/















































































































































































Author: Кораблёва Т.П. Королькова Д.В.
Tags: химия общая и неорганическая химия
ISBN: 5-288-03617-9
Year: 2005




Text
I П. Кораблева ТЕОРИЯ
д в. Корольков ПЕРИОДИЧЕСКОЙ
СИСТЕМЫ
Нс
1 । Be В c N О F No
Na Mg Al Si P S Cl Ar
К i. н Sc Ti V Cr Mn Fe Co Ni
• u Zu (j i Ge As Se Br Kr
HI» Sr Y Zu- Nb Mo Tc Ru Rh Pd
Xr Cd In St! Sb Те I Xe
< ' Вл La* HI Ta W Re Os lr Pt
Xu Hg Tl Pb Bi Po At Rn
1 i R.i Rf Db Sg Bh Hs Mt 110
III 112 113 114 115 116 117 118
В Al C Si N P 0 s F Cl Ne Ar
Ti V Cr Mn Fc Co Ni Cu Zn Ga Ge As Se Br Kr
Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Tc I Xc
Au Hg Tl Pb Bi Po At Rn
•L Но Lr Tm Yb Lu Hf Та W Re Os Ir Pt
f I Fm Md No Lr Rf Db Sg Bh
Hs Mt 110 111 112 113 114 115 116 117 118
САНКТ-ПЕТЕРБУРГСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
Т. П. Кораблева, Д. В. Корольков
ТЕОРИЯ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
Рекомендовано Советом по химии Учебно-методического
объединения по классическому университетскому образованию
Российской Федерации в качестве учебного пособия
для химических факультетов классических университетов
ИЗДАТЕЛЬСТВО С.-ПЕТЕРБУРГСКОГО УНИВЕРСИТЕТА
2005
УДК 541.1 + 530.1
ББК 24.1
К66
Рецензенты: д-р хим. наук, проф. В. Н. Пак (Российский гос. пед. ун-т
им. А. И. Герцена),
д-р хим. наук, проф. И. С. Дмитриев (С.-Петерб. гос. ун-т)
Рекомендовано к изданию
Редакционно-издательским советом
Учебно-научного центра химии
С.-Петербургского государственного университета
Кораблева Т. П., Корольков Д. В.
К66 Теория периодической системы: Учебное пособие. —
СПб.: Издательство С.-Петербургского университета, 2005. —
174 с.
ISBN 5-288-03617-9
В учебном пособии изложена обобщенная теория периодической систе-
мы как суперматрицы в бесконечномерном функциональном пространстве,
отражающей упорядоченное множество химических элементов, и квантово-
механическая теория канонической структуры периодической системы хи-
мических элементов. Теория включает в себя представления о ритмике в
построении периодов системы, о возможном нарушении традиционной пе-
риодичности в изменении атомных свойств (исчезновении менделеевских
групп и подгрупп) в области сверхтяжелых химических элементов, концеп-
цию кайносимметрии, концепцию гипервалентности. Дается теоретическая
интерпретация менделеевского правила четности, вторичной и дополнитель-
ной периодичности. Большое внимание уделяется новым подходам в теории
периодической системы — математическим моделям и теоретико-групповой
интерпретации.
Книга предназначена для студентов, магистров, аспирантов, преподава-
телей химических факультетов классических и педагогических университе-
тов.
ББК 24.1
ISBN 5-288-03617-9
© Т. П. Кораблева,
Д. В. Корольков, 2005
© Издательство
С.-Петербургского
университета, 2005
Светлой памяти Учителя —
профессора Сергея Александровича ЩУКАРЕВА —
посвящаем
От авторов
К 110-летию со дня рождения профессора С. А. Щукарева
Эта книга посвящена теоретическим основам учения о периодич-
ности. Первостепенный вклад в развитие этого учения внес профессор
Санкт-Петербургского государственного университета Сергей Алек-
сандрович Щукарев (1893-1984). Сергей Александрович был нашим
учителем, и в связи со 110-летием со дня его рождения мы считаем
для себя честью сказать благодарственные слова о его творчестве.
Профессор С. А. Щукарев почти полвека, с конца 30-х годов, воз-
главлял кафедру общей и неорганической химии химического факуль-
тета, был одним из самых авторитетных и уважаемых профессоров
и по праву считался университетским патриархом. С. А. Щукарев —
признанный глава крупной научной школы, из которой вышли сотни
кандидатов и докторов наук, профессоров и членов Академии наук.
Сергей Александрович был блестящим преподавателем. Его лекции
неизменно собирали большие аудитории слушателей — студентов, ас-
пирантов, преподавателей, научных сотрудников, профессоров.
Велики заслуги С. А. Щукарева в развитии кафедры неорганиче-
ской химии, которая под его руководством стала сильным научно-
педагогическим коллективом, хорошо известным в отечестве и за рубе-
жом. По его инициативе и при его непосредственном участии на кафед-
ре возникли, сложились и достигли высокого уровня многие направле-
ния неорганической химии, координационной химии, химии растворов,
теоретической химии.
С. А. Щукарев внес большой вклад в развитие химического факуль-
тета и университета, членом Ученого совета которого он был в тече-
ние нескольких десятилетий. Он —один из вдохновителей и главных
инициаторов создания новых учебных дисциплин, новых научных на-
правлений, новых кафедр. Именно ему химический факультет обязан
широким введением сначала термодинамического аспекта в учебный
процесс в 30-40-х годах, а затем — в 60-х годах — внедрением квантово-
3
химического аспекта, что способствовало коренной перестройке всего
химического образования и подняло его на уровень, соответствующий
современному состоянию химической науки.
При самом активном участии С. А. Щукарева на химическом фа-
культете были созданы кафедры коллоидной химии, электрохимии,
химии высокомолекулярных соединений, радиохимии, строения ор-
ганических соединений (впоследствии кафедра физической органи-
ческой химии), технической химии, теории растворов (впоследствии
кафедра химической термодинамики и кинетики), химии природных
соединений, квантовой химии, химии твердого тела. Особо важным
событием стала организация первой отечественной кафедры радиохи-
мии, сыгравшей большую роль в подготовке специалистов для атомной
промышленности, и кафедры квантовой химии, которая остается пока
единственной такой кафедрой в российских классических университе-
тах и во многом определяет высокое положение химического факуль-
тета Санкт-Петербургского университета в системе высшего химиче-
ского образования не только в России, но и в мире. Преподавание и
совершенствование образовательной системы Сергей Александрович
всегда считал одной из главных задач своей многогранной деятель-
ности.
Профессор С. А. Щукарев занимал ведущие позиции во Всесоюз-
ном (теперь снова Российском) химическом обществе им. Д. И. Менде-
леева и был его почетным членом. С. А. Щукарев был Менделеевским
чтецом —этой чести удостоены только лучшие представители отече-
ственной химической науки. В 1954 г. он провел 8-е Менделеевские
чтения «О законах Д. И. Менделеева».
Научное наследие С. А. Щукарева весьма велико —сотни статей и
обзоров, докладов и выступлений, монографий и учебников. Главные
его работы посвящены развитию учения о периодичности. Профес-
сор С. А. Щукарев —признанный в России классик в области Пери-
одического закона и периодической системы химических элементов.
Его вклад в теорию Периодического закона и периодической систе-
мы столь значителен, что одну из трех главных форм периодической
системы элементов иногда называют системой Менделеева — Щука-
рева.
С. А. Щукарев развил идею Д. И. Менделеева о том, что кроме
созданной им периодической системы атомов химических элементов
должны существовать периодические системы простых веществ и хи-
мических соединений, каждая со своими особенностями. Он реально
подтвердил эту мысль, построив периодическую систему оксидов, при
этом выявил и интерпретировал ряд присущих ей закономерностей
(в дальнейшем это послужило основой для построения периодических
систем других соединений).
4
Анализируя закономерности в горизонтальных, вертикальных и
диагональных сечениях периодической системы, он постоянно подчер-
кивал общие, специфические черты каждого сечения и индивидуаль-
ные особенности находящихся на этих сечениях атомов или молекул,
разрабатывая тем самым учение об общих, специфических и индиви-
дуальных характеристиках химических элементов. Существование у
элементов индивидуальных особенностей, как показал С. А. Щукарев,
естественным образом вытекает из общих законов изменения про-
странственных и энергетических характеристик электронных оболо-
чек атомов.
С. А. Щукарев одним из первых утверждал, что наряду с общим за-
коном периодичности свойств химических элементов существует мно-
го более частных законов и закономерностей, основанных также на
электронном строении атомов, и поэтому Периодический закон следу-
ет, безусловно, осознавать как принцип периодичности: «Мы все яснее
понимаем, что имеем закон совершенно особого типа, и даже не закон,
а, скорее, общий принцип периодичности, если понимать под принци-
пом такой общий закон, из которого вытекают как частные случаи
многочисленные обычные законы».
Уже сам Д. И. Менделеев считал, что не известна некая функ-
циональная зависимость, выражающая Периодический закон. Более
того, добавляет С. А. Щукарев, Периодический закон не может, по-
видимому, быть охарактеризован исчерпывающе какой-либо одной ма-
тематической формулой, так как является общим и основным принци-
пом, имеющим бесконечное число частных случаев, к которым толь-
ко и можно надеяться подойти с помощью эксперимента и матема-
тики.
Одновременно С. А. Щукарев приходит к новому представлению
о периодической системе химических элементов. А именно: хотя Пе-
риодический закон, будучи общим принципом, и не поддается опи-
санию некоей математической функцией, у него, безусловно, есть
строгий математический образ. Это — периодическая система, пред-
ставляемая в бесконечномерном функциональном пространстве как
суперматрица, члены которой сами являются матрицами, отражаю-
щими множества изотопов элемента, состояний атомов, образуемых
ими соединений, множества функциональных зависимостей свойств
атомов и свойств соединений от различных параметров. В этом
случае периодическую систему в виде таблицы следует понимать
как проекцию суперматрицы на плоскость, в каждой клетке кото-
рой заключено целое бесконечномерное функциональное простран-
ство отдельного элемента. Такое понимание системы делает в от-
даленной перспективе возможным ее применение как суперматри-
цы для перехода набора функций, характеризующих выбранный
5
элемент, к набору функций, свойственных любому другому эле-
менту.
Для развития учения о периодичности весьма значимыми стали
разработанные С. А. Щукаревым правила изобарной (ядерной) ста-
тистики, учение о кайносимметрии, теория вторичной периодично-
сти, учение о ритмике и стратиграфии периодической системы эле-
ментов.
Еще в 20-х годах С. А. Щукарев первым в мире сформулиро-
вал правила неустойчивости нечетно-массовых и устойчивости четно-
массовых изобар, известные как правила изобарной (ядерной) стати-
стики Щукарева — Маттауха. Эти правила позволили объяснить от-
сутствие устойчивых нечетных элементов 43, 61, 85 и 87 и положили
конец их поискам в природе (технеций, прометий, астат и франций
получены искусственно в 30-40-е годы).
С. А. Щукарев разработал и обосновал представления о так назы-
ваемых ранних и поздних р-, d-, /-элементах и показал, что эффект на-
половину заполненных электронами состояний d5 и /7 лежит в основе
появления дополнительной периодичности в рядах d- и /-элементов.
Большим и концептуально значимым вкладом в развитие теории
периодической системы элементов явилось разработанное С. А. Щука-
ревым учение о кайносимметрии — об эффекте возникновения новой
азимутальной симметрии пространственного распределения электрон-
ной плотности при появлении в атоме очередного внешнего электро-
на. В основе этого учения лежит представление о том, что особые
свойства химических элементов, в атомах которых впервые появля-
ется новая электронная симметрия, т. е. особенности химии водорода
и гелия, родоначальников главных и дополнительных подгрупп систе-
мы, и, наконец, лантаноидов, определяются особыми пространствен-
ными и энергетическими характеристиками валентных 1s-, 2р-, 3d-,
4/-электронов, т. е. валентных электронов в тех состояниях, которые
возникают при первом появлении в атоме соответствующей (s, р, d, /)
симметрии распределения зарядовой плотности и не экранируются
электронами аналогичных по симметрии состояний (по причине от-
сутствия таких электронов).
С. А. Щукарев выполнил обширный цикл работ по вторичной пе-
риодичности, первым разработал теорию вторичной периодичности,
которая отражает особенности химии элементов четных периодов си-
стемы и объясняет, почему вторичная периодичность более присуща
элементам главных (а не дополнительных) подгрупп системы, притом
на их высших ступенях окисления.
С. А. Щукарев не оставлял поисков причин множественности из-
вестных частных законов и закономерностей в русле принципа пери-
одичности, что воплотилось в развиваемом им учении о ритмике и
6
стратиграфии периодической системы химических элементов. Учиты-
вая фермионную природу электронов и их квантовомеханическое по-
ведение в силовом поле ядер, он прослеживает эволюцию строения
электронных оболочек атомов на фоне усложнения ядер по мере дви-
жения к концу периодической системы и приходит к выводу, что в ос-
нове этой множественности (или многообразия) лежат периодичность
стратиграфии, т. е. последовательности появления электронных слоев
и их пространственных и энергетических взаимоотношений, и ритмика
системы, которые налагаются на монотонность изменения целого ряда
характеристик атомов под влиянием растущих зарядов и масс атом-
ных ядер. К таким характеристикам относятся, прежде всего, энергия
электронной корреляции, спин-орбитальное сопряжение, эффект про-
никновения электронов в ядра, релятивистский эффект и некоторые
другие. Постепенно нарастая, они не только влияют на основные зако-
номерности в группах и периодах системы, но ломают саму ритмику
последовательного удлинения периодов, что приводит к трансформи-
рованию традиционной периодичности вплоть до возможного исчезно-
вения традиционных групп системы.
Таким образом, Периодический закон в своей классической форме
описывает, возможно, свойства лишь ограниченного числа химических
элементов. Это может означать, что существует еще более общий прин-
цип, включающий в себя Периодический закон как составную часть.
Здесь уместно привести высказывание С. А. Щукарева в одном из его
докладов: «Менделеев прекрасно понимал неизбежность развития бо-
лее общего принципа, чем принцип периодичности, и писал о нем как о
принципе природной гармонии, господствующем в материальном ми-
ре, т. е. Менделеев употребил даже еще более общее понятие, чем рит-
мика, являющая собой лишь составную часть гармонии».
Особое место в творчестве С. А. Щукарева занимают два труда. В
1962-1964 гг. был издан двухтомник «Лекции по общему курсу хи-
мии», который сразу же стал настольной книгой многих студентов,
аспирантов, преподавателей химических факультетов университетов,
технологических и педагогических институтов.
Это было первое в русской химической литературе изложение ос-
новных понятий и теоретических положений общей химии с позиций
термодинамики, кинетики, статистической термодинамики, квантовой
химии, общей теории химической связи. Большое место в «Лекциях»
занимает учение автора об энергетических характеристиках химиче-
ских соединений, электронодефицитных и электроноизбыточных мо-
лекул, гомонуклеарных и гетеронуклеарных связей в рамках теории
молекулярных орбиталей.
В 1970-1974 гг. были изданы два тома монографии «Неорганиче-
ская химия». В ней изложены современные представления о химиче-
7
ских связях в неорганических соединениях, включая представления о
конфигурационных взаимодействиях, об энергии корреляции, об экс-
травалентных состояниях, а также освещены общетеоретические про-
блемы неорганической химии, в частности проблемы макроэргических
связей и биогенных элементов. Но главное в этой монографии — уче-
ние автора о периодической системе элементов.
Дворянин по происхождению, подлинный аристократ духа и ши-
роко образованный человек, Сергей Александрович владел нескольки-
ми языками (в их числе латынь и древнегреческий), прекрасно знал
мировую литературу, историю, философию. Его друзьями и едино-
мышленниками были известные деятели науки и образования, среди
них академики В. А. Фок, В. Г. Хлопин, Я. К. Сыркин, А. Н. Теренин,
Н. Н. Семенов.
Эта книга явилась результатом наших намерений предоставить в
распоряжение специалистов разного ранга (включая и тех, кто еще
только стремится войти в интеллектуальную элиту адептов химии)
систематизированные и обобщенные представления о не поддающейся
окончательной расшифровке и во многом загадочной картине под на-
званием ПЕРИОДИЧЕСКАЯ СИСТЕМА ХИМИЧЕСКИХ ЭЛЕМЕН-
ТОВ. Возникновению книги во многом способствовали не только соб-
ственные размышления, но и многолетние дискуссии с нашими близки-
ми и едва знакомыми коллегами — всем им мы весьма признательны.
Мы глубоко благодарны нашим рецензентам: доктору химических
наук, профессору Игорю Сергеевичу Дмитриеву — известному в отече-
стве и за рубежом теоретику и историку химии и доктору химических
наук, профессору Вячеславу Николаевичу Паку — известному физико-
химику. Они — тоже ученики Сергея Александровича Щукарева. Ква-
лифицированная, строгая и одновременно доброжелательная критика
рецензентов помогла нам в наших усилиях приблизить рукопись к со-
вершенным очертаниям.
Чем более всеобъемлюще некоторое опре-
деленное положение науки и чем оно более
сложно, тем в более абстрактной форме при-
нуждены мы отображать его главные общие
черты, отбрасывая явные формы функцио-
нальных зависимостей и все бесчисленные их
подробности.
С. А. Щукарев
На каждом новом этапе развития химии
мы снова и снова возвращаемся к периодиче-
ской системе, каждый раз вскрывая новое со-
держание и новые ее закономерности.
Я. К. Съиркин
ВВЕДЕНИЕ
Открытие Д. И. Менделеевым Периодического закона в 1869 г. ста-
ло не только одним из крупнейших событий в истории химии XIX сто-
летия, но и одним из самых выдающихся достижений человеческой
мысли минувшего тысячелетия. И вместе с тем Периодический закон
и периодическая система химических элементов все еще остаются для
нас загадкой. До сих пор не ясны до конца причины (точнее, перво-
причины) периодичности, в частности, причины периодической повто-
ряемости сходных электронных конфигураций атомов, хотя очевидно,
что феномен этот связан с непосредственной динамической симметри-
ей атомных систем. До сих пор не очерчены границы применимости
Периодического закона — продолжается полемика относительно спе-
цифики ядерных и электронных свойств атомов сверхтяжелых эле-
ментов.
Гениальное открытие Менделеева осталось при жизни автора,
несмотря на титанические усилия, без строгой количественной теории.
Процитируем* профессора И. С. Дмитриева [1]: «Менделееву предсто-
яло не просто в один прекрасный день увидеть нужный сон (это леген-
да!), но проделать огромную работу по осмыслению громадной, про-
тиворечивой, не всегда достоверной, фрагментарной информации и
концептуального материала химии. Менделеев, будучи по характеру
и разнообразию своих интересов, по стилю работы, по мегаломании в
постановке задач последним великим натурфилософом XIX столетия,
* В этой книге мы будем неоднократно цитировать не только профессора
С. А. Щукарева, но и профессоров И. С. Дмитриева и Д. Н. Трифонова, внесших
большой вклад в теорию периодической системы химических элементов.
9
не посвятил свою оставшуюся жизнь, главным образом, совершенство-
ванию форм периодической системы химических элементов и поиску
новых корреляций в свете учения о периодичности (что на его месте
сделал бы любой без исключения типичный ученый-исследователь).
Менделеев пошел по пути поиска первопричин периодичности. Три-
умф периодической системы стал прологом трагедии одиночества ее
создателя».
Прежде всего следует отметить, что при поисках строгой коли-
чественной интерпретации Периодического закона необходимо учиты-
вать его своеобразие, а именно: невозможность его выражения в виде
какого-либо аналитического уравнения. Единственный способ матема-
тического выражения этого закона — табличный (скорее, матричный).
И в этом отношении Менделеев сделал, безусловно, самое главное —
предложил и разработал периодическую систему химических элемен-
тов. Большего в учении о периодичности никто и никогда, разумеется,
не сделает. Но тем не менее разработка теории Периодического закона
остается актуальной задачей естествознания.
Есть все основания полагать, что количественная интерпретация
Периодического закона сводится к количественной интерпретации
структуры периодической системы [2]. Теория периодической системы
должна, во-первых, объяснять, почему периодическая система имеет
такую структуру, какую она имеет, и, во-вторых, выявить причины
всех известных и неизвестных еще закономерностей изменения свойств
химических элементов и их соединений в периодической системе.
В теории периодической системы химических элементов есть спе-
циальный аспект, связанный с массами атомов. После открытия
протонно-нейтронной структуры ядра и определения масс протона и
нейтрона стало возможным установить закономерности периодическо-
го изменения содержания нейтронов в ядрах атомов (приращение же
масс атомов за счет протонов в периоде системы равно, естествен-
но, числу химических элементов в этом периоде). Изменение атомных
масс химических элементов в периодической системе строго законо-
мерно, определяется порядком заполнения протонных и нейтронных
уровней и подуровней в нуклонных оболочках ядер и может быть вы-
ражено числовыми рядами; при этом закономерное изменение масс
наблюдается не только для периодов системы, но и для рядов s-, р-,
d-, /-элементов в периодах.
Еще Э. Резерфорд предсказывал наличие ядерной периодичности и
сформулировал следующий тезис: ядра с возрастанием их заряда обна-
руживают периодичность строения, но поскольку при этом существен-
ны два типа частиц (протоны и нейтроны), а не один, периодическая
система ядер может оказаться гораздо более сложной, и для ее иллю-
страции может понадобиться, скорее, поверхность, а не плоскость, как
10
для периодической системы атомов. После работ М. Гепперт-Майер и
И. Йенсена, разработавших теорию распределения квантовых уровней
в атомных ядрах с квантовыми числами, подобными квантовым чис-
лам для электронных оболочек атомов, стало возможным сопоставле-
ние ядерной и электронной (т. е. химической) периодичности. Но мы не
будем останавливаться на этом аспекте теоретической интерпретации
периодической системы.
Глава 1
ПЕРИОДИЧЕСКАЯ СИСТЕМА
ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
КАК УПОРЯДОЧЕННОЕ МНОЖЕСТВО
Формулировка того или иного закона природы совершенна лишь
тогда, когда она выражена в строгой математической форме, количе-
ственно описывающей и предсказывающей подвластные этому зако-
ну явления. Такими математическими формами часто являются ал-
гебраические, дифференциальные, интегральные уравнения и систе-
мы уравнений, другие математические построения с использованием
операторов, матриц, тензоров и т.д. Однако для Периодического за-
кона ни одна из этих форм не подходит по причине его всеобъем-
лющего характера. Ведь это —не закон в привычной терминологии,
это —принцип («Закон законов»), подобный по значимости принци-
пу соответствия, принципу дополнительности, принципу относитель-
ности, принципу неопределенности и другим принципам. Поэтому сам
Д. И. Менделеев предложил очень строгую, весьма лаконичную, эле-
гантную и вместе с тем наиболее действенную и выразительную фор-
му математического выражения принципа периодичности — систему
химических элементов. Именно к идее о системе химических элемен-
тов как к единственному реальному средству строгого математическо-
го описания принципа периодичности обратился Менделеев сразу же
после открытия им Периодического закона. Именно систему предло-
жил Менделеев как основу глубокого по смыслу и строгого по фор-
ме математического выражения Периодического закона. Система —
это строгая математическая категория, означающая упорядоченное
множество. (Для того чтобы не потерять сам смысл менделеевской
идеи, нельзя использовать термин «таблица», так как таблица есть
лишь графическое изображение системы химических элементов на
двумерной поверхности, т. е. на плоскости.)
Следует строго определить используемые понятия [2]: периодиче-
ская система есть упорядоченное, соответствующее принципу перио-
12
дичности множество химических элементов с однозначно фиксируемой
нижней границей и подразделяющееся на подмножества — периоды и
группы элементов, причем выделение периодов и групп основано на
закономерностях последовательного заполнения электронных оболо-
чек атомов с ростом зарядов атомных ядер. Структура периодической
системы — своеобразная топологическая матрица, позволяющая с до-
статочной полнотой передать сущность принципа периодичности.
В отличие от «чистых наук» — логики и математики — резкость по-
ложений и законов не свойственна естественным наукам [3]: подобные
высказывания весьма характерны для Д. И. Менделеева, всегда пытав-
шегося выявить «посредствующие», переходные формы. Подчеркивая
отсутствие в природе «резких границ», Менделеев вместе с тем обра-
щал внимание на существование в ней некоторых предельных форм,
объектов и ситуаций. Эти принципы «предела» и «постепенности»
Менделеев широко использовал при разработке периодической систе-
мы химических элементов [4]. Вместе с тем Менделеев неоднократно
обращался к проблеме аналитического выражения Периодического за-
кона и при этом высказывал надежду на то, что это способствовало
бы выяснению сущности явления периодичности. Но до сих пор Пери-
одический закон не получил какого-либо аналитического выражения,
и единственный способ его отражения — посредством периодической
системы.
Главный вклад в развитие учения о периодической системе хими-
ческих элементов как об упорядоченном множестве внес профессор
С. А. Щукарев, одним из первых подчеркнуто утверждавший, что на-
ряду с общим законом периодичности свойств химических элементов
существует много других, более частных, законов и закономерностей,
и поэтому Периодический закон, безусловно, следует осознавать как
принцип периодичности. Следуя Д. И. Менделееву, полагавшему, что
не существует какой-либо функциональной зависимости, выражающей
Периодический закон, он, более того, заключает, что этот закон, по-
видимому, вообще не может быть охарактеризован исчерпывающим
образом какой-либо одной математической формой, так как является
общим и основным принципом, имеющим бесконечное число частных
случаев, и одновременно приходит к новому представлению о периоди-
ческой системе химических элементов. А именно: хотя Периодический
закон, будучи общим принципом, и не поддается описанию некоей ма-
тематической функцией, у него, безусловно, есть строгий математиче-
ский образ. Это — периодическая система, представляемая в бесконеч-
номерном функциональном пространстве как суперматрица, члены
которой сами являются матрицами, отражающими множества изото-
пов элемента, состояний атомов, образуемых ими соединений, множе-
ства функциональных зависимостей свойств атомов и свойств соеди-
13
нений от различных параметров [5, 6]. В этом случае периодическую
систему в виде таблицы следует понимать как проекцию суперматри-
цы на плоскость, в каждой клетке которой заключено целое беско-
нечномерное функциональное пространство отдельного элемента. Та-
кое понимание системы делает возможным ее применение как супер-
матрицы для перехода от набора функций, характеризующих выбран-
ный элемент, к набору функций, свойственных любому другому эле-
менту.
Таким образом, периодическая система —это суперматрица, кото-
рая в наиболее яркой форме отражает взаимосвязь всех химических
элементов и воплощает идею о том, что химические элементы, являясь
качественно отличными друг от друга индивидуальными субстанция-
ми, в то же время представляют в своей совокупности упорядоченное
множество, подчиненное внутренней взаимосвязи всех элементов меж-
ду собой.
В периодической системе как в упорядоченном множестве, в от-
личие от многочисленных неупорядоченных множеств, каждый хими-
ческий элемент имеет свой строго определенный порядковый — атом-
ный—номер (элементы нельзя менять местами), и этот номер являет-
ся главнейшей характеристикой элемента. Атомный номер непосред-
ственно определяет положение химического элемента в периодической
системе (в периоде, группе, подгруппе). Атомный номер химического
элемента представляет собой его важнейшую характеристику в том
смысле, что с ним связана вся многоликая совокупность свойств хи-
мического элемента и свойств образуемых им соединений. Действи-
тельно, атомный номер химического элемента равен заряду ядра его
атома и, следовательно, суммарному числу электронов в оболочке ато-
ма. Принцип построения электронной оболочки атома (распределение
электронов по атомным орбиталям) позволяет в одноконфигурацион-
ном приближении, без учета конфигурационных взаимодействий и на-
ложения конфигураций, установить валентную электронную конфигу-
рацию атома в основном состоянии исходя только из суммарного числа
его электронов, т. е. на основе одного лишь атомного номера элемен-
та. В свою очередь на основе валентной электронной конфигурации
атома можно установить многие свойства соответствующего химиче-
ского элемента. Таким образом, положение элемента в периодической
системе непосредственно отражает валентную электронную структуру
его атомов, и именно по этой причине между свойствами химического
элемента и его соединений и положением элемента в периодической
системе существует прямая и строгая взаимосвязь.
Периодическая система химических элементов всегда изображает-
ся на плоскости в виде таблицы. Впервые это было сделано самим
Д. И. Менделеевым в момент открытия Периодического закона. В ис-
14
тории химии известна не одна сотня графических изображений пе-
риодической системы на плоскости, но все предложенные варианты
являются лишь повторением главной менделеевской идеи.
При построении таких таблиц желательно обеспечить наглядность
и абсолютно необходимо сохранить безупречный теоретический уро-
вень отражения принципа периодичности, а это почти взаимоисклю-
чающие намерения. Самые полезные формы таблиц — абсолютно пра-
вильные с точки зрения Периодического закона и при этом насколько
возможно наглядные.
Наиболее часто используют лишь три табличные формы пери-
одической системы: короткопериодную (рис. 1), ведущую свое про-
исхождение непосредственно от «Естественной системы элементов»
Д. И. Менделеева (1870 г.) и детально разработанную в дальнейшем
С. А. Щукаревым, длиннопериодную (рис. 2), предложенную Г. Сибор-
гом, и пирамидальную (рис. 3), предложенную Н. Бором и действи-
тельно напоминающую ступенчатую пирамиду. В каждой из этих трех
табличных форм периодической системы есть свои преимущества и
свои недостатки.
В короткопериодной форме системы элементы главной и дополни-
тельной подгрупп одной и той же группы объединены в один верти-
кальный столбец. Это наглядно подчеркивает общность свойств непе-
реходных и переходных элементов одной и той же группы системы.
Это весьма важно, поскольку при всех известных различиях элементов
главной и дополнительной подгрупп одной и той же группы отчетливо
проявляются их сходные черты.
Обе длиннопериодные (вторая и третья) формы системы облада-
ют тем достоинством, что в них периодичность в изменении свойств
химических элементов находит свое ясное графическое отражение,
без каких-либо изломов. Пирамидальная форма периодической систе-
мы, помимо этого, наглядно подчеркивает особенность, уникальность,
неповторимость первых двух химических элементов — водорода и ге-
лия — и отражает тот важный момент, что они не принадлежат ни к
одной из групп системы.
Но, к сожалению, в каждой из этих трех геометрических форм си-
стемы есть свои недостатки. Так, в короткопериодной форме довольно
искусственным выглядит положение лантаноидов и актиноидов (нель-
зя же полагать, что лантаноиды так похожи на лантан, что их можно
условно помещать в одну и ту же клетку периодической системы; то
же самое только в большей мере справедливо и по отношению к ак-
тиноидам). Длиннопериодная и пирамидальная формы лишены этого
недостатка, но ему на смену появляется другой, возможно, еще более
серьезный: исчезает сообщество элементов главной и дополнительной
подгрупп в пределах одной и той же группы системы, обе подгруппы
15
оказываются пространственно разведенными, что сразу же затушевы-
вает общность многих присущих этим элементам свойств.
По-видимому, вообще не может быть совершенной формы двумер-
ного изображения периодической системы химических элементов: рам-
ки плоскости всегда останутся для нее узкими. Слишком сложен и
многогранен Периодический закон, чтобы отражающая его система
могла быть просто и без натяжек передана тем или иным двумерным
изображением.
В своем докладе «Попытка приложения к химии одного из начал
естественной философии Ньютона», прочитанном в Королевском ин-
ституте в Великобритании (Лондон, 1889), Д. И. Менделеев говорил [7]:
«В атомах одновременно открываются и своеобразность индивидуаль-
ности, и беспредельная повторяемость, и подчиненность кажущегося
произвола индивидуумов общему гармоническому порядку природы».
В этих словах — предчувствие великого ученого, его размышления о
том, что периодичность, по крайней мере, в простой, традиционной,
классической форме недостаточна для полного выражения основного
содержания и смысла всего упорядоченного множества химических
элементов. Эти слова говорят о том, что идея о гармонии, види-
мо, казалась Д. И. Менделееву более общей, чем входящие в гармо-
нию важные, но все же более частные понятия симметрии и перио-
дичности, и в перспективе более действенной для истинного и полного
понимания системы химических элементов как упорядоченного мно-
жества.
Универсальная гармония (заложенная в нашем духовном сознании)
столь естественна и существенна для понимания окружающего нас ми-
ра, что ее отражение в системе химических элементов не только не
удивительно, но, напротив, может считаться нормальным приспособ-
лением нашего логического разума и творческой интуиции к понима-
нию и интерпретации химических явлений.
Профессор С. А. Щукарев [8, 9] указывал, что первым шагом пере-
хода к гармонии должно быть обобщение периодичности до ритмики
(с допускаемыми ритмикой свободными вариациями свойств). Ритми-
ка-единство целого, состоящего из внутренне согласованных между
собой, соразмерных по своему объему и стройных по форме частей,
имеющих строго определенное содержание. Ритмика в построении си-
стемы химических элементов проявляется, прежде всего, в структуре
больших (начиная с 4-го) периодов, содержащих два 5-элемента, шесть
p-элементов и ритмично изменяющееся число d-, f-, p-элементов. На-
личие в каждом большом периоде изменяющегося числа d-, f-, д-
элементов правильнее считать проявлением не просто периодичности,
но ритма как более общего понятия, допускающего не простые повто-
рения, но и вариации. Существенно также ритмичное появление но-
16
н He
Li Be В C N о F Ne
Na Mg Al Si P s Cl Ar
К Ca Sc Ti V Cr Mn Fe Co Ni
Си Zn Ga Ge As Se Br Kr
Rb Sr Y Zr Nb Mo Tc Ru Rh Pd
Ag Cd In Sn Sb Те I Xe
Cs Ba La* Hf Ta W Re Os Ir Pt
Au Hg T1 Pb Bi Po At Rn
Fr Ra Ac** Rf Db Sg Bh Hs Mt 110
111 112 113 114 115 116 117 118
* Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
** Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr
Рис. 1. Короткопериодная форма периодической системы химических элементов.
Н Не
Li Be В С N О F Ne
Na Mg Al Si Р S Cl Аг
К Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Вг Kr
Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Те I Xe
Cs Ba La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn
Fr Ra Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr Rf Db Sg Bh Hs Mt 110 111 112 113 114 115 116 117 118
Рис. 3. Пирамидальная форма периодической системы химических элементов.
Н Не
Li Be
В С N О F Ne
Na Mg Al Si P S Cl Ar
К Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr
Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Те I Xe
Cs Ba La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu Hf Ta W Re Os lr Pt Au Hg Tl Pb Bi Po At Rn
Fr Ra Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr Rf Db Sg Bh Hs Mt 110 111 112 113 114 115 116 117 118
Рис. 2. Длиннопериодная форма периодической системы химических элементов.
Н Не
Li Be В С N О F Ne
NaMg Al Si P S Cl Ar
К Ca Sc Ti V Cr MnFeCoNi Cu Zn Ga Ge As Se Br Kr
Rb Sr Y Zr NbMoTcRuRhPd Ag Cd In Sn Sb Те I Xe
Cs Ba La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn
Fr Ra Ac ThPaUNpPuAmCmBkCfEsFmMdNoLr RfDbSgBhHsMt 110 111112 113 114 115 116117118
119 120121 122-139 140----------------------153 154---------------- 162 163 -------- 168
169 170 171 172-189 190---------------------203 204----------------212 213----------218
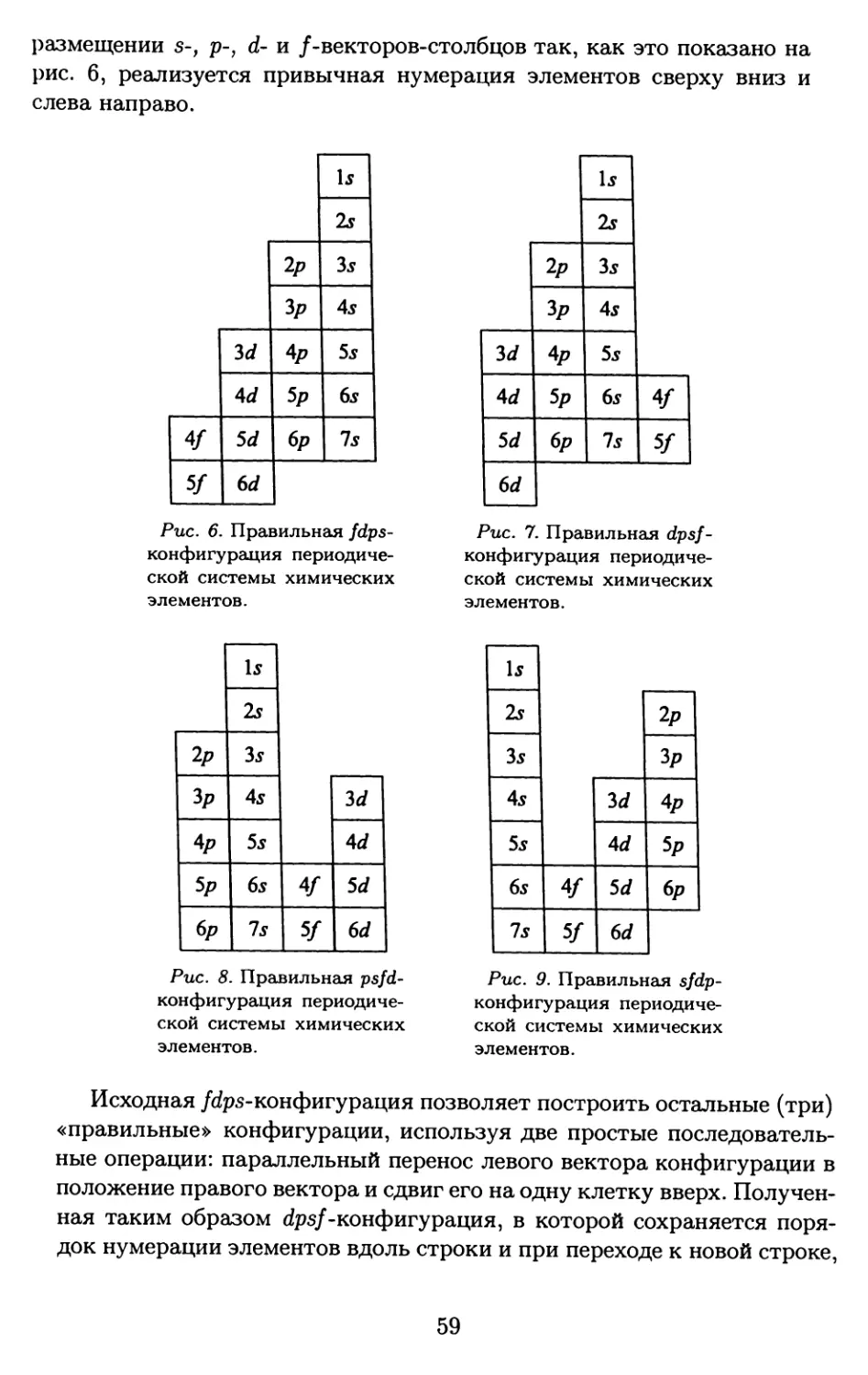
Рис. 10. Периодическая система химических элементов с 8-м и 9-м периодами.
вого типа элементов только в четных периодах системы: 2р — во 2-м,
3d — в 4-м, 4/ — в 6-м, 5д — в 8-м периодах.
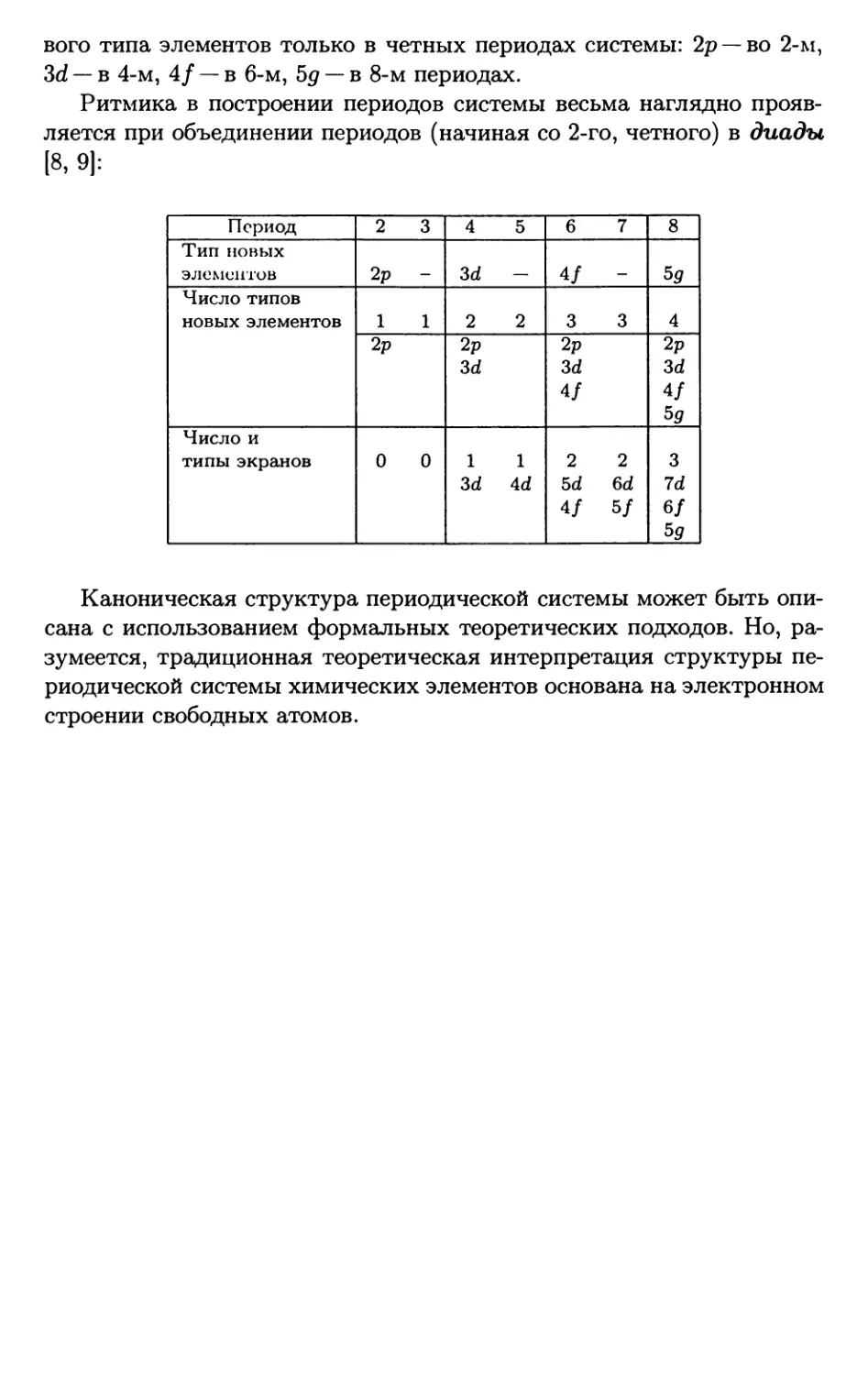
Ритмика в построении периодов системы весьма наглядно прояв-
ляется при объединении периодов (начиная со 2-го, четного) в диады
|8, 9]:
Период 2 3 4 5 6 7 8
Тип новых
элементов 2р - 3d — 4/ — 5g
Число типов
новых элементов 1 1 2 2 3 3 4
2р 2р 3d 2р 3d 2р 3d
4/ If 5g
Число и типы экранов 0 0 1 3d 1 4d 2 5d 2 6d 3 7d
4/ 5/ 6f 5g
Каноническая структура периодической системы может быть опи-
сана с использованием формальных теоретических подходов. Но, ра-
зумеется, традиционная теоретическая интерпретация структуры пе-
риодической системы химических элементов основана на электронном
строении свободных атомов.
Глава 2
КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ
КАНОНИЧЕСКОЙ СТРУКТУРЫ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Обобщенная теория периодической системы как Суперматрицы,
отражающей упорядоченное множество химических элементов, про-
шла в своем развитии несколько исторических этапов.
На первом, и весьма продолжительном, этапе (назовем его клас-
сическим), охватывающем более ста лет, прошедших со дня откры-
тия Периодического закона, главное внимание было уделено верти-
кальному сходству химических элементов в периодической системе.
На доквантовом уровне сходство в свойствах простых веществ и хи-
мических соединений в группах и подгруппах системы описывалось
эмпирически на основе все возрастающей экспериментальной инфор-
мации. Разумеется, и в эмпирическом подходе всегда подчеркива-
лись, во-первых, большее сходство химических элементов в главной
или дополнительной подгруппах, нежели в группе в целом (два типа
элементов-аналогов — вот что самое существенное в классической тео-
рии периодичности), и, во-вторых, постоянно прослеживаемые особен-
ности физико-химического поведения элемента, открывающего глав-
ную или дополнительную подгруппу, т. е. особые характеристики ее
родоначальника, иногда весьма значительно отличающиеся от харак-
теристик всех его последователей в главной или дополнительной под-
группе.
На квантовом уровне вертикальное сходство химических элементов
в периодической системе сразу же получило свое простое и безупреч-
ное объяснение — одинаковое суммарное число валентных электронов
в атомах элементов одной и той же менделеевской группы. Еще боль-
шее сходство элементов подгруппы непринужденно интерпретирова-
лось сходством валентных электронных конфигураций их атомов —
одинаковым набором орбитальных квантовых чисел (одними и теми
18
же типами орбитальной симметрии) валентных электронов при оди-
наковом их числе.
Поэтому на классическом этапе очевиден тезис о главенстве вер-
тикального сходства химических элементов и меньшего их сходства в
рядах, поскольку в рядах последовательно и существенно изменяется
суммарное число валентных электронов в атомах. Разумеется, и здесь
подчеркивалось горизонтальное сходство исходя из того, что в рядах
соседствуют химические элементы, в атомах которых валентные элек-
троны заселяют атомные орбитали, характеризующиеся одинаковыми
(а не регулярно изменяющимися, как в группах и подгруппах) глав-
ными квантовыми числами. И тем не менее на этом этапе главным
оставалось вертикальное сходство химических элементов в периодиче-
ской системе.
Во второй половине, скорее, в последней трети XX века, многое
изменилось в теоретической трактовке периодической системы. На-
чиная с 70-х годов минувшего столетия теория периодической систе-
мы во многих аспектах основывается, с одной стороны, на теоретико-
групповых представлениях, с другой —на представлениях кайносим-
метрии и гипервалентности. На этом этапе исчезает акцент на верти-
кальном сходстве элементов в системе, и сходство и различия в свой-
ствах элементов вместе с причинами, их порождающими, рассматри-
ваются в своей единой общности равноправно во всех направлениях в
периодической системе.
Каноническая структура периодической системы химических эле-
ментов, ее связь с электронной структурой свободных атомов в ос-
новных состояниях, расположение в ней непереходных элементов и
переходных металлов, вертикальные, горизонтальные и диагональные
аналогии в периодической системе —все это неоднократно изложено
во множестве монографий и учебников.
2.1. Структура системы элементов
Периодическая система максимально эффективно отражает взаи-
мосвязь всех химических элементов. Система воплощает главную идею
о том, что химические элементы, являясь качественно отличными друг
от друга индивидуальный субстанциями, в то же время представля-
ют в совокупности упорядоченное множество, подчиненное внутренней
взаимосвязи всех элементов.
В соответствии с периодическим изменением свойств химических
элементов система состоит из нескольких горизонтально расположен-
ных периодов. В длиннопериодных формах системы каждый период
представляет собой один непрерывный ряд элементов. В короткопери-
19
одних формах системы первых три периода (1-й-3-й) также одноряд-
ные (малые периоды), а остальные периоды, называемые большими,
состоят из двух рядов каждый. В современной периодической системе,
включающей в себя к моменту написания этой книги 109 химических
элементов, семь периодов, причем последний (7-й) не завершен, замы-
кающим его элементом является элемент 118 —благородный газ эка-
радон. Только 1-й период уникален, неповторим, остальные попарно
(2-й и 3-й, 4-й и 5-й, б-й и 7-й) аналогичны по структуре.
Первый период системы включает в себя всего два элемента — во-
дород и гелий, которые не имеют даже отдаленных аналогов среди
всех остальных химических элементов. Каждый следующий период
открывается щелочным металлом и завершается благородным газом.
В каждом периоде, кроме 1-го, за щелочным металлом следует щелоч-
ноземельный. В конце каждого периода, кроме 1-го, непосредственно
перед благородным газом располагаются пять химических элементов,
составляющих вместе с благородным газом шестерку основных эле-
ментов периода. Малые периоды (2-й и 3-й) никаких других элементов
не содержат. Во всех больших периодах начиная с 4-го между щелоч-
ным и щелочноземельным металлами, с одной стороны, и основными
шестью элементами, включая благородный газ, — с другой располага-
ются десять переходных элементов, образующих вставную декаду.
В системе элементов в ее нынешнем состоянии четыре вставные де-
кады, четвертая (в 7-м периоде) не завершена и содержит пока лишь
семь (а не десять) элементов; последний из них элемент 109 — майтне-
рий — пока является вообще самым последним химическим элементом
системы. В б-м и 7-м периодах помимо декад переходных элементов со-
держатся еще по 14 элементов, располагающихся между первыми (La,
Ас) и вторыми (Hf, Rf) переходными элементами. Это — лантаноиды
и актиноиды.
