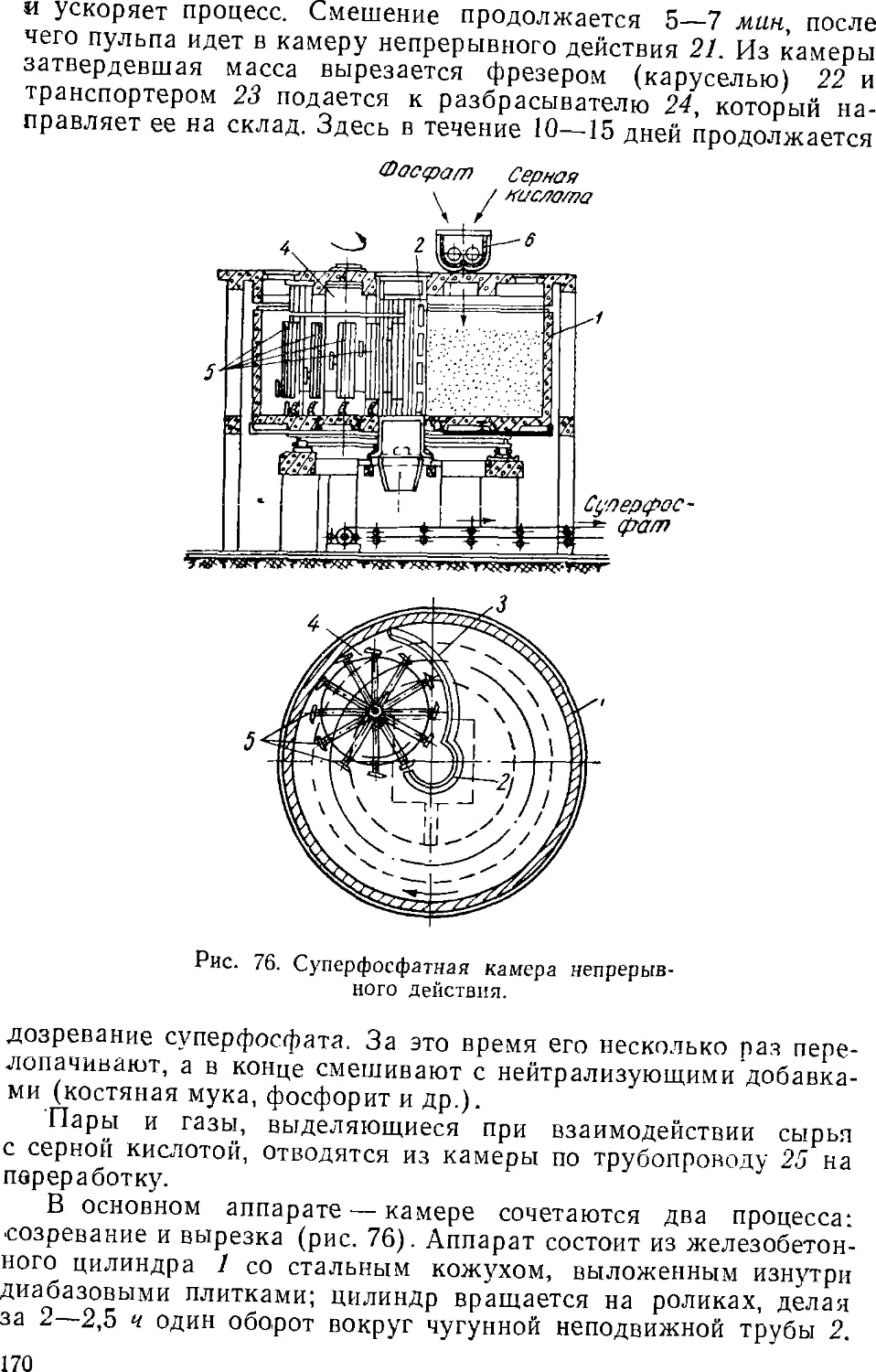
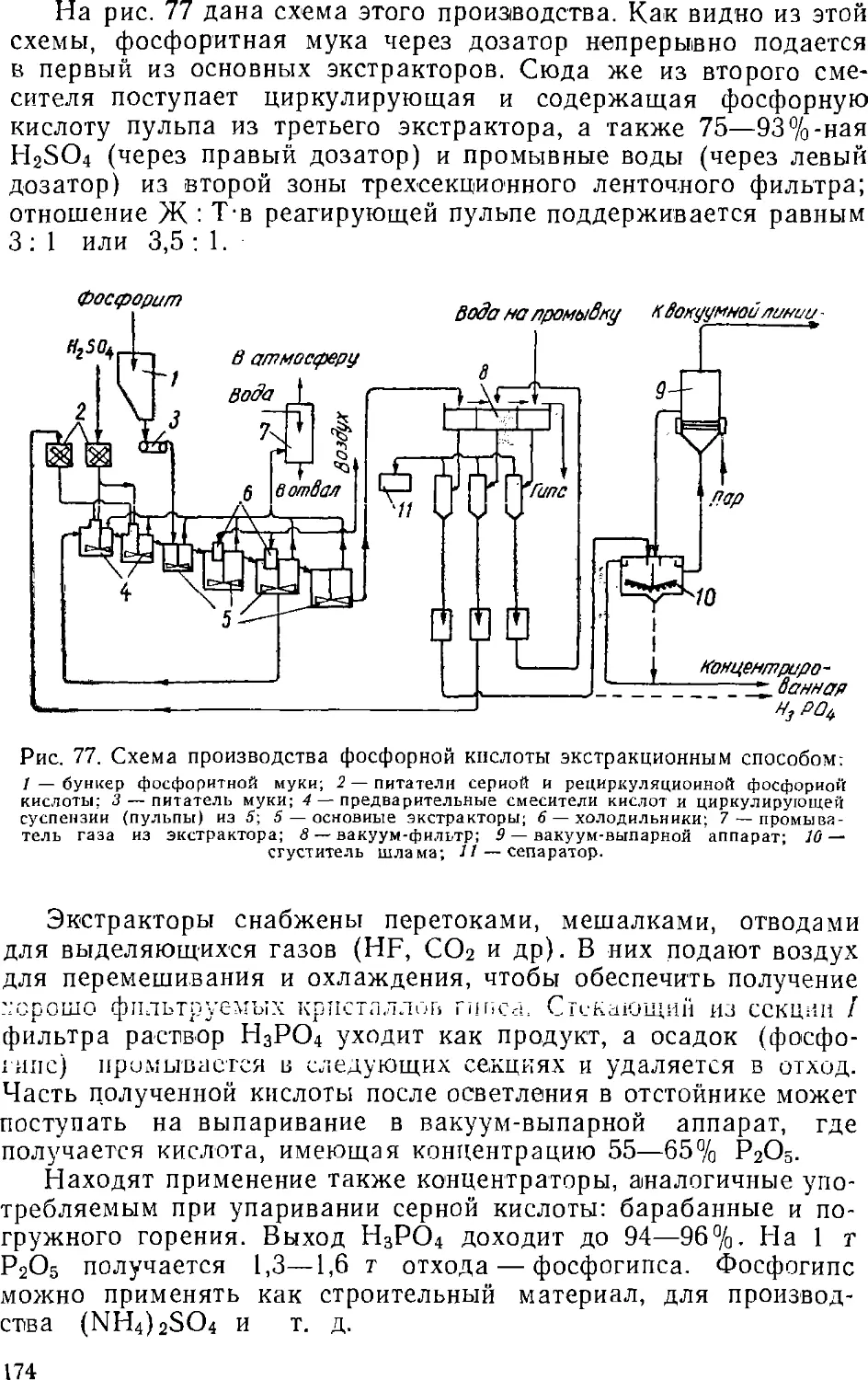
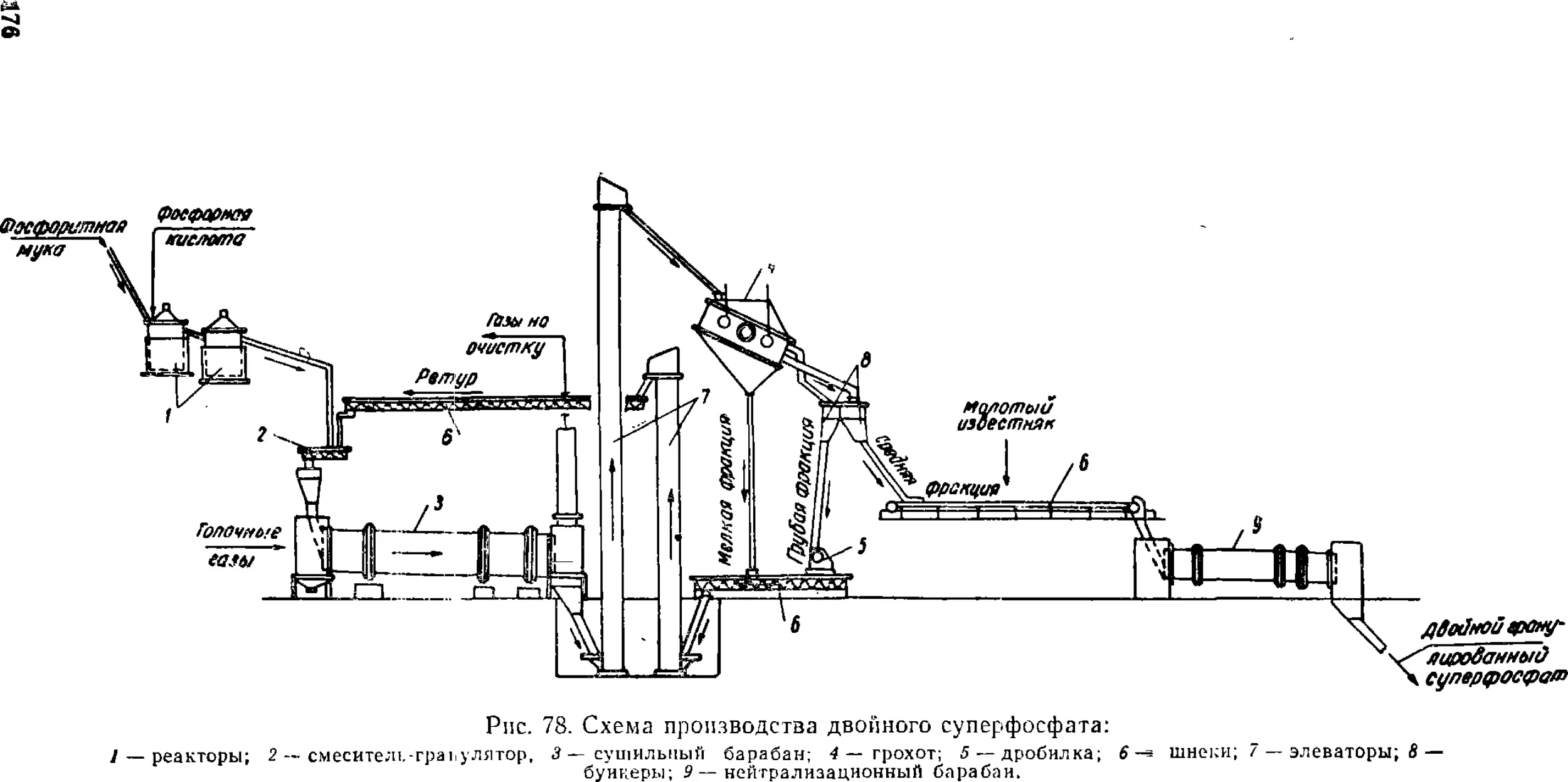
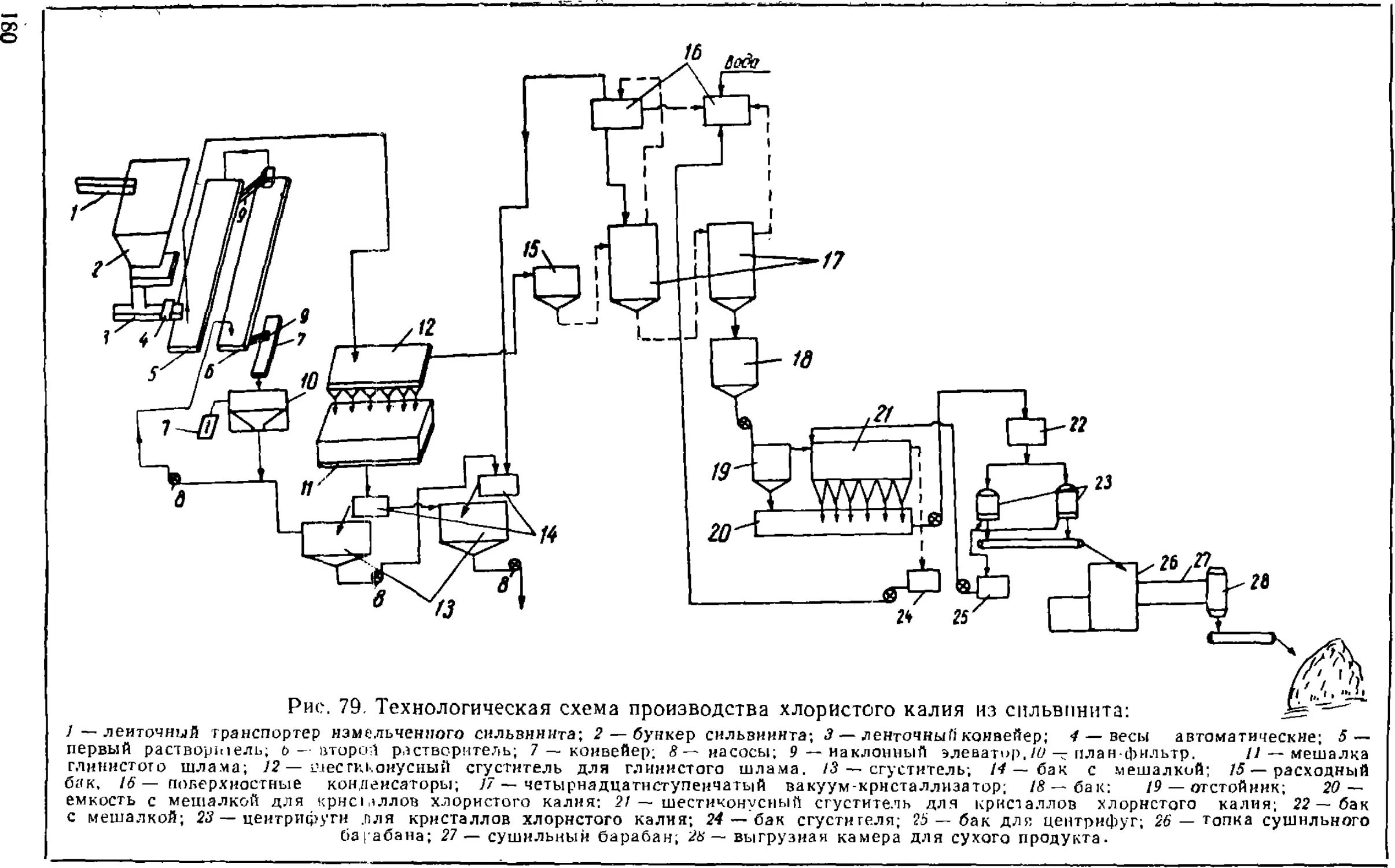
/







































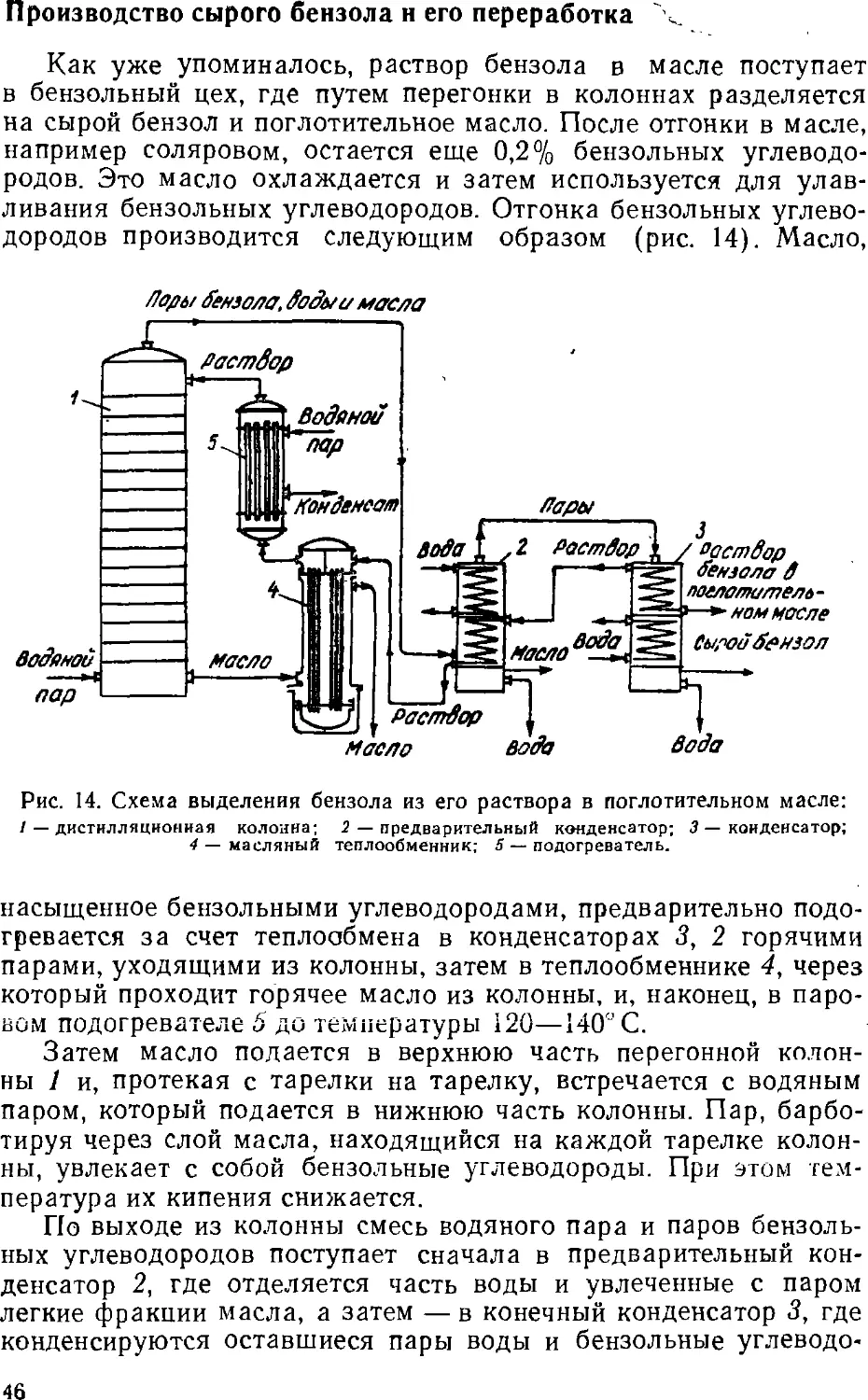


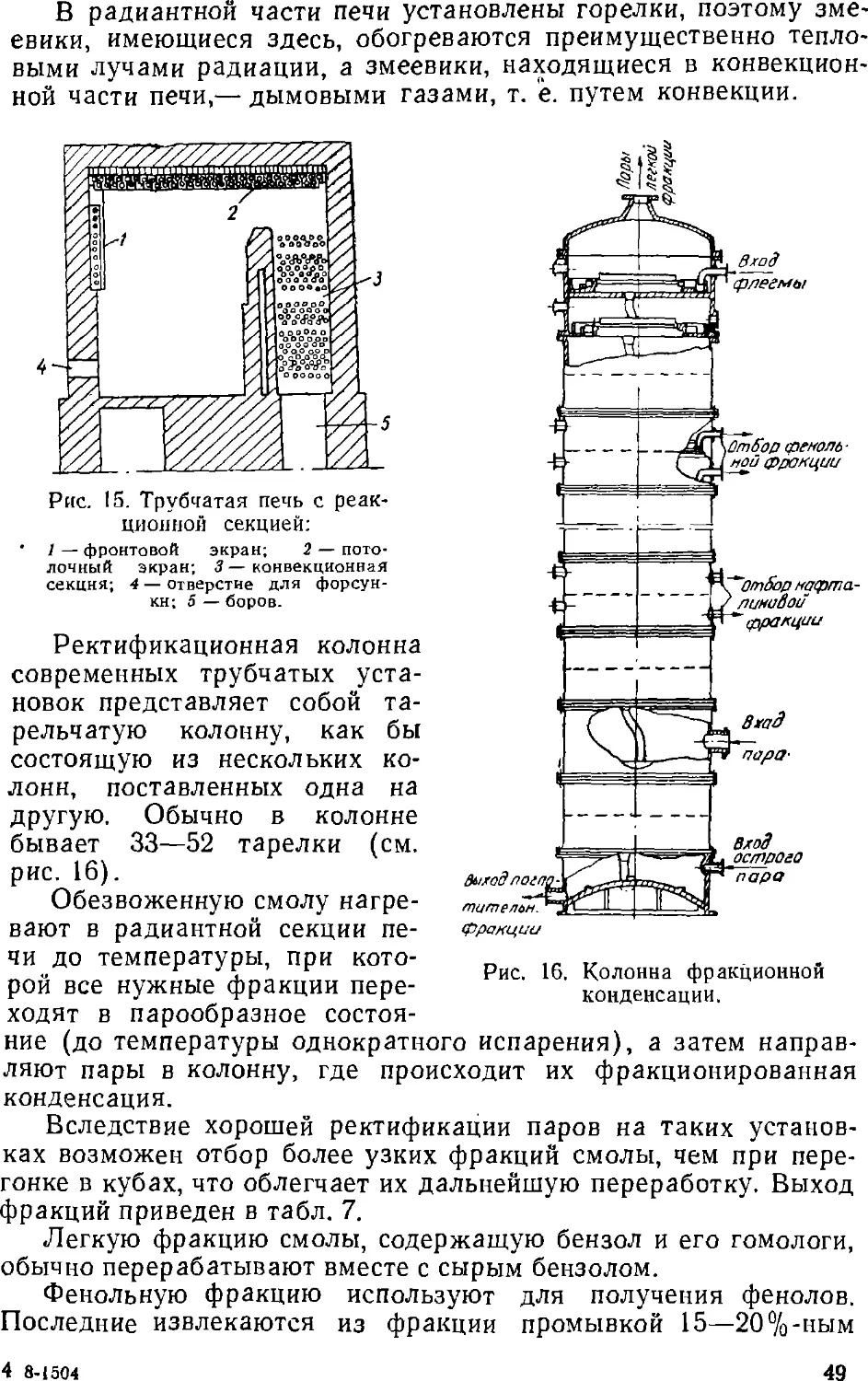




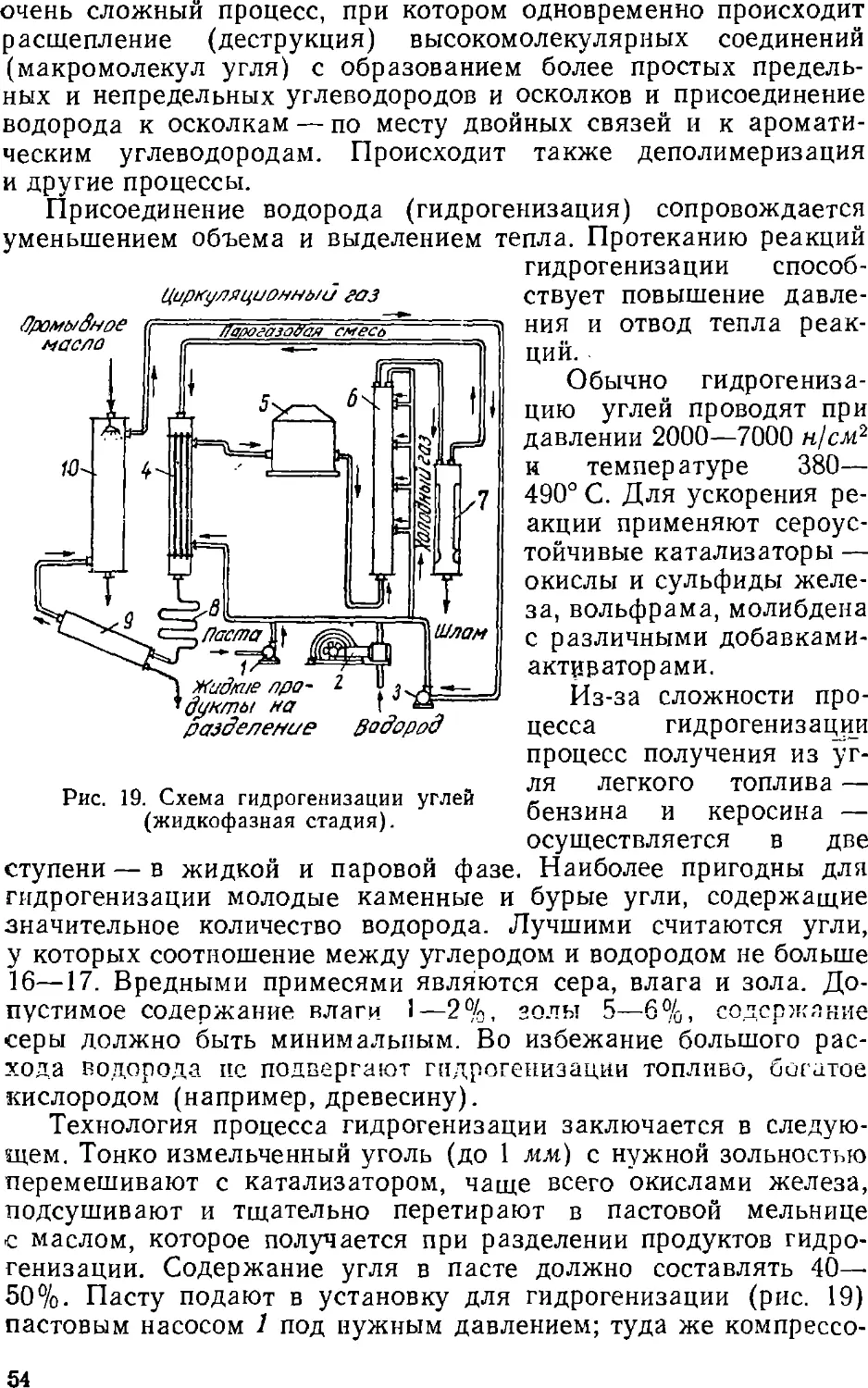


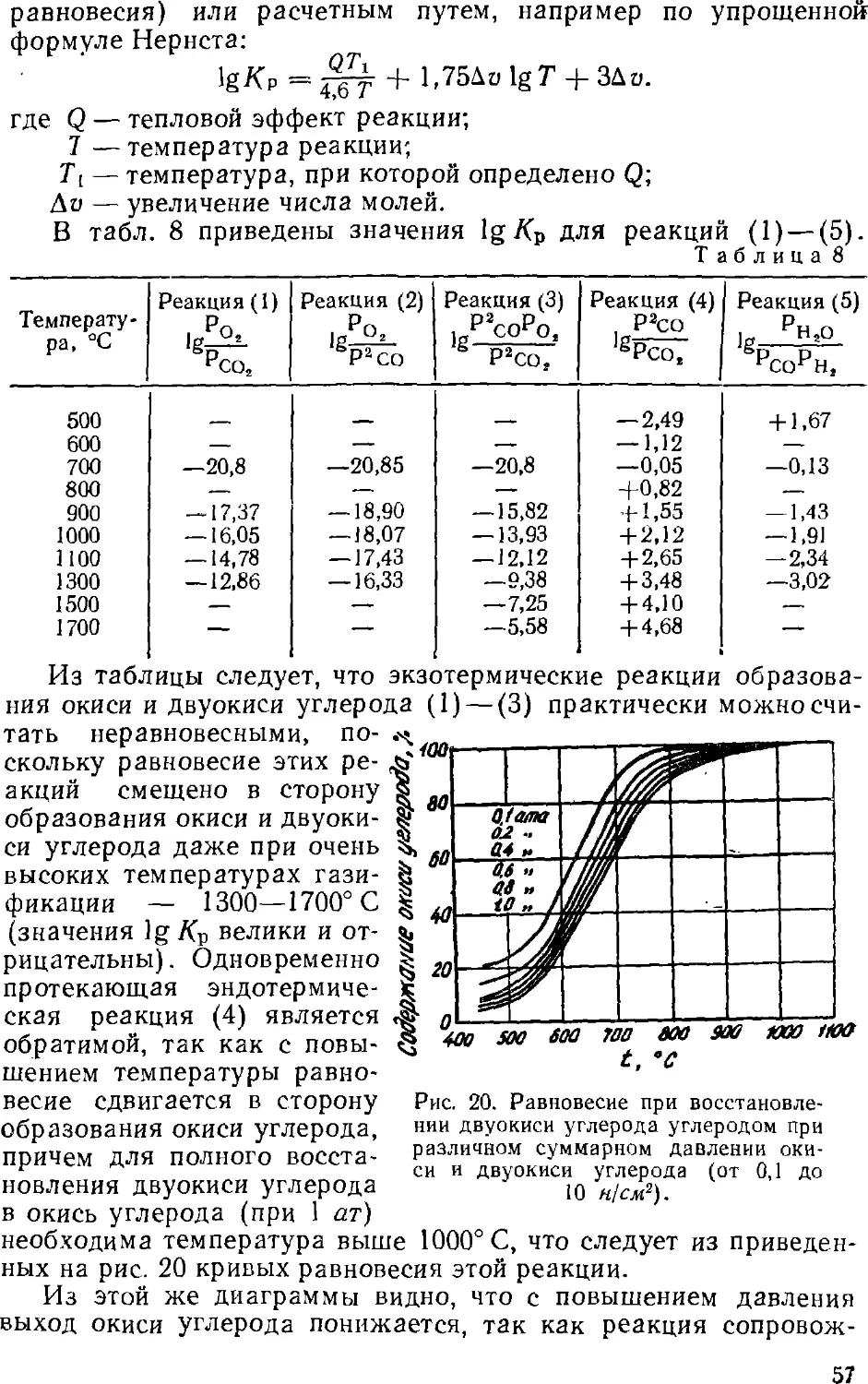




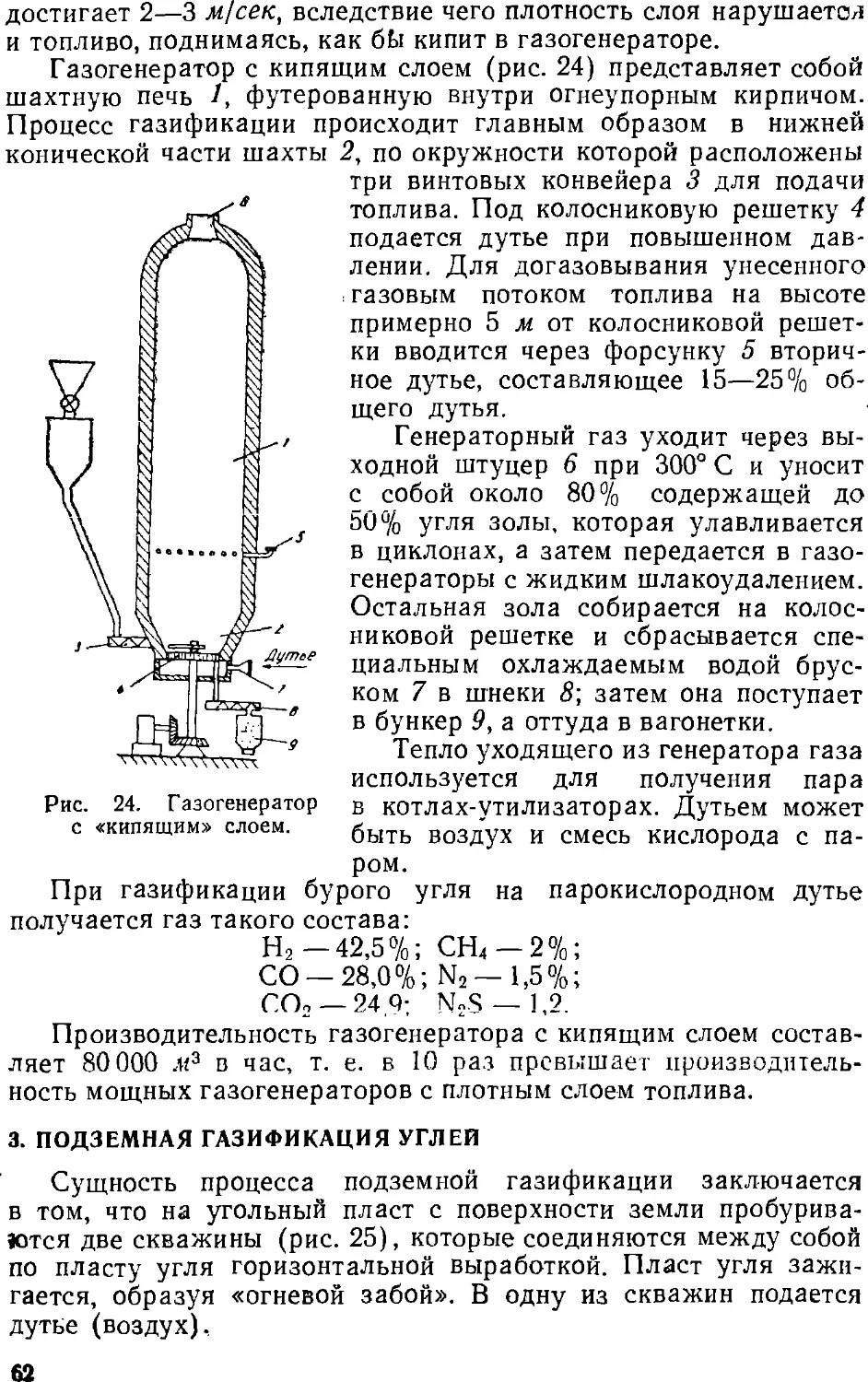



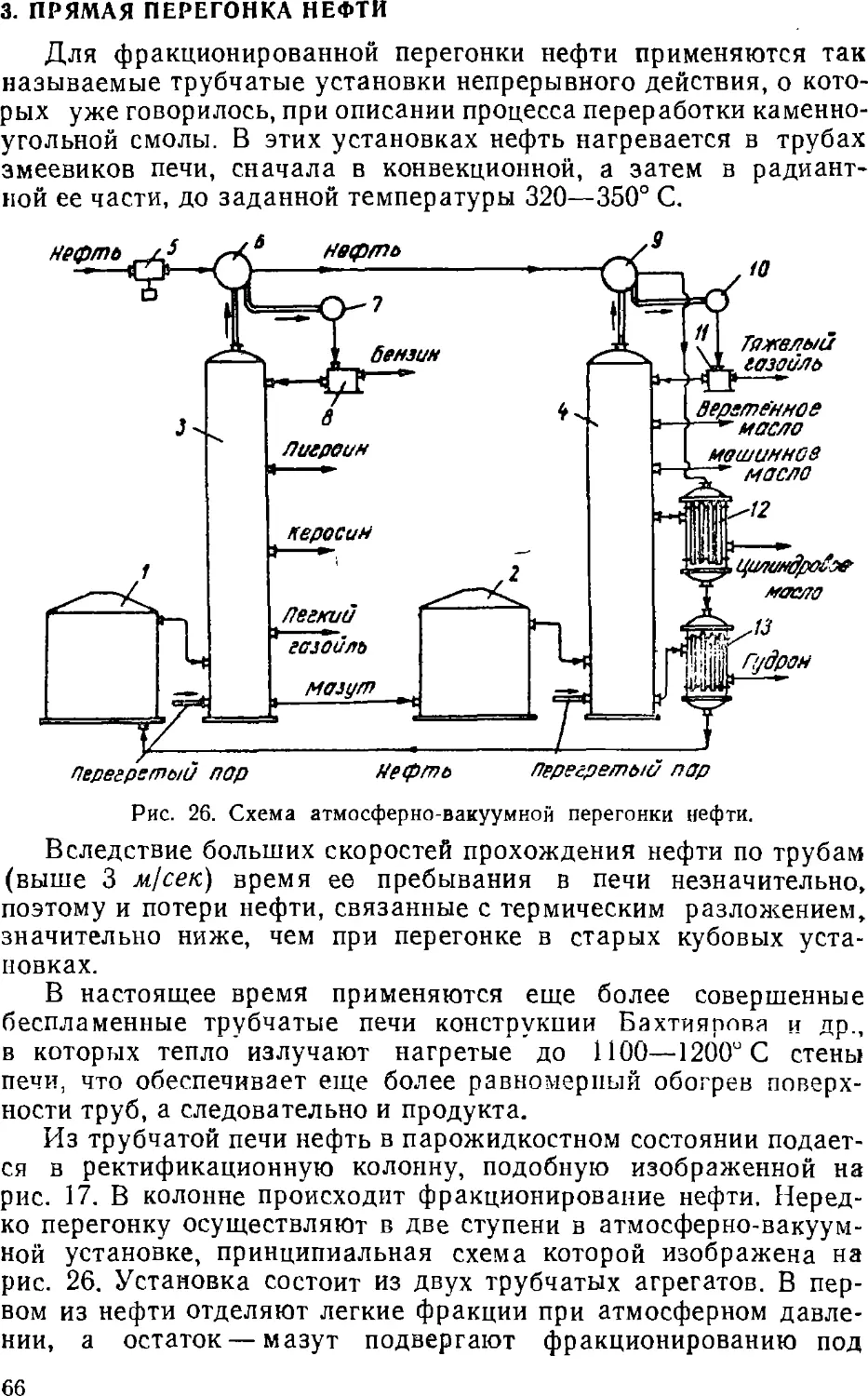
























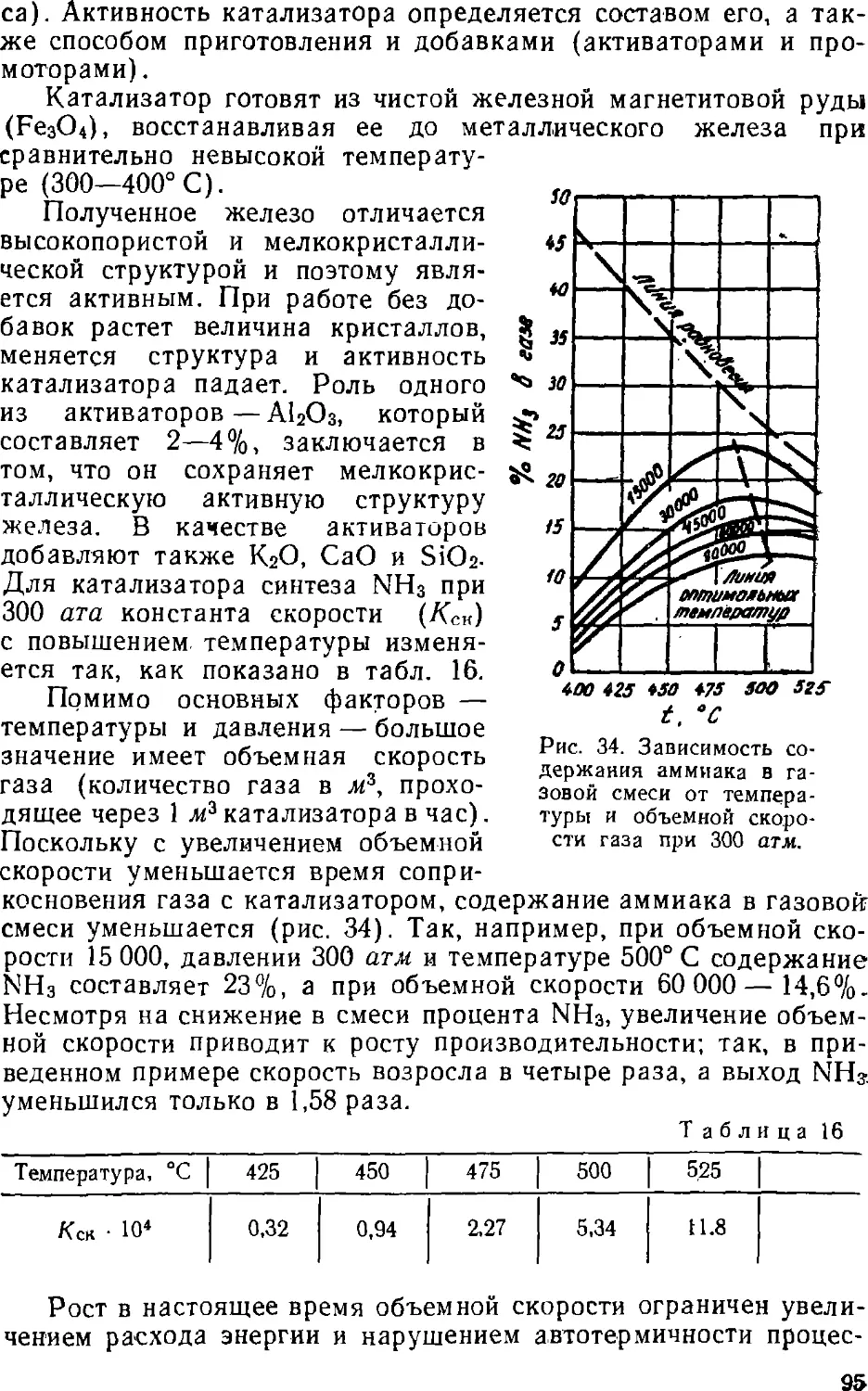


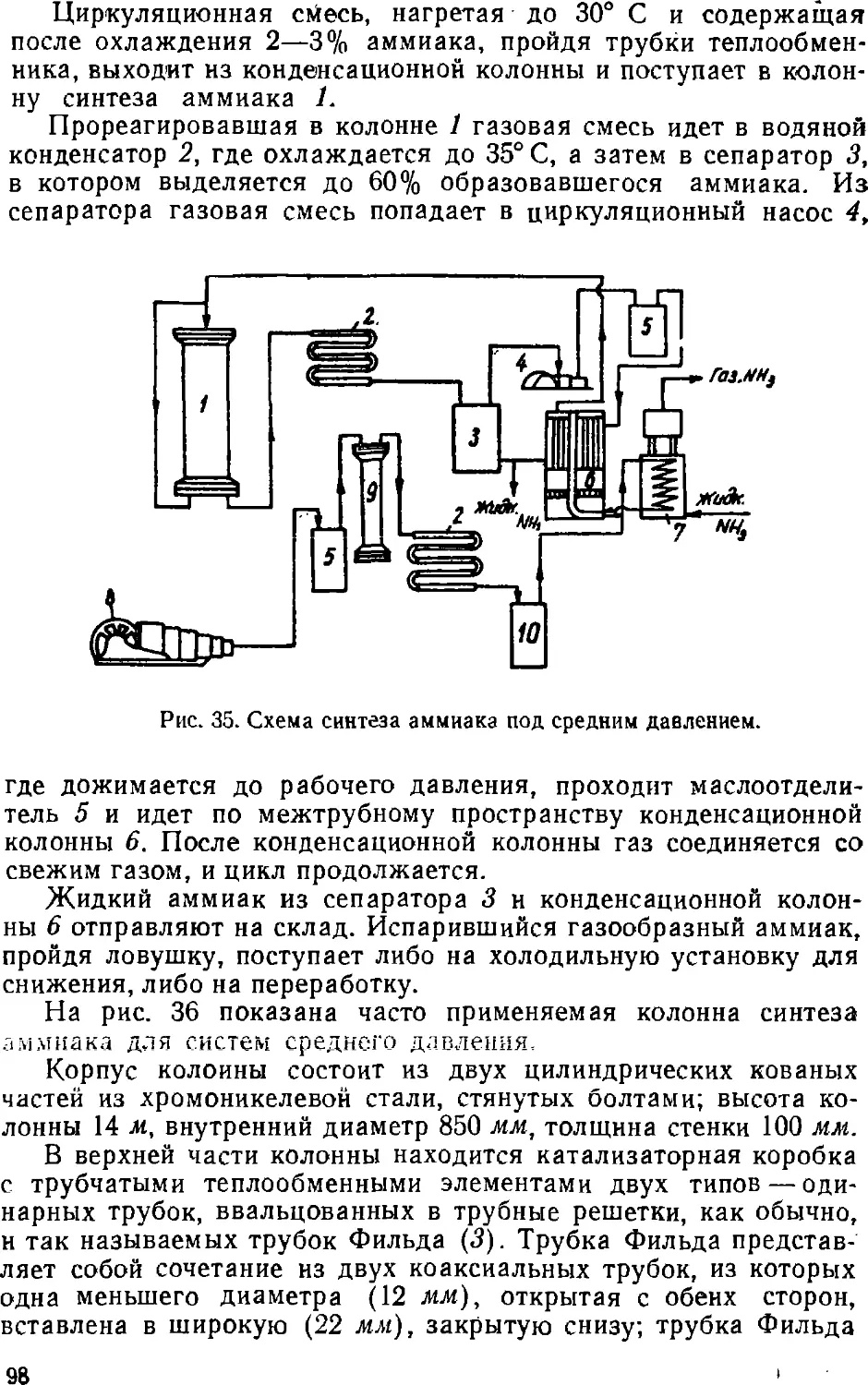













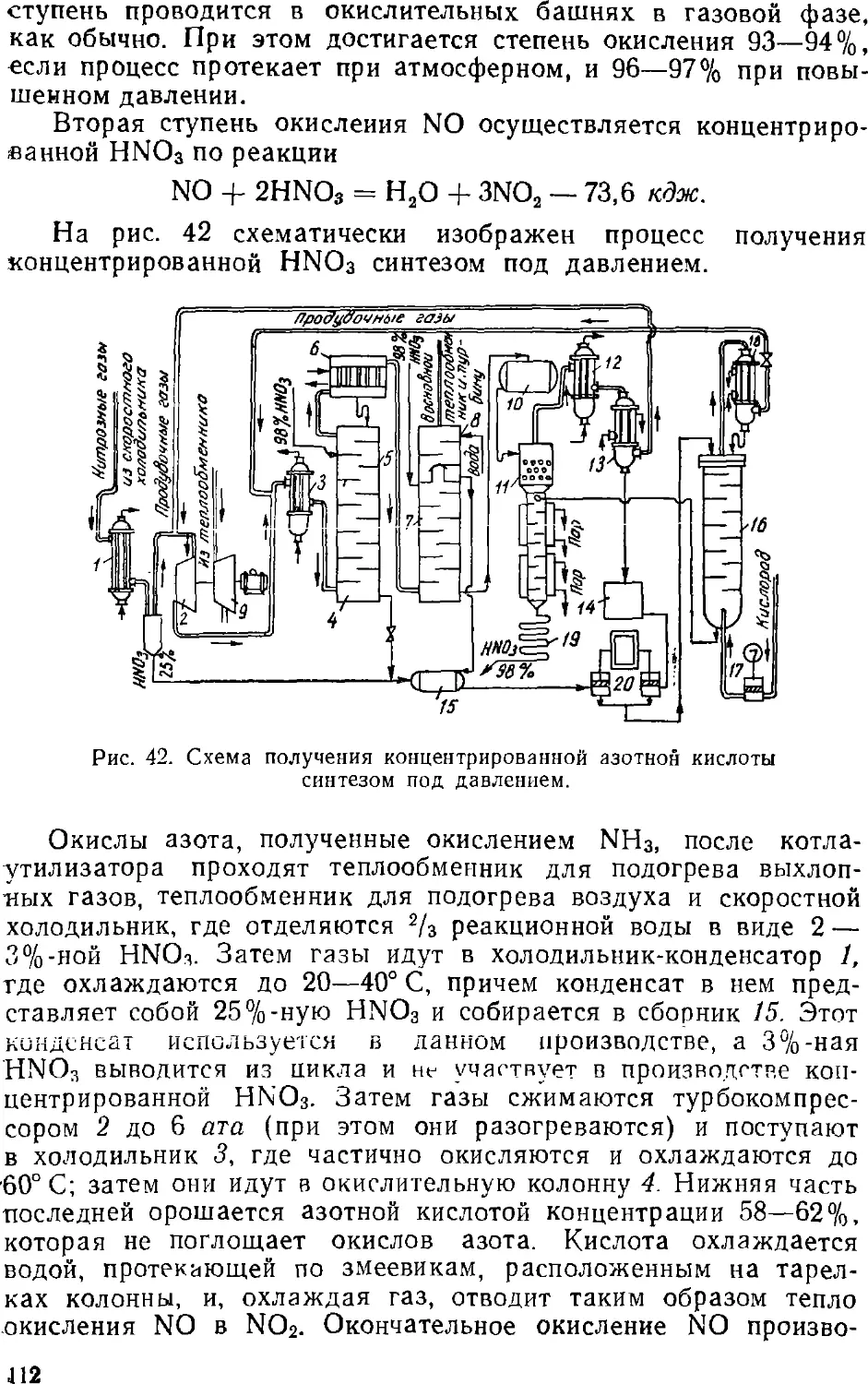
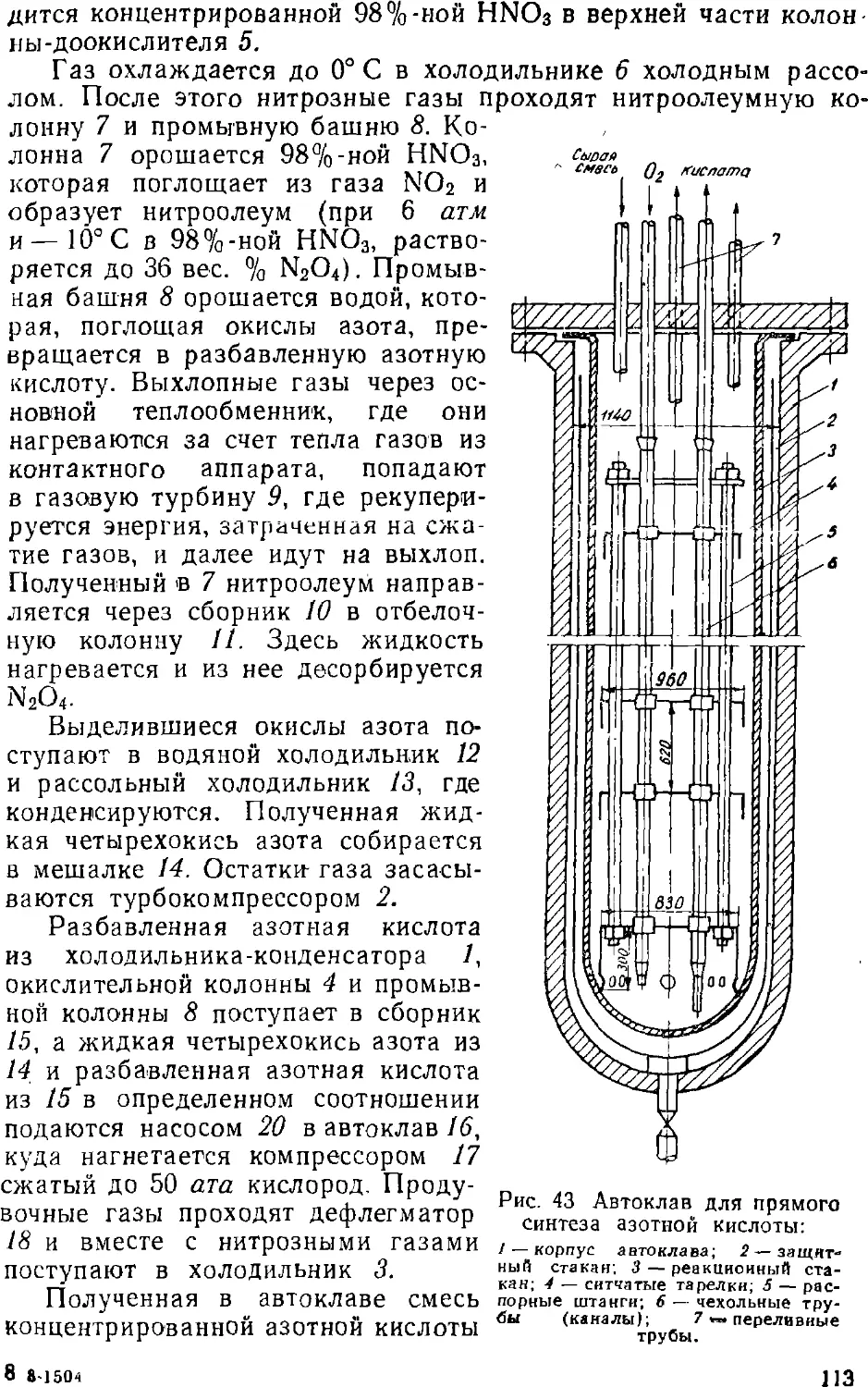

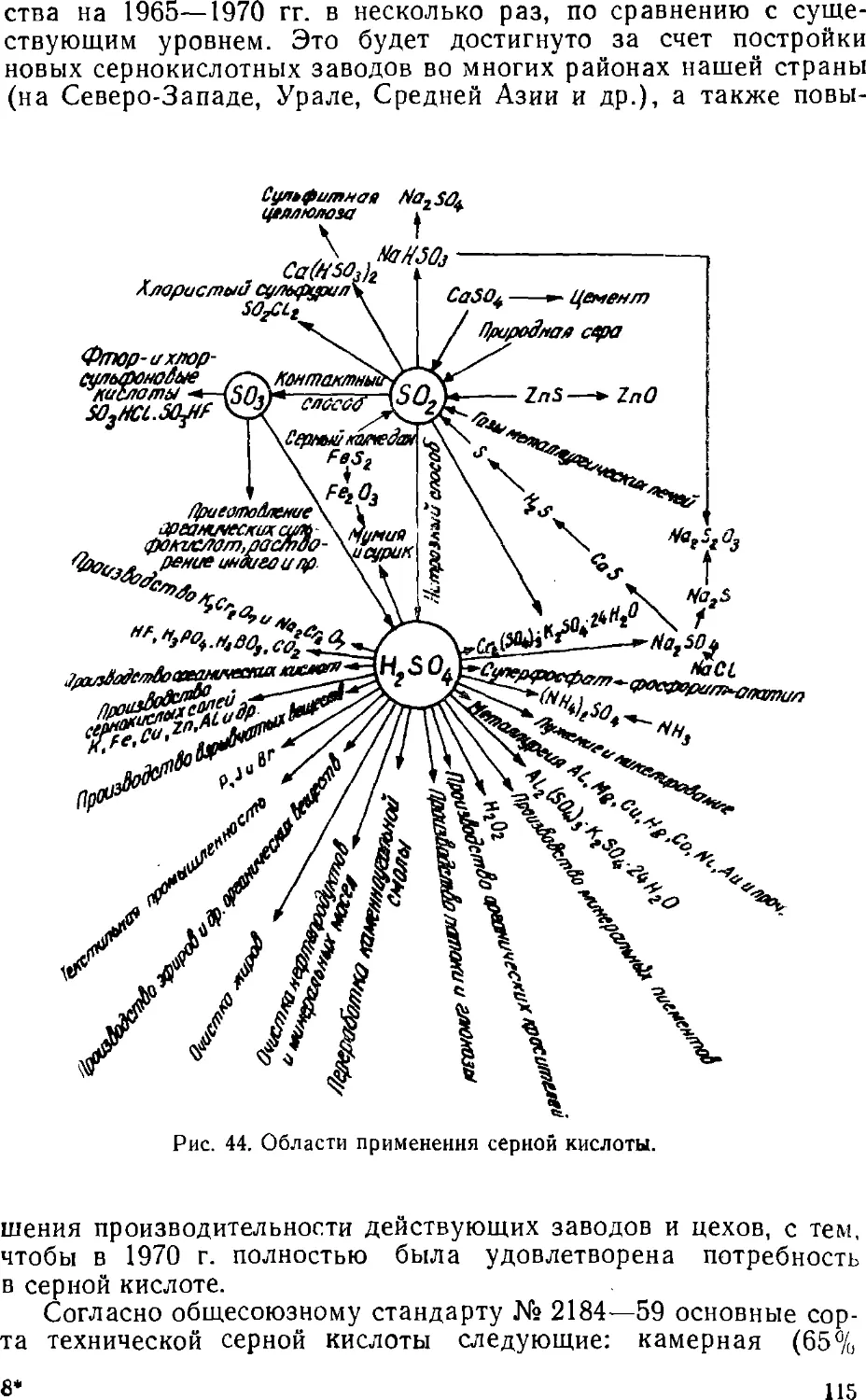





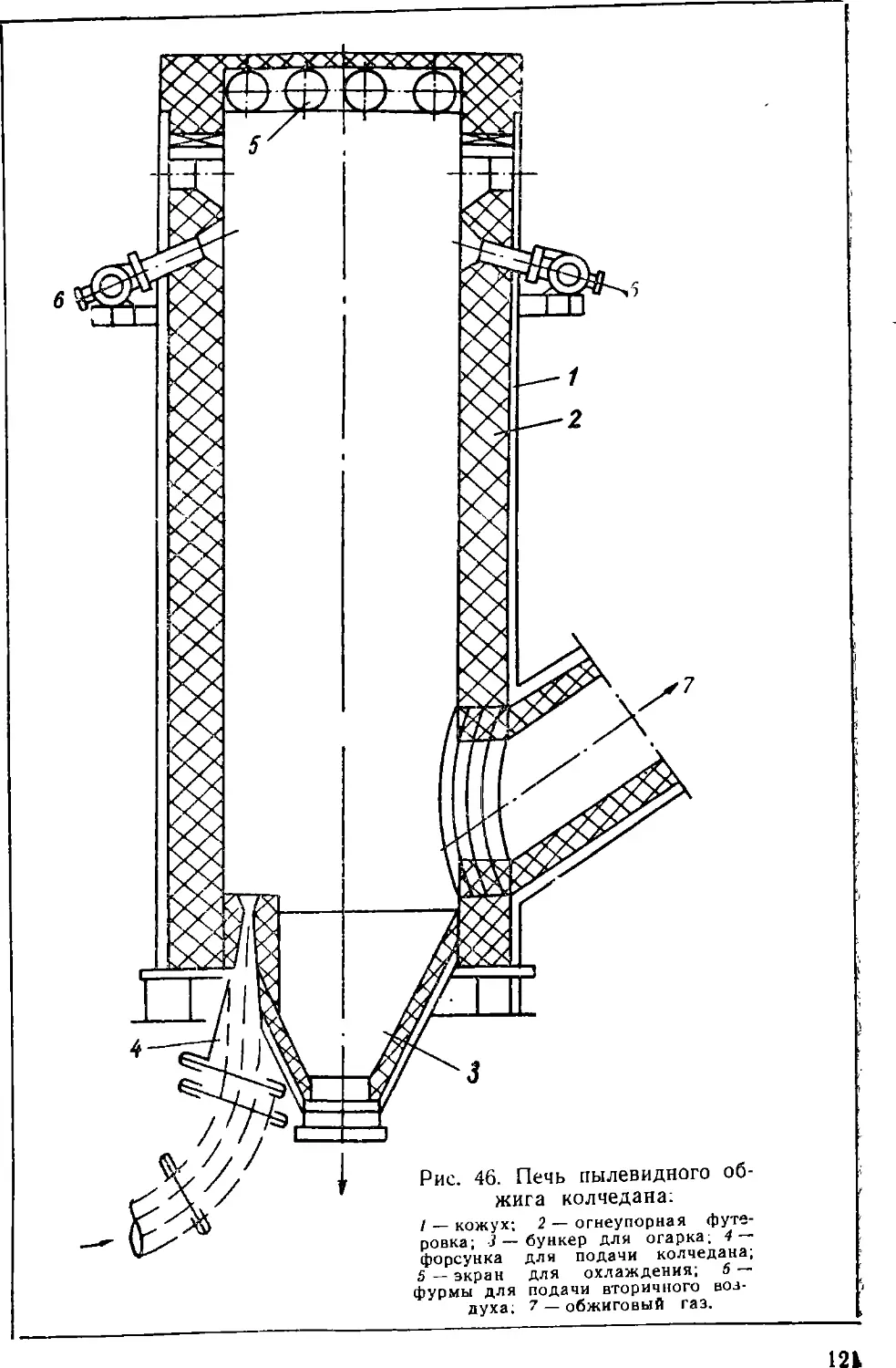
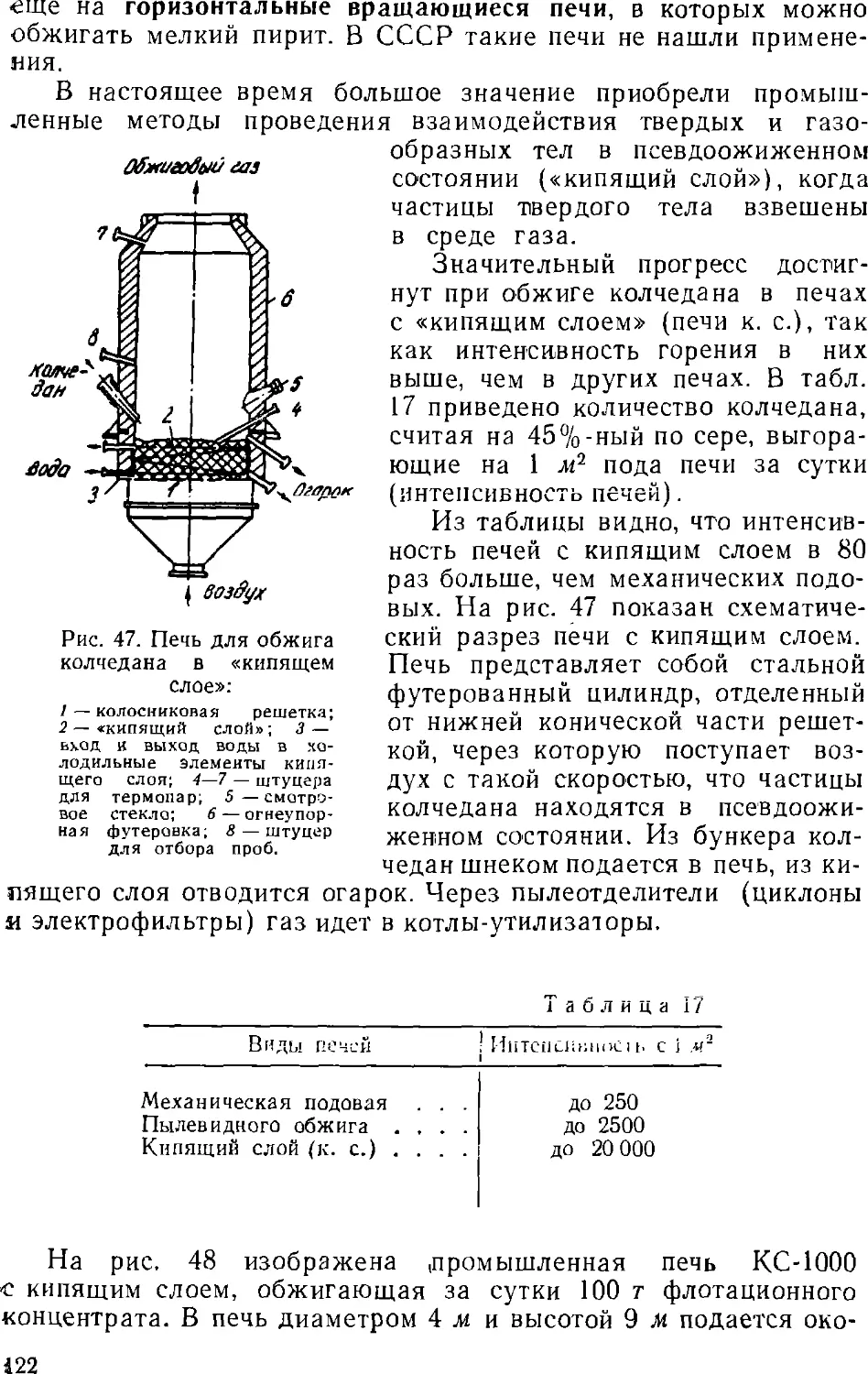
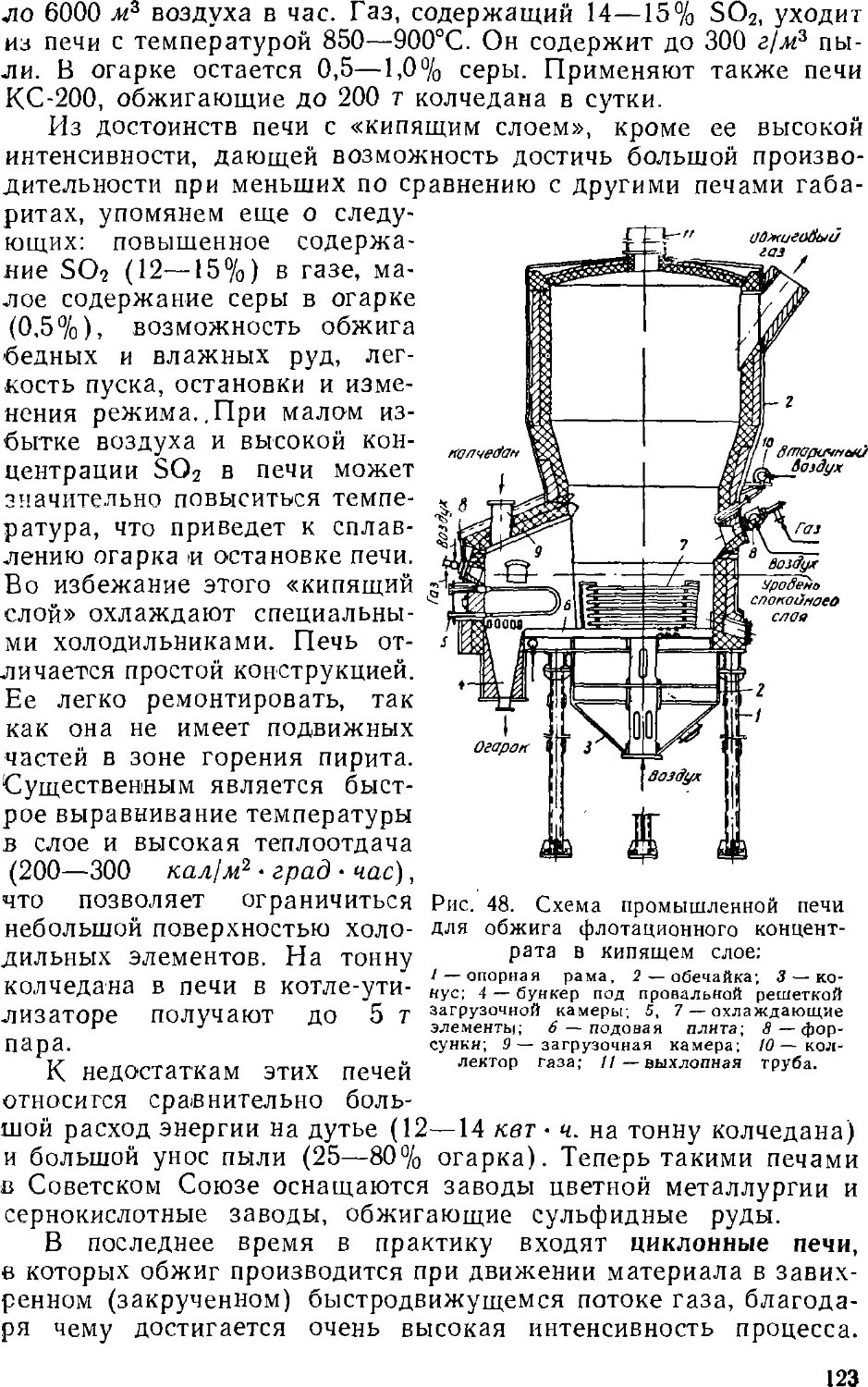


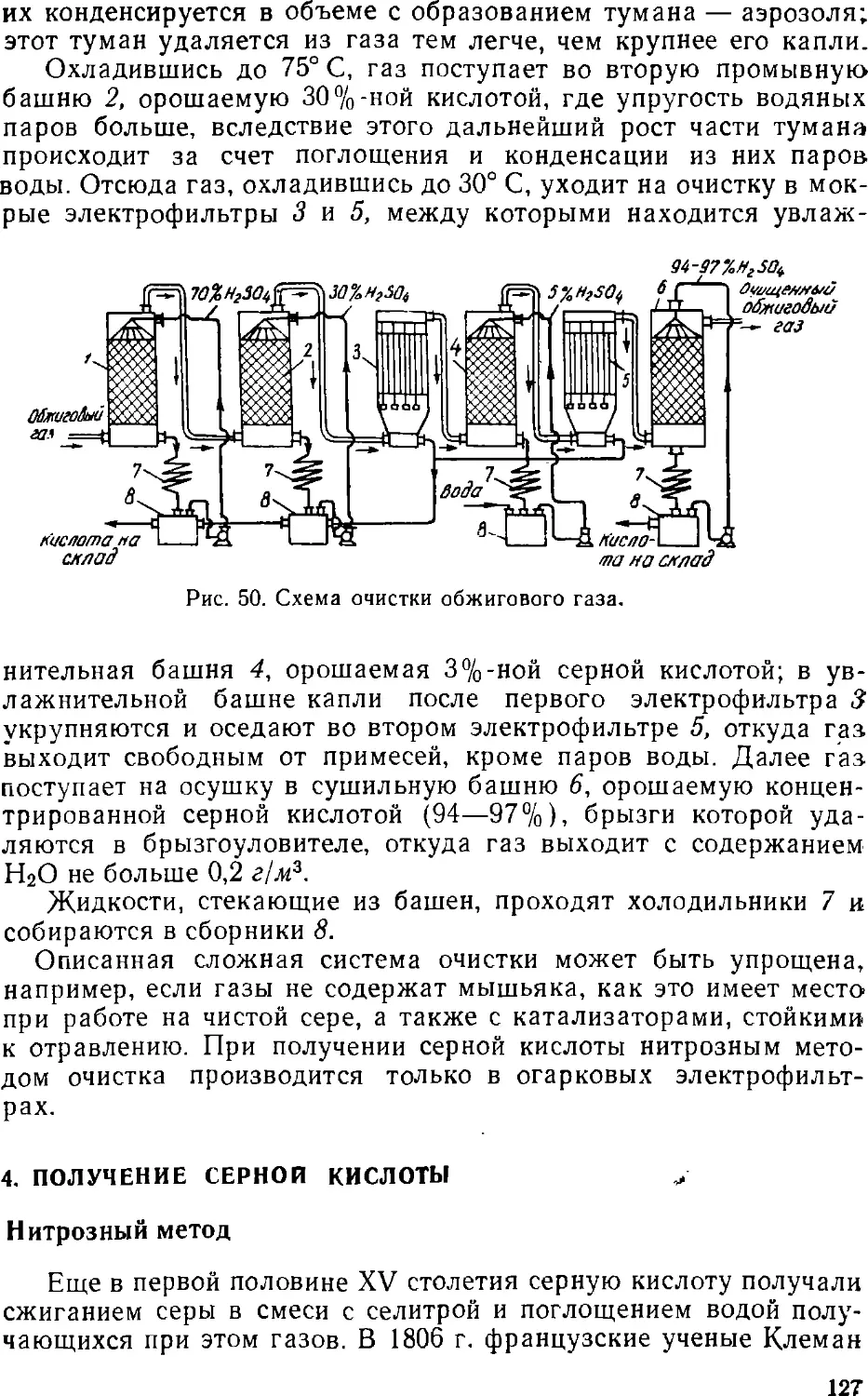

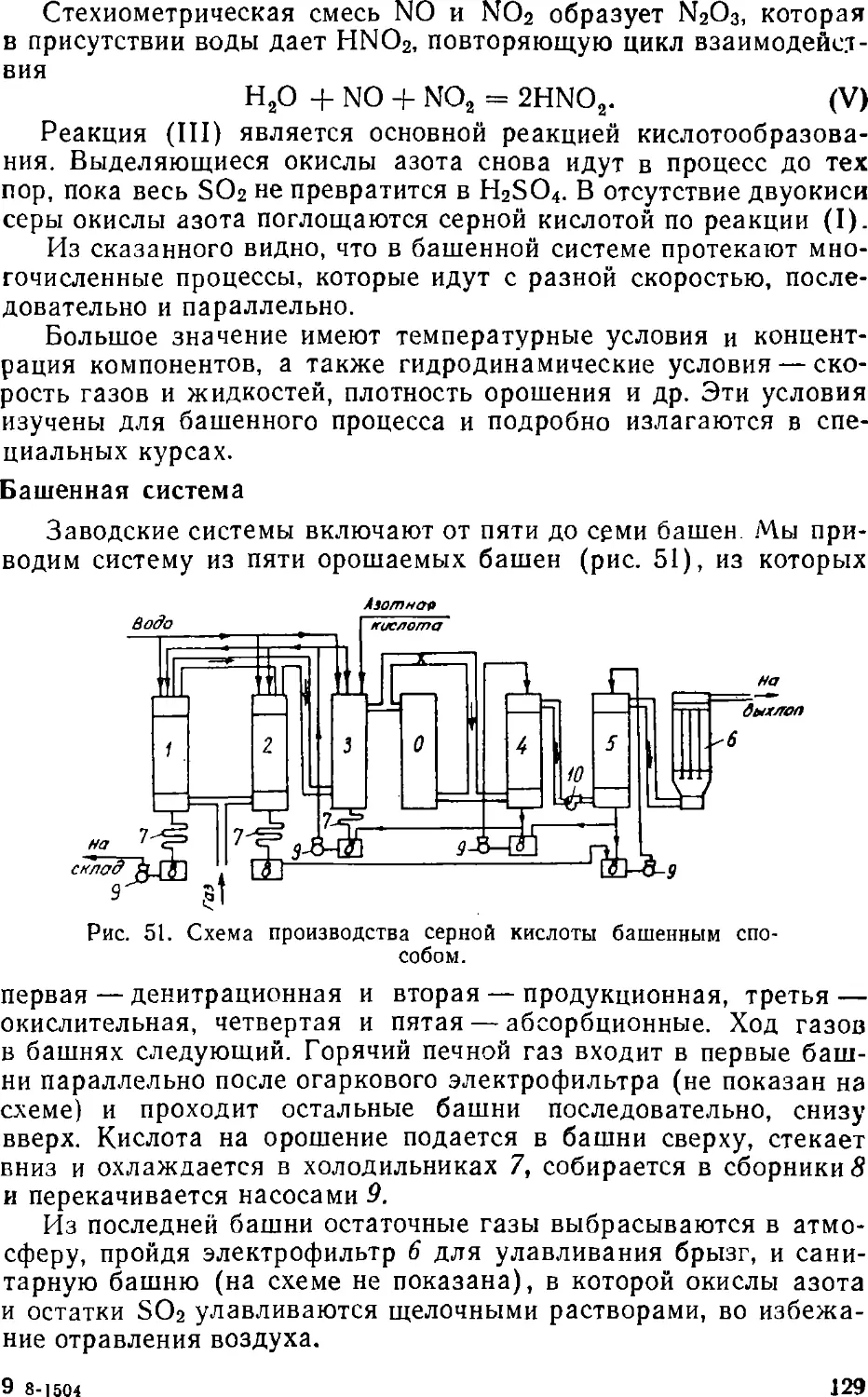

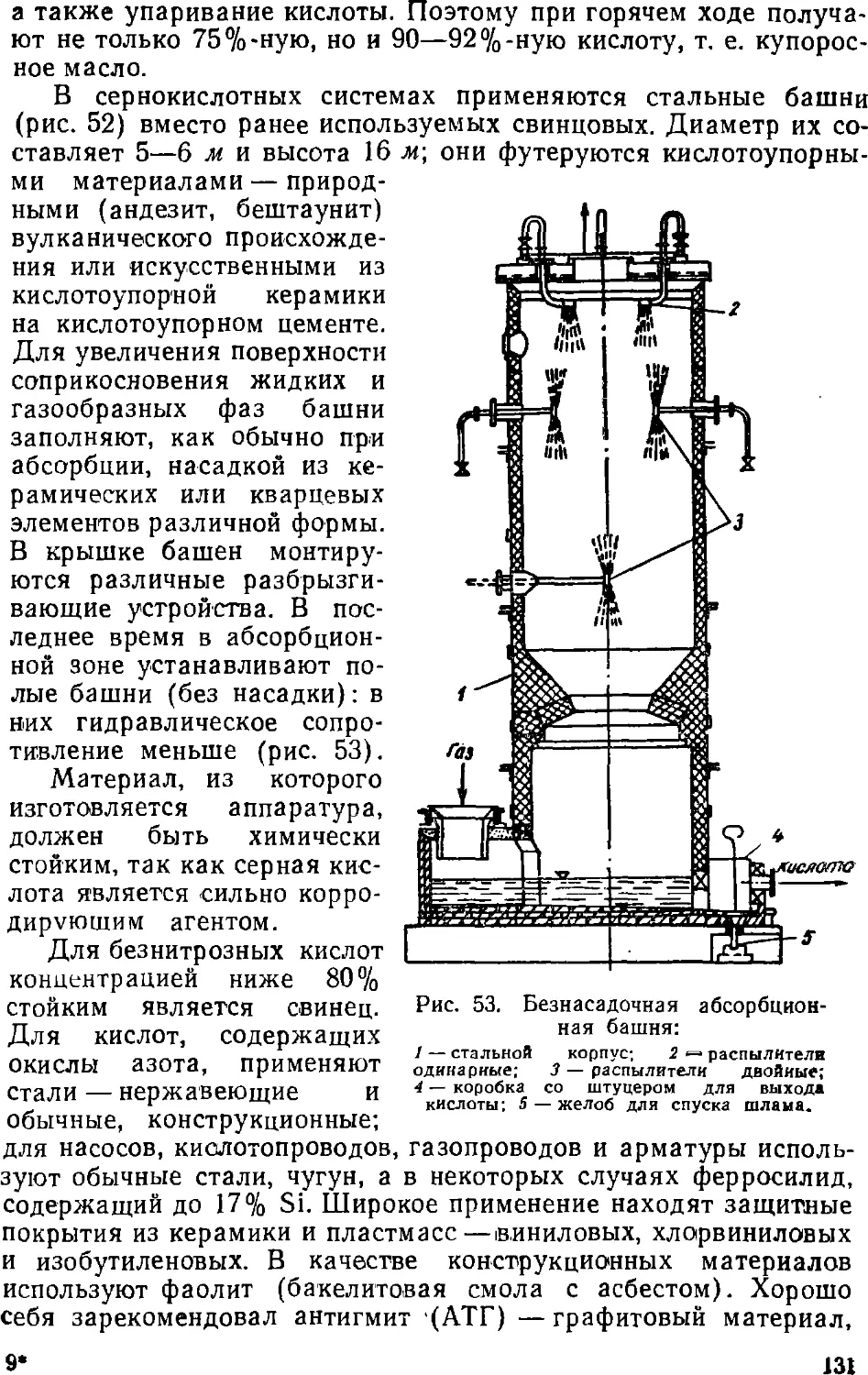


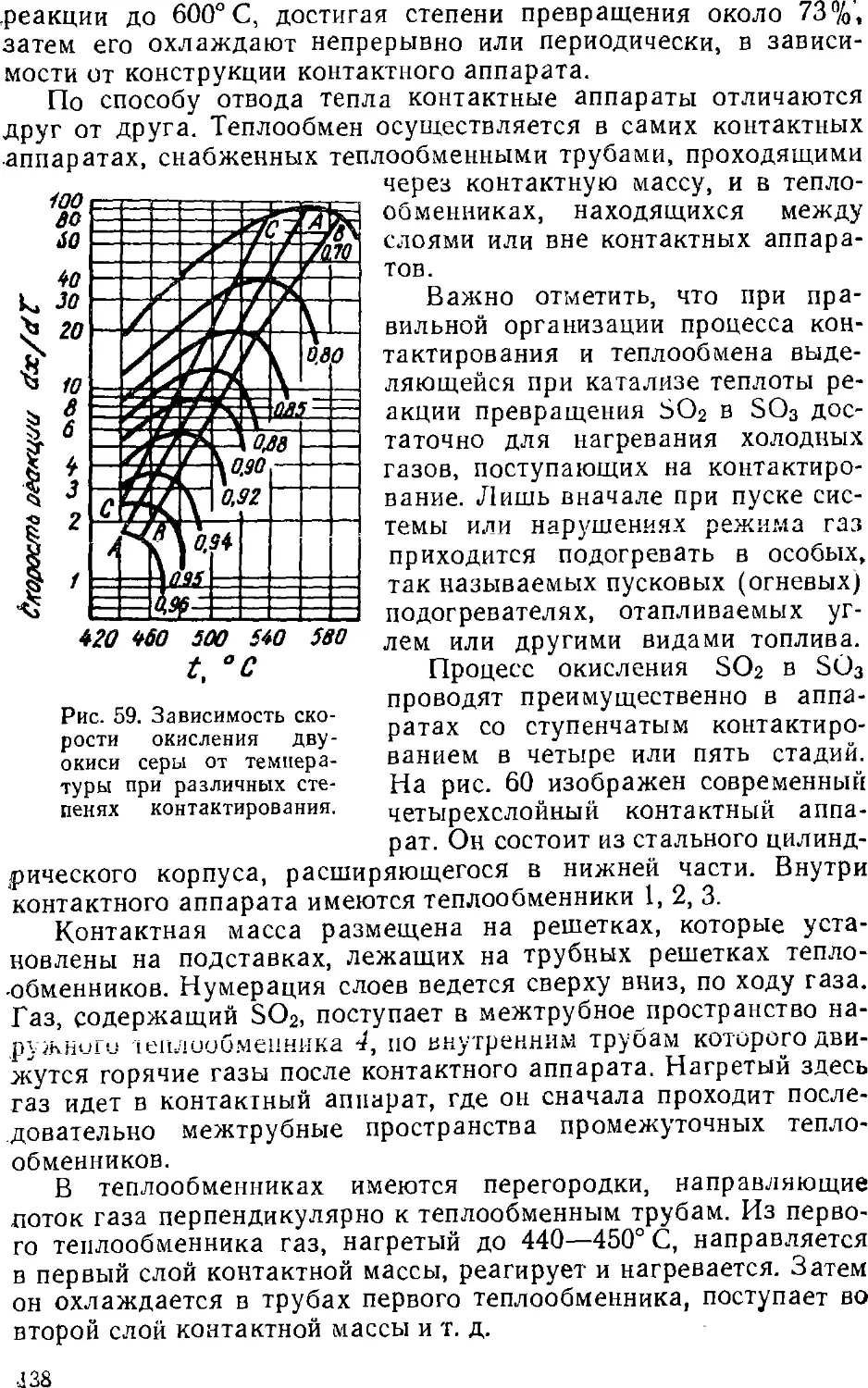


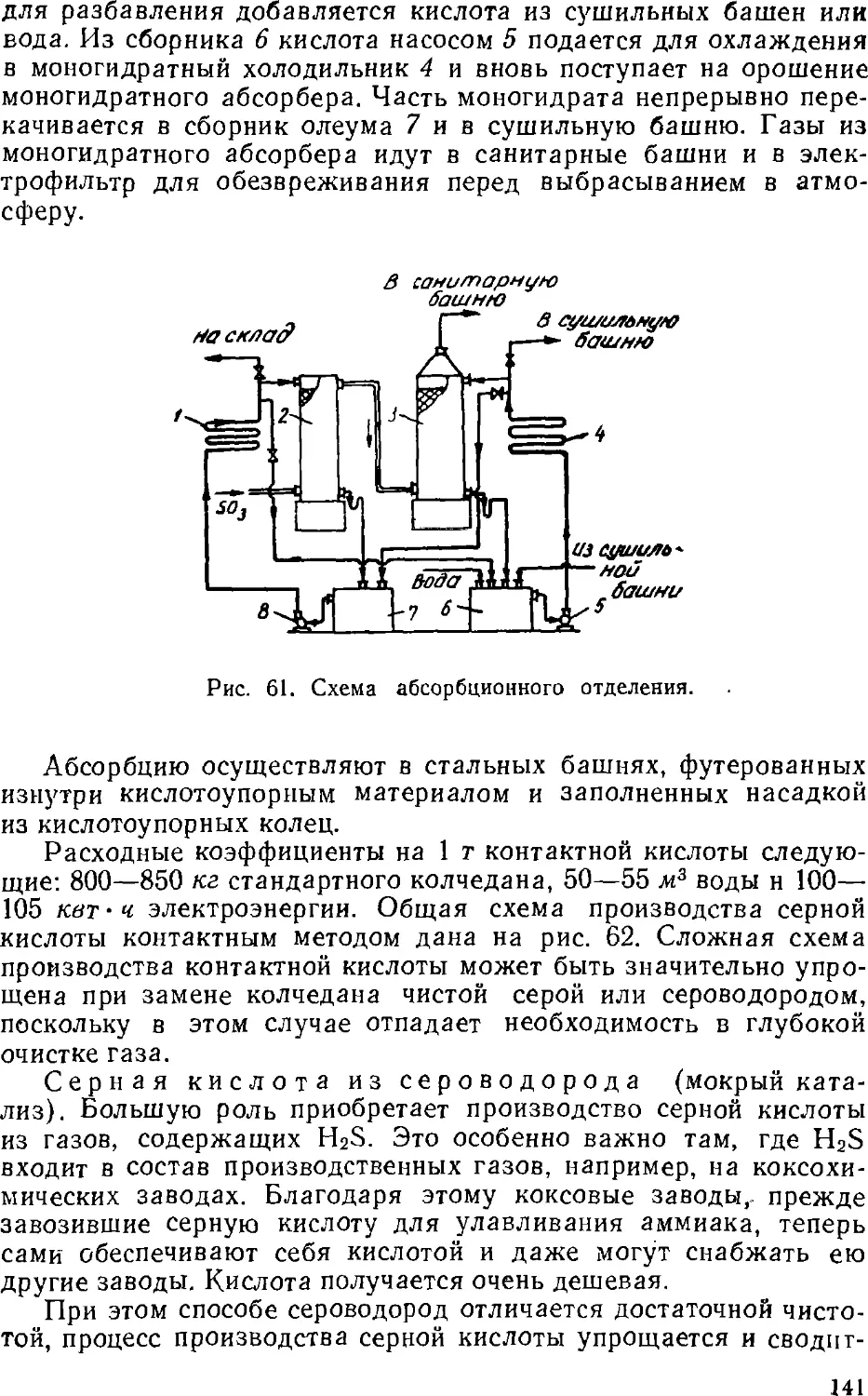
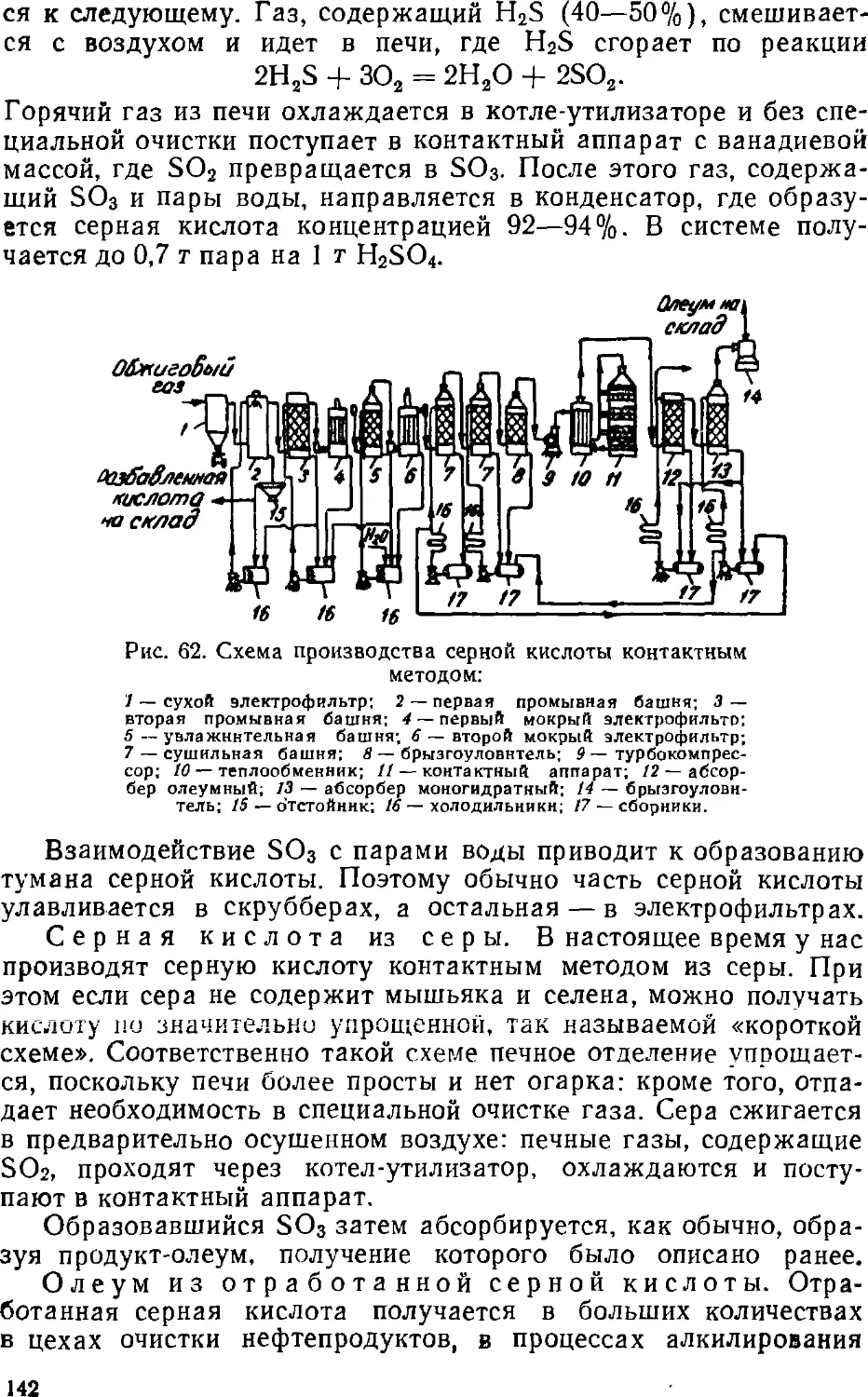




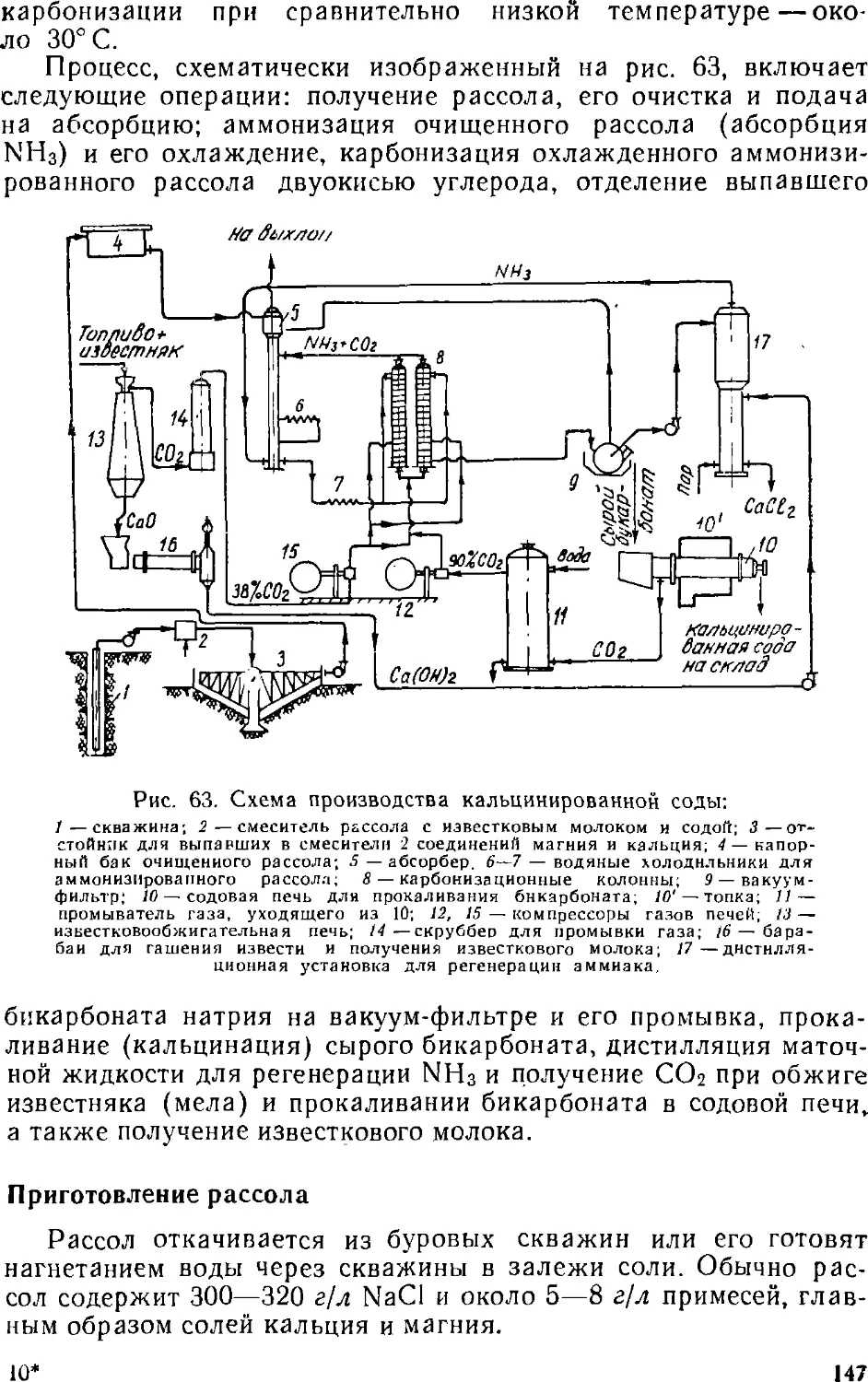

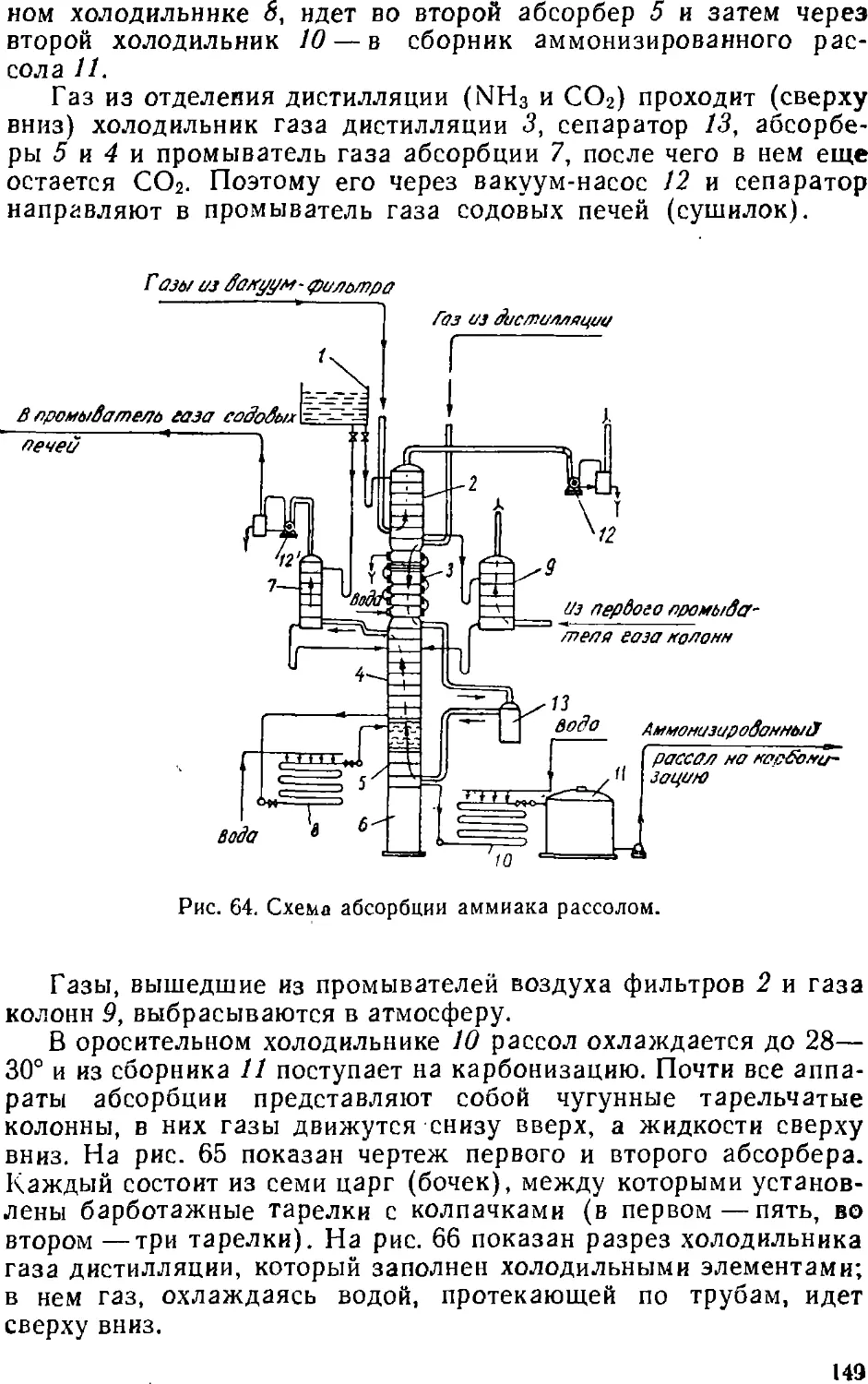


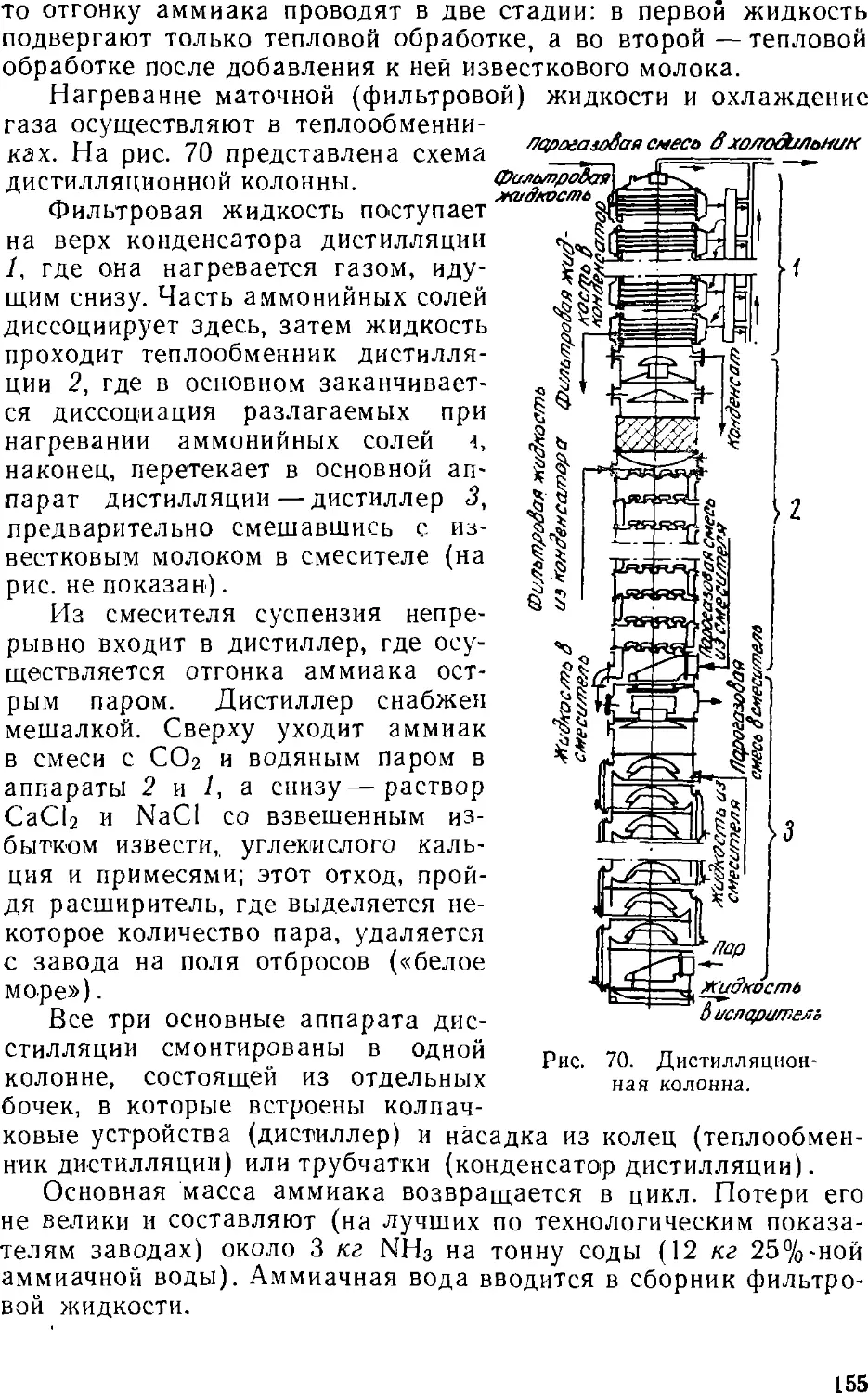



















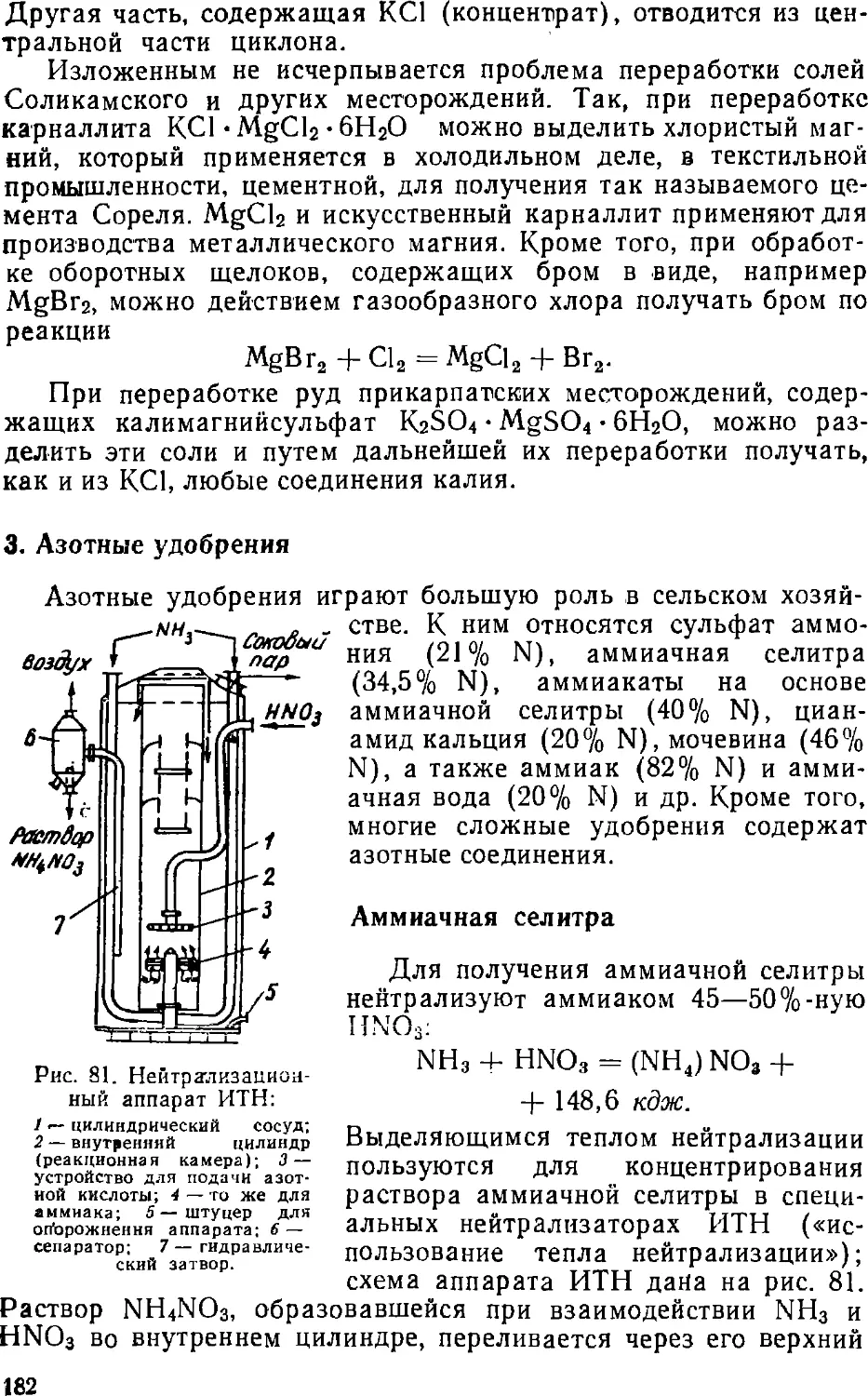
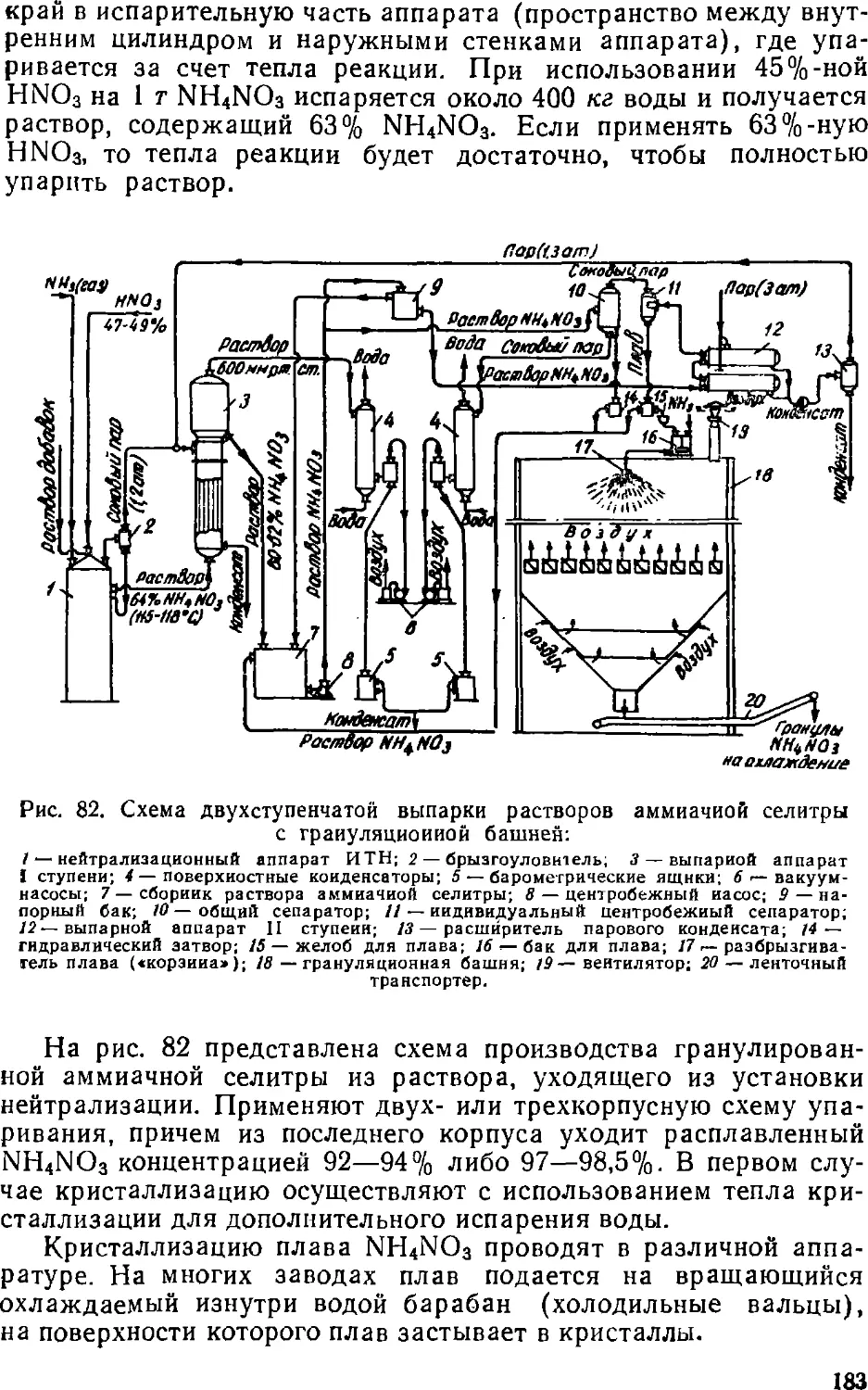














