/

































































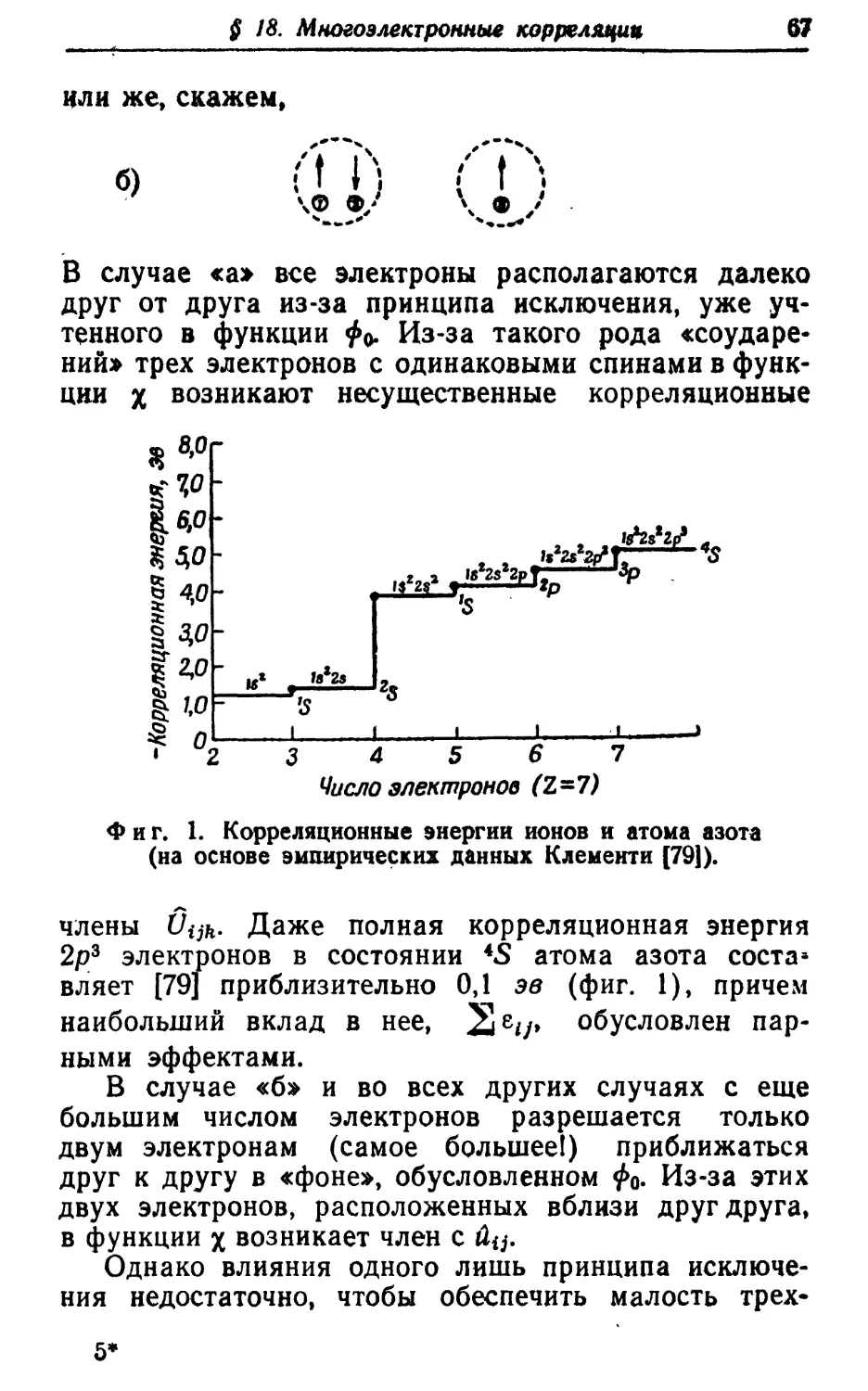





















































































Text
еоретичеспая
MANY-ELECTRON THEORY OF ATOMS,
MOLECULES AND THEIR INTERACTIONS
by
OKTAY SINANOftLU
Sterling Chemistry Laboratory,
Yale University, New Haven, Connecticut
Advances in chemical physics»
volume VI, 315—412, 1964
INTERSCIENCE PUBLISHERS
A DIVISION OF JOHN WILEY AND SONS LTD.,
LONDON-NEW YORK-SYDNEY
ТЕОРЕТИЧЕСКАЯ ФИЗИКА
О. Синаноглу
МНОГОЭЛЕКТРОННАЯ
ТЕОРИЯ
АТОМОВ, МОЛЕКУЛ
И ИХ ВЗАИМОДЕЙСТВИЙ
Перевод с английского
в. я. федянина
Под редакцией
С. В. ТЯБЛИКОВА
ИЗДАТЕЛЬСТВО «МИР»
Москва 1966
Настоящая книга посвящена методам расчета раз-
различных характеристик атомов и молекул (полных энер-
энергий, потенциалов ионизации, энергий диссоциации, теп-
лот образования, сил связи и т. д.). В основу изло-
изложения положен один из вариантов теории систем
многих частиц, разработанный автором в применении
к решению конкретных задач квантовой химии. Особое
внимание уделено последовательному учету корреля-
корреляционных эффектов.
Книга рассчитана на научных работников и. аспи-
аспирантов — физиков и химиков, специалистов в области
теоретической и квантовой химии, теоретической физи-
физики, химической физики и т. п.
Книга может также служить дополнительным по-
пособием для студентов и преподавателей физических
и химических факультетов университетов по курсам
квантовой механики и квантовой химии.
Редакция литературы по физике
ОТ РЕДАКТОРА ПЕРЕВОДА
Для квантовой химии существенное значение
имеет расчет различных характеристик атомов и мо-
молекул. Для этого в первую очередь требуется знать
волновые функции и собственные значения энергии
электронных оболочек атомов и молекул. Однако, ко-
когда число электронов велико, прямое решение соот-
соответствующей квантовомеханической задачи оказы-
оказывается делом весьма сложным, а подчас и невоз-
невозможным. В таких случаях более эффективными
являются методы теории систем многих тел. Сина-
ноглу настойчиво подчеркивает необходимость имен-
именно многоэлектронной трактовки задач квантовой хи-
химии и в связи с этим необходимость более полного
учета эффектов корреляций между электронами.
Автор развивает один из вариантов теории многих
тел и на его основе при расчетах важнейших харак-
характеристик атомов и молекул последовательно прово-
проводит многоэлектронную трактовку. Речь идет главным
образом об определении волновых функций и соб-
собственных энергий и расчете с их помощью следую-
следующих величин:
1) полных энергий, потенциалов ионизации, энер-
энергий диссоциации;
2) теплот образования, изомеризации, изменения
формы для больших молекул;
3) межмолекулярных сил.
Автор отмечает, что расчеты других характери-
характеристик атомов и молекул можно также улучшить при
применении волновых функций данной теории.
В качестве нулевого приближения берется, как
обычно, приближение Хартри — Фока, Однако в
6 От редактора перевода
отличие от многих аналогичных исследований далее
делается попытка включить явным образом корре-
корреляции между комплексами из двух, трех и т. д. элек-
электронов. Такой подход к учету корреляционных эф-
эффектов аналогичен известным разложениям по груп-
групповым интегралам в теории неидеальных газов. По-
Поскольку основная идея метода представляется весьма
интересной, остановимся на этом вопросе несколько
подробнее, чтобы яснее представить себе сильные и
слабые стороны метода (см. § 8—10).
Обозначим через г|) и Е соответственно волновые
функции и собственные значения энергии точного га-
гамильтониана 1) системы из N электронов, находя-
находящихся в поле атомных ядер а:
Яф = ?ф, A)
B)
Здесь gij=l/|*i — *j|—кулоновская энергия взаимо-
взаимодействия /-го и /-го электронов, Щ—оператор одно-
частичной энергии электрона:
hi TV ^_2^, C)
а
где Za —заряд (в единицах е) ядра a, a Ria — рас-
расстояние от i-ro электрона до ядра а.
Рассмотрим предварительно приближение Харт-
ри — Фока для данной задачи. Выпишем обычные
уравнения Хартри — Фока:
h% {Xi) + 2 (Фу (Xj)> g/уФу (*j) ) Ф< (Xt) -
— 2 (Фу (Xj), gijtyj (Xj) ) фу (Xt) =
= e^(^) (/, j=h 2, ..., N); DJ)
величина zt играет роль собственного значения энер-
энергии электрона, находящегося в состоянии /. Обо-
1) Используется система единиц, в которой #=e=*m=l.
2^ Круглые скобки здесь означают скалярное произведение
(Ф (x)t Щх)) ЕЕ J ф*(х)Ъ(хLх и rt д.
От редактора перевода
значим через Vt самосогласованный потенциал для
/-го электрона, включающий в себя кулоновскую и
обменную части:
Vi (xt) = 2 {(Ф/ (•*/)> gijtyj (-*/))—(Ф/ (Xj)i gi$i (Xi))} • E)
j
Составим из функций фДлГ/) антисимметризованное
произведение
Фо (*i. • • •, xN) = А П Ь (*,), (ф09 Фо) = 1, F)
где Л— оператор антисимметризации1) (по коорди-
координатам электронов). Функцию ф0, определенную выра-
выражением F), можно рассматривать как волновую
функцию данной системы в приближении Хартри —
Фока. Далее можно ввести гамильтониан Яо, для ко-
которого ^>о является собственной функцией
Нофо=Еофо, G)
+Ы (8)
где ft? и Vi определяются формулами C) и E). Соб-
Собственное значение Ео энергии электронной системы
равно, как легко показать, сумме энергий е*
?о = (&)> Я0^0) = 2е/. (9)
Сопоставляя выражения A), B) и A0), A1), видим,
что величины Яо, ^о, Ео в приближении Хартри —
Фока можно рассматривать как нулевое приближе-
приближение для точных Я, if, ?, если, конечно, соответствую-
соответствующие отклонения будут малы.
1) Оператор Л имеет вид
где Р — символ перестановки координат электронов; как обычно»
(—l)p=-f-l, если перестановка четная, и (—1)р=—1, если пе-
перестановка нечетная,
8 От редактора перевода
Заметим, что обычно под значением энергии
в приближении Хартри — Фока понимают величину
Бн. р. = (Фо
где
А = (Фо, \И- Щ фо\ Н- Ио = 2 gij ~ S Vt.
Основная идея метода, развиваемого в данной
книге, состоит в том, чтобы в точной волновой функ-
функции N электронов выделить в явном виде слагаемые,
которые представляли бы корреляции комплексов из
двух, трех и т. д. электронов; за основное прибли-
жение~~берется приближение Хартри — Фока. В соот-
соответствии с этой идеей точная волновая функция си-
системы записывается в виде
Ъ=Фо + Ь (Ю)
где ^о — волновая функция в приближении Харт-
Хартри — Фока. Без нарушения общности можно считать,
что функция х ортогональна к ф0 и что ^0 нормиро-
нормирована на единицу. Тогда
(&»Х) = 0, (&>> &>)=!> (Ф> Ф)=1+(Х, X). (И)
Представим функцию х в виде ряда
X = Xi + X2+ ...» A2)
где
N
X"m Ж S (*'!••• 4V *о) иЧ ¦•• 1п (** • • • • *п)-
|.>->|«>1 A3)
Новые неизвестные функции Ui(Xi)y Uij(xiyx2), ...
описывают искомые дополнительные корреляции
комплексов из одного, двух и т. д. электронов. Мож-
Можно заметить, что уже представление волновой функ-
функции в виде A0) — A3) позволяет провести анализ
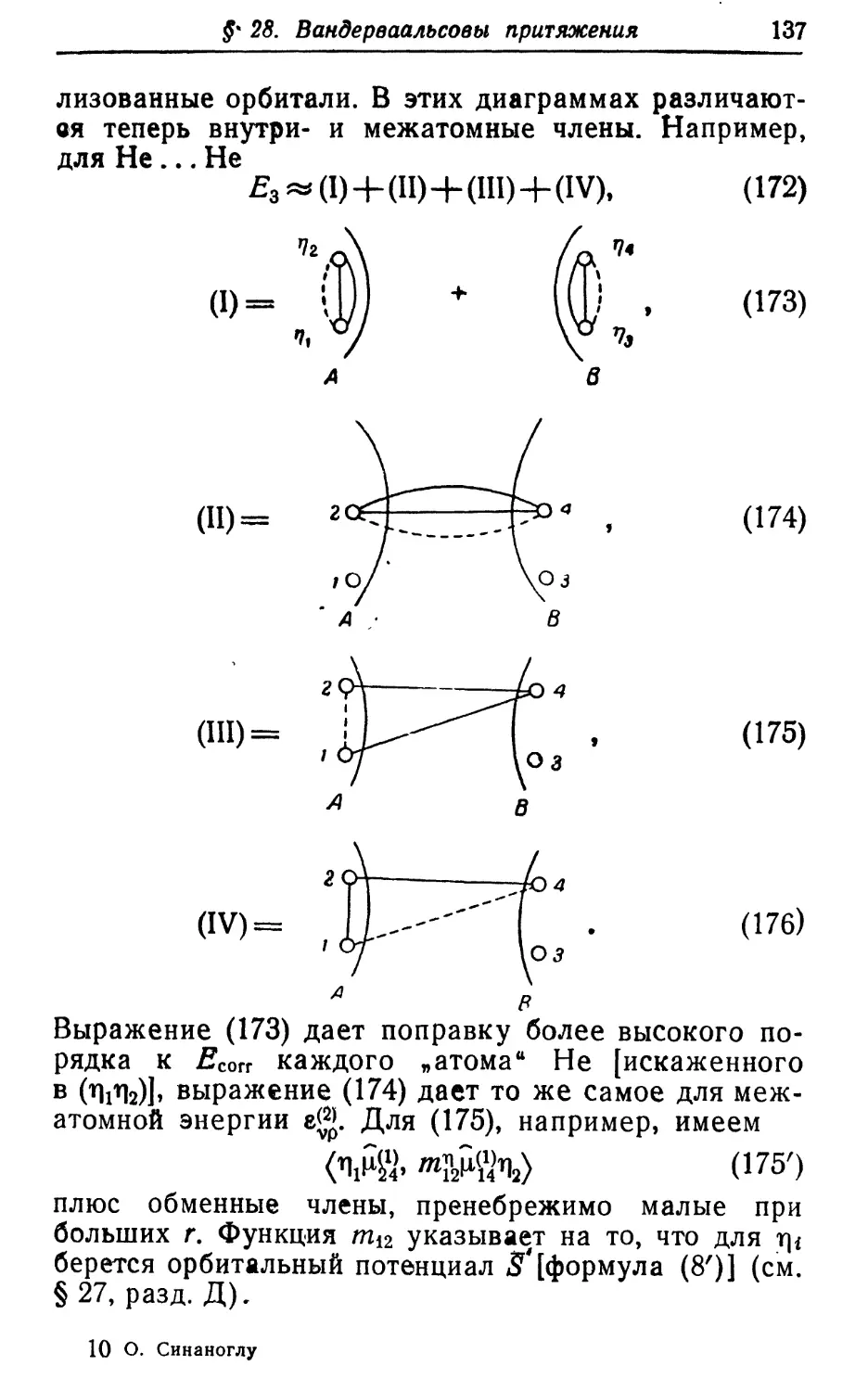
различных корреляционных эффектов в наблюдае-
наблюдаемых величинах. Для функций Uu Иц, • • • можно по-
построить полную систему уравнений, которая будет
эквивалентна исходному уравнению Шредиыгера.
Однако для конкретных расчетов она будет не проще
От редактора перевода 9
исходного волнового уравнения. Поэтому задача со-
состоит в том, чтобы для построения функций Uu U%h • • •
использовать приближенные методы, которые позво-
позволяли бы получать решение достаточно просто и в то
же время обеспечивали необходимую точность. Ав-
Автор обсуждает несколько таких возможностей.
Первая из них состоит в использовании вариаци*
онного принципа (см. § 12) для пробной функции \j),
взятой в виде
$=4>о+Х. A4)
Использование разложений вида A2) и A3) для
пробной функции х позволяет разделить различные
корреляционные эффекты в выражении для энергии
и изучать их порознь (см. § 13). Дальнейшее усо-
усовершенствование этой методики состоит в использо-
использовании представлений теории возмущений наряду
с вариационным методом (см. § 14). Таким путем
оказывается возможным для волновой функции и
собственной энергии построить разложения, анало-
аналогичные соответствующим разложениям теории возму-
возмущений Релея — Шредингера.
Сохраняя в выражении A2) для /ив A3) чле-
члены, содержащие одиночные возмущения {/,} и какую-
то одну парную функцию Vih можно получить путем
минимизации выражения для энергии систему урав-
уравнений для этих величин. Самосогласованные реше-
решения для них будут аналогом решения по методу
Бракнера для одной пары. Если удержать все корре-
корреляции {Uij}, получим аналог соответствующего реше-
решения для всей электронной системы. Как показано,
наибольшие вклады в энергию дают парные корре-
корреляции {Uij}, тогда как влияние одиночных корреля-
корреляций {/г} и более высоких многочастичных корреляций
{Uijk}, {Uijki}, . • • можно рассматривать как возмуще-
возмущение (см. § 15—18). В этом, собственно, состоит основ-
основное упрощение в общей схеме, используемое автором
при построении «многоэлектронной» теории атомов
и молекул.
По существу прогресс, достигнутый автором в его
многоэлектронной теории атомов и молекул, обязан
10 От редактора перевода
учету именно парных корреляций в волновой функ-
функции всей системы. В связи с этим заметим, что пред-
предположение о важности подобных членов в волновой
функции системы не является особенно новым. Впер-
Впервые соображения такого рода, хотя, может быть, и не
в столь явной форме, были высказаны в известной
работе Вигнера 1) по расчету корреляционной энер-
энергии электронного газа, а применительно к теории ато-
атомов они были высказаны в работе Фока, Веселова и
Петрашень2). Заслугой автора является, по-види-
по-видимому, последовательный учет парных корреляций
при исследовании конкретных атомных и молекуляр-
молекулярных систем.
Таким образом, многоэлектронная теория Сина-
ноглу представляет собой метод самосогласованного
поля Хартри — Фока, усовершенствованного введе-
введением поправок на двухэлектронные корреляции в вол-
волновой функции. Простота основной идеи не должна
вводить в заблуждение, поскольку для проведения
конкретных расчетов, как всегда, требуется затрата
значительного труда. Однако это оправдывается, по-
поскольку учет корреляций позволяет провести анализ
различных корреляционных поправок в энергии и по-
получить ряд интересных результатов при рассмотре-
рассмотрении конкретных систем.
Книгу Синаноглу можно рассматривать как хоро-
хороший обзор и краткое руководство по некоторым су-
существенным вопросам квантовой химии. В ней рас?
смотрено большое число конкретных примеров и при-
приведена подробная библиография оригинальных статей,
обзоров и монографий по затронутым вопросам.
По содержанию книга Синаноглу распадается на
три части. В-§ 1—7 обсуждается вопрос о том, на-
насколько необходима многоэлектронная теория ато-
атомов и молекул, и дается постановка задачи. В § 8—17
развивается общий метод многоэлектронной трактов-
трактовки таких систем и рассматривается вопрос о его ме-
1) Wig пег Е. P., Trans. Farad. Soc, 34, 678 A938).
2) Фок В. А, Веселое, Петрашень М, И., ЖЭТФ,
10, 723 A940).
Or редактора перевода 11
сте среди других методов, используемых в квантовой
химии; особо рассмотрены выражения для волновой
функции и энергии. В последующих параграфах кни-*
ги обсуждаются более конкретные вопросы, такие,
как роль многоэлектронных корреляций и локализа-
локализаций связей, влияние ионных бстовов, свойства ато-
атомов и молекул (см. § 18—27), расчет вандервааль-
совых притяжений для случаев внутримолекулярных
и межмолекулярных взаимодействий и, наконец, роль
релятивистских эффектов (см. § 28, 29).
Нужно заметить, что, хотя книга Синаноглу напи-
написана довольно сжато и многие вопросы в ней только
упоминаются, ее можно рекомендовать вниманию
научных работников и аспирантов — физиков и хими-
химиков, интересующихся этими вопросами, и студентов
университетов в качестве дополнительного пособия
по курсам квантовой механики, квантовой химии, хи-
химической физики и т. п. Можно думать, что обзор
Синаноглу будет в равной мере полезен, как специа-
специалистам по теоретической химии, так и физикам-тео-
физикам-теоретикам, занимающимся задачей многих тел в тех
или иных ее аспектах,
С. В. Тябликов
§ 1. ВВЕДЕНИЕ
Эта работа содержит расчет волновых функций
и энергий многоэлектронных атомов и молекул.
Основное внимание уделяется следующим свойствам:
1) полным энергиям, потенциалам ионизации,
электронным спектрам и энергиям диссоциации мо-
молекул;
2) теплотам образования, изомеризации и изме-
изменений формы больших молекул;
3) межмолекулярным силам при различных рас-
расстояниях между молекулами.
Расчет других свойств, как, например, поляри-
зуемостей, магнитной экранировки и т. д., непосред-
непосредственно здесь не производится, однако его можно
улучшить, используя волновые функции данной
теории.
Для расчета большинства из перечисленных харак-
характеристик требуется высокая степень точности, по-
поскольку эти характеристики связаны с малыми разно-
разностями энергий между большими величинами. Пригод-
Пригодная для химии теория должна быть достаточно точной
и в то же время достаточно простой при применении
к великому множеству молекул, содержащих оди-
одинаковые группы. По этой причине мы рассматриваем
и полуэмпирическую *и неэмпирическую схемы. Наша
цель — воспользоваться «химическими» упрощениями
в неэмпирической «молекулярной квантовой механи-
механике» (но делать это не от случая к случаю) и, таким
образом, получить количественную основу для полу-
полуэмпирической «квантовой химии».
§ 2. НЕОБХОДИМОСТЬ «МНОГОЭЛЕКТРОННОЙ ТЕОРИИ»
Квантовая химия основывается на орбиталях: до-
достаточно упомянуть метод Гайтлера — Лондона
(Н. L.) или метод молекулярных орбиталей (МО).
14 § 2. Необходимость «многоэлектронной теории»
Рассматривая какую-либо проблему, можно с рав-
равным правом применять оба метода; и если мы вне-
внесем достаточное количество поправок, то придем
к эквивалентным результатам. Метод Гайтлера —
Лондона оказывается весьма громоздким для боль-
больших молекул, особенно при применении в теории
спектров. Наилучшими молекулярными орбиталями
являются орбитали, полученные с помощью метода
Хартри —Фока [1], которцр дает уравнение для
волновой функции каждого электрона в самосогласо-
самосогласованном среднем поле всех других электронов. В ка-
качестве исходных мы возьмем хартри-фоковские моле-
молекулярные орбитали (H.F. МО) или же аппроксимации
к ним, например молекулярные орбитали — линей-
линейные комбинации атомных орбиталей (LCAO МО).
Это приведет к максимальному упрощению теории
остающихся эффектов.
В орбиталях не учитываются одновременные от-
отталкивания — «столкновения» — между электронами.
Результирующая ошибка, т. е. «разница между точ-
точной нерелятивистской энергией и хартри-фоковской
энергией» многоэлектронной системы, определяется
как «корреляционная энергия» [1]. Как подчеркнули
Лёвдин [1] и Питцер [2], эти'ошибки по величине
сравнимы с энергией, которой мы интересуемся. По
этой причине, по-видимому, нам придется иметь дело
со сложной многоэлектронной проблемой и отка-
отказаться от приближения с помощью орбиталей. Одна-
Однако по двум причинам ситуация не настолько плоха,
как может показаться на первый взгляд. Во-первых,
на некоторые свойства, например на распределения
заряда, корреляция влияет гораздо слабее, нежели
на разности энергий. Во-вторых, принцип исключе-
исключения и влияние атомного остова приводят к тому, что,
несмотря на большой радиус кулоновского отталки-
отталкивания, в каждый момент «сталкиваются» в основ-
основном два электрона. «Многоэлектронная» проблема
в таком случае представляет собой в действитель-
действительности ряд «малоэлектронных» проблем.
Электронная корреляция ответственна за следую-
следующие эффекты;
§ 2. Необходимость € многоэлектронной теории» 15
1. Ошибку приблизительно от 1 до 2 эв на два-
дважды занятое хартри-фоковское состояние. Например,
для ^-состояния в Не ошибка составляет —1,142 эв
[1], для Н2 (при равновесном межатомном расстоянии
г~ге) —1,06 эв [1] и для BpzJ-napbi Ne — около
— 1,6 эв [81]. Относительная ошибка сравнима с энер-
энергией связи, например, в N2 (см. § 25).
2. «Поляризацию остова» валентными электрона-
электронами. Так, например, я-электрон мгновенно поляризует
сигма-остов; подобные же эффекты вызывают сдвиг
уровней в спектрах щелочей. Такого рода (Is)B$)-
корреляция в литии составляет около —0,05 эв [4],
3. Если при вычислении энергии диссоциации ис-
использовать для всех межъядерных расстояйий г по-
потенциальную кривую Aа^J-состояния молекулы Н2,
полученную методом Хартри — Фока, то это приве-
приведет к ошибке, составляющей около 7,74 эв. Если одну
и ту же электронную конфигурацию рассматривать
в рамках метода Хартри — Фока как при больших
г, так и вблизи равновесного значения ге, то ока-
окажется, что большинство молекул будет диссоцииро-
диссоциировать в неверные состояния (см. пример с N2; ра-
работа [5]).
4. Зачастую для электронных спектров, напри-
например, для которых правила Хунда выполняются, харт-
ри-фоковские молекулярные орбитали приводят к
правильной схеме уровней. Каждый уровень, однако,
оказывается сдвинутым вверх на величину ошибок
корреляции («адиабатические» сдвиги уровней).
5. «Почти вырождение», или «резонанс», между
конфигурациями может изменить схему уровней, да-
даваемую методом Хартри — Фока, что проявляется
в нарушении правил Хунда, например, в случае
C53d) -уровней Mg [55]. Такие «неадиабатические»
сдвиги уровней возникают из-за эффектов «конфигу-
«конфигурационного взаимодействия (С. I.) первого порядка»
[б]1). Эффект, описанный в п. 3, такого же типа.
1) Поскольку это не вызывает путаницы, нами везде остав-
оставлен термин оригинала «конфигурационное взаимодействие» вме-
вместо более определенного (введенного Хартри [97]) термина «на-
«наложение конфигураций». — Прим. переви
10 $ ? Полуэмпирические теории
6. Вандерваальсово притяжение лондоновского
дисперси&вного типа, например, между двумя ато-
атомами аргона или между двумя концами п-бутана
является строго корреляционным эффектом [2]. Такие
взаимодействия имеют порядок 10~~3 эв. Корреляцию
между различными оболочками атома или молекулы
(описанные в п. 2) можно также рассматривать как
«вандерваальсово» притяжение.
7. В металлах и высокосопряженных молекулах
красителей многие электроны коррелируют одновре-
одновременно и ведут себя как «электронный газ» с коллек-
коллективными эффектами экранировки и колебаний [7].
8. При обосновании применимости самих орбита-
лей в качественных вопросах квантовой химии важ-
важно выяснить, меняет ли корреляция хартри-фоков-
ские орбитали? То есть являются ли орбитали (полу-
(полученные с учетом корреляционных эффектов) лучшим
приближением к точной волновой функции, нежели,
хартри-фоковские молекулярные орбитали? \
Чтобы разобраться в эффектах, перечисленных
выше, необходимо изучить электронную корреляцию.
Лёвдин [1] сделал обзор состояния теории вплоть
до 1959 г., акцентируя внимание на методах, которые
давали высокую точность для двухэлектронных си-
систем. Питцер [2] рассмотрел корреляцию между не-
неперекрывающимися оболочками электронов и дал
полуэмпирическую оценку меж- и внутримолекуляр-
внутримолекулярных притяжений Лондона — ван дер Ваальса.
Данная работа представляет собой обзор «много-
«многоэлектронной теории», развитой за последние несколь-
несколько лет [8—12]. Сходные приближения обсуждаются
по ходу дела. Предварительное рассмотрение приня-
принятых полу- и неэмпирических схем делают ясным цели
и причины специфического направления атаки, вы-
выбранного «многоэлектронной теорией».
§ 3. ПОЛУЭМПИРИЧЕСКИЕ ТЕОРИИ
Полуэмпирические теории электронных спектров
основываются на орбиталях (хартри-фоковских или
же аппроксимирующих цх), И в случае атомных, и
§ 8. Полуэмпирические теории 17
в случае я-электронных спектров пренебрегают эф-
эффектами остова и эффектами мгновенной «поляриза-
«поляризации остова» внешними электронами. Для я-электро-
нов, как правило, берут эмпирический «П-электрон-
ный гамильтониан» [13, 14] Яп, который зависит от
статического поля сигма-остбва, полагаемого неиз-
неизменным для различных электронных состояний.
Выражения для энергий, получаемые с помощью
теорий, основывающихся на орбиталях, содержат не-
некие параметры. В случае атомйых спектров за полу-
полуэмпирические параметры принимают F и G инте-
интегралы [15], а в случае П-электронного спектра — а,
Р и одно- и двухцентровые кулоновские интегралы.
Эмпирические значения этих интегралов приводят
к вполне удовлетворительному согласию с экспери-
экспериментом, однако согласия уже не будет, если пара-
параметры рассчитать с помощью орбиталей. Их эмпири-
эмпирические значения, несомненно, включают эффекты
электронной корреляции [14, 16, 17].
Относительные энергии насыщенных молекул
можно рассчитать с помощью полуэмпирических тео-
теорий вполне удовлетворительно, если подобрать по-
постоянные энергий связи, учесть нулевые колебаний
и притяжения Лондона — ван дер Ваальса между
несвязанными областями молекулы [2]. Теплоты об-
образования и изомеризации насыщенных углеводоро-
углеводородов, полученные таким способом, составляют около
0,01 эв, или же 0,2 ккал/моль [2].
Общей чертой этих моделей является постулиро-
постулированное разделение электронов на определенные
группы (электроны внутренних и внешних оболочек,
электроны связей и т. д.) и сохранение таких групп
при переходе от одних атомов или молекул к дру-
другим. Эти приближения справедливы (см. ниже) для
орбитальных моделей как таковых, но их следует
обосновывать при рассмотрении корреляционных эф-
эффектов. Полуэмпирические теории должны осно*
вываться на некой теории, которая с самого на*
чала включает электронную корреляцию, что дает в
итоге для параметров вполне определенные выра-
выражения.
2 О. Срцаноглу
18 §4. Неэмпирические теории
§ 4. НЕЭМПИРИЧЕСКИЕ ТЕОРИИ
Точную многоэлектронную волновую функцию
можно разложить в бесконечный ряд по всем «упо-
«упорядоченным» слэтеровским определителям, которые
можно образовать из одноэлектронного базисного
набора. Обычно расчеты для систем, состоящих бо-
более чем из двух электронов, производят, беря в ка-
качестве линейной пробной функции конечное число
подобных конфигураций (метод конфигурационного
взаимодействия [1]). Обобщенную форму обычной
теории возмущений также можно рассматривать с по-
помощью приближений, полученных методом конфигу-
конфигурационного взаимодействия [1, 18].
Метод конфигурационного взаимодействия хо-
хорошо работает при устранении реальных вырожде-
вырождений или при применении к почти вырожденным со-
состояниям [6] (метод конфигурационного взаимодей-
взаимодействия первого порядка). Однако ряды, получаемые
с помощью этого метода, слабо сходятся [19] для дру-
других типов корреляционных эффектов, как, скажем,
в атоме гелия.
Когда число электронов N или же число орбита-
лей М, которые берутся в конечном базисном наборе
в методе конфигурационного взаимодействия, растет,
то число конфигураций, соответствующих виртуаль-
виртуальным возбуждениям трех, четырех, ... электронов,
одновременно растет как Ns, NAy ... или же М3,
М4, ..., т. е. даже для молекулы типа СНЦ оно бы-
быстро становится порядка тысячи.
Точные расчеты двухэлектронных систем, напри-
например Не и Н2, оказались возможными благодаря ис-
использованию непосредственно в волновой функции
межэлектронных координат г12х). Такие методы
«замкнутого вида» обсуждались детально Лёвди-
ным [1].
*) Сравнение метода конфигурационного взаимодействия и
«метода координаты Ги» для Не cms в работе [19], стр, 48,
§ 4. Неэмпирические теории 19
Делались многочисленные попытки [1, 20—22] рас-
распространить метод координаты ri2 на случай N элек-
электронов1). Для этого детерминантную волновую
функцию домножали на корреляционный фактор, со-
содержащий все Гц. Основная трудность была связана
с вычислением интегралов [22], содержащих различ-
различные rij, возникающие от многоэлектронных членов
в энергии.
Помимо вычислительных трудностей, в этих мето-
методах существуют и другие — для каждого атома или
молекулы решается некая особая численная задача.
В них не используется раздельное поведение оболо-
оболочек, связей и т. д., как это делается в полуэмпириче-
полуэмпирических методах, не выясняется существование более
чем одного типа корреляционных эффектов даже
в рамках рассмотрения того же самого атома или
молекулы.
