем абсорбционная спектрофотометрия [20]. При измерении флуоресценции в области 350 нм и возбуждении 260 нм можно определять 8,8—44 мкг оксалата. Реакцию восстановления Ce'v можно применить также для определения мышьяка (III).
Предложен метод, основанный на гашении флуоресценции хелата циркония с 3-гидроксифлавоном в среде разбавленной серной кислоты в присутствии оксалата [21]. Метод очень избирателен, прости чувствителен (1—10 мкг оксалата).
Определению 5 мкг оксалата в 100 мл раствора не мешают следующие ионы [21] (указаны допустимые содержания в мкг): UO? (6000), ВгОз (5000), SiO? (4000), Юз (1000), А13+ (500), NO2 (400), РО?(200), Сг2О? (200), Fe3+(100), WO? (80), МоО? (20), 107(20), Цитрат (20), F-(l).
Электрохимические методы
Для определения оксалата предложены также амперометрический [22, 23] и потенциометрический [10] методы. Оксалат титруют при pH = 4,6 электрогенерированным лантаном (III). В интервале содержаний оксалата 0,5—2,6 мг ошибка составляет 2% [24].
Используют ионоселективные электроды. Для определения $.3—33 мг оксалата при pH — 7 — 11 применяют титрование в присутствии электрода, селективного к кальцию [25]. В работе [26] Ирименен свинец-селектнвный электрод, что позволяет осуществлять потенциометрическое титрование 1—25 мг щавелевой кислоты АВ среде 40%-ного н-диоксана) раствором перхлората свинца. Определению мешают цитраты и другие лиганды, образующие устойчивые комплексы со свинцом (II), а также анионы, образующие *° свинцом (II) нерастворимые соединения.
155
Другие методы определения
Разработаны ферментативные методы, пригодные для анализа образцов биологического происхождения. Для определения оксалатов в плазме крови применен специфический фермент оксалат-декарбоксплаза. Щавелевая кислота декарбоксилируется ферментом с образованием СОг:
(СООН)2 —-> со2 + нсоон
Тот же принцип использован в работе [28] для определения оксалата с высокой чувствительностью с помощью автоматического анализатора Техникой. Минимально определяемое содержание оксалата составляет 5 мкг в 100 мл раствора. Фосфат и сульфат ингибируют ферментативную активность; влияние этих ионов можно учесть, если вводить их в раствор при построении градуировочного графика.
ЛИТЕРАТУРА
1. Dittman J. — J. Chromat., 1968, v. 34, р. 407.
2. Butts U7. C., Rainey W. T. — Analyt. Chem., 1971, v. 43, p. 538.
3. Bock R., Niederauer H.-Т., Behrends K.— Z. analyt. Chem., 1962, v. 190, p. 33.
4. Hodgkinson A— Clin. Chem., 1970, v. 16, p. 547.
5. Sarudi I. — Z. analyt. Chem., 1964, v. 203, p. 106.
6. Sarudi I. — Z. analyt. Chem., 1965, v. 211, p. 281.
7. Sarudi I.— Pharm. Zentralhalle Dtl., 1967, v. 106, p. 217; Analyt. Abstr., 1968, v. 15, p. 2984.
8. Erdey L., Pickering It7. F., Wilson C. L. — Talanta, 1962, v. 9, p. 371.
9. Rao V. P., Rao G. G. — Talanta, 1959, v. 2, p. 370.
10. Rao G. G._ Murty K. S., Rao P. V. K. — Talanta, 1963, v. 10, p. 657.
11. Zielen A. j. — Analyt. Chem., 1968, v. 40, p. 139.
12. Szekeres L., Kardos E., Szekeres G. L.— Chemist Analyst, 1965, v. 54, p. 116.
13. Crisan I. A., Krausz R.— Studia. Univ. Babes-Bolyai, Ser. Chem., 1967, v. 12, p. 19; Analyt. Abstr., 1968, v. 15, p. 6720.
14. Hordgkinson A., Zarembski P. M. — Analyst, 1961, v. 86, p. 16.
15. Kopthoff I. Al., Belcher R. — Volumetric Analysis, Vol. Ill, London, Interscience, 1957, p. 48.
16. Laitinen H. A. — Chemical Analyst, London, McGraw-Hill, 1960, p. 363.
17. Neas R. E., Guyon J. C. — Analyt. Chem., 1972, v. 44, p. 799.
18. Bergerman J., Elliot J. S. — Analyt. Chem., 1955, v. 27, p. 1014.
19. Lee K- S„ Lee D. W., Hwang J. У. — Analyt. Chem, 1968, v. 40, p. 2049.
20. Kirkbright G. F„ West T. S., Woodward C. — Analytica chim. Acta, 1966, v. 36, p. 298.
21. Britton. D. A., Guyon J. C. — Analytica chim. Acta, 1969, v. 44, p. 397.
22. Борк. В. А., Сальникова К. С.—ЖАХ, 1968, т. 23, с. 901; Analyt. Abstr., 1970. v. 18, р. 756.
23. Silverman N. Р„ Skook D. A. — Analyt. Chem., 1963, v. 35, p. 131.
24. Curran D. /., Fletcher K- S. — Analyt. Chem., 1969, v. 41, p. 267.
25. Mukherfi A. K-— Analytica chim. Acta, 1968, v 40, p. 354.
26. Selig W. — Microchem. J., 1970, v. 15, p. 452.
27. Crawhall J. C., Watts R. W. E. — Clin. Sci., 1971, v. 41, p. 213.
28. Knowles C. F., Hodgkinson A. — Analyst, 1972, v. 97, p. 474.
ПЕРМАНГАНАТЫ
Перманганат находит применение в промышленности, например, как окислитель и в предварительных операциях очистки в металлургии.
156
Перманганат давно используют в анализе. Несмотря на применение других окислителей, перманганат до сих пор находит широкое аналитическое применение, например, для определения общей загрязненности воды по потреблению кислорода на окисление примесей. Результат анализа стандартным методом с применением перманганата выражают как химически потребляемый кислород. Методика основана на взаимодействии пробы с подкисленным раствором КМпО4, обычно при 27 °C. Затем титриметрически определяют избыток перманганата [1, 2].
МЕТОДЫ ОТДЕЛЕНИЯ
Известны методы отделения перманганата, основанные на взаимодействии с крупным органическим катионом, например тетра-фениларсонием (C6H5)4As+. Перманганат принадлежит к группе анионов, которые образуют нерастворимые ионные пары с такими катионами. Изучена экстракция таких ионных пар хлороформом [3]. Раствор тетрафениларсония в хлороформе количественно экстрагирует перманганат в интервале pH = 1 — 12 [4]. В широком интервале pH перманганат извлекается тетрафенилфосфонием [5] (С6Н5)4Р+ и трифенилсульфонием (С6Н5)з5+ [6].
Изучена возможность применения некоторых аминов в качестве экстрагентов перманганата [7]. Эффективными экстрагентами являются только дидодециламин и дигексиламин в бензоле. Максимум поглощения экстрактов в области 340 нм сохраняется даже после появления коричневой окраски.
В работах [8, 9] описано соосаждение перманганата с солями ,PbSO4 и BaSO4.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Большинство методов определения перманганата основано на окислительно-восстановительных реакциях.
Титриметрические методы
Редокс-методы
Определение перманганата в отсутствие посторонних ионов не представляет трудностей. Стандартизация растворов перманганата обсуждена подробно и описана в большинстве руководств по аналитической химии [10—12]. Прямое титрование перманганата раствором восстановителя невозможно, поскольку конечная точка маскируется окрашенными продуктами реакции. Прямое титрование возможно, если анализируемый раствор перманганата прили-МТ£> из бюретки в отмеренный объем раствора восстановителя. ток*1*0 Добавлять к анализируемому раствору перманганата избы-избыВ°ССТаН0ВНтеля’ напРимеР> щавелевой кислоты, и титровать к восстановителя стандартным раствором перманганата;
157
Ючность стандартизации растворов перманганата обычно не хуже — 0,1%. Для рутинного анализа приемлема менее высокая точность вследствие способности растворов перманганата к разложению.
Ниже обсуждены некоторые реагенты и методы определения перманганата. Теоретические вопросы рассмотрены в цитированной ранее литературе.
Триоксид мышьяка, который можно получить очень чистым, является, вероятно, наилучшим вторичным стандартом для высокоточного определения перманганата. В определенных условиях взаимодействие мышьяка(III) с перманганатом протекает без побочных реакций, строго по уравнению
4МпО; + 5As2O3 + 12Н+ —► 5As2O5 + 4Мп2+ + 6Н2О
В качестве катализаторов реакции применяют Г, Юз или IC1. Определение проводят визуально или потенциометрически. Обычно перманганат сам служит индикатором конца титрования. Можно применять также ферроин — комплекс о-фенантролина с железом (II). Определение часто проводят по методике, описанной в работе [13]. Метод не селективен, определению мешают многие окислители. Подробная методика приведена ниже.
Для точной стандартизации перманганата обычно используют оксалат натрия:
5С2О|- + 2МпО; + 16Н+ —► ЮСО2 + 2Мп2+ + 8Н2О
Вследствие образования кристаллогидратов щавелевую кислоту обычно не рекомендуют в качестве стандарта. Взаимодействие оксалата с перманганатом протекает по сложному механизму, для точных определений необходим строгий контроль условий анализа. Определению мешают некоторые окислители. Обычно рекомендуют методику анализа, предложенную в работе [14], ее детали описаны ниже.
Перманганат можно определять с помощью стандартного раствора железа (II):
10Fe2+ + 2МпО; + 16Н+ —► 10Fe3+ + 2Мп2+ + 8Н2О
При титровании в среде НС1 ионы железа могут индуцировать окисление хлорида перманганатом до С12 или НОС1. Ионы Мп11 ингибируют эту реакцию. Образование окрашенного в желтый цвет хлорида железа (III) маскирует конечную точку титрования. Хлорид железа (III) можно замаскировать введением орто-фосфорной кислоты, образующей бесцветный фосфатный комплекс железа(III). Раствор, содержащий марганец(П) и фосфат, называемый реагентом Циммерманна — Райнхарта, применяют при титровании перманганата.
В качестве хорошего вторичного стандарта для определения перманганата используют устойчивое соединение железа (II), этилендиамина и сульфата CsH^NHshHsSOrFeSCU^IbO [15]. Это соединение можно получить в высоко чистом виде, оно устой
158
чиво в твердом состоянии в течение длительного времени. Сульфат железа (II)-аммония не применяют для точных определений, поскольку железо(П) подвергается спонтанному окислению при хранении растворов.
Метод титрования железом (II) неизбирателен, поскольку определению мешают некоторые окислители. Описан комплексометрический вариант метода [16]. Образующееся при реакции железо (III) титруют раствором ЭДТА в присутствии салициловой кислоты как индикатора. Этот метод применим также для определения бромата и персульфата.
Для окислительно-восстановительного титрования перманганата применяют также другие восстановители, в том числе соединения Си1, Sn11, W111, L”v, V1V [17], однако преимущества применения этих восстановителей не очевидны. Сравнительно простой окислительно-восстановительный метод основан на взаимодействии перманганата с кислым раствором KI и последующем титровании выделившегося иода стандартным раствором тиосульфата. Избирательность определения часто достигается применением маскировки. Например, в указанном выше методе перманганат (или хромат) можно определять в присутствии железа (III), которое маскируют гексаметафосфатом [18]. При определении 13—65 мг марганца (в виде МпОД в присутствии 10—340 мг Fe3+ ошибка составляет ±0,3%.
Разработаны методы определения перманганата и других окислителей с применением аскорбиновой кислоты [19]. Один из методов основан на добавлении к перманганату избытка стандартного раствора железа (II) и титровании образующегося желе-за(Ш) аскорбиновой кислотой. Конечную точку можно определять потенциометрически.
Предложен интересный метод окислительно-восстановительного титрования, основанный на применении гидрохинона [20]. Метод позволяет определять 10—130 мг перманганата в присутствии Других окислителей, например, бихромата, гексацианоферрата (III) и хлорамина Т. При определении 13—130 мг КМпО4 в присутствии 20—2000 мг других окислителей относительная ошибка определения не превышает 1,8%. Определению мешает ванадий(V). Метод длителен: сначала перманганат восстанавливают до диоксида марганца с помощью формиата натрия в щелочном растворе. Осадок гидратированного диоксида марганца фильтруют, промывают, растворяют в Na2H9P2O7 и образующийся пирофосфат марганца (III) титруют стандартным раствором гидрохинона.
Титриметрическое определение перманганата
гV ГГП анализа- Методика I с применением As2O3 (по Л а н -Buev, ’ БеРУт точную навеску As2O3 (0,2—0.25 г) препарата, предварительно 15 и» оН«?ГО при 105—НО °C (примечание 1) в течение 1—2 ч, и растворяют в 0,002 М аРаСтвоРа NaOH. Добавляют 50 мл воды, 15 мл НС1 (1 : 1) н 1 каплю ими vr» Раств„°ра КЮз или KI. Титруют раствором перманганата до образова-
П п иЧИВОн в течение 30 с ротовой окраски.
Н«НтрацииМ 6 4 Э Н И 6 *’ Взятая наЕС‘С|<а As2O3 соответствует примерно 0.02 М кон-перманганата. Растворы более высокой концентрации можно разба
159
вить перед проведением аначпза. Для титрования более разбавленных растворов перманганата навеску As2O3 следует уменьшить. При разбавлении раствора мышьяка (П1) аликвотную часть раствора следует отбирать с помощью пипетки и резиновой груши. Начальная концентрация НС1 при титровании должна состав тять 0,5—1 И
Методика II с применением оксалата натрия [14]. К точной навеске оксалата натрия (0,3 г высщуенного предварительно препарата при 105—110°С в течение 2 ч) приливают 250 мл разбавленного раствора H2SO4 (15 мл концентрированной H2SO4 разбавляют 285 мл воды, кипятят в течение 10—15 мин и охлаждают) и перемешивают (примечание 2). Устанавливают температуру раствора (27 ± 3)°С, приливают при перемешивании со скоростью 25— 35 мл/мин 9/ю от общего объема раствора перманганата и оставляют до исчезновения розовой окраски раствора. Раствор нагревают до 55—60 °C и завершают титрование после появления не исчезающей в течение 30 с розовой окраски.
Примечание 2. Навеска оксалата натрия соответствует концентрации КМпО4 0,02 М (см. примечание 1).
3. Методика II, как и методика I, характеризуется высокой точностью.
Методика III с применением железа (II) этилендиамин сульфата [15] К точной навеске соединения добавляют H2SO4 до концентрации 1 М и титруют раствором перманганата до устойчивой розовой окраски раствора.
Примечание 4. Синтез железа(П)этилендиамин сульфата приведен в работе [21].
Методика IV — косвенное иодиметрическое определение [22]. К раствору с содержанием перманганата 0,1 г в 40 мл добавляют 3 г KI, растворенного в 10 мл воды*1 приливают H2SO4 до концентрации 0,03— 0,1 М. Оставляют на 10 мнн и титруют выделившийся иод стандартным раствором тиосульфата
2МпО; + ЮГ + 16Н+ —> 2Мп2+ + 512 + 8Н2О
При титровании 1 моль МпО4~ соответствует 5 моль S2Oy'.
Спектроскопические методы
Спектрофотометрический метод
Стандартный метод определения следовых содержаний марганца основан на окислении его перйодатом до марганца (VII) с последующим спектрофотометрированием раствора. Спектры поглощения 10-3 М растворов перманганата и бихромата в среде 1 М H2SO4 и 0,7 М Н3РО4 соответственно приведены на рис. 23. Максимумы поглощения перманганата наблюдаются при 520 и 545 нм. Если в растворе присутствует хром (VI), что часто наблюдается при анализе сталей, перманганат определяют по поглощению в области 545 нм, где поглощение хрома (VI) невелико. Влияние хрома можно устранить добавлением по каплям раствора нитрита калия.
Перманганат окисляет 6-метокси-2-метилтиопиримидин-4-кар-боновую кислоту в сильнощелочном растворе [23]. Поглощение продуктов реакции измеряют при 580 нм. При добавлении раствора перманганата к реагенту образуется раствор сине-зеленого цвета, который в течение нескольких секунд переходит в интенсивный зеленый цвет. Промежуточная сине-зеленая окраска является, вероятно, смесью пурпурной (перманганат-ион) и зеленой
160
Рис. 23. Спектры поглощения 1-10 s U растворов МпО; (/) и Сг,О;' (2) в 1 М H2SO4 и 0,7 М НзРО4 соответственно.
(манганат-ион). Предполагается, что реакция протекает по урав-
нению:
N
H3CS--С'' --ОСН3 2ОН“
I + 2МпО4 --->
I
соон
ОСНз+2МпО4" + Н2О
СООН
Закон Бера соблюдается в интервале концентраций 3,3-10-5— 1-Ю"3 М. Молярный коэффициент поглощения продуктов реакции равен 1,47-103 при 580 нм. Из 59 изученных ионов ни один не образует окрашенные осадки, но некоторые ионы в щелочной среде образуют гидроксиды или гидратированные оксиды.
Разработана группа интересных спектрофотометрических методов определения окислителей, в том числе перманганата, с применением трифенилметановых красителей [24]. Методы основаны на взаимодействии красителя с иодидами или бромидами в присутствии хлорамина В, хлорамина Т или гипохлорита натрия. Установлено, что образующиеся ионы 1+ или Вг+ снижают интенсивность окраски красителя [25, 26]. Возможность определения окислителей связана с их способностью реагировать с KI. Реакцию проводят в присутствии бриллиантового зеленого, кристаллического фиолетового и хлорамина Б при pH = 3—5. Чувствительность определения окислителей обычно не более 0,01 мкг/мл.
Флуориметрический метод
Предложен метод определения марганца после его окисления До перманганата, основанный на реакции Mnvn с силбксаном, сопровождающейся флуоресценцией. Интервал определяемых содержаний Мп (в виде перманганата) 0,1—1 мкг.
Электрохимические методы
Потенциометрический метод
Пд?писан аск°рбиметрический метод определения перманганата пеп ’ й также косвенный потенциометрический метод определения Нол?’анганата (и бихромата), основанный на реакции гексациа-
V Ррата(Ш) с аскорбиновой кислотой. Сначала перманганат
6 Зак- 167 1CI
1D1
взаимодействует с избытком стандартного раствора гексацианофер-рата(П). Образующийся гексацианоферрат (II) титруют стандартным раствором аскорбиновой кислоты с помощью автоматического титратора, потенциометрически устанавливая конечную точку с применением платинового и каломельного электродов. При определении перманганата в интервале 0,1—1 мг коэффициент вариации не превышает 2%. Описан также прямой автоматический микро-метод потенциометрического титрования перманганата с помощью гексацианоферрата(II) [29]. Метод позволяет последовательно титровать перманганат и ванадил путем изменения кислотности раствора.
Амперометрический метод
Амперометрическим методом можно последовательно определять марганец(VII), хром (VI) и ванадий(V) [30], используя в качестве титранта иодид калия. Перманганат титруют в среде 0,1 М H2SO4 и насыщенного раствора K2SO4 при потенциале графитового индикаторного электрода 0,6 В относительно насыщенного каломельного электрода. Метод позволяет определять 10 мкг МпО( в 20 мл раствора. Определению не мешает 50-кратный избыток хрома и 100-кратный — ванадия. Метод применен для анализа сталей.
Кулонометрический метод
Разработан кулонометрический метод, основанный на взаимодействии перманганата с электрогенерированным железом (II) [31]. Использованы результаты работы [32], в которой с целью расширения числа реагентов, применяемых в кулонометрии, применены ионообменные мембраны. Описан метод непрерывного кулонометрического титрования перманганата [33], также основанный на генерировании железа(II) на платиновом электроде.
Существенные достижения в автоматическом определении перманганата (и бихромата) связаны с применением пористого каталитического кислородного электрода [34]. Описана конструкция такого электрода [35]. Метод основан на образовании кислорода при взаимодействии перманганата с Н2О2:
2МпО; + 5Н2О2 + 6Н+ —> 2Мп2+ + 8Н2О + 5О2
Сигнал, регистрируемый кулонометрически, обеспечивается 100%-ным выходом по току. При определении потребления кислорода этот метод может конкурировать с автоматическими спектрофотометрическими методами, основанными на применении перманганата и бихромата.
Вольтамперометрический метод
Вольтамперометрическим методом с использованием вращающегося золотого электрода при потенциале 0,80 В относительно насыщенного каломельного электрода, в среде 1—3 М НСЮ4 МО'
162
жно определять 0,17—8,7 мг перманганата. Вследствие нестабильности электрода в работе используют метод стандартных добавок [36]. Полярографический метод был применен для одновременного определения перманганата и манганата в щелочных растворах [37].
ЛИТЕРАТУРА
I. American Public Health Association, Standard Methods for the Examination of Water and Wastewater, 13th edn, New York, American Public Health Association, 1970.
2. Official, Standardised and Recommended Methods of Analysis, 2nd edn (Compiled and Edited by N. W. Hanson), London, The Society for Analytical Chemistry, 1973.
3. Tribalat S. — Analytica chim. Acta, 1949, v. 3, p. 113.
4. Bock R., Beilstein G. M.— Z. analyt. Chem., 1963, v. 192, p. 44.
5. Neeb R. — Z. analyt. Chem., 1957, v. 154, p. 17.
6. Bock R., Hummel C. — Z. analyt. Chem., 1963, v. 198, p. 176.
7. Шевчук А. И., Никольская H. H., Симонова T. H. — Укр. хим. ж., 1966, т. 32, с. 635; Analyt. Abstr., 1967, v. 14, p. 6112.
8. Hayashi K-, Uzumasa Y. — J. chem. Soc. Japan, pure Chem. Sect., 1960, v. 81, p. 906; Analyt. Abstr., 1961, v. 8, p. 2862.
9 Tanigawa Y., Waki S., Takiyama K.— Japan Analyst, 1965, v. 14, p. 315; Analyt. Abstr., 1966. v. 13, p. 6844.
10. Koithoff I. M, Sandell E. B., Meehan E. J., Bruckenstein S.—Quantitative / Chemical Analysis, 4th edn, London, MacMillan, 1969.
IL Koithoff I. AL, Belcher R. — Volumetric Analysis, Vol. Ill, New York, Interscience, 1957.
12. Laitinen H. A.— Chemical Analysis, New York, McGraw-Hill, 1960.
13. Lang R. — Z. anorg. Chem., 1926, v. 152, p. 197.
14. Fowler R. M., Bright H. A. — J. Res. Nat. Bur. Stand., 1935, v. 15, p. 493.
15. Caraway К. P-, Oesper R. E. — J. Chem. Educ., 1947, v. 24, p. 235.
16. Kellner A., Szekeres L. — Chemist Analyst, 1965, v. 54, p. 75.
17, Berka A., Vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Pergamon Press, 1965.
18. Ripan R., Stanisav C. — Studia. Univ. Babes-Bolyai, 1964, v. 9, p. 77; Analyt. Abstr., 1966, v. 13, p. 6193.
19. Erdey L., Buzas I., Vigh K. — Periodica polytech., 1959, v. 3, p. 1: Analyt. Abstr, 1959, v. 6, p. 4262.
20. Berka A. — Z. analyt. Chem, 1969, v. 246, n. 322; Analyt. Abstr, 1970, v. 19, p. 3033.
21, Belcher R., Wilson C. L. — New Methods in Analytical Chemistry, London, Chapman and Hall, 1955, p. 249.
22. Chariot G„ Bezier D. — Quantitative Inorganic Analysis, London, Methuen, . 1957, p. 58.
23. Chung O. K-, Meloan C. E.~ Analyt. Chem, 1967, v. 39, p. 525.
“4. Раманаускас E. И., Буникене Л. В., Сапрогонене M. С. и dp.—ЖАХ, 1969, t. 24, c. 244; Analyt. Abstr, 1970, v. 19, p. 1022.
25- Буникене JI. В., Раманаускас E. И. — ЖАХ, 1968, t. 23, c. 1364; Analyt Abstr., 1970, v. 18, p. 2396.
26. Раманаускас E. И., Буникене Л. В., Жиленайгe M. B. — Научи, труды высш, учебн. заведений Лит. ССР. Химия и технол, 1968, с. 29; Analyt. Abstr, 1970 v. 18, р. 3962.
27. Бабко А. К, Дубовенко Л. И., Михайлова Л. С. — Укр. хим. ж, 1966, т 32 С; 614; Analyt. Abstr, 1967, v. 14, р. 6111.
Efdey L ’ Svehla G., Weber О. — Z. analyt. Chem, 1968, v. 240, p. 91; Analyt Abstr Щ70, v. 18, p. 12.
• Kiss-Er5ss K., Buzas I., Erdey L. — Z. analyt. Chem, 1971, v. 255, p. 271, Analyt. Abstr, 1972, v. 22, p. 3139.
30. Сангина О. А., Захаров В. А., Бектурова Г. Б.. С.минова Л. И. — ЖАХ, 1974, т. 29, с. 1594.
31. Баринов В. Г., Сангина О. .4., Середа Т Г. — Зав. лаб., 19/2, т. 38, с. 641; Analyt. Abstr., 1973, v. 24, р. 133.
32. Hanselman R. В., Rogers L. В. — Analyt Chem.. 1960, v. 32, p. 1240.
33. Takahashi T., Sukurai H.— Taianta, 1963. v 10, p. 971.
34. Fleet B.. Ho A. Y. W., Tenygl J. — Analyt. Chem., 1972. v. 44. p. 2156.
35. Tenygl J., Fleet B. — Colin Czech, chem. Commun., 1973, v. 38, p. 1714; Analyt. Abstr., 1974, v. 26, p. 112.
36. Huber C. 0. — Analyt. Chem., 1964, v. 36, p. 1873.
37. Landsberg R., Thiele R. — Chem. Tech. Berlin, 1963, v. 15, p. 627; Analyt.
Abstr., 1965, v. 12, p. 1174.
ПЕРРЕНАТ И ПЕРТЕХНАТ
Рений может находиться в степени окисления от +1 ДО +7. Наиболее устойчивы состояния +4, 4-6 и особенно 4-7. Перренат ReOJ является конечным продуктом почти всех окислительных реакций с участием рения и его соединений в растворе. Обычно рений определяют в виде перрената. Перренат устойчив в широком интервале pH, в качестве вторичного стандарта обычно применяют перренат калия.
Перренат не является в отличие от перманганата сильным окислителем. Его трудно восстановить. Аналитическая химия рения и аналогичного ему технеция рассмотрена в работах [1—4], Пертехнат является обычной формой нахождения технеция в растворе, он — более сильный окислитель, чем перренат, и устойчив в широком интервале pH.
Важным аспектом анализа является определение перрената в присутствии молибдата, поскольку эти элементы сопутствуют друг другу в природных объектах.
МЕТОДЫ ОТДЕЛЕНИЯ
Дистилляция
Дистилляция гептаоксидов Re2O7 и Тс2О7 из смеси кислот HBr, НС1, H2SO4, НСЮ4 и Н3РО4 позволяет отделить Re и Тс от многих элементов. Обычно проводят дистилляцию с паром из растворов H2SO4 при 260—270°C. При этом не отделяются As111, SeIV, SeVI, TeIV, Sb111, Sbv и Hg, а также не отделяется Re от Тс,
Осаждение
Рений и технеций во всех степенях окисления, включая -\-7, осаждаются в виде сульфидов, которые затем можно снова окислить до степени окисления 4-7.
Тетрафениларсонийхлорид осаждает перренат из кислых растворов в виде Ph4AsReO4 [5]. Аналогично осаждается технеций. Определению мешают посторонние ионы. Их влияние будет рассмотрено в разделе, посвященном гравиметрическому определению перрената и пертехната.
164
Соосаждение
Микрограммовые количества перрената и пертехната соосаж-даются с перхлоратом тетрафениларсония как носителем из кислых, нейтральных и щелочных растворов [6]. Рений (VII) не осаждается аммиаком, но он соосаждается с осадками гидроксидов из слабощелочных растворов.
Методы бумажной и тонкослойной хроматографии
Способы разделения Re и других ионов методом ТСХ приведены в табл. 8.
Таблица 8. Способы разделения ReO] и других ионов методами бумажной хроматографии и ТСХ
Метод Растворитель Разделяемые ионы Литература
ТСХ (на As2O3) 1-1,16 М Н3РО4 ReO', MoOf, VO3‘, WO*- 7
ТСХ (силикагель, связанный поливиниловым спиртом) 1 М HSCN, или 0,5—0,9 М малоновая кислота, или 1,0—1,8 М СН3СООН, или 1,0-1,8 М NH1SCN ReO[, MoO]', WO42' 8
*ГСХ (листы силикагеля) Смеси метанола с кислотой ReO’, MoO]’, SeO3' 9
ТСХ (силикагель «G») • 26 М н-бутанол НС1 /к • 9^ ReO], MoO42', WO]' 10
Бумага (Ватман Е-20) * 4М НС1 ReO7, Rev, Relv, Sn>v 11
Сильноосновная аниони-»ая бумага 2М HCI ReO[, ReCi]' 11
т • Разделение проводят при 0 °C, чтобы предотвратить разложение Re^,
Метод ионообменной хроматографии
i Перренат и пертехнат можно отделить от посторонних катио-вов методом ионообменной хроматографии [6]. Успешно применяют анионный обмен, особенно для отделения молибдата(VI). В работе [12] с этой целью использован слабоосновной целлюлозный анионит. Анионный обмен в среде азотная кислота — метанол Позволяет разделить перренат и пертехнат [14].
Экстракция
:. Много работ посвящено жидкостной экстракции рения и технеция. Особое внимание уделено разделению перрената и молиб-Дйта. В монографии Колтона [1] приведены подробные сведения ® методах экстракции рения и технеция, опубликованных до 1961 г.
165
Экстракция перрената тетрафениларсония позволяет отделить малые количества рения от больших количеств молибдена. Экстракцию проводят при pH = 8—9 хлороформом, дихлорэтаном и другими растворителями [5]. Перренат тетрафениларсония можно экстрагировать хлороформом из щелочных растворов. Экстракция перрената пиридином из щелочных растворов позволяет отделить 10—100 мкг Re от 100 мг молибдена [13]. Опубликован очень быстрый метод (20 с) отделения технеция от молибдена (основа) и рутения путем экстракции пиридином [14].
Описан метод, включающий образование и экстракцию комплекса перрената с красителем Виктория голубой 4R (С. I. Basic Blue 8) бензолом [15]. Определению мешают Г, SCN', СЮ^ и СЮз. Метод применен для анализа молибденовых руд. Рений отделяют от большого числа примесей путем восстановления ионов жидкой амальгамой цинка в среде 1 М H2SO4 с последующей экстракцией изопентанолом из 3 М H2SO4 [16].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Наибольшее значение имеют гравиметрические и спектрофотометрические методы определения рения и технеция. В последнее время применяют также метод атомно-абсорбционной спектроскопии и ионоселективные электроды.
Гравиметрические методы
Старые гравиметрические методы, основанные на осаждении перрената нитрона или перрената таллия, менее эффективны, чем методы, основанные на образовании малорастворимых солей перрената (или пертехната) тетрафениларсония Ph4AsReO4 [5]. В присутствии хлорида натрия образуется гранулированный осадок. Осадок выдерживают и на следующий день сушат при ПО °C. Определению мешают ионы, осаждаемые тетрафениларсонием, и ионы, образующие нерастворимые хлориды, в том числе МпОГ> С1О4~, SCN~, I-, Br~, F~, Hg2+, Bi3+, Pb2+, Ag+ и Sn4+. Концентрация нитрата должна быть невысокой. Мешающие ионы необходимо предварительно отделять, что снижает ценность метода. Осаждение перрената и пертехната тетрафениларсонием применено также для определения этих ионов методом ИК-спектроскопии.
Спектроскопические методы
Описано большое число спектрофотометрических методов определения перрената и пертехната, включая экстракцию окрашенных комплексов органическими растворителями. Некоторые из них приведены в табл. 9.
Изменение концентрации при хранении разбавленных растворов перрената (5 мкг/мл Rev11) изучено в работе [23]. Путем 166
167
измерения поглощения ионного ассоциата перрената с ориллиан-товым зеленым [21] установлено, что в течение двух месяцев .хранения раствора в сосуде из натриевого стекла или из жесткого полиэтилена потери рения не наблюдается.
ИК-метод использован для доказательства возможности со-осаждения перрената и перте.хната с перхлоратом тетрафенилар-соння как носителем [24]. Осадок промывали, сушили, отбирали 2 мг и смешивали с 300 мг КВг, прессовали прозрачную таблетку, записывали ИК-спектр, измеряли пики поглощения перрената и пертехната при 10,94 и 11,09 мкм соответственно. Вносили поправку на поглощение базовой линии. Преимущество метода состоит в том, что, несмотря на осаждение тетрафенил арсонием ионов SCN', SoCK', СЮз, СЮ[ и Ю[, эти ионы не поглощают в области 11 мкм и не мешают определению.
Описан атомно-абсорбционный метод определения рения. Поглощение измеряют при 346 нм в пламени оксид азота — ацетилен [25]. Большая чувствительность достигнута при использовании аналитических линий 227, 462, 228, 751 и 229,449 нм [26]. Описан метод определения технеция с применением лампы с полым катодом, для измерения используют дублет 261, 423 — 261, 587 нм. Применением обогащенного пламени ацетилен — воздух достигнута чувствительность определения Тс 3,0 мкг/мл [27].
Другие методы определения
Описан электрод с жидкой ионообменной мембраной, чувствительной к перренату и роданиду. Предел обнаружения перрената 10~6 М, потенциал электрода изменяется по уравнению Нернста при увеличении концентрации ионов на несколько порядков величины [28].
Полярографические волны перрената и пертехната необратимы. Перренат в среде 4 М НСЮ4 или HCI подвергается трехэлектронному восстановлению [29]. Изучено также полярографическое поведение пертехната [30, 31].
Изучено каталитическое действие перрената на реакцию восстановления теллурата натрия хлоридом олова (И). Образующийся теллур определяют спектрофотометрически [32].
Микрограммовые содержания технеция можно определять в отсутствие других радионуклидов по их радиоактивности. Вследствие очень низкой энергии p-излучения велико самопоглощение излучения образцом. Метод непригоден для количественных измерений. Для определения малых содержаний технеция применен нейтронный активационный анализ [33].
ЛИТЕРАТУРА
1. Colton R.— The Chemistry of Rhenium and Technetium, Chanter 2 London, Wiley, 1965.
2. Peacock R. D. — The Chemistry of Technetium and Rhenium, Chapter 11. London, Elsevier. 1966.
168
3. Lowenheim F. .4. — Encyclopedia of Industrial Chemical Analysis (Ed.
F. D. Snell and L. S. F.ttre), Vol. 17. New York. Interscience, 1973. p. 515.
4, Magee R. J. — Comprehensive Analytical Chemistry (Ed. C. L. Wilson and D. W. Wilson). Vol. 1C. London. Elsevier. 1962. p. 626.
5. Tribalat S. — Analytica chim. Acta, 1949, v. 3. p. 113.
6. Korkish J. — Modern Methods for the Separation of the Rarer Metal Ions, New York. Pergamon Press. 1969. p. 504.
7. Gaibakiian D. "S. — L'chen. Zap. Erevan, gos. u.tiv. estestv. Nauk, 1971, p. 71; Analyt. Abstr., 1972, v. 23. p. 2293.
8. Shiobara Y. —Japan Analyst, 1968, v. 17. p. 1396; Analvt. Abstr., 1970, v. 19, p. 202.
9. Phipps A. Al. — Analyt. Chem., 1971, v. 43, p. 467.
10. Johri K. N., Menra H. C. — Mikrochim. Acta, 1971, p. 317.
11. Pavlova Л1. — J. Chromat., 1970, v. 51, p. 346.
12. Ishida K-, Kuroda R. —Analyt. Chem.. 1967, v. 39, p. 212.
13 Meshri D. T., Haidar В. C. — J. Sci. Ind. Res., India, 1961, v. 20B, p. 551; Analyt. Abstr., 1962, v. 9, p. 1902.
14. Neirinckx R. D., De Jesus A. S. M. — Analytica chim. Acta, 1972, v. 59, p. 324.
15 Пилипенко A. T. Киш П. П., Желтвай И. И.—Укр. хим. ж., 1971, т. 37, с. 477; Analyt. Abstr., 1972, v. 22, р. 1571.
16. Yatirajaiii V., Kakkar L. R.—Taianta, 1970, v. 17, p. 759.
17. Crouthamel С. E. — Analyt. Chem., 1957, v. 29, p. 1756.
18. Meloche V. IF., Martin R. L., Webb W. H. — Analyt. Chem., 1957, v. 29, p. 527.
19. Fujinaga T., Koyama M., Kanchiku Y., Tsurubo S. — Bull. chem. Soc. Japan, 1967, v. 40, p. 817; Analyt. Abstr., 1968, v. 15, p. 4685.
20. Al-Kayssi M., Magee R. J., Wilson C. L. — Taianta, 1962, v. 9, p. 125.
21. Fogg A. G., Burgess C,, Burns D. T. — Analyst, 1970, v. 95, p. 1012.
22. Okubo T., Kojima AL—Japan Analyst, 1965, v. 14, p. 843; Chem. Abstr., 1965, v. 63, p. 17138.
23. Al-Sibaai A. A., Fogg .4. G. — Analyst, 1973, v. 98, p. 732.
24. Magee R. J., Al-Kayssi Al. — Analytica chim. Acta, 1962, v. 27, p. 469.
25. Biechler D. G, Long С. H. — Atom. Abs. Newsl., 1969, v. 8, p. 56.
26. Bailey B. IF., Rankin J.- AL — Analyt. Chem., 1971, v. 43, p. 936.
27. Hareland IF. A., Ebersole E. R., Ramachandran T. P. — Analyt. Chem., 1972, v. 44, p. 520.
28. Hirsch R. F., Portock J. D. — Analyt. Letters, 1969, v. 2, p. 295.
29. Lingane J. J. — J. Am. chem. Soc., 1942, v. 64, p. 1001.
30. Colton R., Dalziel J., Griffith IF. P., Wilkinson G. — J. chem. Soc., 1960,
• p. 71.
31. Salaria G. B. S., Rulfs C. L., Elving P. J. — J. chem. Soc., 1963, p. 2479.
32. Полуэктов H. С., Кононенко JI. И. — Зав. лаб., 1959, т. 25, с. 548; Analyt. Abstr., 1960, v. 7, p. 998.
33. Boyd G. E., Larson Q. V.—J. phys. Chem.. 1956, v. 60, p. 707.
ПЕРРУТЕНАТЫ
При растворении рутения в 2 М растворе NaOH и обработке раствором K2S2O8 образуется рутенат RuO^. При разбавлении 0>25 М раствором NaOH и окислении гипохлоритом образуется ЯерРУтенат RuO.(.
Кислые растворы перрутената калия неустойчивы и разлагаются с образованием оранжевого рутената и выделением кислорода. -’Кислом растворе рутений присутствует в степени окисления +3 и 4-4. при окислении рутения (IV) .сильными окислителями, апример, K2S2O8, образуется Ruvi" (RuO(), причем промежуточными являются соединения рутения (VI) и (VII). При нагревании
169
•0,29
Рис. 24. Потенциалы кислородных соединений рутения в щелочных растворах,
кислых или щелочных растворов в присутствии сильных окислителей происходит дистилляция RuO4. При поглощении тетраоксида рутения щелочью сначала образуется зеленый перрутенат, который затем восстанавливается до красного рутената. Рутенат и перрутенат значительно более устойчивы в щелочных растворах, чем в кислых. При дистилляции RuO4 из растворов НС1 образуются хлоридные комплексы рутения. Потенциалы кислородных соединений рутения в щелочной среде приведены на рис. 24 [1, 2].
Показано [3], что устойчивые растворы перрутената получаются только при поглощении тетраоксида рутения раствором 1 М NaOH, содержащим гипохлорит. При более высоких концентрациях щелочи (6 М) перрутенат быстро восстанавливается до рутената даже в присутствии гипохлорита. В работе [4] устойчивые растворы перрутената получены поглощением тетраоксида рутения 0,5—1,0 М раствором Na2CO3, содержащим гипохлорит. Содержание перрутената определяли затем по собственному поглощению иона при 315 нм. Определению не мешает широкая полоса поглощения гипохлорита в области 296 нм. Спектры разных ионов рутения в 1 М растворе NaOH приведены на рис. 25 [3].
МЕТОДЫ ОТДЕЛЕНИЯ
Приведенные выше сведения показывают большое значение методов дистилляции для отделения рутения. Дистилляция из
растворов H2SO4, содержащих висмутат натрия, и поглощение паров раствором карбоната натрия, содержащим гипохлорит, позво-
ляет определять перрутенат спектрофотометрически. Определению не мешают многие ионы.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Аналитической химии рутения посвящена работа [5].
Изучены условия спектрофотометрического определения рутения в виде перрутената [3]. Рутений окисляли в среде.
Рис. 25. Спектры RuVI (/), RuVI и Rux 11 (2) И RuVI1 (3) в 1 Л1 растворе NaOH,
170
H2SO4 висмутатом натрия, образующийся тетраоксид отгоняли и поглощали раствором ХаОН, содержащим гипохлорит. Поглощение перрутената измеряли при 385 нм. При определении 200— 400 .мкг рутения с доверительной вероятностью 0,95 ошибка определения составляет ±2%. Значение молярного коэффициента поглощения равно 2150. В работе [4] для 315 нм найдено значение е = 2600.
При длине волны 385 нм е = 2275 для RuOi и 905 для RuOV, при длине волны 465 нм е = 275 для RuO? и 1960 для RuOl'-Измеряя поглощение при указанных длинах волн, можно определить содержание каждой ионной формы рутения [6].
Метод пламенной атомно-абсорбционной спектроскопии имеет невысокую чувствительность 10 мкг/мл. При использовании графитового трубчатого нагревателя чувствительность и избирательность определения рутения значительно улучшаются [7, 8].
ЛИТЕРАТУРА
1. Connick R. Е., Hurley С. R.— J. Am. chem. Soc., 1952, v. 74, р. 5012.
2. Silverman М. D., Levy H. A.—J. Am. chem. Soc., 1954, v. 76, p. 3319.
3. Larsen R. F., Ross L. E.—Analyt. Chem., 1959, v. 31, p. 176.
4. Cotton T. AL, Woolf A. A. — Analyt. Chem., 1962, v. 34, p. 1385.
5. Автократова T. Д. Аналитическая химия рутения. M. 1962, 269 с.
6. Nowogrocki G.. Tridot G. — Bull. Soc. chim. Fr., 1965, p. 684.
7. Everett G. L.— Analyst, 1976, v. 101, p. 348.
8. Megarrity R. G., Siebert B. D. — Analyst, 1977, v. 102, p. 95.
СЕЛЕНАТЫ И СЕЛЕНИТЫ
Селен применяют в электронике при изготовлении выпрямителей и фотосопротивлений, в ксерографии, при изготовлении стекол, керамики и эмалей.
Селен относится к кумулятивным ядам. Повышенные содержания селена в пище и воде приводят к серьезным заболеваниям. Однако в следовых содержаниях селен, вероятно, необходим для нормального питания животных.
В водных растворах селен присутствует в виде селената(У1) или селенита(IV). Описаны методы определения обеих форм селена. При нагревании в растворах НС1 селенат восстанавливается До селенита (IV). Многие восстановители, например, аскорбиновая кислота, SO2 и гидразин восстанавливают соединения селена До элементного селена, что используют для отделения и определения селена.
Установлено, что при хранении водных растворов селена(VI), меченных изотопом 75Se, с общим содержанием селена 100 ррш в посуде из различных материалов (стекло пирекс, флинт и полиэтилен) происходят потери селена [1]. В среде азотной кислоты ;®”воры устойчивы, потери селена составляют 1% в течение 15 при рН=7 потери почти удваиваются. Потери в стекле ™ ннт и пирекс примерно одинаковы, а в полиэтилене, который,
171
вероятно, чаще всего применяют для хранения растворов, потери особенно велики и составляют 8% в течение 15 суток.
Обзор аналитической химии селена приведен в работах [2— 6]. В обзоре [6] рассмотрено определение селена в биологических материалах, включая проблемы хранения и подготовки проб и разрушения органических веществ. Определение селена в пище и биологических пробах встречает трудности, особенно в присутствии жиров. Поэтому иногда анализ проводят методом нейтронной активации, который не требует разрушения проб.
Важным моментом, требующим обсуждения, является определение селена в присутствии теллура.
Для получения стандартных растворов селена(IV) применяют селенит натрия. Навеску селенита натрия растворяют в разбавленной НС1, содержание селена устанавливают иоднметрическим титрованием. Более быстрым является метод, основанный на растворении точной навески металлического селена в минимальном объеме концентрированной НС1, содержащей несколько миллилитров концентрированной HNOa- Смесь нагревают до растворения селена и отгоняют коричневые пары оксидов азота. После охлаждения раствора доводят объем водой до метки, предварительно добавив такой объем НС1, чтобы концентрация кислоты в конечном объеме была примерно 1 М. Растворы селена (VI) обычно получают растворением точных навесок селеновой кислоты или Na2SeO4- ЮН2О.
МЕТОДЫ ОТДЕЛЕНИЯ
Методы отделения селена (и теллура) подробно обсуждены в монографии Коркиша [7].
Дистилляция
Селен можно отделить от многих элементов дистилляцией из растворов НО, или, лучше, НВг. При этом селен и теллур восстанавливаются до степени окисления +4 и отгоняются в виде галогенидов или оксогалогенидов. Концентрация кислоты должна быть выше 6 М, а температура выше 100°С. При дистилляции применяют смеси кислот, например HCI—H2SO4, НВг—H2SO4, НС1О4—НВг. При анализе биологических материалов часто используют смесь Вг2—НВг, причем Вг2 окисляет селен до Se1V. Избыток Вг2 отгоняется при более низкой температуре, чем селен. В присутствии НВг SeVI восстанавливается до Seiv. Отгоняющийся при дистилляции тетрабромид селена поглощают водой. Метод дистилляции применен для определения следов селена [8].
Вместе с селеном частично или полностью отгоняются и другие элементы. Из смеси Вг2—НВг отгоняются As, Sn, Ge, Sb и Те; при дистилляции из растворов H2SO4—HCI вместе с селеном otj гоняются Ge, Sn, Sb, Те, Мо, Re и Hg. Избирательность дистилляции селена изучена в работе [9], в качестве оптимальной рекомендована смесь НВг—Вг2—H2SO4.
172
Рис. 26. При'op для дистилляции селена.
Сведения о дистилляционном разделении селена и теллура противоречивы. В некоторых работах показано, что Se и Те можно разделить в смесях НС1— H2SO4, H2SO4—НВг и НСЮ4— НВг, в других работах утверждается, что теллур отгоняется вместе с селеном. На рис. 26 по
казан аппарат для дистилляции
селена. Детально метод дистилляции описан в работе [10]. После дистилляции из растворов, содержащих НВг, селен (IV) можно восстановить до элементного аскорбиновой кислотой, в результате осаждения селен отделяется от элементов, захваченных при отгонке. Осажденный селен можно определять гравиметрически.
Показано, что при дистилляции из растворов НВг отгоняются флуоресцирующие вещества, мешающие последующему флуори-метрическому определению селена [11].
Ионообменный метод
Селен (IV) не поглощается на сильнокислотных катионитах из 0,1—4 М растворов НС1, поэтому ионным обменом можно отделить мешающие ионы металлов перед определением селена [12]. Однако при анализе растительных материалов этот метод не дал удовлетворительных результатов [13]. При некоторых условиях селен(IV) остается на катионите, что использовано [14] для отделения микроколичеств селена от сульфата и железа(III).
В отличие от селена(IV) теллур (IV) поглощается сравнительно хорошо на катионитах из слабокислых растворов, что можно использовать для разделения селена и теллура.
Селен (IV) не поглощается на сильнокислотных катионитах при концентрации НС1 менее 4 М и слегка задерживается в более кислой среде, что позволяет отделять Se от Те. Селен (IV) элюируют раствором НС1 с концентрацией менее 4 М, при этом Te,v остается в колонке, а затем элюируется 0,1 — I М раствором НС1.
Селен(VI) и теллур (VI) не поглощаются анионитами из растворов HCI любой концентрации, что позволяет отделить большие содержания cexena(VI) от следов сульфата [15]. Ионообменный метод использован для определения следов серы в особо чистом селене. В работе [16] селен(VI) и теллур(VI) отделены те|< же методом от сульфита.
Ионообменная бумага применена для отделения селена(IV) «теллура(IV) [17]. Селен(VI) отделяли от селена(IV) [18] на *В"Ионообменной бумаге SB-2 в хлоридной форме, в качестве рас-.. зрителя использовали 1 Л1 раствор НС1.
173
Экстракция
В последнее десятилетие появилось много работ, посвященных экстракции селена и теллура. В ряде случаев применен экстракционно-спектрофотометрический метод. В табл. 10 приведены экстракционные методы отделения селена(IV) от других элементов и от селена (VI). Другие экстракционные методы рассмотрены в цитированных ранее обзорах и монографии [19].
Таблица 10. Некоторые методы экстракции селена (IV)
Реагент Растворитель Примечание Литература
Толуол-3,4-дитиол Этиленхлорид При концентрации НС1 более 10 М осаждаются только Pd11, TeIV, Au, WVI и Revii 20
Толуол-3,4-дитиол Этилепхло-рид — ССЦ (1 : 1) 6-8 M HC1 11
Диэтилдитнокар-бамат Трибутилфосфат Экстрагируется SeIV, SeVI не экстрагируется. Возможна реэкстракция SeIV раствором 10%-ной НСЮ4-Н2О2 (20: 1) 21
Диэтилдитиофос-форная кислота CCU SeVI не экстрагируется при концентрации НС1 0,1 — 10 М или H2SO4 0,5-7,5 М 22
З.З'-Диаминобен-зидин Бензол, толуол или ксилол Экстракция при pH = 6—7 в присутствии маскирующих лигандов F" и ЭДТА 23
2-Теиоилтрифтор-ацетон Ксилол Экстракция при pH =0,5—4,5. Se можно определять непосредственно в экстракте З.З'-диаминобензидином 24
Дитизон * ссц Экстракция из 6 Л-1 НС1. Один из наиболее чувствительных методов: 6420 = (7—8) • 104 25-28
1,2-Диаминобен-зол Толуол Метод более избирателен, чем с помощью З.З'-диаминобензидина, поскольку комплекс экстрагируется при меньших значениях pH 29
2,3-Диаминонафталин Циклогексан То же 30
• Данные об экстракции дитизоном противоречивы. В работе [22] указано^ что экстрагируется не дитизонат селена (IV), а продукт его окисления. Дитизонат селена (IV) можно выделить в виде красного осадка из водной фазы. Стары и соавторы полагают, что экстрагируется комплекс SeW с дитизоном состава (1 : 4) [27].
Осаждение
Селен восстанавливается в растворах концентрированной ИС1 посредством SO2 или других восстановителен и осаждается в элементном виде. Это позволяет отделить его от большинства элементов, включая теллур, который осаждается в среде 2—4 Л4 HCL Для количественного отделения селена осаждение следует проводить из
174
растворов с концентрацией НС1 выше 9 Л! [30]. Осадок селена сушат при 105 °C и определяют гравиметрически.
При анализе растворов, содержащих селен и теллур, их предварительно выпаривают досуха, однако вследствие летучести хлоридов селена и теллура выпаривать солянокислые растворы нельзя. Потери селена(IV) не наблюдаются при выпариваниип сернокислых растворов до паров серной кислоты. Кипячение с HNO3 также ие приводит к потере селена. Чтобы избежать длительного выпаривания растворов, Se и Те можно осадить, осадки промыть и растворить в минимальном объеме HNO3, а затем упарить досуха на водяной бане [31].
После дистилляции селена (IV) его можно осадить, восстанавливая аскорбиновой кислотой, при этом отделяются элементы, захваченные при дистилляции.
Восстановление с помощью SO2 можно проводить в растворах " НС1 и других средах [43], определение селена заканчивают гравиметрически. Восстановление протекает количественно в среде 6 М НС1, 1,5 М H2SO4 и 3 М НСЮ3. В среде НС1О4 получены наиболее точные результаты.
Селен и теллур можно отделять с помощью тетрафениларсоний-хлорида [32]. Теллур осаждается только в растворе 5 М НС1. Мешают ионы, осаждаемые этим реагентом, в том числе Br , I , F , NO3, Movi и WVI.
Соосаждение
Микроколичества селена соосаждаются с мышьяком. Этот метод применен для анализа сплавов меди и свинца [33], а также биологических материалов после предварительного окисления в кислородной бомбе. Модифицированная методика применена для определения селена в почвах и осадках [34]. При содержании селена
Таблица 11. Методы бумажной (БХ) н тонкослойной (ТСХ) хроматографии, используемые для отделения Selv
Метод Растворитель Отделяемые ионы Литература
БХ - Пиридин — пропанол — НгО SeVI 38
БХ (круговая) БХ (восходящая, с обращенной фазой) (1:2: 1) Ацетон — кислота — вода Te'V 39
Трибутилфосфат, 1 Л1 (неподвижная фаза), 2 М НВг или TeIV, Си11, Ni, Zn, Pb, Fe111, Co, As”1, TiIV, 40
8 М. ХаВг (подвижная фаза)
Б* и ТСХ (силикагель на листках Метанол — 0,6 М НС1 — 10%-ный П„О и др. Th RuO4", Mov1 41
“ иа стекле) ъл (силикагель) Амилацетат в концентрированной НС1 TeIV 42
175
0,1—500 ppm его определяли после отделения спектрофотометрически с помощью З.З'-диаминобензидина. Метод применен для анализа пищевых продуктов [35], геологических и биологических образцов [36]. В работе [36] осадок растворяли в небольшом объеме HNO3 и определяли селен методом ААС.
Изучено осаждение селена (IV) на свежеосажденном гидроксиде железа(III) [37]. Осаждение ведут из ацетатных буферных растворов, при pH < 7; степень соосаждения уменьшается с ростом pH и при pH = 10 составляет всего 10%.
Методы бумажной и тонкослойной хроматографии
Методы бумажной и тонкослойной хроматографии позволяют отделить малые количества селена от ряда ионов. Характеристика некоторых методов приведена в табл. 11.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Гравиметрический метод, основанный на выделении элементного селена, пригоден для определения сравнительно больших содержаний селена. Если в растворе находится селен(VI), его восстанавливают в среде разбавленной НС1 при нагревании. При восстановлении на холоду посредством SO2 селен осаждается в виде осадка красного цвета, при нагревании суспензии образуется черная аморфная модификация селена, способная захватывать примеси. Осадок промывают, сушат при 105 °C и взвешивают. Диоксид серы как восстановитель используют в газообразном виде или в виде водного или метанольного раствора. Применяют также гидроксиламин, гидразин, аскорбиновую кислоту, 1,2,3-триметил-3-пиразолон-5-тион и м-(меркаптоацетамидо) фенол. Установлено, что в отличие от селена (IV) селен(VI) не восстанавливается диоксидом серы [43]. В среде хлорной кислоты SO2 количественно восстанавливает Seiv, не затрагивая SeVI.
Восстановление 1,2,3-триметил-3-пиразолон-5-тионом приводит к повышению селективности [44]. Метод [44] позволяет определять от 20 до 550 мг селена(IV) в среде соляной — лимонной кислот. При нагревании на водяной бане восстановление завершается в течение 1 ч. Мешающее влияние TeIV, Pb'1, Hg11, Sn11, Sb111 и Са можно устранить. Серебро соосаждается, а медь мешает осаждению селена.
Описаны также другие методы осаждения селена, например с помощью 2,3-диаминонафталина [45], применяемого также для спектрофотометрического и флуориметрического определения селена. Этот реагент позволяет осаждать 10—50 мг селена, коэф-1 фициент пересчета на селен равен 0,3385,
176
Гравиметрическое определение селена посредством восстановления диоксидом серы [43]
Ход анализа. К раствору, содержащему селен(1\7), приливают 100 мл 3 М раствора НС1О4. Добавляют небольшими порциями 1,0 г Na2SO3, перемешивают раствор, накрывают и оставляют прп комнатной температуре на 15 мин.
Выдерживают раствор в течение 30 мни при 96 °C для переведения красной модификации селена в черную, фильтруют через доведенный до постоянной массы тигель со стеклянным фильтром. Промывают осадок несколько раз водой, в конце ацетоном и сушат при 110 °C до постоянной массы.
Титриметрические методы
Давно известные титриметрические методы определения селена (IV) основаны на его восстановлении до селена иодидом калия и титровании выделившегося иода стандартным раствором тиосульфата. Другой метод основан на восстановлении селена(IV) избытком стандартного раствора тиосульфата. Избыток SzOj' титруют стандартным раствором иода:
SeO3~ + 4Г + 6Н+ —-> Se + 212 + ЗН2О
4Na2S2O3 + H2SeO3 + 4НС1 —> Na2SeS4O6 + Na2S4O6 + 4NaCI + ЗН2О
Эти методы обсуждаются в монографии Кольтгофа и Белчера [46].
Стандартный метод определения селена(IV) заключается в добавлении избытка стандартного раствора КМпО4 ви титровании избытка раствором железа(II) [47]. Теллур(IV) также окисляется раствором КМпО|, поэтому определяется сумма Selv и TeIv. При использовании бихромата окисляется только TeIV [48], что позволяет по результатам титрования двух аликотных частей раствора определять селен(IV) и теллур(IV) при совместном присутствии. Можно определять Se‘v и Te,v в одной аликвотной части раствора [49]: окисляют Se,v и Te‘v раствором ДМпО4 в среде концентрированной Н3РОз, затем избирательно восстанавливают TeIV мышьяком (III) в присутствии OsO4 в качестве катализатора. Метод прост и экспрессен, определению не мешают Fe111, Asv, Ag, Tl1", Sevl, UVI, Cu", Ni, K. Na, Al и Mg.
Селен(IV) и теллур (IV) можно количественно окислять перйодатом в присутствии буры [50]. Предполагается [50], что в сочетании с методом окисления теллура(IV) биохроматом этот метод позволяет анализировать смесь SeIV и Te,v.
Описан метод восстановления селена(IV) до элементного глюкозой или D-фруктозой. Затем селен окисляют железом (III) в среде 4 М НС1 и образовавшееся Fe" титруют Церием (IV). Определению мешают Ag1, TP, Cun, Hg", Zn'1, Pd", Au"1, Bi"1, TeIV, Zr,v и Thlv [51].
Предложен также иодиметрический метод определения Se,v и Telv при совместном присутствии [52]. К анализируемому кислому Раствору добавляют KI, при этом образуются элементные Se и Те и выделяется эквивалентное количество иода, который титруют тиосульфатом. Другую аликвотную часть раствора нейтрализуют гид-Р°карбонатом натрия перед титрованием, при этом титруется иод,
177
эквивалентный содержанию теллура. Под, эквивалентный содержанию селена, титруют амперометрически гидразином.
Описаны и другие титриметрические методы определения макро-[53] п мнкросодержаннн селена [54].
Титриметрическое определение ce.iena(IV) и (VI)
и теллура(1У) в смеси [49]
Реагенты
Перманганат калия, 0.01 .V раствор.
Л1ышид(П1), 0,0125 Л1 раствор, приготовленный из As2O3.
Осмиевая кислота, 0,1 со-иын раствор.
Концентрированная фосфорная кислота. Нагревают 100 мл 85—90%-ной Н3РО4 в стакане вместимостью 250 мл с 3—4 мл HNO3 (1 : 1) до прекращения выделения оксидов азота. Кислоту хранят в сосуде пз темного стекла.
Ход анализа. К аликвотной части раствора, содержащей селен(IV) и теллур (IV), добавляют 1—5 мл концентрированной фосфорной кислоты и воду до объема 25 мл. Добавляют отмеренный избыток стандартного раствора КМпО4. Через 3—5 мин титруют стандартным раствором железа(II), применяя ферроип в качестве индикатора. При ослаблении окраски индикатора вводят 2—3 мл 10 М раствора H2SO4. Объем раствора КМпО.„ пошедшего на титрование, соответствует содержанию SeIV 11 Telv.
к анализируемому раствору добавляют избыток раствора As’11, 0,4 мл 0,1 %-ного раствора OsO4 и титруют избыток As’11 стандартным раствором КМпО4. Содержание прореагировавшего As’" эквивалентно содержанию теллура (IV).
Спектроскопические методы
Колориметрические и спектрофотометрические методы
Для колориметрического определения селена(IV) использовано его восстановление до устойчивого золя металлического селена. Методика с применением гидразина или аскорбиновой кислоты рекомендована в качестве стандартной [55], в ней не предусмотрена дистилляция селена и применение канцерогенных реагентов, она позволяет определять 10—100 мкг селена.
Метод кольцевой печи использован для отделения и полуколи-чественного определения селена при анализе воздушных загрязнений [56].
Наиболее эффективный спектрофотометрический метод определения селена (IV) основан на его взаимодействии с о-диаминами с образованием пназоселенолов. В качестве такого реагента в 1955 г. предложен 3,3'-диаминобензидин (ДАБ) [57]. Окрашенный в интенсивно желтый цвет комплекс реагента с селеном (IV) образуется в кислой среде при pH <4 и имеет максимум поглощения при 340 и 420 нм. Схема реакции [58]:
178
В работе [59] мешающие ноны предложено маскировать ЭДТА п экстрагировать комплекс толуолом. При этом определению не мешают Fe, Си, Movl, Ti, Сг1”, Ni”, Co”, Те'1 и As, мешают сильные окислители. Предел обнаружения се-
лена при использовании кюветы с толщиной поглощающего слоя 1 см составляет 50 ppb. Молярный коэффициент поглощения равен 2,89-104 при 340 нм. Метод применен для анализа биологических объектов [60, 61]. Минимальное содержание селена — 1 мкг. Серьезным недостатком является канцерогенность ДАБ. Флуориметрический метод определения селена с ДАБ приведен ниже.
Среди о-диампнов более устойчивым реагентом является 2,3-диаминонафталин (ДАН) [62, 45]. Поглощение комплекса измеряют при 380 нм. При экстракции комплекса толуолом закон Бера соблюдается в интервале 0—20 мкг селена. Чувствительность определения селена с ДАН в два раза выше, чем с ДАБ, однако избирательность определения снижается. Мешающие ионы указаны в работе [45]. Мешают окислители (например, гипохлорит окисляет реагент) и восстановители [олово(П) восстанавливает се-лен(1У) до элементного]. Реагент ДАН более эффективен для
флуориметрического определения, чувствительность которого в 10 раз выше; чем фотометрического. Вероятно, ДАН такой же канцерогенный реагент, как и ДАБ.
Другие о-диамины также изучены как спектрофотометрические реагенты, особенно 1,2-диаминобензол [63, 64] и 4-замещенные о-фенилендиамина, например 4-метил-4-хлор- и 4-нитро-о-фенилен-диамин [65]. С помощью 4-метил-о-фенилендиамина определено содержание следов селена в железе и стали [66]. Окраска комплекса развивается при 25 °C в течение 2 ч, однако при 70 °C окраска устанавливается за 5 мин. Железо(III) экстрагируют раствором каприловой кислоты в хлороформе. Некоторые металлы, например Movi, WV1 и Vv, предварительно экстрагируют раствором 8-гидроксихинолина в хлороформе. Затем образующийся при взаимодействии Selv с реагентом 5-метнлпиазоселенол экстрагируют
толуолом и измеряют поглощение при 337 нм. Закон Бера наблюдается в интервале содержаний селена (IV) 1—40 мкг в 10 мл толуола. Определению 19,7 мкг селена не мешают следующие ионы (указано их допустимое содержание в мг): А1111 (200), Asv(5), Ва“ (500), Са” (400), Си” (200), Со” (200), Mg”, (400), Мп’1 (400), Ni^'(200), Сг”1 (100), Vv(2). Окислители, такие, как Fe”’ и Чг *, мешают, образуя окрашенные продукты. Ионы Fe”1 удаляют экстракцией каприлатов или маскируют введением ЭДТА. Ионы От можно восстановить пероксидом водорода, образующийся Сг”1 Не Мешает при содержаниях до 100 мг. Не мешают 10 мг меди(II) ств0МГ ванаДия(А), если толуольный экстракт промыт 0,1 М ра-~-.Д ^Иазоселен°лы при взаимодействии с селеном (IV) образуют и
Реагенты, однако обычно растворы реагентов неустойчивы , ₽еДелению мешают окислители.
179
Другая группа спектрофотометрических методов основана на образовании комплекса селена с серуеолержашим лигандом. К этим реагентам относятся тиоглпколевая кислота [67], 2-мер-каптобензойная кислота [68] и висмутол (II) [69], Обзор этой группы методов дан в работе [68], где приведена чувствительность методов, значения молярных коэффициентов поглощения и мешающие ионы. Комплекс селена с 2-меркаптобензойной кислотой экстрагируют этплацетатом. Молярный коэффициент поглощения комплекса равен 1,56-104, чувствительность определения селена (0,005 мкг'Мл) выше, чем при использовании других реагентов этой группы. Мешающее влияние Hg, Те, Си, Ni, Со, Ag, Ti и Zr можно устранить ионным обменом, маскировкой или предварительной экстракцией. Определению мешает ртуть(II), Интервал определяемых содержаний селена — 0,008—1,5 рргп.
Кроме указанных существуют и другие спектрофотометрические методы. Методы, основанные на каталитической активности Selv, описаны ниже. Некоторые методы основаны на образовании промежуточного соединения селена (IV) с реагентом. Затем промежуточное соединение образует окрашенный продукт. Такой метод, примененный! для анализа загрязнений воздуха [70], основан на описанной ранее [71] капельной реакции, в которой гидроксила мин солянокислый окисляется селенистой кислотой в сильнокислой среде до азотистой кислоты, а возникающий нитрит определяют по образованию азокрасителя методом Грисса. Интервал определяемых содержаний селена 0,01—0,20 мг/л, е= 1,93-105 при 544 нм. Окраска развивается в течение 90 мин. Влияние мешающих ионоз можно устранить.
Предложены спектрофотометрические методы определения анионов с применением трифенилметановых красителей, основанные на окислении иодида и бромида хлорамином Б, хлорамином Т или гипохлоритом. Образующиеся ионы 1+ или Вг+ ослабляют поглощение красителей. При взаимодействии селена (IV) с отмеренным избытком KI образуется иод. Остаточное содержание по-дида определяют описанным выше методом [72] с чувствительностью 8 и 7 нг/мл селена при использовании бриллиантового зеленого и малахитового зеленого соответственно. Определению селена (IV) мешает 10-кратный избыток Sb11’, Hg”, Hg1, Ag, Fe’”, Bi111, Си” и S2~ и 100-кратный избыток Pb" и SnIf.
Флуориметрический метод
Метод, основанный на взаимодействии селена (IV) с о-диами-нами и образовании флуоресцирующих пиазоселенолов, отличается высокой чувствительностью. Метод широко применяют для анализа биохимических объектов, содержащих субмикрограммовые количества селена.
Чувствительность определения селена с применением 2,3-диа-минонафталина (ДАН) выше, чем с помощью 2,3-диаминобензи; дина (ДАБ), однако избирательность определения с помощью ДАБ выше [62]. Комплекс, экстрагированный толуолом, неустойчив [62]-180
Несмотря на высокую летучесть, циклогексан является хорошим растворителем. При прочих равных условиях чувствительность флуорометрического метода выше, чем спектрофотометрического.
При анализе биологических объектов их подвергают мокрому сожжению [73], не проводя предварительного отделения селена. Метод мокрого сожжения растительных материалов, содержащих 0,005 ppm селена, описан в работе [74], посвященной флуорпмет-рическому определению селена с 2,3-диаминонафталпном. Изучено также определение селена с ДАН [75, 76], в том числе при анализе почв [77]. Проведено сравнительное изучение шести методов определения следов селена [78], в частности метода нейтронной активации. Методы очистки и хранения серийно выпускаемого реагента ДАН описаны в работе [74]. Флуориметрический и спектрофотометрический метод определения селена с ДАН рекомендован в работе [79]. Опубликован автоматизированный флуориметрический метод определения селена с ДАН [88].
Нефелометрический и турбидиметрический методы
Реагент ДАН применен для нефелометрического определения селена [80], в интервале содержаний меньших, чем для гравиметрического, но больших, чем для спектрофотометрического метода. Турбидиметрический метод после ионообменного выделения позволяет определять 10—100 мкг селена [55].
Атомно-абсорбционный (ААС)
и атомно-флуоресцентный (АФС) методы
Атомно-абсорбционный метод определения селена имеет недостатки. Наиболее чувствительная линия поглощения атомов селена в пламени расположена в области 196 нм. Для этой линии отношение сигнала к шуму невелико, и предел обнаружения селена со-• ставляет примерно 1 ppm.
Разработан косвенный метод, основанный на экстракции комплекса SeIV с ДАН, взаимодействии комплекса с PdCls и определении палладия методом ААС [81]. Метод более чувствителен, чем основанный на измерении поглощения комплекса Selv с ДАН.
Предложен метод, основанный на генерировании гидрида селена и введении его в аргон-водородное пламя [82]. Улучшенный вариант гидридного генератора, описанный в работе [83], приме-Чен Для анализа пищевых продуктов и фуража. На рис. 27 приведена схема генератора. Достигнута чувствительность определения селена 6- 1СН10 г с относительным стандартным отклонением 5—8%. • ® Другой работе [84] достигнут предел обнаружения селена 0,0018 мкг/мл. Показано, что сигнал селена (VI) ниже, чем селена (IV). В отличие от теллура (VI), который количественно восстанавливается соляной кислотой, выход селена (IV) из селена (VI) оставляет всего 50%. Изучено влияние посторонних ионов при Чределении селена гидридным методом [85] и отмечено, что ме-i.’’ Сложнеел чем ранее предполагалось.
181
Рис. 27. Гпдрпдный генератор для определения селена методом ААС (трубка атомизатора изолирована кварцевой тканью для снижения колебаний температуры):
1 — трубка атомизатора; 2—трубка для ввода кислорода.
Метод непламенной ААС обладает преимуществами [86]. Диапазон определяемых содержаний селена 1,0—50 мкг/л. Мешающие катионы отделяют ионным обменом, для повышения чувствительности и подавления влияния анионов в раствор вводят молибден.
Описан гидридный вариант метода АФС [87]. Предел обнаружения селена, отвечающий удвоению сигнала относительно нулевой линии, составляет 0,0001 мкг.
Метод молекулярной эмиссии в полости
Этим методом можно определять селен и теллур [89]. Методика анализа описана в разделе «Сульфиты и оксиды серы».
Электрохимические методы
Потенциометрический метод
Путем замены жидкого ионообменника в свинец-селективном электроде насыщенным раствором З.З'-диаминобензидина в гексане изготовлен селен(IV)-селективный электрод [90]. Линейность градуировочного графика наблюдается при pH = 2,5 в интервале 10-4—10~3 М. Избирательность определения достаточно велика.
Потенциометрический и амперометрический метод определения селена (IV) и теллура (IV), основанные на взаимодействии их с аскорбиновой кислотой, позволяют определять миллиграммовые содержания элементов [91].
Кулонометрический метод
Кулонометрический метод [92] основан на восстановлении SerV иодидом с образованием свободного иода, который восстанавливают избытком тиосульфата. Тиосульфат титруют электрогенериро-ванным иодом. Микросодержания селена (IV) и теллура (IV) можно титровать электрогенерированным гипобромитом [93] или железом (III) [94]. Окисление SeIV и TeVi проводят раствором КМпО4, а избыток КМпО4 титруют железом(II),
182
Полярографический метод
Первый полярографический метод определения селена опубликован в 1935 г. Проведенные позднее исследования [95,96] .легли в основу метода определения селена в разных электролитах. В зависимости от pH раствора наблюдается три полярографических волны восстановления селена(IV). Определение Se,v рекомендуется проводить в буферном растворе с pH = 2,0 — 2,5. Определению мешает медь, которую, однако, можно экстрагировать раствором дитизона в хлороформе.
Определение следов селена в биологических материалах полярографическим методом (как, впрочем, и другими) встречает трудности. При содержании менее 4 мкг селена рекомендуется его предварительное отделение.
Каталитические методы
Субмикрограммовые содержания селена (IV) можно определять по его каталитическому действию на восстановление 1,4,6,11-тетра-азанафтацена [97]:
N N N NH
N N NH N
Определению не мешает SeVI, мешает 1 мг Ba11, Fe111; 0,1 мг Vv, WVI; 0,01 мг Мп” и Си11. Определению мешают любые содержания Bi1’1, Sn11, Telv и I-. Влияние Sb1” маскируют винной кислотой. Метод относительно прост. Определение заканчивают спектрофо-гбметрированием раствора. Предложен каталитический метод [98]; он основан на каталитическом действии селена (IV) на окисление n-гидразинобензолсульфокислоты до n-диазобензолдиазония, который сочетают с лг-фенилендиамином; при этом образуется азокраситель желтого цвета, который спектрофотометрируют. Значение эффективного молярного коэффициента поглощения равно 1,2- 10s (а при экстракции красителя 2,4-106), предел обнаружения селена (IV) в кювете с толщиной слоя 1 см составляет КУ-7 М. Мешают 100 мкг Си”, Ce'v, MoVI и I-; 10 мкг Fe111, Vv, 'Crvi. Влияние некоторых примесей (кроме Те) можно устранить.
Предложен метод определения селена с чувствительностью 4 нг/мл [99], основанный на каталитическом действии селена(IV) На. реакцию окисления сульфата гидразина посредством КСЮ4. Селен (IV) катализирует восстановление метиленовой сини сульфидом натрия [100]. Метод позволяет определять 0,1 —1,0 мкг седела. Определению мешает медь при содержании более 10 мкг.
Радиохимические методы
В последние годы метод нейтронной активации все больше не-ЯОльзуют для определения селена в биологических образцах. Это у°'Ьясняется возможностью потерь селена на стадиях предвари
183
тельной подготовки проб. Особенности определения селена в биологических материалах обсуждены в обзоре [6].
Радиохимический метод применен для анализа загрязнении воздуха 1Ю1] и почв [102]. Образцы облучали в течение 2 суток в потоке нейтронов 1,5-Ю13 1/(см2-с). у-Активность измеряют германий-литиевым детектором площадью 35 см2, соединенным с многоканальным анализатором. Для определения 0,18—1,6 ppm селена используют пик селена-75 (период полураспада 120 суток) с энергией 265 кэВ.
Газохроматографические методы
В последнее время стала заметной тенденция применения газовой хроматографии в неорганическом субмикроанализе. Комплекс Selv с 2,3-диаминонафталином (ДАН) можно экстрагировать гексаном и определять методом газовой хроматографии [103], используя электронозахватный детектор. Метод применен для анализа крови, мочи и речной воды. Анализ длится в течение 3 ч, включая 2 ч экспозиции для образования комплекса. Подтверждено, что мешающие определению примеси можно замаскировать введением ЭДТА. Электронозахватный детектор очень чувствителен к содержанию комплекса, предел обнаружения селена 0,0005 мкг. Градуировочный график линеен в интервале 0,01—0,1 мкг селена.
Для газохроматографического определения селена применен 1,2-диамино-4-нитробензол [104, 105]. Селен (IV) взаимодействует с реагентом, образуя 5-ннтропиазоселенол, который определяют с помощью электронозахватного детектора.
В работе [104] описано определение селена в молоке, молочных продуктах и альбумине. Метод позволяет определять селен (IV) и сумму Selv и SeVI, поскольку в биологических материалах селен присутствует в двух степенях окисления. Труднее всего определять селен (VI). В работе [104] сначала определяли SeIV, а затем, после восстановления SeIV, суммарное содержание селена. Показано, что 60% селена в молоке находится в виде селена (VI). Практический предел обнаружения селена составляет 0,005 мкг.
В работе [105] описан метод определения селена в высокочистом мышьяке или оксиде мышьяка (III), применяемых для изготовления полупроводников.
ЛИТЕРАТУРА
1. Shendrikar A. D., West Р. W. — Analytica chim. Acta, 1975, v. 74, p. 189.
2. Назаренко И. И., Ермаков А. Н. — Аналитическая химия селена и теллура. М., Наука, 1971, 251 с.
3. Green Т. Е., Turley М. — In: Treatise on Analytical Chemistry (Ed.
I. M. Kolthoff and P. J. Elving), Part II, Vol. 7, New York, John Wiley, 1961, p. 137.
4. Lowenheim F. A. — In: An Encyclopedia of Industrial Chemical Analysis (Ed.
F. D. Snell and L. S. Ettre), Vol. 17, New York, Interscience, 1973, p. 580. 5 Elwell W. T., Saint H. С. J. — In: Comprehensive Analytical Chemistry (Ed.
C. L. and D. W. Wilson), Vol. 1C, Amsterdam, Elsevier, 1962, p. 296.
184
6 Olson О E., Palmer 1 S.. Whitehead E I. — In. Methods of Biochemical Analysis (Ed. D. Glick), Vol. 21, London, John Wiley, 1973. p. 39.
7. Korkisch J. — Modern Methods for the Separation of Rarer Metal Ions, Oxford, Pergamon Press, 1969, p. 357.
8. Barcza L. — Z. analyt. Chem., 1963, v. 199, p. 10.
9. Fogg D. N., Wilkinson N. T. — Analyst, 1956, v. 81, p. 525.
10. Official Methods of Analysis of the AOAS, Washington D C., Association of Official Agricultural Chemists, 10th edn, 1965, p. 109.
jl. Watkinson J. H.— Analyt. Chem., 1960, v. 32, p. 981.
12. Schumann H., Koelling IE. — Z. Chem., 1961, v. 1, p. 371; Chem. Abstr., 1962, v. 57, p. 3076c.
13. Cukor P., Walzyck J., Lott P. F.— Analytica chim. Acta, 1964 v. 30, p. 473.
14. Yamamoto M, Sakai H. — Analytica chim. Acta, 1965, v. 32, p. 370.
15. Miyamoto M.—Japan Analyst, 1961, v. 10, p. 211; Analyt. Abstr., 1963, v. 10, p. 2245.
16. Iguchi A. — Bull chem. Soc. Japan, 1958, v. 31, p. 748, Analyt. Abstr., 1959, v. 6, p. 2567.
17. Lederer M., Kertes S. — Analytica chim. Acta, 1956, v. 15. p. 226.
18 Barooshian A. Lautenschleger M. L, Harris W G. — Atomlight, 1970,
v. 71, p. 5; Analyt. Abstr., 1971, v. 20, p. 3014.
19. De—A. K., Khopkar S. M., Chalmers R. .4. — Solvent Extraction of Metals, London and New York, Van Nostrand Reinhold 1970
20. Clark R. E. D. — Analyst, 1958, v. 83, p. 396.
21. Inarida M. — Bull. chem. Soc. Japan, 1966, v. 39, p. 403; Analyt. Abstr., 1967, v. 14, p. 3148
22. Bode H„ Arnswald W. — Z. analyt. Chem., 1962, v. 185, p. 179.
23. Patrovsky V. — Chemike Listy, 1970, v. 64, p. 715.
24. Akki S. B., Khopkar S. M. — Separation Sci., 1971, v. 6, p. 455.
25. Ramakrishna R. S., Irving H. M. N. H. — Chem. Commun., 1969, p. 1356.
26 Ramakrishna R. S., Irving H. M. N. H. — Analytica chim. Acta, 1970, v. 49, p. 9.
27. Stary J., Marek J., Kratzer K., Sebesta F. — Analytica chim. A^ta, 1971, v. 57, p. 393.
28. Kasterka B., Dobrovolski J. — Chemia Analit., 1970, v. 15, p. 303; Analyt. Abstr., 1971, v. 20, p. 2424.
29. Kunte N. S., Ranade S. N. — Ind. J. Chem., 1970, v. 8, p. 370.
30. Hillebrand W. F„ Lundell G. E. F., Bright H. A., Hoffman J. I. — Applied Inorganic Analysis, 2nd edn, New York, John Wiley, 1953.
31. Ref. 4, p. 590.
32. Bode H. — Z. analyt. Chem., 1951, v. 134, p. 100.
33 Luke C. L. — Analyt. Chem., 1959, v. 31, p. 572.
34. Stanton R. E., McDonald A. /. — Analyst, 1965, v. 90, p. 497.
35. Ewan R. C., Baumann C. A., Pope A. L. — J. agric. Fd Chem., 1968, v. 16, p. 212.
36. Severne В C., Brooks R. R. — Analytica chim. Acta 1972, v. 58. p. 216.
37 Ishibaschi M., Fujinaga T., Kawamoto T., Murai S. — J. chem. Soc. Japan, pure Chem. Sect., 1967, v. 88, p. 76; Analyt. Abstr., 1968, v. 15. p 3636.
38 Kempe G., Denn W. — Z. analyt. Chem., 1966, v. 217, p. 169, Analyt. Abstr., 1967, v. 14, p. 3952.
39. Mao Chih-Hsiang — Acta chim. Sinica, 1965, v 31, p. 432; Chem. Abstr., 1966, .n £ 64, p. 8909f.
40. Hu Zhi-Tei — Acta chim. Sinica, 1964, v. 30 p. 426; Analyt. Abstr., 1965, v. 12, P- 5758.
49 ?hiPPs A. AL —Analyt. Chem., 1971, v 43, p. 467.
' ikai Kukreja V. P. — Chromatographia, 1969, v. 2, p. 18; Analyt. Abstr., d-г £70’ v' 18’ P- 3093-
4? Williams L. R., Haskett P. R — Analyt. Chem., 1969, v. 41, p 1138
naka T., Katagihara H., Iritani N. — Japan Analyst, 1971, v. 20, p. 163;
4c ,П?‘У‘- Abstr., 1972, v. 23, p. 236.
r> iisn’ Cukor P., Moriber G., Soiga J. — Analyt. Chem., 1963, v. 35,
185
46. Koithoff I. M., Belcher R.— Volumetric Analysis, Vol. III. X'ew York, Inter-science. 1957. p. 327.
47. Schrenk W. T., Browning B. L. — J. Am. chem. Soc.. 1926. v. 48. p. 139.
48. Lenher V.. Wakefield H. F.— J. Am. Chem. Soc., 1923. v. 45, p. 1423.
49. Naidu P. P., Rao G. G.— Talanta, 1971, v. 18, p. 112.
50. Kaushik R. L., Prosad R. — J. Ind. chem. Soc., 1969, v. 46, p. 921.
51. Saxena О. C. — J. appl. Chem., 1969, v. 32, p. 89.
52. Захаров В. А., Сангина О. А., Клюева P. И, — Зав. лаб., 1972, т. 38, с. 1066; Analyt. Abstr., 1973, v. 24, р. 2164.
53. Solymosi F. — Chemist Analyst, 1963, v. 52, p. 42.
54. Schulek E., Barcza L.— Talanta, 1959, v. 3, p. 27, 31.
55. Official, Standardised and Recommended Methods of Analysis, 2nd edn (Compiled and Edited by N. W. Hanson), London, The Society for Analytical Chemistry, 1973, p. 440.
56. West P. U7, Cimerman C. — Analyt. Chem., 1964, v. 36, p. 2013.
57. Hoste J.. Gillis J. — Analytica chim. Acta, 1955, v. 12, p. 158.
58. Parker C. A., Harvey L. G. —Analyst, 1961, v. 86, p. 54.
59. Cheng K. L. — Analyt. Chem., 1956, v. 28. p. 1738.
60. Cummins L. M., Marlin J. L., Maag D. D.—Analyt. Chem., 1965, v. 37, p. 430.
61. Cummins L. M., Martin J. L., Maag G. B7., Maag D. D. — Analyt. Chem., 1964, v. 36, p. 382.
62. Parker C. A., Harvey L. G. — Analyst, 1962, v. 87, p. o58.
63. Throop L. J. — Analyt. Chem., 1960, v. 32, p. 1807.
64. Arioshi H., Kiniwa M., Toei K. — Talanta, 1960, v. 5, p. 112.
65. Tanaka M., Kawashima T. — Talanta, 1965, v. 12, p. 211.
66. Kawashima T., Ueno A. — Analytica chim Acta, 1972, v. 58, p. 219.
67. Kirkbright G. F„ Ng W. K- — Analytica chim. Acta, 1966, v. 35, p. 116.
68. Gresser M. S., West T. S. — Analyst, 1968, v. 93, p. 595.
69. Yoshida H„ Taga M., Hikime S. — Japan Analyst, 1967, v. 14, p. 4670.
70. Osburn R. L., Shendrikar A. D., West P. B7.— Analyt. Chem., 1971, v. 43, p. 594.
71. Feigl F., Demant V. — Mikrochim. Acta, 1937, v. 1, p. 134.
72. Раманаускас E. И., Шулюнене A. K.— ЖАХ, 1968, t. 23, c. 1589; Analyt. Abstr., 1970, v. 18, p. 3932.
73. Watkinson J. H. — Analyt. Chem., 1966, v. 38. p. 92.
74. Hall R. J., Gupta P. /.. — Analyst, 1969, v. 94, p. 292.
75. Назаренко И. И., Кислов А. М., Кислова И. В., Малевский А. Ю. — ЖАХ, 1970, т. 25, с. 1135; Analyt, Abstr., 1971, т. 21, с. 3363.
76 Назаренко И. И., Кислова И. В., Гусейнов Т. М. и др. — ЖАХ, 1975, т. 30. с. 733.
77. Hemsted W. R. Т., Sina, Cekicer S. — Analyst, 1972, v. 97, p. 383.
78. Olson О. E.—J. Ass. off. analyt. Chem., 1969, v. 52, p. 627.
79. Ref. 55, p. 288.
80. Smith R. E., Lott P. F. — Microchem. J., 1975, v. 20, p. 519.
81. Lau H. K. Y., Lott P. Л —Talanta, 1971, v. 18, p. 303.
82. Manning D. C. — Atom. Abs. Newsl., 1971, v. 10, n. 123.
83. Sterner D. D., Hagemann L. — Analyt. Lett., 1975, v. 8, p. 323.
84 Thompson К. C., Thomerson D. R. — Analyst, 1974, v, 99, p. 595.
85. Smith A. E. — Analyst, 1975, v. 100, p. 300.
86. Henn E. L. — Analyt. Chem., 1975, v. 47, p. 428.
87. Thompson К. C. — Analyst, 1975, v. 100, p. 307.
88. Brown M. W., Watkinson J. H. — Analytica chim. Acta, 1977, v. 89, p. 29.
89. Belcher R., Kouimtzis T., Townshend A. — Analytica chim. Acta, 1974, v. 68. p. 297.
90. Malone T. L., Christian G. D. — Analyt. Lett.. 1974, v. 7, p. 33.
91. Иванова 3. И., Игнатенко E. Г., Тарасова В. А. — ЖАХ, 1973, т. 28, с. 1980, Analyt. Abstr., 1975, v. 28, 6 В 163
92. Patriarche G. L. — Analyt. Lett., 1972, v. 5, p. 45.
93. Агасян Л. Б. Николаева E. P., Агасян П. К. — Вестник Моск. гос. ун-?а-Сер. хим., 1966, т. 21, с. 96; Analyt. Abstr., 1967, v. 14, р. 7456.
1 or
94. Агасян П. К., Денисова А. И., Агасян Л. Б., Николаева Е. Р.—Зав. лаб., 1968, т. 34, с. 129: Analyt. Abstr., 1969, v. 16, р. 2442.
95. Christian G. D., Knoblock E. C., Purdy W. C. — Analyt. Chem., 1963, v. 35, p. 1128.
96. Chrinstian G. D., Knoblock E. C., Purdy W. C. — Analyt. Chem., 1965, v. 37, p. 425.
97. Kawashima T., Tanaka Al. — Analytica chim. Acta, 1968, v. 40, p. 137.
98. Kawashima T., Kai S., Takashima S. — Analytica chim. Acta, 1977, v. 89, p. 65.
99. Bognar J.. Sarosi S. — Mikrochim. Acta, 1969, p. 361.
100. West P. №.. Ramakrishna T. V. — Analyt. Chem., 1968, y. 40, p. 966.
101. Dams R., Robbins J. A., Rahn K- A., Winchester J. W. — Analyt. Chem, 1970, v. 42, p. 861.
102. Kronborg O. J., Steinnes E. — Analyst, 1975, v 100, p. 835.
103. Young J. №., Christian G. D. — Analytica chim. Acta, 1973, v. 65, p. 127.
104. Shimoishi Y.— Analyst, 1976, v. 101, p. 298.
105. Akiba Al., Shimoishi У., Toei K- — Analyst, 1976, v. 101, p. 644.
СЕЛ ЕНОЦИАНАТЫ
МЕТОДЫ ОТДЕЛЕНИЯ
Дистилляция
Селеноцианат SeCN~ обычно отделяют путем обработки бромом. При этом SeCN- переходит в летучий бромциан:
SeCN" + 4Вг2 + 4Н2О —> CNBr 4- 7Br‘ + SeO4' + 8Н+
Бромциан отгоняют и определяют описанными ниже методами.
Ионообменный метод
Селеноцианат и роданид можно разделить на анионообменной бумаге SB2, используя 6 М раствор LiNO3 как растворитель [1].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
' Титриметрические методы
Иодиметрический метод
Селеноцианат переводят с помощью брома в бромциан, избыток брома связывают фенолом. При взаимодействии BrCN с KI выделяется иод, который титруют стандартным раствором тиосульфата 12]. В работе [3] показано, что определению не мешают сульфат, хлорид и перхлорат, но мешает бромид, вероятно смещая влево равновесие реакции
H2SeO3 4- Вг2 4- Н2О = HSeOi 4- ЗН+ 4- 2Вг'
влияние Вг~ можно устранить дистилляцией BrCN или введением X "аствоР вольфрамата натрия.
187
Иодиметрическое определение селенонианата [2]
Реагенты
п-Этокспхрнзоидин, 0.2%-ный раствор в этаноле.
Фенол. 5%-ный раствор.
Тиосульфат натрия. 0.1 .И раствор.
Соляная кислота. 20%-ный раствор.
Ход анализа. К 5 мл анализируемого раствора добавляют 75 мл воды и I г борной кислоты. Раствор кипятят в течение 10 мин. охлаждают и переносят з колбу Эрленмейера вместимостью 200 мл. Приливают 80 мл воды и нейтрализуют 0.1 31 раствором НС1. добавив 1 каплю раствора индикатора п-этокспхрп-зопдина. Добавляют бромную воду до появления желтой окраски раствора. Через несколько минут приливают 3—5 мл фенола, 2—3 мл 20%-ного раствора НС! п через 5 мпп добавляют 0,2 г KI. Титруют выделившийся иод стандартным раствором тиосульфата, добавляя раствор крахмала вблизи точки эквивалентности. Учитывают, что 1 мл 0,1 Л1 раствора тиосульфата соответствует содержанию 0,5249 мг SeCN-.
Спектроскопический метод
Описан метод определения микросодержаний селеноциапата, основанный па взаимодействии бромциана с пиридином и бензидином [4]. В результате реакции образуются продукты, окрашивающие раствор в оранжевый цвет. Эта реакция была использована ранее [5, 6] для определения цианида и роданида. Поглощение измеряют при 518 нм, закон Бера соблюдается до содержаний SeCN~ 0,5 мкг/мл.
ЛИТЕРАТУРА
1. Lederer М., Martini-Bettolo R. — J. Chromatog, 1968, v. 35, р. 213.
2. Schulek Е., Koros Е. — Z. analyt. Chem., 1953, v. 139, p. 20; Analyt. Abstr., 1954, v. 1, p. 75.
3. Schulek E., Barcza L. — Taianta, 1959, v. 3, r. 23.
4. Vasukl K„ Shankaranarayana M. L., Majumder S. K. — Analytica chim. Acta, 1969, v. 45, p. 353.
5. Aldridge W. Ah—Analyst, 1944, v. 69, p. 262.
6. Aldridge W. hl. — Analyst, 1945, v. 70, p. 474.
СИЛИКАТЫ
Определение силиката в растворах—важная аналитическая задача. Предварительной стадией анализа является переведение всех форм кремния в растворимый силикат. Например, минералы, стекла и другие объекты обычно разлагают сплавлением с NaOH. Плав растворяют и полученный раствор силикатов анализируют.
Определение силикатов проводят при анализе полупроводников, металлургических продуктов, стекол, минералов и промышленных вод. Определение силикатов важно для медицины в связи с силикозом, который развивается при вдыхании кремнезема или кварцевой пыли.
Важной аналитической задачей является определение силиката в присутствии фосфора, мышьяка и германия. Этот аспект анализа будет обсужден ниже. Аналитическая химия кремния рассмотрен3 в работах [1] и [2]. Детальный обзор методов определения креМ-183
ния в керамических материалах и геохимических объектах приведен в работе [3].
В настоящем руководстве методы разложения проб, в том числе при анализе силикатов, не рассматриваются. Методы разложения проб, являющиеся важным этапом силикатного анализа, рассмотрены в монографии Долежала, Повондры и Шулека [4].
При определении мпкросодержаний кремния растворы, особенно стандартные, следует хранить в полиэтиленовой, а не в стеклянной посуде, чтобы избежать внесения примеси кремния из стекла.
Для приготовления стандартного раствора силиката сплавляют точную навеску SiO2 с 10-кратным избытком безводной соды в платиновом тигле. Плав охлаждают и растворяют в воде в полиэтиленовом сосуде. Доводят объем раствора водой до метки и хранят в полиэтиленовом сосуде.
МЕТОДЫ ОТДЕЛЕНИЯ И ПРЕДВАРИТЕЛЬНОГО КОНЦЕНТРИРОВАНИЯ
Осаждение
Силикат можно отделить от фосфата путем осаждения молибдатом аммония [5]. Этот метод применяют в органическом микроанализе для разделения 0,2—2 мг каждого аниона.
Экстракция
Метод экстракции применяют для разделения элементов, образующих гетерополикислоты — Р, As, Si и Ge (см. разд. «Ортофосфат»), Из рассмотрения некоторых схем анализа, приведенных в разделе «Ортофосфат», видно, что в некоторых случаях экстрагируются мешающие ионы, а силикат остается в водной; фазе. Описана также экстракция невосстановленной формы кремнемолибденовой кислоты 2-этилгексанолом [6], изобутилметилкетоном [7], триизооктиламином азотнокислым в 1,2-дихлорэтане [8] и смесью диэтилового эфира с пентанолом [9, 10]. В отличие от восстановленной фосфориомолибденовой' кислоты, которая экстрагируется, восстановленная форма кремнемолибденовой кислоты не экстрагируется органическими растворителями. В работе [И] показано, что восстановленная форма кремнемолибденовой кислоты экстрагируется изоамиловым спиртом при повышенной концентрации кислоты в водной фазе. На этом основан спектрофотометрический метод определения кремния [11]. Описан другой вариант метода, основанный на экстракции кремнемолибденовой кислоты [12] хлороформом в присутствии карбоната пропандиола-1,2 п спектрофо-чометрировании экстракта.
Хроматографический метод
Мет^ЛЯ опРеделения силиката применяют хроматографические слойка В Раб°те [13] силикат и фосфат разделяли методом тонко-нон хроматографии на целлюлозе марки S и S Ecteola с
189
помощью растворителя изопропанол — вода — безводная уксусная кислота (20:5:2). Метод позволяет выделить силикат из 1 мкл при концентрации Ю-4 М.
Описан газохроматографический метод, основанный на разделении производных трнметилснлана [14]. В некоторых случаях возможно ионообменное отделение мешающих ионов, предшествующее спектрофотометрическому определению силиката [15]. Кремневую кислоту отделяют от сопутствующих элементов, включая железо, алюминий и уран, пропуская раствор через колонку с катионитом, а затем со слабоосновным анионитом [16]. При этом а-форма кремневой кислоты не задерживается. Предварительно р- и у-форму кремневой кислоты необходимо перевести в a-форму. Ионный обмен применяют также для предварительного концентрирования силиката. Показано [17], что эффективность разделения повышается в присутствии HF, поскольку образующийся фторсиликат лучше задерживается анионитом. Затем фторсиликат элюируют борной кислотой.
Дистилляция
Отделение кремния методом дистилляции используют при анализе силикатов, в том числе портланд-цемента. Отгонку H2SiFs с паром проводят на глицериновой бане [18], поднимая постепенно температуру до 135 °C.
Устранение влияния мешающих ионов
Приемы, не относящиеся к методам отделения и позволяющие повысить избирательность по отношению к элементам, образующим гетерополикислоты (Р, As и Si), подробно описаны в разделе «Ортофосфаты». Эти приемы основаны на следующих принципах.
1. Лимонная, винная и щавелевая кислоты разрушают фосфор-номолибденовую, но не кремнемолибденовую кислоту. Лимонная кислота разрушает также мышьяковомолибденовую кислоту.
2. Если в раствор вводят НС1О4 перед добавлением молибдата аммония, то кремнемолибденовая кислота не образуется, а соответствующие гетерополикислоты фосфора и мышьяка образуются.
Использование указанных принципов позволяет определять силикат в присутствии фосфата и арсената.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Классический метод определения растворимых силикатов состоит в дегидратации кремневой кислоты путем выпаривания раствора с последующим гравиметрическим определением кремнезема. Для получения растворимых форм кремния пробу разлагают кислотами или сплавляют с карбонатом натрия и образующийся плав обрабатывают кислотой, обычно соляной. Иногда вместо НС1 или H2SO4 рекомендуют применять НСЮ4, поскольку почти все
190
перхлораты металлов растворимы в разбавленной НС1О4. Если в пробе присутствуют восстановители, перед добавлением НС1О4 ее необходимо обработать НХО3. При использовании H2SO4 возможно образование нерастворимых или безводных медленно растворяющихся осадков сульфатов, которые могут захватывать силикаты.
Дегидратация ортокремневой кислоты описывается уравнением H4SiO4 —► SiO2 + 2H2O
Методика дегидратации осложняется вследствие образования коллоидных растворов кремнезема. Подробное описание методики дано в работах [1,2], а также в монографии Кольтгофа [19].
Классический метод позволяет получать точные результаты при соблюдении некоторых методических тонкостей. Метод использован еще Клапротом, который для отделения кремния применял повторное упаривание с соляной кислотой [20]. Несмотря на то, что позднее были предложены другие гравиметрические и титриметрические методы, основанные на образовании молибдосиликата хино-линия, классический метод все еще находит применение.
В методике дегидратации кремневой кислоты предполагается
упаривание раствора досуха и нагревание сухого остатка при 110 °C в течение 1 ч. Затем осадок обрабатывают 10%-ной НС1 в течение 15 мин для извлечения растворимых солей. Растворение кремнезема при этом невелико. Кремнезем отфильтровывают и промывают горячим 5%-ным раствором НС1. Фильтрат упаривают, образующийся кремнезем промывают холодным 1 %-ным раствором HCI. Осадки кремнезема объединяют, прокаливают в платиновой чашке и взвешивают. Несмотря на двукратное упаривание, часть силиката остается в растворе. Содержание растворимых силикатов
молено определить спектрофотометрическим методом и исправить результаты анализа. Осадок кремнезема прокаливают при 1200 °C. Прокаленный осадок может содержать посторонние примеси. Если
осадок обработать смесью HF и H2SO4 и удалить кремний в виде H2SiF6 и SiF4, то, зная массу сухого остатка, можно внести поправку на содержание посторонних примесей. Осадок кремнезема смачивают несколькими каплями воды и концентрированной H2SO4. приливают около 5 мл 60%-нон HF, упаривают досуха и прокаливают осадок при 1200 °C. При этом удаляется кремний и разрушаются сульфаты металлов. Обработку смесью HF и H2SO4 при необходимости повторяют.
Гравиметрический метод определения кремния достаточно избирателен. В присутствии фторидов получаются заниженные результаты вследствие летучести H2SiF6 пли SiFt. Влияние фторида можно устранить добавлением борной кислоты, избыток которой Удаляют в виде метилбората перед дистилляцией. При наличии лова (IV) для его удержания в растворе следует дополнительно вводить серную кислоту
молибД гРави„метРических методов основан на осаждении кремне-деновой кислоты органическими основаниями, например 2,4-
191
диметплхинолином [21], 8-гидроксихинолином [22—24] и хинолином [25, 21, 26]. Изучено 54 органических основания, которые можно применять в качестве осадителей кремнемолибденовой кислоты. Показано, что для определения более 0,1 мг кремнезема наиболее эффективен пиридин.
2,4-Днметилхинолнн, рекомендованный в работе [21], образует с кремнемолибденовой кислотой малорастворимый, но гигроскопичный осадок, поэтому его прокаливают при 500—600 °C до кремнемолибденового ангидрида. В присутствии 8-гидроксихинолина [22] образуется осадок состава (C9H7ON)4H4[SiO4-12Мо03]. Точные условия определения рекомендованы в работе [28]. Метод применен для анализа силикатов, содержащих Р, Ti и Zr. Пробы вскрывают, используя в качестве плавней триоксид бора и карбонат лития; мешающее влияние катионов и анионов устраняют с помощью ЭДТА и ионного обмена Осадок сушат при 140 °C. Коэффициент пересчета массы осадка на содержание SiO2 равен 0,02510.
Хинолин впервые был использован для осаждения кремнемолиб-дата в работе [29]. Осаждение служило предварительной стадией титриметрического метода определения фосфата. Подробно этот метод описан ниже. Разработан гравиметрический метод определения кремния с применением хинолина [21]. Образующийся осадок сушат при 120—150 °C. В работе [26] найдены условия образования а- и p-форм кремиемолибденовой кислоты, а также условия полного осаждения (минимальной растворимости) молибдосили-ката хинолиния. Ранее условия образования а- и [3-форм кремнемолибденовой кислоты были описаны в работе [30]. Осадок, имеющий состав (C9H7N)4H![SiO4-12МоОз], легко отфильтровывается и может быть высушен при 150 °C в течение 1 ч.
Гравиметрический метод определения силиката в виде молиб-досиликата хинолиния был применен после предварительного отделения фосфата в виде молибдофосфата аммония [5]. Метод, пригодный для определения 0,2—2 мг каждого аниона, использован в элементном органическом микроанализе. При определении миллиграммовых содержаний кремнийорганических соединений пробу сплавляют с пероксидом натрия в никелевой бомбе и образующийся силикат определяют гравиметрически в виде молибдосиликата хи-иолиния [31]. Для наиболее эффективного гравиметрического определения силиката применяют хинолин и 8-гидроксихинолин.
Силикат можно также определять гравиметрически в виде K2SiF6 [32]. Этот метод применяют при наличии в пробе фторидов.
Титриметрические методы
Метод кислотно-основного титрования молибдосиликата хинолиния
Метод алкалиметрического титрования молибдосиликата хнно-днния, предложенный в 1949 г. [29], является одним из лучши* для определения микро- и макросодержаний силиката. Ранее [25J
при разработке гравиметрического метода определения силиката с помощью 8-гидроксихииолина была сделана неудачная попытка растворить осадок в избытке стандартного раствора NaOH и оттитровать избыток щелочи кислотой. Этот метод применим, если в качестве основания использовать хинолин. При этом протекаю! реакции
Na2SiO3 + Н2О + 2НС1 —* H4SiO4-f-2NaCl
H4SiO4+ 12 (NH4)2 МоО4 + 24НС1 —> Н4 [SiO4 • 12МоО3] + 24NH4C1 + 12Н2О 4C9H7NHCI + Н4 [SiO4 • 12МоО3] —> (C9H7N)4 Н4 [SiO4 • 12МоО3] + 4НС1
(C9H7N)4H4 [SiO4 • 12МоОзЦ-24МаОН —> 4C9H7N + SiO2+14H7O+ 12Na2MoO4
Метод экспрессен, позволяет определять до 60 мг силиката и дает более точные результаты, чем классический метод, основанный на дегидратации кремнекнслоты. Метод применен для анализа растворимых силикатов и кремнийорганических соединений после сплав ления их с КОН [33].
Титриметрическое определение силиката в виде молибдосиликата хинолиния [29]
Реагенты
Хинолин, 20%-ный раствор, очищенный дистилляцией (отбирают фракцию 230—240°C). К 800 мл горячей воды, содержащей 25 мл концентрированной НС1, при непрерывном перемешивании добавляют 20 мл хинолина. Раствор охлаждают, добавляют бумажную массу и снова интенсивно перемешивают После осаждения хлопьев бумаги раствор фильтруют через бумажную массу и доводят объем раствора до 1 л.
Молибдат аммония, 10%-ный раствор, фильтруют через" 24 ч после приготовления и используют в течение 1 недели.
Тимоловый синий, индикатор. Растворяют 0,4 г тимолового синего в 200 мл этанола, приливают 8,6 мл 0,1 М раствора NaOH и разбавляют водой до 1 л.
Раствор индикаторов крезолового красного и тимолового синего. Встряхивают 0,1 г крезолового красного с 5,3 мл 0,1 М раствора NaOH до растворения, разбавляют водой до 100 мл. Растворяют 0,1 г тимолового синего в 20 мл этанола, добавляют 2,1 мл 0,1 М раствора NaOH и разбавляют водой до 100 мл. Смешивают оба раствора.
Уксусная кислота. Разбавляют 33 мл ледяной уксусной кислоты до 100 мл водой.
Соляная кислота, (1 : 1), (1 :9) и 0,5 М растворы.
Гидроксид натрия, 1 М раствор, ие содержащий карбонатов.
Ход анализа. К раствору, содержащему не более 76 мг SiO , , в сосуде, закрытом пробкой, добавляют 8 капель индикатора тимолового синего, а затем по каплям концентрированную HCI до появления красной окраски. Добавляют о мл разбавленной НС1 (1:9), 5 мл разбавленной уксусной кислоты и затем мл 10%-ного раствора молибдата аммония при непрерывном перемешивании, которое продолжают еще в течение 1 мин после добавления реактива Погру-*10твс°суД « кипяЩую водяную баию на 10—12 мин для достижения темпера-УРЫ °” 90 “С. Затем приливают 40 мл НО (1:1) и немедленно осаждают приМНИЙ’ пРи^авляя из бюретки с большой скоростью 65 мл раствора хинолина гвева,еПРеРЬ1ВН0М пеРемешива(1ии. Сосуд снова помещают в водяную баню, на-РаствгГ РаствоР До 80—90 °C и выдерживают еще 5 мин при перемешивании осесть РЖЫСГР° охлажДают До температуры 15 °C (или ниже) и дают осадку Щуюся в Идкость иад осадком декантируют через бумажную пульпу, содержа-осадок В0Р°нке Диаметром 75 см, и отсасывают жидкость, затем промывают
Д ажды декантацией, приливая по 25—30 мл холодной воды. Переносят
7 Зак. 167
193
осадок на фильтр, смывая его шестью порциями по 30 мл холодной воды (15 °C) п отсасывая воду после каждой промывки.
Тщательно переносят осадок с бумажной массой в сосуд, в котором проводили осаждение, и растворяют в 30,00 мл 1 Л1 раствора NaOH, приливая щелочь по стенке сосуда, чтобы смыть прилипшие к стеклу частицы осадка. Наконец, тщательно промывают вороику минимальным объемом воды. Закрывают сосуд пробкой и встряхивают до полного растворения осадка. Добавляют несколько капель смеси индикаторов крезолового красного и тимолового синего и титруют избыток щелочи 0,5 Л1 раствором НС1 до желтой окраски.
Проводят контрольный опыт (обычно 0,6 мл). Учитывают, что 1 мл 1 Л/ раствора NaOH соответствует 3,182 мг SiO^' или 2,513 мг SiO2
Примечание. В оригинальных работах не проведено систематического изучения влияния посторонних ионов, за исключением фосфатов. В разделе «Ортофосфаты» содержится дополнительная информация о мешающих ионах.
Метод кислотно-основного титрования после осаждения фторсиликата калия
Титриметрический метод, основанный на осаждении фторсиликата калия, часто считают весьма эффективным, сравнимым с классическими гравиметрическими и титриметрическими методами. Этот метод применен Кордоном [34] для анализа металлургических объектов. Ранее Тананаев и Бабко [35] использовали его для анализа стекол. Траверс [36] также одним из первых использовал этот метод.
Осаждение ведут путем добавления избытка КС1 к силЬнокис-лому раствору силиката, содержащему HF:
H2SiOa + 6HF + 2КС1 = K2SiF6 + ЗН2О + 2НС1
Избыток КС1 снижает растворимость K2SiF6 до допустимого уровня. Осадок промывают раствором КС1, переносят в кипящую воду и образовавшуюся HF титруют стандартным раствором NaOH с индикатором фенолфталеином или смесью бромтимоловый синий — феноловый красный. При этом протекает реакция
K2SiFe + 4NaOH = S1O2 + 4NaF + 2KF + 2H2O
Метод значительно экспресснее, чем классический, длительность которого составляет 2 ч. Поскольку метод применяют в анализе сталей, сплавов и других металлургических объектов, важно знать влияние ионов металлов. Влияние Са, Mg и Fe несущественно [37]-В легированных сталях Мп, Сг, Ni и W (до 20%), Со (до 10%). а также Mo, V и Си (до 5%) не мешают определению [38]. Титан и цирконий мешают, но влияние титана подавляют введением кальция. Сведения о влиянии алюминия противоречивы. По данным работы [37], определению силикатов (125 мг SiO2) не мешает 15—30 мг алюминия. Другие авторы указывают на возможное со-осаждение ионов, которое наблюдается, например, в присутствии титана. Для предотвращения соосаждения рекомендуют добавлять хлорид кальция [39,40]. Важно, что определению кремния не мешают фториды.
Метод детально изучен с применением математической статистики [37]. При определении 60—190 мг SiO2 стандартное отклоне-194
иие составляет 0,29 мг, а коэффициент вариации равен 0,15% (при содержании SiO2 190 мг) и 0,48% (60 мг). Статистически значимое влияние оказывает температура осаждения фторсиликата и объем промывных вод.
Метод нашел применение для анализа шлаков [41,37], цемента [42], железной руды [43, 37], сплавов [44, 45, 37], горных пород и минералов [40,37].
Титриметрическое определение силиката после осаждения
в виде KsSiFc [40]
Реагенты
Хлорид кальция, 20%-ный раствор.
Хлорид калия, растворяют 70 г КС1 в 1 л смеси этанол — вода (I : 1).
Гидроксид натрия, 0,15 М раствор.
Фенолфталеин, 1%-ный раствор.
Ход анализа. К анализируемому раствору объемом менее 50 мл с содержанием силиката, эквивалентным 40—100 мг SiO2, добавляют по 10 мл концентрированных кислот НС1 и HNO3. Кипятят раствор 1 мин, охлаждают до 30—40 °C и переносят раствор в полиэтиленовый сосуд вместимостью 200 мл, ополаскивая склянку минимальным объемом дистиллированной воды. Добавляют около 5 мл 20%-ного раствора СаС12 и 1 г NaF. Перемешивают раствор магнитной мешалкой до растворения NaF, затем добавляют твердый КС1 для получения насыщенного раствора и еще 2—3 г (всего около 25 г). Раствор перемешивают еще 2— 3 мии.
Отфильтровывают осадок на воронке Бюхнера, используя фильтровальную бумагу № 42, и промывают его 6—10 раз раствором КС1 порциями по 10 мл для отмывки от избытка кислоты.
Переносят промытый осадок с фильтровальной бумагой и мешалкой в сосуд вместимостью 1 л, содержащий 500 мл прокипяченной горячей воды. Промывают воронку Бюхнера горячей дистиллированной водой, чтобы смыть прилипшие частицы осадка, и промывные воды собирают. Титруют раствор 0,15 М раствором NaOH с индикатором фенолфталеином. Контрольный опыт проводят каждый раз при смене растворов реактивов. Учитывают, что 1 мл 0,15 М NaOH соответствует 2,252 мг SiO2.
Примечание. При анализе горных пород и минералов пробу предварительно сплавляют с Na2O2 в никелевом тигле. Подробности метода даны в работе [40].
Комплексометрический метод
Предложен метод осаждения кремневой кислоты или щелочных силикатов в виде CoSiO3 [46] путем добавления нитрата кобальта к щелочному раствору силиката, содержащему ацетон. Осадок центрифугируют, промывают, растворяют в аммиачном растворе ЭДТА, оттнтровывают избыток ЭДТА раствором магния(II) и рассчитывают содержание кобальта.
Возможно определение Si методом, основанным на образовании кремнемолнбденовой кислоты [47], которую экстрагируют изоами-ловым спиртом и реэкстрагнруют в водную фазу. Затем восстанав-™ают м°либден солянокислым гидразином,* вводят отмеренный цинЫТптСТандаРтного РастВ0Ра ЭДТА и титруют ЭДТА раствором Si с»а * ' Прн анализе искусственных проб, содержащих 40—60% (bon Ши*ка определения не выше 0,6%. Определению мешают фос-’ Мышьяк и германий.
7*
195
Спектроскопические методы
Спектрофотометрические методы
Спектрофотометрические методы основаны на образовании гетерополикислот. Их можно разделить на группы в соответствии с природой комплексов, поглощение которых измеряют: I) кремнемолибденовая кислота (КМК) H4SiMoi204o, 2) продукт восстановления КМК синего цвета; 3) другие комплексы молибдена, образующиеся после разрушения КМК; 4) ионные ассоциаты КМК, например, с родамином В.
В спектрофотометрических методах используют важное обстоятельство— существование а- и p-изомеров КМК [30]. При низкой кислотности устойчива a-форма. При восстановлении обе формы образуют молибденовую синь. Значения е продуктов восстановления а- и p-форм различны, поэтому важно знать условия, в которых эти формы устойчивы, чтобы использовать определенную форму КМК. При восстановлении а- и p-форм КМК образуются соответственно зеленовато-синие и интенсивно-синие растворы [48]. В анализе используют обе формы КМК. Значения е для восстановительной p-формы КМК в два раза выше, чем для а-формы, однако образование p-формы происходит только в сильнокислой среде. В работах [49—51] использовано различие в значениях е и свойствах а- и p-форм КМК. Эти различия, связанные не со свойствами КМК, а с природой изопол и молибдатов, будут рассмотрены ниже.
Описанные ранее спектрофотометрические методы определения кремния детально рассмотрены в монографии Больтца [52]. Ниже описаны некоторые варианты метода.
1. После подкисления раствора силиката и добавления молибдата аммония раствор оставляют для завершения образования КМК- В работе [48] поглощение a-формы КМК измеряли при 390 нм. Можно экстрагировать КМК [9] для отделения ее от избытка молибдена. Экстракцию проводят раствором триизооктилам ина азотнокислого в 1,2-дихлорэтане в соответствии с уравнением, описывающим диссоциацию экстрагируемой частицы в органической фазе:
а-КМКв9 + 4 (RsNHNO8)s = [а-КМК (R3NH)3J* + (R3NH)* + 4 (NO3);7 + хН* где s — 1,2-дихлорэтан.
При определении гетерополикислоты в органической фазе получено значение е = 2,8-104 (при 300 нм) более высокое, чем приведенное в других работах.
Определению кремния мешает ряд ионов. Фторид образует фторсиликатный комплекс, однако его влияние можно устранить, добавив борную кислоту. Цирконий (IV), титан (IV), олово (IV) й вольфрам(IV) осаждают кремний. Эти ионы можно замаскиро* вать фторидом и ввести рассчитанное содержание борной кис* лоты, чтобы разрушить SiFu', не затрагивая комплексов Zr »
196
TiIV, SnIV и WVI. Мешает ванадий, образуя молибдованадатный комплекс, который можно экстрагировать. Некоторые анионы образуют с молибденом не очень устойчивые гетерополикислоты, например, хлориды и цитраты. Наибольшее влияние оказывают Pv, Asv, Ge'v, образуя гетерополикислоты. Влияние этих элементов в последнее время подробно изучено в связи с определением не только силиката, но и Pv, Asv и Ge,v. Взаимное влияние этих
элементов рассмотрено ниже.
В УФ-области (3-форма КМК поглощает значительно сильнее, чем в видимой (еЗОо = 2,0-104). Однако в УФ-области поглощают многие ионы. В работах [53, 54] для определения кремния использовано образование (3-КМК, поскольку (3-форма характеризуется высоким значением е. Стабилизация (3-формы достигается применением в качестве растворителя ацетона. Для повышения избирательности по отношению к фосфору (V) его влияние подавляли введением маннита, лимонной, винной или щавелевой кислоты, которые разрушают фосфориомолибденовую кислоту. Если перед добавлением молибдата в раствор ввести хлорную кислоту, КМК не образуется, а соответствующие гетерополикислоты Pv и Asv образуются.
Кинетика и термодинамика образования гетерополикнслот кремния, мышьяка, фосфора и германия рассмотрены в разделе «Ортофосфаты». Высокая устойчивость КМК в присутствии ацетона позволяет определять кремний в присутствии Pv. Наиболее эффективным реагентом для маскировки фосфатов в широком интервале концентраций является, вероятно, маннит [53, 54]. В результате предложен чувствительный метод определения менее 500 мкг SiO2 в присутствии фосфата. Поглощение измеряют при 370 нм. В случае определения Siiv, Pv, Asv и Ge,v при их со
вместном присутствии повышение селективности достигается также методом жидкостной экстракции. Примеры анализа приведены в разделе «Ортофосфаты», причем силикат определяют в водной фазе после экстракции фосфорномолибденовой кислоты [55]. Другие примеры экстракционно-спектрофотометрического определения силиката даны в табл. 12.
В работах Пунгора и соавторов [49—51] показана возможность определения элементов SiIV, Pv, Asv и GeIV при их совместном присутствии на основе образования гетерополикислот без применения операций отделения или восстановления. Несмотря на близость свойств четырех гетерополикнслот, имеющиеся различия позволяют определять их избирательно путем изменения кислотности, концентрации молибдата, длины волны, использования а-и р-форм кислот [49—51].
Кремнемолибденовая кислота, как и соответствующие гете-Р поликислоты фосфора и мышьяка, легко восстанавливаются до метпН°В’ назь1ваемых молибденовой синью (ГПС). Спектрофото-ствиЧеСКИе методы> основанные на образовании ГПС, более чув-тельны и чаще применяются, чем методы фотометрирования остановленных гетерополикнслот. Максимум поглощения ГПС
197
Таблица 12. Спектрофотометрические методы определения силикатов, основанные на селективной экстракции кремнемолибденовой кислоты
Экстракционная система Конечный этап определения Примечание Литература-
Экстрагируют Si-ГПК 2-этилгексилкетоиом при pH < 0,4 Экстрагируют Si-ГПК смесью диэтиловый эфир—пентанол (5:1) из 2 М НС1 Экстрагируют Si-ГПК смесью диэтиловый эфир — пентанол (5:1) Экстрагируют Si-ГПК изоамиловым спиртом Экстрагируют Si-ГПК хлороформом в присутствии 1,2-пропаидиола карбоната Реэкстракция в Н2О, спектрофотометрическое определение в виде ГПС при 690 нм Спектрофотометрический в УФ-области, по Movl Реэкстракция в Н2О. Переводят МоО2' в Н2МоО6 и определяют спектрофотометрически Спектрофотометрический по ГПС, е= 1,85- 10* Спектрофотометрический, по ГПС е = 2,02 • 10* Не мешает Asv, его определяют в водной фазе, интервал 10— 80 мкг Si Интервал 0,1—0,4 ppm Si Не мешает Pv, его определяют в водной фазе по Movl в УФ-области или методом ААС Интервал 0,25—2,5 ppm Si. Мешает AsOj* (> 2,5 ppm), no;, vo; (> 50 ppm), Cr2O?~ и F" Интервал 0,3—40 мкг Si. Определению 4 мкг Si мешает As (5 мг), P (0,3 мг), Ge (0,1 мкг) Интервал 0,1 —1,4 ppm Si. He мешают определенные содержания AsO,' и РО<", МоО*-, 56 57 10 11 12
no; Вг-> сг, so,2-,
ацетаты и др.
ГПК—гетерополикнслота, ГПС—синяя восстановленная форма ГПК.
лежит в области 800—820 им. Для выбора наиболее эффективного метода восстановления изучено большое число веществ [58, 59], в том числе хлорид олова(II), метол (п-метиламинофенол сернокислый), ионы железа (II), 2,4-диаминофенол, 1-амино-2-нафтол-4-сульфокислота и сульфит натрия. В работе [59] рекомендован метол. Показано [60], что смесь 1-амино-2-нафтол-4-сульфокислоты, однозамещенного сульфита натрия и едкого натра весьма устойчива (по крайней мере, в течение 1 месяца). Кроме того, при использовании этой смеси образуется устойчивая ГПС, что способствует воспроизводимости анализа.
Экстракция невосстановленной кремнемолибденовой кислоты позволяет повысить чувствительность определения. Извлеченную ГПК можно реэкстрагировать в водную фазу и затем восстано
198
вить до ГПС. Примеры определения даны в табл. 12. Метод экстракции применяют при определении силиката в присутствии арсената и фосфата. Восстановленная кремнемолибденовая кислота полностью не экстрагируется. Показано [11], что ГПС извлекается из сильнокислых растворов. В дополнение к данным табл. 12, определению кремния не мешают Sb и Те (5 мг), РЬ и Ti (Ю мг), Sn, Се, V, Bi, Nb, Та, Са, Hg и Ag (25 мг), W (50мг), Сг (75 мг), А1, Мп, Mg, Zn, Си, Со, Ni, Cd и Be (125 мг). Вольфрам, который при подкислении осаждается в виде WO3, можно растворить добавляя HF. Метод применен для анализа высоко-чистых металлов.
Многочисленные методики определения силиката по образованию ГПС отличаются разными условиями анализа: значениями pH, составом и концентрацией раствора молибдата, природой и количеством комплексующего реагента, вводимого для маскировки избытка молибдата, восстановителем и длиной волны при измерении поглощения. Используют также разные методы подготовки пробы, предшествующие фотометрированию, универсальный фотометрический метод [60], основанный на образовании ГПС и включающий три метода предварительной подготовки пробы, пригодный для анализа разных объектов, содержащих кремний. Методика спектрофотометрического определения и влияние посторонних ионов приведена ниже.
Спектрофотометрическое определение микрограммовых содержаний силиката (5—100 мкг Si) по молибденовой сини [60]
Реагенты
Молибдат аммония. Растворяют в воде 50 г гептамолибдата аммония тетрагидрата, фильтруют раствор через мембранный фильтр (0,45 мкм) и разбавляют водой до 500 мл.
Комплексующий реагент, смесь щавелевой и винной кислот с концентрацией 0,75 М каждой кислоты.
Восстановитель. Растворяют 27 г NaHSOs, 2 г NaOH 0,50 г 1-амино-2-иа-фтол-4-сульфокислоты в 225 мл воды, фильтруют и разбавляют до 250 мл водой
Ход анализа. Аликвотную часть анализируемого раствора 75 мл, содержащую 5—100 мкг кремния, помещают в полиэтиленовый сосуд вместимостью 100 мл. С помощью 3 М H2SO4 или 6 М HNO3 устанавливают pH =1,1 ±0,1, добавляют 10 мл раствора молибдата аммония. Устанавливают pH = 1,3 ±0,1 с помощью 3 Al H2SO4 или 6 М NH4OH и оставляют раствор на 10 мин. Добавляют 10 мл комплексующего реагента, перемешивают и через 30 с (ие позднее, чем через 1 мии после добавления комплексующего реагента) приливают 2 мл восстановителя. Оставляют раствор на 20 мин для развития окраски и разбйвляют его до 100 мл водой в мерной колбе. Измеряют поглощение при 815 нм (инфракрасный источник и детектор излучения) или при 705 им (видимый источник и детектор) в кюветах с толщиной слоя 1 или 5 см.
Примечания. 1. Комплексующий реагент вводят для маскировки избытка молибдата и разрушения посторонних комплексов молибдена. При этом происходит также медленное разрушение кремнемолибденовой кислоты, поэтому нтервал времени между введением комплексующего реагента и восстановителя Должен составлять 30—60 с.
та 2- В растворе должны отсутствовать фторид, олово(П), цитрат, оксалат и Опо ЭТ За исключением оксалата и тартрата, которые вводят в ходе анализа), ределеиию кремния не мешают следующие ионы (указан допустимый мольный
199
избыток): Al (950), Со, Сг'п, CrVI, Fe*, лантаноиды**, Мп, Ni п Zn (185). Си (20) (в присутствии Си анализ проводят по другой методике [60], ионы Си2+ поглощают в области 810 нм). В npucjTCTBnii щелочноземельных металлов вместо H2SO4 применяют HNO3. Определению не мешают до 10 ммоль С1-,С1О", NO" и SO*", а также 5-Ю-3 ммоль РО*" в аликвотной части раствора.
3. Кроме приведенной методики в работе [60] даны еще 4 прописи.
Другие методики применяют в зависимости от объекта анализа. Например, одна из методик [60] предназначена для определения суммарного содержания кремния в растворах солей металлов, содержащих фторид.
3. Косвенный ИК-спектрометрический метод определения силиката основан на измерении поглощения хлороформного экстракта зеленого комплекса, образованного при взаимодействии молибдена с 2-амино-4-хлорбензотиолом солянокислым [61]. Сначала экстрагируют кремнемолибденовую кислоту, затем реэкстраги-руют молибден разбавленным раствором аммиака и добавляют 2-амино-4-хлорбензотиол солянокислый. Образующийся комплекс экстрагируют хлороформом и измеряют его поглощение при 715 нм. Предел обнаружения кремния в водном растворе 0,034 мкг/мл. Определению 4 мкг кремния в 50 мл раствора не мешают следующие ионы (в ppm) СГ2О7’, Fe +, УОз(1); F" (5); РО4’, AsOj’(lO); AsOf’, Pb2+(25); Al3*, Zn2+(50). Определению мешает нитрит.
4. Описан спектрофотометрический метод определения кремния, основанный на образовании ионного ассоциата кремнемолибденовой кислоты с родамином В [62, 63]. Интервал определяемых содержаний кремния 5—50 нг/мл. Поглощение измеряют при 590 или 550 нм. Определению кремния не мешает 100-кратный мольный избыток мышьяка и фосфора. Соотношение родамина В и кремния в составе комплекса в водной и органической фазах сохраняется и равно 4 1. Значение е при 550 нм составляет5-105.
Атомно-абсорбционный метод
Использование пламени N2O — ацетилен в ААС позволяет увеличить число определяемых элементов, в том числе определять кремний. В опубликованных за последнее десятилетие работах, посвященных прямому определению кремния методом ААС, рассмотрены две проблемы: методы разложения проб и взаимное влияние элементов. Методы вскрытия силикатов, предшествующие анализу угольной пыли и силикатных пород, обсуждены в работе [64]. В пламени N2O— ацетилен по резонансной линии 251,6 нм можно определять кремний с чувствительностью 3 мкг/мл, оптимальный интервал определяемых содержаний Si 100—1000 мкг/мл.
* При наличии железа в растворе определение проводят по другой методике [60].
** Оксалаты лантаноидов нерастворимы, поэтому осадок нужно отфильтровать или отцентрифугировать.
200
Определение проводят в растворах НС1 или HF—Н3ВО3, однако чувствительность определения существенно от среды не зависит.
Важной проблемой является взаимное влияние элементов [65—67]. В детально проведенном исследовании [65] показано, что Al, Са, Fe, Na и V увеличивают, а РОГ подавляет сигнал кремния. Мешающее влияние можно стабилизировать введением определенной концентрации катионов. Показано, что иногда существенное влияние оказывает среда, например, серная кислота подавляет сигнал кремния. В работе [67] сделана попытка установить, какие ионы при содержании 10 мкг/мл мешают определению 50 мкг/мл кремния методом ААС. Показано, что с ростом концентрации щелочных металлов сигнал кремния непрерывно увеличивается, не достигая насыщения Поэтому следует поддерживать постоянную концентрацию солей щелочных металлов в растворе. Большинство элементов, за исключением никеля, увеличивает сигнал поглощения кремнием; кислоты подавляют сигнал. Влияние меди и цинка невелико.
Наиболее чувствительная методика основана на определении молибдена в кремнемолибденовой кислоте, что в большой мере связано с благоприятным соотношением Мо: Si в гетерополи кислоте, равном 12 : 1.
Селективная экстракция кремнемолибденовой кислоты (или соответствующих кислот фосфора и мышьяка) позволяет определять силикат в присутствии фосфата и арсената. Кремнемолибденовую кислоту можно экстрагировать смесью диэтиловый эфир — пентанол [7]. Затем отмывают экстракт от избытка молибдена, реэкстрагируют молибден в водную фазу и определяют содержание молибдена Метод позволяет определять 0,1 —1,2 ppm кремния. В работе [68] предложено непосредственное определение молибдена в бутанольном экстракте р-кремнемолибденовой кислоты. Интервал определяемых содержаний кремния 0,08—1,2 ppm. Определению не мешают фосфаты, но мышьяк(V) и германий (IV) мешают, завышая результаты анализа. Определению силиката не мешают 100-кратный избыток ионов Al»1, Au111, Bi111, Са11, Cd», Со», Сг»1, Си», Fe»1, Ni», Pb", Mg», Мп", Se'v, Te'v, Zn", Vv, WVI, F’, ЭДТА, NO3 и SO!'.
Предложен метод определения кремния в присутствии мышьяка и фосфора [69], основанный на экстракции силиката (1 — Ю мкг) изобутилметилкетоном с последующим измерением методом ААС. Определению мешает германий.
Интересное применение нашел метод ААС в тптриметрии [70, '![. Раствор, содержащий анион, титруют ионом металла (например Mg"), непрерывно измеряя сигнал поглощения металла. " конечной точке сигнал Mg2+ резко увеличивается. Этот метод, позволяющий определять более 1 мкг/мл кремнезема, применен я одновременного определения силиката, фосфата и сульфата В°ДЫ- Содержание анионов определяют, решая систему
201
Атомно-флуоресцентный метод
Для определения кремния применен метод АФС [72]. В качестве источника возбуждения использована безэлектродная разрядная трубка.
Электрохимические методы
Описано небольшое число электроаналитических методов определения силиката. Силикат можно титровать [73] потенциометрически, с титановым индикаторным электродом, 1 М раствором KF. В другом методе а- и p-формы кремнемолибденовой кислоты совместно титруются потенциометрически раствором оксалата олова (П) или 4-амино-3-гидроксинафталин-1 -сульфокислоты. Во всех случаях объем титранта пропорционален концентрации силиката, однако взаимодействие с 4-амино-З-гидроксинафталин-1-сульфокислотой не связано с целочисленным стехиометрическим коэффициентом [74].
Кинетические методы в
Предложен кинетический метод определения силиката, основанный на измерении скорости образования кремнемолибденовой кислоты (КМК), которая для Р-изомера равна [75]:
d [fl-КМК] [Si (ОН)4] [НМо2]6 dt ki [Н+]7 + k2 [НМо2]5
где НМо2 — димер молибдата.
При низкой кислотности k-i [H+]7<c[HMo2]j, так что кинетическое уравнение упрощается:
= л, [si (ОН)4) [НМо2+]
Методика определения кремния описана в работе [76]. Образование КМК в ходе реакции определяют по оптической плотности раствора. При постоянной исходной концентрации молибдата начальная скорость изменения оптической плотности пропорциональна концентрации силиката, что используют для построения градуировочного графика. Метод обладает высокой чувствительностью и точностью. Поглощение измеряют в определенном интервале времени, когда спонтанное превращение Р формы в a-форму КМК не происходит. Длительность определения 30— 60 с.
Определению 3,36 ppm кремния не мешают следующие ионы (ppm): POjdO), AsOl (25), SO? (2000), Cl (3000), F (50), C2H3O? NOJ, CIO? (>4000).
202
Ионы металлов не препятствуют определению кремния при содержании выше 3000 ppm, за исключением ионов железа (III), которые мешают (> 50 ppm).
Разработан дифференциальный кинетический метод [77] определения 1 — 10 ppm силиката и фосфата (<10 ppm) в смеси с ошибкой менее 3%, основанный на том, что р-кремнемолибде-новая кислота (КМК) образуется значительно медленнее, чем фосфорномолпбденовая кислота. Скорость образования КМК измеряют после завершения реакции фосфата с молибдатом. Длительность определения на автоматическом анализаторе не превышает 5 мин.
Другие методы определения
Описан термометрический метод, основанный на взаимодействии силиката со смесью HF и карбамида [78]. Измеряют рост температуры вследствие образования H2SiF6 и K2SiF6, а также кристаллизации осадка KzSiF6. Метод применен для анализа силикатов натрия и других металлов.
ЛИТЕРАТУРА
I. Shell И. R.— In: Treatise on Analytical Chemistry (Ed. I. M. Kolthoff
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
and
and
P. J. Elving), Part II, Vol. 2, New York, Interscience, 1962, p. 107.
Thomas H. — In: Cemprehensive Analytical Chemistry (Ed. C. L.
D. W. Wilson), Vol. 1C, Amsterdam, Elsevier, 1962, p. 149.
Bennett H. — Analyst, 1977, v. 102, p. 153.
Dolezal J., Povondra P., Sulcek l. — Decomposition Techniques in Inorganic Analysis, London, Iliffe, 1966.
MacDonald A. M. G., Van Der Voort F. H. — Analyst, 1968, v. 93, p. 65.
Paul 1. — Analytica chim. Acta, 1966, v. 35, p 200.
Ramakrishna T. V., Robinson J. W., West P. W. — Analytica chim. Acta, v. 45, p. 43.
Kollar R., Plichon V., Saulnier J. — Analytica chim. Acta, 1970, v. 50, p.
Trudell L. A„ Boltz D. F—Analyt. Lett., 1970, v. 3, p. 465.
Hurford T. R., Boltz D. F. — Analyt. Chem., 1968, v. 40, p. 379.
Kakita Y„ Goto H. — Taianta, 1967, v. 14, p. 543.
Trudell L. A., Boltz D. F. — Analytica chim. Acta, 1970, v. 52, p.
Prison M. E. Q., Fragala R. 1. — Analytica chim. Acta, 1970, v. 52, p. 553.
F. F. H., Gotz /., Jamieson W. D_, Masson C. — J. Chromat., 1970, v. 48, p. 515.
Sussman S., Portnoy I. L. — Analyt. Chem., 1952, v. 24, p. 1644.
то.м°дРУк А. А., Палей П. H., Безрогова E. B. — ЖАХ, 1970, t. 25, c. 319.
p 2662^ —Z’ analyt. Chem., 1959, v. 171, p. 81; Analyt. Abstr., 1960, v. 7, 18. Geyer R.t Muecke H. — Z. Chemie Lpz., 1968, v. 8, p. 388; Analyt. Abstr., 1970, . v. 18, p. 1567.
Kolthoff J. m„ Sandell E. В, Meehan E. J., Bruckenstein S. — Quantitative 20 Analysis, 4th edn., Chapter 31, London, MacMillan, 1969.
1966 р'аГ^2Г—History of Analytical Chemistry. London, Pergamon Press, 22 £ £• palmers R. A. — Analyst, 1953. v. 78. p. 24
23. Bra%n s ! ~ Zav Lab., >936. v. 5, p 162
Analvt п hlattraw H. C., Maxwell G. E., Darrow A., Needharm M. F.— alyt- Chem., 1948, v. 20, p. 504.
1969,
457.
343.
21.
203
24. Ohlweiler О. A., Meditsch J. O., Silveira C. L. P.. Silva S. — Analytica chim Acta, 1972, v. 61, p. 57.
25 Brabson J. A., Duncan R. D., Murphy 1. J. — Analyt. Chem., 1963, v. 35 p. 1102.
26. Armand Al., Berlhoux /. — Analytica chim Acta, 1953, v. 8, p. 510.
27. Harzdorf C. — Z. analyt. Chem., 1967, v. 227, p. 96.
28. Merz J. A. — Svensk. kern. Tidskr., 1941, v. 53. p. 374.
29. Wilson H. N. — Analyst, 1949, v. 74, p. 243
30. Strickland J. D. H. — J. Am. chem. Soc., 1952, v. 74, p. 862.
31. Christopher A. J., Fennell T. R. F. W.— Talanta, 1965, v. 12, p. 1003
32. Hollander M., Rieman W.—Ceram. Age, 1947, v. 50, p. 168.
33. Климова В. А., Виталина M. Д. — Труды комиссии по аналнт. химии АН СССР, 1963, т. 13. с. 7; Analyt. Abstr., 1964. v. 11, р. 5539.
34. Kordon F. — Arch. Eisenhiitt Wes., 1945, v. 18, p. 139.
35. Тананаев H. А., Бабко А. К- — ЖАХ, 1930, r. 82, c. 145.
36. Travers A. — Compt. Rend., 1921, v. 173, p 714.
37. Morris A. G. G. — Analyst, 1965, v. 90, p. 325.
38. Ref. 2, p. 155.
39. Louvrier J., Voinovitch I. A.— Ind. Ceram., 1959, p. 243; Analyt. Abstr., 1960, v. 7, p. 2661.
40. McLaughlin R. J. W., Biskupski V. S. — Analytica chim. Acta, 1965, v. 32. p. 165.
41. Bieber B., Vecera Z. — Huth Listy, 1960, v. 15, p. 397; Analyt. Abstr., 1961. v. 8, p. 70.
42. Calleja J., Bade B. — Rev. Cienc. Apl„ 1961, v. 15, p. 39; Analyt. Abstr., 1962. v. 9, p. 161.
43. Glaso O., Patzauer G. — Analytica chim. Acta, 1961, v. 25, p. 189.
44. Velken S. — J. Iron. Steel Inst., 1958, v. 119
45. Wagner F. — Z. analyt. Chem., 1960, v. 178, p. 34.
46. Jenik J. — Chem. Prumysl., 1961, v. 11, p. 189; Analyt. Abstr., 1961, v. 8, p. 4548.
47. Васильева M. Г., Бешикдашян M. T. — ЖАХ, 1970, t. 25, c. 544; Analyt Abstr., 1971, v. 21, p. 999.
48 Ringbom A., Ahlers P. E., Siitonen S. — Analytica chim. Acta, 1959, v. 20, p. 78.
49. Halasz A, Pungor E. — Talanta, 1971, v. 18, p. 557.
50. Halasz A., Pungor E. — Talanta, 1971, v. 18, p. 569.
51. Halasz A., Pungor E., Polyak K.— Talanta, 1971, v. 18, p. 577.
52. Potter G. V. — In: Colorimetric Determination of Non-Metals (Ed. D. F. Boltz), New York, Interscience, 1958.
53. Chalmers R. A., Sinclair A. G. — Analytica chim Acta, 1965, v. 33, p. 384
54. Chalmers R. A., Sinclair A. G. — Analytica chim. Acta, 1966, v. 34, p. 412.
55. Paul J. — Mikrochim. Acta, 1965, p 836.
56. Paul J. — Analytica chim. Acta, 1966, v. 35, p. 200.
57. Hurjord T. R., Boltz D. F. — Analyt. Chem., 1968, v. 40, p. 379.
58. Jean M. — Chim. Anal., 1956, v. 38, p. 37.
59. Mullin J. B., Riley J. P. — Analytica chim. Acta, 1955, v. 12, p. 162.
60. Duce F. A., Yamamura S. S. — Talanta. 1970, v. 17, p. 143
61. Trudell L. A., Boltz D. F. —Talanta, 1972, v. 19, p. 37.
62. Golkowska A. — Chemia analit., 1970, v. 15, p. 59.
63. Golkowska A., Pszonicki L. — Talanta, 1973, v. 20, p. 749.
64. Boar P. L., Ingram L. K. — Analyst, 1970, v. 95, p 124.
65. Price W. J., Roos J. T. H. — Analyst, 1968, v. 93, p. 709.
66. Guest R. J., Macpherson D. K. — Analytica chim. Acta, 1974, v 71, p. 233.
67. Musil J., Nehasilova M. — Talanta, 1976, v. 23, p. 729.
68. Kirkbright G. F., Smith A. M., West T. S. — Analyst, 1967, v. 92, p. 411-
69. Ramakrishna R. V., Robinson J. W., West P. W. — Analytica chim. Acta, 1969 v. 45, p. 43.
70. Looyenga R. W., Huber С. O.—Analyt. Chem., 1971, v. 43, p. 498.
71. Liu C. /., Huber С. O. — Analyt. Chem., 1972, v. 44, p. 2200.
72. Kirkbright G. F., Rao A. P., West T. S. — Analyt. Lett., 1969, v. 2, p. 465.
9П4
73. Ackerman G. — Talanta, 1971, v. 17, p. 693, 701.
74 Дорохова E. H Опарина .7 И —ЖАХ. 1970, т. 24, с 544
75 Hargis L. G. — Analyt Chem . 1970. v 42, p 1497
76 Hargis L G.— Analytica chim. Acta, 1970, v. 52. p 1
77 Ingle I D Crouch S. R — Analyt Chem 1971. v 43, p 7.
78 Strauss H, Rutkowski R — Plaste Kautsch, 1972, v. 19, p 665; Analyt. Abstr., 1973, v 24. p. 2760.
ТАРТРАТЫ
МЕТОДЫ ОТДЕЛЕНИЯ
Винную кислоту можно отделить от других карбоновых кислот методом ТСХ на целлюлозе [1] (см. разд. «Цитраты»), Для хроматографического отделения 2—30 мкг тартрата использована бумага, пропитанная гидроксидом никеля [2]. Пятно тартрата обнаруживали, обрабатывая хроматограмму дитиооксамидом, вырезали, взвешивали и сравнивали со шкалой стандартов.
Разработаны ионообменные методы отделения тартрата. В работе [3] описано разделение винной, лимонной и малеиновой кислот на Амберлите CG-120 (размер зерен 0,045—0,074 мм, Н+-форма) с помощью элюента ацетон — дихлорметан — вода (160: 100; 9). Метод применен для выделения тартрата из вин [4].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Описаны методы кислотно-основного, редокс- и иодиметриче-ского титрования тартрата. Старый иодиметрический метод, вероятно, широко не применяют.
Метод кислотно-основного титрования
Для алкалиметрического титрования винной кислоты (а также щавелевой и уксусной) в качестве хемилюминесцентного индикатора рекомендована смесь флуоресцеина и люцигенина (ди-метилдиакридиния динитрат) [5]. В щелочном растворе в присутствии пероксида водорода наблюдается зеленая люминесценция люцигенина. Хемилюминесценция заметно увеличивается в присутствии флуоресцеина. Раствор начинают титровать при темпера-тУРе около 60 °C.
Старый метод количественного определения винной кислоты кал°ВаН На осажДении кислого тартрата калия избытком ацетата этан” И Гсусной кислоты в присутствии высоких концентраций ный°Ла Затем осадок растворяют, приливая титрант — стандарт-0азп1.?аСТВор Щелочи (6]. Метод часто применяют для анализа
Р хлителя для выпечки хлеба [7].
205
Редокс-методы
Для титрования тартрата рекомендован ряд окислителей, в том числе манганат и перманганат калия, сульфат церия(IV), гексанитроцеррат(1У) аммония и тетраацетат свинца(IV).
Перед титрованием тартрата щелочным раствором КгМпО4 рекомендуется предварительно обработать пробу раствором КМпО4 в слабокислой среде [8]. Описан метод определения 6 мг винной кислоты путем обработки ее избытком КМпО4 и титрования избытка окислителя гидрохиноном в присутствии дифениламина как индикатора. Для окисления тартрата при комнатной температуре необходимо около 4 ч или 1 ч при 40 °C. В качестве окислителя использовали также тетраацетат свинца [40], избыток которого через 30 мин титруют потенциометрически гидрохиноном.
Церий часто применяют в качестве окислителя органических кислот, в том числе винной. Изучена возможность выбора условий, в которых наблюдается точная стехиометрия при окислении винной кислоты церием (IV). Показано, что (NH4)2Ce(NO3)6 окисляет тартрат в среде азотной кислоты в широком интервале температур и времени реакции, при этом на 1 моль винной кислоты расходуется 6 моль церия(IV). Продуктами реакции является СО2 и вода. Метод, однако, неспецифичен, при введении избытка окислителя определению мешает хлорид.
Определение винной кислоты с помощью КМпО4 [9]
Реагенты
Перманганат калия, 0,02 Л1 раствор.
Буферный раствор (ацетатный), pH = 4,0.
Сульфат марганца, 0,5 М раствор.
Серная кислота, 2 М раствор.
Пирофосфат натрия двузамещенный NasI^PjO?, насыщенный раствор.
Гидрохинон, 0,05 Л1 раствор. Очищенный сублимацией гидрохинон (,5,50 г) растворяют в 500 мл теплой воды и добавляют НС1 до концентрации 1%. Раствор охлаждают и разбавляют водой до 1 л.
Дифениламин, 3%-ный раствор в 96%-ной H2SO4.
Ход анализа. К раствору, содержащему около 6 мг винной кислоты, добавляют 1 мл 2 М ацетатного буферного раствора и 5 мл 0,02 Л1 раствора КМпО4 и оставляют на 4 ч при комнатной температуре или на 1 ч при 40 °C. Добавляют 10 мл 0,5 А1 раствора MnSO4, 4 мл 2 М H2SO4 и 30 мл насыщенного раствора Na2H2P2O7, разбавляют водой до 70—100 мл и титруют 0,05 М раствором гидрохинона до слабой розовой окраски. Добавляют 3 капли раствора дифениламина и продолжают титрование до перехода интенсивной фиолетовой окраски в слабо-желтую. Проводят контрольный опыт.
Комплексометрический метод
Известен метод, основанный на осаждении тартрата (6—18 мг) свинцом и титровании РЬИ раствором ЭДТА с индикатором пирокатехиновым фиолетовым [12]. В работе [13] предложен метод с применением ацетата свинца (IV). Винную кислоту окисляют избытком реагента в присутствии уксусной кислоты и ацетата калия. Избыток ацетата свинца (IV) гидролизуют кипячением водного раствора, образующуюся суспензию фильтруют и опреде
206
ляют содержание свинца в фильтрате косвенным комплексометрическим методом. Определению тартрата описанными методами мешает ряд веществ.
Спектрофотометрические методы
Природа реакций, лежащих в основе спектрофотометрических методов определения низких содержаний тартрата, часто неясна. Описан дифференциальный спектрофотометрический метод определения тартрата с применением хлораниловой кислоты [14], использованный в экспрессном анализе разрыхлителя для выпечки хлеба.
Предложенный в работе [20] метод определения тартрата в присутствии цитрата основан на последовательном протекании реакций
ноос—сн—сн—соон + ю; —> гноос—сно +107 + н2о (1) он он
НООС—СНО +107 —> СО2 + НСООН + ю; (2)
Образующийся иодат определяют спектрофотометрически, по образованию трииодида в присутствии иодида. Избыток перйодата маскируют молибдатом при рН = 3. Тартрат окисляется при 45— 50°С в течение 1 ч. При окислении тартрата образуются 3 моль йодата. Интервал определяемых содержаний тартрата 5—30 мкг. В присутствии цитрата определение проводят при О °C, причем окисление останавливается на стадии образования альдегида по реакции (1) и 1 моль тартрата соответствует образование 1 моль йодата. Определению 15—60 мкг тартрата не мешает до 6 мг цитрата.
Предложены спектрофотометрические методы, основанные на окислении тартрата сульфатом церия (IV) (затем измеряют содержание избыточного CeIV с помощью ферроина [15]), на ослаблении поглощения комплекса Fe11' с 5-нитросалицилатом [16], на взаимодействии винной кислоты с р-нафтолом [17]. Использована также оптическая активность тартрата [18]. Выбраны условия определения тартрата в присутствии других оптически активных веществ. Предложен метод фотометрического титрования тартрата [19].
ЛИТЕРАТУРА
1- Dittman J. — J. Chromat., 1968, v. 34, р. 407.
• svetana 7.—Trudy Vyssh Inst. Narod. Stopanst. Varna, 1961, v. 1, p. 187: 3 e f lyJ' Abstr - 1962, v. 9, p. 4754.
4 S Z Chromat., 1966. v. 22, p. 498.
7756в°виЧ £ —Пищевая техн., 1960, с. 158; Chem. Abstr., 1961, v. 55,
б' Of+i K<PiEkerin8 F- Wilson c L — Taianta, 1962, v. 9, p. 371.
mists ailthedndS1970^nalyS'S °* Assoc’ation of Official Agricultural Che-
207
7. Pearson D.—The Chemical Analysis of Foods, 6th edn., J. and A. Churchill London, 1970.
8. Polak H. L„ Pronk H. F„ Den Boef G. — Z. analyt. Chem., 1962, v. 190, p. 377.
9. Berka A., Htlgard S. — Mikrochim Acta, 1966. p. 164.
10. Berka A. — Analytica chim. Acta, 1961. v 24, p. 171.
11. Rao G. G., Subrahmanyan I., Rao В. M.— Taianta. 1972 v. 19, p. 1083.
12. Crisan I. A., Krausz R. — Studia Univ. Babes Bolyai, Ser. Chem. 1967, v. 12, p. 19, Analyt. Abstr., 1968, v 15, p. 6720.
13. Berka A. — Z. analyt. Chem., 1963, v. 195, p. 263.
14. Johnson A. R. — Analytica chim. Acta., 1965, v. 33, p. 397.
15. Ma T. S., Nazimowitz W. L. — Mikrochim Acta, 1969, p. 345.
16. Lee K- S. — Analyt. Chem., 1968, v. 40, p. 2049.
17. Christian G. D. — Taianta. 1969, v. 16, p. 255.
18. Kristen W. J., Nilsson S. K.— Analytica chim. Acta, 1962, v. 27, p. 345.
19. Pearson К. H., Kirschner S. — Analytica chim. Acta, 1969, v. 48, p. 339
20. Nisli G., Townshend A. — Taianta, 1968, v. 15, p. 1480.
ТЕЛЛУРАТЫ И ТЕЛЛУРИТЫ
Теллур применяют в металлургии в качестве легирующего компонента, например для улучшения кислотостойкост и свинца, используемого для изготовления электрохимических батарей. Теллур применяют также при изготовлении тепло- и износостойких сортов резины. Теллур получают в качестве побочного продукта при электролитическом рафинировании меди. Одновременно возникает задача определения теллура в присутствии ряда других элементов. Микросодержания теллура часто определяют в полупроводниковых монокристаллах.
По химическим свойствам теллур близок к селену. Аналитическую химию этих элементов обычно рассматривают одновременно. В водных растворах теллур присутствует в степени окисления +4 и -J-6. Соединения теллура, как и селена, можно восстановить до металла с помощью восстановителей SO2 SnCl2 и гидразина.
В разделе, посвященном селену, описаны методики разделения и определения селена и теллура. При рассмотрении методов определения теллура будут приведены ссылки на эти методики.
Растворы вторичных стандартов теллура (IV) обычно готовят из чистого металла, который растворяют в концентрированной HCI с небольшим содержанием HNO3. При растворении реакционную смесь слегка подогревают, затем нагревают до удаления коричневых паров, охлаждают и доводят до определенного объема, вводя HCI для создания конечной концентрации ее 1 М.
Растворы теллура (VI) можно приготовить из чистого теллура-та натрия, а затем установить концентрацию теллура.
Аналитическая химия теллура рассмотрена в обзорах [1—4].
МЕТОДЫ ОТДЕЛЕНИЯ
Обзор методов отделения и определения микросодержаний теллура приведен в работе (5]. Подробное обсуждение методов отделения теллура (и селена) дапо в монографии Коркиша [6]. Ниже приведены некоторые методы отделения теллура.
208
Дистилляция
Теллур в отличие от селена обладает малой летучестью при температуре концентрированных солянокислых растворов менее 100°С. При дистилляции из растворов H2SO4—НВг дистиллят содержит селен и лишь следы теллура. Операция дистилляции длительна, ее можно заменить другими, более экспрессными методами, например, экстракцией.
Ионообменный метод
Теллур (IV) в отличие от селена (IV), который не поглощается катионитами при любых концентрациях НС1, довольно хорошо сорбируется при низкой кислотности раствора. На этом основаны методы разделения селена (IV) и теллура (IV). Теллур (VI) не сорбируется при любой кислотности. Некоторые методы ионообменного отделения теллура приведены в разделе «Селенаты и селениты». На катионите можно отделить теллур от Си, Ni, Fe и Pb [7]. В этой же работе приведен метод отделения теллура от хлорид-ных комплексов платиновых металлов.
Теллур (IV) отделяют от иодидов, йодатов и иода на катионите в Sn''-форме [8], при этом теллур восстанавливается до металла и сорбируется, а соединения иода элюируются.
В отличие от селена(IV) теллур (IV) сильнее сорбируется на анионите, сорбция увеличивается с ростом концентрации HCI до 8 М. Это позволяет отделить селен от теллура. Селен (IV) элюируют раствором НС1 с концентрацией менее 4 М. Сорбированный теллур элюируют разбавленным раствором НС1 (0,1 — 1 М). Селен (IV) и теллур (IV) отделяют на анионите от тяжелых металлов [9], используя в качестве комплексующего реагента хлорид лития. При соответствующей концентрации LiCl теллур (IV) можно выделить из бинарной смеси, содержащей Bi111, Pb”. Sb”1, Au111, Fe1” или Selv. Возможно также выделение теллура из многокомпонентной смеси, содержащей Ag’, Ni” и А11П.
Описан метод отделения Telv от TeVI на анионите [10].
Жидкостная экстракция
Экстракцию широко применяют в аналитической химии теллура. Экстракции теллура посвящена обширная литература; обзор методов опубликован в 1973 г. [II]. Ниже рассмотрены некоторые наиболее важные экстракционные системы, в том числе экстракция неполярных соединений, хелатов, ионных ассоциатов и сольватированных соединений.
Теллур (VI) является сильным окислителем, способным окислить некоторые органические растворители и многие неорганиче-кие и°ны. Экстракция часто предшествует спектрофотометриче-кому определению теллура.
ед„ 1этилДитиокаРбамат образует с теллуром (IV) хелатное со-нение, экстрагируемое полярными и неполярными растворите
209
лями с высоким коэффициентом распределения. В присутствии ЭДТА или цианида при pH = 8,5—8,8 вместе с теллуром (IV) экстрагируются только Bi, Sb111 и Те111. Если определение теллура заканчивают методом ААС, то в качестве экстрагентов применяют амилацетат [14] или метилизобутилкетон [15].
При pH = 3,0 висмутол II образует с теллуром (IV) координационное соединение, которое экстрагируется неполярными растворителями. Молярный коэффициент поглощения комплекса в хлороформе равен 3,5-104 [16]. Избирательность определения увеличивается, если предварительно экстрагировать теллур (IV) из растворов НС1. Теллур (VI) остается при этом в водной фазе.
Теллур (IV) можно экстрагировать из 4—6 М НС1 в виде ионного ассоциата алифатическими аминами с длинной цепью LA-1 или Амберлит LA-2 и таким образом отделить его от селена (IV) [17]. Разделение основано на большом различии в устойчивости хлоридных комплексов селена (IV) и теллура (IV). Экстрагируемый ионный ассоциат с бромидным комплексом теллура (IV) образует также краситель виктория голубой 4R, что использовано для спектрофотометрического определения теллура (IV). Ионный ассоциат экстрагируют смесью нитробензол — хлороформ из растворов с концентрацией H2SO4 4,5 М и КВг 0,7 М. Метод чувствителен (e6os = 8 • 104), но не очень избирателен [18]. Для отделения от Zn11, Си", Pb11, Cd11 и SeIV микросодержания теллура можно экстрагировать метилизобутилкетоном из 4—7 М растворов НС1 [19].
Осаждение
Теллур (IV) и селен (IV) в кислой среде восстанавливаются с помощью SO2 до металлов, что позволяет отделить их от других элементов и определить гравиметрически. При определенной кислотности метод осаждения позволяет разделить селен и теллур. Этот метод, а также разделение путем осаждения тетрафенил-арсонийхлоридом описаны в разделе «Селенаты и селениты».
Соосаждение
Теллур (IV) и теллур (VI) полностью соосаждаются с Fe(OH)3 из аммиачных растворов с pH = 9,4—9,7. В этих условиях соосаждается только 1 % селена, содержащегося в исходном растворе [20].
Бумажная и тонкослойная хроматография
Методы хроматографического разделения теллура приведены в разделе «Селенаты и селениты». Описано разделение ТеОз , I и Юз методом ТСХ [21] на силикагеле, кизельгуре и оксиде лантана с применением смеси ацетон— 1 М раствор NH4OH (1: 1) в качестве растворителя.
?IQ
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Гравиметрический метод определения теллура путем восстановления сернистым газом в кислых растворах известен давно. В качестве восстановителей используют также SnCl2 и гидразин. Осаждение теллура (IV) обычно ведут в растворах с концентрацией НС1 1,5—5 Al, при этом селен (IV) также восстанавливается. В среде концентрированной НС1 восстанавливается только селен (IV), теллур переходит в фильтрат и может быть осажден после разбавления раствора. Для коагуляции осадка теллура раствор кипятят. Мелкодисперсные частицы теллура могут окисляться, что является источником ошибок. Чтобы избежать окисления теллура, осадок промывают для освобождения от хлоридов, затем промывают этанолом или изопропанолом и сушат при 90°C. Этот метод подробно обсужден в обзоре [4].
В работе [22] описан гравиметрический метод с применением висмутола II [22]. Теллур количественно осаждается реагентом при pH « 2. Осадок разлагается при нагревании, но может быть высушен на воздухе, весовая форма его Те(С6Н5\2С28з)4, коэффициент пересчета на теллур равен 0,1140. При определении 20 мг теллура ошибка не превышает ±0,5%. Осаждение можно вести в присутствии ЭДТА.
Титриметрические методы
Для титриметрического определения теллура применяют в основном окислительно-восстановительные реакции. Стандартный метод [23], который давно известен, состоит в окислении теллура (IV) избытком стандартного раствора КМпО4, который затем титруют стандартным раствором железа(II). Селен(IV) определяется совместно с Te'v. Для определения теллура в присутствии селена (IV) в качестве окислителя применяют бихромат [24]:
ЗН2ТеО3 + К2Сг2О7 + 8НС1 —> ЗН2ТеО4 + 2СгС13 + 2КС1 + 4Н2О
Проводя титрование двух аликвотных частей раствора посредством КМпО4 и К2СГ2О7, можно определить содержание TeIV и SeIV при их совместном присутствии. Теллур и селен можно определить также в одной аликвотной части раствора [25] (см. разд. «Селенаты и селениты»).
Описан титриметрический метод с применением перйодата [26]:
ТеОз" + ю; —> ТеО4" + IOJ
Для определения микросодержаний теллура в теллурорганических «единениях применен метод, основанный на введении избытка ИзбР°Мата и титровании избытка бихромата железом(II) [27]. чро ЫТОК xP°Ma(VI) можно определить также спектрофотометри-С(-ким методом.
211
Недостатком многих редокс-методов определения телл\ра(1\) является необходимость нагревания или длительного выстаивания растворов. Например, в работе [24] указано, что от приливания избытка бихромата до титрования необходима экспозиция 30 мин. Прл использовании церия(IV) как окислителя это время можно сократить до 3—7 мин, если в качестве катализатора ввести перхлораты серебра (I) и марганца(П) [28]. Избыток церия (IV) титруют железом (II) с индикатором ферроином. При определении 10—105 мг теллура относительное стандартное отклонение равно 0,21 %.