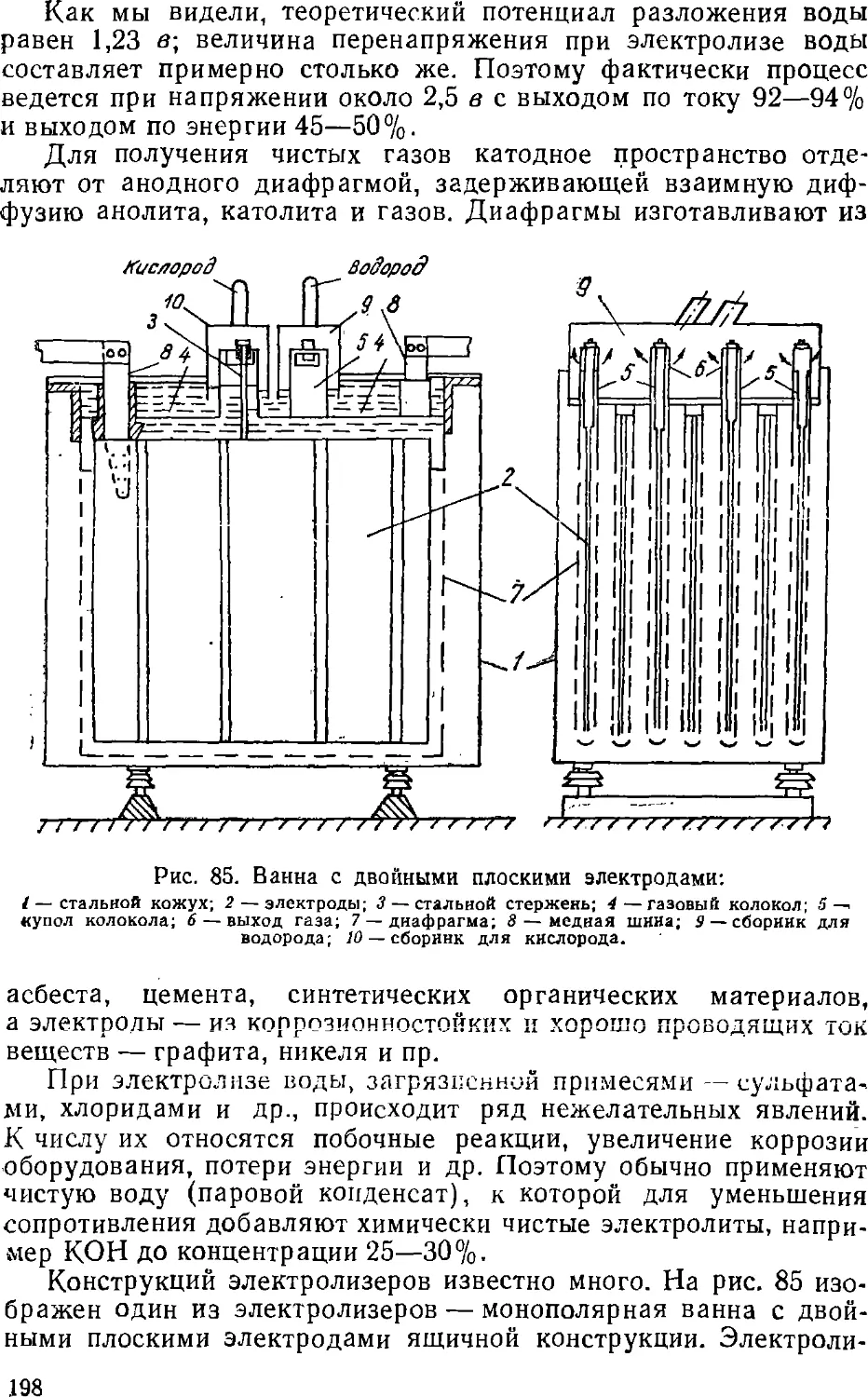


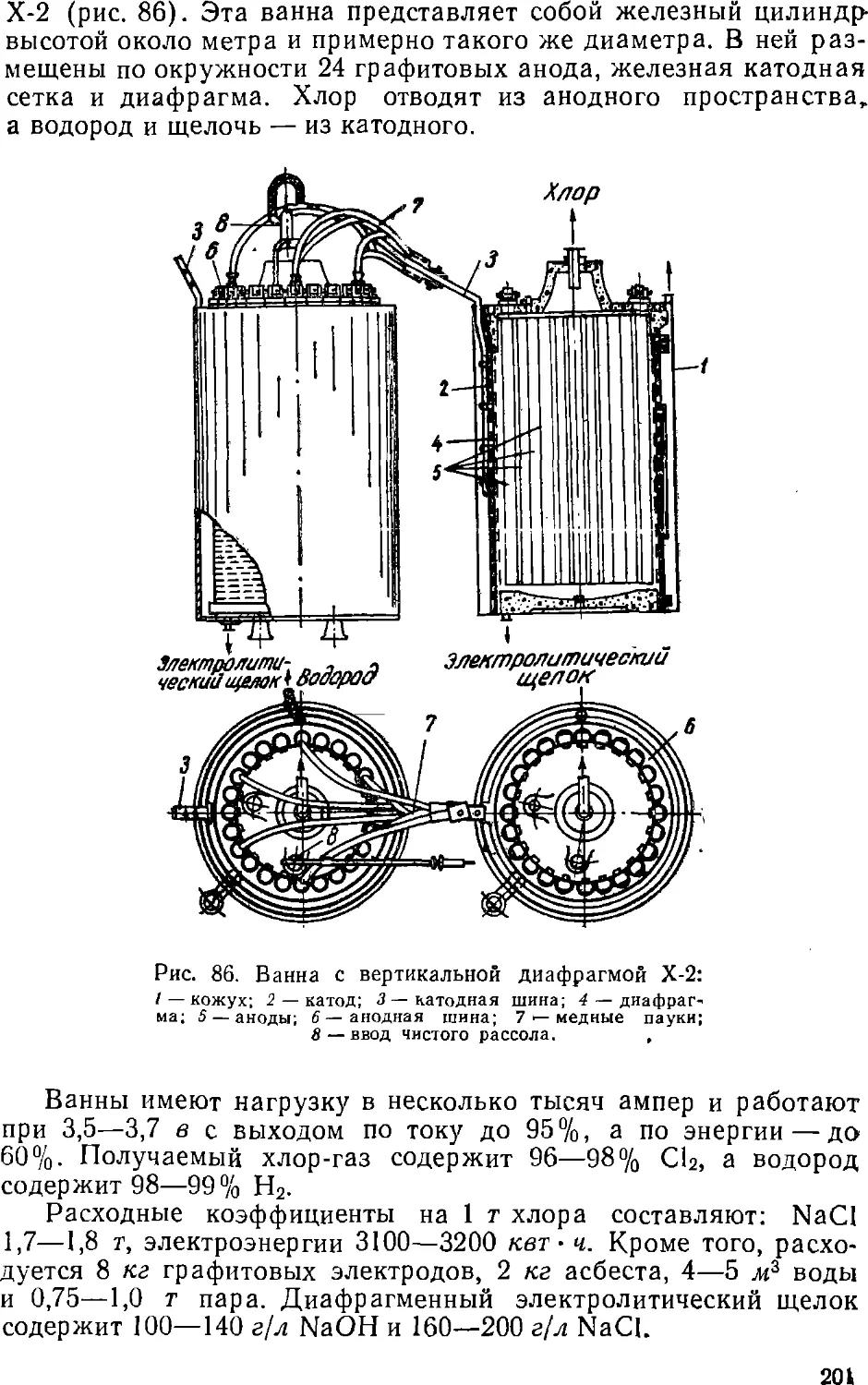

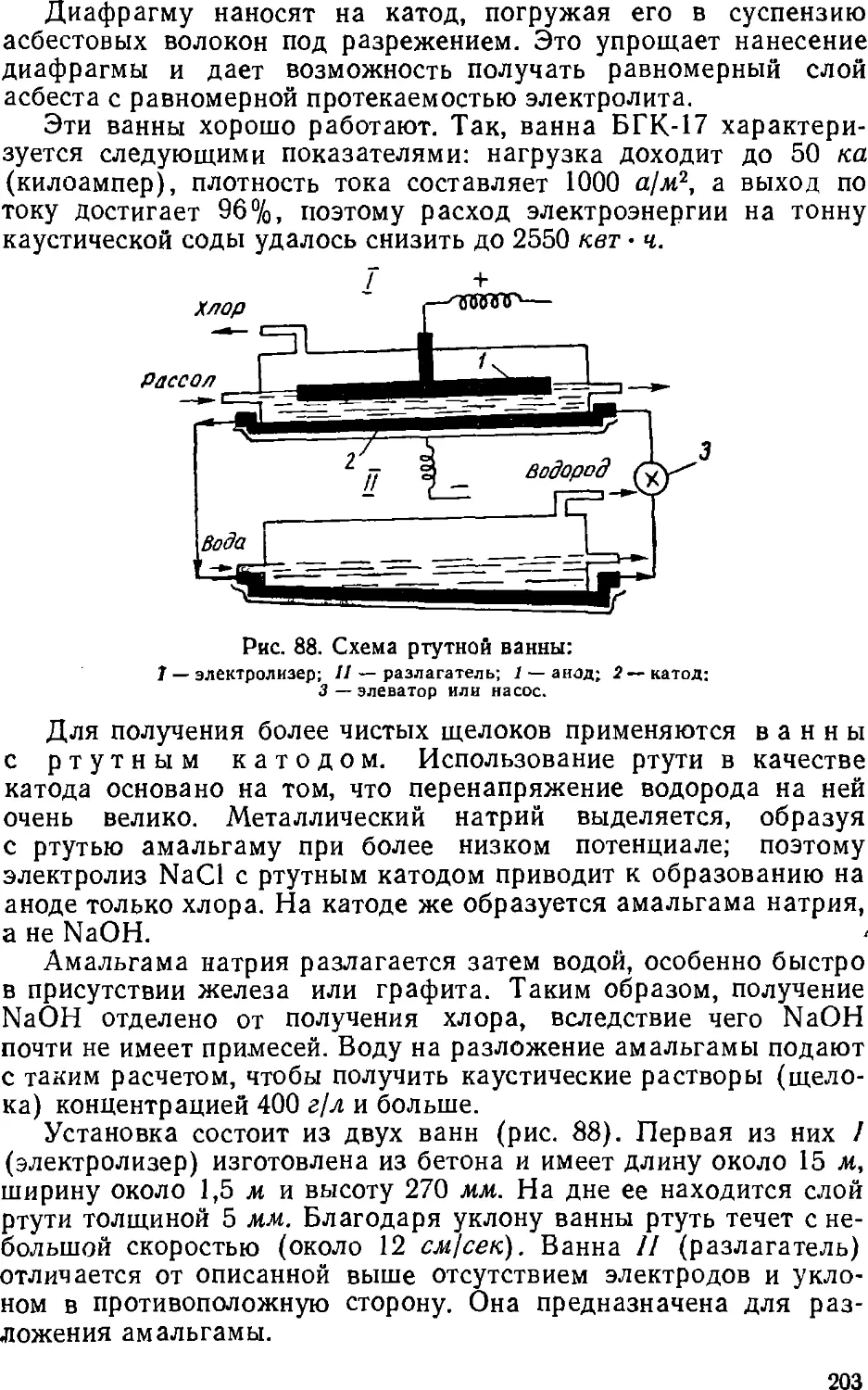

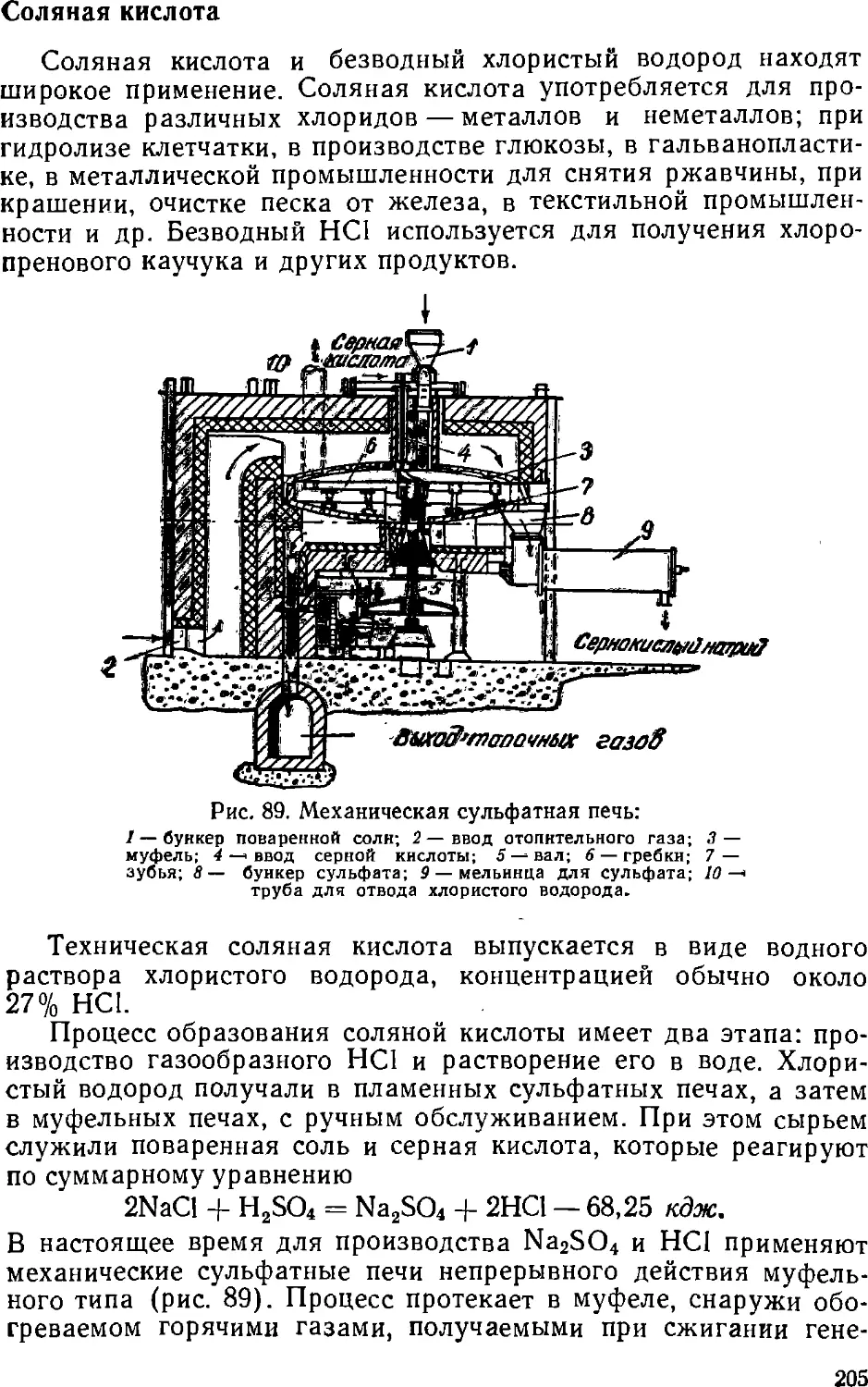


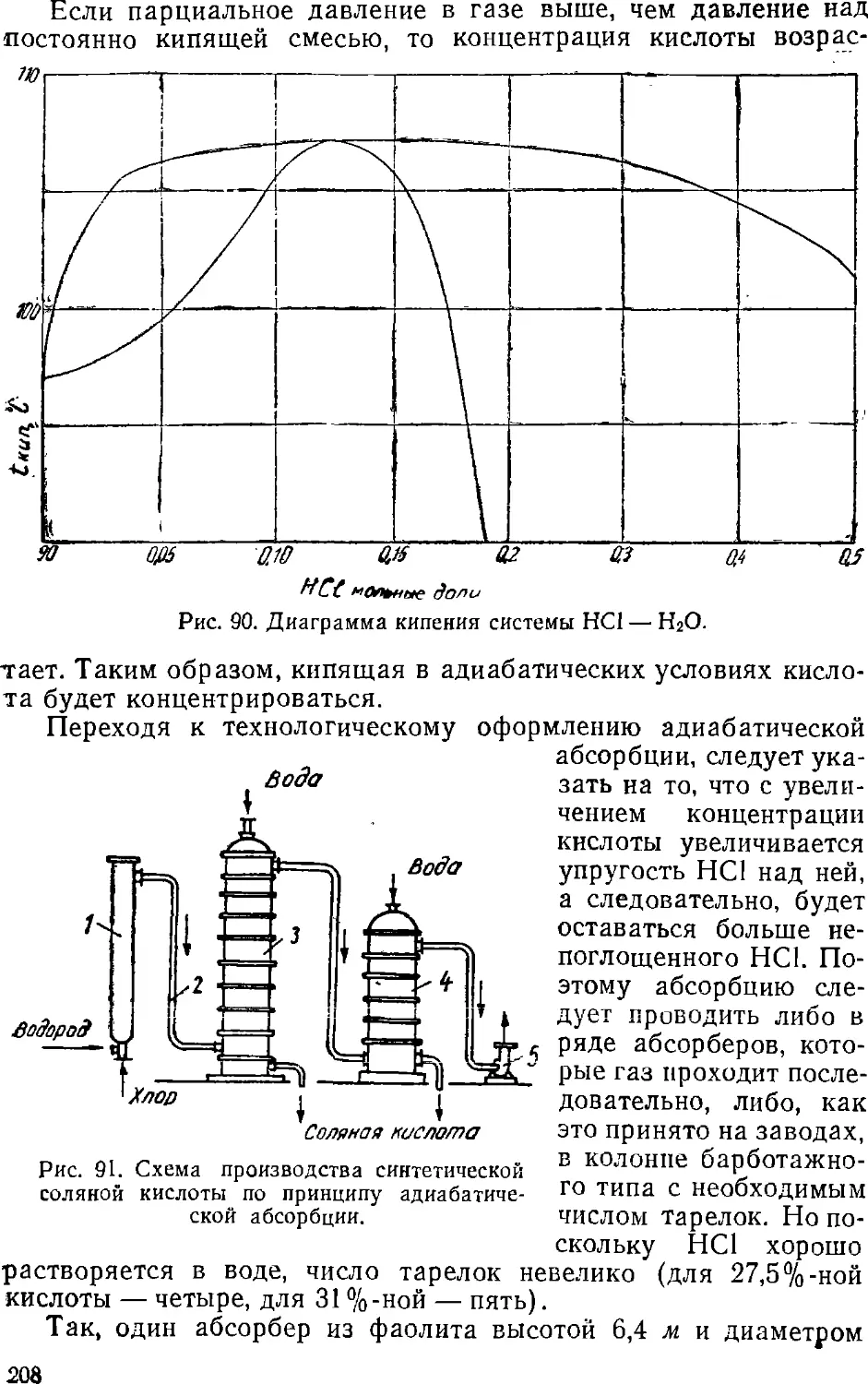


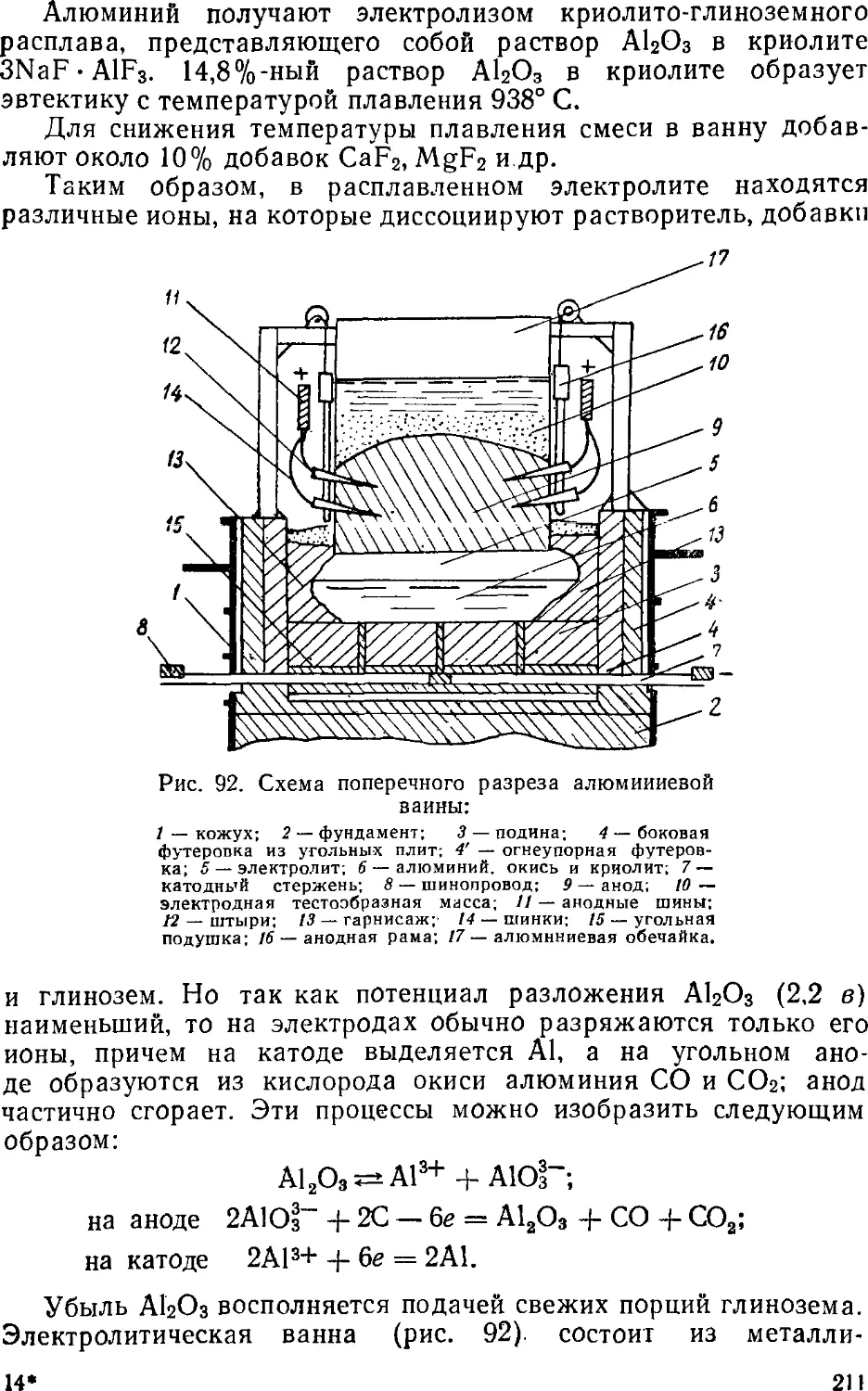



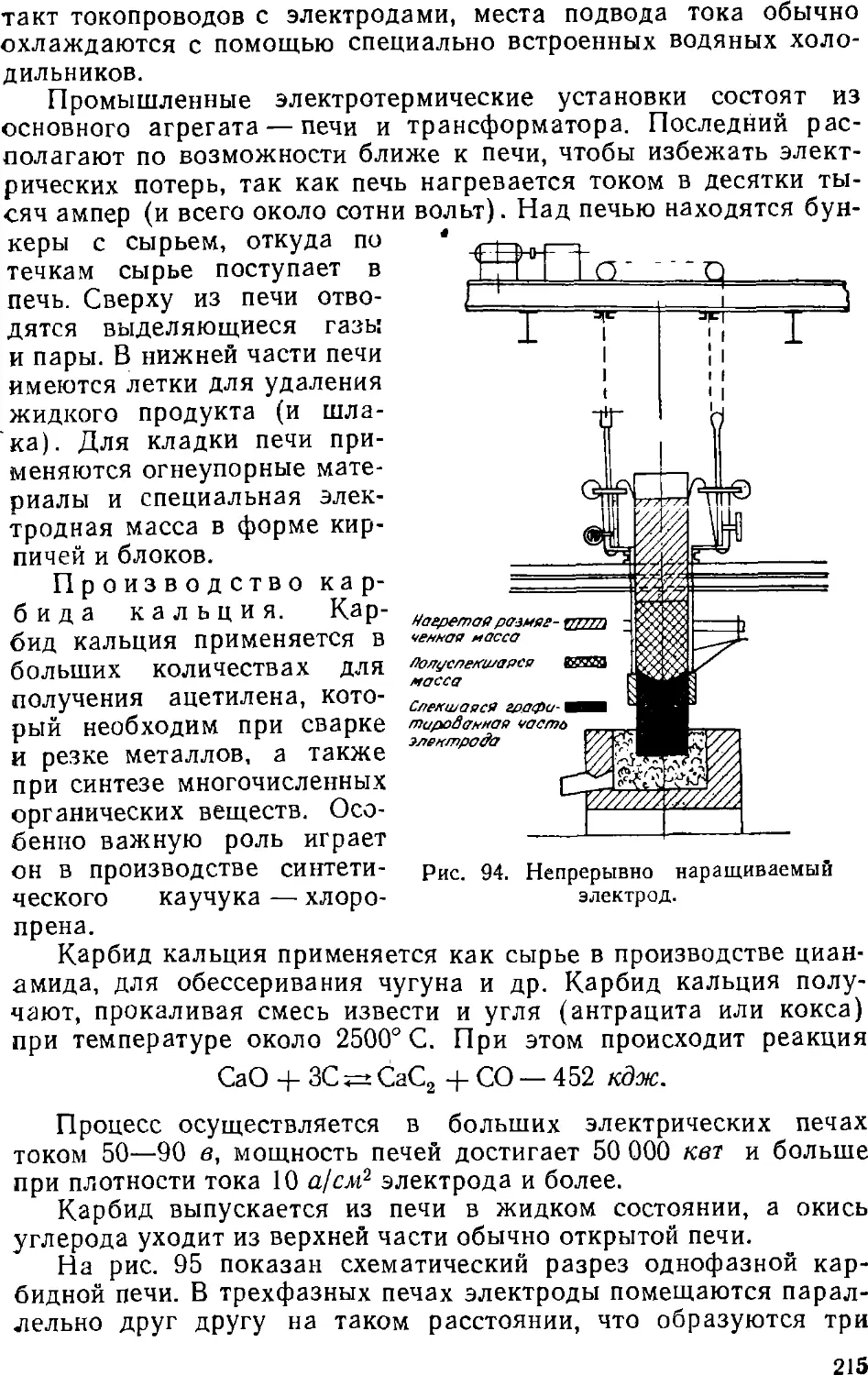
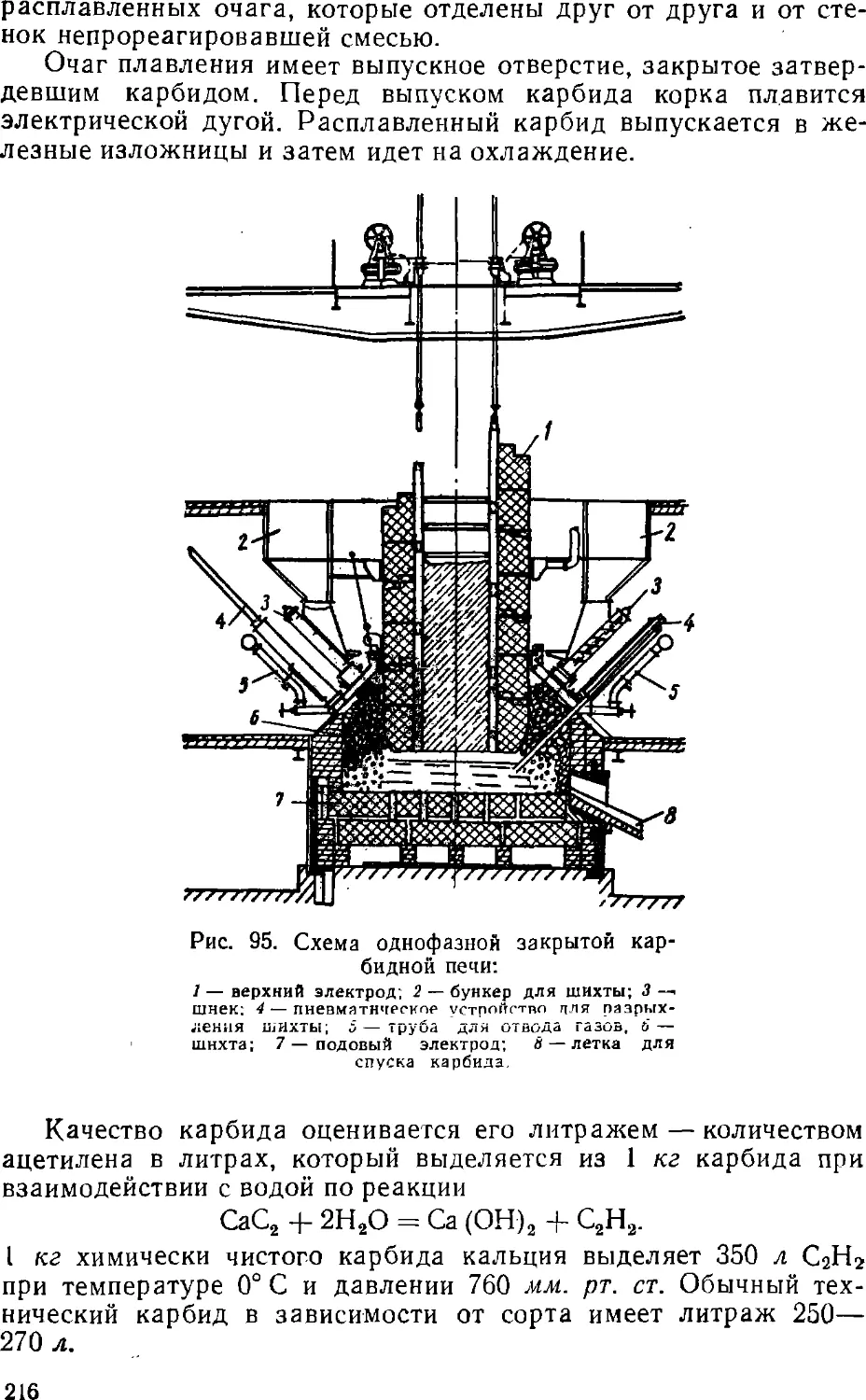

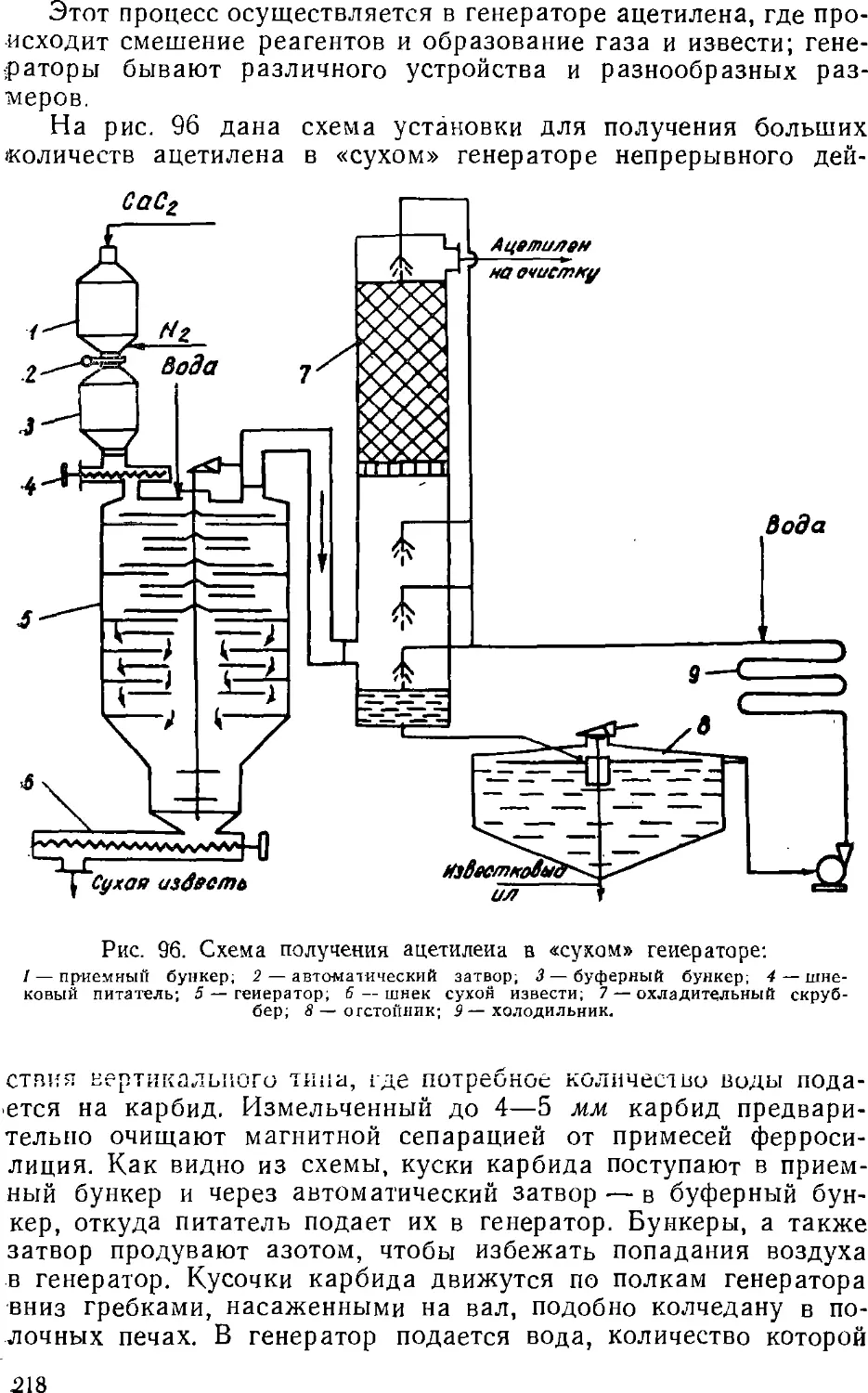

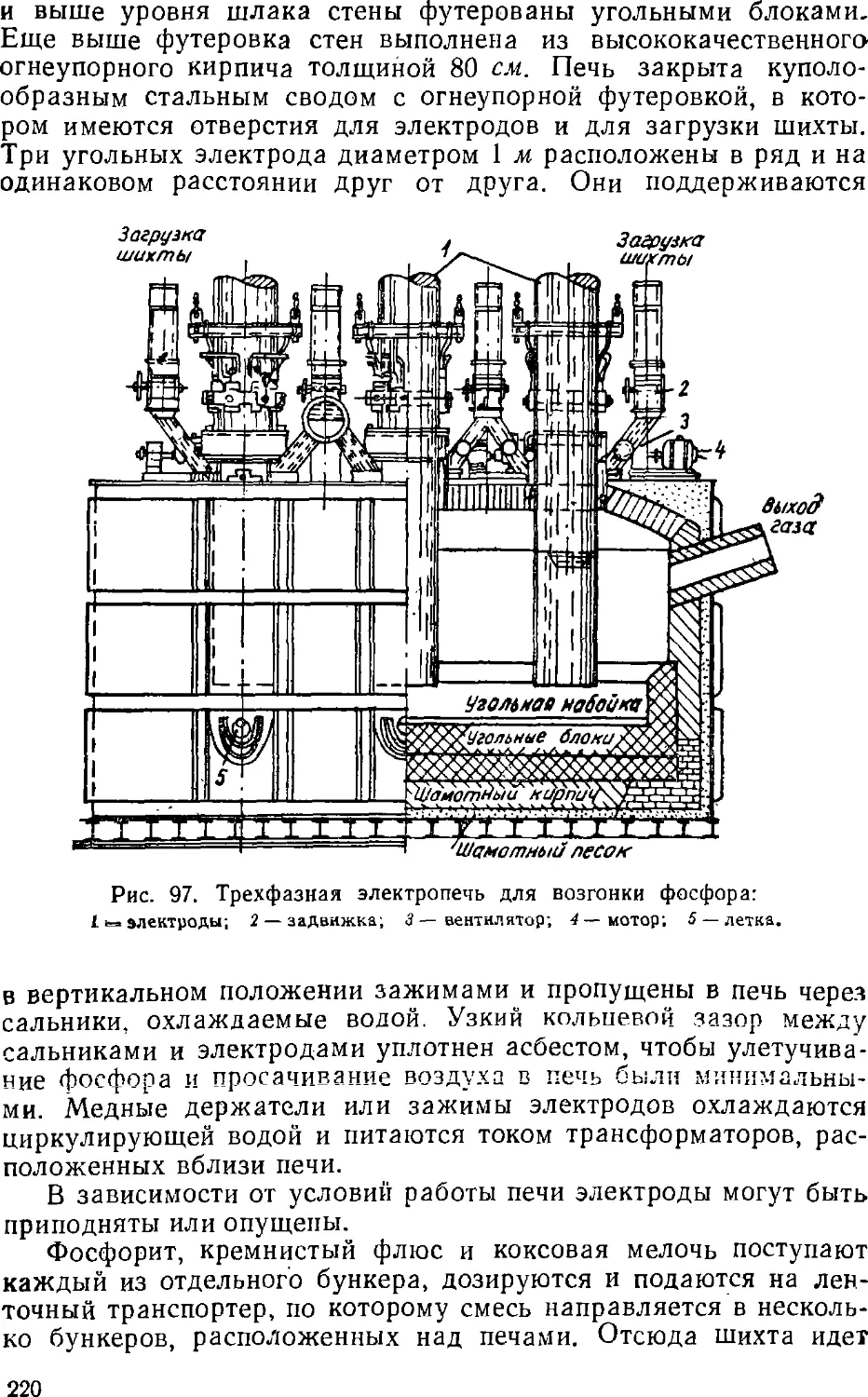
































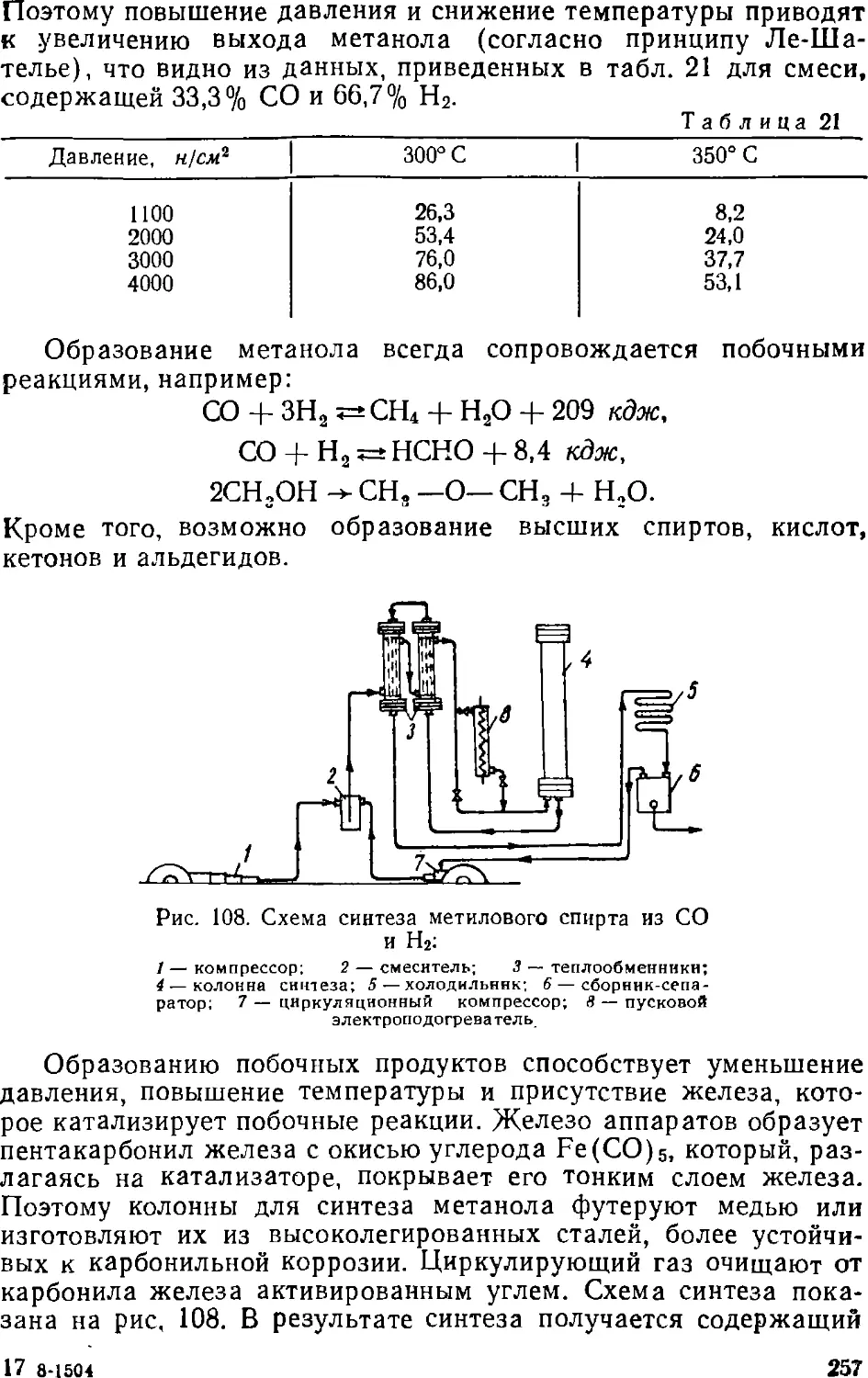
















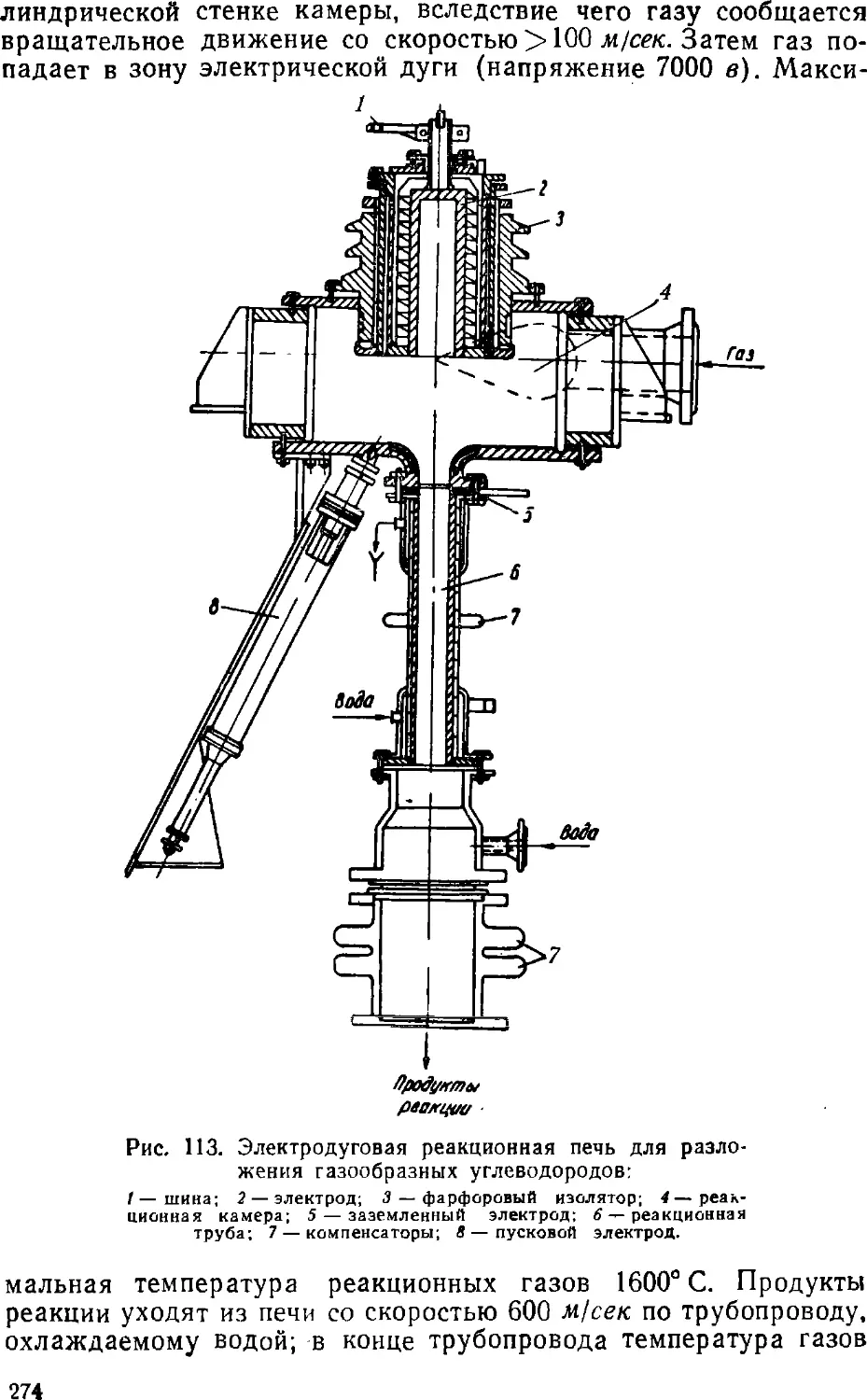
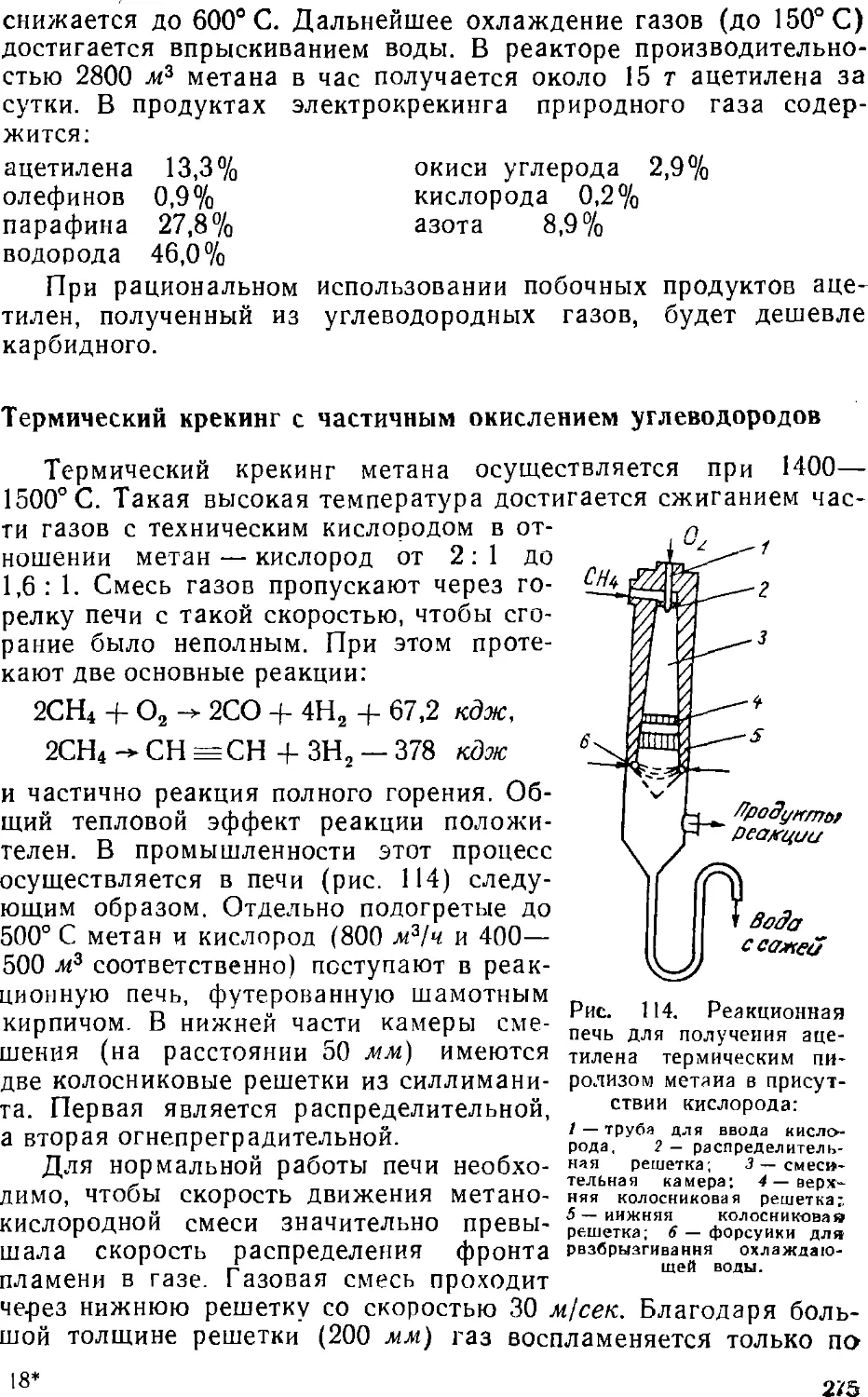

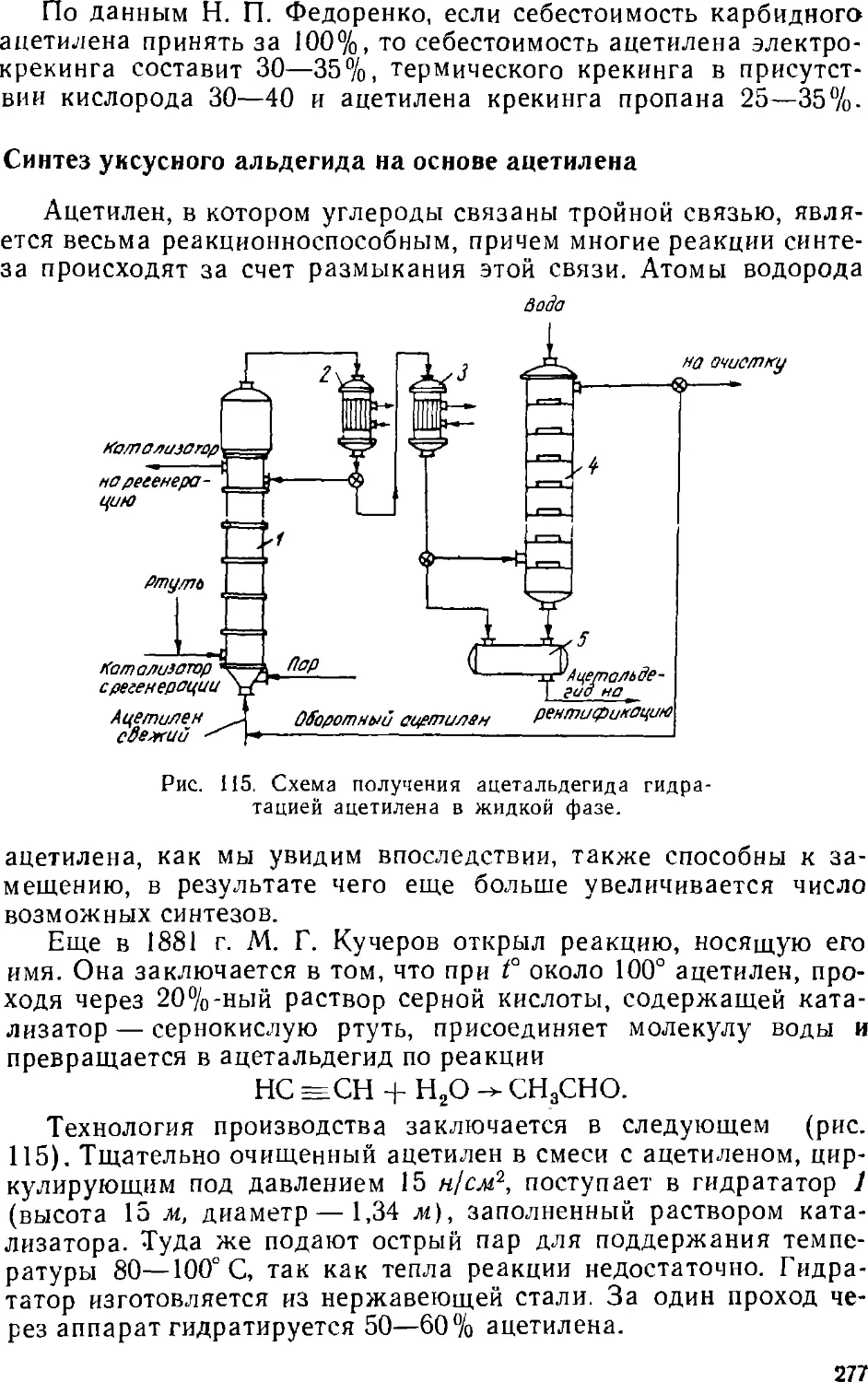

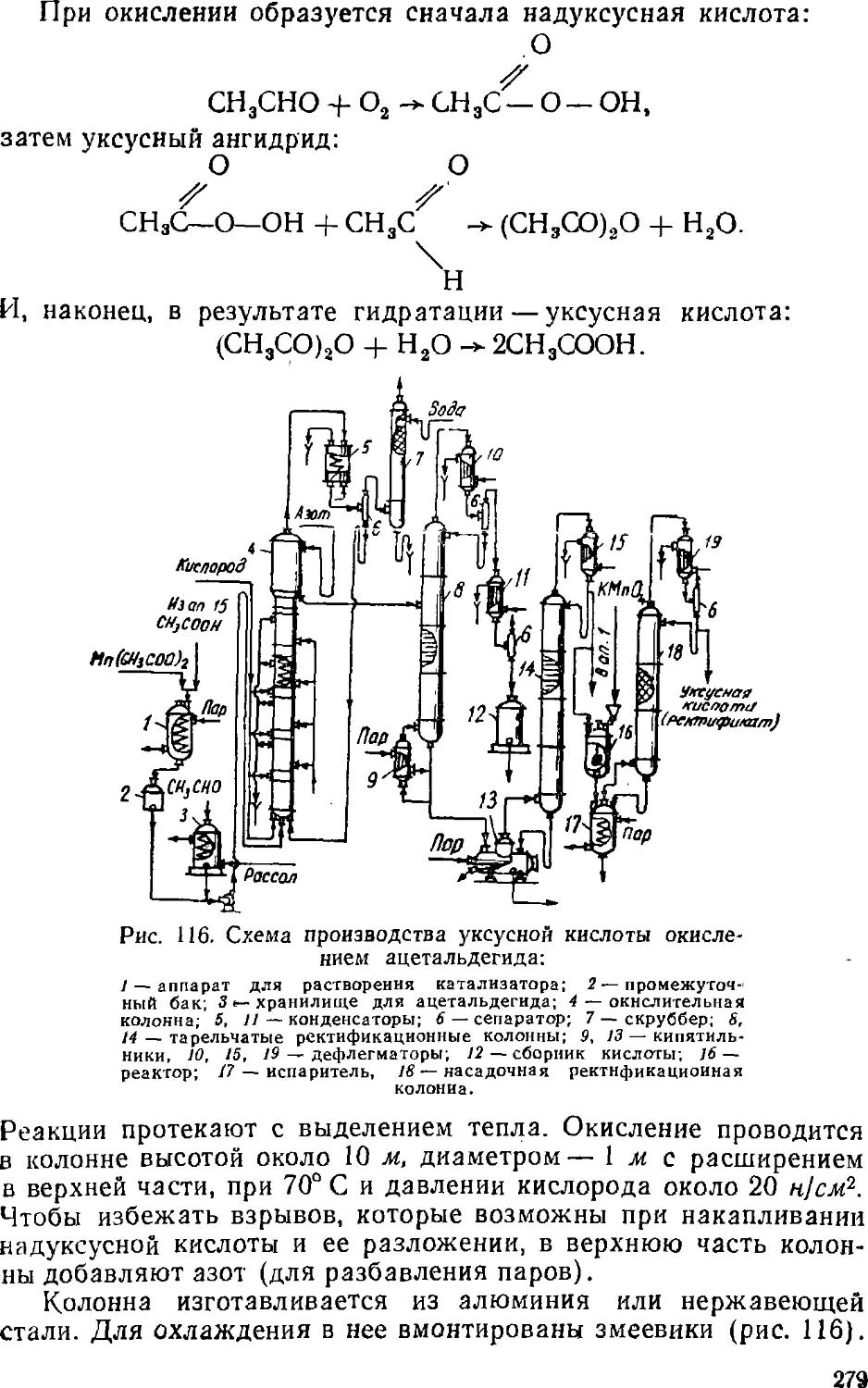

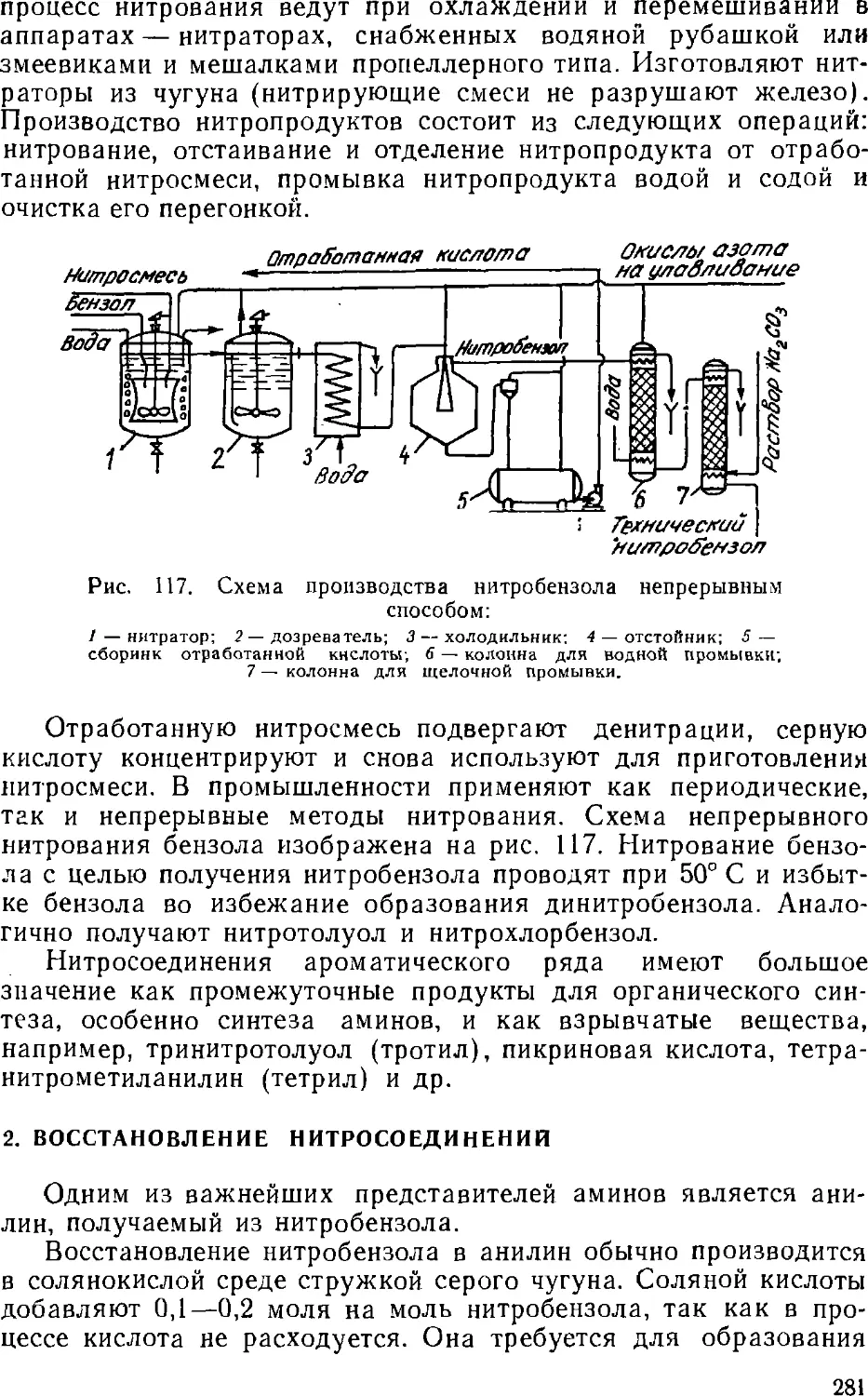




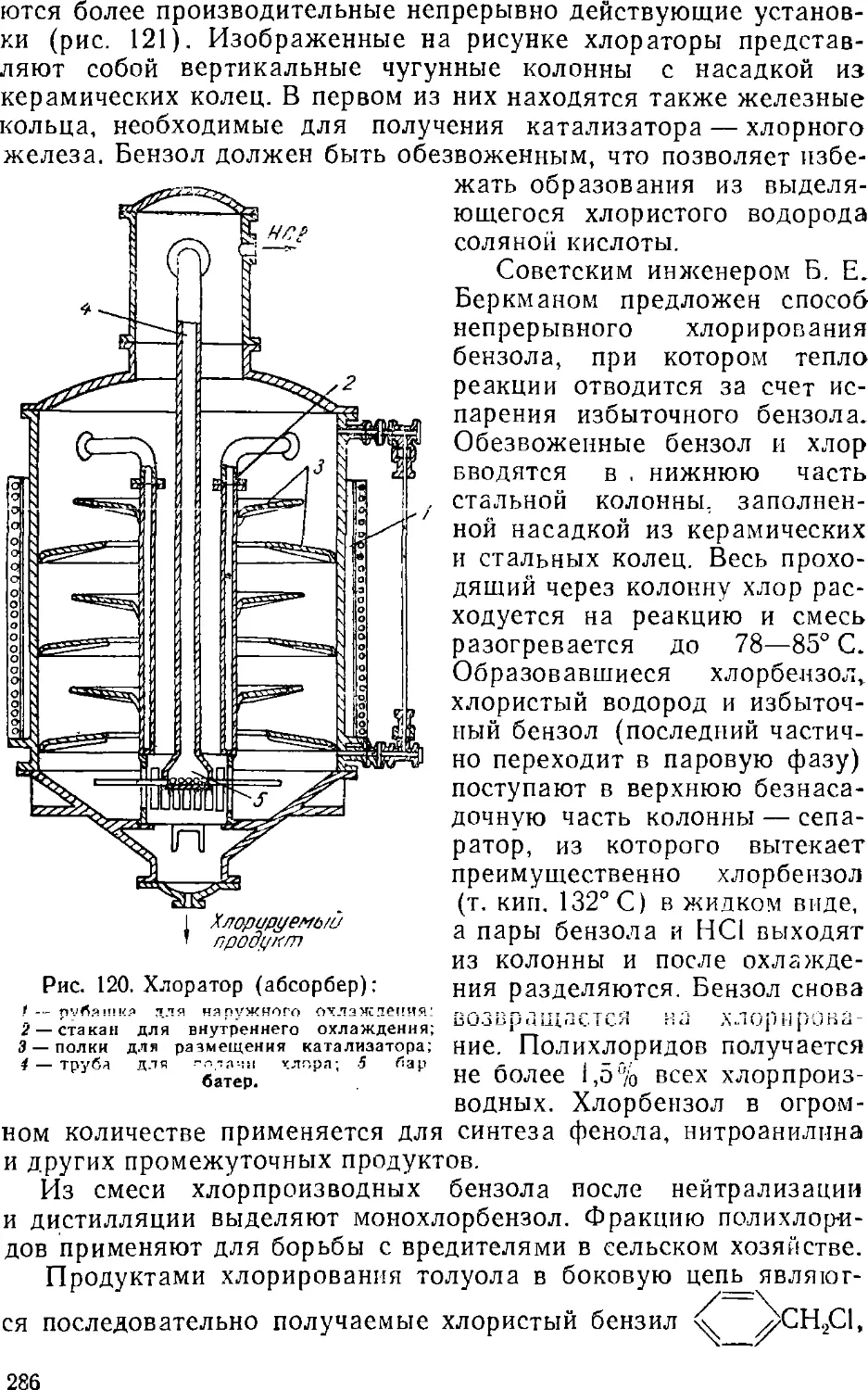
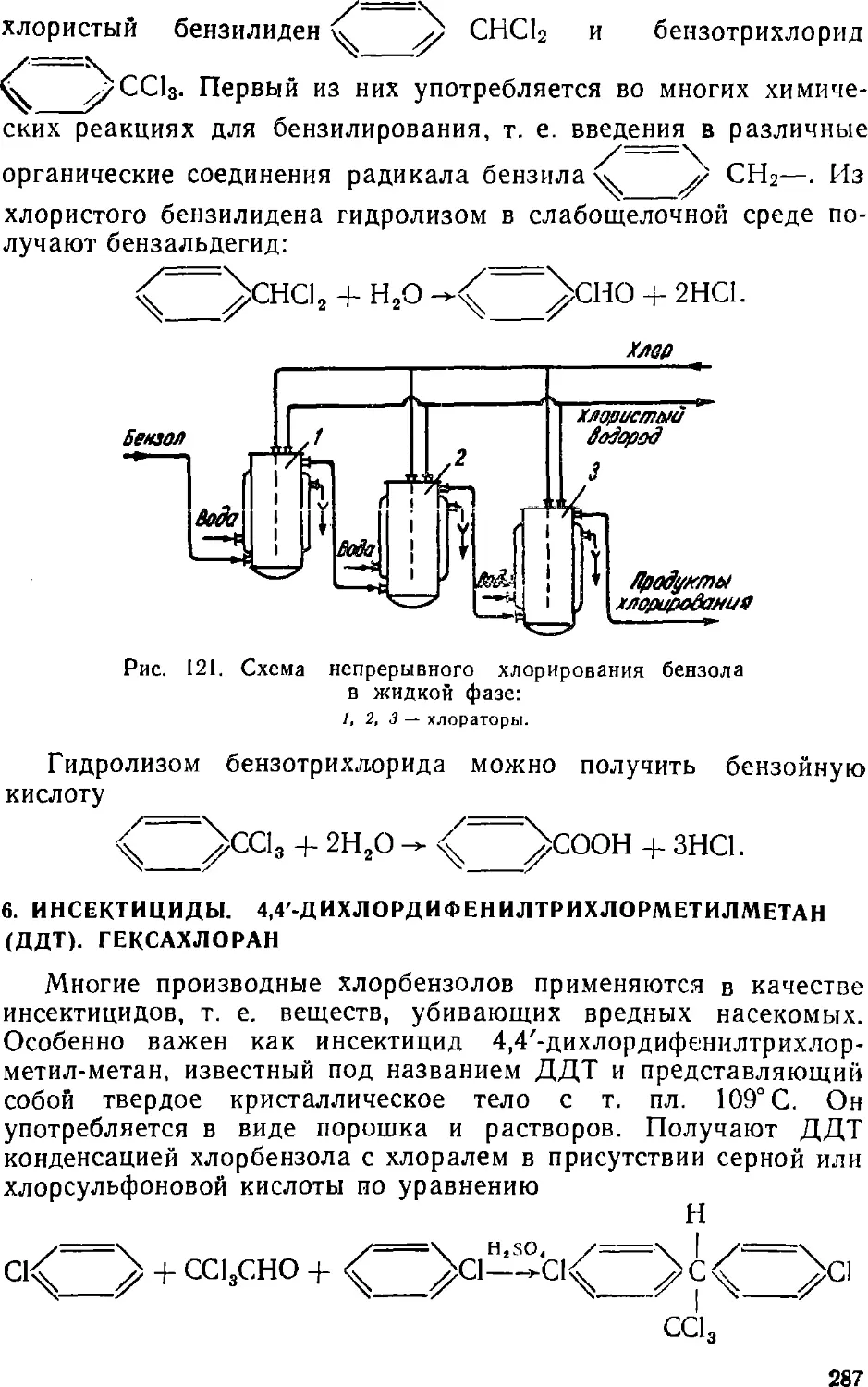
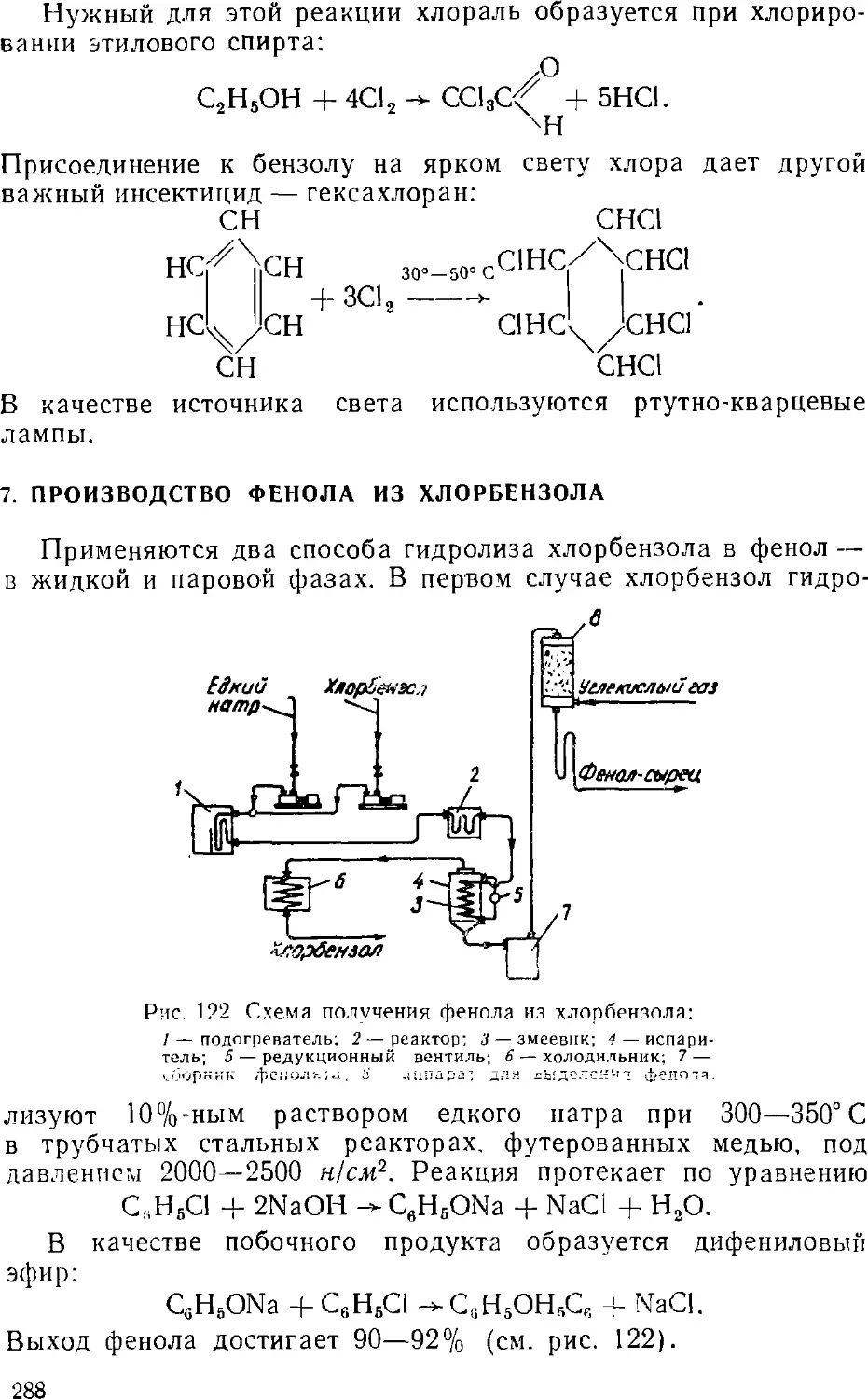
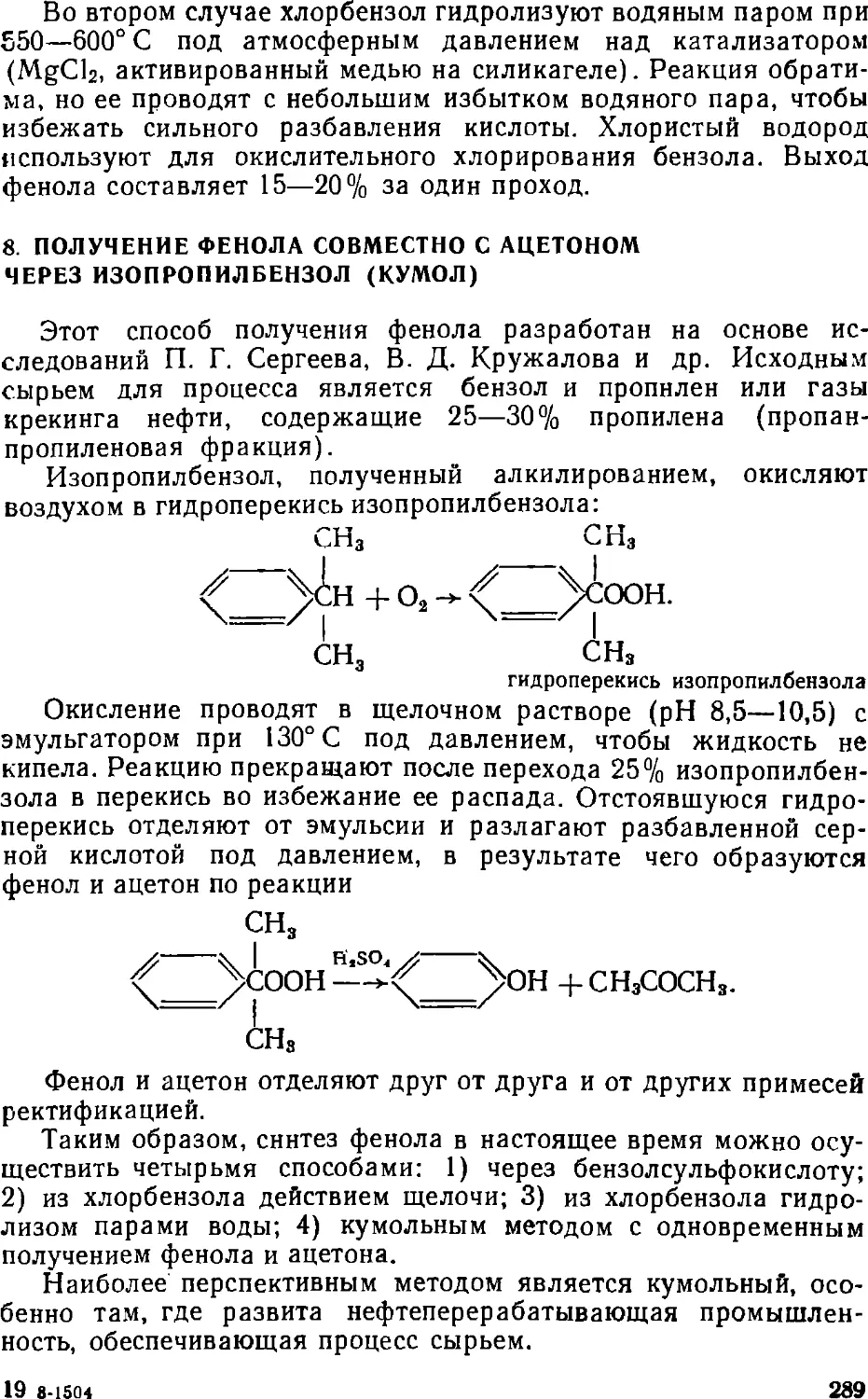
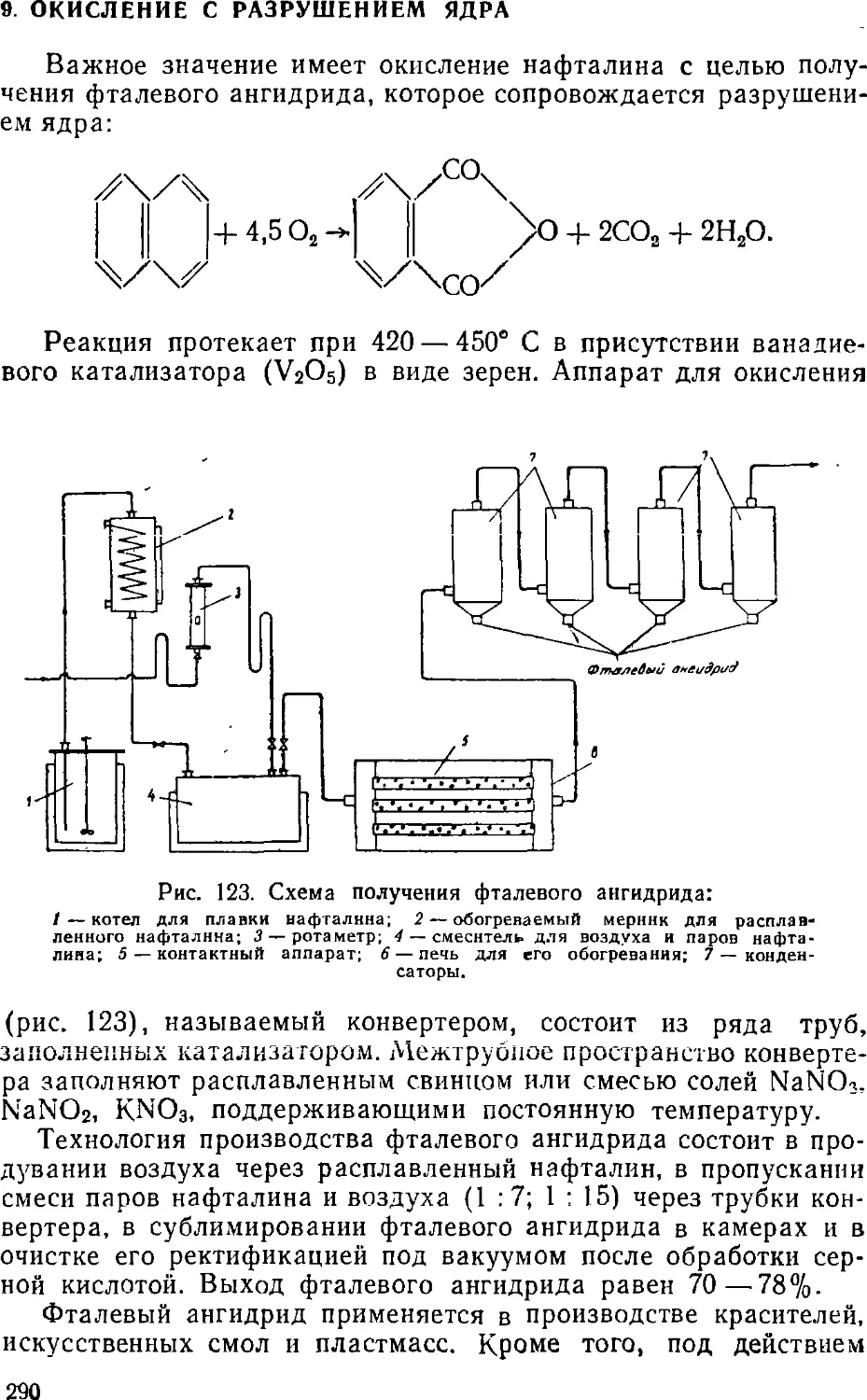