Были сделаны другие попытки [23—25] разделить
электроны на группы и ввести корреляцию в каждой
группе. Полная волновая функция выбиралась в виде
антисимметризованного произведения групп функ-
функций Ла, Лв и т. д. Полагалось, что они удовлетво-
удовлетворяют произвольным и ограничивающим условиям
ортогональности:
<ЛА(/, у, Л, ..., NA), AB(i, го, n, ..., NB))Ki = 0, A)
где ( )Х/ означает интегрирование лишь по координа-
координатам /-го электрона х*. Сигма — пи-разделение также
основывалось на этом приближении [26].
Такой выбор волновых функций и условие A)
в совокупности эквивалентны постулированию желае-
желаемого результата, т. е. разделения электронов на груп-
группы, произведенного с самого начала. Здесь корреляции
между группами, т. е. вандерваальсовы притяже*
ния не включены. По-видимому, включить их не-
невозможно, даже если развить [29, 30] этот метод (см.
1) По сообщению Пайерлса этим вопросом занимался^т
Ленд A929 г.).
20 § 5. Схема построения «многоэлектронной теории»
§ 28) • И для енутриоболочечных аффектов этот фор-
формализм не приводит к успехам, подобным тем, кото-
которые имеют место при применении метода коорди-
координаты ri2. Ограничения его обсуждаются в других ра-
работах [3, 27—31] (см. также § 27).
В течение последних лет были сильно развиты
другие многочастичные методы физики, в частности
теория ядерной материи (теория Бракнера) и теория
электронного газа [1, 32]. Рассматривалась возмож-
возможность применения этих теорий к атомным и молеку-
молекулярным проблемам [1]. Однако оказалось, что рас-
рассмотрение атомов и большинства молекул в рамках
многочастичной задачи физически весьма отлично от
того, что мы имеем в иных проблемах [9].
Теория Бракнера является обобщенным методом
самосогласованного поля (SCF) [32, 33, 59, 60, 65];
орбитали в этом методе «приспосабливаются» к кор-
корреляции. Почему подобное приспосабливание не яв-
является необходимым в случае атомов и молекул, по-
показано в § 15 и 19. В § 15, 18 и 19 дается сравнение
таких теорий с «многоэлектронной теорией», изло-
изложенной в этой работе.
§ 5. СХЕМА ПОСТРОЕНИЯ
сМНОГОЭЛЕКТРОННОЙ ТЕОРИИ»
Обсуждение, проведенное выше, показывает:
1) полу- и неэмпирические модели должны быть
обоснованы в равной мере;
2) в неэмпирической теории атомы или молекулы
также должны постепенно конструироваться, но труд-
трудность такого метода не должна резко возрастать
с увеличением числа электронов N.
Химические упрощения, которые должны быть вве-
введены, были интуитивно очевидны довольно давно.
Сейчас необходимо ввести это химическое описание
строгим и хорошо определенным путем. В действи-
действительности «картина», реально существующая в при-
природе, должна следовать из квантовой механики, а не
вводиться от случая к случаю.
§ 5. Схема построения смногоэлектронной теории» 21
В связи с этим приближения, делаемые ниже, ис-
исходят из точного выражения для волновой функции
ф и энергии Е многоэлектронной системы. Такое вы-
выражение, конечно, эквивалентно исходному уравне-
уравнению Шредингера. Но оно берется в такой детализи-
детализированной форме, что & нем выявляются различные
корреляционные эффекты и т. п. Из этого выражения
не только видны главные корреляционные эффекты,
но также и способ оценки всех членов, которыми
пренебрегают. Полу- и неэмпирические теории, сле-
следовательно, отличаются лишь способом подсчета их
главных частей. Такое приближение позволяет уви-
увидеть также связь между различными теориями, на-
например проследить, какую часть точной функции ф
или Е дает метод Бракнера.
Теория не связана с какими-либо разложениями
в бесконечный ряд, так что там, где необходима
«замкнутая форма», можно применить методы, по-
подобные методу «координаты ri2», или же методу «не-
«незамкнутой оболочки» [1] и т. д. Однако следует при-
признать, что не возникает особых преимуществ при
обобщении [1, 20—22] метода Хиллерааса [1] с по-
помощью прямого введения Гц для всех электронных
корреляций. Так как даже в рамках одного и того
же метода существует много типов корреляционных
эффектов, необходимо использовать различные спо-
способы оценки каждого из них, если только произве-
произведено определенное разделение эффектор.
. Дальнейшее изложение ведется по следующему
общему плану:
1. Исследование различных эффектов в точных -ф
и ?.
2. Методы получения корреляций в оболочках или
электронных парах и между группами.
3. Физический смысл и оценка остающихся оши-
ошибок, обусловленных многоэлектронными членами и
эффектами корреляции в орбиталях.
4. Методы расчета свойств, перечисленных в § 1
для больших атомов и молекул, основанные на об-
общей теории.
Чтобы осуществить все это, мы будем исходить
22 § 6. Орбитали
из молекулярных орбиталей, даже когда будем иметь
дело с большими насыщенными молекулами, подоб-
подобными n-декану. Сначала разделим оболочки на
внутренние и внешние. Для локализованных групп
внутри одной и той же оболочки, Например, таких, как
С—Н- или С—С-связи, отдельные пары Н2О, NH3
и т. д., нам не нужно будет строить заново спе-
специально приспособленную для них теорию. Простое и
строгое преобразование выделяет корреляционные
эффекты и соотносит их с общей теорией, которая
исходит из молекулярно-орбитального описания.
Часть нижеразвитой теории посвящена изучению
малых остающихся эффектов, так что приближения
можно проконтролировать. Те читатели, которые за-
заинтересованы только в практических приложениях и
охотно считают само собой разумеющимися те выра-
выражения, к которым мы приходим в итоге развития на-
нашей теории, могут сразу перейти к § 16, 17, 22 и по-
последующим.
§ 6. ОРБИТАЛИ
Метод Хартри — Фока хорошо описывает зарядо-
зарядовые распределения, потому что в конечных системах
в нем учитывается в основном дальнодействующая
часть кулоновских отталкиваний между электрона-
электронами [9], а это приводит к простым выражениям для
остающихся корреляционных эффектов и к удобной
теории для них. Следовательно, метод Хартри —
Фока является блестящей исходной точкой.
Хартри-фоковские результаты быстро и без осо-
особых усложнений приспосабливают для атомов и не-
небольших молекул [34, 35]. Но получение хартри-фо-
ковских. орбиталей для больших систем все еще пред-
представляет затруднение. Тем не менее теория много-
многоэлектронных систем, исходящая из метода Хартри —
Фока, используется даже для молекул, для которых
еще не доступны точные хартри-фоковсвде резуль-
результаты. Это обусловлено тем обстоятельством, что ме-
метод Хартри — Фока играет роль формальной основы,
§ 6. Орбитали 23
приводящей к простым выражениям для точных г|)
и Е. Общие выводы, например выводы в полуэмпи-
полуэмпирическом случае, основанные на этих формальных
результатах относительно корреляции, оказываются
справедливыми, даже если детальные хартри-фоков-
ские орбитали неизвестны. Далее, на основе выраже-
выражений, которые даются теорией для волновой функции
и энергии, можно исследовать дополнительные эф-
эффекты, возникающие в том случае, если хартри-фо-
ковские орбитали аппроксимируются молекулярными
орбиталями — линейной комбинацией атомных орби-
талей. Корреляционные эффекты оказываются не
слишком чувствительными к тому, насколько хороши
хартри-фоковские орбитали. Однако необходимо
иметь в виду, что в собственно орбитальной части
проблемы применение простых слэтеровских орбита-
лей вместо хартри-фоковских приводит обычно
к ошибкам, сравнимым с корреляцией [36].
Для системы замкнутых оболочек имеется лишь
единственный вариант метода Хартри — Фока. Для
системы незамкнутых оболочек возможны, однако,
различные варианты метода Хартри — Фока. Для од-
одной и той же незамкнутой конфигурации получаются
различные орбитали в зависимости от того, какой
терм используется [например, 3Р или lD, или lS
в случае С (Is22s22p2)]. Следовательно, возможны
различные приближения для устранения недиагональ-
недиагональных энергетических параметров Хц> которые нельзя
убрать с помощью преобразования, как в случае
замкнутой оболочки.
В литературе обсуждалось около девяти вариан-
вариантов подобных методов Хартри — Фока. Перечисление
и обсуждение всех их заведет нас слишком далеко,
поэтому мы отсылаем читателя к другим обзорам,
которые посвящены в основном" методам Хартри —
Фока [19, 37, 38]. К счастью, эти варианты часто
дают энергии, отличающиеся одна от другой менее
чем на 0,01 эв. Заметим, однако, что «корреляцион-
«корреляционная энергия» системы незамкнутых оболочек зависит
от того, какой из вариантов метода Хартри — Фока
применялся.
2# §6. Орбиталй
Различные, орбиталй для каждого мультиплета
с наибольшей точностью получены в работе {34].
Чтобы большие системы, а значит, и формулировка
теории корреляций были простыми, необходимо вы*
полнейте следующих требований:
1. Одна и те же орбиталй должны применяться
во всех возбужденных состояниях, возникающих из
одной и той же конфигурации.
2. Хартри-фоковский потенциал Vi9 действующий
на некий i-й электрон, должен быть одним и тем же
для всех электронов.
3. Орбиталй должны быть ортонормированы.
4. Параметры Хц должны быть опущены в одно-
электронных уравнениях.
Метод [39], который удовлетворяет этим требо-
требованиям, является комбинацией метода Шортли и
Слэтера [15] и метода «симметричных и эквивалент-
эквивалентных ограничений» Несбета [18]. Варьируется средняя
энергия конфигурации [15] [например, средняя энер-
энергия конфигурации (\s22s22p2)]. В получаемых хартри-
фоковских уравнениях потенциалы Vi делаются оди-
одинаковыми путем добавления необходимых долей ку-
лоновского или обменного потенциалов. Эти малые
члены рассматриваются затем как возмущения в кор-
корреляционной части проблемы.
Похожие исходные рассуждения были предло-
предложены Рюденбергом [40] для я-электронных систем.
Существуют также случаи, промежуточные между
замкнутыми и незамкнутыми оболочками. В частно-
частности, существенны два типа таких «почти вырожде-
вырождений»:
а. Для определенности рассмотрим изоэлектрон-
ный ряд Be, В+, С2+, ... Когда атомный номер
Z -* оо, внешняя оболочка 2s2 и оболочка 2р2 стано-
становятся [41] вырожденными.
б. Состояния почти вырожденные возникают
в двухатомных молекулах вблизи диссоциации [1].
Состояния (\ogJ и (lawJ молекулы Н2 вырождают-
вырождаются, когда межъядерное расстояние г->оо.
Каким образом используется «обобщенный» [1]
метод Хартри —Фока и какая часть рассматривается
§ 7. Антисимметрия и электронные распределения 25
в качестве «корреляций» — дел о удобства в таких
промежуточных случаях. Эти вопросы обсуждаются
в § 19—21.
§ 7. АНТИСИММЕТРИЯ И ОТНОСИТЕЛЬНЫЕ
ЭЛЕКТРОННЫЕ РАСПРЕДЕЛЕНИЯ
Ортогональность орбиталей в замкнутых оболоч-
оболочках является следствием антисимметрии. Это объеди-
объединяет электроны в оболочечную структуру («концен-
(«концентрические» оболочки). Однако эффекты действия
оператора антисимметризации <А9 заключающиеся
в локализации электронов, выходят за рамки пра-
правила «два электрона на состояние».
С одной стороны, хартри-фоковские орбитали или
же в молекулах молекулярные орбитали метода само-
самосогласованного поля (SCFMO) пригодны в отдель-
отдельности для описания электронных возбуждений и
ионизации. С другой стороны, одна орбиталь сама по
себе не должна давать вероятности пространствен-
пространственного распределения одного электрона относительно
другого [42—44]. Такое распределение можно полу-
получить лишь с помощью полной хартри-фоковской вол-
волновой функции после перемножения и антисимметри-
антисимметризации спин-орбиталей.
Например, в атоме неона с конфигурацией
(Is22s22p6) нет электронов, распределение которых
выглядело бы подобно 2s или подобно 2pz. Операция
Л в хартри-фоковской волновой функции приводит
к тому, что электроны с одинаковыми спинами рас-
располагаются настолько далеко друг от друга, на-
насколько это возможно. Таким образом; в Ne имеются
два тетраэдра, свободно вращающиеся относительно
друг друга [42—44].
В насыщенных молекулах подобная спиновая
корреляция или эффекты антисимметрии вместе с гео-
геометрией расположения ядер, приводят к локализации
электронов в отдельные пары и электронные парные
связи. Таким образом, хартри-фоковская молекуляр-
26 § 7. Антисимметрия и электронные распределения
ная орбитальная волновая функция неявно передает
локализационные свойства 1).
Теория, излагаемая ниже, показывает, что элек-
электронная корреляция (т. е. кулоновская корреляция,
о которой и идет речь в данной работе, в отличие [1]
от спиновой или же фермиевской корреляции, описан-
описанной выше и содержащейся уже в хартри-фоковской
волновой функции) носит вполне локальный харак-
характер в большинстве молекул, в частности в насы-
насыщенных молекулах. Это означает, что относительные
электронные распределения, а следовательно, и фор-
форма молекул определяются в основном хартри-фоков-
хартри-фоковской частью волновой функции; корреляционная
часть неэффективна (см. также работу [96]). Имеются
в распоряжении хорошие хартри-фоковские расчеты
[45] для Н2О, NH3 и т. д.2). Гипотезы, сформулиро*
ванные выше, были бы подтверждены, если бы они
дали правильные валентные углы [45, 54].
Коулсон [46] и Леннард-Джонс [42, 47] показали,
что относительные зарядовые распределения лучше
передаются с помощью унитарно преобразованного
хартри-фоковского молекулярного 'орбитального де-
детерминанта; унитарное преобразование не меняет де-
1) Эти важные черты теории молекулярных орбиталей изу-
изучались Леннард-Джонсом [42] и Поплом [43]. Линнет [44] иссле-
исследовал относительные зарядовые распределения, отыскивая
максимум квадрата детерминантных волновых функций в зави-
зависимости от изменений относительных положений всех электро-
электронов. В этой работе, однако, использовались голые ядра, т. е. ор-
битали, в которых не учитывалась экранировка. Следовательно,
отсутствие спиновой корреляции между аир спинами в замк-
замкнутой системе оболочек не рассматривалось.
2) Эти молекулы рассматриваются как претерпевшие иска-
искажения основной тетраэдрической формы. Два отдельных те-
тетраэдра в неоново-подобном ионе превращаются в один при
добавлении протонов. Картина эта, однако, представляется
очень упрощенной и не позволяет сделать количественных вы-
выводов, скажем, о причинах, по которым валентный угол при-
приближается к 90° в ряду Н2О, HaS, H2Se и т. д. В ней содержит-
содержится лишь гибридизация, обусловленная принципом исключения
(в Ne), но не учитывается гибридизация поляризационного типа,
обусловленная протонами. Конечно, оба типа неявно содер-
содержатся в расчетах, проводимых с применением полных хартри-
фоковских молекулярных орбиталей самосогласованного поля [54].
§ 7. Антисимметрия и электронные распределения 27
терминант, но позволяет выразить его через локали-
локализованные или «эквивалентные» орбитали. В симмет-
(шчном случае, подобном СН4, это преобразование
46, 47] единственно1). Однако в случае, где все связи
неэквивалентны, как, например, в СгНв, необходим
некий критерий. Унитарное преобразование можно
определить, например, потребовав, чтобы величина
обменных членов была минимальна. Это давало бы
ббльшую часть локализованных и большинство почти
классических связей [49, 50, 57]2).
Для больших молекул теория Хартри — Фока на-
находится все еще в незавершенном виде [52, 53].
Однако должны быть возможны некие упрощения
для больших насыщенных молекул. Электроны дви-
движутся не только в хартри-фоковских (молекулярных
орбитальных) потенциалах всех электронов, но так-
также и в поле ядер. Результирующий потенциал, дей-
действующий на какой-либо электрон в одном конце
молекулы, обусловленный, скажем, электронами
в другой связи, является, таким образом, экраниро-
экранированным, т. е. короткодействующим потенциалом [40].
Например, в этане средний потенциал атома водо-
водорода, находящегося у одного атома углерода, спа-
спадает до нуля на малом расстоянии и, следовательно,
не действует на атомы водорода, расположенные
у другого атома углерода [54]. Это подсказывает
следующую последовательность действий: можно по-
получить С—Н-часть волновой функции С2Н6 из метана,
затем, чтобы получить полную волновую функцию
самосогласованного поля С2Н$, ввести поправки на
самосогласованное поле между группами СН3 и С—С,
возможно, с помощью теории возмущений.
В многоэлектронной теории мы всегда будем ис-
1) Несбет [48] получил приближенные молекулярные орбита-
орбитали самосогласованного поля с небольшим числом базисных функ-
функций для СН4, выбранных в гауссовом виде, а затем преобразо-
преобразовал их в эквивалентные орбитали.
2) Аналогичный критерий, хотя по идее менее удовлетво-
удовлетворительный, использует Бойз [51]. Он разделяет центры тяжести
связей настолько, насколько это возможно,.
2S f & Уравнения Хартри—Фока
ходить из молекулярных орбиталей самосогласован-
самосогласованного поля, ' преобразовывая затем там, где это
требуется, хартри-фоковскую волновую функцию х
локализованному описанию.
| S. УРАВНЕНИЯ ХАРТРИ - ФОКА
Рассмотрим систему из N электронов, описывае-
описываемую точной волновой функцией,
х2, ...
где X/ означает набор пространственных и спиновых
координат /-го электрона. В нерелятивистском слу-
случае имеем уравнение Шредингера:
B)
Dа,
a
,0
где h°i—гамильтониан J-го электрона без учета экра-
экранировки, Rai—'расстояние /-го электрона от ядра а
Для энергии используются атомные единицы
A am. ед.=27,2эв). Теория основывается на уравне-
уравнениях B) и C); релятивистские эффекты обсуждаются
в § 29. Сначала рассмотрим соответствующую хар-
трифоковскую задачу для системы с замкнутыми обо-
оболочками.. В этом случае мы имеем1)
') Далее используются следующие обозначения.
Одноэлектронные функции обозначаются одной буквой:
/»**<*) (/-1,2,3 N).
Произведение N одноэлектронных функций записывается в виде
N
§ 8. Уравнения Хартри—Фока 29
(в)
2 3 ...N); (ба)
—оператор антисиммётризацш! N электронов
Спин-орбитали нумеруются цифровым индексом i,
начиная с наинизшей орбиталп со спином а. В ато-
атомах имеем
l=(Ua), 2=(lsp), 3 = Bsa), .... Fв)
В двухатомных молекулах
i
В больших молекулах индексом i по-прежнему
нумеруются хартри-фоковские молекулярные орби-
тали самосогласованного поля. (По поводу преобра-
преобразования функций <f>0 в эквивалентные орбитали и со-
соответственно уравнений Хартри — Фока см. § 27.)
Таким образом, все нечетные *\ относятся к спи-
спинам а, а все четные *—к спинам р.
В уравнении Fа) до применения оператора <Л по-
полагаем, что спин^орбиталь i занята электроном с ко-
координатами xf. Следовательно,
n))*e*A[\x 2233... NN\% Fr)
так что вообще, если мы специально не подчерки-
подчеркиваем, как в случае (ба), индексом / нумеруются как
а соответствующие произведения, в которых опущена одна,
две, ... одноэлектронные функции, — в виде
Д
Скалярные произведения обозначаются угловыми скобками, на-
например,
Xk) dXk И Т- *¦
—Прим. ред.
30 §8. Уравнения Хартри—Фока
спин-орбиталь, так и соответствующий ей элек-
электрон. Введем
| G)
Здесь Vt — хартри-фоковский потенциал N-электрон-
ной среды (хартри-фоковский „фон44), действующий
на /-й электрон,
;ЗД, (8)
где 5j(i) — сумма кулоновского потенциала Sj(xt)
и обменного потенциала /?у(х^), действующего на
/-й электрон (вычислены по спин-орбитали у-го элек-
электрона):
* (9a)
Sj (/) ЕЕ5 Sj (Xt) = (J (Xj)t giJj (Xj))x/ (96)
R] (X|) / (x,) = (J (xy), fty/ (xy)>Xy у (x,). (9в)
Символ (...)х означает интегрирование лишь по
координатам Xj. Уравнение (8) формально включает
члены с j=i, т. е. „самодействие44.
Сумму Vt в уравнении G) можно переписать как
сумму по парным потенциалам
x*)= S [^
i1
Из уравнения G) имеем
?г19 (И)
где е^ — хартри-фоковская энергия, вычисленная
с помощью орбитали /, приблизительно равная по-
потенциалу ионизации [1] из этого состояния.
Спин-орбитали удовлетворяют уравнению
eii = 0, A2)
где
§ 8. Уравнения Хартри—Фока 31
Заметим, что обычно применяемая полная хартри-
фоковская энергия не равна ?0, а дается выра-
выражением
Ео + Ег, A3а)
- 2 (Jij -Kh\ A36)
где
#1 = 2^-2^, A4)
что следует из уравнений C) и G). Величины Jtj
и Kij являются кулоновскими и обменными инте-
интегралами соответственно
A56)
Комбинируя выражения C), G), A0) — A5), мы опре-
определяем
Н - ?н. F. = (Яо - ?0) + (Я! - ?2) = 2 еь + J m/yf
>j A6)
где
A7)
здесь m*j представляет «мгновенное» отклонение по-
потенциала между электронами i и / от соответствую-
соответствующего среднего значения, даваемого методом Харт-
Хартри — Фока. Следовательно, т^ можно назвать флук-
туационным потенциалом. Потенциал тцу опреде-
определяемый по отклонению от хартри-фоковского потен-
потенциала, отвечает за корреляции.
Для системы с незамкнутыми оболочками вместо
выражения Fа) нужно брать линейную комбинацию
различных вырожденных детерминантов. Уравнения
Хартри — Фока и потенциал V\ в таком случае за-
зависят от того, какой из методов Хартри — Фока (см.
§ 6) используется. Метод «симметричных и'эквива-
32 § 9. Точная многоэлектронная волновал функция
дентнщх Щ|$шчений», основанный на «средней энер-
энергии ковфр$^ации», дает те же самые Яо и Vu что
и уравнение G), для всех мультиплетов, возникаю-,
щих, m той же конфигурации. Это упрощает рас-
рассмотрите коррелйционной части.
Метод Хартри —Фока, примененный к наиниз-
шим замкнутым и незамкнутым оболочечным конфи-
конфигурациям, определяет некую схему электронных
уровней. Весьма часто это взаимно однозначно соот-
соответствует экспериментально наблюдаемой последова-
последовательности состояний [19]. На основе лишь теории
Хартри — Фока можно понять первые два правила
Хунда. Подобные схемы уровней будут относиться
к так называемым адиабатическим схемам уровней.
Описанные выше уравнения Хартри — Фока приме-
применяются к ним непосредственно. В редких случаях,
например в случае MgCs, 3d), упомянутом [55] в §2,
уровни могут пересекаться. Корректная схема уров-
уровней в таком случае должна быть получена или пу-
путем введения корреляции, как это описано в § 22,
или же с помощью первого порядка метода кон-*
фигурационного взаимодействия [6], или же «об-
«обобщением» [1] метода Хартри — Фока на конфигу-
конфигурации, которые очень сильно перемешиваются (см.
§ 19—21). В последнем случае уравнения этого па-
параграфа должны быть соответствующим образом
модифицированы.
Удобно развить многоэлектронную теорию; изла-
излагаемую ниже в основном на базе замкнутых оболо-
оболочек, исходя из уравнений F) — A7). Некоторые моди-
модификации результатов последующих параграфов, ко-
которые необходимо сделать для незамкнутых оболо»
чек, указываются в § 20.
§ 9. ТОЧНАЯ МНОГОЭЛЕКТРОННАЯ ВОЛНОВАЯ
ФУНКЦИЯ
Любую точную волновую функцию можно запи-
записать в виде
A8)
§ 9. Точная многоэлектронная волновая функция Щ
с соответствующим изменением нормировки. При этом
фо может быть хартри-фоковской волновой функцией
для замкнутой или незамкнутой оболочки, а % — все
то, что не учитывается ею. Таким образом,
<ЛиХ>«0. A9а)
{Фо> *>>-!. <¦• *> = 1 + (X, Х>. A96)
Хартри-фоковская волновая функция <f>0 определяется
отталкиваниями между электронами, усредненными
по их орбиталям. В таком случае х определяется
всеми остальными эффектами электрон-электронного
отталкивания. Следовательно, функция х обусло-
обусловливаем флуктуационными потенциалами [урав-
[уравнения A4) и A7)]:
N
Она является точной корреляционной волновой
функцией *).
Для системы с замкнутыми оболочками точная
функция х дается выражением [9—12]
B0а)
где
{|} B06)
B0.)
{U't }=_fL(A 2 3 ... N)^\. B0r)
O[23...N}. ¦ B0д)
!) Заметим, что в волновой функции [1], например, для Не,
подобной $8 A2)(l+afi2), второй член не является корреля-
корреляционной частью. Он не ортогонален к jj?A2) и, следовательно,
все еще содержит какую-нибудь хартри-фоковскую волновую
функцию.
3 О. Синаноглу
34
§ 9. Точная многоэлектронная волновая функция
Хартри-фоковская функция фо описывает некую
«среду», в которой электроны движутся, причем ка-
каждый в своем статическом потенциале W Уравне-
Уравнения B0) корректируют это движение на «столкнове-
«столкновения» между последовательно увеличивающимся чис-
числом электронов в единицу времени в этой «среде».
Мы говорим, что происходит «столкновение», если,
например, электроны / и / входят внутрь области
действия их взаимного флуктуационного потенциа-
потенциала rriiy Движения их в таком случае становятся кор-
коррелированными, и в функцию х вводится соответ-
соответствующая поправка {?///}. По аналогии с теорией
неидеальных газов [56] можно говорить об одно-,
двух-, трех- и т. д. электронных комплексах в урав-
уравнениях B0).
Каждое из ?/' меняет знак при нечетном числе
перестановок:
О'ц (хь Xj) = Olj (/, J) = -
, /), B1)
у, k) = —
и т. д.
Это и обусловливает появление множителя (п\)~ч*
в выражениях B0в) — B0д). Группа перестановок
п электронов является подгруппой группы переста-
перестановок N электронов. Имеем
B2)
где &ij...n — оператор антисимметризации п элек-
электронов. Таким образом,
и т. д. для всех 3Bii,..«.
§ 9. Точная многоэлектронная волновая функция 35
Каждая функция в B0а) ортогональна ко всем
занятым спин-орбиталям в fQi т. е.
о
=h 2, ...,/, ...,у, ..,,Л0,B4)
где, например,
(О'
B5)
Такого рода «орбитальная ортогональность» обозна-
обозначается с помощью значка Л (шляпки); ¦ (звездочка)
означает комплексное сопряжение.
Заметим, что выражения B0) и B4) являются
строгими [8—11]. Они не предполагают каких-либо
ограничений, как это имеет место в A).
Несмотря на то, что выражения B0) все еще
являются формальными, они обладают явными преи-
преимуществами по сравнению, например, с бесконеч-
бесконечными точными рядами, получающимися в методе
конфигурационного взаимодействия:
1) в них не появляется бесконечной суммы, если
система имеет конечное число (N) электронов;
2) они представлены в таком виде, что возможно
рассмотрение каждого члена и проведение система-
систематических приближений, как это будет сделано ниже;
3) функции U' даны в замкнутом виде. Они мо-
могут содержать межэлектронные координаты, «рас-
«расщепление оболочек» и т. п. или же каждую из них
можно по отдельности разложить в ряд по методу
конфигурационного взаимодействия.
Выражения B0) и B4) можно получить [8] в ка-
каждом порядке из волновой функции, найденной по
теории возмущений. Чтобы иметь замкнутые выра-
выражения O'tjt O'tjk и т. д., можно применить опера-
операторную технику. Например [8], в первом порядке
(т. е. для хО» исходя из <j>0 как из невозмущенной
функции, находим лишь uf{} (первый порядок), В %2
$6 § 9. Точная многоэлектронная волновая функция
появляются члены второго порядка функции #//,
т- е* и% * также Jf и О\%* В высших порядках
возникает асе большее и большее число членов. Если
объединить все результаты, например все парные чле-
члены ufp ufr... из всех порядков, то получим
сумму бц. Аналогичное положение имеет место для
Vijk и т. д.
Иначе точную функцию % можно получить с
помощью отвечающего ей бесконечного ряда из ме-
метода конфигурационного взаимодействия. Это разло-
разложение [1] содержит все однородные «упорядоченные»
[1] слэтеровские детерминанты, построенные из пол-
полного одноэлектронного базисного набора. Если функ-
функция ф0 должна быть хартри-фоковской, то такой ба-
базисный набор должен включать N хартри-фоковских
спин-орбиталей, а остальные могут быть произволь-
произвольными [1]. Члены ряда конфигурационного взаимодей-
взаимодействия можно расклассифицировать [3] в одиночные,
двойные и т. д. «возбуждения» из определенных спин-
орбиталей в ^о. Если затем просуммировать каждый
класс [3, 9, 31], то, используя полноту базисного на-
набора, придем в результате к выражениям B0) и B4).
Произвольность выбора базисного набора, исключая
первые N хартри-фоковские орбитали, эквивалентна
тому, что 0' не является еще окончательно опреде-
определенной функцией, а подчиняется соотношениям B4),
Ниже, однако, мы получим для уравнений B0) еще
более детализованную форму и выявим эффекты, не
выявляющиеся в методе конфигурационного взаимо-
взаимодействия.
Выражение B0а) для % можно получить несколь-
несколько иным, чрезвычайно наглядным методом, который
также показывает, каким образом можно найти раз-
различные части х (например, fi или Оц и т. д.) из про-
произвольной функции \|). Метод этот можно назвать
«методом последовательных частичных ортогонализа-
ций».
Будем исходить из точной многоэлектронной вол-
волновой функции ф. Спроектируем ее на ^о и вычтем
§ 9, Точная многоэлектронная волновая функция 37
результат из ф (осуществим так называемую шмид*
товскую ортогонализацию). По определению [см.
уравнение A9а)],
^^-Фо^ B6)
отсюда в силу соотношений A9) имеем (\|>, ?#)»1.
Ортогонализуем далее % по отношению к произведе-
произведениям спин-орбиталей, полученным из функции </>0 вы-
вычеркиванием одной из спин-орбиталей, например по
отношению к произведениям спин-орбиталей B 3...
,.. N), A 3 4 ... N) и т. д,!) В результате приходим
к величине %' — некой малой части х:
здесь det есть УШ Л, а ((х, 123.. .N)/(i)) озна-
означает интегрирование по всем координатам, кроме
Xi — координаты i-й спин-орбитали, опускаемой из
произведения орбиталей. В этот интеграл не входит
оператор антисимметризации, поскольку сама функ-
функция х является антисимметричной [см. соотношения
B3)].
Каждый интеграл в выражении B7) дает неко-
некоторую функцию лишь от Xi и определяет функции
/~ = УЖ<Х, [123... (/-1)(*+1)...JV]>. B8)
Следовательно, пока мы имеем
{A 2 3 ... N)f}+%' B9)
{h *) = 0 (* = 1, 2, 3, .... /, .... N). C0)
Чтобы пояснить, каким образом возникает выра-
выражение C0), рассмотрим для простоты четырехэлек-
тронный случай. Используя определение B8) для fh
1) Как и в B6), «проектируем» % на произведения спин-
орбиталей, получаемые из Фо вычеркиванием i-й спин-орбитали,
и берем разность между зс и суммой по всем проекциям.—
Прим. перев^
38 § 9. Точная многоэлектронная волновая функция
получаем
<flf 1> = /4Г(х, Ii223344) = (x, ^o) = O, C1а)
х, 212Д44>-
О C16)
и аналогичные выражения для fe = 3, 4. В последних
членах равенств C1а) и C16) мы использовали со-
соотношения B2) и тот факт, что Л—эрмитовский
оператор.
Заметим, что «ортогонализации» в стиле выраже-
выражения B7) отличаются от общепринятых тем, что N-
электронная функция ортогонализуется по отноше-
отношению к функции нескольких электронов [8]. Это и
является причиной, по которой в выражении B7)
появляются определители (det).
Выражение B9) расширяется с помощью прове-
проведения шмидтовской «ортогонализации» остающихся
у!\ х"» • • • по отношению к произведениям все мень-
меньшего и меньшего числа спин-орбиталей из функ-
функции ф). Таким образом,
. C2,
Аналогично выражениям B8) — C1) находим
C3)
(О'и, k) = 0 (ft = 1, 2, ...,/,...,/,..., N); C4)
наконец, в обозначениях B0) будем иметь
х". C5)
Этим способом получаются формулы B0), B1) и
B4) с тем дополнительным результатом, что общий
§ 9. Точная многоэлектронная волновая функция 39
член дается в виде
До некоторой степени сходную процедуру Брен-
ниг [58] применил к уравнению Шредингера Hty = Ety.
Умножив одновременно с обеих сторон уравнение на
произведение (N—1), (N — 2), ..., 1 спин-орбита-
лей и проинтегрировав, он получил систему связан-
связанных интегро-дифференциальных уравнений, анало-
аналогичных уравнениям Кирквуда — Боголюбова в тео-
теории жидкости [56]. Уравнение для Ji зависит от O'tj,
0'ijk, •••, уравнение для tf'.j — от 7/, O'ljh и т.д.
Однако эти уравнения не имеют какой-либо практи-
практической ценности. Даже если бы они оказались рас-
расцепленными с помощью приближений (таких, напри-
например, как «суперпозиционное приближение» [56] Кирк-
Кирквуда для жидкостей) [58], то, не говоря о появлении
труднооцениваемых ошибок, их все еще нельзя было
бы непосредственно решить.