Для определения теллура и селена предложены иодиметриче-ские методы, которые позволяют определять селен и теллур при их совместном присутствии (см. разд. «Селенаты и селениты»). Известны методы определения теллура (VI). Описан метод восстановления теллура (VI) избытком KI [29] в цитратном буферном растворе при pH = 2—3:
Н6ТеО6 + ЗГ + ЗН+ —> ТеООН+ + 17 + 4Н2О
После нагревания раствора при 80 °C в течение 30 мин в надежно закрытом сосуде выделившимся иод титруют стандартным раствором тиосульфата Метод позволяет определять 4—80 мг теллура^!) в присутствии Te'v и SeVI, однако селен(IV) окисляет К1 и определяется вместе с TeVI. В работе [30] Tevl обрабатывают раствором арсенита натрия и KI в буферном растворе с pH =5, раствор нагревают и после остывания подщелачивают и титруют избыток As’" стандартным раствором иода. Интервал определяемых содержаний теллура 1—30 мг. Для определения Telv его предварительно окисляют пероксидом водорода до Tevi.
Описан метод прямого фотометрического титрования теллура (IV) [31] раствором бихромата с непрерывным измерением поглощения хрома (VI) в области 380—430 нм, где TeIV и TeVI не поглощают. Определению не мешают Se[V, SeVI, а также хлориды и фосфаты при их концентрации 0,05 и 0,2 М соответственно. Титрование проводят в среде HNO3 или НС1О4, где взаимодействие TeIV с CrVI протекает быстро. Метод позволяет определять 3—30 мг теллура.
Спектроскопические методы
Спектроскопические методы в видимой
и ультрафиолетовой областях
Первые колориметрические методы определения теллура основаны на его восстановлении хлоридом олова (II) или гипофосфор-ной кислотой. Поглощение можно измерять в видимой области при 420 нм или в УФ-области при 240—290 нм [32]. Аналогичным образом определяют селен. Определению мешают некоторые ионы, в том числе окислители. Fe111, Си11, Г и S2O3".
212
Теллур можно определять по острому пику поглощения иодо-теллурита при 285 нм или широкому плато в области 335 нм [33, 34]. Метод применен для определения теллура в арсениде галлия [35].
Большинство реагентов для спектрофотометрического определения теллура является серусодержащими лигандами, в том числе тио-карбамнд, тиогликолевая кислота, диэтилдитиокарбамат натрия и меркаптоуксусная кислота. Обычно окрашенный комплекс теллура экстрагируют. Применение диэтилдитиокарбамата предложено в работе [13]. Тиогликолевую кислоту применяют также для определения селена(IV). Метод не отличается высокой чувствительностью (е2бо = 3300), интервал определяемых содержаний 8—27 ppm. Метод позволяет определять миллиграммовые содержания селена и теллура. Для определения теллура в присутствии больших содержаний селена используют 5-меркапто-З-(2-нафтил)-1,3,4-тиадиазол-2-тион, производное висмутола II. Характеристика этого и других методов определения теллура приведена в табл. 13.
Таблица 13. Характеристика некоторых экстракционно-спектрофотометрических методов определения теллура (IV)
Реагент Условия Молярный коэффициент поглощения Литература
Диэтнлднтиока рба- Экстрагируют СС14. Маскирующие 3,6 • 103 13,
мат реагенты ЭДТА и KCN. Мешают только Bi111, TI111, Hg11, Ag' и Са11 Экстрагируют СНС13. Предварительная экстракция метилизобутил-кетоиом из растворов НС1 (428 нм) 36
Висмутол II 3,5- 104 (335 нм) 16, 37
Виктория голубой R Экстрагируют смесью бензола и нитробензола из 4,5—5,0 Af H2SO4. Мешают In и Hg 8,0 104 (602 им) 18
1-Пирролндинкарбо- Дитионат Экстрагируют СНС13. Мешают Cd”, Со1', Си”, Fe"1, Pb", Мп", Hg", Ni", Т1', и Zn" и некоторые другие катионы 8,12- 104 (257 нм) 38
5-Меркапто-З - (2- иа ф -тил)-1,3,4-тиади-азол)-2-тион (производное висмутола II) Экстрагируют СНС13, С6Н6 или циклогексаном. Закон Бера соблюдается для содержаний 1—50 мкг/мл Те. Не мешает 200-кратный избы- 3,0- 104 (328 нм) 39
1Л-Дифеиилтиосеми-карбазид (в присутствии бромида) ток Se Экстрагируют С6Н6. Закон Бера соблюдается для содержаний Те 10—120 мкг в 5 мл бензола. Мешают Fe1", Bi1", Nb, Zr, Ga, Crvl, Cu", Vv и равные содержания Se « 104 (480 нли 380—390 нм) 40
на
п Теллур(IV) образует окрашенные галогенидные комплексы фотомет^е^Гб В сильнокнслой среде, что используют для спектро-Рического определения теллура. Эти комплексы образуют
213
ионные ассоциаты с некоторыми красителями, например с родамином 4G или виктория голубым R, которые экстрагируют соответствующими органическими растворителями. Показано [40], что наибольшая чувствительность достигается при взаимодействии бромидного комплекса теллура (IV) с 1,4-дифенилтиосемикарбази-дом.
Сведения о некоторых других экстракционно-спектрофотометрических методах содержатся в цитированном ранее обзоре [11].
'Спектрофотометрическое определение теллура(1У)
с помощью диэтилдитиокарбамата [13]
Реагенты
Купферрон, 2 %-ный раствор.
Диэтилдитиокарбамат натрия, 0,5%-ный раствор.
Буферный раствор, pH = 8,6. Растворяют 5 г борной кислоты, 1 г ЭДТА, 1 г однозамещенного фосфата калия в 100 мл воды и устанавливают pH = 8,6.
Ход анализа. Устанавливают pH = 1—2 в анализируемом растворе, содев-жащем 10—15 мкг теллура(1У) в 20 мл, и добавляют 10 мл раствора купферрона. Экстрагируют комплексы металлов хлороформом до получения бесцветного экстракта. Хлороформ извлекает не только купферронаты посторонних металлов. но и избыток купферрона.
К водному раствору добавляют 5 мл буферного раствора и соль KCN, устанавливают pH = 8,5—8,8. Добавляют 1 мл 0,5%-ного раствора диэтилдитиокарбамата натрия и экстрагируют последовательно тремя порциями СС1«. Измеряют поглощение органической фазы при 428 нм.
Флуориметрический метод
Теллур (IV) флуоресцирует красным светом [41] в замороженных 9 М растворах НС1 при —196 °C, что использовано для спек-трофлуориметрического определения 0,02—0,64 ppm теллура. Флуоресценцию измеряют при 586 нм, при возбуждении светом с длиной волны 380 нм [42]. Анализ проводят в пробирках из прозрачного кварцевого стекла длиной 200 мм, диаметром 3 мм (толщина стенок 1 мм). Для анализа достаточно 0,5 мл раствора. Пробирки можно непосредственно погружать в жидкий азот и нагревать до комнатной температуры после измерения, не опасаясь их растрескивания.
Из 50 изученных ионов при их 50-кратном избытке относительно Te'v определению мешает только Fe1", Sn" и I-. Олово(II) можно предварительно окислить, а железо (III) экстрагировать. Описан аналогичный флуориметрический метод [43].
Атомно-абсорбционный и атомно-флуоресцентный методы
В работе [44] показано, что среди аналитических линий теллура 214,3, 225,9 и 238,6 нм наиболее чувствительна линия 214,3 нм. С помощью оптической системы с пятикратным прохождением луча достигнута чувствительность 0,23 ppm и предел обнаружения 0,076 ppm.
214
Определению теллура методом ААС чаще всего предшествует его экстракция, причем важен правильный выбор растворителя. При экстракции теллура (IV) днэтилдитиокарбаматом в качестве растворителя использован амилацетат [14]. Метод применен для определения 0,0005—0,03% теллура в сталях. Чувствительность определения составляет 1,3 ppm в водном растворе и 0,3 ppm в амилацетате.
В последние годы благодаря непрерывному совершенствованию приборов и источников света появилась возможность непосредственного определения теллура в кислых растворах, без экстракции [45].
Обозначилась тенденция в методах ААС и АФС переводить определяемый элемент в летучий гидрид перед введением в пламя. При использовании этого метода достигнут предел обнаружения теллура 0,0015 мкг/мл [46]. Прибор для генерирования гидрида показан на рис. 28. Гидрид получается при введении подкисленного раствора в разбавленный раствор боргидрида натрия. Проведенные исследования показали, что теллур(VI) полностью восстанавливается в среде НО до теллура (IV), но селен (VI) восстанавливается только наполовину.
Описан экспрессный ААС метод определения 5 нг/г теллура в силикатных породах при навеске пробы 0,25 г, основанный на применении гидридного генератора [47].
При использовании гидридного генератора в методе АФС достигнут предел обнаружения теллура 0,0001 мкг[48], однако этот метод имеет ряд ограничений [49].
Метод молекулярной эмиссии в полости
Этот метод анализа появился недавно. Принцип метода и приборное оформление описаны в разделе «Сульфиты и оксиды серы». Метод успешно применен для определения селена и теллура. Измеряя излучение теллура при 780 °C, можно определить с учетом фонового излучения 1 мкг теллура [50].
Электрохимические методы
Электроаналитических методов определения Te,v и Tevi сравнительно мало. При анализе смеси TelV и TeVI потенциометрическим методом определено суммарное содержание теллура [51]. У21У1ед~'1ение основано на описанном ранее классическом методе 12-э]. Теллур(IV) окисляют избытком бихромата, который тит-потенциометрически стандартным раствором железа (II). Тел-yP(VI) определяют амперометрически, по реакции
кипячение
--------ь ТеОз” + Н2О + С12
отгонка
TeOf + 2НС1
Хл
Раз1Р1Л,ВЬ1^ывают потоком
Уюи*иися иод титруют
азота и поглощают раствором KI. Об-амперометрически тиосульфатом.
215
В работе [52] показана возможность амперометрического титрования Telv и Se,v N-гексил- и N-цпклопентнлдитиокарбаматом. При определении 0,1—0,2 мг теллура и селена ошибка не превышает 2%. Определению мешают медь, ртуть и таллий(III). Раздельное титрование селена (IV) и теллура(IV) в смеси можно проводить при разных значениях pH. Для потенциометрического и амперометрического определений миллиграммовых содержаний селена и теллура при их совместном присутствии можно использовать аскорбиновую кислоту [53].
Теллур (IV) и селен (IV) можно титровать кулонометрическим методом, электрогенерированным гипобромитом [54].
Проведено детальное исследование полярографического восстановления теллурита [55, 56] в щелочных растворах. При pH = = 7—II теллур (IV) восстанавливается до металла. При использовании капающего ртутного электрода теллур переходит в ртуть. При pH >11 теллур(IV) восстанавливается до теллурида Те2-. Проведено полярографическое, кулонометрическое и потенциометрическое изучение теллура(IV) в щелочной среде [57].
Методом осциллополярографии можно определять 8—20 мкЛ1 теллура (IV) [58].
Другие методы определения
Чувствительность определения теллура методом нейтронноактивационного анализа составляет 5-10~9 г. Метод применен для определения теллура в арсениде галлия [59] и горных породах [60].
Теллурат можно определять поляриметрически [61]. Интервал определяемых содержаний теллура (IV) 0,15—1,5 мг/мл. Определению не мешают большие содержания TeVI, Se,v и Sevl и умеренные— Asv. Ионы Pb, Cd и Sn можно замаскировать, мешают нитраты.
ЛИТЕРАТУРА
1. Назаренко И. И., Ермаков А. Н. Аналитическая химия селена и теллура. М., Наука, 1971, 251 с.
2. Green Т. Е., Turley М. — In: Treatise on Analytical Chemistry (Ed. I. M. Koithoff and P. J. Living), Part II, Vol. 7, New York, John Wiley, 1961, p. 137.
3. Lowenheim F. A. — In: An Encyclopedia of Industrial Chemical Analysis (Ed. F. D. Snell and L. S. Ettre), Vol 17, New York, Interscience, 1973, p 580.
4. Lyle S. J. — In: Comprehensive Analytical Chemistry (Ed. C. L. and D. W. Wilson), Vol 1C, Avsterdam, Elsevier, 1962, p. 305.
5. Bock R, Tschopel P. — Z. analyt. Chem., 1969, v. 246, p. 81.
6. Korkish J. — Modern Methods for the Separation of Rarer Metal Ions, Oxford, Pergamon Press, 1969, p. 357
7. Стрельникова H. П., Лисцова Г Г. Зав. лаб., 1960, т. 26, с. 142; Analyt-Abstr., 1960, 7, р. 4240.
8. Munze R. — J prakt. Chem., 1959, v. 7, p 262.
9. Бусев А И., Багбанли И Л., Багбанли С. И. и др. — ЖАХ, 1970, т. "э
с. 1374; Analyt. Abstr., 1971, v. 21, р. 4075.
216
10. Khnura К., Ikeda N., Tnarida M. — Japan Analyst, 1958, v 7, p. 174; Analyt. Abstr., 1959, v. 6, p. 140.
11. Havezov 1., Jordanov N. — Talanta, 1974 v 21, p 1013.
12. Bode H. — Z. analyt. Chem.. 1954, v. 143, p. 182.
13. Bode H. — Z. analyt. Chem., 1955, v. 144, p 90.
14 Marcek M. V., Kinson K., Belcher С. B. — Analytica chim. Acta, 1968, v. 41, p. 447.
15. Chakrabarti C. L. — Analytica chim. Acta, 1967. v 39, p 293.
16. Kawamura K-, Ito H., Tanabe T. — Japan Analyst, 1970, v. 19, p. 824; Analyt. Abstr., 1971, v. 21, p 4107.
17. Nakagawa G. — J. chem. Soc. Japan, pure Chem Sect., 1960, v. 81, p. 1258; Analyt. Abstr., 1962, v 9, p. 4073.
18. Киш П. П., Кремнева С Г.—ЖАХ, 1970, т. 25, с. 2200; Analyt. Abstr., 1972,
v. 23, р. 3137.
19 Jordanov N., Havezov I. — Z. analyt. Chem., 1969, v. 248, p. 296; Analyt. Abstr., 1971, v. 20, p. 174.
20. Плотников В. И., ЖАХ, 1959, т, 14, с. 595; Analyt. Abstr., 1960, v. 7, р. 2706.
21. Moghissi А. — J. Chromat., 1964, v. 13, p. 542.
22. Wang J., Cheng K- W.— International Symposium on Analytical Chemistry, Birmingham, 1969.
23. Schrenk W. T, Browning B. L. — J. Am chem. Soc., 1926, v. 48, p. 139
24. Lenher V., Wakefield H. F. — J. Am. chem. Soc., 1923, v. 45, p. 1423.
25. Naidu P. P., Rao G G. — Talanta, 1971, v. 18 p. 112.
26. Kaushik R. L., Prosad R.—J. Ind. Chem. Soc., 1969, v. 46, p. 921.
27. Ma T. S., Zoeilner W. G.—Mikrochim. Acta, 1971, p 329.
28. Guilbault G. G, McCurdy W7. H.— Analytica chim. Acta, 1961, v. 24, p. 214.
29. Beyak R., Jaselskis B.—Analyt. Chem., 1970, v. 42, p. 518.
30. Толстиков В П., Епик П. A.— Укр. хим. ж., 1969, т. 35, с. 1219; Analyt.
Апа-Sons
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49. „„
50- Belch.
51. Со"7
521 с 9'— Analytica chim. Acta, 1971, v 53, p. 325.
P tn.1*-. ’ Barkalov V. S., Usatenko Yu. I. — Zh. analit Khim., 1967, v. 22
Апа-
Abstr., 1970, v. 19, p 4845.
31. Dikshitula L. S. A., Satyanarayana D —Talanta, 1975, v. 22, p. 313
32. Johnson R. A.—Colorimetric Determination of non-Metals (Ed. D. F. Boltz), New York, Interscience, 1958, p. 315
33. Johnson R. A., Kwan F. P. — Analyt. Chem., 1951, v 23, p. 651.
34. Ref. 32, p 323.
35. Roberts J. A., Winwood J., Millett E. J. — Proceedings of the Society for ^^ytical Chemistry Conference at Nottingham, 1965, Cambridge, Heffer and Ltd., 1965, p. 528.
Luke C. L. — Analyt. Chem., 1959, v. 31, p. 572.
Navratil О, Serf a J. — Colin Czech, chem. Commun., 1969, v. 34, p. 975;
lyt. Abstr., 1970, v. 19, p. 196.
Looyenga R. IF., Boltz D. F. — Mikrochim. Acta, 1971, p 507.
Бусев А. И., Симонова Л. H. — /YAX, 1967, т. 22, с. 1850; Analyt. Abstr., 1969, v. 17, p. 832.
Мельчекова 3. E., My розова В И. — ЖАХ, 1970, т. 25, с. 556; Analyt. Abstr., 1971, у. 21, р. 1066. 3
Kirkbright G. F., Saw C. G., West T. S. — Talanta, 1969, v. 16, p. 65.
Kirkbright G. F„ Saw C. G., West T. S. — Analyst, 1969, v 94, p. 457.
Kelyi m. U., Kushnirenko I Ya. — Zh. prikl Spektrosck., 1969, v. 10, p. 810; Analyt. Abstr., 1970, v. 19, p. 2044.
C. L. — Analytica chim. Acta, 1967, v 39, p. 293.
ohb W. D., Foster W. W., Harrison T. S —Analyst, 1976, v. 101, p. 39.
‘nompson К. C„ Thomerson D R — Analyst, 1974, v. 99 p. 595.
reenland L. P., Campbell E. Y — Analytica chim Acta, 1976, v. 87, p. 323.
S„%Ps°n К. С, —Analyst, 1975, v. 100, p. 307.
Bn‘rh —Analyst, 1975 v. 100 p. 300.
P °’’ ^ou*mtzis T- Townshend A.—Analytica chim. Acta, 1974, v. 68, 52. Tulu^n^n' 9' — Analytica chim. Acta, 1971, v 53, p. 325.
> P- Жи- л ’. ’ Barkalov V. S., Usatenko Yu. I. — Zh. analit Khim., 1967, v. 22
S3- Abstr- l969-v- 16- p 122
aa В. И., Игнатенко E Г., Тарасова В А — ЖАХ, 1973, т. 28, с. 1980.
217
54 Агасян П. К., Денисова А. Н., Агасян Л. Б., Николаева Е. Р. — Зав. лаб., 1968 т. 34, с 129.
55. Lingane J. J., Niedrach L. №. — J. Am chem. Soc., 1949, v. 71. p. 196.
56. Schmidt H., Von Steckelberg Af. — J. Polarog. Soc., 1962, v. 8, p. 49.
57. Jamieson R. A., Perone S. P. — J. electroanalyt. Chem., 1969, v. 23, p 441.
58 Чикризова E. T., Копанская Л. C. — ЖАХ, 1968, t. 23, c. 394; Analyt. Abstr., 1969, v. 17. p. 2707.
59 Lloyd K. W. - Ref. 35, p. 180.
60. Lavi N. — Analytica chim. Acta, 1974, v. 70, p. 199.
61. Lanese J. G., Jaselskis B.— Analyt. Chem., 1963, v. 35, p. 1880.
ТЕТРЛФТОРБОРАТЫ
Тетрафторборат применяют в основном при обработке металлов под давлением, например при прокате олова и свинца. Тетрафторборат легко растворяет оксиды металлов, поэтому его применяют при плавке, электрополировке и очистке поверхности металла перед электролитическим нанесением металла.
МЕТОДЫ ОТДЕЛЕНИЯ
Тетрафторборат отделяют, осаждая нитроном или тетрафенил-арсонийхлоридом, однако эти методы обладают рядом недостатков. Предпочтительным является метод экстракции.
В работе [1] проведено систематическое изучение производных тионина для экстракции и прямого фотометрического определения бора в виде BF4". Изучено более 30 органических растворителей, в основном содержащих хлор и бром. Тетрафторборат можно экстрагировать бензолом при pH = 3,4 в виде комплекса с метиловым фиолетовым [2] или тетрафениларсонийхлоридом [3}. При pH = 7—12 тетрафениларсоний образует с тетрафтор-боратом растворимый комплекс, который можно экстрагировать дихлорметаном [3] или хлороформом [4].
Предложен метод экстракции BF4 с помощью ферроина, трис(1,10-фенантролината) железа (II) [5]. Ионный ассоциат ферроина с BF4 экстрагируют при pH = 2—10 н-бутиронитрилом. Этот экстракционно-спектрофотометрический метод обсужден ниже.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Гравиметрический метод определения тетрафторбората осаждением его нитроном в виде C20Hi6N4HBF4 имеет ряд недостатков-Один из них связан с высокой растворимостью осадка, вследствие чего при расчете используют эмпирический коэффициент. Осаждение лучше проводить тетрафениларсонийхлоридом. Реагент! устойчивее, чем нитрон, дает более чистые осадки и позволяв
218
определять меньшие концентрации BF4 [6]. После «контрольной промывки» осадок сушат при 105 °C.
Гравиметрическое определение тетрафторбората
с помощью тетрафениларсоиийхлорида [6]
Реагенты
Аммиак, 15 AI и 1 М растворы.
Тетрафениларсонийхлорид (ТФЛХ), 0,07 М раствор.
Тетрафениларсония тетрафторборат, 250 мл насыщенного раствора с добавлением 10 мл 0,888 М раствора аммиака.
Ход анализа. К 10 мл раствора, содержащего примерно 0,03 г NH4BF4, добавляют 5 мл 15 М раствора NH4OH, 5 мл воды, а затем сразу проводят осаждение. Для этого медленно, при перемешивании приливают такой объем раствора ТФАХ, чтобы его концентрация в конечном растворе была 0,018 М. Раствор выдерживают в течение 1 ч в ледяной воде и фильтруют через пористый стеклянный фильтр со средним размером пор. Осадок промывают 3 мл охлажденного 1 Л1 раствора NH4OH, затем 30—40 мл насыщенного раствора тетрафениларсония тетрафторбората и наконец 5 мл холодного 1 М раствора аммиака. Осадок сушат до постоянной массы при 105 °C
Спектрофотометрические методы
Описан косвенный спектрофотометрический метод определения BF4 с применением тетрафениларсоиийхлорида. Метод основан на определении избытка реагента после осаждения BF4. Поглощение измеряют при 220 нм.
Спектрофотометрическое определение тетрафторбората [6]
К 5 мл раствора, содержащего 0,02—0,04 г NH4BF4, в мерной колбе вместимостью 25 мл приливают 5 мл 0,880 М раствора аммиака и 10,00 мл стандартного раствора ТФАХ (0,06689 М раствор [6]), доводят до метки и оставляют на 1 ч, периодически перемешивая раствор Отбирают 5 мл раствора пипеткой, носик которой обернут фильтровальной бумагой, удерживаемой с помощью резинового колечка, и разбавляют водой до 2 л. Измеряют поглощение раствора при 220 нм относительно раствора сравиеиия, полученного разбавлением 1 мл 0,880 М аммиака до 2 л.
Описан спектрофотометрический метод определения тетрафторбората с помощью ферроина [5], основанный на избирательной экстракции ионного ассоциата н-бутиронитрилом и измерении поглощения при 520 нм. При однократной экстракции извлекается '4 /0 тетрафторбората. Закон Бера соблюдается в широком интервале концентраций. Определению мешают KNO2, КВг, KNO3, AoCN и КНС8Н4О4. Мешают умеренные содержания NH4C1 и значительные концентрации Na2SO4, Na2C2O4, NaH2PO4, 1NaKC4H4O6.4H2O, К2Сг2О7 и Na2SO3.
Другие методы определения
тетрщьСаНк ТитРиметРический метод, основанный на осаждении та, с*Н ЫПрТа В ВИДе цетилтриметиламмоний тетрафторбора-
1» 42NBF4 [7]. Растворимость осадка в 10 раз меньше, чем
219
осадка, полученного при осаждении BF? нитроном. Определению не мешают HF и Н3ВО3. Недостатком метода является необходимость приготовления большого числа растворов.
Нерастворимый в воде осадок ионного ассоциата BFj с тетра-фениларсонием можно экстрагировать дихлорметаном [3], что использовано для экстракционного титрования. Избыток ТФАХ в водной фазе определяют амперометрически или фотометрически при 220 нм. Фторид и борат не мешают определению. Мешают анионы, взаимодействующие с ТФАХ, в том числе хлорат, перхлорат и нитрат.
Для определения тетрафторбората применены ионоселективные электроды. Один из методов основан на титровании BF-t раствором ТФАХ при 2 °C. Конец титрования устанавливают ионоселективным электродом (Орион) с жидким ионообменником, чувствительным к ионам С1ОГ и BF4. Содержания BF4 0,25 ммоль можно определять с ошибкой 1 % [8]. Описан другой аналогичный метод определения BF;[9]. Определению мешают нитраты и иодиды.
ЛИТЕРАТУРА
1. Pasztor L., Bode J. D.— Analytica chim. Acta, 1961, v. 24, p. 467.
2. Полуектов H. С., Кононенко Л. И., Лауер P. С. — ЖАХ, 1958, т. 13, с. 396.
3. Behrends К. — Z. analyt. Chem., 1966, v. 216, р. 13; Analyt. Abstr., 1967, v. 14, p. 4643
4. Coursier J., Hure J., Platzer R.— Analytica chim. Acta, 1955, v. 13, p. 379.
5. Archer V. S.. Doolittle F. G., Young L. M. — Taianta, 1968, v. 15, p 864.
6. Affsprung H. E„ Archer V. S. — Analyt. Chem.. 1964, v. 36, p. 2512.
7. Schaack H. ]., Wagner W. — Z. analyt. Chem., 1955, v. 146, p. 326; Analyt Abstr,. 1955, v. 2, p. 2996.
8. Smith M. J., Manahan S. E. — Analytica chim. Acta, 1969, v. 48, p. 315.
9. Carlson R. M., Paul 1. L. — Analyt. Chem., 1968, v. 40, p. 1292.
ТЕТРАФЕНИЛ БОРАТЫ
Анион тетрафенилборат Ph4B_ обладает уникальным свойством образовывать нерастворимые соли с К+, Rb+, Cs+ и NH<-Эти ионы можно определять путем осаждения тетрафенилбора-том натрия.
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждением тетрафенилбората в виде нерастворимых солей щелочных металлов можно отделить Ph4B~ от других анионов-Применяют также метод жидкостной экстракции. В работе [о описан метод экстракции Ph4B~ хлороформом или хлорбензолом в виде ионного ассопиата с катионным хелатом меди(1) [CuLzjXI XlPihB’], где L — купроин или неокупроин.
220
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Спектрофотометрические методы
Тетрафенилборат можно определять экстракционно-спектрофотометрическим методом, экстрагируя ионный ассоциат окрашенного хелата меди(1) с Ph4B~. Экстракт сушат, встряхивая с безводным сульфатом натрия, и измеряют поглощение при 550 нм.
Нерастворимые тетрафенилбораты щелочных металлов можно обработать концентрированной кислотой и определить выделившуюся борную кислоту, например, с помощью 1,Г-диантримида [7], измеряя поглощение при 620 нм.
Электрохимические методы
Предложен метод определения Ph4B_, основанный на том, что его калиевая соль растворима, а серебряная соль нерастворима в смеси ацетон — вода [2]. Тетрафенилборат титруют кулонометрически серебром (I) [3] в водно-ацетоновой среде. В отличие от результатов работы [4] показано, что взаимодействие протекает строго в соответствии со стехиометрией реакции. Метод применен для определения 0,3—10 мг К+, Rb+ или Cs+ после осаждения их тетрафенилборатом. Фоновым электролитом служил раствор нитрата натрия в нейтральном растворе, содержащем 35—50% ацетона [3].
При титровании тетрафенилбората растворами Hg(NO3)2 или Hg(C104)2 конец титрования устанавливают потенциометрическим или амперометрическим методом [5]. Для потенциометрии применяют покрытый ртутью платиновый и насыщенный каломельный электрод, подсоединенный с помощью агар-агарового мостика. Для амперометрии используют капающий ртутный и насыщенный каломельный электроды.
Описан другой амперометрический метод, основанный на осаждении KPh4B, растворении осадка в смеси ацетонитрил — воды и титровании Рй4В~ раствором нитрата серебра [6]. Ток измеряют с помощью капающего ртутного электрода, неполяризуемым электродом сравнения служит донная ртуть. Метод отличается высокой точностью.
ЛИТЕРАТУРА
I- У., Okamoto N., Tao Е.— Analytica chim. Acta, 1969, v. 47, p. 127.
^Kudorff Zannier H _z analyt chem 1952 v 137 ,
4’ 1 ’ LinSane J. 7, —Analyt. Chem., 1967, v. 39, p 168.
5 Analytica chim. Acta, 1957, v. 16, p. 370.
P 4853 У A ~7- analyt- Chem., 1960 -v- 173- P 301= Analyt. Abstr., 1960, v. 7,
7- Raber SnA-C Vries T De ~ Analyt. Chem , 1956, v. 28, p. 1899.
I , Likussar W.—Mikrochim. Acta, 1972, p. 92.
221
РОДАНИДЫ
Роданиды аммония и щелочных металлов находят применение в разных областях, в том числе в фотографии, катализе, агрохимии (как гербицид), при крашении тканей, как ингибитор коррозии (в красках). Роданид аммония является важным сырьем для получения тиокарбамида. Хотя роданид и не столь токсичен, как цианид, его соединения губительно влияют на жизнь обитателей водной среды. Поэтому необходимы методы определения малых содержаний роданида.
Опубликованы обзоры по аналитической химии роданида [1— 3]. В наиболее детальном обзоре [2] рассмотрены методы отделения и различные методы определения роданида.
МЕТОДЫ ОТДЕЛЕНИЯ
При определении роданидов в большинстве случаев отделение не проводят, селективность достигается другими методами, например маскированием. Так, при анализе смеси роданида и цианида последний маскируют формальдегидом и роданид титруют методом Фольгарда.
Осаждение
Роданид можно осадить в виде роданида серебра из разбавленных растворов азотной кислоты. Если осадок обработать сероводородом, образуется нерастворимый сульфид серебра, а роданид переходит в раствор. Метод позволяет определять микро- и макросодержания роданида [4].
Для отделения роданида можно использовать низкую растворимость его комплекса с медью (I). Для осаждения роданида в раствор вводят сульфат меди и бисульфит, определение заканчивают гравиметрическим методом.
Роданид осаждают растворами меди (II) и пиридина в виде светло-зеленого комплексного соединения [5]. Определению мешают некоторые ионы.
Экстракция
Ион тетрафениларсония (CeHs^As4- и аналогичные катионы используют для отделения роданида от других серусодержащих анионов методом экстракции. При этом роданид экстрагируется хлороформом при pH = I—2 на 97%, в то время как тиосульфат, сульфит и сульфат почти не экстрагируются [6]. При рН==2— 12 роданид полностью экстрагируется хлороформом с помощью тетрафенилфосфонийхлорида. Ионы S2O3 , SO3" и SO4 экстрагируются незначительно [7].
Другие экстракционные системы описаны в монографии [8]«
222
Методы бумажной и тонкослойной хроматографии
Этот метод важен для отделения малых содержаний роданида. Характеристика некоторых методов дана в табл. 14. Другие методы рассмотрены в обзоре [2].
Таблица 14. Отделение роданида методами бумажной и тонкослойной хроматографии
Метод отделения Растворитель Отделяемые иоиы Литература
Бумажная хроматография (Ватман № 1) ТСХ (силикагель) ТСХ (маисовый крахмал) Бутанол — ацетон — NH4OH (3 : 13 : 4) или бутанол — ди-метилформамид — NH4OH (3: 1:1) Ряд растворителей, в том числе бутанол — пропанол — дибутиламин (9:9:2) Ацетон - 3 М NH4OH (1:4) СГ, ВГ, г F', СГ, Вг’, Г, CN’, Fe(CN)|’,Fe(CN)*’ NO’, S2O2’, CrO2’, N’, CN’, BO’’, S2’, AsO3’, NO’, SO2’, PO2’, AsO2’ 9 10 11
ТСХ (силикагель +5% растворимого крахмала) Ацетон — вода (10 : 1) СГ, Вг’, Г, Fe(CN)2’, Fe(CN)j’, C1O3’, BrO’, IO3’, NO’ 12
ТСХ (гельман SA н SG) ТСХ (силикагель на алюминиевой фольге) Ряд растворителей, в том числе бутанол — пиридин — уксусная кис- лота — вода (15 : 10 : 3 : 12) Раствор FeCl3 SO2’, S2O2’, S3O2’, s4oj- CN’, Fe(CN)2’, Fe(CN)*’ 13 14
Газохроматографический метод
Метдды газовой хроматографии рассмотрены в конце этой главы.
методы определения
Бром окисляет роданид количественно до сульфата
SCN' + 4Вг2 + 4Н2О —> SO4’ + 7Br' + BrCN + 8Н+
который можно определить после удаления избытка брома.
223
Гравиметрические методы
Гравиметрические методы определения роданида, вероятно, не находят широкого применения ввиду наличия более простых методов.
Роданид серебра, осажденный из растворов разбавленной HNO3, можно высушить при Н5°С и взвесить. В растворе должны отсутствовать галогениды и цианиды. Гравиметрическое определение SCN- в виде роданида меди(1) было известно давно, но метод чаще применяли для определения меди [15]. Осаждение ведут из нейтральных или слабокислых растворов путем насыщения охлажденного раствора сернистым газом (или добавлением раствора, насыщенного SO2) и введения сульфата меди. Осадок сушат при ПО—120°С и взвешивают. Если к роданиду добавить бромную воду и нагреть раствор на водяной бане в течение 1 ч, роданид окисляется до сульфата, который можно определить в виде BaSO4. Ранее описан метод осаждения комплекса меди с пиридином и роданидом.
Титриметрические методы
Иодиметрические методы
Один из наиболее широко применяемых методов определения роданида основан на его взаимодействии с бромом в слабокис-лой среде с образованием бромциана [16, 18]. Избыток брома удаляют, вводя фенол, гидразин и др., а бромциан определяют иодиметрически, добавляя KI и титруя выделившийся иод тиосульфатом:
BrCN + 2Г —> CN' + L + Вг"
Определению не мешают хлорид, бромид, сульфид, сульфит и тиосульфат. Цианид мешает, его следует удалить или замаскировать.
Другой иодиметрический метод основан на окислении роданида стандартным раствором иода в гидрокарбонатной среде [17, 19]:
SCN'+ 412 + 4Н2О —» SO4'+ 71'+ ICN + 8Н*
Избыток иода оттитровывают стандартным раствором тиосульфата. Иодциан не взаимодействует с тиосульфатом, но реагирует с крахмалом, поэтому конечную точку устанавливают по исчезновению желтой окраски иода.
Роданид можно окислить также в боратном буферном растворе при pH = 9—10. Избыток иода титруют арсенитом, в буфер* j ном растворе восстановление иода протекает количественно:
21г + AS2O3 + 2Н2О < >, 4HI + As2Oj
224
Иодиметрическое определение роданида [16]
Ход анализа. К 0,3—90 мг роданида в склянке для титрования пода приливают 5 мл 20%-ного раствора ортофосфорной кислоты и бромную воду до интен-сивно-жептой окраски раствора. Раствор встряхивают и оставляют на несколько минут. Приливают 2 мл 5%-ного раствора фенола и встряхивают до исчезновения окраски брома.
Добавляют 0,5 г KI, встряхивают до растворения соли н оставляют на несколько минут. Титруют стандартным раствором тиосульфата, используя в качестве индикатора крахмал; 1 моль роданида соответствует 2 моль S2O|".
Примечание. Если присутствует цианид, его необходимо отделить дистилляцией (см. разд. «Цианид»).
Аргентометрические методы
Роданид, как и хлорид, можно титровать методом Фольгарда. К анализируемому раствору добавляют избыток стандартного раствора Ag+ и титруют стандартным раствором роданида в присутствии железоаммонийных квасцов. Конец титрования устанавливают по интенсивной окраске Fe(SX2N)2+. Титрование можно проводить в сравнительно сильнокислой среде, при этом ионы Fe111 не гидролизуются. Мешают цианиды и галогениды. Влияние цианида устраняют введением формальдегида. Если формальдегид не вводить, можно определить сумму роданида и цианида. Цианид можно определить титрованием по Либигу. Содержание роданида определяют по разности.
Роданид можно определять прямым титрованием раствором AgNO3 с адсорбционным индикатором. Недостатком метода, как и при определении галогенидов, является нечеткий конец титрования в присутствии больших концентраций электролитов. Наилучшим индикатором является, вероятно, дихлорфлуоресцеин, а также эозин (тетрабромфлуоресцеин), который можно применять в более кислой среде (при pH = 2).
Титриметрическое определение роданида с помощью AgNO3
и эозина в качестве индикатора
Реагенты
Эозин, 0,1%-ный раствор в 70%-ном этаноле.
Ход анализа. Подкисляют аликвотную часть раствора, содержащего рода-ид, несколькими миллилитрами 6 М уксусной кислоты, добавляют 10 капель ндикатора и тнтруют стандартным раствором AgNO3 до момента, когда осадок внезапно становится красным.
Меркуриметрический метод
рТу?°Данид’ как и хлорид, можно титровать раствором нитрата Тод и В ПРИСУТСТВИИ Дифенилкарбазона как индикатора. Этот ме-мый снользован для определения церия(IV) [21]. В апализируе-В качесСТ°Рвводят SCN-, избыток которого титруют Hg(NO3)a. ве индикатора можно применять также нитропруссид.
8 Зак ley
225
Другие титриметрические методы определения
Роданид можно определять окислительно-восстановительными гитриметрнческими методами [21, 22] с применением разных окислителей, в том числе бромата, гипобромита, гипохлорита, перманганата, йодата, хлорамина, N-бромсукцинимида, гексацианоферрата (III) и церия (IV). Трудно отдать предпочтение какому-нибудь окислителю.
Различные рекомендации по применению церия(IV) в качестве окислителя обобщены в работе [23]. Предложен метод, основанный на применении церия(IV) в сочетании с предварительным окислением роданида монохлоридом иода. Точные и воспроизводимые результаты получены в присутствии НС1 в интервале pH = 1,7—4,5. Низкие результаты, полученные в некоторых рабо тах, объясняются потерями H2S и SO2, что было доказано ме тодом ИК-спектрометрии.
Из методов, предложенных в последнее время, некоторые преимущества имеет метод, основанный на применении N-бромсукцинимида [24]. Этот окислитель отличается устойчивостью и может служить вторичным стандартом, его можно использовать для прямого титрования с применением бордо красного как индикатора. Ранние работы по применению N-бромсукцинимида описаны в монографии [21].
Предложен комплексометрический метод определения роданида в присутствии других анионов [25—27]. При анализе смеси SCN-—CN-—С1_ использован тот факт, что AgCI не растворяется в горячей HNO3, в то время как AgCN и AgSCN разлагаются, причем AgSCN окисляется до сульфата. Осадок AgCI отфильтровывают, промывают, растворяют в тетрацианоникелате калия и выделившийся никель (II) титруют раствором ЭДТА
2AgCl + Ni (CN)f —> 2Ag (CN)J + Ni2++ 2СГ
Сульфат, полученный при окислении роданида, содержащийся в фильтрате, осаждают в виде BaSO4 и определяют титриметриче-ски, с помощью ЭДТА. Цианид определяют из отдельной аликвотной части раствора путем добавления избытка стандартного раствора NiSO4 (образуется тетрацианид никеля) и титрования избытка никеля раствором ЭДТА.
В работе [28] для определения роданида и цианида в смеси SCN~—CN-—1~ [29] использован другой подход, основанный на титровании раствором нитрата ртути (II) в присутствии ЭДТА. Конец титрования устанавливают фотометрически, по поглощению смешанолигандного комплекса SCN~—Hg11 — ЭДТА при 240 несоответствующий комплекс цианида CN-—Hg11 — ЭДТА не поглощает в области 240 нм. Скорость образования цианидного комплекса значительно выше, чем роданидного. При титровании смеси SCN- и CN- конец титрования CN- определяют по подъему поглощения при 240 нм, а конец титрования SCN-—по достижению постоянного значения оптической плотности. Определению мешают 226
иодиды и сульфиды. Бромиды, Ag+ и NH] мешают только при титровании роданидов.
Определение компонентов смеси CN-, SCN- и I- также основано на образовании смешанолигандных комплексов. В качестве визуальных индикаторов применяют метилтимоловый синий или крезолфталеин.
Спектроскопические методы
Колориметрические и спектрофотометрические методы
Роданид образует с ионами Fe111 интенсивно окрашенные красные комплексы. При низких концентрациях роданида преобладают ионы Fe(SCN)2+. Эту реакцию широко применяют для определения роданида и железа [29]. Определению мешают фториды, метафосфаты, пирофосфаты, оксалаты, салицилаты, кетокислоты, фе нолы и таннины, а также умеренные содержания SC1 , РОГ и Мп'1. Ртуть(1) и серебро(1) образуют нерастворимые роданиды, а ртуть(II), Cd, Zn, и Sb111 образуют комплексы с роданидом, вследствие чего снижается поглощение Fe(SCN)2+. По той же причине мешают Си, Bi, Ti, U, Мо, Ru, 1г и Os. Иодид уменьшает устойчивость комплекса Fe(SCN)2+. Важно, что определению не мешает
цианид.
Метод определения роданида по образованию Fe(SCN)2+ отли-'чается простотой, однако окраска комплекса неустойчива и определению мешает ряд ионов. Устойчивость окраски зависит от при-
роды и концентрации кислоты и температуры раствора.
При определении роданидов в сточных водах фенольные соединения предварительно удаляли с помощью анионита в С1_-форме (De-Acidite Е) [30]. Этот метод рекомендован для определения роданида в промышленных сточных водах [31]. В работе [31] при ведены максимально допустимые содержания посторонних ионов.
Для определения роданида предложен бензидин-пиридиновый метод [32,33], недостаток которого связан с канцерогенностью бензидина.
Спектрофотометрический метод, основанный на образовании комплекса меди с пиридином и роданидом Cu(C5H5N)2(SCN)2 [34], не столь чувствителен, как, например, пиридин пиразолоно-ВЫи, но весьма специфичен. Метод прост в исполнении и экспрес-сен- Комплекс можно экстрагировать хлороформом и измерять поглощение при 410 нм. Интервал определяемых содержаний роданида 0,5—20 рргп [34]. Ионы, мешающие определению, приведены в табл. 15.
гхло^НРИДИН ПИРазолоновь1^ метод основан на окислении роданида ЗатеаМИН°М Т Д° ХЛ0Рниа,1а в присутствии Fe111, как катализатора. З.м м х1л?Рциан взаимодействует с пиридином в присутствии T337JS-1 "HPa=°-5-она с образованием синего красителя
1. Закон Бера соблюдается в интервале содержаний 8*
227
роданида 0,4—2,5 мкг. Экстракция н-бутанолом позволяет повы сить чувствительность метода. Поглощение измеряют при 620 нм в водных и при 630 нм в «-бутанольных растворах. Длительность приготовления раствора реагента сравнительно велика, растворы реагента неустойчивы. Определению мешают фенольные соединения, а также восстановители. Детальное изложение метода дано в монографии Сендела [29].
Предложен метод определения роданида, основанный на его взаимодействии с рением в степени окисления (III) — (V), с образованием комплекса, поглощающего при 390 нм [36]; соотношение рения и роданида равно 1 : 2. Закон Бера соблюдается в интервале 0—5 ppm. Определению 9,5 ppm роданида мешают иодат, бромат; окислители Ceiv, СггО? , VO.s, С10з- Ю4, NOj и NO? при их содержании 3 ppm. Влияние других посторонних ионов описано в работе [36].
Показано [37], что ртуть образует с роданидом и хинолином смешанолигандный комплекс Hg(C9H7N)2(SCN)2, который экстрагируется СНС13 при pH = 5,1 —6,5 и поглощает в области 498 нм. Определению мешают Cl~, Br~, I", CN- и значительные содержания NH^ и Си11, Закон Бера соблюдается в интервале содержаний роданида 20-50 мкг.
Роданид экстрагируется в виде тройного комплекса при взаимодействии с трис(1,10-фенантролинатом) Fe11 [38]. Экстракцию проводят нитробензолом и измеряют снижение поглощения водной фазы при 516 нм. Интервал определяемых содержаний роданида 2—40 мкЛ4. Определению мешают некоторые анионы. Влияние катионов можно устранить введением ЭДТА.
Роданид, а также перхлорат, иодид и броммеркурат(П) экстрагируются из водных растворов в виде ионного ассоциата с катионом нейтрального красного, что можно использовать для спектрофотометрического определения роданида [39]. Интенсивность окраски органической фазы пропорциональна содержанию аниона.
Таблица 15. Ионы, мешающие определению роданида по поглощению комплекса медн с пиридином н роданидом
Ион Предельно допустимое содержание (ppm) Ион Предельно допустимое содержание (ppm)
CN', Fe(CN)g' 5* Ацетат, оксалат, тартрат 300
NCO" 50* Цитрат, NOj 500
Вг’ 100 СГ, CrO4", F", lOj- РО’" 1 000
Вго; 200
so,- 10 000
* Цианиды, цианаты и комплексные цианиды можно разложить кипячением с кислотам Иоднды мешают, ио их можно окислить до иода и проэкстрагировать СС1<. Среди катион мешают только ртуть н никель.
Таблица 16. Ионы, мешающие экстракционно-спектрофотометрическому определению роданида с помощью нейтрального красного [39)
Ион • Мольное отношение мешающего иона и SCN” НаВдено SCN", % Ион * Мольное отношение мешающего иона и SCN* Найдено SCN-, %
Са2+ 50 101 SiO* 2" 100 100
Mg2+ 50 100
А13+ 50 100 СГ 200 102
Fe3+ 3 82 ВГ 200 103
30 99** CN" 500 97
NH? 500 101 НСО" 500 98
н2ро; 500 99 no; 5 106
SO2" 500 юГ F" 500 98
СН3СОО 300 103
♦ Катионы вводили в виде сульфатов, анионы —в виде натриевых или калиевых солей Концентрация KSCN равна 8«10—6 М.
•• Введено 10“3 М ЭДТА.
Максимум поглощения экстракта 552 нм. В качестве растворителя применяют нитробензол. Окраска растворов сохраняется в течение
2 ч. Мешающие ионы приведены в табл. 16.
Метод определения цианида с помощью п-фенилендиамина может быть использован для определения роданида в отсутствие цианида и его комплексных ионов [40]. При определении роданидов угол наклона градуировочной кривой в 2,23 раза меньше, чем при определении цианидов.
Роданид образует с метиленовым синим экстрагируемый дихлорэтаном комплекс состава 1 : 1 [41], поглощающий при 657 нм. Определению мешают сравнимые содержания иодида и перхлората. Интервал определяемых содержаний роданида 0,2—10 мкМ.
Атомно-абсорбционный метод
Роданид в интервале концентраций 0,5—2,0 ррш можно опре-(S^N) к^св^ен11С) по содержанию медн в комплексе Си (Руг) 2
Метод молекулярной эмиссии в полости
но ОпРеделение серусодержащих соединений этим методом основа-воля3 ИЗмеРении излучения S2 при 384 нм. Метод чувствителен, поз-литвях определять нанограммовые содержания в нескольких микро-HC1Q рартв°ра- Роданид калия дает ряд пиков, при введении H3Pq4 вабл10дается один более интенсивный пик, а при введении
* нтенсивность пика еще выше не только для роданида но
229
и для других серусодержащпх анионов [43]. Подробное описание методики дано в работе (43], краткое — в разделе «Сульфиты и оксиды серы».
Электрохимические методы
Потенциометрический метод, ионоселективные электроды
Роданид можно титровать потенциометрически раствором Ag1-с серебряным электродом. В раствор вводят Ba(NO3)2 для снижения адсорбционных эффектов. В присутствии желатина возможно последовательное титрование бромида и роданида [44]. В присутствие желатина увеличивается удельная поверхность AgBr, вследствие чего бромид адсорбируется сильнее, чем роданид.
Возможно потенциометрическое титрование роданида раствором ртути с применением серебряного индикаторного электрода, однако точность метода недостаточно высока [45].
Метод дифференциального потенциометрического титрования растворами серебра (I) применен для определения некоторых анионов, в том числе роданида [46] (см. разд. «Хлориды»),
Показано, что перхлоратный ион-селективный электрод на жидком ионообмепнике типа 92—81 (Орион) чувствителен также к роданиду и перренату, что позволяет проводить потенциометрическое титрование роданида растворами Ag+ и Hg2+ [47]. Описан поли-кристаллический мембранный электрод для определения роданида [48], представляющий собой смесь тонкоизмельченных порошков AgS и AgSCN, спрессованных в виде диска. Аналитические характеристики электрода, полученного смешением AgSCN с термопластическим полимером, изучены в работе [49]. Электрод чувствителен к серебру(I) и роданиду. В интервале концентраций 10 мкЛ-1 — 0,1 М при pH = 1 — 13 потенциал электрода изменяется на 59 мВ при увеличении концентрации ионов в 10 раз. В неводных растворах потенциал электрода изменяется в соответствии с уравнением Нернста в интервале концентраций 0,1 мкА!— 0,1 М.
Ионоселективные кристаллические электроды для определения роданида выпускают серийно (Орион 94—58) [50]. Интервал определяемых содержаний роданида 5-Ю*6 — 1 М. Определению мешают сравнимые содержания Вг', NH3, S2O3', CN', Г, S2', не мешает 20-кратный избыток хлорида.
Кулонометрический метод
Роданид можно титровать кулонометрически, генерируя се-ребро(1) со 100%-ным выходом по току с помощью серебряного электрода. Измерение можно проводить при постоянном токе или при постоянном потенциале [51].
Полярографический метод
Обзор методов полярографического определения роданида при‘ веден в работе [2]. Полярографический метод применен для ана^ лиза кислых растворов, содержащих HCN, Н2О2, SO2' и SCN 230
[52]. В работе [52] установлено наличие хорошо выраженной анодной волны, связанной с обратимой деполяризацией капающего ртутного электрода. Удовлетворительным фоновым электролитом является 0,1 М раствор НС1О4. Изучено анодное полярографиче-ческое окисление ртути в растворах, содержащих роданид [53], связанное с образованием комплексного нона типа
[Hg(SCN),]2-/ где / = 2, 3 и 4
Рассчитаны ступенчатые константы устойчивости комплексов.
Другие методы определения
Известно каталитическое действие ионов SCN', S2O3' и S ’ на иод-азидную реакцию [54]:
2Nj 4-12 —> 2Г + ЗЫ2
Показано, что с помощью этой реакции можно определять 8-Ю-11 г/мл и более роданида [55]. Измерение скорости реакции проводят амперометрически.
Роданид и цианид определяют методом газовой хроматографии [56]. При обработке бромом оба аниона переходят в бромциан, который определяют с помощью электронозахватного детектора. Предел обнаружения SCN- и CN- равен 10~8 г. Градуировочный график для определения SCN- линеен до содержания 1,2 ppm. Предел обнаружения снижается до 1 ррЬ, если бромциан экстрагировать диизопропиловым эфиром и затем экстракт вводить в колонку. Комплексные цианиды не мешают, их можно определить после разложения до CN-. Если CN- замаскировать формальдегидом, можно определить роданид в смеси с цианидом.
Роданид (а также CF, CN" и РО4’) при содержании от п мкг до п мг можно определить косвенным рентгенофлуоресцентным методом [57]. От 6 мкг до 1 мг SCN- осаждают раствором AgNO3 вместе с Вг_ и I-, которые вводят в качестве внутренних стандартов Препарат анализируют непосредственно на фильтровальной бумаге.
Для определения SCN-, а также СГ, Вг', Г, AsO4 , СггО? и Fe(CN)e применен метод колориметрии пятен [58]. Определение проводят путем сравнения со шкалой стандартов. Анионы вымывают^ в кольцевую зону и фиксируют там в виде нерастворимых олеи серебра, которые затем переводят в сульфид серебра. Метод зволяет^ определять миллиграммовые содержания роданида. ботртча?1”1 микР°мет°Д определения SCN~ на бумаге описан в ра-поме! 9 Небольшой объем пробы, содержащий 10—150 мкг SCN-ребп^к1 В центР листа бумаги, импрегнированной хроматом се-бумаги центРУ листа снизу подводят полоску фильтровальной зывает’ П° К0Т0Р°^ капиллярными силами поднимается вода и вы-ми1 рацию SCN- от центра листа к его периферии. Роданид
231
взаимодействует с хроматом серебра, образуя белые пятна AgSCN, которые высушивают, вырезают и взвешивают. Метод позволяет определять SCN~ в смеси с С1~ и 1~. В статье [59] описана аппаратура, необходимая для осуществления метода.
Смесь SCN- и 1~ можно анализировать методом термометрического титрования раствором AgNO3 [60]. Прибор для дифференциального термометрического титрования описан в работе [60]. В конце титрования каждого комплекса (при концентрации 0,01 Л1) наблюдается скачок температуры.
ЛИТЕРАТУРА
1. Heinrich В. J.— In: Treatise on Analytical Chemistry (Ed. I M Kolthoff and P. J. Elving), Part II, Vol. 7, New York, Interscience, 1961. n. 87 e
2. Hanley A. V., Czech F. W. — In: The Analytical Chemistry of Sulphur and its Compounds (Ed. J. H. Karchmer), Part I, New York and London. Wileylnterscience, 1970, p 379.
3. Lowenheim F. A.— In: Encyclopedia of Industrial Chemical Analysis (Ed. F. D. Snell and L. S. Ettre), Vol. 18, New York, Interscience, 1973, p. 433.
4. Chariot G., Bezier D. — Quantitative Inorganic Analysis, London, Methuen, 1957, p. 382.
5. Spacu G. — Bui. Soc. Sti. Cluj, 1922, v. 1, p. 284; J. chem. Soc. ii, 1923, p. 40.
6. Bock R., Beilstein G. M.— Z. analyt. Chem., 1963, v. 192, p. 44.
7. Bock R., lainz J. — Z. analyt. Chem., 1963, v 198, p. 315.
8. De A. K-, Khopkar S. M., Chalmers R. A. — Solvent Extraction of Metals, New York, Van Nostrand, 1970.
9. Mao C.-H. — Acta chim. Sinica, 1964. v. 30, p. 496; Analyt. Abstr., 1966, v. 13, p. 533.
10. Gagliardi E., Pokorny G. — Mikrochim Acta, 1965. p 699.
11. Canic V. D., Turcic M. N., Bugaski-Vojinovic M. B., Perisic N. U. — Z. analyt Chem., 1967, v. 229, p. 93; Analyt. Abstr.. 1968, v 15, p. 5211.
12. Kawanabe K., Takitani S., Miyazaki M., Tamura Z.—Japan Analyst, 1964, v. 13, p. 976; Analyt. Abstr., 1964, v. 13, p. 976.
13. Kelly D. P. — J. Chromat., 1970, v. 51, p. 343.
14. Thielemann H.— Z. analyt. Chem., 1974. v. 270, p. 128.
15. Newman E. J.—Analyst, 1963, v. 88, p 500.
16. Schulek E. — Z. analyt. Chem., 1923, v. 62, p. 337; Chem. Abstr. 1923, v. 17, p. 3465.
17. Rupp E., Schied A. — Ber., 1902, v. 35, p. 2191
18. Kolthoff I. M„ Belcher R.—Volumetric Analysis III, New York, Interscience, 1957, p. 303.
19. Ref. 18, p. 305.
20. Sant B. R. — Ber., 1955, v 88, p. 581; Analyt. Abstr., 1955, v. 2, p. 2375.
21. Berka A., Vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Pergamon press, 1965.
22. Kolthoff I. M., Belcher R. — Volumetric Analysis III, New York, Interslience, 1957.
23. Singh S., Siefker J. R. — Analytica chim. Acta, 1962, v. 27, p. 489. „
24. Sarwar M.t Waheed H. A., Chowdhri A., Thibert R. J. — Mikrochim. Acta, 197A
p. 683.
25. De Sousa A. — Taianta, 1961, v 8, p. 782. in
26. De Sousa A. — Inf. Quim. Anal., 1962, v. 16, p. 126; Analyt. Abstr., 1963, v- 1"
p. 2243. ..
27. De Sousa A. — Inf. Quim. Anal., 1963, v. 14 p. 130; Analyt. Abstr., 1964, v ’ p. 5446. •
28. Nomura T. — J chem. Soc. Japan, pure Chem. Sect., 1968, v. 89, p. 580; Anaiy Abstr., 1970, v. 18, p. 114
232
29 Sandell E. В. — Colorimetric Determination of Trace Metals, 3rd edn.. New York, Interscience, 1959.
30. Whiston T. G., Cherry G. W. — Analyst, 1962, v. 87, p. 819.
3b Official, Standardised and Recommended Methods of Analysis, 2nd edn (Compiled and Edited by N. W Hanson). London, The Society for Analytical Chemistry, 1973, p. 452.
32. Aldridge IF. N. — Analyst, 1944, v. 69, p. 262.
33. Aldridge W. A. — Analyst, 1945, v. 70, p 474
34 Kruse J. M., Mellon M. G. — Analyt. Chem., 1953, v. 25, p. 446.
35. Epstein J. — Analyt. Chem., 1947, v. 19, p. 272.
36. Neas R. E., Guyon J. C. — Analyt. Chem., 1969, v. 41, p. 1470.
37. Einaga H., Ishi H., Iwasaki I. — Taianta, 1973, v. 20, p. 1017.
38 Yamamoto У., Tarumoto T., Hanamoto Y. — Bull. chem. Soc. Japan, 1969, v. 42, p. 268; Analyt. Abstr., 1970, v. 18, p. 3050.
39. Tsubouchi M. — Analytica chim. Acta, 1971, v 54, p. 143.
40. Ref. 31, p. 454.
41 Koh T., Iwasaki I. — Bull. chem. Soc. Japan, 1967, v. 40, p. 569; Analyt. Abstr., 1968, v. 15, p. 3866.
42. Danchik R. S., Boltz D. F. —Analyt. Chem., 1968, v 40. p. 2215.
43. Belcher R., Bogdanski S. L., Knowles D. J., Townshend A. — Analytica chim. Acta, 1975, v. 77, p. 53.
44 Scheibitz M. — Z. wiss. Photogr., I960, v. 54, p. 46; Analyt. Abstr., 1961, v. 8, p. 4631.
45. Kolthoff I. M., Lingane J. J. —J. Am. chem. Soc., 1935, v. 57, p. 2377.
46. Bishop E., Dhaneshwar R. G. — Analyst, 1962, v. 87, p. 207
47. Hirsch R. F., Portock J. D. — Analyt. Lett., 1969, v. 2, p. 295.
48. Ross J. W„ Durst R. A. — In: Ion Selective Electrodes. Washington D. C., Special Publication No. 314 NBS, 1969, p. 79.
49. Mascini M. — Analytica chim Acta, 1972, v. 62, p. 29.
50. Orion Research Incorporated, Cambridge, Mass., U. S. Available from MSE-Fisons, Manor Royal, Crawley, West Sussex, U. K.
51. Lingane J. J. — Electroanalytical Chemistry, 2nd edn., New York, Interscience, 1958, p. 514, 599.
52. Plowman R. A., Wilson I. R. — Analyst, 1960, v. 85, p. 222.
53. Nyman C. J., Alberts G. S. — Analyt. Chem., 1960, v. 32, p. 207.
54. Goto H., Shishiokawa T. — J. chim. Soc. Japan, pure Chem. Sect., 1944, v. 65, p. 673; Chem. Abstr., 1947, v. 41, p. 3392c.
55. Mickalski E., Wtorkowska A. — Chemia analit., 1961, v. 6, p. 365; Analyt. Abstr., 1962, v. 9, p. 74.
56. Nota G., Palombari R. — J. Chromat, 1973, v. 84, p. 37.
57. Stork 0., Jung H. — Z. analyt. Chem., 1970, v. 249, p. 161; Analyt. Abstr., 1971, _ v. 20, p. 1523.
58 Celap M. B., Weisz FL — Mikrochim. Acta, 1962, p. 24.
59. Ganchev N., Koev K- — Mikrochim. Acta, 1964, p. 87.
60 Takeuchi T., Yamazaki M. — J. chem. Soc. Japan, pure Chem. Sect-, 1969, v. 72, P- 1263; Chem. Abstr., 1970, v. 72, p. 18094e,
ВОЛЬФРАМАТЫ
Вольфрам широко применяют в производстве легированных алей, а также для получения карбида вольфрама, износостойких лавов и нитей для ламп накаливания и вакуумных ламп.
4-6 Лоедннениях вольфрам находится в степенях окисления 4*2 и ляетс аиб°лее Устойчивой и обычной, особенно для анализа, яв-л0Чн л степень окисления +6. В слабокислых, нейтральных и ще-в аннХ Растворах вольфрам(VI) находится преимущественно иной форме. Химия растворов вольфрама сложна в связи
233
с тенденцией к образованию вольфрамсодержащих поликислот. В растворах с pH = 8 превалирует ион WO}’, а при pH = 4 — 6 анионы гексавольфрамата. При концентрации НС1 выше 4 М вольфрам (VI) образует хлоридные комплексы.
Проблемы аналитической химии вольфрама рассмотрены в монографии Лингана [1]. Стандартные растворы вольфрама (VI) обычно готовят из вольфрамата натрия Na2\VO4-2H2O или чистого триоксида вольфрама.
Опубликован обзор по аналитической химии вольфрама [2—4]. Обзор некоторых аспектов определения вольфрама приведен ниже.
МЕТОДЫ ОТДЕЛЕНИЯ
Вольфрам можно отделять методами осаждения, ионного обмена, жидкостной экстракции, бумажной и тонкослойной хроматографии, эле1 трофореза на бумаге и методом соосаждения. В работе [5] приведен обзор основных методов отделения вольфрама.
В последние годы для отделения следов вольфрама применяют экстракционные методы, которые используют во многих методиках анализа.
Осаждение
Алкалоид цинхонин осаждает вольфрамат из растворов минеральных кислот. Определению мешают мышьяк^) и фосфор(V), образующие комплексы с цинхонином. Осаждается также кремний. Осаждение цинхонином позволяет отделять микросодержания вольфрама, в том числе от молибдена(VI). Этот метод используют для определения вольфрама в стали [6]. "Отделение проводят путем гидролиза с введением цинхонина как дополнительного осадителя.
Рекомендованы также аналогичные осадители, например бензоин а-оксим, но при этом осадок, как правило, экстрагируют органическим растворителем для отделения от других осаждаемых элементов.
Ионообменный метод
Для отделения вольфрама(VI) применяют катионный и анионный обмен. Как и молибден(VI), вольфрам(VI) не поглощается на сильнокислотных катионитах, что позволяет отделить вольфрам (VI) от мешающих катионов металлов, сорбируемых на катионите [7]. Катионный обмен применен для отделения вольфрама от ряда элементов [8] перед флуориметрическим определением.
Вольфрам^!) сорбируется на анионитах как в отсутствие, так и в присутствии комплексующих агентов, например тиогликолевой кислоты. Описан метод разделения VV71, Rev11 и MoVI на слабоос-повном анионите (диэтиламиноэтилцеллюлозе) в роданидных растворах [9]. Метод применен для определения молибдена и вольфрама в металлическом рении [10],
234
Экстракция
Для экстракции вольфрама используют несколько способов. Дитиол (толуол-3,4-дитпол) применен для отделения Мо и W [11]. В среде 10 М НС1 в присутствии восстановителей вольфрам образует интенсивно окрашенный комплекс, который можно экстрагировать эфирами, например этилацетатом. Вольфрам(VI) отделяется от Со, V, Sn, Pb, Ni, Мп и А1, однако соэкстрагируются малые содержания железа, а молибден экстрагируется полностью, как и вольфрам. Метод может быть применен для спектрофотометрического определения вольфрама и молибдена, в частности, при анализе диоксида титана [12].
Другим важным экстрагентом является бензоин а-оксим, который осаждает вольфрам (VI) и молибден (VI). Образующиеся комплексы экстрагируют хлороформом [13,14]. Для полного выделения вольфрама экстракцию проводят несколько раз. Ванадий и хром также экстрагируются, но их можно замаскировать. Как и молибден, вольфрам экстрагируется из кислых растворов в присутствии восстановителей и роданида, но хуже, чем молибден. Этот метод используют для отделения вольфрама, предшествующего его гравиметрическому определению с помощью тетрафениларсоний-хлорида [15]. Вместо обычного восстановителя — хлорида олова (II)— применена ртуть, а комплекс экстрагируют раствором трибензиламина в хлороформе. Вольфрам реэкстрагируют слабощелочным раствором, содержащим пероксид водорода, для разрушения избытка роданида и окисления вольфрама до WVI. Метод позволяет отделять менее 50 мг вольфрама, но не рассчитан на отделение микросодержаний вольфрама.
Описаны другие экстракционные методы отделения вольфрама: с помощью оксида мезитила [16], 8-гидроксихинолина [17] и бензогидроксамовой кислоты [18]. Описан метод отделения вольфрама в виде фосфоровольфрамата [26]. Экстракцию проводят метилизобутил кетоном из 0,1 — 1 М раствора НС1, что позволяет отделить 10—100 мг вольфрама от Fe, Ni, Со, Сг, Мп, Си, Са, U, Th, As, Sb, Bi и Si. Возможность экстракции меньших содержаний зависит от соотношения W и Р.
Методы бумажной и тонкослойной хроматографии
В табл. 17 приведены данные о наиболее важных методах отделения вольфрама.
Соосаждение
Как и молибден(VI), вольфрам (VI) соосаждается на гидро-ВоВДе железа [27]. В интервале pH = 5 — 8 можно разделить воды [28] И Рен1™‘ Метод применен для анализа морской кислРеДЛг^е,,Ь1 11 дРУгие носители, в том числе метаоловянная та [29] и молибдофосфат [30].
235
Таблица 17. Отделение вольфрама(У1) методами тонкослойной (ТСХ) и бумажной хроматографии (БХ)
Метод Растворитель Отделяемые иоиы Литература
БХ Наиболее эффективна смесь Mo, Т1, Th, Mg, uo’+, 19
70%-ный раствор HNO3 — «-бутанол (6; 4) V, Со, Ni, Zn, Al, Fe, nh;
БХ Этанол — СНС13 — 10%-ный раст- MoO’~ 20
вор Н2С2О4 (7:2:1)
ТСХ (алю- 1-1,16 М Н3РО4 ReO,", MoOf, VO" 21
могель) 22
ТСХ (силн- 1-1,8 М NH«SCN ReO" или MoO*"
кагель со
связующим) ТСХ (силн- Метанол — 3 М НС1 (7 : 3) нли ReO* или MoOj" 23
кагель) метанол — 1 М NH4NO3 — 3 М NH4OH (14 : 5 ; 1)
ТСХ Кислый раствор бутанола н амн- MoOf, vo; 24
лового спирта
Примечание. В работе [20] применен также метод электрофореза на бумаге Этот метод применен (25] для разделения вольфрама (VI) н хрома (VI).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
В последнее время гравиметрические методы применяют все реже, за исключением тех случаев, когда необходим высокоточный анализ или при отсутствии других эффективных методов. Вольфрам часто определяют гравиметрически, поскольку титриметрические методы определения вольфрама малоэффективны.
Осаждение вольфрама цинхонином было описано в разделе «Методы отделения». Гравиметрическому определению вольфрама с цинхонином мешают Asv, Pv и Si, не мешает Movl. Комплекс WV! с цинхонином гигроскопичен. Для перевода в WO3 осадок прокаливают при 700—850 °C [6]. Вольфрам в среде HNO3 образует с пероксидом водорода пероксовольфрамат, который при нагревании разлагается с выделением осадка вольфрамовой кислоты. Осадок фильтруют, прокаливают при 700—850°C и взвешивают в виде \VO3 [31]. Для осаждения H2WO4 в методике рекомендуется нагревание раствора до 60 °C. В работе [32] показано, что полное осаждение достигается при 80 °C.
Изучена эффективность применения некоторых органических оснований для осаждения вольфрама в виде фосфоровольфрамата. Показано, что трибутиламин как осадитель обладает рядом преимуществ. Методика заключается в добавлении раствора вольфрама (VI) к смеси Н3РО4 — HCI при pH = 2. Затем приливают раствор три-н-бутиламина солянокислого. Осадок легко фильт
236
руется и не прилипает к стенкам стеклянного сосуда, реагент сравнительно недорог. Если осадок [(C4H9)3NH] P\V12O40 сушить при 105 °C, результаты анализа будут недостаточно точными. Большая точность достигается при температуре сушки 195—220 ’С. Вместо НС1 можно использовать азотную или серную кислоту, однако НгЬОч препятствует осаждению вольфрама. Поэтому осадок с раствором следует оставить на ночь и затем отфильтровать. Хлорная кислота, как и при осаждении молибдена и ванадия, мешает. Не мешают значительные содержания Са, Со, Си, Мп и Ni (хлориды), Pb(NO3)2 и большие концентрации NaCl, NaNO3 и Na2SO4. Метод позволяет определять 10—200 мг вольфрама(VI). Методика определения приведена ниже.
Вольфрамат можно определять гравиметрически в виде вольфрамата серебра путем осаждения из гомогенного раствора [34]. Описан новый метод анализа вольфраматов щелочных металлов, основанный на количественном переведении их в хлориды щелочных металлов в токе НС1 при 500 °C [35]. Содержание вольфрама определяют по разнице весов до и после пропускания НО.
Другие гравиметрические методы основаны на применении 8-гидроксихинолина [36], вариаминового голубого [37] и тетра-фениларсонийхлорида [15]. Последний метод включает предварительную экстракцию трибензиламином и может быть применен для определения вольфрама в присутствии ванадия и титана. Метод рассчитан на определение 5—60 мг вольфрама, он применен для анализа промышленных проб в основном сплавов. Степень экстракции несколько снижается в присутствии хлорида и ацетата и существенно уменьшается в ряду SO4’, РО4', цитрат, тартрат, оксалат.
Другие гравиметрические методы определения вольфрама описаны в обзорах [2,3].
В работе [32] проведено сравнительное изучение трех гравиметрических и одного периметрического метода определения макросодержаний вольфрама (VI). Сравнение гравиметрических методов дано в табл. 18.
Таблица /81. Результаты трех гравиметрических методов определения вольфрама(У1) [32]
Метод Найдено вольфрама, % Стандартное отклоиеиие Число определений
Осаждение три-н-бутиламином [33] азл°жение пероксовольфрамата (модифицированный метод [31]) Осаждение бензидином нитрата^вопил4 а н и я *• Стандартный раство 2 MeV Трама, содержание основного веиц 3. Стяип? пРименены для определения 50-ные определен гное отклоиеиие получено с уче 100,24 99,52 99,96 вольфрама пс ;ства в котором -100 мг вольф р< том всех резу 0,055 0,093 0,070 >лучен из спектр приняли равные 1ма. льтатов, в ключа 4 6 5 альио чистого 100%. я дополнитель-
237
К сожалению, единственным методом, который не дает систематической ошибки, является метод с применением бензидина, который является канцерогенным. Молибден не мешает определению вольфрама только при использовании метода, основанного на разложении пероксовольфрамата.
Определение вольфрамата с помощью три-н-бутиламина [32, 33]
Реагенты
Трибутиламин, очищенный дистилляцией под давлением 4 Па. Отбирают фракцию 56,5—60 °C.
Трибутиламин солянокислый (исходный). Встряхивают 1 мл реагента с 40 мл 0,25 М раствора НС1, получают 2%-ный раствор, годный для анализа в день приготовления.
Промывной 0,1%-ный раствор трнбутиламина солянокислого готовят ежедневно разбавлением исходного раствора в 20 раз.
Ход анализа. К 50 мл раствора, содержащего 10—200 мг вольфрама (VI), добавляют не менее 85 мг фосфата в виде Na2HPO4 и растворяют при перемешивании. Продолжая перемешивание, постепенно приливают раствор НС1 (1:1) до pH — 2 и еще 4 мл избытка кислоты. Разбавляют раствор водой до объема 100 мл.
Нагревают раствор почти до кипения, приливают по каплям, при перемешивании, 10 мл раствора трнбутиламина солянокислого и продолжают нагревать раствор до коагуляции осадка. Раствор с осадком охлаждают и оставляют на ночь, затем фильтруют через мелкопористый стеклянный фильтр и промывают осадок минимальными объемами охлажденного 0,1%-ного раствора трибутил-амина солянокислого, а затем охлажденной водой. Осадок сушат 2 ч при 210 "С, охлаждают в эксикаторе и взвешивают. Коэффициент пересчета на содержание вольфрама равен 0,6424.
Титриметрические методы
Как указано выше, титриметрические методы определения воль фрама, как правило, неэффективны. В одном из обзоров аналитической химии вольфрама [4] обсуждены методы, основанные на осаждении и образовании комплексов вольфрама.
Метод осадительного титрования
Описан метод, основанный на осаждении вольфрамата свинца. В работе [38] сумму вольфрама (VI) и молибдена (VI) определяют титрованием свинцом (II) в гексаминовом буферном растворе в присутствии индикатора ПАР.
Молибден определяют отдельно с помощью ЭДТА. Определению вольфрама(VI) не мешает до 50 мг сульфата и 200 мг фторида [39]. Кроме ПАР можно применять другие индикаторы [40].
В работе [41] сделана попытка улучшить методику [38] путем применения индикатора ксиленолового оранжевого, варьирования pH и введения молибдена (чтобы достичь отношение Movl: : WVI > 1).
Опубликован меркуриметрический метод, в котором конечную точку титрования устанавливают флуориметрически [42].
238
Комплексометрический метод
Вольфрам непосредственно не титруется раствором ЭДТА. В работе [43] к раствору вольфрама в сильнокислой среде добавляли избыток ЭДТА, затем устанавливали pH = 5 с помощью ацетатного буферного раствора и титровали избыток ЭДТА раствором Т1111, применяя в качестве индикатора ксиленоловый оранжевый. Конец титрования плохо индицируется [44].
Описан косвенный метод, основанный на осаждении вольфрама (VI) ионами свинца или кальция. В работе [45] определяли содержание кальция в осажденном CaWO4. Вольфрам (VI) можно осаждать ацетатом свинца(II) [46]. Избыток свинца титруют раствором ЭДТА с помощью ксиленолового оранжевого [46[. Аналогичный метод описан в работе [47].
Другие титриметрические методы
Редокс-методы не нашли широкого применения для титрования вольфрама. Вольфрам(VI) количественно восстанавливается до Wv хромом(II) или ванадием (II) в среде концентрированной НС1 [48]. Вольфрам (V) образуется при взаимодействии WVI и W111 с вероятным промежуточным состоянием W1V. При потенциометрическом титровании вольфрама(VI) раствором W111 образование W[V прямо не индицируется, однако видно, как возникает и исчезает синяя окраска, характерная для WIV. Растворами хрома(П) и ва-надия(П) можно титровать вольфрам^!), определению мешают ортофосфаты.
Вольфрам (VI) можно восстановить до Wv путем облучения раствора светом ртутной лампы в течение 30 мин [49]. Образующийся Wv определяют фотометрически, с помощью роданида или титрованием раствором NH4VO3 с фенилантраниловой кислотой в качестве индикатора. Ошибка определения 7,5—10 мг вольфрама составляет ±2%.
Описан титриметрический метод определения вольфрама(VI), хрома(VI) и молибдена (VI), основанный на образовании ионной пары с Ва2+. В качестве индикатора применяют нитхромазо. При взаимодействии вольфрамата с барием образуется Ba(HWO4)2. Реакция протекает количественно в среде 80—90% ацетона при значении pH = 4,0 — 5,5 (исходный раствор). Метод позволяет определять 10~5—10-3 моль/л вольфрама.
Спектроскопические методы
Спектрофотометрические методы
ОпРедеИб°Лее РаспР0СТРаненный спектрофотометрический метод них к"еНИЯ вольФРама(VI), основанный на образовании роданид-мплексов, описали впервые в 1932 г. Файгль и Крумхольц
239
[51]. Слабощелочной раствор вольфрамата в присутствии роданида и хлорида олова(II) окрашивается в желтый цвет. Молибден (VI) не мешает определению. В последние годы проведены исследования с целью улучшения точности и селективности метода [52].
В некоторых работах указывалось, что этот метод неприемлем В ранних исследованиях рекомендовали с целью получения точных результатов вводить роданид после восстановления вольфрама до вольфрама (V). Более того, отмечалось, что возможно образование нескольких комплексов вольфрама (V) с роданидом с разными значениями е. В детальном исследовании [54] найдены условия достижения удовлетворительных результатов.
В литературе описано три варианта методики определения вольфрама. Поглощение комплекса Wv с роданидом можно измерять в водно-ацетоновой среде. Комплексы можно экстрагировать непосредственно или в виде ониевой соли, например, с тетрафениларсо-нием. Показано [53], что метод экстракции роданидных комплексов некоторых металлов в виде ониевых солей обладает преимуществами по сравнению с экстракцией комплексных роданидных кислот.
Нерастворимую в воде соль тетрафениларсония с вольфрам (V)-роданидным комплексом можно экстрагировать хлороформом [55]. Это использовано в работе [56], где вольфрам восстанавливают до Wv с помощью SnCU и титана (III) и экстрагируют тройной комплекс хлороформом, содержащим гидрохинон для предотвращения окисления вольфрама. Молярный коэффициент поглощения комплекса равен 1,47-104. Метод применен для определения вольфрама в стали. Допустимые содержания молибдена зависят от концентрации железа в растворе.
Опубликован метод, в котором восстановителем является ртуть, а экстракцию проводят 2%-ным раствором трибензиламина в хлороформе [57]. Определению не мешают U, Ti, V, Сг, Fe, Со, Ni, Мп, Al, Pb, Sn, Bi, Pd, Sb и Си. Мешают равные содержания Pt и Мо.
При определении вольфрама роданидным методом применяют разные приемы устранения влияния мешающих ионов. Например, вольфрам и молибден экстрагируют бензоин а-оксимом [58]. Железо экстрагируют метилизобутилкетоном, а другие ионы — купфероном [59].
Несмотря на недостатки, роданидный метод широко применяют для определения вольфрама, особенно в металлургических анализах.
Толуол-3,4-дитиол («дитиол») образует с вольфрамом (VI) слаборастворимый сине-зеленый комплекс [60]. Аналогичный комплекс образует молибден(VI). Окрашенные комплексы с дитиолом дают некоторые металлы, в том числе Си, Sn и Bi; эти комплексы можно отделять экстракцией. Основным недостатком дитиола является его неустойчивость. Описан метод получения более устойчивой формы
240
реагента [61], которую можно хранить в течение нескольких недель в холодильнике.
При определении вольфрамата в присутствии молибдена вольфрам (VI) можно замаскировать лимонной кислотой, отделить комплекс молибдена экстракцией. Затем после демаскирования определяют вольфрам [13].
Для повышения избирательности и устранения влияния молибдена в раствор вводят ЭДТА [62]. Этот реагент применен для определения в сталях и сплавах от 0,02 до 0,25 мг вольфрама (в конечном объеме 50 мл) [63]. В работе [64] предложен метод определения вольфрама в присутствии больших содержаний молибдена (VI) и отмечены преимущества метода по сравнению с роданидным.
Флуориметрические методы
По красной флуоресценции комплекса вольфрама (VI) с карминовой кислотой можно определять вольфрам в интервале содержаний 0,04—0,36 ppm [65]. Максимум спектра флуоресценции комплекса — 585 нм, максимум возбуждения — 515 нм. Определению мешают многие ионы, но влияние некоторых можно устранить введением аскорбиновой кислоты. Мешающие ионы отделяют.
Флаванол при pH = 2,5—5,5 образует с вольфрамом (VI) флуоресцирующий синим светом комплекс, что позволяет определять 6—42 мкг вольфрама в 100 мл раствора [66]. Определению мешают небольшие содержания железа, ванадия и хрома. Не мешают значительные концентрации никеля, марганца и меди. В работе [8] описано ионообменное отделение вольфрама, предшествующее его определению.
Атомно-абсорбционный метод
Чувствительность прямого определения вольфрама методом ААС невелика. Наиболее интенсивными являются линии 255,1, 268,1 и 294,4 нм, но для анализа используют обычно линию 400,9 нм ввиду оптимального соотношения сигнал : фон при этой Длине волны.
Предложен косвенный метод [67], заключающийся в осажде-"ИИ АольФрамата свинцом и определении избытка свинца методом ААС. Метод позволяет определять 3—30 ppm вольфрама. Определению 8 ppm вольфрама мешают А13+, У0з, СгОГ и МоО?'. е мешают 10 ppm Fe2+ и Мп2+.
экс ОльФРам определяют методом атомной абсорбции после весь₽аКЦИИ вольФрам (V) -роданидного комплекса [68]. Метод скихМЭ' из®иРателен‘. нашел применение для анализа геологиче-
24}
Электрохимические методы
Потенциометрические методы
Вольфрам (VI) можно титровать потенциометрически нитратом серебра. При использовании серебряного индикаторного и насыщенного каломельного электродов [69] наблюдается скачок на кривой титрования, соответствующий образованию Ag2\VO4. Для титрования стандартного раствора AgNO3 раствором Na2WO4 применен электрод с мембраной, импрегнированной дитизоном [70]. Обратное титрование вольфрамата нитратом серебра осуществить не удается.
Показано [71], что вольфрамат натрия в присутствии маннита можно титровать потенциометрически 0,05 М раствором H2SO4. После присоединения одного протона к вольфрамату наблюдается резкий скачок потенциала.
Описан метод потенциометрического (а также кондуктометрического и амперометрического) титрования вольфрама (VI) свинцом (II) [72] при строг/) контролируемом интервале pH = 6,5— 7,5. В качестве индикаторного применен вольфрамовый электрод. Минимальная концентрация титруемого вольфрама (VI) составляет 10-4 М. Определению мешают катионы, образующие нерастворимые вольфраматы, например Ag1, Си11, TiIV и Hg11, а также анионы, взаимодействующие со свинцом(II). Мешают также большие концентрации нейтральных солей и комплексообразователи, например ацетат, тартрат, оксалат, цитрат и фосфат.
Амперометрические методы
Вольфрам и молибден при совместном присутствии можно титровать амперометрически раствором Pb(NO3)2 в ацетатном буферном растворе с pH = 4,5. Сначала отделяют вольфрам кислотным гидролизом в присутствии желатина. Применяют платиновый и насыщенный каломельный электроды [73].
Полярографические методы
Ранние работы по полярографическому определению вольфрама описаны в монографии Кольтгофа и Лингана [74]. В среде 12 М НС1 вольфрам (VI) ступенчато восстанавливается до Wv и W'“ [75]. Потенциал второй волны Еу* ——0,52 В (н. к. э.) 11 сдвигается в отрицательную область при снижении концентрации НС1.
Изучено поведение вольфрама (VI) на капающем ртутном электроде в присутствии различных кислот [76]. В среде НС> возможно определение IO-3 М вольфрамата, в сильнокислой сре' де, в присутствии Н3РО4 полярографический ток пропорционален концентрации вольфрамата до содержания 4-Ю3 М.
Вольфрам определяли полярографически после отделения 6
242
виде фосфовольфрамованадатного комплекса [77]. В среде серной кислоты потенциал полуволны равен —0,52 В (н к. э.). Чувствительность определения вольфрама в конечном растворе составляет 0,04 мг/мл.
Вольфрам определяют [78] по кинетической полярографической волне в среде H2O2— щавелевая кислота на капающем ртутном электроде при потенциале 4-0,233 В. Даны условия определения 5—20 и 0,04—0,25 мк.Ч вольфрамата. Определению мешают Crvl, Fe'“, MoVI, Ti,v и Vv,
Другие методы определения
Описан ряд каталиметрических методов определения вольфрама. Реакция окисления иодида пероксидом водорода применена для определения вольфрама методом фиксированной концентрации [79]. Восстановление красителя основного синего К тита-ном(Ш). катализируемое вольфрамом(VI), фиксируют спектрофотометрическим методом при 445 нм [80].
В работе [81] для определения вольфрама в сталях и алюми-* нии применен метод нейтронной активации и изотопного разбавления. Метод изотопного разбавления основан на экстракции вольфрама субстехиометрическими содержаниями толуол-3,4-дитиола в хлороформе. В нейтронно-активационном методе пробу облучают потоком нейтронов 3,6-1012 нейтронов/(см2-с) в течение 4 ч. Затем измеряют у-излучение изотопов 185W и 187W.
and
ЛИТЕРАТУРА
1. Lingane J. J.— Analytical Chemistry of Selected Metallic Elements, New and London, Reinholt, 1966, p. 113.
2. Chalmers R. A. — In: Comprehensive Analytical Chemistry (Ed. C. L.
D. W. Wilson), Vol. 1C, Amsterdam, Elsevier, 1962, p. 598.
3. Elwell W. T., Wood D. F. — Analytical Chemistry of Molybdenum and Tungsten, Oxford, Pergamon Press, 1971.
4. Lowenheim F. A. — In: Encyclopedia of Industrial Chemical Analysis (Ed. F. D. Snell and L. S, Ettre), Vol. 19, New York and London, Interscience, 1974, p. 190.
5. Korkisch J. — Modern Methods for the Separation of Rarer Metal Ions. Oxford, Pergamon Press, 1969, p. 476.
” Bolish Standards Handbook No. 19, Method 1 for tungsten in steel London, British Standards Institute, 1970.
' Gottschalk G. — Z. analyt. Chem., 1962, v. 187, p. 164; Analyt. Abstr., 1962,
8 H 4173
• offei R. s., Trusk B. A. — Analytica chim. Acta, 1967, v. 37, p. 409. 1968, q Y-41, p. 374.
10 3 K ' Kuroda R — Analyt. Chem., 1964, v. 39, p 212.
’ io7q Kasuaki K. — Z. analyt. Chem., 1972, v. 261, p. 394; Analyt. Abstr., П >973, v. 24, p. 2809.
12' H — Analyst, 1940, v. 65, p. 152.
13 Jpftr 1 — Chemist Analyst, 1958, v. 47, p. 68.
14. Plpih>y Pv c —Analyst, 1956, v. 81, p. 104.
15. Yatirnip \,Hecht F-~Z analyt Chem., 1960, v. 177, p. 175
16. Shimko .7 D,hamiia S. - Talanta, 1976, v. 23, p 599.
v. M., Khopkar S. M. — Talanta, 1969, v. 16, p. 525.
York
243
17. Eberle A. R. — Analyt. Chem., 1963, v. 35, p 669.
18 По гуектова E. H., Назаренко В. A.— ЖАХ, 1967, т 22, с. 746; Analyt. Abstr 1969, v. 16, p. 130.
19. Quershi AL, Khan F. — J Chromat., 1968, v. 34. p. 222.
20. Blasius E., Czekay A. — Z. analyt. Chem., 1957, v. 156, p. 81; Analyt. Abstr 1957, v. 4, p. 3312.
21. Гайбакян Д. C. — Ученые записки Ереванск. roc. ун-та. Естеств. науки, 1971 с. 71; Analyt. Abstr., 1972, v. 23, р. 2293.
22. Shiobara Y.—Japan Analyst, 1968, v. 17, p. 1396; Analyt. Abstr., 1970, v. 19 p. 202.
23. Kuroda R., Kawabuchi K-, Ito T.— Taianta, 1968, v. 15, p. 1486.
24. Гайбакян Д. C. — Армянский хим. ж., 1969, т. 22, с. 13; Analyt Abstr., 1970 v. 18, р. 2975.
25. Blum D. — Rev. Chim. Bucharest, 1958, v. 9, p. 28; Analyt. Abstr., 1958, v. 5 p. 3712.
26. Yatirajam V., Dhamija S. — Taianta, 1977, v. 24, p. 52.
27. Новиков А. И.—ЖАХ, 1960, t. 15, c. 742; Analyt. Abstr., 1962, v. 9 p. 1903.
28. Ishibashi M., Fujinaga T., Kuwamoto T., Koyama M., Sugbayashi S.—Japan Analyst, 1960, v. 81, p. 392.
29. NIshida H. — Japan Analyst, 1964, v. 13, p. 760; Analyt. Abstr., 1966, v. 13, p. 4131.
30. Тарасевич H. И., Хлыстова А. Д., Пак E. A. — Зав. лаб., 1959, т. 25, с. 955; Analyt. Abstr., 1960, v. 7, p. 2205.
31. Dams R., Hoste J. — Taianta, 1961, v. 8, p. 664.
32. Crossland B., Fennell T. R. F. W. — Analyst, 1969, v. 94, p. 989.
33. Miller С. C., Thow D. H. — Analyst, 1959, v. 84, p. 440.
34. Varughese K-, Rao K- S.— Analytica chim. Acta. 1971, v. 57, p 219.
35. Lutz C. W„ Conroy L. E. — Analyt. Chem., 1966, v. 38, p. 139.
36. Виноградов А. В., Дронова M. И. — ЖАХ, 1968, t. 23, c. 696; Analyt. Abstr., 1970, t. 18, p. 176.
37. Erdey L., Buzas I., Vigh K.— Taianta, 1967, v. 14, p. 515.
38. Lassner E., Scharf R. — Chemist Analyst, 1960, v. 49, p. 68.
39. Lassner E., Schedle H. — Taianta, 1966, v. 13, p. 326.
40. Brantner H. — Mikrochim. Acta, 1962, p. 125.
41. Cheng K. L., Goydish B. L.—Microchem. J., 1968, v. 13, p. 35.
42. Андрушко Г. С., Максимычева 3. Т., Талипов Ш. Т. — ДАН Узбек. ССР, 1970, с. 26; Analyt. Abstr., 1971, v 21, р. 173.
43. Kinnunen J., Wennerstrand В. — Chemist Analyst, 1958. v. 47, p. 38.
44. Schwarzenbach G., Flaschka H. — Complexometric Titrations, 2nd English edn, London, Methuen, 1969, p. 226.
45. De Sousa A. — Analytica chim. Acta, 1953, v. 9, p 309.
46. Попова О. И., Серая О. Г. — ЖАХ, 1968, т. 23, с. 791; Analyt. Abstr., 1970 v. 18, р. 177.
47. Bourvet Р., Lecuire L-М., Weis С. — Chim. analyt., 1970, v. 52, p. 1114; Analyt Abstr., 1971, v. 21, p. 1807.
48. Geyer R., Henze G. — Wiss. techn. Hochsch. Chem. Leuna-Merseburg, 1960/1. v. 3, p. 261; Chem. Abstr., 1961, v. 57, p. 1542.
49. Немодрук А. А., Безротова E. B. — ЖАХ, 1969, t. 24, c. 404; Analyt. Abstr-1970, v. 19, p. 2221.
50. Кремнев А. П., Кузнецов В. В., Межлумян П. Г. — ЖАХ, 1974, т. 29, с. 1349-Analyt. Abstr., 1975, v. 29, ЗВ 138. ,
51. Feigl F., Krumholz P. — Angew. Chem., 1932, v. 45, p 674; Chem. Abstr., 1933-v. 27, p. 243.
52. Fogg A. G., Marriott D. R., Burns D. T. — Analyst, 1970, v. 95, p. 848.
53. Bowd A. J., Burns D. T., Fogg A. G. — Taianta, 1969, v. 16, p. 719.
54. Crouthamel С. E., Johnson С. E. — Analyt. Chem., 1954, v. 26, p. 1284. .
55. Affsprung H. E., Murphy J. W. — Analytica chim. Acta, 1964. v. p. 401.
56. Fogg A. G., Marriott D. R., Burns D. T. — Analyst, 1970, v. 95, p. 854-
57. Yatirajam V., Dhamija S. — Taianta, 1975, v. 22, p. 760.
244
eg. Peng P. Y., Sandell E. В —Analytica chim. Acta, 1963, v 29, p. 325.
59. Luke C L. — Analyt. Chem., 1964. v. 36, p 1327
gO Sandell E. V. — Colorimetric Determination of Traces of Metals, 3rd edn, New York, Interscience, 1959.
gl. De Silva E. E. AL S.— Analyst. 1975, v 100, p 517.
g2 Бусев А. И., Соколова T. A.—ЖЛХ, 1968, t. 23, c. 1348; Analyt. Abstr., 1970, ’ v. 18, p. 2382
53. Пащенко E. H., Мальцева E. Ф.— Зав. лаб., 1968, т. 34, с. 12.
64. Лебедева Л. И.. Голубцова 3. Г., Янкович Н. Г. — ЖАХ, 1971, т. 26, с. 1962; Analyt. Abstr., 1973. v. 24, р. 1548.
65. Kirkbright G F., West T. S., Woodward C. — Taianta, 1966, v. 13, p. 1637.
66. Bottel R. S., Trusk Br. A. — Analyt. Chem., 1963, v. 35, p. 1910.
67. Chong R. №.. Boltz D. F. — Analyt. Lett.. 1975, v. 8, p 721.
68 Kim С. H., Alexander R. W., Smythe L E. — Taianta, 1976, v. 23, p. 573
69 Shivahare G. C. — Naturwissenschaften, 1965, v. 52, p 157; Analyt. Abstr., ’ 1966, v- 13, p. 4132.
70 Lal S. — Z. analyt. Chem., 1971, v. 255, p. 210; Analyt. Abstr., 1972, v. 22, ’ p. 3121.
71. Sinclair A. G. — Taianta, 1969, v. 16, p. 459.
72. Gupta С. M. — J. Proc. Inst. Chem India, 1966, v. 38, p. 211; Analyt. Abstr., 1968, v. 15, p. 156.
73. Боговина В. И., Новак В. П., Мальцев В. Ф. — ЖАХ, 1965, т. 20, с. 951; Analyt. Abstr., 1967, v. 14, р. 2559
74. Kolthoff I. M„ Lingane J. J. — Polarography, 2nd edn, Vol II, New York and London, Interscience, 1965, p. 461.
75. Lingane J. J., Small L. A. —J. Am. chem. Soc., 1949, v. 71, p. 973
76. Issa R. M, Abd-El-Nabey, Hendawey A. M. — Z. analyt. Chem., 1968, v. 240, p. 9; Analyt. Abstr., 1970, v. 18, p. 178
77. Полотебнова H. A , Данилина JI. M., 1970, t. 36, c. 261.
78. O’Shea T. A., Parker G. A.—Analyt. Chem.. 1972, v. 44, p 184.
79. Hadjiioannou T. P., Valkana C. G. — Hem. Hron., 1967, v. 32, p. 89.
80. Омарова E. С., Сперанская E. Ф., Козловский M. T. — ЖАХ, 1968. t. 23, c. 1826; Analyt. Abstr., 1970, v. 18, p. 3947.
81. Baishya N. K., Heslop R. B. — Analytica chim. Acta, 1971, v. 53, p. 87.
с его вана-
ВАНАДАТЫ
Многие проблемы аналитической химии ванадия связаны применением в черной и цветной металлургии. Соединения Дня используют также в производстве катализаторов, красок и керамических изделий.
В растворах обычно присутствует несколько ионных форм ва-надия, связанных между собой рядом равновесий. В слабокислой среде доминируют полиядерные комплексы. В сильнокислой среде превалирует ион ванадила VO2. Ванадий (V) является окисли-едем, который можно сравнивать с бихроматом. В щелочных Растворах также преобладают полиядерные комплексы, а в силь-^елочной среде доминирует ион VO?'. Обзор химии ванадия, ган3аИт” С его повеДением ПРИ анализе, дан в монографии Лин-(2~_к1 Аналитическая химия ванадия рассмотрена в обзорах та а ’ РаствоРы ванадия (V) можно приготовить из метаванада-V2O5 или ванадата натрия. Для более станл'я Раб°т Растворы, полученные из ванадата натрия, обычно ртизируют с помощью купферона.
245
МЕТОДЫ ОТДЕЛЕНИЯ
Методы отделения ванадия подробно рассмотрены в моногра фин Коркнша [6].
>
Осаждение
Осаждение ванадия (V) купфероном из сильнокислых рас творов позволяет отделить его от Uvl, Сг111, Мп11, AI111 Ni", Zn" и Р. В тех же условиях осаждаются некоторые другие элементы, особенно Fe, Мо, W и Ti.
Используя растворимость ванадия (V) в сильнощелочной сре де, можно отделить его от Fe, Со, Ni, Ti, Zr и других элементов соединения которых нерастворимы в щелочах; И, Мо, Crvi и А1 переходят в раствор вместе с ванадием(V). 8-Гидроксихннолин осаждает ванадий(V), но не хром (VI).
Хроматографические методы
Ванадий(V) можно отделить методом катионного и анионно го обмена. Ванадий (V) отделяют- от Fe1", Со, Ni, лантаноидов Al, Zn, Bi и других ионов на катионите Дауэкс-50, используя в качестве элюента 0,1 М растворы H2SO4 или НСЮ4, содержащие Н2О2. Пероксокомплексы ванадия (V) элюируются, в то время как другие элементы сорбируются [7].
Некоторые методы основаны на адсорбции ванадия и элюиро вании сопутствующих ионов. Для этого восстанавливают вана дий(У) с помощью SO2 до ванадия (IV), который значительно луч ше сорбируется на катионите. Этот метод позволяет отделить ванадий от больших количеств ортофосфата [8].
Описан метод отделения Vv от Mg, Са, Zn, Cd, Со и Ni на анионите в ПО3'-форме [9].
Ванадий (V) отделяют от Movl, WV1 и Fe"1 на колонке, запел ненной А120з. В качестве элюента используют раствор гидрокар боната калия, содержащий Н2О2. Ванадий(V) элюируется, < сопутствующие ионы сорбируются [10]. Показано, что можно разде лить ванадий(V) и (IV) на колонке, заполненной частицами фто ропласта, используя в качестве стационарной фазы насыщенный раствор бензогидроксамовой кислоты в трибутилфосфате [И] Неадсорбированный ванадий (IV) удаляют 5 М раствором Н3РО4 Ванадий (V) элюируют концентрированной Н3РО4.
Тонкослойная хроматография на алюмогеле позволяет разде лить VO3, ReOi, МоО4’[12]. Элюентом служит 1 —1,16 М рас твор Н3РО4. Применение газохроматографических методов описа но ниже.
Экстракция
Жидкостная экстракция является, вероятно, наиболее распр0 страненным методом отделения ванадия (V). Используют экстра* цию хелатов и ионных ассоциатов, часто в комбинации с после
246
дующим спектрофотометрическим или атомно-абсорбционным определением.
Ванадий (V) экстрагируют 8-гидроксихинолином, купфероном, диэтилднтиокарбаматами, N-бензоилфенилгидроксиламином, 4- (2-пиридилазо)-резорцином (ПАР) и бензоин а-оксимом. Ниже будут обсуждены практически наиболее важные экстракционные системы. Детально методы экстракции ванадия описаны в монографии Коркиша [6].
Экстракция комплекса ванадия(V) с 8-гидроксихинолином хлороформом при pH = 4—5 позволяет отделить ванадий (V) от хрома(VI). При анализе почв ванадий отделяют от Movl путем экстракции хлороформом комплекса с 8-гидроксихинолином при pH = 3,5—4,0 [13]. Купферон также является эффективным экстрагентом ванадия [14]. В работе [15] показано, что купферон является лучшим среди реагентов, образующих хелаты, с точки зрения оптимального значения pH и избирательности. Ванадий (V) полностью экстрагируется из 1 М раствора НС1 метилизобутил кетоном. Этот растворитель предпочтителен, если ванадий затем определяют атомно-абсорбционным методом. Определению не мешают ионы любых металлов при их 100-кратном избытке относительно ванадия.
Желтый комплекс ванадия (V) с бензоин а-оксимом можно экстрагировать хлороформом при pH = 2,2 и экстракт фотометри-ровать [16]. Этот спектрофотометрический метод применен для определения ванадия в стали, содержащей хром [17]. Для восстановления Crvl до Сг111 и Vv до Vlv применен Na2S20s, затем Vlv окисляют до Vv с помощью КМпО4, не затрагивая Сг111. Наконец, Vv экстрагируют хлороформом в виде комплекса с бензоин а-ойсимом и фотометрируют. В работе [18] предложена такая же химическая подготовка перед полярографическим определением Vv. При анализе медной руды [19] ванадий (V) экстрагировали бензоин а-оксимом, определение заканчивали спектрофотометрически с помощью ПАР;
Соосаждение
Р ,?.анадий(У)> а также Сг111, Movl, Ti и Sn соосаждаются на дрОН)3. В качестве коллекторов применяют также гидроксиды Zn и Mg. Исследование соосаждения ванадия(V) с молибдо-80^>aTOW аммония показало, что из растворов НС1 выделяется 90% от исходного содержания ванадия [20].
Другие методы отделения
30MB;HaflHft(V) можно отделять от других элементов электроли-творе 3 ргутном катоде. При этом ванадий (V) остается в рас-
247
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Ванадат можно определять гравиметрически с помощью неорганических и органических соосадителей. При осаждении ионами серебра(1), ртути(1) и свинца(II) получается осадок неопределенного состава. Например, при осаждении ртутью (I) осаждаются также галогениды, MoOf, WO4' и СгОГ-
Наиболее эффективными органическими осадителями являются 8-гидроксихинолин (оксин) и купферон. Оксин осаждает ванадий (V) при pH =5—6. Определению мешают многие ионы, в том числе Fe, Ti, Со и Ni (которые, однако, можно замаскировать ЭДТА), а также Мо и W [21]. Метод осаждения ванадия купфероном также неселективен. Определение ванадия заканчивают гравиметрически, в виде V2Og, после прокаливания осадка. Гравиметрические методы определения ванадия (V) описаны в работе [4].
Титри метрические методы
Редокс-методы
Некоторые методы основаны на восстановлении ванадия (V) до ванадия(IV) с последующим титрованием ванадия(IV) стандартным раствором окислителя. Восстановление можно проводить несколькими способами, например с помощью SO2 в среде H2SO4 при кипячении раствора. Избыток SO2 удаляют пропусканием СО2 через раствор. Молибден(VI) не восстанавливается сернистым газом, что позволяет определять ванадий (V) в присутствии MoVI. В качестве восстановителя можно применять также амальгаму висмута, при этом ванадий (V) восстанавливается до V,v.
Ванадий (IV) обычно окисляют стандартным раствором КМпО4. При комнатной температуре реакция протекает медленно, поэтому титруют горячие растворы
5VO2++ МпО; 4-Н2О —> 5VOJ + Мп2+ + 2Н+
Ванадий (V) можно титровать непосредственно стандартным раствором восстановителя. Как правило, применяют растворы Fe1’. которые количественно восстанавливают Vv при pH < 1,5. В качестве индикатора применяют производные дифениламина или N-фенилантраниловую кислоту:
VO2 + Fe2++ 2Н+ —> VO2++ Fe3++ Н2О
Этот метод применен для определения ванадия в железе, стали и феррованадии [22]. Ванадий (IV) окисляют до Vv раствором КМпО4, избыток которого разрушают нитритом натрия, а избыток нитрита связывают сульфаминовой кислотой. Затем ванадий (V) титруют стандартным раствором железа(П).
Опубликованы и другие редокс-методы определения ванадия (V), например, с помощью аскорбиновой кислоты, хрома (II), меди(1), гидразина. Обзор методов дан в монографии [23]. Иногда конечную точку титрования определяют потенциометрически.
Иодиметрический метод основан на восстановлении ванадия (V) избытком KI в среде концентрированной НС1:
2VO34“ + 2Г + 12Н+ —> 2VO2+ + 12 + 6Н2О
Выделившийся иод титруют стандартным раствором тиосульфата [24].
Титрование ванадия(У) железом(П) [25]
Реагенты
Железоаммонийные квасцы, 0.1 М раствор в 0,5 М H2SO4
Дифениламиносульфокислота, бариевая соль, 0,5%-ный водный раствор.
Ход анализа. К раствору ванадия (V) добавляют 5 М H2SO4 до концентрации 1,5 М и затем 15 мл концентрированной Н3РО4 на каждые 100 мл раствора. Добавляют 4—5 капель раствора индикатора и титруют стандартным раствором железа(П) до исчезновения фиолетовой окраски индикатора.
Комплексометрические методы
Метод основан на восстановлении ванадия(V) до ванадия (IV). В работе [26] сначала восстанавливают Vv аскорбиновой кислотой и затем определяют ванадий (IV) титрованием ЭДТА с индикатором — комплексом Си2+ с 4-(2-пиридилазо)-резорцином (ПАР). Определению не мешает вольфрам, мешает молибден. Ванадий (IV) можно определить обратным титрованием избытка ЭДТА нитратом тория с индикатором ксиленоловым оранжевым {27].
В работах [28, 29] конец титрования определяли потенциометрически. Ванадий (V) предварительно восстанавливали до VO2+ с помощью SO3- [28], вводили избыток ЭДТА и титровали потенциометрически раствором ртути(II). Поскольку ванадий^) не определяется совместно с VIV, метод может быть применен для анализа бинарных смесей ванадия с Cd, Со, Ni, Са, Mg, Sr, In и Hg. Для этого сначала титруют раствор, не восстанавливая вана-Awfi(V), и определяют содержание сопутствующего металла. Затем в другой аликвотной части раствора проводят восстановление Vv и определяют сумму ванадия и сопутствующего металла. Содержание ванадия рассчитывают по разности двух титрований.
Разработан новый комплексометрический метод определения ванадия (V) [30], основанный на взаимодействии железа (II) с ванадием (V) в присутствии ЭДТА. Титрование проводят с инди-п1^°£°М ваРиаминовым голубым. Конец титрования отчетлив при Р 1>7—2,0. При определении 10 мг ванадия ошибка состав-
ствет мг- Определению мешают медь и серебро. В присут-ст ”и Ф°сФ0Рн°н, лимонной и винной кислот конец титрования воп1°ВИТся нечетким. Допустимые содержания других ионов приедены в работе [30].
249
Прочие комплексометрические методы описаны в монографии [31].
Другие титриметрические методы
Методы титриметрического определения ванадия с помощью Ag+ и РЬ2+ рассмотрены в обзоре [4].
Спектроскопические методы
Колориметрический и спектрофотометрический методы
Ванадий (V) можно определять спектрофотометрически, с применением пероксида водорода, оксина, ксиленолового оранжевого. ПАР, З.З'-диметилнафтидина, ПАН или в виде фосфовольфра-мованадиевой гетерополикислоты. С точки зрения как чувствительности, так и избирательности, метод с применением ПАР имеет определенные преимущества.
Определению ванадия в виде желтой P-W-V-гетерополпкисло-ты (б4оо = 2,0-103) мешает ряд элементов, особенно те, которые образуют малорастворимые фосфаты. Однако определению не мешают А1 и Сг, что может быть использовано при анализе некоторых специальных объектов.
Ванадий (V) образует с Н2О2 красно-коричневый комплекс, характеризующийся значением е = 300 при 450 нм. Определению мешают Ti, Movl, Fe111, I- и Вг-. Железо (III) можно замаскировать фосфатом или фторидом [32].
Ксиленоловый оранжевый образует с ванадием (V) в слабокислой среде два комплекса с разным соотношением ванадия и реагента. Комплексы характеризуются значениями е59О = 2,0-104 и ваге = 1,3• 104. В присутствии ДЦТА (диаминоциклогексантетраацетат) избирательность определения существенно улучшается.
ПАР является одним из наиболее чувствительных реагентов для определения ванадия(V). Фиолетовый комплекс имеет максимум поглощения в области 550 нм прирН=5—7. Окраска раствора развивается в течение 30 мин и устойчива в последующие 2 ч. Значение е равно 3,6-104 при 550 нм. Установлено образование комплекса состава 1 : 1 [33—35] и 2: 1 [36]. Закон Бугера — Ламберта — Бера соблюдается в интервале концентраций ванадия 0,04—1 мкг/мл, что соответствует содержанию 1—5 мкг ванадия в 25 мл раствора. С пиридилазорезорцином взаимодействует ряд ионов, в том числе Со, Pb, Th, Sc, In, Ga, Pd и U. Определению ванадия не мешают Tilv, Nbv и Zr и умеренные содержания CrVI. Movi, Wvl и NO3. Развитию окраски комплекса мешают тартрат, оксалат, фторид и ЭДТА, но ДЦТА не мешает определению, что можно использовать для маскировки ряда металлов, ионы Ti, Nb, U и Zr при этом не маскируются [33]. Наиболее распространенные анионы не мешают определению ванадия [34]. В последнее время для определения ванадия применены методы, основаН*
250
ные на экстракции ионных ассоциатов, в которых анионом является комплекс ванадия (V) с ПАР. Этот комплекс экстрагируется раствором тетрафениларсоний- или тетрафенилфосфонийхлорида в хлороформе (или других растворителях) и фотометрируется при 560 нм. Метод характеризуется высокой чувствительностью (В560 = 3,9- 104). Определению мешают только Со и Са.
В качестве катионного компонента ионного ассоциата состава (1 : 1 • 1) применен также кристаллический фиолетовый (38]. Ассоциат экстрагируют смесью бензол — изобутил метил кетон (3: 2) и измеряют поглощение при 585 нм. Закон Бугера — Ламберта — Бера соблюдается в интервале содержаний ванадия 0,05— 0,5 мкг/мл, причем е = 1,1 -105. Этот метод применен для определения следов ванадия в растительных материалах после их предварительного озоления. Определению не мешают сульфат и ацетат, мешает ряд анионов, в том числе Cl~, F-, оксалат, цитрат и тартрат при их 100-кратном избытке относительно ванадия.
В работе [39] предложен метод определения ванадия, основанный на образовании при pH =5—6 комплекса V—ПАР — NHjOH (1:1:1), характеризующегося значением е = 3,5-104. Метод чувствителен, но мало избирателен. При повышении кислотности селективность повышается, но чувствительность метода падает. Определению не мешают Re, Th, U, Мо, Мп, Zr и другие элементы.
Спектрофотометрический метод определения ванадия(V) с применением трополона является почти специфичным [40]. Образующийся в сильнокислой среде голубой комплекс экстрагируется хлороформом и характеризуется значением е = 4,6-103 при 590 нм. Большинство анионов не мешает определению. Из 37 изученных катионов мешают только TI111, Ru111, Ptlv, Irlv, Мп11, Tav и Се111, однако их можно замаскировать с помощью ЭДТА.
Уточнены условия определения ванадия с 3,3'-диметилнафти-дином [41] и предложен простой метод определения 0,08—2 ppm ванадия (V). Определению мешают CrVI, Fe11, Fe111, Се111 и
Атомно-абсорбционный метод
При определении ванадия методом атомной абсорбции поглощение измеряют обычно при 318 нм в пламени воздух — ацетилен.
Предложен чувствительный метод с применением графитового атомизатора, использованный для определения ванадия в диоксиде титана [42]. Предел обнаружения — 3,3-10~10 г ванадия в ъеме 1 мкл. Импульсную атомизацию проводят при 2500°C, по-лощение измеряют при 318,4 нм.
Другие спектроскопические методы
Методами ИК-спектроскопии можно идентифицировать и оп-М111ь метаванадат при содержании его 5 мкг на фонеРе(СМ)б’
251
и Fe(CN)^’, Co(CN)e', сульфата и хромата [43]. Методика заключается в прессовании таблеток пробы с КВг и съемке ИК-спектра. Продолжительность определения — около 20 мин.
Электрохимические методы
Потенциометрические методы
Некоторые потенциометрические методы рассмотрены в предыдущем разделе.
Ванадий (V) можно титровать потенциометрически мягкими восстановителями в присутствии Сг"1, UVI, Mov, WVI и TiIv. Более сильными восстановителями можно титровать смесь двух ионов — ванадия (V) и Fe или U, Сг, Ti. Тиосульфат медленно взаимодействует с ванадием(V), однако скорость реакции можно увеличить введением меди(П) как катализатора [44]. Этот принцип использован [45] для потенциометрического титрования ванадия (V) в присутствии Fe, Мп, Сг, Мо и U. Уран (VI) молибден (VI) также взаимодействуют с тиосульфатом, но лишь после восстановления ванадия (V) и железа (III). Ионы Fe111 титруются после ванадия(V), при этом наблюдается второй излом на кривой титрования. Определению ванадия не мешает 10-крЗтный избыток железа (III).
При титровании более сильным восстановителем — раствором железа (II) в среде Н3РО4 [46] — последовательно восстанавливаются ванадий(У) и уран(У1) или ванадий(У) и молибден(VI). При этом наблюдается два скачка. Первый отвечает переходу Vv—V1V, второй —V1V—V" и Uvl—UIV или MoVI—Mov. Марганец (II) и вольфрам (VI) не мешают. Метод имеет преимущества по сравнению с известными методами, основанными на применении Ti111, Sn" и Сг".
В работе [47] предложен аналогичный метод, состоящий в восстановлении ванадия (V) избытком железа(II), титровании избытка раствором бихромата в среде концентрированной Н3РО4. Метод позволяет последовательно титровать молибден (VI) и ванадий (V). Введением избытка железа (II) достигается восстановление до V"1 и Mov. При титровании бихроматом первый скачок потенциала отвечает окислению изытка железа (II). Второй соответствует переходу ванадий(III) — ванадий(IV) и молибден (V)--молибден(VI). Третий скачок соответствует переходу VIV—V*-Описан метод автоматического потенциометрического титрования микроконцентраций ванадия(V) и перманганата раствором гексацианоферрата (II) [48].
Полярографические методы
В среде разбавленных кислот ванадий (V) восстанавливается полярографически до VIV и V11. Поскольку потенциал парь Vv/Vlv более положителен, чем окислительный потенциал ртут*1’
252
диффузионный ток появляется при нулевом потенциале. Потенциал второй полуволны, отвечающий переходу Vlv—Vй, равен __0,98 В (н. к. э ). В аммиачной среде также наблюдаются две волны восстановления, до V1V и V11.
В работе [49] изучено полярографическое восстановление хелатов ванадия с пирокатехином и пирогаллолом. В ацетатном или пиридиновом буферном растворе с pH =5,3, содержащем пирокатехин, Е = —0,53 В, а высота волны пропорциональна концентрации пирокатехина. В этих условиях Vv восстанавливается до Viv, а V111 окисляется кислородом воздуха до V1V. Пирогаллол менее эффективен, чем пирокатехин.
Описан метод осциллополярографического определения 0,5— 6 мкг/мл ванадия [50]. Даны условия, в которых устраняется мешающее влияние Zr, Th, Bi и Си11. Метод применен для анализа руд.
Другие электрохимические методы
Описан метод амперометрического осадительного титрования ванадия (V) раствором AgNO3 [51]. Индикаторный капающий ртутный электрод имел потенциал — 0,3 В (н. к. э.), фоновым электролитом служил раствор нитрата натрия.
Для последовательного определения Мп, Сг и V в среде 6 М H2SO4 в качестве титранта применен раствор K.I [52]. Индикаторным электродом служил графитовый диск с потенциалом 0,6 В (н. к. э). Предел обнаружения составляет 130 мкг ванадия в 20 мл раствора.
Описан ряд методов определения ванадия с использованием кулонометрии при постоянном токе с электрогенерацией Ag, Fe, Sn и Ti. Метод определения миллиграммовых содержаний ванадия основан на генерировании олова(П) [53]. Конечную точку определяли потенциометрически и спектрофотометрически.
Ванадий (V) определяют также методом потенциостатической кулонометрии [54]. Ток интегрировали с помощью высококачественной интегрирующей емкости, изготовленной из полистирола. Ванадий (V) определяют при потенциале —0,128 В в ацетатном УФерном растворе или при +0,247 В в среде 2,0 М H2SO4.
Каталитические методы
°сноОвПИСаНЫ каталитические методы определения ванадия (V), [561 дННЫе на реакции типа реакции Ландольта [55]. В работе ствии СП0льз?вана реакция окисления иодида броматом в присут-цИИ) мск°Рбиновой кислоты (метод фиксированной концентра-нием' ц ^иающее ^влияние Си, Fe, Мо и Ti устраняют примене-ДеляемыхР рГН0Г0 буферного раствора с pH = 2,2. Интервал опре-одержаний ванадия составляет 0—10 мкг/мл.
253
Иодид-броматная реакция применена в работе [57]. Использовано также взаимодействие хлората с хлоридом в присутствии SnCl2 и гидразина [58, 59].
Весьма чувствительный метод определения ванадия основан на окислении красителя бордо броматом в кислой среде [60J. Метод фиксированной концентрации позволяет определять 0,005— 0,2 мкг/мл ванадия. Мешают 10 из 37 изученных ионов, наибольшее влияние оказывают SCN , NG_*. I , Rh и Си.
На основе реакции окисления нидида пероксидом водорода разработан автоматический каталитический метод [61] определения 0,6—12 мкг ванадия с ошибкой около 2%. Продолжительность определения — 15—20 с.
Газохроматографические методы
В работах [62, 63] описано образование производных трпме-тилсилила при взаимодействии с некоторыми анионами, в том числе ванадатом, с последующим определением продуктов реакции методом газовой хроматографии. Наряду с ванаЛатом определяются фосфит, карбонат, оксалат, сульфат, фосфат, арсенат и сульфамат. Газовые хроматограммы, иллюстрирующие метод, приведены в разделе «Сульфаты».
ЛИТЕРАТУРА
1. Lingane J. J.— Analytical Chemistry of Selected Metallic Elements, New York, Reinhold, 1966, p. 123.
2. Grady H. R. — In: Treatise on Analytical Chemistry (Ed. I. M. Koithoff and
P. J. Elving), Part II, Vol. 8, New York, Interscience, 1963, p. 177.
3. Lowenheim F. A. — In: Encyclopedia of Industrial Chemical Analysis (Ed.
F. D. Snell and L. S. Ettre), Vol. 19, New York, Interscience, 1974, p. 320.
4. Chalmers R. A. — In: Comprehensive Analytical Chemistry (Ed C. L. and
D. W. Wison), Vol. 1C, Amsterdam, Elsevier, 1962, p. 542.
5. Svehla G., Tolg G. — Talanta, 1976, v. 23, p. 755.
6. Korkisch J. — Modern Methods for the Separation of Rarer Metal Ions, Oxford, Pergamon Press, 1969, p. 435.
7. Fritz J. S., Abbink J. E. — Analyt. Chem., 1962, v. 34, p. 1080.
8. Hartmann S.—J. analyt. Chem., 1956, v. 151, p. 332; Analyt. Abstr., 1956, v. 3, p. 3314.
9. Федоров П. И., Андреев В. К-, Лиде Т. В. — ЖАХ, 1975, т. 30, с. 379; Analyt Abstr., 1976, v. 30, ЗВ 115.
10. Рязанов И. П., Чистота Л. М. — Сб. научи, трудов Магнитогорск, горной** таллургич. ин-та, 1958, с. 154, Analyt. Abstr., 1959, v. 6, р. 1746.
11. Церковницкая И. А., Лугинин В. А. — Вестник Ленингр. гос. ун-та. Сер. физхимии, 1969, с. 162; Analyt. Abstr., 1970, v. 18, р. 3085. -
12. Гайбакян Д. С.—Ученые записки Ереванск. гос. ун-та. Естеств. науки, 1971» с. 71; Analyt. Abstr., 1972, v. 23, р. 2293 ,
13. Любимова И. Н. — Вестник Московск. ун-та. Биолог, почвовед., 1970, т. “"Я с. 73; Chem. Abstr., 1971, v. 74, 106835.
14. Bock R., Gorbach S. — Mikrochim. Acta, 1958, p. 593.
15. Crump- Wiesner H. J., Purdy W. C. —Talanta, 1969, v. 16, p. 124.
16. Peng P. Y., Sandell E. B. — Analytica chim. Acta, 1963, v. 29, p. 325.
17. Luke C. L. — Analytica chim. Acta, 1967, v. 37, p. 267. -I
18. Inglot J., Panz M.— Chemia analit., 1970, v. 15, p. 965; Analyt. Abstr., 19» 1
v. 21, p. 1115.
254
ig Kozlicka M„ Woitowicz M.— Z. analyt. Chem., 1971, v. 257, p. 191, Analyt. ’ Abstr., 1972, v. 22. p. 3007.
20 Tsyganok L. P-, Chuiko V. T., Reznik В. E. — Zh. analit. “Khim., 1972, v. 27, ' p. 2180.
21 . Pribil R-, Sedlar V —Colin. Czech, chem. Commun., 1951, v. 16, p. 69.
22 British Stnandards Handbook No. 19, London, British Standards Institute, 1970.
2 3^- Berka A., Vulteriri J., Zyka J. — Newer Redox Titrants, Oxford, Pergamon Press, 1965.
24 Vogel A. I. — A Texbook of Quantitative Inorganic Analysis, 3rd edn, London, Longmans, 1961, p. 381.
25 Chariot G., Bezier D. — Quantitative Inorganic Analysis, London, Methuen, 1957, p. 623.
26 . Flaschka H., Abdine H. — Chemist Analyst, 1956, v. 45, p. 58.
27' Kinnunen J., Wennerstrand B. — Chemist Analyst, 1957, v. 46, p. 92.
28 Khalifa H., El-Sirafy A. — Z. analyt. Chem., 1967, v. 227, p. 109; Analyt. Abstr., 1968, v. 15, p. 3294.
29 Gardels M. C., Cornwell J. C. — Analyt. Chem., 1966, v, 38, p. 774
30 Tanaka M., Ishida A. — Analytica chim. Acta, 1966, v. 36, p. 515.
31. Schwarzenbach G., Flaschka H.— Compleximetric Titrations, 2nd English edn, London, Methuen, 1969, p. 213.
32. Sandell E. B. — Colorimetric Determination of Traces of Metals, 3rd edn, New York, Interscience, 1959, p. 923.
33. Budevski 0., Dzhonova L. — Talanta, 1965, v. 12, p. 291
34. Kowahata M., Mochizuki H., Kajiyama R., Ichihashi K- — Japan Analyst, 1965, v. 14, p. 348; Analyt. Abstr., 1966, v. 13, p. 6821
35. Agarwala В. V., Dey A. K. — Current Sci., India, 1967, v. 36, p. 544.
36 Geary W. J., Larsson C. N. — Proceedings of the SAC conference, Nottingham, 1965, Cambridge, Heffer and Sons Ltd, 1965, p. 455.
37. Siroki M., Djordjevic C. — Analytica chim. Acta, 1971, v. 57, p. 301.
38. Minczewski L, Chwastowska J., Pham. Thi Hong Mai — Analyst, 1975, v. 100,
p. 708.
39. Mal'tseva L. S., Shalamova G. G., Gusev S. 1. — Zh. analit. Khim., 1974, v. 29,
p. 2053; Analyt. Abstr., 1975, v. 29, p. 5B 118.
Rizvi G. H., Singh R. P. —Talanta, 1972, v. 19, p. 1198.
Jachson K. W7, West T. S., Balchin L. — Analyt. Chem., 1973, v. 45, p. 240.
Haba F. R., Wilson C. L.— Talanta, 1962, v. 9, p. 841.
Oberhelmann G. — Am. J. Sci., 1915, v. 39, p. 530.
Rao N. P. R.t Sharma В. V. S. — Chemist Analyst, 1965, v. 54, p. 107.
Rao G. G., Dikshitulu S. A. — Talanta, 1963, v. 10, p. 1023.
Muralikrishna U., Rao G. G. — Talanta, 1968, v. 15, p. 143.
Kiss-Eross K-, Buzas L, Erdey L. — Z. analyt. Chem., 1971, v. 255, p. 271; Analyt. Abstr., 1972, v. 22, p. 3139.
Zelinka J., Bartusek M., Okac A. — Colin Czech, chem. Commun., 1974, v. 39, P- 83; Analyt. Abstr., 1974, v. 27, p. 676.
Morales A., Gonzales B. F., Diaz C. — Chemist Analyst, 1967, v. 56, p. 89.
Saxena R. S., Sharma О. P. — Indian J. Chem., 1964, v. 2, p. 502.
Songina O. A., Zakharov V. A., Bekturova G. B., Smirnova L. 1. — Zh. analit. Khim., 1974, v. 29, p. 1594; Analyt. Abstr, 1975, v. 29, p.
Bard A. J.t Lingane J. J.—Analytica chim. Acta, 1959, v. flisftop E„ Hitchcock P. H. — Analyst, 1973, v. 98, p. 572. Svehla G. —Analyst, 1969, v. 94, p 513.
nompson H., Svehla G. — Microchem. J., 1968, v. 13, p. [Ognar J Jellinek O. — Mikrochim. Acta, 1969, p 366.
40. Rizvi G. H., Singh R. P. —Talanta, 1972, v. 19, p. 1198.
41. Bannard L. G., Burton J. D.—Analyst, 1968, v. 93, p. 142.
42. ..................................................—
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
50 n ° ’ ’» *с1**пек v/. — тшгосгпгп. нега, iuov, p. <500.
50' RJnar {-> Sarosi S.— Mikrochim. Acta, 1967, p. 813.
60 Je,lineh O.— Mikrochim. Acta, 1967, p. 193.
61 .' C W ’ Ottaway A M. — Analyst, 1970, v. 95, p. 41.
62 Buihmnir!t ? Sisfeos P. A. — Mikrochim. Acta, 1975, p. 51.
63 - Butt, W Cr~n nalyt- Lett., 1970, v. 3, p. 29
s w. c, Rainey W. T. —Analyt. Chem, 1971, v. 43, p. 538.
3B 14.
20, p. 581.
576.
255
ГАЛОГЕНЫ
БРОМАТЫ
Бромат натрия обычно применяют стве отбеливающего средства.
в промышленности в каче-
МЕТОДЫ ОТДЕЛЕНИЯ
Бромат-, хлорат- и иодат-ионы в миллимолярных концентра циях могут быть разделены методом ионного обмена [1] С по мощью тонкослойной или бумажной хроматографии бромат мож
но отделить от хлората,
йодата,
хлорита,
бромита,
перхлората
фторида, бромида, хлорида и иодида (см. табл. 19, разд. «Хло раты»). Кроме того, броматы можно отделить [2] от йодатов
периодатов и хроматов с помощью тонкослойной хроматографии на А!2О3.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
определения бромата почти полностью основаны на
Методы
его окислительных свойствах. Бромат относится к группе анионов окислителей, способных восстанавливаться под действием SO2 Бромат может быть определен в виде бромида. Обычно для восстановления бромата добавляют 1%-ный раствор гидросульфита натрия и кипячением удаляют избыток SO2. Этот метод пригоден для анализа бромат-бромидных смесей. При обсуждении комплек сометрического и амперометрического методов определения бромата этот метод восстановления будет рассмотрен подробнее.
Титриметрические методы
Титриметрические методы, по существу, являются окислительно-восстановительными методами. Бромат калия широко используют в количественном анализе в качестве сильного окислителя:
ВгО; + 6Н+ + бе" Вг'+ЗН2О
Соль легко очищается перекристаллизацией, стандартные раство
иодид и некоторые
ры готовят по точной навеске. Мышьяк(Ш),
другие ионы можно определять прямым титрованием брома" том. Для определения бромата можно использовать те же реакции, применяя стандартные растворы восстановителей, например мышьяка (III),
256
Иодиметрические методы
Бромат-иодидная реакция проходит в разбавленной минеральной кислоте. По сравнению с иодат-иодидной реакцией, например, эта реакция сравнительно медленная. Более того, она проходит не сразу в соответствии с уравнением
Вго; + ег + бн+ —> вг'+312-рзн2о
а включает в себя промежуточные стадии [3]. Использование НО предпочтительнее серной кислоты. Концентрация кислоты должна быть не меньше 0,5 М, для того чтобы реакция проходила с приемлемой скоростью и строго стехиометрично. В качестве катализатора можно использовать молибдат аммония. Его обычно применяют, если кислотность раствора ниже 0,5 М.
Тот факт, что бромат и хлорат сравнительно медленно взаимодействуют с иодидом, может быть успешно использован при определении йодата в присутствии этих ионов. Во фталатном буферном растворе с pH = 5 иодат быстро реагирует с иодидом с образованием иода, который титруют стандартным раствором тиосульфата. Через 3 мин вторым титрованием определяют иод, выделившийся в результате реакций с броматом и хлоратом. Разработаны методики [4—6] анализа смесей различных анионов, содержащих бромат. Состав смесей может быть следующим:
ВгОз—то;; Юз—ю;—Bro,—сю; и Сго^—AsOj-—ю;—ю;—вго;—сю;
Для каждого случая характерно использование иодиметрического титрования. Иодиметрический метод рекомендован [7] для определения бромата в муке.
Иодиметрическое титрование бромата (примерно
для 50 мг бромата в пробе)
Ход анализа. К 25 мл раствора бромата добавляют 1 г К1 и 5 мл 4 М раствора НС1 (или эквивалентное количество H2SC>4). Тщательно перемешивают раствор. Иод титруют стандартным раствором тиосульфата, добавляя вблизи точки эквивалентности свежеприготовленный крахмал.
Арсенометрические методы
Реакция взаимодействия может быть представлена уравнением
ВгО; + 3As3+ + 6Н+ —> Вг' + 3As5+ + ЗН2О
_ Реакция, в обычных условиях медленная, катализируется хло-м, бромом или чаще всего иодом. В потенциометрическом ва-анте метода в качестве катализатора использовали OsO4 [24].
п Ри Тн^Р°вании, когда реакция проходит полностью, первая ннембромбЬ1ТОЧНОГО бр°мата реагирует с бромидом с образова
ВгО; + 5Вг' + 6Н+ —> ЗВг2 + ЗН2О
8 Зак. 16?
257
Бром разрушает некоторые индикаторы, что приводит к необратимому изменению окраски титруемого раствора. Предлагается несколько индикаторов. Метиловый оранжевый и нафтол синечерный ведут себя как необратимые индикаторы, причем последний оказался предпочтительнее. Обратимый индикатор л-этокси-крезолидин [8], по-видимому, является лучшим индикатором. Методика, по существу, та же, что приведена в разделе «Арсенаты и арсениты» с использованием стандартного раствора бромата. Для определения бромата используют, естественно стандартный раствор мышьяка(III), к которому из бюретки добавляют испытуемый раствор бромата. Предельная концентрация НС1, при которой бромат-арсенитная реакция проходит количественно, была рассчитана для двух методов: титрование с использованием индикатора (в качестве индикатора применяли солянокислый розанилин) и потенциометрическое титрование [9].
Метод с использованием аскорбиновой кислоты
Галогенаты ХОз в присутствии катализаторов реагируют в кислой среде с аскорбиновой кислотой с образованием дегидро-аскорбиновой кислоты и галогенида:
XOJ + ЗС8Н8О8 —> X' + ЗС6н6Ов 4-ЗН2О
Броматы восстанавливаются труднее йодатов, кроме того, необходимо присутствие катализаторов и большая концентрация кислоты. В качестве катализатора обычно используют селенистую кислоту, индикаторами служат сульфат марганца (II) или хлорид ртути(Н) [10]. Метод позволяет определять 10—100 мг бромата. В другом методе используют хорошо изученную систему гексацианоферрат (III)— аскорбиновая кислота:
2 [Fe (CN)t]3‘ + C8H8Oe —> 2 [Fe (CN)c[4‘ + С8Н8О6 + 2Н+ ВгО; + 6 [Fe (CN)£]!‘ + 6Н+ —> Br'4-6[Fe(CN)8|3- + 3H2O
Бромат реагирует с гексацианофферитом (II) и образующийся гексацианоферрат(III) титруют стандартным раствором аскорбиновой кислоты с 2,6-дихлорфенолиндофенолом в качестве индикатора [11]. Метод применим для определения галогенов, гипогало-генитов и других кислородных соединений галогенов. Эрдей [12J использовал производное вариаминового голубого 2-гидрокси-4-амино-4-метоксидифениламин в качестве индикатора для титрования гексацианоферрата (III) аскорбиновой кислотой. Индикатор применяют в виде твердой смеси с NaCL Для определения бР°"_ мата был использован потенциометрический вариант автоматиче ского титрования [13]. Аскорбиметрические методы могут Дава* хорошие результаты, но, по-видимому, они обладают небольШ ми преимуществами перед иодиметрическим или арсенометри4 I ским методами определения бромата.
258
Другие титриметрические методы
Леонард, Шахин и Вильсон [14] нашли, что микроколичества бромата (и йодата) можно определять прямым титрованием хромом (II), используя в качестве редокс-индикатора фенантролинат ванадия(II). Восстановленная форма комплекса окрашена в интенсивный сине-фиолетовый цвет, а окисленная — бесцветна. Для определения бромата применяют ЭДТА. Бромат-бромидную смесь можно проанализировать следующим образом: бромид осаждают в виде бромида серебра, который взаимодействует с K2Ni(CN)4, а освобождающиеся ионы никеля титруют стандартным раствором ЭДТА. Вторую аликвотную часть обрабатывают мышьяком (III), восстанавливая бромат до бромида, и определяют последний, повторяя все операции, описанные выше [15]. Хлориды, иодиды и тяжелые металлы мешают определению.
Кельнер и Зекерес [16] определяли бромат наряду с другими окислителями, добавляя избыток железа (II) и оттитровывая образующееся железо(Ш) ЭДТА, используя салициловую кислоту в качестве индикатора. Метод применим для определения 10— 270 мг бромата. Ни один метод с применением ЭДТА неселективен.
Спектрофотометрические методы
Опубликовано несколько методик определения бромата. Большинство методов основано на редокс-реакциях и неселективно. Бромат в следовых концентрациях дает цветные реакции со стрихнином, метиловым оранжевым, индигокармином и некоторыми другими соединениями. Разработан метод [17], основанный на взаимодействии бромата с о-арсиниловой кислотой с образованием красновато-коричневых продуктов реакции. Закон Бугера — Ламберта — Бера выполняется до содержания бромата 50 ppm, максимум светопоглощения измеряют при 463 нм. Определению 20 ppm бромата мешают 1 ppm иодида, бромида, йодата, нитрита, золота(Ш), церия(1У) и железа(Ш). Интенсивность окраски образующихся продуктов зависит от кислотности растворов, времени от начала реакции до выполнения измерений, температуры, присутствия окислителей и восстановителей, концентрации реагентов и бромата, общей концентрации ионов (ионной силы растворов). Окраска достигает максимума лишь через 2 мин. Чувствительность — 0,05 мкг бромата в 1 мл. В более поздних работах отмечается нестабильность окрашенного соединения [18].
Для определения 20—30 мкг бромата использовали о-амино-ензойную кислоту [19], что позволяет определять броматы до концентраций меньше 3-10-4% (масс.). Определению не мешают
RT’ ХЛ0Рат и органические соединения.
Да 2 АРУГ°М методе [20] используют смесь изониазида и хлори-чае ,'5’5’тРиФенилтетразония в разбавленной НС1. В этом слу-максимальная окраска образуется при взаимодействии
259
с йодатом при 33°С, а с броматом — при 60°С. Можно анализировать смесь, содержащую 10—30 мкг каждого иона в 50 мл.
Одлер (21] разработал метод, пригодный для определения бромата или нитрита, основанный на взаимодействии их с солянокислым 1,2,3-трис(2-диэтиламиноэтокси)-бензолом. В кислом растворе при pH < 1 бромат образует соединение, окрашенное в желтый цвет, а нитрит — в коричневый. Окраска возрастает до максимума, затем уменьшается, поэтому в методике задается строго фиксированное время выдерживания смеси перед измерением светопоглощення при 400 нм. Область определения бромата 0,5—5,0-10-3 А1, влияние посторонних ионов мало.
Сантос и Медич [22] разработали простой спектрофотометрический метод с применением фуксина. Бромат можно определять в области 0,5—2,5 ppm, следовые концентрации хлората и йодата не мешают определению.
При добавлении к бромату антипирина, хлорной кислоты и нитрита натрия образуется красный комплекс с максимумом све-топоглощения при 525 нм и молярным коэффициентом светопо-глощения 2-103 моль-1-см-1-л. Закон Бугера — Ламберта — Бера выполняется в интервале концентраций 25—140 ppm. Мышь-як(Ш), церий(1У), железо(Ш), хромат, бихромат и хлорид не мешают определению [18].
Электрохимические методы
Потенциометрические методы
Бромат можно оттитровать потенциометрически солью Мора в растворе ортофосфорной кислоты [23]. При концентрации кислоты меньше 8 Л1 восстановление идет сразу до бромида, но результаты титрования неточны за счет потери брома. При более высоких концентрациях Н3РО4 на кривой титрования наблюдается два перегиба, соответствующие восстановлению до брома и бромида соответственно. В этих условиях небольшие потери брома оказывают незначительное влияние на положение первого перегиба на кривой титрования. Мешают определению хлорид, иодид, хлорат, иодат, нитрат и перйодат.
Разработай метод потенциометрического определения бромата с использованием мышьяка (III) [24]. Метод заключается в полном анализе смеси ВгО'—ВгОг—ВгОз—Вг' в одном анализируемом растворе. Окислители титруют стандартным раствором мышь-яка(Ш), а бромид —стандартным раствором AgNO3.
По существу, метод аналогичен методу анализа соответствующих хлорсодержащих анионов, основные стадии анализа следующие.
1. Гипобромнт титруют стандартным раствором мышьяка (ПП в сильнощелочном растворе с платиновым и насыщенным каломельным электродами.
260
2. К этому же раствору прибавляют OsO4 и титруют бромит таким же образом (см. п. 1).
3. Бромат титруют прямым или косвенным методом. В прямом титровании раствор подкисляют до 0,25—0,5 Al по H2SO4 и титруют мышьяком (III). В другом варианте добавляют известный точный избыток мышьяка(III) к раствору более чем 0,5 М по H2SO4 и обратное титрование проводят 1\А1пО4. Методика обратного титрования больше подходит при анализе смесей, содержащих значительные количества бромидов. Реакция между КМпО4 и мышьяком(III) также катализируется OsO4.
4. Для определения бромида платиновый электрод заменяют на серебряный и тот же раствор титруют стандартным раствором AgNO3.
Последовательное потенциометрическое определение
ВгО- — BrOJ — Вг03 ~ Вг“ I241
Реагенты
Мышьяк(Ш), 0,025 М (0,1 н. раствор). Раствор готовят из оксида мышья-ка(1П). Методику приготовления раствора см. в разделе «Арсениты н арсенаты».
Катализатор OsO4, 0,1%-ный раствор в 0,3 М растворе NaON.
Нитрат серебра, 0,1 М (0,1 н. раствор).
Перманганат калия, 0,02 М (0,1 н. раствор).
Ход анализа. Определение гипобромита. К аликвотной части аиалнзируемого раствора добавляют 0,5—2 мл 10 М раствора NaOH. Помещают в раствор платиновый и каломельный электроды и медленно растворяют As111, постоянно перемешивая раствор с помошью магнитной мешалки.
Определение бромит а. К этому же раствору добавляют 3—4 капли катализатора OsO4 и проводят титрование как и в предыдущем случае.
Определение бромата. Прямое титрование. Раствор нейтрализуют 5 Af H2SO4 и доводят кислотность раствора той же кислотой до 0,25— 0,5 М. Добавляют раствор As111 до появления неисчезающей желтой окраски и продолжают титрование до скачка потенциала.
Обратноетитрование. К щелочному анализируемому раствору добавляют известный объем 0,025 М раствора As111, затем подкисляют 5 Л1 кислотой, так чтобы кислотность раствора была больше 0,5 М по H2SO4. Обратное титрование проводят 0,02 М раствором КМпО4 до скачка потенциала.
Определение бромида. Аккуратно удаляют платиновый электрод (предварительно промывая его) и заменяют серебряным электродом. Титруют при той же концентрации H2SO4 0,1 М раствором AgNO3 до скачка потенциала, д ni Римечание. При расчете учитывают, что 1 мл точно 0,025 Л1 раствора As соответствует 4,795 мг гипобромита, 2,798 мг бромита и 2,132 мг бромата;
мл точно 0,1 М раствора AgNC)3 соответствует 7,991 мг бромида;
ВгО' + AsOf' —> Br' + AsOV
BrOj + 2AsOi' —> Br' + 2AsO^
BrOJ + 3AsO|" —> Br’ + 3AsO^
^мперометрический метод
pH4eci?MaT'бР°миднУю смесь можно проанализировать амперомет-чески НГт методом I25]- Сначала бромид титруют амперометри-андартным раствором AgNO3 в среде 0,1 Л! раствора
261
KNO3, содержащего 0,02% желатины. В качестве электродов используют вращающуюся платиновую проволоку и насыщенный каломельный электрод. Бромат затем восстанавливают до бромида, используя водный раствор H2SO3, и титрование повторяют. По данным авторов, ошибка метода меньше 0,4%.
Полярографический метод
Бромат и иодат восстанавливаются на капающем ртутном электроде, а хлорат и перйодат — нет. В щелочной среде восстановление бромата проходит по реакции
ВгО3+ бе’ + ЗН2О —> Вг’ + 6ОН’
Благодаря большому значению потенциала перенапряжения бромата, его потенциал восстановления много больше потенциала восстановления йодата. Это позволяет определять бромат на фоне йодата [26].
Другие методы определения
В работе [27] окислители определяли газохроматографическим методом по иоду, выделяющемуся из KI- Хотя бромат специально и не упоминается в этой работе, метод, по-видимому, применим и для определения этого иона. В навеске образца всего 5 мг за 5 мин можно определить содержание окислителей.
ЛИТЕРАТУРА
1. Skloss J. L., Hudson J. A., Cummiskey С. L. — Analyt. Chem., 1965, v. 37, p. 1240.
2. Lederer M., Polcaro C. — J. Chromat., 1973, v. 84, p. 379.
3. Kolthoff I. M., Belcher R. — Volumetric Analysis, Vol. Ill, New York, Interscience, 1957, p. 269.
4. Szekeres L. — Z. analyt. Chem., 1959, v. 165, p. 32; Analyt. Abstr., 1959, v. 6, p. 3502.
5. Szekeres L. — Z. analyt. Chem., 1960, v. 172, p. 256; Analyt. Abstr., 1960, v. 7, p. 3739.
6. Szekeres L. — Annali. Chim., 1961, v. 51, p. 200; Analyt. Abstr., 1961, v. 8, p. 4932.
7. Official Methods of Analysis, 11th edn, Washington, D. C., Association of Official Analytical Chemists, 1970.
8. Schulek E., Rozsa P. — Z. analyt. Chem., 1939, v. 115, p. 185.
9. Ottaway I. M., Bishop E. — Analytica chim. Acta, 1965, v. 33, p. 153.
10. Erdey L., Bodor E., Buzas H.~Z. analyt. Chem., 1952, v. 134, p. 412; Chem-Abstr., 1952, v. 46, p. 3457e.
11. Erdey L., Svehla G. — Z. analyt. Chem., 1959, v. 167, p. 164; Analyt. Abstr., 1960, v. 7, p. 122.
12. Erdey L„ Kasa I. — Taianta, 1963, v. 10, p. 1273.
13. Erdey L., Svehla G., Weber O. — Z. analyt. Chem., 1968, v. 240, p. 91; Analyt-Abstr., 1970, v. 18, p. 12.
14. Leonard M. A., Shahine S A. E. F., Wilson C. L. — Taianta, 1969, v. 16, p 470-
15. De Sousa A. — Z, analyt. Chem.. 1960, v. 174, p 337; Analyt. Abstr., 1961, V. 8« p. 125.
262
16. Kellner A., Szekeres L. — Chemist Analyst, 1965, v. 54, p. 75.
17. Macdonald J. C., Yoe J. H. — Analytica chim. Acta, 1963. v. 28. p. 383.
18. Qureshi Л1., Qureshi S Z., Zehra N. — Mikrochim. Acta, 1970. p 831 (9 Hashmi M. H , Ahmad H, Rashid A. — Analyt. Chem.. 1964, v. 36, p. 2028.
20 Hashmi Л1. H., Ahmad H., Rashid A., Azam F. — Analyt. Chem., J964, v 36, p. 2471
21. Odler /. — Analyt. Chem., 1964. v. 41, p. 1116.
22. Meditsch J. O., Santos S. E. De 4. — Reuta Quim. Ind., Rio de J., 1971, v. 40, p. 14; Analyt. Abstr., 1972, v. 23, p 3853.
23. Vuterin J. — Colin Czech, chem. Commun Engl. Edn., 1967, v. 32, p. 3349; Analyt. Abstr., 1969, v. 15, p. 7255.
24. Норкус П., Стильжене С. ЖАХ, 1969, т. 24, с. 1565. Analyt. Abstr., 1971, т. 20, с. 3029.
25. Ikeda S., Satake Н. — Japan Analyst, 1971, v. 20, p. 721; Analyt. Abstr., 1972, v. 23, p. 2453.
26. Hemala M. — Chemicke Listy, 1953, v. 47. p 1323.
27. Pennington S. N. — J. Chromat., 1968, v. 36, p. 400.
БРОМИДЫ
В разделе «Хлориды» упоминается о близости аналитической химии бромида и хлорида, поэтому там же приведены существенные подробности основных методов определения и разделения галогенидов. Чтобы избежать бесполезного повторения деталей методов, в этом разделе будут приведены лишь ссылки на методы, если не появится необходимость в более подробных комментариях. Особое внимание будет обращено на методы, относящиеся к определению самого бромида.
МЕТОДЫ ОТДЕЛЕНИЯ
Бромид можно отделить, используя возможности различных хроматографических методов, включая и ионный обмен. Методы подробно обсуждаются в разделе «Хлориды». Фоти [1] изучал содержание бромида в морской воде, используя метод ионного обмена с применением радиоактивных индикаторов. Для выделения бромида использовали и метод дистилляционного разделения. Свободный бром может быть выделен из кислых растворов бромидов при использовании жестких окислительных условий. Используют выделение брома в виде цианбромида, который затем поглощается раствором NaOH. Таким методом отделяли 5— 20 ррщ бромида от почти 1000 ppm хлорида. Методом определения была потенциометрия. Общее время, необходимое для приготовления образца, отделения и определения, составляет 15 мии, точность метода 0,1 ppm, чувствительность — 0,5 ppm.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Бромид можно определять гравиметрически в виде бромида ^еребра в тех же условиях, что и хлориды, определению мешают же ноны. Методика, основанная на осаждении из гомогенного Р створа, описана в работе [3]. Методика позволяет гравиметри
263
чески определять хлорид, бромид и иодид последовательным осаждением каждого иона при уменьшающемся значении pH. Это достигается отгонкой аммиака из анализируемого раствора.
Титриметрические методы
Аргентометрические методы
Для определения бромида можно использовать все три метода, названные методами Фольгарда, Мора и методом абсорбционного индикатора (метод Фаянса). Методы Фольгарда и Мора, приведенные в разделе «Хлориды», могут быть использованы без изменений. Для метода абсорбционного индикатора, вероятно, эозин (тетрабромфлуоресцеин) является лучшим индикатором, но флуоресцеин также подходит для проведения определения. Эозин, будучи более сильной кислотой по сравнению с флуоресцеином, позволяет проводить титрование бромида в более кислой среде (pH = 2). Кольтгоф и Берк [4] рекомендовали добавление уксусной кислоты для увеличения контрастности изменения окраски индикатора. Было изучено влияние добавок различных поверхностно-активных веществ на титрование (5]. Найдено, что гидрокси-пропилметилцеллюлоза позволяет получить самый лучший переход окраски в конечной точке титрования и в этом смысле предпочтительнее агар-агара. В этом же исследовании был сделан вывод о том, что эозин является лучшим индикатором для бромида, а флуоресцеин — для смеси галогениДов (хлорид, бромид, иодид).
Определять отдельно каждый из ионов в смеси невозможно.
Титриметрическое определение бромида с использованием абсорбционного индикатора
Реагенты
Нитрат серебра, 0,1 М раствор.
Эозин, 0,1%-ный водный раствор.
Ход анализа. К 50—100 мл нейтрального раствора бромида с концентрацией менее 0,05 М добавляют 10 капель индикатора и 2—3 мл 6 М уксусной кислоты. Титруют 0,1 М раствором AgNOa при постоянном перемешивании и в рассеянном свете. Непосредственно вблизи точки эквивалентности осадок бромида серебра коагулирует, а в точке эквивалентности осадок приобретает интенсивно красную окраску.
Необходимо отметить, что декстрин, который рекомендуют в качестве защитного коллоида при титровании хлоридов, неприменим для бромидов и иодидов.
Для аргентометрического титрования бромида рекомендованы другие индикаторы. Мэлиг [6] нашел, что родизонат натрия пригоден для индикации конечной точки титрования. В этом случае раствор превращается из желтого в нежно-розовый, а осадок становится красно-фиолетовым. Можно использовать ксиленоловый оранжевый [7] при титровании как бромида, так хлорида и иоДИ-да.
264
Меркуриметрические методы
Бромид можно титровать^ меркурпметрически, используя в качестве индикаторов нитропруссид, дифенилкарбазпд и дифенил-карбазон. Вариант метода, предложенный Ченгом, позволяет определять малые концентрации бромида. Детально этот метод описан в разделе «Хлориды». Необходимо отметить, что меркуримет-рический метод нельзя применять для определения иодидов. Не подходит он также и для анализа смеси галогенидов.
Комплексометрический метод
Опубликованы косвенные комплексометрические методы определения бромидов. Гусев и Кожевникова [8] сначала осаждают бромид количественно 2-(2-гидрокси-1-нафтилметиламино)пириди-иом в присутствии свинца, образующийся осадок имеет строение (СиНиОНгЬРЬВг. Свинец, входящий в состав комплекса, титруют стандартным раствором ЭДТА, используя в качестве индикатора ксиленоловый оранжевый; бромид из комплекса перед титрованием удаляют. Синтез реагента описан в аналогичном методе определения хлоридов, где приведена также методика проведения определения.
Метод был применен и для анализа смеси галогенидов [9]. Когда в анализируемом растворе присутствуют бромид и иодид, одну аликвотную часть анализируемого раствора обрабатывают нитратом серебра, а осадок окисляют хромовой кислотой. Иодид окисляется до йодата, а выделяющийся бром удаляют, пропуская через раствор ток воздуха. Затем иодат опять восстанавливают до иодида и осаждают в виде иодида серебра для количественного определения. Во второй части раствора оба галогенида серебра растворяют в тетрацианиде никеля и выделившиеся ионы нике-ля(И) титруют стандартным раствором ЭДТА, получая сумму бромида и иодида. Бромид находят по разности.
Разработана методика определения бромида, иодида и хлорида. И в этом случае для определения общего содержания галогенидов используют тетрацианидный комплекс никеля. Этот метод, по-видимому, неприменим для рутинного анализа [10]. Комплексометрическое определение бромида и бромата при совместном присутствии также можно проводить, используя реакцию с тетрацианидом никеля. В этом случае бромат во второй аликвотной части раствора восстанавливают до бромида мышьяком (III) и определяют общее содержание бромида [11].
Редокс-методы.
б Многочисленные редокс-титриметрические методы определения Р°мида, основанные на окислении его до бромата гипохлорной мртп°ТОп’ нашли широкое применение. Чаще всего ссылаются на Дики Кольтгофа — Ютзи и Кольтгофа — Ютзи — Ван-дср-
265
Мюлеиа. Пригодные как для микро-, так и для макроанализа за счет 6-кратного увеличения чувствительности определения, они нашли широкое применение при определении бромидов, образующихся при сжигании органических веществ в кислороде [12], Окисление бромида проводят, используя горячий щелочной раствор гипохлорита натрия в присутствии буферного раствора дигидрофосфата натрия. Избыток гипохлорита разрушают формиатом натрия и определение заканчивают иоднметрически, используя в качестве катализатора молибдат для ускорения обычно медленной реакции между броматом и иодидом. Окисление, вероятно, проходит в две стадии, суммарной является реакция
Вг' + ЗСЮ' —> ВгОз + ЗСГ
Две другие стадии анализа можно представить следующими реакциями:
ВгОз + 61* + 6Н+ —» 312+Вг* + ЗН2О
2S2O2* + 12 —* S4O|- + 21"
из чего видно, что Вг~ эквивалентен 63гОз". Важной особенностью метода является возможность определять бромид в присутствии большого избытка хлорида. Иодид, если он присутствует в анализируемом растворе, будет реагировать аналогично бромиду. Иодид можно удалить кипячением исходного раствора с нитритом натрия и серной кислотой для переведения иодида в свободный иод. У гипохлоритного метода длительная история развития, большинство исследований касалось выбора типа буферного раствора и pH среды, при которых окисление проходит количественно [13, 14]. Метод нашел применение для определения бромида в пищевых продуктах [15]. В этом случае сначала образец разлагают с применением техники сожжения органических соединений в колбе, заполненной кислородом. Используют другой метод определения конечной точки титрования, основанный на спектрофотометрической индикации.
Редокс-титриметрические методы определения бромида основаны иа окислении его до брома или бромата. Для первого случая предложено несколько окислителей. Образующийся бром можно отделить дистилляцией или экстракцией. Затем бром определяют обычно иодиметрически. В другом варианте метода измеряют количество израсходованного на титрование окислителя. В этом случае конечную точку титрования обычно определяют потенциометрически. Сравнительно простое титрование, основанное на окислении бромида до брома, описано в работе [16]. В качестве селективного окислителя используют сульфат марганца(III):
2Мп3+ 4- 2ВГ —> 2Мпг+ + Вг2
Образующийся бром экстрагируют СС14, затем бром, взаимодействуя с KI, вытесняет эквивалентное количество иода. Иод титруют стандартным раствором тиосульфата:
Вг2 + 2KI —> 12 + 2КВг
266
Сульфат марганца, который легко синтезируется, в течение 5 ч может разложиться, но этот недостаток компенсируется некоторыми достоинствами реагента. Не требуется ни точного установления pH, ни нагревания, метод быстр: на каждое определение необходимо лишь 10 мин, хлориды не мешают определению, результаты хорошо воспроизводимы.
Бромид можно определять, используя церий (IV) или тетраацетат свинца.
Титриметрическое определение бромида, основанное на окислении гипохлоритом
Реагенты
Гипохлорит натрия, 0,5 М (0,1 н.) раствор.
Продажный NaOCI, свободный от бромидов, пригоден для использования [17]. В более ранних работах его синтезировали [14].
Ход анализа. К раствору препарата, содержащего бромид, объемом не более 25 мл добавляют 1 г NaH2PO4-2H2O, 10 г NaCl и 5 мл 0,5 М раствора NaOCI. Нагревают раствор почти до кипения, затем добавляют 5 мл 50%-ного раствора формиата натрия. После охлаждения раствор разбавляют до 150 мл водой, добавляют 1 г КТ, 25 мл 3 М H2SO4 и 1 каплю 0,5 М раствора молибдата аммония. Сразу после этого иод титруют стандартным раствором тиосульфата, добавляя вблизи точки эквивалентности крахмал. В идентичных условиях проводят холостой опыт, заменив анализируемый раствор эквивалентным объемом воды.
Примечания. 1. Добавление большого количества NaCl точно определяется в предварительных опытах и может быть исключено, если количество бромида меньше 2 мг.
2. Хлориды в отличие от иодидов не мешают определению; результатом анализа является содержание суммы бромидов и иодидов.
3. Большие количества Са2+ и Mg2+ могут слегка занижать результаты анализа.
4. Известно, что индикация конечной точки титрования с применением крахмала становится менее четкой при высоких концентрациях электролитов. Медич [18] исследовал некоторые красители, содержащие четвертичные аммониевые группировки с целью замены ими крахмала. Им было найдено, что малахитовый зеленый наиболее эффективен, метиленовый голубой также дает хорошую реакцию. Так как эти индикаторы нечувствительны к высокой концентрации электролитов, они могут служить хорошей заменой крахмалу.
Титриметрическое определение бромида, основанное на окислении марганцем(Ш) [16]
30 мл коицентрироваииой H2SO4, 10 мл К 20 мл этого раствора, содержащего приливают 20—25 мл приблизительно
Реагенты
Сульфат марганца(Ш). Смешивают сиропообразной Н3РО4 и 110 мл воды. г MnSO4-4H2O, медленно о.О2 М (0,1 н.) раствора КМпО4.
Ход анализа. Анализируемый образец, содержащий Вг~ и С1~ (10 или 20 мл),
л"мещ.г^)т в делительную воронку и добавляют 20 мл СС14. В воронку добав-и вс мл раствора сульфата марганца(III). Воронку закрывают пробкой РастТРЯХИВают’ Обрабатываю/ нижний коричневый слой избытком 10%-ного
В0Ра К1’ Экстрагируют, добавляя порциями СС14, до тех пор, пока нижний Эк ие пеРестанет окрашиваться в желтый цвет. Обычно проводят три-четыре польТЦИИ Титруют св°бодный иод стандартным раствором тиосульфата, ис-зовяо в качестве индикатора крахмал, нли до тех пор, пока не исчезнет оо-ая окРаска иода в СС14 слое.
267
Примечания. 1. Без изменения метод может быть использован для определения 0,1—0,005 М раствора бромида.
2. СС14 легко регенерируется следующими способами:
а) СС1< встряхивают с разбавленным раствором тиосульфата для удаления следов иода;
б) СС1« встряхивают с дистиллированной водой до тех пор, пока в промывной воде при добавлении AgNOs не перестанет обнаруживаться нодид.
3. Если присутствует йодид, то в процессе реакции будет образовываться свободный иод, и результатом анализа является содержание суммы Вг_ и 1~.
4. Известно, что простые ионы ие мешают определению. Такне ионы, как сульфит, нитрит и пероксиды, сразу окисляются избытком Мп111.
Другие титриметрические методы
Метод Вибека с использованием оксицианида ртути, описанный в разделе «Хлориды», применим и для определения бромидов, хотя для определения именно бромида, вероятно, лучше применять меркуриметрический метод, модифицированный Ченгом.
Спектроскопические методы
Спектрофотометрический метод в видимой и ультрафиолетовой областях
Многие спектрофотометрические методы определения хлоридов применимы для бромидов, а в некоторых случаях и для иодидов. Это следующие методы:
1. Метод с использованием роданида ртути. Сравнимый с ним метод [19], основанный на применении роданида серебра, применим для определения бромида и иодида в присутствии хлорида.
2. Метод с использованием дифенилкарбазона — дифенилкарб-азида ртути.
3. Метод с использованием бромидных комплексов Fe111. В этом случае в присутствии бромида появляется полоса поглощения при 420 нм.
4. Метод, основанный на вытеснении HCN из Hg(CN)2 галогенидом.
5. Метод, основанный иа образовании фенилртуть(II)-галогенидов и их взаимодействии с диэтилдитиокарбаминатом натрия.
Подробное описание методов и ссылки на оригинальные работы приведены в разделе «Хлориды».