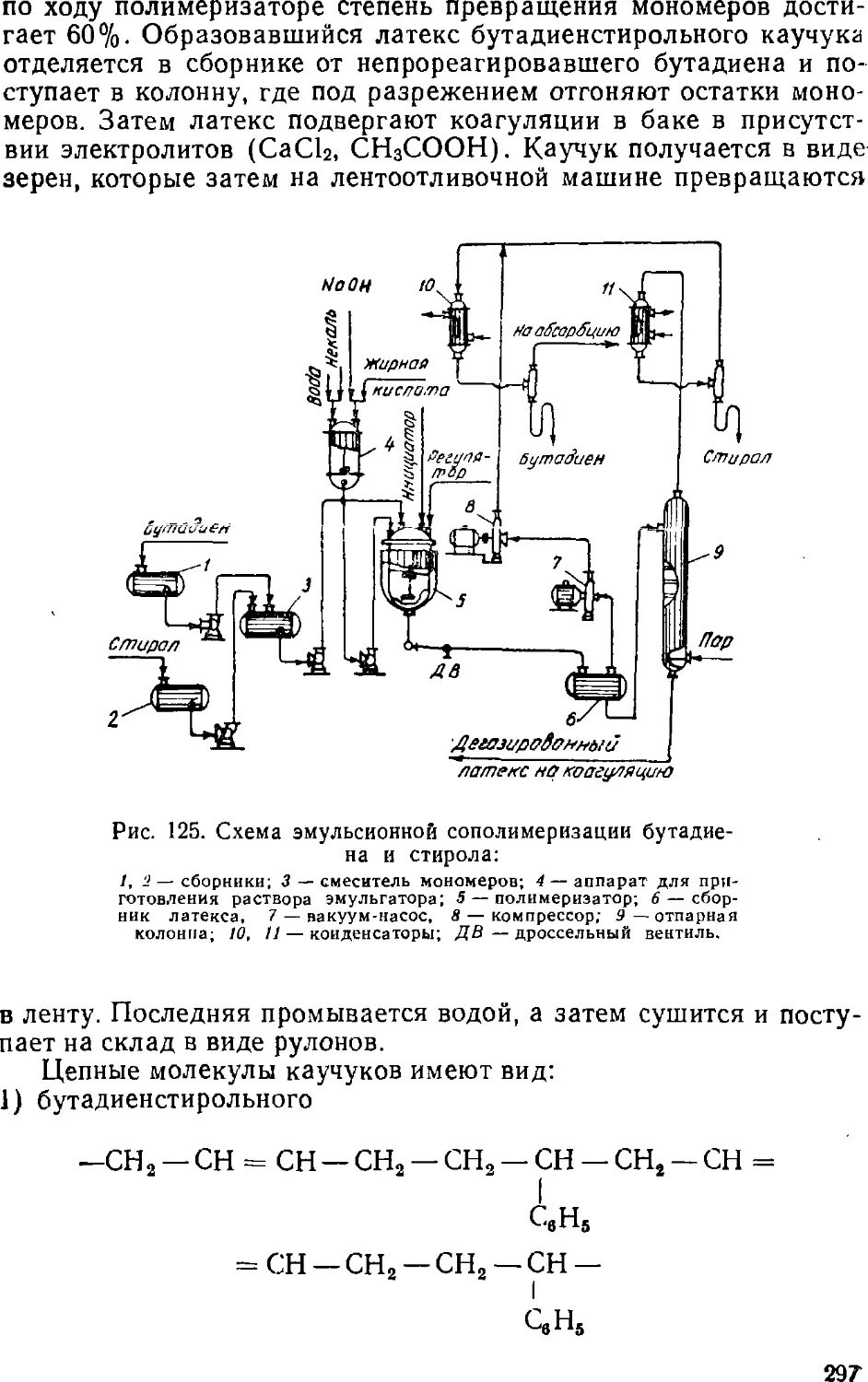

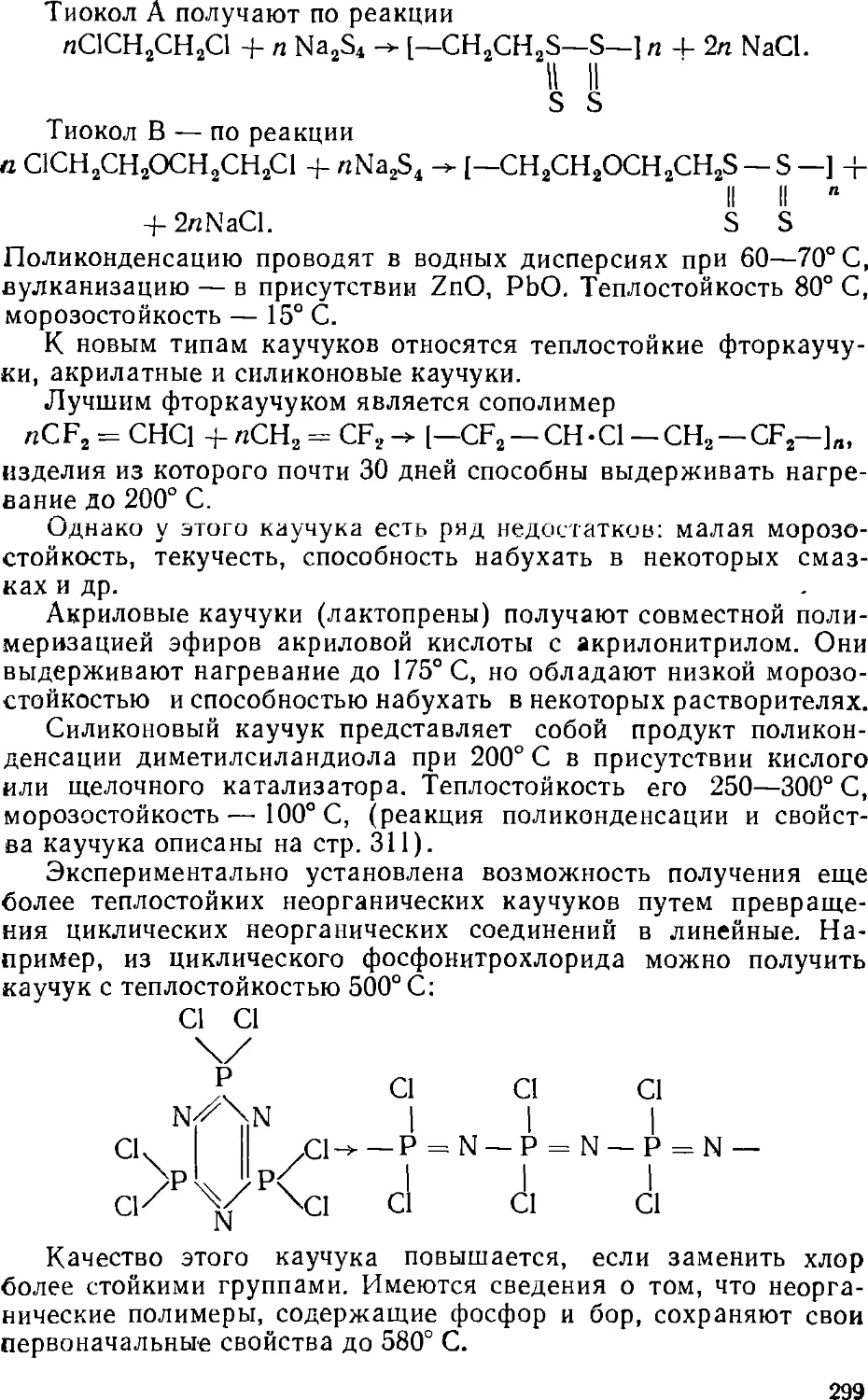







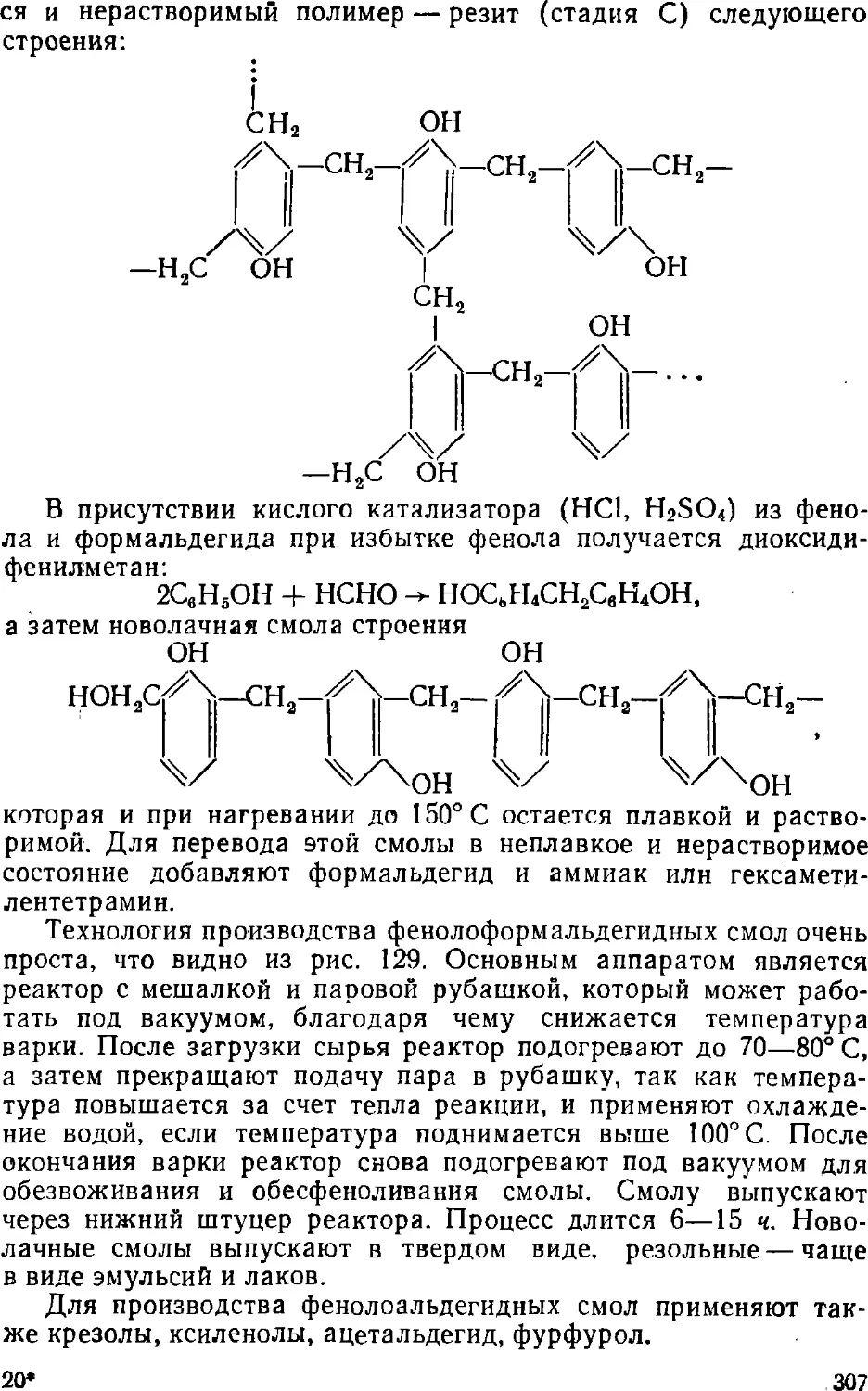






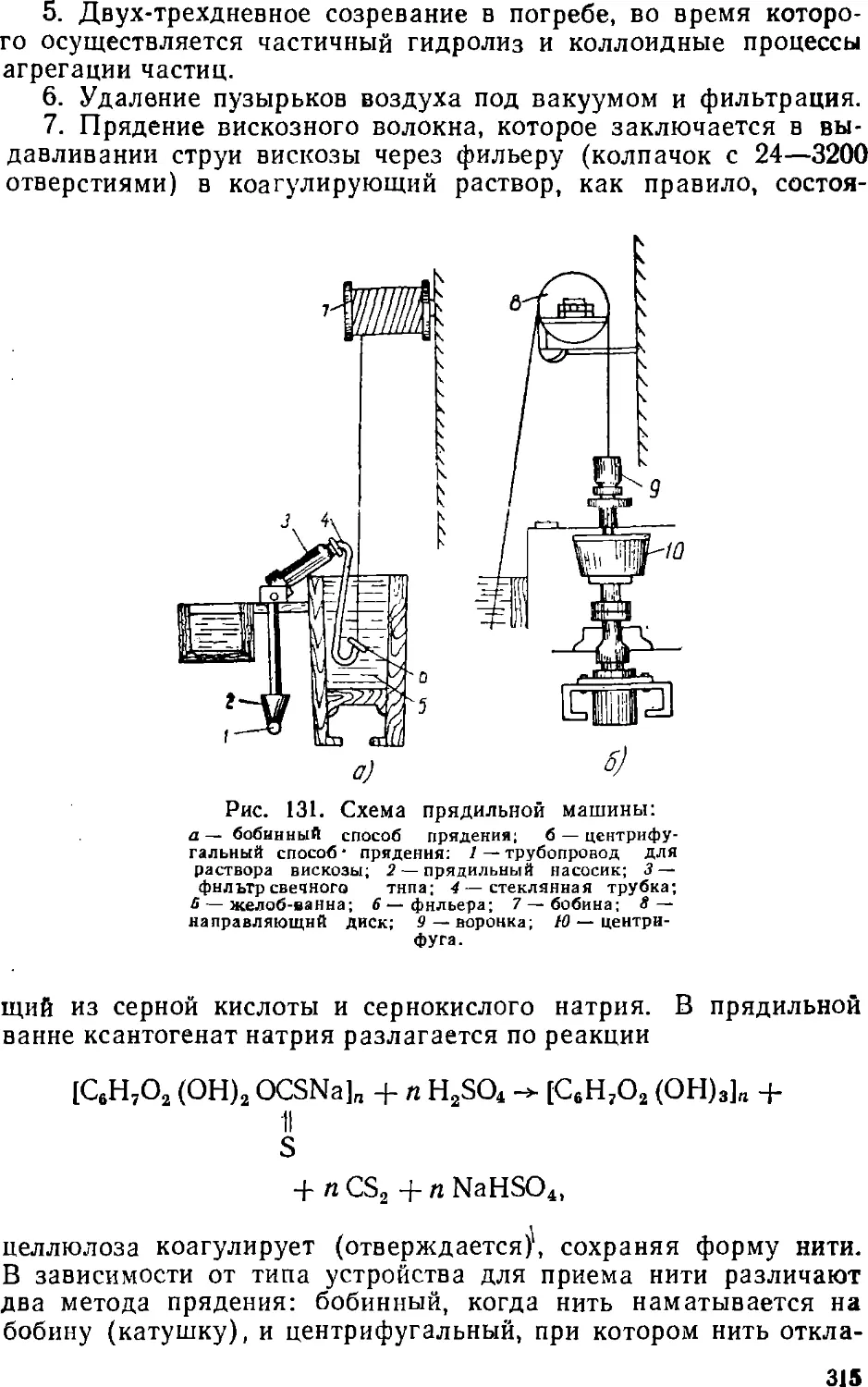


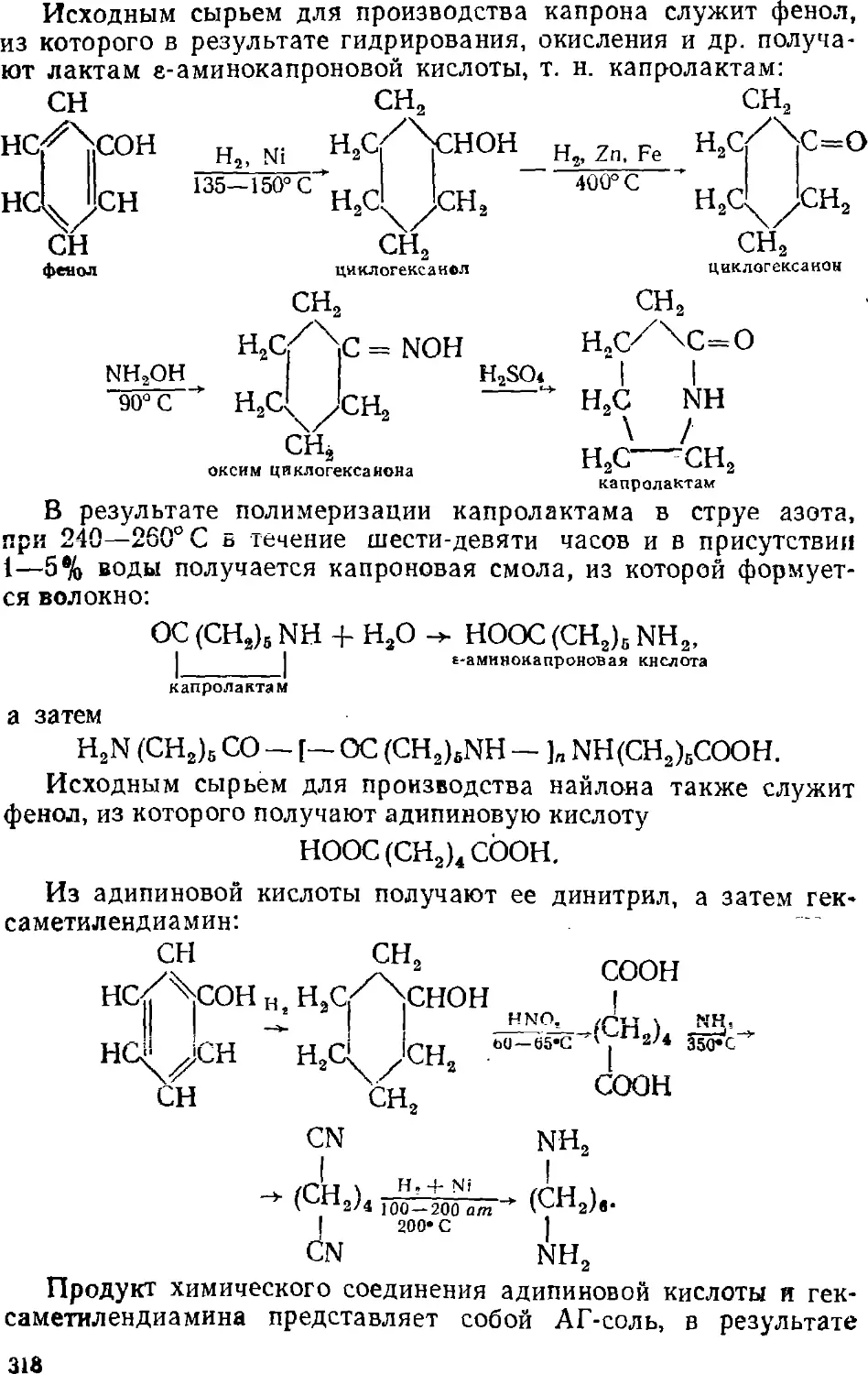




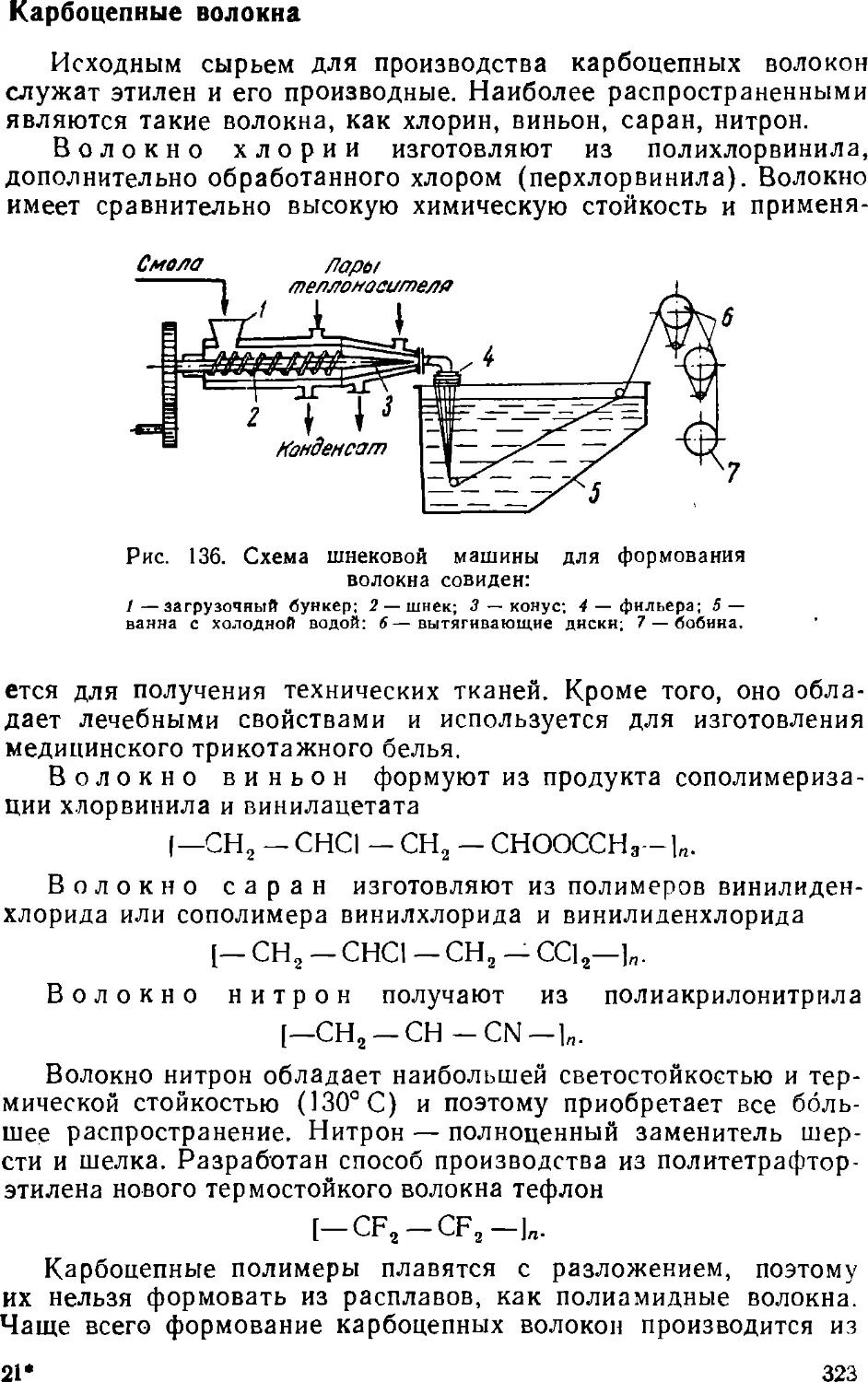

















Text
М. И. НЕКРИЧ,
|М. П. КОВАЛЕВІ,
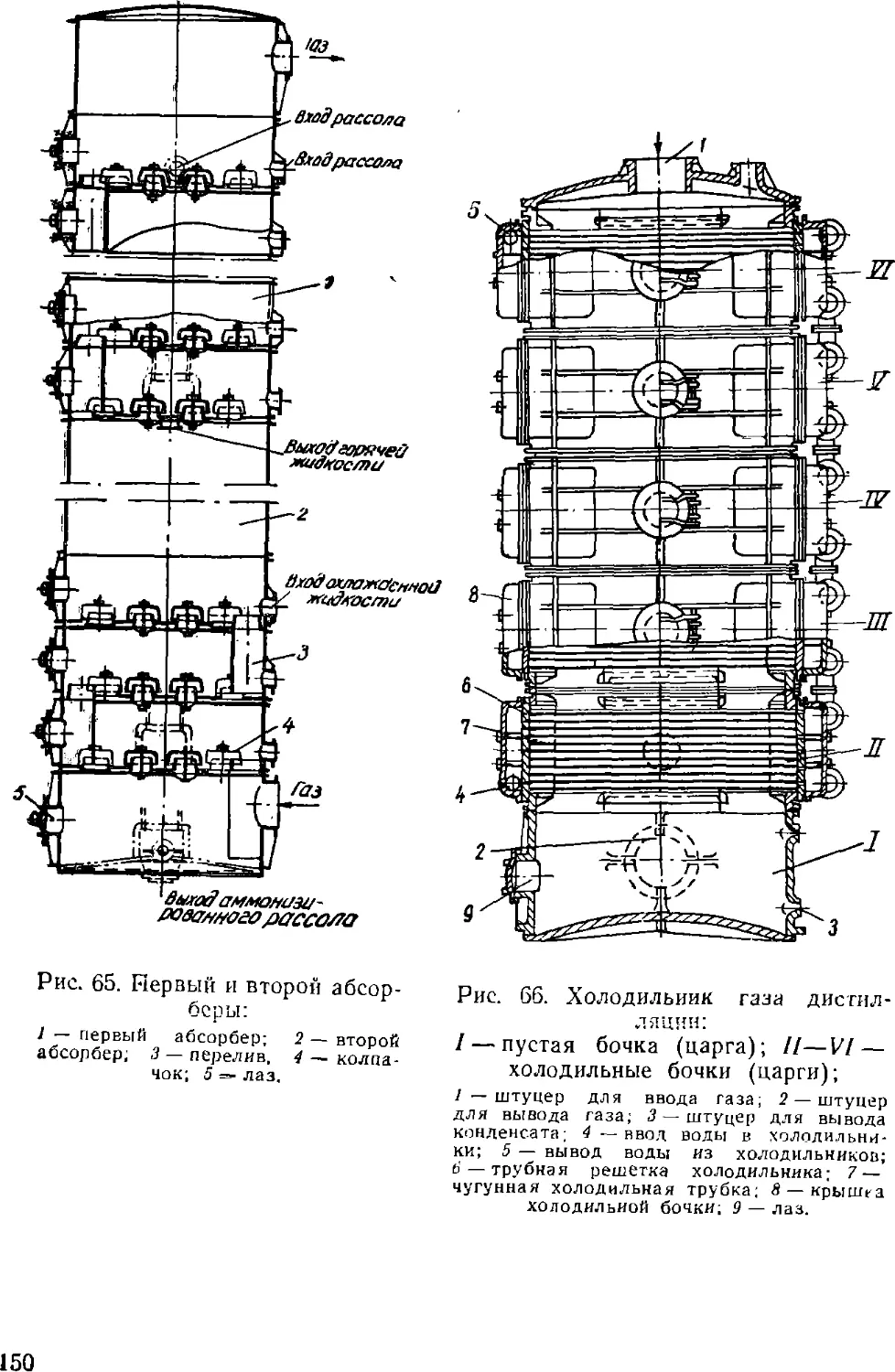
Ю. И- ЧЕРНЯЕВА
ОБЩАЯ
ХИМИЧЕСКАЯ
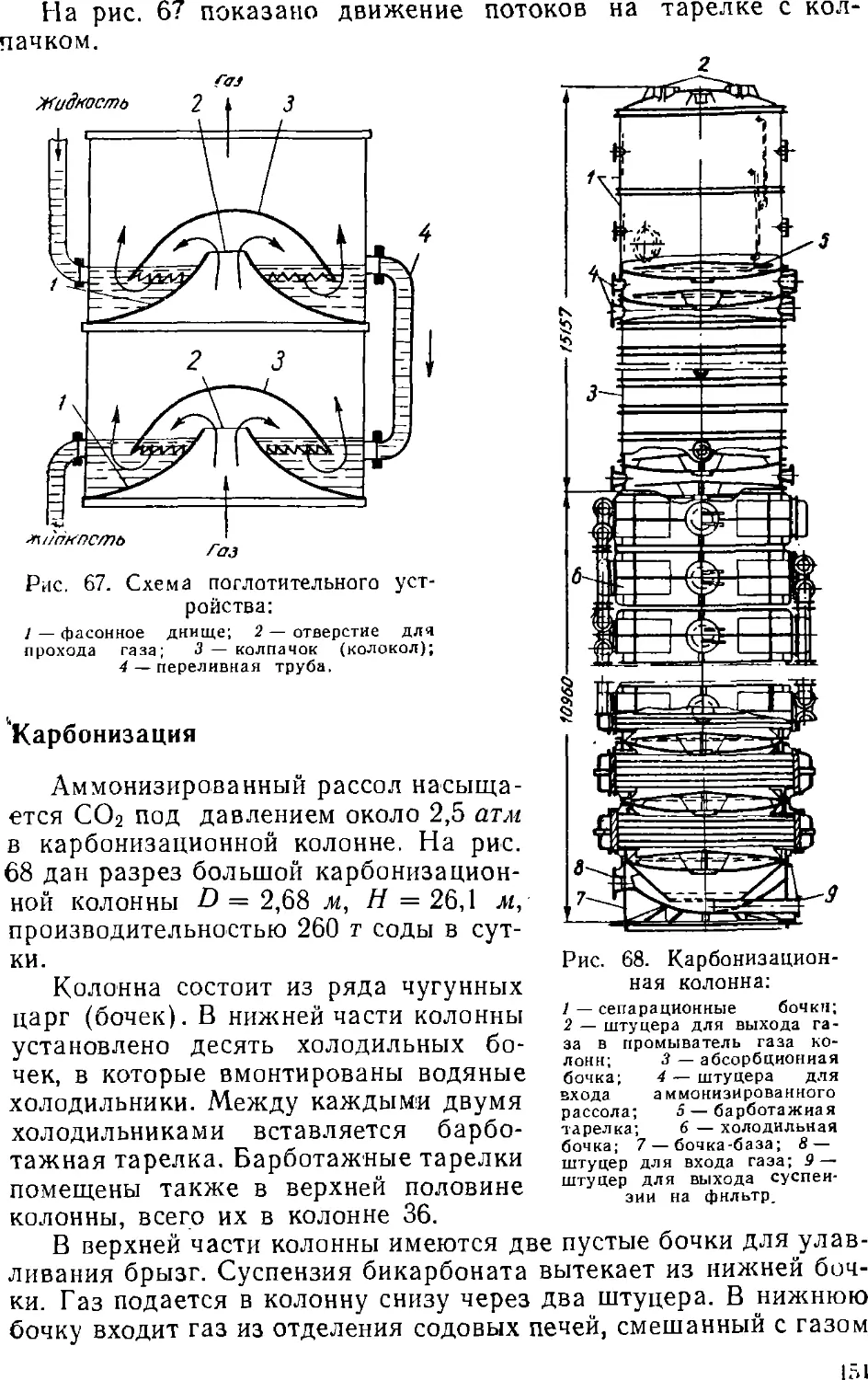
ТЕХНОЛОГИЯ
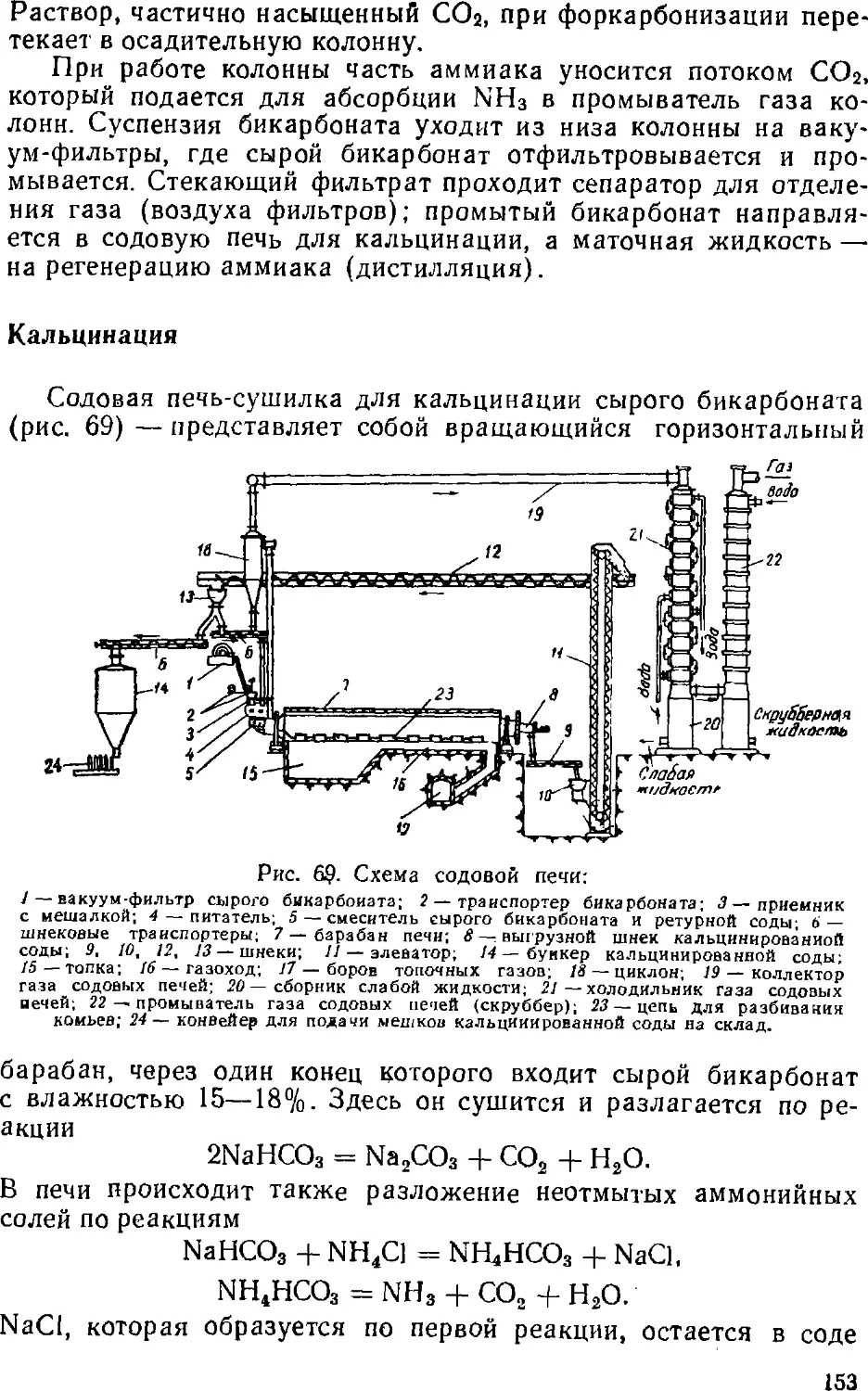
Допущено
Министерством высшего и среднего
специального образования УССР
в начестве учебного пособия для студентов
химико-технологических специальностей вузов
ИЗДАТЕЛЬСТВО
ХАРЬКОВСКОГО
УНИВЕРСИТЕТА
Харьков 1969
6П7.1
H48
Книга представляет собой краткий курс общей
химической технологии, рассчитанный на
студентов высших учебных заведений,
специализирующихся в области химического машиностроения.
В ней приведены теоретические основы и
описание основных технологических схем важнейших
химических производств.
Максим Исидорович Некрич,
^Михаил Павлович Ковалев\,
Юлия Ивановна Черняева
ОБЩАЯ ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ
Редактор И. Л. Базилянская
Тех редактор Г. П. Александрова
Корректоры Р. Е. Дорф, Е. Т. Поступай
Сдано в йабор 13/ІХ 1968 г. Подписано к печати 23/IV 1969 г. БЦ 50139.
Формат 60x90Vie- Объем 21,0 физ. печ. л., 21,0 усл. печ. л., 20,7 уч.-изд. л.
Зак. 8-1504. Тираж 17 500. Цена 73 коп. ТПУ 1968 г. поз. 6.
Типоофсетная фабрика Комитета по печати при Совете Министров УССР.
Харьков, ул. Энгельса, 11.
ПРЕДИСЛОВИЕ
Причиной, побудившей авторов приступить к работе над
предлагаемым курсом общей химической технологии, является
отсутствие достаточно компактного и удобного в пользовании учебника
для студентов химико-технологических и политехнических
институтов. Ее объем соответствует программе для специальностей
химического машиностроения 1967 года.
Курс общей химической технологии под редакцией
акад. С. И. Вольфковича слишком обширен и не под силу
студентам, которые впервые знакомятся с химическими
производствами.
Учебник по общей химической технологии под редакцией
И. П. Мухленова содержит много сведений нз теории, но, к
сожалению, технологической стороне производств в нем уделено
сравнительно мало места.
В настоящем курсе описаны важнейшие производства идаиы
теоретические пояснения к ним, приведены технологические
схемы и описание основной аппаратуры.
Главы I—VI и XVI иапнсаны Ю. И. Черняевой, главы VII—
XIII — М. И. Некричем, главы XIV н XV—|м. П. Ковалевым| и
Ю. И. Черняевой.
В составлении главы XIII принимали участие Б. П. Сахаров и
Е. И. Ведь.
ВВЕДЕНИЕ
Химическая технология представляет собой одну из важных
отраслей современной техники. Содержанием ее является
разработка и совершенствование различных методов
облагораживания и переработки сырья в товарные продукты.
Химическая технология базируется на достижениях таких
наук, как химия, физическая химия, химическая термодинамика
и др. Она охватывает преимущественно те производственные
процессы, в основе которых лежат химические и физические
превращения. Однако нередко в производстве наряду с
химико-технологическими процессами используются и механические,
например в цветной металлургии, промышленности
строительных материалов, бумагоделательной
промышленности и т. п.
В предлагаемом курсе общей химической технологии
рассматриваются общие, наиболее важные, принципиальные
основы производственных процессов. Связь между отдельными
операциями того или иного производства представлена в виде
принципиальных технологических схем.
При характеристике каждого из описанных в курсе
производств учитывается важность получаемого продукта, степень
рационального использования сырья и энергии и возможность
получения максимальных ныходпв продукта при минимальных
расходных коэффициентах.
Вы хол і'отокоіч; продукта вычисляется в процемтлх пт
теоретически возможного выхода в тех случаях, когда
технологический процесс протекает по конкретному химическому уравнению
(например, выход окиси азота при окислении аммиака,
нитробензола из бензола и пр.).
Если технологический процесс нельзя выразить химическим
уравнением, выход продукта определяется в процентах от сырья
или относится к единице затраченного сырья (например, выход
кокса из угля).
Для электрохимических процессов более характерным
является выход не по сырью, а по току. Такой выход определяется
как отношение количества практически полученного продукта к
количеству, теоретически возможному в соответствии с законами
электрохимических процессов (законы Фарадея)-
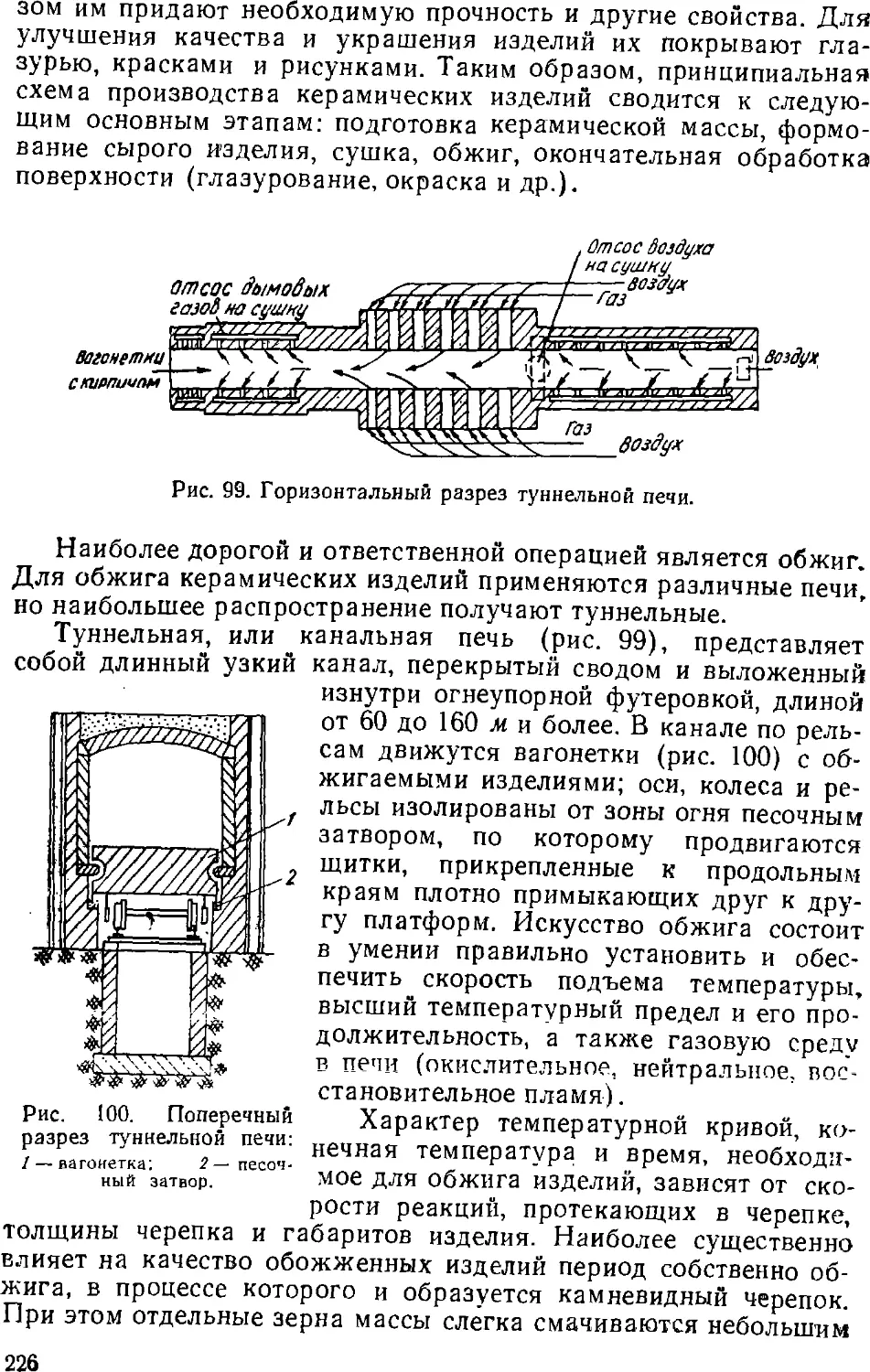
При проектировании какого-либо химического предприятия
необходимо знать производственный рецепт, т. е. соотношение
реагирующих веществ определенного качества, режим
проведения процесса в каждой его стадии и выход продукции по
стадиям и в целом по производству при определенной конструкции
аппаратов, скорости процесса и пр. Все это устанавливается в
ходе предварительных исследований в лабораториях и на
опытных установках. На основании данных исследований
составляются материальные и энергетические балансы (чаще всего
тепловые), а затем производится расчет размеров и количества
аппаратов с учетом заданной мощности завода. Под мощностью
завода подразумевается его максимально возможная проектная
(обычно годовая) производительность.
В основу материального баланса положен закон сохранения
массы. При этом общее количество поступающих в производство
материалов должно быть равно общему количеству получаемой
продукции. Если такого равенства нет, то имеют место потери
в производстве.
Тепловые балансы составляются на основании закона
сохранения энергии.
В качестве примера в табл. 1 приведен упрощенный
материальный и в табл. 2 — тепловой балансы коксования угля (на
1 т рабочей шихты). При этом принимается, чсо в результате
коксования получаются кокс и химические продукты
коксования — смола, водный конденсат и газ.
Таблица 1
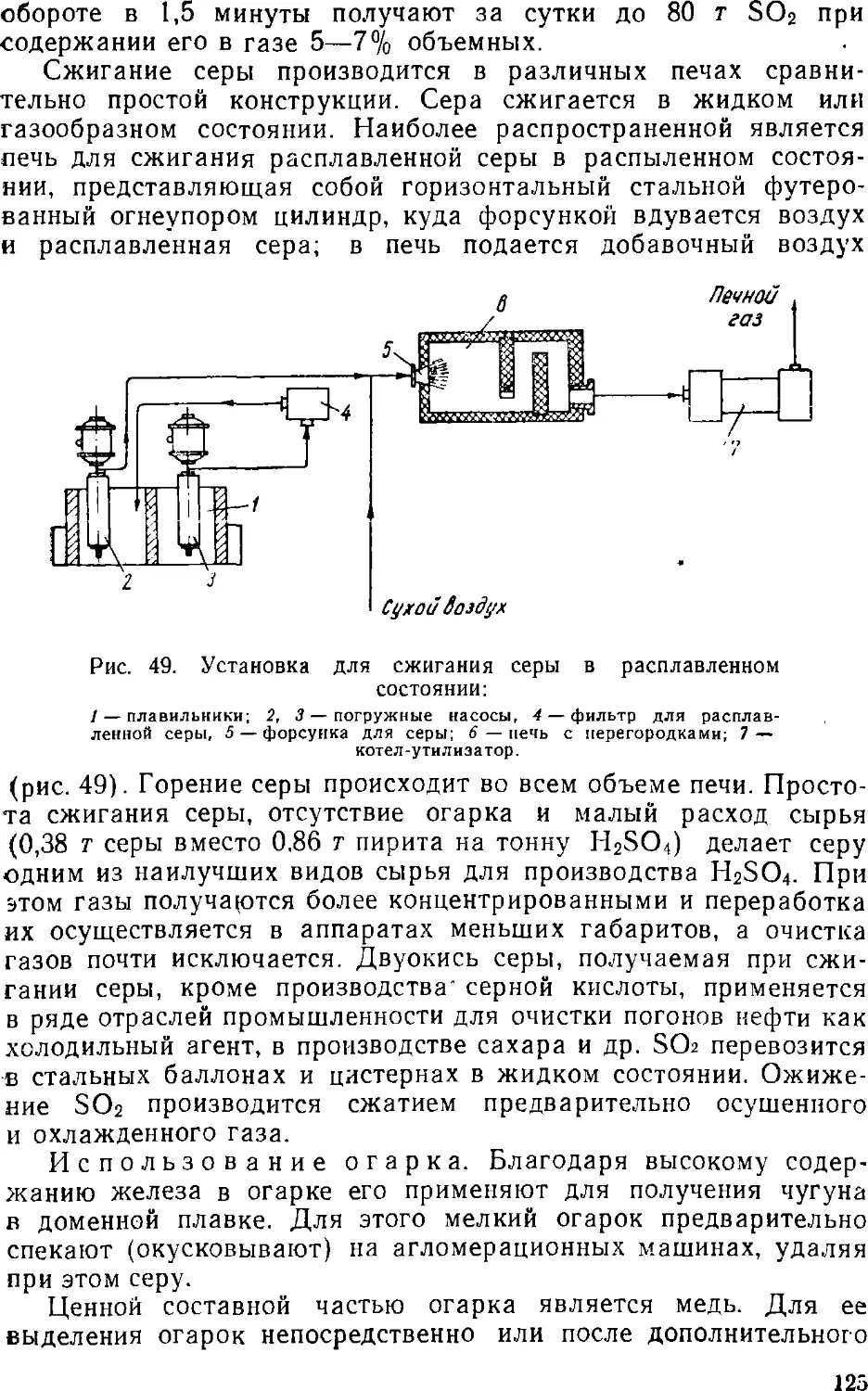
Приход
Загруженный сухой уголь
Влага угля
Всего
кг
920
80
1000
Расход
Кокс валовой
Смола
Сырой бензол
Аммиак
Газ сухой
Сера газа
Влага общая
Потери
Всего
кг
719,6
32 0
9,0
2,9
130,9
2,9
98,4
4,3
1000
Интенсифицировать работающую установку или завод
можно путем усовершенствования технологии процесса, в основном
Таблица 2
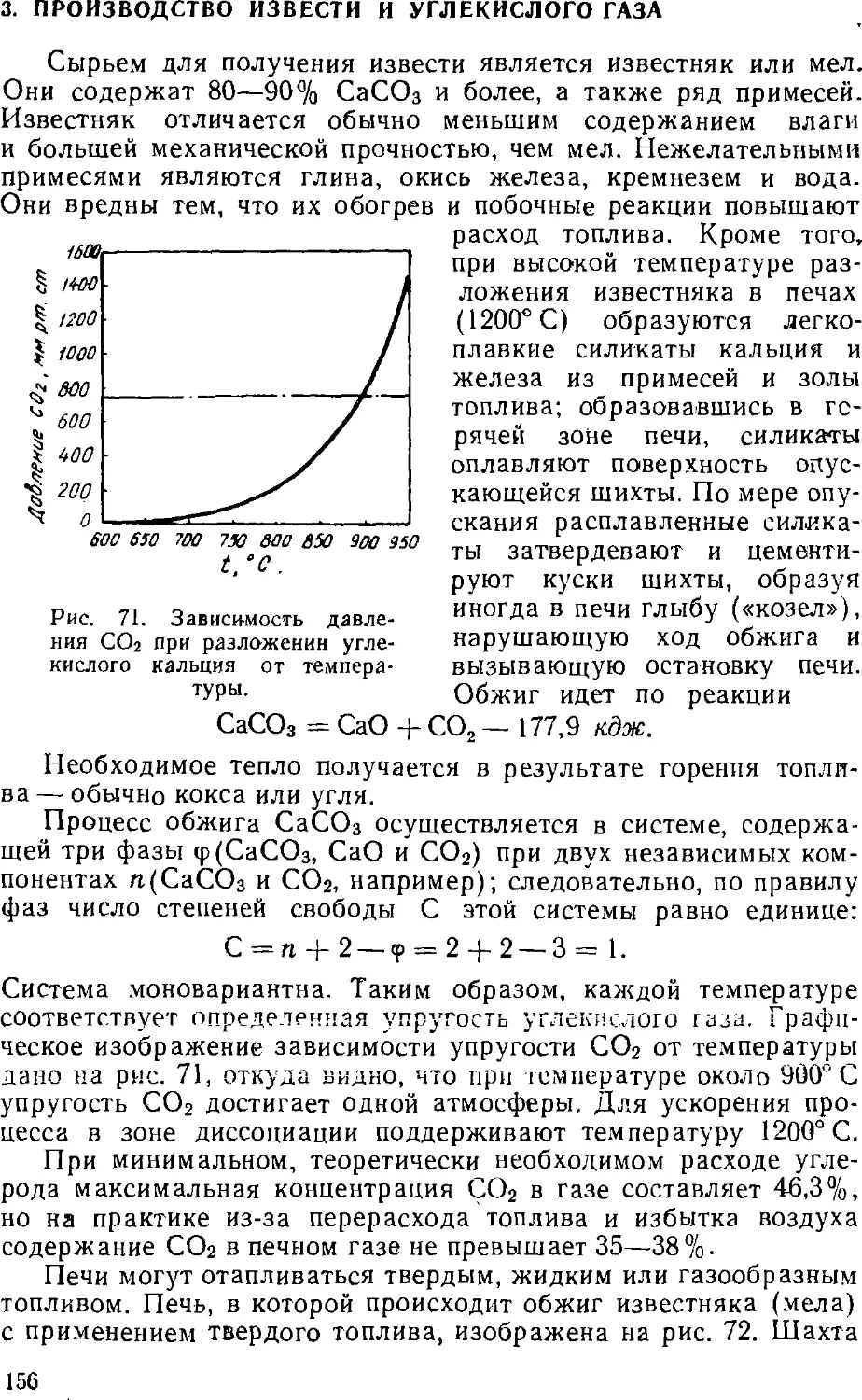
Приход
Тепло горения газа
Физическое тепло
газа и воздуха
Физическое тепло
шихты
Всего
кдж
2 654400
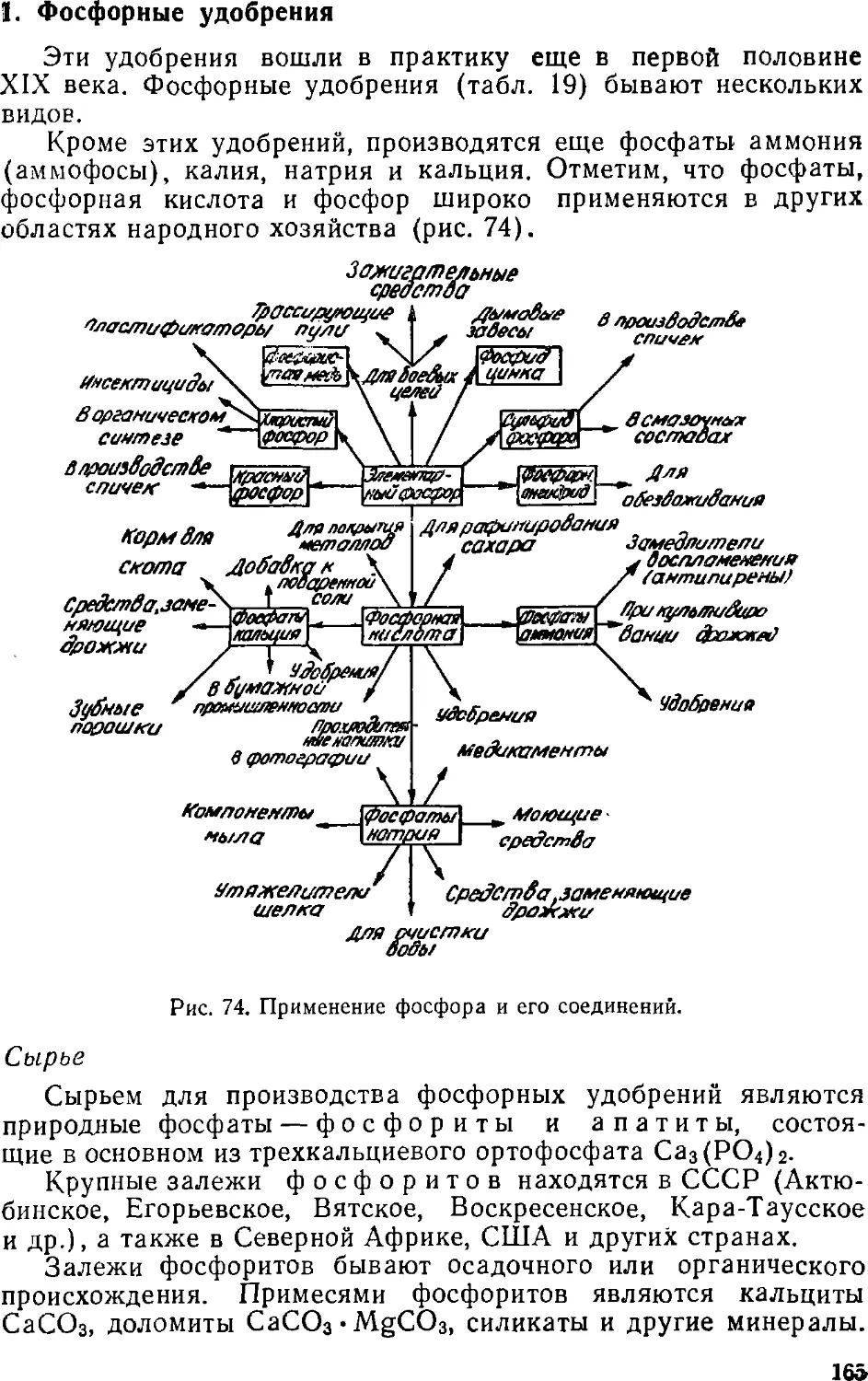
26 376
26 292
2 707 068
%
98
1,0
1,0
100
Расход
Тепло нагрева
кокса
Тепло нагрева
продуктов коксования
Тепло, уносимое
продуктами
горения
Потери тепла в
окружающую среду
Всего
кдж
1 066 800
877 506
399 840
362 922
2 707 068
%
39,4
32,4
14,8
13,3
100
увеличения скорости процесса. Это достигается за счет
повышения температуры, давления, концентрации реагирующих веществ,
увеличения поверхности соприкосновения, перемешивания и пр.
Интенсификация процесса обеспечивается также благодаря
усовершенствованию конструкции аппаратов.
Важнейшими средствами повышения производительности
труда на социалистических предприятиях являются механизация
и автоматизация производственных процессов. Автоматизация
химических производств дает возможность повысить выход и
улучшить качество продукции, так как она позволяет вести
процесс при оптимальном технологическом режиме. За счет
повышения выхода увеличивается выработка каждого рабочего, т. е.
возрастает производительность труда.
Автоматизация производств может привести к уменьшению
числа рабочих, однако не только этим определяется
экономическая эффективность автоматизации. Главное — это снижение
потерь в результате оптимизации и устойчивости
технологического режима. На многих химических производствах уже
автоматизированы отдельные аппараты и узлы, а к 1980 г.
должна быть проведена комплексная автоматизация всех
производств.
Осуществление автоматизации химико-технологических
процессов значительно облегчается в том случае, когда эти
процессы непрерывны. Но, стремясь к распространению непрерывных
методов, необходимо учитывать особенности производства. В
некоторых случаях выгоднее применять периодический метод,
например, для производства малотоннажной продукции:
красителей, лекарственных веществ и др. В высокотоннажных производ-
ствах периодический метод используют лишь при коксовании
угля.
В зависимости от направления, в котором движутся
реагирующие вещества в аппаратах, различают прямоточные, проти-
воточные и перекрестные процессы. В прямоточных процессах
движение материала и, например, теплоносителя происходит в
одном направлении, в противоточных — в противоположных
направлениях, а в перекрестных процессах материал и
теплоноситель движутся под каким-то углом друг к другу.
Обычно принято считать, что реакция заканчивается при
прохождении реагирующих веществ через аппарат. Однако в ряде
производств взаимодействие веществ, особенно газообразных, не
завершается за один проход через аппарат. Не вошедшие в
реакцию газы после отделения продукта реакции снова
возвращаются в аппарат, смешиваясь со свежей порцией газов. Такой
процесс называется циркуляционным или круговым и применяется
при синтезе аммиака, метилового и этилового спиртов и др.
Возможно многократное взаимодействие смеси реагентов в
нескольких аппаратах до полного превращения их в конечный
продукт. Это процессы с открытой цепью. По такой схеме
производится упаривание растворов в многокорпусной
вакуум-выварке, синтез аммиака по некоторым старым схемам и др.
Регенерация материалов и тепла
Для повышения техно-экономических показателей
производства необходимо снижение расхода материалов и энергии,
особенно тепловой. Экономии материалов часто можно достичь
путем регенерации, т. е. восстановления первоначальных свойств
отработанных материалов с целью повторного их использования
(регенерация катализаторов, ионообменных смол, аммиака из
его солей в содовом производстве и др). Под регенерацией
тепла подразумевается использование тепла реакций или тепла
отходящих газов для предварительного нагрева реагирующих
веществ. Наряду с обычными теплообменниками с этой целью
применяются специальные аппараты — регенераторы или
рекуператоры. В некоторых случаях устанавливаются парогенераторы
(котлы-утилизаторы), где тепло отходящих газов используется
для получения пара.
Краткие сведения из истории развития химической
промышленности. Состояние и перспективы развития химической
промышленности в СССР
Основы практической химии были известны человечеству
задолго до нашей эры. Еше в древнем Египте были развиты такие
отрасли производства, как стеклоделие, крашение, обработка
драгоценных металлов, получение соды, уксусной кислоты.
В Киевской Руси также существовали различные промыслы:
выплавка цветных и благородных металлов, железа, выделка
кожевенных товаров, производство пороха, лекарственных
веществ, стекла, керамики. При Петре Первом началось развитие
металлургии и связанной с ней отрасли лесохимии —
смолокурения.
В 1720 г. был построен химический завод возле Москвы,
производивший азотную кислоту, железный купорос, краски,
скипидар, канифоль. В 1805 г. вступил в строй сернокислотный завод,
работавший камерным способом, и др. Становление в России
химии как науки относится ко 2-й половине XIX века, когда
русскими учеными были синтезированы новые органические
соединения. Н. Н. Зинин в 1842 г. получил анилин восстановлением
нитробензола, благодаря чему создались условия для
возникновения анилинокрасочной промышленности. Создатель теории
строения органических соединений акад. А. М. Бутлеров A828—
1886 гг.) разработал методы гидратации олефинов в спирты и
ряд других синтезов.
В. В. Марковников открыл реакции галоидирования
углеводородов; ему же принадлежит открытие в нефти нафтеновых
углеводородов. М. Г. Кучеров A881) открыл реакцию
гидратации ацетилена в присутствии солей ртути, что привело к
производству синтетической уксусной кислоты. Очень много для
развития химической науки и промышленности сделал Д. И.
Менделеев.
Несмотря на высокий уровень химической науки,
дореволюционная Россия по состоянию химической промышленности
значительно отставала от других стран. Это объясняется тем, что
основные отрасли промышленности находились тогда в руках
иностранцев, не заинтересованных в развитии народного
хозяйства. После Октябрьской революции началось бурное развитие
всех отраслей промышленности, в том числе и химической^
Быстрым темпам развития химической промышленности
способствовали решения майского A958 г.) Пленума ЦК КПСС,
положенные в основу семилетнего плана развития народного
хозяйства СССР A959—1965 гг.), в котором особое внимание уделено
развитию химии и технологии полимеров.
Из итогов, подведенных XXIII съездом КПСС A966 г. ),
следует, что семилетний план развития химической
промышленности успешно выполнен. В новом пятилетнем плане A966—
1970 гг.) было снова предусмотрено опережающее
развитие химической и нефтехимической промышленности,
дальнейшее развитие производства синтетических материалов — каучу-
ков, пластических масс, химических волокон, синтетических
моющих средств на основе непищевого сырья, удобрений и т. д.
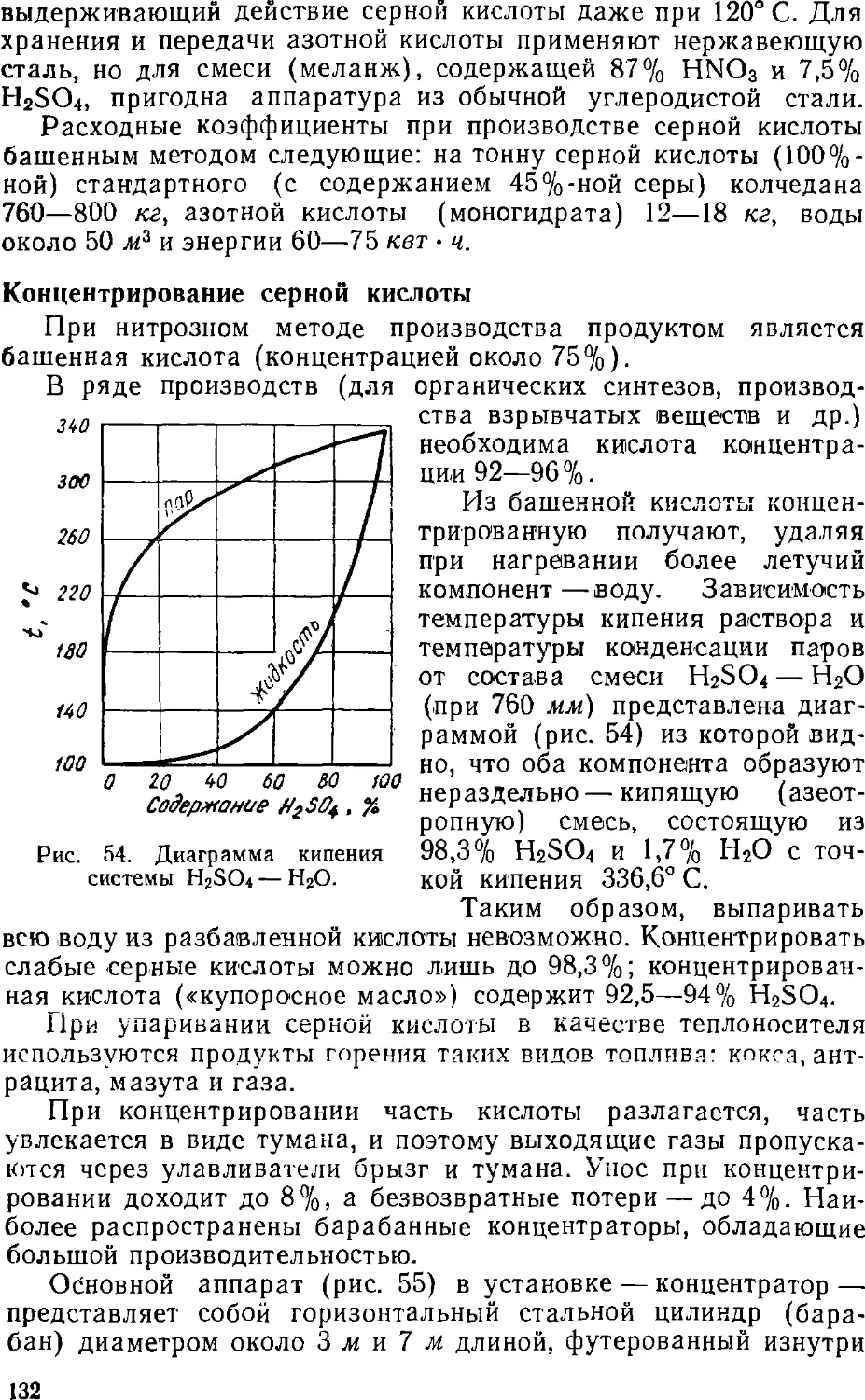
О физико-химических основах химической технологии
Разработке любого технологического процесса должно
предшествовать изучение его теоретических основ. Первостепенное
значение имеет определение обратимости или необратимости
реакций и изучение их скорости, оказывающей решающее влияние
на размеры и производительность производственных установок.
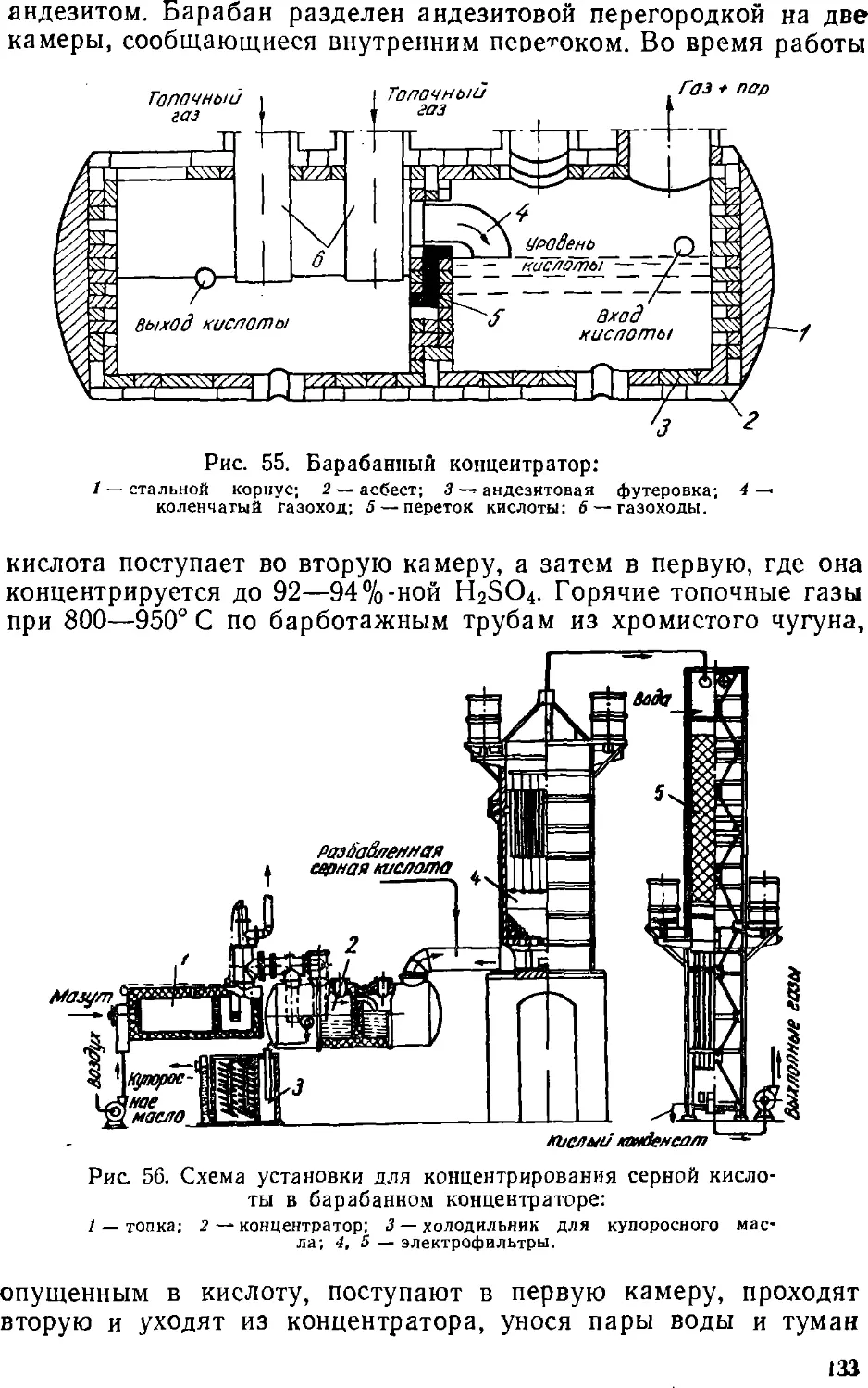
Для ускорения процессов широко используются следующие
физико-химические и технические приемы: а) увеличение
концентрации реагентов; б) удаление конечных продуктов из сферы,
реакции; в) установление оптимального температурного
режима; г) увеличение поверхности соприкосновения реагентов;
д) применение катализаторов. Все эти приемы основаны на
учете существующих физико-химических и термодинамических
закономерностей.
По закону действующих масс скорость реакции в каждый
момент пропорциональна произведению концентраций реагентов к.
этому моменту. В общем виде скорость реакции
аА + ЬВ...ч-rR +sS, A).
о = kC\ X CbB, B>
где а и Ь— количество молей веществ А и В;
С— концентрация;
k — константа скорости.
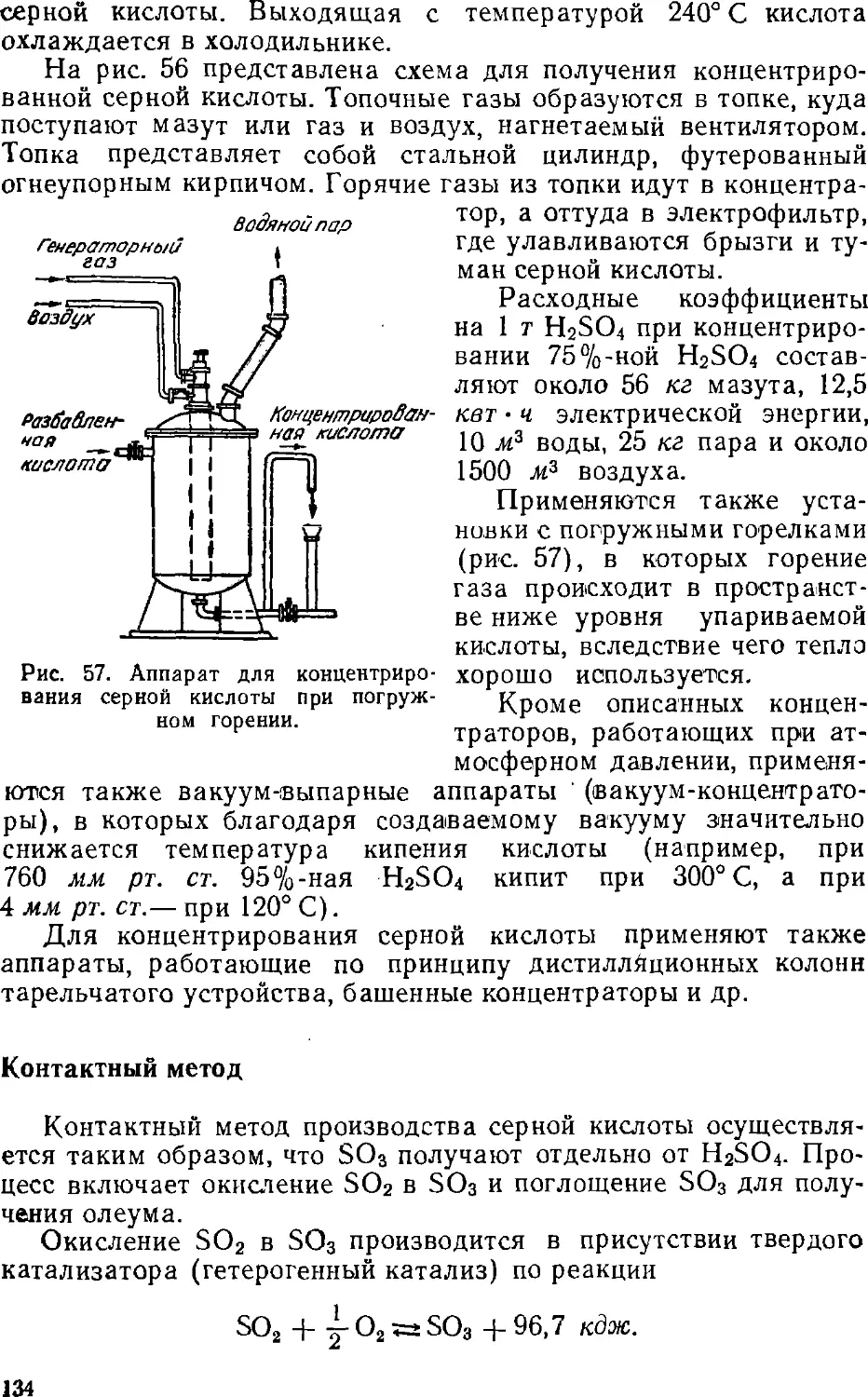
В ходе реакции концентрации исходных веществ и скорость
прямой реакции уменьшаются, а концентрация продуктов
реакции и скорость обратной реакции увеличиваются. Если не
выводить продукты из сферы реакции, наступает состояние
равновесия, при котором скорости прямой и обратной реакций равны
v = vl— о2 = 0 C)
It видимое течение реакции прекращается. Константа
равновесия реакции A) равняется отношению констант скоростей
прямой и обратной реакций
K 4V
A /c2 cacb 14>
2 CACB
и определяется расчетным или опытным путем на основании
анализа продуктов реакции в момент равновесия.
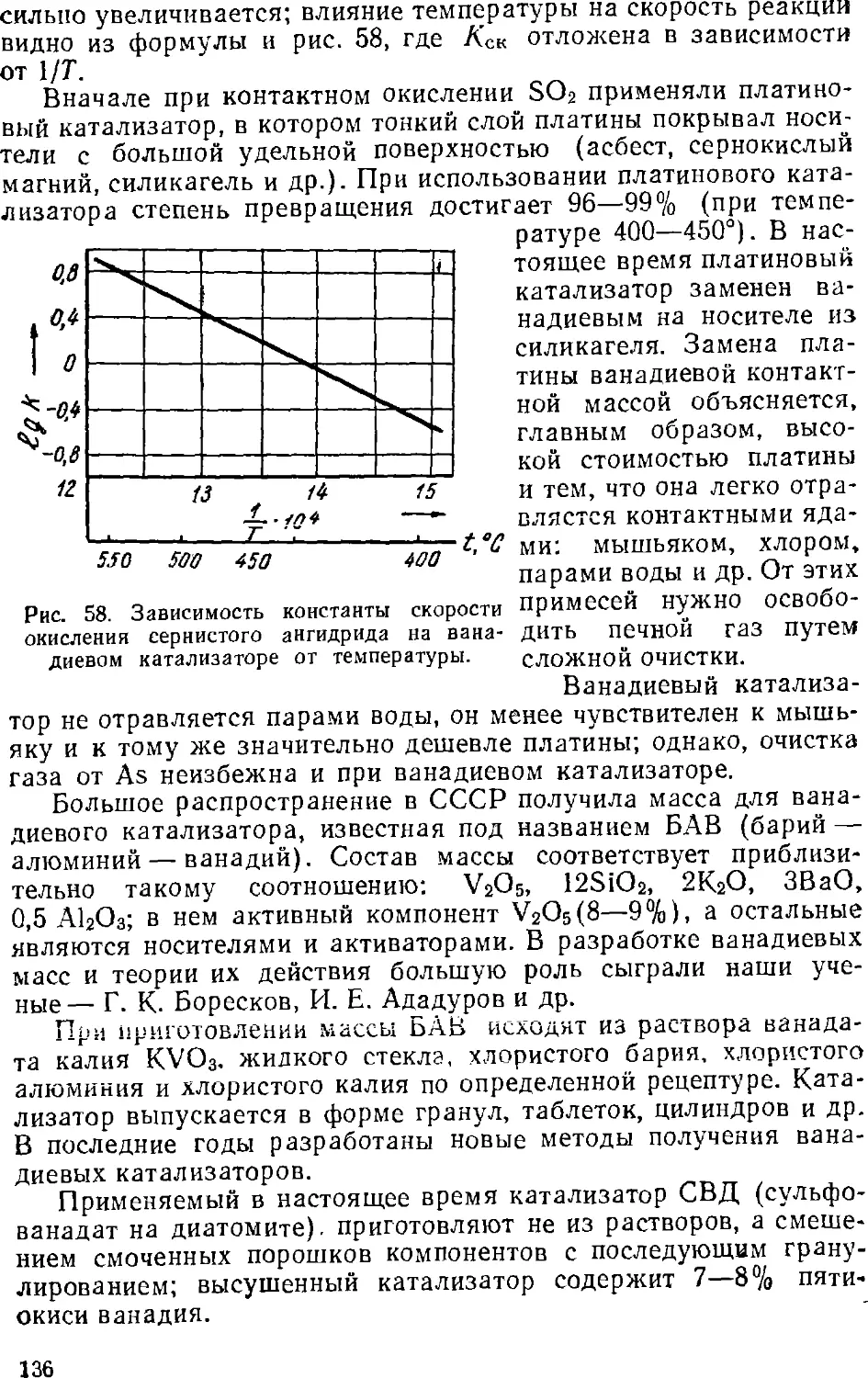
По принципу Ле-Шателье система, выведенная из состояния,
равновесия, стремится ослабить влияние факторов, выводящих
ее из равновесия.
Если реакция протекает по уравнению
ак + ЬВ ->- rR + Q, E)
где а, Ь и г — количество молей веществ А, В и R, Q —
тепловой эффект реакции, и сумма объемов А и В больше объема R,
то согласно принципу Ле-Шателье для увеличения выхода
продукта R необходимо снизить температуру и концентрацию
продукта, т. е. вывести его из сферы реакции и увеличить давление
,н концентрацию исходных веществ А и В. При этом повышение
концентрации А будет увеличивать выход продукта по
отношению к В и наоборот.
Фактором, наиболее сильно ускоряющим реакцию, является
температура. Зависимость константы скорости реакции от
температуры определяется уравнением Аррениуса
k = kae~~ яТ , F)
а после логарифмирования
•где k, kQ, k\ и k2 — константы скорости реакции при
соответствующих температурах Т, Та, Т\, Т2;
е — основание натуральных логарифмов;
Е — энергия активации реагирующих веществ,
дж/моль;
R — газовая постоянная, джімоль • град.
Для большинства реакций применимо правило ВантТоффа,
согласно которому повышение температуры на 10° С увеличивает
скорость реакции в два — четыре раза. При выборе
оптимальной температуры процесса учитывается скорость прямой и
обратной реакции, а также возможность разложения продуктов
реакции и конструктивные особенности аппаратов.
На скорость реакции влияет и увеличение поверхности сопри-
.косновения реагирующих веществ, особенно при медленно
протекающих гетерогенных некаталитических реакциях, т. е.
реакциях между твердыми веществами (Т — Т), между жидким»
и твердыми (Ж — Т) и т. д. ' і
Скорость реакции в этих случаях определяется увеличением
-количества продуктов во времени:
« = ? = К/*С (8)
К—коэффициент массопередачи = г/гдс кг/м2-г/кг/м3-м/ч,
F— поверхность соприкосновения,
ДС—движущая сила реакции.
Для гомогенных реакций (Ж—Ж), (Г—Г)
где и = %=К*С. (9)
К — константа скорости,
ДС—движущая сила (произведение концентраций
реагирующих веществ),
,10
По известному времени х определяется общий объем аппаратов:
v =tV.
где V—общий объем взаимодействующих веществ.
Применение высоких температур в комбинации с высокими
давлениями и при использовании катализаторов позволило
осуществить такие важные технологические процессы, как синтез
аммиака, азотной кислоты, метилового спирта, мочевины,
гидрогенизации угля и др.
Дальнейшей интенсификации производства будут
способствовать повышение активности применяемых в гетерогенном
катализе катализаторов путем введения добавок и улучшения их
микроструктуры, а также получение новых, более активных
катализаторов.
Часть первая
СЫРЬЕ. ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ ТОПЛИВА.
ГЛАВА I
ВИДЫ СЫРЬЯ
И ЕГО ПРЕДВАРИТЕЛЬНАЯ ПОДГОТОВКА
Сырье, перерабатываемое химической промышленностью,
встречается в природе в различном состоянии — твердом,
жидком и газообразном. К твердому сырью относятся различные
руды, твердое топливо, растения; к жидкому — нефть, вода,
соляные рассолы; к газообразному — воздух, нефтяные и природные
газы.
По происхождению сырье подразделяется на три вида —
минеральное, растительное и животное.
1. МИНЕРАЛЬНОЕ СЫРЬЕ
К минеральному сырью относятся различные минералы,
входящие в состав земной коры до глубины 16 км. В минералогии
описано примерно 2500 минералов различного химического
состава, физических свойств и кристаллической структуры.
Условно минеральное сырье подразделяют на рудное,
нерудное и горючее.
Рудное сырье — это горные породы, из которых получают
металлы: черные (Fe, Cr, Мп); цветные, в том числе тяжелые (Си,
Ni, Pb, Zn, Si) и легкие (К, fta, Al, Mg); редкие (W, Mo, V. Co,
Be, Zr); благородные (Pt, Pd, Ir, Os, Ru, Au, Ag); рассеянные
элементы Sc, Hi, Br, Rb, In, Th, Se, Те, Та, Re, Ra и др.),
которые не образуют самостоятельных минералов и месторождений
и добываются вместе с другими металлами, например, рений из
молибденовых руд, гафний из минералов циркония, радий из
урановых руд.
Нерудное сырье—горные породы, которые применяются в
качестве строительных материалов (песок, гравий, глина и др.);
для производства неметаллов, например фосфора, удобрений и
других промышленных продуктов (фосфориты, фторапатиты
и пр.).
12
К горючему ископаемому сырью относят торф, каменные угли,
нефть, горючие сланцы, газы.
Некоторые химические элементы встречаются в самородном
состоянии, например, сера, золото, платина, графит, медь;
многие в виде сернистых соединений (FeS2, HgS, AS2S3, Ag3AsS3
и др.), окислов (SiO2, Fe2O3, FeO, Сг2О3, МиО2), галоидных
соединений (NaCl, КС1 • MgCl2 • 6Н2О, CaF2 и др.), солей —
СаСО3, NaAlSiO4 (нефелин), CaSO4 • 2Н2О, [Са3(РО4J]3 • СаХ2
(апатит) и др.
Еще не все открытые химические элементы добываются и
используются промышленностью. Особенно затруднительна добыча
рассеянных элементов, для извлечения которых необходимо
переработать огромное количество руды. Однако в связи с
расширением области применения редких элементов (атомная
энергетика, радиоэлектроника, ракетная техника) совершенствуется
технология их добычи и увеличивается число используемых
элементов.
Для обеспечения химической промышленности сырьем в
нашей стране успешно ведутся геологические поиски и
горнотехническая разведка полезных ископаемых. В настоящее время по
многим видам разведанного сырья Советский Союз занимает
первое место в мире.
2. ДОБЫЧА И ПЕРВИЧНАЯ ПЕРЕРАБОТКА СЫРЬЯ
Извлечение полезных ископаемых из недр земли
осуществляется в промышленных горных разработках, где в зависимости
от глубины залегания добычу ведут открытым или закрытым
способом. К первому типу разработок относятся карьеры для
добычи известняка, гипса, песчаника; ломки, в которых
добывается строительный камень, гранит и пр.
Ко второму типу разработок принадлежат рудники для
добычи медных, железных, фосфатных и других руд;
каменноугольные и соляные копи и шахты, золотые и платиновые прииски,
нефтяные промыслы и пр.
В связи с трудностью ведения подземных работ там, где это
возможно, стремятся разрабатывать руды и угли открытым
способом. Добытое сырье часто не отвечает требованиям
государственного стандарта (ГОСТ) и нуждается в предварительной
подготовке: размоле, сортировке или классификации,
брикетировании, спекании (агломерации), обогащении, обезвоживании,
пылеотделении и пр.
Предварительная подготовка сырья чаще всего производится
на месте добычи, после чего его доставляют перерабатывающему
предприятию. Для дробления и размола сырья используются
дробилки различных конструкций (щековые, молотковые,
валковые, шаровые, стержневые мельницы и др.). Сортировка или
классификация выполняется либо сухим, либо мокрым способом.
13
Сухой способ заключается в просеивании (грохочении)
сырья через сита (грохоты) с отверстиями различных
размеров. Сита бывают прямоугольными неподвижными,
качающимися, барабанными, разделенными на несколько секций с
отверстиями различной величины. Для грохочения средних и мелких
классов угля и обезвоживания продуктов обогащения широко
применяются полувнбрационные наклонные грохоты с
круговыми качаниями в вертикальной плоскости и вибрационные
грохоты.
Метод воздушной сепарации основан на
различных скоростях падения твердых частиц различного размера н
веса в воздушном потоке.
Мокрый (гидравлический) способ
классификации сырья основан на различных скоростях падения частиц в
потоке воды, в зависимости от их размера и удельного веса.
Обычно методом гидравлической классификации
обрабатывают материал, состоящий из зерен размерами от 1 до 3 мм. Во
многих случаях, например для термических процессов в
металлургии, нужно не измельчать, а укрупнять частицы сырья. К
способам укрупнения частиц относится брикетирование —
прессование порошка, часто со связующим —смолой и др., а также
агломерирование — спекание частиц при высокой температуре.
3. МЕТОДЫ ОБОГАЩЕНИЯ СЫРЬЯ
Обогащением называют процессы, свя-занные с повышением
содержания в сырье полезных составных частей или элементов.
Чем больше в сырье таких элементов, тем экономичнее его
перевозка и переработка.
Таблица 3
Свойства сырья, используемые
при обогащении
Метод обогащения
Цвет и блеск минералов
Величина зерен
Форма минеральных зерен
Скорость движения зерен по
наклонной плоскости
Удельный вес и крупность зерен
Скорость падения зерен в воде
Скорость падения в воздухе
Всплывание материалов в тяжелых
жидкостях и суспензиях
Электропроводность минеральных
зерен
Магнитная проницаемость минералов
Смачиваемость поверхностей граней
минералов жидкостями
14
Ручная рудоразборка
Грохочение, просеивание
Обогащение по форме
Обогащение по треиию
Обогащение промывкой в Желобах,
на концентрационных столах
Мокрый метод обогащения
Воздушное обогащение
Разделение материалов в тяжелых
жидкостях и суспензиях
Сухое и мокрое электростатическое
обогащение
Сухое и мокрое электромагнитное
обогащение
Флотационный метод обогащения
1
-*¦— — і — ^z. > Вода-
Способы обогащения сырья можно подразделить на
механические, основанные на различии в физических свойствах сырья,,
и химические,— основанные на различной растворимости
составных частей сырья в одном или разных растворителях или на
различной способности их окисляться, разлагаться и пр.
Механические способы обогащения и физические свойства сырья, на:
которых они основаны, приведены в табл. 3.
Из мокрых способов обогащения угля наибольшее
распространение получил метод
отсадки в отсадочных машинах.
На рис. 1 схематически
изображена отсадочная
машина, которая состоит из двух
отделений — поршневого и
рабочего. При движении поршня ^^
вверх и вниз в рабочем отделе- ^
нии создается переменная
восходящая и нисходящая струя
воды. Вода, протекая через
отверстия сита, по которому
движется материал, делит
последний на более легкие и
тяжелые частицы. Тяжелые
частицы располагаются на сите
МаШИНЫ, а легкие НахОДЯТСЯ В Рис" '• Схема осадочной машины:
верхних слоях материала и
уносятся вместе с водой через
борт машины. Медленно передвигаясь по ситу, тяжелые частицьь
падают перед бортом машины в специальный бункер для породы.
При обогащении мелких классов углей на сите отсадочной
машины укладывают «постель» — слой тяжелого материала,
например полевого шпата или речной гальки, чтобы предупредить
проскакивание материала через сито. Пульсация воды может
создаваться не поршнем, а сжатым воздухом; такие машины
называются беспоршневыми, или пневматическими.
Методы обогащения сырья в тяжелых средах или суспензиях
довольно широко применяются при обогащении каменных углей.
Для приготовления суспензий в качестве утяжелителя
используются тонкие минеральные порошки, например магнетит и
кварц. Удельный вес тяжелых суспензий должен быть близок к
удельному весу породы; тогда концентраты будут всплывать, а
порода останется в тяжелой жидкости и осядет в ней. При
использовании тяжелых сред разного удельного веса
получаются различные фракции угля. После отделения углей от
жидкости последние регенерируются. Обогащение этими способами
осуществляется в аппаратах различных конструкций: сепараторах,
центрифугах и др.
I
/ — корпус; 2— перегородка; 3 — поршень;
4 — решето.
15-
Электростатическое и электромагнитное обогащение
производится с помощью соответствующих сепараторов.
Электромагнитное обогащение применяется для железных руд, хромистого
железняка FeO • Сг2Оз, пиролюзита МпО2 и пр. Электростатический
метод обогащения, основанный на различной
электропроводности материалов,— для отделения серного и медного колчедана,
свинцового блеска, руд, содержащих самородное золото,
серебро от известняка, гипса, песка, силикатов.
Флотационный метод обогащения
Из всех перечисленных методов обогащения наиболее
распространенным является флотационный (всплывной) метод,
основанный на различной смачиваемости водой содержащихся в руде
минералов. Обогащение
производится следующим
образом. Приготовляется
суспензия из тонкоизмель-
ченной руды и воды во
флотационной машине
(рис. 2), и через эту
взвесь пропускается
воздух в виде мелких
пузырьков. Плохо смачиваемые
водой зерна
прилипают к пузырькам воздуха
и, поднимаясь вместе с
ним вверх, образуют на
поверхности пену,
которую легко удалить из
аппарата. Хорошо
смачивающиеся частицы оседают
на дно аппарата. Таким
образом сырье разделяют
на фракцию, более
богатую полезной
составляющей сырья.— концентрат
и обедненную этой
составляющей — отходы.
Питанив
p;:c 2. Схсмлтії'ієскин разрез ячейки флогя-
цлонной машины с механическим
перемешиванием:
;- колосниковая решетка; 2 - винтовая мешалкп; ьінпггІ_ пг.ш,рит мнпгп
3 — труба для подачи воздуха: 4 — камера для ИНОГДа ПрИМеНЯЮТ МНОГО-
ьыгрузкгс концентрата; 5 — окно; 6—порог; 7 —
промежуточная камера.
кратную флотацию.
Природной плохой
смачиваемостью водой отличаются самородная сера, графит,
блестящий каменный уголь, озокерит и др. Большинство же
минералов обладает хорошей смачиваемостью водой. В таких
случаях флотация возможна только при введении в суспензию
реагентов, снижающих смачиваемость зерен минералов (поверх-
.16
ностно-активные вещества: олеиновая кислота, нафтеновые
кислоты, ксантогенаты и др.)- Эти реагенты называются
собирателями. Действие их регулируется добавкой других реагентов-
регуляторов (известь, сода, серная кислота). Образованию
устойчивой пены способствует добавка пенообразователей
(эфирные масла, керосин, каменноугольная и древесная
смола и др.).
Флотацию называют простой, если в концентрат переходит
смесь веществ, близких по смачиваемости, а в отходы — вся
пустая порода, и избирательной, или селективной, если в
концентрат переходят отдельные минералы, например, свинцовый блеск
PbS — отдельно от цинковой обманки ZnS. Для осуществления
селективной флотации в суспензию вводят различные
химические реагенты из числа названных выше.
При флотации разделение минералов не зависит от их
удельного веса. Часто более тяжелые материалы всплывают, а более
легкие оседают.
4. РАСТИТЕЛЬНОЕ -И ЖИВОТНОЕ СЫРЬЕ
К растительному сырью относятся: древесина, картофель,
сахарная свекла, подсолнух, лен, конопля и пр., к животному —
в основном жиры, кожа и кости.
Многие виды растительного и животного сырья перед
поступлением на завод сортируют, очищают от ботвы (свекла),
скорлупы (орех, подсолнух), измельчают, обеспыливают и т. д.
Чтобы избежать порчи животного и растительного сырья при
хранении, его сушат, стерилизуют, солят, хранят в
холодильниках и т. д.
Другие виды сырья
Многие отрасли химической промышленности в качестве
сырья применяют отходы или продукты других производств. Так,
например, отходы производства окиси цинка служат сырьем
для производства литопона; сера, извлеченная из коксового
газа, используется в производстве серной кислоты.
Для производства органических краоителей, взрывчатых
веществ, пластмасс применяются бензол, толуол, фенол, серная
и азотная кислоты, а также ряд других органических и
неорганических соединений.
В отличие от природного сырья, продукты одного
производства, служащие сырьем для другого, часто называют
полупродуктами.
С развитием техники понятие о сырье, отходах,
полупродуктах и продуктах меняется, так как в производство вовлекаются
новые виды сырья и отходов, и многие из продуктов, имеющие
самостоятельное значение, могут стать сырьем для других
производств.
2 8-1504 17
5. ВОДА
К жидкому природному сырью относится вода.
Электрохимическим путем из нее получают водород и кислород. Кроме того,
вода используется как вспомогательное средство во всех
отраслях химической промышленности — для растворения,
промывания, передачи тепла и холода, получения пара и т. д.
Природная вода никогда не бывает совершенно чистой.
Поверхностная вода — вода рек и озер, морей, а также подземная
вода всегда содержит растворенные соли, газы и иногда
—продукты разложения растительных и животных остатков.
Общее содержание солей в воде определяется величиной
сухого остатка при упаривании ее. Количество растворенных в воде
солей кальция и магния, обусловливающих образование накипи
в котлах и различных аппаратах, характеризует жесткость воды.
Весьма важно знать степень кислотности или щелочности
воды, определяемую величиной рН. Если рН < 6,5, воду
называют кислой, если рН > 7,5 — щелочной, при рН = 6,5—7,5
вода является нейтральной.
При применении воды для технических целей особенно
важным качеством является степень жесткости воды и величина рН.
Жесткость воды в настоящее время выражают в милиграмм-
эквивалентах. 1 мг-экв соответствует 20,04 мг Са или 12,6 мг
Mg на литр воды. Очень жесткая вода содержит 10 мг-экв Са,
средняя вода — 3,6 мг-экв Са, очень мягкая вода — от 0 —
до — 1,5 мг • экв Са.
Очень жесткую воду подвергают очистке. Причем, если
раньше это делали для того, чтобы избежать образования накипи в
паровых котлах и других теплоиспользующих аппаратах, то в
настоящее время приходится очищать воду и регулировать ее
состав для многих химических производств, например, для
электрохимических, солевых, органических, для процесса флотации
и др., так как рН воды и содержание в ней органических и
минеральных примесей могут влиять на протекание химических
реакций.
Методы очистки воды
Полная очистка воды заключается в ряде последовательных
операций: 1) устранение механических примесей отстаиванием
и фильтрацией; 2) удаление коллоидных примесей методом
коагуляции; 3) устранение жесткости: 4) обессоливание; 5)
удаление газов (деаэрация); 6) удаление ядовитых и вредных
примесей; 7) дезинфекция.
Самой важной и распространенной операцией из них
является устранение жесткости воды, или умягчение.
18
В настоящее время в силу своей простоты и эффективности
получил распространение физико-химической способ очистки
воды. Это ионообменный метод, основанный на способности
некоторых труднорастворимых веществ обменивать свой анион или
катион на другие ионы, находящиеся в воде. Такие вещества
называются пермутитами и представляют собой как природные
минералы (цеолит, глауконит и др.), так и синтетически
полученные органические вещества (сульфоуголь и ионообменные
смолы). Если нонит обменивает свой анион, его называют анио-
нитом, если катион — катионитом.
Для ионообменных смол характерно присутствие
карбоксильных и сульфогрупп; водород их обменивается с катионами
солей воды по реакции
2H[R] + [Ca, Mg]Cl2?=tCa, Mg[R2] + 2HCl
В качестве анионитов применяются синтетические смолы,
содержащие группу ОН, которая обменивается на анион солей
воды:
НО [R] -j- НО вCl [R] + Н2О,
где R — составная часть ионита.
Для очистки воды ионообменным способом используются
простые аппараты-фильтры, в которых на слой песка или
гравия, расположенного на колосниковой решетке, помещается
слой ионита в виде зерен размером 0,5—-1,5 мм.
Вода, подлежащая очистке, пропускается через этот слой
сверху вниз и освобождается от жесткости. Степень очистки
воды в промышленных условиях может достигать 94—98 %.
Большие заслуги в области изучения ионного обмена
принадлежат К. К. Гедройцу, Е. Н. Гапону др.
Оборотная вода и очистка сточных вод
Оборотной водой называют воду, многократно используемую
на производстве, чаще всего в тех случаях, когда недостает
свежей воды. Вода, применяемая для охлаждения, перед
возвращением в цикл охлаждается в градирнях или брызгальных
бассейнах.
Градирни представляют собой высокие башни, на верх
которых через специальные разбрызгивающие устройства подается
теплая вода. Стекая по системе реек, вода частично испаряется
и за этот счет охлаждается, чему способствует также
движущийся навстречу воздух. Брызгальные бассейны представляют собой
резервуары, над которыми через специальные трубы с соплами
фонтанами разбрызгивается охлаждаемая вода. Принцип
охлаждения здесь тот же, что и в градирнях. Если оборотная вода
в процессе загрязняется, ее подвергают очистке.
2* ід
Сточные воды различных промышленных предприятий
всегда загрязнены различными вредными примесями,— кислотами,
щелочами, солями металлов и различными органическими
соединениями. Спуск сточных вод в реки и водоемы разрешается
только при допустимых нормах содержания токсических веществ во
избежание отравления всего живого — рыб и растений.
Очистка сточных вод от вредных примесей представляет собой весьма
важную и сложную задачу. К наиболее токсическим веществам
относятся органические, например фенол, и отходы
целлюлозных заводов — сульфитцеллюлозные щелока. Для очистки от
фенолов применяются методы экстрагирования их
растворителями, например бензолом, или отгонка паром. Проблема очистки
сточных вод от сульфитцеллюлозных щелоков еще не совсем
разрешена. Самым эффективным методом очистки сточных вод
от органических примесей является биохимическое окисление их,
которое осуществляется на полях орошения с помощью
соответствующих бактерий. Сточные воды, содержащие ионы
металлов,— меди, никеля, цинка и др.,— очищают с помощью кати-
онитов.
ГЛАВА 11
ВИДЫ И СОСТАВ ТОПЛИВА
Все виды топлива — твердое, жидкое и газообразное —
используются как источник тепловой и других видов энергии
и как сырье для химической переработки.
Значение топлива как химического сырья с каждым годом
возрастает в связи с бурным развитием промышленности
органического синтеза и особенно производства высокомолекулярных
соединений, базирующегося на продуктах химической
переработки топлив.
Кроме того, химическая переработка топлива дает
возможность увеличить ресурсы наиболее ценного легкого моторного
топлива, потребность в котором при современном состоянии
моторной и реактивной техники огромна.
По своему составу и свойствам различные виды топлива
существенно отличаются Друг от друга.
1. ДРЕВЕСИНА
Химический состав древесины сложен и неодинаков для
различных пород дерева. Основными составными частями ее
являются целлюлоза (или клетчатка), лигнин и гемицеллюлозы.
Целлюлоза относится к классу углеводов (полисахаридов)
и представляет собой высокомолекулярное соединение. Обычно
20
ее изображают формулой (СбНюО5)п, где п = 50 000-:-150 000.
Преимущественно из целлюлозы состоят внутренние слои стенок
растительных клеток древесины. Главной составной частью
наружных слоев является лигнин, который сообщает волокнам
жесткость и придает древесине механическую прочность.
Предполагают, что лигнин представляет собой продукт
полимеризации общей формулы (CioHi204)n, где п = 36.
Гемицеллюлозы — спутники целлюлозы. Они также входят
в состав наружных слоев клеток. Это полисахариды: пентозаны
(С5НаО4)п и гексозаны (CeHmOsK- Пентозаны преобладают
в древесине лиственных пород деревьев, а гексозаны — хвойных.
В состав древесины входит также смола, состоящая из
углеводородов терпенового ряда (СюНі6) и смоляных кислот общей
формулы СідНгдСООН.
Кроме того, в древесине содержится незначительное
количество минеральных веществ, образующих при сгорании дерева
золу, некоторые дубильные вещества, фосфор и пр.
Содержание перечисленных составных частей в сухой
древесине колеблется в следующих пределах (см. табл. 4).
Таблица 4
Целлюлоза,
%
40-50
Гемицеллюлозы,
%
17-28
Лигнин,
%
25 — 30
Смола,
%
2 — 8
Минеральные
вещества,
%
0,3—1.0
2. ИСКОПАЕМОЕ ТВЕРДОЕ ТОПЛИВО
К ископаемому твердому топливу относятся торф, бурые
и каменные угли, антрациты, горючие сланцы и сапропелитовые
угли. Состав этого топлива различен и зависит от его
происхождения и условий образования.
Установлено, что ископаемое топливо — продукт очень
длительного разложения высших наземных растений — деревьев,
кустарников и микроводорослей, а также животных остатков,
отмерших и накопившихся на дне болот и водоемов и
постепенно опустившихся в недра земли. При этом считается, что в
первой стадии сложные химические превращения протекали под
влиянием микроорганизмов и воды — торфяная стадия, а затем
под влиянием давления и повышенной температуры без доступа
воздуха — стадия углеобразования.
Различают гумусовые и сапропелитовые образования горючих
ископаемых. Гумусовые—блестящие, сапропелитовые —
матовые. Гумусовое топливо характеризуется наличием гуминовых
кислот или гуминов. Первые извлекают из топлива, обрабатывая
21
его слабыми растворами едкого натра, гумины извлекают
крепкими растворами NaOH при нагревании под давлением. Гумины
образуются из гуминовых кислот в дальнейшей стадии углеобра-
зования. Сапропелитовые образования не содержат гуминовых
кислот и гуминов-
Материнским веществом гумусового топлива являются
преимущественно высшие растения, в состав которых входят
лигнин и целлюлоза (возможность образования гуминовых кислот
из лигнина доказана). К гумусовым образованиям относятся
торфы, бурые и каменные угли, антрациты, представляющие
собой один генетический ряд.
Материнским веществом сапропелитового топлива считают
богатые жирами и восками водоросли (альги). При гниении они
образуют ил (греч. «сапропель»), откуда и произошло название
топлива.
К сапропелитовым образованиям относятся горючие сланцы
и сапропелитовые угли.
Наряду с гуминизацией протекает также процесс
битуминизации, в результате которого жиры, воски и смолы исходных
веществ превращаются в битумы—вторую составную часть
гумусовых топлив. Битумы экстрагируются из угля
растворителями— бензолом, спирто-бензолом и др. Количество битумов
и Тумановых кислот в топливе уменьшается от торфа к
каменным углям, а количество гуминов при этом повышается. Данные
о содержании этих веществ могут характеризовать химический
возраст топлива.
3. ТОРФ
Торф представляет собой самое молодое ископаемое топливо.
Для состава его характерно наличие неразложившихся
морфологических элементов растений. Различают волокнистый,
землистый и смолистый торф. Наиболее старым и ценным из них
является смолистый торф. Обычно торф залегает в открытых
болотах, глубина залегания достигает 2—2,5 м. Торфяные залежи
встречаются по всему Советскому Союзу и составляют около
2,8% общих запасов топлива.
4. БУРЫЙ УГОЛЬ
Бурый уголь является продуктом более глубокого разложения
органической массы растений, чем торф. Самый молодой из
бурых углей называют лигнитом. В лигнитах уже нет
неразложившихся морфологических элементов растений, но заметно
наличие структуры древесины. Цвет бурых углей — от
светло-бурого до черного.
22
Бурые угли представляют собой хорошее сырье для
процессов полукоксования и гидрирования-
В СССР имеются богатые месторождения бурых углей в
Подмосковном угольном бассейне, на Украине, Урале и в Сибири.
5. КАМЕННЫЙ УГОЛЬ
Каменный уголь — продукт дальнейшего превращения
органической массы растений. Это наиболее распространенное
ископаемое твердое топливо. Цвет его черный. Отдельные
разновидности каменного угля по элементарному составу и техническим
свойствам значительно отличаются друг от друга.
По запасам каменных углей СССР занимает первое место
в мире. Они составляют около 8669 миллиардов тонн или '/го
мировых запасов.
Основные угольные месторождения в СССР: Донецкий,
Кузнецкий, Подмосковный, Кизеловский, Челябинский,
Карагандинский, Черемховский бассейны, а также Печорское
месторождение.
6. СЛАНЦЫ
Сланцы относятся к сапропелитовому твердому ископаемому
топливу. Это глинистая или мергелистая слоистая,
раскалывающаяся на пластинки порода. Продукты, получающиеся при
переработке сланцев (при коксовании), очень похожи на продукты
переработки нефти.
В СССР месторождения сланцев находятся в Эстонской ССР,
Ленинградской области, в среднем Поволжье, Казахстане и
Восточной Сибири.
7. СОСТАВ РАЗЛИЧНЫХ ВИДОВ ТОПЛИВА
Состав топлива определяется на основании данных
элементарного и технического анализов.
Обычно содержание углерода, водорода, кислорода и азота
выражают в процентах к органической или условной органичской
массе топлива, или в процентах к горючей массе топлива.
Органическую массу находят как разность между весом угля
и весом минеральных примесей и воды. Так как установить
количество минеральных примесей трудно, определяют золу топлива.
Вес угля за вычетом веса золы и воды называется горючей
массой топлива. Условной органической массой топлива считается
суммарное содержание углерода, водорода, азота и кислорода.
Вся сера топлива условно относится к балласту.
Технический анализ топлива состоит в определении
содержания влаги, золы, а также летучих веществ и кокса.
23
Элементарный состав условной органической массы
различных видов твердого топлива приведен в табл. 5.
Таблица 5
Виды топлива
Гумусовые
образования
Сапропелиты
Древесина
торф,
бурые
угли,
каменные
угли,
антрацит
Горючие
сланцы
одного из
месторождений
Средний состав условной
органической массы,
%
С
50
59
69
82
95
75
Н
6
6
5,5
4,3
2,2
9,0
О
43
33
24
12
2
1'5,7
N
1
2
1,5
1,7
0,8
0,3
Высшая теплотворная
способность
органической массы,
кдж/кг
18 900
22 680
28 140
32 340
34 860
34 020
Как видно из таблицы, для углей одинакового происхождения
содержание углерода растет, а кислорода падает по мере
перехода от торфа через бурые угли к каменным углям. Содержание
водорода меняется мало и лишь при переходе от каменных углей
к антрацитам резко снижается. Параллельно с увеличением
количества углерода возрастает и теплотворная способность углей,
но в некоторых случаях она может быть ниже у антрацитов, чем
у тощих каменных углей (в связи с резким снижением
содержания водорода).
Для сапропелитовых углей и сланцев характерно высокое
содержание водорода в органической массе (до 11%),
промежуточное между содержанием водорода в твердом и жидком
топливе.
По данным технического анализа, гигроскопической влаги
больше всего в древесине (до 40%), торфе (до 45%) и в бурых
углях (по 50%), меньше всего- -б антрацитах A—4%).
Наиболее высокий процент золы в сланцах C8—60), наименьший —
в антрацитах A,5—6) и древесине @,4). Сера находится в
топливе в виде минеральных (FeS2, FeSCu, CaSO4) и органических
соединений. Обычно определяется содержание общей серы.
Малосернистые угли Кузнецкого бассейна содержат 0,5--1,5%
серы, сернистые угли Донбасса 2,6—4%, Кизеловские угли —
высокосернистые — свыше 4% серы. Торф является
малосернистым топливом @,4% серы). Состав топлива характеризуется
также содержанием в нем битумов. Количество битумов А (все, что
извлекается бензолом) з торфах СССР колеблется от 5,6 до
24
28,5%; в бурых углях оно составляет 2,34 — 20,94%, в каменных
углях Кузбасса (жирных) около 0,6%, в Донецких — примерно
0,52%.
Для установления пригодности углей к коксованию особенно
важно определить выход летучих продуктов V и качество
остатка — кокса.
Выход летучих обычно определяют в лабораториях, быстро
нагревая уголь в закрытом тигле в стандартных условиях
(850° С, 7 мин).
Выход летучих и характер остатка положен в основу
технической классификации углей по Грюнеру. В настоящее время
этих показателей считается недостаточно, так как коксуемость
углей определяется также их способностью переходить в
пластическое (размягченное) состояние и давлением вспучивания.
При разработке новой технологической классификации углей
Донбасса и Кузбасса исходными данными послужили выход
летучих и толщина пластического слоя как показателя спекае-
мости углей.
Новая технологическая классификация углей Донбасса
(технические условия ГОСТ 8181—56) приведена в табл. 6.
Таблица 6
Наименование
мяпок углей
Длиннопламен-
ный
Газовый
Жирный
Коксовый
жирный
Коксовый
Отощенный
спекающийся
Тощий
Условное
обозначение
Марка| Группа
Д
Г
Ж
кж
к
ОС
т
г *
Же
Жзі**
ОС,
—
Показатели
V, %
37 и более
35 и более
35 и более
27 и менее
оо
27 и менее
35
18 и менее
27
18 и менее
27
14—22
9—17
у, мм
6—15
16—25
8—20
21 и более
21 и более
14—20
6—13
—
Характеристика нелетучего-
остатка
Порошкообразный слипшийся,
слабоспекши йся
Спекшийся
без порошка
Порошкообразный слипшийся,
слабоспекшийся
Примечание. V — выход летучих в % на горючую массу угля;
у — толщина пластического слоя.
*Прн у, меньшем 6 мм, и спекшемся нелетучем остатке уголь относится
к группе Гв.
При выходе летучих веществ 35% и более и у, большем 21 мм, уголь
относится к группе Жаі.
25
Для коксования применяется смесь углей — шихта из
приведенных в стандарте марок углей, хотя наилучшими для
коксования являются угли марки К. Возможность применения смеси
углей, из которых многие в отдельности не коксуются,
позволила расширить сырьевую базу коксохимической промышленности.
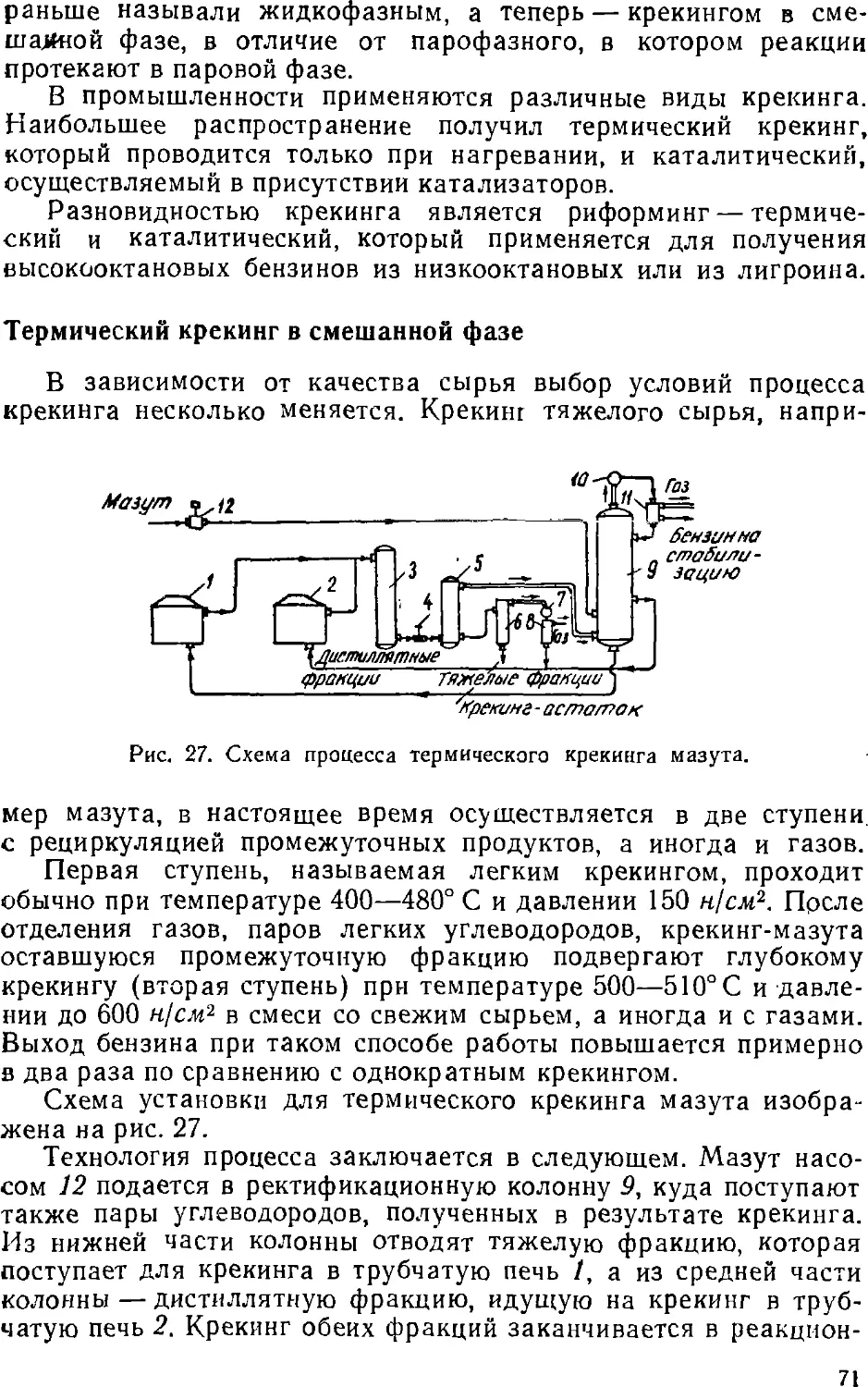
8. НЕФТЬ И ПРИРОДНЫЕ ГАЗЫ
Нефть представляет собой жидкость от светло-желтого до
черного цвета, характерного запаха. Удельный вес нефти
колеблется от 0,75 до 1,0. Более легкие сорта нефти начинают кипеть
ниже 100° С, более тяжелые — выше этой температуры.
Теплотворная способность нефти составляет около 44 100 кдж/кг.
Нефть вращает плоскость поляризации, т. е. является
оптически активной.
По химическому составу нефть представляет собой смесь
углеводородов парафинового (СпН2п+2), нафтенового (СпН%п)
и ароматического (СпНгп-б) ряда с примесью азотистых,
сернистых и кислородсодержащих соединений.
По элементарному составу нефти отличаются незначительно.
Содержание углерода колеблется от 84 до 87%, содержание
водорода— от 11,8 до 14%, содержание S, N и О — до 1 % и выше.
Содержание парафина (смесь твердых парафиновых
углеводородов, растворенная в нефти) колеблется в широких
пределах — от 0,6 до 8,5 %.
Из кислородных соединений в нефти в небольшом количестве
содержатся нафтеновые кислоты и фенолы. Нафтеновые
кислоты, выделенные из нефти, используются для производства
мылонафта, асидола и др. Сера в нефти встречается в виде
сероводорода, меркаптанов, сульфидов, тиофеновых производных и
элементарной серы, азот — в виде аминов и пиридиновых
оснований.
Малосернистые нефти содержат серы не более 0,5% и
сернистые— свыше 0,5%. В башкирской нефти содержание серы дости-»
гает 4%.
Вода всегда сопровождает нефть, часто образуя эмульсию.
В пласте нефть обычно находится под давлением в
равновесии с растворенными в ней газами. При выходе на поверхность
вследствие изменения давления газы выделяются из раствора,
образуя нефтяной газ.
К залежам нефти часто приурочены газовые месторождения.
Газы этих месторождений, в отличие от растворенных в нефти
(нефтяных), называют попутными.
Обладая очень высокой теплотворной способностью C3600—
35700 кдж!мъ), нефтяные и попутные газы применяются как
топливо для бытовых и промышленных нужд. Они представляют
собой также весьма ценное химическое сырье,
26
По запасам нефти СССР занимает первое место в мире.
Помимо крупнейших месторождений нефти (возле Баку, в районе
реки Волги — Второе Баку, на Северном Кавказе и др.).
открыты залежи нефти во многих районах Советского Союза.
Велики и газовые месторождения (на побережье Черного
и Азовского морей, на Украине и др.).
В связи с ростом добычи и переработки нефти и газов в
топливном балансе СССР происходят изменения в сторону
увеличения доли более дешевых и эффективных видов
топлива—нефти и газов.
В 1972 г. доля угля в топливном балансе снизится до 32%,
а нефти и газов возрастет до 61%, в то время как в 1958 г. доля
угля составляла 60%, а нефти и газа — 31 %.
ГЛАВА 111
МЕТОДЫ ПЕРЕРАБОТКИ ТОПЛИВА
Основными методами переработки различных видов
топлива являются термические, к которым относятся: сухая перегонка
дерева; коксование и полукоксование угля, сланцев, торфа;
газификация твердых видов топлива; деструктивная
гидрогенизация углей и различные виды крекинга нефтепродуктов и
газов.
К физическим методам переработки принадлежат
следующие: прямая перегонка нефти с целью получения моторного
топлива и масел; выделение из соответствующих фракций нефти
парафина, церезина, вазелина; экстрагирование воска из углей
и торфа, канифоли — из пневого осмола.
Чисто химическими методами являются: переработка
древесины с целью получения целлюлозы, сахаристых веществ и
спирта.
1. ПЕРЕРАБОТКА ДЕРЕВА
Методом переработки дерева, известным с древних времен,
является термический, который заключается в нагревании
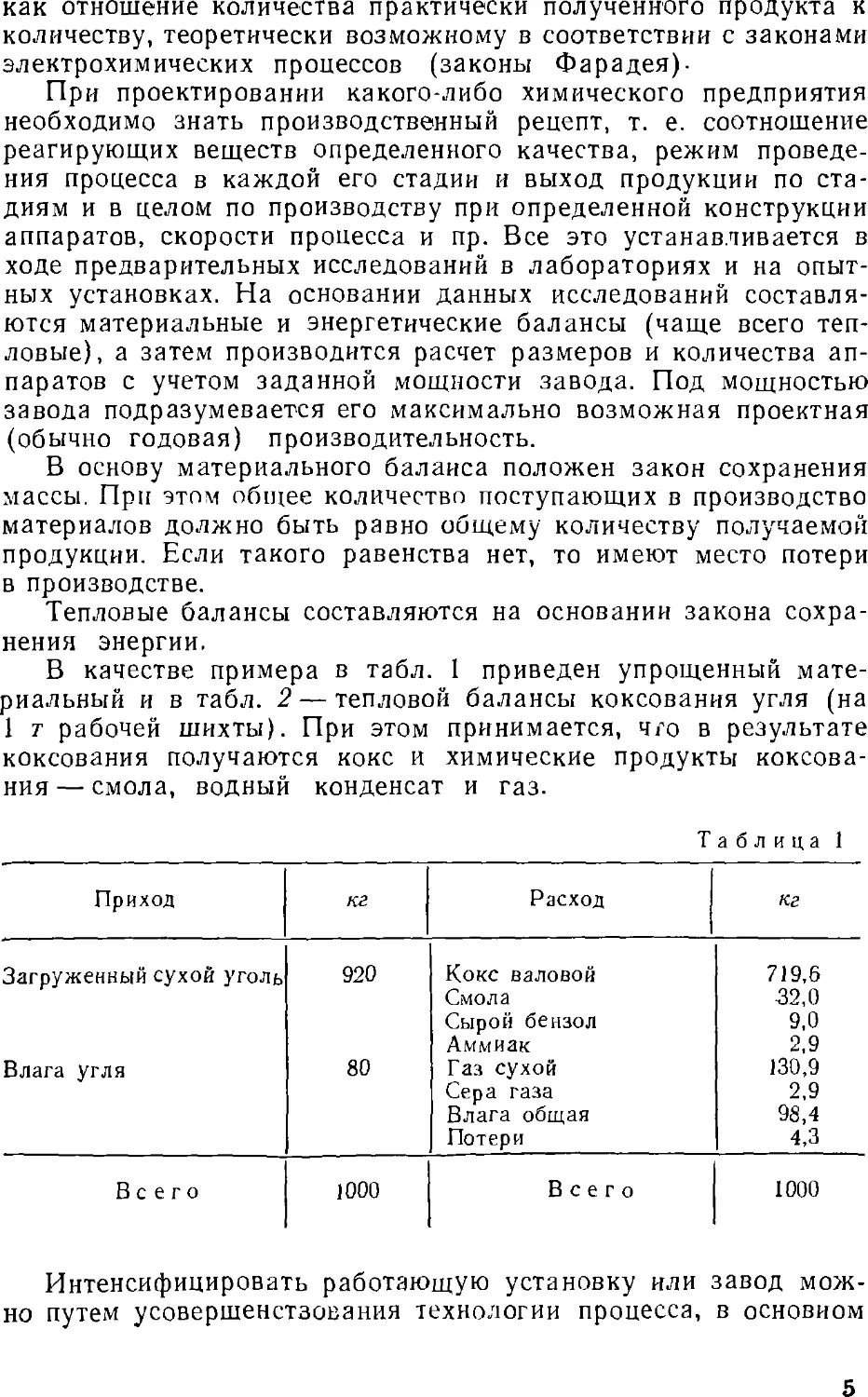
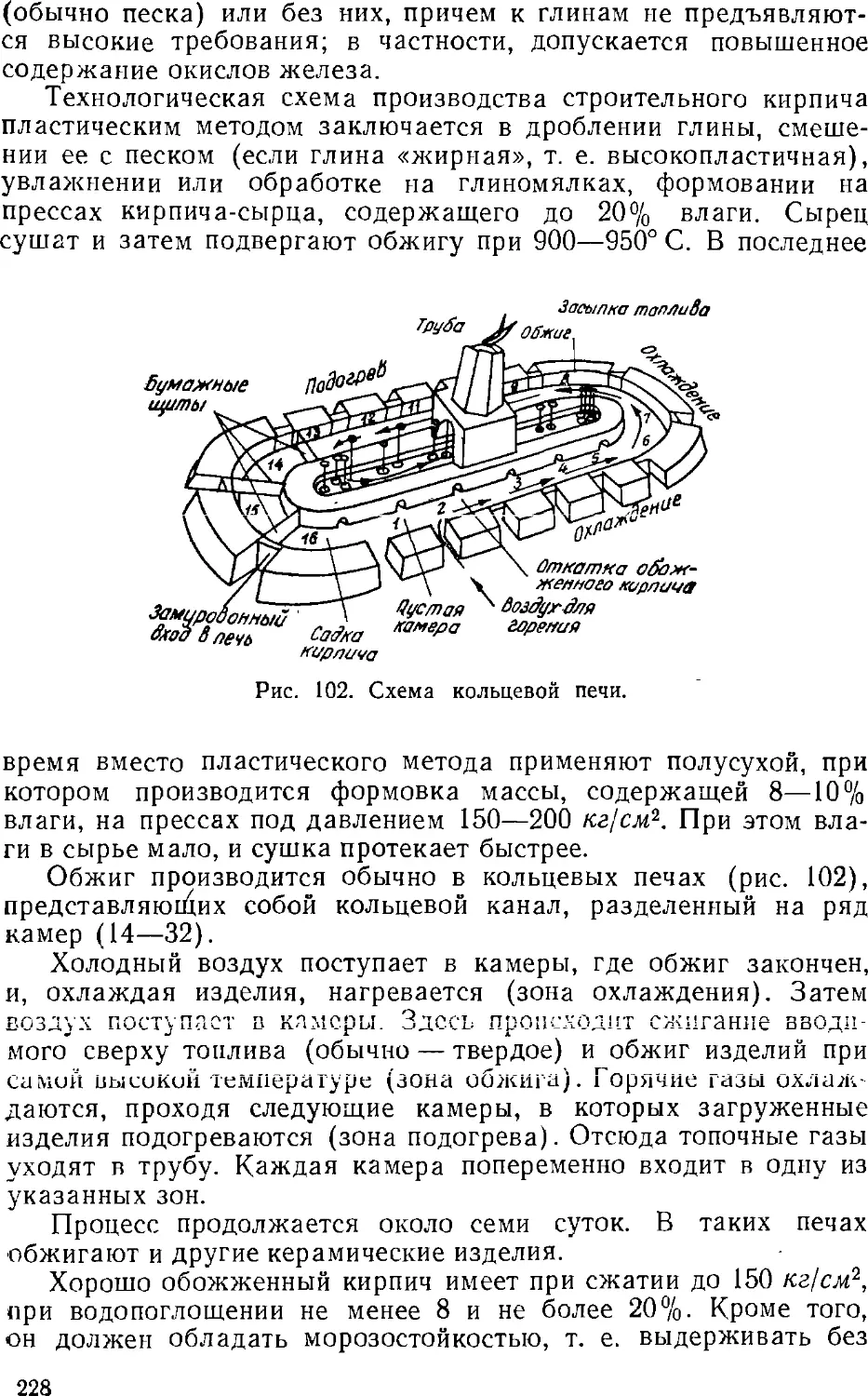
дерева без доступа воздуха до 450—500° С в печах различного
устройства. В настоящее время наиболее совершенными являются
туннельные печи непрерывного действия с циркуляцией
теплоносителя (печи Козлова), в качестве которого применяются
нагретые неконденсирующиеся газы, получающиеся в том же
процессе. Непрерывность работы этих печей достигается
периодическим передвижением в туннеле печи вагонеток с дровами из
камеры сушки в камеру сухой перегонки и затем в камеру
охлаждения (см. рис. 3).
27
• Продуктами термической переработки дерева являются
древесный уголь и парогазовая смесь, из которой после охлаждения
выделяются древесная смола, водный конденсат и газы.
Древесный, уголь до появления в промышленности кокса
использовался для выплавки чугуна.
В настоящее время в металлургии древесный уголь
применяется только для производства стали, не содержащей серы и
фосфора. Кроме того, древесный уголь используют в кузнечном
деле, при восстановительных процессах для производства актив-
Йхимичсскуй
Толочные
газб/
Топочные газы
ИГ]
Топочные
газы
Циркулирующий
І03 U3 XUMUVeC/ґОЄО
цеха
Рис. 3. Схема работы углевыжигательной печи
системы Козлова:
/ — сушилка; 2, 4, 6—шлюзы; 3—9—камера переуглн-
вания; 5 — камера охлаждения; 7 — подогреватель; 8 —
дымовая труба.
ных углей, применяющихся в качестве адсорбентов,
катализаторов. Смола хвойных пород деревьев идет на просмолку
канатов, подводной части судов; из нее получают флотационные
масла, антиокислители и пр.
Смола лиственных пород дерева, особенно бука, содержит
фенол и его производные и является сырьем для производства
ряда фармацевтических препаратов.
Из водного конденсата, содержащего кислоты, спирты, кето-
ны и альдегиды, извлекают главным образом уксусную кислоту,
экстрагируя ее эфиром или маслами, а также метклосий спирт
и аидмон. Выход древесного угля при сухой перегонке дерева
составляет 30—38%, уксусной кислоты 3—7%, метиловою
спирта 0,9—І,Ь"% от веса сухой древесины.
Большая потребность химической промышленности в
уксусной кислоте и метиловом спирте служила некоторое время
стимулом для развития промышленности сухой перегонки дерева.
Однако с появлением синтетических методов производства этих
продуктов сухая перегонка дерева утратила свое значение и
занимает сейчас сравнительно небольшое место в химической
промышленности. К развивающимся в настоящее время отраслям
лесохимической промышленности в СССР относятся: 1) произ-
28
водство целлюлозы из древесины; 2) гидролиз древесины с целью
получения сахаристых веществ и спирта; 3) канифольно-скипи-
дарное производство.
2. ПРОИЗВОДСТВО ДРЕВЕСНОЙ ЦЕЛЛЮЛОЗЫ
Декабрьский Пленум ЦК КПСС A963 г.) отметил большое
народнохозяйственное значение химической переработки
древесины, позволяющей использовать десятки миллионов
кубометров дров и древесных отходов, и постановил довести в 1970 г.
производство целлюлозы до 11,5 миллионов тонн.
Выделение целлюлозы из древесины основано на различии
химических свойств целлюлозы, лигнина и гемицеллюлоз. Под
действием минеральных кислот происходит гидролиз целлюлозы,
степень которого зависит от концентрации кислоты, времени
и температуры.
Гемицеллюлозы растворяются в слабых щелочах и гидроли-
зуются слабыми кислотами. Лигнин не гидролизуется
кислотами, но разрушается под действием окислителей (Cl2, HNO3)
и при повышенной температуре растворяется в растворах
едкого натра, в сернистой кислоте и в растворах ее солей. Это
свойство лигнина, позволяющее отделять его от целлюлозы,
и было положено в основу методов производства целлюлозы из
древесины. Целлюлозу получают в основном тремя способами:
1) кислотным, или сульфитным; 2) щелочным — натронным или
сульфатным; 3) хлорно-щелочным (комбинированный метод).
Производство целлюлозы сульфитным способом состоит
в обработке измельченной и обезвоженной до 20—25% влаги
древесины раствором бисульфита кальция, который образуется
при взаимодействии очищенного сернистого газа с известняком
и водой. Раствор бисульфита кальция называется варочной
кислотой и обычно содержит 1—2% СаО и 2—6% SO2 в виде
Ca[HSO3]2 и H2SO3.
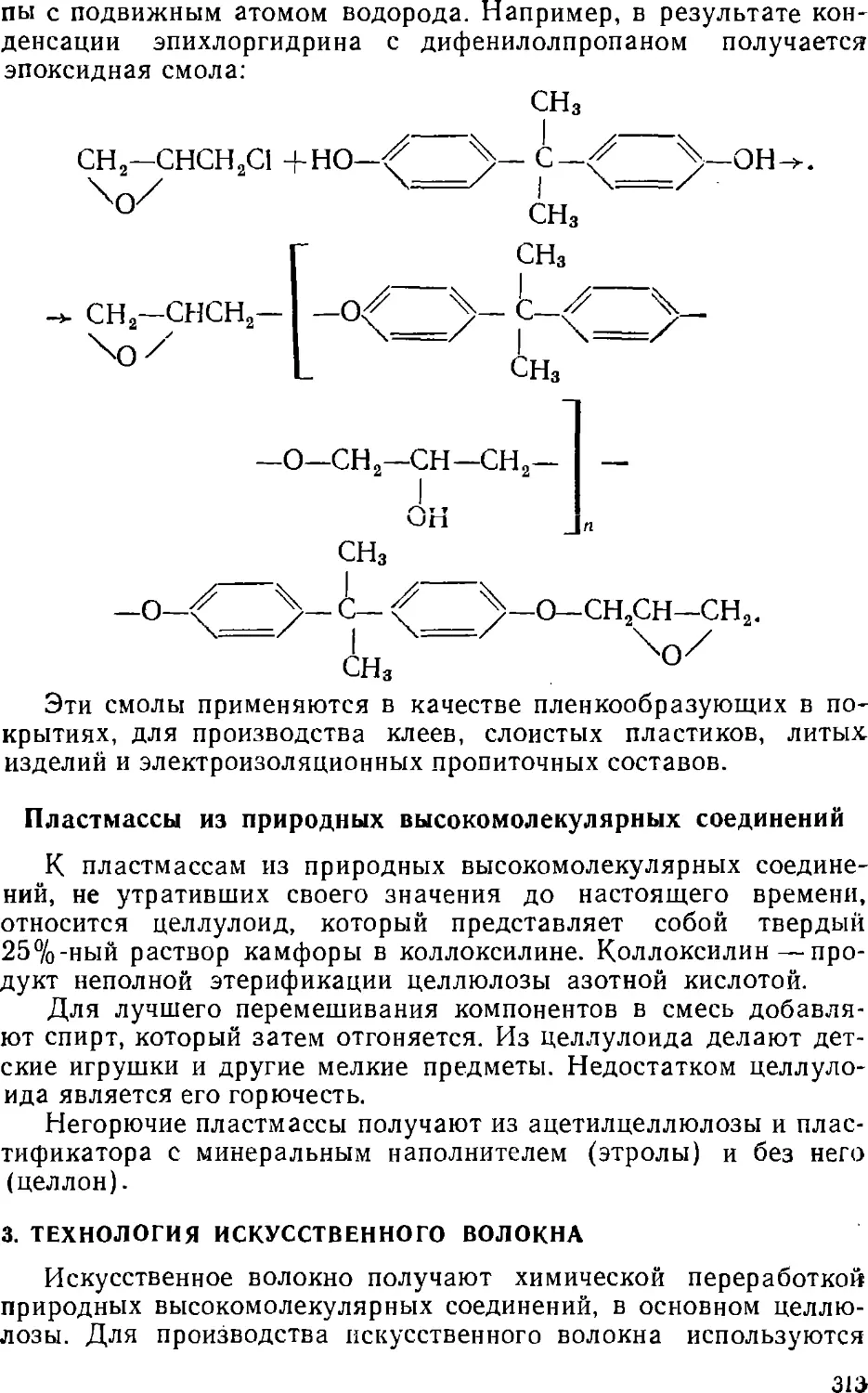
Варка древесины производится в железных котлах емкостью
до 400 м3, футерованных кислотоупорными плитками, при
нагревании острым паром до 140—160° С и давлении 50—70 н/см2.
Продолжительность варки 8—15 часов. В процессе варки в твердой
фазе образуется лигниносульфоновая кислота и ее кальциевая
соль, которые затем гидролизуются и переходят в раствор.
Гемицеллюлозы гидролизуются в сахара, которые тоже
растворяются. Целлюлоза при этом гидролизуется незначительно.
После окончания варки содержимое котла выгружается под
давлением паров через нижнюю трубу в приемник с сетчатым
днищем. Отделенная от отработанной кислоты целлюлоза
разбавляется водой до 1—1,2% сухого вещества и очищается от
сучков, непроваренной щепы и песка в сучкоуловителях и пес-
чаницах. Затем она проходит сгустительные барабаны для отде-
29
ления основного количества воды, после чего отбеливается
окислителями—гипохлоритом кальция или натрия, снова
промывается водой, обезвоживается до содержания влаги 6—12%
и прессуется в листы.
Выход целлюлозного волокна составляет 44—48% от веса
сухой древесины.
Сосна сульфитным способом не перерабатывается, так как
смола препятствует проникновению варочной кислоты внутрь
клеток древесины.
Отходом этого производства являются сульфитные щелока.
Из них можно получить этиловый спирт, дубильные вещества,
метиловый спирт, уксусный альдегид, уксусную кислоту и пр.
Недостатком этого метода является невозможность регенерации
сульфитных щелоков.
Натронный способ переработки древесины
заключается в обработке ее 15—20%-ным раствором едкого натра в котлах
емкостью 125—140 м3 с принудительной циркуляцией и
подогревом щелочи. Продолжительность варки составляет 5—6 часов
при 170—175° С и давлении 80—90 н/см2.
После окончания варки содержимое котла под остаточным
давлением паров выгружается через нижнюю трубу котла
в аппараты для промывки, которая производится по принципу
противотока. Отработанный раствор и промывная жидкость
смешиваются, упариваются досуха и сухой остаток
прокаливается в печах. При прокаливании органические вещества сгорают
в углекислоту, которая с едким натром образует соду:
2NaOH + СО2 = Na2CO3 + Н2О.
Сода снова превращается в едкий натр (каустификация)
взаимодействием с известковым молоком в растворе:
Na2CO3 4- Са (ОН)„ т=> 2NaOH + СаСО3.
Раствор едкого натра после отделения от СаСО3 снова
используется в производстве целлюлозы.
Сульфатный метод получения целлюлозы представляет собой
видоизмененный натронный способ. По этому способу при
регенерации раствора едкого натра в плавильную печь добавляют
рассчитанное количество сульфата натрия для возмещения потерь
едкого натра.
При сгорании обугленного щелока сульфат натрия
восстанавливается в сернистый натрий по реакции
Na2SO4 4- 2С ->- Na2S + 2СО2,
Так же как в натронном способе, плав растворяется в воде
и каустифицируется гашеной известью. При этом получается
раствор едкого натра, содержащий до 17% Na2S, который
возвращается в варочное отделение. Присутствие в растворе серни-
30
стого натрия ускоряет процесс варки древесины и несколько
уменьшает потери целлюлозы, вызванные разрушающим
действием едкого натра.
Едкий натр удаляет из древесины смолы в виде солей.
Поэтому щелочные методы пригодны и для производства сосновой
целлюлозы.
В производстве сульфатной целлюлозы получается ряд
побочных продуктов (скипидар, метиловый спирт, меркаптаны). Кроме
того, из отработанных черных шелоков при охлаждении
выделяется пенообразная масса, которую перерабатывают в продукт,
называемый жидкой канифолью или талловым маслом,
применяющимся в мыловаренной промышленности.
Дальнейшая обработка целлюлозы производится так же, как
и по сульфитному методу.
Выход целлюлозы по сульфитному методу достигает
примерно 40% от веса сухой древесины.
Существует непрерывный способ варки сульфатной
целлюлозы, предложенный проф. Л. П. Жеребовым. Этот способ
позволяет повысить скорость варки в несколько раз.
Хл о р н о-ще л оч н о й способ производства целлюлозы
начали применять с 20-х годов этого столетия преимущественно
для получения целлюлозы из соломы.
Среди новейших способов производства целлюлозы из
древесины наибольший интерес представляют азотнокислотный и гид-
ротропный методы. Гидротролный способ заключается в
обработке измельченной древесины 40—50%-ным водным раствором
ксилолсульфоновокислого натрия (при 160—180° С и рН = 7),
который увеличивает растворимость лигнина и гемицеллюлоз
вводе (гидротропизм).
При этом деструкции целлюлозы не происходит.
Отработанный раствор, содержащий 200—300 г/л лигнина, регенерируете»
(при разбавлении раствора лигнин выпадает в осадок, а раствор
упаривается).
Целлюлоза является чрезвычайно важным химическим
сырьем. Из нее получают бездымный порох, кинопленку,
пластмассы, различные сорта искусственного шелка, лаки и др.
Целлюлоза применяется для производства бумаги и картона.
3. ГИДРОЛИЗ ДРЕВЕСИНЫ
Гидролиз древесины производится с целью осахаривания
целлюлозы и гемицеллюлоз. В дальнейшем сахара сбраживают
для получения этилового спирта.
Гидролизная промышленность является молодой, но весьма
перспективной отраслью, производящей из непищевого сырья
важный продукт — этиловый спирт. Гидролиз древесины можно
проводить концентрированной 25—28%-ной соляной кислотой при
температуре 15—20° С и разбавленными кислотами при нагре-
31
еании. Лигнин при этом не изменяется и используется главным
образом как топливо, а также в качестве наполнителя при
производстве пластмасс, для изготовления салфеточной бумаги и пр.
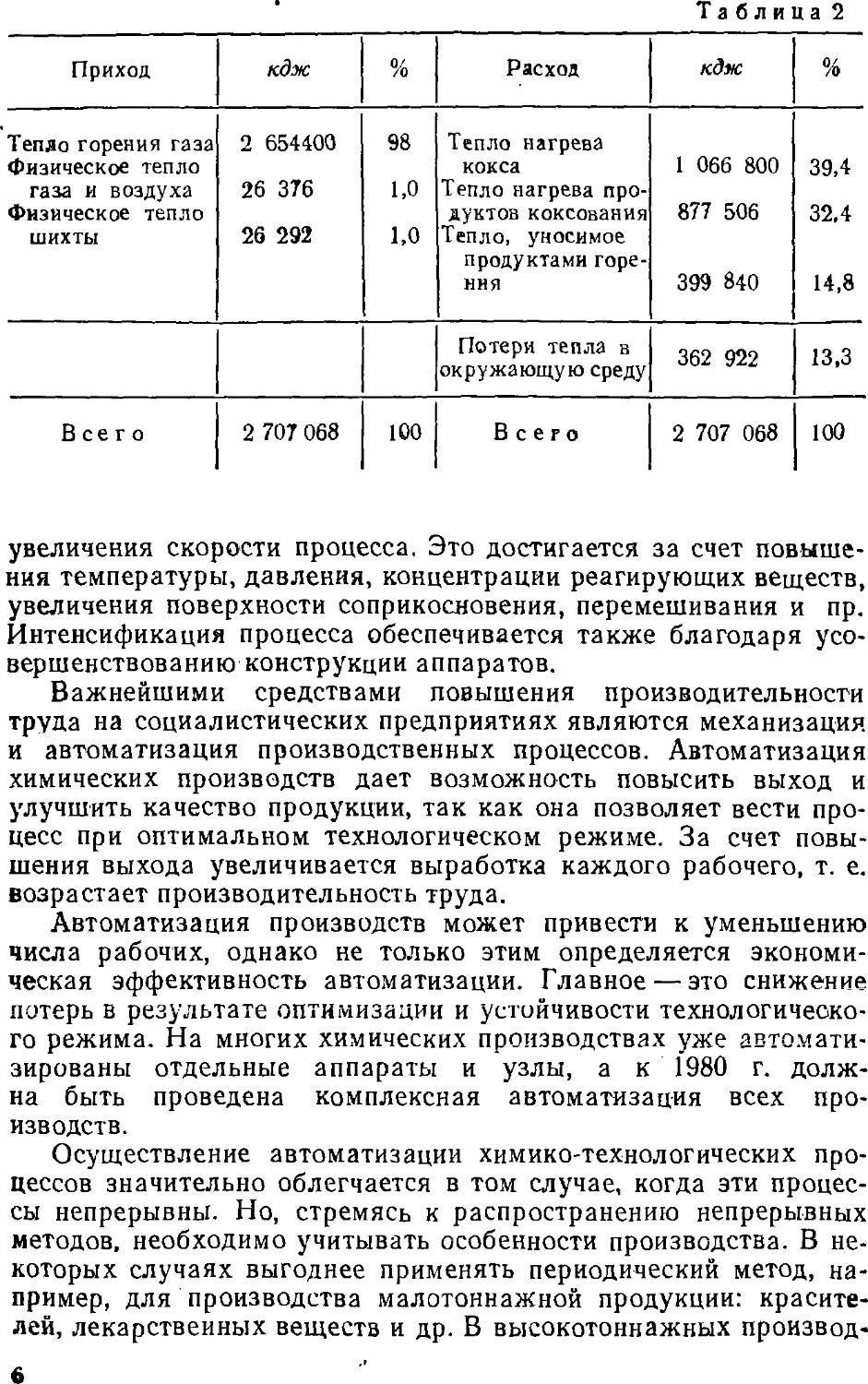
Обычно гидролиз осуществляют в батарейной установке,
состоящей из нескольких вертикальных аппаратов, называемых
перколяторами (рис. 4). Это стальные котлы емкостью 20—35 м3,
выложенные внутри кислотоупорной футеровкой. Перколяторы
заполняют древесной щепой и слабым раствором серной
кислоты @,3—0,5%). Серная кислота передается из котла в котел
Пора/ на
конденсацию
Лиенин
Рис. 4. Установка для гидролиза древесины:
1 — элеватор; 1 — транспортер; 3 — перколяторы; 4 —
теплообменник; 5 — испаритель.
и постепенно обогащается
реакции
сахарами, которые образуются по
H2SO,
/гН2О
»л СвН12О6
(СвН]0О5)я
целлюлоза глюкоза
'Процесс идет при 170—180° С и давлении 70—100 н/см2.
Уходящий из установки раствор содержит до 6% сахара.
Оставшийся после окончания гидролиза лигнин высушивается и
отправляется на переработку. Кислый раствор сахара проходит
теплообменник, где отдает свое тепло, после чего нейтрализуется
известковым молоком. В результате сбраживания охлажденного
и отфильтрованного раствора получается этиловый спирт:
С6Н12О6 ——*4- 2СН3СН2ОН + 2СО2.
Сбраживание осахаренного раствора происходит в
бродильных чанах, куда перед этим добавляют некоторое количество
суперфосфата и сульфата аммония для питания дрожжей. Через
6—18 часов процесс заканчивается.
Оптимальная температура процесса брожения 28—30° С.
При более высокой температуре дрожжи быстро вырождаются.
Концентрация образующегося спирта по той же причине не
должна превышать 12—15%. Реакция брожения экзотермична,
поэтому при недостаточном естественном отводе тепла применяется
искусственное охлаждение. Чтобы избежать потерь спирта с вы-
32
деляющимися газами брожения, на современных больших
заводах применяют закрытые стальные бродильные чаны.
Этиловый спирт выделяют из продуктов брожения
ректификацией. По описанному способу гидролиза из одной тонны сухой
древесины получается около 180 литров спирта. В качестве
побочных продуктов получаются: белковые дрожжи, жидкая
углекислота, фурфурол, лигнин B00—300 кг на 1 тонну
древесины) и пр. Первый завод гидролиза древесины в СССР был
построен в 1933 году. Л
4. КАНИФОЛЬНО-СКИПИДАРНОЕ ПРОИЗВОДСТВО
Канифоль и скипидар получают из естественной смолы
«живицы», которую добывают подсочкой деревьев хвойных пород и из
пневого осмола (пни, которые выкорчевывают пять и больше
лет спустя после рубки деревьев).
Скипидар — это смесь циклических углеводородов —
терпенов, имеющих общую формулу С10Нів.
Канифоль представляет собой смесь твердых изомерных
кислот общей формулы С19Н29СООН. Наиболее изученными из них
являются абиетиновая и пимаровая кислоты. В состав нашей
канифоли входит главным образом абиетиновая кислота.
Производство скипидара и канифоли заключается в
следующем. Живицу расплавляют, фильтруют и отделяют от воды
отстаиванием. Очищенную живицу, подогретую до 150й С,
перегоняют в тарельчатых колоннах паром. Из верхней части колонны
уходит живичный скипидар, который после охлаждения в
холодильниках отделяется от воды. Канифоль вытекает из нижней
части колонны. Применяют также перегонку живицы под
вакуумом глухим паром при температуре 150—160° С.
В пнях накапливается до 30% смолы — их подвергают сухой
перегонке (смолокурение). До 160° С отгоняется скипидар и вода,
а затем идет разложение древесины и выделение смоляных
кислот частично разлагающихся с образованием смоляных и
канифольных масел.
Более совершенным является экстракционный метод.
Воздушно-сухой осмол измельчают, паром отгоняют из него
скипидар, а затем обрабатывают растворителем, например бензолом.
Из раствора, содержащего 4—8% смолы, бензол отгоняют под
пониженным давлением. При этом остается канифоль.
Данный метод дает более высокий выход канифоли, которая
при экстрагировании не разлагается. Оставшаяся щепа
используется для получения целлюлозы.
Для извлечения канифоли применяют и метод
экстрагирования ее растворами едкого натра, а также метод прессования
распаренной щепы под давлением 250 кг/см2; при этом смола
вытекает.
З 8-І504 33
Скипидар широко используется в лаковом производстве,
в малярном деле, типографской технике, для выведения пятен;
в медицине—для приготовлений мазей, для ингаляций. Кроме
того, скипидар является объектом химической переработки, из
него получают терпингидрат С10Н20О2 • Н2О, используемьш
в медицине, камфору СюН|6О, также применяемую в медицине,
для производства целлулоида, кинопленки и в пороховом деле.
Канифоль применяется в мыловарении, бумажной и
резиновой промышленности. В лакокрасочной промышленности из
канифоли вырабатывают резинаты, служащие сиккативами при
изготовлении быстровысыхающих лаков, эфиры, являющиеся
пластификаторами, и т. д. Канифоль используется также при
изготовлении электроизоляционных материалов.
ГЛАВА IV
ПЕРЕРАБОТКА ИСКОПАЕМОГО ТВЕРДОГО ТОПЛИВА
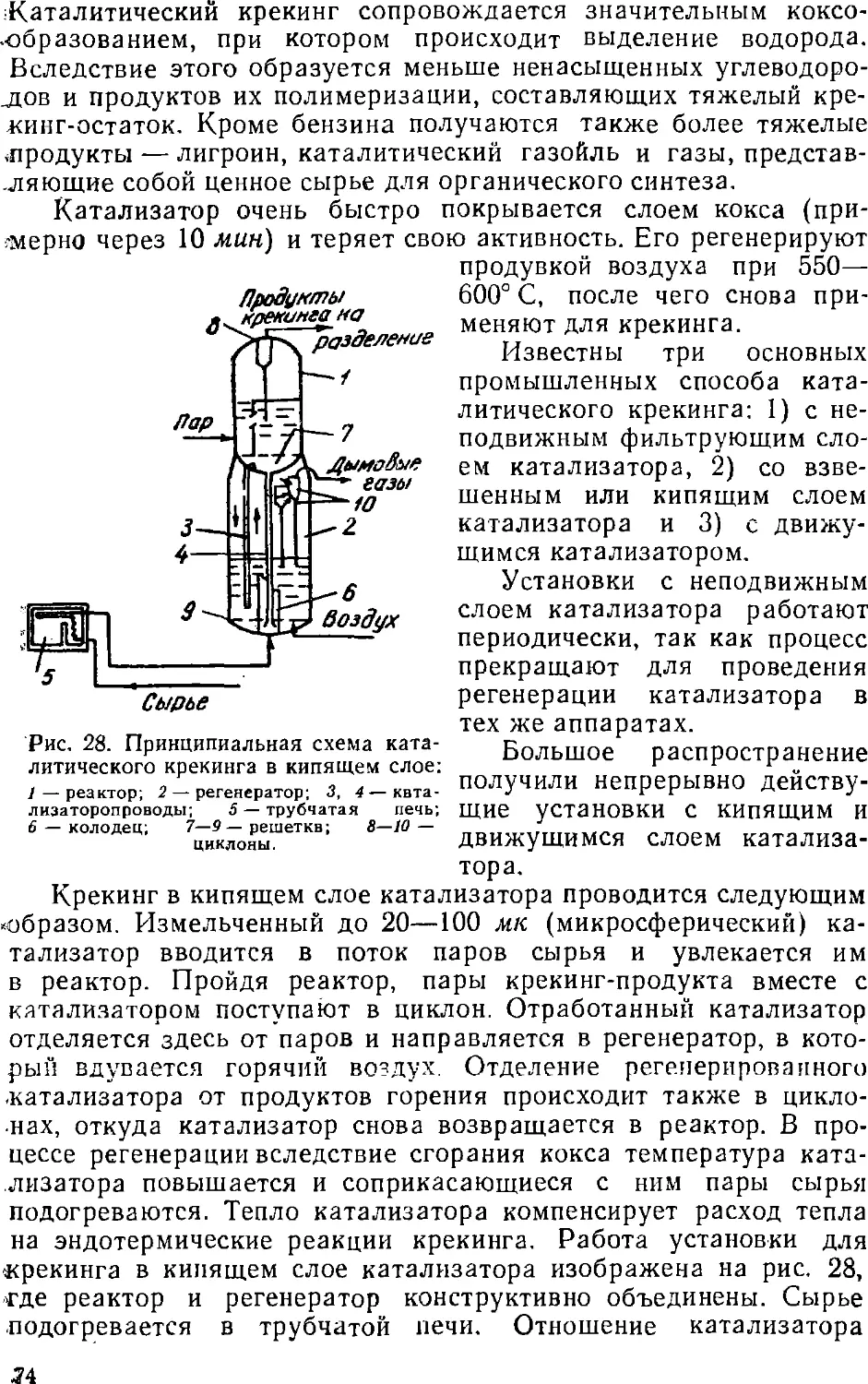
Основными методами химической переработки ископаемого
твердого топлива являются процессы коксования,
полукоксования, газификации и деструктивной гидрогенизации. Из них
наибольшее значение для народного хозяйства имеет коксование
каменного угля.
1. КОКСОВАНИЕ КАМЕННОГО УГЛЯ
Коксованием угля называется процесс нагревания его без
доступа воздуха до 1000—1100°С в камерных печах
периодического действия. Цель коксования — получение прочного и
пористого кокса, необходимого для выплавки чугуна из руд в
металлургии, и химических продуктов, являющихся сырьем для
других отраслей химической промышленности.
Выплавка чугуна на каменноугольном коксе была впервые
осуществлена в 1735 году в Англии. До середины XIX столетия
коксование угля производилось с целью получения кокса или
светильного газа. Образующиеся при это?.? химические продукты
не улавливались и в лучшем случае сжигались в коксовых печах.
В дальнейшем стали строить заводы, оборудованные
аппаратурой для улавливания химических продуктов коксования. Первый
такой завод в России был построен в 1881 году.
Для коксования в настоящее время служат угли различных
марок. Предварительная подготовка углей заключается в
дроблении их до величины зерен менее 3 мм и в шихтовке, т. е.
смешивании пригодных для коксования углей. Если зольность
шихты выше стандартной (8%), шихту подвергают обогащению,
чаще всего мокрым способом в отсадочных машинах.
Подготовленную шихту направляют в башни коксовых печей.
34
Коксовые печи и процесс коксования
Современные коксовые печи представляют собой агрегаты
(батареи), состоящие из ряда горизонтальных камер E8—75)
в общей кладке. Между камерами находятся простенки, в
которых расположены горелочные каналы B5—30). Один простенок
обогревает стенки двух соседних камер. Под простенками
находятся регенераторы, через которые, нагревая насадку
регенераторов, проходят продукты горения, имеющие температуру 700—
750° С. Тепло, отданное регенераторам, используется затем для
8Г~РЇГ я Ч
1 пШҐІІ
gLft KM
ІІШ (li/Iiol 10
« ;| 11
ffl І ІІ
m fflni Rl ill
<6
J ILJ1 -1уЫу ¦
H f «¦
1 1, ." P 9
Lll ii 111 1_
в
Рис. 5. Схема конструкции коксовых печей ПК-2К (с
перекидными каналами).
подогрева воздуха, идущего на горение, и бедных газов,
например доменного, применяемого для обогрева коксовых печей.
Благодаря этому повышается температура горения и
увеличивается теплотехнический коэффициент полезного действия печей.
Различные системы коксовых печей отличаются в основном
устройством обогревательной системы.
В типовых печах ПК-2К, изображенных на рис. 5,
обогревательные каналы одной стороны камер соединены несколькими
перекидными каналами с обогревательными каналами другой
стороны. В то время как в одном простенке происходит горение,
через другой уходят продукты горения. Через 15—20 мин направ-
3* 35
ление обогревающего газа и воздуха и продуктов горения
меняется автоматически на обратное, чем достигается равномерность
обогрева печи.
В печах системы ПВР (рис. 6) обогревательные каналы
одного простенка соединены попарно так, что когда в одном канале
идет горение, через другой уходят продукты горения, а через
15—20 мин направление газов меняется на обратное (кантование).
Камеры типовых печей имеют длину 13,2 м, ширину 0,407 м,
высоту 4 м. Емкость их 18,3 м3. В настоящее время строятся
печи, в которых камеры имеют длину 15 м, высоту 5 м, ширину
0,425—0,475 м, емкость 30 мг. В своде камер имеются
три—четыре загрузочных люка и стояк, через который отводятся летучие
продукты коксования. С торцевых сторон камеры имеют
съемные двери, которые плотно закрыты во время коксования и
снимаются, когда нужно выдать кокс из печи. Шихту загружают
в накаленную камеру через люки с помощью загрузочного
вагона с тремя—четырьмя бункерами.
Нагревание угля в печи происходит быстро от стенок к
середине. Разложение угля начинается около 200° С, для перехода угля
в размягченное (пластическое) состояние требуется температура
выше 350° С. При 480—500° С заканчивается первичное
разложение угля — образуется полукокс, первичная смола и
первичный газ. При температуре— 750° С происходит пирогенетическое
разложение первичных продуктов — образуется кокс,
ароматические углеводороды, коксовый газ.
Химизм разложения органической массы угля при высокой
температуре пиролиза сложен и недостаточно изучен. В нем
сочетаются реакции разложения, поликонденсации,
изомеризации и др. Отщепление отдельных карбоксильных и гидроксиль-
ных групп от макромолекул угля приводит к появлению
свободных валентностей у периферийных атомов углерода, которые,
соединяясь между собой, дают высококонденсированное
вещество — кокс.
В результате отщепления циклов, соединенных с основным
ядром через кислородные, эфирные и полиэфирные мостики,
образуются налс.мольная водя, смола и газ.
Если первичные химические продукты разложения угля
представляют собой смесь преимущественно парафиновых, олефино-
вых, нафтеновых углеводородов, фенола, то продукты коксования
состоят уже из смеси ароматических углеводородов—от
бензола до многокольчатых и гетероциклических углеводородов. Из
серы и азота угля при коксовании образуются сернистые (H2S,
CS2, COS и др.) и азотистые (NH3, HCN и др.) соединения.
Около 65—70% азота и серы остается в коксе. К концу коксования,
через 15—16 часов, коксовый пирог вследствие усадки отходит
от стенок печи, что облегчает выдачу его из печи с помощью
коксовыталкивателя. Образующаяся во время коксования паро-
36
Разрезы
А-А
коксовая сторона
Разрез Г-Г
'*а
Машинная с/порона
Рис. 6. Схема обогрева печей ПВР:
В — воздух; Г—доменный газ; ПГ — продукты горения.
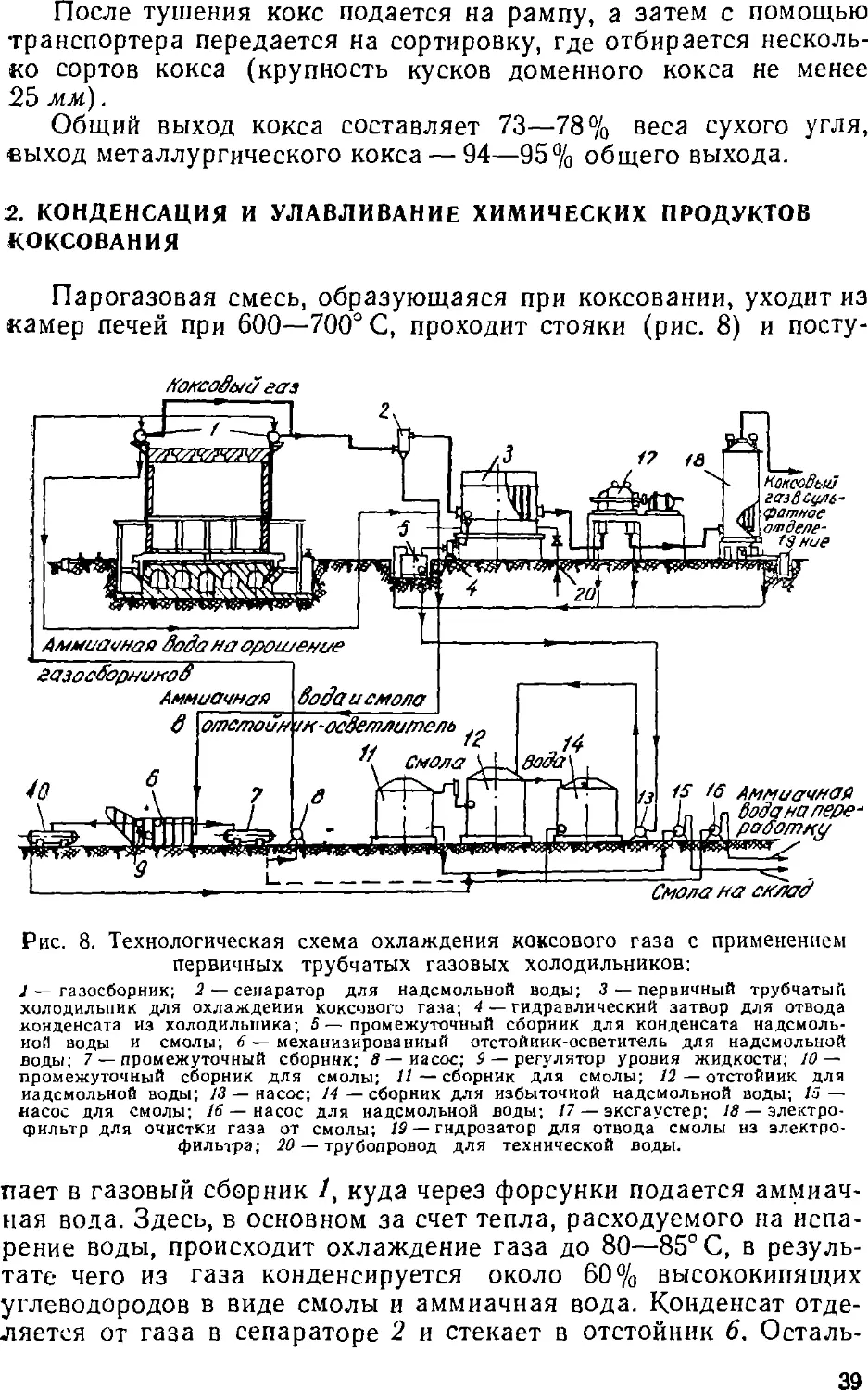
іазовая смесь через стояки камер поступает в общий для всей
батареи газовый сборник.
После окончания коксования камеры печи отключаются от
газового сборника, двери с обеих сторон печи снимаются и
коксовый пирог с помощью коксовыталкивателя выдается из печи
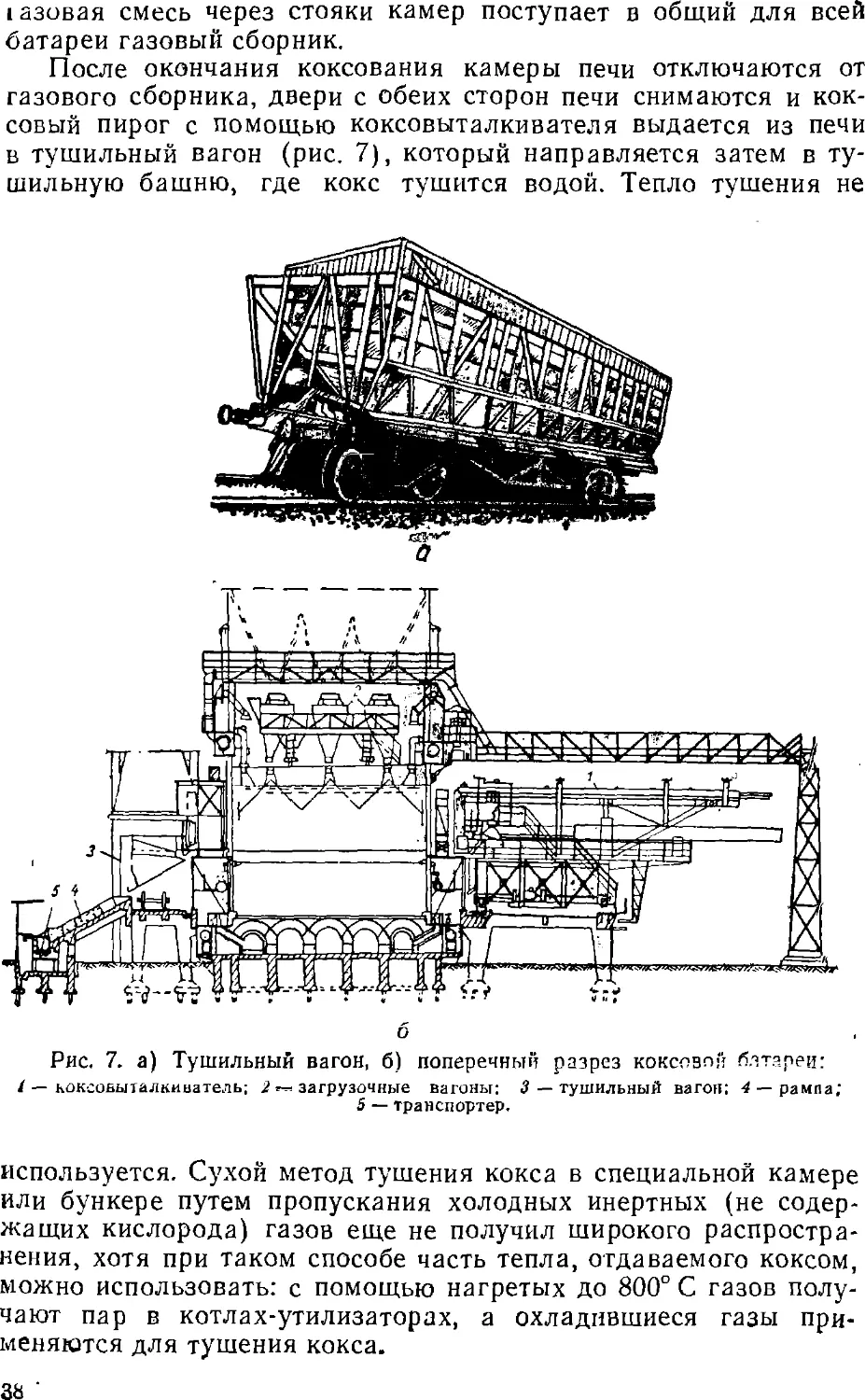
в тушильный вагон (рис. 7), который направляется затем в
тушильную башню, где кокс тушится водой. Тепло тушения не
Рис. 7. а) Тушильный вагон, б) поперечный разрез коксовой блтареи:
/ — коксоБыталкиоатель; 2 *-* загрузочные вагоны: 3 — тушильный вагон; 4 — рампа;
5 — транспортер.
используется. Сухой метод тушения кокса в специальной камере
или бункере путем пропускания холодных инертных (не
содержащих кислорода) газов еще не получил широкого
распространения, хотя при таком способе часть тепла, отдаваемого коксом,
можно использовать: с помощью нагретых до 800° С газов
получают пар в котлах-утилизаторах, а охладившиеся газы
применяются для тушения кокса.
38 '
После тушения кокс подается на рампу, а затем с помощью
транспортера передается на сортировку, где отбирается
несколько сортов кокса (крупность кусков доменного кокса не менее
25 мм).
Общий выход кокса составляет 73—78% веса сухого угля,
выход металлургического кокса — 94—95% общего выхода.
2. КОНДЕНСАЦИЯ И УЛАВЛИВАНИЕ ХИМИЧЕСКИХ ПРОДУКТОВ
КОКСОВАНИЯ
Парогазовая смесь, образующаяся при коксовании, уходит из
камер печей при 600—700° С, проходит стояки (рис. 8) и посту-
ХоксоЗый ааз
Смола на cx/XTd
Рис. 8. Технологическая схема охлаждения коксового газа с применением
первичных трубчатых газовых холодильников:
1 — газосборник; 2 — сепаратор для надсмольной воды; 3 — первичный трубчатьііі
холодильник для охлаждения коксового газа; 4 — гидравлический затвор для отвода
конденсата из холодильника; 5—-промежуточный сборник для конденсата надсмоль-
иоA воды и смолы; 6 — механизированный отстойник-осветитель для надсмольной
воды; 7 — промежуточный сборник; 8 — насос; 9 — регулятор уровня жидкости; 10 —
промежуточный сборник для смолы; // — сборник для смолы; 12 — отстойник для
иадсмольной воды; 13 — насос; 14 — сборник для избыточной надсмольной воды; 1й —
насос для смолы; 16 — насос для надсмольной воды; 17 — эксгаустер; 18 —
электрофильтр для очистки газа от смолы; 19 — гндрозатор для отвода смолы нз
электрофильтра; 20 — трубопровод для технической воды.
ттает в газовый сборник /, куда через форсунки подается
аммиачная вода. Здесь, в основном за счет тепла, расходуемого на
испарение воды, происходит охлаждение газа до 80—85° С, в
результате чего из газа конденсируется около 60% высококипящих
углеводородов в виде смолы и аммиачная вода. Конденсат
отделяется от газа в сепараторе 2 и стекает в отстойник 6. Осталь-
39
ная смола конденсируется при дальнейшем охлаждении газа
(см. ниже). Таким образом, в результате коксования угля
получают кокс, смолу, коксовый газ и аммиачную воду, которые затем
подвергают дальнейшей переработке.
Лары из
колоний*
Пор
Сульфат^
насклад
Рис. 9. Схема улавливания аммиака из газа:
/ — паровой подогреватель; 2—сатуратор; 3—ловушка;
4 — циркуляционная кастрюля; 5 — насос; 6 — площадка
для стока раствора; 7 — кастрюля обратных токов; 8 —
центрифуга; 9 — эжектор.
По выходе из газовых сборников коксовый газ охлаждается
в холодильнике 3 до 25—30° С. Образующийся при этом
конденсат стекает через гидравлический затвор в промежуточный
сборник, а затем в отстойник,
где снизу собирается смола,
а сверху — аммиачная вода
(надсмольная). Затем
коксовый газ газовым насосом
(эксгаустером) подается в
электрофильтр, где практически
полностью освобождается от тумано-
образной смолы.
Выход сырой смолы
достигает 3,5—5,5% от веса сухого
угля.
После освобождения от
смолы газ проходит паровой
, где нагревается
„!гГох™ен^ Т. ?ГР
/-конечный холодильник; 2 - гидравли- ДО 50-60° С, И ПОСТУПаЄТ ЧЄРЄ5
ческий затвор; 3 — нафталиновый отстой- барбОТер В СЭТураТОр ДЛЯ УЛЭВ-
„„к; 4 -^Р«-нр«ля-ГГсосафталина: ливания аммиака кислым F-
8% свободной H2SO4)
насыщенным раствором сульфата аммония (рис.9).
Кристаллы сульфата аммония накапливаются в коническом
днище сатуратора, откуда периодически выдаются насосом или.
эжектором. Содержание NH3 в газе перед сатуратором состав-
40
ляет 6—7, после сатуратора — 0,02—0,03 г/м5. Одновременно
серной кислотой улавливаются из газа и пиридиновые
основания.
Освобожденный от аммиака коксовый газ охлаждается в.
конечных холодильниках, где отделяется нафталин, до 20—25° С
(рис. 10). Затем газ подается в установку для извлечения
бензольных углеводородов, находящихся в газе в виде паров.
Наибольшее распространение в промышленности получил метод
Гаї
Рис. 11. Технологическая схема улавливания
бензольных углеводородов из газа в скрубберах,
орошаемых маслом:
/ —сборник обезбензоленного масла; 2, 3, 4— насосы,
5—сборник насыщенною бензолом масла; 6— насос;
7 — регуляторы уровня масла.
улавливания бензольных углеводородов поглотительными
маслами— каменноугольным или соляровым. Каменноугольное
масло является продуктом переработки каменноугольной смолы,.
соляровое — продуктом переработки нефти.
Установка для извлечения бензольных углеводородов из газа
состоит из трех последовательно соединенных скрубберов с
деревянной хордовой или металлической спиралевидной насадкой
(рис. 11) (диаметр до 5,5 м, высота до 40 м). Газ поступает
в нижнюю часть промывателей, масло — сверху через
разбрызгивающее устройство. Свежее масло подается в последний по ходу
газа скруббер, а затем из последнего в предыдущий и т. д.,
передвигаясь навстречу газу. Таким образом, при извлечении
бензольных углеводородов из газа соблюдается принцип
противотока в каждом скруббере и в системе.
Масло, насыщенное бензольными углеводородами,
отбирается из первого скруббера и передается в бензольное отделение
41
для отгонки из него бензольных углеводородов. После
охлаждения оно снова возвращается на орошение скрубберов.
Содержание бензольных углеводородов в газе до бензольных
скрубберов составляет 30—35 г/м3, а после них — не больше
2 г/м3.
Поглотительное масло, утратившее до некоторой степени
способность извлекать бензол из газа, подвергается регенерации
путем перегонки с водяным паром или в трубчатых установках.
3. ПРИМЕНЕНИЕ КОКСОВОГО ГАЗА И ЕГО ОЧИСТКА
Коксовый газ, выходящий из бензольных скрубберов, имеет
состав:
Н2— 55—60%; СН4 —23—25%; СО — 6—7%; О2-0,3—
0,5 %; С„Н,„ — 2—2,3 %; N2 — 6—7 %; СО2 + H2S — 2—3 %.
Теплотворная способность его 17640—17850 кдж/м3. Выход—300—
330 м3 на тонну сухого угля.
В настоящее время коксовый газ применяется как топливо
на металлургических заводах, в коммунальном хозяйстве и как
химическое сырье. Из коксового газа выделяют водород,
необходимый для синтеза аммиака методом фракционированной
конденсации при низких температурах. Получающаяся при этом
этиленовая фракция служит сырьем для различных синтезов.
Примесь сероводорода в коксовом газе нежелательна и в тех
случаях, когда газ используют в качестве топлива', и тогда, когда
он является химическим сырьем. Поэтому коксовый газ очищают
от сероводорода.
Методы очистки коксового газа от сероводорода
подразделяются на мокрые и сухие. К мокрым методам относятся: 1)
содовый, 2) железо-содовый, 3) поташный, 4) этаноламиновый,
5) мышьяковый и др. Из этих способов в СССР получил
наибольшее распространение вакуум-содовый метод, разработанный
Харьковским углехимическим институтом.
К методам сухой очистки относится метод очистки газа
болотной рудой. Таким методом можно удалить сероводород
практически полностью, до содержания сероводорода 0,02 г/м3. Поэтому
б тех случаях, когда необходима тщательная очистка газа,
например при использовании его в быту, прибегают сначала к
мокрой, а затем к сухой очистке.
Вакуум-содовый метод очистки коксового газа от
сероводорода заключается в следующем. Газ подается в скруббер,
который орошается 4—5%-ным раствором соды при 30° С.
Сероводород, а вместе с ним углекислота и синильная кислота,
присутствующая в газе в небольших количествах @,5—1,2 г/м3),
абсорбируются, образуя соли по следующим обратимым реакциям:
Na2CO3 + HaS5=sNaH0Oa + NaHS, A)
42
Na2COs + C02 + H20 *=t 2NaHCO3, B)
Na2CO3 + HGN ¦=> NaCN + NaHCO3. C)
Раствор этих солей подвергают регенерации, нагревая под
вакуумом 8—8,5 Н/см2 до 60—63° С (температура кипения). При
этом реакции A), B), C) протекают в обратном направлении.
Выделяющийся газ после охлаждения представляет собой смесь
75—80% H2S, 18—2О°/о СО2 и 2—4% HCN. После отмывки
синильной кислоты водой в скруббере, газ направляют в
установку для производства серной кислоты методом мокрого катализа.
mm
Рис. 12. Чугунный очистительный
ящик с гидрозатвором.
Регенерированный раствор соды в смеси со свежим
раствором снова применяют для очистки газа.
Этот метод очистки газов от сероводорода дает возможность
коксохимическим заводам получать серную кислоту,
необходимую для производства сульфата аммония.
После вакуум-содовой очистки в газе остается еще 2—3 г/м3
сероводорода, который в случае необходимости удаляется с
помощью очистки болотной рудой. При этом концентрация H2S
доводится до 1 мг/м3.
Болотная руда содержит 30—75% Fe2O3. Чтобы придать руде
рыхлость, ее смешивают с древесными опилками (около 4%)
и добавляют примерно 0,5% гашеной извести.
Приготовленную таким образом очистительную массу
помещают в ящики или башни, располагая ее слоем на сетчастых
полках (рис 12).
Коксовый газ направляют таким образом, чтобы он проходил
под каждую полку и, пройдя через слой болотной руды, уходил
из нижней части аппарата. Связывание сероводорода в
присутствии щелочи происходит по уравнению
2Fe (ОН), + 3H2S = 6Н2О + Fe2S3 + 52,58 кдж.
Очистительная масса, насыщенная сероводородом,
подвергается регенерации, которую производят, продувая воздух в смеси
с паром через слой массы в тех же аппаратах. Регенерация идет
по уравнению
43
2Fe2S3 + 3O2 -і- 6H2O = 4Fe (OHK + 6S + 1218 кдж.
Очистительную массу считают отработанной, если в ней
накопится до 60% серы; после этого она может служить сырьем для
производства серной кислоты.
Таким образом, сера коксового газа, которая находится в нем
преимущественно в виде сероводорода и представляет собой
вредную примесь, может быть почти целиком использована для
производства необходимой в химической промышленности серной
кислоты.
Поскольку одновременно с сероводородом болотная руда или
масса «Люкс» извлекают из коксового газа также цианистые
соединения, отработанная очистительная масса может служить
сырьем для извлечения цианистых и роданистых соединений
(берлинской лазури, роданистого аммония).
Из мокрых способов очистки газов от сероводорода большое
распространение получили- этаноламиновый и мышьяковосодо-
вый методы.
Взаимодействие этаноламинов с сероводородом протекает по
реакциям
где R =СН2СН2ОН.
Одновременно улавливаются из газа углекислота и синильная
кислота.
При 25° С реакции идут вправо, при нагревании выше 105° —
влево, что используется для регенерации отработанных
растворов. В результате регенерации получается концентрированный
сероводород.
Для поглощения сероводорода по мышьяковосодовому
методу применяется водный раствор окситиомышьяковокислого
натрия. Взаимодействие с сероводородом протекает по уравнению
Na3AsS3O + H2S -* Na3AsS4 + Н2О.
При регенерации раствора продувкой воздухом при 35—40°С
ььідєляєіся сілеменіарная сера:
2Na3AsS4 + О, -+ 2Na3AsS3O + 2S.
Высокая дисперсность серы позволяет с успехом применять
ее для борьбы с вредителями в сельском хозяйстве.
4. ПЕРЕРАБОТКА ХИМИЧЕСКИХ ПРОДУКТОВ КОКСОВАНИЯ
Переработка аммиачной (надсмольной) воды
Содержание аммиака в надсмольной воде достигает 6—
10 г/л, содержание сероводорода 1—2 г/л, содержание СО2
2—3 г/л. Кроме того, в аммиачной воде содержится некоторое
44
количество фенолов, пиридиновых основании, цианистых и
роданистых соединений.
Аммиак находится в воде в виде NH4OH, а также солей,
легко диссоциирующих при нагревании
(NH4JCO3, (NH4JS и NH4CN
и недиссоциирующих
(NH4JSO4, (NH4)C1, NH4CNS.
Аммиачную воду перерабатывают с целью извлечения
наиболее ценного компонента —
аммиака и части фенолов.
Для извлечения аммиака
аммиачную воду подвергают
нагреванию острым паром в
тарельчатых аммиачных колоннах в
присутствии известкового молока.
Известковое молоко необходимо
для разложения прочных
аммонийных солей, например:
(NH4JSO4 + Ca(OHJ =
= 2NH3 + 2Н2О + CaSO4.
УСТРОЙСТВО амМИаЧНОЙ КОЛОН- известно-,
ны видно из рис. 13. Аммиачная
вода подается на тарелки сверху,
пар — снизу, известковое
молоко — в среднюю часть колонны.
Выделяющиеся из колонны пары
аммиака и воды направляются
в дефлегматор, где часть паров
воды конденсируется.
Аммиак направляется в
сатуратор, где получают сульфат
аммония, или в нейтрализатор
пиридиновой установки—для выделения-, легких пиридиновых
оснований из маточного раствора (т. е. из насыщенного
раствора сульфата аммония), выведенного из сатуратора. Пиридиновые
основания связываются в сатураторе серной кислотой в виде
сульфата пиридина и при нейтрализации освобождаются по
реакции
C6HSNHSO4H + 2NH3 ->¦ (NH4J SO4 + CBH6N.
Сульфат пиридина Пиридин
Нейтрализованный раствор после отделения слоя пиридиновых
оснований возвращается в сатуратор.
Пиридин служит сырьем для производства ряда ценных
фармацевтических препаратов.
Сточные
fio'tai
Рис. 13. Аммиачная колонна.
45
Производство сырого бензола н его переработка \_
Как уже упоминалось, раствор бензола в масле поступает
в бензольный цех, где путем перегонки в колоннах разделяется
на сырой бензол и поглотительное масло. После отгонки в масле,
например соляровом, остается еще 0,2% бензольных
углеводородов. Это масло охлаждается и затем используется для
улавливания бензольных углеводородов. Отгонка бензольных
углеводородов производится следующим образом (рис. 14). Масло,
/ІО//6/ бензола, fodt/u масла
Раствор \ / oq
''толаЙ
поглотительном масле
Сыройбрнзол
Растбор
Масло воаЬ
т
Soda
Рис. 14. Схема выделения бензола из его раствора в поглотительном масле:
/ — дистилляциониая колонна; 2 — предварительный конденсатор; 3—конденсатор;
4 — масляный теплообменник; 5 — подогреватель.
насыщенное бензольными углеводородами, предварительно
подогревается за счет теплообмена в конденсаторах 3, 2 горячими
парами, уходящими из колонны, затем в теплообменнике 4, через
который проходит горячее масло из колонны, и, наконец, в
паровом подогревателе 5 до температуры і20—140° С.
Затем масло подается в верхнюю часть перегонной
колонны / и, протекая с тарелки на тарелку, встречается с водяным
паром, который подается в нижнюю часть колонны. Пар, барбо-
тируя через слой масла, находящийся на каждой тарелке
колонны, увлекает с собой бензольные углеводороды. При этом
температура их кипения снижается.
По выходе из колонны смесь водяного пара и паров
бензольных углеводородов поступает сначала в предварительный
конденсатор 2, где отделяется часть воды и увлеченные с паром
легкие фракции масла, а затем — в конечный конденсатор 3, где
конденсируются оставшиеся пары воды и бензольные углеводо-
46
роды. В нижней части конденсатора бензол отделяется от воды
(всплывает вверх) и спускается в сборник.
Обезбензоленное масло стекает из нижней части колонны,,
охлаждается в теплообменнике 4 за счет масла, поступающего
на перегонку, а затем в масляном холодильнике и подается снова
в промыватели бензола (холодильник для масла не указан на
рисунке).
Полученный бензол называют сырым. В нем содержится 60—¦
65% бензола. 10—16% толуола, 5—7% ксилола, 1%
сероуглерода, 0,3% тиофена; 5—8% гомологов бензола: этилбензола,
мезитилена, кумола и пр.; 0,6% фенола, 0,3% пиридина.
Выход сырого бензола составляет 0,9—1,1% от веса сухого
угля.
Переработка сырого бензола заключается в следующем.
Сырой бензол промывают сначала 10—15%-ным раствором
едкого натра для извлечения фенолов, а затем 30%-ной серной
кислотой для извлечения пиридиновых оснований. После
нейтрализации кислоты щелочью бензол промывают водой. Промывка
производится в специальных аппаратах с мешалками
турбинного гипа или в непрерывно действующих промывателях. После-
отстаивания бензол отделяют от промывной жидкости.
Промытый сырой бензол подвергают перегонке в непрерывно-
действующих ректификационных колоннах, получая такие
фракции: 1) сероуглеродную — до 95° С; 2) бензольную— 130°; 3) то-
луольную — 140°; 4) ксилольную—160°; 5) тяжелый бензол—¦¦
200°. Остаток — сольвент-нафта.
Полученные фракции обезвоживают, промывая их 50%-ной
серной кислотой, а затем с помощью 92% -ной серной кислотьі
удаляют из них ненасыщенные углеводороды, сернистые,
азотистые и кислородсодержащие соединения.
При обработке фракций крепкой серной кислотой
происходят реакции сульфирования:
С6НЙ + H2SO4 -*• C6H5SO3H + Н2О;
присоединения серной кислоты по месту двойной связи с
образованием эфиров, а также реакции полимеризации ненасыщенных
соединений.
Сернистые и кислородсодержащие соединения также
участвуют в процессах полимеризации и сульфируются, как,
например, тиофен:
C4H4S + H2SO4 -> C4H3SSO3H + НХ>.
тиофен тиофенсульфо-
кислота
Смесь образующихся при промывке веществ имеет вид темно-
коричневой смолы и называется кислой смолой, так как в ней
содержится свободная серная кислота.
При отстаивании промытых фракций кислая смола
собирается внизу и может быть отделена. Часть ее растворяется в про-
47
мытых фракциях и отделяется при окончательной ректификации
их. Поскольку серная кислота в процессе мойки расходуется
на химические реакции в небольшом количестве, целесообразно
производить ее регенерацию, которая заключается в
разбавлении кислой смолы водой. В процессе разбавления происходит
.гидролиз сульфосоединений и эфиров и отделение собственно
смолы от кислоты.
Промытые фракции сырого бензола подвергают второй
ректификации также в колоннах непрерывного действия. В
результате получают чистый бензол, чистый толуол, чистый ксилол
(смесь о—м—гс-ксилолов), технический сероуглерод,
растворители— сольвент I и сольвент II и кумароновую смолу. Кумаро-
новая смола представляет собой продукт полимеризации кума-
рона
сн
СН
и индена
і
СН
который остается в
О СН2
растворенном виде во фракции тяжелого бензола и отделяется
при ее ректификации.
Кроме того, коксохимические заводы выпускают моторный
и авиабензол, которые представляют собой смеси бензола,
толуола и ксилола, получающиеся из промежуточных фракций при
ректификации.
В настоящее время освоено также производство особых
сортов бензола: для нитрования, бессернистый, бензол для синтеза
и др.
Переработка каменноугольной смолы
Каменноугольная смола представляет собой смесь большого
¦числа органических соединений (около 300) преимущественно
-ароматического ряда.
Переработка смолы на коксохимических заводах обычно
заключается в делении ее на ряд фракций, которые в свою
очередь перерабатываются с целью получения главных
компонентов каменноугольной смолы — бензола, толуола, ксилола,
фенолов, нафталина, пиридиновых оснований, антрацена, масел,
пека (остаток).
Раньше перегонку каменноугольной смолы производили с
кубовых установках периодического действия, но в настоящее
время смола перерабатывается в непрерывнодействующих
установках с трубчатыми печами. Эти печи представляют собой
камеры, разделенные внутри перегородкой на две части — радиант-
ную и конвекционную; в той и в другой части печи проложены
змеевики из труб, по которым движется перерабатываемое сырье
(рис. 15).
48
В радиантнои части печи установлены горелки, поэтому
змеевики, имеющиеся здесь, обогреваются преимущественно
тепловыми лучами радиации, а змеевики, находящиеся в
конвекционной части печи,— дымовыми газами, т. ё. путем конвекции.
вход
флегмы
'отбор
нафталиновой
фракции
,птбор феноль-
ной фракции
j і чгг"*
Рнс. 15. Трубчатая печь с
реакционной секцией:
/ — фронтовой экран; 2 —
потолочный экран; 3—конвекционная
секция; 4 — отверстие для
форсунки; 5 — боров.
Ректификационная колонна
современных трубчатых
установок представляет собой
тарельчатую колонну, как бы
состоящую из нескольких
колонн, поставленных одна на
другую. Обычно в колонне
бывает 33—52 тарелки (см.
рис. 16).
Обезвоженную смолу
нагревают в радиантнои секции
печи до температуры, при
которой все нужные фракции
переходят в парообразное
состояние (до температуры однократного испарения), а затем
направляют пары в колонну, где происходит их фракционированная
конденсация.
Вследствие хорошей ректификации паров на таких
установках возможен отбор более узких фракций смолы, чем при
перегонке в кубах, что облегчает их дальнейшую переработку. Выход
фракций приведен в табл. 7.
Легкую фракцию смолы, содержащую бензол и его гомологи,
обычно перерабатывают вместе с сырым бензолом.
Фенольную фракцию используют для получения фенолов.
Последние извлекаются из фракции промывкой 15—20%-ным
Вход
оапрого
пара
франции
Рис. 16. Колонна фракционной
конденсации.
4 8-1504
49
водным раствором едкого натра; при этом образуются феноляты
натрия по реакции
АгОН -hNaOH -+¦ ArONa + Н2О.
Таблица 7
Название фракций
Легкая
Фенольная
Нафталиновая
Поглотительная
Антраценовая I
Антраценовая II
Антраценовая III
Пек
Выход,
% к
смоле
0,3
4,5
9,64
7,3
9,2
7,3
5,0
51,9
Содержание, % к фракции
фенолов
до 3
36
10
4
5,5
5
нафталина
80
оснований
до 4
6
Феноляты переходят в водный слой, который отделяют от
масла, очищают от углеводородов продувкой паром и затем
разлагают угольной кислотой. В результате реакции выделяются
фенолы:
ArONa + СО2
Н2О
АгОН + NaHCO3.
Под фенолами в данном случае подразумевается смесь фенола,
крезолов, ксиленолов, которая называется черной карболкой.
Путем очистки и дополнительной перегонки получают чистый
фенол, крезолы и ксиленолы.
Промывая все остальные фракции раствором едкого натра,
также извлекают из них фенолы. Промывка 30%-ной серной
кислотой позволяет извлекать основания. Затем кристаллиза-
цией выделяют нафталин
и антоанен
из нафталиновой фракции
с его спутниками -- карбазолом
и фенантреном <^.
из антра-
NH
ценовых фракций.
Кристаллизацию производят в кристаллизаторах ящичного
типа и в механических кристаллизаторах, охлаждаемых водой.
Из кристаллизаторов сырой нафталин или сырой антрацен пода-
50
ют в центрифуги, а затем для более полного удаления масла
прессуют их под давлением 3000—6000 н/см2.
Прессованный нафталин промывают крепкой серной кислотой
и перегоняют или сублимируют, чтобы получить чистый
кристаллический или сублимированный нафталин.
Сырой антрацен подвергают обогащению методами
кристаллизации из растворителей, чтобы повысить содержание более
ценного компонента — антрацена.
В результате промывки фракций смолы серной кислотой
образуются сернокислые соли пиридиновых оснований, которые
отделяют от масел и разлагают аммиаком, получая
пиридиновые основания и сульфат аммония. Из сырых пиридиновых
оснований ректификацией выделяют чистый пиридин, пиридин-
растворитель, а-пиколин и р-пиколин.
Высококипящий остаток смолы — пек вытекает из пековой
колонны при 300—350° С в пековый теплообменник, где
охлаждается до 150° С. Окончательное охлаждение пека происходит
на погруженном в воду металлическом пластинчатом или
ленточном транспортере. К моменту выноса транспортера из воды
пек успевает застыть и в конце транспортера сбрасывается
в виде палочек («макарон»).
Каменноугольный пек применяется в дорожном
строительстве, для производства пекового кокса, используемого в
электродной промышленности, для производства пластмасс и черных
лаков.
Помимо перечисленных главных химических продуктов
коксования, из смолы и сырого бензола выделяются многие
индивидуальные продукты: индол, аценафтен, изомеры крезолов, крио-
скопический бензол, изомеры ксилола, метилнафталин, псевдо-
кумол, мезитилен, хинолин, чистый антрацен, карбазол, фенан-
трен и др.
В настоящее время коксохимическая промышленность
выпускает более 160 наименований индивидуальных продуктов,
применяемых в различных отраслях химической промышленности.
5. КОКСОВАНИЕ СЛАНЦЕВ
Для коксования сланцев применяются вертикальные
камерные печи непрерывного действия с внешним обогревом. Обогрев
обеспечивается сжиганием газа в обогревательных простенках,
соединенных с регенераторами.
Сланец загружается в камеру сверху, кокс непрерывно
выгружается из нее снизу. Парогазовая смесь отводится из зоны
пиролиза F00—700° С) через специальные окна в газовый
сборник, где охлаждается водой до 85° С; при этом конденсируется
смола.
Из 1 г сланца с теплотворной способностью 11 340—
4* 5Ї
11 550 кдж[кг получается 320—380 м5 газа с теплотворной
способностью 18 068 кдж/м3.
После отделения смолы и надсмольной воды из газа
улавливают аммиак, нафталин, очищают газ от сероводорода и затем
под давлением около 50 н/см2 улавливают маслами газовый
бензин, после чего сжимают газ до 500 н/см2, сушат диэтиленглико-
лем и направляют потребителю.
Сланцевый газовый бензин из Кохтла-Ярве содержит около
29,6% непредельных углеводородов, 57% ароматических и 12,5%
нафтеновых и парафиновых углеводородов. В состав
ароматических углеводородов входит до 33% бензола. Смола
коксования сланцев богата фенолами.
s полукоксование
Полукоксованию обычно подвергают молодые каменные угли,
бурые угли, торф и сланцы.
Процесс полукоксования был известен еще в XVIII веке и
использовался для получения бездымного топлива и
осветительных масел. В заводском масштабе полукоксование угля начали
осуществлять с середины XIX века.
Сущность этого процесса сводится к нагреву топлива без
доступа воздуха до 500—600°С в печах различного устройства,
в результате чего получают полукокс, первичную смолу, газ
и водный конденсат. Такие продукты называются первичными,
поскольку они являются продуктами менее глубокого
разложения угля, чем при коксовании.
Полукокс представляет собой кусковой материал или
порошок с 9—10% летучих. Обычно он применяется как местное
высококалорийное топливо.
Смола содержит парафиновые, нафтеновые и ароматические
углеводороды. Гумусовые угли дают смолы, содержащие 35—
50% фенолов, сапропелитовые угли — смолы, не содержащие
фенолов.
Газ полукоксования содержит больше углеводородов, чем
коксовый, и обладает высокой теплотворной способностью (до
25 200—36 540 кдж.'м3 для каменных углей). Вследстппе этого
целесообразно передавать его на далекие расстояния для
использования в промышленных целях (и смеси с низкокалорийным
газом). Кроме того, полукоксовый газ может служить сырьем
для синтезов.
Полукоксование проводят в печах с внешним и внутренним
обогревом. Печь второго типа изображена на рис. 17. Из
образующейся в печах парогазовой смеси выделяют смолу, после
чего улавливают маслами легкие углеводороды — бензин.
Из смол извлекают фенолы, бензиновую и керосиновую
фракции, а полученный тяжелый остаток подвергают крекингу или
гидрогенизации для повышения выхода бензина.
52
Полукоксование является одним из методов переработки
твердого топлива в жидкое.
Особенно большой выход смолы дает полукоксование
сланцев. Так, из сланцев мес-
торождения Кохтла-Ярве
получают 34,4—23,2%
смолы (на сухой сланец).
I Парогазовое
Парогазовая
смесь
і загрузка
\ топлива
Парогазовая
смесь
t
водогретый
газ
Холодный
газ
Рис. 17. Печь для
полукоксования торфа
непрерывного действия с
внутренним обогревом.
Рис. 18. Поперечный разрез
туннельной печи для полукоксования
сланцев:
/ — туннель; 2 — вагонетка; 3 —
обогревательный канал: 4 — калорифер.
Смола представляет собой весьма ценное химическое сырье,
на базе которого создается сейчас «сланцевая химия». Так как
в состав сланцев входит очень много золы, полукокс содержит
всего около 10% углерода. Высокое содержание в полукоксе
извести позволяет использовать его для производства цемента.
Полукоксование сланцев осуществляется главным образом
в туннельных печах (рис. 18). Длится процесс полукоксования
около трех часов.
7. ДЕСТРУКТИВНАЯ ГИДРОГЕНИЗАЦИЯ УГЛЯ
Деструктивная гидрогенизация производится с целью полу-
чени51 из твердого или тяжелого жидкого топлива легкого
жидкого топлива — бензина и керосина. По своему химизму это
53
очень сложный процесс, при котором одновременно происходит
расщепление (деструкция) высокомолекулярных соединений
(макромолекул угля) с образованием более простых
предельных и непредельных углеводородов и осколков и присоединение
водорода к осколкам — по месту двойных связей и к
ароматическим углеводородам. Происходит также деполимеризация
и другие процессы.
Присоединение водорода (гидрогенизация) сопровождается
уменьшением объема и выделением тепла. Протеканию реакций
гидрогенизации способ-
й газ
фомыЗное
масло
ствует повышение давле-
НИЯ И ОТВОД ТеПЛа
^ Ласта Ъ
Жидкие про-
(^А-/77і/ НО
раяЗеление
I
Водород
Рис. 19. Схема гидрогенизации углей
(жидкофазная стадия).
Обычно
гидрогенизацию углей проводят при
давлении 2000—7000 н/см2
и температуре 380—
490° С. Для ускорения
реакции применяют сероус-
тойчивые катализаторы —
окислы и сульфиды
железа, вольфрама, молибдена
с различными добавками-
активаторами.
Из-за сложности
процесса гидрогенизации
процесс получения из
угля легкого топлива —
бензина и керосина —
осуществляется в две
ступени — в жидкой и паровой фазе. Наиболее пригодны для
гидрогенизации молодые каменные и бурые угли, содержащие
значительное количество водорода. Лучшими считаются угли,
у которых соотношение между углеродом и водородом не больше
16—17. Вредными примесями являются сера, влага и зола.
Допустимое содержание влаги 1—2%, золы 5—6%, содержание
серы должно быть минимальным. Во избежание большого
расхода водорода не подвергают гидрогенизации топливо, богатое
кислородом (например, древесину).
Технология процесса гидрогенизации заключается в
следующем. Тонко измельченный уголь (до 1 мм) с нужной зольностью
перемешивают с катализатором, чаще всего окислами железа,
подсушивают и тщательно перетирают в пастовой мельнице
с маслом, которое получается при разделении продуктов
гидрогенизации. Содержание угля в пасте должно составлять 40—
50%. Пасту подают в установку для гидрогенизации (рис. 19)
пастовым насосом 1 под нужным давлением; туда же компрессо-
54
рами 2 и 3 подается свежий и циркулирующий водород. Смесь
предварительно подогревается в теплообменнике 4 за счет тепла
уходящих из колонны гидрогенизации паров и газов, а затем в
трубчатой печи 5 до 440° С и поступает в колонну
гидрогенизации 6, где за счет тепла реакции температура повышается до
480°. После этого продукты реакции разделяются в сепараторе 7,
из верхней части которого уходят пары и газы, а из нижней —¦
шлам. і
Парогазовая смесь охлаждается в теплообменнике 4 и
водяном холодильнике 8 до 50° С и разделяется в газосепараторе 9.
После снятия давления конденсат перегоняют, получая
«широкую фракцию» C00—350°) и тяжелое масло. Широкая фракция
после извлечения из нее фенолов поступает во вторую парофаз-
ную ступень гидрогенизации. Шлам, отделенный в сепараторе 7,
центрифугированием разделяют на тяжелое масдо и твердый
остаток, который подвергают полукоксованию. В результате
образуются тяжелое масло и фракция, которую присоединяют
к широкой. Зольные остатки используются как топливо. Тяжелые
масла идут на приготовление пасты. Газы, отделенные в
сепараторе 9, после абсорбции углеводородов в скруббере 10 маслами
под давлением возвращаются в процесс с помощью
циркуляционного насоса 3.
Гидрогенизацию во второй ступени проводят чаще всего
в присутствии WS2 под давлением 3000 н/см2 при 360—445° С.
Из полученного продукта гидрирования выделяют бензин и
керосин или дизельное топливо. В топливе, полученном
гидрогенизацией, нет непредельных углеводородов, а сера находится в
виде сероводорода, легко удаляемого при промывке щелочью,
а затем водой. Деструктивная гидрогенизация осуществляется
в колоннах, изготовленных из легированных сталей, содержащих
хром, никель, молибден. Толщина стенок — до 200 мм, высота до
18 м и диаметр І м. В колоннах для гидрогенизации в паровой
фазе катализатор помещают на сетчатые полки.
Выход бензина может достигать 50—53% на горючую массу
угля.
ГЛАВА V
ГАЗИФИКАЦИЯ ТВЕРДОГО ТОПЛИВА
Газификацией твердого топлива называется процесс
превращения его в газообразное топливо путем неполного окисления
органической части топлива кислородом, воздухом или другими
соединениями, содержащими кислород, например водяным
паром.
В результате газификации получается генераторный газ —
воздушный, если окислителем является воздух, водяной, если
55
окислитель — водяной пар, смешанный, если в качестве
окислителя используется смесь воздуха с водяным паром.
Генераторные газы применяются не только как топливо, но
и как химическое сырье для производства синтетических
продуктов — аммиака, спиртов, жидкого топлива.
Газифицировать можно все виды топлива — дерево, солому,
торф, бурые и каменные угли, антрацит, горючие сланцы, кокс,
полукокс, древесный уголь.
При газификации топлива воздухом или кислородом
протекают следующие реакции:
С + О2 = СО2 + 400,68 кдж, A)
2С + О2 = 2СО + 233,100 кдж, B)
2СО + О2 = 2СО2 + 568,26 кдж. C)
Одновременно двуокись углерода может восстанавливаться
углеродом до окиси углерода:
СО2 + С = 2С О — 167,58 кдж. D)
При наличии паров воды протекает реакция
С + Н2О = СО + Н2 — 119,19 кдж. E)
Возможна также реакция между окисью углерода и водяным
паром:
СО + Н2О = СО2 + Н2 + 32,75 кдж. F)
В качестве побочного продукта может образоваться и метан по
уравнениям
СО + ЗН2 « СН4 + Н2О + 205,3 кдж, G)
2СО + 2Н2 «=* СН4 + СО2 + 254,562 кдж. (8)
При производстве газов стремятся вести процесс так, чтобы
получались газы, содержащие максимально возможное
количество горючих компонентов, т. е. с возможно большей
теплотворной способностью. Направлять процесс газификации в желаемую
сторону можно, только изучив фтлко-химические ллкоиимер-
ности, которым подчиняется этот сложный гетерогенный процесс.
Основными факторами, влияющими на скорость и равновесие
приведенных выше реакций, являются температура и давление.
Большое значение имеют также скорость дутья, активность
и величина поверхности топлива и другие факторы, поскольку
в гетерогенных процессах на скорость химических реакций
воздействуют скорость диффузии, адсорбционная и десорбционная
способность поверхности топлива.
Определение величины констант равновесия, необходимых
для расчета процесса газификации, производится
экспериментальным (по анализу получающихся продуктов реакции в момент
56
равновесия) или расчетным
формуле Нернста:
путем, например по упрощенной
где Q — тепловой эффект реакции;
7 —температура реакции;
Т[ — температура, при которой определено Q;
До — увеличение числа молей.
В табл. 8 приведены значения lg/Cp для реакций A) — E).
Таблица 8
Температу-
пя °С
500
600
700
800
900
1000
1100
1300
1500
1700
Реакция A)
1 P°2
Чо,
_
—
—20,8
— 17,37
— 16,05
— 14,78
— 12,86
—
—
Реакция B)
І??Р°2
gP2co
_
—
—20,85
—
— 18,90
— 18,07
— 17,43
— 16,33
—
—
Реакция C)
, p2copo,
P2CO,
_
—
—20,8
—
— 15,82
— 13,93
— 12,12
—9,38
—7,25
—5,58
Реакция D)
P2CO
Pco,
— 2,49
— 1,12
—0,05
+0,82
+ 1,55
+ 2.12
+ 2,65
+ 3,48
+ 4,10
+ 4,68
Реакция E)
PH2O
gpcopH,
+ 1,67
—
—0,13
— 1,43
— 1,91
— 2,34
—3,02
—
Из таблицы следует, что экзотермические реакции
образования окиси и двуокиси углерода A) — C) практически можно
считать неравновесными,
поскольку равновесие этих
реакций смещено в сторону
образования окиси и
двуокиси углерода даже при очень
высоких температурах
газификации — 1300—1700° С
(значения Ig/Cp велики и
отрицательны) . Одновременно
протекающая
эндотермическая реакция D) является
обратимой, так как с
повышением температуры
равновесие сдвигается в сторону
образования окиси углерода,
0.1 вою
02 :
0.4 »
t
к
to „ Am
f
w
500 600
TOO 800 SOO
t, V
«00 HOO
Рис. 20. Равновесие при
восстановлении двуокиси углерода углеродом при
различном суммарном давлении
окиси и двуокиси углерода (от 0,1 до
10 ні см2).
причем для полного
восстановления двуокиси углерода
в окись углерода (при 1 ат)