Выражения C5) и C6), с другой стороны, могут
дать определенные результаты, в особенности, ко-
когда они применяются вместе с вариационным прин-
принципом:
ШМ C7)
Предположим, что в нашем распоряжении имеется
хорошая пробная функция \{>. Тогда из уравнения
C6) можно получить простыми интегрированиями
ее компоненты fp O\j, U'ijk и т. д.
В литературе имеется много расчетных схем и
пробных функций, обходящих приближение Хартри —
Фока и не указывающих явно на какие-либо корре-
корреляционные эффекты. Примерами являются расчеты
LiH, Li2 и т. д., проведенные Гаррисом и Тейлором
[83], и метод «альтернантных орбиталей» [84—86].
Можно добиться большего понимания этих проблем
40 § 10+ Ортогональность орбиталеп
с помощью анализа «последовательных частичных
ортого^&рзаций» *). Предположим, что с помощью
такого а*етода мы получили заметную величину для
fi или Uifa, тогда как, согласно результатам § 18 и
19, следовало ожидать малости этих членов в точ-
точной функции х; в таком случае, если судить по энер-
энергии, лучшая пробная функция получилась бы в пре-
пренебрежении ими. Мы въявь проведем подобный ана-
анализ для Не с ri2 в -ф и проверим важность учета ft
для многоэлектронных систем (см. § 19).
§ 10. ОРТОГОНАЛЬНОСТЬ ОРБИТАЛЕЙ
Выражения B4) фактически заключают в себе
два различных эффекта [8]: каждая функция O'iji
является ортогональной
а) к своим собственным орбиталям /, /, /, ...;
б) ко всем другим орбиталям кФг, /, /,..., заня-
занятым в </>о2).
Эффект «а» связан с тем, что в B4) выделяются
корреляции меньшего числа электронов, чем в U [как .
и в уравнениях C2) и C5)]. Это особенно суще-
существенно в Q'lji когда выделены %. Одноэлектрон-
ные члены % описывают влияние корреляций на ор-
битали (см. также § 19).
Чтобы разъяснить этот вопрос, рассмотрим атом
гелия. В этом случае
C8)
есть хартри-фоковский детерминант 1$2-состояния.
Для ф(х1э х2) — точной волновой функции :5-терма —
*) Теперь видно, что этот метод также указывает путь по-
получения хартри-фоковской волновой функции непосредственно
из общей многоэлектронной пробной функции без проведения
отдельного вариационного расчета. См. дополнение автора в
конце этой работы.
2) См. также работы [59, 60]. Авторы в них использовали
лишь принцип исключения, т. е. эффект «б».
§ /0. Ортогональность орбиталей 41
имеем
4>(хьх2)=#A2)+я?2, C9)
где
[из соотношения A8I. Применяя выражения B7)—B9),
получаем
n. D0)
Например,
b x2J(x2))X2 D1)
, 2) = 0. D2)
На языке метода конфигурационного взаимодействия
функции ft являются суммами отдельных возбужде-
возбуждений, а функции О'п — только двойных возбуждений
[3, 8]. С помощью полного базисного набора [к] ряд
в методе конфигурационного взаимодействия запи-
записываем [1, 3] в виде
Ч- S ск1ЖAгк2). D3)
Л/2
Умножая это выражение на 1(хг) и интегрируя,
получаем
оо
К D4)
следовательно,
оо
и
оо
= 2 Сн& (Л^г)- D6)
*>/>2
42 § 11. Несвязанные комплексы
Эти соотношения поясняют, каким образом выра-
выражения B0) и B4) можно проверить, применяя ме-
метод конфигурационного взаимодействия.
Из этого примера видно, что эффект «б», упомя-
упомянутый выше, не имеет места в атоме гелия и оказы-
оказывается возможным лишь в многоэлектронной системе.
Пусть мы имеем
(Gij, ?> = О, k + и j (К k < N). D7)
Это означает, что когда электроны i и / коррели-
коррелируют, т. е. «сталкиваются» друг с другом, то из-за
принципа исключения Паули они не могут оказаться
в состоянии k (k=f=i, /), уже занятом в </>0; отсюда —
термин «эффект запрета» [3, 8]. В методе конфигу-
конфигурационного взаимодействия это означает, что для
случая более чем двух электронов последняя сумма
из выражения D3) начинается с номеров, больших N:
2 сн®AА). D8)
k>l>N
§ П. НЕСВЯЗАННЫЕ КОМПЛЕКСЫ
Точная функция % [выражение B0а)] все еще не-
недостаточно детализирована. Последовательные чле-
члены в этом выражении вносят поправку на эффекты
прогрессивно увеличивающегося числа электронов,
взаимодействующих с собственным флуктуационным
потенциалом. Как и в случае теории неидеального
газа [56], необходимо, однако, различать два типа
«комплексов». Например, четыре электрона могут
«столкнуться» все сразу (четырехчастичное соударе-
соударение) и дать вклад в ?/12з4.Однако в то же самое
время могут иметь место два независимых бинарных
«соударения», происходящих в различных частях ато-
атома или молекулы. Можно говорить о «времени», по-
поскольку теорию можно развить с одинаковым успе-
успехом на основе зависящего от времени формализма,
подобного формализму, использованному Голдстоу-
ном [61]. Индивидуальные бинарные соударения учи-
учитываются членами {0'ij). Наряду с этим такие со-
§ 11. Несвязанные комплексы 43
ударения неявным образом входят также в большие
комплексы. Два независимых члена O\j могут, на-
например, составлять О [-Мм и быть частью О1234.
Действительные многоэлектронные «соударения»
являются «связанными комплексами», которые мы
будем обозначать с помощью Оцъ,... — без штриха.
Произведения независимых, но одновременных «со-
«соударений», например G12()з4 или ?I2#з4#5в и т. д.,
являются «несвязанными комплексами».
Таким образом, каждая точная функция G' в вы-
выражении B0) на самом деле состоит из всевозмож-
всевозможных антисимметризованных произведений величин,
отвечающих предшествующим малоэлектронным чле-
членам и не содержащих некоторых индексов вообще,
и нового члена С/, соответствующего «связанному
комплексу». Для удобства эти члены можно предста-
представить с помощью диаграмм, в которых Д- — малень-
маленькие кружочки (единственные кружочки, которые не
связаны линиями), а связанные комплексы 0—пря-
0—прямые линии, проходящие между связанными электро-
электронами. Например,
O (dU D9)
Аналогично, Uijki представляется так:
~, о о о—о о—о о—о О—о
Uim= + + + \А +11
1 о о оо о—о а о 6—6
E2)
44 § 1L Несвязанные комплексы
Здесь каждая фигура означает все члены, отличаю-
отличающиеся лишь обозначением электронов.
Коэффициенты, подобные ]А2! в выражении E0),
возникают из выражений B1) и B3). Например, в вы-
выражении E2) имеем
+ •¦¦/• E3)
Однако каждая функция О\ в конце концов, анти-
симметризуется при помощи Л. В результате, при-
применяя выражения B06) —B0д), получаем, что \О\\
содержит член
Л Г A23...AQ ^ / Oifiui Wm \\
/47 I (ijki) ®ijkl \VT\V2\ + VT\VT\)) =
Следовательно, в обозначениях B0) имеем
Коэффициент некоего (гХ^Х'Х • - •) — несвязан-
несвязанного комплекса, который является частью я-комп-
лекса (л = г + $4-*+ ...), равен
- да»
Заметим, что более детализированное выражение для
X, получаемое с помощью подстановки соотношений,
подобных D9) — E2), в выражение B0а), является
все еще точным.
Как будет выяснено ниже, основной математиче-
математической функцией «несвязанных комплексов» является
сокращение членрв нормировки в знаменателе выра-
выражения C7). Такое «сокращение несвязанных комп-
комплексов» [61, 62] играет важную роль в многочастич-
многочастичной теории с бесконечно большим числом частиц [32,
61], потому что (х, х)~~*°°> когда N г-юо. В теории
§ It. Несвязанные комплексы 45
возмущений Рэлея — Шредингера [62] сокращение
происходит автоматически, однако его нужно выявить
в вариационных методах путем расширения класса
пробных функций с несвязанными комплексами J9,63].
Каким образом появляются некоторые из несвя-
несвязанных комплексов, можно рассмотреть весьма про-
просто. Возьмем, например, члены f{. Они являются по-
поправками к 1-м хартри-фоковским орбиталям. Пред*
положим поэтому, что каждая i-я орбиталь ь ф0 за-
заменена на (i+Ji) и проведено перемножение. Тогда
имеем
что показывает, каким образом возникает выражение
D9) [ср. выражение D0)].
Эти новые орбитали не нормированы к единице.
Следовательно, (-ф, -ф) содержит множитель A +
+ (Ju Ь))- Однако член (/*, Jj) в волновой функции
дает аналогичный множитель в (ф, #ф). В общем
случае, чем точнее приближает пробная функция точ-
точную функцию %, тем с большей точностью можно со-
сократить из знаменателя A + (х> х)) fCM- точное вы-
выражение E9) для энергии]. Отсюда следует, что проб-
пробную функцию всегда можно улучшить путем вклю-
включения несвязанных комплексов [9].
«Обобщенное произведение групп&вых функций»
[23—30] типа \j> [выражение A)] включает лишь не-
некоторые несвязанные комплексы. Чтобы показать
это, групповую функцию
Л^(/, у, ..., п)
запишем в виде
где
В таком случае произведения
Л (Л0*, AS,...)
46 § 12. Вариационный принцип и точная энергия
содержат лишь несвязанные комплексы, другие ком-
комплексы, которые необходимо учитывать, потеряны.
§ 12. ВАРИАЦИОННЫЙ ПРИНЦИП И ТОЧНАЯ ЭНЕРГИЯ
Предположим, что хартри-фоковская функция ф0
известна и мы должны определить %-ю часть точной
волновой функции ф. Подставляя в выражение C7)
где
и раскрывая знаменатель в Eu.V., получаем вариа-
вариационный принцип [64] для х в виде
О, (Я- ?н, р.) х) + (х,(" -Ен. р.)х)
= Ё. E8)
Если вместо функции х подставить точную функ-
функцию х и использовать наряду с уравнением Шре-
дингера (для х) [64] выражения E), A6) и A9), то
знаменатель сократится и E8) сведется к выражению
(только для точной функции х)-
Для системы замкнутых оболочек, если ^0—хар-
^0—хартри-фоковская волновая функция, уравнение E9)
после подстановки выражений B0) и A4) дает
Е = En. f. + 2 (& Ш eifl'ij) F0)
i> j
(только для точных функций C'ip 5С» ?)•
1) Поясним получение выражения E9). Из уравнения Шре-
дингера для ур, используя E), A6) и A9), имеем
§ 12. Вариационный принцип и тонная энергия 47
Это выражение представляет собой замкнутую
форму «обобщенной теоремы Бриллюэна» [1], обыч-
обычно даваемой в виде разложений метода конфигура-
конфигурационного взаимодействия [1, 32, 38, 63]. Она справед-
справедлива лишь для точной б'ц из выражения B0) и с хар-
три-фоковскими орбиталями. Заметим, что функции
тц свелись к ?^ = l/r^-.
Простота выражения F0) чисто формальная.
С любой пробной функцией %, как показывает под-
подстановка в выражение E8), выражение F0) услож-
усложняется. Его нельзя сделать основой полуэмпириче-
полуэмпирических рассуждений, подобных тем, которые исполь-
используются в методе разделения оболочек, поскольку
парные члены не расцеплены. Каждая из функций
u'tj может весьма сильно зависеть от всех других,
а также от ]r U'ijk и т. д. (Действительно, фор-
формально эта зависимость выражается в том, что эти
функции подчиняются системе связанных интегро-
дифференциальных уравнений, упомянутых выше
[58, 65]О.)
Более детальное выражение для энергии в ва-
вариационном методе можно получить, подставляя B0)
в соотношение E8). Тогда выявятся различные
Домножая на Фо и интегрируя, находим
Е = (ФоНФо) + (Ф0И%) = ?„ F< + (Фо (Но + Нх) у) =
= *н. F. + {Фо (Ео + Нг) Х) = ?н F + {Ф0Н1%).
Далее, используя формулы B0), A4) и выражения (9) и A0)
для Sij (ij), после простых, но довольно громоздких преобразо-
преобразований получаем выражение F0). Формальная простота выра-
выражения F0) обусловлена тем, что взаимодействие электронов ха-
характеризуется суммой «двухчастичных» операторов gfj=l//*ij и
выбором разложения % по корреляционным функциям, удовлет-
удовлетворяющим соотношениям B4). — Прим. перев.
1) В работе [65] приводится бесконечная система связанных
интегро-дифференциальных уравнений, полученных в виде раз-
разложений по методу конфигурационного взаимодействия, а также
делается сравнение метода Хартри—Фока, дополненного мето-
методом конфигурационного взаимодействия, с методом Бракнера.
4S § 13. Различные корреляционные эффекты
эффекты корреляции в энергии, и их можно будет изу-
изучать по отдельности. Это, конечно, необходимо также
для ^шфетяых расчетов.
§ 13. РАЗЛИЧНЫЕ КОРРЕЛЯЦИОННЫЕ ЭФФЕКТЫ
В ТОЧНОЙ ВОЛНОВОЙ ФУНКЦИИ И ЭНЕРГИИ
Точная функция х, определяемая выписанными
выше выражениями B0), B4), D9) — E2) и т. д., со-
содержит:
1) влияние корреляции на орбитали, т. е. чле-
члены fi\
2) двухэлектронные корреляции, т. е. члены Ь^
из D9);
3) многоэлектронные корреляции (N>/z>2),
т. е. члены 6цк, Viikh ...;
4) несвязанные комплексы во всех этих членах.
Хотя каждая из подобных корреляций обусловле-
обусловлена одновременным взаимодействием лишь несколь-
нескольких электронов, определенное влияние оказывают и
все остальные электроны, потому что «столкновения»
происходят не в вакууме, а в «среде» (хартри-фоков-
ском «фоне»), связанной с &> [3, 8—11]. Этот «фон»
действует на комплексы через
а) «принцип исключения», соотношение D7) или
в общем случае
I*1
б) внутренний хартри-фоковский потенциал V,-
[выражение (8)].
Аналогичные корреляционные эффекты возникают
и в энергии. Их можно получить в явном виде, под-
подставив выражение B0) для функции % в выраже-
выражение E8). Сложные алгебраические преобразования
удобно выполнять с помощью диаграмм подобно
тому, как это сделано в выражении E2). Диаграммы
матричных элементов оператора энергии в дополне-
дополнение к сплошным линиям, отвечающим потенциалам
U, содержат пунктирные линии, обозначающие флук-
§ 13. Различные коррелщионные эффекты 49
туационный потенциал гпц, определяемый выраже-
выражением A7). Таким образом, например, имеем
miju-ij). F1)
Браут [63] использовал сходную диаграммную тех-
технику, но в рамках формализма конфигурационного
взаимодействия и в соединении с теорией ядерной
материи Бракнера. Техника, используемая здесь, не-
несколько напоминает обычные методы теории поля
[61] или же методы статистической механики не-
неидеальных систем [56].
Ниже цы не будем выписывать все диаграммы,
которые появляются в точном выражении для энер-
энергии. Для наших целей достаточно отметить, Что не-
некий заданный корреляционный эффект в %, вызы-
вызывает дополнительные эффекты в энергии. Например,
энергия, полученная с помощью функции х> в кото-
которой учитываются лишь члены V^ содержит наряду
с парными и трехэлектронные корреляции [9]. Это
связано с тем обстоятельством, что энергия стацио-
стационарна и ее можно определить с.большей точностью,
нежели точность данной функции х» используемой
для вычисления энергии.
Полные точные % я Е — те же, что и в уравнении
Шредингера, и, следовательно, для получения их не су-
существует разработанной теории, а имеется лишь не-
некая формальная схема. Однако можно выяснить зна-
значение и точность различных приближенных теорий,
если результаты, полученные с их помощью, сравнить
с этими полными формальными выражениями. Мы
покажем в § 15, какую часть точных х и Е дают те
или иные теории. Возможно, что многие формальные
схемы и теории зависят от того, оставили ли мы /* и
&ij или еще и О\)ь и т. д. Многие из таких фор-
формализмов, однако, вводили бы ненужные усложне-
усложнения, если бы в них фигурировали члены, которые
были бы на самом деле физически несущественны.
Таким образом, чтобы прийти к простейшей реали-
4 О. Синаноглу
50 § 14. Вариационный подход
стической теории, должны быть обследованы сле-
следующие эффекты с оценкой их величины в характер-
характерных случаях:
1) парные корреляции Uijt которые являются
основными эффектами;
2) n-электронные корреляции (я>2) 0цк и т. д.;
3) n-электронные корреляции (я>2), которые
возникают в энергии в связи с парными функциями
Uij (эти корреляции будут более существенны, не-
нежели многоэлектронные корреляции в %, поскольку
такие корреляции в х» запутывая больше электронов,
дают эффекты более высокого «порядка» в энергии);
4) ?,.
Упомянутые эффекты будут обсуждаться в § 18-—
20 после того, как будет перечислено, какие эф-
эффекты учитываются в различных теориях. Эти тео-
теории можно развить на базе формальных выражений
для точных х и ? с помощью общего вариационного
подхода, использующего теорию возмущений. Вна-
Вначале мы опишем этот подход, который позволяет так-
также оценить ошибки, обусловленные различными при-
приближениями.
§ 14. ВАРИАЦИОННЫЙ ПОДХОД,
ИСПОЛЬЗУЮЩИЙ ТЕОРИЮ ВОЗМУЩЕНИЙ
Непосредственное использование вариационного
метода, т. е. выражений C7) или E8), связано с ка-
какими-то предположениями относительно пробной
функции. Здесь же предлагается систематически по-
получить реалистическую функцию х-
Условие стационарности или минимальности Е(%)
[выражение E8)] является также условием суще-
существования решения соответствующего уравнения Эй-
Эйлера. Для выражения E8) уравнением Эйлера
является само уравнение Шредингера, переписанное
для х,
ЬЕ=Е-Е0; ( }
§ 14. Вариационный подход 51
его мы не можем решить. Однако можно выделить
большую часть полной энергии, т. е. Ё [уравнение
E8)], причем ей, если потребовать выполнения усло-
условия стационарности, будет соответствовать уравнение
Эйлера, которое зачастую можно решить.
Таким образом, вариационный подход, исполь-
использующий теорию возмущений, вкратце заключается
в следующем:
а) в минимизации большей доли Е (которая
имеет стационарное или же минимальное значение)
[уравнение E8)], причем эта минимизация прово-
проводится с целью получить пробную функцию %;
б) в подстановке % снова в полную энергию Е
для определения верхнего предела ?. Исходная доля
энергии (Е) дает основной энергетический эффект,
а остающаяся часть — оценку ошибки.
Этим способом, например, можно получить пол^-
ное разложение ряда теории возмущений Рэлея —
Шредингера [64]. Также можно получить обобщен-
обобщенные методы самосогласованного поля и другие много-
многочастичные теории [10, 11, 32] (см. § 15).
Для иллюстрации и дальнейших ссылок рассмо-
рассмотрим ниже получение полного разложения ряда тео-
теории возмущений Рэлея — Шредингера.
Большая стационарная часть Е [выражение E8)]
при учете выражения A6) запишется в виде
Это приведет к уравнению
(Н0-Е0)%1 = (Е1-Н1)ф0, F4)
соответствующему первому порядку теории Рэлея —
Шредингера, и к верхнему пределу [66, 67] для
энергии
F5)
1) Выражение в фигурных скобках в F3) варьируем по %. —
Прим. перев.
32 f If. Вариационный подход
где
Процесс можно продолжить дальше, подставив
в выражение E8)
F6)
это позволит расписать знаменатель в членах, вклю-
включающих лишь ф0 и хь и получить новый остаток, вы-
выраженный через Дь Большая часть нового остатка
является стационарной по отношению к дц и дает
уравнение, соответствующее уравнению Рэлея —
Шредингера для поправки второго порядка (для %2),
и ?4, и Е$. Этим способом [64] можно получить пол-
полное разложение ряда теории возмущений Рэлея —
Шредингера.
Можно добавить, что здесь обходятся хорошо из-
известные трудности, связанные с использованием ва-
вариационного метода для возбужденных состояний
т, если хГ ~ 5Ст> т. е. если первый порядок волно-
волновой функции для возбужденного состояния т слу-
служит достаточным приближением к точной волновой
функции этого состояния. Таким образом, здесь мы
все еще имеем дело с задачами на минимум [68],
которые оперируют лишь с известными «невозму-
«невозмущенными» волновыми функциями Ф% состояний бо-
более низких, нежели т.
Первое приближение многоэлектронной теории
атомов и молекул было получено путем решения [8]
уравнения F4), в котором в качестве нулевого при-
приближения использовались хартри-фоковские функ-
функции фо и применялась операторная техника. Развитие
теории Бракнера ядерной материи [62, 32] и других
многочастичных теорий [32] было связано во многих
случаях с использованием формализма теории воз-
возмущений.
Однако полная форма «многоэлектронной тео-
теории», которая является основным содержанием этой
работы, не есть теория возмущений. И многоэлек-
многоэлектронная теория, и теория бракнеровского типа вы-
. $ 15. Различный теории многих частиц 83
водятся теперь из точной функции % и Е при по-
помощи общего вариационного подхода, использующе-
использующего теорию возмущений. Этот подход, который можно
назвать из-за отсутствия лучшего термина «вариа-
«вариационным подходом, использующим теорию возмуще-
возмущений», не следует путать с теорией возмущений.
§ 15. РАЗЛИЧНЫЕ ТЕОРИИ МНОГИХ ЧАСТИЦ
В свете общего приближения, описанного выше,
можно рассмотреть, как соотносятся с точными % и Е
результаты различных теорий многих частиц. (Нами
не будут обсуждаться здесь общеизвестные неэмпи-
неэмпирические схемы, упомянутые в § 4.)
А. Обобщенные методы самосогласованного поля
а. Если оставить в выражении B0а) для функции
% лишь члены {/*} и единственную парную функцию
Oij (отвечающую, например, я-электронной паре
в С2Н4) и минимизировать выражение E8), то полу-
получим систему интегро-дифференциальных уравнений,
связывающих f* с йц. Уравнения эти могут в прин-
принципе решаться методом итераций до тех пор, пока
U и йц не станут самосогласованными [59]. В основ-
основном в этом заключается метод Бракнера, развитый
для случая учета только одной пары.
б. Мы получим метод Бракнера [32, 63, 58], если
удержим в функции г|), приравняв ее фо+%, все {Д},
{Uij} и соответствующие несвязанные комплексы [63].
Функция ? получаемая из выражения E8) для энер-
энергии, дает матричные элементы, содержащие более
чем один сорт 6^ (например, L?tj, V^ и т. д.), а так-
также члены с отдельными дц [см. выражения (82) и
A59)]. Если наряду с fi варьировать лишь эти по-
последние члены, то вновь в результате придем к си-
системе уравнений для самосогласованных f{ и 6ц.
54 § 15. Различные теории многих частиц
Первоначально теория Бракнера для бесконечной
ядерной материи строилась совершенно другим пу-
путем [32]. Орбитали выбирались в виде плоские волн.
Метод Хартри —- Фока нельзя взять в качестве ис-
исходного приближения для описания ядерной материи
по той причине, что матричные элементы, отвечаю-
отвечающие в нем отталкиванию нуклон-нуклонных остовов,
бесконечны. Так как ядерные взаимодействия очень
сильны, то нельзя просто принять, что частицы
в ядре движутся независимо в невозмущаемом
«фоне»1). В теории Бракнера эти бесконечности по
существу сокращаются с помощью добавления не-
некого «корреляционного потенциала» к потенциалу
этого «фона». Однако это было осуществлено более
кружным путем с применением бесконечного базис-
базисного набора (как в методе конфигурационного взаи-
взаимодействия) и усложнено двойной процедурой
самосогласованного поля. Этот метод можно при-
применить для бесконечных систем, для которых, на*
пример, орбитали уже известны (все те же плоские
волны) и суммирования типа суммирований в ме«
тоде конфигурационного взаимодействия можно за-
заменить интегрированиями в пространстве импуль-
импульсов. Однако для конечных систем, скажем для ко«
нечных ядер, такая теория оказывается неудобной
[70-72].
Выражения, получаемые из функции % и приводи-
приводимые здесь, не включают в себя бесконечных сумм.
Используемые в них /г- и й\$, взятые в замкнутой
форме, устраняют по меньшей мере некоторые из
трудностей и их можно применить для конечных
ядер [10], для которых все еще существенна проблема
отталкивания остовов. Для конечных систем весьма
элегантная теория бракнеровского типа или теория,
использующая «оператор реакции», развивалась так-
также Примасом [69], применившим алгебру Ли.
х) Подробно этот вопрос разобран, например, в монографии
К. Бракнера «Теория ядерной материи» (ИЛ, 1964) и моногра-
монографии Д. Киржница «Полевые методы теории многих частиц» (М?|
1963), гл. I, II.-— Прим. перев.
§ 15. Различные теории многих частиц 55
Величины Ji, учитывающие влияние корреляции
на орбитали, конечно, весьма существенны в ядрах.
Они сокращают бесконечности, Ьозникающие в этом
случае при использовании метода Хартри — Фока.
Двойные уравнения самосогласованного поля
упрощаются путем подстановки
* F7)
с помощью которой мы переходим к новым орбита-
лям (бракнеровским орбиталям), и путем замены
взаимодействий между «голыми» нуклонами g{j «кор-
«корреляционными потенциалами» t\$ (или же, на языке
метода конфигурационного взаимодействия, путем
перехода к «матрице реакции»):
1ЦЯ (V) = gu[* (IJ) + Ои]. F8)
Уравнения самосогласованного поля при этом
формально становятся подобными уравнениям Хар-
Хартри— Фока [71]. Заметим, однако, что наибольшая
трудность здесь связана с сильной зависимостью tih
а следовательно, и (VV)bi-. от того, на какие частицы
эти потенциалы действуют [9—11, 65], так как бц
в выражении F8) сильно меняется от пары к паре.
Имеются попытки применить метод Бракнера
к атомам и молекулам [1, 33]. Однако в атомах и мо-
молекулах, где потенциалы взаимодействия между па-
парами, gij^l/rij, хорошо ведут себя, а хартри-фоков-
ское приближение является с определенностью хоро-
хорошим исходным приближением, нет необходимости
использовать подобный метод со всеми присущими
ему трудностями [9—11, 65], достаточно оставить
лишь Ji. В § 19 будет показано, что % в атомах и
молекулах зачастую пренебрежимо малы [11].
Продолжим перечисление обобщенных методов
самосогласованного поля:
в. Метод Лёвдина [33] усложняет выражение
F8): в последнем появляются tijk, т. е., помимо {Д}
и {Oij}, удерживаются бщ и т. д. — многоэлектрон-
многоэлектронные корреляции в %.
56 § 15. Различные теории многих частиц
г. Метод Слэтера [73] сходен с методом Лёвдина;
в нем орбитали V [выражение F7)] самосогласуются
с полной волновой функцией фо+%, которая, однако,
не записывается в таком явном виде, как B0).
Таким образом, методы «в» и «г» с помощью
обобщенной процедуры самосогласованного поля учи-
учитывают многоэлектронные корреляции (и>2) и их
влияние на f{ и в этом смысле превосходят метод
Бракнера. В атомах и молекулах многоэлектронные
корреляции несущественны по сравнению с парными
корреляциями (см. § 18). Следовательно, влияние их
на fi будет еще менее существенным.
Таким образом, обобщенные методы самосогласо-
самосогласованного поля «а»—«г» дают возможность после пре-
преодоления больших теоретических и практических
трудностей получить {/*}, {&*#} и т. д., влияние кото-
которых не настолько существенно, чтобы начинать рас-
рассмотрение корреляционных эффектов именно с них.
Проще рассматривать эти {/г} и {&«#} и т. д. как
малые поправки к теории, основывающейся на учете
лишь u2j, применяя вариационное приближение с ис-
использованием теории возмущений.
Б. Теория возмущений
Выше мы упомянули, что можно получить урав-
уравнения Рэлея — Шредингера первого порядка — урав-
уравнения F4), если минимизировать ту часть Е [соотно-
[соотношение E8)], которая дается выражением F3). Урав-
Уравнение F4) было решено строго [8] путем применения
операторной техники, основанной на хартри-фоков-
ской функции фо, и дало
wl0^}' F9a)
<ЭД, k) = 0 (k= I, 2, ...,/,..., j, ..., TV). F9б)
Здесь эту технику изложить нельзя. Детально она
обсуждается в наших работах [8, 68, 75]. Функции
§ 15. 'Различные теории многих частиц 57
u{ij соответствуют корреляциям первого порядка для
всех хартри-фоковских орбитальных пар.' Они удо-
удовлетворяют уравнениям [8, 10]
{fit + ej) u{Pj + Qgij& (//) = 0, G0)
где Q — проекционный оператор [10], который opmo-
гонализуепг двухэлектронную функцию ко всем k,
если их брать справа от нее [отсюда и выражение
F9 6)]:
Q = 1 - У2 2 Я Щ {к} + 2 Я (*/)> {Я (*. 0, G1)
За исключением эффектов «запрета» и «среды» (Vi)
большего хартри-фоковского «фона», уравнение G0)
аналогично уравнению первого порядка, например
для атома гелия. Мы вернемся ниже к обсуждению
уравнения G0) в связи со свойствами симметрии %i
и к сопоставлению его решения с более «точными па-
парами» «многоэлектронной теории» [9—11].
Заметим, что выражение F9) не содержит {?•},
так как
W> = W> = 0 G2)
[см. соотношения D0) — D2)]. Это справедливо лишь
тогда, когда ф0 — хартри-фоковская волновая функ-
функция1). Если бы орбитали i в ф0 являлись водородо-
подобными орбиталями [А?/ = е// — соотношение
Dа)] или же слэтеровскими орбиталями, или же про-
простыми молекулярными орбиталями — линейной ком-
комбинацией атомных орбиталей — и т. д., то равенства
G2) не были бы более справедливы [68, 11]. Таким
образом, функции fif полученные с помощью D1),
не представляли бы непосредственно влияние «кор-
1) На языке метода конфигурационного взаимодействия это
означает, что отдельные возбуждения не смешиваются с хартри-
фоковской функцией Фо («теорема Бриллюэна»; см., например,
работу [1]).
58 § 15. Различные теории многих частиц
реляции» на орбитали; они в большей степени харак-
характеризовали бы эффекты «орбитальной средней поля-
поляризации» [3, 68], возникающие из-за остающихся
дальнодействующих частей межэлектронных отталки-
отталкиваний, которые полностью учитываются лишь в хар-
три-фоковской волновой функции ф0. Такие Д- в ос-
основном имеют тенденцию модифицировать грубые
орбитали и переводить их в хартри-фоковские орби-
орбитали (см. дополнение автора).
Теорию возмущений можно также применить в ее
обычной форме, т. е. в виде разложения по бесконеч-
бесконечному базисному набору, исходя из хартри-фоковской
волновой функции [18, 3]. Несбет [18] использует
энергию второго порядка ?2, чтобы оценить энерге-
энергетические вклады в методе конфигурационных взаи-
взаимодействий. Однако остается слабая сходимость
[1, 3] разложений метода конфигурационных взаимо-
взаимодействий. Расчеты для молекулы метана [48] с ис-
использованием в качестве базисного набора восьми
гауссовых орбиталей дают лишь 1,95 эв, или же,гру-
же,грубо говоря, 26% корреляционной энергии. Это отно-
относится к грубой хартри-фоковской энергии, которая
сама по себе содержит большие ошибки, однако вы-
вычисленная корреляционная энергия (бралось 20 кон-
конфигураций) не должна быть слишком чувствительна
к грубости этих молекулярных орбиталей </>0. Резуль-
Результаты показывают, что для получения корреляцион-
корреляционной энергии с большей точностью может оказаться
необходимым взять намного больше конфигураций1).
С другой стороны, наш вариант теории возмуще-
возмущений, даваемый выражениями F9) — G1), представлен
в замкнутой форме и не ограничивается бесконеч-
бесконечными разложениями метода конфигурационного вза-
{) Несбет [48] берет лишь эти 20 конфигураций (приблизи-
(приблизительно из нескольких тысяч возможных), каждая из которых
дает вклад, больший 0,0005 ат. ед. Однако учет остальных кон-
конфигураций может просто пояснить большую остающуюся кор-
корреляционную ошибку (около 5 эв). Даже если каждая конфигу-
конфигурация вкладывает около 10~4 ат. ед., то 103 конфигураций долж-
должны бы добавить ~3 эв. Эта слабая сходимость, является глав-
главной трудностью в методе конфигурационного взаимодействия.
§ 15. Различные теории многих частиц
имодействия (приложения к СН4 и т. д. см. в после-
последующих параграфах).
Результаты теории возмущений [8, 68] с самого
начала приводят нас систематическим путем к ва-
варианту «многоэлектронной теории» [9—11] более об-
общего и последовательного вида (см. ниже). Однако,
как уже было нами указано, законченная «много-
«многоэлектронная теория» никоим образом не нуждается
в использовании теории возмущений. Ее можно вы-
вывести непосредственно из соотношений B0). Действи-
Действительно, теорию возмущений можно получить из «мно-
«многоэлектронной теории» в результате последующих
приближений, и зачастую она дает удобное первое
приближение для расчетов [48].
Результаты теории возмущений [см. выражения
F9)] неполны в двух аспектах:
1) парные корреляции включены лишь до первого
порядка (в волновую функцию);
2) не рассмотрено, сколь допустимо пренебреже-
пренебрежение другими корреляционными эффектами Ju ^ijfe>
Oijki, ..., за исключением тех доводов, которые сле-
следуют из сходимости.