Дискуссия о местоположении в периодической системе следующих
за актинием элементов (Th, Ра, U) окончательно завершилась толь-
ко в 1950-е годы. До синтеза первых трансурановых элементов (Np,
Pu, Am, Cm), т. e. до середины 1940-х годов, торий, протактиний и
уран считались, как и актиний, переходными элементами и занимали
вместе с актинием места в дополнительных подгруппах III-VI групп
периодической системы. После успешного синтеза и изучения первых
трансурановых элементов Г. Сиборг (1944 г.) выдвинул гипотезу о том,
что Th, Ра, U, Np, Pu, Am, Cm составляют первую половину ново-
го 14-элементного семейства актиния, аналогичного 14-элементному
семейству лантана. Дальнейший синтез и изучение все более тяже-
лых трансурановых элементов вплоть до Lr, завершающего семей-
ство актиноидов, полностью подтвердили правильность этой кон-
цепции.
20
Распределение элементов по периодам в порядке строжайшего уве-
личения зарядов ядер их атомов приводит к появлению столбцов в
системе, называемых группами и включающих в себя сходные по свой-
ствам элементы. Наличие непереходных и переходных элементов обу-
словливает формирование двух типов подгрупп — главных и дополни-
тельных. Главные подгруппы (их восемь) начинаются с элементов 2-го
периода (водород и гелий из-за их аномальной химической природы не
следует относить определенно ни к одной из групп периодической си-
стемы). Дополнительные подгруппы (по традиции их также восемь)
короче и начинаются с переходных элементов первой вставной дека-
ды, т. е. с переходных элементов первого большого (4-го) периода. К
дополнительным подгруппам относятся все лантаноиды и актиноиды,
формально — к III дополнительной подгруппе.
Важной характеристикой структуры периодической системы, со-
стоящей лишь из элементов главных подгрупп (без переходных ме-
таллов, лантаноидов и актиноидов), является главная диагональ. Эта
диагональ проходит через элементы, которые занимают места с оди-
наковыми номерами группы и периода:
22Ве 33AI ддСе 55Sb ббРо 77эка-At.
Непереходные элементы, расположенные в системе левее и ниже
этой диагонали, проявляют, главным образом, свойства металлов, наи-
более ярко выраженные в поведении Fr, Cs, Ra, располагающихся в
левом нижнем углу периодической системы. Непереходные элементы,
занимающие места правее и выше главной диагонали, являются неме-
таллами, и самые активные среди них —F, О, С1, находящиеся в пра-
вом верхнем углу системы. Непереходные элементы вблизи главной
диагонали обладают двойственной химической природой.
Главный момент в учении о периодической системе атомов* состо-
ит во взаимосвязи ее структуры (расположения элементов) с электрон-
ным строением свободных невозбужденных атомов. Квантовомехани-
* Необходимо строго учитывать, что периодическая система свободных атомов
и периодическая система химических элементов — понятия, отнюдь, не идентич-
ные. Первая лежит в основе второй, которая по своему содержанию шире и глуб-
же первой. Здесь и далее обсуждается теория периодической системы свободных
нейтральных невозбужденных атомов на основе представлений квантовой механи-
ки с целью описания последовательности электронных конфигураций атомов как
функции атомного номера (ядерного заряда) и установления периодической повто-
ряемости определенных типов атомных электронных конфигураций.
Строгая количественная теория периодической системы элементов пока не со-
здана. Решение многих вопросов периодической системы элементов лишь предва-
рительно намечено на основе периодической системы атомов, поскольку даже дос-
кональное знание электронной структуры атома, распределения его электронов по
валентным орбиталям не всегда позволяет однозначно вывести свойства соответ-
ствующего элемента, особенно в области тяжелых и сверхтяжелых элементов.
21
ческая теория электронного строения атомов (и молекул) является
теоретическим обоснованием как самого Периодического закона, так
и отражающей его периодической системы элементов. Поэтому тра-
диционная теория периодической системы исходит из того, что распо-
ложение элементов в системе (в периодах, группах, подгруппах) при
возрастании их атомных номеров однозначно определяется индивиду-
альными особенностями электронного строения атомов, описанного в
рамках так называемого одноэлектронного приближения, и непосред-
ственно отражает энергетическую последовательность атомных орби-
талей 5-, р-, d-, /-типов, заселяющихся электронами при увеличении
их суммарного числа по мере возрастания заряда ядра атома в соот-
ветствии с принципом минимума энергии.* В строгом подходе необ-
ходимо рассматривать полную энергию атома, минимальное значение
которой определяет наиболее стабильное его состояние. Но при исполь-
зовании одноэлектронного приближения дозволен следующий крите-
рий: наиболее устойчиво (и реализуется) то состояние атома, в кото-
ром его электроны находятся на орбиталях с наиболее низкими из всех
возможных энергиями. Для теоретической интерпретации структуры
периодической системы химических элементов в этом приближении
прежде всего следует определить энергетическую последовательность
одноэлектронных состояний в свободных невозбужденных атомах.
Для достижения указанной цели сделаем последовательно «три
шага».
Первый. В гипотетических водородоподобных атомах с одним
лишь электроном при любом заряде ядра межэлектронные взаимо-
действия, естественно, полностью отсутствуют, орбитальная энергия
этого единственного электрона, являющаяся одновременно и полной
электронной энергией, и вообще полной энергией атома Н и любого
водородоподобного атома, определяется только пространственной уда-
ленностью его зарядовой плотности от ядра (главным квантовым чис-
лом п) и не зависит от особенностей движения электрона в поле ядра
(от всех остальных квантовых чисел, в том числе, что особенно важно,
и от орбитального квантового числа I). Поэтому энергетическая после-
довательность одноэлектронных орбиталей водородоподобного атома
выглядит весьма просто:
Is < 2s = 2р < 3s = Зр = 3d < 4s = 4р = 4d = 4/ <
< 5s = 5р = 5d = 5/ = 5д < 6s = 6р = 6d = 6/ < ...
<7s = 7p = 7d< ... <8s = 8р < ... <9s < ...
* Напомним, что каждой атомной орбитали соответствует определенное рас-
пределение электронной плотности.
22
Здесь орбитальная энергия электрона возрастает только по мере
увеличения п и не изменяется при увеличении I: состояния с разными
Z, но с одинаковыми п энергетически эквивалентны, т. е. соответству-
ющие атомные орбитали (АО) пЦ (например, 6s, бр, 6d, 6/) обладают
одинаковой энергией и оказываются энергетически вырожденными.
Второй. В реальных многоэлектронных атомах из-за эффекта
межэлектронных взаимодействий происходит энергетическое расщеп-
ление (расхождение по энергии) орбиталей с одним и тем главным
квантовым числом п, но с разными орбитальными квантовыми чис-
лами I. Если бы это расщепление было сравнительно небольшим и
меньше обязательного во всех случаях расщепления под воздействием
возрастающего главного квантового числа п
< Зу < 4у 5у < бу < 7у 8у 9у < ...
(здесь х — любая разрешенная АО s-, р-, d-, f-, g-типов), то энергетиче-
ская последовательность атомных орбиталей выглядела бы не так, как
в водородоподобных атомах, но все еще достаточно просто, а именно:
Is 2s < 2р 3s < Зр < 3d 4s < 4р < 4d < 4/ <С
5s < 5р < 5d < 5/ < 5g 6s < бр < 6d < 6/ < ...
<С 7s < 7р < 7d < ... 8s < 8р < ... 9s < ...
Третий. В действительности же расщепление по I начиная с
п = 3, т. е. при п > 3, оказывается большим, чем расщепление по
п. Межэлектронные взаимодействия предопределяют сильную зави-
симость орбитальной энергии каждого электрона уже не только от
пространственной удаленности его зарядовой плотности от ядра (от
главного квантового числа п), но и от способа его движения в поле
ядра (от орбитального квантового числа /). Физическая основа этого
явления состоит в различной проникающей способности АО s-, р-, d-,
f-, g-типов. Наличие расположенных близко к ядру внутренних макси-
мумов зарядовой плотности электронов на атомных орбиталях s- (осо-
бенно) и p-типов и отсутствие таких максимумов для электронов на АО
d- и особенно /-, g-типов объясняет, почему при определенных зарядах
ядер атомов одноэлектронная энергия d-, f-, g-орбиталей значительно
выше таковой для s-, р-орбиталей с большими главными квантовыми
числами. Эти различия тем больше, чем больше разница в значениях
орбитальных квантовых чисел.
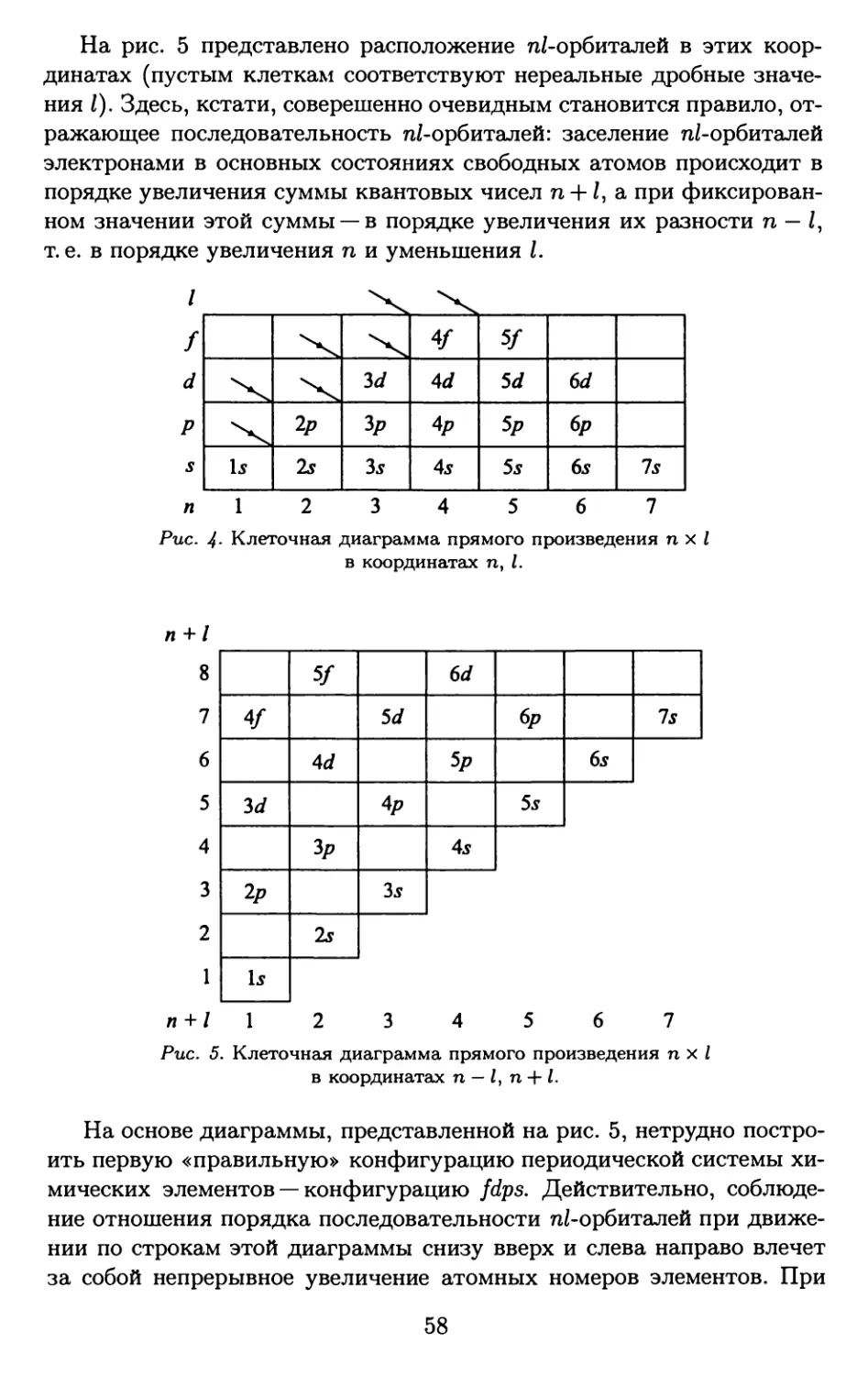
Рассмотрим эти эффекты конкретно. Совершенно очевидно, что
при 0 имеют место энергетические последовательности
23
Is < 25 < 35 < 45 < 5s < 6s < ...,
2p < 3p < 4p < 5p < 6p < ...,
3d < 4d < 5d < 6d < ...,
4/ <5/ <6/ <...,
5g < 6g < ...
При Д/ = 1 наряду с очевидными, уже приведенными ранее ns <
пр, пр < nd, nd < nf, nf < ng, или проще, ns < пр < nd < nf < ng
(например, 5s < 5p < 5d < 5f < 5g), различия в проникающей способ-
ности АО приводят к последовательности (n — 1)р ns, (n — l)d пр,
(п — 1)/ < nd или (п — 1)/ « nd, (п — 1)<? < nf, или (п — 1)р « nf (на-
пример, 2р 3s, 3d < 4р, 4/ < 5d, или 4/ « 5d, 5д < 6f, или 5д & 6/).
При Д/ = 2 увеличивающиеся различия в проникающей способно-
сти АО приводят к ns < (n — l)d, пр < (п — 1)/, nd < (п — 1)д (например,
4s < 3d, 5р < 4f, 6d < 5д).
При Д/ = 3 обсуждаемая тенденция нарастает, и уже ns < (п — 1)/
и даже ns < (п — 2)/, а также пр < (п — 1)р и даже пр < (п — 2)д
(например, 6s < 5/, 6s < 4/, 6р < 5д, 7р < 5д).
Наконец, для последней и самой большой разности Д/ = 4 экстре-
мальные различия в проникающей способности АО обеспечивают ис-
ключительно большое расщепление по I, и реализуются ns < ng,ns <
(п — 1)д, ns < (n — 2)д, ns < (п — 3)д, конкретно 5s < 5д, 6s < 5д, 7s <
5д, 8s < 5д.
Рассмотренные эффекты в своей совокупности приводят для сво-
бодных невозбужденных атомов к следующей приближенной энергети-
ческой последовательности одноэлектронных состояний, заселяющих-
ся электронами при увеличении их суммарного числа по мере возрас-
тания заряда ядра атома:
Is < 2s < 2р < 3s < Зр < 4s < 3d < 4р < 5s < 4d < 5р < 6s < 4/ «
~ 5d < 6р < 7s < 5/ ~ 6d < 7р < 8s < 5g ~ 6/ ~ 7d < 8р < 9s < ...
Эта последовательность близка к действительности, как об этом
свидетельствуют результаты неэмпирических квантовомеханических
расчетов электронного строения атомов и имеющиеся для реально су-
ществующих атомов экспериментальные результаты атомной спектро-
скопии.
Сделаем небольшое, но важное отступление. Осуществление вы-
веденной с учетом всех эффектов электроноядерных взаимодействий
последовательности АО в многоэлектронных атомах
24
15 < 2s < 2p < 3s < 3p < 4s < 3d < 4p < 5s < 4d < 5p < 65 < 4/ «
« 5d < 6p < 7s < 5/ « 6d < 7p < 8s < 5 <7 « 6/ « 7d < Sp < 9s < ...
происходит лишь при некоторых определенных значениях зарядов
ядер атомов. Именно при таких значениях этих зарядов и, следо-
вательно, при таких суммарных числах электронов в атомах, кото-
рые соответствуют заселению обсуждаемых АО электронами, т. е.
когда обсуждаемые АО являются внешними, валентными. При зна-
чительно больших зарядах ядер, и, следовательно, при значительно
больших суммарных числах электронов в атомах, все эффекты элек-
троноядерных и межэлектронных взаимодействий становятся, разу-
меется, иными и по-иному воздействуют на энергетическое распреде-
ление тех же самых атомных орбиталей, которые в этом случае
переходят из внешних, валентных, во внутренние, остовные. Типич-
ность ситуации здесь состоит в том, что в области остовных орбита-
лей атомов их энергетическая последовательность всегда оказывается
«равномерной», квазиводородной:
Is < 2s < 2р < 3s < Зр < 3d < 4s < 4р < 4d < 4/ < 5s < 5р < 5d <
< 5/ < 5р 6s < бр < 6d < б/ < ... 7s < 7р < 7d < ... 8s <
< 8р < ... < 9s < ...
Поясним это на конкретном примере энергетической последова-
тельности АО 3d, 4s, 4р. При зарядах ядер Z = 19 4- 36 и таких же
суммарных числах электронов в атомах элементов с такими же поряд-
ковыми номерами (элементов К, Са, Sc, Ti, V, Cr, Мп, Fe, Со, Ni, Си,
Zn) энергетическая последовательность указанных АО (а они в этих
случаях валентные) такова:
4s < 3d < 4р.
При выходе за этот интервал в сторону больших значений пере-
численных параметров (т. е. для всех остальных элементов начиная
с 37-го, рубидия, и до конца периодической системы) энергетическая
последовательность тех же самых АО (а они теперь остовные) «вос-
станавливается» до «равномерной»:
3d 4s < 4р.
Иными словами, при переходе от одних элементов к другим энерге-
тическое распределение атомных орбиталей закономерно изменяется.
25
Физическая причина этого явления состоит в том, что энергия каждой
одноэлектронной АО зависит от значения ядерного заряда и суммар-
ного числа электронов в атоме. И эти зависимости носят сложный, в
общем случае неодинаковый, несимбатный характер.
Остовные орбитали далеко отстоят по своим энергиям от валент-
ных АО, практически не участвуют в образовании химических связей
в рождающихся молекулах и потому почти не оказывают влияния на
свойства атомов, на свойства химических элементов. Именно поэтому
наиболее важна последовательность заселяющихся электронами ва-
лентных атомных орбиталей. Именно поэтому для теории периодиче-
ской системы, точнее, для теоретической интерпретации ее структу-
ры, важно только то энергетическое распределение АО, которое для
заданного интервала зарядов ядер и суммарных чисел электронов в
атомах характеризует заселяющиеся электронами валентные атомные
орбитали.
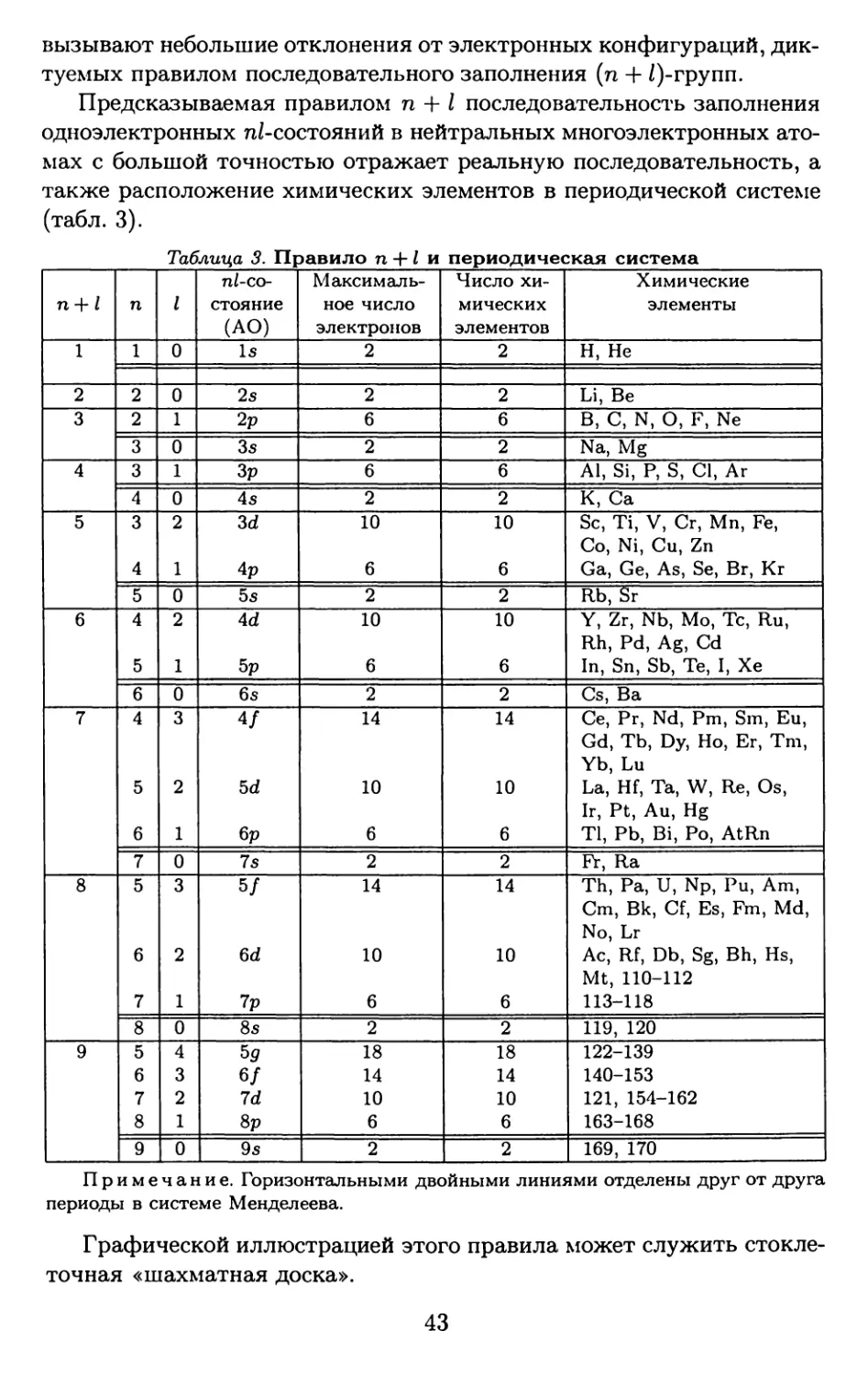
На основе установленной энергетической последовательности за-
селяющихся электронами валентных атомных орбиталей в реальных
атомах
15 < 2s < 2р < 35 < Зр < 45 < 3d < 4р < 5s < 4d < 5р < 6s < 4/ «
w 5d < бр < 7s < 5/ « 6d < 7p < 8s < 5g w 6/ « 7d < Sp < 9s < ...
можно теперь во всех подробностях проследить, как расположение хи-
мических элементов в периодической системе связано с электронным
строением атомов в целом и их валентной электронной структурой в
особенности.
Периодическая система открывается двумя единственными элемен-
тами 1-го периода — водородом и гелием, —в атомах которых содер-
жатся соответственно один или два валентных электрона на орбитали
1s, не экранированной от ядра остовными электронами по причине их
отсутствия. Это последнее обстоятельство вкупе с отсутствием каких-
либо вакантных АО в первом слое электронной оболочки с п = 1
резко отличает электронную структуру атомов Н (1s1) и Не (1s2) от
электронной структуры атомов всех остальных химических элементов.
Уникальная электронная структура атомов Н и Не объясняет, почему
этих два элемента, составляющих 1-й период, стоят в периодической
системе особняком и не относятся ни к одной из ее групп (подгрупп).
Каждый период системы начиная со 2-го открывается элементом, в
атомах которого сверх предшествующей замкнутой электронной обо-
лочки находится лишь один электрон на валентной АО ns. Значение
главного квантового числа всегда совпадает с номером периода. Завер-
шается любой период элементом, атомы которого имеют замкнутую
электронную оболочку с конфигурацией ns2пр6.
26
Во втором и третьем слоях электронной оболочки атомов существу-
ют лишь орбитали ns и пр, заселяемые в соответствии с запретом Па-
ули максимально восемью электронами. Именно поэтому во 2-м и 3-м
(малых) периодах содержится лишь по восемь элементов главных под-
групп. Электронные конфигурации их атомов сверх замкнутых оболо-
чек—от ns1np° до ns2пр6.
В четвертом и во всех последующих слоях электронной оболочки
атомов наборы АО разрастаются, и структура всех больших периодов
начиная с 4-го принципиально иная по сравнению с малыми периода-
ми. В 4-м - 7-м слоях помимо орбиталей ns, пр присутствуют орбитали
(n — l)d, располагающиеся по энергии между ns- и пр-АО; на них спо-
собны разместиться десять электронов. В 4-м - 7-м периодах системы
помимо восьми элементов главных подгрупп (с атомными электронны-
ми конфигурациями от П51пр° до ns2np° и от ns2np* др ns2np6) после
вторых элементов следуют вставные декады, насчитывающие по де-
сять переходных элементов дополнительных подгрупп. Электронные
конфигурации их атомов —от ns2(n — l)^1 до ns2(n — l)d10.
Шестой и седьмой слои электронных оболочек атомов содержат до-
полнительно орбитали (п — 2)/, располагающиеся по энергии между
АО ns и (n — l)d и предоставляющие состояния для 14 электронов. В
6-м и 7-м периодах системы находится дополнительно по 14 элемен-
тов — соответственно лантаноидов и актиноидов с атомными электрон-
ными конфигурациями от ns2(n—l)^1^—2)/1 до ns2(n—Ijd^n—2)/14.
Энергетическая последовательность заселяющихся электронами внеш-
них атомных орбиталей в шестом и седьмом слоях, как было показано,
выглядит следующим образом:
6s < 4/ « 5d < 6р и 7s < 5/ « 6d < 7р.
Близость энергий 4/- и 5б/-орбиталей, а также 5/- и бй-орбиталей не
позволяет сделать однозначные выводы относительно очередности их
заселения электронами. В реальных многоэлектронных атомах после
обязательного заполнения одним и двумя электронами орбитали 6s (Cs
6s1 и Ba 6s2) или 7s (Fr 7s1 и Ra 7s2) третий электрон занимает орби-
таль 5d (La 6s25d1) или 6d (Ac 7s26d1). Следующие 14 электронов засе-
ляют орбиталь 4/ (14 лантаноидов от Се 6s25d14/1 до Lu 6s25d14/14)
или 5/ (14 актиноидов от Th 7s26d15/1 до Lr 7s26d15/14). После это-
го продолжается и завершается заселение двумя-десятью электрона-
ми орбитали 5d (от Hf 6s25d24/14 до Hg 6s25d104/14) или 6d (от Rf
7s26d25/14 до эяа-Hg 7s26d105/14). По этой причине в 6 -м или 7-м пе-
риодах системы лантаноиды или актиноиды располагаются не сразу
за первыми двумя (щелочным Cs или Fr и щелочноземельным Ва или
Ra) элементами, а «пропускают вперед» первый переходный металл
27
(La или Ac) и вклиниваются между ним и остальными девятью пере-
ходными металлами соответствующей (третьей или четвертой) встав-
ной декады.
Седьмой период системы не завершен. Последний пока химический
элемент — майтнерий (109, 7s2 6d7)—находится в четвертой вставной
декаде. До се завершения необходимы еще три элемента — эка-Pt (ПО,
7s26d8), эка-А.и (111, 7s26d9), эка-Hg (112, 7s26d10). Затем должны
последовать шесть элементов от эка-Т1 (113, 7s27p1) до эка-Rn (118,
7s27p6) — благородного газа, который завершит 7-й период.
За исключением только что указанных сложностей в заполнении
электронами валентных АО энергетическая последовательность
1э < 2s < 2р < 3s < Зр < 4s < 3d < 4р < 5s < 4d < 5р < 6s < 4/ «
« 5d < 6р < 7s < 5/ « 6d < 7p < 8s < 5g ~ 6f « 7d < Sp < 9s < ...
в целом верно описывает очередность заполнения электронами ns-,
пр-, nd-, п/-орбиталей в изолированных невозбужденных атомах и слу-
жит основой для интерпретации расположения всех химических эле-
ментов в периодической системе на основе электронного строения их
атомов. Но эта последовательность не отражает специфических осо-
бенностей валентных электронных конфигураций атомов некоторых
элементов в основном состоянии, т. е. в состоянии с наменыпей пол-
ной энергией. Есть только одно регулярное правило касательно этих
специфических особенностей. В квантовой механике атома есть из-
вестное положение о максимальной стабилизирующей энергии внут-
риатомного обмена электронов в случае электронных конфигураций и
соответственно о повышенной энергетической стабильности конфигу-
раций, включающих в себя полностью или наполовину заполненные nl-
состояния ns1, ns2, пр3, пр6, nd5, nd10, nf7, nf 14, ng9, ng18 или вообще
вакантные п/-состояния. Это объясняет, почему следующие атомы име-
ют электронные конфигурации Cr 4s13d5, а не 4s23d4, Mo 5s4d5, а не
5s24d4, Си 4s13d10, а не 4s23d°, Ag 5s4d10, а не 5s24d9, Au Gs^d10, а не
6s25d9, Ce 6s25d24/°, a ne 6s25d14/1, Th 7s26d25/°, а не 7s26d15/1, Eu
6s25d°4/7, а не 6s25d14/6, Am 7s26d°5/7, а не 7s26d15/6, Tb 6s25d24/7,
а не 6s25d14/8, Bk 7s26d25/7, а не 7s26d15/8, Yb 6s25d°4/14, а не
6s25d14/13, No 7s26d°/14, а не 7s26d15/13.
Здесь следует иметь в виду, что подобные «регулярности» в форми-
ровании специфических валентных электронных конфигураций ато-
мов вовсе не обязательны. Так, в отличие от атомов хрома и молиб-
дена невозбужденные атомы их полного аналога — вольфрама имеют
«тривиальную» валентную электронную конфигурацию W 6s25d4, а
не 6s15d5. Далее, следование обсуждаемому правилу не типично для
атомов с валентными ns-, пр-(но не nd-, nf-) орбиталями; для таких
28
атомов характерны «тривиальные» конфигурации. Классический при-
мер — атом углерода с электронной конфигурацией в основном состо-
янии С 2s22p2, а не 2s12p3.
Есть и иные примеры: Nb 4s13d4, а не 4s23d3, Ru бзЧс?7, а не 5s24d6,
Rh бзЧс?8, а не 5s24d7, Pd 5s°4d10, а не 5s24d8 и даже не 5s4d9, Pt
6s15d9, а не 6s25d8. Эти «нерегулярные» специфические особенности
валентных электронных конфигураций невозбужденных атомов неко-
торых элементов объясняются самым общим образом: именно для та-
ких конфигураций минимальна полная энергия атома и (часто, но
не всегда) минимальна дестабилизирующая энергия межэлектронно-
го отталкивания. Этот наиболее общий тезис, разумеется, абсолютно
справедлив и для всех «регулярных» специфических особенностей, и
вообще для всех «тривиальных» электронных конфигураций невоз-
бужденных атомов.
Обсуждаемые эффекты иногда приводят к неодинаковым валент-
ным электронным конфигурациям для атомов элементов-аналогов од-
ной и той же подгруппы системы. Единственный случай, когда эти
конфигурации различны для атомов всех одновременно элементов
одной и той же подгруппы:
Ni Pd Pt
4s23d8 5s°4d10 6s15c?9
До сих пор нет окончательного ответа на вопрос, у атомов ка-
ких лантаноидов и актиноидов впервые появляются п/-электроны.
Отмеченная близость одноэлектронных энергий валентных орбиталей
4/, 5d, и особенно 5/, 6d, затрудняет отнесение (даже при исполь-
зовании атомной спектроскопии) валентных электронов к (п — 2)/-
или (п — 1) d-орбиталям для атомов элементов, стоящих в периодиче-
ской системе сразу после лантана или актиния. Установлено, что, по
крайней мере, в невозбужденных атомах церия и тория, первого лан-
таноида и первого актиноида, п/-электронов нет, и эти атомы имеют
валентные электронные конфигурации Се 6s25d2 и Th 7s26d2 вместо
ожидаемых Се 6525d14/1 и Th 7s26d15/1. Возможно и даже вполне
вероятно, что и атомы вторых элементов этих семейств не содержат
п/-электронов и имеют конфигурации Pr 6s25d3 и Ра 7s26d3 вместо
ожидаемых Pr 6s25cP4/2 и Ра 7526d15/2.*
* Независимо от того, имеются ли в атомах первых и, возможно, вторых ланта-
ноидов (Се, Рг) и актиноидов (Th, Ра) в невозбужденном состоянии п/-электроны,
все 14 лантаноидов от Се до Lu и все 14 актиноидов от Th до Lr являются пол-
ноправными членами семейств лантана и актиния. Причина состоит в том, что
из-за близости орбитальных энергий 4/, 5d, и особенно 5/, 6d, химические свой-
ства всех двадцати восьми лантаноидов и актиноидов являются отражением той
29
Причина всех этих эффектов и явлений — индивидуальность всех
электрон-электронных и электроноядерных взаимодействий для каж-
дого атома, суммарная энергия которых непременно должна быть ми-
нимальной.
Итак, расположение всех (и даже предсказываемых) элементов в
периодической системе, в принципе, полностью соответствует элек-
тронному строению их невозбужденных атомов и, прежде всего, непо-
средственно отражает их валентную электронную структуру. В зави-
симости от того, какого типа (5, р, d или /) орбиталь в атоме засе-
ляется последним электроном, все химические разновидности подраз-
деляются на четыре большие разновидности: 5-элементы, р-элементы,
d-элементы, /-элементы.
В каждом периоде системы находятся два 5-элемента, атомы кото-
рых имеют валентные электронные конфигурации ns1 и ns2.
В каждом периоде системы начиная со 2-го находятся шесть
p-элементов главных подгрупп — непереходных элементов (п52пр1-
П52пр6).
В целом все элементы главных подгрупп представляют собой
5-элементы и р-элементы.
В каждом большом периоде системы начиная с 4-го находятся де-
сять d-элементов дополнительных подгрупп — переходных металлов
(п52(п — l)d1-n52(n — l)d10).
В каждом из двух последних периодов, в б-м и 7-м, находятся
14 /-элементов — актиноидов или лантаноидов (п52(п — l)d1(n — 2)/1-
П52(п — Ijd^n — 2)/14). Все они принадлежат к третьей дополнитель-
ной подгруппе, поскольку электроны на п/-орбиталях формально не
входят в категорию валентных.
В целом все элементы дополнительных подгрупп представляют со-
бой d-элементы и /-элементы.
Каждый период системы представляет собой последовательность
элементов, атомы которых различаются числом валентных электро-
нов. Каждая группа системы является единой совокупностью эле-
ментов (независимо от их принадлежности к главной или допол-
нительной подгруппе), атомы которых обладают одинаковым чис-
лом валентных электронов, равным номеру группы. Исключение со-
ставляет особая группа периодической системы — VIII. Атомы эле-
ментов главной подгруппы VIII имеют 8-электронную конфигурацию
ns2пр6. Из девяти элементов дополнительной подгруппы VIII толь-
ко атомы железа, рутения и осмия имеют 8-электронные конфигура-
ции П52(п — l)d6. Конфигурации атомов кобальта, родия и иридия —
общей особенности электронного строения их атомов, которая отвечает заселению
электронами внешних 4/- или 5/-орбиталей. Никакие другие элементы, кроме этих
двадцати восьми, не обладают такой особенностью.
30
9-электронные ns2(n — l)d7, конфигурации атомов никеля, палладия
и платины — 10-электронные ns2(n— l)d8.
В завершение этого раздела, во многом задающего тон последую-
щему изложению теории периодичности, сделаем несколько важных и
обязательных пояснений.
Выше детально рассмотрена последовательность заполнения элек-
тронами атомных орбиталей нейтральных изолированных атомов в ос-
новных состояниях по мере возрастания ядерного заряда и суммарного
числа электронов на основе относительных энергий (еп/) атомных ор-
биталей. При этом относительное расположение атомных орбиталей по
энергии (т. е. на шкале одноэлектронных энергий eni) объяснено инди-
видуальными особенностями самих атомных орбиталей, в частности
их узловой структурой, а также эффектами экранирования и т. д. Это
возможно лишь в самом простом варианте Хартри, но уже в вариан-
те Хартри — Фока полная энергия атома не равна сумме орбитальных
энергий, и электронная конфигурация атома определяется минимумом
его полной энергии. Поэтому традиционная интерпретация структу-
ры периодической системы на основе последовательности заполнения
электронами атомных орбиталей в соответствии с их относительны-
ми энергиями eni весьма и весьма приближенна, имеет, безусловно,
ряд недостатков и обладает неширокими границами применимости.
Универсальной последовательности орбитальных энергий eni не суще-
ствует, к тому же такая последовательность не определяет полностью
порядок заселения атомных орбиталей электронами, поскольку необ-
ходим учет конфигурационных взаимодействий (наложение конфигу-
раций в многоконфигурационном приближении). И, безусловно, пери-
одичность — это не только и не полностью орбитально-энергетические
эффекты.
В одном из обсуждений с нами учения о периодичности профессор
И. С. Дмитриев, ведущий, по нашему убеждению, современный специ-
алист в этой области, сказал: «Трудности дидактического преподне-
сения читателю столь противоречивого и до сих пор во многом за-
гадочного феномена как принцип периодичности в большой степени
определяются необходимостью постоянного удержания в мысли раз-
граничения между удобными, ясными и красивыми моделями для про-
стых и наглядных качественных пояснений, с одной стороны, и стро-
гой теорией — с другой. Чем строже трактовка, тем яснее ощущение,
что причина повторения сходных электронных конфигураций атомов
в их основных состояниях от нас ускользает, и в рамках одноэлек-
тронного приближения вряд ли вообще может быть выявлена. Более
того, не исключено, что теорию периодичности вообще ждет судьба,
несколько напоминающая судьбу теории планетных ретрогрессий в си-
стеме Птолемея после создания системы Коперника. Вполне возмож-
31
но, что то, что мы называем принципом периодичности, есть результат
непространственных симметрий атома — перестановочной и динами-
ческой».
2,2, Периодичность и симметрия
В пределах одной и той же группы периодической системы элемен-
ты ее главной и дополнительной подгрупп различаются тем, что в их
атомах одно и то же число валентных электронов распределяется по
орбиталям разного типа симметрии распределения зарядовой плотно-
сти: по ns-, пр-орбиталям в атомах элементов главной подгруппы и по
ns-, (n — l)d-, (п — 2)/-орбиталям атомов элементов дополнительной
подгруппы. Это и есть главная причина различий в свойствах непе-
реходных элементов и переходных металлов одной и той же группы,
различий, проявляющихся на фоне известного сходства всех элемен-
тов группы, обусловленного одинаковым числом валентных электро-
нов в их атомах. Этот признак, кстати, очень важен для понимания
физико-химической природы всех элементов в целом, составляющих
группу периодической системы.
Описывающая одноэлектронную атомную орбиталь одноцентровая
пространственная волновая функция (точнее, пространственная часть
этой волновой функции без спинового множителя с нормирующим
множителем 7V)
Xnlm^Pi @ = NRnl (р) Ут ($»
включает в себя не зависящую от главного квантового числа п угло-
вую составляющую У/т(0, <р), которая и отражает симметрию рас-
пределения зарядовой плотности электрона, или просто электронной
плотности, в поле ядра. Это — очень важный параметр валентной элек-
тронной структуры атома с точки зрения образования этим атомом
химических связей в молекуле. Угловая составляющая У/т(0, <р) опре-
деляется орбитальным I и магнитным т квантовыми числами. Маг-
нитное квантовое число т описывает ориентацию соответствующего
электронного облака в поле ядра относительно некоего направления,
тогда как орбитальное квантовое число I описывает собственно сим-
метрию электронного облака, что значительно важнее для описания
природы химических связей в молекулах. Именно поэтому орбиталь-
ные квантовые числа I валентных электронов в атомах, отражающие
симметрию пространственного распределения электронной плотности,
определяют принадлежность соответственных химических элементов
к главной или дополнительной подгруппе данной группы периодиче-
ской системы. Одинаковые по орбитальным квантовым числам I ва-
32
лентные электроны в атомах (например, валентные 2р-, Зр-, 4р-, 5р-,
бр-электроны в атомах элементов какой-либо главной подгруппы или
валентные 3d-, 4d-, 5б/-электроны в атомах элементов некоторой допол-
нительной подгруппы) создают основу для высокого сходства свойств
соответственных химических элементов в пределах той или иной под-
группы системы, несмотря на последовательно изменяющееся главное
квантовое число п валентных электронов в этих атомах.
Значение орбитального квантового числа I определяет не только
тип симметрии изовероятностной поверхности (проще говоря, форму
электронного облака) соответствующей атомной орбитали. Для каж-
дой АО величина I указывает также число узловых плоскостей, про-
ходящих через центр атома и не содержащих зарядовой плотности
электрона (на этих узловых плоскостях электронная плотность соот-
ветствующего состояния равна нулю). Это число узловых плоскостей,
в свою очередь, определяет число узловых поверхностей — тех центри-
рованных на ядре сферических поверхностей в электронной оболочке
атома, на которых зярядовая плотность соответствующего электрон-
ного состояния равна нулю. Число таких узловых поверхностей опре-
деляется соотношением п — I — 1,* где единица символизирует един-
ственную для любого атома, центрированную также на ядре, сфери-
ческую граничную поверхность, за пределами которой электронная
плотность практически отсутствует. Число же указанных узловых по-
верхностей является очень важной характеристикой радиального рас-
пределения зарядовой плотности одноэлектронной валентной атомной
орбитали. Это распределение описывается радиальной составляющей
Rni(p) пространственной волновой функции. Радиальная составляю-
щая Rni (р) определяется главным п и орбитальным I квантовыми чис-
лами и содержит информацию о числе внутренних максимумов элек-
тронной плотности для соответствующей валентной атомной орбита-
ли — очень важной ее характеристике для теории молекулы. Очевид-
но, что число таких внутренних максимумов на единицу больше числа
узловых поверхностей и равно простой разности п — I.
В пределах каждой (главной или дополнительной) подгруппы си-
стемы объединяются элементы, невозбужденные атомы которых об-
ладают количественно (число валентных электронов) и качественно
(их распределение по орбиталям того или иного типа) единообразной
валентной электронной конфигурацией nsanp^ для атомов элементов
* Это соотношение вытекает из следующего канона квантовой механики атома:
число всех математических (с нулевой толщиной) поверхностей для данного одно-
электронного состояния, на которых отсутствует его зарядовая плотность, равно
главному квантовому числу п этого состояния. Граничная поверхность всегда од-
на, число узловых плоскостей равно /, следовательно, число узловых поверхностей,
действительно, равно п — I — 1.
33
главной подгруппы или nsa(n — 1)£*(п — 2)f& для атомов элементов
дополнительной подгруппы. Это означает, что подгруппу периодиче-
ской системы составляют элементы, атомы которых являются пол-
ными электронными аналогами. Эта строгая электронная аналогия
валентных структур свободных атомов отвечает за близость свойств
элементов одной и той же подгруппы, но не исключает и различий в
свойствах, обязанных своим происхождением изменяющимся (в под-
группе) значениям главных квантовых чисел валентных орбиталей и
некоторым другим факторам, в частности наличию или отсутствию
(в валентном слое) вакантных — виртуальных — орбиталей с теми же
главными квантовыми числами, что и для заселяющихся электрона-
ми — реальных — орбиталей.
Главными с позиций общехимического толка следствиями исполь-
зования традиционной теории периодической системы, традиционного
описания ее структуры являются объяснение и всесторонняя интер-
претация первостепенных и более частных закономерностей изменения
свойств химических элементов в группах, подгруппах, периодах.
Периодичность валентных электронных конфигураций свободных
атомов порождает периодичность свойств химических элементов на-
чиная с валентности их атомов и тем самым приводит к появлению
нескольких аналогий в поведении элементов в периодической системе.
Наиболее ярко, безусловно, выражены вертикальные аналогии —
в каждой подгруппе по причине одинакового числа валентных элек-
тронов в атомах элементов одной и той же подгруппы и одинакового
распределения электронов по валентным атомным орбиталям s-, р-, d-,
/-типов. Между тем всегда следует иметь в виду, что элементы одной
и той же подгруппы проявляют не только большое сходство в сво-
их свойствах, но иногда и существенные различия. Иными словами,
элементы одной и той же подгруппы могут быть лишь формальны-
ми аналогами, но не быть функциональными аналогами. Достаточно
сопоставить свойства аммиака и малоустойчивого висмутина или по-
ражающие воображение свойства воды и «дышащего на ладан» ме-
тастабильного дигидрида полония. Вертикальные аналогии в группе
в целом выражены, несомненно, значительно слабее, поскольку при
сохранении одинаковым числа валентных электронов в атомах суще-
ственными оказываются различия в распределении электронов по ва-
лентным атомным орбиталям s-, р-, d-, /-типов.
Несмотря на последовательное изменение числа валентных элек-
тронов в атомах элементов того или иного периода системы и скачкооб-
разное изменение типа валентных атомных орбиталей, заселяющихся
электронами, горизонтальные аналогии в системе — обычное и важное
явление. Приведем два весьма разнохарактерных примера: сходство
типических (по терминологии самого Менделеева) элементов 2-го пе-
34
риода Li, Be, В, C, N, О, F и сходство тяжелых переходных метал-
лов 6-го периода Hf, Та, W, Re, Os, Pt. В горизонтальных аналогиях
первостепенным является, конечно, одинаковость значений главных
квантовых чисел соответственных (по орбитальному квантовому чис-
лу) валентных электронов в атомах в периодах, что не имеет места в
случае вертикальных аналогий в группах и подгруппах системы.
Элементы, находящиеся на одной и той же диагонали (не обя-
зательно на главной) системы, принадлежат, естественно, к разным
группам и разным периодам; их атомы содержат разное число валент-
ных электронов и к тому же на орбиталях, различающихся главными
квантовыми числами. Поэтому в диагональной серии элементов почти
нет сходства по формам образуемых химических соединений, но тем
не менее часто проявляется близость по химической функции. Диаго-
нальные аналогии — это прежде всего аналогии функциональные. Эле-
менты одной и той же диагональной серии сходны по функционально-
му, качественному, химическому содержанию, несмотря на существен-
ные различия в формах образуемых соединений и особенностях их
молекулярного строения. Основная причина проявления диагональ-
ных аналогий в системе заключается в том, что валентные электроны
атомов элементов одной и той же диагональной серии обладают близ-
кими (иногда очень близкими, чуть ли не совпадающими) энергиями.
Примерное постоянство энергий одноэлектронных атомных орбиталей
объясняется, в свою очередь, весьма просто — своеобразной компенса-
цией их разных по знаку градиентов, характерных для изменения этих
энергий в периоде и подгруппе. Отсюда и функциональное сходство
диагонально расположенных элементов.
Аналогии в периодической системе служат надежным инструмен-
том для предсказаний не известных еще свойств и явлений. Теоре-
тический фундамент для предсказания неизвестных свойств химиче-
ских элементов, их простых веществ и химических соединений состоит
в общности квантовомеханических принципов электронного строения
атомов (и молекул). Но, разумеется, масштабность и точность таких
предсказаний во многом зависят от того, насколько верно установлены
закономерности изменения каких-либо свойств элементов и их соеди-
нений в периодах, группах, подгруппах системы.
2.3. Правило последовательного заполнения (п + I)-групп
Важнейшим положением теории периодической системы химиче-
ских элементов является интерпретация зависимости числа элементов
в периоде от его номера, иными словами, концепция, объясняющая
длину и границы периодов.
35
Периоды в системе начиная с 1-го содержат 2, 8, 8, 18, 18, 32, 32,
(50), (50), ...элементов. Первым, кто попытался передать этот ряд
чисто формальным образом, был И. Ридберг. Анализируя структуру
периодической системы, И. Ридберг ввел представление о диадах —
группах парных периодов и предложил ряд для порядковых номеров
элементов, завершающих периоды:
Р = 2(12 + I2 + 22 + 22 + З2 + З2 + 42 + 42 + ...).
Последовательность чисел в ряду Ридберга
2, 2, 8, 8, 18, 18, 32, 32, ...
несколько не соответствует реальности, так как 1-й период не повто-
ряется; последовательность в точности соответствует периодической
системе элементов, если исключить первый член ряда. Истинный ряд
может быть передан предложенным Тюдесом выражением 2[n/2 +1]2,
если при вычислениях брать только целочисленную часть стоящего в
квадратных скобках аргумента.
В классическом, традиционном, подходе число химических элемен-
тов в периоде системы объясняется (для конца системы — предсказы-
вается) максимальным числом электронов, которые могут находиться
в соответствующем слое электронной оболочки атома, т. е. тех, кото-
рые описываются главным квантовым числом п, равным номеру слоя,
а стало быть, и номеру периода.
Квантовая механика атома позволяет на основе принципа исключе-
ния (запрета) Паули установить, какое максимальное число электро-
нов заселяет всю совокупность разрешенных состояний с различными
орбитальными /, магнитными т и спиновыми s квантовыми числами
при заданном главном квантовом числе п. Максимальное число таких
электронов определяется выражением
1=71 — 1
^2 2(2/+ 1) = 2п2,
1=0
где 2(2/ 4-1) — наибольшее число электронов, которые в соответствии с
принципом Паули могут заселять в атоме все разрешенные состояния
с заданным I. Это означает, что в каждом слое электронной оболоч-
ки атома (а каждый такой слой характеризуется строго определен-
ным значением главного квантового числа п) должны размещаться
максимально 2п2 электронов, и распределение электронов по замкну-
тым, т. е. полностью заполненным, слоям в электронной оболочке ато-
ма должно быть таковым:
2,8,18,32,50,...