необходима температура выше 1000° С, что следует из
приведенных на рис. 20 кривых равновесия этой реакции.
Из этой же диаграммы видно, что с повышением давления
выход окиси углерода понижается, так как реакция сопровож-
57
дается увеличением объема (согласно принципу Ле-Шателье).
Эндотермическая реакция взаимодействия углерода с водяным
паром E), которая является основной при производстве
водяного газа,— также обратимая реакция. Равновесие ее смещается
в сторону образования водорода и окиси углерода с повышение^
температуры и понижением давления.
Экзотермические реакции G) и (8) протекают с уменьшением
объема. Поэтому равновесие этих реакций сдвигается в сторону
образования метана с понижением температуры и увеличением
давления, что используется в промышленности для производства
¦газа с высокой теплотворной способностью в газогенераторах,
работающих под давлением.
В идеальном случае образование воздушного генераторного
газа протекает по уравнению B):
2С + О2 + 3,76 N2 = 2СО + 3,76 N2 -f 233,1 кдж.
Состав этого газа
СО= 2+тЫ
Выход воздушного газа должен составить
,, 2 + 3,76 _ „_ ,,
V газа =2.о|536 = 5,37 м3/кг,
12
где 0,536 = 20^4 — количество углерода, кг/м3 СО;
3,76 — количество молей азота на 1 моль кислорода в воздухе.
Теплотворная способность газа равна
Qc= 128,10-34,7 = 4445,0 кдж/м3.
• н
Получаемый на производстве из кокса воздушный
генераторный газ имеет следующий состав:
СО —31,2%; Н2— 0,5%; СО2-1,5%; N2-66,8%; QCH -
4183 кдж їм3.
Производств" воздушного гснорлторного гаяз
характеризуется высокими температурами в зоне газификации (до 1500° С),
что создает большие затруднения, связанные с плавлением золы
и температурным воздействием на обмуровку газогенератора.
Поэтому воздушный процесс ведут в газогенераторах с жидким
шлакоудалением, которые по сеоєй конструкции напоминают
доменные печи.
К недостаткам воздушного генераторного газа относится
также его низкая теплотворная способность. По этим причинам
обычно (если не ставят целью получение газа с высоким
содержанием СО, например для синтезов) получают паровоздушный
генераторный газ. Количество вводимого водяного пара
практически составляет 450—500 г/кг углерода, что дает
возможность снизить температуру газификации примерно до 1000° С
благодаря эндотермичности процесса образования водяного
газа по реакции E). При газификации мелкого кокса
паровоздушной смесью получается газ примерно такого состава:
СО2 —5,5%; H2S —0,2%; СО —27,5%; Н2 — 13,5%; СН4 —
0,5%; N2 —52,6%; О2 — 0,2%; QHC —5166 кдж/м3.
Паровоздушный генераторный газ является наиболее дешевым и
распространенным видом генераторного газа.
1. ПРОИЗВОДСТВО ВОДЯНОГО ГАЗА
Водяной газ получается в результате взаимодействия
водяного пара с раскаленным топливом. В идеальном случае —
реакция E) — получается газ, состоящий из окиси углерода и
водорода A : I). Вследствие эндотермичности реакции производство
водяного газа представляет собой периодический процесс, в
котором чередуются воздушное и паровое дутье.
При воздушном дутье топливо разогревается, образующийся
воздушный генераторный, газ отводится в дожигательную
камеру, где сжигается с воздухом. Тепло продуктов горения
используется для получения пара в котле-утилизаторе. После
вытеснения остатков воздушного генераторного газа паром начинается
собственно процесс получения водяного газа, который состоит
из трех фаз: паровое дутье снизу, сверху и снова снизу слоя
топлива.
Образующийся водяной газ отводится после охлаждения
и промывки в газгольдер водяного газа. Остатки водяного газа
удаляются из газогенератора кратковременной продувкой
воздухом (тоже в магистраль водяного газа) во избежание взрывов
при воздушном дутье. Весь цикл производства водяного газа
занимает 4 минуты при условии автоматического переключения
дутья и газовых потоков и состоит из шести фаз:
1) воздушное дутье для разогрева топлива — 65 сек
2) продувка газогенератора паром — 5 »
3) продувка паром снизу —80 »
4) продувка паром сверху —70 »
5) продувка паром снизу —15 »
6) продувка воздухом — 5 »
Затем цикл повторяется (рис. 21).
При использовании в качестве дутья смеси водяного пара
и кислорода производство водяного газа может стать
непрерывным.
Состав водяного газа из кокса в % по объему следующий:
Н2-50, СО —38; -СО2 —6,0; N2 — 5,0; СН4 — 0,5; H2S — 0,3;
QHC —11 ЗА0кдж/м3.
59
,13
Водяное/
ГО
Лар
Рис. 21. Схема установки для получения водяного газа:
;_ генератор; 2 — водяная рубашка; 3 — паросборник; 4 —
воздуходувка; 5 — топка регенератора; 6 — регенератор; 7 —
пароперегреватели; 8 — котел-утилизатор; 9 — стояк; 10 —
гидравлический затвор; JJ —холодильник: 12— клапан; 13 — дымовая
труба; а, 6. в. г. д — задвижки.
Топливо
Генераторный
газ
Рис. 22. Генератор паровоздушного газа:
/ — кожух; 2 —футеровка; 3 — водяная рубашка;
4 — желоб; 5 — загрузочный барабан;
6—распределительный конус; 7 — колосниковая решетка;
8 — жесткое кольцо; 9 — зольная чаша; 10 —
плужок; ;/ —зольный бункер; 12— центральная
труба; 13 — гидравлический затвор.
60
Водяной газ применяется в основном как сырье для синтеза
аммиака, жидкого топлива, метанола и др. В настоящее время
значение его снизилось, так как имеется природный, более
дешевый газ.
2. ГАЗОГЕНЕРАТОРЫ
Для газификации топлива в плотном слое (крупность кусков
25—100 мм) применяются газогенераторы, представляющие
собой шахтные печи диаметром около 3,6 лі, высотой 4,6 м,
футерованные огнеупорным кирпичом (рис. 22). Нижняя часть
газогенератора,
соответствующая зоне газификации,
охлаждается с помощью
водяной рубашки.
Газогенераторы для
получения паровоздушного
газа снизу открыты и
погружены в чашу с водой,
которая медленно вращается
вместе с колосниковой
решеткой, под которую
подается дутье. При вращении
колосниковой решетки (рис.
23) изогнутые верхние
поверхности колосников то
приподнимают, то опускают слой топлива, благодаря чему
поддерживается равномерная плотность топлива. При этом зола
смещается от центра к стенке, где попадает в кольцевой зазор
между решеткой и жестким кольцом, а затем в чашу с водой,
откуда периодически выгружается плужком. Топливо
загружается сверху газогенератора через питательный механизм.
Образующийся газ охлаждается поступающим топливом до 500—
550° С и отводится через пыльную камеру на охлаждение и
очистку.
Производительность такого газогенератора составляет 6—
7,5 тыс. лг3 газа в час.
Газогенераторы для получения водяного газа снизу закрыты,
потому что при больших скоростях дутья для герметичности
необходимо было бы создавать гидрозатвор большой высоты.
Зола периодически удаляется через специальный бункер.
Газогенераторы для мелкозернистого топлива
Газификация мелкозернистого топлива производится либо
в «кипящем», либо во «взвешенном» слое. В газогенераторах
с кипящим слоем газифицируется мелкозернистый уголь
крупностью до 10 мм и влажностью 8—12%. Скорость газового потока
Рис. 23. Механическая колосниковая
решетка с зольной чашей.
61
достигает 2—3 м/сек, вследствие чего плотность слоя нарушается
и топливо, поднимаясь, как бы кипит в газогенераторе.
Газогенератор с кипящим слоем (рис. 24) представляет собой
шахтную печь /, футерованную внутри огнеупорным кирпичом.
Процесс газификации происходит главным образом в нижней
конической части шахты 2, по окружности которой расположены
три винтовых конвейера 3 для подачи
топлива. Под колосниковую решетку 4
подается дутье при повышенном
давлении. Для догазовывания унесенного
газовым потоком топлива на высоте
примерно 5 м от колосниковой
решетки вводится через форсунку 5
вторичное дутье, составляющее 15—25%
общего дутья.
Генераторный газ уходит через
выходной штуцер 6 при 300° С и уносит
с собой около 80% содержащей до
50% угля золы, которая улавливается
в циклонах, а затем передается в
газогенераторы с жидким шлакоудалением.
Остальная зола собирается на
колосниковой решетке и сбрасывается
специальным охлаждаемым водой
бруском 7 в шнеки 8; затем она поступает
в бункер 9, а оттуда в вагонетки.
Тепло уходящего из генератора газа
используется для получения пара
в котлах-утилизаторах. Дутьем может
быть воздух и смесь кислорода с
паром.
При газификации бурого угля на парокислородном дутье
получается газ такого состава:
Н2-42,5%; СН.,-2%;
СО —28,0%; N2 —1,5%;
ГО2 — 24,9; N«S —1,2.
Производительность газогенератора с кипящим слоем
составляет 80 000 м3 в час, т. е. в 10 раз превышает
производительность мощных газогенераторов с плотным слоем топлива.
3. ПОДЗЕМНАЯ ГАЗИФИКАЦИЯ УГЛЕЙ
Сущность процесса подземной газификации заключается
в том, что на угольный пласт с поверхности земли
пробуриваются две скважины (рис. 25), которые соединяются между собой
по пласту угля горизонтальной выработкой. Пласт угля
зажигается, образуя «огневой забой». В одну из скважин подается
дутье (воздух),
62
Рис. 24. Газогенератор
с «кипящим» слоем.
Омывая поверхность угля, воздух вступает в реакцию с углем,,
в результате чего образуется генераторный газ, который
выводится на поверхность земли через вторую скважину.
При подземной газификации в зависимости от состава дутья
могут быть получены газы, пригодные для энергетического и
технологического использования.
Рис. 25. Схема подземного газогенератора
и газообразования в нем:
/ — дутьевая скважина; 2 — газоотводная
скважина; 3 — пласт угля; 4 — шлак; 5 —первоначальный
разжнговый канал.
Чтобы создать запас газа, устраивают специальные
хранилища — газгольдеры. Газ в них находится под давлением; таким
образом, газгольдеры являются и регуляторами давления газа.
ГЛАВА VI
ПЕРЕРАБОТКА НЕФТИ И ГОРЮЧИХ ГАЗОВ
Перегонка нефти в России впервые была осуществлена в
1745 г. Ф. Прянудовым на реке Ухте, а затем в 18123 г. братьями
Дубиниными вблизи г. Грозного.
До конца XIX века получавшийся при разгонке нефти бензин
считался побочным продуктом и спускался в море или сжигался,
так как не находил себе применения. Основным продуктом
перегонки нефти был керосин, используемый для освещения.
Остающийся после отгонки керосина мазут сжигался как топливо
в форсунках.
С конца XIX века в связи с изобретением двигателей
внутреннего сгорания бензин из отброса превратился в важнейший
продукт. Мазут стал ценным сырьем для производства бензина
и бензола методом крекинга и пиролиза.
Основы процесса крекинга были разработаны в 1880—1890
годах В. Г. Шуховым, спроектировавшим первую в мире крекинг-
установку, построенную, однако, не в России, а в США, в годы
63
первой мировой войны. В первой пятилетке был осуществлен
советский крекинг по методу В. Г. Шухова и М. А. Капелюш-
никова.
В 30-х годах XX столетия были открыты методы
ароматизации нефти (Н. Д. Зелинский, Б. Л. Молдавский, В. И. Коржев).
Под руководством Н. Д. Зелинского разработан метод
каталитической дегидрогенизации углеводородов с получением
бутадиена—сырья для синтеза каучука.
Современные методы переработки нефти подразделяются на
физические, к которым относится метод разделения нефти
обыкновенной перегонкой (прямая гонка), и химические, к которым
принадлежат методы термической переработки нефти и
нефтепродуктов, жидких и газообразных: крекинг, пиролиз,
деструктивная гидрогенизация, полимеризация и затем алкилирование
и изомеризация.
Путем комбинирования различных методов производят
комплексную переработку нефти, получая большое количество
разнообразных продуктов.
В настоящее время нефтехимия представляет собой большую
самостоятельную отрасль химической промышленности и
важнейшими продуктами ее являются различные виды моторного
топлива.
1. СВОЙСТВА МОТОРНЫХ ТОПЛИВ
Моторные бензины, применяемые в карбюраторных
двигателях, где пары топлива и воздуха воспламеняются от искры,
должны обладать хорошими пусковыми свойствами, полностью
испаряться в двигателях, иметь высокую теплотворную способность,
химическую стойкость, высокие антидетонационные свойства,
низкую температуру замерзания, не содержать примесей,
оказывающих корродирующее действие (органических кислот,
сернистых соединений и Др.). Испаряемость бензинов зависит от
температурных границ их кипения. Например, авиационный бензин
(«АБ», ГОСТ 5760—51) перегоняется в пределах 40°—180°С
(97,5%). Давление паров не должно превышать 360 мм рт. ст.
Требуется, что&ы 10% бензина выкипали до /5° С (температуры,
характеризующей пусковые свойства бензина), 90%—до 145° С
(температуры, характеризующей хорошую испаряемость
бензина). Температура начала кристаллизации должна быть не
выше—60° С, а теплотворная способность — около 46 200 кдж/кг.
Чем выше теплотворная способность, тем меньше расход
топлива.
Под антидетонационными свойствами подразумевается
способность бензина воспламеняться в карбюраторном двигателе
без взрыва даже при высокой степени сжатия. Взрывы ведут
к преждевременному выталкиванию поршня, в результате чего
64
нарушается согласованность в работе цилиндров двигателя и
наблюдаются перебои в работе мотора (мотор стучит).
Неправильная работа мотора приводит к снижению его
производительности и более быстрому износу.
Для оценки антидетонационных свойств топлива с 1927 года
применяется определение октановых чисел с помощью эталонных
смесей чистого изооктана B, 2,4-триметилпентан) и
нормального гептана. Изооктан детонирует в двигателе при сжатии 7,7,
а гептан при 2,8. Антидетонационные свойства изооктана
приняты за 100, а гептана за нуль. Если испытуемое топливо детонирует
в стандартных условиях в двигателе так же, как эталонная смесь,
содержащая, например, 80% изооктана, то октановое число
топлива равно 80.
Таким путем установлено, что наиболее стойкими к
детонации являются ароматические углеводороды (бензол—108),
затем следуют изопарафины (изооктан — 100, изопентан—90),
затем нафтеновые (циклогексан — 85,5), ненасыщенные B —
гексилен— 78) и, наконец, жидкие парафиновые с нормальной
цепью (гексан —25).
Для повышения октанового числа к бензинам добавляют
антидетонаторы. Самым распространенным из них является
тетраэтилсвинец; его требуется 2—3 мл на 1 л топлива. Чтобы на
деталях двигателя не отлагался свинец, прибавляют хлористый
или бромистый этил. При этом свинец улетучивается в виде
бромистого или хлористого свинца. Октановые числа
этилированных авиабензинов 98,6; 95; 91; 70 (ГОСТ 1012—54).
2. ПЕРЕРАБОТКА НЕФТИ
Добытую нефть сначала освобождают от растворенных в Heft
газов в сепараторах, снижая давление и скорость движения,
а затем от воды, минеральных солей и механических примесей.
Соли удаляют, промывая нефть водой в специальных
установках. Обезвоживание и освобождение от механических примесей
производят длительным отстаиванием нефти. В случаях
образования эмульсии воды с нефтью простым отстаиванием воду
удалить нельзя. Для разрушения эмульсий применяются методы
центрифугирования, нагревания нефти под давлением до 140° С
и электрический способ, предусматривающий одновременно
и обессоливание нефти. Электрообессоливающие установки,
сокращенно называемые «ЭЛОУ», состоят из системы электро-
дегндраторов, в которые через смесь нефти, умягченной воды
и раствора едкого натра пропускают электрический ток
промышленной частоты C0 000—40 000 в). Эмульсия разрушается,
капли воды соединяются и отделяются отстаиванием. Поступающая
на переработку нефть должна содержать менее 1% воды и не
более 70—100 мг/л солей.
5 8-1504 65
3. ПРЯМАЯ ПЕРЕГОНКА НЕФТИ
Для фракционированной перегонки нефти применяются так
называемые трубчатые установки непрерывного действия, о
которых уже говорилось, при описании процесса переработки
каменноугольной смолы. В этих установках нефть нагревается в трубах
змеевиков печи, сначала в конвекционной, а затем в радиант-
ной ее части, до заданной температуры 320—350° С.
нефть
Тпжвлый
газойль
Веретенное
масло
машинное
масло
Перегретый пор
Нефть
Перегретый пар
Рис. 26. Схема атмосферно-вакуууной перегонки нефти.
Вследствие больших скоростей прохождения нефти по трубам
(выше 3 м/сек) время ее пребывания в печи незначительно,
поэтому и потери нефти, связанные с термическим разложением,
значительно ниже, чем при перегонке в старых кубовых
установках.
В настоящее время применяются еще более совершенные
беспламенные трубчатые печи конструкции Бахтияппва и др.,
в которых тепло излучают нагретые до 1І00—1200й С стены
печи, что обеспечивает еще более равномерный обогрев
поверхности труб, а следовательно и продукта.
Из трубчатой печи нефть в парожидкостном состоянии
подается в ректификационную колонну, подобную изображенной на
рис. 17. В колонне происходит фракционирование нефти.
Нередко перегонку осуществляют в две ступени в атмосферно-вакуум-
ной установке, принципиальная схема которой изображена на
рис. 26. Установка состоит из двух трубчатых агрегатов. В
первом из нефти отделяют легкие фракции при атмосферном
давлении, а остаток — мазут подвергают фракционированию под
66
вакуумом для снижения температуры кипения тяжелых
составных частей и уменьшения потерь от их разложения.
Сырая нефть насосом 5 подается через теплообменники 6,
9, 12, 13, где подогревается до ПО—150° С, а затем в трубчатой
печи / до 320° С, и поступает в среднюю часть нижней секции
первой ректификационной колонны 3. Здесь происходит
испарение и отделение образовавшихся паров от жидкой части —
мазута. Пары, поднимаясь вверх, делятся на фракции, которые
отбираются из соответствующих секций колонны: легкий газойль,
керосин, лигроин. После охлаждения в холодильниках фракции
стекают в сборники. Бензин уходит из колонны в парообразном
состоянии и поступает сначала в теплообменник 6, через который
проходит сырая нефть, а затем в холодильник 7, отстойник, где
отделяется от воды, и в сборник 8. Часть бензина возвращается
в колонну в виде флегмы для конденсации паров высококипящих
фракций, движущихся вверх, что улучшает качество
отбираемого бензина. В нижнюю часть колонны подается острый пар,
который, барботируя через слои жидкости на тарелках,
увлекает с собой легкокипящис углеводороды и приносит тепло,
необходимое для испарения этих фракций (часть тепла выделяется
за счет конденсации более тяжелых фракций). Кроме того,
в присутствии пара снижается температура кипения составных
частей нефти.
Мазут, вытекающий из первой колонны нагретым до 275—
277е С, подается насосом в вакуумную часть трубчатой
установки, подогревается до 425° С в печи 2 и поступает в среднюю
часть нижней секции вакуумной ректификационной колонны 4.
В нижнюю часть колонны подается перегретый пар.
Отделившиеся от жидкой части пары, поднимаясь вверх, делятся на
фракции цилиндрового, машинного и веретенного масел. После
охлаждения эти фракции собираются в соответствующие
сборники. Тяжелый газойль уходит из колонны в виде паров,
проходит теплообменник 9, где охлаждается за счет поступающей
сырой нефти, затем конденсатор 10 и отстойник //, в котором
отделяется вода. Часть газойля возвращается в виде флегмы
в колонну, часть направляется в сборник. Из низа колонны
уходит жидкий остаток — гудрон.
Выход и качество получающихся фракций зависят от
состава перерабатываемой нефти.
В табл. 9 приведены границы кипения и выход фракций для
грозненской парафинистой нефти.
В табл. 10. приведены границы кипения и выход фракций,
полученных при перегонке мазута при отборе фракций под
остаточным давлением 0,7—1,0 ні см2.
Полученные при такой перегонке нефти легкие и тяжелые
фракции подвергают очистке от примесей сернистых,
кислородсодержащих и ненасыщенных соединений, которые обладают
5* 67
корродирующими свойствами и способностью осмоляться,
вследствие чего продукты темнеют.
Таблица 9
Фракции
Температура отбора,
°С
от веса
нефти
1. Бензиновая . . .
2. Лигроиновая . .
3. Керосиновая . .
4. Соляровое масло
5. Остаток — мазут
до 170
160—200
200—300
300—350
14,5
7,5
18,0
5,0
55,0
Таблица 10
Фракции
1. Веретенного масла
2. Машинного »
3. Легкого цилиндрового »
4. Тяжелого цилиндрового »
5. Остаток — гудрон
Температура отбора,
°С
230—250
260—305
315—325
350—370
Выше 370
% от веса
нефти
10—12
5
3
7
27—30
Очищенные дистилляты представляют собой уже товарные
продукты. Легкие дистилляты — различные виды моторного
топлива: 1) для карбюраторных двигателей — бензин, лигроин,
керосин, 2) для дизельных — газойль, соляровые дистилляты;
3) для реактивных двигателей — фракции керосина. Тяжелые
дистилляты, полученные при перегонке мазута, представляют
собой смазочные масла, которые в зависимости от области
применения подразделяются на индустриальные масла —
веретенное, машинное и др.; для двигателей внутреннего сгорания —
авиационные автолы и др.; трансмиссионные; турбинные;
компрессорные; для паровых машин — цилиндровые; масла особого
назначения.
Бензины, полученные прямой перегонкой нефти, в
зависимости от состава имеют октановые числа 50—78. Этилирование
ПОЗВОЧЯЄТ ITORЫСІЇТІі ОКТГІТІОГЇЬІГ* ТЇЇЇ^ТП f^C'ni'TTOr^ Чґї Я 7 - -^Гі
4. КРЕКИНГ НЕФТЕПРОДУКТОВ
Продукты прямой перегонки нефти, чаще всего высококипя-
щие фракции,— мазут, газойль, соляровые дистилляты —
подвергают переработке с целью получения бензина нагреванием до
температур 500—650° С в определенных условиях. Образование
бензина из тяжелых дистиллятов нефти происходит в
результате расщепления сложных углеводородов на более простые.
Вот почему эти процесы получили название крекинга (англ.
to crack — разбивать, расщеплять). При крекинге, однако,
происходят реакции не только расщепления, но циклизации, поли-
68
меризации и конденсации, т. е. образование в результате
взаимодействия продуктов расщепления между собой и
углеводородами исходного сырья новых сложных углеводородов.
Химизм крекинга весьма сложен, так как зависит от
состава исходного сырья и условий процесса — температуры,
давления, времени.
В состав сырья входят углеводороды различной термической
стойкости и разного строения. Наименьшей термической
стойкостью обладают парафиновые углеводороды, а из них менее
стойкими являются высококипящие углеводороды, энергия
активации разложения которых ниже, чем энергия активации,
необходимая для разложения легких углеводородов. Например,
энергия активации фракции 163—215° С грозненской парафиновой
нефти составляет 279,3 кдж/моль, а фракции 380—415° С —
233 кдж/моль.
Распад молекул может протекать с разрывом связи С—С—,
энергия активации которой в среднем равна 297,3 кдж/моль,
и с разрывом более прочной связи С — Н, энергия активации
которой составляет 385,2 кдж/мпль. Разложение с разрывом
связи С—С характерно для высококипящих углеводородов и
протекает по схеме
СгпН4п+2 ->¦ СпНгп-і-2 4- СПН2П — Q.
Разложение с разрывом связи по С—Н более характерно для
легкокипящих углеводородов и может быть представлено схемой
СпНіп+2 ->¦ С„Н2п 4- Н2 — Q,
Таким образом, в результате расщепления тяжелых
углеводородов образуются более простые насыщенные и ненасыщенные
углеводороды, но возможно образование и радикалов
С4Нхо ->¦ СНз + СзН,.
I I •
Эти радикалы способны к перегруппировке в насыщенные и
ненасыщенные углеводороды
СНз 4- СзН, ->¦ СН4 4- СзН6.
I I
Они могут также положить начало цепной реакции, в результате
которой образуются насыщенные и ненасыщенные
углеводороды.
Ненасыщенные углеводороды вступают в реакции между
собой — полимеризуюгся по схеме
ПСпНэп ->¦ (СлН2п)л.
Реакции полимеризации (синтеза) являются вторичными
реакциями процесса крекинга. Они протекают с выделением
тепла. Так как преобладают реакции расщепления —
эндотермические, процесс крекинга связан с затратой тепла
(приблизительно 840 кдж/кг газа и бензина). Нафтеновые углеводороды более
69
устойчивы, чем парафиновые; для них характерны реакции
дегидрогенизации, приводящие к образованию ароматических
углеводородов, например
СН2 СН
н2с/\:н2 нс/^сн
- +ЗН,
Н2СІ yCU2 НС „СН
СН2 СН
Возможны и реакции, сопровождающиеся разрывом кольца
и образованием парафиновых углеводородов.
Наиболее термически стойкими углеводородами являются
ароматические, которые в процессе крекинга склонны скорее не
расщепляться, а конденсироваться с образованием смол, кокса
и газов. Поэтому сырье, богатое ароматическими
углеводородами, не применяется для крекинга.
Влияние температуры и давления на реакции расщепления
углеводородов в крекинг-процессе
С повышением температуры термодинамическая
устойчивость углеводородов снижается, поэтому реакции разложения
углубляются и ускоряются, а выход продукта за один проход
увеличивается.
С повышением температуры меняется также направление
реакции расщепления углеводородов. При более низкой
температуре происходит разрыв цепи углеводорода посередине, где
связь между С—С менее прочная, а при более высокой —
разрыв цепи происходит на конце ее и сопровождается
образованием легких газообразных углеводородов вплоть до метана, т. е.
до более устойчивых термически углеводородов. Повышение
температуры ограничивается коксообразованием.
Давление способствует реакциям конденсации и
полимеризации, подавляет обратимые реакции расщепления,
сопровождающиеся увеличением объема. При более рысоких давлениях
уменьшается образование газообразных продуктов, снижается
содержание в бензинах ненасыщенных углеводородов.
Высокое давление E0—70 атм) в процессе крекинга
способствует удержанию основной массы сырья в жидком состоянии,
что позволяет осуществлять крекинг при более низкой
температуре.
Скорость реакций в жидкой фазе также увеличивается,
теплообмен улучшается, расход тепла уменьшается,
производительность аппаратов возрастает. Таким образом, реакционные
объемы аппаратов, работающих под давлением, можно значи^
тельно уменьшить. Крекинг, осуществляемый под давлением,
70
раньше называли жидкофазным, а теперь — крекингом в сме-
шажюй фазе, в отличие от парофазного, в котором реакции
протекают в паровой фазе.
В промышленности применяются различные виды крекинга.
Наибольшее распространение получил термический крекинг,
который проводится только при нагревании, и каталитический,
осуществляемый в присутствии катализаторов.
Разновидностью крекинга является рнформинг —
термический и каталитический, который применяется для получения
высокооктановых бензинов из низкооктановых или из лигроина.
Термический крекинг в смешанной фазе
В зависимости от качества сырья выбор условий процесса
крекинга несколько меняется. Крекинг тяжелого сырья, напри-
Мазут
бензинна
ста били-
я зоцию
крекинг - остаток
Рис. 27. Схема процесса термического крекинга мазута.
мер мазута, в настоящее время осуществляется в две ступени,
с рециркуляцией промежуточных продуктов, а иногда и газов.
Первая ступень, называемая легким крекингом, проходит
обычно при температуре 400—480° С и давлении 150 ні см2. После
отделения газов, паров легких углеводородов, крекинг-мазута
оставшуюся промежуточную фракцию подвергают глубокому
крекингу (вторая ступень) при температуре 500—510° С и
давлении до 600 н/см2 в смеси со свежим сырьем, а иногда и с газами.
Выход бензина при таком способе работы повышается примерно
в два раза по сравнению с однократным крекингом.
Схема установки для термического крекинга мазута
изображена на рис. 27.
Технология процесса заключается в следующем. Мазут
насосом 12 подается в ректификационную колонну 9, куда поступают
также пары углеводородов, полученных в результате крекинга.
Из нижней части колонны отводят тяжелую фракцию, которая
поступает для крекинга в трубчатую печь /, а из средней части
колонны — дистлллятную фракцию, идущую на крекинг в
трубчатую печь 2. Крекинг обеих фракций заканчивается в реакцион-
71
камере 3 при температуре около 480 С и давлении 200 н/см2
Продукты крекинга проходят через редукционный вентиль 4
в испаритель 5. Затем пары уходят в ректификационную колонну,
а жидкий остаток — в дополнительный испаритель 6. Отсюда
крекинг-остаток направляется после охлаждения в хранилище,
а выделяющиеся пары после охлаждения в конденсаторе 7 и
отделения от газа в газоотделителе возвращаются в процесс. Иа
верхней части колонны отводятся пары бензина, которые
конденсируются в холодильнике 10 и отделяются от газа в сборнике //;
часть бензина идет на орошение колонны. Выход бензина
достигает 30—35 % количества мазута.
Бензин термического крекинга чаще всего применяют как
автомобильное топливо.
В настоящее время часто объединяют установки для
перегонки и крекинга нефти, что дает большую экономию тепла.
Парофазный крекинг
Парофазный крекинг ведется при температуре 600—620° С
и небольшом давлении. Крекингу подвергаются пары, а не
жидкость, что ухудшает условия теплопередачи и увеличивает
расход тепла. Бензин парофазного крекинга содержит больше
ароматических углеводородов и поэтому имеет более высокое
октановое число.
При парофазном крекинге выделяется больше газов с
высоким содержанием непредельных углеводородов. Эти газы служат
сырьем для многих химических производств.
Пиролиз нефтепродуктов
Пиролиз нефтепродуктов (высокотемпературный крекинг
700—750°С), разработанный В. Г. Шуховым, 3. А.
Никифоровым и др. в конце прошлого столетия, использовался для
получения ароматических углеводородов как основы производства
взрывчатых веществ. В годы первой мировой войны этот процесс
широко применялся для получения толуола. До 1940 г. пиролиз
нефтепродуктов осгаьался основным методом производства
толуола.
С появлением более совершенных каталитических методов
получения ароматических углеводородов из нефтепродуктов
(гидроформинг, платформинг) процесс пиролиза, казалось бы,
утратил свое значение. Быстрое возрождение пиролиза связано
с развитием нефтехимии — с ростом потребности в газах,
содержащих непредельные углеводороды,— этилен, пропилен, бутилен.
Содержание этих углеводородов в газах пиролиза в несколько
раз выше, чем в газах коксования, термического и
каталитического крекинга. С целью расширения сырьевой базы для прсцес-
72
са пиролиза кроме жидких нефтепродуктов стали применять
природные и попутные газы, газообразные и жидкие углеводороды
газоконденсатних месторождений.
В ряде европейских стран сырьем для пиролиза служат бен-
зино-лигроиновые фракции прямой перегонки нефти, а также
сырая нефть. В условиях пиролиза, т. е. нагревания сырья до
700—750° С при атмосферном давлении, протекают реакции
между углеводородами сырья по схеме: парафины -> олефины ->-
диолефины і
нафтены К ароматические углеводороды
Наибольший выход ароматических углеводородов дает
дегидрогенизация сырья, богатого нафтеновыми шестичленными
углеводородами. Пиролиз осуществляется непрерывным методом
в установках с трубчатыми печами и ректификационными
колоннами.
Из продуктов пиролиза прежде всего отделяются при
охлаждении тяжелая смола и сажа, а затем в ректификационных
колоннах — масла и газ. Фракционирование легкого масла дает
бензольную (~20%), толуольную (~ 16%) и ксилольную
(— 11%) фракции, после очистки которых повторной
ректификацией получают чистые бензол, толуол, ксилол и пиробензол-
Последний представляет собой смесь ароматических
углеводородов, применяемых как авиационное топливо. В результате
переюнки смолы и масел выделяются зеленое и нафталиновое-
масла и пек, из которого получают беззольный кокс для
электродной промышленности. Выход газа, содержащего до 30%
ненасыщенных углеводородов, достигает 43—48%, выход масел;
45—47% от сырья.
Каталитический крекинг
Крекинг нефтепродуктов в присутствии катализаторов
приобретает все большее значение, так как он позволяет получать
из тяжелого нефтяного сырья большой выход бензинов лучшего.
качества.
В качестве катализаторов применяются алюмосиликаты,,
представляющие собой либо глины, обработанные и
обогащенные присадками окисей никеля, кобальта, меди, марганца и
других металлов, либо специальные синтетические массы.
Каталитический крекинг чаще всего осуществляется в
паровой фазе, при температуре 450—500° С и давлении 6—10 ні см2.
Химизм этого процесса отличается от химизма термического
крекинга. В каталитическом крекинге большое значение имеют
адсорбционные процессы, предшествующие расщеплению
углеводородов.
В результате каталитического крекинга получается бензин,,
содержащий ароматических, нафтеновых и парафиновых
углеводородов изостроения больше, чем бензин термического крекинга.
7»
Продукты
Каталитический крекинг сопровождается значительным коксо-
•образованием, при котором происходит выделение водорода.
Вследствие этого образуется меньше ненасыщенных
углеводородов и продуктов их полимеризации, составляющих тяжелый кре-
*синг-остаток. Кроме бензина получаются также более тяжелые
«продукты — лигроин, каталитический газойль и газы,
представляющие собой ценное сырье для органического синтеза.
Катализатор очень быстро покрывается слоем кокса
(примерно через 10 мин) и теряет свою активность. Его регенерируют
продувкой воздуха при 550—
600° С, после чего снова при-
меняют для крекинга.
Известны три основных
промышленных способа ката-
литического крекинга: 1) с
неподвижным фильтрующим
слоем катализатора, 2) со
взвешенным или кипящим слоем
катализатора и 3) с
движущимся катализатором.
Установки с неподвижным
слоем катализатора работают
периодически, так как процесс
прекращают для проведения
регенерации катализатора в
тех же аппаратах.
Большое распространение
Сырье
Рис. 28. Принципиальная схема
каталитического крекинга в кипящем слое:
/-реактор; 2 - регенератор; 3, 4_квта- "ОЛуЧИЛИ НЄПРЄрЬІВНО ДЄИСТВУ-
лизаторопроводы; S — трубчатая печь; ЩИЄ уСТЭНОВКИ С КИПЯЩИМ И
6 -колодец; 7-,-решеткв; «-« " дВИЖУЩИМСЯ СЛОЄМ КЗТЗЛИЗа-
тора.
Крекинг в кипящем слое катализатора проводится следующим
образом. Измельченный до 20—100 мк (микросферический)
катализатор вводится в поток паров сырья и увлекается им
в реактор. Пройдя реактор, пары крекинг-продукта вместе с
катализатором поступают в циклон. Отработанный катализатор
отделяется здесь от паров и направляется в регенератор, в
который вдувается горячий воздух. Отделение регенерированного
.катализатора от продуктов горения происходит также в
циклонах, откуда катализатор снова возвращается в реактор. В
процессе регенерации вследствие сгорания кокса температура
катализатора повышается и соприкасающиеся с ним пары сырья
подогреваются. Тепло катализатора компенсирует расход тепла
на эндотермические реакции крекинга. Работа установки для
крекинга в кипящем слое катализатора изображена на рис. 28,
где реактор и регенератор конструктивно объединены. Сырье
подогревается в трубчатой печи. Отношение катализатора
и сырья равно примерно 10. Адсорбированные катализатором
углеводороды удаляют, продувая паром нижнюю часть реактора.
При каталитическом крекинге получается около 70% бензина,
12—15% газов, 4—6% кокса от веса сырья, а также лигроин
и газойль, которые частично циркулируют в системе.
Бензин имеет октановое число 78—80; этилированием это
число можно повысить до 90—92.
5. РИФОРМИНГ-ПРОЦЕСС
Ведущим среди процессов получения высокооктановых
компонентов автобензинов является каталитический риформинг. В
настоящее время распространены различные виды риформинга,
путем комбинирования которых стремятся полностью
превратить сырье в высокооктановый компонент автомобильных топлип.
Из новейших процессов только флюид-гидроформинг
осуществляется с применением кипящего слоя пылевидного
катализатора, по схеме каталитического крекинга; в остальных
используется стационарный платиновый катализатор.
Риформинг с платиновым катализатором впервые был
применен в процессе платформинга, который и сейчас остается
наиболее перспективным.
Сырьем для риформинга служат в основном бензиновые
дистилляты, но допускается также переработка лигроиновых
фракций, полученных при прямой перегонке нефти и крекинге.
При каталитическом риформинге протекают
преимущественно реакции дегидрогенизации нафтеновых углеводородов и деги-
дроциклизации парафиновых углеводородов, в результате чего
получаются ароматические углеводороды и водород, например:
СН
нД m
сн2
сан14
но
сн
сн
+зн2
сн
НС!
сн
4Н2.
сн
Происходят, кроме того, реакции изомеризации пятичленных
углеводородов в шестичленные с последующей
дегидрогенизацией в ароматические, а также реакции гидрокрекинга и
изомеризации парафиновых углеводородов. Одновременно протекают
75
реакции расщепления, конденсации и др., в результате которых
образуются газы и кокс.
Для подавления реакций коксообразования процессы рифор-
минга ведут под давлением выделяющегося при
дегидрогенизации водорода. Количество выделяющегося водорода превышает
расход его в процессе. Избыточный водород выводится из цикла
и используется для других целей, а частично для гидроочистки
получающихся при риформинге и других процессах
нефтепродуктов (концентрация водорода — 80—90% об.). В результате
Жидкие продукты
на ректификацию
Рис. 29. Схема процесса ароматизации.
платформинга в зависимости от температуры и давления
образуются разные продукты. При 480—510° С и давлении 150—300
нісм2 получаются ароматические углеводороды, при высоком
давлении, до 500 н/смг,— бензины с октановым числом 98; после
этилирования октановое число бензинов повышается до 100.
Бензины, полученные методом платформинга, отличаются
стабильностью и содержат мало серы, так как в ходе процесса
происходит гидрогенизация ненасыщенных и сернистых соединений.
Газы, которые составляют 5—15% от веса сырья, представляют
собой ценное сырье для синтеза. В СССР получили
распространение разработанные акад. М. Д. Зелинским и др. методы
ароматизации нефтепродуктов с использованием платиновых
катализаторов (платина на активированном угле) и катализаторов из
окиси хрома, окиси молибдена на активной окиси алюминия.
Для ароматизации сырья, богатого парафиновыми
углеводородами, применяется гидроформинг — процесс, идущий при 500° С
и давлении 150—220 н/см2 в парных контактных агрегатах. В то
время как в одном агрегате происходит регенерация
катализатора, другой работает (рис. 29).
76
Сырье подается насосом 1. Циркулирующая в системе
газовая смесь, в состав которой входит водород, подается
компрессором 2 через теплообменники 3 в печь 4. После этого смесь
проходит последовательно первый реакционный аппарат 5, снова
печь 4 и второй реакционный аппарат 6. Получаемый продукт
охлаждается в теплообменниках 3 и конденсаторе 7. В
аппарате 8 отделяется газ, который возвращается в цикл и частично
выводится из системы. Жидкие продукты направляются на
ректификацию.
6. НЕФТЯНЫЕ И ПРИРОДНЫЕ ГАЗЫ И ИХ ПЕРЕРАБОТКА
Нефтяные и природные газы представляют собой смесь
преимущественно предельных углеводородов Сі—Сб с содержанием
метана от 45 до 85% объемных. Теплотворная способность их
достигает 37 300—69 800 кдж/м3. Природные газы чисто газовых
месторождений содержат метана больше G6—98%), и
теплотворная способность их достигает 37 065—39 840 кдж/м3.
Переработка попутных газов нефтедобычи и природных
газов производится физическими и химическими методами.
Улавливание и стабилизация газового бензина
Помимо метана, этана, пропана, бутана и пентана как в
нефтяных и попутных, так и в некоторых природных газах
содержатся в небольших количествах и более тяжелые углеводороды,
которые при передаче газа конденсируются в газопроводах,
образуя легкий газовый бензин.
Поскольку накапливание конденсата в газопроводах
затрудняет их эксплуатацию, а газовый бензин представляет собой
ценный продукт, первоочередной задачей при переработке газов
является извлечение из них газового бензина. Газовый бензин
получают из газов путем фракционированной конденсации их,
поглощением маслами или адсорбцией твердыми поглотителями
с последующей десорбцией бензина. Обычно такой бензин
содержит растворенные в нем легкие углеводороды — метан, этан,
пропан, бутан, которые удаляются из него при нагревании
глухим паром в колоннах. Этот процесс называется стабилизацией,
а бензин, освобожденный от легких углеводородов,—
стабильным.
Удаленные из бензина легкие углеводороды охлаждаются в
холодильниках. При этом конденсируется пропан-бутановая
фракция, а неконденсирующиеся газы отводятся.
Пропан-бутановая фракция отличается высокой теплотворной
способностью (для пропана 98994, для бутана 128982 кдж[м3) и
используется как моторное и коммунальное топливо, а также
как сырье для химической переработки.
77
Газовый бензин в чистом виде не применяется вследствие
образования газовых пробок в двигателях, но в смеси с товарным
автобензином A5—50%) дает хорошее автомобильное топливо.
При обязательном отбензинивании попутных газов и
стабилизации нефти получаются громадные количества сжиженных
газов (пропан-бутановых фракций). Для хранения их
необходимы специальные хранилища — наземные и подземные.
В Советском Союзе с 1954 г. осуществлено сжижение
природного газа с целью получения жидкого метана.
Жидкий метан представляет собой высококачественное мо-
ториое топливо с октановым числом 125. При сгорании его-в
моторах атмосфера не загрязняется отходящими газами, что
особенно важно для больших городов. Жидкий метан весьма
экономичен и удобен для снабжения потребителей, находящихся на
значительном расстоянии (до 100 км) от завода.
Химические методы переработки газов
Химические методы переработки природных газов можно
подразделить на три группы.
1) Методы, позволяющие переводить парафиновые
углеводороды газов непосредственно в ценные химические продукты
путем окисления, хлорирования и нитрования.
2) Методы конверсии парафиновых углеводородов газа
(взаимодействие метана с водяным паром) с целью получения смеси
окиси углерода с водородом — синтез-газа, который является
более дешевым сырьем для получения водорода, необходимого
при синтезе аммиака, чем водяной газ, и применяется для других
синтезов — метанола, жидкого топлива (см. ниже).
3) Методы крекинга углеводородов с целью получения
водорода, сажи, ацетилена и других ненасыщенных углеводородов.
Бутан и пентан природных газов и газов крекинга, а также
пропан-бутановая фракция, получающаяся при стабилизации
газового бензина, перерабатываются с целью получения
высокооктанового топлива путем изомеризации и последующего алки-
лирования:
п"бУтан ШШт^г ізобутан;
, * H2SO4+HF
изобутан+бутилен 20_40 н/см^0°с ~* изооктаны.
Бутан и изопентан служат также сырьем для получения
бутадиеновых и изопреновых каучуков.
Ненасыщенные углеводороды продуктов крекинга нефтяных
и природных газов, а также газы крекинга и пиролиза
нефтепродуктов являются весьма ценным сырьем для органического
синтеза (особенно этилен — см. основной органический синтез).
78
7. ОЧИСТКА НЕФТЕПРОДУКТОВ
Методы очистки нефтепродуктов подразделяются на
химические и физико-химические. Среди последних различают
адсорбционные и абсорбционные (экстракционные) методы.
Из химических способов самым распространенным являете»
сернокислотный, описание которого приведено на стр. 47.
Потери прямогонного бензина при очистке серной кислотой,
составляют приблизительно 1%, крекинг-бензина — до 5%.
Продукты взаимодействия серной кислоты с загрязнениями, в виде
смолы вместе с избыточной кислотой при стоянии отделяются-
от нефтепродукта. Они получили название кислого гудрона.
Серную кислоту из кислых гудронов регенерируют разбавлением
водой. Эфиры при этом расщепляются на углеводороды и кислоту.
Разбавленную серную кислоту концентрируют и снова
используют для очистки. Органическая масса кислого гудрона
применяется для производства асфальта, брикетирования топлив №
других целей.
После промывки серной кислотой нефтепродукты
обрабатывают раствором едкого натра, а затем водой и перегоняют,
получая бесцветные товарные продукты.
Адсорбционные методы очистки
Из физико-химических методов очистки наиболее удобны
адсорбционные. В качестве адсорбентов применяются
естественные глины, бокситы, силикагель. Методы эти основаны на
способности сернистых, кислородсодержащих и азотистых соединений,
а также асфальтов и смол легко адсорбироваться из
углеводородов. Сначала адсорбируются диолефины, затем олефины,
ароматические, нафтеновые и, наконец, предельные углеводороды.
Очистку адсорбентами можно производить в жидкой и
парообразной фазах.
Парофазная очистка удобна для крекинг-бензина. Пары
бензина из колонны поступают в цилиндрический аппарат,
работающий под давлением 100 ні см2, в котором на решетке помещен
адсорбент в виде зерен. Образующиеся продукты полимеризации
диолефинов, имеющие более высокую температуру кипения,
отделяются от паров бензина ректификацией. Ароматические и
непредельные углеводороды в этих условиях не адсорбируются,
поэтому очищенный таким способом бензин имеет более высокое
октановое число. Для удаления сернистых соединений в качестве
адсорбентов применяются бокситы и алюмосиликаты.
Для очистки масел и светлых продуктов прибегают к жидко-
фазной очистке — перемешиванию материалов с адсорбентом
(контактный способ) или фильтрованию их через неподвижный
слой адсорбента.
В первом случае глину смешивают с обрабатываемым мате-
79'
риалом, смесь нагревают, а затем отфильтровывают адсорбент,
получая однородный продукт.
При использовании метода фильтрации по мере загрязнения
адсорбента степень очистки понижается. Адсорбент
регенерируют прокаливанием.
Абсорбционные методы очистки
Эти способы очистки основаны на применении растворителей
селективного действия. При очистке масел получаются продукты
высокого качества; потери растворителей и нефтепродуктов
незначительны. Впервые методы разделения углеводородных
смесей посредством избирательного растворения разработал
А. М. Бутлеров.
Для того чтобы было возможно расслаивание и разделение,
плотность растворителя должна значительно отличаться от
плотности очищаемого продукта.
В качестве растворителей предложено применять жидкую
двуокись серы, фурфурол, фенол, нитробензол, дихлорэтиловый
эфир, смесь пропана с крезолом и фенолом и др.
Для очистки керосина, смазочных, трансформаторных и
медицинских масел используется жидкая двуокись серы. Процесс
очистки осуществляется в цилиндрическом смесителе, в который
снизу поступает очищаемый продукт, а сверху — жидкая
двуокись серы (при —10° С). Сверху из смесителя вытекает «рафи-
нат», т. е. очищенный продукт, содержащий растворенный
сернистый ангидрид. Снизу вытекает экстракт, т. е. сернистый
ангидрид, в котором растворены вредные примеси. Сернистый
ангидрид регенерируют нагреванием, снова охлаждают, сжижают
и возвращают в процесс. Аппарат работает при —10,1° С.
Растворенная в продукте двуокись серы также удаляется
нагреванием.
В настоящее время для очистки масел широко применяется
•фурфурол. Процесс очистки осуществляется аналогично
предыдущему только при 75—100° С.
Гидроочистка
В последнее время получил распространение метод очистки
нефтепродуктов путем гидрпгоппзашш их под давлением 30—
700 н/см2 при 250—420° С в присутствии катализаторов (кобаль-
то-молибденовых). Гидроочистка основана на умеренной
селективной гидрогенизации, в результате которой из органических
соединений серы, кислорода и азота получаются углеводороды
с одновременным выделением сероводорода, аммиака и воды.
Ненасыщенные углеводороды при этом переходят в насыщенные.
Выделяющийся с газами сероводород может быть использован
для получения серы или серной кислоты. При гидроочистке
используется дешевый водород, получаемый в процессах риформинга.
.80
Часть вторая
ТЕХНОЛОГИЯ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
ГЛАВА VII
ПРОИЗВОДСТВО ТЕХНИЧЕСКИХ ГАЗОВ
Технические газы — водород, азот, кислород, окись и
двуокись углерода, углеводороды (метан, этилен и др.) или смеси их:
водород и окись углерода (так называемый синтез-газ), водород
и азот и пр.— применяются для синтеза важнейших продуктов
химической промышленности: аммиака, мочевины, метилового и
других спиртов, углеводородов, синтетического бензина,
цианамида кальция и т. д. Технические газы производятся в огромных
количествах.
1. ПРОИЗВОДСТВО ВОДОРОДА
Наибольшее значение среди технических газов имеет водород.
Он применяется для синтеза аммиака, хлористого водорода,
гидрирования («ожижения») угля, гидрирования (отверждения)
жидких масел, восстановления руд, в производстве капролакта-
ма и других органических соединений, при атомарной сварке,
для охлаждения генераторов электрического тока и т. д.
Наибольшее количество водорода применяется при синтезе
аммиака. Если в 1950 г. мировое производство аммиака (без
СССР) составляло 4270 тыс. г, то в 1960 г. оно превзошло
11,5 млн. т. Водород составляет об. 75% в газах, применяемых
для синтеза аммиака, а стоимость его — 80% себестоимости
аммиака.
По производству соединений связанного азота, главную
массу которых составляют азотные удобрения, СССР занимает
сейчас второе место после США. Успешное выполнение
хозяйственных планов в этой области поставят СССР на первое место в
мире.
Водород получают тремя методами — химическим,
электрохимическим и физическим. К химическим методам относятся:
получение водорода из водяного пара при взаимодействии с
углеродом или с железом, из газообразных углеводородов
6 8-1504 81
(природный газ, попутные газы при добыче нефти и газы
нефтепереработки). Химическими методами получают водород также
из нефти, мазута, крекинг-остатков и пр.
Электрохимический метод используется при электролизе воды
(одновременно получается и кислород) и производстве хлора
электролизом водного раствора поваренной соли.
Физическими методами выделяют водород из коксового или
водяного газа. Водород, и притом чистый образуется как
попутный продукт при дегидрогенизации ароматических нефтяных
погонов и в результате биохимических процессов, например, аце-
тонобутилового брожения кукурузы.
Из перечисленных методов электролиз воды обеспечивает
получение наиболее чистого водорода, этот метод выгоден при
наличии дешевой электроэнергии; -железо-паровой метод сейчас
применяется мало, так как он менее рентабелен, чем другие;
большое значение имеет метод глубокого охлаждения.
Наибольшее распространение получил метод конверсии
природного газа. В табл. 11 показано значение в производстве
аммиака (%) различного вида сырья за 1939—1965 гг.
Виды сырья
Твердое топливо (кокс
и уголь)
Коксовый газ .....
Природный газ ....
Прочие
1939 г.
56,0
40,0
4,0
1953 г.
44 0
43,0
13,0
Тае
1958 г.
46,0
36,0
1,0
20,0
лица 11
1965 г.
15,0
18,0
60,0
7,0
Конверсия окиси углерода
Основным процессом при получении водорода этим методом
является обратимая реакция
ЄО + Н2О^СО2 + Н2+41 кдж.
На рис. 30 приводится зависимость логарифма константы
равновесия, выраженной через парциальные давления
компонентов, от температуры:
•"со ¦ кн,о
Из графика видно, что реакция смещается в желательном
направлении (вправо) при понижении температуры (константа
равновесия увеличивается). Для того чтобы при этом скорость
реакции не падала, конверсию проводят в присутствии
катализаторов.
Используя тепловой эффект реакции, можно вести процесс
автотермично, т. е. за счет тепла продуктов реакции нагреть
82
газы, идущие на реакцию, до необходимого оптимума (около
500° С) без затраты тепла извне.
На основании закона действия масс чтобы добиться
возможно более полного использования СО, нужно увеличить
количество водяного пара сверх стехиометрического. Чем выше
отношение Н2О : СО, тем выше степень конверсии и, следовательно, тем
меньше остаточное содержание окиси углерода в
конвертированном газе. Так как глубокая (98—99%) конверсия СО связана
с расходом большого
количества пара, на практике
ограничиваются меньшей
степенью конверсии (85—
90%). В качестве
катализаторов конверсии окиси
углерода применяются окислы
железа, меди, кобальта,
никеля и др.; наибольшее
распространение получили
окисные катализаторы.
При осуществлении этого
процесса используют
генераторный газ — полуводяной
или смешанный, а также
1.5
-1.0
о.о
-05
-1,0
\
\
\
ч.
ч
400 600 600 1000 I2OO 1400 1600 WOO 2000
t,°K
Рис. 30. График зависимости
константы равновесия конверсии окиси
углерода от температуры.
продукты конверсии
природного газа. Кроме окиси
углерода в них содержатся
водород, азот, двуокись углерода и ряд сернистых соединений:
сероводород, сероуглерод, сероокись углерода. Кроме основной
реакции конверсии СО, может происходить ряд побочных. Так, при
действии водорода на сероуглерод и сероокись углерода
образуются сероводород и двуокись углерода и др. Под действием
сероводорода окись железа превращается в неактивный' FeS.
Подбором условий — температуры, количества пара,
добавок и др.— можно уменьшить выход нежелательных продуктов.
После конверсии содержание СО в газе составляет все же 2,5—
3,5%.
Схема двухступенчатой конверсии окиси углерода приведена
на рис. 31. Содержащий СО газ газодувкой / подается в
орошаемую горячей водой (90э С) сатурационную башню 2, где
насыщается водяным паром, поглощая около 50% необходимого
количества последнего и нагревается до 80—85° С. Отсюда
парогазовая смесь с помощью парового инжектора 3 поступает в
теплообменник 4.
Паровой инжектор создает нужный напор и дает
возможность донасытить газ водяным паром до соотношения пар : газ =
= 1,2—1,4: 1. Далее смесь с температурой 105— 115°С
поступает в межтрубное пространство теплообменника. Здесь она на-
63
гревается до 370—400 С за счет тепла, отдаваемого уходящим
из контактного аппарата (конвертора) 5 горячим газом, и
поступает на контактирование; пройдя слои катализатора, газ
нагревается до 480—520° С. Затем он идет по трубам
теплообменника 4, охлаждается до 160—180° С и уходит в водонагреватель-
ную башню 6, нагревая воду, поступающую из сатурационной
башни с 55—65 до 90° С. При этом газ охлаждается до 85—90° С.
Полцдсдинаи газ
Рис. 31. Схема конверсии окиси углерода.
Окончательное охлаждение газа производится холодной
водой в конденсационной башне 7. Циркулирующая вода из
поддона башни 2 нагнетается насосом 8. Разогрев контактного
аппарата и донагревание его осуществляется с помощью
выносной камеры сжигания, где сжигается часть газа, смешиваемая
с соответствующим количеством воздуха.
Конвертор (рис. 32) состоит из трех царг, в которых
размещены 1-я ступень катализатора, испаритель и 2-я ступень
катализатора, причем в каждой ступени есть три полки с
отверстиями, на которых лежат слои катализатора толщиной 300, 700 и
300 мм. Равномерное распределение газа между полками
достигается с помощью решетки 10. Под третьей полкой верхней
ступени вмонтирована форсунка 7, разбрызгивающая конденсат.
Испаритель заполнен насадкой 8, на поверхности которой
испаряется конденсат, переходящий в газопаровую смесь; избыток
воды стекает по трубкам в гидрозатвор.
Конвертор покрыт теплоизоляцией, его размеры Д = 3,2 м,
И¦ — 7 м, пропускная способность — около 6000 м3 сухого газа в
час; объемная скорость 300 ч~1. Применяют также аппараты с
радиальным размещением контактной массы и тремя ступенями.
Размещение катализатора на трех полках вместо одной
имеет следующие преимущества: уменьшается статическая нагрузка
на каждую полку, сохраняется форма катализатора (так как на
84
и
нижний слои ие давят верхние), верхний, скорее
загрязняющийся при подаче газа сверху слой, легче заменить, лучше
распределение газа по сечеиию аппарата.
В последнее время конверсию окиси углерода проводят
также под давлением 10—30 атм. Повышение давления ие приводит
к смещению равновесия основной
реакции, так как она идет без изменения
объема, но обеспечивает ряд
преимуществ, которые благоприятно
сказываются на техноэкономических
показателях: увеличиваются скорость
конверсии и производительность аппаратуры,
снижаются температура и количество
водяного пара. Поскольку объем
аппаратуры для той же производительности
можно уменьшить, потери тепла
снижаются. К отрицательным сторонам
конверсии под давлением относится
повышенная коррозия аппаратуры,
приводящая к увеличению расхода
легированной стали; при увеличении
парциального давления примесей
ускоряется отравление катализатора.
Один из методов улучшения
конверсии связан с разработкой способов
получения более активных
катализаторов из меди и хрома, дающих
возможность снизить температуру конверсии
до 300° С, т. е. обеспечить более
полную конверсию.
Удешевление конверсии идет по
пути уменьшения числа аппаратов: две
башни — конденсационную и водона-
гревательиую — совмещают в одной
башне с водонагревательной частью
в нижней половине; кроме того
стремятся к увеличению единичной
мощности агрегатов, что значительно
уменьшает затраты на строительство и
эксплуатацию установок конверсии.
В табл. 12 показано (в об. %), как
меняется состав газа, начиная с полу-
водяиого генераторного, и кончая сухим
газом, покидающим систему.
Очистка конвертированного газа от СО2 и СО производится
так, чтобы газ после очистки содержал 75% Н2 и 25% N2. Газ
такого состава необходим для синтеза аммиака.
Рис. 32.
Двухступенчатый конвертор:
/ — крышка; 2 — стальной
корпус; 3, 4, 5 —
катализатор I ступени; в—
форсунка; 7—насадка испариіе-
ля; 8, 9, 10 — катализатор
II ступени; 11 — вход газа
на конверсию; 12 — выход
конвертнрованого газа на
очистку.
конвертированным
85
Таблица 12
Компоненты
СО
Н2О
н2
со2
N2
СН4 + Аг
Полуводяной
газ
34,5
36,6
6,6
22,0
0,5
Газ перед
конвертором
9,8
71,4
10,5
1,9
6,3
0,14
Газ после
конвертора
0,97
62,6
19,5
10,7
6,3
0,44
Газ после
конденсации
паров воды
2,6
51,6
28,6
16,8
0,38
Производство водорода из природного газа
Метод основан на окислении (конверсии) метана парами
воды, двуокисью углерода или кислородом при высокой
температуре. При этом происходят следующие основные реакции:
СН4 + Н2О<^СО + ЗН2 — 206,4 кдж, (I)
(И)
(III)
СН4 + СО2 5=t 2CO + 2Н2 — 248,3 кдж,
СН4 -f- -і О2 s=s CO + 2Н2 -f 34,8 кдж.
Частично СН4 и СО окисляются и до СО2 по реакциям
СН4 + 2О2 вСО2 + 2Н2О + 805,9 кдж. (IV)
СО + Н2О«СО2 +Н2 +41 кдж. (V)
Последовательное проведение реакций A) и E) дает
суммарно
СН4 + 2НЯО«=*СОЯ+4Н„ — 165,4 кдж. (VI)
Гомологи метана, имеющиеся в небольшом количестве в виде
примеси — этан, пропан и др., также окисляются, давая при
неполном окислении СО и Нг, а при полном СО2 и НгО.
Равновесие эндотермической реакции A) сдвигается в
желательную сторону (вправо) при повышении температуры и
избытке водяного пара.
В табл. 13 приводится равновесная степень конверсии метана
(в %) в зависимости от температуры при соотношении метана и
паров воды в исходной смеси 1 : 1 и 1 : 2.
Из таблицы видно, что почти полное превращение метана
достигается при температурах 1100° К (827° С). При получении
водорода выгодно довести до конца вправо как эндотермическую
реакцию A), так и экзотермическую E). Для этого процесс
ведут раздельно, в две ступени, причем вначале при высокой
температуре, а затем увеличивая концентрацию водяного пара
и несколько снижая температуру.
Температура, °к
При
отношении: СН4:
Н2О=1:1
Н2О=1:2
700
14,68
773
35,0
800
40,3
873
65,0
900
70,2
973
92,0
1000
89,84
95,5
1073
Таблиц*
1100
96,58
1173
99,0
і 13
1200
98,72
Реакцию конверсии метана можно осуществить и с
применением катализатора и без него.
Для ускорения реакции при более низких температурах
применяют катализаторы; лучшими из них являются никелевые
катализаторы, активированные добавками алюминия, хрома,
магния и др. В качестве носителя применяют глинозем. Газ, идущий
на конверсию, должен быть очищенным от серы, во всяком
случае, содержать ее не более 2—3 мг/м3; при большем содержании
серы необходима предварительная очистка газа
Рис. 33. Схема каталитической конверсии метана с парокисло-
родовоздушной смесью.
Различают следующие методы конверсии природного газа:
каталитический, высокотемпературный (некаталитический) и
конверсию под давлением. Каталитическая конверсия
подразделяется на одно- и двухступенчатую.
Схема одноступенчатой каталитической конверсии природного
газа со смесью водяного пара и кислорода представлена на рис.
33. Природный газ под давлением 0,7—0,8 ати проходит сатура-
ционную башню 1, орошаемую горячей водой (85—90° С); здесь
достигается степень насыщения водяным паром до соотношения
пар : газ = 0,7 : 1. Затем к газу добавляют пар до отношения
1,1 : 1, после чего газопаровая смесь нагревается в
теплообменнике 2 до 500—600° С и попадает в смеситель 3, куда добавля-
87
ют кислород, если надо получить водород без азота, или
обогащенный воздух, если нужна азотоводородная смесь.
Полученная смесь с температурой 450° С поступает в
конвертор метана 4, заполненный никелевым катализатором; здесь
происходят реакции (I) и (III). Из конвертора газ при 850—900° С
с содержанием остаточного метана 0,5—2% идет в увлажнитель
5, откуда через теплообменник 2 с температурой 400° С
поступает в конвертор 6 с окисножелезным катализатором для
конверсии окиси углерода. Часть газа можно пускать в конвертор 6,
минуя теплообменник 2. После конвертора газ охлаждается до
180° С в котле-утилизаторе 7, а затем идет в водонагревательный
теплообменник 8 и водонагревательную башню 9, где вода
нагревается до температуры 85—90° С и идет на орошение башни 1.
Конвертированный газ окончательно охлаждается до 25—
35° С в конденсационной башне 9, после чего поступает на
очистку от СОг и СО.
Конвертор метана 4 представляет собой аппарат шахтного
типа; он состоит из стального кожуха, выложенного огнеупорным
кирпичом. Для того чтобы остаточное содержание СН4 в
конвертированном газе не превышало 0,5%, температура газа на
выходе из конвертора должна быть не меньше 850° С.
Повышение давления сдвигает рав-новесие при конверсии
природного газа влево и в этом смысле оказывает отрицательное
влияние на процесс. Для достижения необходимой степени
конверсии приходится еще больше повышать температуру. Тем не
менее, в промышленности все чаще осуществляют конверсию под
давлением 16—48 атм, так как при этом увеличивается
производительность установок. Конверсия под давлением
экономически выгодна поскольку природный газ при выходе на поверхность
имеет давление 30 атм и более и поэтому не нуждается в
дополнительном сжатии.
Осуществление конверсии метана с углекислотой, как видно
из реакции (II), менее выгодно для процесса получения
водорода: последнего получается в 1,5 раза меньше, чем при
реакции с водяным паром. Однако этот метод применяется в
производстве метанола и высших спиртов, так как он обеспечивает
требуемое соотношение СО и Нг. Конверсия метана с
кислородом используется при получении ацетилена. В этом случае
одновременно получается газ, содержащий СО и Нг (синтез-газ).
В последнее время получила распространение также беска-
тализаторная конверсия с кислородом при высокой температуре
A400—1500° С); в результате ее образуется до 90% СО + Н2при
30—35 атм и концентрации метана в продуктах реакции 0,3—
0,5%. Наконец, следует указать на «взрывную конверсию»,
которая происходит в условиях неполного сжигания метана в
двигателях внутреннего сгорания.
В этом случае тепло реакций используют для получения энер-
88
гии. Кроме того, одновременно образуется синтез-газ. Для
конверсии метана в одну стадию при атмосферном и высоком
давлении (до 30 атм) применяют также смеси окислителей: О2 +
+ Н2О, О2 + Н2О +СО2.
Очистка конвертированного газа
Газы после конверсии, особенно если они предназначены для
производства аммиака, должны быть тщательно очищены от
примесей до остаточного содержания СО порядка 0,0015%,
СО2 —0,0006%, О2 —0,002%, СН4 —до 0,5%. Очистку ведут в
несколько приемов. Сначала очищают газ от СО2, затем от СО
и других компонентов.
Заводскую очистку газов от СО2 проводят следующими
методами: промывкой водой, растворами щелочей, аминоспиртов,
карбонатов и др. Из-за дешевизны растворителя наиболее
распространена водная очистка. Растворимость СО2 повышается с
ростом давления и понижением температуры. Процесс проводят
в скруббере при 16—30 атм и — 25° С. Тогда остаточное
содержание СО2 составляет 1,5—2,5% при начальном его содержании.
в конвертированном газе 27—30%. При выходе раствора из
скруббера из него нужно удалить растворенные газы, чтобы
снова ввести воду в цикл орошения. Для регенерации энергии
пропускают раствор через турбину, находящуюся на одном
валу с многоступенчатым центробежным насосом высокого
давления и электродвигателем.
Благодаря такой установке мото-турбонасоса регенерируется
50—55% энергии- Выделившиеся при дегазации в расширителе
(экспанзере) и градирне газы содержат 85—90% СО2, 6—10%
Н2, 3—4% N2, около 0,3% СО и 0,5 СН4 (этот СО2 используется,
например, в производстве кальцинированной соды).
Вода после дегазации возвращается в цикл орошения
скруббера. Окончательное удаление СО2 из конвертированного газа
после промывки водой достигается путем промывки щелочами.
Очищенный от СО2 газ идет на очистку от СО. Из-за малой,
растворимости СО в обычных растворителях обработку
газа ведут при высоких давлениях A20—320 ат) и низких
температурах E—20° С). Очистка осуществляется такими методами:.
промывкой медноаммиачным раствором или жидким азотом,,
окислением водяным паром, кислородом, восстановлением до
СН4 и др.
Чаще всего применяется промывка аммиачным раствором
углекислых (реже муравьинокислых) солей, одновалентной
меди, в котором содержится медноаммиачный комплекс
[Cu(NH3J]2CO3.
Химизм процесса растворения СО заключается в образовании,
комплекса по уравнению
[Си (NH3J]2 СОз + 2СО + 2NH3 ^ [Си (NH3K CO]a СО3.
8»
Регенерация сравнительно дорогого растворителя происходит
при нагревании полученного раствора до 45—50° С с
одновременным снижением давления сначала до 1,2—1,3 атм, а затем до
0,1—0,2 атм, по реакции, обратной вышенаписанной. Таким
образом регенерируется абсорбент и освобождается СО, который
используется в процессе.
Насыщенный медноаммиачный раствор из скруббера
проходит рекуперационную машину (жидкостную поршневую
машину), в которой регенерируется часть энергии, необходимой для
подачи раствора обратно в цикл орошения.
Выделяющиеся из раствора газы — СО, СОг, NH3—
используются в производстве.
В последнее время для очистки газов от СО применяется
жидкий азот. Этот прогрессивный метод более эффективен, чем
очистка медноаммиачным раствором, так как одновременно
удаляются из газа и другие примеси — кислород, метан, аргон и др.,
¦благодаря чему отпадает необходимость в дальнейшей тонкой
очистке газа. Содержание, например, кислорода после очистки
жидким азотом составляет всего Я0—40 мл в 1 м3. Поэтому
конверсию можно вести, не добиваясь полного превращения СН4
в СО и Н2; кроме того, упрощается дозирование смеси N2 + Н2
добавочным азотом. Процесс очистки, ведется при давлении
28—30 атм и температуре 83—85° К.
Получение водорода методом глубокого охлаждения
из коксового газа
Метод основан на том, что водород обладает очень низкой
температурой сжижения, а сопровождающие его газы — азот,
кислород и др.— сжижаются при температурах, более высоких
(см. табл. 14). Поэтому, если охладить смесь газов, содержащих
водород, ниже температуры сопровождающих его компонентов,
но выше температуры конденсации водорода, то можно
произвести разделение газовой смеси на газообразный водород и ряд
сжиженных фракций. Так как с увеличением давления
температура конденсации компонентов попытается, для выделения их
из сжатого газа можно обойтись менее низкими температурами,
чю, конечно, выгоднее: метан из коксового газа при 10 ата
конденсируется при —150° С, вместо —161,5° при 1 ата для чистого
СН4. Поэтому коксовый газ, подлежащий разделению, сначала
сжимают до 12—15 атм.
Следует отметить, что образующиеся при охлаждении
коксового газа жидкие фракции (этиленовая, метановая и окись
углеродная) содержат кроме основного компонента и другие, так как
часть несконденсировавшихся компонентов частично
растворяется в конденсате, а часть сконденсировавшихся остается в
паровой фазе.
90
Таблица 14
Компоненты
Водород
Азот
Окись углерода
Аргон
Кислород
Метан
Этилен
Этан
Двуокись углерода
Пропилен
Химическая формула
н2
Ns
СО
Аг
оа
СН4
с2н4
с2нв
СО-
с3нв
Температура кипения,
°С при 760 мм рт. cm
—252,8
— 195,8
—191,5
— 185,9
—183,0
— 161,5
— 103,8
—88,6
—78,5
—47,8
Процесс разделения коксового газа состоит из следующих
операций: 1) очистка коксового газа от нафталина,
сероводорода, двуокиси углерода, окислов азота, бензола, водяных паров
и др.; 2) сжатие газа; 3) промежуточное охлаждение до —45° С;
4) глубокое охлаждение и фракционированная конденсация.
По мере охлаждения из коксового газа последовательно
конденсируются компоненты, отводимые в виде жидких фракций —
пропиленовой, этиленовой, метановой, и фракции окиси углерода
с азотом. Испаряясь, эти фракции охлаждают коксовый газ.
Для более полной очистки газа от остатков СО, СН4 и
кислорода газ промывается жидким азотом, который, растворяет
примеси, является холодильным агентом и служит для увеличения
содержания азота в азотоводородной смеси. Для получения
1 м3 азотоводородной смеси, содержащей 75% Н2 и 25% N2,
расходуется 1,5—1,55 м3 коксового газа и около 0,25 м3 азота.
Извлечение из коксового газа водорода связано, как мы
видели, с получением больших количеств ценных веществ — этилена,
пропилена, метана и других компонентов, находящихся во
фракциях. Сжигание фракций, смешанных после испарения («богатый
газ»), крайне невыгодно (хотя часто имеет место), так как
компоненты газа могут быть использованы в качестве сырья для
целого ряда производств. Так, например, переработка этиленовой
фракции дает этилен, полиэтиленовые смолы, органические хлор-
производные спирты, эфиры; в свою очередь, из фракции окиси
углерода и азота можно синтезировать метанол. Таким образом,
при синтезе аммиака как бы перебрасывается мостик между
технологией неорганических и органических соединений.
2. ПРОИЗВОДСТВО АЗОТА И КИСЛОРОДА
Технический азот и кислород получают из жидкого воздуха,
разделяя последний ректификацией. Воздух используемый для
получения азота, должен быть чистым, не содержащим пыли,
91
водяных паров, двуокиси углерода, окислов азота, ацетилена
и других примесей.
Воздух подвергается постепенному охлаждению до низких
температур для вымораживания паров воды, двуокиси углерода
и сжижения его.
Процесс заканчивается ректификацией жидкого воздуха,
в двойной ректификационной колонне.
В результате ректификации из верха колонны отбирают пары
азота, содержащие 99,8—99,9% N2, а из нижней ее части уходит
жидкий или парообразный кислород, содержащий 90—93% О2.
Аргон и другие инертные газы, содержащиеся в воздухе,
уходят с азотом и кислородом; существуют установки, где эти газы
могут быть выделены.
Таким образом, производство азота из воздуха связано
с получением больших количеств кислорода, который, как
известно, применяется; для сварки и резки металлов, для
интенсификации процессов в металлургической и химической
промышленности и для друпих целей. При получении больших количеств
технического кислорода удешевление производства достигается
путем применения установок низкого давления F—6,5 ат)
с регенераторами и турбодетандерами, причем в настоящее
время строят крупные воздухоразделительные установки.
Так, на современной установке, перерабатывающей 50 000 и
более м3 воздуха в час, расход энергии составляет на 1 м3
газообразного кислорода 0,43—0,47 кет • ч, причем чистота кислорода
доходит до 99,5% О2, азот можно получать чистотой в 99,98%.
Дополнительные колонны могу г обеспечивать извлечение
редких газов (аргона, неона и др.).
Различные схемы разделения газовых смесей воздуха,
коксового газа и др. разбираются в курсе процессов и аппаратов
химической технологии.
ГЛАВА VIII
ТЕХНОЛОГИЯ СВЯЗАННОГО АЗОТА
1. ПРОБЛЕМА ФИКСАЦИИ АЗОТА ВОЗДУХА
Соединения азота играют огромную роль в жизни человека.
Основной частью любого организма — от бактерии до
человека—является белок, содержащий 13—16% азота. На каждый
квадратный километр земли приходится около 8 млн. т азота
воздуха. Но этот, свободный азот растениями не усваивается.
В природе существуют процессы естественного связывания
азота. Так, при электрических разрядах (молнии) в атмосфере азот
воздуха окисляется и в виде азотной и азотистой кислоты выпа-
92
дает с осадками. Таким путем на гектар почвы попадает от 4 до
15 кг связанного азота в год.
Существуют микроорганизмы, усваивающие азот воздуха и
превращающие его в соединения, которыми питаются растения.
Такие микроорганизмы находятся на корнях бобовых растений;
они могут связать до 250 кг азота на гектар в год.
Найдены и свободно живущие бактерии, которые могут дать
до 50 кг азота на гектар. Этот биологический метод фиксации
азота широко используется. Часть азота поступает в почву
с отмирающими растениями и животными остатками, навозом
и пр. Однако часть этого связанного азота в свою очередь
выделяется в свободном состоянии.
До XX века основным видом азотных удобрений была
чилийская и, в значительно меньшей мере, индийская селитра и
сульфат аммония. Роль соединений азота в жизни человека не
ограничивается их участием в питании растений. Соединения азота
используются в производстве красок и пластмасс,
искусственного волокна, взрывчатых веществ, ракетного топлива, серной
кислоты и др.
Поиски методов фиксации атмосферного азота
Промышленное значение впервые приобрел метод фиксации
атмосферного азота в виде цианамида кальция A895 г.). В конце
прошлого и начале нашего столетия была разработана
высокотемпературная фиксация воздуха в виде N0. Этот процесс
основан на реакции
N2 +O2^±2NO— 181,2 кдж.
Для осуществления этой реакции необходимы высокие
температуры. Равновесная концентрация окиси азота в воздухе
составляет около 1% при 2000° С, 3,5% при 3000° и около 10% при
4000° С. Такие температуры дает пламя электрической дуги.
Если полученную окись азота медленно охлаждать, то она будет
разлагаться на азот и кислород. Поэтому необходимо
предусмотреть быстрое охлаждение, «закалку» газа; тогда
разложение не произойдет вследствие падения до нуля скорости этого
процесса.
По этому способу в 1904 году Биркеланд и Эйде
сконструировали е Норвегии электрическую печь для промышленного
получения азотной кислоты и кальциевой селитры из воздуха.
Предложенный ими метод требует больших затрат электроэнергии,
а поэтому нерентабелен в настоящее время.
Разработка способов фиксации атмосферного азота
продолжается. Испытываются методы получения окиси азота в печах,
где сжигается метан, бутан или пропан в токе нагретого до
1500° С воздуха. Температура при этом может быть повышена до
2000—3000° С. Ведутся исследования по окислению азота в так
93
называемом «тихом» разряде. Для получения высоких
температур может быть попользована атомная энергия (в ядерных
реакторах) ; исследуются также возможности применения плазмы для
этой цели. Наиболее экономичным в настоящее время является
метод связывания азота с водородом, при котором образуется
аммиак. Этим методом получают основную массу связанного
азота (85%). Остальной азот используют в виде селитры C%),
цианамида C%) и каменного угля (9%).
В настоящее время СССР по производству азотных
соединений занимает второе место в мире после США. Согласно
пятилетнему плану развития народного хозяйства СССР это
производство должно к 1970 г. возрасти в четыре раза, после чего
наша страна выйдет на первое место в мире.
2. СИНТЕЗ АММИАКА
Синтез аммиака осуществляется по реакции
N2 + 3H2^2NH3 + 55,8 кдж (при 500° и 30 атм).
Так как реакция протекает с уменьшением объема и
выделением тепла, то, согласно принципу Ле-Шателье, для
увеличения выхода аммиака необходимо вести процесс при высоком
давлении и низкой температуре. Реакция идет в присутствии
катализатора.
В табл. 15 приведено процентное содержание аммиака в
равновесной смеси газов при различных давлениях и температуре.
Температура, °С
300
400
500
600
Т а б л
и ца 15
Давление, ата
10
14,73
3,85
1,21
0,49
100
52.04
25,12
10,61
4,52
200
64,24
38,20
19,12
9,15
300
70,96
47,00
26,44
13 77
600
84,21
65,20
42,15
23,10
1000
92,55
79,82
57,47
31,43
В условиях низких температур, несмотря на присутствие
катализатора, скорость мала. Поэтому практически синтез
аммиака ведут при давлении 100—900 ата и температуре 450 -500° С.
Наиболее активным катализатором является железо. При
использовании его выход достигает 50—70% равновесных
концентраций, приведенных в таблице.
Константа скорости реакции, наряду с другими факторами
(соотношение компонентов в газовой смеси, общее давление и
др.), определяет скорость образования аммиака, а следовательно
и производительность установки. Константа скорости зависит от
температуры и качества катализатора. Чем активнее
катализатор, тем больше константа (при прочих равных условиях процес-
94
so
to
35
гд
ca). Активность катализатора определяется составом его, а
также способом приготовления и добавками (активаторами и
промоторами).
Катализатор готовят из чистой железной магнетитовой руды
(Fe3O4), восстанавливая ее до металлического железа при
сравнительно невысокой
температуре C00—400° С).
Полученное железо отличается
высокопористой и
мелкокристаллической структурой и поэтому
является активным. При работе без
добавок растет величина кристаллов,
меняется структура и активность
катализатора падает. Роль одного ** зо
из активаторов—А12Оз, который
составляет 2—4%, заключается в
том, что он сохраняет мелкокрис-
таллическую активную структуру
железа. В качестве активаторов
добавляют также К2О, СаО и SiCb-
Для катализатора синтеза NH3 при
300 ата константа скорости (/Сен)
с повышением температуры
изменяется так, как показано в табл. 16.
Помимо основных факторов —
температуры и давления — большое
значение имеет объемная скорость
газа (количество газа в м3,
проходящее через 1 м3катализатора в час).
Поскольку с увеличением объемной
скорости уменьшается время
соприкосновения газа с катализатором, содержание аммиака в газовой
смеси уменьшается (рис. 34). Так, например, при объемной
скорости 15 000, давлении 300 атм и температуре 500° С содержание
NH3 составляет 23%, а при объемной скорости 60 000—14,6%.
Несмотря на снижение в смеси процента ЫНз, увеличение
объемной скорости приводит к росту производительности; так, в
приведенном примере скорость возросла в четыре раза, а выход NH3.
уменьшился только в 1,58 раза.
Таблица 16
400 425 *SO 475 500 SIS
t. 'С
Рис. 34. Зависимость
содержания аммиака в
газовой смеси от
температуры и объемной
скорости газа при 300 атм.
Температура, °С
Кск • Ю4
425
0,32
450
0,94
475
2,27
500
5,34
525
П.8
Рост в настоящее время объемной скорости ограничен
увеличением расхода энергии и нарушением автотермичности процес-
95
tea. С повышением температуры выход аммиака сначала растет,
.а затем уменьшается. Для каждой объемной скорости
существует оптимальная температура, которой соответствует
максимальный выход аммиака. С повышением объемной скорости
значение оптимальной температуры растет, а выход NH3
уменьшается, как показывает штриховая линия, соединяющая
максимумы на рис. 34.
Для использования непрореагировавшей азотоводородной
смеси нужно возвращать ее в контактный аппарат после удале-
ьния образовавшегося аммиака и восполнения прореагировавшей
части. Таким образом создается циркуляция газа.
Удаление (путем конденсации) образовавшегося аммиака из
газовой смеси неполное; часть NH3 C—5%) остается в нем и
возвращается обратно на катализ. Существенным условием
увеличения производительности контактного аппарата является
минимальное содержание аммиака в циркуляционном газе, так как
по закону действующих масс для того чтобы сместить
равновесие в сторону большего образования аммиака, следует
уменьшить количество его в азотоводородной смеси. Зависимость
производительности аппарата G (кг/час) от объема газа V,
концентрации аммиака в нем а? и концентрации аммиака в выходящем
газе а\, выражается формулой
г _ 0,758 V (аг — а,)
° ~ 100 + а1
Отсюда видно, что выработка аммиака растет с увеличением а.\
м падает с увеличением а^.
В системе синтеза аммиака уменьшение концентрации NH3
в циркуляционном газе достигается конденсацией аммиака при
охлаждении газа после контактного аппарата. Полнота
выделения аммиака зависит от температуры конденсации, увеличиваясь
с понижением ее.
Важное значение имеет поведение «инертов»—компонентов,
химически не меняющихся при прохождении через аппаратуру
(аргон, метан). Поскольку непрореагировавшие газы
возвращаются в колонку синтеза, содержа кие инертов с течением времени
увеличивается. Они снижают концентрацию N2 и Нг в газе,
поступающем на синтез, вследствие чего содержание NHj в
прореагировавшем газе падает. Так, при объемной скорости
30 000 м3 газаДи3 катализатора, давлении 300 атм и
температуре 500°С содержание аммиака в газе после синтеза составляет
18,6%; если же газ, поступающий на синтез, при тех же
условиях содержит 10% метана, то в прореагировавшей смеси будет
всего 14,8% аммиака.
В процессе работы необходимо следить за тем, чтобы газ
содержал возможно меньше инертных примесей. С этой целью
производят продувку, т. е. выпускают часть газовой смеси. В на-
•96
стоящее время предложены методы полезного использования
продувочных газов.
Ядами для железного катализатора являются СО, ССЬ,
О2, Н2О. В присутствии этих газов активность катализатора
уменьшается или совсем исчезает. Однако, если обработать
катализатор чистой азотоводородной смесью, активность его
восстанавливается. Такие яды называют временными, а отравление
обратимым.
Наибольший вред приносят необратимо отравляющие
катализаторы — соединения серы, фосфора, кислород, окись
углерода и др., присутствие которых в газе недопустимо. От
кислорода и окиси углерода освобождаются в колонне предкатализа,
где на отработанном железном катализаторе происходят
реакции
О2 + 2Н2 = 2Н2О,
СО + ЗН2 = СН4 + Н2О.
Масло отделяется с помощью фильтра, а пары воды
конденсируются. Остальные вредные газы также отделяются в процессе
подготовки азотоводородной смеси к синтезу аммиака. Системы
синтеза аммиака в зависимости от величины давления
азотоводородной смеси подразделяются на три типа:
1) системы, работающие при низких давлениях (—100 ати);
2) » » » средних » ( ~300 » );
3) » » » высоких » F00—1000»).
Установки, работающие в условиях низких давлений,
большого промышленного применения не имеют. Наиболее
рентабельными оказались системы, работающие при среднем давлении,—
под этим давлением производится около 70% NH3.
На рис. 35 показана схема, работающая при 300 атм.
Свежая азотоводородная смесь поступает из компрессора 8 в
маслоотделитель 5, затем в колонну предкатализа 9, где происходит
превращение следов окиси углерода и кислорода в СН4 и Н2О.
Затем смесь проходит водяной холодильник 2 и
водоотделитель 10. После этого она смешивается с циркулирующей
азотоводородной смесью, выходящей из конденсатора, и направляется
ь трубки испарителя аммиака 7, где происходит охлаждение
газовой смеси до —5е С за счет аммиака, испаряющегося в
межтрубном пространстве при давлении 2 ати. Из испарителя смесь
попадает в нижнюю часть конденсационной колонны б, где
выделяется аммиак. Пройдя сепараторную часть
конденсационной колонны, газовая смесь поступает в трубное пространство
теплообменника конденсационной колонны. В теплообменнике
конденсационной колонны холодный газ отдает свой холод газу,
идущему противотоком по межтрубному пространству и
направляющемуся в испаритель.
7 8-1504 97
Циркуляционная смесь, нагретая до 30° С и содержащая
после охлаждения 2—3% аммиака, пройдя трубки
теплообменника, выходит нз конденсационной колонны и поступает в
колонну синтеза аммиака /.
Прореагировавшая в колонне 1 газовая смесь идет в водяной
конденсатор 2, где охлаждается до 35° С, а затем в сепаратор 3,
в котором выделяется до 60% образовавшегося аммиака. Из
сепаратора газовая смесь попадает в циркуляционный насос 4,
Рис. 35. Схема синтеза аммиака под средним давлением.
где дожимается до рабочего давления, проходит
маслоотделитель 5 и идет по межтрубному пространству конденсационной
колонны 6. После конденсационной колонны газ соединяется со
свежим газом, и цикл продолжается.
Жидкий аммиак из сепаратора 3 н конденсационной
колонны 6 отправляют на склад. Испарившийся газообразный аммиак,
пройдя ловушку, поступает либо на холодильную установку для
снижения, либо на переработку.
На рис. 36 показана часто применяемая колонна синтеза
аммиака для систем среднего давления.
Корпус колонны состоит из двух цилиндрических кованых
частей из хромоникелевон стали, стянутых болтами; высота
колонны 14 м, внутренний диаметр 850 мм, толщина стенки 100 мм.
В верхней части колонны находится катализаторная коробка
с трубчатыми теплообменными элементами двух типов —
одинарных трубок, ввальцованных в трубные решетки, как обычно,
н так называемых трубок Фильда C). Трубка Фильда
представляет собой сочетание нз двух коаксиальных трубок, из которых
одна меньшего диаметра A2 мм), открытая с обеих сторон,
вставлена в широкую B2 мм), закрытую снизу; трубка Фильда
устанавливается так, чтобы более холодный газ проходил через
внутреннюю трубку сверху вниз, затем кольцевое пространство
между обеими трубками снизу вверх.
Пространство между всеми теплообменными трубками
заполнено зерненым катализатором. Электронагреватель для пуска
колонны расположен на внешней поверхности катализаторной
коробки.
В нижней части колонны находится
теплообменник, состоящий из
железного кожуха с двумя трубными
решетками, в которые ввальцованы
вертикальные трубки. Внутри
теплообменника проходит центральная труба,
соединяющая катализаторную коробку
с теплообменником; в межтрубном
пространстве установлены
горизонтальные перегородки для улучшения
теплообмена.
Основной поток гачя поступает
через крышку в нижнюю часть
колонны, проходит по двойному ходу между
корпусом колонны и кожухом
теплообменника и направляется в трубки
предварительного теплообменника.
Перед входом в одинарные трубки
катализаторной коробки основной поток
смешивается с газом, поступающим по
первому холодному байпасу / через
верхнюю крышку и зазор между
катализаторной коробкой и стенкой
колонны.
Газовый поток по второму
холодному байпасу II подается
непосредственно во внутренние трубки Фильда,
расположенные в катализаторной
коробке. После нагрева газ выходит из
трубок Фильда и смешивается с
потоком, выходящим из одинарных трубок.
Затем смесь газов с температурой
420° С, пройдя через катализатор при температуре 500° С
направляется через центральную трубу в межтрубное пространство
предварительного теплообменника и выходит из колонны, имея
температуру 180—200° С. Такая конструкция обеспечивает
использование тепла реакции для нагрева холодных газов,
поступающих в колонну (автотермичность процесса) и регулирует
температуру катализатора.
Нагрев при пуске и в случае неполадок обеспечивает электро-
Рис. 36. Колонна синтеза
аммиака типа ГИАП-
ДАТЗ:
/ — корпус насадки; 2—4 —¦
простые спротипоточные»
трубки; 3 —- трубки Фильда;
5 — корпус
предварительного теплообменника; 6 —
электроподогреватель-, 7 — кар-
ман для термопары.
нагреватель. Часть тепла может быть использована для
получения пара (в системах с давлением 450 ати и выше).
Технологическая схема синтеза аммиака под давлением
в 600 ати показана на рис. 37. Свежий газ сжимается в шести-
ступенчатом поршневом компрессоре / до давления 600 ати.
В первых трех ступенях ои сжимается до 27 ати, проходит
последовательно маслоотделитель 2 и межтрубное пространство
теплообменника 3, где нагревается до температуры 300° С и
поступает в колонну предкатализа 4. В колонне газ нагревается
электрическим током до 380° С, после чего проходит слой
катализатора, где все
имеющиеся кислородные
соединения превращаются в
воду и метан, затем
газ покидает колонну
предкатализа и,
отдавая свое тепло в
теплообменнике
предкатализа 3, с температурой
100° С поступает в
водяной конденсатор 5,
где конденсируется
вода, отделяемая затем
в сепараторе 6.
Далее газ подается
на четвертую ступень компрессора, сжимается на последних
ступенях и с давлением 600 ати проходит маслоотделитель 7; он
смешивается с газом, выходящим из колонны катализа 8, в
котором содержится 22—24% аммиака, и попадает в водяной
холодильник 9 где охлаждается до 20° С, при этом из него
конденсируется аммиак; затем газ проходит сепаратор 10, где отделяется
жидкий аммиак.
Циркуляционный газ, содержащий около 3% NH3,
направляется в циркуляционный насос //, сжимается здесь с 560 до
600 ати и идет в колонну синтеза 8 по основному и байпасному
ходам.
Из сепаратора 10 аммиак передается в сборник пысокого
давления 12, а отсюда в сборник аммиака низкого давления, на
склад и на переработку.
3. ПРОИЗВОДСТВО АЗОТНОЙ КИСЛОТЫ
Техническая азотная кислота, представляющая собой смесь
HNO3 и воды с очень малым количеством Примесей, выпускается
разбавленной (слабой) с содержанием 50—60% HNO3 и
концентрированной с содержанием 94—98% HNO3.
Прежде (до начала XX в.) азотную кислоту получали из
Рис. 37. Схема синтеза аммиака под
давлением 600 ати.
100
чилийской селитры действием серной кислоты при нагревании.
В этих условиях протекает реакция
NaNO3 + H2SO4 = NaHSO4 + HNO3.
Пары азотной кислоты конденсировались, давая кислоту
высокой концентрации (95% и более). С 1904 г. азотную кислоту
E0%-ную) начали получать дуговым методом. С конца первого
десятилетия XX века распространился метод получения HNO3
с использованием аммиака. В России этот метод был
самостоятельно разработан И. И. Андреевым. На основании материалов,
которые были им получелы в результате большой
исследовательской работы, проведенной совместно с Н. М. Кулепетовым
и А. К- Колосовым, в 1916 г. в г. Юзовке (теперь Донецк)
построили первый завод получения азотной кислоты контактным
окислением аммиака. Такой завод состоит из двух отделений: в
первом происходит окисление аммиака кислородом воздуха по
реакции
4NH3 + 5О2 = 4NO + 6Н2О + 907,3 кдж,
во втором отделении полученная окись азота окисляется до
двуокиси и абсорбируется водой, превращаясь в азотную кислоту.
Окисление аммиака может происходить как в присутствии
катализатора, так и без него. Без катализатора окисление протекает
так:
4NH3 + ЗО2 = 2N2 + 6Н2О + 1269,1 кдж.
Такой ход процесса невыгоден, поскольку при этом происходит
дефиксация азота. Роль правильно подобранного катализатора
состоит в том, что в его присутствии из всех возможных реакций
протекает наиболее желательная. Кроме того, катализатор
ускоряет реакцию. Одним из лучших катализаторов является
металлическая платина-
Свойства катализатора улучшаются, если применяют сплав
платины с родием C%) и палладием D%) или только с родием
(до 10%). При использовании такого катализатора степень
превращения NH3 в N0 достигает 97—99%.
Платинородиевый катализатор отравляется фосфористым
водородом; опытами установлено, что содержание его в
количестве даже 0,00002% уже отравляет платину и снижает степень
контактирования с 95 до 80%; ядом для катализатора являются
также сероводород, пары масла, пыль и др. Поэтому при
окислении 1МНз применяют чистые газы.
Более дешевые неплатиновые катализаторы дают выход до
90%- Процесс при использовании их протекает значительно
медленнее.
На платине процесс протекает быстро: время контакта всего
0,001—0,0001 сек. Поэтому требуется сравнительно небольшое
ее количество. Этим объясняется и своеобразная форма
катализатора— он применяется в виде сетки из проволоки диаметром
101
0,045—0,09 мм с числом петель до 3600 на 1 см2, живое сечение
сетки 50—60%.
Большая скорость процесса достигается при температурах
700—900° С; в условиях более низких температур снижается
скорость процесса и выход окиси азота, высокие температуры могут
быть достигнуты за счет тепла реакции. Каждому проценту NH3,
окисленной в газовой смеси, соответствует повышение
температуры на 70° С. Если к воздуху добавить аммиак из расчета сте-
хиометрического соотношения О2: NH3 = 1,25 моля О2 моль
NH3, то концентрация его составит 14,4%; однако для того чтобы
степень окисления была высокой (97—98%), необходим избыток
кислорода: указанное выше соотношение должно составить 1,7—
2,0. При таком соотношении концентрация NH3 в аммиачно-воз-
душной смеси достигает 9,5—11,5%; чтобы получить достаточно
высокую температуру контактирования при концентрации 9,5%
NH3, приходится подогревать газовую смесь перед поступлением
в контактный аппарат за счет тепла горячих газов, вышедших из
контактного аппарата.
Газовая смесь проходит платиновую сетку при линейной
скорости не более 0,3 м/сек. Если скорость газов больше,
наблюдаются проскоки NH3 через катализатор, что приводит к
снижению производительности установки из-за окисления NH3
окисью азота по реакции
4NH3 + 6NO = 5N2 + 6Н2О.
Из уравнения
NH3 + 2О2 = HNO3 + Н2О
видно, что, используя вместо воздуха кислород (аммиачнокис-
лородную смесь), можно довести содержание аммиака до 33%.
Однако повышение содержания аммиака лимитируется
взрывчатостью подобных смесей. При повышении концентрации
аммиака в смеси с воздухом выше 13% уже имеется опасность
взрыва. Следует учесть, что существуют верхний и нижний
пределы взрывчатости. Эти пределы расширяются с повышением
температуры и зависят от давления, содержания кислорода,
влаги и других примесей; так, совершенно сухая смесь вовсе не
изрывает, а примесь большого количества водяных паров
суживает границы взрывчатости. Пределы взрывчатости
определяются-и другими факторами — формой аппарата, направлением
газового потока: вверх или вниз (опаснее — вверх). Нижняя
граница концентрации NH3 снижается с 15,5% при 1 атм до
10,8% при 10 ата. Взрывов можно избежать, если скорость
газов больше скорости распространения взрывной волны. На
практике снижают содержание NH3 в смеси газов и пользуются
автоматическими устройствами, отключающими газ при
повышении концентрации аммиака сверх нормы.
Для очистки от пыли применяют фильтры, заполненные
стальными кольцами, смоченными густым (висциновым) мас-
102
лом, а также поролитовые фильтры из пористой керамики.
Поверхность платины с течением времени разрыхляется,
вследствие чего увеличивается поверхность и активность катализатора.
При работе происходит унос платины, который на 1 г HNO3
составляет около 0,05 г при атмосферном давлении, доходя до
0,2 г при работе под давлением 8 атм. Это приводит к
увеличению себестоимости продукта, а также к износу платиновых
сеток, которые периодически приходится заменять. Часть
уносимой газами платины улавливается в специальных фильтрах.
Окисление аммиака ведут обычно при атмосферном
давлении, так как реакция сопровождается увеличением объема. Но
в некоторых системах окисление ведут и в условиях
повышенного (до 7—9 ата) давления. При этом увеличиваются скорости
окисления окиси азота и образования HNO3, упрощается схема
и ее обслуживание; концентрация HNO3 также повышается, но
унос платины увеличивается.
Важной проблемой является частичная или полная замена
воздуха кислородом. Такая замена дала бы возможность
увеличить концентрацию аммиака, а затем значительно ускорить
и удешевить стадию процесса — окисление и абсорбцию
окислов азота. Осуществлению этого препятствует опасность взрыва
смеси. Предложено несколько методов устранения указанной
опасности. Один из них заключается в том, что процесс ведется
в присутствии большого количества водяных паров, снижающих
концентрацию аммиака ниже границы взрываемости.
Переработка окислов азота в азотную кислоту
Окись азота, полученная при окислении NH3, после
контактного аппарата окисляется до NO2.
2NO + О2 5=s 2NO2 + 112,3 кдж.
Равновесие этой реакции зависит от температуры и
давления.
Так как реакция идет с уменьшением объема и выделением
тепла, то по правилу Ле-Шателье процесс сдвигается в сторону
образования NO2 с понижением температуры и повышением
давления. При температуре ниже 200° С равновесное
превращение NO в NO2 почти полное, а при температуре выше 700° С
NO2 почти полностью диссоциировано на NO -f O2. Скорость
окисления NO в NO2, как известно, падает при повышении
температуры, так как константа скорости /Сек уменьшается в этих
условиях, что видно из следующих данных:
Температура, °С 0 30 60 100 200 300
А'сК 69,3 42,8 29,2 19,5 8,71 5,13
103
На скорость реакции окисления N0 в N02 оказывает
большое влияние состав газа и его давление. Время, необходимое
для достижения определенной степени превращения, обратно
пропорционально квадрату концентрации окиси азота и
концентрации, кислорода и, следовательно, кубу давления; поэтому
применение повышенного давления ускоряет окисление NO
в N02.
Полученная в результате окисления двуокись азота полиме-
ризуется в четырехокись по реакции
2NO2 N2O4 + 56,9 кдж.
Равновесие этой реакции также сдвигается вправо при
понижении температуры и повышении давления. Ниже показана
зависимость степени полимеризации от температуры
Температура, °С 290 100 70 30 10 О
Полимеризация, % 0,04 12,52 39,01 77,84 87,0 89
Таким образом, в охлажденных иитрозиых газах содержится
преимущественно N2O4.
В результате взаимодействия NO и NO2 образуется и
некоторое количество N2O3:
NO + NO25=tN2O3-f-40,2 кдж.
Равновесие этой реакции при температуре выше 0е С
сдвинуто далеко влево, и концентрация молекул N2O3 в газе очень
мала. Взаимодействие N2O4 с водой идет по реакции
N2O4 + Н2О = HNO3 + HNO2 + 59,0 кдж.
Азотистая кислота нестойка и претерпевает дальнейшее
превращение по реакции
3HNO2 = HNO3 + 2NO + Н2О + 75,8 кдж.
Последние две реакции протекяют быстро. Итог последовав
тельного течения обеих реакций выражается уравнением
3NO2 + Н2О = 2HNO3 + NO + 136,2 кдж.
Таким образом, одна треть поглощений NO2 в виде NO снова
выделяется в газ, и для того чтобы абсорбироваться, NO
должна вновь окислиться до NO2.
Реакции окислов азота с водой, приводящие к образованию
HNO3, протекают быстрее, чем окисление NO в NO2 в газовой
фазе. Это замедляет абсорбцию, особенно к концу процесса.
Поглощение NO2 водным раствором HNO3 увеличивается
с ростом давления и уменьшается с повышением температуры
104
и концентрации HNO3. Процесс абсорбции резко замедляется
при повышении концентрации HNO3 выше 50%. Поэтому
производят примерно 50%-ную HNO3 при атмосферном давлении
и почти 60%-ную при давлении до 7 ата. Таким образом,
условиями, необходимыми для получения концентрированной НМОэ,
являются: повышение давления, понижение температуры,
наличие крепких нитрозных газов и хороший контакт газовой и
жидкой фаз. Для снижения температуры применяют искусственное
охлаждение абсорберов.
Поскольку процесс окисления непрерывно выделяющейся
окиси азота при абсорбции нитрозных газов к концу резка
замедляется, для полного поглощения NO2 требуется
продолжительное время и большой объем аппаратуры. Так, чтобы
поглощение проходило на 98% при атмосферном давлении
и начальной концентрации 9—10% NO2, необходим
абсорбционный объем около 100 м3 на тонну HNO3 в сутки. Для
уменьшения этого объема абсорбцию водным раствором проводят
только на 92—94°/о, а остаток окислов азота поглощают щелочью,
обычно раствором соды. При этом происходят следующие
реакции:
2NO2 + Na2CO3 = NaNO3 + NaNO2 + CO2;
NO2 + NO + Na2CO3 = 2NaNO2 + CO2.
Поглощение щелочью протекает в 1,5—2 раза быстрее водной
абсорбции окислов азота, поскольку окись азота не выделяется,
а для поглощения достаточно, чтобы она была окислена на
50%. Таким путем поглощают еще 5—6% окислов азота, доводя
общую степень абсорбции до 98—98,5%.
Абсорбционный объем уменьшается в этих условиях втрое
C0—35 м3/т). Зато добавляется отделение для переработки
полученных щелоков. Если в газах содержатся окислы азота
в отношении NO/NO2 = 1 и отсутствует кислород, образуется
главным образом NaNO2; обычно, в зависимости от условий,
получаются обе соли — NaNO3 и NaNO2 в варьирующих
количествах.
Щелок, содержащий смесь нитритов и нитратов натрия,
подвергают инверсии действием азотной кислоты. При этом
нитриты превращаются в нитраты
3NaNO2 4- 2HNOa = 3NaNO3 4- 2NO 4- Н3О.
Нитрат натрия (селитру) упаривают и выделяют
кристаллизацией, а окислы азота направляют в абсорбционные башни.
На рис. 38 дана схема производства HNO3 под атмосферным
давлением. Воздух проходит скруббер /, где отмывается от
пыли и газообразных примесей водой или раствором соды,
матерчатый фильтр 2 для удаления механических частиц и
вентилятор 3, куда подается и NH3. Смесь воздуха и аммиака в нуж-
105.
ном соотношении проходит поролитовыи или картонный фильтр
4, затем катализатор (сетки) в контактном аппарате 5 и котел-
утилизатор 6; полученный водяной пар после сепаратора и
пароперегревателя при 450° и 40 атм уходит в паровую сеть.
Горячие нитрозные газы охлаждаются в котле до 160° С,
затем в скоростном холодильнике 7 до 40° С; здесь
конденсируется часть водяного пара, образовавшегося при реакции из
окислов азота. Образуется 3%-ная НЫОз в виде конденсата.
ю
Перегретый паи
40 от
Mi,
іїродухцианноя
Xt/CAO/ThJ
//ff/npt/m pacmfiofl
нитратные Va c0
Рис. 38. Схема установки для получения разбавленной азотной кислоты при
атмосферном давлении.
Во втором холодильнике 8 собирается 25—30%-ная HNO3.
После этого газы с помощью вентилятора подаются в
абсорбционные башни 10. Поглощение производится чистой водой
(конденсатом), поступающей в последнюю башню, а в
последующих башнях—азотной кислотой, образующейся в
предыдущей башне.
Циркуляция жидкости осуществляется центробежными
насосами 14 через водяные холодильники 13. Продукционная
кислота D8—52%-ная) отводится на склад из первой или второй
(по ходу газов) башни.
Нитрозный вентилятор 9 подсасывает воздух так, чтобы
¦и выхлопных газах было до 5% кислорода. Газы, уходящие из
•системы водной абсорбции, проходят пустую окислительную
башню //, а затем щелочные абсорберы 12, куда противотоком
насосы 15 подают раствор соды BПП—250 г/л) или известковое
молоко F0—100 2/л); нитрит-нитратные щелока уходят из
первой башни щелочной абсорбции.
Поскольку большинство реакций в башнях экзотермичны,
низкие температуры благоприятствуют процессу; так, при
снижении температуры с 40 до 20° С производительность по азот-
лой кислоте увеличивается в 1,5 раза, а при снижении с 40э
до 0° С — в два раза,
106
Из аппаратуры, применяемой на заводах, производящих
азотную кислоту, остановимся на контактном аппарате из
нержавеющей стали. Современные аппараты для работы при
атмосферном давлении имеют до 3 м в диаметре (при давлении
7 ата— до 1 м), Аммиачно-воздушная смесь идет сверху вниз.
На входе вмонтирован фильтр для газа. Далее газ проходит
три сетки (при давлении до 8 ата 16—19 сеток), а тепло
уходящего при температуре 700—800° С нитрозного газа
используется для получения пара.
Атмосферне/а
воздух
Вода
Выхлопные
газа
,3
Пар Х&хтобые
та,
Рис. 39. Схема установки для получения разбавленной
азотной кислоты под давлением 8 ата.
¦' Водотрубный котел-утилизатор смонтирован в нижней части
аппарата. На 1 т окисляемого аммиака можно получить 2—3 т
пара. Производительность аппарата определяется
напряженностью катализатора — на каждом кв. м сетки окисляют 600—
800 кг аммиака в сутки. Абсорбция окислов азота
осуществляется при атмосферном давлении в башнях из нержавеющей
стали, заполненных насадкой (керамические кольца).
В настоящее время большинство новых заводов работает по
комбинированной системе, т. е. контактирование
осуществляется при атмосферном, а абсорбция — при повышенном давлении
или же весь процесс осуществляется при повышенном давлении.
В этих условиях абсорбционный объем уменьшается до 2,6—
4 м?/т в зависимости от давления и концентрации. На рис. 39
показано получение азотной кислоты при 8 ата. Примерно по
такой же схеме работают и при давлений 3,5—7 ата.
Воздух сначала промывают водой в башне /, фильтруют
через матерчатый фильтр 2 и сжимают в турбокомпрессоре 4,
находящемся на одном валу с электродвигателем 6 и
редуктором 5. Затем его еще раз фильтруют через асбестовое полотно
107
или фильтровальный картон; применяются также поролитовые
трубки.
Аммиак после испарителя 10, нагретый до 70° С, очищается
в фильтре //. В смесителе 12 аммиак смешивается с воздухом,
имеющем после сжатия температуру 110—120° С, и смесь
поступает в контактный аппарат 13, где на платинородиевом
катализаторе аммиак окисляется до окиси азота при температуре
880° С.
Нижнюю четь контактного аппарата и соединительную
трубу охлаждают водой. Пройдя котел-утилизатор 14, (питаемый
водяньш насосом /5) нитрозные газы охлаждаются до 250° С
и поступают в водяной холодильник-конденсатор 16. На линии
нитрозных газов перед входом в конденсатор устанавливают
фильтр из стеклянной ваты для улавливания платины.
В холодильнике-конденсаторе 16 при охлаждении
нитрозных газов до 40°С образуется 53—56%-ная HNO3 до 50% от
выработки. Нитрозные газы из конденсатора 16 поступают
в поглотительную колонну 18, а кислота, отделившись в
сепараторе и пройдя фильтр для платины 17, может быть либо
направлена в колонну на дальнейшее укрепление, либо выведена
из системы как готовая продукция.
Добавочный воздух в количестве до 20% от основного
вводится перед конденсатором 16 или в нижнюю часть
абсорбционной колонны 18.
Для пуска системы имеется эжектор 9 с вентилями 7.
Для абсорбции под давлением применяют колонну барбо-
тажного типа с колпачковым или более дешевыми и
совершенными ситчатыми тарелками, разработанными в 50-х годах
в СССР.
Колонна диаметром 3 ти из нержавеющей хромоникелевой
стали толщиной 8 мм имеет до 40 тарелок. Высота колонны
45 м; на нижних 20—25 тарелках размещены для отвода тепла
реакции охлаждающие змеевики, по которым проходит вода
или, иногда, холодный рассол из холодильной установки. Сверху
подается очищенная вода (или конденсат); газ входит снизу
и проходит вверх, барботипуя сквозь слой раствора НГ\О3 на
каждой тарелке, а кислота стекает по тарелкам навстречу
газам, поглощая окислы азота и увеличивая концентрацию до
56—6U %.
Концентрация окислов азота в нитрозных газах после
поглотительной колонны снижается до 0.2—0,3%, что
соответствует выходу по кислотному поглощению 97—98%. В отличие
от установок, работающих под атмосферным давлением, газы
не подвергаются здесь щелочной промывке, а подогреваются
либо паром в теплообменнике 8, как показано на схеме, либо
горячими нитрозными газами и поступают в рекуперационную
газовую турбину .3, насаженную на одном валу с турбокомпрес-
108
сором, где отдают до 25—40% энергии, затраченной на сжатие
воздуха.
Из абсорбционной системы уходят газы, содержащие до
0,2% окислов азота, что составляет 2% всего их количества.
Уменьшение потерь и обезвреживание выхлопных газов —
важная задача, для решения которой предложены различные
методы.
Продукционная азотная кислота содержит растворенные
окислы азота в количестве до 2—4% и подвергается продувке
воздухом. Расходные коэффициенты на 1 т HNO3 составляют:
аммиака 0,29—0,295 т, платины 0,15—0,20 г, электроэнергии
350—500 кет, пара 0,20—0,6 т, воды 110—170 ж3.
4. КОНЦЕНТРИРОВАНИЕ АЗОТНОЙ КИСЛОТЫ
Для получения концентрированной азотной кислоты можно
удалять воду из разбавленных кислот кипячением, но система
HNO3 — Н2О образует нераздельнокипящую (азеотропную)
смесь, содержащую 68,4%
HNO3 (рис. 40). На практике
кипячением слабых кислот
получают примерно 65%-ную
азотную кислоту, которая
мало отличается от HNO3,
получаемой после водной
абсорбции под давлением. Поэтому
для получения
концентрированной кислоты применяют во-
доотнимающие средства. '- 0 20 40 б0
Прибавление КОНЦентриро- Содержание
/
Жидис
1 .--1
їсть -А \
V
In
во юо
34
ванной серной кислоты к сме- п .„ „
си HNO3 + Н2О снижает упру- Рис' 40' Ди н^"™ СИСТЄМЬ'
гость водяных паров и
увеличивает упругость паров HNO3. Таким образом, можно отогнать