Некоторые спектрофотометрические методы определения бромида основаны на его предварительном окислении до брома. Такие методы в основном ограниченно селективны. В некоторых условиях хлориды не окисляются. Более того, иодиды могут окисляться Fe111 до иода, который можно удалить кипячением, что позволяет проводить определение бромидов в присутствии иодидов. Хлорамин Т часто используют для окисления бромида Д° брома. После окисления образующийся бром можно определить любым удобным методом. Бром реагирует с метиловым оранЖе' вым с образованием бесцветного продукта реакции. С хлором
268
реакция проходит аналогично. Феноловый красный предложен Стенгером и Кольтгофом [20]. Феноловый красный реагирует с бромом в щелочной среде, требуется жесткий контроль за условиями проведения реакции. Предварительное окисление [21] можно проводить гипохлоритом или хлорамином Т. Подчинения закону Бугера — Ламберта — Бера не наблюдается. Модификацию метода, предложенную Гольманом ц Вейлесом, используют в настоящее время в качестве стандартного метода анализа воды [22]. Минимальная определяемая концентрация бромида — 100 мкг/мл. Крезоловый красный (о-крезолсульфонафтолеин), аналог фенолового красного, по мнению авторов этой работы, является более чувствительным реагентом по сравнению с феноловым красным. Другим применяемым реагентом является флуоресцеин, который при взаимодействии с бромом окисляется до эозина. Следы иода и в этом случае мешают, а следовые количества хлора могут присутствовать, не оказывая мешающего влияния [23]. Эльбай и Эль Сафари [24] окисляли бромид до брома кипячением в смеси (10:1) уксусного ангидрида и 30%-ного пероксида водорода. Хлорид в этих условиях не окисляется. Бром определяют спектрофотометрически, используя хромотроп 2В. Область определения — 30—70 мкг Вг~. Метод дает удовлетворительные результаты при использовании его для анализа природной воды.
В последнее время были исследованы некоторые новые реагенты, в том числе и трифенилметановые красители. Лайкусса [25] использовал оксазиновый краситель — Нильский голубой, который экстрагировали СНС1з, и определяли при 625 нм светопо-глощение экстракта. В этом случае наряду с бромидами определяются и иодиды. Закон Бугера — Ламберта — Бера выполняется для 1—6 ppm Вг~ и 0,5—3 ppm I-.
Бинкин и Раманаускас с сотр. исследовали большое число трифенилметановых красителей (ТФМ). Иодид и бромид реагируют с некоторыми из них в присутствии хлорамина Т, хлорамина В или NaOCl. Установлено, что образующиеся ионы 1+ или Вг+ увеличивают интенсивность окраски красителя. Исследовано И красителей для определения бромида и найдено, что наилуч-шими из них являются бриллиантовый зеленый и кислотный фио-^Т.ОВЬ1Я С [26]. Чувствительность реакции — приблизительно 0>01 мкг/мл Вг~. В более поздних работах отмечено, что кристаллический фиолетовый в присутствии хлорамина Т или NaOCl также можно использовать для определения бромида и иодида [27]. 589ТИЧеСКую плотность измеряют при 590 нм для бромида и при нм для иодида. Медь, марганец, кальций, калий, натрий, хло-Р Д и сульфат не мешают определению, карбонат, гидрокарбонат КаНитрат оказывают мешающее влияние. В последующих публи-ппиИЯХ Те Же исследователи нашли, что малахитовый зеленый °птимДеН ДЛЯ опРеделения бромида и иодида [28]. Найдены бриллаЛЬНЫе условия ДЛЯ определения бромида с использованием иантового зеленого и малахитового зеленого.
269
Спектрофотометрические методы определения бромида основаны на окислении бромида до бромата по методу Кольтгофа — Ютзи. Бромат реагирует с подкисленным KI, и образующийся иод определяют спектрофотометрически при 350 нм. Практическое применение метод нашел при определении брома в пищевых продуктах [15].
Бромид и иодид образуют комплексы с медью(П)—ЭДТА при pH =3,5—7,7 и 5,0—8,3 соответственно. Оптическую плотность измеряют при 250 нм, чувствительность метода 0,05—2,0 мг Вг~. Присутствие цианида, роданида, сульфида и кобальта(II) мешает определению Вг- и I-. Кроме того, NHJ и Ag+ мешают определению иодидов [29].
Бромид в водных растворах реагирует с сульфатом палладия с образованием соединений с характерным спектром поглощения в ультрафиолетовой области. Метод позволяет определять 0,5— 7,0 мкг/мл бромида. Светопоглощение измеряют при 230 нм, такую же реакцию дают хлорид и иодид [30].
Метод молекулярной эмиссии в полости
В присутствии индия бромиды (а также хлориды и иодиды) дают интенсивную молекулярную эмиссию в холодном диффузионном пламени. Это позволяет определять каждый галогенид в присутствии двух других [31]. Метод рассмотрен полнее в разделе «Хлориды».
Электрохимические методы
Потенциометрические методы., ионоселективные электроды
Потенциометрические методы, описанные для хлоридов, в значительной степени применимы и для бромидов. В классическом титровании при использовании стандартного раствора AgNO3 в качестве титранта кривая титрования получается очень четкой. Широкое применение метод нашел в микроанализе бромсодержащих органических соединений после сожжения их в кислороде в закрытой колбе. Для бромидов фактор пересчета меньше, чем для хлоридов, так как для одинаковой навески образца расход титранта AgNO3 ниже за счет большей молекулярной массы бромсодержащих органических соединений. В равной степени этот фактор компенсируется за счет увеличения скачка потенциала в точке эквивалентности. Если бромид находится в смеси с хлоридом, титриметрическое определение суммы галогенидов — несложная задача. Однако определение каждого из галогенидов чрезвычайно сложно за счет образования смешанного осадка галогенидов серебра. При расчете потенциометрических кривых титрования смеси галогенидов Мартин [32] показал, что можно найти конечную точку титрования графическим методом и как можно применить методы коррекции, если мольное соотношение галогени
дов в смеси больше 21 : 1. Проблема определения галогенидов в смеси может быть решена удалением бромида из второй аликвотной части анализируемого раствора и титриметрическим определением в ней хлорида [33].
Дифференциальная электролитическая потенциометрия применима для определения бромидов. Использование метода кулонометрии при постоянном токе позволяет определять 10-11 М бромида, реакцию проводят в ячейке емкостью 0,5 мл в среде 0,01 М HNO3 в смеси 80: 20 метанол — вода [34].
Бромид в присутствии хлорида, иодида и фторида можно оттитровать потенциометрически, используя смешанный титрант AgNO3— Th(NO3)4 [35]. Другие потенциометрические методы, например с использованием ртути(II), применимы для определения бромидов и хлоридов. Они описаны в разделе «Хлориды».
В этом разделе основное внимание будет уделено потенциометрическим методикам, разработанным для определения именно бромида. В работе Вортона [36] показано, что бромид в микро-н полумикроколичествах (1—200 мг в 25—100 мл) можно оттитровать в среде 2 М хлорной кислоты аммонийной солью гексанитрата церия (IV). При использовании платинового и насыщенного каломельного электродов скачок потенциала составляет 300 мВ. Определению не мешают иодиды до 0,02 М и сульфаты при содержании меньше 0,005 М. Хлориды снижают скачок потенциала в конечной точке титрования, поэтому определение бромидов возможно в присутствии не более 0,04 М хлоридов.
Оженко [73] использовал гипохлорит в качестве титранта для определения бромида. Сульфат не мешает определению, хотя в его присутствии скачок потенциала уменьшается. Хлориды при содержании меньшем 15 г в 1 л не мешают определению, большие количества вызывают сдвиг скачка потенциала.
В работе [38] описано прямое потенциометрическое определение бромида в интервале концентраций 1СН—10~4 М при использовании Ag—AgBr-индикаторного электрода и каломельного электрода сравнения. Ацетат, хлорид, перхлорат, сульфат-ионы не влияют на определение на уровне их концентраций 2000 ppm, тогда как цианид, бихромат, иодид, перманганат и тиосульфат мешают при содержании их на уровне 10 ppm. Определению также мешают анионы, которые окисляют бромид до брома, взаимодействуют с Ag+ с образованием менее растворимых по сравнению с AgBr солей или образуют комплексы с ионом серебра. Эти анионы должны быть предварительно отделены
Когда наряду с бромидом присутствует гппобромит, бромит и бромат, можно проводить определение с применением потенциометрического титрования мышьяком(III) в щелочной среде 139]. Титрование в отсутствие OsO4 и при введении его в раствор позволяет определять гипобромит и бромит. Бромат определяют, используя тот же титрант в H2SO4. Наконец, бромид определяют, Т,1ТРУЯ AgNO3 с использованием серебряного индикаторного элек-Р°Да вместо платинового, который применяют при титровании
271
гнпобромита, бромита и бромата. На полное определение всех четырех анионов затрачивается 15 мин.
В качестве редокс-титрантов для потенциометрического определения бромидов рекомендуют также тетраацетат свинца (40], кобальт(Ш) [41] и церий(1¥) |42].
Применение ионоселективных электродов для определения бромидов практически не отличается от использования их для анализа хлоридов, поэтому в этом разделе будут рассмотрены лишь особенности определения бромидов. Твердые электроды фирмы «Орион» (Орион 94-17) более селективны по отношению к бромидам, чем к хлоридам; так, константа селективности для бромидов в присутствии хлоридов составляет 3-102. Соответствующий бромидный электрод марки Орион 94-35 характеризуется электродной функцией [43] в интервале 10°—51 Сг6 А1 бромидов. Мешающие определению бромида иоиы приведены ниже:
Ион.......... S2' CN"
Константа се- ,
лективности Очень 1,2-10
большая
Г NH3 СГ ОН'
5 - 10э 0,5 2,5 • 103 3 • I0-8
Нитраты, фториды, гидрокарбонаты, сульфаты и фосфаты не мешают определению. Очевидно, что электрод более чувствителен к иодиду по сравнению с бромидом, поэтому иодид не мешает определению, если только он присутствует в очень малых концентрациях. Как и хлоридный, твердый Ag+/S2~ электрод, например электрод марки Орион 94-16, пригоден для определения бромида. Максимально допустимое соотношение концентраций хлорида к бромиду в анализируемом растворе задается уравнением
[СГ] Кр (AgCl)
[Br'J Kp(AgBr)
где КР — произведение растворимости соединения.
Это соотношение показывает, что концентрация хлорида, в 20 и более раз превышающая концентрацию бромида, будет вызывать погрешность определения последнего [44]. Опубликованы работы по применению бромидного ионоселективного электрода в органическом микроанализе [45] бромсодержащих органических соединений, сыворотке [46], плазме [47], безалкогольных напитках [48] и атмосферной воде [49].
Кулонометрические методы
Бромид, так же как хлорид и иодид, можно определять кулонометрическим методом. Обычно используют метод с электрогенерацией иона серебра. Смесь галогенидов анализируют, проводя последовательный электролиз при подходящем значении потенциала-Например, в работе [50] 5—100 мкг Вг~ определяли в образце, содержащем хлорид. Более подробно этот метод будет рассмотрен в разделе «Хлориды».
272
Амперометрические методы
Разработаны амперометрические методы определения бромидов. Как и хлорид, бромид можно определять, используя в качестве титранта Cd(NO3)2 в среде безводной уксусной кислоты. Метод, основанный на осаждении CdBr2 [51], может быть применен и для определения роданид-иона. В работе [52] определяли бромид, хлорид и иодид в присутствии цианида и цианата. Метод приведен в разделе «Иодид». В работе [53] описан амперометрический метод определения бромида и бромата при их совместном присутствии. В этой работе используют вращающийся платиновый электрод и AgNO3 в качестве титранта. Содержание бромата находят, обрабатывая аликвотную часть раствора H2SO3 и титруя затем суммарное содержание бромида.
Полярографические методы
Бромид можно определять полярографически. Калзолари, Габ-риэлли и Марлетта [54] использовали экспрессный метод для определения хлорида, бромида и иодида в морских водорослях после их сожжения. Для хлорида и иодида регистрируют анодные волны, для бромида — катодные волны бромата, который получается в результате количественного окисления бромида в контролируемых условиях.
В работе [55] использовали серебряный электрод новой конструкции для осциллополярографического определения бромидов, хлоридов и иодидов. Метод применим для определения 10~3 — — 10-6 М галогенидов. Было опубликовано сообщение об использовании в вольтамперометрии для определения бромида и иодида вращающегося графитового электрода [56]. Катодно-лучевым полярографическим методом Ронея [57] можно определять бромиды, иодиды и некоторые другие ионы.
Каталитические методы
Аммоний окисляется гипохлоритом до трихлорамина по следующей реакции:
NH3 + ЗСЮ' + ЗН+ —> NC13 + 3H2O
В присутствии бромида гипохлорит окисляет бромид до гипобро-мита, который в свою очередь окисляет аммоний до азота:
СЮ' + Вг" —> ВгО' + СГ
ЗВгО* + 2NH3 —> N2 + ЗН2О + ЗВг"
Таким образом, бромид ингибирует реакцию хлорирования аммо-ия до трихлорамина. Это и было использовано [58] в качестве сновы каталитического спектрофотометрического метода определения 0,02-1,20 ppm бромида,
273
Основой другого метода явилось каталитическое влияние бромида на окисление иода до йодата Celv в присутствии К2Сг2О7 в среде HNO3 [59]. Метод применен для определения 0,01—0,1 ppm (10-6— 10~5%) бромида в природных водах. Хлорид в количествах, превышающих 1 ppm, мешает определению, но содержание хлорида можно определить с использованием роданида меди, а затем при определении бромида ввести соответствующую поправку. Этот метод аналогичен известному методу [60], в котором бромид катализирует окисление иода до йодата КМпО4 в среде H2SO4. В этом случае определяют 0,001—0,1 ppm Вг_. Свободный хлор должен отсутствовать. Опубликован [66] кинетический метод определения бромида, основанный на его ингибирующем действии на скорость разрушения метилового оранжевого броматом. Чувствительность метода 5-10-9 М бромида.
Радиохимические методы
Бромид и хлорид можно определять, добавляя к анализируемому раствору 203Hg(IO3)2 и измеряя активность образующихся HgBr2 или HgCl2 [61]. HgBr2 можно экстрагировать диэтиловым эфиром и затем измерять у-активность водной фазы [62]. Метод нейтронной активации применяли для определения бромидов и хлоридов. Выделение радиоактивных изотопов иода и брома из облученных образцов проводили с использованием экстракции и ионного обмена. Разработан метод [63] определения бромидов и йодидов, чувствительность которого равна 0,01 мкг I- и 0,1 мкг Вг~. Нейтронно-активационный анализ был применен для определения бромидов в сигаретах и других материалах [64].
Газохроматографические методы
Разработан быстрый метод разделения и определения малых концентраций галогенидов с использованием метода газовой хроматографии. Некоторые из этих методов приведены в разделе «Хлориды».
Арчером [65] разработан метод определения брома в крови, основанный на окислении бромида в свободном от протеинов фильтрате до брома KMnCU в кислой среде. Бром экстрагируют циклогексаном, содержащим циклогексен, при этом образуется 1,2-дибромгексан. Это соединение определяют газохроматографически, в качестве внутреннего стандарта используют 1,6-диброМ-гексан. Метод применим для определения 0,1—1,0 мг В г- в 1 мл-Газохроматографический метод рекомендован для определения бромида в зерне [67]. В основу метода положены исследования, описанные в работе [68]. Пик 2-бромэтанола измеряют с помощью электроно-захватного детектора. В качестве газа-носителя используют азот, колонку из нержавеющей стали заполняют смесью, с0^ держащей 15% (моль.) полипропиленгликоля и хромосорб В (Ра3*
мер зерен 0,18—0,25 мм), работают при температуре не выше 120 °C.
Другие методы определения
Некоторые методы одинаково пригодны и для хлоридов и для бромидов. К ним относятся полуколичественные методы кольцевой бани [69] и циркуляционной тонкослойной хроматографии [70].
Редклифф [71] разработал экспрессный метод определения бромидов в природной воде, используя метод рентгенофлуоресцен-ции. Бромид концентрировали на круглом фильтре, содержащем ионообменную смолу. Интенсивность флуоресцентной линии Вг/<а1 измеряли с использованием вольфрамовой рентгеновской трубки. Интенсивность линий сравнивали со стандартом, содержащим 10 ppm бромида. Открываемый минимум 0,05 ppm, ошибка — около 5 %.
ЛИТЕРАТУРА
1. Foil S. С. — Rep. Congr. atom. Energy Comm U. S , AD 734383 (1971); Analyt. Abstr., 1973, v. 24, p 511.
2. Winefordner J. D., Maung Tin — Analyt. Chem., 1963, v 35, p. 382.
3. Firsching F. H. — Analyt. Chem., 1960, v. 32, p. 1876.
4. Kolthoff I. M., Berk L. H. — Z. analyt. Chem., 1927, v. 70, p. 369.
5. Kapel M., Fry /. C., Shelton D. R. — Analyst, 1975, v. 100, p. 570.
6. Mehlig J. D. — Chemist Analyst, 1955, v. 44, p. 87.
7. Pribil R., Makrova V. — Chemicke Listy, 1966, v. 60, p. 89; Analyt. Abstr., 1967, v. 14, p. 2544.
8. Гусев С. И., Кожевникова И. А. ЖАХ, 1963, т. 18, с. 366. Analyt. Abstr., 1964, т. 11, с. 968.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
25.' Lik,?? L L M-' El-Sirafy A. A. Ussar U7. — Analvtica chim
9. De Sousa A. — Analytica chim. Acta, 1960, v. 22, p 520.
10. De Sousa A. — Inf. Quim. Anal., 1963, v. 17, p 127; Analyt. Abstr., 1964, v. 11, p. 5476.
И- De Souse A. — Z. analyt. Chem., 1960, v. 174, p. 337; Analyt. Abstr., 1961. v. 8, p. 125.
Fildes J. E., Macdonald A. M. G.— Analytica chim. Acta, 1961, v. 24, p. 121. Kolthoff I. M., Belcher R. — Volumetric Analysis, Vol. Ill, New York, Inter-science, 1957, p. 254.
Belcher R., Wilson C. L. — New Methods in Analytical Chemistry, London, Chapman and Hall, 1955, p. 201.
Dow M. L. — J. Ass. off. analyt. Chem., 1970, v. 53, p. 1040.
Summersgill A'. —Cherny Ind., 1961, p. 782.
E, Moses G.— Analyst, 1950, v. 75, p. 343.
Mediisch J. O. — Analytica chim. Acta, 1964, v. 31, p. 286.
Utsumi S. — J. chem. Soc. Japan, 1953, v. 74, p. 35, Chem. Abstr., 1953, v. 47, P- 6815L
Stenger V. A., Kolthoff I. M. — J. Am. chem. Soc., 1935, v. 57, p. 831.
Houghton G. IL — J Soc. chem. Ind., bond, 1946 v 65 p 277
American Public Health Association, American Waterworks Association and P°**ution Control Federation, Standard Methods for the Examination Water and Wastewater, 13th edn, New York, American Public Health Association, 1970.
Р°208^' A ~Z- апа'У‘- Chem - 1056- v 149’ P- 681 Analyt. Abstr., 1956 v. 3,
----,a.....— Chemist Analyst, 1965, v. 54, p. 8.
— — Analytica chim. Acta, 1970, v. 49, p. 97.
275
26
27.
28.
Буникине Л. В., Раманаускас Е. И ЖАХ. 1968, г 23, с. 1364. Analyt. Abstr. 1970, т. 18. с. 2396.
Раманаускас Е. И., Буникине Л. В., Жиленайт М. Научные Труды Высших учебных заведений Лит. ССР, Химия и техно.т. 1968, с. 29. Analyt. Abstr. 1970, т. 18, с. 3962.
Буникине Л., Раманаукас Е. И., Тамульявичуте М. Научные Труды высших Учебных Заведении Лит. ССР, Химия и хим. технол. 1970. с. 41. Analyt
Abstr., 1972, т. 22, с. 1568.
1967, v. 88
29. Komatsu S., Normura Т. — J. chem. Soc. Japan, Pure Chem. Sect.,
p. 63; Analyt. Abstr., 1968, v. 15, p. 3316.
30. Chapman F. W., Sherwood R. M. — Analyt. Chem., 1957, v. 29, p 172.
31. Dag nail R. M., Thompson К. C, West T. S. — Analyst, 1969, v. 94, p. 643.
32. Martin A. J. — Analyt. Chem., 1958, v. 30, p. 233
33. Prokopov T. S. — Analyt. Chem., 1970, v. 42, p. 1096.
34. Bishop E., Dhaneshwar R G. — Analyt. Chem. 1964, v. 36, p 726
35. Chou D. H., Sams L. C — Microchem. J., 1969, v. 14, p. 507.
36. Wharton H. W. — Taianta, 1966, v. 13, p. 919.
37. Огинко В. С. Ж. прикл. химии, Ленинград, 1960, т. 33, с. 2486.
Analyt.
Abstr.
1812.
1971
Edn
177 552;
i
1962, т. 9, с. 128.
38. Pflaum R. Т., Frohliger J. О., Berge D. G. — Analyt. Chem., 1962, v. 34, p.
39. Норкус П., Стильжене С., ЖАХ, 1969. т. 24, с. 1565. Analyt. Abstr., т. 20, с. 3029.
40. Buzkova V., Muldan В., Zyka J.— Colin. Czech, chem. Commun. Engl.
1965, v. 30, p. 28; Analyt. Abstr., 1966, v. 13, p. 2341.
41. Sramkova B., Zuka J., Dolezal J. — J. electroanal. Chem., 1971, v. 30, p.
42. Mathur N. K., Rao S. P., Gaur J. N.— J. sclent, ind. Res., 1961, v. 20, p. Analyt. Abstr., 1962, v. 9, p. 1896.
43. Orion Technical Information.
44. Bazzelle W. E. — Analytica chim. Acta, 1971, v. 54, p. 29.
45. Potman IF., Dahmen E. A. M. F. — Mikrochim. Acta, 1972, p. 303.
46. Carter R. A. — Proc. Ass. clin Biochem., 1968, v. 5, p. 67.
47. Degenhart H. J., Abein G., Bevaart B., Baks J. — Clinica Chim. Acta, 1972 v. 38, p. 217.
48. Turner D. L. — J. Food Sci., 1972, v. 37, p. 791.
49. Harris R. C., Williams H. H. — J. appl. Met., 1969. v. 8, p. 229.
50. Cadersky I. — Mikrochim. Acta, 1966, p. 401.
51. Крешков A. П., Борк В. А., Швиркова Л. А., Апаршева M. И. ЖАХ, 1965 т. 20, с. 704. Analyt. Abstr., 1967, т. 14, с. 573.
52. Ikeda S., Musha S. — Japan Analyst, 1967, v. 16, p. 445; Analyt. Abstr., 1968 v. 15, p. 6633.
53. Ikeda S., Satake H. — Japan Analyst, 1971, v. 20, p. 721; Analyt. Abstr., 1972 v. 23, p. 2453.
54. Calzolari C., Gabrielli L. F., Marietta G. P. — Analyst., 1969, v. 94, p. 774.
55. Гороховский В. M., Измайлова Ф. К. ЖАХ, 1966, т. 21, с. 87. Analyt. Abstr., 1967 т. 14 с. 5393.
56. Dryhurst G., Elving Р. J. — J. electroanal. Chem., 1966, v. 12, p. 416.
57. Rooney R. C. — J. polarogr. Soc., 1964, v. 10, p. 49.
58. Zitomer F., Lambert J. L. — Analyt. Chem., 1963, v. 35, p. 1731
59. Yonehara N., Utsumi S., Iwasaki I. — Bull. chem. Soc. Japan, 1965, v. 38, p. 1887; Analyt. Abstr., 1967, v. 14, p. 5067.
60. Fishaman M. J, Skovgstad M. IF.—Analyt. Chem., 1963, v. 35, p 146.
61. Banyai E., Szabadvary F., Erdey L. — Mikrochim. Acta, 1962, p. 427.
62. Banyai E., Szabadvary F. — Mikrochim. Acta, 1968, p. 729.
63. Ohno S. — Analyst, 1971, v. 96, p. 423.
64. Jankins R. W., Newman R. H., Ikeda R. M, Carpenter R. D. — Anal Lett,
1970, v. 4, p. 451.
65. Archer A. W. — Analyst., 1972, v. 97, p. 428.
66. Hasty R. A., Lima E. L, Ottaway J. M. — Analyst, 1977, v. 102, p. 313.
67. Committee for Analytical Methods for Residues of Resticides and Veterinary Products in Foodstuffs, Analyst, 1976, v. 101, p. 386.
68. Heuser S. G., Scudamore K- A.— Pestic. Sci., 1970, v. 1, p. 244.
276
69 Celap M. В Weisz Н. — Microchim. Acta, 1962, p. 24.
70 Hashmi M. H., Chughtai N. A. — Mikrochim. Acta, 1968, p. 1040.
71 , Radcliffe D. —Anal Lett., 1970, v. 3, p. 573.
БРОМИТЫ
Бромит и гипобромит очень похожи по химическим свойствам, поэтому при обсуждении методов разделения и определения бро- . мита придется часто ссылаться на методы, описанные в разделе «Гипобромит». В некоторых случаях метод применим лишь для определения одного из ионов.
Растворы гипобромита часто содержат бромит и бромат. При совместном присутствии бромита и гипобромита селективное определение бромита возможно за счет различия в скоростях реакций или за счет удаления гипобромита в результате взаимодействия с фенолом или ионом аммония (некоторые исследователи подвергают сомнению эту возможность).
МЕТОДЫ ОТДЕЛЕНИЯ
Бромит отделяют от перхлората, хлората, хлорита, бромата и йодата методом тонкослойной хроматографии [1]. В качестве неподвижной фазы используют смесь 1 : 1 оксида алюминия и кизельгур, растворителем является смесь бутанола, ацетона, аммиака и воды в соотношении (8:10:2:1).
+ Н2О —> AsOf+ 2Г + 2Н’
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Иодиметрические методы
Наряду с гипобромитом и броматом бромит восстанавливается подкисленным раствором KI, выделяющийся при этом иод можно оттитровать раствором тиосульфата [2]:
ВгО; + 4Г + 4Н+ —> 212 + Вг' + 2Н2О
Для титрования иода можно использовать [3] и стандартный раствор мышьяка (III):
I2 + AsO|
В его обычном варианте иодиметрический метод неселективен по отношению к бромиту. Когда бромит находится в смеси с гипобромитом, последний разрушают фенолом и (NH^zSOi [2,4]. В более поздней работе [3] утверждается, что этот метод неэффективен том случае, когда концентрация бромита значительна, так как фонол и аммонийные соли взаимодействуют не только с гипобро-пт°м, но и с бромитом. Более того, фенол, как найдено в этой ра-
Dofi6’ РеагиРУет с иодом и тем самым мешает определению. Под-но обе методики приведены в разделе «Гипобромит».
277
Арсенометрические методы
Описан [5] метод прямого титрования бромита мышьяком (Ш) с использованием тетрацина (желтый пиразолоновый краситель) в качестве индикатора. В смеси бромита и гипобромита восстановление бромита только начинается, когда восстановление гипобро мита уже завершилось. В этом методе требуется предварительное титрование с использованием внешнего индикатора.
Другой метод основан на добавлении избытка стандартного рас твора мышьяка(III) и обратном титровании стандартным раство ром иода. Эта методика является составной частью метода опре деления [2] гипобромита, бромита и бромата. Введение в раствор избытка мышьяка с последующим обратным титрованием иодом позволяет определять лишь сумму гипобромита и бромита. Тот же метод, используемый после разрушения гипобромита ионом аммония, позволяет определять только бромит. Иодиметрическое титрование дает сумму всех трех анионов.
Предложен метод [3] для анализа тех же трех анионов, в котором применяется только стандартный раствор мышьяка(III), Подробное обсуждение метода и методики анализа приведены в разделе «Гипобромита.
Титриметрическое определение бромита, гипобромита
и бромата в смеси [2]
Реагенты.
Мышьяк(1П), 0,03 М (0,12 н.) раствор. Растворяют ие очень точно взвешенную навеску As2O3 в 10%-ном растворе NaOH, подкисляют по фенолфталеину и добавляют 10 г твердого NaHCO3. Разбавляют до 1 л и титруют стандартным раствором иода (о взвешивании As2O3 см. раздел «Арсенаты и арсениты»).
Ход анализа. 1. К аликвотной части раствора объемом 5 мл добавляют 3 или 4 г твердого KI и доводят объем до 10 мл 2 Л! H2SO4. Раствор разбавляют вдвое. Титруют выделившийся 12 стандартным раствором тиосульфата в присутствии крахмала. Результаты титрования соответствуют содержанию всех трех анионов:
ВгО" + 2Г + 2Н+ —> 12 + Вг' + Н2О
ВгО2 + 4Г + 4Н+ —> 212 + Вт" + 2Н2О
ВгОз + 6Г + 6Н+ —> 312 + Вг' + ЗН2О
2. К 5 мл анализируемого раствора добавляют избыток (NH4)2SO4 и около 1 г NaHCO3. Через 10 мин добавляют 3 или 4 г твердого KI и 10 мл 2 М H2SO4. Через 5 мин разбавляют раствор вдвое и титруют стандартным раствором тиосульфата. Результат титрования соответствует содержанию бромита и бромата:
pH 8-9
4ВгО" + 2NHJ + 2е~ ---------> 4Вг" + N2 + 4Н2О
ВгО2- + 4Г + 4Н+ —» 212 + Вг” + 2Н2О
ВгО3 + 6Г + 6Н+ —> 312 + Вг’ + ЗН2О
3. К 5 мл анализируемого раствора добавляют известное избыточное количество мышьяка(П1). Через 5 мии добавляют 4 или 5 г NaHCO3 и разбавленную уксусную кислоту для нейтрализации NaOH (исходный раствор, содержащий гнпобромит, бромит и бромат, — щелочной и содержит избыток NaOH) до тех пор, пока очередная капля уксусной кислоты не начнет вызывать бурное образование пузырьков на поверхности раствора. Титруют стандартным раствором и>
278
используя в качестве индикатора крахмал. Результаты титрования соответствуют содержанию гипобромита и бромита:
ВгО’ + AsOf —> AsO?' + Вг'
BrOJ + 2AsO|* —> 2AsO?” + Вт'
BrOJ — не реагирует
4. К 5 мл анализируемого раствора добавляют твердый (NH4)2SO4 и около j г NaHCOa- Через 10 мии добавляют известное избыточное количество стандартного раствора As111 и через 5 мин после этого титруют стандартным раствором иода. Результаты титрования соответствуют содержанию только бромита:
ВгО2~ + 2AsO?' —> 2AsO?' + Вг~
Расчет результатов титрования:
(1)-(2) = ВгО-
(2)-[(1)-(3)] = ВгО; (4)
(1)-(3) = ВгОз
Электрохимические методы
Андерсон и Мадсен [6] разработали два потенциометрических метода для определения бромита и гипобромита. Один из этих методов, в котором бромит титруют мышьяком(III) в присутствии катализатора OsOi, также применим для определения одного бромита или бромита в смеси с броматом и бромидом. Метод приведен в разделе «Гипобромит». Норкис и Стильджен |7] распространили метод определения бромата и бромида на определение гипобромита и бромита. Описание методики приведено в разделе «Бромат».
Изучено [9] полярографическое поведение бромита. Волна восстановления бромита лежит при —0,88 В, т. е. достаточно удалена от волн восстановления гипобромита (0,0В) и бромата (—1,7 В). Как было найдено, диффузионный ток линейно изменяется с изменением концентрации гипобромита вплоть до содержания его 2 мг в 10 мл. Исследователи обратили внимание на взаимодействие бромита натрия с ртутью и предлагают капающий ртутный электрод вносить в ячейку непосредственно перед проведением анализа. На электроде в области pH = 7 — 10, по-видимому, проходит следующая реакция:
ВгО2 + 4Н+ + 4е" —> Вг'4-2Н2О
ЛИТЕРАТУРА
Peschke W. _ J. Chromat., 1965, v. 20, р. 573
3 м- н • АУаг А- A. —Analyt. Chem, 1963 v. 35, р. 908.
/ N-’ Claeys A. — Analytica ciim. Acta, 1972, v. 60, p. 377.
5 Am. chem. Soc., 1934, v. 56, p. 2211.
6 АУаг A- A- — Analyst, 1964, v 89, p 147.
7 Hersen Madsen H. E. L. — Analyt. Chem., 1965, v. 37, p. 49.
’ r £fyc Л- А. Стильжене С. П. ЖАХ, 1969, т 24, c. 1565. Analyt. Abstr 1971, 8 T- 20, c. 3029.
9 № Lansberg R. — Analytica chim. Acta, 1969, v. 45, p. 505
A. F., Supp G. R. — Analyt. Chem., 1968, v. 40, p. 2063.
279
ХЛОРАТЫ
Хлорат натрия производится промышленностью в больших ко личествах. Основным его потребителем является производство ди оксида хлора для целлюлозной и бумажной промышленности. Хло рат нашел применение в производстве ядохимикатов, в металлурги ческой промышленности, в производстве красок, в синтезе перхло ратов и различных химических полупродуктов. Хлорат натрия при меняют в производстве спичек и пиротехнических смесей.
МЕТОДЫ ОТДЕЛЕНИЯ
Ионный обмен
Хлорат, иодат и бромат в миллимолярных количествах могут быть разделены [1 ] с помощью анионообменных смол Дауэкс-21К (триметилбензиламмоний) или Дауэкс-2(диметилэтанолбензилам моний) в нитратной форме. Элюируют в каждом случае раствором нитрата натрия, порядок вымывания отличен от порядка вымыва вания из смолы простых галогенидов, а именно: иодат и бромат вымываются после хлората.
Методы бумажной и тонкослойной хроматографии
Результаты разделения представлены в табл. 19.
Таблица 19. Отделение хлората методами бумажной и тонкослойной хроматографии
Ли-
Метод Растворитель Разделяемые ионы тера
тура
Бумажная хрома- Бутанол — пиридин — NH3 сю;—сю-— сю:—сю; 2
тографня н ТСХ (0,880 г/см3) (2:2:1)
(силуфол, UV254) Бумажная хрома- Изобутанол — Н2О — пи- сю:—сю:—сг—сю; 3
тографня ридин — NH3 (0,880 г/см3) (15:2:2:2)
ТСХ Изобутанол — тетрагидро- сю;—сю;—сг—сю; 4
фуран — NH3 (0,880 г/см3) (50 : 30 : 20)
Циркуляционная Бутанол — диметилформа- сю;—вго;—го; 5
бумажная хрома- мнд — NH3 (0,880 г/см3)
тографня (3:1:1) Бутанол — ацетон — NH3
(0,880 г/см3) (1:3:1)
ТСХ [(А12О3) : ки- Бутанол — ацетон — NH3 сю;—сю:—Вго;— 0
зельгур = (1 : 1)] (0,880 г/см3) — Н2О
(8:10:2: 1) го;—сю;
ТСХ (кукурузный Ацетон - 3 М NH3 (7 : 3) сю;—вго;—го;—сю;— 1
крахмал) F*—Вг'—сг—г
ТСХ (силикагель) Ацетон — этилметилке- сю;—вго;—го;—F-— 8
тон — 14%-ный раствор NH3 (6:4:1) Вг —сг—г
280
Экстракция
Хлорат можно экстрагировать хлоридом тетрафениларсония или аналогичными соединениями. Несольватированный ионный ассоциат экстрагируется хлороформом.
Раствором хлорида тетрафениларсония в хлороформе [9] хлорат экстрагируется на 100% в интервале pH от 2 до 12. Йодат и перйодат в этой системе экстрагируются хуже. Раствором хлорида тетрафенилсульфония в СНС13 хлорат и перхлорат экстрагируются на 100% при pH = 2— 12. Бромат экстрагируется на 28%, а иодат и перйодат еще меньше.
Чаще всего для разделения используют хлорид тетрафенилфос-фония. В этом случае хлорат и перхлорат экстрагируются СНС13 при pH = 2— 11 количественно, а бромат лишь на 32% [11].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Аналитические методы для определения хлората, по существу, — окислительно-восстановительные методы. Селективность при определении одних анионов-окислителей в присутствии других достигается часто использованием различия в скоростях редокс-реакций. Например, реакция между хлоратом и иодидом медленнее реакции между броматом или йодатом с иодидом. Другим использующимся в анализе свойством хлората является его способность легко восстанавливаться до хлорида, который затем и определяют. В качестве восстановителей используют SO2 в газообразном виде, бисульфит или нитрит.
Титриметрические методы
Иодиметрические методы
Как упоминалось выше, реакция между хлоратом и подкисленным раствором иодида протекает сравнительно медленно:
СЮз + 6Г + 6Н* —> 312 + СГ + ЗН2О
При увеличении концентрации НС1 скорость реакции увеличивается, но в то же время в сильнокислых растворах происходит окисление иодида кислородом воздуха. Некоторые соединения, такие как железо(П) и молибден(IV), катализируют реакцию. Молибден (IV) также катализирует и окисление иодида кислородом воздуха. Разработаны рекомендации [12], улучшающие титрова-ие. Несмотря на указанные трудности, иодиметрический метод етается одним из основных методов определения хлората. Он ХлНимает важнейшее место среди методов определения диоксида нахРа’ гористой кислоты, хлорита, хлората и хлорида, когда они ири°ДЯТСЯ- В смеси ИЗ]. Описан [14] простой метод титрования, Рост°ДНЬ1В для определения хлората в веществах, подавляющих листьев и сорных трав.
281
Можно вместо KI использовать подкисленный раствор КВг. Образующийся бром реагирует с К1, выделяя иод, который затем титруют по обычной методике стандартным раствором тиосульфата. Этот вариант метода рекомендован в качестве стандартного метода анализа хлората натрия [15].
В некоторых вариантах метода используют реакцию иода с гидразином. Гидразин в качестве редуктометрического титранта хорошо известен [16, 17]:
N2H4 —> N2 + 4Н+ + 4е~
Он реагирует с иодом в кислых растворах медленно, а в растворах, содержащих гидрокарбонатный буфер с pH = 7 — 7,4, — быстрее. Эта особенность была использована [19] следующим образом. В сильносолянокислый раствор вводили хлорат и КВг. Образующийся бром вытеснял иод из KI, иод титровали стандартным раствором хлорида гидразина в гидрокарбонатном буфере:
C1OJ + 6Вг' 4- 6Н+ —> ЗВгг + СГ + ЗН2О
Вг2 4- 2KI —> 2КВг 4- 12
N2H44-2I2 —> N2 4- 4Г 4- 4Н+
Сравнительно медленное восстановление хлората иодидом или бромидом можно использовать в аналитических целях. При титровании смеси IO4—Юз—ВгОз—СЮз сначала иодиметрически определяют все четыре аниона, используя в качестве восстановителей КВг — KI в среде НС1. Вторую аликвотную часть раствора титруют в среде разбавленной серной кислоты после непродолжительного нагревания. В этом случае иод выделяется в количестве, эквивалентном только перйодату, йодату и бромату. При дальнейшем титровании можно определить все четыре аниона [18].
Титриметрическое определение хлората с использованием
хлорида гидразина [19]
Реагенты
Хлорид гидразина. Используют именно хлорид гидразина N2H4-2HC1, потому что он лучше растворим. Его можно очистить перекристаллизацией из водных растворов. Готовят 0,025 М раствор, растворяя 2,6237 г чистой соли в воде и разбавляя раствор до 1 л. Установить титр раствора можно по иоду, тнтр которого, в свою очередь, устанавливают по арсениту натрия.
Ход анализа. К 10 мл раствора хлората добавляют 1 г КВг и 20 мл концентрированной НС1 (конечная концентрация НС1 должна быть 8 Л4). Отставляют колбу и выдерживают раствор 5 мин. Затем добавляют 10 мл 10%-ного раствора KI н гидрокарбонат натрия до pH = 7—7,4 и еще небольшой избыток. Титруют стандартным раствором гидразина, добавляя вблизи точки эквивалентности крахмал или смесь крахмала с гликолатом натрия.
Примечания 1. Добавлять избыток гидрокарбоната натрия необходимо для предотвращения разложения гидразина.
Арсенометрический метод
Хлорат восстанавливается при кипячении с мышьяком(Ш) в среде HCI. Избыток мышьяка (III) оттитровывают стандартны’1 раствором КВгО3. Реакция восстановления катализируется OsOi
282
[20]. Кроме того, можно применять хорошо известную реакцию мышьяк(ш)—церий(1У). Этот метод был использован [21] для определения хлората в присутствии перхлората. Хлорат восстанавливается избытком мышьяка(III) или железа(II). Обратное титрование проводят стандартным раствором церия(IV), используя в качестве индикатора ферроин и в качестве катализатора осмиевую кислоту. В этих условиях перхлорат не восстанавливается. Установлено, что реакцию катализируют ионы Ag+ [22]. Хлорат, гипохлорит, хлорит и хлорид можно определить в одном растворе, проводя потенциометрическое титрование мышьяком (III) в присутствии катализатора OsO4 [23].
Методы восстановления
Хлорат количественно восстанавливается аскорбиновой кислотой в 2 М H2SO4:
С1Оз+ЗС6Н8О6 —> С1' + ЗС6НвОв + ЗН2О
Титрование проводят в горячих растворах в присутствии катализаторов— селенистой кислоты и марганца(II) [24]. О конце титрования судят по появлению оранжевой окраски коллоидного селена. Хлорид ртути(II) также можно использовать в качестве индикатора конца титрования. Метод интересен тем, что в отличие от других редокс-методов при определении хлората в этом случае используют прямое титрование.
Следует отметить, что хлорат можно восстанавливать SO2; избыток реагента удаляют кипячением. В этом заключается важное различие между хлоратом и перхлоратом. Перхлорат не вступает в реакцию с SO2, но его можно восстановить кипячением с титаном (III). В работе [25] описано восстановление хлората до хлорида нитритом натрия и гидросульфитом натрия. Образующийся хлорид определяли титрованием нитратом серебра. Леонард, Шахин и Вильсон [26] определяли ряд окислителей, включая хлорат, обрабатывая анализируемый раствор избытком стандартного раствора хрома(II) и оттитровывая непрореагировавший хром(П) стандартным раствором железа(III). В качестве индикатора применяли 1,10-фенантролинатный комплекс ванадия(II).
Комплексометрические методы
Описаны [27, 28] комплексометрические методы определения ионов смеси С10з—ClOt и СГ—C1OJ—-С1О4. Каждый из методов нован на селективном восстановлении кислородных соединений °Ра до хлорида, удалении хлорида из раствора в виде осадка ни2РИда сеРебра, обработке AgCl аммониевой солью тетрацианида еля и титровании вытесненного из комплекса иона Ni2+ ЭДТА.
283
Спектроскопические методы
Спектрофотометрические методы
Опубликовано несколько спектрофотометрических методов определения хлората.
Хлорат в сильносолянокислом растворе реагирует с о-толидином с образованием окрашенного в желтый цвет продукта реакции. Метод, основанный на этой реакции, позволяет определять 0,05—10 ppm хлората [29, 30]. Нитрит и железо(Ш) мешают определению, но влияние железа аддитивно, поэтому его можно учесть и внести коррективы в результаты анализа. Хлорид и нитрат не мешают определению. Для определения хлората можно использовать и бензидин, но, по последним данным, этот реактив обладает канцерогенными свойствами.
Иорданов и Даев [31] нашли, что М,М,М',М'-тетраметил-о-толи-дин (тетрон) более чувствителен по сравнению с о-толидином. Такую же реакцию дают другие окислители, включая бромат, иодат и перманганат.
Хлорат реагирует в среде H2SO4 с бруцином с образованием окрашенных продуктов реакции с максимумом светопоглощения при 435 нм. Основанный на этой реакции метод можно использовать для определения хлората в присутствии перхлората и применять для анализа взрывчатых веществ и пиротехнических смесей, содержащих хлорат [32]. Влияние хлората на образование окрашенного комплекса рения с а-фурилдиоксимом использовано при разработке нового спектрофотометрического метода [33] определения хлората. Этим методом можно определять до 5 ppm хлората.
Метод инфракрасной спектрофотометрии
Малые количества хлората (0,1—1,0 мг) можно определять в присутствии перхлората, бромата, йодата, перйодата и т. д. по его инфракрасному спектру [34]. Измеряют поглощение пря 490,2 см-1.
Электрохимические методы
Потенциометрические методы, ионоселективные электроды
Хлорат можно оттитровать потенциометрически мышьяком (III). используя в качестве катализатора OsO4 [23]. Предварительно подбирая условия, можно определить последовательно гипохлорит» хлорит, хлорат и хлорид, используя для титрования первых трех ионов мышьяк(III) и для хлорида — AgNO3 или Hg(NO3)2- Основные стадии методики следующие.
1. Сначала определяют гипохлорит. Если хлорит отсутствует» гипохлорит можно оттитровать потенциометрически при pH #
S81 ‘
в среде гидрокарбоната. В присутствии хлорита титруют при pH = 12.
2. Хлорит титруют в слабощелочном растворе при pH = 8— 12 в присутствии OsOi.
3. Хлорат титруют As111 непосредственно в среде H2SO4 тоже в присутствии катализатора OsO4.
4. Хлорид определяют потенциометрическим аргентометрическим или меркуриметрическим методом.
Последовательное потенциометрическое титрование гипохлорита, хлорита, хлората и хлорида [23]
Реагенты
Мышьяк(Ш), 0,025 Л1 (0,1 н.) раствор. Приготовление раствора описано в разделе «Арсенаты и арсениты».
г Оксид осмия OsO4, 0,1%-ный раствор в 3 Л! растворе NaOH.
Нитрат серебра, 1 Л! (0,1 н.) раствор.
Нитрат ртути, 0,05 М (0,1 н.) раствор.
Ход анализа. Определение гипохлорита. К смеси, содержащей 0,25—1,5 ммоль гипохлорита, добавляют NaOH, помещают в раствор платиновый и каломельный электроды и титруют мышьяком (111) при постоянном перемешивании до скачка потенциала (см. примечание 1).
Определение хлорита и хлората. После того, как оттитровали гипохлорит, к раствору добавляют 5 мл насыщенного раствора NaHCOs и несколько капель 0,1-ного водного раствора ализаринового желтого II. По каплям добавляют 5 М H2SO4 до изменения окраски раствора от красно-фиолетовой до желтой. Добавляют 4—5 капель катализатора 0sO4 и титруют, как описано выше.
Определение хлорида. После титрования хлората удаляют платиновый электрод (предварительно его тщательно промывают) и заменяют его серебряным электродом. Титруют при постоянном перемешивании 0,1 М раствором AgNO3.
Примечания. 1. Если присутствует хлорит, NaOH можно заменить 235 мл насыщенного раствора NaHCO3.
2. Рассчитывают результаты титрования на основе того, что 1 мл точно 0,025 М раствора мышьяка (Ill) соответствует 2,573 мг С1О-, 1,686 мг СЮ" и 1.391 мг СЮ".
В результате исследований [37] было установлено, что нитратный ионоселективный электрод можно путем несложной модификации использовать для определения хлората. Эта модификация осуществляется путем переведения жидкого ионообменника из нитратной в хлоратную форму. Предел обнаружения полученного таким образом электрода составляет 3-10~5 М хлората, т. е. становится ниже, чем предел обнаружения нитратного электрода, который тоже чувствителен к хлорату. Изучали также константы ^ективности электрода к другим ионам.
Полярографические методы
в Хлорат не восстанавливается на капающем ртутном электроде *Челочном нейтральном и умеренно кислом растворах. Стандарт-потенциал реакции
С1О£ + 6Н+ + бе" СГ + ЗН2О £0 = + 1,47В
285
велик, поэтому можно предположить, что реакция восстановления проходит медленно. Преодолеть это затрудение можно введением катализатора, сильной кислоты для поддержания высокой ионной силы раствора и превращением хлората в другие соединения, способные давать волны восстановления. Использовали [35] быструю стехиометрическую реакцию между хлоратом и избытком железа (II) в 3 М растворе НС1. Ионы CIO4 и СГ не мешают, сильные окислители, например гипохлорит и хлорит, мешают и должны быть удалены. Окислители удаляют, проводя предварительное восстановление в слабокислом растворе при контролируемом потенциале.
Другие электрохимические методы
Можно анализировать смесь хлората и хлорида амперометрическим методом с использованием вращающегося платинового электрода [36]. Титрантом является 0,1 М раствор AgNO3. После предварительного определения хлорида хлорат восстанавливают кипячением с 0,6%-ным водным раствором SO2 в течение 5 мин и повторяют титрование для определения общего содержания хлорида. Анализ проводят в 0,1 М растворе KNO3, содержащем 4 мл 0,5%-ного раствора желатины.
ЛИТЕРАТУРА
1. Skloss J. L., Hudson J. A., Cummiskey С. L. — Analyt. Chem., 1965, v. 37, p. 1240.
2. Thielemann H. — Mikrochim. Acta, 1971, p. 746.
3. Harrison B. L., Rosenblatt D. L. — J. Chromat., 1964, v. 13, p. 271.
4. Seiler H., Seiler M. — Helv. chim. Acta, 1967, v. 50, p. 2477.
5. Mao C.-H. — Acta chim. sin., 1964, v. 30, p. 412; Analyt. Abstr., 1965, v. 12, p. 5786.
6. Peschke W.— J. Chromat., 1965, v. 20, p. 573.
7. Petrovic S. M., Conic V. D. — Z. analyt. Chem., 1967, v. 228, p. 339; Analyt. Abstr., 1968, v. 15, p. 4674.
8. Takeuchi T., Tsunoda Y.— J. chem. Soc. Japan, pure Chem. Sect., 1966, v. 87, p. 251; Analyt. Abstr., 1967, v. 14, p. 4682.
9. Bock R., Beilsiein G. M. — Z. analyt. Chem., 1963, v. 192, p. 44.
10. Bock R., Hummel C. — Z. analyt. Chem., 1963, v. 198, p. 176.
11. Bock R., Jainz J. — Z. analyt. Chem., 1963, v. 198, p. 315.
12. Kolthoff I. M., Belcher R. — Volumetric Analysis, Vol. Ill, New York, Interscience, 1957, p. 271.
13. Hong С. C., Rapson W. H.—Can. J. Chem., 1968, v. 46, p. 2061.
14. Roth F. J. — J. Ass. off. analyt. Chem., 1969, v. 52, p. 1294.
15. British Standard 4122: 1967, London.
16. Belcher R., Wilson C. L. — New Methods of Analytical Chemistry, 2nd edn, London, Chapman and Hall, 1964, p. 93. «
17. Berka A., vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Pergam°n Press, 1965, p. 180. 7
18. Szekeres L. — Z. analyt. Chem., 1960, v. 172, p. 256; Analyt. Abstr., 1960, v. ' p. 3739.
19. Panwar K. S., Mathur H. K., Rao S. P.— Analytica chim. Acta, 1961, v. ?' p. 541.
20. Van Der Meulen J. H. — Chem. Weekbl., 1931, v. 28, p. 238.
286
21 Smith G F., Veraguth A. J. — Analytica chim. Acta, 1957, v 17, p 386.
22 Csanyi L. J., Szabo M. — Taianta, 1958, v. 1, p. 359.
93 Норкус П К, Стильжене С Л ЖАХ, 1969, т. 24, с. 884. Analyt. Abstr.. 1970. т. 19, с. 4862.
24. Erdey L., Bodor Е. — Z. analyt. Chem., 1951, v. 133, p. 265.
25 Coetzee С. I. — Z. analyt. Chem., 1968, v. 234, p. 245; Analyt. Abstr., 1969, ' v. 16, p. 3009.
26. Leonard M. A., Shahine S. A. E. F., Wilson C. L.— Taianta, 1969, v. 16, p. 470.
27. De Sousa A. — Chemist Analyst, 1960, v. 49, p 18.
28 De Sousa A. — Analytica chim. Acta, 1961, v 24, p. 424.
29 Williams D., Haines G. S. — Ind. Engng Chem. analyt. Edn, 1945, v. 17, p. 538.
30. Urone P., Bonde E.—Analyt. Chem., 1960, v. 32, p. 1666.
31. Jordanov N., Daiev Ch. — Taianta, 1963, v. 10, p. 163.
32. Egers C. — Analyt. Chem., 1955, v. 27, p. 1199.
33. Trautwein N. L., Guyon J. C.— Analytica chim. Acta, 1968, v. 41, p. 275.
34. Miller M. W., Philp R. H. — Taianta, 1963, v. 10, p. 763.
35. Meites L., Hofsass H. — Analyt. Chem., 1959, v. 31, p. 119.
36. Musha S., Ikeda S. —J. chem. Soc. Japan, pure Chem. Sect., 1969, v. 90, p. 180;
Analyt. Abstr., 1970, v. 19, p. 213.
37. Hiiro K., Moody G. J., Thomas J. D. R. — Taianta, 1975, v. 22, p. 918.
ХЛОРИДЫ
Необходимость определять хлориды возникает при анализе самых различных веществ. Кроме того, определение хлоридов является часто конечной стадией определения других соединений, например, в элементном органическом анализе хлорсодержащих органических соединений после окислительного сожжения образца и выделения образующегося хлорида. Некоторые хлорсодержащне анионы, такие, например, как гипохлорит, можно определять после восстановления до хлорида по реакциям, характерным для его определения.
Определять хлорид необходимо на самых различных уровнях его концентраций—от макроопределения в особо чистых веществах до содержания в особо чистой воде на уровне ppb (КН %)'.
МЕТОДЫ ОТДЕЛЕНИЯ
Несмотря на то, что многие новые методы определения хлоридов селективны, выделение его остается важной задачей при анализе хлорида. В литературе можно найти довольно широкий спектр методов. Однако пригодность их для целей разделения хлорида, бромида и иодида в тех случаях, когда требуется высокая точность Результатов, вызывает некоторые сомнения.
Осаждение
в Хлорид можно отделить от многих элементов осаждением его виде хлорида серебра. Мешают соединения, осаждающиеся нитратом серебра; бромиды, иодиды, цианиды и роданиды.
с пр Саждением из гомогенных растворов (ОГР) достигается по-Довательное образование Agl, AgBr и AgCI при определенном
287
значении pH, медленное уменьшение которого достигается постепенным удалением аммиака из анализируемого раствора Все три галогенида можно определить гравиметрически [1], последовательно отделяя образующиеся осадки.
Соосаждение
Разработан метод [2] соосаждения следов хлорида с фосфатом свинца. Осадок затем растворяют в растворе железа (III) и хлорид определяют ртуть (II)-роданидным методом. Метод использован в анализе особо чистой воды.
Швастовска, Марченко и Столярчик [3] соосаждали менее 20 мкг AgCl на BaSO4. После растворения осадка хлорид определяли фототурбидиметрически.
Ионообменный метод
Описано несколько ионнообменных методов, некоторые из них пригодны для отделения хлорида от других галогенидов. Катионообменные смолы используют для отделения хлоридов от тяжелых металлов. Анионообменные методы можно применить для разделения галогенидов. В работе [4] описано разделение смеси хлоридов, бромидов и иодидов, содержащей около 2,6 ммоль каждого галогенида; для полного разделения требуется 3 ч. На колонке длиной 150 см, заполненной ионообменной смолой Вольфатит СБВ, разделили [5] смесь фторидов, хлоридов, бромидов и иодидов, содержащую по 3 ммоль каждого иона. Галогениды вымываются из колонки последовательно, начиная с фторида и кончая иодидом. Для элюирования необходимо 2,6 л 1 М раствора NaNO3, элюирование галогенидов последовательно оксалатом натрия и NaNO3, как было найдено, является более эффективным для некоторых пар галогенидов.
Залевская и Старобинец [6] изучали разделение всех четырех галогенидов и нашли, что лучшие результаты получаются при использовании в качестве сорбента ионообменной смолы Дау-экс-14 и в качестве элюента — 0,045 М раствор КОН. Этот метод применяли для разделения смеси галогенидов, в которую входил роданид. Разделение проводили на колонке длиной 40 см с анионитом А-25 в ОН-форме, элюентом служил раствор NaOH [7]. Жидкие ионообменники также были использованы [8] для разделения хлорида, бромида, иодида и фторида. Как было найдено, метод подходит для разделения смесей галогенидов с общей концентрацией 10~4 моль. Разделение фторида и хлорида оказалось наиболее трудной задачей.
Хроматографические методы
С применением колоночной хроматографии на гидратированном оксиде циркония можно разделить [9] иодид, бромид и хло" рид. Элюируют галогениды последовательно 0,15 Л1, 0,25 М и 1 \ растворами KNO3. Те же исследователи изучали разделение гал°"
288
Таблица 20. Отделение хлорида методами бумажной И тонкослойной хроматографии
Метод Растворитель Разделяемые ионы Литература
Бумажная хроматография Бумажная хроматография (ватман № 1) ТСХ (целлюлоза или бумага) ТСХ ТСХ (силикагель) ТСХ ТСХ (кукурузный крахмал) ТСХ (силикагель) ТСХ (силикагель) ТСХ (силикагель+ +5% кукурузного крахмала) Изопропанол — Н2О — пиридин — NH3 (0,880 г/см3) (15:2:2:2) Бутанол — ацетон — вод-вый NH, (3:13:4) Бутанол — днметилфор-мамид — NH3 (1,5—15Л4) * (3:1:1) Ацетон — вода (в различных соотношениях) Изопропанол — тетрагидрофуран — NH3 (0.880 г/см3) (50 : 30 :20) Бутанол — пропанол — дн-бутнламин (9:9:2) н другие растворители Ацетон — н-бутанол — NH3 (0,880 г/см3) - Н2О (65 : 20 : 10 : 5) Ацетон — 3 М NH3 (7 : 3) Ацетон — этнлметнлке-тон — 14%-ный NH3 (6:4:1) Насыщенный водный изобутанол — 40%-нын раствор ацетата аммония (4:1) Ацетон — Н2О (10 : 1) сг—сю;—сю;—сю; СГ— Вт-—Г—SCN" СГ— Вг"— Г—F" сг—сю;—сю;—сю; СГ— Вг"— Г—F‘—CN"— —SCN"—[Fe (CN)6]3-— - [Fe(CN)6F- СГ—Вг"—Г—F" сг—Вг"—г—F‘—сю;- —сю;—Вго;—ю; с do сг—Вг‘—г—f-ю;— —вго;—сю; СГ—Вг"—Г—РО’" СГ—Вг"—Г—SCN"— -[Fe (CN)e]3- и 12 13 14 15 16 17 18 19 20
* Концентрация не влияет на значение Rf.
генидов с применением оксида алюминия в вариантах колоночной, тонкослойной и бумажной хроматографии. В методике с использованием хроматографической колонки иодид, бромид и хлорид последовательно элюировали 0,2 М раствором KNO3 [10].
Метод бумажной хроматографии сейчас все чаще заменяет метод тонкослойной хроматографии. В табл. 20 приведены некоторые детали наиболее эффективных методов бумажной и тонкослойной хроматографии.
Газохроматографические методы
Этот ОпРеделе Деле.
метод в настоящее время применяют для разделения и ния галогенидов. Он будет рассмотрен полнее в этом раз-
10 Зак. 167
289
Дистилляция
Хотя метод дистилляции и не является основным методом отделения хлоридов, однако он может быть использован для этих целей. Эльшаймер, Джонсон и Кохен [21] выделяли следовые количества С1- из водных растворов, содержащих плутоний и уран; хлорид-ион при этом окисляли церием (IV) до хлора и отгоняли. При использовании этого метода для выделения микроколичеств хлоридов на уровне, не превышающем 50 мкг, открывается около 80% от всего количества хлоридов.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
При выборе метода, пригодного для определения хлорида, аналитики сталкиваются с большим набором методов, в которых достаточно трудно ориентироваться. Выбор того или иного метода зависит от многих причин: уровня содержания хлорида в анализируемом образце, требований точности и чувствительности, числа образцов, природы других ионов, присутствующих в образце и, наконец, требований к аппаратурному оформлению метода.
Если другие компоненты анализируемого раствора не взаимодействуют с серебром или ртутью, для определения миллиграммовых содержаний хлоридов можно применять меркуриметриче-ское или аргентометрическое титрование. Если требуется высокая чувствительность, применяют спектрофотометрический метод с использованием роданида ртути. В некоторых других случаях используют ионоселективные электроды. Очевидное преимущество ионоселективных электродов заключается в экспрессном определении и в возможности автоматизации анализа, что особенно важно, когда необходимо анализировать большое число проб. По-видимому, нет современных обзоров по методам определения хлоридов. Обзор Армстронга, Жиля и Рольфа [22] содержит ценную информацию о методах анализа, разработанных ко времени его публикации. Обзоры, посвященные более узким вопросам, будут упоминаться в соответствующих рубрикациях этого раздела.
Гравиметрические методы
Растворимые хлориды определяют гравиметрически в виде AgCI при добавлении к анализируемой пробе раствора нитрата серебра. Осаждение проводят в присутствии разбавленной HNOs-После фильтрования и промывки осадка его сушат при Ю0^ 200 °C и взвешивают. При строгом соблюдении методических ука' заний можно добиться высокой точности определения хлорида-То, что метод отличается высокой точностью, широко признан и, может быть, лишь за исключением современных кулонометр’ ческих методов, гравиметрический метод остается основным *’ тодом прямого анализа особо чистых хлоридов. Его принципиаД
290
особенности рассмотрены в обзоре Кольтгофа и др. [23]. Пон проведении анализа важно обращать внимание на следующее. 1 Р1 Присутствие разбавленной HNO3 необходимо для предотвращения осаждения карбонатов, фосфатов и других анионов, образующих с серебром нерастворимые соли.
" 2. После образования осадка необходимо выдержать его в том
же растворе в течение нескольких часов для завершения процесса коагуляции и вызревания.
3. Промывание осадка водой вызывает пептизацию коллоида. Для ее предотвращения используют разбавленную HNO3.
4. Воздействие света приводит к медленному разложению осадка на серебро и хлор, которое может стать и значительным. Определение следует проводить при неярком освещении, избегая прямого солнечного света.
5. Растворимость осадка можно снизить до пренебрежимо малой, если при осаждении добавить небольшой избыток реагента. Растворимость увеличивается в присутствии солей щелочных металлов и аммония. Тот же эффект наблюдается в присутствии и других электролитов, не имеющих общего иона с осадком.
6. Соосаждение незначительно сказывается на результатах анализа (в отличие от метода определения сульфата по BaSO4),
если определение выполнено методически правильно.
7. Чтобы исключить нежелательные побочные реакции, осадок отделяют не на бумажном фильтре, а на пористом стеклянном фильтре.
Число мешающих ионов велико, это ионы, образующие труднорастворимые соли серебра (например, цианид и роданид) или осаждающиеся в кипящей HNO3 [например, висмут(Ш), сурьма (Ш)], металлы, образующие галогенидные комплексы [например, ртуть(II), кадмий (II), олово (IV), хром (III)], соединения, восстанавливающие серебро(1) и, наконец, железо(Ш). Бромид и иодид хорошо отделяются при окислении и могут быть определены в виде свободных галогенов.
Гравиметрическое определение хлорида
Ход анализа. Разбавляют анализируемый образец, содержащий 0,1 г хлорида, водой до 150 мл и добавляют 1 мл азотной кислоты (1:1). К холодному рас-0В.°РУ д°бавляют медленно, при постоянном перемешивании, небольшой избыток 0.1 М раствора AgNO3 (избыток составляет 5—10 мл раствора AgNO3). Нагревают раствор с осадком почти до кипения, интенсивно перемешивая его, затем агреванне прекращают. Когда осадок опустится на дно колбы, для проверки олноты осаждения к раствору добавляют несколько капель раствора AgNO3. ставляют* раствор с осадком на 1—2 ч. Фильтруют через предварительно вешен™й пористый стеклянный фильтр, прежде чем переносить на фильтр, док два или три раза декантацией промывают холодной HNO3 (1 : 1000). Не-оса°ДИМ° гаРантиРовать полноту перенесения осадка на фильтр. Промывают в посГ На ФильтРе небольшими порциями разбавленной HNO3 до тех пор, пока Сущ-°МЫВИЬ1х в°Дах при добавлении НС1 не перестанет образовываться AgCl, став.пяет оа24736₽И 200 °C. Фактор пересчета массы осадка на хлорид со-
10»
291
Примечания. 1. Анализ следует проводить в рассеянном свете.
2. После использования стеклянный фильтр следует хорошо вымыть ам-мначным раствором, разбавленной HNO3 и затем дистиллированной водой
Гравиметрический метод определения хлоридов в виде AgCI обеспечивает очень высокую точность, но при этом требования к тщательности проведения всех операций также высоки. Литтль [24] детально разработал еще одну сравнительно простую и приемлемую методику. Используя ее, один образец с тремя параллельными определениями можно проанализировать за 10— 12 ч активного рабочего времени или за 65 ч общего времени. Ниже представлены данные, позволяющие сравнить точность и воспроизводимость методики Литтля [24] и стандартной методики [25]:
Стандартный аргентометрический метод............................ 0,099422
Метод Литтля..................... 0,099401, 0,099394,
0,099398, 0,099399, 0,099398
Среднее.......................... 0,099398
Стандартное отклонение........... 0,000001
Титриметрические методы
Аргентометрические методы
Несмотря на то, что аргентометрические методы были разработаны еще в XIX веке, они занимают ведущее место в определении галогенидов. Прямые титриметрические методы основаны на добавлении небольшого избытка Ag+ после полного осаждения хлорида. Избыточное серебро реагирует с образованием окрашенных комплексов, например, с хроматом. Можно использовать и другой адсорбционный индикатор. Хорошо известен метод Фольгарда, основанный на добавлении избытка стандартного раствора нитрата серебра и титровании непрореагировавшего серебра стандартным раствором роданида.
Для аргентометрического титрования предложено большое число индикаторов. Они подробно описаны в обзоре 1967 г. [26]. В обзоре представлены индикаторы для всех вариантов аргентометрического титрования, включая титрование не только галогенидов, но и других анионов.
По определению хлорида и других галогенидов с использованием AgNO3 имеется обширная литература, в которой основное внимание уделено теории ионного равновесия. Хотя рассмотрение теоретических вопросов, несомненно, чрезвычайно важно, оно остается за пределами настоящего обзора; читатель может обратиться к оригинальным работам [27—29, 32]. Потенциометрическое определение хлорида с использованием AgNO3 также имеет большое значение; оно будет рассмотрено в подразделе, посвященном электроаналитическим методам.
292
Титрование по Мору
Этот прямой метод разработан в 1956 г. н основан на титровании раствора, содержащего хлорид, стандартным раствором ДрЬЮз с использованием г качестве индикатора КгСгО4. Небольшой избыток Ag+ образует красно-коричневый осадок хромата серебра Ag2CrO4. Хромат серебра более растворим по сравнению с AgCI и поэтому не образуется до тех пор, пока полностью не осядет AgCI. Титрование можно проводить в ограниченной области pH (6,5—10,3), оптимальное значение pH = 7. При pH < 6,5 становится заметной растворимость Ag2CrO4. Около pH = 10,5 начинает выпадать в осадок AgOH. Требуемое значение pH достигается насыщением раствора NaHCO3 или Са(НСО3)2.
Исследования Бельчера, Макдональда и Перри [30] позволили внести некоторую ясность в этот метод и отчасти улучшить его. Важной рекомендацией явилось предложение использовать индикатор при нейтрализации (если исходное значение pH анализируемого раствора от 5,5 до 10,0), переход окраски которого наблюдается при pH —7 + 0,1. В более ранних работах при нейтрализации азотной кислотой в качестве индикатора использовали раствор КгСгО4—КгСг2Оу [31].
Точность метода Мора несколько ниже, чем метода Фольгарда. Мешающие ионы приведены в табл. 21.
Мешающее влияние аммония требует дополнительных пояснений. Присутствие этого иона сдвигает конечную точку титрования в сторону завышения результатов титрования. Значение pH = = 7,2, как указано в литературе, является верхним пределом
Таблица 21. Ионы, мешающие определению хлорида методом Мора
Мешающие иоиы Примечание
Br*. Г Fe”1, Bi111, Sn,v NI11, Co’1, Cu11 Pb”, Ba’1 Осаждаются в виде основных солей, мешающее влияние Fe’11 можно устранить введением F' Маскирующая окраска Осаждаются в виде хроматов или сульфатов
s°r. s2-, co^-, c2o2* nh; В нейтральных растворах образуют с серебром труднорастворнмые соли, С2О*- можно осадить в виде оксалата кальция Устранить влияние можно доведением pH до 5,8
оказали^И “ е 4 а и и я. I. Попытки устранить мешающее влияние катионов введением ЭДТА Иивд оп Ье3успешными- 2- В средних и больших количествах фосфор(У) и мышьяк(У) ме-^ьш«рекАоеллие"н“;3: ,±.?а дает лучшие результаты при определении в присутствии
онисац нижеНЧеСТВ злеКт₽олнтов» чем мет°Д адсорбционных индикаторов, который будет
293
pH, когда этот эффект еще незначителен. Блок н Вотерс [33] нашли, что влияние 100 мг аммония сказывается уже при pH = = 6,5—7,2. Позднее в сообщении Ваннинена [34] отмечалось, что ошибка анализа за счет влияния аммония может быть вычислена на основании условных констант равновесия. Дополнения внесены авторами работы [35], изучавшими ионные равновесия в системе с использованием компьютера. Они подтвердили обоснование природы ошибок, сделанное Блоком и Вотерсом, и нашли, что оптимальным значением pH, при котором исключается мешающее влияние аммония, является pH =5,8. В основном метод Мора может быть успешно использован для определения хлорида в количествах, меньше 0,1 мг. Метод рекомендован в качестве стандартного для анализа воды [36].
Титриметрическое определение хлорида методом Мора
Реагенты
Нитрат серебра, 0,1 Л1 раствор. Взвешивают точное количество чистого AgNO3, предварительно высушенного в течение 1—2 ч при ПО °C.
Индикатор для нейтрализации, 4,2 г КгСгОч и 0,7 г К2СГ2О7 растворяют в 100 мл воды.
Ход анализа. К 25 мл раствора, содержащего хлорид, добавляют 1 мг индикатора (см. примечания). Титруют 0,1 М раствором AgNO3. Конечную точку титрования определяют по изменению окраски раствора от желтой до неисчезающей красно-коричневой за счет образования Ag2CrC>4
Примечания. Перед титрованием и введением индикатора pH раствора следует довести до 5,5—10,0. Если pH < 5,5, раствор осторожно нейтрализуют не содержащей хлоридов бурой, NaHCO3 или СаСО3.
Титрование по Фольгарду
Это непрямой метод титрования. К раствору, содержащему хлорид, добавляют избыток стандартного раствора AgNO3, непрореагировавшее серебро определяют титрованием стандартным раствором роданида натрия, который образует с серебром малорастворимый роданид серебра.
Железо(Ш) используют в качестве индикатора, так как оно взаимодействует с избытком роданид-иона с образованием интенсивно окрашенного роданидного комплекса железа [reSCN]2+-Титрование можно проводить в сильнокислых растворах. Раствор
следует подкислять для предотвращения гидролиза индикатора — иона железа (III). В методе Фольгарда меньше число мешаюших ионов, чем в двух других основных аргентометрических методах определения галогенидов. Метод более чувствителен, чем метод Мора. Следует отметить, что окраска в конечной точке титрования за счет образования растворимого соединения появляется быстрее, чем осадок в методе Мора. Недостаток метода, связанный с необходимостью использовать два стандартных раствора вм®сТ одного, используемого в прямых аргентометрических метода • компенсируется его более широкой применимостью и больше чувствительностью. •
294
роданид серебра менее растворим, чем AgCI, поэтому после того, как будет оттитровано серебро, не вступившее в реакцию с хлоридом, избыток роданида может реагировать с AgCI:
SCN" + AgCI AgSCN + СГ
Эта реакция может привести к нечеткой индикации конечной точки титрования и к значительно большему расходу роданида. Одним из решений является отделение осадка AgCI перед обратным титрованием. Если проводить эту операцию, то много времени придется затратить на коагуляцию образовавшегося осадка перед его фильтрованием и на удаление сорбированных осадком ионов Ag+. Шулек, Пунгор и др. [37] рекомендовали добавлять KNO3 и затем 3 мин кипятить раствор с осадком для ускорения коагуляции. Другим способом предотвращения взаимодействия роданида с AgCI является введение несмешивающихся с водой жидкостей, которые будут образовывать вокруг частиц AgCI защитную пленку [38]. Присутствие некоторых соединений мешает определению. Цианид образует осадок с Ag+ в кислых растворах. Мешают соединения, окисляющие роданид, но их влияние можно устранить обработкой раствора железом(II). Даже малые количества диоксида азота (в основном из HNO3) мешают, взаимодействуя с роданидом с образованием окрашенных комплексов. Диоксид азота удаляют кипячением раствора перед анализом. Палладий, ртуть и медь(1) также реагируют с роданидом. Мешающее влияние меди(1) можно устранить предварительным окислением ее до меди(II).
Различные восстановители влияют на индикатор — желе-зо(1П). Ионы, образующие с хлоридом комплексы, например ртуть(П), хром(Ш) и таллий(Ш), следует удалить. Наконец, ионы, обладающие собственной интенсивной окраской, например Со2+ и Ni2+, должны отсутствовать. Важно, что фосфат не мешает определению хлоридов этим методом в отличие от метода Мора, где уже 25 мг фосфата в 1 л оказывает мешающее влияние.
Несмотря на все ограничения, ни один метод определения хлоридов не используют в такой степени, как метод Фольгарда, именно этот метод чаще всего рекомендуется в качестве стандартного метода анализа хлоридов. Объектами анализа методом Фольгарда являются мясо [39], цемент [40] и бетон [41].
Титриметрическое определение хлорида методом Фольгарда (0,05-0,1 мг CI ).
Реагенты
. Нитрат серебра, 0,1 М раствор. Используют как и в методе Мора стандарт-зати Раствор AgNO3. Сухой чистый AgNO3 в течение 1—2 ч при НО °C сушат, ем взвешивают соответствующее количество AgNO3 и растворяют в точном “оъеме воды.
08 /кчгм^ колия, 0,1 М раствор. Готовят раствор, содержащий примерно ного в 1 л. Для установления титра раствора разбавляют 25 мл стандарт-тРчровяСТВ°”₽а д0 100 мл> добавляют 2 мл свежепрокипяченой концен-
до поя НН0Н HNO3 и 1 мл индикатора — железа(Ш). Титруют раствором KSCN влення неисчезающей красно-коричневой окраски.
235
Индикатор, насыщенный раствор железо(III)-аммонийных квасцов, содержащий несколько капель HNO3 в 25 мл.
Ход анализа. Методика с применением фильтрования. Помещают 25 .мл анализируемого раствора в стакан, добавляют 5 мл 6 А! Н\О3, небольшой известный избыток 0,1 М раствора AgNO3 и перемешивают 5 мин' Фильтруют осадок, собирая фильтрат в коническую колбу. Осадок на фильтре промывают 1%-ной HNO3. Добавляют к объединенному фильтрату и промывным водам 1 мл индикатора и титруют стандартным раствором KSCN до появления неисчезающей красно-корнчневой окраски.
Д1етоднка с использованием нитробензола. Помещают 25 мл анализируемого раствора в коническую колбу, добавляют 5 мл 6 Л1 HNO3 и небольшой известный избыток 0,1 М раствора AgNO3. Добавляют 2—3 мл чистого нитробензола (см. примечание 2) и I мл индикатора Для коагуляции осадка раствор перемешивают. Титруют 0,1 М раствором KSCN до образования неисчезающей красно-коричневой окраски.
Примечания. 1. Если применять деппмолярные растворы титрантов, ошибкой титрования за счет введения индикатора можно пренебречь.
2. При работе с нитробензолом необходимо соблюдать правила техники безопасности. Длительное вдыхание паров опасно для жизни. Более того, жидкпй нитробензол всасывается через кожу.
Титрование с адсорбционными индикаторами
Часто этот метод называют по имени его автора методом Фаянса. Действие индикатора можно наблюдать по изменению цвета осадка до точки эквивалентности и сразу после полного осаждения хлорида AgNO3. Это происходит, очевидно, в результате хорошо известного свойства осадков сорбировать ионы. До точки эквивалентности осадок будет сорбировать хлориды из раствора, содержащего избыточное количество свободных хлоридов (как и других ионов), как показано на рис. 28, о. Когда весь хлорид прореагирует с образованием AgCl, осадком будет сорбироваться избыток AgNO3, как показано на рис. 28, б. Анион флуоресцеина прочно сорбируется на положительно заряженной ионами серебра поверхности осадка, вытесняя нитрат и образуя с серебром окрашенный в красный цвет комплекс. Флуоресцеин не взаимодействует с AgNO3 в растворе, но реагирует с AgNO3, сорбированным осадком AgCL Таким образом, флуоресцеин является индикатором ионов, сорбированных кристаллической ре* шеткой осадка.
Рис. 28. Осадок хлорида серебра в присутствии избытка СГ (а) и избытка Ag* ($'
296
Поверхность осадка в процессе титрования меняет знак за-а Анион флуоресцеина поглощается положительно заряженной поверхностью AgCl. Родамин 6Ж, используемый для определения бромида, являясь катионом, сорбируется отрицательно заряженной поверхностью осадка AgBr.
Для определения хлорида предложено несколько индикаторов, из которых дихлорфлуоресцеин является лучшим. Неудачные попытки применения адсорбционных индикаторов при определении бромидов и иодидов объясняются тем, что поверхность осадков AgBr и Agl чувствительна к воздействию светового излучения слоя адсорбированного красителя. По этой причине титрование gr- и I" следует проводить в условиях минимального светового
воздействия.
Область pH, в которой можно проводить титрование, зависит от константы ионизации индикатора; дихлорфлуоресцеин является по сравнению с флуоресцеином более сильной кислотой. Это позволяет титровать СП в присутствии дихлорфлуоресцеина при pH от 4 до 10,5, тогда как с флуоресцеином титруют в области pH от 6,5 до 10,5. Механизм индикации, описанный выше, может быть в какой-то степени применен и для толкования самого процесса образования осадка. Для того чтобы осадок обладал большой поверхностью, необходимо по крайней мере частично диспергировать его в виде золя. Осадок имеет тенденцию коагулировать непосредственно вблизи точки эквивалентности, особен-
но если концентрация галогенида велика или в растворе присутствуют большие количества нейтральных электролитов. Защитные коллоиды, например декстрин, можно добавлять для удержания части осадка в коллоидном состоянии. Капал, Фрай и Шелтон *[42] изучали возможность использования для титрования поверхностно-активных веществ. Они нашли, что гидроксипропилметил-целлюлоза способствует получению более четкой точки эквивалентности, чем агар-агар. Этот реагент пригоден для определения хлорида, бромида или иодида. Два последних иона можно определять только при введении гидроксипропилметилцеллюлозы с флуоресцеином в качестве индикатора. Последовательное определение всех трех галогенидов в смеси невозможно.
Нельзя титровать малые количества хлорида, бромида или иодида. Это объясняется тем, что количество образующегося осадка мало, для того чтобы можно было наблюдать изменение его Цвета. Нижний предел определяемых концентраций хлорида, ко-£да РезУльтаты анализа еще удовлетворительны, составляет о,005 М, т. е. 0,2 мг С1~ в 25 мл раствора.
Титриметрическое определение хлорида с использованием адсорбционного индикатора Реагенты
тсл.Литрат сеРебра, 0,1 М раствор. Готовят так же, как в описаных ранее арген-°метрических методах. F
2.0 г ^opecHeun илн дихлорфлуоресцеин, 0,2 г в 100 мл 70%-ного этанола или натриевой соли в 100 мл воды.
297
Ход анализа. Помещают в коническую колбу 25 мл анализируемого раствора и разбавляют водой до 50 мл. Если необходимо, доводят pH раствора до 7—10 при работе с флуоресцеином и до 4—10 при работе с дихлорфлуоресцеином. Добавляют 5—10 капель индикатора и титруют 0,1 Л1 раствором AgNO3. Вблизи точки эквивалентности осадок коагулирует. Продолжают титрование, интенсивно перемешивая осадок до тех пор, пока он не окрасится в конечной точке титрования в красный цвет.
Примечание. Титровать следует в рассеянном свете.
Рассмотрим принципиальные особенности трех основных методов определения хлоридов. Метод Фольгарда характеризуется минимальным числом мешающих ионов, самой широкой областью применения и максимальной точностью. Для определения этим методом требуется два стандартных раствора, анализ недостаточно экспрессен, так как метод — косвенный. Метод Мора — метод прямого титрования, несколько менее точен по сравнению с методом Фольгарда, и большее число ионов мешает определению. Более того, на результаты титрования оказывают влияние нейтральные электролиты. Метод адсорбционного индикатора позволяет проводить титрование при более низких значениях pH, чем метод Мора, но и в этом методе сильно сказывается влияние электролитов.
Другие аргентометрические методы
Как было найдено [43, 44], родизонат натрия является более чувствительным индикатором, чем хромат и железо(Ш), для титрования галогенидов, роданидов и цианидов. При титровании хлоридов в конечной точке титрования цвет меняется от желтого до красно-фиолетового за счет образования родизоната серебра. Как и хромат, этот индикатор нельзя вводить в кислые растворы. Мешающие ионы — те же, что и в других методах. При сравнении этого метода с методами Мора и Фольгарда оказалось, что результаты расходятся меньше, чем на 0,07%. Обратное титрование серебра хлоридами невозможно, потому что в присутствии избытка галогенида окраска индикатора остается неизменной.
Арчер [45] повторил более раннюю работу Карабаша [46] и показал, что дитизон является эффективным индикатором при титровании галогенидов и цианида. Титрование проводят в присутствии больших количеств ацетона, если в качестве титранта использовать 0,004 М раствор AgNO3, можно определять микро-граммовые количества хлоридов.
Меркуриметрические методы
Прямое титрование хлоридов ртутыо(П) основано на образовании слабодиссоциированного хлорида ртути. Небольшой избыток ртути в конечной точке титрования можно обнаружить, используя подходящий индикатор. Обычно применяют нитропрУс' сид натрия, который образует с Hg2+ труднорастворимое соединение HgFe(CN)sNO, в результате чего в растворе появляется
298
белый коллоидный осадок. В качестве более эффективных инди-яторов рекомендуют дпфенплкарбазид и дефенилкарбазон.
рт (II) с дифенилкарбазоном образует фиолетово-голубой комплекс. В качестве титранта используют нитрат или перхлорат ртути. Достоинством метода является возможность проводить титрование в кислых растворах. Реакция значительно сложнее, чем может показаться сначала: образуется несколько комплексов ртути с хлоридом (HgClt, HgCf, HgCh) ; но HgCI2 — наиболее устойчивый. Реакция в некоторой степени нестехиометрична, поэтому для получения точных результатов определения необходим жесткий контроль за условиями титрования. Для уменьшения индикаторной ошибки ее можно предварительно экспериментально определить. Самой ранней работой, в которой приведен этот метод, является работа Макдональда [47].
Применение в водных средах разбавленных растворов приводит к неудовлетворительным результатам анализа. Были сделаны попытки разработать метод титрования с использованием смесей этанол — вода; наибольших успехов достиг Ченг [48]. Он рекомендует проводить титрование в 80%-ном водно-спиртовом растворе при pH = 3,5 Улучшение контрастности перехода окраски в конечной точке титрования особенно заметно, если этот вариант метода сравнивать с титрованием в чисто водных растворах. Кроме того, титрование становится строго стехиометричным. В дальнейшем обсуждении ссылки будут делаться именно на эту. модификацию метода.
Вайт [49] при обследовании метода и его применимости в органическом микроанализе нашел, что оптимальным является pH = 3,6. В последующей публикации Вайта [50] описано применение метода для многочисленных определений серы и хлорида в органических соединениях. Отклонение pH от оптимального значения является источником двух видов ошибок. При более высоких значениях pH розовая окраска появляется раньше точки эквивалентности, что приводит к занижению результата анализа. При более низких значениях pH (pH 3) интенсивность окраски комплекса уменьшается. В этом случае получаются завышенные результаты титрования. Для регулирования pH применяют разбавленную HNO3.
Щелочные и редкоземельные металлы, средние количества меди и железа(III) не оказывают влияния на результаты анализа. Железо (III), хромат, фосфат, сульфид, бромат и иодид мешают определению. Влияние фосфата связано с его буферирующей спо-ооностью, сульфат ингибирует появление окраски индикатора, вероятно, за счет малой растворимости в водно-спиртовых раство-ВаХ/^ХЛьФата ртути(II). Сульфат легко удаляется введением ₽Щ1О3)2 пеРед доведением pH до нужного значения. Цианид и имя НрД обРазУют с ртутью(II) слабодиссоциированные соедине-с ' влияние фторида и фосфата исследовал Колсон [51] в со»ж С необх°Димостью анализа органических материалов после ения их в кислороде. Как было найдено, эффективным
29Э
оказалось введение нитрата тория, осадки фосфата и фторпда тория отделяли фильтрованием перед титрованием хлорида.
Область нижних значений определяемых концентрацией хлоридов простирается до полумикроопределений. Белчер, Гавернер и Макдональд [52] применили метод в полумикроорганическом анализе, когда оптимальная навеска образца содержала 20-— 25 мкг хлорида. Анализ проводили после сожжения образца в колбе с кислородом. В методе, разработанном Вайтом [49] и описанном выше, в навеске образца содержалось 1 —1,5 мг хлорида. В этом же исследовании было установлено, что в 20 мл водного раствора можно оттитровать с ошибкой 1—2% д0 15 мкг/мл хлорида. Стандартное отклонение быстро увеличивается при уменьшении концентрации хлорида. В другой работе отмечено, что стандартное отклонение при определении 1 мкг/мл С1~ составляет ±40 нг. По мнению авторов, метод может быть применен для определения 5 мкг хлорида.
Чаще всего метод используют для анализа воды. Для этих целей метод обладает преимуществами по сравнению с методами Мора, Фольгарда и Вибека [36, 53]. Метод явился объектом разностороннего изучения несколькими авторами; все они рекомендовали метод для анализа органических соединений после их сожжения.
Титриметрическое определение С1_ (1—1,5 мг) с использованием нитрата ртути(П) [49]
Реагенты
Нитрат ртути, 0,005 М раствор. Растворяют 3,4 г Hg(NO3)2H2O в 570 мл 0,01 М HNO3. Раствор оставляют на 48 ч, затем фильтруют и разбавляют водой до 2 л.
Устанавливают титр раствора. Для этого в колбу помещают 5 мл стандартного раствора NaCl и добавляют 10 мл дистиллированной воды и 2 капли индикатора бромфенолового синего. Нейтрализуют до желтой окраски индикатора. Добавляют 0,5 мл 0,1 М HNO3, доводят раствор до 100 мл 95%-ным этанолом и титруют, как описано в методике. Определяют поправку на индикатор в 15 мл дистиллированной воды [она составлиет 0,035 мл 0,005 М раствора HgfNOsh]-
Хлорид натрия, 0,01 М раствор. Готовят растворением точной навески в известном объеме воды.
Жнфеноловый синий, 0,1%-ный раствор в этаноле. енилкарбазон, 0,1%-ный раствор в этаноле.
Азотная кислота, 0,1 М и 0,01 М.
Ход анализа. К раствору, содержащему 1—1,5 мг хлорида, добавляют 2 капли индикатора бромфенолового синего, 0,1 М HNO3 до появления желтой окраски и еще 0,5 мл 0,1 М HNOa- Доводят объем 95%-ным этанолом до 100 мл; pH этого раствора должен быть 3,6. Добавляют 0,5 мл индикатора дифенилкароа-зона и тнтруют нитратом ртути при постоянном перемешивании до первого появления фиолетовой окраски (до точки эквивалентности раствор не окрашен)-
Комплексометрический метод
Косвенное определение хлорида с использованием ЭДТА возможно после взаимодействия осадка AgCl с КгМЦСП)*:
2AgCl + Ni (CN)l' —> 2Ag (CN)2 ± Ni2+± 2СГ
300
Цоны Ni2+ титруют стандартным раствором ЭДТА с мурекси-дом в качестве индикатора. Отделение осадка, очевидно, является основным недостатком метода, который характерен так-же и для метода, разработанного Гусевым, Соколовой и Кожевниковой (56]. В этом методе хлорид взаимодействует с РЬ2+ и 2-(2-гидроксп-1 -нафтилметнленампно) -пиридином с образованием соединения состава RPbCl2, где R — органический лиганд. Осадок отфильтровывают, растворяют в HNO3 и ионы свинца титруют стандартным раствором ЭДТА, используя в качестве индикатора ксиленоловый оранжевый. Аналогичный метод разработан и для определения бромида.
Разработано несколько методов, в которых хлорид можно определять в присутствии других ионов. Это методы определения хлорида в присутствии бромида и иодида [57], иодида [58], цианида и роданида [59], хлората и перхлората [60]. Комплексометрические методы определения хлоридов, по-видимому, не найдут широкого применения.
Редокс-методы
Хотя редокс-методы не используются для определения одного хлорида, они применимы для анализа смеси галогенидов. Как можно судить по редокс-потенциалу, иодид окисляется легче, чем два другие галогенида:
С12 + 2е" 2СГ Е°= 1,359
Вг2 + 2е" 2Вг’ Е°= 1,064
12 + 2е‘ ^=> 2Г Е° = 0,6197
Окисление иодида проходит довольно легко, при этом хлорид и бромид не окисляются. Определение хлорида и бромида при их совместном присутствии более трудная задача, так как их редокс-потенциалы очень близки. Селективного отделения бромида можно добиться, применяя различные реагенты, хотя точность такого разделения невысока. В этом разделе будут представлены и Другие титриметрические методы последовательного определения хлоридов, бромидов и иодидов.
Другие титриметрические методы
В методе Вибека для определения галогенидов используют гидроксицианид ртути:
2HgOHCN + 2NaX —> Hg (CN)2 + 2NaOH + HgX2
Х = СГ, Вг', Г
Образующийся NaOH титруют стандартным раствором кислоты ти Основной областью использования метода является микро-нидР°рание галогенидов после сожжения органических соедине-• Ьго достоинствами являются высокая точность и селектив
301
ность, возможность использования в присутствии азота или серы. Меркуриметрическии метод, ранее описанный в этом разделе, вероятно, более пригоден для индивидуального определения хлорида или бромида.
Методом Фольгарда можно определять суммарное содержание галогенидов. Метод Вибека также можно использовать для этих целей. Вариант этого метода описан Белчером, Макдональдом и Нюттеном (63]. Следует отметить, что некоторые методы, разработанные для определения хлоридов, неприменимы для анализа смеси галогенидов. К их числу относится меркуриметрическин метод.
Определив общее содержание галогенидов, бромид или иодид можно определить селективным редокс-методом и рассчитать содержание хлорида по разности. Если присутствует иодид, его определяют в присутствии хлорида, используя метод Лайперта. Он основан на окислении иодида до йодата бромной водой, отгонке избытка брома и иодиметрическом окончании с использованием тиосульфата:
Г + ЗВгг + ЗН2О —> Юз' + 6Вг" + 6Н+
IOJ + 5Г + 6Н+ —> 312 + ЗН2О
Подробнее метод описан в разделе «Иодид». Сравнительно большие количества бромида и хлорида не мешают определению. Метод рекомендован для анализа смеси галогенидов, образующихся из галогенсодержащих органических соединений.
Анализ смеси хлорида и бромида классическими титриметриче-скими методами без предварительного разделения провести трудно. Некоторые методы рекомендуют для этих целей, но ни одного простого и эффективного метода так и не разработано. В основном применяют окисление бромида до бромата или брома. Много таких методов приведено в монографии Кольтгофа и Белчера [64]. Бромид селективно окисляется гипохлоритом. Метод, разработанный Кольтгофом, Ютзи и Ван-дер-Мюленом, рассмотрен в разделе «Бромиды».
Окисление бромида до брома используют в селективном методе [65] определения бромида в присутствии хлорида. В качестве окислителя используют сульфат марганца (Ш):
2Мп3+ + 2ВГ —> 2Мп2+ 4- Вг2
Образующийся бромид реагирует с избытком KI, и свободный иод титруют стандартным раствором тиосульфата. Хлорид не мешает определению бромида. Если в смеси присутствует иодид, результат титрования соответствует суммарному содержанию бромида и иодида. Подробно методика описана в разделе «Бромиды». Описаны комплексометрические методы анализа смеси галогенидов. Однако длительность и сложность этих методик делает их непригодными для практического использования.
302
Спектроскопические методы
Спектрофотометрические методы в видимой и ультрафиолетовой областях •
Метод с применением роданида ртути разработан японскими химиками [66, 67] в 1952 г и сейчас считается одним из лучших методов определения следовых концентраций хлорида. Он основан на взаимодействии хлорида с роданидом ртути с образованием слабодиссоциированного хлорида ртути. Освобождающееся в эквивалентном по отношению к хлориду количество роданида реагирует с железом(III) с образованием ярко-красного комплекса Fe(SCN)2+. Реакции могут быть представлены уравнениями
2Cr + Hg(SCN)2 —> HgCl2 + 2SCN' SCN’ + Fe3+ —> Fe(SCN)2*
Метод дает воспроизводимые результаты анализа и применим для определения 1(Н% хлоридов. Жесткого контроля за условиями проведения реакции не требуется. Светопоглощение измеряют при 460 нм. Так как реакция протекает в кислых растворах, число мешающих ионов невелико. Можно использовать азотную или хлорную кислоту, чувствительность в обоих случаях одинакова Серная кислота снижает чувствительность метода. Флоренс и Феррар [68] отдают предпочтение хлорной кислоте, так как в этом случае ниже результат холостого опыта и выше воспроизводимость результатов. Авторы [68] нашли, что азотная кислота дает заниженные результаты и при этом увеличивается разброс параллельных определений, что можно отнести за счет присутствия в HNO3 оксидов азота. Эти же исследователи нашли, что предел обнаружения метода составляет 15 ррЬ (15-10~7%) с вероятностью 95%. Разработан метод определения хлоридов на уровне ррЬ (Ю~7%) с использованием предварительного концентрирования хлоридов из 250 мл раствора образца соосаждением с фосфатом свинца [2]. Хотя метод разработан для определения концентрации 10-7%, его можно использовать и для более высоких концентраций, например 10-5—5-10-3%
Проведенное ранее изучение метода позволяет предполагать, что в процессе анализа возможно поглощение хлоридов из атмосферы лаборатории, что приводит к значительным ошибкам анализа [69]. Было показано, что основные загрязнения попадают в раствор в процессе проведения реакций, а не при проведении измерений [70]. Описан автоматический вариант метода [71, 72].
Кислотность раствора оказывает незначительное влияние на Результаты анализа. Небольшие количества фторида, нитрата, нитрита, сульфата и фосфора допустимы. Большие концентрации сульфата и фосфата мешают определению. Естественно, мешают и иодиды. При определении 100 ррЬ (10~5%) хлоридов аммоний не влияет при содержании до 80 ppm, гидразин—до 1 ppm, а
403
5 ppm его дают ошибку +13%. В присутствии высоких концентраций этанола, изопропанола, винной кислоты и ацетона раствор окрашивается в желтый цвет. В работе [2] исследовали влияние различных соединений на определение 20 ppb (2-10_6%) хлоридов.
Хлориды определяли этим методом в особо чистой воде [2], промышленных водах [68, 73], биологических материалах [74].
Предложена модификация метода, в которой вытесненный роданид экстрагируют нитробензолом в виде ионного ассоциата с 1,10-фенантролинатом железа (III) и определяют спектрофотометрически [75]. Закон Бугера — Ламберта — Бера выполняется в интервале 0,8—5,6-10~5 М СН Окраска стабильна, чувствительность метода выше чувствительности метода, основанного на применении роданида ртути (П)-железа (III). Большие количества сульфата, фосфата, фторида, карбоната, К+, Na\ NHJ и ацетата оказывают незначительное влияние, а бромид, иодид, цианид мешают определению.
Спектрофотометрическое определение хлорида с использованием роданида ртути [76]
Реагенты
Роданид ртути(П), 0,07%-ный водный раствор.
Перхлорат железа(Ш). Растворяют 6 i соли в 100 мл 4 М НС1О4. Реактив можно также приготовить растворением 14,0 г чистой железной проволоки в разбавленной HNO3. После растворения добавляют 120 мл 60 %-ной НС1О4 и нагревают раствор до появления оксидов азота, продолжают нагревать до тех пор, пока раствор не станет пурпурным, затем охлаждают и разбавляют водой до 1 л.
Ход анализа. Помещают 10 мл анализируемого раствора в мерную колбу емкостью 50 мл и добавляют 5 мл 60 %-ной НСЮ4, 1 мл раствора роданида ртути и 2 мл раствора Fe(ClO4)3. Доводят объем раствора до метки и перемешивают. Через 10 мин измеряют оптическую плотность при 460 нм.
Примечания. 1. Определяемая область 0,05—5 ppm (5-10~6—1О~4<,/о) хлоридов.
2. В приведенной методике концентрация НСЮ4—примерно 1,5 М. При этой кислотности чувствительность максимальна, но велик результат холостого опыта. Более низкий результат холостого опыта можно получить, проводя определение в 1,0 М НС1О4, но в этом случае ниже чувствительность и становится заметным отклонение от закона Бугера — Ламберта — Бера.
Избыток ртути после взаимодействия с хлоридом можно определять в виде фиолетового комплекса с дифенилкарбазоном или с дифснилкарбазидом.
Джерлах и Фрайзер [77] отдают предпочтение первому реагенту, но считают при этом необходимым предварительно его очищать, чтобы исключить образование окрашенных комплексов с примесями в реагенте. Раствор реагента следует готовить ежедневно. Определению мешают медь, железо(Ш), бромид, иодид, роданид, ацетат и малые количества оксалата и бихромата. Хотя метод чувствителен (е = 19000 при 520 нм), перечисленные недостатки делают его применение чрезвычайно ограниченным.
Хлоранилат ртути реагирует с хлоридом с образованием сла-бодиссоциированного хлорида ртути и эквивалентного количества пурпурно-красного аниона хлораниловой кислоты.
HgC6CI2O4 + 2СГ + Н+ —> HgCl2 + нс6а2о;
Эта реакция положена в основу спектрофотометрического метода определения хлорида [78, 79] и явилась первым примером использования хлоранилата в аналитической химии. Как было найдено, необходимо введение 0,5 М раствора кислоты, так как спектр поглощения хлораниловой кислоты зависит от pH. Оптимальная длина волны для измерения оптической плотности — 530 нм, при этом молярный коэффициент светопоглощения равняется 0,18-103. Фторид, бромид, иодид, бромат, фосфат и роданид мешают определению. Закон Бугера — Ламберта — Бера выполняется в интервале 0—100 мг/л С1~. Бодэ, Эггелинд и Стайн-брехт [80] определили константу диссоциации хлораниловой кислоты в различных средах и рассмотрели возможность ее использования для определения различных анионов, в том числе хлорида. В более позднем исследовании Хаммера и Грайда [81] рекомендуется измерять светопоглощение при 300 нм, используя 0,05 М НСЮ4. Применять HNO3 нельзя, так как у нее есть полоса поглощения при 300 нм. Те же исследователи нашли, что метод можно улучшить, если использовать 40%-ный водно-этанольный раствор и измерять светопоглощение при 305,5 нм после отделения избытка хлоранилата ртути с применением центрифугирования или фильтрования в вакууме.
Спектрофотометрическое определение хлорида с использованием хлоранилата ртути [78]
Ход анализа. Пропускают раствор, содержащий хлорид, через колонку размен ром 15 X 1,5 см, заполненную катионообменной смолой в Н+-форме (размер перец 0,29—0,83 мм). Доводят pH элюента до 7 разбавленными растворами HNO3 или NH4OH. Аликвотную часть раствора (45 мл), содержащую не более 1 мг хлорида, помещают в мерную колбу емкостью 100 мл, добавляют 5 мл 1 М раствора особо чистой HNO3 и 50 мл метилцеллюлозы. Раствор разбавляют до метки дистиллированной водой, добавляют 0,2 г хлоранилата ртути и в течение 15 мин интенсивно перемешивают. Избыток хлоранилата ртути отфильтровывают и измеряют светопоглощение раствора при 530 нм по отношению к холостому раствору, проведенному через весь ход анализа.
Хлоридные комплексы железа(II), например FeCl2+, поглотают и в видимой области спектра, но светопоглощение в УФ-области значительно выше.
Вест и Колл [82] разработали прямой спектрофотометрический метод, отличающийся простотой и надежностью, который вполне пригоден для рутинного анализа при определении 0— .5 мг/л С1~. Приведены две методики, каждая из которых обладает своими преимуществами. Бромид и иодид мешают опреде-вию, но их влияние можно корректировать.
Другие спектрофотометрические методы основаны на окисле-хлорида до хлора и определении последнего. Первым реаген
305
том, использованным для этих целей, был о-толидин. Его и сейчас используют для анализа канцерогенных веществ. Для селективного окисления бромидов и хлоридов в качестве дифференцирующего окислителя используют КМпО4 [84]. Затем проводят определение с о-толидином. Область определяемых концентраций 1 — 10 мкг для хлоридов и 10—100 мкг для бромидов. Метод применен для анализа воды и продуктов коррозии. Хлор и бром окисляют метилоранж по азо-группе с образованием л-галогенид ди-метиланилина и фенил-п-сульфокислоты, по этой реакции можно определять оба галогенида [83].
Другой метод основан на обменной реакции между хлоридом и йодатом серебра. Образующийся иод определяют спектрофотометрически:
AgIO3 + cr —+ AgClI + IOJ
Юз + 6Н+ + 5Г —> 312 + ЗН2О
Косвенный метод определения галогенидов основан на вытеснении циановодородной кислоты из цианида ртути(II) [85]. Циановодородную кислоту отгоняют, поглощают КОН и определяют:
Hg (CN)2 + 2Х' + 2Н+ —> HgX2 + 2HCN
X = СГ, Вг”, Г или SCN'
При использовании иодиметрического титрования наблюдается линейная зависимость от концентрации хлорида, но реакция неожиданно оказалась нестехиометричной. Другая методика основана на превращении цианида в краситель при использовании пиридин-пирозолонового реагента. Она пригодна для определения 0,014—0,43 мкг/мл хлорида.
В другом методе определения хлорида его связывают в комплекс с фенилртутыо(П), который после экстракции СНС1з реагирует с диэтилдитиокарбаминатом. Образующийся комплекс [фенилртуть(П)-диэтилдитиокарбаминат] характеризуется максимумами светопоглощения при 257 и 297 нм, один из этих максимумов можно использовать для аналитических целей. Соответствующие максимумам молярные коэффициенты поглощения равны 21,3-103 и 6,5-103. В 250 мл образца можно определять 0,04—0,32 ppm С1“ с относительной ошибкой 12%. Число мешающих ионов ограничено, это Ag', Hg1, Hg", бромид, иодид, роданид, цианид и нитрит. Метод можно применять и для определения бромида и иодида, причем чувствительность метода сравнима с методом, в котором используют роданид ртути [86].
Нефелометрические методы
Нефелометрический метод — старейший метод определения хлоридов. Можно определять 4—300-10-6 М хлоридов по суспензии AgCI в 50%-ном метаноле, относительная ошибка определения составляет 3—4%- Тщательная отработка'деталей выполни
305
нпя анализа позволила добиться точности метода на уровне максимальной точности нефелометрического метода [88]. Определению мешают вещества, образующие в разбавленной HNO3 нерастворимые соли, например бромиды и иодиды. Методические особенности метода приведены в монографии Больтца [89]. Титриметрические методы обычно характеризуются более низкой воспроизводимостью по сравнению со спектрофотометрическими методами, так как первые подвержены большему влиянию таких факторов, как температура, время, порядок ввода реагентов и кислотность. Марченко с сотр. [3] использовали метод для определения следовых количеств хлорида в реактивах после концентрирования соосаждением на BaSOj, применяющемся в качестве носителя. Меньше 20 мкг хлорида можно определять в образцах массой от 0,5 до 5,0 г. Разработан автоматический нефелометрический метод дискретного определения хлоридов в воде [90]. Хлориды осаждают в виде AgCI и измеряют концентрацию суспензии при 600 нм. За 48 мин можно проанализировать до 30 образцов, провести холостой опыт и измерить стандартные растворы. Описанный метод был применен для определения 5—250 ppm С1~, но можно его использовать и для больших или меньших концентраций С1_.
Косвенный атомно-абсорбционный метод
Опубликован косвенный ААС метод, основанный на осаждении хлорида с Ag+ и последующем определении или избытка Ag+ в растворе, или Ag+, получающегося после растворения осадка [91, 92].
Другой метод основан на ААС-определении ртути в хлориде фенилртути(П) [93]. Комплекс экстрагируют CHCI3, экстракт упаривают и сухой остаток растворяют в этилацетате. Измеряют линию ртути при 253,65 нм, используя воздушно-ацетиленовое пламя. Можно определять 0,015 ppm хлорида в 250 мл раствора образца. Определению мешают Ag1, Hg1, Hg”, Br~, I-, SCN~ и CN-.
Эмиссионный спектральный анализ
Подобно сере и фосфору хлор, бром и иод имеют свои атом-НЫе резонансные линии в ультрафиолетовой области. Обычные методики определения галогенидов пламенными методами осно-аны на косвенном определении, при этом к раствору, содержащему галогенид, добавляют известное количество ионов серебра, саДОк отфильтровывают и в фильтрате определяют избыток Ag*-Мет°Д°м или пламенной эмиссионной спектроскопии. Этими п одами определяют сумму хлоридов, бромидов и иодидов.
нал, Томпсон и Вест [94], продолжая свои исследования
307
Рис. 29. Эмиссионный спектр InCi [94]:
1 — образец, содержащий 2,94 • 10—3 М индия, 1 • 10™ 2 М хлорида и 7 • 10—2 М нитрата; 2—холостая проба, содержащая 2,94 • 10™3 М иидия н 7 • 10™ 2 М нитрата.
молекулярной эмиссии элементов в холодных азот-водородных пламенах, применили этот метод для анализа серы, фосфора, олова и позднее галогенидов. Как было найдено, индий является самым подходящим элементом для образования интенсивной эмиссии галогенидов. Линии эмиссии InCi и 1пВг находятся в ближней ультрафиолетовой области и плохо видны, эмиссия Ini наблюдается в виде полосы темно-синего цвета в центральной части пламени. Предполагают, что эмиссия вызвана хемилюминесцентной реакцией восстановления соединения 1п|П, приводящей к возбуждению молекул галогенида индия(I). Спектр InCi [94] показан на рис. 29. Для анализа применяют стандартный пламенно-эмиссионный атомно-абсорбционный спектрофотометр (Уникам СП-900А) (Unlearn SP900A), снабженный стандартной воздушно-ацетиле-новой (прямоугольной) горелкой и нестандартной горелкой, которая описана в работе [94]. Данные по определению хлорида, бромида и иодида приведены ниже:
Галогенид.......... СГ Вг"
Оптимальная длина волны, им.......... 360 376
Определяемые кон-центрации..........2-10 —4-10 М 5-10 —10 М
(7,1 — 142 ppm) (1,8—35,5 ppm)
Г
410
4- 10”5—8-(5,1 — 102 ppm)
Хлориды и бромиды можно селективно определять при их совместном присутствии на фоне большого избытка одного из галогенидов. Подробно методика описана в работе [94]
308
Электрохимические методы
Потенциометрические методы,
ионоселективные электроды
Хлориды можно определять с помощью различных потенциометрических методов, начиная от классической потенциометрии и кончая более современными методами, например с использованием ионоселективных электродов. Многократность применения ионоселективных электродов позволяет предполагать, что для анализа хлоридов в определенной области концентраций этот метод заменит все другие методы. В классической потенциометрии основной частью прибора является серебряный индикаторный электрод и каломельный электрод сравнения, последний соединяется с ячейкой солевым мостиком. В качестве титранта в основном используют AgNO3, хотя в некоторых потенциометрических методах применяют ртуть(П). Спиртовая среда предпочтительнее водной, хотя при этом и повышается значение диффузионного потенциала в точке эквивалентности. Потенциал индикаторного электрода зависит по уравнению Нернста от активности титруемого хлорида.
Стандартный вариант метода описан Шинером и Смиттом [95], которые установили, что ошибка определения хлоридов меньше 0,1%. Что касается смеси галогенидов, то определение их
суммы несложно, но определение каждого галогенида в смеси — задача значительно более трудная. Проблема в том, что осаждается смесь галогенидов серебра. Обычный вид кривой потенциометрического титрования показан на рис. 30. Мартин [96] показал, что на таких кривых конечные точки титрования иодилов и
бромидов можно получить в точках пересечения двух касательных и что этот метод может быть распространен на анализ смесей галогенидов при соотношении компонентов Даже превышающем 20: 1. Титрованию хлоридов в смесях, содержащих хлорид и бромид, присущи те же ошибки, что указаны выше, потому что галогениды серебра легко образуют смешанные кристаллы или твердые растворы. Это приводит к соосаждению некоторого количества хлоридов с бромидом, т. е. значительно Раньше осаждается хлорид, ем это должно было бы быть соответствии с кривой ти-т₽°вания.
Рис. 30. Потенциометрическая кривая (идеальная) титрования смеси галогенидов:
1 — конечная точка титрования I-; 2—конечная точка титрования Вг”; 3—конечная точка титрования CI”.
309
Прокопов [97] подошел к решению этой задачи следующим образом; сначала титруют сумму бромида и хлорида, а затем — вт рую аликвотную часть раствора, обрабатывают пероксидом водорода для окисления бромида до брома. Бром удаляют, связывая его с оксином, после чего титруют хлорид:
Н2О2 + 2Вг' + 2Н+ —> Вг2 + 2Н2О
C9H7ON + 2Вг2 —> C9H5Br2ON + 2НВг
Образующийся по реакции бромид повторно окисляют до тех пор, пока весь он не будет связан с оксином. Иодид реагирует аналогичным образом, поэтому методика применима для анализа смеси иодида, бромида и хлорида.
В потенциометрическом титровании галогенидов источником ошибок может быть сорбция ионов поверхностью осадка. Это может происходить в том случае, когда используют серебряный электрод или галогенид-серебряный ионоселективный электрод. Было найдено [98], что введение NaNO3 до концентрации, равной 1 Л1, эффективно снижает эту ошибку. Потенциометрическое титрование нашло широкое применение в элементном органическом микроанализе. Этот аспект использования метода подробно рассматривает Диксон [99]. Для более тщательной изоляции каломельного электрода от анализируемого раствора, содержащего хлорид- • ионы, применяют два нитратных солевых мостика. Хлорид титруют в среде изопропанол — вода 0,002 М раствором AgNO3. Каломельный электрод можно заменить другим электродом сравнения, включая и металлические электроды.
Другой разновидностью потенциометрического метода является потенциометрия с наложением постоянного тока. Фриман [100] использовал этот метод для определения хлорида, применив схему анализа, описанную в более ранней работе [101]. В системе два поляризованных электрода. С помощью прямого потенциометрического титрования хлорида раствором AgNO3 можно определять до 5-10-5 М аниона.
Для определения следовых количеств хлорида применим метод концентрационного элемента [102]. Для этих целей используют два тщательно подобранных одинаковых электрода, изготовленных следующим образом: серебро осаждают на платиновую сетку и анодно поляризуют электрод в разбавленном растворе хлорида,. Сам концентрационный элемент состоит из двух ячеек, в первой ячейке электрод погружают в эталонный раствор хлорида, во второй ячейке другой такой же электрод помещают в анализируе* мый раствор. Концентрация хлоридов пропорциональна разности потенциалов двух электродов. Этим методом определяли [103] концентрацию хлоридов до 70 ppb в растворах с высокой ионнои i силой.
Усовершенствования, введенные Мальмштадтом и ВайнфорД* нером [104, 105] в метод, называемый потенциометрическим титрованием до нулевой точки, позволяет избежать строгого соблюдения постоянства температуры. Этот метод состоит в том, что
310
концентрацию хлорида в исследуемом растворе изменяют, добавляя к нему стандартный раствор хлорида до тех пор, пока сум-
марная концентрация хлорида в исследуемом растворе не станет равной концентрации этого иона в эталонном растворе. При этом обязательным условием является сохранение практически постоянной ионной силы анализируемого раствора. Равенство концентраций соответствует нулевой разности потенциалов в двух ячейках. Установка для титрования представлена на рис. 31.
Другой вариант потенциометрии до нулевой точки разработан [106] Для определения хлоридов в промышленных водах. Хлориды титруют 0,01 М раствором AgNOs, используя одну ячейку с электродом в виде длинной серебряной проволоки, опущенной в раствор с такой концентрацией Ag+, которая должна быть в конечной точке титрования. В другой ячейке второй серебряный электрод помещают в анализируемый раствор. Чувствительный гальванометр (чувствительность 100—200 мм/мЛ) используют для индикации конечной точки титрования. Схема прибора представлена на рис. 32. Чувствительность метода 0,2 мг С1~ в 150 мл анализируемого раствора. Бромид и иодид мешают при определении хлорида. Ионы и вещества, указанные ниже, не влияют на определение хлорида:
Ионы..........
Максимально допустимые содержания, мкг . . . Вещества . . . . Максимально до-
SO2' so2-3 4
1,5-10* 1,5-10*
Нефть
POJ" Cu2+ Fe3+
1-10* 5-Ю3 75
Метафосфат натрия *
пустимые содер-
жания, мкг ... 1-10*
750
• Полимерный метафосфат натрия обычно используют после растворения в воде.
Другим вариантом дифференциальной потенциометрии с использованием серебряного или хлор-серебряного электрода является дифференциальная электролитическая потенциометрия ДЭП-метод, разработанный Бишопом [107—109]. Основным отличием этого варианта потенциометрии является то, что измеряют разность потенциалов двух индикаторных электродов в перемешиваемом растворе при пропускании через него в течение 1 мин постоянного тока небольшой величины. В точке эквивалентности наблюдается резкое падение потенциала. Схема прибора для анализа представлена на рис. 33. Микротитрование хлорида с использованием этого метода позволяет получить результаты анализа с ошибкой ±2% в обычных случаях и ±0,2% при тщательном выполнении анализа. Метод позволяет определить концентрацию боридов, при которой величина тока в соответствии с уравнением врнста не ниже налагаемого извне тока. Для хлоридов нижний Ределяемый предел составляет 10-11 моль [НО]. На проведение ализа затрачивается значительное время. В зависимости от Ла измеряемых на кривой титрования точек все титрование
311
Рис. 31. Схема прибора для определения хлорида методом прецизионной потенциометрии до нулевой точки [104]:
1 — асбестовая пробка; 2—раствор сравнения; 3— пробирка; 4— источник постоянного тока (батарея) 1,5 В; 5—сопротивление; 6—к нуль-детектору; 7—Ag — AgCl электроды; 8— бюретка; S —анализируемый раствор.
Рис. 32. Схема прибора для определения хлорида методом потенциометрии до нулевой точки [106]:
1 — стеклянная пробка; 2 — анализируемый раствор; 3—раствор сравнения; 4—гальванометр;
5 — делитель напряжения; 6—переключатель; 7—мешалка; 8— серебряная проволока.
длится от 1 до 3 ч. Однако это меньше, чем требуется для классической потенциометрии (30—100 ч) при определении концентраций хлоридов, близких к предельным в соответствии с уравнением Нернста. Значительно лучшие результаты можно получить, если вместо металлических серебряных электродов использовать галогенидсеребряные электроды. Однако проводить анализ на пределе чувствительности метода очень трудно. Поэтому определение с применением ДЭП микроколичеств хлоридов меньших 7-Ю-6 М проходит нестабильно [111].
Обычно используемая для микротитрования малых концентраций хлоридов микробюретка в этом методе непригодна. Бишоп [112] рекомендует использовать ячейку с тремя микроэлектродами, применяемую для кулонометрической генерации титранта. Эту разновидность метода можно назвать ДЭП при постоянном токе в кулонометрическом варианте. В статье [НО] описаны подробно конструкция ячейки для титрования и подробная методика. Большим достоинством метода является экспрессность, что
выгодно отличает его от обычного метода ДЭП, описанного выше.
Обратим внимание на некоторые новые аспекты развития прямой потенцио-
Рис. 33. Схема прибора для микротитриметриче-ского определения СГ с использованием AgNOj методом дифференциальной потенциометрии (п° данным Бишопа [107]):
J—ячейка для титрования; 2—рН-метр,
3|2
метрии, описанные в последних работах. Для последовательного определения хлоридов, бромидов, йодидов и фторидов был использован смешанный титрант [113] AgNO3 — Th(ХО3)4. Титрование проводили при pH = 7,2 и О °C. Для индицпрования конечной точки титрования применяли стеклянный натриевый ионоселектив ный электрод [114], использовав при этом данные более ранней работы [115], в которой утверждалось, что стеклянный натриевый ионоселективнып электрод более чувствителен к ионам Ag+, чем к ионам натрия. Следовательно, этот электрод можно применять для определения хлоридов, бромидов и иодидов. Титрант AgNO3 готовят в 80%-ном изопропаноле, титруемый раствор должен содержать не менее 90% ацетона, ионы натрия должны отсутствовать в титруемом растворе. Прокопов [116] использовал платиновый индикаторный электрод вместо традиционного серебряного. Хотя в большинстве потенциометрических титрнметрическнх методов определения хлоридов в качестве титранта используют AgNO3, было показано [117, 118], что для этих целей пригодна Hg(NO3)2. Этот реагент можно использовать при анализе смесей С1-—Вг-—I- —SCN-.
В одной из последних работ [119] было показано, что автоматическим потенциометрическим методом можно определять очень малые концентрации хлоридов Измеряя разность потенциалов Ag/AgCl-электрода и Hg/Hg2SO4 электрода сравнения можно определять 0—150 мкг/л хлоридов. Метод использовали для анализа особо чистой воды на силовых подстанциях. По-видимому, это первый автоматический метод определения таких малых концентраций определяемого нона.
В настоящее время прочное место в аналитической химии заняли ионоселективные электроды [120—122]. Хлориды можно определять с использованием нескольких электродов различного типа. Чаще всего применяют твердые мембранные электроды. Мембраной такого электрода служит кристаллический AgCl или поликри-сталлическая, изготавливаемая прессованием, таблетка. Гомогенные твердые кристаллические электроды выпускают фирмы «Орион» (Орион 94-17) [123], «Корнинг» [124] и «Бекман» [125]. К другому типу, гетерогенным твердым мембранным электродам, относится электрод, разработанный Пунгором (марка ОП-С1-711); мембраной этого электрода является полимерная силиконовая матрица с запрессованным в нее осадком галогенида серебра [126]. Электрод Орион 94-17 характеризуется электродной функцией в интервале от 1 М до 5-10~5 М. Чувствительность можно повысить Д° Уровня ррЬ, если использовать два электрода в дифференциальном варианте. Хлоридный электрод обычно используют в паре с ал°мельным электродом сравнения, схема показана на рис 34. Существует и комбинированный хлоридный электрод, в котором в Дном корпусе смонтирован и хлоридный ионоселективный элек-Мо?Д’ и электрод сравнения. Примером такого типа электродов
>кет служить электрод марки Орион 96-17.
313
тивности. Ее определяют
Рис. 34. Схема прибора с использованием иоиоселектнвного электрода; 1 — иоиоселективный электрод; 2— электрод сравнения; 3 — милливольтметр; 4—анализируемый раствор.
Ионы, образующие с серебром нерастворимые соли или прочные комплексные соединения, мешают определению хлоридов. Для оценки степени их мешающего влияния существует несколько количественных характеристик. Обычно используют константу селек-из эмпирического уравнения
где Е — потенциал иоиоселектнвного электрода, измеренный по отношению к стандартному электроду сравнения; Ео — потенциал электрода в нормальных условиях; Z/, Z, — заряды определяемого и мешающего ионов; а,-, а, — активности определяемого и мешающего ионов; Ki/ — константа селективности по отноше-1 нию к иону i в присутствии иона /.
Если активность анионов можно заранее измерить и мешающий ион одновалентен, приведенное уравнение упрощается:
£ = £'_-^(n(a/ + /<..0/)
Влияние мешающего иона, таким образом, измеряют в виде произведения КцЩ. Чем меньше значение Кц, тем меньше влияние мешающего иона. Если Кц — I, ионоселективный электрод обладает одинаковой селективностью и к иону i, и к иону /. Мешающие ноны и константы селективности К твердого хлоридного электрода марки Орион представлены ниже:
Иовы . . . ОН" NH; S2Of Вг' Г CN- S2*
К.........1,2 10-2 8 IO2 3-102 2 • 106 5.10е Очень*
большое
•S2" должен отсутствовать даже в следовых концентрациях.
Этот электрод можно использовать и для определения бромида и иодида. Электрод нельзя применять в присутствии сильных восстановителей, окислители не влияют на работу электрода-Определению не мешают нитраты, фториды, гидрокарбонаты, сульфаты и фосфаты. Мешающее влияние некоторых анионов можно устранить, например, хлорид можно определять в присуЛ ствии бромида и иодида, если последние предварительно удале' ны введением хромовой кислоты [127].
Еще одним способом выражения селективности ИСЭ являет ся максимально допустимое молярное соотношение мешаюШ^г I и определяемого ионов, которое пропорционально константе с
214
лективности. Для рутинного анализа можно использовать имеющиеся в литературе данные о селективности электродов [122]. Данные о мешающем влиянии посторонних ионов на работу хло-ридного электрода Пунгора отличаются от данных, приведенных для электрода марки Орион.
Не обязательно использовать электроды, выпускаемые промышленностью. Опубликована методика [128] изготовления хло-ридного электрода, мембрану которого получают смешиванием галогенида серебра с термопластичным полимером и нагреванием смеси под давлением. Электродная характеристика такого электрода аналогична характеристикам галогенидных электродов других типов.
Разработано несколько методов определения хлоридов с использованием ионоселективных электродов. В одном из них применяют твердый серебряный ионоселективный электрод, например, Орион 94-16. В этом случае к анализируемому раствору добавляют известное количество раствора соли Ag+ и, используя серебряный электрод, измеряют остаточную концентрацию Ag+. Фрост [129] применил этот метод для анализа очищенных кипячением растворов.
Если нельзя применить твердые хлоридные электроды из-за мешающего влияния посторонних ионов, можно использовать жидкостные мембранные электроды. Таким электродом может быть электрод марки Орион 92-17, константы селективности которого приведены ниже:
Ионы . . . сю; г no; ви он’ oac' нсо; so; f-
К........ 32 17 4,2 1,6 1,0 0,32 0,19 0,14 0,10
С жидкостным электродом хлориды можно определять в присутствии сульфидов, аммония, в восстановительных и окислитель-
ных средах.
Кроме обычно применяющейся методики, основанной на построении градуировочного графика, можно использовать и другую методику. Твердый серебряный электрод применяли [130] в органическом микроанализе для определения конечной точки титрования хлорида и бромида ртутью(П). Определение проводят после сожжения образца. Хотя механизм взаимодействия ионов Hg2+ и Ag2S-элeктpoдa не совсем понятен, известно, что ионы Hg2+ образуют с галогенидами прочные комплексы. Конрад [131] аргентометрически титровал последовательно в одном растворе хлориды и цианиды, для индикации конечной точки титрования был использован Ag2S^eKTpofl (Орион 94-16). Содержа-Ние хлоридов и цианидов рассчитывают по кривой титрования.
Использование ионоселективных электродов в качестве индикаторных электродов при аргентометрическом титровании галоге-ндов, по обще’му мнению, неприемлемо. В работе [132] утвер-^Аается, что по сравнению с хорошо известным металлическим Ребряным индикаторным электродом ионоселективные электро-°бладают существенными недостатками,
315
Анализ с применением ионоселективных электродов может быть непрерывным, что является большим достоинством ионоселективных электродов. Опубликован обзор [133] по непрерывному анализу с применением электродов, разработанных Пунгором твердые электроды другого типа также используют для этих це лей.
Применение хлоридного ионоселективного электрода чрезвы чайно разнообразно, его использовали в различных отраслях про мышленности, в биологии, медицине, агрохимии, геологии, для анализа объектов окружающей среды [134, 135].
Ионоселективные электроды, так называемые «селектроды» описаны в работах Ружички и Лемма [136]. «Селектрод» содер
жит тонкий слой электродно-активного вещества, нанесенного на поверхность гидрофобизированного графита. Линейность Нернстов ской функции для иодидов, бромидов и хлоридов сохраняется до 10~6 М, 10~5 М и 10'4 М соответственно. В этих электродах не используют мембрану и внутренний раствор сравнения. Электроды выпускает промышленность [137].
Кулонометрические методы
Кулонометрический анализ с использованием электрогенера ции ионов серебра давно известен как эффективный метод опре деления хлоридов, бромидов и иодидов. Метод основан на изме рении количества электричества, необходимого для количествен ного протекания реакции по уравнению
Ag + X- —> AgX + e*
где X — СГ, Вг” или Г
Галогенид разряжается на серебряном аноде. Лингейном
[138]
описан вариант метода, в котором точку эквивалентности опре. деляют потенциометрически. Схема ячейки показана на рис. 35
Метод позволяет
определять 0,2—10
мг хлорида,
бромида
или
иодида в 50 мл раствора. Точность метода выше, чем при обыч-
ном титровании галогенидов раство ром AgNO3 с использованием индика
торов. Смесь галогенидов
можно
проанализировать, применяя последовательный электролиз при соответствующем значении потенциала. Для
определения хлоридов после сожжения органических веществ применен [139] автоматический вариант метода-В этом методе, с помощью которого
Рис. 35. Ячейка для кулонометрического ти" троваиия СГ электрогенерироваииым ионом Ag+ [138]:
1—пористый стеклянный диск; 2 —солевой н0СТ!'’]д 3—платиновый катод; 4—серебряный индикатор» электрод; 5—серебряный анод.
316
можно определять также бромид и иодид, используют интегратор для измерения силы тока во время титрования.
В работе [140] изучали кулонометрическую генерацию мак-поколичеств Ag+ и нашли, что метод позволяет с высокой точностью определять хлориды, бромиды и йодиды. Основным источником ошибок при условии тщательного проведения эксперимента является разложение осадка AgCI под действием света, рекомендуется проводить экстремально прецизионное определение (ошибка анализа 0,0012%) вообще в отсутствие света [141]. Экстремально высокая точность была реализована [142] в методах определения поверхностной влаги и чистоты монокристаллов. В этой работе использовали кулонометрию при постоянном токе, время электролиза измеряли с использованием осциллятора частотой 50 Гц на кристалле кварца. О высокой прецизионности метода позволяют судить полученные результаты содержания NaCl в монокристаллах: 99,988%, 99,993%, 99,995%, стандартное отклонение составляло 0,0143%, 0,0138% и 0,124% соответственно.
Кулонометрическое титрование электрогенерированным Ag+-ионом применили [143] для определения 1—200 нмоль С1~ в среде-уксусная кислота — вода (75:25). За 10 мин можно определить 0,01 ppm С1~. Хотя Ag+-HOH легко генерируется из серебряного электрода, присутствие окислителей может затруднить окисление анода независимо от силы тока. Влияние окислителей можно исключить, генерируя А+-ион сквозь проницаемую мембрану из Ag2S; на растворение Ag2S не влияют окислители [144].
Для большинства разновидностей кулонометрического метода требуется дорогостоящее оборудование, однако есть примеры [145] использования и сравнительно простых установок. Применение батарей в качестве источника постоянного тока позволяет определять 0,1—1 ppm хлоридов. Бромиды, йодиды, роданиды и вианиды должны отсутствовать. Индикация конечной точки титрования осуществляется по принципу амперометрического «dead — stops-титрования.
В одной из последних работ Ланге и Борнера [146] описан новый вариант измерения количества электричества в методике °пределения хлоридов. Ион серебра генерируют при постоянном т°ке, а индикацию конечной точки титрования осуществляют с помощью второй пары электродов, помещенной в ту же ячейку. Интегратор объединен со считывающим устройством. Метод использован для определения хлоридов в сыворотке крови.
Изучали [147] применение для кулонометрического титрова-Яия хлоридов электрогенерированных ионов ртути. Авторы этой РЗботы [147] заменили серебряный анод ртутным анодом и нахо-
j. -[* * • J ouvivnil.ill jjljllivil»! ипидиш n пали
Ли точку эквивалентности потенциометрическим методом, при-ПоНяя Ртутный индикаторный электрод. Было найдено, что при ис-^аС1лВанИИ в качестве фонового электролита смеси 0,5 М раствора Xjiod°4 И 0,02 раствора НСЮ4, можно определять содержание
Ридов, бромидов или иодидов с высокой чувствительностью
317
и точностью. Однако при титровании смеси галогенидов неожиданно столкнулись с процессом соосаждения, т. е. с теми же осложнениями, которые были замечены при проведении аргентометрического титрования. В работе (148] изучали применение ртутного анода при титровании галогенидов электрогенерирован-ным ионом ртути.
А мпероме три четкие м етоды
В монографии Штока [149] рассмотрены работы по амперометрическому титрованию хлоридов. В одной из последних работ Крешкова с сотр. [150] сообщается об использовании нитрата кадмия в качестве титранта при титровании хлоридов в среде безводной уксусной кислоты. Метод основан на осаждении хлорида кадмия. С точностью около 2% можно определять 4—6 мг С1~. Бромид или роданид можно определять, осаждая Cd(Br)2 или Cd(SCN)2-
Для определения галогенидов в присутствии цианидов и цианатов использовали AgNO3 в качестве титранта и вращающийся платиновый электрод [151]. Мешающее влияние цианидов до концентрации их, в 1500 раз превышающей концентрацию галогенидов, можно устранить добавлением 1 М раствора формальдегид да, который взаимодействует с цианидом с образованием циан-гидрида. Мешающее влияние избытка формальдегида и цианата при концентрациях их в 100 раз превышающих концентрацию галогенидов можно устранить добавлением HNO3 и подкислением раствора до pH < 3. Титрование 0,1 мМ — 10 мМ хлорида, бромида или иодида проводят в присутствии желатина.
Полярографические методы
Полярографию очень редко используют для определения хлоридов. Впервые изучали возможность применения полярографии для этих целей Кольтгоф и Милнер [152]. Затем эту проблему обсуждал Милнер [153]. При концентрации хлоридов 0,002 М и выше в хорошо изученной анодной области высота волны в 0,1 М растворе KNO3 пропорциональна концентрации хлоридов. Однако анодную область нельзя использовать для определения высоких концентраций хлоридов. Потенциал полуволны процесса зависит от концентрации хлорида. При концентрации 0,001 “ хлоридов его значение составляет 0,25 В.
Полярографию использовали [154] для определения хлор«Да в биологических материалах после осаждения протеинов суль’ фосалициловой кислотой. Сульфосалициловая кислота является также подходящим фоновым электролитом для проведения ана* лиза. Гороховский и Измайлова [155] определяли хлорид, бромид и иодид, используя серебряный электрод специальной кон стр цни, что позволило получать воспроизводимые осциллополяР®! граммы для концентрации хлоридов 10-3—10_6 М, Для опреде^
318
ниЯ галогенидов в морских водорослях после их предварительного сожжения использовали [156] экспрессный полярографический метод. Анализ проводили, регистрируя анодные волны хлоридов и иодидов и катодную волну бромида, которую получали после количественного окисления иона при контролируемом потенциале. Результаты определения хлоридов и бромидов совпадают с результатами, полученными классическими методами, иоднд определяется с большой ошибкой, которая возрастает при уменьшении его концентрации.
Разработан [157] интересный косвенный полярографический метод определения хлоридов и других анионов. Он основан на вытеснении хлоранилат-иона при взаимодействии хлорида с хлор-анилатом ртути. Эта реакция уже упоминалась в связи с использованием ее в спектрофотометрических методах определения хлоридов. Двухэлектронное обратимое восстановление хлораниловой кислоты и хлоранилат-иона [158] нашло практическое применение в работе [157]. Разработанный на этой основе метод позволяет определять 10-5—5-10-3 М хлорида. Авторы отмечают, что преимущество полярографического метода определения хлоридов проявляется при анализе окрашенных соединений. Аналогичные методы описаны для определения цианида, фторида, сульфита и сульфата.
Разработан вольтамперометрический метод [159] определения хлоридов и бромидов при концентрации меньше 10~5 М. Метод применим лишь при сравнимых концентрациях галогенидов. В другой методике [160] хлориды, бромиды и иодиды в виде солей ртути осаждаются на анодно-поляризованной висящей ртутной капле и затем последовательно растворяются при ее катодной поляризации. С точностью ±15% все три аниона можно определять на уровне концентраций 10-3—10-7 М.
Другие электрохимические методы
Для определения каждого галогенида, а также их смесей использовали хронопотенциометрию [161].
Аргентометрическое высокочастотное титрование также применяли для анализа смесей галогенидов и для определения отдельно каждого галогенида. Метод применим и для определения роданида [162].
Радиохимические методы
В радиохимических методах определения хлоридов используют «сколько систем. Шаталова и Мееров [163] применили ll0AgNO3 2Ля осаждения хлорида и измеряли активность ”°AgCl после растворения его в NH4OH. Метод был применен для анализа плазмы Крови, точность метода ±8%. Этот метод используют и в не-Ко°ЛдКО ином варианте: измеряют активность жидкости над осад-
AgCl после отделения последнего центрифугированием [164].
319
Определению не мешают Na, Са, Mg, Al, Fe, сульфат, нитрат, фосфат, карбонат, гидрокарбонат, фенол, пиридин, гумминовые кислоты, лактат натрия, глицин, пирогаллол, пектины, танниновая кислота, додецплсульфонат натрия. Бромид и иодид мешают определению. Метод, разработанный для анализа воды, применим для определения 5—1000 ppm СН.
Опубликован метод с использованием 203Hg(IO3)2, основанный на измерении активности образующихся HgCl2 и HgBr2 [165]. Для определения хлорида к сульфида применяли [166] колонку, содержащую Ag^'CrO-i. При пропускании образца через колонку в элюат выделяется 5|СгОГ- Определению мешают бромид, иодид, цианид, роданид и сульфит. Область определения хлоридов 0,8—80 ммоль.
Иохансон [167] обрабатывал образец, содержащий 1 —10 мкг СН, известным количеством Н36С1, выпаривал досуха и измерял активность сухого осадка. Ошибка определения 1 —10 мкг составила ±1 мкг, в то время как ошибка определения 10—100 мкг СН составляет ±3 мкг. Метод применен для анализа природных вод. Автор утверждает [168], что метод в 10 раз чувствительней по сравнению с субстехиометрическим методом, основанным на осаждении Ag36CL
Газохроматографические методы
Характерной особенностью современного неорганического анализа и анализа металлоорганических соединений является использование газохроматографических методов. Опубликован обзор таких методов [169]; некоторые из них применимы для анализа смеси галогенидов.
Галогениды (хлориды, бромиды или иодиды) сначала переводят в соответствующие кислоты, применяя сильнокислотные ионо-обменники [170]. Затем кислоты реагируют в кислых средах с 1,2-олефиноксидами с образованием галогенсодержащих спиртов. Измеряют пики, образующиеся при концентрации галогенидов меньше 0,5-IO-4 М, с помощью пламенно-ионизационного детектора. В другом методе использовали превращение галогенидов (хлоридов, бормидов м иодидов) с помощью ионного обмена в тетраалкиламмониевые соли. Их пиролитическое разложение приводит к образованию алкиламинов и соответствующих алкилгало-генидов. После газохроматографического разделения определение проводят, используя детектор по теплопроводности [171].
Галогениды (хлориды, бромиды и иодиды) можно экстрагир0" вать [172] из водных растворов карбонатом тетрафениламмоняя в 10%-ном растворе н-деканола в толуоле. Галогениды тетраф6* ниламмония при температуре выше 150 °C подвергаются термич ской диссоциации на 1-галогенгептан и трифениламин. Первь' продукт дает хроматографические пики, которые можно использ вать для количественного определения галогенидов на УРоВ меньше 1 ppm. И
Ч9Л
Белчер с сотр. [173, 174] определяли хлорид в виде хлорида Женилртути(П), который экстрагировали СНС13 перед газохро-матографическнм анализом. С применением пламенно-ионизационного детектора можно обнаруживать около 2,3-10~10 г хлорида. Применение электроно-захватного детектора позволяет повысить чувствительность более чем в 10 раз.
Другие методы определения
Были сделаны попытки разработать количественные и полуко-личественные хроматографические методы определения хлоридов. Хорошо изученный метод кольцевой бани был использован для полумикроопределений хлорида, бромида, иодида, арсената, бихромата и гексацианноферрата (111) с применением стандартной шкалы сульфида серебра [175].
Циркуляционную тонкослойную хроматографию использовали [176] для полуколичественного определения хлорида, бромида, иодида, бромата, хлората, гексацианоферрнта(П), гексацианноферрата (III), роданида, арсенита и сульфита. Точность определения ±5%. Фосфат, сульфид, нитрит, иодат, тиосульфат, перйодат, арсенат и ацетат не мешают определению. Описан еще один метод ТСХ [177], в котором диоксид кремния суспендируют в 3%-ном растворе AgNO3 и наносят на стеклянную пластинку. В качестве растворителей используют смесь насыщенного водой изобутанола и 40%-ного ацетата аммония (4: 1). Длина образующихся полос пропорциональна концентрации хлорида, бромида, иодида и фосфата. В другом аналогичном методе используют хроматографическую бумагу, импрегннрованную AgCI или Ag2O, и измеряют расстояния, на которые мигрируют хлориды, бромиды и иодиды за 30—45 мин на восходящей хроматограмме при использовании в качестве подвижной фазы 12%-ного водного раствора глицерина [178]. Был предложен AgCI для определения бромида и иодида в бинарных смесях или одного из этих ионов в отсутствие другого. Оксид серебра дает лучшие результаты при анализе тройных смесей. Для расчетов результатов анализа используют градуировочные графики. Этим методом можно определять микро-траммовые количества галогенидов и роданидов. В течение 1 ч можно проанализировать 50 образцов.
При использовании рентгенофлуоресцентного метода анализа (стандартами служат бромид и иодид) можно определять 0,7— мг хлорида в присутствии роданида и цианида. Определение д^0Дят [179] после предварительного осаждения хлорида р^ыли сделаны попытки разработать методы определения хло-[18б]В ° использованием принципа умножения. В обзоре Белчера тоды’ Посвященном таким методам, в основном приведены ме-(0 05^озволяющие определять хлориды в интервале содержаний
11 Зак. 16?
Один из методов определения 0,05—10 мг С1_ состоит из следующих этапов [181].
1. Осаждение хлорида в виде AgCl и переведение его в Ag2S обработкой Na2S.
2. Отделение Ag2S фильтрованием; хлориды переходят в фильтрат вместе с избытком сульфида.
3. Окисление S2- в фильтрате до SO4 введением Н2О2, подкисление раствора HNO3 и повторное осаждение хлорида Ag\’O3.
4. Отделение AgCl на фильтре, содержащем Ag2S (см. п. 2).
5. Фильтрат и промывные воды отбрасывают. Хлорид серебра растворяют в минимальном объеме 5 М раствора NH4OH, пропуская его через осадок Ag2S и AgCl.
6. Фильтрат обрабатывают Na2S и образующийся Ag2S опять отделяют на том же фильтре, который использовался в пп. 2 и 4.
Таким образом, возрастающее в арифметической прогрессии количество Ag2S собирают на фильтре. Наконец, суммарное количество Ag2S, полученное из AgCl, взвешивают. Содержание хлорида в исходном образце рассчитывают с учетом числа повторных осаждений хлорида. Хотя на первый взгляд метод может показаться сложным и длительным, использование стеклянного прибора специальной конструкции значительно упрощает проведение анализа.
Опубликован кинетический спектрофотометрический метод определения низких концентраций хлоридов и бромидов [182]. Метод основан на следующих реакциях. Таллий(Ш) количественно окисляет 4,4-дигидроксидифенил (ДГДФ) до дифенохинона (ДФХ).
Т1и1 + но——Г/—ОН —> TI* + о=^ /^\ /==о + 2Н*
Хлорид и бромид реагируют с избытоком таллия(Ш) с образованием прочного комплекса состава 1:1, который уже не окисляет ДГДФ до ДФХ. Этот метод позволяет определять 10~4 — 10~6 М хлорида и бромида. Нитраты и сульфаты в 100-кратном избытке не мешают определению; определение невозможно в присутствии йодидов, роданидов, сульфитов, тиосульфатов и соединений, способных окислять ДГДФ или восстанавливать таллий(III).
Описан метод термометрического титрования для определения галогенидов. Каждый из галогенидов можно оттитровать AgNOa с удовлетворительной точностью, но смесь галогенидов определить этим методом, как и потенциометрическим, невозможно из-за со-осаждения галогенидов серебра [183].
ЛИТЕРАТУРА
1 . Firsching F. Н. — Analyt. Chem., 1960, v. 32, р 1876.
2 . Rodabaugh R. D., Upperman G. T. — Analytica chim. Acta, 1972, v. 60, p-
3 . Chwastowska J., Marczenko Z., Stolarczyk U. — Chemia analit., 1963, v- j p. 517; Analyt. Abstr., 1964, v. 11, p. 3662
322
4 De Geiso R C., Reeman IT., Lindenbaum S. — Analyt. Chem., 1954, v. 26, ’ n. 1840.
к Holzapfel H, Gurtler O. — J. prakt. Chem., 1967. v. 35. p. 113; Analyt Abstr., 1968, v. 15, p 4673.
6 Залевская T. JI., Старобинец Г. JI. ЖАХ, 1969, т. 24, с. 721. Analyt. Abstr., ' 1969, т. 19, с. 3877.
7 Старобинец Г. J1., Захарнева Е. П. Изв. Академии Наук Белорусской ССР, Серия хим. наук, 1970, с. 105. Analyt. Abstr., 1972, т. 22, с. 2264.
я Ramaley L., Holcombe W. А. — Analyt Lett., 1967, v. 1, p. 143.
о Tustanowski S. — J. Chromat., 1967, v. 31, p. 268.
in Tustanowski S. — J Chromat , 1967, v. 31, p 270.
ii Harrison B. L., Rosenblatt D. H. — J. Chromat , 1964, v. 13, p. 271.
12 Mao C.-H. — Acta chim. Sin., 1964, v. 30, p. 496; Analyt. Abstr., 1966, v. 13, p. 533.
13. Bark L. S., Graham R. J. T., McCormick D. — Analytica chim. Acta. 1966, v 35, p 268.
14, Seiler H., Seilei M. — Helv. chim. Acta, 1967, v. 50, p. 2477.
15. Gagliardi E., Pokorny G. — Mikrochim. Acta, 1965, p. 699.
16. Seiler H, Kaffenberger T. — Helv. chim. Acta, 1961, v. 44, p 1282.
17. Petrovic S. M., Canic V. D. — Z. analyt. Chem., 1967, v. 228, p. 339; Analyt. Abstr., 1968, v. 15, p. 4674.
18. Takeuchi T., Tsunoda Y. —-J. chem. Soc. Japan, pure Chem. Sect., 1966, v. 87, p. 251; Analyt. Abstr., 1967, v. 14, p. 4682.
19 Muto M.— J chem. Soc. Japan, pure Chem. Sect., 1964, v. 85, p. 782; Analyt. Abstr., 1966, v. 13, p. 1257.
20. Kawanabe К, Takitani S., Tiyazaki M., Tamura Z. — Japan Analyst, 1964, v. 13, p. 976; Analyt. Abstr., 1966, v. 13, p 5385.
21. Elsheimer H. N., Johnson A. L., Kochen R. L. — Analyt. Chem., 1966, v. 38, p. 1684.
22. Armstrong G. IT., Gill H. H., Rolf R. F.— In: Treatise on Analytical Chemistry (Ed. 1. M. Koithoff and P J. Elving), Part II, Vol. 7, New York, Interscience, 1961.
23. Koithoff I. M., Sandell E. B., Meehan E. J., Bruckenstein S. — Quantitative Chemical Analyst, 4th edn, London, MacMillan, 1969, p. 580.
24. Little K.—Talanta, 1971, v. 18, p. 927.
25. Imperial Chemical Industries Ltd., Analytical Chemists’ Committee — Analyst, 1950, v. 75, p. 577.
26. Desai M. N., Desai К- C., Gandhi M. H. — Lab. Pract., 1969, v. 18, p. 939, 1063.
27. Ref. 23, p. 716.
28. Frieser H., Fernando Q. — Ionic Equilibria in Analytical Chemistry, Chapter XIV, New York, Wiley, 1963.
29. Laitinen H. A. — Chemical Analysis, New York, McGraw-Hill, 1960.
30 Belcher R., Macdonald A. M. G., Parry E.— Analytica chim. Acta, 1957, v. 16, p. 524.
31. Berry A. J., Driver J. E. — Analyst, 1939, v. 64, p. 730.
32. Bishop E., Ayers C. A. — In: Comprehensive Analytical Chemistry (Ed. C. L. and D. W. Wilson), Vol. IB, Chapter VII, London, Elsevier, 1960.
33- Block J., Waters О. B. — Talanta, 1967, v. 14, p. 1130.
34. Wanninen E —Talanta, 1968, v. 15, p. 717.
35. Anfaelt T., Jagner D. — Talanta, 1969, v. 16, p. 555.
36. Standard Methods for the Examination of Water, Sewage and Industrial Waters, The American Public Health Association and the American Waterworks □ Association, 13th edn, New York, American Public Health Association, 1971. 37- Schulek E., Pungor E., Kethelyi J. — Analytica chim. Acta, 1953, v. 8, p. 229. 38- Caldwell J. R., Moyer H. V.— Ind. Engng Chem. analyt. edn., 1935, v. 7, P- 38.
4n £ritish Standard 4401: Pt 6, 1970
41 “ptish Standard 4550 Pt 2, 1970.
''*£₽ I. W. — The Analysis of Concretes, Building Research Station, Department of the Environment, 28, London, H. M. S. 0, 1970.
42. Kapel Л1, Fry J. C., Shelton D. «. — Analyst, 1975, v. 100, p. 570.
43. Ikeda N. — J. chem. Soc. Japan, pure Chem. Sect., 1949, v. 70, p. 329; Chem Abstr., 1951, v. 45, p. 3102.
44 Mehlig J. P.— Chemist Analyst, 1955, v. 44, p. 87.
45. Archer E. E.—Analyst, 1958, v. 83, p. 571.
46. Карабаш А Г. ЖАХ, 1953, t. 8, c. 140.
47. Macdonald A. M. G. — Ind. Chemist, 1960, v. 36, p. 35.
48. Cheng F. W.—Microchem. J., 1959, v. 3, p. 537.
49. White D. C. — Mikrochim. Acta, 1961, p. 449
50. White D. C. — Mikrochim. Acta, 1961, p. 807.
51. Colson A. F. — Analyst, 1965, v. 90, p. 35.
52. Belcher R., Gouverneur D., Macdonald A. M. G. — J. chem. Soc., 1962, p. 1938.
53. Official, Standardised and Recommended Methods of Analysis, London, The Society for Analytical Chemistry, 1973, p. 351.
54. Steyermark A., Lalancette R. A., Contreras E. M. — J. Ass. off. analyt. Chem., 1972, v. 55, p. 680.
55. Flaschka H., Huditz F.— Z. analyt. Chem., 1952, v. 137, p. 104; Chem. Abstr., 1953, v. 47, p. 443a.
56. Гусев С. И., Соколова E. В., Кожевникова И. А. ЖАХ, 1962, т. 17, с. 499. Analyt. Abstr., 1963, т. 10, с. 1795.
57. De Sousa A. — Inf. Quim. Anal., 1963, v. 17, p. 127; Analyt. Abstr., 1964, v. 11, p. 5476.
58. De Sousa A. — Chemist Analyst, 1960, v. 49, p. 45.
59. De Sousa A. — Taianta, 1961, v. 8, p. 782.
60. De Sousa A. — Analytica chim. Acta, 1961, v. 24, p. 424.
61. Viebock F. — Ber., 1932, v. 65B, p. 493, 586.
62. Fildes J. E., Macdonald А. Л1. G. — Analytica chim. Acta, 1961, v. 24, p. 121.
63. Belcher R., Macdonald A. M. G., Kutten A. J. — Mikrochim. Acta, 1954, p. 104.
64. Kolthoff I. M., Belcher R. — Volumetric Analysis, Volume III, New York, Interscience, 1957, p. 254.
65. Summersgill N. — Chem. Inds, Lond., 1961, p. 782.
66. Iwasaki I., Utsumi S., Ozawa T.— Bull. chem. Soc. Japan, 1952, v. 25, p. 226; Chem. Abstr., 1953, v. 47, p. I0407f.
67. Iwasaki J., Utsumi S., Hageno K-, Ozawa T. — Bull. chem. Soc. Japan, 1956, v. 29, p. 860; Analyt. Abstr., 1957, v. 4, p. 3954.
68. Florence T. M., Farrar Y. J. — Analytica chim. Acta, 1971, v. 54, p. 373.
69. Bergman J. G., Sanik J. — Analyt. Chem., 1957, v. 29, p. 241.
70. Elsheimer H. N., Kochen R. L.— Analyt. Chem., 1966, v. 38, p. 145.
71. Britt R. D.— Analyt. Chem., 1962, v. 34, p. 1728.
72. Dojlido J., Bierwagen H. — Chemia. analit., 1969, v. 14, p. 91; Analyt. Abstr, 1970, v. 18, p. 4413.
73. British Standards 2690: Part 6, 1968.
74. Kulhanek V., Fiser C. — Colin Czech, chem. Commun, 1966, v. 31, p. 1890; Analyt. Abstr, 1967, v. 15, p. 4871.
75. Yamamoto Y., Kumamaru T., Tatehata A, Yamada N. — Analytica chim. Acta, 1970, v. 50, p. 433.
76. Zall D. M., Fisher D., Garner M. Q. — Analyt. Chem, 1956, v. 28, p. 1665.
77. Gerlach J. L., Frazier R. G. — Analyt. Chem, 1958, v. 30, p. 1142.
78. Barney J. E., Beriolacini R. J. — Analyt. Chem, 1957, v. 29, p. 1187.
79. Beriolacini R. J., Barney J. E. — Analyt. Chem, 1958, v. 30, p. 202.
80. Bode H., Eggeling W., Steinbrecht V. — Z. analyt. Chem, 1966, v. 216, p. 30; Analyt. Abstr, 1967, v. 14, p. 2409.
81. Hammer C. F., Craig 1. H. — Analyt. Chem, 1970, v. 42, p. 1588.
82. West P. W„ Coll H.— Analyt. Chem, 1956, v. 28, p. 1834.
83. Taras M. — Analyt. Chem, 1947, v. 19, p. 342.
84. Scheubeck E., Ernst O. — Z. analyt. Chem, 1970, v. 249. p. 370; Analyt-Abstr, 1971, v. 20, p. 1655.
85. Bhatty M. K-, Uden P. C. — Taianta, 1971, v. 18, p. 799. .
86. Belcher R., Rodriguez- Vazquez J. A, Stephen W. I. — Analytics chim. Acta‘ 1972, v. 61, p. 223.
324
87 Lamb A. В., Carleton P. IV., Meldrum IV. B. — J Am. chem. Soc., 1920, v. 42, ' p. 251.
88 Johnson C. R. — J. phys. Chem., Ithaca, 1931, v. 35, p. 2237.
89 Boltz D. F., Holland IV. J. — In: Colorimetric Determination of Non-metals (Ed. D. F. Boltz), New York, Interscience, 1958.
on. Ramirez-Munoz J. — Analytica chim. Acta, 1975, v. 74, p. 309.
91 Reichel W., Acs L. — Analyt. Chem., 1969, v. 41, p. 1886.
92. Fuiinuma H., Kasama K-, Takeuchi K-, Hirano S. — Japan Analyst, 1970, v. 19, p. 1487; Analyt. Abstr., 1972, v. 22 p. 2272.
93 Belcher R., Nadjafi A., Rodriguez-V azquez J. A., Stephen IV. /. — Analyst, 1972, v. 97, p. 993.
94. Dagnall R. M., Thompson К. C., West T. S. — Analyst, 1969, v. 94, p. 643.
95. Shiner V. J., Smith M. L. — Analyt. Chem., 1956, v. 28, p. 1043.
96. Martin A. J. — Analyt. Chem., 1958, v. 30, p 233.
97. Prokopov T. S. — Analyt. Chem., 1970, v. 42, p. 1096.
98. Kuttel D., Szabadka O., Czajvari B., Meszaros K., Havas Pungor E. — Magy. kern. Foly, 1969, v. 75, p. 181; Analyt. Abair., 1970, v. 19, p. 3889.
99. Dixon J. P. — Modern Methods in Organic Microanalysis, Chapter 8, London, Van Nostrand, 1968.
100. Freeman R. W. — Analyt. Chem., 1959, v 31, p. 214.
101. Reilley C. N., Cooke IV. D., Furman N. H.—Analyt. Chem., 1951, v. 23, p. 1223.
102. Furman N. H., Low G. IV. — J. Am. chem Soc., 1935, v. 57, p. 1585.
103. Blaedel W. J., Lewis W. B., Thomas J. IV. — Analyt. Chem., 1952, v 24, p 509.
104 Malmstadt H. V., Winejordner J. D. — Analytica chim. Acta, 1959, v. 20, p. 283.
105. Malmstadt H. V., Winejordner J. D. — J. Am. Wat. Wks Ass., 1959, v. 51, p. 733; Analyt. Abstr., 1960, v. 7, p. 1953.
106. British Standard 2690: Part 6, 1968.
107. Bishop E.—Mikrochim. Acta, 1956, p. 619.
108. Bishop E. — Analyst, 1958, v. 83, p. 212.
109. Bishop E., Dhaneshwar R. G. — Analyst, 1962, v. 87, p. 845.
110. Bishop E., Dhaneshwar R. G. — Analyt. Chem., 1964, v. 36, p 726.
111. Florence T. M. — J. electroanal. Chem., 1971, v. 31, p. 77.
112. Bishop E. — Mikrochim. Acta, 1960, p. 803.
113. Chou D. H., Sams L. C. — Microchem. J., 1969, v. 14, p. 507.
114. Hozumi K., Akimoto N. — Analyt. Chem., 1970, v. 42, p. 1312.
115. Mattock G., Uncles R. — Analyst, 1962, v. 87, p. 977.
116. Prokopov T. S. — Mikrochim. Acta, 1968, p. 401.
117. Иванова 3. И., Коваленко П. Н. ЖАХ, 1962, т. 17, с. 739. Analyt. Abstr., 1963, т. 10, с. 2702.
118. Harzdorf С. — Z. analyt. Chem., 1966, v. 215, р. 246; Analyt. Abstr., 1967, v. 14, p. 1966.
119. Tomlinson K-, Torrance K- — Analyst, 1977, v. 102, p. 1.
120. Pungor E., Toth K- — Analyst, 1970, v. 95, p 625.
121. Tenygl J. in Ion-selective Electrode Analysis, MTP International Review of Science, Physical Chemistry, Series one, Vol. 12 (Ed. T. S. West), London Butterworths, 1973, p. 123.
122. Moody G. J., Thomas J. D. R. — Taianta, 1972, v. 19, p. 623.
*23. Orion Research Incorporated, Cambridge, Massachusetts. Also available from MSE Scientific Instruments, Manor Royal, Crawley, W. Sussex, RH10 2QQ
। and E. I. L. Ltd., Hanworth Lane, Chertsey, Surrey KT16 9LF.
*24. Available from D A. Pitman Ltd., Weybridge, Surrey or Corning-Eel Scienti-
**c Instruments, Evans Electroselenium Ltd., Halstead. Essex.
Available from Beckman Instruments Ltd, Sunley House, 4, Bedford Park, 126 nroydon> Surrey.
°- Radelkis Electrochemistry Instruments, Budapest, Hungary, Available from 97 Protech Services Ltd., 40 High Street, Rickmansworth, Herts.
12R л?" Ir00n J- C ~ Analyst, 1968, v. 93, p 788.
124 в asc,ni M., Liberti A. — Analytica chim. Acta. 1949, v. 47, p. 339.
130 DiSt — Analytica chim. Acta, 1969, v 48, p. 321.
otman IV, Dahmen E. A. M. F. — Mikrochim. Acta, 1972, p. 303.
325
131. Cunrad F. J. — Taianta, 1971, v. 18, p. 952.
132. Masson M. R. — Taianta, 1975, v. 22, p. 933.
133. Feher Z., Nagy G., Toth K., Pungor E. — Analyst. 1974. v. 99, p 699.
134. Buck R. P.—Analyt. Chem., 1974, v. 46, p. 28R.
135. Orion Research Incorporated, Analytical Methods Guide, 9th edn., 1978 A]so Orion Newsletters.
136. Ruzicka /., Lamm C. G.—Analytica chim Acta, 1971, v. 54, p. 1.
137. Radiometer, Copenhagen.
138. Lingane J. J. — Analyt. Chem., 1954, v. 26, p. 622.
139. Coulson D. Л1., Cavanagh L. A. — Analyt. Chem , 1960, v. 32, p. 1245
140. Marinenko G., Taylor J. K. — i. Res natn. Bur. Stand., 1963, v 67A, p. 3]
141. Champion С. E., Marinenko C. — Analyt. Chem., 1969, v. 41, p. 205.
142. Yoshimori T., Tanaka T. — Analytica chim. Acta, 1971, v. 55, p. 185.
143. Cedergren A., Johansson G. — Taianta, 1971 v. 18, p. 917.
144. Fletcher K. S., Mannion R. F. — Analyt Chem, 1970. v. 42, p. 285.
145. Jacobsen E., Tandberg G. — Analytica chim. Acta, 1973, v. 64, p. 280.
146. Lange W., Borner K. — Z. klin. Chem. fclin. Biochem., 1972, v. 10, p. 33; Analyt. Abstr., 1972, v. 23, p. 3646.
147. Przybylowicz E. P., Rogers L. B. — Analyt. Chem., 1956, v. 28, p 799.
148. De Ford D., Horn H.— Analyt. Chem., 1956, v. 28, p. 797.
149. Stock J. T. Amperometric Titrations, Chapter 13, New York, Interscience, 1965.
150. Крешков А. П., Борк В. А., Швыркова Л. А., Апаршева M. И. ЖАХ, 1965, t. 20, c. 704. Analyt. Abstr., 1967, t. 14, c. 573.
151. Ikeda S., Musha S. — Japan Analyst, 1967, v. 16, p. 445; Analyt. Abstr., 1968, v. 15, p. 6633.
152. Kolthoff I. M., Miller C. S.—J. Am. chem. Soc., 1941, v 63, p 1405.
153. Milner G. W. C. — Principles and Applications of Polarography, London, Longmans, 1957, p. 286.
154. Bartik M., Hupka J. — Cooln Czech, chem. Comrnun., 1960, v. 25, p. 3391, Analyt. Abstr., 1961, v. 8, p. 2960.
155. Гороховский В. AL, Измайлова Ф. К. ЖАХ, 1966, т. 21, с. 87, Analyt. Abstr., 1967, t. 14, с. 5393.
156. Calzolari С., Gabrielli L. F., Marietta G. P. — Analyst, 1969, v. 94, p. 774.
157. Humphrey R. E., Laird С. E. — Analyt. Chem., 1971, v. 43, p. 1895.
158. Weissbart J., Van Rysselberghe D. — J. phys. Chem. Ithaca, 1957, v. 61, p. 765.
159. Colovos G., Milson G. S., Moyers J. L. — Analyt. Chem., 1954, v 46, p. 1051.
160. Kemula U7., Kublik Z., Taraszewska J. — Chemia. analit., 1963, v. 8, p. 171; Analyt. Abstr., 1964, v. 11, p. 1282.
161. Peters D. G., Kinjd A. — Analyt. Chem., 1969, v. 41, p. 1806.
162. Grey P., Cave G. С. B. — Can. J. Chem., 1964, v. 42, p. 770.
163. Шаталова А. А., Мееров Г. И. Биохимия, 1960, т. 25, с. 769. Analyt Abstr., 1961, т. 8, с. 2551.
