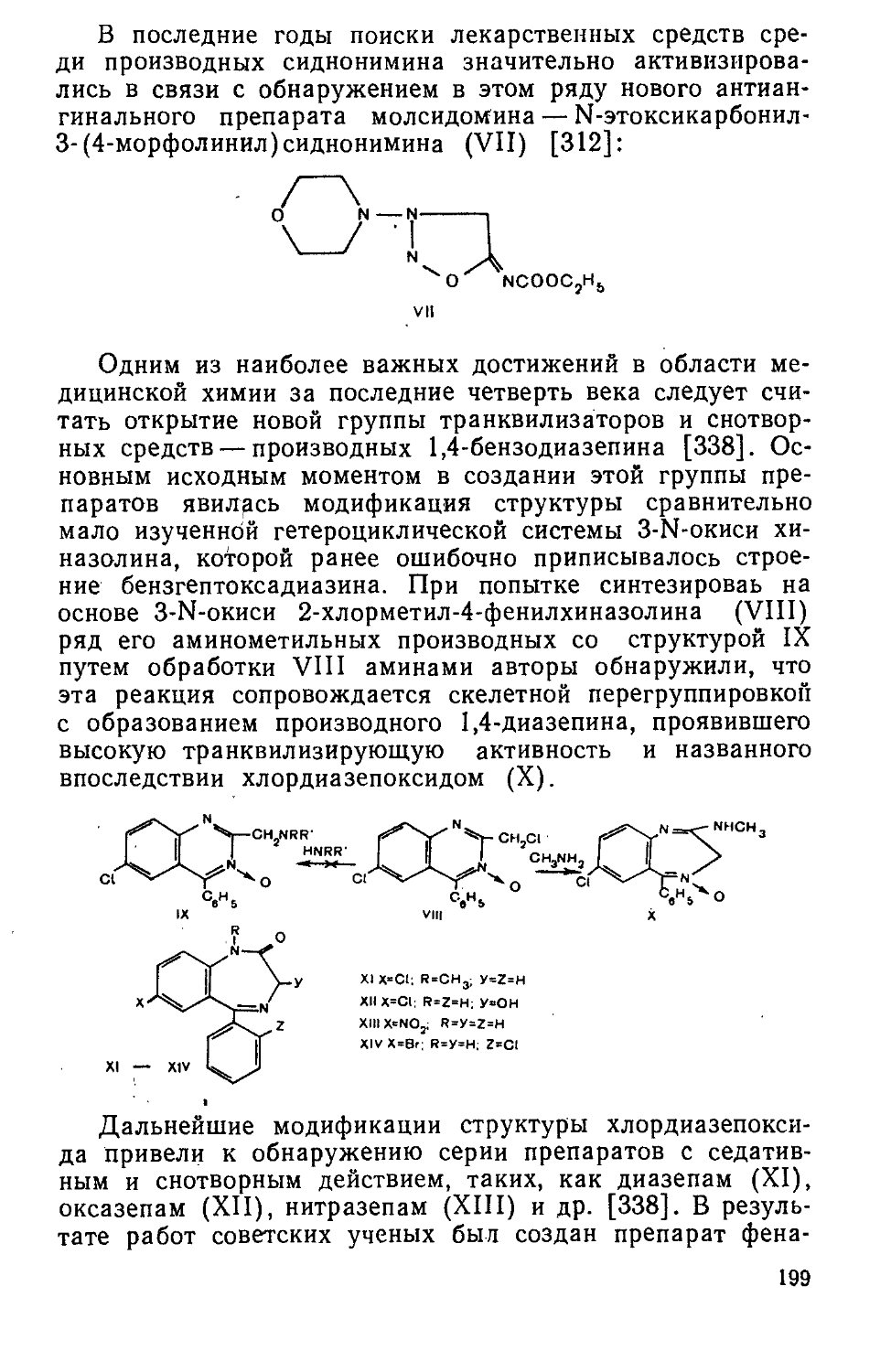
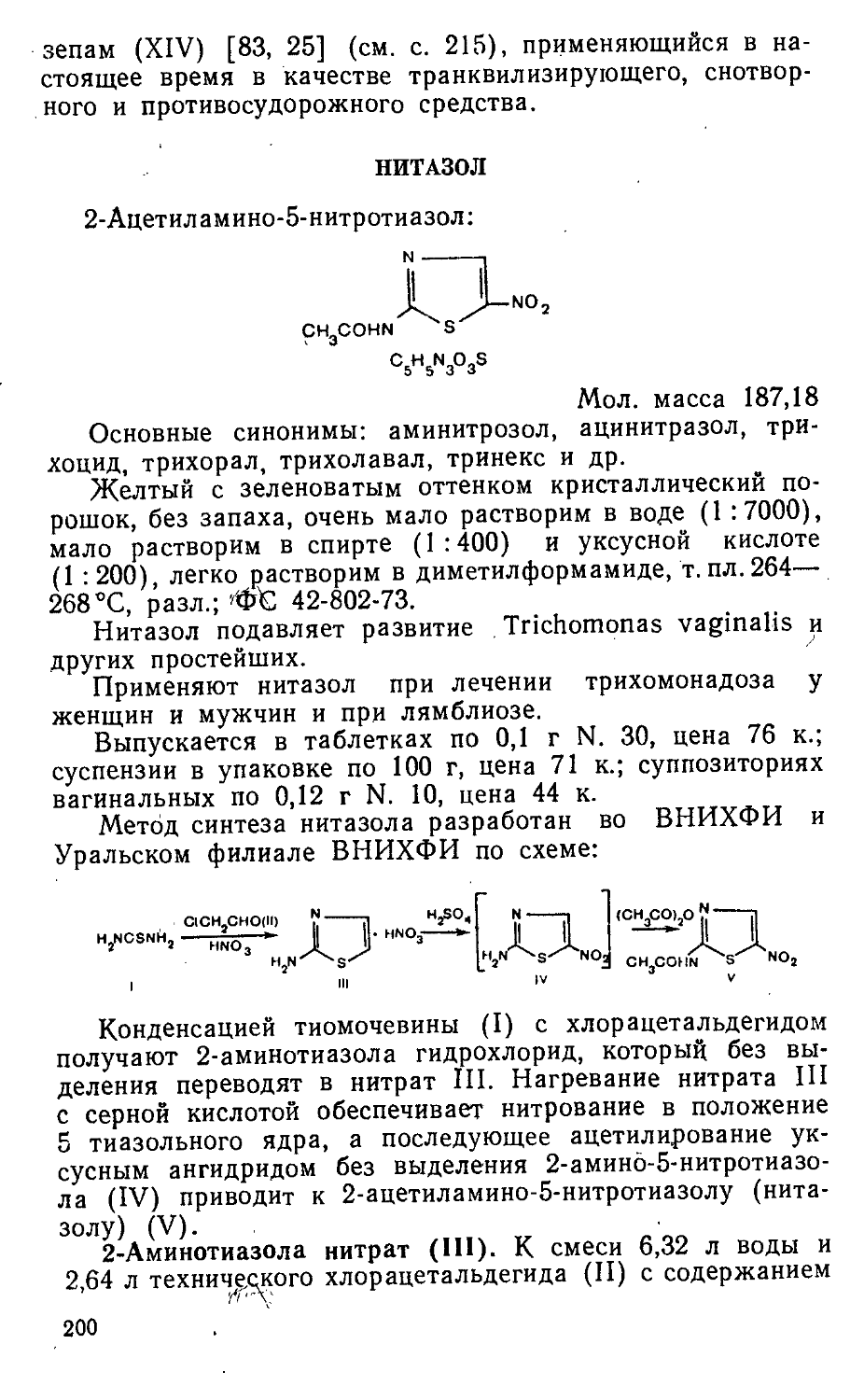
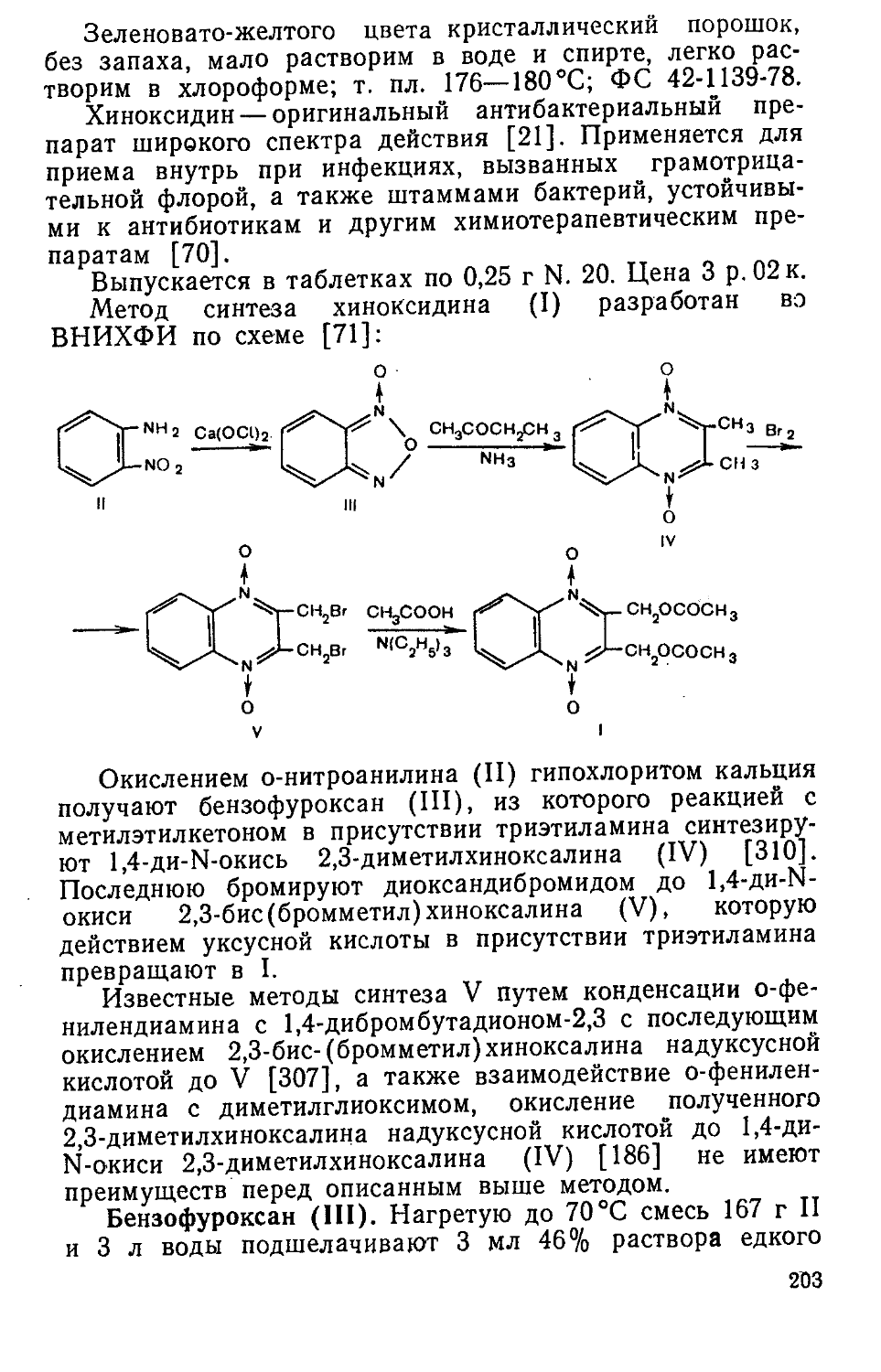
/









































































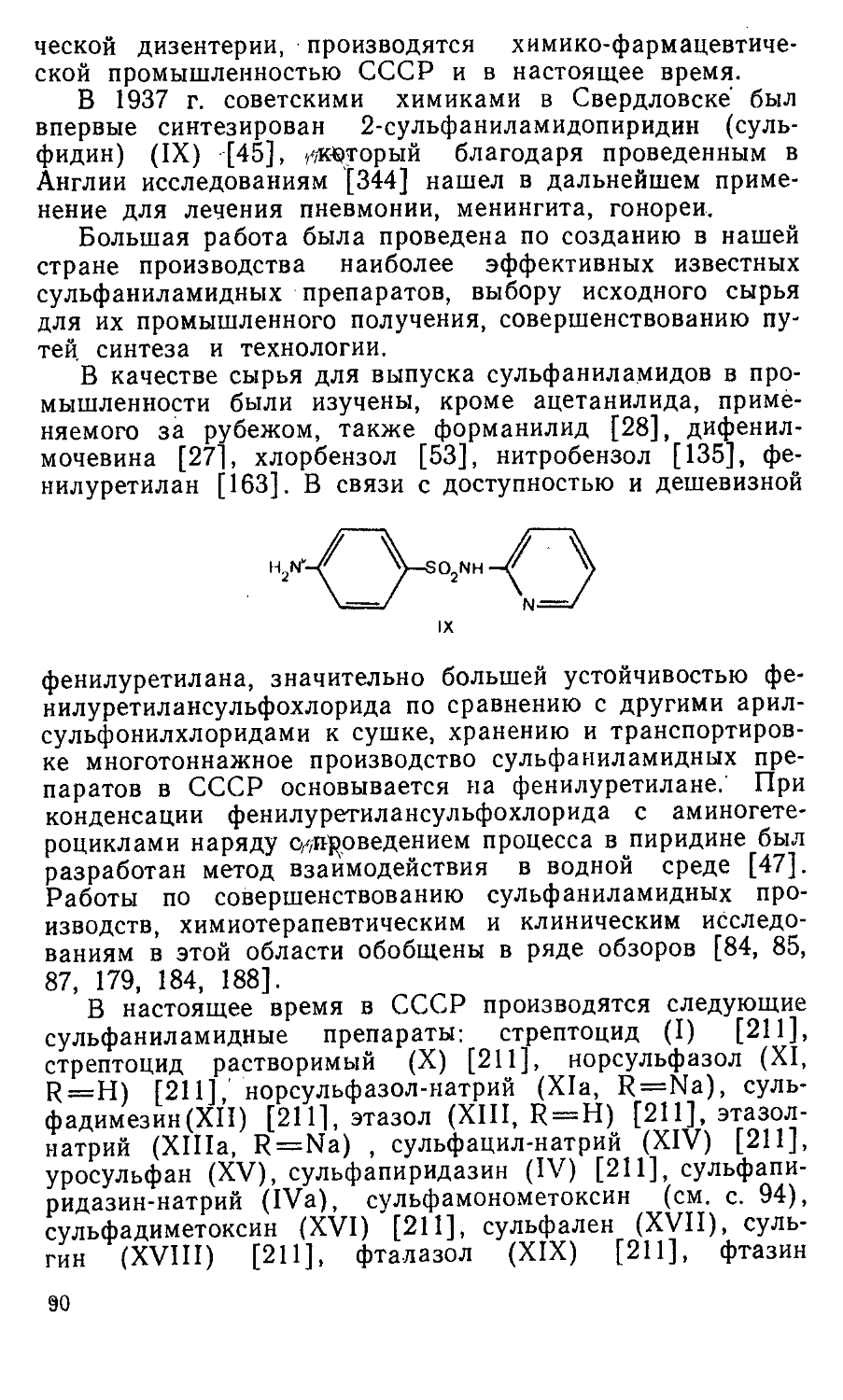
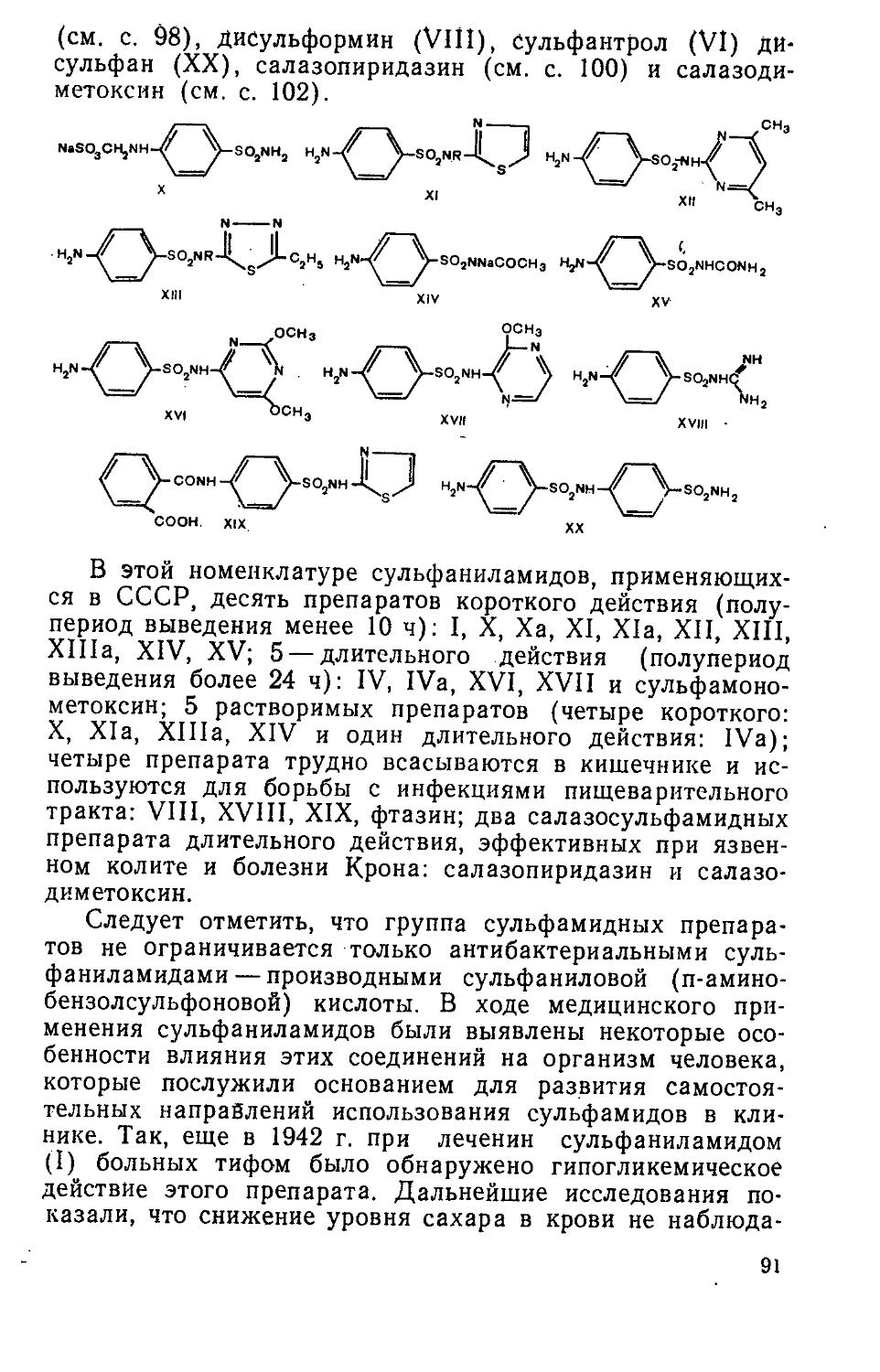



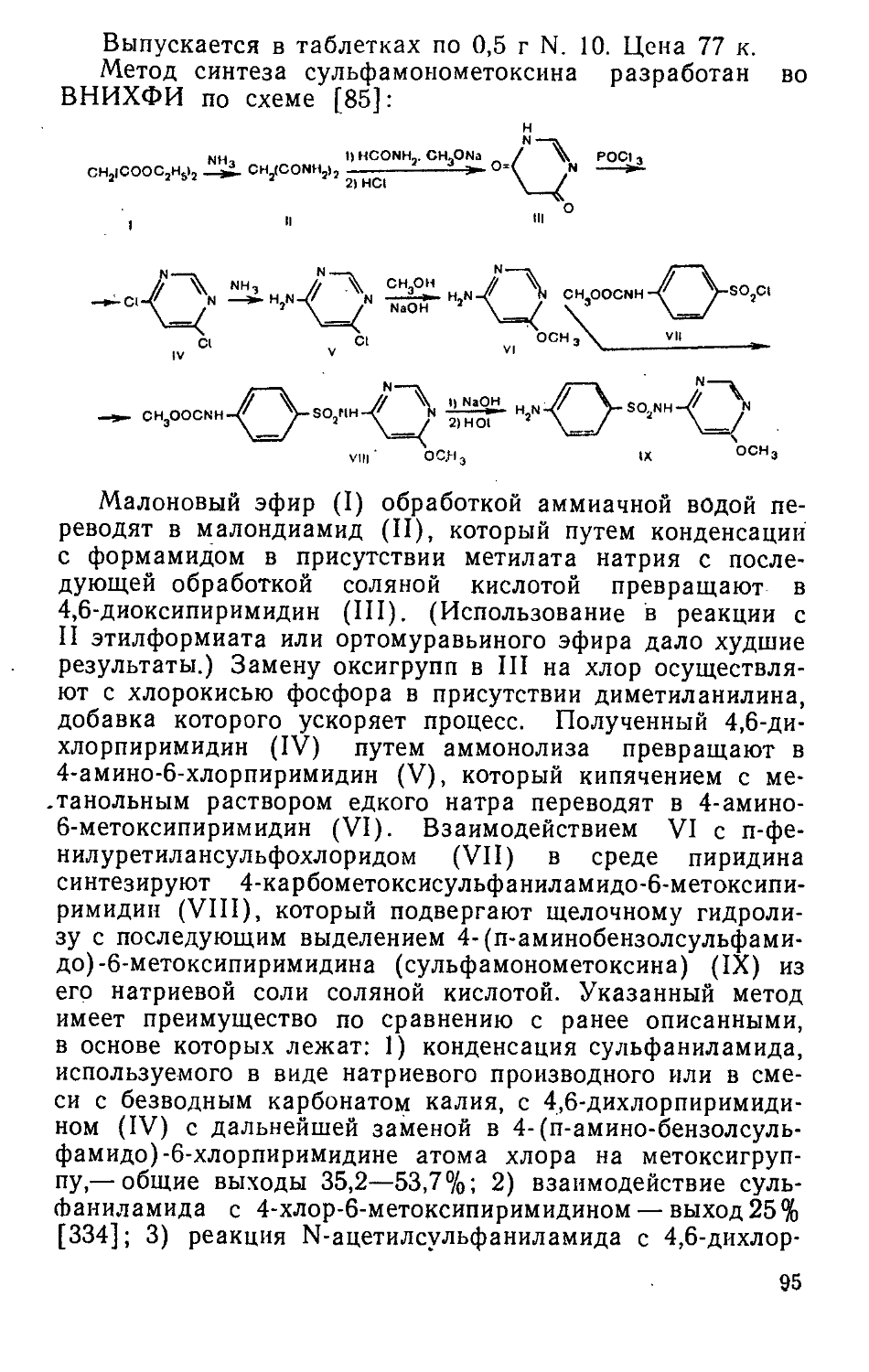












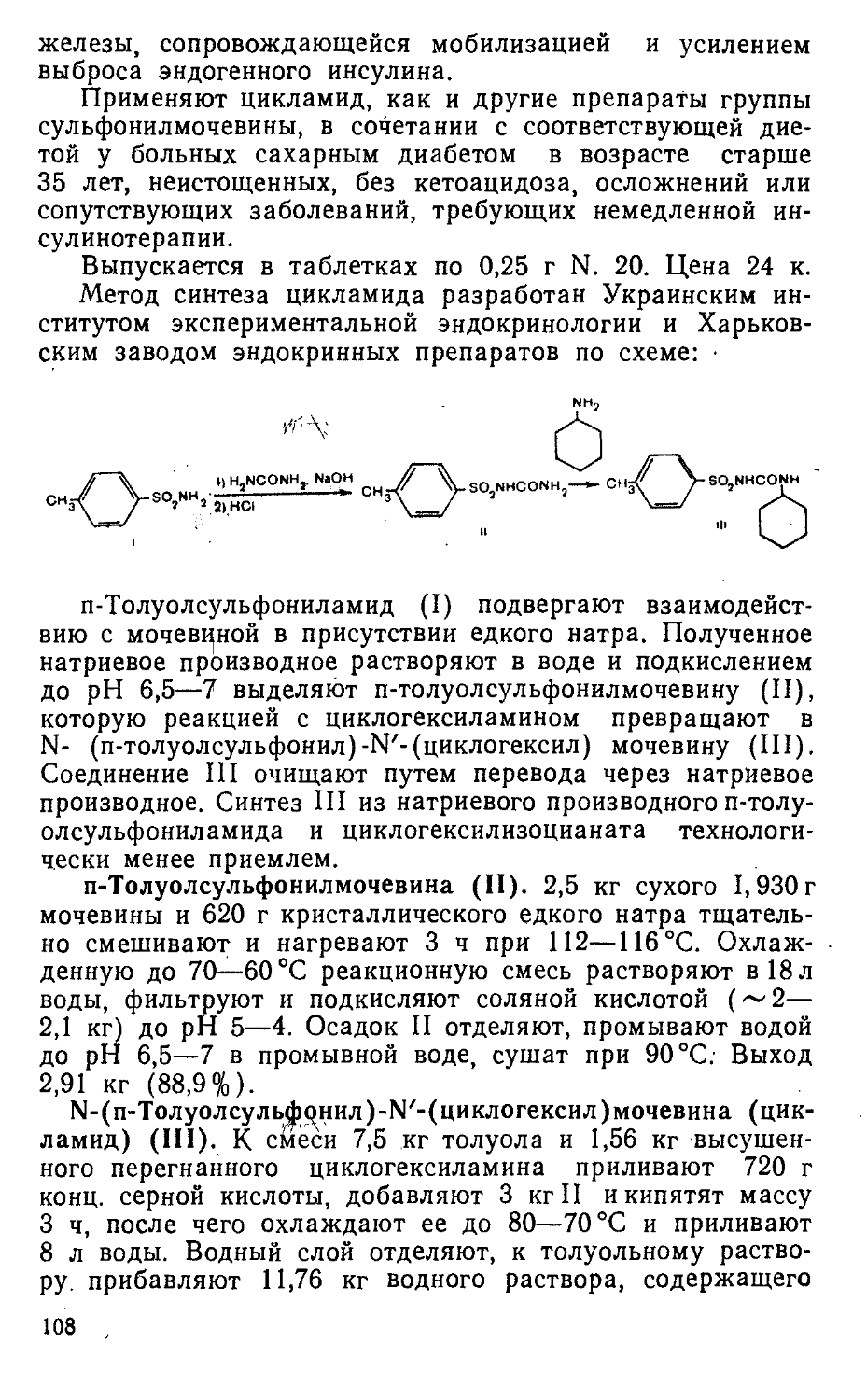
















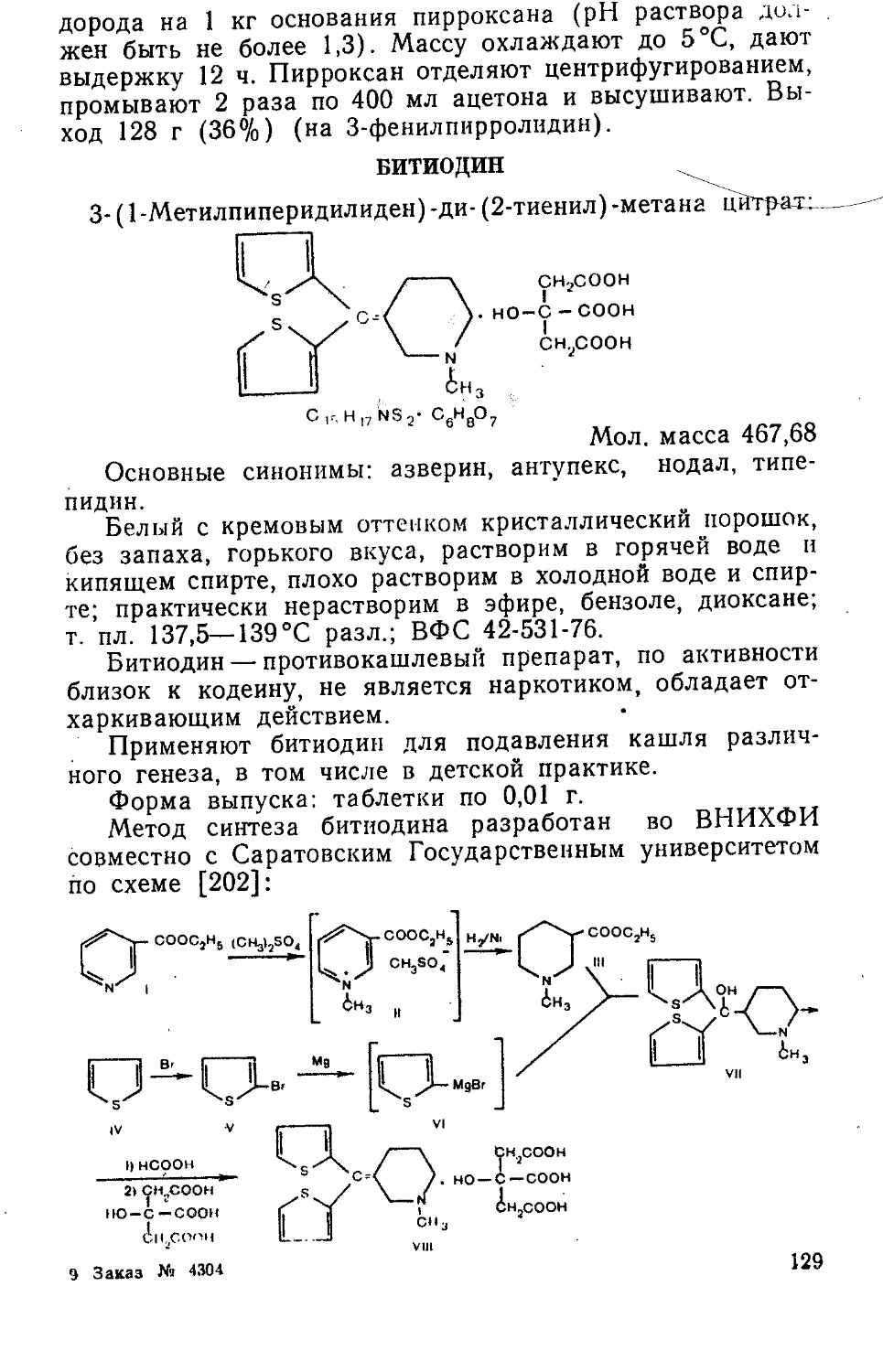































































































































Author: Яхонтов Л.П. Глушков Р.Г.
Tags: лекарственные средства в соответствии с их активным веществом фармакология фармация токсикология
Year: 1983




Text
.1. II. яхонтов
P. Г. ГЛУШКОВ
СИНТЕТИЧЕСКИЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Под редакцией доктора фарм. наук
А. Г. НАТРАДЗЕ
МОСКВА ’МЕДИЦИНА” 1983
ББК 52.81
С 38
УДК 615.2/.3.012.1
ЯХОНТОВ Л. Н„ ГЛУШКОВ Р. Г. Синтетические лекарственные средства./Под ред. А. Г. НАТРАДЗЕ.— М.: Медицина, 1983, 272 с.
Авторы — сотрудники Всесоюзного ордена Трудового Красного Знамени Научно-исследовательского химико-фармацевтического института нм. С. Орджоникидзе.
Л. Н. ЯХОНТОВ — доктор химических наук, профессор, зав. лабораторией синтеза сердечно-сосудистых средств.
Р. Г. ГЛУШКОВ — лауреат Государственной премии СССР, член-корреспондент АМН СССР, директор института, зав. лабораторией химнн нейротропных средств.
В монографии описаны пути поиска, методы синтеза, даны характеристики более 80 новых медикаментов, включенных в Государственный реестр СССР н в основном освоенных отечественной химико-фармацевтической промышленностью. Материал изложен в 8 главах по классам химических соединений. Для каждого лекарственного препарата приведены показатели применения в клинике, номенклатура выпускаемых лекарственных форм. Дается критический анализ известных путей синтеза препарата и его полупродуктов, описаны оптимальные методики получения веществ.
Монография представляет интерес для исследователей, работающих в области биологически активных соединений, а также сотрудников химико-фармацевтической промышленности.
Библиография: 348 названий.
Рецензент профессор Ю. Ф. КРБ1ЛОВ.
Леонид Николаевич Яхонтов, Роберт Георгиевич Глушков
СИНТЕТИЧЕСКИЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА
Зав. редакцией Ю. В. Махотин. Редактор издательства Г. Ф. Крутова.
Художественный редактор М. П. Кузнецова. Художник Г. А. Шипов.
Технический редактор В. П. Сорокина. Корректор 3. П. Бабуева
ИБ № 3617
Сдано в на’бор 23.12.82. Подписано к печати 23.06.83. Т-08586. Формат бумаги 84ХЮ8‘/з2- Бум. тип. № 1. Лит. гарн. Печать высокая. Усл. печ. л. 14.28. Усл. кр. отт. 14,49. Уч.-изд. л. 15,80. Тираж 8300 экз. Заказ 4304. Цена 1 р. 50 к. Ордена Трудового Красного Знамени издательство «Медицина», Москва, Петро-веригскнй пер., 6/8.
Типография им. Смирнова Смоленского облуправлепия издательств, полиграфии и книжной торговли, г. Смоленск, пр. им. Ю. Гагарина. 2.
4108000000—280
039(01)—83
78—83
© Издательство «Медицина» Москва, 1983
ПРЕДИСЛОВИЕ
Коммунистическая партия и Советское правительство уделяют большое внимание здоровью и росту благосостояния людей. В утвержденных XXVI съездом КПСС «Основных направлениях экономического и социального развития СССР на 1981—1985 годы и на период до 1990 года» предусматривается: «Увеличить выпуск продукции медицинской промышленности примерно в 1,4 раза. Предусмотреть создание и освоение производства высокоэффективных лекарственных средств, особенно для лечения сердечно-сосудистых и онкологических заболеваний, болезней эндокринной системы, а также полусин-тетических антибиотиков»1.
В обращении к рабочим, инженерно-техническим работникам и служащим предприятий и организаций Министерства медицинской промышленности 5 декабря 1980 г. Генеральный секретарь ЦК КПСС тов. Л. И. Брежнев писал: «ЦК КПСС выражает уверенность в том, что коллективы предприятий и организаций министерства, эффективно используя накопленный опыт, достижения науки и техники, будут и впредь настойчиво бороться за выполнение решений партии по дальнейшему улучшению обеспечения здравоохранения и советских людей необходимыми лекарственными средствами и изделиями медицинской техники...»2.
Советские ученые, работающие по созданию, внедрению в производство и медицинскую практику новых эффективных лекарственных препаратов, отдают все свои силы выполнению этой благородной задачи.
В монографии обобщены основные результаты исследований советских ученых по созданию новых медикамен
! Материалы XXVI съезда КПСС.— М.: Изд. политической литературы, 1981, с. 183.
2 Брежнев Л. И. Рабочим, инженерно-техническим работникам и служащим предприятий и организаций Министерства медицинской промышленности,—Правда, 1980, 5 дек.
3
тов, большинство которых освоены отечественной хймй-ко-фармацевтической промышленностью за последние 10 лет и вошли в Государственный реестр СССР.
Монография является как бы продолжением известного справочника М. В. Рубцова и А. Г. Банникова «Синтетические химико-фармацевтические препараты», изданного в 1971 г. Как и в упомянутом справочнике, в монографии не рассматриваются биопрепараты, антибиотики, витамины, гормональные и диагностические средства, а также продукты фитохимии. Не включены в монографию и работы по усовершенствованию действующих производств, даже если они связаны с коренными изменениями схем синтеза и технологических решений.
Материал в книге сгруппирован по классам химических соединений. Описанию отдельных лекарственных препаратов в каждой главе предпосланы общая характеристика истории, современное состояние и тенденции развития поиска, закономерности связи между строением и биологической активностью, определено по возможности место новых медикаментов в ряду других лекарственных средств этого типа, используемых в СССР.
Для номенклатуры химико-фармацевтических производств характерна значительная изменчивость. Создание новых эффективных лекарственных средств связано с прекращением выпуска устаревших медикаментов. Из числа описанных в справочнике М. В. Рубцова и А. Г. Байчико-ва 170 препаратов за прошедшее десятилетие по тем или иным причинам были исключены из номенклатуры применяющихся в СССР лекарственных средств 17 препаратов: аллацил, бензацин, галохин, деморфан, димерин, мепанит, метилсульфазин, мефолин, новэмбитол, пара-мион, спиразидин, субехолин, трансамин, трийотраст, фенаком, фентролин, этизин. Тенденция к существенному обновлению номенклатуры медикаментов сохранится, по-видимому, и в дальнейшем.
К сожалению, объем монографии не позволил авторам провести анализ основных направлений поиска новых синтетических химико-фармацевтических препаратов в СССР и других странах, закладывающих фундамент создания будущих медикаментов. Эти исследования весьма многочисленны и разнообразны. Вместе с тем в их проведении наблюдаются некоторые общие тенденции «конструирования лекарств». В частности, можно отметить, что при создании лекарственных 'препаратов, механизм действия которых обусловлен взаимодействием их с био
4
химическими рецепторами, обычно при варьировании структуры в молекуле сохраняются определенные расстояния между реакционными центрами, ответственными за взаимодействие с активными участками рецепторов. Изменения молекул в этих случаях чаще всего направлены на усиление реакционной способности таких центров и варьирование пространственных характеристик молекул. Указанный принцип, в частности, был использован (сознательно или интуитивно) при создании описанных в монографии лекарственных препаратов анатруксония, дитилина, квалидила, метамизила, пиромекаина, темехи-на, тропафена, фенкарола и др.
По иному пути идут «контрукторы лекарств», когда механизм действия препарата связан не с блокадой рецепторов, а с участием соединения в метаболизме путем изменения или блокады ферментативных процессов. В этих случаях обычно при варьировании структуры стремятся сохранить общий объем молекулы, чтобы обеспечить возможность взаимодействия ее с ферментами. При этом меняют характер распределения электронной плотности в молекуле, реакционную способность отдельных участков, вводят, извращают или удаляют реакционные центры, необходимые для процессов нормальных биохимических превращений (переход к аза-, тиа-, окса-, дезаза-, дезоксианалогам и др.). Этот принцип варьирования структуры биологически активных веществ был сознательно или интуитивно использован при создании описанных в монографии лекарственных препаратов асалина, днйодбензотэфа, метотрексата, пиридитола, сульфамоно-метоксина, фенибута, фтазина, фурагина, хлоцикламида и др. Более детальное рассмотрение общих теоретических и методических вопросов поисковых исследований, к сожалению, выходит за пределы монографии и представляет интерес для самостоятельного обсуждения.
При описании отдельных лекарственных препаратов в монографии не приведены качественные реакции подлинности на эти препараты, методы определения чистоты и количественного содержания, но даны ссылки на соответствующие фармакопейные статьи или межреспубликанские технические условия (МРТУ). Приведены только те лекарственные формы препаратов, которые выпускаются химико-фармацевтической промышленностью с указанием розничных цен. В монографию не включены препараты, оригинальные методы синтеза которых не опубликованы в литературе.
5
Для удобства пользования монографией составлены указатели: алфавитный и по медицинскому применению лекарственных средств. (В указатель включены только препараты, разрешенные в СССР. Жирным шрифтом выделены лекарственные препараты, описанные подробно.) В монографии имеются также указатели всех исходных соединений и полупродуктов получения лекарственных препаратов.
Авторы выражают искреннюю благодарность сотрудникам ВНИХФИ С. С. Либерман, Е. Е. Михлиной, Т. А. Гуськовой и Л. С. Блиновой за ценные советы и помощь при написании монографии.
ПРЕПАРАТЫ ГРУППЫ АМИНОКИСЛОТ И ИХ ПРОИЗВОДНЫХ
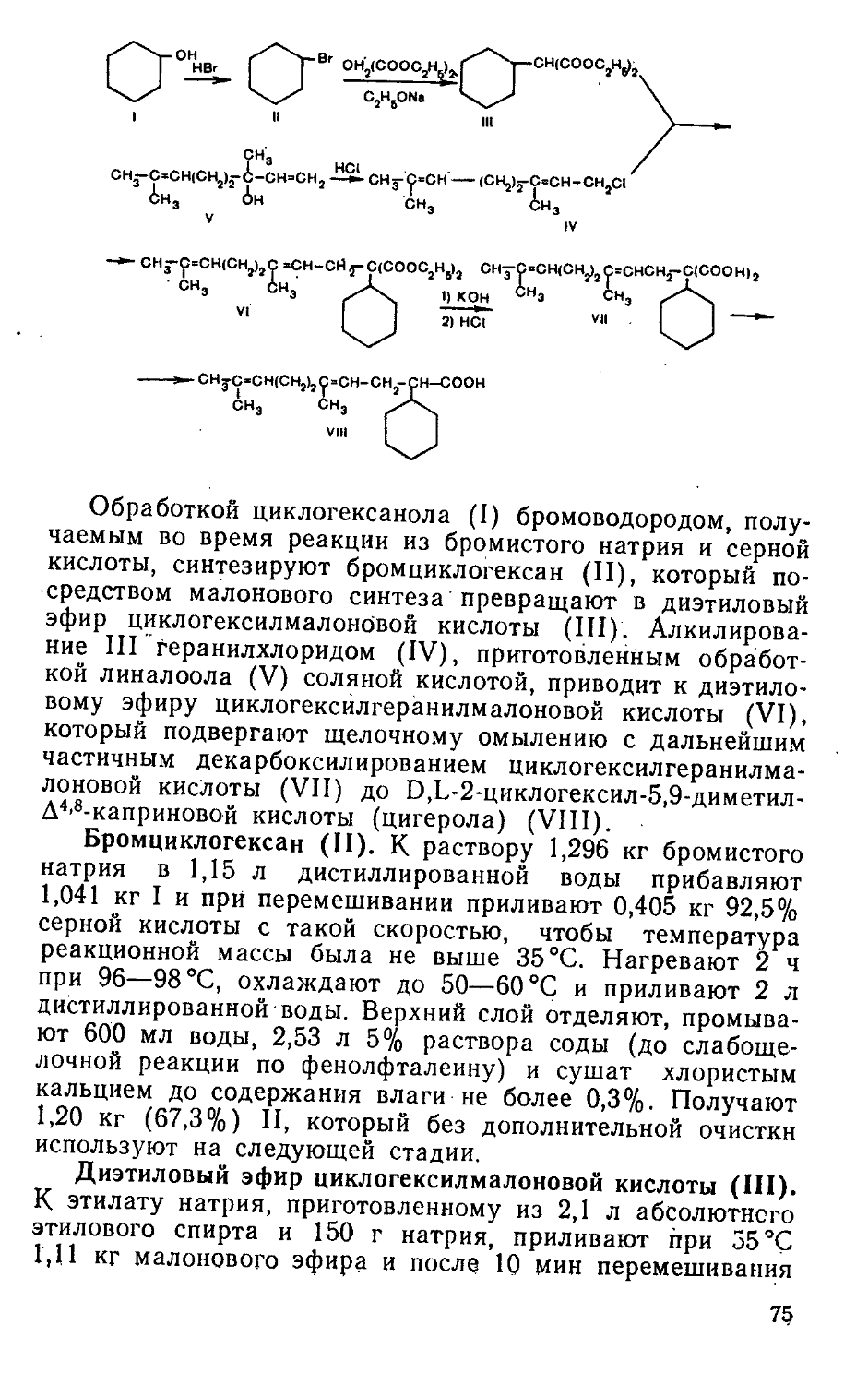
Важная роль аминокислот в процессах жизнедеятельности с давних пор стимулировала исследования по проведению поиска лекарственных средств как среди природных аминокислот, так и их синтетических аналогов. В результате широких фундаментальных исследований такие природные аминокислоты, каю глутаминовая кислота (I), метионин, гистидин, цистеин, а также препараты, являющиеся смесью аминокислот, получаемые из гидролизатов крови и других биологических субстратов, прочно вошли в арсенал лекарственных средств и активно используются в терапии при лечении больных с заболеваниями различной этиологии. Существенное влияние в проблеме направленного поиска новых лекарственных средств среди аминокислот и их производных оказало развитие исследований по биохимии клетки и организма в норме и патологии. Так, изучение метаболических процессов, протекающих в нервных тканях, показало, что первичным продуктом ферментативного расщепления I является у-аминомасляная кислота (II).
HOOC(CH2)2CH(NH2)COOH hooc (Ch2)3nh2
। и
При этом было установлено, что метаболические процессы в мозге нормально функционирующего организма поддерживают концентрацию II на стабильном уровне, а изменение концентрации II приводит к нарушению нервной деятельности [230]. Эти данные послужили основанием для применения II в заместительной терапии с целью лечения различных форм сосудистых заболеваний головного мозга, эндогенных депрессий и алкогольных полиневритов. Использование II для лечения нервно-психических заболеваний открыло перспективы для создания новых эффективных лекарственных средств с избирательным действием на ИНС среди соединений, близких
7
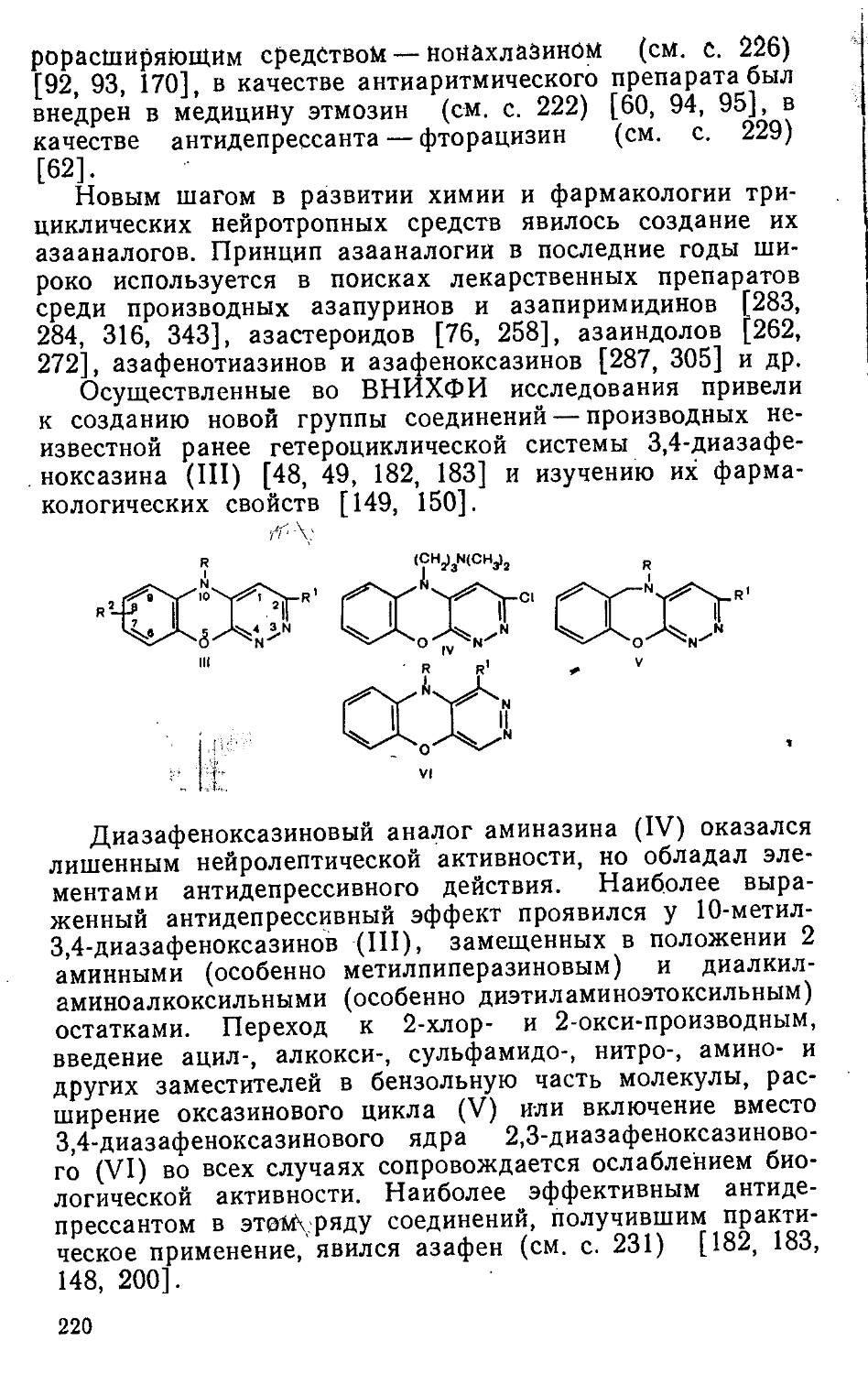
по структуре II, в том числе и среди веществ, являющихся продуктами метаболизма II. Так, р-окси-у-аминомасляная кислота (III) (богамк,лгамибетан) оказалась весьма эффективным средством' при лечении больных эпилепсией и детей с умственной отсталостью; кальциевая соль го-мопантотеновой кислоты (пантогам) (см. с. 10) применяется в качестве противосудорожного средства с седативным компонентом и умеренным гипотензивным эффектом; у-оксимасляная кислота:
H2NCH2C|H(OH|CH2COOH III
HOfCH^COONa IV
CK^CONHj,.
VI
Н NCH СП(СвН5ЮН COOII
V
в виде натриевой соли (IV) [12] оказывает отчетливое успокаивающее действие при неврастенических, неврозоподобных и некоторых психических расстройствах [109]. Исследования по поиску психотропных средств среди синтетических аналогов II привели к открытию оригинального транквилизатора р-фенил-у-аминомасляной кислоты (фенибут, фенигам ) (V) [193, 219], сочетающей в своей структуре фрагменты(,^вух биогенных веществ: у-амино-масляной кислоты и "р-фенилэтиламина — и в отличие от II легко проникающей в мозг. Дальнейшие работы по синтезу структурных аналогов II привели к получению пирролидон-2-ацетамида (пирацетама, ноотропила) (VI) — представителя новой группы лекарственных средств — «ноотропов»— препаратов, действующих на мышление. Низкая токсичность пирацетама, своеобразие его фармакологической активности, связанная с каталитическим действием VI в отношении метаболических процессов, протекающих в мозге, открывают новые перспективы для создания ноотропных лекарственных средств среди структур, родственных II и VI.
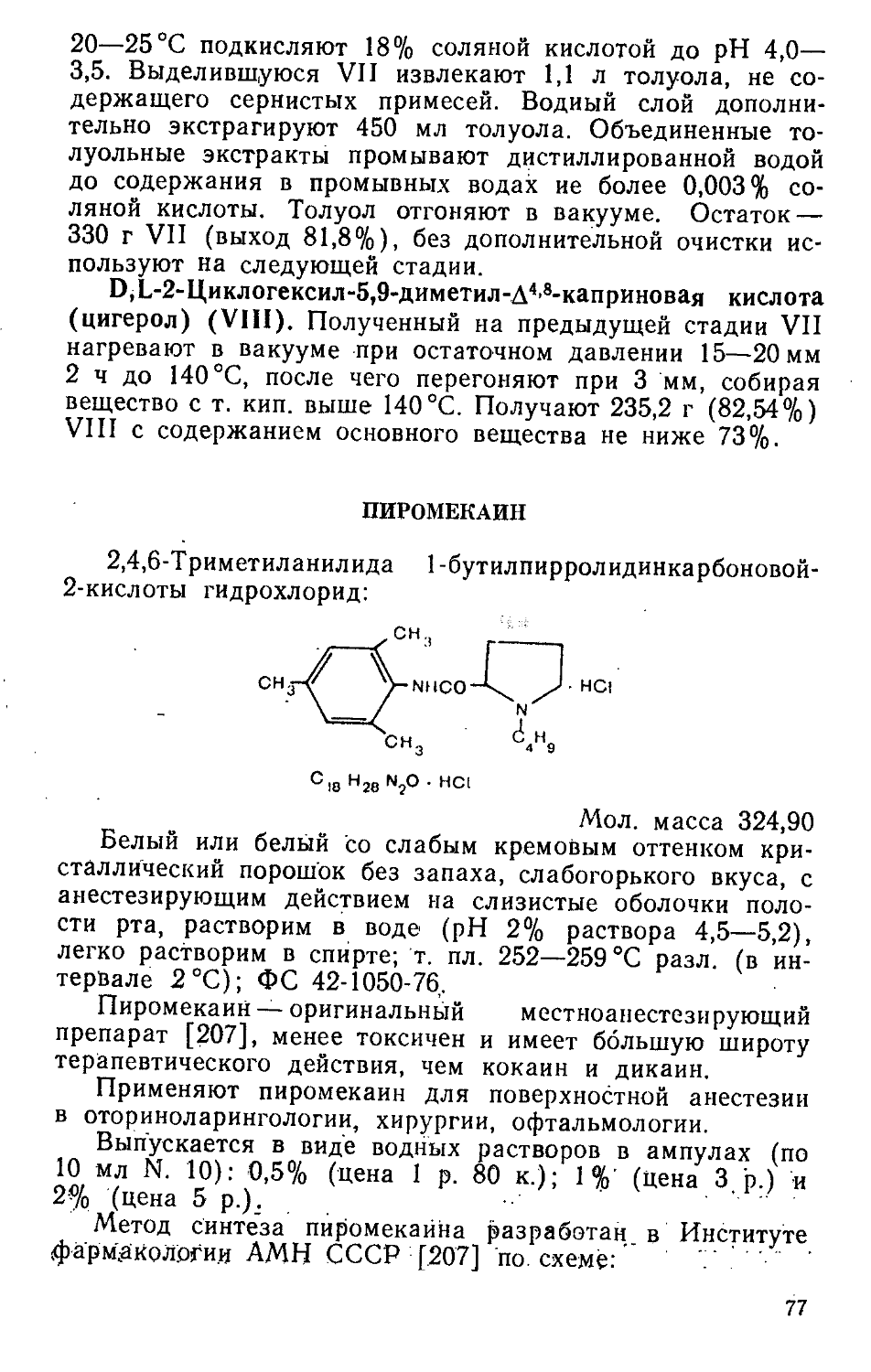
Способность аминокислот как полифункциональных соединений образовывать растворимые в воде комплексы с катионами металлов нашла применение при создании комплексообразующих препаратов, используемых в качестве антидотов при отравлениях тяжелыми металлами.
8
К таким препаратам относятся пеницилламин (VII), тета-цин-кальций (VIII), пентацин (IX) и динатриевая соль этилендиаминтетрауксусной кислоты (X) [273].
4*^3 ,С-СН—СООН NaOOCCHj СНОСН2 CKCOONa
л / I | \ / 2 । Z 2
СН3 SH Nh2
VII бн2 'с» ус,,г
00-0 о — со
CH COON.i VIII / 2 GH,CH.N О
NaOOCGHjN °>Са • nH О
сн!снХ чь CH COONa IX NaOOCCII, CH^COONa
NCH2CH2N\ 2H2O
НО ОС CH, CH?COOH
X
>NH(CH2I5C00h XI x = H: XII >=СОСН;
Среди аминокислот и их производных обнаружены вещества, обладающие антиферментной активностью. Доступная е-аминокапроновая кислота (XI), легко образующаяся при гидролитическом расщеплении е-капролактама, угнетает фибринолиз и используется в качестве кровоостанавливающего средства [140]. N-Ацетил-е-аминокапро-новая кислота (ацемин) (XII) ингибирует действие лизосомальной пептидазы и находит применение в качестве ранозаживляющего препарата (см. с. 15).
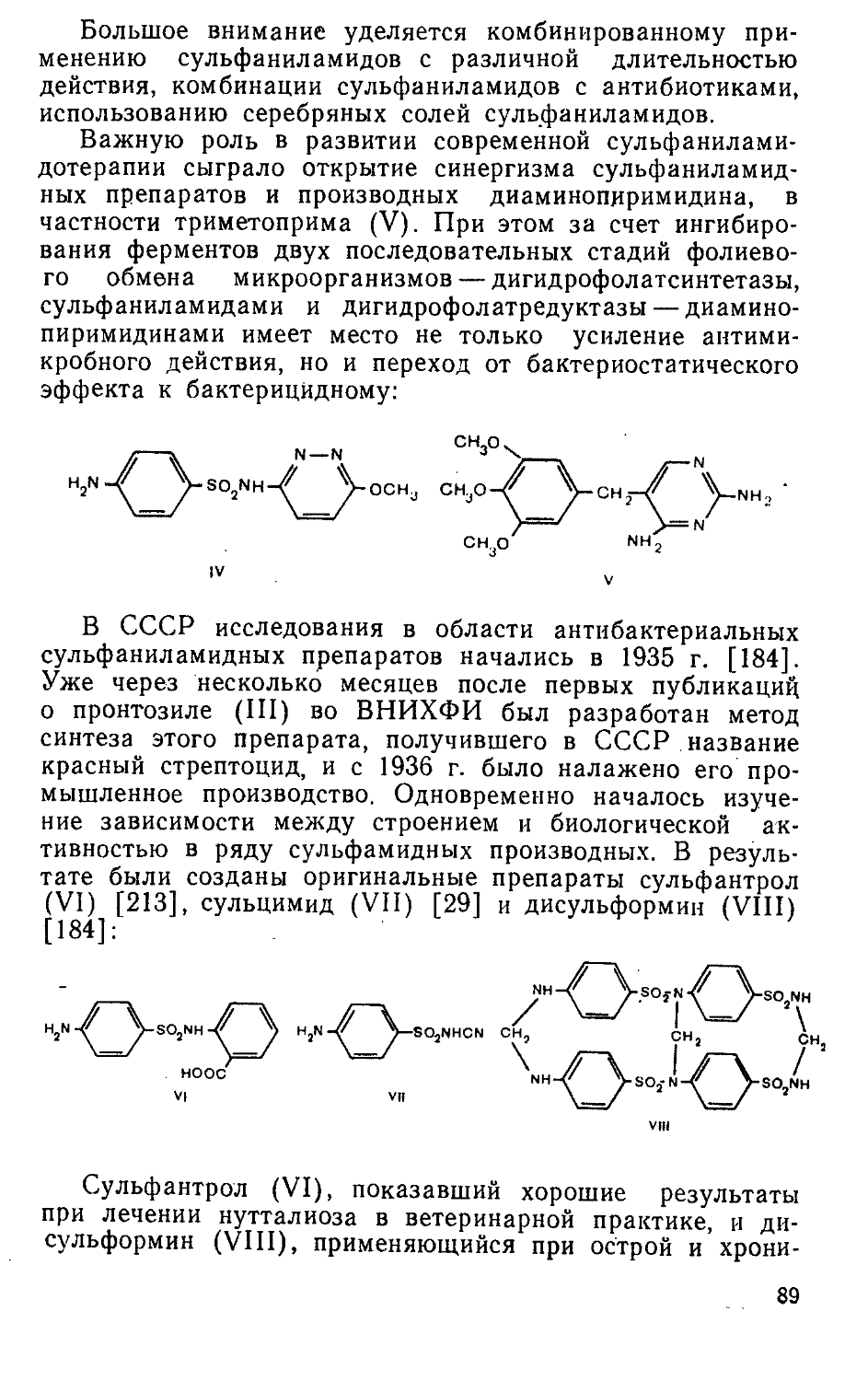
Некоторые производные аминокислот и, в частности, содержащие канцеролитическую бис-(р-хлорэтил) аминную группировку, используются в химиотерапии опухолей [243]. Идея применения аминокислот для создания противоопухолевых препаратов основывается на их способности проникать через мембраны раковых клеток в 4—5 раз быстрее, чем через мембраны нормальных клеток. В данном случае фрагменты аминокислот выполняют транспортные функции для канцеролитических группировок и обеспечивают избирательное накопление лекарственного средства в опухолевой ткани [124]. К подобным
9
препаратам принадлежат (XIV) и асалин (см. с. 12).
сарколизин (XIII), лофенал
XIV
С1СН2СН2
N
с1сн2дн2
nh2
Н^НСООН
XIII
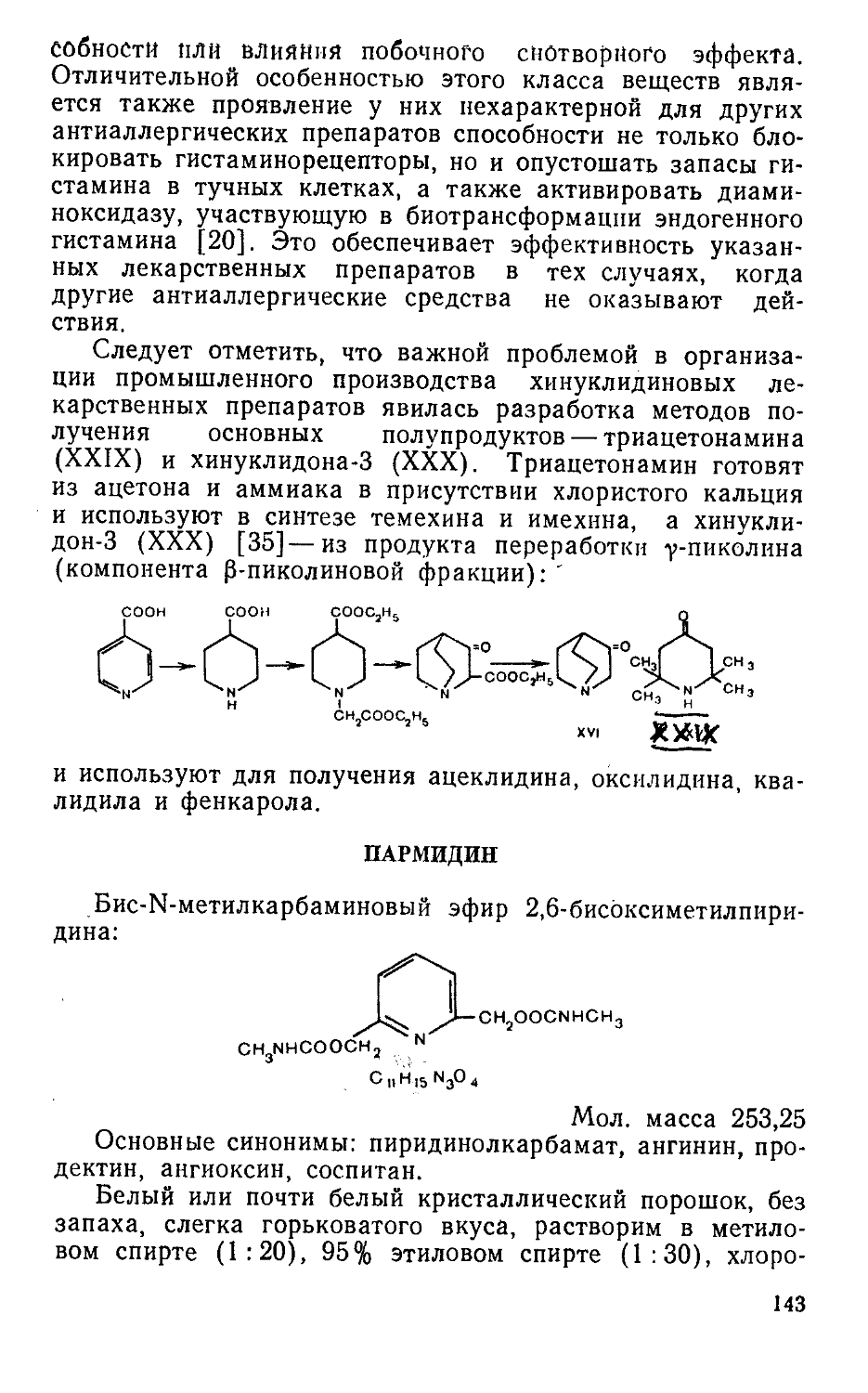
В последние годы интенсивно развивается новое направление по ведению поиска лекарственных средств среди соединений белково-пептидной природы, включающих природные пептиды и их синтетические аналоги. Это направление основывается на результатах фундаментальных биохимических исследований, выявивших роль таких биологически активных пептидов, как энкефалины, эндорфины, в регулировании процессов жизнедеятельности. Этими исследованиями, в частности, было показано, что ренин-ангиотецзцн-альдостероновая система регулирует натриевое и калйевое равновесие в организме и участвует в регуляции артериального давления. Поиск новых лекарственных средств в этом направлении уже привел к первым положительным результатам, и найден биологически активный дипептид — каптоприл (XV) [325].
ноос'
3
xv
Показано, что механизм гипотензивной активности каптоприла связан с его способностью снижать уровень ангиотензина II и аккумулировать содержание в организме брадикинина, обладающего сосудорасширяющим действием.
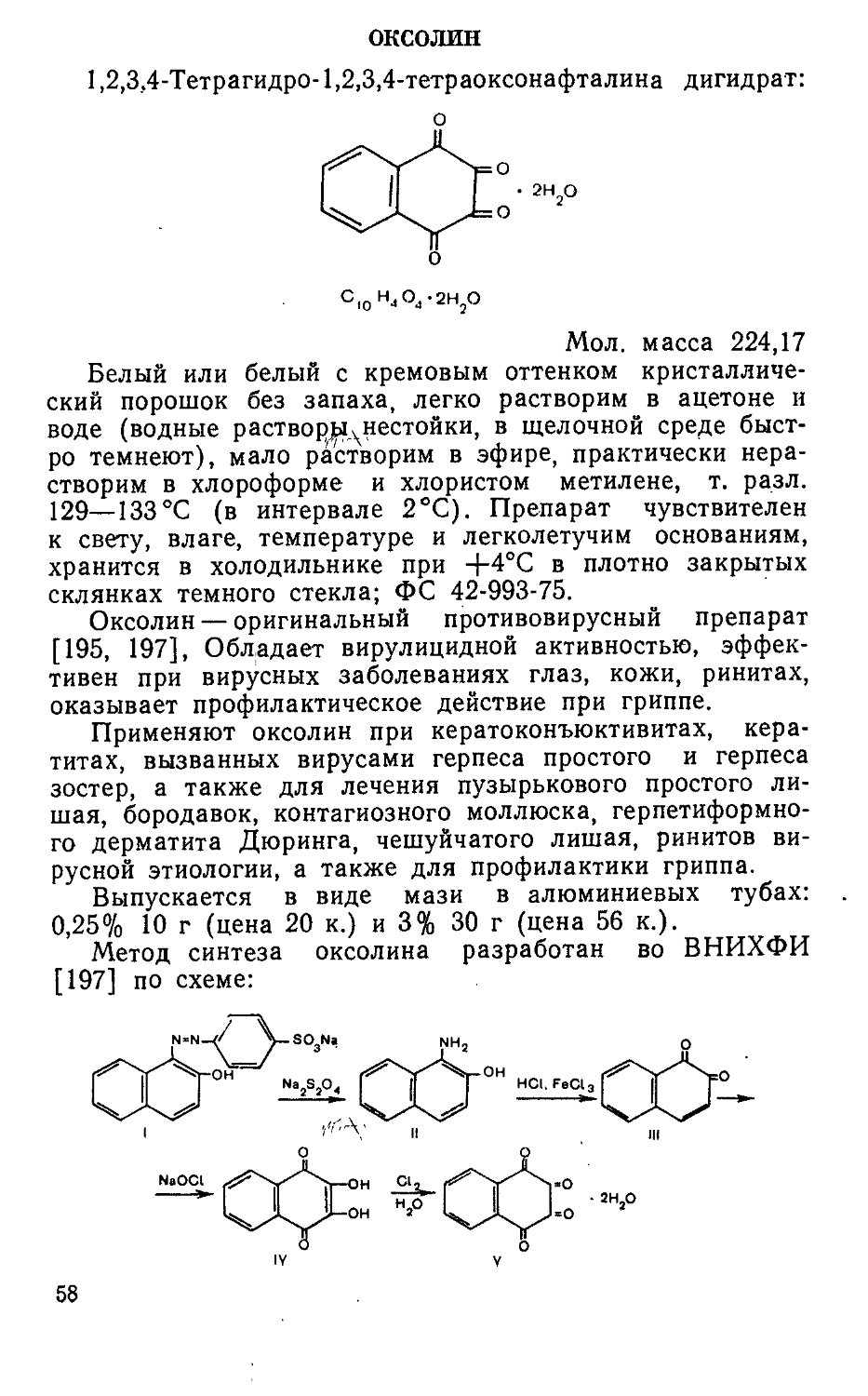
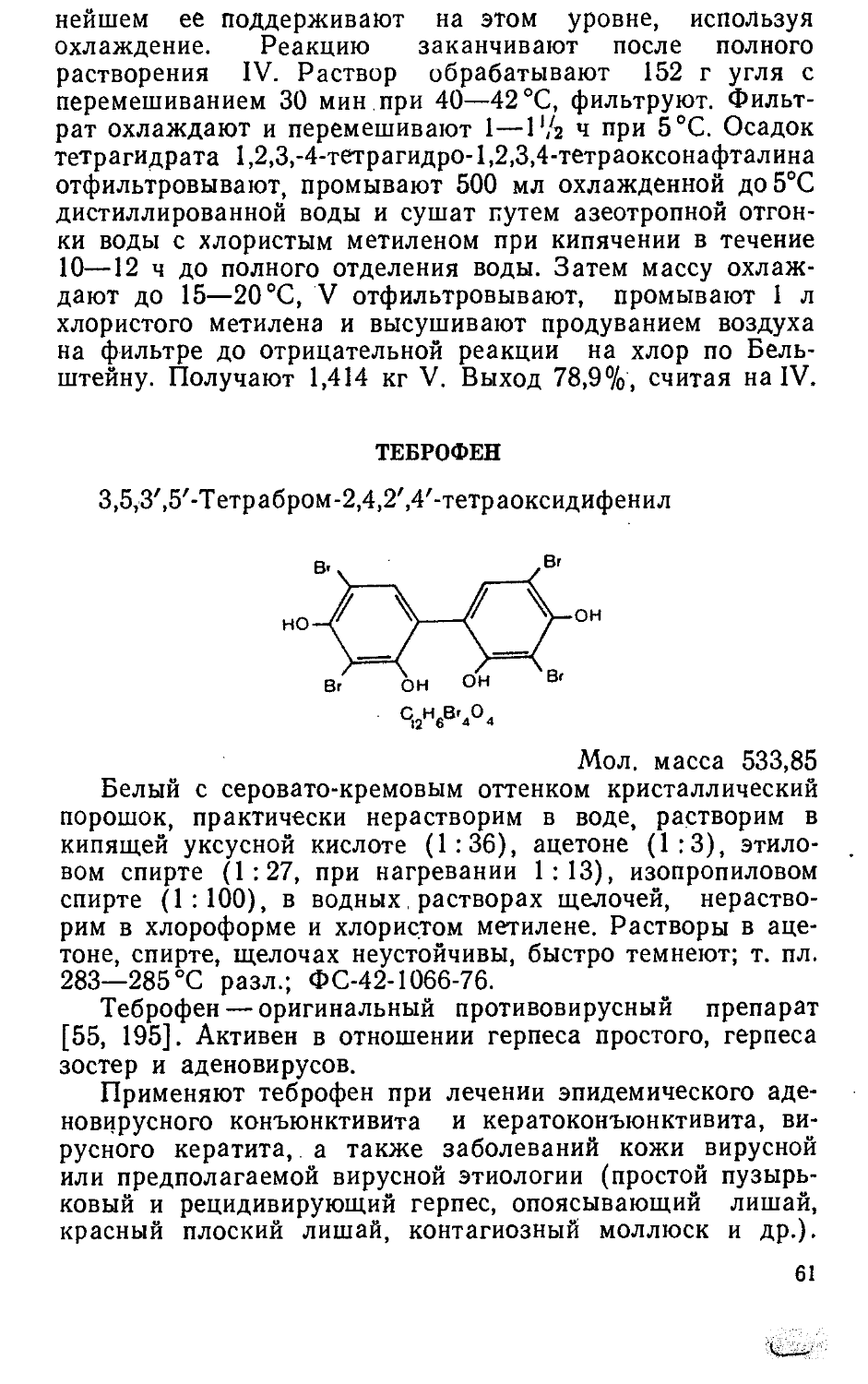
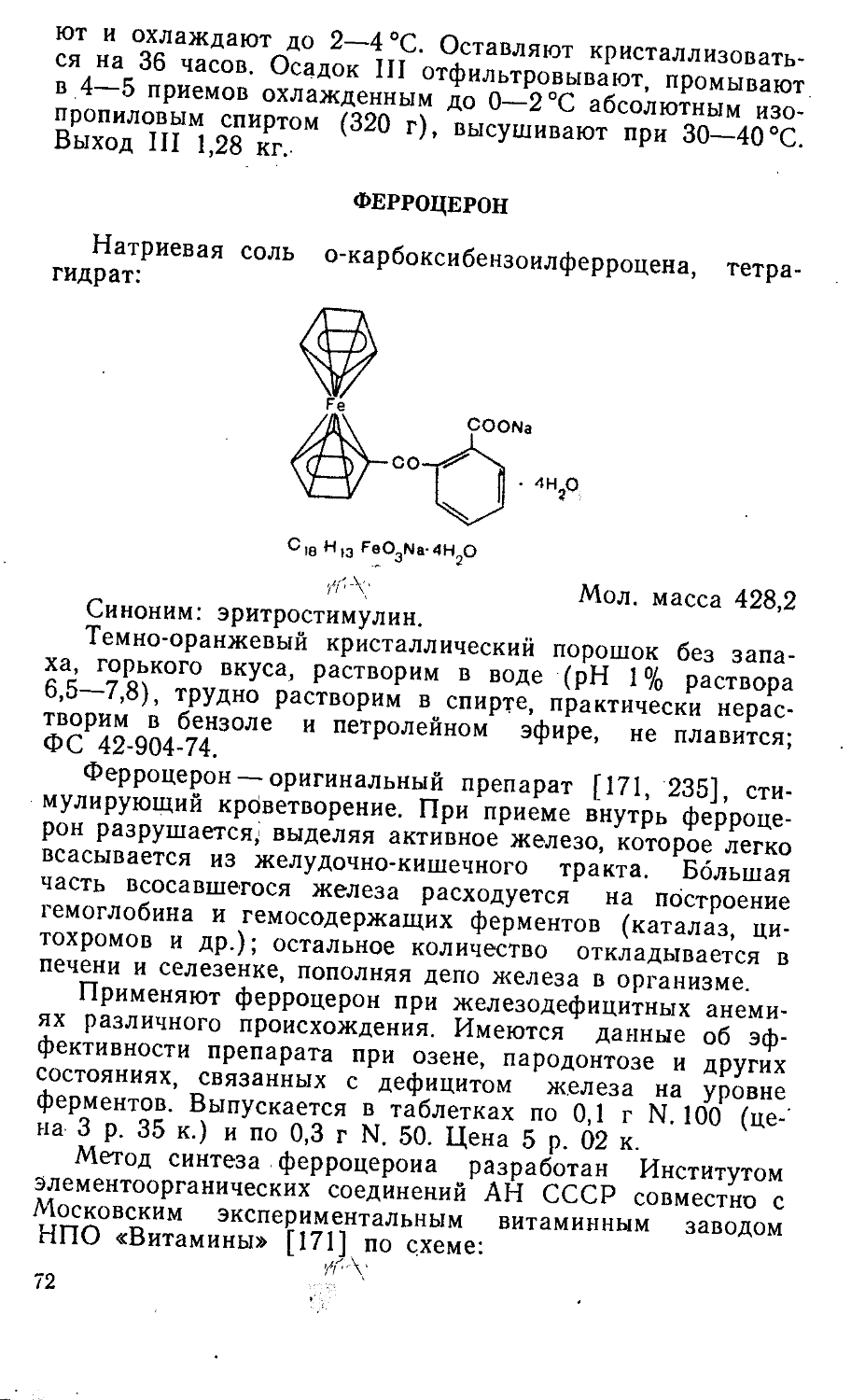
ПАНТОГАМ
Кальциевая соль D-(+)-a, у-диокси-р, р-диметил-бути-рил-у-аминомасляной кислоты:
сн,
(HOCH - С — CHCONIICII Он СН.СОО) Са 2 I I 2 2 ’ 1
C2oH36N2OioCa Мол. масса 504,5
10
Белый кристаллический порошок, без запаха, легко растворим в воде, очень мало растворим в спирте, практически нерастворим в хлороформе; удельное вращение от +22° до 4-25° (5% водный раствор); ВФС 42-834-79.
Пантогам является средством метаболической терапии с психофармакологической активностью [106]. Применяют пантогам при лечении нейролептического экстрапир-амидного синдрома в качестве корректора при побочном действии нейролептических средств, при . умственной недостаточности у детей (при задержке психического развития), олигофрении, задержке речевого развития, а также в комплексной терапии эпилепсии, последствий нейроинфекций, черепно-мозговых травм, церебральной органической недостаточности у больных шизофренией, при подкорковых гиперкинезах различной этиологии.
Выпускаются таблетки по 0,25 г N.50. Цена 5 р. 02 к.
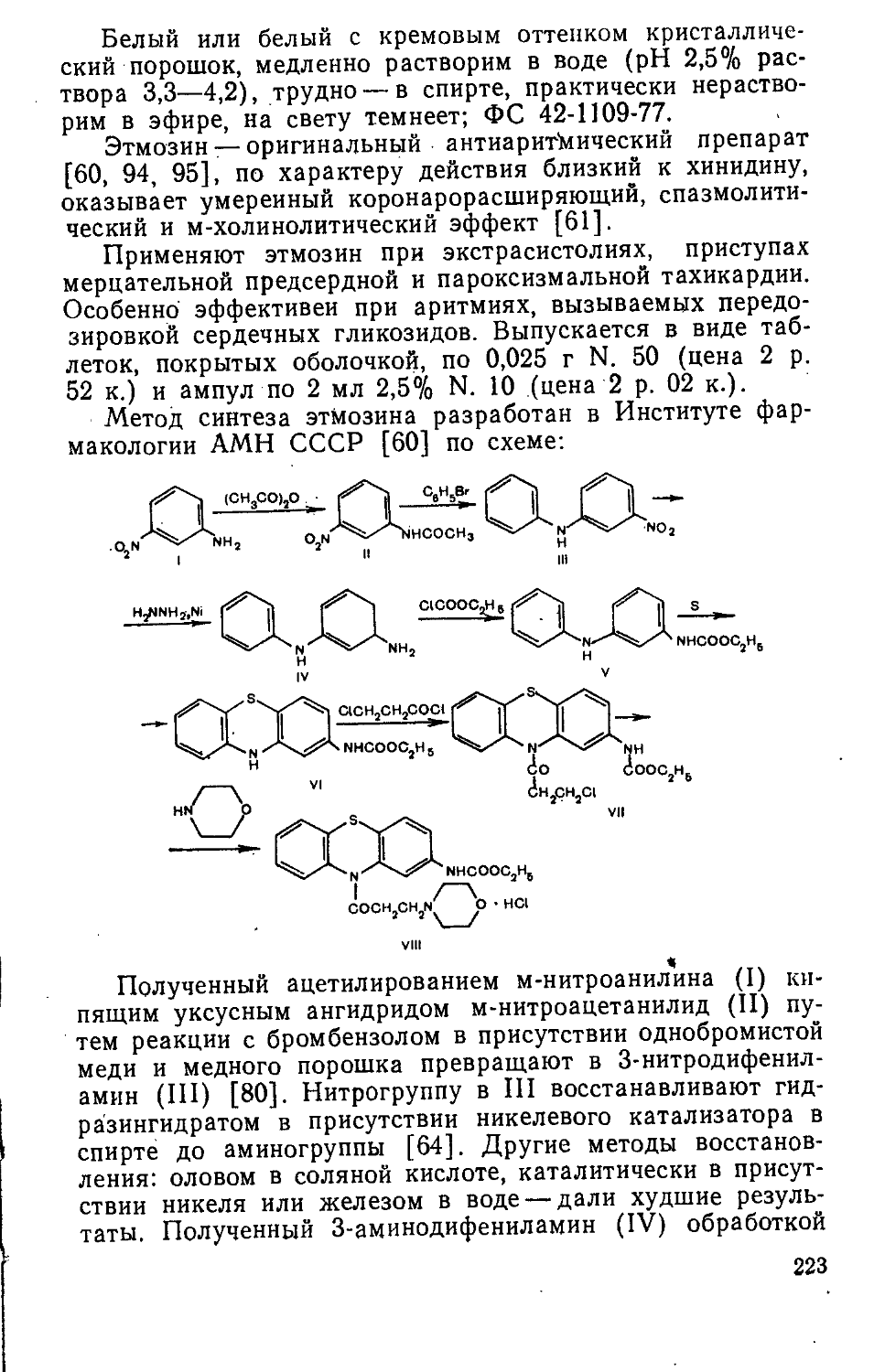
Метод синтеза пантогама (I) из у-аминомасляной кислоты (П) и D-(—)-пантолактона (III) разработан во ВНИВИ по схеме [108]:
(C2H5O)?^J
rv V) Ги
H2N(CH2)3C00H -г.? ?». (Н N(CH„) ООО] Са . J * *• 3 2
сн3
*- (НОСЫ - 6 - СНСОМНСН2СН2СН2СОО)гСа
СИ он л
Обработкой этилатом кальция II превращают в кальциевую соль у-аминомасляной кислоты, из которой при взаимодействии с D-(—)-пантолактоном получают I.
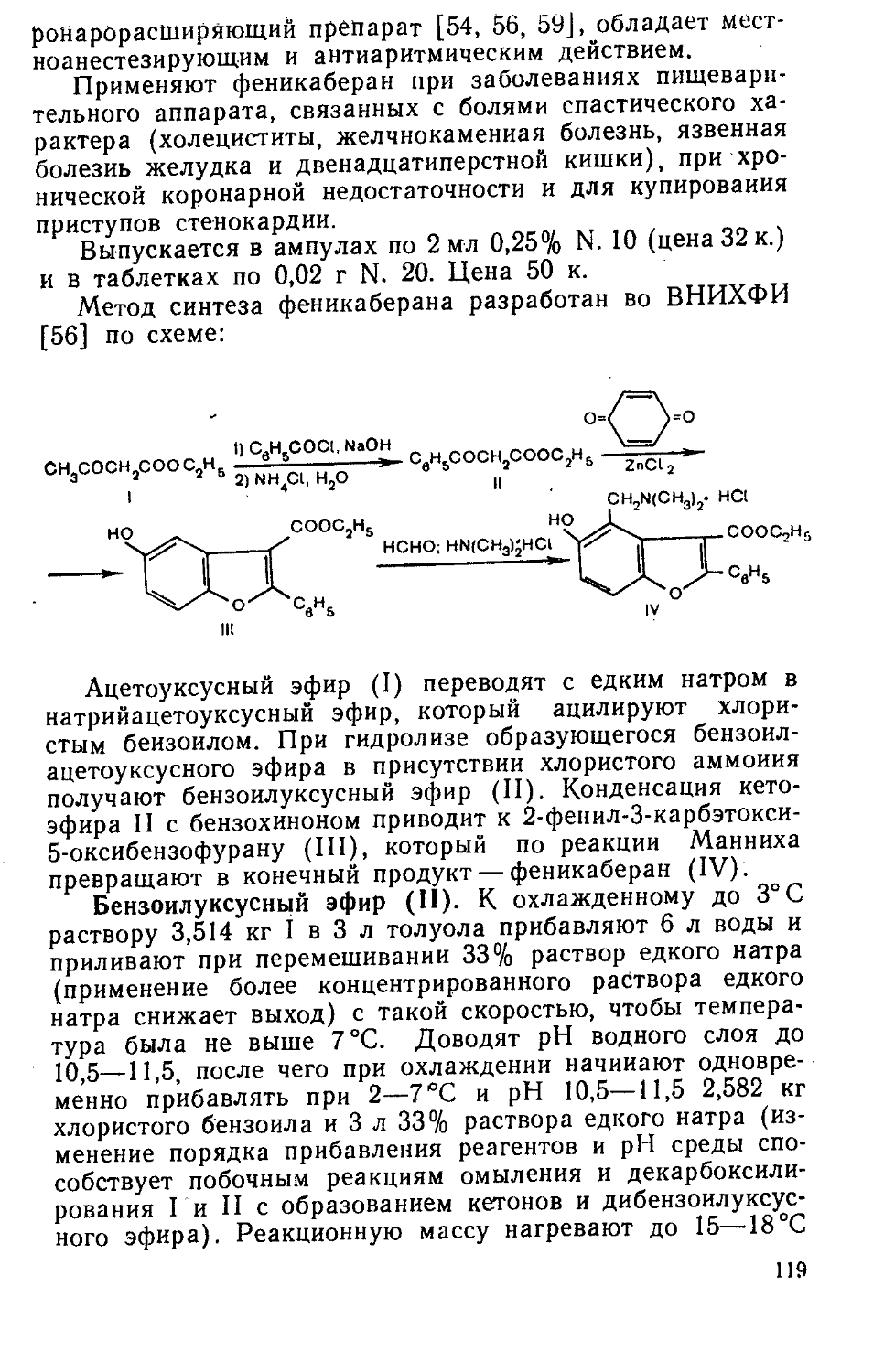
Кальциевая соль D-(-j-)-ct, у-диокси-р, р-диметил-бути-рил-у-аминомасляная кислоты (пантогам) (I). К 0,5 л абсолютного этилового спирта прибавляют 20,4 г металлического кальция, кипятят 3 ч, прибавляют 105 г II и вновь кипятят 1 ч. К массе прибавляют 146 г III, нагревают при 78 °C в течение 1 ч, суспензию при 40—50 °C разбавляют 250 мл абсолютного этилового спирта и
п
фильтруют. Горячий раствор обесцвечивают углем и оставляют на 4—5 сут при 15 °C. Выпавший осадок отфильтровывают, промывают 4 раза по 150 мл абсолютного этилового спирта и сушат при 60—80 °C в вакууме, получают 130 г (47,5%, считая на II) I.
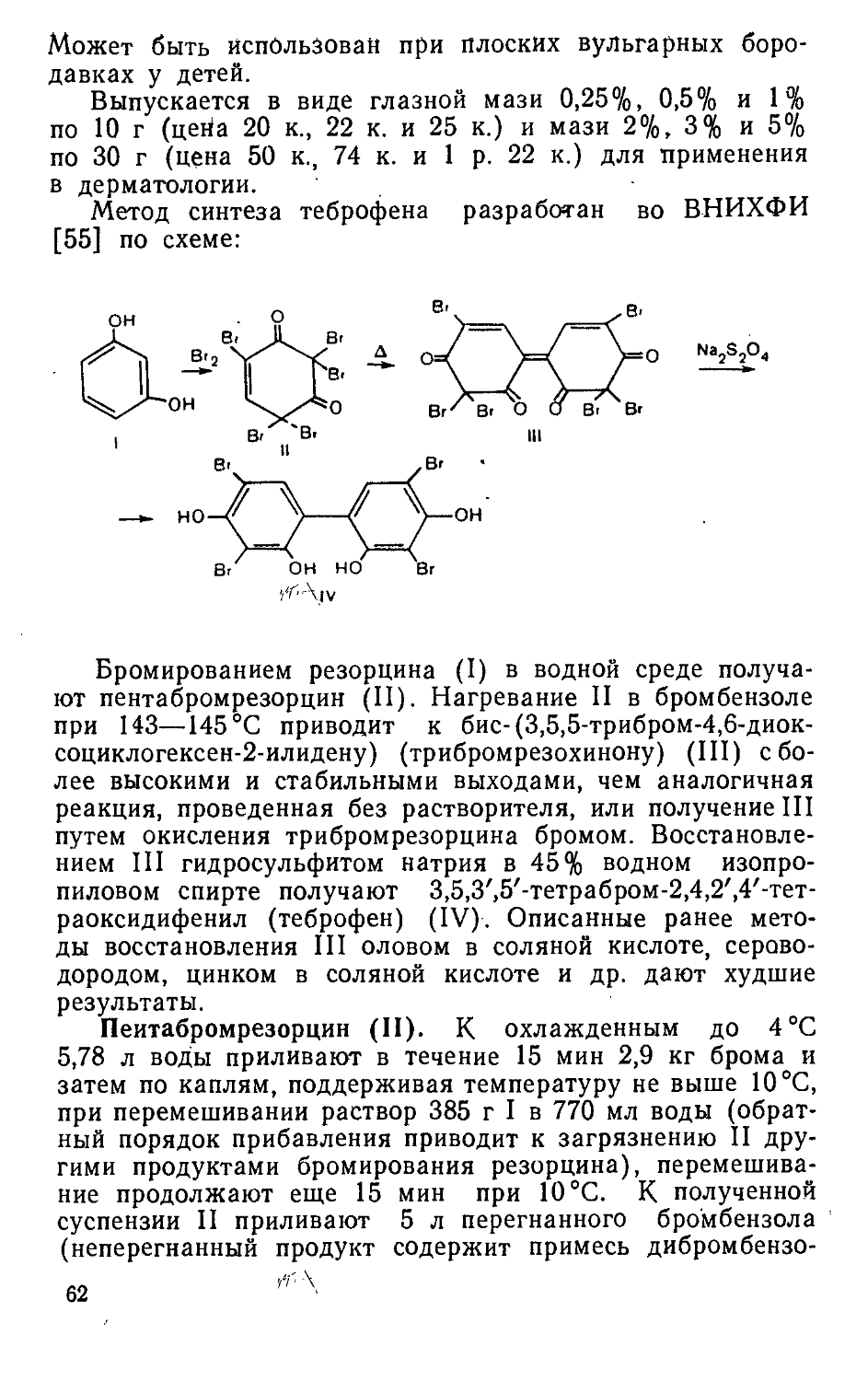
АСАЛИН
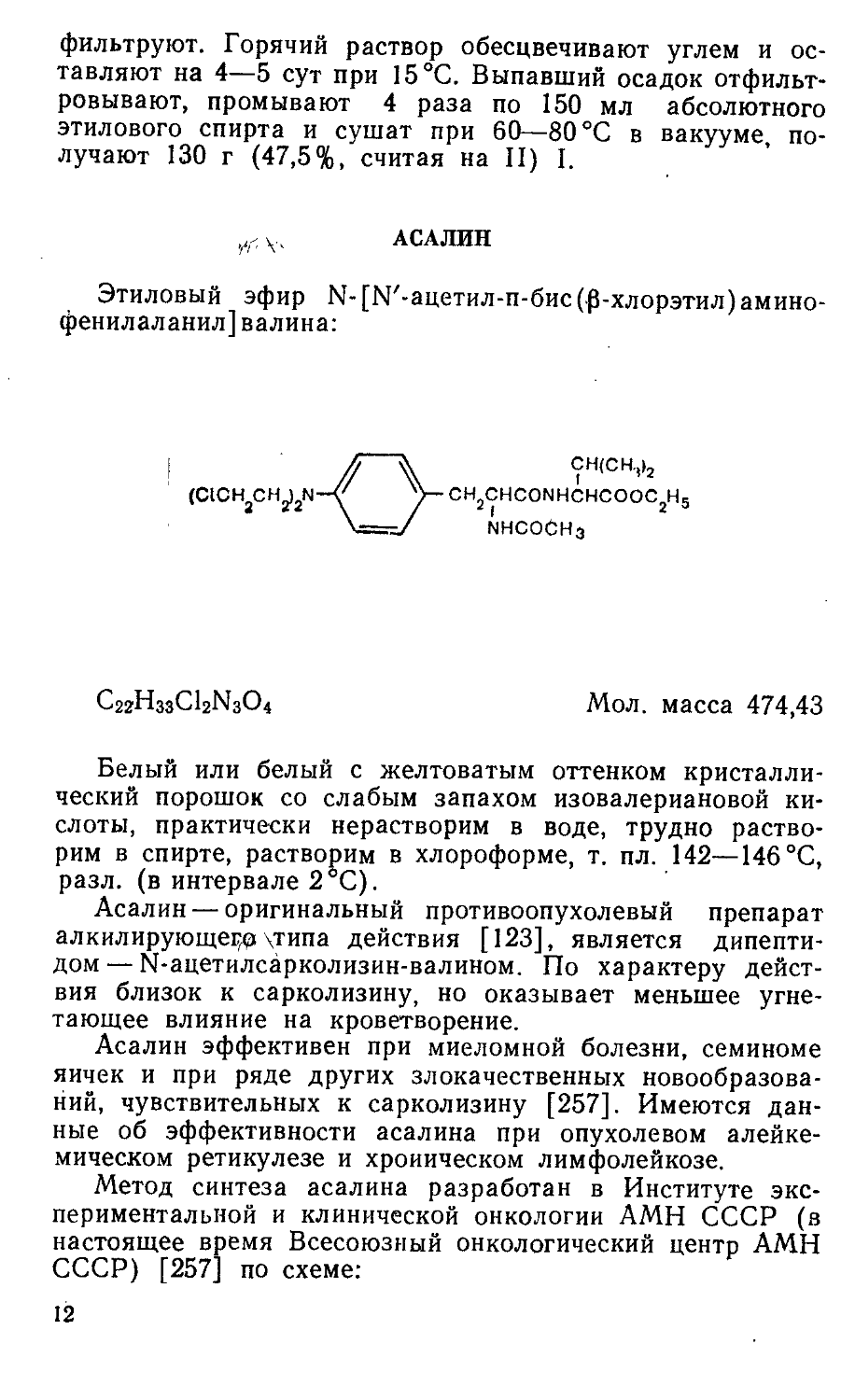
Этиловый эфир М-[М'-ацетил-п-бис(р-хлорэтил)амино-фенилаланил]валина:
(ClCHjCH^N
СН(СН.,)2 ch2chconhchcooc2h5 NHCOCHg
C22H33CI2N3O4 Мол. масса 474,43
Белый или белый с желтоватым оттенком кристаллический порошок со слабым запахом изовалериановой кислоты, практически нерастворим в воде, трудно растворим в спирте, растворим в хлороформе, т. пл. 142—146 °C, разл. (в интервале 2°C).