HNO3 в парах.
Процесс осуществляется в тарельчатых колоннах (или
колоннах с насадкой), в которые сверху подается слабая азотная
кислота и купоросное масло (92—93%-ная серная кислота),
а сверху уходят пары HNO3, которые конденсируются в
водяных холодильниках и дают крепкую HNO3 (98—99%-ную).
На рис. 41 представлена схема концентрирования азотной
кислоты с помощью серной. Разбавленная азотная кислота
направляется в колонну /; часть кислоты проходит через
испаритель 2 и подается в виде жидкости и пара на десятую тарелку
колонны, а другая часть в холодном состоянии поступает на три
тарелки выше.
Купоросное масло подается на шестнадцатую тарелку
109
колонны /. В нижнюю часть колонны (под первую тарелку)
поступает пар для денитрации отработанной серной кислоты.
Пары азотной кислоты уходят через штуцер в крышке
колонны после двадцатой тарелки и конденсируются в
холодильнике 3.
Образующаяся кислота содержит окислы азота и поэтому
возвращается на верхнюю тарелку колонны, где продувается
парами азотной кислоты, идущими из колонны в конденсатор.
Розбавлення»
\
Рис. 41. Схема получения концентрированной
азотной кислоты при помощи серной.
Пройдя две отбелочные тарелки, свободная от окислов азота
кислота выводится из колонны и охлаждается в холодильнике 4,
откуда поступает на склад готового продукта.
Часть паров азотной кислоты, несконденсировавшихся в
холодильнике 3, в смеси с окислами азота и воздухом (попавшим
в систему вследствие неплотности аппаратуры) в виде нитроз-
ных газов уходят на поглощение водой. Отработанная серная
кислота по выходе из колонны 1 направляется в
концентрационные аппараты, а после упаривания возвращается в цикл.
Колонна представляет собой аппарат барботажногп типа из
20 царг; царги и тарелки изготовлены из ферросилиция.
Высота колонны около 9 м при диаметре 1 м. Каждая царга имеет
высоту 360 мм (верхняя и нижняя — по 760 мм). Температура
паров азотной кислоты на выходе из колонны / составляет 70—
85° С, а после конденсатора 3 — около 30°.
В верхней части колонны поддерживают вакуум в пределах
20—50 мм рт. ст. Отработанная 68—70%-ная серная кислота
выходит из колоны с температурой 150—170° С. Давление
перегретого пара на входе в колонну не превышает 0,5 ата; темпе-
110
ратура его около 250 С. Давление насыщенного пара,
поступающего в испаритель 2, составляет 4—6 ата.
Расходные коэффициенты на 1 т HNO3 следующие.
Разбавленная азотная кислота (в пересчете на НЖ)з) 1,01 —
1,015 т. Купоросное масло (91—92% H2SO4): а) при начальной
концентрации азотной кислоты 48—50% 4,0—4,5 т; б) при
начальной концентрации азотной кислоты 58—50% 3,0—3,3 т.
Охлаждающая вода 20—50 мг. Пар перегретый 0,4—0,5 т; пар
насыщенный 0,3 т. Электроэнергия 8—10 кет • ч.
Для концентрации азотной кислоты в последние годы
предложено вместо серной кислоты использовать в качестве водо-
отнимающего средства раствор Mg(NO3J.
При этом вдвое уменьшаются эксплуатационные расходы w
на 30—40% капитальные затраты; HNO3 не загрязняется
серной кислотой, а атмосфера вокруг завода — туманом серной
кислоты. Недостаток этого метода — большой расход греющего-
пара.
Получение концентрированной азотной кислоты
прямым синтезом
Получение HNO3 концентрацией выше 60—70% из
разбавленных нитрозных газов, как мы внделн, затруднительно.
Из жидкой четырехокиси азота, воды и газообразного
кислорода можно получить высококонцентрированную НЖ)з по
реакции
2N2O4 + 2Н2О + О2 = 4ШО3 + 59,5 кдж.
Реакция осуществляется под давлением 50 ата и при
температуре 60—80° С, N2O4 подается с избытком против стехиомет-
рического соотношения. Для процесса требуется чистая N2O4>.
которую нужно выделить из нитрозных газов. Это
осуществляется либо конденсацией ее из газовой смеси при —10° С
(давление 5—10 ата) либо абсорбцией NO2 на холоду из газовой
смеси концентрированной азотной кислоты с последующим
выделением ее при нагревании в виде чистой, 100%-ной N2O4.
Из уравнения видно, что для получения 100%-ной кислоты
на моль NO требуется 0,5 моля воды, что составляет около '/з
воды, имеющейся в нитрозных газах после окисления NH3-
Поэтому из нитрозных газов по выходе из котла-утилизатора
охлаждением отводят около 2/з воды в виде конденсата. Эту
операцию производят быстро (в скоростных холодильниках),.
для того, чтобы NO не успела окислиться до NO2, в значительной
степени (так как NO2, взаимодействуя с водой, образует HNO3
и будет выведена с конденсатом, что приводит к потерям в
производстве) .
К особенностям производства относится метод окисления
NO в NO2. Окисление осуществляется в две ступени. Первая
1П
ступень проводится в окислительных башнях в газовой фазе,
как обычно. При этом достигается степень окисления 93—94%,
если процесс протекает при атмосферном, и 96—97% при
повышенном давлении.
Вторая ступень окисления N0 осуществляется
концентрированной HNO3 по реакции
N0 + 2HNO3 = Н20 + 3NO2 — 73,6 кдж.
На рис. 42 схематически изображен процесс получения
концентрированной HNO3 синтезом под давлением.
is
Рис. 42. Схема получения концентрированной азотной кислоты
синтезом под давлением.
Окислы азота, полученные окислением NH3, после котла-
утилизатора проходят теплообменник для подогрева
выхлопных газов, теплообменник для подогрева воздуха и скоростной
холодильник, где отделяются 2/з реакционной воды в виде 2 —
3%-ной HNCb. Затем газы идут в холодильник-конденсатор /,
где охлаждаются до 20—40° С, причем конденсат в нем
представляет собой 25%-ную HNO3 и собирается в сборник 15. Этот
конденсат используется в данном производстве, а 3%-ная
HNO3 выводится из цикла и не участвует в производстве
концентрированной HNO3. Затем газы сжимаются
турбокомпрессором 2 до 6 ата (при этом они разогреваются) и поступают
в холодильник 3, где частично окисляются и охлаждаются до
¦60° С; затем они идут в окислительную колонну 4. Нижняя часть
последней орошается азотной кислотой концентрации 58—62%,
которая не поглощает окислов азота. Кислота охлаждается
водой, протекающей по змеевикам, расположенным на
тарелках колонны, и, охлаждая газ, отводит таким образом тепло
окисления N0 в NO2. Окончательное окисление N0 произво-
112
дится концентрированной 98%-ной HNO3 в верхней части колон-
ьы-доокислителя 5.
Газ охлаждается до 0° С в холодильнике 6 холодным
рассолом. После этого нитрозные газы проходят нитроолеумную
колонну 7 и промывную башню 8.
Колонна 7 орошается 98% -ной HNOa,
которая поглощает из газа NO2 и
образует нитроолеум {при 6 атм
и—10° С в 98%-ной HNOa,
растворяется до 36 вес. % N2O4).
Промывная башня 8 орошается водой,
которая, поглощая окислы азота,
превращается в разбавленную азотную
кислоту. Выхлопные газы через ос-
новіной теплообменник, где они
нагреваются за счет тепла газов из
контактного аппарата, попадают
в газовую турбину 9, где
рекуперируется энергия, затраченная на
сжатие газов, и далее идут на выхлоп.
Полученный в 7 нитроолеум
направляется через сборник 10 в
отбелочную колонну //. Здесь жидкость
нагревается и из нее десорбируется
N2O4.
Выделившиеся окислы азота
поступают в водяной холодильник 12
и рассольный холодильник 13, где
конденсируются. Полученная
жидкая четырехокись азота собирается
а мешалке 14. Остатки- газа
засасываются турбокомпрессором 2.
Разбавленная азотная кислота
из холодильника-конденсатора 1,
окислительной колонны 4 и
промывной колонны 8 поступает в сборник
15, а жидкая четырехокись азота из
14 и разбавленная азотная кислота
из 15 в определенном соотношении
подаются насосом 20 в автоклав 16,
куда нагнетается компрессором 17
сжатый до 50 ата кислород.
Продувочные газы проходят дефлегматор РИсин~ча^атч°„™а„в. TrZT°r°
. „ 1 * *-ИП I Cod О JU 1 ИиИ KrlLJiUTbl.
18 И ВМеСТе С НИТрОЗНЫМИ ГазаМИ /-корпус автоклава; і-защят-
ПОСТУПЭЮТ В ХОЛОДИЛЬНИК 3. НЬ1Й стакан; 3 — реакционный ста-
_; кан; ¦# — ситчатьге тарелки; 5 — рас-
ІІОЛучеННаЯ В аВТОКЛаве СМеСЬ горные штанги; 6 - чехольные тру-
концентрированной азотной кислоты бы <|миалы^убы.7 ™переливиые
8 8-1504
113
с растворенной в ней двуокисью азота подается в среднюю часть
отбелочной колонны //. Отбеленная концентрирования азотная
кислота проходит холодильник 19, откуда направляется как
готовый продукт на склад; часть ее поступает в холодильник 6, до-
окислитель 5, а также после дополнительного охлаждения в
поглотительную колонну 7.
Автоклав периодического действия состоит из
толстостенного стального цилиндра, в который вставлены два стакана из
чистого алюминия (99,8% А1). В настоящее время входят в
употребление автоклавы непрерывного действия с колпачковыми
или ситчатыми барботажными тарелками. На рис. 48 показан
разрез автоклава с колпачковыми тарелками. В нем имеются
девять тарелок диаметром 830 мм, расположенные на
расстоянии 620 мм друг от друга; тарелка имеет 900 отверстий, d =
= 2 мм.
Кислород подается на нижнюю тарелку под давлением
50 итм, смесь кислот — сверху; готовая продукция отводится по
трубе, опущенной до дна реакционного стакана.
Производительность описанного автоклава в три-пять раз
больше производительности аппарата периодического действия.
Расходные коэффициенты на 1 т концентрированной НМОз,
составляют: аммиак — 0,294 г, кислород—140 м3, вода—190 ж3,
холод — 400 000 кал, электроэнергия — 270 кет • ч, платина —
0,06 г; при этом получается 0,2 т пара.
Прогрессивным направлением в производстве HNO3
является применение газовой турбины для регенерации энергии из
горячих нитрозных газов; таким путем удается свести расход
электроэнергии до минимума и даже получать HNO3 без
дополнительной затраты энергии.
ГЛАВА IX
ПРОИЗВОДСТВО СЕРНОЙ КИСЛОТЫ
1. ПРИМЕНЕНИЕ И СОРТА СЕРНОЙ КИСЛОТЫ
Серная кислота используется при получении ботьгяого
количества продуктов (рис. 44): около 40% ее идет на производство
удобрений, около 35% потребляется химической
промышленностью, 11%—нефтяной и 10% в производстве металлических
изделий.
По данным ЦСУ СССР, в 1967 г. произведено 9,7 млн. г
серной кислоты, что составляет 104% количества ее,
полученного за тот же период 1966 года. Хотя по производству серной
кислоты СССР находится на втором месте после США, планом
развития народного хозяйства намечено увеличение производ-
Л4
ства на 1965—1970 гг. в несколько раз, по сравнению с
существующим уровнем. Это будет достигнуто за счет постройки
новых сернокислотных заводов во многих районах нашей страны
(на Северо-Западе, Урале, Средней Азии и др.), а также повы-
Суяьфи/пнай Mr, SO.
ціялюяоза • *
Л/юристыа сумф($хі
Фтор-ахлор-
фообие
Ыаг&
/SOL
Рис. 44. Области применения серной кислоты.
шения производительности действующих заводов и цехов, с тем,
чтобы в 1970 г. полностью была удовлетворена потребность
в серной кислоте.
Согласно общесоюзному стандарту № 2184—59 основные
сорта технической серной кислоты следующие: камерная F5%
115
H2SO4), башенная G5%), купоросное масло (92% H2SO4) и
олеум B0% свободного SO3). Кроме того, выпускаются чистые
кислоты: химически чистая, аккумуляторная и др.
Процесс производства серной кислоты заключается в
сжигании содержащего серу сырья и получении двуокиси серы,
которая затем окисляется и превращается в H2SO4 с помощью
окислов азота (нитрозный метод в виде камерного или башенного
способов) или твердых катализаторов (контактный метод).
2. СЫРЬЕ
Пириты. Серный колчедан (пирит) применяется в прозвод-
стве серной кислоты с 1835 года. Пирит оценивается по
содержанию в нем серы: чистый пирит (FeS2) состоит из 53,5% S
и 46,5% Fe. Природные пириты содержат от долей процента до
нескольких процентов примесей-сульфидов меди, свинца, цинка,
никеля, мышьяка, соединений селена, теллура и др., окислов
кремния, алюминия, кальция, магния и некоторые соли
(сульфаты и др.); в состав пиритов входят также золото и серебро
в количествах от нескольких граммов до десятков граммов на
тонну.
Присутствие железа, меди и некоторых других металлов
приводит к потере серы, остающейся в огарке в виде сульфатов
этих металлов.
Если содержание цветных металлов — меди, цинка, свинца
превышает один процент, сырье используется для получения
этих металлов. Так, например, для выплавки меди руду
обогащают флотацией. При этом она разделяется на части: медный
концентрат и флотационные хвосты, куда входит серный
колчедан и другие примеси. Содержание серы во флотационных
хвостах составляет 33—38%; флотационные хвосты подвергают
перефлотации с целью удаления «пустой» породы. Полученный пи-
ритный концентрат содержит 45—50% серы.
Медный концентрат идет на производство меди, а
флотационные XDocTLi являются глаиным сырьем для производства серной
кислоты. Так, например, при флотации руды, содержащей
1,3% Cli и 41,5% S, хвосты содержат 48% S и 0,23% Си, а
медный концентрат — 12,0% Си и 43,0% S.
В полученных концентратах и хвостах имеется около 50%
воды; их подвергают отстаиванию, а затем фильтрованию, после
чего остается 10—15% воды. Эту влагу удаляют путем
высушивания топочными газами во вращающихся барабанных
сушилках, выдающих сухой колчедан с 0,5—2% влаги, который
направляется на сернокислотные заводы. Серный колчедан,
содержащий минимальные количества цветных металлов и
достаточное количество серы, называется рядовым и используется только
116
для производства серной кислоты. Кроме флотационного и
рядового может применяться и углистый колчедан, содержащий 35—
45% серы и до 20% угля. Его извлекают при обогащении камен-
іюго угля. Удалив часть угля, можно снизить его содержание
в углистом колчедане до 10—12%. Углистый колчедан не
содержит вредных примесей — мышьяка и селена.
Сера. Сера используется не только в производстве серной
кислоты. Значительные количества серы применяются в
целлюлозно-бумажном производстве, при вулканизации каучука,
в сельском хозяйстве, в военном деле, для производства
искусственного волокна, сероуглерода, получения красителей, в
производстве спичек, фармацевтических препаратов, для
дезинфекции и т. д.
В Советском Союзе выявлены огромные запасы природной
серы — Гаурдагское месторождение и Шорсу в Узбекистане,
Роздольская сера в УССР, а также менее богатые залежи на
средней Волге, в Крыму, на Кавказе и в других местах.
Природная сера залегает главным образом в горных
породах— изверженных и осадочных. Если они залегают неглубоко
под землей, добычу ведут открытыми разработками; применяется
также шахтный способ добычи. Содержание серы в сероносных
породах сильно колеблется. Так, в средневолжских залежах оно
низкое (около 7%), а в залежах Средней Азии—высокое (до
40%). В руде Роздольского месторождения (Западная Украина)
содержится 25—30% серы и выше.
Самородную серу извлекают из руд разными методами в
зависимости от содержания серы. Дробленую серную руду
обрабатывают после предварительного обогащения флотацией. Чаще
всего руду нагревают, чтобы выплавить из нее серу (температура
плавления серы 120° С) огневым способом или паром. Выплав-'
ку серы из богатых руд ведут в автоклавах с помощью водяного
пара при давлении 3,5—5 атм.
В США выплавляют серу под землей перегретой водой
A70° С), которую пускают в сероносные горизонты по трубам,
проложенным в буровых скважинах (метод Фраша).
Расплавленная под землей сера вместе с водой выдается на поверхность,
где она затвердевает в продукт, содержащий 98—99% серы.
Получение серы, содержащейся в углях и выделяющейся
в виде сероводорода при коксовании, изложено при описании
очистки коксового газа.
Значение имеет газовая сера, получаемая из обжиговых газов
при обжиге колчеданов, либо при переработке медистых
колчеданов на медь. Если газ, содержащий серу в виде SO2,
подвергнуть действию восстановителя, то из SO2 выделится сера. В
качестве восстановителя можно применять уголь, окись углерода,
водород, природный и генераторный газы. Основные реакции для
первых двух восстановителей можно изобразить уравнениями
После восстановления газы, содержащие серу, очищаются от
примесей и охлаждаются; сера при этом конденсируется и
затвердевает. Получаемый продукт содержит 96—99,6% S, прочее —
примеси, состав которых зависит от сырья и условий работы
@,2—1% золы, 0,07—0,3% As). При получении меди из
медистых колчеданов, при обжиге руды, а также переработке
полупродукта (штейна) выделяются значительные количества SO2.
Из образовавшегося при производстве 1 т меди SO2 можно
приготовить до 4 г серной кислоты. Большое количество SO2 дает
обжиг сырья на цинко- и свинцовоплавильных заводах. Эти газы
во все возрастающих количествах применяются для
производства H2SO4. Кроме того, SO2 можно получать также из сульфата
кальция — гипса CaSO4 ¦ 2Н2О или ангидрита CaSO4,
прокаливаемых с углем:
CaSO4 + С = СаО + SO2 + CO.
Если к шихте добавить нужное количество глины, то ее
компоненты при 1450—1500° С образуют с СаО клинкер, который
после помола дает портланд-цемент. Таким образом, при
переработке гипса можно получать как серную кислоту, так и цемент.
Однако малое содержание серы в гипсе A8%) мешает пока
широкому использованию его как сырья. Из прочих видов сырья
для производства серной кислоты большое значение имеет
сероводород, извлекаемый из коксовых и других промышленных
газов, кислые гудроны, представляющие собой отходы
нефтеперерабатывающей промышленности. В настоящее время исследуется
возможность использования двуокиси серы, содержащейся в
дымовых газах, получаемых при сжигании угля, серы, входящей
в состав доменных шлаков и др. В СССР для производства
серной кислоты и серы пока применяются главным образом
колчедан F0%), сера A8%), сероводород E%) и газы
металлургических печей A7%). В ближайшие годы при абсолютном росте
всех ьидов применяемого сырья доля колчедана будет
уменьшаться.
3. ПОЛУЧЕНИЕ СЕРНИСТОГО ГАЗА
И ЕГО ОЧИСТКА
Из рядового колчедана двуокись серы получают обжигом
после измельчения до размеров зерен 3—5 мм. Измельчение
происходит в две ступени—дробление в щековых дробилках,
а затем в валковых дробилках. Флотационный колчедан
получается на сернокислотных заводах уже измельченный @,05—
118
0,1 мм). Обжиг производится в пиритных печах; суммарная
реакция обжига выражается уравнением
4FeS2 + 11О2 = 8SO2 + 2Fe2O3 + 3414 кдж.
Окись железа с примесями (огарок) является отходом;
количество огарка составляет около 75% от веса пирита.
Печной газ, уходящий из колчеданной печи, должен
содержать возможно больше SO2. При теоретическом количестве воз-
I
ОбжигоВый
газ
Рис. 45. Вертикальный разрез механической
полочной печи:
/—2 — питатели колчедана; 3 — центральный вал; 4 —
гребки с зубьями, 5 — отверстия в сводах для колчедана;
6 <*-< течка для огарка; 7 — малая шестерня.
духа газ должен содержать 16,2% SO2. Но в печь подается
воздух с избытком, необходимым для того, чтобы обжиг шел
с достаточной скоростью, а сера выгорала с достаточной
полнотой. Газы, полученные в результате работы заводских печей,
имеют сложный состав, так как часть БОг вследствие
контактного действия огарка и материала печи превращается в SO3 (до
10% от SO2). Избыток воздуха зависит также от конструкции
печи. В зависимости от избытка воздуха содержание SO2
составляет 8—12 при 6—11% О2.
119
Распространенным типом пиритных печей является
механическая полочная печь (рис. 45). Печь представляет собой
стальной кожух, футерованный огнеупорным (шамотным) кирпичом,
которым выложены также восемь сводов, из которых верхний,
открытый, служит для подсушки колчедана (сушильный свод),
а семь других—это рабочие своды, где сера колчедана
постепенно выгорает (счет сводов ведется сверху, не считая
сушильного). В центре печи расположен вращающийся полый вал, на
который насажены гребки с прикрепленными к ним зубьями.
Гребки и зубья — съемные, замену можно производить, не
охлаждая печь. Зубья расположены под острым или тупым углом
по отношению к оси гребка. Поэтому при вращении вала пирит
перемещается на нечетных полках от центра к периферии, а на
четных — от периферии к центру.
При производительности 35 г колчедана в сутки печь имеет
высоту 8 м, диаметр 6 м, расстояние между сводами 450 мм,
диаметр стального вала 920 мм. Вал приводится во вращение
электродвигателем, воздух для охлаждения вала нагнетается
в его полость вентилятором. Зубья делаются из огнеупорного
чугуна, легированного хромом. При работе печи измельченный
колчедан подается на сушильный свод через верхний бункер. При
движении гребков (от двух до трех оборотов в минуту) материал
перемещается на сушильном своде к центру печи, откуда через
питатель 2 он подается на первый рабочий свод в печи. Здесь
начинается выгорание серы пирита, который перемещается от
центра к периферии. Попавший на периферию первого свода
материал ссыпается через отверстия на второй свод, где он
передвигается так же, как на сушильном своде.
Из нижнего свода печи высыпается огарок с температурой
около 300сС по течке 6, а печной газ уходит сверху с
температурой 700—800° С. Воздух для горения колчедана подается снизу
через отверстия (воздушники) на VII своде и частично на IV
своде; газы движутся противотоком к колчедану. При этом
осуществляется наилучшее выгорание серы, и газ можно
получить с наибольшим содержанием SO2.
Для обжига флотационного колчедана применяются печи
разных конструкций. Существующие механические печи
приспосабливают к обжигу флотационных хвостов, примешивая
последние к рядовому пириту. Однако для этого сырья наиболее
рационально использовать печи для обжига во взвешенном
состоянии (рис. 46). Такие печи представляют собой стальные
аппараты, обычно цилиндрической формы, с бункером внизу,
футерованные изнутри огнеупорным кирпичом.
Пирит вдувается током сжатого воздуха (первичный
воздух) с помощью форсунки. Кроме того подается добавочный
(вторичный) воздух. Такие печи отличаются высокой
производительностью. Из других типов печей для получения SO2 укажем
120
Рис. 46. Печь пылевидного
обжига колчедана:
/ — кожух; 2 — огнеупорная
футеровка; 3 — бункер для огарка; 4 —
форсунка для подачи колчедана;
5 — экран для охлаждения; 6 —
фурмы для подачи вторичного
воздуха; 7 — обжиговый газ.
121
Обжигодо/й газ
«ще на горизонтальные вращающиеся печи, в которых можно
обжигать мелкий пирит. В СССР такие печи не нашли
применения.
В настоящее время большое значение приобрели
промышленные методы проведения взаимодействия твердых и
газообразных тел в псевдоожиженном
состоянии («кипящий слой»), когда
частицы твердого тела взвешены
в среде газа.
Значительный прогресс
достигнут при обжиге колчедана в печах
с «кипящим слоем» (печи к. с), так
как интенсивность горения в них
выше, чем в других печах. В табл.
17 приведено количество колчедана,
считая на 45%-ный по сере,
выгорающие на 1 м2 пода печи за сутки
(интенсивность печей).
Из таблицы видно, что
интенсивность печей с кипящим слоем в 80
раз больше, чем механических
подовых. На рис. 47 показан
схематический разрез печи с кипящим слоем.
Печь представляет собой стальной
футерованный цилиндр, отделенный
от нижней конической части
решеткой, через которую поступает
воздух с такой скоростью, что частицы
колчедана находятся в
псевдоожиженном состоянии. Из бункера
колчедан шнеком подается в печь, из
кипящего слоя отводится огарок. Через пылеотделители (циклоны
и электрофильтры) газ идет в котлы-утилизаторы.
воздух
Рис. 47. Печь для обжига
колчедана в «кипящем
слое»:
/ — колосниковая решетка;
2 — «кипящий слой» ; 3 —
вход к выход воды в
холодильные элементы
кипящего слоя; 4—7 — штуцера
для термопар;
5—смотровое стекло; 6 —
огнеупорная футеровка; 8 — штуцер
для отбора проб.
Таблица Г/
Виды по чей
' Иптспсніімпсі і. с і
Механическая подовая
Пылевидного обжига . .
Кипящий слой (к. с.) . .
до 250
до 2500
до 20 000
На рис. 48 изображена промышленная печь КС-1000
¦с кипящим слоем, обжигающая за сутки 100 т флотационного
концентрата. В печь диаметром 4 лі и высотой 9 м подается око-
І22
Оймиеобып
газ /
jio 6000 м3 воздуха в час. Газ, содержащий 14—15% SO2, уходит
из печи с температурой 850—900°С. Он содержит до 300 г/м3
пыли. В огарке остается 0,5—1,0% серы. Применяют также печи
КС-200, обжигающие до 200 т колчедана в сутки.
Из достоинств печи с «кипящим слоем», кроме ее высокой
интенсивности, дающей возможность достичь большой
производительности при меньших по сравнению с другими печами
габаритах, упомянем еще о
следующих: повышенное
содержание SO2 A2—15%) в газе,
малое содержание серы в огарке
@,5%), возможность обжига
¦бедных и влажных руд,
легкость пуска, остановки и
изменения режима..При малом
избытке воздуха и высокой
концентрации SO2 в печи может
значительно повыситься
температура, что приведет к
сплавлению огарка їй остановке печи.
Во избежание этого «кипящий
слой» охлаждают
специальными холодильниками. Печь
отличается простой конструкцией.
Ее легко ремонтировать, так
как она не имеет подвижных
частей в зоне горения пирита.
Существенным является
быстрое выравнивание температуры
в слое и высокая теплоотдача
B00—300 кал/м2 • град • час),
ЧТО позволяет ограничиться Рис' 48. Схема промышленной печи
небольшой поверхностью ХОЛО- Для обжига флотационного концент-
дильных элементов. На тонну Рата в кипящем слое:
і^пгшргтяня и прим и vnTip «ти / — опорная рама, 2 — обечайка; 3 — ко-
КОЛЧеДЭНа В пеЧИ В КОТЛе-уТИ- Нус; 4 —бункер под провальной решеткой
лизаторе получают
пара.
К недостаткам этих печей
относится сравнительно
большой расход энергии на дутье A2—14 кет • ч. на тонну колчедана)
и большой унос пыли B5—80% огарка). Теперь такими печами
с Советском Союзе оснащаются заводы цветной металлургии и
сернокислотные заводы, обжигающие сульфидные руды.
В последнее время в практику входят циклонные печи,
в которых обжиг производится при движении материала в
завихренном (закрученном) быстродвижущемся потоке газа,
благодаря чему достигается очень высокая интенсивность процесса.
ДО 5 Т загРУзочной камеры: 5, 7 — охлаждающие
м элементы- 6 — подовая плнта; 8 —
форсунки; 9 — загрузочная камера; 10 —
коллектор газа; //—выхлопная труба.
123
Циклонные топки нашли широкое применение в энергетике,
интенсивность сжигания топлива в них превышает интенсивность
в обычных печах в десятки раз, достигая около 30 млн ккал на
1 м3 печи в час.
Основная часть печи — стальной сварной вертикальный
цилиндр с шипами, футерованный изнутри огнеупорной
замазкой; снаружи цилиндр снабжен водяной рубашкой.
Измельченный колчедан подается в печь сверху из бункера, куда он
вдувается подогретым первичным воздухом. Вторичный воздух
поступает по двум касательным вводам со скоростью 40—
50 м/сек и создает вихревое движение колчедана в печи.
Расплавленный материал (огарок и колчедан) стекает пленкой по
огнеупорной футеровке цилиндра в нижнее отделение печи; отсюда
и удаляется жидкий огарок, содержащий 0,2—0,4% серы.
Благодаря охлаждению цилиндра водой часть
расплавленной массы отверждается и остается на стенке, образуя гарнис-
саж, ограждающий стенку печи от высокой температуры в печи.
Печной газ с высоким содержанием SO2 A2—13%) и
температурой около 1400° С удаляется через нижний газоход. В печи
объемом 10 м3 можно обжигать до 100 т колчедана в сутки. Таким
образом, интенсивность может достигать 10 г с куб. м в сутки.
Используя теплоту газов, можно получать около 0,8 т пара под
давлением 12 атм на тонну серной кислоты.
Для прокаливания гипса применяют вращающиеся
трубчатые печи. Вследствие того, что ось печи наклонена к горизонту
под углом 3—5°, материал передвигается от места загрузки
в верхнем конце к месту выгрузки в нижнем конце барабана.
Реакция разложения безводного сернокислого кальция
следующая:
CaSO4 = СаО + SO3 + -і О2 — 491 кдж.
Диссоциация CaSO4 происходит с поглощением большого
количества тепла. Полное разложение CaSO4 наступает при
температуре около 1400° С. При добавлении угля процесс протекает
с меньшим расходом тепла и при меньшей температуре по
реакции
CaSO4 + С = СаО + SO2 4- СО — 367,1 кдж.
При диссоциации гипса получается известь — около 0,6 г из
1,4 г гипса, нужного для производства 1 г серной кислоты.
Если добавлять к гипсу рассчитанное количество глины
и песка, то при 1400—1450е С смесь спекается и образует
портланд-цементный клинкер. Измельчение клинкера с 2—3%
гипса дает необходимый строительству портланд-цемент. В этом
процессе шихта состоит примерно из 75% ангидрита, 16%
глины, 7% кокса и ряда добавок. Для характеристики установки
укажем, что из печи длиной 60 м и диаметром 3,2 м при одном
124
обороте в 1,5 минуты получают за сутки до 80 г SO2 при
содержании его в газе 5—7% объемных.
Сжигание серы производится в различных печах
сравнительно простой конструкции. Сера сжигается в жидком или
газообразном состоянии. Наиболее распространенной является
печь для сжигания расплавленной серы в распыленном
состоянии, представляющая собой горизонтальный стальной
футерованный огнеупором цилиндр, куда форсункой вдувается воздух
и расплавленная сера; в печь подается добавочный воздух
Печной
газ
СухойЙоздух
Рис. 49. Установка для сжигания серы в расплавленном
состоянии:
/ — плавильники; 2, 3 — погружные насосы, 4 — фильтр для
расплавленной серы, 5 — форсунка для серы; 6 — печь с перегородками; 7 —
котел-утилиэатор.
(рис. 49). Горение серы происходит во всем объеме печи.
Простота сжигания серы, отсутствие огарка и малый расход сырья
@,38 т серы вместо 0,86 т пирита на тонну H2SO4) делает серу
одним из наилучших видов сырья для производства H2SO4. При
этом газы получается более концентрированными и переработка
их осуществляется в аппаратах меньших габаритов, а очистка
газов почти исключается. Двуокись серы, получаемая при
сжигании серы, кроме производства" серной кислоты, применяется
в ряде отраслей промышленности для очистки погонов нефти как
холодильный агент, в производстве сахара и др. SO2 перевозится
в стальных баллонах и цистернах в жидком состоянии.
Ожижение SCb производится сжатием предварительно осушенного
и охлажденного газа.
Использование огарка. Благодаря высокому
содержанию железа в огарке его применяют для получения чугуна
в доменной плавке. Для этого мелкий огарок предварительно
спекают (окусковывают) на агломерационных машинах, удаляя
при этом серу.
Ценной составной частью огарка является медь. Для ее
выделения огарок непосредственно или после дополнительного
125
обжига с сульфатизирующими (с Na2SO4) или хлорирующими
(с NaCl) веществами обрабатывают кислотой. При этом медь
огарка переходит в раствор, из которого можно выделить соли
меди, широко применяемые для борьбы с вредителями в
сельском хозяйстве и для других целей. Из растворов солей мели
можно выделить металлическую медь железом (цементация).
Из огарка готовят также краски —¦ железный сурик и мумию.
Разработаны схемы полной, комплексной переработки огарка:
из него можно получить много ценных веществ — сульфата
натрия, алюминия и др., а также золото, серебро и др.
Очистка печного газа
Газ печей содержит, помимо SO2, ряд примесей, которые
можно разделить на примеси, вредные для дальнейшего
производства настолько, что без освобождения от них нарушается весь
режим дальнейшего процесса, и примеси, прямо не вредящие-
процессу переработки газа, по являющиеся балластом.
При контактном способе получения серной кислоты
соединения мышьяка, попадая в катализатор, отравляют его и
выводят из строй, а при нитрозном методе мышьяк и селен
загрязняют и отравляют кислоту. Так же вредны пыль и кашт
жидкости (воды, кислоты, масла и др.), которые покрывают
катализатор, загрязняют кислоту и забивают аппаратуру.
Азот в печном газе не вреден, но он увеличивает объем
газа, что влечет за собой увеличение мощности вентиляторов,
проталкивающих газ через аппаратуру, объема аппаратуры
и др.; поэтому лучше применять воздух, обогащенный
кислородом, тем более, что он интенсифицирует процесс обжига сырья.
Необходимо подчеркнуть, что примеси нередко и сами по себе
представляют ценность, так как из них можно получить
добавочные продукты. Так, обычно в производстве серной
кислоты выделяют и используют селен, необходимый для
производства фотоэлементов и других целей.
Чтобы получить чистый газ, пригодный для производства
серной кислоты контактным методом, печной газ очищают сначала
от огарковой пыли в іорячем электрофильтре, затем охлаждают
его и очищают от окислов мышьяка, селена, тумана SCb и H?SOt
и, наконец, от паров воды. Очищенный газ проходит компрессор,
очистку от масла и только после такой сложной очистки его
отправляют на катализ для превращения SO2 в SOa.
На рис. 50 представлена схема очистного отделения при
производстве серной кислоты контактным методом. Газ после огар-
кового электрофильтра с температурой 300° С поступает в
первую промывную башню 1, которая орошается серной кислотой
F5—70%-ной); здесь происходит быстрое охлаждение газов и
возникает высокое пересыщение паров, благодаря чему часть
126
их конденсируется в объеме с образованием тумана — аэрозоля;
этот туман удаляется из газа тем легче, чем крупнее его капли.
Охладившись до 75е С, газ поступает во вторую промывную
башню 2, орошаемую 30%-ной кислотой, где упругость водяных
паров больше, вследствие этого дальнейший рост части тумана
происходит за счет поглощения и конденсации из них паров
воды. Отсюда газ, охладившись до 30е С, уходит на очистку в
мокрые электрофильтры 3 и 5, между которыми находится увлаж-
кис пота па
еллад
Кис/ІОЛ.
та на сялад
Рис. 50. Схема очистки обжигового газа.
нительная башня 4, орошаемая 3%-ной серной кислотой; в
увлажнительной башне капли после первого электрофильтра 3
укрупняются и оседают во втором электрофильтре 5, откуда газ.
выходит свободным от примесей, кроме паров воды. Далее газ
поступает на осушку в сушильную башню 6, орошаемую
концентрированной серной кислотой (94—97%), брызги которой
удаляются в брызгоуловителе, откуда газ выходит с содержанием
Н2О не больше 0,2 г/м3.
Жидкости, стекающие из башен, проходят холодильники 7 и
собираются в сборники 8.
Описанная сложная система очистки может быть упрощена,
например, если газы не содержат мышьяка, как это имеет место
при работе на чистой сере, а также с катализаторами, стойкими
к отравлению. При получении серной кислоты нитрозным
методом очистка производится только в огарковых
электрофильтрах.
4. ПОЛУЧЕНИЕ СЕРНОЙ КИСЛОТЫ у
Нитрозный метод
Еще в первой половине XV столетия серную кислоту получали
сжиганием серы в смеси с селитрой и поглощением водой
получающихся при этом газов. В 1806 г. французские ученые Клеман
127
и Дезорм доказали, что при этом имеет место окисление SO2 за
¦счет кислорода воздуха с помощью окислов азота, которые
выделяются из селитры и являются, таким образом, только
передатчиками кислорода (гомогенный катализ).
Более правильный взгляд на процесс обеспечил дальнейшее
развитие сернокислотной промышленности. В XIX веке серная
кислота получалась в системах, состоящих из ряда больших
свинцовых камер (камерный способ) и двух башен.
В настоящее время в СССР не применяются камерные
системы, отличающиеся низкой интенсивностью. Изучение условий
¦работы таких систем показало, что их малая интенсивность
•объясняется следующими причинами: 1) недостаточным
использованием объема; 2) малой концентрацией окислов азота;
3) недостаточным отводом тепла экзотермических реакций
процесса и 4) малой плотностью орошения. Создание камерных
систем, лишенных указанных недостатков, позволило довести
интенсивность систем с 5—6 до 12—15 кг H2SO4 с 1 л3в сутки.
В 20-х годах появились первые установки башенной системы,
съем в которых составлял до 20 кг с 1 мг в сутки. Благодаря
усилиям советских ученых К. М. Малина, В. Н. Шульца, И. Н.
Кузьминых, С. Д. Ступникова и других интенсивность башенных
систем СССР возросла до 200—240 кг с \ м3 в сутки.
Для объяснения процесса образования серной кислоты из SO2,
О2 и окислов азота предложена теория образования H2SO4
через промежуточные соединения, полученные из исходных
компонентов. Промежуточным соединением, которое легко
разрушается, давая конечный продукт, является нитрозилсерная кислота
NOHSO4 (сокращенно HNSO5), образование которой
изображается, например, следующей реакцией:
N2O3 (или NO2 + NO) + 2H2SO4 = 2HNSO5 + H2O. (I)
Нитрозилсерная кислота растворяется в серной кислоте,
образуя более или менее устойчивый раствор (нитрозу); при
разбавлении нитрозы водой происходит гидролиз нитрозилсерной
кислоты —тем в большей степени, чем больше разбавление її
выше температура; при этом выделяются окислы азота, происходит
денитрация нитрозы п. п зависимости от условий (концентрации
кислоты и др.), протекают реакции
HNSOS + Н2О = H2SO4 + HNO,, (II)
SO2 + 2HNO2 =- H2SO4 -I- 2NO. (Ill)
Таким способом образуются H2SO4 и окись азота; NO
частично окисляется кислородом воздуха до NO2:
(IV)
.128
Стехиометрическая смесь NO и NO2 образует N2O3, которая
в присутствии воды дает HNO2, повторяющую цикл
взаимодействия
Н2О
NO + N02 = 2HNO2.
(V)
Реакция (III) является основной реакцией кислотообразова-
ния. Выделяющиеся окислы азота снова идут в процесс до тех
пор, пока весь SO2 не превратится в H2SO4. В отсутствие двуокиси
серы окислы азота поглощаются серной кислотой по реакции (I).
Из сказанного видно, что в башенной системе протекают
многочисленные процессы, которые идут с разной скоростью,
последовательно и параллельно.
Большое значение имеют температурные условия и
концентрация компонентов, а также гидродинамические условия —
скорость газов и жидкостей, плотность орошения и др. Эти условия
изучены для башенного процесса и подробно излагаются в
специальных курсах.
Башенная система
Заводские системы включают от пяти до семи башен Мы
приводим систему из пяти орошаемых башен (рис. 51), из которых
водо
Лютнаи
Рис. 51. Схема производства серной кислоты башенным
способом.
первая — денитрационная и вторая — продукционная, третья —
окислительная, четвертая и пятая — абсорбционные. Ход газов
в башнях следующий. Горячий печной газ входит в первые
башни параллельно после огаркового электрофильтра (не показан на
схеме) и проходит остальные башни последовательно, снизу
вверх. Кислота на орошение подается в башни сверху, стекает
вниз и охлаждается в холодильниках 7, собирается в сборники 5
и перекачивается насосами 9.
Из последней башни остаточные газы выбрасываются в
атмосферу, пройдя электрофильтр 6 для улавливания брызг, и
санитарную башню (на схеме не показана), в которой окислы азота
и остатки S02 улавливаются щелочными растворами, во
избежание отравления воздуха.
9 8-1504
129
из
обеспечивает
улавливание
уходящих
Газы проходят систему благодаря вентилятору 10,
находящемуся между башнями 4 и 5. Орошение башни, как правило,
осуществляется так, что нитрозы, получаемые в башнях 4 и 5
(абсорбционная зона), подаются на башни 1 я 2 (продукционная
зона), откуда денитрированные кислоты поступают на орошение
башен 4 и 5.
Наиболее денитрированная кислота стекает с первой башни-
денитратора; после
охлаждения она идет на склад готовой
продукции. Кислота,
вытекающая из башни 2, после
охлаждения подается на орошение
башни 5, что
более полное
окислов азота
газов.
Башня 3 орошается обычно
«сама на себя». В ней должно
закончиться с максимальной
полнотой превращение SO2
в H2SO4. После башни 3 газ
проходит полую неорошаемую
башню Q (окислительный
объем), включаемую «в шунт»
(т. е. байпасным
трубопроводом), где окись азота
окисляется на 50% до двуокиси, что
является подготовкой к
абсорбции окислов азота. Наконец,
в башнях 4 и 5 окислы азота
поглощаются орошающей
кислотой.
Плотность орошения башен
равна 2—8 м3/м2ч, а кратность
орошения в современных
интенсивных системах составляет 30-50 кг на ! кг получаемой
продукции. Нитрозность орошающих кислот достигает 9% N2O3,
а азотооборот колеблется от 500 до 900 кг окислов азота (в
пересчете на HNO3) на тонну продукта. (Азотооборот — это
количество окислов азота, поглощаемых в башнях 4 и 5). Обычно потеря
окислов азота возмещается тем, что в систему добавляют 10—
15 кг HNO3 иа тонну продукта, но при хорошей работе системы
эти потери снижаются до 7—8 кг. Окислы азота, чаще всего
в виде HNO3, подаются на верх башен 1 и 2, куда поступает
также вода; окислы азота можно подавать на низ этих башен из
установки окисления аммиака, где из NH3 получается NO.
В башне / кроме денитрации происходит очистка газа от пыли.
Рис. 52. Башня с насадкой:
/ и корпус; 2 — кислотоупорная
футеровка из андезита; Л—насадка (кольца):
4 — распылитель; 5 — колонка
колосниковой решетки; 6 — колосники.
130
а также упаривание кислоты. Поэтому при горячем ходе
получают не только 75%-ную, но и 90—92%-ную кислоту, т. е.
купоросное масло.
В сернокислотных системах применяются стальные башни
(рис. 52) вместо ранее используемых свинцовых. Диаметр их
составляет 5—6 м и высота 16 м\ они футеруются
кислотоупорными материалами —
природными (андезит, бештаунит)
вулканического
происхождения или искусственными из
кислотоупорной керамики
на кислотоупорном цементе.
Для увеличения поверхности
соприкосновения жидких и
газообразных фаз башни
заполняют, как обычно при
абсорбции, насадкой из
керамических или кварцевых
элементов различной формы.
В крышке башен
монтируются различные
разбрызгивающие устройства. В
последнее время в
абсорбционной зоне устанавливают
полые башни (без насадки): в
них гидравлическое
сопротивление меньше (рис. 53).
Материал, из которого
изготовляется аппаратура,
должен быть химически
стойким, так как серная
кислота является сильно
корродирующим агентом.
Для безнитрозных кислот
концентрацией ниже 80%
стойким является свинец.
Для кислот, содержащих
окислы азота, применяют
стали — нержавеющие и
обычные, конструкционные;
для насосов, кислотопроводов, газопроводов и арматуры
используют обычные стали, чугун, а в некоторых случаях ферросилид,
содержащий до 17% Si. Широкое применение находят защитные
покрытия из керамики и пластмасс—виниловых, хлорвиниловых
и изобутиленовых. В качестве конструкционных материалов
используют фаолит (бакелитовая смола с асбестом). Хорошо
себя зарекомендовал антигмит (АТГ) — графитовый материал,
Рис. 53. Безнасадочная
абсорбционная башня:
/ — стальной корпус; 2 = распылители
одинарные; 3 — распылители двойные;
4 — коробка со штуцером для выхода
кислоты; 5 — желоб для спуска шлама.
131
выдерживающий действие серной кислоты даже при 120 С. Для
хранения и передачи азотной кислоты применяют нержавеющую
сталь, но для смеси (меланж), содержащей 87% HNO3 и 7,5%
H2SO4, пригодна аппаратура из обычной углеродистой стали.
Расходные коэффициенты при производстве серной кислоты
башенным методом следующие: на тонну серной кислоты A00%-
ной) стандартного (с содержанием 45%-ной серы) колчедана
760—800 кг, азотной кислоты (моногидрата) 12—18 кг, воды
около 50 мг и энергии 60—75 кет • ч.
Концентрирование серной кислоты
При нитрозном методе производства продуктом
башенная кислота (концентрацией около 75%).
является
В
ряде
340
300
260
220
производств (для
180
и о
100
/
г„——
У
//
У
/
f
20 Ь0 60
Содержание
30
100
%
органических синтезов,
производства взрывчатых веществ и др.)
необходима кислота
концентрации 92—96%.
Из башенкой кислоты
концентрированную получают, удаляя
при нагревании более летучий
компонент—воду. Зависимость
температуры кипения раствора и
температуры конденсации паров
от состава смеси H2SO4 — Н2О
(при 760 мм) представлена
диаграммой (рис. 54) из которой
видно, что оба компонента образуют
нераздельно — кипящую
(азеотропную) смесь, состоящую из
98,3% H2SO4 и 1,7% Н2О с
точкой кипения 336,6° С.
Таким образом, выпаривать
всю воду из разбавленной кислоты невозможно. Концентрировать
слабые серные кислоты можно лишь до 98,3%;
концентрированная кислота («купоросное масло») содержит 92,5—94% H2SO4.
При упаривании серной кислоты в качестве теплоносителя
используются продукты горения таких видов топлива: кгжса,
антрацита, мазута и газа.
При концентрировании часть кислоты разлагается, часть
увлекается в виде тумана, и поэтому выходящие газы
пропускаются через улавливатели брызг и тумана. Унос при
концентрировании доходит до 8%. а безвозвратные потери — до 4%.
Наиболее распространены барабанные концентраторы, обладающие
большой производительностью.
Основной аппарат (рис. 55) в установке — концентратор —
представляет собой горизонтальный стальной цилиндр
(барабан) диаметром около 3 м и 7 м длиной, футерованный изнутри
Рис. 54. Диаграмма кипения
системы H2SO4 — Н2О.
132
андезитом. Барабан разделен андезитовои перегородкой на две
камеры, сообщающиеся внутренним перетоком. Во время работы
Газ * пор
Рис. 55. Барабанный концентратор:
/ — стальной корпус; 2— асбест; 3 — андезитовая футеровка; 4 —
коленчатый газоход; 5 — переток кислоты; 6 — газоходы.
кислота поступает во вторую камеру, а затем в первую, где она
концентрируется до 92—94%-ной H2SO4. Горячие топочные газы
при 800—950° С по барботажным трубам из хромистого чугуна,
Мазут
Разбавленная
і серная кислота
Рис 56. Схема установки для концентрирования серной
кислоты в барабанном концентраторе:
/ — топка; 2 — концентратор; 3 — холодильник для купоросного
масла; 4, 5 — электрофильтры.
опущенным в кислоту, поступают в первую камеру, проходят
вторую и уходят из концентратора, унося пары воды и туман
133
серной кислоты. Выходящая с температурой 240° С кислота
охлаждается в холодильнике.
На рис. 56 представлена схема для получения
концентрированной серной кислоты. Топочные газы образуются в топке, куда
поступают мазут или газ и воздух, нагнетаемый вентилятором.
Топка представляет собой стальной цилиндр, футерованный
огнеупорным кирпичом. Горячие газы из топки идут в концентра-
Генераторнь/й
газ
водяной пар
мая /а/слота
кислота
тор, а оттуда в электрофильтр,
где улавливаются брызги и
туман серной кислоты.
Расходные коэффициенты
на 1 т H2SO4 при
концентрировании 75%-ной H2SO4
составляют около 56 кг мазута, 12,5
Концентрирован- кет • ч электрической энергии,
10 м3 воды, 25 кг пара и около
1500 м3 воздуха.
Применяются также
установки с погружными горелками
(рис. 57), в которых горение
газа происходит в
пространстве ниже уровня упариваемой
кислоты, вследствие чего тепло
Рис. 57. Аппарат для концентриро- хорошо используется,
вания серной кислоты при погруж- Кроме описанных концен-
ном горении. траторов, работающих при
атмосферном давлении,
применяются также вакуум-выпарные аппараты '
(вакуум-концентраторы), в которых благодаря создаваемому вакууму значительно
снижается температура кипения кислоты (например, при
760 мм рт. ст. 95%-ная H2SO4 кипит при 300° С, а при
4 мм рт. ст.— при 120° С).
Для концентрирования серной кислоты применяют также
аппараты, работающие по принципу дистилляционных колонн
тарельчатого устройства, башенные концентраторы и др.
Контактный метод
Контактный метод производства серной кислоты
осуществляется таким образом, что SO3 получают отдельно от H2SO4.
Процесс включает окисление SO2 в SO3 и поглощение SO3 для
получения олеума.
Окисление SO2 в SO3 производится в присутствии твердого
катализатора (гетерогенный катализ) по реакции
SO2 + ^-O2«SO3 +96,7 кдж.
134
Так как реакция обратима и сопровождается выделением
больших количеств тепла, понижение температуры повышает
выход SO3, направляя реакцию слева направо, согласно
принципу Ле-Шателье. Это видно из данных по изменению
константы равновесия
p0.5 n
2
Уравнение для расчета Kv имеет вид
4Є05.5 . с.сс
= —f 4,6455,
отсюда найдены следующие значения Кр
Температура, °С 400 450 500 600 1000
К, 440 138 50,2 9,41 0,143
Как видно из этих данных, значение константы равновесия при
повышении температуры быстро падает.
На практике представляет интерес другая величина —
равновесная степень контактирования jcp, представляющая собой
отношение количества SO2, превращенного в SO3, при
равновесии к начальному количеству SCV, выраженная через
парциальные давления она имеет вид
хв =
+ Р
¦ &<;§
Если ввести сюда константу равновесия Кр из приведенной
формулы ее и если начальное содержание в объемных процентах
SO2 обозначить через а, содержание О2 через Ь, общее давление
в атм через Р, то, учитывая, что при окислении одного объема
SO2 расходуется 0,5 объема О2, получим
_
ХР К , -,/ ЮО—0,5хр5~'
р ^ у Р(Ь-0,5ахр)
Из формулы видно, что Хр увеличивается с ростом давления
(знаменатель в подкоренном выражении увеличивается) и падает
с ростом температуры (Кр уменьшается); уменьшение же SO2
в газе (а) приводит к росту Хр, так как разбавление газа
связано с избытком кислорода.
Как известно, константа скорости реакции (Кск) выражается
в зависимости от энергии активации Е и температуры Т так:
_ _?_
Кш=К0-е «г,
где Ко—коэффициент (предэкспоиенциальиый множитель), не
зависящий от температуры и характеризующий катализатор,
ft — газовая постоянная, е = 2,718.
Поскольку катализаторы снижают энергию активации, Кен
13S
сильно увеличивается; влияние температуры на скорость реакции
видно из формулы и рис. 58, где Л'ск отложена в зависимости
от 1/7.
Вначале при контактном окислении SO2 применяли
платиновый катализатор, в котором тонкий слой платины покрывал
носители с большой удельной поверхностью (асбест, сернокислый
магний, силикагель и др.). При использовании платинового
катализатора степень превращения достигает 96—99% (при
температуре 400—450°). В
насй
0.6
1Z
1
ІЗ 1b 15
±¦/04 -—
550 500 450
400
t.'C
рур
тоящее время платиновый
катализатор заменен
ванадиевым на носителе из
силикагеля. Замена
платины ванадиевой
контактной массой объясняется,
главным образом,
высокой стоимостью платины
и тем, что она легко
отравляется контактными яда-
ми: мышьяком, хлором.
окисления сернистого ангидрида на
ванадиевом катализаторе от температуры.
парами воды и др. От этих
Рис 58 Зависимость константы скорости примесей нужно
освободить печной газ путем
сложной очистки.
Ванадиевый
катализатор не отравляется парами воды, он менее чувствителен к
мышьяку и к тому же значительно дешевле платины; однако, очистка
газа от As неизбежна и при ванадиевом катализаторе.
Большое распространение в СССР получила масса для
ванадиевого катализатора, известная под названием БАВ (барий —
алюминий — ванадий). Состав массы соответствует
приблизительно такому соотношению: V2O5, I2S1O2, 2К2О, ЗВаО,
0,5 АЬОз; в нем активный компонент V2O5(8—9%), а остальные
являются' носителями и активаторами. В разработке ванадиевых
масс и теории их действия большую роль сыграли наши
ученые— г. К. Боресков, И. Е. Ададуров и др.
Пі>а приготовлении массы ЬАВ исходит из раствора ванада-
та каГлия KVO3. жидкого стекла, хлористого бария, хлористого
алюминия и хлористого калия по определенной рецептуре.
Катализатор выпускается в форме гранул, таблеток, цилиндров и др.
В последние годы разработаны новые методы получения
ванадиевых катализаторов.
Применяемый в настоящее время катализатор СВД (сульфо-
ванадат на диатомите), приготовляют не из растворов, а
смешением смоченных порошков компонентов с последующим
гранулированием; высушенный катализатор содержит 7—8% пяти-
окиси ванадия.
136
Оптимальные условия контактирования. Контактную массу
заменяют свежей раз в три—пять лет. При точном соблюдении
технологического режима катализатор может служить
значительно дольше. Температура зажигания ванадиевой массы, при
которой начинается контактирование, в зависимости от состава
газа и катализатора колеблется в пределах от 410—440° С.
Зависимость константы скорости окисления на БАВ от
температуры (в координатах Лек, 1/Л выражается прямой (рис. 58).
Отсюда видно (табл. 18), что с повышением температуры
(уменьшение 1/Т) Кск растет.
Таблица 18
Температура,
/Сек
400
0,34
420
0,55
440
0,87
450
1,05
500
2,9
600
15,2
Для правильного выбора температурных условий перехода
SO2 в SO3 важно знать зависимость скорости окисления от
температуры. Приводим предложенную Г. К. Вересковым формулу
для скорости окисления SO2 на ванадиевом катализаторе
273
273 + t '
Здесь х — степень контактирования, хр — равновесная степень
контактирования, а и Ь — начальные концентрации SO2 и О2,
t — температура, °С, Кск — константа скорости.
Проследим на основании приведенной формулы, как
изменяется скорость окисления SO2 от температуры. От температуры
зависят Кск и Хр, причем с повышением температуры Кск также
растет, но хр уменьшается. Поэтому зависимость скорости
окисления SO2 при определенной степени контактирования х от
температуры проходит через максимум, как схематически показано
на рис. 59 для ряда кривых, отличающихся различной степенью-
контактирования (цифры на кривых показывают значение х).
Как видно из рисунка, для увеличивающейся степени
окисления максимумы сдвинуты в сторону уменьшения температуры.
Ход кривых зависит от состава газа. Поэтому оптимальную
температуру надо менять по мере окисления SO2 в SO3, начиная
процесс при более высокой температуре и снижая ее по мере
роста степени контактирования.
На практике в реакцию вводят газ, нагретый немного выше
температуры зажигания; так, например, на ванадиевом
катализаторе газ направляют при 440°С в первый слой катализатора
до достижения оптимальной температуры без отвода
выделяющегося тепла (адиабатически). Газ нагревается здесь теплом
137
.реакции до 600° С, достигая степени превращения около 73%',
затем его охлаждают непрерывно или периодически, в
зависимости от конструкции контактного аппарата.
По способу отвода тепла контактные аппараты отличаются
друг от друга. Теплообмен осуществляется в самих контактных
¦аппаратах, снабженных теплообменными трубами, проходящими
через контактную массу, и в
теплообменниках, находящихся между
слоями или вне контактных
аппаратов.
Важно отметить, что при
правильной организации процесса
контактирования и теплообмена
выделяющейся при катализе теплоты
реакции превращения SO2 в SO3
достаточно для нагревания холодных
газов, поступающих на
контактирование. Лишь вначале при пуске
системы или нарушениях режима газ
приходится подогревать в особых,
так называемых пусковых (огневых)
подогревателях, отапливаемых
углем или другими видами топлива.
Процесс окисления SO2 в SO3
проводят преимущественно в
аппаратах со ступенчатым
контактированием в четыре или пять стадий.
На рис. 60 изображен современный
четырехслойный контактный
аппарат. Он состоит из стального
цилиндрического корпуса, расширяющегося в нижней части. Внутри
контактного аппарата имеются теплообменники 1, 2, 3.
Контактная масса размещена на решетках, которые
установлены на подставках, лежащих на трубных решетках тепло-
-обменников. Нумерация слоев ведется сверху вниз, по ходу газа.
Газ, содержащий SO2, поступает в межтрубное пространство на-
ружниги теплообменника 4, по внутренним трубам которого
движутся горячие газы после контактного аппарата. Нагретый здесь
газ идет в контактный аппарат, где он сначала проходит
последовательно межтрубные пространства промежуточных
теплообменников.
В теплообменниках имеются перегородки, направляющие
лоток газа перпендикулярно к теплообменным трубам. Из
первого теплообменника газ, нагретый до 440—450° С, направляется
в первый слой контактной массы, реагирует и нагревается. Затем
он охлаждается в трубах первого теплообменника, поступает во
второй слой контактной массы и т. д.
420 Ш 500 540
t. "С
Рис. 59. Зависимость
скорости окисления
двуокиси серы от
температуры при различных
степенях контактирования.
¦J38
Для регулирования температуры на входе в каждый слой
контактной массы с помощью задвижек 5 и 6 изменяют
количество газа, поступающего в межтрубное пространство
соответствующего промежуточного теплообменника.
Производительность четырехслойных контактных аппаратов
составляет 240 т серной кислоты в сутки и больше. Эти аппараты
1- слой
2-слой
Из подогревателя
Рис. 60. Схема контактного узла с четырехслойными
контактными аппаратами и промежуточным
теплообменником.
просты в обслуживании, устойчивы в работе, обеспечивают
высокий процент контактирования и занимают меньше места, чем
прежние.
Катализ в «кипящем слое»
Преимущества, связанные с псевдоожижением твердого
компонента при взаимодействии твердой фазы с газообразной,
изложенные ранее, остаются в силе и при гетерогенном катализе.
Впервые «кипящий слой» в каталитических реакциях в
производственном масштабе был применен при каталитическом крекинге
нефтепродуктов, гидрогенизации углеводородов и др.
Остановимся на положительных сторонах процесса
псевдоожижения. Процесс характеризуется высокой интенсивностью
вследствие того, что вся поверхность интенсивно
перемешиваемых зерен катализатора взаимодействует с компонентами
газа. Для уменьшения диффузионных сопротивлений вместо
зерен размером 5—6 мм можно применять более мелкие зерна
размером 0,5—I мм и еще меньше, чем при неподвижном
катализаторе. Это должно привести к уменьшению количества ката-
139
лизатора и поверхностей нагрева. Одновременно появится
возможность использования более активных катализаторов.
Благодаря улучшенной теплопроводности теплопередача и
выравнивание температур проходят быстрее в том случае, когда вместо
катализатора в слое применяют катализатор в псевдоожиженном
состоянии. Подвижность катализатора позволяет вести процесс
с циркуляцией катализатора, выводя его из аппарата. Таким
путем облегчается замена катализатора, которую можно
осуществлять без остановки аппарата.
Отрицательной стороной применения псевдоожиженного
катализатора является истирание зерен и части аппаратуры. При
этом увеличивается унос катализатора в виде пыли, из-за чего
необходимо ставить пылеулавливающие аппараты. Однако,
несмотря на это, на практике применение псевдоожиженного
катализатора оправдывает себя. Важным обстоятельством
является то, что температура зажигания может быть снижена с 430—
450° С до 260—310° С. В то же время возможность легко
поддерживать оптимальные температуры в слое катализатора
позволяет повысить степень превращения SO2 до 98,5%.
Абсорбция SO3
Полученные после катализа газы с температурой около 200° С
направляются на абсорбцию SO3 для получения олеума. Перед
абсорбцией газы охлаждаются водой или воздухом до
температуры 80° С в трубчатых ангидридных холодильниках.
Абсорбция SO3 из газовой смеси происходит обычно в
абсорберах с насадкой, орошаемых олеумом (олеумные абсорберы),
а затем в моногидратных абсорберах, орошаемых техническим
моногидратом, (98%-ной кислотой), так как такая кислота
обладает наибольшей абсорбционной способностью по отношению
к SO3. На рис. 61 показана схема абсорбции (после
ангидридного холодильника).
Из контактного отделения газ поступает в олеумный
абсорбер 2, в котором он орошается 20%-ным олеумом (H2SO4 с
содержанием 20% свободного SO3). Из абсорбера 2 олеум с
содержанием 21% свободного SO3 стекает в сборник 7, куда
добавляется кислота из второго абсорбера в количестве,
достаточном для разбавления до 20% свободного SO3. Из сборника 7
олеум насосом 8 подается в олеумный холодильник /, а из
него — на орошение олеумного абсорбера. В олеумном
абсорбере поглощается до 70% SO3.
Часть олеума из сборника 7 непрерывно откачивается на
склад или в цех, где получают купоросное масло разбавлением.
Из абсорбера 2 газ поступает в моногидратный абсорбер 3,
орошаемый 98%-ной кислотой («моногидратом»); отсюда
моногидрат с содержанием 98,7—99% H2SO4 стекает в сборник 6, куда
но л
для разбавления добавляется кислота из сушильных башен или
вода. Из сборника 6 кислота насосом 5 подается для охлаждения
в моногидратный холодильник 4 и вновь поступает на орошение
моногидратного абсорбера. Часть моногидрата непрерывно
перекачивается в сборник олеума 7 и в сушильную башню. Газы из
моногидратного абсорбера идут в санитарные башни и в
электрофильтр для обезвреживания перед выбрасыванием в
атмосферу.
В санитарную
башню
rtaсклад
Рис. 61. Схема абсорбционного отделения.
Абсорбцию осуществляют в стальных башнях, футерованных
изнутри кислотоупорным материалом и заполненных насадкой
из кислотоупорных колец.
Расходные коэффициенты на 1 г контактной кислоты
следующие: 800—850 кг стандартного колчедана, 50—55 м3 воды н 100—
105 кетч электроэнергии. Общая схема производства серной
кислоты контактным методом дана на рис. 62. Сложная схема
производства контактной кислоты может быть значительно
упрощена при замене колчедана чистой серой или сероводородом,
поскольку в этом случае отпадает необходимость в глубокой
очистке газа.
Серная кислота из сероводорода (мокрый
катализ). Большую роль приобретает производство серной кислоты
из газов, содержащих H2S. Это особенно важно там, где H2S
входит в состав производственных газов, например, на
коксохимических заводах. Благодаря этому коксовые заводы,, прежде
завозившие серную кислоту для улавливания аммиака, теперь
сами обеспечивают себя кислотой и даже могут снабжать ею
другие заводы. Кислота получается очень дешевая.
При этом способе сероводород отличается достаточной
чистотой, процесс производства серной кислоты упрощается и сводпт-
141
ся к следующему. Газ, содержащий H2S D0—50%),
смешивается с воздухом и идет в печи, где H2S сгорает по реакции
2H2S + ЗО2 = 2Н2О + 2SO2.
Горячий газ из печи охлаждается в котле-утилизаторе и без
специальной очистки поступает в контактный аппарат с ванадиевой
массой, где SO2 превращается в SO3. После этого газ,
содержащий SO3 и пары воды, направляется в конденсатор, где
образуется серная кислота концентрацией 92—94%- В системе
получается до 0,7 т пара на 1 т H2SO4.
Олеум *а\
склад
Обхшеобый
еоз
Рис. 62. Схема производства серной кислоты контактным
методом:
1 — сухой электрофильтр; 2 — первая промывная башня; 3 —
вторая промывная башня; 4 — первый мокрый электрофильтр;
S — увлажнительная башня; 6 — второй мокрый электрофильтр;
7 — сушильная башня: 8 — брызгоуловнтель;
9—турбокомпрессор; 10 — теплообменник; // — коитактный аппарат; 12 —
абсорбер олеумный; 13 — абсорбер моногидратный; 14 — брыэгоулови-
тель; 15 — о"тстойннк; 16 — холодильники; 17— сборники.
Взаимодействие SO3 с парами воды приводит к образованию
тумана серной кислоты. Поэтому обычно часть серной кислоты
улавливается в скрубберах, а остальная — в электрофильтрах.
Серная кислота из серы. В настоящее время у нас
производят серную кислоту контактным методом из серы. При
этом если сера не содержит мышьяка и селена, можно получать
кислоту но значительно упрощенной, так называемой «короткой
схеме». Соответственно такой схеме печное отделение
упрощается, поскольку печи более просты и нет огарка: кроме того,
Отпадает необходимость в специальной очистке газа. Сера сжигается
в предварительно осушенном воздухе: печные газы, содержащие
SCb, проходят через котел-утилизатор, охлаждаются и
поступают в контактный аппарат.
Образовавшийся SO3 затем абсорбируется, как обычно,
образуя продукт-олеум, получение которого было описано ранее.
Олеум из отработанной серной кислоты.
Отработанная серная кислота получается в больших количествах
в цехах очистки нефтепродуктов, в процессах алкилирования
142
и др. Регенерация серной кислоты производится таким образом,,
что отходы, содержащие H2SO4, наряду с органическими
остатками, впрыскиваются в печь, где происходит расщепление
кислоты по уравнению
и сжигание органических примесей до СО и СО2. Газ при 800—
850° С уходит в печи дожигания, где СО превращается в СО2.
Затем газы идут через теплообменник, промывные башни,
электрофильтр, направляются в контактный аппарат и далее, как это
описано выше в случае производства олеума.
ГЛАВА X
ПРОИЗВОДСТВО СОДОВЫХ ПРОДУКТОВ
К содовым продуктам относятся кальцинированная сода
Na2CO3, каустическая вода NaOH, очищенный бикарбонат
(пищевая сода) NaHCO3H кристаллическая сода Ыа2СОз • 10Н2О.
Содовые продукты нашли широкое и разнообразное применение
в производстве стекла, мыла, в текстильной промышленности,
ПрОМЫШЛеННОСТИ СИНТеТИЧеСКИХ Материалов, ОСОбеННО ВИСКОЗНОГО'
шелка, в нефтяной, писчебумажной, лакокрасочной,
металлургической, пищевой промышленности, в медицине и др.; сода
широко используется в быту.
По данным ЦСУ СССР в 1967 г. в Советском Союзе
произведено 3169 тыс. т кальцинированной соды и 1525 тыс. г
каустической соды, что соответственно составляет 107 и 109% к
выпуску в 1966 г.
Согласно ГОСТ № 10689—63 и 2263—59, содовые продукты
выпускаются такого качества: в кальцинированной соде должна
быть не менее 95% Na2CO3, в твердом каустике не менее 92%
NaOH, а в пищевой соде не менее 98% NaHCO3.
Так как наибольшее значение для народного хозяйства имеет
кальцинированная сода, мы ознакомимся сначала с
производством этого продукта.
I. СЫРЬЕ И МЕТОДЫ ЕГО ПЕРЕРАБОТКИ
Сырьем для производства кальцинированной соды является
поваренная соль. Ее добывают в виде твердой, так называемой,
каменной соли, или в виде рассола. Меньшее значение имеет
природная сода, которая содержится в некоторых озерах
(например, Петуховские озера в Западной Сибири), сульфат натри*
и другие природные соли.
14»
Источники природного сульфата в СССР огромны.
Наибольшие запасы содержатся в заливе Каспийского моря Кара-Богаз-
Гол. Эти запасы составляют сотни миллионов тонн и могут
служить сырьем для производства соды.
Классический способ получения соды из сульфата был
изобретен еще в конце XVIII века Лебланом. По этому способу
производили соду до конца XIX века. Он заключался в том, что
смесь сульфата, известняка и угля прокаливалась при
температуре около 1000°С. Суммарная реакция следующая:
Na2SO4 + СаСОз + 2С = Na2CO3 + CaS + 2СО2.
Содовый плав выщелачивался водой; содовый раствор (щелок)
отделялся от нерастворимого CaS. При упаривании щелока
выпадал осадок Na2CO3-H2O, который прокаливанием
(кальцинацией) превращали в Na2CO3.
На опытном заводе в районе залива Кара-Богаз-Гол
получали соду по такому варианту: сульфат натрия восстанавливали
углем в печах:
Na2SO4 + 2С = Na2S + 2СО2.
Полученный сульфид Na2S растворялся, и раствор подвергался
карбонизации
Na2S + 2Н2О + 2СО2 = 2NaHCO3 + H2S.
I
Выпавший бикарбонат натрия отфильтровывали и
прокаливанием переводили в соду
2NaHCO3 = Na2CO3 + Н2О + СО2.
Из других возможностей получения щелочей из Na2SO4 следует
указать на опыты по электролизу раствора Na2SO4, в результате
которого образуются NaOH и H2SO4, а также по применению
аммиачного цикла переработки в соответствии с суммарной
реакцией
Na2SO4 + 2NHa + 2СО2 + 2Н2О = (NH4J SO4 + 2NaHCO3.
Пр;; этом одновременно с бикарбонатом натрия образуется
сульфат аммония, используемый как удобрение, а при
прокаливании выпавшего в осадок бикарбоната NaHCO3 получается
кальцинированная сода.
В настоящее время начали получать соду и поташ К2СО3
при переработке нефелинов (или нефелиновых отходов
глиноземных заводов), а также сиенитов, содержащих А12О3, SiO2,
Na2O, К2О и др. Эти руды имеются у нас в огромных
количествах. Нефелин, получаемый в виде отхода при обогащении путем
флотации апатитонефелиновых руд Кольского полуострова,
содержит около 40% SiO2, 30% А12О3 и около 15% Na2O и КгО.
Процесс переработки его заключается в том, что спеканием
144
с известняком получают спек, состоящий в основном из
растворимых алюминатов щелочей и нерастворимого силиката кальция:
(Na, КJО • А12О3 • 2SiO2 + 4СаСО3 = (Na, КJО • А12О3 +
+ 2BCaO-SiO2) + 4CO2.
Спек выщелачивают для перевода алюминатов в раствор
(остается нефелиновый шлам, из которого получают цемент).
Раствор алюминатов после обескремнивания идет на осаждение
гидроокиси глинозема карбонизацией:
(Na, КJО • А12О3 + СО2 + ЗН2О = (Na, К), СО3 + 2А1 (ОН),.
А1(ОНK отфильтровывается, промывается и прокаливается для
получения АЬОз:
2А1 (ОН)з = А12О3 + ЗН2О,
Из фильтрата (маточника), содержащего около 15% Na2CO3
и около 3—4% К2СО3 и K2SO4, выделяют эти соли. Для этого
фильтрат подвергают упариванию и кристаллизации, выделяя
из него карбонат натрия в виде Na2CO3-H2O и карбонат калия
в виде К2СО3 • і-дНгО. После обезвоживания получают
кальцинированную соду и поташ. Особенно примечательно то, что
комплексная переработка нефелинового сырья позволяет получать
А12О3, идущий на производство металлического алюминия,
портланд-цемента, соды и поташа. Потребность народного хозяйства
в глиноземе и цементе огромна, и получаемые при этом сода
и поташ смогут покрыть дефицит щелочей в стране. Из 4,3 т
нефелинового сырья и 7,5 т известняка можно получить 1 т
углекислых щелочей, 1 г АЬОз и 7,5 г цемента.
В СССР строится Ачинский комбинат, который указанным
способом будет производить соду, глинозем и цемент в больших
количествах. Основная масса соды в настоящее время
производится аммиачным методом, разработанным в середине XIX века.
2. АММИАЧНЫЙ СПОСОБ
Еще в 1811 г. во Франции был взят первый патент на
получение соды по реакции
NaCl + NH4HCO3^NaHCO3 + NH.C1.
I
Первый содовый завод по аммиачному способу построен в
Бельгии в 1868 г., а в России — в Березниках и Лисичанске (Донсо-
да) в 1890 году.
Аммиачный способ заключается в карбонизации аммиачного
раствора хлористого натрия в воде; эта основная реакция всего
процесса выражается следующим суммарным уравнением:
NaCl + Н2О + NH3 + СО2 з=* NaHCO3 + NH4C1.
10 8-1504 145
Упрощенная схема процесса, описываемого этим уравнением,
такова. Аммиак реагирует с водой и углекислым газом, образуя
бикарбонат аммония:
NH3 + Н2О = NH4OH; NH4OH + СО2 = NH4HCO3.
В результате реакции обмена при взаимодействии с поваренной
солью образуется бикарбонат натрия, растворимый менее других
солей и выпадающий поэтому в осадок:
NH4HCO3 + NaCl ї=± NaHCO3 + NH4Cl.
I
Эта реакция обратима, поэтому не весь NaCl вступает в реакцию.
Кроме того, бикарбонат натрия не полностью выпадает в
осадок, и поэтому не весь NaCl идет на получение соды.
Образующийся NH4C1 обрабатывают известковым молоком
для выделения из него NH3, вновь идущего в процесс, по реакции
2NH4C1 + Са (ОН), = СаС12 + 2NH3 + 2Н2О.
Необходимая для дистилляции суспензия гидрата окиси
кальция в воде (известковое молоко) образуется при гашении извести
избытком воды:
СаО + Н2О = Са (ОН),.
Негашеная известь СаО получается прокаливанием известняка
(или мела) в печах по реакции
СаСО3 = СаО + СО2,
Углекислый газ используется для карбонизации аммиачно-соле-
вого раствора.
Бикарбонат натрия NaHCO3 отфильтровывается и
прокаливается:
2NaHCO3 = Na2CO3 + СО2 + Н2О.
Образуется кальцинированная сода, а СО2 также используется
для карбонизации аммиачно-солевого раствора. Основную
реакцию изучал П. П. Федотьев, который в 1905 г. опубликовал свои
исследования по растворам, насыщенным солями:
NaCl, NH4HCO3, NaHCO3 и NH4C1.
Этим исследователем построена пространственная диаграмма
равновесия, используя которую можно определить оптимальные
усдопия процесса карбонизации и полможную степень
утилизации сырья (NaCl).
По П. П. Федотьеву, оптимум имеет место при 32° С, когда
f^Na — коэффициент использования натрия равен 84%; на
практике этот коэффициент при карбонизации близок к 70%, а с
учетом всех прочих потерь составляет 65—68%- Основываясь на
результатах ряда опытов, подтвердивших и уточнивших данные
П. П. Федотьева для различных условий, можно прийти к
выводу, что на практике следует придерживаться возможно большей
концентрации NaCl в растворе и СО2 в газе и проводить процесс
146
карбонизации при сравнительно низкой температуре —
около 30° С.
Процесс, схематически изображенный на рис. 63, включает
следующие операции: получение рассола, его очистка и подача
на абсорбцию; аммонизация очищенного рассола (абсорбция
NH3) и его охлаждение, карбонизация охлажденного
аммонизированного рассола двуокисью углерода, отделение выпавшего
Рис. 63. Схема производства кальцинированной соды:
/—скважина; 2 — смеситель рассола с известковым молоком и содоґі- 3 —
отстойник для выпавших в смесители 2 соединении магния и кальция 4— капор-
нып бак очищенного рассола; 5 — абсорбер. 6—7 — водяные холодильники для
аммонизированного рассола; « — карбонизационные колонны- 9 — вакуум-
фильтр; 10 — содовая печь для прокаливания бикарбоната 10' — топка- И
промыватель газа, уходящего из 10; 12, 15 — компрессоры' газов печей: 13 —
изьестковообжигательная печь; И— скруббер для промывки газа- 16 — 'бара-
баи для гашения извести и получения известкового молока; 17— днстнлля-
ционная установка для регенерации аммиака.
бикарбоната натрия на вакуум-фильтре и его промывка,
прокаливание (кальцинация) сырого бикарбоната, дистилляция
маточной жидкости для регенерации NH3 и получение СО2 при обжиге
известняка (мела) и прокаливании бикарбоната в содовой печи,
а также получение известкового молока.
Приготовление рассола
Рассол откачивается из буровых скважин или его готовят
нагнетанием воды через скважины в залежи соли. Обычно
рассол содержит 300—320 г/л NaCl и около 5—8 г/л примесей,
главным образом солей кальция и магния.
10*
147
Рассол подвергают сначала очистке для удаления ионов Са++
и Mg++, так как их соединения загрязняют соду и мешают
производству, образуя осадки в аппаратах. Очистка производится
содово-известковым способом, который применяется также для
умягчения котловой воды в энергетических установках. К
рассолу добавляют растворы Na2CO3 и Са(ОНJ; количество этих
растворов определяется расчетом в соответствии с данными
анализа рассола. Ионы кальция и магния осаждаются в
соответствии с реакциями
Mg++ + 2ОН- = Mg (OHJ |
Степень очистки по Са составляет 99—99,8%, а по Mg 98—99%.
Аммонизация рассола
Очищенный рассол направляют на абсорбцию аммиака, чтобы
получить раствор, содержащий 260—290 гіл NaCl и около 90 г\л
NH3. Насыщение аммиаком производится в несколько стадий
при прохождении рассола через ряд абсорберов, в которых он
встречается с газами, содержащими NH3 и СОг.
Сначала рассол в серии промывателей (промыватель газа
абсорбции, промыватель газа колонн и промыватель воздуха
фильтров) извлекает аммиак из газов, удаляемых на выхлоп
из указанных аппаратов. Затем рассол, содержащий в растворе
некоторое количество NH3 и СО2, поступает на основную
абсорбцию. Этот процесс происходит в двух абсорберах, где
поглощается свыше 80% аммиака из газов, полученных при
дистилляции; начальный состав этих газов 65% NH3 и 12% СО2 при55°С.
Необходимость проведения абсорбции в двух аппаратах
связана с экзотермичностью процессов абсорбции. Поэтому,
чтобы обеспечить растворение возможно большего количества
аммиака в процессе, следует охлаждать либо газ, либо рассол,
нагревавшиеся при абсорбции.
На рис. 64 представлена схема абсорбции современного
содового завода. В большой колонне расположены промыватель
воздуха фильтров 2, холодильник газа дистилляции 3, первый
абсорбер 4, второй абсорбер 5 и резервуар-постамент 6.
Очищенный рассол из напорного бака / идет двумя потоками
в аппаратуру. Большая часть (80%) проходит промыватель
воздуха фильтров 2, (куда снизу поступают содержащие NH3 газы
из вакумм-фильтра) и второй промыватель газа колонн 9.
Второй поток B0%) проходит промыватель газа аисириции 7.
Оба потока из 9 и 7, растворившие NH3 и СОг, поступают
в первый абсорбер 4. Отсюда раствор, охладившись в ороситель-
148
ном холодильнике <§, идет во второй абсорбер 5 и затем через
второй холодильник 10— в сборник аммонизированного
рассола //.
Газ из отделения дистилляции (NH3 и СО2) проходит (сверху
вниз) холодильник газа дистилляции 3, сепаратор 13,
абсорберы 5 и 4 и промыватель газа абсорбции 7, после чего в нем еще
остается СО2. Поэтому его через вакуум-насос 12 и сепаратор
направляют в промыватель газа содовых печей (сушилок).
Газы ш tfa/fyi/м
В промь/батель газа содобш I —-—-—
из первого лоомыда-
/77Є/7Я еоза колонн
¦*— Т водо AiHMOHt/3(/po6oHH6/J
рассол но нисбЬ/г
зоцш
Вода
Рис. 64. Схема абсорбции аммиака рассолом.
Газы, вышедшие из промывателей воздуха фильтров 2 и газа
колонн 9, выбрасываются в атмосферу.
В оросительном холодильнике 10 рассол охлаждается до 28—
30° и из сборника // поступает на карбонизацию. Почти все
аппараты абсорбции представляют собой чугунные тарельчатые
колонны, в них газы движутся снизу вверх, а жидкости сверху
вниз. На рис. 65 показан чертеж первого и второго абсорбера.
Каждый состоит из семи царг (бочек), между которыми
установлены барботажные тарелки с колпачками (в первом—пять, во
втором —три тарелки). На рис. 66 показан разрез холодильника
газа дистилляции, который заполнен холодильными элементами;
в нем газ, охлаждаясь водой, протекающей по трубам, идет
сверху вниз.
149
Рис. 65. Первый и второй
абсорберы:
1 — первый абсорбер; 2 — второй
абсорбер; 3 — перелив, 4 —
колпачок; 5 =» лаз.
Рис. 66. Холодильник газа
дистилляции:
/ — пустая бочка (царга); //—VI —
холодильные бочки (царги);
I — штуцер для ввода газа; 1 — штуцер
для вывода газа; 3 — штуцер для вывода
конденсата; 4 — ввод воды в
холодильники; 5 — вывод воды из холодильников;
в — трубная решетка холодильника; 7 —
чугунная холодильная трубка; 8 — крышга
холодильной бочки; 9 — лаз.
150
На рис. 67 показано движение потоков на тарелке с
колпачком.
г
Жидкость
Рис. 67. Схема поглотительного
устройства:
I — фасонное днище; 2 — отверстие длч
прохода газа; 3— колпачок (колокол);
4 — переливная труба.
Карбонизация
Аммонизированный рассол
насыщается СО2 под давлением около 2,5 атм
в карбонизационной колонне. На рис.
68 дан разрез большой
карбонизационной колонны D = 2,68 м, Н = 26,1 м,
производительностью 260 т соды в
сутки.
Колонна состоит из ряда чугунных
царг (бочек). В нижней части колонны
установлено десять холодильных
бочек, в которые вмонтированы водяные
холодильники. Между каждыми двумя
холодильниками вставляется барбо-
тажная тарелка. Барботажные тарелки
помещены также в верхней половине
колонны, всего их в колонне 36.
В верхней части колонны имеются две пустые бочки для
улавливания брызг. Суспензия бикарбоната вытекает из нижней
бочки. Газ подается в колонну снизу через два штуцера. В нижнюю
бочку входит газ из отделения содовых печей, смешанный с газом
Рис. 68.
Карбонизационная колонна:
/ — сепарационные бочки;
2 — штуцера для выхода
газа в промыватель газа
колонн; 3 — абсорбционная
бочка; 4 — штуцера для
входа аммонизированного
рассола; 5 — барботажиая
тарелка; 6 — холодильная
бочка; 7 — бочка-база; 8 —
штуцер для входа газа; 9 —
штуцер для выхода
суспензии на фильтр.
151
известковых печей,— смесь (I ввод газа) содержит около 60%
СО2; немного выше подается газ известковых печей A1 ввод
газа), содержащий около 35% СО2.
Реакции, происходящие при движении аммонизированного
рассола сверху вниз и газа снизу вверх, могут быть
представлены как монокарбонизация, бикарбонизация и двойной обмен
следующими уравнениями:
2NH3 + Н2О + СО2 = (NH4J СОз,
(NH4JCO3 + СО2 + Н2О = 2NH4HCO3)
NH4HCO3 + NaCl = NH4C1 + NaHCO3.
I
Когда концентрация NaHCO3 в растворе сравняется с его
растворимостью, начнется выделение в осадок кристаллов
NaHCO3. Процессы, происходящие в карбонизационной колонне,
являются экзотермическими. Поэтому для полного протекания
указанных реакций выделяющуюся теплоту отводят в
холодильниках колонны.
Так как ход последующего фильтрования зависит от
размеров кристаллов, чтобы облегчить процесс фильтрования и
промывку осадка бикарбоната, стремятся получить не очень мелкие
его кристаллы.
Первую фазу образования кристаллов — зарождение центров
кристаллизации — ведут при температуре около 62° С, так как
при более низкой температуре образуется большое количество
центров, могущих дать слишком мелкие кристаллы. Зарождение
кристаллов происходит в средней части колонны. Поэтому
отсюда отводят тепло, чтобы смесь не нагревалась выше этой
«критической» температуры. Чтобы обеспечить вторую фазу — рост
кристаллов, следует сильнее охладить раствор, чтобы
вытекающая из колонны жидкость имела температуру 28—30° С. Большая
часть кристаллов выводится из колонн на фильтр, но часть
оседает в ней, так что после нескольких суток колонна засоряется
ими. Прежде ее останавливали и подвергали «пропарке»
горячей водой и паром. В настоящее время процесс ведут, не
прекращая работу колонны. Это осуществляется следующим
образом.
Аммонизированный рассол пускают сначала в
«засорившуюся» колонну. В последнюю подают СО2 в таком количестве,
чтобы бикарбонат еще не осаждался (около 50% всего СО2, нужного
для полной карбонизации). При этом застрявшие кристаллы
бикарбоната переходят в раствор и колонна очищается от осадка.
Процесс в промывной колонне называется форкарбонизацией.
После промывки колонну вновь используют как осадительную,
а на промывку ставят ту, которая работала как осадительная..
Переключая таким образом колонны, мы очищаем их от осадка.
152
Раствор, частично насыщенный СО2, при форкарбонизации
перетекает в осадительную колонну.
При работе колонны часть аммиака уносится потоком ССЬ,
который подается для абсорбции NH3 в промыватель газа
колонн. Суспензия бикарбоната уходит из низа колонны на
вакуум-фильтры, где сырой бикарбонат отфильтровывается и
промывается. Стекающий фильтрат проходит сепаратор для
отделения газа (воздуха фильтров); промытый бикарбонат
направляется в содовую печь для кальцинации, а маточная жидкость —
на регенерацию аммиака (дистилляция).
Кальцинация
Содовая печь-сушилка для кальцинации сырого бикарбоната
(рис. 69)—представляет собой вращающийся горизонтальный
Рис. 69. Схема содовой печи:
/ — вакуум-фильтр сырого бикарбоната; 2 — транспортер бикарбоната- 3 — приемник
с мешалкой; 4 — питатель; 5 — смеситель сырого бикарбоната и ретурной соды- б —
шнековые транспортеры; 7 — барабан печи; 8 — выгрузной шнек кальцинированной
соды; а, 10, 12, 13 — шнеки; 11 — элеватор; 14— бункер кальцинированной соды-
/5 —топка; /6 —газоход; П — боров топочных газов; /« — циклон- 19 — коллектор
газа содовых печей; 20 — сборник слабой жидкости; 21 — холодильник газа содовых
нечей; 22 — промыватель газа содовых печей (скруббер); 23 — цепь для разбиванин
комьев; 24— конвейер для подачи мешков кальцинированной соды на склад.
барабан, через один конец которого входит сырой бикарбонат
с влажностью 15—18%. Здесь он сушится и разлагается по
реакции
2NaHCO3 = Na2CO3 + СО2
Н2О.
2 2
В печи происходит также разложение неотмытых аммонийных
солей по реакциям
NaHCO3 + NH4C] = NH4HCO3 + NaCl,
NH4HCO3 = NH, + CO2 + H2O.
NaCl, которая образуется по первой реакции, остается в соде
153
її ухудшает качество продукта, так как процент Na2CO3
уменьшается (ретроградация соды). Чтобы избежать образования
комьев в печи, нужно уменьшить содержание влаги в сыром
бикарбонате до 8—10%. Этого можно достичь, применяя вместо
вакуум-фильтров центрифуги. На большинстве заводов для
уменьшения концентрации влаги к сырому бикарбонату
добавляют кальцинированную соду, получаемую из печи (ретурная
сода) в количестве, примерно равном весу сырого бикарбоната.
Для разбивания комьев в печь помещают закрепленную на одном
конце тяжелую цепь.
В последнее время применяют подачу сырого бикарбоната
в печь без ретурной соды, распыляя метательным аппаратом
сырой бикарбонат таким образом, чтобы он успел частично
высохнуть в печи «на лету»; печь делает 5 об/мин и
обогревается топочными газами. Температура отходящих из печи
газов — около 250° С.
Выпускаемая сода должна содержать 95—98% Na2CO3,
NaCl от 0,5 до 1%, нерастворимых веществ не более 0,3%.
Потеря б весе при прокаливании составляет от 2,5 до 3,5%, в
зависимости от сорта выпускаемой соды.
Переработка газа содовых печей.
Выделяющиеся из содовой печи газы СО2 и МНз и пары воды, выходя из
передней части сушилки, содержат до 70 кг содовой пыли и до
40 кг СО2 на тонну соды. На рис. 69 дана схема переработки
печного газа. Для отделения содовой пыли газ проходит
циклон 18, а затем холодильник 21 и скруббер 22, где происходит
конденсация паров воды и удаление аммиака в виде так
называемой «слабой жидкости»; она собирается в сборник, а затем
подвергают дистилляции для регенерации 1МНз. Из скруббера
сушилок газ с температурой около 30° С, содержащий около
90% СО2, подается в компрессор. После смешения с газом
известковых печей полученный газ с содержанием 60% СО2
поступает в карбонизационные колонны.
Дистилляция
Маточная жидкость направляется из дистилляцию (реіене-
рация аммиака). В ней содержится I) растворенный аммиак
б виде NII4OH, 2) милусьяааиный аммиак (і\П4JСО3, і\їЦІІСО3;
если в цикл вводят 1\ГНз из коксовых установок, то в нем может
быть также (NH4JS; 3) связанный аммиак, главным образом
в виде NH4C1. Так как соединения «полусвязанного» аммиака
диссоциируют при нагревании, например
(NH4JCO3 = 2NH3 + СО2 + Н2О,
а соединения связанного аммиака разлагаются только при
нагревании с гашеной известью по реакции
2NH4C1 + Са (ОН), = 2NH3 + 2Н2О + СаС12>
154
**>*'**«>»"*>
то отгонку аммиака проводят в две стадии: в первой жидкость
подвергают только тепловой обработке, а во второй — тепловой
обработке после добавления к ней известкового молока.
Нагревание маточной (фильтровой) жидкости и охлаждение
газа осуществляют в
теплообменниках. На рис. 70 представлена схема
ДИСТИЛЛЯЦИОННОЙ КОЛОННЫ.
Фильтровая жидкость поступает
на верх конденсатора дистилляции
/, где она нагревается газом,
идущим снизу. Часть аммонийных солей
диссоциирует здесь, затем жидкость
проходит теплообменник
дистилляции 2, где в основном
заканчивается диссоциация разлагаемых при
нагревании аммонийных солей \,
наконец, перетекает в основной
аппарат дистилляции—дистиллер 3,
предварительно смешавшись с
известковым молоком в смесителе (на
рис. не показан).
Из смесителя суспензия
непрерывно входит в дистиллер, где
осуществляется отгонка аммиака
острым паром. Дистиллер снабжен
мешалкой. Сверху уходит аммиак
в смеси с СОг и водяным паром в
аппараты 2 и /, а снизу—раствор
СаСЬ и NaCl со взвешенным
избытком извести,, углекислого
кальция и примесями; этот отход,
пройдя расширитель, где выделяется
некоторое количество пара, удаляется
с завода на поля отбросов («белое
море»).
Все три основные аппарата дис-
стилляции смонтированы в одной
колонне, состоящей из отдельных
бочек, в которые встроены колпач-
ковые устройства (дистиллер) и насадка из колец
(теплообменник дистилляции) или трубчатки (конденсатор дистилляции).
Основная масса аммиака возвращается в цикл. Потери его
не велики и составляют (на лучших по технологическим
показателям заводах) около 3 кг NH3 на тонну соды A2 кг 25%-ной
аммиачной воды). Аммиачная вода вводится в сборник
фильтровой жидкости.
Рис. 70. Дистилляцион-
ная колонна.
155
3. ПРОИЗВОДСТВО ИЗВЕСТИ И УГЛЕКИСЛОГО ГАЗА
Сырьем для получения извести является известняк или мел.
Они содержат 80—90% СаСОз и более, а также ряд примесей.
Известняк отличается обычно меньшим содержанием влаги
и большей механической прочностью, чем мел. Нежелательными
примесями являются глина, окись железа, кремнезем и вода.
Они вредны тем, что их обогрев и побочные реакции повышают
расход топлива. Кроме того,
при высокой температуре
разложения известняка в печах
A200° С) образуются
легкоплавкие силикаты кальция и
железа из примесей и золы
топлива; образоваівшись в гс-
рячей зоне печи, силикаты
оплавляют поверхность
опускающейся шихты. По іиере
опускания расплавленные
силикаты затвердевают и цементи-
I /т
| 1200
X юоо
$ МО
200
о
600 650 700 7Х 600 650 900 950
Рис. 71. Зависи-мость
давления СОг при разложении
углекислого кальция от
температуры.
СаСОз = СаО -
руют куски шихты, образуя
иногда в печи глыбу («козел»),
нарушающую ход обжига и
вызывающую остановку печи.
Обжиг идет по реакции
СО2—177,9 кдж.
Необходимое тепло получается в результате горения
топлива — обычно кокса или угля.
Процесс обжига СаСОз осуществляется в системе,
содержащей три фазы ф(СаСОз, СаО и СО2) при двух независимых
компонентах п(СаСО3 и СО2, например); следовательно, по правилу
фаз число степеней свободы С этой системы равно единице:
С = п + 2 —9 = 2 + 2 —3=1.
Система моновариантна. Таким образом, каждой температуре
соответствует определенная упругость углекислого газа.
Графическое изображение зависимости упругости СО2 от температуры
дано на рис. 71, откуда видно, что при температуре около 900° С
упругость СО2 достигает одной атмосферы. Для ускорения
процесса в зоне диссоциации поддерживают температуру 1200° С.
При минимальном, теоретически необходимом расходе
углерода максимальная концентрация СО2 в газе составляет 46,3%,
но на практике из-за перерасхода топлива и избытка воздуха
содержание СО2 в печном газе не превышает 35—38%.
Печи могут отапливаться твердым, жидким или газообразным
топливом. Печь, в которой происходит обжиг известняка (мела)
с применением твердого топлива, изображена на рис. 72. Шахта
156
печи покоится на металлических колоннах. На поде печи
укреплено разгрузочное устройство для извести, имеющее форму
спиралевидной ступенчатой круговой вращающейся плиты и
называемое «улитой». В верхней части шахты расположено
загрузочное устройство. Число оборотов «улиты» достигает 1 об/ч.
Улита, подобно винту, ввертывается в лежащий на ней слой,
который выдавливается вниз
и уходит из печи. Шахта
печи имеет
цилиндрическую форму. Наружная
обмуровка состоит из
обычного красного
кирпича с шамотной
футеровкой. Между наружной
обмуровкой и футеровкой
оставляют зазор, который
заполняют1
теплоизоляционной прослойкой. Зазор
позволяет шамотному
кирпичу расширяться при
нагревании. На
кирпичной обмуровке печи
устанавливается загрузочное
устройство.
На современных
содовых заводах
распространены печи диаметром
4,5 м с еьисотой шахты
18 л и общей высотой
печи (до обслуживающей
верхней площадки) 30—
31 м. Строятся и более Рис 72. Схема
мощные печи диаметром
6,2 м.
Чем выше печь, тем
дольше находится в ней
материал, что позволяет применять известняк в виде более
крупных кусков (крупностью до 150 мм). Кроме того, благодаря
лучшему использованию тепла, в более высоких печах удается
понизить расход топлива на обжиг шихты. Топливо подается в
количестве около 75 кг условного G00 ккал на 1 г известняка);
степень обжига известняка доходит до 92—95%.
Газы уходят сверху с температурой около 150° С, а известь
выгружается снизу с температурой около 80° С.
Производительность печи составляет 100—150 т известняка в сутки, в
зависимости от диаметра печи, считая на 85%-ное сырье.
Получаемая в таких печах известь обычно загрязнеиа золой
известково-обжигательной
печи:
I—шахта печи; 2—загрузочная воронка; 3 —
распределительный конус; 4 — разгрузочное
устройство («улита>); 5 — труба для отвода газа;
6—труба для подачн воздуха.
157
топлива. Только при использовании жидкого или еще лучше
газообразного топлива (природный или генераторный газ) можно
получить чистую известь. Однако, применяя твердое зольное
топливо, можно получать чистую известь в шахтных печах с
выносными топками, в которых топочные газы проходят шахту и
нагревают известняк; зола топлива при этом остается вне печи.
В этих печах расход топлива выше, чем в печах со смешанной
шихтой и составляет 12—13% веса карбонатного сырья. Обжиг
мела и известняка в виде мелочи производится в
горизонтальных вращающихся печах. В последнее время стали применять
печи с обжигом в «кипящем» слое.
Печной газ очищают от пыли и других примесей (SO2)
и охлаждают до 30—40° С, промывая водой в скрубберах, и
подают в компрессоры, откуда он направляется в
карбонизационные колонны. Выгружаемая из печи негашеная известь содержит
около 85% СаО и примеси (СаСО3, SiO2 и др.); она поступает
на гашение водой для получения известкового молока в
горизонтальный вращающийся барабан (гаситель); при этом происходит
реакция
СаО -f Н2О = Са (ОНJ + 21,6 кдж.
Для очистки от взвешенных примесей известковое молоко
пропускают через сита, на которых задерживаются куски
необожженного известняка (недопал), куски пережженного известняка
(перепал), песок, зола и др. В 1 л известкового молока для
отгонки NH3 содержится около 350 г СаО.
Расходные коэффициенты на 1 т кальцинированной соды
в среднем составляют: очищенного рассола C10 г/л NaCl) 5 м3;
аммиачной воды B5% NH3) 15—17 кг, известняка или мела
A00% СаСОз), 1,3—1,4 г; воды 80—120 м3; мазута 7000 ккал
ПО—125 кг; пара 1,35—1,45 мгкал, электроэнергии 75—
100 квт-ч.
Следует обратить внимание на отрицательные стороны
процесса. Расход сырья и энергии в производстве соды аммиачным
способом велик; при этом следует учесть, что сырье используется
меньше чем на 50%, так как весь хлор уходит в отбросы в вине
СаСіг, а натрии используется на 65—68%. Поэтому исследуются
иные способы производства соды из Na3SO.,. например, а также
способы получения новых продуктов (стр. 163).
Для удешевления продукта и повышения
производительности труда производится предварительная очистка рассола, фор-
карбопизация, центрифугирование сырого бикарбоната,
исключение из процесса ретурной соды и др.
В настоящее время все содовые заводы обильно оснащены
автоматами контроля и регулирования, что привело к снижению
расходных коэффициентов по сырью, пару и энергии и
повышению производительности труда.
158
4. ПРОИЗВОДСТВО КАУСТИЧЕСКОЙ СОДЫ
Едкий натр получают тремя способами: известковым, феррит-
ным и электролитическим. Эти способы отличаются друг от
друга первой стадией производства — методами получения щелоков
(растворов) едкого натра. Обработка таких растворов для
получения из них твердого едкого натра или концентрированного
каустика во всех способах осуществляется одинаково —
выпариванием.
Известковый способ является наиболее
распространенным. Основная реакция процесса происходит между раствором
соды в воде и известковым молоком по уравнению каустифи-
кации
Na2CO3 + Са (ОНJ = СаСО3 + 2NaOH.
При этом получается раствор едкого натра и осадок
углекислого кальция, который обычно не утилизируется. Константа
реакции может быть представлена выражением
К
Д ~~ [Са (ОНJ] fNa2CO3J "
Поскольку СаСО3 и Са(ОНJ находятся в твердой фазе, их
концентрации в растворе можно считать постоянными. Введя эти
концентрации в выражение для константы, и, заменив
К = -[СаСОз] на Ki, получим
к _ [NaOH]2
Лі [Na2CO3]'
Но NaOH и Na2CO3 диссоциированы на ионы, и поэтому
константу реакции можно выразить так:
[ОН"]2
К' =
-]¦
Из этого выражения видно, что уменьшение концентрации
соды (СО3—), повышает константу равновесия. Поэтому, чтобы
достичь большого выхода едкого натра, применяют
неконцентрированные растворы соды, содержащие 12—15% Na2CO3.
Получение раствора NaOH по этому способу сводится к следующим
операциям: приготовление содового раствора и суспензии
известкового молока, каустификации содового раствора, отделение
и промывка осадков СаСО3, получение «слабого» щелока NaOH,
концентрирование слабого щелока выпариванием.
Для наиболее полного использования соды и извести каусти-
фикацию проводят в несколько ступеней в каустификаторах, где
смесь при помешивании подогревается до 80—90° С.
Достигаемая степень каустификации 92—95%. Образующийся осадок
СаСОз промывают в непрерывно действующих многоярусных
15Э
отстойниках так, чтобы потери NaOH были минимальными; для
этого все промывные воды возвращают в процесс. Промытый
осадок удаляется и не используется в производстве, хотя из него
можно регенерировать СаО и СО2.
Содовый раствор для каустификации часто готовят не
растворением кальцинированной соды, а разложением (мокрая
декарбонизация) сырого бикарбоната, полученного при
карбонизации в процессе получения соды. Декарбонизация
производится в тарельчатых колоннах (декарбонаторах) при 120—150° С.
ҐолочюіО еоз
Рис. 73. Схема производства каустической соды известковым
способом.
Сверху подают суспензию сырого бикарбоната, а снизу — пар;
при этом происходит реакция
2NaHCO3 = Na2CO3 + СО2 + Н2О.
Из декарбонатора вытекает раствор соды нужной
концентрации. Как видно из схемы производства NaOH (рис. 73),
нормальный содовый раствор для каустификации с содержанием 100 г/л
Na2CO3 готовится в смесителе /, где в определенном
соотношении смешиваются три жидкости: а—раствор соды из
декарбонатора; б— промывные воды; в — раствор, полученный из солей
выпарки, осаждающихся в выпарных аппаратах. Через напорный
бак 2 нормальный содовый раствор подается в гаситель — каус-
тификатор 4, куда поступает известь из бункера 3. Здесь
происходит гашение извести с образованием известкового молока и
частичная каустификяция соды. Примеси извести удаляются
в отвал, а полученная суспензия (пульпа) поступает через
сборник 5 в первый каустификатор 6, откуда прореагировавшая
суспензия для отделения выпавшего осадка СаСОз подается в
отстойник 7. Осветленная в 7 жидкость направляется в резервуар
«слабого» щелока 12 для подачи на выпарку, а осадок поступает
на вторую каустификацию в сосуд с мешалкой 8, куда из 11
подаются промывные воды и содовый раствор из а. Выходящая
из второго каустификатора 8 суспензия попадает в многоярусный
сгуститель 9, откуда промывные воды через сборник 10 и
отстойник 11 возвращаются в цикл (через 8, 17, в).
Осадок СаСО3 идет в отвал. Щелок из резервуара 12 через
F0
подогреватель ІЗ поступает на выпарку — сначала в трехкорпус-
ную вакуум-выпарную систему 14. Здесь щелок упаривается,
проходя через три корпуса; при этом выпадают осадки,
содержащие соду. Они отделяются в отстойнике 15 и на вакуум-фильтре
16, а затем направляются в растворитель 17, откуда раствор
подается в смеситель /. После отделения выпавших солей щелока
через подогреватель 18 направляются в однокорпусный
выпарной аппарат 19, откуда раствор концентрацией около 650 г/л
NaOH может быть отправлен на склад как готовый продукт
(жидкий каустик) или на обезвоживание (плавку) в котлы 20,
обогреваемые топочными газами.
Выпаривание растворов, содержащих более 82% NaOH, под
давлением ниже 0,5 ата затруднительно, так как NaOH при
этом затвердевает; поэтому обезвоживание проводят под
атмосферным давлением, обычно в чугунных (с присадкой 1—2% Ni)
котлах, вмурованных в кирпичную кладку печи, где они
обогреваются горячими топочными газами. Обезвоженный каустик
получается в котле в виде расплава при 500—600° С. Примеси,
главным образом гидраты А1(ОНK и Fe(OHK, оседают, а
чистый 92—94%-ный продукт и NaOH откачивается насосом в
железные барабаны, в которых он затвердевает.
Остановимся на аппаратах последней
стадии—обезвоживания (плавки). Так как при непосредственном обогреве пламенем
толстого чугунного котла часто образуются трещины, он редко
выдерживает больше 30—35 плавок. Поэтому применяют также
другие аппараты и методы нагревания. Вместо котлов
(горшков), обогреваемых голым огнем, используют аппараты,
обогреваемые горячей водой, перегретым паром, высококипящими
органическими теплоносителями (ВОТ), как, например, дифенил,
дифенилоксид, минеральные масла. Применяются также вакуум-
аппараты, причем теплоноситель проходит через стальные
трубки, утопленные в чугунных стенках аппарата. Плавка под
вакуумом происходит при 320—330° С, но каустик получается хуже
качеством, так как он не отстаивается. Плавку NaOH
производят также в аппаратах с электрическими обогревательными
элементами.
Кроме того, применяют аппараты для ведения непрерывного
процесса плавки в виде колонок, обогреваемых топочными
газами. Расходные коэффициенты на 1 т 92°/о-ного каустика
следующие: соды(95%)—1,4 г; извести (85% СаО) — 1,6 т, топлива
G000 кал) — 1,7 г; электроэнергии 50—55 кет • ч, причем 1,2 т
топлива расходуется на выпаривание щелоков, а 0,5 т — на
плавку в горшках.
Ферритный способ (способ Левига) основан на
образовании из соды и окиси железа (железной руды) при 1000° мета-
феррита натрия:
Na2CO3 + Fe2O3 = 2NaFeO2 + СО2 — 142,5 кдж.
U 3-1504 161
При разложении феррита водой образуется раствор NaOH и
регенерируется окись железа, возвращаемая в цикл:
2NaFeO2 + Н2О = 2NaOH + Fe2O3.
Основной аппарат — вращающаяся печь (тамбур) имеет уклон
в 3—5° и делает два-три оборота в минуту, благодаря чему смесь
кальцинированной соды и окиси железа, подаваемая через
приподнятый торец, куда вдувается топливо с воздухом. При этом
смесь сушится и прокаливается.
Степень каустификации, т. е. % Na2CO3, перешедшего
в NaOH, составляет 90—92%. По выходе из печи феррит
охлаждается водой («гасится») и отправляется в серию из
восьмидесяти цилиндрических диффузоров, сюда же подается вода для
разложения.
Диффузор представляет собой чугунный цилиндр диаметром
1500 мм и высотой 4600 мм с наклонной чугунной решеткой. Его
заполняют ферритом сверху при закрытой нижней дверце,
служащей для выгрузки окиси. Серия таких аппаратов A0—12)
составляет диффузионную батарею, работающую по принципу
противотока. Отбираемый из диффузионной батареи щелок
содержит до 350—420 г NaOH и около 30 г Na2CO3 в литре.
Во время работы в окиси железа накапливаются примеси,
и когда количество их доходит до 15—20%, окись выводят из
цикла и заменяют свежей рудой. Ферритный метод получения
NaOH в настоящее время применяется редко. Расходные
коэффициенты на одну тонну 92% -ного каустика составляют: соды
(95%) 1,4 т, окиси железа 30—40 кг, топлива G000 кал) 0,4 т,
электроэнергии 70 кет • ч.
5. ДРУГИЕ ПРОДУКТЫ СОДОВОГО ПРОИЗВОДСТВА
Бикарбонат натрия (пищевая сода) представляет собой
чистый продукт, широко применяемый в фармацевтической и
пищевой промышленности. Для его производства готовят раствор
кальцинированной соды концентрацией около 240 г\л NasCO3.
После очистки раствора отстаиванием его карбонизируют в
колоннах очищенным газом известково-обжигятельных пстеґг; пріг
этом выпадают чистые кристаллы бикарбоната:
NaaCO8 і- Н„О -f СО2 - 2NaIICOa.
1
Кристаллы центрифугируют и сушат в барабанных сушилках
при 70—80° С.
Из побочных веществ содового производства остановимся на
нашатыре, карбонатах аммония и хлористом кальцие.
Для получения нашатыря используют жидкость
карбонизационных колонн содового завода после выделения из нее сырого
бикарбоната; ее донасыщают газообразными NH3 и СО2, а также
молотой поваренной солью и выделяют кристаллы NH4C1 охла-
162
ждением до —10 С. Кристаллы отделяют на центрифугах,
промывают и сушат горячим воздухом при 120—140° С. Продукт,
содержащий 97—99% NH4C1, выпускают в виде брикетов или
порошка.
Стекающая маточная жидкость после отделения кристаллов
нашатыря может возвращаться в цикл содового завода. NH4Cl
применяется при пайке металлов, цинковании, лужении, в
текстильной промышленности, гальванотехнике. NH4CI может
использоваться и как удобрение.
Смесь кристаллов NH4CI + КС1 получают, подвергая аммо-
низации и последующей карбонизации растворы, содержащие
КС1 и образующиеся на сильвинитовых фабриках после
отделения КС1; NH4C1 + КСЛ могут применяться как азотно-калиевые
удобрения B0% К2О + 14% N2).
В последнее время NH4C1 начинает перерабатываться на
чистые хлор и хлористый водород, которые находят все большее
применение. Этими мероприятиями по превращению NH4C1
в нужные пртукты повышается степень использования сырья —
NaCl (стр. 146) в аммиачном способе.
Кроме того, уменьшается количество весьма
обременительных отходов содового производства, так как меньше получается
отбросного хлористого кальция при дистилляции. Нерегенери-
рованное количество аммиака возмещается покупным
аммиаком, стоимость которого непрерывно снижается.
Из газов после дистилляции, содержащих NH3, CO2 и пары
воды, можно охлаждением выделять кристаллы карбоната и
бикарбоната аммония, используемые в хлебопечении, при
крашении, для нейтрализации растворов и др.
В отбросной жидкости дистилляции содержатся в
значительных количествах в растворенном виде СаСіг и NaCl, а также-
осадки. После удаления осадка раствор подвергают
двухкратному упариванию так, чтобы удалить сначала выпадающий
NaCl. После промывки NaCl, его можно использовать. Затем
раствор упаривают до содержания 65—70% СаСЬ и сливают
в железные барабаны, где он застывает.
СаС12 применяют в строительстве при затворении цемента
зимой, для обеспыливания дорог, сушки газов, в производстве
металлического кальция, хлористого бария и др.
ГЛАВА XI
ТЕХНОЛОГИЯ ХИМИЧЕСКИХ ПРОДУКТОВ
ДЛЯ СЕЛЬСКОГО ХОЗЯЙСТВА
К продуктам химической промышленности, используемым
в сельском хозяйстве, относятся вещества разнообразного
применения: удобрения, ядохимикаты для уничтожения вредителей'
11* 163
в сельском хозяйстве, а также ряд других продуктов:
дефолианты для уничтожения листвы перед снятием урожая машинами,
химические консервирующие вещества, вещества, повышающие
питательность кормов и т. д. Наиболее важными из них являются
удобрения.
I. УДОБРЕНИЯ
Значение удобрений для сельского хозяйства очень велико.
Они должны восполнить недостаток полезных элементов в
почве. Для интенсификации земледелия в почву вносят естественные
(торф, навоз и др.) и искусственные минеральные удобрения,
главным образом азотные, фосфорные и калийные, а также
микроудобрения, которые применяются в малых количествах,
например бораты, соли меди и др.
По сообщению ЦСУ СССР, в 1967 г. произведено 40,1 млн. т
(в условных единицах) искусственных удобрений, что
составляет 112% к 1966 году. Согласно Директивам XXIII съезда КПСС
по пятилетнему плану развития народного хозяйства СССР на
1966—1970 гг., предстоит довести производство минеральных
удобрений в 1970 г. до 70—80 млн. т, в том числе азотных
удобрении до 30,2, фосфорных до 26,7, калийных до 15,5,
фосфоритной муки до 7,4 и борных до 0,2 млн. т. Кроме того, для
известкования кислых почв нужно производить около 20 млн. т извести
в год.
Если удобрение содержит один полезный элемент, оно
называется простым или однокомпонентним, два — двойным,
двусторонним, и более — тройным, полным,
многокомпонентным, комплексным. Удобрения оцениваются по концентрации
в них полезных веществ Р2О5, N2 и КаО. В зависимости от того,
входят ли компоненты в одну молекулу или в несколько,
удобрения называются сложными, например KNO3 или
смешанными, например КС1 + (NH4JSO4.
Таблица 19
Название
Суперфосфат
Двойной суперфосфат .
Преципитат
Томасшлак
Термофосфат
Фосфоритная мука . . .
Костяная зола
Основные составляющие
Са(Н2РО4J, CaSO4
Са(Н2РО4)а
СаНРО4
4СаО-Р,аО5 и силикаты Са, Mg
2CaO-NaaO-P2O5, CaSiOg
Саа(РО4),
Общее
содержание Р2О5,%
14-20
45-52
30—36
14—18
24—32
18-20
36—39
164
1. Фосфорные удобрения
Эти удобрения вошли в практику еще в первой половине
XIX века. Фосфорные удобрения (табл. 19) бывают нескольких
видов.
Кроме этих удобрений, производятся еще фосфаты аммония
(амнофосы), калия, натрия и кальция. Отметим, что фосфаты,
фосфорная кислота и фосфор широко применяются в других
областях народного хозяйства (рис. 74).
Зажигательные
срдтв
ажигател
средства
в лооаздодслгів
спичек
в органическом^
синтезе
в производстве
спичек
Корм для
скота Добавкд к
Средства>заме-
няющие —-
- Для рафинирования
, сахара
обезвоживания
Замедлители
/антилиремы)
Зубные
порош/си
фсчлвдитт
9 фотографии
Удвбремия
медикаменты
Удобрения
ныла
натрия
моющие ¦
средства
Ул>/гже/?итела I Средства .заменяющие
шелка ¦ ---¦'¦
для очистки
воды
Рис. 74. Применение фосфора и его соединений.
Сырье
Сырьем для производства фосфорных удобрений являются
природные фосфаты — фосфориты и апатиты,
состоящие в основном из трехкальциевого ортофосфата Са3(РО4J.
Крупные залежи фосфоритов находятся в СССР (Актю-
бинское, Егорьевское, Вятское, Воскресенское, Кара-Таусское
и др.), а также в Северной Африке, США и других странах.
Залежи фосфоритов бывают осадочного или органического
происхождения. Примесями фосфоритов являются кальциты
СаСОз, доломиты CaCO3«MgCO3, силикаты и другие минералы.
16»
В процессе обогащения эти примеси следует удалять; наиболее
вредными окислами являются полуторные R2O3 (А12О3 и Fe2O3),
затрудняющие переработку сырья в удобрение.
Большое значение имеют фторапатиты 3Ca3(PO4J* CaF2
или сокращенно Ca5F(PO4K- Они являются изверженными