Сравнивая ряд теории возмущений
- G3)
с выражениями B0) и F9), мы видим, что все эф-
эффекты, отличные от тех, которые описываются функ-
функцией #(/у\ должны выявиться в /г и в более высоких
порядках. Однако %i определяет энергию до третьего
порядка [см. выражение F5)]. Следовательно, 7ь
Uijk и т. д. могут давать вклад в энергию лишь в чет-
четвертом и более высоких порядках.
Оценкой точности [9—11] разложения G3) яв-
является
&$Н<*. Х,)<С1. G4)
В таком случае из выражения F4) мы имеем
G5)
60 § 15. Различные теории многих частиц
Поскольку (хь Xi) составляет обычно около 1%,
если исходить из хартри-фоковской волновой функ-
функции, разложение Рэлея —Шредингера, следователь-
следовательно, сходится быстро и для приложений может ока-
оказаться достаточным взять лишь xi.
Поправки более высокого порядка к парным кор-
корреляциям ufj и т. д. также появляются в Хг, Хзит-Д-
Однако это не означает с необходимостью, что они
дадут вклад того же порядка, что и fu 0цк и т. д.
Все эти члены могут быть формально того же «по-
«порядка» [в выражении G3)], однако величины их мо-
могут все еще различаться, сильно завися от геометрии
молекулы, природы физического эффекта и других
обстоятельств. Следовательно, общие аргументы
о сходимости недостаточны, чтобы установить отно-
относительное значение физически различных корреля-
корреляционных эффектов.
Эти дефекты результата [8], полученного с по-
помощью теории возмущений устраняются в «много-
«многоэлектронной теории» [9—11].
В. «Многоэлектронная теория» [9—11]
Обобщенные методы самосогласованного поля [33,
45, 73] так же, как и другие неэмпирические методы,
подобные методу конфигурационного взаимодействия,
обсужденному в § 4, не дают возможность различить
в х и ? вклад главных эффектов от второстепенных.
До сих пор наибольшие вклады в х [выражение
B0а)] давались парными корреляциями {Vid). По
этой причине они должны быть получаны настолько
точно, насколько это возможно. Остающиеся вклады
должны быть лишь оценены, если они, конечно, малы.
В ближайших параграфах мы получим вклады от
парных корреляций [10] «точно» (т. е. во всех «по-
«порядках»), исходя из главных частей точной функции
X и энергии, в соответствии с «вариационным прибли-
приближением, использующим теорию возмущений», а за-
затем рассмотрим и оценим {/*} [И] и многоэлектрон-
многоэлектронные эффекты [9] (§ 18 и 19). И в идейном, и в прак-
§ 16. Волновая функция € многоэлектронной теории» 61
тическом аспекте это приведет к весьма простым ре-
результатам.
Для удобства мы будем продолжать изложение
на примере систем с замкнутыми оболочками. Дру-
Другие системы обсуждаются в § 20 и 21. Эта теория
все еще основывается на нелокализованных хартри-
фоковских орбиталях и молекулярных орбиталях са-
самосогласованного поля, которые удовлетворяют урав-
уравнениям A2). Результаты преобразуются к локализо-
локализованным орбиталям в § 27 и 28.
§ 16. ВОЛНОВАЯ ФУНКЦИЯ
сМНОГОЭЛЕКТРОННОИ ТЕОРИИ»
Главной частью точной волновой функции % [вы-
[выражения B0), E0) и т. д.] является волновая функ-
функция
| M Ш Gб)
Входящие в это выражение функции й\$ будут по-
получены (см. § 22) минимизацией большей части Е
в соответствии с вариационным приближением, ис-
использующим теорию возмущений. Они позволят нам
также оценить поправочные члены {/4 и т. д. Выра-
Выражение G6) имеет тот же самый вид, что и выраже-
выражение для %ь найденное на основании теории возмуще-
возмущений, однако в нем парные корреляции учитываются
во всех порядках. Соответствующие парные функции
обозначаются строчными буквами й^, чтобы отли-
отличить их от парных функций ?7^-, входящих в точную
волновую функцию [выражения B0)]. Последние за-
зависят неявно от fu Uijh и т. д., в то время как йц
из соотношения G6) не зависят от них.
Если ufj известны, то можно получить лучшую,
нежели %s, волновую функцию, включающую все от-
относящиеся к парным функциям несвязанные комплек-
комплексы. Это обстоятельство существенно, поскольку при
62 § 17. Вариационная энергия €многоэлектранной теории»
использовании только %8, в Е [уравнение E8)] воз*
никают нормировочные члены:
которые сравнимы по величине с эффектом несвязан-
несвязанных комплексов. Как обсуждалось выше, эти два эф-
эффекта имеют тенденцию к взаимному сокращению.
Полная волновая функция многоэлектроннбй тео-
теории [9] для замкнутых оболочек имеет вид
A2 3... *
L
N N ~ ~
+B 0" 2
N N N ~ - -
\1 Ul]Uklttmn
V
U (ijklmn)
t> j k>l m> n
(/ ]ф фк1)
+ •••
G7)
Штрих над %s означает, что в волновую функцию
включены несвязанные комплексы. Отметим, что
в произведениях йц нет общих индексов (т. е. мы
имеем лишь ^йм, ^13^24 и т. д.).
§ 17. ВАРИАЦИОННАЯ ЭНЕРГИЯ
сМНОГОЭЛЕКТРОННОЙ ТЕОРИИ»
Применив диаграммную технику, рассмотренную
в § 9 и 13, легко найти те члены в энергии, которые
возникают из-за выписанных выше членов волновой
функции. Возьмем, например, функцию х* и подста-
подставим ее в выражение E8) для Е. Далее, для каждого
члена используем то обстоятельство, что Л комму-
коммутирует о
Лг N
t и 2
§17. Вариационная энергия «многоэлектронной теории» 63
а также соотношение B2). Сначала нарисуем диа-
диаграмму для каждого члена «без обмена», т. е. не при-
принимая во внимание операцию <Л, затем применим Л
к каждой диаграмме и получим соответствующую об-
обменную часть. Энергия Е была оценена и в случае
волновой функции %s> и в случае х^- Читатель, же-
желающий ознакомиться с диаграммами, которые воз-
возникают при этом, отсылается к работе автора [0].
Функция Xs [уравнение G6)] дает
N
?<?, = Ян.р. + -^ \-§-,. G8а)
ги ss 1 {& (ij), тиии) + («у, (*, + «у + /%) и*у), G86)
Свойства хартри-фоковской функции ^0 приводят
к тому, что многие диаграммы становятся равными
нулю [9]. В случае волновой функции %s остаток R
будет содержать трехэлектронные связанные ком-
комплексы
n
j/ <79>
один из которых при этом частном выборе индексов
приводит, например, для члена „без обмена44 к сле-
следующему выражению:
{iukJy mjtUikj). (80)
Левин, Геллер и Тейлор1) выявили ненулевой
обменный член в одной из диаграмм (выражение
l) Levine H. В., Geller M., Taylor H. S., частное со-
сообщение.
64 § 17. Вариационная энергия «многоэлектронной теории»
(Зба) в работе [9]):
\ °" ° /обм
Этот член обсуждается в следующем параграфе.
Если учитываются несвязанные комплексы, то
возникает еще больше диаграмм [9]. Полная вариа-
вариационная энергия многоэлектронной теории полу-
получается после подстановки выражения G7) для %'s
в уравнение E8) для Ё:
N
V + TF' <82a)
(826)
2 {utJ, и|;) + S S
/ > У i > j k > I
(83)
^=1+ S (««.««>+•••• (84)
* > /
Здесь Dij содержит все пары, за исключением пары
(ij). Это происходит [63, 9] по той причине, что не-
несвязанные комплексы в выражении G7) дают мно-
множители (им, uki), которые умножаются на e*j [урав-
[уравнение G86)].
Многоэлектронные диаграммы, которые сумми-
суммируются в R' в дополнение к тем, которые сумми-
суммируются в R [выражение G9)], приведены в работе [9].
Нет необходимости приводить их здесь.
Снова заметим, что, хотя i's и содержит лишь
парные корреляции, в Ё появляются также трех-,
четырех-, и т. д. электронные эффекты. Их изучение
привело к выводу, что они не имеют большого зна-
значения (см. § 18).
§ 18. Многоэлектронные корреляции 65
Согласно вариационному приближению, исполь-
использующему теорию возмущений, нет необходимости ми-
минимизировать всю энергию Е [уравнение (82)], чтобы
получить йц. Главной частью этой энергии является
ЕъЕн.?.+ 2 г' (85)
где
(/у), тииф + («//, (et + е} + тф иф
) ' (86)
Выражение (86) для е^ получено из (826) после
разложения в последнем знаменателя [10]. Например,
для бериллия имеем
l2. «Х2> + (^34» «34> + («13,
1
1 + («34. «34> '
Выражение (85) является обычно весьма хорошим
«химическим» приближением к Е (т. е. с точностью
приблизительно до 1 ккал/моль). Оно будет исполь-
использовано позже как основа для полуэмпирических при-
приближений. Для неэмпирических расчетов необходимо
минимизировать часть (82) или E8), чтобы полу-
получить uij. Если мы определим u*j, то, подставив об-
обратно в R' и ?>', получим верхнюю границу точной
функции Е и сможем оценить ошибку, которая воз-
возникает при пренебрежении этими малыми членами,
В следующих двух параграфах рассматриваются
приближения, делаемые при получении %'s и E"s
[выражения G7) и (85)] из точных % и Е, определяе-
определяемых выражениями B0), D9) —E2) и E8).
§ 18. МНОГОЭЛЕКТРОННЫЕ КОРРЕЛЯЦИИ
Многоэлектронные корреляции — связанные ком-
комплексы — проявляются
а) в волновой функции % в качестве С^ь* ^i;W
и т. д.;
5 О. Синаноглу
§ IS. Многомектронныё корреляции
б) в энергии (82), возникая даже из одних толь-
только uij. Оба эти эффекта должны быть рассмотрены.
Природа электронной корреляции, которая выше
определилась как то, что получается сверх Хартри —
Фока, обусловливается следующими факторами [9]:
1. Пространственным распределением электронов
относительно друг друга в хартри-фоковском «фоне»,
задаваемом функцией </>0.
2. Парными флуктуационными потенциалами
2 Щ] [см. Н—#0 и выражение A7)], которые от-
ответственны за «столкновения» электронов.
3. Принципом исключения, выражающимся в том,
что слагаемые функции % подчиняются следующему
условию:
{ии, k) = 0 для k + Uj из ф0. (87)
!А. Многоэлектронные эффекты в волновой функции
Первый из перечисленных выше факторов был
нами обсужден в § 7. Относительное распределение
Электронов, задаваемое самой ф0, зависит лишь
от геометрии молекул и от *разности энергий элек-
электронов с одинаковым спином, т. е. от принципа
исключения. Потенциал хартри-фоковского «фона»
V{ обусловливает большую «рыхлость» хартри-фо-
ковских орбиталей по сравнению, например, с водо-
родоподобными орбиталями, однако влияние его на
«соударения» является косвенным и слабым (см.
приводимую ниже зависимость йц от всей «среды»).
Рассмотрим любые три электрона в атоме или мо-
молекуле. По крайней мере два из них могут иметь
одинаковые спины. Может оказаться
а> (t Y t Y t)
§ 18. Многоалектронные корреляции
или же, скажем»
б) A
В случае «а» все электроны располагаются далеко
друг от друга из-за принципа исключения, уже уч-
учтенного в функции #о. Из-за такого рода «соударе-
«соударений» трех электронов с одинаковыми спинами в функ-
функции х возникают несущественные корреляционные
3 4 5 5 7
Число электронов (Z-7)
Фиг. 1. Корреляционные энергии ионов и атома азота
(на основе эмпирических данных Клементи [79]).
члены Uijh. Даже полная корреляционная энергия
2р3 электронов в состоянии 4S атома азота соста*
вляет [79] приблизительно 0,1 эв (фиг. 1), причем
наибольший вклад в нее, 2е</> обусловлен пар-
парными эффектами.
В случае «б» и во всех других случаях с еще
большим числом электронов разрешается только
двум электронам (самое большее!) приближаться
друг к другу в «фоне», обусловленном ^о. Из-за этих
двух электронов, расположенных вблизи друг друга,
в функции х возникает член с йц.
Однако влияния одного лишь принципа исключе-
исключения недостаточно, чтобы обеспечить малость трех-
68 § 18. Многоэлектронные корреляции
или более электронных кулоновских корреляций.
Рассмотрим, например, как далеко простирается дей-
действие флуктуационных потенциалов в случае «б». Бу-
Будет ли потенциал т2з воздействовать, скажем, на
электрон 3? Влияние самого потенциала взаимодей-
взаимодействия 1/ггз из-за его большого радиуса может ска-
сказаться на электроне 3, однако потенциал т2з может
и не оказать воздействия. Обычно флуктуационные
потенциалы обращаются в нуль на расстоянии, мень-
меньшем расстояния до третьего, четвертого и г. д. элек-
электронов [9]. Чем больше локализация электронов, обу-
обусловленная ^о, тем короче оказывается радиус потен-
потенциала Шц, действующего между локализованными по
отдельности электронами.
Это было показано в работе автора [9] для элек-
электронов, расположенных в двух различных концентри-
концентрических оболочках (см. фиг. 1 работы [9]), и электро-
электронов, расположенных в той же самой оболочке, причем
соответствующие им орбитали характеризовались
различной угловой зависимостью (см. фиг. 2 и 3 ра-
работы [9]).
Например, в атоме Be потенциал т^ для ls-элек-
тронов обращается в нуль на расстояниях, меньших
хартри-фоковского радиуса 25-электронов. Это обу-
обусловливает малость межоболочечных (Is2s) парных
корреляций по сравнению с (Is2)-корреляциями и еще
большую малость трех- или же четырехчастичных
корреляций (связанных корреляций), т. е. (Is2)...
ш.. Bs)- или (Is2) .. .Bs2)-корреляций.
Потенциал тц обращается в нуль даже между
электронами, находящимися в состояниях с одним и
тем же главным квантовым числом и различными
другими квантовыми числами. Флуктуационный по-
потенциал между 2/v и 25-электронами в атоме бора
мал как раз в той области, где велика Bs2)-корре-
Bs2)-корреляция [между плоскостями, составляющими ±45°
с плоскостью (ху)]. Читатель, желающий ознако-
ознакомиться-с дальнейшими деталями, касающимися атома
бора, может посмотреть работу [9]. Поведение по-
потенциалов niij, действующих между р-электронами,
скажем, в атомах С, N или Ne или же d-электронами
§ 18. Многоэлектронные корреляции
в атоме Аг и т. д., должно быть качественно сходно
с тем, что мы имеем в случае бора. Оно обусловли-
обусловливает меньшее значение n-электронных корреляций
(п>2) даже в больших оболочках типа 2р6 и 3d10.
Некоторые результаты, полученные методом Хар-
три — Фока и дополненные результатами метода
конфигурационного взаимодействия, естественно, под-
подтвердили эту модель. В атоме бериллия [77] и моле*
куле гидрида лития [78] трехэлектронные корреляции
(тройные возбужденные конфигурации) оказались
целиком несущественными. Позже было показано [9],
что кажущуюся важность четырехэлектронных корре-
Таблица 1
Вклады несвязанных комплексов — четырехэлектронные
корреляции в атоме бериллия
Квадрупольно
возбужденная
конфигурация [771
pi(ls)s](ls)
AQWiQs)
/>MlsKCs)
Вклад в энергию
Коэффициент несвя-
несвязанного четырех-
частичного комплекса,
взятый из полного
разложения (по 37
конфигурациям)
волновой функции
по методу конфигу-
конфигурационного взаимо-
взаимодействия [77]
0,007063
0,005651
0,001585
0,0004639
—0,075 эв3)
Коэффициент несвя-
несвязанного четырех-
частичного комплекса,
подсчитанный с по-
помощью коэффициентов
парных корреля-
корреляций 0,2)
0,0073
0,00647
0,00168
0,000478
—0,074 эв 4)
0 См. формулу (88) настоящей работы, а также работу [9J.
*) Различие между коэффициентами, приведенными во втором и третьем
столбцах, обусловлено четырехэлектронными корреляциями G,2з4. Оно весьма
мало.
ь) Получен из полного разложения по методу конфигурационного взаимо-
взаимодействия [77].
4) Рассчитан по формуле е/у A — DiA/D [ср. выражения G8) и (82)].
Согласие между приведенными значениями вклада в энергию указывает на то,
что корреляционные члены, возникающие в R' из-за четырехэлектронных кор-
корреляций, пренебрежимо малы.
§ 18. Многоэлектронные корреляции
ляций обусловливают несвязанные комплексы. Коэф«
фициенты^х можно получить почти точно, если пере-
перемножить! согласно выражению E5), коэффициенты
независимых парных корреляций. Оказывается, что
О %
2 3 4
(#12^34 + «13^24 + ^14%з} •
(88)
Результаты по Be [9] воспроизведены в табл. 1. Ана-
Аналогичные результаты были найдены для молекулы
ЦН[9] (табл.2).
Таблица 2
Вклады несвязанных комплекеов — четырехэлектронные
корреляции в молекуле Liti
Квадрупольно
возбужденная
конфигурация !)
[3377]
[3357]
13355]
[3456] «)
[3457] •)
Коэффициент несвязанного
четырехчастичного
комплекса, взятый из
полного разложения
волновой функции по
методу конфигурацион-
конфигурационного взаимодействия
[1151
0,00113
—0,00086
0,00062
J -0,00023
\ 0,00006
J 0,00014
\ 0,00007
Коэффициент несвязан- .
ного четырехчастичного
комплекса, подсчитанный
с помощью коэффициен-
коэффициентов парных корреляций 2)
0,00113
—0,00078
0,00052
/-0
~0
Полные трехэлектронные корреляционные энергии меньше
0,01 эв
') Конфигурации даны в обозначениях Иббинга [115].
2) См. формулу (88) настоящей работы, а также работу [9J.
•) Эти конфигурации приводят к двум независимым слэтеровским опре-
определителям.
Решающей неэмпирической проверкой теории дол-
должны явиться расчеты на примере 2/?6-оболочки неона,
которые в настоящее время находятся в стадии вы-
выполнения.
I SB. Многоэлектронные корреляции 71
«Эффекты запрета», выражающиеся для функ-
функции х соотношением (87), также уменьшают вклад
rt-частичных связанных комплексов. Можно ожидать,
что основной причиной малости миогоэлектронных
членов, например, в я-оболочке бензола является
слабая локализация электрона. Выражение (87) пред-»
ставляет собой математическую запись трехэлектрон-
ной «ферми-корреляции» между а-, /- и fc-состояния-
ми электронов. Коррелированные электроны, находя-
находящиеся в i- и /-состояниях (спин-орбиталях), как бы
предохраняются от возможных виртуальных перехо-
переходов в уже занятое А-состояние. Чем больший вклад
дает такое й-состояние в разложении йц по методу
конфигурационного взаимодействия, тем существен-
существеннее этот эффект» Так как электроны становятся де-
локализованными, почти вырожденными, то по этой
причине значение некоторых fe-состояний возрастает.
Это также приводит к тому, что отвечающий им «эф-
«эффект запрета» [3, 8—11] становится более суще-
существенным.
Например, корреляция Aа^J-состояния Н2 дает
вклад в энергию, равный 7,74 эв, вблизи состояния
диссоциации, когда молекулярные орбитали как та-
таковые являются делокализованными и должны быть
поправлены на возможность сильного смешивания
с A<7иJ-состоянием [1]. С другой стороны, корреля-
корреляционная энергия Aа^J-состояния Не+ или (Не—Не)
равна примерно 0,5—1 эв, потому что в этом случае
(la,/)-состояние занято и, таким образом, является
запрещенным. Следовательно, функция (lcruJ уже не
есть главная часть ui2. Этот тип трех- или четырех-
электронных ферми-корреляций является более суще-
существенным, нежели соответствующие кулоновские кор-
корреляции, т. е. появляется ли в функции % член &ия
или же &1234, не столь важно.
Однако подобные аргументы, приводящие к воз-
возрастанию делокализации во все больших и больших
молекулах, не могут использоваться без каких-либо
ограничений, поскольку в итоге мы приходим к ме-
металлам и высокосопряженным молекулам красителей,
72 § 18. Многоэлектронные корреляции
в которых возникают плазменные эффекты и эффекты
коллективной экранировки [32]. В бесконечном элек-
электронном газе [32] (с положительно заряженным
однородным фоном) отсутствие геометрического фак-
фактора компенсирует «эффект запрета». Хартри-фоков-
ские орбитали переходят в плоские волны е/к#г, и
вероятность обнаружить электрон где-либо в выде-
выделенном объеме становится константой. Следова-
Следовательно,
1
когда характеристические размеры объема устрем-
устремляются [9] к бесконечности: L-*oo. Каждый из мно-
многих электронов может теперь «чувствовать» влияние
всех других в один и тот же момент времени и харт-
ри-фоковская функция ф0 становится неадекватным
исходным приближением.
Б. Многоэлектронные эффекты в энергии
Свойства функций </>0, т^ и выражение (87) при-
приводят к тому, что не только члены ?/ijfc...n в волновой
функции %, но также и многоэлектронные члены R'
в энергии [соотношение (82)] становятся малыми.
Рассмотрим, например, треугольную диаграмму [вы-
[выражения G9) и (80)].
Данная функция йц велика лишь в тех точках
пространства, где также велико rtii$&(ij) [см. выра-
выражения (86) и G0); кроме того, следует учесть [10],
что Qgij&(ij)=QmiJ&(ij)]. В треугольной диа-
диаграмме, подобной (80), встречается произведение
корреляционных функций, у которых отличается
один из индексов, а именно произведение ukj и uik.
Однако, как следует из (87) (см., например, случай
бора [9]) и природы тц, когда функция uhj велика,
функция uih мала, и наоборот, что и приводит во
всех случаях к малости произведения, а следова-
следовательно, R.
Эти трехэлектронные вклады были оценены [9] для
Be [77] и LiH [78] на основании результатов, получен-
полученных с помощью метода конфигурационного взаимо-
§ 18. Многоэлектронные корреляции 73
действия. Оказалось [9], что для LiH они составляют
менее 0,01 эв. Для Be было найдено, что единствен-
единственное влияние [9] несвязанных комплексов на энергию
происходит от Dij/D [частичное сокращение норми-
нормировки согласно G8) и (82)]; это означает, что оста-
остаток R' также пренебрежимо мал (см. табл. 1 и 2).
Левин, Геллер и Тейлор нашли наибольший член
из новых ненулевых многоэлектронных членов [см.
(81)]. Для Be он будет иметь вид
(«к. Я34>A4, g1232), (89)
где последний матричный элемент равен A525,
gi225l5). Такие члены очень легко поддаются обсчету.
В матричном элементе {ui2> U34) остаются лишь те
конфигурации (используется метод конфигурацион-
конфигурационного взаимодействия), которые дают вклад и в й12,
и в й34. Перемножив коэффициенты таких конфигу-
конфигураций, взятые из разложения (по 37 конфигурациям)
функции Ватсона [77] по методу конфигурационного
взаимодействия, автор и Туан получили
(«и. %i>-5,3. 10.
С учетом обменного интеграла, найденного из lS- и
35-энергий Ве2+ и равного 0,056 ат. ед., подсчет вы-
выражения (89) дает приблизительно 0,00081 эв, т. е.
полностью пренебрежимую величину.
Природа функции ф0 и принцип исключения, как
мы показали, приводят к тому, что и члены Оцъ„,п
в волновой функции х» и многоэлектронные члены
R' в энергии [выражение (82)], возникающие из uih
становятся малыми. Численные результаты, которыми
мы пока располагаем, подтверждают это и, следова-
следовательно, возможность использования уравнений G7)
и (85) в многоэлектронной теории атомов. Способы
расчета R' для дальнейшего контроля были несколь-
несколько более детально описаны в работах [9, 10]. Было
бы интересно рассмотреть величины этих членов,
в особенности для больших jt-электронных систем.
Основные вклады в энергию от многоэлектронных
эффектов входят в R [уравнения G8а) и G9)]. Члены
Uijk> Uxjhu • • • в волновой функции % должны давать
74
§ IS. Мнагозлектронные корреляций
«w -
Ф
Число электронов (ZSIQ)
и г. 2. Корреляционные энергии ионов и атома неона
(на основе эмпирических данных Клементи [79]).
в энергию меньший вклад; согласно теории возмуще-
возмущений, они дают поправки более высокого порядка. По-
видимому, достаточно, таким образом, оценить
лишь R.
Для неэмпирических расчетов члены R' не обяза*
тельно должны быть полностью пренебрежимыми ве-
величинами. Достаточно, чтобы они были меньше, чем
[ММ
Тогда применима процедура вариационного при-
приближения с использованием теории возмущений.
Большая часть 2 е/у дает "функции йц, которые
в свою очередь определяют /?'.
Однако зачастую можно пренебречь членами /?'¦
Тогда выражение (85) показывает, что имеют место
аддитивные несвязанные парные корреляции. Это су-
существенно для полуэмпирических теорий спектров
(см. § 24 и 26).
§ 19. Влияние корреляции ни орбитали, /< 78
Выражение (85) было подтверждено совсем не*
давно [79] для первого ряда атомов. (Гладней и Ал-
лен [80] позднее получили аналогичные эмпирические
значения.) Клементи [79] вычислил корреляцион-
корреляционные энергии этих атомов и их ионов, используя
полученные им же результаты метода Хартри —
Фока и оценки релятивистских эффектов. Наблюдае-
Наблюдаемая аддитивность корреляций находится в пределах
эмпирической неопределенности данных. Фиг. 1 и 2,
построенные на основании данных Клементи, показы-
показывают, что корреляционная энергия ионов и атомов
азота и неона возрастает по мере того, как туда до-
добавляется все большее и большее число электронов.
§ 19. ВЛИЯНИЕ КОРРЕЛЯЦИИ НА ОРБИТАЛИ, /,
Мы уже упоминали, что в замкнутых оболочках
многоэлектронные корреляции несущественны и что
функции uij имеют резко локальный, короткодей-
короткодействующий характер: они становятся большими лишь
тогда, когда два электрона приближаются друг
к другу. Это означает, что, несмотря на ощутимые
сдвиги в энергии, обусловленные корреляцией, об-
общая электронная плотность не будет сильно ме-
меняться.
Являются ли хартри-фоковские орбитали наилуч-
наилучшими при расчетах или же следует переходить к об-
обобщенному методу самосогласованного поля, зависит
от того, насколько малы fi. Качественные аспекты
квантовой химии основываются на описании с по-
помощью орбиталей, ид таким образом, вопрос, на-
насколько необходимы fu имеет особую важность.
Физический смысл функции fi был рассмотрен
весьма детально, и величина fi оценивалась для не-
некоторых типичных случаев [11].
Корреляционные эффекты можно разделить на
«динамические» и «нединамические» [11, 81]. Дина-
Динамические корреляции возникают при наличии «плот-
«плотных пар» электронов, как, например, в Не или B/72J-
76 § 19. Влияние корреляции на орбитали, Jt
пар в Ne и т. д. В этом случае нет ни одной конфигу-
конфигурации в волновой функции %, которая бы сильно
смешивалась с Фо, и разложения по методу конфигу-
конфигурационного взаимодействия сходятся слабо. Недина-
Нединамические корреляции [81], с другой стороны, возни-
возникают при наличии вырожденных или почти выро-
вырожденных состояний (первый порядок метода конфи-
конфигурационного взаимодействия) [6]. В этом параграфе
мы сконцентрируем наше внимание на Ju возникаю-
возникающих из-за наличия динамических корреляций. Вопрос
о fu обусловленных вырождением, подобным выро-
вырождению в системах с незамкнутыми оболочками, или
же почти вырождением, обсуждается в § 20 и 21.
Можно ожидать, что «динамические» ji будут ма-
малыми [11]. Помимо аргументов теории возмущений,
приведенных в § 15, п. Б, существуют и другие при-
причины для ожиданий такого рода:
1) плотности электронов, наблюдаемые при рас-
рассеянии рентгеновских лучей [82], близки к плотно-
плотностям, получаемым при расчетах, в которых исполь-
используются хартри-фоковские орбитали;
2) расчеты по методу конфигурационного взаимо-
взаимодействия, например расчет Ватсона [77] для Be, по-
показывают, что эффекты, обусловленные отдельными
возбуждениями, пренебрежимо малы [см. выраже-
выражение D5)];
3) дальнодействующие кулоновские отталкивания
дают главный вклад в ф0, а «редко встречающиеся»
эффекты локальных «соударений» йц [см., например,
D1)] во всей области, где соответствующая орбиталь
отлична от нуля, дают вклад в fi\
4) прямые расчеты [11] в замкнутой форме так-
также дают малые f*.
Точные ft определяются выражением B8). Следо-
Следовательно, в принципе все части волновой функции %,
соответствующие двойным корреляциям и корреля-
корреляциям с /г>2, дают вклады в /*. Однако многоэлек-
многоэлектронные члены (я>2) сами по себе слишком малы,
§ 19. Влияние корреляции на орбитали, ft 77
и, следовательно, влияние их на f< будет еще мень-
меньшим.
Выражение B8) показывает, как рассчитать f*.
Предположим, мы имеем хорошую пробную функ-
функцию г|), в которой не выделены ни части от корреля-
корреляционных эффектов, ни даже хартри-фоковская часть.
Такую функцию \j) можно рассмотреть «методом по-
последовательных частичных ортогонализаций» в соот-
соответствии с B6) — C6). После того как из нее будет
выделена часть х> из выражения B8) можно будет
получить fa.
Для атома Не имеют место выражения C9) и
D1). По аналогии, для многоэлектронной системы
выражение B8) можно записать в виде
2,у, />v (90)
где (iiij, k)=0 для k=j=i, /, а я-электронными корреля-
корреляциями (я>2) пренебрегают. Таким образом, необхо-
необходимо просто рассмотреть различные типы пар в мно-
многоэлектронной системе, получить, как это показано
в § 22, соответствующие им й^у отделяя части S3 (ij)
и учитывая влияние эффектов запрета. Вклад, соот-
соответствующий fu определяется по формуле (90).
Для атома Не величины fi были вычислены [11]
с помощью функции г|), содержащей координату г12.
Предположим теперь, что пробная функция дает
непренебрежимые /*. Означает ли это, что в точной
волновой функции х величины fi не малы, или же
означает, что пробная функция была плохо выбрана
и привела к нереальным значениям /*? Если имеет
место последний случай, то необходимо взять луч-
лучшую пробную функцию, т. е. функцию, которой со-
соответствует меньшая энергия за счет исключения
вкладов Ji.
Лёвдин и Редай [87] применяли для расчетов
«взвешенную» [1] волновую функцию Не
(91)
§ 49. Влияние корреляции на орбитали,и
где а — две экспоненциальные аналитические орби-
тали самосогласованного поля, г\ — «масштабный»
[1] параметр, а а — вариационный параметр. Она
2,0
3,0
r{ t am. t
Фиг. 3. Функции // в атоме Не (по работе [11]).
д являются теми модификациями, которые вносят корреляции в хартри-
фоковскую орбиталь 15. Новая орбиталь должна была бы браться в виде
A+Л)/г 1-Н/рЛ>» однако можно пренебречь %. Таким образом, уже метод
хартри-фоковского самосогласованного поля дает нам хорошие орбитали.
Кривая / — функция fi, полученная из шестичленной волновой функции Хил-
дерааса (см. § 19). Кривая 2—функция ?и рассчитанная по взвешенной функ-
функции Лбвдина и Редая в соответствии с выражением (91).
давала 80% корреляционной энергии. Полученные с
ее помощью /г-части [11] отложены на фиг. 3; они
имеют наибольшее значение у ядер (около 5% от
хартри-фоковских орбиталей, отвечающих этим поло-
положениям). Если эти /i-части исключены, так что г|э
§ 19. Влияние корреляции м орбшгали, f< 79
в соответствии с D0) и D9) представляется в виде
*A2) + *1* (92)
то энергия понижается на 0,019 эв, или же на 1,7%
от ?СОГг,
Дальнейшее подтверждение малости U в точной
волновой функции х гелия получается при выборе
лучшей пробной функции. Шестичленная волновая
функция Хиллерааса [88] (дающая 98% корреляцион-
корреляционной энергии) приводит [11] к намного меньшим зна-
значениям fi (см. фиг. 3). Эти ]и видимо, отвечают
действительности, ибо если их опустить, энергия повы-
повышается на 0,24% от ?Согг, что, однако, является пре-
пренебрежимо малой величиной F3 кал/моль). Анало-,
гичные результаты были найдены в работах [91, 92,
11] для молекул Н2, рассматриваемых вблизи поло-
положения равновесия ге. (Рассмотрение Н2 вблизи со-
состояния диссоциации проводится в § 21 [11].)
Динамические корреляции, связанные с другими
парами, например с внутренними оболочками моле-
молекул, BргJ-парами в Ne, и т. д., аналогично должны
приводить к пренебрежимо малым значениям ft. Па-
Парам из внешних оболочек [3, 4], например (Is2s),
соответствуют малые йц, которые очень хорошо по-
получаются по теории возмущений [uffj в F9а)] и,
следовательно, не содержат J{. Несколько других слу-
случаев обсуждалось в нашей работе [11]. .