36
Действительно, это распределение в точности соответствует выве-
денной еще в ранних работах Э. Шредингера последовательности од-
ноэлектронных nZ-состояний в водородоподобных атомах, в которых
присутствует только один электрон, полностью отсутствует межэлек-
тронное взаимодействие, энергия электрона определяется только глав-
ным квантовым числом п и не зависит от орбитального квантового
числа I:
1s 2s 2р 3s Зр 3d 4s 4р 4d 4f 5s 5р 5d 5/ 5д
2 2 6 2 6 10 2 6 10 14 2 6 10 14 18
2е" 8е~ 18е" 32е" 50е“
Теория самосогласованного поля Хартри —Фока для многоэлек-
тронных атомов со строгим учетом межэлектронных взаимодействий
дает следующую приближенную последовательность одноэлектрон-
ных nZ-состояний:
1s 2s 2р 3s Зр 4s 3d 4р 5s 4d 5р
2 2 6 26 2 10 6 2 10 6
2е" 8е" 8е~ 18е“ 18е“
6s 4/ 5d 6р 7s5/6d7p 8з 5р 6/ 7d 8р 9s 6д 7/ 8d9p
2 14 10 6 2 14 10 6 2 18 14 10 6 2 18 14 10 6
32е" 32е" 50е“ 50е"
Это распределение электронов по полностью заселенным слоям в
оболочке многоэлектронного атома
2, 8, 8, 18, 18, 32, 32, 50, 50
уже без малейшего отклонения соответствует числам элементов в пе-
риодах менделеевской системы. Это истинное распределение электро-
нов в отличие от идеализированного (2, 8, 18, 32, 50, ...) отражает за-
висимость энергии электронов в многоэлектронном атоме не только от
значений главного квантового числа п, но уже и от значений орбиталь-
ного квантового числа I — в многоэлектронном атоме по мере увеличе-
ния ядерного заряда и числа электронов (по мере увеличения поряд-
кового номера химического элемента) возрастает отсутствующая в во-
дородоподобном атоме роль углового момента и соответственно умень-
шается относительная роль радиального пространственного распреде-
ления зарядовой плотности в определении энергии одноэлектронного
^/-состояния. В математическом формализме это отвечает возрастаю-
щей роли угловой составляющей и относительному уменьшению роли
радиальной составляющей (протяженность и число узлов) волновой
функции, описывающей это одноэлектронное nZ-состояние.
37
Каноническую структуру периодической системы хорошо передает
известное эмпирическое правило Клечковского — Демкова — Остров-
ского [10-13].
При использовании статистической модели Томаса — Ферми в 60-х
годах было теоретически сформулировано и на основе анализа опти-
ческих и рентгеновских термов многоэлектронных атомов обосновано
общее правило, исходящее из тех главных представлений, что пери-
одическая система химических элементов есть результат последова-
тельности одноэлектронных уровней в атомах и порядка заполнения
атомных орбиталей электронами. Согласно этому правилу заполнение
электронами nZ-состояний в нейтральных невозбужденных атомах с
увеличением заряда ядра, числа электронов, порядкового номера хи-
мического элемента в периодической системе должно происходить по-
следовательными (п + /)-группами, т. е. по возрастающим значениям
суммы главного п и орбитального I квантовых чисел. Под (п + Г)-
группой подразумевают всю совокупность nZ-состояний с одинаковым
значением суммы главного и орбитального квантовых чисел.
В терминах теории атомных орбиталей это правило означает, что
энергетической предпочтительностью (меньшей энергией) обладают
АО с минимальной возможной суммой главного п и орбитального I
квантовых чисел. Согласно этому правилу при соблюдении принци-
па минимума полной энергии нейтрального невозбужденного атома
порядок заполнения электронами его АО определяется минимизацией
суммы n+Z, а при одинаковых значениях этой суммы — минимизацией
главного квантового числа п, а именно: в нейтральных невозбужден-
ных атомах электроны в первую очередь заселяют атомные орбитали,
характеризующиеся минимальным возможным значением суммы п+1,
а при фиксированной сумме — в порядке увеличения главного кванто-
вого числа п или, что то же самое, в порядке уменьшения орбитального
квантового числа I.
Итак, очередность заполнения электронами (п + /)-групп и состав-
ляющих эти группы nZ-состояний по мере возрастания порядково-
го номера элемента отвечает следующему общему правилу, получив-
шему название правила последовательного заполнения (п + 1)-
групп: с увеличением заряда ядра атома, суммарного числа электро-
нов в атоме, атомного номера элемента заполнение одноэлектронных
состояний свободных невозбужденных нейтральных атомов происхо-
дит последовательно от групп с меньшим значением суммы п +1 глав-
ного и орбитального квантовых чисел к группам с большим значением
этой суммы, а в пределах каждой (п + ^-группы —от Ы-состояний
с меньшим значением п и большим значением I к nZ-состояниям с
большим значением п и меньшим значением I. Заполнение каждой
(п + /)-группы завершается двумя s-электронами, после чего проис-
38
ходит переход к заполнению следующей (п 4- /)-группы со значением
п + I, на единицу большим предыдущего.
Несколько переформулируем это правило: последовательное запол-
нение (п 4-0-групп в электронной оболочке нейтрального атома проис-
ходит в порядке возрастания суммы п+1 (для упрощения последующих
операций обозначим ее символом /С), причем при каждом заданном
значении К заполнение идет от больших I и меньших п к меньшим I
и большим п, т. е. с учетом квантовомеханического канона I < (п — 1)
для атомов, в последовательности
1=(К -1)/2(К — 3)/2...О,
п = (К 4- 1)/2(К 4- 3)/2... К (при нечетных К,)
Z = (А7/2 — 1)(А7/2 — 2)... О,
п = (К/2 4-1)(К/2 4- 2)... К (при четных К).
Максимальное число электронов в пределах (п 4- /)-группы в зави-
симости от величны суммы п 4-1 вычисляется по уравнению
Мпах(п + /) = |[(п + /) + Sin2^(n + О]2,
полученному аналогично классической формуле 2п2 для п-группы.
Поэтому число элементов с данным значением К составляет (К+1)2/2
при нечетных К и К2/2 при четных.
При теоретическом выведении правила п 4-1 на основе статистиче-
ской модели Томаса — Ферми были определены также:
1) суммарное число заполнения всех (п4-0-групп для всех значений
суммы п 4-1 от наименьшего (единицы) до некоторого положительного
числа Y:
Wy = U(V + 1)3 + (y + 1) [|Sin2^y-i
( Ь [2 Z О
2) границы интервала порядковых номеров Z химических элемен-
тов, в котором заполняются все nZ-состояния с заданным значением
суммы п 4-1:
l(n + Z)3 + (n + /) [1 2j(n + 0- Ш <z<
о [z z bj J
|(n + / + l)3 + (n + / + l) |sin2^(n + O - i ]
b z z b I
3) максимальное значение порядкового номера элемента, в элек-
тронной оболочке нейтральных невозбужденных атомов которого нет
39
электронов с данным (или превышающим его) значением суммы
п + 1:
°Zn+i = R(n + О3 + (n + 0 [^cos2 J(n + 0 - Ш ;
[о [2 2 bj J
4) значение порядкового номера элемента, отвечающее началу за-
полнения (п + /)-группы с заданным значением суммы п + I:
zn+i =° zn+i +1;
5) наименьшее значение порядкового номера элемента, в электрон-
ной оболочке нейтральных невозбужденных атомов которого заполне-
ны все nZ-состояния, принадлежащие (п-Н)-группам с данным и всеми
меньшими его значениями суммы п + I:
Вычисленные по этим уравнениям значения °Zn+/, %п+1 и п+1%
полностью совпадают с соответственными реальными параметрами на
всем протяжении периодической системы (табл. 1).
Таблица 1. Порядок заполнения (п 0-групп
в периодической системе
п +1 uzn+t Элемент Zn+l Элемент Tl + lZ Элемент
1 0 — 1 H 2 He
2 2 Не 3 Li 4 Be
3 4 Be 5 В 12 Mg
4 12 Mg 13 Al 20 Ca
5 20 Са 21 Sc 38 Sr
6 38 Sr 39 Y 56 Ba
7 56 Ba 57 La 88 Ra
8 88 Ra 89 Ac 120 эка-Ra
В теории периодической системы представления о числе элементов
в периоде, о длинах и границах периодов неразрывно связаны с об-
щими представлениями о закономерности, определяющей последова-
тельность заполнения электронами nZ-состояний с увеличением числа
электронов в многоэлектронных атомах при возрастании порядкового
номера элемента, и, следовательно, с представлениями об электронных
конфигурациях свободных невозбужденных нейтральных атомов.
Для описания электронной конфигурации атомов на основе (п + Г)-
последовательности введена величина конфигурационного индекса 6^,
40
равная суммарному числу вакансий во всех п/-состояниях с данным
значением суммы п + I:
tk — / "Ь "Ь I)3 + (п + / + 1)
-sirr
2
-Z
при условии
|hn + о3 + (п +1) [^cos2 J(n + 0 - |1 ] < Z
[о [2 2 о J J
< I+ 1 + Т)3 + (п + 1 + Т) ^sin2^(n + 0 “ 7
10 2 2 О
где Z — порядковый номер химического элемента в периодической си-
стеме, или суммарное число электронов в его нейтральном невозбуж-
денном атоме.
По мере заполнения электронами nZ-состояний с нечетным К про-
исходит уменьшение 8 к от [(А + 1)2/2 — 1] до нуля с последующим
восстановлением значения 8 к до [(К + 1)2/2 — 1]. При заполнении же
nZ-состояний с четным К величина 8 к уменьшается от [№/2 — 1] до
нуля, после чего с переходом к К + 1 происходит скачкообразное воз-
растание 8к до [(К + 2)2/2 — 1]. Очевидно, что окончанию заполнения
электронами nZ-состояний с разными значениями суммы п+1 отвечают
такие порядковые номера Z химических элементов:
2
4
1
n + l = K 1
Z 2
(^fc)max 1
3
12
7
4
20
7
5
38
17
6
56
17
7
88
31
8
120
31
9
170
49
Величина 8к одинакова для атомов элементов, являющихся элек-
тронными аналогами. Вместе с тем конфигурационный индекс имеет
неодинаковые значения для атомов элементов разных подгрупп (глав-
ной и дополнительной) одной и той же менделеевской группы. С по-
мощью конфигурационного индекса однозначно решается вопрос о ти-
пе электронной конфигурации конкретных атомов, например, атомов
лантана и актиния, с одной стороны, и атомов скандия, иттрия, лю-
теция — с другой. Различия в электронных конфигурациях указанных
атомов обусловлены наличием или отсутствием на периферии их элек-
тронной оболочки пустых /-состояний, принадлежащих к начавшей
заполняться (п + /)-группе: их нет в атомах Sc, Y, Lu и они есть в
атомах La, Ас.
Приведенные в табл. 2 данные свидетельствуют об отсутствии рас-
хождений вычисленных параметров с реальными. Величина 8к инва-
риантна в пределах тех перераспределений электронов между валент-
ными АО ns и (п — l)d, а также между (n — l)d и (п — 2)/, которые
41
Таблица 2. Расположение химических элементов в системе
координат, отражающей взаимозависимость Z, (п + 1) и е^
Ек Элементы Валентная электронная конфигурация атомов
31 La Ac s2/1
30 Ce Th s2f2
29 Pr Pa s'2fA
28 Nd U s2f*
27 Pm Np s'2fb
26 SM Pu s'2fb
25 Eu Am s2f7
24 Gd Cm s'2fa
23 Tb Bk s2fa
22 Dy Cf s2fw
21 Ho Es s2 f11
20 Er Fm s2f12
19 Tm Md s2f13
18 Yb No s2f14
17 Sc Y Lu Lr s2dY
16 Ti Zr Hf Rf s2d2
15 V Nb Ta Db s2dA
14 Cr Mo W Sg s2d4
13 Mn Tc Re Bh s2db
12 Fe Ru Os Hs s2db
11 Co Rh Ir Mt s2d(
10 Ni Pd Pt 110 s2dti
9 Cu Ag Au 111 s2d?
8 Zn Cd Hg 112 s2dlu
7 В Al Ga In Tl 113 s2pL
6 С Si Ge Sn Pb 114 s2p2
5 N P As Sb Bi 115 s2p6
4 О s Se Те Po 116 s2p4
3 F Cl Br I At 117 s2pb
2 Ne Ar Kr Xe Rn 118 s2pb
1 Н Li Na К Rb Cs Fr 119 s1
0 Не Be Mg Ca Sr Ba Ra 120 s2
п 4-1 1 2 3 4 5 6 7 8
Примечание. Валентные электронные конфигурации свободных ней-
тральных невозбужденных атомов приведены в соответствии с правилом п+1
без учета реально существующих отклонений.
42
вызывают небольшие отклонения от электронных конфигураций, дик-
туемых правилом последовательного заполнения (п + /)-групп.
Предсказываемая правилом п + I последовательность заполнения
одноэлектронных nZ-состояний в нейтральных многоэлектронных ато-
мах с большой точностью отражает реальную последовательность, а
также расположение химических элементов в периодической системе
(табл. 3).
Таблица 3. Правило п +1 и периодическая система
п 4-1 п 1 nl-co- стояние (АО) Максималь- ное число электронов Число хи- мических элементов Химические элементы
1 1 0 1s 2 2 Н, Не
2 2 0 2s 2 2 Li, Be
3 2 1 2р 6 6 В, С, N, О, F, Ne
3 0 3s 2 2 Na, Mg
4 3 1 Зр 6 6 Al, Si, P, S, Cl, Ar
4 0 4s 2 2 K, Ca
5 3 2 3d 10 10 Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn
4 1 4р 6 6 Ga, Ge, As, Se, Br, Kr
5 0 5s 2 2 Rb, Sr
6 4 2 4d 10 10 Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd
5 1 5р 6 6 In, Sn, Sb, Те, I, Xe
6 0 6s 2 2 Cs, Ba
7 4 3 4/ 14 14 Ce, Pr, Nd, Pm, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu
5 2 5d 10 10 La, Hf, Ta, W, Re, Os, Ir, Pt, Au, Hg
6 1 6p 6 6 Tl, Pb, Bi, Po, AtRn
7 0 7s 2 2 Fr, Ra
8 5 3 5/ 14 14 Th, Pa, U, Np, Pu, Am, Cm, Bk, Cf, Es, Fm, Md, No, Lr
6 2 6d 10 10 Ac, Rf, Db, Sg, Bh, Hs, Mt, 110-112
7 1 7p 6 6 113-118
8 0 8s 2 2 119, 120
9 5 4 5g 18 18 122-139
6 3 6/ 14 14 140-153
7 2 7d 10 10 121, 154-162
8 1 8p 6 6 163-168
9 0 9s 2 2 169, 170
Примечание. Горизонтальными двойными линиями отделены друг от друга
периоды в системе Менделеева.
Графической иллюстрацией этого правила может служить стокле-
точная «шахматная доска».
43
Заполнение электронами п/-состояний в нейтральных атомах про-
исходит по строкам этой «шахматной доски» слева направо. Грани-
цы между периодами менделеевской системы очерчены идущей сверху
вниз слева направо диагональю.
Это правило не передает лишь нескольких имеющих место в дей-
ствительности отклонений от диктуемой этим правилом последова-
тельности заполнения электронами nZ-состояний. А именно: после за-
полнения двумя электронами орбитали 6s следующий (57-й) электрон
появляется на орбитали 5с/, а не 4/ (рождается атом La с валентной
электронной конфигурацией 6s25(/1). Лишь после этого происходит за-
селение 14 электронами орбиталей 4/ (рождаются атомы 14 лантано-
идов от Се до Ln с электронными конфигурациями 6s25(/14/1'?14) и
вслед за этим продолжается и завершается заселение 10-электронного
состояния 5d. Совершенно аналогичная последовательность заполне-
ния характерна и для соответственных орбиталей 7s, 6d и 5/: после за-
полнения двумя электронами орбитали 7s следующий (89-й) электрон
появляется на орбитали 6с/, а не на 5/ (рождается атом Ас с валентной
электронной конфигурацией 7s26(/1). Лишь после этого происходит за-
селение 14 электронами орбиталей 5/ (рождаются атомы 14 актино-
идов от Th до Lr с электронными конфигурациями 7s26(/15/1“14, что
было предсказано Г. Сиборгом еще в 1944 г. в его актиноидной гипоте-
зе, получившей в дальнейшем блестящее экспериментальное подтвер-
ждение и ставшей одной из важных концепций теории периодической
системы). Вслед за этим продолжается и завершается заселение 10-
электронного состояния 6d. Еще более сложную, многогранную кар-
44
тину заполнения электронами nZ-состояний следует ожидать в атомах
8-го и 9-го периодов при конкуренции орбиталей 7d, 6/, 5д и соответ-
ственно 8(/, 7/, бд.
Таким образом, последовательность заполнения электронами nl-
состояний с увеличением заряда ядра, суммарного числа электронов в
атоме, порядкового номера химического элемента подчиняется строгой
закономерности, которая поддается ясному теоретическому обоснова-
нию и формулируется при помощи совокупности электронных состо-
яний по сумме главного и орбитального квантовых чисел. В оптиче-
ских спектрах нейтральных многоэлектронных атомов при сопостав-
лении энергетической последовательности n-групп с последователь-
ностью (п + /)-групп отчетливо проявляются две области: п-область,
где энергетическая последовательность спектральных термов соответ-
ствует возрастанию значений только главного квантового числа, и
(п + /)-область, где энергетическая последовательность спектральных
термов определяется возрастанием суммы главного и орбитального
квантовых чисел. При этом (п + /)-группы термов в атомных спек-
трах более компактны по сравнению с гораздо менее компактными
n-группами термов, которые взаимно перекрываются и охватывают
чрезвычайно широкий энергетический интервал термов в оптических
спектрах нейтральных атомов.
Существование (п + /)-области атомных явлений наряду с класси-
ческой n-областью, где доминирующую или единственную роль играет
лишь изменение главного квантового числа, составляет сущность кон-
цепции, используемой в разных направлениях физики и химии. Су-
ществование (п + /)-области оправдывает применение представлений
о (п + /)-группах электронных состояний, т. е. состояний, сгруппиро-
ванных по сумме главного и орбитального квантовых чисел, для тео-
ретической интерпретации структуры периодической системы хими-
ческих элементов. Ясно, что (п + /)-область относится прежде всего к
периферическим, внешним, электронам нейтральных многоэлектрон-
ных атомов, к их основному состоянию и близким к нему возбужден-
ным состояниям с не очень высокими значениями орбитального кван-
тового числа. Границей (п + /)-области можно считать значения Z, не
превышающие значения орбитального квантового числа в электрон-
ной оболочке атомного остова, в первом приближении не затронутого
возбуждением. Но ведь это именно такие состояния многоэлектрон-
ных атомов, которые играют преимущественную роль не только при
электронном возбуждении, но и в образовании химических связей в
молекуле за счет валентных электронов. Все эти состояния находятся
в пределах (п+ /)-области или близки к ней. Поэтому (п-Н)-область —
это прежде всего, область химических явлений и эффектов, чем и объ-
ясняется ее важность для теории периодической системы.
45
Использование (n + /)-правила и его следствий в теории периоди-
ческой системы привело к решению многих задач проблемы «распре-
деление электронов в атомах — структура системы Менделеева».
Одной из таких задач как раз и явились установление последо-
вательности заполнения nZ-состояний в электронных оболочках ней-
тральных невозбужденных атомах и, как следствие, строгая интерпре-
тация структуры периодической системы, включая определение числа
химических элементов в ее периодах. Это определение включает в се-
бя следующее положение: в каждом периоде системы вначале проис-
ходит заполнение s-состояния со значением главного квантового числа
п, равного номеру периода, затем — всех, кроме $, состояний со значе-
нием п -Н, на единицу большим номера периода. Такая совокупность
nZ-состояний может быть названа м-группой, отвечающей периоду с
номером м. Тогда
_ ( п при I = О,
М ~ [ п + I — 1 при I > 0.
Для числа nZ-состояний в At-группе, т. е. для числа элементов в
At-периоде системы, справедливо выражение
]/ 7Г \
^ = ^=0 при /тах = - - sin2-мJ ,
I
и окончательно
__ 1 / . о71’ \2
NM = 2 \ 1 + C°S — /
Полученные выражения для зависимости между длиной периода
и его номером в системе, для границ (начала и окончания) каждого
периода, для описания распределения электронов по At-группам как
функции порядкового номера элементов приводят к результатам, во
всех без исключения случаях совпадающих с реальными параметра-
ми. Так, последнее уравнение приводит к последовательности длин
периодов
2, 8, 8, 18, 18, 32, 32,...,
совпадающей с фактической структурой периодической системы.
Правило последовательного заполнения (п+/)-групп устанавливает
не только переход от одной (n + Z)-группы к другой по возрастающим
значениям суммы Z, но и очередность заселения одноэлектронных
s-j р-, d-, /-АО. В пределах (п + /)-группы заселение происходит от АО
с большим значением орбитального квантового числа к АО с мень-
шим его значением, т. е. сначала заселяются АО с большим, а затем с
меньшим I.
46
Вытекающее из правила последовательного заполнения (п + Г)-
групп распределение электронов в атоме по nZ-состояниям в строгой
математической форме как функция n, I и Z в подавляющем боль-
шинстве случаев совпадает с данными атомной спектроскопии. На ос-
нове этого правила в сочетании с правилом Хунда получается вполне
удовлетворительная зависимость между мультиплетностью основного
терма в спектрах нейтральных атомов и порядковым номером Z хи-
мического элемента.
Из правила п +1 следует общий вывод касательно присоединения
электрона к однократно заряженному атомарному катиону: электрон,
присоединяющийся к однократно положительно заряженному атому,
занимает в основном, невозбужденном, состоянии nZ-орбиталь с наи-
меньшей возможной без нарушения запрета Паули суммой главного
и орбитального квантовых чисел. Аналогичный вывод можно сфор-
мулировать для положения основного терма в нейтральных атомах:
основной терм в спектре нейтрального атома с одним излучающим
электроном всегда принадлежит к (п + /)-группе с минимальным зна-
чением суммы главного и орбитального квантовых чисел.
В теории периодической системы важное место занимают представ-
ления о внешних (валентных) и внутренних (остовных) слоях элек-
тронной оболочки атомов, об их участии в химических связях, о вза-
имосвязи между числом валентных электронов и типом электронной
конфигурации атома. Эти представления получают новое освещение
на основе (п + /)-правила и его многочисленных следствий. Здесь на-
ходят свое место известные в атомной спектроскопии перемещения од-
ноэлектронных состояний с высокими значениями углового момента
из внешних положений во внутренние, как это, в частности, проис-
ходит при заполнении электронами /-АО в атомах элементов 6-го и
7-го периодов. Можно предсказать еще более существенные эффекты
для д-АО в атомах октадеканоидов — гипотетических элементов 8-го
периода.
При малых значениях п и I для электронов атомов начала перио-
дической системы последовательность (п + /)-групп и последователь-
ность n-групп отвечают одной и той же (реальной) очередности за-
полнения nZ-состояний. Тем самым в начале периодической системы
(п +^-область и n-область сосуществуют, и это положение аналогично
многим другим известным ситуациям, когда в ограниченной некоторы-
ми определенными рамками области действует одна закономерность,
сменяющаяся при выходе за эти рамки другой, более общей, причем
эта более общая закономерность оказывается справедливой и в преж-
них границах.
Эмпирическое объяснение «нарушений» монотонной зависимости
энергий одноэлектронных атомных орбиталей от главного квантового
47
числа п, т. е. объяснение реализации в многоэлектронном атоме при-
ближенной последовательности Is 2s2p ЗрЗр 4s3d4p 5s4d5p 6s4/5(/6p
7s5f6d7p 8s5g6f7d8p 9s6g7f8d9p ..., а не идеализированной последо-
вательности Is 2s2p 3s3p3d 4s4p4d4f 5s5p5d5f5g ... одноэлектронных
^-состояний, довольно простое [14]. Из известного в квантовой меха-
нике уравнения для квантованных уровней энергии Еп атома водорода
и любого водородоподобного атома в зависимости от заряда eZ атом-
ного ядра
47rme4Z2
2hW
следует, что по мере увеличения главного квантового числа п разность
энергий атомных орбиталей с соседними (отличающимися на единицу)
значениями п уменьшается как 1/п2. В то же время дополнительная
величина, отражающая зависимость энергии атомной орбитали от ор-
битального квантового числа /, увеличивается с ростом I. Это и при-
водит к тому, что состояния с большим п и меньшим I имеют более
низкие одноэлектронные энергии по сравнению с состояниями с мень-
шим п и ббльшим Z, например, Ets < Езл, Els < и т.д.
Но непременно следует иметь в виду, что с увеличением заряда
атомного ядра и соответственного увеличения суммарного числа элек-
тронов в атоме изменяются не только энергии одноэлектронных состо-
яний, но и их относительное расположение. В тяжелых (многоэлек-
тронных) атомах «восстанавливается» монотонная зависимость энер-
гий внутренних (остовных) одноэлектронных атомных орбиталей от
главного квантового числа п: E^s > Bis > B$f и т.д.
Энергетическую последовательность одноэлектронных nZ-состоя-
ний в атомах нельзя однозначно определить на качественном уровне,
исходя из общих положений квантовой механики атома. Для этого
необходимо численное интегрирование релятивистских уравнений Фо-
ка. Вместе с тем существует общий подход, основанный на использова-
нии статистического метода Томаса — Ферми и позволяющий теорети-
чески вычислить число электронов с разными значениями орбитально-
го квантового числа I в нейтральных атомах. В этом методе электроны
в атоме рассматриваются как вырожденный электронный газ, что дает
функцию распределения электронов по скоростям и величинам момен-
та количества движения. Основное уравнение метода, получающееся,
как показал П. Дирак, из уравнений Хартри в пренебрежении некото-
рыми чертами квантовомеханического описания, позволяет вычислить
числа s-, р-, d-, /-электронов в нейтральных атомах. Эти числа пред-
ставляются монотонными плавными функциями порядкового номера
Z атома. При этом первое появление s-, р-, d-, /-электронов следует
ожидать у атомов с Z = l, 5, 21, 55 соответственно, что совпадает с
48
действительностью, за исключением атома, в котором впервые появ-
ляются /-электроны, имеющего порядковый номер Z = 58.
Неэмпирическая интерпретация сформулированного В.М. Клеч-
ковским правила основана на предложенной профессором Ю. Н. Дем-
ковым и профессором В. Н. Островским модели атома, которая непо-
средственно оперирует квантовым числом N = п + I и для частиц с
нулевой энергией использует потенциал Ленца в качестве центрально-
го потенциала с фокусирующими свойствами и кулоновской особенно-
стью в начале координат (т. е. при г = 0):
Z / г \2
+й)
где R = bZ~^3oc, b = (1/2) (Зтг/4)2/3; а — безразмерный параметр, Z —
заряд ядра атома.
Этот потенциал имеет замечательные свойства: 1) он убывает на
бесконечности как г-3, т. е. представляет собой экранированный куло-
новский потенциал, который можно использовать для аппроксимации
усредненного поля многоэлектронного атома ; 2) при Z чуть больших
Z =[(fc/4)7V(7V+l)]3/2 появляется состояние с заданным значением N
и с энергией, близкой к нулю, и, кроме того, при данном N состояния
с меньшим п лежат ниже, что согласуется с правилом п + I.
Происхождение квантового числа N = п + I Ю.Н.Демков и
В. Н. Островский объясняют следующим образом. Можно упростить
известный локальный сферически симметричный потенциал Томаса —
Ферми
(г-\ 2/з
О7Г /
используя приближенное аналитическое выражение для функции
х(х)=
х(х) = 1/(1 + <*х)2
(значение параметра а ~ 0,5 определено с использованием теории воз-
мущений). Отсюда цитируемые авторы вывели приближенный одно-
электронный потенциал
г(1 + r/R)2 ’
R = a~1Z~1/3
2/3
который понижается с возрастанием ядерного заряда Z. При этом
появляются новые уровни {n, Z}. В общем случае появление нового
уровня {n,Z} происходит при некотором значении параметра Z = Zni,
описываемого двумя квантовыми числами п и I. Очень важным свой-
ством вышеприведенного одноэлектронного потенциала является то,
49
что уровни с одной и той же суммой N = п + I появляются одновре-
менно при определенном значении параметра Z = Z^. Это сразу же
показывает, что для этого потенциала реализуется TV-группирование,
а не n-группирование, как это было бы для слабо возмущенного куло-
новского потенциала:
UA(r) « -1/г, Г » Га,
UeJr) « -Zfr, Г < Га.
Для того чтобы вывести вторую часть правила п + I следует рас-
смотреть, как уровни внутри одной и той же TV-группы располагают-
ся для более глубокого потенциала, т. е. для Z > Z#, когда энергии
уровней отличны от нуля и имеет место TV-вырождение. Этот анализ
был успешно выполнен с использованием теории возмущений. Этим
завершается теоретический ab initio (не использующий эмпирической
информации или подгоночных параметров) вывод правила заполнения
п +1.
Таким образом, вышеприведенный одноэлектронный потенциал
обеспечивает объяснение принципа построения. Этот потенциал при-
надлежит к расширенному семейству потенциалов
2 r2R?[(r/R)v + (Я/г)^]2
с силовым параметром у и координатным масштабным параметром
R. Последний потенциал имеет точное решение для Е = 0 и отра-
жает вырождение уровней для одной и той же линейной комбинации
т + (/I-1 — 1)1 квантовых чисел п и I: вырождение непосредственно
определяется параметром д.
В завершение этого раздела сделаем два полезных замечания. Пер-
вое — динамическое происхождение суммы главного п и орбитального
I квантовых чисел остается загадочным и даже таинственным. Эта ли-
нейная комбинация п +1 квантовых чисел никогда не выступала как
результат решения уравнения Шредингера, и правило п +1, как ука-
зывал Левдин, до сих пор не выведено из первых принципов.
Второе — самое раннее представление о принципе построения элек-
тронных оболочек в свободных нейтральных атомах было сформули-
ровано в эмпирической форме Маделунгом (Гоудсмит говорил, что он
получил от Маделунга приватное сообщение об этом в 1926 г.), поэтому
рассматриваемое правило иногда называют правилом Маделунга. Лев-
дин же вообще приписывал авторство этого правила самому Н. Бору.
Наконец, во избежание терминологической путаницы напомним, что
иногда это правило обозначают как n-Н, п или даже n + . Стрел-
ки указывают на возрастание соответствующих параметров.
50
2.4- Исключения из (п + I)-правила
Несмотря на то, что правило п + I обеспечивает принцип постро-
ения, наиболее близкий к реальной последовательности элементов в
периодической системе, размерности TV-групп (TV = n + Z)
2, 2, 8, 8, 18, 18, 32, 32,... (1)
несколько отличаются от длин периодов в этой системе
2, 8, 8, 18, 18, 32, 32,... (2)
Это различие детально показано на следующей схеме:
П + / = 1 n + / = 2 n + / = 3 n + / = 4
13 « 2s < 2p C 3s < 3p < 4s <
dim = 2 dim = 2 dim = 8 dim = 8
п + 1 = 5 n + I = 6 n + I = 7
3d < 4р 5s < 4d < 5p 6s < 4/ < < 5d < 6p < 7s
dim = 18 dim = 18 dim = 32
n + I = 8
< 5/ < 6d < 7p 8s < ., • • j
dim = 32
где размерности (п-Н)-оболочек показаны с учетом электронного спи-
на. Символом обозначены большие энергетические щели между од-
ноэлектронными уровнями. Эти энергетические щели разделяют пе-
риоды в периодической системе, но не совпадают с границами меж-
ду (п + /)-группами уровней. Квантовомеханическая интерпретация
отмеченного различия в двух последовательностях предложена про-
фессором В. Н. Островским [13]: из-за особых свойств слабосвязанных
s-состояний они сдвигаются к более высокой (п + /)-группе и присо-
единяются к ней на энергетической шкале.
При сравнении последовательности (1) с последовательностью, вы-
текающей из водородоподобного правила n, I
п2 = 2,8,18,32,50,..., (3)
видно, что длины периодов одни и те же в обоих случаях, будучи
равными 27V2, где N — некоторое целое число. Однако в случае по-
следовательности (1), вытекающей из (п + /)-правила, каждая длина
удваивается.
Рассматривая электронное строение индивидуальных атомов, мож-
но выявить 18 исключений из (n + /)-правила. В 16 случаях реальная
51
электронная конфигурация атома отличается от электронной конфи-
гурации, предсказываемой (п + /)-правилом, только одним электроном
на занятой орбитали, и лишь в двух случаях отличие определяется
двумя электронами.
Все эти исключения обусловлены несколькими причинами.
Во-первых, для данной электронной конфигурации существует, как
правило, несколько мультиплетных уровней атома, различающихся
полным орбитальным моментом L и полным спином S. Поэтому мно-
гие второстепенные отклонения от (п+/)-правила могут быть отнесены
на счет большой энергетической протяженности совокупности мульти-
плетных уровней для сложных конфигураций. В то время как энерге-
тический центр тяжести мультиплетных уровней может подчиняться
(п + /)-правилу, один из уровней более высокого по энергии состояния
может быть «вытолкнут» большим обменным взаимодействием в энер-
гетическое положение, расположенное ниже более низкого по энергии
состояния [15].
Во-вторых, для тяжелых атомов смешивание разных электронных
конфигураций обычно оказывается более значительным, чем для лег-
ких. Когда число электронов в атомах велико, разные наборы занятых
электронами орбиталей могут характеризоваться близкими величина-
ми полной энергии. Общая тенденция здесь состоит в том, что кон-
фигурации смешиваются сильнее, когда они близки по энергии. Сме-
шивание конфигураций может приводить к нарушению (п + ^-пра-
вила.
В-третьих, в обсуждаемую модель не включены релятивистские
эффекты. Эти эффекты наиболее существенны для внутренних элек-
тронов, но косвенно, посредством формы эффективного потенциала,
создаваемого совместно всеми электронами и ядрами, релятивистские
эффекты могут воздействовать и на внешние электроны атома. Кроме
того, релятивистские дираковские волновые функции для ns- и пру?-
состояний имеют, как известно, сингулярности на кулоновском центре
(ядре), т.е. при г —> 0. Это означает повышение электронной плотно-
сти вблизи атомного ядра, где релятивистские эффекты проявляются
сильнее. Релятивистские эффекты менее значимы, чем непосредствен-
ное динамическое влияние на валентный электрон [16].
Безусловно, важен вопрос: достаточно ли велико число исключе-
ний, чтобы подорвать или даже полностью дискредитировать физиче-
ское объяснение периодической системы в форме (п + /)-правила. Си-
туация здесь отвечает сложности системы и относительной простоте
объяснения, основанного на использованном аналитическом прибли-
жении для одноэлектронного потенциала в атомах:
[fa(r) = -Z[r(l+r/fl)2]-1, /г = а-17-1/3[Зтг(8^2)-1]2/3. (4)
52
Периодический закон весьма сложен, и поэтому вполне можно счи-
тать, что исключения из (п+/)-правила относительно неважны. Кроме
того, следует признать, что в случае тяжелых атомов иногда совсем
нелегко приписать элементам строго определенные места в периоди-
ческой системе на основе чисто химической информации, поскольку
периодическая система содержит многие утонченности и аномалии,
которые представляют непреодолимые трудности для полной редук-
ции. Без строгого отнесения, основанного на величине ядерного за-
ряда, некоторые дискуссии продолжались бы до сих пор. Следова-
тельно, невзирая на исключения ситуация выглядит вполне удовле-
творительной, хотя трудно объяснить, почему исключения касаются
строго определенных элементов. (Имеет смысл еще раз подчеркнуть,
что это — проблема объяснения; квантовомеханическое описание элек-
тронной структуры атома в случае отступления от (п + /)-правила не
представляет какой-либо особой проблемы). Недавно была сделана по-
пытка [17] описать эффективный потенциал, который не приводит к
каким-либо исключениям. Однако цитируемый анализ основан на эм-
пирических данных и не может считаться анализом аЬ initio.
Следует иметь в виду, что для перехода от точного уравнения Шре-
дингера к используемому в формализме (п +/)-правила приближенно-
му одноэлектронному потенциалу Ua(r) (4) вводится длинная цепочка
приближений.
С точки зрения метода Хартри — Фока заполнение орбиталей опре-
деляется, скорее, тонкой, еле уловимой игрой различных эффектов.
Рассмотрим конкуренцию между заполнением 4s- и Зг/-орбиталей, де-
тально изученную в [18]. Правильные электронные конфигурации ато-
мов от Sc до Си были получены при рассмотрении энергий одноэлек-
тронных орбиталей. Однако эти результаты не могут быть получены
в рамках метода Хартри — Фока при использовании дополнительного
приближения — так называемого приближения замороженных орбита-
лей. При вычислениях должно учитывать, что энергия каждой орбита-
ли существенно зависит от электронных заселенностей всех остальных
орбиталей. Следовательно, орбитальные энергии могут изменяться в
результате электронных перемещений. Результаты цитируемых расче-
тов дают объяснение отклонений от (п + /)-правила, которые имеют
место для атомов Сг и Си, и эти результаты свидетельствуют о том,
что потенциал UQ(r) (4) эффективно учитывает некоторые особенно-
сти, которые в действительности лежат за пределами упрощенной од-
ноэлектронной схемы.
Разумеется, правило п + I — приближенное правило. Но академик
В. А. Фок указывал, что только приближения приводят к физическим,
скорее, феноменологическим, представлениям, тогда как из точных
уравнений нередко ничего не видно.
53
2.5. О количественной формулировке
Периодического закона
В аспекте традиционной интерпретации периодической системы це-
лесообразно вновь обратиться к формулировке Периодического за-
кона. Каноническая формулировка — «свойства химических элемен-
тов, а также формы существования и свойства химических соеди-
нений элементов находятся в периодической зависимости от зарядов
ядер атомов элементов» — появилась вскоре после известного закона
Мозли* и стала с тех пор общепринятой.
С первых шагов квантовой механики атомов периодичность
свойств химических элементов была непосредственно связана с перио-
дичностью построения электронных оболочек атомов элементов, или,
точнее, с периодичностью электронной структуры атомов. Принцип
сосуществования электронов в атоме (отраженный в запрете Паули)
обосновывает Периодический закон и периодическую систему элемен-
тов в том смысле, что при последовательном увеличении ядер-
ного заряда и сопутствующем увеличении числа электронов в ато-
мах происходит периодическое образование сходных электронных
структур атомов и, следовательно, сходных по своей природе хими-
ческих элементов. В более простой форме: периодичность в изменении
свойств химических элементов определяется периодичностью возник-
новения подобных электронных структур атомов, подобных электрон-
ных конфигураций атомов. В этом, собственно, и заключается сущ-
ность Периодического закона.
В приведенной формулировке Периодического закона не детали-
зированы представления о сходных электронных структурах атомов.
В этой связи заслуживает внимания предложенная недавно количе-
ственная его формулировка, в которой в качестве конкретного, харак-
терного, периодически повторяющегося свойства химического элемен-
та фигурирует орбитальное квантовое число валентных электронов
атома [19]. Авторы цитируемой работы исходят из обычного представ-
ления электронной конфигурации атома в виде последовательности
* Открытый в 1913 г. английским физиком Г. Мозли закон устанавливает стро-
гое количественное соотношение между частотой у рентгеновского излучения ато-
ма и зарядом Z его ядра: у = [a(Z — 6)]1/2, где а и Ь — константы, очень мало
изменяющиеся с частотой. Этот закон, согласно которому частота рентгеновского
излучения монотонно возрастает с увеличеним ядерного заряда, позволяет «изме-
рять» заряды ядер, точнее, по экспериментальным рентгеновским спектрам атомов
однозначно определять заряды ядер атомов и, стало быть, устанавливать порядко-
вые номера химических элементов. Этот закон сразу же открыл возможность экс-
периментального подтверждения известной гипотезы голландского физика А. Ван
ден Брука (1911 г.) о равенстве порядкового номера химического элемента заряду
ядра его атомов.
54
(п, /)-орбиталей с главными п и орбитальными I квантовыми числами.
Заселение (п, /)-орбиталей электронами происходит в соответствии с
принципом минимума энергии (в порядке возрастания их энергий) и
принципом Паули, ограничивающим максимальное число электронов
на каждой орбитали значением 2(2Z+1). Как уже было сказано, из
атомных спектров и хартри-фоковских расчетов электронной структу-
ры атомов энергетическая последовательность орбиталей в свободных
нейтральных невозбужденных атомах имеет следующий вид:
Is, 2s, 2р, 3s, Зр, 4s, 3d, 4р, 5s, 4d, 5р, 6s, 4/, 5d, 6p, 7s, 5/, 6d, 7p, 8s.
При движении в этой последовательности от 1-й до 20-й орбитали
(N — ее порядковый номер) значения орбитального квантового числа
I периодически повторяются при одновременном увеличении главного
квантового числа п на единицу:
f 4f 5/
d 3d 4d 5d 6d
Р 2р Зр 4р 5p 6p 7p
S 1s 2s 3s 4s 5s 6s 7s 8s
N 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Отчетливо видно, что для каждого конкретного значения I начиная
с (п,/)-орбитали, где п = Z+1, разности порядковых номеров, через
которые повторяются значения /, образуют следующие ряды чисел:
s : 1, 2, 2, 3, 3, 4, 4,
р : 2, 3, 3, 4, 4,
d : 3, 4, 4,
/:4.
На основе этих рядов авторы обсуждаемой работы сформулиро-
вали простое правило, описывающее наблюдаемое регулярное повто-
рение значений I в последовательности атомных орбиталей: разность
порядковых номеров, через которую повторяется данное значение /,
равна
ДМк = [2(fc + 21) + 3 + (—l)fc]/4.
Например, для s-орбиталей (Z = 0) последовательные значения раз-
ностей Д^ь . • образуют ряд чисел, полностью совпадающий
с вышеприведенным, а именно: 1, 2, 2, 3, 3, 4, 4. В общем случае при
любых значениях I первая разность ДАГд, рассчитанная по этому урав-
нению, равна I + 1, вторая и третья разности (ДЛ/2, ДМз) совпадают
и равны I + 2, четвертая и пятая (ДЛм, ДАГ/5) также совпадают друг
с другом и равны I + 3, и т. д.
55
На основе наблюдаемой периодичности повторения значений I при
возрастании порядкового номера атомной орбитали можно дать сле-
дующую формулировку Периодического закона: в последовательно-
сти (n, I)-орбиталей свободных нейтральных невозбужденных ато-
мов химических элементов начиная с (/+1, Z)-орбитали с порядковым
номером N, равным [1(1 + 1) + 1], значение орбитального квантового
числа I периодически повторяется с ростом N через Z +1, Z + 2, Z + 2,
I + 3, Z + 3, Z -Ь 4, Z + 4, ... номеров при одновременном увеличении
главного квантового числа п на единицу.
Эта количественная формулировка Периодического закона содер-
жит полную информацию о том, что и как меняется в электронных
конфигурациях свободных нейтральных невозбужденных атомов по
мере повышения энергии орбиталей.
Периодичность в изменении физико-химических свойств реальных
простых веществ и соединений во многих случаях может быть выра-
жена количественно с использованием периодических функций [20].
Такие периодические функции должны содержать в качестве аргу-
ментов число и качественную характеристику (главное и орбиталь-
ное квантовые числа) электронов атомов, составляющих описываемые
объекты, параметры положения химических элементов в периодиче-
ской системе (в простейшем случае — порядковые номера химических
элементов), конкретные характеристики свойства, вычисляемого для
заданного ряда объектов, параметры эталонного объекта, константы,
характеризующие соответственную периодическую кривую. Такие пе-
риодические функции всегда многочленные. Их составление представ-
ляет собой очень трудную задачу.
2.6. «Векторные» формы периодической системы
Сравнительно недавно Г. Г. Филиппов и А. И. Горбунов ввели поня-
тие конфигурации периодической системы химических элементов как
последовательности s-, р-, d- и /-векторов-столбцов [21, 22] и показали,
что существуют четыре «правильные» (не исключающие, но дополня-
ющие друг друга при описании и объяснении свойств химических эле-
ментов и их соединеий) конфигурации, которые сохраняют порядок
нумерации элементов при движении по строкам конфигурации слева
направо и сверху вниз, при переходе от одной строки к другой. Далее
эти оригинальные представления изложены непосредственно по ука-
занным работам.
Известно, что в соответствии с типом последней заселяемой элек-
тронами атомной орбитали nl все химические элементы можно рас-
пределить по четырем (s, р, d и f) блокам, и периодическую систему
56
можно рассматривать на этой, «блочной», основе. Представим каждый
блок в виде вектора-столбца, например (Is, 2s, 3s, 4s, 5s, 6s, 7s). То-
гда периодическую систему можно представить как набор двумерных
таблиц (конфигураций), состоящих из s-, р-, d- и /-векторов. Эти кон-
фигурации различаются порядком следования векторов и их взаим-
ным расположением. Полное число возможных конфигураций равно
24 —числу перестановок на множестве символов s, р, (/, /. В одних
конфигурациях, так называемых правильных, сохраняется порядок
нумерации химических элементов вдоль отдельной строки конфигу-
рации и при переходе от одной строки к другой, в других конфигу-
рациях этот порядок не сохраняется. В «правильных» конфигураци-
ях при движении по строкам конфигурации слева направо элементы,
входящие в разные векторы-столбцы, можно расположить в порядке
возрастания их атомного номера, в других конфигурациях этого сде-
лать нельзя. Так, к первым, «правильным», конфигурациям относится
s/dp-конфигурация, соответствующая, кстати, длиннопериодному ва-
рианту периодической системы Менделеева, к другим — тривиальная
spdZ-конфигурация. Поскольку расположение химических элементов в
периодической системе в порядке увеличения их атомных номеров яв-
ляется непреложной последовательностью, основанной на фундамен-
тальных свойствах атомов, реалистичные «векторные» формы перио-
дической системы могут быть основаны лишь на «правильных» кон-
фигурациях.
Нахождение и построение «правильных» конфигураций является
комбинаторной задачей. Для ее решения на множестве химических
элементов выделяют четыре (s, р, d и /) подмножества и находят
число конфигураций, сохраняющих порядок нумерации элементов при
движении по строкам конфигурации слева направо и при переходе от
одной строки к другой. При этом используют отношение частичной
упорядоченности на множестве nZ-уровней. Прямое произведение п х I
множеств квантовых чисел {п} и {/} является множеством всех пар
{п/}. На этом множестве задают отношение порядка исходя из после-
довательности заселения nZ-орбиталей электронами в основных состо-
яниях свободных атомов:
Is < 2s < 2р < 3s < Зр < 4s < 3d < 4р < 5s < 4d < 5р < 6s < 4/ <
< 5d < 6p < 7s < 5/ < 6d < ...
Множество nxl и отношение порядка на нем представлено на
рис. 4, где элементам п и I множества поставлены в соответствие nl-
орбитали. Семейству диагоналей, вдоль которых происходит заселение
nZ-орбиталей электронами, отвечает простое уравнение п + I = const.
Поэтому более наглядным отношение порядка на множестве выгля-
дит в иных координатах, а именно, п — I и п +
57
На рис. 5 представлено расположение nZ-орбиталей в этих коор-
динатах (пустым клеткам соответствуют нереальные дробные значе-
ния I). Здесь, кстати, соверешенно очевидным становится правило, от-
ражающее последовательность nZ-орбиталей: заселение п/-орбиталей
электронами в основных состояниях свободных атомов происходит в
порядке увеличения суммы квантовых чисел п + /, а при фиксирован-
ном значении этой суммы —в порядке увеличения их разности п — /,
т. е. в порядке увеличения п и уменьшения I.
Рис. 4- Клеточная диаграмма прямого произведения n х I
в координатах n, I.
Рис. 5. Клеточная диаграмма прямого произведения n х I
в координатах п — I, n + I.
На основе диаграммы, представленной на рис. 5, нетрудно постро-
ить первую «правильную» конфигурацию периодической системы хи-
мических элементов — конфигурацию fdps. Действительно, соблюде-
ние отношения порядка последовательности nZ-орбиталей при движе-
нии по строкам этой диаграммы снизу вверх и слева направо влечет
за собой непрерывное увеличение атомных номеров элементов. При
58
размещении s-, р-, d- и /-векторов-столбцов так, как это показано на
рис. 6, реализуется привычная нумерация элементов сверху вниз и
слева направо.
Рис. 6. Правильная fdps-
конфигурация периодиче-
ской системы химических
элементов.
Рис. 7. Правильная dpsf-
конфигурация периодиче-
ской системы химических
элементов.
конфигурация периодиче-
ской системы химических
элементов.
конфигурация периодиче-
ской системы химических
элементов.
Исходная /dps-конфигурация позволяет построить остальные (три)
«правильные» конфигурации, используя две простые последователь-
ные операции: параллельный перенос левого вектора конфигурации в
положение правого вектора и сдвиг его на одну клетку вверх. Получен-
ная таким образом ^/-конфигурация, в которой сохраняется поря-
док нумерации элементов вдоль строки и при переходе к новой строке,
59
представлена на рис. 7. Аналогичным образом из ^/-конфигурации
получается конфигурация psfd (рис. 8), а из нее — конфигурация sfdp
(рис. 9).
Применяя указанные преобразования к конфигурации sfdp, полу-
чаем исходную конфигурацию fdps. Таким образом, все «правильные»
конфигурации периодической системы химических элементов исчер-
пываются представленными четырьмя. В других конфигурациях ни-
какое взаимное расположение s-, р-, d- и /-векторов-столбцов не может
сохранить порядок нумерации элементов.
Можно ли говорить о существовании «самой правильной» кон-
фигурации? С математической точки зрения все четыре «правиль-
ные» конфигурации равноправны. Однако для описания лишь элек-
тронной структуры свободных атомов предпочтительнее /dps-конфи-
гурация, наиболее ясно отражающая последовательность заселения nl-
орбиталей электронами. Вместе с тем в аспекте периодичности хими-
ческих и физических свойств элементов и их соединений предпочти-
тельнее s/dp-конфигурация, которая отвечает длиннопериодной форме
менделеевской системы и в которой химические и физические свой-
ства элементов и их соединений объясняются во многом двумя тен-
денциями: увеличением атомных потенциалов ионизации и уменьше-
нием атомных размеров при движении слева направо по строке этой
конфигурации. Наконец, при исследовании кристаллических структур
простых веществ и химических соединений предпочтительнее dpsf- и
ps/d-конфигурации. Таким образом, не существует какой-либо одной
«самой правильной» конфигурации. Каждая из четырех указанных
«правильных» конфигураций лишь дополняет другие три, и все они
равно пригодны для «векторного» описания периодической системы
химических элементов.