породами и имеют кристаллическое строение. Огромные количества
фторапатитов залегают на Кольском полуострове вместе с
нефелином (щелочной алюмосиликат[(№, КЬО • А12Оз • 2SiO2] • rtSiO2)
и другими минералами. Апатиты стали применяться у нас
с 1926 г., когда под руководством С. М. Кирова была
организована добыча руды и ее обогащение на флотационной фабрике
в г. Кировске.
Большое значение имеет комплексность этих руд, дающая
возможность получать важные для народного хозяйства
вещества. Для разделения апатито-нефелиновой руды применяется
флотация. При этом получается апатитовый концентрат,
содержащий примерно 39,5% Р2О5, и нефелиновые хвосты.
Обогащению подвергаются фосфориты и апатиты и других
месторождений.
Виды фосфорных удобрений. Фосфор,
содержащийся в природных фосфатах, очень медленно усваивается
растениями, так как нерастворим в воде и мало растворим в слабых
органических кислотах почвы. Тем не менее тонкоизмельченная
фосфоритная мука применяется в качестве длительно
действующего удобрения, особенно на кислых почвах.
Из фосфорных удобрений группа, содержащая, однокальцие-
вый фосфат Са(Н2РО4J— суперфосфаты, растворима в воде
и наиболее эффективна («водорастворимая форма Р2О5»). Менее
эффективна группа удобрений, растворимых в 2% растворе
лимонной кислоты или в аммиачном растворе цитрата аммония,
к ней относятся удобрения, содержащие дикальциевый фосфат
СаНРО4, термофосфаты и др. Эти удобрения нерастворимы в
воде, но растворяются в органических кислотах почвы («лимонно-
растворимая» и «цитратнорастворимая форма Р2О5»). Эти
формы называются усвояемыми. Фосфорные удобрения
оцениваются по содержанию Р2О5,— водорастворимой и усвояемых
форм Р2О5.
Методы переработки природных фосфатов разнообразны.
К ним относятся:
1. Разложение минеральными кислотами: а) для получения
водно- и цитратнорастворимых кислых солей; б) для получения
фосфорной кислоты.
2. Термическая возгонка фосфора с последующим
окислением и гидратацией для получения фосфорной кислоты.
3. Разложение соединения металлов I и II группы при
высокой температуре для получения удобрений с усвояемой формой
Р2О5.
166
4. Гидротермическая обработка водяным паром при 1500° С
для удаления фтора.
5. Прочие методы (обработка двуокисью углерода и др.).
Из перечисленных методов наибольшее значение имеют
первый и второй.
Суперфосфат
Целью производства является превращение Са3(РО4J
природного фосфата в усвояемую форму. Этого достигают,
обрабатывая тонкоизмельченный фосфорит или апатитовый концентрат
серной кислотой. Процесс протекает в две стадии: 1) при
смешении муки с серной кислотой образуется фосфорная кислота
и полугидрат сульфата кальция, 2) образовавшаяся фосфорная
кислота, реагируя с трикальцийфосфатом сырья, дает гидрат
монокальцийфосфата. Полугидрат сульфата кальция при этом
превращается в ангидрит. В результате кристаллизации
сульфата кальция и монокальцийфосфата вся масса затвердевает.
Обе стадии процесса можно изобразить следующими реакциями:
Ca5F (РО4K + 5H2SO4 + 2,5 Н2О = ЗН3РО4 +
+ 5CaSO4-0,5H2O + HF;
Ca5F (РО4K + 7Н3РО4 + 5Н2О = 5Са (Н2РО4J • Н2О + HF;
Примеси сырья: фториды, карбонаты, окислы железа, алюминия
и др. также реагируют с серной кислотой. При разложении
карбонатов выделяется СО2, создающая пористую структуру
суперфосфата. Окислы и соли железа и алюминия, реагируя с
фосфорной кислотой, образуют фосфаты Fe и А1, в которых Р2О5
находятся в плохо усвояемой форме.
Разложение силикатов серной кислотой дает кремневую
кислоту, образующую с выделяющимся HF : SiF4, а затем HaSiFe.
Последний уходит из сферы реакции, a CaSO4 остается в
продукте, поэтому концентрация Р2О5 в суперфосфате меньше, чем
в сырье. Так, из фосфоритов Кара-Тау C0% Р2О5) получается
суперфосфат с 13—14% Р2О5, а из апатитового концентрата
C9,5% Р2О5) суперфосфат с 18—20% Р2О5.
Применяемая серная кислота имеет концентрацию 65—68%
при расходе ее от 680 до 720 кг на 1 г сырья. Расчет ведется по
данным анализа сырья, в соответствии со стехиометрическим
соотношением для указанных выше реакций.
Для ускорения процесса серную кислоту берут обычно с
избытком. Поэтому, а также в связи с тем, что фосфорная кислота
не успевает до конца прореагировать с апатитом, в готовом
продукте содержится свободная фосфорная кислота E—5,5%)
и до 12% воды.
IS7
Кроме химических процессов, в системе протекают и важные
физические процессы. Удобрение должно быть рассыпчатым, беэ
комков, не мажущимся, не гигроскопичным, не
слеживающимся— тогда оно легко поддается рассеву на полях. Физические
свойства суперфосфата ухудшаются с повышением содержания
кислоты и влаги.
Очень важны условия, в которых осуществляется первичный
процесс смешения фосфоритной муки и серной кислоты. Здесь
играет роль тонина помола муки (не больше 15% частиц
размером более 0,15 мм), концентрация и температура E5°—70° С)
серной кислоты, интенсивность и длительность перемешивания,
конструкция аппарата. Все эти условия уточняются
применительно к виду сырья. При выбранном режиме все показатели его
должны соблюдаться очень строго. Этому способствует в
значительной мере автоматизация контроля и регулирования
процесса.
Правильную структуру суперфосфат получает в процессе
созревания. При этом образуются сростки кристаллов
Са(Н2РО4J * Н2О, происходит схватывание суперфосфатного
теста и затвердевание его в рыхлый конгломерат. Процессы
дозревания суперфосфата продолжаются и на складе. Таким
образом, технологический процесс производства суперфосфата
состоит из следующих процессов: 1) смешение тонкоизмельченного
порошка фосфоритной муки с серной кислотой, 2) созревание
суперфосфата, 3) вырезка суперфосфата и 4) дозревание на
складе.
На построенных ранее суперфосфатных заводах
периодического процесса осуществляют модернизацию, заменяя аппараты
периодического действия непрерывными и т. д. Все новые заводы
работают по непрерывному способу.
На рис. 75 показана схема непрерывного производства.
Сырье — фосфатная мука (апатитовая или фосфоритная) с
помощью ленточного транспортера 1 поступает в бункер 2, из
которого через шнек 3, элеватор 4 и шнеки 5—6 идет в бункер
дозатора 7 и на весовой ленточный дозатор 8.
Серная кислота насосом 14 подается ич сборника 13 в
мерник 15, где подогревается паром и в смесителе 16 смешивается
с водой из бака 17. Пройдя сепаратор 18 (где отделяются
окислы азота), кислотомер 19 (здесь определяется концентрация
кислоты) и расходомер 20, кислота направляется в смеситель.
Сюда же поступает сырье из дозатора 8; шнеком 9 избыток
сырья ссыпается через //—12 обратно в приемник элеватора 4.
Смеситель непрерывного действия имеет три мешалки. Его
работа в значительной степени определяет качество продукта.
Важную роль играет то обстоятельство, что компоненты попадают
в пульпу, где уже протекает процесс взаимодействия сырья с
серной кислотой. Это мешает образованию корок на частицах сырья
168
Рис. 75, Схема производства суперфосфата непрерывным способом.
и ускоряет процесс. Смешение продолжается 5—7 мин, после
чего пульпа идет в камеру непрерывного действия 21. Из камеры
затвердевшая масса вырезается фрезером (каруселью) 22 и
транспортером 23 подается к разбрасывателю 24, который
направляет ее на склад. Здесь в течение 10—15 дней продолжается
Фасфа/л серная
\ / xl'с/юта
О z ^
Суперфосфат
Рис. 76. Суперфосфатная камера
непрерывного действия.
дозревание суперфосфата. За это время его несколько рая
перелопачивают, а в конце смешивают с нейтрализующими
добавками (костяная мука, фосфорит и др.).
Пары и газы, выделяющиеся при взаимодействии сырья
с серной кислотой, отводятся из камеры по трубопроводу 25 на
переработку.
В основном аппарате — камере сочетаются два процесса:
созревание и вырезка (рис. 76). Аппарат состоит из
железобетонного цилиндра / со стальным кожухом, выложенным изнутри
диабазовыми плитками; цилиндр вращается на роликах, делая
за 2—2,5 ч один оборот вокруг чугунной неподвижной трубы 2.
170
Камера покрыта неподвижной крышкой с подвешенной к ней
вертикальной чугунной перегородкой 3, которая отделяет зону
загрузки пульпы от зоны выгрузки продукта. Со стороны
выгрузки имеется фрезер (карусель) 4 с ножами 5 для вырезки
суперфосфата, фрезер делает восемь-десять оборотов в минуту в
направлении, противоположном вращению камеры. Из 6 подается
смесь фосфатной муки и серной кислоты, которая во время
вращения камеры схватывается, твердеет и подходит к фрезе;
вырезанный продукт по трубе направляется вниз, на
транспортер, отводящий материал на склад. Эксцентрик 2 создает у
стенки центральной трубы свободный объем для расширяющейся
массы. Газы и пары через крышку отсасываются вентилятором.
При размерах камеры L = 7,1 м и Н = 2,5 м
производительность при одном обороте за 2,5 ч составляет 30 т продукта в час
и больше.
На одну тонну экспедиционного суперфосфата расходуется
0,532 т апатитового концентрата и 0,362 г 100%-ной H2SO4.
Прогресс в производстве суперфосфата, связанный с переходом на
ускоренную и непрерывную работу, привел к значительной
интенсификации производства: с 1 м3 камер при периодическом
процессе получается 85 кг продукта в час, при ускорении
перемешивания и вызревания — 364 кг, а при непрерывном способе —
820 кг и больше.
Использование отходов. Газы, содержащие 13—
30 г/м3 фтора, уносят из камеры около 50% фтора руды. Они
отводятся в орошаемые водой абсорберы, где улавливаются
фтористые соединения. HF реагирует с SiO2 сырья, образуя SiF4
4HF + SiO2 = SiF4 + 2Н2О,
S1F4 претерпевает ряд превращений:
2HF + SiF4 = H2SiF6)
3SiF4 + ЗН2О = H2Si03 + 2H2SiF6.
В результате реакций получаются кремнефтористоводородная
кислота H2SiF6 и гидраты кремневой кислоты. При добавлении
избытка NaCl осаждают плохо растворимый Na2SiF6) его
отфильтровывают, сушат и отправляют потребителям.
На тонну суперфосфата можно получать от 5 до 10 кг
Na2SiF6. Кремнефтористый натрий находит применение в
производстве кислотоупорных цементов и эмалей, для борьбы с
сельскохозяйственными вредителями и т. д.
Из фтористых газов можно получать также и чистые
фтористые соли—фториды натрия, алюминия, магния, бора, криолит
3NaF • AIF3 и фторорганические соединения.
Получаемые в больших количествах нефелиновые хвосты,
которые раньше выбрасывали, теперь используют (стр. 144).
171
Улучшение суперфосфата. Для уменьшения
гигроскопичности и слеживаемости суперфосфата необходимо
снизить содержание в нем влаги и свободной кислоты. Для этого
применяют различные нейтрализующие добавки — костяную
муку или муку легко разлагаемых фосфоритов, молотый
известняк и другие вещества, снижающие кислотность до 1—2,5%.
Сушка и грануляция также уменьшает влажность суперфосфата.
Иногда производят обработку суперфосфата аммиаком
(аммонизация суперфосфата). При этом протекают следующие
реакции:
NH3 + Н3РО4 = (NH4) Н2РО4,
NH3 + Са (Н2РО4J = (NH4) Н2РО4 + СаНРО4.
Избыток NH3 вреден, так как может вызвать ретроградацию
(образование трикальций фосфата):
2СаНРО4 + CaSO4 + 2NH3 = Са3 (РО4J + (NH4JSO4.
Аммонизация осуществляется во вращающемся барабане, где
суперфосфат и аммиак идут прямотоком.
Грануляция суперфосфата. Большое значение
имеет грануляция удобрений. Гранулированное удобрение
меньше слеживается, легко и равномерно рассеивается в поле
рядовыми сеялками, меньше теряется при загрузке, выгрузке,
транспортировке и рассеивании, полнее используется растениями.
Грануляция сопровождается сушкой, в связи с чем гранулы
содержат меньше влаги и больше питательных веществ. Питательные
элементы гранул переходят в почвенные растворы с меньшей
скоростью и равномернее, поэтому действуют они дольше. Кроме
того, при грануляции легко вводить добавки, уменьшающие
кислотность, микроудобрения и др.
Процесс грануляции суперфосфата проводится так.
Суперфосфат с нейтрализующей добавкой подается на вальцы для
измельчения и смешения. Затем масса поступает в гранулятор —
вращающийся барабан, где увлажненная до содержания
примерно 16% воды масса закатывается в гранулы, которые сушатся
топочными газами при температуре не выше 85° С, во избежание
образования мета- и пирофосфата. После охлаждения гранулы
рассеиваются на фракции. Фракция размером 2—4 мм выдается
как продукт.
Улучшенный описанными приемами суперфосфат является
неконцентрированным удобрением, так как в нем содержится
всего около 20% полезного Р2О5, прочее — балласт. Такой
простой (одинарный)суперфосфат заменяют более
концентрированными удобрениями.
Поскольку в суперфосфате основной балласт — это CaSO4,
коренного улучшения можно достичь путем его удаления. Ради-
172
кальным мероприятием является обработка природного фосфата
фосфорной кислотой, действие которой протекает по реакции
Ca6F (РО4)Э + 7НЭРО4 + 5Н2О = 5Са (Н2РО4J • Н2О + HF.
Применяют также обработку фосфата соляной или азотной
кислотой и смесями кислот: Н3РО4 + H2SO4, Н3РО4 + HNO3;
HNO3 + Н2СОз-
Обработка фосфорной кислотой дает удобрение, содержащее до
45—52% Р2О5. т. е. в два—три раза больше, чем простой
суперфосфат. Это так называемый двойной суперфосфат.
Для получения концентрированных удобрений нужна
фосфорная кислота концентрацией от 20 до 55% Р2Об. (Заметим, что
100% Н3РО4 соответствуют 72,6% Р2О5).
Производство фосфорной кислоты
Производство фосфорной кислоты осуществляется двумя
методами: взаимодействием фосфатного сырья с минеральными
кислотами (экстракционный способ) и гидратацией фосфора,
полученного термическим способом из того же сырья.
Экстракционная фосфорная кислота. Чаще
всего для получения фосфорной кислоты фосфорит
экстрагируют серной кислотой. При этом протекает реакция
Саэ (РО4J + 3H2SO4 = 2Н3РО4 + 3CaSO4.
Важна степень гидратации CaSO4 и характер его кристаллов,
находящиеся в прямой зависимости от условий процесса —
температуры, концентрации кислоты, отношения Т : Ж. Сернокислый
кальций может выпасть в виде CaSO4 • 2Н2О (дигидратный
процесс) при 70—80° С и концентрации кислоты 27—35%, в виде
CaSO4-0,5H2O (полугидратный процесс) при 85—95° С и
концентрации 35—40% Р2Об и в виде CaSO4 (ангидритный процесс)
при 100—105° С и концентрации выше 45% P2Os-
Наиболее освоенным является дигидратный процесс, в
результате которого получается фосфорная кислота меньшей
концентрации, но образовавшиеся кристаллы гипса отличаются
большей крупностью, легче фильтруются и лучше промываются.
Так как вместе с H2SO4 циркулирует Н3РО4, процесс можно
изобразить следующим образом:
Са5 (PO4KF + 5H2SO4 + «Н3РО4 + 5/?Н2О =
HSPO4 + 5CaSO4 • р Н2О + HF.
Коэффициент р, как указано, в зависимости от условий,
равен 0, 0,5 или 2.
173
На рис. 77 дана схема этого производства. Как видно из этой
схемы, фосфоритная мука через дозатор непрерывно подается
в первый из основных экстракторов. Сюда же из второго
смесителя поступает циркулирующая и содержащая фосфорную
кислоту пульпа из третьего экстрактора, а также 75—93%-ная
H2SO4 (через правый дозатор) и промывные воды (через левый
дозатор) из второй зоны трехсекционного ленточного фильтра;
отношение Ж : Т-в реагирующей пульпе поддерживается равным
3: 1 или 3,5: 1.
Фосфорит
вода на лромы8ку к вокуумной/гшии -
1 Концентриро-
zz. ванная
Рис. 77. Схема производства фосфорной кислоты экстракционным способом:
/ — бункер фосфоритной муки; 2 — питатели серной и рециркуляционной фосфорной
кислоты; 3 — питатель муки; 4 — предварительные смесители кислот и циркулирующей
суспензии (пульпы) из 5; 5 — основные экстракторы; 6 — холодильники; 7 — промыва -
тель газа из экстрактора; 8 — вакуум-фильтр; 9 — вакуум-выпарной аппарат; 10 —
сгуститель шлама; 11 — сепаратор
одильники; 7 про
ра; 8 вакуумфильтр; 9 — вакуум-выпарной аппарат; 10
сгуститель шлама; 11 — сепаратор.
Экстракторы снабжены перетоками, мешалками, отводами
для выделяющихся газов (HF, СОг и др). В них подают воздух
для перемешивания и охлаждения, чтобы обеспечить получение
';ОрОШО фпЛЬТруЄМЬІХ КрїІСТЛЛЛпіі Пі ПСЛ, СГикіиОіЦИІЇ ИЗ ССКЦііИ /
фильтра раствор Н3РО4 уходит как продукт, а осадок (фосфо-
і'ипс) промывается и следующих секциях и удаляется в отход.
Часть полученной кислоты после осветления в отстойнике может
поступать на выпаривание в вакуум-выпарной аппарат, где
получается кислота, имеющая концентрацию 55—65% РУЭд.
Находят применение также концентраторы, аналогичные
употребляемым при упаривании серной кислоты: барабанные и
погружного горения. Выход Н3РО4 доходит до 94—96°/о- На 1 т
Р2О5 получается 1,3—1,6 т отхода — фосфогипса. Фосфогипс
можно применять как строительный материал, для
производства (NH4hSO4 и т. д.
174
Фосфорная кислота, особенно горячая, сильно корродирует
обычные конструкционные материалы, используемые в
химической промышленности.
Ввиду того, что фосфорная кислота разрушает не только
металлические, но и силикатные материалы, приходится применять
специальные стали, свинец и др. Так, выпарные аппараты
изготовляются из нержавеющей стали; материалом для баков часто
служит дерево, выложенное свинцом; экстракторы делают из
стали, футерованные кислотостойкими диабазовыми или
керамическими плитами на полиизобутилене, углеродистые (карбейт,
антегмит), используются также резина и различные пластмассы.
Лента вакуумфильтра выполняется из резины с прокладками
из синтетической ткани (лавсан, анид и др.).
В заключение добавим, что для получения чистой
экстракционной фосфорной кислоты следует удалить іпримеси,
имеющиеся в природных фосфатах. К примесям относятся соединения
редких элементов цериевой группы, лантанидов, ванадия, урана,
которые можно выделить и использовать. Получение фосфора
электротермическим методом и фосфорной кислоты из него
описаны далее (стр. 219).
Концентрированные фосфорные удобрения
Двойной суперфосфат. На рис. 78 приводится схема
производства по проточному методу. Согласно этой схеме
измельченный фосфорит (или апатит) и фосфорная кислота
подаются в два последовательные реактора, обогреваемые паром;
отсюда масса (пульпа) поступает в смеситель-гранулятор, где
она смешивается с возвратом (ретуром) и проходит в
сушильный барабан, обогреваемый топочными газами; из полученных
в сушильном барабане гранул на грохоте выделяется фракция
(размером от 0,5 до 4 мм), которая после нейтрализации
известняком направляется на склад готовой продукции.
Усложнение схемы заключается в том, что двойной грохот
4 делит материал на три фракции: среднюю, мелкую (меньше
0,5 мм) и грубую (больше 4 мм), причем последняя доизмель-
чается в дробилке 9 и вместе с мелкой и частично средней
фракцией составляет ретур, возвращаемый в смеситель-гранулятор 2.
По этой схеме длительное дозревание в камере или на складе
заменено сушкой; можно употреблять даже экстракционную или
немного более концентрированную фосфорную кислоту B5—35%
Р2О5) вместо термической.
При использовании Н3РО4 + H2SO4 образуется обогащенный
суперфосфат, содержащий P2Os в количестве, ореднем между
его содержанием в простом и двойном суперфосфате.
Преципитат. Преципитат представляет собой дипидрат
дикальцийфосфата СаНРО4*2Н2О. Если при получении его
175-
двойной улну
''яированныО
, суперфосфат
Рис. 78. Схема производства двойного суперфосфата:
1 — реакторы; 2— смеситель-грагулятор, 3— сушильный барабан; 4— грохот; 5 — дробилка; 6-
буикеры; 9—нейтрализационнып барабан.
шнеки; 7 — элеваторы; 8 —
используется экстракционная фосфорная кислота, он содержит
до 35% цитратнорастворимого Р2О5. Если же термическая
кислота,—до 41% Р2О5.
Процесс получения преципитата заключается в
нейтрализации Н3РО4 известковым молоком или измельченным известняком.
При этом происходят реакции:
Н3РО4 + Са (ОН), = СаНРО4 + 2Н2О,
Н3РО4 + СаСО3 + Н2О = СаНРО4 + 2Н2О + СО2.
Процесс осуществляют в реакторах, куда компоненты
добавляют небольшими порциями при усиленном перемешивании,
чтобы избежать покрытия зерен пленкой. Осадок СаНРО4 • 2Н2О
отделяют на вакуум-фильтре, сушат и подают на склад. Тем же
способом готовят н кормовой преципитат, с той разницей, что
сначала дают выпасть части осадков, содержащей примеси
сырья, а затем уже остальному преципитату, который имеет
очень небольшое количество примесей.
Аммофосы. Моноаммонийфосфат (NH4)H2PO4, диаммо-
нийфосфат (NH4bHPO4 и триаммонийфосфат (ЫН4)зРО4, как
аммонийные соли фосфорной кислоты (аммофосы), получают,
нейтрализуя НзРО4 аммиаком. (ЫН4)зРО4 для удобрений не
применяется, так как он легко разлагается с выделением NH3.
Процесс приготовления диаммофоса протекает в ряде
реакторов с мешалками. Суспензия (пульпа) из последнего
реактора смешивается с уже высушенным аммофосом, идет в
барабанную сушилку, гранулируется и отправляется потребителю.
Продукт содержит 48—55% Р2О5 и 11 —13% NH3. Аммофос можно
получать также в результате взаимодействия распыленной
фосфорной кислоты и газообразного NH3. Диаммофос применяется
при производстве смешанных удобрений.
В последнее время аммофосы получают при улавливании
аммиака и на коксобензольных заводах, заменяя серную кислоту
фосфорной.
Термофосфаты. Чтобы обойтись без серной кислоты и
получать удобрения из фосфатов с хорошими физическими
свойствами, прибегают к прокаливанию сырья при 1200—1600° С с
добавками или без них. Если прокаливать сырье в присутствии
паров воды, получаются обесфторенные фосфаты, которые
могут применяться как удобрения, они не ядовиты и добавляются
в корм скоту. Процесс протекает в две фазы — сначала
получается гидроксилапатит, а потом растворяемые в лимонной
кислоте а-трикальцийфосфат и тетракальцийфосфат:
Ca6F (РО4K + Н2О = Са5 (ОН) (РО4K + HF,
2Са5 (ОН) (РО4K = 2Са3 (РО4J + Са4Р2О9 + Н2О.
Добавление песка при высокой температуре способствует
разрушению кристаллической решетки фторапатита, приводя к
12 8-1504 177
образованию фаз переменного состава—оиликофосфатов,
например, по реакции
2Са5 (ОН) (РО4K + 0,5 SiOa = ЗСа3 (РО4)а + Q,5Ca2SiO4 + НаО.
Описанный процесс осуществлен в СССР. Апатитовый
концентрат с 2% SiO2 обжигается во вращающихся печах при
1420—1460° С. В газовой фазе должно быть 10—12 об.% Н2О.
Часто для получения термофосфатов осуществляют
совместное прокаливание природных фосфатов в смеси с солями или
рудами щелочных и щелочноземельных металлов, причем
процесс ведут либо до спекания, либо до получения плавленого
продукта. Таким образом готовят, например, плавленые
магнезиальные или кальциймагнезиальные фосфаты с применением
оливина ((Mg, FeJSi04), серпентина CMgO • 2SiO2 • 2Н2О) и других
минералов. Весь процесс заключается в измельчении сырья,
дозировке составляющих ингредиентов, смешении и прокаливании
шихты с последующим охлаждением и помолом продукта.
Основные аппараты представляют собой печи с обогревом
электроэнергией или топочными газами.
Таким путем удается получить термофосфаты из
фосфоритов Кара-Тау и природного сульфата натрия, имеющихся в
больших количествах в Средней Азии, где термофосфаты широко
применяются в качестве удобрения для хлопка и других культур.
К этой группе удобрений следует отнести отходы металлических
заводов — шлаки. Так, томасовские шлаки содержат 15—20%
РгОб в усвояемой форме. Процесс получения такого удобрения
несложен: шлак после застывания измельчается.
2. Калийные удобрения
Калий и его соединения находят широкое применение не
только в сельском хозяйстве. Соединения калия употребляются
для получения калиевых солей, например бертолетовой соли,
калийной селитры, едкого калия, калийсодержащего стекла, мыла,
красок, фармацевтических препаратов, цианистого калия и т. д.,
металлический калий, получаемый из солей калия электролизом,
используется в военном деле, в производстве. синтетического
каучука и др.
Калийные удобрения выпускаются в виде 1) КС1 E0, 60 в
62% К2О), 2) K2SO4 E0% К2О), 3) смешанной калийной соли
C0—40% К2О) и 4) калимагнезии (K2SO4 + MgSO4) с
содержанием не менее 17% К2О- (Заметим, что 100% КС1
соответствуют 62% КаО).
Сырье. Исходным веществом для получения соединений
калия является хлористый калий и в меньшем количестве —
сульфат калия. Некоторое значение имеют органические отбро-
178
сы, из которых получают соли калия: зола стеблей
подсолнечника, водоросли, отходы сахарных заводов и др.
В природных залежах соли калия смешаны с солями Na
и других металлов, залегающих в виде хлоридов и сульфатов.
Из многочисленных минералов наибольшее значение имеют
карналлит КС1 • MgCla • 6НаО, сильвинит КС1 + NaCl(~~ 12% К2О).
В этих рудах в небольшом количестве содержатся MgBr2; RbCl,
CsCl, а также соединения йода, бора и др. Содержание КгО
колеблется от 9 до 20%.
Наиболее богатые месторождения калийных солей
находятся в СССР и Франции. Богатейший в мире Соликамский район
находится на месте высохшего древнего Пермского моря. Соли-<
камское месторождение калиевых солей было открыто в 1916
году. Большую роль в исследовании Соликамских залежей и
разработке метода их переработки сыграл академик С. Н. Курна-
ков. Вероятное содержание солей во всей толще Соликамского
месторождения — свыше 10 млрд. тонн. В настоящее время
калийные соли найдены также в Средней Азии, в Западной
Украине и Белорусской ССР.
Производство КС1 из сильвинита
Принцип выделения КС1 из сильвинита КС1 + NaCl основав
на том, что растворимость NaCl мало увеличивается с
повышением температуры, а растворимость КС1—сильно. Приводим
процентное содержание NaCl и КС1 в растворах, насыщенных
обеими солями при 25 и 100° С:
NaCl KC1
25° 20,4 11.15
100° 16,8 21,7
Как видно из этих данных, растворимость КС1 при 25° С почти
в два раза меньше, чем при 100° С, между тем как растворимость
NaCl при 25° даже меньше, чем при 100° С. Поэтому если
охлаждать до 25° С раствор, насыщенный при повышенной A00—103°)
температуре обеими солями, то будет кристаллизоваться
только КС1. Оставшийся маточный раствор, насыщенный NaCl, после
подогрева можно снова использовать для растворения КС1 из
смеси его с NaCl. Поваренная соль в этих условиях более не
растворяется.
Получение КС1 из сильвинита сводится к следующим
операциям: 1) измельчение сильвинита; 2) растворение КС1 горя<-
чим раствором, циркулирующим в системе; 3) отделение осадка
NaCl и др.; 4) охлаждение раствора для кристаллизации КС1;
5) отделение кристаллов от маточника; 6) промывка
кристаллов; 7) сушка и 8) упаковка.
Щелок (маточник), полученный после отделения кристаллвв
К.С1, направляют снова на выщелачивание новых порций силь-
12* 179
lodtr
Рис. 79. Технологическая схема производства хлористого калия из сильвинита:
/—ленточный транспортер измельченного сильвинита; 2 — бункер сильвинита; 3 — ленточный конвейер; 4 — весы автоматические; 5 —
первый растворінель; t> — ¦ иторой растворитель; 7 — конвейер; 8— насосы; 9 — наклонный элеаатор./f — план-фильтр. 11 — мешалка
глинистого шлама; 12 — шесп-лоиусный сгуститель для глинистого шлама. 13 — сгуститель; 14— бак с мешалкой; 15 — расходный
бик, 16— поверхностные конденсаторы; 17 — четырнадцатнступеичатый вакуум-кристаллизатор; 18 — бак: 19 — отстойник; 20 —
емкость с мешалкой для крнсі,*ллов хлористого калия: 2! — шестиконуспыи сгуститель для кристаллов хлористого калия; 22—бак
с мешалкой; 23 — центрифуги .пля кристаллов хлористого калия; 24 — бак сгустителя; — бак для центрифуг; 26 — топка сушильного
барабана; 27 — сушильный барабан; й> — выгрузная камера для сухого продукта.
винита. Примеси, имеющиеся в сильвините, постепенно, после
каждой операции выщелачивания, накапливаются в маточнике.
Чтобы предотвратить их выпадение вместе с КС1, часть щелоков
выводят из цикла, заменяя их водой или слабым раствором соли.
Растворение КС1 из сильвинита (рис. 79) происходит в
системе шнековых растворителей; полученный щелок отделяется
от нерастворимого осадка отстаиванием и фильтрованием.
Затем осветленный щелок охлаждается в многокорпусной
вакуум-холодильной установке, в
которой оборотный щелок
нагревается; выпавшие кристаллы КС1
отделяются в отстойнике,
центрифугируются и сушатся в
барабанной сушилке топочными
газами.
Высушенный продукт
содержит около 95% КС1. Остаток,
состоящий из 93—95% NaCl и 5—
7% КС1, очищают от КС! с целью
получения NaCl, используемого
для переработки в соду и другие
продукты. На рис. 80 представлен
один из видов шнекового
растворителя с двумя греющими
камерами. Описанным способом
извлекают 75—89% КС1. На 1 г
95%-ного КС1 расходуется 5 г сильвинита B2%-ного по КС1),
0,75 г пара, 30 кет. ч. энергии, 10 м3 воды и около 20 кг угля
G000 кал).
Можно получать КС1 из сильвинита и без растворения —
флотацией или гидросепарацией. Для флотации измельченный
сырой сильвинит обрабатывают раствором, насыщенным КС1 и
NaCl при соотношении Т : Ж = 1 : 2 в присутствии специальных
флотирующих агентов (алкилсуль'фаты натрия, сосновое
масло и др.).
Кристаллы КС1 всплывают, а кристаллы NaCl и другие
примеси тонут. Степень извлечения КС1 достигает при этом 92 —
97%. КС1 отделяют на центрифугах и сушат. Высушенная соль
содержит 90—98% КС1.
Разделение хлоридов можно производить также по разнице
удельных весов минералов, так как dNaci =2,17, a d ксі =1,98.
Процесс ведут следующим образом. Смесь тонко измельченного
сырого сильвинита помещают в жидкость, служащую
разделительной средой. Процесс разделения кристаллов осуществляется
в гидроциклонах, где под влиянием центробежной силы поток
делится на две части. Одна часть, содержащая главным образом
NaCl (хвосты), отбрасывается к периферии и выводится оттуда.
Рис. 80. Шнековый
растворитель сильвинита.
181
Другая часть, содержащая КС1 (концентрат), отводится из
центральной части циклона.
Изложенным не исчерпывается проблема переработки солей
Соликамского и других месторождений. Так, при переработке
карналлита КС1 • MgCl2 • 6Н2О можно выделить хлористый
магний, который применяется в холодильном деле, в текстильной
промышленности, цементной, для получения так называемого
цемента Сореля. MgCb и искусственный карналлит применяют для
производства металлического магния. Кроме того, при
обработке оборотных щелоков, содержащих бром в виде, например
MgBr2, можно действием газообразного хлора получать бром по
реакции
MgBr2 + СІ 2 = MgCl2 + Вг2.
При переработке руд прикарпатских месторождений,
содержащих калимагнийсульфат K2SO4 • MgSO4 • 6Н2О, можно
разделить эти соли и путем дальнейшей их переработки получать,
как и из КС1, любые соединения калия.
3. Азотные удобрения
Азотные удобрения играют большую роль в сельском хозяй-
нн} .Г/итя - стве. К ним относятся сульфат аммо-
возА/х J парЫи ния B1% N), аммиачная селитра
" '¦'-—*¦ C4,5% N), аммиакаты на основе
аммиачной селитры D0% N),
цианамид кальция B0% N), мочевина D6%
N), а также аммиак (82% N) и
аммиачная вода B0% N) и др. Кроме того,
многие сложные удобрения содержат
азотные соединения.
Аммиачная селитра
Для получения аммиачной селитры
нейтрализуют аммиаком 45—50%-ную
HNO3:
NH3 4- HNO3 = (NH4) NO3 +
+ 148,6 кдж.
Выделяющимся теплом нейтрализации
пользуются для концентрирования
раствора аммиачной селитры в
специальных нейтрализаторах ИТН
(«использование тепла нейтрализации»);
схема аппарата ИТН дана на рис. 81.
Раствор NH4NO3, образовавшейся при взаимодействии NH3 и
HNO3 во внутреннем цилиндре, переливается через его верхний
Раетдср
Рис. 81. Нейтра-лкзаииоа-
нык аппарат ИТН:
/ — цилиндрический сосуд;
2 — внутренний цилиндр
(реакционная камера); Л—
устройство для подачи
азотной кислоты; 4 — то же для
аммиака; 5 — штуцер для
опЪрожнення аппарата; в —
сепаратор; 7 —
гидравлический затвор.
182
край в испарительную часть аппарата (пространство между
внутренним цилиндром и наружными стенками аппарата), где
упаривается за счет тепла реакции. При использовании 45%-ной
HNO3 на 1 т NH4NO3 испаряется около 400 кг воды и получается
раствор, содержащий 63% NH4NO3. Если применять 63%-ную
HNO3, то тепла реакции будет достаточно, чтобы полностью
упарить раствор.
tlaodiam)
пар
Раствор NH^HOj
bQ}
на охлаждение
Рис. 82. Схема двухступенчатой выпарки растворов аммиачной селитры
с грануляционной башней:
На рис. 82 представлена схема производства
гранулированной аммиачной селитры из раствора, уходящего из установки
нейтрализации. Применяют двух- или трехкорпусную схему
упаривания, причем из последнего корпуса уходит расплавленный
NH4NO3 концентрацией 92—94% либо 97—98,5%. В первом
случае кристаллизацию осуществляют с использованием тепла
кристаллизации для дополнительного испарения воды.
Кристаллизацию плава NH4NO3 проводят в различной
аппаратуре. На многих заводах плав подается на вращающийся
охлаждаемый изнутри водой барабан (холодильные вальцы),
на поверхности которого плав застывает в кристаллы.
183
Кристаллизация плава осуществляется также в
грануляционных башнях воздухом. Гранулированная селитра меньше
слеживается и ее удобнее вносить в почву. К недостаткам аммиачной
селитры относятся ее гигроскопичность и способность
слеживаться. Кроме того, наблюдались случаи разложения
аммиачной селитры со взрывом, например по реакции
2NH4NO3 = 2N2 + 4Н2О + О2 + 233,5 кдж
(при детонации или попадании органических и других
примесей). Для устранения этих недостатков аммиачную селитру
получают из пяти возможных кристаллических форм, в наиболее
устойчивой а-ромбической модификации, которая образуется при
температуре ниже +32,1° С. Она обладает наибольшим
удельным весом — 1,725 г/см3. Кроме того, чтобы предупредить сле-
живаемость, аммиачную селитру, высушенную до 0,5—1% влаги,
хранят в битуминизированных мешках. Применяется также
припудривание селитры 2—3% фосфоритной муки, гипса или
каолина, введение добавок, понижающих растворимость селитры
Ca(NO3J, Mg(NO8J.
Взрываемость снижают добавкой стабилизаторов (мочевина,
аммиак, карбонаты Са и Mg, уротропин и др.).
Широко практикуется смешение аммиачной селитры с
другими удобрениями и солями, в результате чего образуются менее
гигроскопичные и меньше слеживающиеся двойные и тройные
удобрения. В некоторых случаях компоненты взаимодействуют,
например, при смешении NH4NO3 с (NH4JSO4 получается суль-
фонитрат аммония состава NH4NO3 ¦ 2(NH4JSO4. Это хорошее
удобрение, содержащее до 27% азота. Его можно получить
также, нейтрализуя газообразным аммиаком смесь кислот
H2SO4 + HNO3.
Сплавление аммиачной селитры с известняком F0% NH4NO3
и 40% СаСОз) дает хорошее удобрение — известково-аммиач-
ную селитру. Применяется также удобрение, полученное
сплавлением аммиачной селитры с фосфатом аммония. Калийно-ам-
мначную селитру получлют in аммиачной селитры и хлористого
калия по реакции
NH4NO3 -f KCI = KNO3 + NH4C1.
Продукты этой реакции почти не слеживаются. На базе нитрата
аммония можно получить смешением или сплавлением
компонентов сложные трехсторонние удобрения — нитрофоски:
смешением нитрата аммония, фосфата аммония и хлористого калия.
KNO3 широко применяется в горном деле. Из нее готовят
взрывчатые вещества — аммоналы, представляющие собой смесь
аммиачной селитры и алюминиевого порошка, и аммониты
(смесь селитры с органическими материалами).
Ж
Нитрат натрия
Нитрат натрия, как известно, добывается из чилийской селлт-
ры. Большое распространение получили заводские способы
производства его взаимодействием растворов NaCl или Na2COs
с азотной кислотой или с окислами азота при щелочной
абсорбции их (стр. 105).
Остановимся на новом способе получения NaNO3 методом
катионного обмена через Ca(NO3J- В этом способе получение
NaNO3, происходит периодически, в одном аппарате. Сначала
раствор Са(ЫОзJ пропускают через катионит. При этом
образуется нитрат натрия (I стадия). После насыщения катионита
кальцием начинается II стадия—регенерация Na-катионита.
Если обозначить Na-катионит через PNa, то реакцию первой
стадии можно изобразить так:
2PNa + Ca (NO3J = Р2Са + 2NaNO3,
а второй стадии —
Р,Са + 2NaCl = 2PNa + СаС12.
Уравнение суммарной реакции изображается уравнением
Ca (NO3J + 2NaCl = 2NaNO3 + СаС12.
Таким образом, процесс получения NaNO3 по этому способу
сводится к следующим операциям:
1) получение раствора Са (ЫОзJ; 2) пропускание этого
раствора через Na-катионит; З) регенерация Na-катионита; 4)
кристаллизация NaNO3; 5) отделение кристаллов; 6) сушка продукта.
Сульфат аммония
Сульфат аммония получают «мокрым» способом, а также
«сухим» из аммиака и серной кислоты. Кроме того, он
образуется из гипса и двуокиси углерода с аммиаком. Процесс
нейтрализации протекает по реакции
2NH3 + H2SO4 = (NH4J SO4 + 280 кдж.
При мокром способе газообразный аммиак вводят в раствор
серной кислоты. Полученный раствор (NH4JSO4 упаривают,
и соль выделяют кристаллизацией.
Сухой способ получения сернокислого аммония состоит во
взаимодействии распыленной серной кислоты с газообразным
аммиаком. Процесс осуществляется в камерах, где сверху
разбрызгивается серная кислота, а снизу подается NH3; в камере
образуются мелкие, сухие, падающие на дно камеры кристаллы,
так как за счет тепла реакции вся избыточная влага испаряется.
Продукт содержит около 0,1% влаги и до 0,2% свободной
H2SO4. Этот способ мало распространён из-за значительной
дисперсности и большого удельного объема продукта
185
Сернокислый аммоний можно получить разложением
суспензии гипса в воде аммиаком и двуокисью углерода по реакции
CaSO4 + 2NH3 + СО2 + Н2О = (NHJ2 SO4 + СаСО3.
После отделения СаСОз фильтрат упаривают в
вакуум-выпарных аппаратах. Выделяющиеся кристаллы отделяют на
центрифуге и сушат.
В последнее время большие количества сульфата аммония
получают из отходов производства капролактама. Так, в
промежуточной стадии этого производства при нейтрализации серной
кислоты, введенной с сульфатом гидроксиламина, аммиаком
образуется раствор сульфата аммония. Так как в дальнейшем
этот раствор не нужен, его упаривают; выпавшие кристаллы
(NH4JSO4 отделяют на центрифуге, сушат и употребляют как
удобрение самостоятельно или в смеои с другими.
4. Жидкие удобрения
Жидкие удобрения дешевле твердых в 1,5—2 раза, так как
для их приготовления не требуется выполнение таких
дорогостоящих операций, как упаривание, кристаллизация, сушка.
Аппаратура для их производства и хранения проще. В США около
50% сложных удобрений применяется в жидком виде.
Жидкие азотные удобрения — аммиакаты представляют собой
растворы солей (нитратов аммония, кальция или мочевины и
др.) в жидком аммиаке или аммиачной воде. Упругость NH3
над этими растворами значительно ниже, чем над аммиаком.
В табл. 20 приводится процентный состав трех аммиакатов.
Таблица 20
NH3
Ca(NO3)i
NH4NO3
НгО
N
Аммиакаты
Первый 1
14—17
64—67
27—і G
34—37,5
Второй
23—26
33—56
24- і 8
37,5—41
Третий
18—20
25—28
27—30
JU— 22
30,5—31,5
Эти аммиакаты характеризуются следующими данными:
упругость аммиака при 40° С равна соответственно 0,1, 1,12 и
1 атм, температура выпадения осадка 9, —25 и —35° С.
Как видно из сказанного, первая смесь лучше других, так как
упругость NH3 составляет всего 0,1 атм и содержание азота
в нем велико, но осадок выпадает уже при +9° С. В качестве
удобрения можно применять также аммиак или аммиачную воду.
Эти виды удобрений требуют применения специальных
хранилищ.
586
5. Многосторонние удобрения
Разнообразные удобрения, содержащие несколько
питательных элементов, подразделяются на: 1) азотно-калийные, 2) фос-
форно-калийные, 3) азотно-фосфорные и 4) азогно-фосфорно-ка-
лийные (полные удобрения).
Они получаются чаще всего смешением различных
удобрений — таким путем можно получить удобрения с любым
отношением N : Р2О5 : КгО (в зависимости от почвы, культуры и
других условий).
Однако следует подчеркнуть, что нельзя смешивать любые
удобрения, так как возможны потери питательных веществ. Если
смешивать аммонийные соли (NH4NO3, NH4C1 и др.) с томас-
шлаком или термофосфатом, то СаО в них реагирует, например,
так:
СаО + (NH4JSO4 = 2NH3 + Н2О + CaSO4.
При этом азот уходит в атмосферу в виде NH3. Кроме того, при
смешении с кислыми удобрениями следует давать
нейтрализующие добавки. Обычно при смешении исходят из односторонних
и сложных двойных, вследствие чего полученные удобрения
называются сложно-смешанными, или комплексными.
К а л и й н о-а ммиачная селитра является азотно-
калийным удобрением и производится смешением
эквимолекулярных количеств NH4NO3 и К.С1. Она содержит 16—16,5% N
и около 25% К2О. Для уменьшения гигроскопичности она
должна выпускаться в гранулированном виде.
В виде сложного двойного удобрения применяется
калиевая селитра K.NO3, содержащая 13,5% азота и 45,6% окиси
калия. KNO3 применяется также в производстве черного
(дымного) пороха, в пиротехнике, в стекольной и пищевой
промышленности.
Его применяют в качестве компонента смешанных удобрений,
например, с фосфорнокислым аммиаком. K.NO3 не гигроскопична
и хорошо рассеивается.
KNO3 готовится конверсионным способом при
взаимодействии NaNO3 и КС1 в растворе при температуре около 100° С:
NaNO3 + КС1 ¦=* KNO3 + NaCl.
После отделения выпавших кристаллов NaCl раствор
охлаждается до 25—35° С; при этом осаждаются кристаллы KNO3,
которые отделяются на центрифуге, сушатся и упаковываются.
Маточный раствор возвращается в процесс.
KNO3 можно также получать при щелочной абсорбции нит-
розных газов в производстве азотной кислоты (стр. 105).
В качестве фосфорно-калийных удобрений предложены
различные смешанные и сложные композиции. К смешанным можно
187
отнести калийный суперфосфат, представляющий
собой смесь суперфосфата с хлористым или сернокислым калием.
Заслуживает внимания упрощенная схема производства
дегидратированного фосфорно-калийного удобрения из апатитового
концентрата, серной кислоты и хлористого калия в аппаратуре
суперфосфатного цеха. При этом происходит реакция
Са5 (PO4KF + 5H2SO4 + ЗКС1 = 5CaSO4 + ЗН3РО4 + ЗКС1 + HF,
в результате которой образуется пульпа.
После затвердевания пульпы ее вырезают, смешивают с рету-
ром и сушат полученные гранулы при 300° С. При этом имеет
место следующая реакция:
5CaSO4 + ЗН3РО4 + ЗКС1 = ЗКРО3 + 5CaSO4 + ЗНС1 + ЗН2О.
Полученное удобрение содержит 17—18% Р2О5 и 12—17%
КгО; выделяющиеся при взаимодействии компонентов HF и НС1
улавливаются.
К сложным удобрениям этой группы относятся фосфаты
калия. Так, метафосфат КРО3, содержащий почти 100%
питательных веществ C9,9% КгО + 60,1% Р2О5), получают
нейтрализацией ортофосфорной кислоты поташом и дегидратацией
ортофосфата калия.
Большую группу двойных удобрений составляют азотнофос-
форные (аммофосы), получаемые нейтрализацией аммиаком
Н3РО4 (стр. 177). Смесь аммофоса с сульфатом аммония —
сульфоаммофос — содержит около 16,2% усвояемой Р2О5
и 18,3% азота, причем фосфаты аммония состоят примерно и-з
75% (NH4)H2PO4 и 25% (NH4JHPO4. Сульфоаммофос
получается также нейтрализацией аммиаком смеси H2SO4 + Н3РО4.
В технологии иолучения двойных и тройных удобрений
наибольшее значение приобретают нитрофосфаты, получаемые
взаимодействием фосфатов с азотной кислотой.
Азот но кислот пая переработка фосфатов
Получение азотно-фосфатных и в особенности тройных азот-
но-фогфорно-калнйны^ удобрений типа нитрофоски еыгг>дпо rccth
разложением природных фосфатов азотной кислотой вместо
серной, так как при этом вместо иона SOJ~ в систему вводили NTO~,
содержащий полезный азот.
Основную реакцию, протекающую между апатитовой мукой
и 50%-ной HNO3, можно изобразить следующим уравнением:
Ca5F (РО4K + 10HNO3 = ЗН3РО4 + 5Са (NO3J + HF.
Реакция протекает при 45—60° С, и за Р/2—2 часа в раствор
переходит более 95% Р2О5 сырья. Небольшой осадок примесей,
состоящий преимущественно из силикатов, отделяют
отстаиванием или фильтрованием. Фтористые соединения выводятся, как
188
обычно, в виде Na2SiF6. Затем из раствора можно получить
соединения редких земель — группы цериевых соединений, ланта-
нидов, ванадия, урана и др.
В оставшемся растворе отношение СаО : Р2О5 примерно
такое же, как и в сырье, и составляет, например, для апатитового
концентрата, 1,32, для фосфорита Кара-Тау — даже 1,59. Если
оставить такое отношение, то может выпасть неусвояемый
Са3(РО4J. Чтобы избежать этого и получить ди- или монокаль-
цийфосфат, надо довести содержание СаО на 1 кг Р2О5 до
0.79 кг или 0,395 кг соответственно. Для этого надо либо удалить
избыточный ион кальция, либодобавить ион РО34~. Удаление
Са++ осуществляется разными путями — вымораживанием
Ca(NO3J • 4Н2О, добавлением к раствору ионов SO'" илиСО23~>
осаждающих ионы кальция в виде CaSO4 или СаСО3.
Кроме того, можно прибавлять стабилизирующие добавки,
препятствующие выпадению Са3(РО4J, — борную кислоту,
лимонную кислоту и другие комплексообразователи; у нас
применяют для стабилизации соли магния — сульфат или фосфат.
Из многих вариантов использования азотнокислого раствора
приводим некоторые.
1. Нейтрализация растворов суспензией СаСО3 по уравнению
ЗН3РО4 + 5Са (Ш3J + ЗСаСОз + ЗН2О = ЗСаНРО4 • 2Н2О +
+ 5Са (Ш3J + ЗСО2.
Здесь Р2О5 находится в цитратно-растворимой форме. После
отделения преципитата фильтрацией выделяют Ca(NO3J
кристаллизацией при упаривании или обрабатывают углекислым
аммонием для получения нитрата аммония, например по реакции
Са (Ш3J + (NH4JCO3 = СаСОз + 2NH4NO3.
2. Нейтрализация раствора аммиака до рН = 5—6 по
уравнению
ЗН3РО4 + 5Са (NO3J+ 6NH3= 3CaHPO4+ 6NH4NO3 + 2Са (NO3J
После отделения преципитата раствор нитрата аммония
упаривают и обрабатывают, как было описано раньше.
3. Получение тройного удобрения обработкой раствора
сернокислым калием и аммиаком:
Н3РО4 '+ Са (NO3J + K2SO4 + 2NH3 = CaSO4 -f-
+ 2KNOS + (NH4J HPCY
Здесь Р2О5 находится в удобрении в водорастворимой форме.
Для получения тройных удобрений обычно смешивают простые.
Разнообразные сорта нитрофоски содержат: Р2О5 от 11 до 30%,
N — от 15 до 18%, К2О — от 15 до 25%.
Перспективным является получение тройного удобрения
упрощенным способом, заключающимся в обработке природного
1S9
фосфата смесями кислот, например H2SO4 + HNO3, без
отделения осадка. Реакцию можно изобразить так:
Ca^F (РО4K + 3HNO3 + 3,5H2SO4 + 7Н2О = ЗН3РО4 +
+ 1,5Са (NO3J + 3,5CaSO4 • 2Н2О + HF.
При усреднении полученной суспензии (пульпы) аммиаком
получается смесь преципитата, аммиачной селитры и
моноаммофоса с примесями:
ЗН3РО4 + 3,5CaSO4 • 2Н2О + 1,5Са (NO3J + 4,5NH3 =
= 3,5CaSO4 • 2Н2О+ 1,5СаНРО4 + 1,5(NH4)H2PO4 + 3NH4NO3.
Распространение получил упрощенный азотно-карбонатный
способ производства тройных удобрений, в которых отношение
N : РгО5 : К2О можно варьировать. Процесс заключается в
обработке азотнокислотной вытяжки при добавлении магнезита как
стабилизатора сначала аммиаком, а потом двуокисью углерода;
суммарно химизм можно выразить следующим уравнением
RCa (NO3), + ЗН3РО4 + 10NH3 -f 2CO2 + 2Н2О =
= ЗСаНРО4 + 10 (NH4) NO3 + 2СаСО3.
Полученную смесь (пульпу) смешивают с КС1 и с ретуром
для уменьшения влаги и сушат до содержания 1% влаги при
250—300°. Удобрение выпускается в виде гранул, отличающихся
малой слеживаемостью, благодаря чему они могут храниться
в складах без тары. P2Os входит в удобрение в цитратнораство-
римой форме. Таким образом, без отделения кальция и
разделения продуктов при сравнительно несложной аппаратуре можно
получать дешевые тройные удобрения, в которых сумма
питательных соединений доходит до 40% и выше
6. Микроудобрения
К микроудобрениям относятся вещества, потребляемые
растениями в малых количествах (несколько килограммов на
гектар). Активными элементами в них являются преимущественна
В, Mg, Mn, Cu, J, Со и другие редкие элементы, главное
назначение которых заключается в том, что они погшптают активность
ферментов, являющихся катализаторами биохимических
процессов в организме растений и животных. Они, активизируют
синтез углеводородов — сахара, крахмала, белков, витаминов и др.,
стимулируют развитие и созревание растений, повышают
устойчивость их при неблагоприятных условиях (холод, засуха,
вредители, болезни). Так, добавка 6—9 кг борной кислоты
повышает урожай клевера на 0,5—1 ц/га, овощей — на 2—5 ц/га,
магний входит в состав хлорофилла, содействуя, таким образом,
фотосинтезу в растениях.
190
Марганец ускоряет окислительно-восстановительные
процессы. При недостатке меди болеют кончики листьев («белая
чума»). Йод обеспечивает деятельность гормона тироксина,
кобальт и молибден облегчают усвоение азота клубеньковыми
растениями (бобовых). Содержание микроэлементов в почвах
различно и часто является недостаточным. Поэтому их
приходится вносить обычно вместе с другими веществами (маргани-
зированный суперфосфат или борный суперфосфат).
Микроудобрения применяют и отдельно (молибдат аммония).
Так, например, в состав одного из полных удобрений с
микроэлементами входят по 12 кг N2, Р2О5 и К2О, 2 кг MgO и по 100 г
бора и марганца, 40 г меди, 20 г цинка и 0,5 г кобальта.
Сырьем для производства микроудобрений являются
минералы, содержащие микроэлементы, например, бура и др., и
отходы разнообразных химических производств, металлургии и др.
Борные удобрения получают из маточных растворов после
выделения борной кислоты из борных руд — ашаритовых
BMgO • В2Оз • Н2О), содержащих 19—21% В2О3, датолитовых
(CaO-B2O3-2SiO2-H2O) с 9—13% В2О3, и другие. Растворы
с содержанием около 1,5% В2О3 по одному из вариантов
нейтрализуются известняком или мелом, полученная суспензия сушится.
Так как в маточниках имеются и соли магния (MgSO4), в
результате нейтрализации образуется боромагниевое удобрение, в
состав которого входит около 10% Н3ВОз и приблизительно 60%
MgO и СаО. Для получения борных удобрений прибегают к
непосредственной обработке руд. Например, датолитовое удобрение
получают путем обработки измельченного датолита 50% -ной
серной кислотой. Вызревание массы дает продукт с 8—10%
Н3ВО3, содержащий гипс и другие примеси. Применяемое в этом
случае оборудование сходно с оборудованием для производства
суперфосфата. Борные удобрения приготавливают также из рапы
озер, содержащих бор, из сопочных грязей, нефтяных вод
и др.
Обычно для производства боросуперфосфата датолитовое
удобрение смешивают с суперфосфатом в определенной
пропорции. Добавление к суперфосфату бората марганца дает
удобрение, содержащее Р2О5, МпО и В2О3.
Магниевые удобрения. В качестве простых
магниевых удобрений можно употреблять измельченные минералы,
содержащие магний: магнезит MgCO3, кизерит MgSO4 • Н2О,
карналлит КС1 • MgCb • 6Н2О и др.; применяют также смешанные
или сложные удобрения, содержащие Mg. Так, азотномагниевое
удобрение получают, смешивая расплав аммиачной селитры с
измельченным доломитом. Так как потребность ряда почв в
магниевых соединениях велика, важное значение приобретает
концентрированное комплексное удобрение магнийаммонийфосфат
MgNH4PO4-H2O, содержащее до 45,7% Р2О5, 10,9% NH3 и 25,9%
191
MgO. Его готовят из Н3РО4, NH3 и MgO или осаждением по
реакции
М?С12 + (NH4JHPO4 ™ NHtOH = Mg\;H4PO4 • Н2О + 2NH4C1.
Добавим, что MgO содержится в ряде отходов. Например,
п состав томасшлака, наряду с Р2О5, входит до 3% MgO.
Марганцевые удобрения применяются чаще всего
в виде MnSO4, получаемого при обработке марганцевых шламов,
содержащих МпО2. Они образуются из отходов, например, при
изготовлении электроэлементов. Для этого шлам смешивают
с опилками и концентрированной H2SO4 и подвергают массу
спеканию. В результате получается спекшаяся или рассыпчатая
масса, содержащая MnSO4. Добавляя к суперфосфату 10—15%
марганцевого шлама или спека, получают марганизированный
суперфосфат 17—18% Р2О5 и 1,5—2,5% Мп.
Медные удобрения приготовляют из руд с малым
содержанием меди, а также из пиритных огарков, которые,
наряду с Си, содержат также Zn, Co и другие элементы.
II. ЯДОХИМИКАТЫ
Большое значение имеют химические средства защиты
растений — ядохимикаты. Они служат для уничтожения вредителей
полевых, овощных и фруктовых культур — саранчи, клещей,
гусениц, а также сорняков и т. п. С каждым годом появляются новые
виды ядохимикатов и быстро растет их производство.
В качестве ядохимикатов используются как неорганические,
так и органические соединения мышьяка, ртути, меди, бария
и др.; последние применяются всё чаще. Наиболее
распространенными и эффективными являются хлорные дериваты
органических соединений, а также фосфорсодержащие яды, гексахлор-
циклогексан, ДДТ, тиофосы и др. Все эти ядохимикаты весьма
эффективны. Назовем несколько групп ядохимикатов.
1. Инсектициды и фунгициды — для борьбы с насекомыми,
клешами, вредящими растениям и запасам
сельскохозяйственных продуктов (органические хлорные дериваты — гексахлор,
линдап, хлортен, токсафсн; фосфороорганические — ткпфос,
метафос; неорганические—хлористый барий).
2. Фунгициды — для борьбы с болезнями растений (хлор-
окись меди, основные сульфаты меди, дитиокарбаматы, фенил-
меркурохлорид, фталимпды).
3. Протравители семян — для дезинфекции и предпосевного
обеззараживания семян (формалин, препараты мышьяка, ртути,
меди, трихлорфенолят меди, гексахлорбензол, гранозан).
4. Зооциды—для борьбы с грызунами (фосфид цинка,
карбонат б
192
5. Репелленты — для отпугивания птиц, кровососущих
комаров, гнуса (диметилфталат, диэтиламид).
6. Гербициды — для борьбы с сорняками (барбан,
тетрахлорбензол).
7. Дефолианты и дессиканты — для подсушки и
предуборочного удаления листвы для облегчения машинной уборки (пен-
тахлорфенолят натрия, хлорат магния, цианамид кальция).
8. Регуляторы роста растений и ускорения созревания
плодов (хлорфеноксиуксусная кислота, окись этилена).
Следует учесть, что многие ядохимикаты имеют комплексное
действие, кроме того, они применяются в смесях, причем в этом
случае воздействие их усиливается (синергизм).
Ядохимикаты используются в различном виде—как
аэрозоли, дусты, растворы и др.
Остановимся на производстве некоторых ядохимикатов.
Хлористый барий получают из тяжелого шпата
BaSO4 (реже из барита ВаСО3), восстанавливаемых углем при
температуре около 1000° С до сульфида BaS по реакции
BaSO4 + 2С = BaS + 2СО2
с последующей обработкой соляной кислотой
BaS +¦ 2НС1 = ВаС12 + H2S,
и получением раствора ВаСЬ. Из последнего кристаллизацией
выделяют продукт. Другой способ заключается в совместном
прокаливании шихты, состоящей из BaSO4 и СаСЬ, с углем
и последующем выщелачивании ВаСЬ, образовавшегося по
реакции
BaSO4 + СаС12 = ВаС12 + CaSO4.
ВаСЬ, как и все растворимые соли бария, является сильным
ядом.
Известков о-с ерные отвары (ИСО) готовят из CaS
и S.
Сернистый кальций производят из гипса аналогично
сернистому барию. Затем суспензию CaS в воде кипятят с тон-
коизмельченной серой. При этом образуется CaSn, где п = 2, 3,
4, 5. Эту суспензию полисульфида применяют против клещей —
вредителей хлопка, цитрусовых и др. Таким же путем готовят
весьма эффективные фунгициды и акарициды.
Фториды и кремнефториды обычно получают
улавливанием и соответствующей обработкой фторсодержащих газов,
выделяющихся при переработке апатитов и фосфоритов в
производстве фосфорных удобрений — суперфосфата и др. (стр. 171).
Медный купорос образуется при действии серной
кислоты на медь или окись меди, его получают, подвергая сульфати-
зирующему обжигу колчеданные огарки.
13 8-1504 193
Бургундскую жидкость готовят, обрабатывая
раствор медного купороса содой. Продукт представляет собой
суспензию основного карбоната меди состава 3Cu(OHJ* CuCO3.
Бордосскую жидкость получают, действуя на
раствор медного купороса гашеной известью; при этом получается
осадок примерного состава 3Cu(OHJ" CuSO4 + CaSO4.
Близкий к этому препарат АБ готовят, заменяя известь более
дешевым мелом, в результате чего образуется основная
углекислая соль по реакции
2CuSO4 + 2СаСО3 + Н2О = СиСО3 • Си (ОНJ + 2CaSO4 + 2СО2.
Препарат содержит около 15% цитратнорастворимой меди.
Парижская зелень представляет собой двойную соль
арсенита натрия и уксуснокислой меди состава, близкого к
2Cu(AsO2J- Cu(CH3COOJ, с содержанием около 51,5% As2O3.
Сырьем является белый мышьяк As2Oa, кальцинированная сода,
медный купорос и уксусная кислота.
Большое значение имеют органические ядохимикаты —
фосфор-, хлорсодержащис и др. Общее количество химических
средств защиты растений, произведенных в 1967 г. в пересчете
на 100% по действующему началу — составляет 123 тыс. т, или
107% к 1966 году.
ГЛАВА XII
ПРОМЫШЛЕННЫЙ ЭЛЕКТРОЛИЗ И ЭЛЕКТРОТЕРМИЯ
Широкое применение электричества в химической
промышленности привело к созданию двух крупных отраслей
производства — электрохимии и электротермии.
1. ОСНОВЫ ЭЛЕКТРОХИМИИ
Основы электрохимии были заложены М. Фарадеєм,
установившим основные законы электролиза. Большая роль в развитии
теоретической и технической электрохимии принадлежит
русским ученым В. В. Петрову, Ф. Гротгусу, Б. С. Якоби, П. П. Фе-
дотьеву, А. Н. Фрумкину, Н. А. Изгарышеву и многим другим.
Б основе электрохимии лежат два закона Фарадея, согласно
которым количества выделенных на электродах веществ прямо
пропорциональны количеству прошедшего через электролит
электричества и прямо пропорциональны эквивалентным весам
выделяющихся веществ.
Для выделения одного грамм-эквивалента любого вещества
необходимо пропустить через электролит 1 фарадей
электричества, равный 96 500 кулонам. Но так как кулон есть количество'
электричества, протекающее в одну секунду через поперечное
сечение проводника при силе тока в один ампер, то 96500
кулонов соответственно составляют 96 500:3600=26,8 ампер-часа
(а-ч).
194
При пропускании 1 а -ч электричества через электролит
выделяется количество вещества, которое называется
электрохимическим эквивалентом.
Электрохимический эквивалент для водорода составляет
- 2б8'= °>0376 Н2! для кислорода -~y = 0,3008 О2; для хлора
35,46 , ОГ)О _.
~2^= 1,323 С12 и т. д.
Вследствие потерь один ампер-час выделяет вещества
меньше, чем должно было быть по расчету.
Производительность аппарата (электролизера) определяется
количеством электричества, пропущенного через электролит. Что
касается второго фактора электрической энергии — напряжения,
то минимальным пределом его является теоретическое
напряжение разложения электролита. Это напряжение равно
алгебраической разности равновесных потенциалов на границе электрод —
раствор, необходимых для выделения катиона и аниона: Е =
= Е, - Ек.
Электродные потенциалы для аниона Еа и катиона Ек могут
быть рассчитаны по формулам Нернста
\па и Ел = Е0 — -^р\па2,
где Eq — нормальный электродный потенциал, п — валентность
иона, аи а2 — активности ионов в растворе электролита, R —
газовая постоянная, Т° К — температура, при которой ведется
электролиз, F — число Фарадея.
Имея, например, нормальный раствор ZnCb и найдя из
таблиц, что Ек = —0,76 в, а Еа = +1,332 в, можно определить
теоретическое напряжение разложения нормального раствора ZnCb
Е = 1,332— (—0,76) = 2,092 в.
Теоретическое напряжение разложения вычисляется также по
формуле Гиббса — Гельмгольца
F- Q 4-T*?
С ~TF + 1 dT'
где: Q—теплота реакции разложения соединения, подлежащего
dE_
dT
электролизу, дж; Е—электродвижущая сила (э. д. с), -т= —
температурный коэффициент э. д. с. Если значение Т~ мало,
можно пользоваться упрощенной формулой Томсона Е = —^.
Так, например, при электролизе воды Q = 239 000 дж п = 2, F =
= 96 500, и Е = 1,24 в, опытное определение дает Е = 1,23 в, что
мало отличается от вычисленного.
Практически электролиз осуществляется при напряжении
большем, чем теоретически рассчитанное, на величину
поляризации и перенапряжения. Кроме того, чтобы преодолеть сопротив-
13* 195
Рис. 83. Схема монополярной
ванны:
/ — аноды; 2 — катоды; 3 — кожух.
ление электролита и проводников первого рода (электродов,
проводов, шин, контактов), требуется дополнительное напряжение.
Электродвижущей силой поляризации называется
возникающая при работе электролизера на электродах э. д. с,
направленная против напряжения, приложенного извне для электролиза.
Причиной этого явления могут быть химические процессы,
протекающие на электродах, изменение концентрации раствора в
+ слое, прилежащем к электроду,
пассивирование последнего и др.
Величина перенапряжения
зависит от условий протекания
процесса электролиза — от
материала электрода, характера его
поверхности, силы тока,
приходящейся на единицу поверхности
электрода (плотности тока),
температуры и т. д. Не
останавливаясь подробно на зависимости
перенапряжения от всех этих
факторов, отметим большое влияние
природы вещества и состояния
его поверхности. Так, если
перенапряжение водорода на гладкой
'платине составляет примерно 0,003 в, то на ртути оно
достигает 0,910 в. Сопротивление электролита зависит от его свойств и
концентрации, от расстояния между электродами, конструкции
электролизера и др.
Чтобы снизить напряжение на ваннах, стремятся уменьшить
сопротивление электролита, прибегая для этого к повышению
концентрации раствора, прибавлению кислот, щелочей и солей
Повышение температуры электролита, как и сближение
электродов, уменьшает его сопротивление.
Увеличение поверхности электродов также уменьшает
напряжение. Однако увеличение поверхности электродов приводит
к уменьшению плотности тока, что не всегда допустимо.
В зависимости от схемы включения электродов в
электрическую сеть различают две группы ванн: монополярные и
биполярные. В монополярных ваннах (рис. 83) одна половина
электродов, параллельно соединенных с положительной шиной, является
анодом, а другая половина — катодом. При таком расположении
электродов напряжение на ванне определяется разностью
потенциалов на одной паре катод—анод, и сила тока пропорциональна
плотности тока и поверхности электродов одного и того же
знака.
В биполярных ваннах (рис. 84) ток подводится к крайним
электродам — аноду и катоду; тогда другие, промежуточные
электроды, например электрод 3, будут включены в электричес-
196
кую цепь последовательно, так что одна сторона электрода
служит катодом, а другая — анодом. Таким образом, промежуточные
электроды биполярны, а крайние — монополярны. Тогда
напряжение на ванне пропорционально числу пар анод—катод, а сила
тока зависит от плотности тока и поверхности только
униполярного электрода. Вследствие этого на эти ванны подается
большее напряжение, чем на монополярные при меньшей силе тока.
/
+ -
+ _
+ _
T7\.
+ _
+ Z.
+ -
+ ~
-F7L
+
+
+
-f
srx
S7X
Рис. 84. Схема биполярной ванны:
/ — анод; 2 — катод; 3 — биполярные электроды.
Промышленный электролиз характеризуется двумя
коэффициентами — коэффициентом использования тока (г]г) и
коэффициентом использования энергии (ті3). гіг представляет собой
отношение количества фактически получаемого на электроде
вещества Ліф к количеству вещества, которое должно было бы
выделиться по закону Фарадея (Мт) в % Ці — дР- ЮО. Значение ц3
получают, умножая ?j, на отношение теоретического
напряжения разложения (Ут) к фактическому напряжению, при
котором происходит электролиз (Уф):
Ф
2. ЭЛЕКТРОЛИЗ ВОДЫ
Электрохимический способ получения водорода и кислорода
заключается в разложении воды. В воде всегда имеются ионы
ОН~ и Н+ в равновесии с Н2О
Электролиз характеризуется следующими электродными
процессами на катоде: Н+ + е = Н; Н + Н = Н2 и на аноде: ОН~ —
— е = ОН; 2ОН = Н2О +-^О2. При этом на один ампер-час
выделяется
рода.
22І9б500 '
0>42 •* в°Д°Р°Да и 0,42/2 = 0,2] л
кисло197
Как мы видели, теоретический потенциал разложения воды
равен 1,23 в; величина перенапряжения при электролизе воды
составляет примерно столько же. Поэтому фактически процесс
ведется при напряжении около 2,5 в с выходом по току 92—94%
и выходом по энергии 45—50%.
Для получения чистых газов катодное пространство
отделяют от анодного диафрагмой, задерживающей взаимную
диффузию анолита, католита и газов. Диафрагмы изготавливают из
Рис. 85. Ванна с двойными плоскими электродами:
/—стальной кожух; 2 — электроды; 3 — стальной стержень; 4—газовый колокол; 5 —.
«упол колокола; 6 — выход газа; 7 —диафрагма; 8 — медная шииа; 9 — сборник для
водорода; 10 — сборник для кислорода.
асбеста, цемента, синтетических органических материалов,
а электроды — из коррознонностойких и хорошо проводящих ток
веществ — графита, никеля и пр.
При электролизе воды, загрязненной примесями —
сульфатами, хлоридами и др., происходит ряд нежелательных явлений.
К числу их относятся побочные реакции, увеличение коррозии
оборудования, потери энергии и др. Поэтому обычно применяют
чистую воду (паровой конденсат), к которой для уменьшения
сопротивления добавляют химически чистые электролиты,
например КОН до концентрации 25—30%.
Конструкций электролизеров известно много. На рис. 85
изображен один из электролизеров — монополярная ванна с
двойными плоскими электродами ящичной конструкции. Электроли-
198
зер представляет собой железный ящик со стальными двойными
электродами, состоящими из двух пластин толщиной 2 мм
каждая. Электроды отстоят друг от друга на расстояние 6 мм,
благодаря чему облегчена циркуляция электролита и газов.
В верхней части ванны укреплены газовые колокола, где
собираются выделяющиеся газы; к верхней части колоколов крепятся
электроды. Колокола имеют отверстия, через которые газы
уходят в общий колпак и в сборники водорода (с катода) и
кислорода (с анода). К газовому колоколу подвешена асбестовая
диафрагма в виде мешка, окружающая электроды. Площадь
каждого электрода составляет 1 лі2. Ток к электродам
подводится по медным шинам. В отводимых газах содержится до
99,6% О2 и до 99,9% Н2. Пространство над католитом и аноли-
том выбирается так, чтобы газы увлекали минимум брызг. Таким
образом экономится дорогой электролит и газы получаются
менее влажными.
В настоящее время распространены ванны так называемого
фильтрпрессного типа. Ванна состоит из большого числа
электролитических ячеек, зажатых стяжными болтами между
концевыми плитами (как в фильтрпрессе). Эти ванны компактны,
занимают мало места и надежны в работе. Напряжение на одной
электролитической ячейке — около 2,2 в. Удельные расходы
материалов и энергии на 1 м3 водорода составляют в среднем: 4,5—
5,5 кет • ч, 0,9 л дистиллированной воды, 0,6 кг щелочи КОН.
Представляет интерес электролиз под давлением, так как
доказано, что потенциал разложения и перенапряжение с
увеличением давления падают. Кроме того, установки для работы под
большим давлением занимают значительно меньше места. На
практике ведут процесс под давлением 8—10 атм.
3. ЭЛЕКТРОЛИЗ ВОДНЫХ РАСТВОРОВ ХЛОРИСТОГО НАТРИЯ
Основные уравнения процесса могут быть выражены
следующим образом:
NaCl зз Na+ + С1~, Н2О = Н+ + ОН~
на аноде С1~ — е = С1; С1+С1=С12;
на катоде Н+ + е = Н; Н + Н = Н2.
В результате процесса получаются три продукта:
газообразный хлор на аноде, газообразный водород на катоде и водный
раствор NaOH у катода.
Минимальное напряжение разложения NaCl в водном
растворе, которое можно найти из теплового эффекта суммарной
реакции процесса, равно 2,172 в. Но фактически напряжение при
разложении NaCl колеблется от 3 до 5 в, а ток используется на
92—95%.
199
Во избежание побочных реакций при электролизе
используется предварительно очищенный от примесей (ионов SO=, Ca++,
Mg++) рассол, почти насыщенный поваренной солью
(концентрация 300 г/л). При этом стремятся избежать перемешивания
анодной и катодной жидкости (анолита и католита).
Остановимся на побочных процессах при электролизе
раствора поваренной соли. Образующиеся в результате электролиза
первичные продукты реагируют с веществами, находящимися
в растворе. Так, хлор растворяется в воде и, реагируя с водой,
дает соляную и хлорноватистую кислоты по реакции
С12 + Н2О = НС1 + НС1О.
Часть ионов гидроксила диффундирует к аноду и реагирует по
реакции ОН- + НС1О = Н2О + СЮ~. Недиссоциированная
НСЮ может давать с ионом СЮ хлораты по реакции
СЮ~ + 2НС1О = СЮГ + 2СГ + 2Н+.
Ион СЮ~ разряжается на аноде, образуя активный кислород по
реакции
6С1О~ + ЗН2О = 2С1ОГ + 4С1- + 6Н+ +3 0 + &.
Выделяющийся кислород разрушает графитовый анод, давая
СО и СО2, загрязняющие хлор-газ.
Таким образом, в зависимости от условий электролиза
конечными продуктами могут являться не только хлор, водород и
едкий натр, но и другие вещества. В присутствии диафрагмы,
отделяющей анодное пространство от катодного, побочные
реакции мало развиваются. Меняя условия, можно направлять
процесс и получать иные продукты. Если для получения хлора,
щелочи и водорода стремятся отделить католит от анолита, то
для производства гипохлоритов NaCIO, КСЮ нужно, наоборот,
обеспечить возможно более полное их перемешивание;
электролиз ведут без диафрагмы при охлаждении.
Если вести процесс при нагревании до 80—90° С с
добавлением хромовокислого натрия, образующего пленку окиси хрома
на катоде, который препятствует катодному восстановлению, то
получается хлорат МаСЮ3 или КСЮз.
Для электрохимического получения перхлората NaClO4 или
КС 104 подвергают электролизу концентрированные растворы
хлорноватокислых солей при 30—35° С с платиновыми анодами
при 5,3 в.
Электролизеры. В качестве анода применяют главным
образом графит, уголь или магнетит (плавленая окись-закись
железа). Катодом служит железо или ртуть. Уменьшения
диффузии достигают введением пористой
перегородки—диафрагмы.
Из различных электролизеров, применяемых в СССР,
остановимся на компактной и легкой для монтажа вертикальной ванне
200
Х-2 (рис. 86). Эта ванна представляет собой железный цилиндр-
высотой около метра и примерно такого же диаметра. В ней
размещены по окружности 24 графитовых анода, железная катодная
сетка и диафрагма. Хлор отводят из анодного пространства,
а водород и щелочь — из катодного.
Хлор
ческшіщемк\Водорад
Электролитический
щело/с
Рис. 86. Ванна с вертикальной диафрагмой Х-2:
' — кожух; 2 — катод; 3— иатодная шина; 4 —
диафрагма; 5 —аноды; 6— анодная шина; 7 — медные пауки;
8 — ввод чистого рассола. ,
Ванны имеют нагрузку в несколько тысяч ампер и работают
при 3,5—3,7 в с выходом по току до 95%, а по энергии — до
60%. Получаемый хлор-газ содержит 96—98% С12) а водород
содержит 98—99% Н2.
Расходные коэффициенты на 1 т хлора составляют: NaCl
1,7—1,8 т, электроэнергии 3100—3200 кет • ч. Кроме того,
расходуется 8 кг графитовых электродов, 2 кг асбеста, 4—5 мг воды
и 0,75—1,0 т пара. Диафрагменный электролитический щелок
содержит 100—140 г/л NaOH и 160—200 г/л NaCl.
201
В связи со значительной потребностью в электролитическом
каустике и хлоре конструкции электролизеров непрерывно
совершенствуют, стремясь работать с большими нагрузками тока при
меньших затратах материалов, энергии и меньшей площади,
занимаемой ванной в цехе.
Внедрение разработанных нашими конструкторами
вертикальных цилиндрических ванн с вертикальным
перфорированным катодом и накладной листовой
НЮ ,9 6
Рис. 87. Схема ванны с осажденной диафрагмой:
1 — бетонное днище; 2 — анодный медный стержень; 3 — сталь-
нан катодная рама; 4 — слой свинца для улучшения контакта;
5—антикоррозионный слой (цемент и смола) для защиты
свинца; 6 — изоляторы; 7 — наружная катодная шина; 8—труба для
отвода щелочи; 9 — теплоизоляция; 10 — бетонная крышка; // —
графитовые анодные шины; 12 — катодные стальные сетки; 13 —
* наружная шниа, приваренная к катодной раме.
диафрагмой позволило увеличить мощность цехов. Но
наиболее экономичными оказались прямоугольные электролизеры
с развитой поверхностью катода и осажденной диафрагмой.
На рис. 87 представлена схема ванны с так называемой
осажденной диафрагмой, состоящей из трех частей:
нижней с анодным блоком, средней с катодами и верхней с
колпаком для газов.
Графитовые аноды, имеющие форму прямоугольных плит,
погружены нижним концом в свинец для улучшения контакта.
Аноды устанавливают так, что они попадают в промежутки
между карманообразными катодами.
В верхней части расположены хлороотвод и труба для
питания ванны рассолом. Такая ванна со 128 анодными плитами
шириной 16 см и высотой 60 см каждая, работая с нормальной
нагрузкой в 20 000 а при плотности тока 640 а/см2, производит
свыше 75 г хлора в сутки. Срок службы ванны — до 15 лет.
?02
Диафрагму наносят на катод, погружая его в суспензию
асбестовых волокон под разрежением. Это упрощает нанесение
диафрагмы и дает возможность получать равномерный слой
асбеста с равномерной протекаемостью электролита.
Эти ванны хорошо работают. Так, ванна БГК.-17
характеризуется следующими показателями: нагрузка доходит до 50 ка
(килоампер), плотность тока составляет 1000 а/ж2, а выход по
току достигает 96%, поэтому расход электроэнергии на тонну
каустической соды удалось снизить до 2550 кет • ч.
хлор
Рассол
Рис. 88. Схема ртутной ванны:
7 — электролизер; И— разлагатель; / — анод; 2—катод:
3 — элеватор или насос.
Для получения более чистых щелоков применяются ванны
с ртутным катодом. Использование ртути в качестве
катода основано на том, что перенапряжение водорода на ней
очень велико. Металлический натрий выделяется, образуя
с ртутью амальгаму при более низком потенциале; поэтому
электролиз NaCl с ртутным катодом приводит к образованию на
аноде только хлора. На катоде же образуется амальгама натрия,
а не NaOH.
Амальгама натрия разлагается затем водой, особенно быстро
в присутствии железа или графита. Таким образом, получение
NaOH отделено от получения хлора, вследствие чего NaOH
почти не имеет примесей. Воду на разложение амальгамы подают
с таким расчетом, чтобы получить каустические растворы
(щелока) концентрацией 400 г/л и больше.
Установка состоит из двух ванн (рис. 88). Первая из них /
(электролизер) изготовлена из бетона и имеет длину около 15 м,
ширину около 1,5 ж и высоту 270 мм. На дне ее находится слой
ртути толщиной 5 мм. Благодаря уклону ванны ртуть течет с
небольшой скоростью (около 12 см/сек). Ванна // (разлагатель)
отличается от описанной выше отсутствием электродов и
уклоном в противоположную сторону. Она предназначена для
разложения амальгамы.
203
Таким образом, ртуть движется непрерывно по дну обеих
ванн и передается из // в / элеватором или насосом.
Образование амальгамы и разложение ее происходит во время движения
ртути.
Полученный в электролизерах щелок обладает высокой
чистотой и содержит 400—600 г/л NaOH и более без упаривания.
Это отличает «ртутный каустик» от «диафрагменного».
В связи с возрастающей потребностью в чистом каустике для
производства искусственного шелка и других целей, мощность
ртутных ванн, например, типа БГК-р-101, удалось повысить до
нагрузки 100 на при катодной плотности тока 5000 а/м2, со
средним напряжением 4,3 в, выходом по току до 97% и расходом
электроэнергии около 3050 кет • ч/т NaOH. Количество ртути
на 1000 а нагрузки составляет 17—20 кг.
В новых ваннах уменьшен расход ртути и мощность насосов,
перекачивающих ее из разлагателя в электролизер.
Электролитический щелок идет на упаривание, а газообразный хлор —
в перерабатывающие цеха или на сушку и сжижение для
транспортировки в стальных баллонах. Сжижение хлора достигается
чаще всего сжатием его до 3—4 атм и охлаждением до минус
10—15° С.
4. ПЕРЕРАБОТКА ХЛОРА
Широкое развитие в нашей стране промышленности
полимерных материалов вызвало большую потребность в хлоре.
Хлор применяется для получения волокон, смол
(полихлорвиниловых и др.) и синтетических каучуков (хлоропренового). Из
полученного с помощью хлора SiCl4 готовят кремнийорганиче-
ские полимеры, а также элементарный кремний.
Большое значение приобретает хлор и в производстве
ядохимикатов (инсектофунгицидов), необходимых для борьбы с
вредителями и болезнями растений.
Хлор находит применение также в производстве
синтетической соляной кислоты, глицерина через аллилхлорид,
хлорбензола, моющих веществ, фреонов и др. В виде хлорной извести
и гипохлоритов хлор используется для беления бумаги, тканей,
дли дезинфекции и дегазации.
Мировое производство хлора (без СССР) составило в 1960 г.
8,2, а в 1961 г.— 9 млн. тонн.
Потребность в хлоре сейчас возросла настолько, что ее нельзя
удовлетворить только путем электролиза растворов поваренной
соли. Кроме того, при этом методе получается едкий натрий,
потребность в котором растет медленнее, чем в хлоре. В
настоящее время возникают методы производства хлора, не
связанные с производством каустической соды. К ним относится
электролиз хлористых солей, дающих в качестве второго продукта
металлы. Таким путем возникла т. н. «хлорная» металлургия.
204
Соляная кислота
Соляная кислота и безводный хлористый водород находят
широкое применение. Соляная кислота употребляется для
производства различных хлоридов — металлов и неметаллов; при
гидролизе клетчатки, в производстве глюкозы, в
гальванопластике, в металлической промышленности для снятия ржавчины, при
крашении, очистке песка от железа, в текстильной
промышленности и др. Безводный НС1 используется для получения
хлоропренового каучука и других продуктов.
газов
Рис. 89. Механическая сульфатная печь:
/ — бункер поваренной солн; 2—ввод отопительного газа; 3 —
муфель; 4 —і ввод серной кислоты; 5^ вал; 6 — гребки; 7 —
зубья; 8 — бункер сульфата; 9 — мельница для сульфата; 10 —<
труба для отвода хлористого водорода.
Техническая соляная кислота выпускается в виде водного
раствора хлористого водорода, концентрацией обычно около
27% НС1.
Процесс образования соляной кислоты имеет два этапа:
производство газообразного НС1 и растворение его в воде.
Хлористый водород получали в пламенных сульфатных печах, а затем
в муфельных печах, с ручным обслуживанием. При этом сырьем
служили поваренная соль и серная кислота, которые реагируют
по суммарному уравнению
2NaCl + H2SO4 = Na2SO4 + 2НС1 — 68,25 кдж.
В настоящее время для производства Na2SO4 и НС1 применяют
механические сульфатные печи непрерывного действия
муфельного типа (рис. 89). Процесс протекает в муфеле, снаружи
обогреваемом горячими газами, получаемыми при сжигании гене-
205
раторного газа. Муфель позволяет избежать проникновения
продуктов горения в НС1.
Изготовлен муфель из кислотоупорной керамики и имеет
вращающийся вал с насаженными гребками и зубьями для
перемешивания сырья и выдачи Na2SO4 у периферии печи. При
диаметре чаши 3 м можно получать до 8 т сульфата в сутки.
Расход топлива на 1 т сульфата составляет около 350 кг каменного-
угля. НС1 после охлаждения поглощается водой. На
производство 1 т соляной кислоты концентрацией 27,5% идет около 450 кг
NaCl и 420 кг H2SO4 (92,5%).
Сульфат натрия применяют в производстве стекла вместо
кальцинированной соды. Восстановлением из него пелучают
сульфид натрия, широко используемый при обработке шкур
в кожевенном деле, в производстве сернистых красителей, при
получении сульфита, гипосульфита и пр.
Несмотря на то, что производство Na2SO4 и НС1
механизировано, выделяющийся газ отличается недостаточной чистотой
и малой концентрацией. Сульфат натрия можно получить
выпариванием также из морской и озерной воды. НС1 получают и
другими методами, например из угля, хлора и воды: при 700—800°
идет реакция
С + Н2О + С12 = СО + 2НС1.
Хлористый водород образуется при хлорировании органических
веществ, содержащих водород, например бензола:
СвНв+С12=СвН8С1+НС1.
Наибольшее распространение получил синтез соляной кислоты
из водорода и хлора.
Синтетическая соляная кислота
Сырьем для производства синтетической соляной кислоты
служат водород и газообразный хлор, которые реагируют по
реакции
Н„ + аг^2НС1 + 184,23 кдж.
В результате реакции получается смесь, состоящая из НС1 + Н2,
так как дается избыток водорода. Температура реакции 2200° С.
Константа равновесия определяется из уравнения
lg/Cp = =р- 0,553 lg T -2,42.
Если х — доля молекул НС1, диссоциированных на Н2 и С12 и
находящихся в равновесии с недиссоциированной частью A—х)
молекул НС1, то
4 A -
Ар = ¦?
206
Для интервала температур от 20 до 2200° х меняется от
2,95-Ю-17 до 1,22-К)-2. Таким образом, концентрация хлора
в синтетическом НС1 крайне мала, а при избытке содержания
водорода — ничтожна.
Употребляя дистиллированную воду и очень стойкую
керамику или кварц, можно синтезировать почти химически чистую
соляную кислоту. Реакция идет в контактной печи,
представляющей собой вертикальную трубу из специальной стали высотой
6 м. В нижнюю часть трубы входит горелка, состоящая из двух
стальных концентрически расположенных труб. Сухой хлор (90—
95% С12) подается снизу во внутреннюю трубу, имеющую в
верхней части отверстия. Через внешнюю трубу поступает
осушенный водород (96—98% Нг). Образовавшийся хлористый водород
(85—90% НС1) уходит из верхней части печи и поглощается
водой.
Производительность таких печей доходит до 30 т хлористого
водорода в сутки и более при следующих расходных
коэффициентах на т НС!: хлора A00%-ного) 0,975 т, водорода
A00%-ного) 375 лі3.
Абсорбция НС1
При поглощении хлористого водорода водой образуется соля-
ная кислота. Выделяющееся тепло используется для получения
концентрированной соляной кислоты по способу адиабатической
абсорбции в башнях с насадкой или отводится путем
поверхностного охлаждения водой в поглотительные сосуды
(керамические туриллы, кварцевые аппараты и др.).
Применяя последний способ, до недавнего времени считали,,
что образующаяся соляная кислота должна соответствовать
азеотропной смеси и поэтому может иметь концентрацию не
выше 20—24%. В связи с этим старались избежать температур,
при которых кислота кипит.
В настоящее время в СССР осуществляется адиабатическая
абсорбция по методу Гаспаряна, значительно упрощающая весь
процесс. Гаспарян показал, что при абсорбции НС1 водой в
адиабатических условиях, когда теплота растворения не отводится,
кислота закипает. Но поскольку при ее кипении расходуется
тепло на испарение воды, концентрация кислоты повышается до
31-33%.
На рис. 90 представлена кривая зависимости температуры
кипения системы НС1 — Н2О от концентрации (мольные доли)
при общем давлении 700 мм рт. ст. Из рисунка видно, что
кислота, содержащая 0,13 мольных долей НС1 B0,4%),
представляет собой азеотропную смесь с максимальной температурой
кипения 105,8° С.
2&7
Если парциальное давление в газе выше, чем давление над
постоянно кипящей смесью, то концентрация кислоты возрас-
W
ofii aw w иг ез at as
"U мольные доли
Рис. 90. Диаграмма кипения системы НС1 — Н2О.
тает. Таким образом, кипящая в адиабатических условиях
кислота будет концентрироваться.
Переходя к технологическому оформлению адиабатической
абсорбции, следует ука-
Вода зать на то, что с увел и-
' чением концентрации
кислоты увеличивается
упругость НС1 над ней,
а следовательно, будет
оставаться больше
непоглощенного НС1.
Поэтому абсорбцию
следует проводить либо в
ряде абсорберов,
которые газ проходит
последовательно, либо, как
это принято на заводах,
в колонне барботажно-
го типа с необходимым
числом тарелок. Но
поскольку НС1 хорошо
растворяется в воде, число тарелок невелико (для 27,5%-ной
кислоты — четыре, для 31 %-ной — пять).
Так, один абсорбер из фаолита высотой 6,4 м и диаметром
208
бодород
Соляная кислота
Рис. 91. Схема производства синтетической
соляной кислоты по принципу
адиабатической абсорбции.
0,45 м обеспечивает получение 30 т 31% -ной кислоты в сутки при
работе на синтетическом НС1, имеющем температуру 250° С.
На рис. 91 дана схема адиабатической абсорбции такого газа
водой. Хлористый водород, образовавшийся в печи / из водорода
и хлора, охладившись в газоотходе 2 до 25° С, поступает на
водную абсорбцию в башню 3, где и образуется соляная кислота.
Неуловленные газы, пройдя башню 4, выбрасываются
эксгаустером 5.
Хлорная известь
Хлорная (белильная) известь представляет собой белый
с желтоватым оттенком сухой порошок, пахнущий хлором; она
широко применяется для отбелки тканей, целлюлозы, бумажной
массы, для дезинфекции питьевой воды, для дегазации, борьбы
с вредителями сельского хозяйства. Эти свойства обусловлены
тем, что в ней содержатся соли хлорноватистой кислоты НСЮ,
легко разлагающейся под действием влаги, кислот, СО2 и др.
с выделением кислорода и хлора.
Хлорная известь образуется при взаимодействии хлора с очень
чистой сухой пушонкой, получаемой при гашении извести СаО
водой так, что в Са(ОНJ остается 1—2% Н2О. Хлорирование
производят в полочных аппаратах, имеющих вид башен,
оборудованных вращающимся валом с гребками и зубьями, как в
полочных печах для обжига пирита. Пушонка подается сверху,
навстречу хлору; продукт выпускается из нижней части башни
и содержит 32—35% активного хлора. Для регулирования
температуры на полках (не выше 35—40° С) служат стальные
змеевики, через которые течет вода или охлаждающий рассол.
Суммарная реакция хлорирования может быть выражена
следующим уравнением:
2Са (ОН)„ + 2С12 = Са (ОС1J + СаС12 + 2Н2О.
Однако получающийся продукт содержит и другие, кроме
Са(ОС1Ь, продукты хлорирования: дветретиосновный
ЗСа(ОС1J • 2Са(ОНJ • 2Н2О, двухосновный Са(ОС1J • 2Са(ОНJ,
а также СаС12, Са(ОНJ, SiO2 и другие компоненты. Поэтому
активного хлора в нем меньше и он разлагается; сильное
каталитическое действие на белильную известь оказывают
соединения никеля, железа и марганца.
Расходные коэффициенты на 1 т хлорной извести,
содержащей 32—35% активного хлора, составляют: 0,5 т СаО;
0,36—0,38 т С12; 11—20 кет ¦ ч и 18—20 м3 воды.
В качестве более ценных отбеливающих и дезинфицирующих
веществ употребляют также другие кислородсодержащие
соединения хлора, например, окись хлора, гипохлорит кальция,
натрия и др.
14 8-1504 209
Гипохлорит кальция (ЗСа (С1ОJ'2Са (ОНJ) вытесняет
хлорную известь, имея ряд преимуществ перед белильной известью:
он содержит почти вдвое больше белящего хлора D7 — 52%
вместо 32 — 35%), менее гигроскопичен, более устойчив, дольше
сохраняется и растворяется в воде почти без остатка. Его
производят хлорированием известкового молока концентрацией
410 г\л Са(ОН)г- Большое значение имеет то обстоятельство, что
относительный вес балласта и тары на единицу активного хлора
при транспортировке и хранении для гипохлорита кальция
меньше, чем для других аналогичных продуктов; если для хлорной
извести этот вес составляет 250 — 300%, а для жидкого хлора
в баллонах— 100—200%, то для гипохлорита — всего 60—90%.
5. ЭЛЕКТРОЛИЗ РАСПЛАВОВ
Металлы первых трех групп периодической системы Na, К, Са,
Mg, A1 и др. получают во все больших масштабах, главным
образом электрохимическим путем. Эти металлы являются
электроотрицательными, и потенциал их выделения на катоде менее
положителен, чем потенциал выделения водорода. Поэтому
такие металлы нельзя выделять электролизом водных растворов.
Их получают электролизом расплавленных соединений — солей
и окислов.
Ванны для электролиза расплавов должны монтироваться
из огнеупорных материалов, причем обогрев производится либо
топочными газами, либо током, вследствие чего расход
электроэнергии обычно велик.
Электролиз расплавов протекает так же, как и электролиз
водных растворов при постоянном токе, но с меньшим выходом
по току и энергии. Это обусловлено тем, что электролит
находится в электролизерах при высокой температуре, вследствие чего
улетучивается некоторое количество сырья и продукта, а часть
последнего растворяется в расплавленном электролите.
Чтобы увеличить выход продукта, температуру плавления
электролита стремятся снизить добавлением веществ,
образующих легкоплавкие эвтектики.
Процесс электролиза расплавов осложняется также
образованием пузырей и неоднородностью среды у электродов.
Особенно вреден анодный эффект, проявляющийся в том, что процесс
электролиза нарушается резким повышением напряжения и
падением силы тока с появлением искровых разрядов; это вызывает
перерасход анерги-и и потерю электролита.
Получение металлического алюминия
Алюминий является наиболее широко применяемым цветным
металлом. Его используют в авиа-, ракето-, машино- и
приборостроении. Широкое применение алюминия и его сплавов в быту
хорошо известно.
210
Алюминий получают электролизом криолито-глиноземного
расплава, представляющего собой раствор А^Оз в криолите
3NaF-AlF3. 14,8%-ный раствор АЬО3 в криолите образует
эвтектику с температурой плавления 938° С.
Для снижения температуры плавления смеси в ванну
добавляют около 10% добавок CaF2, MgF2 и др.
Таким образом, в расплавленном электролите находятся
различные ионы, на которые диссоциируют растворитель, добавки
Рис. 92. Схема поперечного разреза алюминиевой
ваины:
1 — кожух; 2 — фундамент; 3 — подина; 4 — боковая
футеропка из угольных плит; 4' — огнеупорная
футеровка; 5 — электролит; 6 — алюминий, окись и криолит; 7—
катодный стержень; 8 — шинопровод; 9 — анод; 10 —
электродная тестообразная масса; // — анодные шины;
12 — штыри; 13 — гарнисаж; 14 — шинки; 15 — угольная
подушка; 16 — анодная рама; 17—алюминиевая обечайка.
и глинозем. Но так как потенциал разложения А12О3 B,2 в)
наименьший, то на электродах обычно разряжаются только его
ионы, причем на катоде выделяется А1, а на угольном
аноде образуются из кислорода окиси алюминия СО и ССЬ; анод
частично сгорает. Эти процессы можно изобразить следующим
образом:
на аноде 2АЮ|~ + 2С — бе = А12О3 + СО + СО2;
на катоде 2А13+ + бе = 2А1.
Убыль АЬОз восполняется подачей свежих порций глинозема.
Электролитическая ванна (рис. 92). состоит из металли-
14*
211
ческого кожуха, футерованного огнеупором, подины, состоящей
из углеродистых блоков. К блокам подведен шинопровод с
катодными стержнями, на подвижной раме закреплен
самообжигающийся анод. Ток к аноду подводят посредством штырей, которые
по мере обгорання вместе с анодом подаются вниз.
Расстояние между анодом и катодом (межполюсное
расстояние) достигает 4—5 см. Его можно регулировать, перемещая
подвижный анод вверх или вниз. Напряжение составляет
4,5—5 в. Плотность анодного тока 1—0,7 а/см2, температура
расплава около 950° С. Ванны рассчитываются на нагрузки от
десятков до 150 ка и более. Выход по току равен 85—90%, а
выход по энергии около 30%. При этом на 1 т расходуется 17 000 —
20 000 квт-ч электроэнергии.
Получаемый металл содержит около 99,5% А1. Его
рафинируют и переплавляют в чушки, содержащие до 99,9996% А1.
Алюминий, предназначенный для использования в ядерной
и полупроводниковой технике, подвергают особой очистке,
после которой получают «сверхчистый» металл: 99,99999% А1.
Производство металлического магния
Магний принадлежит к распространенным элементам B,35%
земной коры). Его сплавы являются незаменимым материалом
в транспортной промышленности, авиа-
и приборостроении. Он входит в состав
ракетного топлива, используется для сфе-
роидизации углерода чугуна при
выплавке чугуна высокой прочности, в органи-
7 ческом синтезе и т. д.
Магний получают термическим и
электрохимическим способами. Главное
значение имеет последний метод. Электролизу
подвергают чаще всего расплавленный
хлорид MgCl2 или обезвоженный карнал-
Рис. 93. Схема ванны лит KCl-MgCl2. MgCl2 плавится при
для электрохимического 718оС а гмег,и рго с пггявнем шпр ГпР.
производства металличе- ёоло г- г"" * '
ского магния из карнал- и др.) — при 680 С. Еще более низкую
температуру плаьлении имеет металлн-
ЧЄСКИЙ МЭГНИЙ 651°, ПрИЧЄМ бЛЭГОДарЯ
СВОЄМУ МЄНЬШ6МУ уДЄЛЬНОМу Весу ЖИД-
КИЙ Металл ВСПЛЫВаеТ.
При ЗЛЄКТрОЛИЗЄ ПРОИСХОДЯТ ТЭКИе
процессы. на кат()де Mg+ + « = Mg; На
аноде С1-— е = CI; С1 + С1 = С12.
На рис. 93 приведена схема ванны для электролиза
карналлита.
Напряжение между электродами ванны составляет 6—6,5 в.
лнта:
/- железные катоды; 2
теплоилоляиия: 5-огне-
упорная футеровка; 6 — трі1-
ба для отвода газообраз-
ного хлора^-стальной
212
Плотность тока 0,5—0,9 а/см1*. Выход по току 80—85%, а по
энергии около 37%, температура ванны примерно 750°С.
На 1 т магния расходуется электроэнергии 17,5—18 тыс.
кет • ч. Расход MgCb 4,5—4,7 т или карналлита 10 г, расход
анодов 20—25 кг.
в. ЭЛЕКТРОТЕРМИЧЕСКИЕ ПРОЦЕССЫ
Электротермия широко применяется в нашей
промышленности, так как она дает возможность достичь высоких температур,
необходимых для ряда производств. Преимуществом данного
способа является отсутствие золы в электродах, поэтому
продукт загрязняется лишь углеродом из подводящих ток
электродов, а в индукционных печах нет и этого источника примесей.
При электротермии не нужен воздух. Поэтому процесс может
быть осуществлен как в восстановительной, так и в
нейтральной среде, что особенно важно при производстве фосфора,
цианамида, сероуглерода и др.
Широкие возможности применения электротермии в нашей
стране обусловлены вводом в действие новых электростанций
и связанным с этим удешевлением электроэнергии.
В настоящее время на производство 1 г готового продукта
расходуется электроэнергии (в кет. ч): в случае фосфора 15—
22, карбида кальция — от 3 до 3,5, искусственного графита —
от 5 до 7, корунда — от 3 до 4, карборунда — от 8 до 9 и
ферросплавов — от 10 до 14 кет • ч.
В электротермических печах обычно используется
переменный ток низкого напряжения. Количество выделяющегося тепла
определяется по закону Джоуля
Q = El cos <p • х,
где Q — количество тепла, дж; Е — напряжение электрического
тока, в; I — сила тока, a; cos ср — коэффициент мощности @,8—
0,98); ф — угол сдвига фаз; т — время, сек.
Электротермические печи
Используемые в электротермии печи подразделяются на
четыре класса.
1) Дуговые печи, в которых осуществляется прямой или
косвенный нагрев. В дуговых печах прямого нагрева электрическая
дуга помещена в реагирующую систему. Такие печи
применяются при реакциях между газами, например, при окислении азота
воздуха кислородом. В дуговых печах косвенного нагрева шихта
нагревается теплом электрической дуги.
2) Печи сопротивления, которые также бывают различных
видов,
213
В печах непосредственного нагрева электрическим
сопротивлением является шихта. В печах косвенного нагрева ток
проходит через специально установленные в печи сопротивления.
Горизонтальные печи этого класса применяются в производстве
карборунда, карбида бора и др. К печам косвенного нагрева
относятся также многочисленные печи, обогреваемые током,
проходящим через металлические сопротивления,— проволоку,
ленты из нихрома, хромаля и других высокоомных сплавов или
через неметаллические сопротивления — криптол и пр. Такие
печи широко применяются и в лабораторной практике.
3) Комбинированные печи, где нагрев осуществляется дугой
и сопротивлением самой шихты. На практике эти печи
встречаются чаще всего.
4) Индукционные печи. В таких печах производится плавка
чистых металлов. Индукционные печи не имеют электродов,
поэтому их применяют там, где важно избежать загрязнения
продуктов углеродом (от электродов).
Электроды. Электроды должны удовлетворять ряду
требований. Желательно, чтобы они приближались по составу к
чистому углероду, так как при обгорании электрода зола попадает
в электролит (допустимая зольность 0,5%), обладали
химической стойкостью по отношению к содержимому ванны,
механической прочностью, плотностью, большой электропроводностью-
Кроме того, в них не должно быть трещин.
Исходным материалом для электродов служат самые чистые
сорта кокса — нефтяной или пековый кокс. Кокс прокаливают
для удаления летучих веществ, размалывают и классифицируют
по размерам зерен на фракции. Из последних составляется
смесь, к которой добавляют связующие — смолу и пек. Из
полученной массы формуют электроды на прессах под давлением
1000—1500 кг/см2. Затем электроды обжигают при 1400° С
в печах без доступа воздуха в течение 10—12 суток, чтобы
придать им прочность.
После этого в остывшие блоки закладывают чугунные
стержни, посредством которых блоки подвешиваются в ванне. В
последнее время применяют обычно непрерывно самообжнгаю-
щиеся электроды. Такие электроды (рис. 94) состоят из метал-
лическоги кожуха, в котором находится электродная масса. По
мере опускания выгорающего в печи электрода кожух
наращивается сверху и в него забрасывается электродная масса.
Движущаяся вниз масса размягчается в зонах повышенной
температуры и заполняет кожух. По мере опускания в ванну
электроды прокаливаются.
Вес составного электрода в крупных промышленных печах
достигает нескольких тонн. Чтобы удерживать его в
вертикальном положении и подавать вниз по мере обгорання, требуются
специальные приспособления. Важно обеспечить хороший кон-
214
такт токопроводов с электродами, места подвода тока обычно
охлаждаются с помощью специально встроенных водяных
холодильников.
Промышленные электротермические установки состоят из
основного агрегата — печи и трансформатора. Последний
располагают по возможности ближе к печи, чтобы избежать
электрических потерь, так как печь нагревается током в десятки
тысяч ампер (и всего около сотни вольт). Над печью находятся
бункеры с сырьем, откуда по
течкам сырье поступает в
печь. Сверху из печи
отводятся выделяющиеся газы
и пары. В нижней части печи
имеются летки для удаления
жидкого продукта (и
шлака). Для кладки печи
применяются огнеупорные
материалы и специальная
электродная масса в форме
кирпичей и блоков.
Производство
карбида кальция.
Карбид кальция применяется в
больших количествах для
получения ацетилена,
который НеобхОДИМ При СВарКе тированное vacma
и резке металлов, а также
при синтезе многочисленных
органических веществ.
Особенно важную роль играет
он в производстве
синтетического каучука — хлоро-
прена.
Карбид кальция применяется как сырье в производстве
цианамида, для обессеривания чугуна и др. Карбид кальция
получают, прокаливая смесь извести и угля (антрацита или кокса)
при температуре около 2500° С. При этом происходит реакция
СаО + ЗС ї=ї СаС2 + СО — 452 кдж.
Процесс осуществляется в больших электрических печах
током 50—90 в, мощность печей достигает 50 000 кет и больше
при плотности тока 10 а/см2 электрода и более.
Карбид выпускается из печи в жидком состоянии, а окись
углерода уходит из верхней части обычно открытой печи.
На рис. 95 показан схематический разрез однофазной
карбидной печи. В трехфазных печах электроды помещаются
параллельно друг другу на таком расстоянии, что образуются три
Наеретая размие-
ченнаа масса
Лолусле/сшарся
масса
Сткшався графи-
тирсданна*
электрода
Рис. 94. Непрерывно наращиваемый
электрод.
215
расплавленных очага, которые отделены друг от друга и от
стенок непрореагировавшеи смесью.
Очаг плавления имеет выпускное отверстие, закрытое
затвердевшим карбидом. Перед выпуском карбида корка плавится
электрической дугой. Расплавленный карбид выпускается в
железные изложницы и затем идет на охлаждение.
Рис. 95. Схема однофазной закрытой
карбидной печи:
/ — верхний электрод; 2 — бункер для шихты; 3 —.
шнек; 4 — пневматическое устройство пля
разрыхления шихты; 5 — труба для отвода газов, 6 —
шнхта; 7 — подовый электрод; в — летка для
спуска карбида.
Качество карбида оценивается его литражем — количеством
ацетилена в литрах, который выделяется из 1 кг карбида при
взаимодействии с водой по реакции
СаС2 + 2Н2О = Са (ОНJ + С2Н2.
I кг химически чистого карбида кальция выделяет 350 л С2Н2
при температуре 0° С и давлении 760 мм. рт. ст. Обычный
технический карбид в зависимости от сорта имеет литраж 250—
270 л.
216
Расходные коэффициенты на 1 т карбида литражем 270 л-
следующие: электроэнергии 3 000—3 200 кет. ч., извести 900—
960 кг, кокса 600—650 кг, электродов 20—40 кг.
Производство цианамида кальция. Цианамид
кальция применяется в качестве азотного удобрения, а также
как дефолиант, например, для уничтожения листьев
хлопчатника перед сбором коробочек. Из цианамида кальция можно'
получить цианиды, аммиак, а также дицианамид (H2C2N2),
идущий на приготовление меламиновых пластмасс, взрывчатых
веществ, фармацевтических препаратов и пр.
Цианамид кальция получают азотированием измельченного'
карбида кальция в печах по реакции
СаС2 + N2 = CaGN2 -+- С + 293 кдж.
Технический продукт черного цвета содержит, как видно из
уравнения, углерод, выделяющийся в ходе реакции. Процесс
получения цианамида кальция осуществляется в цианамидных
печах разной конструкции, которые подразделяются на печи-
периодического и непрерывного действия. Первые из них
представляют собой цилиндры из огнеупорного материала, в
которые насыпан измельченный карбид.
В центре массы устанавливается электрод для нагрева смеси
до начала реакции при 800° С. Печь герметически закрывается,
и чистый азот подается снизу. Масса сильно разогревается за
счет теплоты реакции и примерно через 24 ч процесс
заканчивается. После остывания печи из нее вынимают горячий
сплавленный черный блок цианамида кальция. Блок охлаждают,
дробят, измельчают, массу обрабатывают водой для
разложения остатка СаСг, а затем маслом и отправляют на склад как
готовый удобрительный тук. Содержание азота в туке — около
19%.
Печи непрерывного действия для азотирования, так
называемые канальные (туннельные), представляют собой длинный
канал с рельсами. По этому каналу в одном направлении
медленно движутся платформы с карбидом, а в противоположном
направлении пускается газообразный азот. В зоне нагрева
канала начинается азотирование, которое заканчивается во время
движения платформы. Платформа выходит из канала с почти
остывшим блоком цианамида.
В последнее время применяется метод фиксации азота в
виде цианидов. Цианиды можно получать также из цианамида
прокаливанием с углем и поваренной солью при 1400—1500° С
по реакции
CaCN2 + С + 2NaCl = CaCL2 + 2NaCN.
Производство ацетилена из карбида
кальция. Ацетилен получают из воды и измельченного карбида
кальция по реакции
СаС, + 2Н2О = Са (ОНJ + С2Н2.
21?
Этот процесс осуществляется в генераторе ацетилена, где
происходит смешение реагентов и образование газа и извести;
генераторы бывают различного устройства и разнообразных
размеров.
На рис. 96 дана схема установки для получения больших
количеств ацетилена в «сухом» генераторе непрерывного дей-
СаСг
Вода
Рис. 96. Схема получения ацетилена в «сухом» генераторе:
/ — приемный бункер; 2 — автоматический затвор; 3 — буферный бункер; 4 — шне-
ковый питатель; 5 — генератор; 6 — шнек сухой извести; 7 — охладительный
скруббер; S — отстойник; 9 — холодильник.
стр.ия вертикального типа, где потребное количество воды
подается на карбид. Измельченный до 4—5 мм карбид
предварительно очищают магнитной сепарацией от примесей
ферросилиция. Как видно из схемы, куски карбида поступают в
приемный бункер и через автоматический затвор — в буферный
бункер, откуда питатель подает их в генератор. Бункеры, а также
затвор продувают азотом, чтобы избежать попадания воздуха
в генератор. Кусочки карбида движутся по полкам генератора
вниз гребками, насаженными на вал, подобно колчедану в
полочных печах. В генератор подается вода, количество которой
регулируют таким образом, чтобы из нижней части генератора
уходила сухая известь, удаляемая шнеком.
Горячий ацетилен проходит скруббер, орошаемый водой,
причем снизу скруббера в отстойник удаляется пыль и
известковое молоко; часть осветленной воды возвращается через
оросительный холодильник в систему. Очистка ацетилена от H2S,
РНз, NH3 и других примесей производится хлорной водой или
другими окислителями.
Генератор, имеющий диаметр 3,5 м и высоту 7,5 м,
производит в час до 2000 мъ и больше ацетилена.
Получение фосфора и фосфорной кислоты
электротермическим способом
Важное место среди электротермических процессов
занимает электротермическая возгонка фосфора из фосфатов, так
как фосфор и его соединения находят широкое применение в
народном хозяйстве.
Этот способ основан на том, что при Ї500—i600"C SiO2
вытесняет РгО5 из его соединений, а присутствующий
восстановитель — уголь восстанавливает P2Os до фосфора.
Нагревание шихты до 1500—1600° С происходит в печах
типа доменных. Однако, при использовании для нагревания
электрической энергии установка может быть упрощена и
фосфор получается более чистый.
Большая часть фосфора перерабатывается в фосфорную
кислоту.
Получение фосфорной кислоты термическим способом
основано на том, что из сырья сначала получают возгонкой
элементарный фосфор, который затем сжигают до Р2О5. Последний,
растворяясь в воде, дает фосфорную кислоту любой
концентрации (до 100%).
Возгонка фосфора осуществляется в электропечах или в
печах типа домен, куда загружают смесь из кусков фосфата,
крупного песка или дробленного кварца и кокса или антрацита;
процесс изображается следующей суммарной реакцией:
Са3 (РО4J + 3SiO2 + 5С = 3CaSiO3 + 5СО2 + Р2 — 1530 кдж.
Применяемые при электровозгонке электрические печи
питаются от трансформатора током напряжения 60—80 в для
однофазных и 220—280 в — для трехфазных печей при плотности тока
отЗ до 12 а/см2.
Приводим схему заводской печи мощностью 15 000 кет и
суточной производительностью 28 т фосфора (рис. 97). Печь
представляет собой охлаждаемый водой стальной кожух длиной
9,75 м, шириной 4,6 м и высотой 3,7 м. Днище печи выложено из
угольных блоков толщиной примерно 1,2 м. Начиная от днища
219
и выше уровня шлака стены футерованы угольными блоками.
Еще выше футеровка стен выполнена из высококачественного
огнеупорного кирпича толщиной 80 см. Печь закрыта
куполообразным стальным сводом с огнеупорной футеровкой, в
котором имеются отверстия для электродов и для загрузки шихты.
Три угольных электрода диаметром 1 м расположены в ряд и на
одинаковом расстоянии друг от друга. Они поддерживаются
Загрузка
шихты
Загрузка
шихты
выход
газа
ШцпОшИЬІи ЛсСОп
Рис. 97. Трехфазная электропечь для возгонки фосфора:
1. ^9 электроды; 2 — задвижка; 3— вентилятор; 4—мотор; 5 — летка.
в вертикальном положении зажимами и пропущены в печь черед
сальники, охлаждаемые водой. Узкий кольцевой зазор между
сальниками и электродами уплотнен асбестом, чтобы
улетучивание фосфора и просачивание воздуха в печь были
минимальными. Медные держатели или зажимы электродов охлаждаются
циркулирующей водой и питаются током трансформаторов,
расположенных вблизи печи.
В зависимости от условий работы печи электроды могут быть
приподняты или опущены.
Фосфорит, кремнистый флюс и коксовая мелочь поступают
каждый из отдельного бункера, дозируются и подаются на
ленточный транспортер, по которому смесь направляется в
несколько бункеров, расположенных над печами. Отсюда шихта идет
220
в закрытые лотки или трубы и равномерно распределяется по
всему сечению печи.
На одном конце печи имеется отверстие, соединенное с
газоходом, по которому газы и пары фосфора направляются в два
электрофильтра для отделения пыли.
Кроме приведенной выше основной реакции, в печи
происходят также другие, например, восстановление Fe2O3:
Fe2O3 + ЗС = 2Fe + ЗСО.
Поэтому в нижней части печи собирается вместе с жидким
шлаком, состоящим в основном из силиката кальция, жидкий
фосфористый чугун (феррофосфор).
Через нижнюю летку жидкий плав периодически выпускается
из печи, а сверху непрерывно отводятся пары фосфора и газы
(СО и др.) После электрофильтров пары фосфора охлаждаются
водой. При этом фосфор конденсируется и под водой собирается
продукт — белый фосфор.
На сдну тонну фосфора расходуется около 8—9 г фосфата,
1,4--1,6 т кокса, 2,5—2,7 т кремниевых материалов, 13- 35 кг
электродной массы и до 15 м3 воды, две-три тонны пара и 13—
20 тыс. кет- ч электроэнергии. При этом получается 1 г фосфора,
8—11 г шлака, около 4000 м3 газа и 0,1—0,5 т феррофосфора.
Образующийся при возгонке жидкий фосфор перекачивают
насосами в хранилище и цистерны для транспортировки или
перерабатывают тут же на заводе в фосфорную кислоту.
Производство термической фосфорной кислоты из фосфора
осуществляется в специальных башнях, выложенных
кислотоупорной керамикой. Расплавленный фосфор подается на
форсунки из хромоникелевой стали и распыляется воздухом,
причем воздух поступает с избытком в два-три раза против
необходимого для реакции
2Р2 + 5О2 = 2Р2О5.
количества, что позволяет избежать образования низших
окислов фосфора.
Количество впрыскиваемой воды определяется с таким