Несущественность /* (см. также § 20 и 21) и чле-
членов, обусловленных многоэлектронными корреляция-
корреляциями, означает:
1. Хартри-фоковские орбитали, например молеку-
молекулярные орбитали самосогласованного поля, позво-
позволяют удовлетворительно описать большинство задач
квантовой химии [54]. Корреляции не меняют каче-
качественных картин, основанных на функциях ^<о (подоб-
(подобных рассматривавшимся Леннард-Джонсом, Поплом
и Линнетом; см. также примечания в § 7) [42—44].
2. Согласно (85), из полного вклада корреляцион-
корреляционных эффектов можно выделить отдельные вклады,
80 § 20. Незамкнутые оболочки
связанные с разными оболочками. Это сделать было
бы нельзя, если бы /г- содержали вклад от дально-
действующих кулоновских эффектов !).
3. Теория весьма сильно упрощается благодаря
исчезновению не только fu но также и отвечающих
им несвязанных комплексов [соотношения D9) —
E5)]. Это приводит к упрощениям, которые дают
возможность не рассматривать обобщенные теории
самосогласованного поля бракнеровского типа. Мож-
Можно по-прежнему оценивать 7ь однако, будучи малыми,
они не должны браться самосогласованными с и,ц.
§ 20. НЕЗАМКНУТЫЕ ОБОЛОЧКИ
В предыдущих параграфах была сформулирована
многоэлектронная теория, а также выписаны выра-
выражения для точных % и Е для систем с замкнутыми
оболочками. Главным образом для них мы рассмо-
рассмотрели роль многоэлектронных эффектов и величин^.
Это было сделано, чтобы избежать чрезмерного обоб-
обобщения и усложнения обозначений. Теория для слу-
случая незамкнутых оболочек требует некоторых моди-
модификаций.
Хартри-фоковская часть волновой функции г|> обсу-
обсуждалась в § 6. Форма точной волновой функции для
системы с незамкнутой оболочкой точно такая же,
как и для системы с замкнутой оболочкой [выраже-
[выражение B0а)]. В качестве функции ф0 — полной хартри-
фоковской части, — вообще говоря, берется линей-
линейная комбинация различных слэтеровских определи-
определителей ф{:
*о *= 2 *,. (93)
/ 1
1) В работе Саса [31] U не выделялись, а рассматривались
вместе с многоэлектронными эффектами на том же основании,
как и iiij. Это, однако, не привело к картине оболочек и не
дало вычислительных и теоретических преимуществ.
§ 20. Незамкнутые оболочки 81
Полное разложение по методу конфигурационного
взаимодействия точной функции г|) содержит все
«упорядоченные», однозначные слэтеровские опреде-
определители, которые можно построить из полного одно-
электронного базисного набора. В случае замкнутых
оболочек, а также для состояний с высокой мульти-
плетностью в случае незамкнутой оболочки един-
единственный определитель является симметричной соб-
собственной функцией (например, в атомах — собствен-
собственной функцией как операторов L2, S2, Lz, Sz, так и Яо).
В этом случае отсутствует вырождение, и этот един-
единственный определитель появляется в разложении по
методу конфигурационного взаимодействия, несо-
несомненно, с наибольшим коэффициентом. Ряд из остав-
оставшихся определителей является слабосходящимся
разложением для «динамической корреляции», обсу-
обсуждавшейся выше. Таким образом, теория, развитая
в предыдущих параграфах, применима как к состоя-
состояниям с замкнутой оболочкой, так и к единственному
определителю для состояний незамкнутой оболочки,
например для состояний 25 в литии, 2Р в боре, 8Р
в углероде, 45 в азоте, 3Р в кислороде, 2Р во фторе
и т. д., если только хартри-фоковские орбитали полу-
получаются путем минимизации энергии этого выделен-
выделенного определителя.
Для других состояний, таких, как lS в С
(\s22s22p2), в полном разложении функции г|) по ме-
методу конфигурационного взаимодействия встречаются
различные вырожденные слэтеровские определители
с большими коэффициентами одного порядка1).
В таких случаях функцию г|) можно разложить на
«хартри-фоковскую» и «корреляционную» части раз-
различными способами, отвечающими разным возмож-
возможным методам Хартри — Фока, упомянутым выше.
Точная функция г|) представляется следующим об-
образом:
М оо
= 2 */#,+ 2 cfo (94)
/>i j>m J J
1) Таблицы и способы конструирования детерминантных соб-
собственных функций атомов см., например, в работе Слэтера [19].
6 О. Синаноглу
82 §20. Незамкнутые оболочки
где f < — М*№рожденнме детерминанты, а & —все
остальные детерминанты, которые входят в точную
функцию х* Первая сумма представляет функцию </>0,
вторая — функцию %. Таким образом, здесь {%, &) =»0.
Функцию х, конечно, не обязательно записывать
в виде разложения по методу конфигурационного
взаимодействия.
В одном из приближений точную функцию ф
[уравнение (94)] представляют следующим образом:
Орбитали хартри-фоковского «фона» получают то-
тогда из единственного определителя фи так же как
в «неограниченном» методе Хартри — Фока [38].
В этом случае «корреляция», кроме «динамических»
эффектов, содержит эффекты вырождения [выраже-
[выражение (93)], которые учитываются, с помощью перво-
первого порядка метода конфигурационного взаимодей-
взаимодействия [6]. Этот метод, примененный Несбетом [5],
обладает определенными вычислительными преиму-
преимуществами, если (93) содержит слишком много опре-
определителей, как, например, в случае молекулы N2
вблизи диссоциации. Однако имеется и недостаток,
заключающийся в том, что физически различные «ди-
«динамическая» и «нединамическая» корреляция более
не разделяются.
Выражение (95), по-видимому, дает большие мно-
многоэлектронные «корреляции» и довольно существен-
существенные fu возникающие на самом деле именно из той
части (93), которую учесть не так трудно, как «ди-
«динамическую» часть.
В другом приближении функцию г|) представляют
так же, как в (94), и получают занятые орбитали из
полного разложения (93) («расширенный» метод
Хартри — Фока) [1, 89]. В таком случае эффекты
первого порядка метода конфигурационного взаимо-
взаимодействия, или же эффекты «вырождения», которые
все ещ^ включают дальнодействующие части куло-
новских отталкиваний и полные зарядовые распреде-
f 20. Незамкнутые оболочки &3
ления, приписывают хартри-фоковской функции ф0.
Если это сделано, то последняя сумма из выражения
(94) будет содержать лишь «динамические корреля-
корреляции», к которым применимы все аргументы, приведен-
приведенные выше для замкнутых оболочек. Тогда «много-
«многоэлектронные корреляции» и Ji должны быть малы.
«Корреляции», определенные таким образом, будут
слагаться из парных корреляций. Результаты Кле-
Клементи [79] для первого ряда атомов, полученные
с учетом состояний незамкнутой оболочки, подтвер-
подтверждают это. Полученные им хартри-фоковские функ-
функции являются функциями «расширенного» типа. Для
каждого состояния незамкнутой оболочки получают-
получаются новые орбитали.
«Расширенный» метод Хартри — Фока можно
рассматривать как обобщенный метод самосогласо-
самосогласованного поля [33], в котором орбитали, например, ^
в выражении (95), должны браться самосогласован-
самосогласованными только с корреляциями вырожденного типа.
Однако в обобщенном методе самосогласованного
поля, подобном методу Бракнера, делается попытка
подогнать орбитали не только к эффектам «первого
порядка метода конфигурационного взаимодействия»,
но и к «динамическим» корреляциям, т. е. к беско-
бесконечным суммам в выражении (94) или (95). Динами-
Динамические корреляции являются корреляциями, которые
приводят к увеличению трудностей в обобщенном
методе самосогласованного поля. С другой стороны,
они являются корреляциями, для которых не нужна
подобная процедура самосогласованного поля, по-
поскольку «динамические» Ji в любых случаях малы.
В этом варианте (расширенный метод Хартри —
Фока+динамические корреляции), хотя и являю-
являющемся точным методом, требуется, чтобы новые ор-
орбитали подсчитывались для каждого состояния не-
незамкнутой оболочки, возникающего, например, из той
же самой атомной конфигурации.
Метод Хартри — Фока, основывающийся на «сред-
«средней энергии конфигурации с ограничениями», яв-
является компромиссным. Функции ty вновь предста-
84 § 21. Почти вырожденные состояния
вляются как в (93) и (94), однако хартри-фоковские
орбитали получаются способом, описанным в § 6,
так что они одни и те же, например, для 3Р-, Ф- и
^-состояний атома С (Is22s22p2). В рамках такого
метода Хартри — Фока большая часть последней
суммы в (94) является «динамической корреляцией»,
т. е. получается в виде G7). В этом случае также
возникают некие fu стремящиеся превратить «сред-
«средние» орбитали этого типа из фо в орбитали «расши-
«расширенного метода Хартри — Фока». Мы упоминали, что
изменения в энергии, обусловленные такими /г-, в ато-
атомах и небольших стабильных молекулах обычно
меньше, нежели 0,01 эв (см. § 6), как об этом можно
судить, если сравнить хартри-фоковские расчеты раз-
различного типа.
Таким образом, для большинства случаев, в ко-
которых рассматриваются незамкнутые оболочки, мож-
можно использовать вариант многоэлектронной теории
в виде, даваемом выражениями G7) и (85), совме-
совместно с методом Хартри — Фока, основывающимся на
«средней энергии незамкнутой конфигурации с огра-
ограничениями». С этими хартри-фоковскими орбиталями
уравнение теории возмущений F4) можно вновь ре-
решить при помощи операторной техники. Поскольку
потенциал Vi один и тот же для всех электронов, это
упрощает суть вопроса. Отметим также, что проек-
проекционные операторы, превращающие, например, фх
в </>о, в соответствии с (93) коммутируют с опера-
операторами (Л, Но и #i. Проекционные операторы Лёв-
дина [89] будут полезны при применении многоэлек-
многоэлектронной теории [9—И] и теории возмущений [8] к не-
незамкнутым оболочкам.
§ 21. ПОЧТИ ВЫРОЖДЕННЫЕ СОСТОЯНИЯ
Мы рассмотрели, каким образом можно приме-
применить многоэлектронную теорию к системам с зам-
замкнутыми и незамкнутыми оболочками. Однако си-
системы с почти вырожденными состояниями попадают
§ 21. Почти вырожденные состояния 85
в разряд, промежуточный по отношению к этим двум
классификациям. В § 6 упоминались две важные при-
причины подобного почти вырождения:
а) в атомах вырождение возникает, когда Z—*oo;
б) почти вырождение имеет место в молекулах
вблизи диссоциации.
В связи с этим возникает вопрос: до каких пор
можно рассматривать такие системы методами, раз-
развитыми для случая замкнутых оболочек, с един-
единственным определителем <j>0 и пренебрежимо ма-
малыми fi?
а. Хорошо изученный [41, 77] пример Z-вырожде-
ния возникает в изоэлектронном ряду Be. Даже в Be
исследование методом конфигурационного взаимодей-
взаимодействия [77] показывает, что корреляционная ошибка во
внешней оболочке появляется в основном из-за силь-
сильного 2s2 — 2р2-смешивания; коэффициент при 2р2-
конфигурации [77] равен —0,297, если принять ко^
эффициент при 2$2-конфигурации, равным 1,000.
В последовательности Be, B+, C2+, N3+, ... этот кор-
корреляционный эффект оказывается [41] линейным
по 2. (Относительно зависимости этой корреляции
от Z в атомах с более чем четырьмя электронами
см. § 24.)
Такую большую примесь 2р2-конфигурации, оче-
очевидно, все еще можно рассматривать как часть %, и
хартри-фоковские расчеты могут быть основаны на
единственной Is22s2-конфигурации, что следует из
возможности полного пренебрежения эффектами оди-
одиночных возбуждений /г, обнаруженной Ватсоном
и др. [77]. Кибартас и др. [90] провели также расчеты
по «расширенному» методу самосогласованного поля,
пользуясь комбинацией Is22s2-, Is22p2- и 2s22/?2-koh-
фигураций для Be. Их результаты отличаются от
результатов, полученных методом Хартри — Фока,
основанным на использовании одной 152252-конфигу-
рации, тем, что в них учитывается вклад попарного
взаимодействия [41] Is22s2- и 1522р2-конфигураций, со-
составляющий около 0,001 ат. ед. @,027 эв). Это ука-
указывает вновь на достаточную малость вклада /*,
88$ 2t. Почти вырожденные
б. Почти вырождение в диссоциирующих двух-
двухатомных молекулах является особенно существенным
в теории межатомных сил. Рассмотрим, например,
молекулу Н2.
Когда в молекуле водорода межъядерное расстоя-
расстояние отвечает положению равновесия г=*ге @,74 А),
то корреляция в Н2 будет «динамического» типа.
Весьма сходная ситуация имеет место в атоме Не.
Метод Хартри — Фока основывается на (^^-кон-
(^^-конфигурации и дает ?согг= —1,06 эв [1]. Давидсон и
Джонс [91] вычислили f< для Н2 при г=ге с помощью
волновой функции, предложенной Колосом — Рута-
ном [92], и нашли, что ими можно пренебречь, как и
в случае Не [11]. (Они рассмотрели разность между
хартри-фоковскими и естественными орбиталями, при
этом fi — первое приближение к этой разности.)
При г = оо, если метод Хартри — Фока все еще
основывать на Aа^J-конфигурации, молекула не дис-
диссоциирует на два атома водорода, которые находи-
находились бы в ls-состояниях. «Корреляционная» ошибка
равна 7,74 эв [1]. Она, однако, целиком связана с эф-
эффектом вырождения. Парное (первого порядка) кон-
конфигурационное взаимодействие между (lagJ- и
AаиJ-состояниями, являющимися вырожденными,
ликвидирует большую часть этой ошибки. Таким об-
образом, при г=оо мы имеем проблему незамкнутой
оболочки с отсутствием динамической корреляции.
Подобная нединамическая корреляция типа выро-
вырождения приводит к неисчезающим fi при г = оо. Этот
результат был найден [11] путем сравнения точной
симметризованной A$а1$б) функции г|? с полученной
Лёвдиным [1] функцией Хартри — Фока, основанной
на Aо^J-состоянии и дополненной по методу конфи-
конфигурационного взаимодействия подмесью (laM) ^со-
^состояния. Вклад в энергию от /*-части равен около
0,4 эв [11].
Фрага и Ранзил [93] изучали молекулы Н2 с по-
помощью хартри-фоковской функции, основанной на
Aа^J-состоянии и дополненной по методу конфигу-
конфигурационного взаимодействия для г в интервале
§ 21. Почта вырожденные состояния
1,0 ат. idO<12 ат. ед. Они применили молекуляр-
молекулярные орбитали, выбрав их как линейную комбинацию
атомных орбиталей самосогласованного поля
(LCAOMOSCF) и взяв в качестве базисного набора
15-, 25- и 2/?-состояния, и учли двенадцать конфигу-
конфигураций. Из них (log2eg)- и (logSog)-конфигурации
были обусловлены одиночными возбуждениями и от-
отвечали первым членам в разложении /t по методу
конфигурационного взаимодействия. С помощью
коэффициентов, данных Фрагой и Ранзилом, и про-
процедуры теории возмущений, обоснованной Кестнером
(см. приложение в работе [11]), вклады в энергию
этих отдельных возбуждений были оценены [11] для
г в интервале 1,1 дг. ed^r^l2fi ат. ед. Полученные
значения приведены в табл. 3; они показывают, как
меняется вклад f< в зависимости от г. Если величину
0,1 эв рассматривать как малую ошибку, то вклад ft
будет пренебрежимо мал для г/ге<2>
Таблица 3
Оценка уменьшения энергии в молекуле Н2 из-за учета
отдельных возбуждений [93], или //
Случай г = оо рассчитан другим способом, нежели остальные;
см. пояснения в тексте и работы [11, 93* 1\.
Межъядерное расстояние г,
am. ед.
оо
12,0
4,0
3,0
2,0
1,4 (~ге)
1Д
Вклад одиночных возбуждений,
ИЛИ }j, ОТ
-4,4
-0,33
-0,32
—0,13
—0,027
—0,0092
—0,0046
Таким образом, при г/ге<2 корреляция носит ди-
динамический характер и волновую функцию можно
представить, как для Не, в виде
88 § 22. Парные корреляции
При ббльших г вклады {/*} возникают, следовательно,
благодаря нединамической корреляции. Их можно
рассматривать либо как дополнительные возмуще-
возмущения, либо с помощью «расширенного» метода Харт-
ри — Фока.
Приведенный пример с молекулой Н2 показы-
показывает, что fi можно рассмотреть и в промежуточных
случаях и выбрать соответствующую исходную функ-
функцию фо- К счастью, эта проблема не возникает при
получении большинства энергий связи (г = ге) и в не-
связывающих взаимодействиях. Например, система
из двух атомов Ne (исходим из молекулярных ор-
биталей) описывается единственным определителем
при всех г благодаря влиянию «принципа исключе-
исключения». Многоэлектронную теорию для замкнутых обо-
оболочек можно применить как к этой системе, так и
в случае несвязывающих взаимодействий между свя-
связями, например к насыщенным углеводородам (см.
§27 и 28).
§ 22. ПАРНЫЕ КОРРЕЛЯЦИИ
В § 16—21 было показано, что после того как
вырождение снято, главными эффектами являются
парные корреляции йг-7-, определяемые выражениями
G7) и (82) или (85). Эти корреляции должны быть
получены с помощью вариационного приближения,
использующего теорию возмущений, путем миними-
минимизации большей части Ё [выражение (82)]. После
того как они определены, мы имеем возможность
оценить ^ и /?'. Свойства парных функций, связь их
со свойствами реальных свободных двухэлектронных
систем и методы для их оценки были даны ранее [10].
Здесь мы подытожим полученные результаты.
Согласно выражению (85), пары являются рас-
расцепленными. Различные йц подчиняются некоторым
соотношениям симметрии, как это будет обсуждено
в § 23, однако в других отношениях они являются
независимыми. Независимая величина г'и имеет вид
§ 22. Парные корреляции
вариационной энергии реальной двухэлектронной си-
системы, например атома Не. Применяя A8) и A9)
или C9) и C7), или E8), получаем, например, для
атома Не:
?(атома Не) « Ец. F.+
, 2 (#A2), т
В § 19 нами было также показано, что
И?2 » «12 (97)
[см. D0), D9)и (92)].
Энергия пары электронов е^ [выражение (86)],
находящихся в N-электронном хартри-фоковском
«фоне» (например, остов Ne в ^-состоянии), отли-
отличается от энергии свободной двухэлектронпсй систе-
системы (например, иона Ne8+) тем, что в первом случае
на пару электронов оказывает влияние принцип
исключения и что потенциал V* обусловлен всеми N
электронами и не равен в точности потенциалу вза-
взаимодействия двух электронов пары (см. также §13).
Эффективное уравнение Шредингера для пары
с учетом «фона» получается варьированием незави-
независимой e'tj no atp на которые, однако, наложены
условия [10]:
(йи, k) = 0 (k=h 2, ...,/,..., у, ..., TV).
Это варьирование проводится без затруднений, если
учесть, что Q [выражение G1)] является эрмитовым
и идемпотентным оператором
Q2=Q, (98)
и если переписать (86) в виде
~, _ 2 {Qmtigf (if), иф + (Q jet + ej + тф щ,, щ,)
В таком случае произвольные вариации butj авто-
автоматически остаются в виде Qbutj, т. е. „орбитально
90 § Ж Парные корреляции
ортогональными* ко всем к из функции <&>:
<Q6ttiy, A>=0. (996)
В результате имеем
(*, + ej)utj+ Qmlf\#(ij) + uu] = Еииф A00а)
где ti} — стационарное значение е/у:
A01)
Уравнение A00) и является эффективным уравне-
уравнением Шредингера [ср. B) и F2)]:
Ни$и = Е1$и, A02а)
где
Eij^Bi + zJ-Jij + K9iJ + nu (Ю2б)
и
Чтобы получить A02а), мы скомбинировали хартри-
фоковскую часть с корреляционной частью [уравне-
[уравнение A00а)].
Если же варьировать часть выражения (86)
и |у>, A03)
то получим члены первого порядка [уравнение G0)]
уравнения A00).
Если варьировать целиком числитель величины
г' , то получим
(et + ej+ Qmtj) % = 0. A006)
Это уравнение подобно уравнению Бете — Голдстоу-
на (в замкнутой форме) [10] для конечных ядер [941.
Последнее, однако, является уравнением для частиц
в бракнеровском «фоне», а не в хартри-фоковском
«фоне», определяемом потенциалом Vi [выраже-
[выражение (8I. Уравнение бете-голдстоуновского типа
в виде, который мог бы быть полезен для реальных
расчетов в случае конечных ядер, а также свойства
§ 2$* Парные корреляции 91
его решений и соответствующий вариационный прин-
принцип даются в приложении к работе [10].
Отметим, что и уравнение G0)» и уравнение A03)
аппроксимируют те решения уравнения A00), для
которых {йц, u<j)<l. Эти виды аппроксимаций спра-
справедливы для решений, отвечающих малым корреля-
корреляционным отклонениям от Я {if) [10].
Уравнение A00), G0) или A03) можно иногда
решить непосредственно, хотя и приближенно, как
для некоторых межоболочечных корреляций в ме-
методе поляризации остова [3, 4] (см. § 24). Однако,
вообще говоря, как и для атома Не, чтобы получить
uijy нам необходимы вариационные методы.
Принципы минимальности. В решениях эффектив-
эффективных уравнений Шредингера A00) или аппроксими-
аппроксимирующих их уравнений G0) н A03) содержится пред-
предположение о стационарности соответствующих е^#
Однако большинство пар, например 2$2-пары в Be»
2р\ -пары в Ne и т. д., размещается во внешних обо-
оболочках и подобно «возбужденным» парным состоя-
состояниям. Трудности применения вариационного метода
к реальным возбужденным состояниям хорошо из-
известны. Внешние оболочки при минимизации обычно
обнаруживают тенденцию приближения к самой глу-
глубокой внутренней оболочке [102]. Однако эта труд-
трудность не возникает в многоэлектронной теории, т. е.
при использовании г'^. Практически вариационный
метод, т. е. принцип минимальности, применим к лю-
любой парной корреляции йц.
Легко показать [10], что каждая величина е^ не
только является стационарной, но и обладает инди-
индивидуальным минимумом. Для этого нужно просто
минимизировать (86) или (99а) или их части, на ко*
торые наложены условия (996), и использовать проб*
ные функции йц для аппроксимации решений соот-
соответствующих дифференциальных уравнений.
Это позволяет рассматривать только внешние обо-
оболочки, как, например, в Na2 или же ВаО или я-элек-
тронной части бензола и т. д., и избавляет нас от
92 § 22. Парные корреляции
затруднений, связанных со стягиванием этих оболо-
оболочек к внутренним оболочкам [10].
Если схема уровней, даваемая хартри-фоковски-
ми орбиталями, приведена во взаимно однозначное
соответствие с экспериментальной схемой, то урав-
уравнения G0) и A03) определяют «адиабатические»
сдвиги, обязанные йц. С другой стороны, если схема
уровней неизвестна, как в случае нарушения правил
Хунда в некоторых незамкнутых оболочках, то не-
необходимо использовать уравнения A00), а не урав-
уравнения G0) или A03). Уравнения A00), например,
автоматически выбирают основное состояние. Орби-
тали дают правильную схему уровней, если минимум
каждой величины г^. имеет место для тех значе-
значений вариационных параметров, которые приводят
к наименьшему по сравнению с единицей значению
{йц, uij). [См. также в работе [11] дискуссию, кото-
которая предшествует уравнению D7).]
Любые методы, применимые к реальным двух-
электронным системам [1], подобным Не или Н2,
можно использовать, чтобы получить пробные функ-
функции для наших парных корреляций uij. Разные пары
из АЛ-электронной системы являются физически
вполне различными, так что не существует ни одного
метода, удобного и годного для рассмотрения всех
пар. Например, метод «координаты ri2» мог бы по-
подойти для внутренней оболочки в ^-состоянии или
для Bр2J-пар в Ne, однако он не нужен для \s2s-
пары. В частности, использование метода «коорди-
«координаты Ti2» для всех пар [57, 60] полной функции %8
может ввести излишние математические трудности.
Типы методов, годных для рассмотрения парных
корреляций, и дальнейшие детали их использова-
использования даны ниже при последовательном рассмо-
рассмотрении первого ряда атомов и их ионов (см.
§24).
Чтобы применить в этом случае многоэлектрон-
многоэлектронную теорию, необходимо, во-первых, волновую функ-
функцию \f>ij из любого двухэлектронного метода (обыч-
§ 23. Свойства симметрии 93
но нормируемую на единицу) записать в виде
4>;у = #(/у) + я?у, <ЯЧ«Л «?у> = 0, A04)
что меняет нормировку. То есть
Это сделано в соответствии с преобразованием
Ф/у = *¦//• с = (Ф/у» ¦#(*/)>»
о _ *и — с#УЛ A06)
Отметим, что ?{ij) являются двумя хартри-фоков-
скими спин-орбиталями для этой конкретной пары,
выбранными из полной N-электронной функции ф0.
Они не являются орбиталями свободной двухэлек-
тронной системы [10].
Величины u°iJt полученные из A06), в дальней-
дальнейшем ортогонализуются ко всем k из функции ф0 пу-
путем применения к ним оператора Q [уравнение G1)].
Интегрирования обычно являются простыми.
Для Л/-электронной системы волновая функция %>
выражения F9а), G6) или G7), содержит
N(N—1)/2 величин uZj. Многие из них, однако, раз-
различаются лишь ориентацией в пространстве или же
своими спинами [например, Bр*J-пары в Ne анало-
аналогичны B/71/J-парам и т. д.]. Число различных [8, 10,
68] величин uij в таком случае определяется поряд-
порядком N. Это можно усмотреть из свойств симметрии
X, обсуждающихся в следующем параграфе. Только
такие характерные пары и необходимо определить.
Некоторые методы их определения даются в § 24.
§ 23. СВОЙСТВА СИММЕТРИИ
Функции х» %s и %'s должны иметь ту же симмет-
симметрию, что и функция ^>о. Например, в Ne они все
должны иметь симметрию функции ^-состояния. Но
удовлетворяют ли функции %i> /s или %s этому
§ 23. Свойства симметрии
требованию и если да, то какие ограничения налагает
оно на Uij?
Функция %8 [уравнение G6)] имеет ту же самую
форму, что и функция &1 [уравнение F9)], и в нее
дают вклады те же самые типы пар, но во всех «по-
«порядках». Напомним, что функция xi была получена
путем строгого решения уравнения F4) [8, 68]. Это
строгое решение гарантирует для функции xi ту же
самую симметрию, что и у функции Фо. Действитель-
Действительно, если ^о описывает ^-состояние в уравнении F4),
то (Яо —?0)xi и (Ei — Hi)f0 также должны обла-
обладать той же самой симметрией ^-состояния, а это
означает, что решение xi имеет симметрию lS-co~
стояния.
Предыдущие рассмотрения [8, 68] проводились
таким образом, что пары сопоставлялись различным
состояниям реальной свободной двухэлектронной си-
системы. Оператор антисимметризации Л позволяет
в уравнении F4) преобразовать 38 Щ) в парные со-
состояния, относящиеся к неприводимым представле-
представлениям группы симметрии для двух электронов [10].
Это означает, например, что, хотя в Be N(N—l)/2
равно 6, там имеется лишь четыре независимых йц%
отличающихся своими пространственными частями:
l5215, 2s21S, (ls2sIS и 35.
Три из шести пар отличаются Sz-компонентой
sS-терма. Аналогично в оболочке Ne в 2р6-состоянии
Л^(Л^— 1)/2=15, однако и здесь различными будут
лишь три пары:
Преобразование пар типа 3} (ij) в такие «непри*
водимые пары» является необходимым, если #</] по-
получаются из первого порядка двухэлектронных урав-
уравнений без учета принципа исключения:
?> = 0 (кФ1ч У).
При применении оператора антисимметризации Л
к конечной функции xt учитывается принцип исклю-
исключения [8, 68]. Поскольку исходные «неприводимые»
§ 28. Свойства симметрии 05
величины a(/j в этом приближения удовлетворяют
уравнениям первого порядка для реальных невыро-
невырожденных двухэлектронных состояний (если отвлечься
от потенциала V<, описывающего больший «фон»),
то их можно применить для полуэмпирических
расчетов.
С другой стороны, для строго неэмпирических рас-
расчетов, можно заранее использовать оператор Л и, не
находя вначале «неприводимых пар» [10], прямо по-
получить уравнения типа G0) для &(ij). Действие опе-
оператора Q (??**', /) в уравнении G0) эквивалентно
действию оператора Jtf и потому уравнение G0)
имеет решение для любого ЯЩ) независимо от того,
будут ли Я (if) и Я(Ы) из функции ф0 соответство-
соответствовать одному и тому же вырожденному состоянию или
нет. Уравнение G0) и относящееся «ко всем поряд-
порядкам» уравнение A00) весьма просты в обращении,
поскольку вначале не требуется отыскивать неприво-
неприводимые парные состояния. Они будут приемлемыми
уравнениями для многоэлектронной теории [10].
Связь между неприводимыми парами и парами типа
Я (if) была описана детально в работе [10]; там же
были приведены результаты теории возмущений [8,
68], в которой использовались проекционные опера-
операторы. Мы не будем повторять их здесь.
С помощью уравнения G0) или (Ю()) легко опре-
определяются различные пары. Например, для 2р6-со-
стояния Ne вместо 3Р-, XD- и *S2p2-napbi можно взять
три единственные пары
(рхару&), (рхарх$) и (рхаруа).
Другие пары отличаются от них лишь измене-
изменением осей или перестановками спинов. Таким обра-
образом, необходимо получить йг-;- лишь для этих един-
единственных пар.
Аргументы, вытекающие из свойств симметрии,
данные выше, применимы как к функции %8у так и
к функции xi- Свойства функции &» даже в более
сложных случаях, например, когда функция ф0
является многодетерминантной функцией, описываю-
описывающей незамкнутую оболочку, всегда можно найти
96 § 23. Свойства симметрии
путем решения уравнения F4) для функции %ь приме-
применив операторную технику [8, 68], а затем выбрав ту
же самую форму для функции %8, как и у функции %i.
Свойства симметрии функции %8 можно также ис-
исследовать и непосредственно; не рассматривая вна-
вначале функцию %i.
Бриксток и Попл [95] сформулировали необходи-
необходимые условия, которым должна удовлетворять про-
пространственная часть волновой функции, если она
является собственной функцией S2 и Sz.
Рассмотрим замкнутую оболочку синглета, напри-
например оболочку в Be (Is22s2 lS). Пусть множитель, за-
зависящий от пространственных координат, при спи-
спиновой функции
аA)аC)рB)рD)
обозначается в полной волновой функции следую-
следующим образом:
Ф(Г1, г3; г2, г4); A07)
тогда условия, сформулированные Брикстоком и
Поплом, приводят к соотношениям
1, г3; г2, г4) = — Ф(г3, гх; г2, г4) = — Ф(г1? г3; г2, г4),
A08)
!, г3; г2, г4) — Ф(г2, г3; rlf г4) — Ф (г4> г3; г2, тг) —
— Ф(г1э г2; г3, г4) —Ф(г1э г4; г2, г3)} =0. A09)
Эти условия были наложены автором и Фу-Тай
Туаном непосредственно на волновую функцию %s
для проверки ранее полученных результатов [8, 68].
Для Be подтверждается, что
2им) = SZJL A25м) - 0,
(Я1234) = Szoi (S1234) = 0
и что только комбинация всех межоболочечных пар
является синглетом:
(Ш24+ 14«23 + Я1423 + и1324) = 0. (Ill)
Все члены функции х5 п0 отдельности являют с
в Be собственными функциями L2 и Lz для 1 = 0.
§ 23. Свойства симметрии 97
Левин, Геллер- и Тейлор получили с помощью
более мощной техники необходимые и достаточные
условия того, чтобы волновая функция являлась
собственной функцией S2 и S^1). Они нашли, что
если
^ = 0, A12)
то
52х; = 0, A13)
где %'s [уравнение G7)] включает несвязанные ком-
комплексы.
Для Be, в дополнение к соотношениям A08), их
условия привели к следующим выражениям:
Ф(г1э г3; г2, г4) — Ф(г2, г4; г1э га) = 0,
Ф(Г1, г3; г2, г4)-Ф(г2, г3; г1э г4)— A14)
— Ф(г4, г3; г2, ^ = 0.
Записав, например, и12A, 2) в виде
и12A, 2)=#{T>12(rlf r2)aip2} A15)
и применив выписанные выше условия к A11), они
нашли, что для пар с параллельными спинами
а для пар с антипараллельными спинами
где vft—антисимметричная, а ^ — симметричная
функции по отношению к обмену координат rt и vi и
VK = V2A — VU = Vfv VSIA = ^23- С118)
Следовательно, имеются лишь две независимые
функции парных корреляций для межоболочечной ча-
части корреляций в Be. Это подтверждает заключения,
!) Мы признательны авторам, ознакомившим нас со своими
результатами до их публикации и Тейлору за обсуждение ряда
вопросов.
7 О. Синаноглу
98 §23. Свойства симметрии
полученные с помощью функции xi [8, 68], так как
vfj отвечает 3S(ls2s)-nape, упомянутой выше, a vfj
отвечает 1S(ls25)-nape. В сумме е^ [уравнение
(85)] любые две линейно независимые комбинации
этих пар могли бы варьироваться по отдельности.