Асалин — оригинальный противоопухолевый препарат алкилирующего\типа действия [123], является дипептидом— N-ацетилсарколизин-валином. По характеру действия близок к сарколизину, но оказывает меньшее угнетающее влияние на кроветворение.
Асалин эффективен при миеломной болезни, семиноме яичек и при ряде других злокачественных новообразований, чувствительных к сарколизину [257]. Имеются данные об эффективности асалина при опухолевом алейкемическом ретикулезе и хроническом лимфолейкозе.
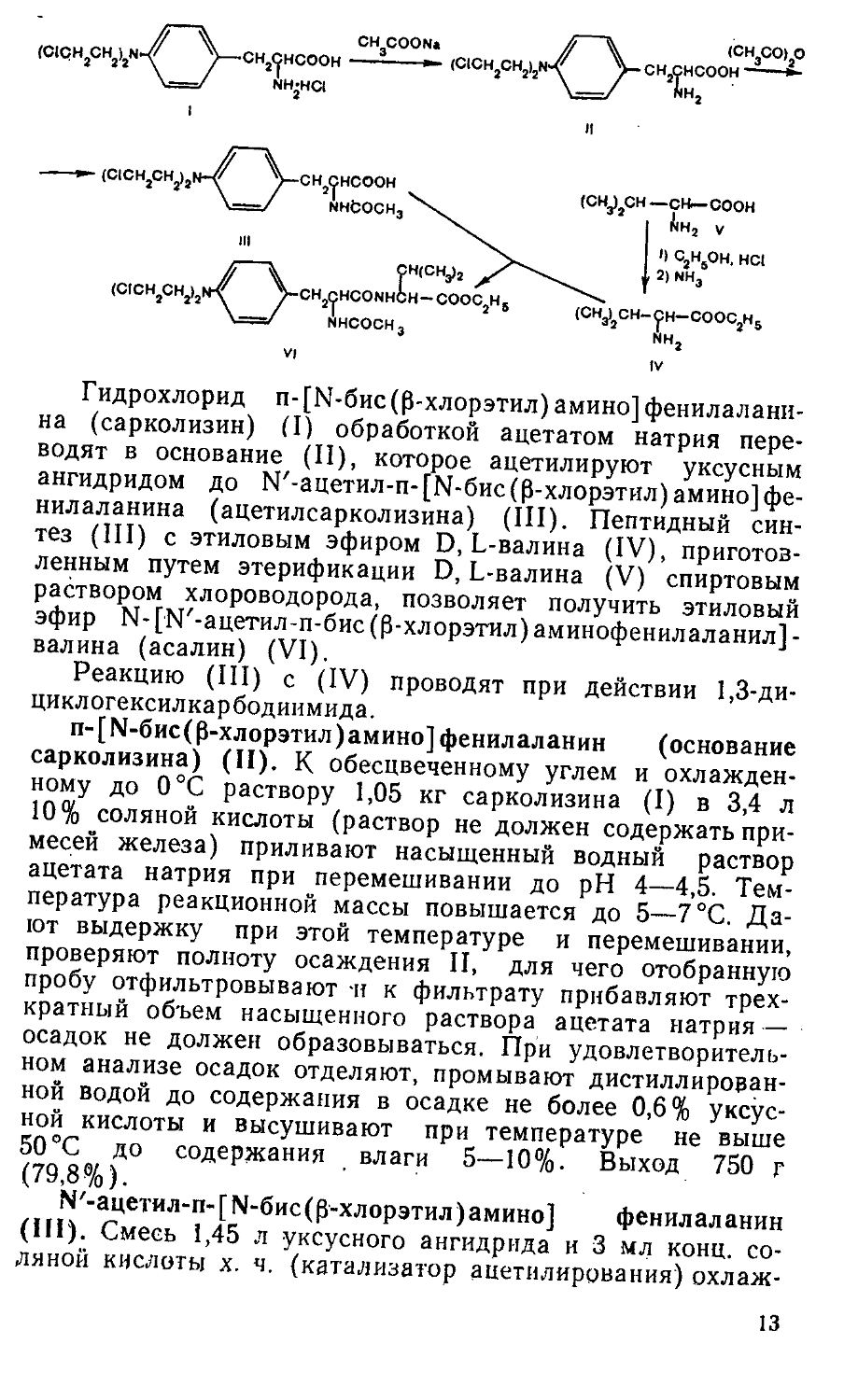
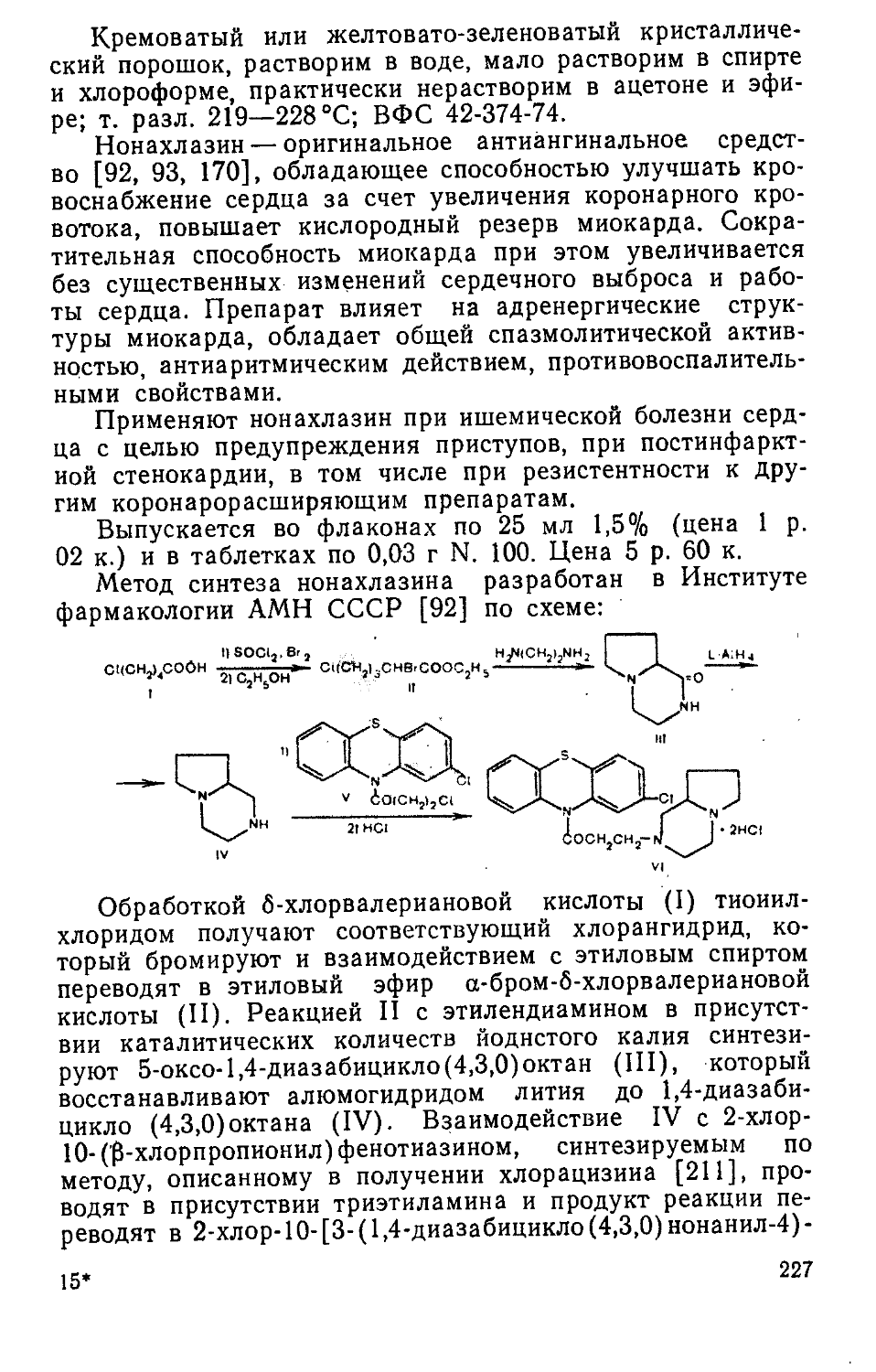
Метод синтеза асалина разработан в Институте экспериментальной и клинической онкологии АМН СССР (в настоящее время Всесоюзный онкологический центр АМН СССР) [257] по схеме:
12
Гидрохлорид п-[М-бис(р-хлорэтил) амино] фенилаланина (сарколизин) (I) обработкой ацетатом натрия переводят в основание (II), которое ацетилируют уксусным ангидридом до М'-ацетил-п-[Ь1-бис(р-хлорэтил) амино] фенилаланина (ацетилсарколизина) (III). Пептидный синтез (III) с этиловым эфиром D, L-валина (IV), приготовленным путем этерификации D, L-валина (V) спиртовым раствором хлороводорода, позволяет получить этиловый эфир N- [N'-ацетил-п-бис (p-хлорэтил) аминофенилаланил] -валина (асалин) (VI).
Реакцию (III) с (IV) проводят при действии 1,3-ди-циклогексилкарбодиимида.
п-[М-бис(£-хлорэтил) амино] фенилаланин (основание сарколизина) (II). К обесцвеченному углем и охлажденному до О °C раствору 1,05 кг сарколизина (I) в 3,4 л 10% соляной кислоты (раствор не должен содержать примесей железа) приливают насыщенный водный раствор ацетата натрия при перемешивании до pH 4—4,5. Температура реакционной массы повышается до 5—7 °C. Дают выдержку при этой температуре и перемешивании, проверяют полноту осаждения II, для чего отобранную пробу отфильтровывают и к фильтрату прибавляют трехкратный объем насыщенного раствора ацетата натрия — осадок не должен образовываться. При удовлетворительном анализе осадок отделяют, промывают дистиллированной водой до содержания в осадке не более 0,6% уксусной кислоты и высушивают при температуре не выше 50°C до содержания влаги 5—10%. Выход 750 г (79,8%).
М'-ацетил-п-[М-бис(р-хлорэтил)амино] фенилаланин (III). Смесь 1,45 л уксусного ангидрида и 3 мл конц. соляной кислоты х. ч. (катализатор ацетилирования) охлаж
13
дают до 10°C и прибавляют при перемешивании 250 г сухого II. При этом температуру доводят до 20°C либо за счет экзотермичности процесса, либо (при необходимости) путем осторожного подогрева (при температуре выше 24°C получается окрашенный III). Масса становится гомогенной, и ее выливают в 10 л охлажденной до 4-2 Т дистиллированной воды (температура массы должна быть не выше 4-5°C). Дают выдержку при температуре 2°C с перемешиванием 10 ч. Осадок отфильтровывают, промывают дистиллированной водой до содержания в осадке уксусной кислоты не более 0,6%, смешивают осадок с 500 мл эфира и 5 мл спирта, отфильтровывают и промывают эфиром (3 раза по 300 мл), после чего сушат над фосфорным ангидридом при комнатной температуре в вакууме до содержания влаги не более 0,3%. Выход 145 г (51,8%).
Этиловый эфир D.L-валина (IV). 500 г D.L-валина (V) и 7,5 л 15% спиртового раствора хлороводорода нагревают 4 ч при 78—80 °C. Массу упаривают в вакууме. К остатку прибавляют 1,9 л абсолютного этилового'спирта и отгоняют его в вакууме. Эту операцию, направленную на удаление х,ортоводорода, повторяют до тех пор, пока pH отгона не станет равным 5. После этого к остатку дважды прибавляют по 1,25 л сухого диэтилового эфира, осадок отфильтровывают, сушат на воздухе, смешивают с 1,75 л хлороформа и приливают в течение 4 ч при 0—5°C и при хорошем перемешивании 6,65 л хлороформа, насыщенного аммиаком. Осадок хлористого аммония отфильтровывают, промывают 1,75 л хлороформа. Объединенные хлороформные растворы упаривают, остаток пере; гоняют при 82—84°C (26 мм рт. ст.). Выход 96,6 г! (15,6%). Вещество неустойчиво при хранении и сразу должно быть использовано для получения асалина.
Этиловый эфир 1Ч-[1Ч'-ацетил-п-бис(р-хлорэтил)амино-фенилаланил]валина (асалин) (VI). К раствору 100 г III в 775 мл сухого хлороформа при интенсивном перемешивании приливают последовательно растворы 41,8 г IV в 320 мл хлороформа и 59,4 г свежеперегнанного дицикло-гексилкарбодиимида в 320 мл хлороформа, поддерживая все время температуру реакционной массы не выше 30 °C. Дают выдержку при 20—25 °C 2 ч при перемешивании и 12 ч без перемешивания, после чего массу охлаждают до 0 °C и дают выдержку 2 ч при перемешивании и температуре 0—5 °C. Осадок дициклогексилмочевины отфильтровывают, промывают охлаждённым до 0—5 °C хлорофор-14
мом (2 раза ио 40 мл). Объединенные хлороформные фильтраты упаривают в вакууме при температуре 30 °C. Остаток перекристаллизовывают из 520 мл этилового спирта с 5 г щелочного активированного угля. Спиртовой раствор охлаждают с перемешиванием 16 ч при 0—5 °C, осадок VI отфильтровывают, промывают 2 раза по 40 мл охлажденного до 0—5 °C спирта-ректификата, затем сухим эфиром (2 раза по 40 мл), высушивают на воздухе до содержания влаги не более 0,5% и вновь перекристаллизовывают из этилового спирта-ректификата. Выход 42,5 г (12,68%, считая на I, или 36,96%, считая на V).
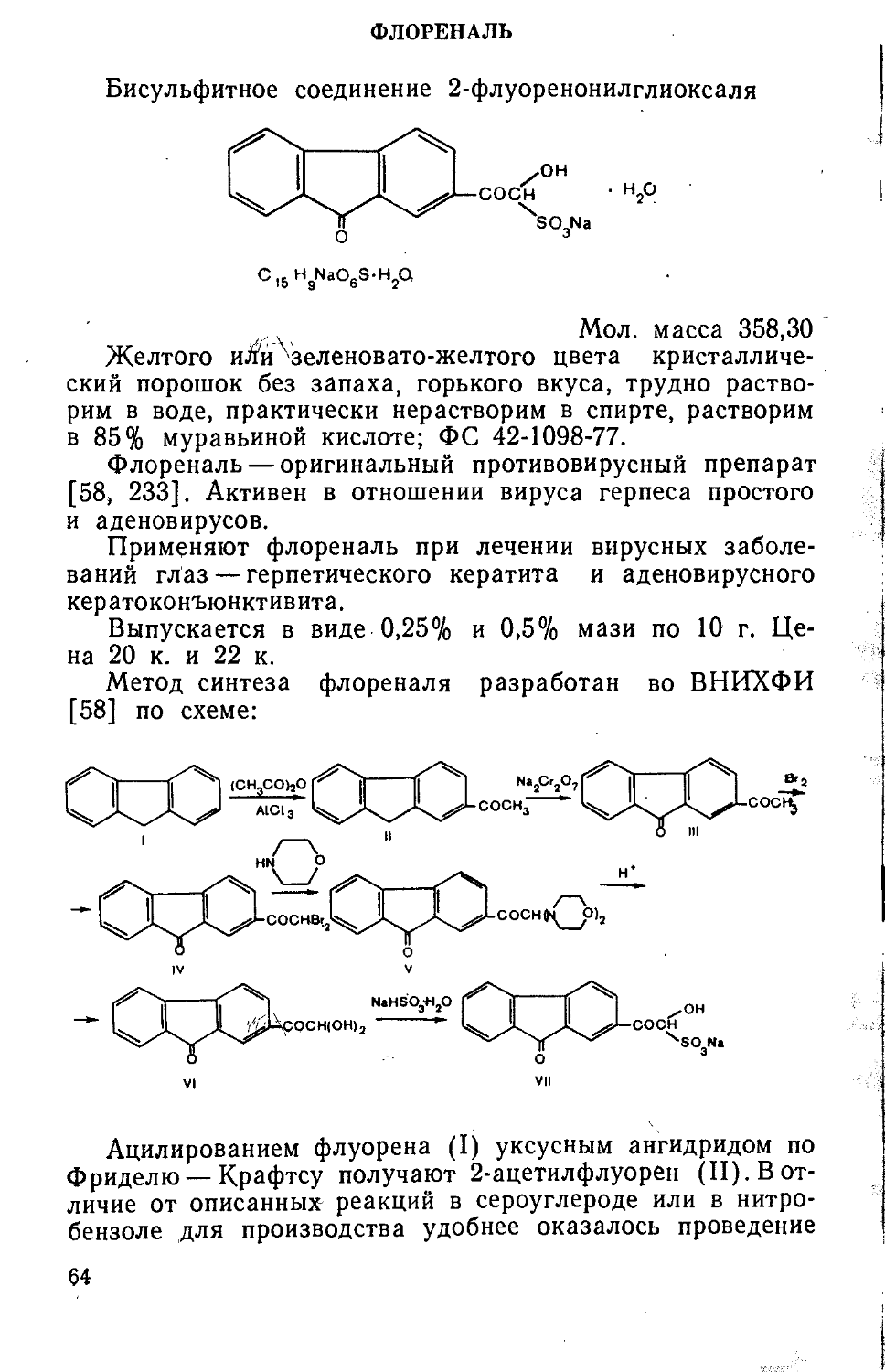
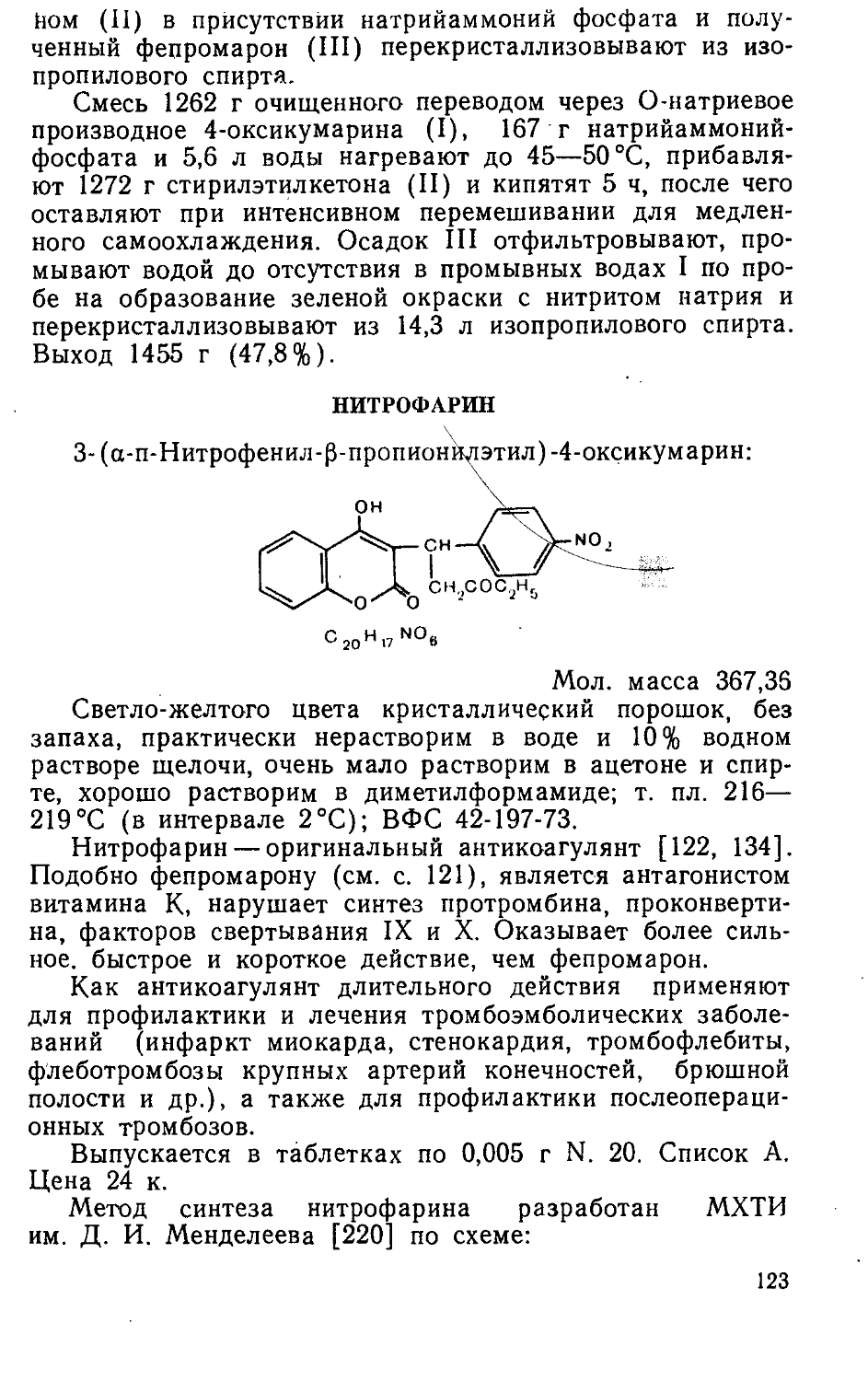
АЦЕМИН
N-Ацетил-е-аминокапроновая кислота:
CH3CONH(CH2)5COOH
С Н NO, в 15 3
Мол. масса 173,21
Основные синонимы: пластенан, ацексампновая кислота.
Белый или белый с желтоватым оттенком кристаллический порошок, легко растворим в спирте, растворим в воде, мало растворим в ацетоне; т. пл. 104—106°C; ВФС 42-837-79.
Ацемин является препаратом, ингибирующим действие лизосомальной пептидазы; ускоряет заживление ран, ожогов, ускоряет .приживление гомотрансплантатов, предупреждает образование гипертрофического рубца, стимулирует срастание костей, уменьшает степень склеровоспалительной реакции в зоне перелома. Применяется при длительно не заживающих ранах, ожогах, закрытых переломах, а также с целью получения послеоперационного косметического рубца [302].
Выпускается в виде 5% мази в тубе по 25 г. Цена 20 к.
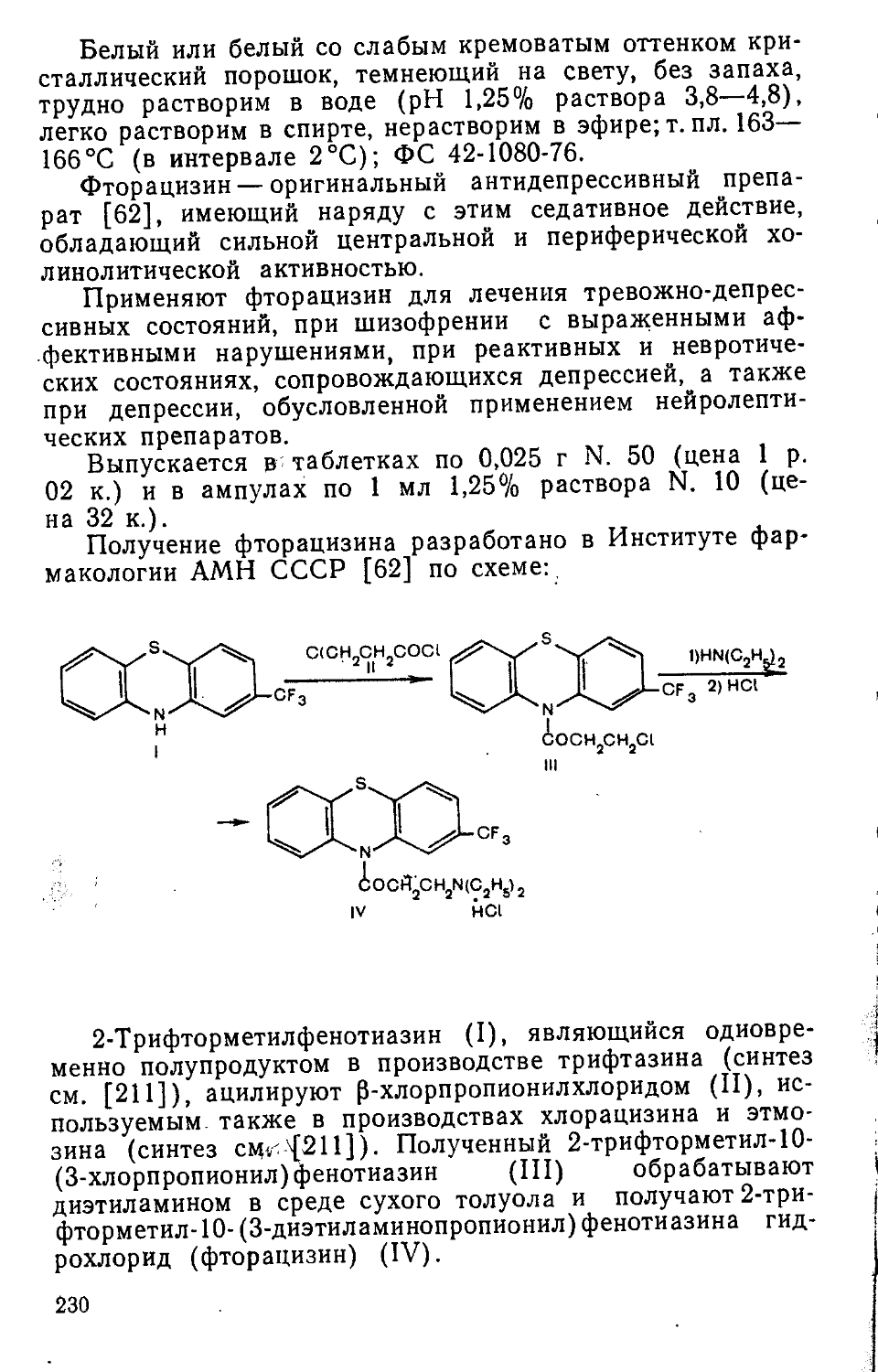
Метод синтеза ацемина (I) основан на ацетилировании е-аминокапроновой кислоты (II) уксусным ангидридом в ацетоне [302] по схеме:
(СН3С0)20
H2N(CH2,aCOOH -------*“ CH3CONH(CH2).COOH
II I
Известен метод получения ацемина путем ацетилирования капролактама уксусным ангидридом с последую
15
щим гидролизом N-ацетилкапролактама до I [261]. Однако этот метод не имеет преимуществ перед описанным выше.
N-Ацетиламинокапроновая кислота (ацемин) (I). К. суспензии 506,7 г II в 1 л ацетона в течение 30—40 мин постепенно прибавляют 400 мл уксусного ангидрида, поддерживая температуру массы 20—30 °C, перемешивают 1 ч, охлаждают при —5—0°С в течение 16 ч, осадок фильтруют и промывают 2 раза по 100 мл ацетона. Технический ацемин кристаллизуют из 770 мл воды с углем и сушат при 70—75 °C, получают 536 г (88%). I.
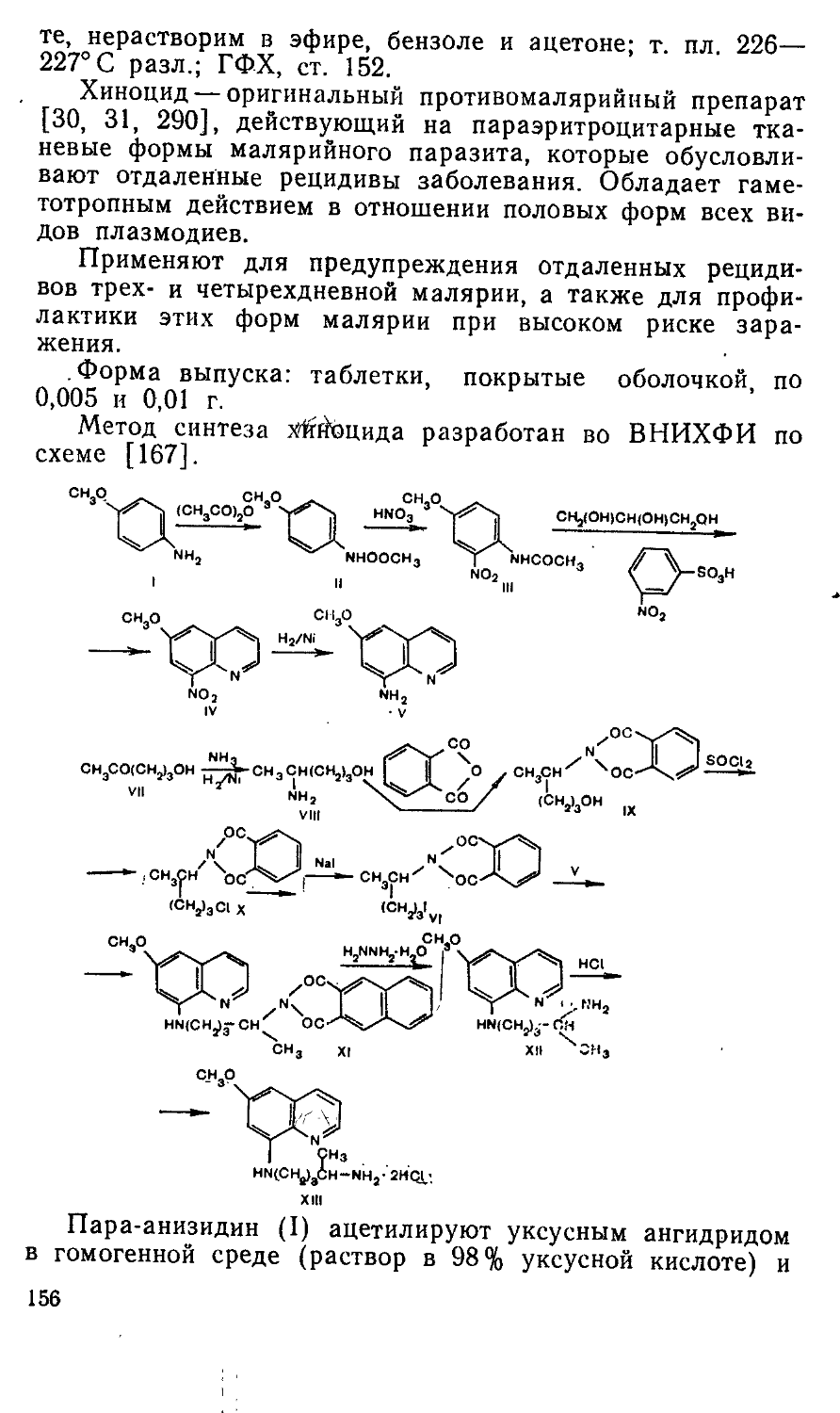
ФОСФОРСОДЕРЖАЩИЕ ПРЕПАРАТЫ
Группа биологически активных фосфорсодержащих органических соединений включает два типа лекарственных препаратов. К первому типу относятся этиленимиды фосфорной и тиофосфорной кислот — эффективные противоопухолевые средства [192, 216, 245]. Интерес к этим соединениям возник в начале 50-х годов в связи с гипотезой о легкой циклизации в водных растворах производных бис- (|3-хлорэтил)амина (азотистых аналогов иприта, обладающих противоопухолевой активностью) в этилен-иммониевые соединения. Последние и считались ответственными за алкилирующее действие препаратов в организме.
Среди большого числа производных этиленимина, испытанных в это время, внимание исследователей привлек триэтиленимид тиофосфорной кислоты (I). Соединение I, использовавшееся ранее в производстве синтетических тканей, оказалось эффективным при лимфогранулематозе, нейробластомах и меланоме. В СССР препарат под названием тиофосфамид был воспроизведен в 1956 г. во ВНИХФИ [246] и в 1958 г. в Институте органического синтеза АН ЛатвССР [226]. Тиофосфамид производится отечественной химико-фармацевтической промышленностью и в настоящее время.
/г ~\'
II III
а) х=О б) x=s
16
В ходе дальнейших поисков веществ, более активных и менее токсичных, чем тиофосфамид, было синтезировано п изучено большое количество соединении общей формулы (II), где R— различные ароматические, гетероциклические или аминокислотные остатки [216, 245]. При этом независимо и практически одновременно в СССР (во ВНИХФИ) и США был получен тетраэтиленимид 1,4-пиперазиндифосфорной кислоты (Ша), названный дипином [211, 246, 336], а затем во ВНИХФИ созданы оригинальные противоопухолевые препараты тиодипин (III6)
и фосфемид (IV) [216, 245]. В Киевском институте фармакологии и токсикологии были синтезированы и исследованы различные бензоиламиды диэтилениминофосфор-ной кислоты общей формулы V. Показано, что характер противоопухолевого действия и уровень токсичности соединений в значительной мере зависят от природы, числа и положения заместителей R в бензоильной части молекулы. Пара-галоген-производные обычно более активны, чем их орто- или мета-аналоги. В той же последовательности меняется и токсичность соединений. Наибольшая эффективность п-йодбензотэфа наблюдается при папилломатозе гортани, п-фторбензотэфа — при гипернефроме и предраковом состоянии полости рта, 2,5-дийодбензотэфа — при раке молочной железы и яичников [206]. Оригинальные противоопухолевые средства бензотэф (Va, R=H) [205, 211], фторбензотэф (V6, R=4-F) [205, 206] и дийодбензотэф (см. с. 19) [205, 206] вошли в медицинскую практику. Оригинальный цитостатический препарат имифос (см. с. 22) создан в Институте органического синтеза АН ЛатвССР [89].
Второй тип биологически активных фосфорсодержащих органических соединений — это вещества общей формулы VI, необратимо ингибирующие холинэстеразу [1, 46, 91] и находящие благодаря этому использование в качестве инсектицидов, пестицидов [157, 259], нервно-паралитических ядов и лекарственных препаратов миоти-
2 Заказ № 4304 17
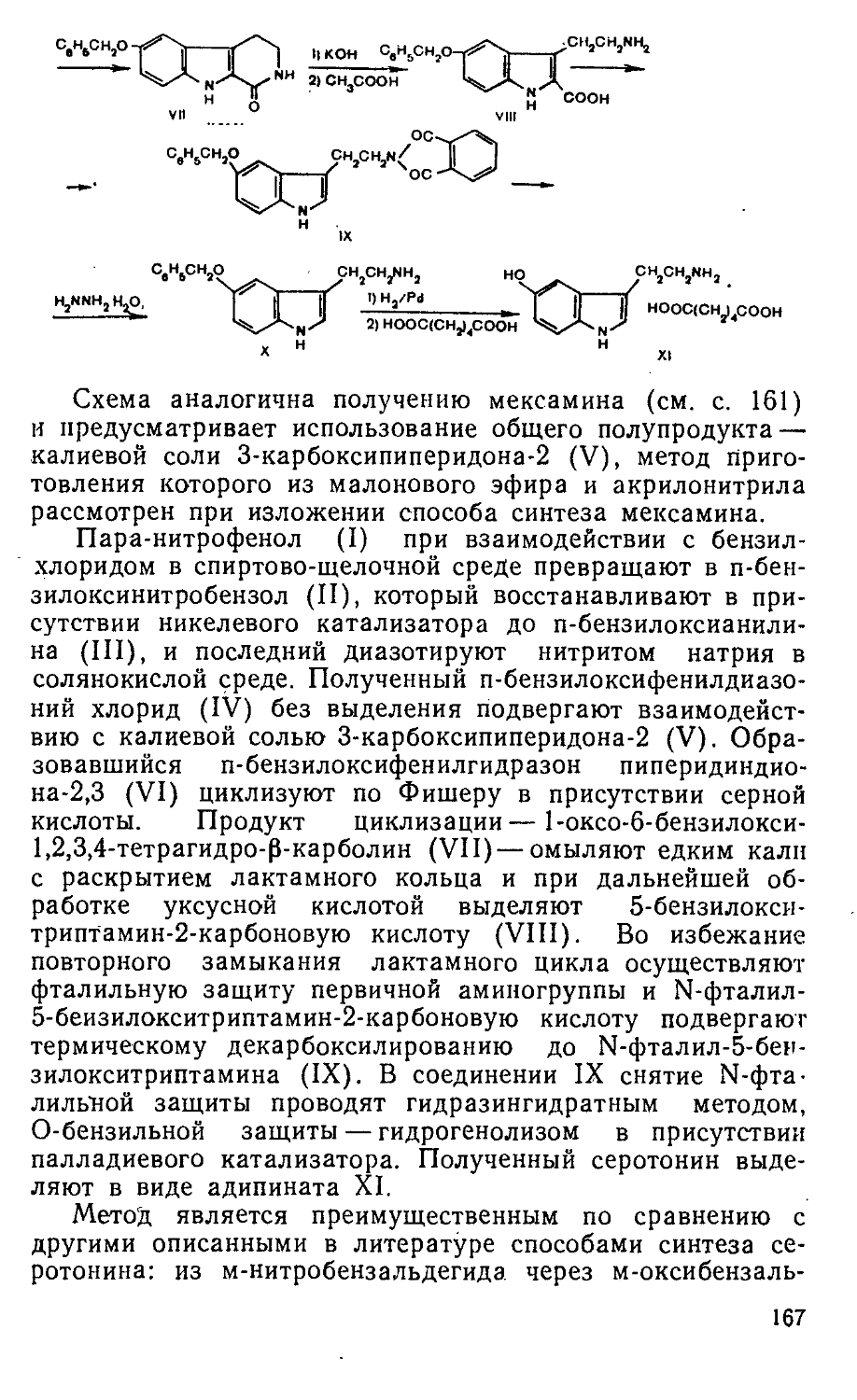
ческого действия. К настоящему времспн синтезировано и изучено более 50 Q0Q веществ этого типа.
R х Р-х l/'O(S)
VI
сн ч
4 9 р-С H ZO
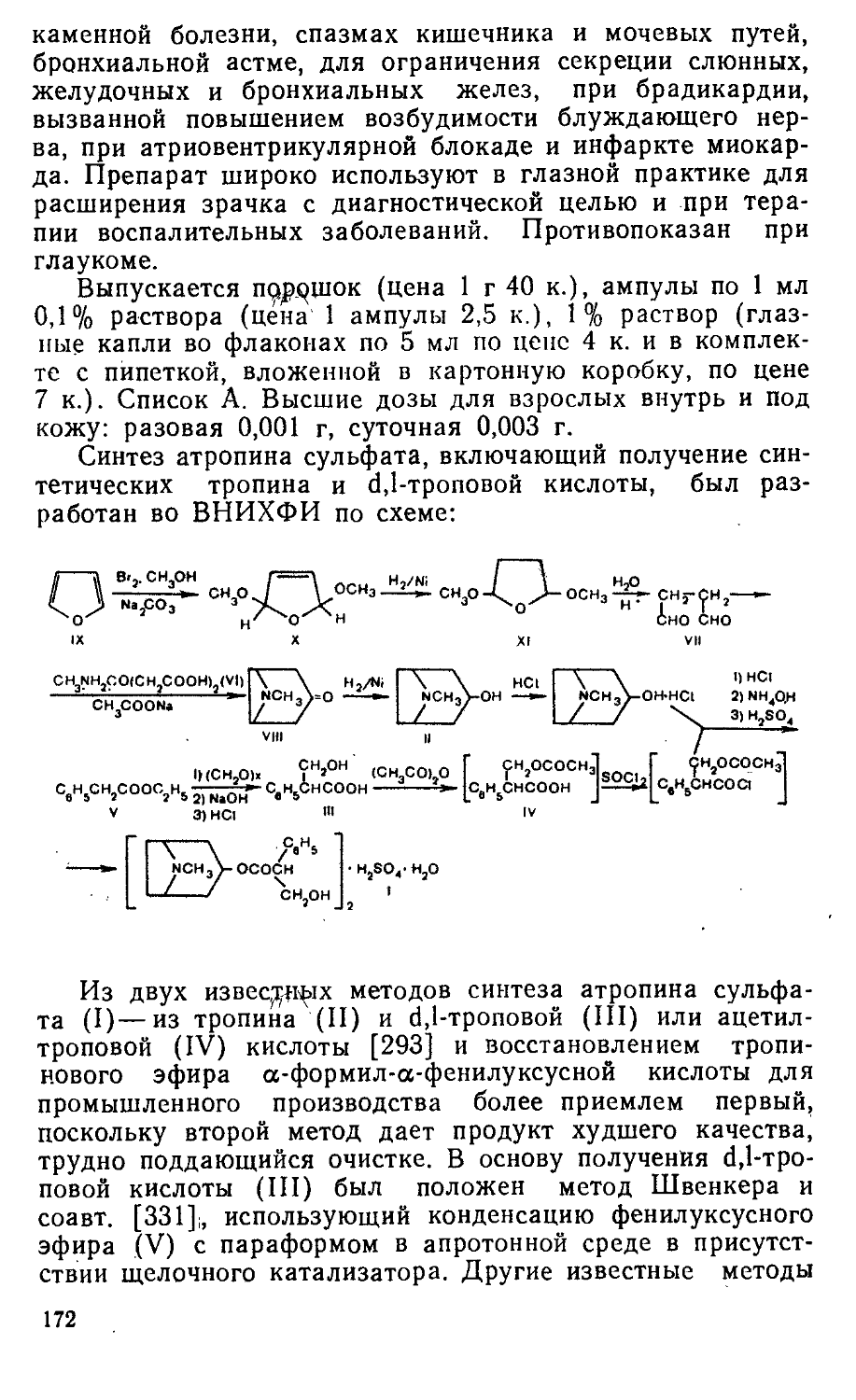
4 У
ОС Н NO-п в 4 2
VIII
IX
Практическое применение в офтальмологии с целью снижения внутриглазного давления при глаукоме и сужения зрачка нашли впервые синтезированный в довоенной Германии [259] и воспроизведенный во ВНИХФИ фос-факол (VII) [101, 211], а также оригинальные отечественные лекарственные препараты нибуфин (VIII) [13, 210, 211], армии (IX) [13, 209, 211] и пирофос [11, 211], созданные в Казанском химико-технологическом институте.
Дальнейшие исследования, проведенные в Институте нефтехимического синтеза им. А. В. Топчиева АН СССР, были связаны с использованием в качестве X у соединений (VI) трихлоралкоксизаместителей [256]. Было показано, что у VI (R'- ^ ^"-алкокси, Х-трихлоралкокси) увеличение длины алкильных цепочек в R' и R" от Ci до С4, в соответствии с общими закономерностями [91], уменьшает антихолинэстеразную активность, а переход от соединений нормального ряда к изо-алкоксипроизводным усиливает ингибирование холинэстеразы. Введение в концевую метильную группу алкоксизаместителя X трех атомов хлора увеличивает эффективность соединений на два порядка, что было объяснено наибольшей склонностью СС1з-группы к гидрофобным взаимодействиям с соответствующим участком холинэстеразы [91]. Наиболее эффективным в этой серии веществ оказался хлофосфол (см. с. 24).
Антихолинэстеразную активность проявил и синтезированный в Институте органической и физической химии им. А. Е. Арбузова Казанского филиала АН СССР оригинальный препарат хлорацетофос (см. с. 26) [177, 178]. Препарат эффективен в отношении различных патогенных грибов, бактерий и нашел применение при лечении 18
грибковых заболеваний гладкой кожи и волосистой части головы. В ходе совместных исследований Всесоюзного научно-исследовательского института химических средств защиты растений и ВНИХФИ фунгицидная и бактерицидная активность обнаружена также у высших аммониевых солей кислых тио- и дитиофосфатов [159]. При этом такая активность в значительной степени зависит от строения фосфорсодержащего аниона, который повышает растворимость веществ в липидах и увеличивает скорость их проникновения в живую клетку [158]. Результатом этих исследований явилось создание оригинального фунгиста-тического и фунгицидного препарата октатиона (см. с. 27), применяемого в практической медицине [158].
ДИЙОДБЕНЗОТЭФ
М-2,5-Дийодбензоил-М', N''-ди (этилен) триампд фосфорной кислоты:
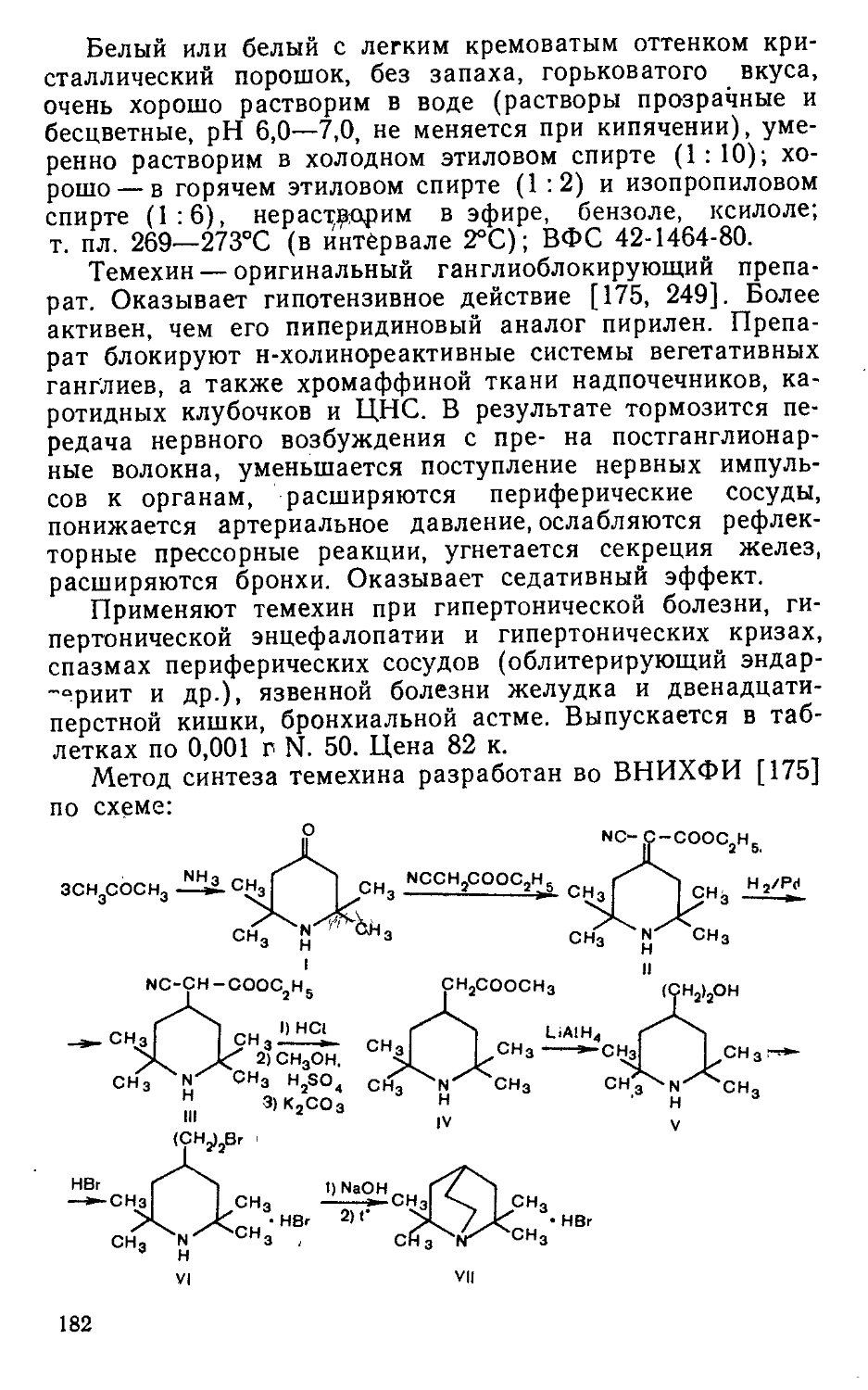
с „и,, у^о,/
Мол. масса 503,0
Белый с кремовым оттенком мелкокристаллический порошок, без запаха, практически нерастворим в воде (pH водной вытяжки 5,5—7), мало растворим в хлороформе и диметилформамиде, очень мало растворим в спирте; ВФС 42-307-74. Дийодбензотэф — оригинальное противоопухолевое средство [206], характеризующееся по сравнению с тиофосфамидом и бензотэфом более широким спектром действия на опухоли и лучшей переносимостью. Относительно редко вызывает в терапевтических дозах угнетение кроветворения. Эффективен при приеме внутрь. Применяют дийодбензотэф при неоперабельных формах рака щитовидной железы, раке молочной железы и мочевого пузыря.
Выпускается в таблетках по 0,05 г N. 60. Список А. Цена 7 р. 52 к.
Метод синтеза дийодбензотэфа разработан Киевским институтом фармакологии и токсикологии [206] по схеме:
2* 19
N
Ъ PCls; 2) н
йодированием антраниловой кислоты (I) получают 2-ами-но-5-йодбензойную кислоту (II), которую подвергают диазотированию нитритом натрия в присутствии йодистого калия, и образующуюся 2,5-дийодбензойную кислоту (III) переводят через хлорангидрид в амид IV. Взаимодействием амида 2,5-дийодбензойной кислоты (IV) с пятихлористым фосфором и этиленимином синтезируют М-2,5-дийодбензоил-Ь1', N''-ди (этилен) триамид фосфорной кислоты (дийодбензотэф) (V).
2-Амино-5-йодбензойная кислота (II). В 4,5 л воды растворяют 280 г едкого кали и затем при 15—20°C 281 г йода. После контроля содержания йода в реакционной массе (не менее 5,9%) добавляют 153 г антраниловой кислоты (I). Полученный раствор охлаждают до 10°С и быстро приливают 312 г уксусной кислоты (возможно повышение температуры до 15—25°C). Дают выдержку при перемешивании 8 ч при 15—20 °C. Контролируют содержание йода в растворе (не более 5 г/л), после чего удаляют избыток йода добавлением 15% раствора сульфита натрия по йодкрахмальной бумажке. Осадок II отфильтровывают и промывают водой до pH промывной воды 6— 7. Получают 550—600 г влажной II (210 г, или 72,97% в пересчете на сухую массу), которую без сушки используют на следующей стадии.
2,5-Дийодбензойная кислота (III). Смешивают 6,5 л воды, 7,74 кг конц. соляной кислоты и 1,78 кг влажного II (650 г в пересчете на сухой продукт). Массу охлаждают до 0—(—3°С) и, поддерживая эту температуру, постелен- -но приливают при перемешивании в течение 2—2’/г ч I раствор 210 г нитри;₽а\натрия в 2 л воды. Дают выдержку 2 ч при 0—(—3)°С; избыток нитрита натрия удаляют добавлением раствора 71 г мочевины в 420 мл воды. Охлажденный ДО 0°С раствор прибавляют к нагретому до
20
55—60° С раствору 1,69 кг йодида калия в 4 л воды. Дают выдержку 2 ч при 55—60 °C, охлаждают до 20 °C и избыток йода связывают прибавлением 15% раствора сульфита натрия. Выделившийся осадок III отфильтровывают, промывают водой до pH 5—6 в промывных водах и сушат при 60—70 °C до содержания воды не более 0,2%. Выход 690 г (74,63%).
Амид 2,5-дийодбензойной кислоты (IV). В 3,17 л бензола с содержанием влаги не более 0,2% прибавляют 150 г III и 87,3 г тионилхлорида. Массу в течение 2 ч tea-гревают до кипения. Кипятят 2 ч, осветляют 20 г угля, фильтруют и прибавляют при перемешивании в течение 30—40 мин к охлажденным до 10—15 °C 2,322 кг водного раствора аммиака. Дают выдержку 2 ч при 15—20 °C, осадок IV отфильтровывают, промывают водой до pH промывной воды 7—8 и перекристаллизовывают из 2,75 кг бутилового спирта с добавлением 20 г угля. Отфильтрованный от угля кипящий бутанольный раствор охлаждают до 0—5 °C и выдерживают при этой температуре 12 ч. Амид IV отфильтровывают, промывают 150 мл ацетона и высушивают при 80—100 °C до содержания влаги не более 0,1%. Выход IV 135 г (54,16%).
М-2,5-Дийодбензоил-1М/, Н"-Ди(этилен)триамид фосфорной кислоты (дийодбензотэф) (V). Смесь 2 л бензола с содержанием влаги не более 0,1%, 300 г пятихлористого фосфора и 480 г IV (содержание железа не более 0,002%) нагревают в течение не более 30 мин до 70— 75 °C с удалением хлороводорода в вакууме (бурное выделение хлороводорода начинается при 45—50 °C). Дают выдержку 30 мин, после чего массу охлаждают до 20 °C и выливают при перемешивании и охлаждении в 15,5 л ацетона, содержащего по данным анализа 20,8 г воды. Раствор осветляют 100 г угля и через 15 мин фильтруют, после чего приливают к охлажденной до 15—20 °C смеси 3 л ацетона, 550 мл триэтиламина и 286 г этиленимина в течение не более 1 ч, поддерживая температуру (реакция экзотермична) в интервале 15—20 °C. Дают выдержку при той же температуре 2 ч. Осадок отфильтровывают, промывают дистиллированной водой до содержания хлоридов не более 0,02%, затем ацетоном (2 раза по500мл) высушивают при комнатной температуре и растворяют в 5,1 л нагретого до 50—55 °C диметилформамида с добавкой 20 г угля. Дают выдержку 15 мин при 50—55 °C, отфильтровывают и фильтрат охлаждают 1 ч при 5—8 °C. Выделившийся кристаллический V отфильтровывают, про
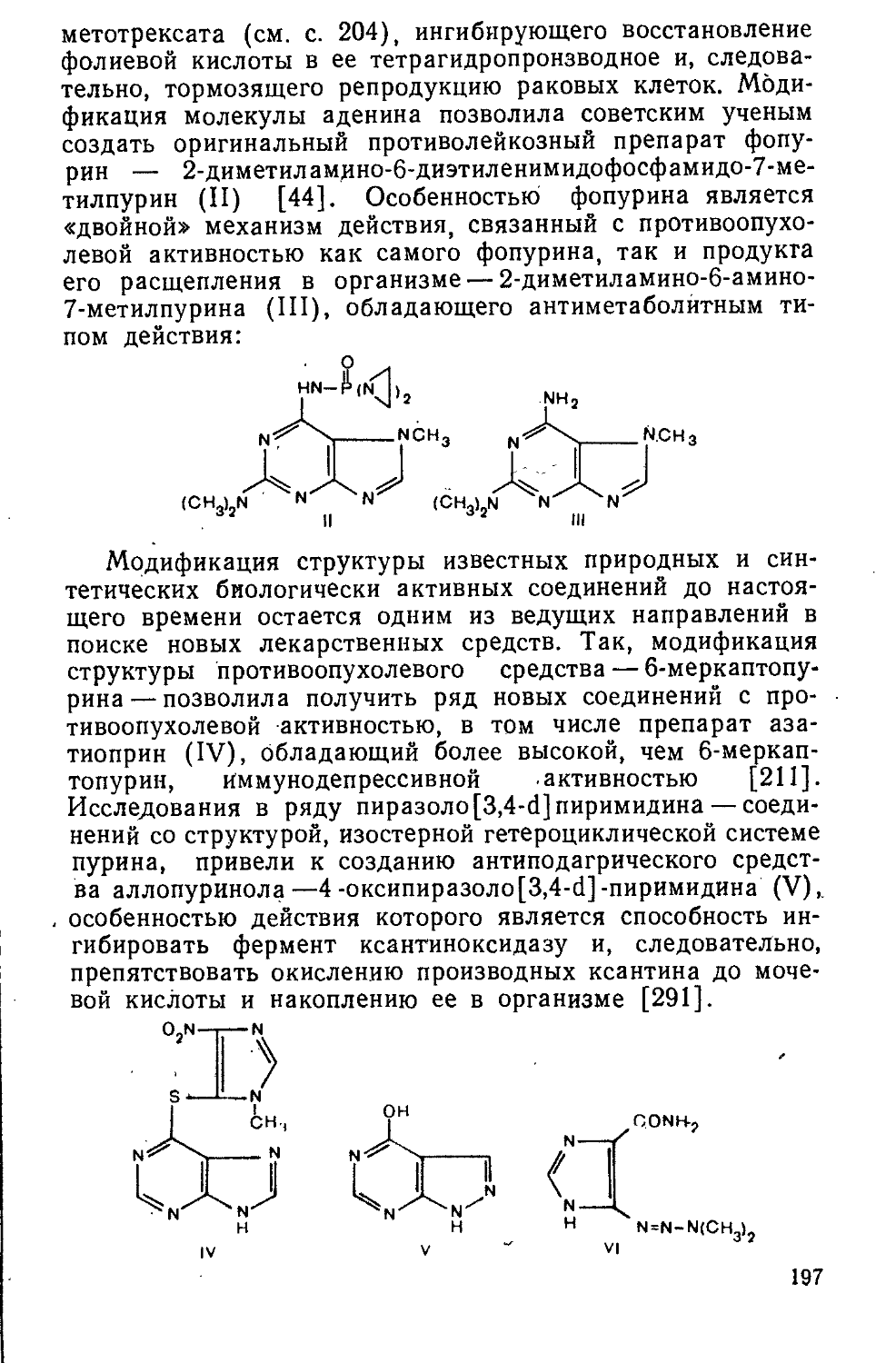
21
мывают водой до содержания хлоридов не более 0,02%, затем 1,1 л ацетона, 300 мл спирта и высушивают при комнатной температуре. Выход 182 г (38,8%).
'ИМИФОС
Диэтиленимид 2-метилтиазолидофосфорной кислоты:
CeHteN3°ps
Мол. масса 233,27
Синоним: маркофан.
Белый кристаллический порошок, очень хорошо растворим в воде, спирте, этилацетате, бензоле, хлороформе, полиэтиленгликоле, трудно — в эфире; т. пл. 58—61 °C (в интервале 2°C); ФС 42-678-73.
Имифос — оригинальный цитостатический препарат [89]. По химиотерапевтической активности сходен с тиофосфамидом, но менее токсичен и обладает меньшими кумулятивными свойствами. Применяют имифос при лечении больных эритремией в развернутой стадии заболевания, протекающей со спленомегалией и панто-цитозом.
Выпускается во флаконах по 0,05 N. 10. Список А. Цена 2 р. 32 к. /ПА;
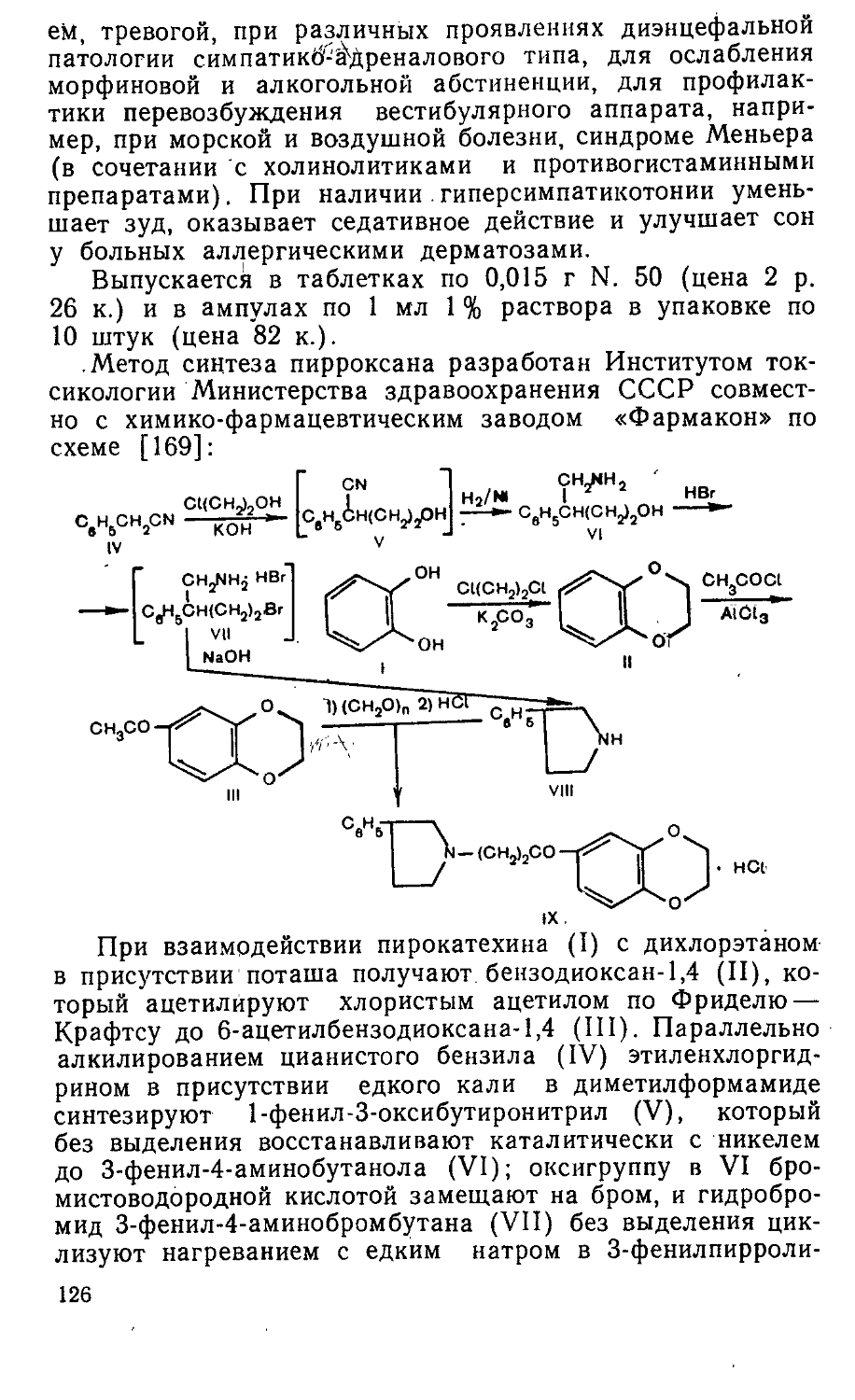
Метод синтеза имифоса разработан в Институте органического синтеза АН ЛатвССР по схеме [89]:
VI
При взаимодействии ацетальдегида (I) с этиленими-ном (II) в метаноле получают 1-этилениминоэтанол (III), который без выделения обрабатывают сероводородом и превращают в 2-метилтиазолидин (IV). Рекция IV с
22
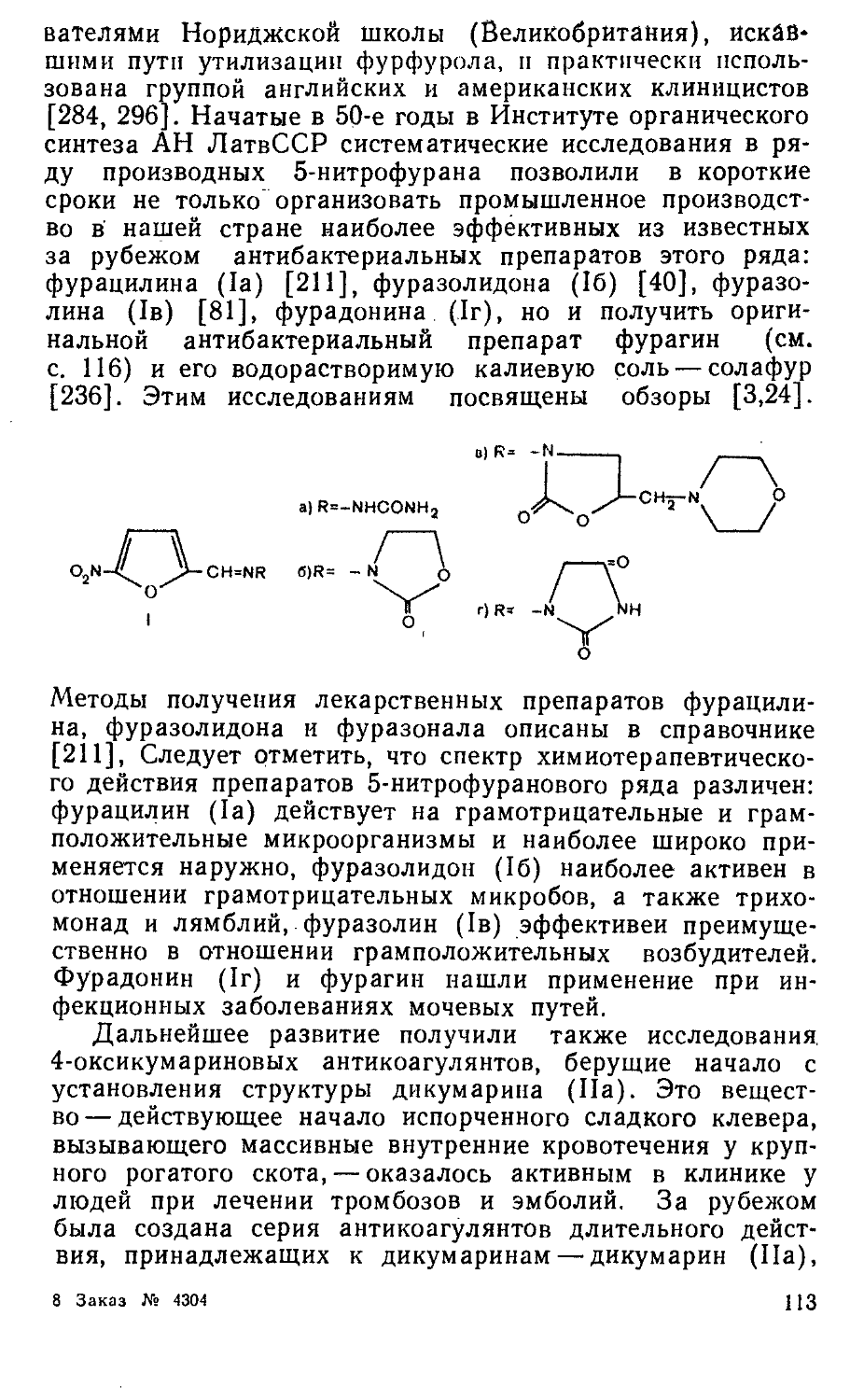
хлорокисью фосфора приводит к дихлориду 2-метилтиазо-лидофосфорной кислоты (V), который со II образует ди-этиленимид 2-метилтиазолидофосфорной кислоты (ими-фос) (VI).
2-Метилтиазолидин (IV). К 320 г безводного метанола прибавляют при перемешивании в течение 15—20 мин 176 г I, поддерживая температуру 10°С, затем при 12,5°C приливают 175 г II. Дают выдержку 30 мин при 12,5±2°С, охлаждают до 4 °C и пропускают в течение 5—6 ч при 8 °C сероводород. Перемешивание без охлаждения продолжают еще 2 ч, метиловый спирт отгоняют, остаток перегоняют в вакууме. Собирают фракцию с т. кип. 72— 80°C (15—20 мм рт. ст.). Получают 200 г IV с 1,5240—1,5260. Выход 47,5%.
Дихлорид 2-метилтиазолидофосфорной кислоты (V). При перемешивании к 200 г IV и 199 г триэтиламина в 1 л безводного бензола в течение Р/2—2 ч, поддерживая температуру 7±3°С, прибавляют 590 г свежеперегнанной хлорокиси фосфора. Дают выдержку при перемешивании и температуре 18±2°С 1 ч. Осадок гидрохлорида триэтиламина отфильтровывают, промывают 250 мл безводного бензола. Бензольные растворы осветляют 8 г активированного угля и упаривают в вакууме (10—30 мм) при температуре в массе не выше 30 °C. Остаток растворяют в 600 мл безводного бензола и передают на следующую стадию. Хранить бензольный раствор можно не более 24 ч при температуре не выше 5 °C.
Диэтиленимид 2-метилтиазолидофосфорной кислоты (имифос) (VI). Полученный на предыдущей стадии бензольный раствор V в течение 2—3 ч прибавляют к 142 г II и 340 г триэтиламина в 600 мл безводного бензола, поддерживая температуру реакционной массы 2,5±2°С. Дают выдержку 1 ч при перемешивании и температуре 16±2°С. Гидрохлорид триэтиламина отфильтровывают и промывают 150 мл безводного бензола. Бензольный раствор осветляют углем и упаривают при остаточном давлении 20—25 мм рт. ст. и температуре в массе не выше 30 °C. Остаток охлаждают до —10° С. Выделившиеся кристаллы растворяют при температуре не выше 30 °C в 500 мл безводного ацетона, ацетоновый раствор обесцвечивают 20 г угля и охлаждают при перемешивании до —18±2°С. Дают выдержку 3 ч и выделившиеся кристаллы VI повторно перекристаллизовывают таким же путем, но в асептических условиях. Получают 190 г стерильного VI. Выход 38,6%, считая на IV.
23
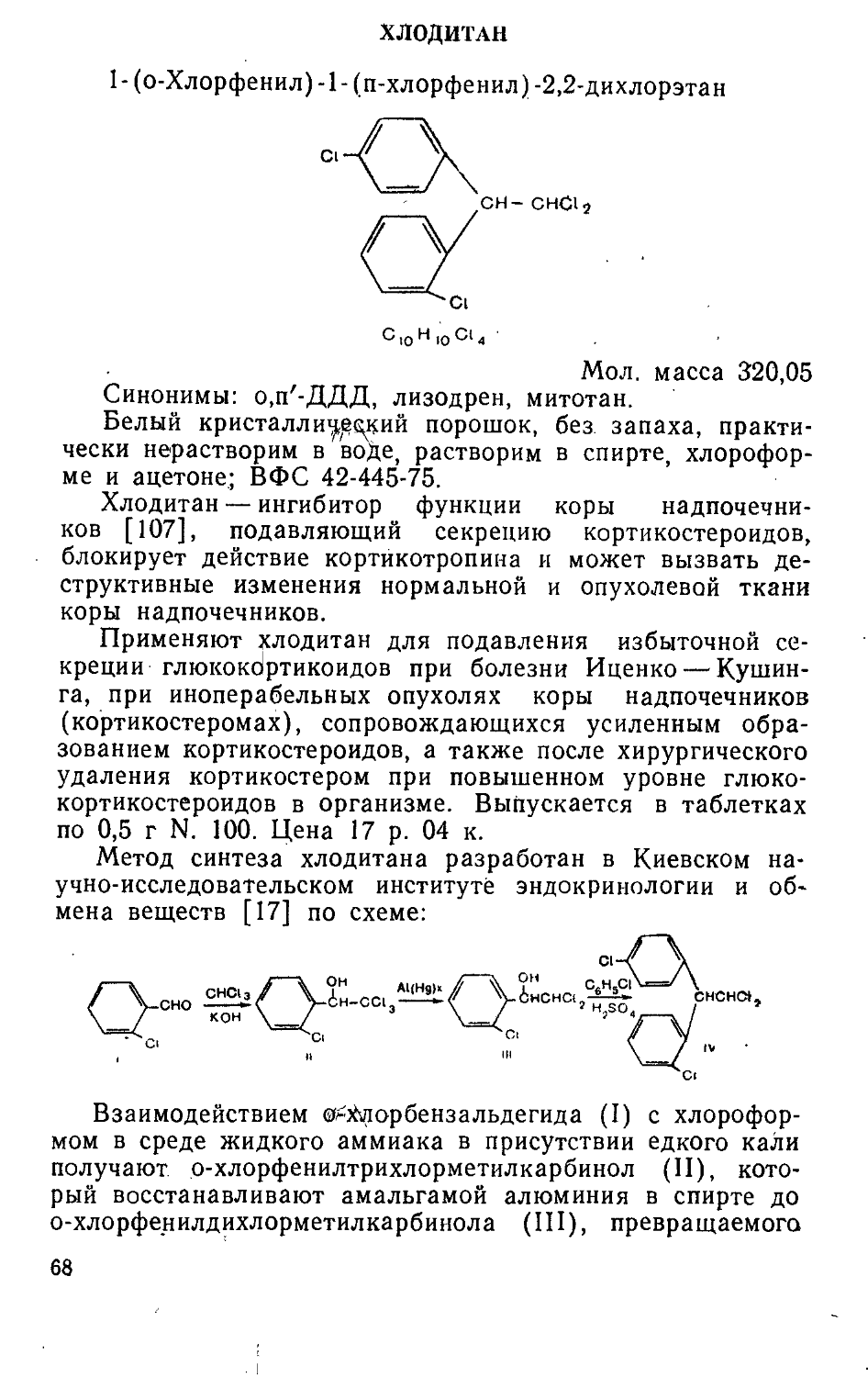
ХЛОФОСФОЛ
О, О-Диэтил-О-трихлорпентнлфосфат:
<с2н5°)2/
О(СН.>).|СС1.|.
С9н18рО,С1з
Мол. масса 327,58 Бесцветная жидкость со слабым ароматическим запахом, легко растворима в обычных органических растворителях, растительных и минеральных маслах, очень мало растворима в воде (1 :2000); МРТУ 42 № 3979-71.
Хлофосфол — оригинальное фосфорорганическое мистическое средство [256], вызывающее необратимое ингибирование холинэстеразы. Применяют хлофосфол для снижения внутриглазного давления при глаукоме, а также в других случаях, например, при травмах глазного яблока, когда необходим длительный мистический эффект.
Форма выпуска: флаконы по 10 мл 5% раствора в персиковом масле. Список А.
Метод получения хлофосфола разработан Институтом нефтехимического синтеза АН СССР совместно с Институтом органического синтеза АН ЛатвССР по схеме [256]:
р«3 С|» НО(СН,)4СС13 о
сЛон —(СЛО12РОН <С2Н5О>2\С[ 'СЛ°1зС(онлсС13
I II III ’4
Взаимодействием этилового спирта с треххлористым фосфором получают О,О-диэтилфосфористую кислоту (I) [208], хлорирование которой приводит к 0,0-диэтилхлор-фосфату (II) [103]. При реакции II с трихлорпентиловым спиртом, получаемым из 1,1,1,5-тетрахлорпентана через трихлорпентилацетат [172], синтезируют О,О-ди-этил-О-трихлорпентилфосфат (хлофосфол) (III).
О,О-Диэтилфосфористая кислота (I). К 55 мл охлажденного до 0 °C абсолютного этилового спирта добавляют по каплям 55,2 г треххлористого фосфора, перемешивают без охлаждения 40 мин, затем подключают вакуум (10— 30 мм рт. ст.) и при 20 °C отгоняют образовавшийся в результате реакции хлористый этил. Остаток перегоняют 24
при 72—73°C (13 мм рт. ст.). Получают 41 г (71,4%) I с 1,4080.
О,О-Диэтилхлорфосфат (II). Через 40 г I, охлажденного до 0—(—5) °C, пропускают сухой хлор до появления в реакционной массе избыточного хлора, который отдувают сухим азотом в слабом вакууме. Остаток перегоняют при 83—84°С (6 мм рт. ст.). Выход II 31 г (76%), n2D° 1,4165.
Трихлорпентилацетат. Смесь 105 г 1,1,1,5-тетрахлор-пентана, 73,5 г безводного ацетата калия, 1,25 г йодистого калия и 200 мл уксусной кислоты кипятят 20 ч при энергичном перемешивании. Массу охлаждают до 20 °C, приливают 200 мл воды и экстрагируют хлороформом (3 раза по 100 мл). Хлороформный экстракт промывают 250 мл 10% водного раствора поташа до нейтральной реакции и 100 мл 10% водного раствора гидросульфита натрия для удаления йода, после чего промывают 2 раза по 150 мл воды, сушат над сульфатом натрия и разгоняют в вакууме. Собирают фракцию с т. кип. 107—109 °C (6 мм рт. ст.). Выход 74 г (63,4%), п™ 1,4675. Содержание основного вещества не ниже 94% [по данным газожидкостной хроматографии (ГЖХ)].
Трихлорпентиловый спирт. Смесь 50 г трихлорпентил-ацетата, 40 мл метанола и 0,6 мл конц. соляной кислоты выдерживают 20 ч при 20 °C. Образовавшийся метилацетат и метанол отгоняют на кипящей водяной бане. К остатку прибавляют 40 мл метанола и 0,6 мл конц. соляной кислоты, кипятят 3 ч, упаривают, после чего вещество растворяют в 50 мл хлороформа. Хлороформный раствор промывают 50 мл 10% раствора соды, 30 мл 10% раствора тиосульфата натрия, 2 раза по 40 мл воды, сушат сульфатом натрия, упаривают. Вещество перегоняют при 102—104°C (7 мм рт. ст.). Получают 30,3 г (80%), п? 1,4880.
О,О-Диэтил-О-трихлорпентилфосфат (хлофосфол) (Ш). К смеси 17,9 г II и 8 г пиридина при 15—20 °C прикапывают 19 г трихлорпентилового спирта. Массу перемешивают 3 ч при 20 °C, гидрохлорид пиридина отфильтровывают. Фильтрат разгоняют в вакууме. Собирают фракцию с т. кип. 155—155,5°C (2 мм рт.ст.). Выход 25 г (73,5%).
ХЛОРАЦЕТОФОС
Диметиловый эфир а-ацетокси-р,р,р-трихлорэтилфос-финовой кислоты:
25
(CH3O,2n — 9НООССН 3 o CCl3
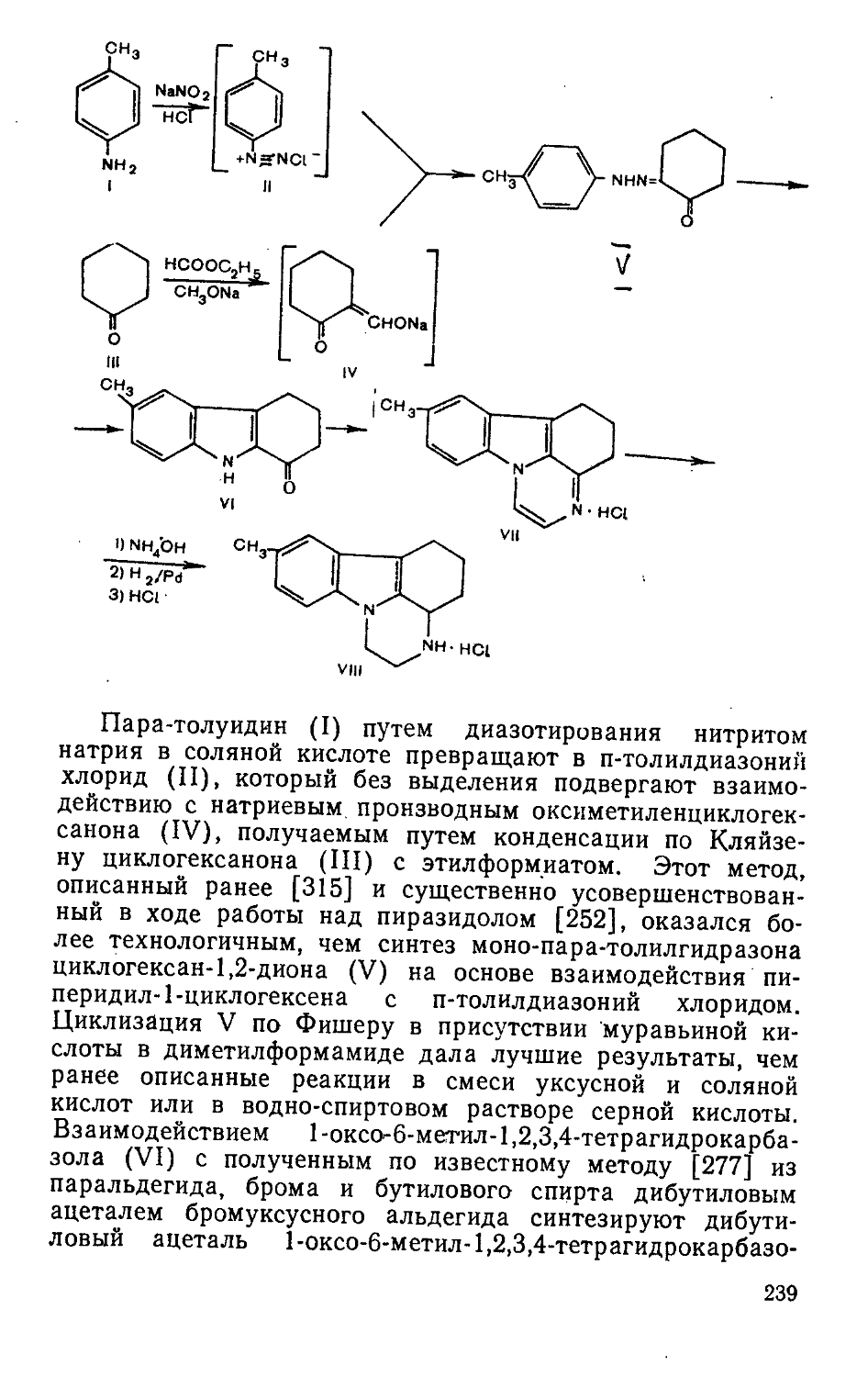
c8Hl0o5PCt3
Мол. масса 299,45
Бесцветная глицериноподобная жидкость, трудно растворима в воде, легко растворима в спирте, эфире, бензоле и ацетоне; т^лкип. 118—120°С/0,05 мм рт. ст.; ВФС 42-567-76. v
Хлорацетофос — оригинальный фунгицидный и бактерицидный препарат [177], активный в отношении различных патогенных грибов — возбудителей дерматомикозов из родов трихофитон, эпидермофитон, микроспорум, кандида и др. Применяют хлорацетофос для лечения грибковых заболеваний гладкой кожи и волосистой части головы. ।
Выпускаемся в виде 7% мази по 30 г. Цена 1 р. 02 к.
Метод синтеза хлорацетофоса разработан в Институте органической и физической химии им. А. Е. Арбузова Казанского филиала АН СССР [178] по схеме:
(СН3О),Р-СН-ОН о cct3
।
(СНзСО)2О HjSO,
/СН3О)2Р-СН-ООССН3 о 6ci3
II
Диметиловый эфир а-окси-$,р,р-трихлорэтилфосфино-вой кислоты (хлорофос) (I) ацетилируют уксусным ангидридом в присутствии катализатора — серной кислоты— до диметилового эфира а-ацетокси-р,р,р-трихлор-этилфосфиновой кислоты (хлорацетофоса) (II). Этот метод [178] имеет преимущество перед первоначально предложенным способ,рм^. получения II путем ацетилирования I хлористым ацетилом в присутствии основания (триэтиламина, пиридина и др.) [177], поскольку в последнем случае конечный продукт' оказывается загрязненным диметиловым эфиром р,р-дихлорвинилфосфиновой кислоты.
К 5,5 кг I приливают 4,8 кг уксусного ангидрида, содержащего 8 г конц. серной кислоты, массу перемешивают 5 ч при 78—80 °C, после чего подвергают разгонке в вакууме. Собирают фракцию с т. кип. 104—120 °C (0,05 мм рт. ст.), которую перегоняют повторно, собирая фракцию с т. кип. 118—120°C (0,05 мм рт. ст.). Выход 4,5 кг (70%).
26
ОКТАТИОЙ
О,О-Диэтилдитиофосфат октадециламмония
S' ch3(ch2)mch2nh3- (c2h5o)2i^
£ 20 ^46 NO 2 Р S 2
Мол. масса 455,8
Белый или белый с сероватым оттенком кристаллический порошок с характерным запахом, практически нерастворим в воде, мало растворим в спирте и эфире, легко растворим в хлороформе и бензоле; т. пл. 58—63°C (в интервале 2°C); ФС 42-970-75.
Октатион — оригинальный фунгистатический и фунгицидный препарат. Применяют октатион при различных дерматомикозах, микробных экземах, осложненных поверхностным кандидамикозом или бластомикозом. Особенно эффективен при межпальцевой интертригинозной эпидермофитии и сквамозно-гиперкератической форме микоза.
Выпускается в виде 3% присыпки по 40 г в упаковке (цена 87 к.).
Метод синтеза октатиона разработан во Всесоюзном научно-исследовательском институте химических средств защиты растений по схеме:
сЛон------|СЛО)2Р' ’ > снэ(сн,)мch2nh3- <С2Н5О),<
I V s
При взаимодействии абсолютного этилового спирта (I) с пятисернистым фосфором (II) получают О,О-диэтилди-тиофосфорную кислоту (III), которая с октадецилами-ном (IV) образует соответствующую соль — 0,0-диэтил-дитиофосфат октадециламмония (октатион) (V).
О,О-Диэтилдитиофосфорная кислота (III). К нагретым до 50—55 °C 2,27 кг I медленно, в течение Р/з ч, присыпают 2,82 кг II, контролируя температуру реакции (процесс экзотермичен). Дают выдержку при перемешивании и температуре 50—55 °C 2 ч с улавливанием выделяющегося сероводорода. Охлаждают до 20 °C и фильтруют с добавкой 200 г угля. Получают 3,1 кг III с содержанием основного вещества не ниже 79%. Выход 53%, считая на I.
О,О-Диэтилдитиофосфат , октадециламмония (октатион) (V). Выдерживают 4,35 кг IV 30 мин при НО—120°C
27
для разрушения карбонатов и удаления углекислого газа, после чего охлаждают до 60 °C, прибавляют 11,4 кг петролейного эфира и к полученной суспензии при 40— 45°C в течение 2 ч приливают 3,1 кг III. Дают выдержку 2 ч при 35—40 °C и оставляют массу при перемешивании охлаждаться до 20—25 °C, после чего охлаждают до (—8) — (—10 °C) 8—10 ч с перемешиванием. Выкристаллизовавшийся технический V отделяют, промывают 2,5 кг охлажденного до 0 °C петролейного эфира (контролируют, чтобы содержание IV в продукте не превышало 1%), перекристаллизовывают из 20,4 кг петролейного эфира (нагревание до 40°C, охлаждение до 8 °C) и высушивают при 30—40°C. Выход 3,5 кг (58,2%, считая на III).
БИСЧЕТВЕРТИЧНЫЕ АММОНИЕВЫЕ СОЕДИНЕНИЯ
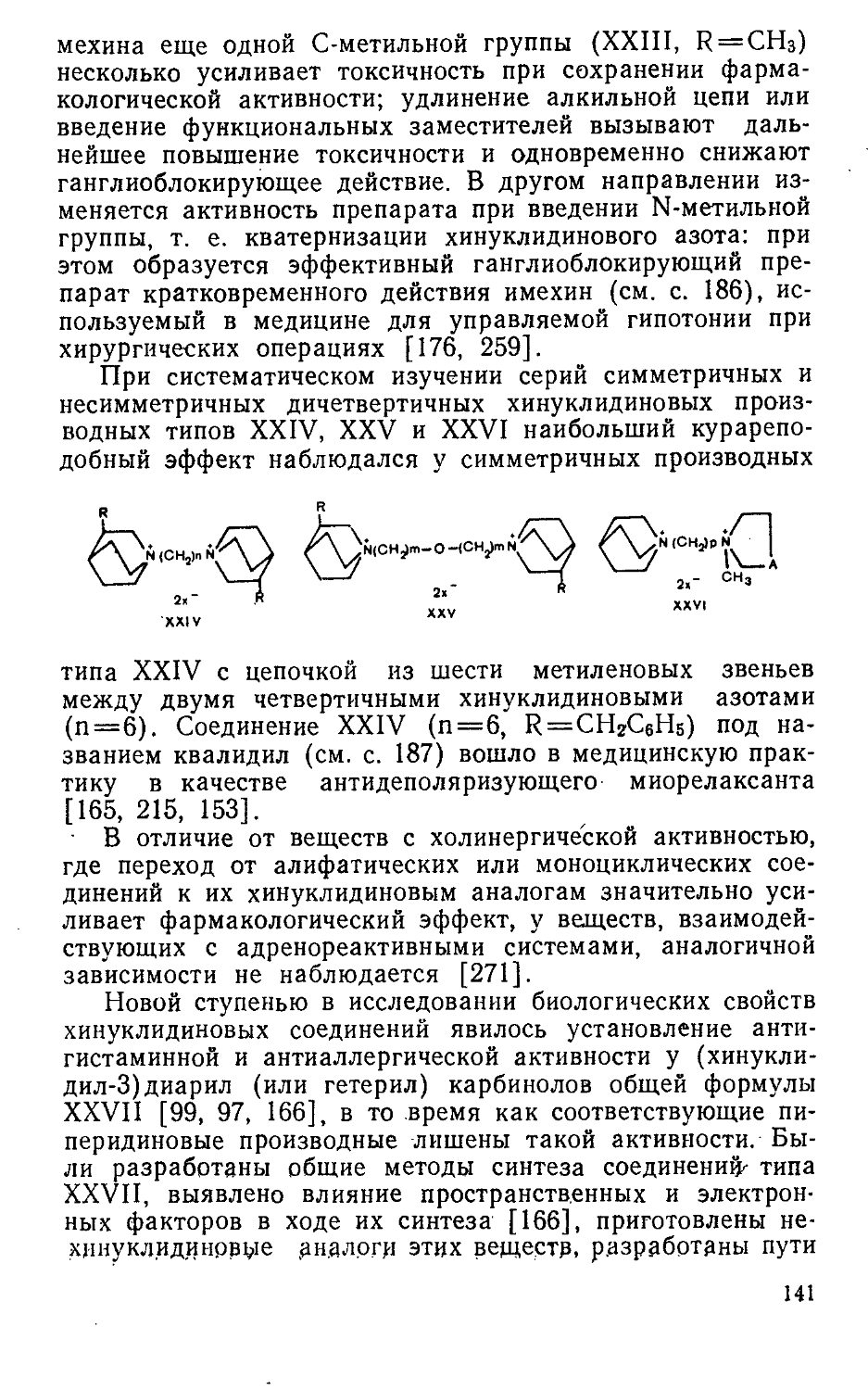
Основная область применения бисчетвертичных аммониевых соединений в медицине — расслабление скелетных мышц (миорелаксация) в результате блокады н-холино-реактивных систем нервно-мышечных синапсов.
История синтетических миорелаксантов начинается с 1935 г., когда была установлена химическая структура алкалоида d-тубокураринхлорида (I), являющегося действующим началом стрельного яда южноамериканских индейцев — кураре.
। а
ОН
Наличие в молекуле d-тубокураринхлорида (1)'двух четвертичных атомов азота привлекло внимание исследо-28
вателей и явилось толчком к поиску курареподобных веществ среди четвертичных производных других алкалоидов (стрихнина, спартеина, хинина, морфина и др.). Советскими учеными во ВНИХФИ на основе кватерниза-ции гелиотридановых алкалоидов создан оригинальный миорелаксант диплацин (II) [117], выпускаемый химикофармацевтической промышленностью СССР. Следует отметить, что исследования позволили обнаружить кураре-подобную активность и у некоторых алкалоидов живокости, являющихся третичными основаниями, а не четвертичными солями. Оригинальные препараты этого типа мелликтин и кондельфин вошли в практику [116, 140]. Курареподобное действие обнаружено также у третичного основания — алкалоида пирролизидинового ряда тезина (III), являющегося сложным эфиром а-изоретронеканола и п,п'-диокси-а-труксилловой кислоты [139].