расчетом, чтобы получалась примерно 90%-пая ортофосфорная
кислота НзРО4:
РА + ЗН2О = 2Н3РО4.
Кислота выводится в гумированные стальные сборники,
выложенные кислотоупорными плитками. Электрофильтр улавливает
брызги и туман кислоты, которая получается весьма чистой
(примеси составляют десятые и сотые доли процента).
Башня высотой 10 м и диаметром 2,5 м дает в сутки ~37,5 г
100%-ной НзРО4, перерабатывая 12 т фосфора. Схема установки
с электропечью приведена на рис. 98.
221
Окисление фосфора можно вести водяным паром по реакции
= Р2О
2О5
5Н2 или
Р2 + 8Н2О = 2Н3РО4 + 5Н2.
Эти процессы протекают при высоких температурах (около 1000J)
в присутствии катализаторов. Получающийся водород может
идти на производство аммиака.
С Л/рае
бода
, содержащий СО
Воздух ^~^
Рис. 98. Схема получения фосфорной кислоты на установке с
электропечью:
/—электропечь; 2—горячие электрофильтры; 3 — конденсатор фосфора: 4 —
приемник для фосфора; 5 — насос для жидкого фосфора; 6 — камера горения
фосфора; 7 —башня гидратации фосфорного ангидрида; 8 — хранилище
фосфорной кислоты; 9 — электрофильтр для улавливания брызг фос-форноЛ
кислоты.
ГЛАВА XIII
ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ СИЛИКАТОВ
Продукты силикатной промышленности имеют чрезвычайно
важное значение для народного хозяйства. К ним относятся
керамические изделия (строительные кирпичи, огнеупорные и
кислотоупорные материалы и др.); вяжущи-е материалы (цемент,
известь, гипс и др.); стеклянные изделия (оконное стекло,
бутылки, эмали и др.).
Продукты силикатной промышленности применяются в
жилищ-ном, промышленном и военном строительстве. Напомним
о широком использовании силикатных изделий в быту
(гончарные, фаянсовые и фарфоровые изделия, хрустальные и
керамические вазы и т. д.). Масштабы производства силикатных изде-
222
лий весьма велики. Так, в 1967 г. производство цемента в СССР
достигло 84,8 млн. г, Превысив производство этого продукта
в США; стекла в том же году изготовлено 205 млн. м2, что
составляет 102% количества, произведенного в 1966 году.
Большая часть силикатных изделий принадлежит к керамическим
материалам. Так, за 1967 г. одних строительных кирпичей (без.
производства колхозами) выпущено 36 млрд. т A03% к 1966 г.).
Основным сырьем для изготовления керамических изделий
являются глинистые материалы (пластичное сырье), кварц,
кварцевые пески, обожженные глины (отощающее сырье), полевые
шпаты, пегматиты (плавни или флюсующее сырье) и ряд
других минералов, в состав которых входят окислы кремния,
магния, кальция, алюминия и щелочных металлов.
По характеру черепка, получаемого после обжига,
различают пористые и плотные керамические изделия, последние
уплотнены благодаря спеканию и плавлению исходных материалов.
Если пористые керамические изделия покрыть глазурью
(стекловидной пленкой), то они также станут водонепроницаемыми.
Пластичное сырье. Все глинистые материалы,
встречающиеся в природе, представляют собой водные
алюмосиликаты с общей формулой дгАЬОз- #SiO2- zH2O. К наиболее
распространенным относятся каолинит А12О3 • 2SiO2 • 2Н2О, галлуазиг
АШз- 2SiO2 • ЗН2О, монтмориллонит А12О3 • 4SiO2 • 2Н2О,
пирофиллит А12О3 • 4SiO2- H2O и др.
Глинистые минералы образовались в природе путем
постепенного разрушения (выветривания) более сложных
алюмосиликатов — полевых шпатов nR2O • *А12О3 • #SiO2 • Н2О,
например ортоклаза К2О • А12О3 • 6SiO2, который под воздействием
углекислых вод при повышенной температуре и давлении
претерпевает реакцию
К,0 ¦ А12О3 ¦ 6SiO2 + 2Н2О + СО2 = К2СО3 + А12О3 ¦ 2SiO2 • 2Н2О +
-j- 4SiO2. каолинит
Основными примесями в глинах являются кварц, полевой
шпат, окислы железа, карбонаты кальция, магния, окись титана,
щелочные окислы и др. Наиболее чистые глины (каолиньт)
состоят преимущественно из каолинита.
1. ГЛИНИСТОЕ СЫРЬЕ И ОСНОВЫ ПРОИЗВОДСТВА
КЕРАМИЧЕСКИХ ИЗДЕЛИИ
Каолины являются ценным сырьем не только в керамике.
Их применяют в производстве бумаги для наполнения, в
фармацевтической, резиновой (как наполнитель) и других отраслях
промышленности. Если каолины загрязнены примесями, их
обогащают.
Качество глины определяется ее химическим, минералогичес-
223
•ким и гранулометрическим составом (размером зерен). Значение
последнего фактора велико, так как от размеров зерен (степени
дисперсности) зависят многие свойства глин.
Разнообразие минералогического состава обусловливает
разнообразие видов и свойств сырья. Полиминеральностью и
наличием примесей объясняется различный цвет глин (от белого до
черного), свойства смесей глины с водой, поведение глин при
сушке и обжиге; по той же причине глины плавятся не при
определенной температуре, а постепенно размягчаются в широком
интервале температур. Последнее обстоятельство используют
,для получения частично оплавленного, спекшегося черепка при
изготовлении керамических изделий.
Затворение глины водой (до 8—10%) дает влажный
порошок, который можно формовать в прессах под давлением. Из
увлажненных до 18—25% глин получают пластичное тесто, спо-
•собное деформироваться без разрыва сплошности. При
нагревании глины до 100° С она высыхает, приобретая значительную
прочность (до 30 кг/см2 и более при разрыве и до 100 кг/см2 при
сжатии). Линейные размеры глины при сушке уменьшаются на
•6—10% (воздушная усадка), что учитывается при
изготовлении изделий. Высохшую глину можно снова превратить в
пластичное тесто, добавив в нее воды. Нагревание воды до более
высоких температур сопровождается потерей способности
образовывать пластичное тесто (около 450—500° С) и огневой
усадкой (около 800—850° С), достигающей 5—8%.
Температура, при которой глина размягчается и изделие
начинает терять свою форму, называется огнеупорностью.
Вести обжиг при такой температуре нельзя, поскольку изделия
в этих условиях деформируются. Глина, обожженная при
температуре на 150—200° С ниже температуры огнеупорности, после
охлаждения приобретает большую механическую прочность: до
400 кг/см2 при сжатии и до 50—60 кг/см2 при растяжении. Эта
способность глин, наряду со способностью образовывать при
затворении водой пластичное тесто, является важнейшим
свойством глин, на котором основано производство керамических
изделий.
Увеличение прочности объясняется частичным оплавлением,
спеканием глины во прсмя обжига. Для количргтг.ешюй оценки
спекания определяют водопоглощение обожженной глины.
Важная характеристика глины — интервал спекания,
т. е. разность между температурой, при которой образец
начинает деформироваться, и температурой полного спекания
(водопоглощение менее 1—2%). Интервал спекания глин 10—250° С.
Чем он больше, тем легче вести обжиг, так как случайное
повышение температуры не вызывает деформации изделий.
По огнеупорности глины делятся на три группы: огнеупорные
(огнеупорность не ниже 1580° С), тугоплавкие (огнеупорность
224
в пределах 1350—1580 С) и легкоплавкие (огнеупорность ниже
1350° С).
По пластичности, определяемой количеством песка, которое
может быть добавлено к глине с водой при условии сохранения
пластичности, глины подразделяются на четыре типа: связующие
(более 50% песка), пластичные B0—50%), тощие (менее 20%
песка), кремнеподобные (не образующие теста при добавлении
песка).
Технические свойства глин можно регулировать. Так,
например, огнеупорность их возрастает при добавлении огнеупорной
глины, содержащей больше А12О3. Глинозем и кремнезем
увеличивают огнеупорность глин, тогда как щелочи (Na2O, КгО)
снижают ее.
Пластичность можно увеличить добавлением электролитов,
коллоидов, вылеживанием и выветриванием, повышением
дисперсности; уменьшение пластичности достигается добавлением
не пластичных (отощающих) материалов (обожженная глина,
кварцит, песок). Спекаемость возрастает при добавлении
легкоплавких флюсующих материалов: полевых шпатов — ортоклаза
К2О • А12Оз • 6SiO2, альбита Na2O • А12О3 • 6SiO2 и др. Чем
меньше частицы, тем выше технические свойства каолина. Степень
дисперсности каолина велика и характеризуется, например, так:
13,8 млрд. частиц в 1 г с общей поверхностью 6400 см2/г, при
среднем диаметре частицы 3,6 мк.
Потеря при прокаливании (п. п. п.) характеризует
огнеупорность. Чем ближе она к п. п. п. каолинита A3,91%), тем выше
огнеупорность сырья.
На основании заранее определенных свойств можно
подобрать для тех или иных целей подходящую глину или смесь глин
и корректировать ее добавкой отощающих, флюсующих или
других материалов. Из смеси всех материалов с добавлением воды
готовится керамическая масса.
Основное требование, предъявляемое к массе: возможно
большая ее однородность. Это достигается измельчением и
тщательным и длительным размешиванием всех материалов.
Из керамической массы установленного состава и
консистенции сырые изделия формуют несколькими способами:
пластичным— из керамических масс, содержащих 16—25% воды,
полусухим из масс влажностью 8—16% и сухим способом — при
влажности 4—8%. Формуют такие массы на прессах.
Применяется также формование литьем из масс, содержащих 30—35%
воды и имеющих консистенцию сливок. В этом случае
используются гипсовые формы, впитывающие избыточную воду.
Отформованное сырое изделие подвергают сушке. Эта
операция длится долго, иногда неделями, на воздухе или в закрытом
помещении (над печами), а также в специальных
механизированных сушилках. Высушенные изделия обжигают; таким обра-
15 а-1504 225
зом им придают необходимую прочность и другие свойства. Для
улучшения качества и украшения изделий их покрывают
глазурью, красками и рисунками. Таким образом, принципиальная
схема производства керамических изделий сводится к
следующим основным этапам: подготовка керамической массы,
формование сырого изделия, сушка, обжиг, окончательная обработка
поверхности (глазурование, окраска и др.).
отсос дымовых
газоб на сушку
. Отсос доздуха
на сушку
*¦ воздух
Рис. 99. Горизонтальный разрез туннельной печи.
Наиболее дорогой и ответственной операцией является обжиг.
Для обжига керамических изделий применяются различные печи,
но наибольшее распространение получают туннельные.
Туннельная, или канальная печь (рис. 99), представляет
собой длинный узкий канал, перекрытый сводом и выложенный
изнутри огнеупорной футеровкой, длиной
от 60 до 160 м и более. В канале по
рельсам движутся вагонетки (рис. 100) с
обжигаемыми изделиями; оси, колеса и
рельсы изолированы от зоны огня песочным
затвором, по которому продвигаются
щитки, прикрепленные к продольным
краям плотно примыкающих друг к
другу платформ. Искусство обжига состоит
в умении правильно установить и
обеспечить скорость подъема температуры,
высший температурный предел и его
продолжительность, а также газовую среду
в печи (окислительное, нейтральное,
восстановительное пламя).
Рис. 100. Поперечный Характер температурной кривой, ко
разрез туннельной^ печи: нечная температура и время, необходи-
/~вагОнЄь!йазатвор. пес°ч мое для обжига изделий, зависят от
скорости реакций, протекающих в черепке,
толщины черепка и габаритов изделия. Наиболее существенно
влияет на качество обожженных изделий период собственно
обжига, в процессе которого и образуется камневидный черепок.
При этом отдельные зерна массы слегка смачиваются небольшим
226
2/00
2000
, 1900
1800
4700
1600
1530
гіао-
гооо-
количеством жидкости, образовавшейся вследствие частичного
расплавления добавляемых в шихту плавней (минерализаторов
и легкоплавких примесей к основным компонентам массы).
Количество этой жидкости зависит как от температуры
нагрева, так и от количества и свойств примесей, образующих
легкоплавкие эвтектики, часто в виде стекловидной фазы.
По мере повышения температуры за счет полного
расплавления легкоплавких примесей увеличивается количество
жидкой фазы, протекает разложение одних и образование других
химических соединений,
перекристаллизация и
другие процессы. В част-
ности, природные
алюмосиликаты типа каолинита,
кианита и другие при
нагреве выше 1200—1400° С
разлагаются и
перекристаллизуются в муллит
ЗА12О3 • 2SiO2, выделяя
при этом избыточный
кремнезем, главным
образом в виде
кремнекислого Стекла. Диаграмма Рис. 101. Диаграмма состояния системы
состояния А12О3 — SiO2 SiO2 —A12O3.
(рис. 101) имеет большое
практическое значение: она приложима к изучению
физико-химических процессов, протекающих в керамических массах
(шамотные изделия, глиноземистые, полукислые).
Чистая окись глинозема плавится при 2050° С, кремнезем —
при 1710° С. Из диаграммы следует, что добавка
незначительных количеств глинозема приводит к быстрому падению
кривой плавкости. При 1545° С образуется эвтектика, состоящая из
92% SiO2 и 8% А12О3. С увеличением содержания А12О3 выше
80% кривая начинает подниматься вверх. В этих условиях
образуется муллит ЗА12О3 • 2SiO2. При 1810° С протекает реакция
разложения муллита:
О 10 20 ЗО 40 50 60 7О)ф0 90 Л*К
А Ог 3Al203-2Si0z 2JU/lj-Si 0г АІг 03
Содержание яомлонентоб, вес %
3Al2O3-2SiO2 = ЗА12О3
2SiO2.
2O3-2SiO2 = ЗА12О3
Процесс муллитообразования протекает при обжиге
шамотных, глиноземистых огнеупоров, а также фарфора. Чем больше
муллита, тем выше качество изделий.
Производство керамических изделий
Строительный кирпич. Обыкновенный красный
кирпич представляет собой искусственный камень размерами
250 X 120 X 65 см, получаемый формовкой глин с добавками
15»
227
(обычно песка) или без них, причем к глинам не
предъявляются высокие требования; в частности, допускается повышенное
содержание окислов железа.
Технологическая схема производства строительного кирпича
пластическим методом заключается в дроблении глины,
смешении ее с песком (если глина «жирная», т. е. высокопластичная),
увлажнении или обработке на глиномялках, формовании на
прессах кирпича-сырца, содержащего до 20% влаги. Сырец
сушат и затем подвергают обжигу при 900—950° С. В последнее
Труба
Засыпка топлива
'Обжиг,
бумажные
Откатка
обожженного кирлацд
горения
Рис. 102. Схема кольцевой печи.
время вместо пластического метода применяют полусухой, при
котором производится формовка массы, содержащей 8—10%
влаги, на прессах под давлением 150—200 кг/см2. При этом
влаги в сырье мало, и сушка протекает быстрее.
Обжиг производится обычно в кольцевых печах (рис. 102),
представляющих собой кольцевой канал, разделенный на ряд
камер A4—32).
Холодный воздух поступает в камеры, где обжиг закончен,
и, охлаждая изделия, нагревается (зона охлаждения). Затем
воздух поступает в камеры. Здесь происходит сжигание
вводимого сверху топлива (обычно — твердое) и обжиг изделий при
самий иышкий температуре (зона обжига). Горячие газы
охлаждаются, проходя следующие камеры, в которых загруженные
изделия подогреваются (зона подогрева). Отсюда топочные газы
уходят в трубу. Каждая камера попеременно входит в одну из
указанных зон.
Процесс продолжается около семи суток. В таких печах
обжигают и другие керамические изделия.
Хорошо обожженный кирпич имеет при сжатии до 150 кг/см2,
при водопоглощении не менее 8 и не более 20%- Кроме того,
он должен обладать морозостойкостью, т. е. выдерживать без
228
разрушения 15 попеременных замораживаний и оттаиваний (теп-
лосмен).
Пористый кирпич готовят так же, как и красный,
но с добавлением выгорающих при обжиге веществ —
древесных опилок и др. С производством красного кирпича сходен
процесс получения ряда других изделий: черепицы, дренажных труб;
терракоты и др. Для их изготовления применяют лучшие сорта
пластичных глин.
Силикатный кирпич. Этот в большом количестве
применяемый строительный материал делают не из глины, а из
тонкоизмельченного кварца (песок) с добавкой 5—8% извести
и пропаркой в автоклаве (стр. 236).
К а м ен н о-к е р а м и ч е с к и е изделия (клинкер).
Клинкером называют изделия, используемые для мощения
дсрог и тротуаров. Это кирпич или плитки с хорошо спекшимся
черепком (водопоглощение, не более 2—6%)- В качестве сырья
для клинкера используют тугоплавкие глины с большим интер-
Балом спекания.
Чтобы улучшить спекание, в конце обжига поддерживают
восстановительную атмосферу для перевода Fe2O3 в FeO,
дающую легкоплавкие силикаты. Температура обжига клинкера
1200—1300° С.
Кислотоупорные керамические изделия. Эти
изделия состоят из плотной спекшейся непрозрачной массы,
покрытой слоем глазури, которая защищает поверхность от
разрушения кислотами. Сырьем служат легкоплавкие пластичные
глины, образующие при обжиге плотный черепок. Они не
должны содержать примесей гипса и окислов железа.
Кислотоупорные изделия в больших масштабах применяются
в химической промышленности для сооружения башен, сосудов,
колец, змеевиков, насосов, труб, кранов и т. д. Эти изделия
должны не пропускать газов и жидкостей и обладать стойкостью
к кислотам. Пористость кислотоупоров обычно не превышает
0,1%. Кислотоупорность определяют, кипятя в серной кислоте
растертое в порошок изделие. При этом потери веса за 1 час не
должны превышать 1—2%.
Для получения щелочестойких материалов в шихту вводят
магнезит, доломит, глинозем. Изделия обжигают при 1250—
1300° С и глазуруют. Часто применяется «соляная» глазурь. Для
этого к концу обжига в печи создают восстановительную
атмосферу, уменьшая количество воздуха, и вводят поваренную соль,
которая под действием водяных паров и газов разлагается на
Na2O и НС1; НС1 уходит с дымовыми газами в трубу, a Na2O
соединяется с силикатами глинозема черепка:
2NaCl + п SiO2 + А12О3 + Н2О -*¦ Na 2О • А12О3 ¦ п SiO2 + 2НС1 \ .
черепок глазурь
229
Каменное литье. В последние десятилетия для
изготовления кислотостойкой химической аппаратуры все шире
применяется каменное литье.
Горные (изверженные) породы — диабаз, базальт и др.—
расплавляют в печах (шахтных или, чаще, пламенных — типа
мартеновских), при 1400—1500° С, а затем отливают в
металлические или земляные формы. Для устранения механических
напряжений и повышения прочности их подвергают затем отжигу
(нагрев до 600—800° С с последующим медленным
охлаждением). Эти изделия отличаются плотностью, прочностью и
высокой кислотоупорностью.
Плавленый кварц. Ассортимент изделий из
плавленого кварца S1O2 постоянно увеличивается. Этот материал
соединяет высокую термостойкость с кислотостойкостью. Изделия
готовят в аппаратуре, сходной с такой, которая используется
для производства стекла. Сырьем служит кварц или кварцевый
песок, содержащий свыше 99,5% SiO2. Температура плавления
1700—1800° С.
Эмали. Керамические изделия по прочности значительно
уступают металлическим, но превосходят их своей химической
стойкостью. Специальные химически стойкие стали дороги.
Поэтому все больше применяются аппараты из обычной
конструкционной стали или серого чугуна, покрытые слоем эмали.
Эмаль — это легкоплавкое стекло, к которому предъявляется
ряд специфических требований. Оно должно быть химически
стойким, прочно соединяться с металлом, не отделяясь при
механических и термических воздействиях и не образуя трещин.
Состав эмалей сложен и разнообразен. В шихту входит глина,
песок, полевые шпаты, криолит, окислы различных металлов.
Все эти компоненты тонко измельчаются и наносятся
равномерным слоем на подготовленную поверхность изделия, которое
затем нагревают. При этом эмаль плавится и тонкий слой ее
остается на поверхности. Рецептура эмалей все время
совершенствуется. Сейчас известны кислотостойкие, щелочестойкие
и жаростойкие эмали.
Технология огнеупоров
Огнеупорными называются материалы, способные
длительное время выдерживать различные физико-химические и
механические воздействия в топочных устройствах. Эти материалы
•подразделяются на огнеупорные (огнеупорность от 1580 до
1770°), высокоогнеупорные A700—2000° С) и материалы высшей
огнеупорности (более 2000° С).
Важнейшими свойствами огнеупоров являются
огнеупорность, температура деформации под нагрузкой, постоянство
объема при высоких температурах, термическая стойкость, шлако-
устойчивость, газонепроницаемость.
230
Наибольшее значение для производства огнеупорных и
высокоогнеупорных ИЗДеЛИЙ ИМеЮТ уГЛерОД, ОКИСЛЫ SiO2, MgO, AI2O3,
СаО, Сг2О3, а также соединения и смеси этих веществ. Для
изготовления изделий высшей огнеупорности применяются карбиды,
нитриды этих же элементов, соединения циркония, титана и др.
Температура плавления некоторых соединений: ВеО B570° С),
MgO B800° С), СаО B570° С), Сг2О3 A990° С), ZrO2 B700° С),
НЮ2 B812° С), ThO2 C050°C), TiC C140°C),W2C B857°С),
SiN B500° С), ZrN C000° С).
Шамотные огнеупоры изготовляются из смеси
огнеупорной глины и шамота; общее содержание в смеси глиноземи
А12Оз должно быть не менее 30 %, а ТЮг— не более 2%.
Шамотом называется огнеупорная глина, обожженная
и размолотая. Шамот добавляется к глине в качестве отощающе-
го вещества для уменьшения ее усадки, повышения
огнеупорности и химической стойкости.
Чтобы получить шамотные огнеупоры, готовят шамот. Для
этого часть глины обжигают в шахтных печах при 1300—1390° С.
После размалывания шамота на бегунах, вальцах или в
шаровых мельницах его рассевают на три фракции по крупности зерен
(от 0 до 5 мм).
Оставшуюся глину подсушивают в барабанах горячими
газами до влажности 6—8%; затем ее размалывают и смешивают
с шамотом при определенном соотношении фракций. Из смеси
формуют сырец, лучше всего способом сухого прессования на
прессах со штампами или на ленточных прессах.
Сырец, сформованный сухим прессованием, может
непосредственно идти в печь для обжига; при мокром способе
производства, когда сырец содержит 17—18% воды, нужно
предварительно высушить его в сушилках. Обжиг производится в туннельных
печах непрерывного действия или в периодических при 1300—
1425°С.
Шамотный огнеупор — наиболее распространенный
огнеупорный материал. Его применяют для кладки доменных, коксовых,
ваграночных и стеклоплавильных печей, для облицовки топок
котлов и т. д.
Различают нормальный шамот, кислый (часть шамота
заменена песком) и глиноземистый (в состав последнего вводят
боксит). Шамотные изделия делятся по огнеупорности на три
класса, а каждый класс на сорта.
Динасов ые огнеупоры. Сырьем для производства
динасовых огнеупорных изделий служат природные кварциты;
содержащие 96—98% SiO2. К ним добавляют 2—5% связующих
и минерализующих добавок — известковое молоко, окислы
железа, шлак и др.
Динасовые огнеупоры широко используются при кладке
печей, так как они сочетают с высокой огнеупорностью большую
231
теплопроводность и меньшую газопроницаемость, чем шамотные
огнеупоры. Их используют при кладке мартеновских,
стеклоплавильных и коксовых печей.
Высокое содержание SiO2 (температура плавления 1710° С)
обусловливает огнеупорность динаса. Однако при нагревании
SiO2 последовательно превращается в ряд модификаций,
отличающихся физическими свойствами — строением, плотностью
и др. Модификации кремнезема переходят одна в другую при
определенных температурах, что видно из следующей схемы
переходов:
870° 1470° 1713°
а-кварцї=?а-тридимит;=іа-кристобалитї=фасплав
573° |1 163° f| _J80° f|
р-кварц р-тридимит ' 270°
117°fj р-кристобалит
7-тридимит
Особенно важно получить а-кварц, а-тридимит, а-кристобалит,
так как соответствующие им модификационные переходы
сопровождаются уменьшением удельного веса. Так, при переходе
а-кварца в а-тридимит удельный вес SiO2 падает с 2,533 до 2,228,
а объем увеличивается на 12—13%. Объемные изменения
являются причиной термической нестойкости изделия: образуются
трещины, нарушается монолитность кладки, что может привести
к разрушению печи. Для получения изделия из кварца,
сохраняющего при высоких температурах свою форму и прочность,
нужно провести предварительную тридимитизацию
кремнезема. Для этого отформованное сырое изделие подвергают
медленному обжигу, длящемуся 9—12 суток, по определенному
режиму с выдержкой при максимальной температуре 1430—
1460° С. Нагрев производится в течение пяти—семи, а
охлаждение четырех—пяти суток.
Чтобы ускорить перерождение кварца, в шихту вводят
плавни-минерализаторы в виде соединений железа, особенно закиси
железа, металлургических шлаков и др.
Производство динасовых огнеупоров заключается в том, что
кварцит очищают от rpv6bix примесей птгевпм и направляют к з
дробилки. Ситованием получают ряд фракций, отличающихся
величиной зерен. Подбор соответствующего количества каждой
фракции дает массу с минимумом пустот. К смеси фракции
добавляют известковое молоко, все перемешивают до полной
однородности и после непродолжительного вылеживания массы
подают ее на пресс, работающий под давлением 200 птм.
Отформованные кирпичи поступают на сушку и обжиг.
Хромитовые огнеупоры получают из породы,
теоретический состав которой соответствует формуле хромита
FeO • Сг2Оз- Природный хромит содержит примеси А12О3, Fe2O^
и другие окислы. Для изготовления хромитовых огнеупоров
232
к хромитовои руде примешиваются предварительно обожженные
магнезит (до 10%) и дунит (около 7%). Температура
размягчения таких огнеупоров 1500—1600° С. Они стойки к кислым и
щелочным материалам.
Магнезитовые огнеупоры. Сырьем для
производства магнезитовых изделий служит магнезит MgCO3, залегающий
на Урале (Сатка). Процесс производства состоит из следующих
операций: грубое дробление магнезита, обжиг его при
температуре 1550—1600° С, когда удаляется СО2, размол, увлажнение до
4—5%, прессование, сушка при 60—75° и обжиг при температуре
1600° С. Такой огнеупор содержит не менее 90% окиси магния
и обладает рядом ценных качеств: огнеупорность не ниже 2000° С,
временное сопротивление сжатию до 300 кг/см2. Однако
термическая устойчивость его невысока. Эти огнеупоры применяются
главным образом для кладки мартеновских печей.
Чистая окись магния плавится при 2800°С в электропечах.
Из нее готовят специальные огнеупоры, имеющие повышенную
термостойкость. К числу магнезитовых огнеупоров можно отнести
также изделия из магнезиально-силикатных материалов
(серпентинит, дунит), которые готовятся в смеси с окисью магния. При
обжиге A600—1700° С) образуется форстерит Mg2Si04. Поэтому
такие изделия называются форстеритов ы ми. Они
содержат 35—55% MgO. Используются также хромомагнезитовые
смеси.
Специальные огнеупоры
Циркониевые огнеупоры состоят из двуокиси
циркония ZrO2, которую извлекают из природных месторождений
путем обогащения. После обжига A800—1900° С) и измельчения
из нее формуют изделия, применяя в качестве связующего
крахмал и другие вещества; сырые изделия сушат и обжигают.
Такие изделия выдерживают температуру до 2000° С.
Корундовые огнеупоры состоят из чистой окиси
алюминия (98—99%) и плавятся при 2050° С. Для некоторых
изделий смешивают корунд с 10—30% огнеупорной глины
(связующее) и обжигают при температуре 1500° С. Огнеупорность таких
изделий 1800—1900° С.
Углеродистые огнеупоры из измельченного
графита или кокса с небольшой добавкой связующего материала
в восстановительной среде выдерживают температуру до 3500° С
(в окислительной среде они сгорают). Наконец, для работы при.
3000—4000° С используют нитриды и карбиды бора, титана,
циркония, таллия, гафния. Так, смесь карбидов таллия и циркония
выдерживает температуру около 4000° С.
Изделия из огнеупорметаллических спеков.
К огнеупорной технологии примыкает производство
огнеупорметаллических спеков, или керметов.
233
Современная техника требует таких материалов, которые
обладали бы высокими механическими свойствами при высоких
температурах (более 1300° С), хорошо сопротивлялись
окислению, не растрескивались при самых резких колебаниях
температуры, имели небольшую плотность и были пригодны для
изготовления сложных деталей. Такое сочетание свойств позволяет
использовать керметы при создании газовых турбин, ракет и ряда
деталей, работающих в тяжелых условиях,— катодов большой
мощности, зажигательных свечей, в ядерной технике и др. Ни
металлы и металлические сплавы, ни огнеупорные материалы,
отдельно взятые, не обладают одновременно всем комплексом
этих свойств.
В настоящее время созданы материалы из комбинации
металла и огнеупорного окисла, получаемые электротермическим
порошковым методом. Физико-химическая сущность этого
метода проявляется в процессе спекания, при котором возможны
следующие случаи: компоненты реагируют без образования жидкой
фазы; компоненты реагируют с образованием жидкой фазы;
компоненты не реагируют друг с другом и не образуют ни
твердых растворов, ни химических соединений.
Спекание такого рода материалов производится при
различных температурах (от 500 до 2500° С) в электрических печах,
где тщательно контролируется атмосфера.
Из опробованных комбинаций огнеупоров с металлами
наибольшего внимания заслуживают А12Оз + Fe или Сг; борид
циркония + №; карбид бора +Fe; карбид титана +№. и др. Такие
композиции можно наносить на поверхность металла, создавая
жаростойкие покрытия, обладающие пластичностью и
устойчивостью к механическим нагрузкам, ударам и пр. Покрытия
получают напылением из газопламенного пистолета тонкоизмельчен-
ного сплава металла с окислом.
В последнее время при нанесении покрытий из
высокоплавких материалов используют режим и аппаратуру
низкотемпературной плазмы.
Изделия из фарфора и фаянса (тонкая керамика)
Эти очень ценные изделия применяются в технике (баки,
аппараты и др.) и в быту (тарелки, чашки и др.). Фарфор имеет
плотный стекловидный черепок, фаянс — пористый. Сырьем для
фарфоровых изделий являются каолиновые глины E0%), кварц
B5%) и полевой шпат B5%)- Из массы пластическим методом
или литьем формуют сырое изделие. Его сушат и обжигают
дважды.
Первый обжиг производят при 900—1000° С в муфелях
(бисквитный обжиг). Остывшие изделия покрывают глазурной массой
234
и обжигают вторично при 1320—1350° С (политой обжиг). При
этом на изделии образуется тонкий слой глазури. Для
украшения под глазурь или на глазурь наносят рисунки. Если рисунки
нанесены на глазурь, то изделие обжигают третий раз для
закрепления рисунка. Обычно третий обжиг —
низкотемпературный, при 900—1000° С.
Производство организовано таким образом, что параллельно
готовят массу для черепка, для глазури и красок. Изделия для
обжига помещают в капсули, чтобы на них не попадала зола.
Аналогично готовят изделия из фаянса, частично заменяя каолин
глиной.
Процессы, происходящие при обжиге фарфоровых масс,
разнообразны и сложны. Суть их сводится к образованию муллита
в среде расплавленного полевого шпата. Таким образом, масса
черепка после остывания представляет собой весьма прочную
систему, состоящую из вязкого стекла, в котором распределены
прочные зерна муллита и кварца, хорошо сопротивляющиеся
внешним воздействиям — нагрузке и высокой температуре,
система химически устойчива, обладает электроизоляционной
способностью и выдерживает быстрые смены температуры.
Все эти замечательные свойства широко используются в быту
и промышленности. В химической промышленности
применяются фарфоровые реакционные сосуды, химическая посуда. Из
фарфора делают изоляторы для высоковольтных линий передач
и др. Для получения специальных сортов фарфора в шихту
вводят окислы алюминия, циркония, бериллия, титана. Эти добавки
повышают огнеупорность, термостойкость и
электроизоляционные свойства фарфора. Фаянсовые изделия применяются в
сантехнике, для изготовления посуды и пр. Почти всегда их
покрывают глазурью. Глазури, представляющие собой тонкий
стекловидный слой на черепке, являются силикатами, состав которых
должен быть таким, чтобы глазурь прочно приставала к
черепку, не давала трещин и не отскакивала при затвердении. Обычно
подбирают глазурь с тем же коэффициентом расширения, что
и у черепка.
Для легкоплавкости глазури в ее состав вводят щелочные
окислы, соли борной кислоты и другие соединения. Окислы
калия и натрия вводят с полевым шпатом, глинозем и
кремнезем — с каолином и кварцем. Например, глазурь для некоторых
видов фарфора готовится из 64% полевого шпата, 27% кварца,
9% каолина. Поскольку глазурная масса наносится на черепок
в виде водной суспензии,— растворимые вещества в
специальных печах переводят в нерастворимые плавлением компонентов,
готовя т. н. фритту. Быстро охлажденную фритту размалывают
и из порошка готовят суспензию, которую наносят на
поверхность изделия различными способами: окунанием изделия,
обливанием, намазыванием кистью, пульверизацией.
235
Для раскраски цветных глазурей и керамических изделий
употребляют различные окислы, которые после обжига
приобретают желаемый цвет, например, окислы и соединения
кобальта (синий цвет), хрома (зеленый цвет), железа (красный цвет)
и т. д. Смешивая различные окислы, можно получить
всевозможные оттенки.
Из новых направлений укажем на усиленно
разрабатываемые в последнее время безобжиговые методы
производства, главным образом керамических изделий. Процесс
получения изделий в этом случае осуществляется
гидротермальным путем. Он заключается в том, что отформованное из
измельченного сырья с водой изделие (кирпич) обрабатывается в
автоклаве («запаривается») при соответствующем режиме.
Так, для приготовления алюмосиликатных огнеупоров
смешивают гидрат глинозема А1(ОНK с кварцевым песком SiO2
тониной не более 1% остатка на сите 10 000 отв/см2 при
влажности 8—10% и отношении А12О3 : SiO2 = 3 : 2. В автоклаве
протекают процессы адсорбции, например А1(ОНK, на поверхности
SiOo, сопровождающиеся образованием гелевидных структур;
такие структуры уплотняются и образуют алюмосиликаты. Эти
процессы интенсифицируются с повышением давления и
температуры. Полученные материалы автоклавного твердения,
например, при режиме 2—8—2 (подъем давления до 8 ата в течение
2 ч, выдержка 8 ч и сброс давления 2 ч), способны заменить
обычные огнеупоры высокотемпературного обжига. Добавим,
что автоклавная обработка в четыре раза экономичнее обжига.
Автоклавный метод применяется уже сравнительно давно при
производстве силикатного (белого) кирпича в строительной
технике (стр. 229).
2. ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ ВЯЖУЩИХ МАТЕРИАЛОВ
Вяжущими материалами называются порошкообразные
вещества, которые при затворении водой приобретают
пластичные свойства, образуя постепенно твердеющее тесто, способное
связывать отдельные куски или массу твердых пород в монолит.
Жилищное и промышленное строительство невозможно без
применения вяжущих. Они необходимы при строительстве дорог,
мостов и т. д.
Вяжущие материалы подразделяются на воздушные (при
смешении с водой они затвердевают и длительное время сохраняют
прочность только на воздухе) и гидравлические (могут
затвердевать на воздухе и в воде). Поэтому воздушные вяжущие
применяются в надземных сооружениях, а гидравлические — как
в надземных, так и в подземных и гидротехнических
сооружениях.
236
Кроме того, известны специальные вяжущие материалы: кис-
лото- и щелочестойкие, минерально-органические и др.
Сырьем для производства вяжущих материалов служат
природные горные породы: известково-глинистые (мергели),
магнезиальные, карбонаты, кремнеземистые, гипс и др., а также
отходы некоторых производств: доменные шлаки, золы, фосфогипс
и др.
Гипсовые вяжущие материалы. Сырьем для их
производства является природный двуводный гипс CaSO4 • 2Н2О,
природный ангидрит CaSO4 и отход производства фосфорной
кислоты — фосфогипс.
Получение строительного гипса основано на химической
реакции
CaSO4-2H2O = CaSO4-0,5H2O + 1,5Н2О.
Гипс обжигают в кусках и порошке в варочных котлах или
вращающихся барабанах. Производится также обжиг гипса во
взвешенном состоянии. Б последнее время совмещают помол
гипса с обжигом во взвешенном состоянии. При этом продукт
обжига— полуводный гипс (алебастр) измельчают до остатка на
сите 900 отв/см2 не более 8%. Смешиваясь с водой, он снова
образует двуводный гипс и твердеет. Алебастр характеризуется
следующими техническими показателями: предел прочности при
сжатии через 3 ч от 75 до 200 кг/см2, начало схватывания —
не ранее 3—7 мин, конец схватывания — не позднее 30 минут.
Схватывание является начальной стадией процесса
твердения вяжущих материалов. В этот период происходит
превращение полужидкой пластичной массы в камневидное тело. При
твердении протекает реакция
CaSO4-0,5H2O + 1,5Н2О = CaSO4-2H2O,
причем на 1 кг CaSO4 • 0,5Н2О выделяется 27 ккал и
температура массы повышается до 40—50° С.
Твердение гипса сопровождается также увеличением объема
примерно на 1%. Это связано и с ростом кристаллов и с
увеличением объема пор. Благодаря такому свойству гипса его
используют в технике для изготовления отливок.
Согласно теории твердения вяжущих материалов,
разработанной А. А. Байковым, процесс делится на три периода:
1. Растворение и образование насыщенного раствора
новообразований, выделение продуктов в коллоидной форме
(студень).
2. Переход коллоидной формы новообразований в
кристаллическую (рост кристаллов в виде иголок и их переплетение). Этот
период соответствует схватыванию вяжущих.
237
3. Высыхание твердеющей массы и дальнейшее повышение
прочности в течение 28 суток и более.
Все три периода протекают во времени, причем длительность
их зависит от многих факторов (вида сырья, температуры
обжига, тонкости помола, количества воды и т. д.).
Для регулирования технических показателей строительного
гипса (ускорение или замедление схватывания, повышение
прочности) применяются многие минеральные и органические
добавки.
При нагреве гипса до 300—450° С он полностью теряет воду
и образует «растворимый ангидрит» CaSO4, который также
твердеет при затворении водой. Но прочность полученного камня
меньше, чем при затворении полуводного гипса. При 600—700° С
образуется намертво обожженный гипс, который не
схватывается при смешении с водой. Он приобретает эту способность, если
его измельчить и добавить катализаторы: известь A—3%) или-
Na2SO4 A %), доломит и некоторые другие добавки. При этом
получают т. н. ангидритовый цемент.
При 800—1000° С получают высокообожженный, или эст-
р и х гипс, который твердеет при затворении водой без
катализатора, так как в нем присутствует примесь СаО,
образовавшейся в результате частичного разложения CaSO4.
В строительной технике гипсовые вяжущие широко
применяются для изготовления строительных деталей: блоков, панелей,
перегородок, гипсобетона, сухой штукатурки, легковесных
теплоизоляционных изделий (газогипсовые). Заполнителями служат
известь, шлаки, мел, пемза, опилки и т. д. Следует отметить, что
все строительные изделия на основе гипса неводостойки и
поэтому их применяют во внутренних элементах сооружений.
Водостойкость повышают органические и минеральные добавки,
а также водоотталкивающие обмазки.
Воздушная известь. Воздушную известь получают
путем обжига известняков, мела, доломитизированных
известняков, содержащих не более 8% примесей в шахтных или
вращающихся печах, где происходит реакция СаСО3 = СаО + СО2.
Негашеную комовую известь — «к и п е л к у» измельчают. При
действии воды образуется гидратная известь —
«п у ш о к к а» в виде тонкого порошка по реакции СаО + Н2О =
= Са(ОНJ. «Пушонка» образует с водой известковое тесто —
продукт пластичной консистенции.
Известковое тесто, смешанное с песком, измельченным
шлаком и т. п., применяют в виде строительных растворов при
кладке стен и для штукатурки. Известь в большом количестве
используется в производстве силикатных изделий автоклавного
твердения.
Известковый раствор на воздухе постепенно отвердевает под
влиянием двух одновременно действующих факторов: удаления
238
свободной влаги и действия СО2. Удаление воды приводит к
выделению и кристаллизации Са(ОНJ. В результате действия СО2
образуется карбонат кальция
Са(ОНJ + СО2 = СаСО3 + Н2О.
Кристаллы срастаются между собой и с зернами
наполнителя, образуя искусственный камень.
Твердение воздушных известковых растворов протекает
медленно. Для ускорения этого процесса к извести добавляют
цемент, гидравлические добавки или гипс.
Гидравлическая известь. Гидравлическая известь,
в отличие от воздушной, начав твердеть на воздухе, может
продолжать твердение в воде. Способность гидравлической извести
сохранять и увеличивать прочность в воде объясняется наличием
в ее составе, кроме свободной СаО, силикатов, алюминатов и
ферритов кальция, которые образуются при обжиге за счет
реакций между глиной и известняком. Для производства
гидравлической извести используются известняки, содержащие глину;
обжиг ведется при 900—1000° С.
Рома н-ц е м є н т. Роман-цемент представляет собой
продукт обжига мергелей при температуре 800—1100° С.
Мергелями называются природные смеси глины и известняка. Для
производства роман-цемента используются мергели, содержание
глины в которых таково, что после обжига почти вся окись
кальция связывается с глиной в силикаты, алюминаты и ферриты. От
гидравлической извести роман-цемент отличается почти полным
отсутствием свободной извести и более высокой прочностью.
Пуццоланов ые цементы. Пуццолановы ми
цементами называют цементы, получаемые в результате совместного
помола вяжущих: цемента, извести и гидравлической добавки.
Гидравлические добавки представляют собой природные или
искусственные материалы, состоящие в основном из активных,
т. е. способных вступать в реакцию с Са(ОНJ, кремнезема,
глинозема и окиси железа. Такими материалами, содержащими
аморфный кремнезем, являются природный вулканический пепел,
пемза, трепел, трасс, пуццоланы, зола и т. д.
Пуццолановые цементы содержат 20—80% гидравлических
добавок. Гидравлические добавки придают цементам
водостойкость, образуя нерастворимые силикаты, алюминаты и
ферриты.
Шлаковые цементы. В производстве вяжущих широко
применяются шлаки. Из различных видов шлаков — доменных,
передельных, топливных и др. — наибольшую ценность
представляют доменные шлаки с высоким содержанием СаО
и А12О3.
Модуль основности таких шлаков больше единицы
(основные шлаки): СаО + MgQ ^
SiO2 + AliAi \
23»
а модуль активности
А1А,
02
SiO2 > K}'z-
Основные доменные шлаки склонны к распаду в мелкий
порошок, что отрицательно сказывается на качестве вяжущего.
Распад шлаков объясняется переходом высокотемпературной
модификации C — 2СаО • SiO2 в низкотемпературную а — 2СаО •
- SiO2, сопровождающимся значительным изменением объема.
Чтобы предотвратить саморассыпание шлака, расплав шлака
подвергают резкому охлаждению — грануляции. Применяют три
вида грануляции: мокрую — сливанием шлака в воду,
полусухую — действием пара и сухую — воздухом.
Доменные шлаки используются для производства
следующих видов вяжущих: шлако-портланд-цемента,
сульфатно-шлакового цемента и известково-шлакового цемента. Их получают
совместным помолом гарнулировэнного шлака с портландт-це-
ментным клинкером и с добавками сульфата кальция и других
веществ.
Добавки, вводимые в шлак, существенно ускоряют
твердение. Так называемый бесклинкерный шлаковый цемент
содержит до 10% ангидрита и около 5% обожженного доломита.
Глиноземистый цемент представляет собой продукт тонкого
томола обожженной до плавления или до спекания сырьевой
смеси, состоящей из боксита и известняка.
Химический состав глиноземистого цемента следующий:
•около 40% СаО, около 40% А12О3, остальное — примеси —
Fe2O3 (нежелательные) и др. Окислы СаО и А12О3 находятся
в глиноземистом цементе главным образом в виде минерала —
однокальциевого алюмината СаО ¦ А12О3. Глиноземистый цемент
отличается высокой прочностью (до 900 кг/см2) и быстрым
твердением при нормальных сроках схватывания. Уже на третий
день твердения прочность его приближается к максимальной.
Получают глиноземистый цемент плавкой в электрических или
.доменных печах, а также спеканием во вращающихся печах
с последующим измельчением.
Магнезиальные цементы. Активным началом магнезиальных
цементов является окись магния. Сырьем служат природные
магнезит MgCO3 и доломит СаСОз • MgCOa. В соответствии с этим
различают два вида магнезиальных цементов — каустический
магнезит, получаемый обжигом до полного удаления СО2 при
800—1000е С и каустический доломит. Доломит обжигают при
температуре 650—800° С (полуобжиг) с тем, чтобы получить
смесь MgO + СаСОз без значительной примеси свободного СаО.
В отличие от других вяжущих магнезиальные цементы
затворяются не водой, а растворами солей MgCl2 или MgSO4, в
некоторых же случаях — серной или соляной кислотой. При
твердении магнезиальных цементов происходит образование
240
Mg(OHJ сначала в коллоидном, а затем в кристаллическом
состоянии; частично же образуется оксихлорид магния mMgO •
• MgCl2 • nH2O.
Магнезиальные цементы применяются для изготовления
искусственного мрамора, литографических камней и др.
Портл анд-цемент
Наибольшее значение как вяжущий материал в
строительстве имеет портланд-цемент. Он представляет собой продукт
помола клинкера, полученного обжигом до спекания смесей из
известняков и глин, встречающихся в природе (мергели), или
искусственно составленных. При помоле к клинкеру
добавляется гипс (до 2%) —для замедления схватывания — и
гидравлические добавки (до 15%), увеличивающие стойкость
портландцемента к разрушающему действию природных вод.
Портландцемент является наиболее распространенным видом вяжущих.
Химический состав портланд-цемента следующий: СаО 62—67%,
SiO2 20—24%, А12О3 4—7%, Fe2O3 2,5%, MgO, SO3 и пр. 1,5—
3,0%. Состав портланд-цемента выражают при помощи модулей
основного или гидравлического Г,
силикатного п и глиноземного р, соответственно равных:
г СаО і о • о 1-
1 SiO2 + А12ОЭ + Fe2O3 ' • ' '
Si°
_ 3
Р ~ Fe2O3 ~ l • °-
Окислы связаны в клинкере в следующие соединения —
минералы:
ЗСаО* SiO2 (сокращенно C3S) — алит, 37—60%;
2СаО-3102@28) —белит; 15—37%;
ЗСаО ' А12Оз(СзА)—трехкальциевый алюминат, 7—15%;
4СаО • АЬОз ¦ Fe2O3(C4AF) — четырехкальциевый алюмофер-
рит, 10—18%.
Эти соединения реагируют при затворении цемента водой
и дают различные гидраты, выделяющиеся в виде студней —
гелей; они образуют пластичное тесто, которое затем схватывается
и упрочняется в цементный камень (стр. 237).
Чтобы судить о количестве алита в клинкере, при расчетах
пользуются коэффициентом насыщения (КН)
СаО - 1,65А12О3 - 0,35Fe2O, а о . n qc
Чем выше КН, тем больше в клинкере алита.
Клинкеры со значительным содержанием алита трудно
обжигать, но они дают быстротвердеющие цементы высоких марок.
16 8-1504 241
Поэтому алит часто называют важнейшим клинкерным
минералом.
Увеличение содержания белита замедляет твердение
цемента, однако в более далекие сроки положительно сказывается на
его прочности. СзА ускоряет и схватывание и твердение цемента.
Аналогично действует и C4AF • СзА и C4AF сравнительно
легкоплавки и способствуют быстрому протеканию обжига.
Свойства минералов клинкера учитываются при вычислении
соотношения между известняком и глиной, которое определяет
минералогический состав портланд-цементов. Для корректировки
состава в смесь известняка и глины добавляют иногда окислы
железа в виде пиритных огарков или руды и кремнезем в виде
трепела. Изменяя минералогический состав клинкера, получают
многие виды специальных цементов, например, сульфат о-
стойкий цемент, не разрушающийся в морской воде. Он
содержит в ограниченном количестве C3S и СзА, содействующие
вредному влиянию сульфат-иона. Подобным же образом
рассчитывают состав тампонажного цемента (много C3S
и строго определенное количество С3А) низкотермичного
(мало C3S и СзА) и дорожного (много C3S и C4AF)
цементов. Белый портланд-цемент, применяемый для
облицовки зданий, готовят из сырья, не загрязненного РегОз.
Схема производства. Принципиальная схема изготовления
портланд-цемента может быть изображена следующим образом:
Известняк Глина Гипс Гидравлическая
| / добавка
Смешение и помол |
| Помол клинкера с добавкой
Обжиг смеси для получения -> |
клинкера Готовая продукция
Помол глины и известняка ведется одновременно в шаровых
мельницах. Схема мокрого способа изображена на рис. 103.
Глину и известняк, поступающие на завод из карьера,
подвергают предварительному дроблению в щековых и молотковых
дробилках: глину дробят также п оалкотшх дробилках.
После этого глина направляется в болтушку, представляющую
собой бетонный бассейн с вращающимися металлическими
граблями. Здесь ее смешивают с водой, и полученный таким образом
глиняный шлам насосом перекачивают в шаровую мельницу,
куда подается известняк. Достаточно измельченную смесь глины
и известняка (шлам) направляют в шламбассейны — большие
бетонные емкости, где перемешивание ведется сжатым воздухом
и мешалками. Шлам поступает во вращающуюся печь,
изображенную на рис. 104. Основной частью ее является футерованный
изнутри огнеупорным материалом металлический кожух
цилиндрической формы. Диаметр кожуха от 3 до 5 м, длина от 51 до
242
Рис. 103. Схема производства портланд-цемента по мокрому способу:
/ — подача известняка; 2 — бункер известняка; 3 — щековая дробилка; 4 —
молотковая пообнлка: 5—бункер нчмольиенногп ""естнякз; 6 вагонетка с глиной; 7 —
болтушка; * — насос для перекачки глнняиого шлама; 9 — бачок; 10 — питатель
известняка; //— шаровая мельница; 12 — вертикальные шламбассейны; 13 —
горизонтальный шламбассейн; 14 — питатель печн; 15 — вращающаяся печь; 16 —
транспортер клинкера; П — конусная дробилка; IS — клинкерный склад; 19 — бункера
клинкера, гнпса и гидравлической добавки; 20 — вагоны с гипсом и гидравлической
добавкой; 21 — склад гнпса и гидравлических добавок; 22 — гидравлическая добавка;
23— бункер; 24 — сушильный барабан; 25 — гипс; 26— шаровая мельница для помола
цемента; 27 — силосы с цементом; 28 —упаковочная машина и приспособления для
погрузки цемента навалом; 29 -~ вагон с цементом.
16*
Рис. 104. Вращающаяся печь для обжига портланд-цемента:
/ — питатель печи; 2 — сливная труба; 3 — шламовый фильтр; 4, 5 —цепная завеса
(навеска цепей гирляндами); « — теплообменник; 7 — холодильник для выхвдящего
клинкера- Я—весы и транспортер клинкера; 9 — форсунка для подачи газа; 10 —
пылевая 'камера; //—электрофильтр; 12 — вентилятор; 13 — труба; 14 — мотор и
венечная шестерня; 15 -^бандаж; 16 — опорный ролик; /7 — опора; U — площадка для
управления печью; 19 — кожух печи.
243
180 лі и более. Печь устанавливается под углом 3—5 град, и
приводится во вращение со скоростью примерно один оборот в
минуту. Вследствие этого материал медленно движется вниз навстречу
дымовым газам. Топливом служат газ, мазут или пылевидное
топливо. Максимальная температура материала в печи 1450е С.
Газы и материалы движутся в ней противотоком. По мере
нагревания глина и известняк реагируют между собой, образуя
клинкер — спекшийся полупродукт. Клинкер вылеживается на
клинкерном складе, где не вступившая в реакцию свободная
известь гасится за счет влаги воздуха. Благодаря увеличению
объема клинкер частично разрыхляется, что облегчает
последующий помол его в клинкерных мельницах. При помоле клинкера
применяют добавки — гипс и др. Выходящий из мельницы
материал представляет собой готовый продукт: портланд-цемент.
После охлаждения на складе он поступает в силосы, отсюда —
на упаковку.
При сухом способе производства материалы перед помолом
высушиваются в сушильных барабанах. Состав смеси
корректируют в силосах — пересыпая материал из одного силоса в другой
или проводя смешение сжатым воздухом. Перед обжигом
сырьевую муку увлажняют до 12—14% и направляют в печи.
По стандарту портланд-цемент делится на марки от 300 до
900. Эти марки соответствуют пределу прочности при сжатии
(в кг/см2) образцов, полученных при затворении с водой смеси
цемента с песком через 28 дней. Начало схватывания при 24—
25%-ном содержании воды наступает не ранее чем через 45 мин,
конец — не позже чем через 12 я от начала затворения.
Некоторые специальные цементы
Расширяющиеся и безусадочные цементы.
Для строителей наибольшую ценность представляет
безусадочный цемент. Кроме того, на некоторых строительных работах
применяется расширяющийся цемент. Чтобы получить такой
цемент, r обычный цемент вводят добавки, способствующие его
расширению (сульфит кальция и алюминат), или стабилизаторы
(доменные шлаки).
Гидрофобный цемент. Впервые в СССР
разработана технология получения гидрофобного цемента, который
образуется при совместном помоле клинкера и органических кислот
(олеиновой, стеариновой, лауриновой и ДР-). Этот цемент можно
хранить длительное время, перевозить навалом без изменений.
При смешении его с наполнителями гидрофобная пленка
разрушается и цемент нормально гидратируется.
Ц е м е н т н о-э м у л ь с и о н ны е смеси получают,
смешивая портланд-цемент с эмульсиями битума, резины и синтетп-
244
ческих смол. Смеси из латекса, песка и глиноземистого цемента
устойчивы к химической коррозии.
Неизвестковые цементы. К таким цементам
относят цинкофосфатный. Для проявления его вяжущих свойств
следует затворять порошок жидкостью, состоящей из 78% Н3РО4,
16% ZnO и 6% А1(ОН)з. Вяжущими свойствами обладают и
другие неизвестковые системы, например, MgO — Р2О5 — Н2О,
CdO — Р2О5 — Н2О.
Кислотостойкие цементы и бетоны. Большой
интерес для химической промышленности представляют мэтериа-
лы из кислотостойких бетонов, применяемых при изготовлении
аппаратуры, например, сосудов большой емкости, башен и др.
Отдельные части или элементы таких конструкций связаны
кислотоупорной замазкой или цементом. В свою очередь, эти части
состоят из мелких камней, скрепленных кислотоупорным
цементом.
Внедрение описанных конструкций экономит дорогостоящие
металлы — свинец, стяль и дп. Кроме того, кислотоупорный
цемент применяют для покрытия поверхностей некислотоупорных
материалов, главным образом металлов.
Заводы, вырабатывающие серную, соляную, азотную,
фосфорную и другие неорганические и органические кислоты, хлор и
различные соли, нуждаются в больших количествах
кислотоупорных изделий и кислотоупорного цемента. Кислотоупорные
цементы готовятся из тонкоизмельченного природного
кислотоупорного минерала, например, андезита, с добавкой Na2SiF6,
затворенного на жидком стекле определенной концентрации.
Компоненты смеси реагируют между собой. Так, жидкое стекло
Na2Si03 и кремнефторид Na2SiFe вступают в следующую
реакцию:
2Na2SiFe + Na2Si03 + 6H2O = 6NaF + 3Si(OHL.
Выделяющийся в результате реакции гидрат окиси кремнезема,
уплотняя массу, приводит к схватыванию цемента.
Бетон и изделия из него
Бетоном называют продукт затвердевания смеси вяжущего
вещества, воды, мелкого и крупного заполнителя. В качестве
вяжущего вещества можно взять любое из рассмотренных
вяжущих. Наиболее часто применяется, однако, портланд-цемент или
его разновидности — особенно быстро твердеющие сорта.
Следует учитывать, что почти весь выпускаемый цемент
перерабатывается в бетонную смесь, а затем — в сборные
железобетонные конструкции.
245
Мелким заполнителем или песком в технологии бетона
называют материалы с размером зерен до 5 мм. Используются как
естественные кварцевые, полевошпатные и другие пески, так
и искусственные, полученные дроблением горных пород или из
шлаков.
В последнее время используется также керамзит,
представляющий собой продукт обжига некоторых глин, легко
вспучивающихся под действием высокой температуры (порядка 1000° С)
и образующих по остывании пористые прочные гранулы.
Керамзит в виде кусков большого размера используется также как
крупный наполнитель бетона. Размер зёрен крупного
заполнителя от 5 до 150 мм. В качестве крупного наполнителя
используются горные породы — гранит, кварцит и т. д., а также
искусственные материалы — битый кирпич, крупные куски шлака и др.
Для изготовления легких бетонов берут пористые разновидности
наполнителей, для тяжелых — наполнитель с большим
удельным весом, без пор.
Состав бетона записывают r виде соотношения, например
1:3: 6; это означает, что на одну часть цемента приходится три
части песка и шесть частей щебня. Количество необходимой
воды выражается водоцементным отношением В/Ц. Расчет состава
бетона ведут по полуэмпирическим формулам и проверяют
экспериментально. Важнейшее свойство бетонной смеси —ее
подвижность. Основная характеристика затвердевшего бетона —•
прочность при сжатии, доходящая до 600 кг/см2. Однако
прочность бетона при растяжении невелика. Сооружение становится
во много раз прочнее, если армировать его сталью. Стальная
арматура располагается в деревянной форме и заливается
бетоном. Такой материал, как известно, называется железобетоном.
Изделия на основе извести
На основе извести готовятся известково-песчаный
(силикатный) кирпич белого цвета, известково-глиняный кирпич, извест-
няково-иесчаные блоки, пеносиликаты^ изделия. Особенно
большой эффект дает применение блоков, которые ускоряют
и удешевляют строительство. Технология изготовления
перечисленных изделий включает: 1) приготовление массы, состоящей
приблизительно из 6—8% извести, 92—94% песка (или глины
и небольшого количества воды (при изготовлении пористых
изделий в массу вводят небольшое количество органических
пенообразователей), 2) формовку изделий и 3) запаривание изделий
при давлении пара 8 ати и температуре 173° С. В результате
запаривания образуется ряд гидросиликатов кальция (в
частности тоберморит — гидросиликат кальция 4СаО • 5SiO2 • 5Н2О),
которые придают прочность силикатным изделиям.
246
Асбестоцементные изделия
К этим изделиям относятся шифер и асбестоцементные трубы,
получаемые в результате твердения смеси: 8—18% асбеста,
остальное — цемент.
Из асбестоцементных масс готовят кровельные волнирован-
ные и плоские листы — шифер, трубы различных диаметров,
электроизоляционные детали, отделочные материалы.
Процесс изготовления асбестоцементных изделий включает
распушку асбеста, подготовку смеси асбеста, цемента и воды,
формовку изделий. Твердение асбестоцемента происходит в
пропарочных камерах. Трубы для окончательного твердения
погружают в бассейны с водой.
3. ПРОИЗВОДСТВО СТЕКЛЯННЫХ ИЗДЕЛИЙ
Основоположником научного стеклоделия является М. В.
Ломоносов, разработавший свыше двух тысяч рецептов различных
сіекил. Б cubticKut ьреми крупные раииты в ииласти стекла
выполнены А. А. Лебедевым, И. В. Гребенщиковым, И. И.
Китайгородским, Н. Н. Качаловым и др.
Стекло применяется очень широко: в жилищном
строительстве (листовые и зеркальные стекла), в быту (посуда, тара,
украшения и пр.), оптике, электротехнике, приборостроении, для
изготовления тканей и т. д.
Строение стекла и его свойства
Стеклом называют аморфное изотропное твердое вещество,
получаемое в результате переохлаждения расплава.
Изотропность стекла связана с тем, что, в отличие от кристаллических
тел, оно состоит из чрезвычайно мелких, имеющих сильно
деформированную решетку кристаллитов.
Характерной особенностью расплавленных стекол (и
металлургических шлаков) является постепенное, а не
скачкообразное, как у кристаллических тел, нарастание вязкости при
понижении температуры. Застывание стекла происходит в
некотором интервале температур. Если этот интервал мал, то стекло
называют коротким, если велик — длинным.
При затвердевании стекла вязкость становится практически
бесконечно большой, что препятствует кристаллизации
компонентов расплава. Таким образом, стекло представляет собой
неравновесную систему. В состав силикатного стекла, кроме
кремнезема SiO2, входят окислы щелочноземельных и щелочных
металлов. Стекло, применяемое в строительстве и быту, имеет
состав, близкий к Na2O • СаО • 6SiO2 (примеси: А12О3, MgO и др.).
Для специальных целей в стекло вводят окислы калия, свинца,
247
бария, цинка, бора и др. Стекло должно обладать
прозрачностью, определенной прочностью, химической стойкостью.
Применяют цветные и непрозрачные, матовые стекла.
Стекломасса имеет склонность к кристаллизации, в
результате которой чаще всего выпадает SiC>2 в виде кристобалита.
Поскольку образование кристаллов приводит к браку, химический
состав стекла и условия варки выбирают таким образом, чтобы
кристаллизации не было. Многокомпонентные стекла меньше
подвержены кристаллизации. Кристаллизацию предотвращает
также замена части SiO2 на А12О3 и ввод MgO вместо СаО.
При охлаждении стекломассы и переходе ее в стекло
происходит некоторое уменьшение размеров стеклоизделия (усадка).
На практике охлаждение изделий идет неравномерно, наружные
слои застывают тогда, когда внутренние еще находятся в
жидком состоянии. В результате этого наружные слои усаживаются
несколько раньше внутренних, что приводит к возникновению
внутренних напряжений в стекле.
Внутренние напряжения могут быть настолько большими,
что стекло самопроизвольно раскалывается на части.
Стеклянные изделия стараются охлаждать таким образом, чтобы
внутренние напряжения как бы армировали стекло и увеличивали его
прочность.
Особое значение имеет равномерность распределения
внутренних напряжений. Чтобы добиться такой равномерности,
изделия из стекла подвергают отжигу, т. е. нагреву до 600—650° С
и медленному равномерному охлаждению. В некоторых случаях
прибегают к быстрому, но также равномерному охлаждению
(закалка стекла). Прочность изделий, подвергнутых закалке,
резко ьозрастает.
Из механических свойств стекла практически важны такие:
прочность при сжатии и растяжении, твердость и хрупкость.
Прочность при сжатии у стекла доходит до 200 кг/мм2 и во много
раз превышает прочность при растяжении, равную обычно 3—
8 кг/мм2. Твердость стекла по шкале Мооса 5—7. Оно легко
режется топазом, корундом и алмазом, твердость которых соотт
ветствеьно ряина 8, 9. 10 Высокая хрупкость стекла в ряде
случаев снижает его технические достоинства, так как уже при
небольшом превышении предела прочности наступает
разрушение стекла. Термостойкость стекла невелика. Только
специальные стекла с высоким содержанием кремнезема обладают
значительной термостойкостью и применяются для изготовления
посуды и некоторых приборов.
Технология стекла
Обычное оконное стекло имеет примерно следующий
химический состав (в %): SiO2 — 71; Na2O — 15,5; СаО — 8,5; MgO—
3,5; А12Оз— 1,5. С целью придания стеклу тех или иных свойств
248
состав его значительно изменяют. Для некоторых нужд готовят
даже чистое кварцевое стекло, состоящее из кремнезема.
Исходные материалы для производства стекла делятся на
стеклообразующие и вспомогательные. К стекло-
образующим относятся кварцевый песок, бура, сульфат натрия
и сода, известняк, доломит, магнезит, а также окислы цинка,
свинца и др.
Вспомогательными материалами являются осветлители
А12О3, NaNO3, NH4C1, способствующие быстрому выделению
пузырьков газа из сваренного стекла, окислители (для
обесцвечивания стекла) NaNO3, MnO2, селен и другие,
красители: Мп2О3 (фиолетовый цвет), СоО (синий цвет), FeO
(зеленый цвет) и др.
Глушители применяются для придания стеклу матовости,
непрозрачности. Это CaF2, Na2SiF6, окислы Zn, Zr, Ті, Sn.
Восстановителями служат уголь, древесные опилки,
металлы Mg, AI, Sn.
В наибольших количествах стекольная промышленность
потребляет песок, соду или сульфат натрия, известняк или мел
и доломит. Приводим состав шихты содово-известкового
оконного стекла: песок 100 вес. ч., доломит — 25,09 вес. ч.,
технический глинозем — 0,49 вес. ч, сода — 39,34 вес. ч., мел — 8,57 вес.
ч. Сырьевые материалы содержат ряд примесей, в том числе
вредных, например, окись железа, двуокись титана,
органические примеси и другие вещества. Поэтому сырье, особенно песок,
промывают соляной кислотой. В оптическом стекле содержание
Fe2O3 не должно превышать 0,012%, в оконном стекле—¦0,2%,.
в бутылочном зеленом стекле содержание железа не
нормируется.
Схема производства стекла. Технологическая
схема изготовления изделий из стекла включает следующие
этапы: приготовление стекольной шихты, варка стекла, формовка
изделий из стекломассы, отжиг изделий, различная обработка
изделий. Приготовление стекольной шихты включает подготовку
сырьевых материалов и смешение их между собой.
Для удешевления стекла часть соды заменяют сульфатом
натрия. В этом случае приходится вводить небольшое
количество угля, который облегчает разложение сульфата натрия.
Исходные материалы сушат, тонко измельчают и тщательно
смешивают. Иногда смесь брикетируют, благодаря чему
увеличивается производительность печи, уменьшается унос пыли,
улучшается качество стекла и удлиняется срок службы печей. Наряду
с шихтой в печь вводят стеклянный бой.
Варка стекла. Варка стекла ведется в ваннах или горш-
ковых печах. Горшковые печи предназначены для варки
небольших партий специальных стекол, например, оптического стекла.
На под такой печи устанавливаются шамотные горшки со сте-
24»
Поперечный разрез
\
Рис. 105. Разрез печи для варки оконного стекла:
/—ванна; 2— регенератор для .подогрева воздуха; 3« регенератор для
подогрева генераторного газа.
250
Рис. 106. План и продольный разрез ванной печи
для варки стекла:
а <—план печи; / — загрузочное отверстие; 2—ванна;
3 —регенераторы; 4 — лодочкн; 5 — выработочный
канал; 6 — продольный разрез,
кольнои шихтой. Как ванные, так и горшковые печи
отапливаются газообразным топливом с применением регенераторов
(стр. 7). Сжигание газа происходит в верхней части печи, под
сводом, над стекольной шихтой и над образующимся стеклом.
Размеры ванны 28 мХб м при глубине 1,5 м. На рис. 105 показан
поперечный разрез ванной печи. Дно и стенки ваины выстланы
огнеупорными брусьями.
На рис. 106 изображен план и продольный разрез
стекловаренной печи. Печь имеет 12 спаренных регенераторов, которые
подогревают воздух за счет тепла отходящих газов.
Загрузка шихты производится специальными загрузчиками,
направляющими шихту в печь непрерывно и тонким слоем. При
нагреве до 1200°С удаляется влага, а затем в стекольной шихте
еще до расплавления начинаются химические реакции
образования силикатов натрия и кальция.
В интервале 400—900° С происходит частичная диссоциация
карбонатов кальция и магния, а также образование двойных
солей, например, NajC^COsh. Карбонаты реагируют с
кремнеземом, образуя силикаты и их сплавы:
Na2Ca(CO3J + 2SiO2 = CaSiO3 + 2СО2 + Na2SiO3,
Na2CO3 + SiO2 = Na2Si03 + CO2,
Na2SiO3 -f CaSiO3 = Na2SiO3-CaSiOs.
Если в шихте присутствуют сульфат натрия и уголь, идут
реакции восстановления сульфата:
Na2SO4 + С = Na2S + СО2 и
2Na2SO4 + С = 2Na2SO3 + СО2.
Продукты этих реакций образуют силикаты:
Na2SO4 + Na2S + 2SiO2 = 2Na2SiO3 + SO2 + S,
Na2SO3 + SiO2 = Na2Si03 + SO2-
Аналогичные реакции протекают и с CaSO4, который
частично образуется в печи за счет взаимодействия
СаСО3 + Na2SO4 = Na2CO3 + CaSO4.
При температуре выше 900 и до 1200° С силикаты
расплавляются и в расплаве растворяется песок. В результате
образуется стекломасса с большим количеством пузырьков газа. Чтобы
облегчить удаление пузырьков (дегазацию), стекломассу
разогревают до температуры 1400—1500° С, при которой вязкость
падает и пузырьки всплывают. Иногда в расплав вводят воду
или сырое дерево. Бурно образующиеся пары воды и газы
перемешивают расплав, состав его выравнивается и газы удаляются.
Заключительная стадия варки стекла — охлаждение его до
температуры 1100—1200°, при которой можно формовать
изделия.
251
Формование изделий осуществляют вытягиванием,
дутьем и прессованием. Методом вытягивания готовят оконное
стекло, стекловолокно и некоторые другие виды изделий.
Вытягивание оконного стекла производится на машинах
вертикального вытягивания (рис. 107). В машину входит подмашинная
камера, являющаяся частью ванной печи, и собственно машина
ВВС (вертикального вытягивания стекла). В подмашинной
камере при помощи металлических штанг удерживается плавающая
б
оэ
сэ
OCkJ
сэ
cjcV
оэ
сэ
сэ
сэ
.3
Рис. 107. Схема машины вертикального
вытягивания стекла.
/—выработочный канал ванной печн; 2 —
подмашинная камера; 3 — лодочка; 4 — машина
вертикального вытягивания стекла; 5—валики; 6 —
лист стекла.
в стекломассе керамическая лодочка со щелью, длина которой
равна ширине вытягиваемого листа. В щель лодочки опускается
металлическая рама. Стекло прилипает к раме и вместе с ней
тянется вверх, в шахту машины ВВС, где остывает и
отжигается. Раму наверху отбивают и дальнейшее вытягивание ведут
уже без нее, при помощи вращающихся валиков машины.
Вытянутое стекло разрезается в верхней части на отдельные листы.
252
Скорость вытягивания стекла достигает 100 и более метров
в час. Методом дутья на специальных машинах готовят
всевозможные пустотелые изделия: бутылки, стеклянные трубки,
электроламповые колбы и пр. Раньше это делали вручную, сейчас
с помощью специальных машин.
Прессование стекла применяют для изготовления
толстостенных изделий — граненых стаканов, кружек, ваз и т. д.
Прессование ведут в разогретых металлических формах. В форму
прямо из ванной печи втекает необходимое количество стекломассы.
Здесь же при помощи пуансона оформляются наружная и
внутренняя поверхности изделия. Разновидностью прессования
является прокатка стекла при помощи валиков. Отформованные тем
или иным способом изделия обязательно идут на отжиг.
Обработка стекла. Изделия из стекла подвергают различной
обработке: шлифовке, полировке, гранению, травлению и т. д.
Специальные виды стекла. Для окон троллейбусов,
автобусов и железнодорожных вагонов часто применяют
трехслойное стекло, представляющее собой два слоя стекла, между
которыми находится слой органической прозрачной прокладки.
При ударе такое стекло не рассыпается. Для автомобильных
окон используется специальным образом закаленное
оконное стекло, имеющее высокую механическую прочность.
Пеностекло представляет собой сильнопористый
строительный материал, пригодный для сооружения строительных
перегородок, теплоизоляции и т. д. Пеностекло получают
спеканием тонкоизмельченного стеклянного боя в смеси с
пенообразователем. В последнее время быстро развивается производство
стекловолокна. Стекловолокно готовят вытягиванием тончайших
стеклянных нитей. Употребляется оно для прядения, изготовления
стеклянных тканей, применяемых как кислотостойкие фильтры,
защитные ткани и т. д. Стекловолокно в композиции
с пластмассой дает материалы, из которых делают корпуса
автомобилей, суда и пр.
Щелочное, главным образом натриевое стекло
Na2O n SiO2, растворимое в воде, применяется для
огнестойкого покрытия древесины, в мыловарении, для получения
силикагеля, как связующее в цементах, как клей в быту и т. д.
Он образуется в результате сплавления при 1300—1500° С
кварцевого песка и соды (или Na2SO4 + С). Обрабатывая
полученный твердый силикат «глыбу» водой в автоклавах при 4—8 ати,
получают раствор жидкого стекла, которое оценивается
по модулю (молярное отношение ЭЮг : NaaO, равное 1-=-4).
Добавление окиси свинца дает хрустальное стекло
с большим коэффициентом лучепреломления. Свинец может
быть частично замещен в хрустальном стекле барием.
Большое значение имеет также химически стойкое
стекло, применяемое для изготовления лабораторной посуды
253
(колонны, колбы, приборы и т. д.). Стойкость к действию
кислот и щелочей достигается введением в состав стекла В2О3, ZnO
и увеличением количества А12О3. В последнее время химически
стойкое стекло применяется в деталях различных промышленных
аппаратов — колоннах, емкостях, трубах, кранах и др. Известно
также термически стойкое огнеупорное стекло, (пирекс),
которое содержит до 80% SiO2 и до 12% ВгО3.
По стойкости к кислотам, огнеупорности и способности
выдерживать резкие смены температур наилучшим является
кварцевое стекло. Из него делают подогреватели, кварцевую
аппаратуру, применяемую в производстве чистой соляной
кислоты и др.
Ситаллы. На смену жаростойким стеклам приходят стекло-
кристаллические материалы, для которых характерны высокая
механическая прочность и повышенная химическая устойчивость.
Эти материалы называют ситаллами, или п и р о к е р а-
мами. Получают их из стекольных расплавов различного
химического состава, либо из расплавленных металлургических
шлаков и горных пород путем введения катализаторов (окислы
титана, хрома, церия, ванадия, циркония, а также сульфиды
тяжелых металлов), которые создают множество центров
кристаллизации. Производя соответствующую термическую
обработку (отжиг), фиксируют мелкую кристаллизацию стекольного
расплава.
Ситаллы имеют прочность при сжатии до 90 000 кг/см2, а при
изгибе — до 1800 кг/см2. Коэффициент теплового расширения их
близок к нулю. Пирокерами устойчивы к действию кислот и
выдерживают резкие (до 1000° С) перепады температур. В
некоторых случаях изготовляют легковесные и пористые ситаллы. Они
являются весьма ценным материалом для различных отраслей
народного хозяйства и используются в строительной технике
(для изготовления панелей, плит, блоков), для термоизоляции
техники и т. д.
Теплоизоляционные материалы
В химической и других отраслях промышленности
необходимы теплоизоляционные материалы, которые часто готовят из
силикатных материалов. Изоляция бывает высокопористой и
обладает малым коэффициентом теплопроводности: 0,07—
0,2 ккал/м • ч • °С. Различают низкотемпературную изоляцию,
которую можно применять при температуре ниже 150° С,
среднего 600° С) и высокотемпературную (до 900° С), а также
огнеупорную (выше 900° С). Для изготовления изоляции используют
стекло- и шлаковату, пеностекло, стеклянные ткани, природные
волокнистые (асбест) и пористые (туф, пемза, ракушечник и др.)
минералы.
Разработаны различные композиции этих материалов с до-»
бавлением связующего компонента (жидкое стекло и др.).
Часть третья
ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ ОРГАНИЧЕСКИХ
ВЕЩЕСТВ
ГЛАВА XIV
ОСНОВНОЙ ОРГАНИЧЕСКИЙ СИНТЕЗ
Под основным органическим синтезом понимают
производство синтетических органических соединений, которые имеют в на-
стояшее время важнейшее значение для химической промыт
ленности.
Продуктами основного органического синтеза чаще всего
являются несложные соединения, используемые как продукты
для дальнейших синтезов, а также в качестве растворителей
ядохимикатов и пр. К ним относятся спирты, углеводороды,
кислоты, альдегиды, кетоны, нитропроизводные, сульфосоединения^
хлорпроизводные и т. д.
1. СИНТЕЗЫ НА ОСНОВЕ ВОДОРОДА И ОКИСИ УГЛЕРОДА
Возможность взаимодействия окиси углерода и водорода
(гидрирования окиси углерода) была установлена в начале
XX века. П. Сабатье в 1902 г. из смеси окиси углерода и
водорода в присутствии мелкораздробленного никеля получил метан.
В 1908 г. Е. И. Орлов из смеси окиси углерода и водорода в
присутствии никеля и палладия синтезировал этилен. Фишер »
Тропш в 1924 г. разработали способ синтеза из окиси углерода
и водорода углеводородов с различной длиной цепи. В
зависимости от условий проведения процесса, т. е. давления,
температуры, катализатора и соотношения между окисью углерода,
и водородом, можно получать самые разнообразные продукты,—
начиная с метана, этилена, метанола и других
кислородсодержащих соединений и кончая высшими углеводородами, вплоть до
парафина, церезина и др. (см. приведенную ниже схему).
Сырьем для этих синтезов служат водяной или смешанный
генераторные газы, либо смеси их с водородом. Смеси окиси
углерода и водорода также получают конверсией метана или
водяного газа.
255
СХЕМА
С0:Н2A : 3), Ni, ThO2, MgO
«CO+H2
250 —500° С, 9,8h/cjk2
Преимущественно метан
CO:H2 A : 1 или 1 : 2) Fe, Cu, Ni, FhO2; MgO _
-5 оПП /-г Парафины и олефины от
i,o — duu н/см метана до твердого па.
рафина
Преимущественно
ароматические
углеводороды
Метанол
—TTqКО
Al2u3, K2U,
СО:Н2A :1)
Сг2О3, ThO2
475 —500° С, 300 н/см*
СО : Н2 A : 2), ZnO, Cu, Сг2О3, МпО
200 — 400° С, 1000 — 10 000 н/см2
СО : На A : 2), ZnO, Си, Сг2О3, МпО, К2О
300 — 450° С, 1000 — 4000 н/см?
СО:Н2 (і :1 у :2І Fe, K2O
'400 -450° С, 1000—1500 н/см*
Метанол
спирты
и высшие
Синтол (смесь спиртов,
альдегидов, эфиров,
кислот)
Очищенная от сернистых соединений и других примесей
-смесь окиси углерода и водорода, применяемая для синтезов,
называется синтез-газом.
Наиболее важным в настоящее время синтезом на базе окиси
углерода и водорода является синтез метанола.
Синтез метилового спирта (метанола)
В СССР первый завод производства синтетического
метанола был пущен в 1933 году. В настоящее время около 90% всего
метанола получают синтетическим путем, остальное — при сухой
перегонке дерева. Значение метанола для химической
промышленности чрезвычайно велико: из него получают формальдегид,
используемый при производстве пластмасс, диметилсульфат,
метиламин, метиловый эфир акриловой кислоты и ряд других
продуктов. Кроме того, метанол применяется как растворитель
и r качестве добавки к моторному топливу.
Сырьем для производства метанола служит синтез-газ,
в котором соотношение СО : Н? достигает 1 : 2—1 : 4.
Метанол образуется по реакции
СО + 2Н2 «=! СН3ОН + 111.3 кдж
при давлении 2500—3000 н/см2 и температуре 350—400° С в
присутствии катализаторов, наилучшим из которых является цинко-
хромовый — 8ZnO • Сг2О3 • СгО3. За один проход через
контактный аппарат в метанол преврашается лишь 5—20% газа,
поэтому процесс ведется по непрерывно-циркуляционному методу, как
и синтез аммиака. Образование метанола, так же как и
аммиака, сопровождается уменьшением объема и выделением тепла.
256
Поэтому повышение давления и снижение температуры приводят
к увеличению выхода метанола (согласно принципу Ле-Ша-
телье), что видно из данных, приведенных в табл. 21 для смеси,
содержащей 33,3% СО и 66,7% Н2.
Таблица 21
Давление, н/см1 |
300° С
350° С
1100
2000
3000
4000
26,3
53,4
76,0
86,0
8,2
24,0
37,7
53,1
Образование метанола всегда сопровождается побочными
реакциями, например:
СО + ЗН2 *=*СН4 + Н2О + 209 кдж,
СО + Н2 в НСНО + 8,4 кдж,
2СН,ОН -* СН. —О— СН, + Н,О.
Кроме того, возможно образование высших спиртов, кислот,
кетонов и альдегидов.
Рис. 108. Схема синтеза метилового спирта из СО
и Н2:
/ — компрессор; 2 — смеситель; 3 — теплообменники;
4— колонна синтеза; 5 — холодильник; 6 —
сборник-сепаратор; 7 — циркуляционный компрессор; 8 — пусковой
электроподогреватель
Образованию побочных продуктов способствует уменьшение
давления, повышение температуры и присутствие железа,
которое катализирует побочные реакции. Железо аппаратов образует
пентакарбонил железа с окисью углерода Fe(COM, который,
разлагаясь на катализаторе, покрывает его тонким слоем железа.
Поэтому колонны для синтеза метанола футеруют медью или
изготовляют их из высоколегированных сталей, более
устойчивых к карбонильной коррозии. Циркулирующий газ очищают от
карбонила железа активированным углем. Схема синтеза
показана на рис, 108. В результате синтеза получается содержащий
17 8-1504
257
92—93% метанола спирт-сырец, который затем подвергают
ректификации и специальной очистке от побочных продуктов.
Выбор объемных скоростей газа при синтезе метанола
определяется теми же условиями, что и для синтеза аммиака.
Обычно работают с объемными скоростями 10 000—30 000 м3/м3
катализатора в час. Процесс должен протекать автотермично.
Колонны синтеза имеют следующие размеры: высота 12—•
18 м; диаметр 0,8—1,2 м, толщина стенок 0,09—0,1 м.
Температура кипения метанола 64,7° С, температура
замерзания — 97,8° С. Метанол является сильным нервным и сосудистым
ядом кумулятивного действия. Пары метанола с воздухом
образуют взрывчатые смеси.
Производство формальдегида
В промышленнбсти формальдегид получают неполным
окислением метана и каталитическим окислением метанола.
Метанол окисляют за счет кислорода воздуха б паровой фазе
при 650—700° С в присутствии катализатора, в качестве
которого применяют медь или серебро в виде сеток, крупных
кристаллов или осажденные на пористом носителе. Серебро активнее
меди в три-четыре раза, поэтому установки, работающие
с серебряным катализатором, более производительны.
Реакция окисления метанола протекает по уравнению
СН3ОН + 0,5О2 -> НСНО + Н2О + 159 кдж.
Обычно процесс проводят при недостатке воздуха, чтобы
частично протекала эндотермическая реакция дегидрирования
метанола
СН3ОН -> НСНО + Н2 — 83,77 кдж.
Вследствие этого снижается суммарный тепловой эффект
реакции, предупреждается разложение формальдегида и удлиняется
срок службы катализатора. Снижает температуру окисления
также присутствие паров воды, поэтому для окисления берут
метанол с примесью 10—12% воды.
Смесь спирта и воздуха получают продувкой очищенного
воздуха через нагретый до 45—50" С метанол. При этом образуется
паровоздушная смесь, содержащая свыше 36,5 об. % паров
спирта (верхний предел взрываемости смеси).
Во избежание конденсации паров спирта паровоздушную
смесь нагревают глухим паром до 110° С и подают в контактный
аппарат, который перед пуском установки подогревают, а затем
процесс протекает автотермично. Продукты реакции по выходе
из контактного аппарата охлаждаются в холодильнике до 100—
130° С и абсорбируются водой в абсорберах. При этом
получается 38—40%-ный раствор формальдегида, называемый форма-
258
лином. Схема установки приведена на рис. 109. Выход
формальдегида составляет 80% от теоретического.
Формальдегид — газ с раздражающим запахом, сжижается
при —21° С, легко поли-
меризуется в параформа-
льдегид. Полимеризации
формальдегида
препятствует примесь 10—12%
метанола.
При взаимодействии
формальдегида с
аммиаком образуется гексаме-
тилентетрамин (СН2)б1^4,
который впервые
синтезировал А- М.
Бутлеров. Гексаметилентетра-
мин, или уротропин, при- рис. Ю9. Схема производства формальде-
МСНЯСТСЯ Б больших КОЛИ- гида (формалина) из метанола:
ЧеСТВЭХ ДЛЯ ПрОИЗВОДСТВа / — воздуходувка; 2—испаритель метанола; 3—
феНОЛОфОрмальдеГИДНЫХ ПЄРЄсГкрЄубберь1': 7 — оросительи^х'омдильни'к. ~~
смол, взрывчатых
веществ, в медицине и как антисептик.
2. СИНТЕЗЫ НА ОСНОВЕ ДВУОКИСИ УГЛЕРОДА.
ПРОИЗВОДСТВО МОЧЕВИНЫ (карбамида)
Синтетическую мочевину получают из двуокиси углерода
и аммиака в две стадии. Сначала образуется карбамат аммония:
2NH3
159,6 кдж,
а затем происходит разложение его с образованием мочевины
и воды:
NH4OCONH2 *a NH2CONH2 + Н2О — 28,56 кдж.
В промышленных условиях (для увеличения выхода) синтез
мочевины проводят под давлением 2000 н/см2, при 185—200° С
и избытке аммиака, достигающем 100%. Выход мочевины при
этом составляет 55—60% от количества образовавшегося карба-
мата аммония.
Избыточный аммиак и не вошедшие в реакцию газы можно
возвращать обратно в колонну (циркуляционный способ) или
использовать для производства аммиачной селитры.
Основным аппаратом в установке для производства
мочевины является колонна синтеза, которую, учитывая агрессивность
среды, изготовляют из специальной легированной стали. Для
защиты корпуса колонны от разрушения в нее вставляют
цилиндр, где в основном и происходит образование карбамата
аммония и мочевины.
17*
259
Установка для производства мочевины изображена на
рис. ПО.
Синтез протекает следующим образом. Очищенная двуокись
углерода сжимается компрессором / до давления 2000 н/см2
и при 30—35° С подается во внутренний цилиндр колонны
синтеза 6. Жидкий аммиак из сборника 2 засасывается трехплун-
жерным насосом 3 и под давлением до 2000 н/см2 через
подогреватель 4 поступает в кольцевое пространство между стенками
12s
Рис. ПО. Схема производства синтетической мочевины.
колонны и цилиндром при 75—80° С. Аммиак, омывая корпус
колонны, предохраняет его от разрушения. Насосом высокого
давления 5 в колонну подается конденсат.
В результате экзотермичности реакции образования карба-
мата аммония температура в колонне повышается до 180—
200° С. Часть карбамата аммония E5—60%) разлагается на
мочевину и вод}".
Плав мочевины дросселируют до 12 н/см2 и направляют в
дистилляционную колонну 7, где температура снижается до 70° С.
Колонна обогревается глухим паром. Здесь происходит
выделение избыточного аммиака, разложение карбамата аммония,
поступившего из колонны синтеза и аммиака, образовавшегося
из мочевины (степень разложения мочевины— 15%)- В колонну
добавляется острый пар, способствующий более полному
отделению газов {NH3, CO2). Газы направляются на производство
аммиачной селитры. Из нижней части колонны вытекает 40—
60%-ный раствор мочевины, который через гидравлический за-
260
твор 8 стекает в сборник-отстойник 9. После отделения от масла,
увлеченного из компрессоров, раствор через фильтр-пресс 10
идет на упаривание в вакуум-выпарку //, присоединенную
к конденсатору 12, а затем в шнековый кристаллизатор 13 и
центрифугу 14. Маточный раствор возвращается в
сборник-отстойник 9. Производительность колонны синтеза составляет около
30 т 98%-ной мочевины в сутки.
Для синтеза мочевины используют двуокись углерода,
получаемую как побочный продукт при производстве конверсионного
водорода для синтеза аммиака. Отсюда следует, что синтез
аммиака и мочевины можно объединить. Это даст возможность
комплексно использовать сырье и несколько сократить расход
электроэнергии.
Мочевина представляет собой наиболее концентрированное
азотное удобрение D6% N2) и применяется в больших
количествах для производства искусственных смол и пластических
масс, а также многих медицинских препаратов, например
бромурала, веронала. Мочевина и ее производные — централиты
применяются как стабилизаторы нитроглицериновых и
пироксилиновых порохов. Нитромочевина NH2CONHNO2 и нитрогуанидин
HNC(NH2JHNO3 взрывчаты и применяются в качестве добавок
к взрывчатым веществам. Кроме того, мочевина является весьма
эффективным кормовым средством для животных.
3. СИНТЕЗЫ НА ОСНОВЕ ПРЕДЕЛЬНЫХ УГЛЕВОДОРОДОВ
Источником получения легких предельных углеводородов —
метана и его гомологов являются природные и нефтяные газы,
отходящие газы нефтеперерабатывающих заводов, коксовый газ.
К сырью, содержащему высшие предельные углеводороды,
относятся нефть и продукты синтеза из окиси углерода и водорода
(стр. 256). Для синтезов на базе предельных углеводородов
применяются реакции хлорирования, нитрования, сульфирования,
окисления, дегидрирования и изомеризации. В данном курсе
рассматриваются только некоторые из синтезов, имеющие
промышленное значение.
Окисление высших предельных углеводородов (парафина)
с целью получения высших жирных кислот
Окисление высших парафиновых углеводородов (Сі7 — С3є)
имеет чрезвычайно важное значение, так как позволяет получать
синтетические высшие жирные кислоты и спирты, необходимые
для производства из непищевого сырья мыла и других моющих
средств.
Такое окисление сопровождается обычно разрывом цепи,
поэтому при окислении парафина до кислот получаются не только
261
кислоты, отвечающие высшим предельным углеводородам, но и
кислоты, имеющие меньшее число углеродных атомов.
Метод окисления парафина впервые разработан акад.
С. С. Наметкиным еще в 90-х годах прошлого столетия, а первый
завод, производящий синтетические жирные кислоты из
парафина, в СССР был построен только после Великой Отечественной
войны.
Перед окислением парафин очищают от механических
примесей, обезвоживают и освобождают от олефинов, изопарафинов,
нафтенов и ароматических углеводородов, обрабатывая его
96%-ной серной кислотой. Все эти углеводороды растворяются
в серной кислоте, образуя частично сульфокислота, которые
удаляются раствором щелочи. Очищенный парафин перегоняют
и отбирают для окисления фракцию, выкипающую при 320—
450° С. Парафин окисляется воздухом в присутствии 0,15%
катализатора (KMnO4, МпОг, соли щелочных металлов). Водный
10—15%-ный раствор катализатора смешивается с
расплавленным парафином и нагревается до 130° С. При этом вода
испаряется, а катализатор равномерно распределяется в
парафине. Окисление ведут при 100—120° С в реакторе из хромо-
никелевой стали или алюминия, вмещающем до 30 г парафина.
В начале процесса парафин подогревается глухим паром
через змеевики, уложенные в реакторе, а затем температура
возрастает за счет тепла реакции окисления, так что для
поддержания температуры 100—120° С реактор приходится охлаж-
дать водой, циркулирующей в змеевиках. Окисление
заканчивают, когда кислотное число реакционной массы достигает 70,
на что требуется 16—28 часов.
Расплавленный окисленный парафин отделяют от
катализатора отстаиванием и затем промывают водой для удаления
низших жирных кислот. Промытый окисленный парафин омыляется
щелочами в две стадии. В первой стадии омыление производится
25%-ным раствором соды при 150—180° С, в результате чего
образуются мыла. После отстаивания парафиновые
углеводороды всплывают и снова направляются на окисление, а нижний
слой солей жирных кислот вторично омыляется 25%-ным
раствором едкого натра под давлением 800—1200 н/'см2 при
температуре 270—300° С. При этом омылению подвергаются
высокомолекулярные жирные кислоты и другие омыляемые продукты
окисления, например
NaOH
RCH2COCH2CH3 -*¦ RCH2COONa + C2He.
После снижения давления температура реакционной массы
падает до 150° С, неомыленные углеводороды, вода, спирты
испаряются, а мыло сливается в аппарат с раствором сульфата
натрия. Полученный 25%-ный мыльный клей разлагают 92—
262
96%-ной серной кислотой, в гуммированных изнутри аппаратах,
чтобы выделить свободные жирные кислоты. Реакционную
массу промывают водой и подвергают перегонке под вакуумом
G57 мм рт. ст.). Получаются три фракции кислот: 16% С<— С<>;
5% Сю — С2о и в остатке — Ci8 — C2s-
Общий выход кислот достигает 75—80% от веса окисленного
парафина.
В качестве побочного продукта образуется сульфат натрия,
который частично используется в производстве.
Синтетические моющие средства
Бурное развитие органического синтеза и поиски путей
замены пищевого сырья непищевым в мыловаренной
промышленности привели к появлению новой отрасли химической
промышленности — производства синтетических моющих средств и
поверхностно-активных веществ.
Обычное жировое мыло, несмотря на свои несомненные
достоинства, имеет ряд недостатков, ограничивающих его
применение в текстильной промышленности и в быту:
1) растворы мыла имеют щелочную реакцию и действуют
разрушающе на шелковое, шерстяное и синтетическое волокно;
2) в жесткой воде мыло образует нерастворимые
кальциевые и магниевые соли, что уменьшает его моющее действие;
3) в кислой среде мыло разлагается;
4) мыло — дорогой продукт, так как на его изготовление
расходуется пищевое сырье.
В Советском Союзе производство жирового твердого мыла
стабилизировалось на современном уровне, а прирост продукции
мыловаренной промышленности на 55—60% будет достигнут за
счет применения поверхностно-активных веществ из непищевого
сырья — преимущественно продуктов химической переработки
нефти и пр.
Новые моющие средства относятся, как и мыло, к
поверхностно-активным веществам и подразделяются на ионогенные и не-
ионогенные. В зависимости от природы полярной группы их
делят на анионо- и катионоактивные. Большинство моющих
средств являются анионоактивными. К ним относятся
преимущественно сульфосоединения с длинной углеводородной цепью
(Сю — Сгб) и группами SO2ONa — сульфонатыи OSO2ONa—
сульфаты.
Сырьем для производства этих веществ служат
высокомолекулярные кислоты и спирты, парафиновые углеводороды,
высшие олефины и алкилпроизводные бензола.
Ближе всего к мылам продукты переработки жирных кислот,
б которых карбоксильные группы блокированы путем этерифи-
263
кации, амидирования и т. д. с одновременным внедрением суль-
фогрупп в конце цепи, например:
RCOOCH2CH2SO3Na — игепон А,
RCON — CH2CH2SO3Na —игепон Т.
Н
Эти производные устойчивы к действию жесткой воды, не
гидролизуются и обладают высоким моющим действием.
Применяются они в быту для стирки тонких тканей из шерсти и
шелка, а также в текстильной промышленности. Подобными
качествами обладают продукты сульфатирования высших жирных
спиртов, первичных или вторичных и их натриевые соли — алкил-
сульфаты:
RCHOSO2ONa; ROSO2ONa.
Ri
Алкилсульфаты выпускаются в продажу в виде 20—40%-ных
водных (коллоидных) растворов или порошкообразных смесей
с Na2SO4, Na2CO3, конденсированными фосфатами и другими
добавками под различными названиями («Новость», «Кристалл»
и др.).
Сульфоэфиры и их натриевые соли обладают моющей
способностью в кислой, нейтральной и щелочной среде. Они снижают
поверхностное натяжение воды в большей степени, чем
жировое мыло.
Для получения моющих средств из парафиновых
углеводородов применяется метод сульфохлорирования, т. е. одновременной
обработки углеводородов хлором и сернистым ангидридом в
жидкой фазе при 40—50° С под действием ультрафиолетовых
лучей. В результате получаются сульфохлориды:
RH + С12 + SO2-^ RSO2C1+ HC1.
Эта цепная реакция начинается с поглощения светового
кванта (фотона) молекулой хлора. Образующиеся ионы хлора
инициируют реакцию:
СЗа -Э-2СЗ-,
RH {-С1- ->Р/ -! НП,
R-+SO2->RSO2\
RSO.2 + C12->RSO2C1 + C1\
RH+C1- ->-R- + HC1 и т. д.
Сырьем для процесса служит когазин — смесь углеводородов
Сіз—'Сів, полученная синтетическим путем (t° кип. 240—340° С).
Во избежание обрыва цепной реакции эту фракцию очищают от
ненасыщенных и кислородсодержащих соединений гидрирова-
264
ниєм, используют и легкие парафины и нафтеновые
углеводороды.
Полученная смесь сульфохлоридов известна под названием
«мерзоли», а натриевые соли их называют «мерзолятами», или
алкилсульфонатами:
C«H2«+,SO2C1 + 2NaOH -*¦ CnH2n+,SO2ONa + NaCl + H2O.
Мерзоляты применяются в производстве грубых и мягких
моющих средств, стиральных порошков и пр., главным образом
в текстильной промышленности.
Производство моющих средств из алкилпроизводных бензола
получило большое развитие в США. Сырьем служит узкая
керосиновая фракция прямой перегонки нефти (т. к. 218—246° С),
которую хлорируют, а затем применяют для алкилирования
бензола в присутствии АІСІз при 80° С. Выделенный и перегнанный
моноалкилбензол сульфируют олеумом при 30—35° С. Сульфо-
кислоты отделяют и нейтрализуют концентрированным NaOH:
C«H2n+iCeH4SO2OH + NaOH ^CnH2n+iCeH4SO2ONa + H2O.
В СССР этот продукт называют сульфонолом. Готовый
продукт (наконол NR), представляющий собой смесь 35% алкил-
бензолсульфонатов с добавкой натриевых солей — фосфатов,
метасиликатов и др.,— обладает довольно хорошим моющим
действием.
При обработке высших олефинов (Сіг — Сів) серной
кислотой с последующей нейтрализацией получаются алкилсульфат-
ные моющие средства:
R\ NaOH R\
RCH = CH2 + H2SO4 - >CHOSO2OH >- >CHOSO2ONa+H2O,
CH/ CH/
представляющие собой преимущественно вторичные алкилсуль-
фаты.
При диссоциации катионоактивных моющих веществ, которые
являются главным образом солями высокомолекулярных
аминов или четвертичных аммонийных оснований общей формулы
R2
R4
+
х-
жировой остаток заряжен положительно.
Эти вещества применяются в основном как вспомогательные
в текстильной промышленности, поскольку они почти не
обладают моющим действием. Достоинством их является
устойчивость к жесткой воде.
26S
К неионогенным моющим средствам относятся вещества с не-
«оногенной гидрофильной группой, например, гидроксильной,
кислородной (в простых эфирах).
Из алкилпроизводных фенола и окиси этилена получают
нейтральные моющие и эмульгирующие средства, называемые
игепалями:
С„Н2„+1С6Н4ОН + тСН2-СН2 -* С„Н2„+1СвН4(ОС8Н1)тОН.
\ /
О
Из производимых в СССР веществ к этой группе относится гли-
колевый эфир алкилфенола, обозначаемый ОП.
Значение поверхностно-активных веществ, используемых в
¦качестве моющих средств и эмульгаторов-вспенивателей, с
каждым годом возрастает. Большой эффект дает применение
поверхностно-активных веществ в нефтяной промышленности — при
деэмульсации нефти и в качестве добавок к глинистым
растворам при бурении.
Поверхностно-активные вещества способствуют более
полному извлечению металлов из руд в процессе флотации. Они
применяются в резиновой промышленности для улучшения
диспергирования наполнителей, пигментов и пр.
Поверхностно-активные вещества нужны также для
приготовления эмульсий с ядохимикатами, для производства бетона,
светочувствительных пластинок и т. д.
Дегидрирование предельных углеводородов
Процесс дегидрирования предельных углеводородов ведет
к получению из них непредельных углеводородов.
Дегидрированием этана, например, может быть получен
этилен:
С2Н6 -*¦ С2Н4 + Н2.
В результате дегидрирования бутана образуется бутилен или
•бутадиен:
CJIiu ^ СНа - СН - СН„-СНа + Н2;
С4Н10 -ч-СН2 - СН — СН = СН2 + 2HS.
Особенно важен процесс дегидрирования бутана с целью
получения бутадиена, который необходим для синтеза каучуков.
По методу Н. Д. Зелинского дегидрирование бутана
производится в присутствии хромовых катализаторов (Сг2О3, нанесен-
ный на AI2O3) при 590° С. Если применяются стационарные
катализаторы, процесс ведется периодическим методом. Через
катализатор пропускают газ, подогретый в трубчатой печи до 600° С.
Вследствие эндотермичности реакции через 7—10 мин насадка
аппарата охлаждается на 25—30° С и покрывается слоем сажи
266
и смолистых веществ. Тогда процесс дегидрирования
прекращают, отсасывают продукты реакции, а сажу и смолу выжигают,
пропуская воздух. Катализатор регенерируется и подогревается
до 590—600° С. После удаления продуктов горения и воздуха
отсасыванием снова проводят дегидрогенизацию.
Применяются также катализаторы в псевдоожиженном
состоянии. Из полученных продуктов дегидрирования бутадиен
извлекают абсорбцией, растворителями, а затем отделяют от
растворителей дистилляцией.
4. СИНТЕЗЫ НА ОСНОВЕ ОЛЕФИНОВЫХ УГЛЕВОДОРОДОВ
Синтезы из олефиновых углеводородов более разнообразны,
чем из предельных углеводородов. Это объясняется большей
реакционной способностью олефиновых углеводородов,
обусловленной наличием в их молекуле двойной связи. Источником оле-
фінов могут служить газы крекинга и пиролиза нефти,
содержащие 30—50% ненасыщенных углеводородов, а также
этиленовые фракции коксового газа, получаемые при выделении из
коксового газа водорода (стр. 91) методом фракцинированной
конденсации в условиях низких температур. В этиленовых
фракциях содержится 25—40% этилена. Олефиновые углеводороды
образуются также в процессе пиролиза предельных
углеводородов природных газов.
Синтез спиртов через моно- и диалкилэфиры серной кислоты
В первую очередь к таким синтезам следует отнести синтез
этилового спирта через моноэтиловый эфир серной кислоты
(этилсерную кислоту) и ее диэтиловый эфир (диэтилсульфат).
Промывая газ, содержащий этилен, 95—98%-ной серной
кислотой, получают эти эфиры и после омыления их — спирт:
НОЧ С2Н6ОЧ
1)СН2=СН2 + >SO2-> >SO2;
НСК НСК
этилсерная кислота
НОЧ С2Н6ОЧ
2) 2СН2 = СН2 + >О2-> >SO2;
НСК С2Н5СК
диэтилсульфат
с2н6оч ноч
3) >SO2 + НОН -> >SO2 + С2Н6ОН;
НСК НСК
с2н6оч на
4) >SO2 + 2НОН -v >SO2 + 2С2Н6ОН.
С2Н6СК НСК
267
Промывка кислотой ведется под давлением, величина
которого зависит от количества этилена в газе. При этом необходимо,
чтобы парциальное давление этилена составляло около 100—
120 н/см2. Образование диэтилсульфата неизбежно, но
нежелательно, поскольку при омылении он реагирует с
образовавшимся спиртом и дает эфир, являющийся побочным продуктом
производства:
с2н5оч с2н5оч
>SO2 + С2Н5ОН -> >SO2 + (С2Н5JО.
с2н5о/ но/
Отходящая серная кислота обычным способом
концентрируется, а затем ее снова возвращают в процесс. К недостаткам
этого способа относятся значительные потери серной кислоты
и необходимость применения химически стойких материалов.
Значение этилового спирта общеизвестно. Крепость его в
процессе ректификации может быть доведена до 95,5%
(азеотропная смесь). •
Абсолютный спирт в технике получают методом азеотропной
дистилляции. При этом 95%-ный спирт смешивают с бензолом
и сначала отгоняют при 64,8° С фракцию бензол — спирт — вода,
а затем повышают температуру до 68,2° С и отгоняют фракцию
спирт — бензол; наконец, при повышении температуры до 78,2° С
отгоняется остаток — абсолютный этиловый спирт.
Прямая гидратация олефинов в спирты
Спирты могут быть получены прямой гидратацией олефинов.
Например,
СН2 = СН2 (газ) + Н2О(пар) -> СН3СН2ОН (пар) + 50,4 кдж/моль.
Реакция эта может быть проведена с парами воды и в жидкой
фазе. Применяя принцип Ле-Шателье, мы видим, что ей должны
благоприятствовать высокие давления и невысокие температуры.
Однако в этих условиях пары воды конденсируются, кроме того,
при низких температурах скорость реакции будет очень малой.
Поэтому приходится вести реакцию при температуре порядка
250—300° С в присутствии катализаторов. В паровой фазе
использовалось давление 600—800 н/см2 и производилось
нагревание до 250° С. В указанных условиях за один проход гидра-
тируется только 4—5% прошедшего катализатор этилена,
поэтому для осуществления процесса требуется многократная
циркуляция. В конечном счете получаются 15—16%-ные растворы
спирта-сырца, из которого получают чистый спирт. Схема
производства изображена на рис. 111.
Свежий и циркулирующий этилен смешиваются в тройнике 6У
подогреваются в теплообменнике 3 до 220—230° С и вместе
с перегретым паром через эжектор 2 поступают в контактный
26»
аппарат /, заполненный фосфорной кислотой на алюмосиликате.
В контактном аппарате за счет тепла реакции поддерживается
температура 280—300° С.
Продукты синтеза по выходе из контактного аппарата
отмываются от фосфорной кислоты в тройнике а щелочью,
подаваемой из сборника // насосом 12, проходят солеотделитель 8,
Рис. 111. Схема производства этилового спирта прямой
гидратацией этилена.
теплообменник 3 и конденсируются в холодильнике 4.
Отделение этилена от спирта происходит в сепараторе высокого
давления 7, откуда этилен забирается циркуляционным
компрессором 5. Концентрацию этилена в циркулирующем газе
поддерживают равной 80—85 об. %. Спирт вытекает из нижней части
аппарата через дроссель ДВ в сепаратор низкого давления 9,
где отделяется еще некоторое количество этилена, который
также возвращается в процесс. Водный раствор спирта, пройдя
теплообменник 10, кипятильник 13 и отпарную колонну 14,
ректифицируется в колонне 15 и, пройдя дефлегматор 16 и
конденсатор 17, стекает в сборник товарного продукта.
Значительно легче идет прямая гидратация высших
гомологов этилена, например пропилена:
СНЗЧ
сн3 • сн = сн2
н2о
сн.
>снон.
269
При пропускании пропилена с парами воды над
катализатором АІ2EО4)з при 105° С и 40—50 н/смя получается спирт с
выходом 82%.
Изопропиловыи спирт является превосходным растворителем
для смол и масел—лучшим, чем этиловый спирт. Но главное
значение его состоит в том, что он служит основным
материалом для приготовления ацетона.
Этиленхлоргидрин, окись этилена, этиленгликоль
и их производные
При присоединеиии к этилену хлорноватистой кислоты
образуется этиленхлоргидрин:
Н2С = СН2 + НС1О -* НОСНаСН,С1.
этиленхлоргидрин
(жидкость)
Обработанный цианистым натрием этиленхлоргидрин дает
широко применяемое в технологии искусственного кяучука,
искусственного волокна и пластических масс соединение — акрило-
нитрил:
С1СН2СН2ОН + NaCN -> СН2 = СН • CN + NaCl + Н2О.
При обработке раствора этиленхлоргидрина слабыми
щелочами, например содой, получается этиленгликоль
2НОСН2СН2С1 + Na2CO3 + НОН -> 2НОСН2СН2ОН + 2NaCl +
+ со2.
этиленгликоль
После обработки этиленхлоргидрина сильными щелочами,
например, известковым молоком или растворами едкого натра,
он превращается в окись этилена:
2НОСН2СН2С1 + Са(ОНJ -> 2Н2С—СН2 + СаС12 + 2Н2О.
\/
О
окись этилена
В настоящее время применяется метод прямого окисления
этилена воздухом в присутствии серебряных порошкообразных
катализаторов в неподвижном или взвешенном слое по реакции
СН2 = СН2 +0,5О2 ->СН2— СН2 + 138 кдж.
V
Частично протекает и реакция полного окисления этилена:
СН2 = СН2 + ЗО2 -> 2СО2 + 2Н2О + 1410 кдж.
Этой реакции способствует присутствие железа, поэтому
аппараты и трубы для катализатора изготовляются из легированной
стали (рис. 112). Очищенный от примесей серы, мышьяка,
ацетилена и других углеводородов этилен в'смеси с воздухом в отно-
270
шении І : 9 подается компрессором в реактор /, где
подогревается в теплообменнике, а затем проходит через катализатор
со скоростью, достаточной для того, чтобы он находился во
взвешенном слое. В межтрубном пространстве теплообменника и в
змеевике 2 циркулирует эвтектическая смесь дифенила и дифе-
нилоксида (даутерм), с помощью которой поддерживается
постоянная температура около 370° С. Для того чтобы катализатор
не уходил из реактора вместе с газами, в верхней части
аппарата сделана насадка из колец Рашига.
ИгО
-+>и-г№*
Рис. 112. Схема производства окиси этилена.
В первом реакторе образуется около 50% окиси этилена,
которая после охлаждения в конденсаторе 3 извлекается водой
в абсорбере 4. Компрессор 5 подает оставшиеся газы в реактор
второй ступени 6, где непрореагировавший этилен окисляется
при 560° С. После охлаждения смеси газов в конденсаторе 7
окись этилена извлекается водой в абсорбере 8. Общая степень
окисления этилена достигает 70—80%.
Водный раствор окиси этилена из сборника 9 через
теплообменник 10 направляется в отпарную колонну //, вода из нижней
части колонны через теплообменник 10 подается на орошение
абсорберов 4 и 8, а парогазовая смесь через дефлегматор 12
поступает в ректификационную колонну 13, снибженную
дефлегматором 14 и кипятильником 15, а затем в колонну 16 для
окончательной очистки. Из нижней части колонны вытекает окись
этилена 99,5%-ной чистоты. Окись этилена кипит при +10,8° С,
и поэтому ее конденсируют с помощью рассолов^ охлажденных
до —5—10° С, а затем хранят в баллонах. Благодаря легкости
размыкания напряженного трехчленного эпоксидного кольца
271
окись этилена относится к весьма реакционноспособным
соединениям. На основе ее можно получать самые разнообразные
синтетические продукты.
Путем гидратации окиси этилена при 50—100° С в
присутствии катализаторов (H2SO4, НзРО4) получают этиленгликоль:
СН2 — СН2 + Н2О -> СН2ОНСН2ОН.
При этом образуются также ди-, три- и полиэтиленгликоли, на-
яример,
СН2—СН2 + НОСН2СН2ОН -*- НОСН2СН2ОСН2 — СН2ОН.
V
Этиленгликоль применяется для приготовления
пластификаторов, алкидных смол, как антифриз и т. д.
Взаимодействие окиси этилена со спиртами в присутствии
серной кислоты дает Простые эфиры, называемые целлозоль-
вами:
H,SO4
Н2С-СН2 + ROH —>¦ HOCH2CH2OR.
\ /
О
Они являются хорошими растворителями эфиров целлюлозы и
применяются в лакокрасочной промышленности.
Реагируя с аммиаком, окись этилена образует этаноламин:
NH3 + Н2С —СН2 ->¦ H2NCH2CH2OH.
Y
При дальнейшем присоединении окиси этилена получаются
ди- и триэтаноламины. Моно- и диэтаноламин используются для
очистки газов от сероводорода и углекислоты, моно- и триэта-
ноламин — в производстве моющих средств и для других целей.
Все большее значение приобретают эпоксидные смолы,
полученные на основе іфоі-мводньіА. икиси а і плена, применяемые
в производстве пленкообразующих вешеств и пластмасс.
Галоидные соединения олефинов. Дихлорэтан.
Хлористый аллнл. Синтез глицерина.
Известно большое количество галоидных соединений
олефинов. Присоединением к этилену хлора получают дихлорэтан
С2Н4СІ2, являющийся одним из наиболее распространенных и
дешевых растворителей, а также материалом для некоторых
органических синтезов. Реакция ведется в закрытом аппарате,
наполненном- дихлорэтаном и снабженном барботажными приспособ-
272
лениями для этилена и хлора, а также змеевиками для
охлаждения водой. Избыточный дихлорэтан отводится в колонну с
насадкой и, опускаясь сквозь подаваемый снизу в колонну раствор
едкой щелочи, отмывается от соляной кислоты и свободного
хлора, а затем направляется на ректификацию.
Хлорирование пропилена в газовой фазе при 400—500° С
приводит к образованию хлористого аллила:
СН2 = СНСНз + С12 -*¦ СН2 = СНСН2С1 + НС1.
Это соединение является основным при синтезе глицерина.
Омыление его (при 160° С и давлении 140 и/см?) раствором едкого
натра дает аллиловый алкоголь:
СН2 = СНСН2ОН (т. кип. 93° С).
После обработки его раствором НС1О получают хлоргидрин