Глава 3
ОБЛАСТЬ СВЕРХТЯЖЕЛЫХ ЭЛЕМЕНТОВ
В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ
Известные в настоящее время — строго идентифицированные — 109
химических элементов, безусловно, не являются окончательной, завер-
шенной совокупностью всех химических элементов. Уже сейчас есть
надежные сведения о синтезе ядер элемента 110, и в будущем ряд
искусственно синтезируемых химических элементов неизбежно будет
увеличиваться. Каков же верхний предел этого ряда?
Разработанные в ядерной физике модели весьма эффективны не
только для развития теории деления ядер, но и для предсказания ста-
бильности сверхтяжелых ядер по отношению к спонтанному делению и
другим видам радиоактивного распада. Согласно этим моделям потен-
циальная энергия ядра является функцией числа нейтронов N, чис-
ла протонов Z и формы ядра. Лежащие в основе этих моделей полу-
эмпирические уравнения позволяют оценивать периоды полураспада
спонтанного деления ядра при заданных Nn Z. Уже в классических
работах нобелевских лауреатов М. Гепперт-Майер и И. Йенсена, разра-
ботавших «оболочечную» теорию ядра, было показано, что ядро как
совокупность составленных из нейтронов и протонов оболочек обла-
дает особой стабильностью, если оно содержит «магическое» число
нейтронов или протонов (еще стабильнее «дважды магические» яд-
ра, в которых «магическими» являются и число нейтронов, и число
протонов одновременно).
Магическими числами нейтронов и протонов называются значения
N и Z, отвечающие целиком заполненным нейтронным и протонным
оболочкам ядра. Обычно магические числа нейтронов и протонов счи-
таются равными 2, 8, 20, 28, 50, 82, 114, 126, 164, 184 (магические
числа 114 и 164 не относятся к нейтронам, а 184 —к протонам). Не
случайно аномально высока стабильность «дважды магических» ядер
свинца 208Pb (N = 126, Z = 82), обладающих замкнутыми ядерными
оболочками и сферической симметрией.
61
Эти «магические числа» получены в результате теоретических рас-
четов, в частности, методом орбиталей Нильсона, позволяющим вы-
числять потенциалы отдельных нуклонов в поле ядра. Такие расчеты
предсказывают существование замкнутых ядерных оболочек при стро-
го определенных значениях N и Z, что означает повышенную устой-
чивость такого ядра по отношению к спонтанному делению. Что же
касается сверхтяжелых ядер, не обладающих замкнутыми оболочка-
ми, то они должны подвергаться самопроизвольному делению с очень
малым периодом полураспада.
Стабилизирующее влияние замкнутой нейтронной или протонной
оболочки означает, что стабильность сверхтяжелых нуклидов обеспе-
чивается дважды магическим сочетанием нейтронов и протонов сфе-
рически симметричного ядра. Можно предвидеть на этой основе, что
весьма стабильным, например, должно быть ядро 294110 (N = 184 и
Z=110), период полураспада которого по отношению к спонтанному
делению и а-распаду может достигать 108 лет и которое, вероятно,
стабильно по отношению к /3-распаду, а также ядра 298114 (N= 184 и
Z=U4) и 310126 (N = 184 и Z = 126).
Любопытно, что реально существующие и теоретически предсказы-
ваемые стабильные ядра с атомными номерами Z, равными 50 (олово),
82 (свинец), 114 (эка-свинец) и 164 (deu-свинец), находятся в одной и
той же (IV главной) подгруппе периодической системы. Это удиви-
тельно, поскольку принципы, определяющие стабильность ядер, по-
видимому, никак не связаны с факторами, определяющими электрон-
ную структуру атома и, стало быть, положение элемента в периодиче-
ской системе.
Представления о стабильности некоторых сверхтяжелых ядер сти-
мулируют поиски соответствующих элементов в природе. Это прежде
всего относится к элементам НО (эка-платина), 111 (эка-золото), 112
(эка-ртуть), 113 (эка-таллий), 114 (эка-свинец). Если период полурас-
пада таких ядер составляет 105 лет, то имеет смысл поискать их даже
в космических лучах. К сожалению, пока нет обнадеживающих све-
дений о присутствии обсуждаемых ядер в природных объектах (их
искали даже в конкрециях, образовавшихся на зубах погибших акул
на очень больших глубинах Мирового океана).
Сверхтяжелые ядра в пределах очерченных «островов стабиль-
ности» могут быть синтезированы искусственно при бомбардиров-
ке в ускорителях тяжелых ядер (мишеней) крупными ядрами (сна-
рядами). Правда, теория предсказывает очень малый выход ядер-
продуктов вследствие преобладания ядерных реакций спонтанного де-
ления над ядерными реакциями синтеза при слиянии ядра-снаряда и
ядра-мишени. Примерами реально осуществимых реакций ядерного
синтеза могут служить (п — осколочный нейтрон):
62
248Стэб +40 Arie -»284 114 + 4п,
244Pu94 +48 Саго -»288 114 4- 4п,
238и92 +50 Ti22 -*284 114 + 4п.
Электронную структуру сверхтяжелых атомов, следующих за ак-
тиноидами, последний из которых — лоуренсий (103) с электронной
конфигурацией атома {[Rn]5f14}7s26d1, — предсказать нетрудно, ес-
ли для такого предсказания использовать известные представления об
энергетической последовательности ni-орбиталей в атомах. Эти пред-
сказания подтверждаются (для Z < 132) результатами квантовоме-
ханических расчетов электронной структуры свободных сверхтяже-
лых атомов в основном состоянии, и в целом их валентные электрон-
ные конфигурации сверх замкнутой электронной оболочки {[Rn] 5f14}
можно представить в следущем виде:
104 7s26d2
105 7s26d3
106 7s26d4
107 7s26d5
108 7s26d6
109 7s26<F
110 7s26d8
111 7s26d9
112 7s26d10
113 7s26d107p1
114 7s26d107p2
115 7s26d107p3
116 7s26d107p4
117 7s26d107p5
118 7s26d107p6
119 7s26d107p68s1
120 7s26d107p68s2
121 7s26d107p68s27d1
122 7s26d107p6 SstJd'bg1 (6/°)
123 7s26d107p6 8s27dl5g2 (6/°)
124 7s26d107p6 8s27d*5g3 (6f°)
125 7s26d107p68s27d15p4 (6/°)
126 7s26d107p6 8s27d15p5 (6/°)
127 7s26d107p6 8s27d15g6 (6/°)
63
128 7s26</107p68s27d15g7 (6/°)
129 7s26</107p68s27d15g8 (6/°)
130 7s26</107p68s27<Z15<?9 (6/°)
139 7s26d107p68s27d15p18 (6/°)
140 7s26d107p68s27d15g186/1
153 7s26d107p68s27d15p186/14
154 7s26d107p68s27d25g186/14
162 7s26<Z107p6 8s27d105p186/14
163 7s26d107p68s27d105p186y148p1
168 7s26d107p68s27d105g186/148p6
Представленные валентные электронные конфигурации сверхтя-
желых атомов 104-168 следует считать тривиальными в том смысле,
что в действительности нельзя однозначно указать валентную элек-
тронную конфигурацию большинства из них, а именно, атомов 122-
161. Близость одноэлектронных энергий 7d-, 5д-, 6/-состояний (точнее,
близость полных энергий свободных атомов для различных распреде-
лений валентных электронов по 7d-, 5 g-, 6/-состояниям) не позволяет
после 8s27(/1 безоговорочно заселять валентными электронами снача-
ла 5д-состояния (18 электронов), затем 6/-состояния (14 электронов)
и только после этого дозаселить 7(/-состояния оставшимися девятью
электронами. Здесь по аналогии с принципиально одинаковой струк-
турой 6-го и 7-го периодов системы (с 32 химическими элементами
каждый) принято, что в атомах третьего элемента 8-го периода систе-
мы (121-го элемента) после заселения двумя электронами состояния 8s
сначала заселяется третьим электроном состояние 7d и только после
этого в атомах четвертого (122-го) и последующих элементов четвер-
тый и последующие электроны заселяют новое состояние 5 g (напом-
ним, что в 6-м и 7-м периодах системы в атомах третьего элемента,
57-го или 89-го, после заселения двумя электронами состояния 6s или
7s сначала заселяется третьим электроном состояние 5d или 6d и толь-
ко после этого в атомах четвертого, 58-го или 90-го, и последующих
элементов четвертый и последующие электроны заселяют состояние
4/ или 5/). Иными словами, здесь принято, что первый электрон но-
вого типа 5g (с новым I = 4) появляется в атомах элемента 122. Но на
этот счет имеются и другие суждения.
Вычисление граничного значения /, при котором в атомах начи-
нается заселение электронами 5(/-состояния, относится к обычной за-
64
даче о первом появлении в атомах электрона с заданным значением
/, здесь —со значением Z = 4. Так, расчеты в рамках статистической
модели атома Томаса — Ферми при использовании соотношения
Z = а(2/ + I)3
с эмпирически подобранным с учетом соответственных данных для
d- и /-электронов множителем а = 0,17ис! = 4 дают значение
Z = 124 для порядкового номера химического элемента, в атомах кото-
рого впервые появляется 5д-электрон [23]. По эмпирическим результа-
там одних авторов первый 5д-электрон появляется существенно рань-
ше — в атомах элемента 121 — в полном соответствии с правилом Клеч-
ковского — Демкова, по результатам релятивистских расчетов других
авторов —в атомах элементов 123 или 125 [24].
Во избежание потери строгости в изложении вопросов, связанных
с электронными конфигурациями свободных атомов в основном со-
стоянии, сделаем отступление самого общего характера. Отсутствие
единых представлений об электронных конфигурациях любых атомов,
отнюдь, не случайно.
В квантовомеханическом описании электронного строения любого
атома представление об одноэлектронных состояниях (одноэлектрон-
ных nZ-орбиталях) не отвечает строгим требованиям квантовой ме-
ханики атома и носит приближенный характер. Применимость этого
приближения различна для разных задач, и часто возникает необхо-
димость более точного описания, требующего выхода за рамки одно-
электронного приближения.
Один из возможных выходов — многоконфигурационное прибли-
жение («наложение конфигураций»), состоящее в том, что волновую
функцию многоэлектронной системы (атома) представляют в виде ли-
нейной комбинации («суммы») функций, каждая из которых отвечает
конкретной электронной конфигурации:
Ф = ЯуСу'фу,
где индекс 7 обозначает конкретную электронную конфигурацию. Ко-
эффициенты Су определяют таким образом, чтобы функция Ф воз-
можно точнее удовлетворяла уравнению Шредингера.
Для большинства атомов среди набора этих коэффициентов один
коэффициент превалирует над всеми остальными. Это означает, что
соответствующая этому коэффициенту конкретная электронная кон-
фигурация достаточно хорошо отражает электронную структуру ато-
ма. Но иногда оказывается, что два или большее число коэффициен-
тов сопоставимы по своим величинам. Тогда достаточно точное описа-
ние электронной структуры атома нельзя получить при помощи одной
65
волновой функции, соответствующей одной определенной электронной
конфигурации, т. е. однозначному набору одноэлектронных состояний
(одноэлектронных nZ-орбиталей). Это имеет место в тех случаях, когда
конфигурации различаются одноэлектронными состояниями с близки-
ми энергиями. Невозможность однозначного определения (т. е. «неод-
нозначное определение») электронной конфигурации атома свойствен-
на тем областям периодической системы, где расположены химические
элементы, в атомах которых заселяются одноэлектронные состояния
f- или g-типов и где могут «смешиваться» энергетически близкие од-
ноэлектронные состояния (п — 3)д, (п — 2)/, (n — l)d и, прежде всего,
4/ и 5d; 5/ и 6d; 5g, 6/ и 7d.
Итак, валентные электронные конфигурации атомов 122-161 из-
за тонких эффектов межэлектронных взаимодействий формируют-
ся нерегулярным образом, их очень условно можно назвать смешан-
ными.
И в этом нет ничего неожиданного. Ведь уже в 6-м периоде бли-
зость одноэлектронных энергий 5d-, 4/-состояний лежит в основе ре-
ализации различной валентной электронной структуры атомов лан-
таноидов. Действительно, после самого La с валентной электронной
конфигурацией 6s25d1 уже у первого лантаноида Се возможна как
конфигурация 6s25d14/1, так и 6s25d24/°. Исключения — срединный
в лантаноидном ряду атом Gd с валентной электронной конфигура-
цией 6s25d14/7, которая включает в себя ровно наполовину заполнен-
ную 4/7-орбиталь и по этой причине (по причине максимальной энер-
гии внутриатомного электронного обмена) весьма стабильна, а так-
же замыкающий лантаноидный ряд атом Lu с валентной электрон-
ной конфигурацией 6s25d14/14, которая содержит полностью запол-
ненную электронами 4/14-орбиталь и очень стабильна по той же при-
чине. Тем более в 7-м периоде, где еще большая близость одноэлек-
тронных энергий 6d-, 5/-состояний обеспечивает возможность суще-
ствования атомов актиноидов с различными валентными электронны-
ми конфигурациями, например, U 7s26d35/1, U7s26d25/2, U 7s26d15/3,
за единственным исключением: атомы последнего актиноида Lr име-
ют только одну и очень стабильную валентную электронную конфи-
гурацию 7s26d15/14 с полностью заполненной электронами 5/14-орби-
талью.
Валентная электронная конфигурация атома 162 определена, по-
видимому, однозначно, поскольку включает в себя полностью за-
полненью электронами состояния 7d105g186/14. Валентные электрон-
ные конфигурации атомов 163-168 тоже можно считать достаточно
определенными, так как в этих атомах происходит последователь-
ное заселение шестью электронами высоколежащего по энергии 8р-со-
стояния.
66
Если не учитывать указанные нерегулярности в распределении
электронов по валентным орбиталям сверхтяжелых атомов, структуру
7-го и 8-го периодов системы можно представить исходя из того, что
электронное строение атома определяет положение соответственного
химического элемента в периодической системе.
В 7-м периоде, открывающемся францием (87) и радием (88), за
третьим элементом — актинием (89) — располагаются 14 актиноидов от
тория (90) до лоуренсия (103), затем —девять переходных металлов от
резерфордия (104), дубния (105), сиборгия (106), бория (107), хассия
(108), майтнерия (109) до эка-ртути (112), которые вместе с актини-
ем составляют четвертую вставную декаду, наконец, — шесть элемен-
тов главных подгрупп от э?са-таллия (113) до благородного э?са-радона
(П8):
Fr Ra | Ас | Th Ра U Np Pu Am Cm Bk Cf Es Fm Md No Lr |
Rf Db SgBh Hs Mt 110 111 112 113 114 115 116 117 118.
В 8-м периоде (здесь уместно лишь высказать предположение)
первыми стоят эка-франций (119) и э?са-радий (120), за ними — эка-
актиний (121), затем 18 элементов принципиально нового ^-семейства
от 122-го до 139-го (по предложению В. И. Гольданского иногда назы-
ваемых октадеканоидами — в их атомах последовательно заселяются
18 электронами кайносимметричные 5^-состояния) и 14 суперактинои-
дов (по терминологии Г. Сиборга) от эка-тория (140) до э?са-лоуренсия
(153), за ними —девять переходных металлов от э?са-резерфордия
(154) до deu-ртути (162), которые вместе с эка-актинием составля-
ют пятую вставную декаду, наконец, — шесть элементов главных под-
групп от deu-таллия (163) до благородного deu-радона (168):
119 120 | 121 | 122-139 | 140-153 | 154-162 | 163-168.
Таким образом, в 8-м периоде менделеевской системы впервые рас-
полагаются 50 химических элементов (8s — 2, 5д —18, 6/ —14, 7d —10,
8р — 6). Девятый период, в принципе, должен быть полностью анало-
гичен 8-му периоду: также 50 химических элементов с Z = 169 — 218
и та же структура периода (9s — 2, 6^ — 18, 7/ —14, Sd — 10, 9р — 6).
На этой основе можно представить структуру гипотетической пери-
одической системы для всех известных и предсказанных химических
элементов (рис. 10).
Но все это слишком упрощенные представления. Структуру пе-
риодической системы не следует считать совершенно неизменной от
первого до последнего (из известных) химического элемента. Уже
сам Д. И. Менделеев указывал, что периодическая правильность на-
рушается в результате изменений в физической и химической при-
роде элементов. По мнению профессора И. С. Дмитриева [1], в этих
67
словах проявилась глубина понимания Д. И. Менделеевым Периоди-
ческого закона и периодической системы химических элементов как
полифонического единства самых разных физико-химических законо-
мерностей (такого понимания сложной игры разнообразных факторов
и тенденций не было ни у одного из его предшественников и совре-
менников). Распределение химических элементов, представленное в
короткой форме периодической системы Менделеева, выявляло слож-
ную полифонию отдельных частных закономерностей, и уже триады
переходных металлов (Fe, Со, Ni в 4-м периоде, Ru, Rh, Pd в 5-м пе-
риоде и Os, lr, Pt в 6-м периоде системы) не вмещались ни в какую
группу.
Теория периодической системы уже в конце минувшего столетия
включает в себя впервые разрабатываемые представления о возмож-
ном нарушении традиционной периодичности в изменении атомных
свойств, о возможном исчезновении менделеевских групп и подгрупп
в области сверхтяжелых химических элементов. Сложность электрон-
ной структуры атомов сверхтяжелых элементов может привести к су-
щественному изменению классических представлений о менделеевских
группах в области химических элементов с большими атомными но-
мерами (в конце 7-го периода, особенно в 8-м периоде и далее) вплоть
до исчезновения, «смешивания» этих групп.
По-видимому, традиционные представления о периодичности в из-
менении свойств химических элементов, основанные на понятиях груп-
пы и подгруппы в системе, охватывают только около 100 элементов и
могут оказаться неадекватными в области сверхтяжелых элементов,
атомы которых включают в себя очень много электронов.
Традиционная периодичность, свойственная элементам первых
пяти-шести периодов системы, может претерпеть в области сверхтя-
желых элементов сильные изменения и приобрести сложный облик.
Возможно, что в области элементов с большими атомными номера-
ми произойдет «размывание периодичности», поскольку в этой обла-
сти нет строго определенных границ между отдельными (Ы)-группами
элементов, как это имеет место между 6d- и 5/-группами элементов.
Не случайно уже в 6-м и 7-м периодах системы наблюдаются откло-
нения от эмпирического правила п +1. Возможно, что эти отклонения
являются предвестниками, точнее, провозвестниками, более глубоких
и серьезных изменений в характере зависимости структуры и свойств
атомов при все большем возрастании числа электронов в них.
Проведенные с учетом релятивистских эффектов квантовомехани-
ческие оценки характеристик атомов некоторых уже синтезированных
и пока еще гипотетических сверхтяжелых элементов 7-го периода при-
водят к выводу о большом своеобразии свойств атомов этих элементов
в сравнении с их известными аналогами по группам системы.
68
Резко отличающиеся друг от друга пространственные характери-
стики атомных орбиталей $, р, с одной стороны, и d, f — c другой
при малых главных квантовых числах затем, при увеличении глав-
ных квантовых чисел, начинают сближаться (например, радиус АО
6р таллия и АО 6d актиния уже совсем близки). Это постепенно вно-
сит относительность в классические формы периодической системы,
которые соблюдаются до 7-го периода, да и то с известными отступ-
лениями.
Далее, при очень большом числе электронов в атоме заселяющи-
еся электронами (т. е. верхние) орбитали имеют близкие друг к дру-
гу энергии, последовательность которых подвержена влиянию тонких
эффектов. И эта энергетическая последовательность верхних атом-
пых орбиталей может подвергаться существенной перестройке под воз-
действием многих и разнообразных факторов, приводя тем самым к
неконтролируемому многообразию электронных конфигураций и соот-
ветствующему вырождению традиционной периодичности, связанной
с понятиями групп и подгрупп сходных по химической природе эле-
ментов.
Поразительно (воистину, уму непостижимо), но сам Д. И. Менде-
леев вскоре после открытия им Периодического закона предвидел
возможность исчезновения идеальных групп, объединяющих действи-
тельно близкие по поведению химические элементы, что, безусловно,
лишь обогащает принцип периодичности, делает его более сложным и
многогранным.
Менделеев прекрасно понимал неизбежность развития более обще-
го принципа, чем принцип периодичности, и писал о нем как о прин-
ципе природной гармонии, господствующем в материальном мире, т. е.
Д. И. Менделеев употребил даже еще более общее понятие, чем ритми-
ка, являющую собой лишь составную часть гармонии.
Учитывая фермионную природу электронов и их квантовомехани-
ческое поведение в поле ядер, можно полагать, что эволюция строения
электронных оболочек атомов на фоне увеличения зарядов атомных
ядер по мере движения к концу периодической системы должна при-
водить к периодичности стратиграфии (как последовательности появ-
ления электронных слоев), налагающейся на монотонность изменения
целого ряда характеристик атомов под влиянием растущих зарядов и
масс атомных ядер.
К таким характеристикам относятся прежде всего энергия элек-
тронной корреляции, спин-орбитальное сопряжение, эффект проник-
новения электронов в ядра, релятивистский эффект и некоторые дру-
гие.
Постепенно нарастая, они не только влияют па основные законо-
мерности в группах и периодах системы, но ломают саму ритмику
сю
последовательного удлинения периодов, что может привести к транс-
формированию традиционной периодичности вплоть до исчезновения
традиционных групп периодической системы [8,9].
Таким образом, Периодический закон в классической форме описы-
вает, возможно, свойства лишь ограниченного числа химических эле-
ментов. Это может означать, что существует еще более общий прин-
цип, включающий в себя Периодический закон как составную часть.
Глава 4
ДВА «ЛАГЕРЯ»
ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Основанная на квантово-химической теории электронного строе-
ния атомов теоретическая интерпретация Периодического закона и пе-
риодической системы изначально учитывает наличие двух «лагерей»
химических элементов — непереходных элементов А (атомы с валент-
ными s-, р-электронами) и переходных металлов М (атомы с валент-
ными s-, d-, /-электронами). Представление о двух «лагерях» химиче-
ских элементов является важнейшим (хотя и очевидным) следствием
традиционного подхода к теории периодической системы.
Проявления принципа периодичности в химии непереходных эле-
ментов и в химии переходных металлов часто оказываются не только
неодинаковыми, но и абсолютно разными. И одна из главных причин
этого —весьма значительные различия в электронном строении ML^
и ALfc. С позиций эмпирической теории полной гибридизации, кото-
рая отражает одну из самых общих по характеру идей в квантово-
химической теории строения молекул, электронная структура ком-
плексов MLfc и ALfc отвечает участию в химических связях с лиганда-
ми не только занятых электронами валентных орбиталей ns, (n — l)d
для атомов переходных металлов и ns, пр для атомов непереходных
элементов, но и виртуальных атомных орбиталей пр или nd соответ-
ственно, не занятых электронами в основном состоянии центрального
атома. В частности, общепринятая в теории молекулярных орбиталей
интерпретация электронной структуры октаэдрических молекул MoFe
и SFe (и, естественно, объяснение всех присущих им свойств) исходит
из следующего представления: в каждой молекуле возникают шесть
делокализованных МО (aig, t\u, ед) сг-типа, образующихся при участии
одной s-, трех р- и двух d-валентных орбиталей центрального атома.
Эти связывающие МО полностью заняты двенадцатью электронами,
соответственные разрыхляющие МО вакантны. Электронную струк-
туру обсуждаемых октаэдрических молекул можно приближенно рас-
сматривать как включающую в себя шесть локализованных (двухцен-
71
тровых) двухэлектронных сг-связей М-F или А-F (в пренебрежении
тг-эффектами), образующихся при участии валентных $р3</2-орбиталей
центрального атома М или А. Это равносильно утверждению, что а-
связи в таких молекулах образуются при непременном появлении элек-
тронов в валентном состоянии центрального атома на ранее вакантных
орбиталях пр (в комплексе переходного металла Мо) или орбиталях nd
(в комплексе непереходного элемента S).
Результаты расчетов электронной структуры комплексов переход-
ных металлов ML^ полуэмпирическими и неэмпирическими методами
ССП МО ЛКАО, а также данные высокоэнергетических (рентгенов-
ских эмиссионных, рентгенофотоэлектронных) спектров этих соедине-
ний систематически и согласованно указывают на большую электрон-
ную заселенность виртуальных пр-орбиталей атомов М [25, 26]. Это
означает, что принципы электронного строения комплексов переход-
ных металлов (молекулярные орбитали, электронная структура хими-
ческих связей M-L и т. д.) отвечают представлениям теории полной
гибридизации, т. е. эффективному участию всех (s, р, d) валентных
орбиталей центральных атомов в связях с лигандами.
Уместно напомнить, что в химии комплексов переходных металлов
весьма распространено эмпирическое правило 18-электронной оболоч-
ки — правило Сиджвика. Оно указывает, что в этих комплексах в ва-
лентной оболочке центрального атома М значительно больше 12 элек-
тронов, отвечающих полной заселенности шести связывающих моле-
кулярных орбиталей с участием одной ns- и пяти (n— 1) d-AO, и тем са-
мым определенно свидетельствует об участии не только ns- и (n — l)d-,
но и пр-орбиталей центрального атома М в этих комплексах в образо-
вании связей с лигандами.
Напротив, аналогичная и дополнительная (из ЯМР- и оже-
спектров) информация для комплексов непереходных элементов AL^
не подтверждает и часто противоречит априорным представлениям
о значительной электронной заселенности виртуальных nd-орбиталей
атомов А [27, 28]. Это означает, что принципы электронного строения
соединений непереходных элементов (молекулярные орбитали, элек-
тронная структура химических связей A-L и т. д.) вовсе не отвечают
положениям теории полной гибридизации, т. е. эффективному участию
всех (s, р, d) валентных орбиталей центральных атомов в связях с ли-
гандами. Симптоматично, что в химии непереходных элементов нет
правила, аналогичного правилу Сиджвика.
Теоретическое обоснование значительного вклада виртуальных
Мпр-орбиталей в валентные МО комплексов переходных металлов и
отсутствия значимого вклада And-орбиталей в валентные МО ком-
плексов непереходных элементов дано Е. М. Шусторовичем [29] исхо-
дя из наиболее общих положений теории молекулярных орбиталей и
72
теории возмущений. Непременным условием эффективного электрон-
ного взаимодействия валентных орбиталей центрального атома и ли-
гандов является близость орбитальных энергий. (Это же самое поз-
воляет понять, почему ковалентные связи, возникающие при взаимо-
действии неспаренных электронов, обычно гораздо прочнее донорно-
акцепторных и дативных связей.) От этого, собственно, зависит сте-
пень смешения взаимодействующих валентных орбиталей центрально-
го атома и лигандов: степень смешения уд и хь тем больше, чем ближе
энергии этих орбиталей. Это означает, что прочные связи A-L образу-
ются лишь при близких значениях энергии взаимодействующих атом-
ных орбиталей уд и дх, тогда как при сильном различии этих энергий
связи с участием таких уд и дх весьма слабы или вообще не обра-
зуются. Во втором порядке теории возмущений энергия стабилизации
связывающей молекулярной орбитали <р = сдуд + clXl составляет
A^al = /Здь / |ад - ох|,
где ад и ах — кулоновские интегралы, выражающие «самосогласован-
ные» энергии орбиталей уд и дх (в тех приближениях теории МО, где
используется эффективный одноэлектронный гамильтониан), /Здь —
резонансный интеграл, выражающий энергию взаимодействия атом-
ных орбиталей уд и уь и пропорциональный интегралу перекрывания
SAL.
В атомах переходных металлов М различия в энергиях валентных
(n — l)d-, ns-, пр-орбиталей сравнительно невелики (5-7 эВ, рис. 11),
и это объясняет участие всех этих валентных орбиталей (естественно,
когда это необходимо), в том числе и вакантных пр-, в связях M-L и
тем самым оправдывает использование представлений гибридизации
валентных орбиталей центрального атома в теории электронного стро-
ения комплексов переходных металлов. Несмотря на некоторые разли-
чия в результатах неэмпирических расчетов электронной структуры
комплексов переходных металлов, а также в результатах рентгеноспек-
трального и рентгенофотоэлектронного исследований, общий тон вы-
водов относительно значительного участия виртуальных пр-орбиталей
центрального атома М в связях с лигандами единообразен.
В атомах непереходных элементов IV-VIII групп периодической си-
стемы энергии nd-орбиталей на 10-15 эВ выше, а энергии ns-орбиталей
на 10-20 эВ ниже энергий пр-орбиталей (рис. 11). Это обстоятельство
указывает на неизбежную неравноценность вкладов ns-, пр- и nd-
орбиталей атома А в связи с лигандами. В типичных случаях энер-
гии валентных орбиталей лигандов лежат несколько ниже энергий
Апр-орбиталей (лиганды более электроотрицательны, чем централь-
ный атом). Поэтому вакантные And-орбитали оказываются слишком
73
a
б
nd
np_
ns
(n-l)d
_CH3
_H
_ I
_ Br ns
— Cl
F
M L A
Puc. 11. Относительное энергетическое расположение валентных орбиталей
атомов переходных металлов (М), непереходных элементов (А) и лигандов (L)
(а) и типичные диаграммы взаимодействия валентных орбиталей атомов М, А и L
в комплексах переходных металлов (MLfc) и непереходных элементов (ALfc) (б).
высоколежащими по энергии (к тому же они весьма диффузны), что-
бы заметно взаимодействовать с заполненными электронами валент-
ными орбиталями лигандов. По этой же причине далеко не всегда в
связывание с лигандами могут вовлекаться занятые Ап$2-орбитали:
часто их энергия оказывается слишком низкой по сравнению с энерги-
ей валентных орбиталей лигандов. В целом большие энергии ns — пр- и
пр — nd-разделения для атомов А обеспечивают решающую роль Апр-
орбиталей в электронной структуре комплексов ALfc и значительно
менее существенные вклады Ans2- и особенно And-орбиталей в ва-
лентные МО. Характеристические свойства соединений непереходных
элементов могут быть получены без участия nd-орбиталей централь-
ного атома А, которые играют роль поляризационных поправок ор-
битального базиса. В целом общетеоретические представления указы-
вают на отсутствие существенного вклада вакантных And-орбиталей
в валентные МО в электронной структуре комплексов непереходных
элементов. Менее ясна роль занятых Ап$2-орбиталей.
При обсуждении проблемы d-орбиталей (точнее, при априорном
утверждении о большом вкладе d-AO в электронную структуру ком-
плекса) часто ссылаются на критерий симметрии, подчеркивая, что
74
для любой точечной группы симметрии комплекса AL^ среди And-
орбиталей всегда найдутся такие, которые принадлежат к тем же
неприводимым представлениям, что и валентные орбитали лигандов.
Поэтому всегда в той или иной степени возможно и даже реально ст-
или тг-взаимодействие And-орбиталей с орбиталями лигандов. В даль-
нейшем этот тезис был уточнен: d-AO вносят реальный вклад в элек-
тронную структуру комплексов тогда, когда они, будучи включенны-
ми в базис, образуют в соответствии со свойствами симметрии новые
связывающие МО при взаимодействии с АО лигандов (например, е
в ML4 или t2g и ед в МЬб); в остальных случаях их следует считать
поляризующими добавками. Но, во-первых, неэмпирические расчеты
показывают, что d-AO участвуют в связях, отнюдь, не всегда, когда
это возможно по условиям симметрии. Во-вторых, результаты иссле-
дования влияния d-функций в различных вариантах расчета (учтены
d-функции центрального атома, необходимые по соображениям сим-
метрии; в базис центрального атома включены d-функции, не соот-
ветствующие критерию симметрии; учтен полный набор d-функций в
сбалансированном базисе) показывают, что d-орбитали, требуемые по
соображениям симметрии, важны не более, чем все остальные d-AO.
Наконец, главное: d-AO совсем не являются необходимыми для постро-
ения заданного числа МО в электронной структуре комплекса AL^ той
или иной симметрии. Так, при наличии только одной ns- и трех пр-
орбиталей центрального атома можно построить шесть эквивалентных
МО, соответствующих каноническим МО ai9, Ziu, ед октаэдрического
комплекса, или только из трех пр-орбиталей центрального атома по-
строить четыре эквивалентные МО, соответствующие каноническим
МО ai, t2 тетраэдрического комплекса. В первом случае МО ед будет
несвязывающей, лигандной орбиталью, во втором таковой оказывает-
ся МО а\.
Проблема d-орбиталей имеет смысл только в тех квантово-
химических методах, которые используют приближение МО ЛКАО,
поэтому участие nd-орбиталей атомов непереходных элементов IV-
VIII групп периодической системы в химических связях с лиганда-
ми в течение длительного времени неоднократно и тщательно об-
суждалось многими авторами на основе экспериментальных резуль-
татов, и, в первую очередь, результатов исследования эмиссионных
рентгеновских спектров соединений этих элементов. При интерпрета-
ции эмиссионных рентгеновских L-спектров соединений серы, хлора и
других непереходных элементов с высокоэлектроотрицательными ли-
гандами (прежде всего, ОН, О, F), а L-спектры дают информацию
о зарядовой плотности именно d-электронов, всегда обращают вни-
мание на коротковолновые максимумы, отвечающие взаимодействи-
ям вида S3d7r-O2p7r, C13d7r-O2p7r, на увеличение интенсивности этих
75
полос по мере увеличения степени окисления центрального атома и
сопутствующего возрастания положительного эффективного заряда
на центральном атоме (например, в рядах С1О21“-С1Оз1“-С1О41“,
SO22"-SO32"-SO42"), что подтверждается экспериментально одно-
временным положительным сдвигом К-линий спектров. Все это, ка-
залось бы, указывает на участие d-орбиталей в валентных взаимодей-
ствиях с лигандами и на увеличение степени этого участия по мере
увеличения степени окисления и эффективного положительного заря-
да центрального атома. Тем более что, как известно, при увеличении
эффективного положительного заряда атома его d-орбитали становят-
ся менее диффузными и понижают свою энергию и, следовательно,
становятся во всех отношениях более пригодными для валентного вза-
имодейстия с лигандными орбиталями [27, 28]. Вместе с тем все эти
обсуждаемые эффекты и параметры описываемых спектров в количе-
ственном отношении весьма невелики, и в целом данные ультрамягкой
рентгеновской спектроскопии (их систематический обзор и детальное
обсуждение приведены в монографиях [27, 28]) не дают оснований счи-
тать значимыми вклады nd-орбиталей атомов непереходных элементов
IV-VIII групп в валентные взаимодействия с лигандами.
Итак, наиболее общие, фундаментальные, представления квантово-
химической теории электронного строения молекулярных систем со-
гласуются с эффективным участием экстравалентных пр-орбиталей
атомов переходных металлов в образовании связей с лигандами в MLfc
и тем самым утверждают теорию полной гибридизации как адекват-
ный подход в теории строения соединений переходных металлов. И те
же самые представления исключают возможность эффективного уча-
стия экстравалентных nd-орбиталей атомов непереходных элементов в
образовании связей с лигандами в ALfc и тем самым отвергают теорию
полной гибридизации (и ее составляющую — концепцию d-орбиталей)
как адекватный подход в теории строения соединений непереходных
элементов.
Результаты неэмпирических расчетов приводят к преимуществен-
ному выводу о том, что nd-орбитали атомов непереходных элементов
не в состоянии внести сколько-нибудь значительный вклад в образова-
ние связей с лигандами. Существуют два ограничения на вовлечение
этих nd-орбиталей в валентные взаимодействия с лигандными орби-
талями: их большая диффузность и высокая энергия по сравнению с
соответствующими валентными ns- и пр-орбиталями того же самого
изолированного атома. Так что в высших соединениях непереходных
элементов nd-орбитали центрального атома практически не участвуют
в связывании с лигандами, и эти связи образуются в основном за счет
валентных ns- и пр-орбиталей центрального атома. Вклады ns-, пр- и
nd-орбиталей атомов непереходных элементов в связывание с лиган-
76
дами чрезвычайно неравноценны, и общая картина здесь такова, что в
образовании химических связей решающую роль играют пр-орбитали
центрального атома, а роль ns- и особенно nd-орбиталей значительно
менее существенна.
Электронная структура соединений непереходных элементов вы-
ражается, главным образом, в валентной неактивности вакантных nd-
орбиталей их атомов из-за резко повышенной энергии вакантных nd-
АО по сравнению с энергиями занятых лигандных орбиталей. Хими-
ческая связь в этих соединениях может быть осуществлена без nd-
орбиталей центрального атома и, главным образом, за счет его ва-
лентных пр-орбиталей, а гибридизация валентных орбиталей атома
А здесь представляется вообще нереалистичной. Особо ненадежным
представляется положение концепции d-орбиталей, требующее такого
числа гибридных орбиталей центрального атома, сколько двухцентро-
вых связей реализуется в комплексе AL^, а также априорное суждение
о кратности связей A-L.
В целом результаты теоретического и экспериментального изуче-
ния электронной структуры наиболее типичных соединений элементов
главных подгрупп приводят к выводу о несостоятельности концепции
гибридизации валентных орбиталей центрального атома (с непремен-
ным участием его виртуальных nd-орбиталей) как общей основы элек-
тронного строения таких комплексов. Для соединений непереходных
элементов типичной является существенная неравноценность вкладов
валентных ns-, пр- и nd-орбиталей центрального атома в его взаимо-
действиях с лигандами: решающую роль играют пр-орбитали, роль
ns-орбиталей во многих случаях существенно приглушена (за исклю-
чением, пожалуй, высших окисленных форм), а роль nd-орбиталей
пренебрежимо мала. Характеристические свойства соединений непе-
реходных элементов могут быть получены без участия nd-орбиталей
центрального атома, которые играют роль поляризационных добавок
орбитального базиса; 5рпМп-гибридизация валентных орбиталей цен-
трального атома представляется вообще нереалистичной. По отноше-
нию к «нормальным» соединениям, содержащим только двухэлектрон-
ные двухцентровые связи, многие соединения непереходных элемен-
тов являются электроноизбыточными, или по-другому, орбитально-
дефицитными с многоцентровыми гипервалентными связями.
Представления о гипервалентных связях появились в теории стро-
ения химических соединений в 70-е годы как стремление теорети-
ков найти замену традиционной идее об участии экстравалентных nd-
орбталей центральных атомов в образовании связей с лигандами, кото-
рая в простой и ясной форме описывала электронную структуру фор-
мально высоковалентных соединений непереходных элементов (и тем
самым их свойства), но не выдержала испытания временем — не вы-
77
держала натиска результатов строгих квантово-химических расчетов
электронной структуры и результатов измерения параметров высоко-
энергетических (рентгеновских эмиссионных и рентгенофотоэлектрон-
ных) спектров этих соединений [27, 28, 30, 31]. Все это стимулировало
поиски новых идей, новых моделей, новых представлений о принци-
пах электронного строения соединений непереходных элементов в ка-
честве альтернативы представлениям, так или иначе связанным с уча-
стием в химических связях виртуальных nd-состояний центрального
атома. Эти новые представления, основанные на идее о трехцентро-
вых четырехэлектронных взаимодействиях [32-34], весьма лаконичны.
В комплексах непереходных элементов связи центрального атома А с
лигандами образуются в основном за счет валентных прт-орбиталей
атома А и иногда за счет его п$2-электронов, тогда как вакантные
nd-орбитали остаются валентно неактивными и в связях А-L участия
не принимают (точнее, участие nd-орбиталей настолько незначитель-
но, что им можно пренебречь). Электронная структура ALfc представ-
ляет собой совокупность двухцентровых двухэлектронных (ковалент-
ных, CV) связей А-L при участии неспаренных пр-электронов атома
А и трехцентровых четырехэлектронных (гипервалентных, HV) свя-
зей L(x)-A-L(2) при участии спаренных пр2-электронов или реже ns2-
электронов атома А [29, 35-38]. Участие Ап$2-электронов в валентных
МО считается обязательным в случае комплексов с максимальной сте-
пенью окисления A (SFe, PF5 и др.).
Согласно этим представлениям:
• электронная структура комплекса ALfc непереходного элемента
не отвечает ни валентному возбуждению центрального атома А (даже
в пределах одного и того же значения главного квантового числа п), ни
участию nd-орбиталей атома А в связях с лигандами, ни гибридизации
валентных орбиталей атома А;
• химические связи в ALfc из-за неактивности nd-орбиталей атома
А не могут быть обычными двухэлектронными двухцентровыми свя-
зями, но наилучшим образом и наиболее просто описываются в терми-
нах локализованных молекулярных орбиталей, использующих только
валентные пр- и ns-орбитали центрального атома;
• электронная структура ALfc включает в себя ковалентные (CV:
А-L) и гипервалентные (HV: L^-A-L^)) связи а- и(или) тг-типа и опи-
сывается совокупностью занятых электронами молекулярных орбита-
лей, отражающих соответствующие CV- и HV-взаимодействия между
центральным атомом и лигандами, и (если таковые имеются) квази-
атомных орбиталей А и L.
Ковалентная двухэлектронная двухцентровая связь образуется
при взаимодействии занятой неспаренным электроном валентной пр-
орбитали атома А с занятой неспаренным электроном валентной орби-
78
талью одного лиганда. Молекулярно-орбитальное описание этого вза-
имодействия включает в себя одну занятую связывающую и одну ва-
кантную разрыхляющую МО (рис. 12, а):
~ <Ра - <Рь(0е ),
<Р1 « <Ра + <рь(2е ).
Рис. 12. Образование молекулярных орбиталей при двухэлектронном
двухцентровом (а), четырехэлектронном трехцентровом (б)
и двухэлектронном трехцентровом (в) взаимодействиях.
Гипервалентная четырехэлектронная трехцентровая связь образу-
ется при взаимодействии занятой электронной парой валентной пр-
(редко п$-)-орбитали атома А с занятыми неспаренными электронами
валентными орбиталями одновременно двух лигандов. Молекулярно-
орбитальное описание этого взаимодействия включает в себя одну за-
нятую связывающую, одну занятую несвязывающую и одну вакант-
ную разрыхляющую МО (рис. 12, б):
<Рз » <PL(1) - <РА + <PL(2)(0e~),
<Р2 «<pL(i) - <РЦ2)(2е~),
<Pi ~ ¥?L(i) + <Ра + <PL(2)(2e-).
Поскольку во фрагменте четыре валентных электрона
могут разместиться на связывающей и несвязывающей ^2 молеку-
лярных орбиталях, не затрагивая антисвязывающей срз*-МО, гиперва-
лентные HV-связи в целом обладают достаточной для их возникнове-
ния энергией, хотя они значительно менее прочны, чем ковалентные
CV-связи. В зависимости от того, участвует в образовании HV-связей
пр2-орбиталь атома А или его пз2-орбиталь, различают соответствен-
но гипервалентные-1 (HV-1: XeF2, ХеРд, XeFe, CIF2”, Вг?4~, IFg“) и
79
гипервалентные-2 (HV-2: PF5, SF6, IF7) связи. Последние реализуют-
ся только в высших соединениях непереходных элементов, в которых
центральный атом обладает высшей формальной валентностью, рав-
ной номеру соответствующей менделеевской группы, и поставляет на
образование связей с лигандами не только все три свои валентные пр-
орбитали, но и занятую неподеленной электронной парой валентную
п$2-орбиталь. Таким образом, гипервалентные-2 связи —это трехцен-
тровые четырехэлектронные связи, упрочненные вкладом ns-орбитали
центрального атома.
При образовании HV-1 связей локализованные связывающие мо-
лекулярные орбитали направлены вдоль «декартовых» пр-орбиталей
центрального атома. В случае же связей HV-2 участие ns-орбитали
атома А обеспечивает наиболее симметричную из доступных геомет-
рию молекулы.
Особенностью гипервалентных связей обеих разновидностей явля-
ется то, что они отвечают заполнению электронами не только связыва-
ющих молекулярных орбиталей, что типично для ковалентных связей,
но и соответствующих несвязывающих МО.
Относительный вклад связывающих молекулярных орбиталей в
полную энергию молекул и, стало быть, в прочность связей увели-
чивается в ряду гипервалентная-1 < гипервалентная-2 < ковалентная
связи (£hv-i < ^hv-2 < #cv), что объясняет все наблюдаемые за-
кономерности в межъядерных расстояниях A-L, энергиях двухцен-
тровых связей A-L и многих других характеристиках этих связей
в обсуждаемых молекулах. Относительная прочность всех этих свя-
зей такова: ковалентная двухцентровая связь прочнее трехцентровой
гипервалентной-1 связи, поскольку первая включает в себя два только
связывающих электрона, тогда как гипервалентная-1 связь включает
в себя два связывающих и два несвязывающих электрона; последние
к тому же уменьшают энергию взаимодействия между центральным
атомом и лигандами из-за дополнительного межэлектронного оттал-
кивания. Прочность гипервалентной-2 связи является промежуточной
из-за участия двух п52-электронов центрального атома [35].
Гипервалентные связи представляют собой орбитально-дефицит-
ные электронные взаимодействия между атомами и образуются в
электронно-избыточных молекулах.
Существует и третий тип связей в молекулярных системах непере-
ходных элементов, а именно: орбитально-избыточные (OR) двухэлек-
тронные трехцентровые связи, в каждой из которых занятая неспа-
ренным электроном валентная орбиталь центрального атома А взаи-
модействует одновременно с двумя валентными орбиталями (заняты-
ми единственным неспаренным электроном) двух лигандов L(x) и Ь(2).
Молекулярно-орбитальное описание этого взаимодействия в трехцен-
80
тровом фрагменте L(i)-A-L(2) включает в себя одну занятую связыва-
ющую, одну вакантную несвязывающую (точнее, слабо связывающую)
и одну вакантную разрыхляющую МО (рис. 12, в):
~ <Ра - (<PL(1) + VL(2))(0e“),
<Р2 « <РА + (<РЦ1) - VL(2))(0e~),
~ VA + (<PL(1) + <PL(2))(2e_).
Орбитально-избыточные OR-связи обладают значительно меньшей
энергией, чем ковалентные CV-связи. Орбитально-избыточные связи
образуются, естественно, в электронодефицитных молекулах. Класси-
ческим примером являются связи в молекулах многочисленных боро-
водородов и их производных. В них основным структурным фрагмен-
том является трехцентровая группировка В-Н-В (центральным ато-
мом является атом Н, а лигандами — атомы В). В электронодефицит-
ной ситуации (2е“) энергетически наиболее выгодной всегда являет-
ся уголковая (C^v) структура трехцентрового фрагмента L(i)-A-L(2),
так как двумя электронами здесь заполнена только самая нижняя по
энергии, связывающая, молекулярная орбиталь у>1, и вакантна уже не
только разрыхляющая <рз*-МО, но и несвязывающая <р2-МО (в отли-
чие от линейных трехцентровых четырехэлектронных HV-связей) [27,
28]. Таким образом, орбитально-избыточные связи всегда стабилизи-
руют трехцентровый фрагмент L(i)-A-L(2) в нелинейной, изогнутой
Сги-форме. Действительно, во всех без единого исключения молеку-
лах боранов группировка В-Н-В всегда изогнута.
Двухцентровые ковалентные взаимодействия являются одинарны-
ми (два связывающих электрона в двухцентровом фрагменте A-L).
Двухцентровые части гипервалентных или орбитально-избыточных
взаимодействий имеют формально половинную кратность, поскольку
здесь два связывающих электрона обеспечивают стабилизацию трех-
центрового фрагмента ЬцрА-Ь^), т. е. двух двухцентровых связей
А-L одновременно. Это объясняет, почему энергия двухцентрового
взаимодействия А-L при образовании CV-связей всегда намного боль-
ше, чем при образовании HV- или OR-связей. Относительную проч-
ность HV- и OR-связей оценить трудно. Предположение, что связи
OR прочнее связей HV из-за присутствия в HV-связях двух несвязы-
вающих электронов и сопутствующего увеличения межэлектронного
отталкивания, нельзя считать достаточно строгим.
Ковалентные CV-связи часто выдерживают (с потерей прочно-
сти, разумеется) не только потерю одного электрона со связывающей
МО <pi, но и появление лишнего электрона на разрыхляющей МО
<Р2*« Трехцентровые связи, имеющие намного меньшую прочность, по-
видимому, всегда разрушаются при заселении разрыхляющей МО <рз*.
81
До сих пор обсуждались особенности CV-, HV- и OR-связей только
сг-типа. В принципе те же самые представления справедливы и по отно-
шению к взаимодействиям тг-типа, но при этом необходимо учитывать
специфику, вносимую в электронную структуру молекулы возникно-
вением в ней кратных связей A-L.
Двухцентровые двухэлектронные (CV) связи могут, как извест-
но, возникать в результате ковалентного (1:1), донорно-акцепторного
(0:2) и дативного (2:0) взаимодействий. Гипервалентные связи в ли-
нейном трехцентровом фрагменте L(i)-A-L(2) содержат четыре ва-
лентных электрона, заселяющих связывающую уч и несвязывающую
<Р2 молекулярные орбитали. Такое заполнение может произойти в ре-
зультате двух разных взаимодействий (рис. 13):
• взаимодействия пары электронов центрального атома (на Ха) и
неспаренных электронов обоих лигандов (на хь(1) и Xl(2))’>
• взаимодействия вакантной орбитали центрального атома (ха) и
электронных пар обоих лигандов (на Хь(1) и Xl(2))-
Н
—4— —I—----------Н-----II -Н—
^Ц1) Xl(2)
— Ха
Рис. 13. Типичная схема образования трехорбитальных
трехцентровых МО (связывающей уч, несвязывающей у>2
и антисвязывающей в линейном фрагменте L(l)-A-L(2) (а)
и относительное расположение орбиталей по энергии,
когда заселенная электронной парой орбиталь донора
лежит выше вакантной орбитали акцептора (б).