164. Beaudet С., Rygaert J.—J. radioanal. Chem., 1968, v. 1, p. 153; Analyt. Abstr., 1969, v. 17, p. 1826.
165. Banyai E., Szabadvary F., Erdey L. — Mikrochim. Acta, 1962, p. 427.
166. Bowen H. J. M. — Radiochem. radioanal. Lett, 1970, v. 3, p. 339.
167. Johannesson J. K. — J. radioanal. Chem., 1970, v. 6, p. 27
168. Johannesson J. K- — Analyst, 1961, v. 86, p. 72.
169. Rodrigues-Vazquez J. A. — Analytica chim. Acta, 1974, v. 73, p. 1.
170. Ruessel H. A. — Angew. Chem. int. Ed. Engl., 1970, v. 9, p. 374.
171. Macgee J., Allen K- G.—Analyt. Chem., 1970, v. 42, p. 1672.
172. Matthews D. R., Shults W. D„ Dean J. A. —Analyt. Lett., 1973. v. o, p. 513. n r
173. Belcher R., Majer J. R., Rodriguez-Vazquez J. A., Stephen IT. /., Vden P-Analytica chim Acta, 1971, v 57, p. 73.
174. Belcher R., Rodriguez-Vazquez J. A, Stephen IV. I. — Unpublished work r-ported in reference 169.
175. Celap M. B., Weisz H. — Mikrochim. Acta, 1962, p. 24.
176. Hashmi M. H., Chughtai N. A. — Mikrochim. Acta, 1968, p. 1040. . »
177. Muto M. — J. chem. Soc. Japan, pure Chem. Sect., 1965, v. 86, p. 91, Ana У I Abstr., 1967, v. 14, p. 3961.
one
Лесковская В Н, Алесковский В. Б ЖАХ, 1969, т. 21, с. 1213. Analyt. Abstr, 1971, т. 20. с. 1657.
i7Q Stork G, 7rmg H.— 7. analyt. Chem., 1970, v 249, p. 161.
80 Belcher R. - Taianta. 1968. v 15, p. 357.
181 Rahim S. A., Abdulahed H, West T. S. — Mikrochim. Acta, 1974, p. Ill
182 Mentasti E, Pelizzetti E.— Analytica chim. Acta, 1975, v 78, p. 227.
183 Earris R. 7. A Quoted in Thermometric Titrimetry by H J. V. Tyrrell and A. E. Beezer, London, Chapman and Hall, 1968, p. 97.
ХЛОРИТЫ
Хлорит сравнительно устойчив в щелочных растворах, но быстро диспропорционнрует в кислых средах с образованием хлорида, хлората и диоксида хлора. Продажный препарат, который выпускают в смеси со свободной щелочью, содержит около 80% хлорита натрия.
Определение хлорита важно, так как связано с анализом питьевой воды. Хлориты нашли применение в производстве отбеливателей. Понятие «активный хлор» включает сумму хлорита, гипохлорита и диоксида хлора.
МЕТОДЫ ОТДЕЛЕНИЯ
Хлорит отделяют от других ионов методами бумажной и тонкослойной хроматографии (табл. 22).
Таблица 22. Отделение хлорита методами бумажной и тонкослойной хроматографии
Метод Растворитель С1О~ отделяется от ионов Литература
Бумажная хроматография ТСХ (оксид алюминия G, кизельгур (1 : 1) Изопропанол — Н2О — пиридин - NH3 (0,880 г/см3) (15:2:2:2) сг, сю;, сю; /
Бутанол — ацетон (0,880 г/см8)—Н2О (8; 10:2: 1) сю;, сю;, Вго;, ю;, Вго; 2
тех Изопропанол — тетрагидрофуран — NH3 (0,880 г/см3) сг, сю;, сю; 3
Бумажная хроматография и ТСХ (силуфол UV 254) Бутанол — пиридин — NHa (0,880 г/см3) скг.с1о;,сю; 3’ 4 4
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Как и некоторые другие хлорсодержащие анионы, хлорит можно восстанавливать SO2 до хлорида и затем определять любым Методом, пригодным для определения хлорида. Например, восстановление можно проводить, добавляя к анализируемому раствору идросульфит натрия и разбавленную H2SO4 и затем удаляя кипя-еннем избыток SO2.
327
Титриметрические методы
Иодиметрические методы
Хлорит в кислой среде восстанавливается иодидом:
С1О; + 4Г+4Н+ —> 212 + СГ + 2Н2О
Свободный иод можно оттитровать стандартным раствором тиосульфата. В первых опубликованных работах использовали H2SO4. Описано [5] применение фосфорной кислоты, при этом для завершения реакции раствор необходимо выдерживать в течение 5 мин.
В присутствии уксусной кислоты реакция проходит очень медленно [6]. Браун [7] нашел, что в присутствии любой из трех кислот получаются совпадающие результаты анализа, но если используют уксусную кислоту, для завершения реакции необходимо выдерживать реакционную смесь в течение 5 мин.
Гипохлорит реагирует аналогично, но его можно удалить введением в щелочной раствор цианида [5]:
C1O' + CN'+H2O —> C1CN + 2OH’
С помощью иод и метрического метода можно определять в смеси хлорит и диоксид хлора [8]. По описанной выше методике находят сумму соединений. После колориметрического определения диоксида хлора с тирозином можно рассчитать содержание хлорита. Хлорид, хлорат и хлорит не мешают определению диоксида хлора. В результате проведения интересного исследования Норкис [9] нашел объяснение, почему арсенит в присутствии осмиевой кислоты в гидрокарбонатной среде ускоряет реакцию взаимодействия хлорита с иодидом. По-видимому, механизм процесса следующий: арсенит восстанавливает OsO4 до Na2OsO4, который в свою очередь восстанавливает хлорит до гипохлорита, а сам окисляется до OsVH1. Гипохлорит окисляет иодид до иода, который и взаимодействует с мышьяком(III). Методика, основанная на описанных выше реакциях, успешно использована для определения диоксида хлора и хлорита [10]. Эти же реакции используют и в потенциометрическом методе определения хлорита, гипохлорита, хлората и хлорида, который будет подробно описан ниже.
Важно определение хлорита в присутствии гипохлорита. Для разрушения гипохлорита рекомендуют фенол, но, по наблюдению некоторых исследователей [И], фенол реагирует и с хлоритом. Гипохлорит в смеси этих двух ионов разрушают введением (NH4)2SO4, который реагирует следующим образом:
рН=8-9
4ВгО” + 2NHJ -----> 4Br” + N2 + 4Н2О
После этого для определения оставшегося хлорита можно применить иодиметрический метод.
828
Арсенометрический метод
Реакция хлорита с мышьяком (III) в щелочной среде происходит медленно, скорость реакции значительно повышается при использовании в качестве катализатора осмиевой кислоты [7]:
„ Кат.
C1OJ + 2AsOj ----> 2AsOt + Cr
Осмиевая кислота служит и катализатором и индикатором, так как в конечной точке титрования окрашивает раствор в синевато-серый цвет. Отсутствие подходящего индикатора оправдывает необычный вариант титрования: в этом случае титруют стандартный раствор арсенита анализируемым раствором хлорита натрия.
Титриметрическое определение хлорита
с использованием мышьяка(Ш) [7]
Ход анализа. К 25 мл раствора NaClOs добавляют 1 г NaHCO3 и 0,1— 0,5 мл 1%-ной осмиевой кислоты. Очень медленно титруют 0,05 М раствором мышьяка(III), вблизи точки эквивалентности добавляя раствор по каплям с интервалом 30 с. Титруют до появления бледной синевато серой окраски, идентичной с окраской раствора сравнения
Примечание. Приготовление раствора мышьяка(Ш) см. раздел «Арсенаты и арсениты».
Метод с применением аскорбиновой кислоты
Один из вариантов этого метода основан на реакции между аскорбиновой кислотой и гексацианоферратом (III) в присутствии индикатора 2,6-дихлорфенол индофенола:
С6Н8О6 + 2 [Fe (CN)6]3- —> С6Н6Ос + 2 [Fe (CN)6]4' + 2Н+
Для определения гипохлорита добавляют избыток гексацианоферрита (II) и титруют образующийся гексацианофферат(Ш) стандартным раствором аскорбиновой кислоты [12]:
C10J + 4 [Fe (СП)бГ’ + 4Н+ —> СГ + 4 [Fe (CN)6]3'+ 2Н2О
Другой косвенный метод состоит в прибавлении к хлориту избытка железа(II) и титровании образующегося железа (III) стандартным раствором аскорбиновой кислоты [13].
Спектроскопические методы
Описан [14] метод определения в сложных смесях ионов С1_, СЮ , СЮг, СЮз, ClOi и СЮг- Часть метода, касающаяся хлорита, основана на фотометрическом определении иода (в виде три-и°Дид-иона), образующегося при добавлении образца к подкислен-иому раствору KI. Метод включает в себя в дополнение к фотометрическому определению и титриметрическое. Для анализа ^еси СЮ2’, НСЮ2, С1О2, СЮз и С12 использовали поглощение лорита при 250 нм. Как и в предыдущем методе, для анализа Рименяют и титриметрию [15].
329
Для определения хлорита и некоторых других анионов используют трифенилметановые красители [16, 17]. Сначала хлорит в сильнокпслой среде окисляет KI, добавляемый в избытке по отношению к хлориту, образующийся трниоднд образует с катионом трифенилметанового красителя (например, малахитовым зеленым) ионную пару, которая экстрагируется органическими растворителями, такими, как бензол. Измеряют оптическую плотность органической фазы. При применении малахитового зеленого молярный коэффициент поглощения равен 162000.
Электрохимические методы
Обычно приводят редокс-метод, в котором используют мы-шьяк(Ш) и осмиевую кислоту как катализатор [10]. Похожий метод, но с потенциометрической индикацией конечной точки титрования позволяет анализировать смеси, содержащие хлорит, гипохлорит, хлорат и хлорид [18]. Хлорит титруют при pH —8 —12 с платиновым индикаторным электродом в присутствии OsO4. Подробно методика описана в разделе «Хлорат».
Потенциометрический метод
Для потенциометрического определения хлорита [19] можно использовать стандартный раствор NaClO:
2C1OJ + СЮ'+ Н++ СГ —> 2С1О2 + 2СГ + ОН*
Для индикации используют платиновый и каломельный электроды, хлорит определяют в присутствии хлората, диоксида хлора и хлорида.
Потенциометрическое определение хлорита
в присутствии диоксида хлора, хлората и хлорида [19]
Реагенты
Гипохлорит натрия, 0,04 М раствор.
Электроды, насыщенный каломельный электрод и пластина Pt.
Ход анализа. Анализируемый раствор, менее 0,01 М no NaClO2, подкисляют 0,1 М раствором НС1. Перемешивая раствор с помощью магнитной мешалки, титруют 0,04 М раствором NaClO Скачок потенциала от 830 мВ почти до 1100 мВ указывает иа конец титрования. Важно, чтобы концентрация НС1 и хлорита не превышала 0,01 М и pH раствора был меньше 3.5.
Полярографические методы
В работе [20] описана необратимая волна восстановления хлорита при pH ~ 4,5. В оптимальной области pH = 4,2—4,5 высота волны диффузионного тока пропорциональна концентрации хлорита в интервале 0,2—2,0 мА4. При pH < 4,2 хлорит очень быстр0 разрушается.
330
Полярографические характеристики хлорита (и диоксида хлора) исследовали с применением вращающегося дискового графитового электрода. Хлорит можно определять при pH = 7—9, а оба соединения при pH = 5,0—5,5, где их устойчивость одинакова [21].
ЛИТЕРАТУРА
I Harrison В L , Rosenblatt D. H. — J. Chromat., 1964 v 13, р. 271.
9 Peschke U7. — J. Chromat., 1965, v. 20, p. 573.
3 Seiler H„ Seiler M. — Helv. chim. Acta, 1967, v. 50, p. 2477.
4 Thielemann H —Mikrochim Acta, 1971, p 746.
5 Schulek E., Endroi P.— Analytica chim. Acta, 1951, v. 5, p. 245.
6. Kolthoff I. M., Sandell E. B., Meehan E. J., Bruckenstein S. — Quantitative Chemical Analysis, 4th edn., London, Macmillan, 1969.
7 Brown E. G — Analytica chim. Acta, 1952, v. 7, p. 474.
8 Туманова T. /1, Пахомова Л. H., Майорова Л. П. Зав. лаборатория, 1970, т 36, с. 1036. Analyt. Abstr., 1971, т. 21, с. 183.
9. Норкус П. К. ЖАХ, 1964, т. 19, с. 518. Analyt. Abstr., 1965, т. 12, с. 3875.
10 Норкус П. К ЖАХ, 1965, т. 20, с. 612. Analyt. Abstr., 1967, т. 14, с. 142.
11. Hashmi М Н., Ayaz А. А.—Analyt. Chem., 1963, v. 35, р. 2194.
12. Erdey L., Svehla G.— Z. analyt. Chem., 1959, v. 167, p. 164; Analyt. Abstr., 1960, v. 7, p. 122.
13. Erdey L., Bazas I., Vlgh K-—Periodica polytech , 1959, v. 3, p 1; Analyt. Abstr., 1959, v. 6, p. 4262.
14. Prince L. A. — Analyt. Chem., 1964, v. 36, p. 613.
15. Hong С. C., Rapson W. H. — Can J. Chem., 1968, v. 46, p. 2061; Analyt. Abstr., 1970, v. 17, p 2096.
16. Раманаускас E. И, Буникине Л. В., Шулюнене А. К-, Жиленайте М. В. ЖАХ, 1969, т. 24, с. 244 Analyt. Abstr., 1970, т. 19, с 1022.
17. Сапрагонине М. С., Раманаускас Е. И. Научи. Труды высших учебных зав. Лит. ССР, Химия и хим. технол., 1971, т. 13, с. 15, 1973, т. 24, с. 131.
18. Норкус П. К-, Стильжене С. П. ЖАХ, 1969, т. 24, с. 884. Analyt. Abstr., 1970, т. 19, с. 4862.
19. Kepinski К L, Blaszkiewicz В. G. — Taianta, 1966, v. 13, р. 357.
20. Hartley А. М., Adams А. С. — J. electroanal. Chem., 1963, v. 6, р. 460.
21. Schwarzer О., Landsberg R. — J. electroanal. Chem., 1967, v. 14, p. 339.
ФТОРИДЫ
Литература но анализу фторсодержащих соединений, вероятно,
самая многочисленная, превосходящая по числу ссылок литературу по анализу любого другого элемента. Причина этого кроется как в важности информации о содержании фтора в воде, воздухе, биологических материалах, инсектицидах, удобрениях, полимерах и Т- Д., так и в характерных особенностях химии фтора, отличной l®' химии других галогенов и делающих анализ фтора чрезвычайно сложной задачей.
Большинство фторидов в различной степени труднорастворимы, что затрудняет вскрытие фторсодержащих материалов. Фтор характеризуется максимальным сродством к электрону и по сравнению с другими элементами наиболее реакционноспособен Разно-оразно влияние других ионов, присутствующих в анализируемом Растворе: они или осаждаются в виде фторидов, или образуют Рочные металлфторидные комплексы. По этой причине чрезвы
331
чайно важной стадией анализа является предварительное выделение фтора перед его определением. Выделение фтора дистилляцией длительно, поэтому в некоторых современных методах анализа эту стадию заменяют.
Важным принципиальным источником ошибок в анализе фторида является его взаимодействие со стеклянной посудой. Выпаривание щелочных фторидных растворов в стекле может привести к потере фторида, поэтому такие операции следует исключать. Это явление впервые было обнаружено Рейнольдсом и Хиллом [1] и подтверждено Ринком [2], который рекомендовал вместо боросиликатного стекла использовать платину или йенское стекло и строго контролировать pH от 6 до 8. Было найдено, что потери фторида происходят не за счет его отгонки, а скорее за счет адсорбции его на поверхности или на границах раздела кристаллической решетки материала посуды [3].
Опубликованы подробные обзоры [4—8] по методам разделения и определения фторида, проведенная в этих обзорах систематизация методов делает сами обзоры чрезвычайно полезным источником информации. Важно отметить, что замечания, сделанные в обзоре Макдональда [5] и касающиеся утверждений авторов обзора [4], не всегда правильны и в этих случаях следует обращаться к оригинальным работам. В упомянутых выше обзорах рассматриваются работы, опубликованные до 1950 г. Опубликован обзор по определению фтора в органических материалах [9] и сборник микроаналитических методик [10].
Особую трудность представляет собой анализ фтора в биологических материалах, так как его содержание в них обычно меньше < 5 ppm в пересчете на сухую массу образца. Эта проблема обсуждалась Холлом [11, 12]. Растения значительно более чувствительны к фториду, чем к другим вредным примесям в воздухе, поэтому содержание фтора в растениях широко используют в качестве эталона для оценки содержания вредных загрязнений в воздухе. Опубликован обзор по современным методам анализа фтора в растениях [13].
В связи с появлением в 1958 г. нового реагента — ализарин-комплексона [14] и в 1966 г. — фторидного ионоселективного электрода [15] некоторые известные методы определения фторида устарели и в настоящее время представляют лишь академический интерес.
МЕТОДЫ ОТДЕЛЕНИЯ
Отгонка кремнефтористоводородной кислоты
Первые методы отделения фторида были основаны на летучести SiF4. Этот метод потерял свою ценность при введении Виллар* дом и Винтером [16] в 1933 г. и независимо от них ТананаевыМ [17] дистилляции с паром кремнефтористоводородной кислоты
332
H2SiFe* из хлорной пли серной кислоты при 135 °C в присутствии стеклянных шариков.
Этот метод использовали многие авторы, большинство из них рассматривали проблему полноты выделения фторида из определенного анализируемого образца. Прибор в простейшем варианте (рис. 36) состоит из колбы емкостью 100—150 мл из боросиликатного стекла, соединенной с холодильником трубкой. В горло колбы почти до ее дна вставляют термометр. Такой прибор был рекомендован для определения фтора в продуктах питания [19].
Метод отгонки модифицировали как усовершенствованием прибора для отгонки, так и оптимизацией выбора кислоты. При дистилляции с паром из хлорной кислоты, как было найдено, устраняется влияние фосфата, сульфата и арсената на последующее определение фторида. Особенно важно предотвратить попадание сульфата в конденсат в том случае, когда фторид определяют ти-триметрически нитратом тория. Фосфорную кислоту также можно использовать в качестве среды при дистилляции, но в этом случае вероятность попадания ее следов в конденсат очень велика.
Мешающее влияние хлорида и бромида можно устранить, добавляя Ag2SO4 или AgC104 для переведения галогенидов в нелетучие соединения. Ни один ион металла с паром не перегоняется.
Тот факт, что кремний, алюминий и железо снижают летучесть фторида, был отмечен в нескольких работах. Для снижения этого влияния рекомендовали [20] проводить отгонку из серной кислоты при 150 °C, так как при отгонке из хлорной кислоты при температуре выше 139 °C идет ее разложение, хлорная кислота отгоняется и конденсируется вместе с паром и мешает при титровании фторида нитратом тория. Отгонка из смеси серной и фосфорной кислот происходит лучше, но перед титрованием фторида нитратом тория необходимо из конденсата удалить следы этих кислот введением солей бария и серебра [21].
При определении кремния и фторида во фторсиликатах применяли [22] плавленый оксид цинка, полученный осаждением ZnO из аммиачного раствора оксида цинка. В этом же исследовании было обнаружено положительное влияние присутствия борной кислоты. Было отмечено не полное выделение фторида, отнесенное за счет образования недиссоциированной тетрафторборной кислоты или ее солей. Выпаривание дистиллята и затем сплавление позволяют добиться количественного выделения фторида.
Влияние кремния и алюминия при определении фторида полностью исключается [23] при сплавлении образца с Na2CO3 и ^пО, растворении плава, отфильтровывании осадка и дистилляционном выделении фторида из смеси фосфорной и хлорной кислот.
Пэк и Смит [24] подтвердили ранее обнаруженный эффект с°рбции фторида стеклом аппаратуры для перегонки Дистилляция становится более полной, если прибор промывают раствором
Фторид перегоняется в виде SiF4 [18].
333
Рис. 36. Простейший прибор для отделения фторида методом перегонки с паром.
Рис. 37. Прибор для микродистилляцин фторидов:
1— пробирка с пробой: 2—устройство для ввода азота и воды; 3—холодильник; 4—трубка, внешний диаметр 5 мм, внутренний диаметр 1 мм, на конце оттянута в виде капилляра [29Ц
карбоната натрия. Было найдено, что вследствие такого рода потерь удается отделить лишь 98% фторида, даже если прибор изготовлен из борсиликатного стекла и отгонку проводят из H2SO4 [25]. Многие недостатки дистилляции как метода выделения фторида можно свести к минимуму или даже устранить.
Длительность отгонки исключает возможность применения этого метода для анализа большого числа образцов или в рутинном анализе, хотя описаны и автоматические варианты метода [26—28].
При определении микро- и полумикроколичеств фторида большой объем водного конденсата, необходимый для количественной отгонки фторида, приводит к сильному разбавлению анализируемого раствора. Это, безусловно, затрудняет определение, а упаривание конденсата до малых объемов с целью концентрирования может привести к потере фторида. Описан метод [29], пригодный для использования в таких случая?. Для отгонки фторида из микродистилляционного прибора используют азот; метод применим для определения 0—3,0 мкг фторида. Для этого варианта метода разработан [30] усовершенствованный прибор, с применением которого удается свести к минимуму попадание в дистиллят фосфата и сульфата. Авторы [30] нашли, что в сочетании с определением фторида с ализаринкомплексоном определению мешают только борат и силикат, остальные исследованные 42 иона в молярном отношении к фториду 500 : 1 не мешают анализу.
Прибор для микродистилляции фторида представлен на рис. 37. Этот вариант метода широко рекомендуется для использования в органическом анализе [31, 32].
334
Выделение фторида отгонкой с паром
Методика [31, 32] приведена с некоторыми сокращениями.
Аппаратура
Прибор для дистилляции. В верхней части прибора (рис. 38) помещены дистилляционная колба н конденсатор. Его нижняя часть соединяется стеклянной трубкой с иижией частью прибора, в которой находится генератор пара — пло-скОДониая колба вместимостью 1 л из боросиликатного стекла. Эта колба снабжена соединительной трубкой и устройством, приспособленным для выпуска лишнего пара.
Пар проходит через две концентрические трубки 4 и 5 и попадает в реактор 6. После прохождения через раствор, из которого отгоняют фторид, пар, содержащий летучие примеси, попадает в конденсатор, который состоит из трех концентрических трубок. В трубках 7 и 8 пар конденсируется, а в трубке 9 циркулирует холодная вода. Стеклянная трубка 10 служит для введения раствора, содержащего фторид, в нее же во время дистилляции помещают стеклянный термометр (шкала до 150°C); 3—дополнительный нагревательный элемент из никелевой проволоки 2 (сопротивление 7660 Ом/м) длиной 600 см. Оптимальная длина проволоки 420 см; остальную часть проволоки используют для закрепления ее на клеммах. Дополнительный нагреватель представляет собой стеклянный цилиндр диаметром 48 мм, покрытый алюминиевой фольгой и листом асбеста.
Желательно вместо никелевой проволоки использовать нихромовую, которую следует изолировать слоем цемента. Дополнительный нагреватель питается от источника постоянного тока, генератор пара нагревается электроплиткой мощностью 500 Вт.
Ход выделения. Образец (содержащий 0,1—0,5 мг F) растворяют, используя, например, микроавтоклав. Раствор количественно переносят в прибор для дистилляции через трубку 10. Чашку, в которой проводили растворение, ополаскивают 20 мл 70—72%-ной хлорной кислоты, этот раствор также помещают в прибор для дистилляции. Туда же вводят 2 мл 25%-ного водного раствора перхлората серебра и 8—10 стеклянных шариков. В трубку 10 помещают термометр.
Горячую электроплитку, переключатель которой переведен в положение, соответствующее максимальному нагреву, помещают под колбу-генератор пара. В дополнительный нагреватель подают ток 1,5 А, напряженке 45 В, температура при дистилляции поддерживается 135± ±2 °C. За 45 мин в полиэтиленовом приемнике собирается около 250 мл конденсата.
Для того чтобы процесс дистилляции прекратить, отключают дополнительный нагреватель, и когда температура заметно снизится, отставляют электроплитку и открывают устройство для вы пуска лишнего пара из колбы генератора
Перед дистилляцией фторида из ьЛЛНзнРУем°Г0 образца весь прибор про-ЛЯ.Вают в условиях проведения дистил-
Рис. 38. Верхняя часть прибора для отделения фторидов перегонкой с паром [31].
/ — шлиф; 2—сопротивление; 3—корпус дополнительного нагревателя; 4, 5, 7,
8 — концентрические трубки; 6—реактор;
9 —холодильник; 10—трубка для ввода пробы.
335
Микродиффузия
Диффузия как метод отделения фторида чрезвычайно важна при определении фторида в биологических материалах. Фторид-ион вытесняют минеральными кислотами, превращая его в HF который поглощают раствором щелочи. Фторид обычно определяют спектрофотометрическим методом. Этим методом можно определять < 10 мкг фторида. Процесс диффузии проходит особенно успешно при невысоких температурах. Предложенный в 1954 г. [33] метод можно осуществлять в нескольких вариантах. Используют [34] полипропиленовую ячейку Конвея (рис. 39). Такую же ячейку применяли [35—37] для определения фторида в воздухе, при этом использовали чашку Петри из полистирола с неплотно закрывающейся крышкой, на внутреннюю сторону которой нанесен слой щелочи. В отличие от других исследователей авторы работ [35—37] не нашли нужным герметизировать крышку. Диффузионный процесс можно осуществлять и в полиэтиленовых бутылях; было показано [11, 38—40], что этот вариант обладает преимуществами по сравнению с диффузионной методикой по Конвею [34]. Для выделения фторида этим методом требуется сравнительно много времени (в некоторых случаях 24 ч), время, затрачиваемое на проведение единичного анализа, можно снизить [41], если использовать сразу несколько ячеек, которые можно оставить на ночь.
В более поздних работах основное внимание уделяли влиянию силиконовой смазки на процесс диффузии. Найдено [42, 43], что диффузия фторида значительно возрастает в присутствии силиконовой смазки, используемой для герметизации в микродиффузион-ных ячейках (рис. 40). В присутствии простейшего силикона — гексаметилдисилоксана фторид выделяется из образца за 1 ч при комнатной температуре, т. е. за время в 10 раз меньшее, чем в предыдущей диффузионной методике. Предполагают, что образуется триметилфторсилан
Образец*
/+нсю4
NaOH
Изолирующий слой
Рис. 39. Схема микродиффу-зноииой ячейки, иа которой показано относительное расположение реактивов для проведения диффузии фторида (34].
по реакциям
HF + (СНз)з SiOSi (СНз)3 —> (CH3)3SiF + + (СН3)з SiOH
HF + (СНз)з SiOH —> (СНз)з SiF+ Н2О
Сульфат и фосфат в этих условиях не диффундируют.
В работе [44] объединили микродиффузию с применением гексаметилдисилоксана и определение с помощью фторидного электрода. Метод использовали для анализа зубных паст. Для снижения времени полной диффузии было проведено систематическое исследование [45], целью которого было выяснение влияния Ра3"
336
Рис. 40. Диффузия радиоактивного фторида при комнатной температуре в присутствии силикона и в его отсутствие [42]:
/—силикон введен с кислотой; 2 —в изолирующий слой введена силиконовая смазка; 3—ко.ч трольный опыт (без силикона) [421-
Рис. 41. Насадка прибора для отделения фторида микродиффузией [38].
/—фильтровальная бумага, свернутая в виде цилиндра; 2—стеклянный стержень; 5—цолн-этнленовая трубка; 4—плотно надевающаяся пробка.
личных факторов на скорость диффузии. Это позволило рассчитать для данной стандартизированной ячейки время, необходимое для полной диффузии.
Отделение фторида с использованием микродиффузии
Метод, в общих чертах описанный Холлом, был рекомендован [46] для анализа кормов.
Аппаратура и реагенты
Прибор для отделения фторида микродиффузией (рис. 41).
Фтористоводородная кислота поглощается импрегнированиой бумагой, закрепленной в полимерной крышке полиэтиленовой банки вместимостью 20 мл
Гидроксид натрия, 0,5 М раствор, содержащий 50% по объему пропилеи-гликоля.
Сульфат серебра в HClOt, приблизительно 0,25 М раствор Ag2SO4 в 70%-ной 79«/Массе НСЮ4. Смешивают 4 г мелкодисперсного AgsSOi с 2 мл воды и 23 мл '•‘л-иой НС1О4 в колбе Эрлеимейера емкостью 250 мл и нагревают до 80 °C для Растворения AgjSOi. Затем добавляют 75 мл 72 %-ной НС1О4 и охлаждают. . Х°Д анализа. [38, 39, 11]. Помещают в бутыль прибора для проведения диффузии 1 мл анализируемого раствора, на верхнюю часть фильтровальной бумаги омещают 1 каплю 0,5 М раствора NaOH. содержащего 50% по объему поли-уН^нленгликоля. Немедленно добавляют пипеткой 2 мл раствора Ag2SO4 в ты "'и°й HCIO4, стараясь не дотрагиваться до внутренней стороны горла бу-TjflH. Внутреннюю сторону горла бутыли протирают фильтровальной бумагой, тщательно удалить капли HCIO4.
Раб ЬыстР° закрывают бутыль пластмассовой крышкой с помешенной в нее об-таиной фильтровальной бумагой и тщательно ее завинчивают. Затем горло
337
бутыли запечатывают разогретым воском следующим образом. Бутыль поддержи, вают в горизонтальном положении так, чтобы ее горло помещалось над сосудом с горячим воском, и заливают жидким воском соединение, добавляя воск по каплям из пипетки.
Объем жидкости в диффузионной бутыли ие должен превышать 3 мл. Бу. тыль выдерживают 24 ч при 60 °C. Затем бутыль открывают, пинцетом вынимают бумагу и определяют содержание фторида.
Примечания. 1. Для определения фторида используют ализарпиком-плексон. Если содержание фторида около 0,2 ppm, измеряют светоплоглощение раствора при 615 им. Для определения меньших концентраций фторида или при необходимости более точных определений следует проводить экстракцию раствором амина в изобутиловом спирте.
2. Бумагу пропитывают полипропилеигликолем для того, чтобы замедлить процесс испарения воды.
Пирогидролиз
При анализе некоторых горных пород и тугоплавких материалов, в которых отсутствуют органические вещества, самым удобным методом отделения фторида является пирогидролиз. Он состоит из двух стадий: нагревание образца перегретым паром и поглощение образующейся HF.
Процесс описывается следующим уравнением
MF2n + лН>0 (пар) 4=*: ЧО„ + 2-iHF (газ)
Так как вместо 1 моль пара образуется 2 моль HF, то реакция в целом проходит с увеличением энтропии.
Пирогидролиз — не новый метод, основные принципы этого метода опубликованы в 1856 г. Но только в результате систематического изучения метода авторами работы [47], давшими методу название «пирогидролиз», он приобрел практическую значимость. Было найдено [47], что при 1000°C фториды довольно быстро гидролизуются. Гидролиз фторидов легких металлов проходит медленнее, но при добавлении к ним катализатора U3O8 реакция проходит полностью за сравнительно короткое время.
Усовершенствование метода было направлено [43] на концентрирование конденсата, содержащего фторид, который обычно получают очень разбавленным. Вместо перегретого пара применяют влажный кислород. В течение 20 мин можно отделить миллиграммовые количества фторида. Пирогидролиз используют для определения простых и сложных фторидов в радиоактивных материалах, горных породах, рудах, минералах, шлаках, стекле, катализаторах и т. д. Этот метод экспресснее дистилляции, поэтому он был рекомендован [49] вместо дистилляционного концентрирования для рутинного анализа горных пород и минералов. Аппаратурное оформление метода показано на рис. 42.
Выделенный фторид определяют спектрофотометрически с алй-заринкомплексоном или с использованием фторидного- ионоселективного электрода. Фторид можно определять с хорошей точностью до 50 ppm.
Метод пирогидролиза можно несколько ционная камера может быть платиновой, из
видоизменить. Реак кремниевых сплав0
338
рис. 42. Прибор для отделения фторидов методом пирогидролпза [49]: сжатый воздух; 2—фильтр с ватой;
•—ротаметр; 4— никелевая лодочка с во-
5—кварцевая лодочка с образцом и Сносом; 6—кварцевая трубка; 7—горелки; ^.полиэтиленовый абсорбер.
йли никелевой, а гидролиз можно осуществлять, пропу-ская перегретый пар или кислород. Можно варьировать также температуру и длительность пирогидролпза, природу добавок катализаторов и метод определения фторида.
При пирогидролнзе микрограммовых количеств фторида особенно сильно сказываются механические потери фторида [50]. Это было выяснено [51] при изучении оптимальных условий отделения микрограммовых количеств фторида с применением многофакторного планирования эксперимента для выявления вклада каждой переменной в результат анализа. При отделении 2—20 мкг фторида обычно получают несколько заниженные результаты. В этом исследовании определение фторида проводили с анализаринкомп-лексоном.
Хроматографические методы
Все виды хроматографии используют для отделения мешающих определению катионов и анионов. Если, например, отделение проводят с ализаринкомплексоном, то необходимо отделить ионы, взаимодействующие с лантаном и тем самым затрудняющие его реакцию с ализаринкомплексоном, и те ионы, которые образуют комплексы с фторидом, например, Pb2+, Fn2+, АР+, Fe3+ и др. Ионы металлов-комплексообразователей можно отделить, используя ионный обмен. Этот метод был применен [52] при определении фторида в стандартных образцах.
В микроанализе, если предварительное разложение образца проводили сплавлением, избыток щелочи также можно удалить, используя катионный обмен [53]. Естественно, металлы должны находиться в катионной форме.
С помощью анионообменных смол фторид можно отделить от вешающих анионов, особенно от фосфата и сульфата. Отделение Фосфата чрезвычайно важно при анализе биологических материалов. Отделение фторида от анионов может осложниться присут-вием фторидных комплексных ионов, таких, например, как г*^б , A1F1’, SiFe' и др., которые не будут элюироваться вместе ^фторидом, не связанным в комплекс. Описано [54] отделение п0°РИДа (25 мкг) от фосфата (12,5 мг Р) на смоле Дауэкс 1 с ис-
Ьз°ванием 0,5 М раствора NaOH в качестве элюента.
339
Обычно мешающие катионы и анионы в анализируемом рас. творе присутствуют одновременно, и их отделение с применением ионного обмена может увеличить ошибку анализа и сушественнс увеличит время анализа. Был разработан метод отделения и анионов и катионов за одну операцию, при этом катионы связывали в прочные отрицательно заряженные комплексы с ЭДТЛ, которые хорошо удерживаются анионообменной смолой, в то время кап фториды, слабо сорбирующиеся той же смолой, количественно элюировали (55]. Метод был успешно применен для анализа фос. фатных горных пород, определение фторида проводили титрованием нитратом тория. В работе [56] показано, что фторид можно определять с помощью фторидного ионоселективного электрода в концентрированной фосфорной кислоте даже в присутствии алюминия [57], что позволило разработать метод прямого (без предварительного разделения) определения фторида в фосфатных горных породах.
При разработке экспрессного метода определения фторида в промышленных водах использовали ионообменное отделение фторида в виде комплексного тетрафторида бериллия BeF*' [58].
Разделение фторида и сульфата можно проводить также с применением ионного обмена [59]; метод описан в книге по аналитическому применению ионного обмена [60]. Разработана методика [61] ионообменного разделения фторида, фосфата, сульфата и некоторых других анионов.
Для отделения фторида используют и другие хроматографические методы. Было найдено [54], что методом бумажной хроматографии можно легко и количественно разделить фториды и фосфаты. С помощью тонкослойной хроматографии на целлюлозе [62] или силикагеле [63] можно отделить галогенид-ионы (включая фториды). В бумажной и тонкослойной хроматографии галогенидов, как правило, значение Rf увеличивается с увеличением атомной массы. В случае ионообменных материалов последовательность сорбции хлоридов, бромидов и иодидов обычно обратная.
Газовую хроматографию используют для выделения фторида. С применением селективной экстракции раствором хлортриметил-силана в бензоле определяли [64] 0,01—10 мкг фторида в 1 мл мочи, сыворотке и других биологических материалах. Фторид реагирует с образованием фторотриметилсилана, который переходит в бензол. Прямое введение экстракта (1—3 мл) в стальную газохроматографическую колонку, содержащую 20% силиконового масла и хромосорб Р, использование азота в качестве газа-носителя и пламенно-ионизационного детектора позволяют получать хроматографический пик, пропорциональный концентрации фтор' гриметилсилана. Этот принцип был положен в основу более позД' ней методики определения общего и растворимого фторида в 3У ной пасте [65].
340
Соосаждение
Шапиро и Колесникова [66] отделяли фторид на суспензии |4g(OHh, которую растворяли в разбавленной HNO3 и определяли фторид фотометрически.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Осаждение в виде фторида кальция
Методу гравиметрического определения фторида в виде фторида кальция уже более 150 лет. К недостаткам метода, которые обычны и для других методов, основанных на осаждении фторидов металлов, следует отнести склонность к образованию коллоидных осадков и большое число мешающих ионов. История развития метода, его титриметрические варианты описаны в литературе [4-6].
Кажется маловероятным, чтобы в настоящее время метод нашел практическое применение.
Осаждение в виде хлорида-фторида свинца
Осаждение в виде хлорида-фторида свинца — один из самых ранних методов определения фторида. Несмотря на сравнительно высокую растворимость осадка в воде (325 мг/л 18°C), преимуществом метода является то, что практически независимо от условий осаждения образуется крупный кристаллический осадок. Метод широко использовали для макро- и полумикроопределений фторида.
Определение можно закончить гравиметрически и одним из титриметрических методов: определение содержания хлорида в осадке или избытка хлорида в растворе по методу Фольгарда, комплексометрическое определение свинца. Арсенат, фосфат, сульфат, сульфид, малые концентрации железа и алюминия мешают определению. Арсенит, небольшие концентрации бората и аммо-Ния, большие количества ацетата, перхлората, нитрата, бромида, и°Дида, натрия и калия не мешают определению фторида.
проведено детальное изучение [67] осаждения хлорида-свинца и показана сложность процессов образования
°СаДКа, которые могут привести к завышению или занижению Результатов анализа. Можно получить хорошие результаты, про-°Дя осаждение в строго контролируемых условиях при более низ-их значениях pH в присутствии буферов (формиата натрия или я^Рацетата натРия)- Оптимальным значением pH, по-видимому, вдяется pH == 2, дальнейшее снижение pH приводит к занижению
иЫЛО Фторида
341
результатов анализа. По мнению авторов работы [68], проведение осаждения при pH = 4,6 позволяет уменьшить ошибку анализа до нескольких процентов.
Осаждение в виде фторида лития
Для определения 20—200 мт F в растворах был разработан метод осаждения фторида в виде фторида лития [69]. Так как в водных растворах фторид лития осаждается не полностью, необходимо к раствору добавлять какой-нибудь смешивающийся с водой органический растворитель. Удобной средой является 50%-ный по объему раствор этанола в воде, более высокие его концентрации ухудшают качество осадка. При определении более низких концентраций фторида рекомендуется уменьшать объем раствора, чтобы уменьшить ошибку анализа за счет растворимости осадка в 50 %-ном этаноле.
Определение фторида в виде фторида лития [69]
Реагент
Хлорид лития, 30 г/л в 95%-ном этаноле.
Ход анализа. Доводят объем анализируемого раствора, содержащего 20— 200 мг F, до 15—40 мл в зависимости от концентрации фторида. Нагревают до 70 "С и медленно, при перемешивании, добавляют равный объем раствора LiCl. Оставляют раствор стоять до тех пор, пока он не остынет и не станет прозрачным. Фильтруют осадок через фильтр со средним размером пор и промывают 50%-ным этанолом, насыщенным LiF до отрицательной реакции на хлорид. Промывают 5 мл 95%-ного этанола и сушат в сушильном шкафу в тигле 1 ч при НО °C.
Примечания. 1. В методике для точного определения фторида нельзя использовать полиэтиленовые колбы, гак как осадок сорбируется на стенках колбы и очень трудно визуально определить полноту перенесения осадка на фильтр. Было найдено, что посуда из боросиликатного стекла не вносит в анализ значительных ошибок.
2. Мешающее влияние сульфатов, являющееся трудной проблемой в других гравиметрических методах определения фторида, незначительно для малых количеств фторидов и сульфатов, при определении же больших количеств фторида ошибка становится существенной, вероятно, за счет сэосаждения NajSC^ или Li2SO4 растворимость которых в спиртовых средах понижается. При необходимости можно вводить поправочный коэффициент, учитывающий влияние сульфата
3. Мешающее влияние других анионов не исследовали, но можно предположить, что в этом методе оно будет сказываться меньше, чем в методах определения фторида в виде фторида кальция или хлорнда-фторида свинца, за счет большей растворимости литиевых солей.
Другие гравиметрические методы
Описаны методы гравиметрического определения фторида в виде LaF3 [70] и в виде трифенилфторида [71]. Для осаждения [71] используют раствор трифенилхлорида при pH = 4—9. Д0' стоинством метода является то, что фосфат, борат, ионы желе^ алюминия и циркония не мешают определению. На искусственны смесях было показано, что чувствительность метода — 1—^0 < ’ ошибка определения фторидов в минералах составляет 342
Титриметрические методы
Ториметрический метод
В очень разбавленных растворах торий (IV) реагирует с фторидом с образованием бесцветного комплексного иона (ThF6)2- по реакции
Th (\О3)4 + 6F" —► (ThF6)2’ + 4NO7
Реакция не строго стехиометрична за счет образования менее устойчивых низших комплексов тория. При добавлении раствора тория (IV) к фториду в присутствии ализаринового красного С (ализаринсульфонат натрия) сначала образуется комплекс (ThFe)2-, затем избыток тория взаимодействует с ализариновым красным С с образованием розового комплекса. Развитие розовой окраски проходит во времени и зависит от таких факторов, как объем раствора, концентрация индикатора, pH и др. При титровании необходим строгий контроль за pH, так как ализариновый красный С является еще и кислотно-основным индикатором (при pH = 3,7желтый, при pH = 5,2 — фиолетовый). Метод прямого титриметрического определения фторида торием (IV) в присутствии ализаринового красного С пригоден для точного определения фторидов, если выполняется строгий контроль за условиями проведения анализа. Методом обратного титрования можно определять 25—350 мкг F с точностью ±1 мкг, правда, для достижения таких результатов требуются специальные навыки [72].
При определении больших содержаний фторида (около 400 мкг) образуется осадок фторида тория, что приводит к новым трудностям. Индикатор сорбируется осадком, окрашивая его в розовый цвет, что не позволяет четко фиксировать точку эквивалентности. Вблизи точки эквивалентности в присутствии малых количеств фторида на результатах титрования сказываются конкурентные реакции Th — фторид и Th — индикатор.
Мешающими ионами, как и в большинстве методов определения фторидов, являются ионы металлов, образующие с фторидом комплексные соединения или труднорастворимые соединения: Ва11, Fe11, Al111, Be11, ZrIV, Pv, Asv, As111; сульфат и сульфид-ионы должны отсутствовать.
Несмотря на присущие методу недостатки, титрование то-Рием(1У) является основным методом определения фторида, наведшим применение в анализе широкого круга фторидсодержа-Щих материалов. По-видимому, самым интересным применением Метода было определение фторида в ископаемом черепе, что по-°гло археологам определить возраст находки [73]. Метод реко-енДован для определения фторида в пищевых продуктах [74].
В последнее время метод был видоизменен [75], в качестве ВиДИкатора был предложен метилтимоловый синий, а для созда-Hbiflp^ = ^’35 в качестве буфера использовали глицин-перхлорат-°Уферный раствор. Точка эквивалентности становится четкой,
343
кроме того, буфер обеспечивает строгую стехиометрию комплексов при определении 1—10 мг F. Метод был всесторонне исследован [76], а его модифицированный вариант рекомендован для титрования миллиграммовых количеств фторида.
Описан метод кондуктометрической индикации конечной точки титрования фторида ThIV [77]. Этот метод применен для анализа горных пород после отгонки из них фторида.
Титриметрическое определение фторида с помощью нитрата tophh(IV) [75]
Реагенты
Буферный раствор. 6,7 г глицина и 11 г NaCIO< растворяют в воде, добавляют 11 мл 1 М НС1О< и разбавляют до 100 мл водой.
Нитрат тория. 0,02 М раствор 11,044 г четырехводного нитрата тория растворяют в 0,001 Н HNO3 и разбавляют раствор до 1 л 0,001 и М HNO3.
Метилтимоловый синий, 0,2%-ный по массе раствор.
Ход анализа. Образец может содержать в объеме не более 15 мл до 10 мг фторнда. Доводят pH раствора 1 М НС1О< до 3,35 ± 0,10 и добавляют 2 мл буферного раствора. К раствору добавляют 3 капли индикатора н титруют нитратом тория до появления неисчезающей голубой окраски. Титр нитрата тория устанавливают по стандартному раствору фторида.
Примечания. 1. Определению мешают катионы, взаимодействующие с метилтимоловым синим: Си11, Mg, Zn, Cd, Hg11, Pb”, Мп11, Fe11, Co" и Ni.
2. Сульфаты, сульфиды и фосфаты мешают определению. Фосфат можно удалить введением Ag2O [78]. Сульфиды можно отделить осаждением бензидином.
Комплексометрический метод
Комплексометрический метод обычно используют после осаждения фторида каким-нибудь металлом. Бельчер и Кларк [79] осаждали фторид стандартным раствором СаСЬ и после отделения осадка (раствор с осадком выдерживали около 12 ч) титровали избыток Са2+ стандартным раствором ЭДТА в присутствии индикатора эриохром черного Т. Достоинством метода является то, что 10-кратный избыток сульфатов и фосфатов не мешает определению, а существенным недостатком метода является его длительность Метод комплексометрического титрования с предварительным осаждением фторида в виде PbCIF обладает теми же недостатками; кроме того, при использовании солей свинца для получения строго стехиометрического осадка необходим контроль за условиями проведения реакции.
Метод Леонарда [80] определения 2—20 мг F основан на предварительном осаждении PbCIF избытком стандартного раствора нитрата свинца. После отфильтровывания и промывки осадка избыток осадителя титруют ЭДТА. Из 16 параллельных анализов 30—45 мг образца чистого NaF нашли, что содержание в не^ основного вещества составляет 100,0 + 0,19%. Мешают анализу ионы, образующие со свинцом или фторидом комплексные и I труднорастворимые соединения, например фосфаты, которые титровываются практически количественно.
344
Разработан метод определения фторидов в неорганических образцах, содержащих значительные количества металл-ионов и нитратов, хлоридов и перхлоратов [81]. После пиролиза фторид осаждают при pH = 1,75 известным избыточным количеством церия (III)» избыток последнего оттитровывают ЭДТА с индикатором арсеназо. Церий(III) образует труднорастворимый фторид, произведение растворимости которого 10~15. Большие количества хлорида и перхлората и небольшие количества бората и силиката не мешают определению. Сульфат и фосфат мешают анализу, но фосфат не перегоняется с фторидом при пиролизе.
Описан метод определения фторида, основанный на осаждении фторида самария и обратном титровании избытка самария ЭДТА [82]. При определении 1—4 мг фторида в качестве индикатора использовали метилтимоловый синий. В отличие от более простых анионов фосфат и сульфат мешают определению, образуя с самарием комплексные ноны.
Комплексометрическое титрование фторида [80]
Ход анализа. К 20 мл воды, содержащей 2—20 мг F~, добавляют 8,0 мл 0,2 М раствора NaCl и 13 мл 95%-ного этанола. Нагревают раствор почти до кипения и из бюретки добавляют 25,00 мл стандартного 0,05 М раствора Pb(NO3)2, сначала медленно, по каплям, затем, когда процесс осаждения стабилизируется, быстрее. Раствор постоянно перемешивают. Раствор с осадком выдерживают 1 мин при температуре, близкой к температуре кипения, для завершения коагуляции осадка, затем охлаждают до комнатной температуры, фильтруют и промывают осадок 4 раза порциями по 15 мл 0,02 М раствором NaCl в 20%-ном по объему этаноле. Фильтрат и промывные воды титруют стандартным 0,05 М раствором ЭДТА в присутствии индикатора ксиленолового оранжевого и твердого гексамина для поддержания pH раствора около 6. 1 мл 0,0500 М раствора Pb(NO3)i соответствует 2,10 мг NaF.
Другие титриметрические методы
Можно использовать для экстракционного титрования фторида сульфат тетрафенилсурьмы [83]. Фторид образует ионный ассоциат (C6H5)4SbF, экстрагирующийся хлороформом, остаточное содержание фторид-иона в водной фазе можно постоянно контролировать с помощью фторидного ионоселективного электрода. Фосфаты, мышьяк (III), мышьяк (V) и сульфаты не мешают определению, а сульфиты и нитриты перед определением следует Удалить. Фторид можно титровать электрогенерированным ланта-Ном(Ш) [84]. При титровании фторида лантаном (III), церием ли иттрием конечную точку титрования можно определить фото-прически с использованием в качестве индикатора 4-(2-пириди-ззо) -резорцина (ПАР) [85]. При определении 0,1—9 мг F~ стан-с Ртн°е отклонение составляет ±5 мкг. Церий (III) по сравнению лантаном и иттрием является оптимальным титрантом.
Д1р£ анализе фторида реализована высокая прочность комплекса стве ’ ХЛоРиД или нитрат алюминия можно использовать в каче-титранта для определения фторида. Описан метод [86]
345
титрования фторида нитратом алюминия в присутствии хромазуро. ла5 в качестве индикатора. Фторид титруют после его дистилляционного отделения при pH = 6 в 50%-ном метаноле. При опре-делении 1,5—120 мг фторида стандартное отклонение составляет 0,007—0,034 мг.
Спектрофотометрические методы
В обзоре [87] представлены ранние работы (до 1955 г.) по спектрофотометрическим методам определения фторида.
За исключением метода с ализаринкомплексоном, который основан на образовании тройного (1:1:1) комплекса, содержащего фторид-ион, все спектрофотометрические методы являются косвенными методами, в которых используется способность фто-рид-иона вытеснять катионы (Zriv, А13+ и другие) из их интенсивно окрашенных комплексов.
В основном все косвенные методы недостаточно селективны.
Метод с использованием алиоаринкомплексона
В водных растворах ализаринкомплексон (ализариновый фтор-голубой, 1,2-дигидроксиантрахинон-3-метиламин-Н,Ь1 диуксусная кислота, 3-аминоализарин-М,М-диуксусная кислота) реагирует с церием, лантаном или празеодимом с образованием красно-фиолетового комплекса.
Вхождение фторид-иона в координационную сферу металла с вытеснением одной молекулы воды приводит к депротонизации одной из гидроксигрупп в молекуле ализаринкомплексона, при этом возникает характерная сине-фиолетовая окраска [88].
Механизм образования окрашенного комплекса изучали фотометрическими и потенциостатическими методами [89]. Реакция, впервые предложенная Белчером, Леонардом и Вестом [90], очень стабильна и высокоселективна, чувствительность метода позволяет использовать его для анализа следовых количеств фторида.
Обычно к анализируемому раствору, содержащему фторид, добавляют реагент, затем буфер и наконец раствор соли лантана; чаще всего используют нитрат. Через некоторое время измеряют светопоглощение раствора при 625 нм.
В ряду лантаноидов чувствительность возрастает от празеодима к лантану: La > Се > Рг.
Как было найдено [91, 92], чувствительность увеличивается, если реакцию проводить в водно-ацетоновых растворах, оптимальным является 20%-ный по объему водно-апетоновый раствор-Варьировали некоторые параметры методики, такие, как pH. ТЧ буфера и концентрацию реагентов, отмечено их влияние на чУВ ствительность и воспроизводимость метода. Например, Холл отметил, что при замене обычного ацетатного буфера на сук11 натный увеличивается и чувствительность метода, и скорость пр текания реакции.
346
Опубликованы результаты всестороннего исследования метода определения 40 мкг F с ализаринкомплексоном [93]. Методика, рекомендованная в этой работе, будет описана ниже.
Другим важным исследованием является сравнительная аттестация пяти спектрофотометрических методик определения фторида в воде [94]. В этой работе сделан вывод о том, что метод с использованием ализаринкомплексона имеет преимущества по сравнению с косвенными методами, хотя метод с использованием фторидного ионоселектпвного электрода, который также исследовали в этой работе, превосходит все колориметрические методы по экспрессности, точности и может быть вполне использован для анализа воды.
Важным достоинством метода является возможность его применения для определения микроконцентраций фторида, что особенно важно при анализе биологических объектов. Обычно для этих целей используют микродиффузионный метод отделения и спектрофотометрическое определение фторида Холлом опубликована серия работ [И, 12, 39] по определению следов фторида. Как было найдено, при определении 0,1 —1,0 мкг F в 2 мл образца лучше всего использовать экстракцию хелатов лантана изобутанолом, содержащим солянокислый гидроксиламин, при этом фторид экстрагируется на 95%. Методика, однако, чрезвычайно сложна, предложена [95] более простая методика, позволяющая вдвое повысить чувствительность: определяют 0,1 —1,0 мкг F в 4 мл образца.
Опубликованы фундаментальные исследования по синтезу ализаринкомплексона [96] и определению констант его диссоциации [97] в растворах с ионной силой 0,1: Ki = 1,28 ± 0,30-10~5; =
= 2,82 ±0,24-10-8; К3 = 3,72 ± 0,19-10-"; = 6,39 ± 0,12-10“12.
Этот реагент нашел очень широкое применение. Его используют также для анализа воздуха [98—100], морской воды (автоматическое определение) [101, 102], мочи (автоматическое определение) [103], фосфатных горных пород [104], силикатных горных пород [105] и органических соединений [106].
Сульфонат ализаринкомплексона, по-видимому, обладает некоторыми преимуществами по сравнению с ализаринкомплексоном. Растворимость его натриевой соли выше, комплекс с фторидом прочнее и область определения фторида шире [107].
Определение фторида (2—40 мкг) с использованием ализаринкомплексона [93]
реагенты
нце“0^а- Дистиллированную воду очищают, пропуская ее через сильнокислот-катионообменные и сильноосновные анионообменные смолы.
ВовойкЧинатный буферный раствор, 0,1 М pH = 4,6. Растворяют 5,9 г сукни-«а 4 g «слоты в 300 мл воды и нейтрализуют 0.5 М раствором NaOH до pH = п°Ль'зо П° метру; разбавляют водой до 500 мл. Раствор готовят перед ис-
347
Ализаринкомплексон, 0,002 М раствор. Растирают 0,44 г ализаринкомплек-сона с 5 мл 0,5 М раствора NaOH и разбавляют до 200 мл водой. Добавляют 50 мл 0,1 М сукцинатного буферного раствора (pH = 4,6), pH раствора измеряют по pH-метру и, если необходимо, доводят pH до 4,5—4,8. Разбавляют рас. твор водой до 500 мл и, если раствор мутный, фильтруют. Хранят раствор в холодильнике.
Нитрат лантана, 0,002 М раствор. Растворяют 0,43 г La(NO3)3 в 500 мл воды.
Лантан-ализаринкомплексон. Смешивают равные объемы 0,0022 М раствора ализариикомплексона и 0,002 М раствора La(NO3)3- Эту смесь следует готовить ежедневно.
Ацетон.
Типовой раствор фторида. 2,2103 г NaF, предварительно высушенного в течение 1 ч прн 140°C, растворяют в 1 л воды; 1 мл раствора содержит 1,00 мг F.
Разбавленный стандартный раствор фторида. 10,0 мл типового раствора фторида разбавляют водой до 1 л непосредственно перед использованием; 1 мл этого раствора содержит 10,0 мкг F.
Построение градуировочного графика. В мерные колбы вместимостью 50 мл помещают точно измеренные объемы разбавленного стандартного раствора фторида, содержащие 0—40 мкг фторид-иона, и в каждую колбу добавляют воду до 10 мл, 5,0 мл 0,1 М сукцинатного буферного раствора (pH = 4,6), 10,0 мл реагента ланта-ализаринкомплексона и 10 мл ацетона, раствор тщательно перемешивают и отставляют на 30 мин. Светопоглощение каждого раствора измеряют в, кюветах с толщиной слоя 10 мм при длине волны 625 нм, используя в качестве раствора сравнения раствор, не содержащий фторида. По градуировочному графику или любым иным путем находят зависимость между оптической плотностью раствора и содержанием в нем фторида (см. примечание 2).
Ход анализа. Анализируемый раствор, содержащий не более 40 мкг фторида в объеме ^20 мл, нейтрализуют до pH = 4—5 и помещают в мерную колбу вместимостью 50 мл, добавляют 5,0 мл 0,1 М сукцинатного буферного раствора pH = 4,6 и далее выполняют все операции, описанные выше. Одновременно проводят холостой опыт, используя вместо анализируемого раствора воду. Измеряют светопоглощение анализируемого раствора по отношению к холостому раствору, как описано при построении градуировочного графика. По градуировочному графику рассчитывают содержание фторида в анализируемом образце.
Примечания. 1. Порядок введения реагентов оказывает влияние на скорость развития окраски, поэтому необходимо строго соблюдать установлений порядок ввода растворов. I
2. При проведении рутинного анализа можно пользоваться градуировочным графиком, построенным с применением тех же реагентов. Новый градуировочный график необходимо строить только при замене реагентов или в целях повышения точности анализа.
Мешающее влияние различных ионов изучали, вводя их в анализируемый раствор. При определении 2—40 мкг F- в растворе могут присутствовать следующие соли и ионы в количествах, не превышающих указанные ниже: 1,0 г КС1, 5,0 г ЫаСЮгНгО; 100 мг бромата, бромида, иодида, нитрата, нитрита, селената и тетрабората, 10 мг сульфата, 1 мг ацетата, цитрата, силиката и тартрата, 100 мкг оксалата и фосфата, 10 мкг карбоната и суль-фида; 1 мг аммония, бария, кальция, лития и магния, 200 мкг хромата, 100 мкг меди(II), марганца(П) и молибдена(VI), 50 мкг хрО' ма(П1), 20 мкг бериллия, 10 мкг церия(1У), серебра, титана(1*1 и цинка, 5 мкг алюминия, кобальта(П), ртути(П) и никеля, 3 мк железа(II, III), 2 мкг ванадия(V). в
Более подробная информация о влиянии посторонних ион дана в литературе [93]. 4
348
Метод с использованием комплекса циркония с солохромцианином Р
Этот метод основан [108] на обесцвечивании фторидом красного комплекса Zr с солохромцианином за счет образования [ZrOF2] -
Так как фторид и циркониевый комплекс реагируют не в строго определенном стехиометрическом соотношении, для достижения воспроизводимых результатов анализа необходимо контролировать условия его проведения. Концентрацию фторида измеряют по уменьшению светопоглощения циркониевого комплекса при 540 нм.
Опубликованы работы по использованию метода для определения 0—2,5 мкг фторида [109, ПО]. Однако в работе [37] отмечали, что холостой раствор содержит 1,0 мкг F-.
Несомненно, метод обладает достоинствами. Чувствительность, простота выполнения, экспрессность, возможность выполнять анализ в сильнокислых растворах без жесткого контроля pH создают предпосылки для широкого использования метода. Одним из недостатков метода является постепенное уменьшение окраски циркониевого комплекса, явление, обнаруженное при тщательном изучении [93]. Как было найдено, необходимо точно устанавливать время полного протекания реакции и время, в течение которого раствор находится в кювете перед измерением. Ошибки, связанные с флуктуацией времени от момента сливания растворов до завершения измерения, становятся особенно значительными при использовании метода для анализа партии образцов. Однако этот недостаток не сказывается на популярности метода, по спектру применения он уступает лишь методу с использованием ализарин-комплексона.
Мешающие ионы можно разделить на три группы:
1) ионы металлов, образующие с солохромцианином Р окрашенные комплексы, в сильнокислых средах ни один из обычных металлов не образует с этим реагентом окрашенных комплексов; 2) катионы, образующие с фторидом комплексы более прочные, чем с Zr, например алюминий, железо(III); 3) анионы, образующие комплексы с цирконием, например фосфат, сульфат, цитрат, тартрат.
Метод можно использовать для определения фторида без его предварительного выделения, если упомянутые выше ионы в анализируемом растворе находятся в количествах, меньших указан-вЫх ниже:
Мешающие ионы и ха- 3+ ракгер ошибки . . . . AI (—) Содержание ионов, дающих ошибку 10% при определении 2 мкг F, мг 0,4 решающее влияние алюминия , . _
Дец^Ни^1 ^еагента раствор подщелачить. Сульфаты удаляют вве-
Fe3+ (-)
>з-
РО3' (+) SOf (+)
0,8
100
"1( !веде|
можно
устранить, если перед
5
349
Метод применен для анализа минералов и горных пород после отделения фторидов дистилляцией [111], силикатных и фосфатных горных пород без предварительного выделения фторида [112], дЛя определения HF и других неорганических фторидов в воздухе [113, 114], в органических соединениях [115] и воде [116]. Метод использован для определения фторида после отделения его микродиффузией [35].
Спектрофотометрическое определение фторида [ПО] (0—2,5 мкг)
с использованием солохромцианина Р
Реагенты
Солохромцианин Р. Растворяют в воде 80 мг очищенного солохромцианина Р (см. примечание 1), добавляют 1 ми 1 Л1 раствора НС1 и разбавляют водой до 1 л. Приблизительно 0,001 М раствор НС1 пригоден в течение года.
Раствор циркония. Твердый хлороксид циркония ZrOCl2-8H2O, 180 mi помещают в мерную колбу вместимостью 100 мл, растворяют в нескольких миллилитрах воды, добавляют 5 мл 2 М раствора HCI и разбавляют до метки водой. Переносят пипеткой 10,0 мл раствора в мерную колбу вместимостью 1 л, добавляют 53 ±1 мл концентрированной НС1 (плотность 1,18 г/см3), перемешивают и разбавляют водой до метки. Концентрация этого раствора (0,62 М по НС1) — 5 мкг/мл циркония. Раствор выдерживают 48 ч перед использованием Он стабилен в течение года (см. примечание 2).
Ход анализа. В мерную колбу вместимостью 25 мл помещают образец или стандартный раствор, содержащий 2,5 мкг F- в 20 мл воды, н 2,00 мл реагента солохромцианина Р. Раствор перемешивают и доводят водой до метки. Через 5 мин измеряют в течение 2 мин светопоглощение в кювете с толщиной слоя 2 см при 540 нм по отношению к воде.
Примечания. 1. Лучшие продажные образцы солохромцианина Р содержат около 40% Na2SO4 или NaCl. Рекомендуется следующая методика очистки препарата. Растворяют 4 г препарата в 60 мл воды и при перемешивании добавляют 40 мл НС1 (плотностью 1,18 г/см3) для выделения свободной кислоты. На воронке Бюхнера осадок отфильтровывают и промывают 6 М раствором HCI. Осадок растворяют в 100 мл СНС13 и раствор фильтруют для отделения водной фазы. Раствор в СНС13 промывают двумя порциями до 100 мл дистиллированной воды и выпаривают досуха. Измельченную свободную кислоту сушат 1 « при 60 °C.
2. Стабильность рабочих растворов зависит от методики их приготовления, | поэтому очень важно точно выполнять условия приготовления растворов.
3. Вся посуда, особенно мерные колбы и кюветы, не должна содержать фторида. Кислые растворы циркония пригодны для тщательной промывки поверхности стеклянной посуды.
4. Объемы добавляемых реагентов должны быть обязательно одинаковыми, хотя и не обязательно очень точными; обычные пипетки подходят для этих целей.
Метод с использованием комплекса циркония с СФАНДС
Был изучен [117, 34, 41] метол определения фторида с применением комплекса Zr с СФАНДС; причем в двух последних ра’’° тах его использовали после отделения фторида микродиффУзие Метод аналогичен методу с применением солохромцианина Р-
Хотя чувствительность этого метода ниже чувствительное двух других описанных ранее методов, он нашел применение в честве стандартного для анализа воды [118].
350
Метод с использованием комплекса циркония с ализариновым красным С
Комплекс циркония с ализариновым красным С (ализарин сульфонат натрия) в кислых растворах имеет красно-коричневый цвет в присутствии избытка ализаринового красного С и фиолетовый цвет в присутствии избытка циркония. Комплекс обесцвечивается в присутствии фторида, фосфата, арсената, сульфата, тиосульфата, оксалата и других органических оксикислот. Чувствительность метода ниже чувствительности двух первых описанных выше методов, однако простота выполнения анализа этим методом делает его пригодным для целей рутинного анализа. Метод был использован для анализа воды [119—122]. Так как примеси в воде влияют на результаты анализа, требуется предварительная обработка анализируемых проб. Метод применим для определения 0,1-1,0 мг F-.
Метод с использованием хлоранилатов
Для определения фторида, хлорида, фосфата и сульфата применяют труднорастворимые хлоранилаты. Метод основан на следующем взаимодействии:
Анион”' -|-М-хлоранилат + Н+ —> Хлораниловая кислота + М-анион фиолетовая осадок
После окончания реакции избыток хлоранилата металла и нерастворимую соль металла отфильтровывают, а интенсивность окраски фильтрата зимеряют спектрофотометрически.
Для определения фторида рекомендованы косвенные методы с использованием хлоранилата стронция [123], лантана [124] и тория [125]. При использовании соли лантана [124] можно определять 2—200 ppm F- с точностью до ±2%, определению не мешают сульфат, хлорид, нитрат в количестве, меньше 200 ppm.
. При использовании хлоранилата тория можно определять до 0,2 ppm фторида, но воспроизводимость результатов существенно зависит от тщательности приготовления черно-фиолетовой соли тория.
Спектрофотометрическое определение фторида с хлоранилата-ми требует тщательного контроля за условиями проведения реакции, недостатком метода является высокая оптическая плотность холостой пробы опыта. Трудно предположить, что метод обладает какими-либо преимуществами по сравнению с рассмотренными выше.
Электрохимические методы
Метод с использованием фторидного ионоселективного электрода
Предложенный в 1966 г. [15] фторидный ионоселектпвный ектрод оказал существенное влияние почти на все области ана-Лиза фторидов.
Описаны два вида фторидных электродов: гомогенный твердый мембранный электрод, или монокристаллический мембранный электрод, и гетерогенный твердый мембранный электрод, содержащий труднорастворимую фторидную соль, запрессованную в силиконовую резину [126].
Преимущественно используют гомогенный ЬаР3-электрод (рис. 43). Выпускают 4 модели электродов*, из которых модель Орион 94-09 большинством исследователей признана лучшей. Этот электрод был подробно изучен [127] и проведено сравнение его с другими типами электродов.
Выпущен электрод модели 94-09 А с мембраной из монокристалла LaF3, укрепленной в конце пластмассовой трубки. По конструкции электрод подобен стеклянному электроду, только вместо стеклянной мембраны в нем использована мембрана из монокристалла фторида лантана, легированного европием. Так же как с помощью стеклянного электрода измеряют активность ионов водорода, фторидным электродом измеряют активность ионов фторида. В обоих электродах измеряют разность потенциалов двух поверхностей тонкой мембраны, характеризующейся ионной проводимостью, но в отличие от стеклянного электрода мембрану фторидного электрода не обязательно выдерживать в воде перед использованием. Потенциал Ag/AgCl-электрода определяется активностью хлорид-ионов во внутреннем растворе, активность фто-рид-ионов в том же растворе определяет потенциал внутренней поверхности кристаллической мембраны фторида лантана.
При погружении электрода в исследуемый раствор между внешней и внутренней поверхностями мембраны возникает разность потенциалов, значение которой зависит от отношения активностей во внутреннем и внешнем растворах. Потенциал электрода измеряют обычно по отношению к потенциалу насыщенного каломельного электрода сравнения, электролитическую ячейку можно представить в виде
Ag/AgCl, СГ (0, 1), F" (0, l)/LaFgTB (исследуемый раствор) НКЭ
Франт и Росс [15] нашли, что потенциал электрода подчиняется уравнению Нернста, если мембрана фторидного электрода проницаема лишь для фторид-ионов:
RT Явиутр
Е = const -J—=— In---
г авнешн где а — активность фторид-ионов.
Так как активность фторид-ионов во внутреннем растворе постоянна, уравнение принимает вид
RT
Е = const---=— In (а)внеШн = const + 0,0591 бра при 25 °C
Г
гдС/0л =-log(a).
Постоянный член в уравнении зависит от активности фторид и хлорид-ионов во внутреннем растворе и от потенциала внутрен*
* Орион 94-09, Бекман 39600, Колеман 2-803, Корнинг 47604.
352
Рис. 43. Схема твердого ЬаР3-электрода:
/—Ag/AgCI-электрод; 2—0,1 At растворы NaCl и NaF; 3—мембрана из монокристалла LaFj.
Рис. 44. Зависимость потенциала фторидного ионоселективного электрода фирмы Орион от концентрации фторид-иона (градуировочная кривая снималась я присутствии универсального буферного раствора).
него электрода сравнения, помещенного во фторидный электрод. На рис. 44 представлена типичная зависимость потенциала электрода от концентрации фторид-иона. При десятикратном изменении активности фторида (или концентрации при постоянной ионной силе растворов) потенциал электрода изменяется на 59,16 мВ. Следует отметить, что электродная функция линейна до 10-5 М концентрации фторида (0,19 ppm), в растворах с меньшей концентрацией наблюдается отклонение от линейности. В водных растворах потенциал электрода является также функцией pH, так как подвижности гидроксил- и фторид-ионов в фазе мембраны близки. При низких значениях pH активность фторида изменяется
за счет образования недиссоциированных HF и HF2, при высоких значениях pH сказывается мешающее влияние гидроксил-иона [127]. Чувствительность электрода к фториду в десять раз выше чувствительности его к гидроксил-иону. Поэтому во всех случаях, кроме измерений высоких концентраций фторида, не следует измерять фторид-ион в растворах с высокими значениями pH. В даль-кейшем мы еще вернемся к рассмотрению этого вопроса. В очень Разбавленных растворах фторида оптимальным рабочим интервалом является pH = 6—8. Это показано на рис. 45, где представлена зависимость потенциала фторидного электрода от pH растра для трех концентраций фторид-иона [15].
Ионоселективные электроды реагируют на изменение активно-дТи ионов, поэтому электродная функция зависит от ионной силы Раствора. Необходимо при измерении ионные силы анализируемого р стандартного растворов поддерживать одинаковыми. Франт и ^28] рекомендовали применять для поддержания постоян-и Ионной силы буфер, содержащий уксусную кислоту и цитрат рия (TISAB); pH раствора доводят до 5—5,5 добавлением
12 Зак 167
QK i
Рис. 45. Зависимость потенциала фторидного иоиоселектнвного электрода от pH [15].
Рис. 46. Экспериментальные кривые титрования стандартных концентраций фторида нитратом лантана с использованием фторидного ноноселектнвного электрода (133).
NaOH. Этот буфер был использован для определения фторида в
воде при концентрации его на уровне ppm.
В работе [129] найдено, что буферные растворы, содержащие органические кислоты, непригодны для использования при работе с ЬаР3-электролами, так как образование комплексов типа [Lag-xAcx] приводит к замедленному установлению равновесного потенциала и отклонениям от уравнения Нернста. В более позднем исследовании тех же авторов [130] сообщается, что хелатообразующие лиганды, такие, как молочная, малоновая, лимонная кислоты, и другие влияют на электрод; генерировать электрод можно лишь полируя кристаллическую поверхность мембраны алмазной пастой.
Первоначальная оценка фторидного электрода привела к возникновению противоположных мнений относительно его эксплуатационных качеств, что было вызвано значительными различиями в условиях практического использования электрода. Например, в 10~3 М растворах фторида потенциал устанавливается практически мгновенно, тогда как в 10-5 М растворах время установления потенциала составляет 3 мин. Лингейн изучал [131] влияние перемешивания и других важных факторов на потенциал фторидного
электрода.
Описано два метода использования электрода: прямой потенциометрический метод, когда концентрацию фторида в анализируемом растворе определяют по градуировочному графику, и п тенциометрическое титрование фторида нитратами лантана и тория, когда электрод используют для индикации точки эквив лентности. Последний метод подробно изучен Лингейном I1 ’ 132], который в качестве титрантов применял соли лантана, т0Р и кальция. Как было найдено, нитрат лантана дает минималь
ошибки определения при проведении титрования в нейтральном растворе в присутствии буфера. Но многих более поздних работах этот метод был воспроизведен и использован для анализа достаточно широкого круга объектов, например для определения фторида в органических препаратах. Органические вещества разлагали и образующийся фторид титровали стандартным раствором лантана или тория в 60%-ных по объему водно-изопропанольных смесях [133]. Кривые титрования различных количеств фторида представлены на рис. 46.
Для получения удовлетворительных результатов анализа необходимо выполнять одинаковые операции и с анализируемым раствором и со стандартными растворами, используемыми для построения градуировочного графика.
В более поздней работе [134] определены оптимальные условия потенциометрического титрования фторида солями лантана или тория в небуферных растворах. Было найдено, что конечная точка титрования не совпадает с точкой эквивалентности, так как кривые титрования асимметричны и положение этих двух точек на кривой титрования зависит от скорости перемешивания.
Изучали условия [135] потенциометрического титрования фторида нитратом тория в водно-органических растворах. В результате было найдено, что оптимальными условиями являются pH — = 5—7 и 50%-ный раствор диоксана в воде, ошибка титрования при этом составляет ±3%.
Бауман [57] изучал применение электрода в методе прямой потенциометрии и нашел, что с помощью фторидного электрода можно определять фторид в интервале его концентраций от 10-5 до 10~4 М.
Эти выводы были использованы для разработки метода стандартных добавок, согласно которому стандартный раствор NaF добавляют к анализируемому раствору и измеряют потенциал фторидного электрода после введения каждой добавки. При сведении к минимуму изменения факторов, оказывающих влияние на коэффициент активности фторида, например, ионной силы раствора и диффузионного потенциала, зависимость потенциала электрода от концентрации фторида подчиняется уравнению Нернста. В кислых растворах можно определять общую концентрацию фто-Рида (свободного и связанного в недиссоциированные частицы) при условии, если активность ионов водорода остается неизменной, в растворе отсутствуют другие фторидные комплексы, кроме соединений фторида с водородом, и концентрация несвязанного фторида достаточна для его определения. Метод подробно описан н оригинальной работе. Рассмотрена возможность определения фторида в растворах, содержащих различные неорганические °ны. В присутствии металлов, мешающих определению фторида, скольку они связывают последний в прочные комплексы Ннпример, Al3+, UO2 , Fe3+, Th4+), введение Н3РО4 оказывается
355
Рнс. 47. Зависимость потенциала фторидного ноноселективного электрода от концентрации фторид-иона [128]:
1 — чистый ра твор NaF; 2 — NaF в присутствии уНи-вереального буферного раствора TISAB.
эффективным, так как позволяет разрушить фторидные комплексы за счет образования более прочных фосфатных комплексов.
Метод стандартных добавок был использован Селигом [136], который для расчета результатов анализа применил метод Грана. Метод Грана заключается в пересчете концентрации отделяемого иона на объем титранта в каждой точке титрования. Это до
вольно сложно. Задачу можно упростить, если использовать специально выпускаемую фирмой «Орион» диаграммную бумагу. На графике получают линейную зависимость между потенциалом электрода и объемом добавляемого NaF. Этот прием позволяет точнее рассчитывать концентрацию фторида, чем в случае использования S-образной кривой титрования.
Прямой потенциометрический метод определения фторидов был использован [127] для анализа воды, проведено сравнение потенциометрического и спектрофотометрического метода с применением комплекса Zr—СФАНДС. Фторидные комплексы А13+ или Fe3+ разрушали введением цитрата, являющегося составной частью универсальной буферной смеси (TISAB). График показан на рис. 47.
Хорошие результаты получены [137] при использовании метода прямой потенциометрии в органическом микроанализе. Авторы [137] полагают, что большое значение в снижении ошибки анализа имеет ежедневное построение градуировочного графика. Наклон графика постоянен и соответствует величине, найденной по уравнению Нернста, однако небольшое перемещение всего графика не позволяет использовать один график в течение длительного времени без снижения точности анализа. Достаточно ежедневно стандартизировать электрод по одной концентрации фторида, чтобы с удовлетворительной точностью определять содержание фторида в анализируемом растворе. При определении 0,4— 2,0 мг F- в серии из 18 растворов NaF ошибка определения составляла 0,17%.
Исходя из произведения растворимости фторида лантана, электродная функция F-электрода должна быть линейна до Ю~6 М г • J Бауман [138] изучал зависимость нижнего предела чувствител^ ности электрода от введения катионов, связывающих Фт0РиД1’1% J в комплекс, и по константам устойчивости соответствующих Ф I ридных комплексов рассчитывал равновесную концентрацию Ф рид-ионов. Результаты исследований показали, что подчине
уравнению Нернста сохраняется даже тогда, когда концентрация фторид-ионов на три порядка меньше 10-6 Л1. Определение малых концентраций фторидов, как правило, связано с проведением анализа в малых объемах. Дарст [139] использовал модифицированный электрод и метод концентрационных ячеек для определения 0,38 нг фторида в 10 мкл с ошибкой 0,002 нг F.
В работе [140] изучали влияние различных ионов на электродную функцию F-электрода. Неионные соединения практически не оказывают влияния на потенциал электрода, а введение NaCI или MgSO4 приводит к существенному изменению потенциала. Железо, борат и силикат не оказывают заметного влияния; Al, Ве, Zr и Ti мешают определению, так как образуют прочные фторидные комплексы.
При использовании метода стандартных добавок для анализа чая, в котором может содержаться до 17000 мкг/г алюминия, было найдено [141], что присутствие алюминия (и железа) приводит к значительным ошибкам определения фторид-ионов. Использование цитрата натрия в качестве маскирующего агента приводит к количественному выделению фторида из комплексов с алюминием, железом, магнием и кремнием.
Без сомнения, использование фторидного ионоселективного электрода широко и многообразно. Его можно применить также для анализа фосфатных горных пород [142], зубных паст [143, 144], растений [144], крови [145], мочи [146], фторированных углеводородов [147], силикатных горных пород [148], костей [149], оксидов [150], воздуха [151], чая [141]. Фирмой «Орион» выпущено методическое руководство [152] и обзоры по использованию фторидного электрода.
Радиохимические методы
Радиохимические методы могут быть использованы для анализа материалов, содержащих фтор в виде фторида. Опубликован обзор [153] по радиометрическим методам определения фтора. Известно 5 изотопов фтора: стабильный изотоп фтор-19 и четыре короткоживущих изотопа; самый большой период полураспада (109,8 мин) у l8F. В активационных методах применяют тепловые [154] и быстрые [155] нейтроны, а-частицы [156], протоны [157], ядра гелия-3 [158] и у-протоны [159, 160]. Цитируемые работы позволяют оценить эту область анализа фторида, но они составляют лишь малую часть публикуемых работ.
Несомненный интерес представляет определение фторида в °Рных породах и особенно в образцах лунного грунта, достав-енного на Землю экипажем Апполон II [159]. Применяли метод меТОядеРн°й активации, пригодный для определения легких эле-ИспТ0В’ В Том числе и ФТ0Ра- По реакции l9F(y, п)—18F последний с ,„УСКает позитроны, при аннигиляции которых выделяются у-лучи тоце Гие® 0’51 Мэв. Период полураспада 18F (109,8 мин) доста-н для проведения его химического выделения из нерадио
357
активной матрицы. Во всех упомянутых выше исследованиях i8F выделяли дистилляцией и затем определяли на у-спектрометре [детектор Nal(TI)]. Минимальное определяемое количество фтора 0,002 мкг. Полученные результаты хорошо совпадают с результатами спектрофотометрических определений.
Фторид можно определять также после его взаимодействия с радиоизотопом другого элемента, активность полученного соединения пропорциональна содержанию фторида. С использованием этого принципа определяли 20—100 мкг F- [161]. Раствор, содержащий фторид, пропускали через колонку, содержащую селенид циркония, который в течение 12 ч облучали нейтронным потоком 1,2-1012 нейтрон-см~2-с-1. Реакция протекает по следующему уравнению:
Zr (SeO3)2 + 6HF —> ZrFr + 2SeOf
Измеряют у-активность выделяющегося 75Se. Определению мешают фосфат и алюминий.
Еще одним радиохимическим методом определения фторида является метод изотопного разбавления; 18F применяют для определения субмикрограммовых количеств фторида в образцах зубной эмали массой менее 1 мг.
Каталитические методы
При определении очень малых количеств фторида используют каталитические реакции, и в том числе реакции, катализируемые энзимами. Наряду с высокой чувствительностью методы характеризуются также высокой селективностью и сравнительно недороги.
Один из методов основан на зависимости скорости обесцвечивания фторид-ионом комплекса алюминия с гематоксилином за' счет образования прочного фторида алюминия от концентрации фторида [163].
А1 — гематоксилин + 6F" —> гематоксилин + A1F|" (желтый)
Этим методом определяли 0—15 ppm F~ в 100 мл образца с точностью 0,05 ppm. Способность фторида ингибировать энзимный гидролиз этилбутирата была использована [164] для определения 0,1 мкг/мл F~ в крови, моче и воде, а также для определения нескольких нанограммов фторида в присутствии больших количеств фосфата [165].
Фторид 0,5—5 мкг в 100 мл раствора определяли по его каталитическому действию на реакцию взаимодействия оксихлорида циркония, содержащего полимерные частицы, с ксиленоловым оранжевым [166]. Этот прием называют кинетохромной спектр®" фотометрией. Эта реакция сама тоже представляет интерес, та, I как она основана на известной способности ионов циркония °®Р® J зовывать в слабокислых растворах полимерные частицы. В ®в жеприготовленном растворе оксихлорида циркония быстро обр зуется красный комплекс с ксиленоловым оранжевым, через Z
868
комплекс образуется медленно, а в растворе, хранившемся в течение недели, комплекс не образуется. В растворах, хранившихся 24 ч, скорость образования комплекса прямо пропорциональна концентрации фторида. В течение 90 мин интенсивность окраски достигает максимума и в течение 60 мин остается постоянной: молярный коэффициент поглощения в пересчете на фторид равен 2,0-105. Из исследованных катионов только алюминий и желе-зо (Ш) в 10-кратном по отношению к фториду избытке мешают определению. Гораздо более значительное влияние на определение фторида оказывают анионы. Фосфат, арсенат, сульфат, цитрат и оксалат мешают определению, так как тоже катализируют реакцию. Поэтому, если в растворе присутствуют перечисленные анионы, необходимо проводить отделение фторида. Следует отметить, что, хотя метод отличается высокой чувствительностью, он менее стабилен по сравнению с обычными спектрофотометрическими методами, так как самые различные факторы влияют на воспроизводимость результатов. Для улучшения стабильности метода необходимо очень тщательно ежедневно строить градуировочный график.
Еще один аналогичный метод [167] позволяет определить до 0,02 мкг F~ Реакция циркония с метилтимоловым синим также катализируется фторидом. Этот метод обладает преимуществами по сравнению с методом, основанным на реакции циркония с ксиленоловым оранжевым [168]. Он более чувствителен, сульфат в равных с фторидом концентрациях не мешает определению фторида.
Описан [169] каталитический метод определения фторида на уровне ррЬ. Метод основан на ингибирующем действии фторида на реакцию взаимодействия пербората и иодида, катализируемую цирконием; 19—190 нг F- (в 50 мл) можно определять с ошибкой ±7,4 нг.
Методы термометрического титрования
В последнее время оживился интерес к методам термометрического титрования. Эти методы наряду с полезной информацией о протекании и природе реакций взаимодействия металлов с фторидом интересны и с аналитической точки зрения.
Как было найдено, соли тория (IV), церия (III), алюминия и кальция являются лучшими титрантами. Разумный выбор ти-тРанта позволяет избежать мешающего влияния средних концен-гРаций сульфата, бората или силиката. Мешающее влияние большинства ионов можно было бы устранить, отделив фторид дистилляцией, но метод термометрического титрования неприменим для Разбавленных растворов, которые образуются после отделения фТоРидов.
±0пПИСан 1*711 метод определения 6—8 мг F- с ошибкой .04 мг по реакции
Pb2t±F'+Cr —> РЬБСЦ
359
Реакция протекает достаточно быстро и сопровождается значительным тепловым эффектом. Присутствие фосфатов, сульфатов, нитратов и хлоридов влияет на энтальпию реакции. Описан метод их отделения с использованием ионного обмена.
Другие методы определения
В биологических материалах фторид определяют методом газовой хроматографии [64]. Сначала фторид экстрагируют раствором хлортриметилсилана в бензоле. Фторид взаимодействует с экстрагентом с образованием фтортриметилсилана, который переходит в бензольный экстракт. Экстракт используют для прямого введения в колонку. Метод применим для определения 0,01— 10 мкг фторида в 1 мл анализируемого образца.
Описан атомно-абсорбционный метод определения 0,2— 20 мкг/мл фторида [172]. Фторид снижает абсорбционные характеристики магния в воздушно-ацетиленовых пламенах, изменение интенсивности поглощения линии магния (285,2 нм) пропорционально концентрации фторидов. Основными мешающими ионами являются сульфат и фосфат; алюминий, лантан, оксалат и ацетат также мешают определению фторидов. Этот метод экспресснее двух упомянутых выше и может быть использован без предварительного отделения фторида. Одни из вариантов этого метода основан на увеличении поглощения циркония в пламени оксида азота (I)—ацетилен в присутствии фторида (5—200 мкг/мл), число мешающих ионов невелико. В аналогичном методе, основанном на увеличении поглощения титана в том же пламени в присутствии фторида (40—400 мкг/мл), присутствие фосфата и сульфата не мешает определению фторида.
Предложено несколько полярографических методов определения фторида. Один из методов [173] определения 2—10 мкг F" основан на взаимодействии фторида с торий-о-нитробензоларсо-ниевой кислотой. Измеряют полярографическую волну свободного органического лиганда, концентрация которого пропорциональна концентрации фторцда.
Для потенциометрического титрования фторида (или оксалата) можно использовать La3+, электрогенерируемый из анода гексаборида лантана [84]. При pH = 5,0 можно определять 0,5—2,0 мг F- в 100 мл с точностью до нескольких частей на тысячу.
В гравиметрическом методе [174] также используют лантан (III). Фторид осаждают в виде LaF3, вводя известное количество La(NO3)3, избыток его после фильтрования определяют гравиметрически с купферроном. По мнению авторов работы [12ш’ LaF3 образует коллоидный осадок, однако в работе [159] 1-а ’ используют для выделения F- при его у-активационном опреДеЛ нии. тп В
Для определения конечной точки титрования фторида Th(N^s/_ предпринимали попытки использовать в качестве индикатора а I заринсульфонат; разработан метод кондуктометрического °nr I
деления конечной точки титрования [175]. До точки эквивалентности практически не происходит изменения электропроводности раствора, так как фторид-ион (X = 55,4-1 см2-экв) замещается на нитрат (Х° = 71,4-1 см2-экв) и наблюдается лишь незначительный ее рост. После точки эквивалентности добавление нитрата тория вызывает резкое увеличение электропроводности. Этим методом можно титровать 0,3—5 мг F- с относительной ошибкой 2%. Метод используют после дистилляционного отделения фторидов.
Описан новый метод определения фторида в биологических материалах [176], объединяющий диффузионный метод Холла, описанный выше, с поглощением фторида фосфатом кальция. По существу, метод заключается в кипячении раствора, содержащего фторид, в течение 5—10 мин с 20 мг фосфата кильция, не содержащего фторид. Раствор охлаждают, центрифугируют для отделения фосфата кальция, содержащего сорбированный фторид, и затем выделяют из осадка фторид диффузионным методом Холла. Метод применен для анализа воды, мочи, экстрактов растений и плазмы.
Разработан новый флуориметрический метод [177] определения 0,01 ppm F-. Он основан на образовании тройного комплекса цирконий — кальционовый голубой — фторид (1:1:1).
Образование комплекса усиливает флуоресценцию. Определению мешает лишь пятикратное по отношению к фториду количество фосфата, очень мало влияют вольфрамат-, ацетат-, тартрат-ионы. В 50-кратном по отношению к фториду соотношении молибдат занижает, а сульфат завышает результаты анализа. Катионы оказывают на реакцию чрезвычайно малое влияние: 5-кратные количества алюминия, марганца и серебра слегка влияют на реакцию, а 50-кратные количества мышьяка(У), сурьмы(Ш), бериллия и кобальта занижают результаты анализа. Ошибка определения фторида в концентрациях, соответствующих средней части градуировочного графика, составляет +3%.
ЛИТЕРАТУРА
1. Reynolds D. S., Hill W. L. — Tnd. Engng Chem analyt. Edn, 1939, v. 11, p. 21.
2- Rinck G. — Bull. Soc. chim. Fr., 1948, v. 15, p. 305.
Specht R. C. — Analyt. Chem., 1956, v. 28, p. 1015.
4. Elving P. J.t Horton C A., Willard H. H. — Fluorine Chemistry (Ed. J. H. Si-mons). Vol. II, New York, Academic Press, 1954, p. 51.
°- Macdonald A. M. G. — Comprehensive Analytical Chemistry (Ed. C. L. Wil-son and D. W Wilson), Vol 1C, Amsterdam, Elsevier, 1962, p. 319.
°- Horton C. A. — Treatise on Analytical Chemistry (Ed. I. M. Kolthoff, P. J. El-7 A'HJ’)’ Vol. 7, New York, Interscience, 1961, p. 207.
Rodama K. — Methods of Quantitative Inorganic Analysis — an Encyclopedia of Gravimetric, Titrimetric and Colorimetric Methods, New York, Interscience, 8 L963; P- «8.
norton C. A. — Advances in Analytical Chemistry and Instrumentation (Ed.
9 M N- Reilley), Vol. 1. New York, Interscience, 1960, p. 151.
p“/- — Treatise on Analytical Chemistry (Ed. I. M. Kolthoff and
v. J. Elving), Part II, Vol. 12, New York, Interscience, 1965, p. 117.
361
10. Dixon J. P. — Modern Methods of Organic Microanalysis, London, Van Nostrand, 1968.
11. Hall R. /. — Analyst, 1968, v. 93, p. 461.
12. Hall G. J. — Proc. Soc. analyt. Chem., 1966, v. 3, p. 162.
13. Cooke J. A., Johnson M. S., Davison A. IV. — Environ. Pollut., 1976, p. 257.
14. Belcher R, Leonard M. A., West T. S. — J. chem. Soc., 1958, p. 2390
15. Front M. S., Ross J. W. Science, 1966, v. 154, p. 1553.
16. Willard H. H., Winter О. B. — Ind. Engng. Chem. analyt. Edn, 1933, v. 5,
17. Тананаев И. Журнал прикладной химии, Ленинград, 1932, т. 5, с. 834.
18. Fox Е. J. — Analyt. Chem., 1960, v. 32, р. 1530.
19. Official, Standardised and Recommended Methods of Analysis, Cambridge, England, The Society for Analytical Chemistry, W. Heffer, 1963. p. 123.
20. Smith O. D., Parks T. D. — Analyt. Chem., 1955, v. 27, p. 998
21. Murty G. V. L. N., Yiswanathan T. S., Ramakrishna V. — Analytica chim. Acta, 1957, v. 16, p. 213.
22. Shell H. R., Craig R. L. — Analyt. Chem., 1954, v. 26, p. 996.
23. Grimaldi F. S., Ingram B., Cuttitta F. — Analyt. Chem., 1955, v. 27, p. 918.
24. Peck L. C., Smith V. C. — Taianta, 1964, v. 11, p. 1343.
25. Haff L. V., Butler С. P., Bisso J. D. — Analyt. Chem., 1958, v. 30, p. 984.
26. Estill W. B., Mosier L. C. — Analyt. Chem., 1955, v. 27, p. 1669.
27. Schall E. D., Williamson H. G.—J. Ass. off. agric. Chem., 1955, v. 38, p. 452.
28. Devonshire L. N-, Rowley H. H. — Proc. Okla. Acad. Sci., 1953, v. 34, p. 155.
29. Singer L. S., Armstrong W. D. — Analyt. Chem., 1959, v. 31, p. 105.
30. Wade M. A., Yamamura S. S. — Analyt. Chem., 1965, v. 37, p. 1276.
31. Ma T. S., Gwirtsman J. — Analyt. Chem., 1957, v. 29, p. 140.
32. Official Methods of Analysis of the Association of Official Analytical Chemists, 12th edn, Washington, А. О. A. C., 1975, p. 927.
33. Singer L., Armstrong W. D. — Analyt. Chem., 1954, v. 26, p. 904.
34. Wharton H. W. — Analyt. Chem., 1962, v. 34, p. 1296.
35. Greenland L. — Analytica chim. Acta, 1962, v. 27, p. 386
36. Frere F. /. — Analyt. Chem., 1961, v. 33, p. 645.
37. Marshall B. S., Wood R. — Analyst, 1969, v. 94, p. 493.
38. Hall R. J.—Analyst, 1960, v. 85, p. 560.
39. Hall R. J. —Analyst, 1963, v. 88, p. 76.
40. Hall R. /. — Analyst, 1963, v. 88, p. 899.
41. Nicholson C. R. — Analyt. Chem., 1966, v. 38, p. 1966.
42. Taves D. R. — Analyt. Chem., 1968, v. 40, p. 204.
43. Taves D. R.—Taianta, 1968, v. 15, p. 969.
44. Sara R-, Wanninen E. — Taianta, 1975, v. 22, p. 1033.
45. Hanoco M. — Mikrochim. Acta, 1973, p. 729.
46. Official, Standardised and Recommended Methods of Analysis, Cambridge, England, The Society for Analytical Chemistry, W Heffer, 1963, p. 404.
47. Warf 1. C., Cline IV. D., Telebaugh R. D. — Analyt. Chem., 1954, v. 26, p. 342.
48. Powell R. H., Menis O.—Analyt. Chem., 1958, v. 30, p. 1546.
49. Clements R. L., Sergeant G. A., Webb P. J. — Analyst, 1971, v. 96, p. 51.
50. Valach R. — Taianta, 1961, v. 8, p. 629.
51. Davies A. G., Foreman J. /(. — Proceedings of the Society for Analytical Chemistry Conf., Nottingham, Cambridge, England, W. Heffer, 1965, p. 165.
52. Jeffery P. G., Wiliiams D. — Analyst, 1961, v. 86, p. 590.
53. Eger C., Yarden A. — Analyt. Chem., 1956, v. 28, p. 512.
54. Zipkin I., Armstrong W. D., Singer L.— Analyt. Chem., 1957, v. 29, p. 310.
55. Newman A. C. D. — Analytica chim. Acta, 1958, v. 19, p 471.
56. Edmond C. R. — Analyt. Chem., 1969, v. 41, p. 1327.
57. Baumann E. W. — Analytica chim. Acta, 1968, v. 42, p. 127.
58. Kelso F. S., Mattews J. M., Kramer H. P. — Analyt. Chem., 1964 v. л p. 577. и
59. Fresen J. A., Van Gogh H. Van Pinxteren J. A. C. — Pharm. Weekbl Ne I 1950, v. 95, p. 33. oll
60 Jnczidy J. — Analytical Applications of Ion Exchangers, Oxford, Pregan Press, 1966, p. 210.
362
*j Funasaka W, Kavane M., Kujima T., Matsuda Y —Japan Analyst. 1955. v 4. p. 514; Analyt. Abstr., 1956, v. 3, p. 2079.
62 Gagliardi E., Pokorny G. — Mikrochim. Acta, 1965, p 099
63. Bark L. S., Graham R. J. T., McCormick D. — Analytica chim. Acta, 1966, v. 35, p. 268.
64. Fresen I. A., Cox F. H., Witter M. J. — Pharm. Weekbl. Ned., 1968, v. 103, p. 909; Analyt. Abstr., 1969, v. 17, p. 3619.
65. Cropper E., Puttnam N. A. — J. Soc cosmet. Chem., 1970, v. 21, p. 533.
66 Шапиро M. Я-, Колесникова В. Г. ЖАХ, 1963, т. 18, с. 507, Analyt. Abstr., 1964, т. 11, с. 1982.
67. Bournique R. A., Dahmer L. Н. — Analyt. Chem., 1964, v. 36, р. 1786
68. Hoffman F., Lundell G. E. — J. Res. natn Bur Stand., 1929, v. 3, p. 589.
69. Caley E. R., Kahle G. R. — Analyt. Chem., 1959, v. 31, p. 1880.
70. Popov A. /., Knudson G. E. — Analyt. Chem., 1954, v. 26, p. 892.
71. Ballczo H„ Schiffпег H. — Z. analyt. Chem., 1956, v. 152, p. 3.
72. Dahle D., Bonnar R. U., Wichmann H. J. — J. Ass. off. Agric. Chem., 1938, v. 21, p 459.
73. Hoskins C. R., Fryd C. F. M. — J. appl. Chem., 1955, v. 5, p. 85.
74. Official Methods of Analysis of the Association of Official Analytical Chemists, 12th edn, Washington, А. О. A. C., 1975, p. 436.
75. Selig W.— Analyst, 1968, v. 93, p. 118.
76. Analytical Methods Committee of the Society for Analytical Chemistry — Analyst, 1972, v. 97, p. 734.
77. Israel Y., Bernas B., Yahalom A. — Analytica chim. Acta, 1966. v. 36, p. 526. 78. Colson A. F. — Analyst, 1963, v. 88, p. 26.
79. Belcher R., Clark S. J. — Analytica chim. Acta, 1953, v. 8, p. 222
80. Leonard M. A. — Analyst, 1963, v. 88, p. 404.
81. Yamamura S. S., Kussy M. E., Rein J. E. — Analyt. Chem., 1961, v. 33, p. 1655.
82. Combs H. F., Grove E. L.—Taianta, 1970, v. 17, p. 599.
83. Orenberg J. B., Morris M. D. — Analyt. Chem., 1967, v. 39, p. 1776.
84. Curran D. J.. Fletcher K- S. — Analyt. Chem., 1969, v. 41, p. 267.
85. Harzdorf C. — Z. analyt. Chem., 1968, v. 233, p. 348.
86. Harzdorf C. — Z. analyt. Chem., 1967, v. 227, p. 161.
87. Megregian S. — In: Colorimetric Determination of Non-metals (Ed. D. F. Boltz), New York, Interscience, 1958
88. Leonard M. A., West T. S. — J. chem. Soc., I960, p. 4477.
89. Anfalt T., Jagner D. — Analytica chim. Acta, 1974, v. 70, p. 365.
90. Belcher R., Leonard M. A., West T. S. — J. chem. Soc., 1958, p. 2390.
91. Greenhalgh R., Riley J. P. — Analytica chim. Acta, 1961, v. 25, p 179.
92. Belcher R., West T. S. — Taianta, 1961, v. 8, p. 863.
93. Report prepared by the Fluorine sub committee of the Analyrical Methods Committee of the S. A. C. — Analyst, 1971, v. 96, p. 384.
94. Crosby N. T., Dennis A. L., Stephens J. G. — Analyst, 1968, v. 93, p. 643.
95. Haarsma J. P. S., Agterdenbos J. — Taianta, 1971, v. 18, p. 747
96 Al-Ani K., Leonard M. A. — Analyst, 1970, v. 95, p. 1039.
97. Laird С. K., Leonard M. A. — Taianta, 1970, v. 17, p. 173.
98. West P. W., Lyles G. R., Miller J. L. — Environ. Sci. a. Technol., 1970, v. 4, on p 487-
99. Bailey D. L. R., Killick С. M., Trott P. E. — Report Warren Springs Labora-irm LR 129 <AP)’ 197°-
"fonteriolo S. C., Pepe A. — Pure appl. Chem., 1970, v. 24, p. 707 ink A A-1., Riley J. P. — Analytica chim. Acta. 1966, v. 35, p. 365. in»' R- A., Richards F. A. — Analyt. Chem., 1970, v. 42, p. 1435. l<u‘ "larEreaves J. A., Ingram G. S., Cox D. L. — Analyst, 1970, v. 95, p. 177. Ite uhcafer H- N- s — Analyt. Chem., 1963, v. 35, p. 53.
Kv. 7°^ A-> Walsh J. N. —Analytica chim. Acta. 1969, v. 45, p 341 toy Johnson C. A., Leonard M. A. — Analyst, 1961, v. 86, p. 101.
Leonard M. A — Symposium on the Determination of Anion-forming Ele-108 ^ents. Analytical Division of the Chemical Society, Swansea, September, 1976.
megregian S. — Analyt. Chem., 1954, v. 26, p. 1161.
ала
109. Sarma P. L. — Analyt. Chem., 1964, v. 36, p 1684.
110. Dixon E. J. — Analyst, 1970, v. 95, p. 272.
111. Huang P. M., Jackson Л1. L.—Am. Miner., 1967, v. 52, p. 1503.
112. Sen Gupta J. G. — Analytica chim. Acta, 1968, v. 42, p. 119.
113. Methods for Detection of Toxic Substances in Air. H. M. Factory Inspectorate No. 19. London, H. M. S. O., 1970.
114. Adams D. F., Корре R. K., Matzek N. E. — Analyt. Chem., 1961, v. 33, p. 117,
115 Ferrari H. J., Geronimo F. C., Brancone L. AL — Microchem. J., 1961, v. 5, p. 617.
116. Manual on Industrial Water and Industrial Waste Water. A. S. T. Al. Special publication No. 148-F, 2nd edn, 1962 printing.
117. Bellack E„ Schouboe P. 7,—Analyt. Chem., 1958, v. 30, p. 2032.
118. Standard Methods for the Examination of Water and Waste Water, New York, American Public Health Association, 12th edn, 1965, p. 144—146.
119. Meyling A. H., Meyling J. — Analyst, 1963, v. 88, p. 84.
120. British Standards Institute, B. S. 2690: Pt 4 (1967).
121. Palin A. T. — J. Am. Wat. Wks Ass., 1967, v. 59, p. 255.
122. Ref. 118, p. 142
123. Bertolacini R. J., Barney J. E. — Analyt. Chem., 1958, v. 30, p. 202
124. Hayashi K., Danzuka T., Ueno K-—Talanta, 1960, v. 4, p. 126.
125. Hutchinson K. A —Diss. Austr., 1964, v. 24, p. 4372.
126. Macdonald A. M. G., Toth K. — Analytica chim. Acta, 1968, v. 41, p. 99.
127. Evans P. A.. Moody G. J., Thomas J. D. R —Lab. Pract., 1971, v. 20, p. 644.
128. Frant M. S., Ross J. W.—Analyt. Chem., 1968, v. 40, p. 1169.
129. Anfalt T., Jagner D. — Analytica chim. Acta, 1969, v. 47, p. 483.
130 Anfalt T., Jagner D. — Analytica chim. Acta, 1970, v. 50, p. 23.
131. Lingane J. J. — Analyt. Chem., 1968, v. 40, p. 935.
132. Lingane J. J. — Analyt. Chem., 1967, v. 39, p. 881.
133. Francis H. J., Deonarine J. H., Persing D. D. — Microchem. J., 1969, v. 14, p. 581.
134. Eriksson T., Johansson G. — Analytica chim. Acta, 1970, v. 52, p. 465.
135. Hassan S. S. M. — Mikrochim. Acta, 1974, p. 889.
136 Selig U?— Mikrochim. Acta, 1973, p. 87.
137. Pavel J., Kuebler R., Wagner H. — Microchem. J., 1970, v. 15, p. 192.
138. Baumann E. W. — Analytica chim Acta, 1971, v. 54, p. 189.
139. Durst R. A. — Analyt. Chem., 1968, v. 40, p. 931.
140. Bock R., Strecker S. — Z. analyt Chem., 1968, v. 235, p. 322.
141. Vickery B., Vickery M. L.—Analyst, 1976, v. 101, p. 445.
142. Evans L., Hoyle R. D., Macaskill J. B. — N. Z. Ji Sci., 1970, v. 13, p. 143
143. Shane N., Miele D. — J. Pharm. Sci., 1968, v. 57, p. 1260.
144. Levaggi D. A., Oyung W., Feldstein M. — J. Air Pollut. Control Ass., 1971, v. 21, p. 277.
145. Singer L., Armstrong W. D. — Arch. Oral. ВЫ., 1969, v. 14, p. 1343.
146. Tusl J. — Clin. chim. Acta, 1970, v. 27, p. 216
147. Woodward B., Taylor N. F., Brunt R. V.—Analyt. Biochem., 1970, v 36, p. 303.
148. Jagner D., Pavlova V. — Analytica chim. Acta, 1972, v. 60, p. 153.
149. Singer L., Armstrong W. D. — Analyt. Chem., 1968, v. 40, p. 613.
150. Peters M. A., Ladd D. M. — Talanta, 1971, v. 18, p. 655.
151. Hrabeczy-Pall A., Toth K., Pungor E., Valid F. — Analytica chim. Acta, 1975, v. 77, p. 278.
152. Orion Research Incorp., Analytical Methods Guide. 9th edn, 1978.
153. Foreman J. K- — Analyst, 1969, v 94, p. 425.
154. Yule H. P. — Analyt. Chem., 1965, v. 37, p. 129; 1966, v. 38, p. 818.
155. England E. A. M., Honsby J. B., Jones W. T., Terrey D. R. — Analytica chini-Acta, 1968, v. 40, p. 365. , e
156. Plaskin I. N., Belyakov M. A., Starchik L. P.—Atomn. Energ., 1962, v.
p. 374.
157. Bewers J. M., Flack F. C. — Analyst, 1969, v. 94, p. 7.
158 Markowitz S. S., Mahoney J. D. — Analyt. Chem., 1962, v. 34, p. 399.
159. Hislop J. S., Pratchett A. G., Williams D. R. — Analyst, 1971, v. 96, p. I*'"
ifiO Kosta L., Slunecko J. — Analyt Chem., 1970 v. 42, p. 831.
161. Carmichael 1. 4., Whitley J. E. — Analyst, 1969, v. 94, p. 737.
162 Kudahl J. N , Fremlin J H., Hardwick J. L. — Radioisotopes in the Physical Sciences and Industry, Vol. II, p. 317.
163- Price M. Walker O. J. — Analyt. Chem., 1952, v. 24, p. 1593.
164. Linde H. II7. -Analyt. Chem., 1959, v. 31, p. 2092.
165- McGaughey C., Stowell E. C. — Analyt. Chem, 1964, v. 36, p. 2344.
166. Cabello-Tomas M. L., West T. S. — Talanta, 1969, v. 16, p. 781.
167 Knapp G. — Mikrochim. Acta, 1970, p. 467.
168 Hems R. V, Kirkbright G. F„ West T. S. — Talanta, 1970, v. 7, p. 433.
169 Klockow D., Ludwig H., Giraudo M. A. — Analyt. Chem., 1970, v. 42, p. 1682. 170. Everson W. L., Ramirez E. M. — Analyt. Chem., 1967, v. 39, p. 1771.
171 Johansson С. E. — Talanta, 1970, v. 17, p 739
172. Bond A. M., O’Donnell T. A. — Analyt. Chem., 1968, v. 40, p. 560.
173. Wallis С. P. — Talanta, 1960, v. 5, p. 61.
174. Popov A. I., Knudson G E. — Analyt. Chem., 1954, v. 26, p 892
175. Israel Y., Bernas B., Yaholom A. — Analyt. chim. Acta, 1966, v. 36, p. 526.
176. Vznkateswarlu P., Sita P. — Analyt. Chem., 1971, v. 43, p. 758.
177. Tan Lay Har, West T. S. — Analyt. Chem., 1971, v. 43, p. 136.
ГИПОБРОМИТЫ
Растворы гипобромита заметно менее устойчивы, чем растворы гипохлорита. Стабильность растворов гипобромитов явилась предметом разносторонних исследований [1—3]. Изучали влияние различных факторов на устойчивость растворов: концентрации гипобромита и гидроксил-иона, света, температуры, присутствия посторонних ионов. Термодинамическая неустойчивость приводит к диспропорционированию гипобромита в щелочных растворах на бромид и бромат:
ЗВгО" —> 2Вг" + ВгОз
Возможно разложение гипобромита в кислых и щелочных растворах и по следующим реакциям:
2НВгО —> 2НВг + О2
2ВгО' —> 2Вг" + О2
Исследовали устойчивость 10_|—10~3 М растворов гипобромита, хранившихся в различных условиях при комнатной температуре 13]. Рекомендуется хранить растворы в темных склянках и желательно в темном месте, особенно это важно для растворов с концентрацией меньше 10~2 М. Эти условия особенно необходимо соблюдать при хранении растворов гипобромита, применяющихся в качестве стандартных растворов или редокс-титрантов, и образцов для анализа, содержащих гипобромит. Растворы гипобромита часто содержат различные количества бромита, бромата и бромида. Очень важен полный анализ таких растворов, и при обсуждении методов анализа гипобромита такие методики будут рассмотрены.
Растворы гипоиодита еще менее устойчивы [2], чем растворы ипобромита, гипоиодит диспропорционирует на иодид и иодат:
310’ —> 2Г + Юз
365
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Количественные методы определения гипобромита — в основ ном редокс-методы с визуальной или потенциометрической индикацией конечной точки титрования.
Гипобромит можно определять, используя некоторые методы, описанные в разделе «Гипохлорит». При определении гипохлорита добавляют бромид натрия, который превращается в гипобромит
это делают для того, чтобы использовать свойство гипобромита быстро взаимодействовать с восстановителями:
СЮ* + Вг' —> ВгО’ + СГ
Требование, чтобы именно гипобромит разрушался по реакции с фенолом или ионом аммония, становится особенно важным, когда необходимо определять в присутствии гипобромита бромит и бро мат. В более поздних работах было показано, что эти реакции недостаточно специфичны для использования их в количественном анализе. Подробнее этот вопрос будет обсуждаться ниже.
Как и некоторые другие бромсодержащие анионы, гипобромит можно восстановить SO2 до бромида и затем определять. Восстановление обычно проводят, добавляя к анализируемому раствору бисульфит натрия и разбавленную H2SO4, кипячением удаляют избыток SO2.
Редокс-методы
Йодометрический метод
Этот метод достаточно селективен, но он вытесняется другими редокс-методами, более экспрессными.
Метод с применением аскорбиновой кислоты
Метод с использованием аскорбиновой кислоты является одним из основных косвенных методов определения гипобромита [4].
В одной из методик к гипобромиту добавляют избыток стандартного раствора сульфата таллия и непрореагировавший Т13+ титруют стандартным раствором аскорбиновой кислоты с вариами-новым голубым в качестве индикатора:
Т13++ С6Н6О6 —♦ Т1* + С6Н6О6 + 2Н+
Подробно метод описан в разделе «Гипохлориты». На основании детального изучения метода [5] был сделан вывод о том, что реакция TI.+—ВгО~ идет до образования брома и что определяется сумма бромита и бромата. Поэтому метод так же неселективен, как и иодиметрический метод.
Другой метод основан на реакции аскорбиновой кислоты й гексацианоферрат(П1)-пона [6]. Гипобромит, как и гипохлорит 366
реагирует с K«Fe(CN)6 с образованием эквивалентного количества K3Fe(CN)6, который титруют стандартным раствором аскорбиновой кислоты:
С6Н.Ов + 2 [Fe (CN)6)3' —> С6Н6О6 + 2 [Fe (CN)6]‘' + 2Н+
Арсенометрический метод
Разработан метод прямого титрования [5] гипобромита мышьяком (III) в присутствии индикатора тартразина. Присутствующий в смеси бромит не взаимодействует с мышьяком (III) до тех пор, пока гипобромит не прореагирует полностью. Индикатор обесцвечивается [7] гипобромитом, но не взаимодействует с бромидом и броматом. В последующих работах отмечали нечеткую конечную точку титрования, для обнаружения которой необходимо пользоваться внешним индикатором [8].
В разделе «Электрохимические методы» приведена методика потенциометрической индикации конечной точки титрования [10].
Мышьяк используется в методике [9] анализа растворов, содержащих гипобромит, бромит и бромат. Методика включает в себя следующие стадии.
1. В первой аликвотной части иодиметрическим титрованием определяют ВгО' + ВгСЬ’ 4- ВгОз, выделяющийся из KI иод титруют стандартным раствором тиосульфата.
2. Во второй аликвотной части раствора введением сульфата аммония разрушают гипобромит и иодиметрически титруют сумму BrOjJ + ВгОз:
4BrO'+2NHj —> 4Br'+N2 + 4H2O
3. В третьей аликвотной части анализируемого раствора селективно восстанавливают гипобромит и бромит избытком мышьяка (111), непрореагировавший As111 титруют стандартным раствором иода.
4. В четвертой аликвотной части добавлением сульфата аммония разрушают гипобромит и по п. 3 находят содержание Гипобромита. Эта методика приведена в разделе «Бромиты», по мнению авторов работы [18], методика чрезвычайно длительна, реакция взаимодействия бромита и аммония не проходит количественно (это отмечалось и в более ранних работах) и требуется применение трех титрованных растворов. В работе [10] отмечалось, что метод применим лишь при большом соотношении гипобромита и бромита и неприменим в тех случаях, когда бромит Находится в избытке по отношению к гипобромиту.
Описан [8] альтернативный приведенному выше метод анализа смеси гипобромита, бромита и бромата, основанный на применении только раствора мышьяка(III). Методика заключается » следующем.
367
1. Определение гипобромита основано на том, что скорость его взаимодействия с мышьяком в щелочных растворах велика:
ВгО' + AsO’" —> Вг' + AsOf
В этой среде бромат не реагирует с мышьяком. Бромит лишь медленно взаимодействует с As”1 в соответствии с уравнением реакции
BrOJ + 2AsO|' —> Вг" + 2AsO5‘
Этой реакцией можно пренебречь, если быстро титровать гипобро-мит. Необходимо использовать обратимый трудноразрушающийся индикатор. Для этих целей пригодны бромтимоловый синий, хинолиновый желтый или эпсилон голубой.
2. Бромит определяют после титрования гипобромита мышьяком (III). Поскольку это реакция медленная, бромит обычно определяют, добавляя избыток мышьяка(III) и затем оттитровы-вая As111, не вступивший в реакцию. В работе [8] найдено, что взаимодействие бромита с большим избытком иодида происходит значительно быстрее, образующийся иод можно оттитровать мышьяком (III) в слабощелочном растворе:
ВгОj + 4Г + 2Н2О —> Вг'+212 + 4ОН'
AsOl’ + 12 + Н2О —> AsO’* + 2Г + 2Н+
3. Титрование всех трех анионов мышьяком(III) проводят в среде разбавленной HCI в присутствии обратимого индикатора хинолинового желтого. Титровать следует раствор мышьяка(III) анализируемым раствором (содержащим гипобромит, бромит и бромат), так как при подкислении анализируемого раствора, содержащего гипобромит, образуется легколетучий бром.
Титриметрнческий анализ растворов [8], содержащих гипобромит, бромит и бромат.
Реагенты
Стандартный раствор мышьяка(1П), 0,025 М (0,1 н.) Готовят раствор из As2O2; подробно методика описана в разделе «Арсенаты и арсениты».
Боратный буферный раствор, pH = 8,3.
Бромтимоловый синий, 0,05%-ный раствор. Растворяют 0,05 г препарата в 100 мл воды или 0,04 М раствора NaOH.
Хинолиновый желтый, готовят так же, как и бромтимоловый синий.
Ход анализа. Определение гипобромита. Титруют 25 мл анализируемого раствора 0,025 М раствором мышьяка(Ш); титрование прекращают, не добавив 0,2 мл титранта до конечной точки титрования. Добавляют 25 мл буферного раствора с 0,5 мл 0,05%-ного раствора бромтимолового синего. Продолжают титрование до перехода окраски индикатора от желто-зеленой до си-
не-зеленой.
Определение бромита. К реакционной смеси после определения ВгО~ добавляют 5 мл 20%-ного раствора KJ и титруют выделившийся « 0,025 М раствором мышьяка(III), добавив перед концом титрования крахмал При титровании окраска раствора изменяется от розово-лиловой до сине-зеленою
Определение гипобромита, бромита и бромата. К 25 мл 0,025 М раствора мышьяка(Ш) в конической колбе добавляют 12 мл 6 М И
365
К 0,5 мл хинолинового желтого Титруют анализируемым раствором до перехода окр'аски от желтой до бесцветной. Содержание бромата находят, вычитая из суммарного содержания трех анионов, найденного по результатам последнего титрования, содержание гипобромита и бромита.
Другие редокс-методы
Для редокс-титрования гипобромита используют пероксид водорода в качестве титранта и люцигенин в качестве индикатора [Ц], причем метод оказался лучшим среди редокс-методов с применением пероксида водорода [12]. Описан редокс-метод косвенного определения гипобромита с использованием ванадия(IV) и железа(II) [13].
Электрохимические методы
Опубликован [10] потенциометрический метод селективного определения гипобромита и бромита. Селективность метода основана на каталитическом влиянии тетраоксида осмия на реакцию бромита с As,H. Приведены две методики, одна основана на том, что реакция бромита и K.4Fe(CN)6 очень медленная, что позволяет в присутствии бромита определять гипобромит. Другая методика основана на ускорении реакции бромита с As111, тетраоксидом осмия. В отсутствие катализатора реакция протекает чрезвычайно медленно и можно определять один гипобромит.
Описан полный вариант методики, в которой определяются также бромат и бромид, для определения которых используют перманганат и нитрат серебра [14]. Методика приведена в разделе «Броматы».
Рассматривались факторы, влияющие на определение гипобромита и бромита при их совместном присутствии, описаны методы их одновременного определения с использованием хронопотенциометрии с золотым электродом и полярографии [15].
Последовательное микроопределение [10] гипобромита и бромита
Реагенты
Мьииьяк(Ш), 0,0025 М раствор. Точную навеску As2O3 растворяют в растворе NaOH (4 г в 100 мл Н2О). Подкисляют по ализарину разбавлеииой H2SO<-Добавляют 100 мл насыщенного раствора гидрокарбоната натрия и разбавляют 40 1 л водой.
Тетраоксид осмия-. 0,1%-ный раствор в 0,3 М растворе NaOH.
Электроды: платиновый и насыщенный каломельный.
Ход анализа. К 1 мл смеси ВгО и ВгО2 добавляют 8 мл 0,3 М раствора гид-Р карбоната натрия и титруют раствором мышьяка(1П) По результатам титро-। Ия находят содержание ВгО-. Добавляют 1 мл 1,5 М раствора NaOH н каплю 0,1%-ного раствора OsO« и продолжают титрование. Общий объем ти-* ^а, израсходованного на титрование, пропорционален содержанию ВгО’ + + BrOj.
₽авн^7МеЧаИНЯ- 1' ®птимальное значение для титрования бромита
369
2. Введение OsO« в раствор бромита, содержащим пшобромит, искажает ре. зультаты титрования за счет каталитического влияния OsO4 на разложение гн-побромита.
3. Если содержание гипобромита мало по сравнению с содержанием бро-мита. необходимо повторить определение для уточнения точки эквивалентности.
4. Метод можно применять в присутствии бромата и бромида, не мешающих определению.
Определение гипобромита в присутствии других галогенсодержащих анионов
Некоторые методы определения гипобромита, рассмотренные в этом разделе, можно применить в присутствии ВгОг [3, 4, 7, 8 — - 10, 14, 15], ВгОз [8—10, 14], С10‘ [4] и Вг [10, 14].
ЛИТЕРАТУРА
1. Polak Н. L., Feenstra G., Slagman I.—Taianta, 1966, v. 13, p. 715.
2. Hashmi M. H., Ayaz A. A., Rashid A., AH E. — Analyt. Chem., 1964, v. 36 p. 1379.
3. Velghe H., Claeys A. — Taianta, 1972, v. 19, p. 1555.
4. Erdey L., Vigh K- — Taianta, 1963, v. 10, p. 439.
5. Hashmi M. H., Rashid A., Ayaz A. A., Chughtai N. A. — Analyt. Chem., 1966, v. 38, p. 507.
6. Erdey L., Svehla G. — Z. analyt. Chem., 1959, v. 167, p. 164; Analyt. Abstr, 1960, v. 7, p. 122.
7. Hashmi M. H., Ayaz A. A. —Analyst, 1964, v. 89, p 147.
8. Velghe N., Claeys A. — Analytica chim. Acta, 1972, v. 60, p. 377.
9. Hashmi M. N., Ayaz A. A. — Analyt. Chem., 1963, v. 35, p. 908.
10. Andersen T., Madsen H. E. L. — Analyt Chem, 1965, v. 37, p. 49
11. Erdey L., Buzas I. — Acta chim. hung, 1955, v. 6, p 77.
12. Berka A., Vulterin J., Zyka J. — New Redox Titrants, Oxford, Pergamon Press, 1965, p. 102.
13. Tandon J. P., Chawia K. L. — Bull. chem. Soc. Japan, 1966, v. 39, p. 254; Analyt. Abstr, 1968, v. 15, p. 670.
14. Норкус П. К., Стильжене С. П. ЖАХ, 1969, т 24, с. 1565. Analyt. Abstr, 1971, т. 20, с. 3029.
15. Fuchs Н., Landsberg R. — Analytica chim. Acta, 1969, v. 45, p. 505.
ГИПОХЛОРИТЫ
Гипохлорит определяют в отбеливателях и в водах. При анализе воды в понятие общего «активного хлора» включено наряду с содержанием гипохлорита содержание хлора и хлорамина. В настоящее время основным методом анализа гипохлорита является классическая редокс-титриметрия. Наблюдается тенденция развития автоматизированных методов
МЕТОДЫ ОТДЕЛЕНИЯ
Гипохлорит, хлорит, хлорат и перхлорат можно разделить М тодами тонкослойной или бумажной хроматографии [24]. В ка стве неподвижной фазы использовали Silufol UV 254, растворит ?7Q
лем служила смесь бутанола, пиридина и воды в соотношении (2:2: 1); длительность метода 30 мин.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гипохлорит — один из самых сильных окислителен, о чем можно судить по его стандартному потенциалу в щелочных растворах. Для сравнения потенциалы MnOi'/MnCh и MnOj/MnOi* в щелочных растворах составляют 0,60 и 0,56 В соответственно; потенциал С1О_/С1_ составляет 0,89 В.
Титриметрические методы
Для определения гипохлорита предложен большой набор ре-докс-реагентов, многие из которых представляют лишь академический интерес. В двух монографиях [1, 2] приведен подробный обзор редокс-методов определения гипохлорита, поэтому из них можно почерпнуть дополнительную информацию
Как и некоторые другие хлорсодержащие анионы, гипохлорит восстанавливают SO2 до хлорида и затем определяют. Восстановление проводят обычно, добавляя !%-ный раствор гидросульфита натрия и разбавленную H2SO4, избыток SO2 удаляют кипячением.
Нами будут рассмотрены наиболее важные классические методы, которые в значительной степени связаны с современными методами анализа.
Иодиметрический и арсенометрический методы
Гипохлорит в кислых растворах окисляет иодид до иода, который можно оттитровать стандартным раствором тиосульфата, используя для индикации конца титрования крахмал:
СЮ" + 2Г + 2Н* —> СГ + 12 + Н2О
Эта методика часто рекомендуется для установки титра раствора гипохлорита, который используют в качестве редокс-титранта. Метод применим для определения активного хлорида в порошках-отбеливателях, которые имеют неопределенный состав, примерно саС1(ОС1). Содержание активного хлора эквивалентно выделяющемуся по реакции иоду (в присутствии уксусной или серной кислоты) и выражается в процентах от массы порошка-отбеливателя.
Прямое визуальное титрование гипохлорита стандартным раствором мышьяка (HI) невозможно, так как гипохлорит разрушает идикаторы. Мышьяк(III) можно использовать для титрования оделяющегося иода вместо тиосульфата, в этом случае для инди-аДйи конца титрования тоже используют крахмал.
Лучший метод основан на добавлении к гипохлориту избытка н СТв°ра мышьяка(III) в среде гидрокарбоната натрия и титрова-а Раствором иода непрореагировавшего мышьяка(III):
H3AsO3 + I2 + Н2О —> HAsOV + 4Н+ + 2Г
371
Определению не мешают хлораты, хлориты и перхлораты, методика рекомендована для определения гипохлорита в растворах пестицидов [3].
Используя стандартный раствор гипохлорита, можно проводить обратное титрование мышьяка(III). В качестве индикаторов можно использовать бордо — необратимый индикатор [4] или тартразин — обратимый индикатор (в последнем случае предварительно к раствору добавляют бромид для превращения гипохлорита в гипобромит) [5].
Титрование стандартного раствора мышьяка(III) гипохлоритом можно применить для определения гипохлорита. Эффективны в титровании два обратимых индикатора: бромкрезоловый красный [6] и бромтимоловый синий [7]. Оба индикатора используются в боратном буферном растворе в присутствии катализатора — осмиевой кислоты.
При титровании восстановителя, такого, как мышьяк(Ш), гипохлоритом обычно добавляют бромид, который, взаимодействуя с гипохлоритом, окисляется до гипобромита. Это делают потому, что, хотя гипохлорит и более сильный окислитель по сравнению с гипобромитом, скорость взаимодействия гипобромита с восстановителями выше.
Уже отмечалось, что прямое визуальное титрование гипохлорита стандартным раствором мышьяка (III) невозможно, так как гипохлорит — очень сильный окислитель, разрушает индикаторы. Однако можно использовать внешний индикатор — иод-крахмальную бумагу. К анализируемому раствору гипохлорита добавляют гидрокарбонат натрия и титруют стандартным раствором мышьяка (III). Титрование заканчивают, когда при нанесении на иод-крахмальную бумагу капли титруемого раствора не образуется синего пятна. Метод рекомендован в качестве стандартного для анализа гипохлорита натрия [8].
Иодиметрическое определение гипохлорита
Ход анализа. К 25 мл анализируемого раствора гипохлорита добавляют 1 г KI и 10 мл 2 М H2SO4. Тнтруют выделившийся иод стандартным раствором тиосульфата, вводя вблизи точки эквивалентности крахмал.
Метод с применением аскорбиновой кислоты
Аналитические методы, основанные на применении аскорбиновой кислоты, в течение многих лет разрабатывали Эрдей с сотр-[9—11]. Косвенный метод определения гипохлорита основан на окислении гексацианоферрата(II) до гексацианоферрата(III), к°' торый затем титруют стандартным раствором аскорбиновой кислоты. В качестве индикатора используют дихлорфенолиндофеноЛ-
СЮ’+ 2 [Fe (CN)6f-+ 2Н+ —> СГ + 2 [Fe (CN)6]3'+ Н2О-
С6Н8ОЬ + 2 [Fe (CN)6J3“ —> С6Н6О6 + 2 [Fe (CN)6]4'+ 2Н+
£72
Другая методика основана на окислении гипохлоритом тал-лня(1) до та.тлия(Ш) с последующим титрованием таллня(Ш) стандартным раствором аскорбиновой кислоты:
С1О' + Т1+ + 2Н+ —> Т13+ + СГ + Н2О
Т13+ + С6Н3О6 —> Т1+ + С6Н6О6 4- 2ЬГ
В качестве индикатора применяют вариаминовый голубой. Метод можно использовать и для определения гипобромита. Основным достоинством системы является то, что можно проводить определение и не в атмосфере СО2. Было установлено [9—11], что хлорит не оказывает мешающего влияния на определение гипохлорита при содержании хлорита равном или меньшем, чем содержание гипохлорита. Выяснен механизм реакции [12].
Титриметрическое определение гипохлорита с использованием аскорбиновой кислоты [11]
Реагенты
Аскорбиновая кислота, 0,05 М (0,1 и. раствор). Растворяют 9,0 г чистой аскорбиновой кислоты в I л дистиллированной воды. •
Титр раствора устанавливают по йодату натрия или гексацианоферрату (III).
Для этого в конической колбе взвешивают 0,6—0,7 г КзРе(СМ)6 н растворяют в 15—20 мл воды, добавляют 2 г КНСО3 и 0,5 г смеси (1 : 500 по массе) 2,6-дихлорфенолиндофенола и NaCl. Титруют раствор аскорбиновой кислотой до исчезновения голубой окраски. Расчет титра основан на уравнениях реакций, приведенных выше.
На воздухе ежедневно концентрация аскорбиновой кислоты за счет разложения уменьшается на 1%. Процесс можно замедлить, если растворы хранить в атмосфере СО2.
Стандартный раствор таллия(1), 0,025 М (0,1 н.) раствор. Растворяют 12,622 г сульфата таллия(I) в воде и разбавляют до 1 л.
Ацетатный буферный раствор. Смешивают 1 объем 2 М раствора ацетата натрия с 2 объемами 2 М уксусной кислоты.
Вариаминовый голубой, индикаторная смесь. Одну часть вариаминового голубого (солянокислый, 4-амнно-4-метоксндифениламин) смешивают с 400 частями NaCl. На каждое титрование используют около 0,2 г.
Ход анализа. К анализируемому раствору, содержащему 25—250 мг CIO-, Добавляют избыток (10—55 мл) 0,025 М раствора T12SO4, перемешивают, добавляют 15 мл 2 М раствор НС1 и опять перемешивают. Добавляют 2 М раствор NaOH до начала образования осадка и затем вводят 10 мл буферного распора. После добавления около 0,2 г вариаминового голубого титруют 0,05 М Раствором аскорбиновой кислоты до исчезновения голубой окраски индикатора. Для определения гипохлорита на уровне концентраций 0,005 М используют ту Же методику.
Примечание. Метод пригоден для определения активного хлора в хлор-в°и извести.
Спектроскопические методы
Гипохлорит можно определять по образованию соединения ин-енсивно желтой окраски при взаимодействии его с бензидином в Разбавленной НС1. Но в связи с тем, что в последнее время обнаружены канцерогенные свойства бензидина, этот метод нельзя комендовать для использования [13].
373
Максимум поглощения гипохлорита находится в ультрафиолетовой области при 290 нм. Однако максимум поглощения сравнительно невелик и перекрывается линиями других оксигалогенидов (хлорита и оксида хлора), что не позволяет использовать метод для определения гипохлорита в их присутствии.
Разработан метод, основанный на взаимодействии гипохлорита с о-толидином, светопоглощение продуктов реакции измеряют при 436 нм. Как и некоторые другие реагенты, например 3,3-диметил-нафтидин и М,М-диэтил-п-фенилендпамин, о-толидин неселективен, его взаимодействие с сильными окислителями зависит только от редокс-потенциалов последних Оба упомянутых выше реагента используют для определения общего активного хлора в воде. о-Толидиновый метод был применен для определения гипохлорита в стоках, содержащих также цианиды, хроматы и нитриты. Гипохлорит определяли [14] на уровне, соответствующем 10 мкг хлора в 1 л, измеряя светопоглощение растворов при 436 нм.
В последнее время появился более селективный реагент — си-рингалдазин [бис -(4-гидрокси-3,5-диметоксибензилиден)гидразин]. Его можно использовать для определения свободного хлора в воде, реагент не взаимодействует с хлораминами [15]. Стехиометрия реакции (1:1), оптимальные условия — pH = 7 и 530 нм. Закон Бера выполняется до 1 ppm CU-
Бабко, Терлецкая и Дубовенко [16] нашли, что СЮ~ в 10 раз увеличивает флуоресценцию смеси пероксида водорода и люминола (5-амино-2,3-дигидрофталазин-1,4-дион). Чувствительность метода, основанного на этой реакции, составляет 0,5 г С1О~ в 10 мл раствора.
Электрохимические методы
Гипохлорит можно титровать хлоридом меди(1), это первый потенциометрический метод определения гипохлорита [17].
В другом методе [18] используют последовательное потенциометрическое титрование гипохлорита, хлорита, хлората и хлорида. Гипохлорит титруют 0,025 М раствором мышьяка (III) с платиновым индикаторным электродом при pH ~8. Хлорит и хлорат титруют соответственно при pH = 8—12 и в разбавленной H2SO4. причем в каждом случае используют в качестве катализатора тетраоксид осмия. Точность титрования растворов гипохлорита, содержащих малые концентрации хлорита, выше при использовании иодиметрического титрования. Подробно метод описан в разделе «Хлораты».
Для потенциометрического титрования гипохлорита исгюльзо^-вали железо(П), индикаторным электродом служил электрод, а электродом сравнения — насыщенный
[19]. Хлорид, хлорат, иодат, сульфат и нитрат мешают только в больших концентрациях, хлорит титруется одновременно с гипО" хлоритом, а бромид, иодид, и фосфат необходимо отделять перед титрованием.
платиново,,‘ каломельный
374
Гипохлорит необратимо восстанавливается на ртутном капающем электроде в интервале pH от 3,6 до 11, потенциал полуволны в нейтральных растворах равен 4-0,08 В. Дроздецкая и Ильин [20] опубликовали полярографический метод определения гипохлорита, в котором используют платиновый микроэлектрод и 4 Л1 раствор NaCI в качестве фонового электролита. Хлорит не мешает определению.
Разработано несколько кулонометрических методов. Гипохлорит титруют [21] 0,1 М раствором Fe11, генерируемым в ацетатном буфере при pH = 4,3—4,7 при пропускании через раствор тока азота. Хлорит, хлорат, перхлорат, сульфат и 10-кратное количество нитрата не мешают определению гипохлорита. Хлорит титруется одновременно с гипохлоритом. Разработан полностью автоматизированный метод [22] определения гипохлорита натрия (и пероксида водорода) в отбеливателях с использованием пористого серебряного электрода. В каждом случае выделяется кислород, который определяют кулонометрически. Гипохлорит реагирует с пероксидом водорода в щелочной среде с выделением кислорода:
NaClO + Н2О2 —> NaCI + Н2О + О3
на серебряном индикаторном электроде:
О2 + 2Н2О + 4е" —> 4ОН"
на Pt электроде:
4ОН' —> О2 + 2Н2О + 4е'
Другие методы определения
I Предложено длительное термометрическое титрование гипохлорита в отбеливателях, в качестве реагентов используют 10%-ный раствор Na2SO3 или 5%-ный раствор Na2S20s [23]. Теплоты реакций 5708 и 4024 Дж/г соответственно, ошибка определения составляет 2,3%.
ЛИТЕРАТУРА
1 Koithoff 1. М, Belcher R.— Volumetric Analysis, III New York, Interscience, 1957.
2. Berka A., Vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Pergamon Press, 1965. x
3- Official Methods of Analysis of the Association of Official Analytical Che-mists, 11th edn, 1970, p. 97, 257.
4- Koithoff I. M., Stenger V. A. — Ind. Engng Chem. analyt. Edn, 1935, v. 7,
R ?,79
°- Belcher R —Analytica chim. Acta, 1950, v. 4, p. 468.
b. Belcher R, El Khiami I., Stephen W. I. — Talanta, 1965, v. 12, p. 775
' Cerana L. A. — Rev. Facultad. Ing. Quim. Univ. Nac. del Littoral, Argentina. I960, v. 29. p. 105.
° British Standard 4426, 1969.
in v'-1 Svehla G. — Chemist Analyst, 1963, v. 52, p. 24
,/dey L., Svehla G. — Z. analyt. Chem., 1959, v. 167, p. 164; Analyt. Abstr,
*960, v. 7, p. 122.
375
11. Erdey L., Vigh K- — Taianta, 1963, v. 10, p. 439.
12. Hashmi M. H., Rashid A., Ayaz А. A., Chughtai N. A. — Analyt. Chem., 1966, v. 38, p. 507.
13. Klut H.— Kleine Mitt. Mital. Ver. Wass Versog., 1927, v. 3, p. 184.
14. Zuse M. — Galvanotechnik, 1966, v. 57, p. 500; Analyt. Abstr., 1967, v. 14. p. 7933.
15. Bauser R., Rupe С. O. — Analyt. Chem., 1971, v. 43, p. 421.
16. Бабко А. K-, Терлецкая А. В., Дубовенко JI. И. — Укр. хим. ж., 1966, т. 32, с. 728.
17. Касаткина JI. А., Шапиро Я. М. ЖАХ, 1967, т. 22, с. 953. Analyt. Abstr., 1969, v. 16, р. 1253.
18. Норкус П. К-, Стильжене С. П. ЖАУ, 1969, т. 24, с. 884. Analyt. Abstr., 1970, v. 19, р. 4862.
19. Vulterin I., Hovorka J.— Colin Czech, chem. Commun. Engl. Edn, 1967, v. 32, p. 4063; Analyt. Abstr., 1969, v. 16, p. 1254.
20. Дрозорецкая E. П., Ильин К. Г. ЖАХ, 1972, т. 27, с. 200. Analyt. Abstr., 1973, v. 25, р. 177.
21. Gruendler Р., Holzapfel Н. — Taianta, 1970, v. 17, р. 246.
22. Fleet В., Но А. У. W., Tenygl 1. — Taianta, 1972, v. 19, р. 317.
23. Strafelda F., Kroftova J. — Colin Czech, chem. Commun. Engl. Edn, 1968, v. 33, p. 4171; Analyt. Abstr., 1970, v. 19, p. 2226.
24. Thielemann H. — Mikrochim. Acta, 1971, p. 746.
ИОДАТЫ
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение
Иодат можно отделить осаждением его в виде труднорастворимой бариевой соли. Метод применим для анализа смеси хлорид— иодат, в которой хлорид определяют с применением нитрата серебра. В качестве осадителя применяют крупный катион тетрафениларсония (C6H5)4As+. В виде ионного ассоциата в этом случае осаждаются также перманганат, перренат, перхлорат, перйодат и вольфрамат. Тетрафениларсоний можно применять и для отделения йодата и для его определения.
Ионообменный метод
Иодаты, хлораты и броматы в миллимолярных концентрациях можно разделять с использованием ионного обмена Подробно методика приведена в разделе «Хлораты».
Бумажная и тонкослойная хроматография
В табл. 19 (см. раздел «Хлораты») приведены некоторые данные о выделении йодата методами бумажной и тонкослойной хроматографии. Кроме того, применяют еще следующие методы разделения.
Иодат, иодид и перйодат разделяли [1] на силикагеле с использованием смеси метанола (25%-ный в воде), NH3 водного, воды и 10%-ной уксусной кислоты (18:2:2:1). Иодид, иодат, 376
теллурит, фосфат и сульфат разделяли на силикагеле, кизельгуре или оксиде лантана, в качестве растворителя применяли смесь ацетона и 1 Л1 раствора NH4OH (1:1) [2]. Позднее в методе ТСХ стали применять оксид алюминия [3] для разделения йодата, перйодата, бромата и хромата.
Бумажная хроматография была применена для разделения иодида, йодата и перйодата [4]. Использовали бумагу ватман № 1 и растворитель амиловый спирт — пиридин — NH4OH (3:7:10). Хроматографию на стекловолокне применяли с различными растворителями, содержащими изопропанол и 1,5 М раствор NH3 (7:3) для разделения иодида, йодата и перйодата [5]. По мнению автора работы [5], бумажную хроматографию в этом случае нельзя было применить из-за восстановления перйодата до йодата.
Соосаждение
Новиков опубликовал метод [6] отделения йодата от перйодата соосаждением последнего на гидроксиде железа. Повторяя соосаждение несколько раз, удается более чем на 99% провести разделение этих двух ионов.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Как и некоторые другие галогенсодержащие анионы, иодат восстанавливается SO2. Определение образующегося иодида позволяет находить содержание йодата в анализируемом образце.
Титриметрические методы
В основном титриметрические методы — это редокс-методы. Необходимо применять специальные приемы для того, чтобы сделать эти методы более селективными, особенно если иодат находится в смеси с броматом, хлоратом и перйодатом.
Иодат калия в чистом виде в аналитической химии широко ис-пользуют в качестве стандартного вещества. Поэтому его реакции с рядом восстановителей были подробно изучены. Во многих случаях эти редокс-реакции можно применять для точного определения восстановителей, используя стандартный раствор К1О3. Те же Реакции можно использовать и для определения йодата.
Иодиметрические методы являются, по-видимому, почти единственными методами, которые могут обеспечить требуемую селективность определения йодата.
Иодиметрические методы
В разбавленных минеральных кислотах иодат реагирует с и°Дидом с образованием иода, который титруют стандартным раствором тиосульфата.
377
Реакция проходит быстро, что позволяет определять иодат с высокой точностью. Обычно эту реакцию используют для установки титра растворов тиосульфата, который рассчитывают, исходя из точной навески чистого КЮз. При определении йодата, наоборот, используют точный раствор тиосульфата.
Титриметрическое определение йодата с помощью тиосульфата натрия
Ход анализа. К 25 мл раствора йодата, содержащего 0,1—0,15 г Ю3, добавляют 2 г KI и перемешивают до полного растворения. Добавляют 10 мл 1 Af раствора НС1 и титруют стандартным раствором тиосульфата, применяя для инти-кации конечной точки титрования крахмал.
Примечание. Титр тиосульфата можно устанавливать по этой же методике, применяя точную навеску чистого, высушенного при 150 °C КЮ3.
Методика пригодна для анализа смеси КЮ3 и NaIO3 или их растворов; если Ю“ находится в смесн с другими окислителями, в методику необходимо вносить изменения.
При анализе смеси йодата и перйодата перйодат обычно восстанавливают до йодата в буферном растворе, вводя иодид, мышьяк(Ш), марганец(П), сульфит, бромид и т. д. В работах [7, 8] для этих целей использовали бромид. В части раствора определяют сумму йодата и перйодата иодиметрическим титрованием по обычной методике с тиосульфатом натрия. В другой части раствора перйодат восстанавливают до йодата добавлением КВг и НС1. Раствор подщелачивают КНСО3 и образующийся NaOBr восстанавливают до NaBr кипячением с этанолом. Оставшийся иодат титруют иодиметрически, как описано выше. В более поздних публикациях вместо этанола использовали карбамид. Белчер с сотр. [9] разработали метод определения смеси анионов, в котором перйодат связывается молибдатом с образованием 6-мо-либдопериодата. Затем перйодат выделяют, вводя щавелевую кислоту. По этому методу можно проводить полный анализ смеси иодат-периодат в одной аликвотной части анализируемого раствора.
Анализ смеси иодат — бромат можно проводить несколькими методами. В одной аликвотной части раствора сумму анионов определяют иодиметрическим титрованием. В другой аликвотной части оба аниона восстанавливают до иодида и бромида кипячением с !%-ным раствором KHSO3. Иодид можно определить в присутствии бромида, так как у иодида ниже редокс-потенциал-Подробно это определение описано в разделе «Иодиды».
Еще один метод определения йодата в присутствии бромата и хлората основан на восстановлении во фталатном буферном растворе с pH — 5. Через 3 мин образующийся иод титруют стандарт* ным раствором тиосульфата. Бромат и хлорат в этих условиях не восстанавливаются, бромат восстанавливается лишь в присутствии катализатора — молибдата [10].
Описан метод [11], в котором смесь йодата, перйодата, бро* мата и хлората определяют иодиметрически, постепенно изменяя условия реакции.
378
Можно определять иодат иодиметрически восстановлением до 1+ в сильнокислых средах. В присутствии сильной кислоты (4—6 М НС1) иодат реагирует с иодидом следующим образом:
КЮ3 + 2KI + 6НС1 —> 3IC1 + ЗКС1 + ЗН2О
этой реакции основан хорошо известный метод определения иодида. Эта же реакция может быть использована для определения йодата, если к раствору добавить известное количество иодида. Подробно методика описана в разделе «Иодиды».
Арсенометрический метод
Титрование мышьяком(III) йодата хорошо изучено и является одним из основных методов определения мышьяка. Точный стандартный раствор мышьяка можно приготовить из чистой Аз20з (см. раздел «Арсенаты и арсениты») и с его помощью определять иодат.
Метод с применением гексацианоферг>ата( 11)
Иодат (как бромат и хлорат) реагирует с гексацианоферратом (II), окисляя его до гексацианоферрата (III). Последний можно оттитровать стандартным раствором аскорбиновой кислоты [12]:
Юз + 6 [Fe (CN)6)4' + 6Н+ —> Г+6[Fe(CN)6]3'+ ЗН2О
Галогены, гипогалогены и галогениты также можно определить с применением этой реакции. Метод видоизменяли, улучшая индикаторы [13], был разработан автоматический потенциометрический вариант метода [14].
Другие редокс-методы
Описан метод прямого титрования йодата хромом(II) синдикатором 1,10-фенантролинатом ванадия (II) [15]. Метод может быть использован также для определения хлората, бромата и нитрата.
Комплексометрический метод
Обычно комплексометрические методы основаны на образовании металлом с определяемым анионом труднорастворимых соединений.
Вильборг и Дроздов [16] определяли иодат, осаждая его в ВиДе йодата свинца, который затем растворяли в избытке стандартного раствора ЭДТА в присутствии аммиачного буферного Раствора. Избыток ЭДТА оттитровывали сульфатом цинка в при-Утствии кислотного хрома черно-синего. Этот метод может быть епользован для определения хромата и гексацианоферрата(III).
379
Спектрофотометрические методы
Спектрофотометрическое определение иода, выделяющегося при взаимодействии йодата с иодидом в кислых растворах, можно применить как метод определения йодата. Ламберт с сотр. [17, 18] разработали кадмий-иодидкрахмальный реагент, в котором Cdl, выступает в качестве источника иодид-ионов. Этот реагент использовали и в более поздних работах [19]. Как было найдено, нитрит мешает определению, но его можно удалить введением сульфаминовой кислоты. Для определения окислителей в воде плавательных бассейнов применили реагент лейкооснование кристаллического фиолетового [4,4',4"-метилидентрис(М,М-диметиланилин)] и хлорид ртути [20]; метод можно применить для определения иода, выделяющегося по реакции йодата с иодидом. Хлорид ртути(II) является катализатором реакции.
Разработан метод [21] одновременного определения йодата и бромата с применением смеси изониазида и хлорида 2,3,5-трифенил-тетразолия и разбавленной НС1. Реагент образует с йодатом розовое соединение при 33 °C, брома г взаимодействует с этим реагентом лишь при 60 °C. Область определяемых концентраций каждого из ионов 10—300 мкг в 50 мл; светопоглощение измеряют при 480 нм или при 308 нм. Хлорат не мешает определению.
Иодат в количестве 30 мкг и выше можно определять с п-ами-нофенолом. Окислителями средней силы этот реагент окисляется с образованием голубого индамина. Иодат окисляет солянокислый и-аминофенол до хинонимина, который образует с непрореагировавшим п-аминофенолом синее соединение [22]:
Хлорат и бромат не мешают определению, хлорид, бромид, фосфат, Fe3+, V®+ должны отсутствовать.
Пославска [23] разработала метод определения следовых количеств йодата (и иодида) с применением N,N'-6nc (2-гидроксипропил) -о-фенилендиамина. Окисленная (йодатом) форма реагента— красного цвета. Иодид, присутствующий в смеси с йодатом, определяют после его окисления бромом, избыток которого удаляют, используя фенол. Область определяемых концентраций йодата (и иодида) — 0,1—3 мкг/мл.
Электрохимические методы
Потенциометрические методы
Тетраоксид осмия катализирует реакцию йодата с мышьяком или сульфатом гидразина в щелочных средах. Иодат определяли [24] потенциометрическим титрованием с платиновым и каломель-
380
flbIM электродами. Ошибка определения 20 мг КЮ3 составляла -4-0,5%- Железо(П) использовали в качестве титранта в фосфорнокислых средах в присутствии катализатора тетраоксида осмия [25]- Иодат восстанавливается при этом до иодида. Иодат определяли потенциометрически, проводя реакцию иодата с избытком стандартного раствора иодида, избыток которого оттитровывали нитратом ртути(П) [26]-
Для определения йодата использовали ионоселективные электроды. Нагреванием при 60°С и pH = 9 с алюминиевой фольгой иодат восстанавливали до иодида, который определяли с помощью иодидного электрода [27]. Смесь йодата и иодида можно анализировать при содержании ионов 10'4—10~5 г/мл.
Полярографические методы
Бромат и иодат дают одну полярографическую волну восстановления. Реакцию восстановления йодата в щелочных средах можно представить уравнением
Юз + бе" + ЗН2О —> Г + 6ОН"
Бромат также восстанавливается, принимая 6 электронов. Так как ни бромат, ни иодат не взаимодействует с сульфитом в щелочных растворах, их можно определять полярографически в простых негерметичных ячейках в атмосфере воздуха в присутствии сульфита. Сульфит не только удаляет растворенный кислород, но и служит фоновым электролитом.
Перенапряжение бромата значительно больше перенапряжения йодата, что является причиной того, что его потенциал восстановления лежит в более отрицательной области. Поэтому бромат и иодат не оказывают взаимного влияния при полярографическом определении их смеси. Подробно метод описан в монографии [28]. Хлорат и перйодат не восстанавливаются в катодной области.
Другие электрохимические методы
Иодат и иодид можно определять амперометрическим титрованием. Метод основан на различии в определенных условиях растворимости йодата и иодида серебра [29].
Кинетические методы
Японские исследователи разработали кинетический метод определения микроколичеств йодата и иодида, основанный на их каталитическом действии на восстановление роданидного комплекса ^леза (III) [30]. Сначала определяют сумму йодата и иодида. °том иодид окисляют до иода, который удаляют экстракцией етыреххлористым углеродом. Остающийся в водной фазе иодат
381
определяют тем же методом. Метод использован для анализа мор. ской воды. Чувствительность метода — 0,001—0,01 ppm Ю3 4-1~
Для определения йодата использовали (31] реакцию це-рий(IV)—мышьяк(Ш), катализируемую иодидом и осмием. Иодат не является катализатором реакции, но его можно определять после восстановления до каталитически активной формы Иодид и иодат можно определять при совместном присутствии в концентрациях до 10-8 Л1. Относительная ошибка — около 2%. Разработан автоматический вариант этого метода [32], который применен для анализа речной воды.
Другие методы определения
В аналитической химии йодата большое значение имеет реакция образования в кислых растворах свободного иода по реакции Юз — Г. Пеннингтон [33] применил газовую хроматографию для определения иода. Метод применим для определения миллиграммовых количеств окислителей, количественно выделяющих иод из KI- Все определение занимает 5 мин.
Радиометрический метод основан на использовании хлорида 45Са. Когда реакция заканчивается, в водной фазе появляется радиоактивный кальций. Оксалаты и карбонаты реагируют так же.
Можно проводить термометрическое титрование КЮ3 в силыю-кислых средах 2 М раствором роданида натрия. В точке эквивалентности, как было найдено, молярное соотношение SCN : КЮ3= = 5:7, что соответствует уравнению реакций
5SCN'+ 6Юз + 2Н2О + Н+ —> 5SOf + 5HCN + 312
5HCN + 2I2 + IOJ + Н+ —> 5ICN + ЗН2О
Стандартное отклонение, найденное из 12 параллельных определений, составляет 0,4%.
ЛИТЕРАТУРА
1. Palagyi S., Zaduban М. — Chemicke. Zvesti, 1969. v. 23, p. 876, Analyt. Abst', 1971, v 20, p. 1660.
2. Moghissi A. — J. Chromat., 1964, v. 13, p 542
3. Lederer M.,Polcaro C. —J. Chromat., 1973, v. 84, p. 379.
4. Obrenovic-Paligoric I. D., Cvoric J. D. — Bull. Boris Kidrich Inst. nucl. Sci, 1963, v. 14, p. 95; Chem. Abstr., 1963, v. 59, p. 14555b.
5. Naeumann R. — J. Chromat., 1965, v. 18, p. 385.
6. Новиков А. И. ЖАХ, 1964, r. 19, c. 541. Analyt. Abstr., 1965, v. 12, p. 4534.
7. Szekeres L., Kardos E, Rady M. — Z. anal Chem, 1958, v. 160, p 401; Analyt-Abstr, 1958, v. 5, p. 4111
8. Szekeres L, Kardos E, Rady M. — Z. analyt Chem., 1958, v. 162, p. 430; Analyt Abstr., 1959, v. 6, p 1730
9. Belcher R., Tomnshend A. — Analytica chim. Acta, 1968, v. 41, p. 395 e 10. Kolthoff I. M., Hume D. N. — Ind. Engng Chem. analyt. Edn., 1943, v. 101
p. 174. 7
11 . Szekeres L. — Z. analyt. Chem., 1960, v. 172, p. 256; Analyt. Abstr., I960, V- « p. 3739.
382
12 Erdey L., Svehla G. — Z. analyt. Chem., 1959. v. 167, p. 164: Analyt. Abstr., 1960, v. 7, p. 122.
13 Erdey L., Kasa I. — Taianta, 1963, v. 10, p. 1273.
14 Erdey L., Svehla G., Weber O. — Z. analyt. Chem., 1968, v. 240, p. 91; Analyt. ' Abstr., 1970, v. 18, p. 12.
15 . Leonard M. A., Shanine S. A. E. F., Wilson C. L. — Taianta, 1969, v. 16, p. 470.
1б ' Вильборг С. С., Дроздов В. А. Научные доклады Высшей школы. Химия и химическая технология, 1958, с. 721. Analyt. Abstr., 1960, v. 7, р. 865.
17 . Lambert J. L., Arthur P., Moore T. E.— Analyt. Chem., 1951, v. 23, p. 1101.
18 , Lambert J. L.— Analyt. Chem., 1951, v. 23, p. 1247.
19 Schnepfe M. M. — Analytica chim. Acta, 1972, v. 58, p. 83.
2Q Black A. P., Whittle G. P. — J. Am. Wat. Wks Ass., 1967, v. 59, p. 471.
21 Hashmi M. H., Rashid H. A. A., Azam F. — Analyt. Chem., 1964, v. 36, ' ' p. 2471.
22. Fuchs J., Jungreis E., Ben-Dor L. — Analytica chim. Acta. 1964, v. 31, p. 187.
23. Paslawska S. — Chemia. analit., 1971, v. 16, p. 961; Analyt. Abstr., 1972, v. 22, p. 3135.
24. Норкус П. К. ЖАХ, 1965, т. 20. с. 88. Analyt. Abstr., 1966, v. 13, p. 3553.
25. Vulterin J. — Colin Czech. Chem Commun. Engl. Edn., 1966, v. 31, p. 2501; Analyt. Abstr., 1967, v. 14, p. 5398.
26. Khalifa H., Ateya B. — Microchem. J., 1969, v. 31, p. 147.
27. Paletta B. — Microchem. J., 1969, v. 31, p. 1210.
28 Heyrovsky J., Zuman P. — Practical Polarography, London, Academic Press, 1968.
29. Musha S., Ikeda S., Nishida G. — Japan Analyst, 1968, v. 17, p. 332; Analyt. Г Abstr., 1970, v. 18, p. 919.
30. Iwasaki I., Utsumi S., Yonehara N. — Bull. Chem. Soc. Japan, Pure chem.
I Sect., 1964, v. 85, p. 36; Analyt. Abstr., 1965, v. 12, p. 5789.
31. Rodriguez P. A., Pardue H. L. — Analyt. Chem., 1969, v. 41, p. 1376.
32. Truesdale V. W., Smith P. J.—Analyst, 1975, v. 100, p. 111.
33. Pennington S. N. — J. Chromat., 1968, v. 36, p. 400.
34. Omboly C., Szarvas T., Horvath L. — Radiochem. radioanalyt. Lett., 1969, v. 1 p. 49.
35. Harries R. J. N. — reported in Thermometric titrimentry by Tyrrell, H. J. V. and Beezer A. E., Chapman and Hall, London, 1968.
ИОДИДЫ
Йодиды применяют, главным образом, в фотографии и фармакологии. Следовые количества иодидов играют важную роль в Жизни животных и растений. Поэтому иодид применяют в очень малых концентрациях в качестве различных добавок.
, Основные методы разделения и определения галогенидов изложены в разделе «Хлориды». Чтобы избежать повторений, в этом Разделе мы будем лишь ссылаться на эти методы, обращая основ-в°е внимание на методики, касающиеся собственно иодида.
МЕТОДЫ ОТДЕЛЕНИЯ
Йодиды можно отделить самыми различными хроматографи-Ческими методами, включая ионный обмен. Эти методы рассматриваются в разделе «Хлориды». Описано экстракционное отделе-_Не иодида [1]. Иодид связывают в комплекс с кадмием и экстра-Руют смесью трибутилфосфата и метилбутилкетона. Цианид и ** Данид так же экстрагируются, в меньшей степени извлекаются
383
хлорид, бромид, оксалат и нитрат. Фторид, тиосульфат, сульфат, фосфат, ацетат, сульфид, гексацианоферрат(II) и борат или слабо экстрагируются, или совсем не экстрагируются. Известно несколько экстракционно-спектрофотометрических методов, в которых иодид экстрагируется в качестве аниона ионной пары. Описано разделение иодидов, йодатов и перйодатов методом бумаж-ной хроматографии на стекловолокне [2]. Применяли различные комбинации растворителей. Эти же ионы можно разделить на ватмане № 4, используя в качестве растворителя амиловый спирт— пиридин—NH4OH (3:7:10) [3].
При определении иодида в присутствии хлорида и бромида его можно окислить до йодата, при этом хлорид и бромид не окисляются, что создает предпосылки для селективного аналитического метода определения иодида. Старым методом разделения этих ионов является их предварительное осаждение в виде солен серебра с последующей обработкой смеси серной и хромовой кислотами. Хлорид и бромид серебра разрушаются, выделяя хлор и бром, которые могут быть легко удалены. Иоднд окисляется до йодистой кислоты, которую затем восстанавливают до иодида и осаждают в виде иодида серебра.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Иодид можно определять в виде иодида серебра, используя методику, описанную в разделе «Хлорид». Мешают определению те же ионы. Осадок имеет тенденцию сорбировать на своей поверхности различные соединения, что осложняет его отмывку. Описана [5] методика полного анализа смеси CI- — Вг-—I-.
В присутствии хлорида и бромида иодид можно селективно осадить в виде соли палладия.
Титриметрические методы
Меркуриметрнческий метод определения хлорида и бромида неприменим для анализа иодида из-за сравнительно низкой растворимости иодида ртути(II). Сначала образуется комплексный тетраиодид ртути(II):
4Г + Hg!+ «=> Hgl?
Затем осаждается красный иодид ртути (II):
Hg2+ + Hgir 2HgI2
За счет того, что тетраиодид диссоциирует непосредственно вблизи конечной точки титрования и это происходит слишком быстро» конечная точка титрования и точка эквивалентности не совпадают*
Hglf Hgl2 4- 21*
384
Аргентометрические методы
Иодиды можно определять методом Фольгарда, используя методику, описанную в разделе «Хлорид». Титрование по Мору непригодно для определения иодида, так как хромат сорбируется осадком, что затрудняет индикацию точки эквивалентности.