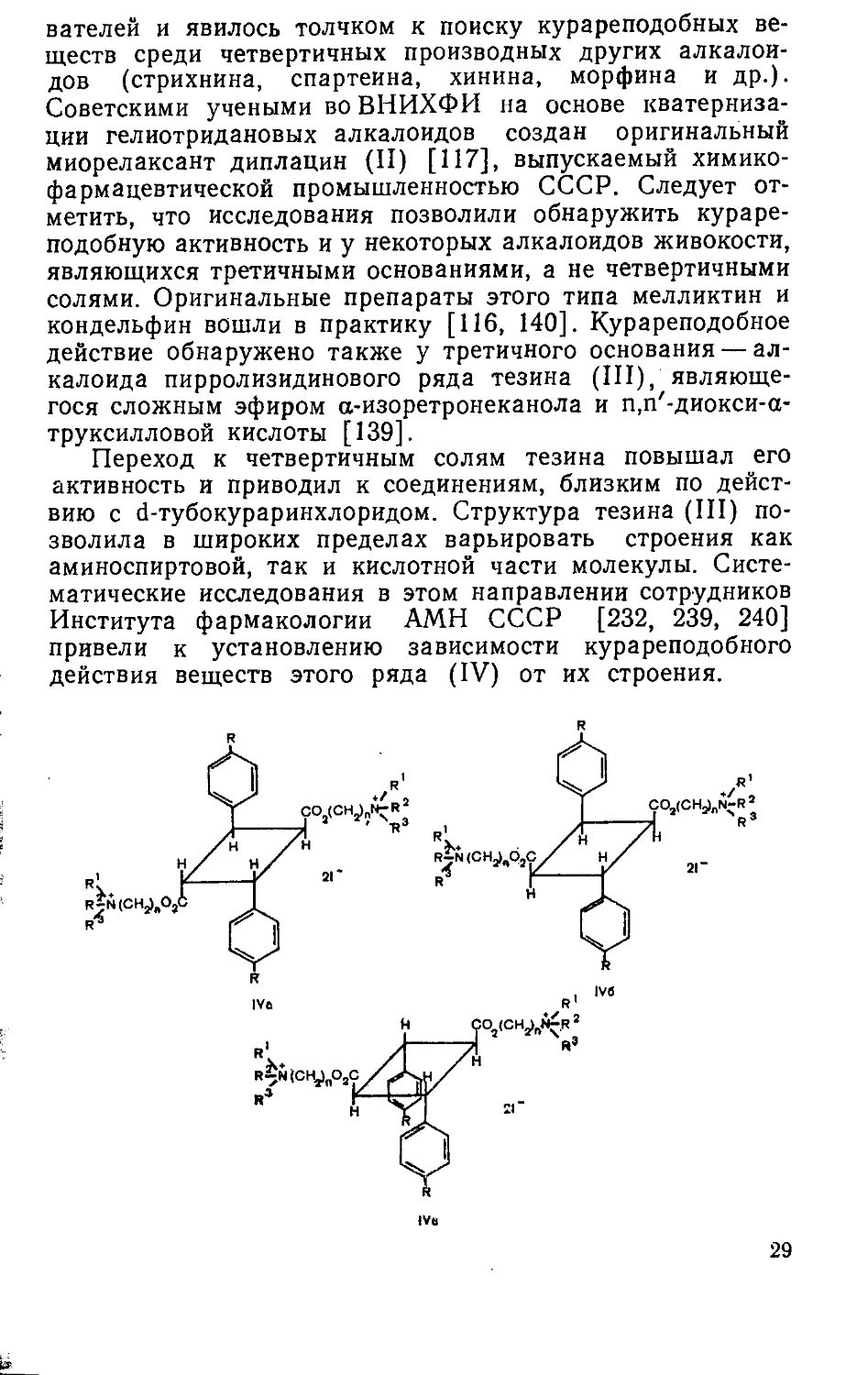
Переход к четвертичным солям тезина повышал его активность и приводил к соединениям, близким по действию с d-тубокураринхлоридом. Структура тезина (III) позволила в широких пределах варьировать строения как аминоспиртовой, так и кислотной части молекулы. Систематические исследования в этом направлении сотрудников Института фармакологии АМН СССР [232, 239, 240] привели к установлению зависимости курареподобного действия веществ этого ряда (IV) от их строения.
IV©
29
Было найдено, что у производных а-труксилловой кислоты (IVa) миорелаксантная активность выше, чем у аналогичных соединений у-труксилловой (IV6) и е-тру-ксилловой (IVb) кислот— пространственных изомеров веществ (IVa). При варьировании расстояния между оние-выми центрами максимум активности наблюдается при п=3, увеличение или уменьшение числа п снижает ку-рареподобное действие веществ. Триэтиламмониевые соли в этом ряду оказались активнее, чем триметиламмоние-вые, но наиболее эффективны биспиперидиниевые и бис-пирролидиниевые производные. Бисморфолиниевые соединения относительно малоактивны. Введение любых заместителей в п-положения фенильных ядер также ослабляет миорелаксантные свойства веществ. Наиболее эффективные оригинальные соединения этого ряда — анатруксо-ний (см. с. 33) [242] и циклобутоний (см. с. 36) [241] вошли в медицинскую практику.
По механизму действия рассмотренные лекарственные препараты: d-тубокураринхлорид, диплацин, мелликтин, кондельфин, анатруксоний, циклобутоний, а также оригинальный миорелаксант хинуклидинового ряда квалидил (см. с. 187) — относятся к группе недеполяризующих (или антидеполяризующих) миорелаксантов (пахикураре). Вещества этой группы парализуют нервно-мышечную передачу путем уменьшения чувствительности к ацетилхолину н-холинореактивных систем синаптической области, за счет чего исключается возможность деполяризации концевой пластинки и возбуждение мышечного волокна. Их фармакологическими ,,<д^тагонистами являются антихолнн-эстеразные вещества, которые, угнетая холинэстеразу, приводят к накоплению в области синапсов ацетилхолина. Последний ослабляет взаимодействие курареподобных веществ с н-холинореактивными системами и, следовательно, восстанавливает нервно-мышечную проводимость.
Механизм действия курареподобных лекарственных средств другой группы — деполяризующих препаратов (лептокураре) —включает !холиномиметический эффект, сопровождающийся стойкой деполяризацией. При этом нарушение передачи возбуждения с нерва на мышцу имеет такой же характер, как и при накоплении в синапсе большого избытка ацетилхолина. Препараты этой группы относительно быстро гидролизуются холинэстеразой и при однократном введении оказывают кратковременное действие, которое усиливается антихолинэстеразными препаратами. К деполяризующим средствам из применяющихся 30
в СССР миорелаксантов относится дитилин (см. с. 38), описанный за рубежом [322] и синтезированный в Институте тонкого органического синтеза им. А. Л. Мнджояна АН АрмССР [68].
Оригинальным миорелаксантом смешанного типа действия является лекарственный препарат диоксоний (см. с. 40), созданный в Институте органического синтеза АН ЛатвССР [222]. Исследования в этом направлении были начаты с изучения циклических диацеталей янтарного, фумарового и ацетиленового диальдегидов [221], аналогичных описаннсму за рубежом 2-метил-2-н-амил-4-ок-симетилдиоксалану, применявшемуся в клинике под названием гликеталь в качестве противосудорожного и миорелаксирующего средства. Дальнейший переход к дичетвертичным аммониевым производным с расстоянием между атомами азота 1,3—1,5 нм, характерным для других миорелаксантов, позволил получить препарат диоксоний (см. с. 40), представляющий практический интерес [222, 224]. Диоксоний сначала вызывает деполяризацию постсинаптической мембраны, а затем начинает действовать по антидеполяризующему механизму. Даже многократное введение прозерина как антихолинэстеразного средства не всегда оказывается эффективным для восстановления нервно-мышечной проводимости. На примере диметиламинного аналога диоксония (V), имеющего так же, как и диоксоний (см. с. 40), четыре асимметрических центра, было показано, что цис D-(—)-изомер является недеполяризующим миорелаксантом, а цис L-(-]-) характеризуется деполяризующим действием [223].
<°h8>8nch2-ch-o^Sh(ch 5н-г'о-снсн25(сн8)8 я/ V
32 >о-6н, 2|_ \___/\____/\___/
Поскольку применяющийся в медицине препарат диоксоний является смесью диастереоизомеров, смешанный тип действия этого средства, возможно, связан с различным характером миорелаксантной активности его пространственных изомеров.
Особое место в ряду бисчетвертичных аммониевых соединений занимает найденный во ВНИХФИ оригинальный класс противоопухолевых средств — производные дис-пиротрипиперазиния (VI) [161]. Как известно, введенные в 40-е годы в онкологическую практику алкилирующие противоопухолевые средства — производные бис-(р-хлор-
31
этил)-амина (соединения ряда азотистого иприта) малоустойчивы. Предполагалось, что эти соединения разлагаются с отщеплением молекулы хлороводорода и образованием винил (p-хлорэтил) аминов, которые легко полимеризуются. При изучении советскими исследователями [161] взаимодействия бис-(p-хлорэтил) амина с а-окисями п-нитростирола и пропилена наряду с другими продуктами были получены и производные диспиротрипиперазиния (VI). Образование этих соединений было объяснено с позиций циклоцепной таутомерии р-галогенэтиламинов. Одно из полученных в ходе исследования веществ—дихлорид М,М"-ди(р-хлор-этил)-М',]Ч"'-диспиротриииперазиния (VI, R = CH2CH2C1)— проявило противоопухолевую активность при раке гортани и легких, опухолях носоглотки, лимфогранулематозе и под названием спиразидин было разрешено в СССР для медицинского применения [160, 162, 211, 225]. Однако спиразидин оказался неустойчивым препаратом и вскоре был вытеснен более стабильным и эффективным производным диспиротрипиперазиниевого ряда, получившим название проспидин (VI, R = CH2CH(OH)CH2C1) (см. с. 43 [164, 204].
Бисчетвертичные аммониевые соединения, содержащие у атомов азота большие углеродные остатки, являются катионными детергентами, обладающими противогрибковыми и антибактериальными свойствами. Их создание явилось результатом начатых еще в 1908 г. исследований по антисептической активности четвертичных аммониевых соединений, вошедших затем в медицинскую практику [284]. В 30—50-е годы было синтезировано и изучено большое количество соединений этого типа [308]. Лекарственные препараты декамин (VII) и цетилпиридиний хлорид (VIII) воспроизведены во ВНИХФИ и выпускаются химико-фармацевтической промышленностью СССР [211].
Препарат .VIII является моночетвертичной, а препарат VII — бисчетвертичной аммониевыми солями. К новым
32
бисчетвертичным аммониевым солям группы катионных детергентов относится и разработанный в Институте органической химии АН УССР совместно с Черновицким университетом лекарственный препарат этоний (см. с. 46) [199].
АНАТРУКСОНИЙ
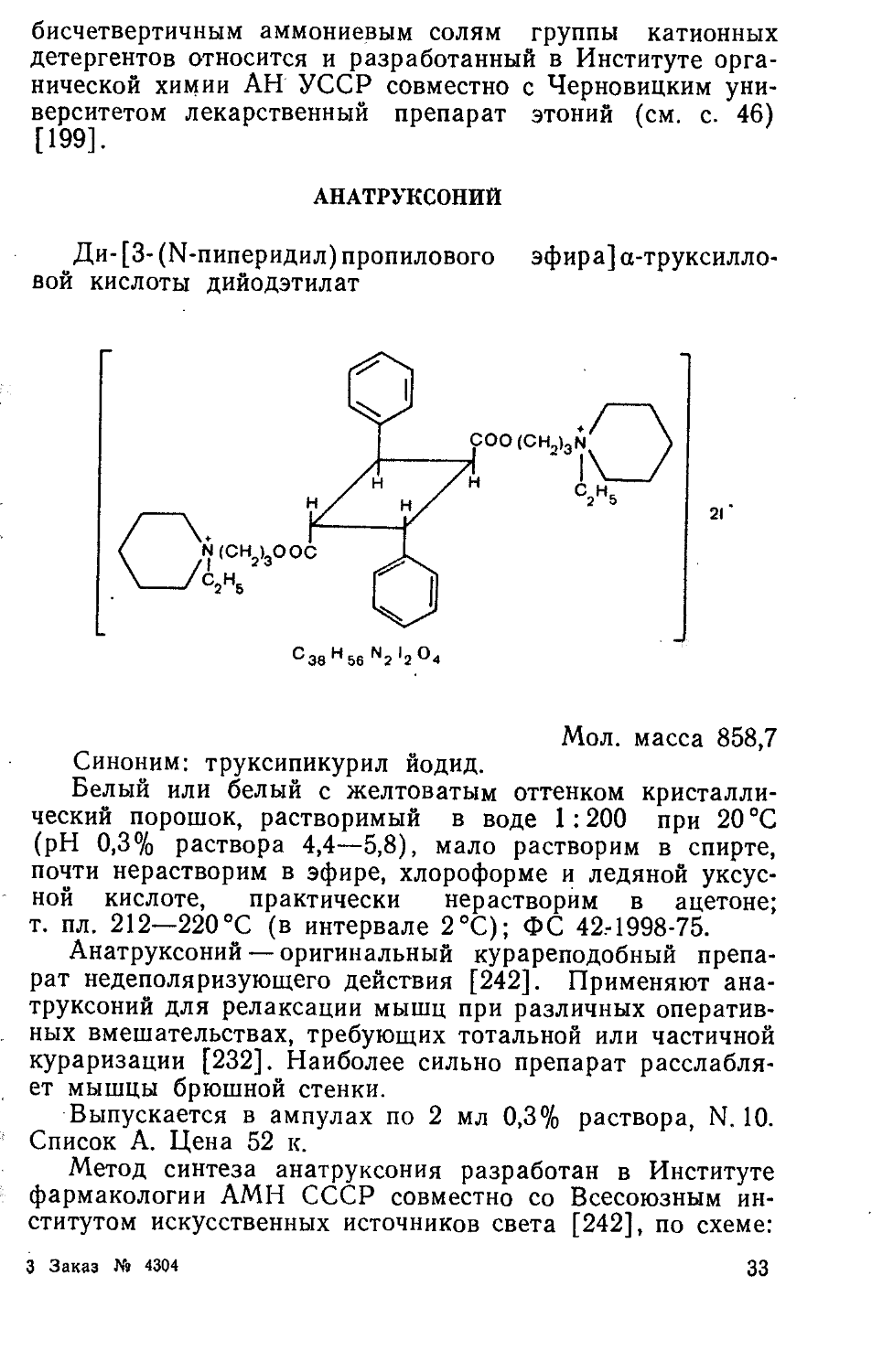
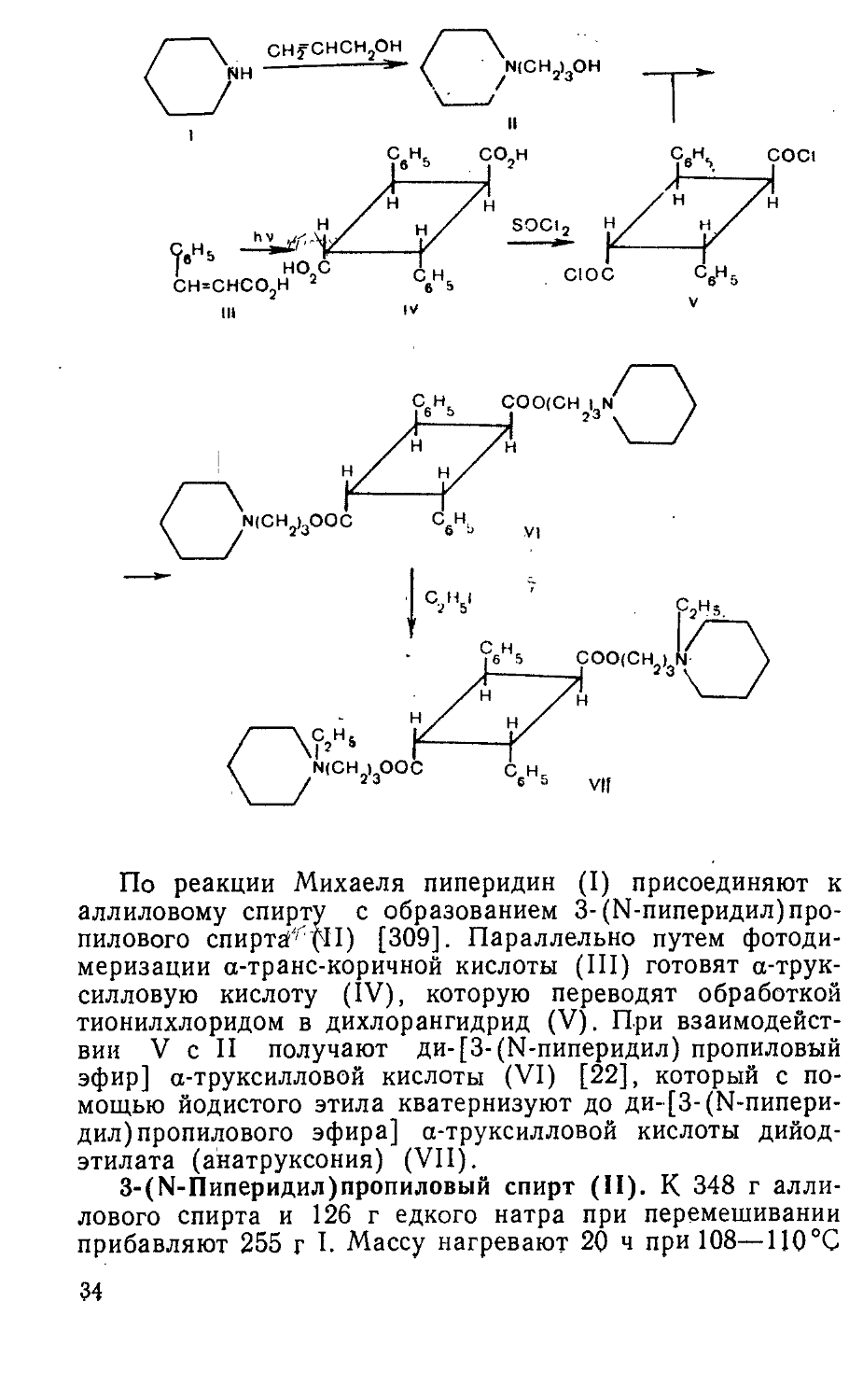
Ди- [3- (N-пиперидил) пропилового эфира] а-труксилловой кислоты дийодэтилат
Мол. масса 858,7 Синоним: труксипикурил йодид.
Белый или белый с желтоватым оттенком кристаллический порошок, растворимый в воде 1:200 при 20 °C (pH 0,3% раствора 4,4—5,8), мало растворим в спирте, почти нерастворим в эфире, хлороформе и ледяной уксусной кислоте, практически нерастворим в ацетоне; т. пл. 212—220°C (в интервале 2°C); ФС 42.-1998-75.
Анатруксоний — оригинальный курареподобный препарат недеполяризующего действия [242]. Применяют анатруксоний для релаксации мышц при различных оперативных вмешательствах, требующих тотальной или частичной кураризации [232]. Наиболее сильно препарат расслабляет мышцы брюшной стенки.
Выпускается в ампулах по 2 мл 0,3% раствора, N. 10. Список А. Цена 52 к.
Метод синтеза анатруксония разработан в Институте фармакологии АМН СССР совместно со Всесоюзным институтом искусственных источников света [242], по схеме:
3 Заказ № 4304 33
По реакции Михаеля пиперидин (I) присоединяют к аллиловому спирту с образованием 3-(N-пиперидил)пропилового спирта^ (Н) [309]. Параллельно путем фотодимеризации а-транс-коричной кислоты (III) готовят а-трук-силловую кислоту (IV), которую переводят обработкой тионилхлоридом в дихлорангидрид (V). При взаимодействии V с II получают ди-[3-(N-пиперидил) пропиловый эфир] а-труксилловой кислоты (VI) [22], который с помощью йодистого этила кватернизуют до ди-[3-(N-пиперидил) пропилового эфира] а-труксилловой кислоты дийод -этилата (анатруксония) (VII).
3-(М-Пиперидил)пропиловый спирт (II). К 348 г аллилового спирта и 126 г едкого натра при перемешивании прибавляют 255 г I. Массу нагревают 20 ч при 108—ПО °C 34
с повышением температуры в конце реакции (не менее 5 ч) до 118—123 °C. Затем охлаждают до 20 °C и приливают 600 мл воды. Водный слой отделяют и экстрагируют 3 раза по 400 мл бензола. Объединенные бензольные экстракты сушат поташом, упаривают. Продукт перегоняют в вакууме, собирая фракцию с т. кип. 98—100 °C (10 мм рт. ст.). Получают 247 г (57,8%) II с п£° 1,4745— 1,4772.
а-Труксилловая кислота (IV). Водную суспензию, получаемую перемешиванием 100 г III с 2 л дистиллированной воды в течение 58—60 ч, разбавляют дистиллированной водой до 3 л и при поддержании путем охлаждения температуры около 60 °C облучают ее светом лампы с in=0,3 нм, ир=180В в течение 60 ч при перемешивании. После этого осадок (90 г) отфильтровывают, высушивают и кипятят 1 ч с 300 мл дихлорэтана для отделения III. Нерастворившуюся часть промывают 2 раза по 25 мл горячего (60—70 °C) дихлорэтана и снова кипятят со 100 мл дихлорэтана. Осадок IV отфильтровывают, промывают 2 раза по 25 мл горячего дихлорэтана и высушивают. Получают 60 г (60%) IV с т. пл. 275—285 °C. Из дихлорэтановых растворов при охлаждении выкристаллизовывается III, которую вновь используют в синтезе.
Ди-[3-(1Ч-пиперидил) пропиловый эфир] а-труксилловой кислоты (VI). Смесь 59,2 г IV, 400 мл дихлорэтана, 163 г тионилхлорида и 1,2 г пиридина нагревают 1 ч при кипении. Гомогенный раствор упаривают в вакууме, прибавляют 400 мл безводного дихлорэтана с последующей отгонкой в вакууме. Контролируют температуру плавления полученного V (не ниже ПО °C), и поскольку он неустойчив при хранении, его сразу растворяют в 400 мл безводного дихлорэтана и приливают к охлажденному до 5 °C раствору 60 г II в 300 мл безводного дихлорэтана с такой скоростью, чтобы температура реакционной массы была не выше 10 °C. Дают выдержку 2 ч при 10 °C и 15 ч при 20 °C, вновь охлаждают до 10 °C, приливают 200 мл воды и перемешивают 10 мин. Водный слой отделяют, дихлорэтановый— экстрагируют 3 раза по 100 мл 5% серной кислоты и 100 мл воды. Кислые и водные экстракты объединяют, осветляют 10 г угля, охлаждают до 5 °C, подщелачивают 25% водным раствором аммиака до pH 9, дают затравку VI и перемешивают 1 ч. Осадок отфильтровывают, промывают 200 мл охлажденной до 5 °C воды до отсутствия в промывной воде С!~ и SO4~_ и высушивают. Получают 95 г (87%) VI с т. пл. 56—58°C.
3*
35
Ди-[3-(М-пиперидил)пропилового эфира] а-труксилло-вой кислоты дийодэтилат (анатруксоний) (VII). Нагревают при кипении смесь 90 г IV, 176 г перегнанного йодистого этила и 200 мл метанола 10—.13 ч до отсутствия щелочной реакции; после этого охлаждают до 0°С и дают выдержку 15 ч. Выпавшие кристаллы отфильтровывают, промывают 300 мл ацетона, высушивают и перекристаллизовывают из смеси 925 мл спирта и 46 мл воды с добавкой 6 г угля. Для кристаллизации фильтрат после отделения угля оставляют при 0-f-5°C на 18—20 ч в темном месте. Вещество отфильтровывают, промывают 200 мл этилового спирта и сушат в вакууме при 40—50 °C. Выход 89 г (63,1%).
ЦИКЛОБУТОНИЙ
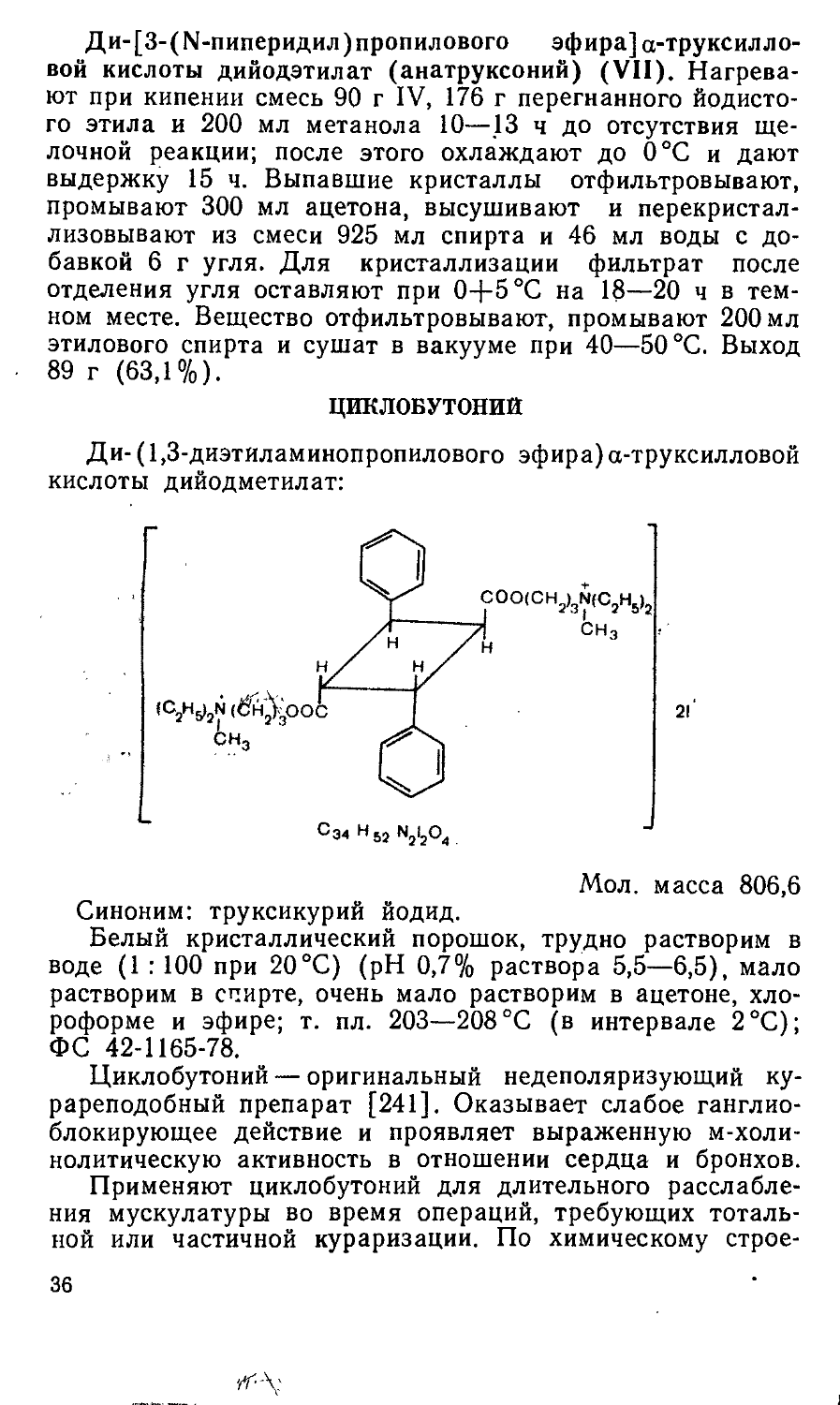
Ди- (1,3-диэтиламинопропилового эфира) а-труксилловой кислоты дийодметилат:
Мол. масса 806,6 Синоним: труксикурий йодид.
Белый кристаллический порошок, трудно растворим в воде (1 : 100 при 20°С) (pH 0,7% раствора 5,5—6,5), мало растворим в спирте, очень мало растворим в ацетоне, хлороформе и эфире; т. пл. 203—208°C (в интервале 2°C); ФС 42-1165-78.
Циклобутоний — оригинальный недеполяризующий ку-рареподобный препарат [241]. Оказывает слабое ганглиоблокирующее действие и проявляет выраженную м-холи-нолитическую активность в отношении сердца и бронхов.
Применяют циклобутоний для длительного расслабления мускулатуры во время операций, требующих тотальной или частичной кураризации. По химическому строе
36
нию и фармакологическим свойствам близок к анатрук-сонию.
Выпускается в ампулах по 2 мл 0,7% раствора, N. 10.
Список А. Цена 92 к.
Метод синтеза циклобутония разработан в Институте фармакологии АМН СССР [241] по схеме:
сн3 ’3"(С2Н5>5
2Г
Полученную фотодимеризацией коричной кислоты, как это описано в методе синтеза анатруксония (см. с. 35), а-труксилловую кислоту (I) обработкой тионилхлоридом также по методу, описанному в случае анатруксония (см. с. 35), переводят в дихлорангидрид (II), который при взаимодействии с 1,3-диэтиламинопропанолом образует ди-(1,3-диэтиламинопропиловый эфир) а-труксилловой кислоты (III). Последний кватернизуют с помощью йодистого метила до ди-(1,3-диэтиламинопропилового эфира) а-труксилловой кислоты дийодметилата (циклобутония) (IV).
Ди-(1,3-диэтиламинопропиловый эфир) а-труксилловой кислоты (Ш). Дихлорангидрид II, приготовленный из 59,2 г а-труксилловой кислоты (I) по методу, описанному на стадии получения ди [3-(N-пиперидил) пропилового эфи-
37
ра]а-труксилловой кислоты в синтезе анатруксония (см. с. 35), в виде дихлорэтанового раствора приливают к охлажденному до 5 °C раствору 52,6 г 1,3-диэтиламинопро-панола в 300 мл безводного дихлорэтана с такой скоростью, чтобы температура реакционной массы была не выше 10 °C. Дают выдержку 2 ч при 10 °C и 15 ч при 20 °C, вновь охлаждают до 10 °C, приливают 200 мл воды, перемешивают 10 мин. Водный слой отделяют, дихлорэтановый экстрагируют 3. раза по 100 мл 5% серной кислоты и 100 мл воды. Кислые и водные экстракты объединяют, ответляют 10 г угля, охлаждают до 5 °C, подщелачивают 25% водным раствором аммиака до pH 9 и выделившееся основание экстрагируют 4 раза по 250 мл бензола. Высушенные сульфатом магния бензольные экстракты упаривают в вакууме. Изучают 88 г (84%) III в виде густого вязкого маслообразного вещества, очень трудно растворимого в воде.
Ди-( 1,3-диэтиламинопропилового эфира)а-труксилловой кислоты дийодметилат (циклобутоний) (IV). Нагревают при кипении смесь 88 г III, 136,8 г перегнанного йодистого метила и 200 мл метанола 5—8 ч до отсутствия щелочной реакции. После этого охлаждают до 0°С и дают выдержку 18—20 ч. Выпавшие кристаллы отфильтровывают, промывают 200 мл ацетона, высушивают в вакууме при 50 °C и перекристаллизовывают из 880 мл этилового спирта с добавкой 0,1 г гидросульфита натрия и 6 г угля. Для кристаллизации фильтрат после отделения угля оставляют при 0+5 °C на 18—20 ч в темном месте. Вещество отфильтровывают, промывают 200 мл этилового спирта и повторно перекристаллизовывают в тех же условиях из 840 мл этилового спирта с добавкой 0,1 г гидросульфита натрия. Получают 99 г (73%) IV.
дитилин
Р-Диметиламиноэтилового эфира янтарной кислоты дийодметилат:
[(CH3)3N(CH2)2OOC(CH2)2COO(CH2l2N(CH3)3] 21 -c,4h30i2n2o4
Мол. масса 544,2 Синонимы: сукцаметониум йодид, анектин, сколин, бре-ведил и др. Аналогичные дихлорметилаты и диброммети-латы выпускаются за рубежом под названиями: листенон, 38
миорелаксин, сукцаметонил хлорид, анектин, бревидил, целокаин, целокурин, курахолин, диацетилхолин, лепто-сукцин, пантолакс, сукцинилхолин хлорид, синкурор, ку-рацит, куралест, квелицин и др.
Белый мелкокристаллический порошок, легко растворим в воде, очень мало в спирте и ацетоне, практически нерастворим в эфире; т. пл. 247—252°C (в интервале 2 °C); ГФХ, ст. 233.
Дитилин — деполяризующий мышечный релаксант [68], при внутривенном введении нарушает проведение нервно-мышечного возбуждения и вызывает расслабление скелетных мышц. Препарат оказывает быстрое, но кратковременное действие, так как расщепляется псевдохолинэстеразой До холина и янтарной кислоты. Растворы дитилина нельзя смешивать с растворами барбитуратов (образуется осадок и происходит гемолиз крови).
Применяют дитилин при эндоскопических процедурах (бронхо- и эзофагоскопия, цистоскопия и др.), кратковременных операциях (наложение швов на брюшную стенку, вправление костных отломков, вывихов и др.). Препарат может быть также использован для устранения судорог при столбняке.
Выпускается в ампулах по 5 мл 2% раствора N. 10. Список А. Цена 1 р. 70 к.
Метод синтеза дитилина разработан Институтом тонкой органической химии им. А. Л. Мнджояна АН АрмССР [68] по схеме:
ин2соон
CHjCOOH ,<C^hN<CH2>2OH<»> (CH3i2N(CH2)2OOC(CH.)2COO<CH;!)2N(CH^i-*-
1 снз' + 111 ♦
—*- [(CH^NtCHjPjOOCfCH^COOfCHjlj N(CH^3] 21
IV
Этерификацией янтарной кислоты (I) р-диметиламино-этанолом (II) в толуоле получают р-диметиламиноэтило-вый эфир янтарной кислоты (III), который обработкой йодистым метилом в ацетоне переводят в р-диметиламино-этилового эфира янтарной кислоты дийодметилат (дитилин) (IV).
Р-Диметиламиноэтиловый эфир янтарной кислоты (III). Смесь 120 г II, 60,5 г I и 67 мл толуола кипятят с водоотделителем 45—50 ч до прекращения отгонки воды, затем реакционную массу промывают 40 мл 50% раствора по
39
таша. Водный слой экстрагируют толуолом (2 раза по 65 мл). Толуольный раствор сушат, упаривают, III перегоняют при 147—148°C (1 мм рт. ст.). Выход 58,65 г (43%, считая на I).
Р-Диметиламино^тилового эфира янтарной кислоты дийодметилат (дитилин) (IV). К охлажденному до 12— 15 °C раствору 19,26 г III в 110 мл абсолютного ацетона при интенсивном перемешивании приливают 33,1 г йодистого метила. Реакция экзотермична и скорость прибавления йодистого метила регулируют так, чтобы температура массы была не выше 20 °C. Затем охлаждают до 12—15°C, осадок IV отделяют, промывают 60 мл абсолютного ацетона и перекристаллизовывают из 20 мл дистиллированной воды (нагревание до 60—65 °C, охлаждение до 14— 16°C), промывают 2 раза по 10 мл абсолютного ацетона и сушат сначала при 40 °C, затем при ПО °C в вакууме. Выход 34 г (80,8%).
ДИОКСОНИЙ
1,2-Бис- (4'-пирролидинометил- 1',3'-диоксоланил-2') -эта- \ на дийодметилат:
с 20 н з8 I 2 N 2 О 4
Мол. масса 624,3
Желтоватого цвета мелкокристаллический порошок, слегка гигроскопичный, очень легко растворим в воде (водные растворы не меняются при стерилизации), труднее — в этиловом спирте, практически нерастворим в эфире, ацетоне, хлороформе; ВФС 42-556-76.
Диоксоний — оригинальный миорелаксант смешанного типа действия [222]. Сначала вызывает фазу деполяризации, а затем действует по антидеполяризующему механизму. Применяют диоксоний для расслабления мускулатуры и выключения спонтанного дыхания как самостоятельно, так и после предварительного введения дитилина [223].
Выпускается в ампулах по 5 мл 0,1% раствора N. 10. Список А. Цена 1 р.
40
Метод синтеза диоксония разработан Институтом органического синтеза АН ЛатвССР [222] по схеме:
СП Вг2 СН3ОН (СН^О) jCH|_CHmCH_CH(0CH3)2 (CHjOjCHICHjJjCHIOCHjJj
г СН он
7
С1СНГСН-ОЧ zO-CH-CH2Cl
СНОН(СН.ОН), ----*- НОСНГСН(ОН)СН,С1— CH-(CHj>5-CH
’ 2 онсоон сн-о о-сн2
IV V ’ VI
асн31
1---С 1-----f <5нтол ^о-сн3
VII
Путем бромирования фурана (I) в метаноле получают диметилацеталь фумарового диальдегида (II) [15], который каталитически в присутствии никеля восстанавливают до диметилацеталя янтарного диальдегида (III) [15]. Эти стадии синтеза III аналогичны первым стадиям получения тропина в производстве атропина (см. с. 173). Параллельно из глицерина (IV) с соляной кислотой в присутствии каталитических количеств уксусной кислоты получают З-хлорпропандиол-1,2 [218], который при взаимодействии с III образует 1,2-бис- (4'-хлорметил-1', 3'-диоксоланил-2')-этан (VI) [221]. Нагревание VI с пирролидином по общему методу [224] приводит к 1,2-(4'-пирролидинометил-1',3'-диоксоланил-2') -этану (VII), который превращают обработкой йодистым метилом в дийодметилат VIII — препарат диоксоний.
Диметилацеталь фумарового диальдегида (II). В охлажденный до —45 °C раствор 210 г I в 1,55 л метанола в течение Р/г ч прибавляют при перемешивании раствор 500 г брома в 1,25 л метанола (растворение проводить осторожно при охлаждении!) с такой скоростью, чтобы температура в массе оставалась —45—(—35°C). Затем дают реакционной смеси нагреться до —10 °C, выдерживают при этой температуре Р/2 ч, вновь охлаждают до —40 °C и при перемешивании и температуре не выше —30 °C пропускают газообразный аммиак до щелочной реакции по универсальному индикатору. Осадок бромистого аммония отфильтровывают и промывают 250 мл эфира. Фильтрат выдерживают 3 ч над 190 г прокаленного пота
41
ша, фильтруют и упаривают при остаточном давлении 300—400 мм. Осадок бромистого аммония повторно отфильтровывают и промывают 120 мл эфира. Операцию отделения бромида аммония повторяют до прекращения выделения его в осадок. После этого вещество перегоняют в вакууме, собирая фракцию с т. кип. 85—92 °C (13 мм рт. ст.), и для дополнительной очистки (примеси отравляют катализатор на следующей стадии) вторично перегоняют, собирая фракцию в интервале 85—92°C (13 мм рт.ст.). Получают 350 г II, выход 64,5%.
Днметилацеталь янтарного диальдегида (III). Смесь 350 г II, 350 мл метанола и 18 г скелетного никелевого катализатора гидрируют в автоклаве при 50 °C и начальном давлении водорода 60 атм. Катализатор отфильтровывают, метанольный раствор упаривают. Остаток перегоняют в вакууме, собирая фракцию с т. кип. 77—79°C (9 мм рт. ст.). Выход III 332 г (93,8%).
З-Хлорпропандиол-1,2 (V). Нагревают смесь 810 г глицерина, 70 мл дистиллированной воды, 1,81 л конц. соля^ ной кислоты и 50 мл уксусной кислоты 10 ч, сначала осторожно (выделяется большое количество хлороводорода), затем при интенсивном кипячении. Реакционную массу упаривают до температуры 140 °C, после чего подвергают фракционной разгонке в вакууме. Собирают вещество с т. кип. 115—117°C (И мм рт.ст.), которое подвергают вторичной разгонке. Получают 520 г (55%) V.
1,2-Бис- (4'-хлорметил-1',3'-диоксоланил-2')-этан (VI). 330 г III, 430 г V и 1—2 мл конц. соляной кислоты кипятят с отгонкой метанола, контролируя кислотность среды (должна быть не более 4,0). После отгонки 220 мл метанола массу подвергают фракционной перегонке в вакууме. Собирают фракцию с т. кип. 149—155°C (2 мм рт. ст.). Получают 380 г VI с п*) 1,484—1,485, d420 1,295—1,296. Выход 75%.
1,2-Бис-(4'-пирролидииометил-1 ',3'-диоксоланил-2') -этан (VII). Смесь 380 г VI и 410 г пирролидина нагревают 6 ч, равномерно повышая температуру от 80 °C до 150 °C, после чего оставляют медленно (в течение 10 ч) охлаждаться. Смесь продуктов сливают с закристаллизовавшегося пирролидина гидрохлорида и перегоняют в вакууме. Собирают фракцию с т. кип. 195—210°С (1 мм рт. ст.). Получают 400 г VII с п“ 1,490. Выход 83,5%.
1,2-Бис-(4'-пирролидинометил-1/,3,-диоксоланил-2')-эта-на дийодметилат (диоксоний) (VIII). К раствору 400 г VII в 2,2 л безводного ацетона при 15 °C и интенсивном 42
перемешивании в течение 2 ч прибавляют 376 г йодистого метила. Кристаллический осадок VIII отфильтровывают, промывают 400 мл безводного ацетона и перекристаллизовывают из 1,38 л абсолютного этилового спирта. Сушат при температуре не выше 60 °C. Получают 400 г VIII. Под-париванием маточного раствора и дополнительной перекристаллизацией выделившегося продукта из абсолютного этилового спирта получают дополнительно 130 г VIII. Общий выход вещества с т. пл. не ниже 165°C 530 г (72,5%)•
ПРОСПИДИН
Ь1,Ь1"'-Бис- (у-хлор-р-оксипропил) -М',М"-диспиротрипипе-разиния дихлорид:
2сГ. н2о
СШ H36Cl4N4O2H2O
Мол. масса 500,3
Белый кристаллический порошок, легко растворим в воде (pH 2% раствора 6,0—7,0), практически нерастворим в спирте, хлороформе и эфире; в водных растворах нестоек (гидролизуется с отщеплением хлороводорода); ФС 42-966-75.
Проспидин — оригинальный противоопухолевый препарат [164, 204], в лечебных дозах не оказывающий значительного угнетающего действия на кроветворение. Наиболее чувствительны к препарату экзофитно растущие и гистологически малодифференцированные новообразования.
Применяют проспидин при раке гортани и злокачественных новообразованиях глотки независимо от стадии, формы роста и локализации, а также при папилломатозе верхних дыхательных путей, раке легких, лимфогранулематозе, ретикулезах кожи (при ангиоретикулезе Капоши, грибовидном микозе, саркоматозе кожи); ретинобластомах. Препарат можно сочетать с лучевой терапией.
Выпускается лиофилизированный для инъекций по 0,1 г во флаконе. Цена 36 к.
Метод синтеза проспидина разработан во ВНИХФИ [162, 164] по схеме:
43
Из N-бензоилпиперазина гидрохлорида (I) получают соответствующее основание, которое без выделения алкилируют этиленхлоридгидрином до 1-бензоил-4-(р-окси-этил)пиперазина (II) и обработкой тионилхлори-дом переводят в 1-бензоил-4-(р-хлорэтил)-пиперазина гидрохлорид (III). Взаимодействие III с три-этиламином позволяет получить М,М/"-дибензоил-М',М"-диспиротрипиперазиния дихлорид (IV), в котором N-бен-зоильные группы удаляют нагреванием с соляной кислотой, а из Й',Ы"-диспиротрипиперазиния дихлорида дигидрохлорида (V) свободный М'.ЬГ-диспиротрипиперази-ния дихлорид (VI) выделяют с помощью гидроокиси лития. Алкилированием VI эпихлоргидрином получают проспидин (VII). Строение проспидииа явилось предметом специальных исследований методом ЯМР13С [66].
1-Бензоил-4-(р-хлорэтил)пиперазина гидрохлорид (III). К раствору 255 г I в 280 мл воды при 20 °C приливают 184 мл 20% раствора едкого натра с такой скоростью, чтобы температура оставалась 20—25 °C, после чего прибавляют 113,3 г этиленхлоргидрина и в течение 10—12 ч приливают постепенно при 20—25 °C, перемешивании и постоянном контроле pH среды (pH должен быть в интервале 9—11, в конце процесса —до 11,5) 452 мл 11,4% раствора едкого натраЛПосле дополнительной выдержки 10—11 ч при 20—25 °C контролируют содержание N-бензоилпиперазина (должно быть не более 0,6%) и II экстрагируют 5 раз по 400 мл дихлорэтана (в последнем экстракте должно быть менее 0,5% плотного остатка).Дихлорэтановый раствор сушат поташом до содержания влаги менее 0,2%, приливают к нему при температуре не выше 40 °C 141 мл тионилхлорида и нагревают ступенчато: 40— 45 мин при 62—65 °C; 1—Р/г ч при 70—75 °C; 30 мин до 80—83 °C и 3 ч при 80—83 °C (к концу реакции выделение
44
газообразных продуктов практически прекращается, т. пл. выделенного из пробы образца III должна быть не ниже 210°C). Реакционную массу охлаждают до 6—8°C, дают выдержку 2 ч, осадок III отфильтровывают, промывают 2 раза по 250 мл охлажденного до 6—8 °C дихлорэтана и высушивают при 80—100°C. Выход 221,4 г (68,1%).
М,М///-Дибензоил-М',М"-диспиротрипиперазиния дихлорид (IV). К охлаждаемым 194,5 г III в 1,07 л бензола прибавляют при температуре не выше 40 °C (процесс экзотер-мичен) 82 г триэтиламина, после чего массу кипятят 30 мин, охлаждают до 50—55 °C, добавляют 20 г угля и при 20 °C фильтруют. Бензол отгоняют при 80—85 °C, к остатку прибавляют 155 г этиленгликоля и массу нагревают 8 ч при 100—105 °C; температуру доводят до 25— 30 °C и разбавляют 715 мл ацетона. После выдержки 6 ч при 3—5 °C выделившийся осадок IV отфильтровывают, промывают 75 мл охлажденного до. 3—5 °C ацетона и высушивают при 70—80°C. Выход 94 г (50,29%), содержание основного вещества не менее 92%.
N',N"^HcnHpOTpHnHnepa3HHHH дихлорида дигидрохлорид (V). 350 г 1Укипятят2 ч с 1,5 л 13% соляной кислоты (обязательно отсутствие железа), охлаждают до 85—90°C и прибавляют 650 мл толуола. Перемешивают 5 мин и при температуре не ниже 75 °C (при более низкой температуре начинают выделяться кристаллы бензойной кислоты) водный слой отделяют, охлаждают до 2—5 °C и дают выдержку при этой температуре 15 ч. Кристаллы V отделяют центрифугированием, промывают 2 раза по 500 мл абсолютного изопропилового спирта (для отмывки остатков бензойной кислоты) и высушивают при 50—60 °C. Выход V в виде дигидрата 264 г (90%). Содержание основного вещества с учетом кристаллизационной воды не менее 98,6%.
М',№'-Диспиротрипиперазиния дихлорид (VI). К раствору 187 г дигидрата V (168 г безводный V) в 260 мл воды в течение 15—20 мин прибавляют 41,5 г моногидрата гидроокиси лития. Через 10—15 мин проверяют pH среды (8,5—9,0), обрабатывают 40 г нейтрального угля и фильтруют. Фильтрат выливают в 2,72 л охлажденного до 8— 10 °C метанола, дают выдержку при этой температуре и перемешивании 5 ч. Осадок отделяют, промывают охлажденным до 8—10°С метанолом (2—3 раза по 150 мл) и высушивают без обогрева. Выход VI 109,3 г (81 %).
М,М"'-Бис-(у-хлор-р-оксипропил)-М',М"-диспиротрипипе-разиния дихлорид (проспидин) (VII). 756 г VI растворя
.45
ют в 1 л воды и пр» 26—28 °C приливают 450 мл эпихлоргидрина (температуру поддерживают скоростью прибавления эпихлоргидрина и охлаждением массы). Дают выдержку 3 ч при 26—28 °C (при более высокой температуре происходит отщепление хлороводорода с образованием р-, у-эпоксипропильного производного), прибавляют 250 мл этилового спирта, 50 г угля, выдерживают еще 30 мин, фильтруют через угольную подушку. Уголь промывают этиловым спиртом и водой из расчета соотношения их 2,5:1. Общий объем фильтрата 4,5 л, pH раствора 6—7, плотность 0,97 г/мл. Срок хранения водноспиртового раствора проспидина не более 5 ч. Полученный раствор медленно выливают в 10 л ацетона, выдерживают 2 ч при 18—22°С и 7—10 ч при 4—6°С с периодическим перемешиванием. Осадок отделяют центрифугированием, промывают 2 раза по 500 мл охлажденного до 4—6°C ацетона и высушивают при 40—45 °C. Выход VII 827 г (65%).
ЭТОНИЙ
1,2-Этилен-бис-(Ы-диметилкарбодецилоксиметил)-аммония дихлорид:
•сн3 сн3
С,он2| ООССН,— N-(CH2)2— N-CH.COOC Н, 2С1* /Г’Х' I I 10 21
сн3 сн3
С30 He2Cl2N2O4Cl2
Мол. масса 585,74 Белый кристаллический порошок со специфическим запахом, слегка горький на вкус, вызывает чувство онемения языка, легко растворим в воде (pH 1% водного раствора 4,5—6,5), растворим в спирте, очень мало растворим в эфире, ацетоне, и хлороформе, т. пл. 161—165°С (в интервале 2°C); ФС 42-1006-75.
Этоний — катионный детергент, оказывающий бактериостатическое и бактерицидное действие [199]. Эффективен в отношении стрептококков, стафилококков и других микроорганизмов, подавляет действие стафилококкового токсина.
Применяется этоний наружно при стоматитах, гингивитах, глосситах, язвах роговицы, кератитах, отитах, пульпитах, кариесе зубов, язвенных поражениях кожи, а также при трофических язвах и лучевых поражениях кожи.
Выпускается в виде порошка. Цена 1 г 1 р.
46
Метод синтеза этония разработан в Институте органической химии АН УССР [199] по схеме:
HNICHJ, CICH2COOC|0H3I .1 3 *| 3
ClCHjCHjCl -(CH3)2N(CH2)jN(CH3>2-1----^CHjN-(CH2)2-N-CH3 2С1
I NaOH II CHj CH2
соос;0н2£оос|(н2,
III
При взаимодействии дихлорэтана (I) с водным раствором днметиламина и едким натром при нагревании под давлением получают тетраметилэтилендиамин (II), который обработкой дециловым эфиром хлоруксусной кислоты переводят в 1,2-этилен-бис-(N-диметилкарбодецилоксиме-тил)-аммония дихлорид (этоний) (III).
Тетраметилэтилендиамин (II). Смесь 4,8 л I, 12,7 л 42% водного раствора едкого натра и 27,5 л 33% водного раствора диметиламина нагревают при перемешивании в автоклаве 6 ч при 120—130 °C (давление 10—16 атм). После снижения давления ниже 10 атм реакционную массу охлаждают до 25 °C, выгружают и обрабатывают твердым едким натром до полного разделения слоев. II отделяют, сушат твердой щелочью и перегоняют на колонке. Собирают фракцию с т. кип. 118—123 °C. Выход 1,97 кг (35%).
1,2-Этилен-бис-(Ы-диметилкарбодецилоксиметил)-аммо-ния дихлорид (этоний) (III). Нагревают 45 л децилового эфира хлоруксусной кислоты, 12,7 л II и 25 л бензола 5 ч при 75—85 °C с перемешиванием. Затем'охлаждаютдо20°С, осадок отфильтровывают и перекристаллизовывают из 40 л метанола. Охлаждают до 0 (+5°C). Кристаллы отделяют, промывают 15 л эфира, высушивают при 45—50 °C в атмосфере азота. Выход 19,3 кг (33%).
КАРБОЦИКЛИЧЕСКИЕ И АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Важным направлением рассматриваемого раздела медицинской химии явилось создание и внедрение в производство за последнее время ряда противовирусных лекарственных препаратов [284, 296].
Первые достижения в этой области были связаны с исследованием производных адамантана (I) — углеводорода с жесткой, но ненапряженной структурой, включающего четыре конденсированных циклогексановых кольца в
47
конформации кресла, в котором атомы углерода имеют такое же расположение в пространстве, как и в ячейке кристаллической решетки алмаза (греч. абацаотаю— алмаз).
।
В 1964—1966 гг. появились первые сообщения о противовирусной активности 1-аминоадамантана гидрохлорида (мидантана, амантадина) (см. с. 53), эффективного в отношении вирусов гриппа типа А. Препарат нашел применение в качестве противовирусного средства при лечении гриппа (см. обзбры [38, 104, 194, 195]) и был воспроизведен в СССР в Институте органического синтеза АН ЛатвССР. Однако прн лечении азиатского гриппа в клиниках США вскоре было случайно обнаружено, что мидантан влияет на основные симптомы паркинсонизма, улучшая состояние больных [330]. Дальнейшие исследования подтвердили эффективность мидантана как средства для лечения паркинсонизма, влияющего на дофаминергические системы мозга, что существенно ограничило использование этого препарата в качестве противовирусного средства и мидантан в настоящее время назначают в основном при болезни Паркинсона и паркинсонизме различной этиологии [100, 102]. Вместе с тем высокая противовирусная активность мидантана послужила основанием для развертывания дальнейших поисковых работ в ряду различных производных адамантана.
Среди исследованных соединений наибольшая противовирусная активность в отношении вируса группы А была обнаружена у ремантадина (см. с. 54) [329]. Ремантадин воспроизведен в Институте органического синтеза АН ЛатвССР и выпускается химико-фармацевтической промышленностью СССР.
Систематические поиски противовирусных средств в новых направлениях проведены во ВНИХФИ. Теоретической основой этих оригинальных исследований явилась гипотеза об активности веществ, функциональные группы которых способны взаимодействовать с аминокислотными остатками белковой оболочки вируса [112]. В качестве таких веществ были предложены полнкарбонильные соеди
48
нения, причем по аналогии с другими химиотерапевтическими средствами предполагалось, что при введении в их молекулы атомов галогена противовирусное действие должно повыситься.
Экспериментальные исследования подтвердили эту гипотезу [196]. В результате .систематического изучения больших рядов разнообразных поликарбонильных и полиоксисоединений в медицинскую практику и химнко-фарма-цевтическое производство СССР внедрены оригинальные противовирусные препараты местного действия: оксолин (см. с. 57) [197], теброфен (см. с. 61) [58] и флоре-наль (см. с. 64) [58, 233].
Другое направление исследований в рассматриваемом ряду соединений — это получение веществ, оказывающих влияние на различные системы гомеостаза (функцию коры надпочечников, обмен в организме железа, липидов, холестерина и др.). Среди внедренных в химико-фармацевтическую промышленность СССР за последнее время лекарственных препаратов к этой группе относятся разнообразные медикаменты. Хлодитан (см. с. 67)—воспроизведенный Киевским НИИ эндокринологии и обмена веществ Министерства здравоохранения УССР ингибитор секреции кортикостероидов [107]. История создания препарата связана с изучением токсичности известных инсектицидов ДДТ (II), ДДД (III) и их аналогов. В ходе этих исследований было обнаружено значительное угнетающее действие о,п-изомера ДДД на функцию коры надпочечников, что и послужило основанием для изучения вещества в качестве ингибитора секреции глюкокортикоидов..
Цетамифен (см. с. 70)—воспроизведенный на производственном химико-фармацевтическом объединении «Здоровье» с участием Харьковского НИИ эндокринологии и химии гормонов гипохолестеринемический препарат [137]. Подобно другим производным фенилуксусной кислоты, он,
4 Заказ № 4304
49
по-видимому, тормозит биосинтез холестерина из эфиров уксусной кислоты, превращая часть коэнзима А в фенил-этилкоэнзим А, вместо оксиметилглутарилкоэнзима А, т. е. играет роль «ложного метаболита». Следует отметить, что при организации производства цетамифена в СССР был разработан метод егб.синтеза, основанный на использовании отходов от промышленного получения снотворного и противосудорожного препарата — фенобарбитала (IV).
К этой же группе относится оригинальное лекарственное средство ферроцерон (см. с. 72), разработанное в Институте элементоорганических соединений АН СССР и Ленинградском институте гематологии и переливания крови, предложенное для лечения железодефицитных анемий [171]. В отличие от других препаратов железа, широко применяющихся при анемиях, ферроцерон представляет собой индивидуальное водорастворимое железоорганическое соединение, производное ферроцена (V). Ферроцерон’ практически не вызывает побочных явлений и улучшает кроветворение даже в тех случаях, когда другие препараты железа не оказывают положительного действия.
История создания другого оригинального лекарственного препарата этой группы—цигерола (см. с. 74), разработанного в Институте органической химии АН СССР им. Н. Д. Зелинского [127], связана с поисками химиотерапевтических препаратов. Длительное время для лечения проказы использовалось хаульмугровое масло, действующим началом которого является хаульмугровая кислота (VI). Исследования показали, что аналогичной активностью обладают и другие циклоалканжирные кислоты, преимущественно С16—Ct8 ряда. При этом оказалось, что синтетические аналоги по действию на возбудителей лепры и туберкулеза в ’ряде случаев превосходят природные продукты. С намерением получить еще более активные вещества синтезировали циклогексилалкенкарбоновые кислоты, содержащие непредельные и разветвленные боковые цепи [127]. Соединение этого ряда — цигерол — оказалось малотоксичным и активным в отношении возбудителей туберкулеза, проказы, красной волчанки. При восстановлении боковой цепи активность уменьшалась. Дальнейшие исследования показали, что цигерол характеризуется не только антисептическим, но и ранозаживляющим действием. Он способствует регенерации нормальных тканей, ускоряет заживление ран, язв, ожогов и др., что дало основание для организации промышленного производства этого препарата.
50
VI
(CH2,I2C.OOH
Поиски синтетических местноанестезирующих средств, начавшиеся с варьирования структуры кокаина (VII),привели к выявлению активности у различных типов веществ, из которых практическое значение приобрели два класса соединений: диалкиламиноалкиловые эфиры ароматических кислот (VIII) й диалкиламиноациланилиды (IX) [244]. Из местных анестетиков первого типа химико-фармацевтическая промышленность СССР выпускает новокаин (VIII, R=NH2, R'=C2H5) [211], дикаин (VIII, R = h-C4H9NH; R'=CH3) [211], а также близкие к ним по структуре анёстезин (X) и совкаин (XI); из препаратов второго типа —ксикаин (IX, R = C2H5, Р'=2,6-ди-СН3) ’211] и тримекаин (IX, R=C2H5) Р'=2,4,6-три-СН3) i211].
Проведенные в. Институте фармакологии АМН СССР систематические исследования ариламидов N-замещенных азациклоалкан-2-карбоновых кислот (XII) [232] позволили выявить определенные закономерности связи между строением и местноанестезирующим действием. Как и в ряду других N-аминоациланилидов, активность соединений возрастает при последовательном введении метильных групп в положения 2,6 и 4 ароматического ядра. Переход от гексаметилениминовых (XII, и — 5) к пиперидиновым (XII, п=4) и далее к пирролидиновым (XII, п=3) производным сопровождается увеличением активности при инфильтрационной анестезии и снижением ее при поверхностной анестезии. В случае R = H вещества XII слабо
51
активны, введение И=СНз существенно повышает эффективность, которая возрастает с удлинением алкильной цепи от СНз до С4Н9. Усиление местноанестезирующего дей- ствия наблюдается и при дальнейшем увеличении алкильного заместителя R. Однако в этом случае резко возрастает разражающее действие соединений и ухудшается их растворимость в воде. Менее активны вещества XII с ( R-бензил или циклогексил. Эффективность соединений нормального и изорядов мало различается между собой. На основании этих исследований был отобран для медицинского применения новый оригинальный местный анестетик— пирромекаин (см. с. 77) [207], производство
которого освоено химико-фармацевтической промышленностью СССР.
Разносторонние исследования алкаминовых эфиров 4 диарилжирных кислот, синтезированных по принципу j
«утяжеления» кислотной части молекулы ацетилхолина,
на протяжении ряда лет проводились во ВНИХФИ [129, 133], Институте тонкого органического синтеза АН :
АрмССР [2], Институте экспериментальной медицины |
АМН СССР [67]. Результатом ранее выполненных работ явилось создание оригинальных отечественных холино- и спазмолитиков — тифенаГ' (XIII), апрофена (XIV), дипрофена (XV), метацина (XVI), арпенала (XVII), которые наряду с воспроизведенными препаратами этого типа — спазмолитином (XVIII) и амизилом (XIX) выпускаются химико-фармацевтической промышленностью СССР [211].
с«з ’
<CeH5>2CHCOS <CH2>2n<C2H5>2 н°1 • (СеН5)2ССОО(СН2) N(C Н )2. НС1 XIH 1 ! XIV
1 он
(C^CHCOS (CH2i2N(C Н ) - HCl (С Н ) А-coo (CH ) N(CH. )3Г '
xv XVI
(CVVjCHCOO(CH2)3r;i<C2H5>2’ HC1 (C6H512CHCOO<CH2>2N<C2H5>2 ,НС‘ xvii xviii
он >
(СвН5’2ССО° (СН2’2Н(С2Нь’2' HCt I
XIX I
Дальнейшие исследования во ВНИХФИ привели к со- '
зданию анальгезирующего и противокашлевого препарата !
эстоцина (см. с. 79) [130, 269], а работы в Институте i экспериментальной медицины АМН СССР — оригинально- |
52
го центрального холинолитика метамизила (см. с. 83) [67, 114, 115], производство которых организовано на заводах химико-фармацевтической промышленности СССР.
МИДАНТАН
1-Аминоадамантана гидрохлорид
Ci0H17N HC1
Мол. масса 187,71
Синонимы: амантадина гидрохлорид, амандин, аман-тан, антадин, атарин, вирегит, виросол, вирофрал, габи-рол, десфакс, инфлуенол, мантадан, мантадикс, мантидан, парамантин, профил, симметрел, фиелтон, флувиатол.
Белый кристаллический порошок, горького вкуса, с легким запахом, растворим в воде и хлороформе, легко растворим в спирте, практически нерастворим в эфире; т. разл. 370 °C; ФС 42-506-72.
Мидантан — противопаркинсонический препарат, влияющий на дофаминергические системы мозга [100, 102, 330]. Наряду со стимулированием выделения дофамина из нейрональных депо и повышением чувствительности дофаминергических рецепторов препарат, по-видимому, тормозит генерацию импульсов в. моторных нейронах ЦНС. Действие мидантана развивается быстро, и лечебный эффект проявляется уже в течение первых дней. Препарат эффективен в отношении вирусов гриппа типа А. Однако высокая активность в отношении дофаминергических систем существенно ограничила использование его в качестве противовирусного средства.
Применяют мидантан при болезни Паркинсона и паркинсонизме разной этиологии. Препарат эффективен главным образом в отношении акинеторигидного синдрома, значительно меньше влияет на гиперкинетический синдром (тремор).
Выпускается в таблетках, покрытых оболочкой, по 0,1 г N. 100. Цена 8 р. 02 к.
Метод синтеза мидантана разработан в Институте органического синтеза АН ЛатвССР [100] по схеме:
53
При взаимодействии 1-бромадамантана (I) с формамидом по реакции Лейкарта получают 1-формиламиноадамантан (II) [297], который гидролизом с соляной кислотой превращают в 1-аминоадамантана гидрохлорид (мидантан) (III) [337].
1-ФормиламиноадаМантан (II). Смесь 1 кг I и 3,2 л формамида нагревают 1 ч при 175—185 °C и оставляют без нагревания на 30 мин. Охлажденную массу прибавляют к 9,65 л воды с температурой 5 °C, дают выдержку 3 ч при 5—10 °C, осадок отделяют, промывают 5 л воды, охлажденной до 5°С, и влажный продукт II (1,5—1,7 кг с содержанием влаги 60—65%) направляют на следующую стадию.
1-Аминоадамантана гидрохлорид (мидантан) (III). К полученному на предыдущей стадии II приливают соляную кислоту из расчета 4 кг 16—20% соляной кислоты на 1 кг 100% II (при использовании соляной кислоты большей концентрации образуется значительное количество 1-хлорадамантана, при снижении концентрации соляной кислоты увеличиваются потери III за счет его растворимости) и смесь кипятят 1 ч (при этом осадок II полностью растворяется). Массу охлаждают до 0°С и дают выдержку 1 ч. Осадок технического III отделяют, сушат при 80°C до содержания влаги 5% и перекристаллизовывают из десятикратного количества абсолютного изопропилового спирта с добавкой активированного угля. Изопропаноль-ный раствор охлаждают до 0—(—5°C), дают выдержку 2 ч, осадок отфильтровывают, промывают изопропиловым спиртом (2 раза по 100 мл) и высушивают при 80 °C до содержания влаги не выше 0,5%. Выход III 395,5 г (45,4%, считая на I). \
РЕМАНТАДИН
1 (а-Аминоэтил)-адамантана гидрохлорид, [а-метил-1-адамантилметиламина гидрохлорид]:
Л><Ч<-л“ <?н~снз
I NH2 HCt
ci2H2iN'HCl Мол. масса 215,77
54
Основные синонимы: римантадин, мерадан.
Белый кристаллический порошок без запаха, горького вкуса, трудно растворим в воде, растворим в спирте, легко растворим в хлороформе; т. пл. не ниже 360 °C; ВФС 42-501-76.
Ремантадин — противовирусный препарат [201, 329], активный в отношении штаммов вирусов гриппа типа А, особенно при лечении в первые 1—2 дня болезни. Применяют ремантадин в качестве химиотерапевтического средства при гриппе типа А, особенно А2, у взрослых.
Выпускается в таблетках по 0,05 г N. 20. Цена 1 р. 52 к.
Метод получения ремантадина разработан в Институте органического синтеза АН ЛатвССР [201] по схеме:
Бромированием адамантана (I) бромом в четыреххлористом углероде в присутствии катализатора — меди — получают 1-бромадамантан (II), из которого по реакции Коха-Хааера, т. е. путем карбоксилирования окисью углерода, образующейся прн взаимодействии олеума с муравьиной кислотой, синтезируют 1-адамантанкарбоновую кислоту (Ш). Кислоту III обработкой тионилхлоридом переводят в хлорангидрид, который без выделения вводят во взаимодействие с этоксимагниймалоновым эфиром с последующим гидролизом и декарбоксилированием. Синтезированный таким путем 1-ацетиладамантан (V) подвергают восстановительному аминированию по Лейкарту— Валлаху формамидом с муравьиной кислотой и в условиях кислотного гидролиза с соляной кислотой переводят в 1-(а-аминоэтил)-адамантана гидрохлорид — ремантадин (VI).
Рассматриваемый метод выгодно отличается от ранее описанного в литературе, где перевод соединения V
55
в ремантадин осуществляется через соответствующий оксим с восстановлением $tq. алюмогидридом лития в тетрагидрофуране.
1-Бромадамантан (II). К 220 г высушенного I прибавляют 6,47 г медного порошка, 120 мл четыреххлористого углерода и при перемешивании прикапывают 1035 г брома; масса разогревается до 56—60 °C. Дают выдержку при этой температуре 6 ч, после чего массу упаривают. Остатки брома удаляют прибавлением 30 мл четыреххлористого углерода с последующей отгонкой. Остаток растворяют в 170 мл кипящего изопропилового спирта, следы брома удаляют добавлением 6 г сульфита натрия, массу охлаждают до 0—(—10 °C) и оставляют кристаллизоваться при этой температуре на 12 ч. Продукт отфильтровывают, промывают охлажденным изопропиловым спиртом (3 раза по 230 мл), затем 0,5% водным раствором трилона Б (до отсутствия тяжелых металлов) и высушивают при 35—45°C. Выход 222,1 г (63,4%).
1-Адамантанкарбоновая кислота (III). К охлажденной до 20 °C смеси 600 мл конц. серной кислоты и 600 мл олеума прибавляют 420 г II и медленно при 30—32°C приливают 350 мл муравьиной кислоты (при температуре выше 34 °C образуются побочные продукты — адамантанполикарбоновые кислоты, оксикислоты и смолистые продукты, при температуре ниже 20°C реакция не идет). Масса вспенивается, особенно в начале процесса. После выдержки Г ч при 30—Й2 газообразные продукты отдувают и массу выливают в 2,4 л охлажденной до 5 °C воды, поддерживая температуру не выше 60 °C (процесс экзотермичен и сопровождается вспениванием). После выдержки 1 ч при 18—20 °C осадок III отфильтровывают, промывают 2 л воды, растворяют при нагревании до 80— 90°C в 7 л водного раствора, содержащего 260 г едкого натра. Контролируют pH (8—9) и горячий раствор отфильтровывают (при pH выше 9 масса при фильтрации может закристаллизоваться). Профильтрованный раствор при 70 °C подкисляют конц. серной кислотой до pH 3, охлаждают до 18—20°C. Осадок III отфильтровывают, промывают дистиллированной водой до нейтральной реакции и сушат при 40—50°C до содержания влаги не более 1%. Продукт должен иметь т. пл. в интервале 165—175 °C. Выход 273 г (77,5%).
1-Ацетиладамантан (V). К смеси 537,5 г III, 750 мл бензола (влаги не более 0,02%) и 21,2 мл пиридина (вла
56
ги не более 0,2%) при перемешивании приливают 700мл тионилхлорида. Массу осторожно нагревают до кипения и кипятят 4 ч, затем избыток тионилхлорида отгоняют с бензолом до температуры в массе 85 °C, охлаждают до 50 °C и приливают еще 2 раза по 600 мл сухого бензола с последующей его отгонкой (для более полного удаления остатков тионилхлорида, примеси которого в ходе дальнейшего синтеза способны разлагать магнийорганические соединения, поэтому содержание тионилхлорида в продукте реакции не должно превышать 2 г/л). Полученный IV растворяют в 1 л сухого бензола и прибавляют при 70— 75 °C постепенно к этоксимагниймалоновому эфиру, приготовленному по обычному методу из 80 г магния, 240 г четыреххлористого углерода, 120 мл малонового эфира, 400 мл абсолютного этилового спирта и 1,54 л бензола. Кипятят 2 ч, охлаждают до 20 °C и выливают в охлажденные до 5—10°С_3 л воды, после чего приливают 2,5 л 50% серной кислоты. Водный слой отделяют и дополнительно экстрагируют 750 мл бензола. Объединенные бензольные экстракты промывают 1,5 л воды, бензол отгоняют. К остатку приливают 2,6 л уксусной кислоты, 1,15 л воды и 250 мл конц. серной кислоты. Массу кипятят 5 ч, охлаждают до "20 °C и экстрагируют 3 раза по 2,2 л бензола. Бензольные экстракты промывают 1,5 л воды. Бензол отгоняют, остаток закристаллизовывается. Продукт содержит 50—65% основного вещества V и 10—15% примеси кислоты III. Выход V 638,4 г (50,5%).
1-(а-Аминоэтил)-адамантана гидрохлорид (ремантадин (VI) . Смесь 980 г формамида, 414 мл 99% муравьиной кислоты и 300 г технического V нагревают 6 ч при 170—175°С, охлаждают до ПО °C и выливают на 2 л горячей воды. Экстрагируют 1,8 л и 100 мл бензола. Бензольные экстракты упаривают; к остатку приливают 522 мл конц. соляной кислоты и 480 мл воды, кипятят 5 ч. Охлаждают до 20 °C,. прибавляют 480 мл бензола, осадок отфильтровывают, высушивают при 40—50 °C до содержания влаги не более 1% и перекристаллизовывают из 13,6 л абсолютного изопропилового спирта с добавлением угля. Выделение и фильтрацию ремантадина ведут при —2— (—4°C). Продукт сушат при температуре не выше 50°C. Выход 248 г (45%).
57
оксолин
1,2,3,4-Тетрагидро-1,2,3,4-тетраоксонафталина дигидрат:
Мол. масса 224,17
Белый или белый с кремовым оттенком кристаллический порошок без запаха, легко растворим в ацетоне и воде (водные растворщ^нестойки, в щелочной среде быстро темнеют), мало растворим в эфире, практически нерастворим в хлороформе и хлористом метилене, т. разл. 129—133°C (в интервале 2°С). Препарат чувствителен к свету, влаге, температуре и легколетучим основаниям, хранится в холодильнике при +4°С в плотно закрытых склянках темного стекла; ФС 42-993-75.
Оксолин — оригинальный противовирусный препарат [195, 197], Обладает вирулицидной активностью, эффективен при вирусных заболеваниях глаз, кожи, ринитах, оказывает профилактическое действие при гриппе.
Применяют оксолин при кератоконъюктивитах, кератитах, вызванных вирусами герпеса простого и герпеса зостер, а также для лечения пузырькового простого лишая, бородавок, контагиозного моллюска, герпетиформного дерматита Дюринга, чешуйчатого лишая, ринитов вирусной этиологии, а также для профилактики гриппа.
Выпускается в виде мази в алюминиевых тубах: 0,25% Ю г (цена 20 к.) и 3% 30 г (цена 56 к.).
Метод синтеза оксолина разработан во ВНИХФИ [197] по схеме:
58
Восстановлением Натриевой соли 2-оксинафтил-1-азо-бензол-4-сульфокислоты (красителя оранжевого кислотного) (I) гидросульфитом натрия в водной среде получают 1-амино-2-нафтол (II) [218], который в виде гидрохлорида окисляют хлорным железом в водной среде до ₽-наф-тохинона (III) [218]. Дальнейшее окисление III гипохлоритом натрия приводит к изонафтазарину (IV), а окисление IV хлором — к 1,2,3,4-тетрагидро-1,2,3,4-тетраоксо-нафталину дигидрату (оксолину) (V). Использованный в синтезе метод [218] восстановления I в 1-амино-2-нафтол (II) имеет ряд преимуществ для промышленности по сравнению с другими описанными методами, где в качестве восстановителей для превращения I в IJ применяют цинковую пыль в соляной кислоте, сернистый натрий в нейтральной или щелочной среде, каталитические или электрохимические методы. Хлорное железо для окисления II в III имеет преимущество по сравнению с другими окислителями, например с хромовой кислотой, поскольку даже при избытке хлорного железа не происходит дальнейшего окисления 'Р-нафтохинона (III) до фталевой кислоты. Используемый в синтезе метод превращения р-нафтохинона (III) в изонафтазарин (IV) с гипохлоритом натрия аналогичен описанному ранее процессу с применением хлорной извести, но исключает необходимость фильтрования раствора хлорной извести и позволяет точнее дозировать количество окислителя. Другие описанные в литературе способы получения изонафтазарина (IV) из 2,3-диоксинафталина через 2,3-диокси-1,4-диаминонафта-лин, из 2-окси-3-амино-1,4-нафтохинона, из 4,4-дихлор-1,2,3-триоксотетрагидронафталина и нз 2-окси-1,4-нафто-хинона являются более сложными и не имеют преимуществ. Недостатком описанных в литературе способов получения 1,2,3,4-тетрагидро-1,2,3,4-тетраоксонафталина дигидрата (V) является применение в качестве окислителя вместе с хлором азотной кислоты и проведение процесса в среде уксусной кислоты.
1-Амино-2-нафтол (II). Растворяют 1,648 кг I в 16 л воды при перемешивании и нагревании до 50 °C. К полученному раствору оранжево-красного цвета прибавляют в один прием 1,68 кг гидросульфита натрия, перемешивают 30 мин при 50 °C. Конец реакции контролируют по изменению окраски раствора до темно-розового и выпадению II в осадок. Вещество отфильтровывают, промывают водой до отсутствия в промывной воде сульфаниловокислого натрия и полученную пасту направляют на сле
59
дующую стадию. П неустойчив, на воздухе легко окисляется, поэтому процессы фильтрации и промывки необходимо проводить быстро.
0-Нафтохинон (Ш). К смеси 16 л воды и 460 мл технической соляной кислоты прибавляют полученную На предыдущей стадии пасту II (обратный порядок прибавления приводит к осмолению) и 75 г угля. Массу нагревают 15 мин при 80 °C и фильтруют. Фильтрат охлаждают до 14 °C и в один прием быстро приливают к нему при перемешивании охлажденный до 14 °C раствор 2,28 кг хлорного железа в 1,68 л воды (в случае медленного приливания окислителя образующийся III взаимодействует с гидрохлоридом II, превращаясь в побочные окрашенные соединения). При этом сразу же выпадает желто-зеленого цвета осадок III, который после перемешивания в течение не более 6 мин отфильтровывают и промывают на фильтре водой до pH промывных вод 6—7. Влажную пасту III сразу же передают на следующую стадию, поскольку при хранении вещество разлагается.
Изонафтазарин (IV). Влажную пасту III, полученную на предыдущей стадии, смешивают с 8 л воды и к однородной суспензии прикапывают в течение 5—6 мин 5 л водного раствора гипохлорита натрия с содержанием активного хлора 52 г/л и pH 9,3—10,5. Дают выдержку 10 мин и раствор отфильтровывают от примесей. К фильтрату при перемешивании прибавляют 475 мл ледяной уксусной кислоты и 85 мл технической соляной кислоты. Реакционную массу с pH 2 нагревают 10 мин при 80°С, охлаждают до 60 °C. Осадок IV отфильтровывают, растворяют в 4,8 л воды с добавкой 100 мл 42% раствора технического едкого натра при 80—85 °C. Темно-синий интенсивно окрашенный раствор чистят углем (28 г), фильтруют и к горячему (78—80°C) фильтрату приливают при перемешивании техническую соляную кислоту (~ 172 мл) до pH 3,0—2,5. Выпавший ярко-красного цвета осадок IV отфильтровывают после охлаждения суспензии до 18— 20°С, промывают 200 мл воды и сушат при 60—80°. Получают 260 г IV с т. разл. 276—277 °C. Выход 68,4%, считая на I. В виде пасты вещество неустойчиво и должно быть сразу использовано на следующей стадии.
1,2,3,4-тетрагидро-1,2,3,4-тетраоксонафталина дигидрат (оксолин) (V). Во взвесь 1,52 кг IV в 6,72 л дистиллированной воды при 18°С и перемешивании пропускают хлор со скоростью около 2 л/мин. Температура массы постепенно повышается до 40—42°С и в даль
60
нейшем ее поддерживают на этом уровне, используя охлаждение. Реакцию заканчивают после полного растворения IV. Раствор обрабатывают 152 г угля с перемешиванием 30 мин при 40—42 °C, фильтруют. Фильтрат охлаждают и перемешивают 1—1 ’/а ч при 5 °C. Осадок тетрагидрата 1,2,3,-4-тетрагидро-1,2,3,4-тетраоксонафталина отфильтровывают, промывают 500 мл охлажденной до5°С дистиллированной воды и сушат путем азеотропной отгонки воды с хлористым метиленом при кипячении в течение 10—12 ч до полного отделения воды. Затем массу охлаждают до 15—20 °C, V отфильтровывают, промывают 1 л хлористого метилена и высушивают продуванием воздуха на фильтре до отрицательной реакции на хлор по Бель-штейну. Получают 1,414 кг V. Выход 78,9%, считая на IV.
ТЕБРОФЕН
3,5,3',5'-Тетрабром-2,4,2',4'-тетраоксидифенил
Мол. масса 533,85
Белый с серовато-кремовым оттенком кристаллический порошок, практически нерастворим в воде, растворим в кипящей уксусной кислоте (1:36), ацетоне (1:3), этиловом спирте (1:27, при нагревании 1:13), изопропиловом спирте (1:100), в водных растворах щелочей, нерастворим в хлороформе и хлористом метилене. Растворы в ацетоне, спирте, щелочах неустойчивы, быстро темнеют; т. пл. 283—285 °C разл.; ФС-42-1066-76.
Теброфен — оригинальный противовирусный препарат [55, 195]. Активен в отношении герпеса простого, герпеса зостер и аденовирусов.
Применяют теброфен при лечении эпидемического аденовирусного конъюнктивита и кератоконъюнктивита, вирусного кератита, а также заболеваний кожи вирусной или предполагаемой вирусной этиологии (простой пузырьковый и рецидивирующий герпес, опоясывающий лишай, красный плоский лишай, контагиозный моллюск и др.).
61
Может быть использован при плоских вульгарных бородавках у детей.
Выпускается в виде глазной мази 0,25%, 0,5% и 1% по 10 г (цейа 20 к., 22 к. и 25 к.) и мази 2%, 3% и 5% по 30 г (цена 50 к., 74 к. и 1 р. 22 к.) для применения в дерматологии.
Метод синтеза теброфена разработан во ВНИХФИ [55] по схеме:
Бромированием резорцина (I) в водной среде получают пентабромрезорцин (II). Нагревание II в бромбензоле при 143—145 °C приводит к бис-(3,5,5-трибром-4,6-диок-социклогексен-2-илидену) (трибромрезохинону) (III) с более высокими и стабильными выходами, чем аналогичная реакция, проведенная без растворителя, или получение III путем окисления трибромрезорцина бромом. Восстановлением III гидросульфитом натрия в 45% водном изопропиловом спирте получают 3,5,3',5'-тетрабром-2,4,2',4'-тет-раоксидифенил (теброфен) (IV). Описанные ранее методы восстановления III оловом в соляной кислоте, сероводородом, цинком в соляной кислоте и др. дают худшие результаты.
Пентабромрезорцин (II). К охлажденным до 4 °C 5,78 л воды приливают в течение 15 мин 2,9 кг брома и затем по каплям, поддерживая температуру не выше 10 °C, при перемешивании раствор 385 г I в 770 мл воды (обратный порядок прибавления приводит к загрязнению II другими продуктами бромирования резорцина), перемешивание продолжают еще 15 мин при 10 °C. К полученной суспензии II приливают 5 л перегнанного бромбензола (неперегнанный продукт содержит примесь дибромбензо-
ла, которая загрязнит II) и перемешивают 20 мин. Бромбензольный слой, содержащий II, отделяют, промывают 2 раза по 1 л воды, чистят 50 г угля и передают на следующую стадию без выделения II.
Бис-(3, 5„ 5-трибром-4, 6-диоксоциклогексен-2-илиден) (трибромрезохинон) (Ш). Бромбензольный раствор II, полученный на предыдущей стадии, нагревают 30 мин при перемешивании и температуре 143—145°C (реакция сопровождается выделением брома), после чего растворитель полностью отгоняют в вакууме (сначала при остаточном давлении 400 мм, затем при 20—25 мм). К охлажденному остатку приливают 4,5 л хлористого метилена, раствор перемешивают и охлаждают 30 мин при —5°С. Осадок III отфильтровывают, промывают 2 раза по 0,5 п и 0,35 л охлажденного до >— 5 °C хлористого метилена, высушивают при 20°C. Получают 550 г III с содержанием основного вещества 99,89%; т. пл. 197—200 °C. Выход 45,7%, считая на I.
3,5,3',5'-Тетрабром-2,4,2',4'-тетраоксидифенил (тебро-фен) (IV). К суспензии 500 г III в 3,31 л 44,5% водного изопропилового спирта при 24 °C и энергичном перемешивании прибавляют в один прием 473 г гидросульфита натрия (прибавление восстановителя порциями приводит к получению некачественного IV); обратный порядок прибавления вызывает образование вместо IV другого вещества неустановленного строения. Реакция экзотермична и температура повышается до 43—45 °C. Дают выдержку при этой температуре с перемешиванием 15 мин (выделяется сернистый газ, масса из оранжево-красной становится сначала темно-зеленой, затем белой) и выливают при перемешивании в 14,5 л воды. Через 5 мин осадок IV отфильтровывают, промывают водой (5 раз по 0,5 л) до отсутствия кислой реакции в промывной воде и растворяют в смеси 2,5 л ацетона и 1,5 л воды с добавкой 35 г угля и 8 г гидросульфита натрия. Раствор фильтруют и выливают в 12,5 л, нагретой до 45 °C воды, при этом IV выпадает в осадок. Массу перемешивают 10 мин, осадок IV отфильтровывают, промывают 2 раза по 0,25 л дистиллированной воды, снова растворяют в 2,5 л ацетона. Раствор обрабатывают 20 г угля, фильтруют и выливают в 14 л нагретой до 45 °C дистиллированной воды. Суспензию перемешивают 10 мин. Осадок IV отфильтровывают, промывают 2 раза по 0,25 л дистиллированной воды и сушат при 80°С. Выход 320 г (82%, считая на III).
63
ФЛОРЕНАЛЬ
Бисульфитное соединение 2-флуоренонилглиоксаля
Мол. масса 358,30
Желтого или'зеленовато-желтого цвета кристаллический порошок без запаха, горького вкуса, трудно растворим в воде, практически нерастворим в спирте, растворим в 85% муравьиной кислоте; ФС 42-1098-77.
Флореналь — оригинальный противовирусный препарат [58, 233]. Активен в отношении вируса герпеса простого и аденовирусов.
Применяют флореналь при лечении вирусных заболеваний глаз — герпетического кератита и аденовирусного кератоконъюнктивита.
Выпускается в виде 0,25% и 0,5% мази по 10 г. Цена 20 к. и 22 к.
Метод синтеза флореналя разработан во ВНИХФИ [58] по схеме:
Ацилированием флуорена (I) уксусным ангидридом по Фриделю — Крафтсу получают 2-ацетилфлуорен (II). В отличие от описанных реакций в сероуглероде или в нитробензоле для производства удобнее оказалось проведение
64
процесса в хлорбензоле. Окисление II до 2-ацетилфлуоре-нона (Ш) ведут бихроматом натрия в уксусной кислоте (в отличие от описанного ранее метода лучшие результаты дало осуществление реакции не при 20 °C, а при 90°C). Бромированием III в хлороформе при освещении получают 2-(©-дибромацетил)флуоренон (IV). Ацилирование I по Фриделю — Крафтсу дихлорацетилхлоридом позволяет получить с высоким выходом 2-(©-дихлораце-тил) флуорен, который может быть окислен бихроматом натрия в 2-(и-дихлорацетил) флуоренон, пригодный для дальнейшего синтеза флореналя, только при использовании дихлоруксусной кислоты высокой степени чистоты, приготовление которой в промышленном производстве оказалось нерентабельным. Нагреванием дибромацетильного производного IV с морфолином при 50 °C, получают 2-(©-диморфолиноацетил) флуоренон (V), а кислотным гидролизом V — 2-флуоренонилглиоксальгидрат (VI), что имеет преимущество перед ранее описанным . получением VI окислением III двуокисью селена. Опыты превращения IV в VI прямым гидролизом с серной кислотой, использованием реакций с едким кали в спирте, алкоголятов в спиртах или в диметилформамиде, . ацетата калия или анилина в различных условиях не дали положительных J результатов. Не дало хороших результатов и превращение III через монохлорпроизводное в 2-(©-морфолиноацетил) флуоренон с последующим окислением солями двухвалентной ртути. Обработка VI бисульфитом натрия приводит к бисульфитному производному 2-флуоренонйлгли-оксаля (флореналю) (VII).
2-Ацетилфлуорен (II). В раствор 664 г I в 11,3 л све-жеперегнанного хлорбензола загружают 1,08 кг безводного хлористого алюминия и постепенно при перемешивании прикапывают 420 мл уксусного ангидрида с такой скоростью, чтобы температура реакционной смеси не превышала 52 °C. После выдержки 1 ч при 48—52 °C охлаждают до комнатной температуры и образовавшийся комплекс разлагают путем выливания на смесь 2 л воды и 3 л 3,5% соляной кислоты, регулируя скорость прибавления так, чтоб температура массы не поднималась выше 50 °C. При более высокой температуре происходит частичное осмоление. Хлорбензольный слой отделяют, растворитель отгоняют при 50—55°C (100 мм рт. ст.). Остатки хлорбензола удаляют отгонкой с водой. II без дополнительной очистки используют на следующей стадии.
2-Ацетилфлуоренон (111). Продукт II, полученный на
5я Заказ- № 4304-
65
предыдущей стадии, растворяют в 11,6 л уксусной кислоты при 85—90°С и При перемешивании добавляют порциями 3,7 кг бихромата натрия в течение 40—50 мин при 85—90 °C. Дают выдержку при этой температуре 4 ч и отгоняют при 50—55 °C (100 мм рт. ст.) 7 л уксусной кислоты, после чего остаток разбавляют 16 л нагретой до 60 °C воды. Образовавшуюся суспензию перемешивают 20 мин и охлаждают до 20°C. Осадок III отфильтровывают, промывают 2°/о серной кислотой до бесцветного промывного раствора и водой до отсутствия кислой реакции. Для отделения примеси побочного продукта — флуоренон-2-карбоновой кислоты — осадок перемешивают 30 мин при 85—90°C с 400 мл 42% раствора едкого натра и 4,7 л воды, отфильтровывают, промывают водой и высушивают, после чего для отделения II и неорганических солей перекристаллизовывают из 9 л бензола с добавкой 60 г угля. Получают 430 г III. Подпариванием маточных бензольных растворов дополнительно получают 100 г III. Общий выход 530 г (59,6%, считая на I).
2-(©-Дибромацетил)флуоренон (IV). К раствору 530г III в 7,1 л хлороформа при нагревании до 45 °C в течение 37г ч прикапывают при перемешивании и освещении (лампа 150 Вт) раствор 890 г брома в 21 л хлороформа. После выдержки 37г ч в тех же условиях массу охлаждают до 20 °C и дают выдержку еще 3 ч. Осадок IV отфильтровывают, промывают 2 раза по 0,5 л хлороформа и высушивают при температуре не выше 40°C. Получают 805 г IV с т. пл. 207,5—208,5 °C, содержащего не более 3% 2-(ю-монобромацетил)флуоренона. Выход 86%.
2-(©-Диморфолиноацетил)флуореион (V). К 2,4 л морфолина прибавляют порциями 805 г IV при перемешивании в течение 1 ч с такой скоростью, чтобы температура не поднималась выше 50 °C (при более высокой температуре возможно осмоление). Дают выдержку 37г ч при 50 °C, контролируя методом ТСХ содержание IV (должно остаться не более 0,5%), затем 1 ч при 5 °C. Реакционную массу разбавляют 1,6 л охлажденного до 5 °C метанола, перемешивают 15 мин и осадок отфильтровывают. Продукт для отделения от смолистых примесей промывают путем перемешивания с 900 мл метанола в течение 30 мин, отфильтровывают и для отделения гидробромида морфолина промывают перемешиванием 30 мин с 4 л воды при 20°C. Вновь отфильтровывают, промывают на фильтре 2 раза по 0,5 л воды и без высушивания (вещество лабильно) используют на следующей стадии.
66
/г '
2-Флуоренонилглиоксальгидрат (VI). Влажную пастуУ от предыдущей стадии смешивают с 2,2 л воды в течение 20—25 мии и к полученной суспензии при 20°C прибавляют по каплям 2,7 л 20% раствора серной кислоты с такой скоростью, чтобы температура реакционной смеси не была выше 60 °C (реакция экзотермична). Затем массу перемешивают еще 30 мин при 55—60 °C и 3 ч при 20 °C. Осадок отфильтровывают, промывают водой до отсутствия в промывной воде кислой реакции на конго и сушат при 80 °C. Получают 445 г технического VI с содержанием основного вещества 98,4%, который очищают переводом через бисульфитное производное VII. Для этого продукт кипятят 30 мин с 8,65 л 60% этилового спирта и 23 г угля. Раствор отфильтровывают и обрабатывают при перемешивании и температуре 70—75°C 400 мл 50% раствора бисульфита натрия. Массу перемешивают еще 10 мин и охлаждают до 20 °C. После выдержки в течение 2 ч осадок отфильтровывают, промывают 450 мл спирта, высушивают при 35 °C и растворяют в течение 10 мин (увеличение времени вызывает частичное разложение) в предварительно нагретых до 80 °C 5,7 л воды с добавкой 28 г угля. Горячий раствор фильтруют и при 65—70 °C прибавляют 1,14 л 40% серной кислоты. Массу нагревают до 95 °C, дают выдержку 6 ч и охлаждают до 20 °C. Осадок VI отфильтровывают, промывают водой до отсутствия кислой реакции по конго и высушивают при 80°C. Получают 355 г VI с содержанием основного вещества 100% и с т. пл. 193—195°C.’Выход 68,3%, считая на IV.
Бисульфитное производное 2-флуоренонилглиоксаля (флореналь) (VII). Растворяют 355 г VI в 7 л 60% этилового спирта при кипении в течение 20 мин. Раствор обесцвечивают 18 г угля и при 70—75°С прибавляют к нему с перемешиванием 317 мл 50% раствора бисульфита натрия. Реакционную массу перемешивают при 70—75°C 15 мин, охлаждают до 20 °C и оставляют на 2 ч. Выпавший VII отфильтровывают, промывают 350 мл этилового спирта, 350 мл ацетона и высушивают при температуре не выше 35°С. Получают 460 г VIII. Выход 91,8%. •
5*
ХЛОДИТАН
1- (о-Хлорфенил) -1- (п-хлорфенил) -2,2-дихлорэтан
Мол. масса 320,05 Синонимы: о,п'-ДДД, лизодрен, митотан.
Белый кристаллический порошок, без запаха, практически нерастворим в воДе, растворим в спирте, хлороформе и ацетоне; ВФС 42-445-75.
Хлодитан — ингибитор функции коры надпочечников [107], подавляющий секрецию кортикостероидов, блокирует действие кортикотропина и может вызвать деструктивные изменения нормальной и опухолевой ткани коры надпочечников.
Применяют хлодитан для подавления избыточной секреции глюкокортикоидов при болезни Иценко — Кушинга, при иноперабельных опухолях коры надпочечников (кортикостеромах), сопровождающихся усиленным образованием кортикостероидов, а также после хирургического удаления кортикостером при повышенном уровне глюкокортикостероидов в организме. Выпускается в таблетках по 0,5 г N. 100. Цена 17 р. 04 к.