Поскольку несвязывающая у?2-МО представляет собой чистую
групповую орбиталь лигандов, гипервалентное связывание во фраг-
менте L(x)-A-L(2) осуществляется очевидно в результате или датив-
ного А—> (L(1), L(2)), или донорно-акцепторного А <— (Ьц), Ь(2)) вза-
имодействий — случаи «а» и «б» соответственно, отвечающие связям
центрального атома с ацидными или донорными лигандами [39]. В
принципе аналогичные различия можно, по-видимому, найти и в ме-
ханизме образования двухэлектронных трехцентровых (OR) связей.
В ряду рассмотренных взаимодействий четырехэлектронные трех-
центровые (HV) связи занимают особое место.
При возникновении четырехэлектронных гипервалентных связей в
трехцентровом фрагменте L(i)-A-L(2) одна электронная пара заселяет
несвязывающую молекулярную орбиталь у?2 (в исходном состоянии это
были два неспаренных электрона X1 L( 1) и Х1!^)» по одному на двух ли-
гандах). В образовании этой у?2-МО валентная пр2- или пз2-орбиталь
82
центрального атома А участия не принимает, и тем самым электрон-
ный заряд <р2-МО остается локализованным на лигандах. Другая элек-
тронная пара занимает связывающую молекулярную орбиталь <pi и
тем самым делокализована между всеми тремя атомами фрагмента (в
о
исходном состоянии эта электронная пара Хд целиком принадлежала
центральному атому).
Анализ заселенностей этих двух МО
<Р2 = С12<РД + C22<Pb(l) - С32<РЬ(2),
= Сц<рд + С21 <PL(1) + C31<PL(2)
при Ci2= 0 (если Хь(1) = Хь(2) и группа симметрии Do^) и при любых
соотношениях не равных нулю коэффициентов Си, С21 и С31 приво-
дит к однозначному выводу, что заселенность орбитали центрального
атома в L(i)-A-L(2) меньше двух электронов, а суммарная заселен-
ность лигандных орбиталей больше двух электронов. Таким образом,
заселение четырьмя электронами молекулярных орбиталей <pi и <р2
во фрагменте L(i)-A-L(2) отвечает перетеканию валентной электрон-
ной плотности с центрального атома на лиганды. Иначе говоря, обра-
зование гипервалентных связей L^-A-L^) происходит по механизму
дативного взаимодействия L^)*— A—>L(2), где донором является непо-
деленная электронная пара центрального атома А, а акцептором —
вакантная групповая орбиталь ацидных лигандов. Поэтому в роли по-
следних могут выступать только сильно электроотрицательные атомы
(F, О и т.д.). Поскольку центральный атом выступает в роли донора
электронов, прочность и энергия диссоциации гипервалентных связей
должны в общем случае уменьшаться по мере увеличения потенциа-
ла ионизации центрального атома. Это, кстати, объясняет существо-
вание большого семейства (около 200 соединений) устойчивых фто-
ридов, хлоридов, оксогалогенидов, оксидов ксенона, ксенатов, перк-
сенатов, тогда как для криптона известно лишь несколько фторидов
(KrF2, [KrF]+[SbFe]“ и т.д.), да и то на грани устойчивости, а для
аргона, неона и гелия до сих пор не получены устойчивые химические
соединения, и есть серьезные сомнения в том, что они вообще могут
существовать.
Особенно важно требование высокой электроотрицательности ли-
гандов в тех комплексах непереходных элементов AL^, где централь-
ный атом имеет высшую (равную номеру группы в периодической си-
стеме) формальную валентность. Здесь образуются гипервалентные-2
связи L(!)-A-L(2), в которых участвуют пз2-орбитали атома А, и су-
щественную роль играет относительное энергетическое расположение
валентной ns-орбитали атома А и сг-орбитали лигандов: как прави-
ло, устойчивы только те высшие комплексы AL^, для которых энер-
гия сг-орбиталей лигандов меньше (лежит ниже) энергии ns-орбитали
83
атома А. Поэтому неустойчивы гипервалентные-2 молекулы типа РН5
или Р(СН3)5, и их стабилизация требует введения более электроот-
рицательных лигандов; действительно, устойчивыми оказываются не
только PF5, но уже и (СРз)2Р(СНз)3, (СР3)зР(СНз)2 и т. д.
Эта же причина лежит в основе очень важного явления в химии
элементов главных подгрупп — отсутствия высших соединений у азота,
кислорода, фтора даже при их взаимодействиях с наиболее электро-
отрицательными лигандами. Нестабильность NF5, OFg, F2O? объяс-
няется тем, что п$2-электронная пара атомов N, О или F имеет столь
низкую энергию, что гипервалентные-2 связи с ее участием не могут
быть достаточно прочными даже при взаимодействии с валентными
2ра-орбиталями атомов F или О, являющихся сильнейшими по элек-
троотрицательности лигандами. Стабильность же гипервалентных-2
молекул PF5, SFe, IF7 обусловлена прежде всего более донорным ха-
рактером атомов Р, S, I (по сравнению с N, О, F).
Гипервалентные связи, как указано, орбитально-дефицитны. По-
этому в электронной структуре молекул с такими связями валентные
электроны должны занимать высоколежащие по энергии несвязываю-
щие молекулярные орбитали, локализованные исключительно на ли-
гандах. Именно по этой причине взаимодействия А-L в молекулах с
HV-связями (SFe, SF4, PF5, CIF5, CIF3) слабее, чем в соответствую-
щих молекулах с более низкой валентностью центрального атома и
исключительно CV-связями (SF2, PF3, C1F). В терминах теории моле-
кулярных орбиталей связи в орбитально-дефицитных молекулах обя-
заны своим происхождением расщеплению между соответствующими
связывающей и несвязывающей молекулярными орбиталями и требу-
ют перераспределения валентного электронного заряда с центрального
атома на лиганды и именно поэтому — высокой электроотрицательно-
сти лигандов (для притяжения валентных электронных пар централь-
ного атома) и достаточно низкой электроотрицательности централь-
ного атома (для предоставления пр2- и даже п$2-электронных пар на
связывание с лигандами). Существенный критерий устойчивости ги-
первалентных связей состоит в том, что лиганды должны быть более
электроотрицательны, чем центральный атом. Но при этом следует
учитывать, что речь идет исключительно об относительных характе-
ристиках и что общий перенос электронного заряда в комплексе AL^
при возникновении таких связей не всегда должен идти в указанном
направлении (A—>L).
Таким образом, для образования гипервалентных связей необходи-
мо выполнение ряда условий:
1) центральный атом А должен взаимодействовать одновременно с
двумя лигандами и трехцентровый фрагмент L(i)-A-L(2) при участии
валентной пр2-электронной пары атома А должен быть линейным;
84
2) лиганды должны иметь высокую электроотрицательность;
3) орбитальные энергии (потенциалы ионизации) валентных ns- и
пр-электронов атома А должны быть небольшими; вместе с требова-
нием п. 2 это означает, что необходима значительная разница в элек-
троотрицательностях центрального атома и лигандов.
Гипервалентные связи универсальны, но реально возникают лишь
в тех случаях, когда природа центрального атома и лигандов удовле-
творяет всем этим требованиям. Гипервалентные связи в молекулах
непереходных элементов V-VIII групп периодической системы возни-
кают, по-видимому, всякий раз, когда центральный атом образует с
лигандами больше связей по сравнению с числом имеющихся у него
неспаренных валентных пр-электронов, доступных для образования
ковалентных связей; и, конечно, гипервалентные связи возникают в
тех случаях, когда центральный атом развивает (повышает) свою ва-
лентность, образуя связи с наибольшим числом лигандов за счет своих
валентных орбиталей ns-типа.
Роль, которую в молекулах с гипервалентными связями играют
О
валентные ns -электроны центрального атома, состоит в понижении
полной молекулярной энергии, когда занятая двумя электронами ns2-
орбиталь центрального атома смешивается с подходящей по симмет-
рии несвязывающей орбиталью лигандов, так что электроны заселя-
ют результирующие связывающую и несвязывающую молекулярные
орбитали. Однако даже качественная природа такого взаимодействия
зависит от конкретной ситуации: п52-орбиталь центрального атома мо-
жет смешиваться сильно и играть при этом решающую роль в молеку-
лярной устойчивости (PF5, SFe, IF7); вместе с тем П52-орбиталь цен-
трального атома может смешиваться слабо и не давать значительного
вклада в связывание с лигандами; так, в молекулах XeFe, IF5, SF4
нет связывающих молекулярных орбиталей с участием п52-орбитали
(соответствующие МО носят существенно несвязывающий характер).
Таким образом, есть радикальная разница между двумя разновид-
ностями гипервалентных молекул. Молекулярно-орбитальный подход,
который «смешивает» все валентные орбитали центрального атома, в
том числе и ns, без достаточных оснований, лишь бы только учиты-
валась симметрия, часто дает обманчивую картину важности участия
п52-электронов центрального атома в образовании связей с лигандами
в комплексах непереходных элементов.
В отличие от идеи об участии экстравалентных d-орбиталей цен-
тральных атомов в связях с лигандами представления о HV-связях бо-
лее обоснованы с общетеоретических позиций и находят подтвержде-
ние в результатах квантово-химических расчетов электронной струк-
туры комплексов непереходных элементов и в параметрах их высо-
коэнергетических спектров. (Хотя и эти представления по являются
85
абсолютно строгими, поскольку нет окончательного ответа на вопрос
о том, какие валентные электроны (орбитали) атомов непереходных
элементов реально участвуют в связях с лигандами. И, несмотря на
то, что общий тон выводов теоретических и экспериментальных работ
свидетельствует о весьма малой роли экстравалентных вакантных nd-
орбиталей центрального атома в формировании электронной структу-
ры комплексов непереходных элементов, обсуждаемая проблема тре-
бует дальнейших исследований, тем более что здесь часто большая
роль приписывается пр2-электронам центрального атома в образова-
нии связей с лигандами в высших комплексах, но эффективное участие
п$2-электронов в связях никогда не было, по крайней мере, повсемест-
но, убедительно доказано.
Тем не менее представления о HV-связях в учении о принци-
пах электронного строения соединений непереходных элементов, несо-
мненно, универсальны в том смысле, что на их основе можно дать
единообразную интерпретацию электронного строения весьма раз-
ных комплексов, можно на единой основе, а именно, на основе
представлений об образовании гипервалентных связей в результате
трехцентрового четырехэлектронного взаимодействия, описать при-
роду таких разнохарактерных соединений, как фториды благород-
ных газов, интергалогены и их комплексы, высшие соединения непе-
реходных элементов (прежде всего, подгрупп фосфора, серы, хло-
ра), бороводороды и их производные, комплексы с водородной свя-
зью; далее, на той же единой основе можно объяснить многие важ-
ные аспекты химии элементов главных подгрупп: вторичную перио-
дичность (вместе с явлением кайносимметрии и эффектом инертной
пары), менделеевское правило четности, геометрию молекул непере-
ходных элементов, взаимное влияние лигандов в комплексах, особен-
ности химии фтора и кислорода, реакционную способность соединений
и т. д.
Итак, электронную структуру любого комплекса непереходного
элемента в терминах локализованных молекулярных орбиталей мож-
но приближенно представить как единую систему МО, являющуюся
индивидуальной совокупностью трех перечисленных выше типов элек-
тронных взаимодействий (т. е. молекулярно-орбитальных фрагментов
трех типов: CV, HV и OR). Одни комплексы включают в себя лишь
CV-связи (например, SF2), другие — только HV-связи (XeF4, IFe“); су-
ществуют многочисленные комплексы, в электронной структуре кото-
рых одновременно реализуются CV- и HV-связи (SF4, CIF3, PF5) или
CV- и OR-связи (ВгНб). Возможно, что названными категориями ком-
плексов непереходных элементов не исчерпываются все известные мо-
лекулярные системы, но иные разновидности электронной структуры
для обсуждаемых комплексов мало характерны.
86
Все вышесказанное приводит к общехимической концепции кова-
лентных и гипервалентных взаимодействий, приложения которой
весьма многочисленны и касаются важных положений общей химии
(general chemistry): с позиций этой концепции можно адекватно и с
успехом объяснять многие закономерности, связанные с периодиче-
ской системой и проблемой валентности.
При описании электронной структуры многоатомной молекулы ме-
тодом МО можно основываться как на представлениях о делокали-
зованных многоцентровых связях, так и на представлениях о лока-
лизованных электронных взаимодействиях. Для комплексов переход-
ных металлов чаще всего необходим многоцентровый подход. Для ком-
плексов непереходных элементов наряду с многоцентровым подходом
часто используют идеи локализации, при этом в большинстве случа-
ев электронные взаимодействия в многоатомных комплексах непере-
ходных элементов можно свести к локализованным двух- и трехцент-
ровым.
Электронную структуру комплексов непереходных элементов мож-
но описывать в общем виде с помощью теории молекулярных орбита-
лей, используя делокализованные МО, но не содержащие nd-орбиталей
центрального атома. Поскольку одновременно все связи между цен-
тральным атомом и лигандами в комплексах непереходных элемен-
тов ALfc (особенно при высокой степени окисления атома А) не мо-
гут быть обычными двухцентровыми двухэлектронными связями из-
за неактивности nd-орбиталей центрального атома, они лучшим об-
разом описываются в терминах локализованных орбиталей (исполь-
зующих только валентные пр- и ns-электроны центрального атома)
взаимодействиями трех типов: ковалентными CV, гипервалентными
HV, орбитально-избыточными OR.
Изложенные соображения позволяют в теории строения соедине-
ний непереходных элементов отказаться от концепции гибридизации
валентных spmdn-орбиталей центрального атома с эффективным во-
влечением его вакантных nd-орбиталей и использовать лишь теорию
трехцентровых четырехэлектронных связей (и ее более сложные мо-
дификации для пяти- и шестикоординационных комплексов), которая
оставляет решающую роль за np-валентными электронами централь-
ного атома в его взаимодействиях с лигандами в высших (орбитально-
дефицитных) комплексах.
Представление об орбитально-дефицитных молекулах с трехцен-
тровыми четырехэлектронными гипервалентными связями более обос-
новано по сравнению с традиционным представлением о двухцентро-
вых ковалентных связях центрального атома с лигандами. Оно отве-
чает экспериментально определенным и теоретически рассчитанным
характеристикам электронной структуры этих молекул.
87
Все вышесказанное оправдывает использование концепции гипер-
валентных связей применительно к высшим соединениям непереход-
ных элементов, где степень окисления, или формальная валентность,
превышает число неспаренных валентных пр-электронов в основном
(невозбужденном) состоянии изолированного центрального атома. Об-
щий вывод в пользу концепции гипервалентных связей, поэтому лишь
на ее основе целесообразно описывать принципы и особенности элек-
тронного строения комплексов непереходных элементов (причем воз-
можно использование как делокализованных канонических МО, так
и локализованных эквивалентных МО, отражающих двух- и трехцен-
тровые связи), а также интерпретировать принцип периодичности в
химии элементов главных подгрупп.
Концепция гипервалентных связей позволяет надежно и без натя-
жек интерпретировать очень важный для всей химии главных под-
групп факт: непереходные элементы V-VIII групп периодической си-
стемы на высших ступенях окисления образуют устойчивые соедине-
ния только с сильно электроотрицательными лигандами. Это прямо
следует из того, что высшие соединения непереходных элементов V-
VIII групп должны быть структурами с гипервалентными связями, об-
разованными при участии валентной п$2-электронной пары централь-
ного атома. Эта п$2-орбиталь имеет весьма низкую энергию, поэтому
гипервалентные связи с ее участием могут быть достаточно прочны-
ми (а без этого высшее соединение не может быть стабильным) только
при условии очень низкой энергии валентных орбиталей лигандов, т. е.
при условии особо высокой электроотрицательности лигандов. Именно
поэтому, например, существуют SFe, SO3, SO2CI2 и нестабильны хло-
рид, бромид, иодид формально шестивалентной серы, а также не суще-
ствует соединений серы на высоких ступенях окисления с водородом,
металлами, углеродом, азотом, с атомами многих других химических
элементов и многоатомными группами.
Сказанное ранее, во-первых, объясняет различия в электронном
строении соединений непереходных элементов и переходных металлов:
в электронной структуре последних и пространственный, и энергети-
ческий факторы благоприятствуют эффективному вовлечению в де-
локализованные МО всех типов валентных орбиталей атома М (ns,
(n — l)d, пр) в соответствии с симметрией комплексов ML^, в то вре-
мя как в электронной структуре соединений AL& возможно вовлече-
ние в делокализованные МО лишь валентных ns- и пр-орбиталей. Во-
вторых, подводит к той новой точке зрения, что химия переходных
металлов —это химия структур, построенных в основном за счет ва-
лентных (n—1)d-орбиталей (с участием, но без решающей роли пр-АО)
центрального атома, тогда как химия непереходных элементов вклю-
чает в себя, главным образом, соединения, образованные за счет ва-
88
лснтных пр-орбиталей (в предельном выражении — вообще без nd-АО)
центрального атома. В-третьих, позволяет для теоретической интер-
претации принципа периодичности широко использовать концепцию
ковалентных и гипервалентных взаимодействий в теории канониче-
ских делокализованных МО.
С позиций этой концепции значительные различия в электронном
строении MLfc и AL^ позволяют обособить мир соединений переходных
металлов почти исключительно с ковалентными структурами от мира
соединений непереходных элементов как с ковалентными, так и с ги-
первалентными структурами. При этом химию непереходных элемен-
тов можно с некоторой условностью разделить на две части. Первую
составляют соединения с самыми разнообразными лигандами и срав-
нительно невысокими степенями окисления центральных атомов: по-
давляющее большинство соединений элементов I-IV групп, низкова-
лентные соединения V, VI и VII групп (их число резко уменьшается
при переходе от элементов V группы к элементам VI и тем более VII
групп). Речь идет о соединениях с ковалентными структурами.
В этом отношении инертные газы, действительно, инертны и прак-
тически не вступают в химические взаимодействия. Для них почти
полностью «закрыта» эта часть химии —химия бромидов, иодидов,
сульфидов, селенидов, теллуридов, нитридов, фосфидов, карбидов,
силицидов, боридов, гидридов и многих других соединений, типич-
ных для остальных низковалентных непереходных элементов. Другую
часть составляют, главным образом, высшие соединения непереходных
элементов V-VII групп и все соединения инертных газов (VIII груп-
па); здесь лигандами являются атомы фтора, кислорода, реже хлора,
серы, азота и некоторые другие частицы (скажем, ОН-группы). Это
соединения с гипервалентными структурами.
Если представить прямоугольную матрицу периодической систе-
мы только непереходных элементов и если одновременно использовать
концепцию гипервалентных связей как парадигму теории химического
строения соединений непереходных элементов, химия этих элементов
приобретает образ двух воображаемых конусов: один, символизирую-
щий ковалентные структуры, сужается при движении в системе слева
направо (от щелочных и щелочноземельных металлов к халькогенам и
галогенам) и не достигает группы инертных газов; другой, символизи-
рующий гипервалентные структуры, сужается в обратном направле-
нии, охватывает все соединения инертных газов, большую часть химии
галогенов и халькогенов (все их соединения, кроме одновалентных и
двухвалентных соответственно), значительную часть химии элементов
подгруппы фосфора и не достигает щелочных и щелочноземельных
металлов. Такой переход ковалентных соединений в гипервалентные
представляет собой ведущий мотив в соотношении свойств непереход-
89
пых элементов в периодической системе и тем самым отражает доми-
нирующее проявление принципа периодичности в химии непереходных
элементов.
Для гносеологической оценки концепции гипервалентных связей в
учении о периодичности добавим, что эта концепция, во-первых, да-
ет простую и надежную основу для интерпретации горизонтального
сходства химических элементов в рядах периодической системы наря-
ду с вертикальными сходством в подгруппах, во-вторых, рационально
и непротиворечиво объясняет, как будет показано далее, такие важ-
ные «составные части» принципа периодичности, как менделеевское
правило четности и вторичная периодичность, которые по-настоящему
начинают проявляться лишь с появлением гипервалентных структур
в химии непереходных элементов.
Глава 5
МЕНДЕЛЕЕВСКОЕ ПРАВИЛО ЧЕТНОСТИ
И ГРАНИЦЫ ЕГО ПРИМЕНИМОСТИ
Конкретным следствием значительных различий в электронном
строении ALfc и MLfc являются правило четности в химии непереход-
ных элементов и отсутствие аналогичного правила в химии переход-
ных металлов.
Менделеевское правило четности является одной из важных зако-
номерностей в химии непереходных элементов и тем самым важной
составляющей учения о Периодическом законе и периодической систе-
ме. Это правило относительно большей энергетической стабильности
тех форм соединений элемента главной подгруппы (эти формы иногда
оказываются единственно существующими), в которых центральный
атом имеет степень окисления той же четности, что и четность но-
мера соответствующей группы в периодической системе. Использова-
ние правила четности является действенным и простым приемом для
качественной оценки относительной устойчивости и реакционной спо-
собности различных окисленных форм непереходного элемента. Пока-
зательной иллюстрацией правила четности служит система фторидов
непереходных элементов V-VIII групп:
V группа PF pf2 PF3 ^^4) PFS
VI группа SF sf2 sf4 (SFj) SF6
VII группа C1F C1F3 (C1F4) cif5 (C1F6) C1F7
VIII группа XeF2 (XeF3) XeF4 (XeFs) XeF6 (XeF7) XeF,
Здесь в скобках приведены малоустойчивые или несуществующие
фториды. Хорошо видно, что правило четности не охватывает всю
систему фторидов и выполняется в ней лишь правее черты. Тради-
91
ционная интерпретация правила четности, основанная на концепции
d-орбиталей, была в определенной мере противоречивой и не объясня-
ла этого важного момента.
Правило четности легко объяснимо в рамках концепции гиперва-
лентных связей. На основе представлений о гипервалентных связях
правило четности находит последовательное и непротиворечивое объ-
яснение. Далее становится возможным уточнение этого правила.
Действительно, в соответствии с концепцией гипервалентных свя-
зей можно предвидеть, что оно не должно охватывать все существую-
щие соединения непереходного элемента, т. е. все его возможные сте-
пени окисления, но должно выполняться только начиная с некоторой
определенной степени окисления центрального атома, а именно, с мо-
мента возникновения первых гипервалентных связей. До тех пор пока
на валентных пр-орбиталях центрального атома имеются неспаренные
электроны, образуется непрерывный ряд достаточно устойчивых фто-
ридов с молекулярными структурами, включающими в себя только
ковалентные связи. Как только в этих связях оказываются заняты-
ми все неспаренные пр-электроны, исчезает возможность дальнейше-
го образования фторидов только с ковалентными связями А-F. В этот
момент степень окисления центрального атома по своей четности со-
ответствует четности номера его группы в периодической системе (это
очевидно, так как число неспаренных валентных пр-электронов в ос-
новном состоянии атома четное для непереходного элемента четной
группы и нечетное —для нечетной).
Дальнейшее увеличение степени окисления центрального атома
(и его координационного числа) сверх этой характеристичной, ре-
гламентированной числом неспаренных электронов на валентных пр-
орбиталях центрального атома величины становится невозможным за
счет ковалентных связей и поэтому происходит за счет образования
гипервалентных связей F(1)-A-F(2). Эти связи с присущей им трех-
центровой природой возникают только при одновременном присоеди-
нении к центральному атому сразу двух новых одновалентных ли-
гандов, т. е. степень окисления центрального атома возрастает сразу
на две единицы, что и определяет правило четности.
Итак, «скачки» в изменении степени окисления центрального ато-
ма в ряду его устойчивых соединений начинаются лишь с момента
возникновения гиперструктур. Эта граница в вышеприведенной систе-
ме фторидов элементов главных подгрупп проходит через XeF, C1F2,
SF3, PF4. Естественно, чем раньше (т. е. на меньшей ступени окис-
ления центрального атома) появляются гиперструктуры, тем в боль-
шей мере правило четности охватывает соединения непереходных эле-
ментов данной группы периодической системы. Это происходит при
уменьшении числа неспаренных пр-электронов, т. е. при увеличении
92
суммарного числа валентных электронов в атомах непереходных эле-
ментов, по мере возрастания номера группы в периодической системе.
Соединения инертных газов, в которых ковалентные связи отсутству-
ют и гиперструктуры появляются на самых первых ступенях окисле-
ния, являют собой пример соединений, полностью следующих правилу
четности.
Глава 6
ОСОБЕННОСТИ ХИМИИ ЭЛЕМЕНТОВ-
РОДОНАЧАЛЬНИКОВ d-, /-СЕМЕЙСТВ
В теории периодической системы аксиоматическими являются раз-
биение всех химических элементов на четыре семейства (по признаку
наличия электронов s-. р-, d-, /-типов в валентной структуре их ато-
мов) и констатация индивидуальности и особенностей свойств, имма-
нентно присущих элементам каждого такого семейства. Показательно,
что наиболее наглядное оформление любого варианта таблицы, отра-
жающей периодическую систему, всегда четырехцветное.
Одно из главных, если не самое главное, положение теории перио-
дической системы заключается в утверждении и аргументированном,
безусловно, концептуальном, объяснении особенностей химии элемен-
тов, открывающих каждое из этих четырех семейств.
Действительно, среди 109 известных (к моменту написания этой
книги) химических элементов выделяются своими особыми свойства-
ми 32 (s2, р6, d10, /14) элемента, являющихся родоначальниками s-
семейства (водород и гелий), р-семейства (элементы 2-го периода), d-
семейства (металлы первой вставной декады) и /-семейства (лантано-
иды). Перечисленные элементы отличаются от своих последователей
в периодической системе многими признаками. Эти отличия проявля-
ются прежде всего в том, что атомы элементов-родоначальников се-
мейств обладают ограниченными (по сравнению с соответствующими
атомами-аналогами) валентными возможностями.
Нет нужды сколько-нибудь подробно останавливаться на совершен-
но особых свойствах водорода и гелия, не имеющих даже отдаленных
аналогов среди всех химических элементов. Валентная (она же пол-
ная) электронная структура атомов Н и Не представляет собой един-
ственную 1 s-орбиталь, занятую одним или двумя электронами соот-
ветственно. Это объясняет сугубо одновалентное состояние атомов Н
и Не и их отчетливо выраженную химическую уникальность.
Химическое поведение p-элементов 2-го периода системы —В, С,
N, О, F, Ne — характеризуется ярко выраженными особенностями, не
94
присущими остальным p-элементам. Принципиальные отличия родо-
начальников главных подгрупп от их последователей проявляются (и
притом в отчетливой и строгой форме) в большей или меньшей степе-
ни в свойствах любых образуемых ими неорганических соединений, на
любых ступенях окисления центрального атома. Но наиболее сильно
эти отличия проявляются в свойствах высших соединений, где степень
окисления центрального атома равна номеру соответствующей группы
в периодической системе, и выражаются в пониженной устойчивости
(вплоть до отсутствия) высших соединений родоначальника главной
подгруппы по сравнению с аналогичными соединениями остальных ее
элементов. При этом важно подчеркнуть, что отличия типических эле-
ментов от всех остальных элементов главных подгрупп наиболее силь-
но выражены в группах с большим порядковым номером (V-VII), т. е.
в химии непереходных элементов правой половины периодической си-
стемы.
В этом отношении полезно деление типических элементов (равно
как и элементов любого другого периода системы) на ранние и позд-
ние. В атомах ранних типических элементов (элементов первой поло-
вины ряда—Li, Be, В, С) число валентных электронов не превыша-
ет числа (4) имеющихся валентных орбиталей. Эти атомы образуют
с лигандами связи за счет своих неспаренных электронов и вакант-
ных АО. Особое положение занимает углерод, в атомах которого чис-
ло валентных электронов равно числу валентных орбиталей. В ато-
мах поздних типических элементов (вторая половина ряда —N, О, F,
Ne) число валентных электронов всегда больше числа (4) имеющих-
ся валентных орбиталей. Эти атомы образуют с лигандами связи за
счет своих неспаренных электронов и неподеленных электронных пар.
Особое положение занимает неон, в атомах которого все валентные
электроны спарены.
Именно в химии поздних непереходных элементов наиболее сильны
различия в свойствах типического элемента и всех его последователей
в главной подгруппе. Здесь прежде всего наиболее отчетливо проявля-
ется снижение предела окисления типического элемента. В частности,
ни фтор, ни кислород не образуют соединений на высоких ступенях
окисления, приближающихся или тем более достигающих значений но-
мера группы периодической системы. В то же время последователи
фтора и кислорода по их подгруппам — галогены и халькогены — об-
разуют такие соединения во множестве, и эта способность галогенов и
халькогенов образовывать соединения на самой высокой ступени окис-
ления, значение которой равно номеру группы, является весьма харак-
терной чертой всей их химии.
Основой обсуждаемых отличий типических элементов от осталь-
ных непереходных элементов является пониженная валентность атома
95
типического элемента по сравнению с его последователями в главной
подгруппе, которые всегда в той или иной мере проявляют тенденцию
к увеличению валентности своих атомов. Эта тенденция прослежива-
ется в химии элементов любой главной подгруппы, и если в случае
ранних непереходных элементов она выражена слабо, но, отнюдь, не
исчезает, то в случае поздних непереходных элементов она выражена
весьма отчетливо.
Химическое поведение переходных металлов первой вставной де-
кады (3d) существенно отличается от переходных металлов второй и
третьей декад (4d, 5d). Сопоставление форм существования соедине-
ний Зй-элементов, с одной стороны, и соединений 4d- и 5й-элементов, с
другой, неизбежно приводит к общему выводу о пониженной валент-
ности Зс/-атомов по сравнению с их последователями 4d, 5d в перио-
дической системе. Один из ярких примеров: последователи железа —
рутений и осмий — образуют в отличие от железа много стабильных
соединений на высоких ступенях окисления. В химии этих тяжелых
переходных металлов далеко не единичны соединения, в которых ато-
мы Ru и Os используют в связях с лигандами большинство или даже
все свои валентные орбитали и проявляют, по-видимому, очень высо-
кую валентность (находятся в шести-, семи-, восьмивалентных состо-
яниях). Это подчеркивает ограниченность валентных возможностей
атомов железа как элемента первого переходного ряда в сравнении
с его последователями — элементами второго и третьего переходных
рядов.
Аналогичными примерами богата химия переходных металлов и
других дополнительных подгрупп периодической системы.
Большинство сведений об особенностях элементов-родоначальни-
ков дополнительных подгрупп касается поздних переходных металлов,
т. е. элементов вторых половин вставных декад. Объясняется это тем,
что обычно подчеркивают резкое отличие Зй-элементов от их после-
дователей в подгруппах в смысле максимально достижимых значений
формальной валентности. В этом отношении ранние Зй-элементы (ме-
таллы первой половины переходного ряда), казалось бы, не отличают-
ся принципиально от своих последователей. В самом деле, титан, вана-
дий, хром и марганец не могут в этом смысле считаться отличными по
своей природе от соответственно циркония, ниобия, молибдена и техне-
ция и их более тяжелых аналогов (Hf, Та, W, Re), так как в этих допол-
нительных подгруппах максимальное значение формальной валентно-
сти одинаково для всех элементов подгруппы и равно номеру соответ-
ствующей менделеевской группы. Но если обратиться к более строгим
характеристикам молекулярной структуры, и в том числе к координа-
ционному числу и истинной валентности центрального атома, то сразу
же наиболее важные особенности химии элементов-родоначальников
96
дополнительных подгрупп ярко проявятся и в случае ранних переход-
ных металлов. Подтверждающих это примеров очень много, и один из
них, притом весьма общего характера, состоит в том, что кластерные
соединения (в них валентность атомов М намного больше формально-
го значения) образуются в основном тяжелыми (4d, 5d) переходными
металлами.
Разительно отличаются друг от друга значениями достигаемых сте-
пеней окисления лантаноиды (4/) и актиноиды (5/). Так, в соединени-
ях лантаноидов степень окисления металла, превышающая значение
3, крайне редка, и известные примеры исчерпываются производны-
ми четырехвалентного церия, а также несколькими кислородными и
фтористыми соединениями четырехвалентных тербия, неодима и пра-
зеодима, существующими только в твердом состоянии и неустойчивы-
ми в водных растворах из-за высокой окислительной активности PrIV
и Tbiv. Напротив, в соединениях актиноидов высокие ступени окис-
ления металла достигаются очень часто и оказываются вполне ста-
бильными не только в твердом состоянии, но и в водных растворах.
Недаром до открытия и последующего изучения трансурановых эле-
ментов три самых тяжелых в те времена элемента —торий, протакти-
ний и уран — считались родственными гафнию, танталу и вольфраму
и располагались в периодической системе соответственно в IV, V и VI
дополнительных подгруппах. Тогда же предполагалось, что элементы
93, 94, 95 и 96 по свойствам должны быть похожи на рений, осмий,
иридий и платину.
Многочисленные систематизированные примеры, иллюстрирую-
щие глубокие и часто проявляющиеся отличия элементов-родоначаль-
ников s-, р-, d-, /-семейств от их последователей в периодической си-
стеме, приведены в [40, 41].
Таким образом, взяв за основу главную характеристику химиче-
ского элемента — валентность его атомов, которая отвечает не только
за формы существования соединений, их устойчивость и реакционную
способность, но и за весь комплекс присущих им свойств, нетрудно
убедиться в уникальной природе водорода и гелия (1s), в особой при-
роде типических элементов (2р), элементов первого переходного ряда
(3d) и редкоземельных элементов (4/) по отношению ко всем осталь-
ным элементам периодической системы. Перечисленные элементы от-
личаются от своих последователей в периодической системе многими
признаками. Но эти отличия наиболее ярко проявляются в том, что
родоначальники соответствующих семейств обладают ограниченными
(по сравнению со своими аналогами) валентными возможностями, что
их атомы характеризуются пониженной валентностью.
Несомненно, что основополагающей причиной особенностей химии
всех перечисленных ранее элементов является валентная электронная
97
структура их атомов со всеми присущими ей отличительными черта-
ми и своеобразием. В атомах этих особенных по химической приро-
де элементов валентными являются электроны на 1s-, 2р-, 3d- и 47-
орбиталях, и поэтому причину специфичности химической природы
этих элементов надлежит искать в исключительных особенностях ва-
лентных электронов в 1s-, 2р-, 3d- и 4/-состояниях. Существенно, что
особенности химии 1s-, 2р-, 3d- и 4/-элементов имеют общую причину,
так как отвечают общему поведению валентных электронов в состоя-
ниях 1s, 2р, 3d и 4/.
Особые и общие в своей особенности характеристики Is-, 2р-, 3d-
и 4/-электронов непринужденно объясняются тем, что, несмотря на
все различия электронов в 1s-, 2р-, 3d- и 4/-состояниях, они являются
первыми (и это их объединяет) носителями новой симметрии распре-
деления электронной плотности в атоме.
Заранее подчеркнем, что электроны 1s-, 2р-, 3d- и 4/-орбиталей
имеют наименьшие радиусы пространственного распределения заря-
довой плотности и наибольшие энергии связи с ядром, т. е. наиболь-
шие потенциалы ионизации, по сравнению с электронами соответству-
ющих ns-, np-, nd- и п/-орбиталей с большими квантовыми числами.
Этими свойствами обладают электронные конфигурации с минималь-
ным значением главного квантового числа п при впервые появляю-
щемся новом орбитальном квантовом числе I (здесь и далее исполь-
зуются приближенные представления об электронных конфигураци-
ях основных и иногда возбужденных состояниях многоэлектронных
атомов).
По-видимому, К. Йоргенсен бы первым, кто обратил внимание на
то, что «электроны 2р и 3d имеют весьма малые радиусы и большие
энергии связи с ядром; выглядит так, что этими свойствами обладают
электронные конфигурации в тех случаях, когда впервые появляются
п, для которых данное I уже возможно; такое представление опре-
деленным образом связано с отсутствием у этих орбиталей нодаль-
ных мод» [42]. Здесь под радиусом электрона К. Йоргенсен имеет в
виду расстояние от ядра атома до внешнего максимума его зарядовой
плотности, а под отсутствием нодальных мод — отсутствие внутренних
максимумов и узлов соответственной волновой функции.
Это было, безусловно, крайне важным представлением об особых
свойствах электронов, впервые в атоме характеризующихся новым
значением орбитального квантового числа I. Но К. Йоргенсен не сфор-
мулировал никаких следствий и не обобщил эти свои представления в
том смысле, что не распространил их на электроны 1s, 4/, 5д.
Такое обобщение впервые сделал С. А. Щукарев [5, 6, 8, 9, 43], он же
ввел и термин кайпосимметрия (от древнегреч. кайнос — новый)
для описания состояний 1s, 2р, 3d и 4/, осуществляющихся в атомах
98
при первом появлении соответствующих симметрий пространствен-
ного распределения электронного заряда.
В целом особенности химии 1s-, 2р-, 3d- и 4/-элементов объединены
и объяснены в концепции кайносимметрии [41].
Нет ничего неожиданного в том, что именно симметрия, а не иная
характеристика, описывающая поведение электрона в атомах и мо-
лекулах, лежит в основе фундаментальных особенностей химической
природы обсуждаемых элементов и что именно симметрия по-новому
освещает первостепенные аспекты учения о Периодическом законе и
системе элементов.
Симметрия является фундаментальным свойством структуры ма-
териальной системы на любом уровне организации материи. Теория
относительности и квантовая механика, установление связей между
симметрией пространства-времени и законами сохранения, открытие
античастиц и установление тем самым симметрии зарядового сопря-
жения, несохранение четности при слабых взаимодействиях, инвари-
антность важнейших уравнений физики относительно пространствен-
ных, временных и зарядовых преобразований, электронное строение
молекул и их стереохимия, природа кристаллов, достижения молеку-
лярной биологии и генетики — все это указывает на то, что симметрия
действительно является фундаментальным свойством, присущим лю-
бой материальной системе.
Симметрия представляет собой совершенно особую разновидность
структурных закономерностей. Это проявляется прежде всего во все-
общности явления симметрии в структурах самых различных матери-
альных систем, включающих в себя симметрию как фундаментальную
черту, часто определяющую все другие их особенности.
По словам Ньюмена, «симметрия устанавливает удивительное род-
ство между предметами, явлениями и теориями, внешне никак не
связанными: земным магнетизмом, женской вуалью, поляризованным
светом, естественным отбором, теорией групп, инвариантами и преоб-
разованиями, рабочими привычками пчел в улье, строением простран-
ства, рисунками ваз, квантовой физикой, скарабеями, лепестками цве-
тов, интерференционной картиной рентгеновских лучей, делением кле-
ток морских ежей, равновесными конфигурациями кристаллов, ро-
манскими соборами, снежинками, музыкой, теорией относительнос-
ти. ..».
Симметрия отражает наиболее общие структурные закономерности
материальных систем, и ее законы, воплотившиеся в теории групп,
являются наиболее общими структурными законами. Особый харак-
тер симметрии как структурной закономерности подчеркивается еще
и тем, что ее математическая интерпретация не связана с конкретной
природой системы и поэтому допускает широкие абстракции.
99
Далее мы будем использовать терминологию, связанную с теорией
симметрии лишь условно, вкладывая в термин «кайносимметрии» (не
часто, кстати, используемый в химической литературе) весьма про-
стой смысл, а именно: первые орбитальные состояния электронов в
атоме с заданным значением квантового числа I в одноэлектронном
приближении.
Электроны в состояниях 1s, 2р, 3d, 4/, 5</, которые впервые вносят
в атом новую симметрию распределения электронной плотности и для
каждого из которых соответственное орбитальное квантовое число (О,
1, 2, 3 или 4) впервые характеризует одноэлектронную атомную ор-
биталь, можно называть кайносимметричными электронами. Из-
вестно, что кайносимметричным 1s-, 2р-, 3d- и 47-электронам в сво-
бодных атомах присущи характеристики, общие для столь, казалось
бы, различных по свойствам электронов и одновременно отличные от
аналогичных (по симметрии пространственного распределения заря-
довой плотности) состояний с теми же самыми орбитальными кван-
товыми числами Z, но с большими главными квантовыми числами п.
Их можно называть палинсимметричными (древнегреч. палин —
повторный) электронами.
Прежде всего для кайносимметричных электронов равно нулю чис-
ло узловых поверхностей, характеризующих распределение зарядовой
плотности электрона в атоме как функцию расстояния от ядра. Этот
результат давно известен в квантовой механике атома: он непосред-
ственно вытекает из известного канона квантовой механики атома,
согласно которому число узлов атомной одноэлектронной волновой
функции хП1 равно п — I — 1. Этому же выражению равно число сфери-
ческих узловых поверхностей (математических, с нулевой толщиной),
где зарядовая плотность электрона, описываемого указанной волновой
функцией Xnh равна нулю. Далее приведены числа таких узловых по-
верхностей для атомных орбиталей при последовательном увеличении
значений главных п и орбитальных I квантовых чисел:
Is 2s 3s 4s 5s 6s 7s 8s
0 1 2 3 4 5 6 7
2p 3p 4p 5p 6p 7p Sp
0 1 2 3 4 5 6
3d 4d 5d6d7d
0 12 3 4
4/5/6/
0 1 2
5g 6g
0 1
100
Вполне очевидно, что число узловых поверхностей для кайносим-
метричных электронов 1s, 2р, 3d, 4/, 5д действительно равно нулю.
Отсутствие узловых поверхностей означает отсутствие внутренних
(в околоядерной области пространства, в остове атома) максимумов в
радиальном распределении зарядовой плотности любого кайносиммет-
ричного электрона. В самом деле, раз нет узловых поверхностей, то
нет и внутренних максимумов радиального распределения зарядовой
плотности этих электронов. Для всех остальных — палинсимметрич-
ных — электронов начиная с 2s, Зр, 4d, 5/, бд число узловых поверх-
ностей «больше нуля», они существуют и, следовательно, существуют
(точно в таком же количестве) внутренние максимумы радиального
распределения электронной плотности, причем число таких максиму-
мов последовательно возрастает по мере увеличения главного кванто-
вого числа п.
Представление об отсутствии или наличии (притом строго опре-
деленное число) внутренних максимумов радиального распределения
зарядовой плотности у электронов с заданными значениями главно-
го п и орбитального I квантовых чисел является одним из важнейших
факторов, определяющих свойства атомов и образуемых ими молекул.
В теории самосогласованного поля Хартри — Фока для атомов кай-
носимметричные электроны описываются самыми простыми волновы-
ми функциями. Но главное заключается в том, что орбитальные ради-
усы кайносимметричных электронов меньше орбитальных радиусов
всех остальных электронов с тем же значением главного квантового
числа п. Орбитальный радиус является важной характеристикой про-
странственного распределения электронной плотности той или иной
атомной орбитали. Величину орбитального радиуса в теории элек-
тронного строения атома определяют как расстояние от центра ато-
ма (ядра) до главного максимума радиальной электронной плотности
соответствующей атомной орбитали. Значения орбитальных радиусов
внешних (валентных) электронов для основных состояний атомов и
атомарных ионов большинства известных химических элементов бы-
ли вычислены при решении уравнения Дирака, учитывающего реля-
тивистские эффекты.
Результаты этих вычислений позволили строго охарактеризовать
радиальное распределение зарядовой плотности внешних электронов
в терминах положения главного максимума электронной плотности
для атомов почти всех элементов периодической системы. Эти резуль-
таты показали, что только в атомах элементов-кайносимметриков ва-
лентные электроны в кайносимметричных состояниях имеют меньшие
орбитальные радиусы по сравнению с другими валентными электро-
нами с тем же главным квантовым числом: 2р меньше 2s, 3d меньше
3s и Зр, 4/ меньше 4s, 4р и 4d (табл. 5, рис. 14); 1s электроны имеют,
101
Рис. 14- Соотношение орбитальных радиусов внешних электронов с одним
и тем же главным квантовым числом в атомах элементов-кайносимметриков (а)
и в атомах их последователей (б).
естественно, наименьшие орбитальные радиусы —их просто не с чем
сравнивать.
Таблица 4- Расстояния (А) от центра атома
до главного максимума радиальной
электронной плотности
внешних орбиталей свободных
нейтральных атомов в основном состоянии
N Р As (р-элементы) Sb Bi 2s 0,521 3s 0,803 4s 0,826 5s 0,969 6s 0,963 2pi/2 0,487 ЗР1/2 0,916 4Pl/2 0,982 5Pi/2 1,140 6P1/2 1,130 2P3/2 0,488 Зрз/2 0,919 4РЗ/2 1,001 5рз/2 1,193 6рз/2 1,295
Со Rh (d-элементы) Ir Зрз/2 0,355 4Рз/2 0,496 5Рз/2 0,536 3^3/2 0,342 4^з/2 0,584 5^з/2 0,655 3d5/2 0,344 4^5/2 0,589 5^5/2 0,676
В атомах остальных химических элементов последовательность ор-
битальных радиусов валентных электронов всегда соответствует по-
следовательности увеличения орбитального квантового числа при со-
102
хранении главного квантового числа постоянным (ns < пр < nd < nf).
Это легко объясняется уменьшением числа внутренних максимумов
электронной плотности и соответственным падением проникающей
способности электронов с одним и тем же главным квантовым чис-
лом, т. е. теми двумя эффектами, которые сопутствуют возрастанию
орбитального квантового числа.
Таким образом, отличительная особенность, общая для всех кайно-
симметричных электронов в валентном состоянии атомов (и не толь-
ко нейтральных атомов, но и атомарных ионов), состоит в том, что
эти электроны, описываемые «безузловыми» волновыми функциями
(здесь не учитываются приходящиеся на центр атома узлы волновых
функций пр, nd, nf) и обладающие только одним (главным) макси-
мумом в радиальном распределении зарядовой плотности, заселяют
атомные орбитали наименьшей протяженности среди всех ор-
биталей данного главного квантового числа.
Далее, кайносимметричные электроны обладают наибольшими
значениями орбитальных потенциалов ионизации среди всех электро-
нов данного орбитального квантового числа. Это особенно ярко вид-
но на примере np-элементов: орбитальный потенциал ионизации 2р-
кайносимметрика (В, С, N, О, F, Ne), т. е. энергия удаления из ней-
трального атома кайносимметричного 2р-электрона, резко отличает-
ся от соответствующих значений энергии удаления Зр-, 4р-, 5р- и 6р-
электронов из атомов элементов, следующих за кайносимметриком в
той или иной главной подгруппе периодической системы. Орбиталь-
ный потенциал ионизации 2р-кайносимметрика в сравнении с осталь-
ными np-элементами данной группы очень высок. После резкого скач-
ка значения орбитального потенциала ионизации при переходе от 2р-
кайносимметрика к Зр-элементу дальнейшее его уменьшение замедля-
ет темп, приводя тем самым к сравнительно узкому интервалу зна-
чений орбитальных потенциалов ионизации для всех остальных пр-
элементов одной и той же подгруппы. Аналогичная картина наблюда-
ется в случаях Is-, 2s-, 3s-, 4s- ... и 3d-, 4d-, 5d- ... элементов.
Причину резко повышенных орбитальных потенциалов ионизации
атомов в тех случаях, когда происходит удаление валентного кайно-
симметричного электрона 1s, 2р, 3d или 4/, следует, безусловно, видеть
в том, что этим электронам в атомах не предшествуют электроны с той
же пространственной симметрией. Отталкивательное действие, оказы-
ваемое на внешние электроны внутренними, остовными, электронами
той же самой орбитальной симметрией называют псевдопотенциалом.
Понятие о псевдопотенциале, действующем на электрон подобно
силе, отталкивающей его от ядра в сторону периферии электронной
оболочки атома, первоначально было развито в 30-х годах в работах
В. А. Фока, М. Г. Веселова, М. И. Петрашень и Г. Г. Геллмана [44]. Для
103
кайносимметричных электронов псевдопотенциал равен нулю, и это
одно из самых важных, по сути, определений кайносимметрии, по-
могающее в простой форме понять причину аномально высоких ор-
битальных потенциалов ионизации кайносимметричных электронов в
валентном слое атома. Именно из-за нулевого псевдопотенциала кай-
носимметричные электроны 1s, 2р, 3d, 4/, 5д «проваливаются» глубо-
ко в близкую к ядру область атомного пространства — имеют меньший
радиус и большую энергию связи с ядром (больший орбитальный по-
тенциал ионизации), чем для всех остальных электронов данного глав-
ного квантового числа. В случае же палинсимметричных электронов
псевдопотенциал всегда больше нуля.
Причина высоких значений орбитальных потенциалов ионизации
кайносимметричных электронов становится понятной и при теорети-
ческом вычислении одноэлектронных энергий атомных орбиталей ме-
тодом самосогласованного поля Хартри — Фока. При решении вариа-
ционной задачи на собственные значения энергии валентных кайно-
симметричных электронов вклад в межэлектронное отталкивание за
счет взаимодействия этих электронов с электронами с той же орби-
тальной симметрией, но с меньшими главными квантовыми числами
равен нулю (поскольку таких электронов просто нет). Это обеспечива-
ет низкие значения одноэлектронной энергии валентных кайносиммет-
ричных атомных орбиталей, т. е. высокую прочность связи кайносим-
метричных электронов валентного слоя с ядром (или остовом атома), в
сравнении с любыми другими электронами (валентными электронами
некайносимметричных атомов), энергетически дестабилизированными
отталкивательным взаимодействием с предшествующими им внутрен-
ними электронами с той же орбитальной симметрией. Феноменологи-
чески это означает, что весьма низкие 1s-, 2р-, 3d- и 4/-орбитальные
энергии в соответствующих атомах и сильная сжатость этих валент-
ных атомных орбиталей имеют своей главной причиной отсутствие в
атомных остовах электронов с теми же значениями орбитальных кван-
товых чисел Z = 0, 1, 2 или I = 3, т. е. отсутствие в атомных остовах
р-электронов с п < 2, d-электронов с п < 3, /-электронов с п < 4; что
же касается ls-электронов, то им вообще ничто не предшествует.