Для титрования иодидов можно использовать метод адсорбционных индикаторов. Иодид нельзя титровать в присутствии хлорида и бромида, так как эти ионы сорбируются поверхностью осадка, что приводит к завышению результатов определения иодидов- Декстрин, который обычно рекомендуют в качестве защитного коллоида в аналогичном методе определения хлоридов, для определения иодида непригоден. Был исследован [6] ряд поверхностно-активных соединений для использования в этом методе. Как было найдено, гидроксипропилметилцеллюлоза позволяет получить наиболее четкие конечные точки титрования при определении всех трех галогенидов (хлорида, бромида и иодида), для определения нодида лучшим индикатором является эозин. Исследователи подчеркнули, что невозможно титриметрически определять три галогенида в смеси.
В качестве индикаторов для аргентометрического определения иодидов были рекомендованы также родизонат калия [7], ксиленоловый оранжевый [8] и п-этоксихризоидин [9]. При использовании ксиленолового оранжевого можно определять иодид в присутствии 104-кратного избытка хлорида, n-этоксихризоидин в присутствии избытка Ag+ сорбируется в виде нейтральных молекул, которые окрашивают осадок в желтый цвет.
Комплексометрический метод
Разработано несколько комплексометрических методик определения иодида в присутствии бромида и хлорида. Они описаны в разделе «Хлорид».
Опубликован метод [10] титрования иодида комплексом Ртуть(Ц)—ЭДТА в присутствии комплекса Hg(II) — метилтимоловый синий в качестве индикатора. Определение основано на образовании комплекса состава (1:1) комплексоната ртути и иодида (Цианид и роданид образуют аналогичные комплексы). Иодид ОпРеделяют по градуировочному графику, который линеен при содержании иодида 0,3—32 мг.
Редокс-методы
Л Иодид можно определять, окисляя его до иода, 1+ или йодата, этих целей рекомендовано несколько реагентов.
Деление д0 иода' В кислой среде проходит реакция с ни-
2NO24-2I4-4H+ —> 2NO + 12 + 2Н2О
13 Зак 167
385
Выделяющийся иод титруют стандартным раствором тиосульфата. Избыток нитрита и оксиды азота удаляют введением карбамида. Карбамид можно вводить в подкисленный раствор перед добавлением нитрита, так как реакция между карбамидом и нитритом или оксидами азота проходит медленно, в то время как иодид быстро реагирует с нитритом. Хлорид не мешает определению, если присутствует в равных или больших по сравнению с иодидом концентрациях. Метод, по-видимому, не нашел широкого применения.
Титриметрическое определение иодида окислением его до иода [11]
Ход анализа. В коническую колбу из темного стекла вместимостью 250 мл помещают 25 мл анализируемого раствора иодида, содержащего 0,15—0,65 г иоднда, добавляют 1 г карбамида, 5 мл 0,5 М раствора нитрита натрия и 5 мл 2 М H2SO4. Раствор закрывают и оставляют на 10 мнн, изредка встряхивая. Охлаждают в ледяной бане, добавляют 1—2 г KI для растворения образующегося иода и титруют стандартным раствором тиосульфата, добавляя вблизи конечной точки титрования крахмал.
Окисление до 1+. Окисление иодида йодатом калия до монохлорида часто используют для определения иодида. Применение йодата калия в титровании [12, 13] основано на восстановлении его до монохлорида по реакции
ю; + 6Н+ + 4е~ —> 1*+ЗН2О
Используя эту реакцию, можно определять ряд соединений. При определении иодида проходит следующая реакция:
Юз + 2Г + 6Н++ ЗСГ —> 31С1 + ЗН2О состоящая из двух стадий:
Юз + 5Г + 6Н+ —> 312 + ЗН2О
ю; + 21а + 5СГ + 6Н* —> 5IC1 + ЗН2О
Последнее уравнение упрощено, монохлориодид превращается в ICG по уравнению
ICl + СГ 1С12-
Для этой реакции требуется сравнительно высокая концентрация кислоты, метод селективен в присутствии хлорид-иона [14]. При титровании кислотность должна быть не ниже 3 М по НС1. ДлЯ нахождения конечной точки титрования раствор встряхивают с несколькими миллилитрами четыреххлористого углерода. СвобоД' ный иод, образующийся на первой стадии реакции, окрашивает органическую фазу в фиолетовый цвет. Конечную точку титров3 ния замечают по исчезновению следов фиолетовой окраски орг3 нической фазы, что указывает на полное превращение иода в м нохлорид иода. Бромид не мешает в концентрации, меньше 30 в 100 мл. Влияние больших концентраций бромида можно устр I нить [15] добавлением большого избытка бромида калия.
386
При титровании других восстановителей, например мышьяка (Ш), сурьмы(Ш) и S2Of, использовали красители (для практического использования рекомендовали амарант и п-этоксихри-зоидин). В присутствии цианида иодид количественно окисляется до цианиода:
IOZ + 2Г + 3HCN + ЗН+ —> 3ICN + ЗН2О
Этот вариант метода обладает тем преимуществом, что реакцию можно проводить в менее кислых растворах; очень большим недостатком метода является чрезвычайная токсичность цианистоводородной кислоты.
Титриметрическое определение иода с помощью
йодата калия [12]
Ход анализа. В коническую колбу из темного стекла емкостью 250 мл помещают 25 мл анализируемого раствора, содержащего около 0,2 г I-, добавляют 60 мл концентрированной НС1 и 5 мл четыреххлористого углерода. Титруют 0,025 М раствором К1О3 до тех пор, пока раствор, сначала интенсивно окрашенный иодом, не станет светло-коричневым. Титрование прекращают н встряхивают колбу. Слой четыреххлористого углерода становится за счет экстракции иода фиолетовым. Продолжают титрование, встряхивая раствор после прибавления очередной порции титранта, до тех пор, пока не исчезнут последние следы фиолетовой окраски в органической фазе.
1 мл 0,025 М раствора КЮ3 эквивалентен 0,006345 г I-.
Окисление до йодата. Одним из лучших методов определения небольших количеств иодида (около 1 мг) является метод, основанный на количественном окислении иодида до йодата бромной водой:
Г + ЗВг2 + ЗН2О —> 10з+6Вг' + 6Н+
Избыток брома разрушают муравьиной кислотой:
Вг2 + НСООН —> СО2 + 2Н+ + 2Вг”
Добавляют избыток KI и выделившийся иод титруют стандартным раствором тиосульфата:
1О3" + 5Г + 6Н+ —♦ 312 + ЗН2О
I2 + 2S2or —> 21" + S4Og"
Из уравнений видно, что превращение иодида в свободный иод сопровождается 6-кратным увеличением его содержания [16—18). Наряду с очень удачным эффектом умножения, этот метод обладает еще и тем преимуществом, что бромид и хлорид не мешают °пределению, если их содержание не очень велико, так же как и нитрат, сульфат и аммоний. Метод широко применяют в органическом микроанализе галогенсодержащих соединений и в анализе алкоксигрупп.
Титриметрическое определение иодида по методу Лайперта [16, 17]
сОд ХоД анализа. В коническую колбу помещают 25 мл анализируемого раствора, вед РЖащего до 2 мг иодида, разбавляют раствор водой до 100 мл и добавляют °лько капель метилового оранжевого. Нейтрализуют 1 М H2SO4 и добав
ляют еще 2 мл избытка кислоты. Добавляют насыщенную бро.миую воду до тех пор, пока раствор при перемешивании не останется желтым. Затем раствор кипятят до исчезновения желтой окраски и еще 2 мин после этого. Охлаждают, отмывают стенкн колбы водой, добавляют 2 мл 10%-ного раствора KI а титруют свободный нод 0,02 М стандартным раствором тиосульфата из бюретки емкостью 10 мл, добавтяя в качестве индикатора крахмал.
Примечания. 1. Бромид в 20-кратном избытке не мешает определению.
2. В растворе может присутствовать до 5 г хлорида.
3. Большие концентрации электролитов снижают чувствительность определения конечной точки титрования с крахмалом. Вместо крахмала в тех же условиях рекомендуется применять малахитовый зеленый.
Метод с использованием оксицианида ртути
Метод с использованием оксицианида ртути (метод Вибека), разработанный для определения хлорида и бромида, может быть применен и для определения иодида, но, по-видимому, по сравнению с другими методами не обладает никакими преимуществами. Подробно метод описан в разделе «Хлориды».
Спектроскопические методы
Спектрофотометрические методы в видимой
и ультрафиолетовой областях
В монографии Больтца [19] приведен обзор спектрофотометрических методов определения иодида до 1951 г. Многие спектрофотометрические методы определения хлоридов и бромидов можно использовать и для определения иодидов. К ним относятся следующие методы.
1. Меркуриметрический роданидный! метод. Сравнимый с ним метод [20], основанный на применении роданида серебра, также применим для определения бромида и иодида.
2. Метод с применением комплексов ртути с дифенилкарбазо-ном и дифенилкарбазидом.
3. Метод, основанный на вытеснении галогенидом HCN из цианида ртути.
4. Метод с использованием комплексоната ртути(II) (для бромида).
5. Метод, основанный на образовании галогенидов фенилртути и их взаимодействии с диэтилдитиокарбаминатом натрия.
6. Методы с использованием трифенилметановых красителей (для бромида).
В методе 4 иодид образует комплекс с комплексонатом ртУ' ти(П) при pH = 5,0—8,3. Светопоглощение измеряют при 260 нМ> область определения иодида 0,05—2,0 мг. Метод с применение* трифенилметановых красителей описан в серии статей [21-Бромид и иодид реагируют с этим красителями в присут хлорамина Б, хлорамина Т или гипохлорита натрия. Бриллиант
383
вый зеленый, кислый фиолетовый С, кристаллический фиолетовый и малахитовый зеленый применимы для определения иодида (и бромида). Чувствительность определения иодида около 0,01 мкг/мл. При использовании кристаллического фиолетового максимум светопоглощения лежит в области 582 нм.
Медь, магний, кальций, натрий, калий, хлорид и сульфат не мешают определению, тогда как карбонат, гидрокарбонат и нитрат мешают проведению анализа. Механизм этой реакции состоит в окислении иодида или бромида до 1+ или Вг+, которые и вызывают уменьшение интенсивности окраски основных красителей.
Были использованы также малахитовый зеленый, бриллиантовый зеленый и основной бирюзовый в присутствии хлорамина Б, в этом случае реакция была основана на окислении иодида до 1з.
Взаимодействие 1з с катионом красителя приводит к образованию ионного ассоциата, экстрагирующегося бензолом. Для трех упомянутых красителей закон Бера соблюдается в интервале 0,032—5,54, 0,031—1,52 и 0,023—2,54 мкг/мл I-. Не мешают определению большие концентрации хлорида, сульфата, нитрата, фосфата, Са2+, Си2+, Mn2+, Pb2+, Zn2+, Со2+, А13+, ацетата и 10-кратный избыток Fe3+. Мешают бромид, сульфид и бихромат [24].
Многие спектрофотометрические методы определения иодида основаны на его предварительном окислении. В качестве окислителей используют бром, азотную кислоту и Fe3+. Образующийся иод можно определять, используя крахмал, хотя а-нафтофлавон более чувствителен 125]. Можно экстрагировать иод хлороформом и измерять светопоглощение органической фазы. Так как экстра-гируемость иода зависит от присутствия в водном растворе галогенидов, которые образуют с иодом комплексные анионы, градуировочный график следует строить в тех же условиях, в каких проводят определение [26].
Можно применять кроме прямых методов окисления до иода реакции окисления до йодата (см. титриметрические методы). После разрушения избытка окислителя иодат реагирует в кислых растворах с KI с образованием свободного иода.
Комплекс Hg"—ЭДТА используют для спектрофотометрического определения бромида и иодида; кроме того, было найдено [27], что иодид реагирует с комплексом Hg”—1,2-диаминоцикло-гексан—тетрауксусная кислота. Иодидный комплекс образуется за 10 мин при pH = 8,9—9,8, светопоглощение измеряют при 255 нм. Этим методом можно определять 0,03—1,5 мг иодида. Определению мешают роданид, цианид, аммоний, серебро, медь, сульфид, тио-сУльфат, железо и ртуть. При концентрации, превышающей 15 мкг 6 50 мл, бромид мешает определению.
Многие спектрофотометрические методы определения галогени-Д°в основаны на вытеснении окрашенных соединений из их комплексов с металлами. Вытесненный из дитизоната ртути дитизон Экстрагируют толуолом, измеряют оптическую плотность экстракта пРи 620 нм. Этот вариант метода пригоден для определения
гзэ
2—25 мкг иодида в 10 мл раствора. В другом методе экстрагируют иодид ртути, а оставшуюся ртуть определяют с помощью дитизона.
С использованием хлорида палладия можно определять 2—8 мкмоль I- в присутствии 1000-кратного избытка хлорида и бромида [29]. Этот метод не нов, он был изучен Мартенсом [30]. Реакция между иодидом (бромидом) и сульфатом палладия в водных растворах была исследована Чапменом и Шервудом [31], которые рекомендовали ее для определения иодида и бромида. Можно определять 1,0—6,0 мкг/мл иодида; оптимальная длина - волны 390 нм.
Эмиссионный спектральный метод
Для иодида (бромида и хлорида) характерно образование интенсивных молекулярных эмиссионных полос в присутствии индия в холодном водородно-азотном диффузионном пламени [32]. Метод рассмотрен подробно в разделе «Хлориды».
Флуориметрический метод
Для флуориметрического определения 20 мкг иодида используют ацетат уранила [33]. Флуоресценцию измеряют при 520 нм, длина возбуждающего излучения света 365 нм. На определение 10 мкг иодида не влияет 10 мг следующих ионов: СГ, ВгОз, NO3', SOt, СЮз, СЮ;, Sr2*, Mg2+, Ва2+, Са2+, Ni2*, Zn2*, Cd2+, NH4*. К*, Na+ и СНзСОО'. На определение 10 мкг иодида оказывают влияние следующие ионы (в скобках указано содержание в мкг): Вг’(150), 104(110), F"(80), NOa(60), IO; (50), оксалат(40), SCN’(2), Al3* (20), WOt, Мп2+ и Со2* (2000), РЬ2*(500), Си2* (400) и МоО|"(200). Другой метод основан на реакции иодида с 2',7'-бис(ацетоксимеркури) флуоресцеина [34]. Флуоресценция реагента уменьшается за счет образования иодидного комплекса ртути. Этим методом можно определять микрограммо-вые количества иодида и цианида, концентрации хлорида и роданида больше 0,005 М мешают определению, бромид мешает в еще меньших концентрациях.
Атомно-абсорбционный метод
Киркбрайт, Вест и Вильсон [35] определяли иодид по резонансной линии 183,0 нм в пламени оксид азота(1)—ацетилен. Предел определения— 12 ppm для йодата, иодида или перйодата. Разработан косвенный ААС-метод определения иодида [36], сравнимый по характеристикам с упомянутым методом. Экстрarnpyi°T ионный ассоциат иодида и 1,10-фенантролина или его производного и измеряют поглощение экстракта. Еще один ААС-метоД 390
основан на экстракции комплекса бис(1,10-фенантролинат) кадмия (II) —иодид нитробензолом и определении кадмия, используют полосу кадмия 228,8 нм.
Электрохимические методы
Потенциометрические методы, ионоселективные электроды
Метод аргентометрического и меркуриметрического потенциометрического титрования иодида аналогичен таковым для бромида или хлорида, в этом разделе мы будем на них ссылаться. Суммарное содержание галогенидов в смеси определяют сравнительно просто. Определение каждого галогенида в смеси с другими значительно осложняется из-за образования смешанных кристаллов. Одним из путей решения задачи при анализе бинарных смесей является отделение бромида или иодида из второй аликвотной порции раствора и определение оставшегося хлорида [37].
Можно определять микроконцентрации иодидов на фоне 2'К^-кратного избытка хлорида методом потенциометрии до нулевой точки. На анализ 5—100 ppm иодидов требуется 3 мин. Применяют электроды серебро—иодид серебра и платина [38].
Для определения субмикроколичеств бромида и хлорида применяют дифференциальную электролитическую потенциометрию (ДЭП) в сочетании с кулонометрией при постоянном токе. Этот метод непригоден для определения иодида, так как выделяющаяся на генерирующем аноде пленка иодида легко разлагается.
Раньше было отмечено, что благодаря более низкому, чем у бромида и хлорида, редокс-потенциалу иодида его можно селективно окислять в присутствии этих ионов. Этот прием используют в визуальной редокс-титриметрии, например, в методе Лайперта.
Для редокс-определения иодида предложено большое число окислителей. Использовали [39] пероксомолибдат натрия Na2MoOs, синтезированный окислением раствора молибдата натрия в разбавленной серной кислоте Н2О2. Хлорит можно использовать для точного определения иодида в присутствии бромида и хлорида [40]. Максимальный скачок потенциала, как было найдено, удается получить в 5,5 М растворе НС1.
Сильные окислительные свойства кобальта(III) делают его потенциальным титрантом в редокс-методах. Основным препятствием Для более широкого использования Со"1 является относительная Нестабильность его водных растворов [41, 42]. Будезинский с сотр. [43] описали электролитический метод синтеза ацетата кобаль-Та(1П), который позволяет получать сравнительно стабильные Растворы. Этот реагент был применен для анализа некоторых неорганических восстановителей [44], в частности, иодида. Процесс окисления иодида стехиометричен в 1,5—2,5 М H2SO4 в отличие °т процесса с использованием 1+ в 4 М НС1. Хотя окисление про-°Дит значительно быстрее в 2,5 М H2SO4, на титрование затрачи-ается 20—25 мин. Ошибка 15 параллельных определений 150 мг
391
иодида, как было найдено, составляет 4-0,36%. Не мешают определению 10-кратный избыток бромида и хлорида, большие их концентрации несколько снижают скачок потенциала в конечной точке титрования. Это происходит за счет образования смешанных га-логенидных соединений бромида иода или монохлорида иода.
Общие вопросы определения галогенидов с ионоселективными электродами рассматриваются в разделе «Хлориды». Иодидный ионоселективный электрод, разработанный в 1961 г„ характеризуется лучшей селективностью по сравнению с другими анионоселективными электродами [45]. Пунгором с сотрудниками было проведено подробное изучение твердых иодидных электродов [46, 47J, их свойства оказались сравнимыми со свойствами аналогичных иодидных электродов [48J. Константы селективности электрода Пунгора ОР-1-711 показаны ниже:
Ион . . CN' Вг SCN’ [Fe(CN)J‘- SO’f СГ
К . . 1 1,8- 10-4 3- 10“’ 3,5 • 10““ 5,5-10“’ 3,7- 10“’
Ион . SO?- 4 ОН' AsO]' SrOf РО]' no;
К . . . 1 io-’ 9,1 • 10“9 2,6- 10“10 6,6- кг" 2,5-10““ < 10“8
Низкие значения констант селективности свидетельствуют о том, что с помощью ионоселективного электрода иодид можно определять в присутствии больших концентраций всех приведенных ионов [49].
Наряду с выпуском пунгоровских гетерогенных электродов осадочного типа налажен промышленный выпуск монокристаллических иодидных электродов типа Орион 94-53. Как было найдено, электродная функция этого типа электродов линейна в области концентраций иодида 10—10“7 М [50]. Определению иодида не мешают 1000-кратные концентрации фторида, хлорида и бромида; цианид и сульфид мешают определению.
Опубликованы результаты практического использования иоднд-ного электрода. Определение иодида в продуктах питания и растениях с иодидным электродом значительно экспресснее стандартного метода его определения [51]. Электрод использовали для анализа органических и биологических объектов для определения 10-7—10-8 г/мл иодидов [52]. Высокая селективность иодиД-ного электрода позволяет использовать его для прямого определения иодида в минеральных водах [53]. Интересной областью использования электрода является прямое определение иодида в присутствии йодата [54].
Кулонометрические методы
Йодиды, так же как бромиды и хлориды, можно определять кулонометрическим методом. Чаще всего титруют электрогенерИ' рованным ионом серебра. Можно определять каждый из галогенидов, кроме фторида, в смеси. Методика описана в разделе «Хлорид».
392
Для титрования микроконцетраций иодида использовали элек-трогенерированный бром, который окисляет иодид до йодата [55]. Ошибка определения 50 мкг иодида меньше 1%. Хлорид не мешает определению.
Амперометрические методы
С применением амперометрической аргентометрии можно определять иодид, хлорид и бромид в присутствии цианида и цианата [56]. Мешающее влияние цианида предварительно устраняют введением формальдегида, избыток формальдегида и OCN- разрушают добавлением HNO3 до pH = 3. Для индикации точки эквивалентности используют платиновый электрод.
Область определения галогенидов — 0,1—10 мМ.
Милуанова и Сонгина [57] разработали амперометрический метод определения иодида с использованием К2СГ2О7. Титрование проводят в 2 М H2SO4. Не мешают определению 1000-кратный избыток хлорида и 2000-кратный избыток бромида. Относительная ошибка определения 2—3 мг иодида ниже 0,4%.
Полярографические методы
На вращающемся графитовом электроде можно определять [58] иодид в присутствии 100-кратного избытка бромида и хлорида. Плиска [59] с помощью полярографического метода изучал окисление иодида в присутствии избытка хлорида и бромида. При этом наблюдается лишь одна волна окисления иодида до иода.
Кинетические методы
В 1934 г. Кольтгоф и Сэндел опубликовали [60] кинетический метод определения 0,01—1 мкг иодида, основанный на его каталитическом действии на реакцию церий(IV)—мышьяк(III). После этого в течение нескольких лет длился инкубационный период, после которого кинетические и каталиметрические методы стали вновь интенсивно развиваться и в настоящее время заняли значительное место среди методов анализа следовых концентраций ионов. Что касается анализа иодидов, то каталиметрические методы особенно важны, так как способны обеспечить определение ультрамалых его концентраций в присутствии различных ионов. Высокая чувствительность метода позволяет проводить непосредственное определение иодида, минуя длительную стадию его предварительного концентрирования.
Кроме системы церий(ГУ)—мышьяк(Ш) исследовали и другие системы. Был найден 61 каталитический метод, обладающий существенными преимуществами. Наиболее удачные методы приведены в табл. 23.
Ь Уместно прокомментировать некоторые из методов, представленных в таблице. Например, метод Тороповой и Тамаренко [71]
393
селективен по отношению к хлоридам и бромидам: определению иодида не мешают 100-кратный избыток бромида и 1000-кратный избыток хлорида.
Некоторые методы основаны на разложении монороданидных комплексов (см. табл. 23) и [69, 70). Было найдено, что с применением этого метода [80] можно анализировать растворы, содержащие 0,001—10 ppm иодидов, практически не изменяя методики. Более концентрированные растворы разбавляют до требуемой концентрации иодидов. Метод, основанный на реакции це-рий(1У)—мышьяк(Ш), рекомендован в качестве стандартного метода анализа воды [81]. Разработан автоматический вариант этого метода. Для определения концентрации иодида регистрируют скорость реакции в интервале 10—100 с [63]. Для анализа образцов речной воды можно применить автоматический анализатор [82]. Ошибка определения 20—100 мкг/л иодида составляет по 6 параллельным определениям ±0,36%.
Очень чувствительный каталитический метод основан на реакции, описанной Файглем и Юнграйсом [83]. В реакции используют каталитическое влияние иода или иодида на реакцию хлорамина Т с ацетатом М,М-тетраметилдиаминодифенилметана (тетра). Хлорамин Т в воде гидролизуется с образованием гипохлорита, концентрация которого недостаточна для окисления тетра до голубого хиноидного производного. Очень малые концентрации I-/1° катализируют реакцию. Влияние иодида объясняется превращением его в свободный иод, который восстанавливается до иодида при образовании голубого производного реактива:
[НзС——SO2NClj ±Н2О НзС—^2y~S°2NH2 ± С1(Г
С1О' + 2Г±2Н+ —> СГ + Н2О±21°
(ch3)2n ——СНг——к(СНз)2 + 21°
Н
(СН3)2 N
Разработан метод определения иодида в питьевой воде [84], в дальнейшем усовершенствованный Раманаускасом [85]. Нижний предел определяемых концентраций иодида — 0,04 мкг/л. Бромид при содержании ниже 0,5 мг/л и хлорнд не мешают определению-Интенсивность голубой окраски достигает максимума в течение 40—50 с и затем уменьшается, поэтому измерение оптической плотности следует проводить через точные промежутки времени после начала реакции.
Используют и энзимные каталитические реакции [79], которые основаны на следующих реакциях. Присутствие некоторых ионов металлов, таких, например, как Ag+ и Hg2+, ингибирует каталитическое действие энзима fj-фруктофураноксидазы на гидролиз не-
S94
Таблица 23. Кинетические методы определения иодида
Реакция Метод * Способ измерения Чувствительность Литература
2Се° + AsO3' + Ф. к. Визуальный 5 • ,0-8 60
+ Н2О —> 2Се3++ AsO3" + Ф. в. Спектрофотометрический 1•10~8 62
+ 2Н+ Автоматический То же I • 10~8 63
Н. с. > 1 • 10“я 64
О. к. Визуальный 1 • 10", 65
Ф. в. Спектрофотометрический 1 • 10 66
Н. с. То же 1 • ю-9 67
Амперометрический 1 • 10 68
2NO2 + 2SCN* + Н. с. Спектрофотомет- 1 • 10"9 69
+ 4Н* —> 2NO + (SCH)2 + + 2Н2О рический То же » 9 1 • 10", 2 • 10, 70 73
2Мп3* + AsO3’ + + Н2О —> 2Мп2+ + AsO3" + Ф. к. » 2- 10 74
4-2Н*
2Се4+ + Sb3+ —> 2Се3+ + Sb5+ О. к. Визуальный 1-10 65
3AsO3" + ю; —> 3AsOf + r Ф. к. Амперометриче- 1 • Ю"9 71
ский
3,3'-Диметилнафтндин + H2O2 О. к. Визуальный 1 • ю“ ° 72
Ф. к. Спектрофотометрический 2- 10 75
п,п-Тетраметилдиаминодифенил-метан + хлорамин Т Н. с. То же 1-10"’4 76
2NO" + 2S2O2" + + 4Н+ —> 2NO + S4O2" + + 2Н2О Визуальный 77
Замещение лнганда в комплексе Hg11 — ПАР на 1,2-циклогексан-Днамнн N.N.N'.N'-TeTpayKcycHyio Ф. в. 1 • 10"9 78
кислоту Деингибироваиие реакций, ката- Ф. в. Поляриметриче- 79
лизируемых энзимами ский
* Примечание. Ф. к.—метод фиксированных концентраций, Ф. в. — метод фиксированного времени, Н. с. — метод начальных скоростей, О. к. метод одновременного компарнро-вания.
которых веществ, например, сахарозы. Малые концентрации некоторых ионов, которые образуют очень прочные комплексы с Ag+ и Hg2+, могут вытеснять энзимы из их комплексов с металлами. Такие анионы, как цианид, сульфид и иодид, снижают ингибиторное действие металлов.
Знание механизма каталитических реакций необходимо для Разработки новых кинетических методов. Опубликован обзор
395
работ по определению механизма реакций [86]. Одна из наиболее широко применяющихся каталитических реакций, а именно взаимодействие церия (IV) с мышьяком (III), проходит по следующим стадиям:
Г + CeIV —> Се,п + 1°
1° + As111 —> промежуточный комплекс
Промежуточный комплекс 4-Celv —> Се111 4- Asv + I"
Радиохимические методы
Нейтронно-активационный метод, как было найдено, может быть применен для определения иодида и других галогенидов. Иодид и бромид можно определять по их радиоактивности после проведения селективной экстракции каждого галогенида [87]. Метод позволяет определять 0,01 мкг иодида и 0,1 мкг бромида. Морган, Блэк и Митчел [88] использовали нейтронную активацию для определения стабильного неорганического иодида в жидкостях. Осложняющим фактором является то, что кроме 1281 при активации тепловыми нейтронами образуются другие у-излучаю-щие изотопы. В этом исследовании [88] после облучения отделяли иодид на ионообменной колонке. Метод позволяет определять 20 мкг иодида в 100 мл.
При анализе морской воды после нейтронной актив'ации для выделения иодида применяли сильноосновные ионообменные смолы [89]. Элюируемый иодид концентрировали соосаждением в виде иодида палладия(II) в присутствии избытка палладия(II) на металлическом палладии. Образцы облучали в течение 5 мин нейтронным потоком плотностью 4-Ю12 нейтрон-см-2-с-1. Можно определять до 0,006 мкмоль I-.
Опубликован метод изотопного обмена для определения иодида. Рихтер [90] вводил известное количество ,3Ч в анализируемый образец, окислял иодид до иода и экстрагировал последний бензолом. Активный иод распределялся между двумя фазами пропорционально содержанию иода и иодида в каждой фазе. Метод был применен для анализа природной воды; чувствительность определения 1 мкг/л иодида.
Разработан метод определения 25—550 нг иодида [91], основанный на реакции
CH3I + Na,3lI СНз l3,I + Nal
При использовании экстракционного радиохимического титрования хлоридом 203Hg можно определять 12—2000 нг I- [92].
Газохроматографические методы
Этим методом можно отделять и определять малые количества галогенидов, включая и иодид. Подробно метод описан в разделах «Хлориды» и «Бромиды».
396
Другие методы определения
Некоторые методы не соответствуют принятым в этом разделе рубрикациям. Часть из них применяют также для анализа хлорида и бромида и описана в разделах, посвященных этим галогенидам. К ним относятся методы полуколпчественного анализа: метод кольцевой бани [93] и циркуляционная ТСХ [94]. К таким методам принадлежит и бумажная хроматография, использующаяся для макроопределения хлорида, роданида и иодида в смеси [95]. После разделения пятна проявляют, затем выделяют и взвешивают. Авторы работы [95] нашли, что с ошибкой ±1,5% можно определять 15—100 мкг 1~
ЛИТЕРАТУРА
1. West Р. W, Lorica A. S. — Analytica Chim. Acta, 1961, v. 25, p. 28.
2. Naeumann R. — J Chromat., 1965, v. 18, p. 385.
3. Obrenovic-Paligoric I. D, Covoric J. D. — Bull. Boris Kidrich Inst. nucl. Sci , 1963, v. 4, p. 95; Chem. Abstr., 1963, v 59, p. 14555b.
4. Bekk J. — Chemiker Zeitung, 1915, v. 39, p. 405; Chem. Abstr., 1915, v. 9, p. 2042.
5. Firsching F H. — Analyt. Chem., 1960, v. 32, p. 1876.
6. Kapel M., Fry J. C., Shelton D. R. — Analyst, 1975, v. 100, p 570.
7. Mehlig J. P. — Chemist Analyst, 1955, v. 44, p. 87.
& Pribil R., Markova V. — Chemike Listy, 1966, v. 60, p. 89; Analyt. Abstr., 1967, v. 14, p. 2544.
9. Schulek E., Pungor E. — Analytica chim. Acta, 1950, v. 4, p. 109, 213.
10. Nomura T. — Bull. Chem. Soc. Japan, 1969, v. 42, p. 952; Analyt. Abstr., 1970, v. 19, p. 218.
11. Abeledo C. A., Kolthoff 1 M. — J. Am. chem. Soc., 1931, v. 53, p. 2893
12. Andrews L. W. — J. Am. chem. Soc., 1903, v. 25, p. 756.
13. Jamieson G. S. — Volumetric Iodate Methods, New York, Reinhold, 1926
14. Kipling J. J., Grimes G.— Taianta, 1960, v 5, p 278.
15. Mutschin A.— Z. analyt. Chem., 1936, Bd. 106, S. 1.
16. Leipert T. — Mikrochem. Pregl. Festschr., 1929, p. 266.
17. Leipert T.—Mikrochim. Acta, 1938, v. 3, p. 73, 147.
18. Belcher R. — Taianta, 1968, v. 15, p. 357.
19. Boltz D. F. (Ed.) Colorimetric Determination of Nono-metals, New York, Interscience, 1958.
20. Utsumi S. — J. chem. Soc. Japan, 1953, v. 74, p 35; Chem. Abstr., 1953, v. 47, p. 6815f.
21. Буникине Л. В., Раманаускас E. И. ЖАХ, 1968, t. 23, c. 1364. Analyt. Abstr., 1970, t. 18, c. 2396.
22 Раманаускас E. И., Буникине Л. В., ^Киленайте M. Научные Труды высших Учебных заведений Лит. ССР, Химия и хим. техиол. 1968, с. 29. Analyt
_ Abstr., 1970, т. 18, с. 3962.
Буникине Л. В., Раманаускас Е. И., Тамульявичуте М., Научн. Труды высших учебных заведений Лит. ССР, Химия и хим. технол. 1970, с. 41. Analyt.
, Abstr., 1972, t. 22, с. 1568.
*4. Невердаускине 3., Раманаускас Е., Буникине Л. Научные Труды высших
I Учебных заведений Лит. ССР, Химия и хим. технол., 1972, с. 25. Analyt. Abstr 1974, т. 26, с. 2644.
й- Lambert J. F — Analyt. Chem., 1951, v. 23, p 1247, 1251.
to ^,Venslow T. C., Rees W. T.— Analytica di m Acta, 1951, v. 5, p. 123.
Noniura T. — J. chim. Soc Japan, pure Chem. Sect., 1967, v. 88, p. 199; Analyt.
Abstr., 1968 ,v. 15, p 2619.
397
28. Agterdenbos J., Juette В. A. H. G., Elberse P. .4. — Talanta, 1970, v. 17, p. 1085.
29. Novak J., Slama I. — Colin Czech, chem. Common. Engl. Edn., 1972, v. 37, p. 2907, Analyt. Abstr., 1973, v. 24, p. 2174.
30. Martens F. F.— Verb, dt phys. Ges., 1930, v. 4. p. 138
31. Chapman F. W., Sherwood R. M.—Analyt. Chem., 1957, v. 29, p. 172.
32. Dagnall R. M., Thompson К- C., West T. S. — Analyst, 1969, v. 94, p. 643.
33. Britton D. A., Guyon J. C. — Mikrochem. J., 1969, v 14, p. 1
34. Colvos G., Haro At , Freiser H. — Talanta, 1970, v. 17, p. 273.
35. Kirkbright G. F., West T. S„ Wilson P. I. — Atom. Abs. Newsl, 1972, v. 11, p. 53.
36. Yamamoto K, Kumamura T., Hayashi Y., Otani K.— Japan Analyst, 1968, v. 17, p. 92; Analyt. Abstr., 1969, v. 17, p 2101.
37. Prokopov T. S. — Analyt. Chem., 1970, v. 42, p. 1096.
38. Malmstadt H. V., Winefordner J. D.— Analytica chim. Acta, 1961. v. 24, p. 91.
39. Kotkowski S., Lassocinska A. — Chemia analit., 1966, v. 11, p. 789, Analyt. Abstr., 1967, v. 14, p. 6774.
40. Minczewski J., Glabisz U, — Acta chim. hung., 1962, v. 32, p. 133; Analyt. Abstr., 1963, v. 10, p. 1424.
41. Bricker С. E., Loefler L J. — Analyt. Chem., 1955, v. 27, p. 1419.
42. Minczewski J., Pszonika M.— Chemia analit., 1964, v. 9, p. 785, 947.
43 Budesinsky J., Dolezal J., Sramkova B., Zyka J. — Microchem. J., 1971, v. 16, p. 121.
44. Hanif M., Dolezal J., Zyka J. — Microchem. J., 1971, v. 16, p. 291.
45. Pungor E., Hollos-Rokosinyi E. — Acta chim. hung., 1961, v. 27, p. 63; Analyt. Abstr., 1961, v. 8, p. 3561.
46. Pungor E., Havas J., Toth K-— Acta chim. hung., 1964, v. 41, p. 239.
47. Pungor E., Havas J., Toth K. — Instrums Control Syst., 1965, v. 38, p. 105; Analyt. Abstr, 1966, v. 13, p. 7236.
48. Rechnitz G. A., Kresz M. R., Zamochnick S. B. — Analyt. Chem, 1966, v. 38, p. 973.
49. Pungor E., Toth К —Analyst, 1970, v. 95, p. 625.
50. Orion Research Incorporated, Technical Information.
51. Hoover W. L., Melton J. R., Howard P. A. — J. Ass. off. analyt. Chem, 1971, v. 54, p. 760.
52. Paletta B., Panzenbeck K. — Clin. chim. Acta, 1969, v. 26, p. 11.
53. Pungor E. — Analyt. Chem, 1967, v. 39, p. 28A.
54. Paletta B. — Mikrochim. Acta, 1969, p. 1210.
55. Berraz G., Delago O. Rev. Facultad. Ing. Quim. Univ. Nac. del Littoral Argentina, 1959, v. 28, p. 53; Analyt. Abstr, 1961, v. 8, p. 3275.
56. Ikeda S, Musha S.— Japan Analyst, 1967, v. 16, p. 445, Analyt. Abstr, 1968, v. 15, p. 6633.
57. Милянова H. M., Сангина О. А. ЖАХ, 1969, т. 24, с. 1794. Analyt. Abstr, 1971, т. 21, с. 187.
58. Dryhurst G.. Elving P. J. — J. electroanal. Chem, 1966, v. 12, p. 416.
59. Pliska V. — Z. analyt. chem, 1961, v. 184, p. 17; Analyt. Abstr, 1962, v. 9, p. 1898.
60. Sandell E. B., Koithoff I. M.—J. Am. chem. Soc, 1934, v. 56, p. 1426.
61. Svehla G. — Proc. Soc. analyt. Chem, 1971, v. 8, p. 80.
62. Stoic К—Mikrochim. Acta, 1961, p. 710.
63. Malmstadt H. V, Hadjiioannou T. P. — Analyt Chem, 1963, v. 35, p. 2157.
64 Lein A., Schwartz N. — Analyt. Chem, 1951, v. 23, p 1507.
65. Bognar J., Sarosi SZ. — Mikrochim. Acta, 1965, p. 1004.
66. Rogina B., Dubravcic M. — Analyst, 1953, v. 78, p. 594.
67. Rodriguez P. A., Pardue H. L.— Analyt. Chem, 1969, v. 41, p. 1376.
68. Czarnecki K. — Chemia analit, 1960, v. 5, p. 875; Analyt. Abstr, 1961, v-p. 2858.
69. Яцимирский К. В., Бударин Л. И., Благовещенская Н. А., Смирнова П-Федорова А. П., Яцимирский В. К. ЖАХ, 1963, т. 18, с, 103. я.
70. Iwasaki I., Ursumi S., Ozawa T. — Bull. chem. Soc. Japan, 1953, v. 26, p I"01 Chem. Abstr, 1953, v 47, p. 12123h.
398
71. Торопова В Ф., Тамарченко Л. М ЖАХ, 1967, т. 22, с. 234.
72- Bognar J., Sarosi SZ. — Mikrochim. Acta. 1969, p. 463.
73. Проскурякова Г. Ф. ЖАХ, 1967, т. 22, с. 802. Analyt. Abstr., 1969, т. 16, с. 136.
74. Bogurth W., Schaeg W. — Mikrochim. Acta, 1967, p 658.
75. Bognar J., Nagy L. — Mikrochim. Acta, 1969, p. 108
76. Ballzo H. — Z. analyt. Chem., 1969, v. 245, p. 20.
77. Baines H.—J. Soc. chem. Ind.. Lond., 1930, v. 49, p. 481.
78 Funahashi S., Tabata M, Tanaka M.— Analytica chim. Acta, 1971, v. 57, p. 311
79. Mealor D„ Townshend A.— Talanta, 1968. v. 15, p. 1477.
gO. Ottaway J. M., Fuller C. №.. Rowston W. B. — Analytica chim. Acta, 1969, v. 45, p. 541.
81. American Public Heath Association, American Waterworks Association and Water Pollution Control Federation, Standard Methods for the Examination of Water and Wastewater, 14th Edition, New York, American Public Health Assoc., 1975. t
82. Truesdale V. W., Smith P. J.—Analyst, 1975, v. 100, p. 111.
83. Feigl F., Jungreis E. — Z. analyt. Chem., 1958, v. 161, p. 87.
84. Jungreis E., Gedalia I. — Mikrochim. Acta, 1960, p. 145.
85. Ramanauskas E. Lietuvos TSR Aukstuju Mokyklu Mokslo Darbai, Chem. ir Chem. Technol., 1964, v. 5, p. 9; Chem Abstr., 1964, v. 61, p. 10450b.
86- Bontchev P. R. — Talanta, 1970, v. 17, p. 499.
87. Ohno S. — Analyst, 1971. v. 96, p. 423.
88. Morgan D. J., Black A., Mitchell G. R. — Analyst, 1969, v. 94, p. 740.
89. Wong G. T. F., Brewer P. G. — Analytica chim. Acta, 1976, v 81, p. 81
90. Richter H. G. — Analyt. Chem., 1966, v. 38, p. 772.
91. Gabrielsson A.-B., Beronius P. — Analytica chim. Acta, 1972, v. 61, p. 123.
92. Juette В. A. H. G., Agterdenbos J., Elberse P. A. — Talanta, 1970, v. 17, p. 1130. 93. Celap M. B., Weisz H. — Mikrochim. Acta, 1962, p. 24.
94. Hashmi M. H., Chughtai N. A. — Mikrochim. Acta, 1968, p. 1040.
95. Ganchev N., Koev K. — Mikrochim. Acta, 1964, p. 87.
ПЕРБРОМАТЫ
Существование пербромат-иона и пербромной кислоты было обнаружено в 1968 г. [1]. Был опубликован [2] метод синтеза граммовых количеств пербромата калия. Так как этот ион сравнительно новый, следует упомянуть о некоторых особенностях его синтеза и свойств. Сначала синтез проводили, окисляя бромат электролитически или дифторксеноном. Ни один из этих методов не пригоден для синтеза больших количеств иона [1]. Затем был найден еще один метод, в котором использовали окисление фтором в щелочных растворах |2]. Водные растворы пербромной кислоты устойчивы до концентрации приблизительно 6 М (55% НВгО4), при более высоких концентрациях кислота разрушается. Пербромная кислота — сильный окислитель, по окислительной способности она занимает промежуточное положение между хлорной и периодной кислотами. Результаты титрования позволяют сделать Вывод о том, что содержащая 7-валентный бром пербромная кислота в отличие от периодной кислоты — сильная одноосновная кислота, как и хлорная.
Щелочные соли пербромата готовят, нейтрализуя кислоту соответствующим раствором гидроксида; КВгО4, отделяют охлажде-нВем нейтрального раствора в бане со льдом, фильтруют и сушат.
399
Сушить можно в вакуум-эксикаторе при комнатной температуре или на воздухе при 110 °C. Бесцветную соль можно перекристаллизовать из горячей воды.Термогравиметрическим методом с использованием дифференциального термического анализа было найдено, что пербромат калия экзотермически разрушается при 275—280 °C до бромата, который в свою очередь при 390—395 °C разрушается до бромида калия.
МЕТОДЫ ОТДЕЛЕНИЯ
При изучении [3] хроматографических методов разделения было найдено.
1. При разделении пербромат переходит с высокими значениями Rf в такие растворители, как этанол — NH4OH или бутанол — пиридин — NH4OH. При использовании бумажной хроматографии наблюдается во время эксперимента восстановление пербромата; его разделение на силикагеле проходит, вероятно, без разложения.
2. Методом ионообменной бумажной хроматографии можно добиться прочной сорбции пербромата, Rf для пербромата очень близки к значениям Rf для перхлората и перйодата.
3. Разделить перхлорат, пербромат и перйодат можно с применением электрофореза на бумаге. Бромат и пербромат легко разделить с помощью ионного обмена.
4. Применяя некоторые хроматографические системы, можно разделить и идентифицировать пербромат в присутствии всех галогенидов, галогенатов и пергалогенатов.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Разработали два редокс-титриметрическнх метода [2]. В первом пербромат восстанавливают в 12 М НВг до бромата. Бромат реагирует с КБ образующийся свободный иод титруют стандартным раствором тиосульфата в присутствии буферного раствора фосфорная кислота — фосфат. Анализируемый раствор разбавляют в 5 раз насыщенной НВг и оставляют на 10 мин в закрытой колбе. Затем его приливают к 2%-ному раствору иодада натрия, содержащему такое количество фосфата натрия, которое необходим0 для нейтрализации кислоты, содержащейся в анализируемом рас-творе. Объем этого раствора составляет 1/10 часть от приливаемого к нему исследуемого раствора. После сливания раствор сразу титруют стандартным раствором тиосульфата. Концентрация НВГ должна быть определенной. Для того чтобы восстановление про* шло количественно за 10 мин, концентрация НВг должна быть не меньше 11,5 М:
ВгОз + 6Г + 6Н+ —> Вг’+ 312 + ЗН2О
40Q
Второй метод заключается в восстановлении бромата до бромида хлоридом олова (И) в 6 М растворе НС1 в присутствии катализатора молибдена(VI). Образующийся бромид окисляется в нейтральном растворе цианида до цианбромида, который затем взаимодействует с иодидом и окисляет его до иода. Иод титруют стандартным раствором тиосульфата. Если в растворе пербромата присутствуют значительные концентрации брома в низких степенях окисления, их можно восстановить разбавленной НВт; образующийся бром отгоняют пропусканием через раствор какого-нибудь инертного газа. В работе [2] для этих целей использовали аргон.
В более поздних работах отмечалось, что оба метода достаточно точны, но очень длительны и сложны [4].
Спектрофотометрические методы
Экстракционно-спектрофотометрический метод определения перхлората [4] с кристаллическим фиолетовым, как было найдено, можно использовать и для определения пербромата. Можно опре-дглить спектрофотометрически (1 — 10) 10-6 М BrO4, измеряя светопоглощение хлорбензольного экстракта при 596 нм; 1000-кратный избыток бромата и бромида не мешают определению [5].
Электрохимические методы
Перхлоратный ионоселективный электрод чувствителен к пербромату и может быть использован для его определения [6]. В титриметрическом варианте этого метода использовали хлорид тетрафениларсония, конечную точку титрования определяли с индикаторным электродом марки Орион 92-81, чувствительным к пербромату в концентрациях от 1£Н до 10-4 М. Чувствительность этого электрода к пергалатам можно расположить в следующем порядке: ВгО^ > Ю4 > С1О^. Коэффициент селективности, соответствующий отношению пербромат: перхлорат, равен 1,4. Определению перхлората с ионоселективным электродом не мешают хлорид, хлорат, бромит, бромат, фторид, сульфат, нитрат и хромат. Эти же ионы не будут мешать определению пербромата [6].
При полярографическом изучении пербромата было обнару-ено, что он подобно перйодату необратимо восстанавливается на ртутном капающем электроде [7]. При pH = 3 восстановление пербромата до бромида проходит в две стадии, как и при восстановлении бромата. В сильнокислых растворах проходит прямое восстановление до бромида. Был разработан полярографический Метод определения пербромата или его смеси с небольшими количествами бромата в среде 0,05 М НС1О4. В присутствии больших концентраций бромата добавляют гидрокарбонат натрия до тех ПоР, пока раствор, содержащий метиловый красный, не окрасится в Желтый цвет. В этих условиях пербромат восстанавливается
401
только до бромата и поэтому можно селективно определять пербромат на фоне бромата. Ошибка определения пербромата в присутствии бромата меньше 2%.
ЛИТЕРАТУРА
1. Appelman Е. Н. —J. Am. chem Soc., 1968, v. 90, р. 1900.
2. Appelman Е. Н. — Inorg. Chem., 1969, v. 8, p. 223.
3. Lederer M., Sinibaldi M. —J. Chromat., 1971, v. 60, p. 275.
4. Brown L. C., Boyd G. E. — Analyt. Chem , 1970, v. 42, p. 291
5. Uchikawa S. — Bull. chem. Soc. Japan, 1967, v. 40, p. 798.
6. Brand J. R., Smith M. L. — Analyt. Chem., 1971, v. 43, p 1105.
7. Jaselskis B., Huston J. L. — Analyt. Chem., 1971, v. 43, p. 581.
ПЕРХЛОРАТЫ
Перхлорат применяют для различных целей, например в производстве взрывчатых веществ и ракетного топлива. Опубликованы обзоры [1, 2] по аналитической химии перхлората.
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение
Перхлорат количественно осаждается хлоридом тетрафениларсония (ТФА). С помощью этого реагента перхлорат можно не только выделять, но и гравиметрически определять. Иодид, перйодат, бромид, хлорат, роданид и перманганат в этих условиях также осаждаются.
Хлорид 2,4,6-трифенилпиридилия, как полагают, имеет некоторые преимущества по сравнению с ТФА [3]. Его химическое поведение аналогично поведению ТФА. Подробно этот реагент будет рассмотрен в связи с гравиметрическим определением перхлората. С использованием ТФА перхлорат выделяли из морской воды на коллекторе — ионном ассоциате ТФА с перренатом [4].
Экстракция
Некоторые экстракционные системы позволяют количественно отделить перхлорат. В некоторых случаях отделение можно сочетать с одновременным спектрофотометрическим определением перхлората. В виде ионного ассоциата с ТФА перхлорат на 100% экстрагируется хлороформом при pH = 2—12 [5J. В такой же степени перхлорат экстрагируется хлороформом с хлоридом тетра-фенилфосфония (ТФФ) при pH = 2—11 [6] и с хлоридом трй-фенилсульфония (ТФС) при pH = 2—12 [7]. В отличие от перхлората перйодат плохо экстрагируется с ТФА и с ТФС. Бромат с ТФФ экстрагируется на 32%, а ТФС на 28%.
Перхлорат на 100% экстрагируется при pH = 5—10 с комплексным ионом Fe (РЬеп)з+ п-бутиронитрилом [8]. Можно прово-
402
дпть спектрофотометрическое измерение оптической плотности раствора, молярный коэффициент поглощения при 510 нм равен 1,08* Ю4. В качестве растворителя можно использовать и нитробензол [9]. Ионный ассоциат Fe(Dip)s* с перхлоратом экстрагируется нитробензолом при pH = 3,5—8,5 [10]. Другие экстракционные системы будут рассмотрены в подразделе, посвященном спектрофотометрическим методам.
Методы бумажной и тонкослойной хроматографии
Для выделения малых концентраций перхлората можно успешно использовать хроматографические методы, подробно представленные в табл. 24.
Таблица 24. Отделение перхлората методами бумажной и тонкослойной хроматографии
. Метод Растворитель Разделяемые иоиы Литература
Бумажная хроматография и ТСХ (силуфол UV 254) Бутанол — пиридин — NH3, 0,880 г/см3 (2:2:2) сю:—сю-—сю;—сю; 11
Бумажная хроматография Изопропанол — Н2О — пиридин — NH3 (0,880 г/см3) (15:2:2:2) сю;—сю;—с г—сю; 12
ТСХ Изопропанол — тетрагнд-рофу ран — NH3 (0,880 г/см3) (50 : 30 : 20) с 1 о;—с ю;—ci '—с i о; 13
ТСХ (А12О3 + G кизельгур 1:1) Бутанол — ацетон — NH3 (0,880 г/см3) — Н2О (8:10:2:1) сю;—сю;—Вго;— —ю;—сю; 14
ТСХ (кукурузный крахмал) Ацетон — 3 М раствор NH3 (7:3) сю;—вго;—ю;— —cio;-f—Вг’—сг—г 4 15
Электрофорез
Электрофорез на ионообменной бумаге использовали [ 16] для Разделения смеси хлорид—гипохлорит—хлорат—перхлорат. Раз-Деление проходит эффективно при pH = 7 в фосфатном буферном растворе на слабоосновной бумаге амберлит-\¥В-2. При использовании силы тока 20 мА на полосе бумаги 2,5 см разделение осуществляется за 60 мин.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Перхлорат сравнительно устойчив. Он не восстанавливается Д° хлорида под действием SO2, но его можно восстановить до Хлорида при использовании более сильного восстановителя, такого,
403
например, как титан (III). Другой способ восстановления до хло
рида — сухое сожжение с безводным сульфитом
натрия, нитритом
натрия или пероксидом натрия.
Гравиметрические методы
Перхлорат количественно осаждается хлоридом тетрафенил — арсония в виде Phen4AsCIO4 [17]. Оптимальные условия осаждения перхлората по этой реакции изучали Гловер и Розе [18]
и нашли, что
полнота осаждения зависит от соотношения
Р’г’4 XsCl
сю;.
Для 0,006 А1 раствора
перхлората в 0,8 А1
растворе
НО выделение будет не полным, если отношение Ph4AsCl : С1О4 меньше 2:1. При соотношении большем 5:1 получаются завышенные результаты. Перхлорат осаждается на 98,8% при соотношении компонентов большем 2: 1 и меньшем 5 . 1 Хлорат и бро-
мид не мешают осаждению, если примесь этих ионов в перхло рате аммония не превышает 0,4%. Осадок сушат при 110сС. Фактор пересчета для перхлората 0,2433.
Позже были изучены новые реагенты хлорид 2,4,6-трнфенил пиридилия (ТФП) и нитрон [19]. ТФП (2%-ный раствор) образует в 0,2 А4 растворе HCI осадки с иодидом, роданидом, ниграгом, перхлоратом, перманганатом, бихроматом, гексацианоферритом (II) и хлоридными комплексами цинка, свинца, кадмия, оло-ва(П), платины(1У) и золота(Ш). Осадки не образуют фторид, бромид, иодат, хлорат, сульфат, оксалат и хлоридный комплекс железа (Ш). Реагент можно использовать для гравиметрического определения 40—160 мг перхлората-
(Свн6)з сБн2о+ + сю; —► (с6н6)3 сБню • сю4
Хлорид тетрафенилфосфония тоже количественно осаждает перхлорат и может быть использован [20] для определения СЮ4 на уровне ppm в присутствии 3000-кратного избытка хлората. Хуттон и Стефен [21] использовали ТФФ для анализа смеси перхлората с нитратом. Сначала ТФФ осаждает перхлорат, который определяют количественно. Оставшийся в фильтрате нитрат осаждают М-(4-хлорбензол)-1-нафтилметиламином и определяют гравиметрически или титриметрически. При соотношении нитрата и перхлората от 1 :4,8 до 8,3- 1 получаются удовлетворительные ре-
зультаты определения каждого иона.
Акимов, Емельянова и Бусев разработали гравиметрический метод определения перхлората (и перйодата) с антипирином и его производными [22]. Образующиеся труднорастворимые ионные ассоциаты можно использовать непосредственно для гравиметри ческого окончания или, после отделения, определение можно закончить методом потенциометрического титрования 0,1 М раствором NaOH. Определению не мешают хлорид-, хлорат-, сульфат-ионы, обычно встречающиеся в смеси с перхлоратом. Этим методом можно определять 4—50 мг перхлората.
404
Титриметрические методы
Перхлорат можно восстановить стандартным раствором тита-ва(1П), избыток которого оттнтровывают железо(III)-аммонийными квасцами [23]. Подробно метод описан в монографии [24].
Перхлорат можно восстановить сульфатом железа(II) в сильнокислой среде при нагревании до 150—155°С в течение 15 мин [25]. После охлаждения и разбавления раствора непрореагировав-niee железо(II) титруют КМпО4. Точность метода — около 0,3%.
Амальгама цинка в присутствии молибдена (VI) в качестве катализатора [26] восстанавливает перхлорат до хлорида, который можно определить по методу Фольгарда. Мешающее влияние нитрата и хлората можно устранить, если проводить восстановление при более низкой температуре и концентрации H2SO4. В этих условиях нитрат и хлорат восстанавливаются количественно, тогда как для восстановления перхлората требуются более жесткие условия.
Спектроскопические методы
Спектрофотометрические методы
Перхлорат образует малорастворимый фиолетовый ионный ассоциат [27] с метиленовым голубым. Система была использована в различных спектрофотометрических методиках, некоторые из них являются очень громоздкими и многостадийными [28]. В работе [1] применен для осаждения метиленовый голубой, избыток которого потом определяют в фильтрате.
Ионный ассоциат можно экстрагировать СНС13 или другими растворителями. Экстракция 1,2-дихлорэтаном [29] позволяет снизить ошибку определения за счет растворимости осадка. Закон Бера выполняется до 1 ppm. В значительной мере мешают определению хлорат, перйодат, нитрат, нитрит, тиосульфат, вольфрамат, хромат и магний. Недостатком метода определения перхлората с метиленовым голубым является большое значение холостого опыта, причиной чего является экстракция различными растворителями не только интересующего нас ионного ассоциата.
Кроме метиленового голубого применяли и некоторые другие красители. Кристаллический фиолетовый образует с перхлоратом соединение [30], экстрагирующееся хлорбензолом, максимум све-Топоглощения лежит при 595 нм. Определению не мешают большие содержания сульфата, йодата и хлорида. Подробно метод Hl] описан в разделе «Перйодат».
Голосницкая и Петрашень [32, 33] использовали два трифенил-^становых красителя бриллиантовый зеленый и малахитовый зевный.
| В каждом случае ионные ассоциаты экстрагировали.
405
Для экстракционно-спектрофотометрического определения пер. хлората можно использовать pH-индикатор нейтральный красный [34]. Катион нейтрального красного образует с многими ионами, включая и роданид, ионные ассоциаты. Ассоциат с перхлоратом экстрагируют нитробензолом при pH = 1—3 (цитратный буфер, ный раствор). Поглощение экстракта измеряют при 552 нм. График линеен для 10-6—10-5 М перхлората. Большие концентрации роданида и иодида мешают определению, но их можно отделить добавлением сульфата серебра.
Разработан новый [35] спектрофотометрический метод определения следовых концентраций перхлората в хлорате калия. Сначала перхлорат в виде ионного ассоциата с катионом тетрабу-тилфосфония экстрагируют о-дихлорбензолом. Затем экстракт встряхивают с водным раствором, содержащим роданидный комплекс железа, в результате чего в органической фазе оказывается ионный ассоциат тетрабутилфосфония и роданида железа (HI), светопоглощение которого измеряют при 510 нм. Метод применим для определения до 0,003% перхлората в хлорате
натрия.
Описан метод [36], основанный на редокс-реакции, в котором перхлорат восстанавливают до хлорида ванадием (III) в присутствии тетраоксида осмия. Образующийся ванадий (IV) определяют
спектрофотометрически. Другим вариантом метода является потенциометрическое титрование образующегося хлорида.
В некоторых работах экстрагируют ионные ассоциаты перхлората и хелатов металлов. Так, можно экстрагировать п-бутиро-нитрилом перхлорат три(1,10-фенантролината) железа(П) и проводить затем спектрофотометрическое измерение экстракта [8]. Этот же комплекс можно экстрагировать нитробензолом [9]. Экстракция проходит количественно при pH = 1,5—10,0, максимум светопоглощения лежит при 516 нм, график линеен от 10-6 до 4-Ю-5 М перхлората. Определению не мешают хлорид, сульфат и фосфат (104-кратный избыток каждого иона), но некоторые ионы мешают анализу. Был изучен хелат три-2,2'-дипиридила с желе
зом (II) [10]. Ионный ассоциат хелата с перхлоратом экстрагируют нитробензолом, максимум светопоглощения — при 524 нм-Закон Бера выполняется для 0,4—4 ррш перхлората. Определению
не мешают хлорид, бромид и сульфат.
Изучали возможность применения катионных хелатов меди(П с купроином (2,2-дихинолин) и неокупроином для спектрофотометрического • определения некоторых анионов, включая перхлорат [37]. Комплексы экстрагировали метилэтилкетоном. Светопогло; щение измеряли при 456 нм. График линеен для концентрации ClOi от 10~6 до 10-5 М. Определению мешают хлорид, нитрат> роданид, бромид и иодид, но их легко можно устранить обработ'
кой раствора солями серебра или ртути(II).
В большинстве рассмотренных методов предполагается пере^ измерением осаждение или экстракция. Разработан простой меТ° определения СЮ; [38], в котором исключены эти предварител
406
нЫе операции. Метод—косвенный, основанный на мешающем влиянии перхлората на образование окрашенного комплекса рения с фурилдиоксимом, реакция комплексообразования проходит ври восстановлении перрената оловом в присутствии а-фурилдиок-сима. Отклонения от рекомендованных условий влияют на окраску. ПреДУсмотРено Два периода по 30 мин, необходимых для завершения реакций. Мешают определению тиосульфат, медь(II), уранил, ванадий (V), нитрат и хлорат. Хлорат можно количественно отделить нагреванием раствора с НО. Не приводятся данные о точности метода.
Другой новый метод предложен Ирвингом и Дамодараном [39]. Метод основывается на более ранней работе Клиффорда и Ирвинга [40]. В нем используют окрашенный жидкий ионообмен-ник, приготовленный экстракцией из водного раствора соли Эрдманна раствором иодида тетрагексиламмония и подходящим растворителем (NHt [Со (NH3)2(NO2)4]'). Молярный коэффициент светопоглощения окрашенного жидкого ионообменника равен 104. Различные анионы могут замещать окрашенный эрдманнат иона Е" в соответствии с реакцией
{(NR4)+Е-)Орг + СЮ; ч=ь {(NR4)+ СЮ4-)орг + Е-
В результате этой реакции уменьшается оптическая плотность органической фазы и увеличивается интенсивность окраски водной фазы. Одна из двух рекомендованных методик основана на построении градуировочного графика для определения перхлората при измерении оптической плотности только водной фазы. Хлорат, гипохлорит, хлорит, нитрит и нитрат, мешающие определению, можно удалить обработкой НС1. Образующийся хлорид-ион, как и многие другие ионы, не мешает определению.
I Атомно-абсорбционный метод
I Косвенный атомно-абсорбционный метод основан на экстракции [41] перхлората ди(2,9-диметил-1,10-фенантролината)меди(1). Для измерений используют резонансную линию меди 324,7 нм. Этим методом можно определять 0,5—5 ррш перхлората.
I Метод ИК-спектроскопии
В При обсуждении спектрофотометрических методов часто упоминается определение следовых концентраций перхлората в хло-Рате калия. Еще одним методом является спектроскопия в инфракрасной области [42]. Хлорат калия после добавления НС1 упа-Ривают почти досуха. При этом хлорат восстанавливается до Дорида. Для определения перхлората применяют интенсивную °лосу поглощения при 628 см-1.
407
Электрохимические методы
Потенциометрические методы, ионоселективные электроды
Два метода, упоминавшихся выше [22, 36], можно использовать и в варианте потенциометрического титрования. Потенциометрическое титрование применяли для анализа смеси хлорная — азотная кислоты [43]. Последовательное титрование проводили в среде метилэтилкетон — метанол (1 : 1) раствором КОН со стеклянным индикаторным электродом и каломельным электродом сравнения. Относительная ошибка определения — меньше 1%.
Что касается ионоселективных электродов, то наблюдается тенденция создания новых электродов с использованием новых электродно-активных материалов [44]. Для аналитических целей можно применять выпускаемые промышленностью жидкостные ионоселективные электроды; описано несколько твердых электродов. Для потенциометрического титрования перхлората хлоридом тетрафениларсония использовали [45] жидкостной электрод типа Орион 93-81. При выполнении титрования при 2 °C удалось в 5 раз снизить предел определяемых концентраций (до 0,05 мМ в 50 мл раствора) перхлората [46]. Метод прост и довольно селективен. В одном из выпускаемых промышленностью жидкостных электродов в качестве жидкого ионообменника используют комплекс железа (III) с производным 1,10-фенантролина [47]. Шарп [48] сообщил о твердом ионоселективном электроде, который обладал близкой к нернстовской электродной функцией в интервале концентраций СЮ< от 10~' до 3 • 10-4 М.
Кулонометрические методы
Описан метод определения перхлората, основанный на его сожжении и последующем кулонометрическом определении хлорида [49].
Другие методы определения
Реакцию перхлората с хлоридом тетрафениларсония применяют и в термометрической методике [50]. Обычные ионы, включая галогениды и нитраты, не мешают определению. Присутствие перманганата, хлората, борфторнда вносит в определение значительные ошибки. Хлорат мешает определению, а присутствие бромата вносит в анализ отрицательную ошибку.
ЛИТЕРАТУРА
1 . Nabar G. М. Ramachand'an С. R —Analyt. Chem., 1959, v. 31, р 263. |
2 . Schumacher J С. — Perhclorates, their Properties, Manufacture and Use, Monograph No 146, New York, Reinhold, I960,
408
3 . Chadwick T С. — Analyt. Chem., 1973, v. 45 p 985
4 Greenhalgh R., Riley J. P. — J. mar. biol. Ass. U. K-, 1961, v 41, p. 175.
5 Bock R., Beilstein G. M.— Z. analyt. Chem., 1963, v. 192. p 44
g Bock E., Jainz J — Z analyt. Chem , 1963, v 198, p 315.
7 Bock R , Hummel C. — Z. analyt. Chem., 1963, v. 198, p. 176.
8 Fritz J. S., Abbink J. E., Campbell P. A. — Analyt. Chem., 1964, v. 36,
' p. 2123.
9 Yamamoto ¥., Kotsuji K., Kinuwaki S, Sawamura H.—J chem. Soc lapan, pure Chem. Sect., 1964, v. 85, p. 869; Analyt. Abstr., 1966, v. 13, p 3551
Ю Yamamoto Y., Kotsuji K. — Bull. chem. Soc. Japan, 1964, v 37, p. 785, Analyt.
' Abstr, 1965, v 12, p. 3307.
Ц. Thielemann H. — Mikrochim. Acta, 1971, p. 746.
12. Harrison B. L., Rosenblatt D. L. — J. Chromat., 1964, v. 13, p. 27',
13. Seiler H., Seiler M. — Helv. chim. Acta, 1967, v. 50, p. 2477.
14. Peschke W. — J. Chromat., 1965, v 20, p. 573
15. Petrovic S. M., Canic V. D. — Z. analyt. Chem., 1967, v. 228, p. 339; Analyt. Abstr., 1968, v. 15, p. 4674.
16. Tokutomi M., Kamiya S. — Japan Analyst, 1972, v 21, p. 81; Analyst, Anstr., 1974, v. 26, p 31.
17. Smith G. M., Willard H. — Ind. Engng Chem. analyt. Edn., 1939, v. Il, p. 186.
18. Glover D. J., Rosey J. M. — Analyt. Chem., 1965, v. 37, p. 306.
19. Chadwick T C. — Analyt. Chem., 1973, v. 45, p 985.
20. Funrman D. L. — Taianta, 1969, v. 16, p. 121
21. Hutton R. C., Stephen W. I. — Analyst, 1967, v. 92, p. 501.
22. Акимов В. К., Емельянова И. А., Бусев А. И. ЖАХ, 1971, т. 26, с. 956. Analyt. Abstr, 1972, т 23, с. 3148.
23. Burns Е. A., Muraca R. F. — Analyt. Chem., 1960, v. 32, р. 1316.
24. Belcher R., Wilson C. L. — New Methods of Analytical Chemistry, 2nd edn, London, Chapman and Hall, 1964, p. 320.
25. Aravamudan G., Krishnam V. — Taianta, 1966, v. 13, p. 519.
26. Haight G. P. — Analyt. Chem., 1953. v. 25, p. 642.
27. Hahn F. L. — Z. angew. Chem., 1926, v. 39, p. 451; Chem. Abstr., 1926, v. 20, p. 1967
28. Boltz D. F, Holland W. J. — In: Colorimetric Determination of Non-metals (Ed. D. F. Boltz), New York, Interscience, 1958, p. 176.
29. Iwasaki I., Utsumi S., Kang C. — Bull. chem. Soc. Japan, 1963, v. 36. p. 325; Analyt. Abstr., 1964, v. 11, p 1979.
30. Uchikawa S — Bull. chem. Soc. Japan, 1967, v. 40, p. 798; Analyt. Abstr., 1968 v. 15, p. 4678.
31. Hendrick С. E., Berger B. A. — Analyt. Chem., 1966, v. 38, p. 791.
32. Голосницкая В. А., Петрашень В. И ЖАХ, 1962, т 17, с. 878. Analyt. Abstr. 1963, т. 10, с. 2264.
33. Голосницкая В А., Петрашень В. И. Труды Новочеркасского Полнтехнич. института, 1964, т. 141, с. 73. Analyt. Abstr., 1966, т. 64, с. 1351е.
34. Tsubouchi М.— Analytica chim Acta, 1971, v. 54, p 143.
35. Fogg A. G., Burns D. T., Yeowart E. H. — Mikrochim. Acta, 1970, p. 974.
36. Zatko D. A., Kratochvil B. — Analyt. Chem., 1965. v. 37, p. 1560.
37. Yamamoto Y., Okamoto, Tao E. — Analytica chim. Acta, 1969, v. 47, p. 127.
38- Trautwein N. L., Guyon J. C. — Analyt. Chem, 1968, v. 40, p 639.
39. Irving H. M. N. H., Damodaran A. D. — Analyst, 1965, v. 90, p. 443.
40. Clifford W. E., Irving H. M. N. H.—Analytica chim. Acta, 1964, v. 31, p. 1.
41. Collinson W. Boltz D. F. — Analyt. Chem., 1968, v 40, p 1896.
42. Briggs A. G., Hayes W. P, Howling P. A., Burns D T.— Mikrochim. Acta, 1970, p. 888.
43. Денисова H. И., Голубева А. А., ЖАХ, 1972, t. 27, c. 1221. Analyt. Abstr., 1973, t. 25, c. 2254.
44. Covington A. K. — CRC crit. Rev. analyt. Chem. 1974 v. 3, p. 355.
5- Baczuk R. J., Dubois R. J. — Analyt. Chem. 1968, v. 40, p. 685
°- Smith M. J., Manahan S. E. — Analytica chim. Acta, 1969, v. 48, p 315.
•Ross J. W. — In: Ion Selective Electrodes (Ed. R. A. Durst), Publn. No. 311, Washington, Nat Bur Stands, 1969, p. 73.
409
48. Sharp M. — Analytica chim. Acta, 1972. v. 61, p. 91.
49. Secor G. E., Ricci B. A , White L. M. — Microchim. J., 1968, v. 13, p. 273.
50. Carr P. W., Jordan J. — Analyt. Chem., 1972, v. 44, p. 1278.
ПЕРЙОДАТЫ
Аналитические методы определения периодата — это в основном редокс-методы. Определение периодата в отсутствии других окислителей — довольно простая задача, которая существенно усложняется при определении периодата в смесях окислителей.
В работе [1] утверждалось, что растворы периодата нестабильны. Если авторы работы правы, то возникают трудности аналитического использования периодата, например, при хранении типовых растворов периодата калия или при его использовании в качестве аналитического реагента. Есть достаточно оснований не доверять этому утверждению. В более поздних работах отмечалось, что в кислых растворах периодата содержатся два соединения: тетраэдрическая и октаэдрическая модификации Н5Ю6. Равновесие между этими двумя формами достигается очень быстро, растворы устойчивы в течение длительного времени, если их хранить в емкостях из темного стекла [2].
МЕТОДЫ ОТДЕЛЕНИЯ
Методы бумажной и тонкослойной хроматографии
Некоторые примеры разделения смесей, содержащих перйодат, приведены в табл. 25. Для разделения нужно применять бумагу из стекловолокна [5]. При параллельном проведении разделения на обычной бумаге получаются неудовлетворительные результаты за счет восстановления периодата, возможно, до йодата.
Таблица 25. Отделение периодата методами бумажной и тонкослойной хроматографии
Метод Растворитель Разделяемые иоиы Литера тура
ТСХ (силикагель) Метанол — 25%-ный NH3—Н2О—10%-ная ук- сусная кислота(18: 2:2:1) Ю'—г—ю; 3
Бумажная хрома- Амиловый спирт — пирй- ю:—г—ю; 4
тографня (ватман № 4) дин — NH3 (0,880 г/см3) (3:7: 10)
Бумажная хрома- Смеси нзопропанола — NH3 юг—г—ю; э
тографня (стекловолокно) (7:3) £
ТСХ (А12О3) Различные растворы неорганических веществ ю;—го;—вго;—сю3' V
410
Электрофорез
Опубликована методика [7] количественного разделения перио-дата и йодата с использованием электрофореза в тонком слое.
Соосаждение
Гидроксид железа(III) является эффективным коллектором для соосаждения периодата [8]. Было показано, что перйодат можно отделить от йодата, при этом иодат остается в растворе. При повторении операции соосаждения эти два иона можно разделить на 99%- На гидроксиде алюминия перйодат соосаждается в присутствии больших концентраций йодата [9]. После отделения перйодат можно определять спектрофотометрическими методами.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Диоксид серы восстанавливает перйодат до йодата, который затем определяют. Восстановление можно проводить газообразным SO2 или разбавленным раствором гидросульфита натрия. Избыток SO2 удаляют кипячением.
Титриметрические методы
Иодиметрические методы
В щелочных буферных растворах перйодат восстанавливается до йодата. В качестве восстановителей можно использовать также иодид, мышьяк(Ш), сульфат марганца(II). С иодидом реакция протекает по уравнению
ю; + 2Г + н2о —> ю; + 12 + 2ОН‘
В качестве буферного раствора рекомендовали смесь буры и борной кислоты [10], для титрования иода применяли мышьяк(Ш).
В кислых растворах перйодат восстанавливается до иода:
Ю; + 7Г-|-8Н+ —> 412 + 4Н2О
Иод можно титровать либо стандартным раствором тиосульфата, либо мышьяком (III) [11]. Используя обе эти реакции, можно анализировать смесь периодата и йодата. Сначала в щелочном рас-Творе определяют перйодат. В кислых средах титруют сумму пе-Рйодата и йодата.
Предложен модифицированный иодиметрический метод анализа периодата и йодата. Перйодат селективно осаждают введе-Вием ацетата цинка [12]. Осадок Zns(IO6)2 отфильтровывают, растворяют в разбавленной кислоте и иодиметрически титруют, фильтрате иодат определяют иодиметрическим титрованием. Белчер и Тауншенд [13] нашли более изящное решение этой аДачи. Перйодат связывают молибдатом при pH = 3, образуется
411
6-молибдопериодат [I (МоО4)6]5~. Иодат титруют иодмметрически с использованием тиосульфата. При введении щавелевой кислоты комплекс разрушается, по реакции йодата с иодидом выделяется иод, который затем титруют тиосульфатом. Весь анализ проводят в одной аликвотной части анализируемого раствора.
Опубликованы методики иодиметрического анализа смесей иодат—перйодат—бромат—хлорат [14] и хромат—арсенат—перйодат—иодат—бромат—хлорат [15].
Метод с применением различных восстановителей
В разбавленной H2SO4 возможно прямое титрование перйодата мышьяком (III), в качестве индикатора используют ферроин [16].
Для определения перйодата использовали реакцию аскорбиновой кислоты с гексацианоферратом(III) [17]. К анализируемому образцу в присутствии фосфорной кислоты добавляют избыток раствора гексацианоферрата(II):
Ю; + 2 [Fe (CN)6J4- + Н2О —> lOj + 2 [Fe (CN)e]3' + 2 ОН"
Создают необходимую среду, добавляя NaHCO3, и образовавшийся гексацианоферрат(Ш) титруют стандартным раствором аскорбиновой кислоты, применяя в качестве индикатора 2,4-ди-хлорфенолиндофенол. Йодат не мешает определению.
Йериодат можно оттитровать сульфатом гидразина в присутствии йодата [18]. Для косвенного титрования перйодата можно применять хлорамин Т.
В других редокс-системах использовали потенциометрическую индикацию конечной точки титрования, эти методы будут рассмотрены ниже, г
Иодиметрическое титрование перйодата
Ход анализа. Образец растворяют в 50 мл воды, добавляют 3 г KI н 3 мл концентрированной НС1. Раствор оставляют на 5 мнн, затем разбавляют до 150 мл водой и тнтруют стандартным раствором тиосульфата в присутствии крахмала. Для точных определений необходимо проводить холостой опыт:
104’==412=852ОГ
Спектроскопические методы
Спектрофотометрия в видимой и ультрафиолетовой областях
Для определения перйодата существует несколько спектрофотометрических методов, большинство из них оригинально.
Был изучен спектр поглощения водных растворов перйодат0 при различных значениях pH [20]. Перйодат можно определять в слабокислых или щелочных растворах, измеряя его светопоглО' щение при 222 нм. Было найдено [9], что закон Бера выполняете0 для 1-10-5—18-10-5 М растворов КЮ4, молярный коэффициент светопоглощения равен 2,87-103.
412
Для определения перйодата изучали экстракцию различными растворителями ионного ассоциата периодат-нона с некоторыми трифенилметановыми красителями [21]. Лучшие результаты получаются при использовании кристаллического фиолетового, оптимальным растворителем является бензол. Этим методом можно определить 0,1 — 1,0- 10-'1 М 1О4- Светопоглощение измеряют при 615 нм. Определению не мешают 10-3 М галатов и IO-2 М IOj. Нитрат, гидрокарбонат и ацетат в этих условиях не экстрагируются. При концентрации выше 10“4 М роданид мешает анализу, сульфит и цианид взаимодействуют с красителями.
В качестве реагента на перйодат изучали бензгидразид [22]. Его же использовали в качестве титранта для определения перйодата в очень разбавленной H2SO4 [23]. Градуировочный график линеен в интервале 6,0 — 14,0 • 10 4 М 1О4. Определению мешают [Fe(CN)6]4’, [Fe(CN)r,]3 , СгО2’, Г, S2’, S2O2’, AsOt, Sn2+, Hg2>, Pb2+, Ag+ и Bi3+. Определение можно проводить в присутствии йодата, бромата, хлората и перхлората, если их содержание не более чем в 2,5 раза превышает содержание перйодата.
Перйодат можно определять с применением о-дианизидина [24] или 1,2-бис(4-диметиламинофенил)этан-1,2-диола [25]. Следует отметить, что в настоящее время изучается канцерогенность этого реактива [26].
Инфракрасная спектроскопия
Интенсивная и узкая полоса поглощения, характерная для перйодата тетрафениларсония, лежит в области 856 см-1. Хотя некоторые анионы, например, перманганат и хлорат, также образуют осадки с ТФА, в этой области длин волн их ионные ассоциаты не поглощают. На этом принципе основан метод определения микро-граммовых концентраций перйодата [27]. Перйодат осаждают с ТФА в слабокислых растворах в присутствии коллектора перхлората. Осадок помещают на пластину бромида калия и измеряют поглощение при 856 см-1. Перманганат, перхлорат, хлорат, бромат, йодат и перренат не мешают определению. Методика длительна, перед фильтрованием осадки выдерживают 24 ч на ледяной бане.
Электрохимические методы
Потенциометрические методы, ионоселективные электроды
Перйодат (0,09—70 мг) можно потенциометрически титровать в среде Н3РО4 железо(П)-аммонийными квасцами в присутствии Катализатора тетраоксида осмия [28]. В больших концентрациях Фторид, хлорид, бромид, иодид, сульфат, фосфат, борат и иодат Решают определению. Для потенциометрического титрования Использовали мышьяк(Ш) [29, 30]. В качестве катализатора
413
применяли K2RuO4, перйодат восстанавливали до йодата в сильнощелочных средах. Ошибка определения 5—200 мг перйодата меньше 2%. Катализатором реакции может быть также тетраоксид осмия.
ЛИТЕРАТУРА
1. Head F. S. Н., Hughes G.—J. chem. Soc., 1952, р. 2046.
2. Galliford D. J. В., Nuttali R. H., Ottaway J. M.— Taianta, 1972, v. 19, p. 871.
3. Palagyi S., Zaduban M.— Chemicke Zvesti, 1969, v. 23, p. 876; Analyt. Abstr., 1971, v. 20, p. 1660.
4. Obrenovic-Paligoric I. D., Cvoric J. D. — Bull. Boris Kidrich Inst. nucl. Sci., 1963, v. 14, p. 95; Chem. Abstr., 1963, v. 59, p. 14555b.
5. Naeumann R —J. Chromat, 1965, v. 18, p. 385.
6. Lederer M., Polcaro C. — J. Chromat., 1973, v. 84, p. 379.
7. Dobici F., Grassini G. — J. Chromat, 1963, v. 10, p. 98.
8. Новиков А. И., Финкельштейн E. И. ЖАХ, 1964, t. 19, c. 541. Analyt. Abstr., 1965, t. 12, c. 4534.
9. Bhattacharyya S. N., Chetia P. K. — Analyt. Chem., 1967, v. 39, p. 369.
10. Willard H. H„ Greathouse L. H. — J. Am. chem. Soc., 1938, v. 60, p. 2869.
11. Furman N. H. (Ed.) Standard Methods of Chemical Analysis, 6th edn, Vol. 1, Princeton, N. J., Van Nostrand, p. 522.
12. Kahane E. — Bull. Soc. chim. Fr., 1948, p. 70
13. Belcher R., Townshend A. — Analytica chim. Acta, 1968, v. 41, p. 395.
14. Szekeres L. — Z. analyt. Chem., 1960, v. 172, p. 256; Analyt. Abstr., 1960, v. 7, p. 3739.
15. Szekeres L. — Annali Chim., 1961, v. 51, p. 200; Analyt. Abstr., 1961, v. 8, p. 4932.
16. Glen K., Katthan W. — Ber., 1953, v. 86, p. 1077; Chem. Abstr., 1954, v. 48, p. 3834b.
17. Kaushik L., Rajendra P.— J. indian chem. Soc., 1970, v. 47, p. 1199; Analyt. Abstr., 1971, v. 21, p. 2567.
18 Berka A., Zyka J. — Cska Farm.. 1959. v. 8, p. 136; Analyt. Abstr., 1959, v. 6. p. 4769.
19. Singh B., Sood К. C. — Analytica chim. Acta, 1955, v. 13, p. 305.
20. Crouthamel С. E., Meek H. V., Martin D. S., Banks С. V. — J. Am. chem. Soc., 1949, v. 71, p. 3031.
21. Hendrick С. E., Berger B. A.— Analyt. Chem., 1966, v. 38, p. 791.
22. Escarrilla A. M., Maloney P. F., Maloney P. M. — Analytica chim. Acta, 1969, v. 45, p. 199.
23. Vulterin J., Zyka J. — Taianta, 1963, v. 10, p. 891.
24. Guernet M. — Bull. Soc. chim. Fr., 1964, p. 478; Analyt. Abstr., 1965, v. 12, p. 3308.
25. Fields R., Dixon H. B. F. — Biochem. J., 1968, v. 108, p. 883.
26. The Carcinogenic Substances Regulations, London, H. M. S. O., 1967.
27. Al-Kayssi M„ Magee R. J. — Analytica chim. Acta, 1963, v. 28, p. 176.
28. Vulterin J. — Colin Czech, chem. Conimun. Engl. Edn, 1966, v. 31, p. 3529; Analyt. Abstr., 1967, v. 14, p. 7468.
29. Норкус П. К, Симкевичуте Г. С., Янкаускас Я. ЖАХ, 1970, т. 25, с. 1673. Analyt Abstr., 1972, т. 22, с. 1569.
30. Норкус П. К., Симкевичуте Г. С. ЖАХ, 1971, т. 26, с. 1076. Analyt. Abstr., 1972, т. 23, с. 3149.
ЧАСТЬ III
ОКСИАНИОНЫ ФОСФОРА
Перед детальным рассмотрением аналитической химии каждого из анионов, входящих в эту группу, следует остановиться на их номенклатуре. Устаревшая в настоящее время номенклатура была разработана в 1833 г., в соответствии с ней фосфаты делились на орто-, пиро- и метафосфаты, сейчас эта номенклатура постепенно отмирает, но пока еще используется в литературе, посвященной технологическим вопросам. Однако современные классификации не всегда однозначны. Например, по одной классификации [1] «метафосфаты» — это линейные или циклические структуры, а по другой [2] — только циклические образования, для описания линейных структур предлагается использовать термин «линейные полифосфаты».
Термины «полифосфаты» и «конденсированные фосфаты» обычно используют как синонимы, хотя по решению ИЮПАК к поли-фосфорным кислотам относятся соединения общей формулы Нл+гРпОзп+ь в то время как метафосфорные кислоты, которые, по ИЮПАК, относятся к «конденсированным фосфатам», имеют общую формулу (НРОз)л-
Подробно номенклатура соединений фосфора обсуждается в двух работах [1, 2].
ЛИТЕРАТУРА
1. Van Wazer J. R. — Phosphorus and its Compounds, Vol. 1, Chapter 8, New York, Interscience, 1964.
2. Thilo E. — Condensed phosphates and arsenates. Adv. Chem. Radiochem., 1962, v. 4, p. 1.
ДИФОСФАТЫ (ПИРОФОСФАТЫ)
Дифосфат часто используется в аналитической химии в методах разделения ионов. Строение иона:
О’ о-
I I
'О—Р—О—Р—О' 4 4
Щелочных металлов устойчивы в нейтральных’и ще-ворах. При уменьшении pH их гидролиз ускоряется, нашли применение в пищевой и текстильной промыш
ленности, в водоподготовке, производстве детергентов и отбеливателей.
Дифосфаты [очных рас Дифосфаты
415
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение
Хлорид три (этилендиамин) кобальта (III) при pH — 7,5 селективно осаждает [1, 2] дифосфат в присутствии монофосфага и некоторых полифосфатов. Однако, как показали радиохимические исследования с применением 32Р, процесс отделения осложнен явлениями соосаждения и неполного осаждения, которые становятся особенно заметными при низких концентрациях дифосфата. Осадок дифосфата соответствует формуле Со(еп)3НР2О7. Вызывает сомнение эффективность этого метода по сравнению с методом ионообменной хроматографии.
Преимущества метода отделения дифосфата с использованием методов осаждения проявляются в некоторых комплексометрических методах определения дифосфата. Например, дифосфат осаждают при pH = 3,8 ацетатом цинка [3], при этом ортофосфаты остаются в растворе, их осаждают затем после отделения дифосфата цинка магнезиальной смесью; оба аниона косвенно, по металлу, определяют комплексометрическим титрованием. Тот же прием можно использовать при анализе смеси дифосфата и метафосфата, в этом случае дифосфат осаждают в виде нерастворимой соли цинка, а метафосфат определяют в фильтрате.
Методы бумажной и тонкослойной хроматографии
Некоторые методики, основанные на использовании этих методов, представлены в разделе «Полифосфаты». Дополнительные методы разделения приведены в табл. 26.
Таблица 26. Отделение дифосфата методами бумажной и тонкослойной хроматографии
Метод Растворитель Ионы, от которых отделяется дифосфат Литература
Бумажная хроматография (ватман № 31, ДТ) Диоксан — раствор NH3 — трихлоруксусная кислота — Н2О Орто- и трнфОс-фаты 4
ТСХ (силикагель) Метанол NH3 (0,880 г/см3) — 10%-ная трихлор- уксусная кислота — Н2О (10 :,3 : 1 : 10) Ортофосфат, фосфит, гипофосфит 5
ТСХ (целлюлозная масса) Дноксан — NH3 (0,880 г/см3) — трихлоруксусная кислота — Н2О (65 мл : 0,25 мл : 5 г : 27,5 мл) Ортофосфат 6
Преимущества бумажной и тонкослойной хроматографии при разделении в основном конденсированных фосфатов критически рассмотрены Поллардом и др. [7].
416
Метод ионообменной хроматографии
Вероятно, это наиболее эффективный метод разделения миллиграммовых концентраций фосфатов. Подробно метод обсуждается в разделе линейных и циклических полифосфатов. В табл. 27 приведены примеры отделения дифосфата от других фосфорных соединений.
С помощью гель-хроматографии можно отделить дифосфаты от линейных фосфатов [15] со степенью полимеризации до 13. Таблица 27. Отделение дифосфата от фосфорных анионов методом ионообменной хроматографии
Примечание
Последовательное элюирование
То же
Автоматический метод
Разделяются и другие соединения Р, включая низшие Р-анионы
Отделяются также высшне линейные Р-анионы
Литература
8
9
10
11
12
13
14
• Элюируются вместе.
МЕТОДЫ ОТДЕЛЕНИЯ
Гравиметрические методы
Разработаны методы определения дифосфата с использованием хлорида три (этилендиамин) кобальта (Ш) [1, 2]. При их использовании были обнаружены эффекты соосаждения и неполного осаждения, особенно при низких концентрациях дифосфата. В аналогичном методе [16] для осаждения дифосфата в виде Na[Co(C2H8N2)2P2O7] используют хлорид цис-дихлорбис(этилен-Диаминкобальт(Ш). Осаждают 20—80 мг дифосфата при pH = = 11—12 и сушат при 100—120 °C. Ортофосфат, трнфосфат, три-Метафосфат и тетраметафосфат в 10-кратном избытке не мешают определению. Другие мешающие ионы, кроме Ag+, Cu2+, VO2*, ^g2+, As3+, Br3+, Ce4+ и Th4+, можно маскировать.
Титриметрические методы
В горячих сильных кислотах дифосфат гидролизуется до °Ртофосфата, который можно определять титриметрически. Другим вариантом определения является титрование кислоты, об-
14 Зак. 167
417
разующейся при осаждении дифосфатов сульфатом цинка: Na2H2P2O7 + 2ZnSO< —> Zn2P2O7 + Na2SO4 + H2SO4
Разработанный в 1927 г. Брицке и Драгуновым [17] метод впоследствии был улучшен. Белл рекомендовал доводить pH до 3,8 перед добавлением сульфата цинка и титровать выделяющийся водород до pH = 3,8 [18]. Было замечено, что при постоянной концентрации сульфата цинка кислотность раствора после его введения является также функцией концентрации трифосфата. В отсутствие дифосфата методику можно использовать для определения трифосфата, для этих же целей методику можно использовать в том случае, если дифосфат определить другим методом. В работе [19] рекомендуют pH добавляемого раствора сульфата цинка также доводить до pH = 3,8.
Титриметрическое определение дифосфата 119]
Ход анализа. Растворяют в 100 мл воды 0,5 г дифосфата, не содержащего полифосфатов или «гексаметафосфата», перемешивая раствор 2 мин и добавляют 0,1 М раствор НС1 нли 0,1 М раствор NaOH до pH = 3,8. Добавляют 70 мл 12,5%-ного раствора ZnSO4-7H2O, pH которого предварительно тоже доводят до 3,8. Перемешивают 2 мин и затем титруют 0,1 М раствором NaOH до pH = 3,8; 1 мл 0,1 М раствора NaOH соответствует 13,23 мг Na4P2O7.
Результаты анализа несколько хуже при использовании раствора хлорида цинка. Дифосфат количественно осаждали ацетатом цинка при pH = 3,8, осадок растворяли в аммиачном буферном растворе и титровали цинк при pH = 10 раствором ЭДТА с индикатором эриохром черным Т [3]. Ортофосфат затем осаждали в виде MgNH4PO4 магнезиальной смесью из аммиачного раствора, осадок растворяли в HCI, добавляли ЭДТА, избыток которого титровали стандартным раствором Mg2+ в присутствии эриохром черного Т.
Дифосфат можно осаждать [20] в виде МпР2О7 при pH = 4,1, добавляя к анализируемому раствору избыток стандартного раствора Мп2+. Через 16 ч осадок отфильтровывали и избыток Мп2+ в фильтрате титровали при pH = 10 с эриохром черным Т. Осаждение, как было найдено, проходит количественно, если концентрация дифосфата не меньше 0,2 мг Р2О7” в 1 л, избыток Мп2+ должен быть не менее пятикратного. Ортофосфат мешает определению и должен отсутствовать.
Спектрофотометрические методы
Это, как правило, косвенные методы, некоторые из них основаны на уменьшении интенсивности окраски комплексных соединений. Метод, описанный в работе [21], основан на определении следовых концентраций дифосфата в ортофосфате по уменьшению интенсивности окраски роданидного комплекса железа(III), которое происходит за счет образования дифосфатного комплекса же-
418
деза (III). Светопоглощение измеряют при 435 нм. В аналогичном методе [22] определяют содержание дифосфата и трифосфата, измеряя оптическую плотность раствора при двух различных значениях pH.
Для определения дифосфата применяют комплекс железа(II) с 1,10-фенантролином [23]. Как было найдено, железо и алюминий, часто присутствующие в реактивных фосфатах натрия, мешают определению и должны быть удалены.
Для определения дифосфата в ортофосфате были использованы различные приемы [24]. К смеси добавляли железо(Ш) и дифосфат железа(III) экстрагировали раствором додециламина в смеси ацетона и СНС13. Железо в экстракте (а следовательно, содержание дифосфата) определяли после реэкстракции его гидроксидом аммония с сульфосалициловой кислотой. Светопоглощение водных растворов измеряли при 440—460 нм.
Амперометрический метод
Этот метод, на определение с помощью которого не влияют орто- и трифосфаты, основан на образовании при pH = 4,0 в смеси вода — этанол (2,5:1) нерастворимого дифосфата кадмия [25].
Ферментативные методы
Неорганические фосфаты образуются в результате некоторых процессов биосинтеза. Джонсон с сотр. [26] разработали метод их определения, в котором используется проба на ДНК-полиме-разу. Ни ортофосфат, ни органические фосфаты не дают такой же реакции, а следовательно, не мешают определению.
ЛИТЕРАТУРА
1. МсКипе Н. W., Arquette G. /. — Analyt. Chem., 1955, v. 27, p. 401.
2. Weiser H. J. — Analyt. Chem., 1956, v. 28, p. 477.
3. Kato T., Hagiwara Z., Shinozawa S., Tsukada S. — Technol. Reports Tohuka Univ., 1954, v. 19, p. 93; Analyt. Abstr., 1955, v. 2, p. 2697; Japan Analyst, 1955, v. 4, p. 84; Analyt. Abstr. 1955, v, 2, p. 3025.
4. Kolloff R. H. — Analyt. Chem., 1961, v. 33, p. 373.
5. Seiler H. — Helv. chim. Acta, 1961, v. 44, p. 1753.
6. Clesceri N. L., Lee G. F. — Analyt. Chem., 1964, v. 36, p. 2207.
7. Pollard F. H., Nickless G., Burton K., Hubbard J.— Microchem. J., 1966, v. 10, p. 131.
8. Lindenbaum S., Peters T. V., Riernan W.— Analytica chim. Acta, 1954, v. 11, p. 530.
9. Peters T. V., Rieman W.— Analytica chim. Acta, 1956, v. 14, p. 131.
Ю. Beaukenkamp J., Rieman IT., Lindenbaum S. — Analyt. Chem, 1954, v. 26, p. 505.
11. Lundgren D. P., Loeb N. P.—Analyt. Chem., 1961, v. 33, p. 366.
12. Grande J. E., Beaukenkamp J. — Analyt. Chem., 1956, v. 28, p. 1497.
13. Pollard F. H., Nickless G., Rogers D. E., Rothwell M. T. — J. Chromat., 1965, v. 17, p. 157; Pollard F. H., Nickless G., Rogers D. E., Crone D. L. — Proc. Soc. analyt. Chem. Conference, Nottingham, 1965, Cambridge, W. Heffer and Sons Ltd., 1965, p. 481.
14*
419
14. Rothbart H. L., Weymouth H. W., Rieman W.— Taianta, 1964. v 11, p. 33.
15. Ueno У. K, Yoza N. N., Ohashi S. — J. Chromat., 1970, v. 52, p. 481.
16. Sharma C. L. — Z. analyt. Chem., 1971, v. 257, p. 133; Analyt, Abstr., 1972, v. 23, p. 1393.
17. Братске E. В., Драгунов С. С. Журн. хим. пром СССР, 1972, т. 4, с. 49.
18. Bell R. — Analyt. Chem., 1947, v. 19, p. 97.
19. Dewaid W., Schmidt H.— Z. analyt. Chem, 1951, v. 134, p. 17.
20. Nietsch W., Giefer L. — Z. analyt. Chem., 1955, v. 142. p. 323.
21. Kolloff R. H., Ward H. K-, Ziemba V. F. — Analyt. Chem., 1960, v. 32, p. 1687.
22. Maurice J. — Bull. Soc. chim. Fr., 1959, p. 819.
23. Chess W. B., Bernhart D. N. — Analyt. Chem., 1958, v. 30, p. 111.
24. Шевчук И. А., Скрипник H. А.. Еналева Л. Зав. лаборатория, 1967, т. 33, с. 288. Analyt. Abstr., 1968, т. 15, с. 3289.
25. Сангина О. А., Близняк В М., Омаркулова Г. О. Зав. лаборатория, 197], т. 37, с. 271. Analyt. Abstr., 1972, т. 22, с. 106.
26. Johnson 1. С., Snanoff М., Bass S. Т., Boezi J. A., Hansen R. G. — Analyt. Biochem , 1968, v. 26, p. 137.
ГЕКСАФТОРФОСФАТЫ
Гексафторфосфат применяют в промышленности, например, в качестве катализатора и в гальванических ваннах при нанесении электропокрытий на сплавы. Описан синтез [1] гексафторфосфатов натрия, калия и аммония.
МЕТОДЫ ОТДЕЛЕНИЯ
От большого числа ионов гексафторфосфат можно отделить осаждением с нитроном или хлоридом тетрафениларсония. На этом принципе основан и метод его определения.
Разработан [2] экстракционный метод отделения, в котором н-бутиронитрилом экстрагируют гексафторфосфат 1,10-фенантро-лината железа (II). Экстракция очень селективна, на ее применении основан метод спектрофотометрического определения гексафторфосфата.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
До 1963 г. существовало ограниченное число методов определения гексафторфосфата. Их разработка началась с использования солей тетрафениларсония (ТФА) [3, 4] для гравиметрического и амперометрического методов определения этого иона.
Гравиметрические методы
До 1963 г. стандартным методом определения было гравиметрическое определение с нитроном. Этот реагент, впервые предложенный для определения гексафторфосфата Ланге и Мюллером [5], широко использовали для определения нитрата, но результаты определения анионов с его применением были неудовлетворительными вследствие нестабильности реагента.
Введение хлорида тетрафениларсония вместо нитрона значИ* тельно улучшило точность метода. Методика выполнения опре' 420
деления с ТФЛ довольно простая. Мешают определению все анионы, образующие с хлоридом тетрафениларсония труднорастворимые соли: перманганат, перренат, перхлорат, бромид, иодид, иодат и роданид. Чаще всего в смеси с гексафторфосфатом находятся ортофосфат и монофторфосфат. Эти ионы не образуют осадков с ТФА, в то время как дифторфосфаты уже в некоторой степени мешают определению. Их можно удалить из смеси, для этого раствор подщелачивают и в течение нескольких минут кипятят, за это время происходит гидролиз дифторфосфата до монофторфосфата. Во время этой обработки гексафторфосфат не разрушается.
Гравиметрическое определение гексафторфосфата с использованием хлорида тетрафениларсония [3]
Ход анализа. К анализируемому раствору, содержащему 36—55 мг гексафторфосфата калия, добавляют до щелочной реакции гидроксид аммония. Конечная концентрация аммония должна быть в интервале 5—11 Л1, общий объем раствора — 50 мл. Раствор нагревают до 50 °C и медленно, при постоянном перемешивании, добавляют двойной избыток 0,015 Л1 раствора хлорида тетрафенил-арсоння. Через 30 мин осадок отфильтровывают через пористый стеклянный фильтр и промывают 50 мл разбавленного раствора гидроксида аммония порциями по 5—10 мл. Осадок высушивают до постоянной массы при 105—115 °C и взвешивают (C6H5)4AsPF6.
Амперометрический метод
Амперометрическое титрование гексафторфосфата основано на образовании (CeHshAsPFg и восстановлении на ртути избыточного тетрафениларсония [4]. Определению не мешают 10-кратный избыток фторида, гидроксил-иона, сульфата, хлорида, ортофосфата и дифторфосфата, мешают ионы, образующие с (C6H5)4As+ осадки. Метод можно использовать для определения 0,0014—0,01 Л1 PFe. Относительная стандартная ошибка определения 0,037 г KPF6, найденная из 11 параллельных определений составляет ±0,66%.
Спектрофотометрические методы
В подразделе «Разделение» уже упоминалось об экстракции н-бутиронитрилом гексафторфосфата 1,10-фенантролината желе-за(Ц) [2].
Спектрофотометрический метод, основанный на экстракции этого ионного ассоциата, ограниченно селективен, определению мешают фталат, бромид, монофторфосфат (слегка), нитрат, нитрит, перйодат, роданид. Ацетат натрия до концентрации 0,5 мА! не мешает определению, а ацетат аммония мешает даже в малых концентрациях.
Светопоглощение измеряют при 505 нм.
Титриметрические методы с одновременным разделением
Титриметрическое определение с использованием гидроксида тетрафениларсония в качестве титранта объединено с одновременной экстракцией образующегося соединения 1,2-дихлорэтаном
421
[6]. Для фотометрического или визуального определения конечной точки титрования в качестве индикатора используют КМпО4> экстрагирующийся избытком титранта.
ЛИТЕРАТУРА
1. Woyski М. М.— Inorganic Syntheses (Ed L. F. Audreth) Volume 111, McGraw-Hill, 1950, p. 111.
2. Archer V. S., Doolittle F. G. — Analyt. Chem., 1967, v. 39, .p. 371.
3. Affsprung H. E., Archer V. S. — Analyt. Chem., 1963, v. 35, p. 1912.
4. Affsprung H. E., Archer V. S —Analyt. Chem., 1963, v. 35, p. 976
5. Lange Iv?, Muller E —Chem. Ber., 1930, v. 63B, p. 1058
6. Behrends K. — Z analvt. Chem., 1970, v 250, p. 246; Analyt. Abstr., 1971, v. 20, p. 2996.
ГИПОФОСФАТЫ
Гипофосфат-ион сравнительно устойчив к действию окислителей. Его окисляют при нагревании в кислых растворах бихроматом или перманганатом, окисление проходит за счет предварительного гидролиза с образованием фосфорных кислот. Гипофосфаты, за исключением гипофосфатов щелочных металлов, плохо растворимы в воде. Разбавленные растворы чистой кислоты можно хранить в течение длительного времени и даже кипятить; концентрированные растворы разлагаются при нагревании выше 30 °C.
Метод синтеза четырехзамещенного гипофосфага натрия и аналитическая химия гипофосфатов описаны в обзоре Палмера [1]. Опубликован синтез двузамещенного гипофосфата натрия [2].
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение
Гипофосфаты можно полностью отделить от орто-, ди-, три- и триметафосфатов осаждением в виде гипофосфата серебра из 0,5 М азотной кислоты [1]. Метод осаждения в виде Ag4P2O6 более селективен по сравнению с осаждением в виде гипофосфата цинка, мешают из обычно присутствующих в смеси ионов только дифосфаты.
Методы бумажной и тонкослойной хроматографии
В других разделах приведены примеры использования методов бумажной и тонкослойной хроматографии для разделения фосфор* ных оксикислот. Гипофосфат можно отделить, используя метод бумажной хроматографии (табл. 28),
422
Габмца 28. Отделение гипофосфата методом бумажной хроматографии
Методика Растворитель Соединения, от которых отделяют гипофосфит Литература
Ватман № 1, предварительно промытый НС1 Ватман № 4, восходящая хроматография (ци- линдр) Ватман № 3 (ленты) Кислотный Изобутанол — ацетилацетон — трихлоруксусная кислота — NH3 (0,880 г/см3) - Н2О (70 мл : 6 мл : 2,5 г : 0,2 мл : 23,8 мл) Основной Изобутанол — нзопропанол — NH3 (0,880 г/см3) - Н2О (20 мл : 40 мл : 1 мл : 39 мл) Изопропанол — трихлоруксусная кислота — NH3 (0,880 г/см3) — Н2О (75 мл : 5 г : 0,3 мл : 25 мл) Изопропанол-изобутанол — NH3 (0,880 г/см3) - Н2О (40 мл : 39 мл : 1 мл : 20 мл) и другие растворители Изопропанол — трнхлоруксусная кислота или изопропанол — изобутанол — водный NH3 Ортофосфат, дифосфат, As111 Asv Ортофосфат, дифосфат Трифосфат, триметафосфат, тетраметафосфат, фосфит, гипофосфит, пирофосфит h2so;, soj-, фосфаты, гипофосфит 3 4 4 5
Метод ионообменной хроматографии
Применение этого метода для разделения фосфорных анионов подробно обсуждается в разделах, посвященных полифосфатам, фосфиту и дифосфату. Гипофосфат можно отделить от орто- и дифосфата, фосфита, гипофосфита и дифосфита.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Гравиметрические методы разделения [1 ], упомянутые выше, Можно использовать и для определения гипофосфатов. Гипофосфат серебра осторожно сушат при 110°С и затем нагревают до Тех пор, пока кремовая окраска осадка не станет однородно-серой:
Ag4P2Oe —*• 2Ag + 2AgPO3
Переход происходит быстро при 300°C. Если требуется более Высокая селективность анализа, сначала гипофосфат осаждают £Ри pH == 4 в виде гипофосфата цинка, а затем переводят его гипофосфат серебра.
423
Титриметрические методы
Гравиметрический метод можно перевести в тнтрнмегрическпй, если к анализируемому раствору добавить избыток раствора нитрата серебра, который затем можно оттитровать [6] стандартным раствором роданида. Палмер [1] для определения гипофосфата рекомендовал использовать методы, основанные на окислении последнего иодистой кислотой при 100 °C в 40%-ной H2SO4 или водным раствором брома при pH = 6—9. Первый метод довольно прост, но определению мешают все окисляющиеся оксианионы фосфора. В методе окисления бромом не мешают фосфит и гипофосфит, которые окисляются в более кислых растворах.
Титриметрическое определение гипофосфита (0,2—0,25 г)
с помощью водного раствора брома [1]
Реагенты
Бромат калия, 0,3 г КВгО3 растворяют в 1 л воды.
Молибдат аммония, 2%-ный раствор.
Тиосульфат натрия, 0,1 М раствор.
Ход анализа. В колбу с притертой пробкой помещают следующие растворы: анализируемый раствор, полученный растворением гипофосфата в 20 мл 1 М раствора НС1, 25 мл раствора КВгО3, 55 мл воды, содержащей 5 капель раствора (NH4)2MoO4 и 1 г КВг. Сразу после сливания растворов колбу закрывают и тщательно перемешивают ее содержимое. Во вторую такую же колбу помещают все те же растворы, но не вводят гипофосфат. Через 30 мин открывают обе колбы, добавляют 4,5 г буры н опять закрывают колбы пробкой. После растворения буры растворы выдерживают еще 1 ч. Добавляют К1 и разбавленную H2SO4, при этом выделяется нод, который титруют стандартным раствором тиосульфата. Ошибка единичного определения — около 0,5%.
Спектрофотометрические методы
Гипофосфат в присутствии гипофосфита, фосфита и фосфата можно определять спектрофотометрически с использованием реагента молибден (V) — молибден (VI) [7].
В отсутствие окисляющихся в этих же условиях других фосфорных анионов гипофосфит можно окислять до ортофосфата и определять его с помощью молибденовой сини или по методу образования ванадомолибдофосфорной кислоты. Первый метод был использован Поллардом при автоматическом определении гипофосфата и других фосфорных анионов после их разделения методом ионообменной хроматографии [8].
ЛИТЕРАТУРА
1. Palmer В7. G.—J. chem. Soc., 1961, р. 1079. „
2. Bailar J. С. (Ed) — Inorganic Synthesis, Vol. IV, New York, McGraw-Hill. 195* p. 68.
3. D’Amore G— Ann. Chim. Rome, 1956, v. 46, p. 517.
4. Ebel J. P. — Mikrochim. Acta, 1954, p. 679. Q
5. Rudnicki R. — Chemia. analit., 1961, v. 6, p. 761; Analyt. Abstr., 1962, v. y* p. 2731.
6 Moeller T., Quinty G. H. — Analyt. Chem., 1952, v. 24. p. 1354.
1 Yoza N., Ohashi S. — Bull. chem. Soc. Japan, 1964, v. 37, p. 33; Analyt. Abstr., ' 1965, v. 12, p. 2196.
g Pollard F. H., Nickless G., Rogers D. E., Rothwell Л1. T. — J. Chromat., 1965, v. 17, p. 157.
ГИПОФОСФИТЫ
Гипофосфит H>PO2 настолько сильный восстановитель, что для его определения преимущественно используют редокс-методы. Гипофосфиты применяют, например, в фармакологии, где их использование, по-видимому, уменьшается, и в технологии нанесения «безэлектродных» металлических покрытий.
МЕТОДЫ ОТДЕЛЕНИЯ
Методы бумажной и тонкослойной хроматографии
Методы представлены в табл. 29.
Таблица 29. Отделение гипофосфита методами бумажной и тонкослойной хроматографии
Метод Растворитель Соединения, от которых отделяют гипофосфит Литература
Бумажная хроматография (ватман № 4) Кислотный растворитель Эбеля и др. Изопропа-иол — трихлоруксусная кислота — NH3 (0,880 г/см3) - Н2О (75 мл : 5 г : 0,3 мл : 25 мл) Фосфит, гипофосфит, пирофосфат, орто-, ДН-, три-, тримета- и тетраметафосфаты /
Бумажная хроматография (ватман № 3, ленты) Изопропанол — хлоруксусная кислота или Изопропанол — изобутанол — этанол — водный NH3 Сульфат, H2SO4, фосфит, гипофосфат, полнфосфаты 2
ТСХ (силика- гель + крахмал) Метанол — NH3 (0,880 г/см3) хлоруксусная кислота 10%-чая — Н2О (10 мл : 3 мл : 1 г : 10 мл) Ортофосфат, дифосфат и фосфит 3
ТСХ (MN поли-грам на ленте CEL 300) Изопропанол — трихлоруксусная кислота — тетра-этиламмоний гидро- ксид — Н2О (750 мл : 50 г : 16 мл (20% объемн): 250 мл) Изопропанол — изобутанол — NH3 (0,880 г/см3) (400 мл ; 200 мл: 10 мл) Ортофосфат, дифосфат, три-, тримета-, тетраметафосфат, фосфит, полифосфаты (п = 20—30) Продажные циклические и линейные фосфаты 4
425
Метод ионообменной хроматографии
Как было показано [5], ионообменная хроматография — наиболее эффективный метод разделения гипофосфита, фосфита и фосфата. В некоторых работах предлагаются автоматические хроматографические методы, в которых гипофосфит можно отделять от ортофосфата, фосфита, гипофосфата, дифосфата, дифосфита, монотиофосфата, трифосфата и триметафосфата [6, 7]. Эти работы подробно описаны в разделе «Полифосфаты».
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Для определения гипофосфита применяют методы определения фосфита. Подробно методики описаны в работах [8, 9]. В табл. 34 представлены методы определения гипофосфита в присутствии фосфита. Дополнительные методы помещены в табл. 30.
Описаны комплексометрические методы определения гипофосфита. В одном из них гипофосфит окисляют HNO3. Образующийся фосфит обрабатывают избытком нитрата висмута. Пойле отделения осадка в фильтрате избыток висмута титруют ЭДТА с пирокатехином в качестве индикатора [14].
Другой метод основан на реакции гипофосфита с комплексо-натом ртути [15]. При кипячении Hg11 восстанавливается до металла, при этом в растворе оказывается свободная ЭДТА в количестве, эквивалентном содержанию гипофосфита. ЭДТА титруют медью(11) с ПАН в качестве индикатора.
Таблица 30. Редокс-методы определения гипофосфита
Окислитель Основы метода Примечание Литература
Fe”1 боратный избыток Fe111 в 1,6 М растворе НС1 кипятят около 20 мин Не мешает фосфит 10
AgC104 и85%-ная Н3РО4 Кипятят с реагентом 3—3,5 мин. Титруют избыток Ag+ потенциометрически хлоридом То же 11
CrVI Реагент добавляют к сильнокислому раствору (H2SO4), разбавляют, избыток Crvl титруют Fe11 Фосфит также окисляется 12
Celv Нагревают с реагентом до 100 °C 7 мин в присутствии катализатора Мп” —Ag1 То же 13
426
рис. 48. Градуировочные графики для определения Н2РО” и Н2РО; (кювета I см: светопоглощение Н,РО" измеряют через 30 мин в 2,5 М НС1; светопоглощение Н2РО~ измеряют через 30 мин в 0,14 М HCI [18]: I—Н2РО2, 400 нм; 2—Н2РО4, 670 нм.
Спектрофотометрические методы
Гипофосфит в присутствии серной кислоты образует с молибдатом аммония синее соединение, которое можно использовать для спектрофотометрического определения гипофосфита [16]. Фосфит в отличие от фосфата не мешает определению. Вербицкой и Романовой [17] независимо от авторов работы [16] эта реакция была обна-
ружена и использована для разработки метода определения гипофосфита. Энтон метод устранения мешающего влияния фосфата.
[18] предложил В методах, опи-
санных в работах [16] и [17], измеряют светопоглощение при 470 нм. В модифицированном методе [18] измеряют светопогло-
щение при 400 нм, изменяя кислотность растворов, что позволяет определять гипофосфит в присутствии фосфата. Если необходимо, то можно определять и содержание фосфата.
Метод основан на том, что фосфат не образует гетерополикислоту в 1,6 М растворе НС1. Комплекс гипофосфита поглощает при 400 нм в широкой области кислотности растворов. Если нужно определять фосфат, кислотность раствора во второй аликвотной части доводят до 0,14 М по НС1 и измеряют светопоглощение при 670 нм. Если известна концентрация гипофосфита и оптическая плотность его раствора при 670 нм, можно учесть ее вклад в суммарное светопоглощение при этой длине волны. Разность оптических плотностей соответствует поглощению фосфата, по этой величине можно найти его содержание. Градуировочные графики Для определения гипофосфита и монофосфата показаны на рис. 48.
Спектрофотометрическое определение гипофосфита и ортофосфата при их совместном присутствии [18]
Реагенты
Молибдат аммония, 10 г (NH4)6Mo7O24-4H2O растворяют в 200 мл дистиллированной воды, содержащей 60 мл концентрированной НС1. Раствор разбавляют водой до 1 л.
Сульфит натрия, 20 г Na2SO3 растворяют в 100 мл дистиллированной воды.
Ход анализа. В мерную колбу вместимостью 25 мл помещают аликвотную пас.Ть анализируемого раствора (не более 15 мл), содержащую не более 5 мг ги-г°Фосфнта. Добавляют 10 мл 6 М раствора НС1, 5 мл раствора молибдата ам-Ония и 5 мл раствора сульфита натрия. Через 30 мин измеряют светопоглощение Створа в кювете с толщиной слоя 1 см по отношению к холостому раствору.
Содержание фосфата в другой аликвотной части раствора можно определить, не вводя 10 мл 6 М раствора НС1 н измеряя светопоглощение при 670 нм через 30 мин.
Градуировочный график для определения гипофосфита при 400 нм линеен при содержании его 1—5 мг/25 мл.
Другие методы определения
Можно проводить окисление гипофосфита до ортофосфата и затем определять его, хотя при этом определению мешают другие фосфорные анионы, так как при такой обработке они тоже превращаются в ортофосфат.
Окисление фосфорных анионов (включая гипофосфит) является первой стадией метода автоматического определения, который упоминается в этом разделе при обсуждении ионообменного разделения [6, 7]. Различные ионы разделяют с применением ионообменной хроматографии и затем переводят в ортофосфат для колориметрического определения с помощью молибденовой сини. Низшие оксианионы фосфора при окислении переходят в ортофосфат.
Два новых метода определения гипофосфита основаны на выделении водорода. Первый метод был разработан для автоматического анализа растворов, используемых для «безэлектродных» покрытий металлов *; для этих растворов обычные методы неприменимы [19].
Палладиевая чернь служит катализатором разложения гипофосфита с выделением водорода:
Pd°
Н(Н2РО2) - > НРО2 + Н2
НРО2 + Н2О —> Н2(НРО3)
Выделяющийся водород определяют, измеряя температуру пламени при его сгорании.
В другом методе выделяющийся водород определяли волюмо-метрически [20]. Анализируемую смесь, содержащую палладий (II), помещали в шприц для подкожных инъекций и вытесняли из него избыток воздуха. Реакционный объем доводили до требуемого уровня, шприц закрывали и нагревали на кипящей бане 10 мин. После охлаждения, используя деления на шприце, измеряли объем выделившегося водорода, который пропорционален содержанию гипофосфита. Реакции проходят в соответствии с уравнениями
Pd2++ Н2РО2‘+ Н2О —> Pd° + Н2РО3" 4- 2Н+
Pd°
н2ро2' + н2о —> н2ро; + н2|
Первая реакция проходит лишь в незначительной степени. Полный анализ длится 15 мин.
* Автокаталитический процесс химического восстановления для непрерЫВ' него осаждения металла из растворов, содержащих соль металлу и восстав0' витель.
4X3
ЛИТЕРАТУРА
J Ebel J. P. — Mikrochim Acta, 1954, p. 679.
'2 Rudnicki R.— Chemia. analit., 1961, v. 6, p. 761; Analyt. Abstr., 1962, v. 9, p. 2731.
3 Seiler H. — Helv. chim. Acta, 1961, v. 44, p. 1753.
4. Burns D. T., Lee J. D. — Mikrochim. Acta, 1969, p. 206.
5. Pollard F. H., Rogers D. E., Rothwell Л1. T., Nickless G. — J. Chiomat., 1962, v. 9, p. 227.
6. Pollard F. H., Nickless G.t Rogers D. E., Rothwell M. T. — J. Chromat., 1965, v. 17, p. 157.
7. Pollard F. H, Nickless G., Rogers D. E, Crone D. L. — Proc. Soc. Analyt. Chem. Conference, Nottingham, 1965, Cambridge, W. Heffer and Sons Ltd, 1965, p. 481.
8. Kolthoff I. M., Belcher R. — Vilurpetric Analysis, Volume 111, New York, Interscience, 1957
9. Berka A., Vulterin J., Zyka 1. — Newer Redox Titrants, Oxford, Pergamon Press, 1965.
10. Kin’Ya Ogawa—Bull. chem. Soc. Japan, 1969, v. 42, p. 1449; Analyt. Abstr., 1970, v. 19, p 1192.
11 Ackermann G., Mende A. — Z. analyt. Chem., 1967, v. 232, p. 97; Analyt. Abstr., 1969, v. 16, p. 101.
12. Rao V. R. S. — Z. analyt. Chem., 1969, v. 246, p. 384; Analyt. Abstr., 1970, v. 19, p. 2997.
13. Guilbault G. G., McCurdy W. H. — Analytica chim. Acta, 1961, v. 24, p. 214.
14. Ciogolea G., Morait G., Nguyen H. B. — Farmacia, Bucharest, 1962, v. 10, p. 331; Chem. Abstr., 1963, v. 58, p. 6189b.
15. Fulop L., Blazek A. — Farmacia, Bucharest, 1962, v. 10, p. 525; Chem. Abstr., 1963, v. 58, p. 7370b.
16. Scanzillo A. P. — Analyt. Chem., 1954, v. 26, p. 411.
17. Вербицкая T Д., Романова И. К- Зав. лаборатория, 1960, т. 26, с. 818. Analyt. Abstr., 1961, т. 8, с. 994.
18. Anton А.—Analyt. Chem., 1965, v. 37, p. 1422.
19. Greenfield S., Cooper R. M. — Taianta, 1962, v. 9, p. 483.
20. Tischer T. N., Baitscholts A. D., Pryzbylowicz E. P. — Analytica chim. Acta, 1966, v. 34, p. 101.
МЕТАФОСФАТЫ: ТРИМЕТАФОСФАТ,
ТЕТРАМЕТАФОСФАТ, ГЕКСАМЕТАФОСФАТ
Триметафосфат
О о*
II I
О—Р—О—Р=О
I I
о о
I I
О=Р—О—Р—О'
I I
О' о
Тетраметафосфат
Иногда эти ионы называют циклотрифосфатом и циклотетрафосфатом. Соли щелочных металлов растворимы, соли щелочноземельных металлов менее растворимы, однако их растворимость выше, чем растворимость таких же солей линейных полифосфатов, Которые осаждаются барием из кислых растворов. Растворы метафосфатов сильных оснований имеют нейтральную реакцию.
429
В обычных условиях растворы триметафосфатов щелочных металлов очень устойчивы.
При низких значениях pH проходит гидролиз, конечной стадией которого является образование монофосфата:
(Р3О9)3- H2P3O1J —> Н2Р2ОГ —> н2ро;
тримета- Триполи- ди- моно-
В присутствии катионов-катализаторов гидролиз ускоряется. Метафосфаты также важны, но значительно меньше, чем линейные полифосфаты. Как и некоторые полифосфаты, триметафосфат натрия используют для модификации крахмала при приготовлении фосфатного крахмала. Триметафосфат натрия используют в процессах дубления, при pH = 2—3 с его помощью осаждают про теины.
Описан метод приготовления растворов, содержащих смесь различных метафосфатов [1]. Известен синтез [2] циклического гексаметафосфата натрия.
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение »•
Поскольку метафосфаты с трудом осаждаются солями Ва2+, разработан метод осаждения всех других фосфатов в виде бариевых солей, при этом циклические фосфаты остаются в растворе. Недостатком метода является неполное осаждение. Например, было показано [3], что основанный на этом принципе метод определения триметафосфата в присутствии орто-, ди- и трифосфатов дает большие ошибки. В работе [4] было найдено, что в то время как орто-, ди- и трифосфаты не полностью осаждаются барием, высшие фосфаты осаждаются количественно, и в растворе остаются только циклические фосфаты. Таким образом, в отсутствие орто-, ди- и трифосфатов метафосфаты можно определять в фильтрате после отделения высших полифосфатов.
Методы бумажной и тонкослойной хроматографии
Бумажная и тонкослойная хроматография — наиболее эффективные методы разделения как линейных полифосфатов, так и циклических фосфатов. Наиболее интересные методы разделения приведены в табл. 35. Основные успехи бумажной и тонкослойной хроматографии рассмотрены в обзоре Полларда и др. J5],
Метод аниоиообмеиной хроматографии
Чаще всего именно этот метод используют для разделения миллиграммовых количеств линейных и циклических фосфатов. Метод разделения тримета- и тетраметафосфатов описан в работе Римана с сотр. [6, 7]. В этих работах использовали моди-
430
фнцированную сшитую полпстирольную смолу, содержащую .—СН2К+(СНз)з-ионные группировки. Опубликована работа, посвященная теоретическому рассмотрению процессов ионного обмена [8].
Поллард с сотр. разработали автоматический ионообменный метод, основанный на применении анализатора, для разделения смеси, содержащей триметафосфат, линейные фосфаты, низшие фосфорные анионы и другие фосфорсодержащие ионы. Подробно методика приведена в разделе «Полифосфаты».
С использованием анионообменной смолы IRA-400 отделяли три циклических фосфата от ортофосфата и различных его линейных полимеров до тридекафосфата [9].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Спектрофотометрические методы
После выделения индивидуальных циклических фосфатов можно провести их гидролиз до ортофосфата, который затем определяют спектрофотометрически или титриметрически, в зависимости от его содержания. Для спектрофотометрического определения используют молибдованадатный метод [6, 8, 4] или молибдофосфат-ный метод [10].
Метод ИК-спектроскопии
Опубликован метод инфракрасной спектрофотометрии с использованием прессованного диска КВг для количественного определения фосфатов. Анализировали в виде сухих порошков четырехкомпонентные и трехкомпонетные смеси, при анализе трех-Компонентных смесей ошибка определения каждого компонента составляла ±0,5% [П]. Метод, по-видимому, нашел широкое применение.
ЛИТЕРАТУРА
I Thilo Е., Schulke U. — Z. Anorg. Allgem Chem., 1965, v. 297, p. 431
2. Griffith E. J., Buxton R. L. — Inorganic Chemistry, 1965, v. 4, p. 549.
3. Dewaid W., Schmidt H. Z. — Z. analyt. Chem., 1952, v. 137, p. 178.
4. Rothbart H. L., Rieman V7. — Talanta, 1964, v. 11, p. 43.
5. Pollard F. H., Nickless G., Burton K., Hubbard J. — Microchem. J., 1966, v. 10, p. 131
6. Lindenbaum S., Peters T. V., Rieman B7. — Analytica chim. Acta, 1954, v. 11, p. 530.
7. Peters T. V., Rieman W. — Analytica chim. Acta, 1956, v. 14, p. 131.
8. Beukenkamp J., Rieman W., Lindenbaum S. — Analyt. Chem., 1954, v. 26, p. 505.
9. Rothbart H. L., Weymouth H. W., Rieman B7.— Talanta, 1964, v. 11, p. 33.
•0. Pollard F. H., Nickless G., Rogers D. E., Rothwell M. T. — J. Chromat., 1965, v. 17, p. 157.
И. Corbridge D. E. C., Lowe E. J. — Analyt. Chem., 1955, v. 27, p. 1383.
431
МОНОФТОРФОСФАТЫ
Опубликован обзор по химии монофторфосфат-иона (PO3F)2-[1]. Нет реакций, специфичных для ортофосфата, фторида или иона (PO3F)2- в целом, его химия очень похожа на химию сульфата. Это становится понятным, если обратить внимание на идентичность радиусов S6+ и Р5+. Кроме того, фторид-ион и гидроксил-ион тоже очень близки по стерическим характеристикам.
Применение монофторфосфата связано в основном с использованием монофторфосфата натрия в качестве добавки к некоторым зубным пастам для предотвращения разрушения зубной эмали. В качестве добавок к пастам используют и фториды олова и натрия. Однако эффективность монофторфосфата натрия выше. Если зубной порошок или паста содержат карбонат кальция, то при добавлении к ним фторида натрия может образовываться нерастворимый фторид кальция, связывающий фторид-ион в неактивную форму, что в значительной степени снижает действие фторид-иона. Из монофторфосфата натрия, который обычно присутствует в пастах в концентрациях меньше 1%, свободный фторид-ион образуется в результате гидролиза, медленно проходящего в щелочных средах и быстро — в кислых растворах:
H2PO3F + H2O h3po4 + hf
При определении монофторфосфата проводят гидролиз и затем определяют содержание образующегося фторида или фосфата. Сложность определения в том, что фторид и фосфат мешают определению друг друга. Эта задача удовлетворительно решается с применением современных методов анализа.
' Описан синтез монофторфосфата натрия [2], но методом ТСХ было показано, что в продукте находится постоянное количество примесей [3].
МЕТОДЫ ОТДЕЛЕНИЯ
Метод электрофореза
Монофторфосфат можно отделить от дифторфосфата (РОгРг)" и ортофосфата, используя электрофорез [4].
Осаждение
Монофторфосфат осаждается в виде соли серебра Ag2PO3F при добавлении нитрата серебра в 80%-ные водно-спиртовые растворы; содержание серебра в осадке выше стехиометрического за счет соосаждения Ag+ [5]. Монофторфосфат можно затем определить, выделив из него фторид дистилляцией из растворов серной или хлорной кислоты, фторид титруют нитратом тория (IV), как описано в разделе «Фториды». Свободный фторид не влияет на осаждение монофторфосфата серебра, так как фторид серебра хорошо растворим в 80%-ных водно-спиртовых средах. Метод длителен и, вероятно, в настоящее время не нашел широкого приме' нения в рутинном анализе,
432
Рис. 49. Схема прибора для автоматического определения монофторфосфата [6]: /—баии (95 СС); 2—смесители; 3—стандартная мембрана; 4—слив и отвод пузырьков воздуха; 5—ионообменная колонка; 6—сосуд с 0.1 М раствором КС1; 7 — сосуд с 0,25 М раствором КС1; 8 — самописец; 9—колориметрическая проточная ячейка (15 мм, 660 нм).
Д—образец; Б— воздух; В—ЗМ H2SO4; Г — дистиллированная вода; Д—сульфат гидразина; Е— молибдат аммония; Ж—элюент.
Хроматографический метод
Монофторфосфат отделяют от органических веществ и других соединений фосфора, используя различие в их сорбции ионообменными смолами. Бенц и Келлей [6] разработали автоматический вариант метода. После отделения проводят гидролиз, образующийся ортофосфат определяют колориметрически с молибдатом. Разделение проводят на смоле Дауэкс-1-Х8 в хлоридной форме, в качестве элюента используют раствор хлорида калия. На рис. 49 приведена схема прибора для осуществления этого метода. Концентрацию монофторфосфата вычисляют, измеряя площадь индивидуального пика. На рис. 50 показана хроматограмма монофторфосфата натрия.
50. Хроматограмма монофторфосфата [6J: ^«андарт; 2 — ортофосфат; 3— монофторфосфат.
433
Рис. 51. Типичная хроматограмма, полу, ченная при определении общего и ра. створимого фтора (в виде фторнда в зуб-ных пастах с добавками фторида натрия и монофторфосфата) [9J:
I—триметилфторсилан; 2—и-пеитаи; 3—избы, ток кремиийсодсржащего реагента н раствора, теля (бензол).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Аналитические методы определения монофторфосфата основаны на определении ортофосфата или фторида, образующихся после гидролиза монофторфосфата.
Метод с использованием фторидного ионоселективного электрода
Свойства этого электрода рассмотрены в разделе «Фторид». Электрод применяют для определения монофторфосфата в водных суспензиях зубных паст, при этом можно определять сразу
два важных параметра, а именно «свободный» фторид и содержание монофторфрсфата. «Свобод-
ный» фторид попадает в пасты в виде примеси в исходных компонентах паст и как продукт частичного гидролиза монофторфосфата. Содержание монофторфосфата рассчитывают по разности между общим содержанием фторида, которое находят после полного гидролиза образца, и содержанием «свободного» фторида.
Описан метод [7] анализа зубных паст с применением фторидного электрода. Электрод использовали для измерения «свободного» фторида. При добавлении к раствору НС1 до сильнокислой реакции гидролиз монофторфосфата завершается за 30 мин. Повторное измерение с фторидным электродом позволяет рассчитать общее содержание фторида. Для того чтобы измерение в анализируемом и стандартном растворах выполнялось в одинаковых условиях, необходимо поддерживать во всех измеряемых растворах
постоянную ионную силу.
Спектрофотометрический метод
Метод основан на спектрофотометрическом определении оРт° фосфата, образующегося при гидролизе монофторфосфата. Эт метод использовали для определения монофторфосфата после ° 1
деления его с применением ионного обмена [6]. Для определения фосфата обычно используют молибденовую синь.
Газохроматографический метод
В 1968 г. был опубликован метод быстрого синтеза триметил-фторсилана из триметилхлорсилана в кислых растворах [8]. Эту реакцию использовали для разработки быстрого метода определения общего и растворимого («свободного») фторида в смесях, содержащих монофторфосфат [9]. Триметилфторсилан экстрагировали бензолом, содержащим н-пентан в качестве внутреннего стандарта. Разделение осуществляли в стеклянных колонках, содержащих 10% силикона OVI на целите AWDMCS с размером зерен 0,15—0,18 мм, при 60°C; газом-носителем служил азот. На анализ затрачивается меньше 30 мин, это лучший метод определения монофторфосфата. Типичная хроматограмма [9] показана на рис. 51.
ЛИТЕРАТУРА
1. Schmutzler R.— In: Advances in Fluorine Chemistry (Ed. M. Stacey, J. C. Tat-low and A. G. Sharpe), Vol. 5, Butterworths, London, 1965, p. 134.
2. Hill O. F., Audrieth L. F. — Inorganic Syntheses (Ed. L. F. Audrieth), Vol. 3, McGraw-Hill,, 1950, p. 106.
3. Williams W. J., Thompson К. H., unpublished work.
4. Element R., Knollmuller К. O. — L. analyt. Chem., 1959, v. 166, p. 193; Chem. Abstr., 1959, p. 16805h.
5. Hill H. J., Reynolds C. A. — Analyt. Chem., 1950, v. 22, p. 448.
6. Benz C., Kelley R. M. — Analytica chim. Acta, 1969, v. 46, p. 83.
7. Shane N., Miele D. — J. pharm. Sci., 1968, v. 57, p. 1260.
8. Fresen J. A., Cox F. H., Witter M. J. — Pharm. Weekblad, 1968, v. 103, p. 909; Analyt. Abstr., 1969, v. 17, p. 3619.
9. Cropper E, Puttnarn N. A. — J. Soc. cosmet. Chem., 1970, v. 21, p. 533.
ОРТОФОСФАТЫ
Определение ортофосфатов проводят в самых различных областях: в металлургии, фармакологии, биохимии, агрохимии, при производстве пищевых продуктов и при анализе воды. Поэтому Понятно, что регулярно публикуются обзоры по методам его определения. Самыми последними были опубликованы обзоры Грин-Филда [1] и Римана и Байненкампа [2].
Многие методы определения фосфата основаны на его способ-н°сти образовывать молибдофосфорную гетерополикислоту. По-сКольку этот метод будет довольно подробно рассмотрен, кажется Уместным выделить различные методики, основанные на образовании этой гетерополикислоты. Они представлены на схеме. Хотя ®Ся схема основана на молибдофосфорной кислоте, следует учиты-ать, что некоторые элементы также образуют гетерополикислоты. А°гда эти элементы присутствуют в смеси с фосфором, для се-еКТивного определения последнего необходимо вводить в мето-АИкУ дополнительные стадии разделения. Например, мешающие
435
Схема g
Селективная экстракция с последующим спектрофотометриче: кин определением
Граоиметрическое оп -ределение в виде NH4-солей или солей органических оснований
Алкалиметрическое титрование
Спектрофотометрическое определение гете -рополикислоты с восстановлением или дез него
Спектрофлуорометра -ческое определение РО4; например в виде молибдофосфата родамина В
Селективное подавление образования гете-рололикислот с последующим определением устойчивых в этих условиях гетерополи -кислот
Нолибдофосфорная кислота в присутствии _ гетерополикислот St,Ge и As
Дтомно -абсорбционная спектроскопия, определение Мо, входящего в состав гетеро поликислоты
Спектрототометричес кое определение гло* после разрушения гетерополикислоты
Выделение Мо из гетерополикислоты, восстановление его до Мо1 и его периметрическое титрование
Превращение гетеро-поликислоты в комплекс Мо с оксином. Броматометрическое титрование оксина, входящего в состав комплекса с Мог
ионы маскируют введением оксикислот перед спектрофотЬметри-ческим определением или предварительно проводят селективную экстракцию.
Растворы, содержащие меньше 20 мкг/л фосфата, следует хранить в условиях, исключающих сорбцию фосфата. Было найдено [3], что промытые предварительно водой стеклянные колбы сорбируют до 2О°/о фосфата за 1—6 ч хранения. Аналогичным образом ведут себя емкости из полипропилена и полиэтилена. Предварительная обработка полимерных емкостей фосфатом улучшает условия хранения. Например, при хранении воды в такой емкости в течение 6 ч сорбции фосфата не наблюдалось.
МЕТОДЫ ОТДЕЛЕНИЯ
Отделение фосфата необходимо проводить или перед определением, или в том случае, если его присутствие мешает определению других ионов.
В анализе следовых концентраций предварительное концентрирование становится такой же важной проблемой, как и отделение. Описан метод концентрирования и отделения [50], который был использован для определения оксида фосфора в воздухе.
Осаждение
Превращение фосфата в молибдофосфорную кислоту имееТ большое значение и в методах его отделения осаждением.
Некоторые элементы, например висмут(Ш), титан(1У) и цир* копий (IV), осаждаются в виде фосфатов, затрудняя выделенИ. фосфора в виде гетерополикислоты. Другими потенциально М
436
тающими ионами являются мышьяк(У), вольфрам(VI), молибден (VI), ванадий(V), теллур, кремний, селен, но мешающее влияние большинства из этих ионов можно устранить, проводя осаждение из достаточно концентрированных азотнокислых растворов.
Использование хинолина и других органических оснований дает некоторые преимущества при осаждении. Так, можно разделить фосфор и мышьяк, если осаждать молибдофосфат лютидиния [4]. Фосфор и кремний можно разделить, если проводить осаждение молибдофосфата аммония в присутствии цитрата [5].
Осаждение в виде MgNH4PO4-6H2O можно использовать и для определения фосфата, и для его отделения, хотя этот метод менее эффективен для выделения фосфора из растворов, содержащих щелочноземельные металлы, которые тоже осаждаются в аммиачных средах. В присутствии лимонной кислоты фосфаты можно отделить от W, Мо, Мп, V, Fe111, Al, Sn, Zr, Ti, Ca, Zn, F~ и следовых концентраций кремния. Отделение от мышьяка в этих условиях не происходит, так как он образует магний-аммоний арсенат. Разработан метод [6] отделения фосфата при микротитровании сульфата нитратом свинца.
При титровании сульфата перхлоратом бария фосфат отделяли осаждением карбонатом магния [7]; Колсон [8) нашел, что лучшие результаты удается получить, отделяя фосфат в виде труднорастворимой соли серебра.
Для отделения применяют и труднорастворимый фосфат тория. Колсон [9] использовал этот метод для отделения фосфата и фторида при меркуриметрическом титровании хлорида. Было найдено, что 1 —100 мг РО4" можно количественно осадить при pH = = 2,0—3,5 в присутствии 5 г ацетата аммония в 100 мл раствора [10]. Меньше 10 мг магния или кальция в этих условиях не осаждается; они могут быть определены в фильтрате.
Считая классические методы отделения фосфата слишком трудоемкими, авторы работы [11] осаждали фосфат избытком бериллия в присутствии хлорида аммония и ЭДТА, последний добавляли для предотвращения осаждения катионов. В слабоаммиач-ном растворе осаждается BeNH4PO4, избыток бериллия образует гидроксид бериллия. Необходимо обратить внимание на токсичность соединений бериллия, которые используют в этой методике.
Фосфат можно отделить от некоторых элементов, особенно от Щелочноземельных металлов, если осаждение фосфата железа Проводить при pH = 5.
Экстракция
Давно и хорошо известна экстракция гетерополикислот органическими растворителями, например, об экстракции фосфора упоминается в работе 1920 г. [12]. Хотя наряду с этой экстракционной системой исследовали и другие методики, основанные на
4.47
438
Таблица 31. Методы определения фосфора, мышьяка, кремния и германия, основанные на экстракции гетерополикислот
Состав смеси Экстрагент Метод определения Примечание I Литература
р As Изобутилацётат Восстановление Р-ГПК в органической фазе до молибденовой сини. Спектрофотометрия Восстановление в водной фазе As-ГПК до молибденовой сини. Спектрофотометрия 13
Р As Si Изобутилацетат Восстановление Р-ГПК в органической фазе. Спектрофотометрия Восстановление в водной фазе As-ГПК до молибденовой сини. Спектрофотометрия Восстановление в водной фазе Si-ГПК до молибденовой сини. Спектрофотометрия Введение НСЮ, перед добавлением молибдата для предотвращения образования молибденокремневой кислоты Введение лимонной кислоты во вторую аликвотную часть для разрушения Р- и As-ГПК 14
Р Si Ge As Sb Хлороформ — бутанол (4:1) Спектрофотометрическое определение фосфора по молибденовой сини после разрушения гетерополикислоты Вольфрам(У1) маскируют. 100-кратный избыток Si, Ge, As и Sb ие мешает определению. Определяют 1,25—5 мкг Р 15
P As Ge Изобутилацетат, pH = 0,8-1,0 2-Этилгексаиол, pH = 0,4 Восстановление Р-ГПК в органической фазе, спектрофотометрическое определение Восстановление As-ГПК в водной фазе после экстракции Р и Ge. Спектрофотометрическое определение As по молибденовой сини Реэкстракция в Н2О и спектрофотометрическое определение Ge по молибденовой сини 16
As Si 2-Этилгексанол pH < 0,4 Спектрофотометрическое определение As в водной фазе по молибденовой сини Реэкстракция в Н2О и спектрофотометрическое определение по молибденовой сини 16
P As 1 н-Бутилацетат (очень \ плохо экстрагируется) Гравиметрическое определение Метод применен для определения Р в стали 17
Р Si 1 Изобутилацетат 1 Бутанол Определение методом ААС молибдена в Р-ГПК в органической фазе Метод аналогичный Определяют 0,08—1 ppm Р Определяют 0,08—1,2 ppm Si, As и Ge мешают, завышая результаты анализа 18
Р Si Диэтиловый эфир, 1 М по НС1 Диэтнловый эфир — пентанол (5 : 1), 2 М по НС1 Спектрофотометрическое определение по молибдену^!) в УФ-области или определение молибдена методом ААС Метод аналогичный Определяют 0,1—0,4 ppm Р или Si (УФ) 0,1 —1,2 ppm Р или Si (ААС) 19
Р As Sb Ge Si Изобутилацетат Гетерополикислоту реэкстрагируют и разрушают NaOH. MoVI Mov Титруют церием(1У) As, Sb, Ge, Si не мешают определению 20
P As Si Изобутилацетат. Этилацетат — бутанол — изоамиловый спирт Метилизобутилке-тон — цитрат Определение методом ААС Мо в органической фазе Определяют 1 — 10 мкг Р Определяют 2—20 мкг As Определяют 1 — 10 мкг Si 21
Р Пропиленкарбонатхлоро-форм Спектрофотометрическое определение ГПК в органической фазе (е = 22 300 при 308 им) или по молибденовой сини в органической фазе (е = 28 500 при 790 нм) Условия более жесткие при определении по молибденовой сини, чем по ГПК. Определяют 0,4—1,2 ppm Р. При определении 0,5 ppm Р допустимые концентрации As111, Asv н SiIV составляют 10,0 и 50 ppm 22
As Si
CJ Р As Si Этил, пропил или бутилацетат Спектрофотометрическое определение в виде ионного ассоциата с кристаллическим фиолетовым Определяют 0,03—0,3 мкг/мл Р. Насыщенный раствор по силикату н 10-кратный избыток As не мешают определению 23
образовании гетерополпкислоты, обладают по сравнению с ними существенными преимуществами.
Наиболее эффективными экстрагентами являются алкилацетаты и их смеси с другими растворителями. Селективную экстракцию гетерополикнслот обычно используют для разделения микроколичеств Р, Si, As и Ge. Существует несколько вариантов определения экстрагируемых соединений непосредственно в экстрактах. В табл. 31 приведены методы определения Р, As, Si и Ge. Исследовали экстракцию большого числа анионов производными трифе-нилсульфония [24]. Фосфат с большой группой анионов остается в водной фазе, при этом происходит отделение его от перхлората, перманганата и перрената.
Хроматографические методы
С помощью ионообменной хроматографии фосфат можно отделить и от анионов, и от катионов. Фосфат можно отделить практически от большинства катионов [25, 26]. Ванадий отделяют после восстановления до V3+ или VO2+. В присутствии больших концентраций железа (III) перед ионообменным разделением его восстанавливают до железа(II), так как Fe1" может осадить в ионообменной колонке некоторое количество фосфата.
Описаны ионообменные методы отделения фосфата от смеси пиро- и трифосфата [27], пирофосфата [28], смесей гипофосфита и фосфита [29] и пиро-, три-, тримета- и тетраметафосфата [30]. Поллард с сотр. [31] разработали автоматический метод разделения и количественного определения большого числа фосфорсодержащих анионов, включая и ортофосфат.
Большое значение имеют методы разделения фосфата и фторида, причем фосфат в значительно большей степени мешает при определении фторида, чем фторид при определении фосфата. Описаны две методики ионообменного разделения этих ионов [32, 33]. Разработан быстрый метод их разделения [34], основанный на поглощении фосфата суспензией основных смол. Фосфат в виде 0,3 мЛ1 раствора двузамещенного фосфата натрия сорбируется полностью за 2 мин. Ортофосфат и пирофосфат можно отделить от аденозинтрифосфата по методике, основанной на сорбции последнего древесным углем [35].
Бумажная и тонкослойная хроматография являются обычными методами отделения фосфата. В табл. 32 представлены некоторые наиболее важные методики.
Газовую хроматографию используют также для отделения фосфата. Аммонийные соли фосфата, ванадата, арсената, оксалата и т. д. реагируют с бис(триметилсилил)три-фторацетамидом с образованием соединений, которые можно разделить на газожидкостной хроматографической колонке. Используют пламенно-ионизационный детектор, в случае фосфата образуется соединение (TMS)3PO4 [45].
440
графин
441
Электролиз на ртутном катоде
Эгот метод разработан для отделения фосфата от железа, кобальта, никеля, молибдена и т. д., фосфат при этом остается в растворе. Методика была использована [46] для отделения малых концентраций фосфора от железа при анализе особо чистого железа. Опубликован обзор по применению электролиза на ртутном катоде [47].
Соосаждение
Соосаждение фосфата на гидроксиде алюминия оказалось эффективным методом для отделения следовых концентраций фосфата от германия(V), кремния(IV) и мышьяка (V). Германий, кремний и мышьяк можно отогнать с HF, НС1 и НВг, фосфор при этом остается [48].
Селективное подавление влияния нежелательных соединений
Известно, что лимонная, винная и щавелевая кислоты разрушают молибдофосфорную кислоту, но не разрушают молибдокре’м-невую кислоту. Лимонная кислота разрушает молибдомышьяко-вую кислоту [14], затем, после добавления хлорной кислоты и молибдата аммония, образуется преимущественно молибдофосфор-ная кислота, при этом молибдокремневая кислота не образуется. В этой же работе ссылаются на предыдущие работы по изучению влияния оксикислот на гетерополикислоты. Этот вопрос изучался и в работе [49].
Этот прием можно с успехом использовать для анализа смесей Р, Si, As и Ge. Селективное образование гетерополикислот позволяет проводить спектрофотометрическое определение одного иона в присутствии других. Подробнее эта методика обсуждается при рассмотрении спектрофотометрифеских методов определения ортофосфата.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
Метод с осаждением в виде магний-аммоний фосфата
В течение уже половины столетия метод определения фосфора в виде MgNH4PO4-6H2O является основным:
РО?‘ + NHJ + Mg2+ + 6Н2О —> MgNH4PO4 6Н2О
2MgNH4PO4 • 6Н2О —> Mg2P2O7 + 2NH3 + 7Н2О
Эти же реакции положены в основу стандартного макроопределения магния, несмотря на многочисленные потенциальные источники ошибок.
142
Метод подробно описан в многочисленных оригинальных работах и в обзорах [1, 51, 52]. Он основан на добавлении к подкисленному анализируемому раствору фосфата магнезиальной смеси * и аммиака до небольшого избытка, после чего раствор над осадком выдерживают 4 ч или оставляют на ночь. После этого осадок растворяют и повторяют осаждение. Наконец, после фильтрации и промывания осадка магний-аммоний фосфат переводят в пирофосфат при 900 °C и взвешивают. Эта кажущаяся простой методика заключает в себе ряд потенциальных ошибок.
Магний-аммоний фосфат — одно из наиболее растворимых соединений, применяющихся в количественном гравиметрическом анализе. Это следует учитывать при выполнении аналитических операций. Необходимо сравнительно длительное время для осаждения, чтобы получить осадок постоянного состава. Обычно осадок с раствором оставляют на 4 ч или даже на ночь. Золотавин, Сана-тина и Нестеренко [53] изучали возможность ускорения этого процесса, так же как и процессов образования осадков сульфата бария и оксалата тория и нашли, что замораживание осадка до —18—78 °C и затем оттаивание его в горячей воде (60—70 °C) приводит к образованию крупного кристаллического осадка, значительно более удобного для гравиметрических целей по сравнению с мелкокристаллическим. Результаты определения MgNH4PO4-6H2O показали, что благодаря этому приему продолжительность анализа можно сократить с 7 до 3 ч, при этом точность анализа не уменьшается.
Процессы соосаждения в этом методе могут приводить и к отрицательным и к положительным ошибкам, в зависимости от соосаждающихся соединений. Например, сорбция фосфата аммония вызывает отрицательные ошибки, в то время как соосаждение фосфата магния является источником положительных ошибок, которые устраняют переведением фосфата при повышенных температурах в пирофосфат. Чистый MgNH4PO4-6H2O разлагается при 380—480 °C [54], температура 900 °C необходима для превращения в пирофосфаты некоторый сорбированных фосфатов. Однако некоторые соединения, например, Mg(H2PO4)2, теряют оксид фосфора^) в заметной степени лишь при 1150—1200°С.
Повторное осаждение необходимо для удаления избытка ионов магния и аммония. В одной из работ Линделла и Хофмана [55] отмечается, что при переосаждении происходит потеря фосфора. За счет проведения осаждения из аммиачных растворов можно ожидать мешающего влияния большого числа катионов, в том числе и ионов щелочноземельных металлов. Для маскирования Железа успешно используют лимонную кислоту [56]. При введении достаточного количества лимонной кислоты можно проводить определение в присутствии заметных концентраций Са, Fe, Al, Sn, V, Ti и Zr. Мышьяк мешает определению, так как образует аналогичную магниевую соль. Было найдено [57], что железо и алюми-
• Раствор MgCl26H2O и NH«C1, содержащий небольшой избыток NH3.
443
нпй при концентрации больше 1% можно маскировать введением ЭДТА, кальций при этом также связывается в комплексонат и не мешает определению. Этим приемом достигается более полное устранение мешающих ионов, чем в случае отделения фосфора в виде гетерополикислоты. Те же выводы сделаны и в работе [58].
Тайрон (пирокатехол-3,5 дисульфокислота) также используют в качестве маскирующего агента при определении фосфата в присутствии А1, Сг, Bi, Sb, Sn, Fe111, U, Ti или Be. Для отделения катионов перед проведением определения можно использовать и ионообменную хроматографию.
Для определения фосфата в виде MgNH4PO4-6H2O был разработан метод осаждения из гомогенной среды [59]. Медленное образование NH3 по реакции между хлоридом аммония и этилами-ном приводит к медленному образованию в присутствии иона магния осадка. Мешающее влияние кальция устраняют введением молочной кислоты; Fe, Ti и Al маскируют ЭДТА. При использовании этого варианта гравиметрического метода ошибка определения составляет ±0,4%.
Метод с осаждением в виде молибдофосфата аммония
При добавлении избытка молибдата аммония к раствору фосфата в разбавленной HNO3 образуется желтый труднорастворимый осадок молибдофосфата аммония *:
РО^ + 12Н2МоО4 + 3NIС —> (NH4)3 [РО4 • 12МоО3] • 12Н2О
Точное строение гетерополикислоты (ГПК) явилось предметом споров. Каннон [60] предполагал, что строение ГПК выражается формулой (NH4)2HPO4-i2MoO3-12H2O. В работе [61] пришли к выводу, что строение осадка — среднее между теоретическим и приведенным в работе [60]. В более поздних исследованиях были сделаны попытки обобщить имеющиеся в литературе результаты исследования осадков.
С 1826 г. известна способность фосфорной кислоты образовывать гетерополикислоты. Многие аналитические методы определения фосфора основаны на предварительном осаждении ГПК-Осажденный молибдофосфат аммония содержит HNO3 и воду. Избыток реагента удаляют, промывая осадок разбавленной HNO3, которую затем удаляют, промывая осадок раствором нитрата аммония. Осадок водой разрушается. Образование осадка точного состава зависит от различных факторов, в частности, от концентрации растворов молибдата и HNO3 и температуры осаждения. При осаждении рекомендуется вводить нитрат аммония для ускорения образования осадка и снижения его растворимости.
Изучали условия, необходимые для количественного определения фосфата [62] с использованием 32Р и "Мо, определяли [63]
• Правильнее эту соль называть триаммоний додекамолибдофосфат (ИЮПАК).
411
растворимость осадка в разбавленной HNO3. Найдены по результатам работ [62, 63] оптимальные условия определения. Фосфат можно количественно осаждать из азотнокислых растворов молибдатом при 50—80 °C. Смесь выдерживают не менее 30 мин при этой температуре и 30 мин при комнатной температуре, каждые 15 мин раствор перемешивают. Анализ осадка, полученного при 80 °C, показал соответствие с соотношением компонентов в формуле, приведенной в работе [61]. Двукратный по сравнению со стехиометрическим избыток молибдатного реагента позволяет при 70°С выделить 99,9% фосфата. Присутствие HNO3 необходимо, образующийся нитрат аммония не влияет на отделение фосфата. Хлорная кислота в огличие от НС1 и H2SO4 не мешает определению.
Осадок можно сушить при 210—410 °C и взвешивать в виде (NH3) [РО4- 12МоО3], хотя это соединение гигроскопично. Повышение температуры до 500—700 °C приводит к образованию мо-либдофосфорного ангидрида Р2О5-24МоО3, рекомендованного в разработанном ранее стандартном методе в качестве весовой формы.
Присутствие хлорида, фторида и сульфата аммония тормозит образование осадка, такое же действие оказывают HCI, HF и H2SO4. Мышьяк, селен, кремний, теллур, вольфрам, ванадий, титан и цирконий мешают определению, их мешающее влияние можно устранить, если осаждение проводить в более концентрированных растворах HNO3. Изучали [64] влияние мышьяка и ионов переходных металлов на осаждение, результаты исследований показали, что фосфат можно количественно осадить при 50—70°C 3,5-кратным по сравнению со стехиометрическим избытком молибдата аммония даже в присутствии эквивалентных количеств мышьяка. С помощью радиоактивных изотопов было показано, что количество осаждающегося As зависит от избытка молибдата аммония и температуры, при которой проводят осаждение. В этой же работе было найдено, что нитрат железа ингибирует осаждение фосфата (так же, как и арсената), нитраты хрома (III), никеля (II) и марганца (II) оказывают меньше влияния на скорость образования осадка.
Следовательно, точность гравиметрического определения фосфата этим методом зависит от точного соблюдения условий реакции. Недостаток метода, связанный с нестехиометричностыо состава осадка, можно устранить введением эмпирического фактора пересчета.
Разработано несколько вариантов определения. Например, мо-либдофосфат аммония можно растворить в растворе NH3 и содержание молибдена в образующемся растворе определить гравиметрически в виде молибдата свинца — соединения, являющегося более удобной весовой формой. В другой методике применяли амперометрическое титрование осадка. Эти методы будут рассмотрены в подразделе, посвященном титриметрическнм методам.
445
Метод с осаждением молибдофосфата органических оснований, в том числе хинолина
Изучали некоторые органические соединения с целью замены ими иона NH-. Ранние работы рассмотрены в обзоре [65] и монографии Белчера и Вилсона [66]. Макдональд и Риверо [67] изучили большое число органических оснований и нашли, что а-пц. колин (2-метплпиридин) обладает некоторыми преимуществами по сравнению с хорошо известным хинолином, хотя его использование ограничивается тнтриметрией.
Хеслоп и Пирсон [4] применили 32Р для изучения возмож-ности использования молибдатов лютилиния и хинолиния в качестве реагентов для осаждения фосфата. При использовании последнего в оптимальных условиях количественного осаждения фосфата осаждается и мышьяк, при использовании молибдата лютидиния мышьяк остается в растворе.
С целью увеличения массы осадка изучали ион иодиния (С6Н5)21+ [68]. Получили те же результаты, что и в случае применения хинолиния, с теми же мешающими ионами.
Среди всех изученных оснований хинолин, впервые предложенный для этих целей Вилсоном [69], оказался оптимальным:
Н3РО4 + 12Na2MoO4 + 24НС1 —> Н3 [РО4 • 12MoOs] + 24NaCl + 12Н2О
3C9H7NHC1 + Н3 [РО4 • 12Мо03] ► (C9H7N)3 Н3 [РО4 • 12МоО3] + ЗНС1
В методике, предложенной Вилсоном, раствор солянокислого хинолина медленно добавляют к кипящему раствору фосфата, содержащему молибдат натрия и подкисленному НС1. По этой методике получается крупный кристаллический осадок, удобный для фильтрования. Методика рекомендуется для гравиметрического определения средних концентраций фосфата или для алкалиметрического титрования осадка. Этот вариант метода очень популярен и нашел чрезвычайно широкое применение. О методе Вилсона
будет упомянуто в этом разделе и в связи с титриметрическими методами определения фосфата.
По сравнению с методом осаждения молибдофосфата аммония метод Вилсона обладает следующими преимуществами:
1) осадок менее растворим, 2) состав осадка ближе к теоретическому, 3) метод относительно прост и свободен от процессов соосаждения, 4) меньше число мешающих ионов, 5) менее критичными являются сами условия осаждения, такие, как температур3 и избыток реагентов. Перрин [70] нашел, что повышение температуры до 200—250 °C приводит к превращению осадка в соединение строго стехиометричного состава (С9Н7М)3-Нз[РО4- 12МоОзЬ а при повышении температуры до 550 °C получается молибдофосф°Р' ный ангидрид, менее удобная весовая форма. При термограви метрическом исследовании [17] было найдено, что стабильная без* водная форма образуется при 155—370°C.
Мешают определению те же соединения, что мешают в меТд„ осаждения молибдофосфата аммония. Мешающее влияние мыШь
446
ка, кремния, H2SO4, HF и ионов аммония можно устранить, если рвести некоторые дополнительные операции. Удалось, введя модификацию метода, устранить влияние Fe, As, Zr, Nb, Та и W при титриметрпческом и гравиметрическом определении фосфора в железе и стали [72]. Мышьяк отделяли отгонкой с НВг. Соосажде-ние железа предотвращается при введении большего количества реагента или при выдерживании раствора при температуре, близкой к температуре кипения. Мешающее влияние кремния устраняли введением цитрата [73], который предотвращает образование цолибдокремневой кислоты, однако при определении микроконцентраций фосфата более 100 мг лимонной кислоты влияет на осаждение молибдофосфата [74]. Добавлением цитрата можно устранить мешающее влияние ионов аммония, так как молибдофосфат аммония в этих условиях растворим. Устранение мешающего влияния иона аммония особенно важно при анализе удобрений, где метод применяется очень интенсивно. Для тех же целей можно использовать ацетон, этот растворитель включен в состав реагентов для определения фосфора, куда наряду с ацетоном входят хи-
нолин, молибдат натрия, лимонная и азотная кислоты [75].
Влияние фторида, связанное с выщелачиванием кремния из стекла, можно устранить добавлением борной кислоты [74]. Влияние железа на определение очень малых -концентраций фосфата мало. На определение не влияют кальций, магний, алюминий, щелочные металлы и цитрат, 18-кратные по отношению к фосфату концентрации хрома и 3,5-кратные концентрации титана не мешают, содержание ванадия должно быть не больше 0,2 от содержания фосфата.
Метод определения в виде молибдофосфата хинолина удовлетворителен по своей точности, воспроизводимости и сложности. Он рекомендован после всестороннего исследования в качестве возможного стандартного метода анализа удобрений [76, 77]. Гравиметрическое определение 2—3 мг фосфора применяют в полу-микроорганическом элементном анализе; осадок взвешивают после высушивания при 160 °C, чтобы ускорить методику, осадок промывают диоксаном или эфиром [78].
Метод с осаждением фосфата серебра
Фосфат серебра можно осаждать из гомогенных растворов при постепенном поступлении в раствор ионов серебра, образующихся в результате разрушения аминов серебра [79]. Как и в случае постепенного удаления NH3 при нагревании растворов в методе Осаждения MgNH4PO4-6H2O, по этой методике получаются круп-вые, легко фильтрующиеся кристаллы, содержащие 7,4% Р.