Наиболее удобным из возможных выборов является
выбор пар Ui3 и Ли (линейные комбинации 3S- и
*S-nap [68]), отвечающих (lsa2sa)* и (lsa2sp)-корреля-
(lsa2sp)-корреляциям, поскольку в этом случае не нужно вначале
отыскивать неприводимые парные состояния. Эти
пары и являются теми функциями, которые исполь-
использованы в F9), G7) и (86). Поскольку различные й^
определены, а набор эквивалентных им функций оди-
одинаков с ними, автоматически получается, что функ-
функция %'s имеет соответствующую симметрию. Анало-
Аналогично 1Асвойства х^ можно рассмотреть непосред-
непосредственно. Однако форма функции хь полученная выше,
вновь указывает, что функция %s является собствен-
собственной функцией L2, поскольку различные йц опреде-
определяются так же, как для Ne, и находятся независимо.
Бриксток и Попл [96] провели модельные расчеты
на Ne, исследуя корреляции восьми Bs22p6) электро-
электронов на поверхности сферы. Они рассмотрели проб-
пробную функцию вида [95]
Ф=*оЛ (И9а)
где
F=l + ^Kjf(ri9Tj) 0*96)
— «корреляционный множитель» [1].
Они использовали соотношения A08) и A09) и
нашли, что функция if является синглетом, если все
вариационные параметры Я^ и функции f(ru г,) одни
и те же [например, функции для (РхОсруР)- и
(рзсСфяР)-пары одинаковы]. Это требование отли-
отличается от требования, найденного для функций Xs, где
&ц из (рхару$)- и (рхаРхР)-пар должны быть неза-
независимыми. Отметим, однако, что вид функции A19)
весьма отличен от вида функции %s [выражение G6)].
§ 23. Свойства симметрии 99
В выражении A196) оператор Л применяется лишь
к функции ^о и не применяется к Лчасти!). Это
весьма ограничивает класс функций и налагает до-
дополнительные условия A09) на парные корреляции.
На основании материала, изложенного в этом
параграфе, мы приходим к следующим выводам:
1. Функция %'s [выражение G7)] имеет ту же
симметрию, что и функция ^-0. И эта величина и энер-
энергия [уравнения (85) и (86)] содержат пары йц для
каждого произведения спин-орбиталей <%(*'/), обра-
образованного из k в функции <j>0.
2. Число различных пар й^ которое должно быть
определено, обычно находят с помощью предвари-
предварительного рассмотрения. Оно порядка N. Многие из
этих пар отличаются друг от друга лишь своей ори-
ориентацией в пространстве или перестановкой спинов.
В сомнительных случаях, различные пары можно
найти сведением [8, 68] уравнения F4) к паре урав-
уравнений или же путем применения соотношений [95],
аналогичных A28) и A14).
Это рассмотрение применимо к молекулам, если
основываться на функции фо, представляющей собой
молекулярные орбитали самосогласованного поля.
В таком случае даже для больших насыщенных мо-
молекул будут иметься пары й^ для каждой молеку-
молекулярной орбитальной пары &(ij). Однако эта теория
будет преобразована в § 27 для описания с помощью
локализованных орбиталей. Тогда молекулярные ор-
орбитали uij будут сопоставлены локализованным кор-
корреляционным функциям H-vp-
Многоэлектронная теория [8, 9—11], описанная
в § 16, 17 и 22, позволяет теперь построить последо-
последовательную теорию атомов и молекул (по мере их
усложнения) с помощью нескольких парных функций.
Следующие три параграфа содержат применения
]) Краткие обозначения Фо (йцЩ), используемые в урав-
уравнении C) работы [12], были бы ошибочны для уравнения B0)
этой работы. В наших уравнениях оператор <А применяется так-
также к йч.
100 § 24. Атомы
результатов многоэлектронной теории к первому ряду
атомов, к малым молекулам и к я-электронным си-
системам. Для перечисленных случаев будут обсуждены
также полуэмпирические выводы из этой теории.
§ 24. АТОМЫ
Проиллюстрируем теперь применение многоэлек-
многоэлектронной теории на примере последовательного рас-
рассмотрения первого ряда атомов, их ионов и наиниз-
наинизших возбужденных состояний. В этом случае тре-
требуется лишь одиннадцать парных функций для всего
первого ряда. Некоторые из функций используются
повторно при переходе от одной системы к другой.
И в упрощенных неэмпирических расчетах, и
в полуэмпирических расчетах [68] желательно знать,
например, чем именно отличается атомный остов
для Li от свободного иона Li+ или от остова
Li2!). Хартри-фоковская часть в этих расчетах хо-
хорошо изучена [97]. Например известно, что метод
Хартри —Фока, примененный к Na+ или Na, приво-
приводит к мало отличающимся орбиталям остова. Здесь
мы рассмотрим корреляционную часть волновой функ-
функции.
Начиная с гелиевого изоэлектронного ряда и доба-
добавляя последовательно по одному электрону, мы вы-
вынуждены вводить новые парные функции. Эта про-
1) Чтобы полуэмпирически сопоставить первый ряд атомов
с двухэлектронными состояниями, Бечер и Гоудсмит [98] ис-
использовали теорию возмущений, взяв в качестве Фо орбитали
голых ядер. В этом приближении не возникает различия между
малыми многоэлектронными эффектами, например /?' из (82),
более существенными эффектами, обусловленными сильной «ор-
«орбитальной поляризацией» водородоподобных орбиталей (выбрано
плохое исходное зарядовое распределение) и эффектами «прин-
«принципа исключения» [уравнение (87)]. Хорак (в печати) исполь-
использует улучшенную форму процедуры такого типа. Он все еще
берет в качестве невозмущенной функции Фо орбитали голых
ядер и рассматривает %i и Е2. Однако он точно подсчитывает
«эффекты орбитальной поляризации» (одиночные виртуальные
возбуждения), рассматривая эмпирически лишь двойные воз-
возбуждения.
§ 24. Атомы 101
цедура продолжается до тех пор, пока мы не придем
к Ne. При добавлении каждого электрона вводятся
не только новые йц, но и происходит модификация
старых.
Qs2)*S: He, Li+, Be2*, ..., Ne»*
Конфигурация Is2 является «плотной парой»
с сильной «динамической» корреляцией, поэтому для
этой пары наиболее подходящим методом исследо-
исследования является метод [1] координаты ri2. Возьмем,
например, шестичленную волновую функцию Хилле-
рааса [88] со следующими независимыми перемен-
переменными:
s — rx + r29 t = r2 — rx и u — rl2.
Эту волновую функцию при помощи соотношений
A04) —A06) необходимо переписать в виде .$> A2) +
4~#i2. Функции и\2 следовало бы сделать, как
обычно, ортогональными к хартри-фоковским ls-op-
биталям, чтобы устранить /*• Однако /* были подсчи-
подсчитаны выше [см. замечания после соотношения (92)]
и было найдено, что для Не они дают около
0,0025 эв. Таким образом, этим эффектом можно
пренебречь.
(ls22sJS: Lit Ве+, В2* Ne7*
В этом случае используются те же парные функ-
функции ui2, что и для Li+, Ве2+ и т. д. Функция и12 в Li,
Ве+ и т. д. отличается от соответствующей функции
предыдущего ряда лишь модификацией потенциала
Vu наличием хартри-фоковского потенциала «среды»
и учетом принципа исключения для электрона
в 25-состоянии [см. (81), (86) и (87)]. Эта функция и12
должна быть совершенно нечувствительной к сла-
слабому изменению потенциала Vu потому что даже при
изменении ядерного заряда, например при переходе
от Не к Li+, корреляционная энергия в (^^-состоя-
(^^-состоянии изменяется лишь на 0,04 эв [1], а при переходе
от Li+ к Ве2+ она меняется на 0,012 эв [1] (табл. 4).
Влияние на функцию и& принципа исключения для
102
§ 24. Атомы
электрона в 25-состоянии сводится по грубой оценке
к внесению вклада порядка 0,004 эв в корреляцион-
корреляционную энергию для Li (табл. 4). Изменения, напри-
например, при переходе от Li+ к Li очень малы и нет не-
необходимости вновь менять параметры функции ui2y
чтобы минимизировать выражение (86) для ряда Li
[68]. Функции можно непосредственно взять из ряда
Li+, ... и ортогонализовать к 25-состоянию.
Таблица 4
Корреляционные эффекты в Li и Li2
Все перечисленные в таблице величины даны в 9в.
Энергия поляризации остова [4]1) Ер
атом Li
а) хартри-фоковская орбиталь са-
самосогласованного поля Bs)
б) ортогональная атомная орби-
орбиталь B5°)
Эффекты «проникновения» [3]
j' а') молекулярная орбиталь само-
самомолекула 1 согласованного поля Bog)
Li2 1 б') ортогональная молекулярная
1 орбиталь Bog)°
Вклад Ер в энергию связи (De) для Li2
(а')-2Х(а)
(б') -2Х(б)
Энергия связи De для Li2 [116]
Влияние принципа исключения для 2$-состояния
на корреляционную энергию Li+ [3]
Изменение ?согг в A$2)-состоянии [1] при пере-
переходе от Не к Li+
Изменение Есш в A$2)-состоянии [1] при пере-
переходе от Li+ к Ве2+
Дисперсионное притяжение остов — остов в Li2
-ОД018
-0,1066
<0,03
-0,2473
—0,2520
—0,044
-0,039
1,04
— 0,004
0,040
0,012
< 0,001
») См. § 24.
Когда добавляется третий электрон, то необхо
димы две новые парные функции: й13 для (lsa2sa)
конфигурации и й2з Аля U5fr2sa)-конфигурации. "-
§ 24. AfoMbi 103
этих - парных функций нет необходимости пользо-
пользоваться вариационным методом. По порядку величины
межоболочечные корреляции *) будут меньше, чем
внутриоболочечнЫе. Они хорошо аппроксимируются
по теории возмущений [уравнение G0)].
Когда два электрона отличаются своими глав-
главными квантовыми числами, приближенные решения
уравнения G0) можно получить методами «поляри-
«поляризации остова»2). Эти методы были критически разо-
разобраны и была дана более полная теория, которая
включает эффекты проникновения и др. [3].
В Li внешний электрон (с координатой г3) менее
прочно связан и, следовательно, он движется медлен-
медленнее, чем внутренний электрон (с координатой г2).
В этом случае можно использовать «адиабатическое
приближение». При этом полагают, что внешний
электрон покоится в каждый фиксированный момент
времени и энергия остова определяется для различ-
различных фиксированных значений г3. Эта энергия зависит
от г3 параметрически и в свою очередь играет роль
потенциальной энергии [потенциала «поляризован-
«поляризованного остова» Vp(r3)] движущегося внешнего элек-
электрона с координатой г3. В этом приближении вол-
волновая функция дается выражением
1|>23 (х2, х3) = % [vc (г2, г3) v0 (г3) Р2аз); A20)
здесь vc(r2] r3) —орбиталь «остова» с координатой
г2, параметрически зависящая от г3; ?>о(г3)—орби-
?>о(г3)—орбиталь внешнего электрона, определяемая хартри-фо-
ковским потенциалом «остова», добавленным к по-
потенциалу «поляризованного остова» Vp(r3). По-
Поскольку выражение A20) включает хартри-фоков-
скую часть ^B3), то при помощи соотношений A05),
A06) и G1) оно должно быть записано в виде A04).
В первом приближении в выражении A20)
1) Корреляции, рассмотренные Клементи [79], оказываются
слишком большими для Z=8, 9 и 10. Маккой указал, что вели-
величина их, по-видимому, возрастает как Z4. Это означает, что ре-
релятивистские эффекты, которые также пропорциональны Z4, не
были должным образом отделены.
2) Для полного ознакомления с литературой см. работу [3].
104 § 24. Атомы
заменяют vo(r3) на хартри-фоковскую орбиталь &.
В таком случае корреляционная энергия1) е2з стано*
вится равной просто величине математического ожи-
ожидания потенциала поляризованного остова Ур(г3), вы-
вычисленного с помощью 3(х3),
е23 = C;КрЗ>. A21)
Корреляционная энергия ei3 имеет такой же вид, но
в потенциал Vp включается обменный член. Кола*
вей [99] получил численные значения потенциала Vp
для Li+/ Na+ и К+ в дипольном приближении и ис-
использовал эти результаты для соответствующих ме-
металлов. Тот же потенциал Vp был использован [4]
для Li и Li2, а эффекты «проникновения» были уч-
учтены с использованием оценок Колавея [3, 4]
(см. табл. 4). Табл. 4 указывает на нечувствительность
(813+823) к точности выбора используемой внешней
орбитали. Значение математического ожидания
A21), подсчитанное с помощью 25°-орбитали Слэте-
ра, ортогональной к орбитали остова, отличается от
значения, подсчитанного с помощью хартри-фоков-
ской 2я-орбитали, всего лишь на 0,005 эв.
S: Bet B+, С2+, ...
Здесь снова парные функции щ2 в (Is2)-состоянии
можно взять непосредственно из ряда Ве+, ... В этом
случае не возникает нового эффекта запрета и по-
потенциал Vi меняется несущественно.
Межоболочечная корреляция равна
е14 = 2(е13 + е2з). A22)
Величина ее, будучи не чувствительной к тому, на-
насколько аккуратно выбраны хартри-фоковские орби-
орбитали (см. табл. 4) и потенциал Уи может быть
1) Отметим, что «поляризация остова» описывает здесь пол-
полный эффект корреляции (поляризацию внешним электроном, рас-
рассматриваемым в каждый момент времени покоящимся). Этот
эффект не будет маскироваться среднеорбитальной «поляриза-
«поляризацией» [3, 18], т. е. сферическим потенциалом полной 2s-op6nTa-
ли; последний исчезает, если для Li полностью провести хартри-
фоковские расчеты.
§ 24. Атомы 105
взята равной удвоенному значению соответствующей
величины для ряда Ве+, В2\ ...
Из-за почти вырождения [41, 77] главная часть
функции йз4, т. е. корреляция в 2$2-оболочке, равна
» Y B/>J = Y {det
)}; A23)
здесь у — вариационный параметр, определяемый ми-
минимизацией е^ [выражение (86)]. В примененном
Ватсоном [77] разложении (по 37 конфигурациям) по
методу конфигурационного взаимодействия, Is2BpJ-
конфигурация вносила вклад около 1,2 эв (учтены
несвязанные комплексы) [10]. Отметим, что в много-
многоэлектронной теории даже в том случае, когда необ-
необходимо использовать метод конфигурационного взаи-
взаимодействия, он применяется лишь для одной пары.
Это требует расчета очень немногих матричных эле-
элементов и приводит к весьма компактному секуляр-
ному уравнению по сравнению с тем, которое полу-
получается при рассмотрении всех функций г|э и Н с по-
помощью метода конфигурационного взаимодействия
в обычной формулировке.
Из выражения A23) для функции им получаем
оценку корреляционной энергии для ряда Be, B+,
С2+ ... [41]
e34~Z. A24)
(ls22s22pzJP: В, О, N2*, ..., Ne*»
Парная функция й12 в ^-состоянии из ряда
В+, ... должна быть теперь ортогонализована [урав-
[уравнение G1)] по отношению к орбиталям E)=2/?0а.
«Эффекты запрета» на таких «динамических» uijy во-
вообще говоря, должны сказываться мало.
Однако влияние принципа исключения на функ-
функцию й34 в 2рг-состоянии [выражение A23)] весьма
существенно [9]. Оно приводит к исчезновению
det (/7оссрор), т. е. к исчезновению части выражения
A23); в результате имеем
ам « yf {det(Р№_ха)— det(pxap^)}. A25)
ИЛ § 24. Атомы
Этот эффект возрастает по мере добавления элек-
электронов в 2р-состоянни (см. С, N, . ¦., Ne, описанные
ниже). Для изоэлектронного ряда с (Is22s22pz)-кон-
(Is22s22pz)-конфигурацией корреляционная энергия ез4 все еще мо-
может оставаться линейной по Z, однако при переходе
от В+ к В, т. е. от выражения A23) к A25), \г3ь\
меняется от 1,2 эв до примерно 0,82 эв 1). Из-за прин-
принципа исключения функция й34 [выражение A25)] ло-
локализуется в плоскости, перпендикулярной 2рг-орбя-
тали. Это приводит к малости трехэлектронных
{2s22pz) -корреляций. Флуктуационный потенциал,
действующий между электронами в 2s- и 2р2-состоя-
ниях, мал именно тогда, когда функция йи велика
(см. фиг. 1 и 2 работы [9]).
Новые парные функции й15 и й25 для (Isoc2pza)- и
(ls$2pza)-конфигураций легко можно получить, вы-
вычислив по хартри-фоковским Bpz)-состояниям, мате-
математическое ожидание того же потенциала поляризо-
поляризованного остова в Н^-состоянии, что и для В2+ или В+.
Этот результат можно бы улучшить, если сделать
соответствующие функции иц из A20) ортогональ-
ортогональными к 2р2-орбиталям.
Парные функции Й35, йкЪ для 2$2/?-конфигурации
можно получить с помощью метода конфигурацион-
конфигурационного взаимодействия, метода координаты ri2 или ка-
какого-либо другого метода, используемого в связи
с соотношением (86).
(ls22s22p2KP: 1Dt *S; С, N+, O2+, F3+, Ne4+
Большинство парных функций в этих ионах по-
подобно парным функциям, обсуждавшимся для преды-
1) Глембоцкий, Кибартас и Юцис [100] рассмотрели «расши-
«расширенный метод Хартри — Фока» также для двух конфигураций.
Однако результаты их незначительно отличаются от результатов
метода Хартри — Фока, в основе которого лежит функция
0o(ls22s22pz) и который дополнен с помощью метода конфигура-
конфигурационного взаимодействия. Результаты «расширенного метода
Хартри — Фока» для трех конфигураций в Be отличаются от ре-
результатов метода Хартри—Фока, дополненного с помощью ме-
метода конфигурационного взаимодействия, приблизительно на
0,03 э$.
§ 24. Атомы 107
дущего ряда. Теперь нединамическая часть функции
^34 для 3Р-состояния приводится к виду
A26)
а 1834I уменьшается примерно до 0,5 эв [100]. Стано-
Становятся ли теперь существенными динамические эф-
эффекты в функции #34, неясно (см. ниже).
Эмпирически полученная функция й5в, т. е.
(/?iap-ia) -корреляция в 3Р-состоянии, составляет по
грубой оценке 0,05 эв [79]. Любой из методов, обсу-
обсуждаемых ниже для Ne, можно использовать для
2/?2-пар в 3Р-, XD- или ^-состояниях.
(ls22s22p3LS: 2D, 2P; Nt Of, F2+, Ne3*
Парные функции этого ряда аналогичны парным
функциям предыдущего ряда. Эмпирически получен-
полученная BрK-кор^еляция [79] (см. фиг. 1) в азоте DS)
примерно равна
г5б + е67 + е75^0,1 эв. A27)
Эффект запрета для электронов в 2р-состоянии
сводит к нулю для ^-состояния нединамическую кор-
корреляцию функции Й34 предыдущего ряда. Эти части
#34 и ?з4 равны нулю в атомах О, F и Ne, поскольку
в них 2px-t 2py-, 2р2-орбитали уже заняты.
Было найдено, что в ряду Be, ... корреляционная
энергия 8з4 линейно зависит от Z [41]. С другой сто-
стороны, выше было показано, что по мере добавления
электронов, т. е. при переходе от Be к Ne, эту линей-
линейную зависимость функции е34 больше ожидать нельзя.
Однако недавние экспериментальные данные [79] все
еще указывают на рост в этих атомах корреляцион-
корреляционных энергий с увеличением Z. Это несоответствие
означает, что либо экспериментальные результаты
нуждаются в пересмотре, возможно в оценках реля-
релятивистских эффектов, либо же динамическая часть
функции й34, которая была мала в Be, становится су-
существенной по мере приближения к Ne (фиг. 4)«,
Этот вопрос изучается Маккоем в лаборатории ав-
автора.
108
§ 24. Атомы
: F-t Ne, Na+, ...
Парные функции для внутренних оболочек и
пары, ответственные за поляризацию остова, можно
взять из предыдущего ряда. Для BрN-состояния
a.z
2.8
2.4
2,0
Ч 1,6
ff.z
0.8
0,4
0
_ у
\ •
\
1
1*1 1 \ J
-
\
N
\
\
\>. о —« -<
till
9 10
Фиг. 4. Влияние эффекта запрета 2/?-электронов на корреля-
корреляционную энергию е34 B$2)-пары (см. § 24).
А —эмпирические Bs2) корреляционные энергии в атомах Be, В, С, N, F
и Ne (на основе данных Клементи [79J). 7 —уменьшение энергии в ионах
Be, B+, С2+, ..., Nee+, обусловленное 2s2 — 2р2-смвшиванием (почти выро-
вырождением) (по Линденбергу и Шуллу [41J); 2—часть e»4 близ вырождения в ато-
мах от Be до Ne. Подмесь 2р* к 2s2 уменьшается из-за эффекта запрета для
электронов, уже занимающих 2/т-орбитали.
необходимо ввести три новые парные функции, соот-
соответствующие (АкОсрхР)-, (р*аруР)- и (рхосрусс) -конфи-
-конфигурациям. Их можно получить методом координаты
ri2 или методом конфигурационного взаимодействия,
используя соотношение (86), но, по-видимому, более
практичным здесь будет метод «незамкнутой обо-
оболочки» или метод «различных орбиталей для разных
§ 25. Малые молекулы 109
спинов» [1]. Расчеты для Ne и других атомов из ука-
указанного ряда делаются в лаборатории автора.
Обсуждение, проведенное выше, показывает, что
с помощью одиннадцати парных функций, шесть из
которых существенны, можно построить весь первый
ряд атомов и ионов [3, 8—11]. Многие из этих пар-
парных функций используются повторно и новые инте-
интеграции выполняются только для дополнительных эф-
эффектов запрета.
Зная функции и^, можно также подсчитать U и R'
[выражение (82)], чтобы оценить ошибки, связанные
с пренебрежением этих величин в нашей теории. От-
Отметим, что метод координаты ri2 годится лишь для
нескольких пар. Нет твердых гарантий, что, исполь-
используя его для всех пар, удастся получить лучшие ре-
результаты. Применение всего лишь нескольких Гц
в функции %'s упрощает [9—11] интегралы, нужные для
вычисления R\ по сравнению с тем случаем, когда
вводятся Гц во все функции u*j [22, 31, 60].
Было бы интересно проверить R'\ например, для
Ne, хотя это, по-видимому, не является больше ре-
решающим моментом, поскольку аддитивность, пред-
предсказанная теорией [выражение (85)], в настоящее
время подтверждена на опыте [79].
Выражение (85) и проведенное выше обсуждение
характерных пар дает некоторое обоснование для
полуэмпирических теорий, применяемых в случае
атомных спектров. Атомные остовы мало подверг
жены влияниям внешних электронов, и межоболочеч-
ную корреляцию можно просто учесть с помощью
эмпирического потенциала остова. Выражение (85)
имеет тот же самый вид, как сама хартри-фоковская
часть энергии [см. A1) и A3)]. Таким образом, эм-
эмпирические параметры F и G [15] включают корреля-
корреляцию.
§ 25. МАЛЫЕ МОЛЕКУЛЫ
Процедура, описанная выше для атомов, приме-
применима также к небольшим молекулам, например к мо-
молекулам LiH, Li2, N2, HF, F2 и т. д. Для них теория
110 § 25. Малые молекулы
основываемся на молекулярных орбиталях хартри*фо«
ковского ^самосогласованного поля {34, 35]; utj —
парные функции для молекулярных орбиталей. Од-
Однако внутренние оболочки свободных атомов можно
использовать непосредственно после того, как сделано
эквивалентное орбитальное преобразование, напри-
например, к Aа^JAаиJ-части Li2 (см. § 27). Эффекты
«запрета» и «среды» для наиболее внутренних обо-
оболочек опять оказываются малыми. Так, например,
ожидается, что эффекты остова для Li2 отличаются
от эффектов остовов для Li или Li+ менее чем на
0,01 эв (см. в табл. 4 изменения при переходе от
Li+ к Li). Это означает, что изменениями в энергиях
корреляции самих остовов можно спокойно прене-
пренебречь, если речь идет об энергиях связи молекул.
Если мы располагаем результатами метода Хар-
три — Фока, то Ёсотт и, следовательно, энергии связи
можно легко оценить, используя выражение (85) для
молекулы и для диссоциированных атомов. Рассма-
Рассматривая образование двухатомной молекулы из ато-
атомов А и В, полагая при этом eij«l-bl,5 эв для два-
дважды занятых пар и пренебрегая всем прочим, полу-
получаем
А + В = АВ, Д?Согг ~ (е/у) Ал,
где {ец) — средняя корреляционная энергия пары,
а А/г — приращение числа дважды занятых пар при
образовании молекулы. Например, на основании та-
таких представлений имеем
2N D5) = N2, A?corr » — 3,9 + 0,6 эв;
2F BР) = F2, A?corr» — 1,3 Т 0,2 эв.
Стантон [101] обсудил полуэмпирические оценки та-
такого рода.
Разделение валентных электронов в (85) и отсут-
отсутствие «кошмара от внутренних оболочек» [102] при
минимизации именно внешних е^ [выражение (86)]
позволяет теперь рассмотреть более тяжелые си-
системы, подобные Na2 и ВаО. Энергии связи и другие
свойства даже таких молекул можно теперь рассчи-
# 26. л-электронные системы 111
тывать путем минимизации энергий е^ валентных
электронов, причем эти энергии комбинируются с их
хартри-фоковскими энергиями. Пробные функции
следовало бы выбирать ортогональными к атомным
хартри-фоковским орбйталям остова.
§ 26. я-ЭЛЕКТРОННЫЕ СИСТЕМЫ
Многоэлектронная теория подтверждает «я-элек-
тронное приближение» [13, 14], упоминавшееся в § 3.
В выражении (85) отделены 2- и П-оболочки. Запи-
Записывая хартри-фоковскую часть энергии [выражение
A3)] и комбинируя ее с корреляционной частью, из
соотношения (85) получаем
N N
A28)
Выражение это имеет тот же вид, что и сама
энергия ?н. f.. Точное выражение для энергии F0)
имело такой же вид. Заметим, что для оправдания
раздельного рассмотрения оболочек величины e*j не-
необходимо представить в расцепленном виде, как это
сделано в A28) и (85). Вместе с тем, в точной энер-
энергии Е [выражение F0)] парные энергии и U\j могли
бы в принципе зависеть весьма сильно друг от друга
и от % и Um и т. д. [ср. B0) и G6) или G7)].
Часть R, связывающая 2- и П-электроны в со-
соотношении (82), будет малой по тем же причинам,
что и в § 18, в частности из-за локализации отдель-
отдельных оболочек. В то же время многоэлектронные чле-
члены /?', возникающие из-за учета самих я-электронов,
должны бы быть малы в основном из-за условия
(87). Было бы интересно проверить значение вели-
величины R', например, для бензола.
Как и в атомных спектрах, выражение A28) по-
поясняет, почему полуэмпирические параметры я-элек-
тронной теории [14] имеют тенденцию включать в себя
корреляцию. При определенных условиях («нулевое
частичное перекрывание») оказывается возможным
112 § 26. л-электронные системы
разложить интегралы /<j в хартри-фоковской части
энергии [выражение A28)] на одно- и двухцентровые
кулоновские интегралы. Большая разница между их
неэмпирическими и полуэмпирическими значениями
объясняется введением [16, 17] корреляции в BpzJ-
состояние. Последнее оправдывается тем, что нулевое
частичное перекрывание можно в равной мере хо-
хорошо провести для ец, сводя их к одно- и двухцен-
тровым кулоновским корреляционным энергиям.
Для я-электронных спектров можно исходить, по
крайней мере формально, из хартри-фбковских выра-
выражений, основанных на «средней энергии конфигура-
конфигурации» [40], учитывая ограничения, разобранные в § 6.
Как указывают также результаты и для атомов, ве-
величины e*j 2-остова, равно как потенциалы поляри-
поляризации остова, не сильно меняются при взаимодей-
взаимодействии с валентными электронами, и потому мы имеем
дело лишь с П-частью выражения A28).
Выражение A28) можно записать в виде [10]:
?'; = ?2-|-?п + еая, A29)
я*= 2 <*.*!/>+ 2 G,, + Зу). A30)
i^1 / > у ^ 1
* > п„ 1>к>п„
•w-S S в;*. A32)
где па и пл — числа а- и я-электронов, Е^ — энер-
энергия остова, включающая корреляционную энергию.
Предполагается, что она не зависит от того, какие
я-орбитали заняты; Еп—энергия П-оболочки в йоле
статического хартри-фоковского остова; еоя—энергия
межоболочечйой корреляции. Ее можно добавить
либо как небольшую поправку при использовании
теории возмущений, либо оценить в адиабатическом
§ 26. п-электронные системы 113
приближении [3], согласно выражению A20). Тогда
*ол~ 2 <*. Vp{xk)k\ A33)
k>na
где /г — я-молекулярные орбитали самосогласован-
самосогласованного поля, a Vp — потенциал поляризации остова, ко-
который определяется лишь свойствами остова; Vp
является одним и тем же независимо от того, на ка-
какое «&» он действует.
Формальное обоснование теории Хюккеля было
дано Симпсоном [103].
Используя формулы (8), A6) и (86), энергию Ец
[выражение A31)], например, для этилена, можно за-
записать в виде
?п<?п = ?п " +
2 (& (fe/)t тнин) + (Пи
1 -f- (ukh uki)
где k, l относятся к двум jt-электронам. Однако для
(€k-\-ei-\-mki) мы имеем [см. A2) и A7)]
Vk-t-Vi — Sk — Si = Vcm(x*) + VeOTe(xt\ A35)
т. е. имеем часть хартри-фоковского потенциала Vi9
зависящую от „остова". Далее запишем [10] функ-
функцию $н в виде
Мы имеем
Это — я-часть, которую нужно минимизировать от-
отдельно при условии {ukh /) = 0, чтобы получить фи.
Отметим, что
ЖГ Г* ± A37)
и является „я-электронным гамильтонианом", кото-
который был ранее постулирован на интуитивной основе
8 О. Синаноглу
114$ 27. Связи — локализация, отдельные пары, ионные остовы
[13, 14]. „Потенциалы поляризации остова" Vр в A33)
можно также включить в 3@Тте [3].
В случае более чем двух я-электронов имеем
п> Фп)
+ 2) ... пП) + Хп, A39)
п ' A40>
k > na k > I
Функция Хп Дается в виде G7); однако она может
также включать многоэлектронные корреляции 0цк,
Uijki> • ¦ •» как и точная функция % [выражение B0 а)]
(например, если мы имеем дело с молекулой почти
«металлического» красителя).
§ 27. СВЯЗИ-ЛОКАЛИЗАЦИЯ, ОТДЕЛЬНЫЕ ПАРЫ,
ИОННЫЕ ОСТОВЫ
Изложенная до сих пор теория основывалась на
молекулярно-орбитальном описании, в котором ис-
использовались функции йц для молекулярно-орбиталь-
ных пар, даже если мы имели дело с большими на-
насыщенными молекулами с сильно локализованными
электронами. В такой теории оболочки разделяются
на внутренние и внешние. Однако для насыщенных
молекул мы еще должны рассмотреть, каким обра-
образом возникают локализованные связи и отдельные
пары, почему энергии связи совершенно не зависят
от молекулярного окружения и какие силы действуют
между связями. К счастью, нет необходимости за-
заново начинать рассмотрение таких систем с помощью
другой теории. Простое преобразование переводит
молекулярные орбитали, на которых основывается
многоэлектронная теория, в локализованные орби-
орбитали.
В этой задаче полная функция и полная энергия
также имеют орбитальную и корреляционную части-.
§ 27. Связи — локализация, отдельные пары, ионные остовы 115
Орбитальная часть была получена Леннардом-Джон-
сом [42, 46, 47], показавшим, что в определителе ф0>
построенном на хартри-фоковских молекулярных ор-
биталях самосогласованного поля, электроны уже ло-
локализованы относительно друг друга (например,
в СН4, Н2О, Ne, ...), как упоминалось в § 7. Это рас-
распределение электронов относительно друг друга
лучше всего описывать с помощью унитарного «экви-
«эквивалентного» или «локализованного» орбитального
преобразования t, которое не меняет ф0:
N
2 kvr\v, A41a)
где А = 1, 2, 3, ..., N — хартри-фоковские молеку-
молекулярные орбитали самосогласованного поля со спи-
спином, t]v—новые локализованные спин-орбитали,.
a {lvk} — матрица, эрмитово сопряженная матрице!.
Отметим, что выражение A416) в действитель-
действительности содержит два независимых преобразования:
одно для нечетных k, т. е. а-спин-орбиталей, а дру-
другое для четных k, т. е. р-спин-орбиталей.
Если взять
то Ео [выражение (И)] и Ег [выражение A3)] сохра-
сохраняют свой вид
?о= 2 %, A43)
v>l
?1 = -S {7Pv-<v}, A44)
Р> V
однако формула A2) больше не применима. Вместо
нее имеем
A45)
Vv — ev,
116 I' Sf, Связи ~~ локализация, отдельные пары, ионные остовы
где Yvp—-недйагойальные хартри-фоковские орбиталь-
орбитальные энергии, а ?у —диагональная орбитальная
энергий; hi и Vv — те же самые величины, что и
раньше [определяются формулами Dа) и (8), но х*
заменено на xv]!).
В СН* эквивалентные С—Н-орбитали [46—48]
определяются при помощи условий симметрии. Для
других молекул с меньшей симметрией, например для
молекул НгО, СгНв'и т. д. преобразование t можно
определить как преобразование, минимизирующее
величины обменных членов [49, 50, 57] в A44), или
же с помощью аналогичных критериев [51].
Локализованные орбитали были подробно изу-
изучены Поплом [43], Линнетом [44] и Холлом [50]. Со-
Совсем недавно Рюденберг и Эдмистон развили мето-
методику для их расчета, основанную на критерии «ми-
«минимального обмена» [49, 50].