В случае 2р-элементов специфика электроноядерных и межэлек-
тронных взаимодействий в их атомах, содержащих в качестве нева-
лентного остова всего оболочку Не (1s2), обеспечивает особо низкие
орбитальные энергии не только для 2р-, но и для 25-состояний.
Итак, в общих чертах ясны и причинно обоснованы особые и об-
щие в своей особенности пространственные и энергетические харак-
теристики валентных кайносимметричных электронов, и важно лишь
проследить, как все эти особенности проявляются в химии элементов
кайносимметриков. Главное заключается в том, что аномально низкие
104
энергии валентных Is-, 2р-, 3d-, 4/-орбиталей (аномально высокие ор-
битальные потенциалы ионизации) вкупе с малой пространственной
протяженностью этих валентных атомных орбиталей являются реша-
ющей причиной пониженной валентности соответствующих атомов.
Проявления кайносимметрии многообразны и, отнюдь, не исчерпы-
ваются специфичностью химии элементов-родоначальников соответ-
ствующих семейств, специфичностью, имеющей в основе ограничен-
ные валентные возможности атомов этих элементов. Проблема кайно-
симметрии включает в себя многие аспекты принципа периодичности
и может служить основой для обобщений и более глубокого понима-
ния многочисленных и давно известных экспериментальных фактов,
воспринимающихся до сих пор как результат эксперимента без ясной
связи с порождающими его причинами.
Повторим, что под кайносимметриками понимают, естественно, хи-
мические элементы, атомы которых содержат валентные электроны в
состояниях 1s, 2р, 3d или 4/. К этим элементам относятся водород
и гелий со своими валентными 1 s-электронами, типические элемен-
ты от В до Ne с валентными 2р-электронами, переходные металлы
первой вставной декады (Sc-Zn) с Зd-элeктpoнaми в валентном слое
и редкоземельные элементы, в атомах которых происходит заселение
электронами состояния 4/. К кайносимметрикам непременно следует
относить и оставшихся два типических элемента — литий и бериллий.
Хотя их атомы в свободном состоянии и не содержат 2р-электронов
(основное состояние несвязанных атомов Li и Be описывается валент-
ными электронными конфигурациями 2s1 и 2s2), образование многих
соединений лития и всех без исключения соединений бериллия проис-
ходит с участием валентных 2р-орбиталей их атомов в возникающих
химических связях.
В отношении бериллия это утверждение тривиально, так как обра-
зование, по меньшей мере, двух связей в молекулярной структуре лю-
бого бериллиевого соединения невозможно без валентного возбужде-
ния одного из двух спаренных электронов на вакантную 2р-орбиталь.
Кроме того, атомы Li и Be вполне могут использовать свои вакантные
орбитали для образования донорно-акцепторных связей, что и проис-
ходит во многих известных их соединениях. Последнее находит под-
тверждение в том, что в структурах таких соединений координаци-
онное число атома Li или Be больше 1 или 2 соответственно, а это
возможно лишь при участии в образовании химических связей с ли-
гандами вакантных 2р-орбиталей этих атомов. Таким образом, кайно-
симметриками оказываются все типические элементы от Li до Ne.
По причинам, во многом схожим с только что указанными, к кай-
носимметрикам надлежит причислять не только те редкоземельные
элементы, атомы которых в основном состоянии включают в себя хо-
105
тя бы один 4/-электрон, но и те металлы этого ряда, атомы которых
характеризуются отсутствием электронов на 4/-орбиталях, т. е. все без
исключения лантаноиды. Правда, лантаноиды — самый сложный слу-
чай.
Дело в том, что до сих пор окончательно не решен вопрос об уча-
стии 4/-электронов атомов этих элементов в образовании связей с ли-
гандами, и веские аргументы говорят о сохранении 4/-электронов в
псевдоатомном состоянии и их неспособности (по орбитальным энерги-
ям и пространственным характеристикам) эффективно участвовать в
связывании с разнообразными лигандами. В таком случае особенности
химии редкоземельных элементов нельзя прямо связывать с особыми
характеристиками кайносимметричных 4/-электронов, но можно объ-
яснять опосредованным влиянием 4/-электронов, появление которых
в атомах лантаноидов резко изменяет состояние их валентных 6s- и
5й-электронов в результате межэлектронных взаимодействий.
Важный аспект концепции кайносимметрии состоит в том, что в ее
рамках (на фоне традиционных для Периодического закона представ-
лений о вертикальном сходстве свойств химических элементов в груп-
пах) находит надежную и аргументированную интерпретацию уже и
горизонтальное сходство —в рядах и периодах системы. Напомним,
что первым, кто заметил и постоянно подчеркивал резкие отличия,
характерные для физико-химических свойств элементов определенных
строк периодической системы, в сравнении с остальными элементами
в пределах одной и той же главной подгруппы системы, был сам ав-
тор Периодического закона. Д. И. Менделеев даже использовал специ-
альные термины, назвав нынешние 1s-, 2р-элементы типическими
элементами, а нынешние 3d-, 4/-элементы — элементами чет-
ных периодов. Д. И. Менделеев приписывал всем им особые свойства.
В отличие от большинства своих последователей Менделеев придавал
строкам (рядам, периодам) системы не меньшее значение, чем столб-
цам (группам, подгруппам). Интуиция и на этот раз не обманула ве-
ликого ученого, и мы сейчас возвращаемся к этим его идеям, вводя
понятие кайносимметрии и называя кайносимметриками элементы, со-
держащие в валентном слое своих атомов электроны 1s, 2р, 3d, 4/,
впервые (в порядке возрастания порядкового номера) обладающие но-
вой орбитальной симметрией, новым значением орбитального кванто-
вого числа I.
Итак, атомы элементов-кайносимметриков, содержащие в своих ва-
лентных слоях кайносимметричные 1s-, 2р-, 3d-, 4/-электроны, име-
ют ярко выраженные специфические особенности. Классический при-
мер — биогенность атомов Н, С, N, О, содержащих в своей валентной
оболочке кайносимметричные 1s-, 2р-электроны (многообразные мо-
лекулы, составленные легкими кайносимметричными атомами, весьма
106
прочны и проявляют высокую тенденцию к сохранению своих струк-
тур, что весьма важно в аспекте памяти, наследственности и т.д.).
В атомах же элементов-палинсимметриков ни один из их валентных
электронов не обладает новой симметрией, отсюда их большее сход-
ство друг с другом и отличие от кайносимметриков — родоначальников
главных или дополнительных подгрупп.
Глава 7
ВТОРИЧНАЯ ПЕРИОДИЧНОСТЬ
В единой менделеевской группе периодической системы ни о какой
монотонности в изменении свойств химических элементов не может
быть и речи, поскольку при переходе в группе (начиная с 3-го пери-
ода) от одного элемента к другому происходит поочередное обраще-
ние то к элементу главной подгруппы (непереходному элементу), то
к элементу дополнительной подгруппы (переходному металлу). Это
утверждение тривиально в свете деления всех химических элементов
на два «лагеря». Что же касается характера главных закономерностей
изменения свойств химических элементов в пределах конкретной под-
группы, то эта проблема уже скоро столетие является одной из самых
значительных проблем общей химии.
В 1915 г. российский физико-химик Е. В. Бирон опубликовал в
«Журнале Российского физико-химического общества» большую и об-
стоятельную статью «Явления вторичной периодичности» (эта статья
полностью воспроизведена в книге [45]) со следующим заключением:
«В подгруппах периодической системы элементов многие свойства эле-
ментов и их соединений изменяются при последовательном увеличе-
нии атомного веса элемента не последовательно тоже, а периодиче-
ски. Эту своеобразную периодичность, как бы накладывающуюся на
основную периодичность элементов Д. И. Менделеева, я предлагаю на-
зывать вторичной периодичностью».
Вторичная периодичность была открыта эмпирически, но Е. В. Би-
рон, безусловно, связывал явление вторичной периодичности с осо-
бенностями электронного строения атомов и пытался подтвердить это
на примере вторичпопериодического изменения некоторых характери-
стик атомных спектральных линий.
В упомянутой книге представлена история развития представле-
ний о вторичной и других видах дополнительной периодичности, дан
анализ современных моделей их теоретического объяснения.
Новые достижения экспериментальной и теоретической химии не
оставляют сомнений в существовании эффекта вторичной периодич-
108
пости, и накопленный обширный эмпирический материал позволил пе-
рейти к теоретическим обобщениям. Первая концептуальная теорети-
ческая интерпретация явления вторичной периодичности разработана
профессором С. А. Щукаревым, и его вклад в учение о вторичной пери-
одичности общепризнан [6]. Эта теория отражает особенности химии
элементов четных периодов системы и объясняет, почему вторичная
периодичность более присуща элементам главных (а не дополнитель-
ных) подгрупп системы, притом на их высших ступенях окисления.
Вторичная периодичность, несомненно, должна быть интерпрети-
рована как важнейшая закономерная периодическая немонотонность
изменения свойств химических элементов (от атомных до молекуляр-
ных), составляющих подгруппы системы.
Фактор немонотонности проявляется уже в самой структуре пери-
одической системы химических элементов, если иметь в виду последо-
вательное изменение их числа в ее периодах. Числа элементов в после-
довательных периодах увеличиваются немонотонно, а начиная со 2-го
периода, попарно повторяются: 2, 8, 8, 18, 18, 32, 32, ... Эта немонотон-
ность является важнейшей, определяющей особенностью структуры
периодической системы.
Периодическое повторение числа элементов в последовательных
периодах системы основано в конечном счете на реальной схеме фор-
мирования электронных конфигураций свободных атомов по мере ро-
ста их ядерных зарядов и соответствует изменению числа электронов
в последовательных слоях электронной оболочки атома. Эта немоно-
тонность — своеобразный предшественник вторичной периодичности.
Профессор Д. Н. Трифонов [45] указывает на следующую, хотя и оче-
видную, но важную закономерность, существенную для интерпрета-
ции эффекта вторичной периодичности. Суть этой закономерности
становится вполне понятной при рассмотрении характера распределе-
ния электронов по слоям в оболочках атомов щелочноземельных ме-
таллов (табл. 5).
Таблица 5. Распределение электронов по слоям
в оболочках атомов щелочноземельных металлов
Заряд ядра, Z Элемент, период К L М N О Р Q
4 Ве (2) 2 2
12 Mg (3) 2 8 2
20 Са (4) 2 8 8 2
38 Sr (5) 2 8 18 8 2
56 Ва (6) 2 8 18 18 8 2
88 Ra(7) 2 8 18 32 18 8 2
109
Числа слоев в электронных оболочках атомов равны номерам пе-
риодов системы, в которых расположены соответствующие элементы.
Видно, что в атомах четных периодов (2-го, 4-го, 6-го) число запол-
ненных электронами слоев и число неполностью заполненных слоев
одинаковы, причем количество и распределение электронов в непол-
ностью заполненных слоях являются зеркальным отражением количе-
ства и распределения электронов в заполненных электронами слоях,
т. е. атомы четных периодов характеризуются своеобразной «зеркаль-
ной симметрией» своих электронных конфигураций. В атомах нечет-
ных периодов (3-го, 5-го, 7-го) подобная «зеркальная симметрия» ока-
зывается неполной, так как число слоев в таких атомах нечетное.
Таким образом, в рассматриваемых атомах наблюдается весьма
четкая немонотонность в построении электронных конфигураций, за-
ключающаяся в том, что по мере возрастания Z периодически реализу-
ются электронные конфигурации, характеризующиеся полной (четные
периоды) и неполной (нечетные периоды) «зеркальной симметрией».
Это периодическое чередование важно не только для общей интер-
претации структуры периодической системы, но и для обсуждения за-
кономерностей изменения свойств элементов в ее четных и нечетных
периодах.
Прежде всего очевидна следующая закономерность: в четных пе-
риодах после реализации электронных конфигураций с полной «зер-
кальной симметрией» при дальнейшем увеличении Z в атомах после-
дующих элементов появляются валентные электроны с новыми зна-
чениями орбитального квантового числа I (кайносимметричные 2р-
электроны во 2-м периоде, Зс/-электроны в 4-м, 4/-электроны в 6-
м). В нечетных периодах после возникновения электронных конфи-
гураций с неполной «зеркальной симметрией» при дальнейшем уве-
личении Z в атомах последующих элементов появляются валентные
электроны с повторными значениями орбитального квантового числа
I (палинсимметричные Зр-электроны в 3-м периоде, 4й-электроны в
5-м, 5/-электроны в 7-м). Такое периодическое чередование появляю-
щихся в атомах кайносимметричных и палинсимметричных валентных
электронов лежит в основе характерных закономерностей изменения
свойств химических элементов в главных подгруппах периодической
системы, проявляющихся в эффекте вторичной периодичности.
Теорию вторичной периодичности составляют убедительные и ар-
гументированные качественные модели, объясняющие это явление [6,
27, 28, 40, 41]. На подробном описании этих моделей мы сейчас и оста-
новимся.
110
7.1. Вторичная периодичность
в химии непереходных элементов
Вторичная периодичность является важнейшей закономерностью
изменения свойств непереходных элементов в пределах каждой глав-
ной подгруппы: она проявляется в свойствах изовалентных химиче-
ских соединений непереходных элементов одной и той же главной под-
группы (непереходных элементов-аналогов) и отражает особый, немо-
нотонный, характер изменения физико-химических свойств непереход-
ных элементов и их соединений в зависимости от положения элементов
в главной подгруппе, т. е. от их принадлежности к тому или иному пе-
риоду системы.
Вторичная периодичность обусловлена индивидуальными особен-
ностями центральных атомов. В каждой главной подгруппе валентная
электронная структура атомов непереходных элементов А существен-
но изменяется в зависимости от номера периода:
2: 2s22pn,
3: 3s23pn,
4: 3d104s24pn,
5: 4d105s25pn,
6: 4/145d106s26pn (здесь везде n = 1 4-6).
Поэтому в каждой главной подгруппе закономерности изменения
свойств принадлежащих к ней непереходных элементов отчетливо
немонотонны, и эта немонотонность проявляется как истинная вто-
ричная периодичность.
Основополагающим фактором, определяющим немонотонный, вто-
ричнопериодический, характер изменения свойств элементов в глав-
ных подгруппах (и одновременно вторичнопериодическую индиви-
дуальность непереходного элемента), являются эффекты взаимодей-
ствий валентных электронов со сложным остовом многоэлектронно-
го атома. Вторичная периодичность представляет собой проявление
особенностей химии непереходных элементов четных (2-го, 4-го, 6-го)
периодов системы, а эти особенности, в свою очередь, являются от-
ражением особых свойств валентной электронной структуры атомов
непереходных элементов именно четных периодов. Поясним это на ос-
нове вышеприведенных электронных конфигураций свободных невоз-
бужденных атомов непереходных элементов.
Особыми свойствами валентной электронной структуры обладают
атомы непереходных элементов:
1) 2-го периода: причина состоит в том, что главные валентные
электроны этих атомов — 2р-электроны — не имеют аналогичных (по
значению орбитального квантового числа /) предшественников в нева-
111
лентных оболочках атомов, характеризуются нулевым псевдопотенци-
алом и поэтому обладают резкой сжатостью в пространственном рас-
пределении своих зарядовых облаков и аномально высокими значени-
ями орбитальных потенциалов ионизации;
2) 4-го периода: причина состоит в появлении в атомах этих эле-
ментов внутреннего ЗсР°-электронного слоя, отсутствующего в атомах
непереходных элементов предшествующих периодов;
3) 6-го периода: причина состоит в появлении в атомах этих эле-
ментов внутреннего 4/14-электронного слоя, отсутствующего в атомах
непереходных элементов предшествующих периодов.
Вторичная периодичность проявляется уже на вертикальных ор-
битальных потенциалах ионизации валентных ns- и пр-электронов и
на формируемых ими первых адиабатических ионизационных потен-
циалах свободных невозбужденных атомов элементов той или иной
главной подгруппы. Конкретный вид этих зависимостей для любой
главной подгруппы всегда указывает на повышенные значения этих
потенциалов для атомов элементов 2-го (особенно), 4-го и 6-го пери-
одов системы по сравнению со значениями, ожидаемыми в предполо-
жении плавного снижения обсуждаемых потенциалов при движении в
подгруппе сверху вниз, т. е. при последовательном увеличении главных
квантовых чисел валентных ns- и пр-электронов и при соответствую-
щем повышении их орбитальных энергий.
Итак, вторичная периодичность, будучи в целом отражением осо-
бенностей свойств непереходных элементов четных (2-го, 4-го, 6-го)
периодов системы, особенностей, вытекающих из особых электронных
характеристик их атомов, исходит, главным образом, из соотноше-
ний энергетических характеристик валентных ns- и пр-электронов. В
этом состоит основа интерпретации вторичной периодичности в тео-
рии электронного строения молекул.
Особые свойства непереходных элементов 2-го периода, понижен-
ные валентности их атомов, обусловленные повышенными значениями
орбитальных потенциалов ионизации валентных 2s- и 2р-электронов
для свободных атомов этих элементов, уже были рассмотрены ранее
(явление кайносимметрии).
Особые свойства непереходных элементов 4-го периода, понижен-
ные валентности их атомов также обусловлены повышенными значе-
ниями орбитальных потенциалов ионизации валентных 4s- и 4р-элек-
тронов для свободных атомов этих элементов. Причина повышенных
значений орбитальных потенциалов ионизации валентных 4s- и 4р-
электронов в атомах непереходных элементов 4-го периода по сравне-
нию с соответствующими величинами валентных 3s- и Зр-электронов
в атомах непереходных элементов 3-го периода, точнее, по сравнению
с ожидаемыми значениями, заключается, естественно, в принципиаль-
112
ном отличии электронной структуры атомов непереходных элементов
4-го периода от электронной структуры атомов элементов предше-
ствующего, 3-го, периода. Это отличие состоит в появлении в атомах
элементов 4-го периода (сверх замкнутой электронной оболочки ато-
ма соответствующего инертного газа) внутреннего Зс/10-электронного
слоя, который двояким образом формирует энергетические и про-
странственные характеристики валентных 4s- и 4р-электронов.
Присутствие этого слоя увеличивает межэлектронное отталкива-
ние и тем самым повышает орбитальные энергии валентных 4s- и
4р-электронов. Но этот слой имеет низкую орбитальную энергию и
глубоко расположен в электронных оболочках атомов непереходных
элементов 4-го периода, и поэтому его дестабилизирующее воздей-
ствие на валентные 4s- и 4р-электроны этих атомов относительно неве-
лико.
Другим и противоположно действующим фактором, более чем ком-
пенсирующим дестабилизирующее воздействие межэлектронного от-
талкивания с участием Зс/10-электронов, является относительное воз-
растание (на 10 единиц) положительного заряда ядер атомов непере-
ходных элементов 4-го периода. Этот второй фактор является решаю-
щим в понижении орбитальных энергий валентных 4s- и 4р-электронов
в атомах непереходных элементов 4-го периода. Решающая роль этого
второго фактора объясняется тем, что 4s-(особенно) и 4р-электроны
являются глубоко проникающими, и существенная доля их зарядо-
вой плотности расположена в атоме в близких к ядру областях про-
странства, перед Зс/10-слоем, что и позволяет 4s- и 4р-электронам особо
сильно воспринимать стабилизирующее действие относительного воз-
растания (на 10 единиц) заряда атомного ядра при появлении в атоме
Зй10-электронного слоя. Оценки показывают, что из 10 дополнитель-
ных единиц положительного заряда ядра Зс/10-электроны экранируют
всего 2,5 единицы для 45-электронов и пять единиц для 4р-электронов,
т. е. Зс/10-электронный слой весьма «прозрачен» для нарастающего (на
10 единиц) положительного заряда ядра, что создает повышенный за-
ряд остова для валентных электронов атомов непереходных элементов
4-го периода.
Таким образом, воздействие слоя Зй10-электронов на энергетиче-
ское состояние валентных в 4s- и 4р-электронов в атомах непереход-
ных элементов 4-го периода приводит в целом к повышенной энерге-
тической стабильности электронов на валентных 4s- и 4р-орбиталях
в этих атомах и к повышенным значениям их ионизационных потен-
циалов. Стабилизирующий эффект Зй10-слоя в значительно большей
мере сказывается на энергетических характеристиках валентных 4s-, а
не 4р-электронов. Объясняется это тем, что проникающая способность
45-электронов с тремя внутренними максимумами зарядовой плотно-
113
сти, первый из которых находится в непосредственной близости от
ядра, существенно выше, чем у 4р-электронов с двумя такими макси-
мумами при отсутствии зарядовой плотности в примыкающей к ядру
области пространства. Именно поэтому особо высокими значениями
орбитальных потенциалов ионизации характеризуются валентные 4s-,
а не 4р-электроны, так же как и в парном сочетании валентных 2s- и
2р-электронов.
Соединения непереходных элементов 6-го периода, замыкающих
главные подгруппы и называемых иногда тяжелыми непереходными
элементами, значительно отличаются по свойствам от соответствую-
щих соединений непереходных элементов предшествующих периодов.
Сильнее всего эти отличия выражены на высших ступенях окисления,
и самая важная из особенностей химического поведения Tl, Pb, Bi, Ро,
At, Rn состоит в пониженной устойчивости их высших окисленных
форм в сравнении с соответствующими соединениями непереходных
элементов 3-го, 4-го и 5-го периодов. Часто высшая окисленная форма
тяжелого непереходного элемента вообще отсутствует.
В основе особенностей непереходных элементов 6-го периода ле-
жит, в сущности, то же самое явление, которое объясняет особенности
химического поведения непереходных элементов 4-го периода. Особые
свойства тяжелых непереходных элементов, пониженные валентности
их атомов определяются повышенными значениями орбитальных по-
тенциалов ионизации валентных 6р- и 65-электронов для свободных
атомов этих элементов. Причину повышенных орбитальных иониза-
ционных потенциалов для атомов Tl, Pb, Bi, Po, At, Rn и связанных
с этим особенностей химии тяжелых непереходных элементов следует,
разумеется, видеть в принципиальном отличии электронной структу-
ры этих атомов от атомов-предшественников в соответствующих под-
группах (In, Sn, Sb, Те, I, Хе). В атомах Tl, Pb, Bi, Po, At, Rn появ-
ляется внутренний 14-электронный слой 4/14 (сверх замкнутой элек-
тронной оболочки атома соответствующего инертного газа), которого
нет в атомах In, Sn, Sb, Те, I, Хе.
Как и в случае Зй10-слоя, внутренний слой 4/14-электронов двоя-
ким образом формирует энергетические и пространственные характе-
ристики валентных 6s- и бр-электронов атомов непереходных элемен-
тов 6-го периода. Присутствие этого слоя увеличивает межэлектронное
отталкивание и тем самым повышает орбитальные энергии 6s- и 6р-
электронов. Но поскольку 4/14-слой имеет очень низкую орбитальную
энергию и глубоко расположен в электронных оболочках атомов Т1,
Pb, Bi, Po, At, Rn, его дестабилизирующее воздействие на валентные
6s- и бр-электроны оказывается сравнительно небольшим..,
Другим и противоположно действующим фактором, более чем ком-
пенсирующим дестабилизирующее воздействие межэлектронного от-
114
талкивания с участием 4/14-электронов, является относительное воз-
растание положительного заряда ядер атомов на 14 единиц, без чего
немыслимо появление 4/-слоя в атомах Tl, Pb, Bi, Ро, At, Rn. По-
прежнему, как и случае Зб/10-слоя, этот второй фактор оказывается
решающим в понижении орбитальных энергий валентных 6s- и 6р-
электронов в атомах тяжелых непереходных элементов. Решающая
роль этого второго фактора объясняется тем, что 6s-(особенно) и 6р-
электроны являются глубоко проникающими, и существенная доля их
зарядовой плотности расположена в атоме в близких к ядру областях
пространства, перед 4/14-слоем, что и позволяет 6s- и бр-электронам
особенно сильно воспринимать стабилизирующее действие относитель-
ного возрастания (на 14 единиц) заряда атомного ядра при появлении
в атоме 4/14-электронного слоя.
Простые вычисления показывают, что из 14 дополнительных еди-
ниц положительного заряда ядра 4/14-электроны экранируют только
3,5 единицы для 65-электронов и семь единиц для бр-электронов. След-
ствием такой высокой «прозрачности» 4/14-электронного слоя для уве-
личивающегося (на 14 единиц) положительного заряда ядра является
повышенный заряд остова для валентных электронов атомов непере-
ходных элементов 6-го периода.
Таким образом, как и в случае Зй10-слоя, воздействие слоя
4/14-электронов на энергетическое состояние валентных 6s- и 6р-
электронов в атомах непереходных элементов 6-го периода приводит в
целом к повышенной энергетической стабильности валентных электро-
нов на 6s- и бр-орбиталях в этих атомах и к повышенным значениям
их ионизационных потенциалов.
По-прежнему в случае тяжелых непереходных элементов Tl, РЬ,
Bi, Ро, At, Rn стабилизирующий эффект 4/-слоя (точнее, стабилизи-
рующее воздействие возрастающего на 14 единиц ядерного заряда) в
значительно большей мере сказывается на энергетических характери-
стиках валентных 6s- (а не 6р-) электронов в их атомах. Объясняется
это тем, что проникающая способность 65-электронов (пять внутрен-
них максимумов зарядовой плотности, первый из которых в непосред-
ственной близости от ядра) существенно выше, чем у бр-электронов
(четыре таких максимума и отсутствие зарядовой плотности в примы-
кающей к ядру области пространства).
Именно поэтому особенности химии непереходных элементов 6-го
периода, отличия их химических свойств от свойств всех их предше-
ственников в главных подгруппах, вообще говоря, имеющие место при
всех возможных формальных валентностях, наиболее ярко проявля-
ются на высших, равных номеру группы, ступенях окисления и выра-
жаются в резко пониженной устойчивости высших окисленных форм
тяжелых непереходных элементов по сравнению со всеми предшеству-
115
ющими (кроме кайносимметриков 2-го периода) элементами соответ-
ствующих главных подгрупп.
Феноменологическая причина сниженной устойчивости высших
окисленных форм тяжелых непереходных элементов, как и в случае
элементов 4-го периода, состоит в том, что их образование предпола-
гает участие валентных 6$2-электронов атомов Tl, Pb, Bi, Ро, At, Rn,
а именно, эти электроны наиболее стабилизированы (их орбитальные
энергии понижены) под воздействием факторов, отвечающих наличию
в атомах данных элементов внутренних 4/14-электронных слоев. Такое
упрочение связи валентных 6s2-электронов с остовом в атомах Tl, РЬ,
Bi, Ро, At, Rn выражено настолько сильно,* что носит название эф-
фекта инертной пары. Именно эффектом инертной пары принято
объяснять частое снижение максимально достижимой степени окисле-
ния тяжелых непереходных элементов на две единицы по сравнению с
высшей формальной валентностью, равной номеру группы (Т11, РЬ11,
Bi111, PoIV, Atv, RnVI вместо Tl111, PbIV, Biv, PoVI, AtVI1, Rnvln).
Подчеркнем, что пониженная валентность атомов, пониженная
устойчивость высших соединений и другие родственные явления в хи-
мии непереходных элементов четных периодов (2-го — кайносиммет-
рия, 4-го и 6-го —эффект инертной пары) наиболее ярко выражены
у поздних элементов —V, VI и особенно VII, VIII групп. И причина
нарастания этих эффектов заключается в том, что преимущественная
энергетическая стабилизация валентных 2s2-, 4s2-, б52-электронных
пар в атомах тем больше, чем больше заряд ядра атома.
Вторичная периодичность, будучи в химии этих элементов явле-
нием всеобщим, затрагивающим в той или иной степени все возмож-
ные ступени окисления, проявляется, вообще говоря, в свойствах всех
соединений элементов той или иной главной подгруппы. Но наибо-
лее ярко она выражена в свойствах высших соединений непереходных
элементов, включающих в себя центральный атом на максимальной,
равной номеру группы в периодической системе, ступени окисления.
Это обусловлено тем, что вышеописанные изменения в электронной
* Упрочнение связи 6з2-электронов с остовом в атомах Tl, Pb, Bi, Ро, At, Rn
выражено намного сильнее, чем аналогичное упрочнение связи 4з2-электронов в
атомах Ga, Ge, As, Se, Вг, Кг. И причин тому, очевидно, две: во-первых, эндовклад
в энергетические характеристики 6з2-электронов за счет межэлектронного оттал-
кивания с участием 4/14-электронного слоя меньше аналогичного эндовклада в
случае межэлектронных взаимодействий 4з2-электронов с ЗсР°-электронным слоем
(в первом случае главные квантовые числа отличаются на 2, что свидетельствует о
большем пространственном разделении зарядовых плотностей 6з- и 4/-состояний,
во втором случае — только на 1); во-вторых, стабилизация 6з2-электронов за счет
относительного возрастания ядерного заряда на 14 единиц, естественно, выше ста-
билизации 4з2-электронов за счет относительного увеличения заряда ядра на 10
единиц (то же, в принципе, относится и к большему упрочнению связи с остовом
6р-, нежели 4р-электронов).
116
структуре атомов, происходящие при увеличении порядкового номера
элемента в главной подгруппе, наиболее сильно сказываются на состо-
янии валентных ns-электронов, являющихся наиболее проникающими.
Поэтому из всех валентных электронов атома непереходного элемен-
та немонотонное, вторичнопериодическое, изменение характеристик (в
частности, орбитальных энергий) с увеличением главного квантового
числа наиболее сильно проявляется именно в случае валентных ns-
электронов, на фоне весьма умеренного, но также вторичнопериоди-
ческого изменения этих величин для валентных пр-электронов. Уча-
стие же этих электронов в связях с лигандами единственно приводит,
как полагают, к образованию высших окисленных форм. Иначе гово-
ря, поскольку высшие окисленные формы образуются при участии ва-
лентных ns-электронов центрального атома, характеристики которых
в наибольшей мере подвержены вторичнопериодическому изменению
в пределах главной подгруппы, именно в свойствах высших соедине-
ний непереходных элементов вторичная периодичность проявляется в
наиболее яркой форме.
В случае высших соединений вторичнопериодический характер из-
менения свойств проявляется, как правило, в виде экстремальных за-
висимостей. Для других соединений непереходных элементов немоно-
тонность в изменении свойств в подгруппе проявляется в общем в зна-
чительно более затушеванной форме и иногда обнаруживается лишь
по изменению величины (но не знака) градиента, т. е. по перегибам
линии, описывающей изменение того или иного свойства в главной
подгруппе.
Валентные 2s-, 4s-, es-электроны в атомах непереходных элемен-
тов четных периодов по указанным причинам относительно более ста-
билизированы, чем 3s- и Ss-электроны в атомах элементов нечетных
периодов. Следовательно, феноменологически можно утверждать, что
2s-, 4s-, 65-электроны испытывают наибольшие энергетические затруд-
нения в образовании прочных связей центральных атомов с лиганда-
ми. Именно поэтому главным проявлением вторичной периодичности
в свойствах непереходных элементов той или иной подгруппы оказы-
вается относительно малая устойчивость высших соединений, образу-
емых элементами 2-го, 4-го и 6-го периодов.
Теоретическая интерпретация вторичной периодичности в целом
как отражения особенностей непереходных элементов четных перио-
дов системы и составляющих ее моментов в отдельности (кайносим-
метрия, особенности химии непереходных элементов 4-го периода, эф-
фект инертной пары) разработана с использованием концепции гипер-
валентных связей [27, 28, 40, 41]. Повышенные орбитальные энергии
валентных 2s-, 2р-, 4s-, 4р-, 6s-, бр-электронов в атомах непереход-
ных элементов четных периодов системы обеспечивают сравнительно
117
небольшие энергии гипервалентных связей центрального атома с ли-
гандами, т. е. трехцентровых четырехэлектронных связей Lh)-A-L(2),
образуемых за счет валентных электронных пар ns2 или пр2 атома А.
В свою очередь это приводит к сравнительно небольшим усредненным
энергиям всех (ковалентных и гипервалентных) связей в молекулах и,
следовательно, к пониженной устойчивости (или просто к отсутствию)
соответствующих соединений.
Из-за преимущественной стабилизации валентной ns-орбитали цен-
трального атома особенно слабыми оказываются гипервалентные свя-
зи L(i)-A-L(2) на основе п$2-электронной пары атома А. Именно поэто-
му наименее прочными, имеющими наименьшие усредненные энергии
связей между центральным атомом и лигандами, оказываются выс-
шие соединения обсуждаемых элементов, где предполагается участие
валентной п$2-электронной пары центрального атома в связях с ли-
гандами.
Количественной мерой стабилизации ns-орбитали атома А может
служить разность епр—епз энергий его валентных пр- и ns-орбиталей
(так называемая энергия пр — ns-разделения). Чем больше стабилизи-
рована ns-орбиталь атома А, тем, с одной стороны, больше эта энер-
гетическая разность, а с другой —тем меньше усредненная энергия
связей A-L в молекулах высших соединений, где существуют гипер-
валентные связи L(i)-A(ns2)-L(2). Поэтому ясно, что характеристики
таких молекул, и, в первую очередь, суммарная энергия связей в них,
зависят от величины энергии пр — ns-разделения валентных орбиталей
центрального атома. И можно предвидеть, что с увеличением (умень-
шением) энергии пр—ns-разделения в атомах непереходных элементов
будет уменьшаться (увеличиваться) усредненная энергия связей A-L
в образуемых этими атомами однотипных молекулах. Иными слова-
ми, будет все более затрудняться участие валентных п52-электронов
центрального атома в гипервалентном связывании с лигандами в тех
молекулах, где центральный атом характеризуется повышенной вели-
чиной разности епр — ens.
Поскольку в пределах главной подгруппы энергия np-ns-
разделения изменяется (по причинам, подробно рассмотренным ранее)
вторичнопериодически, усредненная энергия связей A-L в молекулах
(и вместе с ней прочность высших соединений и их реакционная спо-
собность) должна изменяться в главной подгруппе вторичнопериоди-
чески. Ниже приведены значения энергии Ев двухцентровых связей
для гексафторидов AFe кислорода и халькогенов вместе с характер-
ными для свободных атомов А величинами энергии пр—ns-разделения:
118
0F6 SF6
Ев, ккал/моль He существует 73
£np-£ns, эВ 14,9 9,8
SeFe TeFe PoFe
69 79 57
10,4 8,8 12
В тех случаях, когда электронная структура высших соединений
непереходных элементов не является гипервалентной и отвечает кон-
цепции 5ртп-гибридизации (атом А относится к III или IV менделеев-
ской группе), вторичная периодичность также обусловлена величина-
ми энергии пр—ns-разделения, поскольку уменьшение энергии пр—ns-
разделения способствует гибридизации В АО, а увеличение — препят-
ствует.
В новой теории вторичной периодичности для химии непереходных
элементов кайносимметрия как явление и как концепция фигурирует
и непосредственно (особенности валентных 2р-электронов), и опосре-
дованно, объясняя особенности валентных 4s, 4р- и 6s-, бр-электронов
появлением в атомах кайносимметричных субвалентных 3d10- и 4/14-
электронных слоев, слабо экранирующих кулоновское поле ядра в
сравнении с s2- и р6-слоями.
7.2. Квантовомеханические модели вторичной
периодичности в химии непереходных элементов.
Теоретике-групповая интерпретация
Вторичная периодичность проявляется, разумеется, не только в
свойствах химических соединений элементов главной подгруппы, но
и в определяющих эти свойства характеристиках свободных атомов и,
прежде всего, в потенциалах ионизации, в энергиях валентных атом-
ных орбиталей (ВАО), в орбитальных радиусах, в энергиях валентных
состояний атомов.
Сначала остановимся на квантовомеханической интерпретации
вторичной периодичности на основе обсуждения характера измене-
ния орбитальных энергий (еп/) и орбитальных потенциалов иониза-
ции (Ini) ns- и np-ВАО атомов непереходных элементов. Эта интер-
претация дана в уже упоминавшихся наших работах [34, 35, 40, 41] и
профессором И. С. Дмитриевым в заключительной главе монографии
[45]. Величины еп1 и Ini с достаточной точностью определены экспери-
ментально из атомных спектров и теоретически рассчитаны методом
самосогласованного поля Хартри —Фока (табл. 6-8).
Орбитальные энергии, орбитальные потенциалы ионизации и осо-
бенно энергии ns — пр-разделения атомов непереходных элементов в
119
пределах главной подгруппы изменяются вторичнопериодически (если
не всегда вторичнопериодически изменяется знак градиента той или
иной величины, то этой закономерности всегда следует темп ее изме-
нения). Наиболее отчетливо вторичная периодичность проявляется в
характеристиках ns-BAO.
Таблица 6. Орбитальные потенциалы ионизации ns-BAO
(Ins, эВ) атомов непереходных элементов
Период Группа
II III IV V VI VII
2 9,32 12,93 16,59 20,39 28,46 37,84
3 7,64 10,61 13,45 16,14 20,20 24,55
4 6,11 11,87 14,27 16,60 20,15 23,79
5 5,69 11,03 13,10 15,15 17,84 20,61
6 5,19 12,24 14,60 18,17 - -
Таблица 7. Орбитальные потенциалы ионизации
np-ВАО (/пр, эВ)
атомов непереходных элементов
Период Группа
III IV V VI VII
2 8,30 11,26 14,54 13,61 17,42
3 5,98 8,15 10,48 10,36 13,00
4 6,00 7,88 9,81 9,75 11,84
5 5,79 7,34 8,64 9,01 10,45
6 6,11 7,42 7,29 8,46 9,06
Таблица 8. Энергии пр — ns-разделения
(^np €ns, эВ)
для атомов непереходных элементов
Период Группа
III IV V VI VII
2 4,63 5,33 5,85 14,85 20,42
3 4,63 5,30 5,66 9,84 11,15
4 5,87 6,39 6,79 10,40 11,95
5 5,24 5,76 6,51 8,83 10,16
6 6,13 7,18 10,88 - -
В тех случаях, когда изменение энергии валентной атомной nl (ns-
или пр-)-орбитали оказывается сглаженным, картину проясняет раз-
ложение этой энергии на кинетическую и потенциальную со-
ставляющие. При этом оказывается, что в пределах подгруппы обе
составляющие изменяются вторичнопериодически, но трудно (если во-
обще возможно) найти какие-либо универсальные правила, регулиру-
ющие соотношения между е™" и е™т в суммарной eni.
120
Так или иначе весь большой массив обсуждаемой информации ука-
зывает на особые характеристики атомов непереходных элементов чет-
ных периодов как на основу эффекта вторичной периодичности. Эти
их особенности действительно определяются воздействием субвалент-
ных экранов 3d10 и 4/14 на энергию и пространственную протяжен-
ность ns- и пр-ВАО (в отсутствие экранов 3d10 и 4/14 энергии ns- и
пр-ВАО и энергии ns — пр-разделения атомов непереходных элемен-
тов изменяются в пределах главной подгруппы монотонно. Это под-
тверждается характеристиками атомов непереходных элементов I и II
групп, в электронной структуре которых указанных экранов нет, и,
следовательно, валентные ns- и пр-электроны не претерпевают непол-
ного экранирования от ядра электронами заполненных состояний 3d10
и 4/14).
Следует иметь в виду, что подробно рассмотренная в предыду-
щем разделе радиально-узловая структура валентных атомных ns- и
пр-орбиталей (внутренние локальные максимумы зарядовой плотно-
сти электронов на ns- и пр-ВАО) обусловлена или ортогональностью
атомных орбиталей с различными орбитальными квантовыми числа-
ми I («ортогональность по симметрии»), или ортогональностью атом-
ных орбиталей с одинаковыми Z, но с разными главными квантовыми
числами п («принудительная ортогональность» как дополнительное
условие). Именно в силу ортогональности второго рода АО имеют уз-
лы, а следовательно, и локальные максимумы электронной плотно-
сти [4].
Кайносимметричные АО не имеют узлов, а стало быть, и локаль-
ных максимумов. Не будь условий ортогональности, все некайносим-
метричные атомные орбитали превратились бы в безузловые АО (как
и кайносимметричные) с одним большим максимумом электронной
плотности и «провалились бы в остов». Учет условий ортогональности
для безузловых орбиталей потребовал бы соответствующей замены по-
тенциала эффективного поля, действующего на описываемые этой ор-
биталью электроны, псевдопотенциалом, отличающимся от исходного
некоторой положительной добавкой. Иными словами, условия ор-
тогональности реальных АО предопределяют увеличение их орбиталь-
ных энергий, что являет собой одну из компонент, обусловливающих
вторичнопериодическое изменение энергий ВАО в пределах главной
подгруппы.
Обсуждаемые в теории вторичной периодичности специфические
особенности валентных 6s- и бр-АО неправомерно сводить только к
одной причине, а именно, к действию экрана 4/14, поскольку важ-
ную роль играют и релятивистские эффекты. При большом ядерном
заряде уже нельзя пренебрегать одноэлектронным спин-орбитальным
взаимодействием, имеющим релятивистскую природу, и следует учи-
121
тывать расщепление состояния nl на два состояния с разными значе-
ниями спин-орбитального квантового числа j:
£n/j(i)> J*(l) = I 1/2,
£nl \
J (2) = I — 1/2.
При этом конфигурация атома характеризуется распределением элек-
тронов по n/J-состояниям.
Релятивистскими эффектами объясняется тот факт, что первые ор-
битальные р-потенциалы ионизации в ряду бр-элементов изменяются
нерегулярно: при переходе от атома Т1 (6ру2 6р^2 : 6,11 эВ) к атому
РЬ (бр2у2 6рз/2 : 7,42 эВ) возрастают, а при переходе от атома РЬ (бр2/2
6рз/2 : 7,42 эВ) к атому Bi (6р^2 6р^2 : 7,29 эВ) падают, тогда как в
аналогичных рядах 2р-, Зр-, 4р-, 5р-элементов происходит лишь после-
довательное увеличение первых орбитальных р-потенциалов иониза-
ции. Вышесказанное, кстати, заставляет при выявлении причин вто-
ричной периодичности учитывать не только «вертикальные» (в под-
группах), но и «горизонтальные» (в рядах) закономерности, например,
монотонное возрастание величин Ins во всех периодах и немонотонное
изменение величин 1пр во 2-м - 5-м периодах.
Еще раз обратимся к пониженной устойчивости или даже отсут-
ствию высших окисленных форм непереходных элементов б-го пери-
ода. Причина снижения максимально достижимой степени окисления
тяжелых непереходных элементов на две единицы по сравнению с выс-
шей формальной валентностью, равной номеру менделеевской группы
(эффект инертной пары Сиджвика), объясняется разным релятивист-
ским сжатием остовных и валентных ns-состояний, в результате кото-
рого орбитальная энергия 6s-BAO значительно понижается по сравне-
нию с орбитальной энергией 5s-BAO:
e5s(In) = —11,03,
e5s(Sn) = -13,10,
MSb) = -15,15,
e6s(Tl) = -12,24,
66s(Pb) =-14,60,
66s(Bi) = -18,17 эВ,
и можно говорить о наличии в атомах Tl, Pb, Bi, Po, At, Rn па-
ры (бз2!/2)-электронов, стабилизированной эффектом релятивистско-
го сжатия. В то же время для валентных 6pi/2- и особенно брз/2-
орбиталей релятивистское сжатие менее характерно, и при переходе
от 5р-орбиталей к бр-орбиталям энергии изменяются незначительно
(~ 0,1 — 0,3 эВ). Поэтому величина энергии ns — пр-разделения в
б-м периоде существенно возрастает по сравнению с предыдущими пе-
риодами, что, в свою очередь, препятствует гибридизации 6s- и бр-
122
В АО (происходит «релятивистская дегибридизация») и соответствен-
но ограничивает образование высших окисленных форм. Таким об-
разом, химическое различие между непереходными элементами 5-го и
б-го периодов, по-видимому, обусловлено значительно большим и даже
доминирующим вкладом релятивистских эффектов, которые, однако,
проявляются по-разному в различных главных подгруппах и для раз-
личных степеней окисления элементов [46].
Разработана и теоретико-групповая интерпретация явления вто-
ричной периодичности. Исходным положением является предложен-
ная академиком В. А. Фоком [47] дополнительная симметрия кулонов-
ского поля, отличающая это поле от других полей сферической сим-
метрии. Известно, что так называемая стереографическая проекция
позволяет взаимно однозначным образом сопоставить каждой точке
поверхности шара точку на плоскости, и наоборот. Аналогично каж-
дой точке трехмерного пространства соответствует точка на поверх-
ности четырехмерного шара. Уравнение Шредингера для электрона,
движущегося в кулоновском поле, может быть представлено с помо-
щью преобразования Фурье в переменных импульсного пространства
в форме интегрального уравнения для гиперсферических функций на
поверхности четырехмерного шара, стереографической поверхностью
которого служит трехмерное импульсное пространство. Таким обра-
зом, группа вращений 0(4) этого 4-шара в импульсном пространстве
представляет симметрию кулоновского поля.
Если на четырехмерный шар стереографически спроектировать не
импульсное, а трехмерное координатное пространство [48], то положе-
ние точки на 4-шаре с постоянным радиусом R может быть задано
единичным 4-вектором £ = {£i, £2, £з, £4}, компоненты которого вы-
ражаются через обычные трехмерные координаты г = {x,yfz} следу-
ющим образом:
Ci =2Ях/(Л2 + г2),
е2 = 2Лу/(Л2 + г2),
<з = 2Rz/{R2 + г2),
е4 = (Я2 -г2)/(Я2 + г2),
Stife2) = 1-
Далее, при обсуждении свойств симметрии атомных волновых
функций являющихся решениями уравнения Шредингера на
4-шаре с потенциалом Ленца [/(г) = —Z/r(l + r/Н)2, целесообразно
ввести новое квантовое число I/, удовлетворяющее следующим усло-
виям:
123
у = (ДГ + 1)Iе! для нечетных TV,
у = 7V/2 для четных N.
Для любого нечетного N и последующего четного N 4- 1 квантовое
число д/ принимает одно и то же значение:
N 1,2 3,4 5,6 7,8
у 1 2 3 4
Такое «удвоение» значений у можно описать дополнительным
квантовым числом В
В = 1 для нечетных N,
В = — 1 для четных N.
Тогда гильбертово пространство Н квантовых состояний разделя-
ется на два подпространства Н± и 7У_, задаваемых соответственно
функциями <р^1т = ^ЛГнечетн 1т И <p~lm = <PNчетн 1т, МвЖДу КОТОРЫМИ
существует взаимно однозначное соответствие. Существуют операто-
ры, связывающие эти подпространства:
= уЛ,
= О,
Т*р+= <р~,
Т\р~ = О,
/2 есть оператор отражения координаты £4 относительно
начала координат, т.е. £2, £4} = (1/2){£1, £2, £з, ~ ^41-
Функции ipuim образуют базис неприводимого представления ди-
намической группы (9(4, 2). При этом функция <р+/тп инвариантна (с
точностью до постоянного множителя) относительно операции а
функция изменяет знак. Поэтому собственное значение операто-
ра Т£ равно В/2\
± ___
73
4>±im
Поскольку N = п 4- I = пг + 2/ + 1 (пг — радиальное квантовое
число), состояния с четным пг, т. е. с четным числом радиальных узлов
в волновой функции, характеризуются нечетным N и В = 4-1, тогда
124
как состояния с нечетным пг отвечают четному N и В = — 1. Таким
образом, квантовое число В выражает четность волновой функции
относительно операции Т£.
Главное в теоретико-групповой интерпретации вторичной перио-
дичности состоит в том, что множество атомов элементов каждой глав-
ной подгруппы естественным образом разбивается на два подмноже-
ства, отвечающих четным и нечетным значениям числа N = п +1, и
в пределах каждого такого подмножества закономерности изменения
атомных свойств с увеличением заряда ядра различны. Тем самым
устанавливается строгая зависимость между характером изменения
некоторого свойства атома и четностью числа 7V, или, что то же са-
мое, четностью числа радиальных узлов его валентных АО. Валентные
ns- и пр-АО, характеризуемые одним и тем же значением квантового
числа В, повторяются в главных подгруппах через период, причем чет-
ности ns- и np-состояний с одинаковым главным квантовым числом п
противоположны:
ns-BAO Значение В np-BAO Значение В
2s - 1 2p + 1
3s + 1 3p - 1
4s - 1 4p + 1
5s + 1 5p - 1
6s - 1 6p + 1
В теоретико-групповом подходе эффект вторичной периодичности
проявляется, правда, в значительно ослабленном виде, и в тех главных
подгруппах периодической системы, где нет экранирующего действия
d- или /-электронов, т. е. в тех подгруппах, где этот эффект выражен
крайне слабо или вообще не отмечается.
7.3. Вторичная периодичность
в химии переходных металлов
В каждой дополнительной подгруппе валентная электронная
структура атомов переходных металлов М во всех периодах совершен-
но однотипна:
4: 4s23dn,
5: 5s24dn,
б: 6s25dn,
7: 7s26dn (здесь везде п = 1 4-10).
Поэтому априори ясно, что в каждой дополнительной подгруппе зако-
номерности изменения свойств находящихся в ней переходных метал-
125
лов не аналогичны и не должны быть аналогичными строгой вторич-
нопериодической закономерности в соответствующей главной подгруп-
пе. К тому же дополнительные подгруппы включают в себя лишь по
три переходных металла (а не по пять или иногда даже по шесть непе-
реходных элементов в главных подгруппах). В столь коротких сериях
вторичнопериодические закономерности гораздо менее очевидны. Не
изменяют общей ситуации четыре переходных металла (Sc, Y, La, Ac)
в третьей дополнительной подгруппе, а также практически не изучен-
ные сверхтяжелые элементы Rf (104), Db (105), Sg (106), Bh (107), Hs
(108), Mt (109), завершающие свои дополнительные подгруппы. Тем
не менее необходимо выявить принципиальный, главный вид законо-
мерности изменения свойств переходных металлов в дополнительной
подгруппе и по возможности выяснить его причину.