Можно определять 3—300 мг РО/ с ошибкой, меньше 0,13 мг. нРи изучении влияния посторонних ионов было найдено, что ка-Лии- аммоний, нитрат и ацетат не оказывают существенного влия-ия на определение, большие концентрации натрия и сульфата не-к°лько завышают результаты анализа. Силикат в значительной
447
степени мешает определению, арсенат, хромат, вольфрамат и ванадат также влияют на определение. Многозарядные катионы мешают определению, но их можно перед определением отделить с помощью ионного обмена. Хлорид, иодид, бромид, цианид, роданид и оксалат, образующие с серебром труднорастворимые соединения, следует предварительно отогнать с HNO3. После выпаривания раствора почти досуха удается добиться полного отделения этих анионов. Для разрушения полифосфатов, которые могут образовываться в результате предварительной обработки анализируемого раствора, необходимо проводить гидролиз в течение 4 ч в горячем растворе HNO3.
Другие гравиметрические методы
Фосфат можно осадить в виде серебро-таллий(I) фосфата [80] при добавлении к раствору фосфата ацетата таллия и нитрата серебра. Осадок устойчив при 20—720°C. Можно использовать титриметрическую методику, основанную на методе Фольгарда.
Фосфат висмута — еще одна весовая форма, предложенная для определения фосфата [81]. В 1 М HNO3 определению не мешают Mg, Zn, Мп, Ni и Со.
Гравиметрическое определение фосфата в виде магний-алюмииий фосфата [82]
Реагенты х , 1О
Нейтральная магнезиальная смесь. Растворяют 102 г MgC12-6H2O и 53 г NH4CI в дистиллированной воде, добавляют 5 мл NH4OH (плотность 0,880 г/см3) и разбавляют дистиллированной водой до 1 л. Оставляют стоять сутки и фильтруют.
Хлористоводородная кислота, 30%-ный раствор. Разбавляют 30 мп НС1 (плотность 1,180 г/см3) дистиллированной водой до 100 мл.
Гидроксид аммония, 10%-, 1%- и 3%-ный растворы. Разбавляют 10 мл NH4OH (плотность 0,880 г/см3) дистиллированной водой до 100 мл (10%-ный раствор); 1- н 3%-иый растворы готовят разбавлением 10%-ного раствора.
Лимонная кислота, твердая.
Метиловый красный. Растворяют 0,20 г метилового красного в воде, содержащей 1 мл 1 М раствора NaOH, и разбавляют до 1 мл дистиллированной водой.
Ход анализа. К 100—200 мл холодного раствора, содержащего 10 мл раствора НС1 (плотность 1,18 г/см3) и 0,5—0,75 г Р2О5, добавляют 2 г лимонной кислоты и несколько капель индикатора метилового красного, затем добавляют NH4OH (плотность 0,880 г/см3) до тех пор, пока индикатор не станет желтым-Проводят осаждение (при постоянном перемешивании раствора), добавляя по каплям 100 мл магнезиальной смеси и затем 10%-иый раствор NH4OH до тех пор, пока суммарная концентрация NH<OH ие станет 2,5—3 0% по объему. РаС' твор с осадком оставляют стоять сутки.
Прозрачный раствор декантируют и отфильтровывают на воронке Бюхнера, используя в качестве фильтра фильтровальную бумагу ватман № 31. Фильтрат отбрасывают, небольшое количество осадка, попавшее иа фильтр, растворяют пропуская через фильтр 30 мл горячей 30%-иой НС1. Этот раствор собирают в колбу, содержащую основное количество осадка. Фильтровальную бумагу пр*^ мывают несколько раз дистиллированной водой. К раствору добавляют 2 г л g монной кислоты, 10 мл нейтральной магнезиальной смеси (см примечание О ’ по каплям, при постоянном перемешивании раствора, вводят NH4OH (плотное
448
л 880 г/см3) до тех пор, пока не перестанет выпадать осадок. Если объем осадка пёлнк, основную его часть опять растворяют, добавляя по каплям 30%-ную НС1; этот прием довольно эффективен.
’ Общий объем раствора доводят до 100 мл, охлаждают и по каплям, при постоянном перемешивании, добавляют 1%-ный раствор NH4OH и затем 90 мл Ю >-ного раствора NH4OH, так чтобы суммарная его концентрация в растворе составляла 3% по объему (в пересчете на концентрированный NH4OH с плотностью 0,880 г/см3). Оставляют раствор стоять не меиее 4 ч, лучше — на сутки.
фильтруют через бумажный фильтр ватман № 31, тщательно перенося на фильтр кристаллы, которые осели на мешалку и на стенки колбы, с помощью стеклянной палочки с надетой на ее конец резиновой трубкой Фильтр и осадок шесть раз промывают 3%-иым раствором NH4OH. Фильтр с осадком помещают во взвешенный платиновый тигель. Добавляют не более 0,2 г NH4NO3 и обугливают бумажный фильтр, не давая образоваться пламени. Углерод выжигают при температуре около 900 °C и доводят тигель до постоянной массы в муфель-у ной печи при 1050—1100 °C (см. примечание 2).
фактор пересчета с Mg2P2O7 на Р2О5 равен 0,6377, а на Р — 0,2784.
Примечания. 1. Рекомендованные количества лимоииой кислоты н избыток магнезиальной смеси нельзя менять, недостаток магнезиальной смеси может привести к заниженным результатам анализа.
2. Обычно с магний-аммоний фосфатом соосаждаются небольшие количества цитрата магния, которые сжигают до углерода и удаляются только при точном соблюдении методики.
3. Точность метода 0,5%.
Титриметрические методы
Метод кислотно-основного титрования молибдофосфатов аммония и хинолина
Мы уже говорили о трудностях гравиметрического определения фосфата в виде молибдофосфата аммония. Одним из возможных вариантов этого метода является алкалиметрическое титрование осадка в присутствии фенолфталеина:
(ЫН4)3 [РО4 • 12МоО3] + 26NaOH —> 12Na2MoO4 + Na2HPO4 + 3NH3 + 14H2O
В результате реакции выделяется NH3, что делает конечную точку титрования нечеткой. Применение молибдофосфата хинолина имеет ряд преимуществ перед аммонийной солью, некоторые преимущества уже отмечались в связи с обсуждением гравиметрического метода определения фосфата. Достоинства метода отмечались в работе Вилсона [69] и полностью подтверждены в более поздних работах. Этот метод в настоящее время является стандартным методом определения макро- и микроконцентраций фосфата, если., от результатов анализа требуется высокая точность. Уравнение реакции можно представить следующим образом:
(C.H7N)3 Hs [РО4 • 12МоО3] + 26NaOH —»
—» Na2HPO4 + 12Na2Mo04 + 3CaH7N + 14H2O
методику Вилсон применил для анализа удобрений и некоторых других веществ. Осадок отфильтровывали на бумажном Фильтре, затем его промывали до полного удаления кислоты и
15 Зак 167 449
растворяли в избытке стандартного раствора NaOH. Не вступив-шую в реакцию NaOH оттитровывали стандартным растворов НС1, в качестве индикатора использовали смесь фенолфталеина ц тимолового синего. Изучали влияние на определение веществ, обычно присутствующих в удобрениях. Было найдено, что все эти вещества ведут себя так же, как и в методе определения ортофосфата в виде молибдофосфата аммония. При определении низких концентраций фосфата влияние Fe3+. невелико. Мышьяк и кремний I в этих условиях осаждаются количественно, мешающее влияние кремния можно устранить введением цитрата [73].
Некоторые ионы являются ингибиторами осаждения, как и в гравиметрическохм методе. Хеслоп и Пирсон [4] нашли, что увеличение избытка реагента (что не влияет на результаты определения ортофосфата) и выдерживание осадка с раствором в течение 2 ч устраняет ингибирующее действие некоторых ионов, особенно железа (III).
Существенное улучшение метода было сделано Фернлундом и Цехнером [83], которые предложили сложный осадитель, устойчивый в течение месяца (реагент, описанный Вилсоном, устойчив лишь в течение недели). Титриметрические методы, основанные на образовании осадка молибдофосфата аммония или хинолина^ применяют для анализа удобрений. В своем первоначальном варианте алкалиметрический метод неприменим в присутствии больших концентраций аммония, мешающих анализу нейтральных или щелочных цитратных экстрактов удобрений. Для того чтобы снизить возможность перехода аммония в экстракт в состав реагента [75], был введен ацетон (реагент: хинолин, молибдат натрия, лимонная и азотная кислоты и ацетон), как уже отмечалось раньше.
Одним из недостатков метода, который ограничивает его использование, является способность осадка образовывать плотные комки, которые впоследствии трудно растворяются. Как было найдено [84], дополнительное введение лимонной кислоты способствует образованию крупнокристаллического осадка, который при фильтровании не образует агломератов и легко растворяется в щелочи.
Авторы этой работы [84] нашли, что метод вполне надежен и по точности сравним со стандартным гравиметрическим методом. После разностороннего изучения метод рекомендован [85] для определения в удобрениях общего фосфора (прямое определение) и фосфора, нерастворимого в цитратном растворе.
Были сделаны попытки улучшить титриметрический метод, основанный на образовании молибдофосфата аммония, но положительный эффект довольно сомнителен, а точность этого варианта метода значительно ниже, чем в методе с применением молибдо* фосфата хинолина [84]. Последний метод нашел многочисленны® области применения, например, в анализе железа и стали [72]. в микро- и полумикроорганическом анализе [74, 76].
450
Титриметрическое определение фосфата в виде молибдофосфата хинолина [83]
Реагент
Раствор реактива. Растворяют 250 г Na2MoO4-2H2O в 500 мл воды, добавляют 460 мл концентрированной HCI н 1 каплю 30%-кого Н2О2. К 660 мл (1:1) HCI добавляют 28 мл хинолина. Этн два раствора смешивают и нагревают до кипения. Охлаждают, выдерживают не менее 24 ч и фильтруют.
Ход анализа (до 25 мг Р). К анализируемому раствору фосфата добавляют раствор НС1 до 0,9—1,6 М и несколько капель 6%-ного раствора КСЮз- Нагревают до кипения и добавляют 10 мл реагента (при содержании фосфата больше |5 мг добавляют 20 мл реагента). Раствор оставляют на несколько минут, фильтруют через бумажный фильтр н промывают сначала 1 М НС1, затем водой до нейтральной реакции промывных вод. Осадок смывают в колбу, в которой проводили осаждение (колбу предварительно отмывают от кислоты) и добавляют к нему избыток стандартного (0,1 Л1) раствора NaOH. Избыток щелочи титруют 0,1 или 0,5 М НС1 по фенолфталеину.
Примечание. 1. Реагент, использующийся в этой методике, можно хранить в течение месяца, тогда как реагент, предложенный Вильсоном [69], хранится лишь в течение недели.
Другие титриметрические методы, основанные на предварительном образовании молибдофосфорной кислоты
Описан метод [88] определения следовых концентраций фосфора, в котором дважды используют прием умножения. Сначала фосфат переводят в 12-молибдофосфорную кислоту, а молибден, входящий в ее состав, после разрушения кислоты переводят в комплекс с оксихинолином. Комплекс после добавления КВг титруют КВгО3.
Так как каждый моль молибдата реагирует с двумя молями органического реагента, а на каждый моль оксина расходуется четыре эквивалента стандартного раствора бромата, в результате анализа получается 96-кратное умножение исходной концентрации фосфата.
РОГ —> Н2 [РО4 • 12МоО3] X 12 (Мо)
MoVI —> МоО2 [CeHeON]2 X 2 (оксии)
С8О7ОН СвН6Вг2ОН 4-2HBr X 4 (бром)
Разработана еще одна методика [20], в которой используется принцип умножения. Молибдофосфорную кислоту селективно экстрагируют изобутилацетатом, реэкстрагируют в водную фазу и разрушают щелочью. Образующийся молибден(VI) восстанавливают до молибдена (V) в серебряном редукторе и определяют це-Риметрическим титрованием в присутствии индикатора ферроина. Серебряный редуктор, который используют в этой методике, был описан в работе [89] Определению ортофосфата этим методом не мешают элементы, образующие гетерополикислоты (As, Sb, Ge и Si), и многие другие ионы. За счет эффекта умножения можно
15* 451
определить 10~6 г фосфата, используя обычную бюретку, на титрование расходуется несколько миллилитров 10~3 Л1 раствора титранта, точка эквивалентности определяется визуально.
Определение 3—30 мкг фосфора методом, основанным на образовании молибдофосфорнон кислоты и использующим эффект умножения [20]
Аппаратура и реагенты
Восстановительная колонка (редуктор) размером 10X1 см, заполненная серебром на высоту 7 см Серебро готовят следующим образом: 30 г нитрата серебра растворяют в 400 мл воды, добавляют несколько капель HNO3, пО|ружают в раствор лист электролитической меди’ размером 10 см2 и перемешивают до тех пор, пока серебро не осядет полностью. Осадок промывают декантацией разбавленной H2SO< до тех пор пока не удалят большую часть меди, затем переносят осадок серебра в редуктор водой так, чтобы в колонку не попадал воздух, и промывают разбавленной H2SO« до тех пор, пока полностью не удалят медь. Постоянно колонку хранят заполненной 2 М раствором НС1, а непосредственно перед использованием — горячей (60—80 °C) 2 М НС1.
Стандартный раствор ортофосфата. Растворяют 0,1098 г однозамещенного ортофосфата калия в дистиллированной воде н разбавляют раствор до 1 л; 1 мл раствора эквивалентен 25 мкг фосфора.
Молибдатный реагент. Растворяют 10,69 г тетрагидрата молибдата аммония (NN4)6Mo7O24-4H2O в 1 л дистиллированной воды.
Раствор сульфата церия(1У). Исходный 0,05 М раствор разбавляют I М H2SO4 до IO-3 М. <
Ферроин, 5-Ю-3 М водный раствор. В каждый раствор при титровании добавляют 2 капли этого раствора.
Ход анализа. Анализируемый раствор (^5 мл), содержащий 3—30 мкг Р, помещают в делительную воронку вместимостью 100 мл. содержащую 10 мл мо-либдатиого реагента и 10 мл воды. Добавляют концентрированную НС1 так, чтобы раствор стал 0,96 М по НС1, и выдерживают смесь 5 мин Добавляют 10 мл изобутилацетата и 1 мин встряхивают воронку.
Когда фазы полностью разделятся, водную фазу отбрасывают, а органическую фазу промывают 10 мл 2 М HCI. Затем органическую фазу встряхивают с 5 мл 4 М раствора NH4OH, сливают изобутилацетат, оставшийся щелочной раствор содержит молибден (VI).
Водную фазу переносят из делительной воронки в колбу вместимостью 50 мл, добавляют 6 М НС1 для нейтрализации NH4OH и доводят раствор НС1 до 2 М концентрации по НС1. Раствор количественно переносят в редуктор, используя для этого 2 М НС1. В колоике-редукторе проходит восстановление молибдена (VI) до молибдена (V), который элюируют, добавляя в колонку три раза порциями по 5 мл горячую 2 М НС1 и три раза порциями по 5 мл холодную 2 М НС1. Элюат собирают в колбу для титрования, быстро нагревают его до кипения, охлаждают почти до комнатной температуры и молибден (V) титруют стандартным 10~5 М раствором сульфата церия (IV) в присутствии ферроииа. Точку эквивалентности (после определенного навыка) можно определять визуально по исчезновению последних следов розовой окраски.
Примечания. 1. Определению ие мешают I—6 мг следующих иоиов: SeIV, Te,v, Ba”, Мп”, SiIV, Al"1, Pb”, Co”, Ni", Asv, GeIV, Ca", Fe"1, Zn"r Cd”, Cu”, Sbv.
2. Метод можно использовать для полумикроопределеиия фосфора в органических соединениях.
Комплексометрический метод
Разработано несколько косвенных титриметрическнх методов определения фосфора с использованием ЭДТА. Все они основаны ffS образовании труднорастворимых фосфатов металлов, в одних
452
методах осадок отделяют и после его растворения находят содержание металла в осадке, в других методах в фильтрате после отделения осадка определяют избыток металла-осадителя.
В более ранних работах использовали MgNH4PO4-6H2O, Mg2+ титровали комплексонометрически в аммиачном буферном растворе в присутствии индикатора эриохром черного Т [58]:
poi + nh; + Mg2++ брьо — MgNH4po4 • бн2о
Интересной особенностью этой работы является применение ЭДТА в качестве маскирующего агента при осаждении MgNH4PO4-6H2O. ЭДТА, добавленный перед осаждением, комплексует все поливалентные катионы. При введении реагента-осадителя, содержащего хлориды магния и аммония, Mg2+ сначала реагирует с избытком ЭДТА, а затем осаждается в виде MgNH4PO4-6H2O. Сравнительно небольшая константа устойчивости комплекса Mg — ЭДТА не позволяет магнию вытеснять поливалентные катионы из их комп-лексонатов, исключение составляют железо, алюминий, таллий и бериллий. Для маскирования железа, алюминия и таллия вводят лимонную кислоту, бериллий связывают сульфосалициловой кислотой.
Методика, основанная на использовании MgNH4PO4-6H2O, обладает недостатками, например, осадок сравнительно хорошо растворим, что может привести к неполному осаждению фосфата и ошибке анализа, особенно это относится к определению малых концентраций фосфата. Соосаждение также является потенциальным источником ошибок анализа. Этот фактор можно исключить практически полностью, используя переосаждение. Более серьезным источником ошибок является непостоянство состава осадка. Осадок постоянного состава можно получить, лишь выдерживая осадок с раствором в течение длительного времени, точность результатов зависит от мольного отношения Mg: РО4, которое должно быть точно 1: 1. В методике [90J использовали цинк-аммоний фосфат, преимущество метода в том, что при титровании цинка конечная точка титрования четче, чем при титровании магния, недостатком метода с применением ZnNH4PO4 является необходимость проводить титрование при pH = 10.
Достоинством методов осаждения фосфата из кислых растворов является их большая селективность. Это преимущество реализуется при использовании висмута [91], циркония [92, 93], лантана [94] и тория [95, 96], которые были предложены для осаждения фосфата. При использовании висмута необходимо тщательно поддерживать кислотность растворов: если добавлен большой избыток кислоты, — растворяется осадок фосфата висмута, если кислоты недостаточно, — образуется хлопьевидный флоккулентный осадок. Ридел [91] выпаривал досуха анализируемый раствор и затем доводил кислотность раствора до оптимального значения 0,2 М по НС1О4. Добавлял раствор висмута и проводил обратное Титрование раствором ЭДТА с пирокатехиновым фиолетовым в
453
качестве индикатора. Перед анализом, если это необходимо, сульфаты отделяли в виде сульфата бария, а хлориды (которые тоже могут мешать определению) удаляются при выпаривании образца. Метод был использован [97] для определения суммы фосфата и арсената в смеси, кроме того, арсенат затем определяли иодиметрически. Мешающие катионы (Pb2+, Fe3+) отделяли с помощью ионного обмена, хлориды и сульфаты—нитратом серебра и нитратом бария соответственно.
Разработан косвенный комплексометрический метод, основанный на осаждении фосфата циркония известным избытком цирко-ния(1У) в 15—20%-ной H2SO4. Затем титруют цирконий(1У), не вступивший в реакцию с ортофосфатом, ЭДТА с ксиленоловым оранжевым в качестве индикатора. Метод характеризуется высокой селективностью, поскольку осаждение и титрование проводят в сильнокислых растворах. Авторы работы [92] полагают, что соотношение Zr: Р в осадке равно 1 :2 (в отличие от авторов работы [93]), которые нашли, что соотношение равно 1 : 1. В дальнейших работах был сделан вывод на основании рентгенографических данных о том, что осаждается, вероятно, фосфат цирконила ZrO(HPO4), а не Zr(HPO4)2-
Методика, описанная в работе [93], может быть использована для определения 0,95—118 мг РО^ > тогда как методика, приведенная в работе [92], позволяет определять лишь 35—55 мг РО^Т определению не мешают железо(III), титан(IV), торий(IV) и впсмут(Ш). Если в растворе присутствует больше 1,4 мг фторида, его следует отделить. В качестве индикатора применяют ксиленоловый оранжевый, титрование проводят при 90 °C.
Описан [98] косвенный комплексометрический метод, основанный на осаждении фосфата из нейтральных или слабощелочных растворов нитратом серебра. Осадок взаимодействует с KzNi(CN)4, вытесняя эквивалентное количество Ni2+, которое титруют стандартным раствором ЭДТА. На этом же принципе основан и метод определения микроконцентраций фосфата.
Быстрый комплексометрический метод, основанный на осаждении при pH = 5,0 фосфата лантана, описан в работе [94]. Можно определить в присутствии хлорида, сульфата, ионов щелочных и щелочноземельных металлов 50—300 мг РО?'. Методика заключается в введении в анализируемый подкисленный раствор, содержащий фосфат, известного объема стандартного раствора хлорида лантана и, после образования осадка фосфата лантана, титровании избытка лантана ЭДТА в присутствии ксиленолового оранжевого. В присутствии сульфата в методику следует ввести некоторые изменения. С использованием 0,01 М растворов хлорида лантана и ЭДТА можно определять 1—10 мг фосфата. Определению не мешают 1930 ppm хлорида, 480 ppm Mg2+, 800 ppm Са2+, 1100 ppm Na+, 1920 ppm сульфата и 2740 ppm Ba2+. В отличие от метода с использованием циркония, не требуется отделять осадок.
454
Описан быстрый титриметрический метод [95] определения очень малых концентраций фосфора (10—50 мкг), в котором осаждают фосфат тория. И в этом случае не нужно отделять осадок. В анализируемом растворе могут присутствовать большие концентрации хлорида, бромида, иодида, нитрата, сульфата, перхлората и силиката; фторид, борат и арсенат мешают определению. Осаждение проводят нитратом тория, титруют избыток тория при pH = 3 в присутствии ксиленолового оранжевого.
Комплексометрическое определение фосфата [93]
Реагенты
Нитрат циркония, 0,05 М раствор. Растворяют 13,5 г нитрата цирконила (ZrO(NO3)2-2H2O) в 140 мл 8 М HNO3. Для деполимеризации полимерных образований цирконил-иона раствор кипятят 5 мии и затем разбавляют до 1 л.
ЭДТА, 0,05 М раствор. Растворяют 18,6 г двунатриевой соли ЭДТА в 1 л дистиллированной воды. Точную молярную концентрацию раствора определяют по одному из стандартных веществ, например по оксиду цинка. Из этого пас-твора готовят разбавлением 0,02 М раствор.
Ксиленоловый оранжевый, 0,2%-ный раствор.
Ход анализа. Анализируемый раствор, содержащий 0,95—118 мгРО®',помещают в мерную колбу вместимостью 250 мл, добавляют 125 мл 4 М HNO3 и разбавляют до 200 мл. Добавляют известный избыточный объем 0,05 М раствора нитрата циркония (10—30 мл), нагревают 30 мин иа водяной баие, охлаждают, доводят объем 1 М HNO3 до метки и перемешивают.
Фильтруют раствор через сухой бумажный фильтр, первую порцию фильтрата отбрасывают. Переносят 100 мл фильтрата в коническую колбу вместимостью 500 мл и разбавляют до 200 мл водой. Нагревают раствор до кипения и кипятят 1 мии для удаления оксидов азота, затем цирконий (IV) медленно титруют стандартным раствором ЭДТА, используя в качестве индикатора ксиленоловый оранжевый. Температура раствора при титровании должна быть 90 °C или выше, раствор ЭДТА вблизи точки эквивалентности следует добавлять со скоростью 1 капля в 1 с. Конец титрования определяют по переходу окраски раствора от розово-красиой до лимонио-желтой.
Точную молярную концентрацию раствора циркония определяют титрованием ЭДТА в этих же условиях.
Поскольку осаждение фосфата циркония проходит в 2—3,5 М HNO3, число мешающих ионов мало, титан (IV), висмут (III), железо (Ш) и торий(IV) не мешают определению. Присутствие фторида завышает результаты анализа, вероятно, за счет образования прочного комплекса с цирконием (IV). При содержании фторида больше 1,425 мг следует образец предварительно обрабатывать смесью НСЮ4 и борной кислоты для удаления фторидов.
Метод был с успехом применен для анализа удобрений.
Другие титриметрические методы
Фосфат можно определять титрованием растворами висмута (III) [99, 100). В работе [100] 50 мг РО1~ титровали с применением в качестве индикатора метилтимолового синего. Переход окраски индикатора от зеленой до красновато фиолетовой легче наблюдать, если к раствору добавить хлороформ и при титровании раствор встряхивать.
455
Для быстрого титриметрического определения 5—15 мг Р рекомендовали церий(III) и эриохром черный Т в качестве индикатора [101].
Мешающее влияние железа (III), алюминия, магния и кальция можно устранить, применяя перед проведением анализа ионообменное отделение. Сульфат в концентрациях, равных или больших концентрации фосфата, мешает определению. В более поздней работе для титрования 2—300 мг Р рекомендовали лантан, индикацию конечной точки титрования осуществляли электрохимическим методом [102].
Спектроскопические методы
Спектрофотометрические методы
Фосфат можно определять в виде молибдофосфорной кислоты ее восстановленной формы — молибденовой сини и в виде желтого ванадомолибдофосфорного комплекса. Кроме того, существуют косвенные методы, основанные на применении хлоранилатов или на определении молибдата, входящего в состав молибдофосфорной кислоты.
Литература по определению фосфата в виде гетерополикислотк обширна. Риманом и Бойненкампом [103] опубликован обзор ранних работ, посвященных изучению спектров поглощения молибдо фосфорной кислоты и ее восстановленных форм в водных и орга нических средах. Чувствительность спектрофотометрического метода, основанного на образовании гетерополикислоты, невелика и зависит, как было показано, от условий проведения реакции и от природы экстрагентов. Гетерополикислоту можно сначала восстановить до молибденовой сини, а затем экстрагировать, или сначала можно провести экстракцию, а восстановление осуществлять в органической фазе. Экстракция — один из способов повышения селективности определения фосфата. Другой подход основан на подавлении образования гетерополисоединений другими ионами. Опубликовано несколько обзоров, посвященных этим вопросам ]49, 104, 105]. В некоторых работах обсуждаются проблемы аналитического использования додека молибдатов, существующих в двух различных формах. Хеслоп и Пирсон [17] особенно подчеркивали это свойство гетерополимолибдата и утверждали, что восстановленная молибденовая синь также содержит два соединения: НзРО4- 10МоО3-Мо2О5 и Н3РО4-8МоОз-2Мо2О5. Этим и объясняются противоречивые данные ранних работ по спектрофотометрическим методам определения фосфора; требуются определенные условия, чтобы получить единственную форму гетерополикислоты.
Условия, неблагоприятные для образования нежелательных гетерополисоединений, следует рассмотреть подробнее. Известно, что лимонная, винная и щавелевая кислоты разрушают молибдо-фосфорную кислоту, тогда как молибдокремневая кислота в их
456
присутствии не разрушается [106]. Пауль [14] нашел, что лимонная кислота разрушает молибдомышьяковую кислоту и, более того, если перед введением молибдата аммония к раствору добавить НС1О4, молибдокремневая кислота не образуется. Аналогичных этим реакциям известно еще две — это скорее парадоксальные реакции.
1. Если к смеси молибдофосфорной и молибдокремневой кислот добавить винную кислоту, разрушается только молибдофос-форная кислота.
2. Если к смеси фосфата и силиката добавить смешанный реагент, содержащий молибдат и винную кислоту, образуется только молибдофосфорная кислота.
Для объяснения этих реакций были привлечены константы устойчивости реакций и кинетические характеристики реакционных систем:
быстро Молибдофосфорная кислота + Виииая кислота t г Фосфат + Молибдотартрат
медленно Молибдокремневая кислота + Виииая кислота <-.—
ч=± Силикат + Молибдотартрат
Первая реакция проходит быстро с образованием молибдо-тартратного комплекса. Молибдосиликат разрушается значительно медленнее. Так, если винную кислоту добавляют после завершения образования гетерополикислоты, молибдосиликат сохраняется в растворе еще в течение длительного времени и его концентрацию можно измерить или провести восстановление кислоты до молибденовой сини.
Если винная кислота добавлена к смеси фосфата и силиката одновременно с молибдатом аммония, образуется молибдотартраг-ный комплекс, в присутствии которого молибдофосфат образуется не полностью в соответствии с константами равновесия этих дв\ х реакций. При восстановлении молибдофосфата до молибденовой сини в присутствии восстановителей равновесие реакции сдвигается в сторону образования Р-ГПК до тех пор, пока весь фосфор не будет связан в молибдофосфорную гетерополикислоту. При этом молибдосиликат, образующийся значительно медленнее, не мешает определению фосфата. Так были объяснены экспериментальные данные. На этих реакциях был основан метод определения фосфата и силиката [105] при совместном присутствии.
Наличие двух форм (а и 0) гетерополикислоты было постулировано Стриклендом [107]. При высоких соотношениях [Н+]/[МоОГ] образуется нестабильная 0-Форма. Она спонтанно переходит в стабильную а-Форму, которая существует в менее кислых растворах с более низким соотношением [Н+]/[Мо02']. 0-Форма характеризуется более высоким молярным коэффициентом поглощения, этим и объясняются противоречия в спектрофотометрических методиках, в которых, вероятно, условия выполнения
457
анализа создавали возможность одновременного существования в растворе сразу двух форм гетерополикислоты. 0-Форма стабилизируется ацетоном [104, 105]. На гравиметрические методы, основанные на применении гетерополикислоты, наличие ее двух модификаций не оказывает влияния.
Пунгором с сотр. [108—110] была получена информация по химии и модификациям гетерополикнслот. Было найдено, что различие в молярном коэффициенте поглощения и другие проявления нестабильности свойств следует отнести не за счет изменения самой гетерополикислоты, а за счет влияния на спектрофотометрические характеристики растворов образующихся изополимолибдатов. Несмотря на значительную аналогичность свойств четырех гетерополикнслот (Р, As, Si и Ge), авторы нашли, что некоторые различия в их свойствах позволяют определять гетерополикислоты в смеси без их предварительного разделения. Для этого необхо-димо изменять концентрацию кислоты и молибдата, длину волны при измерении, использовать преимущественно а- или 0-модификацию кислот и подбирать среду для проведения реакции. Например, при 427 нм а-молибдомышьяковая кислота практически не поглощает, а три другие гетерополикислоты в этой области длин волн интенсивно поглощают. В методике не используют ни восстановителей, ни экстракции.
Следует обратить внимание на возможность извлечения кремния из стекла химической посуды, весьма существенный источник ошибок при определении малых концентраций фосфора.
При добавлении к подкисленному раствору фосфата раствора молибдата аммония образуется окрашенное в желтый цвет соединение, поглощение которого измеряют при 380—420 нм. Этот метод был использован для определения фосфата [111]. Определению мешают мышьяк, кремний, вольфрам, ванадат, висмут и значительные концентрации никеля, меди и фторида.
Опубликована модифицированная методика определения 1— 10 мкг фосфора [112], в которой светопоглощение измеряют при 362 нм. В работах [104, 105] обсуждается влияние на образование гетерополикислоты оксикарбоновых кислот. Описана методика определения 0,2—1,0 мг Р и 0,1—1,0 мг SiO2 в одном растворе [105]. К одной части раствора добавляют винную кислоту для предотвращения образования молибдокремневой кислоты. Методика сравнительно проста. Молибдофосфорную кислоту (или ее синюю восстановленную форму) экстрагировали раствором про-пиленкарбоната в СНС13 [113]. Молярный коэффициент поглощения в органической фазе при 308 нм составляет 22 300. По этой методике можно определять до 7,5 мкг растворимого фосфата.
Выше упоминалось о работах Пунгора с сотр. [108—110]-В этих работах, посвященных определению Р, As, Si и Ge, приведены методики анализа бинарных смесей этих элементов и смесей, содержащих Р, As и Si или Р, As и Ge. Определяли 2—17 мкг/мл фосфора, 3—40 мкг/мл As, 2—19 мкг/мл кремния и 60—50 мкг/мл германия.
458
При восстановлении молибдофосфорной кислоты образуются синие полимерные соединения, эта реакция положена в основу многочисленных колориметрических методик. Впервые предложенный в 1940 г. Дикманом и Брейем [114], метод впоследствии был модифицирован для улучшения его точности, чувствительности и селективности. Несколько ссылок на эти методики было сделано в подразделе, посвященном методам отделения фосфата. Синие модификации гетерополикислоты можно экстрагировать кислородсодержащими органическими растворителями, можно сначала проводить экстракцию фосфата из водных .растворов, а затем восстанавливать гетерополикислоту в органической фазе.
Очень важен выбор восстановителя, его природа влияет как на время, необходимое для восстановления, так и на молярный коэффициент поглощения синих продуктов. В качестве восстановителей рекомендуется железо(П) [111, 115], гидрохинон [116], хлорид олова(П) [117], сульфат гидразина [111, 118], 1-амино-2-нафтол-4-сульфокислота [119] и аскорбиновая кислота [120, 121].
Большое значение имеет кислотность растворов. Рекомендуется использовать растворы 0,4—0,6 М по H2SO4. Изучали стехиометрию реакции, ее механизм и влияние кислотности [122]. Для определения фосфата по синей форме гетерополикислоты необходимо контролировать кислотность растворов, делая ее возможно более высокой для предотвращения восстановления изополимолибдатов (VI), которое проходит при pH = 0,7. Так как при более низкой кислотности скорость реакции ниже, рекомендуется проводить определение в среде 0,15—0,25 М по H2SO4 или 0,3—0,5 М по HNO3—это минимально допустимая кислотность растворов.
Применение катализаторов снижает время, необходимое для проведения анализа. Описана [123] методика определения фосфора в коксе после сожжения его в колбе с кислородом. В качестве восстановителя применяли аскорбиновую кислоту, катализатором служил антимонил-тартрат калия, оба соединения добавляют в виде одного сложного реагента. Этот прием использовали в работах [124, 127] для анализа морской воды.
Если в методе молибденовой сини не применять экстракцию, определению мешает большое число ионов, в частности, кремний (IV), германий (IV) и мышьяк(V). Мешающее влияние кремния можно устранить увеличением кислотности растворов или введением цитрата. Ниобий^), тантал^), олово (IV), вольфрам^!), титан (IV), цирконий (IV) и висмут мешают определению, так как в условиях анализа образуют осадки, сорбирующие фосфат. Барий(II), стронций(II) и свинец(П) в сульфатных растворах осаждаются. Большие концентрации меди(П), никеля(П) и хрома(III), образующие окрашенные растворы, искажают результаты определения фосфата. Ванадий^) мешает, так как образует ванадомолибдофосфатный комплекс. Влияние ванадия можно устранить, если его восстановить до ванадия(IV) перед введением молибдата аммония. Железо можно перевести в ион железа(II). Мешающее влияние нитрата устраняют при
459
восстановлении его до NH3 металлическим алюминием в растворе NaOH [125].
Изучали влияние ртути(II) на определение фосфора [126, 127J. Ртуть вносит в определение значительную положительную ошибку. Было найдено, что ее влияние можно устранить обработкой раствора хлоридом или смесью метабисульфита и тиосульфата, которые комплексуют ртуть. Смешанный реагент (метабисульфит — тиосульфат) устраняет также мешающее влияние мышьяка (V).
При определении фосфора с применением экстракции значительно уменьшается число мешающих ионов. В методе, разработанном Люком и Больтцем [128], которые впервые проводили экстракцию изобутиловым спиртом и затем восстанавливали мо-либдофосфорную кислоту хлоридом олова (II), было показано, что определению 0,6 ppm фосфора существенно мешают только мышьяк^), церий(1У), германий(1У), золото(Ш), вольфрам(У1), ва-надий(У), олово(П) и тиосульфат. Допустимо присутствие следующих ионов в концентрации вплоть до указанной в скобках (в ppm): As111 (60), I~ (60), Hg1 (20), SiIV (30) и SnIV (40) и тиосульфат (60).
Чувствительность метода зависит от ряда факторов, в число которых входит природа восстановителя. Максимальная чувствительность характеризует методы, в которых в качестве восстановителя применяют 1-амино-2-нафтол-4-сульфокислоту (е = 26600) [119] и сульфат гидразина (е = 26800) [129].
Метод широко используют. Его применяют также для определения фосфора в полумикроорганическом анализе [130], микро-органическом анализе [131] и для анализа портланд-цемента [132].
Для целей рутинного анализа были разработаны автоматические методы определения фосфора по молибденовой сини. В автоматическом варианте применяют методы, в которых в качестве восстановителя используют олово(II) или аскорбиновую кислоту. С помощью автоматического анализатора, выпускаемого фирмой «Beckman», в течение 1 ч можно проанализировать 60 образцов. Градуировочный график линеен в интервале 0,1—0,001 ppm, относительное стандартное отклонение ^3%.
Метод был использован для определения РО<’ в присутствии полифосфатов.
Многие методики определения фосфата по молибденовой сини рекомендованы в качестве стандартных колориметрических методов анализа [135]. В публикации [135] приводятся подробные методики и обсуждается влияние посторонних ионов.
Есть работы, посвященные определению фосфата в морской воде. Следует обратить внимание на то, что можно столкнуться с изменением концентрации фосфата в образцах при хранении их до проведения анализа. Например, планктонные морские водоросли могут содержать фосфор, который переходит в раствор после отбора пробы или даже во время проведения анализа. Наблюдается уменьшение концентрации фосфора при хранении
460
растворов, что связано с жизнедеятельностью бактерий, а не с сорбцией фосфата полиэтиленовой емкостью, в которой находится образец [136].
В присутствии ванадия (V) и молибдена (VI) в кислых растворах фосфЗт образует желтую комплексную ванадомолибдофосфор-ную кислоту. Эта реакция положена в основу колориметрического метода определения фосфата [137]. С точки зрения рутинного анализа, этот метод обладает преимуществами по сравнению с ме-k тодом молибденовой сини. Например, не очень строго нужно контролировать условия проведения реакции.
Комплекс можно экстрагировать, что приводит к заметному [^увеличению селективности метода. Максимум светопоглощения лежит в области 315 нм, окраска очень стабильна. Оптимальная для развития окраски кислотность 0,25 М по H2SO4 (0,5 М по HNO3 или НСЮ4), верхний предел — 0.75 М по H2SO4 (1,5 М по HNO3 или НСЮ4). Дальнейшее увеличение кислотности препятствует развитию окраски комплекса.
Мешают определению (без экстракции комплексной кислоты) следующие ионы: кремний в больших концентрациях, железо(III) в присутствии хлорида или сульфата, восстановители, хром (VI), •мышьяк(У) и цитрат. Висмут(Ш), торий(1У), хлорид и фторид влияют на развитие окраски. Кремний можно удалить при кипя-Г чении раствора с концентрированной НСЮ4. Железо(III) можно связать в комплекс с фторидом, избыток которого удаляют введением борной кислоты. Борную кислоту можно использовать и для связывания фторидов, присутствующих в исходном анализируемом растворе. С использованием экстракции комплексной гетерополикислоты был разработан метод определения фосфора. Метод был применен для анализа практически всех фосфорсодержащих материалов: стали [139, 140], железных руд [141], алюминиевых, медных и никелевых сплавов с белыми металлами [142], воды [143, 144] и удобрений [145—147]. Работы по анализу удобрений [145—147] посвящены автоматизации очень точного метода определения фосфора с применением автоматических анализаторов. В анализаторы был заложен метод прямого измерения светопоглощения, а не дифференциальный вариант, который обычно используют для повышения точности определения. Полученные результаты позволяют заключить, что абсолютная ошибка измерения оптической плотности в интервале 0—1,2 единицы не выше ошибки самого измерительного прибора (0,001 единицы поглощения). Следует отметить, что описанный метод по точности превосходит метод с применением молибдофосфата хинолина и, кроме того, обладает еще одним преимуществом — простотой выполнения определения. В биохимии метод применяли для определения фос-I фата в присутствии неустойчивых органических фосфатов [148] и Е неорганического фосфата в аденозинтрифосфате [149]. Метод был использован для анализа фосфатных горных пород [150]. В органическом микроанализе метод применяют после сожжения органических соединений в колбе с кислородом [151, 131].
461
Спектрофотометрическое определение фосфата в виде ванадомолибдофосфата
Метод 1. Определение фосфата в воде[143].
Реагенты
Следует применять реагенты только реактивных квалификаций. .
Ванадий-молибдатный раствор. Растворяют 0,5 г ванадата аммония в горячен воде, охлаждают н добавляют 125 мл HNO3 плотностью 1,42 г/см3. Добавляют раствор 10,0 г молибдата аммония в воде и разбавляют до 1 л.
Азотная кислота, приблизительно 2,0 М раствор.
Стандартный раствор фосфата. Растворяют в воде 0,716 г однозамещенного фосфата калия (высушенного при 150 °C) и разбавляют раствор до 1 л. В день анализа 20 мл этого раствора разбавляют до 1 л, концентрация фосфата соответствует 10 мкг/мл.
Построение градуировочного графика. Стандартные растворы фосфата, содержащие 0—200 мкг фосфора в виде фосфата, разбавляют до 50 мл. К каждому раствору добавляют 1 мл ванадий-молибдатного раствора, перемешивают и выдерживают прн комнатной температуре 10 мин. Светопоглощение каждого стандартного раствора измеряют в кюветах с толщиной слоя 4 см по отношению к воде при длине волны 370 нм. График зависимости оптической плотности от концентрации фосфата должен быть линейным.
Ход анализа. Отбирают 50 мл образца и выполняют все операции, описанные выше. Если образец окрашен перед введением ванадий-молибдатного раствора добавляют 1 мл 2 М HNO3
Метод 2. Определение фосфора в алюминиевых, медных и никелевых сплавах с белыми металлами [142].
Методика иллюстрирует применение метода в присутствии больших осадков металлов, когда необходимо использовать экстракцию.
Реагенты
Стандартный раствор фосфора. Исходный раствор, содержащий 0,1 мг/мл Р, готовят растворением 0,4393 г однозамещенного фосфата калия в 1 л воды. Рабочий раствор (0,04 мг/мл) готовят разбавлением 100 мл исходного раствора до 250 мл.
Молибдат аммония, 15%-ный раствор. Готовят из тетрагидрата молибдата аммония и хранят в полиэтиленовых емкостях.
Ванадат аммония, 0,25%-ный раствор. Растворяют 2,5 г метаванадата аммония в 500 мл воды при нагревании, охлаждают и разбавляют до 1 л.
Ход анализа. Помешают 0,5 г образца во фторопластовую чашку, добавляют 20 мл воды, 10 мл 15 М HNO3 и 3 мл. 40%-ной HF После растворения разбавляют водой до 35 мл и кипятят Добавляют 5 мл 1 % ного раствора перманганата калия, кипятят 2 мин, затем добавляют 2 мл 5%-ного раствора нитрита калия и кипятят еще 3 мии. Охлаждают, добавляют 10 мл раствора ванадата аммония, 65 мл раствора молибдата аммония и оставляют стоять в течение 15 мин. Затем добавляют 30 мл воды, переносят раствор в делительную воронку и разбавляют водой до 150 мл.
Добавляют 15 мл 50%-ной лимонной кислоты, перемешивают, сразу добавляют 40 мл метилизобутилкетона и встряхивают 30 с. После расслаивания фаз водную фазу отбрасывают. Слой кетона быстро фильтруют через плотный двойной бумажный фильтр и очень быстро измеряют оптическую плотность прн длине волны 425 нм в кювете с толщиной слоя 2 см, если в препарате содержится 50—400 мкг фосфора, и при 400 нм в кюветах с толщиной слоя 4 см, если в препарате содержится 0—50 мкг фосфора. В качестве раствора сравнения используют воду, учитывают содержание фосфора в холостом рас-творе
Для определения содержания фосфора в холостом растворе и построения градуировочного графика для 0—400 мкг фосфора к аликвотной части стандарт' ного раствора фосфора добавляют 15 мл HNO3 и затем выполняют все операции, опис'нные в методике.
Примечания: 1. Сурьма и селен должны находиться в своих высши* степенях окисления. При определении Р в металлических Sb и Se к 0,5 г об" разца добавляют 20 мл 1 %-ного раствора КМпО<, так чтобы раствор окрасился
462
в розовый цвет от избытка перманганата. Для обесцвечивания раствора вводят 2 мл 5%-ного раствора нитрита натрия.
2. Можно измерять оптическую плотность в интервале длин волн от 400 до 480 нм. Оптические плотности экстрактов холостой пробы и комплекса увели-ияваются при уменьшении длины волны. Прн содержании фосфора ниже 0,1% следует проводить измерения в кюветах с толщиной слоя 4 см при 400 нм, в этом случае чувствительность почти вдвое выше, чем получающаяся при измерении прн 425 нм.
~ 3. Молярный коэффициент поглощения комплекса в органическом растворителе равен 1820 при 425 нм и 3110 прн 400 нм. Закон Бера выполняется при содержании фосфора до 38 мкг/мл. Окраска устойчива в течение 1 ч и не зависит от температуры в интервале 19—25 °C. Если растворы стояли 15 мин, необходимо вводить дополнительную операцию — фильтрование.
4. Определение 200 мкг Р не мешают 1 мг As и 0,5 г следующих элементов- Ag, Bi, Cd, Се, Со, Си, Fe, Hg, La, Мп. Мо, Nb, Ni, Pb, Sb, Se Sn, Th, Ti, U, W и Zn.
5. До 0,5 г Al не мешает определению, но поскольку алюминий образует очень прочные фторидные комплексы, он вытесняет кремний из его фторидных комплексов и поэтому мешает определению даже в миллиграммовых концентрациях. Большие концентрации хрома слегка мешают определению (3% на каждые 50 мг хрома). Более 1 г германия вносят в анализ положительную ошибку. Ванадий мешает незначительно. До 0,5 г циркония, за исключением того случая, когда он присутствует совместно с кремнием, не мешают определению.
6. Как было найдено, стандартное отклонение из 10 параллельных определений составляет ±0,5% при определении 5 мкг/мл и ±1% при определении 1,25 мкг/мл Р.
Методы определения некоторых ионов основаны на выделении при реакциях осаждения фиолетового хлоранилат-иона. При определении фосфата [152] можно использовать хлоранилаты лантана или тория, реакции проходят по уравнениям
2РОГ ± La2 (СвС12О4)3 ± ЗН+ —> 3HCeCl2O4‘ ± 2LaPO<
4РОГ + ЗТЬ(СвС12О4)2 + 6Н+ —> 6НСвС12О; + Th3 (РО4)4
Осаждаются нерастворимые фосфаты лантана или тория, содержание выделяющегося кислого хлоранилат-иона в каждом случае пропорционально содержанию фосфата в анализируемом растворе. Оба метода — косвенные. Оптическую плотность растворов измеряют при 530 нм; 3—300 ppm фосфата можно определять с ошибкой ±2%, до 400 ppm хлорида и нитрата не мешают определению. Мешающее влияние сульфата можно компенсировать добавлением к анализируемому и стандартному растворам больших количеств сульфата.
Бодэ [153] изучал константы диссоциации хлораниловой кислоты в различных средах и разработал метод определения фосфатов, основанный на описанном выше принципе. Трудно предположить, что метод с использованием хлоранилатов найдет широкое применение, так как метод характеризуется большими результатами холостого опыта, что отмечается многими исследователями.
При переведении фосфата в молибдофосфорную кислоту образуется 12 моль молибдена на каждый моль исходного фосфата. Понятно, что если разрушать образовавшуюся молибдофосфорную кислоту и спектрофотометрически определять содержание молибдена» то мы получим чувствительный косвенный метод определения
463
фосфата. Для определения молибдена использовали раствор 2-амино-4-хлорбензолтиола в хлороформе [15]. В зависимости от методики эффективный молярный коэффициент поглощения для ( фосфора составляет 96900 или 359 000, что позволяет определять 0,2 мкг (0,008 ppm) фосфора даже в присутствии больших концентраций Si, Ge, As и Sb. Определению мешают 30-кратные концентрации вольфрама (VI), но его влияние можно устранить простым маскированием. После разрушения гетерополикислоты щелочью зеленый комплекс молибдена с 2-амино-4-хлорбензолтиолом экстрагируют смесью хлороформа и бутанола и измеряют оптическую плотность при 710 нм.
Харфорд и Больтц [19] впервые предложили метод разделения фосфора и кремния, основанный на селективной экстракции их ГПК и последующем измерении поглощения молибдата в УФ области при 230 нм. По этой методике можно определять 0,1— ।
0,4 ppm фосфора и кремния.
Другая методика основана на взаимодействии гетерополикислоты, содержащей фосфор, с различными органическими красителями с образованием ионных ассоциатов. Бабко с сотр. {154, 23] исследовали ряд красителей и для экстракционно-спектрофотометрического определения фосфата рекомендовали иодидный зеленый и кристаллический фиолетовый. Опубликованы методы с'при-менением малахитового зеленого [155] и сафранина [156].
Появились новые методы анализа биологических, фармакологических и пищевых продуктов [157]. Сначала фосфат осаждают в виде магний-уранил фосфата, который растворяют в H2SO4. Выделяющийся ПОГ реагирует с гексацианоферратом (II) по реакции |
2Na3PO, 4- 2Mg (UO2) (СН3СО2)Ч —>
—> Mg (UO2)2 (PO4)2 + 6NaCH3CO2 + Mg (CH,CO2)2
2UO?+ + K4 [Fe (CN)e] + 2SO?' —> (UO2)2 [Fe (CN)„] + 2K2SO4
< Чувствительность спектрофотометрического метода 0,01 мг Р, ошибка ±1%.
Фосфат в воздухе можно определить не совсем обычным колориметрическим методом — по сравнению интенсивности окраски пятен [158]. Образец (1 мкл) пропускают через нагретую зону кольцевой бани, после высушивания пятно опрыскивают реагентом, содержащим о-дианизидин, безводную уксусную кислоту и молибдат натрия. Образующееся коричневое пятно сравнивают со стандартами, приготовленными из стандартного раствора однозамещенного фосфата калия. Обычно присутствующие в воздухе примеси не мешают определению.
Метод пламенной эмиссионной спектроскопии
Как и сера, фосфор имеет характерные атомные резонансны6 линии в далекой УФ-области спектра (177,5, 178,3, 178,8 нм)-Однако из-за поглощения в этой области воздуха, газового пла
464
мени и кварцевой оптики нельзя применять обычные варианты методов атомно-абсорбционной и атомно флуоресцентной спектрофотометрии.
Разработаны косвенные эмиссионно-спектроскопические методы. Брикер и Фурман [159] использовали эффект подавления фосфатом эмиссии кальция и магния. Этот метод характеризуется ограниченной областью определяемых концентраций фосфора. Использовали еще одну систему, только с другим вторым металлом, например барием(II) [160], который частично замешает кальций в невозбуждающихся соединениях. Измеряли эмиссию кальция и бария в растворах, содержащих фосфат, при этом общая концентрация металлов была постоянной. Эмиссия кальция при 630 нм линейно зависит от концентрации кальция, наклон этой прямой пропорционален концентрации фосфата. Очевидно, метод пригоден для быстрого определения фосфата. Существенно мешает определению 10-кратный по отношению к фосфату избыток А1, Сг, Со, Fe, Sr, Sn и NOL В некоторой степени влияют на определение эквимолекулярные концентрации цинка и ртути, 10-кратные концентрации Мп, К и Na.
Разработан [162] прямой эмиссионный метод, основанный на известном газохроматографическом методе определения фосфора в фосфорсодержащих органических соединениях [161]. В этой работе использовали холодное азотоводородное диффузионное пламя, дающее некоторые преимущества Фоновая эмиссия ниже, чем в
других смешанных пламенах, относительно низкая температура пламени приводит к очень малому возбуждению даже щелочных металлов. Пламя обладает восстановительными свойствами. Более
того, за счет низкой температуры и ограниченной подачи кислорода можно наблюдать эмиссию соединений, которые в обычных смешанных пламенах не проявляются. В случае фосфора наблюдают интенсивную зеленую эмиссионную полосу НРО. Излучение следует отнести за счет частицы НРО, переходящей в невозбужденное состояние за счет хемилюминесцентной реакции, возвращающей электроны на их низшие энергетические уровни. На рис. 52 показан эмиссионный спектр НРО, полученный распылением 1,2-10-2 М раствора ортофосфорной кислоты в азотоводо-
родное диффузионное пламя.
Интенсивность поглощения при 528 нм линейно зависит от концентрации ортофосфорной кислоты при содержании фосфора 0,2—500 ppm. Фосфаты металлов дают слабый сигнал, так как в сравнительно холодном
₽ис. 52. Эмиссионный спектр НРО, полученный распылением 1,2-10 М раствора ортофосфорной кислоты в диффузионное азото-водородное пламя [1Ы].
пламени не проходит их диссоциация. Депрессивное влияние катионов, присутствующих в анализируемом образце, можно устранить их предварительным отделением методом ионного обмена. На определение не влияют 15-кратный по отношению к фосфату избыток уксусной, бромистоводородной, хлористоводородной, азотной, щавелевой, серной и винной кислот.
Этот метод был применен для анализа фосфорсодержащих моющих средств после отделения катионов с помощью ионного обмена. Ошибка определения фосфора в материалах, содержащих до 20% фосфата (в пересчете на Р2О5), составляет 2—4%. Метод прост и экспрессен. При автоматической регистрации полосы эмиссии НРО в узкой полосе спектра чувствительность метода снижается. Метод был существенно улучшен [164] за счет использования модифицированного фотометра, содержащего широкополосный фильтр и усовершенствованную горелку. Нижний предел определения фосфора снижен в 100 раз до 0,007 рргп, метод применен для определения фосфора в органических веществах и воде после отделения катионов с помощью ионного обмена.
Флуорометрические методы
Фосфат в флуориметрических методах давно известен в качестве мешающего иона, это его свойство было использовано для аналитических целей. В работе [165] использовали свойство фосфора гасить люминесценцию комплекса алюминия с морином. Многие ионы мешают определению, некоторые из них можно отделить предварительным выпариванием анализируемого раствора с хлорной кислотой или с помощью ионного обмена. Киркбрайт, На-райянасвари и Вест [166] попытались реализовать потенциально высокую чувствительность спектрофлуориметрии, оставив при этом селективность определения фосфата, достигнутую в более ранних работах. Им удалось этого добиться следующим образом. Фосфат превращают в молибдофосфорную кислоту, которая, в свою очередь, взаимодействует с основным красителем родамином Б с образованием ионного ассоциата. После экстракции избытка красителя хлороформом ионный ассоциат молибдофосфата и родамина Б экстрагируют смесью 4:1 по объему хлороформа и бутанола и измеряют флуоресценцию этого раствора при 575 нм, длина волны возбуждающего света 350 нм. Изучение влияния на определение фосфора [37] посторонних ионов показало, что метод отличается высокой селективностью. Не мешают определению большие концентрации силиката. Мышьяк(1П) и ванадий(У) могут присутствовать в 25- и 59-кратном избытке по отношению к фосфору. Метод применим для определения 0,04—0,6 мкг Р. При изучении природы комплекса было показано, что соотношение родамина Б и молибдофосфата в ионном ассоциате составляет 3 моля на 1 моль. Это позволяет предполагать, что образуется незаряженный комплекс типа [RhB+]3[PMO3-].
Те же исследователи [167] предложили еще один аналогичный метод, в котором используют в качестве органического основания
466
хинин. Преимущества этого метода в том, что хинин характеризуется высокой интенсивностью флуоресценции и стабилен в растворах H2SO4; хинин часто применяют в качестве стандарта для калибровки спектрофлуориметров. Чувствительность этого метода 0,02—1,2 мкг фосфора, а селективность сравнима с селективностью метода, описанного выше [166].
Рентгенолюминесценция (или рентгеноэмиссионная спектроскопия) может рассматриваться как особый вариант флуориметрии. Поэтому уместно именно здесь упомянуть о применении этого метода для определения фосфата. Малые концентрации фосфата (а также хлорида, роданида, цианида, бромида и иодида) можно определять этим методом после осаждения или соосаждения его со сравнительно тяжелыми элементами [168]; 1—10 мг фосфата соосаждаются с таллием (I) и нитратом серебра. Смешанный осадок анализируют непосредственно на фильтре.
Атомно-абсорбционный, метод
Атомно-абсорбционную спектроскопию используют для анализа многих элементов, но ее нельзя применить в прямом варианте метода для определения следовых концентраций фосфора, мышьяка или кремния из-за того, что резонансные линии этих элементов лежат в далекой ультрафиолетовой области, или за-за образования тугоплавких соединений, которые полностью подавляют диссоциацию в пламени. Прямой метод был описан Киркбрайтом и Маршаллом [169], которые использовали пламя азот — оксид азота (I)—ацетилен и микроволновый безэлектродный незаряженный источник возбуждения фосфора. Чувствительность определения 4,8 и 5,4 мкг/мл Р при длинах волн 177,5 и 178,3 соответственно. Атомно-абсорбционную спектроскопию можно использовать для косвенного определения фосфора по молибдену, входящему в состав молибдофосфорной кислоты. Описан основанный на этом принципе метод анализа [170], но определению мешают мышьяк и кремний.
Описан метод последовательного определения фосфора и кремния в пламени оксид азота (I) — ацетилен [18]. Фосфорномолиб-Деновую гетерополикислоту количественно экстрагировали изобу-тилацетатом, а после подкисления раствора кремний экстрагировали бутанолом. В каждом случае атомно-абсорбционным методом определяли молибден. Можно определять 0,08—1,0 ppm фосфора. За счет экстракции метод очень селективен. Мышьяк(V) и германий (IV) не мешают определению фосфора, но влияют на определение кремния, завышая результаты анализа; 10-кратный избы-, ток вольфрама не мешает определению фосфора, а 100-кратный — уже мешает.
Описаны и другие методики, в которых определяют фосфор [21, 19]. В работе [21] определяли фосфор, мышьяк и кремний, методика приведена ниже. В работе [19] после селективной экстракции определяли фосфор и кремний.
467
Определение 1—10 мкг фосфата, арсената и силиката с использованием метода атомно-абсорбционной спектроскопии [21]
Реагенты
Раствор молибдата Растворяют 10 г тетрагндрата молибдата аммония в 100 мл дистиллированной воды.
Раствор цитрата. Растворяют 100 г лимонной кислоты в 800 мл дистиллированной воды, доводят pH раствора NaOH до 3,2 и разбавляют до i л.
Ход анализа. В делительную воронку переносят 10 мл анализируемого раствора (или аликвотную часть этого раствора), содержащего не более 10 мкг фосфора, 10 мкг кремния и 20 мкг мышьяка. Если необходимо, разбавляют до 10 мл дистиллированной водой. Добавляют 1 мл раствора молибдата и 2 мл разбавленной НЙОз (или такое количество кислоты, чтобы довести pH раствора до 0.7) и перемешивают. Содержимое делительной воронки выдерживают 20 мин для полного образования молибдофосфорной, молибдокремневой и молибдо-мышьяковой кислот.
Определение фосфора. Добавляют 10 мл изобутилацетата и встряхивают воронку 1 мин. После расслаивания фаз нижнюю водную фазу (Л) переносят в другую делительную воронку для определения мышьяка и (или) кремния Измеряют поглощение молибдена в изобутилацетатном экстракте, как описано в примечаниях, используя в качестве нулевого раствора экстракт холостого опыта.
Определение мышьяка. К водной фазе (А) или в отсутствие фосфора к анализируемому раствору, обработанному как описано в начале методики, добавляют 5 мл смеси (1:1) этилацетата и бутанола и встряхивают 1smiih Добавляют 5 мл изоамилацетата и экстрагируют еще раз 30 с. Отставляют воронку до полного расслаивания фаз и затем водную фазу (В) переносят в другую детительную воронку для определения кремния. Органическую фазу дважды по 1 мин встряхивают с 10 мл промывной жидкости для удаления свободного молибдатного реагента. Измеряют поглощение молибдена в органической фазе, как описано в методике при определении фосфора.
Определение кремния. К водной фазе (А + В) или к анализируемому раствору добавляют 2 мл раствора цитрата, перемешивают и оставляют на 2 мин Молибдокремиевую кислоту экстагнруют 10 мл метплизобутилкетона, переворачивая делительную вооонку 30 с, и измеряют поглощение молибдена, как и в первых двух случаях по отношению к экстракту холостого опыта.
Концентрацию фосфора, мышьяка и кремния определяют по градуировочному графику, построенному по серин стандартных растворов, содержащих 1 — 10 мкг Р, 2—20 мкг As и 1—10 мкг Si.
Примечания 1. Определению 2,5 мкг фосфора и кремния и 5 мкг мышьяка не мешает 200 мкг каждого из следующих ионов:
группа 1 — Li*, Na*, К*, Cs*, Си2*, Ag*-
группа 2 - Be2*, Са2*, Sr2*, Mg2*, Zn2*, Cd2*, Hg2*, Hg22*;
группа 3—B2O2*, Ai3*, Ce4*;
группа 4 — Sn4*, Pb2*‘
группа 5—VOj, Sb5*, Bi3*;
группа 6 - SO2’, SO2’, Cr3*, SeO2’, TeO23’, WO2’, UO2’;
группа 7— F’, Cl', Вг', I', Mn’*-
группа 8 - Fe’*, Co2*, Ni2*. Pd2*.
2 Титан(1У) и цирконий(1У) ухудшают отделение фосфора. Германий не влияет на определение фосфора или мышьяка, но мешает определению кремния.
3 Условия инструментального определения молибдена методом ААС следующие: длина волны 313,2 нм, ширина щели 0,3 мм, сила тока 40 мА, оксид азо-та(1) 15 пси, ацетилен — ток газа устанавливают таким, чтобы образовалось
468
кРасно-розовое внутри пламя высотой 1—2 см, формирующееся при попадании в jgro органической фазы.
Г 4. Все растворы хранят в полиэтиленовых емкостях, чтобы исключить попа-дание в них кремния.
' Электрохимические методы
Г Потенциометрические методы
t Описано несколько потенциометрических методов определения фосфата. Содержание общего ортофосфата в стиральных порошках проводили [171] следующим образом: фосфат переводили в форму однозамещенного фосфата, затем осаждали в виде Ag3PO4 и потенциометрическим титрованием определяли выделяющуюся по реакции кислоту:
NaH2PO, + 3AgNO3 —> Ag3PO<; -|- NaNO3 + 2HNO3
•Борат, карбонат, силикат и сульфат не мешают определению.
г В другом методе проводят потенциометрическое титрование с серебряным электродом в боратном буферном растворе, осаждая фосфат в виде Ag3PO4 [172]. Хотя реакция не идеально стехио-•Метрична, этим методом можно получить результаты с удовлетворительной точностью. Нитрат, сульфат и ацетат не мешают определению, не мешает и фторид, который в большинстве методов определения фосфата является мешающим ионом.
р Можно проводить прямое определение фосфата титрованием раствором нитрата серебра или электрогенерируемым (кулонометрический вариант) из серебряного анода (в 80%-ном этаноле, 0,1 М по ацетату натрия) ионом Ag+ [173]. Конечную точку титрования определяют потенциометрически до 2-Ю-4 М фосфата или амперометрически до 1,7-10-3 М фосфата. Галогениды, со-осаждаясь с фосфатом серебра, мешают определению. Эквимолярные концентрации сульфата могут присутствовать в анализируемом растворе, ионы кальция (II), алюминия (III) и железа (III) должны отсутствовать.
F Описан метод [174] потенциометрического микроопределения фосфата. Ортофосфат образует одну из наименее растворимых солей свинца (—1g ПР = 42,1 при 25°C). В методе в качестве титранта используют перхлорат свинца и свинцовый ионоселективный электрод марки Орион 94-82 для индикации конечной точки титрования. С помощью буферных растворов поддерживают == 8,25—8,75. Кривые потенциометрического титрования раз-Пйчных количеств ортофосфата показаны на рис. 53.
I Сульфат и нитрат не мешают определению, хлорид и фторид немного завышают результаты анализа. Содержание силиката не Должно превышать содержание фосфата. Анионы, образующие со свинцом труднорастворимые соли, менее растворимые, чем сульфит свинца (—1g ПР-8), должны отсутствовать [молибден(VI), вольфрам (VI) и хром (VI)]. Мешающее влияние карбоната и
469
день дополнительные сведения
Рис. 53 Кривые потенциометрического микротнтрования различных концентра, цнй ортофосфата 0,005 М раствором перхлората свинца с использование^ свинцового ионоселективного электро, да [174].
гидрокарбоната устраняют под. кислением раствора до pH =4 кипячением его и затем повторным доведением pH до 8,25—8,75.
Для определения 0,5—1,5 мг Р используют перхлорат свинца. Для меньших концентраций фосфора вплоть до 0,01 мг Р рекомендуют 0,005 М растворы перхлората свинца. В более поздней работе Селига [175] приве-о мешающем влиянии посторон-
них ионов и зависимости нижнего предела определяемых концентраций фосфора от чувствительности используемого свинцового ионоселективного электрода. Сульфат до соотношения сера ; фосфор = 320 не мешает определению, фторид не мешает до соотношения фторид : фосфат = 133. При использовании 0,001 н. раствора перхлората свинца нижний определяемый предел для фосфора составляет 10 мкг. Тот же исследователь сообщил, что в случае использования в качестве титранта нитрата серебра и серебряного А£/52--ионоселективного электрода, боратный буферный раствор предпочтительнее ацетатно-метанольного,
если титрование проводить в водно-органических растворах.
Попытки создания фосфатных ионоселективных электродов, включая фосфат висмута, были безуспешны, так как все электроды характеризовались дрейфом потенциала и низкой селективностью. Разработан жидкостной ионоселективный электрод [176], основанный на применении в качестве жидких ионообменников четвертичных аммониевых, арсониевых и фосфониевых солей, а также солей трифенилолова. Электроды обладали хорошей чувствительностью, но низкой селективностью. Эти же исследователи [177] использовали энзимные электроды. Энзим селективно катализирует реакцию, в которой участвует фосфат. В реакциях участвуют два энзима, взаимодействие проходит по следующим схемам:
_ . , , Щелочная фосфатаза _ , . .
Глюкоза-6-фосфат------------------► I люкоза + Фосфат
_ Глюкоза оксидаза _ , ,, _
Глюкоза ---------------» Глюконовая кислота + Н2О2
О2
Амперометрические методы
Фосфат можно амперометрически титровать железом(П).
тод может быть рекомендован для определения 2—55 ppm Р [17°г Ошибка колеблется от —7 до +8%, относительная ошибка 4 /о-В качестве фона-электролита использовали хлоруксусную кислоту-
470
Радиохимические методы
При добавлении вольфрама к молибдофосфорной кислоте молибден частично замещается на вольфрам. Концентрация вольфрама (VI) > экстрагирующегося с гетерополикислотой, пропорциональна содержанию геиерополикислоты, а следовательно, содержанию фосфора. Для образования вольфрамомолибдофосфорной кислоты использовали [179] 185W. Кислоты экстрагировали и по p-излучению определяли активность вольфрама. Мышьяк, кремний и ниобий мешают определению, германий не мешает. Метод применяли для определения 0—0,54 ppm РО« в воде.
Для анализа фосфатных пород использовали [180] Mg2P2O7 в методе изотопного разбавления 32Р и метод нейтронной активации фосфора для его определения по p-излучению в силикатных горных породах.
Кинетические методы
В последние годы существенные успехи в области кинетических методов анализа привлекают к ним все большее внимание. Это происходит потому, что кинетические методы отличаются высокой чувствительностью и дешевы. Хотя большинство методов посвящено определению металлов, появляются и методы определения анионов.
Без сомнения, сама идея кинетического метода пока ассоциируется с продолжительным периодом набора данных и проходящими в течение длительного промежутка времени реакциями. Во многих опубликованных в последнее время методах применяют автоматические системы, способные снизить время проведения анализа до секунд. Разработан метод [181], на основании которого изучали механизм образования молибденовой сини [122]. В контролируемых условиях скорость образования молибденовой сини из фосфата, молибдата и аскорбиновой кислоты пропорциональна концентрации фосфата. Использовано автоматическое считывающее устройство, позволяющее связать определяемую функцию пропорциональной зависимостью с концентрацией фосфата; с ошибкой 1—2% можно определять 1—12 ppm Р. Для анализа каждого образца продолжительность протекания реакции составляет всего лишь 20—30 с. Используют автоматическую систему регистрации скорости реакции, которая выдает данные о концентрации (0,1—5,00 ppm Р) фосфора в сыворотке крови за 100 мс [182].
Разработан метод последовательного определения силиката (I—10 ppm) и фосфата (1—10 ppm). В этой работе использовали автоматический дифференциальный кинетический вариант метода. Крисс, Руденко и Курбатова [184] показали, что метод можно Использовать для определения малых концентраций фосфата в Присутствии полифосфатов.
471
В кинетических методах определения фосфора используют и другие реакции. Например, скорость взаимодействия KI и молибдата аммония в присутствии крахмала при pH = 0,6 пропорциональна концентрации фосфата, являющегося катализатором реакции [185].
Другие методы определения
Газовая хроматография является новым методом, нашедшим применение в анализе фосфата. Аммонийные соли фосфата и некоторых других анионов (ванадат, арсенат, оксалат и т. д.) реагируют с бис(триметилсилил)трифторацетамидом, триметилсилил-производное определяют газохроматографически с пламенно-ионизационным детектором. В случае фосфата образуется соединение (ТМ5)зРО4. Натриевые и калиевые соли сначала переводят в аммонийные с помощью ионного обмена [45].
ЛИТЕРАТУРА
1. Greenfield S. — In: Comprehensive Analytical Chemistry (Ed. C L. Wilson and D W. Wilson), Vol. 1C, Amsterdam, Elsevier, 1962, p. 220
2. Rieman W., Beukenkamp J. — In: Treatise on Analytical Chemistry (Ed.
I. M. Kolthoff and P. J. Elving), Part II, Vol. 5, New York, Academic,.Press, 1961, p. 317.
3. Ryden J. C., Syers J. K., Harris R. F.— Analyst, 1972, v. 97, p. 903.
4. Heslop R. B., Pearson E. F. — Analytica chim. Acta, 1967, v. 37, p. 516.
5. Macdonald A. M. G., Van Der Voort F. H. — Analyst, 1968, v. 93, p. 65.
6. White D. C. — Mikrochim Acta, 1959, p. 254.
7. Fritz J S , Yamamura S S —Analyt. Chem, 1955, v. 27, p 1461
8. Colson A. F. — Analyst, 1963, v. 88, p. 26.
9. Colson A. F. — Analyst, 1965, v. 90, p. 35.
10 Furuya M., Tajiri M.— Japan Analyst, 1963, v. 12, p. 1139; Analyt. Abstr., 1965, v. 12, p. 1141.
11 Kinnunen J.; Wennerstrand B. — Chemist Analyst, 1955, v. 44, p. 51.
12. Wu H. — J. biol. Chem., 1920, v. 43, p 189.
13. Paul J.— Mikrochim. Acta, 1965, p. 830.
14. Paul J. — Mikrochim. Acta, 1965, p. 836.
15 Djurkin V., Kirkbright G F., West T. S. — Analyst, 1966, v 91, p 89.
16. Paul J. — Analytica chim Acta, 1966, v. 35, p. 200.
17. Heslop R. B., Pearson E. F. — Analytica chim. Acta, 1967, v. 39, p. 209.
18. Kirkbright G. F., Smith A. M., West T. S. — Analyst, 1967, v. 92, p. 411.
19. Hurford T. R., Boltz D. F. — Analyt. Chem., 1968, v. 40, p. 379.
20. Kirkbright G. F., Smith A. M., West T. S. — Analyst, 1968, v. 93, p. 224.
21. Ramakrishna T. V., Robinson J. W., West P. W. — Analytica chim. Acta, 19®, v 45 p 43.
22. Jakubiec R. J., Boltz D. F. — Mikrochim. Acta, 1970, p. 1199.
23 Бабко A. К, Шкаравский Ю. Ф., Ивашкович E. M. ЖАХ, 1971, т 26, с. 851 Analyt. Abstr., 1972, т. 23, с. 2420.
24. Bock R., Hummel С. — Z. analyt. Chem., 1963, v. 198, p. 176.
25. Yoshino Y. — Japan Analyst, 1954, v. 3, p. 121; Chem. Abstr., 1954, v. 48, p. 9867. .
26 Kindt В. H., Balis E. W., Liebhafsky H. A. — Analyt. Chem.. 1952, v. 24, p. 1501 , .
27. Ohashi S Takada S. — Bull. chem. Soc. Japan, 1961, v. 34, p. 1516; Analyt-Abstr, 1962, v. 9, p. 1866.
28 Winand L. — J. Chromat., 1962, v. 7, p. 400. 7
29 Pollard F. H., Rogers D. E., Rothwell M. Г. — J. Chromat., 1962, v. 9, p. 22'-30. Grande J. A., Beukenkamp J.— Analyt. Chem., 1956, v. 28, p. 1497.
473
3]_ Pollard F. H Nickless G., Rogers D E, Crone D L. — Proc. Soc. analyt. Chem Conference. Nottingham, 1965, Cambridge, Heffer and Sons Ltd.. 1965, p. 481
32. Zipkin I., Armstrong W. D., Singer L. — Analyt Chem., 1957, v 29, p. 310.
33. Newmann A. C. D. — Analytica chim. Acta, 1958, v. 19, p. 471.
34. Pinfold T. A., Karger B. L. — Separation Sci., 1970, v. 5, p. 183.
35. Huang К. P — Analyt. Biochem, 1970, v. 38, p. 383.
36. Kawanabe K, Takitani S., Miyazaki M., Tamura Z.— Japan Analyst, 1964, v. 13, p. 976, Analyt. Abstr., 1966, v. 13, p 5385.
37. Ионова Л. А., Постников H. H. Проблемы химии, 1969, с. 198. Analyt. Abstr., 1970, т. 18. c 2344.
38. Wagner E. F — Seifen-Ole-Fette-Wachse, 1968, v. 94, p. 27, Analyt. Abstr., l 1969, v 16. p. 2969.
39. Seiler H. — Helv. chim. Acta, 1961, v. 44, p. 1753.
40. Clesceri N. L„ Lee G. F. — Analyt. Chem., 1964, v 36, p. 2207.
41. Moghissi A. — J Chromat., 19G4, v. 13, p. 542.
42 Catiic V. D., Turcic M. N., Burgarski-Vojinovic M. B. — Z. analyt Chem., 1967, v. 229, p. 93
43 Muto M. — J chem Soc. Japan, pure Chem Sect., 1964, v. 85, p. 782, Analyt. Abstr., 1966, v 13 p 1257.
44 Canic V. D., Turcic M. N., Petrovic S M., Petrovic S E. — Analyt. Chem., 1965, v. 37, p 1576
45. Butts W. C — Analyt. Lett., 1970, v 3, p. 29
46. Gates O. R. — Analyt. Chem.. 1954 v. 26, p. 730
47. Page I. A., Maxwell J. A., Graham R. P. — Analyst, 1962, v. 87, p. 245.
48. Levine H„ Rowe J. J., Grimaldi F. S. — Analyt. Chem., 1955, v. 27, p. 258.
49. Chalmers R. A. — Proc. Soc. analyt. Chem., 1956, v. 3, p. 157.
50. Rayner L. W. W. — Mikrochim. Acta, 1970 p. 214.
51. Kolthoff I. M„ Sandell E. B., Meehan E. J., Bruckenstein S. — Quantitative Inorganic Analysis. 4th edn, London, Macmillan, 1969, p 642.
52. Belcher R., Nutten A J., Macdonald A M. G. — Quantitative Inorganic Analy-Г sis, 3rd edn, London Batterworths, 1970, p. 107.
S3- Золотавин В. И., Сонатина В. Н., Нестеренко В. П., ЖАХ, 1966, т. 21, с. 1289.
54. Duval С. — Inorganic Thermogravimetric Analysis, 2nd edn, New York, Elsevier, 1963.
55. Lundell G. E. F., Hoffman J. I. — J. Ass. off. agric. Chem., 1924, v. 8, p. 188.
56. Lundell G. E. F., Hoffman J. I. — Ind. Engng Chem. analyt Edn, 1923, v. 15, p. 44, 171.
57. Schulek E., Endroi A. — Magy. kfem. Foly, 1960, v. 66, p. 139; Analyt Abstr., 1961, v. 8, p 505.
58. Huditz F., Flaschka H, Petzold I. — Z analyt. Chem., 1952, v. 135, p. 333.
59. De Saint Chamant H., Vigier R. — Bull. Soc. chim. Fr., 1954, v. 21, p. 180; Analyt. Abstr., 1954, v. 1, p. 1232.
60. Cannon P. — Taianta, 1960, v. 3. p. 219.
61. Thistlethwaite W. P. — Analyst, 1947, v. 72, p. 531.
62. Archer D. W., Heslop R. B., Kirby R. — Analytica chim. Acta, 1964, v. 30, p. 450.
63. Archer D. W„ Heslop R. B. — Analytica chim. Acta, 1964, v. 30, p. 582.
64. Heslop R. B., Pearson E. F. — Analytica chim. Acta, 1965, v. 33, p. 522.
65. Macdonald A. M. G. — Ind. chem. Manuf, London, 1959, v 35 p 88
66. New Methods of Analytical Chemistry, Belcher, R. and Wilson C. L. in as-
R sociation with West T. S., 2nd edn, London, Chapman and Hall, 1964. p. 323.
67. Macdonald A. M G, Rivero A.-M. — Analytica chim. Acta, 1967, v. 37, p. 525.
68. Bowd A. J., Burns D 7. — Mikrochim. Acta, 1967, p. 560.
69 Wilson H. N —Analyst, 1951, v. 76, p. 65.
JO. Perrin C. N. — J. Ass. off. Agric. Chem., 1958, v 41, p. 758; 1959, v. 42, p 567. 'I. Wendlandt W. W., Hoffmann W. M. — Analyt Chem.. 1960, v. 32. p 1011. '2. Lench A. —Analyt Chem., 1963, v 35, p. 1695.
'3 Wilson H. N. — Analyst, 1954, v. 79, p. 535.
‘4. Belcher R., Macdonald A. M. G. — Taianta, 1958, v. 1, p. 185
473
75. Dahlgren S. E. — Z. analyt. Chem., 1962, v. 189. p. 243; Analyt. Abstr., 1963 v. 10, p. 1230.
76. Hoffman W M., Breen H. J. — J Ass. off. agric. Client, 1964, v. 47, p. 413.
77. J Ass. off. Agric. Chem., 1965, v. 48, p. 210
78. Fennell T. R. F., Webb J. R. — Taianta, 1959, v. 2, p. 105.
79. Firsching F. H. — Analyt. Chem., 1961, v. 33, p. 873.
80. Spacu G., Dima L. — Z. analyt. Chem., 1940, v. 120, p. 317.
81. Kesans A — Z. analyt. Chem., 1948, v. 128, p 215.
82. Greenfield S — In: Comprehensive Analytical Chemistry (Ed C L. Wilson and D. W. Wilson), Vol. 1C, Amsterdam, Elsevier, 1962, p. 225.
83. Fernlund U., Zechner S.—Z. analyt. Chem., 1955, v- 146, p. Ill; Analyt Abstr., 1955, v. 2, p. 2695.
84. Duncan R. D, Brabson J. A.— J Ass. off. agric. Chem, 1966, v. 49, p. 1201
85. Caudill P. R. — J. Ass off. agric. Chem., 1969, v. 52, p. 587.
86. Belcher R., Macdonald A. M. G., Phang S. E, West T S.—J. chem. Soc
1965, p. 2044.
87. Belcher R. — Taianta, 1968, v. 15, p. 357.
88. Belcher R., Uden P. C —Analytica chim. Acta, 1968, v 42, p. 180
89. Salam Khan M A., Stephen W. I. — Analyst, 1968, v. 93, p 26.
90. Buss H. e. a. — Z. analyt. Chem., 1963, v. 193, p. 264.
91. Riedel K.— Z. analyt. Chem., 1959, v. 168, p. 106.
92. Budevski O., Pencheva L., Rusinova R., Ruseva E. — Taianta, 1964, v. 11 p 1225.
93. Sinha В. C, Das Gupta S., Kumar S. — Analyst, 1968, v. 93, p. 409
94. Yofe J., Rappart B. R. — Analytica chim. Acta, 1968, v. 43, p. 346.
95. Kirkbright G. F„ Smith A. M., West T. S. — Analyst, 1969, v. 94, p. 321.
96. Kinnunen J., Wennerstand B. — Chemist Analyst, 1957, v. 46, p. 92.
97. Bassett J. — Analyst, 1963, v. 88, p. 238.
98. De Sousa A — Analytica chim. Acta, 1973, v 67, p. 234.
99. Salmon I. E„ Terry H. — J. chem. Soc., 1950, p. 2813.
100. Bakacs E. — Z. analyt. Chem., 1962, v. 190, p. 373.
101. Tavlli T. A., Irani R. R. — Analyt. Chem., 1963, v. 35, г. 1060.
102. Griepink B., Slanina J. — Mikrochim. Acta, 1967, p. 241.
103. Ref. 2, p. 348
104. Chelmers R. A., Sinclair A. G. — Analytica chim. Acta, 1965, v. 33, p. 384.
105 Chalmers R. A., Sinclair A. G. — Analytica chim. Acta, 1966, v. 34, p. 412.
106. Alimarin I. P., Sverev V. S. — Mikrochemie, 1937, v. 22, p. 89.
107. Strickland J. D. H. — J. Am. chem. Soc., 1952, v. 74, p. 862, 868, 872.
108. Halasz A., Pungor E. — Taianta, 1971, v. 18, p. 557.
109, Halasz A., Pungor E. — Taianta, 1971, v. 18, p. 569.
110. Halasz A., Pungor E., Polyak K. — Taianta, 1971, v. 18, p. 577.
111. Boltz D. F., Mellon M. G. — Analyt. Chem., 1948, v. 20, p. 749.
112. Kennedy J. F., Weetman D. A. — Analytica chim. Acta, 1971, v. 55, p. 448.
113. Jakubiec R. J., Boltz D. F. — Mikrochim. Acta, 1970, p. 1199
114. Dickman S. R., Bray R. H.—Ing. Engng. Chem. analyt. Edn, 1940, v 12, p. 665
115. Chalmers R. A., Thomson D. A. —Analytica chim. Acta, 1958, v. 18, p. 575.
116. Ging N. S. — Analyt. Chem., 1956, v. 28, p. 1330.
117. Laws E. Q., Webley D. J. — Analyst, 1959, v. 84, p. 28.
118. Hague ]. L., Bright H. A. — J. Res. natn. Bur. Stand., 1941, v. 26, p- 405.
119. Griswold B. L., Humoller F. L., McIntyre A. R. — Analyt. Chem., 1951, v 23, p 192-
120. Chen P. S„ Toribara T. У., Warner H. — Analyt. Chem., 1956, v. 28, p. 175°
121. Fogg D. N., Wilkinson N. T. — Analyst, 1958, v. 83, p. 406.
122. Crouch S. R., Malmstadt H. V. — Analyt. Chem., 1967, v. 39, p. 1084-
123. Kirk В. P., Wilkinson H C. — Taianta, 1972, v 19, p 80
124. Шафран И. Г., Павлова М. В., Титова С А , Якушева Л. Д. Труды Всес-НИИ Хим. реакт., 1966, с. 66. Analyt. Abstr., 1967, т. 14, с. 6767.
125. Duff Е. J., Struart J. L. — Analyst, 1971, v. 96, р. 802.
126. Tillma R. W., Syers J. K- — Analyst, 1975, v. 100, p. 322.
127. Murphy J, Riley J. P. — Analytica chim. Acta, 1962, v. 27, p. 31
128. Lueck С. H, Boltz D F. — Analyt Chem., 1956, v. 28, p 1168.
129. Boltz D. F., Mellon M. C. — Analyt. Chem., 1947, v. 19, p. 873.
474
130 Belcher R., Macdonald A. M. G., Phang S. E., West T. S. — J. chem. Soc., j 1965. p. 2044.
131. Saliman P. M.— Analyt. Chem., 1964, v. 36, p. 112.
[32. Nestoridis A —Analyst, 1970, v. 95, p 51.
133 Ramirez Munos J. — Analytica chim Acta, 1975 v. 78, p 431.
[34 Lucci G. C. — Annali Chim., 1966, v 56, p. 1343, Analyt. Abstr., 1968, v. 15, p. 737.
35. I. U. P. A, C., Spectrometric Data for Colorimetric Analysis, London, Butterworths, 1963.
|36 Herron J. — Limmol. and Oceanogr., 1962, v 7, p. 316; Analyt. Abstr., 1964, v. 11, p. 5764.
137 Kitson R. E , Mellon M G. — Ind Engng Chem. analyt. Edn, 1944, v. 16, p. 379-
138. Heslop R. B„ Ramsey A. C. — Analytica chim. Acta, 1969, v. 47, p 305.
139. Lindley G. — Analytica chim. Acta, 1961, v 25, p. 334.
140 B. S. 1121: Pt 45: (1966), British Standards Institute, London.
141 B. S. 4158: Pt. 2: (1970), British Standards Institute, London.
142. Pakalns P —Analytica chim. Acta, 1970, v 51, p. 497.
143 Abbott D. C, Emsden G. E., Harris J. R.—Analyst, 1963, v. 88, p. 814.
144. B. S. 2690: Pt 3: (1966), British Standards Institute, London
145. Docherty A. C. — Lab. Pract., 1969, v. 18, p. 47.
146. Jordan D. E. — J. Ass. off. agric. Chem., 1969, v. 52, p. 581.
147. Docherty A. C., Farrow S. G., Skinner J. M. — Analyst, 1972. v. 97, p 36.
148. Parvin R , Smith R. A. — Analyt Biochem., 1969, v. 27, p. 65.
149. Ueda 1, Wada T.— Analyt. Biochem., 1970, v 37, p. 169.
150. Schafer H N. S. — Analyt. Chem., 1963, v. 35, p. 53.
151. Dixon J. P. — Modern Methods in Organic Microanalysis, London, Van Nostrand, 1968, p. 160.
152. Hayashi K., Danzuka T., Ueno K.— Taianta, 1960, v. 4, p 244.
153. Bode H, Eggeling W, Steinbrechi V. — Z. analyt Chem., 1966, v. 216, p. 30.
154. Бабко А. К., Шкаравский Ю. Ф, Кулик В. И., ЖАХ, 1966, т. 21, с. 196.
155. Itaya К., Ui М. — Clin. chim. Acta, 1966, v. 14, p. 361.
156. Ducret L., Drouillas M. — Analytica chim. Acta, 1959, v. 21, p. 86.
157. Ghimicesuc G. H., Dorneanu V. — Taianta, 1972, v. 19, p. 261.
158. Sachdev S. L., West P. W. — Atmos. Envir., 1968, v. 2, p. 331
159. Dippel W. A., Bricker С. E., Furman N. H — Analyt. Chem., 1958, v. 26, p. 553.
160. Czebenyi-Gyory E., Slevin P. J., Svehla G., Erdey L.— Taianta, 1970, v. 17, p. 1167.
161. Dagnail R. M., Thompson К. C., West T. S.—Analyst, 1968, v. 93, p. 72.
162. Brody S. S., Chaney J. E. — J. Gas Chromat., 1966, v. 4, p. 42.
163. Elliott W. N., Mostyn R. A.— Analyst, 1971, v 96, p. 452.
164. Aldous К. M., Dagnail R. Af., West T. S. — Analyst, 1970, v. 95, p. 417.
165. Land D. B., Edmunds S. Af. — Mikrochim. Acta, 1966, p. 1013.
166. Kirkbright G. F., Narayanaswary R., West T. S. — Analyt. Chem., 1971, v. 43, p. 1434.
167. Kirkbright G. F., Narayanaswary R, West T. S. — Analyst, 1972, v. 97, p 174.
168. Stork G., Jung H. — Z analyt. Chem., 1970, v 249, p. 161; Analyt Abstr., 1971, v. 20, p. 1523.
169. Kirkbright G. F., Marshall M. — Analyt. Chem., 1973, v. 45, p. 16(0.
170. Zaugg W. S., Knox R. J. — Analyt. Chem., 1966, v. 38, p. 1759.
171. Cullum D. C., Thomas D. B. — Analytica chim. Acta, 1961, v. 24, p. 205.
172. McColl D. H., O’Donnell T. A. — Analyt. Chem., 1964, v. 36, p. 848.
3. Christian G. D., Knoblock E. C., Purdy W. C. — Analyt. Chem., 1963, v. 35,
I p. 1869.
174. Selig W. — Mikrochim. Acta, 1970, p. 564.
*75. Selig W. — Mikrochim. Acta, II, 1976, p. 9.
176. Nanjo Af., Rohm T. J., Guilbault G. G. — Analytica chim. Acta, 1975, v. 77, p. 19.
77. Guilbault G. G., Nanjo M. —Analytica chim Acta, 1975, v. 78, p 69.
}78. Bixler J W, Colwell L. F. — Analytica chim. Acta, 1976, v 85, p. 185.
}'9. Hahn R. B., Schmitt T. M. — Analyt Chem., 1969, v. 41, p. 359.
PO- Rijkheer J. — J. South. Afr. Chem. Inst, 1960, v. 13, p. 1; Analyt. Abstr., 1961, v. 8, p. 86.
475
181 . Crouch S. R-, Malmstadt H. V. — Analyt. Chem., 1967, v. 39, p. 1090.
182 . Javier A. C., Crouch S. R., Malmstadt H. V. — Analyt. Chem., 1969, v 41, p. 239.
183 Ingle J. D., Crouch S. R. — Analyt. Chem., 1971, v. 43, p. 7.
184 Крисс E. E., Руденко В. К- н Курбатова Г. Т., ЖАХ, 1971, т. 26, с. 1000. 185. Алексеева И. И., Немзер И. И. и Толстых Е. А. Зав. лаборатория. 1969, т. 35, с. 1305. Analyt. Abstr., 1970, т. 19, с. 3835.
ФОСФИТЫ
Фосфит НРОз’ — сильный восстановитель, хотя и обладает меньшими восстановительными свойствами по сравнению с гипофосфитом, с которым фосфит часто сравнивают в анализе. При определении фосфита важное место занимают редокс-титриметри-ческие методы.
МЕТОДЫ ОТДЕЛЕНИЯ
Бумажная и тонкослойная хроматография
В табл. 33 представлены характеристики четырех методик разделения с применением этих методов. %
Таблица 33. Хроматографическое отделение фосфита
Метод Растворитель Фосфит отделяется Литература
Бумажная хроматография (ватман Xs 3) Бумажная хроматография (восходящая, ватман Xs 4) ТСХ (MN поли-грам, полосы CEL 300) ТСХ (силикагель и крахмал) Изопропанол — хлоруксусная кислота или Изопропанол — изобутанол — этанол водный NH3 Изопропанол — трихлоруксусная кислота — NH3 (0,880 г/см3) (75 мл : 5 г в 25 мл: 0,3 мл) Кислотный растворитель Эбеля Другие растворители, включая основные Изопропаиол — трихлоруксусная кислота — тетраэтилам моиин гидро- ксид — Н2О (750 мл : 50 г : 16 мл 20%-ного по объ- ему :250 мл) Изопропанол — нзобута-нол — NH3 (0,880 г/см3) (400 мл : 200 мл : 10 мл) Метанол — NH3 (0,880 г/см3) 10%-ная хлоруксусная кислота — Н2О (10:3: 1 : 10) H2SO4, SOj", гипофосфат, гипофосфит, полифосфаты Гипофоефнт, пирофосфит, гипофосфат Орто-, ди- три-, тримета-, тетраметафосфат Орто-, ди-, трнфосфат, тримета-, тетраметафосфат, гипофосфит, полифосфат (л =20—30), продажные циклические и линейные фосфаты Орто-, дифосфат, гипофосфит 1 2 4
476
Анионообменная хроматография
Когда в смеси содержится в основном фосфит, анионообменная хроматография дает хорошие результаты при разделении. Поллард с сотр. [5] использовали последовательное элюирование раствором KCI и колонку со смолой Дауэкс-1 Х8. В двух изученных системах, при pH = 6,8 и при pH = 11,4, удалось добиться очень хороших результатов при отделении фосфита. Продолжением этой работы явился автоматический ионообменный метод анализа смеси фосфорных анионов с использованием автоматических анализаторов [6, 7]. В этом методе анионы (включая фосфит) после разделения превращали в ортофосфат и автоматически определяли последний, используя колориметрический метод, основанный на образовании молибденовой сини. Подробно метод описан в разделе «Полифосфаты».
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Редокс-методы
Почти все окислители, использующиеся в количественном анализе, можно применять для определения фосфита. К ним относятся перманганат, церий (IV), иод, ванадат, хромат, гипохлорит и гипобромит. Для того чтобы получить удовлетворительные результаты, необходимо проводить измерения через строго определенный интервал времени. Можно упростить анализ, если раствор нагревать в течение длительного времени. Подробное описание методов приведено в обзорах [8, 9].
Методы определения фосфита и гипофосфита аналогичны. Реакции этих анионов с различными окислителями проходят медленно. В современных методах существенно снижено время, необходимое для количественного окисления фосфита. Использована [10] реакция окисления фосфита (и гипофосфита) церием (IV), катализируемая ионами серебра(I) [11], рекомендован смешанный катализатор, содержащий нитраты марганца(II) и серебра(I). Со смешанным катализатором время окисления фосфита и гипофосфита уменьшается до 10 и 7 мнн соответственно.
Гипохлорит в качестве окислителя фосфита был изучен Масаловичем и др. [12]. При добавлении к фосфатному буферному раствору с pH = 7 гипохлорита фосфит окисляется количественно за 3 мин. Заканчивают определение, добавляя к анализируемому раствору KI и титруя выделившийся иод. Разработан быстрый титриметрический метод [13], основанный на окислении фосфита I в насыщенном гидрокарбонатом натрия, двузамещенным фосфатом натрия или бурой водном растворе. Двукратный избыток иода в 1,7% KI окисляет фосфит за 5 мин. Избыток иода титруют Мышьяком (III).
На основании фундаментальных исследований Масаловича и др. [14] становятся понятными оптимальные условия окисления
477
фосфита иодом. Многофакторный эксперимент показал, что оптимальными являются следующие условия: время окисления — 10,6 мин, температура — 22,5 °C, содержание KI —4,7%, pH = 7,28, избыток иода — 180,3%. Температура и pH оказывают самое большое влияние на реакцию.
Для определения в одном растворе фосфита и гипофосфита можно использовать несколько методик (табл. 34).
Таблица 34. Редокс-методы определения фосфита и гипофосфита в растворах
Фосфит и гипоф >сфит Фосфит Гипофосфит Литература
Кипятят с избытком CeIV, избыток CeIV ж- И титруют ге По разности Окисляют до фосфита избытком Celv в H2SO4 1,5 ч при комнатной температуре, избыток CeIV титруют Fe11 15
Выдерживают с ванадатом 20 мин или с хроматом 1 ч, избыток г- н То же Кипятят с избытком Fe111 в 1,5—2,0 М HCI, Fe11 титруют ванадатом 16
ванадата титруют be Окисляют до ортофосфата в 2-6 М НС1 Окисляют до фосфита Си11 в 6 М НС1 при комнатной температуре 2—3 мин, потенциометрически или визуально избыток Fe11 эквивалентный Си1 титруют различными окислителями 17
Окисляют до ортофосфата ванадатом с катализатором Ag1, потенциометрический метод По разности В отсутствие катализатора Ag1 до ортофосфата окисляется только гипофосфит 18
Определение фосфита редокс-методом [10]
Реагенты
Сульфат железа(П), стандартный раствор.
Сульфат церия(1У), стандартный раствор.
Катализатор, 1,2 мл 50%-ного раствора нитрата марганца(П) и 5,0 г нитрата серебра (I) растворяют в 100 мл 1 М НС1О4.
Ферроин.
Ход анализа. К раствору, содержащему 10—100 мг фосфита, добавляют 2 мл раствора катализатора, 5,5 мл концентрированной H2SO4 на каждые 100 мл раствора и избыток (70—150%) стандартного раствора сульфата церня(1У). Нагревают на горячей плитке 10 мин до 10 °C, останавливают реакцию, охлаждая раствор, добавляют 10 мл концентрированной H2SO< и тнтруют стандартным раствором сульфата железа(П), используя ферроин в качестве индикатора.
Примечание. Стандартное отклонение при определении 15—100 мг фосфита составляет ±0,17.
Спектрофотометрические методы
Как и некоторые другие фосфорсодержащие анионы, фосфит можно окислить до ортофосфата и определить спектрофотометрически по молибденовой сини или с использованием фосфорнована-
478
диевомолибденового метода. Последний метод Поллард с сотр. [5] использовали в автоматическом методе определения фосфита, после его ионообменного отделения и элюирования. Метод молибденовой сини также применяли в автоматическом методе определения фосфита [6, 7].
ЛИТЕРАТУРА
1. Rudnicki R. — Chemia analit., 1961, v. 6, p. 761; Analyt. Abstr., 19627 v. 9, p. 2731.
2. Ebel J. P. — Mikrochim. Acta, 1954, p. 679.
3. Burns D. T., Lee J. D. — Mikrochim. Acta, 1969, p. 206.
4. Seiler H. — Helv. chim. Acta, 1961, v. 44, p. 1753.
5. Pollard F. H., Rogers D. E., Rothwell M. T., Nickless G. — J. Chromat., 1962, v. 9, p. 227.
6. Pollard F. H., Nickless G., Rogers D. E., Rothwell M. T. — J. Chromat., 1965, v. 17, p. 157.
7. Pollard F. H.. Nickless G., Rogers D. E., Crone D. L. — Proc. Soc. Analyt. Chem. Conference, Nottingham, 1965, Cambridge, W. Heffer and Sons Ltd., 1965, p. 481.
8. Kolthoff I. M., Belcher R. — Volumetric Analysis, Vol. Ill, London, Inter-science, 1957.
9. Berka A., Vulterin J., Zyka /. — Newer Redox Titrants, London. Pergamon Press, 1965.
10. Guilbault G. G., McCurdy W. H. — Analytica chim. Acta, 1961, v. 24, p. 214.
II. Rao К- B., Rao G. G. — Z. analyt. Chem., 1955, v. 147, p. 274, 279.
12. Масалович В. M., Агасян П. К., Масалович Н. С., Николаева Е. Р. Зав. лаборатория. 1969, т. 35, с. 257. Analyt. Abstr., 1970, т. 18, с. 3898.
13. Норкус П. К-, Лунъятскас А. М., Царанкуте С. П. ЖАХ, 1965, т. 20. с. 753 Analyt. Abstr., 1967, т. 14, с 662
14. Масалович Н. С., Агасян П К-, Масалович В. М. и Николаева Е. Р. Зав. лаборатория, 1967, т. 33, с. 1053. Analyt. Abstr., 1968, т. 15, с. 7236.
15. Bernhart D. N. — Analyt. Chem., 1954, v. 26, p. 1798.
16. Вербицкая T. Д., Романова Н. К. Зав. лаборатория, 1960, т. 26, с. 818. Analyt. Abstr., 1961, т. 8, с. 994.
17. Норкус П. К. и Маркевичине Р. М. ЖАХ, 1967, т. 22, с. 1527. Analyt. Abstr., 1969, т. 17, с. 126.
18. Matsuo Т., Shida J., Kudo К- — Japan Analyst, 1969, v. 18, p. 488; Analyt Abstr., 1970, v. 19, p. 2998.
ПОЛИФОСФАТЫ: ТРИФОСФАТЫ, ТЕТРАФОСФАТЫ И ВЫСШИЕ ФОСФАТЫ
Полифосфаты — соединения, описываемые общей формулой ^п+гРлОзп+ь Это название часто используют как синоним названия «конденсированные фосфаты». В этом разделе мы в основном ограничимся рассмотрением трифосфата и тетрафосфата
Трифосфат
О’ О’ О’ О’
Тетрафосфат
479
Дифосфат (пирофосфат) в основном рассматривается в подразделе, посвященном методам разделения, хотя при обсуждении некоторых методов определения он тоже будет упоминаться.
Полифосфаты нашли широкое применение для умягчения воды (за счет его комплексующего действия, особенно это касается щелочноземельных металлов), в производстве пищевых продуктов, удобрений, поверхностно-активных моющих веществ, фармацевтических препаратов, а также в текстильной и бумажной промышленности.
Наибольшее значение среди линейных полифосфатов имеют тетрафосфат натрия и калгон (соль Грахамса — смесь высших полифосфа гов, содержащих в молекуле 13—25 атомов фосфора, часто ошибочно называемая «гексаметафосфат», как можно называть только циклические фосфаты). Общая химия конденсированных фосфатов была подробно описана в обзорах [1, 2].
В водных растворах при комнатной температуре и при pH = = 7—9 полифосфаты устойчивы в течение практически неограниченного времени. При повышении кислотности растворов и температуры гидролиз ускоряется, в результате образуются моно- и дифосфат
[РзО|0]5- [НР2О7]3- + [НРО4]2-
Полнфосфаты осаждаются из кислых растворов ионами бария. Это свойство использовано при создании основного метода отделения попифосфатов от более растворимых метафосфатов.
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение
Уже упоминалось, что осаждение Ва2+ — способ разделения линейных и циклических фосфатов; этот метод не всегда позволяет добиться количественного отделения линейных фосфатов. Трудность отделения заключается в неполном осаждении и соосажде-нии, особенно при малых концентрациях полифосфатов. Для разделения сейчас успешнее используют хроматографию. В качестве осадителей полифосфатов изучали комплексные соли кобальта, в некоторых случаях были достигнуты определенные успехи [3]. С помощью хлорида три (этилендиамин) кобальта (II) можно разделить трифосфат и дифосфат [4, 5], если проводить осаждение при pH = 3,6; с помощью 32Р было показано, что процессы неполного осаждения и соосаждения проходят даже при тщательно контролируемых условиях осаждения.
Методы бумажной и тонкослойной хроматографии
Впервые для разделения неорганических фосфатов метод бумажной хроматографии был применен Эбелем [6, 7]. Его большая статья [8], посвященная разделению фосфорных кислородсодер-
480
экащих кислот, послужила основой для последующих работ, в которых использовали этот метод разделения. В табл. 35 представлены наиболее важные работы по разделению, включая разделение линейных и циклических фосфатов, с применением метода бумажной и тонкослойной хроматографии.
Таблица 35. Разделение линейных и циклических фосфатов методами бумажной и тонкослойной хроматографии
Отделяемый ион
Ортофосфат Дифосфат Трифосфат Тетрафосфат Пентафосфат Гексафосфат Гептафосфат Октафосфат Триметафосфат Тетраметафосфат Гипофосфит Гипофосфат Фосфит Пирофосфит Литература
Примечание, а—Изопропанол—трихлоруксусная кислота — аммиак пл. 0,880 г/см3 (75 мл : 5 г в 25 мл воды : 0,3 мл) — кислотный реагент Эбеля. б—Изопропанол —изобута-иол — НгО— аммиак пл. 0,880 г/см3 (400 мл : 200 мл : 390 мл : 10 мл), в — Изопропанол—20%-иая трихлоруксусная кислота — НгО— аммиак пл. 0.880 г/см3 (70 мд : 20 мл : 10 мл : 0,3 мл), г — Изопропанол— изобутанол — Н2О—25%-ный аммиак (40 : 20 : 39: 1). д—Для отделения линейных фосфатов методом циркуляционной ТСХ. Изопропанол—трихлоруксусная кислота—0,1 М ЭДТА—НгО—25%-ный аммиак (8 мл : 5 г : I мл : 39 мл : 0,3 мл).
Для отделения циклических фосфатов от линейных
Этанол — изобутанол—0,1 М ЭДТА — НгО—25%-иый аммиак (30 : 30 : 1 : 38 ; 1)
Сорбент: кукурузный крахмал, е — Изопропанол—монохлоруксусная кислота — НгО — аммиак пл. 0,880 г/см3 (250 мл : 12,5 г : 350 мл : 15 мл), лс —Изопропанол—трихлоруксусная кислота—20%-иый тетраэтила ммоний гидроксид—НгО (750 мл :50г: 16 мл : 250 мл). Пластина Для ТСХ—MN полиграм, CEL 300. з— Изопропанол — изобутаиол — аммиак пл. 0,880 г/см3 (400 мл : 200 мл : 10 мл). Пластина для ТСХ — MN полиграм, CEL 300. и — Изомеры бутанола— НгО — муравьиная кислота (80 мл : 20 мл : 5 мл), к — Изопропанол — изобутанол—три-Хлоруксусная кислота — НгО — аммиак (21 различное сочетание). Сорбент—авицел.
Три основных фактора влияют на степень гидролиза полифосфатов при хроматографическом разделении: время, температура и pH растворителя. В работе [14] выполнено сравнительное изучение влияния растворителя. Преимущества методов бумажной и тонкослойной хроматографии в разделении фосфатов обсуждались Поллардом с сотр. [18].
Метод бумажной хроматографии был применен [19] для разделения длинноцепочечных полифосфатов (до п = 5000). В этой Работе был использован кислотный растворитель, предложенный Абелем.
Для линейных фосфатов со степенью полимеризации от 1 до 13 был применен метод гель-хроматографии [20]. Использовали
16 Зак. 167 4Н1
колонну с сафадексом G = 25 и 0,1 М раствор КС1 в качестве элюента.
Метод ионообменной хроматографии
Это, по-видимому, наиболее удачный метод разделения как линейных, так н циклических фосфатов. С его помощью можно разделить и затем определить даже соседние члены ряда фосфатов, что не удается сделать классическими химическими методами даже для гомологов, отличающихся на п — 3.
В то время как с помощью бумажной и тонкослойной хроматографии удается отделить микрограммовые концентрации фосфатов, с помощью ионообменной хроматографии можно выделить миллиграммовые концентрации.
Некоторые полифосфаты разделяли [21, 22] на модифицированных сшитых полистирольных смолах, содержащих ионные группировки СН2К+(СНз)з. Теория ионообменного поведения полифосфатов обсуждается в работе [23].
Описан [24] автоматический вариант ионообменного метода, предложенного в работе [25]. В системе с последовательным элюированием выходящий из колонки элюент, содержащий конденсированные полифосфаты, подвергали гидролизу при 95 °C после введения 3,4 М H2SO« для превращения фосфатных соединений в ортофосфат. Последний непрерывно определяли колориметрическим методом, основанным на образовании молибденовой сини. Поскольку все присутствующие фосфаты переводили в единственную форму — ортофосфат, система не нуждается в калибровке по каждому фосфорсодержащему аниону.
Поллард с сотр. [26, 27] применили эту работу в автоматическом варианте для анализа смесей, содержащих как низшие фосфорные анионы, так и конденсированные полифосфаты. Метод состоит в следующем. Смесь фосфатов последовательно элюируют из колонки, содержащей Дауэкс-1 X и 10% дивинилбензола. Элюируемые анионы, как показано на рис. 54, пропускались через анализатор «Technicon Auto Analyser». Типичная хроматограмма фосфатов показана на рис. 55.
Количественный расчет кривых вымывания основан на том, что концентрация фосфора в колонке является функцией времени, в этом случае площадь под кривой пропорциональна высоте пика и ширина пика составляет */2 от максимальной ширины. Другой метод расчета заключается в делении пика на узкие полосы равной ширины и последовательном вычислении и суммировании оптической плотности.
t-t,
Содержание фосфора = А 1g -у-
где К — постоянная, в которую входит ширина отдельной полосы; /2 и 6 с0* ответственно время конца и начала пика.
482
Рис. 54. Схема разделения различных фосфорсодержащих анионов с применением автоматического анализатора (Technicon AutoAnalyzer) [27]:
1—самописец; 2— колориметр (801 нм); 3—слив; 4—холодильник; 5—нагреватели (95 °C);
6—смесители; 7—устройство, замедляющее скорость потока;
Л—-образец; Б— воздух; В — окислитель; Г — сериая кислота; Д — молибдат аммония; Е — сульфат гидразина.
Исследования включали в себя и разработку системы трансформации данных элюирования непосредственно в сигнал, поступающий на компьютер, рассчитывающий результат анализа. Чтобы получить более подробную информацию о методе, следует обращаться к оригинальным статьям [26, 27].
7
Время, ч
Рис. 55. Хроматограмма простых и сложных фосфорсодержащих анионов, полученная в автоматическом варианте метода аиионообменной хроматографии [26]: гипофосфит; 5—фосфит; 3—фосфат; 4 — монотиофосфат; 5—дифосфат; 6—трифосфат;
* “—три метафосфат.
16*
483
Разделение смесей, содержащих ортофосфат, линейные и циклические фосфаты, выполнено в работе [28], в которой использовали смолу IRA-400 и в качестве элюента — раствор КС1. Удалось добиться почти идеального разделения орто-, ди-, линейных полимеров до тридекафосфатов, трех циклических соединений: тримета-, тетраметафосфатов и одного неидентифицирэванного фосфата.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Метод рН-метрнческого титрования
Этот метод основан на различии констант диссоциации моно- и полифосфорных кислот, обусловленных положением иона водорода в молекуле [29, 30]. Рассмотрим приведенные ниже формулы:
О II (осл) НО—Р—ОН (ел)
I
ОН
(с)
(сл)
О
II НО—Р—О—
I он
Г ° 1 °
—Р—О----Р—ОН (сл)
- ОН Jn ОН
(с) (с) (с)
О% /ОН (с) Р
О/ \о
(с)НОх| |/О о^РхЬхР\он (с)
с - сильная кислота; сл — слабая кислота; осл — очень слабая кислота.
На концевых группах линейных полифосфатов находятся два слабодиссоциирующих иона водорода.
Для ортофосфорной кислоты p/<i = 2,23; р/<2 = 7,20; рАз = = 12,40. При титровании гидроксидом натрия при pH = 4,5 от-титровывается первый ион водорода, а при pH = 9 — второй.
В результате полного гидролиза полифосфатов (линейных и циклических) образуется только ортофосфат. Третий ион водорода в ортофосфате обычно не титруется, но при добавлении нитрата серебра осаждается ортофосфат, в этом случае после осаждения можно оттитровать и третий ион водорода.
Обычно метод pH-метрического титрования имеет следующие этапы. К смеси фосфатов добавляют разбавленную НС1, чтобы перевести их в соответствующие кислоты. Затем раствор титруют стандартным раствором NaOH. На кривой титрования наблюдают
484
Рис. 56. Кривая титрования слабокислотных групп в ортофосфате и линейных фосфатах.
Рис. 57. Кривая титрования фосфорной кислоты с использованием метода осаждения фосфата серебром.
два скачка, первый — при pH = 4,5, второй — при pH = 9 (рис. 56).
Объем титранта А соответствует содержанию слабокислотных групп в ортофосфате и линейных фосфатах.
Тот же анализируемый раствор обрабатывают избытком НС1 для переведения нейтрализованных фосфатов в соответствующие кислоты. Избыток НС1 и сильнокислотные группы фосфатов опять оттитровывают стандартным раствором NaOH до pH = 4,5 и добавляют избыток нитрата серебра. Продолжают титровать до pH = 5. Кривые титрования показаны на рис. 57.
Объем титранта В соответствует содержанию оттитрованных слабокислотных групп и косвенно оттитрованным слабокислотным группам ортофосфата, которые нельзя оттитровать при прямом
алкалиметрическом титровании.
Наконец, во второй аликвотной части анализируемого раствора проходит полный гидролиз. По оригинальной методике предполагалась [29] обработка раствора НС1 в течение почти 8 ч. Улучшенный вариант метода позволяет проводить полный гидролиз за 1 ч. Вновь проводят pH-метрическое титрование образующегося ортофосфата, как описано выше, получая скачки на кривой титрования при pH = 4,5 и 9,0. Кривая титрования показана на рис. 58.
Объем титранта С соответствует общему содержанию фосфора. В этом случае для анализа орто- и полифосфатов можно провести три титрования (до и после гидролиза и после введения нитрата серебра). Очевидно, что присутствие метафосфатов, содержащих
одну сильнокислотную группу на 1 атом Р, существенно усложнит методику. Поэтому метафосфаты должны отсутствовать.
Р1С. 58. Кривая титрования фосфорной кислоты после полного гидролиза линейных и циклических фосфатов до ортофосфата.
485
Рис. 59. Градуировочный график для определения трнполифосфата (чувствительность — вся шкала прибора—1,5 мкА, время жизни капли — 1,0 с) (31].
Рис. 60. Зависимость высоты полярографической волны 3-10 М раствора моиооктилолова в 50%-ном по объему растворе 2-пропаиола в воде, 1 М по НСЮ< н 0,25 М по НС1 от концентрации фосфата (условия те же, что на рис. 59) [31]:
1—ортофосфат; 2— пирофосфат; 3—трнметафосфат; 4—тетраметафосфат; 5—триполифосфат.
Если присутствуют только линейные полифосфаты, среднее значение Н рассчитывают по уравнению
(общее содержание Р) содержание концевых групп Р pH-Титрование можно использовать для отнесения неизвестного фосфата к группе линейных или циклических фосфатов.
В работе [28] методом ионного обмена был выделен неизвестный фосфат и проведено его потенциометрическое микротитрование гидроксидом натрия. Один скачок на кривой титрования свидетельствует о том, что соединение не содержит слабокислотного водорода и, таким образом, является циклическим фосфатом. Этот результат был подтвержден двумерной хроматографией.
Следует заметить, что рН-тнтриметрический метод применим только в отсутствие других слабокислотных фосфорных анионов.
Метод бумажной хроматографии
Метод бумажной хроматографии с последующим колориметрическим определением общего содержания фосфора в каждом пятне после гидролиза всех соединений фосфора до ортофосфата является одним из наиболее точных методов определения полифосфатов в смесях [12]. Чаще всего для колориметрического определения применяют метод молибденовой сини.
Полярографический метод
Описан [31] косвенный полярографический метод определения трифосфат-иона в присутствии других полифосфатов. Сами фосфа-486
ты и полифосфаты не восстанавливаются на ртутном капающем электроде. В работе [31] указывается, что триполпфосфат-ионы образ}Тот растворимый комплекс с катионом монооктилолова и вызывают снижение высоты полярографической волны октилолова. Можно определять 2-Ю-4 — 4-10-3 триполифосфата в присутствии 2-10_3Л1 орто-, ди-, триметафосфатов и 10-3 М тетрафосфата в смеси или в любых комбинациях. На рис. 59 приведен градуировочный график для определения триполифосфата. На рис. 60 показано влияние различных фосфатов на высоту полярографической волны.
Метод — простой, быстрый, характеризуется удовлетворительной точностью. То, что определению не мешают ортофосфат, сульфат, ионы кальция и магния, позволяет применить метод для анализа моющих препаратов и стоков.
Другие методы определения
Разработан метод осаждения триполифосфата цинком с последующим комплексометрическим определением цинка [32]. Другой метод анализа полифосфатов основан на изучении спектров, полученных методом ядерного магнитного резонанса [33, 34]. С удовлетворительной точностью метод позволяет определять соотношение ортофосфата, концевых и промежуточных групп и, вероятно, разветвленность цепочек полимеров.
ЛИТЕРАТУРА
1. Wazer J. R Van — Phosphorus and its Compounds, Vol. 1, New York, Interscience, 1964
2. Thilo E.— Adv. Inorg. Chem. Radiochem., Vol. 4, New York, Academic Press, 1962, p. 1.
3. McCune H. W., Arquette G. J. — Analyt. Chem., 1955, v. 27, p. 401.
4. Голубчик E M. Зав. лаборатория, 1969, т. 35, с. 926. Analyt. Abstr., 1970,
5. Weiser H. J. — Analyt. Chem., 1956, v. 28, p. 477.
6. Ebel J. P. — Bull. Soc. chim. Fr., 1953, v. 20, p 991, 998.
7. Ebel J. P. — Compt. rend., 1952, v. 234, p. 621.
8. Ebel I. P. — Mikrochim. Acta, 1954, p. 679.
9. Karl-Kroupa E. — Analyt. Chem., 1956, v. 28, p. 1091.
10. Thilo E., Grunze H —Die Papierchromatographie der kondensierten Phosphate, Berlin, Akademie-Verlag, 1955
11. Mehrotra R. C., Gupta V. S. — J. Polymer Sci., Pt. A2, 1964, p. 3959.
12. Woodis T. C., Trimm J. R., Duncan R. D. — Analytica chim. Acta, 1973, v. 65, p. 469.
13. Lederer M., Majani C. — Chromatog. Rev., 1970, v. 12, p 239.
14. lida T., Yatnabe T. — J. Chromat., 1971, v. 54, p. 413.
15. Ионова Л. А., Постников H. H. Хим. пром. 1969, с. 198. Analyt. Abstr., 1970, т. 18, c. 2344.
16. Canic V. D., Turcic M. N., Petrovic S. M., Petrovic S. E.—Analyt. Chem., 1965, v. 37, p. 1576.
io Qurns D. T., Lee 1. D. — Mikrochim. Acta, 1969, p. 206.
18 Pollard F. H., Nickless G., Burton K, Hubbard J. — Microchem. J, 1966, v 10, p. 131.
19- Ohashi S., Van Wazer J. R. — Analyt. Chem., 1963, v. 35, p. 1984.
487
20. Ueno Y., Yoza N., Ohashi S. — J. Chromat., 1970, v. 52, p. 481.
21 Lindenbaum S, Peters T. V., Rieman W. — Analytica chim. Acta 1954, v 11, p 530
22. Peters T. V., Rieman W.— Analytica chim. Acta, 1956, v. 14, p. 131.
23. Beukenkamp J., Rieman W., Lindenbaum S.— Analyt. Chem., 1954, v. 26, p. 505.
24. Lundgren D P, Loeb N. P. — Analyt. Chem., 1961, v. 33, p. 366
25. Grande J. E Beukenkamp L — Analyt. Chem., 1956, v. 28, p. 1497
26. Pollard F. H., Nickless G., Rogers Ь. E., Rothwell M. T. — J. Chromat, 1965, v. 17, p. 157.
27. Pollard F. H, Nickless G., Rogers D. E, Crone D L. — Proc. Soc. Analyt. Chem. Conference, Nottingham, 1965, Cambridge, W. Helfer and Sons Ltd, 1965, p. 481.
28. Rothbart H. L., Weymouth H. W., Rieman W.— Talanta, 1964, v. 11, p. 33.
29. Van Wazer J. R., Griffith E. J., McCullough J. F. — Analyt. Chem., 1954, v. 26, p 1755.
30. Griffith E. Л — Analyt Chem., 1956, v. 28, p 525.
31. Shaw S., Townshend A. — Talanta, 1973, v. 20, p. 332.
32. Akiyama T., Fujiwara M., Okamoto H., Fusaka K. — Bull. Kyoto Coll. Pharm., 1955, p. 19.
33. Callis C. F, Van Wazer J. R, Shoolery L N. — Analyt. Chem, 1956, v. 28, p 269
34. Van Wazer 1. R., Callis C. F., Shoolery J. N. — J. Am. chem. Soc., 1955, v. 77, p. 4945.
ЧАСТЬ IV
АНИОНЫ СЕРЫ
ПИРОСУЛЬФИТЫ
Пиросульфит-анион S2O5' существует только в твердом со* стоянии и имеет структуру
\s—S—О’
При растворении в воде пиросульфит гидролизуется до гидросульфита:
Na2S2O5 4- Н2О 2NaHSO3
При осторожном выпаривании растворов гидросульфита образуется безводный пиросульфит натрия. Кислоты средней силы превращают пиросульфит в H2SO3. Основания или соли слабых кислот превращают пиросульфиты в сульфиты.
Таким образом, аналитическая химия водных растворов пиросульфита в значительной степени является аналитической химией гидросульфита и сульфита.
Титрование раствором иода является стандартным методом, например анализ фотографического метабисульфита натрия (этот метод рекомендуется Британскими стандартами [1].
При определении сульфита, образованного из пиросульфита, важно добавлять его к избытку иода, а не наоборот, так как в последнем случае получаются заниженные результаты. Неизрасходованный иод титруют стандартным раствором тиосульфата [2]:
SOf + Ia + HaO —> SO?’+ 2Н++ 2Г
При определении днсульфита в присутствии тиосульфата и тетратионата используют [3] их различное поведение при окислении галогенами. С иодом реагирует пиросульфит и тиосульфат, а с бромом — все три аниона. Если окисление иодом проводят в присутствии формальдегида, пиросульфит маскируется и с иодом реагирует только сульфит.
ЛИТЕРАТУРА
1. British Standards 3834: London, 1964.
2. Standard Methods of Chemical Analysis. Ed. N. H. Furman, 6th Edition, Vol. I, 1016, Van Nostrand, Princeton, N Jersey, 1962.
2 Szekeres L., Sugar E. Magyar Kem. Lapja, 1961, v. 16, p. 434, Analyt. Abstr., 1962, v. 9, p 1453.
489
ДИТИОНАТЫ
Дитионат 5гОе' относительно устойчив к окислению, поэтому удобно обсудить его определение и определение политионатов раздельно. Дитионаты устойчивы к гидролизу, к окисляющим агентам на холоду и многим другим реагентам. Ввиду этого обстоятельства число методов определения дитионатов ограничено.
Синтез дитионата описан ранее [1]. Методы определения дитионата включены в несколько обзоров по аналитической химии соединений серы [2—4].
МЕТОДЫ ОТДЕЛЕНИЯ
Дитионат можно отделить от других серусодержащих анионов методами хроматографии на бумаге [5]. Для отделения его от сульфита, пероксодисульфата, дисульфата и тетратионата в качестве растворителя использована смесь н-бутанола и воды (3: 1). Отделение дитионата от сульфата, тиосульфата, сульфида, дисульфита и гидросульфата достигается применением смеси изопропанола с водой (3: 1).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Окисление дитионата до сульфата служит основой большинства титриметрических методов его определения. Исследованы методы окисления дитионата как в твердой, так и в жидкой фазе. В кислой среде при повышенной температуре дитионаты разлагаются в соответствии с уравнением
S2Of —> sor + so2
На этой реакции основан титриметрический метод определения дитионата. Для лучшего отделения SO2 от раствора используют газ-носитель (СО2), выделенный SO2 определяют иодометрически.
Метод с использованием смеси бихромата
с серной кислотой
Дитионат окисляется до сульфата стандартным раствором бихромата в среде концентрированной H2SO4 [6, 7]:
8S2O|” + Сг2ОГ 4- 2Н+ —> 2Cr3+ + 6SOf + Н2О
Избыток бихромата определяют иодометрически.
Описан метод [8] определения дитионата в присутствии сульфида, сульфита, тиосульфата и политионата. Пробу кипятят с щелочным раствором перманганата калия для окисления сульфида, сульфита, тиосульфата и политпоната. Избыток перманганата калия разрушают кипячением с сульфатом марганца В
490
нейтральной среде. Осажденный диоксид марганца фильтруют, в раствор добавляют Н2БО4 до ) становления ее концентрации 5 Л1 и обрабатывают его бихроматом для окисления дитпоната.
Для титриметрического определения дитионата также рекомендованы другие окислители, но имеются сомнения в их эффективности. Правильные результаты определения дитионата получаются только при использовании окислительной смеси бихромата с серной кислотой [3].
Иодиметрический метод
Описан метод анализа смесей S О]’—БОз"—БОГ> основанный на обработке пробы НС1 [9]. Выделенный SO2 пропускают в охлажденный избыток стандартного раствора иода (газ-носитель СО2). Образующийся из сульфита SO2 выделяют до определения дитионата. Дигионат определяют иодометрически после маскирования мешающих катионов раствором ЭДТА. Сульфат определяют комплексометрически в виде сульфата бария после удаления мешающих катионов ионообменным методом. Описанный метод был использован для анализа жидкостей, содержащих 3—70 г/л дитионата, менее 11 г/л суммы суль'фита и SO2 и ПО—240 г/л БОГ.
Редокс-методы
Окисление дитионата ванадатом предложено [10J проводить в среде H2SO4. Этот метод является модификацией метода, предложенного в работе [11], и отличается от последнего большей экспрессностью. Образующийся ванадий(IV) титруют стандартным раствором КМпО4.
В качестве окислителя предложен также церий (IV) [12]. Сульфат церия(IV) в растворах кислоты при кипячении окисляет дитионат, как и три- и тетратионаты, до сульфата. Дитионат количественно окисляется в 1 М Н2БО4 в течение 30 мин при температуре кипения раствора. Такой же результат получается в 6 М или в 9 М Н2БО4. Три- и тетратионаты полностью окисляются церием (IV) при кипячении лишь в среде 6 М H2SO4.
В качестве окислителя был исследован [13] гексацианофер-рат(Ш). Избытком гексацианоферрата(III) в 4—5 М щелочном растворе при 50—60 °C в присутствии осмиевой кислоты в качестве катализатора окисляются также сульфит, гидросульфит, сульфид, тиосульфат, дисульфит и тетратионат.
Колориметрические и спектрофотометрические методы
Предложен колориметрический метод определения дитионата [14], основанный на его окислении смесью бихромата с серной кислотой. Образец, смешанный с известным избытком бихромата калия в 5Л1 H2SO4, нагревают на водяной бане в течение 15 мин.
491
Затем измеряют светопоглощение относительно раствора реагента. Избыток бихромата можно определять также полярографически.
Метод, основанный на окислении дитионата ванадием (V), позволяет определять до 80 мг дитионата в присутствии сульфита после предварительного окисления сульфита КМпО4 в щелочной среде.
Для определения дитионата описан [15] спектрофотометрический метод, основанный на превращении дитионага в сульфат упариванием анализируемого раствора с 1 М раствором карбоната натрия и последующем сплавлении остатка при 900—950°C в течение 5 мин. Необходимо предварительное удаление сульфата из образца.
ЛИТЕРАТУРА
1. Fernelius W. С. (Ed.), Inorganic Synthesis, Vol. 2, New York, McGraw-Hill, 1946, p. 170.
2. Blasius E., Horn G., Knochel A., Munch J., Wagner //. — In: Inorganic Sulphur Chemistry (Ed. G. Nickless), Amsterdam, Elsevier, 1968, p. 221.
3. Szekeres L.— Talanta, 1974, v. 21, p. 2.
4. Haff L. V. — In: The Analytical Chemistry of Sulphur and its Compounds (Ed.
J. H. Karchmer), Part 1, New York, Wiley-Interscience, 1970, p. 237.
5. Servigne K, Duval C. — Compt. Rend., 1957, v. 245, p. 1803; Analyt. Abstr., 1958, v. 5, p. 1833.
6. Yoshida J. — J. chem. Soc. Japan, pure Chem. Sect., 1942, v. 63, p. 1533; Chem. Abstr., 1947, v. 41, p. 3016
7. Glasstone S., Hickling A. — J. chem. Soc., 1933, p. 5.
8. Murthy A. R. V. — Current Sci, India, 1953, v 22, p. 371; Chem Abstr., 1954, v. 48, P. 6912g.
9. Softer N.— Analyst, 1961, v. 86, p. 843.
10. Crossland H., Hofmann-Bang A.—Acta. Chem. Scand., 1961, v. 15, p. 1064.
11. Lang R., Kurtenacker H. — Z. analyt. Chem., 1942, v. 123, p. 81.
12. Nair V. R., Nair C. G. R. — Talanta, 1971, v. 18, p. 432.
13. Solymosi F., Varga A. — Analytica chim. Acta, 1957, v. 17, p. 608.
14. Verek-Siska J., Eckschlager K., Wagnerova D. W. —Chemikfe Listy, 1965, v. 59, p. 1479; Analyt. Abstr., 1967, v. 14, p. 1952.
15. Ozawa T. — J. chem. Soc. Japan, pure Chem. Sect., 1966, v. 87, p. 859; Analyt Abstr., 1967, v. 14, p. 7454.
ДИТИОНИТЫ
Дитионит натрия Na2S2O4 находит применение в качестве отбеливающего агента в текстильной и бумажной промышленности Для крашения целлюлозных волокон.
Дитиониты — очень сильные восстанавливающие агенты, более сильные, чем гидросульфиты, до которых дитиониты окисляются в водных растворах.
Безводные щелочные дитиониты стабильны. При нагревании до 190 °C они превращаются в сульфит и тиосульфат с выделением SO2. Гидратированные соли и водные растворы дитионитов быстро окисляются на воздухе до сульфата и гидросульфита. В отсутствие
491Г
воздуха растворы также нестабильны и диспропорционируют в гидросульфит и тиосульфат:
2\a2S2O4 + H2O —» 2NaHSO3 + Na2S2Os
Скорость диспропорционирования зависит [1] от температуры, концентрации дитионита и pH. Исследования диспропорционирования водных растворов дитионита были продолжены авторами работы [2].
Методы определения дитионита описаны в нескольких руководствах по общей аналитической химии соединений серы [3—5].
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Титриметрические методы
Йодометрические методы
Дитионит окисляется иодом до сульфата:
S2O^‘+ 312 + 4Н2О —» 2SO<" + 6Г + 8Н+ (1)
Дитнонит вступает также в реакцию с формальдегидом, в которой образуются гидросульфитное и сульфоксилатное производные формальдегида:
S2Oj” + 2НСНО + Н2О —> HCHO-HSOJ+ HCHO-HSO; (2) Гидросульфит формальдегида не реагирует с иодом, сульфоксилат формальдегида окисляется иодом:
НСНО • HSOJ + 212 + 2Н2О —> SO*’+ 4Г + БН++ НСНО (3)
Интересно, что формальдегид маскирует присутствующие гидросульфиты и только сульфоксилаты реагируют с иодом.
Опубликован [6] метод определения дитионита, основанный на приведенных реакциях, который был впоследствии рекомендован Американской ассоциацией химиков-текстильщиков и специалистов по крашению [7]. Этот метод неприменим при значительных содержаниях тиосульфатов, сульфитов и других окисляемых примесей, не маскируемых формальдегидом.
Для определения дитионита, гидросульфита и тиосульфата при их совместном присутствии предложен метод [8], в котором проводят три йодометрических титрования.
1. Избыток стандартного раствора иода прибавляют к образцу в присутствии формальдегида. Неизрасходованный иод титруют стандартным раствором тиосульфата. Дитионит при этом участвует в реакциях (2) и (3).
2. К другой аликвотной части раствора прибавляют определенное количество иода, а затем вводят в раствор избыток суль-
493
фита; сульфит реагирует с тетратионатом, образовавшимся при взаимодействии тиосульфата с иодом:
S4O62-+SOr —> S8or + Ssor (4)
Избыток сульфита маскируют формальдегидом, а тиосульфат, образовавшийся по реакции (4), титруют иодом.
3. Третью аликвотную часть прибавляют к известному избытку стандартного раствора иода, а неизрасходованный избыток иода титруют стандартным раствором тиосульфата. Дитионит реагирует с иодом в соответствии с уравнением (1), а гидросульфит — по уравнению (5):
hso;4-i2 + h2o —> hso; + 2hi (5)
Концентрации трех анализируемых компонентов можно определить с помощью соотношений, приведенных ниже.
Описан вариант метода [8] с использованием хлорамина Т в качестве окислителя [9].
Для определения дитионита использованы и другие варианты йодометрического метода. Например, был разработан метод, основанный на диспропорционировании водных растворов дитионита до гидросульфита и тиосульфата [10], при этом авторами работы отмечено, что 1 моль дитионита реагирует с шестью эквивалентами иода, а продукты диспропорционирования дитионита — только с пятью.
Для анализа смесей S2O4’—S2O3’—SO3' была использована реакция разложения дитионита в присутствии НС1 [11].
Титриметрическое определение дитионита, гидросульфита и тиосульфата в образцах дитионита натрия [28]
Реагенты
Раствор формальдегида, приблизительно 37%-ный.
Уксусная кислота, 20%-иый раствор.
Иод, приблизительно 0,1 М раствор (стандартный).
Тиосульфат натрия, приблизительно 0,1 М раствор (стандартный).
Раствор сульфита натрия, 25 г/л безводного Na2SOs
Ход анализа. Прибавляют 35 мл раствора формальдегида к 50 мл кипяченой и охлажденной дистиллированной воды, добавляют 0,3 г Па2СОз-10Н2О К этому раствору прибавляют взвешенный твердый образец или аликвотную часть образца и перемешивают. Прибавляют 25 мл воды, 5 мл 20 %-ной уксусной кислоты, перемешивают и титруют раствор стандартным раствором иода (объем израсходованного раствора иода — 4).
Прибавляют аликвотную часть анализируемого раствора к избытку стандартного раствора иода (для 0,2 г Na2S2O< достаточно около 75 мл раствора иода), в котором растворено около 1 г кристаллического CH3COONa. Медленно прибавляют раствор сульфита натрия до тех пор, пока раствор иода не обесцветится, и затем добавляют еще 30 мл раствора сульфита иатрня. Прибавляют 2 капли раствора фенолфталеина и нейтрализуют полученный раствор 1 М раствором NaOH. Через 5 мин прибавляют 10 мл раствора формальдегида н 10 мл 20%-ной уксусной кислоты, раствор перемешивают и титруют стандартным раствором иода (объем израсходованного раствора иода — В).
Прибавляют аликвотную часть образца дитиовита к известному объему стандартного раствора нода, в котором растворено около 1 г кристаллического 494
CH3COONa. Титруют избыток иода стандартным раствором тиосульфата (объем израсходованного раствора иода — С).
Расчет.
мМоль S2O|~ = 2В
мМоль S2O«~ =
„ Т1ОЛЛ_ 2С-ЗД + 2В мМоль HSO3 ----------т------
4
Примечание. Вывод этих формул приведен в оригинальных работах [2.8].
Редокс-методы
Для определения дитионитов обычно применяют иодиметриче-скпе методы, однако предложены методы, основанные и на других окислительно-восстановительных реакциях. В этих методах используют гексацианоферрат(Ш), железо(Ш), иодомеркурат(П) калия (реактив Несслера), аммиачные растворы нитрата серебра и сульфата меди и метиленовый синий. В некоторых случаях конец титрования регистрируют не визуально, а потенциометрически.
В первой феррометрической методике использован нейтральный раствор хлорида железа (III) в смеси с гексацианоферра-том (III) и тиоцианатом в качестве индикатора [12]. В точке эквивалентности окраска раствора меняется с Глубокой красной на синюю. Погрешность определения дитионита описанным методом составляет менее 0,1%.
При изучении кинетики окисления водных растворов дитионита воздухом найдено [13], что титрование дитионита аммиачным раствором сульфата меди является наиболее удовлетворительным аналитическим методом
2Cu (NHs)f + S2or + 2Н2О —> 2Cu+ + 280^ + 4NHJ + 4NH3
Этот реагент был исследован [14], и установлено, что надежные результаты определения дитионита этим методом достигаются даже в присутствии больших количеств смеси гидросульфита с сульфитом.
Удовлетворительные результаты определения дитионита достигаются при использовании метиленового синего в качестве титранта [15, 16]. В процессе титрования анализируемого раствора метиленовым синим интенсивная синяя окраска реагента уменьшается до почти бесцветной окраски лейкоформы. Техника проведения эксперимента описана недостаточно, но приведено указание, что гидросульфит, сульфат и тиосульфат не мешают определению дитионита.
При использовании перйодата [17] селективность определения дитионита достигается благодаря применению формальдегида, так как сульфоксилат формальдегида, образуемый из дитионита по уравнению (2), реагирует с перйодатом, а гидросульфит формальдегида с ним не реагирует.
495
Титрование дитионита гексацианоферратом (III) является одним из наиболее изученных методов
S2OJ- + 2 [Fe (CN)6J3- + 4 ОН' —> 2SO?'+ 2 [Fe (CN)6]4'+ 2Н2О
В ранних работах [18], посвященных этому методу, предложено железо(П) в качестве индикатора. Позднее в качестве индикатора были использованы диметилглиоксимат железа(II) [19] и метиленовый синий [20].
Титриметрическое определение дитионита в техническом дитиоиите натрия [20]
Ход анализа. К 90 мл воды, помещенным в колбу для титрования, прибавляют 10 мл примерно 0,1 М раствора NaOH, 10 мл метанола и 1 мл 0,2%-ного раствора метиленового синего Пропускают через раствор азот, свободный от кислорода. После удаления кислорода из раствора прибавляют небольшое количество дитионита натрия и затем 0,02 М раствор KsFe(CN)e до появления голубой окраски. Прибавляют образец дитионита, содержащий около 250 mi Na2S2O4, и титруют раствор 0,1 М раствором K3Fe(CN)6 до нового появления синей окраски.
Примечание. Определению дитионита мешает сульфид, его необходимо предварительно удалить. Определению дитноннта не мешают следующие вещества: Na3PO4 (0,2 г), Na2P2O7 (0,2 г), Na5P3O10 (0,2 г), Na2SO4 (0,2 г), Na2S2O3 (0,5 г), ЭДТА (0,5 г), Na2SO3 (1 г), Na2S3O6 (0,1 г). Не мешают определению дитионита также NaCl, Na2CO3, карбамид и древесная мука.
Спектроскопические методы
Колориметрические методы основаны на способности дитионита восстанавливать некоторые окрашенные вещества до бесцветных соединений, например инантренового желтого G (С.1.1118) [21], азорубина (С.1.179), амаранта [21]. Для полуколичественного колориметрического определения дитионита был использован нафтоловый желтый [22]. При добавлении образца дитионита к красителю цвет его мгновенно превращается из желтого в красный. Время, необходимое для восстановления желтой окраски, является полуколичественной оценкой содержания присутствующего дитионита. Определению дитионита не мешают сульфит, сульфат, сульфид, хлорид, гидроксил и алкоксид, а также SO2.
Косвенный метод определения дитионита [23] основан на образовании соединения хромата с о-дианизидином. Дитионит обрабатывают известным избытком хромата, а непрореагировавший хромат определяют по реакции с о-дианизидином (светопоглощение измеряют при 470 нм). Предложенный метод экспрессен, прост и позволяет определять 0,1—4 мкг/мл Дитионита. При использовании метода необходимо учитывать канцерогенность о-дианизидина.
Электрохимические методы
Потенциометрические методы
Потенциометрические методы определения дитионита основаны на его окислении гексацианоферратом (III), железом (III) и таллием (Ш),
496
Дитионит окисляется до сульфита в мягких условиях гексацианоферратом (III). Если образец кроме дитионита содержит сульфит, дальнейшее окисление в сильнощелочном растворе прн 50—60°C в присутствии OsO4 в качестве катализатора превращает эти анионы в сульфаты. Метод применим для одновременного определения дитионита, сульфата и тиосульфата с погрешностью до 0,4% [24, 25].
Описан метод определения дитионита, основанный на его окислении железом (III) [26].
При титровании дитионита хлоридом таллия(III) с использованием платинового и каломельного электродов было найдено [27], что в точке эквивалентности потенциал изменяется не менее чем на 300 мВ. Такое же изменение потенциала наблюдается при титровании Na2SO3 и Na2S2O5 этим титрантом. Использование иодид-крахмальной смеси позволяет визуально установить точку эквивалентности при титровании дитионита таллием (III).
Полярографические методы
Предложен автоматический полярографический метод определения дитионита в присутствии NaOH [28]. Этот метод применен для анализа восстановительных растворов в производстве красителей. В отсутствие кислорода дитионит дает полярографическую волну при —0,45 В относительно насыщенного каломельного электрода. Определению дитионита предложенным методом не мешают тиосульфаты, сульфаты, гидросульфаты, а также красители индантренового ряда при концентрации, не превышающей 0,01%.
Непрерывное полярографическое определение дитионита проведено в основном электролите 0,5 М по (NH4)2HPO4 и 0.5 М по аммиаку [29]. Описан также метод амперометрического титрования дитионита с использованием 0,2 н. раствора рубина (С. I. Acid Red 14).
ЛИТЕРАТУРА
1. lellinek К.. Jellinek Е. — 1 phys. Chem., 1919, v. 93, p. 325.
2. Danehy J. P., Zubritsky C. W. — Analyt. Chem., 1974, v. 46, p. 391.
3. Szekeres L.— Taianta, 1974, v. 21, p. 2.
4. Blasius E., Horn G., Knockel A., Munch J., Wagner H. — In: Inorganic Sulphur Chemistry (Ed. G. Nickless), Amsterdam, Elsevier, 1968, p. 220.
5. Haff L. V. — In: The Analytical Chemistry of Sulphur and its Compounds (Ed.
J. H. Karchmer), Part 1, New York, Wiley-Interscience, 1970, p. 244.
6. Merriman R. W. — Chem. Ind, Lond., 1923, v. 42, p. 290.
7. Am. Dyestuff Reptr, 1957, v. 46, p. 443; Analyt Abstr., 1958, v 5, p. 1498.
8. Wollak R.— Z. analyt. Chem., 1930, v. 80, p. 1.
9. Nair V. R„ Nair C. 6. R. — Res. Ind., 1971, v. 16, p. 47; Chem. Abstr., 1972, v. 76, p. 54069s.
10. Schulek E.t Maros L. — Acta chim. hung., 1958, v. 17, p. 273; Analyt. Abstr., 1959, v. 6, p. 1279.
11 CZ51306S апа1У‘- Chem., 1964, v. 203, p. 178; Analyt. Abstr., 1965, v. 12,
497
12. Hahn F. L. — Analytica chim. Acta, 1949, v. 3, p. 62.
13. Spencer M. S. — Trans. Faraday Soc., 1967, v. 63, p. 2510.
14. Patel С. C., Rao M. R. A. — Proc. natn. Inst. Sci. India, 1949, v. 15, p. 115.
15. Rinker R. G., Gordon T. P., Mason D. M., Sakaida R. R., Corcoran W. H________
J. phys. Chem, Ithaca, 1960, v. 64, p. 573
16 Rinker R. G, Lynn S., Mason D. M, Corcoran IF. H. — IEC Fundam., 1965, v. 4, p. 282.
17. Kaushik R. L., Rajendra P.—J. Indian chem. Soc., 1969, v. 46, p. 405.
18. Ciba Rev., 1941, v. 4, p. 1391.
19. Wawzryczek W., Rychcik W. — Chemia analit., 1963, v. 8, p. 539; Analyt. Abstr., 1964, v. 11, p. 3639
20. De Groot D. C. — 7. analyt. Chem., 1967, v. 229, p. 335; Analyt. Abstr., 1968, v. 15, p. 6122.
21. Goebel E. F., Roseira A. — Rev. Quim. Ind. Rio de Janeiro, 1957, v. 26, p. 20; Analyt. Abstr., 1958, v. 5, p. 1832.
22. Whaley T. P., Gyan J. A. — Analyt. chem., 1957, v. 29, p. 1499.
23. Buscarons F, Artigas A, Rodriguez-Roda C. — Analytica chim. Acta, 1960, v 23, p. 214.
24. Solymosi F., Varga A. — Analytica chim. Acta, 1957, v. 17, p. 608.
25. Solumosi F., Varga A. — Magy. kern. Foly, 1959, v. 65, p. 52; Analyt. Abstr., 1959, v. 6, p. 4746.
26. Eerteroile E Guiffre L. — Tinctoria, 1954, v. 51, p 364.
27. Wawrzczek W, Tylzanowska D., Jacyk A. — Chemia analit., 1968, v. 13, p. 575; Analyt. Abstr., 1969, v. 17, p. 2635.
28. Oka S. — Japan Analyst, 1961, v. 10, p. 774; Analyt. Abstr., 1963, v. 10, p. 3193.
29. Lem W. J., Wayman M. — J. Soc. Dyers Colour., 1967, p. 277; Analyt. Abstr., 1968, v. 15, p. 5957.
ПЕРОКСОДИСУЛЬФАТЫ
Пероксодисульфатами называют производные надсерной кислоты (кислоты Маршалла). Ион S2O8- известен также под назва-
О О
II II
О—S —О—О—S—о
II II
о о
Пероксодисульфат 8гО|“
нием персульфат. Щелочные пероксодисульфаты в сухом и свободном от кристаллизационной воды виде совершенно стабильны. В процессе гидролиза пероксодисульфатов образуется пероксид водорода, пероксодисульфаты обычно содержат это соединение в виде примеси. В литературе описано несколько методов анализа смесей, содержащих оба компонента.
Будучи сильным окислителем, пероксодисульфат находит применение для обесцвечивания и окрашивания различных материалов, кроме того, его используют для приготовления медицинских и косметических препаратов. Пероксодисульфаты аммония и калия часто прибавляют в малых количествах (около НО ppm) к пудрам.
Аналитическая химия пероксодисульфата изложена в нескольких обзорах, посвященных анализу соединений серы [1—3]»
498
g статье [4] описаны гравиметрические и спектрофотометрические методы определения пероксодисульфата.
МЕТОДЫ ОТДЕЛЕНИЯ
Пероксодисульфат можно отделить от серной кислоты, сульфата, гипофосфита, фосфита, гипофосфата и конденсированных фосфатов методами хроматографии на бумаге [5]. В качестве подвижных фаз используют смеси изопропанола с хлоруксусной кислотой и изопропанола, изобутанола, этанола и водного аммиака.
Для разделения сульфата, тиосульфата, сульфита и пероксодисульфата предложена [6] тонкослойная хроматография на силикагеле, смешанном с крахмалом, с подвижной фазой метанол — пропанол — 0,88 М аммиак — вода (10: 10: 1 :2).
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Методы определения пероксодисульфата основаны на окислительно-восстановительных реакциях.
Гравиметрические методы
В качестве осаждающих реагентов для пероксодисульфатов предложены [4] соединения типа R3P(CH2)nPR3+ где R — арил или аралкил. При определении 20—70 мг пероксодисульфата использован [(С6Н5)зР(СН2)4Р(С6Н5)з]Вг2. Образующийся в этом случае осадок высушивают при 150 °C и взвешивают.
Титриметрические методы
Иодиметрические методы
Авторы ранних работ по окислению иодида пероксодисульфатом считают, что реакция
8аОГ + 2Г —> 2sor + b
слишком медленна для аналитических целей. В качестве катализаторов этой реакции предложены хлорид железа(III) [7] и следы Железа при большом избытке иодида калия [8].
Иной подход к определению пероксодисульфата [9, 10] основан иа его разложении концентрированной серной кислотой и последующем титровании образующегося пероксида водорода Иодом или перманганатом калия.
Последующее изучение реакции пероксодисульфата с иодидом показало [11], что скорость этой реакции сильно зависит от pH и что в нейтральном растворе иодид количественно окисляется до иода быстро, без нагревания и катализаторов. Определение микроколичеств пероксодисульфатов с использованием иодида можно
499
проводить и спектрофотометрическим методом. Методика тптри-метрического подпметрического определения макроколпчеств пероксодисульфата приведена ниже.
Иодиметрический метод использован для анализа смесей пероксодисульфата с пероксидом водорода. Сначала пероксид селективно восстанавливают гипохлоритом, а избыток гипохлорита удаляют цианидом. После удаления избытка цианида в токе СО2 пероксодисульфат определяют иодиметрически [12].
Иодиметрическое титрование пероксодисульфата [11]
Реагенты
Иодид калия, 6 М раствор.
Тиосульфат натрия, 0,1 М раствор
Фосфатный буферный раствор, pH = 6,85; смесь 0,05 М растворов КН2РОЧ и К^НРО4.
Ход анализа. Точно взвешенный образец, содержащий 1—2 моль пероксодисульфата, растворяют в 20 мл буфервого раствора. Прибавляют 10 мл 6 М раствора KI и титруют стандартным раствором тиосульфата до тех пор, пока раствор почти не обесцветится. При необходимости прибавляют индикатор — крахмал и титруют до исчезновения синей окраски.
Метод с использованием железа(П)
В кислой среде пероксодисульфат восстанавливается сульфатом железа (II):
S2O|' + 2Fe2+ —> 2SOf + 2Fe3+
Методы определения пероксодисульфата, основанные на этой реакции, очень просты, правильны и точны [13]. Избыток железа (III) можно определить цериметрически или с помощью аскорбиновой кислоты [14].
Метод с использованием разнообразных восстановителей
Для количественного восстановления пероксодисульфата использован мышьяк(Ш). Для определения избытка мышьяка(Ш) применяют иодиметрическое [15] и перманганатометрическое [16] окончания.
Для определения пероксодисульфата и пероксида водорода и их смесей использована аскорбиновая кислота в качестве восстановителя, а ее избыток титруют гексацианоферратом (III) [17].
Комплексометрические методы
Один из вариантов комплексометрического определения пероксодисульфата основан на окислении им избытка железа(II) и титровании образующегося железа(III) стандартным раствором ЭДТА с салициловой кислотой в качестве индикатора [18]. Метод применен для определения 10—270 мг пероксодисульфата.
500
Другой вариант определения пероксодисульфата основан на его разложении до сульфата [19]. Разложение происходит при кипячении водного раствора пероксодисульфата. Для осаждения образованного сульфата прибавляют избыток хлорида бария и избыток бария оттитровывают стандартным раствором ЭДТА.
Спектроскопические методы
Колориметрические и спектрофотометрические методы
Для спектрофотометрического определения пероксодисульфата при его микросодержаниях используют йодометрическую методику [11], а светопоглощение растворов измеряют при pH = 6,85 и длине волны 355 нм. Метод применен для определения 0,01— 0,15 мкмоль пероксодисульфатов
Описан метод определения пероксодисульфата, основанный на его реакции с катионом тетрафениларсония [4]. Образующийся бис(тетрафениларсоний) пероксодисульфат экстрагируют 1,2-дихлорэтаном, а затем обрабатывают иодидом и измеряют светопоглощение при 366 нм. Пероксодисульфат, пероксосульфат и пероксид водорода можно экспрессно определить в смесях, применяя растворы сульфатов церия(IV) и железа (II) [20]. В случае присутствия всех трех компонентов к аликвотной части анализируемого раствора прибавляют мышьяк(Ш) и определяют пероксид водорода периметрически:
2Се4+ + Н2О2 —> 2Се3+4-2Н+ + О!
Содержание пероксодисульфата определяют следующим образом. Прибавляют к этой аликвотной части анализируемого раствора избыток железа(II) и определяют содержание образовавшегося железа (III). Общее содержание окислителей находят после прибавления железа(II) к другой аликвотной части анализируемого раствора и определения образующегося железа(III) спектрофотометрически. Пероксосульфат находят по разности.
Для определения пероксодисульфата предложено использовать краситель Альциан синий (С. 1.74240) [21]. Степень необратимого окисления и обесцвечивания красителя определяется содержанием пероксодисульфата. Определению пероксодисульфата этим мето дом не мешает 8-кратный избыток бромата калия.
Очень чувствительный метод косвенного определения пероксо Дисульфата основан на использовании трифенилметановых красителей [22]. Эти красители можно применить для определения и некоторых других окислителей. Чувствительность определения пероксодисульфата описанным методом — около 0,01 мкг/мл.
Другие спектроскопические методы
Описан флуориметрический метод определения пероксодисуль-Фата, основанный на окислении им лейкооснования флуоресцеина [23]. Интенсивность флуоресценции растворов пропорциональна
501
содержанию пероксодисульфата. Метод использован для определения 5—10 ppm пероксо дисульфата в пудрах. Определению пероксодисульфата предложенным методом мешает бромат.
Для идентификации пероксодисульфата использованы методы инфракрасной спектроскопии [24, 25].
Для определения некоторых серусодержащих анионов, в том числе пероксодисульфатов, применяют метод молекулярной эмиссии в полости. Разработанная методика [26] позволяет определять нанограммовые количества пероксодисульфатов в образцах объемом в несколько микролитров. Краткое описание этой методики приведено в разделе, посвященном сульфиду.
Электрохимические методы
Потенциометрические методы
Пероксодисульфат титруют в сильнощелочном растворе с использованием арсенит-иодидной системы [27]. К образцу прибавляют избыток мышьяка (III) и титруют раствор стандартным раствором иода при 60—70°C с платиновым и стандартным каломельным электродами.
Для определения пероксодисульфата использованы фотосенсп-билизированные каталитические системы [28]. Пероксодисульфат титруют железом(II) в присутствии иода, серебра(I) и эритрозина В при освещении лампой мощностью 150 Вт. Другая фотохимическая система основана на использовании нитрата ртути(I) в качестве титранта [29].
Полярографические методы
Для раздельного определения H2S2O8, H2SO5 и Н2О2 предложен полярографический метод с платиновым микроэлектродом в сочетании с эталонным электродом — Hg2SO4 [30]. В I М растворах НСЮ4, HNO3 и H2SO4 три определяемых окислителя дают три различные полярографические волны в интервале между —1,4 и —0,2 В. Волны соответствуют восстановлению H2SO5, Н2О2 и H2S20e.
Смеси, содержащие пероксодисульфат калия и галогениды щелочных металлов, изучены методами переменнотоковой полярографии с синусоидальной волной [31]. Высота максимума, вызванного взаимодействием пероксодисульфата и галогенида, пропорциональна концентрации пероксодисульфата при условии, что концентрация галогенид-иона поддерживается в определенном интервале.
502
Газохроматографические методы
Пероксодисульфат можно определить простым газохроматографическим методом [32] после восстановления его до сероводорода. В качестве восстановителя используют 20%-ный раствор хлорида олова(II) в концентрированной фосфорной кислоте.
ЛИТЕРАТУРА
1. Szekeres L. — Taianta, 1974, v. 21, р. 2.
2. Blasius Е.. Horn G., Knochel A., Munch J., Wagner H. — Inorganic Sulphur Chemistry (Ed. G. Nickless), Amsterdam, Elsevier, 1968, p. 199.
3. Haff L. V. — In: The Analytical Chemistry of Sulphur and its Compounds (Ed. J. H. Karchmer), Part I, New York, Wiley-Interscience, 1970, p. 255.
4 Behrends K.-—Z. analyt. Chem., 1967, v. 226, p. 1; Analyt. Abstr., 1968, v. 15, p. 2598.
5 Rudnicki R.— Chemia analit., 1961, v. 6, p. 761; Analyt. Abstr., 1962, v. 9, p. 2731.
6. Seiler H., Erlenmeyer H. — Helv. chim. Acta, 1964, v. 47, p. 264.
7 Wahba N., El Asmar M. F., El Sadr M. M. —- Analyt. Chem., 1959, v. 31, p. 1870.
8. Indelli A., Prue J. E. — J. chem. Soc., 1959, p. 107.
9. Gupta У. K-— Analytica chim. Acta, 1961, v. 24, p. 415.
10. Gupta У. K. — Z. analyt. Chem., 1961, v. 180, p. 260; Analyt. Abstr., 1961, v. 8. p. 4604.
11. Frigerio N. A. —Analyt. Chem., 1963, v. 35, p. 412.
12. Schulek E., Pungor E„ Prompter /. — Acta. chim. hung., 1954, v. 4, p. 393; Analyt. Abstr., 1954, v. 2, p. 612.
13. Kolthoff I. N., Carr E. M. — Analyt. Chem., 1953, v. 25, p. 298.
14. Erdey L., Buzas I., Vigh K'.— Periodica Polytech., 1959, v. 3, p. 1; Analyt. Abstr., 1959, v. 6, p. 4262.
15. Gupta У. K., Ghosh S. — Analytica chim. Acta, 1957, v. 17, p. 379.
16 Prasad G„ Gupta У. K- — Z. analyt. Chem., 1963, v. 198, p. 173; Analyt. Abstr., 1964, v. 11, p. 4827.
17. Erdey L., Svehla G. — Analytica chim. Acta, 1962, v. 27, p. 164.
18. Kellner A., Szekeres L. — Chemist Analyst, 1965, v. 54, p. 75.
19. De Sousa A. — Inf. Quim. Anal., 1961, v. 15, p. 91; Analyt. Abstr., 1962, v. 9, p. 1028.
20. Mariano M. H. — Analyt. Chem., 1968, v. 40, p. 1662.
21. Villegas E., Pomeranz У., Shellenberger J. A.— Analytica chim. Acta, 1963, v. 29, p. 145.
22. Раманаускас E. И., Буникиене Л. В., Сапрогонене M. С. и др. ЖАХ, т. 24, с. 244 (1969); Analyt. Abstr., 1970, v. 19, р. 1022.
23. Auerbach М. Е„ Eckert W., Angell E. — Cereal. Chem., 1949, v. 26, p. 490; Chem. Abstr., 1950, v. 44, p. 2135c.
24. Al-Kayssi M., Magee R. L.— Taianta, 1962, v. 9, p. 667.
25. Kyrti J. R. — Acta chem. fenn., 1965, v. 38B, p. 51; Chem. Abstr., 1965, v. 63, p. 16g.
26. Belcher R., Bogdanski S. L., Knowles D. J., Townshend A. — Analytica chim. Acta, 1975, v. 77, p. 53-
27. Норкус П. К., Шемкоявичюте Г. С. — ЖАХ, 1972, т. 27, с. 1419.
28. Sierra J. F., Monzon Е. — An. R. Soc. esp. Fis Quim. В, 1967, v. 63, p. 51; Analyt. Abstr., 1968, v. 15, p. 3303.
29. Sierra J. F., Sanchez-Pedreno C. — An. R. Soc. esp. Fis Quim. B, 1967, v. 63, p. 1111; Analyt. Abstr., 1969, v. 16, p. 1845
30. Raspi G., Venturini M. — Chimica Ind. Milano, 1968, v. 50, p. 536; Chem. Abstr., 1968, v. 69, p. 49018a,
5Q3
31. Hakoila E. — Taianta, 1968, v. 15, p. 55.
32. Ito S. — J. chem. Soc. Japan, pure Chem. Sect., 1969, v. 90, p. 1027: Analyt. Abstr., 1970, v. 19, p. 4842.
ПОЛИСУЛЬФИДЫ
Полисульфид не является анионом в общепринятом смысле этого слова, но рассмотрение его свойств в данной части оправдано в связи с широким использованием полисульфидов. Полисульфиды щелочных металлов и аммония имеют общую формулу M2Sn, где п> 1 (М — щелочной металл или аммоний). Известны полисульфиды натрия Na2S2, Na2S4 и Na2S5, аналогичные калиевые соединения известны вплоть до КгЗб-
Полисульфиды имеют важное значение в производстве целлюлозы. При сульфатной обработке древесины ее обрабатывают так называемой «белой жидкостью», включающей гидроксид и сульфид натрия с добавками малых количеств карбоната, сульфата, тиосульфата и сульфита натрия. Регенерация жидкости для варки древесины включает выделение из пульпы «белой жидкости», которую потом выпаривают и подвергают окислению для отделения неорганических соединений. Раствор, который обработан окислителем (так называемая «зеленая жидкость»), содержит МпО2, FeS и другие соединения. Этот раствор последовательно превращают в «белую жидкость», которую затем используют для варки древесины [1].
Полисульфиды используют как средства защиты растений. Определение полисульфидов, как и других серусодержащих ионов, необходимо бывает проводить в сточных водах предприятий металлургической промышленности.
Полисульфиды неизменно сопровождаются другими серусодер-жащими анионами, как правило, сульфидом (моносульфидом), тиосульфатом и сульфитом. Поэтому основное внимание уделено определению полисульфидов в таких смесях. При анализе поли-сульфидов целесообразна дифференциация форм серы — общей, моносульфидной и полисульфидной.
Определение полисульфидов проводят гравиметрическим и ти-триметрическим методами.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Аналитическая химия полисульфидов рассмотрена в обзоре [2]. Кроме того, методы определения полисульфидов обсуждены в нескольких обзорах по аналитической химии серусодержащих соединений [3—5].
Многие методы определения полисульфидов и в особенности методы разделения смесей, содержащих полисульфиды, основаны на определенных реакциях, наиболее важные из которых приведены ниже.