Метод синтеза хлодитана разработан в Киевском научно-исследовательском институте эндокринологии и обмена веществ [17] по схеме:
Взаимодействием о-Хлорбензальдегида (I) с хлороформом в среде жидкого аммиака в присутствии едкого кали получают о-хлорфенилтрихлорметилкарбинол (II), который восстанавливают амальгамой алюминия в спирте до о-хлорфенилдихлорметилкарбинола (III), превращаемого
68
конденсацией с хлорбензолом в присутствии 4 % олеума в 1- (о-хлорфенил) -1- (п-хлорфенил) -2,2-дихлорэтан (хлоди-тан) (IV). Этот метод имеет существенные преимущества по сравнению с первыми способами получения IV, основанными на использовании IV, содержащимся в количествах 5—15% в техническом 2,2-бис-(п-хлорфенил)-1,1-дихлорэтане (ДДД). Извлечение IV из технического ДДД методом тонкослойной хроматографии или по избирательной растворимости его в органических растворителях позволяло получать только небольшие количества IV, обычно загрязненного примесью ДДД. Синтез II ранее был осуществлен путем взаимодействия о-хлорфенилмагнийбро-мида с хлоралем [298], восстановления о-хлорфенилтри-хлорметилкетона [278] и присоединения хлороформа к I в присутствии оснований [286]. Однако выходы II по этим методам не превышали 30%. Исследование условий конденсации I с хлороформом в присутствии едкого кали показало, что при молярном соотношении этих реагентов 1 :8:6 и проведении процесса в жидком аммиаке выходII достигает 93% [17].
Синтез III с выходами 25—42% был описан из о-хлор-фенилмагний бромида и дихлорацетальдегида и восстановлением II амальгамой алюминия в спирте. Подбор условий последней реакции позволил увеличить выход III до 92% [16, 17]. Получение IV путем конденсации III с хлорбензолом ранее проводилось в присутствии 100% серной, борфтористоводородной кислот или полифосфор-ной кислоты, насыщенной фтороводородом. Оптимальные результаты дало проведение процесса при 30—35 °C в присутствии 4% олеума [17].
о-Хлорфеиилтрихлорметилкарбинол (II). К смеси 336 г порошкообразного едкого кали и 3 л жидкого аммиака при —75 °C постепенно приливают раствор 140,5 г I в 500 мл хлороформа с такой скоростью, чтобы температура не поднималась выше —65 °C. Массу перемешивают 30 мин и затем прибавляют небольшими порциями 320 г хлористого аммония, контролируя, чтобы температура не поднималась выше —60 °C. Дают выдержку при перемешивании 1 ч. Прекращают охлаждение и после того, как аммиак улетучится, остаток выливают в 1 л воды. Органический слой отделяют, водный экстрагируют хлороформом (3 раза по 100 мл). Объединенные хлороформный и органический слой промывают водой до нейтральной реакции, сушат сульфатом натрия и упаривают. Остаток кристаллизуют из гексана. Выход II 242 г (93%).
69
,о-Хлорфенилдихлорметилкарбинол (111). Смесь 130 гП и 65 г амальгамы алюминия в 1250 мл 90% этилового спирта кипятят до полного растворения амальгамы алюминия (10—12 ч). Раствор отделяют, к остатку добавляют воду и экстрагируют хлороформом (4 раза по 100 мл). Спирт и хлороформ объединяют и упаривают. Получают 107 г (92%) III.
1-(о-Хлорфенил)-1-(п-хлорфенил)-2,2-дихлорэтан (хло-дитан) (IV). К ИЗ г III и 79 г хлорбензола при 30°C и энергичном перемешивании прибавляют по каплям 135 мл 4% олеума. Дают выдержку 6 ч при 30—35°С и перемешивании. После охлаждения закристаллизовавшийся IV отфильтровывают, промывают водой, сушат и перекристаллизовывают из гексана. Выход 114 г (65%); т. пл. 77— 78 °C.
ЦЕТАМИФЕН
Р-Этаноламинная соль фенилэтилуксусной кислоты:
ОН,- CH — GOOH • H„NGH„CH„OH
Ан С2Н5
С10Н О2- С^Н-? NO
Мол. масса 225,29
Основные синонимы: фенэксол и др.
Белый кристаллический порошок без запаха, горьковатого вкуса. Легко растворим в воде и спирте, трудно — в ацетоне, практически нерастворим в эфире и бензоле; т. пл. 102—106°С (в интервале 2°С); ФС 42-1247-79.
Цетамифен — препарате умеренным гипохолестерине-мическим действием [137], механизм которого связан, по-видимому, с торможением в качестве «ложного метаболита» синтеза холестерина в организме из эфиров уксусной кислоты. Не исключено, однако, что в случае цетамифена участвуют и другие факторы, в частности, превращение этаноламина (коламина) в холин, обладающий липотропными свойствами (уменьшение холестерина в сыворотке крови, повышение содержания фосфолипидов и уменьшение фракции p-липоПротеидов, обладающей атерогенными свойствами. Препарат усиливает тиреотропную функцию гипофиза, проявляет умеренный гипотензивный эффект, повышает желчевыделительную функцию печени.
Применяют цетамифен при гиперхолестеринемии различной этиологии, атеросклерозе, церебросклерозе, кардиосклерозе с явлениями хронической коронарной недо-70
статочности, гипертонической болезни, липоидном нефрозе, микседеме, ксантоматозе, сахарном диабете.
Выпускается в таблетках по 0,25 г N. 50. Цена 21 к.
Метод синтеза цетамифена на основе отходов производства фенобарбитала разработан на производственном химико-фармацевтическом объединении «Здоровье» с участием Харьковского научно-исследовательского института эндокринологии и химии гормонов [137] по схеме:
1) NaOH H,NCH,CH,OH
CHCH-CN -----► CHCH-COOH —-------L». с н сн-соон. h2ngh2ch,oh.
в 6Ан 2) Hot о»Ан 6н.
С2Н6 2 6 2 6
I II III
Отход производства фенобарбитала — «масло фенобарбитала», содержащее фенилэтилацетонитрил (I), подвергают омылению нагреванием с водной щелочью. Фенил-этилуксусную кислоту (II) выделяют подкислением щелочного водного раствора соляной кислотой. р-Этанол-аминную соль III выделяют в среде абсолютного изопропилового спирта, что дает лучшие результаты, чем проведение процесса в воде.
Фенилэтилуксусная кислота (II). 1,45 кг «Масла фенобарбитала»— отхода от производства фенобарбитала, 3,6 л воды и 770 г едкого натра нагревают при перемешивании 48 ч. Массу охлаждают до 36—40°C и добавляют соляную кислоту (~2,3 кг) до pH 2. При дальнейшем охлаждении до 20 °C и перемешивании в течение 3—4 ч выпадают кристаллы технической II. Массу оставляют на 10 ч, после чего дополнительно охлаждают до 4—6°C, осадок отфильтровывают, промывают 600 мл воды, растворяют при 55—60°C в 4 л воды с добавкой 770 г 25% водного аммиака. Проверяют pH (должен быть 8,0) и раствор фильтруют с углем. К фильтрату прибавляют 2 л воды и 1,2 кг соляной кислоты (до pH 2), охлаждают при перемешивании 3—4 ч/ дают выдержку 10 ч и доводят температуру до 4—6 °C. Осадок отфильтровывают, промывают 1,6 л горячей (80—90 °C) воды (при этом II расплавляется) и после охлаждения до 40 °C еще 1,5 л воды при перемешивании. Фильтруют при 4—6 °C, сушат при 26—28 °C. Получают 1,66 кг II.
р-Этаноламинная соль фенилэтилуксусной кислоты (цетамифен) (III). 1,1 кг II растворяют в 1,84 кг изопропанола при 24—26 °C и прибавляют 420 г моноэтанолами-на (до pH 8). Перемешивают 4 ч, контролируют pH (8,0), нагревают до 65°C (полное растворение осадка), фильтру
71
ют и охлаждают до 2—4 °C. Оставляют кристаллизоваться на 36 часов. Осадок III отфильтровывают, промывают в 4—5 приемов охлажденным до 0—2 °C абсолютным изопропиловым спиртом (320 г), высушивают при 30—40°C. Выход III 1,28 кг.
ФЕРРОЦЕРОН
Натриевая соль о-карбоксибензоилферроцена, тетрагидрат:
С да Н,з Fe03Na-4H2O
Ч Мол. масса 428,2
Синоним: эритростимулин.
Темно-оранжевый кристаллический порошок без запаха, горького вкуса, растворим в воде (pH 1% раствора 6,5—7,8), трудно растворим в спирте, практически нерастворим в бензоле и петролейном эфире, не плавится; ФС 42-904-74.
Ферроцерон — оригинальный препарат [171, 235], стимулирующий крбветворение. При приеме внутрь ферроцерон разрушается; выделяя активное железо, которое легко всасывается из желудочно-кишечного тракта. Большая часть всосавшегося железа расходуется на построение гемоглобина и гемосодержащих ферментов (каталаз, цитохромов и др.); остальное количество откладывается в печени и селезенке, пополняя депо железа в организме.
Применяют ферроцерон при железодефицитных анемиях различного происхождения. Имеются данные об эффективности препарата при озене, пародонтозе и других состояниях, связанных с дефицитом железа на уровне ферментов. Выпускается в таблетках по 0,1 г N. 100 (це-' на 3 р. 35 к.) и по 0,3 г N. 50. Цена 5 р. 02 к.
Метод синтеза ферроцероиа разработан Институтом элементоорганических соединений АН СССР совместно с Московским экспериментальным витаминным заводом НПО «Витамины» [171] по схеме:
72
Ферроцен (дициклопентадиенилжелезо) (I) ацилируют фталевым ангидридом (II) в среде безводного хлористого метилена в присутствии хлористого алюминия. Полученный о-карбоксибензоилферроцен (III) очищают через натриевую соль с последующим подкислением и выделяют натриевую соль о-карбоксибензоилферроцена, тетрагидрат (ферроцерон) (IV). Этот метод является предпочтительным по сравнению с первоначально предложенным [171] ацилированием ферроцена хлорангидридом этилового эфира о-фталевой кислоты с дальнейшим омылением образующегося о-карбалкоксибензоилферроцена и выделением натриевой соли о-карбоксибензоилферроцена, тетрагидрата (IV).
о-Карбоксибензоилферроцен (П1). Смесь 148 г II, 220 г хлористого алюминия и 4 л хлористого метилена с содержанием влаги не более 0,05% выдерживают при перемешивании и температуре 20—23 °C в атмосфере азота 1 ч, после чего приливают раствор 186 г I в 1 л хлористого метилена. Массу перемешивают 3 ч при 38—40 °C, охлаждают до 5—6°C и прибавляют 500 мл 9,5% соляной кислоты с такой скоростью, чтобы температура была не выше 20 °C. Затем прибавляют 2 л воды, перемешивают 10 мин, органический слой отделяют, водный — дополнительно экстрагируют 800 мл и 400 мл хлористого метилена. Органический слой промывают 2 раза по 2 л воды, фильтруют от примесей и упаривают, завершая процесс отгонки при остаточном давлении 400—500 мм. Вещество растворяют в 2,5 л 4,6% водного раствора едкого натра при 70—75 °C. Горячий раствор фильтруют и после охлаждения до 4—6 °C подкисляют при перемешивании 500 мл 20,9% соляной кислотой до pH 1,0—2,0. Перемешивание прекращают, дают выдержку 15—20 мин при 4—6 °C, осадок отделяют, промывают 500 мл охлажденной до 15—18 °C воды и высушивают. Получают 125,4 г (28%) III.
73
Натриевая соль о-карбоксибензоилферроцена, тетрагидрат (ферроцерон) (IV). К нагретому до 60 °C 1 л 12,6% раствора едкого натра прибавляют 532,8 г III, перемешивают 30 мин при 80—85 °C и охлаждают до 5—10 °C, дают выдержку 3 ч с периодическим перемешиванием. Кристаллический осадок отфильтровывают, промывают 3 раза по 300 мл охлажденной до 4—6 °C дистиллированной воды, 180 мл охлажденного до 4—6°C этилового спирта, высушивают и перекристаллизовывают из 1,6 л дистиллированной воды (нагревание'5—7 мин при 88—90 °C, охлаждение 3 ч при 3—5°C с периодическим перемешиванием). Полученный кристаллический IV промывают 2 раза по 500 мл охлажденной до 3—5 °C воды (до pH промывных вод 7,1—7,4), 280 мл охлажденного до 3—5°C этилового спирта и высушивают при 20—25 °C. Выход 460 г (65%).
ЦИГЕРОЛ
П,Ь-2-Циклогексил-5,9-диметил-Д4>8-каприновая кислота
СН--С?СН-(С1! ,»„-С»СН- 011,-СН-сс-эи
Ан3 226н3 X
Мол. масса 278,44
Высококипящая прозрачная вязкая жидкость (консистенция глицерина) светло-желтого цвета со слабым характерным запахом, горьковатого вкуса, практически нерастворима в воде, растворима в спирте, хлороформе, растительных маслах ив других органических растворителях, Иц0 1,4890—1,4950.dgl 0,9770—0,9794; ФС 42-1449-80.
Цигерол — оригинальное антисептическое и ранозаживляющее средство [127]. Способствует регенерации нормальных тканей, ускоряет заживление ран, уменьшает воспалительную реакцию.
Применяют цигерол наружно при лечении гранулирующих ран, трофических язв, ожоговых поверхностей и др.
Выпускается во флаконах по 50 г. Цена 10 р. 07 к.
Метод синтеза цигерола разработан в Институте органической химии АН СССР им. Н. Д. Зелинского совместно с химико-фармацевтическим заводом «Акрихин» по схеме [127]:
Обработкой циклогексанола (I) бромоводородом, получаемым во время реакции из бромистого натрия и серной кислоты, синтезируют бромциклогексан (II), который посредством малонового синтеза превращают в диэтиловый эфир циклогексилмалоновой кислоты (III). Алкилирование III геранилхлоридом (IV), приготовленным обработкой линалоола (V) соляной кислотой, приводит к диэтиловому эфиру циклогексйлгеранилмалоновой кислоты (VI), который подвергают щелочному омылению с дальнейшим частичным декарбоксилированием циклогексилгеранилма-лоновой кислоты (VII) до О,Ь-2-циклогексил-5,9-диметил-Д4’8-каприновой кислоты (цигерола) (VIII).
Бромциклогексан (II). К раствору 1,296 кг бромистого натрия в 1,15 л дистиллированной воды прибавляют 1,041 кг I и при перемешивании приливают 0,405 кг 92,5% серной кислоты с такой скоростью, чтобы температура реакционной массы была не выше 35°C. Нагревают 2 ч при 96—98 °C, охлаждают до 50—60 °C и приливают 2 л дистиллированной воды. Верхний слой отделяют, промывают 600 мл воды, 2,53 л 5% раствора соды (до слабощелочной реакции по фенолфталеину) и сушат хлористым кальцием до содержания влаги не более 0,3%. Получают 1,20 кг (67,3%) II, который без дополнительной очистки используют на следующей стадии.
Диэтиловый эфир циклогексилмалоновой кислоты (Ш). К этилату натрия, приготовленному из 2,1 л абсолютного этилового спирта и 150 г натрия, приливают при 55 °C 1,11 кг малонового эфира и после 10 мин перемешивания
75
тонкой струей 1,116 кг II, поддерживая температуру массы 40—45°C. Затем кипятят (76—84°C) 80—90 ч при перемешивании, контролируя конец реакции по отсутствию в пробе щелочной среды с фенолфталеином. Этиловый спирт отгоняют; к остатку приливают 3 л дистиллированной воды и 1 л четыреххлористого углерода, перемешивают 10 мин, нижний слой отделяют, верхний дополнительно экстрагируют 2 раза по 0,63 л четыреххлористого углерода. Экстракты объединяют, упаривают в вакууме, перегоняют. Собирают фракцию с т. кип. 134—170 °C (20 мм рт. ст.). Выход III с по 1,438—1,4520 705 г (42,5%).
Гераиилхлорид (IV). К охлажденным до 12—15°С 2,1 кг 36% соляной кислоты приливают медленно 1,13 кг V, перемешивают 1 ч при 15—20 °C, прибавляют 1,2 л толуола и перемешивание продолжают еще 20 мин. Толуольный слой отделяют, промывают 1 л дистиллированной воды, 3 л 5% раствора гидрокарбоната натрия (до значения pH 7 в водном слое), сушат хлористым кальцием до содержания влаги не более 0,1% и определяют содержание геранилхлорида. Получают 2,04 кг 44% толуольного раствора IV. Выход 79,4%.
Диэтиловый эфир циклогексилгеранилмалоиовой кислоты (VI). Распыляют 120 г натрия в 1,37 кг толуола с содержанием влаги не более 0,1% й к нему в течение 5— 6 I медленно приливают 1,26 кг III с такой скоростью, чтобы масса слабо кипела. Кипячение продолжают еще 6 ч (до полного исчезновения натрия). Охлаждают до 40— 45 °C и медленно приливают полученный на предыдущей стадии толуольный раствор IV с такой скоростью, чтобы масса слабо кипела (температура 102—112°C). Дают выдержку 15 ч при кипении с перемешиванием, контролируют содержание натрия (не более 0,2%) и добавляют 1 л дистиллированной воды. Водный слой отделяют и контролируют на отсутствие VI. Толуольный раствор промывают 1 л дистиллированной воды, упаривают в вакууме. Остаток перегоняют, собирая фракцию с т. кип. 150—160 °C (10 мм рт. ст.). Получают 947,2 г VI (48,12%).
Циклогексилгеранилмалоновая кислота (VII). К раствору 404 г едкого кали в 835 мл абсолютного этилового спирта приливают 473,6 г VI. Загустевшую массу кипятят с перемешиванием 2 ч, охлаждают до 70 °C, контролируют полноту омыления и отгоняют этиловый спирт. К остатку при 35—40 °C приливают 1,1 л дистиллированной воды, перемешивают до образования гомогенного раствора и при 76
М-,\ .
20—25 °C подкисляют 18% соляной кислотой до pH 4,0— 3,5. Выделившуюся VII извлекают 1,1 л толуола, не содержащего сернистых примесей. Водный слой дополнительно экстрагируют 450 мл толуола. Объединенные толуольные экстракты промывают дистиллированной водой до содержания в промывных водах ие более 0,003% соляной кислоты. Толуол отгоняют в вакууме. Остаток — 330 г VII (выход 81,8%), без дополнительной очистки используют на следующей стадии.
D.L-2-Циклогексил-5,9-диметил-Д48-каприновая кислота (цигерол) (VIII). Полученный на предыдущей стадии VII нагревают в вакууме при остаточном давлении 15—20 мм 2 ч до 140°C, после чего перегоняют при 3 мм, собирая вещество с т. кип. выше 140°C. Получают 235,2 г (82,54%) VIII с содержанием основного вещества не ниже 73%.
ПИРОМЕКАИН
2,4,6-Триметиланилида 1-бутилпирролидинкарбоновой-2-кислоты гидрохлорид:
С 13 Hjg N?O • HCt
Мол. масса 324,90
Белый или белый со слабым кремовым оттенком кристаллический порошок без запаха, слабогорького вкуса, с анестезирующим действием на слизистые оболочки полости рта, растворим в воде (pH 2% раствора 4,5—5,2), легко растворим в спирте; т. пл. 252—259 °C разл. (в интервале 2°C); ФС 42-1050-76.
Пиромекаин — оригинальный местноанестезирующий препарат [207], менее токсичен и имеет большую широту терапевтического действия, чем кокаин и дикаин.
Применяют пиромекаин для поверхностной анестезии в оториноларингологии, хирургии, офтальмологии.
Выпускается в виде водных растворов в ампулах (по 10 мл N. 10): 0,5% (цена 1 р. 80 к.); 1%’ (цена 3 р.) и 2% (цена 5 р.).
Метод синтеза пиромекайна разработан в Институте фармакологии АМН СССР [207] по схеме:
77
СЦСН^СООН S'»- [CKCH^CHBrCOOMI S0C.1? CI|CH2I3CHB> COCI —» I PC‘3 II “I
! IV
6-Хлорвалериановую кислоту (I) под действием брома в присутствии каталитических количеств треххлористого фосфора превращают в а-бром-6-хлорвалериановую кислоту (II), которую без выделения переводят с тионилхлори-дом в хлорангидрид (III). При взаимодействии хлоран-гидрида III с 2,4,6-три^етиланилином в среде хлороформа получают амид IV. ЗаМыкание пирролидинового цикла осуществляют при кипячении хлорбромамида IV с н-бутил-амином в спирте в присутствии каталитических количеств йодистого калия. Полученный продукт переводят обработкой спиртовым раствором хлороводорода в гидрохлорид — препарат пиромекаин (V).
Хлорангидрид а-бром-6-хлорвалериановой кислоты (III). К смеси 218 г I и 327 г брома прикапывают при температуре не выше 35,°С 1,9 мл треххлористого фосфора, температуру медленно повышают до 80 °C и дают выдержку 20 ч, после чего нагревают 2 ч при 100 °C (конец реакции определяется прекращением выделения бромоводорода). Массу охлаждают до 20—25 °C, промывают водой 3 раза по 100 мл. Водные промывки экстрагируют 100 мл бензола и объединенные органические слои сушат хлористым кальцием. Бензол отгоняют. К остатку медленно при температуре в массе 20°C приливают 120 мл тионилхлорида и кипятят 4 ч. Избыток тионилхлорида отгоняют в вакууме при 95—100°C (50—60 ммрт.ст.). Остатки тионилхлорида удаляют в виде азеотропа с сухим хлороформом (30,мм рт.ст.), контролируя конец отгонки по содержанию в массе тионилхлорида (должно быть не более 0,1%, пВ” в интервале 1,5060—1,5070). Выход III 128 г (71 %).'Продукт нестабилен и должен быть непосредственно использован в дальнейшем .^цнтезе.
2,4,6-ТриметиланилиД а-бром-6-хлорвалериановой кислоты (IV). К раствору 620 г 2,4,6-триметиланилина в 1 л хлороформа, охлажденному до 5 °C, медленно прибавляют, поддерживая температуру массы 5°С, 543 г III в 1,5 л хлороформа. В ходе реакции выпадает осадок гидрохло
78
рида 2,4,6-триметиланилина. После 4 ч выдержки без нагревания массу кипятят 1 ч, охлаждают до 18—20°C, гидрохлорид 2,4,6-триметиланилина отфильтровывают, промывают 500 мл хлороформа. Объединенные хлороформные растворы промывают 3 раза по 400 мл 3% раствора соляной кислоты, затем водой до нейтральной реакции. Хлороформ отгоняют (в конце отгонки при остаточном давлении 50—60 мм рт. ст.) и температуру доводят до 90—95°C. Проба остатка после отгонки хлороформа и охлаждения должна становиться сухой и рассыпчатой. Вещество перекристаллизовывают из 1,5 л абсолютного изопропилового спирта с углем и высушивают при 60—70 °C. Выход 319 г (44%).
2,4,6,-Триметилаиилида 1-бутилпирролидинкарбоно-вой-2 кислоты гидрохлорид (пиромекаин) (V). К раствору 500 г IV в 1,5 л абсолютного этилового спирта приливают небольшими порциями раствор 440 г н-бутиламина в 1,5 л этилового спирта, вносят 4,4 г йодистого калия, кипятят при перемешивании 25 ч, охлаждают до 20 °C, фильтруют и выливают в 1,5 л воды при перемешивании и температуре 3—5 °C. Перемешивают 3—4 ч, выделившийся продукт закристаллизовывается, его отделяют центрифугированием, промывают охлажденной до 3—5 °C водой (550 мл), растворяют в 3 л бензола. Раствор сушат сульфатом магния до содержания влаги менее 0,3% и фильтруют. Гидрохлорид осаждают прибавлением спиртового раствора хлороводорода при 5—10°C до pH 1. Дают выдержку 24 ч, осадок отфильтровывают, промывают 400 мл охлажденного до 5—7 °C абсолютного этилового спирта, сушат при 60 °C до содержания влаги менее 1% и перекристаллизовывают из 1,4 л абсолютного изопропилового спирта с углем и добавкой изопропанольного раствора хлороводорода. Вещество сушат при температуре не выше 60°C. Выход 247 г (48,2%).
эстоцин
р-Диметиламиноэтилового эфира а,а-дифенил-а-этоксп-уксусной кислоты гидрохлорид:
СвН Ч 9С2Н5
C-COOCH,CH,N(CH ), HCt
•С6н5^ 2 • 3 31
®гон25м®з‘н<*'
Мол. масса 363,8"
79
Основные синонимы: дименоксадол, локарин, про-палгил.
Белый кристаллический порошок, без запаха, горького вкуса, растворимый в воде (pH 2% раствора 4—5,5), этиловом и других спиртах, хуже растворим в хлороформе, плохо растворим в ацетоне, практически нерастворим в эфире, этилацетате, бензоле; т. пл. 170—173 °C (в интервале 2°C); ФС 42-1166-78.
Эстоцин— анальгезирующий и противокашлевой препарат [130, 269], обладающий также умеренным спазмолитическим и противовоспалительным действием [131]. По анальгезпрующей активности уступает промедолу, но менее токсичен, слабее угнетает дыхание и в отличие от промедола и морфина не вызывает запоров; оказывает умеренное спазмолитическое (в том числе бронхолитическое) и холинолитическое действие, уменьшает спазмы кишечника и бронхов. По имеющимся клиническим наблюдениям, привыкания и пристрастия к эстоцину не развивается.
Применяют как болеутоляющее средство при болях, связанных со спазмами гладкой мускулатуры, для премедикации и в послеоперационном периоде, при обезболивании родов. Препарат хорошо переносится детьми, не дает побочных эффектов, вызываемых промедолом. Используется как противокашлевое средство, в том числе при явлениях бронхоспазма.
Выпускается в таблетках по 0,015 г N. 6. Список А. Цена 19 к.
Метод синтеза эстоцина разработан во ВНИХФИ по схеме [269]:
СвН^ 1С2Нб 1)ClC^CHaN(CH3)5-HCt свнь ОС2Н5 ^0-СООНа —--------------------------- >С-СООСН2СН^(СНз)г. НС!
|у 6 о у
Бензиловая кислота (I), которая используется химико-фармацевтической промышленностью в качестве полупродукта в производстве лекарственных препаратов бен-зацина, тифена, метацина и амизила, при обработке тио-нилхлоридом превращается в а,а-дифенил-а-хлоруксусную
80
кислоту (II), которую нагреванием с абсолютным этиловым спиртом переводят в а,а-дифенил-а-этоксиуксусную кислоту (III). Натриевая соль последней (IV) при взаимодействии с Р'-диметиламиноэтилхлорида гидрохлоридом превращается в р-диметиламиноэтиловый эфир а,а-дифе-нил-а-этоксиуксусной кислоты, гидрохлорид которого V и выпускается под названием эстоцин.
В литературе известны несколько методов превращения бензиловой кислоты (I) в V [269], отличающихся последовательностью введения сложноэфирного алкаминового и этоксильного остатков. Первая группа методов предусматривает синтез через диметиламиноэтиловый эфир бензиловой или а,а-дифенил-а-хлоруксусной кислоты , с дальнейшим введением этоксигруппы. Существенный недостаток этих методов — использование диметиламино-этилхлорида на начальных стадиях синтеза, что увеличивает расход алкаминовой цепочки и способствует образованию трудно отделяемых побочных продуктов. Это ведет к снижению качества получаемого эстоцина и к существенному уменьшению его выхода. Более перспективна группа методов, основанных на использовании в качестве полупродукта а,а-дифенил-а-этоксиуксусной кислоты или ее этилового эфира. Превращение бензиловой кислоты (I) в этиловый эфир а,а-дцфенил-а-этоксиуксусной кислоты путем обработки I пятихлористым фосфором с дальнейшим замещением хлора на этокси-группу протекает неоднозначно и сопровождается образованием значительного количества (до 65%) этилового эфира а,а-дифенил-а-хлоруксусной кислоты. Не имеет существенных преимуществ и синтез по схеме: бензиловая кислота (I), ее серебряная соль, этиловый эфир бензиловой кислоты, этиловый эфир а,а-дифенил-а-бромуксусной кислоты, этиловый эфир а,а-дифенил-а-этоксиуксусной кислоты. Переэтерификация этилового эфира а,а-дифенил-а-этоксиук-сусной кислоты с 0-диметиламиноэтанолом требует применения металлического натрия. Наибольший практический интерес представляет синтез эстоцина (V) из бензиловой кислоты (I) через а,а-дифенил-а-хлоруксусную кислоту (II) и а,а-дифенил-а-этоксиуксусную кислоту (III). Замена оксигруппы в I на хлор может быть осуществлена с хлорокисью фосфора (выход 65%), ацетил-хлоридом (выход 55%), через хлорангидрид и этиловый эфир а,а-дифенил-а-хлоруксусной кислоты с последующим омылением (выход не указан) и с тионилхлоридом. Последний вариант является предпочтительным. Замена га
6 Заказ № 4304
81
Логена На этоксйгруппу во 11 может быть проведена С этилатом натрия при взаимодействии со спиртом в присутствии алкоголятов, карбонатов или пиридина, а также просто при нагревании со спиртом. Получающаяся этоксикислота (III) устойчива в щелочной среде, но легко гидролизуется до бензиловой кислоты (I) при нагревании с водой. Использование на последней стадии синтеза очищенной этоксикислоты III и проведение процесса в гетерогенной среде обеспечивает хороший выход и высокую степень чистоты конечного продукта [269].
а,а-Дифенил-а-хлоруксусная кислота (II). 296,7 г I, 240 мл хлористого метилена и 113 мл тионилхлорида перемешивают 96 ч при 20—25 °C. Конец реакции определяют методом ТСХ (содержание I не должно превышать 1 %). Затем массу охлаждают до 5—8°C, дают выдержку 2 ч. Осадок кислоты II отфильтровывают и высушивают при 20—25°C. Получают 247,2 г II. Выход 72,4%. Вещество имеет т. пл, 117—118°C и содержание основного продукта не ниже 93%.
а,а,-Дифенил-а-эток^иуксусная кислота (III). К нагретым до 70—72 °C 465 мл абсолютного этилового спирта приливают по каплям раствор 117 г II в 100 мл абсолютного этилового спирта. Массу нагревают 1 ч при 78 °C, охлаждают до 60 °C и при наружном охлаждении прикапывают 343 мл 10% водного раствора едкого натра (до pH 10). Этиловый спирт отгоняют при атмосферном давлении, поднимая в конце отгонки температуру в реакционной массе до 98—100 °C. Оставшийся раствор IV разбавляют 430 мл воды, контролируют, чтобы pH оставался не ниже 10, и экстрагируют 2 раза по 200 мл хлористого метилена. Затем водный слой подкисляют технической-соляной кислотой по конго при температуре 20—25 °C с экстракцией выделяющейся III хлористым метиленом (5 раз по 200 мл). Экстракты сушат сульфатом натрия и упаривают. К остатку прибавляют 380 мл абсолютного этилового спирта, после чего для более полного удаления хлористого метилена отгоняют около 90 мл этилового спирта, образующего азеотроп с хлористым метиленом и водой. К остатку прибавляют активированный уголь, нагревают до кипения, отфильтровывают, разбавляют 278 мл воды и охлаждают Р/г ч при 5—8 °C. Выпавшие кристаллы III отфильтровывают, промывают 50 мл охлажденного до 5—8°C 50% этилового спирта и сушат при 45—50°С. Получают 100,3 г III с содержанием основного вещества 99,5%. Выход 87%, т. 114—115°C.
82
р-Диметиламиноэтилового эфира а,а-дифенил-а-этокси-уксусной кислоты гидрохлорид (эстоцин) (V). Смесь 812 г III, 434 г сухой кальцинированной соды и 16,24 л абсолютного изопропилового спирта выдерживают 2 ч при 20—25 °C. Контролируют полноту образования натриевой соли а,а-дифенил-а-этоксиуксусной кислоты (IV) (должно оставаться не более 2% свободной кислоты III) И прибавляют 552 г гидрохлорида р-диметиламиноэтилхлорида. Массу нагревают 1 ч при 80—82 °C, контролируя образование сложного эфира по отсутствию натриевой соли IV. По окончании процесса изопропиловый спирт отгоняют. Для более полного удаления его остатки отгоняют до температуры в парах 98—100 °C после прибавления 2 л воды от предыдущей операции перекристаллизации эстоцина. Затем водный раствор охлаждают до 20 °C и экстрагируют 12 л этилацетата. Этилацетатный экстракт промывают 3 раза по 600 мл дистиллированной воды, сушат сульфатом магния, ответляют углем и обрабатывают при 20°C спиртовым раствором хлороводорода до кислой реакции по конго. Массу охлаждают 1 ч при 10—12°С. Осадок отфильтровывают, промывают 400 мл охлажденного до 10— 12 °C этилацетата, высушивают при 65—70 °C и перекристаллизовывают из 2 л дистиллированной воды (нагревание до 80°C, охлаждение 1 ч при 10—12°C). Получают 943 г V, который дополнительно перекристаллизовывают из 2 л абсолютного этилового спирта. Выход 915 г (79,9%).
МЕТАМИЗИЛ
Р-Диэтиламинопропилового эфира бензиловой кислоты гидрохлорид:
гм ОН Свн5\|
C-COOCH?CH-N (С-Н.) . HCI о U х I * 0 2
%Н5' СН3
с21 H27NO3- HCI
Мол. масса 377,92
Синонимы: ИЭМ-275, метамицил, метилдиацил, метил-бенактизин. Белый мелкокристаллический порошок, растворим в воде (pH 2% раствора 4,0—5,0), спирте и хлороформе, практически нерастворим в эфире; т. пл. 156— 159°C (в интервале 2°C); ФС 42-1403-80.
Метамизил — оригинальный центральный холинолитик [67, 114, 115] с преимущественным влиянием на м-холино-
6*
83
реактивные системы. Препарат оказывает успокаивающее (транквилизирующее), холинолитическое и спазмолитическое действие, усиливает эффекты анальгетиков и снотворных, является антидотом при отравлениях антихолин-эстеразными и холиномиметическими веществами.
Применяют метамизил при невротических, фобических и депрессивных состояниях, психомоторном и эмоциональном возбуждении, при детских церебральных параличах и гиперкинетическом синдроме, при сосудистых заболеваниях мозга и повышенном внутричерепном давлении, при язве желудка и двенадцатиперстной кишки, пилороспазме, кишечных и почечных коликах, бронхиальной астме, а также как средство пре- и постмедикации.
Выпускается в таблетках по 0,001 г N. 50 (цена 16 к.) и в ампулах по 1 мл 0,25% раствора N. 10 (цена 32 к.). Список А.
Метод синтеза метамизила разработан в Институте экспериментальной медицины АМН СССР совместно с Ленинградским! производственным химико-фармацевтическим объединением «Фармакон» [115] по схеме:
НСНО <?Н2ОН H2/Ni ?н2он с Н5Вг CH3CH2NO2-----*- CH3CH-NO2----- CH3tH-NH2------>-
I II III
OH ppi _ Cl l_j Cl
CeH-&;C-COOH-^sCe ^-COCl—► 6 -GOOCH
CeH^ CeH5 CeH5
V VI VII
<j!H2OH CH3CH-N(C2H5)2
IV
_CH-N(C2H6)2—-»-
CHg
l)H2° СвН5 Г
—*- 6 3C-CQOCHjCH-N(C,Hi • HCl
2)HCt К JbS
VIII
Конденсацией нитроэтана (I) с формалинов в метанольном растворе едкого натра получают 2-нитропропа-нол (II). Нитрогруппу восстанавливают каталитически и полученный 2-аминогфойанол (III) алкилируют бромистым этилом до 2-диэтиламинопропанола (IV). Параллельно обработкой бензиловой кислоты пятихлористым фосфором синтезируют хлорангидрид а,а-дифенил-а-хлор-уксусной кислоты (VI), который при взаимодействии с IV образует p-диэтиламинопропиловый эфир а,а-дифенил-а-хлоруксусной кислоты (VII). Дальнейший гидролиз VII и обработка хдороводородом позволяют получить р-ди-этиламинопропиЬового эфира бензиловой кислоты гидрохлорид (метамизил) (VIII). Метод является предпр-
84
чтительным перед описанной ранее конденсацией бензиловой кислоты с 2-диэтиламинопропилхлоридом или синтезом VIII из пропионовой кислоты через ее хлоран-гидрид с дальнейшим бромированием, этерификацией этиловым спиртом, обработкой диэтиламином, восстановлением этилового эфира 2-диэтиламинопропионовой кислоты алюмогидридом лития и переэтерификацией получающимся при этом 2-диэтиламинопропанолом метилового эфира бензиловой кислоты [115].
2-Нитропропанол (II). К смеси 2 кг I и 960 мл метанола прибавляют 70 г едкого натра, после чего приливают 2,43 л формалина с такой скоростью, чтобы температура массы находилась в интервале 18—25 °C. Дают выдержку при перемешивании 1 ч при 20—25 °C и 2 ч дри 35—40 °C периодически проверяя, чтобы pH среды был не ниже 8,0. Затем массу охлаждают до 15°С, загружают 170 г соляной кислоты (до кислой реакции по конго) и отгоняют в вакууме при 50—60 °C летучие продукты. Остаток перегоняют, собирая фракцию с т. кип. 88—100 °C (5—10 мм рт. ст.). Выход II 1,115 кг (50,4%).
2-Аминопропанол (Ш). 950 г II в 11,1 л метанола гидрируют с 950 г нейтрального никелевого катализатора при 20—70 °C (процесс экзотермичен) и давлении 5 атм, контролируя процесс по определению содержания II (не более 4%). Катализатор отфильтровывают, метанол отгоняют в вакууме при 40—50 °C. Оставшийся технический III с содержанием основного вещества 55—65 % без дополнительной очистки используют на следующей стадии.
2-Диэтиламинопропанол (IV). Полученный на предыдущей стадии III приливают к раствору 385 г едкого натра в 600 мл воды и при перемешивании и температуре не. выше 35 °C прибавляют порциями 1,08 кг бромистого этила. Дают выдержку 2 ч при 40—50°C и 2 72 ч при 60 °C, после чего охлаждают до 20 °C и экстрагируют IV эфиром (2 раза по 1,2 л). Эфирный экстракт сушат поташом и упаривают. Остаток перегоняют в вакууме. Собирают фракцию с т. кип. 70—75°C (10—15 мм рт.ст.). Выход IV 398 г (33,8%, считая на II).
Р-Диэтиламинопропилового эфира бензиловой кислоты гидрохлорид (метамизил) (VIII). К 1 кг V в 2,5 л безводного толуола прибавляют 1 кг пятихлористого фосфора. Температура реакционной массы за счет экзотермического процесса повышается до 30—50 °C. Дают выдержку 1 ч при 30—50 °C, охлаждают до 20 °C, прибавляют еще 1 кг пятихлористого фосфора и кипятят при перемешивании
85
Р/г ч. Охлаждают до 50°C и отгоняют в вакууме при 80— 90 °C хлорокись фосфора с толуолом. Добавляют свежие порции толуола (по 2 л) с последующей отгонкой до тех пор, пока содержание хлорокиси фосфора в отгоне не будет менее 1%. Остаток растворяют в 2 л толуола и выливают на 2,5 кг смеси льда и воды. Перемешивают 30 мин при 20—25 °C, толуольный слой отделяют, сушат 800 г прокаленного хлористого кальция и полученный толуольный раствор VI приливают к охлажденному до +2— (—2)°С раствору 455 г IV и 333 г триэтиламина в 650 мл толуола, содержащему не более 0,2 °/о влаги. Дают выдержку 4 ч при +2—(—2) °C, контролируя, чтобы pH раствора был все время в интервале 5,0—6,0. Осадок гидрохлорида триэтиламина отфильтровывают, к толуольному фильтрату приливают 13 л воды и 363 г соляной кислоты до pH 3. Перемешивают 15 мин, контролируют, чтобы pH водного слоя был не менее 3,0. Водный слой отделяют и дополнительно промывают 850 мл толуола. Объединенные водные кислые растворы осветляют 110 г активированного угля, смешивают с 3,3 л бензола и подщелачивают поташом до pH 10,0. Бензольный слой отделяют, водный экстрагируют бензолом (2 раза по 1,65 л). Объединенные бензольные экстракты сушат поташом, фильтруют и обрабатывают изопропанольным раствором хлороводорода до фН 3,0. (Прибавление изопропанольно-го раствора хлороводорода ведут медленно 2—3 ч при 10—15 °C, не допуская добавления его избытка, так как это приводит к снижению выхода VIII.) Дают выдержку 10 ч при 5—8 °C, контролируя, чтобы pH все время был 3,0. Осадок VIII отфильтровывают, промывают 500 мл ацетона на фильтре и затем кипячением при перемешивании с 600 мл ацетона в течение 30 мин с последующим охлаждением и выдержкой 1 ч при 15—20 °C и перекристаллизовывают из 300 мл изопропилового спирта с 4 г активированного угля. Охлаждают 6 ч при 5—10°С, фильтруют, промывают 30 мл охлажденного до 5—10 °C изопропилового спирта, сушат при 40—50 °C. Получают 200 г VIII (20,8%, считая на IV).
СУЛЬФАМИДНЫЕ ПРЕПАРАТЫ
Сульфамидные препараты — эффективные химиотерапевтические средства, которые первыми стали систематически использоваться для предупреждения и лечения бактериальных инфекций у человека.
86
В развитии этой области медицинской химии можйо выделить несколько периодов [284, 312, 296].
Первоначально, с момента синтеза в 1908 г. сульфаниламида (I), и до 1932 г. исследования этого класса соединений были связаны с химией красителей. Для увеличения стойкости коричневого красителя хризоидина (II) в конце 20-х — начале 30-х годов специалисты фирмы И. Г. Фарбениндустри (Германия) синтезировали в числе других его производных и сульфаниламидный аналог хризоидина (III). Полученные вещества, помимо прямого назначения, были подвергнуты скринингу на антибактериальное действие in vitro и оказались неактивными. Однако в экспериментах на мышах, зараженных смертельной дозой гемолитического стрептококка, соединение III, получившее название пронтозил, спасло от гибели более 80% животных, а в клинике проявило высокую эффективность при таких тяжелых стрептококковых инфекциях, как рожа и родовой сепсис. Такая особенность биологического действия оказалась связана с ферментативным расщеплением III в организме млекопитающих по Ы=М-связи с образованием сульфаниламида (I), ответственного за химиотерапевтическое действие препарата, и второго вещества, неактивного в отношении микроорганизмов, но токсичного для человека и животных,— 1,2,4-триаминобензола. Синтетический сульфаниламид (I) показал высокий терапевтический эффект в опытах in vivo и подавлял развитие микроорганизмов in vitro. Положительные результаты лечения с помощью I и III общего сепсиса и менингококковых инфекций послужили толчком к широкому использованию сульфамидов в клинике.
В этот период, продолжавшийся до первой половины 40-х годов, было синтезировано и изучено несколько тысяч разнообразных аналогов I и III. Сформулирована общая гипотеза о механизме действия на микробную клетку сульфаниламида как конкурентного антагониста п-аминобен-зойной кислоты, необходимого компонента нормального фолиевого обмена. В дальнейшем было установлено, что сульфаниламидные препараты ингибируют дигидрофолат-синтетазу. Выяснены определенные закономерности связи
87
Строения и биологической активности в ряду сульфаниламидов: показана необходимость наличия в их структуре сульфамидной группы, связанной через атом серы с бензольным кольцом, в п-положении которого находится свободная NHz-rpynna. Последняя может быть замещена только такими остатками (ациламино-, азо- и др.), которые в организме способны превращаться в NH2-rpynny. Другие варианты замещения первичной ароматической аминогруппы, ее перемещение в иные положения бензольного ядра, а также введение в это ядро любых дополнительных заместителей приводят к потере антибактериальных свойств. Заместители у азота сульфамидного остатка, не подавляя химиотерапевтической активности, влияют на спектр, продолжительность и силу антимикробного действия. Это влияние подробно рассмотрено в ряде обзоров [279, 340], й’ которых, в частности, подчеркивается наибольшая эффективность сульфаниламидодиазинов [279] и пролонгирование их действия при введении в диазиновую часть молекулы метокси-групп [340].
Наряду с сульфаниламидами общего действия были созданы препараты, эффект которых локализуется в определенных органах и системах. Найдены вещества с уменьшенной абсорбируемостью из желудочно-кишечного тракта для лечения Инфекционных заболеваний пищеварительных органов, препараты, хорошо растворимые в моче с малой ренальной токсичностью для борьбы с инфекциями мочевых путей и др.
Появление к середине 40-х годов большого числа сульфаниламидоустойчивых штаммов микроорганизмов, широкое внедрение в медицинскую практику высокоэффективных антибиотиков вызвало определенное снижение интереса к сульфаниламидам, «вытеснение» их антибиотиками. Этот период продолжался до середины 50-х годов, когда недостатки лечения антибиотиками (побочные реакции, необходимость частых введений препаратов парентерально, появление антибиотикоустойчивых штаммов) привели к «ренессансу» сульфаниламидотерапии.
Особенно возрос интерес к сульфаниламидам в связи с созданием первого пролонгированного препарата этого класса — сульфапиридазина (кинекса) (IV). За короткий срок (с 1956 по 1965й К) были внедрены в медицинскую практику 12 сульфаниламидов с полупериодом выведения из организма до 60—120 ч, что создало возможность принимать эти препараты от 1 раза в сутки до 1 раза в неделю.
88
Большое внимание уделяется комбинированному применению сульфаниламидов с различной длительностью действия, комбинации сульфаниламидов с антибиотиками, использованию серебряных солей сульфаниламидов.
Важную роль в развитии современной сульфаниламидотерапии сыграло открытие синергизма сульфаниламидных препаратов и производных диаминопиримидина, в частности триметоприма (V). При этом за счет ингибирования ферментов двух последовательных стадий фолиевого обмена микроорганизмов — дигидрофолатсинтетазы, сульфаниламидами и дигидрофолатредуктазы — диаминопиримидинами имеет место не только усиление антимикробного действия, но и переход от бактериостатического эффекта к бактерицидному:
В СССР исследования в области антибактериальных сульфаниламидных препаратов начались в 1935 г. [184]. Уже через несколько месяцев после первых публикаций о пронтозиле (III) во ВНИХФИ был разработан метод синтеза этого препарата, получившего в СССР название красный стрептоцид, и с 1936 г. было налажено его промышленное производство. Одновременно началось изучение зависимости между строением и биологической активностью в ряду сульфамидных производных. В результате были созданы оригинальные препараты сульфантрол (VI) [213], сульцимид (VII) [29] и дисульформин (VIII) [184]:
VIII
Сульфантрол (VI), показавший хорошие результаты при лечении нутталиоза в ветеринарной практике, и дисульформин (VIII), применяющийся при острой и хрони
89
ческой дизентерии, производятся химико-фармацевтической промышленностью СССР и в настоящее время.
В 1937 г. советскими химиками в Свердловске был впервые синтезирован 2-сульфаниламидопиридин (сульфидин) (IX) [45], //который благодаря проведенным в Англии исследованиям [344] нашел в дальнейшем применение для лечения пневмонии, менингита, гонореи.
Большая работа была проведена по созданию в нашей стране производства наиболее эффективных известных сульфаниламидных препаратов, выбору исходного сырья для их промышленного получения, совершенствованию путей. синтеза и технологии,
В качестве сырья для выпуска сульфаниламидов в промышленности были изучены, кроме ацетанилида, применяемого за рубежом, также форманилид [28], дифенил-мочевина [27], хлорбензол [53], нитробензол [135], фе-нилуретилан [163]. В связи с доступностью и дешевизной
IX
фенилуретилана, значительно большей устойчивостью фе-нилуретилансульфохлорида по сравнению с другими арил-сульфонилхлоридами к сушке, хранению и транспортировке многотоннажное производство сульфаниламидных препаратов в СССР основывается на фенилуретилане.' При конденсации фенилуретилансульфохлорида с аминогетероциклами наряду с/дроведением процесса в пиридине был разработан метод взаимодействия в водной среде [47]. Работы по совершенствованию сульфаниламидных производств, химиотерапевтическим и клиническим исследованиям в этой области обобщены в ряде обзоров [84, 85, 87, 179, 184, 188].
В настоящее время в СССР производятся следующие сульфаниламидные препараты: стрептоцид (I) [211],
стрептоцид растворимый (X) [211], норсульфазол (XI, R=H) [211], норсульфазол-натрий (Xia, R=Na), суль-фадимезин(ХП) [211],этазол (XIII, R = H) [211], этазол-натрий (ХШа, R=Na) , сульфацил-натрий (XIV) [211], уросульфан (XV), сульфапиридазин (IV) [211], сульфапи-ридазин-натрий (IVa), сульфамонометоксин (см. с. 94), сульфадиметоксин (XVI) [211], сульфален (XVII), сульгин (XVIII) [211], фталазол (XIX) [211], фтазин
90
(см. с. 98), Дисульформин (VIII), Сульфантрол (VI) дисульфан (XX), салазопиридазин (см. с. 100) и салазоди-метоксин (см. с. 102).
В этой номенклатуре сульфаниламидов, применяющихся в СССР, десять препаратов короткого действия (полупериод выведения менее 10 ч): I, X, Ха, XI, Xia, XII, XIII, ХШа, XIV, XV; 5 — длительного действия (полупериод выведения более 24 ч): IV, IVa, XVI, XVII и сульфамоно-метоксин; 5 растворимых препаратов (четыре короткого: X, Xia, ХШа, XIV и один длительного действия: IVa); четыре препарата трудно всасываются в кишечнике и используются для борьбы с инфекциями пищеварительного тракта: VIII, XVIII, XIX, фтазин; два салазосульфамидных препарата длительного действия, эффективных при язвенном колите и болезни Крона: салазопиридазин и салазо-диметоксин.
Следует отметить, что группа сульфамидных препаратов не ограничивается только антибактериальными сульфаниламидами— производными сульфаниловой (п-амино-бензолсульфоновой) кислоты. В ходе медицинского применения сульфаниламидов были выявлены некоторые особенности влияния этих соединений на организм человека, которые послужили основанием для развития самостоятельных направлений использования сульфамидов в клинике. Так, еще в 1942 г. при лечении сульфаниламидом (I) больных тифом было обнаружено гипогликемическое действие этого препарата. Дальнейшие исследования показали, что снижение уровня сахара в крови не наблюда
91
ется у панкреатоэктомированных животных, что дало основание связать гипогликемическую активность I со стимуляцией им выделения инсулина поджелудочной железой. Это позволило использовать сульфаниламидный препарат карбутамид (надизан) (XXI) в качестве орального гипогликемического средства для лечения больных сахарным диабетом. Варьирование структуры XXI показало, что в отличие от сульфаниламидов, характеризующихся антибактериальным действием, для гипогликемической активности замещенных сульфонилмочевин совершенно не обязательно наличие в бензольном кольце NHz-группы в п-положении к сульфамидному остатку. Переход от XXI к его толильному аналогу XXII позволил получить соединение, лишенное антибактериальных свойств, но проявляющее гипогликемическую активность и менее токсичное, чем карбутамид. Вещество XXII вошло в практическую медицину под названием толбутамид. В СССР этот препарат получил название бутамид [86, 211, 238]. Еще более активными гипогликемическими препаратами оказались воспроизведенные в СССР хлорпропамид (см. с. 105) [86, 238/]л цикламид (см. с. 107) и хло-цикламид (см. с. 109) [37, 138]:
N-----N
XXVII
Другое направление в развитии сульфамидотерапии берет свое начало с обнаруженного в клиниках при лечении инфекционных заболеваний сульфаниламидными препаратами метаболического ацидоза, связанного с ингибированием карбоангидразы. Дальнейшее исследование процесса подкисления мочи при использовании сульфаниламидов позволило выявить роль карбоангидразы в «ренальном транспорте», установить требования к структуре сульфамидов, ингибирующих этот фермент. Сульфамидное производное тиадиазола под названием фонурит (в СССР диакарб) (XXIII) нашло практическое применение в ме
дицине как диуретик, избирательно ингибирующий карбо-ангидразу [203]. Установление зависимости между строением и антикарбоангидразной активностью показало, что указанное действие бензолсульфониламидов возрастает при введении в м-положение бензольного ядра второй сульфониламидной группы. Циклизация такого типа соединений привела к весьма сильным диуретикам бензтиа-диазинового ряда общей формулы XXIV. Диуретический эффект соединений XXIV сопровождался значительным увеличением в моче количества хлоридов и не коррелировал с ингибированием карбоангидразы. Дальнейшие исследования показали, что бензтиадиазины оказывают прямой эффект на реабсорбцию ионов натрия и хлора в извитых канальцах почек независимо от инактивации карбоангидразы. Сильное увеличение натрийуреза при одновременном обильном выделении с мочой хлор-иона (са-люретическое действие) бензтиадиазинов XXIV обеспечило этим препаратам широкое использование в практике.. Изучение связи между строением и биологической активностью в ряду бензтиадиазинов XXIV показало, что любые устойчивые заместители у Ц7-сульфамидной группы ликвидируют ингибирующее влияние на карбоангидразу и значительно слабее влияют на хлоруретическое действие, Ыт-ацилпроизводные дезацилируются in vivo. Введение галогенов в положение 6, различных заместителей в положение 3 и восстановление Д-3,4-двойной связи усиливают диуретическую и салуретическую активность. Введение' каких-либо заместителей в положения 1,4 или 5 приводит: к ослаблению биологического эффекта. Один из наиболее активных препаратов этой группы циклометиазид, (XXV) выпускается отечественной химико-фармацевтической промышленностью [211].
С влиянием на процессы реабсорбции в извитых канальцах почек связано и действие сульфамидного препарата этамида (см. с. ПО). Его создание было вызвано развернувшимся в 50-е годы поиском веществ, выделяющихся, подобно пенициллину, путем канальцевой секреции и за счет конкурентных взаимодействий с пенициллином, пролонгирующих его пребывание в организме. Эти исследования привели к получению бенемида (XXVI) [280], задерживающего выделение пенициллина почками. В дальнейшем был получен низший гомолог бенемида — препарат этамид, который продлевает действие пенициллина в организме и способствует выведению через почки мочевой кислоты. Этамид нашел применение в отечественной
93
Медицине как средство, регулирующее пуриновый обмен при подагре, полиартритах, мочекаменной болезни [217], н выпускается химико-фармацевтической промышленностью СССР.
К сульфамидным производным непосредственно примыкает и созданный во Львовском политехническом институте в результате исследования эфиров тиосульфаниловой кислоты (XXVII) оригинальный лекарственный препарат эсулан (XXVII, R = C2H5), используемый как фун-гистатическое и фунги^йдное средство [198].
СУЛЬФАМОНОМЕТОКСИН
4- (п-Аминобензолсульфамидо) -6-метоксипиримидин:
Мол. масса 280,29 Основные синонимы: дайметон и др.
Белый или белый с кремовым оттенком кристаллический порошок, без запаха, очень мало растворим в воде, мало — в спирте и ацетоне, легко — в разбавленных минеральных кислотах и в водных растворах едких и углекислых щелочей; т. пл. 203^—208°C (в интервале 2°C); ФС 42-1178-78.
Сульфамонометоксин— антимикробный препарат широкого спектра действия [285, 334], активный при бактериальных инфекциях, вызванных стрептококком, стафилококком, кишечной палочкой, дизентерийной палочкой, палочкой Фридлендера. Относится к группе длительно действующих сульфанилаяййдов. Быстро всасывается, проникает через гематоэнцефалический барьер. Относительно малотоксичен.
Применяется сульфамонометоксин при инфекциях дыхательных путей, гнойных заболеваниях уха, горла, носа, дизентерии, энтероколитах, инфекциях желчевыводящих и мочевыводящих путей, гнойничковых заболеваниях кожи, раневой инфекции, генерализованной менингококковой инфекции, гнойных менингитах, гонорее, для профилактики гнойных бактериальных инфекций в послеоперационном периоде [188], Высшая разовая и суточная дозы 2 г.-94
Выпускается в таблетках по 0,5 г N. 10. Цена 77 к.
Метод синтеза сульфамонометоксина разработан во
ВНИХФИ по схеме [85]:
POQ3
II ««
NH, I) HCONH,. СНдОМа
сн2|соос2н5>2 CH2(CONH2>, ——------------>- О-
Малоновый эфир (I) обработкой аммиачной водой переводят в малондиамид (II), который путем конденсации с формамидом в присутствии метилата натрия с последующей обработкой соляной кислотой превращают в 4,6-диоксипиримидин (III). (Использование в реакции с II этилформиата или ортомуравьиного эфира дало худшие результаты.) Замену оксигрупп в III на хлор осуществляют с хлорокисью фосфора в присутствии диметиланилина, добавка которого ускоряет процесс. Полученный 4,6-ди-хлорпиримидин (IV) путем аммонолиза превращают в 4-амино-6-хлорпиримидин (V), который кипячением с ме-.танольным раствором едкого натра переводят в 4-амино-6-метоксипиримидин (VI). Взаимодействием VI с п-фе-нилуретилансульфохлоридом (VII) в среде пиридина синтезируют 4-карбометоксисульфаниламидо-6-метоксипи-римидин (VIII), который подвергают щелочному гидролизу с последующим выделением 4-(п-аминобензолсульфами-до)-6-метоксипиримидина (сульфамонометоксина) (IX) из его натриевой соли соляной кислотой. Указанный метод имеет преимущество по сравнению с ранее описанными, в основе которых лежат: 1) конденсация сульфаниламида, используемого в виде натриевого производного или в смеси с безводным карбонатом калия, с 4,6-дихлорпиримиди-ном (IV) с дальнейшей заменой в 4-(п-амино-бензолсуль-фамидо)-6-хлорпиримидине атома хлора на метоксигруппу,— общие выходы 35,2—53,7%; 2) взаимодействие сульфаниламида с 4-хлор-6-метоксипиримидином — выход 25 % [334]; 3) реакция N-ацетилсульфаниламида с 4,6-дихлор-
95
пиримидином (IV) в диметилформамиде с последующей обработкой метилатом' натрия и гидролизом N-ацетильной группы нагреванием с 8% раствором едкого натра — выход не приводится; 4) конденсация N-ацетилсульфанил-амида с обладающим сильным кожно-нарывным действием 2,4,6-трихлорпиримидином, при которой образуется смесь 2-ацетилсульфаниламидо-4,6-дихлорпиримидина и 4-ацетилсульфаниламидо-2,6-дихлорпиримидина; обработка последней метилатом натрия, дальнейшие дегалогенирование с палладиевым катализатором и дезацетилирование, приводящие к смеси 4-сульфаниламидо-2-метокси- и 4-сульфанил-амидо-6-метоксипиримидинов, разделенных методом дробной перекристаллизации.
Малондиамид (II). К 1,9 л 25% водного раствора аммиака, охлажденным до 8—10°С, приливают 800 г I; массу перемешивают 2 ч при 8—15°С (уменьшение концентрации аммиака или повышение температуры до 26—30°C приводит к снижению выхода II на 10—15%). Дают выдержку без перемешивания 3 ч при 8—12 °C и 1 ч с перемешиванием при 0—(+3°C). Осадок II отделяют, промыва-вают 200 мл охлажденного до 0—3°С этилового спирта, сушат при 65—70°C. Получают 380 г II. Метанольные растворы упаривают до объема 300 мл, охлаждают 1 ч при 0—(4-5°С). Осадок II отфильтровывают, промывают 2 раза по 15 мл охлажденного до 0°С этилового спирта, сушат при 65—70°C. Общий выход II 421 г (84%).
4,6-Диоксипиримид'ин'(П1). Смесь 1,955 кг 19% раствора метилата натрия в метаноле, 220 г II и 234 г формамида в течение 2 ч постепенно при перемешивании нагревают, до кипения и кипятят 2 ч. Охлаждают до 10—15°С, прибавляют 1,14 л воды и полученный раствор подкисляют конц. соляной кислотой до pH 2,8—3,1. Дают выдержку 1 ч при 5—10°C с перемешиванием. Осадок отделяют, промывают 3 раза по 200 мл воды и сушат при 60—70 °C. Получают 182 г III с содержанием основного вещества 95%. Выход 72%, считая на II.
4,6-Дихлорпиримидин (IV). К 2,21 кг III и 6,44 л хлорокиси фосфора постепенно в течение 30 мин при перемешивании и температуре не выше 35 °C приливают 1,28 л диметиланилина. Затем массу постепенно в течение Р/г— 2 ч нагревают до кипения, кипятят З’/г ч, охлаждают до 20—25°C и в течение 2’/г ч при перемешивании выливают в охлажденные до 10 °C 38 л воды. Перемешивание продолжают без охлаждения еще 1 ч; при этом температура массы повышается до 19—21 °C. Осадок IV отфильтровы-
эа
вают, промывают 2 раза по 1 л воды. Из водного фильтрата дихлорэтаном (2 раза по 3 л) дополнительно экстрагируют IV, который объединяют с отфильтрованным продуктом (IV легко возгоняется, поэтому отгонку дихлорэтана ведут при температуре не выше 60°C). Общий выход IV 2,094 кг (75%).
4-Амино-6-хлорпиримидин (V). Смесь 510 мл водного аммиака и 216 г IV нагревают в закрытой системе 1 ч до 50°C и 4’/г ч при 50—60°C (давление 1—2 атм), охлаждают до 20 °C. Контролируют методом ТСХ содержание в реакционной массе IV (должно быть менее 0,5%), охлаждают до 8—12 °C и дают выдержку 1 ч. Осадок V отфильтровывают, высушивают при 65—70 °C. Выход V 116 г (83%).
4-Амино-6-метоксипиримйдин (VI). К раствору 513 г едкого натра в 6,21 л метанола прибавляют 1,161 кг V. Массу кипятят 7 ч, охлаждают до 20 °C, контролируют методом ТСХ отсутствие V, отгоняют 5,16 л метанола. К остатку приливают 2,61 л воды и отгоняют водный метанол до температуры в парах 98—100°С. Массу охлаждают до 0—3°С с перемешиванием, дают выдержку 1 ч. Осадок отфильтровывают, промывают 1,3 л охлажденной до 1—3°С воды (VI достаточно хорошо растворим в воде, поэтому полностью отмывать едкий натр и хлористый аммоний нецелесообразно). Слабощелочная реакция (pH 8) не является препятствием для проведения следующей стадии; сушат при 60—70°C. Выход 847 г (80%).
4-Карбометоксисульфаниламидо-6-метоксипиримидин (V111). К смеси 4,4 л пиридина и 1,103 кг VI при 18— 20 °C (охлаждение) прибавляют порциями в течение 30— 35 мин 2,512 кг перекристаллизованного из дихлорэтана VII (содержание влаги во всех продуктах не должно превышать 0,1%). Массу перемешивают 2 ч при 18—20°C. При этом происходит образование 4,4-ди-(карбометоксисульфаниламидо)-6-метоксипиримидина), затем постепенно нагревают до 55°С и дают выдержку 2 ч при перемешивании и температуре 55—60 °C [4,4-ди-(карбометоксисульфаниламидо)-6-метоксипиримидин реагирует с оставшимся VI, превращаясь в VIII]. Охлаждают до 20°C и выливают в 5,1 л воды, после чего постепенно в течение 2 ч при 18—23 °C добавляют 14,8 л 10% соляной кислоты до pH- 2,8—3,1. Осадок VIII отфильтровывают, промывают 3 раза по 1,5 л воды и сушат при 65—70 °C. Выход 3,227 кг (93%). Технический продукт с содержанием основного вещества 84,9% без дополнительной очистки используют на следующей стадии.
7 Заказ № 4304
97
4-(п-Аминобензолсульфамидо)-6-метоксипиримидин (сульфамонометоксин) (IX). К смеси 23,9 л воды и 1,606 л 44% раствора едкого натра прибавляют 3,227 кг VIII. Массу нагревают 1 ч при 88—90°C. Контролируют конец реакции по полному растворению отобранной пробы в избытке разбавленной соляной кислоты, после чего охлаждают до 50—60 °C, присыпают 274 г угля, перемешивают 30 мин при 50—60 °C и фильтруют. К фильтрату при 60— 70°C постепенно (вспенивание за счет выделения СОг!) приливают 10% соляную кислоту до pH 5,3—6, охлаждают до 20°C (выдержка 30 мин при перемешивании). Осадок отделяют, промывают водой 3 раза по 1 л, растворяют в смеси 18,93 л воды и 3,118 л 10% раствора едкого натра, осветляют 218 г угля при 50—60°C (30 мин), фильтруют. К фильтрату при 50—60°C приливают 10% соляную кислотулдо pH 5,3—6,0. Суспензию охлаждают до 20°C (выдержка при перемешивании 30мин). Осадок отделяют, промывают 80 л дистиллированной воды, высушивают при 70—75°C. Выход 1,939 кг (85,5%).
ФТАЗИН
6- (п-Фталиламинобензолсульфамидо) -3-метоксипирида-зин:
Мол. масса 428,41
Белый или белый со слегка желтоватым оттенком кристаллический порошок, без запаха, практически нерастворим в воде, спирте и разведенных кислотах, растворим в горячей ледяной уксусной кислоте, в растворах щелочей и гидрокарбоната натрия, в диметилформамиде; т. пл. 180 °C. При нагревании отщепляет воду, превращаясь в фталимидное производное с т. пл. 250—252 °C; при длительном кипячении с водой гидролизуется; ФС 42-475-72.'
Фтазин — оригинальный сульфамидный препарат [51, 187, 190] продотированного действия, используемый при лечении кишечных заболеваний. Являясь N-фталоильным производным сульфапиридазина — сульфаниламидного препарата длительного действия,, фтазин плохо всасывается и постепенно гидролизуется с образованием сульфа-
98
пириДазина, накапливающегося в высоких концентрациях в толстом кишечнике. Препарат действует дольше, чем фталазол, но и обладает несколько большим резорбтивным действием.
Применяется фтазин при лечении дизентерии, гастроэнтеритов, энтероколитов, колитов.
Выпускается в таблетках по 0,5 г N. 10. Цена 41 к.
Метод синтеза фтазина разработан во ВНИХФИ [34, 51] по схеме:
Сульфапиридазин (I) [211] подвергают конденсации с фталевым ангидридом (II) в водной среде [34] по аналогии с синтезом фталазола [211] и очищают III переводом через натриевую соль с последующим выделением соляной кислотой. Этот метод имеет преимущества по сравнению с ранее описанной конденсацией I и II в абсолютном этиловом или в изопропиловом спирте [51].
Смесь 534 г II и 6,3 л воды кипятят 1 ч. При этом фталевый ангидрид переходит во фталевую кислоту. Массу охлаждают до 80 °C и при перемешивании добавляют 630 г I. Дают выдержку 6 ч, после чего постепенно в течение Р/г ч охлаждают до 50 °C, выдерживают 1 ч при этой температуре и осадок III отделяют. (При более высокой температуре выход снижается за счет растворения фтазина; фильтрация при более низкой температуре приводит к загрязнению продукта выпадающей в осадок фталевой кислотой.) Осадок на фильтре тщательно отжимают и промывают 2 раза по 6 л нагретой до 50°С воды с целью наиболее полно удалить примеси фталевой кислоты. Полученную пасту растворяют в нагретом до 40 °C растворе 192 г едкого натра в 9,76 л воды. Дают выдержку 30 мин при 50 °C. В этих условиях примесь фталимидного производного также превращается в натриевую соль фтазина. Повышение концентрации щелочи может привести к N-дезацилированию. Раствор осветляют 90 г угля (выдержка 10 мин при 50°C), фильтруют и при 40°C под-7* 99
кйсляют 18% соляной кйслбтой до pH 5,0. Реакционную массу перемешивают еще 1 ч, контролируют pH, охлаждают до 15 °C (выдержка 2 ч) и фильтруют. Осадок промывают 5 л холодной водопроводной воды и 1 л дистиллированной воды до отсутствия в промывных водах хлор-ионов, сушат при 70—100°C. Выход 875 г (90,5%).
САЛАЗОПИРИДАЗИН
5- (п- [N- (З-Метоксипиридазинил-6) -сульфамидо] -фенил-аЗо)-салициловая кислота:
Мол. масса 429,42
Мелкокристаллический порошок оранжевого цвета, практически нерастворим в воде, очень мало растворим в хлороформе, мало растворим в спирте, ацетоне и ледяной уксусной кислоте, легко растворим в диметилформамиде и растворах едкого натра; т. пл. 200—210 °C разл. (в интервале 2°C); ВФС 42-202-73.
Салазопиридазин — оригинальный сульфаниламидный препарат [125, 189] с противовоспалительным и иммунодепрессивным действием. Относится к группе салазо-сульфамидов, представитель которой сульфасалазин (са-лазосульфидин), предложенный шведскими авторами [299], уже ранее нашел использование в клинике неспецифического язвенного колита. Однако применение для этих целей салазопроизв^^йых сульфамидов длительного действия, в том числе и сульфапиридазина, известно не было. В литературе имелось сообщение об определении в крови и моче людей салицилазосульфамонометоксипиридазина и продуктов его расщепления [282], но отсутствовали какие-либо сведения об антимикробных свойствах этого соединения, его химиотерапевтической активности при бактериальных инфекциях и лечебном действии при язвенных коликах.
Применяют салазопиридазин при всех формах неспецифического язвенного колита как средство, более эффективное, чем сульфасалазин.
Выпускается в таблетках по 0,5 г N. 50. Цена 3 р. 02 к.
100
Метод синтеза салазопиридазина разработан во ВНИХФИ по схеме:
соон Vi
Сульфапиридазин (I) [211] переводят обработкой едким натром в водорастворимую натриевую соль (II), которую диазотируют нитритом натрия в соляной кислоте (нитрита натрия берут эквимолярное количество, так как при его избытке происходит нитрозирование по азоту сульфамидного фрагмента; соляной кислоты применяют до 4,5 молей на 1 моль I, чтобы воспрепятствовать образованию в слабокислой среде диазоаминосоединения за счет реакции III с I). Полученную диазониевую соль III вводят в реакцию азосочетания с динатриевой солью салициловой кислоты IV в присутствии едкого натра (экспериментально установлено, что оптимальные молярные соотношения III и IV составляют 1 :2; уменьшение избытка IV замедляет реакцию и ухудшает качество VI; прибавление IV к III ведут одномоментно, так как в противном случае азосочетание будет проходить при разных pH с образованием различных соединений, в том числе продуктов, лишенных карбоксильной группы). Из тринатриевой соли азосоединения V 5-(п-[Ы-(3-метоксипиридазинил-6)-сульфамидо] -фенилазо)-салициловую кислоту (салазопиридазин) (VI) выделяют соляной кислотой. Все операции проводят последовательно без получения индивидуальных промежуточных продуктов II, III и V. Очистку салазопиридазина осуществляют путем перевода его с гидрокарбонатом натрия в водорастворимую мононатриевую соль и последующего осаждения VI соляной кислотой. В основу выбора метода синтеза салазопиридазина положен анализ общих способов азосочетания сульфамидных препаратов и салициловой кислоты.
101
К раствору 62 г едкого натра в 1,37 л воды прибавляют 421 г I, перемешивают при 20—30 °C до полного растворения осадка (5—10 мин), после чего прибавляют 108г нитрита натрия. Полученный раствор в течение 20—30 мин выливают в охлажденную до 0°С смесь 720 мл соляной кислоты и 1^5 л воды. Реакция диазотирования протекает быстро и в осадок выделяется ярко-желтого цвета соль диазония III. Дают выдержку 1 ч при 0°С—(+5°C), определяют конец реакции по йодкрахмальной пробе. Массу охлаждают до —6—(—8 °C) и в охлажденную суспензию при перемешивании быстро, в один прием приливают охлажденный до —6°С—(—10°С) раствор 415 г салициловой кислоты и 460 г едкого натра в 1,95 л воды. Образуется раствор V красно-бурой окраски. При этом нельзя допускать, чтобы наблюдающееся за счет экзотермич-ности реакции нейтрализации повышение температуры реакционной массы превысило 10 °C. Дают выдержку 1 ч при 3—5 °C, контролируют окончание процесса по пробе с Н-кислотой. Полученный раствор разбавляют 21 л воды, осветляют 60 г угля и при 5—10 °C к нему в течение 2 ч приливают 18% соляную кислоту до pH 5—4,5 (осторожно! масса пенится). После выдержки 3 ч проверяют pH среды, осадок отделяют, промывают 1 л воды и влажную пасту растворяют в течение Р/г ч в 20 л 2% раствора гидрокарбоната натрия при 30—35°C (при температуре выше 40°C происходят побочные реакции). Проверяют полноту растворения продукта, массу осветляют 60 г угля, фильтруют и раствор подкисляют 18% соляной кислотой до pH 1—2 (осторожно! масса пенится) (хранение щелочного раствора VII недопустимо, так как при этом происходят побочные реакции с образованием веществ неустановленного строения). Осадок VI отделяют, промывают 21 л воды (до отрицательной реакции в промывной воде на хлор-ион), сушат при 70—80°C. Выход 548 г (85%).
САЛАЗОДИМЕТОКСИН
5- (п- [N- (2,4-Диметоксипиримидинил-6) -сульфамидо] фенилазо)-салициловая кислота:
102
Буровато-оранжевого цвета мелкокристаллический порошок, без запаха, практически нерастворим в хлороформе и воде, мало растворим в спирте и ледяной уксусной кислоте, легко растворим в диметилформамиде и растворах едкого натра; т. пл. 215—219 °C в интервале 2 °C; ВФС 42-202-73.
Салазодиметоксин — оригинальный сульфаниламидный препарат [188], активный в отношении диплококков, стрептококков, гонококков, кишечной палочки. Подобно сала-зопиридазину (см. с. 100) и сульфосалазину, распадается в кишечнике с образованием 5-аминосалициловой кислоты и невключенного в азосоединение сульфаниламидного производного (в данном случае сульфадиметоксина), которые оказывают противовоспалительное и антибактериальное действие.
Препарат малотоксичен и действует в меньших дозах, чем салазосульфидин.
Применяют салазодиметоксин при лечении неспецифического язвенного колита в легких и среднетяжелых формах заболевания, а также при болезни Крона.
Выпускается в таблетках по 0,5 г N. 50. Цена 3 р. 02 к.
Метод синтеза салазодиметоксина разработан во ВНИХФИ по схеме [85].
103
Сульфадиметоксин (I) переводят обработкой едким натром в водорастворимую натриевую соль (II), которую диазотируют нитритом натрия в соляной кислоте. (Диазотирование I гидрохлорида приводит к получению VI, загрязненного I.) Диазониевую соль III вводят в реакцию азосочетания с динатриевой солью салициловой кислоты (IV) в присутствии едкого натра и из образовавшейся тринатриевой соли азосоединения (V) обработкой соляной кислотой получают 5-(п-[Ь1-(2,4-диметоксипиримидинил-6)-сульфамидо] фенилазо)-салициловую кислоту (салазоди-метоксин) (VI). Все Операции проводят без выделения промежуточных продуктов. Очистку готового салазодиме-токсина осуществляют переводом с гидрокарбонатом натрия в водорастворимую мононатриевую соль с последующим осаждением VI соляной кислотой. В основе метода лежит общий способ азосочетания сульфамидных препаратов и салициловой кислоты.
К раствору 41 г едкого натра в 1,12 л воды прибавляют 313 г I, перемешивают при 20—30 °C до полного растворения осадка (5—10 мин), после чего прибавляют 75 г нитрита натрия (уменьшение или увеличение количества нитрита натрия ухудшает качество VI). Полученный раствор в течение 45 мин—1 ч выливают в охлажденные до 2 °C 1,29 л соляной кислоты (при меньшем количестве соляной кислоты образующийся III выпадает в осадок, что затрудняет дальнейшее получение V; кроме того, избыток соляной кислоты предотвращает побочные реакции, в частности образование аминоазосоединений). Дают выдержку 1 ч при 2°C—(+5°C), контролируют окончание процесса по йодкрахмальной пробе и полученный раствор III приливают в течение 40—50 мин к охлажденному до 2 °C раствору 150 г салициловой кислоты в 2,6 л водного раствора едкого натра, содержащего 667 г едкого натра (избыток щелочи необходим для нейтрализации кислого раствора диазониевой соли и создания необходимого pH среды при азосочетанЙй; дальнейшее повышение концентрации едкого натра способствует вытеснению карбоксильной группы в салициловой кислоте, а также образованию малореакционноспособного диазотат-иона). Дают выдержку 1 ч при 2—6 °C, проверяют окончание реакции (по пробе с Н-кислотой) и полученный раствор выливают в 12 л воды (при меньшем количестве воды VI оказывается загрязненным салициловой кислотой). Дают выдержку 20 мин при 15—20 °C, осветляют 30 г угля и в течение Р/г ч приливают 18% соляную кислоту до pH 2,3—2,6
Г04
(осторожно! наблюдается вспенивание). После выдержки 1 ч проверяют pH среды, осадок VI отделяют, промывают 3 л воды и влажную пасту растворяют в 15 л 2% раствора гидрокарбоната натрия, осветляют углем (25 г), раствор подкисляют 18% соляной кислотой до pH 2,3—2,6. Осадок VI отделяют, промывают 12 л воды и высушивают при 70—80°C. Выход 391 г (85%).
ХЛОРПРОПАМИД
N- (п-Хлорбензолсульфонил) -N'-пропилмочевина:
ci
so2nhconhg3h7
с ю н ]3 ctN2oas
Мол. масса 276,74
Основные синонимы: биоглюмин, галирон, диабамид, диабарил, диабет, диабексан, диабенезе, катанил, мелине-зе, продиабен и др. Белый кристаллический порошок, без запаха и вкуса, практически нерастворим в воде, мало растворим в эфире, растворим в спирте, ацетоне, бензоле и хлороформе; т. пл. 126—130°C (в интервале 2°C); ГФХ, ст. 162.
Хлорпропамид— противодиабетический сульфамидный препарат, не оказывающий антибактериального действия [86, 238], более активный, чем бутамид и цикламид (см. с. 107), быстро всасывается из желудочно-кишечного тракта: обнаруживается в крови в течение 1-го часа после приема, пик концентрации в крови достигается через 2—4 ч. Медленно выделяется через почки, главным образом в неизмененном виде. Длительность действия после однократного приема 24—36 ч.
Применяют хлорпропамид при сахарном диабете легкой и средней тяжести. Препарат эффективен у ряда больных, слабо реагирующих на бутамид и цикламид, а также при развитии устойчивости к последним во время лечения. Иногда назначают вместе с инсулином для усиления его действия. Во избежание гипогликемии препарат применяют обычно 1 раз в сутки — до или во время завтрака: высшая суточная доза 1 г.
Выпускается в таблетках по 0,25 г N. 20. Цена 24 к.
105
Метод синтеза хлорпропамида разработан Харьковским заводом эндокринных препаратов на основе известного метода, описанного в литературе, по схеме [86, 238]:
Путем конденсации п-хлорбензолсульфониламида (I) с мочевиной получают п-хлорбензолсульфонилмочевину (II). Параллельно из масляной кислоты (III) через амид (IV) с дальнейшим использованием перегруппировки по Гофману готовят сульфат н-пропиламина (V). При взаимодействии II с V получают Ы-(п-хлорбензолсульфонил)-М'-пропилмочевину (хлорпропамид) (VI). Менее удобны для этой цели другие описанные в литературе методы синтеза VI, основывающиеся на использовании в реакции с I н-пропилизоцианата или веществ, которые в процессе синтеза легко образуют н-пропилизоцианат.
п-Хлорбензолсульфонилмочевина (II). Смесь 3 кг I, 1,04 кг мочевины и 180 г кристаллического едкого натра нагревают 2 ч при Ю^-Х-116 °C, охлаждают до 40—50°C и растворяют в 20 л воды. Раствор осветляют 75 г угля, фильтруют, подкисляют ~2,05 кг соляной кислоты до pH 4—5. Осадок отделяют, промывают 15 л воды до pH 6,5—7 и высушивают при 85—90°C. Выход 2,66 кг (74,9%).
н-Пропиламина сульфат (V). В 5 кг III пропускают газообразный аммиак с такой скоростью, чтобы через массу проходили (на выходе) только следы аммиака, а температура в реакционной среде не была выше 135 °C (реакция экзотермична). Затем отгоняют с использованием колонны образовавшуюся воду при температуре в массе до 115 °C. Температуру повышают до 180—190 °C и дают выдержку 24 ч с продувкой аммиака. Контролируют, чтобы содержание III в массе не превышало 20%', после чего дают смеси охладиться до 70—80 °C, приливают 4 л воды и размешивают 10 мин. Полученный раствор приливают к смеси раствора 3,67 кг едкого натра в 5 л воды, 3,88 кг гипохлорита кальция и 4,65 кг льда. Дают выдержку 1 ч при 45—50 °C и 1 ч при 60—65 РС, после чего н-пропиламин отгоняют с паром в раствор 1,08 кг серной кислоты в 2 л воды. Получают 11 л раствора, содержа
106
щего 2,09 кг V (67,2%, считая на III), который без дополнительной обработки используют на следующей стадии.
Н-(п-Хлорбензолсульфонил)-М'-пропилмочевина (хлорпропамид) (VI). От приготовленного на предыдущей стадии водного раствора V отгоняют воду сначала без вакуума, затем в вакууме, остатки воды — в виде азеотропной смеси с толуолом. К оставшейся смеси V с толуолом при 50—60°C добавляют 4 кг II и массу кипятят 2 ч. После охлаждения до 60 °C прибавляют 8 л воды, перемешивают, водный слой отделяют, к толуольному приливают раствор 920 г едкого натра в 15 л воды, перемешивают. Водный раствор натриевой соли хлорпропамида отделяют, осветляют 120 г угля, отдувают остатки толуола нагреванием 10 мин при 70—80 °C с продуванием воздуха, фильтруют. К фильтрату приливают 12 кг изопропилового спирта, охлаждают до 20 °C и добавлением конц. серной кислоты доводят при температуре не выше 24 °C pH раствора до 5—5,5. Массу охлаждают до 15—20 °C. Осадок VI отфильтровывают, промывают 4,5 кг 50% изопропилового спирта, затем водой до нейтральной реакции и отсутствия сульфат-ионов в промывной воде. Выход 3,56 кг (74,8%).
ЦИКЛАМИД
Ц-(п-Толуолсульфонил)-М'-(циклогексил) мочевина:
С 14 Н 20 N2°3S
Мол. масса 296,39 Основные синонимы: адабил, аглирал, грироксил, цик-лорал, диаборал, толикламид, толгексамид.
Белый кристаллический порошок, без запаха, практически нерастворим в воде, мало растворим в эфире, растворим в спирте, легко растворим в хлороформе; т. пл. 171— 173 °C; ФС 42-1282-79.
Цикламид*—противодиабет'ический препарат, менее токсичный и несколько более активный, чем бутамид. Быстро всасывается и медленно выделяется из организма. Не обладает антибактериальной активностью. Механизм действия связан со стимуляцией 0-клеток поджелудочной
107
железы, сопровождающейся мобилизацией и усилением выброса эндогенного инсулина.
Применяют цикламид, как и другие препараты группы сульфонилмочевины, в сочетании с соответствующей диетой у больных сахарным диабетом в возрасте старше 35 лет, неистощенных, без кетоацидоза, осложнений или сопутствующих заболеваний, требующих немедленной ин-сулинотерапии.
Выпускается в таблетках по 0,25 г N. 20. Цена 24 к.
Метод синтеза цикламида разработан Украинским институтом экспериментальной эндокринологии и Харьковским заводом эндокринных препаратов по схеме: •
NH,
п-Толуолсульфониламид (I) подвергают взаимодействию с мочевиной в присутствии едкого натра. Полученное натриевое производное растворяют в воде и подкислением до pH 6,5—7 выделяют п-толуолсульфонилмочевину (II), которую реакцией с циклогексиламином превращают в N- (п-толуолсульфонил)-М'- (циклогексил) мочевину (III). Соединение III очищают путем перевода через натриевое производное. Синтез III из натриевого производного п-толу-олсульфониламида и циклогексилизоцианата технологически менее приемлем.
п-Толуолсульфонилмочевина (II). 2,5 кг сухого 1,930г мочевины и 620 г кристаллического едкого натра тщательно смешивают и нагревают 3 ч при 112—116 °C. Охлажденную до 70—60 °C реакционную смесь растворяют в 18 л воды, фильтруют и подкисляют соляной кислотой (~ 2— 2,1 кг) до pH 5—4. Осадок II отделяют, промывают водой до pH 6,5—7 в промывной воде, сушат при 90 °C; Выход 2,91 кг (88,9%).
М-(п-Толуолсульфонил)-М'-(циклогексил)мочевина (цикламид) (III). К смеси 7,5 кг толуола и 1,56 кг высушенного перегнанного циклогексиламина приливают 720 г конц. серной кислоты, добавляют 3 кг II и кипятят массу 3 ч, после чего охлаждают ее до 80—70 °C и приливают 8 л воды. Водный слой отделяют, к толуольному раствору. прибавляют 11,76 кг водного раствора, содержащего 108 .
700 г едкого натра. Массу нагревают 10 мин при 50— 55 °C, водный раствор натриевого производного циклами-да отделяют, добавляют 180 г угля и отдувают воздухом остатки толуола (30 мин при 80—90°C). Затем уголь отделяют, к фильтрату приливают 13,5 кг изопропилового спирта и конц. соляную кислоту (~1,49 кг) до pH 5,5—5. Охлаждают до 30—25°С, дают выдержку 1 ч, III отфильтровывают, промывают 6,5 кг 45% водного изопропилового спирта, затем 15 л воды (до pH промывных вод 6,5—7), 12. л дистиллированной воды (до отсутствия в промывной воде реакции на сульфат-ион с ВаС1г) и высушивают при температуре не выше 90°C. Выход 2,68 кг (67,5%).
ХЛОЦИКЛАМИД
М-(п-Хлорбензолсульфонил)-М'- (циклогексил) мочевина:
so2nhconh
c)3hi7cin2o3s
Мол. масса 316,80
Основные синонимы: орадиан, хлоргексамид.
Белый кристаллический порошок, без запаха, практически нерастворим в воде, мало растворим в толуоле, растворим в спирте, легко растворим в ацетоне; т.пл. 169— 173°C разл. (в интервале 2°C); ФС 42-924-74.
Хлоцикламид — противодиабетический препарат [37], несколько более активный, чем цикламид; в некоторых случаях эффективен при резистентности к другим пероральным гипогликемическим средствам.
Показания к применению такие же, как у цикламида (см. с. 106) [37].
Выпускается в таблетках по 0,25 г. N. 20. Цена 24 к.
Метод синтеза хлоцикламида разработан Харьковским заводом эндокринных препаратов [138] по схеме:
I) HjNCONH,. NaOH
SO.NHCONH.
-SO NHCONH
109
которая аналогична схеме синтеза цикламида. Путем взаимодействия п-хлорбензолсульфониламнда (I) с мочевиной в присутствии едкого иатра с последующей обработкой соляной кислотой пбйучают п-хлор-бензолсульфонилмоче-вину (II) (как описано в синтезе хлорпропамида, см. с. 106). II реакцией с циклогексиламином переводят в N- (п-хлорбензолсульфонил) -N'- (циклогексил) мочевину (III). Этот метод является преимущественным по сравнению с описанным получением III из п-хлорбензолсульфо-ниламида натрия и циклогёксилизоцианата или десульфуризацией N- (п-хлорбензолсульфонил) -N'- (циклогексил) тиомочевины при обработке гидроокисями серебра и трехвалентного железа.
К смеси 6,4 кг толуола и 1,15 кг перегнанного цикло-гексиламина приливают 610 г конц. серной кислоты, добавляют 2,5 кг II; массу кипятят З’/г ч, охлаждают до 80°C и приливают 6 л воды. Водный слой отделяют; к толуольному раствору прибавляют 13 кг водного раствора, содержащего 500 г едкого натра; массу нагревают 5— 8 мин при 80—85 °C. Водный слой отделяют, обрабатывают 65 г угля, отдувают воздухом остатки толуола (10 мин при 80—85°C), прибавляют 6,8 кг изопропилового спирта, фильтруют, разбавляют еще 4,2 кг изопропилового спирта и добавляют конц. серную кислоту до pH 5,5—5, охлаждают до 25—30°C (3 ч), фильтруют, осадок III промывают изопропиловым спиртом, водой (пробы на нейтральные промывные воды—pH 7 и отсутствие сульфат-иона с ВаС1?) и высушивают.
Выход 2,45 кг ('74^7%).
ЭТАМИД
N.N-Диэтил-п-карбоксибензолсульфониламид
ноос
C„Hl5NO4S
so2n(c2h5)2
Мол. масса 257,30
Основные синонимы: этебенецид, лонгацид, урелим.
Белый кристаллический порошок, без запаха и вкуса, практически нерастворим в воде, трудно растворим в спирте и эфире, растворим в разбавленных растворах едких
110
н углекислых щелочей; т. пл. 192—197°С (в интервале 2°C); ФС 42-1229-79.
Этамид тормозит реабсорбцию мочевой кислоты в почечных канальцах, способствует выведению ее с мочой и уменьшению содержания в крови, обладает способностью задерживать выделение почками пенициллина и некоторых других веществ, пролонгируя их действие в организме [217].
Применяется при хронической подагре, полиартритах с нарушением пуринового обмена, мочекаменной болезни с образованием уратов, для пролонгирования действия пенициллина [217].
Выпускается в таблетках по 0,35 г. N. 50. Цена 72 к.
Метод синтеза этамида разработай на кафедре химии Военно-медицинской академии им. С. М. Кирова по схеме [90]:
и усовершенствован во ВНИХФИ без изменения общей схемы синтеза.
Окислением п-толуолсульфонилхлорнда (I) бихроматом натрия получают п-карбоксибензолсульфонилхлорид (II), который обработкой диэтиламином превращают в N.N-диэтил-п-карбоксибензолсульфониламид (этамид) (III). Этот метод дает лучшие результаты, чем окисление п-толуолсульфониламида бихроматом калия или перманганатом калия с последующим алкилированием п-карбок-сибензолсульфониламида бромистым этилом. Преимущественным он является и перед методом, включающим окисление щелочным перманганатом калия п-толуолсуль-фокислоты с последующей обработкой хлорсульфоновой кислотой и диэтиламином, а также методом, основанным на диазотировании п-аминобензойной кислоты, обработке образующейся соли диазония сернистым ангидридом и взаимодействии получающегося п-карбоксибензолсульфо-нилхлорида с диэтиламином.
п-Карбоксибензолсульфонилхлорид (И). К смеси240мл 15% раствора I в уксусной кислоте и 56,3 г бихромата натрия при перемешивании постепенно в течение 1 ч прибавляют 94,5 мл 92,5% серной кислоты, поддерживая температуру массы (внешнее охлаждение) в интервале 55— 60 °C. По мере прибавления серной кислоты масса густеет
П1
и в осадок выпадает II. После добавления всего количества кислоты массу перемешивают еще 30 мин, затем приливают 500 мл охлажденной до 10 °C воды и охлаждают до 15 °C с перемешиванием. Дают выдержку 30 мин. Выпавший осадок отделяют, промывают 3 раза по 250 мл воды (до pH 5—6). Влажную пасту используют на следующей стадии. Выход II с содержанием влаги ~30%— 56,6 г (94,1%).
М,М-Диэтил-п-карбоксибензолсульфониламид (этамид) (П1). К охлажденным до 5 °C 86,4 мл 8% раствора едкого натра прибавляют 21,6 мл технического диэтиламина и затем при 5 °C 56,6 г пасты II, полученной на предыдущей стадии. Прибавление ведут с такой скоростью, чтобы температура не была выше 7 °C. Приливают еще 86,4 мл 8% раствора щелочи, перемешивают 2 ч, в течение которых температура самопроизвольно повышается до 15— 18°С. Затем к раствору приливают 114 мл 20% уксусной кислоты; температура повышается до 30 °C и выпадает осадок. Массу охлаждают до 18—20 °C, дают выдержку при перемешивании 2 ч. Осадок отфильтровывают, промывают 300 мл вода и растворяют в нагретых до 60 °C 457 мл 1,5% раствора едкого натра. Раствор осветляют 2,7 г угля (30 мин при 60°C с перемешиванием), фильтруют и при 60°C к фильтрату прибавляют 119 мл 20% уксусной кислоты (до pH 4). Дают выдержку 1 ч при перемешивании и 60 °C, охлаждают до 16—20 °C. Осадок отфильтровывают, промывают 360 мл воды и сушат при 80°C. Выход III 35,4 г (77,6%).
КИСЛОРОД- ИЛИ СЕРОСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Изыскание новых лекарственных средств в этом классе веществ проводилось путем варьирования структуры известных медицинских препаратов и выявления закономерностей связи между строением и биологической активностью. Практическим результатом этих исследований явилось создание ряда оригинальных медикаментов антимикробного и сердечно-сосудистого действия. Кроме того, был воспроизведен известный за рубежом противокашле-вой препарат битиодин (см. с. 129), лишенный наркотических свойств [202].