Типичным видом зависимости изменения той или иной характери-
стики в серии однотипных химических соединений переходных метал-
лов 3d — 4d — 5d любой дополнительной подгруппы является немоното-
ная зависимость с отчетливо (иногда весьма резко) выраженным из-
ломом в точке, соответствующей 4й-элементу. Часто в этой точке гра-
диент изменения рассматриваемой характеристики меняет знак, часто
его знак остается неизменным, но в таком случае значительно изме-
няется темп уменьшения или увеличения рассматриваемой характери-
стики. В большинстве случаев разница в величинах рассматриваемой
характеристики в парном сочетании 4d— 5d значительно меньше, чем в
парном сочетании 3d—4d, т. е. в большинстве случаев различия в свой-
ствах однотипных соединений 4d- и 5d-элeмeнтoв существенно меньше
различий в свойствах 3d- и 4d-элeмeнтoв («4d- и 5d-элeмeнты больше
похожи друг на друга, чем каждый из них в отдельности на соответ-
ствующий Зd-элeмeнт »).
Не может быть ни малейших сомнений в том, что за наблюдаемые
закономерности в изменении макроскопических свойств химических
соединений переходных металлов в пределах дополнительной подгруп-
пы ответственно строение электронных оболочек их атомов. В прин-
ципе ясно, что немонотонный («с изломом посредине дополнительной
подгруппы») характер зависимостей каких угодно свойств в серии од-
нотипных химических соединений переходных металлов любой допол-
нительной подгруппы, безусловно, определяется тем, что при переходе
от 3d- к 4d-элeмeнтy электронная структура их атомов совершенно од-
нотипна не только в отношении валентных орбиталей (4s23dn-5s24dn),
но и в отношении всех остовных уровней. В то же время при переходе
от 4d-элeмeнтa к 5d-элeмeнтy полностью аналогичны лишь валентные
орбитали атомов (5s24dn-6s25dn), тогда как совокупности остовных
уровней неодинаковы, а именно, в атомах 5d-элeмeнтa появляется пол-
ностью заполненный остовный уровень 4/14, отсутствующий в атомах
126
4й-элемента. Немонотонные, напоминающие вторичнопериодические
закономерности проявляются уже в величинах орбитальных радиу-
сов свободных атомов переходных металлов в любой дополнительной
подгруппе, а также в величинах суммы двух первых ионизационных
потенциалов свободных атомов [45].
Таким образом, в химии непереходных элементов вторичная перио-
дичность определяется особенностями валентной области электронной
структуры атомов, тогда как в химии переходных металлов вторич-
ная периодичность (здесь и сам термин можно использовать лишь с
большой натяжкой) определяется различиями в электронных остовах
атомов.
В целом же можно заключить, что в любой дополнительной под-
группе закономерности изменения свойств переходных металлов хо-
тя и носят нелинейный характер, но определяются в основном лишь
особенностями родоначальника (Зй-кайносимметрия) и большой схо-
жестью свойств обоих его последователей, т. е. 4d- и 5с?-элементов.
Глава 8
ДОПОЛНИТЕЛЬНАЯ ПЕРИОДИЧНОСТЬ
Однотипным проявлением Периодического закона как в химии
непереходных элементов, так и в химии переходных металлов, без-
условно, является отчетливое деление химических элементов одной и
той же разновидности в одном и том же периоде системы на ранние
и поздние. Например, деление всех (s, р) элементов 2-го периода на
ранние (Li, Be, В, С) и поздние (N, О, F, Ne), аналогичное деление
d-элементов 4-го периода (Sc, Ti, V, Cr, Мп — Fe, Со, Ni, Си, Zn) и f-
элементов 7-го периода (Th, Ра, U, Np, Pu, Am, Cm — Bk, Cf, Es, Fm,
Md, No, Lr). Физической основой такого деления является достиже-
ние «срединной» валентной электронной конфигурации атома (s!p3,
d5, /7), в которой все имеющиеся орбитали соответствующего типа за-
няты неспаренными электронами, и нет ни вакансий, ни электронных
пар.
В атомах ранних элементов валентная электронная структура на-
ряду с неспаренными электронами содержит вакантные орбитали, а
в атомах поздних элементов — орбитали, занятые электронными пара-
ми. Такое деление, отнюдь, не формально. Оно отражает важнейшие
закономерности изменения свойств соответствующих элементов в пе-
риоде системы. В частности, в химии d- и /-элементов к этим законо-
мерностям относятся:
• пониженная валентность атомов поздних элементов по сравнению
с атомами ранних элементов;
• пониженная кратность связей атомов поздних элементов с 7Г-
электронодонорными лигандами и вытекающая отсюда малая стабиль-
ность или отсутствие оксоионов и аналогичных систем в противопо-
ложность высокой стабильности оксоионов ранних элементов (напри-
мер, ТЮ2+, СгО22+, UO22+, NpO2+ и др.);
• повышенная энергия связей атомов поздних элементов с 7г-элект-
роноакцепторными лигандами и определяемая этим высокая стабиль-
ность комплексов с 7Г-акцепторными лигандами (например, карбони-
лов) и тг-комплексов (например, ферроцена (тг-СбЩ^Ее и др.);
128
• повышенная устойчивость низкозарядных гидратированных ка-
тионов поздних элементов в водных растворах (например, Со2+, Ni2+,
Cu+, Cm3+, Bk3+ и др.) в противоположность нестабильности анало-
гичных катионов ранних элементов.
Есть и другие, аналогичные «по духу», закономерности. Все это
позволяет говорить о наличии некоей внутренней периодичности в ря-
дах периодической системы, называемая дополнительной [45]. Эта
дополнительная периодичность не всегда проявляется достаточно от-
четливо, тем не менее некоторые ее проявления охватывают столь зна-
чительные части химии металлов, что заслуживают подробного об-
суждения.
В теории поля лигандов уже давно установлено, что переходные ме-
таллы первой вставной декады (Зс/-элементы) образуют с лигандами
слабого поля комплексы, многие свойства которых (магнитные, спек-
троскопические, энергия экстрастабилизации полем лигандов и т. п.)
изменяются строго периодически в том смысле, что характеристики
комплексов элемента, атомы которого имеют валентную электронную
конфигурацию d5+T?, «повторяют» характеристики (точнее, разумеет-
ся, близки характеристикам) комплексов элемента, атомы которого
имеют валентную электронную конфигурацию dn; например, Co(d7)-
Ti(rf2), Ni(d8)-V(d3) и т.д.
Притчей во языцех стала близость характеристик комплексов (с ли-
гандами слабого поля) двухвалентного цинка с валентной электронной
конфигурацией атомов Zn d10 и двухвалентного марганца с валентной
электронной конфигурацией атомов Mn d5. В отличие от однотипных
комплексов всех остальных Зd-элeмeнтoв эти комплексы не обладают
энергией экстрастабилизации полем лигандов и бесцветны. Отсутствие
окраски у этих комплексов единообразно объясняется запрещенностью
(d-d)-пepexoдoв, хотя и на несколько различной основе, а именно: в
случае Mn d5 на указанные электронные переходы налагается жест-
кий «запрет по спину» (малая вероятность электронных переходов, в
результате которых должен был бы измениться суммарный спин си-
стемы), в случае же Zn d10 — абсолютный запрет Паули.
Но наиболее ярко дополнительная периодичность проявляется в
ряду редкоземельных (4/) элементов. По-прежнему здесь постоянно
отмечается схожесть свойств лантаноидов с электронными конфигура-
циями атомов и /7+г?, например, Се f1 и ТЬ /8, Рг /2 и Dy /9, Ей /6
и Yb /13, Gd f7 и Lu /14. Всегда обращают на себя внимание особен-
ности химических свойств первого лантаноида — церия и восьмого —
тербия. По причине повышенной стабильности электронных конфи-
гураций /°, /7 и /14 (из квантовой механики атома хорошо извест-
но, что именно для этих электронных конфигураций энергия внутри-
атомного электронного обмена максимальна) атомы этих лантаноидов
129
способны находиться не только в состояниях с тривиальными валент-
ными электронными конфигурациями Се 6s25d14f1 и Tb 6s25d14f8,
но и в состояниях с валентными электронными конфигурациями Се
6s25d24f° и Tb 6s25d24f7. Из-за валентной неактивности кайносим-
метричных атомных 4/-орбиталей в первом случае церий и тербий об-
разуют трехвалентные, во втором случае — весьма малохарактерные
для лантаноидов четырехвалентные соединения. В меньшей мере то
же самое справедливо и по отношению к общехимическому поведению
второго лантаноида — празеодима и девятого —диспрозия.
Аналогично показательны особенности химических свойств шесто-
го лантаноида — европия и тринадцатого — иттербия. По той же при-
чине (имеется в виду повышенная стабильность электронных конфи-
гураций /°, /7 и /14) атомы этих лантаноидов способны находить-
ся не только в состояниях с тривиальными валентными электронны-
ми конфигурациями Eu 6s25d14f6 и Yb 6s25d14f13, но и в состояни-
ях с валентными электронными конфигурациями Eu 6s25d°4f7 и ТЬ
6s25d°4f14. Валентная неактивность кайносимметричных атомных 4/-
орбиталей приводит к тому, что в первом случае европий и иттербий
образуют трехвалентные, во втором случае — аномальные для ланта-
ноидов двухвалентные соединения. Опять-таки в меньшей мере то же
самое относится и к общехимическому поведению пятого лантаноида —
самария и двенадцатого —тулия.
Что же касается актиноидов (5/-элементов), то здесь поздние эле-
менты (элементы второй семерки) не так отчетливо повторяют свой-
ства ранних элементов — элементов первой семерки. В ряду актинои-
дов дополнительная периодичность фактически не имеет места, она
«размывается»; по крайней мере, имеющиеся данные не свидетель-
ствуют о каком-либо закономерном (в смысле дополнительной перио-
дичности) чередовании валентностей атомов в соединениях актинои-
дов. И причина этого прежде всего в том, что в противоположность
валентной электронной структуре атома лантаноида Ln 6s25d14/n,
где электроны на кайносимметричных 4/-орбиталях характеризуются
очень низкими энергиями, весьма сжатым зарядовым распределени-
ем (малой протяженностью) в пространстве и потому заметного уча-
стия во взаимодействиях с валентными лигандными орбиталями (т. е.
в образовании связей с лигандами) не принимают, в-валентной элек-
тронной структуре атома актиноида An 6s25d14/n орбитали 5/ уже не
являются кайносимметричными, они достаточно диффузны, их энер-
гии близки энергиям валентных лигандных орбиталей, и потому они
эффективно взаимодействуют с валентными лигандными орбиталями
(т. е. участвуют в образовании связей с лигандами).
В наибольшей мере все вышесказанное справедливо для ранних ак-
тиноидов, в наименьшей — для поздних. Поэтому многие актиноиды
130
первой половины ряда весьма часто высоковалентны в своих моле-
кулярных структурах в отличие от большинства актиноидов второй
половины ряда, для которых типичны низковалентные состояния. Но
и в химии актиноидов, безусловно, есть, хотя и нечастые, примеры
большого сходства общехимического поведения двух соответственных
элементов — раннего /г/ и позднего /7+т?.
Глава 9
ИНДИВИДУАЛЬНОСТЬ
ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Каждый химический элемент индивидуален, обладает большими
или меньшими одному ему присущими особенностями. Безусловно,
индивидуальность химического элемента, точнее, мера этой индиви-
дуальности, есть функция его положения в периодической системе. В
этой главе проследим проявление индивидуальности на примере двух
элементов — фтора и кислорода — в наиболее яркой форме олицетво-
ряющих химию элементов, занимающих самые верхние (в столбцах)
и самые правые (в рядах) положения. Особенности любого элемента
надлежит выявлять и очерчивать на фоне общих свойств, присущих
элементам одной с ним группы (подгруппы) или одного < ним пери-
ода системы. В случае фтора и кислорода специфические свойства
должны быть выявлены, с одной стороны, в сравнении со свойствами
типических элементов (Li, Be, В, С, N, Ne), с другой —в сравнении со
свойствами галогенов (Cl, Br, I, At) и халькогенов (S, Se, Те, Ро). При
этом химическое поведение фтора и кислорода следует характеризо-
вать и в том случае, когда они выступают в роли центральных атомов,
и в том случае, когда они играют роль лигандов.
9.1. Атомы фтора и кислорода
в роли центральных атомов
В сравнении с галогенами (Cl, Br, I, At) особые свойства фтора как
центрального атома заключаются, как известно, в том, что фтор ни с
какими лигандами не образует соединений на высоких ступенях окис-
ления. Так, не существует соединений фтора, подобных оксидам, оксо-
анионам и другим соединениям хлора с высокими (вплоть до номера
менделеевской группы) значениями его степени окисления. В сравне-
нии с элементами своего периода (Li, Be, В, С, N, Ne) особые свойства
фтора (принципиальные отличия этого элемента, прежде всего, от В,
132
C, N) состоят опять-таки в резко пониженной валентности его атомов.
Атомы F не могут использовать все свои четыре валентные орбитали
(2s, 2pz, 2ру, 2pz} в химических связях, и это контрастирует с четы-
рехвалентным поведением атомов бора, углерода, азота. Помимо одной
ковалентной (1:1) a-связи за счет своего единственного неспаренного
2ра-электрона атом F, в принципе, мог бы образовывать с лигандами
еще и дативные (2:0) связи за счет своих валентных электронных пар
(а их у него три) или предоставлять эти пары на образование гипер-
валентных связей в трехцентровых фрагментах L(l)-F-L(2). Но и в
том, и в другом случае решающим фактором, резко ограничивающим
реальную валентность атомов фтора, является очень высокий потен-
циал ионизации атома F, не допускающий значительного участия его
валентных электронных пар как в дативных, так так и в гипервалент-
ных связях.
Действительно, известные примеры всегда указывают на малую
способность атомов F образовывать дативные связи даже с такими
атомами или многоатомными группами, которые обладают весьма зна-
чительной электроотрицательностью. Образование дативных связей
F —> У облегчается предварительным накоплением некоторого эффек-
тивного отрицательного заряда на атоме F, снижающего потенциал
ионизации атома F и повышающего тем самым его электронодонорную
способность. Что же касается гипервалентных связей L(l) - F - L(2), то
они, по-видимому, вообще не могут быть образованы. И причина это-
го в отсутствии таких лигандов, которые обладали бы орбиталями,
заселенными неспаренными электронами и имеющими более низкую
энергию по сравнению с энергией заселенных электронными парами
валентных 2ра-электронов атомов F, как этого требует природа трех-
центровых четырехэлектронных связей.
В целом ясно, что из четырех валентных орбиталей атома фто-
ра далеко не все участвуют в химических связях, и реальная валент-
ность атомов фтора никогда не достигает максимального (принципи-
ально допустимого) значения 4, свойственного атомам срединных ти-
пических элементов — атомам бора, углерода, азота. Можно, вероятно,
утверждать, что часто состояние атомов фтора может быть описано
как одновалентное, в большинстве своих соединений атомы фтора име-
ют валентность немного выше единичной и, наконец, в редких случаях
валентность атомов фтора заметно приближается к 2. Яркими приме-
рами последних являются молекулярный катион [H2F]1+ и молекула
BF.
Повышение валентности атома фтора сверх единичной возможно
(и происходит) в таких молекулярных системах, где атом-партнер по-
мимо неспаренных валентных электронов имеет вакантную валент-
ную орбиталь и малую электроотрицательность, так что образуемая
133
им «первичная» ковалентная (1:1) сг-связь с атомом F обладает до-
статочно большой степенью ионности, что обеспечивает значитель-
ный эффективный отрицательный заряд на атоме F, соответствую-
щий эффективный положительный заряд на атоме-партнере, повы-
шает тг-донорную способность атома F и соответственно увеличивает
7г-акцепторную способность атома-партнера. Все это способствует об-
разованию «дополнительной» дативной (2:0) тг-связи атома F с его
партнером (F —> У) и тем самым приводит к повышенной валентности
атома F сверх единичной.
Особенности химических свойств кислорода как центрального ато-
ма несложно описать в тех же самых терминах, что и в случае фтора.
Кислород весьма похож на фтор, и различия между этими двумя эле-
ментами обусловлены, главным образом, тем, что атомы кислорода
содержат большее число неспаренных валентных электронов (два, а
не один) и электроотрицательность кислорода несколько ниже, чем у
фтора, хотя и продолжает оставаться очень высокой. Как и в случае
фтора, следует прежде всего отметить пониженную валентность кис-
лородных атомов в сравнении как с халькогенами (S, Se, Те, Ро), так
и с элементами 2-го периода (Li, Be, В, С, N, Ne). Многие примеры
иллюстрируют отличия кислорода от серы и более тяжелых халько-
генов. Кислород не образует соединений на высоких ступенях окисле-
ния (и тем более на высшей, равной номеру группы в периодической
системе, ступени окисления), что весьма характерно для халькогенов.
Достаточно в этом отношении указать на отсутствие в химии кислоро-
да соединений, подобных тетра- и гексагалогенидам халькогенов. При
описании же отличий кислорода от бора, углерода, азота в первую
очередь следует отметить отсутствие в химии кислорода четырехва-
лентных соединений, столь свойственных В, С, N. В этом отношении
кислород «повторяет» свойства фтора.
Но, несмотря на то, что кислород остается подобным фтору позд-
ним типическим элементом, он уже заметно отличается от фтора. При
обсуждении валентных свойств кислородных атомов следует иметь в
виду, что их валентность существенно выше, чем у атомов фтора. И
дело здесь не только в том, что кислородный атом имеет два неспа-
ренных валентных электрона (по сравнению с одним в атоме фтора),
позволяющих ему образовывать уже две ковалентные (1:1) связи и
тем проявлять двухвалентность. Дело в том, что по сравнению с ато-
мом фтора кислородный атом имеет меньший потенциал ионизации,
меньшую электроотрицательность. По этой причине валентные пары
кислородных атомов принимают более эффективное участие в образо-
вании дативных (2:0) связей с атомами-партнерами за счет вакантных
валентных орбиталей последних. Поэтому кислородный атом с двумя
неспаренными электронами значительно чаще проявляет валентность
134
выше двух, чем атом фтора с одним неспаренным электроном валент-
ность выше единицы. Что же касается гипервалентных связей L(l)-
0-L(2), то, как и в случае фтора, их образование маловероятно в
молекулярных системах кислорода.
В противоположность атомам фтора кислородные атомы уже спо-
собны образовывать ковалентные и дативные связи за счет трех своих
валентных орбиталей и, по-видимому, даже (в редчайших случаях)
за счет всех четырех своих BAO: 2s, 2pz, 2ру, 2pz. Близко к трехва-
лентному состояние кислородных атомов в структурах [НзО]1+, С=О,
[O=U=O]2+. Формально четырехвалентны кислородные атомы (это
пока единственный случай) в структурах экзотических координаци-
онных соединений K^RugOClio] и K^OsgOClio], включающих в се-
бя линейные трехатомные группы [Ru = О = Ru] и [Os = О = Os]. Необ-
ходимые предпосылки осуществления столь высокого валентного со-
стояния кислородных атомов состоят в следующем: атомы-партнеры
помимо неспаренных валентных электронов должны иметь вакант-
ные валентные орбитали с малой электроотрицательностью; образу-
емые «первичные» ковалентные (1:1) сг-связи кислородных атомов с
атомами-партнерами должны обладать достаточно большой степенью
ионности, что должно приводить к значительным эффективным от-
рицательным зарядам на кислородных атомах и соответствующим
эффективным положительным зарядам на атомах-партнерах и, как
следствие, к повышенной тг-донорной способности кислородных ато-
мов и соответственно увеличенной тг-акцепторной способности атомов-
партнеров. Именно это способствует образованию «дополнительных»
дативных (2:0) тг-связей кислородных атомов с их партнерами (О
—> У) и тем самым приводит к значительно повышенной валентно-
сти кислородных атомов по сравнению с «классической» двухвалент-
ностью.
9.2. Атомы фтора и кислорода в роли лигандов
Не менее важная особенность химии фтора и кислорода, также свя-
занная с их индивидуальными атомными характеристиками и опреде-
ляемая параметрами электронной структуры их атомов, состоит в том,
что, имея наивысшую электроотрицательность, атомы F и О выступа-
ют в качестве наиболее эффективных модифицирующих лигандов и
способствуют образованию наибольшего (в сравнении со всеми осталь-
ными лигандами) числа и многообразия ковалентных и гипервалент-
ных соединений того или иного химического элемента. Таким образом,
атомы F и О способствуют проявлению тем или иным химическим эле-
ментом наибольшего числа устойчивых ступеней окисления, включая
135
высшие окисленные формы, причем это относится как к элементам
главных, так и к элементам дополнительных подгрупп.
Устойчивость высших окисленных форм непереходного элемента
или переходного металла именно с атомами F или О в качестве ли-
гандов проявляется либо в том, что высшие фториды и оксиды этого
элемента имеют значительно большую прочность, чем его аналогич-
ные соединения с любыми другими лигандами, либо в том, что высшие
фториды и оксиды просто существуют (устойчивы), тогда как высшие
соединения с любыми другими лигандами отсутствуют (нестабильны).
Достаточно упомянуть стабильность SFe, SO3, SO42-, CrFe, СгОз,
CrO42", BiFs, BigOs, ВЮ31”, IF7, IO41”, XeFe, ХеОз, где степень окис-
ления центрального атома равна номеру менделеевской группы, и от-
сутствие иных соединений формально шестивалентной серы, шести-
валентного хрома, пятивалентного висмута, семивалентного иода, ше-
стивалентного ксенона. При этом стабильность фторидов, как прави-
ло, выше стабильности соответствующих оксидов; частичное замеще-
ние атомов О атомами F в молекулярной структуре повышает ее ста-
бильность (устойчивость оксифторидов ксенона ХеОдВг, XeOF4 вы-
ше устойчивости соответствующего оксида ХеОз). Правда, в некото-
рых случаях кислород, занимая следующее после фтора место в этой
«табели о рангах», способствует образованию таких высших окислен-
ных форм, которые неустойчивы даже со фтором. Например, октафто-
рид ксенона XeFs, по-видимому, неустойчив (по крайней мере, до сих
пор не получен), в то время как кислородные соединения формально
восьмивалентного ксенона известны: это сам тетраоксид ксенона ХеО4
и его гидратные формы — ЩХеОв, НзХеОб1”, HgXeOe2", НХеОб3”,
ХеОб4”. Не известны CIF7, FeFs и им подобные молекулярные струк-
туры, тогда как существуют CI2O7, FeO4. Во всех таких примерах
преимущественная стабильность высших окисленных форм оксосоеди-
нений в сравнении со фторидами объясняется тем, что при одной и
той же формальной степени окисления центрального атома реальное
число координированных им лигандов в оксосоединении значительно
(формально вдвое) меньше, чем во фториде, и решающим фактором
оказывается эндоэффект межлигандного отталкивания. Этот эндоэф-
фект в случае фторокомплексов из-за большого числа координиро-
ванных лигандов становится непомерно большим, не допускающим их
образования в сколько-нибудь устойчивом состоянии.
В качестве лигандов атомы кислорода, и особенно атомы фтора,
способствуют образованию таких стереохимических форм с высокими
координационными числами центральных атомов, которых с други-
ми лигандами не существует вовсе или они оказываются очень ред-
кими. Так, стереохимия фторсодержащих молекул и молекулярных
анионов включает в себя многочисленные гепта-, окта-, нонакомплек-
136
сы типа IF7, ReF7, NbF72~, TaF72~, ZrF73~, HfF73", WF71-, ХеР^-,
PuF72~, PaF72", TaF83-, TeF82~, UF82", ReF81-, MoF82~, WF82"
и даже OsFg1-, в то время как аналогичные комплексы непереход-
ных элементов и переходных металлов с другими лигандами весь-
ма редки: Mo(CN)83-, W(CN)83-, U(NCS)84“, Re(CN)82", ReHg2',
(Tr-CsHs^WHa14-, (тг-СбЩ^ТаНз3” и некоторые другие.
Наконец, в качестве лигандов атомы фтора являются единствен-
ными партнерами, обеспечивающими некоторым элементам возмож-
ность химического связывания, возможность образования молекуляр-
ных структур. Так, аргон и криптон могут, по-видимому, образовы-
вать соединения лишь со фтором или при непременном участии ато-
мов фтора (KrF2, KrFg • 2SbF5).
С позиций теории гипервалентных связей специфика атомов фто-
ра и кислорода в роли лигандов в соединениях непереходных элемен-
тов находит простое и ясное объяснение. Будучи наиболее электроот-
рицательными, с очень низкими энергиями и малой протяженностью
валентных пр-орбиталей, атомы фтора или кислорода способны в ка-
честве лигандов образовывать с центральным атомом любого непе-
реходного элемента наиболее прочные гипервалентные четырехэлек-
тронные трехцентровые связи Lpj-A-L^), единственно ответствен-
ные за устойчивость (а иногда просто за сам факт существования)
высших соединений этого элемента. Причина этого заключается в том,
что при прочих равных условиях именно при атомах фтора или кисло-
рода в четырехэлектронной трехцентровой связи L^-A-L^) проис-
ходит наибольшее перераспределение зарядовой плотности валентных
электронов центрального атома А на лиганды, приводящее к наиболь-
шей энергетической стабилизации единственной связывающей моле-
куляной орбитали трехцентрового трехорбитального фрагмента L^) -
А-Ь(2) и тем самым к наибольшей прочности этих гипервалентных
связей между центральным атомом и лигандами.
Особая роль атомов фтора и кислорода как лигандов в соединени-
ях переходных металлов состоит в обеспечении наибольшей энергети-
ческой стабилизации высших окисленных форм переходных металлов
(подчас с уникальной стереохимией, с высокими (от пяти до девяти)
координационными числами атома М) по причине вовлечения в связи
M-L валентных пр-орбиталей центральных атомов М, виртуальных в
основном состоянии свободного атома М. Действительно, атомы F или
О со своей наивысшей электроотрицательностью обусловливают появ-
ление на центральном атоме М достаточно большого положительного
эффективного заряда уже при образовании первых ковалентных свя-
зей M-L (М-F или М-О). Этим они и обеспечивают наиболее выгодные
(в сравнении с другими лигандами при прочих одинаковых факторах)
условия для вовлечения в связывание валентных пр-орбиталей цен-
137
трального атома М, которые были вакантными в невозбужденном со-
стоянии свободного атома М. Эти экстравалентные орбитали атома М
необходимы для образования молекулярных структур с кратными свя-
зями М-L или с высокими координационными числами атома М. Без
высоких координационных чисел атома М невозможно образование та-
ких высших соединений переходного металла, в которых лиганды не
могут образовать с центральным атомом существенно кратных связей.
Таким образом, именно при атомах F или О в качестве лигандов со-
здаются наиболее благоприятные условия для образования возможно
более прочных связей при их повышенной кратности или при высо-
ких координационных числах атома М и тем самым для наибольшей
энергетической стабилизации высших окисленных форм переходных
металлов.
Итак, по причине своих особых электроноядерных характеристик
атомы F и О в качестве лигандов могут наиболее значительным об-
разом модифицировать электронную структуру любого центрально-
го атома и «заставить» его образовывать соединения таких форм, на
таких ступенях окисления, какие с другими лигандами просто невоз-
можны.
9.3. Окислительные функции молекулярных фтора
и кислорода
Остановимся еще на одной отличительной черте химии этих двух
элементов — их важной особенностью является то, что во всех процес-
сах молекулярные фтор и кислород всегда оказываются окислителя-
ми (подробно это изложено в [40]). Исключительно окислительную, и
притом очень высокую, способность проявляет молекулярный фтор, в
несколько меньшей мере это присуще молекулярному кислороду.
В двухатомной молекуле F2 и связывающие, и разрыхляющие мо-
лекулярные тг-орбитали полностью заселены электронами, поэтому тг-
связи в ней нацело отсутствуют. По существу электронная структура
включает в себя только одинарную сг-связь (занятая двумя электрона-
ми связывающая МО а и вакантная разрыхляющая МО сг*), характе-
ризующуюся небольшой энергией и значительным межъядерным рас-
стоянием (табл. 9). Малая прочность связи в молекуле F2 объясняется
не только ее единичной кратностью, но и большим дестабилизирую-
щим эффектом тг-электронов.
Несколько прочнее молекула О2, в электронной структуре которой
лишь два электрона, и притом в неспаренном состоянии, заселяют две
разрыхляющие МО тг* (а не четыре электрона, как в молекуле F2), и
поэтому в молекуле О2 существует уже двойная (сг + тг)-связь с уко-
138
роченным (по сравнению с молекулой F2) межъядерным расстоянием
и повышенной энергией (табл. 9).
Таблица 9. Характеристики двухатомных молекул А2 фтора
и кислорода
а2 Пе Электронная конфигурация „в пе Яа-а #А-А #А-А
^2+ 13 o-?(aj)2 тг2 тг2 (?rj)2 (тг^)1 3 1,5 (<т + 0,5тг) - 59
f2 14 ст1(ст1)2 ст2 *1 к2 ("Г)2 (7Гг)2 2 1,0 (а) 1,44 37
IL 15 <72(<’’;)2 °2 A О^)2 (7Гг)2 (^г)1 1 0,5 (а) - 23
О2+ 11 <*i (CTi)2 °2 7Г1 ^2 «)' 5 2,5 (а + 1,5тг) 1Д2 153
О2 12 <7?(<rJ)2 itj 7г^ (irj)1 (ttJ)1 4 2,0 (ст + 7г) 1,21 118
О2~ 13 CTi(o’i)2 *1 (*;)2 (7г2)1 3 1,5 (<т + 0,5тг) 1,28 94
Примечание. пе — суммарное число валентных электронов; п® — эффектив-
ное число связывающих электронов; /?д-а — межъядерное расстояние (А), Ед_д —
энергия связи (ккал), Кд-А — кратность связи.
Разумеется, и молекула F2, и молекула О2 в элементарном акте
той или иной химической реакции ведет себя, равно как и любая дру-
гая молекула, так, чтобы энергия связи понизилась, а следовательно,
понизилась энергия активации реакции. Для этого есть только один
путь, а именно, «приобрести» лишний электронный заряд. Распреде-
ление этой, дополнительной по сравнению с нейтральной молекулой
F2 или О2, электронной плотности неминуемо приводит к заселению
вакансии на сильно разрыхляющей молекулярной орбитали сгг* (F2)
или 7г| (О2). Это сразу же приводит к уменьшению кратности свя-
зи, увеличению межъядерного расстояния и понижению энергии свя-
зи в образующемся молекулярном анионе F21- или О2" (табл. 9), что,
безусловно, способствует активации F2 или О2. Движущей же силой
активирующей анионизации F2 или О2 является понижение полной
энергии при переходе нейтральной молекулы F2(O2) в молекулярный
анион F^“ (О2") (рис. 15).
Напротив, любая катионизация молекулы F2 или О2 в элементар-
ном акте химической реакции невыгодна, поскольку приводит к повы-
шению энергии активации по следующей вполне понятной причине.
Потеря электронного заряда молекулой F2(C>2) приводит к уменьше-
нию электронной заселенности сильно разрыхляющей молекулярной
орбитали 7г*-типа. Это сразу же сказывается на возрастании кратно-
сти связи, увличении ее энергии и уменьшении межъядерного рассто-
яния в образующемся молекулярном катионе F^ (О^+) (табл. 9). И
такое упрочнение молекулярного катиона F^"1- (О^4-) по сравнению с
139
нейтральной молекулой Рг(О2) происходит несмотря на то, что полная
энергия молекулярной системы в процессе ее катионизации возрастает
(рис. 15), поскольку нейтральная молекула Р2(Ог) имеет значитель-
ный эндоэнергетический потенциал ионизации.
Таким образом, локализация лишнего электронного заряда на раз-
рыхляющих МО молекулы Рг(О2) снижает ее прочность, а следова-
тельно, энергию активации любой реакции с участием этой молекулы.
В то же время потеря электрона молекулой Рг(Ог) приводила бы к
увеличению ее прочности и повышала бы энергию активации такой
реакции. Именно поэтому молекулярные фтор и кислород в своих ре-
акциях выступают в роли окислителей.
Такое поведение характерно вообще для молекул с избытком ва-
лентных электронов, т. е. для таких молекулярных систем, в кото-
рых электроны заселяют не только связывающие, но и разрыхляющие
МО. Молекулы же с недостатком валентных электронов, где не полно-
стью заняты связывающие МО, должны проявлять восстановительные
свойства.
Все эти представления в обобщенном виде отражает рис. 15. На
приведенных кривых потенциальной энергии различия в энергиях си-
стем (А + А) и (А + А+) или различия в энергиях систем (А + А) и
(А + А-) равны потенциалам ионизации атомов А или соответствен-
но
но энергиям сродства к электрону атомов А. Энергетические разли-
чия между минимумами А 2 и Aj или энергетические различия меж-
ду минимумами А2 и А^ отвечают потенциалам ионизации молекул
А2 или соответственно энергиям сродства молекул А 2 к электрону. Из
представленных диаграмм хорошо видно, как влияет изменение сум-
марного числа валентных электронов в двухатомной молекуле А 2 на
глубину ее потенциальной ямы, т. е. на прочность молекулы, при этом
становятся вполне ясны путь активации молекулы в химической реак-
ции и отвечающее этому пути окислительное поведение молекулярных
фтора и кислорода.
Глава 10
НОВЫЕ ПОДХОДЫ В ТЕОРИИ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
Несмотря на почти полуторавековую историю учения о Периоди-
ческом законе и периодической системе химических элементов, мно-
гие фундаментальные вопросы этого учения до сих пор не получи-
ли окончательного объяснения. В 30-е годы XX столетия казалось,
что это учение близко к завершению на основе стремительного раз-
вития квантовой механики атомов и молекул. Хорошо известны слова
Э. Резерфорда, сказанные им в 1934 г. в сообщении «Периодический
закон и его интерпретация» на заседании Британского химического
общества, посвященного столетию со дня рождения Д. И. Менделеева:
«Развитие волновой механики столь совершенно, что Периодический
закон химических элементов теперь может быть выведен исходя из ее
основных принципов. Любой действительно очень компетентный ма-
тематик, знакомый с законами квантовой механики атома и с одним
дополнительным постулатом — принципом Паули, даже в том случае,
если бы он никогда не слышал о Периодическом законе, был бы в
состоянии построить периодическую систему химических элементов.
Ему, конечно, понадобилось бы громадное количество времени и по-
мощь вычислителей, и, если только он не отличается совершенно ис-
ключительным умением, время от времени в некоторых критических
точках периодической системы ему пришлось бы делать свои шаги
очень осторожно». Но тридцать пять лет спустя, в 1969 г., известный
и тоже английский теоретик Ч. Коулсон в докладе «Волновая меха-
ника и периодическая система», представленном к столетию Перио-
дического закона, сказал: «Современная волновая механика не внес-
ла достаточной ясности в детали периодической системы. Однако в
этот вывод нужно внести поправки: хотя квантовая механика не мо-
жет углубить наше знание периодической системы, она в то же время
в состоянии дать качественную, а иногда и количественную информа-
цию по некоторым вопросам, имеющим отношение к периодической
системе элементов» [49].
142
Внимание теоретиков к математическому описанию структуры пе-
риодической системы не ослабевает и в самое последнее время. Попыт-
ки чисто математического вывода периодической системы предприни-
маются постоянно, притом с использованием таких фундаментальных
теорий, как теория чисел, теория множеств, теория групп. Ниже мы
остановимся на этих новых подходах, используя, главным образом,
статью профессоров И. С. Дмитриева и Д. Н. Трифонова [2].
10.1. Математические модели
Основоположником чисто математического моделирования струк-
туры периодической системы можно считать И. Ридберга, который в
1913-1914 гг. впервые представил математическую модель периодиче-
ской системы и при этом указывал, что моделирование действительной
системы элементов сопряжено с рядом проблем, решение которых воз-
можно посредством изучения эмпирически заданных численных зна-
чений без принятия какой-либо гипотезы о природе материи.
Математические модели основаны на общепринятой структуре пе-
риодической системы и используют представления квантовой меха-
ники атома о реальной схеме построения электронных конфигураций
атомов по мере увеличения заряда ядра Z. Эти закономерности пере-
водятся в математическую форму для определения граничных значе-
ний Z, в частности, Zn и nZ — порядковые номера химических элемен-
тов, отвечающих границам заполнения электронной оболочки с дан-
ным значением главного кантового числа n; Zt и iZ — границы перио-
дов системы; Zn+/ и n+/Z — границы (п-М)-групп; и n>/Z —грани-
цы п/-подгрупп; Z/ — порядковые номера элементов, в атомах которых
впервые появляется электрон с данным значением орбитального кван-
тового числа I. Совокупности соответствующих аналитических выра-
жений (алгебраических уравнений) дают математическое описание за-
кономерностей построения реальных электронных оболочек атомов и
общих принципов структуры периодической системы, в целом дают
функциональные зависимости граничных значений Z от обоих кван-
товых чисел п и I или от одного из них. Наиболее важным и значимым
примером обсуждаемых моделей является уже рассмотренное правило
Клечковского — Демкова.
Своеобразная математическая модель периодической системы хи-
мических элементов исходит из того, что многие граничные значения
Z получаются в результате четырехкратного суммирования последо-
вательностей некоторых чисел Fa (табл. 10), которые индивидуальны
для каждого граничного Z и сумма которых в каждом случае равна
восьми.
143
Таким образом, общее выражение для граничных Z имеет вид
Zrp =4SFa,
где индекс а соответствует индексам п, /, t, n + I.
Таблица 10. Последовательности Fa для соответствующих
последовательностей Zrp
^гр Последовательности
Zrp Fa
Zi 1, 5, 21, 57, 121, ... 1, 1, 7, -1, 0, ...
nz 2, 10, 30, 70, 138, ... 2, 2, 2, 2, 0, ...
^нечетн 1, 11, 37, 87, 169, ... 1, 7, -1, 1, 0, ...
^четн 3, 19, 55, 119, 219, ... 3, 7, -3, 1, 0, ...
^нечетн 2, 18, 54, 118, 218, ... 2, 10, -6, 2, 0, ...
tZ4eTH 10, 36, 86, 168, ... 10, -4, 2, 0, ...
учетн Zn+/ 3, 13, 39, 89, 171, ... 3, 1, 5, -1, 0, ...
п+/^нечетн 2, 12, 38, 88, 170, ... 2, 4, 2, 0, 0, ...
п+^ЧеТН 4, 20, 56, 120, 220, ... 4, 4, 0, 0, 0, ...
Поскольку общая структура периодической системы определяется
значениями Zt и tZ, то, взяв от каждой из соответствующих последо-
вательностей по четыре первых члена, можно составить определитель
четвертого порядка:
1 7 -1 1
2 10 -6 2
Д = 3 7 -3 1
10 —4 2 0
который правомерно рассматривать как математическую модель пе-
риодической системы (с 1-го по 8-й период включительно). Вычитая
последовательно из чисел третьей строки соответственные числа пер-
вой, а из чисел четвертой — числа второй, получаем последовательно-
сти 2, 0, -2, 0 и 8, -14, 8, -2, четырехкратное суммирование которых
дает длины четных и нечетных периодов: 2, 8, 18, 32 и 8, 18, 32, 50
соответственно.
Оперирование двумя квантовыми числами — главным п и орби-
тальным I — для отражения реальных электронных конфигураций сво-
бодных невозбужденных атомов позволяет вывести разнообразные
аналитические выражения, моделирующие ту или иную особенность
электронных конфигураций. В работе профессора Д. Н. Трифонова
[50] при использовании параметра Я, отражающего порядковый номер
п /-подгруппы и являющегося функцией значений квантовых чисел п
144
и /, выведено уравнение для определения значения Z, при котором в
атоме впервые появляется электрон с данным I:
Zi = (1/2)/(37?г - 1) + 1.
Для Ri, равных 1 (1s), 3 (2р), 7 (3d), 13 (4/), 21 (5<?), получаем Zi = 1, 5,
21, 58, 125 соответственно, т. е. значения, вычисляемые квантовомеха-
нически и точно соответствующие действительным и прогнозируемым
(для I = 4). Последовательность Ri = 1, 3, 7, 13, 21 получается при од-
нократном суммировании числовой последовательности ti = 1, 2, 4, б,
8, где ti — порядковый номер периода, в котором содержится элемент,
отвечающий Zi. Таким образом, простой математический прием позво-
ляет передать весьма важную особенность периодической системы —
порядковый номер химического элемента, в атомах которого впервые
появляются 1s-, 2р-, 3d-, 4/-, 5(/-электроны.
Даже простейшие математические модели дают ожидаемые резуль-
таты. Так, в работе [51] показано, что из последовательности натураль-
ных чисел t = 1, 2, 3, 4, 5, б, 7, 8,... можно легко получить последо-
вательность М— ^2 (t+1)2, если t нечетное, или М= ^2 (£+2)2, если
t четное, а именно, М= 2, 8, 8, 18, 18, 32, 32, 50, ..., которая точ-
но соответствует последовательности чисел химических элементов в
периодах системы. Далее, исходя из двух простых последовательно-
стей 1, 2, 4, б, 8 (номера периодов, включающих в себя 1s-, 2р-, 3d-,
4/-, 5^-элементы) и 0, 1, 2, 3, 4 (соответствующие значения орбиталь-
ного квантового числа /) при выполнении последовательных опера-
ций почленного сложения, суммирования полученного ряда, почлен-
ного перемножения с прибавлением единицы легко можно получить
последовательность 1, 5, 21, 58, 125, которая представляет собой очень
важную в теории периодической системы последовательность зарядов
ядер атомов Z(Zi), в которых впервые появляется электрон с новым
значением орбитального квантового числа I (0, 1, 2, 3, 4), т. е. после-
довательность порядковых номеров химических элементов, к которым
приурочено появление 1s-, 2р-, 3d-, 4/-, 5р-элементов в периодической
системе.
Как отмечает профессор Д. Н. Трифонов, эти тривиальные матема-
тические операции, позволяющие построить числовую модель перио-
дической системы и вычислить очень важные «граничные» значения
Z(Zi), не претендуют на обобщения, но они подчеркивают удивитель-
ную гармоничность структуры периодической системы.
Еще один подход к математическому моделированию структуры
периодической системы состоит в следующем. На основе представ-
лений теории множеств или теории чисел и посредством определен-
ных математических операций производится построение некоторой аб-
145
страктной математической модели. Это может быть либо теоретико-
числовая модель, отражающая распределение чисел натурального ря-
да или простых чисел, либо модель, описывающая совокупность неко-
торых дискретных состояний. Затем при отождествлении чисел или
дискретных состояний математических моделей с параметрами упоря-
доченного множества химических элементов производится поиск фи-
зических аналогов построенных моделей. Среди найденных аналогов
может оказаться структура периодической системы или отдельных ее
фрагментов, и тогда можно говорить о количественной интерпретации
периодической системы на основе чисто математических представле-
ний.
При разработке теоретико-множественных или теоретике-число-
вых моделей периодической системы показано, что в зависимости от
выбора условий разбиения множества элементов на собственные под-
множества удается воспроизвести каноническую структуру периоди-
ческой системы. Далее, при определенных условиях разбиения мно-
жества натуральных чисел на подмножества распределение простых
чисел в интервале 1 — 120 по этим подмножествам оказывается зако-
номерным и может быть представлено в аналитической форме.
Реальным физическим аналогом такой числовой модели являет-
ся совокупность химических элементов, в атомах которых происходит
заселение одноэлектронных состояний с заданным значением суммы
квантовых чисел пи/. Но при Z > 120 модель простых чисел не рабо-
тает, что, между прочим, вполне согласуется с одновременным резким
изменением (и даже, скорее, нарушением) закономерной периодично-
сти формирования электронных конфигураций атомов. И вполне до-
пустимо констатировать, что в этой области задаваемая моделью ха-
отичность распределения простых чисел согласуется с нерегулярным
заполнением одноэлектронных состояний в атомах с зарядами ядер
больше 120.
10-2. Теоретика-групповая интерпретация
Особое место в теории периодической системы занимают теорети-
ко-групповые методы.
К недостаткам традиционной интерпретации структуры периоди-
ческой системы относится отсутствие строгого обоснования последова-
тельности заполнения одноэлектронных состояний (Is, 2s, 2р, 3s, Зр,
4s, 3d, 4р, 5s, ...), что и явилось стимулом для поиска новых кван-
товомеханических подходов к объяснению явления периодичности. В
теории периодической системы в последнее время успешно использу-
ются теоретико-групповые методы.
146
В квантовой механике используются в основном четыре типа групп
симметрии:
1. Группы пространственной (геометрической) симметрии, напри-
мер, точечные группы, характеризующие симметрию ядерного скелета
молекул, группа трехмерных вращений и др.
2. Группы «динамической» («скрытой», «высшей») симметрии,
способные дать полное описание квантовомеханической системы,
включая энергетический спектр.
3. Группы «неинвариантности» — это такие группы, часть элемен-
тов которых не коммутирует с гамильтонианом системы, а действу-
ют как понижающие или повышающие операторы на волновую функ-
цию системы; в качестве группы неинвариантности обычно фигуриру-
ет некоторая некомпактная группа, содержащая динамическую группу
в качестве подгруппы; наиболее известным примером является группа
де Ситтера О (4,1) атома водорода.
4. Симметрические (перестановочные) группы, используемые в тео-
рии многочастичных систем; например, многофермионные волновые
функции преобразуются по одномерному неприводимому представле-
нию вида [/N] группы 5дг (АГ —число фермионов в системе), что от-
ражает содержание принципа Паули.
В основном рассмотрим динамическую симметрию периодической
системы. Но сначала обратимся к группе симметрии атома водорода.
Атом водорода представляет собой квантовомеханическую систему,
для описания всех свойств которой недостаточно чисто геометриче-
ских преобразований, образующих ортогональную компактную груп-
пу 0(3). Эта группа является прямым произведением группы 50(3)
ортогональных преобразований трехмерного координатного простран-
ства на группу Ci инверсии пространства относительно начала коор-
динат: 0(3) = 50(3) xCf. Координатные волновые функции электрона
при этом классифицируются по (2/ + 1)-мерным неприводимым пред-
ставлениям группы 50(3), что соответствует заданию квантовых чи-
сел / и т, и по одномерным неприводимым представлениям группы
Of, что соответствует заданию четности состояния. Квантовое число
/, определяющее величину квадрата орбитального момента количества
движения, нумерует неприводимые представления группы 0(3).
Каждому значению I соответствует 21 + 1 различных значений кванто-
вого числа т, определяющего проекцию орбитального момента на ось
квантования. Число возможных значений т, т. е. 21 + 1, равно размер-
ности неприводимого представления Таким образом, с помощью
группы пространственных преобразований 0(3) можно объяснить вы-
рождение спектра атома водорода по т, но эта группа не объясняет
случайного вырождения по /, характерного для водородоподобных
систем.
147
В 1935 г. один из основоположников квантовой механики академик
В. А. Фок показал [47], что полная группа симметрии атома водорода,
объясняющая оба типа вырождения (по т и /), изоморфна группе 0(4)
вращений четырехмерного шара. Собственные функции водородного
атома, отвечающие главному квантовому числу п, образуют базис ко-
нечномерного неприводимого представления группы 0(4). Эта группа,
в свою очередь, изоморфна прямому произведению SU(2) х 51/(2), где
5(/(2) — группа унитарных унимодулярных преобразований двумерно-
го векторного пространства.
Генераторами группы 0(4) являются векторные оператор орби-
тального момента количества движения и оператор, соответствую-
щий классическому вектору Лапласа — Рунге — Ленца. В случае мно-
гоэлектронных атомов группа 0(4) уже не является их динамической
группой симметрии. Тем не менее в одноэлектронном приближении со-
стояния электронов по-прежнему задаются с помощью квантовых чи-
сел, полученных при решении задачи для атома водорода. В качестве
приближенной динамической симметрии периодической системы эле-
ментов можно [52] использовать группу G = SU(2) х 5(/(2) х 5(/(2).
Тогда периодичность свойств химических элементов можно свя-
зать с нарушением G-симметрии, обусловленным увеличением меж-
электронного отталкивания и релятивистских эффектов с возрас-
танием атомного номера (т. е. числа электронов в атоме) элемента.
Остановимся на этом подробнее.
В каждой составной физической системе (в том числе в атоме) су-
ществуют ряд взаимодействий и соответствующая ему иерархия сим-
метрий. Эта иерархия симметрий определяет редукцию представле-
ний высших групп симметрии на представления их подгрупп, т. е. кон-
кретную реализацию базиса пространства состояний этой физической
системы. В случае атома схема обсуждаемой редукции определяется
последовательностью учета различных поправок к приближению цен-
трального поля и может быть представлена редукционной цепочкой
[52, 53]
G = SU(2) х SU(2) х 50(2) D 0(4) D 50(3).
Последовательное «сужение» группы G на ее подгруппы 0(4) и
50(3) сопровождается тем, что неприводимые представления груп-
пы G распадаются на неприводимые представления ее подгрупп
0(4) и 50(3). Неприводимые представления группы G задаются тре-
мя квантовыми числами p,q,r, принимающими значения 0, 1/2, 1,
3/2, ... Неприводимые представления подгруппы 0(4) задаются дву-
мя квантовыми числами р = р, и = г + q, г + q — 1,..., |г — д|, кото-
рые могут быть либо целыми, либо полу целыми, но оба одновременно.