504
Полисульфиды реагируют с сульфитами в нейтральном растворе при 50 СС с образованием тиосульфата:
Na2Sn-|-i + nNa2SO3 —> Na2S + nNa2S2O3 (1)
Элементная и полисульфндная сера гидролизуются горячей водой, образуя сероводород и тиосульфат. Эта реакция быстро протекает в щелочном растворе:
4S + ЗН2О —> S2O?' + 2H2S + 2Н* (2)
Иод в кислой среде окисляет только моносульфидную часть полисульфида:
H2Sn + I2 —> 2H* + 2r + nS (3)
Тиосульфат окисляется аналогичным образом:
2S2O|- + Ij —* S4O62' +21-
Содержание общей серы в полисульфиде можно определить, окисляя его в щелочном растворе пероксидом водорода. Избыток щелочи титруют стандартным раствором серной кислоты:
Na2S„ + (2п — 2) NaOH + (Зп - 1) Н2О2 —> nNa2SO4 + 4пН2О (4)
Полисульфид реагирует С цианидами щелочных металлов (без доступа воздуха) с образованием тиоцианата, при этом на каждый полисульфидный атом серы образуется по одной молекуле тиоцианата:
Na2Sn+i + nCN' —> Na2S + /iSCN* (5)
В среде борной кислоты из раствора, полученного по реакции (5), образуется сероводород, который, как и избыток цианистого водорода, удаляется при кипячении в растворе борной кислоты. Тиоцианат и тиосульфат остаются неизменными при указанной обработке.
Бром (смесь бромата с бромидом) и гипобромит окисляют моно- и полисульфидную серу, как и тиосульфат, до сульфата:
S2" + 4Н2О + 4Вг2 —> SO4-+ 8Н++ 8Вг' (6)
S + 4Н2О + ЗВг2 —> SO4‘ + 8Н+ + 6Вг" (7)
В смеси сульфита и полисульфида сульфит можно замаскировать формальдегидом и определить полисульфид без помех:
hcho + so5’ + h+ —> ch2oh-so; (8)
Тиоцианат окисляется бромом (но не иодом) в кислом растворе:
SCN’ + 4Н2О + 4Вг2 —> SOV + 8Н+ + 7ВГ + BrCN (9)
Гравиметрические методы
Полисульфндная сера легко превращается в элементную, которую можно определить гравиметрически [6]. Это происходит при нейтрализации водного раствора полисульфидов с индикато
605
ром метиловым оранжевым или при нагревании раствора поли-сульфидов в бензоле или сероуглероде с последующим выпариванием растворителя.
С помощью другой гравиметрической методики определяют суммарное содержание серы. Для превращения всех форм серы в сульфаты окисляют образец пероксидами щелочных металлов или бромом и образующиеся сульфаты определяют гравиметрически в виде сульфата бария. Эта методика не позволяет учесть содержания различных форм серы. Иногда гравиметрическую методику используют в сочетании с йодометрическим титрованием, в этом случае можно определить моносульфпдпую часть серы. Моносульфид можно отделить и определить в виде сульфида кадмия.
В настоящее время стремятся заменить продолжительные гравиметрические методы анализа более экспрессными.
Титриметрические методы
Иодиметрические методы
Стандартный метод определения полисульфида был разработан в 1925 г. [7].
Анализируемый раствор кипятят с цианидом щелочного металла в среде борной кислоты. При этом образуется сероводород и по одной молекуле тиоцианата на каждый полисульфидный атом серы (уравнение 5). Избыток цианида удаляют кипячением в кислой среде. Присутствующие в растворе тиоцианат и тиосульфат при этой обработке не изменяются. Тиоцианат окисляют бромом до BrCN, а избыток брома связывают фенолом. К раствору добавляют иодид калия и образующийся иод титруют стандартным раствором тиосульфата:
BrCN + 21" —> CN' + h+Br'
1 мл 0,1 М раствора тиосульфата соответствует 1,603 мг поли-сульфидной серы.
В присутствии тиосульфата раствор после кипячения с цианидом щелочного металла переносят в мерную колбу и определяют полисульфидную серу в одной аликвотной части анализируемого раствора, используя вышеприведенный метод. Тиосульфат определяют во второй аликвотной части следующим образом: подкисляют ее раствором НС1, добавляют избыток стандартного раствора иода и оттитровывают непрореагировавший иод стандартным раствором тиосульфата.
1 мл 0,05 М раствора иода соответствует 6,40 мг тиосульфатной серы.
Предложен другой иодиметрический метод определения полисульфида [8]. В соответствии с этим методом полисульфиды обрабатывают нейтральным раствором сульфита натрия при 50 °C, при этом образуется тиосульфат (реакция 1). Сульфид, который
506
также образуется в этой реакции, осаждают ацетатом цинка и затем отфильтровывают ZnS. Избыток сульфита связывают формальдегидом (реакция 8). Раствор подкисляют уксусной кислотой и тиосульфат титруют стандартным раствором иода. Если анализируемый раствор содержит тиосульфат, что очень вероятно в полисульфидных растворах, необходимо сделать контрольный опыт (провести определение по всем стадиям анализа без добавления сульфита).
Анализ смесей, содержащих полисульфиды и другие соединения серы, очень важен. Описан метод определения полисульфида, сульфида и тиосульфата при их совместном присутствии [9]. Предложенный метод использует то обстоятельство, что иод окисляет в кислой среде только моносульфидную серу и тиосульфат, а бром окисляет еще и полисульфидную серу (реакции 3 и 6 соответственно). В одной аликвотной части анализируемого раствора окисление проводят избытком иода и непрореагировавший иод определяют титриметрическн с помощью тиосульфата. Это титрование определяет сумму моносулъфидной серы и тиосульфата. Вторую аликвотную часть окисляют бромом, в результате титрования этой части определяют сумму полисульфидной и моносульфид-ной серы и тиосульфата. Третью аликвотную часть обрабатывают цианидом щелочного металла и затем — борной кислотой (реакция 5). Эта обработка превращает полисульфидную серу в тиоцианат. При нагревании анализируемого раствора с борной кислотой моносульфидная сера удаляется в виде сероводорода. Четвертую аликвотную часть кипятят с борной кислотой для удаления сероводорода, а дальнейшая обработка этой аликвотной части аналогична обработке второй аликвотной части.
Сходные методы использованы для анализа сточных вод металлургической промышленности [10]. Образцы этих вод могут содержать сульфат, тиосульфат, сульфид, полисульфид, сульфит и тритионат.
Предложен метод анализа смесей сульфида, полисульфида и тиосульфата [11], основанный на превращении полисульфидной серы в сульфид и его осаждении в виде сульфида кадмия.
Метод, основанный на сочетании иодиметрических, комплексометрических и ацидиметрических методик, позволяет определять полисульфиды, тиосульфаты и общую сульфидную серу [12]. Сначала полисульфиды осаждают в виде ZnSx, используя аммиачный раствор сульфата цинка. Осадок фильтруют и затем кипятят в НС1. Образующуюся элементную серу окисляют пероксидом щелочного металла, избыток которого титруют стандартным раствором кислоты (1 мл 1 М НС1 соответствует 0,016033 г полисульфидной серы). Тиосульфат остается в растворе после осаждения ZnSx и затем переходит в фильтрат, после чего его можно определить иодиметрически. Общую сульфидную серу определяют после окисления ее до сульфата бромом. Сульфат осаждают в виде BaSO4, а непрореагировавший барий определяют комплексометрически. Появление в некоторых случаях заниженных резуль
507
татов определения полисульфидной серы вызвано тем, что некоторая часть ее может превратиться в сульфид при кипячении с НС1 [4].
Описано несколько методов определения моносульфидной части полисульфидов [13]. Анализируемый образец обрабатывают сульфитом натрия (реакция 1) и образующийся моносульфид титруют о-оксимеркурибензойной кислотой. Этот метод приведен в разделе, посвященном сульфиду.
Метод кислотно-основного титрования
Определение общей серы полисульфида возможно после ее окисления пероксидом щелочного металла. На этом принципе основана титриметрическая методика [14], в соответствии с которой избыток пероксида щелочного металла титруют стандартным раствором H2SO4.
Электрохимические методы
Для анализа смесей полисульфида, сульфида и тиосульфата аммония использован «метод конечной мертвой точки» с платиновыми электродами [15]. В одной аликвотной части анализируемого раствора сульфид титруют аммиачным раствором нитрата серебра, тиосульфат не мешает этому определению. Во второй аликвотной части сульфид удаляют кипячением с борной кислотой и тиосульфат титруют стандартным раствором HgCl2. Третью аликвотную часть обрабатывают сульфитом для превращения полисульфида в тиосульфат и определяют тиосульфат после маскирования избытка сульфита формальдегидом.
Для анализа смесей серусодержащих анионов, включая полисульфид, предложено несколько потенциометрических методов. В «белой», «зеленой» и «черной» жидкостях для обработки древесины определяют полисульфид, тиосульфат, сульфит и содержание активной щелочи [16]. Избытком сульфита превращают полисульфид в сульфид и тиосульфат. После осаждения сульфида в виде ZnS и маскирования непрореагировавшего сульфита формальдегидом (реакция 8) тиосульфат титруют потенциометрически стандартным раствором HgCl2.
Для анализа аналогичных смесей описан автоматический микрометод [17]. Более поздние работы по потенциометрии включают использование сульфидного ионоселективного электрода [18, 19]. Для определения полисульфидов использованы вышеописанные реакции и HgCl2 в качестве титранта. Для индикации конечной точки титрования применен сульфидный ионоселективный электрод «Radelkis OP-S-711». Предложенный метод использован для анализа «белой», «зеленой» и «черной» жидкостей для обработки древесины, содержащих полисульфиды, сульфиды, тиосульфаты, сульфиты и тиолы.
608
ЛИТЕРАТУРА
1. Tomlinson G. H. — In: «Pulp», Kirk-Othmer Encyclopaedia of Chemical Technology, 2nd edn.. Vol. 16, New York, Wiley, 1968, p. 708.
2 Ahlgren P., Harter N. — Svensk. kern. Tidskr.. 1966, v. 78, p. 404; Chem. Abstr., 1967, v. 66, p. 25679d.
3. Blasius E., Horn G., Knochel A., Munch J., Wagner H. — Inorganic Sulphur Chemistry (Ed. G. Nickless), Amsterdam, Elsevier, 1968, p. 199.
4. Szekeres L. — Taianta, 1974, v. 21, p. 2.
6. Haff L. V. — In: The Analytical Chemistry of Sulphur and its Compounds (Ed. J. H. Karchmer), Part 1, New York, Wiley-Interscience, 1970, p. 347.
6. Griffin R. C. — Technical Methods of Analysis, New York, McGraw-Hill, 1921, p. 60.
7. Schulek E. — Z. analyt. Chem., 1925, v. 65, p. 352.
8. Kurtenacker A., Bittner K. — Z. analyt. Chem., 1925, v. 142, p. 115; Chem. Abstr., 1925, v. 19, p. 2000.
9. Szekeres L.— Z. analyt. Chem., I960, v. 178, p. 81; Analyt. Abstr., 1961, v. 8, p. 2387.
10. Blasius E., Wagner H., Ziegler K-— Arch. Eisenhiitt Wes., 1971, v. 42, p. 473; Analyt. Abstr., 1972, v. 22, p. 3164.
11. Szekeres L. — Pharm. Zentralhalle Dtl., 1963, v. 102, p. 6; Analyt. Abstr., 1963, v. 10, p. 5137.
12. Legradi L. — Analyst, 1961, v. 86, p. 854.
13. Wronski M. — Analyt. Chem., 1960, v. 32, p. 133
14. Feher F., Berthold H. J. — Z. analyt. Chem., 1953, v. 138, p. 245.
15. Kiss S. A. — Z. analyt. Chem., 1962, v. 188, p. 341; Analyt Abstr., 1963, v. 10, p. 112.
16. Danielsen A. J., Johnsen K-, Landmark P. — Norsk Skogind., 1969, v. 23, p. 378; Analyt. Abstr., 1971, v. 21, p. 1252.
17. Danielsen A. J., Johnsen K., Landmark P. — Meddr. Pap Ind. Forsk. Inst., No. 232 (1969); Norsk Skogind., 1969, v. 23, p. 77; Analyt. Abstr., 1970, v. 19, p. 395.
18. Papp J., Havas J. — Mady kern. Foly, 1970, v. 76, p. 307; Analyt. Abstr., 1971, v. 20, p. 4439.
19. Papp J. — Cellulose Chem. Technol., 1971, v. 5, p. 147; Analyt. Abstr., 1972, v. 22, p. 2404.
ПОЛИТИОНАТЫ
При взаимодействии раствора сероводорода с раствором сульфита в течение суток или более образуется смесь продуктов, известных как раствор Вакенродера. Этот раствор содержит тритионат 5зОб", тетратионат 340б’ и пентатионат SsOe", а также тиосульфат. Такие смеси встречаются в различных отраслях промышленности, например в деревообрабатывающей. Поэтому определение как общего содержания политионатов, так и отдельных компонентов представляет важную задачу.
Большое значение имеет определение политионатов в присутствии других серусодержащих анионов. Для смесей, содержащих политионаты с тремя — шестью атомами серы вместе с другими серусодержащими кислотами, предложено несколько схем анализа. В одном исследовании [1] предполагается присутствие политионатов с числом атомов серы в молекуле большим шести. Однако существование таких форм политионатов маловероятно.
509
Определение индивидуальных политионатов в чистом виде выполняется довольно просто, тогда как анализ смесей более сложен. Названия публикаций не всегда отражают их содержание и, например, некоторые ссылки на сопределение тетратионата» подразумевают определение и других политионатов рассматриваемым методом.
Дитионат БгОе’ отличается от других политионатов, например, окисляется с трудом. Поэтому дитионат рассмотрен отдельно и в этом разделе обсуждается лишь в общих чертах.
В литературе описаны способы получения тритионата [2], а также тетра- и пентатионатов калия [3, 4].
Аналитическая химия соединений серы, включая и политионаты, приобрела большое значение в последние годы и описана в монографиях [5, 6]. Анализу политионатов уделено место в обзоре по аналитической химии серусодержащих кислот [7].
МЕТОДЫ ОТДЕЛЕНИЯ
Политионаты так сходны по своим химическим свойствам, что анализ их смесей классическими методами очень длителен и может привести к неправильным результатам. Поэтому необходимо предварительное разделение компонентов смеси. В литературе [8] показана перспективность использования хроматографического разделения, предшествующего определению политионатов.
Разделение, основанное на фракционной кристаллизации
Описаны методы разделения политионатов, основанные на фракционной кристаллизации соединений политионатов с бензидином [1] или пиридинокупратами [9].
Разделение, основанное на различии в растворимости
Описан метод разделения политионатов, основанный на разной растворимости тионатов свинца [10].
Методы хроматографии на бумаге и тонкослойной хроматографии
Сводка экспериментальных данных по использованию методов тонкослойной хроматографии представлена в табл. 37. Длительность разделения этим методом приводит к появлению вторичных реакций между компоиеитами на хроматограмме [8]. Поэтому метод применим при низких концентрациях компонентов. В некоторых случаях возможен и количественный анализ больших концентраций компонентов методом хроматографии на бумаге,
610
Таблица 37. Разделение политионатов методом тонкослойной хроматографии
Неподвижная фаза Подвижная фаза Разделяемые анноны Литература
Силикагель — Метанол — диоксан — кон- S,O«~, S,O*-, S,O*-, 11
крахмал Силикагель центрированный раствор NH3—Н2О (3 : 6 : 1 : 1) Этанол — бутанол — кон- S5°r S,O*-, S,Oj?‘, SsO’’, 12
Пластины Gelman центрированный раствор NH3—Н2О (75 : 75 : 8-10:10-30) Бутанол — пиридин — ук- s o2- °2V3 S3O*-, S4O*-, sof, 13
SA и SG сусная кислота — Н2О (15 : 10 : 3 : 12) S20f, SCN-
Метод анионообменной хроматографии
Ди-, три-, тетра- и пентатионаты разделяют [14] на анионообменной смоле Dowex 1-Х2 в хлоридной форме. Дитионат элюируют 1 М раствором НС1, тритионат — 3 М раствором НС1, тетратионат— 6М раствором НС1 и, наконец, пентатионат — 9 М раствором НС1. Политионаты в сильнокислых средах мало стабильны, в качестве элюента предложено [15] использовать гидрофталат калия. Этот метод позволяет отделить политионаты от тиосульфата и сульфита. Тритионат не отделяется от гексатионата на ионообменных смолах из-за разложения .гексатионата [16]. Однако смеси ЭОз’—S2O3’—ЭзОб' и ЗзОб'—840б’—SsOi'. без труда разделяются при использовании в качестве элюентов NaCI или НС1.
Метод высокоэффективной жидкостной хроматографии
При разделении политионатов методом высокоскоростной жидкостной хроматографии получены хорошие результаты [17]. Политионаты (три-, тетра-, пента-) и тиосульфаты разделяют этим методом и затем определяют их в течение 15 мин.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Можно рассмотреть три аспекта анализа политионатов: определение индивидуальных компонентов, определение общего содержания политионатов и анализ их смесей.
Определение индивидуальных политионатов несложно, однако анализ их смесей представляет большие трудности. Поэтому было отмечено [18], что удовлетворительный метод определения индивидуальных политионатов в их смеси отсутствует. Предложено решение этой задачи с помощью высокоскоростной жидкостной
511
хроматографии. Существующие методы определения политионатов неэкспрессны и, кроме того, возможно разложение этих анионов в процессе анализа.
Рассмотрим некоторые важные реакции, на которых основаны методы определения политионатов.
Сульфиты щелочных металлов взаимодействуют с высшими тионатами (тетра-, пента- и гексатионатом), образуя тритионат и тиосульфат:
8л+3ОГ + xSO|- —> S3O62’+ xS2O2’ (1)
Цианиды щелочных металлов реагируют с высшими тионатами (реакция цианолиза) в соответствии с уравнением
Sx+зОГ + (х + 2) СКГ + Н2О —> SO2’+ xSCN" + 2HCN + S2O|' (2) Тетра-, пента- и гексатионаты быстро вступают в реакцию цианолиза, тритионаты с цианидами при комнатной температуре реагируют довольно медленно.
Сульфиды щелочных металлов восстанавливают политионаты:
SXO|’+ S2" —> (х — 3) S + 2S2O|" (3)
Тритионаты медленно реагируют с сульфидами при комнатной температуре. Высшие тионаты (тетра-, пента- и гексатионаты) гидролизуются в разбавленных щелочных растворах при комнатной температуре:
4S4O2’+6OH’ —> 5S20f-+ 2S3O62’+ ЗН2О (4)
2S50r + 6OH’ —> 5S2O|'+ЗН2О (5)
S6O2- —> SsOf’+S (6)
Образующийся по реакции (6) пентатионат затем гидролизуется по уравнению (5). Тритионаты гидролизуются лишь в растворах более концентрированных щелочей:
2S3O|' + 6OH’ —> S2O3' + 4SO3' + ЗН2О (7)
Вышеприведенные реакции служат основой аналитических методов определения политионатов. Другие методы анализа основаны на реакциях окисления, на количественной сканирующей хроматографии и полярографии.
Гравиметрические методы
Один из наиболее старых методов определения политионатов основан на окислении их до сульфатов, которые затем определяют гравиметрически. Другой метод, предложенный для определения тритионата, основан на обработке анализируемого раствора сульфатом меди(II), при этом образуется CuS и H2SO4. Тритионат можно определить в виде оксида меди (после сожжения CuS), алкалиметрически или гравиметрически в виде BaSO4 [19]. Этот метод, вероятно, не найдет широкого применения в практике анализа.
512
Титриметрические методы
Метод Куртенакера — Геринга
Метод Куртенакера — Геринга — классический метод анализа политионатов [20]. Три аликвотные части анализируемого раствора обрабатывают сульфитом, цианидом и сульфидом соответственно (уравнения 1—3). В каждой аликвотной части титруют выделяющийся тиосульфат стандартным раствором иода. По результатам этих трех титрований можно определить содержание три-, тетра- и пентатионатов. Однако в реакциях (1) — (3) участвуют также и гексатионаты. В дальнейшем этот метод был модифицирован [21]; помимо ранее упомянутых операций проводят гидролиз политионатов при контролируемых условиях (реакции 4—7).
Метод Куртенакера—Геринга обычно приводит к завышенным результатам. Недостоверность его может быть связана со следующими причинами. Тритионат медленно реагирует с сульфидом, и эта реакция происходит с нарушениями стехиометрии. Сульфиды щелочных металлов обычно содержат примеси тиосульфата и других серусодержащих соединений, которые титруются иодом. Кроме того, коллоидная сера, часто присутствующая в виде примеси в политионатах, реагирует с сульфитом, образуя тиосульфат. Это приводит к завышенным результатам. Некоторые авторы вычисляют среднее число атомов серы в молекуле, но эта информация имеет ограниченное значение.
Метод кислотно-основного титрования
Алкалиметрический метод определения политионатов [22] основан на выделении кислоты при обработке анализируемого раствора хлоридом ртути(П):
2SXO62’ + 3HgCl2 + 4Н2О —> HgCl2 • 2HgS + 8Н++ 4СГ + 4SO<'+ (2х—6) S
Три-, тетра- и пентатионаты реагируют, образуя 4 моль кислоты на 1 моль политионата. Дитионат не реагирует с HgCl2. Свободную кислоту титруют в присутствии КГ необходимого для связывания избытка HgCh- Определению политионатов этим методом мешают тиосульфат и сульфид.
Редокс-методы
Описано несколько титриметрических методов определения политионатов, основанных на их окислении. Политионаты можно окислять избытком стандартного раствора гипохлорита [23] в кипящем щелочном растворе. Непрореагировавший гипохлорит определяют затем иодиметрически. Полученные результаты показывают, что дитионат в этих условиях не окисляется, тритионат
17 Зак. 167
513
окисляется на 30—40%, тетратионат окисляется количественно ц пентатионат — почти количественно (на 98%).
В качестве окислителя при определении политионатов предложен хлорамин Т [24]. Этот метод не применим для определения дитионата, гепта- и октатионаты окисляются при 45—50 °C.
Перйодат по характеру окислительного действия по отношению к политионатам сходен с гипохлоритом [25]. Метод, основанный на применении периодата калия, использован для определения три- и тетратионатов, гидросульфида HS~ и полисульфпда. Установлено, что этот метод позволяет анализировать различные бинарные смеси, но детали экспериментов не описаны.
Раствор церия (IV) в 2 М растворе НС1О4 количественно окисляет тиосульфат, три-, тетра- и пентатионаты до сульфатов [26]. Определению политионатов этим методом мешают ионы галогенидов, дитионаты и окрашенные ионы. Этот же окислитель был использован в сернокислой среде [27]. В 1 М растворе H2SO4 количественно окисляется дитионат. Неизрасходованный церий (IV) титруют стандартным раствором железа(II) с ферроином в качестве индикатора. Количественное окисление три- и тетратионата происходит при кипячении в 6 М H2SO4 в течение 30 мин. Для окисления 1 моль дитионата необходимо 2 моль церия (IV):
S20f + 2Н2О + 2CeIV :₽=► 2SO^ + 4Н+ + 2СеП1
Расход окислителя для три- и тетратионата составляет 8 и 14 моль соответственно:
S3O62- + 6Н2О + 8CeIV ч=> 3SO2" + 12Н+ 4- 8СеП1
S4O|-+ 10Н2О+ 14CeIV ^=±: 4SOr + 2 0H++14Сеи*
Метод пригоден для определения индивидуальных политионатов и дитионата. Так как метод, основанный на использовании хлорамина Т, не учитывает содержания дитионата, сочетание этого метода с цериметрическим позволяет определить содержание дитионата и политионатов в смесях. Определению политионатов цериметрическим методом мешает присутствие других восстановителей, например сульфида, сульфита и тиосульфата.
Периметрическое титрование политионатов
(три- и тетратибиатов) [27]
Реагенты
Сульфат церия-аммония, стандартизован раствором железа(П).
Индикатор ферроин, растворяют 0,468 г моногидрата фенантролина в 100 мл воды, содержащей 0,278 г FeS04-7H2O.
Ход анализа. К 40 мл раствора церия (IV) в колбе вместимостью 50 мл прибавляют столько H2SO<, чтобы общая кислотность составляла 6 М. Прибавляют раствор политноната, содержащий около 0,25 ммоль трн- нлн около 0,12 ммоль тетратноната и кипятят 30 мин. Охлаждают его и определяют неизрасходованный церий(1У), тнтруют его стандартным раствором железа(П) с каплей ферроина в качестве индикатора.
Пр имечання. 1. Контрольного опыта ие требуется. 2. Определению поли-тнонатов мешают сульфид, сульфит и тиосульфат, нх определяют нодиметриче-ски. 3. При определенной кислотности можно оттитровать дитноиат [27].
514
Спектрофотометрические методы
Спектрофотометрические методы можно применить для определения индивидуальных политионатов, однако они могут быть использованы и для анализа смесей политионатов.
Спектрофотометрические методы могут быть основаны на образовании роданида при цианолизе политионатов (реакция 2) или тиосульфата при взаимодействии политионатов с сульфитом (реакция 1). Образующийся при цианолизе роданид определяют с помощью железа(II) [28]. Этот метод применяют для анализа пента- и гексатионатов. Этот же метод был использован [29] для определения тритионата в смеси с тиосульфатом и тетратионатом. В работе использована медь(II) в качестве катализатора реакции цианолиза тиосульфата, что приводит к быстрому протеканию реакции при комнатной температуре. Об использовании меди(II) в качестве катализатора этой реакции было известно ранее [30]. В дополнение к этому авторы работы [29] использовали тот факт, что цианолиз тритионата происходит только при температурах, близких к 100 °C. Протекающие реакции могут быть описаны уравнениями (8) — (Ю):
S4Oc'+ 3CN'+ Н2О —> S2O|'+ SO<" + 2HCN + SCN" (8) Си
S2O|--|-CN' --> SO|4-SCN' (9)
S3O|" + 3CN” + Н2О —> so23' +sot+ 2HCN +SCN' (10) Данные, получаемые при выполнении указанных условий, позволяют рассчитать содержание каждого компонента.
Японские авторы [31—33] опубликовали несколько работ по спектрофотометрическим методам определения политионатов, основанным на образовании роданида. Этими методами можно определить отдельно тетра-, пента- и гексатионаты. Эти же авторы [34] предложили метод микроопределения двух компонентов в случае совместного присутствия тетра-, пента- и гексатионата. Авторы используют медь(II) в качестве катализатора превращения тиосульфата в роданид. В работе [35] чувствительность определений политионатов была повышена в 60 раз, что позволило определять 3,3-10-7—1,0-10_5М тетратионата, 1,7-10-7—5,0-10~6М пентатионата и 1,0-10 7—3,3-10 6 М гексатиоиата. Такое повышение чувствительности достигнуто экстракцией 1,2-дихлорэтаном комплексного соединения роданида (эквивалентного политионату) с метиленовым синим. После экстракции проводят спектрофотометрическое определение, основанное на измерении светопоглоще-ния при 657 нм. Определению полптионатов описанным методом мешают медь(П), бромид, иодид, нитрат, перхлорат и сульфид.
Предложен быстрый и чувствительный метод определения (раздельного или суммарного) тетратионата и тиосульфата [36]. Методика анализа включает цианолиз в отсутствие катализатора и последующее определение тиоцианата в виде комплексного соединения с железом(Ш). Метод использован для анализа почвенных
17*
515
вытяжек. Большие количества сульфата не мешают анализу. Определению 100 мкг тетратионата или тиосульфата не мешают до 2 мг нитрата натрия, сульфата натрия, нитрита натрия, гидрофосфата натрия, гидрокарбоната натрия, хлорида магния, хлорида кальция, хлорида лития, хлорида алюминия, ацетата калия, цистина, метионина, сульфаминовой и сульфаниловой кислот.
Основная реакция цианолиза (уравнение 2) приводит к образованию из каждого моля политионата одного моля тиосульфата. Предложен метод [37], основанный на прибавлении избытка иода к образованному тиосульфату и на измерении неизрасходованного избытка иода спектрофотометрически. Этот метод позволяет определять суммарное содержание политионатов (тетра-, пента- и гексатионатов) в их смесях. Модифицированная методика дает возможность определять сумму политионатов в присутствии тиосульфата и сульфита.
Для определения политионатов можно использовать их реакцию с сульфитом [38]. К анализируемому раствору прибавляют избыток сульфита натрия. После окончания реакции неизрасходованный сульфит маскируют формальдегидом и прибавляют избыток иода. Непрореагировавший иод определяют спектрофотометрически, измеряя светопоглощение при 372 или 440 нм. Реакция политионатов с сульфитом также была применена [39] для микроопределения гексатионата в смесях, содержащих тиосульфат и сульфит; при содержании политионата 0,5 мкмоль относительное стандартное отклонение определений гексатионата составляет 2,2%.
Электрохимические методы
Для определения политионатов можно использовать прямое потенциометрическое титрование гексацианоферратом (III) с катализатором OsO4 [40]. Титрование проводят в сильнощелочной среде при 50—60 °C. Сульфит, сульфид и тиосульфат также окисляются в этих условиях.
Предложен быстрый кулонометрический метод микроопределения политионатов [41]. Этот метод основан на обработке анализируемого раствора политионата сульфитом или цианидом (реакции 1 и 2) с последующим кулонометрическим титрованием образующегося тиосульфата. Описанный метод, использованный для определения тетра-, пента- и гексатионатов, как сообщают авторы, позволяет проводить определение политионатов с большей точностью, чем другие методы.
Для определения индивидуальных политионатов предложено несколько полярографических методов, основанных на различии потенциалов полуволн политионатов. Однако практическое использование предложенных методов затруднено, так как высоты волн, образуемых политионатами, недостаточны. Для определения политионатов предложен метод катодно-лучевой полярографии [421-
516
Рис. 61. Хроматограмма тионатов в растворе Вакенродера, полученная методом высокоэффективной жидкостной хроматографии [17]:
J—SjOa" (25 мг); 2—SjO^ (125 мг); 3—S4O*
(50 мг); 4—S5OJ (39 мг).
Хроматографические методы
Для разделения политионатов предложено [43] использовать хроматографию на бумаге в сочетании со сканированием хроматограммы. Предложенный метод основан на образовании
однородного слоя, состоящего из 2 4 Б 8 10 12 /4 16
сульфида серебра и серы, обра- Время, мин
зуемых при разложении тиоцианатов серебра, возникающих при проявлении хроматограммы раствором нитрата серебра. Светопоглощение хроматографических слоев измеряют с помощью сканирующего устройства. Описанный метод использован для анализа смесей, содержащих три-, тетра-, пеита- и гексатионаты.
Для разделения и определения политионатов, составляющих раствор Вакенродера, использован метод высокоскоростной жидкостной хроматографии. Разделение проведено в четырехфутовой колонке из нержавеющей стали, заполненной активным углем с размером зерен 53—74 мкм. Определение политионатов проводят спектрофотометрически при длине волны детектирования 254 нм. Для уменьшения времени удерживания использован тетрагидрофуран. Хроматограмма представлена на рис. 61. Слабый положительный дрейф базисной линии, обозначенной пунктиром, не влияет на результаты анализа, если площади под пиками измеряют с помощью цифровых приборов. Точность определения различных компонентов следующая [17]:
Соединение............Na2S2O3 K2S3O6
Относительное стандартное отклонение, % 2,5 3,6
Введено соединения, мкг/50 мл............. 50 100
KaS^Oe KzSsOe • 1,5Н2О
3,0 2,3
50 39
ЛИТЕРАТУРА
1 Weitz F., Spohn А. — Вег., 1956, v. 89, р. 2332.
2 Krause R. А„ Busch D. Я. — Analyt. Chem., 1958, v. 30, p. 1817.
3. Stamm H., Goehring M., Feldmann U. — Z. anorg. allg. Chem., 1942, v. 250, p. 226.
4. Goehring M, Feldmann U. — Z. anorg allg. Chem., 1948, v. 257, p. 223.
517
5. Haff L. V. — In: The Analysis of Sulphur and its Compounds (Ed. J. H. Karch-mer), Part 1, New York, Wiley-Interscience, 1970, p. 238.
6. Blasius E., Horn G., Knochel A., Munch J., Wagner H. — Inorganic Sulphur Chemistry (Ed. G. Nickless), Amsterdam, Elsevier, 1968, p. 199.
7. Szekeres L. — Taianta, 1974, v. 21, p. 2.
8. Pollard F. H., Jones D. J., Nickless C — J. Chromat., 1964, v. 15, p. 393.
9. Heinze G. — Z. anorg. allg. Chem, 1954, v. 276, p. 146.
10. Mangan J L —N. Z. J. Sci. Technol, 1949, v. 30B, p 323, Chem. Abstr., 1951, v. 45, p. 5069h.
11 Seiler H, Erlenmeyer H. — Helv. chim Acta, 1964, v. 47, p. 264, Analyt. Abstr., 1965, v. 12, p 1703.
12. Naito K., Takei S., Okabe T. — Bull. chem. Soc. Japan, 1970, v. 43, p. 1360; Analyt. Abstr., 1971, v. 20, p. 3012. •
13. Kelly D. P.— J. Chromat., 1970, v. 51, p. 343.
14. Iguchi A. — Bull, chem Soc. Japan, 1958, v. 31, p. 597; Analyt. Abstr., 1959, v. 6, p. 1706.
15. Pollard F. H., Nickless G., Glover R. B. — J. Chromat., 1964, v. 15, p. 533.
16. Schmidt M, Sand T. — Z. anorg Chem., 1964, v. 330, p 188, Analyt. Abstr., 1965, v. 12, p. 5754.
17. Chapman J. N., Beard H. R. — Analyt Chem, 1973, v. 45, p. 2268.
18. Reference 5, p 241.
19. Josephy E.— Z. anorg. allg. Chem., 1924, v. 135, p. 21.
20. Kurtenacker A, Goldbach E. — Z. anorg. allg. Chem., 1927, v. 166, p. 177.
21. Goehring M., Feldmann U., Helbing W. — Z. analyt. Chem., 1949, v. 129, p. 346.
22. Jay R. R. — Analyt. Chem., 1953, v. 25, p. 288.
23. Hofman-Bang N., Christiansen M. T. — Acta chem. Scand., 1961, v. 15, p. 2061; Analyt. Abstr., 1962, v. 9, p. 1881.
24. Sharada K., Murthy A. R. V.— Z. analyt. Chem., I960, v. 177, p. 401; Analyt. Abstr., 1961, v 8, p. 2388.
25. Kaushik R. L., Prosad R.— Indian J. Chem., 1970, v. 8, p. 462, Analyt. Abstr., 1971, v 20, p 3775.
26. Krause R. A., Busch D. H. — Analyt Chem., 1958, v. 30, p. 1817.
27. Nair V. R„ Nair C. G. R. — Taianta, 1971, v. 18, p. 432.
28. Nietzel O. A., De Sesa M. A. — Analyt. Chem., 1955, v. 27, p. 1839.
29. Kelly D. P., Chambers L. A., Trudinger P. A. — Analyt. Chem., 1969, v. 41, p. 898.
30. Urban P. J. — Z. analyt. Chem., 1961, v. 179, p. 422; Analyt. Abstr., 1961, v. 8, p. 4149.
31. Koh T. — Bull. chem. Soc. Japan, 1965, v. 38, p. 1510; Analyt. Abstr., 1967, v. 14, p 685
32. Koh T., Iwasaki I. — Bull. chem. Soc. Japan, 1965, v. 38, p. 2135; Analyt. Abstr., 1967, v. 14, p 1953.
33. Koh T, Iwasaki I. — Bull. chem. Soc. Japan, 1966, v. 39, p 352; Analyt. Abstr., 1967, v. 14, p. 3144.
34. Koh T., Iwasaki I. — Bull. chem. Soc. Japan, 1966, v. 39, p. 703; Analyt. Abstr., 1967, v. 14, p. 4667.
35. Koh T., Saito N., Iwasaki I. — Analytica chim. Acta, 1972, v. 61, p. 449.
36. Nor У. AL, Tabatabai M. A.—Analyt. Lett., 1975, v. 8, p. 537.
37. Koh I., Taniguchi K- — Analyt. Chem., 1973, v. 45, p. 2018.
38. Iwasaki I., Suzuki S. — Bull. chem. Soc. Japan, 1966, v. 39, p. 576; Analyt. Abstr., 1967, v. 14, p. 3951.
39. Koh I., Taniguchi K. — Analyt. Chem., 1974, v. 46, p. 1679
40. Solymosi F, Varga A. — Acta chim hung., 1959, v. 20, p 295; Analyt. Abstr., 1959, v. 6, p 2938.
41 Blasius E, Muench J. — Z. analyt. Chem., 1972, v. 261, p 198. Analyt. Abstr., 1973, v. 24, p. 2163.
42. Schmidt M., Sand T.—J. inorg. nucl. Chem., 1964, v. 26, p. 1185.
43. Pollard F. H-, Nickless G., Jones D. J., Glover R. B. — J. Chromat., 1964, v. 18, p. 407.
518
СУЛЬФАТЫ
Сульфат — один из важнейших анионов, используемых в химической и других отраслях промышленности. Естественно, что аналитическая химия сульфатов явилась предметом глубокого исследования, и это привело к появлению обширной литературы по данному вопросу. Обзоры [1—5], опубликованные в течение последних двадцати лет, посвящены анализу широкого круга серусодержащих соединений и, в частности, анализу сульфатов.
МЕТОДЫ ОТДЕЛЕНИЯ
Метод ионообменной хроматографии
В простейшем случае анализируемый раствор пропускают через катионообменную колонку перед анализом [6]. Описан косвенный метод [7], в котором раствор, содержащий сульфат и нитрат, пропускают через колонку, заполненную катионитом в РЬ-форме. Сульфат свинца осаждается и остается в колонке, а неосажденный нитрат свинца титруют раствором ЭДТА.
Предложено несколько методик анионообменного разделения, включающих разделение сульфата и хромата, сульфата и больших количеств фосфата и смеси сульфата, сульфита, тиосульфата и сульфида [8—10]. При пропускании анализируемого раствора через колонку, содержащую анионит в Cl-форме, выделяется хлорид, который затем титруют по методу Мора или Фольгарда [11].
Метод адсорбционной хроматографии
В колонке, заполненной оксидом алюминия в кислой форме, можно отделить сульфат от различных катионов и большинства анионов [12, 13]. Этот метод может быть использован [14] как предварительная стадия перед титрованием сульфата перхлоратом бария.
Метод тонкослойной хроматографии
В табл. 38 представлены варианты тонкослойной хроматографии, применяемые для разделения сульфатов и других анионов.
Газохроматографический метод
Использование газовой хроматографии для анализа описано в разделе определения сульфата.
519
Другие методы разделения
Отделение сульфата в виде сульфата бария почти всегда возможно с некоторыми ограничениями, описанными в разделе, посвященном гравиметрическому определению сульфата.
Таблица 38. Варианты метода тонкослойной хроматографии, применяемые для разделения сульфатов я других анионов
Неподвижная фаза Подвижная фаза Анионы, от которых отделяют сульфат Литература
Снлнкагель — крахмал Метанол — пропанол — 0,88 М раствор аммиака — вода (10:10: 1 :2) S2O*', SO|', S2O*’ /5
Пластины SA и SG Силикагель, кизельгур нли La2O3 Кукурузный крахмал Несколько; но преимущественно следующая: бутанол — пиридин — СНзСООН—Н2О (15: 10:3: 12) Ацетон — 1 М раствор аммиака (1 : 1) Ацетон — 3 М раствор аммиака (1 :4) S,O32', S3O2', S.O?-, SCN' POf NO2', S2O|', CrO2', N3, CN', SCN', BO*', s? , AsO*', AsOf NO', POf 16 17 18
Силикагель — 5%-ный пшеничный крахмал Метанол — «-бутанол — вода (3:1:1) F', NO", S2O|', CrO2'. PO*', AsO]', AsO]' 19
От сульфата можно отделить ряд элементов, включая железо, с помощью гидроксида аммония, а также посредством электролиза с ртутным катодом.
Кипячение анализируемого образца с концентрированной НС1 разрушает сульфиты и сульфиды. Этот метод был использован для отделения сульфатов от сульфитов и сульфидов [20, 21].
Возможно отделение сульфата после его восстановления до сероводорода. Образующийся сероводород абсорбируют поглотительным раствором и затем определяют, например, спектрофотометрически с применением метиленового синего. Этот высокоселективный метод обсужден в разделе, посвященном спектрофотометрическому определению сульфатов.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Большинство используемых в настоящее время методов определения сульфата основано на нерастворимости его бариевой соли. Ранее наиболее применяемым был гравиметрический метод В последние годы предпочтение оказывают тшриметрнческим методам, особенно в тех случаях, когда требуется высокая правильность определений.
520
Гравиметрические методы
Определение сульфата в виде BaSOi
Гравиметрическое определение сульфата осуществляют, прибавляя хлорид бария к анализируемому раствору, содержащему сульфат:
SOf + BaClj —> BaSO4 + 2Cl’
Белый осадок фильтруют, промывают, прокаливают и взвешивают. Основной источник возможных ошибок — соосаждение. Правильность результатов зависит от условий эксперимента, а именно концентрации и скорости прибавления реагентов, температуры, условий созревания и прокаливания осадка, а также природы присутствующих катионов и анионов. В данной монографии не рассмотрены детально различные аспекты определения, они подробно описаны в учебных руководствах [22—24]. В книге [22] содержится несколько ссылок на ранние работы, посвященные этому методу.
При соосаждении катионов и анионов загрязнения из раствора концентрируются на поверхности зародышей осадка. Это приводит к окклюзии, при которой происходит внедрение в кристаллическую решетку образующегося осадка, ионов или молекул, адсорбированных в процессе роста зародышей. Аналогично может внедряться в кристаллическую решетку осадка вода, которая не удаляется затем количественно при нагревании до 100 °C. При соосаждении может протекать также адсорбция ионов раствора на поверхности кристаллических частиц. На кристаллической поверхности осадка могут адсорбироваться соли, имеющие общий с сульфатом бария ион. В этом случае происходящие процессы описываются, хотя и с некоторыми исключениями, адсорбционным правилом Панета — Фаянса — Гана. Соли, не имеющие с осадком общего иона, подвергаются обменной адсорбции. Другими факторами, которые существенно дополняют правило Панета — Фаянса—Гана, являются способность адсорбированных соединений к электролитической диссоциации и деформируемость адсорбированных ионов.
Растворимость сульфата бария мала, но ее необходимо учитывать. Его растворимость в воде увеличивается с ростом температуры, но погрешность, связанная с использованием горячей воды, невелика. Обычно осаждение проводят в разбавленном растворе НС1. В присутствии соляной кислоты образуется более плотный, легче фильтрующийся осадок. Применение соляной кислоты предотвращает осаждение других бариевых солей, таких, как карбонаты и фосфаты. Присутствие соляной (или азотной) кислоты приводит к увеличению растворимости сульфата бария, однако раствор, содержащий около 0,5 Л4 НС1, можно использовать для проведения осаждения без значительных потерь осадка.
521
Катионы соосаждаются с сульфатом бария в виде сульфатов или гидросульфатов. Когда в качестве осадителя используют раствор соли бария, обычно соосаждение катионов преобладает над соосажденнем анионов. Но осаждение анионов доминирует в случае прибавления щелочного раствора сульфата к подкисленному раствору хлорида бария.
Соосаждение солей щеточных металлов и сульфата аммония увеличивается с увеличением их концентраций и уменьшается с увеличением скорости осаждения и времени созревания осадка. Присутствие ионов натрия в больших концентрациях мешает определению, ионы калия даже при относительно небольших содержаниях вызывают ошибки вследствие их соосаждения. Соосаждение сульфата аммония приводит к значительным отрицательным ошибкам вследствие его разложения при прокаливании осадка (950—1100°C). В случае присутствия ионов натрия быстрое прибавление хлорида бария уменьшает соосаждение сульфата натрия.
Все двухвалентные катионы соосаждаются с сульфатом бария в большей или меньшей степени. Поведение солей, имеющих общие с осадком ионы, описывается вышеупомянутым правилом Панета— Фаянса — Гана. Например, ионы кальция адсорбируются осадком в большей степени, чем ионы магния, поскольку растворимость сульфата кальция значительно ниже, чем сульфата магния. Из двухвалентных катионов наиболее значительные ошибки при гравиметрическом определении сульфата в виде BaSO4 вызывает кальций.
Присутствие железа (III) вызывает ошибки при определении. Эти ошибки связаны с протеканием гидролиза железа (III) и образованием во время этого процесса положительно заряженных основных соединений железа(III), которые сильно адсорбируются на отрицательно заряженной поверхности сульфата бария. В присутствии хрома(III) наблюдаются ошибки из-за неполного осаждения сульфата в виде бариевой соли, поскольку хром частично связывает сульфат в комплексное соединение.
Для устранения мешающего действия катионов можно предложить два метода. Двух- и трехвалентные катионы можно связать в комплексное соединение с помощью ЭДТА [25]. Можно анализируемый раствор перед определением сульфатов пропустите через колонку, заполненную катионообменной смолой.
Некоторые анионы вызывают ошибки из-за окклюзии. Для оценки возможного мешающего влияния анионов необходимо руководствоваться правилом Панета — Фаянса — Гана, в соответствии с которым большая тенденция к окклюзии наблюдается у тех анионов, которые образуют менее растворимые бариевые соли. Например, нитрат бария менее растворим, чем хлорид, и поэтому нитрат сильнее хлорида соосаждается на сульфате бария. Нитрат, а также хлорат вызывают ошибки при определении, и поэтому их необходимо предварительно удалять. Удаление их обычно дости
522
гается двух- пли трехкратным выпариванием раствора досуха с концентрированной НО:
NOJ + ЗСГ + 4Н+ —> С1а + NOC1 + 2НаО
СЮз + 5СГ + 6Н* —> зсь + зн2о
Для фильтрования осадка необходимо использовать очищенный бумажный фильтр или, лучше, фильтрующую бумажную массу. Отфильтрованный осадок промывают горячей водой, высушивают при 110°C и прокаливают при 800—1000°С. Необходимо следить, чтобы бумажный фильтр не загорелся. Окончательное прокаливание нужно выполнять при достаточном доступе воздуха, чтобы предотвратить возможное восстановление сульфата до сульфида углеродом.
Несмотря на обширную литературу, посвященную описываемому методу анализа, исследование метода гравиметрического определения сульфата в виде его бариевой соли продолжается. Например, вместо прокаливания и других операций, связанных с нагреванием осадка, предложено [26] высушивать осадок, помещенный в керамический тигель, промывая его этанолом или эфиром. Промытый осадок подсушивают воздухом 20 мин, выдерживают в эксикаторе 5—10 мин и затем взвешивают. Значительное сокращение продолжительности анализа приводит к повышению воспроизводимости определений. Результаты анализа, полученные описываемым методом, хорошо согласуются с данными, полученными при использовании обычной гравиметрической методики. Например, для образца пирита получено среднее содержание серы 54,1% по методу [26] и 54,0% с помощью гравиметрической методики, включающей прокаливание осадка.
В других исследованиях основное внимание было уделено процессу старения осадка. В общепринятой методике продолжительность созревания осадка составляет несколько часов, а лучше — ночь. Это значительно увеличивает продолжительность анализа. Естественно, были сделаны попытки найти способы химической обработки осадка, которые обеспечили бы большую экспрессность анализа. Например [27], было найдено, что замораживание осадка смесью твердого СО2 с ацетоном в течение 15 мин и последующее оттаивание его водой с температурой 60—70°C в течение 15 мин оказывает такое же действие, как и продолжительное созревание осадка при комнатной температуре. Этот метод применим для ускоренного созревания не только сульфата бария, но и MgNH4PO4 и оксалата тория. Использование предложенного метода позволяет уменьшить продолжительность гравиметрического определения сульфата с 10 до 2 ч.
Большое влияние на скорость образования и роста кристаллов BaSO4, а также на скорость седиментации осадка оказывают pH, избыток осадителя — соли бария и концентрации ионов бария и сульфата [28]. В исследовании установлено, что кристаллы
523
максимального размера с наибольшей скоростью образуются при pH = 3,0—3,5.
Изучение влияния посторонних солей на размер частиц сульфата бария показало [29], что высокие концентрации NaCl, NaNO3, КВг, KNO3 и других солей мешают росту кристаллов сульфата бария даже при высокой степени пересыщения.
Сульфат бария из гомогенного раствора осаждается сульфаминовой кислотой или диметилсульфатом [30]:
NH2SO3H + н2о —> so?- + nh; + н* (СН3)2 SO4 + 2Н2О —* SO?- + 2СН3ОН + 2Н+
Гравиметрическое определение сульфата в виде сульфата бария
Примечания. 1. Предполагается, что мешающие ионы удалены с помощью указанных в тексте методов. При использовании катионного обмена можно взять колонку диаметром 2 см и длиной 10 см с снльнокислым катионитом (например, марки Zeocarb 225), промытым 4—6 М раствором НС1. После медленного пропускания анализируемого раствора сульфата колонку промывают 200 мл деионизованной воды Если в растворе присутствуют ионы щелочных металлов, разбавляют раствор хлорида бария, используемого в качестве осадителя ' (10 мл 10%-ного раствора разбавляют до 100 мл), нагревают его до кипения и в процессе осаждения быстро прибавляют его к анализируемому раствору. Это приводит к уменьшению соосаждения щелочных металлов.
2. Если раствор сульфата подкислен соляной кислотой, доводят его кислотность до 0,05 М.
Ход анализа. Разбавляют анализируемый раствор сульфата, содержащий около 0,3 г сульфатов, до 250 мл и прибавляют 2 мл концентрированной соляной кислоты. Нагревают раствор почти до кипения и, помешивая, медленно добавляют к нему 10 мл 10%-ного раствора хлорида бария. Позволяют осадку седиментировать и проверяют полноту осаждения, добавляя небольшой объем осадителя. Оставляют осадок на горячей плите по крайней мере на 2 ч, но при возможности осадок лучше оставлять на ночь.
Фильтруют раствор через чистый бумажный фильтр, фильтрующую бумажную массу или чистый фарфоровый фильтрующий тигель. Промывают осадок горячей водой, отбирая пробы фильтрата на хлорид-ион (реакция с нитратом серебра). Промывание осадка продолжают, пока пробы не дадут отрицательной реакции на хлорид-ион. Если для фильтрования применяют бумажный фильтр (или фильтрующую бумажную массу), то его подсушивают и затем осторожно прокаливают, не допуская загорания бумаги. При использовании фарфорового фильтрующего тигля осадок можно помещать в печь без высушивания Прокаливают осадок при 900 °C до постоянной массы и взвешивают.
Методы с использованием органических реагентов
Слабое основание бензидин (4,4/-диаминобифенил) предложено в качестве гравиметрического реагента для определения сульфата еще в 1896 г. [31]. Растворимый в воде гидрохлорид бензидина осаждает сульфат с образованием нерастворимого соединения CiaHnzNzH^SOl'» которое можно взвесить. Растворимость этого соединения в воде составляет 98 мг/л и увеличивается при использовании раствора НС1, что приводит к потерям при проведении определения сульфата этим методом, и поэтому этот метод не на-524
ходит широкого применения. Этому содействует канцерогенность бензидина Позднее была проведена окончательная оценка этого реагента в качестве осадителя сульфатов [32]. ।
Методы с использованием смешанных осадителей
В качестве осадителя сульфатов были предложены тригалогениды гексааммннкобальта(Ш) [33]. Лучшие результаты были получены при использовании бромида, в этом случае образуется осадок (NH3)6CoBrSO4. Его взвешивают после высушивания при 80 °C. Растворимость этого соединения можно уменьшить добавлением этанола или ацетона. В этой же работе описаны титриметрические и спектрофотометрические методы определения сульфатов.
Для гравиметрического определения сульфатов предложено [34] использовать комплекс Вернера р-амино-р-нитрооктааммин-дикобальта(Ш).
Растворимость образующегося осадка в воде в этом случае составляет 22,4 мг/л при комнатной температуре, но значительно уменьшается в присутствии избытка реагента в 25%-ном органическом растворителе. Этот метод позволяет определять от 1 до 100 мг сульфатов. Значительные концентрации фосфатов (при pH ^5), фторидов, нитратов и пероксидов не мешают определению сульфатов.
Титриметрические методы
Методы с использованием солей бария в качестве титранта
Для создания быстрых титриметрпческих методов определения сульфата можно использовать нерастворимость сульфата бария. Большинство ранних методов основано на обратном титровании избытка соли бария. Одним из лучших индикаторов для такого титрования считалась родизоновая кислота. Но воспроизводимость и правильность этого метода все же не совсем удовлетворительны.
Большинство современных методов определения сульфата основано на исследованиях 1954—1957 гг. [35—37]. Предложен метод, основанный на титровании сульфатов перхлоратом бария в растворе, содержащем 30—40% этанола с эффективным значением pH = 2,3—3,7. Если сульфаты присутствуют в макро- или полумикроколичествах, то в качестве адсорбционного индикатора можно применить ализариновый красный S. При мнкросодержа-ниях сульфатов в качестве индикатора титрования используют торон [1-(о-арсонофеиилазо)-2-нафтол-3,6-дисульфокислоту]. В качестве осадителя хлориду бария предпочитают перхлорат вследствие его значительно меньшего соосаждения.
Найдено, что соосаждение анионов на осадке сульфата бария уменьшается в ряду NOj > СГ > Вг > С10-Г» в этом же ряду
525
увеличивается растворимость бариевых солей, что соответствует адсорбционному правилу Панета — Фаянса — Гана. Тем не менее в несколы^х недавно опубликованных статьях рекомендовано использовать хлорид бария.
При определениях необходимо применять спиртовую среду, наилучший эффект достигается при концентрации этанола 30— 40%. При меньшей концентрации спирта конечная точка титрования становится менее отчетливой, при более высокой — наблюдается значительное замедление реакций и возникают «ложные» конечные точки титрования, в которых раствор медленно обесцвечивается, что вызывает большие трудности при проведении определений. Присутствие спирта также влияет на характер осадка. Из водных растворов сульфат бария осаждается обычно чистым и кристаллическим, тогда как в спиртовых средах осадок получается легким, хлопьевидным, а иногда и студенистым.
Очень неудовлетворительной особенностью осадка сульфата бария является его сильная склонность к соосаждению катионов и анионов. Для удаления катионов рекомендуется использовать ионный обмен.
Перед осаждением сульфата необходимо раствор довести до pH = 2,3—3,7. При более низких значениях pH конечная точка титрования получается размытой, что приводит к заниженным результатам. При проведении анализа для установления необходимого значения pH используют хлоруксусную кислоту или пиридин. Если в качестве предварительной стадии перед анализом использован ионный обмен, то анализируемый раствор перед титрованием можно частично нейтрализовать ацетатом магния.
Ализариновый красный S (ализаринсульфонат натрия) является кислотно-основным индикатором с интервалом перехода окраски при pH = 3,7—5,2. В предложенном методе титриметриче-ского определения сульфата это соединение выполняет функции адсорбционного индикатора, механизм действия которого можно описать следующим образом. В процессе титрования осадок сорбирует сульфат-ноны на поверхности, что приводит к отрицательному заряду осадка. При появлении избытка бария в растворе осадок приобретает положительный заряд вследствие адсорбции ионов бария. После достижения точки эквивалентности отрицательно заряженный ион ализаринсульфоната притягивается к положительно заряженной поверхности, при этом образуется окрашенный в розовый цвет бариевый комплекс ализарннсуль-фоната.
При микросодержаниях сульфата массы осадка недостаточно для использования многих индикаторов. Например, окраска торона изменяется при титровании с желтой до розовой. Такой переход часто бывает трудно наблюдать, особенно при искусственном освещении. Для лучшего наблюдения за переходом-окраски предложено применять метиленовый синий [38]. Смесь торона с метиленовым синим используют в качестве индикатора в некоторых современных вариантах описываемого метода.
526
рис. 62. Прибор для хроматографического отделения сульфатов в колонке с активным оксидом я1Юминия [37J:
- колонка с оксидом алюминия (размер зерен 0.074 — 0 149 мм)! 2—стекловолокно нлн стеклянная вата; •’—колба вместимостью 1 л со срезанным дном; 4—стакан вместимостью 159 мл; 5—короткая стеклянная палочка; 6—толстая стеклянная пластина.
Фосфат вызывает значительные помехи при титриметрическом определении сульфата, и его необходимо предварительно удалить. Для осаждения фосфата рекомендуют использовать карбонат магния [36]. Устранения мешающего действия фосфата можно достичь осаждением фосфата серебра после прибавления оксида серебра к анализируемому раствору [39]. Образующийся сульфат серебра разлагают на ионообменной смоле
и выделяющуюся серную кислоту титруют перхлоратом бария. В работе [39] описано микроопределенне серы в органических соединениях после сожжения их в кислороде.
Мешающее действие сульфита при определении сульфата можно учесть, титруя сульфит стандартным раствором иода и вычитая затем содержание сульфата, эквивалентное иоду, из общего содержания сульфата.
Если описываемый метод используют для определения микроколичеств сульфатов (0,12—12 мг сульфата в 10 мл), в методику необходимо внести некоторые изменения. Титрование в этом случае проводят в 80 %-ном этаноле при эффективном значении pH = 2,5—4,0, которое автоматически достигается после пропускания раствора образца через катионообменную колонку. Образец должен содержать не слишком высокие концентрации посторонних солей, максимальная молярная концентрация солей должна быть не более 5—10-кратной концентрации сульфатов. В описываемом варианте используют раствор титранта, приготовленный на 80 %-ном этаноле.
Описан хроматографический метод отделения сульфатов от других ионов в колонке (рис. 62), заполненной оксидом алюминия [36]. Метод позволяет выделить до 0,5 ppm сульфатов из растворов, содержащих значительные концентрации хлоридов, нитратов, перхлоратов и большинства ионов металлов. Сульфат элюируют из колонки разбавленным раствором аммиака, пропущенным предварительно через катионообменную колонку, и затем титруют его Раствором соли бария. Описываемый метод не позволяет отделить сульфаты от фторидов и фосфатов. Мешающее действие фторида устраняют добавлением борной кислоты к анализируемому раствору [40]. Удаление фосфата описано выше.
Сульфат плохо отделяется от хрома(III), циркония(IV) и тория (IV) из-за образования инертных сульфатных комплексов этих
527
528
Таблица 39. Индикаторы титрования сульфатов солями бария
Общепринятое название Наименование по номенклатуре 1UPAC Характеристика Литература
Торон 1-(о-Арсонофенилазо)-2-наф-тол-3,6-дисульфокислота Переход окраски: желтая — розовая 36, 38
Карбоксиарсеназо 3-(о-Арсонофенилазо)-6-(о-карбокси-фенилазо)-4,5-диоксинафталин-2,7-дисульфокислота Переход окраски: фиолетовая — синяя. Среда: ацетон — буферный раствор пиридина в НС1О4 с pH = 5,8 (2: 1). Определению мешают фосфаты 41—43
Нитхромазо (нит-роортаниловый S, динитросульфоназо III) 3,6-Бис(4-нитро-2-сульфофенил-азо)-4,5-Диокси нафталин-2,7-дисульфокислота Переход окраски' фиолетовая — синяя. Допустимо присутствие 5-кратного избытка фосфатов и 4-кратного — арсенатов. Определению ие мешают фториды, хлориды, нитраты н перхлораты 44—47
Арсоназо III 2,7-Бис(2-арсонофенилазо)-1,8-ди-оксинафталин-3,6-дисульфокислота Переход окраски: синяя — красная 48, 49
Сульфоназо III 3,6-Бис(2-сульфофеиилазо)-4,5-ди-оксиндфталии-2,7-дисульфокислота Переход окраски: пурпурно-красная — синяя. Среда-слабо подкисленная вода — ацетон (1 : 1). Точка эквивалентности при титровании достигается медленно [43] 50—52, 43
Диметилсульфо-назо III 3,6-Бис(4-сульфо-п-толилазо)-4,5-ди-оксинафталин-2,7-дисульфокислота Переход окраски: фиолетово-синяя — сине-серая. Мешают определению соли при концентра- . 53, 54, 43
цни > 0,1 мМ. Минимальные помехи вызывают фосфаты, арсенаты, ЭДТА и NaCI. Точка эквивалентности при титровании достигается медленно [43]
Ортанил К (орта-ниловый К, карбоксисульфоназо Ш) 3-(2-Карбоксифсиилазо)-6-(2-сульфо-фенил азо)-4,5-диоксинафта -лии-2,7-дисульфокислота Почти все катионы необходимо предварительно отделять (с помощью ионного обмена). Индикатор очень эффективен при определении сульфатов в присутствии фторидов и ацетатов. Среда: вода — ацетон (1:4) 55, 56
Хлорофосфоиазо III 3,6-Бис(4-хлоро-2-фосфонофенил-азо)-4,5-Диоксин афта лин-2,7-дисульфокислота Переход окраски: винно-красиая — синяя. Из исследованных 21 катиона и 10 анионов мешают определению только V, La, Zr, Hg, Th, UIV и UVI 57
Дибромосульфо-иазо III 3,6-Бис(4-бромо-2-сульфофе-нил-азо)-4,5-Диоксинафталнн-2,7-дисульфокислота Переход окраски: красная — синяя 53 9
Карбоксиазо III 3,6-Бис(2-карбоксифенилазо)-4,5-ди-оксинафталин-2,7-дисульфокислота Переход окраски: пурпурно-розовая — зеленовато-синяя. Рекомендован для титримстрических и фотометрических (685 нм) определений. Среда: 75%-иый этанол, pH = 2,55. Рекомендовано обратное титрование 58
Карбоксисульфоназо 3-(2-Карбоксифеннлазо)-6-(2-сульфо-нилазо)-4,5-Диоксинафталин-2,7-дисульфокислота Переход окраски: пурпурно-розовая — зелеиовато-си-ияя. Рекомендован для титримстрических и фотометрических (645 им) определений. Среда: 75%-ный этанол, pH = 2,1. Рекомендовано обратное титрование 58
металлов. Устранение мешающего действия ионов этих металлов достигается предварительной обработкой образца N-оксиэтилен-диаминтриуксусной кислотой, которая образует прочные комплексные соединения с указанными ионами. Полученный раствор затем можно пропустить через хроматографическую колонку с оксидом алюминия. Нельзя использовать ЭДТА вместо N-оксиэтилендиамин-триуксусной кислоты, так как комплексные соединения этих металлов с ЭДТА нестабильны на оксиде алюминия.
В дополнение к торону как индикатору для титриметрического определения сульфата предложен ряд производных хромотроповой кислоты и проведено их сравнительное изучение. В этих исследованиях рассмотрены различные критерии выбора индикатора: контрастность конечной точки титрования, время, необходимое для достижения равновесия, правильность и селективность определений. В табл. 39 описаны свойства некоторых индикаторов, включая торон.
Тщательное сравнительное изучение индикаторов [54] показало, что лучшим индикатором для визуального и фотометрического титрования является диметилсульфоназо III. При использовании этого индикатора минимальные помехи определению оказывают фосфаты, арсенаты, ЭДТА и хлорид натрия.
Правильность определения сульфатов при использовании различных индикаторов представлена в табл. 40. Из таблицы видно, что торон снижает правильность определений по сравнению с другими индикаторами, изученными в этой работе. Эти результаты находятся в соответствии с молярными коэффициентами поглощения индикаторов.
Таблица 40. Правильность титриметрического определения сульфата калия при применении различных индикаторов
Индикатор Найдено сульфатов •, % Стандартное отклонение определений, %
Торон 99,4 ±0,15
Карбоксиарсеназо 99,6 ±0,15
Сульфоназо III 100,4 ±0,10
Динитросульфоназо III 100,5 ±0,12
Диметилсульфоназо III 99,8 ±0,08
Дибромсульфоназо III 100,2 ±0,10
* Среднее из десяти определений.
Авторы работы [43] предпочли для титрования сульфата использовать карбоксиарсеназо и нашли, что применение диметилсульфоназо III приводит к медленному достижению равновесия при титровании. По мнению авторов работы [48], использование арсеназо III в качестве индикатора предпочтительней, нежели сульфоназо III или торона.
53Q
Титрование сульфата ионами бария используют в ряде важных областей аналитической химии: для определения серы в органических соединениях при полумикро-, микро- и субмикроконцентра-цпях ее после сожжения образца в кислороде [40]. Описано одновременное определение серы и хлора в органических соединениях [59]. Титрование сульфата ионами бария использовано для определений 10—30 мкг серы в органических соединениях [60]. В другом исследовании достигнут предел определения сульфатов 0,5 мкг [61].
Этот метод используют также для анализа масел [62], углей [63], пищевых продуктов [64] и воды [65].
Титриметрическое определение сульфата с использованием перхлората бария
Реагенты
Перхлорат бария, 0,1 М раствор. Готовят примерно 0,1 М раствор, доводят его до pH = 3,0—3,5 и титруют стандартным раствором H2SO«.
Перхлорат бария, 0,01 М раствор. Растворяют 3,9 г Ва(С1О4)2-ЗН2О в 200 мл воды. Прибавляют 800 мл изопропанола. Стандартизируют с помощью 0,005 М H2SO4. Вместо изопропанола можно использовать этанол или метанол, но изопропанол медленней испаряется.
Ализариновый красный S, 0,2%-ный водный раствор.
Торон, 0,2 %-ный водный раствор.
Ход анализа. Макроопределение. Методику используют для определения 2—4 ммоль сульфата в 45 мл воды. К анализируемому раствору прибавляют 40 мл метанола и доводят раствор до pH = 3,0—3,5 при помощи разбавленного («0,25 Л1) раствора ацетата магния нли хлорной кислоты. Быстро прибавляют около 90% требуемого количества перхлората бария, 5 капель индикатора ализаринового красного S и титруют до появления розовой окраски, не исчезающей в течение 3—5 с после прибавления последней капли титранта.
Примечание. При полумикросодержаннях (0,2—0,8 ммоль сульфата) к анализируемому раствору прибавляют 10 мл воды, 10 мл метанола н одну каплю индикатора.
Микроопределение. К 10 мл анализируемого раствора прибавляют 40 мл этанола и одну каплю индикатора торона. Раствор доводят до эффективного значения pH = 2,5—4,0. Титруют 0,01 А1 раствором перхлората бария до появления неисчезающей окраски. Почти весь титрант необходимо прибавить быстро, а к концу титрования капли титранта следует вводить с интервалом 2—3 с.
Микроопределенне с предварительным отделением сульфата Раствор анализируемого образца, содержащий 0,12—12 мг сульфата, доводят до pH = 0,5—1,0 с помощью разбавленной НС1 нли HCIO4. Пропускают раствор через колонку, заполненную оксидом алюминия, со скоростью 120 капель в 1 мин. Промывают колонку, пропуская через нее 10 мл 5%-ного раствора НС1 и 25 мл воды. Сульфат элюируют, прибавляя раствор аммиака: последовательно 5 мл I М, 20 мл 0,1 М и 20 мл 0,1 М (в виде пяти порций) и, наконец, около 20 мл воды. Элюент, содержащий сульфат, пропускают через ионообменную колонку и собирают раствор на выходе из колонки в мерную колбу емкостью 100 мл. Колонку промывают водой, используя такое ее количество, чтобы довести раствор в колбе до метки.
Аликвотную часть полученного раствора (10 мл) титруют в соответствии с приведенной выше методикой микроопределения.
Примечания: 1. Раствор торона нельзя хранить длительное время, и его необходимо заменять не реже одного раза в месяц.
2 , Можно использовать смешанный индикатор, прибавляя каплю 0,05%-ного водного раствора метиленового синего к капле торона.
531
3 Ионообменная колонка должна нметь диаметр 2,5 см и быть заполнена слоем кагнонообменной смолы (Dowex 50-Х8 с размером зерен 297—841 мкм в Н-форме) высотой 7—8 см.
4 Колонку с оксидом алюминия подготавливают следующим образом: хроматографический окснд алюминия (74—177 мкм) промывают в мензурке и позволяют взвеси седиментировать. Осветленную жидкость удаляют декантацией и обработку повторяют, чтобы удалить очень мелкие частицы. После переведения оксида алюминия в колонку промывают ее, прибавляя 50 мл, а затем несколько порций по 5 мл 1 М раствора аммиака, несколько порций 0,1 М раствора аммиака п около 50 мл воды. Затем промывают колонку 10 мл НС1 или НС1О< с pH — 0,5—1,0. Нельзя допускать высыхания колонки.
5 . Если в анализируемом растворе присутствуют хром(Ш), торнн(1У) или цнрконий(1У), необходима следующая подготовка образца. Прибавляют избыток 0,5 Л1 раствора N-оксиэтилендиаминтриуксусной кислоты (растворяют 69,5 г кислоты в воде, прибавляя гранулированного NaOH). Для увеличения растворимости, раствор должен иметь pH 5—6, раствор разбавляют до объема 1 л) и доводят pH до 5—6 Нагревают раствор до кипения н затем охлаждают. Если присутствует цирконий, раствор необходимо подкислить. Если в анализируемом растворе присутствуют хром или торий, то нужно кипятить раствор в течение 10—15 мнн, после чего охладить н подкислить ею, необходимость кипячения связана с медленным протеканием реакции комплексообразования.
Метод с использованием нитрата свинца
в качестве титранта
Титриметрическое определение сульфата с использованием пн-трата свинца как титранта основано на образовании при титровании сульфата свинца, практически нерастворимого в водно-ацетоновых смесях [66, 67]. Небольшой избыток реагента вызывает резкий переход окраски от зеленой к пурпурно-красной, связанной с образованием дитизоната свинца.
Дальнейшее изучение позволило расширить применение этого метода в области микро- и субмикроконцентраций сульфатов и найти оптимальные условия для проведения определений [68, 69].
Ионы тяжелых металлов, образующие комплексные соединения с дитизоном и поэтому мешающие определению сульфата, можно удалить на катионообменной смоле. Калий мешает определению, если присутствует в концентрации, эквивалентной концентрации сульфата. Это связано с низкой растворимостью сульфата калия в водно-ацетоновых смесях. Если предварительное ионообменное отделение тяжелых металлов не проводят, то при определении 0,25—50 мг сульфатов допустимы следующие концентрации примесей: Hg и Ag — по 0,1 мг/мл; Со — 5 мг/мл; Zn — 20 мг/мл; Ni—40 мг/мл; Fe и А1 — по 100 мг/мл; Si — 500 мг/мл и Мп — 2000 мг/мл (погрешность определения ±1,5%) [70].
Фосфат мешает определению и должен быть предварительно удален осаждением MgNH4PO4. Хлорид при небольших содержаниях мешает определению сульфатов, и его рекомендуется удалять [68, 69]. Другие авторы полагают, что хлорид мешает при семикратном избытке относительно концентрации сульфата [67]. Мешающее действие нитрита, связанное с окислением индикатора, устраняют введением карбамида в анализируемый раствор до ти-
532
тровання сульфата. Нитрат при больших содержаниях не мешает определению сульфата, также не мешает и перхлорат.
В серии статей [71—74] описано использование дифенилкар-базона вместо дитизона в качестве индикатора при титровании сульфатов. Комплексное соединение свинца с дифенилкарбазоном менее стабильно, чем дитнзонат свинца. Определение рекомен-д ют проводить в водно-этанольной среде. Определению сульфатов с этим индикатором мешают ионы, указанные выше, необходимо ионообменное удаление катионов. Погрешность определения ие превышают 2 и 3% при определении 25 и 10 мкг сульфатов соответственно.
Метод пригоден для одновременного определения следовых концентраций сульфатов и хлоридов в водопроводной воде. Сначала титруют сульфат- стандартным раствором нитрата свинца, раствор слегка подкисляют и титруют хлорид стандартным раствором нитрата ртути(II). Титриметрическое определение сульфата дополнено его предварительным концентрированием в колонке, заполненной оксидом алюминия [37]. Описанный метод использован для анализа дождевых вод и вод буровых скважин [75]. Метод экспрессен и обеспечивает получение правильных результатов [76]. Кальций мешает определению сульфатов как при использовании дитизона, так и с дифенилкарбазоном, поскольку образуется сульфат кальция, нерастворимый в водно-спиртовых средах.
Для индикации конечной точки титрования в варианте с использованием дитизона предложено применять фотометрический метод [77]. В этом случае можно прямо оттитровать до 20 ppm сульфатов.
Макроопределение сульфата с использованием
нитрата свинца [68, 69]
Реагенты
Нитрат свинца, 0,005 М раствор. Кислотность раствора не должна превышать кислотность, соответствующую зеленой окраске индикатора бромфенолового синего.
Смесь ацетона с уксусной кислотой, 0,5%-нын раствор ледяной уксусной кислоты в ацетоне. Ацетон должен быть очищен от следов тяжелых металлов.
Дитизон, 0,05%-ный раствор в ацетоне; приготовляют ежедневно.
Аммиак, 0.02 М раствор.
Азотная кислота. 0.02 М раствор, приготовляют из 2 М раствора HNO3. Необходимо, чтобы 2 М раствор постоял в течение некоторого времени (эта стадия необходима для предотвращения появления следов азотистой кислоты при прямом разбавлении концентрированной азотной кислоты; азотистая кислота быстро окисляет дитизон).
Ход анализа. Раствор образца, содержащего не более 5 мг сульфатов, выпаривают досуха на водяной бане. Растворяют остаток в 1 мл воды, прибавляют одну каплю бромфенолового синею и, осторожно добавляя раствор аммиака или азотной кислоты, доводят pH раствор до pH. соответствующего зеленой окраске индикатор?.. Раствор не должен иметь голубого оттенка. Прибавляют '8 мл смеси уксусной кислоты с ацетоном и затем около 0,4 мл раствора дитизона.
533
Быстро тнтруют полученный раствор стандартным раствором нитрата свннна, используя механическую мешалку Вблизи конечной точки титрования прибавляют дополнительно 2 мл смеси уксусной кислоты с ацетоном и продолжают титрование по каплям до изменения цвета раствора от изумрудно-зеленого через синий н пурпурный к неисчезаюшему пурпурно-красному, который указывает на достижение конечной точки титрования. Прибавление избытка титранта приводит к красному окрашиванию раствора.
Для большей правильности определения проводят контрольный опыт.
Примечания. 1. Уксусную кислоту добавляют к ацетону для простоты титриметрпческого определения.
2. Слишком большое содержание воды в титруемом растворе ухудшает контрастность окраски в конечной точке, что приводит к увеличению ошибки определения. Поэтому вблизи конечной точки прибавляют смесь ацетона с уксусной кислотой, что обеспечивает поддержание концентрации воды вблизи оптимального значения («25%).
3. Появление розовой нли красной окраски при добавлении первых капель дитизона к раствору образца указывает на присутствие примесей тяжелых металлов. Избыток дитизона маскирует малые концентрации этих примесей. Если содержание примесей велико, необходимо провести ионообменное отделение сульфатов.
Комплексометрический метод
После разработки первого комплексометрического метода определения сульфата [78] предложены методы, часть из которых рассмотрена в монографии [79]. Большая часть комплексометрических методов определения сульфатов основана на их осаждении стандартным раствором хлорида бария, фильтровании осадка и обратном титровании избытка иона бария стандартным раствором ЭДТА. Очевидно, что стадия осаждения в этой методике имеет очень важное значение и требует такого же внимания, как и при гравиметрическом определении сульфата. Необходим такой выбор условий осаждения, чтобы осадок получался с соотношением компонентов, очень близким к стехиометрическому.
При осаждении сульфата бария в присутствии ЭДТА предотвращается соосаждение многих ионов. Кроме того, если осадок содержит посторонние ионы, то после его растворения в аммиачном растворе ЭДТА и переосаждении при подкислении осадок получается чистым [80].
Растворимость сульфата бария можно уменьшить добавлением этанола или другого смешивающегося с водой растворителя. Другая трудность, с которой приходится сталкиваться при комплексометрическом определении сульфатов, связана с тем, что при обратном титровании избытка соли бария титруются и другие присутствующие ионы металлов. Одним из путей преодоления этой трудности служит использование метода катионного обмена, хотя в этом случае увеличение объема титруемого раствора приводит к осложнениям при титровании.
Другой вариант комплексометрического определения сульфата основан на растворении сульфата бария в аммиачном растворе ЭДТА и последующем титровании избытка ЭДТА [81]. Обратное титрование ЭДТА проводят стандартным раствором хлорида маг-
534
нпя с эрнохромом черным Т в качестве индикатора. Этот метод был использован для определения серы в стали [82].
При больших содержаниях сульфата возникают проблемы, связанные с трудностью полного растворения сульфата бария в аммиачном растворе ЭДТА и поэтому для растворения вместо аммиака были предложены 5%-ные растворы моно- и триэтаноламина [83]. Была сделана попытка использовать раствор NaOH [84] вместо аммиака, однако в этом случае возможно появление помех из-за частичного выщелачивания ионов из стекла.
Предложен еще один вариант комплексометрического определения сульфата, основанный на первоначальном осаждении сульфата свинца. Этот осадок обладает значительно лучшими свойствами, чем BaSO4, а его относительно высокую растворимость можно уменьшить, добавляя этанол. Для проведения определения анализируемый образец, содержащий 5—350 мкг сульфата, обрабатывают известным избытком раствора нитрата свинца в среде, содержащей уксусную кислоту и 25—30% этанола [85]. После фильтрования осадка фильтрат титруют стандартным раствором ЭДТА, используя комплексное соединение медь(П)—ПАН[1-(2-пиридплазо)-2-нафтол] в качестве индикатора. Определению сульфатов описанным методом не мешают ноны калия, натрия, аммония, магния, хлорида, а также СО2; однако кальций и фосфат соосаждаются на сульфате свинца и мешают определению сульфатов.
Осажденный сульфат свинца можно растворить в аммиачном растворе ЭДТА с известной концентрацией ЭДТА и затем оттитровать избыток ЭДТА. Осаждение сульфата свинца проводят в кислой среде, для увеличения полноты осаждения к раствору добавляют изопропанол. Избыток ЭДТА титруют стандартным раствором хлорида цинка, используя эриохром черный Т в качестве индикатора [86]. Продолжительность анализа составляет 30—45 мин, в том числе 15 мин, необходимых для растворения осадка. Этот метод применен для определения сульфатов в макроинтервале концентраций. Описан вариант этого метода [87], в соответствии с которым осадок выдерживают 3 ч и только после этого фильтруют. Для фильтрования применяют два бумажных фильтра (Ватман № 3) вместо пористой стекломассы, использованной в работе [86]. Эта замена приводит к большей правильности и воспроизводимости определений. Присутствие фосфатов, молибдатов и селе-натов приводит к завышению результатов определения сульфатов. Определению не мешают мышьяк и сурьма.
Для растворения сульфата свинца можно использовать специфический комплексообразующий агент, например ацетат натрия или тартрат аммония, после чего оттитровать свинец(П) стандартным раствором ЭДТА.
Для растворения применяли также ацетат аммония [88]. После растворения осадка доводят pH раствора до 5 и титруют свинец(II), используя ксиленоловый оранжевый в качестве индикатора. При определении 1, 10 и 50 мг сульфатов коэффициенты
535
вариации составляют 2%, 0,5 и менее 0,2% соответственно. Определению сульфатов мешают фториды, иодиды, хроматы, фосфаты, оксалаты и арсенаты, а также хлориды [89].
Описано несколько простых методик определения сульфатов и других серусодержащих соединений при их совместном присутствии. Сульфат, сульфид и сульфит определяют следующим образом [90]. Сульфид осаждают раствором нитрата меди, а неизрасходованную медь(П) титруют стандартным раствором ЭДТА. Во второй аликвотной части анализируемого раствора определяют сульфат, осаждая его в виде сульфата бария с последующим титрованием избытка Ва2+ стандартным раствором ЭДТА. В третьей аликвотной части сульфид и сульфит окисляют бромом до сульфатов и затем определяют сульфат, как описано выше. Состав анализируемого раствора можно определить на основании результатов, полученных при титровании трех аликвотных частей.
Для определения сульфатов и сульфидов при их совместном присутствии предложена другая методика [91]. Одну аликвотную часть анализируемого раствора кипятят с соляной кислотой для удаления сульфидов в виде H2S. В этой аликвотной части определяют сульфат, используя ЭДТА. Вторую аликвотную часть окисляют бромом, при этом сульфид превращается в сульфат. Определяют сульфат выше описанным методом, а содержание сульфида находят по разности двух титрований.
Аналогичный метод предложен для анализа смесей сульфатов и сульфитов [92]. Описанный метод включает удаление сульфита в виде SO2, а также окисление сульфита бромом до сульфатов.
Для определения сульфата и фосфата при их совместном присутствии можно использовать комплексометрический метод [93]. Первоначально осаждают фосфат в виде MgNH4PO4, осадок отфильтровывают и неизрасходованный Mg24 титруют ЭДТА. В фильтрате осаждают сульфат в виде сульфата свинца и снова титруют раствор ЭДТА.
Комплексометрический метод определения сульфатов широко используют для анализа цемента [94], удобрений [95], стали [82] и химических реагентов [96]. Описаны и другие применения этого метода [79].
Метод титрования сульфида, образованного
при восстановлении сульфата
Восстановление сульфата с последующим выделением H2S служит основой методов отделения и определения сульфата. Для восстановления сульфатов предложены смеси, содержащие титан с фосфорной кислотой [97] и иодистоводородную кислоту [98]. Образующийся H2S поглощают растворами солей цинка или кадмия или раствором NaOH и определяют затем титриметрически или спектрофотометрически. В качестве титранта можно использовать ацетат ртути(II) с индикатором дитизоном [99].
536
Модификация этого метода была применена для определения следовых содержаний сульфатов в химических реактивах [97]. Сульфид, образованный при восстановлении сульфата смесью титана с фосфорной кислотой, титруют ацетатом ртути(II) с индикатором дифенилтиокарбаминатом. Описанный метод позволяет определять до 10 мкг сульфатов в химических реактивах.
Восстановление сульфата было использовано для определения серы в пищевых продуктах [100]. Первоначальное разложение образцов проводят в кислороде. Результаты, полученные для 26 различных типов пищи, указывают, что предложенный метод характеризуется большей воспроизводимостью и правильностью, чем турбидиметрический; описанный метод особенно пригоден для определения серы в образцах с большим содержанием неорганических компонентов: недостатком является его значительная продолжительность (3 ч на выполнение одного анализа).
Титрование сульфидов солями ртути с индикатором дитизоном
проходит малоудовлетворительно из-за маскирования окраски индикатора осаждающимся сульфидом ртути [101]. Лучшие результаты получаются при использовании в качестве титранта 2-(оксимеркури)-бензойной кислоты с индикатором дитизоном [102]:
асосг ^х^СОО' 'ООС^-.
+ S2’ ч=± Г jT ]| + 2ОН"
HgOH S—Hg^4^
Авторы работы [101] применили этот метод для определения серы в скальных породах при ее содержании 5—2000 мг/кг. Метод основан на окислении серы до сульфата, восстановлении его до сульфида смесью, содержащей пять компонентов, поглощения H2S раствором КОН и титровании этого раствора вышеуказанным реагентом.
Другие титриметрические методы
Несколько титриметрических методов определения сульфатов основано на реакции сульфата с бензидином. Однако ввиду канцерогенных свойств бензидина эти методы не находят больше применения.
Одним из наиболее чувствительных реагентов для определения •сульфата является 4-амино-4'-хлоробифенил. Этот реагент образует осадок с 0,0005 М H2SO4 (в виде мути) [103]. Осаждение сульфата этим реагентом положено в основу титриметрического метода прямого определения 2,5—100 мг сульфатов. Реагент прибавляют к анализируемому раствору при pH = 1,0—2,0, осадок кипятят 1 мин и титруют раствор стандартным раствором NaOH, используя смесь фенолового красного и бромтимолового синего в качестве индикатора. Нитрат не мешает определению сульфатов. Окислители должны отсутствовать, так как они окисляют реагент.
537
Алюминий образует нерастворимую комплексную соль, содержащую сульфат-ионы, однако его мешающее действие можно устранить, добавляя винную кислоту. Нерастворимые соединения с реагентом образуют фосфаты, оксалаты, селенаты и теллуриты. Помехи, вызываемые фосфатом, можно устранять, связав его в MgNH4PO4. Описанный метод использован для определения серы в углях [104]. Можно заметить, что 4-амино-4/-хлорбифенил не обладает канцерогенными свойствами. Недостаток этого реагента— его относительно высокая растворимость [105].
Для определения сульфата используют методы умножения [106, 107]. Например, проводят реакцию обмена сульфата с йодатом бария в среде, содержащей ацетонитрил, и иодат затем определяют, используя стандартную титриметрическую методику [108]. Другой метод [109] основан на прибавлении избытка бромата бария к анализируемому раствору сульфата. При этом осаждается сульфат бария, затем, после прибавления ацетона, осаждается избыток бромата бария. Осадки фильтруют, фильтрат обрабатывают иодидом калия и образующийся иод титруют стандартным раствором тиосульфата [109]. Описанный метод предложен для микроопределения серы в органических соединениях
Сульфат можно определить, используя в качестве титранта раствор НС1 в диметилсульфоксиде (фотометрическая индикация точки эквивалентности) [110]. Этот метод применяют для определения сульфата в морской воде. Большинство общепринятых методов определения сульфатов не применимо для анализа морской воды из-за высокого содержания солей в ней. Для определения сульфатов в этом объекте используют гравиметрическую методику, однако в этом случае наблюдаются ошибки, связанные с соосаждением солей щелочных металлов и кальция. В соответствии с вышеупомянутым методом [ПО] сульфат титруют до H2SO4, используя в качестве индикатора бромкрезоловый зеленый. Конечную точку в этом титровании находят графически.
Спектроскопические методы
Спектрофотометрические методы в видимой и ультрафиолетовой областях спектра
Сульфат-ион образует несколько окрашенных соединений с различными реагентами. Это положено в основу прямых спектрофотометрических методов определения сульфатов. Косвенные методы часто бывают связаны с замещением сульфат-ионом каких-либо хромофорных соединений. Другая возможность косвенного определения сульфата связана с определением избытка иона (или соединения), реагирующего с сульфатом с образованием нерастворимых форм. Такие методы используют нечасто, так как их чувствительность и селективность обычно невысоки. Здесь не рассматриваются методы определения сульфата, основанные на его реакции с бепзидийом, поскольку этот реагент канцерогенен.
538
Для осаждения сульфат-ионов описано применение 4-амино-4'-хлорбифенила [103], однако позднее [112] этот реагент был использован для спектрофотометрического определения субмикро-концентраций сульфатов. Катион этого амииа обладает высоким молярным коэффициентом поглощения (около 23 000 при 254 нм), методика определения сульфата с использованием этого реагента включает определение концентрации амина после осаждения и удаления сульфата этого реагента. Удаление осадка лучше выполнять с помощью центрифуги (осадок обладает относительно высокой растворимостью и поэтому фильтрование и промывание осадка могут приводить к ошибкам при определении). Описанный метод применен для анализа сульфатов в миллиграммовом интервале содержаний [ИЗ], это позволило проводить определение серы в углях и некоторых других материалах. Погрешности определения при содержаниях сульфатов 3—7,5 мг не превышают 1%.
Сходный метод для определения сульфатов основан на использовании 2-аминопиримедина [114], позднее этот реагент был применен для нефелометрического определения сульфата [115]. Это соединение обладает сильным светопоглощением в ультрафиолетовой части спектра и имеет двойной пик при 200—230 нм и широкий максимум при 305 нм. Для аналитических целей более пригоден второй максимум. Методика определения заключается в следующем: к анализируемому раствору сульфата прибавляют известное количество реагента, выдерживают смесь 30 мин для образования сульфата 2-аминоперимидина и измеряют светопоглощение при 305 нм после осветления раствора центрифугированием. Описанная методика позволяет определять 4—120 ppm сульфатов. Мешающее действие 100 ppm фторидов, нитратов и фосфатов при определении 50 ppm сульфатов выражено в слабой степени.
Как и 4-амино-4'-хлоробифенил, 2-аминопиримидин рекомендован для определения сульфатов в дождевых и поверхностных водах. Для синтеза реагента можно воспользоваться методикой [Н4] или ее улучшенным вариантом [116].
Для колориметрического определения сульфата предложен хлоранилат бария (этот реагент можно применять также для определения хлоридов и фосфатов) [117]. Метод основан на следующей реакции:
Y + МА (тв) —» А" + MY (тв) где Y — определяемый нон; А" — хлоранилат-ион
При определении сульфата нерастворимый хлоранилат бария реагирует с сульфатом, образуя сульфат бария и анион хлоранилата в количестве, эквивалентном содержанию сульфата. Анион хлораниловой кислоты (2,5-дихлоро-3,6-диокси-л-хинона) обладает интенсивной окраской с широким максимумом светопоглощения при 530 нм:
SOV + BaQClA + Н+ —> НС5С12О; 4- BaSO,
539
Реакцию проводят в 50%-ном этаноле с буферным раствором с pH = 4. Буферный раствор необходим, поскольку хлораниловая кислота обладает pH-индикаторными свойствами. В присутствии этанола чувствительность определения сульфатов увеличивается до 2 ppm.
Определению сульфатов описанным методом мешают катионы, образующие нерастворимые хлораннлаты, и поэтому их предварительно удаляют катионообменным методом. Не мешает определению сульфатов до 100 ррш хлоридов, нитратов, фосфатов и оксалатов. Дальнейшими исследованиями [118] установлено, что верхний предел содержаний фторидов и фосфатов при определении сульфата составляет 10 мг. Предложенный метод имеет ряд недостатков. Неприемлемо высокие результаты контрольного опыта отмечались, например, при анализе пластиков [119]. Рекомендованный фталатный буферный раствор заменен другими растворами, так как он не обеспечивает требуемого значения pH после операции катионного обмена [146, 147]. На воспроизводимости методики сильно сказывается способ приготовления реагента [120, 121]. Быт выполнен математический анализ имеющихся в этой методике равновесий [122], и это вызвало дальнейшие дискуссии [120, 123].
Молярный коэффициент поглощения хлораниловой кислоты при 280—350 нм значительно выше, чем при 530 нм [124]. Другим возможным источником ошибок при измерениях в ультрафиолетовой части спектра являются изменения ионной силы растворов [125].
Несмотря на приведенные критические замечания, метод находит широкое применение. Его используют для определения сульфатов в почвах [126], моче |127], пищевых продуктах [64] и различных водах [128]. Разработан автоматический метод определения 5—400 ррш сульфатов с использованием хлоранилата бария в качестве реагента [129].
Для определения сульфата предложен бариевый комплекс 3,6-диокси-2-метил-п-бензохинона, который сходен с хлораниловой кислотой. В этом случае выделяющийся анион определяют по его светопоглощению при 515 нм; метод позволяет определять 5— 70 мкг сульфатов |130].
Описан метод определения сульфата, основанный на экстракции хлоранилатного иона в присутствии трис (1,10 фенантролина) железа(II) нитробензолом [131]. Светопоглощение при 516 нм пропорционально концентрации хлоранилат-иона, что позволяет определять сульфаты в интервале концентраций 4-Ю-6 — 4-Ю-5 М. Определению сульфатов предложенным методом не мешают фосфаты, фториды, гексацианоферраты(II), нитриты, сульфиты и ацетаты. Ионы металлов, за исключением ионов щелочных металлов, мешают определению сульфатов этим методом. Хлоранилаты можно применять и для определения других анионов [132].
Восстановление сульфата до сульфида с последующим определением последнего в виде метиленового синего служит основой
540
одного из наиболее чувствительных косвенных методов определения сульфата. Образование метиленового синего происходит при реакции H2S с кислым раствором n-аминодиметиланилина в присутствии ионов железа(Ш). Для восстановления сульфата предложено несколько восстановителей: смесь титана с фосфорной кислотой, иодистоводородная кислота и раствор фосфорнова-тистой кислоты в уксусной кислоте [97, 98]. Различные аспекты этого метода рассмотрены в обзорах и монографиях [98, 133, 134].
При анализе азотсодержащих материалов описанным методом результаты определения сульфатов часто бывают заниженными. Это связано с образованием легколетучих соединений, мешающих реакции H2S с п-аминодиметиланилином [98, 135]. Мешающее действие азотсодержащих соединений можно удовлетворительно устранить с помощью обработки анализируемого раствора ацетатом цинка [136]. Обработанный раствор упаривают досуха и остаток прокаливают при 320 °C в течение 1 ч. Обработка основана на том, что нитрат цинка разлагается в процессе прокаливания, а сульфат остается неизменным.
Приведенный метод определения сульфатов очень чувствителен (молярный коэффициент поглощения метиленового синего 34 000), но образующаяся окраска нестабильна и быстро бледнеет на дневном свете. Возможная область применения метода очень ограничена, к тому же он ненадежен [135]. Аналитические характеристики этого метода можно улучшить, применив пиридин [137]. -Несомненно, восстановление сульфата до сульфида служит хорошей основой для разработки простого, селективного и чувствительного метода определения сульфата. Количественная отгонка сероводорода и его поглощение щелочными растворами легко осуществима. Помимо метода, основанного на образовании метиленового синего, для определения образующихся сульфидов предложены и другие спектрофотометрические методы, например основанный на использовании трис(1,10-фенантролина)железа(П) [138].
Несколько методов определения сульфатов основано на реакциях замещения лигандов в координационной сфере ионов бария, тория и циркония. При реакции сульфата с растворами или суспензиями комплексных соединений этих металлов происходит замещение окрашенных лигандов в них и эти лиганды можно определить спектрофотометрически. До недавнего времени не было разработано прямых методик, основанных на этом принципе. Описан метод определения сульфатов, основанный на измерении све-топоглошепия комплекса FeSOl при 325—360 нм [139]. Этот метод позволяет экспрессно определять сульфаты в интервале концентраций 10—500 мг/л, низкие концентрации фосфатов, фторидов, железа (III) и других ионов не мешают определению сульфатов, что позволяет использовать предполагаемый метод для анализа природных вод. При анализе рассолов полученные результаты нуждаются в корректировке.
6-11
Для определения сульфатов предложен косвенный метод, основанный на применении молибдата бария [140]:
SOJ- + ВаМоО, —> BaSO4 + MoOf
Образующийся молибдат связывают тиогликолевой кислотой и измеряют светопоглощение при 365 нм. Для уменьшения растворимости сульфата бария к раствору добавляют этанол. Калибровочный график линеен при концентрации сульфата ниже 25 мкг/мл.
Описан аналогичный метод определения сульфатов, основанный на использовании хромата бария [141], метод применен для анализа почв. Фосфаты мешают определению сульфата, но, если соосадить их с карбонатом кальция, можно определять до 1 мг сульфата в присутствии 500 ppm фосфатов [142].
Ионы бария образуют комплекс (1:2) с анионом родизоната. Этот комплекс имеет максимум светопоглощения при 480 нм. На образовании этого комплекса основан метод определения сульфата [143]. Сначала проводят реакцию сульфата с ярко-красной суспензией родизоната бария при pH = 2—3,8. В этой реакции осаждается сульфат бария с выделением эквивалентного количества родизоновой кислоты. Чувствительность этого метода 0,5— 0,8 мкг/мл сульфата.
Восстановление сульфатов до сульфидов с последующим определением сульфидов уже обсуждено ранее. Начальные стадии определения сульфатов, основанного на этой процедуре, протекают вполне удовлетворительно. Однако следующее за восстановлением и отделением H2S спектрофотометрическое окончание обладает рядом недостатков. Поэтому предложено [138] заменить эту стадию восстановлением железа(III) сероводородом в присутствии 1,10-фенантролина. Образующееся при восстановлении железо(П) дает стабильные комплексы с реагентом. Спектрофотометрическое определение интенсивно окрашенных комплексов позволяет проводить определение образованного H2S и, следовательно, первоначально присутствовавших сульфатов с высокой чувствительностью (молярный коэффициент поглощения 11000). Определению сульфатов мешают только нитриты и другие ионы-восстановители (из более чем 20 наиболее распространенных ионов).
Предложен метод определения сульфатов, основанный на использовании ионообменной смолы для обмена сульфата с тиоцианатом и последующем определении тиоцианата экстракцией его комплекса с метиленовым синим 1,2-дихлорэтаном при pH = 1 [144];
2SCN' + SOf <=* SO2/ + 2SCN" (смола) (раствор) (смола) (раствор)
Для анализа концентрированной (85—99%) серной кислоты предложен быстрый и простой спектрофотометрический метод [145]. В качестве реагента в этом методе используют хинализарин (1,2,5,8-тетраоксиантрахинон). Концентрацию кисдоты определяют
542
по соотношению светопоглощенпя комплекса прн 535 и 630 нм, правильность определения ±0,3%.
Для определения сульфата в индустриальных водах используют фотометрическое титрование сульфата хлоридом бария с тороном в качестве индикатора [65].
Спектрофотометрическое определение сульфата с использованием хлоранилата бария [117]
Реагенты
Хлоранилат бария, торговый продукт. Может быть приготовлен следующим образом: смешивают 1 л 0,1%-ного раствора хлораниловой кислоты с 1 л 5%-ного раствора хлорида бария и оставляют смесь на ночь (прн комнатной температуре). Промывают осадок на фильтре водой до полного удаления хлорид-нонов. Промывают осадок трижды этанолом н одни раз — днэтиловым эфиром, отделяя его каждый раз от жидкости центрифугированием. Высушивают остаток прн 60 °C в течение 1 ч прн пониженном давлении.
Буферный раствор, pH = 4,0, 0,05 М раствор гндрофталата калия реактивной квалификации.
Смола Dowex 50X8 с размером частиц-297—841 мкм в Н-форме.
Ход анализа. Пропускают анализируемый водный раствор, содержащий сульфат, через колонку диаметром 1,5 см н длиной 15 см, содержащую катнонооб-менную смолу. Доводят pH раствора, прошедшего через колонку, до 4 при помощи разбавленной НС1 или NH«OH
Аликвотную часть анализируемого раствора, объемом ие более 40 мл и содержащую не больше 40 мг сульфатов, помещают в мерную колбу емкостью 100 мл, прибавляют 10 мл буферного раствора и 50 мл 95%-ного этанола. Разбавляют раствор до метки дистиллированной водой, прибавляют 0,3 г хлоранн-лата бария и встряхивают содержимое колбы в течение 10 мни. Удаляют избыток хлоранилата бария н осажденный сульфат бария фильтрованием или с по-| мощью центрифуги.
Светопоглощение раствора измеряют при 530 нм относительно контрольного раствора, приготовленного по выше приведенной прописи, но не содержащего сульфатов. Для построения калибровочного графика используют сульфат калия.
Примечание. Растворы подчиняются закону Бугера — Ламберта — Бера вплоть до концентрации сульфатов 400 мкг/мл. Для измерения светопоглоще-ння применяют кювету с толщиной поглощающего слоя 1 см. Для кювет с толщиной 5 см чувствительность определения 2 ppm.
Турбидиметрические и нефелометрические методы
Отсутствие совершенно удовлетворительных методов определения малых концентраций сульфатов дает основания для широкого исследования в области нефелометрических и турбидиметрических методов анализа. В ранних работах в этой области были исследованы суспензии сульфата бария, но в последнее время разработаны лучшие методики с использованием органических реагентов. Описан турбидиметрический метод, основанный на прибавлении раствора хлорида бария к подкисленному раствору сульфата. Для получения однородного мелкого осадка применяют различные стабилизирующие агенты, например крахмал. При сравнении турбидиметрического метода с другими методами определения серы в пищевых продуктах, основанных на окислении серы в кислороде,
543
установлено следующее [64]: турбидиметрический метод определения сульфатов можно применить, если не требуется высокая точность определений и можно пренебречь помехами, вызываемыми фосфатами, железом, кальцием и магнием. Сообщается [148], что результаты, достигаемые турбидиметрическим методом, можно оценить как хорошие.
Турбидиметрический метод можно применить для определения сульфатов в различных материалах. Помимо определения сульфатов в пищевых продуктах метод можно использовать для анализа заводских материалов 1149] и вод, для анализа которых турбидиметрический метод остается стандартным рутинным методом [150].
При турбидиметрическом анализе интенсивность света измеряют в направлении падающего луча. В нефелометрии, которая характеризуется, в общем, большей чувствительностью, чем турбидиметрия, рассеянный осадком свет измеряют под углом (обычно 90°) к направлению падающего света. Как и в турбидиметрии, в нефелометрическом анализе характеристики методик зависят от физических свойств частиц осадка. Предложено несколько методов определения сульфатов, основанных на низкой растворимости сульфата бария, особенно в смешанных растворителях (вода и спирты).
Проведено всестороннее изучение методов анализа, основанных на осаждении сульфатов ионами бария [151]. В более поздних работах рассмотрены возможности использования других осадителей сульфата. Например, высокую чувствительность определения сульфатов обеспечивает применение 4-амино-4'-хлоробифенила [103, 112]. В этом случае образуется высокодисперсный осадок, обладающий хорошими характеристиками как для нефелометрического, так и турбидиметрического анализа. Полуколичественная методика применена для анализа пластических материалов [152]. Более детальное изучение системы позволило предложить нефелометрический метод для определения 2,5—25 ppm сульфатов в 10 мл раствора [153]. Гуммигут —полисахарид, содержащий L-арабинозу, D-ксилозу, D-галактозу, D-маннозу и глюкуроновую кислоту, предложен в этой работе в качестве стабилизатора осадка. Присутствующие в анализируемом растворе фосфаты необходимо удалить, например, в виде MgNH4PO4. Результаты серии определений 5,0—30,0 ppm сульфатов показывают, что средняя погрешность определения составляет ±2,8%. В работе [153] описан также способ приготовления из гуммигута улучшенного стабилизатора осадка. Использование этого стабилизатора позволяет улучшить чувствительность определений и определять сульфат при содержаниях 1—25 ppm. Описан метод определения сульфатов в присутствии бария, в соответствии с этим методом маскируют барий при помощи ЭДТА [154].
Изучение 4-амино-4'-хлоробифенила не подтвердило предположений о канцерогенности этого соединения, и поэтому его можно использовать в аналитической практике без ограничений.
544
Для нефелометрического определения сульфатов предложен новый реагент — 2-ампноперпмпдин [114]. Этот реагент был упомянут в разделе, посвященном спектрофотометрическому определению сульфатов. Гетероциклическое соединение перпмпдпн образует слаборастворимые соли с некоторыми кислотами, однако это явление более характерно для его 2-аминопроизводных. Осаждая сульфат 2-аминоперимидина, можно обнаружить до 0,05 рргп сульфата. Суспензии осадка очень стабильны и прибавление стабилизаторов не требуется.
Подобно другим солеобразующим реагентам 2-аминоперими-дин не позволяет определять сульфаты с высокой селективностью, поскольку при высоких концентрациях многие ионы образуют осадки с этим реагентом. Однако в условиях определения сульфата большинство наиболее распространенных анионов может присутствовать в анализируемом растворе по крайней мере в 10-кратном избытке по отношению к концентрации сульфата. Этот метод особенно пригоден для определения микросодержаний сульфатов в природных водах [153].
Растворимость сульфата 2-аминоперимидина составляет 0,020 г/л, тогда как растворимость сульфатов бензидина и 4-ами-но-4' хлоробифенила составляет 0,098 и 0,155 г/л соответственно. Помимо снижения растворимости сульфата органического основания на увеличении чувствительности определений в еще большей степени сказывается увеличение молярного коэффициента поглощения реагента.
Способы синтеза реагента и его применения для определения сульфата описаны в литературе [153], опубликованы материалы по улучшенному варианту синтеза 2-аминоперимидина [116].
Нефелометрическое определение сульфата
с применением 2-аминоперимидина [114]
Реагенты и аппаратура
Раствор гидрохлорида 2-аминоперимидина, растворяют 0,5 г гидрохлорида 2-аминопернмндина в 100 мл теплой дистиллированной волы, фильтруют теплый раствор и помещают его в склянку из темного стекла. Реагент чувствителен к окислителям и прн хранении его растворов на свету и при доступе воздуха в течение нескольких дней происходит частичное окисление реагента. Если раствор реагента хранят в закрытой склянке и темного стекла, то окисление резко замедляется и реагент можно использовать по крайней мере в течение четырех суток, при этом в точности сохраняется калибровочный график.
Раствор сульфата калия, 10 ppm. Растворяют 18,14 мг свежеподсушенной соли реактивной степени чистоты в 1 л свежеперегнаннон дистиллированной воды
Нефелометр EEL ("Evans Electroselenium Ltd.”, Essex, England). Круглые кюветы с внутренним диаметром 10 мм и емкостью 12 мл.
I '• Ход анализа. При определении 0—5 ppm сульфатов в пять мерных колб емкостью 10 мл помешают 1—5 мл стандартного раствора сульфата Разбавляют содержимое колб до 5 мл дистиллированной водой, прибавляют по 4 мл раствора гидрохлорида 2-аминопернмиднна н разбавляют содержимое колб до метки. Перемешивают содержимое колб н оставляют стоять суспензии в течение 5—10 мин. Помещают в кювету нефелометра контрольный раствор, не содержащий сульфатов, и устанавливают барабан нефелометра на нуль. Помещают в
18 Зак. Г67
545
кювету раствор с максимальным содержанием сульфата и подбирают чувствительность прибора таким образом, чтобы показания его составляли 80% от верхнего значения шкалы. Помещают в кювету нефелометра каждый нз приготовленных растворов н измеряют его светорассеяние При низкой чувствительности прибора дистиллированная вода н контрольный раствор (раствор реагента) должны давать одинаковые показания. Строят калибровочный график (показания гальванометра— содержание сульфата в 10 мл раствора).
Прн определении 0—I н 0—0,5 ppm сульфатов предусмотрена точно такая же методика, но с более высокой чувствительностью прибора.
Примечание. Реагент можно регенерировать из использованных растворов следующим образом. В емкость для отстаивания, содержащую 5%-ный раствор сульфата натрия, сливают все растворы и суспензии, содержащие реагент. Осадок сульфата 2-амннопернмнднна седиментирует, а образовавшуюся осветленную жидкость периодически декантируют. Прибавляют дополнительно раствор сульфата, чтобы поддержать его концентрацию в водной фазе на достаточном уровне. Когда накапливаются значительные количества осадка, его фильтруют через большую стеклянную фильтрующую воронку, промывают водой, после чего суспендируют осадок в воде. К суспензии прибавляют 4 М раствор NaOH, смесь тщательно встряхивают и фильтруют выделившееся основание 2-аминопернмндина. Растворяют его в минимальном количестве горячего ацетона. Ацетоновый раствор обрабатывают значительным объемом 4 М раствора НС1 для полного осаждения гидрохлорида 2-аминопернмидина. Осадок фильтруют, высушивают, промывают на фильтре небольшим объемом ацетона и затем днэтнлового эфира и наконец высушивают на фильтре.
Продукт можно дополнительно очистить следующим образом. Растворяют гидрохлорид 2-амннопернмнднна в 40-кратном (по массе) количестве горячей воды, фильтруют горячий раствор и обрабатывают его насыщенным раствором хлорида калия вплоть до достижения полного осаждения очищаемого продукта. Осадок фильтруют, промывают разбавленным раствором хлорида калия и затем ацетоном н днэтнловым эфиром, как описано выше. Гидрохлорид получается в виде серовато-белого кристаллического порошка, легко растворяющегося в теплой воде. Раствор реагента при взаимодействии с 0,05%-ным раствором сульфата калия дает шелковистый белый осадок.
Спектрофлуориметрический метод
Описано несколько флуориметрических методов определения сульфатов. Предложен метод, основанный на ингибирующем действии сульфатов на флуоресценцию комплексного соединения тория с морином в 80%-ном этанольном растворе при pH = 2,35 [155]. Чувствительность метода высока, однако серьезные помехи определению оказывают некоторые ионы, в том числе фториды, фосфаты, вольфраматы, молибдаты, мышьяк(III), железо(III) и алюминий(III). Другой спектрофлуориметрический метод определения сульфатов основан на усилении ими флуоресценции комплексного соединения циркония с кальцеином. Флуоресценцию возбуждают при 350 нм и измеряют ее при 410 нм. Описанный метод применен для определения 0,2—12 мг сульфатов [156]. Определению сульфатов мешают фториды, фосфаты и некоторые другие ионы.
Метод ИК-спектроскопии
Некоторые многоатомные анионы, и в том числе сульфат, имеют характеристические полосы в ИК-спектре, которые позволяют идентифицировать и определять эти анионы. Предложен
546
метод определения до 20 мкг сульфатов в присутствии гексацианоферрата (II) и (III), гексацнанокобальтата(1П), метаванадата и хромата с использованием дисков из бромида калия толщиной 1 мм [157].
Изучение правильности и селективности анализа водных растворов, содержащих нитрат, нитрит и сульфат, методом ИК-спек-троскопии показало [158], что при определении 1—50 мг/мл сульфатов относительная ошибка определения составляет около 3%. Значительные помехи определению сульфатов оказывают органические соединения, соли аммония, бораты, карбонаты, фосфаты и перхлораты. Применение этого метода весьма ограничено. Для определения сульфатов бария, стронция, натрия и калия используют диски из бромида калия [159]. Высоты пиков BaSO4 и SrSO4 измеряют при 983 и 993 см-1 соответственно, пики Na2SO4 и Кг5О4 измеряют при 619 см-1. Сульфаты бария и стронция можно определить в их смесях при соотношении компонентов от 20:1 до 1 :20. Карбонаты и нитраты не мешают определению, но присутствие гигроскопичных соединений натрия и калия приводит к помутнению дисков, что ухудшает правильность получаемых результатов. Описан метод определения сульфатов в присутствии 400-кратного избытка нитратов и 60-кратного избытка нитритов [160].
Атомно-абсорбционный метод
Определение элементов, присутствующих в водных растворах в виде анионов, методами пламенной спектроскопии затруднено. Это связано с тем, что длины волн как абсорбционных, так и эмиссионных линий этих элементов соответствуют области вакуумного ультрафиолета. В руководстве [161] по пламенной спектроскопии рассмотрено определение следующих неметаллов: S, В, Si, Р, As, Se, Те, Cl, Вг и I. Поскольку прямое определение неметаллов связано с трудностями, часто используют косвенные методы анализа. В этом случае для определения сульфатов применяют обычно барий (II).
Пламенную фотометрию для определения сульфатов используют обычно в двух вариантах: с помощью определения избытка бария после осаждения его сульфата или определения ионов бария, образуемых осадком. Первый вариант применяют для определения до 200 ppm сульфатов (применено измерение интенсивности линии бария 499,3 нм) [162]. Такая же методика использована для анализа воды и почвенных экстрактов [163, 167], некоторые образцы в этом случае содержали до 1000 мг/л ионов натрия, калия и магния. Описаны методы, основанные на определении бария в осадке сульфата бария, например, отделяют осадок от раствора с помощью центрифуги, сульфат бария суспендируют в 1%-ном растворе крахмала и анализируют суспензию методом пламенной фотометрии [164]. В модифицированной методике
18*
547
[165] осадок растворяют в растворе ЭДТА, преимущество этой методики связано с тем, что здесь отсутствует тенденция взвеси к седиментации. Описанные методы определения сульфатов использованы для анализа детергентов [165, 166] и других объектов |168].
Ранние работы по определению сульфатов методом пламенной фотометрии были осложнены наличием большого числа помех и относительно низкой чувствительностью определения бария. В работе [166] предложено использовать пламя ацетилена с оксидом азота(I) для определения 0,5—10 ppm сульфатов; определению сульфатов мешает только кальций. Методика определения очень проста и включает осаждение сульфата бария в 50%-ном растворе пропан-2-ола, что снижает растворимость осадка до приемлемого уровня и обеспечивает более стабильное введение образца в пламя.
Для определения сульфата можно использовать метод, основанный на осаждении сульфата свинца в этанольном растворе [169]. При последующем атомно-абсорбционном определении измеряют светопоглощение при резонансной линии свинца 283,3 нм.
Новое важное направление в прямом определении сульфатов методом атомно-абсорбционной спектроскопии связано с применением микроволновых безэлектродных разрядных ламп и пламени оксида азота(I) с ацетиленом, изолированного от азота [170]. С помощью этого метода можно определить 50—700 ppm сульфатов, измеряя светопоглощение при резонансной линии серы 180,7 нм. На сигнал 200 ppm серы не оказывает никакого влияния присутствие 50-кратного избытка Al3+, Cu2+, К\ Mg2+, Мп2+, MoVl, Na*, Ni2*, Zn2*, F", POt, СГ, Вг и Г.
Метод молекулярной эмиссии в полости
Интерес вызвало обнаружение молекулярной эмиссии ассоциата S2 в области 394 нм. Для генерации этого ассоциата использовано холодное диффузное пламя [171]. Это пламя возникает при разбавлении диффузного пламени водорода в воздухе азотом или при горении водородно-воздушного пламени внутри объема с охлаждаемой поверхностью. Применение азота (или аргона) не только уменьшает температуру пламени, но и вызывает значительное снижение эмиссии фона вследствие понижения содержания таких радикалов, как ОН’. Для превращения серусодержащих соединений, включая сульфаты, в S2 наиболее благоприятной является температура 390°C. На рис. 63 представлен эмиссионный спектр S2. При использовании монохроматоров чувствительность определений значительно снижается и поэтому для определения используют обычные фотометры со светофильтрами. Такая замена приводит к повышению чувствительности определений в 100 раз, и значение ее достигает 0,08 ppm серы в водных растворах [172]. Большинство катионов мешает
548
Рис. 63. Эмиссионный спектр S2, полученный распылением 5-10 3 М раствора диоксида серы в водород-азотное диффузное пламя [171].
определению (депрессирующее влияние, вызванное матричным эффектом). Для преодоления этой трудности предварительно используют ионообменное отделение мешающих примесей.
Серусодержащие соединения, включая сульфаты, можно определять методом молекулярного эмиссионного резонанса [173— 175]. Экспериментальные подробности, посвященные этому методу, приведены в разделе, описывающем определение сульфидов.
Наблюдаемый сигнал сульфата зависит от природы присутствующего катиона. Если сульфаты присутствуют в виде соединений с катионами Na*, NH4, Fe2*, Мп2* и Н*> то интервал времени между введением образца в пламя и достижением максимальной эмиссии (/макс) увеличивается с ростом термической стабильности соединений:
Na* > Fe2* > Мп2* > NH4* > H*
Интенсивность эмиссии уменьшается с ростом /Макс Найдено, что прибавление Н3РО4 к анализируемым сульфатам металлов усиливает сигнал S2, вероятно, из-за образования присутствующими катионами трудно разлагаемых в пламени солей. Максимальное значение сигнала сульфатов наблюдается в среде 0,10 М Н3РО4. Сульфат и сульфид дают хорошо разрешаемые сигналы, что позволяет определять их при совместном присутствии. Метод молекулярного эмиссионного резонанса позволяет определять нано-граммовые содержания сульфатов при объеме образца несколько микролитров.
549
Электрохимические методы
Потенциометрические методы
Достижения аналитической химии в последние годы во многом связаны с внедрением в аналитическую практику ионоселектив-ньгх электродов [176]. Для определения сульфатов можно предложить по крайней мере четыре различных типа ионоселективных электродов: 1) свинцовый селективный электрод, обеспечивающий потенциометрическую индикацию конечной точки титрования сульфатов стандартными растворами солей свинца; 2) мембранный электрод, импрегнированный сульфатом бария; 3) мембранные электроды, в состав которых входят компоненты: сульфат свинца, сульфиды свинца, серебра и меди; 4) электроды, действие которых основано на равновесии в системах, содержащих железо(Ш) и сульфат. Электроды первых двух типов применяли довольно широко, тогда как электроды третьего и четвертого типа предложены совсем недавно.
Для потенциометрического титрования сульфатов использован свинцовый селективный электрод [177] типа ORION 94-82 Pb в сочетании со стандартным электродом ORION 90-02. Для понижения растворимости сульфата свинца применяли диоксан, который в этом отношении оказался более эффективным, чем этанол. Кривая титрования сульфата в 50%-ном диоксановом растворе перхлоратом свинца изображена на рис. 64. Чувствительность определения сульфатов этим методом ограничена растворимостью сульфата свинца и составляет 5-Ю-5 М.
Определению сульфатов с использованием свинцового селективного электрода мешают Cu2+, Hg2+, Ag+ й 10-кратный (молярный) избыток хлоридов, нитратов и СО2. Мешающее влияние СО2 можно устранить доведением pH образца до 4. При изменении pH в интервале 3,0—6,5 в отсутствие карбонатов кривые титрования сульфата перхлоратом свинца остаются неизменными.
Свинцовый селективный электрод применен для титрования сульфатов в минеральных и морских водах [178]. Образцы первоначально пропускают через ионообменную колонку с катионитом в Ag-форме для удаления хлорид-ионов, а затем через такую же колонку, заполненную катионитом в Н-форме для удаления избытка серебра. Этот метод позволяет определить 20—3000 ppm сульфатов. Упомянутый электрод использован для микро- и полумикроопределений серы в органических соединениях [179]. В этой работе присутствующий фторид отделяют либо прибавлением борной кислоты, либо кипячением с хлорной кислотой. Другие галогены и азот не мешают определению серы, но фосфор должен отсутствовать. Результаты анализа 10 различных соединений показывают, что погрешность определения не превышает ±0,3% (абсолютные значения). Свинцовый селективный электрод применен для определения серы в нефти [180] и в растворах, используемых для никелирования [181].
550
Рнс. 64. Кривая потенциометрического титрования мВс сульфатов в 50%-ном растгоре дноксаиа перхлоратом свинца с применением свинцового ноно- -W -селективного электрода; и
I—титрование 100 мл 10~3 Л1 раствора NajSOi Ю~2 М раствором PbfCIOpj; 2 — титрование 100 мл 5 • 10~5 М раствора NazSO, 5 • I0~ М раствором РЬ(С1О4>2 [177]. -ДО
Мембранный электрод, импрегниро- 1Z0 ванный сульфатом бария, первоначально был предложен в работе [182], затем до было проведено тщательное изучение его характеристик [183]. Калибровочный график для этого электрода линеен в интервале 101 — 10 4 М сульфата, воспроизводимость достигает ±5%, однако при содержании хлоридов выше 10 3 М наблюдаются помехи, поэтому электрод может иметь ограниченное исполь
160
-zoo
210
IW 0
2 6 6 I 9 И ft О(пем тиглранто., мл
зование.
Проведенные исследования показали, что для описанного электрода трудно достичь высокой селективности [184], и поэтому работы по поиску селективных электродов для определения сульфатов были продолжены. Описаны электроды, в состав которых входят сульфат свинца, сульфиды свинца, серебра и меди [184, 185]. В статье [125] приведены сведения об изготовлении и свойствах электрода. Электрод позволяет получить линейный калибровочный график и обеспечивает селективное определение сульфатов. Следует отметить, что механизм действия электрода пока
остается неясным.
Электрод четвертого типа предложен недавно, в этой монографии он описан, поскольку работа его основана на новом принципе. Этот электрод изготовляют из стекла Fe-1173 (халькогенидное
стекло состава Ge28Sbi2Se6o, легированное железом). Его работа основана на равновесии комплексообразования железа(III) с сульфатом. Сигнал электрода определяется концентрацией железа (III). При наличии ионов бария в растворе комплексы железа (III) с сульфатом разрушаются из-за осаждения BaSO4 и возникает электродный сигнал, пропорциональный содержанию ионов бария. Этот электрод можно использовать для индикации конечной точки при титровании анализируемого раствора сульфата стандартным раствором соли бария [186, 187].
Помимо методов, основанных на использовании ионоселективных электродов, для определения сульфатов предложены и другие потенциометрические методы. Описан быстрый метод определения сульфатов, нитратов и хлоридов в смесях [111]. Первоначально анионы превращают в соответствующие кислоты с помощью ионного обмена. Все три кислоты титруют потенциометрически гидроксидом бария. Затем, титруя раствор пальмитатом калия, определяют суммарное содержание нитрата и хлорида. Наконец,
551
титруя раствор нитратом серебра, определяют содержание в нем хлорида.
Для определения 1—6 мг сульфатов описан следующий метод [188]. К анализируемому раствору прибавляют избыток стандартного раствора ацетата бария, после чего титруют неизрасходованный ацетат хлорной кислотой в среде ледяной уксусной кислоты. Для устранения мешающего действия воды к титруемому раствору прибавляют уксусный ангидрид. Эта методика использована для определения сульфата в реакторном топливе, определению сульфатов с ее помощью не мешает UO1+ и Fe3+ и другие ионы.
Предложен метод титрования сульфатов смесью нитратов бария и свинца с поляризованным висмутовым электродом [189]. Титрование сульфатов этой смесью можно сочетать с рН-метрпче-ской индикацией конечной точки титрования (использованы стеклянный и каломельный электроды), при титровании pH изменяется на 1—3 единицы [190]. Определению сульфатов этим методом мешают фосфаты, дифосфаты, сульфиды, сульфиты, тиосульфаты и ацетаты.
Амперометрические методы
Первоначально определение сульфата амперометрическим методом проводили, титруя анализируемый раствор сульфата раствором нитрата свинца [191]. Правильность определения составляет ±0,3%, определению мешают высокие концентрации калия, кальция и хлорид-иона. Для автоматического амперометрического определения сульфата предложен метод [192], основанный на использовании стационарного ртутного электрода (в качестве электрода сравнения использован хлорсеребряный электрод). Метод позволяет титровать 0,001 М растворы сульфатов (титрант — 0,002 М раствор нитрата свинца).
Полярографические методы
Сульфат непосредственно не восстанавливается на ртутном капельном электроде, но описано несколько косвенных полярографических методов определения сульфатов, основанных на использовании сульфата свинца [193—195]. В одном из этих методов [195] осаждают 0,2—2 мМ сульфата нитратом свинца в смешанной среде вода — ацетон или вода — диоксан (последнюю смесь используют в случае присутствия хлоридов в анализируемом растворе) . Осадок сульфата свинца растворяют в насыщенном растворе хлорида аммония и ионы свинца в растворе определяют полярографически.
Аналогичный метод можно использовать, определяя избыток бария после осаждения его сульфата. Метод применен для определения сульфата после сожжения органических серусодержащих соединений в кислороде [196].
552
в полярографических методах определения сульфатов часто требуется их предварительное концентрирование. Избежать этой стадии можно, например, с помощью метода, основанного на обмене сульфата на хромат [197]. Этот обмен осуществляется с использованием хромата бария в качестве реагента. Метод применен для анализа вод, содержащих такие компоненты, как железо (II) и сульфиды.
Интересная полярографическая методика [198] основана на замещении сульфата эквивалентным количеством хлоранилат-иона, который можно определить полярографически.
Для определения сульфата предложена осциллографическая полярография [199].
Кондуктометрические методы
Для определения сульфата при его содержании 2—5 мг можно применить кондуктометрический метод [200]. Метод основан на осаждении сульфата бария избытком стандартного раствора ацетата бария в среде уксусной кислоты и последующем кондуктометрическом титровании неизрасходованного ацетата стандартным раствором хлорной кислоты. Кондуктометрическое титрование сульфатов можно применять в разных областях науки и техники. Например, этот метод использован для анализа клинкера портландцемента (после предварительного пропускания раствора образца через катионообменную колонку) [201]. Сульфат в виде H2SO4 титруют стандартным раствором КОН, используя высокочастотное кондуктометрическое титрование.
Кинетические методы
Реакция между ионами циркония и метилтимоловым синим в кислом растворе протекает очень медленно. Некоторые анионы, например, сульфаты, фториды и фосфаты катализируют эту реакцию, вероятно, благодаря лабилизации координационной сферы циркония. На этом принципе основан каталитический метод определения сульфатов при их содержании 0,1—2,4 ppm [202]. Важно заметить, что раствор соли циркония можно использовать в течение ограниченного периода времени. Определению сульфатов мешают многие катионы, но предварительное пропускание анализируемого раствора через колонку, заполненную катионообменной смолой, устраняет эти помехи. Мешающее действие эквимолярных концентраций мышьяка(У), фторида и фосфата устраняется предварительной обработкой анализируемого раствора оксидом магния.
Радиохимические методы
Описан [203] метод определения сульфата, основанный на использовании хромата бария, меченного изотопом 51Сг. Для определения 2—100 мкмоль (60—3200 мкг) сульфатов предложен
553
метод, основанный на пропускании анализируемого раствора через колонку, заполненную йодатом бария, меченным изотопом ,3Ч и последующем измерении у-активности фильтрата [204]. Описанный метод прост, экспрессен и обеспечивает воспроизводимость ±2,5%. Помех при определении сульфатов этим методом не обнаружено. Сульфат можно определить методом субстехиометрического изотопного разбавления [205]. При предварительной ионообменной обработке превращают сульфат-ионы в H2SO4, к которой прибавляют H2SO4, меченную изотопом 35S. Осаждают сульфат нитратом бария и измеряют 0-активность. Метод позволяет определять 10—100 мкг сульфатов.
Другие методы определения
В последние годы вновь появился интерес к термометрическому титрованию. Его использование при определении сульфата основано на экзотермическом характере реакции осаждения сульфата бария. Термометрический метод определения сульфатов был предложен более 50 лет назад [206]. Для прямого термометрического титрования сульфата можно применять перхлорат бария [207]. Этот метод позволяет определять 1—25 мг сульфатов, отличается экспрессностью и селективностью, однако результаты, полученные с его помощью, в значительной степени зависят от содержания этанола в титруемом растворе, кроме того, правильность описанного метода ниже правильности других инструментальных методов определения сульфатов. Другой термометрический метод определения сульфатов также основан на титровании анализируемого раствора ионами бария(II), точность определения не хуже 0,5% [208].
Для определения сульфатов применяют метод газовой хроматографии. Предложен метод определения сульфата, основанный на его восстановлении до сульфида при помощи 20%-ного раствора ZnCb в концентрированной ортофосфорной кислоте при 250—317 °C [209]. Образующийся H2S направляется газом-носителем (водородом) в газовый хроматограф на колонку длиной 2 м, заполненную силикагелем.
Для определения некоторых анионов, в том числе сульфатов, использовано образование ими триметилсилиловых производных [210, 211]. Метод синтеза этих производных основан на реакции аммонийных солей определяемых анионов с трифтор-бис (триме-тилсилил) ацетамидом в среде диметилформамида при 25°C. Для разделения анионов используют колонку длиной около 2 м, содержащую SE-30 Хромосорб W(HP) при 70—150°С. Установлено, что упомянутые производные не образуются натриевыми и калиевыми солями определяемых анионов, однако эти соли легко превращаются в аммонийные ионообменным методом. Гидросульфат и сульфит, который, вероятно, в условиях определения окисляется до сульфата, дают те же хроматографические пики, что и сульфат.
554
Время, мин
5:---'---1----1---1____L—. । । ।
7Z7 80 90 100 110 120 130ШО 150
Температура, °C
Рнс. 65. Хроматограмма трнметилсилнловых производных семи различных анионов, включая сульфаты [211]:
1 — фосфит; 2—карбонат; 3—оксалат; 4—сульфат; 5—ванадат; 6—фосфат; 7—арсенат.
На рис. 65 представлен пример разделения 7 анионов, в том числе сульфата, описанным методом.
ЛИТЕРАТУРА
1 Heinrich В. /., Grimes М. D., Puckett 1. Е. — In: Treatise on Analytical Chemistry (Ed. I. M. Kolthoff and P. J. Elving), Part If, Vol. 11, New York, Interscience, 1961, p. 1—35.
2 Haddock L. A. — In: Comprehensive Analytical Chemistry (Ed. C. L. Wilson and D. W Wilson), Vol. 1c, Amsterdam, Elsevier, 1962, p. 282.
3. Haff L. V. — In: Analytical Chemistry of Sulphur and its Compounds (Ed. J. H. Karchmer), Part 1, New York, Wiley-Interscience, 1970, p. 183.
4. Blasius E., Horn G., Knochel A., Munch J., Wagner H. — In: Inorganic Sulphur Chemistry (Ed. G. Nickless), Amsterdam, Elsevier, 1968.
5. Szekeres L. — Taianta, 1974, v. 21, p. 2.
6. Inczedy J. — Analytical Applications of Ion-exchange, Oxford, Pergamon, 1966, p. 135.
7. Рябинин А. И., Богатырев В. Л. ЖАХ, 1968, т. 23, с. 894; Analyt. Abstr., 1970, v. 18, р. 875.
8. Субботина А. И., Архангельская Е А., Петров А М. — Труды по химии и хим. техиол. (Горький), 1963, с. 118; Analyt. Abstr., 1965, v. 12, р. 1149.
9. Shiraishi N., Iba T., Morishige T.—Japan Analyst, 1965, v. 14, p. 450; Analyt. Abstr., 1967, v. 14, p. 126.
10. Iguchi A. — Bull. chem. Soc. Japan, 1958, v. 31, p. 600, Analyt. Abstr., 1969, v. 6, p. 1705.
11 Mikes J. A., Szanto J. — Taianta, 1959, v. 3, p. 105.
12. Nydahl E., Gustafsson L. — Acta chem. scand., 1953, v. 7, p. 143.
13. Nydahl E. — Analyt. Chem., 1954, v. 26, p. 580.
14 Fritz J. S., Yamamura S. S., Richard M. J. — Analyt. Chem., 1957, v." 29, p. 158.
15 Seiler H„ Erlenmeyer H. — Helv. chim. Acta, 1964, v. 47, p. 264.
16. Kelly D. P. — 3. Chromat., 1970, v. 51, p. 343
17. Moghissl A. J. — J. Chromat., 1964, v. 13, p. 542.
555
18 Canic V. Г) , Turcic M. N.. Bugarski-Vojinovic M. B. — Z. analyt Chem. 1967, v. 229, p. 93, Analyt. Abstr., 1968, v. 15, p. 5211
19. Kawanabe K., Takitani S., Miyazaki M, Tamura Z.—Japan Analyst, 1964, v. 13, p. 976; Analyt. Abstr., 1966, v. 13, p. 5385.
20. De Sousa A.— Inf. quim. Anal., Madrid, 1961, v. 15, p. 121; Analyt. Abstr., 1962, v. 9, p. 1454.
21. De Sousa A.— Inf. quim. Anal., Madrid, 1962, v. 16, p. 177; Chem. Abstr., 1964, v. 60, p. 22g.
22. Kolthoff I. M., Sandell E. B., Meehan E. J., Bruckenstein S. — Quantitative Chemical Analysis 4th edn., London, MacMillan Co., 1969.
23. Belcher R., Nutten A. J. — Quantitative Inorganic Analysis, 3rd edn.. London, Butterworths, 1970.
24. Laitinen H. A. — Chemical Analysis, New York, McGraw-Hill, 1960.
25. Pribil R., Marincova D. — Chemike Listy, 1952, v. 46, p. 542.
26. Nishimura M. — Analytica chim. Acta 1966, v. 34, p. 246.
27. Золотавин В. Л., Сонатина В. Н., Нестеренко В. П.—ЖАХ, 1966, т. 21, с. 1289; Analyt. Abstr., 1968, v. 15, р. 4513.
28. Плескач Л. И., Чиркова Г. Д—ЖАХ, 1970, т. 26, с. 2290; Analyt. Abstr., 1973, v. 24, р. 3362.
29. Liteanu С., Lingner Н.— Taianta, 1972, v. 19, р. 945.
30. Wagner W. F., Wuellner J. A. — Analyt. Chem., 1952, v. 24, p. 1031.
31. Vaubel W.— Z. analyt. Chem., 1896, v. 35, p 163.
32. Stephen W. I. — Ind. Chemist, 1961, v. 37, p. 551.
33. Mahr C., Kraus K. — Z. analyt. Chem., 1948, v. 128, p. 477.
34. Belcher R., Gibbons D. — J. chem. Soc., 1952, p. 4216.
35. Fritz J. S., Freeland M. Q. — Analyt. Chem., 1954, v. 26, p. 1593.
36. Fritz J. S., Yamamura S. S. — Analyt. Chem., 1955, v. 27, p. 1461.
37. Fritz J. S., Yamamura S. S., Richard M. J. — Analyt. Chem., 1957, v. 29, p 158.
38. Wagner H. — Mikrochim. Acta, 1957, p. 19.
39 Colson A. F. — Analyst, 1963, v. 88, p. 26.
40 Macdonald A. M. G. — Analyst, 1961, v. 86, p. 3.
41. Новикова К- Ф., Басаргин Н. Н., Цыганкова М. Ф.—ЖАХ, 1961, т. 16, с. 348; Analyt. Abstr., 1962, v. 9, р. 175.
42. Новикова К. Ф., Басаргин Н. п. —Труды комиссии по аналит. химии АН СССР, 1963, т. 13, с. 27; Analyt. Abstr , 1964, v. 11. р. 2175.
43 Archer Е. Е., White D. С., Mackison R. — Analyst, 1971, v. 96, p. 879.
44. Кузнецов В. И., Басаргин Н. Н. — Заводск. лаборатория, 1965, т. 31, с. 538; Analyt. Abstr., 1966, v. 13, р. 4801.
45. Акимов В. К-, Брагина С. И., Бусев А. И. — Вестник МГУ, Сер. хим., 1968, с. 70; Analyt. Abstr., 1970, v. 18, р. 1043.
46. Басаргин Н. Н., Новикова К- Ф-— ЖАХ, 1966, т. 21, с. 473; Analyt. Abstr., 1968, v. 15, р. 1143.
47. Басаргин Н. Н., Ногина А. А. — ЖАХ, 1967, т. 22, с. 394; Analyt. Abstr., 1968, v. 15, р. 6998.
48 Hozumi К., Umemoto К. — Microchem. J., 1967, v. 12, p. 46.
49 Umemoto K„ Inoue K., Nishii A.— Japan Analyst, 1970, v. 19, p. 450; Analyt. Abstr., 1971, v. 21, p. 3362.
50. Budesinsky B. — Analyt. Chem., 1965, v. 37, p. 1159.
51. Budesinsky B, Vrzalova D. — Z. analyt. Chem. 1965, v 210, p. 161.
52. Sedlak M., Cowell W. H., Milner О. I — Microchem. J., 1971, v. 16, p. 59.
53. Budesinsky B., Vrzalova D — Chemist Analyst, 1966, v 55, p. 110.
54. Budesinsky B., Krumlova L. — Analytica Chim Acta, 1967, v. 39. p. 375.
55. Саввин С. Б., Акимова T. Г., Дедкова В. П., Варшал Г. М. — ЖАХ, 1969, т. 24, с. 1868; Analyt. Abstr., 1971, v. 21 р 669
56. Петрова Е. И., ЖАХ, 1971, т. 26, с. 402; Analyt. Abstr., 1972, v. 22, р. 3641.
57. Budesinsky В. — Microchem. J., 1969, v. 14, p 242.
58. 'Armeanu V., Dragusln I—Revue rumm. Chim., 1969, v. 14, p. 1511; Analyt. Abstr., 1971, v. 20, p. 74.
59. White D. C. — Mikrochim. Acta, 1962, p. 807.
60. Belcher R., Campbell A. D., Gouverneur P., Macdonald A. M. G.—J. chem. Soc., 1962, p. 3033.
556
61. McGillivray R., Woodger S. C. — Analyst, 1966. v. 91, p. 611.
62. Lonsdale M. — Proc. Soc analyt Chem., 1970, v. 7, p. 12.
63. Nellist G. R, Edwards A. H — Proc. Soc. analyt. Chem., 1965, v 2, p. 144.
64. Bestaick G., Johnson R. Л1. — Taianta, 1970, v. 17, p. 709.
65. British Standards 2690: Part 6, 1968, London.
66. Archer E. E. — Analyst, 1957, v. 82, p. 208.
67 Нечипоренко Г. H.—Изв. АН СССР, Отд. хим. наук, 1958, с. 359; Analyt. Abstr., 1959, v. 6, р. 135.
68. White D. С. — Mikrochim. Acta, 1959, p. 254.
69 White D. C Mikrochim. Acta, 19(50, p. 280.
70 Нечипоренко Г. H. — Гидрохпм. материалы, 1961, т. 33, с. 143; Analyt. Abstr., 163, v. 10, р. 2673.
71 Нечипоренко Г. Н.— Гидрохнм. материалы, 1959, т. 29, с. 214; Analyt. Abstr., 1961, v. 8, р. 1500.
72. Нечипоренко Г. //. — Гидрохпм. материалы, 1957, т. 26, с. 207; Analyt. Abstr., 1958, v. 5, с. 2008.
73. Матвеев А. А., Нечипоренко Г. Н.— Гндрохнм. материалы, 1961; т. 33, с. 134; Analyt. Abstr., 1963, v. 10, р. 2934.
74. Нечипоренко Г. Н. — Гндрохнм. материалы, 1961, т. 33, с. 151; Analyt. Abstr., 1963, v. 10, р. 2937.
75. Lewis W. М. — Proc. Soc. Wat. Treat. Exam., 1967, v. 16, p. 287.
76. Parker S. — Proc. Soc. analyt. Chem., 1967, v. 4, p. 102.
77 Niwa U., Parry E. P. — Chemist Analyst, 1960, v. 49, p. 102.
78. Munger J. R., Nippier R. W., Ingols R. S. — Analyt. Chem., 1950, v. 22, p. 1455.
79. Schwarzenbach G, Flaschka H. — Complexometric Titrations, 2nd edn., London, Methuen, 1969
80. Pribil R., Maricova D. —Chemice Listy, 1952, v. 46, p. 542.
81. Belcher R., Gibbons D., West T. S. — Chem. Inds, London, 1954, p. 127.
82. Belcher R., Gibbons G., West T. S. — Analyst, 1955, v. 80, p. 751.
83. Rumler F., Herbolsheimer R., Wolf G. — Z. analyt. Chem., 1959, v. 166, p. 23;
Analyt. Abstr., 1959, v. 6, p. 3953.
84. Morris A. G. C. — Chemist Analyst, 1959, v. 48, p 76.
85. Iritani N., Tanaka T., Oishi H. — Japan Analyst, 1959, v. 8, p. 30; Analyt. Abstr., 1960, v. 7, p. 102.
86. Sporek K. F. — Analyt. Chem., 1958, v. 30, p 1032.
87. Ashbrook A. W., Ritcey G. M. — Analyst, 1961, v. 86, p. 740.
88. Tanaka T., Tanabe H.—Japan Analyst, 1959, v. 8, p. 826; Analyt. Abstr., 1961, v. 8, p. 192.
89. Odler I., Gebauer J. — Chemike Zvesti, 1961, v. 15, p. 563; Analyt. Abstr., 1961, v. 9, p. 1450.
90. Burriel-Marti F., Alvarez-Herrero C. — Quim. analit. pura apl. Ind., 1964, v. 18, p. 142; Analyt. Abstr., 1966, v. 13, p. 132.
91. De Sousa A. — Inf. quim. Anal., 1961, v. 15, p. 121; Analyt. Abstr., 1962, v. 9, p. 1452.
92. De Sousa A. — Inf. quim. Anal., 1962, v. 16, p. 177; Analyt. Abstr., 1963, v. 10, p. 3667.
93. Iritani N., Tanaka T. — Japan Analyst, 1960, v. 9. p. 1; Analyt. Abstr., 1961, v. 8, p. 4603.
94 Nestoridis A. — Analyst, 1970, v. 95, p. 51.
95. Campbell A. D., Hubbard D. P., Tioh N. //. — Analyst, 1974, v. 99, p. 519.
96. Jangida B. L., Varde M. S., Venkatasubramanian V. — Indian J. Chem., 1964, v. 2, p. 149; Analyt. Abstr., 1965, v. 12, p. 3847.
97. Quartermain P. G., Hill A. G. — Analyst, 1960, v. 85, p. 211.
98. Gustafsson L. — Taianta, 1960, v. 4, p. 227, 236.
99 Archer E. E. — Analyst, 1956, v. 81, p. 181.
100. Beswick G , Johnson R. M. — J. Sci. Fd Agric., 1970, v. 21, p. 565.
101. Murphy J. M„ Sergeant G. A. — Analyst, 1974, v 99, p. 515.
102. Wronski M. — Analyst, 1958, v. 83, p. 314.
103. Belcher R., Nutten A. J., Stephen W. I. — J. chem. Soc., 1953, p. 1334.
104. Wilkinson H. C. — Analyst, 1956, v. 81, p. 9.
557
105. Bengtsson T. A.— Analytica chim. Acta, 1958, v. 18, p. 353.
106. Belcher R. — Talanta, 1968, v. 15, p. 357.
107. Koithoff /. AL, Belcher R. — Volumetric Analysis, Vol. Ill, New York, Interscience, 1957, p 339
108. IFeisz H., Fritsche U. — Mikrochim. Acta, 1970, p. 638.
109. Gawargius Y. A., Farag A. B.—Talanta, 1972, v. 19, p. 641.
110. Jagner D. — Analytica chim. Acta, 1970, v. 52, p. 483.
111. Wagner A. — AVitt. Ver. Grosskesselbesitzer, 1958, p. 50; Analyt. Abstr, 1959, v. 6, p. 4187.
112. Jones A. S., Letham D. S. — Analyst, 1956, v. 81, p. 15.
113. Ahmed M. N., Lawson G. I. — Talanta, 1958, v 1, p. 142.
114. Stephen W. I. — Analytica chim. Acta, 1970, v. 50, p. 413.
115. Jones P. A., Stephen W. /. — Analytica chim. Acta, 1973, v. 64, p. 85.
116. McClure G. L. — Analytica chim. Acta, 1973, v. 64, p. 289
117. Bertolacini R. J., Barney J. E. — Analytica Chem., 1957, v 29, p. 281.
118. Jenemann H., Zimmermann G. — Glastech. Ber., 1961, v. 34, p. 191; Analyt.
Abstr., 1963, v. 10, p. 1441.
119. Haslam J., Hamilton J. B, Squirrell D. С. M. — Analyst, 1961, v. 86, p 239.
120. Agterdenbos J, Martinius N.— Talanta, 1965, v 12, p. 426
121. Prochazkova L. — Z. analyt. Chem., 1961, v. 182, p. 103; Analyt. Abstr., 1962, v. 9, p. 885.
122. Agterdenbos J., Martinius N.— Talanta, 1964, v. 11, p. 878.
123. Barney J. E. — Talanta, 1965, v. 12, p. 425.
124. Spencer B. — Biochem. J., 1960, v. 75, p 435.
125. Sutherland /. T. — Clinica chim. Acta, 1966, v. 14, p. 554.
126. Magar W. ¥., Pollard A. G. — Chem. Inds, London, 1961, p. 505.
127. Wainer A., Koch A. L. — Analyt. Biochem., 1962, v. 3, p. 457.
128. Carlson R. M., Rosell R. A , Vallejos IT. — Analyt Chem., 1967, v. 39 p. 688.
129. Gales M. E., Kaylor W. H, Longbottom J. E. — Analyst, 1968, v. 93, p 97.
130. Kudo H., Tanaka Y.— Japan Analyst, 1970, v. 19, p. 99; Analyt. Abstr., 1971, v. 21, p. 1061.
131. Yamamoto K, Hirro K., Tanaka T.—Japan Analyst, 1968, v. 17, p. 206; Analyt. Abstr., 1969, v 17, p. 2440
132. Bark L. S. — Ind. Chem., 1964, v. 40, p. 153.
133. Beswick G., Johnson R. M.—J. Sci. Fd Agric., 1970, v. 21, p. 565.
134. Boltz D. F. (Ed.), Colorimetric Determination of Non-metals, New York, In-
terscience, 1958.
135. Johnson С. M., Nishita H. — Analyt. Chem., 1952, v. 24, p 736.
136. Sinclair A., Thorburn Burns D., Hall R. D., Hayes W P. — Talanta, 1971,
v. 18, p. 972.
137. Kirsten W. J., Patel V. J. — Microchem. J., 1972, v. 17, p. 277.
138. Davis I. B., Lindstrom F. — Analyt. Chem., 1972, v. 44, p. 524.
139. Goguel R. — Analyt Chem., 1969, v. 41, p. 1034.
140. Kanno S. — Japan Analyst, 1959, v. 8, p. 180, Analyt. Abstr., 1960, v. 7, p. 469.
141. Nemeth K. — Z. PflErnahr Dung. Bodenk, 1963, v. 103, p. 193; Analyt. Abstr., 1965, v. 12, p. 3069.
142. Ozawa T. — J. chem. Soc. Japan, pure Chem. Sect, 1966, v 87, p. 855; Analyt. Abstr., 1967, v. 14, p. 7451.
143. Бабко А. К., Литвиненко В. A.—ЖАХ, 1963, т. 18, с. 237; Analyt. Abstr., 1964, v. 11, p. 948.
144. Ducret L., Ratouis M. — Analytica chim. Acta, 1959, v. 21, p. 91.
145. Zimmerman E., Brandt W. W.— Talanta, 1958, v 1, p. 374.
146. Staffyn P., Keane W. — Analyt. Chem., 1964, v. 36, p. 397.
147. Barney J. E. — Talanta, 1965, v. 12, p. 425.
148. Wimberley J. W. — Analytica chim. Acta, 1968, v. 42, p. 327.
149. Garrido M. L.— Analyst, 1964, v. 89, p. 61.
150. Standard Methods for the Examination of Water and Waste Water, 13th edn., Washington D C, American Public Health Association, 1971, p. 334
151. Toennies G., Bakay B. — Analyt. Chem., 1953, v. 25, p. 160
152. Haslam J., Hamilton J. B., Squirrell D. С. M. — Analyst, 1961, v. 86, p. 239.
658
153. Martin J. M„ Stephen IF. /. — Analvtica chim. Acta, 1967, v. 39, p. 175, 525
154. Mendes-Bezerra A. E, Uden P C.— Analyst, 1969, v 94, p. 308
155. Guyon J. C, Lorah E. J. — Analyt. Chem., 1966, v. 38, p. 155.
156. Tan L. H., West T. S. — Analyst, 1971, v. 96, p. 281.
157. Haba F. R., Wilson C. L. — Talanta, 1962, v. 9, p. 841.
158 Underwood A L., Miller M. W., Howe L. H. — Analytica chim. Acta, 1963, v. 29, p. 79
159. Chasan D. E., Norwitz G. — Talanta, 1971, v. 18, p. 499.
160. Meehan B. J., Tario S. A.—Talanta, 1973, v. 20. p. 1215.
161. Gilbert P. T. — In: Analytical Flame Spectroscopy (Ed. R. Mavrodineanu), London, Macmillan, 1970, p. 181.
162. Плескач JI. И., Чиркова Г. Д —Заводск. лаборатория, 1971, т. 37, с. 168.
163. Shaw W. М. — Analyt. Chem., 1958, v. 30, р. 1682.
164. Cullum D. C., Thomas D. В. — Analyst, 1959, v. 84, p. 113.
165. Cullum D. C., Thomas D. B. — Analyst, 1960, v. 85, p. 688.
166. Forbes E. A. — Analyst, 1973, v 98, p. 506
167. Varley ]. A., Chin P. Y. — Analyst, 1970, v. 95, p. 592.
168. Wollin A. — Atom. Abs. Newsl., 1970, v. 9, p. 43.
169. Rose S. A., Boltz D. F. — Analytica chim. Acta, 1969, v. 44, p. 239.
170. Kirkbright G. F., Marshall M. — Analyt. Chem., 1972 v. 44, p. 1288.
171. Dagnall R M., Thompson К. C., West T. S. — Analyst, 1967, v. 92, p. 506.
172. Aldous К- M., Dagnall R. M., West T. S. — Analyst, 1970, v. 95, p. 417.
173. Belcher R., Bogdanski S. L., Townshend A. — Analytica chim. Acta, 1973, v. 67, p. 1.
174. Belcher R., Bogdanski S. L., Knowles D. J., Townshend A. — Analytica chim. Acta, 1975, v. 77, p. 53.
175. Belcher R., Bogdanski S. L., Knowles D. J., Townshend A. — Analytica chim. Acta, 1975, v. 79, p. 292.
176. Tenygl J.— MTP International Reviews of Science, Physical Chemistry, Senes one, Vol 12 (Ed T. S. West), London, Butterworths, 1973, p. 123.
177. Ross J. W, Frant M. S. — Analyt. Chem., 1969, v. 41, p. 967.
178. Mascini M. — Analyst, 1973, v. 98, p. 325.
179. Selig W. — Mikrochim. Acta, 1970, p. 168.
180. Heistand R. N., Blake С. T. — Mikrochim. Acta, 1972, p. 212.
181. Frant M. S.— Plating, 1971, p 686.
182. Pungor E., Havas 1. — Acta chim. hung., 1966, v. 50, p. 77; Analyt. Abstr., 1968, v. 15, p. 2422.
183. Rechnitz G. A, Lin Z. F., Zomochnick S. B. — Analyt. Lett., 1967, v. 1, p. 29; Analyt. Abstr, 1969, v 16, p. 118.
184. Rechnitz G. A., Fricke G. H., Mohan M S. — Analyt. Chem, 1972, v. 44, p. 1098.
185. Mohan M. S., Rechnitz G. A. — Analyt. Chem., 1972, v. 45, p. 1323.
186. Jasinski R, Trachtenberg I. — Analyt. Chem, 1972, v. 44, p. 2373.
187. Jasinski R, Trachtenberg I. — Analyt. Chem, 1973, v. 45, p. 1277
188. Goldstein G, Menis O, Manning D. L. — Analyt. Chem., 1961, v. 33, p. 266.
189. Jovanovic M. S., Gaa’l F. F., Bjelica L. I. — Mikrochim. Acta, 1971, p. 778.
190. Baudisch J, Beilstein G, Nevenhausen H. E. — Z analyt. Chem., 1968, v. 235, p 231; Analyt. Abstr, 1969, v. 17, p. 143.
191. Koithoff I. M, Pan Y. D. — J. Am chem. Soc., 1940, v. 62, p. 3332.
192. Myers S. A, Swann W. B. — Talanta, 1965, v. 12, p. 133.
193. Ohlweiler O. A. — Analytica chim. Acta, 1953, v. 9, p. 476.
194. Long M. I E. — Analyst, 1968, v 93, p. 339
195. Baranowski R, Gregorowicz Z — Chemia analit., 1965, v. 10, p 125; Analyt. Abstr, 1966, v. 13, p. 2323.
196. Bishara S. W. — Microchem. J, 1970, v. 15, p. 211.
197. Mayer J., Hluchan E., Abel E. — Analyt. Chem, 1967, v. 39, p. 1460.
198 Humphrey R. E., Laird С. E. — Analyt. Chem, 1971, v. 43, p 1895.
•99. Гладышев В. П —Заводск. лаборатория, 1962, т. 28, с. 1063; Analyt. Abstr, 1963, v. 10, р. 1774.
559
200. Goldstein G., Manning D. L., Zittel H. E. — Analyt. Chem., 1962, v. 34 p. 1169
201. Bowley M. J. — Analyst, 1969, v. 94, p. 787.
202. Hems R. V.. Kirkbright G. F., West T. S. — Tai anta, 1969, v. 16, p. 789.
203. Armenia W. J., Larson С. E. — Analyt Chem., 1963, v. 35, p. 918.
204 Bowen H. J. Af. — Analyst, 1970, v. 95, p 665.
205. Johannesson J. K- — Analyst, 1967, v. 92, p. 766.
206. Dean P M., Watts О. O. — J. Am chem Soc., 1924, v. 46, p. 855.
207. Williams Af. B., Janata J. — Taianta, 1969 v. 17, p. 548
208 Perchec H, Gilot B. — Bull. Soc. chim. France, 1964, p. 619; Analyt. Abstr 1965, v. 12, p. 3610.
209 Ito S., Hara T. — J. chem. Soc. Japan, pure Chem. Sect., 1969, v. 90, p. 1027;
Analyt. Abstr., 1970, v. 19, p. 4842.
210. Butts W. C. — Analyt. Lett., 1970, v. 3, p. 29.
211. Butts W. C., Rainey W. T. — Analyt. Chem., 1971, v. 43, p. 538.
СУЛЬФИДЫ
Определение сульфидов является очень важной аналитической операцией по нескольким причинам. Одной из причин является высокая токсичность сероводорода, а также его органолептические свойства (неприятный запах растворов сероводорода наблюдается при содержании его в растворе на уровне нескольких десятых миллиграмма в литре). Образование сероводорода обычно вызывается разрушением органических веществ под действием микробов в анаэробных условиях.
Сульфиды часто присутствуют в промышленных сточных водах и поэтому очень важны автоматические методы для непрерывного контроля. Определение сульфида необходимо в некоторых отраслях промышленности, особенно в отраслях, связанных с обогащением руд и минералов. В некоторых технологических процессах необходимо гарантировать отсутствие сульфидов и сероводорода, по той, например, причине, что они являются сильными каталитическими ядами.
В качестве общепринятого стандарта сульфидов используют обычно Na2S-9H2O. Недавно [1] в качестве стандарта сульфидов предложен диизопропилфосфородитиоат калия, в этой работе описан способ приготовления чистого соединения.
Аналитическая химия сульфидов рассмотрена в нескольких обзорах по общей аналитической химии серусодержащих соединений [2—5].
МЕТОДЫ ОТДЕЛЕНИЯ
Отделение газообразного сероводорода
Наиболее широко используемый метод отделения сероводорода в газообразной форме основан на подкислении анализируемого раствора, содержащего сульфиды, и отгонке выделенного сероводорода в токе воздуха, диоксида углерода или, что наиболее предпочтительно, азота. Газ пропускают через подходящий абсорбент.
560
Обычно в качестве абсорбентов используют растворы ацетатов цинка или кадмия, из которых выделяются соответствующие сульфиды. Этот метод особенно пригоден при малых содержаниях сульфидов [6, 7].
Сульфиды часто определяют иодометрически после их отделения выше описанным методом. Изложенный метод отделения сульфидов находит применение и при определении сульфатов после их восстановления нодистоводородной и фосфорноватистой кислотами [8,9].
Осаждение сульфидов кадмия или цинка
В методе отделения сульфидов, основанном на осаждении сульфидов кадмия или цинка, не требуется выделения сероводорода. Этот метод наиболее целесообразен в случае присутствия значительных содержаний сульфидов. Для отделения сульфидов в виде сульфида цинка к анализируемому раствору предложено прибавлять ацетат цинка и затем гидроксид натрия [10]. Осадок отделяют от аккуратно декантируемой осветленной жидкости фильтрованием.
Осаждение сульфидов используют для их отделения от сульфитов или тиосульфатов, так как эти два аниона не осаждаются в этих условиях. Следует учитывать, однако, возможность соосаж-дения тиосульфата на объемистых осадках сульфидов цинка или кадмия. Осадок с меньшей поверхностью и, следовательно, обеспечивающий меньше возможностей для соосаждения, образуется при прибавлении свежей суспензии карбонатов цинка или кадмия к анализируемому раствору [11].
Иодиметрическая методика, включающая первоначальное отделение сульфида в виде сульфида кадмия, описана ниже в этом разделе.
Ионообменные методы
Для разделения смесей сульфата, сульфита, тиосульфата и сульфида применяют апионообменную смолу в NOi-форме [12]. Сульфит и сульфид элюируют 0,1 М раствором нитрата аммония в 30%-ном ацетоне при pH = 9,7.
Ультрамикроколичества сероводорода в газовой фазе или в воде можно отделить на анионообменной смоле Амберлит IRA-400 с последующим элюированием 4 М раствором NaOH [13]. Смола удерживает сульфид примерно 10 дней без потерь.
Метод тонкослойной хроматографии
Тонкослойная хроматография с использованием кукурузного крахмала в качестве неподвижной фазы ацетона с 3 Л1 раствором NH4OH (1:4) в качестве растворителя позволяет отделять сульфид
561
I-----------1_
0 20
_1_____________|
00 пн
Рис. 66. Полиэтиленовые чашки, вкладываемые в ячейки для микродиффу. зии [15].
от нитрита, тиосульфата, хромата, азида, цианида, тиоцианата, бората, арсенита, арсената, нитрата, сульфата и фосфата [14].
Микродиффузия
Определение следов различных компонентов в биологических образцах часто вызывает затруднения. Для выделения сульфидов из таких материалов можно использовать микродиффузию, применив стандартные ячейки [15]. Чтобы предотвратить травление внутренних поверхностей ячейки 40%-ным раствором NaOH, используемым для поглощения H2S, в ячейку помещают маленькие полиэтиленовые чашки с поглотительным раствором (рис. 66). Для полного отделения H2S необходимо 3 ч, затем полиэтиленовые чашки переносят в пробирки и определяют содержание сульфидов.
Ускоренный микродиффузионный метод, также использующий стандартные ячейки, позволяет провести полную микродиффузию в течение 15 мин [16].
Другие методы отделения
Сероводород при низких концентрациях извлекают из воздуха бумажными фильтрами, импрегнированными нитратом серебра [17]. Метод применим при содержании сероводорода около 5 частей на триллион (10-12). Для определения сульфида используют флуориметрический метод.
Для отделения сульфидов предложен газохроматографический метод, его описание приведено в разделе, посвященном определению сульфидов.
Описан экстракционный метод отделения H2S в виде (С2Н5)3РЬ—S—РЬ(С2Н6)3 (экстрагент — н-гексанол). Этот метод разделения является составной частью метода микроопределения сероводорода в природных водах с использованием о-оксимеркури-бензойной кислоты. Более полное обсуждение этого метода приведено ниже.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Гравиметрические методы
При гравиметрическом определении сульфида его можно окислить до сульфата и определить в виде сульфата бария. Окисление выполняют щелочными растворами пероксида водорода, гипобро-
562
Митом или раствором брома в HCI. Хотя сульфид количественно осаждается в виде сульфида кадмия, однако осадок получается нестехпометрическим и поэтому его можно применять для отделения, но не для определения сульфидов.
Титриметрические методы
Иодиметрические методы
Иодиметрия является наиболее применяемым титриметрическим методом определения сульфидов, хотя она и содержит несколько возможных источников ошибок. Основная реакция метода следующая:
H2S + I2 s; + 2r + 2H+
Прямое титрование может привести к значительным погрешностям. В общепринятой методике прибавляют избыток стандартного раствора иода к анализируемому раствору, содержащему сульфид, затем титруют неизрасходованный иод стандартным раствором тиосульфата. Если сульфид предварительно отделен (выделение H2S и последующее поглощение его ацетатом цинка), то избыток иода можно вводить непосредственно в подкисленную суспензию и затем титровать избыток иода.
Большое значение имеет pH. В слабощелочных средах, например в разбавленных растворах гидрокарбонатов, происходит частичное окисление сульфидов до сульфатов:
S2’ + 41а + 8ОН" —> SO42'+ 8Г + 4Н2О
Если в подкисленном растворе сульфид титровать прямо раствором иода, то могут происходить потери сероводорода из-за его летучести. Если титровать разбавленные растворы сульфида, то образующаяся сера не мешает проведению титрования. Однако в более концентрированных растворах наблюдается окклюзия иода на осадке.