Важным классом антимикробных химиотерапевтических средств являются производные 5-нитрофурана, эффективность которых впервые была обнаружена исследо-112
вателями Нориджской школы (Великобритания), искавшими пути утилизации фурфурола, и практически использована группой английских и американских клиницистов [284, 296]. Начатые в 50-е годы в Институте органического синтеза АН ЛатвССР систематические исследования в ряду производных 5-нитрофурана позволили в короткие сроки не только’организовать промышленное производство в нашей стране наиболее эффективных из известных за рубежом антибактериальных препаратов этого ряда: фурацилина (1а) [211], фуразолидона (16) [40], фуразо-лина (1в) [81], фурадонина (1г), но и получить оригинальной антибактериальный препарат фурагин (см. с. 116) и его водорастворимую калиевую соль — солафур [236]. Этим исследованиям посвящены обзоры [3,24].
Методы получения лекарственных препаратов фурацилина, фуразолидона и фуразонала описаны в справочнике [211], Следует отметить, что спектр химиотерапевтического действия препаратов 5-нитрофуранового ряда различен: фурацилин (1а) действует на грамотрицательные и грам-положительные микроорганизмы и наиболее широко применяется наружно, фуразолидон (16) наиболее активен в отношении грамотрицательных микробов, а также трихо-монад и лямблий, фуразолин (1в) эффективен преимущественно в отношении грамположительных возбудителей. Фурадонин (1г) и фурагин нашли применение при инфекционных заболеваниях мочевых путей.
Дальнейшее развитие получили также исследования. 4-оксикумариновых антикоагулянтов, берущие начало с установления структуры дикумарина (Па). Это вещество— действующее начало испорченного сладкого клевера, вызывающего массивные внутренние кровотечения у крупного рогатого скота, — оказалось активным в клинике у людей при лечении тромбозов и эмболий. За рубежом была создана серия антикоагулянтов длительного действия, принадлежащих к дикумаринам — дикумарин (Па),
8 Заказ № 4304 113
неодикумарин (Пб) [294] и монокумаринам — варфа-рии (111а) [319, 339], синкумар (Шб) [339], маркумар (Шв) [296]:
II
a)R*H; 6)R«COOC,H5
ОН
III
CH R1 I ' CH—<
R
a|R’?COCH,. R?-H 6)R'-C0CH.t R’^NO^ h) R’=CHT R?=H
'‘r \'
В МХТИ им. Д. И. Менделеева исследования в этой области, предпринятые первоначально с целью поиска новых средств для борьбы с грызунами — ратицидных препаратов [220], привели к получению не более, а менее токсичных веществ. Исходя из этого указанные исследования были переориентированы и совместно со II МОЛГМИ им. Н. И. Пирогова были направлены на изыскание новых, более эффективных и менее токсичных, лекарственных средств, выявление новых закономерностей связи строения и антикоагулянтных свойств в этом ряду соединений [121, 122]. Ранее работами английских ученых было показано [319], что для антикоагулянтной активности необходимо наличие незамещенной в фенильном ядре кумариновой системы со свободной оксигруппой в положении 4 и углеводородным остатком в положении 3. Советские исследователи [122, 220] установили, что при удлинении ацильного заместителя в соединении Ша на одну метиленовую группу сохраняется высокая антикоагулянтная активность, скорость ее проявления и продолжительность действия, ио значительно уменьшается токсичность. Дальнейшее удлинение ацильного остатка резко снижает антикоагулянтное действие, а разветвление боковой цепи сопровождается появлением коагулянтных свойств. Замена в соединениях III фенильного остатка в a-положении боковой цепи на бифенильный или 0-наф-тильный не меняет антикоагулянтных свойств, алкильные заместители у а-углеродного атома резко снижают активность соединений. Введение нитрогруппы в п-положение фенильного ядра 3-(а-фенил-р-пропионилэтил)-4-оксику-марииа (III, Р'=СОСН2СНз) значительно повышает антикоагулянтное действие, делает более быстрым его проявление и сокращает продолжительность эффекта. Введение нитрогруппы в боковую цепь резко снижает антикоагулянтную активность. Все это позволило создать и внедрить в медицинскую практику новые оригинальные
114
лекарственные препараты — фепромарон (HI, R’ = = СОСНгСНз; R2=H; см. с. 122) и нитрофарин (III, R1 = COCH2CH3; R2 = NO2; см. с. 123).
В результате совместных исследований во ВНИХФИ и Винницком медицинском институте большой серии разнообразных производных бензофурана и индола' был создан оригинальный коронарорасширяющий и спазмолитический бензофурановый препарат — феникаберан (см. с. 118) [54, 56, 59].
6) R=(CH3)nAr
в) R=CH3CH3COAr
г) R=CHjCH3CO-f
a) R=H
О
О
Оригинальный а-адреноблокатор 1,4-бензодиоксаиового ряда — пирроксан (Vr) (см. с. 125) появился в результате систематических исследований в Институте токсикологии Министерства здравоохранения СССР влияния на адренергические системы циклических аналогов р-фенил-алкиламинов (IV)—производных 3-фенилпирролиди-на (V) [73].
Соединение Va проявило в эксперименте адреномиметическое действие. Введение к азоту арилалкильных заместителей обусловило появление у веществ V6 адреноли-тических свойств. Аналогичные 2-фенилпирролидиновые производные были лишены этой активности [73]. Ослабление адренолитических свойств наблюдалось и у 3-фенил-пиперидиновых аналогов активных соединений V6, что авторы связали с большей конформационной подвижностью пиперидинового цикла по сравнению с пирролидиновым [74]. Активность исследованных производных N-арилал-кил-3-фенилпирролидинов (V) оказалась существенно зависящей от длины алкильной цепи в R и характера заместителей в ней [73,74,248]. Наиболее активны вещества с кетогруппой в боковой цепи — производные ₽-(3-феиилпирролидил-1) пропиофенона (Vb) [248]. Дальнейшее удлинение боковой цепи повышало активность, но параллельно возрастала и токсичность [248], поэтому бутирофеноновые соединения этого типа практического интереса не представили. Замена в Vb кетогруппы на спиртовую или водород, а также удаление ее от арильного остатка приводили к снижению фармакологической активности [248]. Четвертичные сПиропирролидиниевые
8*
115
соединения были лишены адреноблокирующих свойств, а по некоторым тестам характеризовались даже адреномиметической активностью. В связи с интересом к аминопроизводным бензодиоксана-1,4, как адреноблокирующим веществам бензодиоксановый фрагмент был введен в качестве арильного заместителя и в N-арилкетоалкил 3-фенил-пирролидины [69]. Полученный препарат Vr в виде гидрохлорида показал высокую а-адреноблокирующую активность и вошел в медицинскую практику [113].
ФУРАГИН
1 - [р- (5-Нитрофурил-2) аллилиденамино] -гидантоин
С ю Н8 N4O5
Мол. масса 264,21
Желтого или оранжево-желтого цвета мелкокристаллический порошок, без запаха, горьковатого вкуса, очень мало растворим в воде (4 мг/л) и спирте (0,38 мг/л); в ацетоне растворяется 2,9 мг/л, в диметилформамиде — 23 г/л; т. пл. 260—266 °C разл.; ФС 42-685-73.
Фурагин — оригинальный антибактериальный препарат широкого спектра действия в отношении грамположитель-ных и грамотрицательных микроорганизмов [236]. Для ряда возбудителей инфекционных заболеваний обладает большей бактериостатической активностью, чем другие нитрофурановые производные и левомицетин; действует на штаммы, устойчивые к антибиотикам и сульфаниламидам, с мочой выделяется около 6% от введенного коли
чества препарата.
Применяют фурагин внутрь главным образом при лечении заболеваний мочевых путей (острые и хронические пиелонефриты, циститы, уретриты, инфекции после оперативных вмешательств на органах мочеполовой системы и др.), местно (1 : 13 000 в изотоническом растворе натрия хлорида)> для промываний и спринцеваний в хирургической и акушерско-гинекологической практике, для лечения гнойных ран, ожогов, промываний свищей и др.; в офтальмологии в виде глазных капель (по 2 капли водного
116
раствора 1 : 13 000) при конъюнктивитах, кератитах, в послеоперационном периоде и т. п.
Выпускаются таблетки по 0,05 г N. 100. Цена 1 р. 52 к.
В виде калиевой соли, растворимой в воде 1 :500, под названиями фурагин растворимый, фурагииа калиевая соль или солафур [236] препарат используют для внутривенных инъекций при тяжелых формах заболеваний, вызванных стафилококками, стрептококками, кишечной палочкой и некоторыми другими возбудителями (при сепсисе, раневых и гнойных инфекциях, пневмонии, воспалительных заболеваниях мочевых путей, анаэробных инфекциях); в сочетании с другими препаратами солафур применяется при лечении брюшного тифа и паратифа.
Выпускается в порошках. Цена 1 г — 60 к.
Метод синтеза фурагина разработан Институтом органического синтеза АН ЛатвССР по схеме [236]:
5-Нитрофурфуролдиацетат (I) гидролизуют в кислой среде до 5-нитрофурфурола (II), который конденсацией с уксусным альдегидом в изопропиловом спирте в присутствии ацетата морфолина превращают в р-(5-нитрофурил-2)-акролеин (Ш). Последний подвергают конденсации с 1-аминогидантоином (V), полученным путем кипячения 2-семикарбазидуксусной кислоты (IV) с разбавленной серной кислотой. Фурагин (VI) перекристаллизовывают из 80% изопропилового спирта.
5-Нитрофурфурол (II). К смеси 8,4 л воды и 2,02 кг серной кислоты прибавляют 4,5 кг I. Массу нагреваю! 40 мин при 96 °C, фильтруют и охлаждают до 2 °C, дают выдержку 2 ч. Осадок отфильтровывают, промывают охлажденной до 2°C водой (4 л), отжимают и без высушивания направляют на следующую стадию. Выход II 2,38 кг (содержание основного вещества 90%).
р-(5-Нитрофурил-2)-акролеии (III). К 2,38 кг II, полученного на предыдущей стадии, растворенным в 9 л 80%
117
изопропилового спирта, прибавляют 28 г уксусной кислоты и 34 г морфолина, охлаждают до 0—1 °C и приливают 1,592 кг охлажденного до +1 °C уксусного альдегида. Реакционную смесь нагревают под Дзотом в закрытой системе 3 ч при 80°C (давление повышается до 3 атм). Затем охлаждают до —7 °C, летучие продукты отделяют пропусканием в течение 30 мин тока азота, дают выдержку 2 ч при —7 °C, осадок отфильтровывают и промывают 1 л 80% изопропилового спирта. Выход III 2,54 кг (содержание влаги 12—15%), или 58,3%.
1-[р-(5-Нитрофурил-2)-аллилидеиамино]гидантоин (фурагин) (VI). Смесь 5 л воды, 640 мл серной кислоты и 936 г IV нагревают 2 ч при 98°C, обесцвечивают углем и полученный раствор 1-аминогидантоина (V) приливают при 52°C к обесцвеченному углем раствору 712 г III в 15 л 80% изопропилового спирта, pH которого предварительно доведен серной кислотой до 4,5. Реакционную массу выдерживают 20 мин при 52°C, охлаждают до 38°C и после выдержки в течение 1 ч осадок фурагина отфильтровывают, промывают 23 л нагретого до 52°C 80% изопропилового спирта и 25 л дистиллированной воды (до отсутствия SO4__ в промывной воде), сушат в вакууме при 65°C. Выход VI 744 г (80,5% на III).
ФЕНИКАБЕРАН
2-Фенил-3-карбэтокси-4-диметиламинометил-5-оксибензо-фурана гидрохлорид:
Мол. масса 375,9
Белый или почти белый кристаллический порошок, без запаха, горького вкуса, растворим в воде (1:8), метиловом (1:5), этиловбму (1 : 7), абсолютном изопропиловом (1 :75), спиртах и диметилформамиде (1:10); т. пл. 198— 203°C (в интервале 2°С); в УФ-спектре в 0,1 н. растворе едкого натра имеется максимум поглощения при 300 нм; ВФС 42-712-78.
Феникаберан — оригинальный спазмолитический и ко-
118
ронарбрасширяющий препарат [54, 56, 59], обладает местноанестезирующим и антиаритмическим действием.
Применяют феникаберан при заболеваниях пищеварительного аппарата, связанных с болями спастического характера (холециститы, желчнокаменная болезнь, язвенная болезнь желудка и двенадцатиперстной кишки), при хронической коронарной недостаточности и для купирования приступов стенокардии.
Выпускается в ампулах по 2 мл 0,25% N. 10 (цена 32 к.) и в таблетках по 0,02 г N. 20. Цена 50 к.
Метод синтеза феникаберана разработан во ВНИХФИ [56] по схеме:
Ацетоуксусный эфир (I) переводят с едким натром в натрийацетоуксусный эфир, который ацилируют хлористым бензоилом. При гидролизе образующегося бензоилацетоуксусного эфира в присутствии хлористого аммония получают бензоилуксусный эфир (II). Конденсация кетоэфира II с бензохиноном приводит к 2-фенил-З-карбэтокси-5-оксибензофурану (III), который по реакции Манниха превращают в конечный продукт — феникаберан (IV).
Бензоилуксусный эфир (II). К охлажденному до 3°С раствору 3,514 кг I в 3 л толуола прибавляют 6 л воды и приливают при перемешивании 33% раствор едкого натра (применение более концентрированного раствора едкого натра снижает выход) с такой скоростью, чтобы температура была не выше 7 °C. Доводят pH водного слоя до 10,5—11,5, после чего при охлаждении начинают одновременно прибавлять при 2—7°С и pH 10,5—11,5 2,582 кг хлористого бензоила и 3 л 33% раствора едкого натра (изменение порядка прибавления реагентов и pH среды способствует побочным реакциям омыления и декарбоксилирования Ги II с образованием кетонов и дибензоилуксусного эфира). Реакционную массу нагревают до 15—18°С
119
И Дают выдержку Р/г ч, после чего нагревают 1 ч При 27—33 °C, прибавляют 973 г хлористого аммония и проводят расщепление натрий-бензоилацетоуксусного эфира при перемешивании, контролируя pH водного слоя. Реакцию считают законченной при pH 8,7—9,6 (~ через Г5 ч). Прибавляют 800 г хлористого натрия, перемешивают (~30мин) до ет полного растворения (высаливание II). Толуольный слои Отделяют, водный экстрагируют 600 мл толуола. Объединенные толуольные экстракты промывают 1,85 л насыщенного раствора хлористого натрия, содержащего 1% соляной кислоты (перемешивают 30 мин, pH нижнего слоя должен быть 4—5), затем 1,2 л насыщенного водного раствора хлористого натрия (pH водного слоя должен быть 7). Промытый толуольный раствор сушат 350 г прокаленного сульфата магния, упаривают в вакууме, остаток подвергают фракционной разгонке (II — термолабильное соединение и его перегонку нужно вести при остаточном давлении не более 1—2 мм, присутствие кислых и щелочных примесей способствует разложению II; процесс катализируется железом, поэтому разгонку необходимо проводить в стекле). Собирают фракцию с т.кип. 127—128°С (1—2 мм рт.ст.). Выход II 2,814 кг (80,5%), содержание основного вещества не менее 97%.
2-Фенил-3-карбэтокси-5-оксибензофуран (III). 1,638 кг пасты п-хинона растворяют в 6,5 л хлористого метилена. Раствор отфильтровывают от примеси хингидрона, высушивают азеотропной отгонкой воды и прибавляют равномерно в течение 10 ч со скоростью 0,79 л/ч при 68—73 °C (с одновременной отгонкой хлористого метилена) к нагретому до 68—79°C раствору 2,627кг II и 1,877 кг хлористого цинка в 2,43 л абсолютного изопропилового спирта (проведение ^процесса без хлористого метилена не позволяет очистить пасту п-хинона от примесей хингидрона и воды, увеличивает растворимость конечных продуктов реакции и снижает выход III; избыток п-хинона увеличивает образование побочного продукта — производного бензодифурана, в том же направлении сдвигает реакцию и повышение температуры;. снижение температуры увеличивает количество побочных продуктов с незамкнутым фурановым циклом). По окончании прибавления п-хинона массу охлаждают до 18—20 °C, разбавляют 1 л хлористого метилена (для растворения примеси производного бен-зодйфурана). Осадок III отфильтровывают, промывают 2 раза по 650 мл охлажденного до 10—15 °C абсолютного изопропилового спирта, затем 3 л воды, содержащей 15 мл
120
31% соляной кислоты (для отделения цинковых солей) и 2 раза по 500 мл воды. Выход III 2,406 кг (60,5%), содержание основного вещества не ниже 95%, хлористого цинка не более 0,5%.
2-Фенил-3-карбэтокси-4-диметиламинометил-5-оксибен-зофурана гидрохлорид (феникаберан) (IV). Раствор 2,086 кг III, 586 г диметиламина гидрохлорида и 503 г 38,5% формалина в 7,1 л диметилформамида.’нагревают 4 ч при 94—97 °C (при снижении температуры до 80— ___82 °C реакция не идет), контролируя конец реакции по определению содержания в массе исходного III методом ТСХ (должно остаться не более 4% от взятого в реакцию III). Массу охлаждают до 18—20°C, отфильтровывают от примесей, диметилформамид отгоняют при остаточном давлении 20 мм до 7 л отгона, остаток разбавляют при 20—25 °C 1 л ацетона, охлаждают 30 мин при 3—7 °C. Осадок отфильтровывают, промывают 1 л и 0,4 л ацетона, охлажденного до 3—7 °C (конец промывки — по отсутствию окраски смолистых примесей в ацетоновом растворе). Полученный продукт растворяют в 14,4 л воды при 55— 60 °C. Водный раствор отфильтровывают от примесей. Фильтрат охлаждают до 3—7 °C, прибавляют 7,2 л хлористого метилена и приливают 8,64% раствор едкого натра до pH 7 с такой скоростью, чтобы температура не была выше 10 °C. Раствор основания феникаберана в хлористом метилене отделяют. Водный слой дополнительно экстрагируют 7,2 л хлористого метилена. Объединенные и профильтрованные хлористометиленовые растворы упаривают; для более полного удаления хлористого метилена прибавляют 1,5 л абсолютного изопропилового спирта с последующей отгонкой. Остаток растворяют в 13,09 л абсолютного изопропилового спирта и при 22—35 °C приливают раствор хлороводорода в изопропиловом спирте до pH 3. Выпавший в осадок феникаберан переводят в раствор нагреванием массы до кипения, раствор обесцвечивают углем (0,1 кг), охлаждают до 20—25°C, а затем при работающей мешалке 1 ч при 10—15 °C. Феникаберан (IV) отфильтровывают, промывают 2 раза по 400 мл охлажденного до 10—15 °C абсолютного изопропилового спирта и еще раз перекристаллизовывают из 12,46 л абсолютного изопропилового спирта. Выход 1,333 кг (50%).
121
ФЕПРОМАРОН
3- (а-Фенил-р-пропионилэтил) -4-оксикумарин:
Мол. масса 322,36
Белый или белый со слегка кремоватым оттенком кристаллический порошок, без запаха, растворим в ацетоне, мало растворим в этиловом и изопропиловом спиртах, практически нерастворим в воде; т. пл. 149—151 °C; ФС 42-958-75.
Фепромарон — оригинальный антикоагулянт непрямого действия [220]. Является антагонистом витамина К, необходимого для образования в печени протромбина. Нарушает синтез протромбина, проконвертина, факторов свертывания IX и X. Бол^е активен, чем дикумарин и неодику-марин, обладает мёньшим кумулятивным эффектом, чем дикумарин. По активности близок к варфарину, но менее токсичен.
Применяют для профилактики и лечения тромбозов, тромбофлебитов, эмболий, тромбоэмболических осложнений при инфаркте миокарда. При коронарной недостаточности не только понижает протромбиновый показатель, но и уменьшает загрудинные боли, что связывают с непосредственным сосудорасширяющим действием.
Выпускается в таблетках по 0,01 г N. 20. Список А. Цена 14 к.
Метод синтеза фепромарона разработан МХТИ им. Д. И,- Менделеева совместно с Харьковским химикофармацевтическим заводом «Красная Звезда» по схеме [220]:
Очищенный через О-натриевое производное 4-о.ксику-марин (I) подвергают конденсации со стирилэтилкето-
122
йом (II) в присутствии натрийаммоний фосфата и полученный фепромарон (III) перекристаллизовывают из изопропилового спирта.
Смесь 1262 г очищенного переводом через О-натриевое производное 4-оксикумарина (I), 167 г натрийаммоний-фосфата и 5,6 л воды нагревают до 45—50 °C, прибавляют 1272 г стирилэтилкетона (II) и кипятят 5 ч, после чего оставляют при интенсивном перемешивании для медленного самоохлаждения. Осадок III отфильтровывают, промывают водой до отсутствия в промывных водах I по пробе на образование зеленой окраски с нитритом натрия и перекристаллизовывают из 14,3 л изопропилового спирта. Выход 1455 г (47,8%).
НИТРОФАРИН
с20н„ NO6
Мол. масса 367,36 Светло-желтого цвета кристаллический порошок, без запаха, практически нерастворим в воде и 10% водном растворе щелочи, очень мало растворим в ацетоне и спирте, хорошо растворим в диметилформамиде; т. пл. 216— 219°C (в интервале 2°C); ВФС 42-197-73.
Нитрофарин — оригинальный антикоагулянт [122, 134]. Подобно фепромарону (см. с. 121), является антагонистом витамина К, нарушает синтез протромбина, проконверти-на, факторов свертывания IX и X. Оказывает более сильное. быстрое и короткое действие, чем фепромарон.
Как антикоагулянт длительного действия применяют для профилактики и лечения тромбоэмболических заболеваний (инфаркт миокарда, стенокардия, тромбофлебиты, флеботромбозы крупных артерий конечностей, брюшной полости и др.), а также для профилактики послеоперационных тромбозов.
Выпускается в таблетках по 0,005 г N. 20. Список А. Цена 24 к.
Метод синтеза нитрофарина разработан МХТИ им. Д. И. Менделеева [220] по схеме:
123
Путем конденсации п-нитробензальдегида (I) с метил-этилкетоном (II) в присутствии щелочи получают 1-(п-нитрофенил)-1-оксипентанон-3 (III). В кислой среде реакция идет с преимущественным образованием изомерного 1 - (п-нитрофенил)йФхркси-2-метилбутанона-3. Дегидратацию III проводят нагреванием с п-толуолсульфохлоридом в толуоле. Использование для этих целей п-толуолсульфо-кислоты не имело преимуществ. Полученный п-нитрости-рилэтилкетон (IV) вводят в конденсацию с 4-оксикума-рином (V) в присутствии натрийаммонийфосфата в условиях, аналогичных описанным для синтеза фепромарона (см. с. 123). Синтезы VI по другим схемам, исходя из незамещенного бензальдегида с нитрованием продуктов на стадии стирилэтилкетона или путем нитрования фепромарона, дали худшие результаты.
1-(п-Нитрофенил)-1-оксипентанон-3 (III). К раствору 200 г I в 2380 г II при 18—20°C с перемешиванием приливают в течение 2 ч .180 мл 6% водного раствора едкого натра с такой скоростью, чтобы температура стала не выше 26°C (используют охлаждение реакционной массы). Затем дают выдержку 2 ч при комнатной температуре, после чего pH массы доводят прибавлением 15% соляной кислоты до 5—6. Органический слой отделяют и упаривают при остаточном давлении 25—30 мм рт. ст. и температуре 20—24 °C. Остатки II отгоняют в виде азеотропа с водой, для чего приливают 200 мл воды с последующей отгонкой при остаточном давлении 25—30 мм рт. ст. (отгонку II с водой необходимо проводить сразу после окончания реакции, не допуская стояния образовавшихся продуктов с мет^лртилкетоном, так как это может привести к побочным реакциям и ухудшению качества III). Из остатка выкристаллизовывается III, который отфильтровывают и перекристаллизовывают из 200 мл 67% изопропилового спирта. Выход 89 г (30%); т. пл. в пределах 85—88 °C.
п-Нитростирилэтилкетон (IV). Раствор 89 г III и 27 г п-толуолсульфохлорида в 1,2 л толуола кипятят 4 ч. То-124
луол отгоняют в вакууме. Кристаллический остаток отжимают от маслообразных примесей и перекристаллизовывают из 125 мл4И>% водного изопропилового спирта. ВыходГ60^т473,^%); т. пл. не ниже 101 °C. Вещество раздражает слизистые оболочки и кожу.
3-(а-п-Нитрофенил-р-пропионилэтил)-4-оксикумарин (нитрофарин) (VI). Смесь 60 г IV, 55,2 г V, 4,8 г натрий-аммонийфосфата и 400 мл воды кипятят 5 ч при интенсивном перемешивании, охлаждают до комнатной температуры. Образовавшийся твердый продукт отфильтровывают, промывают водой, высушивают 6 ч при 100 °C, растворяют при нагревании в 7,25 л ацетона и к горячему раствору добавляют воду (~6,75 л) до появления неисчезающей мути. Выпавшие после охлаждения кристаллы отфильтровывают, промывают 5,8 л воды и высушивают при 100 °C. Получают 64,3 г (60%) VI.
ПИРРОКСАН
6-[р- (З-Фенилпирролидил-1) -пропионил] -бензодиокса-па-1,4 гидрохлорид:
С 2, H2.,NO3- НС1
Мол. масса 373,88
Белый или белый со слегка желтоватым оттенком кристаллический порошок, без запаха, трудно растворим в воде, растворим в спирте, легко растворим в хлороформе, практически нерастворим в эфире; т. пл. 138—144 °C (в интервале 2°C); ФС 42-870-74.
Пирроксан — оригинальный а-адреноблокирующий препарат [ИЗ], влияет на периферические и центральные ад-ренореактивные системы; в отличие от других адреноблокаторов (галоперидола, аминазина) не нарушает условно-рефлекторной деятельности и не влияет на биоэлектрическую активность мозга.
Применяют пирроксан при лечении и профилактике различных заболеваний, в основе которых лежит патологическое повышение симпатического тонуса, в том числе при диэнцефальных и гипертонических кризах, гиперсим-патикотонии, сопровождающейся психическим напряжени
125
ем, тревогой, при различных проявлениях диэнцефальной патологии симпатикб-'а'дреналового типа, для ослабления морфиновой и алкогольной абстиненции, для профилактики перевозбуждения вестибулярного аппарата, например, при морской и воздушной болезни, синдроме Меньера (в сочетании с холинолитиками и противогистаминными препаратами). При наличии . гиперсимпатикотонии уменьшает зуд, оказывает седативное действие и улучшает сон у больных аллергическими дерматозами.
Выпускается в таблетках по 0,015 г N. 50 (цена 2 р. 26 к.) и в ампулах по 1 мл 1 % раствора в упаковке по 10 штук (цена 82 к.).
.Метод синтеза пирроксана разработан Институтом токсикологии Министерства здравоохранения СССР совместно с химико-фармацевтическим заводом «Фармакон» по схеме [169]:
с.н CH CN вО *
оценкой кон
CN 1 н /М '
7^-*- с^снюн^он
HBr
При взаимодействии пирокатехина (I) с дихлорэтаном в присутствии поташа получают бензодиоксан-1,4 (II), который ацетилируют хлористым ацетилом по Фриделю — Крафтсу до 6-ацетилбензодиоксана-1,4 (III). Параллельно алкилированием цианистого бензила (IV) этиленхлоргид-рином в присутствии едкого кали в диметилформамиде синтезируют 1-фенил-З-оксибутиронитрил (V), который без выделения восстанавливают каталитически с никелем до З-фенил-4-аминобутанола (VI); оксигруппу в VI бромистоводородной кислотой замещают на бром, и гидробромид З-фенил-4-аминобромбутана (VII) без выделения циклизуют нагреванием с едким натром в 3-фенилпирроли-
126
дин (VIII). Соединения III, VIII и параформ вводят в реакцию Манниха, продукт который выделяют ввидегидро-хлорида— препарата пирроксан (IX). Другие методы синтеза 3-фецилпирролидина (VIII): восстановление алюмогид-ридом лития 4-фенилпирролидона-2 [73] или имида фенилянтарной кислоты [247]—являются технологически менее приемлемыми.
Бензодиоксан-1,4 (II). Смесь 450 мл дихлорэтана, 507 г этиленгликоля, 294 г I и 638 г прокаленного поташа нагревают с перемешиванием 2 ч при 80—85 °C, затем 2 ч при 123—125 °C, дают охладиться до 80 °C, приливают 1,5 л воды, экстрагируют бензолом (3 раза по 800 мл). Бензольный экстракт промывают водой, упаривают, остаток перегоняют в вакууме. Собирают фракцию с т. кип. 84—86°C (12 мм рт. ст.). Выход 207 г (57%), содержание основного вещества не ниже 99%.
6-Ацетилбензодиоксан-1,4 (III). К нагретой до 30—32°C смеси 780 мл дихлорэтана (с содержанием влаги не более 0,2%) и 430 г хлористого алюминия приливают 418 г II и 250 г хлористого ацетила с такой скоростью, чтобы температура реакционной массы оставалась 38—40 °C. После выдержки 1 ч при 38—40 °C нагревают 2 ч при 80 °C, охлаждают до 20—25 °C, прибавляют 1,56 л воды и 350 г соляной кислоты с такой скоростью, чтобы температура массы была не выше 25°C. Экстрагируют дихлорэтаном (500 мл и 3 раза по 300 мл), экстракт сушат безводным сульфатом натрия, обесцвечивают углем, упаривают в вакууме (0,6—0,7 атм) до температуры в массе 60 °C. Продукту III дают возможность закристаллизоваться (2—3 ч при 20—25°C). Кристаллы отфильтровывают, промывают изопропиловым спиртом (150—200 мл) и высушивают. Получают 330 г III (62,2%).
З-Фенил-4-аминобутанол (VI). К 975 г сухого диметил -формамида (влаги.не более 0,2%) и 342 г гранулированного едкого кали при 20—30 °C в токе азота, регулируя температуру скоростью прибавления реагента, приливают 151 г свежеперегнанного цианистого бензила (IV). Перемешивают 4 ч при 25—30 °C и затем медленно в токе азота прибавляют 126 г этиленхлоргидрина, дают выдержку 5 ч при 40—50 °C, охлаждают до 20 °C, приливают 800 мл бензола и 1,65 л воды, охлажденной до 1—3°С. Бензольный слой отделяют, промывают водой; бензол отгоняют, остаток растворяют в 300 мл этилового спирта. Полученный спиртовой раствор V смешивают с 950 мл спирта, насыщенного аммиаком, прибавляют 339 г скелетного нике-
127
левого катализатора и проводят гидрирование при 20— 30 °C и давлении водорода 5 атм. Катализатор отфильтровывают, этиловый спирт отгоняют. Получают 300 г VI (32,8% на цианистый бензил).
З-Фенилпирролидин (VIII). К бромистоводородной кислоте с концентрацией не ниже 40% (1,04 кг в пересчете на 100%) прибавляют 300 г VI при температуре не выше 40 °C. Полученный раствор промывают экстракцией 300 мл бензола, после чего водный кислый слой нагревают с отгонкой до тех пор, пока содержание бромоводорода в отгоне не составит 40% (температура в массе 115°C). Затем, постепенно повышая температуру в массе до 125°С, продолжают отгонять бромистоводородную кислоту, после чего массу охлаждают до 70—80 °C, подключают вакуум (0,6—0,8 атм) и отгоняют бромистоводородную кислоту до прекращения погона при температуре в парах 100 °C. Массу охлаждают, приливают 2,7 л воды и 300 мл бензола. Бензольный слой отделяют, водный — экстрагируют бензолом (2 раза по 300 мл), осветляют углем и подщелачивают твердым едким натром из расчета 100 г едкого натра на 400 г водного раствора. Кипятят (100—105°С) 3 ч, охлаждают до 20—25°C и экстрагируют бензолом (900 мл). Бензольный раствор упаривают, остаток перегоняют в вакууме; собирают фракцию с т. кип. 105— 107°С (10 мм рт. ст.). Получают 153 г (31,8%) VIII (содержание основного вещества не менее 90% и до 10% р-фенилэтиламина).
6- [р-(3-Фенилпирролидил-1)пропионил]-бензодиокса-на-1,4 гидрохлорид (пирроксан) (IX). К раствору 161 г VIII в 550 мл абсолютного этилового спирта приливают при 20—30 °C 176 мл 30% раствора хлороводорода в абсолютном этиловом спирте до pH 2—3, после чего прибавляют 196 г сухого III и 838 г параформа. Массу перемешивают 12 ч при 78—80 °C, охлаждают до 20 °C и прибавляют 300—350 мл 20% раствора едкого натра до pH 9,4—10 (по тимолфталеину — синее окрашивание) и экстрагируют бензолом (1 л и 300 мл). Бензольные экстракты промывают водой, сушат прокаленным поташом, упаривают в вакууме. Остаток закристаллизовывают путем добавки 500—600 мл диэтилового эфира и охлаждения до 0 °C (6 ч). Выделенное после центрифугирования основание пи^ррксана высушивают (масса 155 г), растворяют в 1,5 л ацетона, раствор осветляют углем и при 20 °C быстро обрабатывают 30% раствором хлороводорода в абсолютном этиловом спирте из расчета 137 г хлорово-128
дорода на 1 кг основания пирроксана (pH раствора должен быть не более 1,3). Массу охлаждают до 5 °C, дают выдержку 12 ч. Пирроксан отделяют центрифугированием, промывают 2 раза по 400 мл ацетона и высушивают. Выход 128 г (36%) (на 3-фенилпирролидин).
БИТИОДИН
3- (1 -Метилпиперидилиден) -ди- (2-тиенил) -метана цитрат:
C,.-HI7NS2- с6нво7
Мол. масса 467,68 Основные синонимы: азверин, антупекс, нодал, типе-пидин.
Белый с кремовым оттенком кристаллический порошок, без запаха, горького вкуса, растворим в горячей воде и кипящем спирте, плохо растворим в холодной воде и спирте; практически нерастворим в эфире, бензоле, диоксане; т. пл. 137,5—139°С разл.; ВФС 42-531-76.
Битиодин — противокашлевый препарат, по активности близок к кодеину, не является наркотиком, обладает отхаркивающим действием.
Применяют битиодин для подавления кашля различного генеза, в том числе в детской практике.
Форма выпуска: таблетки по 0,01 г.
Метод синтеза битиодина разработан во ВНИХФИ совместно с Саратовским Государственным университетом по схеме [202]:
9 Заказ № 4304
129
Этиловый эфир никотиновой кислоты (I) обработкой диметилсульфатом превращают в четвертичную соль II, которую без выделения восстанавливают каталитически в присутствии никеля до 1-метил-З-карбоэтоксипиперидина (III), параллельно бромированием тиофена (IV) в четыреххлористом углеррде получают 2-бромтиофен (V), который переводят с магнием в реактив Гриньяра — 2-тиенил-магнийбромид (VI). Взаимодействием сложного эфира III с реактивом Гриньяра (VI) получают 3-(1-метилпипери-дил)-ди-(2-тиенил)-карбинол (VII), который подвергают дегидратации муравьиной кислотой и получившееся непредельное соединение переводят в цитрат — препарат битио-дин (VIII). Этот метод имеет преимущества перед ранее описанным получением III путем гидрирования этилового эфира никотиновой кислоты (I) на никелевом или платиновом катализаторе с последующим метилированием формалином с муравьиной кислотой, а также получением 2-бромтиофена (V) путем бромирования тиофена бромом в ледяной уксусной кислоте или дорогостоящим N-бромсук-цинимидом.
1-Метил-З-карбэтоксипиперидин (HI). К 470 мл I прикапывают при перемешивании 393 мл диметилсульфата, поддерживая температуру реакционной массы не выше 75°С. Затем дают выдержку 2 ч при 78—80°C, контролируют конец реакции методом ТСХ (содержание I в массе не более 2,5%) и приливают при перемешивании и температуре 40—50 °C 750 мл абсолютного изопропилового спирта. Раствор четвертичной соли II разбавляют равным объемом абсолютного изопропилового спирта, прибавляют 140 г промытого абсолютным изопропиловым спиртом скелетного никелевогс^л^тализатора Ренея и массу гидрируют при начальном давлении водорода 70 атмосфер и температуре 58—60 °C. После прекращения гидрирования катализатор отфильтровывают, изопропиловый спирт отгоняют, остаток растворяют в 500 мл воды и при 20— 25°C подщелачивают 22% водным раствором аммиака до pH 10. Выделившееся основание III извлекают бензолом (3 раза по 500 мл). Бензольные экстракты сушат сульфатом натрия (100 г) и упаривают при температуре в парах 30—40 °C и остаточном давлении 100—120 мм. Остаток перегоняют в вакууме; т. кип. 87—90°C (10 мм). Выход 444 г (76,5%), содержание основного вещества не ниже 99%.
2-Бромтиофен (V). К раствору 1 л тиофена в 4,75 л четыреххлористого углерода при 4-2—(—2 °C) медленно, 130
в течение 6 ч, прикапывают 700 мл брома. Дают выдержку 30 мин при 2—5 °C, 1 ч при 20—25 °C, нагревают до кипения и медленно (0,6 л погона/ч) отгоняют продукт с т. кип. 75—80°C (температура в массе до 115°C), после чего остаток подвергают фракционированию на ректификационной колонне. Собирают фракцию с т. кип. 80— 180 °C (1,316 кг, содержание V ~70%). Ее нейтрализуют при 20—25 °C твердой кальцинированной содой (50 г) [нейтрализуется примесь бромоводорода и в присутствии соды подвергают вновь разгонке на ректификационной колонне (1 650 мм, d 24 мм)].
Получают 852 г (51,8%) V, содержание основного вещества не ниже 98%.
3-(1-Метилпиперидил)-ди-(2-тиенил)-карбинол (VII). К 125,1 г промытых сухим бензолом магниевых стружек, 0,7 г йода и 800 мл сухого эфира (содержание влаги менее 0,1%), нагретым до 25°C, постепенно прибавляют раствор 838 г V в 800 мл сухого эфира с такой скоростью, чтобы поддерживать равномерное слабое кипение реакционной массы. После 2 ч кипячения (магний полностью вступает в реакцию) к полученному эфирному раствору VI приливают 400 мл сухого бензола, кипятят 10—20 мин, охлаждают до 10—14 °C и в течение IV2 ч прибавляют раствор 440 г III в 703 мл сухого бензола. Смесь нагревают за 30 мин до 20—25°C, кипятят 3 ч и оставляют на 10 ч при 20 °C. Разложение гриньяровского комплекса ведут путем постепенного прибавления при температуре не выше 20 °C раствора 940 г хлористого аммония в 1,5 л воды. Массу перемешивают 2 ч при 20—25 °C, органические растворители отгоняют. Остаток перемешивают 1 ч при 20 °C. Технический VII отфильтровывают, промывают 1,5 л воды (до бесцветной промывной воды) и перекристаллизовывают из 6 л изопропилового спирта с 60 г угля. Выход 446 г (59,1%, считая на эфир III).
3- (1 -Метил пиперидил идеи )-ди-( 2-тиенил )-метана цитрат (битиодин) (VIII). Нагревают смесь 485 г VII с 950 мл 88% муравьиной кислоты до 110°С, при этом осадок VII полностью растворяется и наблюдается слабое кипение, которое поддерживают 15 мин. Затем массу охлаждают до 50 °C и при остаточном давлении 35—40 мм отгоняют муравьиную кислоту. К остатку при перемешивании прибавляют 2 л 30% раствора поташа до pH 9,5—10, поддерживая температуру массы 20—25 °C. Выделившееся основание извлекают бензолом (3 раза по 850 мл). Бензольные экстракты сушат 100 г сульфата натрия с добавкой гид
9!
131
рохинона (10 г), упаривают, вещество перегоняют при 158—161 °C (2,5—3 мм) (в холодильник необходимо подавать теплую воду, так как при 35°C основание VIII кристаллизуется). Поскольку основание при хранении темнеет, его рекомендуется сразу переводить в цитрат. Перегнанное основание (412 г) растворяют в 1,25 л изопропилового спирта при нагревании до 50—52 °C и приливают при перемешивании в течение 3—5 мин раствор 289 г лимонной кислоты в 662 мл изопропилового спирта. Температура массы снижается на 2—3°С (процесс солеобразо-вания эндотермичен). Поддерживая температуру 50—55 °C, перемешивают еще 15 мин. Появляются кристаллы битио-дина, и масса быстро кристаллизуется. Перемешивание при самоохлаждении продолжают 1 ч, после чего дают выдержку 3 ч при +5 °C. Осадок отфильтровывают и многократно промывают до получения бесцветного осадка охлажденным до 4-5 °C изопропиловым спиртом (примерно 662 мл). Продукт перекристаллизовывают из 2,5 л изопропилового спирта с добавлением 30 г угля, сушат 4— 5 ч при 40—50 °C, затем 6—8 ч при 70—80 °C. Выход 511г (73%).
/г '
МОНО- И БИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ С ОДНИМ ГЕТЕРОАТОМОМ
Одной из важных сырьевых проблем, решенных отечественной химико-фармацевтической промышленностью за последние 10 лет, явилась организация комплексного использования р-пиколиновой фракции. Как известно [266], основным природным источником пиридиновых соединений являются продукты коксования каменного угля. В процессе коксования производные пиридина концентрируются главным образом в коксовом газе, где их содержание в зависимости от типа угля, режима коксования, а также условий улавливания и разделения продуктов колеблется от 0,3 до 0,1 г/м3. Отделение так называемых легких пиридиновых оснований, включающих по данным ГЖХ не менее 23 соединений, и их первичная ректификация позволяют выделить р-пиколиновую фракцию с т. кип. 138—146°С, главными компонентами которой являются у-пиколин (I) (37,5—41%), р-пиколин (II) (29,5—31,5%), и 2,6-лутидин (III) (18,0—23,5%).
132
До 50-х годов наиболее ценным компонентом р-пико-линовой фракции считался 0-пиколин (II), который легко превращается в никотиновую кислоту (IV) и ее амид (V)—витамин РР. В связи с этим в химико-фармацевтическом производстве р-пиколиновую фракцию подвергали конденсации в жестких условиях под давлением с формалином, Компоненты фракции I и III при этом осмолялись, оставшийся неизмененным р-пиколин отгоняли и превращали в витамин РР и кордиамин (VI)—стимулятор ЦНС, возбуждающий дыхательный и сосудодвигательный центры. Начиная с 50-х годов никотинамид стал использоваться также для производства никодина (VII) — желчегонного средства, обладающего, кроме того, бактериостатическими и бактерицидными свойствами [264].
Выявление в 50-е годы высокой антитуберкулезной активности гидразида изоникотиновой кислоты (изониазида VIII, R = H) и его производных: фтивазида (IX, R=H, R-СбНз-п-ОН-м-ОСНз) [10, 11, 6], салюзида (IX, R=H, Р'-С6Н2-о-СООН-м-ОСНз-п-ОСНз) [9, 11, 6], метазида (VIII, R-CH2NHNHOCC5H4N) — заставило обратить внимание на использование в химико-фармацевтическом производстве у-пиколина (I). В связи с этим в СССР были изучены различные варианты избирательного и наиболее полного окисления до изоникотиновой кислоты (X) у-пико-лина (I), содержащегося в р-пиколиновой фракции [266]. В результате был разработан и вот уже почти 30 лет используется в промышленности способ получения изоникотиновой кислоты (X), основанный на обработке неразде-
133
ленной фракции формалином с последующей отгонкой от образующейся смеси моно-, ди- и три-метилольных производных у-пиколина, не взаимодействующих в этих условиях с формальдегидом р-пиколина (II) и 2,6-лутиди-на (III), и окислении смеси метилол-у-пиколинов азотной кислотой [211]. Выделенный из отогнанных оснований Р-пиколин (II) был использован для превращения в никотиновую кислоту (IV). В дальнейшем гидразид изоникотиновой кислоты явился полупродуктом в промышленном получении ипразида (VIII, R-CH(CH3)2) и ниаламида — воспроизведенных в СССР антидепрессантов — ингибиторов моноамирюксидазы [211, 266]. На основе выделенного из р-пиколинЬвой фракции чистого у-пиколина (I) был разработан контактный метод синтеза 4-формилпириди-на [120] и превращения его в известный реактиватор холинэстеразы—дипиридоксим (XI) [128], Получаемая из р-пиколиновой фракции изоникотиновая кислота (X) явилась также исходным сырьем для приготовления хинукли-дона-3 (XII) [35] — основного полупродукта в производстве большинства хинуклидиновых лекарственных препаратов (см. с. 137).
Более сложной оставалась проблема утилизации 2,6-лути-дина (III) [266]. Проведенные в этом направлении первоначальные исследования [212] не дали практических результатов, а использование III в производстве оригинальных гипотензивных ганглиоблокирующих препаратов ди-меколина (XIII, R-CH3), диколина (XIII, R-C2H5) и нано-фина (XIV) [211] не могло поглотить больших количеств 2,6-лутидина — отхода от производства препаратов группы никотиновой и изоникотиновой кислот [266].
Положение изменилось в связи с созданием японскими авторами эффективного антисклеротического препарата ангинина (пармидина, продектина), характеризующегося антибрадикининовым действием. Разработка метода синтеза пармидина (см. с. 143) и внедрение его в производство [265] позволили перейти к комплексному использованию в химико-фармацевтической промышленности СССР всех компонентов р-пиколиновой фракции. При этом наряду с расширением арсенала лекарственных средств, применяемых в медицине, были созданы условия охраны окружающей среды от загрязнения ее высокотоксичными пиридиновыми основаниями [266].
Пиридиновые производные другого типа—тиоаналоги витамина Вв — первоначально привлекли внимание в связи с поиском радиопротекторов. В дальнейшем у этих сое-134
динений было обнаружено гипнотическое действие [304], а дисульфидный аналог пиридоксина, получивший название пиридитол (см. с. 146), нашел применение в клинике в качестве психотропного препарата. Синтез пиридитола был воспроизведен в СССР; во ВНИВИ разработан метод его получения и организован выпуск препарата [77, 106].
В области гидрированных пиридиновых соединений — производных пиперидина — следует отметить воспроизводство психостимулирующего препарата меридила (см. с. 149), для которого во ВНИХФИ была изучена сравнительная активность трео- и эритро-изомеров [270] и впервые разработан стереоспецифический синтез, позволяющий получать чистый трео-изомер, характеризующийся психостимулирующей активностью [267].
В филиале ВНИХФИ был воспроизведен хинолиновый антибактериальный препарат с широким спектром химиотерапевтического действия — нитроксолин (см. с. 153) [36], а во ВНИХФИ существенно усовершенствован метод получения разработанного в Институте паразитологии и тропической медицины им. Е. И. Марциновского оригинального антималярийного препарата хиноцида (см. с. 155) [30, 31, 290] и создан метод, пригодный для его промышленного производства [167].
Высокая радиозащитная активность индолилалкил-аминов — триптамина и серотонина — была обнаружена еще в 1952 г. и в дальнейшем изучена достаточно подробно. Однако за рубежом индолилалкиламины в целом не привлекли серьезного внимания и в радиозащитном отношении было изучено всего около 15 веществ этого класса. Советские ученые [75, 229] на протяжении почти 25 лет детально исследовали этот класс радиопротекторов и выявили в нем определенные закономерности связи между структурой и биологической активностью. Показано, что в ряду индолилалкиламинов общей формулы XV для проявления радиозащитных свойств необходимо наличие аминоэтильной группы в положении 3 индольного кольца.
135
Перемещение этой группы в положения 1 или 2, ее укорочение, удлинение или разветвление неблагоприятно сказываются на активности. Снижает радиозащитное действие и введение в боковую цепь дополнительных окси, кето, карбоксильной и других функциональных групп. Дополнительное введение меркаптогруппы усиливает активность. Наибольшим радиозащитным действием обладают соединения XV, у которых аминогруппа в боковой цепи является первичной (XV, X=Y=H) (за исключением некоторых монометилзамещенных соединений) или ацилирована аминокислотными, а также небольшими пептидными остатками. Неблагоприятно сказывается на радиозащит-ном действии введение любых заместителей в пиррольную часть молекулы (R1 и R2 в XV), а также в положения 4, 6 или 7 бензольного фрагмента (R4, R6 и R7 в XV). Наличие заместителей в положении 5 (R5 в XV), напротив, нередко повышает активность веществ, причем это не коррелируется ни со сродством к электронам, ни с пространственными факторами заместителей. Оригинальный радиозащитный препарат мексамин (см. с. 161), относящийся к ряду 5-замещенных триптаминов, нашел практическое применение в медицине [33, 96, 229, 260]. На основе общего метода синтеза индолиалкиламинов по Абрамовичу и Шапиро, включающего взаимодействие акрилонитрила с малоновым эфиром, восстановительную циклизацию образующегося продукта, конденсацию с арил-диазонием по Джеппу — Клингеману, замыкание тетра-гидро-р-карболиновой системы по Фишеру и дальнейшее размыкание пиперидинового цикла с декарбоксилированием, были разработаны методы получения серотонина адипината (см. с. 166) и мексамина (см. с. 161), пригодные для промышленного производства [229].
Одной из общих тенденций развития химико-фармацевтической промышленности является все больший переход от выделения индивидуальных лекарственных веществ из труднодоступных природных объектов к их полному синтезу. Вслед за полным промышленным синтезом папаверина, левомицетина, кофеина, теофиллина, теобромина, эфедрина [211] в СССР было осуществлено на основе полного синтеза промышленное получение алкалоида атропина (см. с. 171), а также гоматропина гидробромида (см. с. 176).
Способ получения синтетического тропина — аминоспирта, лежащего в основе обоих указанных продуктов,— базируется на существенно усовершенствованной во
136
ВНИХФИ известной реакции Робинсона — Шэпфа, заключающейся в трехкомпонентной конденсации ацетоиднкар-боновой кислоты, метиламина и янтарного альдегида с последующим восстановлением образующегося тропинона [325, 328]. Появление промышленного синтетического тропина способствовало внедрению в производство и атропиноподобных веществ, поиски которых в различных странах интенсивно ведутся в течение уже нескольких десятилетий [115]. Проведенные советскими авторами во ВНИХФИ исследования впервые показали. [136, 184, 25], что в зависимости от характера ацилирующего остатка жирноароматические эфиры тропина общей формулы XVI
XVI
обладают не только м- или н-холинолнтической, но и а-адреноблокирующей активностью, которая ранее для эфиров тропина не была известна. Показано, что м- и н-холинолитические свойства характерны для соединений XVI, где R1 и R2— ароматические остатки, а а-адренобло-кирующие свойства проявляются у веществ XVI, где R1— ароматический заместитель, a R2—замещенный бензильный остаток, т. е. у тропиновых эфиров а, р-диарилпропио-новых кислот. Наряду с оригинальным атропиноподобным препаратом — тропацином, гидрохлоридом тропинового эфира дифенилуксусной кислоты (XVI, RI = R2 = C6H5) [136, 184], применяемым для лечения паркинсонизма и других заболеваний, сопровождающихся повышением мышечного тонуса, ранее внедрены в медицинскую практику и в производство [211], советскими учеными [234, 251] был создан также оригинальный а-адреноблокирую-щий препарат тропафен (см. с. 179), нашедший практическое применение в медицине при патологических процессах, связанных с нарушением периферического кровообращения.
Одним из оригинальных направлений поиска новых лекарственных средств, развиваемых советскими учеными во ВНИХФИ в течение последних четырех десятилетий, являются исследования в ряду производных хпиуклндпна (1-азабицикло [2,2,2] октана) (XVII) [263, 313, 346].
137
X V| f
В ходе этих исследований были выявлены определенные химические особенности хинуклидиновых соединений, представляющих собой достаточно жестко закрепленную бициклическую систему с узловым атомом азота, где каждый из шестичленных циклов зафиксирован в форме «ванны», обычно нехарактерной для насыщенных шестичленных систем. В отличие от алифатических и моноцикличе-ских аминов, а также от других 1-азабициклоалканов у хинуклидиновых соединений существенно ограничены конформационные изменения, связанные со свободным вращением атомов вокруг осей связей. И хотя при ваннообразной форме колец возникающие заслоненные конформации несколько повышают энергию взаимодействия несвязанных атомов, что ведет к отклонению симметрии молекул от Сзу, эти конформационные искажения незначительны и даже для 2,3-цис-дизамещенных соединений не превышают 12°. Одним из проявлений жесткой фиксации хинуклидиновых молекул является специфический характер свободной электронной пары узлового атома азота, которая практически не испытывает экранирующего влияния соседних атомов водорода, стерически направлена по оси симметрии молекулы и практически ортогональна л-электронам кратных связей, если последние являются семициклическими в положении 2, эндоциклическими в положениях 2, ^'или включены в конденсированные с хи-нуклидином ароматические системы. Указанные особенности строения исключают в соединениях такого типа возможность перекрывания р- и л-орбиталей и возникновения мезомерных эффектов. В результате у хинуклидиновых производных влияние узлового атома азота на соседние ароматические и непредельные группы оказывается чисто индукционным, без участия сопряжения, что не наблюдается в других классах веществ. Это приводит к ряду химических особенностей хинуклидиновых производных [высокая нуклеофильность хинуклидинового азота, кетон-ный, а не амидный характер хипуклидонов-2, отсутствие енаминных свойств у Д2-дегидрохинуклидинов и 2-илиден-хинуклидинов, ароматического сопряжения с азотом в бенз(Ь)хинуклидинах и др.].
138
Особенности структуры и реакционной способности хинуклидиновых молекул открывают широкие перспективы для направленного поиска новых биологически активных веществ [271].
Закрепленная структура бицикла, ограничивающая его конформационные изменения, позволяет фиксировать расстояния между реакционными центрами, исключить внутримолекулярные взаимодействия этих центров с образованием, например, водородных связей; подавить некоторые таутомерные превращения, изменяющие реакционную способность веществ в отношении биохимических рецепторов. Дезэкранирование и связанная с этим высокая нуклеофильность узлового хинуклидинового азота обеспечивает легкость подхода молекул к электрофильным центрам биохимических рецепторов, более прочное взаимодействие с ними. Структурная аналогия 3-ацилоксихинукли-динов (XVIII) со сложными эфирами холина (XIX) и в первую очередь с медиатором нервной системы ацетилхолином (XIX, П=СНз), а также с соответствующими алифатическими и моноциклическими соединениями, способными к различным конформационным изменениям, позволила синтезировать вещества с фиксированной структурой, имеющей закрепленные расстояния между сложно-эфирными центрами и атомами азота, при более высокой реакционной способности и стерической доступности обоих центров. В результате были созданы оригинальные лекарственные препараты ацеклидин — салицилат 3-ацето-ксихинуклидина (XVIII, R=CH3), применяемый при атонии внутренних органов и при глаукоме [211], оксили-дин — гидрохлорид 3-бензоилоксихинуклидина (XVIII, R = C6H5), центральное гипотензивное и седативное средство [211].
Ch2ocor снзснз сн2
<^3 х-
XIX
Следует отметить, что эти и другие сложные эфиры 3-оксихинуклиднна проявляют значительно большую активность, чем аналогичные алифатические, моноцикличе-ские и другие азабициклические соединения [271]. Дальнейшие исследования показали, что переход от 3-ацило-
139
ксихинуклидинов к их бенз(Ь)хинуклидиновым аналогам, связанный с изменением полярности веществ, их гидрофобности, размеров молекул, экранирования и реакционной способности функциональных групп, а также введение вместо кислорода в положении 3 хинуклидинового ядра атома азота приводят к существенному снижению фармакологического эффекта соединения на холинореактивные системы. При этом наиболее интересными с теоретической точки зрения оказались избирательная антиарит-мическая активность анти-изомера З-бензоилокси-6-мето-ксибенз(Ь)хинуклидина (XX), а также некоторых 3-амино-и 3-аминометил-хинуклидипов и пейтротропная активность, сходная по силе действия с триоксазином, у ЗЧЗ'Д'.б'-три-метоксибензоилокси) бенз(Ь)хинуклидина (XXI).
XXII ххш
Принцип увеличения биологической активности в отношении холинореактивных систем при переходе от алифатических и моноциклических соединений к хинуклиди-новым производным был использован во ВНИХФИ и при создании нового ганглиоблокирующего лекарственного препарата темехина (см. с. 181) [175, 249]. По сравнению с его пиперидиновым аналогом, лекарственным препаратом пириленом (пемпидином) (XXII) [211], соответствующее хинуклидиновое соединение (XXIII, R = H) — темехин характеризуется значительно большей избирательной активностью в отношении н-холинореактивных систем и терапевтической широтой [249]. Введение в молекулу те-
140
мехина еще одной С-метильной группы (XXIII, К = СНз) несколько усиливает токсичность при сохранении фармакологической активности; удлинение алкильной цепи или введение функциональных заместителей вызывают дальнейшее повышение токсичности и одновременно снижают ганглиоблокирующее действие. В другом направлении изменяется активность препарата при введении N-метильной группы, т. е. кватернизации хинуклидинового азота: при этом образуется эффективный ганглиоблокирующий препарат кратковременного действия имехин (см. с. 186), используемый в медицине для управляемой гипотонии при хирургических операциях [176, 259].
При систематическом изучении серий симметричных и несимметричных дичетвертичных хинуклидиновых производных типов XXIV, XXV и XXVI наибольший курарепо-добный эффект наблюдался у симметричных производных
XXVI
типа XXIV с цепочкой из шести метиленовых звеньев между двумя четвертичными хинуклидиновыми азотами (п=6). Соединение XXIV (п=6, К=СНгС6Нз) под названием квалидил (см. с. 187) вошло в медицинскую практику в качестве антидеполяризующего миорелаксанта [165, 215, 153].
В отличие от веществ с холинергической активностью, где переход от алифатических или моноциклических соединений к их хинуклидиновым аналогам значительно усиливает фармакологический эффект, у веществ, взаимодействующих с адренореактивными системами, аналогичной зависимости не наблюдается [271].
Новой ступенью в исследовании биологических свойств хинуклидиновых соединений явилось установление антигистаминной и антиаллергической активности у (хинуклидил-3) диарил (или гетерил) карбинолов общей формулы XXVII [99, 97, 166], в то время как соответствующие пиперидиновые производные лишены такой активности. Были разработаны общие методы синтеза соединений- типа XXVII, выявлено влияние пространственных и электронных факторов в ходе их синтеза [166], приготовлены не-хннуклидинорце .аналоги этих вещестр, разработаны пути
141
получения веществ, меченных тритием в хинуклидиновом и арильных ядрах. Гидрохлорид (хинуклидил-3)-дифенил-карбинола (XXVII, R1 = R2=C6H5) нашел практическое
XXVIII
применение в медицине для лечения различных аллергических заболеваний под названием фенкарол (см. с. 192) [97, 99, 166]. Показано, что увеличение расстояния между хинуклидиновым ядром и дифенилкарбинольным остатком за счет введения между ними мртиленовых групп, а также уменьшение этого расстояния путем перемещения заместителя в положение 2 хинуклидинового ядра (XXVIII), частичное дегидрирование хинуклидинового фрагмента — в положениях 2, 3, а также переход от третичных спиртов ко вторичным (XXVIII, R'=H) значительно снижают антигистаминную активность. (Недавно американские авторы установили, что соединение XXVIII, Ri = R2==C6H5 обладает в эксперименте достаточно сильным местноанестезирующим и антиаритмическим действием [317].) Неактивны вещества, в которых хинуклидино-вое ядро замещено другими гетероциклическими остатками, значительно убывает антиаллергическая активность при R1 и R2-aflKHfl независимо от длины алкильных цепей. Интересно, что переход от (хинуклидил-3) дигексилкарби-нола (XXVII, R1 = R2=C6Hi3) к его циклогексильному аналогу повышает антиаллергическое действие вещества. Введение в арильные остатки R1 и R2 мета- и пара-заме-стителей понижает, ортозаместителей усиливает активность. Основным напрай'йе'нием биотрансформации соединений XXVII в организме животных и человека является их инактивация и детоксикация путем превращения в соответствующие N-окиси [98].
Особенностями (хинуклидил-3)-диарил (или гетерил)-карбинолов как нового класса антигистаминных антиал-лергических средств являются высокая эффективность и низкая токсичность. Отсутствие стимулирующего или угнетающего влияния на ЦНС, что позволяет назначать их широкому кругу амбулаторных больных, в том числе людям активных профессий, не опасаясь потери работоспо-
142
собностй пли влияний побочного сйОтворйого эффекта. Отличительной особенностью этого класса веществ является также проявление у них нехарактерной для других антиаллергических препаратов способности не только блокировать гистаминорецепторы, но и опустошать запасы гистамина в тучных клетках, а также активировать диами-ноксидазу, участвующую в биотрансформацпи эндогенного гистамина [20]. Это обеспечивает эффективность указанных лекарственных препаратов в тех случаях, когда другие антиаллергические средства не оказывают действия.
Следует отметить, что важной проблемой в организации промышленного производства хинуклидиновых лекарственных препаратов явилась разработка методов получения основных полупродуктов — триацетонамина (XXIX) и хинуклидона-3 (XXX). Триацетонамин готовят из ацетона и аммиака в присутствии хлористого кальция и используют в синтезе темехина и имехнна, а хинукли-дон-3 (XXX) [35]—из продукта переработки у-пиколина (компонента р-пиколиновой фракции): '
и используют для получения ацеклидина, оксилидина, ква-лидила и фенкарола.
ПАРМИДИН
Бис-М-метилкарбаминовый эфир 2,6-бисбксиметилпири-дина:
ch2oocnhch3
CH3NHCOOCH3 "
С п Н |5 NjO
Мол. масса 253,25
Основные синонимы: пиридинолкарбамат, ангинин, про-дектин, ангиоксин, соспитан.
Белый или почти белый кристаллический порошок, без запаха, слегка горьковатого вкуса, растворим в метиловом спирте (1:20), 95% этиловом спирте (1:30), хлоро
143
форме (1:20), мадю^ растворим в воде (1:700), негигроскопичен; т. пл. 139—141 °C; ВФС 42-1465-80.
Пармидин — антисклеротический препарат [265].'В отличие от других антисклеротических средств, действие которых направлено на регуляцию липидного обмена, эффект пармидина связан главным образом с его антибрадикини-новой активностью [154]; он препятствует повышению проницаемости стенок сосудов и предупреждает проникновение в стенки сосудов атерогенных липидов. Препарат уменьшает или предотвращает изменения эндотелия артерий, уменьшает агрегацию тромбоцитов, улучшает микроциркуляцию, ослабляет воспалительные реакции, способствует улучшению кровообращения в органах, пораженных склеротическим процессом.
Применяют пармидин для лечения атеросклероза сосудов мозга, сердца, конечностей, при атеросклеротической и диабетической ретинопатии, тромбозе вен сетчатки, при облитерирующем эндартериите, трофических язвах конечностей. Препарат хорошо переносится, не вызывает выраженных побочных явлений, оказывает как лечебное, так и профилактическое действие и может применяться при лечении лиц пожилого возраста, когда другие антисклеротические средства малоэффективны.
Выпускается в таблетках по 0,25 г N. 100. Цена 8 р. 61 к.
Метод синтеза^ Апармидина в СССР разработан ВНИХФИ и ХФЗ «Акрихин» [265] по схеме:
2,6-Лутидин (I) окисляют до дипиколиновой кислоты (II), восстановлением дибутилового эфира которой (III) получают 2,6-бисоксиметилпиридин (IV), образующий при взаимодействии с метилизоцианатом пармидин (V).
Этот метод имеет преимущества перед другими способами, предусматривающими окисление 2,6-лутидина до 2,6-диформилпиридина, превращение 2,6-лутидина в его 144
N-окись с Двукратным повторением реакции БекеЛьхайде, синтез через 2,6-бисгалогенметилпиридины.
При окислении I во II перманганат калия дал лучшие результаты, чем двуокись селена и ее комплексы, окислы азота в присутствии двуокиси селена или бихромат натрия при высокой температуре под давлением. Разработка полярографического метода анализа и изучение кинетики превращения 2,6-лутидина в 6-метилпиколиновую, дипиколиновую, щавелевую и угольную кислоты позволили оптимизировать процесс [268].
Этерификация кислоты II бутанолом в присутствии катионитов с последующим восстановлением III (без его выделения) боргидридом натрия [126] дали наилучшие результаты по сравнению с осуществлением процессов этерификации через дихлорангидрид, калиевые или серебряные соли, спиртом с конц. серной кислотой или диазометаном, а также восстановлением III алюмогидридом лития или смесью боргидрида натрия с хлористым кальцием.
Реакция с метилизоцианатом в дихлорэтане в присутствии каталитических количеств октоата олова оказалась преимущественной по сравнению с обработкой IV фосгеном или его фтораналогами и затем метиламином, превращением IV с хлоругольными эфирами в карбонаты с дальнейшей обработкой метиламином или через азидоформиаты, а также с использованием в качестве ацилирующих агентов различных производных N-метилкарбами-новой кислоты.
Дипиколиновая кислота (II). К 76,3 г 40,2% 2,6-лутидина (30,6 г 100% I), растворенного в 690 мл воды, при 70—75 °C и интенсивном перемешивании добавляют 9 порций перманганата калия (по 23,5 г), внося каждую следующую порцию после полного раскисления предыдущей. Температуру реакционной массы поддерживают 70—75 °C. Двуокись марганца отфильтровывают при 70—75 °C, промывают нагретой до 80° С водой. Объединенные фильтраты подкисляют конц. соляной кислотой. Для более полного перевода монокалиевой соли дипиколиновой кислоты в свободную кислоту первоначально выпавший осадок растворяют при нагревании реакционной массы до 90 °C, после чего раствор с pH от —0,2 до —0,25 медленно охлаждают и дают выдержку 1 ч при 10—12 °C. Осадок дипиколиновой кислоты отфильтровывают, высушивают и определяют процентное содержание титрованием едким натром (по фенолфталеину) и полярографически (по обоим методам содержание II не ниже 98%). Щавелевая кислота
10 Заказ № 4304
145
(перманганаТоМетрйчески) и несгораемый остаток (примесь монокалиевыхл'ёолей дипиколиновой и щавелевой кислот) отсутствуют. Выход 29 г (60%); т. пл. 243— 244 °C разл.
2,6-Бисоксиметилпиридин (IV). К 440 мл безводного бутилового спирта (влаги не более 0,2%) прибавляют 35 г II и 35 г сухого катионита КУ 2X8 в Н+ форме. Кипятят с отгонкой воды по принципу Дина — Старка, контролируя конец реакции по определению титрометрически содержания в рёакционной массе монобутилового эфира дипиколиновой кислоты (конец реакции — при его содержании 0,007 г/мл). Массу оставляют охлаждаться до 90— 95 °C, декантируют с катионита. Катионит промывают 3 раза по 85 мл безводного бутанола и используют на следующей операции этерификации (катионит без регенерации может быть использован не менее 10 раз). К объединенным бутанольным растворам III прибавляют порциями 13,45 г боргидрида натрия, поддерживая температуру массы 35—40 °C. Дают выдержку 5 ч. Проверяют по ТСХ отсутствие в растворе бутилового эфира 6-оксиметилпико-линовой кислоты, после чего прибавляют 220 мл воды. Бутанольный слой отделяют, из водного слоя диол IV дополнительно извлекают бутанолом. Объединенные бутанольные экстракты упаривают в вакууме. Остаток кристаллизуют из воды в соотношении 1 : 1,55 с углем, используя маточный раствор на следующих операциях: при кристаллизации дибл'а IV и при разложении комплекса после восстановления боргидридом натрия диэфира III. Выход 16,85 г (57,8% на II).
Бис-М-метилкарбаминовый эфир 2,6-бисоксиметилпири-дина (V). К взвеси 54,49 г IV в 450 мл безводного дихлорэтана прибавляют несколько капель 21% раствора октоата олова в дихлорэтане и приливают 49 г метилизоцианата. Перемешивают 1 ч при кипении, контролируя течение процесса по ТСХ (конец реакции — при содержании монокар-баминового эфира не более 0,5 мг/мл). Реакционную массу охлаждают до 15—20 °C. Осадок V отфильтровывают, промывают 45 мл дихлоэтана и перекристаллизовывают из 1900 мл воды с углем. Выход 90,8 г (91%).
ПИРИДИТОЛ
Бис- (2-метил-3-окси-4-оксиметил-пиридил-5-метил) -дисульфида дигидрохлорид, моногидрат:
146
CI8 H2ON2°4S2'2HC1H2°
Мол. масса 459,41
Основные синонимы: энцефабол, энербол, пиритинол, пиритиоксин, бонифен, бонтиол, когитан, динерфен, энце-форт, цервиталин, цефалоген и др.
Белый со слегка желтоватым оттенком кристаллический порошок, легко растворим в воде, трудно растворим в спирте, практически нерастворим в хлороформе; ВФС 42-858-79.
Пиридитол — ноотропный препарат [77]. Снижает уровень у-аминомасляной кислоты (ГАМК) в нервной ткани, не изменяя концентрации ее предшественника — глутаминовой кислоты, подавляет активность пиридоксалевых ферментов и, в частности, ГАМК-трансаминазы, не влияя на активность глутаматдекарбоксилазы. Нормализует обмен в тканях головного мозга, ускоряет проникновение через гематоэнцефалический барьер аминокислот, жирных кислот, ионов натрия и глюкозы, способствует экономному использованию глюкозы, снижает образование избыточной молочной кислоты.
За счет влияния на обмен катехоламинов повышает содержание в ЦНС серотонина, увеличивает устойчивость тканей мозга к гипоксии, оказывает аитидепрессивное и седативное действие.
Применяют пиридитол при заторможенности и неглубоких депрессивных состояниях независимо от нозологической принадлежности, сочетающихся с сосудистой и другой органической недостаточнотью ЦНС, при шизофрении, астенических состояниях, адинамии, неврозоподобных расстройствах органического генеза, вегетоангиодистонии, последствиях нарушения мозгового кровообращения, а также в качестве корректора нейролептической терапии.
Выпускают в таблетках: по 0,05 г N. 50, цена 4 р. 97 к.; по 0,1 г N. 60, цена 9 р. 92 к.
Метод синтеза пиридитола разработан во ВНИВИ [77] по схеме:
10
147
Нагреванием 2-мет.ил-3-окси-4,5-диоксиметилпиридина гидрохлорида (пиридоксина гидрохлорида) (I) с тионил-хлоридом в хлористом метилене с добавкой диметилформ-амида получают 2-метил-3-окси-4,5-дихлорметилпиридина гидрохлорид (II), который путем частичного гидролиза водой при 45—50 °C превращают в 2-метил-3-окси-4-окси-метил-5-хлорметил пиридина гидрохлорид (III). Последний без выделения обработкой дисульфидом натрия с дальнейшей перекристаллизацией из смеси соляной кислоты со спиртом превращают в 2-метил-3-окси-4-оксиметил-пиридил-5-метил)-дисульфида дигидрохлорид, моногидрат (пиридитол) (IV). Этот метод имеет преимущества по сравнению с описанным синтезом по той же схеме, где II получают нагреванием I в избытке тионихлорида, а превращение II в IV осуществляют с выделением III в виде неустойчивого основания. Технологически менее удобным оказалось также получение IV из 4,5-ди-О-метилового, 4-О-этилового, 4,5-О-этилиденового или 4,5-О-изопропи-лиденового эфиров путем нагревания с бромистоводородной или соляной кислотами с последующим гидролизом 2-метил-3-окси-4,5-дибромметилпиридина гидробромида или II до 2-метил-3-окси-4-оксиметил-5-брометилпиридина гидробромида и /ббртветственно III, с дальнейшей обработкой дисульфидом натрия.
2-Метил-3-окси-4,5-дихлорметилпиридина гидрохлорид (II). К 200 г I в смеси 2 л хлористого метилена и 50 мл диметилформамида приливают при 20—25 °C в течение 30 мин 240 мл тионилхлорида, массу кипятят 12 ч, контролируют конец реакции хроматографически или полярографически. Осадок II отфильтровывают, промывают 1 л хлористого метилена и высушивают при 45—50 °C и остаточном давлении 25—30 мм рт. ст, Выхрд II 225 г (95%); т. пл. не ниже 195 °C.
14?
Бис- (2-метил-3-окси-4-оксиметил пиридил-5-метил ^дисульфида дигидрохлорид, моногидрат (пиридитол) (IV). К нагретым до 45 °C 1,65 л дистиллированной воды прибавляют при перемешивании НО г II, дают выдержку 1’/2 ч при 45—47 °C с перемешиванием. Раствор осветляют 10 г угля при 38—40°C (15—20 мин), фильтруют и приливают в течение 1 ’/2—2 ч к нагретому до 40—42 °C раствору дисульфида натрия, полученному путем обработки 182 г сульфида натрия в 290 мл дистиллированной воды, 24,5 г серы 30—40 мин при 80 °C. Реакционную массу нагревают 2 ч при 40—42 °C, охлаждают до 15—20 °C, контролируют, чтобы pH среды был 7. Осадок основания IV отделяют, промывают 10 л дистиллированной воды, кипятят 10 мин с 330 мл 10% соляной кислоты и 10 г угля, фильтруют. Фильтрат охлаждают до +1+3 °C и нейтрализуют 10% раствором едкого натра до pH 7. Выпавший осадок основания IV отфильтровывают, промывают 10—12 л дистиллированной воды, высушивают и смешивают с 120 мл спирта и 32,5 мл 35% соляной кислоты. Массу нагревают до 70—80 °C при перемешивании до полного растворения осадка, добавляют 4,5 г угля и дают выдержку 20 мин при 70—80°С и работающей мешалке. Горячий раствор фильтруют, охлаждают до —4 °C—(—6 °C) и дают выдержку 18—20 ч при периодическом размешивании. Выделившиеся кристаллы IV отфильтровывают, промывают 60 мл охлажденного до 0°С абсолютного спирта и высушивают при 40—50 °C и остаточном давлении 20—25 мм. Получают 61 г технического IV. Из маточных растворов после упаривания до ]/ю первоначального объема выделяют дополнительно 4 г технического IV. Технический IV перекристаллизовывают из 65 мл 10% соляной кислоты с добавкой 5 г угля (нагревание до 70 °C, охлаждение до —6 °C, 18—20 ч). Кристаллический IV отфильтровывают, промывают 50 мл охлажденного до — 4°С абсолютного этилового спирта и высушивают. Получают53,2г (57,4%) IV.
МЕРИДИЛ
Метилового эфира трео-а-фенил-а- (пиперидил-2) -уксусной кислоты гидрохлорид:
I I соосн3
N CH НС1
п X
СвН5
С и Н 19 no2 • HCI Мол- Macca 269-62
149
Основные синонимы: метилфенидата гидрохлорид, цен-тедрин, риталин и др.
Белый кристаллический порошок, легко растворим в воде и метиловом спирте, растворим в этиловом спирте и хлороформе, мало растворим в ацетоне; т. пл. 206—208 °C.
Меридил — психостимулятор [270], оказывает менее сильное возбуждающее действие на ЦНС, чем фенамин, мало влияет на периферические адренореактивные системы, не вызывает повышения артериального давления. В отличие от меридила его диастереомер — метилового эфира эритро-а-фенил-а-(пиперидил-2)уксусной кислоты гидрохлорид малоактивен в отношении ЦНС, но проявляет периферическое симпатомиметическое действие.
Применяют меридил в психиатрической практике и клинике нервных болезней при легких формах депрессий, повышенной утомляемости, астенических состояниях.
Форма выпуска: таблетки по 0,01 г. Список А. Высшая разовая доза 0,015‘'г;суточная— 0,03 г.
Метод синтеза меридила разработан во ВНИХФИ [267, 270] по схеме:
При конденсации 2-хлорпиридина (I) с цианистым бензилом (II) под действием гидрида лития получают а-фе-нил-а-(пиридил-2)-ацетонитрил (III), который омыляют водно-спиртовым раствором едкого кали до калиевой соли а-фенил-а-(пиридил-2)-уксусной кислоты и без выделения гидрируют с никелевым катализатором при 70 °C и давлении водорода 50 атм. Продукты гидрирования нагревают в сильно щелочной среде, что приводит к частичной изомеризации эритро-формы в трео-, и при дальнейшем подкислении осаждакй* v трео-а-фенил-а-(пиперидил-2)-уксусную кислоту (V), которую этерифицируют метанолом и серной кислотой. Последующее выделение гидрохлорида
150
позволяет получить индивидуальный диастереомер — .Метилового эфира трео-а-фенил-а-(пиперидил-2)-уксусной кислоты гидрохлорид (VI). Превращение III в IV проводят без выделения в чистом виде продуктов IV и V. Преимуществом этого метода по сравнению с описанными ранее двумя схемами синтеза из 2-хлорпиридина (I) и цианистого бензила (II) через а-фенил-а-(пиридил-2)-ацетонитрил (III) является возможность получения индивидуального трео-изомера конечного продукта (VI). По первой из ранее предложенных схем III с конц. серной кислотой превращают в а-фенил-а-(пиридил-2)-ацетамид, который подвергают алкоголизу до метилового эфира а-фенил-а-(пиридил-2)-уксусной кислоты с дальнейшим восстановлением в присутствии платинового катализатора. По второй схеме восстановление в присутствии платины пиридинового ядра осуществляют на стадии а-фенил-а-(пиридил-2)-ацетамида с дальнейшим омылением а-фенил-а-(пиперидил-2)-ацетамида до свободной кислоты и ее этерификацией метанолом. По обеим этим схемам, используемым за рубежом в промышленных условиях, удается получить только смесь эритро- и трео-изомеров, причем их соотношение варьирует в широких пределах. Кроме того, платиновый катализатор является дорогостоящим и при гидрировании в его присутствии обычно наряду с восстановлением пиридинового ядра наблюдается частичное гидрирование фенильной группы до циклогексильной, что приводит к загрязнению целевого продукта трудно отделимой примесью метилового эфира а-циклогексил-а-(пиперидил-2)-уксусной кислоты гидрохлорида. Применение на первой стадии, при конденсации I с II, вместо амида натрия гидрида лития (или натрия) позволило сделать процесс более безопасным и на 20% повысить выход продукта конденсации III. При использовании в качестве конденсирующих средств фениллития, этилата калия или натрия, третичного бутилата калия, трилона Б в 50% растворе едкого натра выходы III существенно понижались. Опыты конденсации 2-хлорпиридина (I) с фенилуксусным эфиром или а-цианфенилуксусным эфиром не дали положительных результатов. При гидрировании в присутствии никелевого катализатора метилового эфира а-фенил-а-(пиридил-2)-уксусной кислоты образуется только неактивный эритро-изомер.
а-Фенил-а-(пиридил-2)-ацетонитрил (III). К нагретой до кипения суспензии 120 г гидрида лития (диаметр частиц 0,2—0,3 мм) в 2,6 л толуола в течение 3чприбавля-
151
ют 568 г 2-хлорпиридйна (i) и 910 г Цианистого бензила (II) в 750 мл толуола. Массу кипятят 7 ч, охлаждают до 20—25 °C и постепенно приливают 1 л метанола, кипятят 1 ч, охлаждают до 20—25 °C и прибавляют 3,5 л воды. Толуольный слой отделяют, водный — экстрагируют толуолом (3 раза по 1 л). Объединенные толуольные экстракты упаривают, остаток перегоняют в вакууме, собирают фракцию с т. кип. 150—160°С (3—4 мм рт. ст.); т. пл. 89°C. Выход III 88,4% (с учетом возвращенных в первой фракции I и II, которые вновь используются в последующих загрузках).
Метилового эфира трео-а-фенил-а-(пиперидил-2)-уксус-ной кислоты гидрохлорид (меридил) (IV). Раствор 190 г а-фенил-а-(пиридил-2)-а-ацетонитрила (III) и 380 г едкого кали в 4 л 50% водного метанола кипятят 7 ч. Метанол и часть воды отгоняют, уменьшая объем реакционной массы до 2 л. pH раствора доводят до 8,0 добавлением конц. соляной кислоты, прибавляют 200 г скелетного никелевого катализатора и гидрируют при 70 °C и начальном давлении водорода 50 атм. Катализатор отфильтровывают, промывают водой и из фильтрата хлороформом (3 раза по 200 мл) извлекают побочный продукт — 2-бензилпипе-ридин (19,5 кг). К водному раствору прибавляют 400 г твердого едкого кали и отгоняют постепенно в течение Зч воду, доводя объем реакционной массы до 1,6 л (в про-
цессе упарки проходит частичная изомеризация эритроизомера в трео-изомер), после чего раствор охлаждают до 20—25 °C и подкисляют конц. соляной кислотой до
pH 6,0. Выпавший осадок трео-а-фенил-а-(пиперидил-2)-уксусной кислоты (VII) отфильтровывают. К фильтрату добавляют 400 г едкого кали, массу кипятят 5 ч, охлаждают до 20—25 °C и прибавляют коиц. соляной кислоты до pH 6,0. Выделившийся осадок VII отфильтровывают. Вод
ный маточный раствор, оставшийся после отделения трео-кислоты VII, упаривают, остатки воды отгоняют в виде азеотропа с бензолом, добавляют 1 л метанола и 150 мл
конц. серной кислоты, кипятят 6 ч, подщелачивают при 2—5°C 50% водным раствором поташа и экстрагируют хлороформом 44 г метилового эфира эритро-а-фенил-а-(пиперидил-2)-уксусной кислоты, содержащего примесь трео-изомера. К остатку после отгонки хлороформа прибавляют раствор 44 г едкого кали в 100 мл воды, кипятят 5 ч, охлаждают до 20 °C и подкисляют конц. соляной кислотой до pH 5,0. Выделившийся осадок трео-кисло-ты VII отфильтровывают. К маточному раствору прибав
152
ляют 88 г едкого кали и 100 мл воды, кипятят 5 ч и снова подкисляют до pH 6,0. Выделяют трео-кислоту VII. Все выделенные порции трео-кислоты VII объединяют, приливают к ним 1 л метанола и 150 мл конц. серной кислоты. Массу кипятят при перемешивании 6 ч, охлаждают до 2— 5 °C и подщелачивают при этой температуре 50% водным раствором поташа. Метиловый эфир трео-а-фенил-а-(пиперидил-2)-уксусной кислоты экстрагируют хлороформом (3 раза по 500 мл). Экстракт высушивают поташом, хлороформ отгоняют. Остаток растворяют в 400 мл эфира, обесцвечивают углем и подкисляют спиртовым раствором хлороводорода. Выделившийся осадок меридила перекристаллизовывают из изопропилового спирта. Выход VII83 г (31,4% на III). По данным ТСХ примеси эритро-изомера и метилового эфира а-циклогексил-а- (пиперидил-2) -уксусной кислоты в препарате отсутствуют.
НИТРОКСОЛИН
5-Нитро-8-оксихинолин
с8нви3оэ
он
Hojb
Мол. масса 190,16 Основные синонимы: 5-НОК, нибиол, ниурон, ноксибиол, изинок, никинол, никопет, 5-нитро-оксин, ноксигур, урит-рол, уролин и др.
Мелкокристаллический порошок от желтого до зеленовато-желтого цвета, без запаха, горького вкуса, практически нерастворим в воде, мало растворим в спирте, бензоле, ацетоне, хлороформе, растворим в диметилформами-де, разбавленных минеральных кислотах и щелочах; т. пл. 177—181 °C (в интервале 2°C); с солями большинства металлов дает нерастворимые хелатные комплексы; ВФС 42-578-78.
Нитроксолин — химиотерапевтическое средство с широким спектром действия. Эффективен против грамполо-жительных и грамоТрицательных бактерий, а также некоторых грибов (рода кандида и др.). В отличие от других производных 8-оксихинолина быстро всасывается из же-
153
лудочно-кишечного тракта и выделяется в неизмененном виде через почки, что приводит к высокой концентрации препарата в моче.
Применяют нитроксолин при инфекциях урогенитального тракта (пиелонефрит, цистит, уретрит, простатит и др.). Препарат часто эффективен в случае резистентности микрофлоры к антибиотикам и другим антибактериальным средствам. Назначают внутрь во время еды, при почечной недостаточности возможна кумуляция препарата. Выпускается в виде драже по 0,05 г N. 50 во флаконах. Цена 2 р. 02 к.
Метод синтеза нитроксолина разработан филиалом ВНИХФИ в поселке Купавна совместно с ХФЗ «Акрихин» [36] на основе схемы, предложенной В. Петровым и Б. Сте-геоном [321] и усовершенствованной Ю. Прэтом и Н. Дрейком [323].
По этому методу 8-оксихинолин (I) нитрозируют нитритом натрия в сернокислой среде до 5-нитрозо-8-оксихино-лина (II), который окисляют азотной кислотой до нитроксолина— 5-нитро-8-оксихинолина (III). Окисление II феррицианидом калия в щелочной среде или перекисью водорода в присутствии щелочей дает худшие результаты. Прямое нитрование 8-оксихинолина (I) сопровождается образованием значительных количеств 7-нитро- и 5,7-ди-нитро-8-оксихинолинов, а синтез III по Скраупу из 2,4-динитрофенола или 2-амино-4-нитрофенола приводит к очень низкому (~2%) выходу целевого продукта.
5-Нитрозо-8-оксихинолин (II). В охлажденной до 15— 18 °C смеси 667 мл дистиллированной воды и 30 мл конц. серной кислоты растворяют при интенсивном перемешивании 73,43 г I, после чего при температуре не выше 20 °C прикапывают раствор 36,7 г нитрита натрия в 68 мл воды. Контролируют pH среды (pH 1—2), наличие избытка нитрит-иона и выдерживают при перемешивании и температуре 18—20 °C еще 3 ч (каждый час контроль pH и NO?-), затем охлаждают до 10—15°C и подщелачивают 42% раствором едкого натра (~34 мл) до pH 7,5—8, после чего сразу же подкисляют (для более полного вы
154
Деления II) ледяной уксусной кислотой до pH 3—4. Перемешивают 20 мин, осадок II отфильтровывают, промывают дистиллированной водой (~50 мл) и пасту с влажностью около 48% без сушки (высушивание II взрывоопасно!) направляют на окисление.
5-Нитро-8-оксихинолин (нитроксолин) (III). Полученную на предыдущей стадии пасту 5-нитрозо-8-оксихиноли-на смешивают с 226 мл дистиллированной воды и к полученной смеси по каплям при 35—40 °C и энергичном перемешивании прибавляют 107 мл конц. азотной кислоты (при более высокой температуре образуется преимущественно 5,7-динитро-8-оксихинолин). Перемешивают при 35—40 °C еще 2 ч, контролируя конец окисления по определению в пробах соотношения продуктов III: II (не должно быть ниже 20:1). Массу охлаждают до 5—10°С и прибавляют постепенно при хорошем перемешивании и температуре не выше 25 °C 42% раствор едкого натра до pH 7,5—8 (раствор приобретает красно-оранжевое окрашивание), после чего подкисляют при 15—20 °C ледяной уксусной кислотой до pH 3—4, перемешивают 20 мин, осадок III отфильтровывают и промывают водой (4 раза по 200 мл) до полного удаления нитрит-, сульфат- и аце-тат-ионов (аналитические пробы), затем промывают 100мл ацетона и перекристаллизовывают из 1840 мл ацетона с добавкой 5 г активированного угля (5-нитрозо-8-оксихи-нолин и 5,7-динитро-8-оксихинолин, содержащиеся в техническом продукте, в кипящем ацетойе практически не растворяются и отходят в смеси с осветляющим углем). Готовый продукт промывают 250 мл дистиллированной воды и сушат при 70°С. Выход III 65,79 г (68,7%, считая на I).
хиноцид
6-Метокси-8- (4'-аминопентиламино) хинолина дигидро-хлорид
сн3о
NH(CH,),CHNH,- 2НС1
Ан
С is Н 20 N3O-2HCl
Мол. масса 332,27
Кристаллический порошок желто-оранжевого цвета, очень хорошо растворим в воде, трудно — в этиловом спир
155
те, нерастворим в эфире, бензоле и ацетоне; т. пл. 226— 227° С разл.; ГФХ, ст. 152.
Хиноцид — оригинальный противомалярийный препарат [30, 31, 290], действующий на параэритроцитарные тканевые формы малярийного паразита, которые обусловливают отдаленные рецидивы заболевания. Обладает гаметотропным действием в отношении половых форм всех видов плазмодиев.
Применяют для предупреждения отдаленных рецидивов трех- и четырехдневной малярии, а также для профилактики этих форм малярии при высоком риске заражения.
.Форма выпуска: таблетки, покрытые оболочкой, по 0,005 и 0,01 г.
Метод синтеза х^йоцида разработан во ВНИХФИ по схеме [167].
Пара-анизидин (I) ацетилируют уксусным ангидридом в гомогенной среде (раствор в 98% уксусной кислоте) и
156
[ N-ацетилпройзводное (Ц) без выделения нитруют 12% | азотной кислотой в орто-положение к ацетиламиногруппе. I Полученный 2-нитро-4-метоксиацетапплид (III) в виде пасты вводят с глицерином в присутствии мета-нитробензолсульфокислоты в реакцию Скраупа и продукт «скраупирова-ния»—6-метокси-8-нитрохинолин (IV)—восстанавливают
каталитически до 6-метокси-8-аминохинолина (V). Параллельно синтезируют 4-фталимидо-1-йодпентан (VI), для чего очищенный у-ацетопропиловый спирт (VII) подвергают восстановительному аминированию до 4-аминопентано-, ла-1 (VIII), в который вводят фталимидную защиту аминной функции, окси-группу в 4-фталимидопеитаноле-1 (IX) с тионилхлоридом замещают на хлор и 4,-фталимидо-1-хлорпентан (X) с йодистым натрием превращают в более реакционноспособное йодпроизводное VI. При взаимодействии VI с метоксиаминохинолином V получают 6-метокси-8- (4'-фталимидопентиламино) хинолин (XI), в котором фталильную защиту снимают с помощью гидразингидрата и едкого кали. Полученный 6-метокси-8-(4'-аминопентил-амино)хинолин (XII) переводят в гидрохлорид—препарат хиноцид (XIII). В основу этого метода легла описанная ранее схема [31, 290]. Другая известная схема [30], базирующаяся на конденсации 6-метокси-8-аминохинолина с 4-амино-1-бромпентаном, без использования фталильной защиты хотя и отличается меньшим количеством стадий, но дает существенно более низкие выходы XIII, поскольку имеют место побочные процессы циклизации незащищенного аминоалкилбромида с образованием значительных количеств 2-метилпирролидина. Применение на стадии «скраупирования» в качестве окислителя мета-нитробензолсульфокислоты [167] ' обеспечивает более спокойное, без экзотерм и выбросов течение реакции по сравнению с ранее использовавшейся в этом процессе высокотоксичной мышьяковистой кислотой [30]. Восстановление 6-метокси-8-нитрохинолина (IV) в присутствии скелетного никелевого катализатора технологически более приемлемо, чем другие известные методы восстановления IV: двухлористым оловом, смесью железа и хлористого железа в соляной кислоте, сернистым аммонием в спиртовом растворе, сернистым натрием и хлористым аммонием в спирте, каталитически в присутствии платины или палладия, а также никеля при более высоком давлении, когда частичному восстановлению подвергается и хинолиновое ядро. Предложенный в ходе разработки промышленного синтеза хиноцида метод очистки у-ацетопропилового спирта (VII)
157
от сопутствующих циклических продуктов 2-метил-4,5-ди-гидрофурана и 2-метил-2-(у-ацетопропокси) -тетрагидрофурана — путем превращения их в у-ацетопропиловый спирт в присутствии разбавленной соляной кислоты [168] — обеспечил более однозначное течение процесса восстановительного аминирования VII.
2-Нитро-4-метоксиацетанилид (III). Смесь 500 мл технической уксусной кислоты и 200 г технического п-анизи-дина нагревают 30 мин при 50—60 °C и к полученному раствору при 50—60 °C в течение 30 мин приливают 200 мл технического уксусного ангидрида. Массу нагревают 1 ч при 60 °C, охлаждают до 40—45 °C и приливают за 15— 20 мин 120 мл 12% азотной кислоты. Перемешивают 30 мин при 45—50° С, охлаждают до 15—18 °C и дают выдержку 1 ч. Выделившийся осадок III отфильтровывают, промывают 4 раза по 250 мл воды. Получают 256 г (76%) III в пересчете на 100% сухое вещество. Продукт без высушивания в виде пасты с 61,6% содержанием основного вещества и более 35% влаги направляют на следующую стадию.