Неприводимые представления подгруппы 50(3) задаются одним кван-
148
товым числом I = p+v, p+v— 1,..., \р—1/|, которое всегда целое. Ниже
указаны неприводимые представления группы G и ее подгрупп 0(4) и
50(3) для вышеприведенной редукционной цепочки и даны собствен-
ные значения Е = (1/n2) Ry (в относительных единицах) оператора
полной энергии атома (табл. 11).
Таблица 10. Значения квантовых чисел для группы G
и ее подгрупп и собственные значения Е
G (Р> 9. г) 0(4) (м- у} SO(3) (0 Е
0, 0, 0 0, 0 0 -1
1 1 2’ 2 о, 1 _ 1 4
1 0 1 1 1 0, 1 1
2 ’ 2 2’ 2 5,5
1, 1, о 1, 1 о, 1, 2 _1 9
1. 0, 1 1, 1 0, 1, 2 13
3 3 о 2 ’ 2’ U з з 2’ 2 0, 1, 2, 3 _ 1 16
3 о 2 2 ’ U’ 2 з з 2’ 2 0, 1, 2, 3 1 23,5
2, 2, 0 2, 2 0, 1, 2, 3, 4 _ 1 25
Из данных таблицы легко может быть найдено значение главного
квантового числа , соответствующее первому появлению одноэлек-
тронных состояний с заданным значением орбитального квантового
числа I. Это значение вычисляется по формуле
n(i) = 2м+ 1.
Рассмотрим в качестве примера d-состояние, т. е. состояние с чис-
лом 1 = 2. Впервые d-орбиталь появляется в неприводимом представ-
лении (1, 1, 0) G-группы, что соответствует /i = 1, и, следовательно,
— з, стало быть, первая d-орбиталь в свободном невозбужденном
атоме появляется при п = 3, это действительно 3d-op6imuib.
Приведенные ранее значения Е являются собственными значения-
ми гамильтониана, относящегося к некоторой многоэлектронной систе-
ме, учитывающего межэлектронные взаимодействия и имеющего до-
статочно сложное выражение через три квантовых числа p,q,r непри-
водимых представлений динамической группы симметрии G (она яв-
ляется группой такого гамильтониана). Эти численные значения Е
следует понимать только как некоторые условные, формальные, точ-
нее, относительные величины и принимать во внимание лишь характер
их изменения.
В рамках развиваемых представлений легко убедиться в том, что
последовательность заполнения nZ-электронных оболочек в свободных
атомах, получаемая в терминах нарушения G-симметрии, хорошо сов-
149
падает с реальной. Для иллюстрации рассмотрим знаменитый пример,
а именно: последовательность состояний Зр, 3d, 4s. Атомной орбитали
Зр отвечает неприводимое представление РД О, V2), и для конфигура-
ции с занятыми Зр-орбиталями собственное значение Е = -1/6,4 Ry.
Атомной орбитали 3d отвечает неприводимое представление (1, 1, 0),
и для конфигурации с занятыми Зd-opбитaлями собственное значение
Е = -1/9,4 Ry. Атомной орбитали 4s отвечает неприводимое пред-
ставление (1, 1, 0), и для конфигурации с занятыми 45-орбиталями
собственное значение Е = -1/9 Ry.
Сопоставляя собственные значения оператора полной энергии ато-
ма для этих трех состояний, получаем (с учетом принципа миниму-
ма энергии, разумеется) следующий порядок заполнения электронами
рассматриваемых орбиталей: ... Зр, 4s, 3d, ..., что полностью совпа-
дает с имеющим место в действительности.
Теоретико-групповая интерпретация принципа заполнения элек-
тронных оболочек в атомах может быть дана и с помощью групп
инвариантности. В этом случае основные состояния атомов всех эле-
ментов образуют базис специального неприводимого представления
некомпактной группы 50(4,2).
Среди новейших исследований выделяется изящная работа [54], в
которой использованы теоретико-групповые методы и методы алгеб-
ры Ли, играющие, как известно, важную роль в классификации ато-
мов и молекул. В рамках этих методов на основе обобщенного прави-
ла Маделунга для D-размерности и принципа построения, т. е. одно-
временного учета принципа минимума энергии и запрета Паули, со-
здана модель для построения D-размерной периодической системы (в
частности, двумерной, на плоскости) и для репродуцирования порядка
атомных оболочек.
Остановимся подробно на главных результатах этой работы. Ав-
торы [54] разработали алгоритм для конструирования D-размерной
периодической системы и модель для воспроизведения порядка атом-
ных оболочек для D-размерности. Отправным моментом для генери-
рования атомных оболочек служит в этой модели кулоновская система
D-размерности. Атомные оболочки располагаются в порядке, который
соответствует D-размерному правилу Маделунга. Это правило вместе
с принципом построения применено для выведения D-размерной пе-
риодической системы.
Для теоретического объяснения того порядка следования атомных
оболочек, который представляет собой обобщенное правило Маделун-
га, предложена оригинальная модель, основанная на введении специ-
ального гамильтониана, что приводит к разрушению динамической
симметрии SO(D + 1) водородного атома для D-размерности. Всегда
особое внимание уделено случаю с D= 2. В этом случае в соответствии
150
с представлениями однопараметрической алгебры Ли динамическая
симметрия 50(3) должна быть замещена симметрией 50(3) D 50(2).
Тогда для дискретных собственных значений энергии можно получить
уравнение
Е =----------2-^-------------,
/ 1 \2 / с; \
АГ+-) + 3--9 ([V
\ " / \ о J
где
p(Ze2)2
Е° N = n~1'
fi — приведенная масса водородоподобного атома с ядерным зарядом
Ze; е —заряд электрона; h — постоянная Планка; п — главное кванто-
вое число; I — орбитальное квантовое число;
[/]g = q1'1 4- ql~3 4- ... 4- для I 0; q — параметр модели.
Для того чтобы найти порядок следования атомных оболочек n, I в
соответствии с вышеприведенным уравнением для Е, достаточно рас-
смотреть безразмерную величину
/"/ 1 \ 2 / 5 \
е(п, 0 = 1/ АГ + -) + I 3 —-g ([I],)2, / = 0,1,..-Л,
V \ " / \ о /
которая дает энергетическое положение каждой атомной оболочки п I.
В результате оптимизации этого уравнения для q = 0,8 было найдено,
что энергии Е воспроизводят с умеренной точностью порядок следо-
вания
(Is) < (2s) < (2р) < (3s) < (Зр) < (4s) < (3d) < (4р) < (5s) <
< (4d) < (5р) < (6s) < (4/) < (5d) < (6p) < (7s) < (5/) < (6d) < (7p).
Таким образом, обсуждаемая модель согласуется с обобщенным
правилом п 4- I для двумерного случая, в частности, даваемая этой
моделью последовательность
e(ls) = 0,5 < e(2s) = 1,5 < е(2р) = 2,0 < e(3s) = 2,5 <
< е(3р) = 2,8 < б(4з) = 3,5 < e(3d) = 3,6 < е(4р) = 3,7
полностью аналогична последовательности, найденной из хартри-
фоковских вычислений с использованием базиса гауссиана и при опи-
сании кулоновских взаимодействий в форме 1/г [55].
Итак, традиционная квантовомеханическая интерпретация пери-
одической системы химических элементов на основе орбитальной
п/-оболочечной модели атома в определенной степени противоречи-
ва. Для детального анализа «принципа построения» конструктивны
формально-математические и особенно теоретико-групповые модели
151
периодичности на уровне свободных атомов. И в настоящее время пе-
риодичность правомернее всего связывать непосредственно с симмет-
рией атома, установленной академиком В. А. Фоком для атома водо-
рода.
В своей работе [56] он поставил главный для учения о принципе пе-
риодичности и теории периодической системы вопрос: вмещаются ли
свойства атомов и их составных частей в рамки чисто пространствен-
ных представлений или необходимо расширить понятия пространства
и пространственной симметрии, чтобы вместить присущие атомам и
их составным частям степени свободы?
В своей теоретической интерпретации Периодического закона и пе-
риодической системы элементов Н. Бор на основе своей же теории
атомных электронных оболочек и с использованием первоначальной
формулировки принципа Паули (как требования присутствия не более
двух электронов на каждой орбите, характеризуемой тремя опреде-
ленными квантовыми числами) ввел понятие замкнутых электронных
оболочек атомов и сопоставил их с периодами менделеевской систе-
мы. Это был первый серьезный успех в теоретической трактовке за-
кономерностей, выражаемых периодической системой элементов. Но,
несмотря на этот значительный успех, задача полного описания пери-
одической системы Менделеева была далека от решения.
При всей глубине и радикальности этих новых идей они еще укла-
дывались в рамки тривиальных пространственных представлений.
Дальнейший важный шаг связан с открытием внутренней, не про-
странственной, степени свободы электрона — спина, представляюще-
го собой, отнюдь, не механическое понятие, и разработкой волновой
механики. В волновой механике принцип Паули имеет более совер-
шенную формулировку и связан со свойствами симметрии волновой
функции. И успехи в теоретической интерпретации периодической си-
стемы в рамках более строгой квантовомеханической теории электрон-
ного строения атомов основаны на непространственном обобщении и
последовательном применении понятия симметрии, само же понятие
симметрии родственно понятию периодичности в том смысле, как его
применял Д. И. Менделеев.
Ответ академика В. А. Фока на им же поставленный вопрос без-
упречен и ясен: «Чисто пространственных степеней свободы электро-
на недостаточно для описания свойств электронной оболочки атома
и нужно выйти за пределы чисто пространственных понятий, чтобы
выразить те законы, которые лежат в основе периодической систе-
мы. Новая степень свободы электрона — его спин — позволяет описать
чуждые классическим представлениям свойства физических систем.
Эта внутренняя степень свободы электрона существенно необходима
для формулировки свойств многоэлектронных систем, а тем самым
152
и для теоретического обоснования периодической системы Менделее-
ва».
Периодичность свойств атомов обусловлена фермионной природой
(полуцелым спином) электронов. Объкты, составленные из бозонов,
изменяют свои свойства достаточно плавно, последовательно, даже
(правда, редко) линейно. Можно полагать, что если бы мир состоял
из бозонных объектов, он был бы скучнее.
Глава 11
НОВЫЕ АТОМЫ И ИХ ПОЛОЖЕНИЕ
В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ
В этом, заключительном, разделе в краткой форме изложены ос-
новные представления о новых атомах — именно о новых атомах, а не о
новых химических элементах. Основные сведения взяты из известной
работы академика В. И. Гольданского [57].
Все атомы, размещенные в строгой и стройной последовательности
в менделеевской системе, характеризуются общностью носителей элек-
трического заряда: сосредоточенный в атомных ядрах положительный
заряд принадлежит протонам (р), носителями отрицательного заряда
в атомах являются электроны (е“).
Во второй половине XX века обнаружены и искусственно получены
так называемые новые атомы, в составе которых протоны и(или) элек-
троны заменены другими одноименно заряженными элементарными
частицами. Число уже сейчас известных элементарных частиц состав-
ляет несколько десятков, и разные их комбинации могут, в принципе,
дать сотни разных новых атомов.
Замена электронов другими отрицательными элементарными ча-
стицами, например, /z~- или 7г~-мезонами, никак не связана с изме-
нением заряда ядра и, стало быть, не приводит к изменению индиви-
дуальности химического элемента. Замена протонов другими положи-
тельными элементарными частицами, например, позитронами е+ или
/1+-мезонами, также не изменяет индивидуальности химического эле-
мента. Такая замена приводит к образованию новых атомов, которые
в химическом смысле являются изотопами по отношению к «незаме-
щенному».
Простейшими примерами новых атомов являются открытый в
1951 г. позитроний (Ps) — атом, состоящий из позитрона и электрона, и
мюоний (Ми), состоящий из /1+-мезона и электрона. Это — легчайшие
«изотопы водорода». Позитроний вообще наилегчайший из всех но-
вых атомов — он в 920 раз легче атома водорода. Максимальное время
жизни позитрония в триплетном состоянии *Ps (3Si) с параллельными
154
спинами электрона и позитрона и в отсутствие каких бы то ни было
химических взаимодействий (т. е. собственное время жизни) не пре-
вышает 1,4-10“7 с. Собственное время жизни синглетного позитрония
sPs (%), в котором спины электрона и позитрона антипараллельны,
еще меньше —всего 1,3-10“10 с. Время жизни мюония определяется
скоростью распада /1+-мезона: > е+ + и + и~(у и и~ — нейтрино
и антинейтрино) и составляет 2,2-10“6с. Из-за столь малого времени
жизни позитрония и мюония их стационарные концентрации в реаль-
ных системах ничтожно малы — всего несколько атомов в кубическом
сантиметре. Ни о каких химических методах наблюдения за этими ато-
мами не может быть и речи. Исследования возможны лишь методами
ядерной физики и физики элементарных частиц. Малость собственно-
го времени жизни позитрония и мюония позволяют использовать это
время в качестве своеобразного стандарта при изучении быстрых хи-
мических реакций с участием этих водородоподобных атомов, в част-
ности изучать роль диффузионных и квантовых факторов в кинетике
таких реакций, роль туннельного эффекта в химической кинетике.
В свойствах водорода, позитрония («сверхсверхлегкого изотопа во-
дорода») и мюония много общего, но есть и различия, обусловленные
большой разницей в массах этих атомов. Эти различия приводят к
новым эффектам и определяют совершенно особые свойства молекул,
в которых атом водорода замещен новым атомом — позитронием или
мюонием. Обычная молекула водорода Щ — двухцентровая молекула,
в которой два электрона движутся в поле двух относительно мало-
подвижных протонов. Молекулу же гидрида позитрония PsH можно
рассматривать в действительности как одноцентровую молекулу, в ко-
торой два электрона и позитрон образуют своеобразную электронно-
позитронную оболочку. Здесь протон окружен подвижным облаком
двух электронов и одного позитрона, и молекула PsH напоминает, ско-
рее, трехэлектронный атом лития. Из-за очень малой массы позитрона
по сравнению с массой протона частоты нулевых колебаний в молеку-
лах PsX (и соответственно энергии их диссоциации) должны быть зна-
чительно меньше, чем для аналогичных молекул НХ. Все химические
связи атомов позитрония Ps-X должны быть гораздо слабее аналогич-
ных связей атомов водорода Н-Х, а все реакции замещения атомов во-
дорода атомами позитрония — эндотермическими. Напротив, посколь-
ку ионизационный потенциал атома позитрония (6,8 эВ) ровно вдвое
меньше ионизационного потенциала атома водорода (13,6 эВ), реак-
ции окисления позитрония в газовой фазе энергетически значительно
выгоднее (сопровождаются значительно большим экзоэнергетическим
эффектом, в идеале на 6,8 эВ), чем аналогичные реакции окисления
водорода. Вообще реакции с участием позитронийсодержащих моле-
кул должны быть более экзотермическими (менее эндотермическими)
155
по сравнению с аналогичными реакциями водородсодержащих моле-
кул. Наряду с энергетическими существуют и кинетические различия
в поведении производных позитрония и водорода.
Таким образом, сопоставление свойств нового атома позитрония и
обычного атома водорода, занимающих по величине «ядерного» заря-
да одно и то же место в периодической системе, ярко выявляет разли-
чия, обусловленные именно массой, — в духе первоначального подхода
Менделеева к периодизации физико-химических свойств.
При взаимодействии //“-мезонов с веществом происходит их за-
хват. В конечном счете //“-мезоны захватываются имеющимися в ве-
ществе атомами на свои (атомные) орбитали, в результате образуются
мезоатомы. Первоначально захват происходит на атомные орбитали
с большими главными квантовыми числами (п = 10 — 15), затем в
ходе каскадных переходов с испусканием //-мезорентгеновского излу-
чения //“-мезон «спускается» на внутренние атомные уровни вплоть
до К(1з)-слоя электронной оболочки данного атома, т. е. до основного
энергетического состояния — состояния с минимальной полной энер-
гией. Радиус каждой мезоорбитали примерно в 200 раз (соотношение
масс //“-мезона и электрона е~) меньше радиуса соответствующей
электронной орбитали. Например, в атоме мезоводорода Н//“ (или,
точнее, р//“), в котором единственный электрон атома водорода заме-
щен //“-мезоном, равен 2,5-10“11 см. При столь малом размере атома
мезоводорода, близком уже к ядерным размерам, этот атом ведет себя
как единая электронейтральная частица, может чуть ли не вплотную
сближаться с разными атомами (Y) и взаимодействовать с ними без
значительных активационных барьеров, которые, как правило, значи-
тельны для обычного атома водорода Н (ре“) из-за большого электро-
статического отталкивания протона атома Н от ядер тех атомов (Y),
с которыми взаимодействует атом Н. В частности, мезоводород может
служить квазикатализатором холодного, мезоядерного, синтеза гелия
из водорода.
Медленные 7г“-мезоны также захватываются атомами некоторых
молекул, прежде всего водородсодержащих. Закономерности такого
захвата описываются так называемой моделью больших мезомолекул.
Согласно этой модели захват мезонов происходит вначале не на ме-
зоатомные. а на обобщенные мезомолекулярные уровни, квантовые
числа которых столь велики, что отвечают обычным межъядерным
расеIояииям (~ 10-8 см). Вероятности захвата 7г"-мезонов разными
атомами одной и той же молекулы различны. Эти вероятности
количественно описываются Z-законом Ферми Геллера
= ntZ,
' ^niZC
156
где ni — «концентрация» г-го атома в молекуле; Zt — его атомный но-
мер.
Для общехимических концепций еще важнее то, что обсуждаемые
вероятности различны для одних и тех же атомов в разных молекулах.
Так, существенно различны вероятности захвата 7г~-мезонов атомами
/И
водорода в связях С—Н в молекуле этилена H2C=Cf ив молекуле
\н
ацетилена НС=С—Н, что связано, по-видимому, с различием в энерги-
ях связей С—Н в молекуле этилена и в молекуле ацетилена. Влияние
тонких различий в характеристиках однотипных химических связей на
процессы захвата мезонов открывают большие перспективы использо-
вания мезохимических методов в структурной химии.
Глава 12
ПЕРИОДИЧЕСКАЯ СИСТЕМА
И ВАЛЕНТНОСТЬ АТОМОВ В МОЛЕКУЛАХ
Периодическая система химических элементов в своей канониче-
ской форме характеризует свободные невозбужденные атомы всех эле-
ментов, конкретнее, характеризует свободные невозбужденные атомы
любого химического элемента по его положению в системе. Основой
для этого является квантовомеханическая теория электронного строе-
ния атомов. Но помимо «периодической системы атомов» должны су-
ществовать и «периодические системы молекул». Как в общем случае
перейти от периодической системы атомов к периодической системе
молекул? Несомненно, такой переход осуществим лишь при использо-
вании квантовомеханической теории электронного строения молекул
и, прежде всего, ее главного раздела — теории валентности.
Напомним, что в теории молекулы валентность Уд атома А в моле-
куле определена как сумма кратностей Кдв всех двухцентровых свя-
зей A-В, образуемых в молекуле атомом А со всеми остальными ее
атомами В [27, 41]:
Va = АГав-
Здесь использовано ключевое представление о соотношении крат-
ностей связей и валентности атома, точнее, о распределении валентно-
сти атома по его связям со всеми остальными атомами молекулы. При
этом кратность связи — вещественное число (два атома А и В не свя-
заны, если КАВ = 0), для гомонуклеарных двухатомных молекул она
равна половине разности занятых связывающих и занятых разрых-
ляющих одноэлектронных молекулярных спин-орбиталей и, наконец,
обязательно инвариантна относительно унитарных преобразований ба-
зисных волновых функций (спин-орбиталей) любого атома, входящего
в молекулу. Всему этому удовлетворяет следующее определение крат-
ности двухцентровой связи:
А'аВ = 2 J^aGA SbGB ™аЬ,
158
где А и В — атомные центры; а и b — соответствующие базисные атом-
ные спин-орбитали; wab — обобщенные индексы Вайберга для про-
странства этих спин-орбиталей:
Wab = ^i^j^ai^bi^aj^bj >
cai — а-й коэффициент разложения г-й молекулярной спин-орбитали
по атомным спин-орбиталям левдинского базиса.
Итоговое определение кратности двухцентровой связи A-В в моле-
куле
АГдв = =
= Y}iYjj2(Yja^^caiCaj)(YjbEBcbicbj)
имеет смысл в любых вариантах теории ССП МО ЛКАО (например,
в методе Рутаана), где базисными функциями являются именно лев-
динские орбитали, т. е. «ортогонализованные атомные орбитали».
В ортонормированном базисе валентность атома А в молекуле
VA = 2 (Раа У^а/GA | Раа/|^)
определяется заселенностями и внутриатомными порядками связей
спин-орбиталей
Paa =Е ?СС Cai C*ai И Раа, = Cai ^afi,
т. е. матричными элементами Раа и Раап относящимися лишь к атому
А. Это означает, что валентность атома А в молекуле определяется
валентным состоянием атома А, зависящим от всех остальных атомов
молекулы и изменяющимся от молекулы к молекуле.
В гомонуклеарных и гетеронуклеарных двухатомных молекулах
валентность атомов совпадает, естественно, с кратностью связи.
В формировании валентности атома наиболее важную роль играют
суммарное число его валентных орбиталей и их электронная заселен-
ность. Последнее связано с тем, что в зависимости от электронной засе-
ленности (0, 1 или 2 е~) орбитали, т. е. в зависимости от того, вакантна
она, занята неспаренным электроном или неподеленной электронной
парой, степень ее участия в образовании химических связей, ее вклад
в кратности образуемых атомом двухцентровых связей существенно
меняются.
Как правило, этот вклад максимален для орбитали с неспаренным
электроном (ковалентные взаимодействия 1:1) и существенно меньше
для вакантной орбитали (донорно-акцепторные взаимодействия 0:2)
и особенно для орбитали, занятой неподеленной электронной парой
(дативные взаимодействия 2:0).
159
Интерпретация атомной валентности с позиций представлений об
электронной структуре свободных атомов создана, безусловно, на ос-
нове периодической системы [49, 58, 59]. Рассмотрим в феноменоло-
гическом аспекте валентности атомов химических элементов первых
шести периодов системы, не затрагивая при этом располагающегося
в 6-м периоде семейства лантаноидов по причине сложности валент-
ного поведения электронов, локализованных на кайносимметричных
4/-орбиталях.
В неповторимом во всех отношениях 1-м периоде атомы обоих эле-
ментов (Н и Не) исключительно одновалентны. В каждом из осталь-
ных периодов в ряду соответствующих непереходных элементов мак-
симально высокая валентность присуща срединным элементам. Клас-
сический пример — четырехвалентность атомов В, С, N и понижен-
ная по сравнению со значением 4 валентность атомов всех остальных
непереходных элементов 2-го периода (Li, Be, О, F, Ne). При перехо-
де от срединных непереходных элементов влево валентность атомов
снижается из-за уменьшения суммарного числа валентных электро-
нов, сопутствующего увеличения числа вакантных валентных орбита-
лей и одновременного уменьшения электроноакцепторной способности
атома, сдерживающего образование донорно-акцепторных связей меж-
ду центральным атомом и его партнерами (лигандами) А<— L за счет
вакантных орбиталей атома А. Валентность атомов снижается так-
же при переходе от срединных непереходных элементов вправо из-за
увеличения суммарного числа валентных электронов, сопутствующего
увеличения числа занятых электронными парами валентных атомных
орбиталей и одновременного уменьшения электронодонорной способ-
ности атома, сдерживающего образование дативных связей A—>L за
счет занятых электронными парами орбиталей атома А.
Аналогично в каждом большом периоде в ряду соответствующих
переходных элементов наибольшую валентность проявляют атомы
центральных по своему положению во вставной декаде переходных
металлов. Яркий пример тому атомы Mo, Тс, Ru в 5-м периоде или
W, Re, Os в 6-м периоде. Слева от них — атомы с меньшим суммар-
ным числом валентных электронов, с вакантными валентными орби-
талями, но со слабыми электроноакцепторными свойствами; но лишь
высокие электроноакцепторные свойства необходимы для образования
донорно-акцепторных связей, без которых, в свою очередь, невозмож-
но повышение валентности. Справа от них — атомы с большим числом
валентных электронов, с электронными парами на валентных орбита-
лях, но со слабыми электронодонорными свойствами, тогда как, на-
против, сильная электронодонорная способность необходима для об-
разования дативных связей, без которых не может быть повышения
валентности.
160
Таким образом, главный тезис чрезвычайно прост и важен: валент-
ность атомов химических элементов в системе изменяется периодиче-
ски.
Но валентности атомов в молекуле определяют ее устойчивость и
реакционую способность (в принципе ясно, что с повышением валент-
ности атомов, с увеличением числа и (или) кратности связей в моле-
кулярной системе растет ее стабильность и уменьшается реакционная
способность). Поэтому и эти наиглавнейшие характеристики молекул
изменяются периодически, а вслед за ними если и не все остальные,
то многие другие молекулярные свойства. Все это и служит основой,
позволяющей вести речь о периодических системах молекул.
Система атомов отражает периодическое образование сходных од-
ноцентровых электронных структур при возрастании заряда ядра, уве-
личении суммарного числа электронов, т. е. при увеличении атомного
номера химического элемента в системе. Системы молекул должны,
естественно, отражать периодическое образование сходных по элек-
тронному строению многоцентровых структур. В простейших случаях
двухатомных (гомо- или гетероядерных) молекул для этого можно, по-
видимому, использовать так называемую оболочечную модель и прин-
цип изоэлектронности. Поясним это подробно.
Согласно теории МО делокализованные молекулярные орбитали
составляют валентную оболочку в электронной структуре молекулы,
легко «отделимую» от локализованных внутренних или остовных обо-
лочек той же молекулы (их число равно числу атомов). Теоретические
и экспериментальные критерии такого «отделения» вполне очевидны.
Это многоцентровый характер одноэлектронных волновых функций
для каждого валентного электрона на фоне практически одноцентро-
вого, близкого к сферическому, распределения зарядовой плотности
всех остовных электронов. Это и критически большие разности энер-
гий валентных электронов, с одной стороны, и остовных электронов —
с другой, о чем надежно свидетельствуют данные высокоэнергетиче-
ских (рентгеновских эмиссионных и рентгенофотоэлектронных) спек-
тров.
Роль каждой делокализованной МО в валентной оболочке регули-
руется распределением всех МО по трем типам: связывающие, несвя-
зывающие (неподеленные электронные пары, вакансии) и разрыхляю-
щие.
Теория МО указывает на существование молекул с принципиаль-
но близкими параметрами электронной структуры. Такие молекулы
содержат одно и то же суммарное число валентных электронов и по-
этому называются изоэлектронными.
Принципиальное сходство электронного строения, в свою очередь,
обеспечивает и большее сходство (часто даже количественную бли-
161
зость) многих свойств изоэлектронных молекул, включая их энерге-
тическую устойчивость и реакционную способность. В этом главное
содержание принципа изоэлектронности, который являет собой одну
из важнейших концепций теоретической химии, непосредственно отра-
жает парадигму молекулярных орбиталей и используется, как прави-
ло, в рамках валентного приближения теории электронного строения
молекул.
В своей общей форме принцип изоэлектронности [41] гласит: изо-
электронные молекулы с одинаковой геометрией обладают сходны-
ми молекулярными орбиталями. Основу принципа изоэлектронности
нетрудно уяснить при помощи так называемых корреляционных диа-
грамм.
Корреляционные диаграммы отражают связь молекулярных орби-
талей молекулы, с одной стороны, с атомными орбиталями, в которые
переходят рассматриваемые МО при бесконечном увеличении межъ-
ядерных расстояний (т. е. при образовании предельной конфигурации
разъединенных атомов), а с другой —с атомными орбиталями, в кото-
рые переходят рассматриваемые МО при уменьшении межъядерных
расстояний до нуля (т. е. при слиянии ядер молекулы с образованием
другой предельной конфигурации — объединенного атома). Эти диа-
граммы отвечают правилу непересечения Неймана — Вигнера [60, 61],
согласно которому «энергетические кривые МО с одинаковой симмет-
рией не пересекаются» и даже взаимно отталкиваются. Именно с по-
мощью этого правила можно коррелировать энергии орбиталей в двух
предельных случаях — объединенного атома и изолированных атомов.
Корреляционные диаграммы, показывающие изменение энергий мо-
лекулярных орбиталей молекулы с изменением межъядерных рассто-
яний, исключительно важны и, по словам Дж. Ван-Флека, «вполне до-
стойны находиться рядом с периодической системой элементов Менде-
леева. Точно так же как последняя позволяет понять строение атомов,
так эти диаграммы позволяют понять строение молекул».
Корреляционные диаграммы для гомо- и гетероядерных молекул
несколько различны, но между ними существует единство, отвечающее
принципу изоэлектронности.
Рассмотрим в качестве примера молекулы N2 и СО: представим се-
бе, что N2 превращается в СО в результате постепенного «удаления»
единичного положительного заряда от одного ядра азота и «добав-
ления» его ко второму ядру азота. Если это «проделать» достаточно
постепенно, то можно ожидать, что орбитали молекулы N2 постепен-
но, без резких изменений (такого рода изменения называют адиаба-
тическими) превратятся в орбитали молекулы СО. Орбитали обеих
молекул коррелируют друг с другом. При одинаковой электронной
заселенности таких однотипных МО в двух изоструктурных молеку-
162
лярных системах эти молекулы должны обладать сходными наборами
физико-химических характеристик. В этом и состоит смысл принципа
изоэл ектронности.
Вышесказанное поясняет, почему концепция изоэлектронности,
утверждающая близость свойств молекул со сходными ядерными кон-
фигурациями и одинаковым числом электронов и не требующая уче-
та атомного происхождения валентных электронов в молекуле, может
быть положена в основу построения периодических систем молекул.
ВМЕСТО ЗАКЛЮЧЕНИЯ
Периодический закон — квинтэссенция химии, основа всей хими-
ческой систематики. Отражающая Закон периодическая система хи-
мических элементов открывает в отношении химической систематики
практически неограниченные возможности.
Единство фундаментальных принципов электроноядерной струк-
туры соединений непереходных элементов (AL^) и соединений пере-
ходных металлов (ML^) приводит к общности их физико-химических
и химико-физических свойств и, как следствие, однотипным прояв-
лениям Периодического закона как в химии непереходных элементов,
так и в химии переходных металлов. Это прежде всего особенности хи-
мии Is-, 2р-, 3d- и 4/-элементов, являющихся родоначальниками своих
(главных или дополнительных) подгрупп. Далее — схожесть свойств
их последователей, например, Зр-, 4р-, 5р-, бр-элементов, как антите-
за 2р-кайносимметрии или тяжелых (4d, 5d) переходных металлов как
антитеза Зс?-кайносимметрии и одновременное нарастание обсуждае-
мой схожести свойств элементов главных и дополнительных подгрупп
в периодах по мере увеличения номера периода.
Несмотря на известные различия в проявлении принципа периодич-
ности в М-химии и A-химии (правило четности, вторичная периодич-
ность, биогенность, кластерообразование и т.д.), принцип периодич-
ности, безусловно, един. И в химии переходных металлов, и в химии
непереходных элементов на основном фоне сходства химического об-
лика элементов в пределах одной и той же (главной или дополнитель-
ной) подгруппы со всей отчетливостью проявляется сходство свойств
М- или А- элементов одного и того же периода.
Все это составляет основу Т-образной (в рамках периодической си-
стемы химических элементов) интерпретации принципа периодично-
сти и в М-, и в А-химии.
Учение о периодической системе — важнейший раздел химической
науки и химической дисциплины. Недаром десять Нобелевских пре-
мий по химии (из 94 за все время со дня учреждения премии за пер-
востепенные открытия во всех областях химии) присуждены авторам
исследований именно в этой области.
164
Результаты обсуждения современных подходов к количественной
интерпретации структуры периодической системы химических элемен-
тов несколько пессимистичны. Они показывают, что создание адекват-
ной модели периодичности в мире химических элементов — намного
более сложная, более трудная задача, чем это представлялось первона-
чально. До конца она, естественно, не решена, и наиболее перспектив-
ной для дальнейшего продвижения к окончательному решению пред-
ставляется теоретико-групповая интерпретация принципа периодич-
ности. Эту интерпретацию следует признать дающей наиболее глубо-
кое понимание принципа периодичности, поскольку она сопоставляет
периодичность с обобщенной (динамической) симметрией.
Вышесказанное позволяет предвидеть, что дальнейшее развитие
теории периодической системы будет происходить по двум направ-
лениям: все более точные и полные квантовомеханические расчеты
электронной структуры свободных атомов и теоретико-групповые под-
ходы; вероятно, и формально-математические модели также получат
определенную теоретико-групповую интерпретацию. При этом совре-
менная формулировка принципа периодичности («свойства атомов,
простых веществ, а также свойства и формы химических соеди-
нений находятся в периодической зависимости от зарядов атом-
ных ядер») постоянно требует строгого выяснения того, о каком ти-
пе свойств — коллективных, одноэлектронных или вообще иных — и о
каком топологическом типе соединений идет речь.
Стократно повторены слова Д. И. Менделеева о том, что будущее
Периодическому закону не грозит разрушением, а только развитие и
надстройку обещает. Но развитие теории этого фундаментального за-
кона Природы должно непременно сопровождаться четким установле-
нием области ее применимости во избежание превращения этой теории
в формальную схему, что, кстати, отчасти присуще современной тео-
рии периодичности в силу нерешенности ряда фундаментальных про-
блем электронного строения атомов и молекул. Поэтому при изучении
закономерностей изменения свойств всегда необходимо однозначно и
теоретически обоснованно устанавливать, когда действительно наблю-
дается периодичность в изменении молекулярных свойств (т. е. между
изменением молекулярных и атомных свойств существует причинно-
следственная связь), а когда наблюдаемые корреляции носят случай-
ный характер.
И, наконец, еще раз подчеркнем, что теория периодической систе-
мы химических элементов, безусловно, находится в развитии, и наибо-
лее вероятно, что в дальнейшем она будет построена на основе общих
представлений о пространстве и симметрии.
Завершая эту книгу, предназначенную, главным образом, для мо-
лодых естествоиспытателей, авторы не могут не отдать дань безгра-
165
ничного уважения и почитания великому профессору нашего универ-
ситета — автору Периодического закона.
Периодическая система химических элементов — бессмертная за-
слуга Д. И. Менделеева. Гениальная работа в науке характеризуется
двумя непременными чертами. Во-первых, в ней проповедуется о боль-
шем, чем известно в данное время; во-вторых, ее положения плодо-
творно развиваются в направлениях, которые нельзя было предвидеть.
Оба этих признака в полной мере присущи открытию Д. И. Менде-
леева — принципу периодичности. Периодическая систематика могла
быть разработана лишь таким титаном, который был в состоянии пре-
одолеть противоречия, связанные не только с недостатком и неточно-
стью имевшихся данных, но и — главное — с несовершенством логики,
который сумел бы выяснить причины отклонений от установленных
им закономерностей и внести необходимые уточнения. Все это блестя-
ще сделал Д. И. Менделеев.
На X юбилейном международном Менделеевском съезде, состояв-
шемся в Ленинграде в сентябре 1969 г. и посвященном столетию Пе-
риодического закона химических элементов, автор актиноидной ги-
потезы Нобелевский лауреат Глен Сиборг, синтезировавший со сво-
ими сотрудниками наряду со многими сверхтяжелыми элементами
первый элемент второй сотни — 101-й — и давший ему имя менделее-
вий, сказал: «Наиболее сильное впечатление производит то, что Дмит-
рий Менделеев создал периодическую систему элементов, хотя ему
не были известны такие общепринятые теперь понятия, как ядерная
структура и электронное строение атома, порядковые номера и ва-
лентность. .. Несмотря на огромные достижения науки, за прошедшее
столетие периодическая система Менделеева не претерпела сколько-
нибудь значительных изменений». На том же съезде английский тео-
ретик Чарльз Коулсон так подытожил свой доклад: «Химическая пе-
риодичность — совершенно оригинальное явление, и она не может быть
понята по аналогии с механической периодичностью, как, например,
смена дня и ночи. Периодичность элементов, по существу, является
квантовой периодичностью, лучшим образом она может быть понята
в математической форме. Устанавливая периодическую зависимость
свойств элементов, Менделеев высказал больше, чем он мог знать в то
время. Менделеев опередил свое время на 60 лет. Идея периодической
системы является работой гения. Система не только внесла порядок в
химию, но и сказала значительно больше, чем можно было даже отда-
ленно себе представить. Система привела к новым фундаментальным
открытиям, и сделано это было с величайшим мастерством. Совер-
шенно заслуженно периодическая система элементов навсегда стала
основной частью всемирной химии».
Литература
1. Дмитриев И. С. // ВИЕТ. 2001. №1. С. 31-82.
2. Дмитриев И. С., Трифонов Д. Н. Современные методы количествен-
ной интерпретации периодической системы // Вопросы истории и методо-
логии химии. Вып. 2 / Под ред. Р. Б. Добротина. Л., 1978. С. 24-52.
3. МенделеевД. И. Сочинения: В 25 т. Л.; М., 1949. Т. 15. С. 154.
4. Дмитриев И. С., Семенов С. Г. Квантовая химия, ее прошлое и насто-
ящее. М.: Атомиздат, 1980.
5. Щукарев С. А. Неорганическая химия: В 2 т. М.: Высшая школа, 1970.
Т. 1. 352 с.
6. Щукарев С. А. Там же. 1974. Т. 2.
7. МенделеевД. И. Сочинения: В 25 т. Л.; М., 1947. Т. 8. С. 637-659.
8. Щукарев С. А. // Журн. общей химии. 1977. Т. 47. С. 246-259.
9. Щукарев С. А. // Там же. С. 489-501.
10. Клечковский В. М. Распределение атомных электронов и правило по-
следовательного заполнения (п 4- /)-групп. М.: Наука, 1968.
11. Клечковский В. М. Развитие некоторых теоретических проблем пе-
риодической системы Д. И. Менделеева // Сто лет Периодического закона
химических элементов. М.: Наука, 1971. С. 54-67.
12. ДемковЮ. Н., Островский В. Н. // Журн. эксп. и теор. физики. 1972.
Т. 62. С. 125-132.
13. Ostrovsky V. N. // Foundations of Chemistry. 2001. Vol. 3. N 2. P. 145-
182.
14. Веселов M. Г. Периодический закон и квантовая механика // Сто лет
Периодического закона химических элементов. М.: Наука, 1969. С. 210-221.
15. GoudsmithS. A., Richards Р. I. // Proc. Nat. Acad. Sci. 1964. Vol. 51.
P. 664-671.
16. PyyokkoP. // Chem. Revs. 1988. Vol. 88. P. 563-594.
17. ТарбеевЮ. В., ТруновH. Н., Лобашев А. А. и др. // Журн. эксп. и
теор. физики. 1997. Т. 112. С. 1226-1238.
18. Melrose М., ScerriE. R. // J.Chem. Educ. 1996. Vol. 73. P. 498-503.
19. Филиппов Г. Г., Горбунов А. И. // Журн. физич. химии. 1998. Т. 72.
№7. С. 1334-1336.
20. СабоЗ. Периодическая система и периодические функции // Сто лет
Периодического закона химических элементов. М.: Наука, 1969. С. 244-255.
21. Филиппов Г Г, Горбунов А. И. // Журн. физич. химии. 1993. Т. 67.
№9. С. 1809-1812.
167
22. Филиппов Г. Г., Горбунов А. И. // Докл. РАН. 1993. Т. 329. №4. С. 462-
464.
23. Ландау Л. Д., Лифшиц Е. М. Квантовая механика. М.: Изд-во АН
СССР, 1963. Гл. 10.
24. Трифонов Д. Н. Тяжелые элементы и периодическая система // Пе-
риодический закон и строение атома. М.: Атомиздат, 1971. С. 204-238.
25. Мазалов Л. Н. Рентгеновские спектры и химическая связь. Новоси-
бирск: Наука, 1982. С. 11.
26. Нефедов В. И., ВовнаВ. Н. Электронная структура химических со-
единений. М.: Наука, 1987. С. 347.
27. Корольков Д. В. Электронное строение и свойства соединений непе-
реходных элементов. СПб.: Химия, 1992. С. 312.
28. Korolkov D. V. Electronic structure and properties of non-transition ele-
ment compounds. New York: NOVA Sci. Publ. Inc., 1996.
29. Шусторович E. M. Химическая связь в координационных соединени-
ях. М.: Знание, 1975.
30. Корольков Д. В. Развитие учения о валентности // Под ред. В. И. Куз-
нецова. М.: Химия, 1977. С. 194.
31. KorolkovD. V. Theory of valency in progress // Ed. V. I. Kuznetsov. M.:
Mir, 1980. P. 210.
32. PimentelG. C. // .J.Chem. Phys. 1951. Vol. 19. P. 446.
33. Rundle R. E. // Rec. Chem. Progr. 1962. Vol. 23. P. 195.
34. Пиментел E, СпратлиР. Как квантовая механика объясняет хими-
ческую связь. М.: Мир, 1973. С. 331.
35. Musher J. I. // Angew. Chem. Int. Ed. 1969. Vol. 8. P. 54.
36. Koutecky V. B., Musher J. I. // Theor. Chim. Acta. 1974. Vol. 33. P. 227.
37. Шусторович E. Af. // Коорд. химия. 1976. T. 2. С. 435.
38. Корольков Д. В. Эволюция идей Д. И. Менделеева в современной хи-
мии // Под ред. Б. П. Никольского, Л.С.Лилича. Л.: Наука, 1984. С. 50.
39. ШусторовичЕ. М., Буслаев Ю. А. // Коорд. химия. 1975. Т. 1. С. 740.
40. Корольков Д. В. Принцип периодичности в химии непереходных эле-
ментов. СПб.: Изд-во С.-Петерб. ун-та, 1992.
41. Корольков Д. В., Скоробогатов Г. А. Теоретическая химия. СПб.: Изд-
во С.-Петерб. ун-та, 2001.
42. Jorgensen К. Oxidation numbers and oxidation states. Berlin: Springer
Verlag, 1969. P. 49.
43. Щукарев С. A. // Сто лет Периодического закона химических элемен-
тов. М.: Наука, 1971. С. 40-53.
44. Абаренков И. В., Братцев В. Ф., Тулу б А. В. Начала квантовой хи-
мии. М.: Высшая школа, 1989.
45. Мельников В. П., Дмитриев И. С. Дополнительные виды периодич-
ности в периодической системе Д. И. Менделеева. М.: Наука, 1988.
46. Pyykko Р., Desclaux J.-P. // Accounts Chem. Res. 1979. Vol. 12. N8.
P. 276-281.
47. ФокВ. А. Атом водорода и неевклидова геометрия // Изв. АН СССР.
Сер. 7. 1935. №2. С. 169-179.
168
48. Ostrovsky V. N. // J. Phys. B: Atom, and Mol. Phys. 1981. Vol. 14.
P. 4425-4439.
49. Коулсон Ч. Волновая механика и периодическая система // Сто лет
Периодического закона химических элементов / Под ред. Н. Н. Семенова.
М.: Наука, 1969. С. 201-209.
50. Трифонов Д. Н. О количественной интерпретации периодичности. М.:
Наука, 1975.
51. Трифонов Д. Н. // Химия в России. 2001. №4. С. 12-13.
52. Berondo М., Novaro О. А. // J. Phys. В: Ftom. Molec. Phys. 1973. Vol. 6.
P. 761-769.
53. Barut A. O. Group structure of the periodical table // Rutherford Cen-
tenial Lectures. New Zeeland, 1972.
54. Negadi T., Kibler M. // Int. J. Quant. Chem. 1996. Vol. 57. P. 53-61.
55. PyykkoP., Zhao Y. // Int. J. Quant. Chem. 1991. Vol. 40. P. 527.
56. ФокВ.А. Вмещаются ли химические свойства атомов в рамки чи-
сто пространственных представлений? // Периодический закон и строение
атома. М.: Атомиздат, 1971. С. 107-117.
57. Рольданский В. И. Периодическая система и проблемы ядерной хи-
мии // Сто лет Периодического закона химических элементов. М.: Наука,
1969. С. 158-177.
58. Сыркин Я. К. Периодическая система и проблема валентности // Сто
лет Периодического закона химических элементов. М.: Наука, 1971. С. 85-
102.
59. ЛиннетДж. У. Валентность и система Менделеева // Сто лет Пери-
одического закона химических элементов. М.: Наука, 1971. С. 103-112.
60. Коулсон Ч. Валентность. М.: Мир, 1965.
61. Фудзинага С. Метод молекулярных орбиталей. М.: Мир, 1983.
предметный указатель
Аналогии вертикальные 18, 106
-горизонтальные 19, 106
-диагональные 19, 35
(п 4- /)-группа 38
д-группа 46
Гипотеза Ван-ден-Брука 54
Группа динамической симметрии
146
- неинвариантности 147
- пространственной (геометриче-
ской) симметрии 146
-симметрическая (перестановоч-
ная) 147
Диаграммы корреляционные 161
Диады периодов 17
Закон Мозли 54
- Ферми — Теллера 155
Индекс Вайберга 158
- конфигурационный 40
Конфигурация атома электронная
валентная периодической систе-
мы 42, 56
-правильная 57
Концепция кайносимметрии 99
-связей гипервалентных 87
— ковалентных 87
Кратность двухцентровой связи
157
Кривые потенциальные 140
Множества упорядоченные 12
Молекулы изоэлектронные 160
Мюоний 153
Октадеканоиды 47, 67
Орбитали nd экстравалентные 95
«Острова стабильности» 62
Позитроний 153
Последовательность Ридберга 36
Потенциал Демкова — Островско-
го 49, 50
-ионизации орбитальный 119, 120
- кулоновский 49
- Ленца 49
- Томаса — Ферми 49
Правило Сиджвика 72
- непересечения Неймана — Вигне-
-ра 161
Приближение многоконфигураци-
онные 65
Принцип построения изоэлектрон-
ности 161
Псевдопотенциал 103
Ряд Тюдеса 36
Связи гипервалентные четырех-
электронные трехцентровые 79
- ковалентные двухэлектронные
двухцентровые 79
- орбитально-избыточные двух-
электронные трехцентровые 80
Симметрия орбитальная 32, 33
Система 12
Суперактиноиды 67
Суперматрица 13
Теория полной гибридизации 72,
87
Уровни атома одноэлектронные 37
-- мультиплетные 52
Форма периодической системы
длиннопериодная 15
---короткопериодная 15
---пирамидальная 15
170
Функции периодические 56
Числа магические нейтронов 61
-- протонов 61
Число квантовое орбитальное 22,
33
---граничное значение 143
Электроны кайносимметричные 100
- палинсимметричные 100
Элементы поздние 128
-ранние 128
-сверхтяжелые 62
Энергия разделения 118, 120
Эффект инертной пары 116
ОГЛАВЛЕНИЕ
От авторов. К 110-летию со дня рождения профессора С. А. Щу-
карева....................................................... 3
Введение..................................................... 9
. Глава 1. Периодическая система химических элементов как упо-
рядоченное множество........................................ 12
Глава 2. Квантовомеханическая теория канонической структуры
периодической системы химических элементов............... 18
2.1. Структура системы элементов................... 19
2.2. Периодичность и симметрия..................... 32
2.3. Правило последовательного заполнения (п + /)-
групп........................................... 35
2.4. Исключения из (п + /)-правила................. 51
2.5. О количественной формулировке Периодического за-
кона............................................ 54
2.6. «Векторные» формы периодической системы.... 56
Глава 3. Область сверхтяжелых элементов в периодической си-
стеме....................................................... 61
Глава 4. Два «лагеря» химических элементов................ 71
Глава 5. Менделеевское правило четности и границы его приме-
нимости............................................ 91
Глава 6. Особенности химии элементов-родоначальников s-, р-,
d-, /-семейств................................... 94
Глава 7. Вторичная периодичность.......................... 108
7.1. Вторичная периодичность в химии непереходных
элементов..................................... 111
7.2. Квантовомеханические модели вторичной периодич-
ности в химии непереходных элементов. Теоретико-
групповая интерпретация........................... 119
7.3. Вторичная периодичность в химии переходных ме-
таллов ........................................... 125
Глава 8. Дополнительная периодичность...................... 128
Глава 9. Индивидуальность химических элементов............. 132
9.1. Атомы фтора и кислорода в роли центральных ато-
мов ................................................. —
9.2. Атомы фтора и кислорода в роли лигандов.... 135
172
9.3. Окислительные функции молекулярных фтора и
кислорода..................................... 138
Глава 10. Новые подходы в теории периодической системы.. 142
10.1. Математические модели..................... 143
10.2. Теоретико-групповая интерпретация.......... 146
Глава 11. Новые атомы и их положение в периодической системе 154
Глава 12. Периодическая система и валентность атомов в моле-
кулах ................................................... 158
Вместо заключения........................................ 164
Литература............................................... 167
Предметный указатель..................................... 170
Учебное издание
Татьяна Петровна Кораблева,
Дмитрий Васильевич Корольков
Теория периодической системы
Учебное пособие
Директор Издательства СПбГУ
проф. Р. В. Светлов
Главный редактор Т. Н. Пескова
Художественный редактор Е. И. Егорова
Технический редактор А. В. Борщева
Верстка И. М. Беловой
Лицензия ИД №05679 от 24.08.2001
Подписано в печать 13.05.2005. Формат 60x90 1/i6-
Бумага офсетная. Печать офсетная.
Усл. печ. л. 11 -Ьвкл. 0,25. Заказ №98
Издательство СПбГУ. 199034, С.-Петербург, Университетская наб., 7/9
Тел. (812) 328-96-17; факс (812) 328-44-22
E-mail: editor@unipress.ru
www.unipress.ru
По вопросам реализации обращаться по адресу:
С.-Петербург, 6-я линия В. О., д. 11/21, к. 21
Телефоны: 328-77-63, 325-31-76
E-mail: post@unipress.ru
Типография Издательства СПбГУ.
199061, С.-Петербург, Средний пр., 41