При титровании иодом вместе с сульфидом титруются сульфит и тиосульфат. Можно заметить, что растворы сульфидов часто бывают загрязнены продуктами их окисления, которые также реагируют с иодом. Для анализа смесей серусодержащих анионов, включая сульфид, предложено несколько методов, в том числе иодиметрических. Сульфид и сульфит определяют в присутствии сульфата следующим образом [18]. Обрабатывают анализируемый раствор хлорной кислотой в присутствии хлорида ртути (11), при этой обработке сульфит разлагается с выделением SO2, а сульфид осаждается в виде сульфида ртути. Выделенный SO2 поглощают водным раствором пероксида щелочного металла, и раствор затем можно оттитровать иодиметрически. После удаления SO2 сульфид ртути обрабатывают хлоридом олова(II) и НС1 для выделения сероводорода, который затем поглощают ацетатом кадмия и определяют иодиметрически.
Б63
Для анализа смеси сульфида, сульфита и тиосульфата предложен другой иодпметрнческпй метод (19]. В публикациях [20— 22] описан анализ смесей сульфида, тиосульфата п полисульфидов. Подробное описание анализа такой смеси приведено в разделе, описывающем определение полисульфидов.
Иодиметрическое титрование сульфидов в промышленных
сточных водах [23]
Реагенты
Раствор ацетата кадмия, 250 г двуводного ацетата кадмия в I л воды.
Раствор иодида калия, 100 г KI в 1 л воды. Хранить в посуде нз темного стекла.
Серная кислота, разбавленная; I объем 98%-ной кислоты осторожно разбавляют, добавляя 3 объема воды.
Раствор иода, 0,0625 Л1. Растворяют 25 г KI в 50 мл воды в мерной колбе емкостью 1 л Взвешивают 15,9 г пода н переносят его в колбу, закрывают колбу и встряхивают ее до тех пор, пока весь иод полностью не растворится, разбавляют раствор до метки. Стандартизируют раствор тиосульфатом (0,125 Л1), как описано ниже.
Иодат калия, 0,020 М раствор. Высушивают 5 г йодата калия при 120 °C, аккуратно взвешивают около 4,3 г сухой соли, растворяют в воде и разбавляют раствор до 1 л.
Тиосульфат натрия, 0,125 М раствор. Растворяют 31.2 г пентагидрата тиосульфата натрия в 1 л свежепрокипяченной и охлажденной воды и прибавляют 3 капли хлороформа для стабилизации раствора. Раствор необходимо хранить в склянке нз темного стекла. До использования раствор оставляют стоять в течение нескольких суток. Стандартизируют раствор йодатом калия, как описано ниже.
Раствор крахмала. Смешивают 5 г водорастворимого крахмала с небольшим количеством воды в однородную массу и вливают смесь в I л кипящей воды при непрерывном помешивании. Кипятят раствор 1 мин и охлаждают.
Стандартизация раствора тиосульфата. В коническую колбу для титрования помещают 5 мл раствора KI и 100 мл воды. Отбирают пипеткой 25,0 мл 0,020 М раствора КЮ3 и переносят его в колбу. Прибавляют в колбу 10 мл раствора сериой кислоты и немедленно титруют раствором тиосульфата, прибавляя индикатор вблизи конечной точки.
Стандартизация раствора иода. Отбирают пипеткой 25,0 мл раствора нода и переносят его в колбу для титрования, разбавляют раствор в колбе и добавляя 100 мл воды. Титруют раствор стандартным раствором тиосульфата, прибавляя индикатор вблизи конечной точки.
Ход анализа. Прибавляют 10,0 мл анализируемого раствора (см. примечание 1) к 100 мл воды и добавляют 10 мл раствора ацетата кадмия. Позволяют осадку седиментировать и сливают осветленную жидкость над осадком на бумажный фильтр Переносят осадок на фильтр н промывают его водой. В широ-когорлую коническую колбу емкостью 250 мл помешают 100 мл воды, 10 мл разбавленной H2SO< и 25 мл 0,0625 М раствора иода. Переносят бумажный фильтр в колбу и быстро закрывают ее. Тщательно встряхивают содержимое колбы для полного растворения осадка сульфида. Титруют неизрасходованный иод тиосульфатом натрия, прибавляя раствор крахмала вблизи конечной точки.
Расчет. Содержание сульфида (х, в мг/л) вычисляют по уравнению
0,0625 • 32 V,
Х~ V2
где — объем 0,0625 М раствора нода, мл; V2— объем образца, мл.
Примечания 1. Рекомендуется объем образца 10 мл Если приблизительное содержание сульфидов известно, то оптимальный объем образца можно рассчитать по формуле 16000/с, где с — концентрация сульфида, мг/л.
564
2. Если искомая концентрация сульфидов должна быть выражена в концен-тоацпн сероводорода, то в формуле для вычисления содержания сульфида вме-с!ро фактора 0,0625-32 — 2000 используют значение 2125.
Титриметрический метод определения сульфидов с использованием металлорганических соединений
Разработано несколько титриметрических методик определения сульфидов с использованием о-оксимеркурибензойной кислоты, первоначально предложенной в качестве титранта в 1958 г. 124]. Основная реакция для определения сульфидов, протекающая в щелочной среде, описывается уравнением
СОО'
+ S2’
HgOH
‘ООС
^'"^'Hg—S— Hg
+ 2ОН'
Для титрования предложено несколько индикаторов. Тиофлуоресцеин обладает синей окраской в щелочной среде, эта окраска исчезает от прибавления первой избыточной капли титранта. Кроме него в качестве индикаторов предложены дитизон, нитропруссид натрия, дифенилкарбазон и мономеркурифенолфталеин. Обширная литература, посвященная использованию описываемого метода, отражена в обзорах [2, 25]. Большим достоинством о-окси-меркурибензойной кислоты является то обстоятельство, что определению сульфидов с ее помощью не мешают сульфиты и тиосульфаты. Мешающие влияния, в общем, определяются типом индикатора. В некоторых случаях индикатор влияет на протекание основной реакции. Например, при использовании нитропруссида титр кислоты оказывается соответствующим следующей реакции:
4-ОН"
Титрование сульфида в интервале макроконцентраций проводят 0,05 М раствором реагента.
Большое аналитическое значение имеет тушение флуоресценции меркурипроизводного флуоресцеина сульфид-ионом. На этом принципе основаны два варианта аналитических методик, одна включает построение калибровочного графика в присутствии избытка меркурипроизводного, другая—прямое флуориметрическое титрование. Первый вариант использован для определения сероводорода в воздухе при содержании порядка 1 ч. на млрд ч. (т. е. около 10—7%) [26]. Воздух пропускают через 0,1 —1,0 М раствор NaOH в течение 60 мин и затем анализируют полученный раствор. Описан автоматический вариант предложенного метода [27]. Флуориметрическое титрование имеет ряд преимуществ [28]. На результатах титрования не сказывается присутствие тиолов, ксантатов, цианида и других анионов, которые влияют на флуоресценцию
565
Рис. 67. Кривые флуори метрического титрования сероводорода в виде судь. фида трнэтклсвинца 2 • 10~5 и. раство. ром тетрартутьацетатфлуоресцеина (ра. створитель — смесь этанол — гексанол): / — введено На** 0 мкг, найдено HaS 0 МКг. 2—введено H2S —0.0514 мкг, найдено НоЧ 0,0510 мкг; 3—введено H2S—0,120 мкг. найдено HaS 0,119 мкг [25].
меркурипроизводного флуоресцеина. Кроме того, все вычисления в этом случае основаны на содержании ртути в меркури-производном и не требуют стандартизации растворов.
Описап экстракционный метод отделения сульфида в виде сульфида триэтилсвинца (С2Н5)3РЬ—S—РЬ(С2Н5)3 [25] перед его титриметрическим определением с тетрартутьацетатфлуоресцеином. Кривые флуориметрического титрования представлены на рис. 67. Подробности этого метода описаны в оригинальной работе.
Другие титриметрические методы
Описаны методы определения сульфидов, основанные па использовании ЭДТА, однако они, видимо, не находят широкого применения. Предложен косвенный метод [29] определения сульфидов, основанный на том, что при смешении щелочного раствора анализируемого сульфида с нейтральным или слабокислым раствором перхлората меди(П) образуется сульфид меди стехиометрического состава. Избыток меди(II) в фильтрате титруют раствором ЭДТА, используя мурексид в качестве индикатора.
Метод комплексометрического титрования предложен для анализа смесей сульфидов, сульфитов и сульфатов [30]. Сначала к анализируемому раствору прибавляют избыток нитрата меди, при этом осаждается сульфид меди, а неизрасходованную медь(II) определяют с помощью ЭДТА. Во второй аликвотной части анализируемого раствора осаждают сульфат, прибавляя избыток хлорида бария, и неизрасходованный барий (II) титруют раствором ЭДТА. В третьей аликвотной части анализируемого раствора окисляют сульфид и сульфит до сульфата и определяют сульфат, как описано выше. На основании результатов, полученных при титровании этих трех аликвотных частей, можно определить содержание всех трех компонентов.
Для определения сульфида и сульфита в их смесях предложен другой метод [31]. Сульфид отделяют от сульфита экстракцией этанолом, а остающийся в водной фазе сульфит окисляют бромом до сульфата. Сульфат определяют, прибавляя избыток хлорида бария к анализируемому раствору и титруя затем неизрасходованный барий(II) ЭДТА. Во второй аликвотной части анализи
Ь66
руемого раствора определяют сумму сульфида и сульфита, окисляя их до сульфатов и определяя последние, как указано выше.
Описан комплексометрический анализ смесей сульфидов и сульфатов [32]. Первоначально удаляют сульфид в виде сероводорода кипячением анализируемого раствора с НС1. Остающийся в растворе сульфат определяют комплексометрически (с использованием хлорида бария). Во второй аликвотной части сульфид окисляют бромом до сульфата и определяют сульфат выше указанным методом, в этом случае определяется сумма сульфида и сульфата.
Для определения сульфида кроме иода рекомендованы и другие окисляющие агенты: бром, гипохлорит, иодат, гексацианофер-рат(Ш), перманганат и перйодат. В большинстве случаев никаких преимуществ по сравнению с иодом они не имеют. Большинство указанных реагентов должно быть стандартизировано каким-либо независимым методом. Исключение составляет N-бромсук-цинимид [33]. Этот реагент окисляет сульфид до элементной серы. Раствор реагента устойчив в течение нескольких дней при хранении в прохладном темном месте. Анализируемый раствор с добавкой раствора иодида калия титруют раствором N-бромсукцинимида, используя в качестве индикатора крахмал. Механизм действия этого реагента описывается следующим образом. Реагент селективно окисляет иодид до иода, который в свою очередь реагирует с сульфидом:
Вг* + 2Г —> 12 + Вг'
H2S + I» —> 2Н+ + 2Г + S
Взаимодействие N-бромсукцинимида с иодидом, вероятно, проходит через стадию промежуточного гидролиза реагента с образованием бромноватистой кислоты:
О О
II II
СН2—С. сн2—с.
I ^NBr + Н2О —> | + НВгО
СН2—С/ СН,—с/
Описанный метод позволяет определять 0,6—13 мг сульфидов, некоторые подробности аналитической методики приведены ниже.
Описан [34] метод определения сульфидов, основанный на титровании по методу осаждения солями цинка. Довольно значительная растворимость сульфида цинка в водной среде приводит к появлению ошибок при определениях, и поэтому титрование рекомендовано проводить в присутствии этанола с индикатором метилтимоловым синим. При необходимости точку эквивалентности определяют потенциометрически. Определению сульфидов не мешают сульфаты, сульфиты, тиосульфаты, карбонаты и фосфаты; мешающее действие ионов кальция и магния устраняют их маски
567
рованием с помощью ЭДТА. Описываемый метод дает слегка завышенные результаты по сравнению с иодиметрическим. Метод можно использовать для определения сульфидной части в растворах полисульфидов. В этом случае анализируемый раствор первоначально обрабатывают раствором сульфита натрия:
NasSx + (х - 1) Na.SO3 —> Na2S + (х - 1) Na2S2O3
Образующийся сульфид титруют, используя описанный выше метод [35]. Для анализа смесей сульфида, полисульфида и тиосульфата предложено несколько методов, в том числе и иодиметриче-ских. Они представлены в разделе, описывающем полисульфиды.
Титриметрическое определение сульфида с использованием N-бромсукцинимида [33]
•
Реагент
N-бромсукцинимид, 0,005 М раствор. Растворяют 0,2225 г перекристаллизованного в воде А-бромсукцинимида и разбавляют до 250 мл.
Ход анализа. Отбирают 0,5—100,0 мл анализируемого раствора в колбу емкостью 250 мл, прибавляют 1,0 мл 10%-ного раствора иодида калия и несколько капель раствора крахмала. Кислотность создают несколькими каплями 2 М раствора НС1 и затем титруют раствор N-бромсукцинимидом с постоянным перемешиванием. На достижение точки эквивалентности указывает появление синей окраски, не исчезающей в течение 30 с.
Спектроскопические методы
Спектрофотометрические методы в видимой и ультрафиолетовой областях спектра
Определение сульфидов в виде метиленового синего остается до настоящего времени наиболее распространенным спектрофотометрическим методом, несмотря на то, что реакция, положенная в основу этого метода, известна с 1883 г. [36]. Эта реакция между сульфидом и кислым раствором М,М-диметил-п-фенилендиамина (известным также под названием n-аминодиметиланилина) протекает в присутствии железа(Ш). Первоначальный красный раствор при взаимодействии с сульфидом превращается в синий краситель. Реакция может быть описана следующим образом:
N
N (СН3)2
S + 6FeCb 4- NH4C1 + 4НС1
N (СН3)а С1
568
Эта реакция определения сульфида очень чувствительна и селективна. Светопоглощение растворов метиленового синего обычно измеряют прн 670 нм, но положение этого максимума сильно зависит от условий проведения эксперимента [37]. Для получения воспроизводимых результатов необходимо строго соблюдать условия, например температура при проведении определений не должна отличаться более чем на 2СС. Для полного развития окраски метиленового синего требуется не менее 3 ч [38].
Описываемый метод определения сульфидов был изучен несколькими исследователями и наиболее детально рассмотрен в работе [39]. В названной работе метод использован для определения следов сероводорода в воздухе после поглощения его подходящим раствором. Установлены оптимальные условия определения сероводорода в объектах окружающей среды [37]. Метод использован для определения 0,5—100 мкг сульфидов в образцах различных вод (молярный коэффициент поглощения продуктов при 670 нм составляет 3,4-104) [40]. Применяя анионный обмен для выделения и предварительного концентрирования сульфидов, можно определить до 7-10—6% сероводорода в воздухе и 10~80/о — в воде [13].
Метод определения сульфидов очень селективен. Сульфит и тиосульфат мешают определению при их содержании 10 мг/л, определению сульфидов мешают сильные восстановители.
Описываемый метод широко используют в аналитической практике, особенно при анализе воды и воздуха. При определении сероводорода в воде метод, основанный на образовании метиленового синего, рекомендован как стандартный [6, 40]. Он предложен для определения сульфатов после их предварительного восстановления до сульфидов, описаны автоматические варианты этого метода. В частности, было установлено [41], что из-за его очень большой чувствительности необходимо предварительное разбавление анализируемого раствора.
При определении следов сульфидов в конденсированном водяном паре найдено, что вместо 1Ч,М-диметил-п-фениленди-амина лучше применять М,М-диэтил-п-фенилендиамип [42] (методика, основанная на использовании этого реагента, приведена ниже).
Описаны другие спектрофотометрические методы определения сульфидов. Нитропруссидный метод основан на образовании промежуточного продукта с фиолетовой окраской прн взаимодействии нитропруссида с сульфидом в щелочном растворе [43]. Эта реакция применена для автоматического определения 1—10 ppm сульфидов [44]. Допустимые содержания сульфитов, тиосульфатов, фенола и формальдегида при определении сульфидов составляют 500, 2000, 100 и 100 ppm соответственно. При автоматическом определении сульфидов [44] наблюдается значительное ослабление окраски. В связи с этим рекомендована модификация метода [45], которая позволила достичь хороших результатов
В69
при определениях сульфидов. Закон Бугера — Ламберта — Бера в этом случае выполняется вплоть до 40 мкг/мл сульфидов.
Хлоранилатный метод, первоначально использованный для определения хлоридов, фторидов и фосфатов, был применен для определения сульфидов [46]. К анализируемому раствору сульфида прибавляют хлоранилат ртути(П) и выделяющуюся хлораниловую кислоту определяют спектрофотометрически. Для повышения чувствительности определения светопоглощение хлораниловой кислоты лучше измерять при 330 нм, а не при 525 нм [47]. Кажущийся молярный коэффициент поглощения сульфидов в этом случае составляет 3,2-104.
Для определения сульфидов предложены трифенилметановые красители. Для определения 0—6,0 ppm сульфидов в качестве реагента используют ассоциат кристаллического фиолетового с тетранодмеркурат(П)-ионом [48]. Высокая чувствительность достигается при использовании кристаллического фиолетового в присутствии иодида калия и хлорамина В [49]. Закон Бугера —Ламберта — Бера справедлив в интервале концентраций сульфидов 0,023—0,082 мкг/мл, чувствительность определений (с предварительной экстракцией бензолом) составляет 2 нг сульфида (измерение светопоглощения проведено при 610 нм).
Для определения сульфидов в индустриальных сточных водах рекомендуют его отделение в виде сероводорода [7]. Выделяющийся сероводород пропускают через подкисленный раствор молибдата аммония, при этом образуется молибденовая синь, концентрацию которой и определяют спектрофотометрически при 700 нм.
Прямой анализ смесей серусодержащих анионов можно провести методом ультрафиолетовой спектроскопии [50]. Водные растворы сульфидов, тиосульфатов, сульфитов и сульфатов обладают максимумами светопоглощения при 230, 220, 230 и 210 нм соответственно. Измеряя оптическую плотность растворов в максимумах (для сульфита измерение проводят при 240 нм, чтобы устранить мешающее влияние сульфида) и решая систему соответствующих уравнений, можно рассчитать концентрацию каждого компонента. Чувствительность определения сульфида этим методом составляет 35 нг в 50 мл раствора.
Новый метод определения сульфидов основан на их взаимодействии в аммиачном растворе с железом (III) и нитрилотриук-сусной кислотой [51]. Этот метод позволяет определять 8— 1200 ррш сульфидов. Из 17 изученных анионов серьезные помехи определению оказывает только селен (IV). Определению сульфидов мешает также 100-кратный избыток оксалата, цитрата, тартрата и хромата, но меньшие содержания этих анионов допустимы Светопоглощение растворов измеряют через 2—12 мин после смешения компонентов. Был предложен метод определения сульфидов, основанный на их реакции с железом (III) в присутствии 1.10-фенантролина [52],
Б70
Спектрофотометрическое определение сульфида (в конденсированном водяном паре), основанное на образовании метиленового синего [40]
Реагенты
, Обескислороженная вода-, обрабатывают воду азотом.
I Раствор сульфата аммония-железа(Ш), концентрация 180 г/л; фильтруют при необходимости удаления взвеси.
Й.И-Диэтил-п-фенилендиамин (ДПД), растворяют 2,0 г сульфата N.N-диэтил-я-фенилендиамина в 100 мл 9 М раствора H2SO4. Приготовленный раствор стабилен в течение, по крайней мере, одного месяца при хранении его в темноте.
Серная кислота, 9 М раствор; разбавляют концентрированную H2SO4 (98%-ную) равным объемом воды.
Раствор дигидрата ацетата цинка, концентрация 25 г/л.
Иод, 0,00313 М стандартный раствор. Отмеряют микробюреткой 6,25 мл 005 М раствора иода в мерную колбу емкостью 100 мл и разбавляют раствор водой до метки. Раствор следует использовать свежеприготовленным.
Сульфид, стандартные растворы Раствор А. Растворяют 2—3 г Na2S-9Н2О в обескислороженной воде и разбавляют раствор приблизительно до 1 л. Определяют содержание сульфидов в растворе прямым титрованием стандартным 0,00313 М раствором иода с индикатором крахмалом. Для титрования 1 мл раствора сульфида требуется около 3 мл раствора иода.
Отбирают объем этого раствора, содержащий 10 мг сульфида, и разбавляют его обескислороженной водой немедленно до объема 500 мл. Прибавляют 20 мл раствора ацетата цинка и доводят объем раствора до 1000 мл обескислороженной водой.
Стандартизируют полученный раствор стандартным 0,00313 М раствором иода и при необходимости корректируют его концентрацию.
Стандартный раствор А, содержит в 1 мл 10 мкг сульфидной серы.
Раствор Б. Разбавляют 10 мл стандартного раствора А, добавляя 50 мл обескислороженной воды и около 2 мл раствора ацетата цинка, и затем разбавляют раствор до 100 мл обескислороженной водой, содержащей 0,5 г/л дигидрата ацетата цинка.
Стандартный раствор Б содержит в 1 мл 1,0 мкг сульфидной серы.
Оба стандартных раствора сульфида необходимо использовать в течение 1 ч с момента приготовления.
Раствор крахмала-, готовят обычным методом.
Построение калибровочного графика. Для каждого заново приготовленного раствора ДПД и сульфата аммония-железа(Ш) необходимо построение нового калибровочного графика. Для построения калибровочного графика в интервале 0,5—10 мкг сульфидов в семи градуированных мерных цилиндрах емкостью 100 мл смешивают по 50 мл обескислороженной воды с 2 мл раствора ацетата Цинка. Вводят в мерные цилиндры 0; 1,0, 2,0; 5,0; 7,0; 9,0 и 10 мл стандартного раствора сульфида Б Разбавляют содержимое каждого цилиндра до 97 мл водой, после чего содержимое цилиндров аккуратно перемешивают; прибавляют в каждый цилиндр по 1,0 мл раствора ДПД и вновь перемешивают раствор. Оставляют каждый раствор стоять в течение 60 ± 30 с, прибавляют в них по 1.0 мл раствора сульфата аммония-железа, перемешивают растворы и разбавляют их до 100 мл.
Растворы оставляют стоять в течение, по крайней мере, 10 мин н затем измеряют светопоглощение каждого раствора в кюветах с толщиной поглощаю-Чего слоя 40 мм прн температуре 20—25 °C прн длине волны, соответствующей никсимальиому светопоглощенню (примерно 670 нм, но точная длина волны Должна быть выбрана индивидуально для каждого спектрофотометра). В качестве раствора сравнения используют воду.
Вычитают оптическую плотность контрольного раствора (раствора, не содержащего сульфида) из оптических плотностей, полученных для стандартных Растворов, и строят калибровочный график в координатах: исправленное значение оптической плотности — содержание сульфидов, мкг.
571
Примечание. Светопоглощение раствора, содержащего 10 мкг сульфц. ной серы в приведенном объеме раствора, составляет примерно 0,6. Для по' строения калибровочного графика в интервале 10—100 мкг сульфида исподь. зуют выше приведенный метод, но в цилиндры вводят стандартный раствор сульфида А и светопоглощение измеряют в кюветах с толщиной поглощающей слоя 5 мм.
Отбор проб для анализа. Температура анализируемого образца не должна превышать 20 °C, при температуре выше 20 °C необходимо использовать охлаждающий змеевик из нержавеющей стали. Если пробы отбирают из потока, то скорость течения должна быть 20—50 мл/мин.
Ход анализа. Определение необходимого обьема образца проводят следую, щим образом. В градуированный мерный цилиндр емкостью 100 мл вводят 2,0 мл раствора нода и 1 мл раствора индикатора. Отбирают образец воды погруженной в нее трубкой, другой конец которой опушен ниже уровня реагентов в цилиндре. Отмечают объем анализируемого раствора, при котором синяя окраска реагентов исчезает. Этот объем содержит 200 мкг сульфидной и сульфитной серы и для аналитических испытаний необходимо взять не более этого объема. Если окраска не исчезает от прибавления 95 мл раствора образца, то этот объем используют для анализа.
Отбирают подходящий объем раствора образца в градуированный цилиндр емкостью 100 мл, содержащий 2 мл раствора ацетата цинка. Для пробоотбора применяют стеклянную погружную трубку, конец которой опущен ниже поверхности ацетата цннка. В случае необходимости доводят объем раствора до 97 мл водой и осторожно перемешивают. Прибавляют в цилиндр 1,0 мл раствора ДПД и вновь перемешивают содержимое. Оставляют стоять раствор 60 ± 30 с и затем
прибавляют 1,0 мл раствора сульфата аммония-железа. Перемешивают раствор н разбавляют его до 100 мл.
Раствор оставляют стоять, по крайней мере, в течение 10 мин, после чего
измеряют светопоглощение раствора, используя кювету с толщиной поглощающего слоя 40 или 5 мм в зависимости от содержания сульфида. Измерение проводят в спектрофотометре при той же длине волны и температуре (с точностью до 1 °C), что и при построении калибровочного графика. В качестве рас,нора
сравнения используют воду.
Контрольный раствор реагента готовят следующим образом. В градуированный мерный цилиндр емкостью 100 мл вводят 2 мл раствора ацетата циика, 95 мл воды, 1 мл раствора ДПД и 1 мл раствора сульфата аммония-железа. Разбавляют содержимое водой до метки и перемешивают раствор.
Приготавливают окрашенный контрольный раствор в другом градуированном цилиндре емкостью 100 мл. В этом цилиндре к 2 мл раствора ацетата цинка прибавляют выбранный объем анализируемого раствора и при необходимости разбавляют раствор до 97 мл водой. Прибавляют 1 мл раствора H2SO4, разбавляют раствор до 100 мл и перемешивают содержимое цилиндра.
Измеряют светопоглощение двух контрольных растворов при тех же условиях (длине волны, толщине поглощающего слоя и температуре), что и при измерении оптической плотности образца. В качестве раствора сравнения исполь-
зуют воду.
Расчет. Вычитают оптические плотности, полученные для контрольного раствора реагента и окрашенного контрольного раствора, из оптической плотности, полученной для образца, и вычисляют содержание сульфида в образце по калибровочному графику.
Сульфидная сера, ppm
найдено серы, мкг объем образца, мл
Флуориметрические методы
Сульфиды реагируют с соединением ртути(II) с 2,2/-пиридил-бензимидазолом с образованием сульфида ртутн и выделением флуоресцирующего органического основания в количестве, эквива-
672
рис- 6®- Прибор для измерения молеку-дрной эмиссии в полости (помещение Образца в резонатор и введение резонатора в пламя) [54]:
|_ввод образца; 2—детектор.
лентном сульфиду. На этой реакции основан высокочувствительный метод определения сульфидов [53]. Помимо высокой чувствительности достоинствами реагента являются его устойчивость и возможность измерения флуоресценции в течение 48 ч.
Предложен косвенный флуо-риметрический метод определения сульфидов с использованием 2-(о-оксифенил) бензоксазола в качестве реагента. При взаимо-
действии сульфидов с этим реа-
гентом в ацетоновой среде наблюдается ярко-зеленая флуоресценция, определению сульфидов этим методом мешает медь(П), ослабляющая флуоресценцию из-за образования комплексного соединения с реагентом. Метод применяют для определения О—100 нг/мл сульфидов с пределом обнаружения 1 нг [54].
Метод молекулярной эмиссии в полости
Метод молекулярного эмиссионного резонанса предложен для определения некоторых серусодержащих анионов, включая сульфиды [55]. Этот метод основан на измерении интенсивности эмиссии ассоциата S2 при 384 нм.
t Прибор для измерения молекулярной эмиссии показан на рис. 68. Излучение из резонатора проходит через щель шириной 8,5 нм и монохроматор в фотоумножитель с присоединенным к нему самопишущим потенциометром с быстрым срабатыванием. Размер пиков измеряют геометрически или с помощью электронного интегратора. В работе [55], включающей описание техники метода, указано, что сульфид натрия дает широкий пик эмиссии в течение очень короткого промежутка времени. Прибавление к анализируемому раствору фосфорной кислоты, что дает положительный эффект при анализе других серусодержащих ионов, при определении сульфидов не оказывает такого действия из-за потерь сероводорода. Использование аммонийно-фосфатного буферного Раствора при pH = 7 не влияет на положение максимума эмиссии, но зато приводит к значительному сужению пика. С применением выше приведенного буферного раствора калибровочный гРафик для определения сульфида линеен вплоть до 0,65 мкг этого аниона.
573
Электрохимические методы
Потенциометрические методы
Титриметрический метод определения сульфида титрованием его раствором цинка (II) в аммиачной среде с металлохромнымц индикаторами приведен выше. Это титрование можно провести потенциометрически [56].
Гексацианоферрат(Ш) реагирует с сульфидом в сильнощелоч-ной среде в присутствии тетраоксида осмия как катализатора:
S2- + 2 [Fe (CN)6]3‘ —* S + 2 [Fe (CN)6]4-
Для индикации точки эквивалентности можно использовать метод «конечной мертвой точки» или обычное потенциометрическое титрование [57].
Для анализа сульфидсодержащих смесей потенциометрическим методом в качестве титранта предложено серебро(1), этот же титрант используют для анализа смесей сульфида, полисульфида и тиосульфата [58]. В этом случае сульфид аммония титруют аммиачным раствором нитрата серебра. Тиосульфат титруют хлоридом ртути(II) после удаления сульфида кипячением анализируемого раствора с борной кислотой. В последнюю очередь определяют полисульфид, для чего анализируемый раствор обрабатывают сульфитом (для превращения полисульфида в тиосульфат) и титруют образовавшийся тиосульфат. Титрование растворов ведут с платиновыми электродами, индикацию конечной точки осуществляют по методу «конечной мертвой точки». В этом варианте метод скорее можно назвать амперометрическим, чем потенциометрическим.
Для прямого определения сульфидов щелочных металлов в качестве титранта можно использовать раствор ацетата кадмия с индикацией точки эквивалентности методом «конечной мертвой точки» [59]. Определению сульфидов в , этом случае мешают цианиды и ионы гидроксила, не мешают определению азотистая, соляная и серная кислоты.
Детально изучено потенциометрическое титрование сульфидов ионами серебра в сильнощелочном аммиачном растворе [60]. В качестве индикаторного электрода при титровании предложено использовать вращающийся серебряный (или сульфидно-серебря-ный) электрод, а в качестве стандартного — нормальный каломельный. Авторы отмечают быстрое достижение равновесия при титровании. Описанный метод позволяет определять 0,2—4 мг сульфидов.
Для анализа смесей сульфидов с цианидами рекомендован раствор иода в сильно щелочной среде [61]. Если сульфид находится в смесях с полисульфидами, сульфитом и тиосульфатом, то наиболее подходящим титрантом является хлорид ртути(II) [62].
674
^Подробно этот метод дан в разделе, посвященном определению Ьлнсудьфидов.
г Для определения сульфидов (или ионов серебра) вплоть до Ю 7 М можно испотьзовать ионоселективный электрод на основе сульфида серебра [63]. Определению сульфидов с помощью этого электрода мешает ртуть(II), и она должна быть предварительно удалена. Мешающее действие цианидов сказывается прн их концентрации выше 10-3 М [64]. Работа ионоселективного электрода ORION 94-16 была подвергнута всестороннему анализу [65]. Сульфид при низких содержаниях (10-5—1(Н М) удовлетворительно мржно определить только в том случае, если окисление его пренебрежимо мало, и в связи с этим при определении малых концентраций сульфидов к анализируемому раствору следует до-'бавлять аскорбиновую кислоту [66].
Определению сульфидов с помощью ионоселективных электродов не мешают фториды, хлориды, сульфаты, карбонаты, фосфаты и ионы гидроксила. Отсутствие мешающего влияния галогенидов делает эти электроды особенно ценными при потенциометрическом титровании смесей галогенидов и сульфидов, например, в фотографических материалах.
Йоноселективные электроды предложены для анализа смесей серусодержащих анионов, в том числе сульфидов. Например, при анализе смесей полисульфида, сульфида, сульфита и тиосульфата используют титрование раствора хлоридом ртути(II), а сульфидный ионоселективный электрод применяют для индикации рту-ти(П) [67, 68]. Более подробно метод описан в разделе «Полисульфид».
Потенциометрическое титрование сульфидов нитратом серебра при низких содержаниях сульфидов неосуществимо из-за их гидролиза и образования гидроксида серебра. Применяя плюмбат(П) натрия в качестве титранта, можно определить до 1 ppm сульфидов в присутствии 105—106-кратного избытка хлоридов, бромидов, иодидов, сульфитов, тиосульфатов или тиоцианатов. Цианид при определении сульфидов описываемым методом должен отсутствовать [69]. Соли свинца(II) предложено использовать как титрант при автоматическом потенциометрическом титровании на-нограммовых количеств сульфидов [70]. Стандартное отклонение определений составляет ±2% (при уровне содержания сульфидов 90 нг). Определению сульфидов этим методом не мешают галогениды, ацетат, сульфат, цианид, нитрат, фосфат и ионы аммония, рписываемый метод использован для определения серы в органических соединениях [71]. После сожжения образца серу восстанавливают в токе водорода над платиновым катализатором при 900 °C и образующийся сероводород поглощают в специальном сосуде. Автоматически титруют сульфиды стандартным раствором свинца(II) с фиксацией конечной точки сульфидным ионоселек-тивным электродом.
675
Кулонометрические методы
Для определения 4—85 мкг сульфидов предложено кулонометрическое титрование с внутренней .генерацией ионов серебра в основном цианидном растворе [72]. Точку эквивалентности титрования индицируют потенциометрически или амперометрически. Определению сульфидов описываемым методом не мешают 100-кратные избытки хлоридов, бромидов, иодидов, тиоцианатов и тиосульфатов. Следовые содержания сульфидов можно определить автоматически, используя электрогенерацию иода [73]. Этот метод позволяет определять 1—50 мкг серы в виде сероводорода при объеме образца 10 мл.
Полярографические методы
Изучение [74] прямого полярографического определения сульфидов показало, что они образуют четко выраженную волну на
Рис. 69. Традиционная н экспрессная переменно-токовые полярограммы 1,5 10" раствора сульфидов в 1 М растворе перхлората натрия: а—период капания 2,9 с; б —период капания 0,16 с [75].
ртутном капающем электроде. Однако результаты, получаемые методами анодной полярографии, не совсем удовлетворительны. Экспрессные полярографические методы, в которых используют короткие периоды капания ртутных капель, часто имеют преимущества перед традиционными методами. Экспрессные полярографические методы использованы для определения сульфидов [75]-На рис. 69 сопоставлены полярограммы, полученные экспрессным и традиционным полярографическими методами (в обоих случаях использованы одинаковые капилляры с равными по размеру каплями ртути, различались лишь периоды капания и скорости ска-
576
пирования). Большим достоинством описанного метода является отсутствие мешающего действия цианидов, неизбежного при определении сульфидов многими другими методами.
Описаны косвенные методы определения сульфидов с использованием полярографии. Один из методов основан на реакции сульфидов с иодидом метилртути(П) [76]. Метод позволяет определять менее 1 ppm сульфидов с относительной ошибкой, не превышающей ±5%. Определению сульфидов не мешает несколько распространенных анионов, однако цианид мешает определению. Для определения сульфидов использована реакция образования метиленового синего по реакции сульфидов с М,М-диметил-п-фени-лендиамином в присутствии железа(III) [77]. В этом случае измеряют высоту полярографического пика метиленового синего при —0,2 В. Калибровочный график определения сульфидов линеен в интервале 10—90 мкМ сульфидов, определению мешают галогениды.
Кинетические и каталитические методы
Каталитический метод определения сульфидов основан на реакции, катализируемой сульфидом
Fe2+4-Ag+ —> Fe3t + Ag
Для определения используют спектрофотометрическую регистрацию скорости реакции (вариант фиксированного времени). Чувствительность определения сульфидов этим методом составляет 1-Ю-13 г/мл [78].
Иод-азидная реакция, также катализируемая сульфидом, положена в основу другого метода определения сульфидов [79]:
2Мз+Ь —> 3N24-2I’
Для контроля скорости реакции применяют амперометрию (вращающийся платиновый электрод). Содержание сульфидов этим методом можно определить на трех различных уровнях: 10-1Б, 10-14 и 10~13 г/мл. Эту реакцию также используют для определения тиоцианатов и тиосульфатов.
Для определения сульфидов, цианидов и иодидов предложен энзиматический метод [80], основанный на ингибировании некоторыми ионами металлов каталитического действия энзима Р-фруктофуранозидазы (инвертазы) в реакциях гидролиза таких субстратов, как сахароза. Если в анализируемом растворе содержатся анионы, например сульфиды, образующие прочные комплексные соединения с этими ионами металлов, то эти анионы замещают энзим в координационной сфере металла, что приводит к уменьшению эффекта ингибирования. Этот метод позволяет определять (1—2,5) -10—7 М сульфидов. Скорость реакции контролируют по вращению плоскости поляризации. Калибровочный график для определения сульфидов необходимо строить ежедневно.
19 Зак. 167
677
Другие методы определения
Нанограммовые содержания сульфидов можно определять по реакции
2Ag,s,I + S2- —> Ag2S + 2,3II’
Для определения предложены две методики (81]. В соответствии с первой анализируемый раствор сульфида встряхивают с кружком фильтровальной бумаги, на которую нанесен меченый иодид серебра. По второй методике пропускают анализируемый раствор через фильтр с нанесенным меченым иодидом серебра. Для фиксации у-излучения используют одноканальный анализатор с детектором Nal.
Для определения сульфидов (и хлоридов) применяют колонку, содержащую хромат серебра, который мечен изотопом 51Сг [82]. При использовании колонки 25ХЮ мм2 можно определять 0,4— 40 мкМ сульфидов в водных растворах. Определению сульфидов мешают бромиды, иодиды, йодаты, цианиды, тиоцианаты и сульфиты. Метод пригоден для анализа водных растворов с pH 4,0.
Газовая хроматография также находит применение для определения сульфидов [83]. Например, после кислотной обработки анализируемого раствора можно газохроматографически анализировать смеси сульфидов, сульфитов и карбонатов [84]. Для определения используют газовый хроматограф с термокопдукто-метрическим детектором, чувствительность определения составляет 1—10 мг сульфидов и 2—18 мг сульфитов.
Для определения этих анионов предложен другой газохроматографический метод [28], устройство для подкисления в этом случае присоединено к входу хроматографа.
ЛИТЕРАТУРА
1. Blair J. S. — Analyt Chem., 1969, v. 41, p. 1497.
2. Szekeres L. — Talanta, 1974, v. 21, p. 2.
3. Blasius E., Horn G., Knochel A., Munch J., Wagner H. — In: Inorganic Sulphur Chemistry (Ed. G. Nickless), Amsterdam, Elsevier. 1968, p. 199.
4. Hanley A. V., Czech F. №. — In: The Analytical Chemistry of Sulphur and its Compounds, Part 1 (Ed. J. H. Karchmer), New York, Wiley-Interscience, 1970, p. 285.
5. Lowenheim F. A. — In: Encyclopedia of Industrial Chemical Analysis (Ed F. D. Snell and L. S. Ettre), Vot. 18, New York, Interscience, 1973, p. 392.
6. American Public Health Association, American Waterworks Association, and Water Pollution Control Federation, Standard Methods for the Examination of Water and Wastewater, 13th edn., New York, American Public Health Association, 1970.
7. Official, Standardised and Recommended Methods of Analysis, Compiled and Edited by N W. Hanson, 2nd edn., London, The Society for Analytical Chemistry, 1973, p 462.
8. Beswick G., Johnson R. M. — J. Sci. Fd Agric., 1970, v. 21, p. 565.
9. Gustafsson J. — Talanta, 1960, v. 4, p. 236.
10. Pomeroy R. — Analyt. Chem., 1954, v. 26, p. 571.
11. Koithoff I. M., Belcher R. — Volumetric Analysis, Vol. Ill, New York, Interscience, 1957, p. 295.
578
12. Iguchi A. — Bull. chem. Soc. Japan, 1958, v. 31, p. 600; Analyt. Abstr., 1959, v. 6, p. 1705.
13. Paez D. M., Guagnini O. A. — Mikrochim. Acta, 1971, p. 220.
14. Canic V. D., Turcic M. N., Gubarski-Vojinovic M. B., Perisic N. U. — Z. analyt. Chem., 1967, v. 229, p. 93; Analyt. Abstr., 1968, v. 15, p. 5211.
15. Patel S. S., Spencer С. P. — Analytica chim. Acta, 1962, v. 27, p. 278.
16. Debevere J. M., Voets 1. P. — Lab. Pract., 1972, p. 713.
17. Natusch D. F. S., Klonis H. B., Axelrod H. D., Teck R. J., Lodge J. P. — Analyt. Chem., 1972, v. 44, p. 2067.
18. Umland F., Janssen A. — Z. analyt. Chem., 1967. v. 224, p. 413; Analyt. Abstr., 1968, v. 15, p. 2600.
19. Szekeres L. — Analytica chim. Roma, 1962, v. 52, p. 67; Analyt. Abstr., 1962, v. 9, p. 5140.
20. Szekeres L.— Z. analyt. Chem., 1960, v. 178, p. 81; Analyt. Abstr., 1961, v. 8, p. 2387.
21. Szekeres L. — Acta chim. hung., 1961, v. 26, p. 167.
22. Szekeres L. — Pharm. Zentralhalle Dtl., 1963, v. 102, p. 6; Analyt. Abstr., 1963, v. 10, p. 5137.
23. Official, Standardised and Recommended Methods of Analysis, Compiled and Edited by N. W. Hanson, 2nd edn., London, The Society for Analytical Chemistry, 1973, p. 460.
24. Wronski M. — Analyst, 1958, v. 83, p. 314.
25. Wronski M.— Analyt. Chem., 1971, v. 43, p. 606.
26. Axelrod H. D., Cary J. H., Bonelli J. E., Lodge J. P. — Analyt. Chem., 1969, v. 41, p. 1856.
27. Andrews T. R., Nichols P. N. R. — Analyst, 1965, v. 90, p. 367.
28. McDonald K. L.— Analyt. Chem., 1972, v. 44, p. 1298.
29. Kivalo P. — Analyt. Chem.. 1955, v. 27, p. 1809.
30. Burriel-Marti F., Alvarez Herrero C. — Inf. quim. analit. pura apl. Ind., 1964, v. 18, p. 142; Analyt. Abstr., 1966, v. 13, p. 132.
31. De Sousa A. — Inf. quim. analit., 1963, v. 17, p. 51; Analyt. Abstr., 1964, v. 11, p. 1266.
32. De Sousa A. — Inf. quim. analit., 1961, v. 15, p. 121; Analyt. Abstr., 1962, v. 9, p. 1452.
33. Sarwar M., Elahi M., Anwar M., Hanif M. — Mikrochim. Acta, 1970, p. 846.
34. Szekeres L., Kardos E., Szekeres G. L. — Chemist Analyst, 1964, v. 53, p. 115.
35. Szekeres L., Kardos E., Szekeres G. L. — Chemist Analyst, 1965, v. 54, p. 70.
36. Fischer E.— Ber., 1883, v. 16, p. 2234.
37. Zuishi P. K; Mahadevan T. N. — Talanta, 1970, v. 17, p. 1014.
38. Mecklenburg M., Rosenkranzer F. — Z. anorg. Chem., 1914, v. 86, p. 143.
39. Gusiafsson L. — Talanta, 1960, v. 4, p. 227.
40. British Standards 2690: Part 14, 1972, London.
41. Grasshoff К. M., Chan К. M. — Analytica chim. Acta, 1971, v. 53, p. 442.
42. Rees T. D., Gyllenspetz Л. B., Docherty A. C. — Analyst. 1971, v. 96, p. 201.
43. Bernstein V. N., Biliker V. G. — Russian chem. Revs., 1961, v. 30, p. 227.
44. Casapieri P., Scott R., Simpson E. A. — Analytica chim. Acta, 1969, v. 45, p. 547.
45. Bethea N. J., Bethea R. M. — Analytica chim. Acta, 1972, v. 61, p. 311.
46. Hoffman E. — Z. analyt. Chem., 1962. v. 185, p. 372; Analyt. Abstr., 1962, v. 9,
p. 3016.
47. Humphrey R. E., Hinze W. — Analyt. Chem., 1971, v. 43. p. 1100.
48. Lambert J. L., Manzo D. J. — Analytica chim. Acta, 1969, v. 48, p. 185.
49. Раманаускас E., Григониене К. — Научи, труды высш, учебн. заведений Лит. ССР. Химия и хим. техиол., 1971, т. 13, с. 21; Analyt. Abstr., 1973, 24, 114.
50. Еремин Ю. Г., Киселева К. С. —ЖАХ, 1969; т. 24, с. 1201; Analyt. Abstr., 1971, v. 20, р. 978.
61. Rahim S. A., West Т. S. — Talanta, 1970, v. 17, p. 851.
62. Rahim S. A., Salim A. K, Shereef S. — Analyst, 1973, v. 98, p. 851.
53. Bark L. S., Rixon A. — Analyst, 1970, v 95, p. 786.
54. Vernon F., Whitham P. — Analytica chim. Acta, 1972, v. 59, p. 155.
19*
579
55. Belcher R„ Bogdsnski S. L., Knowles D. J., Townshend A. — Analytica chim. Acta, 1975, v. 77, p. 53.
56. Gallus J. — Chemia analit., 1957, v. 2, p. 249.
57. Solymosi F., Varga A. — Analytica chim. Acta, 1957, v. 17, p. 608.
58. Kiss S. A. — Z. analyt. Chem., 1962, v. 188, p. 341; Analyt. Abstr., 1963, v. 10 p. 112.
59. Kiss S. A. — Taianta, 1961, v. 8. p. 726.
60. Liu С. H., Shen S. — Analyt. Chem., 1964, v. 36, p. 1652.
61. Норкус П., Шемкявичюте Г. ЖАХ, 1971, т. 26, с. 39; Analyt. Abstr., 1972 v. 22, р. 2234.
62. Danielsen A. J., Johnsen К., Landmark Р. — Meddr. Pap Ind. Forsk Inst., No. 232 (1969); Norsk skogind., 1969, v. 23, p. 77; Analyt. Abstr., 1970, v. 19 p. 395.
63. Orion 94-16 electrode, Orion Research Inc U. S. Available from E. I. L. Ltd, Chertsey, England. Radelkis OP-S-711, Radelkis, Hungary.
64. Vesely J., Jensen O. J., Nicolaisen B. — Analytica chim. Acta, 1972, v. 62, p. 1.
65. Light T. S., Swartz J. L. — Analyt. Lett., 1968, v. 1, p. 825.
66. Bock R., Puff H.-J. — Z. analyt. Chem., 1968, v. 240, p. 381; Analyt. Abstr., 1970, v. 18, p. 159.
67. Papp J. — Cellulose Chem. Technol., 1971. v. 5, p. 147: Analyt. Abstr., 1972, v. 22, p. 2404.
68. Papp J., Havas J. — Magy. кёт. Foly, 1970, v. 76, p. 307; Analyt. Abstr., 1971, v. 20, p. 4439.
69. Naumann R., Webber C. — Z. analyt. Chem., 1971. v. 253, p. Ill; Analyt. Abstr., 1971, v. 21. p. 4071.
70. Slanina J., Buysman E., Agterdenbos J., Griepink B. F. A. — Mikrochim. Acta, 1971, p. 657.
71. Slanina J., Agterdenbos H., Griepink B. F. A. — Mikrochim. Acta, 1970, p. 1225.
72. Cadersky I. — Z. analyt. Chem., 1968, v. 239, p. 14; Analyt. Abstr., 1969, v. 17, p. 3420.
73. Pribyl M., Slovak Z. — Mikrochim. Acta, 1963, p 119.
74. Kolthoff I. M., Miller C. S. — J. Am. chem Soc., 1941, v. 63, p. 1405.
75. Canterford D. R. — Analyt. Chem., 1973, v. 45, p. 2414.
76. Gruen L. C. —Analytica chim. Acta, 1970, v. 52, p. 123.
77. Hasebe K., Kambara T. — Bull. chem. Soc. Japan, 1970, v. 43, p. 2110; Analyt. Abstr., 1971, v. 20, p. 3772.
78. Бабко А. К., Максименко T. C. — ЖАХ, 1967, t. 22, c. 570; Analyt. Abstr., 1968, v. 15, p. 6621.
79. Michalski E., Wtorkowska A. — Chemia analit., 1962, v. 7, p. 691; Analyt. Abstr., 1963, v. 10, p. 2244.
80. Mealor D., Townshend A. — Taianta, 1968, v. 15, p. 1477.
81. Krivan V., Pahlke S., Tolg G. — Taianta, 1973, v. 20, p. 391.
82. Bowen H. J. M. — Radiochem. radioanalyt Lett., 1970, v. 3, p. 339.
83. Rodriguez-Vazquez J. A. — Analytica chim. Acta, 1974, v. 73, p. 1.
84. Birk J. R., Larsen С. M., Wilbourn R. G. — Analyt. Chem., 1970, v. 42, p. 273.
СУЛЬФИТЫ И ДИОКСИД СЕРЫ
Сульфит находит применение в различных областях деятельности человека. В металлургической промышленности его использование евязано с контролем за процессом обжига сульфидных руд, а также с контролем сточных жидкостей предприятий металлургической промышленности. В теплосетях сульфит часто прибавляют к бойлерной жидкости для снижения содержания растворенного кислорода в ней. Сульфит используют как восстановительный агент в процессах отбелки. Хорошо известно его применение при консервировании пищевых продуктов. Большие ко
680
личества сульфита применяют в процессе приготовления бумажной пульпы.
Сульфит часто образуется как промежуточное соединение при анализе серусодержащих материалов, например, при определении серы в нефти [1] или пищевых продуктах [2] после сожжения в кислороде. При этих анализах образующийся сульфит окисляют, обычно пероксидом водорода, и получающийся сульфат определяют титрованием перхлоратом бария. Окисление до сульфата с последующим определением последнего часто используют для определения сульфита. Подходящим окислителем является, например, водный раствор брома. Этот метод зачастую применяют, если сульфит находится в смеси с другими серусодержащими анионами.
Для маскирования сульфита рекомендован формальдегид:
Na2SO3 + НСНО + Н2О —> СН2 (ОН) SO3 Na + NaOH
Несколько обзоров, описывающих аналитическую химию серусодержащих соединений, включают определение сульфита [3—5]. Аналитическая химия сульфитов во многом определяется химией диоксида серы, и поэтому в этом разделе уместно рассмотреть методы определения SO2. Определение этого соединения тем более необходимо описать, что диоксид серы является агентом, вызывающим значительное загрязнение окружающей среды. Подробное обсуждение методов определения SO2 дано в конце этого раздела, хотя некоторые ссылки сделаны ранее.
МЕТОДЫ ОТДЕЛЕНИЯ
Отделение в виде диоксида серы
При подкислении растворов сульфитов выделяется SO2, который можно перенести инертным газом-носителем и затем поглотить щелочным раствором. Диоксид серы, отделенный этим методом, определяют иодиметрически или, в случае следовых содержаний сульфитов, — спектрофотометрически. Для поглощения следов диоксида серы предложен раствор хлорида ртути (II) [6].
Если тиосульфат присутствует вместе с сульфитом, то при действии иа анализируемый раствор минеральной кислоты происходит разрушение обоих анионов с выделением SO2. Тиосульфат, однако, не разрушается в уксуснокислом буферном растворе.
Метод ионообменной хроматографии
Смеси сульфитов, сульфатов, тиосульфатов и сульфидов можно разделить анионообменным методом, используя для элюирования растворы нитратов различной концентрации [7]. Сульфиты и сульфиды элюируются смесью 0,1 М раствора нитрата аммония с ацетоном (30%) при pH = 9,7.
Для разделения смеси сульфитов, тиосульфатов и низших по
581
литионатов предложена анионообменная хроматография с использованием смолы De-Acidite FF [8]. Сульфиты и тиосульфаты элюируются и разделяются 2 М раствором гидрофталата калия.
Метод тонкослойной хроматографии
Смеси сульфитов, сульфатов, тиосульфатов и пероксодисульфатов разделяют методом тонкослойной хроматографии на неподвижной фазе силикагель + крахмал, в качестве подвижной фазы используют смесь метанол — пропанол — концентрированный NH4OH—Н2О (10: 10:1:2) (9]. Другой метод предложен для разделения смеси сульфида, сульфита, сульфата и тиосульфата [10]. В качестве неподвижной фазы авторы используют микрокристаллическую целлюлозу, в качестве подвижной — смесь «-пропанола— 1 М раствор NH4OH — ацетон (30:20:2). Количественная оценка содержания разделяемых анионов проведена с применением кольцевых печей.
Другие методы отделения
Для отделения сульфитов от мешающих ионов можно предложить гравиметрический метод. Этот метод используют для отделения сульфитов от сульфидов. Когда оба аниона присутствуют в смеси, сульфид можно осадить карбонатом цинка или кадмия. Для предотвращения окисления сульфитов в процессе фильтрования осадка сульфидов рекомендуют добавление к анализируемому раствору глицерина.
Об использовании формальдегида в качестве маскирующего агента для сульфита упомянуто ранее. Маскирование формальдегидом часто используют для анализа смесей, например тиосульфата и сульфита. При анализе такой смеси в одной аликвотной части анализируемого раствора сумму обоих ионов определяют иодиметрически. Вторую аликвотную часть обрабатывают формальдегидом и иодиметрически определяют один тиосульфат. Содержание сульфита рассчитывают по разности.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Существует мнение [11], что удовлетворительные методы определения макросодержаиий сульфитов отсутствуют. Наиболее приемлемым, вероятно, является иодиметрический метод.
Гравиметрические методы
Сульфиты легко превращаются в сульфаты, которые затем можно определить в виде сульфата бария. Удовлетворительные результаты достигаются при окислении сульфитов несколькими окислителями: бромной водой, аммиачным раствором пероксида водорода и гипобромитом натрия. Избыток окислителя затем раз
662
рушают кипячением растворов. Метод приводит к значительным погрешностям, и поэтому его можно рекомендовать лишь при отсутствии возможностей титриметрического определения.
Титриметрические методы
Иодиметрические методы
Иодиметрический метод определения сульфитов известен уже более 100 лет и тем не менее остается, вероятно, наиболее используемым методом определения при макросодержаниях сульфитов:
SOi' + h + HjO —> SOf+ 2Г+2Н*
Потенциальный источник погрешности этого метода связан с частичным окислением сульфитов атмосферным кислородом. Для минимизации этой погрешности рекомендуют следующий прием. К анализируемому образцу прибавляют избыток стандартного раствора иода и титруют неизрасходованный иод тиосульфатом. Другой предложенный вариант заключается в следующем: известный объем стандартного раствора иода титруют раствором сульфита [12]. Этот метод значительно менее удобен. Для уменьшения окисления сульфитов рекомендуют вводить анализируемый раствор пипеткой в колбу для титрования, погружая кончик пипетки в глубь раствора иода. Более поздние исследования показали, что эта рекомендация излишняя.
В качестве индикатора иодиметрического титрования предложен люминол, хемилюминесценция которого наблюдается в присутствии иода в щелочных растворах; сульфит гасит хемилюминесценцию индикатора, восстанавливая иод до иодида [13]. С использованием люминола можно определить вплоть до 0,1 мг сульфита натрия в присутствии интенсивно окрашенных веществ. При определении 25 мг сульфита натрия ошибки определения
' составляют 0,01—0,06 мг.
Иодиметрический метод определения сульфитов находит широкое применение. Для определения сульфита в промышленных водах Британские стандарты рекомендуют подкисление анализируемого раствора, прибавление избытка стандартного раствора КЮ—KI и титрование неизрасходованного иода стандартным раствором тиосульфата [14]. Фактически такая же методика предложена Обществом аналитической химии [15]. Стандартные методы ’исследования вод [16] рекомендуют аналогичную методику с тем лишь исключением, что предлагается прямое титрование анализируемого раствора смесью КЮ3 с KI.
Иодиметрический метод можно применить для определения атмосферного SO2. Определенный объем исследуемого воздуха ^пропускают через раствор NaOH, который затем подкисляют и титруют стандартным раствором иода. Погрешности в этом случае ' вызываются мешающим действием диоксида азота, озона и восстановителей, таких, как сероводород.
683
Сульфит можно определять в присутствии других серусодер-жащих анионов, используя иодиметрический метод. Этим методом анализируют смеси сульфита с сульфидом и тиосульфатом [17] и сульфита с сульфидом и сульфатом [18]. Обе методики приведены в разделе, посвященном определению сульфида. Анализ смеси сульфита с дитионатом и сульфатом [19] описан в разделе «Дитионит». Описаны анализ дитионита натрия, содержащего сульфиты и тиосульфаты, и анализ смеси сульфита, тиосульфата и тетратионата [20, 21]. При анализе последней смеси использовано то обстоятельство, что сульфит и тиосульфат окисляются иодом, тогда как бром окисляет все три аниона. Содержание сульфита и тиосульфата определяют раздельно после маскирования сульфита формальдегидом.
В 1930 г. был предложен простой иодиметрический метод определения дитионита, бисульфита и тиосульфата при их совместном присутствии [22]. Критический анализ предложенного метода [23] показал его несомненные достоинства.
Иодиметрическое титрование сульфита
Ход анализа. Избыток стандартного раствора иода (в соответствии с предполагаемым содержанием сульфита) помещают в колбу, прибавляют туда анализируемый образец (предпочтительно в твердом виде), закрывают колбу пробкой и перемешивают до растворения образца. Неизрасходованный избыток стандартного раствора иода титруют стандартным раствором тиосульфата, применяя крахмал в качестве индикатора вблизи конечной точки титрования.
Примечания 1. Определению сульфита мешают железо(П), ртуть(1) и мышьяк(П1), а также несколько других анионов.
2. Для определения сульфита в присутствии других восстановителей первую аликвотную часть анализируемого раствора титруют выше приведенным способом. Этот результат соответствует содержанию всех присутствующих восстановителей. Ко второй аликвотной части анализируемого раствора до титрования прибавляют формальдегид. В этом случае формальдегид маскирует сульфиты и разность полученных результатов соответствует содержанию сульфитов [24].
Другие редокс-методы
Для титриметрического определения сульфитов предложены окислители: перманганат калия, бихромат калия, сульфат церия (IV), хлорамин Т, хлорит натрия, перхлорат таллия(III) и гексацианоферрат(Ш) калия [25, 26]. Однако ни один из этих реагентов не имеет преимущества по сравнению с иодом.
Перхлорат таллия(III) является слабым окислителем, быстро реагирующим с сульфитом, тиосульфатом и тиокарбамидом [27]. Эти восстановители можно определить прямым титрованием перхлоратом таллия (III) в присутствии индикатора п-этоксихризо-идина или косвенным методом:
Т1,п + SO?" + Н2О —> Т11 + 2Н+ + SOf
Определению мешают хлориды и бромиды, образующие прочные комплексные соединения с таллием(III). При косвенном опреде-
684
ленип мешают медь(П) и железо(П). Этот титрант нашел применение для анализа смесей сульфид — сульфит — тиосульфат с индигокармином в качестве индикатора [28]. Использование этого титранта для анализа приведенной смеси анионов позволяет получить приемлемые результаты, одиако замена таллием(III) иода в более старых методиках приводит к появлению значительных ошибок. Преимущества перхлората таллия(III) связаны с тем, что он сохраняет свои окислительные свойства в течение длительного периода времени и не подвергается влиянию воздуха и света.
Комплексометрические методы
Для определения сульфитов предложен комплексометрический метод, основанный на том, что ЭДТА образует устойчивые комплексные соединения с таллием(III), но не с таллием (I). При определении к анализируемому раствору прибавляют избыток стандартного раствора таллия(III) и его неизрасходованный избыток титруют стандартным раствором ЭДТА при pH = 6,0 с ксиленоловым оранжевым в качестве индикатора [29].
Другие комплексометрические методы определения сульфитов основаны на том обстоятельстве, что сульфаты легко определяются комплексометрически после осаждения сульфата бария. Такой метод использован’ при анализе смесей сульфатов, сульфидов и сульфитов [30]. Этот метод описан в разделе «Сульфат».
Аналогичный метод нашел применение для анализа смесей сульфидов и сульфитов [31]. Сульфиты окисляют водным раствором брома и образующийся сульфат определяют комплексометрически после осаждения сульфата бария. Сходный метод предложен для анализа смесей сульфитов и сульфатов [32]. В одной аликвотной части анализируемого раствора разрушают сульфиты, а остающиеся сульфаты определяют комплексометрически. Во второй аликвотной части окисляют сульфит до сульфата, после чего определяют суммарное содержание сульфатов и сульфитов. Содержание сульфитов определяют по разности.
Спектроскопические методы
Спектрофотометрические методы в видимой
и ультрафиолетовой областях спектра
Сульфит реагирует с гидрохлоридом парафуксина и формальдегидом, образуя пурпурно-красный продукт — парафуксинметил-сульфоновую кислоту. Эта реакция находит применение для определения диоксида серы в атмосферном воздухе. Этот метод основан на ранее предложенном способе селективного определения гидросульфита [33] с помощью гидрохлорида фуксина. При определениях применяют подкисленный (для обесцвечивания красителя) раствор реагента. Фуксин также применен для определения SO2 в воздухе [34]. Для поглощения диоксида серы из воздуха
685
предложено использовать раствор тетрахлормеркурата(П), а вместо фуксина рекомендованы замещенные трпаминотрнфенилмета-новые красители [35]:
SO2 + Н2О + [HgCl4]2' —> [HgCLSOs]2* + 2Н* + 2СГ Образующийся дихлоросульфитомеркурат(П) очень стабилен и это позволяет избежать окисления сульфитов.
Осложнения, возникающие при использовании триаминотрифенилметановых красителей, обусловлены загрязнениями реагентов, что отрицательно сказывается на чувствительности и воспроизводимости метода. Для решения задачи о целесообразности применения красителя проверяют его спектральные характеристики [36]; описаны методы очистки парафуксина [37], которые дают возможность улучшить чувствительность и воспроизводимость метода, а также расширить интервал определяемых концентраций по сравнению с данными работы [35]. Кроме того, очистка реагента минимизирует мешающие влияния оксидов азота, озона и тяжелых металлов. Парафуксинсульфоновая кислота обладает следующими максимумами светопоглощения: 575 нм при pH = 1,2 ± 0,1 и 548 нм при pH = 1,6 ± 0,1.
Сравнение аналитических характеристик фуксина и парафуксина проведено после очистки обоих реагентов методом хроматографии на бумаге [38]. В результате анализа сульфита, выполненного очищенными реагентами, лучшие результаты получены при использовании парафуксина, что объясняется меньшим результатом контрольного опыта. При проведении реакции сульфита с парафуксином и формальдегидом в разбавленном растворе НС1 (pH =1,1) калибровочный график для определения сульфитов линеен вплоть до 80 мкг SO2. Однако авторы работы [38] нашли, что очистку реагента методом хроматографии на бумаге трудно использовать в повседневной аналитической работе. Они установили, что основание парафуксина, которое образуется при обработке кислого раствора гидрохлорида парафуксина раствором гидроксида натрия, обладает преимуществами перед другими красителями триаминотрифенилметанового ряда. Это основание можно очистить перекристаллизацией из водного раствора метанола, при этом образуется чистый и стабильный реагент.
Определению сульфитов с помощью этого реагента мешают оксиды азота. Их влияние устраняют прибавлением сульфаминовой кислоты к поглотительному раствору.
При определении сульфитов с помощью бихромата прибавляют избыток окислителя к анализируемому раствору и затем измеряют уменьшение светопоглощения [39]; определение сульфитов этим методом неселективно. Предложен метод определения сульфитов, основанный на восстановлении ими железа(III) в растворе, содержащем 2,4,6-три (пиридил)-1,3,5-триазол и пропиленкарбонат [40]. Образующееся железо (П) реагирует с триазолом, давая комплексное соединение — бис [2,4,6-три- (2-пиридил) 1,3,5-триазин]-
В86
железо(II), обладающее светопоглощением с максимумом прн 593 нм. Закон Бугера — Ламберта — Бера справедлив в интервале 3—57 мкг диоксида серы. Мешают определению сероводород и марганец(П).
Сходный метод определения сульфитов основан на восстановлении ими железа(III) в присутствии 1,10-фенантролина [41]. Этот метод является модификацией ранее предложенного метода [42].
Метод определения сульфитов с хлоранилатом ртути(II) основан на их взаимодействии с образованием недиссоциированного сульфита ртути (II) и интенсивно окрашенного хлоранилат-иона:
HgCh + SOi’ —> HgSO3 + Ch2'
Светопоглощение растворов измеряют при 525 или 330 нм, что дает возможность определять сульфиты в интервале 5—100 мкг/мл и 0,5—8,0 мкг/мл [43].
При определении сульфитов с 4,4'-дитиопиридином или 5,5'-ди-тиобис (2-нитробензойной кислотой) калибровочный график строят в интервале концентрации диоксида серы 0,2—6,0 мкг/мл [44]. Определение сульфита с тиоцианатом ртути(II) проводят в метанольной или этанольной среде (в водном растворе реакция не протекает), в этом случае при реакции сульфитов с реагентом тиоцианат выделяется в количестве, эквивалентном содержанию сульфитов. Образующийся тиоцианат определяют в виде его комплексного соединения с железом (III) [45].
Для анализа смесей сульфита, тиосульфата и политионатов (тетра-, пента- и гекса-) предложен простой спектрофотометрический метод [46], описанный в разделе «Тиосульфат».
Для анализа смесей сульфита, сульфата, сульфида и тиосульфата предложен прямой метод анализа, основанный на измерении светопоглощения растворов в ультрафиолетовой части спектра [47]. Детали этого метода описаны в разделе «Сульфид». Определение сульфита и диоксида серы можно провести по другой методике, также основанной на измерениях в ультрафиолетовой области [48]. В соответствии с этой методикой анализируемый раствор подкисляют раствором серной кислоты и затем измеряют светопоглощение растворов при 276 нм. Чувствительность определения сульфитов при этой длине волны низка, и для ее увеличения измерения лучше проводить при 198 нм [49]. Эта замена позволяет увеличить чувствительность определения в 4 раза и определять до 1 мкг/мл диоксида серы. Помехи определению сульфитов при 276 нм оказывают сульфиды и нитриты. При 198 нм определению сульфитов не мешают фториды, сульфаты, фосфаты, хлориды и цианиды, тогда как сульфиды, нитриты, тиосульфаты, бромиды, нитраты, тиоцианаты и иодиды имеют светопоглощение в этой области и мешают определению сульфитов. Мешающее действие этих анионов можно устранить выделением диоксида серы из подкисленного анализируемого раствора с последующим
687
поглощением его щелочным раствором ЭДТА (в качестве газа-носителя используют азот) [48].
Описан метод определения сульфитов, основанный на образовании ими комплексного соединения с ртутью (II) состава 1 :2 в среде 0,05—1,0 Л1 H2SO< [50]. Этот комплекс имеет максимум светопоглощения при 237 нм.
Спектрофотометрическое определение диоксида серы с помощью гидрохлорида парафуксина [38]
Реагенты
Парафуксин, обесцвеченный раствор. Реагент очищают следующим образом Растворяют 1 г гидрохлорида парафуксииа в 250 мл 2,5 М раствора НС1. Раствор выдерживают 2 ч и фильтруют осветленный раствор через фильтровальную бумагу (ватман № 1). Прибавляют к фильтрату небольшой избыток 2,5 Л1 раствора NaOH для осаждения основания. Фильтруют раствор через стеклянную фильтрующую пластину (пористость Ns 3) и тщательно промывают осадок на фильтре дистиллированной водой для удаления хлорида и избытка гидроксида натрия. Основание перекристаллизовывают, растворяя его в 70 мл кипящего метанола и прибавляя затем 300 мл воды при 80 °C. Охлаждают полученный раствор до комнатной температуры. Выход перекристаллизованного продукта составляет около 64%. Растворяют 1,0 г очищенного основания парафуксина в 60 мл концентрированной HCI и разбавляют раствор до 1 л водой.
Раствор реагента устойчив в течение 6 недель при хранении его в темном месте.
Тетрахлормеркурат калия, раствор 27,2 г хлорида ртути(П) и 14,9 г хлорида калия разбавляют водой до 1 л.
Формальдегид, 0,2%-ный раствор. Разбавляют 5 мл 40%-ного раствора формальдегида до 1 л.
Стандартный раствор сульфита, приготовляют раствор 0,4 i безводного сульфита натрия в 500 мл воды, что соответствует концентрации диоксида серы 300—400 мкг/мл. Точную концентрацию сульфита определяют иодиметрическим методом; 1 мл 0,05 М раствора иода соответствует 3,2030 мг диоксида серы.
Непосредственно после устаиовлеиия титра раствора сульфита этот раствор стабилизируют, для чего 10 мл раствора сульфита разбавляют раствором тетра-хлормеркурата калия до объема 500 мл, полученный раствор имеет концентрацию 6—8 мкг/мл диоксида серы.
Ход анализа. Переносят аликвотную часть анализируемого раствора сульфита, содержащего не более 80 мкг диоксида серы, в мерную колбу емкостью 50 мл, прибавляют по 10 мл растворов реагента и формальдегида. Через 20 мин измеряют оптическую плотность раствора в кювете с толщиной поглощающего слоя 1 см при 572 нм относительно раствора реагента.
Флуорометрические методы
Флуориметрический метод определения сульфитов основан на реакции гидросульфитного производного формальдегида с 5-ами-нофлуоресцеином [51], при которой образуется нефлуоресцирующий продукт. Метод применим для определения диоксида серы, выделенного с помощью тетрахлормеркурата(П). Определению не мешают КН2 М растворы нитратов, сульфатов, пероксида водорода, железа (II) и аммония. В присутствии 10~2 М растворов калия, кальция, магния, меди(П), ацетата и нитрита погрешность определения достигает 5%. Такая же ошибка наблюдается в присут
Б88
ствии Ю“3 М раствора иодида. Железо(III) при его содержании 10~2 М приводит к появлению ошибки 45%. С помощью описанного метода можно определять до 0,02 мкг/мл диоксида серы.
Атомно-абсорбционный метод
Определение сульфитов атомно-абсорбционным методом проводят косвенно. В одном из вариантов к анализируемому раствору сульфита прибавляют суспензию оксида ртути(II), что вызывает перенос ионов ртути в жидкую фазу из твердой в связи с образованием очень прочных комплексных соединений [Hg(SO3)2]2-[52]. Аналогично сульфиту с оксидом ртути реагируют иодиды, тиосульфаты и тиоцианаты; эти анионы мешают определению сульфитов. Не мешают определению ионы аммония, сульфаты, нитриты, бромиды и цианиды. Этот метод можно использовать для определения до 83 ppm сульфитов.
Другой метод определения сульфитов методом атомной абсорбции основан на определении избытка ионов свинца в растворе после осаждения сульфата свинца [53]. Предварительное окисление сульфита до сульфата проводят пероксидом водорода. Этот метод позволяет определять 2—20 ppm диоксида серы. Определению сульфитов при их содержании 5 ppm не мешает 500 ppm хлоридов, нитритов, нитратов, перхлоратов, Na*, К*, NHJ, Са2+ и Mg2*-Допустимо присутствие следующих ионов (в ppm): ацетата — 250, ЭДТА — 20, Ва2+ и А13+—10. При определении сульфитов должны отсутствовать железо(III) и фосфаты.
Метод молекулярной эмиссии в полости
Метод молекулярного эмиссионного резонанса применяют для определения сульфитов, также как и для определения других серу-содержащих анионов [54, 55]. Техника этого метода, основанного на измерении эмиссии ассоциата серы S2 при 384 нм, более подробно описана в разделе «Сульфид». Сульфиты и сульфаты обладают хорошо разрешимыми сигналами и поэтому могут быть определены одновременно. Сульфиты и тиосульфаты дают одинаковые сигналы и поэтому не определяются одновременно. Метод позволяет определять 3—300 ppm сульфитов в образцах объемом 3—5 мкл.
Электрохимические методы
Потенциометрические методы
Для прямого потенциометрического титрования сульфитов применяют хлорит натрия [56]. Оптимальное значение pH при титровании 4—4,6. При pH < 4 наблюдаются потери SO2 вследствие разрушения сульфитов; при pH > 5 реакция сульфитов с
589
хлоритом протекает медленно и неколичественно. Даже при тщательном соблюдении требуемого значения pH степень открытия сульфита составляет 94,0—94,2%, так что требуется эмпирическая стандартизация реагента.
Для потенциометрического титрования сульфитов предложено использовать раствор иода в 2—10 М растворе NaOH с платиновым и каломельным электродами [57]. Смеси сульфита, полисульфида и тиосульфата, а также сульфита, полисульфида, тиосульфата и сульфида можно анализировать потенциометрически, применяя хлорид ртути(II) в качестве титранта [58, 59] (методы описаны в разделе «Полисульфиды»).
Для определения серусодержащих соединений применяют ионоселективные электроды. В частности, при анализе смеси сульфидов, тиосульфатов, сульфитов и полисульфидов проводят потенциометрическое титрование анализируемого раствора хлоридом ртути(II) с сульфидным электродом в качестве индикаторного [60, 61] (см. раздел «Полисульфиды»)..
Для определения диоксида серы в выхлопных газах предложено использовать свинцовый ионоселективный электрод [62]. В этом случае анализируемый газ пропускают через 3%-ный раствор пероксида водорода и затем потенциометрически титруют образующийся сульфат стандартным 0,1 М раствором свинца(II). Результаты, получаемые этим методом, сопоставимы с результатами, полученными титриметрически с использованием бария(II) и торона. Для определения сульфитов можно применять сульфитный ионоселективный электрод [63, 64], область применения этого электрода—10-6—10~2 М растворы сульфитов. Определению сульфитов этим методом при их концентрации 10-4 М мешают З-Ю^Л! фтористоводородная кислота, 5-10~3М уксусная кислота и >1М НС1 [64].
Полярографический метод
Полярографический метод позволяет определять сульфиты с высокой чувствительностью, однако этому определению мешают тиосульфаты и некоторые другие серусодержащие ионы. В кислом растворе сульфиты восстанавливаются одностадийно или в несколько промежуточных стадий в зависимости от pH электролита. В 1 М растворе НС1 волна сульфита соответствует его восстановлению до дитионита, тогда как при pH = 6 сульфит восстанавливается до тиосульфата [65].
Для устранения мешающего действия посторонних ионов при анализе сточных вод предложен следующий метод [66]. Через нейтральный или щелочной анализируемый раствор пропускают ток азота, при этой операции из раствора удаляется кислород, но без потерь диоксида серы. Затем анализируемый раствор подкисляют и вновь пропускают азот для удаления образующегося диоксида серы. Разность между диффузионными токами, измеренными
690
непосредственно после подкисления раствора и после удаления SO2, пропорциональна концентрации сульфитов.
Предложен полярографический метод определения диоксида серы, основанный на поглощении газообразного SO2 раствором' Na2HgCi4 с образованием комплексного иона [HgCl2SO3]2- [67]. На волну восстановления этого иона накладывается волна иона [HgCh]2-. Эту помеху устраняют восстановлением мешающего иона до элементной ртути в щелочной среде с последующим удалением ртути. После установления pH = 1,0 измеряют волну восстановления SO2.
Описан автоматический полярографический метод анализа концентрированных растворов сульфитов, образующихся при поглощении промышленных выхлопных газов водным раствором аммиака [68]. В качестве фонового электролита в этом случае применяют 3,5 М H2SO4. Метод позволяет определять вплоть до 0,1 М сульфитов.
Спектрофотометрические методы определения хлоридов, цианидов, фторидов, сульфатов и сульфитов с использованием хлор-анилатов металлов находят широкое применение. Как альтернативный к спектрофотометрическому методу предложено полярографическое определение этих анионов, основанное на замещении ими хлоранилат-ионов [69]. В этом методе использовано обратимое двухэлектронное восстановление хлоранилат-иона на ртутном капающем электроде [70]. Полярографический ток пропорционален концентрации анализируемых ионов, для сульфитов такая зависимость соблюдается в интервале их концентраций 5-Ю-5— 5-10~3 М. В качестве реагента используют хлоранилат ртути. Для замедления окисления сульфита к анализируемому раствору прибавляют глицерин. Метод применяют, если анализируемый раствор содержит окрашенные ионы.
Кулонометрические методы
Кулонометрия является одним из наиболее используемых в настоящее время методов определения серусодержаших соединений, особенно в случае их низких содержаний. Микрокулонометрический метод применяют, например, для определения следовых концентраций серы в легких фракциях нефти [71]. Диоксид серы, образуемый при сожжении анализируемого образца, поступает в ячейку, где он окисляется электрогенерированным иодом:
i; + so2 + h2o —> so3 + 3i + 2н+
31 —> I3 -f~2e
Ток, требуемый для регенерации 13> подают через группу точных сопротивлений на самописец и дисплей. Углеводороды при
691
сожжении образуют СО2 и Н2О и не мешают определению серы. Результаты анализа вычисляют из закона Фарадея:
® = о^--|0Э УОтУоМ
где w — масса титруемого вещества, нг; Q — количество электричества, Кл; М — молекулярная масса титруемого вещества; п — число электронов, обмениваемых в реакции.
Количество электричества определяют по формуле
Q = It = 4” Ю3-60£Д А
где /— ток, A; t—время, с; v — чувствительность самописца, мВ/см; R — сопротивление группы резисторов; S—обратная скорость движения диаграммной бумаги, мин/см; А — площадь под пиком сигнала.
Описаны другие кулонометрические методы определения серы в нефтепродуктах [72, 73]. В одном из этих методов, как и в ранее упомянутых, используют переменнотоковую кулонометрию. В другом применен более простой метод постоянно-токовой кулонометрии. Методы позволяют определять 0,01—1,0% серы в нефтепродуктах после сожжения образцов до SO2.
Для индикации конечной точки при кулонометрическом титровании SO2 применена дифференциальная потенциометрия [74].
Другие методы определения
Методы определения сульфитов и сульфидов в их смесях часто сложны и неэкспрессны. Для анализа таких смесей предложен газохроматографический метод [75], основанный на образовании H2S, SO2 и СО2 при подкислении анализируемого раствора из сульфидов, сульфитов и карбонатов соответственно. Метод можно применять для определения миллиграммовых количеств сульфитов.
Сульфит и гидросульфит натрия можно определять методом термометрического титрования [76].
Определение атмосферного SO2
Сведения о методах определения атмосферного SO2 приведены в работах, цитируемых в данном разделе.
В качестве поглотительного раствора для газообразного SO2 применяют тетрахлормеркурат(П) натрия, этот поглотитель рекомендован исследователями США [77]. Чувствительность метода в зависимости от объема образца воздуха составляет 0,005— 0,2 ppm диоксида серы.
Реакция диоксида серы с тетрахлормеркуратами(П) изучена разными исследователями [37, 38]. Эта реакция применена для параллельного фотометрического анализа [79] с использованием фотометрического анализатора [80]. В этом приборе анализируют одновременно 15 образцов, находящихся в 15 кюветах, рас
592
положенных в роторе анализатора. При вращении ротора каждая кювета проходит через луч света и вторичный прибор (осциллоскоп) регистрирует светопоглощение растворов в каждой кювете. Анализатор позволяет значительно увеличить экспрессность определения сульфитов, что особенно необходимо при рутинных определениях SO2. Область определяемых концентраций SO2 с помощью этого метода составляет 0,25—2,5 ppm.
При иодиметрическом определении SO2 в воздухе в качестве поглощающего раствора применяют раствор NaOH. После подкисления этого раствора определяют SO2, титруя его стандартным раствором иода. Некоторые газы мешают определению диоксида серы, особенно диоксид азота, сероводород и озон.
Относительно простой метод определения диоксида серы основан на пропускании анализируемого газа через разбавленный раствор пероксида водорода при pH == 5. Образующуюся при этом H2SO4 титруют стандартным раствором NaOH. Некоторые ионы мешают определению.
Описан пламенно-фотометрический детектор для определения SO2 [81]. Этот детектор находит применение для определения серы в органических соединениях на уровне ниже 1 ppm. В детекторе используют фотометрическое детектирование эмиссии серусодержащих соединений в пламени водород — воздух. Детектор применен для непрерывной регистрации содержания SO2 в атмосфере на уровне ниже 0,06 ppm [82].
ЛИТЕРАТУРА
1. Lonsdale М.— Proc. Soc. analyt. Chem., 1970, v. 7, p. 12.
2. Beswick G., Johnson R. M.— Taianta, 1970, v 17, p. 709
3. Szekeres L. — Taianta, 1974, v. 21, p. 2.
4. Blasius E., Horn G., Knochel A., Munch J., Wagner H. — In: Inorganic Sulphur Chemistry (Ed. G. Nickless), Amsterdam, Elsevier, 1968, p. 199.
5. Haff L. V. — In: The Analytical Chemistry of Sulphur and its Compounds (Ed. J. H. Karchmer), Part 1, New York, Wiley lnterscience, 1970, p. 223.
6. West P. W., Gaeke G. C. — Analyt. Chem., 1956, v. 28, p. 1816.
7. Iguchi A. — Bull. chem. Soc. Japan. 1958, v. 31, p. 600; Analyt. Abstr., 1959, v. 6, p. 1705
8. Pollard F. H., Nickless G., Glover R. B.—J. Chromat., 1964, v. 15, p. 533.
9. Seiler H., Erlenmeyer H. — Helv. chim. Acta, 1964, v. 47, p. 264; Analyt. Abstr., 1965, v. 12, p. 1703.
10. Handa A. C., Johri K. N. — Taianta, 1973, v. 20, p. 219.
11. Sengar H. G. S., Gupta Y. K. — Taianta, 1965, v. 12, p. 185.
12. Kolthoff I. M.,— Z. analyt. Chem., 1921, v. 60, p. 448.
13. Луковская H. M., Маркова JI. B. — ЖАХ, 1969, t. 24, c. 1893. Analyt. Abstr., 1971, v. 21, p 162.
14. British Standards 2690: Part 2 (1965), London.
15. Hanson N. W. (Ed.), Official, Standardised and Recommended Methods of Analysis, London, The Society for Analytical Chemistry, 1973, p. 463.
16. American Public Health Association, American Waterworks Association, Water Pollution Control Federation, Standard Methods for the Examination of Water and Wastewater, 13th edn., New York, American Public Health Association, 1971, p. 337.
17. Szekeres L. — Ann. chim. Roma, 1962, v. 52, p. 67; Analyt. Abstr., 1962, v. 9, p. 5140.
20 Зак. № 167
593
18. Umland F.— Z. analyt. Chem., 1967, v. 224, p. 413; Analyt. Abstr., 1968, v. 15, p. 2600.
19. Soffer N.— Analyst, 1961, v. 86, p. 843.
20. Szekeres L. — Z. analit. Chem., 1964, v. 203, p. 178; Analyt. Abstr., 1965, v. 12, p. 5130.
21 Szekeres L., Sugar E. — Magy. Kem. Lap., 1961, v. 16, p. 434; Analyt. Abstr., 1962, v. 9, p. 1453.
22. Woliak R. — Z. analyt. Chem., 1930, v. 80, p. 1.
23. Danehy J. P., Zubritsky C. W.— Analyt. Chem., 1974, v. 46, p. 391.
24. Prater A. N., Johnson C. AL, Poole M. F., Mackinney G. — Ind. Engng Chem. analyt. Edn., 1944, v. 16, p. 153.
25. Berka A., Vulterin J., Zyka J. — Newer Redox Titrants, Oxford, Pergamon Press 1965
26. Kolthoff I. M„ Belcher R. — Volumetric Analysis, Vol. Ill, New York, Inter-science, 1957.
27. Sharma P. D., Gupta У. K-— Taianta, 1974, v. 21, p. 168.
28. Sharma D. N., Sharma P. D., Gupta У. K. —Taianta, 1976, v. 23, p. 326.
29. Szekeres L., Kellner A. — Chemist Analyst, 1966, v. 55, p. 77.
30. Burriel-Marti F., Alvarez-Herrero C.— Inf. quim. analit., 1964, v. 18, p. 142; Analyt. Abstr., 1966, v. 13, p. 132.
31. De Sousa A. — Inf. quim. analit., 1963, v. 17, p. 51; Analyt. Abstr., 1964, v. 11, p. 1266.
32. De Sousa A. — Inf. quim. analit., 1962, v. 16, p. 177; Analyt. Abstr., 1963, v. 10, p. 3667.
33. Steigmann A. — Analyt. Chem., 1950, v. 22, p. 492.
34. Urotie P., Boggs W. E., Noyes С. M. — Analyt.* Chem., 1951, v. 23, p. 1517.
35. West P. W., Gaeke G. C. — Analyt. Chem., 1956, v. 28, p 1816.
36. Pate J. B., Lodge J. P., Wartburg A. F. — Analyt. Chem., 1962, v. 34, p. 1660.
37. Scaringelli F. P., Saltzman В. E., Frey S. A. — Analyt. Chem., 1967, v. 39, p. 1709.
38. King H. G. C„ Pruden G. — Analyst., 1969, v. 94, p 43.
39. Sussman S„ Portnoy I. L. — Analyt. Chem., 1952, v. 24, p. 1652.
40. Stephens B. G., Suddeth H. A. — Analyst, 1970, v. 95, p. 70.
41. Attari A., Igilski T. P., Jaselkis B. — Analyt. Chem., 1970, v. 42, p. 1282.
42. Stephens B. G., Lindstrom F. — Analyt. Chem., 1964, v. 36, p. 1308.
43. Humphrey R. E., Hinze W. — Analyt. Chem., 1971. v. 43, p. 1100.
44. Humphrey R. E., Ward M. H., Hinze W.— Analyt. Chem., 1970, v. 42, p. 698.
45. Hinze W., Elliott J., Humphrey R. E. — Analyt. Chem., 1972, v. 44, p. 1511.
46. Koh T., Taniguchi K- — Analyt. Chem., 1973, v. 45, p. 2018.
47. Еремин Ю. Г., Киселева К. С. — ЖАХ, 1969, т. 24, с. 1201; Analyt. Abstr., 1971, v. 20, с. 978.
48. Scoggins М. W. — Analyt. Chem., 1970, v. 42, p. 1091.
49. Bhatty M. K., Townshend A. — Analytica chim Acta, 1971, v. 55, p. 263.
50. Okutanl T., Ito S., Utsumi S. — J. chem. Soc. Japan, pure Chem. Sect., 1967, v. 88, p. 1296; Analyt. Abstr., 1969, v. 16, p. 1849.
51. Axelrod H. D., Bonelli J. E., Lodge J. P. — Analyt. Chem., 1970, v. 42, p. 512, 743.
52. Jungreis E., Anavi Z. — Analytica chim. Acta, 1969, v. 45, p. 190.
53. Rose S. A., Boltz D. F. — Analytica chim. Acta, 1969, v. 44, p. 239.
54. Belcher R., Bogdanski S. L., Knowles D. J., Townshend A. — Analytica chim. Acta, 1975, v. 77, p. 53.
55. Belcher R., Bogdanski S. L., Knowles D. J., Townshend A. — Analytica chim. Acta, 1975, v. 79, p. 292.
56. Minczewski J., Glabisz U.— Taianta, 1960, v. 5, p. 179.
57. Норкус П., Шемкявичюте Г. — ЖАХ, 1971, т. 26, с. 39; Analyt. Abstr., 1972. v. 22, р. 2234.
58. Danielsen А. Л, Johnsen К-, Landmark Р. — Norsk. Skogind., 1969, v. 23, p. 378. Analyt. Abstr., 1971, v. 21, p. 1252.
59. Danielsen A. J., Johnsen K., Landmark P. — Meddr. Pap Ind Forsk Inst.. No. 232; Norsk. Skogind., 1969, v. 23, p. 77; Analyt. Abstr., 1970, v. 19, p. 395.
594
60. Papp J-, Havas J. — Magv кёт. Foly, 1970, v. 76, p. 307; Analyt. Abstr., 1971, v. 20, p. 4439.
61. Papp J. — Cellulose Chem. Technol., 1971, v. 5, p. 147; Analyt. Abstr., 1972, v. 22, p. 2404.
62. Foung M„ Driscoll J. N., Mahoney K. — Analyt. Chem., 1973, v. 45, p. 2283.
63. E. I. L. Electrode No. 8010, E. I. L. Hanworth Lane, Chertsey, Surrey, England.
64. Orion Electrode No. 95-64, MSE Scientific Instruments, Manor Royal, Crawley, West Sussex, England.
65. Kolthoff I. M., Miller C. S. — J. Am. chem. Soc., 1941, v. 63, p. 1405.
66. Aulenbach D. B., Balmat J. L. — Analyt. Chem., 1955, v. 27, p. 562.
67. Ciaccio L. L., Cotsis T. — Analyt. Chem., 1967, v 39, p. 260.
68. Strafelda F., Dolezal J. — Colin Czech, chem. Commun. Engl. Edn., 1967, v. 32, p. 2707; Analyt. Abstr., 1968, v. 15, p. 6622.
69. Humphrey R. E., Laird С. E.— Analyt. Chem., 1971, v. 43, p. 1895.
70. Wessbart J., Rysselberghe D. — J. phys. Chem., Ithaca, 1957, v. 61, p. 765.
71. Killer F. C. A., Underhill К E. — Analyst, 1970, v. 95, p. 505.
72. Dixon J. P. — Analyst, 1972, v. 97, p. 612.
73. Carter J. M. — Analyst, 1972, v. 97, p. 929.
74. Bailey P. L., Bishop E. — Analyst, 1972, v. 97, p. 311.
75. Birk J. R., Larsen С. M., Wilbourne R. G. — Analyt. Chem., 1970, v. 42, p. 273. 76. Priestley P. T. — Analyst, 1963, v. 88, p. 194.
77. ASTM D2914-70T, American Society for Testing and Materials, Philadelphia, Pa. (1972).
78. Scarangelli F. P., Elfers L., Norris D., Hochheiser S.—Analyt. Chem., 1970, v. 42, p. 1818.
79. Coleman R. L., Shults W. D., Kelley M. T., Dean J. A. — Analyt. Lett., 1972, v. 5, p. 169.
80. Anderson N. G. — Analyt. Biochem., 1969, v. 28, p. 545.
81. Brody S. S., Chaney J. E. — J. Gas Chromat., 1966, v. 4, p. 42.
82. Stevens R. K, O’Keeffe A. E. — Analyt. Chem., 1970, v. 42, p. 143A (Feb.).
ТИОСУЛЬФАТЫ
Наибольшее применение тиосульфаты находят в фотографии, кроме того, их восстановительные свойства используют для отбеливания.
Следует подчеркнуть, что тиосульфат играет очень большую роль в аналитической химии. Несмотря на введение новейших реагентов, титрование иода тиосульфатом остается одной из наиболее применяемых аналитических операций. Тиосульфат является более сильным восстановителем, чем сульфит. В нейтральных растворах мягкие окислители, такие как иод, окисляют тиосульфат до тетратионата. Пероксид водорода, бром и хлор окисляют тиосульфат до сульфата. В слабокислых растворах тиосульфат неустойчив и распадается на сульфит, политионат и элементную серу.
МЕТОДЫ ОТДЕЛЕНИЯ
Осаждение ионами серебра
В разбавленных азотнокислых растворах ионы серебра осаждают тиосульфаты. Это можно использовать при отделении тиосульфатов от ионов, которые не образуют осадков в данных условиях.
20*
595
Ионообменный метод
Для отделения тиосульфатов используют ионный обмен. Для разделения сульфатов, сульфитов, тиосульфатов и сульфидов применяют анионообменные смолы в нитратной форме [1]. В дальнейшем используют различные элюенты. Так, для смеси сульфатов, сульфитов и тиосульфатов предложено первоначальное элюирование аммиачным раствором нитрата натрия для удаления сульфата и сульфита. Затем нейтральным раствором нитрата натрия элюируется тиосульфат.
Для разделения смеси тиосульфата, сульфита и тритионата можно применить анионообменную смолу Dowex 1-XZ и растворы NaCI и НС1 в качестве элюентов [2].
Анионный обмен использован для разделения смеси тиосульфатов, сульфитов и низших политионатов (тритионата, тетратионата и пентатионата) [3].
Метод тонкослойной хроматографии
Для отделения тиосульфата от других ионов предложено несколько методик, использующих тонкослойную хроматографию (табл. 41).
Таблица 41. Отделение тиосульфатов методом тонкослойной хроматографии
Неподвижная фаза Подвижная фаза Анионы» от которых отделяют тиосульфат Литера тура
Силикагель — крахмал Кукурузный крахмал Метанол — пропанол — 0,88 М аммиак—вода (10 : 10 : 1 : 2) Метанол — диоксан — 0,88 М аммиак — вода (3 : 6 : 1 : 1) Ацетон — 3 М четвертичные аммониевые основания SO*-, SO*-, Занизите политионаты no;, scn', его*-, n;, CN', BO|', S*', AsO*', AsO*', NO3', SO*', PO*’ 4 5
Пластины SA и SG Несколько, в том числе бутанол — пиридин — уксусная кислота — вода (15 : 10:3: 12) SO*', SCN', S,O*', 4 r 9 3 6’ s4o*- 6
Силикагель Этанол — бутанол — 0,88 М аммиак — 2 М ацетат аммония — вода (75:75:8-10:20-40:10-30) S3O*', S4O*', S5o*- 7
Экстракция
Для отделения тиосульфатов предложено несколько экстракционных методик. Описана экстракция тиосульфатов с помощью 596
основных красителей с последующим определением тиосульфатов [45]. Этот метод приведен в разделе «Спектрофотометрические методы».
Высокоскоростная жидкостная хроматография
Высокоскоростная жидкостная хроматография находит применение для разделения тиосульфатов и политионатов [66]. Этот метод приведен в разделе, посвященном определению политионатов.
МЕТОДЫ ОПРЕДЕЛЕНИЯ
Аналитическая химия тиосульфатов описана в нескольких обзорах по определению серусодержащих соединений [8—11].
Гравиметрические методы
Основной гравиметрический метод определения тиосульфатов основан на их окислении пероксидом водорода или бромом до сульфата с последующим гравиметрическим определением сульфатов в виде их бариевой соли. Определению тиосульфатов этим методом мешают другие серусодержащие соединения, методу присущи общие недостатки, характерные для гравиметрических методов.
Титриметрические методы
Иодиметрические методы
Из всех редокс-методов иодиметрическое определение тиосульфатов является наиболее важным и широко распространенным:
2S2O’- + I2 —> S4Oe2' + 2I-
Прежде чем обсуждать применение этой реакции для определения тиосульфатов, рассмотрим некоторые ее особенности. Детальное изучение этой реакции дает возможность установить условия ее проведения, позволяющие получить правильные результаты определений. Эти условия в основном приведены, в монографиях [12, 13]. При прямом титровании, т. е. при добавлении тиосульфата к иоду, раствор иода должен иметь нейтральную или слабокислую реакцию. В щелочных растворах иод реагирует с гидроксильными ионами, образуя иодноватистую кислоту:
12 + ОН" НЮ+Г
или гипоиодиты:
I2 + 20 Ы"
1О' + Г + Н2О
697
Образующаяся иодноватистая кислота окисляет тиосульфаты до сульфатов:
4HIO + S2Og“ + 6ОН" 4Г + 2SO*'+ 5Н2О
Ситуация осложняется в связи с протеканием реакции диспропорционирования иодноватистой кислоты в йодаты и иодиды. Проведение титрования иода тиосульфатом даже в слабощелочной среде ведет к частичному окислению тиосульфата гипоиодитом или йодатом до сульфата, это приводит к отрицательным ошибкам при определениях тиосульфата.
Если раствор иода подкислен слишком сильно, то возможны ошибки другого рода. Появление одной из ошибок связано с возможностью частичного разложения тиосульфата. Некоторые литературные источники указывают, что титрование довольно сильно кислых растворов иода возможно при условии, что раствор титранта прибавляют медленно, непрерывно перемешивая титруемый раствор.
Второй источник ошибок связан с окислением иодидов кислородом воздуха, которое ускоряется в кислых растворах. Таким образом, при титровании необходимо, чтобы титруемый раствор был нейтральным иди слабокислым. При обратном титровании, т. е. когда прибавляют раствор иода, тиосульфат должен быть подкислен в слабой степени для предотвращения его разложения. Если титровать тиосульфат иодом в щелочной среде, то окисление тиосульфата идет дальше тетратирната и это приводит к ошибкам. При определении тиосульфата йодометрическим методом рекомендуют титровать стандартный раствор иода анализируемым раствором тиосульфата.
Во многих случаях, например при анализе продажного тиосульфата натрия, образец должен иметь нейтральную реакцию, тогда упомянутые источники ошибок не могут возникнуть. Аналогичная ситуация возникает при анализе безводного тиосульфата натрия для фотографии [14]. Продажные образцы тиосульфата часто содержат примеси сульфитов. Для их маскирования применяют формальдегид и после этого тиосульфат титруют иодом без помех. Точку эквивалентности при титровании устанавливают визуально по появлению или исчезновению окраски иода. Однако обычно при титровании вблизи точки эквивалентности вводят раствор крахмала, который образует окрашенный в интенсивно синий цвет адсорбционный комплекс с иодом.
Полисахариды крахмала разделяются на две основные фракции: амилозу, имеющую неразветвленную или слабо разветвленную структуру, и амилопектин, обладающий разветвленной структурой. Эти фракции образуют крахмал, находясь в его составе в переменных соотношениях в зависимости от происхождения крахмала. Например, картофельный крахмал содержит значительные количества амилозы. Именно амилоза образует синий комплекс с иодом, тогда как амилопектин с иодом дает красноватопурпурный комплекс. При титровании целесообразно применять 598
свежеприготовленные растворы крахмала, поскольку это позволяет получить очень резко выраженную конечную точку титрования. В ботее старых растворах крахмала появляется красноватый оттенок при реакции с иодом, что приводит к ухудшению контрастности при определении. Кинетическое изучение [15] реакции амилозы с подом показало, что лимитирующей стадией процесса является образование полииодной цепи In внутри структуры амилозы.
Раствор крахмала готовят следующим образом. Смешивают около 2 г крахмала с небольшим количеством воды и выливают суспензию в 200 мл кипящей дистиллированной воды. Продолжают нагревание, непрерывно перемешивая раствор, до тех пбр, пока он не станет прозрачным. Полученный раствор охлаждают и разбавляют дистиллированной водой до 1 л.
Следует заметить, что при использовании раствора крахмала необходимо принимать меры для подавления побочных реакций. Один из возможных путей преодоления этой трудности состоит в применении твердых крахмальных растворов. Например, сообщено [16],. что 5%-ный твердый раствор крахмала в карбамиде непосредственно растворяется в водной среде. Этот твердый раствор обладает высокой степенью устойчивости и обеспечивает получение четкой конечной точки титрования, его выпускают в виде продажного продукта «Thyodene».
Стандартный раствор иода для титрования тиосульфатов можно приготовить, растворив подходящее взвешенное количество чистого кристаллического иода. Однако эта операция неудобна, ее редко используют. Лучший в практическом отношении метод связан с приготовлением раствора иода примерной концентрации и стандартизации его с помощью оксида мышьяка (III):
As2O3 + 2L, + 5Н2О 2HAsO3'+ 4Г + 8Н+
Эта методика приведена ниже.
Наиболее удобный и используемый метод приготовления стандартного раствора заключается в получении известного количества иода по реакции подкисленного избытка иодида с точно взвешенным количеством йодата:
1О3’ + 5Г + 6Н* —> 312 + ЗН2О
Образующийся в реакции иод титруют стандартным раствором тиосульфата. Вместо йодата для окисления иодида можно применять бихромат калия, но эта реакция при низкой кислотности раствора протекает медленно:
Сг2О2" + 6Г + 14Н+ 7Z* 2Сг3+ + 312 + 7Н,0
При повышенной кислотности, однако, протекание реакции окисления иодида кислородом воздуха приводит к ошибкам. Таким образом, в рассматриваемом отношении иодат обладает преимуществами по сравнению с бихроматом. Необходимо заметить, что
59Э
использование йодата может привести к погрешностям в связи с малой навеской. Например, при титровании 25 мл 0,1 М раствора тиосульфата масса йодата калия, которая соответствует иоду, эквивалентному рассматриваемому объему тиосульфата, составляет 0,0892 г. Взятие такой навески на аналитических весах (точность взвешивания ±0,1 мг) приводит к значительной погрешности. Поэтому обычно рекомендуют готовить 250 или 500 мл раствора йодата калия и для стандартизации отбирать аликвотные части этого раствора.
Для титрования тиосульфата предложен раствор церия (IV) [29]. Это титрование проводят в присутствии иодида калия, который прибавляют до титрования в анализируемый раствор:
2Се,у±2Г —> 12 + 2Сеш
I2 + 2S2O^- —> 2Г ± S4O|-
Точку эквивалентности при титровании находят визуально с помощью крахмала. Для титрования микросодержаний тиосульфатов необходимо применять модифицированную методику [30].
На иодиметрическом титровании тиосульфата основано несколько методов анализа смесей серусодержащих анионов, например метод анализа смеси дитионита, гидросульфита и тиосульфата, который описан в разделе «Дитионит». Установлено, что применение этого метода в модифицированной форме позволяет определять тиосульфаты непосредственно в смесях, содержащих окисляемые иодом соединения серы [17].
Многие иодиметрические методы анализа смесей, содержащих тиосульфаты и другие серусодержащие анионы, приведены в других разделах. Для удобства эти методы представлены в табл. 42.
Таблица 42. Иодиметрические методы определения тиосульфатов в смесях с другими серусодержащими анионами
Смесь Примечание Литература
Полисульфид — SCN-—S2O2' Методика дана в разделе «Поли- 18
сульфид»
s2o2-—so2*—s2o|* Методика дана в разделе «Ди- 19-21
ТИОНИТ»
S2O2'— SO2-— S2O2’ Бромометрическая методика 22
S2"—SO2"—S2°2" Методика дана в разделе «Суль- 23
фид»
Полнсульфнд— S2'—S,O2' Методика дана в разделе «Поли- 24-26
сульфид». Рассмотрена бромометрическая методика
Полисульфид — S2"—S2O2' 27
Полисульфид — SO2-—S2-—S2O2' Аналогичен методу, приведенному в работах [24—26] 28
600
Иодиметрическое определение тиосульфатов
Ход анализа. Прямое титрование. Прибавляют 2 мл крахмала к раствору тиосульфата. Титруют стандартным раствором иода до появления не-исчезаюшей синей окраски.
Обратное титрование. Титруют аликвотную часть стандартного раствора иода тиосульфатом до появления бледно-желтой окраски иода. Разбавляют раствор примерно до 200 мл водой, прибавляют 2 мл раствора крахмала и продолжают титрование до обесцвечивания синей окраски.
Примечание. Раствор иода, используемый в приведенной методике, стандартизируют оксидом мышьяка(Ш). Для получения правильных результатов AS2O3 необходимо высушить, либо нагревая его при 110 °C в течение 1 ч, либо оставляя его на ночь в эксикаторе иад концентрированной H2SO4. Количества AS2O3, приведенные ниже, соответствуют примерно 0,1 М растворам иода.
Тщательно взвешивают около 2,5 г высушенного А520з, переносят навеску в стакан и растворяют ее в 10 мл 2 М раствора NaOH. Прибавляют по каплям разбавленную НС1 или H2SO4 до кислой реакции раствора (проверяют лакмусовой бумагой). Раствор разбавляют водой в мерной колбе емкостью 500 мл до метки. Отбирают 25 мл этого раствора (обязательно используют резиновую грушу) и переносят в коническую колбу для титрования. Добавляют к раствору 1 г гидрокарбоната натрия и 2—3 мл индикатора (крахмала). Титруют раствором иода до появления неисчезающего синего окрашивания.
Использование йодата калия в качестве стандарта. Высушивают иодат калия, нагревая его I ч при 180 °C. Тщательно взвешивают около 0,90 г йодата калия, растворяют иавеску в воде и разбавляют растнор до 250 мл в мерной колбе. Отбирают аликвотную часть раствора (25 мл), прибавляют 1 г KI и 5 мл 1 М H2SO4. Титруют раствором тиосульфата до появления бледно-желтой окраски иода, прибавляют 2—3 мл раствора крахмала и продолжают титрование до обесцвечивания раствора:
КЮ3=312=682Оз’
Примечание. Приведенная в методике масса йодата калия соответствует 0,1 М раствору тиосульфата.
Другие редокс-методы
Для титрования тиосульфата использованы следующие окислители: перманганат и манганат калия, медь(Ш), гипобромиты, гипохлориты, ванадий(V), монохлорид иода, хлорамин Т, гексациа-ноферрат(Ш), таллий(Ш), перхлораты, церий(1У), перйодат натрия, тетраацетат свинца [8, 9, 31]. В некоторых случаях методики, разработанные с применением этих окислителей, сложны или связаны с критическими ограничениями. Довольно часто в качестве окислителя используют церий (IV). Тиосульфаты (как и отдельные политионаты) быстро окисляются сульфатом церия (IV) в 2 М НСЮ4 при 85 °C [32]. Избыток церия (IV) титруют стандартным раствором оксалата натрия. Для титрования неизрасходованного церия применяют также стандартный раствор железа(II) с ферроином в качестве индикатора [33].
Большими достоинствами обладает таллий (III) [34]. Его соли стабильны, негигроскопичны и на них не влияют воздух и свет. Окисление тиосульфата солями таллия(III) происходит очень быстро;
TI111 + 2S2O32’ —* Т1‘ + s«°e'
601
Прямое титрование тиосульфата солями таллия (III) проводят с индикатором и-этоксихризоидином, возможен и косвенный вариант этого титрования. Прямому определению тиосульфатов с помощью таллия(III) мешают хлориды и бромиды, образующие устойчивые комплексные соединения с таллием (III) и препятствующие его реакции с тиосульфатом. При косвенном определении мешают медь(II) и железо (II), тогда как хлориды и бромиды не мешают определению.
Другие титриметрические методы
Для определения тиосульфата в его смеси с сульфидами предложен комплексометрический метод [34, 35]. Оба аниона окисляют бромом до сульфатов, которые затем определяют с помощью ЭДТА обычным методом. Во второй аликвотной части анализируемого раствора сульфид удаляют, осаждая его в виде сульфида кадмия, после чего определяют тиосульфат выше приведенным методом. Определению тиосульфатов (и сульфидов) мешают сульфаты и сульфиты.
Для титрования тиосульфатов и сульфидов в присутствии ди-тионитов предложена о-оксимеркурибензойная кислота [36]. Для экспресСнбго прямого титрования тиосульфатов в присутствии сульфитов используют тот факт, что тиосульфат в виде тиосульфата бария окисляется пероксидом водорода, при этом образуется H2SO4, которую титруют стандартным раствором NaOH; сульфит в этих условиях не окисляется. Если анализируемые анионы присутствуют в виде натриевых солей, то необходимо первоначальное превращение их в нерастворимые бариевые соли.
Спектроскопические методы
Спектрофотометрические методы в видимой и ультрафиолетовой областях спектра
Тиосульфат реагирует с цианидом с образованием тиоцианата, при взаимодействии которого с железом (III) получается интенсивно красный тиоцианатный комплекс железа(Ш) [37]. Светопоглощение растворов измеряют при 496 нм. Мешающее действие политионатов устраняют, поддерживая pH раствора около 5 [38]. Дальнейшие исследования [39] показали, что тиосульфат (и те-Тратионат) можно определить спектрофотометрически при условии, что в анализируемом растворе отсутствует медь(П). Этот метод экспрессен, чувствителен и приводит к получению правильных результатов. Метод позволяет определять микрограммовые содержания анализируемых ионов в присутствии очень больших концентраций сульфатов. При определении тиосульфатов при их содержании 100 мкг в анализируемом растворе могут присутствовать до 2 мг NaNO3, Na2SO4, NaNO2, Na2HPO4, NaHCO3, MgCl2, CaCl2, LiCl, AICI3, CH3COOK, цистина, метионина, сульфаминовой
602
и сульфаниловой кислот. Этот метод нашел применение для анализа почв.
Описан спектрофотометрический метод определения тиосульфата, основанный на окислении его избытком иода с последующим определением неизрасходованного пода по его светопоглощению при 365 нм. Такой метод использован для определения тиосульфатов при их концентрации ниже 10-3 М [40], описан метод устранения возможного мешающего действия сульфитов и нитритов. Метод анализа смесей, содержащих политионаты (тетра-, пента- и гекса-), тиосульфаты и сульфиты, также основан на спектрофотометрическом определении избытка иода [41]. Политионаты превращают в тиосульфат, используя их цианолиз по уравнению
Sx°|’+ (х — 1) CN" + Н2О —► SO*’+ 2HCN + (х - 3) SCN’+ S20j-где х =5 4,5 или 6.
Методика включает определения тиосульфата до и после реакции цианолиза и маскирование присутствующего сульфита формальдегидом.
Для спектрофотометрического определения тиосульфата используют реакцию ртути(II) и дифенилкарбазона [42, 43]. В простейшем варианте анализируемый раствор, содержащий 0,1—1 ррш тиосульфатов, встряхивают со смесью, содержащей раствор Hg(NO3)2 в HNO3, KI, дифенилкарбазон и бензол, после чего измеряют оптическую плотность при 562 нм. В работе [43] приведен модифицированный вариант этого метода, использованный для определения тиосульфата и сульфида в присутствии больших концентраций хлоридов.
Описано несколько других методов определения тиосульфатов с применением хлоранилатов [44] и трифенилметановых красителей [45]. В работе [45] использована экстракционная методика, это позволяет определять тиосульфаты в присутствии больших концентраций ацетатов, сульфатов и сульфитов, а также при небольших содержаниях сульфидов и хлоридов. Минимально обнаруживаемая концентрация тиосульфата составляет 0,02 мкг/мл.
Смеси, содержащие сульфиды, сульфаты, сульфиты и тиосульфаты, можно анализировать с помощью прямых измерений свеТо-поглощения в ультрафиолетовой части спектра [46]. Подробности этого метода описаны в разделе «Сульфид».
Метод молекулярной эмиссии в полости
Тиосульфат натрия дает сигнал молекулярного эмиссионного резонанса в виде ряда пиков [47]. Этот метод позволяет определять нанограммовые содержания тиосульфатов. Обычная процедура увеличения интенсивности эмиссии (прибавление хлорной или фосфорной кислот) неприменима из-за разложения тиосульфатов в кислых растворах. Для увеличения интенсивности в этом случае можно использовать буферный раствор фосфатов аммония. Сигнал тиосульфата идентичен сигналу сульфита, и
603
поэтому раздельное определение этих анионов методом молекулярного эмиссионного резонанса невозможно. Этот метод описан в разделе, посвященном определению сульфидов.
Электрохимические методы
Потенциометрические методы
Для определения тиосульфатов предложено несколько потенциометрических методов, некоторые из которых позволяют определять тиосульфаты в присутствии других серусодержащих ионов. Например, иодиметрическое титрование тиосульфатов можно провести потенциометрически. Титрование иодом ведут в среде 2—10 М раствора NaOH, используя платиновый и каломельный электроды [48].
Разработаны прямые и косвенные потенциометрические методики определения тиосульфатов с применением ацетата свинца (IV) в качестве титранта [49]. При косвенном определении к анализируемому раствору тиосульфата прибавляют избыток ацетата свинца(IV) и неизрасходованный окислитель титруют гидрохиноном. Косвенное определение необходимо проводить быстро, так как в противном случае происходит гидролиз тетраацетата свинца (продолжительность определения не должна превышать 35 мин). При использовании 0,02—0,05 М раствора титранта в точке эквивалентности потенциал изменяется примерно на 70 мВ.
Потенциометрическое титрование тиосульфатов гексацианоферратом (III) исследовано различными авторами [50, 51]. Этот титоант применен для анализа смесей дитионита, тиосульфата и сульфита. Точку эквивалентности при титровании находят методом «конечной .мертвой точки». В качестве катализатора реакции используют тетраоксид осмия, а титрование проводят в среде NaOH. В другом варианте [52] тиосульфат титруют гексацианоферратом (III) в кислой среде в присутствии цинка(II) и меди(II) в качестве катализатора.
Для потенциометрического титрования тиосульфата применен ацетат кобальта(Ш) [53]. Приготовление этого титранта описано в литературе [54]. Титрование выполняют в атмосфере азота с платиновым и каломельным электродами, оно происходит количественно только в насыщенном растворе ацетата натрия. Окисление тиосульфата в тетратионат требует около 40 мин. Средняя ошибка определения 70—240 мг тиосульфатов составляет —0,15%.
Для титрования тиосульфата предложен хлорид ртути(II) [55]. Достоинством этого титранта является то, что сульфат, сульфит и нитрит не мешают определению. Этот титрант был применен для анализа смеси тиосульфата, сульфида и полисульфидов по методу «конечной мертвой точки» [56]. Более подробно этот метод описан в разделах «Полисульфид» и «Сульфид».
604
Хлорид ртути(II) использован для анализа смеси тиосульфата, сульфида, сульфита н полисульфпда, а также смеси тиосульфата, сульфита и полисульфида [57, 58]. Эти методы приведены в раз,-деле «Полисульфиды».
При титровании хлоридом ртути(II) можно использовать сульфидный ионоселективный электрод для индикации конечной точки титрования [59, 60]. Метод применен для анализа смесей, содержащих тиосульфаты, сульфиты, сульфиды, полисульфиды и тиолы.
Амперометрические методы
Для амперометрического титрования тиосульфатов в качестве титрантов предложены КЮ3, таллий(III), КВгО3, К2СГ2О7 и КМпО4 [61]. При титровании таллием (III) и КМпО4 определению не мешают сульфаты, нитраты, хлориды, карбонаты, ацетаты, тартраты, Н2РО3 и Н3ВО3, значительные помехи наблюдаются в присутствии высоких содержаний молибдатов. Описан автоматический вариант амперометрического титрования тиосульфатов [62].
Каталитические методы
Иод-азидная реакция
2N' +12 —> 3N24-2I-
положена в основу каталитического метода определения ультрамикроконцентраций тиосульфатов (10~п—10-13 г/мл) [63]. Скорость реакции индицируют амперометрически с вращающимся платиновым и каломельным электродами. Аналогичные методы предложены для определения тиоцианатов и сульфидов. Для измерения скорости реакции можно применить спектрофотометрический метод [64], в этом случае тиосульфат определяют в интервале 0,01—0,15 ppm, однако определению мешают железо(Ш), медь(П), сульфиты и цианиды.
Другие методы определения
Раствор Вакенродера, образованный по реакции сероводорода с растворами сульфитов, содержит тиосульфат, тритионат, тетратионат и пентатионат. До недавнего времени анализ таких смесей представлял крайне трудную задачу [65]. Использование высокоскоростной жидкостной хроматографии привело к созданию замечательного метода анализа упомянутых смесей [66]. Полное разделение и определение компонентов выполняется за 15 мин. Метод приведен в разделе «Политионаты».
Для определения тиосульфатов предложена газовая хроматография [67]. Метод основан на первоначальном превращении тиосульфатов в сульфат бария под действием гидроксида бария и брома. Это позволяет отделить тиосульфат от ацетатов, нитратов и хлоридов, которые мешают последующему газохроматографическому
605
определению. В дальнейшем сульфат восстанавливают до сероводорода с помощью 20%-ного раствора хлорида олова(II) в концентрированной Н3РО4, после чего определяют сероводород методом газовой хроматографии.
Тиосульфат можно определить микрогравпметрическим методом [68]. Переносят 0,01 мл анализируемого раствора, содержащего 20—200 мкг тиосульфата, на узкую полоску бумажного фильтра, импрегнированного хлоридом серебра. Помещают фильтр в специальный аппарат и обрабатывают его водой, в результате происходит миграция ионов. После высушивания фильтра белые пятна вырезают и взвешивают. Сравнение со стандартами позволяет определять тиосульфат с точностью ±1,5%. Определению тиосульфатов описываемым методом мешают бромиды, иодиды и цианиды; не мешают хлориды, сульфаты, карбонаты, нитраты, сульфиты, хроматы, тетратионаты и оксалаты.
Описано термометрическое титрование тиосульфатов церием (IV) с помощью цифрового термометрического титратора [69]. Конструкция титратора приведена в этой же статье.
ЛИТЕРАТУРА
1. Iguchl А.— Bull. chem. Soc. Japan, 1958, v. 31, p. 600; Analyt. Abstr., 1959, v. 6, p. 1705.
2. Schmidt M., Sand T. — Z. anorg. Chem., 1964, v. 330, p. 188; Analyt. Abstr., 1965, v. 12, p. 5754.
3. Pollard F. ti.. Nickless G., Glover R. B. — J. Chromat, 1964, v. 15, p. 533.
4. Seiler H., Erlenmeyer H. — Helv. chim. Acta, 1964, v. 47, p. 264; Analyt. Abstr., 1965, v. 12, p. 1703.
5. Canic V. D., Turcic M. N., Bugarski-Vojinovic M. B., Perisic N. U. — Z. analyt. Chem., 1967, v. 229, p. 93; Analyt. Abstr., 1968, v. 15, p. 5211.
6. Kelly D. P. — J. Chromat, 1970, v. 51, p. 343.
7. Naito K., Takel S., Okabe T. — Bull. chem. Soc. Japan, 1970, v. 43, p. 1360; Analyt. Abstr., 1971, v. 20, p. 3012.
8. Szekeres L. — Taianta, 1974, v. 21, p. 2.
9. Blasius E., Horn G., Knochel A., Munch I., Wagner H. — In: Inorganic Sulphur Chemistry (Ed. G. Nickless), Chapter 6, Amsterdam, Elsevier, 1968.
10. Lowenheim F. A. — In: Encyclopedia of Industrial Chemical Analysis (Ed. F. D. Snell and L. S. Ettre), Vol. 18, New York, Interscience, 1973, p. 429.
11. Heinrich B. J. — In: Treatise on Analytical Chemistry (Ed. I. M. Kolthoff and P. J. Elving), Part II. Vol. 7, New York, Interscience, 1961, p. 84
12. Kolthoff I. M., Belcher R. — Volumetric Analysis, Vol. Ill, New York and London, Interscience, 1957, p. 213.
13. Laitinen H. A., Harris W. E. — Chemical Analysis. 2nd edn. New York, McGraw-Hill, 1975.
14. British Standards 3302; (1960).
15. Thompson J. C., Hamori E. — S. phys. Chem., Ithaca, 1971, v. 75, p. 272.
16. Clark E. D. — Nature, 1951, v. 168, p. 876.
17. Latimer G. W. — Taianta, 1966, v. 13, p. 321.
18. Schulek E. — Z. analyt. Chem., 1925, v. 65, p. 352.
19 Wollack R. — Z. analyt. Chem., 1930, v. 80, p. 1.
20. Goehring M. — Z. analyt. Chem., 1949, v. 128, p. 341.
21. Danehy J. P., Zubritsky C. W. — Analyt. Chem, 1974, v. 46, p. 391.
22. Szekeres L. — Z. analyt. Chem., 1964, v. 203, p. 178; Analyt. Abstr., 1965, v. 12, p. 5130.
606
23. Szekeres L. — Ann. chim. Roma, 1962, v. 52, p. 67; Analyt. Abstr.. 1962, v. 9, p. 5140.
24. Szekeres L. — Z. analyt. Chem., 1960, v. 178, p. 81; Analyt. Abstr., 1961, v 8, p. 2387.
25. Szekeres L. — Acta chim. hung., 1961, p. 167.
26. Szekeres L. — Pharm. Zentralhalle Dtl., 1963, v- 102, p. 6; Analyt. Abstr., 1963, v. 10, p. 5137.
27. Legradi L. — Analyst, 1961, v. 86, p. 854.
28. Blasius E., Wagner H., Ziegler K.— Arch. Eisenhiitt Wes., 1971, v. 42, p. 473; Analyt. Abstr., 1972, v. 22, p. 3164.
29. Furman N. H., Wallace J. H.— J. Am. chem. Soc., 1931, v. 53, p. 1283.
30. Griepink B. F. A., Cysouw H. A. — Mikrochim. Acta, 1963, p. 1033.
31. Berka A., Vulterin S. J., Zyka J. — Newer Redox Titrants, England, Pergamon Press, Oxford, 1965.
32. Krause R. A., Busch D. H.— Analyt. Chem., 1958, v. 30, p. 1817.
33. Singh B., Singh S. — Analytica chim. Acta, 1956, v. 14, p. 505.
34. De Sousa A. — Chemist Analyst, 1961, v. 50, p. 76.
35. De Sousa A. — Inf. quim. analit., 1963, v. 17, p. 139; Analyt. Abstr., 1964, v. 11, p. 3070.
36. Wronski M. — Z. analyt. Chem., 1961, v. 179, p. 350; Analyt. Abstr., 1961, v. 8, p. 4151.
37. Sorbo В. H.— Biochim. Biophys. Acta, 1957, v. 23, p. 412.
38 Urban P. J. — Z. analyt. Chem., 1961, v. 179, p. 415; Analyt. Abstr., 1961, v. 8, p. 4191.
39. Nor У. M., Tabatabai M. A. — Analyt. Lett., 1975, v. 8, p. 537.
40. Ozawa T. — J. chem. Soc. Japan, Pure Chem. Sect., 1966, v. 87, p. 576; Analyt. Abstr., 1967, v. 14, p. 6084.
41. Koh T., Taniguchi K. — Analyt. Chem., 1973, v. 45, p. 2018.
42. Okutani T., Utsumi S., Shibaia K., Iwasaki I. — J. chem. Soc. Japan, Pure Chem. Sect., 1965, v. 86, p. 831; Analyt. Abstr., 1967, v. 14, p. 3949.
43. Okutani T., Utsumi S. — J. chem. Soc. Japan, Pure Chem. Sect., 1965, v. 86, p. 1149; Analyt. Abstr., 1967, v. 14, p. 3950.
44. Hoffmann E. — Z. analyt. Chem., 1962, v. 185, p. 372; Analyt. Abstr., 1962, v. 9, p. 3016.
45. Григониене К., Раманаускас E., Буткевичу с И. ЖАХ, 1972, т. 27, о. 2028; Analyt. Abstr., 1974, v. 26, р. 2629.
46 Еремин Ю. Г., Киселева К- С. — ЖАХ, 1969, т. 24, с. 1201. Analyt. Abstr., 1971, v. 20, р. 978.
47 Belcher R., Bogdanski S. L., Knowles D. J., Townshend A. — Analytica Chim. Acta, 1975, v. 77, p. 53; 1975, v. 79, n. 292.
48. Норкус П., Шемкявичюте Г.—ЖАХ, 1971, т. 26, с. 39; Analyt. Abstr., 1972, v. 22, р. 2234.
49. Rencova J., Zyka J. — Chemist Analyst, 1967, v. 56, p. 27.
50. Solymosi F„ Varga A. — Analytica chim. Acta, 1957, v. 17, p. 608.
51. Solymosi F., Varga A. — Magy. kem. Foly, 1959, v. 65, p. 52; Analyt. Abstr., 1959, v. 6, p. 4746.
52. Rao V. P. R., Sarma В. V. S.— Chemist Analyst, 1966, v. 55, p. 110.
53. Hanif M., Dolezal J., Zyka J. — Microchem. J., 1971, v. 16, p. 291.
54 Budesinsky J., Dolezal J., Scramkova B., Zyka J. — Microchem. J., 1971, v. 16, p. 121.
55 Aiss S. A. — Z. analyt. Chem., 1961, v. 182, p. 251; Analyt. Abstr., 1962, v. 9, p. 1029.
56. Kiss S. A. — Z. analyt. Chem., 1962, v. 188, p. 341; Analyt. Abstr., 1963, v. 10, p. 112.
57. Danielsen A. I., Johnsen K., Landmark P. — Norsk. Skogind., 1969, v. 23, p. 378; Analvt. Abstr., 1971, v. 21, p. 1252.
58. Danielsen A. J., Johnsen K., Landmark P. — Meddr. Pap Ind. Forsk. Inst., No. 232; Norsk. Skogind., 1969, v. 23, p. 77; Analyt. Abstr., 1970, v. 19, p. 395.
59. Papp J., Havas J. — Magy. кёт. Foly., 1970, v, 76, p. 307; Analyt. Abstr., 1971, v. 20, p. 4439.
607
60. Papp J. — Cellulose Chem. Technol., 1971, v. 5, p. 147; Analyt. Abstr., 1972, v. 22, p. 2404.
61. Жданов A. K-, Ахмедов Г. — Узбекский хим. ж., 1971, с. 6; Analyt Abstr., 1973, v. 24, р. 771.
62. Myers S. A., Swann W. В. — Talanta, 1965, v. 12, p. 133.
63. Michalski E., Wtorkowska A. — Chem. Anal. (Warsaw), 1962, v. 7, p. 783, Analyt. Abstr.. 1963, v. 10, p. 1773.
64. Uisumi S., Okutani T. — J. Chem. Soc. Japan, Pure Chem. Sect., 1973, p. 75; Chem. Abstr., 1973, v. 78, p. 92122c.
65. Haff L. V.— In: The Analytical Chemistry of Sulphur and its Compounds (Ed. J. H. Karchmer), Part 1, New York, Wiley-Interscience, 1970, p. 241.
66. Chapman J. N.. Beard H. R. — Analyt. Chem., 1973, v. 45, p. 2268.
67. Ho S., Hara T. — J. chem. Soc. Japan, Pure Chem. Sect., 1969, v. 90, p. 1027; Analyt. Abstr., 1970, v. 19, p. 4842.
68. Ganchev N., Koev K. — Mikrochim. Acta, 1964, p. 97.
69. Priestley P. T. —Analyst, 1963, v. 88, p. 194.
предметный указатель
'Альбумин, определение селена 184 Аммиак, определение по Несслеру 129
Арсенат и арсенит 12—29 определение 16—27 — активационным методом 27 — атомио-абсорбционное 24, 468 — гравиметрическое 16 сл. — кинетическое 26 — колориметрическое 22 — кулонометрическое 25 сл. — методом кольцевой печи 321 — полярографическое 25 — радиохимическое 27 — спектрофотометрическое 23 сл. — титриметрическое 17—22 — ТСХ 321 — флуориметрическое 25 — электрохимические методы 25 отделение и концентрирование 12—16 — адсорбцией 16
' — дистилляцией 12 сл.
— осаждением 15 сл.
— хроматографическое 14 сл.
— экстракцией 13 сл.
Ацетаты 9—11 определение 10 сл. отделение 9
Бетон, определение хлорида 295 Биологические материалы определение бора 29, 37 — иодида 392 — молибдата 106 — оксалата 152 — селена 172, 181, 183 сл. — формиата 90 — фосфата 464 — фторида 331, 336, 347," 360, 361 — хлорида 304 цианида 304, 73, 74, 85 отгонка муравьиной кислоты 90
Бихроматы см. Хроматы и бихрома-
ты . . —
Бораты 29—43
определение 32—43 .
— атомио-абсорбциоииое 39
— гравиметрическое 32
— кулонометрическое 40
— масс-спектрометрическое 41
— потенциометрическое 40
— спектроскопическое 39
— спектрофотометрическое 34—
39
— термометрическое 40 сл.
— титриметрическое 32—34
— флуориметрическое 39
— фотометрией пламени 40
— хроматографическое 41
— электрохимическими методами
40
отделение 30—32
— дистилляцией 30 сл.
— ионообменное 31
— ТСХ 32
— экстракцией 31
— электролизом 32
Борная кислота 33
Броматы 256—263
определение 256—262
— амперометрическое 261
— газохроматографическое 262
— полярографическое 262
— потенциометрическое 260 сл.
— спектрофотометрическое 259
— титриметрическое 256—259
— ТСХ 321
— электрохимическими методами 260—262
отделение 256
Бромиды 263—277
определение 263—275
— амперометрическое 273
— вольтамперометрическое 273
— газохроматографическое 274, 320
— гравиметрическое 263
— с иоиоселективиым электродом 272
— каталитическое 273 сл.
609
Бромиды
определение кулонометрическое
272
— методом кольцевой печи 275, 321
---молекулярной эмиссии в полости 270
— нейтронно-активационное 274
— полярографическое 273
— потенциометрическое 270—272
— радиохимическое 274
— рентгеиофлуоресцентное 275
— спектроскопическое 268—270
— спектрофотометрическое 268—
270
— титриметрическое 264—268
— ТСХ 275, 321
— электрохимическое 270—273
— эмиссионным спектральным
анализом 307
отделение 263
Бромиты 277—280
определение 277—279
— полярографическое 279
— потенциометрическое 279
— титриметрическое 277—278
— электрохимическое 279
отделение 277
Ванадаты 245—255
определение 248—254
— амперометрическое 253
— атомио-абсорбциоииое 251
— газохроматографическое 254
— гравиметрическое 248
— ИК-спектроскопическое 251 сл.
— каталитическое 253 сл.
— колориметрическое 250
— микроколичеств 259
— полярографическое 252 сл.
— потенциометрическое 252
— спектроскопическое 250—252
— спектрофотометрическое 250
— титриметрическое 248
— электрохимическими методами
252 сл.
отделение 246 сл.
— осаждением 246, 248
— соосаждением 247
Ванадаты определение хроматографическое 246
— экстракцией 246
— электролизом 247
Взрывные смеси, определение хлората 284
Виииая кислота 205 сл.
Вода
определение бора 37
— бромида 272, 275
— гипохлорита 370, 374
— йодата 382
— иодида 392, 394
— карбоната 48
— молибдата 107, 116
— мышьяка 24
— нитрата 127, 129, 130, 135, 137, 138
— нитрата и нитрита 146
— нитрита 146, 147
— полисульфида 507, 509
— селеиата и селенита 184
— силиката 188, 201
— сульфата 531, 538, 540, 547, 553, 583
— сульфида 569
— сульфита 583
— сульфита и диоксида серы 580
— формиата 92
— фосфата 460, 461, 487 фторида 331, 347, 356, 358, 361
— хлорида 294, 300, 304, 311, 320
— цианида 75, 84, 97
Воздух определение диоксида серы 585, 592
— нитрата 130, 136
— нитрита 147
— оксидов азота 136
— селена 178, 180, 184
— сероводорода 565, 569
— фторида 331, 347, 357
— цианидов 82
Вольфраматы 233—243 определение 236—243 •— амперометрическое 242
610
Вольфраматы определение атомно-абсорбционное 241
— гравиметрическое 236—238 — каталитическое 243
— кондуктометрическое 242
— методом кольцевой печи 243 — иейтроиио-активационное 243 — полярографическое 242 — потенциометрическое 242 — спектроскопическое 239—241 — спектрофотометрическое 239— 241
— титриметрическое 238 сл.
— флуориметрнческое 241
— электрохимическими методами 242 отделение 234—236 — ионообменное 234
— осаждением 234, 236 сл., 238 — соосаждеиием 235
— ТСХ и БХ 235 сл.
— экстракцией 235
Газы, смеси, определение СОг 49 Гексаметафосфат см. Метафосфаты Гексафторсиликаты см. Фторсиликаты Гексафторфосфаты 420—422 определение 420—422 — амперометрическое 421 — гравиметрическое 420 — спектрофотометрическое 421 — титриметрическое 421 ел. отделение 420
Гексацнаноферраты (II) и (III) 93— 101
определите 95—100 — амперометрическое 100 — биамперометрическое 100 — вольтамперометрическое 100 — гравиметрическое 95 — методом кольцевой печи 321 — полумикрометод 100 — потенциометрическое 99 — спектроскопическое 97—99 — спектрофотометрическое 97 сл. — титриметрическое 95—97 — ТСХ 321 — турбидиметрическое 98
Гексациа иоферраты определение хроматографическое 100
— электрохимическими методами 99 сл.
отделение 94 сл.
Германий, определение спектрофотометрическое 438 сл.
Гидрокарбонаты см. Карбонаты и гндрокарбоиаты
Гипобромиты 365—370
диспропорционирование 277, 365 определение 368—370
— микроколичеств 369 сл.
— потенциометрическое 369
— в присутствии других гало-геисодержащих анионов 370
— титриметрическое 278, 366— 369
— электрохимическими методами 369 сл.
Гипонитриты 102 сл.
Гнпофосфаты 422—425
определение 423 сл.
— гравиметрическое 423
— спектрофотометрическое 424
— титриметрическое 424 отделение 422 сл.
Гипофосфиты 425—429 определение 426—428, 477 — автоматическое 428 — при совместном присутствии с ортофосфатом 427 сл.
— спектрофотометрическое 427 сл.
— титриметрическое 426 отделение 425 сл.
Гипохлориты 370—376 определение 371—375 — кулонометрическое 375 — потенциометрическое 374 — спектроскопическое 373 сл. — термометрическое 375 — титриметрическое 371—373 — электрохимическими методами 374 сл отделение 370 сл
Глины, определение карбонатов 50
Грсусамра соль 480
611
Гудцайта метод 22
Детергенты
определение сульфата 548
— фосфата 466, 487
Дитиоиаты 490—492
определение 490—492
— колориметрическое 491 сл.
— спектрофотометрическое 491 сл.
— титриметрическое 490 сл.
отделение 490
Дитиониты 492—498
диспропорционирование 493
определение 493—497
— полярографическое 497
— потенциометрическое 496 сл.
— спектроскопическое 496
— титриметрическое 493—496
— электрохимическими методами 496 сл.
Дифосфаты 415—420
определение 417—419
— амперометрическое 419
— гравиметрическое 417
— спектрофотометрическое 418 сл
— титриметрическое 417 сл.
— ферментативное 419
отделение 416 сл.
Зерно, определение бромидов 274 Зубные пасты, определение фторида 336, 340, 357
Инсектициды, определение фторида 331
Иодаты 376—383
определение 377—382
— амперометрическое 381
— газохроматографическое 382
— кинетическое 381 сл.
— микроколичеств 380
— полярографическое 381
— потенциометрическое 380 сл.
— радиометрическое 382
— спектрофотометрическое 380 — титриметрическое 377—379 термометрическое 382
— электрохимическими методами 380 сл.
Иодаты
отделение 376 сл.
Йодиды 383—399
определение 384—397
— амперометрическое 393
— атомно-абсорбииоиное 390 сл.
— газохроматографическое 320
396
— гравиметрическое 384
— с ионоселективным электродом 391 сл.
— кинетическое 393—396
— кулонометрическое 392 сл.
— методом изотопного разбавления 396
-----кольцевой печи 321, 397
— нейтронно-активационное 396
— полярографическое 393
— потенциометрическое 391 сл.
— радиометрическое 396
— спектроскопическое 388—391
— спектрофотометрическое 388— 390
— титриметрическое 384—388
— ТСХ 321, 397
— флуориметрическое 390
— электрохимическими методами 391—393
— эмиссионно-спектроскопическое 390
отделение 383 сл.
экстракция 383
Карбонаты и гидрокарбоиаты 43—51
динамическая сорбция 50
определение 43—50
— вакуумной дистилляцией 48
— волюмометрическое 44 сл.
— газохроматографическое 49 сл.
— гравиметрическое 43 сл.
— манометрическое 44 сл.
— спектрофотометрическое 48 сл.
— титриметрическое 45—48
--термометрическое 50
— электрохимическое 49
Керамические материалы, определе-
ние силиката 189
Конвея ячейка 47
612
Корма определение нитрата 127 — фторида 337
Кости, определение фторида 357 Кремнефтор истоводородна я кислота, отгонка 332—335 Кровь определение бромида 272 — гексацианоферратов 74 — диоксида углерода 49 — нитрита 147 — оксалата 156 — селена 184 — фторида 340, 357 сл., 361 — хлорида 319 — цианида 79 — цитрата 69
Магнезиты, определение бора 36
Маршалла кислота 498
Масло, определение серы 531 Металлургические продукты определение силиката 188, 194 — фосфата 461
Метафосфаты 429—431 определение 431 — ИК-спектроскопнческое 431 — спектрофотометрическое 431 отделение 430 сл.
Молибдаты 103—118 определение 108— 116 — амперометрическое 115 — атомно-абсорбционное 113 сл. — вольтамперометрическое 114 сл. — гравиметрическое 108 сл. — каталнметрнческое 115 сл. — кулонометрическое 115 — методом ЭПР 116 — полярографическое 114 сл. — потенциометрическое 114 — спектроскопическое 111—114 — спектрофотометрическое 111— 113 — титриметрическое 109 сл. — флуориметрическое 113 — электрохимическими методами 114 сл. отделение 104—107
Молибдаты отделение адсорбцноиио-коллоид-иой флотацией 107 — ионообменное 104 сл. — осаждением 104—105, 108 — соосаждением 106 сл. — хроматографическое 105 — экстракцией 105 сл. — электрофорезом 105
Молоко определение селена 184 — цитрата 68
Монофторфосфаты 432—435 определение 434 сл. — автоматическое 433 — газохроматографическое 435 — с иоиоселектнвным электродом 434 — спектрофотометрическое 434 сл. отделение 432—434 — осаждением 432 — хроматографическое 433 — электрофорезом 432
Моча определение сульфата 540 — фторида 340, 347, 357, 361
Мука, определение бромата 257 Мышьяк и его оксиды, высокочистые, определение селена 184 Мясо определение нитрата 138 — нитрита 147, 149 — хлорида 295
Нефть, определение сульфита и ди оксида серы 581
Нитраты 118—142 восстановление 125, 127 метод Деварда 120 сл. -------- Кьельдаля 121 концентрирование 119 сл. определение 122—139 — амперометрическое 136 сл. — атомно-абсорбционное 133 — вольтамперометрическое 136 сл.
— газометрическое 138 — гравиметрическое 122—127
613
Нитраты
определение ИК-спектроскопиче-ское 133
— с ионоселектнвиым электродом 134—136
— кулонометрическое 137 сл.
— методом капельным 126
----- кольцевой печи 139
-----Кьельдаля 125
-----Лейте 124
— микрометодом 123
— полярографическое 137
— потенциометрическое 134
— совместно с нитритом 138 сл. — спектроскопическое 126—133 — спектрофотометрическое 128 — титриметрическое 124—126 — ферментативное 139 — флуорнметрнческое 132 сл. — электрохимическими методами 134—138 отгонка в виде NH3 120 отделение 119—121 — гравиметрическое 119 — ионообменное 119 сл. — микродиффузией 121 — ТСХ 121 — экстракцией 121
Нитриты 142—150
определение 143—150 — автоматическое 146 — атомио-абсорбционное 149 — газометрическое 150
— с ионоселектнвиым электродом 149
— кулонометрическое 149
— методом Грисса (Грисса — Илосвая) 146—148 ----- Зальцмана 143 -----кольцевой печи 150 — потенциометрическое 149 — совместно с нитратом 150 — спектроскопическое 145—149 — спектрофотометрическое 145— 148
— титриметрическое 143—145 — флуорнметрнческое 149
Нитриты
определение электрохимическим], методами 149 сл.
отделение 143
Оксалаты 151—156
определение 152—156
— амперометрическое 155
— гравиметрическое 152
— с ионоселективным электро дом 155
— потенциометрическое 155
— спектроскопическое 154 сл.
— спектрофотометрическое 154 сл.
— титриметрическое 152—154
— ферментативное 156
— флуоресцентное 155
— электрохимическими методами 155 сл.
отделение 151 сл.
Ортофосфаты 435—476
определение 442—472
— автоматическое 460
— амперометрическое 470 сл.
— атомно-абсорбцноиное 467— 469
— газохроматографическое 472 — гравиметрическое 442—449 — с ионоселектнвиым электродом 470
— кинетическое 471 сл.
— колориметрическое 464
— методом пламенной спектроскопии 464—466
— микроколичеств 469
— потенциометрическое 469 сл.
— радиометрическое 471
— селективное подавление мешающего влияния 442
— спектроскопическое 456—469 — спектрофотометрическое 438 сл., 456—464
— титриметрическое 449—457
— флуорнметрнческое 466 сл.
— электрохимическими методами 469—471
отделение 436—442
614
Оксалаты
отделение осаждением 436 сл.
— соосаждением 442
— хроматографическое 440 сл.
— экстракцией 437—440
— электролизом 442
Отбеливатели, определение гипохлорита 375
Перброматы 399—402
определение 400—402
— с ионоселектнвиым электродом 401
— полярографическое 401
— спектрофотометрическое 401
— титриметрическое 400 сл.
— электрохимическими методами 401 сл.
отделение 400
Пербромная кислота 399
Перйодаты 410—414
определение 411—414
— ИК-спектроскопическое 413
— с ионоселектнвиым электродом 413 сл.
— потенциометрическое 413
— спектроскопическое 412 сл.
— спектрофотометрическое 412
— титриметрическое 411 сл.
— электрохимическими методами 413 сл.
отделение 410 сл.
Перманганаты 156—164
определение 157—163
— амперометрическое 162
— вольтамперометрическое 162
— кулонометрическое 162
— полярографическое 163
— потенциометрическое 161 сл.
— совместно с манганатом 163
— спектроскопическое 160 сл.
— спектрофотометрическое 160 сл.
— титриметрическое 157—160
— флуорнметрнческое 161
— электрохимическими методами 161 сл.
отделение 157
Пероксодисульфаты 498—502 определение 499—501 — газохроматографическое 503 — гравиметрическое 499 — ИК-спектроскопическое 502 — колориметрическое 501 — методом молекулярной эмиссии в полость 502 — полярографическое 502 — потенциометрическое 502 — спектроскопическое 501 сл. — спектрофотометрическое 501 сл.
— титриметрическое 499—501 — флуорнметрнческое 501 сл. — электрохимическими методами 502 отделение 499 Перреиаты 164—169 определение 166—168 — атомно-абсорбционное 168 — гравиметрическое 166 — ИК-спектроскопическое 168 — с ионоселектнвиым электродом 168 — каталиметрическое 168 — полярографическое 168 — спектроскопическое 166—168 — спектрофотометрическое 166— 168 отделение 164—166 — дистилляцией 164 — соосаждением 165 — ионообменное 165 — от молибдена и нольфрама 234 — осаждением 164 — соосаждением 165 — ТСХ 165
— экстракцией 165 сл.
Перрутеиаты 169—171 определение 170 сл. — атомно-абсорбционное 171 — спектрофотометрическое 170 сл. отделение 170
Персульфаты см. Пероксодисульфаты Пертехиаты 164—169 определение 166—168 — атомно-абсорбциоииое 168 — гравиметрическое 166
616
Пертехиаты
определение ИК-спектроскопиче-ское 168
— нейтроиио-актнвациоиное 168
— полярографическое 168
— спектроскопическое 166—168
'• — спектрофотометрическое 166—
168
отделение 164—166
— дистилляцией 164 сл.
— ионообменное 165
— осаждением 164
— соосаждением 165
— ТСХ и БХ 165
— экстракцией 166
Перхлораты 402—410
определение 403—408
— атомно-абсорбционное 407
— гравиметрическое 404
— ИК-спектроскопическое 407
— с ионоселективиым электро-
дом 408
— кулонометрическое 408
— потенциометрическое 408
— спектроскопическое 405—407
— спектрофотометрическое 405—
407
— термометрическое 408
— титриметрическое 405
— электрохимическими методами
408
отделение 402 сл.
— осаждением 402
— ТСХ и БХ 403
— экстракцией 402
— электрофорезом 403
Пестициды, определение мышьяка 12
Пиросульфиты 489
Пиротехнические смеси, определение хлората 284
Пирофосфаты см. Дифосфаты
Пищевые продукты
определение бора 29, 37, 38
— бромида 270, 274
— мышьяка 12, 22, 23
— нитрата 121, 138'
— нитрита 142, 147
— селена 171
— серы 531, 537, 540, 544, 581
Пищевые продукты
определение сульфита и диоксида серы 581
— формиата 90
— фосфата 464
— фторида 333, 343
— цианида 84
Полимеры, определение фторида 331 Полисульфиды 504—509
определение 504—508
— автоматическое 508
— амперометрическое 508
— гравиметрическое 505 сл.
— с ионоселективиым электродом 508
— потенциометрическое 508
— титриметрическое 506—508
— электрохимическими методами 508
Политионаты 509—518 определение 511—517 — гравиметрическое 512 — кулонометрическое 516 — полярографическое 516 — потенциометрическое 516 — спектрофотометрическое 515 сл.
— титриметрическое 513 сл.
— хроматографическое 517
— электрохимическими методами 516 сл
отделение 510 сл.
— ионообменное 511
— методом высокоэффективной жидкостной хроматографии 511 -------кристаллического фракционирования 510 разделение, основанное на различие в растворимости 510 Полифосфаты 479—487
гидролиз 480
определение 484—487
— автоматическое 482—484
— БХ 486
— методом ЯМР 487
— полярографическое 486 сл.
— титриметрическое 484—486 отделение 480—484
— БХ и ТСХ 480 сл.
616
Полифосфаты
отделение гель-хроматографней
481 сл.
— осаждением 480
разделение 482
Полупроводники, определение кремния 188
Почвы
определение бора 29, 38
— карбонатов 47, 48, 50
— молибдата 103
— нитрата 121, 128, 135 сл
— нитрита 146
— селена 184
— сульфата 540, 547
Растения
определение бора 38
— молибдата 103, 116
— селена 181
— фторида 357, 361
— цианида 73
Реактивы, определение следов суль-
фата 536, 537
Роданиды 222—233
определение 223—232
— атомио-абсорбцнониое 229
— газохроматографнческое 231
— гравиметрическое 224
— с ионоселективиым электродом 230
— каталиметрическое 231
— колориметрией пятен 231
— колориметрическое 227
— кулонометрическое 230
— методом молекулярной эмиссии в полость 229 сл.
— микроколичеств 231 сл.
— полярографическое 230
— потенциометрическое 230
— реитгеиофлуоресцеитиое 231
— спектроскопическое 227—230
— спектрофотометрическое 227
— титриметрическое 224—227
---термометрическое 232
— ТСХ 321
— электрохимическими методами
230 сл.
отделение 222 сл.
— осаждением 222
— экстракцией 222
Руды и минералы определение бора 29 — ванадата 253 — карбоната 50 — кремния 188, 195, 200 — сульфата 537 — фосфата 461 — фторида 338, 347, 357
Селеиаты и селениты 171—187 определение 176—184 — амперометрическое 182 — атомио-абсорбциоииое 181 сл. — атомно-флуоресцеитное 182 — газохроматографическое 184 — гравиметрическое 176 сл. — каталитическое 183 — колориметрическое 178 — кулонометрическое 182 — методом кольцевой печи 178 -------- молекулярной эмиссии в полость 182 — микроколичеств 182 — нефелометрическое 181 — полярографическое 183 — потенциометрическое 182 — в присутствии теллура 172 — радиохимическое 183 сл. — спектроскопическое 178—182 — спектрофотометрическое 178— 180 — титриметрическое 177 сл. — турбидиметрическое 181 — флуориметрнческое 180 сл — электрохимическими методами 182 отделение 172—176 — дистилляцией 172 сл. — ионообменное 173 — осаждением 174 сл. — соосаждением 175 сл — ТСХ и БХ 175 сл. — экстракцией 174
Селеиоциаиаты 187 сл. определение 187 сл. — микроколичеств 188 — спектрофотометрическое 188 — титриметрическое 187 сл. отделение 187 — дистилляцией 187 — ионообменное 187
617
Серы диоксид см. также Сульфиты определение в воздухе 592 сл. — в выхлопных газах 590
Сигареты, дым
определение бромидов 274
— нитритов 149
Силикаты 188—203
концентрирование 189 сл. определение 190—203
— атомио-абсорбциоииое 200 сл,. 468
— атомно-флуоресцеитиое 202
— гравиметрическое 190—192
— ИК-спектроскопнческое 200
— кинетическое 202
— микроколичеств 189, 199
— в присутствии As и Р 201
-------As, Р, Ge 442
— спектроскопическое 196—202
— спектрофотометрическое 196— 202, 438 сл.
— термометрическое 203
— титриметрическое 192—195
— устранение мешаюших влияний 190
— электрохимическими методами 202
отделение 189 сл.
— дистилляцией 190
— осаждением 189
— хроматографическое 189 сл.
— экстракцией 189
Сплавы
определение бора 29
— силиката 195
— фосфата 461
отделение Fe 15
Сталь
определение вольфрамата 234, 241
— молибдата 106, 111, 115
— мышьяка 14, 23
— перманганата 162
— сульфата 535, 536
— углерода 47
— фосфата 450, 461
— хромата 55, 57, 58, 61
Стекло, определение силиката 188
Сульфаты 519—560
определение 520 555
— амперометрическое 552
— атомно-абсорбциоииое 547 сл.
— газохроматографическое 554
— гравиметрическое 521—525
----в виде BaSO* 521
---с органическими осадителями 524
---со смешанным осадителем 525
— ИК-спектроскопическое 546 сл.
— с иоиоселектнвным электродом 550 сл.
— кинетическое 553
— кондуктометрическое 553
— методом молекулярной эмиссии в полость 548 сл.
— нефелометрическое 545 сл.
— полярографическое 552 сл.
— потенциометрическое 550—552
— радиохимическое 553 сл.
— спектроскопическое 538—549
— спектрофлуорнметрическое
546
— спектрофотометрическое 538— 543
— титриметрическое 525—538 ----в виде сульфида 536 сл.
----комплексометрическое
534—536
----солями бария 525—532
----солями свинца 532—534
----термометрическое 554
— турбидиметрическое 543 сл.
— хроматографическое 527
— электрохимическими методами 550—553
отделение 519 с л.
Сульфиды 560—680
определение 562—578
— газохроматографическое 578 — гравиметрическое 562 сл.
— с иоиоселектнвным электродом 575
— каталитическое 577
— кинетическое 577
— кулонометрическое 576
— методом молекулярной эмис-
618
Сульфиды
сии в полость 573
определение полярографическое
576
— потенциометрическое 574
— радиохимическое 578
— спектроскопическое 568—573
— спектрофотометрическое 568—
572
— титриметрическое 563—568
— ферментативное 577
— флуориметрическое 572 сл.
— электрохимическими методами 574—577
отделение 560—562
— газохроматографическое 562
— в виде HsS 560
— ионообменное 561
— микродиффузией 562
— осаждением 561
— ТСХ 561 сл.
Сульфиты 580—595
определение 582—593
— автоматическое 591
— атомно-абсорбционное 589
— газохроматографическое 592
— гравиметрическое 582
— кулонометрическое 591 сл.
— методом молекулярной эмис-
сии 589
— микроколичеств 592
— полярографическое 590 сл.
— потенциометрическое 589
— спектроскопическое 585—589
— спектрофотометрическое 585—
588
— ТСХ 321
— турбидиметрическое 583—585
— флуориметрическое 588
— электрохимическими методами
589 сл.
отделение 581 сл.
— в виде SO2 581
— ионообменное 581 сл.
— осаждением 582
— ТСХ 582
[Тартраты 205—208
определение 205—207
Тартраты определение спектрофотометрическое 207 сл.
— титриметрическое 205—207 отделение 205
Теллураты и теллуриты 208—218 определение 211—216 — амперометрическое 216 — атомио-абсорбциоииое 214 — атомио-флуоресцеитное 215 — гравиметрическое 211 — кулонометрическое 216 — методом молекулярной эмиссии в полость 215 — микроколичеств 182, 211 — иейтроиио-активационисе 216 — осциллополярографическое 216 — поляриметрическое 216 — полярографическое 182, 216 — потенциометрическое 182, 216 — спектроскопическое 212—215 — спектрофотометрическое 214 — титриметрическое 211 сл. — флуориметрическое 214 — электрохимическими методами 215 сл. отделение 208—210 — дистилляцией 209 — ионообменное 209 — осаждением 210 — от селена 209 — соосаждением 210 — ТСХ и БХ 210 — экстракцией 209 сл. разделение 209
Тетраметафосфат см. Метафосфаты Тетрафеиилбораты 220 сл. определение 221 отделение 220
Тетрафосфаты см. Полифосфаты Тетрафторбораты 218—220 определение 218—220 — гравиметрическое 218 сл. — с иоиоселектнвным электродом 220 — спектрофотометрическое 219 титриметрическое 219 сл. отделение 218
619
Тиосульфаты 595—606 определение 597—606 — амперометрическое 605 — высокоскоростной жидкостной хроматографией 605 — газохроматографическое 605 — гравиметрическое 597 — каталитическое 605 — методом молекулярной эмиссии в полость 603 сл. — микроколичеств 606 — потенциометрическое 604 сл. — спектроскопическое 602—604 — спектрофотометрическое 602 сл.
— титриметрическое 597—602
•---термометрическое 606
— ультрамикроколичеств 605
— электрохимическими методами 604 сл.
отделение 595—597
— высокоскоростной жидкостной хроматографией 597 — ионообменное 596
— осаждением 595
— ТСХ 596
— экстракцией 596
Триметафосфаты см. Метафосфаты
Трифосфаты см. Полифосфаты
Углн
определение карбоната 47
— серы 531, 538
Угольная пыль, определение силиката 200
Удобрения
анализ автоматический 461 определение бора 29, 39 сл. — нитрата 129, 137 — сульфата 536
— фосфата 461
— фторида 331
Уксусная кислота 9 сл.
Фармацевтические препараты определение бора 29, 40 — фосфатов 464 — цианидов 84
Формиаты 90—93
определение 90—93
— газохроматографическое 92
— методом ЯМР 93
— спектрофотометрическое 92
— титриметрическое 90—92
отделение 90
Фосфаты
высшие см. Полифосфаты
конденсированные см. Полифос-фаты
линейные и циклические, разделение 481
Фосфиты 476—479
определение 477—479
— автоматическое 479
— спектрофотометрическое
478 сл.
— титриметрическое 477 сл отделение 476 сл.
Фрукты и продукты нз них
определение нитрата 138
— цитрата 64, 68
Фториды 331—365
пирогидролиз 338 сл.
определение 341—361
— атомно-абсорбцнонное 360
— газохроматографическое 360
— гравиметрическое 341
— с ионоселектнвиым электродом 351—357
— каталитическое 358 сл.
— полярографическое 360
— потенциометрическое 360
— радиохимическое 357 сл.
— спектрофотометрическое 346— 351
— титриметрическое 343—346 ---термометрическое 359 сл.
— флуорнметрнческое 361
— хроматографическое 339 сл.
— электрохимическими методами 351—357
отгонка 332—335
отделение 332—341
— ионообменное 339
— микродиффузией 336—338
— микроколичеств 334
— осаждением 341 сл.
620
Фториды
определение соосаждением 341
— хроматографическое 339
Фторсиликаты 88—90
Хлораты 280—286
восстановление 283 определение 281—286 — амперометрическое 286 — ИК-спектроскопическое 284 — с ноноселектнвным электродом 284
— полярографическое 285 сл.
— потенциометрическое 284 сл.
— спектрофотометрическое 284
— титриметрическое 281—283
— электрохимическими методами 281—283 отделение 280 сл.
Хлориды 287—322
определение 290—322
— амперометрическое 318
— атомно-абсорбционное 307
— газохроматографическое 320 сл.
— гравиметрическое 290—292
— с ионоселективным электродом 313—316
— кинетическое 322
— кулонометрическое 316—318
— методом кольцевой печи 321
— микроколичеств 310
— нефелометрическое 306 сл.
— полярографическое 318
— потенциометрическое 309—313
— радиохимическое 319 сл.
— рентгенофлуоресцентное 321
— спектроскопическое 303—308
— спектрофотометрическое 303— 306
— титриметрическое 292—302
---с адсорбционным индикатором 296—298
---аргентометрическое 292—
298
--- высокочастотное 319
---комплексонометрическое
300 сл.
Хлориды
определение титриметрическое меркуриметрическое 298—300 ----методом Вибека 301 сл.
-------Мора 293 сл.
-------Фольгарда 294—296
--- редоксметодом 301
— — смеси галогенидов 301
----термометрическое 322
— ТСХ 321
— хронопотенциометрическое 319
— электрохимическими методами 309—319
— эмиссионным спектральным анализом 307 сл.
отделение 287—290
— газохроматографическое 289 — дистилляцией 290 — ионообменное 288 — осаждением 287 сл.
— соосаждением 288
— хроматографическое 288 сл.
Хлориты 327—331
определение 327—331
— полярографическое 330
— потенциометрическое 330
— спектроскопическое 329 сл. — титриметрическое 328 сл. — электрохимическими методами 330
отделение 327
Хроматы и бихроматы 51—63 определение 54—61 амперометрическое 59 — атомно-абсорбционное 58 — гравиметрическое 54 — ИК-спектроскопическое 58 — каталитическое 61
— колориметрическое н спектрофотометрическое 57 сл.
— кулонометрическое 59 сл.
— методом кольцевой печи 321
— полярографическое 61
— потенциометрическое 59
— радиохимическое 61
— спектроскопическое 57—58
— титриметрическое 54—57
621
Хроматы и бихроматы
— электрохимическими методами 59—61
отделение 52 сл.
— ионообменное 53
— осаждением 52
— соосаждением 53
— ТСХ и БХ 53
— экстракцией 52 сл.
— электрофорезом 53
Цемент
определение силиката 190, 195
— хлорида 295
Цианаты 69—71 определение 70 сл.
— амперометрическое 71
— гравиметрическое 70
— спектрофотометрическое 70 сл. отделение 69 сл.
Цианиды 72—88
определение 74—86
— автоматическое 78 с л.
— амперометрическое 84
— атомно-абсорбционное 82
— газохроматографическое 85
— гравиметрическое 74
— каталитическое 84 сл.
— колориметрическое 77 сл.
— полярографическое 84
— потенциометрическое 82—84
— радиохимическое 85
— рентгенофлуоресцентное 86
— спектроскопическое 77—82 — спектрофлуориметрическое 81 сл.
Цианиды
определение спектрофотометрическое 77—81
— титриметрическое 75—77
— ТСХ 86
— электрохимическими методами 82—84
отделение 72—74
— дистилляцией 72 сл.
— микродиффузией 73 сл.
— хроматографическое 74
— экстракцией 74
Цитраты 63—69
определение 64—69
— автоматическое 68
— гравиметрическое 64 сл.
— колориметрическое 67 сл.
— радиохимическое 69
— титриметрическое 65—67
— флуоресцентное 68
отделение 63
— ионообменное 63
— ТСХ 63
— экстракцией 64
Чай, определение фторида 357
Шлаки, определение силиката 195
Шреётера алкалиметр 44
Щавелевая кислота 152
Электролиты хромирования, определение фторсиликатов 89
У. ДЖ. УИЛЬЯМС
ОПРЕДЕЛЕНИЕ АНИОНОВ
Редактор Абрамова В. Л.
Художник Бекетов Е. В.
Художественный редактор Носов Н. В.
Техническвй редактор Косачева Г. И., Скитина В. М.
Корректор Черниховская М. В.
ИБ № 1251
Сдано в наб. 19.04.82. Подп. в печ. 24.09.82. Формат бумаги бОХЭО’/и, Бумага тнп. № I. Гарн. литературная. Печать высокая. Усл. печ. л. 39,0. Усл. кр.-отт. 39.0. Уч.-изд. л. 46,98. Тираж 12 300 экз. Заказ № 167. Цена 3 р. 60 к. Изд. № 2353.
Ордена .Знак Почета" издательство „Химия", 107076, Москва, . Стромынка, 13
Ленинградская типография № 2 головное предприятие ордена Трудового Красного Знамени Ленинградского объединения «Техническая книга» им. Евгении Соколовой Союзполнграфпрома прн Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
198052, г. Ленинград, Л-52, Измайловский проспект, 29