Холл [50] скоррелировал потенциалы ионизации
углеводородов от этана до декана, полагая неизмен-
неизменными локализованные орбитали и энергии ev и ypv#
Потенциалы ионизации в небольшой степени под-
подвержены воздействию корреляции электронов («тео-
(«теорема Купмена» [1]). Однако корреляция весьма важ-
важна для теплот образования и конфигурационных из-
изменений [2].
Харли, Леннард-Джонс и Попл [23] ввели корре-
корреляцию в локализованных группах с помощью подо-
подобранной для этого случая волновой функции
Л{АА(р, v)AB(X,r) ...}, A46)
и произвольного условия A). Ограничения этого
приближения [24—26] обсуждаются после формулы
A) в § 4 и в ряде работ [3, 10, 23, 27—31] (см. при-
1) Инвариантность всех операторов в Л|** при преобразо-
преобразовании t очевидна, исключая, быть может, потенциал Vu который
зависит от орбиталей. Потенциал Vi [выражение (8)] можно
выразить через матрицу плотности р(хг, Xj) Фока—Дирака [1].
Суммирование в p(xf, Xj) проводится по набору молекулярных
орбиталей, которые образуют базис для определенных непри-
неприводимых представлений. Такая запись инвариантна при любом
унитарном преобразовании [46, 47].
§ 27. Связи — локализация, отдельные пары, ионные остовы 117
мечание 29 в работе [10]). Помимо остальных огра-
ограничений, выражение A46) не позволяет переходить
от локальных свойств в основном состоянии к свой-
свойствам делокализованных электронов, например при
рассмотрении электронных спектров.
К счастью, нет необходимости использовать раз-
различные теории для локализованных групп и для де-
делокализованных электронов. Оба случая охваты-
охватываются одной и той же «многоэлектронной теорией».
Необходимо лишь выяснить, каким образом преоб-
преобразуются [104] функции % и ?согг при действии того
же самого оператора t, который преобразует функ-
функцию ^о. Зная это, можно переходить от делокализо-
ванного описания к локализованному, и наоборот [12].
А. Преобразование точной энергии
Точная энергия определяется выражением F0)
через молекулярные спин-орбитали /, /. Чтобы
усмотреть, каким образом преобразуется корреля-
корреляционная энергия, если преобразование, заданное
A41а), применено к хартри-фоковским молекуляр-
молекулярным орбиталям, подставим
N
<$(ij)= 2 tivtjQ&ОуПр) A47)
v> р
в выражение F0)
где
(pfipk) = Q (k=l, 2, 3, ..., /, у, ..., N). (бО'б)
Меняя порядок суммирования и замечая, что опе-
оператор одинаков для всех электронов, получаем
N
Е = ЕН.?.+ S (^OV1P)' ?vP!%>> О48)
v>p
'r'ih A49)
118 § 27. Связи — локализация, отдельные пары, ионные остовы
Кроме того, имеем
<jlvpf *) = 0, J150)
где k — все еще молекулярные спин-орбитали, jo,vp—
корреляционные волновые функции между локализо-
локализованными спин-орбиталями. Они также локализо-
локализованы. Данная функция jo,vp, если v и р отличаются
лишь своим спином, является корреляционной вол-
волновой функцией для этой связи, отдельной пары или
пары внутренней оболочки, подобной (Is2) в Li2.
С другой стороны, если v и р относятся к локализо-
локализованным орбиталям с различными пространственными
частями (межоболочечная корреляция), соответ-
соответствующая функция jlxvp определяет вандерваальсово
притяжение между локализованными орбиталями от-
отдельных групп.
Когда в распоряжении имеются молекулярно-ор-
битальные парные функции Vih скажем, в виде раз-
разложений по методу конфигурационного взаимодей-
взаимодействия или в виде зависимости от ri2 и т. д., формула
A49) дает немедленно корреляции в локализованном
описании. Например, формула A49) могла бы быть
применена к результатам Несбета [48] для молекулы
СН4 (метод хартри-фоковских молекулярных орбита-
лей, дополненный методом конфигурационного взаи-
взаимодействия). Тем самым можно было бы извлечь
из этих результатов корреляционную энергию С—Н-
связи и «вандерваальсово притяжение» между двумя
различными С—Н-связями.
Выражение A49) показывает, что энергия ЕСОтг
насыщенной молекулы в точности равна сумме кор-
корреляционных энергий связей, отдельных пар и ион-
ионного остова и внутримолекулярного вандерваальсова
притяжения между этими группами. Результат этот
точен настолько же, насколько справедливо само
уравнение Шредингера, однако пока он чисто фор-
формальный. Точные функции |ivp, которые входят в вы-
выражение A48), сопоставляются с точной функцией %
[выражение B0)] таким же образом, как это было
сделано для 0ц [выражение F0)]; в принципе они
могут зависеть от всех других членов функции %.
§ 27. Связи — локализация, отдельные пары, ионные остовы 119
Остаются следующие вопросы:
1) каким образом можно непосредственно полу-
получить функции fxvp (поскольку это было бы более
удобно, нежели сначала находить функции u<j)?
2) Какие подразумеваются эффекты окружения в
juivp и локализованных корреляционных энергиях?
?vp?vp>- О51)
Б. Преобразования приближенных выражений
для энергии
Выражения для энергии, даваемые теорией воз-
возмущений и многоэлектронной теорией, имеют один и
тот же вид F0').
Если учесть парную функцию первого порядка
#№, удовлетворяющую G0), то соотношение F9)
даст нам следующее выражение для энергии с точ-
точностью до второго порядка:
Е « Ей. ?. + Е2 = ?н. f. + .2 {Я Ш gift)). A52а)
В многоэлектронной теории функция г'., переходит
в Eij [выражение A01)], как только каждая из энер-
энергий е*7 [выражение (86)] минимизирована так, что
пары «во всех порядках» удовлетворяют уравнению
A00), при этом (85) вновь принимает вид
Е = Ен. f. + ^2. {ЯШ gij»ij)- A53a)
Отметим, что выражении F0), A52а) и A53а) выгля-
выглядят одинаково. Однако в каждом из них парная кор-
корреляционная функция uij различна. Выражение F0)
получено из точной функции %> а в выражениях A52а)
и A53а) подразумевается, что пары являются несвя-
несвязанными между собой и соответствующие парные
корреляционные функции не зависят явно от ]г или
ОТ 0щ, Uijkl И Т. Д,
13Q § 27. Связи-— локализация, отдельные пары, ионные остовы
Так как формы этих выражений одинаковы, то
преобразования A47) и A49) с равным основанием
применимы % A52а) и A53а). Результирующие выра-
выражения, подобны A48), но содержат теперь либо кор-
корреляционную функцию первого порядка ji^» либо
«точные» (бее порядки) парные корреляционные
функции jxvp «многоэлектронной теории». Имеем
Д ty A526)
N
E°s = En. f. + 2 {Я (T|v4P), gvp^vp). A536)
{ |v4P)
v>p
Различие между fxvp в выражении A536) и |ягр в вы-
выражении A48) аналогично тому, которое имеет ме-
место между функцией u*j и функцией 0ц, полученной
из полной точной функции %.
В. Преобразования вариационных энергий
Выражения A52а) и A53а) в предыдущем разделе
справедливы только для таких парных корреляцион-
корреляционных функций, которые удовлетворяют отвечающим
им эффективным уравнениям Шредингера G0) или
A00а). Чтобы получить парные корреляционные
функции tiij в случае, когда исходят из молекуляр-
молекулярных орбиталей, необходимо минимизировать вариа-
вариационные выражения вида (86). Выражения A526) и
A536) имеют силу лишь для оптимальных функций
jlvp. Чтобы найти их непосредственно, необходимо
располагать вариационным принципом для каж-
каждого |ivp.
Пригодность концепции «энергии связи» указы-
указывает на то, что, когда рассматривается большая мо-
молекула, гораздо легче получить непосредственно те
или иные jlvp для корреляций локализованного и
вандерваальсова типа, чем получить вначале йц. Это
обстоятельство имеет большую практическую цен-
ценность. Чтобы рассчитать, к примеру, a priori энергию
§ 27. Связи <~- локализация, отдельны* пары, ионный остовы 121
S—Н-связи, не нужно получать молекулярные орби-
тали, скажем, из рассмотрения всей молекулы про-
протеина или полностью проводить вначале расчеты по
методу Хартри —Фока, дополненному методом кон-
конфигурационного взаимодействия.
Чтобы понять, каким образом ^ можно непо-
непосредственно подсчитать, обратимся вначале к теории
возмущений. Вариационная форма для выражения
A52а) [см. F3), F4) и "A03)] имеет вид
Ен. f. Ч~ E<i ^ ?н. f. Ч~ Еъ% A54а)
?2= 2 г% A546)
>, AMb)
А> = 0 (Л=- 1, 2, 3 ifj, ..., N).
A54г)
Функции uffj являются пробными функциями для по-
получения парных корреляционных функций первого
порядка в молекулярном орбитальном описании. Они
стремятся к u[l)J9 когда ej[2j->efj.
Первая сумма в Е2 — та же самая, что и в A52а);
она преобразуется путем подстановки A47) и A49).
Величина ei^rei содержит [см. выражения G) и A2)
и примечание на стр. 116] одну и ту же часть для
всех электронов:
Следовательно* обращение соотношения A49) дает
1J
N
Отметим, что изменено лишь обозначение перемен-
переменных суммирования в Л?*** Во всех других отноше*
9 О. Синаноглу
122 § 27. Связи — локализация, отдельные пары, ионные остовы
ниях величины Л?фф и Л*фф совершенно аналогичны;
они обе включают в себя потенциал Vt всей моле-
молекулы, определяемый формулой (8) с заменой спин-
орбиталей молекулярными орбиталями.
Величина e^ej содержит часть энергии (е^ + ву),
которую можно вычислить при помощи молекуляр-
молекулярных орбиталей; она различается для каждой пары
орбиталей; так, используя преобразование A49)
дважды (и слева и справа), получаем
N
N N [ N
v > р а > т \ l> j
Тильда над а и \i дл5
выражение A54) дает
еB) = 2 (<%? (r|vr|), gv uW\
N N
д<2) — 2 S
v, p=^a, т
В выражении A56)
и|47|
i простоты опущена.
N
v>p Vp
\«VOT ' • pt/ \^vp' r~Ox/'
мы использовали
tl=l
•>• 056)
В итоге
A57а)
A576)
A57в)
A58а)
A586)
где г — матрица орбитальных энергий, вычисляемых
при помощи молекулярных орбиталей, av — матрица
орбитальных энергий локализованных состояний
[см. A43) и A45)]. В выражении A576)
Yvv = ev. A58в)
Теперь в соответствии с методикой вариацион-
вариационного приближения, использующего теорию возму-
возмущений, необходимо было бы минимизировать каждую
§ 27. Связи — локализация, отдельные пары, ионные остовы 123
***
функцию е^, взяв пробные функции м$ и потребо-
потребовав, чтобы
<Й, *> = 0 (А = 1, 2, 3, ..., N), A57г)
где k — все еще полные молекулярные орбитали. Эта
процедура и эффекты окружения, которые внесены
орбиталями k и полной хартри-фоковской функцией
Фо в потенциалы Vv в A576), будут обсуждаться в
следующем разделе.
Члены Д<2> легко оцениваются. Они намного мень-
меньше, чем член 2 812п> так что применимо вариацион-
ное приближение с использованием теории возмуще-
возмущений (см. § 14). Те же самые величины м^р» которые
найдены из A576), можно использовать, чтобы под-
подсчитать Д<2).
Холл [50] получил эмпирические значения для не-
недиагональных хартри-фоковских энергий yVQ от угле-
углеводородов метана до декана. Применяя выражение
A49) к несбетовой [48] функции для СН4, представ-
представляющей собой хартри-фоковскую молекулярную ор-
биталь, исправленную с помощью метода конфигу-
конфигурационного взаимодействия, получаем [105] грубую
оценку для (?$М&). При Yvv=^—13,75 величина
Ypv= — 1,75 эв; в результате для метана получаем
эв, <158г)
а для полной корреляционной энергии мы имели
N
*** vp
v>p
Величина —7 эв является эмпирической оценкой
для полной корреляционной энергии СН4; она не рав-
равна величине, полученной Несбетом [48] и составляю-
составляющей 1,95 эв.
Если ограничиваться ?2> то функцию \xvp можно
аппроксимировать функцией |лЯ>, полученной мини-
минимизацией A576),
9*
124 § 27. Связи — локализация, отдельные пары, ионные остовы
Рассмотрим теперь полные верхние пределы, да-
даваемые для Е волновыми функциями вида F9а) и
G6). Энергия [выражение F5)], полученная с по-
помощью хь содержит ?3. Выражение F5), записанное
более детально, имеет ту же самую форму, что и Е8
[соотношение G8)], однако оно содержит функции
#(Д\ полученные минимизацией меньшей доли от e*j
[выражение A54в)]. Далее, сравнение F5), A54в) и
G8) показывает, что Ег включает поправки третьего
порядка к парным функциям, а также трехэлектрон-
ные члены
J A59)
где /?<3> определяется формулами G9) — (81), однако
там вместо функций йг-;- надо подставить иЭД.
Полная функция %t не меняется при преобразова-
преобразовании к локализованным орбиталям [выражение A41)],
поскольку величины, входящие в F4), все еще инва-
инвариантны при таком преобразовании. Таким образом,
значения ?2 и ?3 по отдельности остаются неизмен-
неизменными. С другой стороны, ожидается возрастание
вклада пар в Es до сравнению с трехэлектронными
корреляциями RW [Ю]. Локализованные пары (кото-
(которые, однако, не обязательно совпадают с вандерва-
альсовыми членами) должны быть в большей мере
расцепленными (независимыми друг от друга) по
сравнению с тем, что имело место для парных кор-
корреляционных функций, основанных на молекулярных
орбиталях. Тот же аргумент [10] применяют к Е8 и
Es [выражение (82)]. Из этого следует, что лучшая
энергия и волновая функция должны бы получаться
путем минимизации большей части энергии, содер-
содержащей вариационные энергии локализованных пар
во «всех порядках» (т. е. включая (|Ivp, /rcvpM<Vp)), a
не до второго порядка, как в A576). Полученные в
результате этого функции \ivp должны бы были дать
меньшие значения для остаточных членов в R.
§ 27. Связи — локализация, отдельные пары, ионные остовы 125
Подробности таких преобразований, применяемых
в полной форме (82), пока еще в стадии изучения
[105]. По аналогии с (86) и из рассмотрения влияний
окружения на связь, влияния ионных остовов и т. д.
можно предположить, что вариационное выражение
для локализованных пар во «всех порядках» дается
приведенным ниже выражением A62). В любом слу-
случае может оказаться достаточным учет только Е2, так
что можно работать с выражением A57).
Г. Влияние молекулярного окружения на связь
Экспериментальные энергии связи мало меняются
от молекулы к молекуле, например в насыщенных уг-
углеводородах эти изменения лежат в пределах 0,01 эв
[2]. Возникает вопрос о том, совместимы ли эффекты
молекулярного окружения, подразумеваемые в дан-
данных выше выражениях с такого рода постоянством
энергий связи.
Проблему молекулярного окружения можно под-
подразделить на части: молекулярное окружение в хар-
три-фоковском приближении и корреляционная часть.
Локализованные орбитали из A45) подвержены
действию потенциала всего хартри-фоковского «фо-
«фона» Vv, т. е. потенциала молекулы; кроме того, на
них оказывает влияние принцип исключения, приво-
приводящий к тому, что они выбираются ортогональными
ко всем другим riv, а также приводящий к обменным
членам. Потенциал l/v, включающий притяжения ядер,
вызывает поляризацию определенных групп у отри-
отрицательных ионов. Обмен и ортогональность приводят
к несвязывающим отталкиваниям [106, 107]. И эти
два последних эффекта и потенциал Vv вследствие
экранировки ядер имеют малый радиус действия. Та-
Таким образом, на орбитали r\v в основном воздействует
их непосредственное окружение. Эта орбитальная
часть проблемы была исследована [42—44, 49, 50],
хотя, по-видимому, необходимо провести еще значи-
значительную работу, чтобы рассмотреть, каким образом
локализованы i\v и насколько они постоянны.
126 § 27. Связи — локализация, отдельные пары, ионные остовы
Корреляционная часть функции связи, назовем ее
Д$ (v и р имеют те же самые пространственные ча-
части), также подвержена действию полного молеку-
молекулярного хартри-фоковского «фона» фо, хотя \ivp в
A576) являются почти несвязанными друг с другом.
Как и в случае атома (см. § 24), влияние окру-
окружения на M<vp через фо проявляется двояким обра-
образом:
1. Потенциал Vv, входящий в формулы
(^%^ Ух-гч, A2')
Vv(xv) = Д {k, gkv(\-Pkv)kL = 2S*(v), (8')
включает в себя кулоновский и обменный потенциалы
всех k молекулярных орбиталей. Однако, применяя
A41а), потенциал Vv можно записать через кулонов-
кулоновский и обменный потенциалы для t)v. Далее видно,
что действие Z/rIVj например С—Н-связи (Лтат1тР) в
в этане, экранировано кулоновской частью потен-
потенциала ST(v) этой связи. Этот экранированный потен-
потенциал обращается в нуль на расстоянии до С—Н-свя-
зей других метальных групп. Несомненно, С—Н-связь
весьма слабо экранирована С—С электронами от за-
заряда своего собственного атома углерода; однако в
§ 24 мы упоминали, насколько нечувствительны «ди-
«динамические» корреляции к изменениям заряда ядра.
Судя по Не, Li+, Be2+ (см. табл. 4) и значениям кор-
корреляции Н2 [1], следует ожидать, что влияние этих
эффектов окружения (Vv — Z/r)-типа, скажем, на
С—Н-связь будет не больше 0,01 эв, т. е. в пределах
точности экспериментальных энергий связи.
2. Функции fjtvp ортогональны к полным молеку-
молекулярным орбиталям k [выражение A57г)]. Согласно
A41а), это ведет к следующим результатам:
A60)
§ 27. Связи—-локализация, отдельные пары, ионные остовы 127
Таким образом, как и парная корреляционная функ*
ция (§ 10), функция jxvp ортогональна не только
к своим собственным t]v, rip (аналогично влиянию ft),
но также ко всем другим функциям tj0 (o=f=v, p). По-
Последний «эффект запрета» теперь представляет дру-
другой тип влияния окружения. Можно ожидать, что
влияние его на С—Н-связи меньше 0,01 эв. Даже в
атоме лития (см. табл. 4), где электроны располо-
расположены концентрически и, следовательно, сильнее за-
запрет, 25-ортогональность изменяет ei2 для Li+ лишь
примерно на 0,004 эв. Случай остова Li2c (logJ(louJ'
конфигурацией (см. § 25) аналогичен. Соотношения
A416) переводят эту конфигурацию в (lsaJ(lSbJ,
как в случае Не—Не, рассмотренном ниже. Энергии
evp этих локализованных остовов должны быть по-
подобны ei2 для Li+ (—1,18 эв) [1] и располагаться, ве-
вероятно, внутри интервала 0,01-5-0,02 эв.
Из этих рассмотрений, по-видимому, следует, что
влияние окружения во.всяком случае, пока речь идет
о корреляционной части, находится в пределах точ-
точности- экспериментальных энергий связи. Корреля-
Корреляционные энергии отдельных пар и ионных остовов
также, по-видимому, не меняются в пределах указан*
ной выше точности (~0,01-f-0,02 эв).
Д. Расчет энергии связей
Так как эффекты окружения весьма малы, корре-
корреляционные энергии связей или отдельных пар можно
подсчитать порознь, как это делалось, например, для
молекулы Н2.
При расчетах с точностью до второго порядка ми-
минимизируем A576), однако в выражении для ?v за-
заменяем 2 ^i/riv и ^v соответствующими частями из
этой связи. Таким образом, вместо A57г) можно на-
написать
Й<Ит> = 0 (* = v, р), A61)
где v и р различаются лишь своими спинами.
128 § 27. Связи — локализация, отдельные пары, ионные остовы
По аналогии с уравнением (86) можно предполо-
предположить, что во всех порядках
IЧр>» A62>
где функция 5vp в /rcvp включает в себя лишь потен-
потенциалы, вычисленные с помощью r\v и г]р. Однако,
чтобы подтвердить это выражение, если не удовле-
удовлетворится A576), которое сейчас представляется до-
достаточно обоснованным, необходимо дальнейшее ис-
исследование.
Пробные функции ^ipv могут зависеть от коорди-
координат ri2 аналогично функции Джеймса — Кулиджа
[108, 92] для Нг. При использовании таких функций
часть 38 (ту1р) расщепляется вначале в соответствии
с A04) —A06)..
Чтобы рассчитать a priori энергию связи, необхо-
необходимо обратиться к хартри-фоковской части именно
этой связи. Метод Хартри — Фока для локализован-
локализованных групп находится пока еще в стадии развития
[52, 53].
Как следует из A2), A43) и A44), вариационная
форма Ел, f. имеет вид
?н. F. < Ён. F. =
N
+ 2 {я йЛ). gvp^fivi)}. A63)
>p
v>p
а пробные орбитали r]v подчинены следующему тре-
требованию:
(ii)=V A64)
Снова комбинируя члены экранировки и пренебрегая
взаимодействиями между группами, получаем
N/2
Ён. f. « 2 <Я (W), (Ag + А°. Я
§ 28, Вандерваальсовы притяжения 129
где Р и ($' отличаются лишь спинами, hi и А^ —
оставшиеся части А?фф именно для этой связи.
Вероятно, можно рассмотреть хартри-фоковскую про-
проблему двухцентровой связи при помощи минимиза-
минимизации отдельной пары в A65), сохраняя ортогональ-
ортогональность функции Tjo к внутренним оболочкам и выби-
выбирая для С—Н потенциал остова Л° углеродного иона.
Этот потенциал затем можно улучшить, используя
функцию Sv из, скажем, связанной орбитали С—С
(например в СгНе); или же для расчета С—С-связи
в СгНв, СзНв и т. д. можно взять С—Н часть из СН4.
Необходимо рассмотреть, насколько практически вы-
выполнимы и хорошо определены подобные действия.
Здесь они упомянуты лишь как возможные.
Если A65) является работающей формулой, то ее
можно скомбинировать с корреляционной частью и
получить
причем
ФЭр, = SB (ripV) + ?«*' • Феэ'' V = °-
Хотя в настоящее время мы располагаем теорией
корреляционной части, для самих расчетов полной
энергии связи из выражений вида A66) требуется
дальнейшее развитие молекулярной (локализован-
(локализованной) теории Хартри — Фока.
§ 28. ВАНДЕРВААЛЬСОВЫ ПРИТЯЖЕНИЯ
Общепринято рассматривать вопрос о вандерва-
альсовых притяжениях между двумя атомами, на-
например Не ... Не, с помощью теории Гайтлера —
Лондона. Притяжение при минимальном расстоянии
между атомами отчасти обусловлено искажением,
гайтлер-лондоновских атомных орбиталей (эффектом
«орбитальной средней поляризации») [3], а при боль-
больших мёжъядерных расстояниях г оно возникает в ос-
основном из-за одновременных возбуждений в обоих
ISO § 28. Вандерваальсовы притяжения
атомах (т. е» из-за обычных лондоновских дисперсион-
дисперсионных сил). Эти два эффекта сравнивались [109] для
Н2 в триплетном состоянии при различных значе-
значениях п
Эффект искажения для системы взаимодействую-
взаимодействующих атомов автоматически учитывается в хартри-фо-
ковских расчетах, основывающихся на молекулярных
орбиталях самосогласованного поля. Расчет Ранзила
[110] с помощью «наилучшим образом ограниченных»
хартри-фоковских молекулярных орбиталей самосо-
самосогласованного поля хорошо передавал взаимодействие
Не ... Не, если межатомное расстояние бралось рав-
равным приблизительно ге (т. е. было минимальным) >
однако он приводил к слишком малому притяжению
для г/ге больших, чем примерно 1,2*). Притяжение
при больших г является «межоболочечным» корреля-
корреляционным эффектом. Этот эффект до сих пор рассма-
рассматривали с помощью теории лондоновских дисперсион-
дисперсионных сил. Его считали ответственным за несвязывающие
притяжения между различными частями насыщенной
молекулы для того, чтобы получить теплоты образо-
образования и изомеризации [2].
Теория Лондона использует второй порядок тео-
теории возмущений в ее обычном (рэлей-шредингеров-
ском) варианте, т. е. в виде бесконечной суммы по
полному базисному набору. Этот набор для системы
из двух взаимодействующих молекул а и b состоит
из всех произведений 'ф^ собственных функций для
систем а и Ъ по отдельности. Таким образом, в тео-
теории Лондона предполагается не только отсутствие
перекрывания между основными состояниями ато-
атомов, но также и отсутствие перекрывания между лю-
любыми виртуальными атомными возбужденными со-
состояниями [3].
Рассмотрим, например, два атома водорода, имею-
имеющие одинаковые спины и расположенные на расстоя-
1) Однако оказалось, что этот расчет дал случайные ре-
результаты; возможно, что использование только одного метода
Хартри—Фока не приведет к минимуму потенциала. Более точ-
точные хартри-фоковские расчеты выполняются в настоящее время
Рутаном и сотр. в Чикагском университет^»
§ 28. Вандерваальсовы притяжения 131
нии ЗА [12J. При таком разделении атомов пере-
перекрывание между орбиталями основных (lsa) (ls&)-со-
(ls&)-состояний фактически пренебрежимо мало. Однако
Боровский радиус состояния пропорционален п2, так
что даже следующее виртуальное возбужденное
Bрг)аBр2)ь-состояние, которое должно бы давать
вклад в лондоновские силы, обнаруживает большое
перекрывание. Следовательно, непосредственное рас-
рассмотрение не подтверждает применимость лондонов-
ской формулы даже для вполне разумных межъядер-
межъядерных расстояний г. Таким образом, использование этой
формулы применительно к внутренним оболочкам мо-
молекулы также требует подтверждения [3, 104].
Экспериментальное значение коэффициента при
члене с г~6 меньше (более отрицательно) [2], чем его
лондоновское значение на множитель 2,2 для Ne, Ar,
Кг и Хе, но оно отличается лишь на множитель 1,2
для Не и 1,09 для Н2. Донаф и Питцер [2] приписали
это корреляциям более высокого порядка, нежели
второй, которые становятся существенными в боль-
больших атомах (см. ниже).
Мак Вини [30] распространил применение группо-
групповых функций [выражение A46)] на межмолекулярные
силы, добавляя к волновым функциям, рассчитанным
во втором порядке, набор многоэлектронных функций
Ла, Лв для каждого атома А и В соответственно.
Функции, отвечающие атому А, предполагались ор-
ортогональными в смысле уравнения A) не только друг
к другу, но также и к функциям, отвечающим ато-
атому В. Нет рецептов для получения этих функций. Та-
Таким образом, слишком жесткое требование A) отно-
относительно ортогональности между группами функций
А и В эквивалентно [3] постулату отсутствия пере-
перекрывания в теории Лондона. Эти ограничения изуча-
изучались применительно к взаимодействию Не... Не [29].
Многоэлектронная теория [3, 104] дает вандер-
вандерваальсовы притяжения для всех г, как для межмо-
межмолекулярных, так и для внутримолекулярных случаев,
без предположений относительно перекрывания. Нет
необходимости различать исходные приближения или
же строить различные теории: одну для больших г,
132 § 28. Вандерваальсовы притяЬюения
другую для f, близких ге. Многоэлектронная теория
хорошо покрывает трудную [111] промежуточную об-
область.
А. Внутримолекулярные притяжения
Члены evp и ?vp с v и р, относящимися к про-
пространственно различным локализованным спин-орби-
талям, существенны для межгрупповых корреляций.
Они описывают обобщенные притяжения Лондона —
ван дер Ваальса. Однако jxvp представлено в замкну-
замкнутом виде, не разложенном по атомюым базисным на-
наборам.
Соотношения A48) и A49) определяют вандер-
вандерваальсовы притяжения между локализованными груп-
группами при использовании любой функции ^о+Х
(такой, как хартри-фоковская, или из метода конфи-
конфигурационного взаимодействия) в молекулярно-орби-
тальном описании. На этом пути, т. е. при переходе
к локализованному описанию, можно получить, напри-
например, корреляционную энергию между двумя С—Н-свя-
зями в СН4.
Членов второго порядка Е2 [выражение A526)] ока-
оказывается достаточно, чтобы рассчитать, скажем, энер-
энергию изомеризации с притяжениями между связями,
например (С—Н), (С—Н) и т. д. Каждая группа
имеет всего несколько электронов, как в случае взаи-
взаимодействия Не ... Не, и поправки высших порядков,
упоминавшиеся выше [2], например, для Ne...Ne
здесь не нужны. Использование лондоновских сил в
углеводородах [2] можно обосновать [3] с помощью
выражения A526) по крайней мере до того, как мы
коснемся проблемы перекрывания базисных функций
и эффектов запрета.
Вандерваальсовы члены можно получить отдельно
без проведения расчета для полной молекулы. Меж-
Межгрупповые пары нужно было бы минимизировать в
A576), используя пробные функции jivp. Связь ме-
между различными nvp, которые нужны, чтобы функ-
§ 28. Вандерваальсовы притяжения 133
ция % обладала соответствующей симметрией (как
обсуждалось для атомов в § 23), нуждается в даль-
дальнейшем исследовании [105].
Индивидуальные молекулярно-орбитальные пар-
парные корреляции гц можно сопоставить с локализо-
локализованными корреляциями evp, прямо применяя выра-
выражение A49). Этим можно обосновать полуэмпириче-
полуэмпирический метод, с помощью которого из легко оценивае-
оцениваемых локализованных корреляций получаются моле-
молекулярно-орбитальные парные корреляции. Подобный
метод может оказаться полезным в расчете ультра-
ультрафиолетовых спектральных распределений, основанном
на простой теории молекулярных орбиталей, и дать
оценки сдвигов, обусловленных корреляциями. Было
бы практически интересно рассмотреть п — л*-пере-
ходы.
Б. Межмолекулярные притяжения
При рассмотрении межмолекулярных сил системы
взаимодействующих молекул многоэлектронная тео-
теория вновь исходит из хартри-фоковских молекуляр-
молекулярных орбиталей самосогласованного поля. В Не... Не
[ПО] хартри-фоковская часть объясняет постепенное
искажение атомных орбиталей ls?, ls?, lsg, ls? и за-
замену их эквивалентными орбиталям-и (rji, ri2, Цз, щ)
при меньших г. Ранзиловские [НО] хартри-фоковские
функции фо, будучи преобразованы [57] к различным г,
позволяют получить эти орбитали. Поведение моле-
молекулы N2 вплоть до диссоциации Несбет рассмотрел
с помощью метода Хартри — Фока, непосредственно
используя в нем эквивалентные орбитали [5].
То же самое преобразование к эквивалентным ор-
биталям, которое применимо к функции фо, пригодно
также для корреляционных частей [выражения G7)
и A53а)] волновой функции и энергии и приводит
к обобщенным лондон-вандерваальсовым членам
при всех г. Последние являются теми парными функ-
функциями в A526), которые имеют две локализованные
орбитали riv и т]р с пространственными частями,
134 § 2В. Вандерваальсовы притяжения
относящимися к двум различным молекулам. Обозна-
Обозначая сумму всех таких межмолекулярных пар через
?*?, имеем
067)
где со означает межмолекулярную вандерваальсову
часть; атом А содержит па электронов, а атом В — пьл
причем Па+пь—N, так что в выражении A67) имеет-
имеется папь членов. Даже на малых расстояниях г мы
можем говорить об индивидуальных «молекулах» или
«атомах», если будем рассматривать локализованные
орбитали как искаженные формы орбиталей отдель-
отдельных атомов или молекул.
В молекулярно-орбитальном парном описании вну-
внутриатомные корреляции не отделены от межатомных
корреляций. Следовательно, при расчетах по методу
Хартри — Фока, дополненному методом конфигура-
конфигурационного взаимодействия, или при расчетах йг-;- боль-
большая часть усилий тратится на получение больших
внутриатомных корреляций1). При описании с по-
помощью эквивалентных орбиталей рассматриваются
как раз межатомные пары [выражение A67)] с при-
применением A576) [Е$ мало по сравнению с полной
величиной Е2 и поэтому необходимо рассчитать также
Д<2) [выражение A57в)]. Внутриатомные корреляции
(как в Не) не чувствительны к детальному виду атом-
атомных орбиталей и потенциалам V{. Следовательно,
даже при меньших г атомные орбитали искажаются
в орбитали т). Полагают, что они сократят чисто
атомные вклады для г= оо.
Межатомные парные функции в A67), отвечаю-
отвечающие парам с одинаковыми спинами, можно иногда
1) Это было обнаружено Моффитом [112] и привело его к
теории «атомов в молекулах». Он пытался получить даже кор-
корреляции валентных оболочек свободных атомов. Поскольку ва»
лентные электроны в молекуле не сохраняют свой атомный
характер, идея эмпирических внутриатомных корреляционных по-
поправок должным образом применяется лишь к внут-ренним обо*
лочкам (см. также § 25),
§ 28. Вандерваальсовы притяжения 135
связать с определенной одной или несколькими пар-
парными функциями йц. Например, в Не...Не
«13 = — Даз A68а)
соответственно
Я A(ух1(ух) = — Я (lsaalsba) A686)
(rj->ls, когда R~+oo). Это обусловлено тем, что в
выражении A41) а спин-орбитали преобразуются не-
независимо от р спин-орбиталей. Особенно прост Не...