6-Метокси-8-нитрохинолин (IV). К 334 мл воды при перемешивании приливают ПО мл конц. серной кислоты, поддерживая температуру 50—60 °C. Затем при той же температуре приливают 360 мл 28% сернокислого раствора м-нитробензолсульфокислоты, полученного прибавлением к 280 мл 20% олеума при 45—50 °C и перемешивании в течение 30—40 мин 92,5 г нитробензола с последующим нагреванием 2 ч при 120—125 °C (контроль конца реакции получения м-нитрдбензолсульфокислоты— полное растворение отобранной пробы в воде при отсутствии запаха нитробензола). Затем прибавляют 132 мл глицерина и 125 г 2-нитро-4-метоксиацетанилида (III). Смесь нагревают 4 ч при 132—136°C, охлаждают до 18—20°С и выливают при перемешивании в 2,5 л воды. Обесцвечивают 40 г угля, отфильтровывают. Фильтрат при перемешивании и охлаждении рбрабатывают водным раствором аммиака (1,1 л) до pH 4—5, поддерживая температуру 30—35 °C. Водный маточный раствор сливают с выделившегося смолистого осадка. К осадку добавляют 1,8 л технического дихлорэтана и 300 мл воды. Массу кипятят 1 ч, охлаждают до 25—30 °C. Дихлорэтановый слой, содержащий. IV, отделяют, обесцвечивают углем и упаривают. К остатку при 35—40 °C приливают 100 мл метанола и охлаждают при перемешивании до 15—18 °C. Осадок IV отфильтровывают, промывают на фильтре 2 раза по 25 мл метанола.
158
Выход IV 65 г (56,5%); т. пл. 158—160°С, содержание основного вещества 99—100%.
6-Метокси-8-аминохинолин (V). 609 г 6-метокси-8-нит-рохинолина (IV), 90 г никелевого катализатора и 2,4 л метанола гидрируют в автоклаве при 20—25 °C и давлении водорода 50 атм. Катализатор отфильтровывают, а метанольный раствор используют без выделения V на стадии взаимодействия с 4-фталимидо-1-йодпентаном (VI).
4-Аминопеитанол-1 (VIII). Заливают 300 мл технического у-ацетопропилового спирта (VII) 180 мл воды и прибавляют соляную кислоту х. ч. до pH 2,5—3,0 (по рН-метру), после чего реакционную смесь перемешивают 18 ч при 20—25 °C и без дополнительной обработки подвергают восстановительному аминированию, для чего охлажденный до —3°С раствор VII приливают в течение 1 ч при перемешивании к охлажденным до —3°С 670 мл 25%' водного раствора аммиака. После дополнительной выдержки 1 ч при —3—0 °C загружают 30 г никелевого катализатора и массу гидрируют при 70°C и давлении водорода 50 атм. Катализатор отфильтровывают. Из фильтрата азеотропной отгонкой с ксилолом удаляют воду, после чего отгоняют ксилол, а VIII перегоняют в вакууме, собирая фракцию с т. кип. 86—105°C (7 мм рт. ст.). Выход 263 г (88,5%).
4-Фталимидо-1-йодпентан (VI). К смеси 108 г VIII и 700 мл ксилола прибавляют при перемешивании 148 г фталевого ангидрида, кипятят 1—1 ’А ч с азеотропной отгонкой воды. Для завершения процесса после прекращения отгонки воды реакционную смесь дополнительно кипятят 3 ч, ксилол отгоняют в вакууме. К остатку прибавляют 500 мл бензола, охлаждают до 10 °C и при перемешивании, поддерживая температуру 10—15 °C, приливают 117 мл тионилхлорида, кипятят 6 ч. Бензол с избытком тионилхлорида отгоняют, сначала при нормальном давлении и температуре в массе 80—85 °C, затем для полноты отгонки при 90 °C и давлении 300 мм рт. ст. Остаток растворяют в 800 мл бензола и промывают 200 мл 10% раствора! соды. Бензол отгоняют, вещество растворяют в 1 л ацетона, нагревают при кипении и перемешивании 40 ч с 244 г йодистого натрия, охлаждают до 15—18 °C, неорганические соли отфильтровывают, фильтрат обесцвечивают углем и полученный ацетоновый раствор, содержащий 23% VI, 2,3% X, около 0,46% йодистого натрия и следы хлористого натрия, без дополнительной переработки направляют на следующую стадию.
159
6-Метокси-8-(4'-фталимидопентиламино)хинолин (XI). Ацетоновый раствор 4-фталимидо-1-йодпентана (VI) от предыдущей стадии упаривают (отгонку завершают при давлении 300 мм рт. ст. и температуре в массе 70—75°C). К остатку при 50—55 °C и перемешивании прибавляют 61,5 г кальцинированной соды и 364 мл метанольного раствора, содержащего 48 г 6-метокси-8-аминохинолина (VI). Метанол отгоняют в вакууме (60—75°C, 300 мм рт. ст.). К остатку приливают 400 мл ксилола, часть которого отгоняют с целью удаления в виде азеотропа следов воды, пока температура в массе не достигнет 118—120 °C, после чего кипятят 20 ч, отфильтровывают от неорганических солей и упаривают в вакууме (90—100°C, 300 мм рт. ст.). К охлажденному до 40—50 °C остатку приливают 1,5 л метанола и подкисляют по конго метанольным раствором хлороводорода. Массу перемешивают 10 ч. Гидрохлорид XI отфильтровывают и промывают метанолом. Выход 67 г (55%, считая на V и VI).
6-Метокси-8-/(Д/-аминопентиламино)хинолин (XII). Смесь 67 г гидрохлорида XI, 17,6 мл гидразингидрата и 640 мл этилового спирта кипятят 3 ч при перемешивании. Спирт отгоняют в вакууме (60—70 °C в массе, 300 мм рт.ст.), к остатку прибавляют 410 мл 35% водного раствора едкого кали и 400 мл бензола, перемешивают 30 мин при 40—50 °C, охлаждают до 18—20 °C. Бензольный слой отделяют, водный слой экстрагируют бензолом (2 раза по 100 мл). Объединенные бензольные растворы промывают водой (3—4 раза по 50 мл) до исчезновения в промывных водах щелочной реакции на фенолфталеин, после чего обесцвечивают углем и упаривают. Остаток разгоняют в вакууме; собирают фракцию с т. кип. 212— 220°C (4 мм рт. ст.). Выход XII 36 г (87%).
6-Метокси-8-(4'-аминопентиламино)хинолина дигидрохлорид (хиноцид) (XIII). Основание хиноцида (XII) от предыдущей стадии растворяют в 360 мл абсолютного этилового спирта, спиртовой раствор обесцвечивают 4 г угля, отфильтровывают и к фильтрату при 20 °C и хорошо работающей мешалке в один прием приливают 100 мл 10,4% (по объему) спиртового раствора хлороводорода (среда должна быть кислая по конго). Температура массы повышается до 30—35°С и через 10—15 мин из прозрачного раствора начинает выпадать ярко-оранжевого цвета осадок хииоцида.
Реакционнуюусмссь охлаждают до 5—10°C и перемешивают 2 ч при этой температуре. Хиноцид (XIII) от-160
фильтровывают, промывают абсолютным этиловым спиртом (3 раза по 30 мл) и высушивают при 50—60°C. Выход 41,5 г (90%).
МЕКСАМИН
5-Метокситриптамина гидрохлорид:
н
с„ hi4n2o hci
Мол. масса 226,5
Белый или белый с серовато-кремовым оттенком кристаллический порошок, растворим в воде, метиловом и этиловом спиртах, нерастворим в эфире, бензоле, хлористом метилене; в УФ-спектре имеет характерный максимум при 280 нм, не смещающийся при изменении pH раствора; т. пл. 242—244 °C разл.; МРТУ 42 № 3585-68.
Мексамин — оригинальный радиопротектор [33, 75, 96, 229, 260], близкий по фармакологическим свойствам к серотонину (см. с. 166). Препарат вызывает сокращение гладкой мускулатуры, сужение кровеносных сосудов, уменьшение диуреза.
Применяют мексамин для профилактики лучевой реакции, в том числе при рентгенотерапии злокачественных новообразований. В отличие от серотонина используется перорально.
Форма выпуска: таблетки по 0,05 г, покрытые оболочкой, Ц. 50. Цена 5 р. 02 к.
Метод синтеза мексамина разработан во ВНИХФИ на основе схемы, предложенной Абрамовичем и Шапиро [276].
Ц Заказ Xs 4304
151
XI
При конденсации по Михаелю малонового эфира (I) с акрилонитрилом (II) получают диэтиловый эфир р-циан-этилмалоновой кислоты (III), который путем каталитического восстановления в присутствии никеля превращают в З-карбэтоксйпиперидон-2 (IV). Сложноэфирную группу в оксоэфире IV омыляют и полученную калиевую соль З-карбоксипиперидона-2 (V) без выделения подвергают взаимодействию с п-метоксифенилдиазоний хлоридом (VI), образующимся при диазотировании нитритом натрия в солянокислой среде п-анизидина (VII). Возникающий 3-п-метоксифенилгидразон пиперидиндиона-2,3 (VIII) подвергают циклизации нагреванием в водно-спиртовой среде с кислотным катализатором. Продукт циклизации по Фишеру — 1-оксо-6-метокси-1, 2, 3, 4-тетрагидро-р-карболин (IX)—омыляют едким кали с раскрытием шестичленного лактамного кольца и при дальнейшей обработке уксусной кислотой выделяют 5-метокситриптамин-2-карбоно-вую кислоту (X), которую подвергают декарбоксилированию с дальнейшим выделением 5-метокситриптамина гидрохлорида [мексамина (XI)]. Приведенный выше метод имеет преимущество перед другими опубликованными в литературе способами синтеза 5-метокситриптамина: из п-метоксифенилгидразина и диэтилацеталя у-аминомасля-ного альдегида с циклизацией по Фишеру соответствующего гидразона, основанного на использовании труднодоступного у-аминомасляного альдегида; из п-метоксифенилгидразина и труднодоступного у-хлормасляного альдегида [52], из 5-метоксииндола через магиийорганическое производное с дальнейшей конденсацией его с хлорацетонитрилом и восстановлением 5-метоксииндолил-З-ацетони-трила натрием в кипящем спирте [214]; через полученный по реакции Вильсмайера 5-метоксииндол-З-альдегид и 3-(р-нитровинил)-5-метоксииндол с восстановлением последнего алюмогидоидом лития в тетрагидрофуране; из
162
п-метоксифенилгидразона fj-формилпропионовой кислоты с циклизацией по Фишеру в этиловый эфир 5-метоксиин-долил-3-уксусной кислоты и далее через гидразид и амид с восстановлением последнего алюмогидридом лития [228]; из п-метоксифенилдиазоний хлорида и этилового эфира а-ацетил-6-фталимидовалериановой кислоты с циклизацией получающегося гидразона по Фишеру (в варианте Абрамовича и Шапиро), омылением сложноэфирной группы в 2-карбэтокси-3-(р-фталимидоэтил)-5-метоксииндоле, декарбоксилированием и снятием фталильной защиты [227]; из 5-метоксииндола через 5-метоксиграмин и 5-метоксиин-долилацетонитрил с восстановлением нитрильной группы по методу А. П. Терентьева, М. Н. Преображенской и Бан-Лун-Ге [231] — действием гидразингидрата в присутствии никеля Ренея.
Диэтиловый эфир р-цианэтилмалоновой кислоты (Ш). К 1137 мл I прибавляют 47,2 г натрия. Когда он полностью прореагирует, массу нагревают до 80 °C и приливают 496 мл II с такой скоростью, чтобы температура оставалась в пределах 85—90 °C. Перемешивают до тех пор, пока температура не понизится до 70°C, и охлаждают до 20 °C. Затем приливают 20 мл 98% уксусной кислоты (pH достигает 5—6), перемешивают 15 мин, проверяют pH раствора, добавляют 680 мл дихлорэтана и 480 мл воды. В водный раствор переходят ацетат натрия и побочные продукты полимеризации акрилонитрила. Дихлорэтановый слой отделяют, промывают водой 2 раза по 200 мл и упаривают при давлении 100—150 мм рт.‘ст. Остаток перегоняют в вакууме, собирают фракцию с т. кип. 164—168°С (8 мм рт. ст.). Выход 1002 г (65,5%), считая на II.
З-Карбоэтоксипиперидон-2 (IV). Раствор 1 кг цианэфи-ра III в 800 мл абсолютного этилового спирта гидрируют в присутствии 75 г никелевого катализатора при 50 °C и давлении водорода 50—60 атм. Конец гидрирования определяют по методу ГЖХ, анализом отобранной пробы (содержание III не должно превышать 2%). Катализатор отфильтровывают. От фильтрата продуванием азотом в течение 6 ч отделяют летучие амины, образовавшиеся в качестве побочных продуктов при гидрировании и могущие без отдувки при последующем упаривании вызвать сильное вспенивание. Затем спирт отгоняют (для более полного удаления спирта, препятствующего дальнейшей перекристаллизации из фреона-113, в конце отгонки подключают вакуум с остаточным давлением 150 —
11
163
200 мм рт. ст. й поднимают температуру в массе до 00— 95°C). Остаток перекристаллизовывают из 1000 мл фреона-113 [вещество растворяют при 45—47°С, кристаллизуют при 0—(4-3°C)]. Осадок отфильтровывают, промывают 200 мл фреона-113 и высушивают в вакууме. Выход 443 г (55,5%).
З-п-Метоксифенилгидразон пиперидиндиона-2,3 (VIII). “ К раствору 94 г едкого кали в 2,5 л воды прибавляют L, 251 г IV, нагревают 3 ч при 27—30 °C с перемешиванием и к охлажденному до —5 °C раствору калиевой соли З-карбоксипиперидона-2 (V) приливают охлажденный до —2°С водный раствор п-метоксифенилдиазоний хлорида, приготовленного путем диазотирования 196 г перекристаллизованного из этилового спирта п-анизидина (VII) [диазотирование проводят постепенным добавлением к VII, растворенному в смеси 1,85 л воды и 706 мл 27% соляной кислоты, 485 мл раствора нитрита натрия, содержащего 20 г/л основного вещества, поддерживая pH раствора 1,0, температуру — 2 — (4-2 °C) и контролируя конец процесса по йодкрахмальной бумажке; избыток нитрита натрия разлагают приливанием 50% раствора мочевины при той же температуре до отрицательной реакции на нитрит-ион по йодкрахмальной бумажке; затем избыток соляной кислоты нейтрализуют ,раствором поташа до pH 6,0]. Взаимодействие VI с V проводят в уксуснокислой среде при pH 4,0, для чего в течение 2—3 мин к реакционной смеси приливают 2,13 л уксусной кислоты при температуре не выше .4-2 °C. Для завершения реакции массу перемешивают 30 ч при —2—(4-2°C) и 24 ч при 0°С—(4-4°C). Осадок VIII отфильтровывают, промывают водой (2 раза по 250 мл) и высушивают при 20—25 °C в вакууме. Выход 288 г (86%, считая на IV).
1 -Оксо-6-метокси-1,2,3,4-тетрагидро-р-карболин (IX).
К 1,24 л 50% этилового спирта прибавляют кислотный катализатор и 295 г 3-п-метоксифенилгидразона пипери-диндиона-2,3 (VIII) и кипятят 3 ч, охлаждают до 0— (4-3°C), перемешивают при этой температуре еще 2 ч (снижение температуры ниже 0°С приводит к загрязнению выкристаллизовывающегося продукта смолистыми примесями). Осадок IX отфильтровывают, отмывают водой до нейтральной реакции (расходуется ~ 1,68 л воды) и высушивают в вакууме при 60—70 °C. Выход 192,5 г (70%).
5-Метокситриптамин-2-карбоновая кислота (X). К раствору 119 г едкого кали в 950 мл 60% этилового спирта
164
прибавляют 61,5 г IX и массу кипятят 8 ч, после чего отгоняют часть этилового спирта, повышая в течение 3 ч температуру в парах до 95°С. К остатку приливают 100 мл воды, прибавляют 12 г угля и перемешивают 15 мин при 30—35°C; уголь отфильтровывают и к фильтрату, охлажденному до 8—10 °C, медленно приливают 96% уксусную кислоту до pH 6 с такой скоростью, чтобы температура реакционной массы не превышала 15 °C. Затем массу охлаждают до 0°С и дают выдержку 5 ч. Выделившийся осадок X отфильтровывают, промывают 50 мл воды и в виде пасты, содержащей 32% основного вещества, 14% ацетата калия и около 54% воды, используют на следующей стадии.
5-Метокситриптамина гидрохлорид (мексамин) (XI). Пасту X, полученную на предыдущей стадии, смешивают С 442 мл воды и 102 мл 28% соляной кислоты. Медленно нагревают до кипения (имеет место вспенивание за счет выделения углекислого газа). Когда масса закипает, ток углекислого газа сильно замедляется и для завершения процесса и защиты от окисления кислородом воздуха в реакционную массу через барбатер начинают пропускать азот. Кипячение в токе азота продолжают 4 ч, затем массу охлаждают до 40—50 °C, обесцвечивают углем и обрабатывают при 25—35 °C 150 мл 42% раствора едкого натра, охлаждают до 0—(+5°C) и дают выдержку 2 ч. Выделившийся осадок основания 5-метокситриптамина отфильтровывают, промывают 30 мл холодной (от 10 до 12 °C) воды, высушивают и растворяют в 1,8 л хлористого метилена. Раствор высушивают прокаленным поташом, разбавляют 40 мл абсолютного этилового спирта и очищают пропусканием через «угольную подушку», приготовленную из 80 г нейтрального активированного угля, промытого хлористым метиленом и этиловым спиртом. К фильтрату прибавляют 30% раствор хлороводорода в этиловом спирте до pH 4—5. Осадок XI отфильтровывают, промывают смесью хлористого метилена с этиловым спиртом (45:1), высушивают при 20—22 °C и остаточном давлении 300— •400 мм и перекристаллизовывают из абсолютного этилового спирта в соотношении 1:20 с добавкой 2,5% нейтрального активированного угля. Процесс ведут в атмосфере азота. Выход XI (с учетом выделенного из маточных растворов) 22,1 г (34,4% на IX).
165
СЕРОТОНИНА АДИПИНАТ
5-Окситриптамина адипинат:
CH2CHjNH2- НООС(СН2)4СООН
С 10 Н ,5 N2O-CeHt04>4
Мол. масса 322,3
Белый или белый с кремоватым оттенком кристаллический порошок, без запаха, растворим в воде, трудно растворим в спирте, 'йерастворим в эфире; т. пл. 175— 179°С (в интервале 2°С); МРТУ 42 № 3851-70.
Серотонин — биогенный амин [65, 320], обладающий разнообразными физиологическими свойствами. Характерной его особенностью является способность вызывать сокращение гладкой мускулатуры и сужение кровеносных сосудов, укорачивать время кровотечения, повышать количество тромбоцитов в периферической крови и их агрегацию, увеличивать устойчивость капилляров [96]. За рубежом серотонин используется в виде комплекса с креатининсульфатом, в СССР — в виде более доступного, устойчивого и дешевого адипината, который по фармакологическим свойствам не отличается от серотонина креатининсульфата [147].
Применяют серотонина адипинат для лечения геморрагического синдрома при различных патологических состояниях, в том числе при болезни Верльгофа, гипо- и апластической анемии, тромбастении, геморрагическом васкулите, после лечения злокачественных новообразований цитостатическими средствами.
Выпускается в ампулах по 1 мл 1 % раствора N. 10. Цена 2 р. 32 к.
Метод синтеза серотонина адипината разработан во ВНИХФИ на основе схемы, предложенной Абрамовичем и Шапиро [276]: ,
166
Схема аналогична получению мексамина (см. с. 161) и предусматривает использование общего полупродукта — калиевой соли З-карбоксипиперидона-2 (V), метод приготовления которого из малонового эфира и акрилонитрила рассмотрен при изложении способа синтеза мексамина.
Пара-нитрофенол (I) при взаимодействии с бензил-хлоридом в спиртово-щелочной среде превращают в п-бен-зилоксинитробензол (II), который восстанавливают в присутствии никелевого катализатора до п-бензилоксианили-на (III), и последний диазотируют нитритом натрия в солянокислой среде. Полученный п-бензилоксифенилдиазо-ний хлорид (IV) без выделения подвергают взаимодействию с калиевой солью З-карбоксипиперидона-2 (V). Образовавшийся п-бензилоксифенилгидразон пиперидиндио-на-2,3 (VI) циклизуют по Фишеру в присутствии серной кислоты. Продукт циклизации—1-оксо-6-бензилокси-1,2,3,4-тетрагидро-р-карболин (VII) — омыляют едким кали с раскрытием лактамного кольца и при дальнейшей обработке уксусной кислотой выделяют 5-бензилокси-триптамин-2-карбоновую кислоту (VIII). Во избежание повторного замыкания лактамного цикла осуществляют фталильную защиту первичной аминогруппы и N-фталил-5-беизилокситриптамин-2-карбоновую кислоту подвергают термическому декарбоксилированию до М-фталил-5-бен-зилокситриптамина (IX). В соединении IX снятие N-фта-лильной защиты проводят гидразингидратным методом, О-бензильной защиты — гидрогенолизом в присутствии палладиевого катализатора. Полученный серотонин выделяют в виде адипината XI.
Метод является преимущественным по сравнению с другими описанными в литературе способами синтеза серотонина: из м-нитробензальдегида через м-оксибензаль-
167
дегид, 2-нитро-5-оксибензальдегид, 2-нитро-5-бензилокси-₽-нитростирол и 5-бензилоксииндол с последующей конденсацией с оксалилхлоридом, обработкой дибензиламином, восстановлением(длюмогидридом лития и каталитическим снятием О-бензйльной защиты (11 стадий с общим выходом 11%); из о-хлорбензойной кислоты через 2-хлор-5-нит-робензойную кислоту, 2-хлор-5-аминобензойную кислоту, 2-хлор-5-бензилоксибензойную кислоту, 2-М-карбоксиме-тил-М-ацетиламино-5-бензилоксибензойную кислоту, 1-аце-тил-З-ацетокси-5-бензилоксииндол, 1-ацетил-5-бензилокси-индоксил с последующей конденсацией с циануксусной кислотой, .восстановлением нитрильной группы алюмогид-ридом лития и каталитическим снятием О-бензильной защиты (11 стадий с общим выходом 11%); по реакции Фишера из п-бензилоксифенилгидразина и труднодоступного ацеталя у-аминомасляного альдегида, из диэтилового эфира янтарной кислоты и этилформиата через 0-формил-пропионовую кислоту и ее п-бензилоксифенилгидразон с применением реакции Фишера, превращением 3-карбэто-ксиметил-5-бензилоксиндола через гидразид в амид и восстановление последнего алюмогидридом лития (8 стадий с общим выходом 10,5%); из п-бензилоксианилина через п-бензилоксифенилдиазоний хлорид и продукт взаимодействия последнего с этиловым эфиром а-ацетил-6-фталими-довалериановой кислоты с дальнейшей циклизацией по Фишеру и снятием N-фталильной и О-бензильной защитных групп (девять стадий с общим выходом ~ 11 %).
п-Бензилоксинитробензол (II). Смесь 252 г п-нитрофе-нола (I), 1,92 ^изопропилового спирта, 134 мл 42% раствора едкого натра и 250 мл хлористого бензила кипятят 6 ч, охлаждают до 15 °C и оставляют при этой температуре на 2 ч. Осадок II отфильтровывают, промывают изопропиловым спиртом (2 раза по 120 мл) и водой (3 раза по 500 мл). Получают 387,7 г (93%) II с т. пл. 106—107°C (содержание основного вещества 99%).
п-Бензилоксианилина гидрохлорид (III). Раствор 840 г II в 4,2 л этилового спирта гидрируют в присутствии никелевого катализатора при 20—25 °C и давлении водорода 20 атм. Катализатор отфильтровывают, промывают 500 мл этилового спирта. К объединенным спиртовым растворам при температуре не выше 16 °C приливают постепенно при перемешивании ~400 мл конц. соляной кислоты до pH 3,5. Выделяются кристаллы III; для полноты кристаллизации дают выдержку l’/г ч при +5 °C, периодически проверяя pH. Осадок отфильтровывают. Спирто
168
вой маточный раствор упаривают до V13 первоначального объема, остаток охлаждают до 10—15°С и дополнительно выделившееся количество Ш отфильтровывают. Общий выход 786 г (91%); т. пл. 214—216°C, содержание основного вещества не ниже 99%.
З-п-Бензилоксифенилгидразон пиперидиндиона-2,3 (VI). В смеси 900 мл воды и 195 мл соляной кислоты растворяют 141,4 г III. Раствор охлаждают до 10—15°С и к нему под поверхность постепенно приливают раствор 46,3 г нитрита натрия в ПО мл воды, поддерживая температуру реакционной Массы 10—15 °C за счет охлаждения извне. Дают дополнительно выдержку 30 мин и контролируют конец реакции йодокрахмальной бумажкой (по наличию свободной азотистой кислоты). Азотистую кислоту разлагают прибавлением 5 г мочевины. К диазораствору приливают раствор 140 г ацетата натрия в 140 мл воды до pH 4,5—5. Массу охлаждают до 0°С, фильтруют и при перемешивании прибавляют к охлажденному до 0°С и подкисленному уксусной кислотой до pH 4,5—5 раствору V, приготовленному, как описано в синтезе мексамина (см. с. 164), из 105,8 г З-карбэтоксипиперидона-2. Внешнее охлаждение снимают и массу перемешивают 6 ч (температура постепенно повышается до комнатной), после чего перемешивание продолжают еще 24 ч при 20—25°С. Осадок VI отфильтровывают, промывают водой (5 раз по 100 мл), спиртом (2 раза по 50 мл) и высушивают в вакууме при 40°C. Выход 177,8 г (86,9%).
1-Оксо-6-бензилокси-1,2,3,4-тетрагидро-0-карболин (VII). Смесь 540 мл этилового спирта, 27,6 мл серной кислоты и 177,8 г гидразона VI кипятят при перемешивании 2 ч. При нагревании взвесь гидразона VI растворяется и примерно через 1 ч появляется осадок VII. Массу охлаждают до 8—10 °C в течение 2 ч. Осадок VII отфильтровывают, промывают 60 мл этилового спирта и 3 раза по 100 мл роды (до отсутствия кислой реакции на конго). Получают 190 г сырого VII (127 г в пересчете на сухой, выход 80,7%), который без высушивания направляют на следующую стадию.
5-Бензилокситриптамин-2-карбоновая кислота (VIII). К раствору 216 г едкого кали в 1,3 л воды прибавляют 100 г карболина VII (в пересчете на сухой) и 1,3 л этилового спи^а. Кипятят при перемешивании 6 ч, подкисляют уксусной кислотой ('-—-216 мл) до pH 6, охлаждают до 20—30 °C, дают выдержку 1 ч. Осадок отфильтровывают, промывают 200 мл 50% этилового спирта и 5 раз по200мл
169
горячей воды. Сушат при 100°C. Выход 122 г (86,5%); т. пл. 212—214 °C, содержание основного вещества не менее 86%.
5-Бензилокситриптамин (X). Смешивают 100 г мелко растертой кислоты VIII с 50,2 г фталевого ангидрида и нагревают без перемешивания до 180 °C. При этом происходит образование М-фталил-5-бензилокситриптамин-2-карбоновой кислоты и отгонка воды. Далее температуру постепенно поднимают в течение 1 ч до 240 °C. Начинается процесс декарбоксилирования и масса разжижается. Пускают мешалку и перемешивание при 235—240 °C продолжают 2 ч (конец декарбоксилирования контролируют по прекращении выделения углекислого газа). Затем массу охлаждают до 100 °C и постепенно приливают 700 мл этилового спирта и 45 мл гидразингидрата. Кипятят при перемешивании 3 ч, охлаждают до 20 °C, осадок отфильтровывают, промывают спиртом (3 раза по 50 мл). Объединенные спиртовые растворы охлаждают до 5 °C и подкисляют по конго соляцой кислотой (~65 мл), перемешивание при 0+5 °C продолжают 3 ч. Выделившийся осадок 5-бензилокситриптамина гидрохлорида отфильтровывают, промывают 2 раза по 25 мл этилового спирта и растворяют в 1,2 л кипящей воды с добавкой 8 г угля. К отфильтрованному водному раствору при 70—80 °C и перемешивании постепенно приливают 10% раствор едкого натра до устойчивой сильнощелочной реакции по фенолфталеину. Массу охлаждают до 10 °C, выделившийся осадок основания X отфильтровывают, промывают водой (3 раза по 20 мл) до отсутствия щелочной реакции и сушат при 50— 60 °C, после чего перекристаллизовывают из 200 мл бензола с добавкой 2 г активированного угля. Выпавшие при охлаждении до 10 °C кристаллы X отфильтровывают, промывают 2 раза по 20 мл холодного бензола. Бензольные фильтраты упаривают до 'А первоначального объема, охлаждают до 10 °C и отфильтровывают дополнительное количество X. Общий выход 27,5 г (37,7%); т. пл. 90— 92°C; содержание основного вещества не ниже 98%.
Серотонина адипинат (XI). 150 г X, 3 л этилового спирта и 30 г палладиевого катализатора, содержащего 5% окиси палладия на карбонате калия, гидрируют при 18—25 °C и давлении водорода 3 атм. Катализатор отфильтровывают, промывают 150 мл этилового сйирта и к спиртовому фильтрату приливают горячий раствор 82,5 г адипиновой кислоты в 450 мл этилового спирта. (Фильтрацию растворов, содержащих серотонин, и получение се-170
ротонина адипината необходимо проводить быстро, так как серотонин на воздухе темнеет.) При охлаждении выделяется осадок серотонина адипината (выделение кристаллов ускоряется затравкой). Массу охлаждают 1чпри О °C, продукт отфильтровывают, промывают 150 мл этилового спирта. Спиртовые маточные растворы упаривают в вакууме до ’/в первоначального объема и выделяют дополнительное количество XI. Весь полученный продукт перекристаллизовывают из 85% этилового спирта с добавлением активированного угля. Спиртовой раствор охлаждают до 0 °C, XI отфильтровывают, промывают холодным (0°С) этиловым спиртом и сушат при 50—60 °C. Получают 143 г (82,4%) XI.
АТРОПИНА СУЛЬФАТ
Тропинового эфира d.l-троповой кислоты сульфат, моногидрат:
рсл /с6н5 •
(МСНзу—ОСОСН . н SO4*
L—I- / ^CHjOH
L -1 2
Мол. масса 694,8
Белый кристаллический или зернистый порошок, без запаха, легко растворим в воде и спирте, практически нерастворим в хлороформе и эфире.
Растворы имеют нейтральную реакцию; для стабилизации инъекционных растворов добавляют соляную кислоту до pH 3,0—4,5. Безводный атропина сульфат очень гигроскопичен и на воздухе превращается в кристаллогидрат; т. пл. 187—191 °C (в пределах 2°C). Оптически неактивен. ГФХ, ст. 76.
При выделении из растений содержащегося в них левовращающего изомера гиосциамина последний легко ра-цемизуется в атропин. Фармакологически гиосциамин в 2 раза активнее атропина.
Основной фармакологической особенностью атропина сульфата является его способность блокировать м-холино-реактивные системы организма: на н-холинорецепторы препарат действует слабо [114].
Применяется при язвенной болезни желудка и двенадцатиперстной кишки, пилороспазме, холецистите, желчно
171
каменной болезни, спазмах кишечника и мочевых путей, бронхиальной астме, для ограничения секреции слюнных, желудочных и бронхиальных желез, при брадикардии, вызванной повышением возбудимости блуждающего нерва, при атриовентрикулярной блокаде и инфаркте миокарда. Препарат широко используют в глазной практике для расширения зрачка с диагностической целью и при терапии воспалительных заболеваний. Противопоказан при глаукоме.
Выпускается порошок (цена 1 г 40 к.), ампулы по 1 мл 0,1% раствора (цена 1 ампулы 2,5 к.), 1% раствор (глазные капли во флаконах по 5 мл по цене 4 к. и в комплекте с пипеткой, вложенной в картонную коробку, по цене 7 к.). Список А. Высшие дозы для взрослых внутрь и под кожу: разовая 0,001 г, суточная 0,003 г.
Синтез атропина сульфата, включающий получение синтетических тропина и d.l-троповой кислоты, был разработан во ВНИХФИ по схеме:
снэУна9О(СН2СООН)2(У|)
CH COONa
l)HCl
2) NH4QH
3) h2so4
H2/Ni
ОН-НС1
ш
IV
(CHaCOhO fH2OCOCH
—----* |сйм снсоон
ОСОСН; chcoci
1>(СН2О)«
Свн5сн2соос,нбЗП=5Г v 3)НС|
Из двух известных методов синтеза атропина сульфата (I)—из тропина (II) и d.l-троповой (III) или ацетил-троповой (IV) кислоты [293] и восстановлением тропинового эфира а-формил-а-фенилуксусной кислоты для промышленного производства более приемлем первый, поскольку второй метод дает продукт худшего качества, трудно поддающийся очистке. В основу получения d.l-тро-повой кислоты (III) был положен метод Швенкера и соавт. [331], использующий конденсацию фенилуксусного эфира (V) с параформом в апротонной среде в присутст-ствии щелочного катализатора. Другие известные методы
172
синтеза d.l-троповой кислоты — из атроповой кислоты, восстановлением формилфенилуксусной кислоты и ее низших эфиров, а также путем взаимодействия формальдегида с реактивом Иванова, приготовленным из фенилуксусной кислоты или натриевой соли фенилуксусной кислоты и изопропилмагнийхлорида,— дают худшие результаты.
В основе получения синтетического тропина (II) лежит реакция Робинсона — Шэпфа, заключающаяся в конденсации ацетондикарбоновой кислоты (VI), метиламина и янтарного альдегида (VII) в тропинон (VIII) с его последующим восстановлением [14, 325, 328]. Другие методы получения тропина (II)—из циклогептанона, а также из лимонной кислоты через эфиры N-метилпирролидинук-сусной кислоты — промышленного значения не имеют. Янтарный альдегид (VII) синтезируют в ходе производства из фурана (IX) [327], который обработкой бромом в метаноле переводят в 2,5-диметокси-2,5-дигидрофуран (X) с восстановлением X до 2,5-диметокситетрагидрофурана (Xi) и гидролизом последнего до VII.
2,5-Диметокси-2,5-дигидрофуран (X). К охлажденной до —8 °C—(—10 °C) суспензии 205 г кальцинированной соды в 1410 мл метанола прибавляют 66 мл фурана (IX) и, поддерживая температуру не выше —5°C, прикапывают при перемешивании 47 мл брома. Осадок неорганических солей отфильтровывают. От фильтрата медленно отгоняют метанол при температуре в парах 66—69 °C, прибавляют 210 мл воды и перемешивают при 55—60 °C до полного растворения остатков неорганических солей. Раствор охлаждают до 20 °C и экстрагируют хлористым метиленом (3 раза по 150 мл). Хлористометиленовый экстракт сушат сульфатом натрия, упаривают при атмосферном давлении (к концу упарки температура в массе 100°C). Остаток подвергают фракционной перегонке в вакууме. Собирают фракцию с т. кип. 70—71 °C (30 мм рт. ст.). Выход X 107 г (90%). I фракцию, содержащую смесь X и хлористого метилена, а также метанольный отгон, содержащий ~0,3% IX и ~2% X, используют при последующих операциях.
2,5-Диметокситетрагидрофуран (XI). Смесь 107 г X и 6,4 г скелетного никелевого катализатора гидрируют в автоклаве при 35 °C и давлении 35—10 атм до полного прекращения поглощения водорода. Катализатор отфильтровывают. Получают 106 г (93,5%) XI, который без' дополнительной очистки направляют на следующую стадию.
173
Тропинон (VIII). Раствор 104 мл XI в 1 л воды и 8,5 мл 36% соляной кислоты кипятят 30 мин, охлаждают до 18— 20 °C и анализируют на содержание янтарного альдегида (VII) (58,5 г/л). К массе прибавляют 2 н. раствор едкого натра до pH 5—6, 237 г кристаллического ацетата натрия, 117 г 97% ацетондикарбоновой кислоты (VI) и 53,5 г 98,5% метиламина гидрохлорида. После смешивания всех реагентов раствор обычно имеет pH 4—4,5. Массу перемешивают 30 мин при 42—43 °C (наблюдается выделение углекислого газа и вспенивание), охлаждают до 30°C, прибавляют 251 г технического поташа, охлаждают до 18—20 °C и экстрагируют хлористым метиленом (900 мл, 600 мл и 200 мл). Хлористометнленовый раствор сушат сульфатом натрия до содержания влаги 0,3% и упаривают при атмосферном давлении (остатки растворителя удаляют в вакууме водоструйного насоса при 80—100°C). Для более полного удаления летучих примесей остаток растворяют в 30 мл сухого бензола с последующей отгонкой в вакууме. Тропинон перегоняют при 83—85°С (5 мм рт. ст.). Перегнанный продукт (выход 75,5 г, или 72%, считая на XI) растворяют в 170 мл метанола и передают на восстановление до тропина (II) (при хранении продукт краснеет). А;
Тропин (П) К раствору 75,5 г VIII в 170 мл метанола от предыдущей стадии прибавляют 11,2 г промытой метанолом пасты никелевого катализатора. Гидрируют при 20—25 °C и давлении 60—30 атм до прекращения поглощения водорода. Катализатор отфильтровывают. Метанол отгоняют в вакууме при остаточном давлении 100— 150 мм рт. ст. и температуре 21—25°С (температура в массе 60—70°C, к концу отгонки — до 100°C). К остатку прибавляют 20 мл сухого бензола (для удаления следов воды) с последующей отгонкой в вакууме при 100— 150 мм рт. ст. и температуре 26—30 °C. К концу отгонки для более полного удаления бензола температуру в массе повышают до 105—НО °C. Горячий остаток тропина выливают в тару, где он закристаллизовывается. Выход 75 г (98%); т. пл. 61—63 °C. В ИК-спектре характерны полосы поглощения при 958 см-1 и отсутствует поглощение при 1020 см-1 (псевдотропин) и 1720~1 (тропинон).
Атропина сульфат (I). Смесь 138,5 г б,1-троповой кислоты (III) и 118 мл свежеперегианного хлористого ацетила нагревают 30 мин при 30°C. Реакция сопровождается бурным выделением хлороводорода. Для завершения процес-174
Г/A'
са массу нагревают 1 ч при 60°C, после чего в вакууме при остаточном давлении 360—400 мм рт. ст. и температуре в массе 57—60° отгоняют избыточный хлористый ацетил (к концу отгонки вакуум повышают при той же температуре до 100 мм рт. ст.). Оставшуюся ацетилтропо-вую кислоту (IV) охлаждают до 25—30 °C и прибавляют 95,5 мл тионилхлорида. Массу медленно нагревают до 68—70°C (выделяется хлороводород) и выдерживают при этой температуре Р/г—2 ч, после чего в вакууме при остаточном давлении 360—400 мм рт. ст. и при температуре в массе 70—75 °C отгоняют тионилхлорид, повышая в конце отгонки вакуум до 100 мм. Остатки тионилхлорида удаляют азеотропной отгонкой с бензолом (50, 50 и 20 мл). Полученный хлорангидрид ацетилтроповой кислоты охлаждают до 30—35 °C и приливают к нему 490 мл хлороформного раствора тропина гидрохлорида, содержащего 133 г основного вещества и приготовленного пропусканием в раствор основания тропина в промытом от спирта и высушенном хлороформе газообразного сухого хлороводорода до pH 3. Реакционную массу кипятят 6 ч, охлаждают до 45—50°C, прибавляют 463 мл 2 н. соляной кислоты и 7г активированного угля; смесь нагревают З’/г ч при 45— 50 °C, охлаждают до 20 °C. Уголь отфильтровывают, водный кислый слой отделяют, хлороформный слой промывают водой. От объединенных водных растворов атропина гидрохлорида отдувают остатки хлороформа, после чего при охлаждении до 0°С—(4-2°С) при перемешивании прибавляют 12% водный раствор аммиака до pH 8 с такой скоростью, чтобы температура реакционной массы не превышала 6 °C. Массу перемешивают 1—1 ’/г ч при 2 °C— (+6 °C) и оставляют при той же температуре на 10—12 ч без перемешивания. Основание атропина отфильтровывают, промывают водой, высушивают до содержания' влаги не более 0,1% и перекристаллизовывают из 680 мл ацетона с добавкой 8 г активированного угля. Полученное основание атропина белого цвета с содержанием не ниже 99%, т. пл. 115—117°C и содержанием апоатропина не более 0,2% растворяют в 500 мл этилового спирта, очищают, путем обработки с углем и при охлаждении до 2—5°C медленно подкисляют 20% серной кислотой (~146 мл) до pH 7—7,5. Затем прибавляют 5 г нейтрального активированного угля и при атмосферном давлении отгоняют этиловый спирт, уголь отфильтровывают. Атропина сульфат (I) осаждают прибавлением 1000 мл ацетона. Выход ПО г (54% на тропин и 50% на троповую кислоту).
175
ГОМАТРОПИНА ГИДРОБРОМИД
Тропинового эфира миндальной кислоты гидробромид:
С is Н 21 NO3- НВг
Мол. масса 356,26 Белый кристаллический порошок, без запаха, горького вкуса, легко растворим в воде (1:6) (растворы бесцветные, прозрачные, нейтральные), трудно — в спирте, очень мало растворим д ^лороформе, практически нерастворим в эфире; т. пл. 21'6—214 °C (в пределах 2 °C с частичным разложением); ГФХ, ст. 336.
По фармакологическим свойствам гоматропина гидробромид близок к атропина сульфату (см. 171), является холинолитическим средством с менее продолжительным действием. Применяется главным образом в глазной практике для расширения зрачка и паралича аккомодации. Противопоказан при глаукоме.
Выпускается во флаконах по 5 мл 0,25% раствора с метилцеллюлозой. Список А. Цена 3 к.
Существуют два метода синтеза гоматропина гидробромида, пригодные для промышленного производства:
а) Оригинальный метод, разработанный во ВНИХФИ [43] по схеме:
l)NaBH4
2) НВг
VX /С.н5
МСНэУ-ОСОСН . нВ.
Z____/ чон
IV
осососвн6—»-
Тропин (I) переводят в гидрохлорид (II) и последний обрабатывают хлорангидридом фенилглиоксиловой кислоты. Полученный тропиновый эфир фенилглиоксиловой кислоты (III) вб&станавливают боргидридом натрия и обработкой бромистоводородной кислотой переводят в гоматропина гидробромид (IV);
б) метод, разработанный на алкалоидном заводе и основанный на опубликованной ранее схеме этерификации тропина миндальной кислотой в присутствии минеральных кислот [255]:
176
—“А ОН
\ \ I l) HC(
NCH.-Л—OH fC.HCHCOntt -----7-*-
/ . / " ' 2|K2CO3
3) HBr
OCOCH • HBr
чон
IV
V
По этому методу через расплав тропина (I) и миндальной кислоты (V) пропускают ток сухого хлороводорода и полученный гоматропина гидрохлорид через основание переводят в гидробромид (IV).
Второй метод проще технологически, но выход конечного продукта IV на тропин по нему 48,5%, в то время как по первому методу выход 74%.
а) Калиевая соль этилового эфира щавелевой кислоты (этоксалат калия) [335]. Смесь 146,5 г диэтилового эфира щавелевой кислоты, 107,1 г ацетата калия, 300 мл абсолютного этилового спирта и 15 мл воды кипятят при перемешивании 2 ч. Калиевая соль этилового эфира щавелевой кислоты выпадает в осадок из кипящего раствора через 20—30 мин после начала реакции. Для полноты выделения продукта реакционную смесь оставляют на 18—20 ч при 20°C. Выход 123 г (79%).
Хлорангидрид этилового эфира щавелевой кислоты (этоксалилхлорид) [335]. К 114 г этоксалата калия в течение 30—40 мин при перемешивании прибавляют 55 мл тионилхлорида (температура реакционной массы повышается до 35—40°C). Затем массу нагревают 2 ч при 95— 98 °C (температура наружного обогрева) и полученный этоксалилхлорид отгоняют из реакционной смеси при остаточном давлении 80 мм и температуре в парах 73—75 °C, охлаждая приемник ледяной водой (для полноты отгонки обогрев массы в конце доводят до 160—165°C). Выход 87 г (86,7%). Этоксалилхлорид хранят не более 7 дней при температуре не выше 8 °C. Методы получения этокс-алилхлорида взаимодействием диэтилоксалата с PCU и последующим разложением образующегося дихлорэтокси-этилацетата термически в присутствии платины и обработкой этоксалата калия хлорокисью фосфора технологически менее приемлемы.
Этиловый эфир фенилглиоксиловой кислоты [303]. К охлажденной до 4—5 °C суспензии 60,6 г безводного хлористого алюминия в 200 мл бензола (содержание влаги не более 0,05%) при интенсивном перемешивании прибавляют по каплям раствор 54 г этоксалилхлорида в 100 мл бензола. Перемешивание продолжают 4*/г ч при 4—6°С, после чего при наружном охлаждении порциями
12 Заказ № 4304
177
прибавляют 300 г измельченного льда так, чтобы температура массы не превышала 25—30 °C. Бензольный слой отделяют, водный — экстрагируют бензолом 2 раза по 50 мл. Объединенные бензольные экстракты промывают 8% раствором гидрркарбоната натрия до исчезновения кислой реакции, затем водой и подвергают разгонке. Собирают фракцию с т. кип. 127—128°C (8 мм). Выход 48,9 г (68,6%). Другие методы получения производных фенилглиоксиловой кислоты, основанные на окислении соответствующих производных фенилуксусной кислоты кислородом в присутствии бензоата кобальта, перманганатом калия, тетрацетатом свинца, озоном, хромовым ангидридом, а также на окислении перманганатом калия стирола, этилбензола, ацетофенона, двуокисью селена а-бромаце-тофенона, кислородом в присутствии щелочных катализаторов производных а-фенил-а-циануксусной кислоты, не имеют преимуществ, поскольку фенилглиоксиловая кислота и ее производные, получаемые окислительными методами, всегда содержат трудно отделимые примеси бензойной кислоты. По той же причине не дают хороших результатов и методы получения фенилглиоксиловой кислоты из бензоилцианида и из п-нитрозо-М,Г4-диметиланилина.
Хлорангидрид фенилглиоксиловой кислоты [43]. К раствору 35,9 г карбоната натрия в 325 мл воды прибавляют 36 г этилового эфира фенилглиокснловой кислоты, кипятят 5 ч, охлаждают, подкисляют 60 мл конц. серной кислоты и извлекают бензолом 4 раза по 50 мл. Бензольный экстракт упаривающ ее объема 80 мл, прибавляют 1,6 г сухого диметилформамида, нагревают до 60—65 °C, по каплям прибавляют 15 мл тионилхлорида, выдерживают 30 мин при 60—65°C, упаривают при остаточном давлении 70 мм рт. ст. и температуре 50—60°C (наружный обогрев). Получают 28,7 г (85,2%) технического хлорангидрида фенилглиоксиловой кислоты, который без дополнительной очистки сразу же используют в дальнейшем синтезе. Хранить продукт не рекомендуется из-за его способности легко гидролизоваться.
Гоматропина гидробромид (IV). В охлажденный до 7—10°С раствор 18,59 г тропина (I) в 72 мл хлороформа пропускают ток сухого хлороводорода с такой скоростью, чтобы он весь поглощался тропином, до pH раствора ~4, затем вносят 34,16 г технического хлорангидрида фенилглиоксиловой кислоты в 40 мл хлороформа и смесь кипятят 5 ч. К охлажденному раствору при перемешивании и температуре не выше 3°С постепенно добавляют 178
66 мл 8% водного раствора аммиака до pH 8,0. Слои разделяют и водный слой дополнительно экстрагируют 40 мл хлороформа. К объединенным хлороформным растворам основания тропинового эфира фенилглиоксиловой кислоты (III) добавляют 35 мл этилового спирта и при интенсивном перемешивании и температуре — 5 °C—(—10 °C) вносят тремя порциями с интервалом в 40 мин 2,49 г бор-гидрида натрия. Затем приливают 200 мл воды, перемешивают 2 ч. Хлороформный слой отделяют, водный — промывают 40 мл хлороформа. Объединенный хлороформный раствор сушат сульфатом натрия и упаривают. Остаток растворяют в 200 мл ацетона, раствор осветляют активированным углем, охлаждают до 5—10 °C и постепенно при температуре не выше 20°С добавляют 40% бромистоводородную кислоту до pH 3—4 (~19 мл). Выпавший продукт перекристаллизовывают с обесцвечиванием раствора углем из 80% этилового спирта и промывают ацетоном. Выход 34,89 г (74,38%, считая на I).
б) Гоматропина гидробромид (IV). Смесь 200 г тропина (I), 305 г миндальной кислоты (V) и 40 мл воды расплавляют при 110°C и пропускают через расплав при НО—120 °C ток сухого хлороводорода 8—9 ч с такой скоростью, чтобы ои полностью поглощался до pH 5—6. Плав охлаждают до 80 °C, растворяют в 300 мл воды, подщелачивают 50% раствором поташа и экстрагируют толуолом (4 раза по 1,6 л). После отгонки толуола получают 290 г основания гоматропина. Из водных маточных растворов экстракцией хлороформом (3 раза по 1,5 л) выделяют 5,5 г тропина. К 290 г основания гоматропина в 290 г абсолютного этилового спирта прибавляют бромистоводородную кислоту плотностью 1,64—1,65 до кислой реакции по конго. Массу выдерживают 10—12 ч при 10 °C. Продукт отфильтровывают и перекристаллизовывают из этилового спирта с углем. Выход 220 г (48,5%, считая на I).
ТРОПАФЕН
Тропинового эфира а-фенил-р-(п-ацетоксифенил)-пропионовой кислоты гидрохлорид:
Мол. масса 443,98
12*
179
Белый или белый со слегка серовато-кремовым оттенком кристаллический порошок, без запаха, легко растворим в воде (pH раствора 5,0—6,5), 95% спирте и хлороформе, трудно растворим в ацетоне, нерастворим в бензоле, толуоле, эфире, этилацетате; т. пл. 190—198°С (в пределах 4 °C); ВФС 42-972-80.
Тропафен — оригинальный а-адреноблокатор [136, 184, 251], уменьшает или полностью снимает а-адренергиче-ские эффекты адреналина, норадреналина и адреномиметических веществ. Сильно расширяет периферические сосуды и вызывает понижение артериального давления, обладает антисеротониновой активностью и слабыми холинолитическими свойствами.
Применяют тропафен при лечении заболеваний, связанных с нарушением периферического кровообращения (эндартериит, болезнь Рейно, акроцианоз, трофические язвы конечностей), вяло заживающих ран, для устранения симпатомиметических' реакций при оперативных вмешательствах, при диагностике и лечении феохромоцитомы. Выпускается лиофилизированный для инъекций по 0,02 г в ампулах N. 10. Цена 2 р. 60 к.
Метод синтеза тропафена разработан совместно ЛПХФО «Фармакон» и ВНИХФИ по схеме [184]:
Взаимодействием а-фенил-0- (п-оксифенил) -пропионовой кислоты (I) с уксусным ангидридом получают а-фе-нил-р-(п-ацетоксифенил)-пропионовую кислоту (II), которую переводят в хлорангидрид (III), используемый без выделения для ацилирования тропина (IV). Указанная схема технологически более целесообразна, чем второй описанный [184] вариант получения тропафена (V) путем перевода кислоты I в ее метиловый эфир, переэтерификации этого эфира с тропином в присутствии алкоголя-та натрия или металлического натрия и ацетилирования п-оксифенильной группы на последней стадии синтеза.
а-Фенил-р-(п-ацетоксифенил)-пропионовая кислота (II). Смесь 362 г уксусной кислоты, 212,6 г уксусного ангидри-180
да и 290 г а-фенил-ф-(п-оксифенил) -пропионовой кислоты нагревают 3 ч при 123—125°C (в массе). При содержании исходного соединения I не более 0,2% реакцию прекращают, дают охладиться до 50—55 °C и выливают порциями на смесь 900 г льда и 300 г воды, после чего для разложения избытка уксусного ангидрида нагревают до 20 °C и оставляют при этой температуре на 12 ч при перемешивании. Выделившуюся а-фенил-'р-(п-ацетоксифенил)-пропионовую кислоту отфильтровывают, промывают 960 мл воды, сушат сначала при 30—40 °C (12 ч), затем при 80— 85°C (12 ч) и перекристаллизовывают из 1560 мл сухого бензола. Получают 281 г (82,2%) II, т. пл. 141—145 °C, процентное содержание 98,5—100,5%.
Тропафен (V). К раствору 390 г тионилхлорида в 1000 мл сухого бензола прибавляют 462 г II и смесь постепенно нагревают сначала 30 мин от 20 °C до 60 °C, затем 2 ч — от 60 °C до 65 °C, 2 ч — от 65 °C до 75 °C и 1 ч от 75 °C до 82°С, после чего дают выдержку 2 ч при 82°C (кипение бензола). В отобранной пробе проверяют т. пл. хлорангидрида а-фенил-р-(п-ацетоксифенил)-пропионовой кислоты. При положительном результате анализа (т. пл. 90—94°C) реакционную массу упаривают в вакууме, остатки тионилхлорида отгоняют в виде азеотропа с бензолом. Продукт растворяют в 2 л толуола, обесцвечивают углем и прибавляют к нему при 55—60 °C единовременно 580 г 27,5% толуольного раствора тропина. Реакционную массу кипятят 3 ч, анализируют на содержание тропина (не более 0,1%), охлаждают до 50—55°C. Выделившийся осадок тропафена отфильтровывают, промывают нагретым до 50—55 °C сухим толуолом и перекристаллизовывают из абсолютного изопропилового спирта с углем. Выход тропафена (V) 310 г (58,87% на I, 28,95% на а-фенил-₽-(п-оксифенил)-пропионовую кислоту).
ТЕМЕХИН
2,2,6,6-Тетраметилхинуклидина гидробромид
CBH2I N-HBr
Мол. масса 248,21
181
Белый или белый с легким кремоватым оттенком кристаллический порошок, без запаха, горьковатого вкуса, очень хорошо растворим в воде (растворы прозрачные и бесцветные, pH 6,0—7,0, не меняется при кипячении), умеренно растворим в холодном этиловом спирте (1:10); хорошо— в горячем этиловом спирте (1:2) и изопропиловом спирте (1:6), нерастворим в эфире, бензоле, ксилоле; т. пл. 269—273°С (в интервале 2°С); ВФС 42-1464-80.
Темехин — оригинальный ганглиоблокирующий препарат. Оказывает гипотензивное действие [175, 249]. Более активен, чем его пиперидиновый аналог пирилен. Препарат блокируют н-холинореактивные системы вегетативных ганглиев, а также хромаффиной ткани надпочечников, каротидных клубочков и ЦНС. В результате тормозится передача нервного возбуждения с пре- на постганглионарные волокна, уменьшается поступление нервных импульсов к органам, расширяются периферические сосуды, понижается артериальное давление, ослабляются рефлекторные прессорные реакции, угнетается секреция желез, расширяются бронхи. Оказывает седативный эффект.
Применяют темехин при гипертонической болезни, гипертонической энцефалопатии и гипертонических кризах, спазмах периферических сосудов (облитерирующий эндар-~°риит и др.), язвенной болезни желудка и двенадцатиперстной кишки, бронхиальной астме. Выпускается в таблетках по 0,001 г N. 50. Цена 82 к.
Метод синтеза темехина разработан во ВНИХФИ [175]
182
Триацетонамин (2,2,6,6-тетраметилп11перпдон-4) (ij, получаемый путем конденсации трех молекул ацетона с аммиаком, подвергают взаимодействию с этиловым эфиром циануксусной кислоты. Продукт конденсации (II) восстанавливают каталитически и соединение III без выделения гидролизуют с частичным декарбоксилированием и последующей этерификацией монокарбоновой кислоты. Основание эфира IV восстанавливают алюмогидридом лития и полученный 2,2,6,6-тетраметил-4- (р-оксиэтил) пиперидин (V) переводят кипячением с бромистоводородной кислотой в 2,2,6,6-тетраметил-4-(р-бромэтил) пиперидина гидробромид (VI). При внутримолекулярной циклизации выделенного из гидробромида VI основания получают темехин (VII).
В указанной схеме ранее был описан только синтез триацетонамина (I) [23]. Конденсация I с этиловым эфиром циануксусной кислоты наиболее полно (с выходом 97%) протекает в условиях реакции Кневенагеля под действием ацетата аммония в кипящем бензоле с азеотропной отгонкой образующей воды в течение I ч. Более длительное нагревание реакционной смеси или использование вы-сококипящих растворителей (толуол, ксилол) приводят к дальнейшим процессам N-ацетилирования II и перегруппировки образующегося N-ацетильного производного в З-циан-4- (р,р-диметилвинил) -6,6-диметил-Д3-дегидропипери-дон-2. Экзоциклическое положение двойной связи в соединении II позволило легко и избирательно восстановить ее в присутствии палладия на носителе в нейтральной среде. Гидрирование с никелевым катализатором, а также в кислой среде с платиной или с палладием сопровождается восстановлением нитрильной группы. Гидролиз в III нит-чнльной и сложноэфирной групп, а также частичное декарбоксилирование образующейся замещенной малоновой кислоты удается осуществить в одну операцию при кипячении 10 ч с 20% соляной кислотой. Дальнейшая этерификация лучше проходит в присутствии серной кислоты, чем со спиртовым раствором хлороводорода. Высокие выходы II, III и IV позволили провести все указанные процессы без выделения и очистки продуктов с общим выходом эфира IV 61,3%, считая на триацетонамин (I). При восстановлении IV алюмогидридом лития в связи с бурным течением процесса в эфире, особенно на операции разложения литиевоалюминиевого комплекса водой, возникла необходимость осуществления реакции в бензоле с добавкой эфира.
183
Другой путь синтеза оксипроизводного V, основанный на конденсации триацетонамина (I) с бромуксусным эфиром в условиях реакции Реформатского с последующей дегидратацией, восстановлением алюмогидридом лития и каталитическим гидрированием дал худшие результаты [173, 174]. Замещение оксигруппы в V на бром было осуществлено двумя путями: кипячением с 40% бромистоводородной кислотой при одновременной отгонке получающейся в ходе реакции более разбавленной бромистоводородной кислоты и обработкой оксипроизводного V трехбромистым фосфором в кипящем бензоле. Первый метод оказался предпочтительным [175]. Основание бромпро-изводного VI легко циклизуется при кипячении в ксилоле с образованием VII. Общий выход темехина (VII) на триацетонамин (I) 34,4%.
2,2,6,6-Тетраметилпиперидон-4 (триацетонамин) (I). В смесь 420 мл ацетона и 107,5 г прокаленного измельченного хлористого кальция пропускают в течение 30 мин газообразный аммиак, поддерживая температуру в массе 23—27 °C. Затем с интервалами 2'/г—3 ч проводят еще 12—15 15-минутных пропусканий аммиака. Процесс насыщения заканчивают, когда плотность ацетонового раствора достигает 0,87—0,91 при 20 °C. Затем реакционную массу нагревают 7 ч прй\ кипении с перемешиванием. После охлаждения и отстаивания ацетоновый слой отделяют, промывают 100 мл 40% раствора поташа. Прозрачный раствор сушат прокаленным поташом и подвергают разгонке в вакууме с дефлегматором. Получают 100 г триацетонамина (I) с т. кип. 75—85°C (5 мм рт. ст.) и содержанием 85%. Выход 27,8%, считая на ацетон. При комнатной температуре триацетонамин неустойчив, темнеет и осмоляет-ся, поэтому желательно по мере получения его использовать или хранить при температуре не выше 5 °C.
2Д6,6-Тетраметил-4-карбометоксиметилпиперидин (IV). Смесь 100 г I, 77,5 г циануксусного эфира и 20 г ацетата аммония кипятят в 200 мл бензола 1 ч с азеотропной отгонкой воды, охлаждают, обрабатывают 150 мл 40% раствора поташа, водный слой отделяют, экстрагируют бензолом. Объединенные бензольные растворы упаривают в вакууме. Остаток (160 г) растворяют в 480 мл абсолютного изопропилового спирта и гидрируют в присутствии 20 г палладия на карбонате кальция с содержанием окиси палладия около 5%. Гидрирование проводят в автоклаве с перемешиванием при давлении водорода 15 атм и комнатной температуре. Затем катализатор отфильтровыва-184
ют, фильтрат упаривают в вакууме, остаток (157 г) растворяют в 1180 мл 23% соляной кислоты, полученный раствор кипятят 10 ч и упаривают в вакууме. Остатки воды удаляют азеотропной отгонкой с бензолом. К полученному продукту прибавляют 200 мл безводного метанола (с содержанием влаги не более 0,25%), охлаждают до 10°C и постепенно при перемешивании прикапывают 76 мл конц. серной кислоты, поддерживая температуру 10—15 °C. Затем реакционную массу кипятят 8 ч, охлаждают и выливают в смесь 220 мл дихлорэтана и 220 г льда, подщелачивают 40% раствором поташа. Продукт экстрагируют дихлорэтаном. Дихлорэтановый раствор сушат поташом, упаривают, остаток перегоняют в вакууме. Собирают фракцию с т. кип. ПО—115°С (10—12 мм рт. ст.). Выход IV 72,9 г (содержание 98,1 %), т. е. 61,3%, считая на I. Без дополнительной очистки вещество используют на следующей стадии.
2,2,6,6-Тетраметил-4-(р-оксиэтил)пиперидин (V). 72,9 г соединения IV в 530 мл бензола прибавляют к 13,2 г алюмогидрида лития в 270 мл эфира с такой скоростью, чтобы температура в реакционной массе была не выше 38 °C. Кипятят 3 ч, охлаждают до 10 °C и прибавляют 27 мл воды (температура в массе не выше 20°C). Осадок неорганических оснований отфильтровывают, промывают бензолом, фильтраты упаривают в вакууме. Получают 58,9 г (содержание 98,7%) оксипроизводного V. Выход 93,6%; т. пл. 59—62 °C (аналитически чистый образец имеет т. пл. 67—68°C). Без дополнительной очистки вещество используют на следующей стадии.
2,2,6,6-Тетраметил-4-(р-бромэтил)пиперидина гидробро-мид (VI). Растворяют 58,9 г оксипроизводного V в 220 мл 40% бромистоводородной кислоты, нагревают до НО °C и, постепенно повышая температуру в парах до 121 °C (в реакционной массе 128—129°С), отгоняют разбавленную бромистоводородную кислоту. К остатку, охлажденному до 70—75°C, при интенсивном перемешивании приливают 60 мл нагретой до 95—100 °C воды и оставляют массу на 8—10 ч для кристаллизации. Вещество VI отфильтровывают, промывают 20 мл холодной воды. Получают 93,2 г пасты с содержанием влаги 7,4%, которую направляют на получение темехина. Выход 83,5%, считая на VI.
2,2,6,6-Тетраметилхинуклидина гидробромид (темехин) (VII). К смеси 93,2 г VI, 80 мл воды и 980 мл ксилола приливают 24 мл 42% раствора едкого натра, перемешивают 1 'Ъ ч. Ксилольный слой отделяют, водный, имеющий
185
pH 10, дополнительно экстрагируют 30 мл ксилола. Объединенные ксилольные экстракты сушат сульфатом магния, обесцвечивают углем и кипятят 6 ч, затем охлаждают до 18—20°C, после 2-часовой выдержки осадок VII отфильтровывают и перекристаллизовывают из абсолютного этилового спирта. Получают 47 г темехина. Выход 72%.
ИМЕХИН
2,2,6,6,-Тетраметилхинуклидина йодметилат:
Мол. масса 309,22
Белый кристаллический порошок, горького вкуса, легко растворим в воде (pH 1 % раствора 6,0—7,0, не меняется при кипячении; растворы прозрачны и бесцветны), растворим в хлороформе и этиловом спирте, трудно растворим в ацетоне, практически нерастворим в эфире и бензоле; т. пл. 238—242°C (в интервале 2°C); ВФС 42-211-73.
Имехин — оригинальный ганглиоблокатор кратковременного действия [176], применяется в анестезиологической практике для управляемой гипотонии [250] при различных хирургических вмешательствах. Более активен и менее токсичен, чем используемый для тех же целей ар-фонад. По сравнению с арфонадом удобнее для практического применения, так как является более управляемым средством, не оказывает гистаминоподобного и непосредственного сосудорасширяющего действия. Имехин применяется также для предупреждения и купирования гипертонических кризов и при отеках легких и головного мозга. Вводят имехин внутривенно.
Выпускается в ампулах по 1 и 2 мл 1% раствора N. 10. Цена 1 р;§2 к и 2 р. 52 к.
Метод синте^ имехина разработан во ВНИХФИ [176] на основе темехина (см. с. 181) по схеме:
in
186
Из 2,2,6,6-тетраметиЛхинуклидйна гидробромида (препарата темехин) (I) путем обработки едким натром выделяют свободное основание II, которое с йодистым метилом переводят в йодметилат III.
К раствору 470 г I в 640 мл воды при перемешивании прибавляют 174 мл 42% раствора едкого натра с такой скоростью, чтобы температура в реакционной массе не превышала 25 °C. Выделившееся основание II экстрагируют 4 раза по 700 мл бензола. Объединенные бензольные экстракты сушат поташом, бензол отгоняют при остаточном давлении 200—250 мм рт. ст. и температуре 44—50 °C (основание II — очень летучее соединение), повышая в конце отгонки температуру до 80 °C в течение 5 мин. Остаток (329 г) растворяют в 2 л ацетона (содержание влаги не более 0,5%) в течение 40—50 мин. Прибавляют 287 г йодистого метила в 280 мл ацетона, поддерживая температуру не выше 40 °C. Массу перемешивают еще 2 ч и оставляют для полного выпадения кристаллов на 5—7 ч, защищая от воздействия света. Выход имехина (III) 545 г (92,5%).
КВАЛИДИЛ
1,6-Г ексаметилен-бис-.(3-бензилхинуклидиний хлорид), тетрагидрат:
Мол. масса 629,00
Белый кристаллический порошок без запаха, гигроскопичен, легко растворим в воде, растворим в спирте, хлороформе, нерастворим в ацетоне, этилацетате, бензоле, эфире; ФС 42-1428-80.
Квалидил— оригинальный антидеполяризующий миорелаксант [165, 153]. Применяют квалидил внутривенно для расслабления мышц и управляемого дыхания при наркозе. Вывод из состояния кураризации происходит постепенно: релаксация мускулатуры туловища сохраняется в течение 15—20 мин; после появления самостоятельных дыхательных движений через 25—30 мин мышечный тонус и дыхание полностью восстанавливаются. Препарат не
187
Оказывает влияния на уровень артериального давления, но вызывает умеренную тахикардию.
Выпускается в виде 2% раствора в ампулах по 2 мл в упаковке по 10 штук. Список А. Цена 1 р. 42 к.
Метод синтеза квалидила разработан во ВНИХФИ [165] по схеме:
=0 СвНьСНаМ8С1
сн,
ОН
socu
>свн. —*3
«СНСвНБ Н д/Pd
СНаСвН8
III СжН.ОН L.AIH4 SOCU
HOOCfCH^COOH C^OOCfCH^COOCjH^—*-НО(СНд)вОН -------•2-СцСНа)вС1
V 2 * VI VII VIII
2С1*4НаО
Хинуклидон-3 (I) с бензилмагнийхлоридом переводят в З-окси-З-бензилхинуклидин (II), который подвергают дегидратации нагреванием с тионилхлориДом в хлороформе до 3-бензилиденхинуклидина (III). (Строение III как 3-бензилиденхинуклидина, а не 3-бензил-Д2-дегидрохину-клидина установлено методом спектроскопии ПМР.) Другие способы дегидратации II — нагреванием с уксусным ангидриром, в ксилоле с катализаторами — п-толуолсуль-фокислотой или йодом — с азеотропной отгонкой Воды положительных результатов не дали. При дегидратации нагреванием с 70°/о,л<дсерной кислотой III получен с более низким выходом (~50%). Восстановлением III с палладиевым катализатором получают 3-бензилхинуклидин (IV). Одновременно из адипиновой кисЛоты (V) готовят 1,6-дихлоргексан. Кислоту V переводят в диэтиловый эфир VI нагреванием с этиловым спиртом в толуоле с каталитическими количествами серной кислоты и азеотропной отгонкой воды [311]. Восстановление VI алюмо-гидридом лития и последующая обработка диола VII тио-нилхлоридом приводят к 1,6-дихлоргексану (VIII) [301], который с 3-бензилхинуклидином образует квалидил (IX). Наиболее успешно последняя стадия синтеза проходит в водном ацетоне с содержанием воды ~23% [165]. (В безводных кетонах и в спиртах процесс протекает менее однозначно.)
З-Окси-З-бензилхинуклидин (II). К 195 г свежеприготовленных магниевых стружек (содержание 99,9%, тол
188
щина не более 0,2 мм) в 2 л безводного эфира (влаги менее 0,05%) и 2,5 г сублимированного йода при перемешивании прибавляют эфирный раствор хлористого бензила (всего 920 мл) с такой скоростью, чтобы масса равномерно кипела. Для завершения процесса реакционную смесь кипятят еще 1 ч, охлаждают до 2 °C и в течение 2—3 ч при температуре 2—4 °C прибавляют раствор 400 г хинуклидона-3 (I) в 4,3 л безводного эфира. Массу перемешивают 5—6 ч для нагревания ее до комнатной температуры и продолжают процесс еще 18 ч при комнатной температуре. Полученный гриньяровский комплекс охлаждают до 0 °C и медленно приливают сначала 990 мл воды, затем 3 л 42% серной кислоты, поддерживая темпе-' ратуру массы 0—6 °C. Затем приливают еще 2 л воды. Отделяют эфирный слой, содержащий примеси хлорпро-изводных и смолы, водный слой промывают 2,48 л эфира и подщелачивают твердым поташом (вспенивание!) по фенолфталеину. Перемешивают 1 ч, проверяют щелочность среды и отфильтровывают выделившийся в осадок II вместе с неорганическими продуктами. Осадок высушивают при 70—80 °C, измельчают на шаровой мельнице и экстрагируют 5,9 л абсолютного этилового спирта при кипении в течение 2 ч. Горячий спиртовой раствор отфильтровывают от неорганических солей, которые дополнительно экстрагируют 2 раза по 4,9 л кипящего абсолютного этилового спирта. Спиртовые растворы объединяют, обесцвечивают углем, упаривают до ’/з первоначального объема, охлаждают до 4—5°С (выдержка 18—20 ч). Выпавший в осадок II отфильтровывают и высушивают при 70— 80°C в вакууме (300—400 мм рт. ст.). Выход 550 г (81,1% на I).
З-Бензилиденхинуклидин (III). К раствору 550 г II в 1,39 л хлороформа, поддерживая температуру 28—35 °C, медленно приливают 875 г тионилхлорида (реакция экзо-термична). Затем массу медленно в течение 4 ч нагревают до кипения (скорость нагрева контролируют по интенсивности, выделения газов) и кипятят 4 ч, охлаждают до 40 °C и при этой температуре в вакууме (150—200 мм рт. ст.) отгоняют смесь тионилхлорида и хлороформа (в конце отгонки температура 55 °C, остаточное давление 640 мм рт. ст.). К остатку при 30°C приливают 930 мл воды. Образовавшийся раствор гидрохлорида 3-бензил-иденхинуклидина обрабатывают 590 мл 45% раствора едкого натра до сильнощелочной реакции по фенолфталеину и экстрагируют бензолом (2 и 0,8 л). Объединенные бен-
189
зоЛьйые экстракты, содержащие 3-бензиЛиденхйнуклйдйн (III) и З-хлор-З-бензилхинуклидин (X), сушат поташом и упаривают в вакууме (80 °C в массе, 300— 350 мм рт. ст.). К остатку приливают 3,32 л спиртового раствора, содержащего 63 г/л едкого натра, и массу кипятят 5 ч для дегидрохлорирования X, после чего этиловый спирт отгоняют в вакууме (до 58 °C, остаточное давление 300—650 мм рт. ст.). Вещество растворяют в 745 мл воды и экстрагируют бензолом (1,27 л и 2 раза по 675 мл). Бензольные экстракты сушат поташом, упаривают в вакууме; остаток перегоняют, собирают фракцию с т. кип. 155— 160°C (7 мм рт. ст.). Выход III 403 г (78,85%).
З-Бензилхинуклидин (IV). 400 г III гидрируют при 25— 30 °C и давлении водорода 3—5 атм в 4 л этилового спирта с добавкой раствора 25 г хлористого палладия в 250 мл 36% соляной кислоты. Процесс считают законченным, когда содержание III Подобранной пробе составляет не более 0,2 г/100 мл. После отделения катализатора и отгонки спирта остаток растворяют в 300 мл воды, подщелачивают едким натром по фенолфталеину и основание IV экстрагируют бензолом (2 л и 2 раза по 0,5 л). Бензольные растворы промывают водой, бензол отгоняют; 3-бензилхи-нуклидин перегоняют в вакууме; т. кип. 123—125°С (1— 2 мм рт. ст.). Выход IV 365 г (89,76%), содержание основного вещества 99,5%.
Диэтиловый эфир адипиновой кислоты (VI). Смесь 700 г адипиновой кислоты (V), 2,65 л толуола (содержание влаги менее 0,1%), 4 мл конц. серной кислоты и 1,23л абсолютного этилового спирта кипятят с азеотропной отгонкой тройной смеси (толуол — спирт—вода) при температуре в парах 75 °C. Процесс продолжают 7—8 ч, наблюдая, чтобы отгонка шла равномерно. В конце реакции температура в парах повышается до 85—87 °C (температура кипения двойной азеотропной смеси толуол — спирт). Отбирают пробу реакционной массы и определяют методом ТСХ содержание V (не более 0,1 г/100 мл). К охлажденной до 30°C массе прибавляют 5% раствор гидрокарбоната натрия (~200 мл) до pH 7. Толуольный слой отделяют, водный экстрагируют толуолом (2 раза по 325 мл). Объединенные толуольные растворы промывают водой (2 раза по 500 мл), упаривают, VI перегоняют в вакууме, т. кип. 120—123 °CТО мм рт. ст.). Выход 820 г (83%).
1,6-Диоксигексан (VII). К 180 г алюмогидрида лития в 3,2 л безводного эфира при энергичном перемешивании приливают постепенно раствор 480 г VI в 2,4 л сухого
190
бензола, регулируя скорость прибавления так, чтобы масса слабо кипела. Кипячение продолжают еще 2 ч, охлаждают до 4-3 °C и при этой температуре медленно прибавляют 400 мл воды, перемешивают 2 ч при 3—8°С. Осадок гидроокисей лития и алюминия отфильтровывают, высушивают при 50—65°C в вакууме (300—400 мм рт. ст.) и подвергают дополнительной экстракции 3 раза по 2,3 л кипящего абсолютного этилового спирта (время кипячения каждый раз 1 Vs ч). Эфирно-бензольные и спиртовые растворы упаривают. Получают 254 г технического 1,6-ди-оксигексана (VII) с содержанием основного вещества 97,8%, застывающего при 30 °C, который без дополнительной очистки используют на следующей стадии. Выход близок к количественному.
1,6-Дихлоргексан (VIII). К смеси 254 г VII, полученного на предыдущей стадии, и 28 г пиридина, используемого в качестве катализатора реакции и как вещество, улучшающее растворимость диола VII, медленно прибавляют 638 мл тионилхлорида. Реакция экзотермична, и за счет скорости прибавления тионилхлорида поддерживают температуру 45—50 °C. После прибавления первых 53 мл тио-нилхлррида масса становится прозрачной; включают мешалку, охлаждают до 25—30 °C и дальнейшее прибавление тионилхлорида проводят при этой температуре с внешним охлаждением. Затем массу в течение 2‘/2—3 ч нагревают до кипения (75 °C) (более быстрый нагрев может привести к бурному вспениванию), кипятят 2 ч и упаривают в вакууме (до 90°C в массе при 100 мм рт. ст.). Остаток растворяют при 20—25°C в смеси 370 мл бензола и 990 мл воды. Бензольный слой отделяют, водный экстрагируют бензолом (2 раза по 370 мл). Объединенные бензольные растворы промывают водой (3 раза по 370 мл), 6% раствором гидрокарбоната натрия (345 мл) и снова водой (370 мл) для полного удаления кислых и щелочных продуктов (pH последней промывной воды должен быть около 7). Бензол отгоняют, вещество перегоняют в вакууме, собирая фракцию с т. кип. 95—98 °C (23мм рт.ст.). Выход VIII 257 г (77%), содержание основного вещества 97,3%.
1,6-Гексаметилен-бис-(3-бензилхинуклидиний хлорид), тетрагидрат (квалидил) (IX). Раствор 598 г 3-бензилхи-нуклидйна (IV) и 235 г 1,6-дихлоргексана (VIII) в смеси 1,68 л ацетона и 388 мл дистиллированной воды кипятят 10 ч. Конец реакции определяют по анализу пробы методом ТСХ (содержание IV не более 0,5 г/100 мл). Рас
191
твор обесцвечивают углем (27 г) и выделяют IX выливанием реакционной массы в 6,4 л ацетона с затравкой кристаллов квалидила (без затравки IX выделяется в виде маслообразного вещества, плохо кристаллизующегося). Для полного выпадения кристаллов IX суспензию перемешивают 18 ч при 20—25 °C. Квалидил отфильтровывают, промывают ацетоном и высушивают при 20—25°С. (При повышении температуры препарат теряет кристаллизационную воду.) Выход 750 г (80,1 % на IV и 78,6% на VIII).
ФЕНКАРОЛ
Гидрохлорид (хинуклидил-3) -дифенилкарбинола:
С 2о Н 23 NO’HCl
Мол. масса 329,87
Белый кристаллический порошок без запаха, горьковатого вкуса, мало растворим в воде (1:500), 95% этиловом и метиловом спиртах (1:350), практически нерастворим в хлороформе, негигроскопичен; ВФС 42-561-76.
Фенкарол — оригинальный противогистаминный, ан-тиаллергический препарат [97, 99]. Характеризуется малой токсичностью и хорошей переносимостью. В отличие от димедрола, дипразина и ряда других применяющихся в медицине противогистаминных препаратов фенкарол не оказывает выраженного угнетающего действия на ЦНС, не обладает адренолитической активностью, лишен холинолитических свойств. Отсутствие выраженного седативного эффекта ^Ьдволяет применять фенкарол в амбулаторных условиях, а также лицам активных профессий, работа которых требует быстрых физических или психических реакций (водители транспорта, операторы и др.) и которым противопоказано применение во время работы димедрола, дипразина и других противогистаминных препаратов, угнетающих ЦНС. Фенкарол эффективен у больных с развившимся привыканием к другим антиаллерги-ческим средствам.
Применяют фенкарол при поллинозах, острой и хронической крапивницах, ангионевротическом отеке Квинке, сенной лихорадке, аллергических ренитах, дерматозах —
192
экземе, нейродермитах, кожном зуде и др., пищевой и карственной аллергии.
Выпускается в таблетках по 0,025 г N. 50. Цепа
2 р. 52 к.
Метод синтеза фенкарола разработан во ВНИХФИ и осуществляется по схеме [99, 166]:
Хинуклидон-3 (I) [35] обработкой ацетонциангидрином в водной среде превращают в З-окси-З-цианохинукли-дин (II), который подвергают алкоголизу. Образующийся З-окси-З-карбэтоксихинуклидин (III) без выделения переводят нагреванием с тионилхлоридом в хлороформе в 3-карбэтокси-Д2-дегидрохинуклидин (IV). Соединение IV также без выделения гидрируют в присутствии никелевого катализатора до 3-карбэтоксихинуклидина (V), который путем взаимодействия с фениллитием превращают в (хинуклидил-3) -дифенилкарбинол и переводят в гидрохлорид VI. Использование при синтезе З-окси-З-цианохинукли-дина (II) ацетонциангидрина является более безопасным и технологически приемлемым, чем описанное ранее [303] взаимодействие хинуклидона-3 (I) с цианистым калием. Алкоголиз оксицианпроизводного (И) [166] оказался более удобным способом получения оксиэфира (III) по сравнению с описанным омылением II до 3-оксихинукли-дин-3-карбоновой кислоты с последующей ее этерификацией [295]. В связи с легкостью превращения метилового эфира З-оксихинуклидин-З-карбоновой кислоты в бетаин [295] более целесообразным оказалось использование не метилового, а этилового эфира [166]. (Хинуклидил-3)-дифенилкарбинол был впервые получен при взаимодействии 3-карбометоксихинуклидина с фенилмагнийброми-дом [341]. Однако, как показали дальнейшие исследования [99, 166], синтезируемый по этому методу продукт содержал значительные количества трудно отделяемого (хинуклидил-3)-фенилкетона. Лучшие результаты дает взаимодействие 3-карбэтоксихинуклидина (V) с фенилли-
13 Заказ № 4304 193
тием, когда получаемый (хинуклидил-3)-дифенилкарбинол (VI) не содержит примеси побочного кетона [166].
З-Окси-З-цианохинуклидин (II). ..К охлажденному до 5°C раствору 200 г хинуклидона-3 (I) в 200 мл воды постепенно приливают 136 г ацетонциангидрина. После выдержки 4 ч при 5—7 °C контролируют конец реакции по содержанию хинуклидона-3 (I) в водном растворе методом полярографии (его должно быть менее 3,5 г/100 мл), осадок II отфильтровывают и высушивают до содержания влаги не более 0,2%. Выход II 211 г (86,5%).
З-Карбэтоксихинуклидин (V). З-Окси-З-карбэтоксихину-клидин (III), полученный путем алкоголиза 150 г 3-окси-3-цианохинуклидина (II), экстрагируют хлороформом 6 раз по 210 мл. Хлороформный экстракт сушат сульфатом магния, анализируют на содержание III методом ГЖХ, после чего хлороформ частично отгоняют (до концентрации III около 60%) и при температуре не выше 30°C приливают 450 мл тионилхлорида. Реакционную смесь кипятят 10 ч при 72—74 °C. Конец дегидратации III определяют анализом по ГЖХ (содержание III не более 2%). Затем смесь упаривают при 150—200 мм рт. ст. и 40— 60 °C. Остаток растворяют в 300 мл воды, охлажденной до 5°C, подщелачивают 25% раствором аммиака до pH 9,5—10 и эк&грагируют хлороформом 4 раза по 150 мл. Хлороформные экстракты, содержащие З-карбэтокси-Д2-дегидрохинуклидин с некоторым количеством З-хлор-З-карбэтоксихинуклидина, сушат сульфатом магния, упаривают. Остаток растворяют в 1300 мл абсолютного спирта, раствор обесцвечивают углем и подвергают гидрированию в присутствии 9,8 г никеля Ренея при 50 атм и 50 °C в течение 10 ч. После анализа на конец восстановления пблярографически и методом ТСХ (содержание IV не более 0,2 г/100 мл) катализатор отфильтровывают. Раствор упаривают в вакууме. Остаток подщелачивают водным аммиаком до pH 10—10,4 и экстрагируют хлороформом 5 раз по 260 мл. Хлороформные экстракты сушат сульфатом магния, упаривают и остаток перегоняют при 105—110°С (2—3 мм рт. ст.). Получают 11.6 г (60,9%) V.
Гидрохлорид (хинуклидил-З)-дифенилкарбинола (VI). Раствор фениллития, полученный при взаимодействии 9,3 г лития с 107,5 г бромбензола в 400 мл абсолютного эфира, охлаждают до 0°С и прибавляют 260 мл смеси 3-карбэтоксихинуклидина (V) с эфиром, содержащей 35,3 г V, поддерживая температуру 0—5 °C. Затем охлаждение прекращают и массу перемешивают 4 ч при 18— 194
25 °C. Конец реакции определяют методом ТСХ (содержание V не должно превышать 0,05 г/100 мл), после чего при охлаждении до 5 °C постепенно приливают 380 мл воды. Эфир отгоняют без вакуума. Осадок отфильтровывают, промывают на фильтре водой 4—5 раз по 100 мл до pH осадка 7,5—8 и затем 210 мл кипящего ацетона. Получают 50,4 г (хинуклидил-3)-дифенилкарбинола, который переводят в гидрохлорид. Выход 44,8 г (70,55%).
ПРЕПАРАТЫ ГРУППЫ МОНО- И БИЦИКЛИЧЕСКИХ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ
С НЕСКОЛЬКИМИ ГЕТЕРОАТОМАМИ
Эта группа объединяет большое число лекарственных средств с различным спектром биологического действия. К ней, в частности, относятся сульфаниламиды с антибактериальной активностью (см. с. 90), включающие в свою структуру ядра тиазола (норсульфазол, фталазол), тиадиазола (этазол), изоксазола (сульфаметоксазол), пиримидина (сульфазин, сульфадимезин, сульфамонометок-син, сульфадиметоксин, салазодиметоксин), пиридазина (сульфапиридазин, салазопиридазин) и пиразина (сульфален). К этой группе следует отнести также комбинированный антибактериальный препарат бактрим, состоящий из сульфаметоксазола и триметоприма (см. с. 89).
Наряду с сульфаниламидами важную роль в современной химиотерапии инфекционных заболеваний играют препараты, относящиеся к нитропроизводным гетероциклического ряда. Исследованиям по поиску лекарственных средств в классе нитрогетероциклов положила начало обнаруженная в середине 40-х годов XX века высокая антимикробная активность производных 5-нитрофурфурола (см. с. 113). Интерес к нитросоединениям как к потенциальным антибактериальным средствам в значительной степени усилился после открытия антибиотика левомицетина, относящегося к нитропроизводным ароматического ряда. В результате широких исследований был получен ряд высокоэффективных препаратов, в том числе нитазол, обладающий противотрихомонадным и антиамебным действием (см. с. 200), а также серия нитропроизводных имидазола с противотрихомонадной активностью, из которых наиболее широкое применение в медицине нашел 1-(0-оксиэтил)-2-метил-5-нитроимидазол — метронидазол (флагил, трихо-пол) [211]. В определенной степени родственными нитр1о-соединениями по химической структуре являются гетеро
13*
195
Циклические соединения, содержащие N-окисные группировки. Наибольший интерес с точки зрения создания новых антибактериальных средств представили N-оки-си производных хиноксалина. Подробные исследования этой группы соединений, проведенные советскими учеными [170], выявили высокую антибактериальную активность у 1,4-ди-Ы-окисей 2,3-бисоксиметил- и 2,3-бис-ацето-ксиметилхиноксалинов, которые под названиями диоксидин (см. с. 201) и хиноксидин (см. с. 202) в настоящее время применяются для лечения различных форм бактериальных инфекций и в первую очередь инфекций, устойчивых к действию антибиотиков.
Большая группа производных пиримидина и конденсированных пиримидинов в настоящее время используется в медицине в качестве противоопухолевых средств. Предполагается, что эти соединения являются антиметаболитами природных пиримидинов и пуринов, входящих в состав нуклеиновых кислот. Так, противоопухолевая активность 5-фторурацила (1а) [289] связана с его способностью подвергаться в организме рибозидированию с образованием 5-фтор-2-дезоксиуридин-5-монофосфата, который ингибирует фермент тимидинсинтетазу, участвующий в синтезе дезоксирибонуклеиновой кислоты [300]. Советскими учеными создан фуранидиновый аналог 5-фторурацила — ЬГ-(2-фуранидил)-5-фторурацил (16)—фторафур [39], который менее токсичен и лучше переносится, чем 5-фторура-цил. К ряду производных пиримидана с противоопухолевой активностью относится l-p-D-арабинофуранозилцито-зин (цитарабин) (1в), обладающий также противовирусной й иммунодепрессивной активностью [312].
С середины %^-х годов XX века проводился широкий поиск противоопухолевых средств среди конденсированных пиримидинов и прежде всего среди производных птеридина и пурина. В частности, структурные модификации фолиевой кислоты привели к созданию ее антагониста —
196
метотрексата (см. с. 204), ингибирующего восстановление фолиевой кислоты в ее тетрагидропронзводное и, следовательно, тормозящего репродукцию раковых клеток. Модификация молекулы аденина позволила советским ученым создать оригинальный противолейкозный препарат фопу-рин — 2-диметиламино-6-диэтиленимидофосфамидо-7-ме-тилпурин (II) [44]. Особенностью фопурина является «двойной» механизм действия, связанный с противоопухолевой активностью как самого фопурина, так и продукта его расщепления в организме — 2-диметиламино-6-амино-7-метилпурина (III), обладающего антиметаболйтным типом действия:
Модификация структуры известных природных и синтетических биологически активных соединений до настоящего времени остается одним из ведущих направлений в поиске новых лекарственных средств. Так, модификация структуры противоопухолевого средства — 6-меркаптопу-рина — позволила получить ряд новых соединений с противоопухолевой активностью, в том числе препарат аза-тиоприн (IV), обладающий более высокой, чем 6-меркап-топурин, иммунодепрессивной активностью [211]. Исследования в ряду пиразоло[3,4-с1] пиримидина — соединений со структурой, изостерной гетероциклической системе пурина, привели к созданию антиподагрического средства аллопуринола—4-оксипиразоло[3,4-(1]-пиримидина (V),. особенностью действия которого является способность ингибировать фермент ксантиноксидазу и, следовательно, препятствовать окислению производных ксантина до мочевой кислоты и накоплению ее в организме [291].
197
Неожиданные результаты были получены при модификации структуры адреномиметических средств имидазолинового ряда: нафтизина и галазолина. Как известно, эти препараты вызывают сужение периферических кровеносных сосудов и 'ЙёМут к повышению артериального давления. С целью получения аналогов препаратов подобного типа был синтезирован ряд соединений, также включающих в свою структуру имидазолиновый цикл, но отличающихся тем, что ароматический фрагмент у них связан с положением 2 имидазолина не СНг-звеном, а через NH-группу. Эта структурная модификация привела к существенному изменению биологической а§тивности— полученный ряд веществ обладал гипотензивным действием, а одно из наиболее активных соединений этого ряда — 2- (2,6-дихлорфениламино) имидазолина гидрохлорид — кло-фелин (см. с. 208) нашел применение в практик^ лечения различных форм гипертонической болезни.
Производные имидазола представляют большой интерес с точки зрения поиска новых лекарственных средств. К этой группе гетероциклов наряду с такими природными соединениями, как гистидин и гистамин, играющими важную роль в процессах жизнедеятельности, относятся также эффективные лекарственные средства с разнообразным спектром биологической активности: клофелин, метронидазол, этимизол, мебикар (см. с. 211). При модификации структуры одного из ключевых веществ в биосинтезе пуринов — амида 4-аминоимидазол-5-карбоноцой кислоты— получен новый противоопухолевый препарат диме-тилтриазеноимидазолкарбоксамид (VI) [333], обладающий активностью в отношении некоторых видов меланом и сарком.
Весьма интересными с точки зрения поиска новых лекарственных средств оказались производные сидноними-на — гетероцикла, имеющего мезоионную структуру. Фундаментальные работы советских ученых в этом ряду соединений [274] первоначально привели к получению препарата сиднофен (см. с. 212), сочетающего психостимулирующее действие с антидепрессивной активностью. Дальнейшими исследованиями было показано, что введение арилкарбамоильного остатка в экзоциклическую иминогруппу синдофена приводит к существенному повышению психостимулирующей активности, а одно из соединений этого ряда — сиднокарб (см. с. 213) — по некоторым показателям имеет определенные преимущества перед известным психостимулятором фенамином.
198
В последние годы поиски лекарственных средств среди производных сиднонимина значительно активизировались в связи с обнаружением в этом ряду нового антиан-гинального препарата молсидомина — N-этоксикарбонил-3-(4-морфолинил) сиднонимина (VII) [312]:
VII
Одним из наиболее важных достижений в области медицинской химии за последние четверть века следует считать открытие новой группы транквилизаторов и снотворных средств — производных 1,4-бензодиазепина [338]. Основным исходным моментом в создании этой группы препаратов явилась модификация структуры сравнительно мало изученной гетероциклической системы S-N-окиси хи-назолина, которой ранее ошибочно приписывалось строение бензгептоксадиазина. При попытке синтезирован на основе S-N-окиси 2-хлорметил-4-фенилхиназолина (VIII) ряд его аминометильных производных со структурой IX путем обработки VIII аминами авторы обнаружили, что эта реакция сопровождается скелетной перегруппировкой с образованием производного 1,4-диазепина, проявившего высокую транквилизирующую активность и названного впоследствии хлордиазепоксидом (X).
Дальнейшие модификации структуры хлордиазепокси-да привели к обнаружению серии препаратов с седативным и снотворным действием, таких, как диазепам (XI), оксазепам (XII), нитразепам (XIII) и др. [338]. В результате работ советских ученых был создан препарат фена-
199
зепам (XIV) [83, 25] (см. с. 215), применяющийся в настоящее время в качестве транквилизирующего, снотворного и противосудорожного средства.
НИТ АЗОЛ
2-Ацетиламино-5-нитротиазол:
C5HSN303S □ □ О о
Мол. масса 187,18
Основные синонимы: аминитрозол, ацинитразол, три-хоцид, трихорал, трихолавал, тринекс и др.
Желтый с зеленоватым оттенком кристаллический порошок, без запаха, очень мало растворим в воде (1 :7000), мало растворим в спирте (1:400) и уксусной кислоте (1 : 200), легко растворим в диметилформамиде, т. пл. 264— 268°C, разл.; 42-802-73.
Нитазол подавляет развитие Trichomonas vaginalis и других простейших.