... Не случай, в котором не будет внутриатомных пар
аа. Расчет функций ei3 и 824 в таком случае совпа-
совпадает с расчетом корреляционной энергии Н2 в ^-со-
^-состоянии [109]. При больших г, пренебрегая разницей
между оса и ар парами, имеем
Е? « 4ei3. A69)
Использование йц исключает проблему перекрыва-
перекрывания.
Уже применение ?f дает вандерваальсовы притя-
притяжения Не ... Не или же Нг ... Нг с достаточной
точностью. Это можно показать, сравнивая резуль-
результаты, полученные с помощью лондоновской формы
(r->oo) E® и экспериментальных данных [2].'Однако
для больших атомов (Ne, Аг, Кг...) использование
только ?г приводит [2] к ошибочному множителю по-
порядка 2,2. Чтобы объяснить множитель 2, мы рассмо-
рассмотрим ниже отношения между результатами многоэлек-
многоэлектронной теории и результатами Донафа и Пит-
цера [2]. Это покажет также, каким образом можно
подсчитать поправки при всех г, не обращаясь к ап-
аппроксимациям лондоновского типа [2].
Ошибки, по-видимому, возникают не из-за каких-
либо поправок, которые обычно появляются для ка-
каждого парного члена evp в A67) подобно поправкам
от непрерывного спектра к формуле Лондона. Они
были бы в таком случае пропорциональны паХп>ь и
давали бы аналогичные поправочные множители как
для Не и Н2, так и для Ne, Аг и т. д.
Полная энергия, даваемая многоэлектронной тео-
теорией, включает корреляции R [соотношения G8) —
135 § 28. Вандерваальсовы притяжения
(80)]. Эти многоэлектронные корреляции пренебре-
пренебрежимо малы по сравнению с полной энергией атома
?согг или с энергией связи стабильной молекулы
(см. § 18). С другой стороны, корреляционная энер-
гия A67) между двумя несвязанными атомами весь-
весьма мала, чтобы стать основой нашего подхода. Лон-
доновское притяжение между двумя атомами неона
[2], расположенными на расстоянии 4,0 А (ге=3,08 А),
равно всего лишь —1,3-10""8 эв по сравнению с
Есогт^ — И эв отдельного атома неона [1]. В таком
случае члены R могли бы оказаться сравнимыми с
??. Следовательно, рассмотрение этих многоэлек-
многоэлектронных членов R становится особенно существенным
в связи с межмолекулярными силами.
Предположим1), что, использовав соотношения
A41) и A45), мы преобразовали F9а):
N~na+nb
^{^} A70>
аналогичные результаты получим и для х, и %'s [вы-
[выражения G6) и G7)]. В таком случае энергию можно
вновь оценить при помощи диаграмм, как это дела-
делалось в § 18 с применением молекулярных орбиталей.
За исключением Д<2> [выражение A57в)], формула для
энергии обычно имеет тот же вид; что и A59) и
G9) — (81), но в ней t, /, k заменены на i)v> Лр» Лт-
С помощью молекулярно-орбитальных пар мы
имели бы [пренебрегая (81)]:
A71)
Аналогичные диаграммы можно нарисовать для энер-
энергии, полученной для функции A70), используя лока-
х) В отличие от случая, когда рассматриваются молекуляр-
молекулярные орбитали, здесь могли бы также возникнуть одноэлектрон-
ные члены {fv}, как поправки к T|v.
§* 28. Вандерваальсовы притяжения
137
лизованные орбитали. В этих диаграммах различают-
различаются теперь внутри- и межатомные члены. Например,
для Не... Не
A72)
ъ
(II)=
(Ш) =
(IV) =
оз
A74)
A75)
A76)
Выражение A73) дает поправку более высокого по-
порядка к Есоп каждого „атома" Не [искаженного
в (%%)], выражение A74) дает то же самое для меж-
межатомной энергии е^>. Для A75), например, имеем
\ 'г 24' 12^ 14 '2/ 11/О )
плюс обменные члены, пренебрежимо малые при
больших г. Функция т12 указывает на то, что для гц
берется орбитальный потенциал 3' [формула (8')] (см.
§27, разд. Д).
10 О. Синаноглу
138 §28. Вандерваальсовы притяжения
Аналогично выглядит выражение для энергии, по-
полученное не по теории возмущений, т. е. с помощью
Я*. Функции и$ в A72), появляющиеся там согласно
A75'), заменяются на |xvp, которые минимизируют
полную энергию пар [выражение A62), включая A73)
или A74), но отнюдь не A576)].
Члены, выбранные Донафом и Питцером [2] [см,
их уравнения B1) и B2)], соответствуют Е% [выра-
[выражение A67)] и A75) в гайтлер-лондоновском прибли-
приближении (г-*оо; перекрыванием пренебрегают). Их
волновая функция не содержит внутриатомные jivp>
так что не появляются члены (I) — (IV). В дополни-
дополнительном диполь-дипольном приближении исчезает
член (II) [но не (IV) или (III)].
Поправочный множитель [2] ^exp/fbond равен 1,2
в Не и 2,2 в Ne, Ar, Кг, Хе, N2 и СН4, где
Число диаграмм типа (III) равно
Mm = na{na— \)nb A77)
по сравнению с числом пар папь в ??. Поэтому
в Не ... Не
а если взять лишь наиболее поляризуемые (npf
электроны в Ne, Ar, Кг и Хе, то получим
36
Считая, что для отдельных диаграмм приблизи-
приблизительно справедливо то же самое отношение, можно
показать, что поплавочный множитель примерно в
5 раз больше в тяжелых атомах, нежели в гелии. От-
Отношение наблюдаемых поправок примерно равно
§ 28. Вандерваальсовы притяжения 139
1,2/0,2, т. е. б1). Однако трудно понять, почему этот
множитель.остается примерно тем же самым в N2 и
СН4.
Подобные поправки к ?? [выражение A67)]
даются многоэлектронной теорией без аппроксима-
аппроксимаций гайтлер-лондоновского типа, и, следовательно,
они пригодны для всех г. Например, член Rui [выра-
[выражение A75')] можно подсчитать с помощью ц и jxvp
в замкнутом виде.
Эти члены можно также вычислить из йц в моле-
кулярно-орбитальном описании. Как упоминалось в
§ 27 разд. В, относительная величина вклада пар
[выражение A73)] по сравнению с частью, даваемой
треугольными диаграммами A75) и A76), должна
быть больше в rj-описании, нежели в молекулярно-
орбитальном описании [выражение A71)]. Тем не ме-
менее благодаря простым преобразованиям отдельных
1) Члены Rin — положительны, так что Е2+Е$ или E2+R111
дает меньшее притяжение, чем сама по себе функция ?<¦?, Это
применяется к формулам Лондона до введения эксперименталь-
экспериментальных поляризуемостей. При этих предположениях наши уравне-
уравнения становятся уравнениями типа B4) и B6) работы [2]
(стр. 65):
в то время как
После того как сделаны диполь-дипольные приближения в пред-
предположении отсутствия перекрывания, формула A67) для Е®
переходит в выражение, содержащее г*6.
Таким образом, в формулах, не учитывающих поляризуемо-
поляризуемостей, поправочный множитель возникает от //?/« 1/2,2 тогда как
после подстановки поляризуемостей, которым также отвечают
Ui (ai^2h2i/UiI этот множитель равен [///«2,2. Формулы, со-
содержащие h], дают/?„,/?? « 0,2 в Не и Rlu/E% = 1,2 в Ne, Ar
и т. д. Эмпирические формулы Лондона [2] (в ?bond вместо /
везде подставлены U) отличаются от формулы для Е$.
10*
140 § 29. Релятивистские эффекты
пар, таких, как в A68), некоторые члены из треуголь-
треугольных диаграмм в молекулярно-орбитальноад описании
можно непосредственно сопоставить с /?щ и возмож-
возможно упростить расчеты этой величины. Необходима
дальнейшая работа в этом направлении (см. допол-
дополнение автора в конце данной работы).
Члены (I) и (III), по-видимому, остаются основ-
основными членами в A72) при меньших г. Как член (II),
так и член (III) содержат m^iW^HmH**)» однако
Щь<^ИЩг- Следовательно, можно ожидать, что
член (II) значительно меньше члена (III). Можно
оценить величину члена (IV) (влияние атомной кор-
корреляции на вандерваальсово притяжение). Она, по-
видимому, мала, поскольку ц12 и jx24 почти ортого-
ортогональны, а п№ невелико. Величину А<2) в A57в) также
необходимо рассчитать при меньших г (см. дополне-
дополнение автора).
В заключении этого раздела отметим, что много-
многоэлектронная теория дает возможность рассчитывать
непосредственно межмолекулярные притяжения при
всех г, хотя они и представляют собой по абсолютной
величине малые корреляционные эффекты.
Расчеты по методу Хартри — Фока, например, для
BpfaBpfb-части составной системы Ne—Ne, остают-
остаются решающей предпосылкой для рассмотрения по-
подобных больших систем. Для дальнодействующей ча-
части притяжения необходимы лишь хартри-фоковские
функции отдельных атомов; поправки, которые обыч-
обычно возникают от использования точных атомных
функций, по-видимому, несущественны.
§ 29. РЕЛЯТИВИСТСКИЕ ЭФФЕКТЫ
Выше мы пользовались нерелятивистским урав-
уравнением Шредингера B) и определяли корреляцион-
корреляционную энергию как разность между точным значением,
даваемым этим уравнением, и хартри-фоковской
энергией [1]. Однако с увеличением заряда ядра Z,
начиная с Z^IO, как известно, релятивистские эф-
§ 29. Релятивистские эффекты 141
фекты [113] становятся существенными и фактически
сравнимыми с корреляционными энергиями.
Основные релятивистские эффекты можно учесть
не с помощью членов, которые добавляются для того,
чтобы сделать потенциалы 1/г^ лоренц-инвариант-
ными, а с помощью одноэлектронных членов (#t и
Я4), входящих в уравнение B) работы [113] (см.
также работу [111]). Увеличение массы электрона
при более высоких скоростях приводит здесь, как и
в мезоатомах
1) к более интенсивной связи с ядрами,
2) к сжатию орбит.
Эти эффекты, возникающие из-за одноэлектронных
членов, оказываются учтенными в релятивистском
обобщении метода Хартри — Фока [76, 97].
Для тяжелых атомов и молекул можно, исходя
из релятивистских уравнений, развить многоэлектрон-
многоэлектронную теорию. Хотя точный релятивистский гамильто-
гамильтониан неизвестен, хорошей основой для аппроксима-
аппроксимации, по-видимому, будет релятивистский [76] хартри-
фоковский гамильтониан #0, дополненный нереляти-
нерелятивистскими \\гц членами.
Релятивистскую многоэлектронную теорию можно
тогда сформулировать точно таким же образом, как
и нерелятивистскую, рассмотренную выше: можнр
получить «релятивистскую» функцию % и в ней можно
выделить различные оболочки и электронные группы.
Из-за своей сильной зависимости от Z* (эффектив-
(эффективного заряда ядра) релятивистские эффекты в основ-
основном сказываются на внутренних оболочках [76] и вы-
выпадают из расчетов энергий молекулярной связи и
других свойств валентных электронов. Чтобы прийти
к нерелятивистским выражениям, исходя из этой ра-
работы, можно сделать дальнейшие аппроксимации в
формальной релятивистской теории для частей Xrei и
jferei, относящихся к внешним оболочкам.
Хотя релятивистские эффекты существенны глав-
главным образом для внутренних оболочек, они должны
оказывать непрямое влияние и на внешние электроны.
Поясним это.
142 § Ж Заключение
Можно минимизировать энергию именно валент-
валентной оболочки, чтобы получить отвечающую ей вол-
волновую функцию (и хартри-фоковскую часть, и Лу)
при условии, чтобы пробные функции оставались ор-
ортогональными к внутренним орбиталям [см. (86) и
(996)]. При этом релятивистское сжатие внутренних
оболочек и оболочечной структуры приводит также
к стягиванию внешних электронов. Следовательно,
даже для расчета нерелятивистской валентной обо-
оболочки необходимо знать внутренние орбитали. По-
Последние, по-видимому, можно аппроксимировать ре-
релятивистскими хартри-фоковскими орбиталями, полу-
полученными для реальных свободных ионов, отвечающих
этим остовам. Они, а следовательно, и релятивист-
релятивистские эффекты оказывают влияние также на потен-
потенциал «среды» Vi. Однако й^-части внешней волновой
функции не чувствительны к изменениям Vi.
Исходный релятивистский формализм позволил бы
оценить ошибки, которые возникали бы при дальней-
дальнейших аппроксимациях для нерелятивистских внешних
оболочек.
§ 30. ЗАКЛЮЧЕНИЕ
Мы исходили из точной волновой функции и энер-
энергии многоэлектронной системы, не детализируя ор-
орбитали базисного набора и не обращаясь к разло-
разложениям по теории возмущений. Рассматриваемые
нами величины состояли из хартри-фоковской части
и остающихся корреляций* между последовательно
увеличивающимся числом электронов. Были найдены
главные корреляционные эффекты, появляющиеся
для всех пар электронов. Они получаются при по-
помощи любого из имеющихся в нашем распоряжении
двухэлектронных методов. Например, корреляции в
Не или Нг зависят от природы пары, как это обсу-
обсуждалось в § 22. Остающиеся эффекты, например из-
изменение орбиталей из-за корреляции и я-электронные
корреляции (п>2), оцениваются, согласно (90) и
G9), как малые поправки.
§ 80. Заключение 143
Можно также использовать метод «последователь-
«последовательных частичных ортогонализаций» (см. § 9), привод
дящий нас к конкретной форме точной волновой
функции. В этом случае можно в качестве исходной
функции использовать любую вариационную много-
электронную пробную функцию, которая может и не
поддаваться какой-либо физической или химической
интерпретации. Этот метод частичных ортогонализа-
ортогонализаций не только ясно указывает, что вносится этой
пробной функцией в орбитали и различные корреля-
корреляционные эффекты, но также позволяет весьма просто
установить, каким образом улучшить пробную функ-
функцию.
«Многоэлектронная теория» позволяет раздельно
рассматривать:
1) внутренние — внешние оболочки, например
(а — я)-оболочки;
2) локализованные связи, отдельные пары и ион-
ионные остовы, причем на строгой основе как в рамках
полуэмпирических расчетов, так и неэмпирических.
Показано, каким образом при учете соответствующей
электронной корреляции можно рассчитать с^ хими-
химической точностью следующие величины:
1) молекулярные энергии связи;
2) энергию отдельной связи внутри большой мо-
молекулы;
3) внутримолекулярные вандерваальсовы притя-
притяжения, а тем самым и изменения энергии образова-
образования в молекулах;
4) межмолекулярные вандерваальсовы силы во
всей области межъядерных расстояний;
5) электронные спектры.
Методы не слишком сильно усложняются по мере
того, как растет число электронов или же размер
молекулы.
Развитая теория должна бы дать возможность
вычислить энергию связи тяжелых молекул, подоб-
подобных Na2, при помощи рассмотрения лишь их валент-
валентных электронов. Это можно было бы осуществить
почти точно таким же образом, как в расчете типа
Джеймса — Кулиджа для Н2. Мы уверены, что
144 Дополнение автора
такие расчеты для больших систем возможны сейчас
при вычислительных усилиях, не больших чем те,
что в настоящее время тратятся во многих лабора-
лабораториях на расчеты молекул первого ряда.
Необходима дальнейшая работа по применению
методов Хартри — Фока для рассмотрения частей
больших систем, многоэлектроиной теории незамкну-
незамкнутых оболочек и внутримолекулярных вандерваальсо-
вых сил.
Я благодарен многочисленным коллегам за их
ценные замечания и обсуждения ряда вопросов, явив-
явившиеся большим вкладом в эту работу. Я чрезвычай-
чрезвычайно благодарен Кестнеру, Маккою и Туану за по-
помощь при окончательном обсуждении работы. Дан-
Данная работа была частично выполнена в течение моего
пребывания на химическом факультете Гарвардского
университета и в университете техники в Анкаре. Ав-
Автор глубоко признателен персоналу обоих универси-
университетов за их гостеприимство.
ДОПОЛНЕНИЕ АВТОРА
В то время, когда эта работа находилась в пе-
печати, появился ряд работ, развивающих многоэлек-
многоэлектронную теорию.
1. Было показано, что с помощью «метода частич-
частичных ортогонализаций» (см. § 9) можно получить не
только различные члены из функции %, но также и
сами хартри-фоковские орбитали [117] *). Таким об-
образом, с помощью произвольной пробной функции tj>,
если она выбрана достаточно точно, можно выделить
хартри-фоковские орбитали, не проводя отдельного
вариационного расчета. Этот метод был применен
к трехчленной волновой функции гелия хиллераасов-
ского гатила и были получены точные хартри-фоков-
1) Работа автора по этому вопросу была включена в вы-
луск, посвященный симпозиуму в Санибел Айленд в честь Хил-
лерааса, январь 1963.
Дополнение автора 145
ские орбитали с помощью настольной вычислитель-
вычислительной машины [118].
2. Для демонстрации и дальнейшей проверки тео-
теории были проведены подробные численные расчеты
на машинах для атома Be [74]. Предварительные ре-
результаты дают
е12 = —1,175 эв, ?34 = — *^2 эв.
Оценены межоболочечная корреляция 4eis2s и
вклад треугольных диаграмм в R. Прежние значения
составляли [4, 79] приблизительно —0,2 эв; ожидает-
ожидается, что R будет меньше 0,01 эв [9]. Наша энергия
?согг при учете только ei2 и ез4 составляет лишь
89,16% полной корреляционной энергии. Расчеты Ват-
сона [77] с функцией, взятой в разложении (по 37 кон-
конфигурациям), по методу конфигурационного взаимо-
взаимодействия (учитывающей все эффекты), приводят к
89,36% полной корреляционной энергии.
3. Было рассчитано в широкой области изменения
межатомных расстояний (г > 2 А) взаимодействие
Не... Не. Один расчет дал чрезвычайно точное не-
неэмпирическое значение коэффициента при члене дальт
нодействия г~6. Это значение с= — 1,540 аг. ед. срав-
сравнимо с «экспериментальным» значением —1,50±10%
Далгарно и Кингстауна [114]. Многоэлектронные эф-
эффекты (описанные в § 28) дают вклад в с порядка
6,7%. В менее точных расчетах, где ограничивались
имеющимися хартри-фоковскими результатами, были
получены в функции г относительные величины раз-
различных эффектов и диаграмм, обсуждавшихся в § 28.
Для расстояния Го, отвечающего минимуму потен-
потенциала, вклад многоэлектронных эффектов и искаже-
искажения составлял 9%. В этих первых контрольных рас-
расчетах использовались орбитали Слэтера для грубой
оценки хартри-фоковской части и наипростейших
парных функций. Был получен [119] минимум потен-
потенциала, равный 5,24 ° К при 3,08 А. «Эксперименталь-
«Экспериментальные» значения отличаются; «лучшие значения» Бу-
кингейма составляют 9,63° К при 2,88 А. Результаты,
несомненно, могли бы быть улучшены путем приме-
146 Дополнение автора
нения точных хартри-фоковских молекулярных орби-
орбиталей или же даже молекулярных орбиталей, взятых
как линейные комбинации атомных орбиталей, по-
построенных из атомных хартри-фоковских орбиталей,
и более полных парных пробных функций. Добавляя
некоторые поправки к рассчитанным результатам,
Кестнер получил новый потенциал, равный 9,92° К
(±10%) при 2,98 А.
ЛИТЕРАТУРА
1. Low din P. О., Adv. Chem. Phys., H, 207 A959).
2. Pitzer K. S., Adv. Chem. Phys., II, 59 A959).
3. Sinanoglu O., Journ. Chem. Phys., 33, 1212 A960).
4. Sinanoglu О., Мог ten sen E. M., Journ. Chem. Phys*
34, 1078 A961).
5. Nesbet R. K., Phys. Rev., 122, 1497 A961).
6. Moffitt W., Journ. Chem. Phys., 22, 320 A954).
7. Pines D., в сборнике «The Many-Body Problem., New York,
1962. (Имеется перевод: Д. Пайнс, Проблема многих
тел, ИЛ, 1963.)
8. Sinanoglu О., Ргос. Roy. Soc, A260, 379 A961).
9. Sinanoglu О., Journ. Chem. Phys., 36, 706 A962).
10. Sinanoglu O., Journ. Chem. Phys., 36, 3198 A962).
11. Sinanoglu O., Tuan D. F., Journ. Chem. Phys., 38, 1726
A963).
12. Sinanoglu O., Journ. Phys. Chem., 66, 2283 A962).
13. Mayer M. G., Sklar A. L., Journ. Chem. Phys., 6, 645
A938).
14. Pariser R., Parr R. G., Journ. Chem. Phys., 21, 466
A953).
15. Slater J. C, Quantum Theory of Atomic Structure, Vol. 1,
New York, 1960.
16. Kolos W., Journ. Chem. Phys., 27, 591 A957).
17. Dewar M. J. S., Wulfman С. Е., Journ. Chem. Phys.,
29, 158 A958).
18. Nesbet R. K., Proc. Roy. Soc, A230, 312 A955).
19. Slater J. C, Quantum Theory of Atomic Structure, Vol II,
New York, 1961.
20. Jastrow R., Phys. Rev., 98, 1479 A955).
21. Peirls R., 1958, Boulder Lectures in Theoretical Physics,
Vol. I, New Jork, 1959, p. 238.
22. Szasz L., Journ. Chem. Phys., 35, 1072 A961).
23. Hurley A. C, L e n n a r d - J о n e s J. E., P о p 1 e J. A,,
Proc. Roy. Soc, A220, 446 A953).
24. Allen T. L., Shu 11 H., Journ. Chem. Phys., 35, 1644 A961),
25. Parr R. G., Quantum Theory of Molecular Electronic Stru-
Structure, New York, 1963.
26. Lykos P. G., Parr R. G., Journ. Chem. Phys., 24, 1166
A956); 25f 1301 A956).
27. Low din P. O., Journ. Chem. Phys., 35, 78 A961),
148 Литература
28. Lykos P. G., Parr R. G., Journ. Chem. Phys., 25, 1301
A956).
29. McWeeny R., Preprint No. 59, Quantum Chemistry Group,
Uppsala, 1961.
30. McWeeny R., Proc. Roy. Soc, A253, 242 A959).
31. Szasz L., Phys. Rev., 126, 169 A962).
3t2. De Witt C, Nozieres P., в сборнике «The Many-Body
Problem», New York, 1959.
33. Low din P. O., Journ. Math. Phys., 3, 1171 A962).
34. Clement i E., Roothaan C. *C. J., Yoshimine M.,
Journ. Chem. Phys., 127, 1618 A962).
35. Nesbet R. K. Rev. Mod. Phys., 32, 272 A960); Journ.
Chem. Phys., 36, 1518 A962).
.36. Allen L С, Каго А. М., Rev. Mod. Phys., 32, 275 A960).
37. Roothaan С. С. J., Rev. Mod. Phys., 32, 179 A960).
38. Nesbet R. K., Rev. Mod. Phys., 33, 28 A961).
39. Freeman A. J., Rev. Mod. Phys., 32, 273 A960).
40. Ruedenberg K-, Journ. Chem. Phys., 34, 1861 A961).
41. L i n d e n b e r g J., S h u 11 H., Journ. Mol. Spectr., 5, 1
A960).
42. Lennar d-Jones J. E., Ann. Rev. Phys. Chem., 4, 167
A953).
43. Pople J. A., Quart, Revs., 11, 291 A957).
44. L i n n e 11 J. W., Wave Mechanics and Valency, London,
1960.
45. Мое ci a R., Journ. Chem. Phys., 37, 910 A962).
46. Coulson C. A., Trans. Farad. Soc, 38, 433 A942).
47. Lennar d-Jones J. E., Proc. Roy. Soc, A198, 1, 14
A949).
48. Nesbet R. K., Journ. Chem. Phys., 32, 1114 A960).
49. L e n n a r d - J о n e s J. E., Pople J. A., Proc. Roy. Soc,
A202, 166 A950).
50. H a 11 G. G., Proc Roy. Soc, A205, 541 A951).
51. Boys S. F., Rev. Mod. Phys., 32, 296 A960).
52. Adams W. H., Journ. Chem. Phys., 37, 2009 A962).
53. G i 1 b e r tJT. L., в печати.
54. S i n a n о g 1 u O., Electronic Structure of Atoms and Molecu-
Molecules, Harvard Lecture Notes, Spring, 1962.
55. В acher R. F., Phys. Rev., 43, 264 A933).
56. Hill T. L., Statistical Mechanics, New York, 1956. (Имеется
перевод: Т. Хил л, Статистическая механика, ИЛ, 1960.)
57. Е d m i s t о n С, Ruedenberg К., Rev. Mod. Phys., 35,
457 A963).
58. BrenigW., Nucl. Phys., 4, 363 A957).
59. Фок В., Веселое М., Петрашень М., ЖЭТФ, 10,
723 A940).
60. Szasz L., Journ. Chem. Phys., 37, 193 A962).
61. Gold stone J., Proc Roy. Soc, A239, 267 A957).
62. Brueckner K. A., Phys. Rev., 100, 36 A955).
63. В rout R., Phys. Rev., Ill, 1324 A958).
64. Sinanoglu O., Journ. Chem. Phys., 34, 1237 A961).
Литература 149
65. Nesbet R. К., Phys. Rev., 109, 1632 A958).
66. H у 11 e г a a s E. A., Zs. Phys., 65, 209 A930).
67. Be the H. A., Sal peter E. E., Quantum Mechanics of
One- and Two- Electron Atoms, New York, 1957. (Имеется
перевод: Г. Бете, Э. С о л п и т е р, Квантовая механика
атомов с одним и двумя электронами, ИЛ, 1960.)
68. Sinanoglu О., Phys. Rev., 122, 491, 493 A961).
69. Primas H., Helv. Phys. Acta, 34, 331 A961).
70. Rodberg L. S., Ann. Phys., 2, 199 A957).
71. Eden R. J., в книге «Nuclear Reactions», Vol. I, Amsterdam,
1958.
72. В r u e с k n e г К. A., L о с k e 11 A. M., R о t e n b e r g M.,
Phys. Rev., 121, 255 A961).
73. Slater J. C, Phys. Rev., 91, 528 A953).
74. T u a n D. F., Sinanoglu O., Journ. Chem. Phys., в печати.
75. Sinanoglu О., Journ. Chem. Phys., 36, 564 A962).
76. С a 11 a w а у J., Woods R. D., S i г о u n i a n V., Phys. Rev.,
107,934 A957).
77. Watson R. W., Phys. Rev., 119, 170 A960).
78. Ebbing D. D., Journ. Chem. Phys., 36, 1361 A962).
79. С 1 e m e n t i E., Journ. Chem. Phys., в печати.
80. Allen L. С, С 1 e m e n t i E., G 1 a d n e у Н. M., Rev. Mod.
Phys., 35,465 A963).
81. Kotani M., et al., Encyclopedia of Physics, Vol. 37/2 (Mo-
(Molecules II), Berlin, 1961.
82. D a u d e 1 R., L e F e b v r e R., M о s e r C, Quantum Chemi-
Chemistry, New York, 1959.
83. На г г i s F., T а у 1 о г Н. S., будет опубликовано.
84. P a u n с z R., D e H e e r J., L б w d i n P. O., Journ. Chem.
Phys., 36, 2242,2257 A962).
85. DeHeer J., Journ. Chem. Phys., 37, 2078 A962).
86. PaunczR., Journ. Chem. Phys., 37, 2739 A962).
87. Low din P. O., Rede i L., Phys. Rev., 114, 852 A959).
88. Ну Пега as E. A., Zs. Phys., 54, 347 A929).
89. Low din P. O., Phys. Rev., 97, 1509 A955).
90. Ки б а рта с В. В., Каветскис В. Н., Юцис А. П.,
ЖЭТФ, 29, 623 A955).
91. Davidson E. R., Jones L. L. Journ. Chem. Phys., 37,'
2966 A962).
92. К о 1 о s W., R о о t h a a n С. С. J., Rev. Mod. Phys., 32, 205
A960).
93. F г a g a S., R a n s i 1 B. J., Journ. Chem. Phys., 35, 1967
A961).
94. В e t h e H. A,, G о 1 d s t о n e J., Proc. Roy. Soc, A238, 551
A956).
95. Brickstock A., Pople J. A., Phil. Mag., 44, 697 A953).
96. Brickstock A., Pople J. A., Phil. Mag., 44, 705 A953).
97. H a r t r e e D. R., Calculation of Atomic Structures, New
York, 1957. (Имеется перевод: Д, X a p т р и, Расчеты
атомных структур, ИЛ, 1960.)
150 Литература
98. Bacher R. R, Goudsmit S., Phys. Rev., 46, 948 A934).
99. С a 11 a w а у J., Phys. Rev., 106, 868 A957).
100. Г л е м б о ц к и й И. И., К и б а р т а с В. В., Ю ц и с А. П#|
ЖЭТФ, 2, 476 A956).
101. Stan ton R. E., Journ. Chem. Phys, 36, 1298 A962).
102. Van Vleck J. H, Sherman A., Rev. Mod. Phys., 7,
167 A935).
103. Simpson W. Т., Journ. Chem. Phys., 28, 972 A958); The-
Theories of^Electrons in Molecules, New Jersey, 1962.
104. Sinanoglu O,, Journ. Chem. Phys., 37, 191 A962).
105. McKoy V., Sinanoglu О., не опубликовано.
106. Salem L, Longuet-Higgins H. C, Proc. Roy., Soc.»
A259, 433 A961).
107. Salem L, Proc. Roy. Soc, A264, 379 A961); 433 A961),
108. James H. M., С о о 1 i d g e A. S., Journ. Chem. Phys., 1,
825 A933).
109. D о n a t h W. E., P i t z e г К. S., Journ. Am. Chem. Soc., 81,
3213 A959).
110. Ransil B. J., Journ. Chem. Phys., 34, 2109 A961).
111. Hirschfelder J. O, Curtis С F., Bird R. В., Molecu-
Molecular Theory of Gases and Liquids, New York, 1954. (Имеет-
(Имеется перевод: Д. О. Гиршфельдер, К. Ф. Кэртис*
Р. Б. Б и р д, Молекулярная теория газов и жидкостей,
ИЛ, 1960.)
112. Mof f itt W., Proc. Roy. Soc, A210, 245 A951).
113. From a n A., Rev. Mod. Phys., 32, 317 A960).
114. Dal gar no, Kingston, Proc. Phys. Soc, 73, 455 A959);
78, 607 A961).
115. Ebbing D. D., Ph. D. Thesis, Dept. of. Chem., Indiana
Univ., June 1960.
116. Varshni Y. P., Rev. Mod. Phys., 29, 664 A957).
117. S i n a n о g 1 u O., Rev. Mod. Phys., 35, 517 A963).
118. Kestner N. R., Journ. Chem. Phys., 39, 1361 A963).
119. Bull. Amer. Phys. Soc, 8, 535 A963).
СОДЕРЖАНИЕ
От редактора перевода 5
§ 1. Введение 13
§ 2. Необходимость «многоэлектронной теории» 13
§ 3. Полуэмпирические теории 16
§ 4. Неэмпирические теории .18
§ 5. Схема построения «многоэлектронной теории» .... 20
§ 6. Орбитали 22
§ 7. Антисимметрия и относительные электронные распреде-
распределения . , 25
§ 8. Уравнения Хартри—Фока 28
§ 9. Точная многоэлектронная волновая функция .... 32
§ 10.. Ортогональность орбиталей 40
§ 11. Несвязанные комплексы 42
§ 12. Вариационный принцип и точная энергия 46
§ 13. Различные корреляционные эффекты в точной волно-
волновой функции и энергии 48
§ 14. Вариационный подход, использующий теорию возму-
возмущений 50
§ 15. Различные теории многих частиц 53
A. Обобщенные методы самосогласованного поля . . 53
Б. Теория возмущений 56
B. «Многоэлектронная теория» 60
§ 16. Волновая функция «многоэлектронной теори^» ... 61
§ 17. Вариационная энергия «многоэлектронной теории» . . 62
§ 18. Многоэлектронные корреляции 65
А. Многоэлектронные эффекты в волновой функции 66
Б. Многоэлектронные эффекты в энергии 72
§ 19. Влияние корреляции на орбитали fi 75
§ 20. Незамкнутые оболочки 80
§ 21. Почти вырожденные состояния 84
§ 22. Парные корреляции 88
152 Содержание
§ 23. Свойства симметрии 93
§ 24. Атомы 100
§ 25. Малые молекулы 109
§ 26. я-злектрониые системы Ш
§ 27. Связи — локализация, отдельные пары, ионные остовы 114
A. Преобразование точной энергии 117
Б. Преобразования приближенных выражений для
энергии 119
B. Преобразования вариационных энергий .... 120
Г. Влияние молекулярного окружения на связь . . . 125
Д. Расчет энергии связей 127*
§ 28. Вандерваальсовы притяжения 129
А. Внутримолекулярные притяжения 132
Б. Межмолекулярные притяжения 133
§ 29. Релятивистские эффекты 140
§ 30. Заключение ,142
Дополнение автора 144
Литература 147
О. СИНАНОГЛУ
Многоэлектронная
теория
атомов, молекул
и их взаимодействий
Редактор Е. И. МАЙКОВА
Художник Ф. Л. Лейн
Технический редактор Н. А. Иовлева
Сдано в производство 10/1 1966 г. Подписано к печати 25/V 1966 г.
Бумага 84xl08VM=2,38 бум. л. 7,98 печ. л. Уч.-изд. л. 6,91 Изд. № 2/3529
Цена 50 коп. Зак. № 26
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2
Ленинградская типография № 2 имени Евгении Соколовой
Главполиграфпрома Комитета по печати при Совете Министров СССР-
Измайловский проспект, 29