Применяют нитазол при лечении трихомонадоза у женщин и мужчин и при лямблиозе.
Выпускается в таблетках по 0,1 г N. 30, цена 76 к.; суспензии в упаковке по 100 г, цена 71 к.; суппозиториях вагинальных по 0,12 г N. 10, цена 44 к.
Метод синтеза нитазола разработан во ВНИХФИ и Уральском филиале ВНИХФИ по схеме:
(CH COLO
CH COHN
no2
Конденсацией тиомочевины (I) с хлор ацетальдегидом получают 2-аминотиазола гидрохлорид, который без выделения переводят в нитрат III. Нагревание нитрата III с серной кислотой обеспечивает нитрование в положение 5 тиазольного ядра, а последующее ацетилирование уксусным ангидридом без выделения 2-амино-5-нитротиазо-ла (IV) приводит к 2-ацетиламино-5-нитротиазолу (нита-золу) (V).
2-Аминотиазола нитрат (III). К смеси 6,32 л воды и 2,64 л технического хлорацетальдегида (II) с содержанием
200
основного продукта 860 г при перемешивании добавляют 654 г тиомочевины (I), нагревают 2 ч при 60°C, охлаждают до (—5) — (+5°C), приливают при этой температуре 1,46 л 55% азотной кислоты и дают выдержку 6 ч при 0—-5°С. Осадок отфильтровывают, промывают 400 мл этилового спирта и высушивают. Выход III 810 г (55%).
2-Ацетиламиио-5-нитротиазол (нитазол) (V). К 1,21 кг 93% серной кислоты, охлажденной до 3—5 °C, прибавляют 350 г III с такой скоростью, чтобы температура массы поддерживалась 8—10 °C, затем дают выдержку 9 ч при 18—20 °C, нагревают до 40—45 °C и приливают 870 мл уксусного ангидрида при перемешивании и температуре (реакция экзотермична) 55—60°C. Затем дают выдержку 30 мин при 70—75 °C, охлаждают до 25—30 °C и выливают в 8,6 л воды. Выпавший осадок отделяют, промывают 5 л воды и высушивают при 60—75р. Выход V 338 г (89%).
диоксидин
1,4-Ди-М-окись 2,3-бис (оксиметил) хиноксалина:
Мол. масса 222,20 Зеленовато-желтого цвета кристаллический порошок, без запаха, мало растворим в воде, спирте, хлороформе; т. пл. 169—172°С; ФС 42-1232-79.
Диоксидин — оригинальный антибактериальный препарат широкого спектра действия. Он активен при инфекциях, вызванных штаммами бактерий, устойчивыми к антибиотикам или препаратам других химических групп [185, 186].
Используют для лечения различных форм гнойных бактериальных инфекций, вызванных грамотрицательны-ми бактериями, в частности синегнойной палочкой, протеем и клебсиеллами, а также стафилококками или флорой, устойчивой к антибиотикам; применяют местно, в полости и внутривенно.
201-
Выпускают ампулы по 16 мл 1% раствора N. 10. Цена 2 р. 50 к.
Метод синтеза диоксидина (I) из 1,4-ди-М-окси 2,3-бис-(ацетоксиметил) хиноксалина (см. хиноксидин) (II), разработанный во ВНИХФИ [72], заключается в гидролизе II метанолом в присутствии каталитического количества едкого натра.
Описанный в литературе метод получения I [307], заключающийся в гидролизе II разбавленной соляной кислотой, менее удобен, так как в этих условиях протекают окислительно-восстановительные процессы, сопровождающиеся образованием побочных продуктов.
К суспензии 220 г II в 2,17 л метанола приливают 12,5 мл 8% раствора едкого натра в метаноле, перемешивают при 20—22 °C в течение 3 ч, затем 2 ч при 15—?0°С, фильтруют, осадок промывают 2 раза по 170 мл метанола и сушат при 20 °C, получают 145 г технического I, который прибавляют к нагретым до 60 °C 1,3 л воды, перемешивают до растворения (30—40 мин), раствор обесцвечивают углем, охлаждают при 8—10 °C в течение 3 ч, фильтруют, осадок промывают 2 раза по 60 мл воды, 115 мл спирта и сушат при 50—55 °C. Выход .1 12,3 г (78%).
/л\ хиноксидин
1,4-Ди-ЬЬокись 2,3-бис (ацетоксиметил) хиноксалина: о I
СН2ОСОСН3
СН2ОСОСН3
о
CMH>^2Oe
Мол. масса 306,28
202
Зеленовато-желтого цвета кристаллический порошок, без запаха, мало растворим в воде и спирте, легко растворим в хлороформе; т. пл. 176—180°С; ФС 42-1139-78.
Хиноксидин — оригинальный антибактериальный препарат широкого спектра действия [21]. Применяется для приема внутрь при инфекциях, вызванных грамотрица-тельной флорой, а также штаммами бактерий, устойчивыми к антибиотикам и другим химиотерапевтическим препаратам [70].
Выпускается в таблетках по 0,25 г N. 20. Цена 3 р. 02 к.
Метод синтеза хиноксидина (I) разработан во ВНИХФИ по схеме [71]:
Окислением о-нитроанилина (II) гипохлоритом кальция получают бензофуроксан (III), из которого реакцией с метилэтилкетоном в присутствии триэтиламина синтезируют 1,4-ди-И-окись 2,3-диметилхиноксалина (IV) [310]. Последнюю бронируют диоксандибромидом до 1,4-ди-М-окиси 2,3-бис (бромметил) хиноксалина (V), которую действием уксусной кислоты в присутствии триэтиламина превращают в I.
Известные методы синтеза V путем конденсации о-фе-нилендиамина с 1,4-дибромбутадионом-2,3 с последующим окислением 2,3-бис-(бромметил) хиноксалина надуксусной кислотой до V [307], а также взаимодействие о-фенилен-диамина с диметилглиоксимом, окисление полученного 2,3-диметилхиноксалина надуксусной кислотой до 1,4-ди-N-окиси 2,3-диметилхиноксалина (IV) [186] не имеют преимуществ перед описанным выше методом.
Бензофуроксан (III). Нагретую до 70°C смесь 167 г II и 3 л воды подщелачивают 3 мл 46% раствора едкого
203
натра, прибавляют в течение 40 мин 1,48 л 6% водного раствора гипохлорита кальция, нагревают при 60—65 °C в течение 30 мин, поддерживая pH 10—12 прибавлением едкого натра, постепенно охлаждают, осадок промывают водой, получают 126 г пасты III, выход 90%.
1,4-Ди-М-окись 2,3-диметилхиноксалина (IV). К охлажденному раствору 109 г аммиака в 605 мл метанола в течение 15 мин прибавляют 118 мл метилэтилкетона, поддерживая температуру 8—10 °C охлаждением массы водой. Образующийся раствор енамина метилэтилкетона в течение 3 ч при 26—30°C прибавляют к раствору 126 г III в. 260 мл метанола, нагревают при 30—34 °C в течение Зч, фильтруют, осадок промывают 2 раза по 50 мл метанола и сушат при 50—60 °C. Выход 144 г (90%).
1,4-Ди-М-окись 2,3-бис(бромметил)хиноксалина (V). К 375 мл безводного диоксана постепенно прибавляют 145 г брома, 57 г IV и нагревают при перемешивании 3 ч при 80—85 °C. Выпавший по охлаждении гиДробромид V отфильтровывают, прибавляют его к 25 мл насыщенного раствора ЫаНСОз, осадок фильтруют, промывают водой и сушат, выход 83 г (80%).
1,4-Ди-1Ч-окись 2,3-бис(ацетоксиметил)хиноксалина (хи-ноксидин) (I). К смеси 848 мл ацетона и 108 мл уксусной кислоты при 20—35 \С постепенно приливают 123 мл три-этиламина. К полученному раствору прибавляют 99 г V, кипятят 3 ч, охлаждают до 15—18 °C, перемешивают 1’/а ч и фильтруют. Осадок I промывают ацетоном, затем водой, сушат при 70—75°C, кристаллизуют из 30 объемов 80% изопропанола и сушат при 70—75 °C в вакууме 300— 400 мм. Выход 70 г (79%).
МЕТОТРЕКСАТ
4-Дезокси-4-амино-М-метилфолиевая кислота:
Мол. масса 454,5
Основные синонимы: аметоптерин, метоптерин, метиламиноптерин. Желтого или оранжево-желтого цвета мелкокристаллический порошок, практически нерастворим в
204
воде и спирте, легко растворим в растворах едких щелочей и карбонатов щелочных металлов; ФС 42-479-75.
Метотрексат — противоопухолевое средство, являюще-ется структурным аналогом и антагонистом фолиевой кислоты (витамин Вс). Под влиянием метотрексата ингибируется активность фермента дигидрофолатреДуктазы, участвующей в превращении фолиевой кислоты в тетрагидро-фолиевую, которая играет важную роль в процессе биосинтеза нуклеиновых кислот и репродукции клеток [243].
Применяют метотрексат для лечения хорионэпителиомы матки, острых лейкозов, терминальной стадии хронического миелолейкоза и псориаза. Выпускается в таблетках по 0,0025 г N. 50. Цена 3 р. 55 к. В амп. по 2 мл 2,5% раствора, N. 5, цена 7 р. 77 к.
Метод синтеза метотрексата (I) разработан во ВНИВИ [22] по схеме:
NH,
1)Н2.Р4С
‘ ’°4
NH
NaNOg
СН-СООН nh2 3
CHJCNUIII)
H2NC(«NH)NH^ hNO3 — 11
CHjNHj Ba Cig
ооон
CONHCH X
СН2СН2СООН
Конденсацией нитрата гуанидина (II) с динитрилом малоновой кислоты (III) получают 2,4,6-триаминопирими-дин (IV), который нитрозированием нитритом натрия в уксусной кислоте превращают в 2,4,6-триамино-5-нитрозо-
205
пиримидин (V). Восстановлением над Pd/C и последующей обработкой серной кислотой V превращают в 2,4,5,6-тетра-аминопиримидина сульфат (VI). Второй компонент для получения I — бариевую соль п-метиламинобензоил-Ь-глу-таминовой кислоты (VII)—синтезируют из п-аминобен-зоил-Ь-глутаминовой кислоты (VIII) путем ее диазотирования с последующей обработкой диазониевой соли раствором йодистого калия и конденсацией образующейся п-йодбензоил-Ь-глутаминовой кислоты (IX) с метиламином в присутствии хлористого бария. При трехкомпонентной конденсации VI, 1,1,3-трихлорацетона (X) и VII получают I. Впервые синтез I осуществлен путем трехкомпонентной конденсации VI, динатриевой соли п-метиламинобен-зоил-Ь-глутаминовоц.кислоты и 2,3-дибромпропионового альдегида [332]. В этом случае процесс протекает с образованием большого числа побочных продуктов. Впоследствии был опубликован ряд других методов синтеза I, включая двухкомпонентные конденсации с использованием в качестве исходных веществ 6-хлор-2,4-диамино-5-нитро-пиримидина, l-N-окиси 2-амино-3-циано-5-хлорметилпира-зина, 2,4-диамино-6-галогеноптеридинов; однако эти методы, как правило, многостадийны и основаны на использовании малодоступного и дорогостоящего сырья.
2,4,6-Триамино-5-нитрозопиримидин (V). К 0,5 л 10% раствора едкого натра в спирте прибавляют 120 г II, нагревают при 75—78 °C в течение 20—25 мин, охлаждают до 50—60 °C и постепенно в течение 10—15 мин приливают 157 мл 37,5% раствора III в спирте. Массу кипятят 2 ч, охлаждают до 5—7 °C, выпавшие кристаллы отфильтровывают, промывают 200 мл охлажденного до 0°С абсолютного этанола и сушат на воздухе в течение 6—7 ч, получают 125—135 г IV. Последний суспендируют в 0,5 л дистиллированной воды, нагревают при 75—80 °C до растворения и прибавляют -~15 мл уксусной кислоты до pH 6,9—7,2. К охлажденному до 65—70 °C раствору в течение 10—15 мин приливают 130 мл 55% раствора нитрита натрия в воде и затем 50 мл уксусной кислоты. По окончании нитрозирования pH среды составляет 5,2—5,7. Массу нагревают прр^О—75°C в течение 1—1’А ч, горячую суспензию фильтругбт, осадок промывают 2 разд по 0,5 л нагретой до 70—75°С воды, затем 0,5 л холодной дистиллированной воды и сушат при 50—60 °C в вакууме 40— 50 мм рт. ст., получают 87,8 г (58,2%) V.
2,4,5,6-Тетрааминопиримидина сульфат, дигидрат (VI). В автоклав последовательно загружают 2,4 л 1% раство
206,
ра ёДкОгб натра, 150 г V и катализатор (приготовлен пу* тем смешения 3 г хлористого палладия с 6 г активированного угля и последующего добавления небольшого количества 1% раствора едкого натра). Массу гидрируют при 45—50 °C и давлении 1,45—1,5 атм в течение 55—65 мин до поглощения 0,065—0,07 м3 водорода, фильтруют. К раствору в токе азота приливает 470 мл 60% серной кислоты, охлаждают при 4—5 °C в течение 12 ч, фильтруют, осадок промывают 3 л холодной воды и сушат при 50—60 °C, получают 191 г (73,6%) VI.
п-Йодбензоилглутаминовая кислота (IX). К 732 мл 9% соляной кислоты прибавляют 100 г VIII, перемешивают при 20°C до полного растворения VIII, охлаждают до 3°С и в течение 35—45 мин прибавляют 160 мл 14,4% водного раствора нитрита натрия. Массу перемешивают 10—15 мин и при 3—4 °C в течение 55—60 мин прибавляют раствор 100 г йодистого калия в 180 мл дистиллированной воды (к концу добавления допускается повышение температуры до 14—17°C). Массу перемешивают 1’/г—2 ч, фильтруют, осадок промывают 6 раз по 1 л нагретой до 50 °C воды и перекристаллизовывают из 600 мл 50% спирта. Выход 72 г (52,9%); т. пл. 170—172 °C.
Бариевая соль п-метиламинобензоил-Ь-глутаминовой кислоты (VII). Смесь 280 мл 25% водного раствора метиламина, 100 г IX и 0,5 г медного катализатора нагревают в автоклаве при 130—135 °C в течение 6 ч, охлаждают до 40—50 °C, фильтруют, раствор разбавляют 0,5 л этилового спирта и избыток метиламина отгоняют со спиртом. Остаток растворяют в 380 мл этилового спирта, прибавляют раствор 28 г хлористого бария в 130 мл дистиллированной воды и охлаждают при 1—2 °C в течение 18— 20 ч. Осадок фильтруют, промывают 2 раза по 150 мл этилового спирта и сушат при 40—50 °C в вакууме 30— 40 мм рт. ст., получают 74,3 г (37,2%) VII.
4-Дезокси-4-амино-М-метилфолиевая кислота (метотрексат) (I). к нагретым до 88—89°C 0,5 л дистиллированной воды последовательно прибавляют раствор 85 г VII в 100 мл воды, раствор 8 г VI в 50 мл воды и раствор 8,5 г метабисульфита натрия в 50 мл воды. К смеси при pH не ниже 3,5 прибавляют 60 г 12% водного раствора X, нагревают при 88—89 °C в течение Р/г ч, поддерживая pH 3—3,5 прибавлением 6% раствора едкого натра, нагревают при 70—72 °C в течение 2 ч, горячую массу фильтруют и осадок промывают 50 мл воды. К раствору при 48—50 °C в течение 55—60 мин приливают 40 мл 6% рас
207
твора едкого натра до pH 5, охлаждают при 2—3°С в течение 10—12 ч, фильтруют, осадок промывают 100 мл холодной воды, 40 мл спирта и сушат при 30—35 °C в вакууме 40—50 м рт. ст. в течение 7—9 ч. Полученный технический I очищают хроматографированием на целлюлозе с использованием боратного буфера. Выход 72,7%, считая на VII.
КЛОФЕЛИН
2- (2,6-Дихлорфениламино) имидазолина гидрохлорид:
• HCt
Мол. масса 266,57 Основные синонимы: гемитон, катапресан, хлофазолин, клонидин. Белый кристаллический порошок, без запаха,, горького вкуса, растворим при 20 °C в воде 1 : 14, в кипящей воде — 1:2, в кипящем этиловом спирте — 1:5, мало растворим в хлороформе, плохо растворим в других органических растворителях; т. пл. 314—316 °C, разл. ВФС 42-647-77.
Клофелин — высокоэффективный гипотензивный препарат с центральным механизмом действия [156, 348],уменьшает тонус симпатической нервной системы и одновременно усиливает активность блуждающего нерва, в результате чего понижается артериальное давление и урежаются сердечные сокращения, оказывает седативное действие.
Применяют клофелин при разных формах гипертонической болезни, в том числе при гипертонии, обусловленной хронической почечной недостаточностью, а также при лечении глаукомы.
Выпускается в таблетках по 0,075 мг N. 50, цена 25 к.; таблетках по 0,15 Mr N. 50, цена 47 к.; ампулах по 1 мл 0,01% раствора N. 10, цена 32 к.; глазных каплях в тюбиках-капельницах 0,125% раствор по 1,5 мл N. 2, цена 18 к.; глазных каплях в тюбиках-капельницах 0,25%. раствор по 1,5 мл N. 2, цена 34 к.
Оригинальный синтез клофелина (I) из тетраметил-тиурамдисульфида (II) разработан во ВНИХФИ по схеме [42]: , . ‘
208
Cl
Тетраметилтиурамдисульфид (II, тиурам Д) хлорируют в сухом дихлорэтане или хлористом метилене сухим газообразным хлором до М,Ы-диметил-Ы-дихлорметилен-иммоний хлорида (III) [342], используя эквимолярное количество хлора, так как при избытке хлора образуется молекулярный комплекс III с хлором, который может быть разрушен с высвобождением III лишь при нагревании до 80 °C или при кристаллизации из ацетонитрила. Хлорирование тиурама является экзотермическим процессом, поэтому его проводят при охлаждении, поддерживая температуру 20—30 °C. При хлорировании тиурама образуется двухлористая сера, что не позволяет получить V, и ее удаляют отгонкой с растворителем й промывкой осадка сухим хлористым метиленом. Поскольку III гигроскопичен, его без выделения превращают в V взаимодействием с IV и избытком триэтиламина. При реакции V с этилендиамином в мягких условиях (18—23 °C) образуется замещенный гуанидин VI, который можно выделить. Однако более удобно получать I без выделения VI — ступенчатым нагреванием при 18—23 °C и 100—105 °C. I синтезируют также из других тетраалкилтиурамдисульфидов, например, из этильного аналога II — тетурама [42].
2,6-Дихлоранилин (IV) получают хлорированием сульфаниламида смесью соляной кислоты и перекиси водорода с последующим гидролизом образующегося 3,5-дихлор-сульфаниламида водной H2SO4 [288]:
Cl Cl
В литературе описано несколько вариантов синтеза клофелина из IV [42], которые не имеют преимуществ
14 Заказ № 4304 209"
Пё£еД описанным выше. Один из методов получения 1 основан на формилировании IV смесью НСООН и (СНзСО)гО, хлорировании М-формил-2,6-дихлоранилина смесью тионилхлорида и сульфурилхлорида с последующей обработкой N-(2,6-дихлорфенил) изоцианиддихлорида этилендиамином. Выход I, считая на IV, не превышает 25%. Согласно другому методу мочевину сплавляют с этилен-
диамином или с этиленгликолем, полученную этиленмочеви-ну (VII) ацилируют бензоилхлоридом, М-бензоилЛП! последовательно обрабатывают РОС13, IV и образующийся 1 -бензоил-2- (2,6гдихлорфениламино) им,идазолйн гидролизуют до I. Метод технологически сложен, так как получение VII протекает в жестких условиях (~250°).
М,1Ч-Диметил-1Ч'-(2,6-дихлорфенил)-С-хлорформамидин
гидрохлорид (V). Через раствор 96 г II в 1,2 л сухого CH2CI2 в течение 30—40 мин пропускают 200 г сухого хлора, поддерживая температуру 20—30 °C охлаждением массы холодной:?'вбдой; смесь CH2CI2 и SCI2 отгоняют,
приливают 0,6 л сухого CH2CI2, снова отгоняют; операцию отгонки повторяют еще раз. К остатку дважды приливают по 400 мл сухого CH2CI2, каждый раз смесь перемешивают и растворитель отсасывают через грибок с пористым
стеклянным фильтром. К остатку приливают 330 мл сухого CH2CI2, прибавляют при 5—10 °C раствор 98 г IV и 184 г М(СгНз)з в 70 мл сухого CH2CI2, перемешивают 2'/г чпри 20—23 °C и отфильтровывают (С2Н5)зН-НС1. Раствор упаривают в вакууме, остаток растворяют в 0,5 л ацетона, прибавляют Спиртовой раствор НС1 и охлаждают во льду. Осадок V фильтруют, промывают 3 раза по 50 мл СН2С12, сушат в вакууме в течение 1 ч. Выход 150 г (85,7%).
2-(2,6-Дихлорфениламино)имидазолина гидрохлорид (кл^фелин) (I). К 120 мл этиленгликоля, охлажденным до 5 °C, прибавляют 116,6 г V. К суспензии при 5—10 °C постепенно приливают 68 мл 72,8% этилендиамина, перемешивают при 18—23 °C в течение 3 ч, поддерживая температуру охлаждением массы холодной водой, нагревают при 100—105°C 3 ч, охлаждают до 40—50°С и в вакууме 10—20 мм рт. ст. отгоняют смесь этиленгликоля и IV. Остаток охлаждают до 40—50 °C, разбавляют 60 мл воды,
перемешивают 15—20 мин, приливают конц. . соляную кислоту (~28 мл) до pH 4, перемешивают 1 ч при 18— 23 °C, поддерживая pH 4 прибавлением НС1, фильтруют, осадок промывайУгЗ раза по 12 мл воды, затем 3 раза по 40 мл ацетона и сушат сначала на воздухе, а затем при 60—80 °C. Получают 71,9 г I, который кристаллизуют из
210
165 мл кипящей дистиллированной воды с обесцвечиванием углем. Выход 57 г (56,6%).
МЕБИКАР
2,4,6,8-Тетраметил-2,4,6,8-тетраазабицикло (3.3.0) октан-дион-3,7.
сн3 сн3
с В Н И Н4О 2
Мол. масса 198,25
Белый кристаллический порошок, без запаха, легко растворим в воде (допускается слабая опалесценция), хлороформе; растворим в спирте, очень мало растворим в эфире; т. пл. 224—228 °C; ВФС 42-735-78.
Мебикар-^—оригинальный психотропный препарат, сочетающий транквилизирующий и антипсихотический эффекты [88]. Он не вызывает миорелаксацию и вялость. Применяют мебикар при лечении неврозов и неврозоподобных состояний.
Выпускаются таблетки по 0,3 г N. 10. Цена 40 к.
Синтез мебикара (I) разработан в Институте органической химии им. Н. Д. Зелинского АН СССР путем конденсации Ь1,М'-диметилмочевины (II) с глиоксалем (III) в кислой среде [180, 318] по схеме:
сн3 сн3
НС1 / Т \ , 2CH3NHCONHCH3 * (СНО)2->-О=( >=О
N
ОН "1
(CH3)2SO4
Na ОН
IV
II
III
N
2H2NCONHJ
НС1
H N
N
3 I
H N
H
Мебикар может быть также получен конденсацией мочевины с глиоксалем в присутствии НС1 с последующим метилированием IV диметилсульфатом в щелочной среде [318].
14* 211
К 26, 75 кг 40% водного раствора III прибавляют 31,25 кг II, нагревают при 45 °C и перемешивании до растворения осадка, приливают конц. соляную кислоту (~0,625 л) до pH 1—2. При этом температура реакционной массы повышается до 80 °C. Смесь нагревают при слабом кипении 1 ч, охлаждают до 55°C и прибавляют 10% раствор NaOH (2,9 л) до pH 7—8. К реакционной i, массе при 35—50 °C приливают 137,4 л н-бутанола, отгоняют азеотропйую смесь н-бутанола с водой; раствор нагревают 30 мин с углем, фильтруют, охлаждают в течение 1—2 ч при 4 °C и полученный I кристаллизуют (1:6) из изопропилового спирта. Выход И кг (36,6%, считая на II).
СИДНОФЕН
З-(Р-Фенилизопропил)сиднонимина гидрохлорид: .
СНз c6h5ch2Ah-n---сн
L 1 Hci
x0/^nh
и Н|з N3O HCI
Мол. масса 239,71
Белый или белый с легким желтоватым оттенком мелкокристаллический порошок, без запаха, горького вкуса, легко растворим в воде, растворим в спирте, практически нерастворим в эфире, ацетоне, бензоле; т. пл. 158—162 °C разл.; ФС 42-1060-76.
Сиднофен — оригинальное психотропное средство, сочетающее психостимулирующее действие и умеренную ан- | тидепрессивную активность [4,5]. Применяют сиднофен I при астенических состояниях различного происхождения, 1 при адинамищ . вялости, подавленности, апатии в связи с '*
неврозами. ' . I
Выпускается в таблетках по 5 мг N. 50. Цена 30 к.
Метод синтеза сиднофена (I) из р-фенилизопропил-амина (II) разработан во ВНИХФИ по схеме [4]j
ОН
СНз 1)СН2О,(СН3)2С^ <j=H3 NaN02
C6H5CH2tHNH2 -----——--------^CeH CH2CHNHCH£N;HCl -----------*
О Q * . 2} nvl
I II III
' ' с н э
С HuCH,in-N-CH,CN
> - 6-: .Э * . I ‘
' NO
HCI
=NH-HC1
212
Взаимодействием формальдегида с ацетонциангидрином получают нитрил гликолевой кислоты, который вводят в реакцию с ₽-фенилизопропиламином и в виде гидрохлорида выделяют p-фенилизопропиламиноацетонитрил (III); последний нитрозируют нитритом натрия и образующийся N-нитрозо-Ш без выделения действием хлороводорода в изопропаноле превращают в I.
p-Фенилизопропиламиноацетонитрил (III). К смеси 303 г ацетонциангидрина и 284 г формалина приливают 30 мл 10% водного раствора поташа, охлаждают до 10— 12 °C и перемешивают 4 ч, поддерживая pH 8—9 прибавлением 10% раствора поташа. К массе при 10—12°C в течение 2—2'/2 ч прибавляют 543 г р-фенилизопропилами-на, перемешивают 2 ч, органический слой отделяют, растворяют в 2,14 л абс. изопропанола; к раствору при 18— 20 °C постепенно приливают 360 мл 30% раствора хлороводорода в изопропаноле до pH 3—3,5, перемешивают при 18—20 °C в течение 2 ч, поддерживая pH 3—3,5 добавлением раствора хлороводорода в изопропаноле. Осадок фильтруют, промывают изопропанолом и сушат при температуре не выше 60 °C в течение 20—24 ч, получают 520 г (77,6%) III.
3-(р-Фенилизопропил)сиднонимина гидрохлорид (сид-нофен) (I). К смеси 520 г III, 2,2 л воды и 1,32 л этилацетата, охлажденной до 3—5 °C, в течение Р/г—2 ч приливают раствор 205 г NaNO2 в 265 мл воды, поддерживая pH 1 прибавлением конц. соляной кислоты. Смесь перемешивают 2 ч; при этом температура в массе повышается до 22—23 °C. Органический слой отделяют, водный экстрагируют этилацетатом (2 раза по 220 мл). Объединенные растворы I в этилацетате сушат прокаленным Na2SO4 и обесцвечивают углем; к раствору при 3—5 °C в течение Р/2—2 ч приливают 102 г 30% раствора хлороводорода в изопропаноле, перемешивают при 6—8 °C в течение 2 ч, фильтруют, осадок промывают этилацетатом, охлажденным до 8—10 °C, и сушат при 40 °C в течение 20—24 ч, получают технический I, который кристаллизуют из 1,3 л абсолютного изопропанола. Выход 324 г (42,5%, считая на 0-фенилизопропиламин).
213
СИДНОКАРБ
N-Фенилкарбамоил-З- (p-фенилизопропил) сиднонимин:
си3 с-н-сн-дн- n---сн
6 Э л | I
N ^J.=NCONHC0H5
I с 18 Н 18
Мол. масса 322,37
Белый с желтовато-зеленоватым оттенком кристаллический порошок, без запаха, легко растворим в хлороформе и ацетоне, трудно растворим в спирте, практически нерастворим в воде; т. пл. 132—138 °C разл.; ФС 42-992-75.
Сиднокарб — оригинальный, стимулятор ЦНС [155]. В отличие от фенамина не вызывает эйфории, двигательного возбуждения, тахикардии и повышения артериального давления. Оказывает более длительное стимулирующее действие, чем фенамин.
Применяют сиднокарб при лечении астенических и неврастенических расстройств, сопровождающихся вялостью, заторможенностью, апатией, сонливостью, пониженной работоспособностью; астении, обусловленной применением нейролептических средств.
Выпускается в таблетках по 5 мг N. 50, цена 52 к.; в таблетках по 10 мг N. 50, цена 1 р. 02 к.
Метод синтеза сиднокарба (I) из 3-(р-фенилизопро-пил)сиднонимина гидрохлорида (II) (см. Сиднофен), разработанный во ВНИХФИ, заключается в ацилировании II фенилизоцианатом в изопропаноле в присутствии ацетата натрия по схеме [6]:
сн3 с6н6сн2<Ьн-ы
N
сн =
----CH c6h5nco c6h5ch2ch-n
• HCl CH-COONa NH J
N
NCONHC.H. О О
II
Ы-Фенилкарбамоил-3-(р-фенил изопропил )сиднонимин (сиднокарб) (I). К суспензии 130 г II в 0,7 л абсолютного изопропанола при 0° С прибавляют 448 г ацетата натрия, перемешивают до получения однородной массы и при температуре 0—5 °C постепенно приливают 64,6 г фенилизоцианата., перемешивают 1 ч при 12—15 °C, фильтруют,
21.4
промывают 2 раза по 25 мл абсол1бтного изопропаноЛа. Осадок влажного I суспендируют в 740 мл изопропанола, нагревают до кипения, фильтруют от хлористого натрия и ацетата натрия; раствор перемешивают при 18—20 °C в течение 5—6 ч, фильтруют, промывают изопропанолом (2 раза по 50 мл), затем 100 мл эфира и сушат на воздухе. Выход 121 г (69,2%).
ФЕНАЗЕПАМ
7-Бром-5- (о-хлорфенил) -1,2,-дигидро-ЗН, 1,4-бензодиазе-пин-2-он:
Мол. масса 349,62
Белый или белый с кремоватым оттенком кристаллический порошок, практически нерастворим в воде, трудно растворим в эфире, спирте, хлороформе, растворим в бензоле, толуоле, диоксане, диметилформамиде; т. пл. 225— 230°C; ВФС 42-671-77.
Феназепам — оригинальное транквилизирующее, снотворное и противосудорожное средство [26, 214].
Препарат применяют при лечении различных психопатологических и невротических состояний, сопровождающихся тревогой, страхом, эмоциональным напряжением, расстройством сна. Выраженная противосудорожная, седативная. и миорелаксирующая активность феназепама позволяет применять его у больных эпилепсией, при вегетативных кризах, болевых синдромах и психопатологических нарушениях в результате органических поражений ЦНС.
Выпускается в таблетках по 0,5 мг N. 20, цена 27 к.; по 1 мг N. 50, цена 52 к.; в амп. по 1 мл 3% раствора, N. 10, цена 3 р, 02 к.
Метод синтеза феназепама (I) разработан в Физико-химическом институте АН УССР по схеме [26]:
215
VII
VIII
Ацилированием п-броманилина (II) хлорангидридом о-хлорбензойноД кислоты (III) в присутствии катализатора— хлористого цинка — получают 2-(о-хлорбензоилами-но)-5-бром-2-хлорбензофенон (IV), который без выделения гидролизуют водной H2SO4 до 2-амино-5-бром-2-хлорбен-зофенона (V). Последний без выделения промежуточных веществ превращают в феназепам следующим образом: V ацилируют гидрохлоридом хлорангидрида аминоуксус-ной кислоты (VI) в хлороформе, образующийся 2-(ами-номет^илкарбониламино)-5-бром-2-хлорбензофенона гидрохлорид (VII) действием водного NH3 переводят в основание (VIII), которое термически циклизуют в феназепам. VI получают обработкой глицина PCI5 в хлороформе.
2-Амино-5-бром-2-хлорбензофенон (V). Смесь 276 г II и 672 г III нагревают 1 ч при 100—ПО °C, повышают температуру до 150—160 °C, прибавляют 264 г ZnCl2, нагревают 2 ч при 190—198 °C и охлаждают. К массе приливают 0,6 л 14% соляной кислоты, нагревают до 100 °C, водный слой отделяют, органический слой промывают путем кипячения с 0,3 л воды. Операцию промывки повторяют трижды. К остатку после промывки приливают 0,6 л 72% серной кислоты, нагревают 2 ч при 160 °C и выливают на смесь льда и воды. Осадок фильтруют, промывают водой, размешивают 1 Чг(\20% водным раствором NaOH, фильтруют, промывают водой до pH 7 и сушат на воздухе. Выход технического V 228 г с содержанием основного вещества 70%.
7-Бром-5- (о-хлор фенил) -1,2-дигидро-З Н, 1,4-бензодиазе-пин-2-он (феназепам) (I). Раствор 850 г V в 2 л хлороформа сушат азеотропной отгонкой смеси хлороформа и воды, прибавляют 496 г гидрохлорида хлорангидрида ами-216
ноуксусной кислоты (VI), кипятят 3 ч и охлаждают. К смеси приливают разбавленный водный раствор аммиака до pH 8, органический слой промывают водой и упаривают досуха. К остатку приливают 2 л толуола, нагревают до кипения, отгоняют азеотропную смесь толуола с водой, охлаждают и фильтруют. Технический I кристаллизуют (1:60) из толуола, фильтруют, осадок промывают толуолом, спиртом и сушат при 100—НО °C. Выход 250 г (26%).
КОНДЕНСИРОВАННЫЕ ТРИ-И ТЕТРАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ГЕТЕРОАТОМЫ
Исследования в ряду конденсированных три- и тетрациклических соединений, содержащих гетероатомы, были направлены в основном на поиск новых психотропных лекарственных препаратов.
Синтез в 1952 г. хлорпромазина (аминазина, ларгак-тила) (1а) и выявление , его высокой нейролептической активности [284, 296] произвели настоящую революцию в современной психиатрии и стимулировали широкое изучение 10-аминоалкилфенотиазиновых производных. В результате развернувшихся во всем мире исследований было получено несколько тысяч новых фенотиазиновых соединений, установлены определенные закономерности связи строения и биологической активности для этого ряда веществ, найдено большое число высокоэффективных .психотропных препаратов.
5 a) R=Cl; R'=(CH2)3N(C>y2 • HCI
6) R=H; R'=(CH2)2N(CH3)a- HCI e) R=H, R'^CHjIjNICj^j. HCI r) R=H; R'^CHjf^fCH^j- HCI
д) R=H; R'=CH2CH(CH3)N(CH3)2- HCI
e) R=Cl; R'^CHjjV NCHjCHjOH-HCl
ж) R=CF3; R'=(CH2)3N NCH3 2HCI
3) R=Cl; R'^CH^N NCH3- HOOCCH'CHCOOH
и) R=CF3 R =(CHj)3N NCH2 CHjOH • 2HCI
217
Разностороннее изучение 10-аминоалкилфенотиази-иов (I) показало их многогранную биологическую активность. Еще в 19*45 г. были обнаружены выраженные про-тивогистаминные свойства у Ю-(гР-диметиламинопропил)-фенотиазйна гидрохлорида (1д), получившего в практической медицине название фенерган (дипразин, пипольфен, прометазин). Противогистаминная активность выявлена и у других 10-аминоалкилфенотиазинов (I). В дальнейшем было показано, что соединения этого ряда обладают также адренолитическими, седативными, противорвотными и гипотермическими свойствами, потенцируют действие наркотиков, снотворных, болеутоляющих и местноанестезирующих веществ, оказывают холинолитическое и проти-воаритмическое действие. У незамещенных в бензольных ядрах 10-аминоацилфенотиазинов шведские исследователи обнаружили значительную местноанестезирующую активность, спазмолитические свойства и слабый нейролептический эффект. В ряду 10-аминоалкилфенотиазинов значительное внимание было уделено влиянию на нейролептическую активность характера заместителей в положении 2 трициклической системы и строения аминоалкильной группы в положении 10. Показано, что заместители в положении 2 обычно увеличивают нейролептический эффект, а в зависимости от характера 10-аминоалкильной группы все нейролептики фенотиазинового ряда могут быть разделены на три типа: 1) диалкиламиноалкилпроизводные, характеризующиеся наряду с антипсихотическим действием наличием тормозного компонента, способностью вызывать вялость, интеллектуальную и моторную заторможенность, вплоть до акинетического синдрома; 2) пиперази-ноалкильные производные, обладающие наряду с антипсихотическим действием также стимулирующим, активирующим компонентом; 3) пиперидиноалкильные производные, проявляющие менее сильное антипсихотическое действие, слабый гипноседативный эффект и редко вызывающие экстрапирамидные расстройства.
Обширная литература, посвященная химии, фармакологии и медицинскому применению 10-аминоалкилфенотиазинов, обобщена в ряде обзоров [32, 82, 281, 284, 314, 326].
Благодаря работам, проведенным во ВНИХФИ [184] и Институте фармакологии АМН СССР [232], наиболее ценные 10-аминоалкилфенотиазиновые препараты были воспроизведены в СССР. Разработаны методы получения этизина (16), динезина (1в), пропазина (1г), дипрази-на (1д), аминазина (1а), этапиразина (1е), трифтазина 218
(1ж), метеразина (1з), фторфеназнна (1и) и для большинства из них организовано промышленное производство [2Н].
Новым этапом в исследовании фенотиазиновых соединений явилось изучение 2-замещенных 10-аминоацилпроиз-водных этого ряда (II) и обнаружение у них антианги-нальных свойств.
COR и
a)R=Cl: R'=CH2CH2N(C2H5)2-HCl
Синтезированные первоначально с целью поиска нейролептических и противогистаминных средств [78], эти вещества оказались малоинтересными в указанных направлениях, но показали значительную холино- и адрено-литическую активность, проявили выраженное антианги-нальное и антиаритмическое действие [181]. Первым лекарственным препаратом этого ряда, получившим практическое применение, явился хлорацизин (Па) [63, 211]. Дальнейшие исследования [61, 62, 82, 232] позволили выявить определенные закономерности связи между структурой и биологическими свойствами для 10-аминоацил-фенотиазинов. Было установлено, что наиболее активны 10-аминопропионильные производные, а удлинение и укорочение ацильной цепи снижает активность. Введение вместо диэтиламинной группы диметиламинного или пиперазинового остатков уменьшает, а морфолинового или 1,4-диазобицикло (4,3,0) нонанового в ряде случаев существенно усиливает антиангинальные н антиаритмические свойства. Переход к 1,4-диазабицикло (4,3,0) ундекано-ацильному производному приводит к появлению угнетающего действия на миокард [170]. Значительное влияние на антиангинальные свойства оказывает характер заместителя в положении 2 фенотиазинового ядра. Атом хлора или остаток этилового эфира карбаминовой кислоты, введенные в положение 2, увеличивают активность. Трифторметильная группа обычно, напротив, подавляет антиангинальные свойства, но усиливает психотропную активность. Однако при этом вещества характеризуются уже антиде-прессивным действием.
В результате этих исследований хлорацизин (Па) в медицинской практике был заменен более активным корона-
219
рорасширяющим средством — нонахлазином (см. с. 226) [92, 93, 170], в качестве антиаритмического препарата был внедрен в медицину этмозин (см. с. 222) [60, 94, 95], в качестве антидепрессанта — фторацизин (см. с. 229) [62].
Новым шагом в развитии химии и фармакологии трициклических нейротропных средств явилось создание их азааналогов. Принцип азааналогии в последние годы широко используется в поисках лекарственных препаратов среди производных азапуринов и азапиримидинов [283, 284, 316, 343], азастероидов [76, 258], азаиндолов [262, 272], азафенотиазинов и азафеноксазинов [287, 305] и др.
Осуществленные во ВНИХФИ исследования привели к созданию новой группы соединений — производных неизвестной ранее гетероциклической системы 3,4-диазафе-ноксазина (III) [48, 49, 182, 183] и изучению их фармакологических свойств [149, 150].
Л?
Диазафеноксазиновый аналог аминазина (IV) оказался лишенным нейролептической активности, но обладал элементами антидепрессивного действия. Наиболее выраженный антидепрессивный эффект проявился у 10-метил-3,4-диазафеноксазинов (III), замещенных в положении 2 аминными (особенно метилпиперазиновым) и диалкил-аминоалкоксильными (особенно диэтиламиноэтоксильным) остатками. Переход к 2-хлор- и 2-окси-производным, введение ацил-, алкокси-, сульфамидо-, нитро-, амино- и других заместителей в бензольную часть молекулы, расширение оксазинового цикла (V) или включение вместо 3,4-диазафеноксазинового ядра 2,3-диазафеноксазиново-го (VI) во всех случаях сопровождается ослаблением биологической активности. Наиболее эффективным антидепрессантом в этоМ\ ряду соединений, получившим практическое применение, явился азафен (см. с. 231) [182, 183, 148, 200].
220
Проводимые й течение ряда лет в Институте фармакологии АМН СССР исследования тетра-(VII) и гекса-(VIII) гидропроизводиых у-карболина [110, 111, 118, 119] позволили выявить в этом классе триклических соединений вещества, угнетающие ЦНС, для которых характерно сочетание нейролептической и антидепрессивной активности:
Из 26 веществ с различными заместителями R1—R5 и степенью насыщенности индольного фрагмента молекулы [232] наиболее эффективным оказался нейролептический препарат карбидин (см. с. 234) [18, ПО].
Осуществленный во ВНИХФИ переход от трициклических конденсированных индольных систем типа карболи-нов к тетрациклическим соединениям дал принципиально новые результаты.
Производные конденсированной системы пиразинокар-базола (IX) [254] обладали в ряде случаев антисеротони-новой активностью [10], а некоторые соединения проявили свойства антидепрессантов [8, 144]. Спектр терапевтического действия этих соединений оказался достаточно широким: по проявлению и механизму вызываемые ими эффекты имели сходство одновременно как с действием трициклических антидепрессантов, так и антидепрессантов— ингибиторов моноаминоксидазы [8, 142, 150]. Наиболее активным препаратом этой группы, получившим практическое применение, явился гаиразидол (см. с. 238). В отличие от антидепрессантов типа имизина и амитриптилина, оказывающих на фоне антидепрессивного эффекта
221
одностороннее стимулирующее или седативное действие, пиразидол сочетает с тимолептической активностью регулирующее влияние на ЦНС: стимулирующее при заторможенных депрессиях или седативное при тревожных. Препарат действует на оба основных пути инактивации биогенных моноаминов: тормозит их нейрональный захват и обратимо угнетает окислительное дезаминирование [7, 151].
Раскрытие в молекуле пиразидола кольца D приводит к потере активности в отношении нейронального захвата моноаминов при сохранении действия на окислительное дезаминирование; раскрытие кольца С при сохранении кольца D сопровождается практически полной утратой психотропной активности. Удаление метильной группы (R1) в положении 8 тетрациклической системы, а также введение вместо нее хлора или брома снижает фармакологический эффект препарата [7, 57]; уменьшение активности наблюдается также при введении к азоту в положе- < нии 2 алкильных групп (R4) и при введении атомов хлора в положения 6 или 7 (R2 и R) [57].
Производное другой тетрациклической системы — пи-разино-р-карболина (X) — лекарственный препарат инка-зан (см. с. 243) [41] явился антидепрессантом со стимулирующим влиянием на ЦНС. Инказан в отличие от трициклических антидепрессантов имизина и амитриптилина не показывает холинолитических свойств, не усиливает действия гексенала и анальгетиков [149, 150].
Замена в инказане 8-метокси группы (R1) на водород, удлинение заместителя в положении 2 (R2), расширение или сужение кольца С сопровождается ослаблением психотропного, действия, а размыкание цикла D приводит к полной потере антидепрессивной активности.
этмозин
Этилового эфира 10-(3-морфолилпропионил)фенотиа-зинкарбаминовой-2-кислоты гидрохлорид:
NHCOOC2H5
<^ОСН2СН2М\ О . HCI
c22h2Sn3o4s i:ci •
Мол. масса 463,95
222
Белый или белый с кремовым оттенком кристаллический порошок, медленно растворим в воде (pH 2,5% раствора 3,3—4,2), трудно — в спирте, практически нерастворим в эфире, на свету темнеет; ФС 42-1109-77.
Этмозин — оригинальный антиарит'мический препарат [60, 94, 95], по характеру действия близкий к хинидину, оказывает умеренный коронарорасширяющий, спазмолитический и м-холинолитический эффект [61].
Применяют этмозин при экстрасистолиях, приступах мерцательной предсердной и пароксизмальной тахикардии. Особенно эффективен при аритмиях, вызываемых передозировкой сердечных гликозидов. Выпускается в виде таблеток, покрытых оболочкой, по 0,025 г N. 50 (цена 2 р. 52 к.) и ампул по 2 мл 2,5% N. 10 (цена 2 р. 02 к.).
Метод синтеза этМозина разработан в Институте фармакологии АМН СССР [60] по схеме:
IV V
УШ
Полученный ацетилированием м-нитроанилина (I) кипящим уксусным ангидридом м-нитроацетанилид (II) путем реакции с бромбензолом в присутствии однобромистой меди и медного порошка превращают в 3-нитродифениламин (III) [80]. Нитрогруппу в III восстанавливают гид-разингидратом в присутствии никелевого катализатора в спирте до аминогруппы [64]. Другие методы восстановления: оловом в соляной кислоте, каталитически в присутствии никеля или железом в воде — дали худшие результаты. Полученный 3-аминодифениламин (IV) обработкой
223
хлоругольным эфиром превращают в этиловый эфир ди-фениламийокарбамииовой-3-кислоты (V) [60, 79, 80].
Тионированием V серой в толуоле в присутствии йода синтезируют этиловый эфир фенотиазинкарбаминовой-2-кислоты (VI) [60, 79, 80], который ацилированием (3-хлор-пропионилхлоридом переводят в этиловый эфир 10-(3-хлор-пропионил)фенотиазинкарбаминовой-2-кислоты (VII) и дальнейшей обработкой морфолином — в этиловый эфир Ю-(З-морфолилпропионил)' фенотиазинкарбаминовой-2-ки-слоты, выделяемой в виде гидрохлорида (этмозина) (VIII).
м-Нитроацетанилид (II). 276 г м-нитроанилина (I) и 306 г уксусного ангидрида кипятят при перемешивании 3 ч, охлаждают, не выключая мешалки, до 5—10 °C. Выпавший в осадок II отфильтровывают, промывают 1 л бензола, высушивают при 60—80°С. Получают322г (89%) II.
3-Нитродифениламин (III). Смесь 660 г II, 1149 г перегнанного бромбензола, 363 г прокаленного мелко растертого карбоната калия, 7,2 г одиобромистой меди и 7,5 г медного порошка нагревают при хорошем перемешивании 24 ч сначала при 185—195 °C, а к концу реакции при 200—210 °C. Количество отгоняющегося с водой бромбензола замеряют и соответствующий объем этого продукта добавляют в реакционную массу. После окончания реакции смесь охлаждают, приливают 600 мл воды и избыток бромбензола отгоняют с водяным паром. Оставшийся маслянистый продукт промывают для отделения неорганических солей горячей водой (2 раза по 450 мл), отделя-. ют от водного слоя, растворяют в 5820 мл этилового спирта, отфильтровывают от медного порошка, разбавляют 3100 мл 8% водного раствора едкого натра, кипятят 2 ч и оставляют для кристаллизации. Красный кристаллический осадок отфильтровывают, промывают нагретой до 20—25°C водой до нейтральной реакции и высушивают. Получают 585 г (75%) III.
3-Аминодифениламин (IV). К раствору 321 г III в 1,4 л этилового спирта прибавляют 158 г влажного никелевого катализатора и в течение 2’/г ч при перемешивании по каплям (наблюдается вспенивание) приливают смесь 229 г гидразингидрата и 200 мл этилового спирта с такой скоростью, чтобы температура не была выше 40.°С. Конец процесса контролируют по отсутствию IV на ТСХ. Затем для разложения избытка гидразина массу кипятят при перемешивании 1 ч, охлаждают до 20 °C, никелевый катализатор отфильтровывают; к фильтрату медленно в течение 2 ч приливают 5 л холодной воды. Выпавший оса-224
док IV отфильтровывают, промывают водой до нейтральной реакции, сушат при температуре не выше 50°C. Выход 248 г (90%).
Этиловый эфир дифениламинокарбаминовой-3-кисло-ты (V). К охлажденному до 5—7 °C раствору 184,23 г IV в 1 л этилового- спирта прикапывают при перемешивании и температуре не выше 4-10 °C 75,75 г этилового эфира хлоругольной кислоты. Затем при той же температуре одновременно прибавляют раствор 93 г карбоната натрия в 450 мл воды и 81,55 г этилового эфира хлоругольной кислоты. Реакционную массу перемешивают 1 ч при 18—20°C, приливают 600 мл воды и перемешивают еще 30 мин. Осадок V отфильтровывают, промывают 2 раза по 500 мл воды (до нейтральной реакции), отжимают и без высушивания перекристаллизовывают из смеси 1500 мл этилового .спирта и 900 мл воды с добавкой 25. г активированного угля. При охлаждении отфильтрованного горячего раствора выпадает смолистый осадок, с которого теплый спиртовой раствор сливают декантацией и оставляют для выделения кристаллического V при перемешивании и температуре 5—10 °C. Получают 150 г V. Из смолистого осадка (60 г) путем повторной кристаллизации по описанному выше методу выделяют дополнительно 42 г V. Общий выход 190—200 г (75—80%).
Этиловый эфир фенотиазинкарбаминовой-2-кисло-ты (VI). Смесь 400 г V, 116 г серы, 6,8 йода и 800 мл толуола кипятят при хорошем перемешивании 10 ч. Затем приливают 400 мл толуола и для растворения осадка кипятят с перемешиванием 30 мин, после чего массу охлаждают до 3—5 °C. Выпавший осадок VI отфильтровывают, промывают 150 мл толуола и высушивают при 80 °C. Получают 344,04 г (77%) VI.
Этиловый эфир 10-(3-хлорпропионил)фенотиазинкарба-миновой-2-кислоты (VII). Смесь 350 г VI, 185,5 г р-хлор-пропионилхлорида и 1,75 л высушенного над хлористым кальцием толуола кипятят 4 ч. Осветляют кипячением 15 мин с 35 г активированного угля, фильтруют, охлаждают при перемешивании до 3—5 °C. Осадок VII отфильтровывают, промывают 550 мл охлажденного до 3—5 °C толуола, сушат при 60—80°C. Выход 348,3 г (75,6%).
Этилового эфира 10-(3-морфолилпропионил)фенотиазин-карбаминовой-2-кислоты гидрохлорид (этмозин) (VIII). К нагретому до 30—35°C раствору 420 г VII в 2 л сухого толуола при перемешивании в течение 30 мин прибавляют 214 г морфолина. Массу кипятят 30 мин и после охлаж-
15 Заказ № 4304 2 25
дения до комнатной температуры сразу (при стоянии из реакционной массы может выпасть в осадок также основание этмозииа; в этом случае массу разогревают и фильтруют теплой) отфильтровывают осадок гидрохлорида морфолина, который промывают иа фильтре 200 мл толуола, объединенные толуольные растворы промывают водой до нейтральной реакции и экстрагируют 6% соляной кислотой (2 раза по 460 мл и 1 раз 300 мл). Объединенные кислые растворы разбавляют 800 мл воды, осветляют 25 г активированного угля (50—55°C, 15 мин), фильтруют и подщелачивают 10% раствором едкого натра до pH 11—12. Масст охлаждают до 12—15°С. Осадок основания этмозииа отфильтровывают, промывают водой до отсутствия щелочной реакции и растворяют в 2 л 70% этилового спирта. Спиртовой раствор осветляют кипячением 15 мин с 20 г активированного угля, фильтруют, охлаждают до 5—7 °C. Выпавший осадок отфильтровывают, сушат при 60—80°C и растворяют при нагревании (50— 70°C) в 3,8 л этилацетата. Раствор охлаждают до 20°C и при перемешивании приливают 20—25% спиртовой раствор хлороводорода (~ 160—200 мл) до pH 2—3. Перемешивают еще 30 мин, контролируют pH и оставляют кристаллизоваться при 5—7 °C на 3—4 ч. Осадок отфильтровывают, промывают 400 мл этилацетата, высушивают при 50—60°C. Получают 372 г VIII, который перекристаллизовывают из смеси 560 мл этилацетата и 1400 мл абсолютного этилового спирта с добавкой 25 г активированного угля. После отделения угля массу оставляют для кристаллизации на 8 ч при 5—10 °C. Продукт отфильтровывают, промывают 300 мл этилацетата и высушивают при 50—70°C. Получают 226 г VIII. Из маточного раствора после упаривания до '/з первоначального объема выделяют еще 62 г VIII. Выход 288 г (58%).
НОНАХЛАЗИН
2-Хлор-10- [3- (1,4-диазабицикло (4,3,0) нонанил-4) пропионил] фенотиазинЙ'’,Нигидрохлорид:
226
Кремоватый или желтовато-зеленоватый кристаллический порошок, растворим в воде, мало растворим в спирте и хлороформе, практически нерастворим в ацетоне и эфире; т. разл. 219—228 °C; ВФС 42-374-74.
Нонахлазин — оригинальное антиангинальное средство [92, 93, 170], обладающее способностью улучшать кровоснабжение сердца за счет увеличения коронарного кровотока, повышает кислородный резерв миокарда. Сократительная способность миокарда при этом увеличивается без существенных изменений сердечного выброса и работы сердца. Препарат влияет на адренергические структуры миокарда, обладает общей спазмолитической активностью, антиаритмическим действием, противовоспалительными свойствами.
Применяют нонахлазин при ишемической болезни сердца с целью предупреждения приступов, при постинфаркт-иой стенокардии, в том числе при резистентности к другим коронарорасширяющим препаратам.
Выпускается во флаконах по 25 мл 1,5% (цена 1 р. 02 к.) и в таблетках по 0,03 г N. 100. Цена 5 р. 60 к.
Метод синтеза нонахлазина разработан в Институте фармакологии АМН СССР [92] по схеме:
Обработкой б-хлорвалериановой кислоты (I) тиоиил-хлоридом получают соответствующий хлорангидрид, который бромируют и взаимодействием с этиловым спиртом переводят в этиловый эфир а-бром-б-хлорвалериановой кислоты (II). Реакцией II с этилендиамином в присутствии каталитических количеств йодистого калия синтезируют 5-оксо-1,4-диазабицикло(4,3,0)октан (III), который восстанавливают алюмогидридом лития до 1,4-диазаби-цикло (4,3,0) октана (IV). Взаимодействие IV с 2-хлор-10- (р-хлорпропионил) фенотиазином, синтезируемым по методу, описанному в получении хлорацизииа [211], проводят в присутствии триэтиламина и продукт реакции переводят в 2-хлор-10-[3-(1,4-диазабицикло (4,3,0) нонанил-4)-
15*
227
пропионил!] фенотиазина дигидрохлорид (нонахла-зии) (VI). \
Этиловый эфир а-бром-б-хлорвалериановой кислоты (II). К раствору 1,09 кг перегнанной б-хлорвалериано- I вой кислоты (I) в 500 мл хлороформа приливают 1,19 кг тионилхлорида с такой скоростью, чтобы температура была не выше 25—30°C. Затем массу нагревают 3 ч при 80—85 °C с перемешиванием. Избыток тионилхлорида и хлороформ отгоняют в вакууме до содержания в остатке не более 2% тионилхлорида. К полученной массе, охлаж- ! денной до 20 °C, медленно приливают 1,55. кг брома, нагревают в течение 1 ч до 40—45 °C и дают выдержку при ’ перемешивании 1 ч. Дальнейшее повышение температуры проводят постепенно, давая при нагреве на каждые 10 °C выдержку по 30 мин. По достижении температуры 85 °C реакционную массу выдерживают в течение 3’/2 ч, затем охлаждают до 30 °C, избыток брома отгоняют в вакууме и к охлажденному до 20 °C остатку постепенно приливают ; 1,4 л абсолютного этилового спирта с такой скоростью, |
чтобы температура была не выше 65 °C. Реакционную ]
смесь медленно в течение 3—З’/г ч нагревают до 82 °C и дают выдержку 2’/г ч, после чего охлаждают до 20 °C и приливают 2 л воды. Нижний слой эфира II отделяют, . водный раствор дополнительно экстрагируют хлороформом (1 л-)-600 мл). Объединенные с основной порцией II | хлороформные экстракты промывают 1 л 10% раствора ! гидрокарбоната, натрия, сушат хлористым кальцием, упа- ! ривают в вакууме. Продукт II перегоняют, собирая фракцию с т. кип. 115—125°С (10 мм рт. ст.). Выход 1,5 кг (77,3%).
5-Оксо-1,4-диазабицикло(4,3,0)нонан (III). Смесь 755 г II, 2,5 л этилового спирта, 600 мл 70% этилендиамина и 3 г йодистого калия медленно в течение 3 ч нагревают до 67 °C, затем в течение 1 ч до 83 °C и выдерживают 15 ч при этой температуре. Затем массу охлаждают до 60 °C, прибавляют 60 г активированного угля, выдерживают 30 мин, охлаждают до 20°C и отфильтровывают от угля и гидробромида этилендиамина, которые промывают 920 мл абсолютного этилового спирта. Фильтрат упаривают в вакууме. Остаток для удаления следов гидробромида этилендиамина растворяют в 250 мл воды и III экстрагируют хлороформом (3 раза по 1 л и 3 раза по 650 мл). Хлороформные растворы фильтруют через уголь (150 г) и упаривают в вакууме. Остаток растворяют в 900 мл сухого бензола с содержанием влаги не более 0,05%. Бен
228
зольный раствор анализируют на содержание основного вещества, влаги и без дополнительной обработки используют на следующей стадии.
1,4-Диазабицикло(4,3,0)нонан (IV). К смеси 400 г алюмогидрида лития и 10 л абсолютного эфира при перемешивании приливают полученный на предыдущей стадии бензольный раствор III с такой скоростью, чтобы не прекращалось кипение реакционной массы. Затем дают выдержку 15 ч при 40°С. Образовавшийся комплекс и избыток алюмогидрида лития разлагают прибавлением последовательно 500 мл воды, 300 мл 20% раствора едкого натра; 1,1 л воды и 1,2 л 40% раствора едкого натра. Вещество IV извлекают из водного слоя кипящим эфиром (3 раза по 9 л) и после отгонки растворителей перегоняют в вакууме при 75—ПО °C (10 мм рт. ст.). Выход 510 г (29,6%).
2-Хлор-10-[3-(1,4-диазабицикло(4,3,0)нонанил-4)пропио-нил]фенотиазина дигидрохлорид (нонахлазин) (VI). Смесь 19,4 л сухого толуола, 850 г 1,4-диазабицикло(4,3,0) октана (IV), 640 г триэтиламина с содержанием влаги не более 0,2% и 2,1 кг 2-хлор-10-(р-хлорпропионил)феиотиа-зина (V) перемешивают З'Д ч при 78 °C, осветляют 600 г активированного угля и после охлаждения до 20 °C фильтруют от угля и осадка гидрохлорида триэтиламина. К толуольному фильтрату при 10°C и перемешивании прибавляют 2,8 л 15% раствора хлороводорода в абсолютном этиловом спирте до pH 1, после чего массу перемешивают еще 10 ч при 10 °C. Осадок VI отфильтровывают, промывают 3 л толуола, 3 л эфира и перекристаллизовывают из 15 л абсолютного этилового спирта. Перекристаллизованный продукт промывают 3 л эфира, затем 3 л ацетона и высушивают при 55°C. Получают 1,85 кг VI. Выход 58,6%, считая на IV, и 58,7%, считая на V.
ФТОРАЦИЗИН
2-Трифтормет'ил-10-(З-диэУиламинопропионил) фенотиазина гидрохлорид:
ioCH2CH2N(C2H5)2- HCI
С ол Н Of F-hLOS • HCI .Df\
20 21 3 2 Мол. масса 430,92
229
Белый или белый со слабым кремоватым оттенком кристаллический порошок, темнеющий на свету, без запаха, трудно растворим в воде (pH 1,25% раствора 3,8—4,8), легко растворим в спирте, нерастворим в эфире; т. пл. 163— 166°C (в интервале 2°C); ФС 42-1080-76.
Фторацизин— оригинальный антидепрессивный препарат [62], имеющий наряду с этим седативное действие, обладающий сильной центральной и периферической холинолитической активностью.
Применяют фторацизин для лечения тревожно-депрессивных состояний, при шизофрении с выраженными аффективными нарушениями, при реактивных и невротических состояниях, сопровождающихся депрессией, а также при депрессии, обусловленной применением нейролептических препаратов.
Выпускается в' таблетках по 0,025 г N. 50 (цена 1 р. 02 к.) и в ампулах по 1 мл 1,25% раствора N. 10 (цена 32 к.).
Получение фторацизина разработано в Институте фармакологии АМН СССР [62] по схеме:
2-Трифторметилфенотиазин (I), являющийся одновременно полупродуктом в производстве трифтазина (синтез см. [211]), ацилируют p-хлорпропионилхлоридом (II), используемым. также в производствах хлорацизина и этмо-зина (синтез см« ^[211]). Полученный 2-трифторметил-10-(3-хлорпропионил) фенотиазин (III) обрабатывают диэтиламином в среде сухого толуола и получают 2-три-фторметил-Ю-(З-диэтиламинопропионил) фенотиазина гидрохлорид (фторацизин) (IV).
230
2-Трифторметил-10-(3-хлорпропионил) фенотиазин (III). Смесь 240,3 г I, 148,5 г II и 1,5 л безводного толуола кипятят 3 ч, толуол и избыток II отгоняют при остаточном давлении 60—80 мм рт. ст. и температуре не выше 75 °C (при более высокой температуре возможно осмоление). Остаток растворяют в смеси 1,89 л изопропилового спирта и 0,63 л воды при нагревании, добавляют 30 г угля. Горячий раствор фильтруют и охлаждают до —5 °C. Выпавший осадок отфильтровывают, промывают 100 мл охлажденного до 4—5 °C 80% изопропилового спирта и высушивают при 40—50 °C. Получают 272 г III с т. пл. 79— 81 °C. При упаривании маточного раствора до */з первоначального объема, охлаждении до —5°C, фильтрации осадка и перекристаллизации его из 70% изопропилового спирта дополнительно получают 17,2 г III с т. пл. 79— 81 °C. Выход III 289,2 г (90,3%).
2-Трифторметил-10-(3-диэтиламинопропионил)фенотиа-зина гидрохлорид (фторацизин) (IV). К смеси 188 г III с 1,65 л толуола прибавляют 82,5 г диэтиламина, реакционную массу кипятят 3 ч и охлаждают до 20—25 °C. Выпавший осадок гидрохлорида диэтиламина отфильтровывают, промывают 200 мл толуола. Объединенные толуольные растворы промывают водой до pH промывных вод 8—9, после чего экстрагируют 1,8 л 5% соляной кислоты (1,2 л+0,6 л). Объединенные кислые растворы подщелачивают 20% раствором едкого натра до pH 11—12 и выделившееся основание экстрагируют толуолом (0,6 л, 0,4 л и 0,2 л). К высушенным сульфатом магния толуольным экстрактам приливают раствор хлороводорода в спирте до pH 3—3,5 и массу охлаждают при перемешивании до +5 °C. Выпавший осадок отфильтровывают, получают 156 г технического IV. При упаривании маточного раствора до ’А первоначального объема выделяют еще 52 г технического IV. Весь полученный продукт растворяют в 620 мл кипящего абсолютного изопропилового спирта с добавкой 25 г активированного угля, фильтруют. Фильтрат охлаждают до +5 °C, дают выдержку 5 ч. Выпавший в осадок IV отделяют, промывают 100 мл охлажденного до +5 °C абсолютного изопропилового спирта и высушивают при 60—70°C. Выход IV 188 г (83%).
АЗАФЕН
2-(4-Метил-1-пиперазинил)-10-метил-3,4-диазафенокса-зина дигидрохлорид, моногидрат:
231
is N50.2HC(-H5O
Мол. масса 388,30 '
Кристаллический порошок желтовато-зеленоватого
цвета, горько-кислого вкуса, легко растворим в воде (pH 1,25% раствора 2,5—3), практически нерастворим в спирте, эфире и хлороформе; т. пл. 310—315 °C с разл., в заплавленном капилляре, в интервале 2 °C; не является характерной. При нагревании до 110°С препарат теряет кристаллизационную воду, но на воздухе притягивает ее снова; ФС 42-994-75. ,
Азафен — оригинальный препарат антидепрессивного действия, у которого тимолептический эффект сочетается с седативным [148, 152, 182, 183, 200]. Препарат улучшает функциональное состояние ЦНС, нормализует сон, ( не оказывает ингибирующего влияния на МАО и в отличие от имизина не обладает холинолитической активностью, характеризуется хорошей переносимостью.
Применяют азафен при нерезко выраженных депрессивных состояниях различного характера: депрессивной стадии маниакально-депрессивного психоза, инволюционной меланхолии, депрессиях органического генеза, сома-тогенно обусловленных депрессиях, депрессивных состояниях, развивающихся при длительном лечении нейролеп- j тическими препаратами, при астенодепрессивных состояниях невротического, .характера, а также в качестве «долечивающего» средства после лечения другими препаратами.
Выпускается в таблетках по 0,025 г N. 250. Цена 3 р. 1 02 к.
Метод синтеза азафена разработан во ВНИХФИ [50] по схеме:
232
Нагреванием о-аминофенола (I) с мочевиной (II) в присутствии соляной кислоты получают бензоксазолон (III). В отсутствие соляной кислоты для осуществления этой реакции необходима более высокая температура — сплавление при 180—200°C. Метилированием III диметилсульфатом в щелочной среде получают N-метилбензокса-золон (IV), который путем конденсации с 3,4,6-трихлорпи-ридазином (V) превращают в 2-хлор-10-метил-3,4,-диаза-феноксазин (VI). Синтез через 2-хлор-3,4-диазафеноксазин с последующим метилированием по азоту дает менее чистый продукт за счет неоднозначности реакции N-метили-рования. При взаимодействии 2-хлор-10-метил-3,4-диаза-феноксазина (VI) с N-метилпиперазина дигидрохлоридом (VII) получают азафен (VIII). Использование основания N-метилпиперазина дает худшие результаты.
Бензоксазолон (III). Смесь 1,637 кг о-аминофенола (I), 1,08 кг мочевины (II) и 1,875 л 36% соляной кислоты нагревают 1 ч при 80—90 °C, повышают температуру массы до кипения (118—120°С) и кипятят 3 ч, прибавляют в течение 30—40 мин 2 л воды, затем для растворения примесей основного характера — 375 мл 36% соляной кислоты и еще 1,75 л воды. Реакционную массу медленно охлаждают до 20 °C и дают выдержку 2 ч. Осадок отфильтровывают и промывают водой до pH 7,0 в промывной воде. Получают 2,451 кг пасты III с содержанием влаги около 31%, которую без высушивания используют на следующей стадии. Выход в пересчете на 100% вещество III 83% (на I).
N-Метилбензоксазолон (IV). К раствору, содержащему 0,93 л 42% едкого натра и 7,85 л воды, прибавляют 2,532 кг пасты III. Перемешивают 1 ч до полного превращения III в натриевое производное. К полученному раствору при 20—25 °C в течение 1 ‘А ч приливают 1,34 л диметилсульфата. Перемешивают при той же температуре еще 1 ч, выделившийся осадок отфильтровывают, промывают водой до нейтральной реакции промывных вод и сушат при 35—45 °C. Получают 1,625 кг IV с содержанием вещества не ниже 98%; т. пл. 83—85°C. Выход 83%.
2-Хлор-10-метил-3,4-диазафеноксазин (VI). К раствору 250 г 96% едкого натра в 2 л этилового спирта прибавляют 304 г 98% IV и кипятят 45 мин. Происходит омыление N-метилбензоксазолона (IV) с образованием натриевого производного N-метил-б-аминофенола, к кипящему раствору которого приливают при перемешивании в течение 30—40 мин 1,8 л спиртового раствора V, содержа
233
щего 367 г V. (Соединение V обладает кожно-нарывным действием и его в процессе производства не выделяют. Описание одного из способов получения V см. [50].) Реакционную массу кипятят 5 ч, отгоняют 1,8 л спирта; остаток охлаждают до 5 °C и дают выдержку 3 ч. Осадок VI отфильтровывают, промывают 300 мл этилового спирта, смешивают с 7 л воды и прибавляют соляную кислоту до pH 6,0—7,0, перемешивают 4 ч, контролируют pH. Осадок VI отфильтровывают, промывают 3 л воды и сушат до содержания влаги не более 1%. Получают 404 г VI с содержанием основного вещества не ниже 90%. Выход 78% на IV или V.
2-(4-Метил-1-пиперазинил)-10-метил-3,4-диазафеноксазина дигидрохлорид (азафен) (VIII); Из смеси 4 л цикло-гексананола и 1,112 л 48% водного раствора N-метилпи-перазина дигидрохлорида (VII) азеотропно отгоняют около 0,6 л воды (при этом температура в массе повышается от 105°C до 140°C). Охлаждают до 80°C и прибавляют 537 г VI, кипятят 5 ч, охлаждают до 70 °C и смесь дигидрохлоридов VII и VIII растворяют путем прибавления 7,5 л воды. Водйыи слой отделяют от циклогексанольного, обесцвечивают 60 г угля, охлаждают до 10°C и приливают 677 мл 42% раствора едкого натра до pH 12. После выдержки 9 ч при 5°C выделившееся основание VIII отфильтровывают, промывают --'ll л воды до pH 7—7,5 в промывной воде и перекристаллизовывают из 2 л ацетона (с учетом влаги в пасте ацетон получается 50%) с 56 г угля. Полученные 470 г основания VIII растворяют в 3,525 л этилового спирта и при перемешивании и температуре 40—45 °C приливают 271 мл 36% соляной кислоты. Реакционную массу охлаждают до 5 °C и дают выдержку 5 ч. Осадок отфильтровывают, промывают 1,1 л охлажденного до 5 °C этилового спирта до pH в промывном спирте 6,5—7,0 и высушивают при 80 °C. Получают 564 г VIII. Выход 70%.
КАРБИДИН
3,6-Диметил-1,2,3,4,4а,9а-гексагидро-у-карболина дигидрохлорид:
C,3H18N2- 2НС1
Мол. масса 275,22
234
Белый или белый с кремовым оттенком кристаллический порошок, темнеющий на свету и от влаги, легко растворим в воде (pH 2% раствора 2,0—2,5), очень мало — в спирте, практически нерастворим в хлороформе и эфире; т. разл. 268—274°С (начало вспенивания); ФС 42-1146-78.
Карбидин — оригинальный психотропный препарат [18, ПО], обладающий нейролептической, антипсихотической активностью и оказывающий одновременно антидепрессив-ное действие. Характеризуется центральным адренолити-ческим эффектом, усиливает действие наркотиков и анальгетиков [19, 191].
Применяют карбидин при периодической депрессивнопараноидной шизофрении, при циркулярной шизофрении с депрессивно-параноидной структурой приступов, при других формах шизофрении с преобладанием депрессивнобредовых расстройств, а также при простой шизофрении с аффективными расстройствами и при алкогольных психозах. Выпускается в таблетках, покрытых оболочкой, по 0,025 г N. 50 (цена 3 р. 02 к.) и в ампулах по 2 мл 1,25 % раствора N. 10 (цена 1 р. 52 к.).
Метод синтеза карбидина [232] разработан Институтом фармакологии АМН СССР совместно с Новокузнецким научно-исследовательским химико-фармацевтическим институтом и Новокузнецким производственным химикофармацевтическим объединением «Органика» по схеме:
Путем циклизации по Дикману ди-(0-карбметокси-этил)метиламина (I) с этилатом натрия получают 1-ме-тилпиперидон-4 (II). Параллельно диазотированием п-то-луидина (III) нитритом натрия и восстановлением образующейся соли диазония сульфитом натрия с последующей обработкой соляной кислотой синтезируют п-толил-гидразина гидрохлорид (IV). Возникающий при взаимо-
235
действии II с IV п-толилгидразон 1-метилпиперидона-4 циклизуют по Фишеру [107] с помощью спиртового раствора хлороводорода и образующийся 3,6-диметил-1,2,3,4-тетрагидро-у-карболин (V) восстанавливают цинком в соляной кислоте до 3,6-диметил-1,2,3,4,4а,9а-гексагидро-у-карболина дигидрохлорида (карбидина) (VI).
1-Метилпиперидон-4 (II). От смеси 1,5 л 25% спиртового раствора этилата натрия и 2,23 л ксилола отгоняют в виде азеотропа спирт до температуры в реакционной массе 95 °C, после чего в течение 1 ч при 80—90 °C прибавляют 852 г/IV одновременной азеотропной отгонкой смеси спиртов и ксилола. Продолжая отгонку, температуру в массе повышают до 100—103 °C и дают выдержку 30 мин. Затем массу охлаждают до 30 °C и приливают при перемешивании 3,46 л 18% соляной кислоты. Через 30 мин кислый водный слой отделяют и кипятят 3 ч. После окончания процесса декарбоксилирования массу охлаждают до 10—20 °C; подщелачивают 50% раствором едкого кали до pH 9—10 и экстрагируют хлороформом (4 раза по 1,2 л). Хлороформ упаривают, остаток перегоняют в вакууме. Собирают фракцию с т. кип. 53—55°C (9 мм). Получают 190 г (38%, считая на I) II с содержанием основного вещества не менее 93%. Продукт темнеет на свету и его хранят в темном холодном месте не более 7 дней.
п-Толилгидразина гидрохлорид (IV). К 519 мл охлажденной до 0—(+5°C) 36% соляной кислоты прибавляют 184 г III, перемешивают 15 мин, приливают 1,31 л воды, охлаждают до (—5 °C) и добавляют при этой температуре и перемешивании 16,8% водный раствор нитрата натрия до положительной реакции на йодкрахмальную бумагу. После этого массу выливают в раствор 1,24 кг сульфита натрия в 1,26 л воды при перемешивании, поддерживая температуру массы 78—83 °C, с такой скоростью, чтобы 2/з всего количества прибавить за первые 15 мин (при более медленной' 'прибавлении возможно образование азокраски). Дают выдержку 30 мин, контролируют, чтобы pH среды был 7—8, и приливают в течение 15—20 мин 66 мл конц. соляной кислоты, поддерживая температуру реакционной массы 80—85 °C. После 30 мин выдержки проверяют, чтобы pH среды был 1, добавляют 18 г угля, нагревают до кипения и фильтруют. Фильтрат охлаждают 2 ч при 10—15 °C с перемешиванием. Осадок отфильтровывают. Получают 375 г (52,1%) пасты IV с содержанием основного вещества не менее 60% и влаги не более 236
40%. Продукт без дополнительной очистки используют на следующей стадии. (При необходимости хранят в. прохладном темном месте; на свету и воздухе вещество разлагается.)
3,6-Диметил-1,2,3,4-тетрагидро-у-карболин (V). К смеси 1,67 л этилового спирта, 578 г IV и 344 мл конц. соляной кислоты прибавляют при перемешивании и охлаждении 266 г II с такой скоростью, чтобы температура реакционной массы была не выше 35 °C. Дают выдержку 15 мин, после чего кипятят 40 мин, охлаждают до 70 °C, прибавляют 1,07 л воды и 73 г угля. Кипятят 10 мин, фильтруют, охлаждают до 20—25 °C и подщелачивают насыщенным водным раствором поташа по фенолфталеину. Осадок V отделяют, промывают водой до нейтральной реакции и сушат при 50—60 °C. Получают 304 г (68%, считая на IV) V с содержанием основного вещества не менее 98% и т. пл. 149—150°С.
3,6-Диметил-1,2,3,4,4а,9а-гексагидро-у-карболина дигидрохлорид (карбидин) (VI). К нагретому до 70°С раствору 700 г V в 6,8 л изопропилового спирта прибавляют при перемешивании 8,3 г хлорной ртути и 3 кг цинковой пыли. Дают выдержку 10 мин, после чего при 70—78 °C в течение 2 мин приливают 23,3 л конц. соляной кислоты со скоростью сначала 150 мл/мин, а затем 2,9 л/ч (проведение процесса при температуре ниже 70 °C снижает его скорость, а при температуре выше 80 °C масса темнеет. В обоих случаях выход и качество продукта ухудшаются). Цинковую пыль прибавляют порциями по 1 кг через каждый час (всего загружают 9,24 кг). Дополнительно дают выдержку 1 ч при 70—78 °C, контролируют конец реакции по содержанию V (должно быть не более 10%), охлаждают до 60 °C, отфильтровывают от цинкового шлака. Фильтрат охлаждают до 10—15 °C и подщелачивают 25% водным раствором аммиака до pH 9—10. Дают выдержку 30 мин, контролируют pH и экстрагируют основание VI хлороформом (4 л и 3 раза по 2 л). Хлороформные экстракты промывают водой (2 раза по 2 л), сушат сульфатом магния, фильтруют и добавлением 400 мл спиртового раствора хлороводорода (до pH 1—2) выделяют дигидрохлорид VI, который отфильтровывают, промывают 220 мл абсолютного этилового спирта, высушивают при 60—80 °C и перекристаллизовывают из 2,21 л дистиллированной воды с 40 г активированного угля (при выделении кристаллов дают выдержку 1 ч при 7—10°C). Выход VI 348,6 г (45,6%).
237
ПИРАЗИДОЛ
1,10-Триметилен-8-метил-1,2,3,4-тетрагидропиразино [1,2-а] индола гидрохлорид:
Синоним: пириндол. Мол. масса 262,79
Светло-желтого цвета со слабым зеленоватым оттенком кристаллический порошок, мало растворим в воде, метиловом и этиловом спиртах, очень мало растворим в хлороформе; т. пл. 260—266 °C (в интервале 2 °C с разложением в запаянном капилляре); ВФС 42-1436-80.
Пиразидол — оригинальный антидепрессивный препарат [142, 144] с широким спектром терапевтического действия и регулирующим эффектом на ЦНС, где влияет на содержание и обмен катехоламина и других биогенных аминов [143], но не оказывает холинолитического действия.
Применяют пиразидол у больных с маниакально-депрессивным психозом, шизофренией с аффективными расстройствами и инволюционным психозом, соматогенным психозом и другими психическими заболеваниями, протекающими с депрессией. Препарат назначают при депрессиях с психомоторной заторможенностью, а также при депрессиях, сопровождающихся тревожно-депрессивными и тревожно-бредовыми компонентами, анестетической, ипохондрической и неврозоподобной симптоматикой, при лечении депрессивных и депрессивно-бредовых состояний у больных алкоголизмом.
Выпускается в таблетках по 0,025 г N. 50 (цена 1 р. 27 к.) и по 0,05 г N. 50 (цена 2 р. 52 к.).
Метод синтеза пиразидола разработан во ВНИХФИ [144] по схеме:
238
Пара-толуидин (I) путем диазотирования нитритом натрия в соляной кислоте превращают в п-толилдиазоний хлорид (II), который без выделения подвергают взаимодействию с натриевым производным оксиметиленциклогексанона (IV), получаемым путем конденсации по Кляйзе-ну циклогексанона (III) с этилформиатом. Этот метод, описанный ранее [315] и существенно усовершенствованный в ходе работы над пиразидолом [252], оказался более технологичным, чем синтез моно-пара-толилгидразона циклогексан-1,2-диона (V) на основе взаимодействия пиперидил-1-циклогексена с п-толилдиазоний хлоридом. Циклизация V по Фишеру в присутствии муравьиной кислоты в диметилформамиде дала лучшие результаты, чем ранее описанные реакции в смеси уксусной и соляной кислот или в водно-спиртовом растворе серной кислоты. Взаимодействием 1-оксо-6-метил-1,2,3,4-тетрагидрокарба-зола (VI) с полученным по известному методу [277] из паральдегида, брома и бутилового спирта дибутиловым ацеталем бромуксусного альдегида синтезируют дибутиловый ацеталь 1-оксо-6-метил-1,2,3,4-тетрагидрокарбазо-
239
лил-9-уксусного альдегида, который без выделения и очистки циклизуют под действием ацетата аммония в 1,10-триметилен-8-метилПиразино [1,2-а] индол, выделяемый в виде гидрохлорида VII [254]. В основании соединения VII пиразиновое ядро восстанавливают каталитически до тетрагидропиразинового [252], что оказалось для промышленности более приемлемым, чем первоначально предложенное -.[254] восстановление натрием в спирте. Другие методы наращивания тетрагидропиразинового кольца, основанные на конденсации VI с хлорацетонитрилом или хлорацетамидом и последующей восстановительной циклизации, дают худшие результаты [252].
Моно-п-толилгидразон циклогексан-1,2,-диона (V). К нагретым до 80°С 2,06 л воды прибавляют 858 г паратолуидина (I), перемешивают 20 мин при 80°C до полного расплавления I и''образования эмульсии, к которой приливают 2,84 л соляной кислоты. Массу охлаждают до О °C, суспензию разбавляют 2 л воды и при охлаждении (температура массы не выше 5 °C) добавляют по каплям 2,06 л охлажденного 23,1% раствора нитрита натрия, контролируя конец диазотирования по йодкрахмальной пробе. К полученному раствору соли диазония II, имеющему pH 1—2, добавляют ацетат натрия до pH 5—6 и сразу же (при pH 5—6 диазосоединение неустойчиво) при 0—10 °C приливают 8,75 л водно-спиртового раствора натриевого производного оксиметиленциклогексанона (IV), приготовленного путем приливания в течение 3 ч к смеси 985 г циклогексанона (III) и 1110 г этилформиата при температуре не выше 35 °C 1,89 л метанольного раствора метилата натрия, содержащего 29,5% (допускается колебание в интервале 26—31%) основного вещества, с последующей выдержкой 4 ч при 30—35 °C (когда содержание III в реакционной массе не будет превышать 3%) и разбавлением 5 л воды при температуре не выше 35 °C. Прибавление охлажденного водно-спиртового раствора IV- ведут в два приема: первую порцию ( — 2 л) приливают медленно в течение 40—50 мин, оставшиеся 6,75 л прибавляют значительно быстрее — примерно за 1 ч. Для завершения процесса дают выдержку 1 ч при 0-]-10оС. Выпавший осадок V отфильтровывают, промывают 1 л воды, высушивают и Перекристаллизовывают из смеси диметил-формамида с метанолом (4: 1). Получают 1448 г моногидразона V с содержанием основного вещества не ниже 98%; т. пл. 185—193°C (с разложением в интервале 2°C). Выход 82,1%, считая на пара-толуидин.
240
]-Оксо-6-метил-1,2,3,4-тетрагидрок арб азол (VI). Смесь 2,68 л 95% муравьиной кислоты, 1000 г V, 1,03 л диметил -формамида и 435 мл воды медленно, в течение 40—50 мин, нагревают до 80 °C. Нагревание прекращают. Температура в реакционной массе самопроизвольно повышается до 112—114°С. (При проведении процесса без диметил-формамида реакция протекает настолько бурно, что происходит выброс.) При 112—114 °C (если самопроизвольное нагревание не достигло этой температуры, массу осторожно подогревают до указанного температурного интервала) дают выдержку 30 мин. Охлаждают до 0—5 °C и перемешивают при этой температуре Р/г ч. Осадок VI отфильтровывают, промывают 2,5 л воды, 1 л метанола и высушивают при 70—80 °C. Получают 828 г VI с содержанием основного вещества 96,1%. Выход 86,4%.
Дибутиловый ацеталь бромуксусного альдегида. К смеси-3,2 л бутилового спирта и 661 г свежеперегнанного паральдегида (т. кип. 123—125 °C) с «затравкой» кристалликом йода при 20°С и перемешивании приливают в один прием 100 мл брома. Температура повышается до 28— 30°C. Перемешивают 40 мин, раствор обесцвечивается. После этого массу охлаждают до 22—24 °C и прикапывают еще 670 мл брома с такой скоростью, чтобы температура была не выше 30 °C. (Время прибавления брома 11/2—2 ч.) Дают выдержку при перемешивании 3 ч, массу охлаждают до +2 °C и прикапывают 3,65 л воды за Р/з—2 ч с такой скоростью, чтобы температура была не выше +4 °C. Нейтрализуют кальцинированной содой (~1000 г) при температуре 4—5 °C до pH 7 (время нейтрализации 2—2'/г ч). Верхний бутанольный слой, содержащий дибутиловый ацеталь бромуксусного альдегида, отделяют и отгоняют в вакууме бутанол до температуры 75 °C в парах при давлении 80—90 мм рт. ст. Остаток перегоняют, собирают фракцию с т. кип. 115—119 °C (10 мм рт. ст.). Получают 2394 г дибутилового ацеталя бромуксусного альдегида с содержанием основного вещества не ниже 96%. Выход 60,6%, считая на паральдегид.
1,10-Триметилен-8-метилпиразино [1,2-а] иидола гидрохлорид (VII). От смеси 828 г VI, 2,4 л диметилформамида и 1111 г метанольного раствора метилата натрия (с содержанием 19,6% алкоголята) после перемешивания в течение 15 мин отгоняют метанол при остаточном давлении 80 мм рт. ст. до тех пор, пока температура в парах не достигнет 77 °C. К полученному натриевому производ
16 Заказ № 4304
241
ному 1-оксо-6-метил-1,2,3,4-тетрагидрокарбазола в диме-тилформамиде без охлаждения приливают 922 мл дибутилового ацеталя бромуксусного альдегида с содержанием основного вещества 96%. Кипятят 1 ч, охлаждают до 70 °C и выливают в смесь 9 л воды и 1,5 л бензола. Перемешивают, бензольный слой отделяют, водный — экстрагируют бензолом (1 раз 1,5 л; 2 раза по 1 л). Объединенные бензольные экстракты упаривают, остатки влаги удаляют добавлением 200 мл сухого бензола с последующей отгонкой в вакууме. Затем прибавляют 3,6 л уксусной кислоты и 2,4 кг ацетата аммония. Циклизацию ведут при перемешивании и кипячении (124—126 °C в массе) в течение 2 ч. Массу охлаждают, разбавляют 9,6 л воды и продукт реакции извлекают четыреххлористым углеродом (2 раза по 2 л; 3 раза по 1,5 л; 1 раз 1 л). К объединенным экстрактам при 20—25°С прикапывают 21% соляную кислоту до кислой реакции по конго. Осадок VII отфильтровывают, промывают в три приема 1,6 л ацетона и сушат при 40—80°C. Выход VII (с содержанием основного вещества 96,3%) 707 г (65,8%).
1,10-Триметилен-8-метил-1,2,3,4-тетрагидропиразино [1,2-а]индола гидрохлорид (пиразидол) (VIII). Растворяют 268 г VII в 4,5 л воды при кипячении, горячий раствор обесцвечивают 27 г активированного угля, фильтруют в токе азота, фильтрат охлаждают до 30—40 °C и при дальнейшем охлаждении приливают водный раствор аммиака до pH 9—10. Выделившееся в виде масла основание извлекают бензолом (2 раза по 1,5 л; 2 раза по 1 л). Объединенные бензольные растворы обесцвечивают при 20—25 °C 27 г активированного угля и фильтруют в токе азота. Бензол отгоняют, остаток растворяют в 2 л метанола и гидрируют с 40 г палладиевого катализатора на карбонате кальция (содержание палладия 4,35%) в автоклаве при температуре не выше 50 °C и давлении 70—60 атм. Катализатор отфильтровывают, метанол отгоняют при остаточном давлении 500—600 мм рт. ст. Вещество растворяют в 1 л бензола и при 20—25 °C прибавляют 0,1 л 35% соляной кислоты. Выделившийся продукт отфильтровывают, промывают 500 мл ацетона (в три приема) и перекристаллизовывают с добавкой 43 г угля из метанола в расчете 28 л метанола на 1 кг продукта. Выход пиразидо-ла (VIII) 175 г (66,6%). Из маточного раствора путем подпаривания дополнительно выделяют 80 г технического пиразидола, который используют на следующей загрузке при перекристаллизации готового продукта,
242
ИНКАЗАН
З-Метил-8-метокси-ЗН, 1,2,5,6-тетрагидропиразино [1,2, 3-аЬ]-р-карболина гидрохлорид.
(CI5H17 N3O • HCI
Мол. масса 291,70
Белый или белый с кремоватым оттенком кристаллический порошок, без запаха, горького вкуса, растворим в воде и метиловом спирте, трудно растворим в этиловом спирте, мало растворим в хлороформе, практически нерастворим в эфире; т. пл. 306—309°C (в интервале 2°C); ВФС 42-731-78.
Инказан — оригинальный препарат антидепрессивного действия [9, 41], обладает противорезфпиновой, а также центральной и периферической адрено- и серотонинопозитивной. активностью, сочетает тимолептический эффект со стимулирующим действием на ЦНС и при лечении непсихотических депрессий оказывает более быстрое и мягкое действие, чем имизин и амитриптилин. Препарат лишен побочного холинолитического действия и хорошо сочетается с антидепрессантами трициклического строения, транквилизаторами и нейролептиками.
Применяют инказан при депрессивных состояниях различного генеза (шизофрения, маниакально-депрессивный психоз, органические заболевания головного мозга, циклотимия и др.) с преобладанием гипо- и анергических расстройств. Наиболее показан препарат при вялоапатических, адинамических депрессиях, депрессиях с заторможенностью, а также при неглубоких депрессиях с неврозоподобной и невыраженной гипохондрической симптоматикой.
Форма выпуска: таблетки по 0,025 г: N. 150 — цена 9 р. 02 к., N. 250 — цена 15 р. 02 к.
Метод синтеза инказана разработан во ВНИХФИ [9, 41] по схеме:
16*
243
1-Оксо-6-метокси-1,2,3,4-тетрагидро-р-карболин (I), получаемый так же, как описано в синтезе мексамина (см. с. 161), алкилируют p-диметиламиноэтилхлоридом в сухом диметилформамиде в присутствии гидрида натрия с последующим переводом продукта в гидрохлорид II добавлением к спиртовому раствору основания спиртового раствора хлороводорода. Гидрохлорид II обрабатывают хлорокисью фосфора с последующим алкоголизом промежуточного дихлорфосфоната III в инказан (IV).
1-Оксо-6-метокси-9-(р-диметил аминоэтил)-1,2,3,4-тетра-гидро-р-карболина-гидрохлорид (II). К суспензии 432 г 1-оксо-6-метокси-1,2,3,4-тетрагидро-р-карболина (I) в 2,5 л сухого диметилформамида при 25 °C порциями прибавляют 60 г гидрида натрия и нагревают сначала 2 ч при 30 °C, затем 30 мин при 50°С. К нагретой до 50—55°С реакционной массе по каплям прибавляют 225 г диметиламино-этилхлорида и нагревают при 50—55 °C в течение 4 ч. По охлаждении реакционную массу выливают в 2 л 50% уксусной кислоты, фильтруют, раствор подщелачивают 15% водным аммиаком и экстрагируют бензолом (2 раза по 2,5 л). Объединенные бензольные экстракты тщательно промывают водой (4 раза по 2 л) и экстрагируют 2 раза по 2,5 л 6% соляной кислотой. Водный кислый слой обесцвечивают активированным углем (40 г) и подщелачивают 10% раствором едкого натра до pH 10. Основание II экстрагирует^ бензолом (3 раза по 2 л), бензольные экстракты тщательно промывают водой (3 раза по 2 л) и упаривают в вакууме. Остаток растворяют в 2,5 л кипящего абсолютного этилового спирта, обесцвечивают 40 г активированного угля, охлаждают до 10 °C и прибавляют 23% спиртовой раствор хлороводорода до pH 1,0. Суспензию гидрохлорида II в спирте охлаждают до 0°С, осадок отфильтровывают, промывают 0,2 л абсолютного этилового спирта и высушивают при 60—70 °C. Получают 305 г 244
II с содержанием основного вещества не ниже 95%, выход 45%; т. пл. 243—245 °C.
3-Метил-8-метокси-ЗН,1,2,5,6-тетрагидропиразино [1,2, 3-аЬ]-р-карболина гидрохлорид (инказан) (IV). Смесь 580 г II и 2,14 л хлорокиси фосфора кипятят 2 ч, охлаждают до 80°C и разбавляют 2,14 л бензола, после чего охлаждают до 10°C в течение 1 Vs—2 ч. Осадок дихлорфос-фоната III отфильтровывают, промывают 1 л бензола и высушивают в вакууме (1—2 ч при 20—25 °C, 500— 550 мм рт. ст.). Полученный продукт (около 600 г) нестабилен при хранении, его смешивают с 2,14 л абсолютного этилового спирта и нагревают 1 ч при кипении. Происходит алкоголиз, осадок переходит в раствор и выпадает новый осадок — осадок технического инказана. Для полноты выделения последнего массу охлаждают и выдерживают 1 г/з—2 ч при температуре от 0°С до +5°C. Осадок отфильтровывают, промывают 400 мл абсолютного этилового спирта и перекристаллизовывают из 9,1 л абсолютного этилового спирта с добавлением 90 г нейтрального активированного угля, регулируя pH спиртового раствора после фильтрации от угля (pH должен быть около 4) добавкой небольшого количества спиртового раствора хлороводорода. Для полноты выделения инказана массу охлаждают 2 ч при температуре от 0°С до +5 °C, осадок отфильтровывают, промывают 300 мл охлажденного до температуры от 0°С до +5 °C абсолютного этилового спирта и высушивают в вакууме. Получают 269 г инказана (IV). Путем подпаривания маточного спиртового раствора и перекристаллизации выпавшего в осадок продукта дополнительно получают 41 г инказана. Общий выход IV 64,2%.
АЛФАВИТНЫЙ УКАЗАТЕЛЬ
Азатиоприн 197
Азафен 220, 231—234
Аллопуринол 197 Амнзил 52
Аминазин 125, 217, 218, 220
Амнналон 7
Аминокапроновая кислота 9
Анатруксоний 5, 30, 33—36, 37, 38
Анестезин 51
Апрофен 52
Армин 18
Арпенал 52
Асалин 5, 10, 12—15
Атропина сульфат 41, 136, 171— 175
Ацеклидин 139, 143
Ацемин 9, 15—16
Бензотэф 17, 19
Бити один 112, 129—132
Бутамнд 92
Варфарнн 114, 122 Галазолин 198 Гистидин 7, 198 Глутаминовая кислота 7, 147 Гоматропина гидробромид 136, 176—179
Декамин 32 Диазепам 199 Диакарб 92 Дийодбензотэф 5, 17, 19—22 Дикани 51, 77 Диколнн 134 Дикумарин 113, 122 Димеколин 134 Динатриевая соль Зтилен — днаминтетрауксусной кислоты
Дннезнн 218
Диоксидин 196, 201—202 Диоксоний 31, 40—43 Днпнн 17 Днпнридокснм 134 Диплацнн 29, 30 Дипразнн 218 Дипрофен 52 Дисульфан 91 Дисульформнн 89, 91 Дитилин 5, 31, 38—40 Изониазид 133 Имехин 141, 143, 186—187
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Имифос 17, 22—23 Инказан 222, 243—245 Ипразид 134 Карбидин 221, 234—237 Квалидил 5, 30, 141, 143,187—192 Клофелин 198, 208—211 Кокаин 51, 77 Кондельфин 29, 30 Кордиамин 133 Ксиканн 51 Левомицетин 195 Лофенал 10 Маркумар 114 Мебикар 198, 211—212 Мексамин 136, 161—165, 244 Мелликтнн 29, 30 ' Меридил 135, 149—153 Меркапопурин 197 Метазид 133 Метамизил 5, 53, 83—86 Метацин 52 Метеразин 219 Метионин 7 Метотрексат 5, 197, 204—208 Метронидазол 195, 198 Мидантан 48, 53—54 Нанофнн 134 Нафтизин 198 Неоднкумарин 114, 122 Ниаламид 134 Ннбуфин 18 Нико дин 133 Никотиновая кислота 133, 134 Нитазол 195, 200—201 Нитразепам 199 Нитроксолин 135, 153—155
9 Нитрофарин 115, 123—125 Новокаин 51
Нонахлазин 220, 226—229 Норсульфазол 90, 91, 195 Норсульфазол-натрий 90, 91 Оксазепам 199
Оксолин 49, 57—61 Октатион 19, 27—28 Оксилидин 139, 143 Пантагам 8, 10—12 Пармидин 134, 143—146 Пеницилламин 9 Пентацнн 9
Пиразидол 221, 222, 238—242
246
Пирацетам 8
Пиридитол 5, 135, 146—149
Пирнлен 140, 182
Пиромекаин 5, 52, 77—79
Пирофос 18
Пирроксан 115, 125—129
Пропазин 218
Проспидин 32, 43—46 Ремантадин 48, 54—57 Салазодиметоксин 91, 101, 102—
105, 195
Салазопиридазин 91, 100—102,
103, 195
Сарколизии 10
Серотонина адипинат 135, 136, 166—171
Сиднокарб 198, 213—215
Сиднофен 198, 212—213
Синкумар 114
Совкаин 51
Солафур 113, 117
Спазмолитин 52
Стрептоцид 87, 90, 91, 92, 95
Стрептоцид растворимый 90, 91
Сульгин 90, 91
Сульфадимезин 90, 91, 195
Сульфадиметокснн 90, 91, 103
Сульфален 90, 91, 195
Сульфамонометоксин 5, 90, 91, 94—98, 195
Сульфантрол 89, 91
Сульфапиридазин 88, 90, 98, 99, 101, 195
Сульфапнридазнн-натрий 90
Сульфацил-натрий 90, 91
Теброфен 49, 61—63
Тетацин-кальций 9
Темехин 140, 143, 181—186, 187
Тиоднпин 17
Тиофосфамид 16, 17, 19, 22
Тифен 52
Тримеканн 51
Триметоприм 89
Трифтазин 218, 230
Тропафен 137, 179—181
Тропацин 137
Тубокурарин-хлорид 28, 29, 30
Уросульфан 90, 91
Феназепам 199, 200, 215—217
Фенамин 150, 198, 214 Фенибут 5, 8 Феникаберан 115, 118—121 Фенкарол 5, 142, 143, 192—195 Фенобарбитал 50, 71 Фепромарон 115, 121—123, 124 Ферроцерон 50, 72—74 Флореналь 49, 64—67 Фопурин 197 Фосфакол 18 Фосфемид 17
Фтазии 5, 90, 91, 98—100 Фталазол 90, 91, 99, 195 Фтивазид 133
Фторацизин 220, 229—231 Фторафур 196 Фторбензотэф 17 Фторурацил 196 Фторфеназнн 219 Фурагин 5, ИЗ, 116—118 Фуразолидон 113 Фуразолнн 113 Фурацилин 113 Хиноксидин 196, 202—204 Хиноцид 135, 155—161 Хлодитан 49, 67—70 Хлорацетофос 18, 26 Хлорацизин 219, 227, 230 Хлордиазепоксид 119 Хлорпропамид 92, 105—107 Хлофосфол 18, 24—25 Хлоцикламид 5, 92, 109—ПО Цетамифен 49, 50, 70—72 Цетилпиридиний хлорид 32 Цигерол 50, 74—77
Цикламид 92, 103, 107—109, 110 Циклобутоний 30, 36—40 Циклометиазид 93 Цистеин 7 Цитарабин 196 Эстоцин 52, 79—83 Эсулан 94
Этазол 90, 91, 195 Этазол-натрнй 90, 91 Этамид 93, 110—112 Этаперазин 218 Этимизол 198 Этмозин 220, 222—226, 230 Этоний 33, 46—47
247
УКАЗАТЕЛЬ ПО МЕДИЦИНСКОМУ ПРИМЕНЕНИЮ
лекарственных препаратов
Средства, применяющиеся при лечении неинфекционных заболеваний
Препараты для лечения заболеваний ЦНС
Азафен 220, 231—234
Амизнл 52
Аминазин 125, 217, 218, 220
Аминалон 7 ,/,< \
Диазепам 199
Динезин 218
Инказан 222, 243—245
Ипразнд 134
Карбидин 221, 234—237
Мебикар 198, 211—212
Меридил 135, 149—153
Метамизил 5, 53, 83—86
Метеразин 219
Мидантан! 48, 53—54
Ниаламнд 134
Нитразепам 199
Оксазепам 199
Оксилндин 139, 143
Пантагам 8, 10—12
Пиразидол 221, 222, 238—242
Пирацетам 8
Пиридитол 5, 135, 146—149
Пирроксан 115, 125—129
Пропазин 218
Сиднокарб 198, 213—215
Сиднофен 198, 212—213
Трифтазин 218, 230
Тропацнн 137
Феназепам 199, 200, 215—217
Фенамин 150, 198, 214
Фенибут 5, 8
Фторацизин 220, 229, 231
Фторфеназин 219 ,
Хлордиазепоксид 199 ।
Этаперазин 218 5
Средства, понижающие тонус скелетной мускулатуры
Анатруксоний 5, 30, 33—36, 37,
38
Динезин 218
Диоксоний 196, 201—202
Диплацин 29, 30
Дитилин 5, 31, 38—40
248
Квалидил 5, 30, 141, 143, 187—192
Коидельфин 29, 30 Меллнктин 29, 30 ’ Мидантан 48, 53—54 Тропацин 137
Тубокурарин-хлорнд 28, 29, 30 Циклобутоний 30, 36—40
Сердечно-сосудистые средства
Аминалон 7
Апрофен 52
Арпенал 52
Атропина сульфат 41, 136, 171—175
Варфарин 114, 122
Галазолин 198 Диколнн 134 Дикумарнн 113, 122 Димеколнн 134 Дипрофен 52 Имехин 141, 143, 186—187 Клофелин 198, 208—211 Кордиамин 133
Ксикаин 51 Маркумар 114 Нанофин 134 Нафтизин 198 Неодикумарнн 114, 122 Никотиновая кислота 133, 134 Нитрофарин 115, 123—125 Новокаин 51 Нонахлазии 220, 226—229
, Окснлидин 139, 143
। Пармидин 134, 143—146
Пирилен 140, 182
Пирроксан 115, 125—129 Синкумар 114
Спазмолитин 52
Темехии 140, 143, 181—186, 187 Тифен 52
' Тропафен 137, 179—181 Феникаберан 115, 118—121 Фепромарон 115, 121—123, 124 Цнклометназнд 93
Этмозин 220, 222—226, 230
Средства, применяющиеся при лечении других внутренних органов
Амизил 52
Апрофен 52
Арпенал 52
Атропина сульфат 41, 136, 171—175
Ацеклиднн 139, 143
Битиодин 112, 129—132
Гоматропина гидробромид 136, 176—179
Диколин 134
Димеколии 134
Дипрофеи 52
Метацин 52
Метамизил 5, 53, 83—86
Нанофин 134
Пирилеи 140, 182
Спазмолнтии 52
Темехии 140, 143, 181—186, 187
Тропацин 137
Эстоцин 52, 79—83
Антиглаукоматозные средства
Армин 18
Ацеклидин 139, 143
Клофелин 198, 208—211
Нибуфнн 18
Пирофос 18
Хлофосфол 18, 24—25
Местные анестетики
Анестезин 51
Днкаин 51, 77
Кокаин 51, 77
Ксикаин 51
Новокаин 51
Пиромекаин 5, 52, 77—79
Совкаип 51
Тримекаин 51
Средства, влияющие на тканевый обмен, и противоядия а)
Аминокислоты
Аминокапроиовая кислота 9
Гистидин 7, 198
Глутаминовая кислота 7, 147
Метионин 7
Пеницилламин 9
Цистеин 7 б)
Антиаллергические и иммунодепрессивные препараты
Азатиоприн 197
Дипразин 218
Феикарол 5, 142, 143, 192—195 в)
Антидиабетические средства
Бутамид 92
Хлорпропамид 92, 105—107
Хлоцикламид 5, 92, 109—110 г)
Противоядия и другие препараты
Аллопуринол 197
Динатриевая соль этиленднамин-тетрауксусной кислоты 9
Дипиридоксим 134
Мексамин 136, 161—165, 244
Серотонина адипинат 135, 136, 166—171
Тетацин-кальций 9
Ферроцерон 50, 72—74
Цигерол 50, 74—77
Средства, применяющиеся при лечении инфекционных заболеваний
Антибактериальные препараты
Дскамин 32
Диоксидин 196, 201—202
Дисульфан 91
Днсульформнн 89, 91
Левомицетин 195
Ннкодин 133
Нитроксолин 135, 153—155
Норсульфазол 90, 91, 195
Норсульфазол-натрий 90, 91
Салазодиметоксин 91, 101, 102—105, 195
Салазопиридазин 91, 100—102, 103, 195
Солафур ИЗ, 117
Стрептоцид 87, 90, 91, 92, 95
Стрептоцид растворимый 90, 91
Сульгин 90, 91
Сульфадимезин 90, 91, 195
Сульфадиметокснн 90, 91, 103
Сульфален 90, 91, 195
Сульфамонометоксин 5, 90, 91 94—98, 195
Сульфантрол 89, 91
249
Сульфапнридазнн 80, 90, 98, 99, 101, 195
Сульфапиридазии-натрий 90
Триметоприм 89
Уросульфан 90, 91 .
Фтазин 5, 90, 91, 98—100
Фталазол 90, 91, 99, 195
Фурагин 5, 113, 116—118
Фуразолидой 113
Фуразолнн 113
Фурацнлин 113
Хиноксидин 196, 202—204
Цетилпнридииий хлорид 32
Этазол 90, 91, 195
Этазол-натрий 90, 91
Этоний 33, 46—47
Противотуберкулезные средства
Изониазид 133
Метазид 133
Фтивазид 133
Антипротозойные и противогрибковые препараты
Метронидазол 195, 198 Нитазол 195, 200—201 Октатион 19, 27—28 Хиноцид 135, 155—161
Хлорацетофос 18, 26
Эсулан 94
Противовирусные препараты
Оксолин 49, 57—61 Ремантадин 48, 54—57 Теброфен 49, 61—63 Флореналь 49, 64—67
Средства, применяющиеся при лечении злокачественных новообразований
Асалин 5, 10, 12—15
Бензотэф 17, 19
Дийодбензотэф 5, 17, 19—22
Имифос 17, 22—23
Лофенал 10
Меркаптопурин 197
Метотрексат 5, 197, 204—208
Проспидин 32, 43—46
Сарколизин 10
Тиодипнн 17
Тиофосфамнд 16, 17, 19, 22
Фопурнн 197
Фосфемид 17
Фторафур 196
Фторбензотэф 17
Фторурацил 196
Хлодитан 49, 67—70
Цетарабин 196
указатель исходных веществ*
Гидроксилпроизводиые (спирты и фенолы)
Аллиловый спирт 34 Глицерин 42, 158 Ацетопропнловый спирт 159 Линалоол 76
Циклогексанол 75
Пирокатехин 127
Резорцин 62 п-Нитрофенол 168 4-Оксикумарин 123, 125 8-Оксихинолин 154 Пиридоксин 148
Г алогенопроизводные
Йодистый метил 38, 40, 43
Бромистый этил 85
йодистый этил 36
Хлорбензол 70
Бромбензол 194, 224
Хлористый бензил 168
Эпихлоргидрин 46
Этнленхлоргндрин 44, 127
Трнхлорпентнловын спирт 25
1,1,1 -Трихлор-5-ацетокснпентан 25
1,1,1,5-Тетрахлорпентан 25
Хлорофос 26
* В указатель не включены вещества, широко используемые в химико-фармацевтической промышленности для многих производств, типа хлороформа, этилового спирта, толуола и др.
250
2-Хлордиридии 152 3,4,6-Трихлорпиридазин 233 2-Хлор-10- (р-хлорпропионил) фенотиазин 229
Амины и их производные
Метиламин 174, 207 Диметиламии 47, 121 Диэтиламин 112, 231 Триэтиламин 21, 23, 45, 229 БутилЭмин 79 Октадециламин 27 Этаиоламин 71
Диметиламииоэтилхлорид 83, 244 Этилендиамии 210, 228 Диэтиламиноэтанол 39 Диэтиламииопропаиол 38 Циклогексиламии 108, 110 (З-Фенилизопропиламии 213 п-Толуидии 236, 240 о-Нитроаиилин 203 м-Нитроанилин 224 о-Амннофенол 233 п-Анизидин 158, 164 п-Броманилин 216 2,6-Дихлоранилин 210 2,4,6-Триметиланилин 78 Этиленимин 21, 23 0-Пиколии 132 у-Пиколин 132 2,6-Л ути дин 132, 145 Пирролидин 42 Пиперидин 34 Морфолин 66, 117, 225 N-Метилпиперазин 234 N-Бензоилпиперазии 44
Альдегиды, кетоиы и их производные
Ацетальдегид 23, 117 Хлорацетальдегид 200 Глиоксаль 212 о-Хлорбензальдегид 69 п-Нитробензальдегид 124 5-Нитрофурфуролди ацетат 117 Метилэтилкетон 124, 204 Циклогексанон 240 Этилстирилкетон 123 Хинуклидон-3 188, 194 п-Хинои 120 1,1,3-Трихлорацетон 207 Ацетонциангидрин 194, 213
Кислоты и их производные
Муравьиная кислота 58, 241
Пропионовая кислота 106 б-Хлорвалериаиовая кислота 78, 228
Янтарная кислота 39
Адипиновая кислота 171, 190
Коричная кислота 35
Салициловая кислота 102, 104
Бензиловая кислота 82, 85
Ацетондикарбоновая кислота 174
Валин 13
у-Аминомасляиая кислота 11 е-Аминокапроиовая кислота 16
Антраниловая кислота 20
Сарколизин 13
п-Аминобензоил-Б-глутами-новая кислота 207
Семикарбазидоуксусиая кислота 118
Миндальная кислота 179 м-Нитробензолсульфокислота 158 2-Оксинафтил-1 -азобензол--4-сульфокислота 59
Хлоругольиый эфир 225
Этилформиат 240
Хлорускусиой кислоты дециловый эфир 47
Ацетоуксусный эфир 119
Циаиуксусный эфир 184
Малоновый эфир 57, 75, 96, 163
Диэтилоксалат 177
Фенилуксусной кислоты этиловый эфир 172
Никотиновой кислоты этиловый эфир 130
Ди (р-карбометоксиметил-этил)метиламин 236
D(—)паитолактон И
Фталевый ангидрид 73, 99, 159, 170
Ацетил хлористый 127
Р-Хлорпропиоиилхлорид 225, 231
Амииоацетилхлорид, гидрохлорид 216
Беизоил хлористый 119 о-Хлорбензоилхлорид 216 п-Толуолсульфонилхлорид 111, 124
Фенилуретилаисульфохлорид 97
Формамид 57, 96
п-Толуолсульфониламид 108 п-Хлорбеизолсульфоииламид 106,
НО
'51
Мочевина 106, 108, 233 1,3-Диметидмочевина 212
Тиомочевииа 201
Тетраметилтиурамдисульфид 210
Акрилонитрил 163
Малондинитрил 206
Беизил цианистый 127, 152
Фенилэтилацетонитрил 71
Гуанидин нитрат 206
Метилизоциаиат 146
Фенилизоциаиат 214
Дициклогексилкарбодиимид 14
Другие соединения
Фуран 41, 173
Тиофен 130
Флуорен 65
Ферроцен 73
Нитроэтан 85
указатель полупродуктов, методы синтеза которых ОПИСАНЫ в монографии
Алифатические соединения
Тетраметилэтилендиамин 47 Бромуксусного альдегида дибу-
тилацеталь 241 '''' 'V
Щавелевой кислоты моиоэтилрвый эфир 177
его калиевая соль 177
его хлорангидрид 177
и-Пропиламин 106
З-Хлорпропандиол-1,2 42
2-Аминопропаиол 85
2-Днэтиламинопропанол 85 2-Нитропропанол 85
Малондиамид 96
Фумарового диальдегида диме-
тилацеталь 41
Янтарного диальдегида диметилацеталь 42
Янтарной кислоты диметиламииоэтиловый эфир 39
D.L-Валииа этиловый эфир 14 4-Аминопентаиол-1 159 4-Фталимидо-1-хлорпентан 159 4-Фталимидо-1-йодпеитан 159 а-Бром-6-хлорвалериановая кисло-
та 228, 78
ее хлораигидрид 78, 228
ее этиловый эфир 228
ее 2,4,6-триметиланилид 78 Р-Цианэтилмалоновой кислоты
диэтиловый эфир 163 1,6-Диоксигексаи 190'' \ 1,6-Дихлоргексан 191 Адипиновой кислоты диэтиловый
эфир 190
Геранилхлорид 76
Карбоциклические и ароматические соединения м-Нитроацетаиилид 224 п-Толилгидразин 236 2-Нитро-4-метоксиацетаиилид 158 3-Нитродифениламин 224 3-Аминодифениламин 224
его N-карбаминовый эфир 225 М,М-Диметил-М'- (2,6-дихлорфе-нил)хлорформамидииа гидрохлорид 210
Р-Фенилизопропиламииоацето-нитрил гидрохлорид 213
З-Фенил-4-аминобутанол 127 п-Беизилоксинитробензол 168 п-Бензилоксианилии 168 о-Хлорфенилдихлорметилкарби-иол 69
о-Хлорфенилтрихлорметилкарби-нол 69
(п-Нитростирил)этилкетои 124 2-Амино-5-бром-2-хлорбензофе-нои 216
Пентабромрезорцин 62 1 - (о-Хлорфенил) -1 - (п-хлорфе-нил)-2,2-дихлорэтан 70
Трибромрезохинон 63 2-Амино-5-йодбензойная кислота 20
2,5-Дийодбензойная кислота 20 ее амид 21
Феиилглиоксиловой кислоты хлорангндрид 178 диэтиловый эфир 177
Бензоилуксусный эфир 119 1 - (п-Нитрофенил) -1 -оксипеита-нон-3 124
Фенилэтилуксусная кислота 71
252
а,а-Дифенил-а-хлоруксусная кислота 82
а,а-Дифенил-а-этоксиуксусиая кислота 82
Троповая кислота, ее О-ацетилпроизводное 174 а-Фенил-Р-(п-оксифенил)-пропионовая кислота 180
п- [N-Бис (0-хлорэтил) амино] фенилаланин 13
его N-ацетилпроизводное 13 о-Карбоксибензоилферроцеи 73 п-Метиламинобеизоил-Б-глутами-новая кислота, бариевая соль 207
п-Иодбеизоилглутамииовая кислота 207
|ц-Толуолсульфоиилмочевииа 108, ПО
П-Хлорбензолсульфонилмоче-вина 106
п-Карбоксибензолсульфонил-хлорид 111
а-Труксилловая кислота 35 ее эфиры 35, 36, 37, 38
Бромциклогексан 75
Циклогексилмалоиовой кислоты диэтиловый эфир 75
Циклогексаи-1,2-дион, моно п-толилгидразои 240 Циклогексилгер анилм алоновой кислоты диэтиловый эфир 76
Моиоядерные гетероциклические соединения
5-Нитрофурфурол 117
0-(5-Нитрофурил-2)-акролеин 117
2,5-Диметокси-2,5-дигидрофу-ран 173
2,5-Диметокситетрагидрофу-раи 173
2-Бромтиофен 130
3- (1 -Метнлпиперидил) -ди--(2-тисиил)-карбинол 131
З-Фенилпирролидин 128 2,6-Бис(оксиметнл)пиридин 146 Дипиколиновая кислота 145 ее дибутиловый эфир 146 а-Фенил-а- (пиридил-2) -уксусная кислота 151 ее нитрил 151
2-Метил-3-окси-4,5-дихлорметил-пиридин 148
3- (N-Пиперидил) -пропиловый спирт 34
1-Метилпиперидон-4 236
2,2,6,6,-Тетраметилпиперидон-4 (триацетонамии) 184
1 -Метил-З-карбэтоксипипери-дин 130
а-Фенил-а-(пиперидил-2)-уксусная кислота 152
З-Карбэтоксипиперидон-2 163, 169 2,2,6,6-Тетраметил-4- (0-окси-
этил)пиперидин 185
2,2,6,6-Тетраметил-4-(Р-хлор-этил)пиперидин 185
2,2,6,6-Тетраметил-4-карбэтокси-метилпиперидии 184
Пиперидиндиона-2,3
3-п-метоксифеиилгидразон 164 3-п-бензилоксифеиилгидра-
зон 169
1,2-Бис (4'-хлорметил-Г,З'-ди-оксалаиил-2') -этаи 42
1,2-Бис (4'-пирролидииометил--Г,3'-диоксаланил-2')-этан 42
2-Аминотиазол 200
2-Метилтиазолидин 23
2-Метилти азоли дофосфорной
кислоты дихлорид 23
4,6-Диоксипиримидин 96
4,6-Днхлорпиримидин 96 4-Амино-6-хлорпиримидин 97 4-Амино-6-метоксипиримидин 97 2,4,5,6-Тетрааминопиримидии 206 2,4,6-Триамино-5-нитрозопири-
мидин 206
1 -Бензоил-4- Р-оксиэтил) пиперазин 44
1-Бензоил-4-р-хлорэтил) пиперазин 44
Бициклические соединения
1-Амино-2-нафтол 59
Р-Нафтохиноп 60
Изоиафтазарин 60
1-Бромадамаитан 54, 56
1-Аминоадамаитаи 54
его N-формилпроизводное 54 1-Адамаитаикарбоиовая кислота 56
ее хлорангидрид 56
1-Ацетиладамаитаи 56
1-(а-Аминоэтил) адамантан 57 2-Феиил-3-карбэтокси-5-оксибен-
зофурап 120
253
5-Метокситриптамии-2-карбоио-вая кислота 165
5-Беизилокситриптамии-2-карбо-новаи кислота 169
Бензоксазолои 233
его N-метилпроизводное 233
Бензофуроксан 203 \
Бензодиоксан-1,4, 127
6-Ацетилбензодиоксан-1,4 127
5-Нитрозо-8-оксихииолин 154 5-Нитро-8-оксихииолин 155 6-Метокси-8-нитрохинолии 158
6-Метокси-8-амииохинолин 159
6-Метокси-8- (4'-аминопеитил-
амино) хинолин 160
его фталимидное производное 160
1,4-Ди-М-окись-2,3-диметил-хиноксалина 204
1,4-Ди-М-окись-2,3-дибромме-тилхиноксалииа 204
Тропиион 174
Тропин 174, 181
его эфиры 174, 181
З-Карбэтокснхииуклидин 194 2,2,6,6-Тетраметилхинуклидин 185, 187
З-Окси-З-беизилхинуклидин 188
З-Окси-З-цианохииуклидии 194
З-Беизилиденхинуклидии 189
З-Беизилхинуклидии 190 (Хииуклидил-3) -дифеиилкарби-нол 194
1,4-диазабицикло(4,3,0)иоиаи 229 его 5-оксопроизводное 228
Трициклические соединения
2-Ацетилфлуорен 65
2-Ацетилфлуореион 65
2-(ы-Дибромацетил)-флуореиои 66
2- (<о-Диморфолииоацетил) -флуоренон 66
1 -Оксо-6-метил-1,2,3,4-тетра-гидрокарбазол 241
1 -Оксо-6-бензилокси-1,2,3,4--тетрагидро-р-карболин 169
3,5-Диметил-у-карболииа
1,2,3,4-тетрагидропроизводное 237
1,2,3,4,4а,9а-гексагидропро-изводное 237
1-Оксо-6-мётокси-1,2,3,4-тетра-гидро-(3-карболии 164, 244 его Э-(р-диметиламииоэтил)-производиое 244
1,10-триметилен-8-метилпирази-ио (1,2-а) индол 243
2-хлор-10-Метил-3,4-диазафе-иоксазии 233
2-Трифторметилфеиотиазин 231 его N-ацилпроизводиые 231
2-Амииофеиотиазииа карбаминовый эфир 225
его N-ацилпроизводные 225
N',N"-его
водные 45
Диспиротрипиперазииий 45 N,N -ацил и алкилпроиз-
СПИСОК ЛИТЕРАТУРЫ
1. Абувахабов А. А., Годовиков Н. Н., Кабачник М. И. Химия органических соединений фосфора.— Л.: Наука, 1967.
2. Авакян В. М. К вопросу об изменении фармакологических свойств ацетилхолина путем утяжеления кислотной части его молекулы.— Фармакол. и токсикол., 1959, № 1, с. 20—27.
3. Алексеева Л. Н. Антибактериальные препараты — производные 5-нитрофураиа.— Рига: Изд-во АН Латв. ССР, 1963.
4. Альтшулер Р. А., Волжина О. Н., Лейбельман Ф. Я. и др. Сиднофен— новый психостимулятор.— Хим.-фарм. журн., 1971, № 4, с. 59—62.
5. Альтшулер Р. А., Машковский М. Д., Рощина Л. Ф. Фармакологические свойства синдофена.— Фармакол. и токсикол,, 1972, № 4, с. 406—410.
6. Альтшулер Р. А., Машковский М. Д., Холодов Л. Е., Яшун-ский В. Г. Лекарственный препарат.— А. с. № 222370. (СССР) Бюлл. изобретений, 1973, № 21, с. 211.
7. Андреева Н. И., Алтухова Л. В., Асина В. В. и др. Сравнительное изучение некоторых аналогов пиразидола.— Хим.-фарм. журн., 1976, № 11, с. 46—49.
8. Андреева Н. И., Алтухова Л. Б-, Гринев А. Н. и др. Исследование психотропной активности в ряду пиперазино[1, 2-а]индо-ла.— Фармакол., и токсикол., 1974, № 5, с. 524—530.
9. Андреева Н. И., Глушков Р. Г., Машковский М. Д. Новый антидепрессант ииказан.— Хим.-фарм. журн., 1979, № 4, с. 111—114.
10. Андреева Н. И., Машковский М. Д. Исследование некоторых производных 1,10-триметиленпиразино [1, 2-а]индола.— Фармакол. и токсикол., 1968, № 3, с. 326—329.
11. Арбузов Б. А., Алимов П. И., Зверева М. А. и др. О синтезе некоторых органических производных тиопирофосфориой кислоты.—Изв. АН СССР. Сер. хнм., 1954, № 6, с. 1038—1046.
12. Арендарук А. П., Серебряков Л. А., Сколдинов А. П. Синтез и некоторые вопросы , фармакодинамики у-аминомасляной и у-окси-масляной кислот.—Мед. пром. СССР, 1963, № 5, с. 6—8.
13. Афонская Л. С., Заиконникова И. В., Ржевская Г. Ф., Студенцо-ва И. А. О механизме действия армина и ннбуфнпа. Фармакол. и токсикол., 1963, № 2, с. 184—189.
14. Базилевская Г. И., Гура Д. В., Байнова М. С. и др. Синтез тропан-За-ола, тропина.— Журн. общ. хим., 1958, т. 28, с. 1097—1105.
15. Байнова М. С., Базилевская Г. И., Дюмаев К. М., Преображенский Н. А. Синтез метилового эфира тропанои-З-карбоновой-2 кислоты.— Журн. общ. хим., 1960, т. 30, с. 1120—1123.
16. Бальон Я, Г., Шульман М. Д. Синтез и некоторые превращения арилдихлорметилкарбинолов.— Жури. орг. хим., 1976, т. 12, с. 1973—1979.
255
17. Бальон Я. Г., Шульман М. Д. Синтез 2-о-хлорфеиил-2-п‘-хлорфе-иил-1,1-дихлорэтана.— Хим.-фирм, жури., 1975, № 2, с. 36.
18. Барков Н. К. О фармакологических свойствах карбидина.— Фармакол. и токсикол., 1971, № 6, с. 647—650.
19. Барков Н. К. О механизме действия карбидина.— Фармакол. и токсикол., 1973, № 2, с. 154—157.
20. Баумание Э. А., Горкин В. 3., Калниня И. Э. и др. Влияние фенкарола на активность тканевых аминоксидаз.— Фармакол. и токсикол., 1980, № 1, с. 36—41. 1
21. Белозерова К. А., Елина А. С., Магидсон О. Ю. и др. Антибактериальное средство.— А. с. № 428626 (СССР), Бюлл. изобр., 1957, № 18, с. 151.
22. Березовский В. М., Биринберг Е. М., Глебова Г. Д. Способ получения 4-амино-(дезокси)-10-метилптероилглутаминовой кислоты.— А. с. (СССР), № 245115. Бюлл. изобрет., 1969, № 19, с. 32.
23. Бикова Н., Желязков Л. Изучение синтеза ганглиоблокирующего препарата пемпидина.— Фармация (Болг.), 1961, т. И, № 5, с. 13—17.
24. Блюгер А. Ф. Нитрофураны и их применение в медицине.— Рига. Изд. АН Латв. ССР, 1958.
25. Богатский А. В., Андронати С. А. Современное состояние химии 1,5-беиздиазепииов.— Успехи хим., 1970, т. 39, с. 2217—2255.
26. Богатский А. В., Андронати С. А., Вихляев Ю. И. н др. Вещество, обладающее транквилизирующей, снотворной и противосудорожной активностью. А. с. (СССР), № 484873. Бюлл. изобрет., 1975, № 35, с. 12.
27. Враз Г. И. Получение N'-ацетилсульфаииламида.— Журн. прикл. хнм., 1944, т. 1'7,''С. 508—513.
28. Враз Г. И., Альбицкая О. П., Шкраль М. Л. Сульфаниламид из форманилида.— Журн. прикл. хим., 1944, т. 17, с. 502—505.
29. Враз Г. И., Лизгунова М. В., Чемерисская А. А. Сульфаниламид из дифеиилмочевины.— Журн. прикл. хим., 1946, т. 19, с. 379—384.
30. Брауде М. Б., Ставровская В. И. Синтез 6-метокси-8-(4*-амино-пентил)-аминохинолииа. Журн. общ. хим., 1956, т. 26, с. 878—881.
31. Брауде М. Б., Ставровская В. И. Новый вариант синтеза хино-цида.— Мед. пром. СССР, 1957, № 7, с. 19—23.
32. Вихляев Ю. И. Фармакология и химия.— М.: Медицина, 1965.
33. Войнович Н. Д., Палыга Г. Ф. Противолучевая эффективность мексамина.— Мед. радиол., 1974, № 1, с. 74—86.
34. Волжина О. Н„ Гортинская Т. В., Городецкий Л. Ш. и др. Промышленный метод получения фтазина.— Хим.-фарм. журн., 1971, № 1, с. 54—55.
35. Воробьева В. Я., Янина А. Д., Михлина Е. Е. и др. Получение и некоторые превращения хниуклидона-3.— Хим.-фарм., журн., 1979, Кг 7, с. 86—90.
36. Воронин В. Г., Петрова И. Д., Лексин А. Н., Шемерянкин Б. В. Синтез 5-нитро-8-оксихииолина.— Хим.-фарм. журн., 1976, № 9, с. 82—84.
37. Ворона М. И., Генес С. Г., Гринченко Г. С. и др. Терапевтическая эффективность хлорцикламида.— Клин, мед., 1968, № 9, с. 114— 118.
38. Галегов Г. А. Современное состояние и перспективы развития химиотерапии вирусных инфекций.— Журн. Всесоюз. хим. о-во, 1973, т. 18, № 2, с. 200—207.
39. Гиллер С. А., Жук Р. А., Лидак М. Ю. Аналоги пиримидиниуклео-тидов,— Докл. АН СССР, 1967, т. 176, № 2, с. 332—335.
256
40. Гиллер С. А-, Калнберг Р. 10. ФураЗолИдои.— Рига: Изд. АН Латв. ССР., 1962.
41. Глушков Р. Г., Васильевых Л. Г., Коган 3. С., Горкин В. 3. Синтез и антимоноамииооксидазная активность инказана и его аналогов.— Хим.-фарм. журн., 1981, № 5, с. 58—62.
42. Глушков Р. Г., Дронова Л. Н., Николаева Л. А., Костикова Ю. И. Новый синтез клофелина.— Хим.-фарм. журн., 1978, № 1, с. 93—96.
43. Глушков Р. Г., Корецкая Н. И., Домбровская К- И. и др. Новый метод получения гоматропина.— Хим.-фарм. журн., 1977, № 7, с. 30—35.
44. Глушков Р. Г., Николаева Л. А., Головчинская Е. С. и др. Синтез и противоопухолевая активность 6-диэтилеиимидофосфамидо-и 6-триэтиленнмииофосфазопуринов.— Хим.-фарм. журн., 1981, № 3, с. 16—20.
45. Голдырев Л. Н<, Постовский И. Я. К строению и фармакологическому действию веществ, содержащих группу SOjNHR.— Журн. прикл. хим. 1938, т. 11, с. 316—330.
46. Голиков С. Н., Розенгарт В. И. Холинэстеразы и аитихолинэсте-разные вещества.— Л.: Медицина, 1964.
47. Головчинская Е. С. К синтезу сульфамидных препаратов.— Журн. прикл. хим., 1945, т. 18, с. 647—652.
48. Гортинская Т. В., Ныркова В. Г., Савицкая Н. В., Щукина М. Н.— Синтез замещенных 3,4-диазафеноксазина.— Хим. гетероцикл, соед., 1971, № 6, с. 750—754.
49. Гортинская Т. В., Ныркова В. Г., Савицкая Н. В., Щукина М. Н. Синтез 2- и 2,10-замещенных 3,4-диазафеноксазииа.— Хим.
гетероцикл, соед., 1971, К» 8, с. 1033—1035.
50. Гортинская Т. В., Ныркова В. Г., Савицкая Н. В. и др. Метод получения азафена.— Хим.-фарм. журн., 1973, № 6, с. 21—22.
51. Гортинская Т. В., Шеина Н. П., Щукина М. Н. Некоторые производные З-метокси-6-(сульфанила мндо)пирндазина.— Мед. пром. СССР, 1960, № 9, с. 23—25.
52. Грандберг И. И., Афонина Н. И., Зуянова Т. И. Новый путь синтеза 1-замещенных триптаминов.— Хим. гетероцикл, соед., 1958, № 6, с. 1038—1040.
53. Григоровский А. М., Гангрский П. А. Получение стрептоцида из п-хлорбеизосульфокислоты.— Мед. пром. СССР, 1955, № 3, с. 23—26.
54. Гринев А. Н., Веневцева Н. К. Основания Манниха в ряду производных 5-оксибензофурана —Журн. общ. хим., 1963, т. 33, с. 820—824.
55. Гринев А. Н., Крафт М. Я., Бородина Г. М. и др. Новый противовирусный препарат теброфен.— Хим.-фарм. журн., 1970, № 9, с. 61—62.
56. Гринев А. Н., Панишева Е. К-, Столярчук А. А., и др. Новый лекарственный препарат феникаберан.— Хим.-фарм. жури., 1979, № 11, с. 118—121.
57. Гринев А. Н., Романова О. Б., Кричевский Э. С. и др. Синтез и психотропная активность новых производных 1,10-триметилен-1,2,3,4-тетрагидропиразино[1,2-а] нидола.— Хим.-фарм. журн.,
1980, № 8, с. 32—37.
58. Гринев А. Н., Урецкая Г. Я., Рябова С. Ю. и др. Новый противовирусный препарат флореиаль.— Хим.-фарм. жури., 1976, № 10, с. 115—119.
59. Гринев А. Н., Шведов В. И., Столярчук А. А. и др. Феникаберан.— Хим.-фарм. журн., 1977, № 6, с. 142—143.
17 Заказ № 4304
257
60. Гриценко А. Н., Ермакова 3. И., Журавлев С. В. СпнТеЗ этмо-зина.— Хим.-фарм. жури., 1972, № 9, с. 17—19.
61. Гриценко А. Н., Ермакова 3. И., Журавлев С. В. и др. Синтезы в ряду фентиазина.— Хнм.-фарм. журн., 1971, № 7, с. 10—14.
62. Гриценко А. И., Ермакова 3. И., Журавлев С. В. и др. Фтораци-зин и его аналоги.— Хим.-фарм., журн., 1971, № 6, с. 18—20.
63. Гриценко А. Н., Журавлев С. В. Получение хлорацизина.— Мед. пром. СССР, 1960, № 7, с. 25—27.
64. Гриценко А. Н., Журавлев С. В., Скородумов В. А. Методы получения химических реактивов и препаратов. М., 1964, вып. 10, с. 5—7.
65. Громова Е. А. Серотонин и его роль в организме.— М.: Медицина, 1966.
66. Дворянцева ГЖГ'.} Луценко А. И., Ульянова Т. Н. н др. Спектры ЯМР 13С противоопухолевого препарата проспидин и родственных соединений. Докл. АН СССР, 1979, т. 245, № 6, с. 1410^—1414.
67. Денисенко П. П. Центральные холииолитики.— Л.: Медицина, 1965.
68. Дитилин и опыт его клинического применеиия/Под ред. А. М. Мнджоян.— Ереван: Изд-во АН Арм. ССР, 1957.
69. Добрина В. А., Иоффе Д. В., Кузнецов С. Г., Чигарев А. Г. Пирроксан и родственные соединения.— Хим.-фарм. журн., 1974, Кг 6, с. 14—17.
70. Елина А. С., Падейская Е. Н. N-Окнси производных хиноксалина и их химиотерапевтическое действие.— В кн.: Сборник трудов Всесоюзн. науч.-исслед. хим.-фарм. ин-та. М., 1971, вып. 2, с. 197—209.
71. Елина А. С., Титкова Р. М., Цырульникова Л. Г., Филипенко Т. Я. Синтез биологически активных N-окисей ацетокси- и оксиметил-хииоксалинов.— Хим.-фарм. журн., 1976, № 1, с. 44—48.
72. Елина А. С., Цырульникова Л. Г., Магидсон О. Ю. Способ получения 1,4-ди-М-окиси-2,3-бис (оксиметил) хиноксалина.— А. с.
№ 207917, Бюлл. изобретений, 1968, № 3, с. 19.
73. Ельцов А. В., Чигарев А. Г., Старых Н. Т. N-аралкильные производные фенилпирролидинов.— Журн. общ. хим., 1964, Т. 34, с. 3344—3351.
74. Ельцов А. В., Чигарев А. Г., Старых Н. Т. Биологически активные соединения.— Л.: Медицина, 1965.
75. Жеребченко П. Г. Противолучевые свойства нндолилалкилами-нов.— М.: Атомиздат, 1971.
76. Жунгиету Г. И., Дорофеенко Г. Н. Успехи в области стероидных гетероциклов.— Успехи химии, 1967, т. 36, с. 48—76.
77. Жукова 3. Н., Балякина М. В., Гунар В. И. Синтез пиридито-ла.— Хим.-фарм. журн., 1978, № 11, с. 77—80.
78. Журавлев С. В., Гриценко А. Н. Синтез диалкиламиноацил-2-хлорфептиазинов.— Журн. общ. хим., 1956, т. 26, с. 3385—3391.
79. Журавлев С. В., Гриценко А. Н, Синтез эфиров фентиазинкарба-миновой-2-кислоты.— Журн. общ. хим., 1963, т. 33, с. 1596—1601.
80. Журавлев С. В., Скородумов В. А. Амины ряда фентиазииа.— Журн. общ. хим., 1961, т. 31, с. 3129—3132.
81. Заева С. П., Гиллер С. А., Германе С. К. и др. Экспериментальное изучение нового препарата — фуразолнна. Журн. микробпол., 1961, № 10, с. 17—20.
82. Закусав В. В. Вопросы психофармакологии.— М.: Медицина, 1967.
83. Закусов В. В. Новый психотропный препарат феиазепам.— Хим.-фарм. журн., 1979, № 10, с. ’108—112.
258
84. Засосов В. А. Современные сульфамидные препараты.— Журн. Всесоюз. хим. о-ва, 1965, т. 10, Кв 6, с. 671—678.
85. Засосов В. А.— В кн.: Сборник трудов ВНИХФИ. М.: Медицина, 1971, вып. 2, с. 105—128.
86. Засосов В. А., Акифьева Т. Н„ Веселовская Т. А. К синтезу производных сульфонилбутилмочевииы.— Мед. пром. СССР, i960, № 1, с. 7—12.
87. Засосов В. А., Блинова Л. С., Сычева В. Н., Соколова Г. Н. Упрощенный способ получения карбометоксисульфанилмочеии-иы.— Хим.-фарм. жури., 1981, № 1, с. 42—43.
88. Зимакова И. Е. Фармакология мебикара.— Фармакол. и токсикол., 1977, Кв 6, с. 684—687.
89. Имифос:—Сборник/Под ред. С. А. Гиллера. Рига: Зинатне, 1968.
90. Иоффе И. С., Забоев С. А., Сухомлинов Ф. К., Антипович А. И. Синтез этамида.— Труды Воен.-мед. акад. им. С. М. Кирова, 1958, т. 83, с. 26—29.
91. Кабачник М. И., Абдувахабов А. А., Агибекова И. И. и др. Гидрофобные области активной поверхности холинэстераз.— Успехи хим., 1970, т. 39, с. 1050—1073.
92. Каверина Н. В., Климова Н. В., Лихошерстов А. М. и др. Нона-хлазии — новый препарат для лечения ишемической болезни сердца.— Хим.-фарм. журн., 1979, № 5, с. 115—121.
93. Каверина Н. В., Маркова Г. А., Чичканов Г. Г. и др. Синтез и аитиаигинальиые свойства нонахлазииа и иоиафтазина.— Хим.-фарм. журн., 1978, К» 7, с. 97—101.
94. Каверина Н. В., Сенова 3. П., Вихляев Ю. И., Ульянова О. В. О противоаритмических свойствах этмозина.— Фармакол. н
токсикол., 1970, № 6, с. 693—698.
95. Каверина И. В., Сенова 3. П-, Митрофанов В. С., Рунова М. Ф. К фармакологии эмтозина.— Фармакол. и токсикол., 1972, № 2, с. 182—185.
96. Каминка М. Э. Препараты серотонина.— Хим.-фарм. жури., 1971, Кв 5, с. 51—54.
97. Каминка М. Э. Фенкарол — противогистаминный препарат.— Фармакол. и токсикол., 1977, Кв 2, с. 158—162.
98. Каминка М. Э., Воробьева В. Я., Голованова Е. А. и др. Фармакокинетика и метаболизм лекарственных препаратов.— М.: ВНИХФИ. В кн.: Труды ВНИХФИ, 1978, вып. 7, с. 109—119.
99. Каминка М. Э., Михлина Е. Е., Воробьева В. Я. и др. Синтез и изучение нового противогистаминиого препарата фенкарола.—
Хим.-фарм. журн., 1976, № 6, с. 48—53.
100. Камянов И. М., Полис Я. Ю. Куписк А. Г. Мидантан и его применение в профилактике нейролептического синдрома.— Рига: Зи-иатне, 1973.
101. Катышкина В. В., Крафт М. Я. О получении хлорангидридов н эфиров арилфосфиновых кислот.— Жури. общ. хим., 1956, т. 26, с. 3060—3066.
102. Кименис А. А., Витолинь Р. О., Клуша В. Е., Камянов И. М. Мидантан как средство для лечения паркинсонизма.— Хим.-фарм. журн., 1976, № 10, с. 144—146.
103. Кирби А., Уоррен С. Органическая химия фосфора.— М.: Мир, 1971.
104. Ковалев И. Е. Биологическая активность адамаитансодержащих веществ.— Хим.-фарм. журн., 1977, № 3, с. 19—27.
17*
259
105. Ковлер М. А., Абакумов В. М., Кругликова-Львова Р. П. и др. Пантогам — новое 'психофармакологическое средство.— Хим.-фарм. жури., 1980, № 9, с. 118—121.
106. Ковлер М. А., Абакумов В. М., Кругликова-Львова Р. П. и др. Пиридитол — препарат метаболической терапии для нервно-психической практики.— Хим.-фарм. журн., 1980, № 7. с. 117—121.
107. Комиссаренко В. П„ Резников А. Г., Комиссаренко И. В. и др. Хлодитан.— Хим.-фарм. жури., 1977, Кз 9, с. 146—149.
108. Копелевич В. М., Евдокимова Г. С., Мариева Т. Д„ Шмуйло-вич Л. М. Синтез D-гомопаитотеновой кислоты.— Хим.-фарм. журн. 1971, № 9, с. 21—22.
109. Копелевич В. М., Сытинский И. А., Гунар В. И. Современный подход к созданию ноотропных средств на основе у-аминомасля-ной кислоты.— Хим.-фарм. журн., 1981, № 5, с. 27—39.
ПО. Кочетков Н. К, Кучерова Н. Ф., Жукова И. Г. Синтез некоторых производных 1,2,3,4,4а,9а-гексагидро-у-карболина.— Жури. общ. хим., 1961, т. 31, с. 924—930.
111. Кочетков Н. К, Кучерова Н. Ф„ Пронина Л. П., Петручен-ко М. И. 6-Замещенные 1,2,3,4-тетрагидро-у-карболинов.— Жури, общ. хим., 1959, т. 29, с. 3620—3625.
112. Крафт М. Я. Перспективы химиотерапии вирусных заболеваний.— Журн. Всесоюзи. хим. о-ва, 1965, т. 10, № 6, с. 630—636.
113. Крылов С. С., Старых Н. Т. Фармакологическая характеристика пирроксаиа.— Фармакол. и токсикол., 1973, № 4, с. 396—399.
114. Кузнецов С. Г., Голиков С. Н. Синтетические атропиноподобные вещества.— Л.: Медицина, 1962.
115. Кузнецов С. Г., Ельцов А. В. Некоторые новые амииоалкиловые эфиры бензиловой кислоты.— Журн. общ. хим., 1962, т. 32, с. 511—515.
116. Кузовков А. Д., Массагетов П. С., Рабинович М. С. Исследование аконитовых алкалоидов.— Журн. общ. хим., 1955, т. 25, с. 157—161.
117. Кузовков А. Д'('Г Машковский М. Д., Данилова А. В., Меньшиков Г. П. Получение курареподобиых препаратов.— Докл. АН СССР, 1955, т. 103, № 2, с. 251—252.
118. Кучерова Н. Ф., Жукова И. Г., Комзолова Н. Н. и др. 9-Ацил-1,2,3,4,4а,9а-гексагидро-у-карболины.— Жури. общ. хим., 1961, т. 31, с. 930-936.
119. Кучерова Н. Ф., Кочетков Н. К. Синтез некоторых производных 1,2,3,4-тетрагидро-у-карболина.— Журн. общ. хим., 1956, т. 26, с. 3149—3154.
120. Лаздиньш Л. Я., Авотс А. А., У ласте В. К- и др. Оптимизация синтеза дипИроксима.— Хим.-фарм. жури., 1973, № 2, с. 38—41.
121. Лакин К. М. Антикоагулянтная активность нафарина.— Фармакол. и токсикол., 1965, № 3, с. 329—333.
122. Лакин К. М., Смирнова Т. В., Карасева М. Г., Фельдбаум В. А. Действие нитропроизводных ва'рфарина на свертывание крови.— Фармакол. и токсикол., 1967, № 4, с. 456—460.
123. Ларионов Л. Ф. О некоторых направлениях в создании противоопухолевых препаратов.— Вести. АМН СССР, 1965, № 4, с. 26—34.
124. Ларионов Л. Ф-, Софьина 3. П. Вопросы экспериментальной и клинической онкологии/Под ред. Н. Н. Блохина.— М.: Медицина, 1972.
125. Левитан М. X. Сравнительная оценка салазопиридазина и сульфасалазина.— Хим.-фарм. жури., 1976, № 12, с. 128—132.
260
126. Левхоева Е. И., Мастафанова Л. И., Краснокутская Д. М. и др. Этерификация пиридннкарбоновых кислот в присутствии ионитов.— Хим. гетероцикл, соед., 1976, № 2, с. 233—237.
127. Ледер-Пакендорф Л. С. О циклогексил-геранил-уксусной кислоте.— Докл. АН СССР, 1941, т. 31, № 8, с. 756—759.
128. Лейтис Л. Я., Сколмейстере Р. А., Голендер Л. О., Шиманская М. В. Каталитический синтез и превращения гетероциклических соединений.— Рига: Зинатне, 1976.
129. Либерман С. С. Поиски новых холинолитических и спазмолитических средств.— Фармакол. и токсикол., 1956, № 6, с. 10—17.
130. Либерман С. С. Анальгезирующая активность эстоцина.— Фармакол. и токсикол., 1968, № 6, с. 668—671.
131. Либерман С. С., Кутчак С. Н„ Рудзит Э. А., Шахназарова Н. Г. Противовоспалительная активность эстоцина.— Фармакол.. и токсикол., 1972, № 3, с. 333—335.
132. Либерман С. С., Полежаева А. И. Аналгезирующее и противокаш-левое действие некоторых эфиров дифенилалкоксиуксусной кислоты.— Фармакол. и токсикол., 1963, № 6, с. 656—659.
133. Либерман С. С., Яхонтов Л. Н. Зависимость фармакологической активности от строения производных диарилжирных кислот.— Хим.-фарм. журн., 1971, № 2, с. 7—19.
134. Лукомский П. Е„ Казьмина П. В. Лечение антикоагулянтными и фибринолитическими средствами. Каунас, 1967, с. 115—119.
135. Лукьянов А. В., Оноприенко В. С., Бородина К. С. и др. п-Нитро-бензолсульфохлорид как сырье для производства сульфамидных препаратов.— Хим.-фарм. журн., 1980, № 9, с. 87—91.
136. Магидсон О. Ю., Федосова В. М. Химия и медицина. Новые средства для лечения паркинсонизма и других заболеваний центральной нервной системы.—М.: Медгиз, 1956, с. 14—21.
137. Максимов С. В., Симон Й. Б. О некоторых средствах с гипохо-лестеринемическим действием.— Врач, дело, 1959, №2, с. 119—123.
138. Махиенко Н. И., Сысоева Т. Ф. Хлоцикламид как антидиабетическое средство.— Труды/Украин. ин-т экспериментальной эндокринологии, 1961, т. 18, с. 333—338.
139. Машковский М. Д. О курареподобных свойствах алкалоида тезина и его дийодметилата.— Фармакол. и токсикол., 1955, № 6, с. 3—9.
140. Машковский М. Д. Лекарственные средства.— М.: Медицина, 1977.
141. Машковский М. Д., Альтшулер Р. А., Авруцкий Г. Я. и др. Сиднокарб и синдофеи — новые психотропные препараты.— Жури, невропатол. и психиатр., 1971, № 11, с. 1704—1708.
142. Машковский М. Д., Андреева Н. И. Экспериментальное изучение психотропного действия пиразидола.— Фармакол. и токсикол., 1975, № 1, с. 5—10.
143. Машковский М. Д., Горкин В. 3., Андреева Н. И. и др. Исследование механизма действия пиразидола.— Фармакол. и токсикол., 1975, № 5, с. 531—536.
144. Машковский М. Д., Гринев А. Н., Андреева И. И. и др. Новый антидепрессивный препарат пиразидол.— Хим.-фарм. жури., 1974, № 3, с. 60—63. .
145. Машковский М. Д., Гринев А. Н., Андреева Н. И. и др. Исследование психотропного действия производных 1,10-триметиленпира-зино[1,2-а] индола.— Фармакол. и токсикол., 1971, № 4,
с. 387—391.
146. Машковский М. Д., Зайцева К. А. Андренолитическне свойства эфиров тропина.— Бюлл. экспер, биол., 1956, № 1, с. 56—61.
261
147. Машковский М. Д-, Каминка М. Э. Фармакологические свойства некоторых солей 5-окситриптамина.— Фармакол. И токсикол.,
1970, № 6, с. 673—675.
148. Машковский М. Д., Полежаева Л. И. Фармакологические свойства нового аитидепрессивиого препарата азафена.— Фармакол. и токсикол., 1969, № 6, с. 656—662.
149. Машковский М. Д., Полежаева А. И., Андреева Н. И. Антидепрессанты (обзор).— Хим.-фарм. жури., 1979, № 6, с. 19—29.
150. Машковский М. Д., Полежаева А. И., Андреева Н. И. и др. Поиск новых аитидепрессивных препаратов и изучение механизма их действия.— Хим.-фарм. жури., 1978, № 2, с. 38—50.
151. Машковский М. Д., Раевский К. С., Майсов Н. И. и др. Влияние пиразидола и сидиокарба иа захиат нейромедиаторов сииаптосо-мами мозга.— Хим.-фарм. жури., 1976, № 8, с. 3—6.
152. Машковский М. Д., Рощина Л. Ф. Влияние азафена на биоэлектрическую активность головного мозга.— Фармакол. и токсикол., 1971, № 2, с. 144—148.
153. Машковский М. Д., Садритдинов Ф. Курареподобные свойства квалидила.— Фармакол. и токсикол., 1962, № 6, с. 685—691.
154. Машковский М.' Д., Шварц Г. Я., Либерман С. С. и др. Фармакологическое и токсикологическое исследование парамидина.— Хим.-фарм. жури., 1981, № 2, с. 117—121.
155. Машковский М. Д., Яшунский В. Г., Альтшулер Р. А. и др. Лекарственное средство.— А. с. (СССР), № 329890. Бюлл. изобретений, 1972, № 8, с. 8.
156. Медведев Б. А. Новый гипотензивный препарат клофелин.— Хим.-фарм. жури., 1980, № 1, с. 115—117.
157. Мельников Н. Н. Химия пестицидов.— М.: Химия, 1968.
158. Мельников Н. Н., Хаскин Б. А., Першин Г. Н. и др. Октатион — новый препарат для лечения дерматомикозов.— Хим.-фарм. жури., 1973, № 3, с. 59—62.
159. Мельников Н. Н., Хаскин Б. А., Швецова-Шиловская К- Д-О взаимодействии эфиров тио- и дитиофосфориых кислот с первичными аминами.— Жури. общ. хим., 1962, т. 32, с. 1836—1838.
160. Михалев В. А., Дорохова М. И., Смолина Н. Е. Синтез производных диспиротрипиперазииия, обладающих каицеролитической активностью.— Мед. пром. СССР, 1963, № 1, с. 17—20.
161. Михалев В. А., Дорохова М. И., Смолина Н. Е., Тихонова О. Я-О взаимодействии бис (Д-хлорэтил) амина с а-окисями.— Журн. общ. хим., 1964, т. 34, с. 3716—3719.
162. Михалев В. А., Дорохова М. И., Смолина Н. Е„ Тихонова О. Я. Производные N', N''-диспиротрипиперазиния.— Жури, орг. хим., 1965, т. 1, с. 460—464.
163. Михалев В. А., Скалдинов А. П. Феннлуретиланы как сырье для получения сульфаниламида и его производных.— Жури, прикл. хим., 1946, т. 19, с. 1373—1380.
164. Михалев В. А., Чернов В. А., Дорохова М. И. и др. .Спиразидин, проспидии и другие производные диспиротрипиперазииия.— Хим.-фарм. журн., 1972, № 6, с. 9—14.
165. Михлина Е. Е!,' Воробьева В. Я-, Рубцов М. В. Синтез галогенидов полиметилен-бис-хииуклидииия.— Жури. общ. хим., 1961, т. 31, с. 2609—2613.
166. Михлина Е. Е„ Янина А. Д., Воробьева В. Я. и др. Синтез и свойство (хинуклидил-З)-диарилкарбинолов.— Хим. гетероцикл,
соед., 1976, № 7, с. 935—939.
.262
167. Михлина Е. Е., Янина А. Д„ Воробьева В. Я. и др. Синтез хино-цнда.— Хим.-фарм. журн., 1973, № 10, с. 3—6.
168. Михлина Е. Е„ Янина А. Д., Яхонтов Л. Н. О свойствах и превращениях у-ацетопропилового спирта.— Хим.-фарм. жури., 1970, № 6, с. 26—28.
169. Мостова М. И., Иоффе Д. В., Малько И. Л. и др. Изучение окси-этилироваиия фенилацетоиитрила.— Хим.-фарм. жури., 1980, № 6, с. 58—61.
170. Назарова Л. С., Лихошерстов А. М., Маркова Г. А. и др. Аза-циклоалкаиы.— Хим.-фарм. журн., 1978, № 6, с. 84—89.
171. Несмеянов А. Н., Богомолова Л. Г., Андрианова И. Г. и др. Новый препарат ферроцерои.— Хим.-фарм. жури., 1972, X» 4, с. 61—62.
172. Несмеянов А. Н., Захаркин Л. И., Фрейд лина Р. X. Получение и константы диссоциации карбоновых кислот строения СС1Э(СН2)„СООН и СС12=СН(СН2)пСООН.
Изв. АН СССР. Сер. хим., 1955, Ns 1, с. 40-47.
173. Никитская Е. С., Левкоева Е. И., Пучков В. А. и др. Изучение продуктов взаимодействия триацетонамина и циапуксусиого эфира.— Хим. гетероцикл, соед., 1970, № 5, с. 642—646.
174. Никитская Е. С., Левкоева Е. И., Усовская В. С. и др. Синтез 2,2,6,6-тетраметилхииуклидина.— Хим. гетероцикл, соед., 1971, № 2, с. 230-234.
175. Никитская Е. С., Шарапов И. М„ Левкоева Е. И. и др. Синтез нового ганглиоблокирующего препарата темехииа.— Хим.-фарм.
журн., 1970, № 10, с. 58—61.
176. Никитская Е. С., Шарапов И. М., Машковский М. Д. та др. Синтез и фармакологические свойства имехииа.— Хим.-фарм. жури., 1974, № 8, с. 59—60.
177. Никоноров К. В. Синтез некоторых эфиров диалкилфосфонтри-хлорэтилфосфориой кислоты.— Изв. АН СССР. Серия хим., 1958, № 11, с.. 1340—1344.
178. Никоноров К. В., Никоненко В. А. Синтез некоторых эфиров ацетокситрихлор-этилфосфиновой кислоты,— Изв. АН СССР. Сер. хим., 1962, № 10, с. 1882—1884.
179. Никулина Т. Н., Оноприенко В. С., Бушуева К. С., Засосов В. А. Об усовершенствовании способа получения этазола.— Хим.-фарм. жури., 1971, № 9, с. 32—35.
180. Новиков С. С-, Хмельницкий Л. И., Лебедев О. Л. и др. Способ получения 2,4,6,8-тетраметил-2,4,6,8-тетраазабицикло-3,3,0-октадио-на-3,7. А. с. № 419619.— Бюлл. изобр., 1975, К» 42, с. 59.
181. Новые данные по фармакологии и клинике производных феио-тиазинового ряда.— М.: Медицина, 1958.
182. Ныркова В. Г., Гортинская Т. В., Щукина М. Н. Синтез 2-хлор-3,4-диазафеноксазина.— Жури. общ. хим., 1964, т. 34, с. 3132— 3135.
183. Ныркова В. Г-, Гортинская Т. В., Щукина М. Н. Синтез новой гетероциклической системы 3,4-дназафеиоксазииа,— Жури. орг. хим., 1965, т. 1, с. 1688—1691.
184. Основные направления работ ВНИХФИ.— М.: Медицина, 1959.
185. Падейская Е. Н. Поиски новых химиотерапевтических препаратов в ряду хиноксалина и их N-окисей.— В ки.: Новые химиотерапевтические препараты для лечения больных инфекционными заболеваниями. М., 1976, с. 89—103.
263
186. Падейская Е. Н., Елина А. С., Першин Г. Н. и др. Химиотерапевтическая активность производных хиноксалина и N-окисей хиноксалина при острых бактериальных инфекциях.— Фармакол. и токсикол., 1967, № 5, с. 617—626.
1S7. Падейская Е. Н., Полухина Л. М. Фтазин —новый сульфаниламидный препарат для лечения кишечных инфекций.— Мед. пром. СССР, № 6, с. §5х-57.
188. Падейская Ё. И'.', Полухина Л. М. Новые сульфаниламидные препараты длительного действия для лечения инфекционных заболеваний.— М.: Медицина, 1974.
189. Падейская Е. Н., Полухина Л. М., Першин Г. Н. Экспериментальное изучение салазопиридазииа.— Фармакол. и токсикол., 1969, № 4, с. 467—472.
190. Падейская Е. Н., Полухина Л. М., Першин Г. Н. Экспериментальное изучение фаиазила и метофазина.— Фармакол. и токсикол., 1971, №> 5, с. 609—612.
191. Парин В. В. Влияние карбидина на продолжительность и силу действия наркотических веществ.— Фармакол. н токсикол., 1973, № 2, с. 157—161.
192. Переводчикова И. И. Клиническая химиотерапия опухолевых заболеваний.— М.: Медицина, 1976.
193. Перекалин В. В., Сопова А. С„ Зобачева Н. М. и др. Лекарственное средство.— А. с. № 236479 (СССР).—Бюлл. изобр., 1970, № 34, с. 24.
194. Першин Г. Н, Химиотерапия и химиопрофилактика гриппа и других острых респираторных заболеваний.— Фармакол. и токсикол., 1974, № 5, с. 517—524.
195. Першин Г. Н., Богданова Н. С. Химиотерапия вирусных инфекций.— М.: Медицина, 1973.
196. Першин Г. Н., Богданова И. С., Знаева К. И., Крафт М. Я-Некоторые закономерности в подавлении -размножения вируса гриппа синтетическими соединениями.— Фармакол. и токсикол., 1961, Ns 6, с. 690—694.
197. Першин Г. Н., Крафт М. Я., Богданова Н. С. и др. Новый противовирусный препарат оксолин.— Хим.-фарм. пром., 1970, № 11, с. 56—58.
198. Першин Г. И., Кукушкина Т. С., Гуськова Т. А. Эсулаи.— Хим.-фарм. журн., 19,76, № 11, с. 146—150.
199. Писько Г. Т., Гладышевская Л. И., Кириенко Ю. А.— В кн.: Развитие медицинской науки и здравоохранения на Северной Буковине за годы Советской власти.— Киев, Изд-во АН УССР, 1969, с. 170—177.
200. Полежаева А. И., Ветроградова О. П., Багреева М. Р. Новый антидепрессивиый препарат азафен.— Хим.-фарм. жури., 1970, № 2, с. 59—61.
201. Полис Я. Ю. Современные аспекты исследований в области фармации.— Рига: Зннатне, 1977.
202. Пономарев А. А., Мартемьянова И. И. Синтез битиодииа,—Хим. гетероцикл, соед., 1967, № 1, с. 174—175.
203. Предвадителева Г. С., Щукина М. И. Новый вариант синтеза диакарба,—Мед. пром. СССР, 1959, № 9, с. 24—26.
204. Проспидин — новое противоопухолевое средство: Сборник ста-тей/Под ред. В. А. Чернова.—М.: Медицина, вып. 3, 1973.
205. Проценко Л. Д., Корнев К. А. Синтез и изучение дийодбензотэ-фа.— Укр. хим. жури., 1958, т. 24, с. 636—639.
264
206. Проценко Л. Д., Радионов П. В. Пути синтеза и изыскания противоопухолевых препаратов.— Рига: Зииатне, 1970, вып. 3, с. 69—76.
207. Прянишникова Н. Т., Лебедева А. С., Лихошерстов А. М. и др. Пиромекаин — новый препарат для поверхностной анестезии.— Хим.-фарм. жури., 1975, № 5, с. 59—62.
208. Пурдела Д., Вылчану Р. Химия органических соединений фосфора.— Пер. с рум.— М.: Химия, 1972.
209. Разумов А. И., Маркович Е. А., Решетникова А. Д. Смешанные эфиры первичных фосфиновых кислот.— Жури. общ. хим., 1957, т. 27, с. 2394—2396.
210. Разумов А. И., Мухачева О. А., Заиконникова И. В. Эфиры и амиды диалкилфосфиновых кислот и их биологическая активность.— Жури. общ. хим., 1957, т. 27, с. 754—757.
211. Рубцов М. В., Байчиков А. Г. Синтетические химико-фармацевтические препараты.— М.: Медицина, 1971.
212. Рубцов М. В., Никитская Е. С. Синтетические диаза- и азаби-циклические системы с азотом не в узловой точке.— Успехи хим., 1965, т. 34, с. 1040—1070.
213. Рубцов М. В., Федосова В. М. Производные сульфаниламида,— Жури. общ. хим., 1944, с. 14, с. 848—856.
214. Руденко Г. М. Новые психотропные средства пиразидол и феназепам.— Хим.-фарм. жури., 1979, К? 4, с. 114—120.
215. Садритдинов Ф. Влияние бисчетвертичных соединений хинуклиди-на иа передачу нервного возбуждения в симпатических ганглиях и на нервно-мышечную проводимость.— Фармакол. и токсикол., 1962, № 3, с. 327—335.
216. Сафонова Т. С. Химия алкилирующих препаратов.— Журн. Все-союзн. хим. о-ва, 1973, т. 18, № 6, с. 657—669.
217. Сергель Е. В. Результаты применения этамида как пролоигато-ра действия пенициллина.— Сов. мед., 1961, № 11, с. 103—106.
218. Синтезы органических препаратов: Пер. с англ. М.: Иностр, лит., 1949—1959.
219. Смирнова А. А., Перекалин В. В. Синтез у-амииокислот и а-пир-ролидоиов.— Жури. орг. хим., 1968, т. 4, с. 2245—2255.
220. Смирнова Т. В., Дукельская Н. М., Горбунова В. П. и др. Исследования кумариновых антикоагулянтов.— Химия и хим.
технол., 1962, т. 5, с. 107—ПО.
221. Соколов Г. П., Воронков М. Г., Гиллер С. А. Синтез циклических диацеталей янтарного диальдегида.— Изв. АН ЛатвССР. Сер. хим., 1964, № 6, с. 667—672.
222. Соколов Г. П., Гиллер С. А., Кименис А. А. и др. Диоксоний— новое кура реподобное средство.— Хим.-фарм. журн., 1974, № 2, с. 60.
223. Соколов Г. П., Данилов А. Ф-, Михельсон М. Я., Протас Л. Л. Бисчетве'ртичиые соли стереоизомеров циклических бисацеталей.— Хим.-фарм. жури., 1977, № 2, с. 47—5k
224. Соколов Г. П., Клуша В. Е., Кименис А. А., Гиллер С. А. Синтез и курарезующее действие четвертичных солей бис-аминометпл-диоксоланилэтана.— Хим.-фарм. журн., 1968, № 3, с. 3—7.
225. Соркина Ю. А., Чернов В. А. О противоопухолевой активности некоторых производных пиперазина.— Вопр. оикол., 1962, № 1, с. 77—81.
226. Страдынь П. И., Цеплите Р. К. Опыт лечения онкологических больных тиофосфорамидами.— Клин, мед., 1958, т. 36, № 7,
с. 135—137.
265
227. Суворов Н. Н., Гордеев Е. Н., Васин М. В. Синтез и биологическая активность некоторых триптаминов.— Хим. гетероцикл, соед., 1974, № II, с. 1496—1501.
228. Суворов Н. Н., Мурашова В. С. Новый синтез триптаминов.— Мед. пром. СССР, 1961, № 1, с. 6—11.
229. Суворов Н. Н., Шашков В. С. Химия и фармакология средств профилактики радиационных поражений.—М.: Атомиздат, 1975.
230. Сытинский И. А. Средства для лечения заболеваний головного мозга, воздействующие на метаболизм у-амииомасляиой кислоты.— Жури. Всесоюзн. хим. о-ва, 1973, т. 18, № 2, с. 182—192.
231. Терентьев А. П„ Преображенская М. Н., Бан-Лун-Ге. Восстановление нитрилов гидразиигидратом в присутствии никеля Ренея.— Хим, наука и хим. пром., 1959, Т. 4, с. 281—282.
232. Успехи в создании новых лекарственных средств/Под ред. Д. А. Харкевича.— М.: Медицина, 1973.
233. Урецкая Г. Я., Крафт М. Я. Карбонильные производные ряда флуорена.— Жу"ри. общ. хим., 1963, т. 33, с. 3053—3056.
234. Федосова В. М., Магидсон О. К>. Производные 0- (4-метоксифе-иил)-а-фенилпропиоиовой кислоты.— Журн. общ. хим., 1956, т. 26, с. 3475—3479.
235. Филатов А. Н., Андрианова И. Г. Препараты для лечения железо-дефицитиой анемии.— Хим.-фарм. журн., 1970, № 4, с. 45—48.
236. Фурагин и солафур/Под ред. С. А. Гиллера.— Рига: Зииатие, 1968.
237. Фурациллин и опыт его применения.Под ред. С. А. Гиллера.— Рига: Зииатне, IQ53.
238. Хараг И. М., Явлинский М. Д., Савин Б. М. Усовершенствованный синтез бутамида.— Мед. пром. СССР, 1962, № 2, с. 39—43.
239. Харкевич Д. А. Фармакология курареподобных средств.— М.: Медицина, 1969.
240. Харкевич Д. А., Сколдинов А. П. Новые антагонисты ацетилхолина.— Жури. Всесоюз. хим. о-ва, 1970, т. 15, № 2, с. 145—155.
241. Харкевич Д. А., Сколдинов А. П„ Арендарук А. П. и др. Циклобутоний — новое отечественное кура реподобное средство.— Хим.-фарм. жури., 1977, № 2, с. 145—150.
242. Харкевич Д. А., Сколдинов А. П., Арендарук А. П. и др. Анатруксоний — новое кура реподобное средство недеполя’ризующего действия.— Хим.-фарм. жури., 1974, № 4, с. 59—62.
243. Химиотерапия злокачественных опухолей/Под ред. Н. Н. Блохина.— М.: Медицина, 1977.
244. Черкасова Е. М„ Прянишникова Н. Т., Богатков С. В., Еркомай-швили Г. С Успехи в химии анестетиков.— Успехи хим., 1973, т. 42, с. 1892—1919.
245. Чернов В. А. Цитостатические вещества в химиотерапии злокачественных новообразований.— М.: Медицина, 1964.
246. Чернов В. А., Сергиевская С. И., Кропачева А. А. О противоопухолевой активности некоторых новых фосфорамидов.— Фармакол. и токсикол., 1956, № 4, с. 30—36.
247. Чигарев А. Г., Иоффе Д. В. Синтез и превращения 0-фенил-0-цнаипропионовой кислоты.— Жури. орг. хим., 1967, т. 3, с. 85—88.
248. Чигарев А. Г., Иоффе Д. В. Синтез адренолитических веществ.— Хим.-фарм. журн., 1968, We 8, с. 25—30.
249. Шарапов И. М. К фармакологии темехина.— Фармакол. и токсикол., 1972, № 6, с. 687—690.
250 Шарапов И. М. К фармакологии имехина.— Фармакол. и токсикол., 1974, № 1, ^с. у25—29.
266
251. Шварц Г. Я. АиТисеротоиииОвая активность тропафеиа.— Хим.-фарм. жури., 1977, № 1, с. 89—90.
252. Шведов В. И., Алтухова Л. Б., Андреева II. И. и др. Производные пиразино- и пииеразиио [1,2-а] индола.— Хим.-фарм. жури., 1972, № 10, с. 14—17.
253. Шведов В. И., Алтухова Л. Б., Комиссарова Е. К-, Гринев А. Н. Производные тетрагидрокарбазола.— Хим. гетероцикл, соед., 1965, № 3, с. 365—369.
254. Шведов В. И., Алтухова Л. Б., Гринев А. Н. Синтез 1,10-триме-тиленпиразино [1,2-а] индолов.— Хим.-фарм. журн., 1967, № 3, с. 25—30.
255. Швицер Ю. Производство химико-фармацевтических препаратов.— М., 1931.
256. Шепелева Е. С., Бородач М. С., Санин П, И. и др. Препарат хлофосфол.— Докл. АН СССР, 1972, т. 203, № 4, с. 856—859.
257. Шкодинская Е. Н. Вопросы экспериментальной и клинической онкологии.— М.: Медицина, 1972.
258. Шнер В. Ф., Рулин В. А., Суворов Н. Н. Синтез и биологическая активность стероидов, содержащих гетероатом в кольце А.— Хим.-фарм. жури., 1978, № 4, с. 22—37.
259. Шрадер Г. Новые фосфорсодержащие инсектициды.— М.: Мир, 1965.
260. Ярмоненко С. П., Суворов Н. Н., Карочкин Б. Б. и др. Количественная характеристика радиозащитиого эффекта мексамина.— Радиобиология, 1970, т. 10, с. 78—82.
261. Ясницкий Б. Г., Дольберг Е. Б., Спивак А. Д. N-Ацетил-е-амино-капроновая кислота. — Хим.-фарм. жури., 1979, № 4, с. 78.
262. Яхонтов Л. Н. Химия азаиндолов.— Успехи хим., 1968, т. 37, с. 1258—1287.
263. Яхонтов Л. Н. Химия хинуклидина.— Успехи хим., 1969, т. 38, с. 1038—1071.
264. Яхонтов Л. Н. Успехи химии гетероциклов в хим.-фарм. Промышленности СССР.— Хим. гетероцикл, соед. , 1977, № 11, с. 1443—1454.
265. Яхонтов Л. Н., Волжина О. Н., Глушаков С. И. и др. Разработка метода получения пармидина.— Хим.-фарм. жури, 1980, № И, с. 77—82.
266. Яхонтов Л. Н., Карпман Я. С. Коксохимическая р-пиколнновая фракция как комплексное сырье в производстве гетероциклических лекарственных препаратов.— Хим. гетероцикл, соед., 1981, № 4, с. 435—447.
267. Яхонтов Л, Н., Левкоева Е. И. Новый путь синтеза метилового эфира трео-а-фенил-а-(пиперидил-2)-уксусной кислоты.— Хим.-
фарм. журн., 1975, № 10, с. 19—23.
268. Яхонтов Л. Н., Левкоева Е. И., Мастафанова Л. И. и др. Изучение процесса окисления 2,6-лутидина перманганатом калия.— Хим.-фарм. журн., 1979, К» 4, с. 72—77.
269. Яхонтов Л. И., Мастафанова Л. И., Либерман С. С. и др. Проти-вокашлевый препарат эстоцин.— Хим.-фарм. журн., 1974, № 3, с. 56—59.
270. Яхонтов Л. Н., Машковский М. Д., Левкоева Е. И. и др. О трео-и эритро-изомерах меридила.— Хим.-фарм. журн., 1974, As 1, с. 3—6.
271. Яхонтов Л. Н., Машковский М. Д., Михлина Е. Е. Исследования в ряду хинуклидина по изысканию новых лекарственных препаратов.—Хим.-фарм. журн., 1980, № 12, с. 23—32.
267
272. Яхонтов Л. Н., Прокопов А. А. Успехи химии азаиндолов.—Успехи хим., 1980, т. 49, с. 814—847.
273. Яшунский В. Г. Комплексообразователн и применение их в медицине.— Жури. Всссоюз. хим. о-ва, 1965, т. 10, К» 6, с. 679—683.
274. Яшунский В. Г., Холодов Л. Е. Химия сиднониминов.— Успехи хим., 1980, т. 49, с. 54—91.
275. Abramovitch R. A. 1-Methyl- and l.N-Dimethyltryptamines.— J. chem. Soc., 1956, p. 4593—4603.
276. Abramovitch R. A., Shapiro D. A convenient synthesis of tripta-mines and p-carbolines.— J. chem. Soc., 1956, p. 4589—4592.
277. Baganz H., Mit% C. Ober 1,2-Dialkoxy-athane.— Chem. Ber., 1953, Bd 86, S. 398—400.
278. Begmann E. D., Ginsburg D., Lavie D. Aryltrichlormethylcarbinols.— J. Amer. chem. Soc., 1950, vol. 72, p. 5012—5014.
279. Bell P. H., Roblin R. O. A theory of the relation of structure to activity of sulfanilamide type compounds.— J. Amer. chem. Soc. 1942, vol. 64, p. 2905—2917.
280. Beyer R. H., Russo H. F., Tillson E. et al. Benemid its renal affinity and its elimination.— Amer. J. Physiol., 1951, vol. 166, p. 625—640.
281. Bodea\ C., Silberg I. Recent advances in the chemistry of pheno-thiaziries.— In: Advances in heterocyclic chemistry/Ed. A. R. Kat-ritzky. New York. 1968, vol. 9, p. 321—460.
282. Bottinger L. E., Mollerberg H. Bestimmung der Sulfanylamide im Blut.— Acta med. Scand., 1959, Bd 165, S. 241—248.
283. Broughton B. J., Chaplen P., Rnowles P. et al. Antiallergic activity of 2-Phenyl-8-azapurin-6-ones.—J. med. Chem., 1975, vol. 18, p. 1117—1122.
284. Burger A. Medicinal chemistry.— New York, London: Sydney — Toronto, Wiley—Interscience, 1970.
285. Clarkson R., Martin A. R. 4-Sulfanilamido-6-methoxy and 6-ethoxy Pyrimidine: two new long-acting Sulphonamides.— Nature, 1961, N 4802, p. 523—524.
286. Counsell R. E., Willete R. E. Synthesis of dichlorodiphenyldichloroethanes (DDD).— J. Pharm. Sci. 1966, vol. 9, p. 1012—1016.
287. Craig P. N., Maxuvell G., Lafferty J. 10-Aminoalky Derivatives of 2-substituted Phenothiazines and 2-Azaphothiazines.— J. org. Chem., 1960, vol. 25, p. 944—947.
288. Drake N. L., Ed. 2,6-Dichloroaniline.— Org. Synth. New York, London, J. Wiley, 1944, vol. 24, p. 47—48.
289. Duschinsky R.^Rleven E., Heidelberger C. The syndthesis of 5-Flu-oropyrimidines.—J. Amer. Chem. Soc., 1957, vol. 79, p. 4559—4560.
290. Elderfield R. C., Claflin E. F., Mertel H. E. et al. Further syntheses of primaquine Analogs.—J. Amer. chem. Soc., 1955, vol. 77, p. 4819—4822.
291. Elion G. B., Rovensky A., Hitchings G. H. et al. Metabolic studies of allopurinol, an inhibitor of xanthine oxidase.— Biochem. Pharmacol., 1966, vol. 15, p. 863—880.
292. Field J. B., Larsen E. G., Spero L., Link R. P. Hyper prothrombinemia induced by methylxanthines and its effect on the action of 3,3'-methylene bis-(4-hydroxycoumarin).— J. biol. Chem., 1944, vol. 156, p. 725—737.
293. Fodor G., Janzso G., Otvos L., Banfi D. Synthese von Hyoscya-min.— Chem. Ber., 1960, Bd 93, S. 2681—2685.
268
)4 . Fucik К., Prochazka Z„ Cechova V. Sur quelques derives de 1’acide di-(hydroxy-4)-coumarinyl-(3)-acetique.— Bull. soc. chim. Franc.,
1949, p. 99—103.
295. Grob C., Renk E. 3-Chibuclidin-carbonsaure.— Helv. chiin. Acta, 1954, v-:. 37, p. 1689—1698.
!96. Goodman L. S., Gilman A. The pharmacological basis of therapeutics.— New York, Toronto, London: Macmillan Publishing, 1970.
297. Haaj W. Untersuchungen fiber die Ritter-Reaktion.— Chem. Ber., 1963, Bd 96, S. 3367—3369.
298. Haller H. L., Bartlett P. D., Drake N. L. The chemical composition of technical DDT. J. Amer. chem. Soc., 1945, vol. 67, p. 1591—1602.
299. Hanngren A., Svartz N., Hansson E., Ullberg S. Ober Sulphasala-zin.— Acta med. Scand., 1963, Bd 173, S. 1—17.
300. Hartmann K- U., Heidelberaer C. Studies on fluorinated pyrimidines, inhibition of thymidylate synthetase.— J. biol. Chem:, 1961, v. 236, p. 3006—3013.
301. Goffmann F. W. w, w'-Difluoroalkanes. — J. org. Chem., 1949, vol. 14, p. 105—110.
302. Inacio M. M. L., Manuela L., Travares A. S., Leal A. M. Preparation of acexamic acid.—Rev. Port, farm., 1969, v. 19, N. 4, p. 223—226.
303. Kindler K-, Metzendorf W„ Dschi-Yin-Kwok. Synthese der substi-tuierten Phenylacetic-Sauren.— Chem. Ber., 1943, Bd 76, S. 310— 317.
304. Koch R., Klemm D., Setter I. Zur Toxikilogie schwefelhaltiger Pyri-doxin-Derivate.— Arzneimittelforsch., 1960, Bd 10, S. 683—696.
305. Kopp E., Strell M. Ober 2,7-Diazaphenothiazin.— Arch. Pharmazie, 1962, Bd 295, S. 99—106.
306. Kuemmerle H. P., Garrett E. R., Spitzy К. H. Klinische Pharma-kologie und Pharmakotherapie.— Miinchen, Berlin, Wien: Urban u. Schwarzenberg, 1976.
307. Landquist J. K-, Siek J. A. —J. Chem. Soc., 1956, p. 2052—2058.
308. Lawrence C. A. Desinfection, sterilisation, and preservation.— Philadelphia: Lea and Febiger, 1968.
309. Lespagnol A., Deprey J.— Bull. Soc. Chem. Franc., 1962, p. 1117.
310. Ley K„ Seng F., Eholzer U. et al. Neue Synthesen von Chinoxalin-und Phenazin-di-N-oxiden.— Angew. Chem., 1969, Bd 81, S. 569— 570.
311. Locoquin R., Elghozy F. Preparation de 1’adipate d’ethyle et des ethers seis en general.— Bull. soc. chim. Franc., 1927, Bd 41, p. 445—448:
312. Martindal W. The extra pharmacopoeia, 27-th.— London: Pharmaceutical press, 1977.
313. Mashkovsky M. D., Yakhontov L. N. Relationships between the chemical structure and pharmacological activity in a series of synthetic quinuclidine derivatives.— In: Progress in drug research/ Ed. E. Jiicker, Basel, 1969, vol. 13, p. 293—339.
314. Massie S. P. The chemistry of phenothiazine.— Chem. Rev., 1954, vol. 54, p. 797—833.
* 315. Mears A. J., Dakeshott S. H., Plant S. G. Some reactions of 1-Keto-1,2,3,4-tetrahydrocarbazole.— J. chem. Soc., 1934, p. 274—276.
316. Montgomery J. A., Laseter A. G., Shortnacy A. T. et al. Nucleosi-des of 2-Azapurines.—J. med. Chem., 1975, vol. 18, p. 564—567.
317. Nelson P. H., Strosberg A. M., Untch K. G. Mono- and dia zyl-2-quinuclidylcarbinols with local anestetic and antiarrhitmic activity.—J. med. Chem., 1980, vol. 23, p. 180—193.
269
318. Nematollahi J., Kercham ft. The reaction of ureas with g Tetrahydroimidazo/4,5-d/imidazole-2,5-diones.— J. org. Chem., vol. 28, p. 2378—2380.
319. Overman ft. S., Stahmann M. A., Huebner C. F. et al. Antic' ' lant activity and structure in the 4-Hydroxycouni.r'n gro J. biol. Chem., 1944, vol. 153, p. 5—24.
320. Page J. Serotonin.— Chicago: Year Book Med. Publ., 1966.
321. Petrov V., Sturgeon B. Some Quinoline-5,8-quinones.—J. chem. Soc., 1954, p. 570—574.
322. Phillips A. P. Synthetic curare substitutes from aliphatic dicarboxylic acid aminoethyl esters.— J. Amer. chem. Soc., 1949, vol. 71, p. 3264—3264.
323. Pratt Y. T., Drake N. L. Quinolinequinones.— J. Amer. chem. Soc., . 1960, vol. 82, p. 1155—1161.
324. Roberts P. J. Captopril. Drugs of the Future, 1978, N 11, p. 795— 799.
325. Robinson R. Synthesis of tropinone.— J. chem. Soc., 1917, p. 762—768.
326. Schenker E., Herbst H. Phenothiazine und Azaphenothiazine als Arnzeimittel.— In: Progress in drug research/Ed. E. Jucker, Basel, 1963, vol. 5, p. 269—627.
327. Schmidt С. H. Neuer Entwicklung der Furan-Chemie.— Angew. Chem, 1956, Bd 68, S. 175—176.
328. Schopf C., Lehman G. Die Synthese des Tropinons.— Lieb. Ann., 1935, Bd 518, S. 1—37.
329. Schulman J. L. Effect of amantadine and rimantadine on transmission of influenza''virus infection.— Proc. Soc. exp. Biol. Med. (N. Y.), 1968, vol. 128, p. 1173—1178.
330. Schwab R. S., Poskanzer D. C., England A. C. Amantadine in Parkinson’s disease.—J. A. M. A., 1972, vol. 222, p. 792—801.
331. Schwenker G., Prenntzell W„ Gassner U., Gerber R. Notiz fiber eine neue allgemein anwendbare Tropansaureestar-Synthese.— Chem. Ber., 1966, Bd 99, S. 2407—2408.
332. Seeger D. R„ Cosulich D. B., Smith J. M., Hultquist M. E. Analogs of pteroylglutamic acid.—J. Amer. Chem. Soc., 1949, vol. 71, p. 1753—1758.
333. Shealy J. F., Struck R. F., Holum L. B., Montgomery J. A. 5-Diazo-imidazole-4-carboxamide and 5-Diazo- v-triazole-4-carboxamide.—J. org. Chem., 1961, vol. 26, p. 2396—2401.
334. Shepherd R. G., Taft W. E., Rrazinski U. M. Sulfamlamidopyrimi-dines.—J. org. Chem., 1961, vol. 26, p. 2764—2769.
335. Southwick P. L„ Seivard L. L. The ring closure of N-Alkoxalyl-0-anilinopropionic acids.—J. Amer. chem. Soc., 1949, vol. 71, p. 2535—2538.
336. Sparks S. ]., James E., Landes M. N. et al. 1,4-Piperazinedipho-sphoricamides.— Proc. Amer. Ass. Cancer, Res., 1956, vol. 2, p. 149—153.
337. Stetter H., Mayer J., Schwarz M., Wulf< K. Beitrage zur Chemie der Adamantyl-(1)-Derivate.—Chem. Ber.,’ 1960, Bd 93, S. 226—230.
338. Sternbach L. H. The benzodiazepine story.— J. med. Chem., 1979, vol. 22, p. 1—7.
339. Stoll IF. G„ Litvan S. Thrombose und Embolie.— Basel, 1954.
340. Struller Th. Progress in sulfonamid research.— In: Progress in drug research/Ed. E. Jucker. Basel, 1968, vol. 12, p. 389—457.
341. Tondeur R., Urbain M. New quinuclidine derivatives.—Chem. Therap., 1966, vol. 19, p. 207—208.
СОДЕРЖАНИЕ .mt
<1?дасловие..........................................: 3
• Ь>Чйараты группы аминокислот и их производных .... 7
Пантогам............................................10
Асалин.........................................: . . 12
Ацемин.................................................15
Фосфорсодержащие препараты................................16
Дийодбензотэф..........................................19
Имифос.................................................22
Хлофосфол..............................................24
Хлорацетофос...........................................25
Октатион...............................................27
Бисчетвертичные аммониевые соединения.....................28
Анатруксоиий ......................................... 33
Циклобутоний...........................................36
Дитилин................................................38
Диоксоний..............................................40
Проспидин..............................................43
Этоний.................................................46
Карбоциклические и ароматические соединения...............47
Мидантан...............................................53
Ремантадин.............................................54
Оксолин................................................58
Теброфен...............................................61
Флореналь..............................................64
Хлодитан...............................................68
Цетамифен..............................................70
Ферроцерон.............................................72
Цигерол.............................................. 74
Пиромекаии.............................................77
Эстонии.....................................' . . 79
Метамизил..............................................83
Сульфамидные препараты....................................86
Сульфамонометоксин.....................................94
Фтазин.................................................98
Салазопиридазин ..................................... 100
Салазодиметоксин......................................102
Хлорпропамид ........................................ 105
Цикла мид.............................................107
Хлоцикламид...........................................109
Этамид...............................................,110
Кислород- или серосодержащие гетероциклические соединения . Ц2
Фурагии.....................................• • • • 116
Феникаберан...........................................118
Фепромарон............................................122
Нитрофарин............................................123
Пирроксаи . . ....................... • 125
Битиодин..............................................129
Моно- и бициклические азотсодержащие соединения с одним гетероатомом.............................................132
Пармндип...........................................: 143
Пиридитол . . ..........................: 146
Мер и ди л............................................149
Нитроксолин , ................................153
271
Хиноцид...............................................: 155
Мекса мин.............................................161
Серотонина адипинат ................................. 166
Атропина сульфат .....................................171
Гоматропина гидробромид ............................. 176
Тропафен............................................ 179
Темехин ..............................................181
Имехин...............................................186
Квалидил..............................'..............187
Фенкарол.............................................192
Препараты группы моио- и бициклических азотсодержащих соединений с несколькими гетероатомами...................195
Нитазол......................................... . . 200
Диоксидин.............................................201
Хиноксидии ...........................................202
Метотрексат \.........................................204
Клаф ели н ...........................................208
Мебикар 211
Сиднофен..............................................212
Сидиокарб ........................................... 214
Феназепам.............................................215
Конденсированные три- и тетрациклические соединения, содержащие гетероатомы......................................217
Этмозии................................................222
Ноиахлазии.............................................226
Фторацизин . -...........................................229
Азафен.................................................231
Карбидии...............................................234
Пиразидол..............................................238
Инказаи : . ................ . . . 243
Алфавитный указатель лекарственных препаратов . . . . 246
Указатели по медицинскому применению лекарственных препаратов ...................л.............................248
Указатель исходных веществ..................................250
Указатель полупродуктов, методы синтеза которых описаны в монографии.............................................252
Список литературы...............................• . . . 255