глицерина СН2ОНСНОНСН2С1, в результате омыления которого
образуется глицерин СН2ОНСНОНСН2ОН. Это один из главных
способов получения глицерина.
Глицерин применяется в лакокрасочной промышленности для
производства эфиров — глифталей и др., в медицине,
кондитерском деле (благодаря своей вязкости и сладкому вкусу),
в производстве взрывчатых веществ (тринитроглицерин,
динамит).
5. СИНТЕЗЫ НА ОСНОВЕ АЦЕТИЛЕНА
На стр. 217 была описана технология производства ацетилена
карбидным способом. Этим методом получают ацетилен высокой
концентрации с небольшим количеством примесей. Недостатком
метода является громоздкость установок и большой расход
сырья и энергии. В настоящее время более перспективными
являются методы производства ацетилена из углеводородов — метана,
этана, пропана, бутана и их смесей, в качестве которых могут
быть использованы природный газ и жидкие нефтяные фракции.
Для получения ацетилена из газов применяются следующие
методы: 1) электрокрекинг; 2) термический крекинг с частичным
сжиганием углеводородов; 3) высокотемпературный крекинг
пропана и др.
Электрокрекинг углеводородов
Сущность этого способа заключается в быстром пропускании
углеводородов через электродуговую реакционную печь (рис.
113). Роль электрической энергии в данном случае сводится
к созданию высокой температуры. Процесс осуществляется
следующим образом. В камеру диаметром 1000 мм, Н 400 мм под
давлением 15 н/см2 подают смесь примерно равных объемов
свежего и оборотного газов, направленных по касательной к ци-
1? 8-1504 273
линдрической стенке камеры, вследствие чего газу сообщается
вращательное движение со скоростью > 100 м/сек. Затем газ
попадает в зону электрической дуги (напряжение 7000 в). Макси-
1
Рис. 113. Электродуговая реакционная печь для
разложения газообразных углеводородов:
I— шина; 2 — электрод; 3 — фарфоровый изолятор;
4—реакционная камера; 5 — заземленный электрод; 6 ^- реакционная
труба; 7 — компенсаторы; 8 — пусковой электрод.
мальная температура реакционных газов 1600° С. Продукты
реакции уходят из печи со скоростью 600 м/сек по трубопроводу,
охлаждаемому водой; в конце трубопровода температура газов
274
снижается до 600° С. Дальнейшее охлаждение газов (до 150° С)
достигается впрыскиванием воды. В реакторе
производительностью 2800 м3 метана в час получается около 15 т ацетилена за
сутки. В продуктах электрокрекинга природного газа
содержится:
ацетилена 13,3% окиси углерода 2,9%
олефинов 0,9% кислорода 0,2%
парафина 27,8% азота 8,9%
водорода 46,0%
При рациональном использовании побочных продуктов
ацетилен, полученный из углеводородных газов, будет дешевле
карбидного.
Термический крекинг с частичным окислением углеводородов
Термический крекинг метана осуществляется при 1400—
1500° С. Такая высокая температура достигается сжиганием
части газов с техническим кислородом в
отношении метан — кислород от 2:1 до
1,6: 1. Смесь газов пропускают через
горелку печи с такой скоростью, чтобы
сгорание было неполным. При этом
протекают две основные реакции:
2СН4 + О2 -> 2СО + 4Н2 + 67,2 кдж,
2СН4->СН=СН + ЗН2 — 378 кдж
и частично реакция полного горения.
Общий тепловой эффект реакции
положителен. В промышленности этот процесс
осуществляется в печи (рис. 114)
следующим образом. Отдельно подогретые до
500° С метан и кислород (800 м3/ч и 400—
500 мъ соответственно) поступают в
реакционную печь, футерованную шамотным
кирпичом. В нижней части камеры сме- пе? для ^^ЗяТие
шения (на расстоянии оО мм) имеются тш,ена термическим пи-
две колосниковые решетки из силлимани- ролизом метяиа в присут-
та. Первая является распределительной, ствии кислорода:
аптппяа пгиаппогпа питопі.или ' — труба для ввода кисло-
ВТОраЯ ОГНепреГраДИТеЛЬНОИ. рода 2 - распределитель-
ДЛЯ НОрмалЬНОЙ работы ПЄЧИ НЄО6ХО- ная решетка, Л-смеси-
, г тельная камера: 4 — верх-
ДИМО, ЧТОбЫ СКОРОСТЬ ДВИЖеНИЯ МеТаНО- няя колосниковая решетка;.
кислородной смеси значительно превы- ре~ётк*ГЯ« - фКоТуСиНкиКОдл»
Шала СКОРОСТЬ распределения фронта Разбрызгивания охлаждаю-
г г^ шей воды.
пламени в газе. I азовая смесь проходит
через нижнюю решетку со скоростью 30 м/сек. Благодаря
большой толщине решетки B00 мм) газ воспламеняется только по
*?
выходе из решетки. Для поддержания непрерывного горения
под нижние колосники вводят дополнительное количество
кислорода, нагретого до 700—800° С.
Выйдя из реакционной камеры, продукты реакции быстро
охлаждаются до 80° С в водной завесе, создаваемой форсунками.
В нижней части печи осаждается сажа. Баланс процесса
выражается уравнением
45СН4 + 30,25О2 -> 8СН = СН + 3,5СО2 + 25,5СО +
+ 54Н2 + 28Н2О.
В печи образуется около 1500 лг3/ч газа следующего состава:
ацетилен 8,9%, двуокись углерода 3—4%,
водород 54—65%. метан 4—6%.
окись углерода 24—26%, кислород 0,4%.
Высокотемпературный крекинг пропана
Крекинг высших углеводородов, более легко расщепляемых,
чем метан, проводят в трубчатых цилиндрических аппаратах. Для
подавления вторичных реакций парциальное давление
углеводорода снижают, добавляя перегретый водяной пар, а затем
смесь с большой скоростью пропускают через трубки,
обогреваемые топочными газами. Время пребывания смеси в зоне
высоких температур — меньше 1 сек.. При 1300° С из смеси Н2О :
: С3Н8 = 6,5 : 1 выход ацетилена и этилена составляет 58%.
Состав газообразных продуктов реакции:
ацетилен 16,4% водород 49,8%,
этилен 8,9% ¦ окись углерода 0,7%,
метан 17,0%, двуокись углерода 0,6%,
этан 0,2% кислород 0,1%,
пропилен 0,9%, азот 0,4%.
Ацетилен выделяют из сложных газовых смесей, получаемых
этими методами, либо водой, либо другими растворителями,
например диметилформамидом, под повышенным давлением.
Извлечение видой производят под даьлинисм 200 н/см2.
Оставшиеся непоглощенными газы (Н2. СН4, С2Н6. С2Н4. N2
и др.) разделяют методами глубокого охлаждения. Водород
и этилен передают потребителям, а метан-этановую фракцию
возвращают на крекинг.
Ацетилен выделяют из водных растворов, постепенно снижая
давление, а затем для освобождения от примесей промывают
в скрубберах минеральным маслом, серной кислотой и,
наконец, очищают от СО2 раствором щелочи. В результате
получается 97—98%-ный ацетилен, содержащий до 1% СО2 и 1—2%
других газов.
276
По данным Н. П. Федоренко, если себестоимость карбидного
ацетилена принять за 100%, то себестоимость ацетилена
электрокрекинга составит 30—35%, термического крекинга в
присутствии кислорода 30—40 и ацетилена крекинга пропана 25—35%.
Синтез уксусного альдегида на основе ацетилена
Ацетилен, в котором углероды связаны тройной связью,
является весьма реакционноспособным, причем многие реакции
синтеза происходят за счет размыкания этой связи. Атомы водорода
вода
на очие/пку
т
норееенера-
цию
Катализатор
срегенероции
Ацетилен
сдехсий -
Otopoi
альде-
' на
рентификаци/о
Рис. 115, Схема получения ацетальдегида
гидратацией ацетилена в жидкой фазе.
ацетилена, как мы увидим впоследствии, также способны к
замещению, в результате чего еще больше увеличивается число
возможных синтезов.
Еще в 1881 г. М. Г. Кучеров открыл реакцию, носящую его
имя. Она заключается в том, что при f около 100° ацетилен,
проходя через 20%-ный раствор серной кислоты, содержащей
катализатор — сернокислую ртуть, присоединяет молекулу воды и
превращается в ацетальдегид по реакции
НС =СН + Н2О -> СНдСНО.
Технология производства заключается в следующем (рис.
115). Тщательно очищенный ацетилен в смеси с ацетиленом,
циркулирующим под давлением 15 ні см2, поступает в гидрататор 1
(высота 15 м, диаметр — 1,34 м), заполненный раствором
катализатора. Туда же подают острый пар для поддержания
температуры 80—100° С, так как тепла реакции недостаточно.
Гидрататор изготовляется из нержавеющей стали. За один проход
через аппарат гидратируется 50—60% ацетилена.
277
Выходящая из гидрататора парогазовая смесь поступает в
холодильник 2, где конденсируются в основном пары воды.
Конденсат стекает в гидрататор, а пары ацетальдегида и оставшиеся
пары воды конденсируются в холодильнике 3 и стекают в
сборник 5. Неконденсирующиеся газы отводятся в колонну 4, где
остатки ацетальдегида отмывают водой, а ацетилен возвращают
с процесс. Часть газа направляют на очистку от азота и двуокиси
углерода, чтобы избежать накапливания их в циркулирующей
смеси. Раствор ацетальдегида подвергают ректификации. Выход
ацетальдегида составляет 96% от теории. В качестве побочного
продукта получается кротоновый альдегид.
Катализатор периодически подвергают регенерации, которая
заключается в добавлении к нему раствора окисной соли
железа. Последняя, восстанавливаясь до закисной соли, переводит
образовавшуюся металлическую ртуть в сернокислую:
Hg -f- Fe2(SO4K -> HgSO4 + 2FeSO4.
Известен и другой, открытый А. Е. Фаворским способ
получения ацетальдегида через виниловые эфиры спиртов.
Катализатором этой реакции является КОН (раствор в спирте):
кон
НС =СН + ROH -> ROCH = СН2.
Полученный эфир гидролизуется в присутствии разбавленных
кислот, образуя ацетальдегид и регенерируя спирт:
HjSO.
ROCH = СН2 + Н2О > ROH + СН3СНО.
На основе ацетальдегида осуществляется значительное
количество синтезов, из которых наибольшее значение имеет
производство бутадиена и уксусной кислоты.
Производство уксусной кислоты
С развитием органического синтеза потребность в уксусной
кислоте, которая необходима для производство красителей,
лекарственных и душистых веществ, искусственного шелка,
пищевых продуктов и т. д.. настолько возросла, что уксусной кислоты,
образующейся при сухой перегонке дерева и окислении
этилового спирта, стало недостаточно. Поэтому примерно с 1920 г.
начали применять синтетические методы получения уксусной
кислоты, которые занимают ведущее место в промышленности
органического синтеза.
Производство уксусной кислоты из ацетальдегида
заключается в окислении его кислородом в присутствии катализаторов
(соли Мп, Со, Fe, V). Обычно применяют уксуснокислый
марганец или кобальтомарганцевый катализатор.
278
При окислении образуется сначала надуксусная кислота:
О
СН3СНО + 02 -*¦ СН3С— О — ОН,
затем уксусный ангидрид:
О О
f /-
CH3G-O—ОН + СН3С -^ (СН3СОJО + Н2О.
\
\
И, наконец, в результате гидратации — уксусная кислота:
(СН3СО)аО + Н2О -^ 2СН3СООН.
Упусти
vuoomtj
(Pejmn/фи/ахт)
Рис. 116. Схема производства уксусной кислоты
окислением ацетальдегида:
/ — аппарат для растворения катализатора; 2 —
промежуточный бак; Зі- хранилище для ацетальдегида; 4 — окислительная
колонна; 5, // — конденсаторы; 6 — сепаратор; 7 — скруббер; S,
14 — тарельчатые ректификационные колонны; 9, 13 —
кипятильники, 10, 15, 19 — дефлегматоры; 12 — сборник кислоты; 16 —
реактор; /7 — испаритель, 18 — насадочная ректификационная
колонна.
Реакции протекают с выделением тепла. Окисление проводится
в колонне высотой около 10 м, диаметром— 1 м с расширением
в верхней части, при 70° С и давлении кислорода около 20 ні см2.
Чтобы избежать взрывов, которые возможны при накапливании
надуксусной кислоты и ее разложении, в верхнюю часть
колонны добавляют азот (для разбавления паров).
Колонна изготавливается из алюминия или нержавеющей
стали. Для охлаждения в нее вмонтированы змеевики (рис. 116).
279
Снизу в колонну поступает ацетальдегид и раствор
уксуснокислого марганца в уксусной кислоте. Кислород подается во все
царги колонны, за исключением верхней, которая служит брыз-
гоуловителем. Пары ацетальдегида и уксусной кислоты,
выходящие из верхней части этой царги, охлаждаются в холодильнике
до —10° С и возвращаются обратно на окисление. Уксусная
кислота отводится из нижней части царги. Небольшая часть сырой
уксусной кислоты расходуется для приготовления катализатора,
а остальная подвергается дистилляции. Получается техническая
97—99%-ная уксусная кислота, выход которой составляет
около 90% от теоретического.
Для получения пищевой кислоты техническую уксусную
кислоту подвергают химической очистке и повторной дистилляции.
Разработан также синтез уксусной кислоты, основанный на
термическом разложении ацетона и гидратации образующегося
кетена:
650 —700° С
СНзСОСНз *¦ СН4 + СН2 = СО,
СН2 = СО + Н2О -^ СНзСООН.
ГЛАВА XV
СИНТЕЗЫ НА ОСНОВЕ АРОМАТИЧЕСКИХ
УГЛЕВОДОРОДОВ
Методами синтеза на основе ароматических углеводородов
можно получать самые разнообразные продукты, имеющие
чрезвычайно важное значение для народного хозяйства, — красители,
медицинские препараты, взрывчатые вещества и вещества,
используемые в парфюмерной промышленности, инсектициды и др.
В основу этих синтезов положены классические реакции
нитрования, восстановления, сульфирования, галоидирования,
окисления, гидрирования, алкилирования. Наибольшее значение имеют
синтезы из бензола, толуола, нафталина и др.
I. НИТРОВАНИЕ
Нитрованием называют процесс замещения атома водорода
ядра нитрогруппой —NO2. Нитрующим средством служат смеси,
азотной и серной кислот — нитросмеси. Серную кислоту
прибавляют для связывания выделяющейся при реакции воды:
АгН + HNO3 -*¦ ArNO2 + Н2О.
Соотношение между серной и азотной кислотами зависит от
вещества, которое нитруется, и от того, какое количество нитро-
групп нужно ввести в ядро. Реакция нитрования необратима и
экзотермична. Для поддержания определенной температуры,
280
процесс нитрования ведут при охлаждении и перемешивании в
аппаратах — нитраторах, снабженных водяной рубашкой или
змеевиками и мешалками пропеллерного типа. Изготовляют нит-
раторы из чугуна (нитрирующие смеси не разрушают железо).
Производство нитропродуктов состоит из следующих операций:
нитрование, отстаивание и отделение нитропродукта от
отработанной нитросмеси, промывка нитропродукта водой и содой и
очистка его перегонкой.
нитросмееь
нзал
Отработанная кис/гота
Окисла/ азота
на улаЗлидание
Технический
'нитробензол
Рис. 117. Схема производства нитробензола непрерывным
способом:
/ — нитратор; 2 — дозреватель; 3 — холодильник: 4 — отстойник- 5 —
сборинк отработанной кислоты; 6 ~ колонна для водной промывки-
7 — колонна дли щелочной промывки.
Отработанную нитросмесь подвергают денитрации, серную
кислоту концентрируют и снова используют для приготовления
нитросмеси. В промышленности применяют как периодические,
так и непрерывные методы нитрования. Схема непрерывного
нитрования бензола изображена на рис. 117. Нитрование
бензола с целью получения нитробензола проводят при 50° С и
избытке бензола во избежание образования динитробензола.
Аналогично получают нитротолуол и нитрохлорбензол.
Нитросоединения ароматического ряда имеют большое
значение как промежуточные продукты для органического
синтеза, особенно синтеза аминов, и как взрывчатые вещества,
например, тринитротолуол (тротил), пикриновая кислота, тетра-
нитрометиланилин (тетрил) и др.
2. ВОССТАНОВЛЕНИЕ НИТРОСОЕДИНЕНИЙ
Одним из важнейших представителей аминов является
анилин, получаемый из нитробензола.
Восстановление нитробензола в анилин обычно производится
в солянокислой среде стружкой серого чугуна. Соляной кислоты
добавляют 0,1—0,2 моля на моль нитробензола, так как в
процессе кислота не расходуется. Она требуется для образования
281
соли двухвалентного железа, раствор которой играет роль
электролита в процессе окисления железа кислородом воды,
а водород восстанавливает нитробензол.
Суммарная реакция восстановления нитробензола в
слабокислой среде соответствует уравнению
4C6HSNO2 + 9Fe + 4Н2О -> 4CeH6NH2 + 3Fe3O4-
Восстановление проводят в стальном аппарате — редукторе
(рис. 118) при энергичном перемешивании. Сначала в редуктор
загружают горячую воду, железные опилки и соляную кислоту.
Bodo
водо
Рис. 118. Схема восстановления нитробензола:
.' — редуктор; 2 — обратный холодильник; 3 —
холодильник для легких углеводородов; 4 — отводная труба; 5 —
загрузочная труба для стружкя; 6 —задвижка.
а затем, поддерживая температуру 100° С, добавляют порциями
• пиробензол. При этом холодильник 2 работает как обратный.
После окончания восстановления анилин высаливают из
водного раствора повареішиіі солью. Всплывший слой анилина
выдают из редуктора, а анилин, оставшийся в растворе,
отгоняют с паром, переключив холодильник 2 на прямой.
Полученную при этом анилиновую воду экстрагируют нитробензолом,
который извлекает анилин из воды и используется в
последующей операции.
Очищают анилин перегонкой под вакуумом (/° кип. 184,4°С).
Подобным же образом получают из соответствующих нитросое-
динений орто- и паратолуидины, а-нафтиламин и др.
В качестве восстановителя нитрогрупп применяются и суль-
282
фиды в виде растворов Na2S, (NH4bS; для нитробензола
перспективным является восстановление водородом в
присутствии катализаторов, например Pd, при 200—400° С.
3. СУЛЬФИРОВАНИЕ
Под сульфированием понимают процесс введения сульфо-
групп SO3H в ядро. В зависимости от перерабатываемого
продукта сульфирующим агентом может служить 92—95%-ная
серная кислота, олеум C—65%-ный) и хлорсульфоновая
кислота. Реакция сульфирования сопровождается выделением воды:
АгН + H2SO4 ч- ArSO3H + Н2О.
Поэтому процесс сульфирования проводят с избытком серной
кислоты, обеспечивающим достаточную скорость реакции.
Сульфосоединения представляют собой весьма важные
промежуточные продукты органического синтеза. Они растворимы в
воде и поэтому применяются для производства растворимых
красителей, обладающих благодаря наличию сульфогрупп,
сродством к животным волокнам.
Из ароматических сульфосоединений получают фенолы.
Сульфирование жирноароматических соединений с длинной
цепью дает поверхностно-активные вещества, обладающие
моющей способностью. С помощью сульфокислот можно разделять
изомеры, например, орто-, мета- и параксилолы.
При сульфировании выделяется небольшое количество
тепла, поэтому для проведения этого процесса нужны стальные
аппараты — сульфураторы, снабженные паровой рубашкой. Так
как среда гетерогенна, необходимо также производить
перемешивание смеси.
Бензол сульфируют в жидкой и паровой фазах.
Сульфирование перегретых паров бензола осуществляется при 150° С.
При этом часть бензола сульфируется, а избыток его уходит из
аппарата вместе с парами реакционной воды. После конденсации
бензол отделяют от воды и возращают в процесс. Схема
сульфирования бензола в парах изображена на рис. 119.
При таком способе сульфирования концентрация кислоты
остается высокой и почти вся кислота используется. Получаемая
сульфомасса содержит до 95% сульфокислоты, нейтрализуя
которую сульфитом натрия выделяют бензолсульфокислоту в
виде менее растворимой натриевой соли.
Если получена дисульфокислота, избыточную серную
кислоту осаждают в виде гипса, нейтрализуя мелом, а
образующиеся кальциевые соли сульфокислот переводят в натриевые и
высаливают, прибавляя раствор поваренной соли.
Большое значение имеют сульфопроизводные нафталина.
Моносульфокислоты нафталина служат исходным материалом
283
для получения а- и р-нафтолов. В результате сульфирования
нафталина, р-нафтола, р-нафтиламина получаются многие поли-
сульфокислоты, используемые при синтезе красителей, например
Г-кислота
Рис. 119. Схема сульфирования бензола в парах:
1 — кипятильник бензола; 2— перегреватель паров
бензола; 3 — сульфуратор; 4 — трубопровод к холодильнику; 5 —
холодильник; 6 — сепаратор; 7 — сифон для спуска воды;
8 — сушитель; 9 — трубопровод; 10 — сифон; И — мерник
для серной кислоты; 12 — сифон для отвода из сульфу-
ратора реакционной массы.
4. ПОЛУЧЕНИЕ ФЕНОЛОВ, 0 -НАФТОЛА
Фенолы получают из соответствующих сульфокислот путем
замены сульфогруппы гидроксилом с помощью сплавления
с едким натром по реакции
ArSO3Na -f 2NaOH -^ ArONa + Na^SC, \- Н2О.
Образующийся сульфит натрия применяется для нейтрализации
сульфокислот:
2ArSO3H
Na2SO3 -* 2ArSO3Na + SO2 + H2O.
Щелочное плавление проводят с избытком едкого натра B—3
моля на один моль арилсульфокислого натра). Чтобы придать
плаву большую подвижность и снизить температуру плавки
(вместо 327,6—270—280° С), к едкому натру прибавляют
5—10% воды, а затем вводят сульфосоединение, порциями при
284
размешивании. Продолжая размешивание, повышают
температуру до 315° С, после чего выпускают плав в гаситель, где он
растворяется в воде. Полученный в гасителе щелочной раствор
фенолята или нафтолята через очистной фильтр-пресс
передается на дальнейшую обработку, которая состоит в том, что
фенолят или нафтолят обрабатывают сернистым газом,
образовавшимся при получении сульфосоли. Например, в случае
нафтолята происходит реакция
+ SO, + H2O ->- 2
Na2SO3.
Суспензия р-нафтола идет в аппарат (делительную воронку),
где нагревается до расплавления р-нафтола, который всплывает
в виде верхнего слоя над раствором сульфита. Для очистки
Р-нафтол подвергают перегонке под вакуумом.
5. ХЛОРИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Хлорпроизводные применяются главным образом в
органическом синтезе, так как хлор легко заменяется другими
заместителями.
Хлорирование ароматических соединений может происходить
как в ядре, так и в боковой цепи. Хлорирование с замещением
в ароматическом ядре проводится в жидкой фазе при невысоких
температурах в присутствии катализаторов (Fe, FeCb, АІСЬ).
Хлорирование ароматических углеводородов с замещением
в боковой цепи протекает под воздействием света
(фотохимическая реакция).
Хлорирование бензола проводят в жидкой фазе в
присутствии хлорного или металлического железа в аппаратах,
называемых хлораторами (рис. 120). При этом, наряду с основной
реакцией образования монохлорбензола, протекает реакция
дальнейшего хлорирования с образованием дихлорбензола и
полихлоридов.
Реакция хлорирования — экзотермическая A17 кдж на
1 г моль хлора, вступившего в реакцию), поэтому для
поддержания определенной температуры хлораторы должны быть
снабжены водяными рубашками. Раньше хлорирование
проводили при 35° С, считая, что в условиях более высоких температур
образуется больше полихлоридов. Затем было доказано, что до
80° С заметного увеличения выхода полихлоридов не
наблюдается. Чтобы уменьшить возможность образования полихлоридов,
реакцию заканчивают, когда в смеси остается еще около 50%
непрореагировавшего бензола.
С развитием органического синтеза потребность в хлор-
производных увеличилась, поэтому в настоящее время применя-
285
НС?
ются более производительные непрерывно действующие
установки (рис. 121). Изображенные на рисунке хлораторы
представляют собой вертикальные чугунные колонны с насадкой из
керамических колец. В первом из них находятся также железные
кольца, необходимые для получения катализатора — хлорного
железа. Бензол должен быть обезвоженным, что позволяет
избежать образования из
выделяющегося хлористого водорода
соляной кислоты.
Советским инженером Б. Е.
Беркманом предложен способ
непрерывного хлорирования
бензола, при котором тепло
реакции отводится за счет
испарения избыточного бензола.
Обезвоженные бензол и хлор
вводятся в , нижнюю часть
стальной колонны,
заполненной насадкой из керамических
и стальных колец. Весь
проходящий через колонну хлор
расходуется на реакцию и смесь
разогревается до 78—85° С.
Образовавшиеся хлорбензол
хлористый водород и
избыточный бензол (последний
частично переходит в паровую фазу)
поступают в верхнюю безнаса-
дочную часть колонны —
сепаратор, из которого вытекает
преимущественно хлорбензол
(т. кип. 132° С) в жидком виде,
а пары бензола и НС1 выходят
из колонны и после
охлаждения разделяются. Бензол снова
возвращается на хлорнрова-
Хлорируемьш
продукт
Рис. 120. Хлоратор (абсорбер):
/-- руЛяткя г!ля наружного ох.чзжлепия:
Ї — стакан для внутреннего охлаждения;
3 — полки
4 — труба
для размещения катализатора; НИЄ. ПоЛИХЛОрИДОВ ПОЛуЧЭеТСЯ
для "п.тп--!!! хлпря; 5 flap
батер.
не более 1,о% всех хлорпроиз-
водных. Хлорбензол в
огромном количестве применяется для синтеза фенола, нитроанилмна
и других промежуточных продуктов.
Из смеси хлорпроизводных бензола после нейтрализации
и дистилляции выделяют монохлорбензол. Фракцию полихлор-и-
дов применяют для борьбы с вредителями в сельском хозяйстве.
Продуктами хлорирования толуола в боковую
цепь_являются последовательно получаемые хлористый бензил \\ у/СН2С1,
286
хлористый бензилиден
\х
СНС12 и бензотрихлорид
;СС13. Первый из них употребляется во многих
химических реакциях для бензилирования, т. е. введения в различные
органические соединения радикала бензила \ч j/ СН2—. Из
хлористого бензилидена гидролизом в слабощелочной среде
получают бензальдегид:
Бензол
морцроданив
Рис. 121. Схема непрерывного хлорирования бензола
в жидкой фазе:
1, 2, 3 — хлораторы.
Гидролизом бензотрихлорида можно получить бензойную
кислоту
^ ^ 2Н2О ^ ^ ^СООН + ЗНС1.
6. ИНСЕКТИЦИДЫ. 4,4-ДИХЛОРДИФЕНИЛТРИХЛОРМЕТИЛМЕТАН
(ДДТ). ГЕКСАХЛОРАН
Многие производные хлорбензолов применяются в качестве
инсектицидов, т. е. веществ, убивающих вредных насекомых.
Особенно важен как инсектицид 4,4'-дихлордифенилтрихлор-
метил-метан, известный под названием ДДТ и представляющий
собой твердое кристаллическое тело с т. пл. 109° С. Он
употребляется в виде порошка и растворов. Получают ДДТ
конденсацией хлорбензола с хлоралем в присутствии серной или
хлорсульфоновой кислоты по уравнению
Н
СС13СНО
ссц
287
Нужный для этой реакции хлораль образуется при
хлорировании этилового спирта:
°
С2Н5ОН + 4С12
5НС1.
Присоединение к бензолу на ярком свету хлора дает другой
важный инсектицид — гексахлоран:
СН СНС1
С1НС/\СНС!
НС
НС
СН 30°—50° С
+ ЗС1, >-
СН С1НС
/
Сна
СН СНС1
В качестве источника света используются ртутно-кварцевые
лампы.
7. ПРОИЗВОДСТВО ФЕНОЛА ИЗ ХЛОРБЕНЗОЛА
Применяются два способа гидролиза хлорбензола в фенол—
в жидкой и паровой фазах. В первом случае хлорбензол гидро-
Едкий
натр
:¦¦:] Углекислый газ
Рис. 122 Схема получения фенола из хлорбензола:
/— подогреватель; 2—реактор; 3 — змеевик; 4 —
испаритель; 5 — редукционный вентиль; 6 — холодильник; 7 —
wjopnUK фсі!о,-і:-.і-;, 'd -uiuapj^ дан ?Ыд?ле'-'т (Ь^иптч.
лизуют 10%-ным раствором едкого натра при 300—350° С
в трубчатых стальных реакторах, футерованных медью, под
давлением 2000—2500 н/см2. Реакция протекает по уравнению
С„Н5С1 + 2NaOH -CeH5ONa + NaCi + Н2О.
В качестве побочного продукта образуется дифениловьш
эфир:
CeH6ONa +С6Н5С! ->СAН5ОН5С,, + NaCl.
Выход фенола достигает 90—92% (см. рис. 122).
288
Во втором случае хлорбензол гидролизуют водяным паром при
550—600° С под атмосферным давлением над катализатором
(MgCl2, активированный медью на силикагеле). Реакция
обратима, но ее проводят с небольшим избытком водяного пара, чтобы
избежать сильного разбавления кислоты. Хлористый водород
используют для окислительного хлорирования бензола. Выход
фенола составляет 15—20% за один проход.
8. ПОЛУЧЕНИЕ ФЕНОЛА СОВМЕСТНО С АЦЕТОНОМ
ЧЕРЕЗ ИЗОПРОПИЛБЕНЗОЛ (КУМОЛ)
Этот способ получения фенола разработан на основе
исследований П. Г. Сергеева, В. Д. Кружалова и др. Исходным
сырьем для процесса является бензол и пропилен или газы
крекинга нефти, содержащие 25—30% пропилена (пропан-
пропиленовая фракция).
Изопропилбензол, полученный алкилированием, окисляют
воздухом в гидроперекись изопропилбензола:
СН3 С
Чсн
+ о2
| |
сн3 сн3
гидроперекись изопропилбензола
Окисление проводят в щелочном растворе (рН 8,5—10,5) с
эмульгатором при 130° С под давлением, чтобы жидкость не
кипела. Реакцию прекращают после перехода 25%
изопропилбензола в перекись во избежание ее распада. Отстоявшуюся
гидроперекись отделяют от эмульсии и разлагают разбавленной
серной кислотой под давлением, в результате чего образуются
фенол и ацетон по реакции
СН3
VXX)H—-€ >ОН+СНзСОСН3.
Фенол и ацетон отделяют друг от друга и от других примесей
ректификацией.
Таким образом, синтез фенола в настоящее время можно
осуществить четырьмя способами: 1) через бензолсульфокислоту;
2) из хлорбензола действием щелочи; 3) из хлорбензола
гидролизом парами воды; 4) кумольным методом с одновременным
получением фенола и ацетона.
Наиболее перспективным методом является кумольный,
особенно там, где развита нефтеперерабатывающая
промышленность, обеспечивающая процесс сырьем.
19 8-1504 289
9. ОКИСЛЕНИЕ С РАЗРУШЕНИЕМ ЯДРА
Важное значение имеет окисление нафталина с целью
получения фталевого ангидрида, которое сопровождается
разрушением ядра:
4,5О2
2СОа + 2Н2О.
Реакция протекает при 420 — 450е С в присутствии
ванадиевого катализатора (УгО5) в виде зерен. Аппарат для окисления
Рис. 123. Схема получения фталевого ангидрида:
/ — котел для плавки нафталина; 2 — обогреваемый мерник для
расплавленного нафталина; 3 ~- ротаметр; 4 — смеситель, для воздуха и паров
нафталина; 5 — контактный аппарат; 6 — печь для его обогревания; 7—
конденсаторы.
(рис. 123), называемый конвертером, состоит из ряда труб,
заполненных катализатором. Межтрубное пространство
конвертера заполняют расплавленным свинцом или смесью солей NaNO3,
NaNCb, KNO3, поддерживающими постоянную температуру.
Технология производства фталевого ангидрида состоит в
продувании воздуха через расплавленный нафталин, в пропускании
смеси паров нафталина и воздуха A :7; 1 : 15) через трубки
конвертера, в сублимировании фталевого ангидрида в камерах и в
очистке его ректификацией под вакуумом после обработки
серной кислотой. Выход фталевого ангидрида равен 70—78%.
Фталевый ангидрид применяется в производстве красителей,
искусственных смол и пластмасс. Кроме того, под действием
290
водяного пара при 400 С в присутствии катализатора СаСОз,
АЬОз, РегОз из него получают бензойную кислоту. Из смеси паров
фталевого ангидрида и бензола в присутствии силикагеля при
370—380° С синтезируют антрахинон, а из смеси паров фталевого
ангидрида с хлорбензолом — р-хлорантрахинон, применяемые
в производстве красителей.
ГЛАВА XVI
ТЕХНОЛОГИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
К высокомолекулярным соединениям, или полимерам,
относятся вещества, состоящие из многократно повторяющихся
звеньев одинаковой структуры. Число звеньев, образующих и
определяющих длину цепи полимера, называется степенью
полимеризации п.
Благодаря своим свойствам (небольшой удельный вес,
прочность, легкая обрабатываемость, достаточная теплостойкость,
антикоррозионная и электроизоляционная способность,
пластичность и эластичность) высокомолекулярные соединения получили
значительное распространение в технике и в быту.
Успехи современной химии и технологии позволяют
синтезировать различные полимерные молекулы и создавать из них
вещества с заданными свойствами.
Классификация высокомолекулярных соединений основана
на различных признаках. Прежде всего, их подразделяют на
природные высокомолекулярные соединения — целлюлоза, белки,
крахмал, натуральный каучук, силикаты — и синтетически
полученные полимеры. Большинство полимеров относится к
органическим соединениям. По химическому составу главной цепи
полимеры делятся на три группы:
а) карбоцепные, у которых главная цепь состоит из
углеродных атомов (полиэтилен и др.);
б) гетероцепные, главная цепь которых содержит, кроме
атомов углерода, атомы кислорода, азота и др. (целлюлоза,
полиамидные синтетические смолы);
в) элементоорганические полимеры с основной цепью,
состоящей из неорганических элементов — кремния, фосфора и др.
По строению различают полимеры линейной, разветвленной
и трехмерной структуры. Полимеры с пространственной
структурой могут образовываться из полимеров линейной или
разветвленной структуры при определенных условиях, в результате
химической связи между линейными молекулами — сшивания
молекул (рис. 124). Появление химической связи резко изменяет
свойства полимера.
По физическим свойствам полимеры можно разделить на
пластомеры и эластомеры. Пластомеры обладают высоким
19* 291
модулем упругости и слабой растяжимостью (менее 25%). К ним
относятся пластмассы. Эластомеры имеют меньший модуль
упругости и большую растяжимость (до 2000%). К ним
принадлежат каучуки и др.
Рабочие температуры пластомеров лежат всегда ниже
температуры стеклования, т. е. перехода в аморфное состояние (для
них это температура теплостойкости); рабочие температуры
эластомеров всегда выше температуры стеклования (для них это
температура морозостойкости).
В зависимости от способа
получения известны полимери-
зационные и
поликонденсационные полимеры.
Полимеризацией
называется процесс соединения
молекул мономера в молекулу
полимера того же
элементарного состава, что и мономер.
Обычно полимеризации
подвергают ненасыщенные
углеводороды — этилен и его гомо-
Рис. 124. Связи между мономерами логи- бутадиен. Соединение мо-
в молекуле полимера: лекул мономера друг с другом
а- мономеры; б-полимер с линейными ОСуЩЄСТВЛЯЄТСЯ за СЧЄТ разрЫ-
связями; в - полимер с пространственными ва ДВОЙНЫХ СВЯЗЄЙ, ПРОИСХОДЯ-
щего под действием различных
активаторов: света, температуры, радиационных лучей,
катализаторов, инициаторов и др. В качестве инициаторов применяют
перекиси водорода и бензоила, персульфат аммония и др.
Инициатор в определенных условиях распадается и образует
свободный радикал, который возбуждает мономер, давая с ним новый
радикал и т. д. (цепной радикальный процесс). Процесс
полимеризации заканчивается, когда происходит обрыв цепи в
результате взаимного насыщения полимерного или свободного
радикалов или образования устойчивого соединения.
В качестве катализаторов чаще всего применяют
А1С13, SnCl4, ВР3,А1(Алк)з -{-ТіСЦ или ПС13.
В присутствии катализаторов происходит катионная и
анионная полимеризация по схеме
+
-•— R2C — CR2
R2C = CR2 -f катализатор .
-*_R2C-CR2
Катализатор входит в состав только промежуточных
продуктов. В присутствии металлоорганических катализаторов
А1(АлкK + ТіСЦ или ТіСіз получаются стереорегулярные (изо-
292
тактические) полимеры строго линейной структуры, у которых
расположение боковых групп вдоль оси макромолекулы
упорядочено и симметрично.
Стереорегулярные полимеры отличаются повышенной
прочностью, теплостойкостью, более высоким удельным весом и
образуются чаще всего по анионному механизму. В присутствии
других катализаторов получаются нестереорегулярные полимеры
(атактические).
В настоящее время широко применяется метод совместной
полимеризации разных мономеров, т. н. сополимеризация. Этим
методом получают бутадиен стирольный, бутадиен нитрильный,
каучуки и пр. Применяется также привитая, или графтполимери-
зация. В результате ее образуются полимеры, в основной цепи
которых содержится один тип элементарных звеньев, а в
боковой — другой по схеме:
д д д д д д д
І І
Б Б
к і
и блокполимеризация. К блокполимерам относятся природные
или синтетические полимеры, в макромолекулах которых
чередуются блоки разных полимеров, полученных при разрыве цепи
исходных полимеров методом перетирания и др. В основной
цепи блокполимера чередуются разные блоки (куски) по схеме
_А—А—А—Б—Б—Б—А—А—А—Б—Б—Б-...
Блокполимеризация дает полимеры, обладающие новыми
свойствами. Полимеризацию полимеров проводят без
растворителя в массе — блочная полимеризация; в растворителе и в
суспензии — капельная полимеризация — ив эмульсии (последний
способ наиболее распространенный).
Поликонденсацией называют процесс взаимодействия
двух (или более) разных мономеров, в результате которого,
наряду с полимером, образуются такие простые вещества, как
вода, аммиак, спирт. Элементарный состав продукта
поликонденсации отличается от элементарного состава мономеров. Методом
поликонденсации получают фенолоформальдегидные, мочевино-
формальдегидные, полиамидные и другие смолы. Для ускорения
реакции применяются катализаторы, причем в зависимости от
условий образования можно получить термопластичную
(плавкую и растворимую) и термореактивную (неплавкую и
нерастворимую) смолу.
293
Свойства высокомолекулярных соединений зависят от
молекулярного веса, химического состава и строения, формы
макромолекул, ориентации и релаксации (релаксация — снятие
напряжений в материале при нагревании), а также упорядоченности
структуры макромолекулы. С увеличением молекулярного веса
до известного предела улучшаются физико-механические свойства
полимеров. Химический состав и строение оказывают большое
влияние на тепло-, морозостойкость и химическую стойкость
полимеров. Полимеры, имеющие менее разветвленное
(асимметричное) строение макромолекулы, отличаются большей
вязкостью, меньшей растворимостью и большей прочностью. От
правильной ориентации макромолекул во многом зависит качество
искусственного и синтетического волокон.
Любой полимер представляет собой смесь макромолекул
различной величины, кристаллического и аморфного строения, что
затрудняет их исследование. Физико-химические свойства
полимеров улучшаются с увеличением кристаллической фазы.
Технология высокомолекулярных соединений включает методы
производства каучука и резины, пластмасс, искусственного и
синтетического волокон, пленкообразующих.
1. КАУЧУК
Натуральный каучук (НК) впервые стали добывать из
млечного сока (латекса) тропического дерева — бразильской гевеи.
Е XVI веке в Европу были впервые привезены грубые изделия
из каучука, которые в жаркое время плавились, а в холодное
становились хрупкими и растрескивались. Промышленное
значение каучук приобрел лишь после того как в 50-х годах XIX
века был изобретен метод вулканизации, позволяющий длительное
время сохранять первоначальные эластичные свойства
каучуковых изделий и их устойчивость к изменению температуры.
Латекс представляет собой 30—40%-ный коллоидный раствор
каучука в воде с небольшой примесью белков, Сахаров и смол.
При подкислении уксусной или муравьиной кислотами до рН
4,4—4.9 каучук выпаляет (коагулирует) в виде сгустка и после
промывки водой затвердевает. Чтобы придать каучуку форму
листов, его пропускают чере^ ьальцы, а затем сушат при
20—50° С и консервируют.
В зависимости от способа предварительной обработки,
предохраняющей от гниения, получают разные сорта сырого
каучука— «смокед-шигс», «светлый креп».
Вулканизированный каучук применяется для изготовления
различных резиновых изделий, необходимых в промышленности
и в быту. Особенно важным является производство резиновых
шин для автомобилей и самолетов. После того как каучук
превратился в важное стратегическое сырье, в странах тропического
294
пояса стали разводить гевею на плантациях, а в странах
умеренного климата начались поиски других каучуконосов. В СССР
культивируется каучуконосное растение кок-сагыз.
В результате исследований состава и строения НК
установлено, что он представляет собой полимер изопрена со степенью
полимеризации выше 2000 и молекулярным весом 136 000—
340 000. Структура НК регулярна — образование цепи
происходит только в положении 1—4 и макромолекулы имеют только
цис-строение:
...—СН2 СН2— СН2 СН2—
>< Ж
>< Ж
СНз Н СНа Н
Спектрографические исследования показали, что растянутый
НК имеет кристаллическое строение.
Процесс вулканизации, в результате которого каучук
делается более эластичным, неломким на холоду и нелипким при
повышении температуры, представляет собой реакцию присоединения
серы к молекулам каучука пс месту двойных связей Атомы
серы образуют мостики между двумя цепными молекулами,
соединяя их между собой, как показано на схеме а и б:
СНа Н СНа Н СНа Н
II II II
—СН2—С = С —СН2 СНа-С — С— СН2 СН2—С—С —СН2
і і
—СН,— С = С — СН2 СН2—С —С—СН2 СНа—С —С —СН2
II II II
СНаН СНаН СН3 Н
а б
Свойства полученной резины зависят от количества
присоединенной к ней серы. В обычные мягкие сорта резины вводят
2—3%, а в эластичную резину — до 8% серы. Если
содержание серы превышает 14—15%> образуется твердое вещество
эбонит, употребляемый главным образом в качестве
электроизоляции.
Синтетические каучуки (СК)
Первые попытки синтеза каучука из изопрена относятся к
концу XIX в. (Бушарда, 1879 г.; Тильден, 1882—1884 гг.). В
1902 г. И. Л. Кондаков полимеризацией диметилбутадиена
получил белую каучукоподобную массу, что послужило толчком для
дальнейших теоретических и практических исследований. В 1909 г.
Остромысленский, С. В. Лебедев и др. впервые синтезировали
изопреновый и бутадиеновый каучук.
295
Однако промышленный способ синтеза натрийдивинилового
каучука был разработан С. В. Лебедевым лишь в 1928 г. — в
годы первой пятилетки, когда для удовлетворения нужд растущей
промышленности нашей страны понадобились большие
количества резины и создание отечественного каучука стало
первоочередной необходимостью.
Первый завод синтетического каучука в СССР начал работать
в 1932 году. В Германии синтетический каучук начали
производить в 1938 г., а в США — в 1942 году.
Натрийдивиниловый каучук С. В. Лебедева
Сырьем для производства каучука по методу С. В. Лебедева
служит этиловый спирт. Пропуская его пары при 450° С над
дегидратирующим и дегидрирующим катализаторами (А12Оз +
.+ ZnO), получают дивинил (бутадиен) по реакции
2C2HSOH -* СН2 = СН — СН = СН2 + Н2 + 2Н2О.
После очистки дивинил подвергают полимеризации в
автоклавах под давлением в присутствии металлического натрия. При
этом образуется натрийдивиниловый каучук по реакции
СН2 = СН — СН = СН2 — 2Na -* NaCH2 — СН = СН — CH2Na!
NaCHtCH = CHCH2Na + СН2 = СН — СН = СН2 -*
-* NaCH2CH = СНСН2 — СН2 — СН = СН — CH2Na и т.д.
Молекулы дивинила в этом каучуке соединяются в
положениях 1—4 и 1—2. Таким образом, в отличие от натурального, он
имеет боковые цепи, вследствие чего менее прочен, чем
натуральный каучук (как и резина на его основе).
Сополимерные бутадиеновые каучуки
Под этим названием известны бутадиенстирольный и бута-
диеннитрильный каучуки, получаемые совместной
полимеризацией бутадиена со стиролом и бутадиена с акрилонитрнлом.
Количество стирола и акрилонитрила может колебаться от 10
до 50%.
Бутадиенстирольный каучук (СКС) является универсальным
типом С К. Выпуск его в СССР составляет 70% всех СК- Сопо-
лимеризацию бутадиена и стирола проводят в эмульсии (схема
рис. 125). Бутадиен и стирол смешивают и вместе с водным
раствором эмульгаторов (олеат натрия и дибутилнафталинсульфо-
кислота — некаль) подают в батарею полимеризаторов,
соединенных трубами. Туда же при непрерывном перемешивании
добавляют инициатор (персульфат калия) и регулятор. В последнем
296
по ходу полимеризаторе степень превращения мономеров
достигает 60%- Образовавшийся латекс бутадиенстирольного каучука
отделяется в сборнике от непрореагировавшего бутадиена и
поступает в колонну, где под разрежением отгоняют остатки
мономеров. Затем латекс подвергают коагуляции в баке в
присутствии электролитов (СаСіг, СНзСООН). Каучук получается в виде
зерен, которые затем на лентоотливочнои машине превращаются
ЫаОн Ю,
Стирол
латекс но коагуляцию
Рис. 125. Схема эмульсионной сополимеризации
бутадиена и стирола:
/, '1 — сборники; 3 — смеситель мономеров; 4 — аппарат для
приготовления раствора эмульгатора; 5 — полимеризатор; 6 —
сборник латекса, 7 — вакуум-насос, 8 — компрессор; 9— отпарная
колонна; 10, П — конденсаторы; ДВ — дроссельный вентиль.
в ленту. Последняя промывается водой, а затем сушится и
поступает на склад в виде рулонов.
Цепные молекулы каучуков имеют вид:
J) бутадиенстирольного
—СН2 — СН = СН — СН2 — СН2 — СН — СН2 — СН =
С,Н.
= СН —СН, —СН, —СН —
с6н5
297
2) бутадиеннитрильного
—СН2—СН = СН — СН2 — СН2 — СН — СН2 — СН =
CN
= СН —СНЯ —СН, —СН —
CN
Каучуки эти после вулканизации имеют почти то же
сопротивление разрыву и удлинение, что и натуральный каучук. Изделия
из бутадиенстирольного каучука отличаются значительной
прочностью к механическим воздействиям и морозостойкостью. Бута-
диеннитрильный каучук стоек к растворяющему действию масел
и нефтяных продуктов.
Хлорпреновый каучук
Хлорпреноиый каучук синтезируют из ацетилена через винил-
ацетилен в присутствии Си2С12 и NH4C1 по схеме
СН2=--С —
СІ
Хлорпрен легко и быстро полимеризуется в водной эмульсии
.при 40° С, образуя каучук малоразветвленного строения,
сходный с натуральным. Этот каучук вулканизируется в присутствии
окисей цинка, магния или ртути.
Хлорпреновый каучук негорюч, химически стоек, теплостоек,
светостоек, но имеет низкую морозостойкость. Применяется в
кабельной промышленности и для изготовления масло- и бензо-
стойких изделий.
Другие виды каучуков
Полимеризацией изобутилена в присутствии BF3 при —100° С
получают каучукоподобный материал полиизобутилен, который
не содержит двойных связей и применяется как
антикоррозионный и электроизоляционный материал. Совместная
полимеризация изопрена с изобутнленом при —100° С в присутствии АІСІз
или BF3 в жидком этилене дает бутилкаучук, используемый для
изготовления шин, электроизоляционных резин и пр.
При поликонденсации дихлорпроизводных углеводородов
с полисульфидами натрия или калия образуются особо масло-
и бензостойкие каучуки — тиоколы.
298
Тиокол А получают по реакции
яС1СН2СН2С1 -f- л Na2S4 -* [—CH2CH2S—S—] п + In NaCl.
11 II
s s
Тиокол В — по реакции
а С1СН2СН2ОСН2СН2С1 -j- «Na2S4 -* [—CH2CH2OCH2CH2S —S —] +
II II "
-f- 2«NaCl. S S
Поликонденсацию проводят в водных дисперсиях при 60—70° С,
вулканизацию — в присутствии ZnO, PbO. Теплостойкость 80° С,
морозостойкость — 15° С.
К новым типам каучуков относятся теплостойкие фторкаучу-
ки, акрилатные и силиконовые каучуки.
Лучшим фторкаучуком является сополимер
nLr2 = СНС1 -j- «CH2 = Cr2 -*¦ I—Cr2 — СН*(_Л —CH2 — Cr2—]„,
изделия из которого почти 30 дней способны выдерживать
нагревание до 200° С.
Однако у этого каучука есть ряд недостатков: малая
морозостойкость, текучесть, способность набухать в некоторых
смазках и др.
Акриловые каучуки (лактопрены) получают совместной
полимеризацией эфиров акриловой кислоты с акрилонитрилом. Они
выдерживают нагревание до 175° С, но обладают низкой
морозостойкостью и способностью набухать в некоторых растворителях.
Силиконовый каучук представляет собой продукт
поликонденсации диметилсиландиола при 200° С в присутствии кислого
или щелочного катализатора. Теплостойкость его 250—300° С,
морозостойкость—100° С, (реакция поликонденсации и
свойства каучука описаны на стр. 311).
Экспериментально установлена возможность получения еще
более теплостойких неорганических каучуков путем
превращения циклических неорганических соединений в линейные.
Например, из циклического фосфонитрохлорида можно получить
каучук с теплостойкостью 500° С:
С1 С1
\/
сі сі сі
III
СК Х1->— P = N — P = N — P = N —
/Р\\/Р\ I I I
a/ y CI C1 CI C1
Качество этого каучука повышается, если заменить хлор
более стойкими группами. Имеются сведения о том, что
неорганические полимеры, содержащие фосфор и бор, сохраняют свои
первоначальные свойства до 580° С.
299
Производство резиновых изделий
Поступивший в производство натуральный каучук и
некоторые синтетические каучуки подвергают мойке, а затем сушке
при 50—60° С. После этого высушенный каучук проходит
пластикацию, приобретая пластичность и мягкость. Пластикация
заключается в многократном вальцевании полученной
каучуковой ленты. Большинство синтетических каучуков, например, на-
трийдивиниловый каучук, в пластикации не нуждается.
Улучшая механические свойства и пластичность каучука,
такая обработка ухудшает прочность каучука и уменьшает его
молекулярный вес; поэтому она не должна продолжаться
слишком долго.
Для изготовления резины составляются смеси, компоненты
которых могут меняться в зависимости от назначения резины.
Приводим примерный состав такой смеси в весовых частях.
Каучук 100 Активные наполнители
Вулканизующие вещества: 10—50
сера 0,5—3 Противостарители 0,5—1,6
ускорители 0,1—3 Пластификаторы 2—10
активаторы 1—3 Красители 0,05—5
В качестве ускорителей вулканизации применяют различные
органические соединения, в присутствии которых процесс
протекает быстрее и при более низкой температуре. Например, ульт-
н с\ с
раускоритель диметилдитиокарбаматнатрия 3 yN — S—Na
позволяет проводить вулканизацию при 100° С в течение
нескольких минут.
Противостарители, например неозон Д (фенил-р-нафтил-
амин),
Ч
предупреждают окисление резины воздухом, сохраняя ее
первоначальные эластичные свойства.
Пластификаторы повышают пластичность резиновой смеси,
благодаря чему облегчается формование изделий. В качестве
пластификаторов применяются высшие кислоты, нефтяные и
каменноугольные смолы и другие вещества, способные вступать
в реакцию с активными группами каучука.
Различают активные и неактивные наполнители. Первые
улучшают физико-химические свойства резины (газовая сажа,
коллоидная кремнекислота или белая сажа). Вторые только
уменьшают расход каучука (мел, каолин и др.).
300
Количество активного наполнителя в резиновой смеси
зависит от типа каучука.
Однородную резиновую смесь из перечисленных выше
компонентов получают в специальных закрытых смесителях, иногда
с помощью вальцевания. Сначала каучук пластифицируется,
а затем к нему добавляют остальные компоненты, за
исключением серы (во избежание преждевременной вулканизации, так как
перемешивание сопровождается разогреванием до 100—120°С).
После окончания
размешивания добавляют серу.
С целью экономии
каучука в резиновую смесь
иногда добавляют
регенерат (пластический
материал, полученный
переработкой старой резины)
или фактис (суррогат
каучука, полученный при
обработке высыхающих
растительных масел
серой).
Приготовленную
резиновую смесь (клей)
слегка подогревают и
формуют. Метод формования Рис- 126- Трехвалковый каландр:
1ЯПИГИТ пт пиля ичлйпип ' — фундаментная плита; 2 — приводная шестер-
ЗаВИСИТ ОТ ВИДа ИЗДеЛИЯ. ня. з-вапкя; ./-траверса; 5_станина; 6 -
ДЛЯ ИЗГОТОВЛЄНИЯ ГЛаДКИХ передаточные, переключающие н фрикционные
ИЛИ Профилированных ре- їаЄиияРНзазора ""между "алк"^"* 8 — труб^воды
ЗИНОВЫХ ЛИСТОВ прИМеНЯ- для пояачн паРа нлн В°ДЫ ВИУТРЬ валков.
ются трехвалковые и с
большим количеством валков машины — каландры (рис. 126).
Прорезиненную ткань получают на фрикционных каландрах, т. е.
каландрах, в которых валки вращаются с различной скоростью
(фрикция 1,1—1,2), благодаря чему резиновая смесь лучше
втирается в ткань.
Резиновые трубки, шнуры, ленты, протекторы для шин,
автомобильные камеры получают методом шприцевания на
червячных прессах (шприц-машинах). Схема такой машины
изображена на рис. 127.
Головка шприц-машины, которую можно менять, придает
определенную форму изделию. Применяется также метод литья
под давлением, при котором резиновая масса под давлением
поршня машины заполняет разъемную форму.
Пропитывают также латексом или клеем корд,
изготовляемый из капрона. Специальные изделия штампуются на особых
прессах и идут на вулканизацию. Вулканизацию каучука серой
производят при нагревании до 125—160°С. Вулканизация рези-
301
новых изделий в прессах или вулканизационных
котлах-автоклавах осуществляется в атмосфере горячего воздуха или
насыщенного водяного пара. Разработаны также новые методы
вулканизации, например, токами высокой частоты, радиационная
вулканизация и др. Эти методы позволяют быстрее и лучше
прогревать изделия, улучшить их качество. Вулканизацию
полухлористой серой производят при комнатной температуре, но такой
вид вулканизации применяется редко.
Рис. 127. Червячный пресс (шприц-машина):
/ — полый цилиндр; 2—¦ червяк; 3 — полости для нагрева или охлаждения;
і — загрузочное отверстие; 5 — приводная шестерня; 6 — упругая муфта; 7 —
электродвигатель.
2. ПРОИЗВОДСТВО СМОЛ И ПЛАСТМАСС
Органические пластмассы представляю г собой
высокомолекулярные соединения или композиции таких соединений с
другими веществами (наполнителями, красителями,
пластификаторами и др.). Изделия из них получают пластическими методами:
прессованием, литьем под давлением, штамповкой, вытяжкой
Производство пластмасс с каждым годом увеличивается,
причем в СССР эта отрасль химической промышленности
развивается особенно интенсивно. Если в 1965 г. была выпущена
821 тыс. т пластмасс и синтетических смол, то в 1970 г.
Директивами XXIII съезда КПСС намечено увеличить их выпуск до
2100—2300 тыс. т. Пластмассы, производимые нашей
промышленностью, делятся на четыре класса:
302
A — пластические массы на основе полимеров, полученных*
цепной полимеризацией;
Б—пластические массы на основе полимеров,
образовавшихся в результате поликонденсации;
В — пластмассы на основе природных полимеров;
Г — пластмассы на основе природных и нефтяных асфальтов,
а также смол, полученных деструкцией различных органических
веществ.
В свою очередь, смолы, из которых изготовляются
пластмассы, подразделяются на термопластичные и термореактивные.
Полимеризационные смолы (термопластичные)
и пластмассы на их основе
Наиболее важными из полимеризационных смол являются
полиэтилен, полипропилен, поливинилхлорид, сополимеры винил-
хлорида с другими мономерами, полистирол, полиакрилаты,_
МепрореагироЗавший зтилен
Рис. 128. Принципиальная схема получения
полиэтилена под высоким давлением.
фторсодержащие полимеры и пр. Особенностями этих
полимеров являются их химическая стойкость, гидрофобность, высокие
диэлектрические свойства, эластичность и др.
Полиэтилен. Известны три способа производства
полиэтилена: 1) под высоким давлением A0000—20 000 н/см2) при
нагревании до 200° С в присутствии небольших количеств
кислорода как катализатора; 2) при атмосферном давлении и
нагревании до 50—60° С в растворе в присутствии металлооргани-
ческих соединений A% А1(С2Н5)з + ТІСЦ) — метод Циглера;
3) под давлением 300—500 н]см2 в присутствии катализатора
(окислы хрома на алюмосиликате) при нагревании до 130—
170° С.
Схема производства полиэтилена высокого давления
изображена на рис. 128. Этилен в смеси с 0,002% кислорода засасывается
зоз
из газгольдера 1 и сжимается в компрессорах 2 и 3 до 3600—
4 000 н/см2, а затем до конечного давления 15 000 н/см2. Затем
он подается в змеевиковый реактор 4, где циркулирующей в
рубашке водой под давлением поддерживается температура 200° С,
и поступает в приемник 5, где давление снижается до
атмосферного и расплавленный полиэтилен отделяется от невошедшего
в реакцию газа и стекает в приемник 6. Газ возвращается на
полимеризацию в смеси со свежим этиленом. Степень
превращения за один проход составляет 16—25%. Полученный таким
способом полиэтилен пСН2 = СН2 -*¦ [—СНг — СНг—]п
характеризуется линейным строением с небольшим количеством
разветвлений (около 1 г СНз на 100 г СН2). Вследствие этого он имеет
более низкую температуру размягчения— 110° С и меньший уд.
вес — 9200 н/м3 @,92 г/см3), чем полиэтилены среднего и низкого
давления, имеющие строго линейное строение, температуру
размягчения 135—138°С и уд. вес 9700 н/м3 @,94—0,97 г/см3).
Из полиэтилена высокого давления, как более эластичного
материала, делают различные сосуды, пленки, шланги; из поли-
этиленов среднего и низкого давления — трубы. Шприцеванием
можно изготовить полиэтиленовые трубы любой длины
непосредственно на месте их применения, например, на полях
орошения. Из полиэтилена выполняют детали аппаратуры,
работающей в агрессивных средах, электроизоляционные материалы,
в частности, изоляцию подводных кабелей, пенопласты.
Полиэтилен легко склеивается и сваривается. После
непродолжительного облучения полиэтилена радиационными лучами
теплостойкость его повышается до 240—250° С.
Полипропилен. Метод полимеризации полипропилена
разработан недавно итальянским химиком Натта, который
получил его в стереорегулярной форме. Такой полипропилен имеет
ряд преимуществ перед полиэтиленом — более высокую
температуру размягчения A64—165°С), большую химическую и
механическую стойкость, меньшие газопроницаемость и уд. вес
(9000 н/м3, или 0,9 г/см3). К. недостаткам полипропилена
относится более легкая окисляемость и меньшая светостойкость, чем
у полиэтилена.
Поливинилхлорид. Поливинилхлорид Г—СНо—СНС1—
—]п является одним из наиболее распространенных пластиков.
Сырьем для производства его служит хлорвинил, который
получают из дихлорэтана, омыляя последний спиртовым раствором
щелочи. Хлорвинил можно получить также пиролизом
дихлорэтана при 400—500° С над пористыми материалами или из
ацетилена, присоединяя к нему хлористый водород. Хлорвинил
в обычных условиях — газ, температура кипения которого
13,9° С.
Полимеризацию хлорвинила проводят в эмульсии в
автоклавах под давлением 60—100 н/см2 при 40—50° С (инициато-
304
ры — 0,3—0,7% перекиси бензоила или 0,1—0,3% азодинитрил-
диизомасляной кислоты).
Поскольку поливинилхлорид является одним из самых деше-
еых пластиков, области применения его самые разнообразные.
Из него изготовляют предметы широкого потребления,
электроизоляцию, различные пленки, упаковочный материал, трубы
и листы.
Изделия производят прессованием, каландрованием,
экструзией гомогенной массы, полученной из смеси хлорвинила,
пластификатора и стабилизатора (связывающего НС1).
Полистирол. Полистирол получают полимеризацией
стирола блочным или эмульсионным методом. Он имеет
следующее строение:
... —СН2СН —СН2 —СН —...
С6Н6 С6Н6
Полимеризацию в блоке производят в присутствии азота
сначала при 80—82° С в реакторах из алюминия или
эмалированной стали, снабженных мешалками и змеевиками. Затем жидкий
продукт, содержащий 32—35% полимера, идет в колонну, где
полимеризация осуществляется в шесть ступеней с постепенным
повышением температуры от 100 до 200° С. Расплавленный
полистирол поступает в шнек-пресс, выходит из него в виде стержня,
охлаждается на ленточном транспортере и режется на куски.
Молекулярный вес полистирола достигает 180 000.
Полистирольные пластические массы обладают высокими
электроизоляционными свойствами, значительной прочностью,
прозрачностью, светостойкостью, морозостойкостью и особенно
характерной для них гидрофобностью.
Наряду с этим они имеют низкую теплостойкость (80—90° С).
Однако в настоящее время разработаны способы получения сте-
реорегулярного полистирола, теплостойкость которого
составляет 220—230° С.
Полистирол широко применяется как электроизоляционный
материал, для изготовления галантерейных изделий,
облицовочных плит, для производства пенопластов и пр.
Акриловые смолы. Акриловые смолы получают в
основном полимеризацией метилового эфира метилакриловой
кислоты в блоке (от 45 до 125° С). При этом образуется
прозрачная смола, которая идет на производство бесцветного
органического стекла (плексигласа). Полиметилакрилат имеет
следующее строение:
СН3 СНз СНз
-СН2 —С —СН2 —С —СН2—С—...
СООСНз СООСН, СООСН,
20 8-1504 305
Благодаря оптическим свойствам органического стекла,
высокой прочности и электроизоляционной способности, его
широко применяют в машиностроении, приборостроении,
электротехнике и медицине.
Фторсодержащие полимеры. Фторопласты —
политетрафторэтилен и политрифторхлорэтилен — обладают
большой стойкостью к химическим реагентам — кислотам,
окислителям, высокой термической стойкостью (выдерживают
температуру 250—350° С), низкой гигроскопичностью. Удельный вес
фторопластов 21 000—22 000 н/м3 B,1—2,2 г/см3).
Политетрафторэтилен — фторопласт-4 (тефлон) получают
полимеризацией тетрафторэтилена в эмульсии:
п [CFa = CF2] -* [-CF, - CF2 -] „.
Вследствие высокой вязкости фторопласт-4 перерабатывают
в изделия холодным прессованием порошка, спеканием
заготовки в печи с последующей механической обработкой.
Применяется он для изготовления деталей химической аппаратуры,
используемой в агрессивных средах, и в качестве диэлектрика,
работающего в широком диапазоне температур A30—280° С)
Политрифторэтилен [—CF2—CFC1—]п— фторопласт-3
термопластичен и поэтому формуется литьем под давлением и
прессованием при 250—350° С. Фторопласт-3 применяется как
диэлектрик и для химически стойких покрытий, прокладок и др.
Пластмассы на основе поликонденсацион-
йых смол. Из поликонденсационных смол наибольшее
распространение получили фенолоформальдегидные, мочевинофор-
мальдегидные (карбамидные), полиэфирные и др. Все эти
полимеры являются термореактивными.
Производство фенолоальдегидных смол в России началось
с 1917 года. В настоящее время оно занимает значительное
место в общем производстве смол. Свойства фенолоальдегидных
смол зависят от условий получения и сырья.
Из фенола и альдегида в присутствии щелочного
катализатора NH3, NaOH и избытка формальдегида образуется
термореактивная смола — резол (бакелит), а із кислой среде при избытке
фенола — термопластичная — новолак.
В щелочной среде в первой стадии получается оксибензило-
вый спирт (стадия А):
CeHsOH + НСНО -> НОСвН4СН2ОН,
а затем при нагревании происходит конденсация этих молекул
с частичным образованием сетчатых структур — получается уже
неплавкий, но размягчающийся продукт — резитол (стадия В),
а при дальнейшем нагревании выше 150° С — неразмягчающий-
306
ся и нерастворимый полимер — резит (стадия С) следующего
строения:
-Н2С ОН
—Н2С ОН
В присутствии кислого катализатора (НС1, H2SO4) из
фенола и формальдегида при избытке фенола получается диоксиди-
фенилметан:
2СвН5ОН + НСНО ->¦ НОСь
а затем новолачная смола строения
ОН ОН
которая и при нагревании до 150° С остается плавкой и
растворимой. Для перевода этой смолы в неплавкое и нерастворимое
состояние добавляют формальдегид и аммиак илн гексамети-
лентетрамин.
Технология производства фенолоформальдегидных смол очень
проста, что видно из рис. 129. Основным аппаратом является
реактор с мешалкой и паровой рубашкой, который может
работать под вакуумом, благодаря чему снижается температура
варки. После загрузки сырья реактор подогревают до 70—80° С,
а затем прекращают подачу пара в рубашку, так как
температура повышается за счет тепла реакции, и применяют
охлаждение водой, если температура поднимается выше 100° С. После
окончания варки реактор снова подогревают под вакуумом для
обезвоживания и обесфеноливания смолы. Смолу выпускают
через нижний штуцер реактора. Процесс длится 6—15 ч. Ново-
лачные смолы выпускают в твердом виде, резольные—чаще
в виде эмульсий и лаков.
Для производства фенолоальдегидных смол применяют
также крезолы, ксиленолы, ацетальдегид, фурфурол.
20*
307
На основе фенолоформальдегидных смол приготовляют
различные пластмассы — фенопласты. Формование изделий из
пластмасс основано на пластичности последних при повышенной
температуре.
Наиболее распространенным методом изготовления изделий
является прессование пресспорошков, которые представляют
собой смесь из 40—50% смолы, 35—50% наполнителя,
например древесной муки. Кроме того, в состав их входят краситель,
пластификатор и другие добавки.
ґа/ла/>і/з а/лор
Воды но обесфено/п/-
вание
Рис. 129. Схема установки для получения фенолоформальдегидных смол:
/ — вакуум-варочный аппарат: 2 — холодильник; 3 — приемник
воды; 4 — смотровые фонари
для надсмольной
Прессование ведется в формах (рис. 130) под давлением
около 1470 н/см2 при нагревании до 160—170° С. Если в качестве
наполнителя применяются ткяни. бумага, древесный шпон и др.,
образуются слоистые пластики, производство которых
заключается в пропитывании ткани смолой пли эмульсией,
высушивании и прессовании нескольких слоев этой ткани под давлением
при нагревании.
Для приготовления пресспорошков чаще применяют новола-
ки с добавлением гексаметилентетрамина в качестве отверждаю-
щего вещества, а для получения слоистых пластиков —
преимущественно резолы.
В зависимости от наполнителя меняются свойства материала
и его название. Слоистый пластик, в котором наполнителем
служит бумага, называется «гетинакс», бумажная ткань — «тексто-
308
лит», асбестовая ткань — «асботекстолит», стеклоткань —
«стеклотекстолит», древесный шпон — «древеснослоистый
пластик». Наиболее прочными являются стеклопластики. Из слоистых
пластиков делают трубы, плиты, различные фасонные
резервуары. Асботекстолит применяется для изготовления
кислотостойкой химической аппаратуры.
Профильные изделия — трубки, шланги, бруски и др. —
формуются выдавливанием (экструзией) с
помощью машин поршневого или
червячного типа.
Мочевиноформальдег и-
дные с молы — аминопла с ты.
Мочевиноформальдегидные смолы
образуются в результате конденсации
мочевины или тиомочевины, а также меламина
с формальдегидом. Они могут быть
полупрозрачными и даже совершенно
прозрачными, но склонны к образованию
трещин, слишком мягки и быстро
царапаются, вследствие чего их нельзя использовать
в качестве заменителя стекла. Смолы,
обладающие большой крепостью, получают
из смесей мочевины с тиомочевиной.
Большинство смол для прессовальных
порошков производится конденсацией
двух молей мочевины с тремя молями
формальдегида в виде 40%-ного
формалина.
Первоначальными продуктами
конденсации в щелочной среде являются мо-
но- и диметилолмочевина:
Рис. 130. Прессформа для
обычного прессования:
/ — пуансон; 2 —
оформляющая полость (заполнена
материалом); 3 — матрица;
4 — выталкиватель.
С=О +НСНО^-С=О
.NHCHaOH
о
монометилол мочевина
,NH2 xNHCH2OH
С=О +2НСНО^-С=О
диметилолмочевина
В кислой среде образуется метиленмочевина:
NH2 .NH
С=О
309
или
* N=CH
С=О +Н2О.
В отличие от фенолоальдегидных смол, нерастворимые кар-
бамидные смолы образуются в присутствии кислых
катализаторов, например щавелевой кислоты. Чтобы предотвратить
самопроизвольное образование геля, в процессе поддерживается рН
раствора в пределах 6,2—7,4 (соотношение мочевины и
формальдегида 1 : 1,7—2). Пресспорошки из этих смол и изделия из
них получают так же, как из фенолоальдегидных смол
(прессование при 140—165° С). Предполагают, что в результате
нагревания образуются пространственные молекулы из гетероциклов
углерод — азот.
Меламиноформальдегидные смолы идентичны мочевинофор-
мальдегидным, но обладают более высокой теплостойкостью
и водостойкостью, лучшими механическими и диэлектрическими
свойствами.
Алкидные смолы. Глифталевые смолы. Ал-
кидные смолы образуются при конденсации многоатомных
спиртов с многоосновными кислотами. Наиболее распространены
глифталевые смолы — из глицерина и фталевого ангидрида.
Глифталевые смолы получают нагреванием смеси фталевого
ангидрида (три моля) с глицерином (два моля). При этом
сначала происходит этерификация глицерина по уравнению
СН2ОН
/СО
СНОН +2С6нЛо->СН2ООСС6Н4СООН.
А:
' \
Н2ОН ^ СНОН
СН2ООССвН4СООН
2
т. е. этерифицируются только а-оксигруппы глицерина. В
дальнейшем образуется цепной (линейный.) полимер, а при
нагревании до более высокой температуры р-оксигруппы глицерина
этерифицируются, что сопровождается соединением линейных
молекул в пространственные молекулы термореактивной смолы.
Глифталевые смолы используются главным образом для
нанесения покрытий, подобных лаковым, но выдерживающих
высокую температуру.
Кремний органические полимеры. Кремний-
органические полимеры относятся к конденсационным смолам,
метод получения которых был разработан в 1936—1941 гг.
К. А. Андриановым.
310
Мономерами являются кремнийорганические соединения,
например диметилхлорсилан, который образуется, если
пропускать СН3С1 над сплавом кремния с порошком меди при 300—
360° С.
Путем гидролиза сначала получают алкилсиландиолы,
конденсирующиеся затем при 200° С в течение 4—8 часов:
(CH3JSiCla Н- 2НОН -*- (CH3JSi (ОН), + 2НС1,
СН
з
п НО—Si—ОН -
СНз
СНз СНз
_Si_O—Si—О—
НаО.
а
СНз СНз
В зависимости от степени поликонденсации и строения
органического радикала можно получать различные продукты
конденсации силандиолов.
Полимеры со степенью поликонденсации 20—30 — жидкости,
применяемые в качестве смазочных масел с низкой температурой
замерзания.
Полимеры со степенью поликонденсации 800—1500 идут на
изготовление пластмасс и лаков, полимеры со степенью
поликонденсации 10 000—30 000 представляют собой синтетические
каучуки (см. стр. 299). Все эти продукты обладают высокой
термической стойкостью, морозо- и влагостойкостью. Они являются
хорошими диэлектриками, но отличаются небольшой
прочностью. Пластмассы, изготовленные на основе этих полимеров
с наполнителями, имеют более высокую прочность и
теплостойкость.
Армированные стеклопластики. Пластмассы на
основе термореактивных смол с 45—60% наполнителя из
стеклянного волокна называются армированными стеклопластиками
и отличаются механической прочностью, в некоторых случаях
превышающей прочность стали. Получают их, пропитывая
стеклянное волокно или ткань жидким полимером или его раствором
с отвердителем. Пропитанную ткань или стекловолокно режут
на куски и прессуют в специальных формах при нагревании до
80—100° С в течение 30—60 мин. Полимер при этом отверждает-
ся в монолитный материал. Применяют также вакуумное
формование, сущность которого состоит в том, что размягченный
лист материала, прикрепленный к форме, прижимается к ней
вследствие выкачивания воздуха из пространства между формой
и листом через множество отверстий в форме. В качестве
термореактивных полимеров применяют фенолоформальдегидные
смолы, полиэфиры сетчатого строения и другие полимеры. Из
армированных стеклопластиков изготовляют детали самолетов,
трубы для нефтепродуктов и химических веществ, кузова
автомобилей, корпуса судов и пр.
311
Легкие и сверхлегкие полимерные
материалы. Легкие пластмассы (объемный вес 200—2000 hjm3)
представляют собой пористые полимеры, ячейки которых заполнены
воздухом, азотом или другим газом. Получают их такими
методами:
1. Вспенивание эмульсии, раствора полимера или мономера
воздухом или другим газом и последующее отверждение этой
пены.
2. Вспенивание вязко-жидких композиций полимера
газообразными веществами, образующимися при отверждении
полимера или разложении газообразователя.
3. Вспенивание полимера в высокоэластичном состоянии
газом под давлением, приводящее к получению пенопласте»
с объемным весом 600—2200 н/м3 @,06—0,2 г/см*).
Процесс вспенивания осуществляется следующим образом.
Смесь термопластичного полимера с газообразователем (поро-
фором) прессуют под давлением 1000—2500 н/см2 при
температуре 140—160° С. При этом порофор разлагается, а
выделяющиеся газы сорбируются полимером. В качестве порофора
применяют диазоаминобензол (температура разложения 100—110°С),
азодинитрилдиизомасляную кислоту, которая разлагается при
105—И0" С по реакции
СНз СНз СНз
I
сн
г- С— N=N—С—СНз-^СНз—С— С—CH3+N2.
II II
CN CN CN CN
Подбирая порофор, способный одновременно служить
инициатором полимеризации, например, азодинитрилдиизомасляной
кислоты, можно сочетать два процесса — получения пенопласта
и полимеризации мономера. Таким путем получают пенопласт
из стирола. В настоящее время пенопласты широко
применяются как материалы для тепло- и звукоизоляции в строительстве,
холодильной технике, в химической промышленности, для
изготовления спасательных плиюв, кругов, незатопляемых катероа
и т. д. Из эластичных пенопластов делают амортизационные
прокладки, матрацы и пр.
Температура эксплуатации пенопластов из термопластичных
полимеров 60—80°, а из термореактивных— 150° С.
Эпоксидные смолы. В последние годы непрерывно
возрастает промышленное значение эпоксидных смол. Эти смо-
—С—С—
лы содержат эпоксигруппу \ / и образуются при
взаимодействии производных окиси этилена с фенолами, спиртами,
аминами и другими веществами, в состав которых входят груп-
312
пы с подвижным атомом водорода. Например, в результате кон-
денсации эпихлоргидрина с дифенилолпропаном получается
эпоксидная смола:
СН2—СНСН2С1 + Н0—<
сн.—снсн.—
СНз
СНз
СНз
"Ч
-он-
¦\=
СНз
—О—СН,— СН— СН,—
—о—
—О—СН2СН—СН2.
Эти смолы применяются в качестве пленкообразующих в
покрытиях, для производства клеев, слоистых пластиков, литых.
изделий и электроизоляционных пропиточных составов.
Пластмассы из природных высокомолекулярных соединений
К пластмассам из природных высокомолекулярных
соединений, не утративших своего значения до настоящего времени,
относится целлулоид, который представляет собой твердый
25%-ный раствор камфоры в коллоксилине. Коллоксилин —
продукт неполной этерификации целлюлозы азотной кислотой.
Для лучшего перемешивания компонентов в смесь
добавляют спирт, который затем отгоняется. Из целлулоида делают
детские игрушки и другие мелкие предметы. Недостатком
целлулоида является его горючесть.
Негорючие пластмассы получают из ацетилцеллюлозы и
пластификатора с минеральным наполнителем (этролы) и без него
(целлон).
3. ТЕХНОЛОГИЯ ИСКУССТВЕННОГО ВОЛОКНА
Искусственное волокно получают химической переработкой
природных высокомолекулярных соединений, в основном
целлюлозы. Для производства искусственного волокна используются
313
жоротковолокнистые материалы (линт, древесная целлюлоза),
которые непригодны для непосредственного прядения.
Изготовление искусственного целлюлозного волокна состоит в
получении растворимых производных целлюлозы и в вытягивании
нитей из их растворов.
Прядение ведут мокрым и сухим способами. При первом
способе прядильный раствор продавливается через микроотверстия
фильеры (элемент прядильной машины) в осадительную ванну
-с определенными гидролизирующими реагентами, где и
происходит образование нити или пучка нитей. По сухому способу
производные целлюлозы растворяются в каком-либо
растворителе (в спирте и ацетоне), а затем раствор продавливается через
¦фильеру в нагретую камеру. Растворитель испаряется, волокно
¦отверждается. Если производное целлюлозы не растворяется
в растворителях, прядение осуществляется путем продавливания
через фильеру размягченного при нагревании соединения.
Многие виды искусственного волокна по внешнему виду
напоминают шелк. Поэтому их называют искусственным
шелком. Известны следующие виды (сорта) искусственного шелка:
вискозный, ацетатный, медноаммиачный, нитрошелк. Вискозный
шелк занимает главное место в производстве искусственного
волокна, что объясняется дешевизной его изготовления. Лучшим
по качеству, но в то же время самым дорогим является
ацетатный шелк.
Производство вискозного шелка
Основным сырьем для производства вискозного шелка
служит сульфитная целлюлоза. Производство шелка состоит из
следующих операций:
1. Обработка целлюлозы 18%-ным раствором едкого
натра — мерсеризация, повышающая реакционную способность
целлюлозы:
(СвН10О6)„ + п NaOH -»¦ (CcH10OsNaOH)n.
2. Предсозр^ізание отжатой алкалицеллюлозы а камере C0—
50 ч при 20—22° С), приводящее к уменьшению ее вязкости.
3. Ксантогенирование алкалицеллюлозы в течение двух—трех
часов сероуглеродом C3—38% отвеса целлюлозы):
.{CeH7Q2 (OHK-NaOHb + nCSa — [СвН7О2 (ОН), OCSNaln + п Н2О
Ксантогенат — комковатая масса оранжевого цвета.
4. Растворение ксантогената в 4—5%-ном растворе едкого
натра с образованием вискозы, содержащей около 7%
целлюлозы и 7% едкого натра.
.314
5. Двух-трехдневное созревание в погребе, во время
которого осуществляется частичный гидролиз и коллоидные процессы
агрегации частиц.
6. Удаление пузырьков воздуха под вакуумом и фильтрация.
7. Прядение вискозного волокна, которое заключается в
выдавливании струи вискозы через фильеру (колпачок с 24—3200
отверстиями) в коагулирующий раствор, как правило, состоя-
6)
Рис. 131. Схема прядильной машины:
а — бобннный способ прядения; б —
центрифугальний способ- прядения: / — трубопровод для
раствора вискозы; 2 — прядильный насосик; 3 —
фнлътр свечного тнпа; 4—стеклянная трубка;
Л — желоб-ванна; 6—фнльера; 7 — бобина; Я —
направляющий диск; 9 —воронка; /0 —
центрифуга.
щий из серной кислоты и сернокислого натрия. В прядильной
ванне ксантогенат натрия разлагается по реакции
[СвН7О2 (ОН), OCSNa]n + n H2SO4 -> [С6Н7О2 (ОН),]„ +
11
S
+ п CS2 + n NaHSO4l
целлюлоза коагулирует (отверждается), сохраняя форму нити.
В зависимости от типа устройства для приема нити различают
два метода прядения: бобинный, когда нить наматывается на
бобину (катушку), и центрнфугальный, при котором нить откла-
315
дывается на стенках центрифуги, образуя «кулич» (рис. 131, а
и б). Скорость прядения составляет 47—70 м/мин.
8. Обработка волокна 1—2,5%-ным раствором сульфита
натрия, сернистого или едкого натра для удаления серы,
последующее отбеливание гипохлоритом натрия, обработка
раствором олеинового мыла и сушка при 60—70° С.
Вискозный шелк обладает большей теплопроводностью, чем
натуральный, и меньшей, прочностью во влажном состоянии.
Производство вискозного шелка относится к вредным, так как
при разложении ксантогената целлюлозы и побочных продуктов
выделяются сероуглерод и сероводород.
Кроме шелка, из вискозы получают целлофан. С этой целью
ее продавливают через щель фильеры шириной до 2 л в осади-
тельный раствор, а затем подвергают отбелке в ванне с
глицерином, после чего она приобретает пластичность. Высушенный
целлофан представляет собой прозрачную желтоватую пленку
толщиной до 0,01—0,06 мм.
Вискоза служит также материалом для изготовления
коробочек, колпачков для укупорки бутылок, пробок, искусственной
соломки и пр.
Ацетатный шелк. Исходным сырьем для получения
ацетатного шелка служит ацетилцеллюлоза, которую получают
ацетилированием целлюлозы уксусным ангидридом в
присутствии серной кислоты как катализатора и уксусной кислоты как
растворителя. Ацетилированню подвергаются очесы хлопка
(линти делинт).
Конечный продукт близок к триацетату:
[СвН,О2 (ОН)«]„ + Зл (СН3СОJО »[С6Н7О2 (ОСОСНзK]п +
+ Зл СНзСООН.
Триацетат целлюлозы ие растворяется в ацетоне и сложных
эфирах. Поэтому производится частичное омыление его
уксусной кислотой, в результате чего получается ацетилцеллюлоза,
приблизительно соответствующая формуле
[С6Н7О2 (ОСОСНзЬ (ОН)о,5]»
и растворяющаяся в этих растворителях.
Для получения прядильного раствора из триацетата
целлюлозы в настоящее время применяют смесь метиленхлорида и
спирта.
Прядение волокна осуществляется сухим методом. При этом
нагретый до 40—50° С прядильный раствор продавливается через
фильеру в закрытую вертикальную камеру высотой 3—4 м,
через которую продувают нагретый до 55—70° С воздух (рис.
132). Растворитель легко испаряется, а ацетилцеллюлоза затвер-
316
девает, образуя волокна. По выходе из машины волокно
наматывается на бобину.
Выходящая из камеры смесь воздуха с растворителями
направляется в абсорберы с активированным углем, где
растворители поглощаются, а затем отгоняются из угля и
возвращаются в производство. Ацетатное
волокно, в отличие от вискозного,
представляет собой эфир целлюлозы. Ацетатный
шелк более эластичен и красив.
Недостатком его является малая прочность по
отношению к щелочам и высокой
температуре.
Вся аппаратура выполняется из
фосфористой бронзы или легированных
сталей.
Медно-аммиачный шелк и нитрошелк
в настоящее время почти не
производятся.
4. ТЕХНОЛОГИЯ СИНТЕТИЧЕСКОГО
ВОЛОКНА
Синтетическое волокно получают из
высокомолекулярных соединений — смол
{молекулярный вес 15 000—100 000).
Промышленность синтетического волокна
очень молода — она существует всего
15—25 лет.
Все известные синтетические волокна
делятся на две группы: гетероцепные и
карбоцепные волокна.
Гетероцепные волокна фор- —
муюгся из смол, скелет молекул которых
состоит не только из углеродных атомов,
НО И ИЗ Других элементов — азота, КИСЛО- Рис. 132. Схема машины
рода. Группа гетероцепных волокон под- для прядения ацетатного
разделяется на такие подгруппы: поли- , . шелка.
ґ , FJU / — фильтр; 2 —фнльера;
амидные волокна (капрон, найлон, энант а —дырчатые трубы для ог-
и др.), полиэфирные (лавсан, терилен и соса паров Раств°Рителя: *-
др.) и полиуретановые волокна.
Карбоцепные волокна
формуются из смол, скелет молекул которых
состоит только из углеродных атомов. К этой группе относятся
производные этилена — волокна хлорин, саран, виньон, орлон
и др.
Гетероцепные волокна. Из полиамидных волокон
наибольшее распространение получили капрон и найлон.
коллектор для отвода
отсасываемых паров; 5 —
обогревающее приспособление; 6 —
бобина; 7—иитеводнтель.
317
Исходным сырьем для производства капрона служит фенол,
из которого в результате гидрирования, окисления и др.
получают лактам е-аминокапроновой кислоты, т. н. капролактам:
сн
нс/>.сон
сн2
сн2
Ni
И.,, Zn. Fe
400° С
сн2
циклогексанел
112^\ /03.2
сн2
циклогексанон
NH2OH
сн2
= NOH
H2SO<
2
I
Н2С
I
NH
оксим циклогексаиона
капролактам
В результате полимеризации капролактама в струе азота,
яри 240—260° С в течение шести-девяти часов и в присутствии
1—5% воды получается капроновая смола, из которой
формуется волокно:
ОС (СН2M NH + Н2О -+ НООС (СН2M NH2,
І І Е-амннокапроновая кислота
капролактам
а затем
H2N (СН2M СО — [— ОС (CH2)fiNH — ]„ NH(CH2MCOOH.
Исходным сырьем для производства найлона также служит
фенол, из которого получают адипиновую кислоту
НООС(СН2LСООН.
Из адипиновой кислоты получают ее динитрил, а затем гек-
саметилендиамин:
сн2 соон
Хсн
ьо-65'С
CN
NH,
I
C
ЮО-200 І7
200* С
N
NH2
2
Продукт химического соединения адипиновой кислоты и гек-
саметилендиамина представляет собой АГ-соль, в результате
318
поликонденсации которой получается полиамидная смола
найлон. Из этой смолы формуется найлоновое волокно:
СООН П Г NH,
п
(СН2)а
LCOOHJ NH2 J
: п [НООС (СН2L СООН] [NH2 (CH2N NH2]
260—280° С
АГ—соль
-[— NH (CH2N NH-OC(CH2L CO—]„ + In H2O.
10-12ч
Температура плавления волокна 250—254° С. Общепринятая-
формула найлона
H2N (СН2)а NH — [—ОС (CH2LCONH (CH2)eNH—]„ОС(СН2LСООН.
В настоящее время вырабатывают новое полиамидное
волокно энант из ш-аминоэнантовой кислоты ЫНг(СН2)бСООН,
которую получают синтетическим путем из этилена методом теломе-
пизятгии. Синтез ro-аминсзнантоБОй кислоты разработан в СССР'
академиком А. Н. Несмеяновым с сотрудниками. Теломериза-
ция — новый цепной радикальный процесс полимеризации.
Характерным признаком его является образование продуктов
присоединения частей предельного соединения, например ССЦ,.
к концам молекулы мономера, например СНэ = СН2 в
присутствии инициатора, который образует радикал R". Этот радикал дает
с ССЦ новый радикал:
Полученный радикал после реакции с этиленом образует
более сложный радикал:
п СН2 = СН2 + СС1з -+ СС13 (СН2 — СН2—)„.
Обрыв цепи наступает в том случае, когда радикалы с
молекулой CC1.J дают тетрахлоралканы:
СС1з (СН2 — СН2—)п + СС14 -+ СС1з (СН2—СН2)П С1 + СС1з.
После того как весь инициатор израсходован, передача цепи
прекращается, так как образуется нейтральная молекула:
2СС13 -*¦ С2С16.
На рис. 133 изображены кривые, характеризующие состаа
продуктов процесса теломеризации в зависимости от соотношения
между этиленом и четыреххлористым углеродом. С увеличением
количества этилена в смеси выход высших теломеров
повышается.
Реакция теломеризации этилена с участием ССІ4, целью
которой является получение аминоэнантовой кислоты и других
кислот, проводится непрерывно в трубчатом реакторе при 90—
319-
100° С и давлении 120—150 н/см2. Молекулярное соотношение
СгН4: ССЦ равно 2—3,5. Время пребывания смеси в реакторе
.30—40 мин. Инициатор — азодинитрилдиизомасляной кислоты.
В результате реакции получается жидкость, содержащая после
отгонки ССІ4 35—40% тетрахлоралканов, которые разделяют
перегонкой под вакуумом.
Для получения (ц-аминоэнантовой кислоты выделяют тетра-
хлоралкан СС13(СН2)йС1. В дальнейшем при нагревании с сер-
60
50
оЧ
40
I JO
і'
г
\
\
/
\
/
с7
***
с,
^—
Си і
C7*Cs*C/t
u —
-—
— —
¦ —
— ¦¦
- ¦
f 2 3 4
Моль. С2
5 6 7
/молб
д 9 /О
Рис. 133. Зависимость состава смесей
тетрахлоралканов от концентрации этилена.
иой кислотой он подвергается гидролизу, в результате чего
образуется ы-хлорэнантовая кислота С1(СНг)б'СООН. Нагревание
ее с раствором аммиака дает ш-аминоэнантовую кислоту:
С1 (СН2NСООН + 2NH3 -^-* NH2 (CH2)e СООН + NH4C1.
-С помощью ионообменных смол (ш-) аминоэнантовую кислоту
отделяют от хлористого аммония. Таким путем получают раствор
ш-аминоэнантовой кислоты, который упариванием превращают
в кристаллическую кислоту, плавящуюся при 195° С.
Как и в случае капрона, ш-аминоэнантовую кислоту
превращают в полимер нагреванием концешрировашшл растворов
в лвтоклаве (в атмосфере азота) под давлением 134—147 н/см2.
Пары воды постепенно выпускают из автоклава, затем
выдерживают при той же температуре несколько часов, снизив
предварительно давление до атмосферного. После этого
расплавленную массу выдавливают через узкие ліели в воду, где полимер
затвердевает в виде узкой ленты, которую режут на куски,
получая таким образом крошку полимера. Крошка энанта не
содержит мономера и включает не более 1 % примесей. Поэтому
переработку энанта можно производить, исключив операции
экстрагирования и сушки.
Волокна полиамидных смол в пять раз прочнее шерсти, в три
раза — шелка, в два—три раза — хлопка.
Волокно лавсан (терилен) формуется из
продукта поликонденсации диметилтерефталата и этиленгликоля в
присутствии небольших количеств CH3ONa или (CH3OJMg:
п СНзООС
\ СООСНз + п ОНСН,— СН,ОН
f—ОС
/ \ — СООСН2— СН2О —
2л СН3ОН.
Лавсан (терилен)
Лавсан — это полиэфирное волокно.
Температура плавления его 250—255° С. Общепринятая
формула его
НОСН,СН,ООС;
\ —
ЧсОО —[—СНаСН„ООС/
СН2СН.2ОН.
Схема производства лавсана изображена на рис. 134. В
аппарате / происходит
расплавление
диметилтерефталата в заранее подо- і і
гретом этиленгликоле. Эта JjB|J
смесь при 150° С
поступает в автоклав 2 для
переэтерификации. Здесь
в присутствии катализа- ч/
тора диметиловый эфир Т
превращается в диглико-
левый эфир терефталевой
кислоты. Процесс
сопровождается выделением
метилового спирта. После Рис. 134. Схема производства волокна
отгонки рассчитанного лавсан,
количества метилового
спирта расплав передается в аппарат 3, где при температуре
275° С происходит поликонденсация в вакууме, соответствующем
остаточному давлению ниже 1 мм рт. ст.
Образующееся высокомолекулярное соединение — полиэти
лентерефталат, обладающий вязкостью 2 000—3 000 пз (при
275° С), передавливается в виде широкой ленты на барабан 4,
с которого затвердевший продукт поступает в рубильную
машину 5. Полученная крошка после высушивания идет в
прядильную машину 6, где сначала плавится, а затем выдавливается
ч ерез отверстия фильеры в шахту 7. По выходе из шахты затвер-
21 8-1904
321
девшие нитн наматываются на бобины 8. В дальнейшем из них
делают шелк и штапельное волокно. Чтобы получить прочные
волокна, нить вытягивают в пять раз.
Прядение гетероцепных волокон
Прядение этого вида волокон осуществляется сухим
способом. Крошку капроновой смолы подвергают предварительно
экстрагированию горячей водой для удаления незаполимеризовав-
шегося капролактама, сушат до
содержания воды менее 0,1% и направляют на
прядение. Крошка смол — иайлона, энан-
та, терилена и полиуретановой — идет на
прядение без предварительной обработки.
Процесс прядения осуществляется в
прядильной машине, имеющей высоту
до 8,5 м (рис. 135). Сухую крошку
загружают в верхний стальной буикер
емкостью 300 л, во избежание окисления
предварительно наполненный азотом. В
нижней части бункера расположена
электроплавильная решетка. Здесь смола
превращается в расплав, который с помо-
Рис 135. Фильерная го- щью нагнетающего и дозирующего
ловка машины для пря- шестеренчатых насосов подается в
дения гетероцепного во- фильериый комплект, где фильтруется
локна из расплава: через сетку и кварц И продавливается
/ — плавильная решетка; 2— иапаа Аигткрпи
прядильный насос; 3 - поп- чеРЄЗ фИЛЬеру.
емный конус; 4 — фильера. ФиЛЬера ДЛЯ КЭПрОИа ПреДСТЭВЛЯеТ
собой пластинку из хромовоникелевой
стали толщиной 5—6 мм с отверстиями диаметром 0,25 мм.
Жидкие струйки расплава проходят через обдувочные щитки,
где охлаждаются в среде азота. Окончательное охлаждение
осуществляется.в воздушной камере, после чего волокна
проходят через замасливатель и наматываются на бобину.
Прядение волокон производится со скоростью до 1000 м/мин.
Волокна шелка подвергают вытягиванию в 3,5—4,5 раза, после-
чего прочность их значительно повышается благодаря
ориентации беспорядочно расположенных макромолекул вдоль оси
волокна. Крутка производится несколько раз, а затем волокно
идет на отделку.
Обладающие высокой прочностью, эластичностью и
термостойкостью гетероцепные волокна широко применяются в
технике и в быту.
Из гетероцепных волокон делают шелк для трикотажных
изделий и легких верхних тканей, а также штапельное волокно,
которое смешивают с шерстью и хлопком, получая ткани,
превышающие по прочности чисто шерстяные.
322
Карбоцепные волокна
Исходным сырьем для производства карбоцепных волокон
служат этилен и его производные. Наиболее распространенными
являются такие волокна, как хлорин, виньон, саран, нитрон.
Волокно хлории изготовляют из полихлорвинила,
дополнительно обработанного хлором (перхлорвинила). Волокно
имеет сравнительно высокую химическую стойкость и применя-
Смвла
Лары
теплоносителя
Рис. 136. Схема шнековой машины для формования
волокна совиден:
/—загрузочный бункер; 2 — шнек; 3 — конус; 4 — фнльера; 5 —
ванна с холодной водой: 6—вытягивающие днскн; 7 — бабина.
ется для получения технических тканей. Кроме того, оно
обладает лечебными свойствами и используется для изготовления
медицинского трикотажного белья.
Волокно виньон формуют из продукта сополимериза-
ции хлорвинила и винилацетата
|—СН2 — CHCI — СН2 — СНООССН,—1„.
Волокно саран изготовляют из полимеров винилиден-
хлорида или сополимера винилхлорида и винилиденхлорида
[— СН2 — CHCI — СН2 — СС12—]„.
Волокно нитрон получают из полиакрилонитрила
[—СН2 — CH-CN—1„.
Волокно нитрон обладает наибольшей светостойкостью и
термической стойкостью A30° С) и поэтому приобретает все
большее распространение. Нитрон — полноценный заменитель
шерсти и шелка. Разработан способ производства из
политетрафторэтилена нового термостойкого волокна тефлон
[-CF.-CF.-L,.
Карбоцепные полимеры плавятся с разложением, поэтому
их нельзя формовать из расплавов, как полиамидные волокна.
Чаще всего формование карбоцепных волокон производится из
21*
323
растворов их в ацетоне или других растворителях (диметилфор-
мамиде). Если же полимер не растворяется и не плавится,
а лишь размягчается (например, саран), прядение волокна
осуществляется методом шприцевания через профилированное
отверстие с охлаждением в ванне с водой (рис. 136). Получается
одиночное волокно неориентированной структуры, которое затем
вытягивают в два — три раза. Саран применяется для
изготовления обивочных тканей, рыболовных снастей и пр.
Разработан новый метод прядения волокна тефлон из водной
суспензии, так как полимерная смола не растворяется в
растворителях и не размягчается при нагревании.
ЛИТЕРАТУРА
1. Сб. 50 лет. «Советская химическая наука и промышленность». Главн.
ред. Л. А. Костандов, изд-во «Химия», 1967.
2. П. М. Лукьянов. Краткая история химической промышленности
СССР, Изд-во АН СССР, М., 1959.
3. А. И. Бродский. Физическая химия, т. II, Госхимиздат, 1948.
4. В. А. Киреев. Краткий курс физической химии, Госхимиздат, М.,
1962.
о. С. 11, Б о л ьф к о в и ч, А. П. Егоров, Д. А. Эпштейн и др.
Общая химическая технология, Госхимиздат, М.—Л., т. I, 1953 г., т. II, 1959.
К ГЛА BE I
1. С. М. Я с ю к е в и ч. Обогащение руд, Металл ургнздат, 1947.
2. И. Л. Гордон. Водоприготовление в теплоэнергетическом хозяйстве,
нертичдат, !940.
рд,
3. И. Э. А п е л ь ц и н, В. А. К л я ч к о, Ю. Ю. Лурье, А. С.
Смирнов. Иониты и их применение, Стандартгиз, 1949.
4. Л. А. К у л ь с к и й. Химия н технология обработки воды, Изд-во
АН УССР, К.. 1960.
К ГЛАВАМ II и Ш
1. H. И. Никитин. Химия древесины, Изд-во АН СССР, 1951.
2. Н. И. Н и к и т и н, Н. И. С о л е ч н и к, Ф. П. Комаров.
Химическая технология дерева, изд. Ленхимсектора, 1931.
3. К. И. Ногнн. Сухая перегонка дерева, Гослестехнздат, 1936.
4. Общая химическая технология топлива. Под ред. С. В. Кафтанова,
Госхнмиздат, 1947.
5. В. С. Кры м. Химия твердого топлива, ГОНТИ Украины, 1936.
6. А. А. А г р о с к и н. Химия н технология угля, Госгортехиздат, М.,
1961.
7. А. Ф. Засядь ко. Топливноэнергегическая промышленность, Гос-
планиздат, 1959.
8. И. П. My х л е н о в и др. Общая химическая технология, изд-во
«Высшая школа», 1964.
9. С. Г. Аронов, Л. Л. Нестеренко. Химия твердых горючих
ископаемых, Изд-во ХГУ, 1960.
325
К ГЛАВЕ IV
1. К. А. Белов. Переработка химических продуктов коксования, Ме-
таллургиздат, 1949.
2. Я. А. Б р о н. Аппаратчики трубчатого смолоперегонного агрегата.
Металлу ргиздат, 1956.
3. Л. Я. Коляндр. Улавливание и переработка химических продуктов
коксования, Металлургиздат, 1962.
4. С. О. Федосеев, А. В. Чернышев. Полукоксование и
газификация твердого топлива, Гостоптехиздат, М., 1960.
5. Г. И. Д е ш а л и т. Курс технологии коксохимического производства,
ОНТИ, 1947.
6. М. М. Борщевский и др. Справочник по переработке горючих
сланцев, Гостоптехиздат, 1963.
К ГЛАВЕ V
1. Д. Б. Г и н с б у р г. Газификация топлива и газогенераторные
установки, Гизлегпром, 1938 .
2. Г. О. Н у с и к о в. Подземная газификации углей, ГОНТИ, 1938.
3. А. Р и х е. Основы технологии органических веществ, Госхимиздат,
І959.
К ГЛАВЕ VI
1. С. С. Наметкин. Хнмня нефти, ОНТИ, 1939.
2. С. Н. Обрядчиков. Технология нефти, ч. II, Гостоптехиздат, 1947.
3. А. Ф. Добр я некий. Научные основы крекинга нефти, ОНТИ,
і 935.
4. В. Е. Пархоменко. Технология переработки нефти н газов, Гос-
їоптехиздат, М., 1959.
5. А. П. П и ч у г и н. Переработка нефти, Гостопиздат, 1960.
6. Е. М. Зел ьк инт. Нефтеперерабатывающая промышленность США,
ГОНТИ, 1959.
7. П. И. Лукьянов нА. Г. Басистое. Пиролиз нефтяного сырья,
Гостоптехиздат, М., 1962.
8. А. Я. Г а н ч и к о в а. Новые процессы переработки нефти н газа за
рубежом, Гостоптехиздат, 1S60.
9. С. Ф. Гудков. Переработка углеводородов н попутных газов,
Гостоптехиздат, М.—Л., 1962.
К ГЛАВЕ VII
1. В. Б. Иоффе. Основы производства водорода, Гостоптехиздат, 1960,
2. С. Я. 1 с р ш. Глубокое охлаждение, ч, I, Госэнергоиздат, 1957; ч. II,
1960.
К ГЛАВЕ VIII
1. В. И. А трощен к о и др. Технология связанного азота, Изд-во X ГУ,
1962.
2. В. И. Атрощенкон др. Технология азотной кислоты,
Госхимиздат, 1949.
3. Ф. А. А н д р е е в и др. Технология связанного азота, Госхимиздат, 1966.
4. Е. Б л а с я к и др. Технология связанного азота. Синтетический аммиак,
Госхимиздат, 1961.
326
К ГЛАВЕ IX
1. А. Г. Амелии. Производство серной кислоты, ¦ изд-во «Химия», М.,
J967.
2. Г. К. Бор ее к о в. Катализ в производстве серной кислоты,
Госхимиздат, 1954.
3. А. В. Авдеева. Газовая сера, Госхимиздат, 1950.
4. М. А. Меиковскнй. Природная сера, Госхнмиздат, 1949.
К ГЛАВЕ X
1. М. Е. Позии. Технология минеральных солей, Госхнмиздат, Л., 1961.
2. В. Ф. Чернов. Производство кальцинированной соды,
Госхимиздат, 1956.
3. М. Б. Зелик и н. Производство каустической соды химическими
способами, Госхимиздат, 1961.
4. В. Ф. Чернов. Производство кальцинированной соды,
Госхимиздат, 1956.
5. И. И. Колмаиовский. Производство двууглекислого натрия, иэд-во
•«Химия», 1964.
К ГЛАВЕ XI
1. А. А. Соколовский. Технология минеральных удобрений, М., 1966.
2. А. Н. Андреичев и др. Добыча и переработка калийных солей,
Госхимиздат, 1960.
3. М. В. К а та л ым о в, Т. П. У н а и я н ц. Производство и прнмеие-
нне микроудобрений в СССР и за рубежом, Госхнмиздат, 1960.
4. Н. Н. Мельников. Химия пестицидов. Госхимиздат, 1966.
5. Л. Д. Стонов. Дефолианты и десиканты, Госхимиздат, 1961.
6. М. Г. Г а б р и е л о в а и др. Производство неорганических
ядохимикатов, М. — Л., 1964.
7. В. В а г г а м а и. Фосфорная кислота, фосфаты и фосфорные удобрения,
Госхимиздат, М., 1956.
КГЛАВЕХП
1. Н. П. Федотьев. Прикладная электрохимия. Госхимиздат, Л.,
1962.
2. Л. С. Геинн. Электролиз растворов поваренной соли. Госхимиздат,
Л., 1960.
3. Л. Я. Марковский и др. Химическая электротермия.
Госхнмиздат, Л., 1952.
4. Л. А. Кузнецов. Производство карбида кальция. Госхимиздат,
Л., 1954.
5. А. М. Лайнер. Производство глинозема. Металлургиздат, М., 1961.
К ГЛАВЕ ХШ
1. П. П. Б у дни ков. Технология керамики и огнеупоров, Госстрой-
нэдат, 1962.
2. Ю. М. Бутт и др. Общая технология силикатов, Госстройнздат, 1962.
3. Ю. М. Бут т. Технология цемента и других вяжущих материалов,
Промстройиэдат, 1956.
4. И. И. Китайгородский и др. Технология стекла, Госстройиз-
дат, 1961.
327
5. Л. Н. Усов и др. Применение плазмы для получения
высокотемпературных покрытий, изд-во «Наука», 1965.
6. Г. В. Кукол ев. Химия кремния и физико-химия силикатов, изд-во
«Высшая школа», М., 1966.
К ГЛАВЕ XIV
1. Б. М. Т ю т ю н н н к о в, Н. В. Н а у м е н к о и др. Технология пере-
работкя жнров, Пищепромиздат, 1963.
2. И. Н. Юкельсон. Технология основного органического синтеза,
Госхимиздат, 1959.
3. С. Н. К а з а р н о в с к н й, В. Н. К о з л о в. Технологические схемы
процессов основного органического синтеза, ч. I и II. Труды по химии и
химической технологии Горьковского ун-та, 1958—1960.
4. Б. А. Крен цель. Хлорирование парафиновых углеводородов, изд-
во «Наука», М., 1964.
5. Б. А. Кренцель. Основы синтеза алифатических спиртов из
нефтяных углеводородов. Изд-во АН СССР, М., 1954.
К ГЛАВЕ XVI
1. Е. И. Бар г. Технология синтетических пластических масс, Госхим-
издзт, 1954.
2. Н. И. Смирнов. Синтетические каучуки, Госхнмнздат, 1954.
3. Неметаллические материалы и их применение в авиастроении. Под
ред. И. П. Лосева и Е. Б. Тростянской, Оборониздат, 1958.
4. В. А. К а р гн и, Г. Л. Слонимский. Краткие очерки физико-
хнмии полимеров. Изд-во МГУ, 1960.
5. А. А. С т р е п и х е е в, В. А. Д ер е в и ц к а я. Основы химии
высокомолекулярных соединении, Госхнмиздат, М., 1961.
6. И. П. Лосев, Е. Б. Тростя некая. Химия синтетических
полимеров, Госхимнздат, М., 1960.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абиетиновая кислота —33
Абсорбция серного ангидрида —140
Агломерация —13, 14
АГ-соль (гексаматнленднаминади-
пат) —318
Аднпиновая кислота —318
Адсорбенты — 79
Азодинитрил диизомасляной кислоты
-320
Азеотропная смесь —109
Азот —91
» получение нз воздуха —91
Азотная кислота —100
» » получение под
атмосферным давлением—106
Азотная кислота под давлением —107
» » концентрирование —
109
» » прямой синтез —111
Акрилонитрил —270
Активные угли —28
Алкилирование —78, 289
Алкидные смолы — 310
Алкилсиландиолы —311
Алкилсульфаты —264
Алкнлсульфонаты —265
Альдегид муравьиный —258
» уксусный —277
Алюминий —210
Амальгама натрня —203
Е-амннокапроновая кислота —318
ш-аминоэнантовая кислота —320
Аммиак —94
Аммиачная вода — 39
Аммиачная селитра —182
Аммиачносодовое производство —145
Аммоний сернокислый —185
Аммофосы —177, 188
Аминопласты — 309
Ангидрид уксусный —379
Ангидрид фталевый —290
Андезит —131
Анилин —281
Анионит —19
Антидетонатор —65
Антидетонация —64
Антрацен —50
Антрацит —22, 24
Апатит — 165
Армированные пластики —311
Ароматизация —76
Ароматические углеводороды —36
Асбест —280
АсботєкстолЦт —309
Атмосферновакуумная перегонка
нефти —66
Ацетилен —273
Ацетилцеллюлоза —316
Ацетон —28, 289
Атактические полимеры —293
Баланс материальный —5
» тепловой —6
Бактерии —20, 93
Барий хлористый —193
Барабанный грохот —14
Башенная система —129
Белильная известь — 209
Бензин авиационный —64
» газовый — 77
» крекинга — 72
Бензол —41
» извлечение из коксового гак
за —41
Бензол сырой —46, 47
» чистый —48
» моторный — 48
Бетон —245
Бикарбонат натрия —143
Биохимические процессы —20
Биполярные ванны — 196
Битумы —22
Бобинное прядение —317
Болотная руда —43
Брожение —32
Бурые угли —22, 24
Бутадиен —296
329
Бутадиенстирольный каучук —296
Бутадиеннитрильный каучук —297
Брызгальные бассейны —19
Вагон тушильный —38
Вакуумсодовый метод очистки газов
от сероводорода —42
Ванадий, пятиокись —136
Винилиденхлорид —304
Вода природная —18
» аммиачная —39
Водород (методы получения) — 81
Воздух (разделение) —91
Волокна искусственные —313
» синтетические —317
Высокомолекулярные соединения —
291
Таз водяной —59
» воздушный —58
» коксовый —42
» паровоздушный —58Л
» природный —26
Газгольдеры —63
Газификация твердого топлива —55
Газойль —67
Гемицеллюлозы —21
Гексахлоран —288
Гетерогенный —10
Гетероциклические соединения —317
Гексаметнлендиамнн —318
Гексаметилентетрамнн —259
Гексозани —10
н-Гептан —65
Гидравлические вяжущие вещества —
236
Гидрататоры —277
Гидрирование (гидрогенизация)—53
Гидромодуль цемента — 241
Гидроперекись изопропилбензола—289
Гидротропное выделение целлюлозы —
31
Гидрофильная группа —266
Гидроформинг-процесс —76
Глины —223
Глифталевые смолы —310
Глиняные изделия (см. керамика)
Глицерин —272
Глюкоза —32
Гравитационное обогащение — 14
Градирни —19
Грохоты —14
Гудрон —67
Гумнны —21
Гуминовые кислоты —21
Даутерм —271
Дегидрогенизация —70
Декарбонизация
бикарбоната —7, 160
Древесина —20
Деструктивная гидрогенизация —53
Детонация (см. антидетонация)
Днвиннл (см. бутадиен)
Дизельное топливо —68
Динас —231
Динитробензол —281
Диссоциация ангидрита —124
» бикарбоната натрия —160
Дихлорэтан —272
Дробилки —13
Дуговой метод —93
Едкий натр —
» » производство
известковым способом —159
» » производство ферритныы
способом —161
» » производство
электрохимическим способом —199
Живица —33
Животное сырье —17
Жидкофазная очистка масел —79
Игепаль —266
Игепон —264
Известковое молоко —45
Известняк —154
Известь гашеная —154
» негашеная — 158
» хлорная (см. известь белнль-
ная) —209
Изомеризация —78
Ииден —48
Интенсивность системы —128
Ионообменные смолы —19
Каландрование —30І
Калий азотнокислый —187
» хлористый —179
Кальций азотнокислый —93
» сернистый —193
» фосфорнокислый (см.
фосфорит)
Кальций хлористый —163
Каменный уголь —23
Каменноугольная смола —40, 48
Камерная кислота —180
» система —128
Камеры коксовых печей —36
330
Камфора —34
Канифоль —33
Каолин — 223
Кара-Богаз-Гол —144
Карбазол —50
Карбид кальция —215
Карбонизация аммиачного рассола -
151
Карналлит —179
Катионоактнвные моющие средства ¦
265
Катионовая очистка —19
Каустификация —159
Кварц —230
Керамика безобжиговая —236
Керамика (изделия) —227
Керамика кислотоупорная —229
Керметы —23.3
Кнрпнчи динасовые —231
» шамотные —231
Кислород —91
Клинкер —229, 241
Когазин —264
Кокссва;;ие каменных углей —34
э сланцев —51
» торфа —52
Коксовые печи —35
Кокс —39
Коллоксилин —313
Колчедан —116
Конверсия —82, 86
Контактный способ —134
Концентрирование серной кислоты -
132
» азотной кислоты —
111
Костяная зола —170
Крекинг в смешанной фазе —71
» » паровой фазе —72
Кремнефторнстоводородный натрий —
Криолит —211
Ксилолы —48
Кумароновая смола —48
Кумарон —48
Кумол —289
Купорос медный —193
Лигннн —21
Лигнит —22
Люкс (очистительная масса) —44
Магнезиальный цемент Сореля —182
Магний металлический —212
» хлористый —212
Мазут —66, 68, 71
Масла каменноугольные —50
» смазочные —68
Медный купорос —193
Мел —156
Мергель —241
Мерзоли —265
Мерзоляты—265
Металлы черные —12
» цветные —12
» тяжелые —12
» легкие —12
» редкие —12
» рассеянные —12
Металлоорганнческие катализаторы —
303
Метилметакрилат—305
Метиловый спирт —28, 256
Модуль гидравлический —256, 241
Мочевина (карбамид) — 260
Мочевиноформальдегидные смолы —309
Моющие средства —263
Муллнт —227
Муфель (печь муфельная) —234
Мышьяк (см. очистка газа)
Надсмольная вода (см. аммиачная
вода)
Найлон —318
Наконол —265
Наполнители —300
Натр едкнй —159
Натрнйбутадненовый каучук —296
Натронный способ производства
целлюлозы —30
Нафталин —50
р-Нафтол —284
Некаль (дибутилнафталинсульфокис-
лота) —296
Неозой —300
Нефелин —166
Нефть —26
Нефтяной газ —27
Нитробензол —281
Нитрование —280
Нитрогуанидин —261
Ннтроза —128
Нитрознлсерная кислота — 128
Нитрофоска —189
Новолачные смолы —306
Норвежская селитра (кальциевая)
—93
Носители —136
Обжиг керамических изделий —225
Обогащение — 14
Огарок — 125
Огнеупорные глины —224
Окисление аммиака —101
Окислы азота —101, 128
331
Октановое число —65
Отсадочные машины —15
Очистка газов —42, 127
» нефтепродуктов —79
» сырого бензола —47
Паста (см. деструктивная
гидрогенизация) —54
Парафин (окисление) — 261
Пенообразователи —17
Пенопласта — 312
Пентан —77
Перегонка каменноугольной смолы —
48
» сырого бензола —47
» иефти —66
Перколяторы —32
Печн циклонные —123
» вращающиеся —124, 243
» туннельные — 226
» известковс -обжигательные —157
» кольцевые —228
» колчеданные—118
а-Пиколнн —51
р-Пиколнн —51
Пиридиновые основания —51
Пирнт (см. колчедан)
Пнролнз —72
Плавка едкого натра —161
Пластикация каучука —300
Пластмассы —302
Платформннг-процесс —75
Пневый осмол —33
Подземная газификация —62
Полимеризация —292
Полиэтилен —303
Полукоксование углей —52
» сланцев —53
» торфа —52
Портланд-цемент (см. цемент)
Предкатализ —97
Преципитат —175
Природные газы —77, 8G
Противостарителн —300
Пушонка (см. известь іаііичіая)
Равновесие в процессах газификации
топлива —56, 57
Равновесие прн синтезе аммиака —94
Рассол —143
Растворимое стекло —253
Регенераторы —7
332
Регенерация тепла — 35
» катализаторов при
крекинге —74
Резина —300
Резитол —306
Резит —307
Резол —307, 306
Ректификация воздуха (см. азот)
Ретроградация —154, 172
Ртутный способ —203
Сажа —300
Сапропелитовое топливо —22
Саран —323
Сгустители —29
Сера —117
Селитра аммиачная —182
» калиевая —187
» конверсионная (см. селитра
калиевая) 184
Селитра чилийская (селитра
натриевая) — 185
Сероводород (очистка от него) — 42
» использование —43
» для получения серы —44
Силикаты (см. глина и каолины)
Сильвинит —179
Синтез аммиака —94
Синтез-газ —81, 256
Синтол —256
Синтетические волокна —317
Синтетические каучуки —295
Ситаллы —254
Сланцы —23, 51
Смолы — 48, 53, 302
Сода кальцинированная —143
» каустическая —143
» кристаллическая —143
Содовая печь —153
Соляная кислота —205
Соляровое масло —41, 68
Сольвент I и II —48
Стабилизация бензина —77
Сополимеры бутадиена с акрилонит-
рилом —298
Сополимеры бутадиена со
стиролом —297
Стекло —247
Стереорегулярные полимеры — 293
Сульфат аммония (см. аммоний
сернокислый)
Сульфирование — 283
Сульфитная целлюлоза —314
Сульфохлорирование —264
Суперфосфат —167
Сшивание полимерных цепей —295
Талловое масло —31
Текстолит —308
Теломеризация —319
Теплоносители —161
Терилен —321
Термопластические смолы —303
Термореактивные смолы —306
Термофосфат —177
Тетраэтилсвинец — 65
Тефлон —306
Тиофен — 47
Толуол —45. 72
Томасшлак —178
Топливо —20—27
Триацетилцеллюлоза —316
Уголь активированный —28
» древесный —28
Удобрения азотные —182
» калийные —178
» фосфорные —165
» микро —190
Уксусная кислота —278
Уксусный альдегид —279
» ангидрид —279
Уротропин —259
Фарфор —234
Фаянс —234
Фенантрен —50
Фенол —50
Фенопласты —306
Феррит — 161
Фиксация азота воздуха — 93
Флотация —16
Фильеры —315
Формалин —258
Формование —301, 308
Фосфор (возгонка) —220
Фосфорит —165
Фосфогипс —174
Фталевый ангидрид —209
Фторкаучуки —299
Фторапатиты —165
Фурфурол —33, 80
Хлор —204
Хлораль —287
Хлораммоний —163
Хлорбензол —285
Хлорин —323
Хлорирование —285
Хлористый водород —206
Хлорнощелочной способ выделения
целлюлозы —31
Хлорпреновый каучук —298
Целлюлоза —29
Целлулоид —313
Цемент портландский —241
» кислотоупорный —245
Цианамид кальция —217
Циклогексан —65, 70, 75
Циклогексанол —318
Циклогексанон —318
Четыреххлористый углерод —319
Шамот —231
Шахтные печи —157
Шихта —26
Экстракция фосфорной кислоты —173
» уксусной кислоты —28
Электролиз —194
Электромагнитное обогащение —14,16
Электростатическое обогащение —14,16
Электрофильтры —40, 126
Этаиоламины —44, 272
Эмалирование —230
Энантовое волокно —319
Этилен —267—272, 319
Этиленгликоли —272
Яд каталитический —97, 126
Ядохимикаты —192, 287
СОДЕРЖАНИЕ
Стр.
Предисловие 3
Введение 4
Часть первая. СЫРЬЕ. ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ ТОПЛИВА
Глава I. Виды сырья и его предварительная подготовка 12
!. .Минеральное сырье 12
2. Добыча и первичная переработка сырья 13
3. Методы обогащения сырья 14
4. Растительное и животное сырье 17
5. Вода 18
Глава II. Виды и состав топлива 20
1. Древесина 20
2. Ископаемое твердое топливо 21
3. Торф 22
4. Бурый уголь 22
6. Каменный уголь 23
6. Сланцы > 23
7. Состав различных видов топлива 23
8. Нефть и природные газы 26
Глава 111. Методы переработки топлива 27
1. Переработка дерева 27
2. Производство древесной целлюлозы 29
3. Гидролиз древесины 31
4. Канифольно-скипндарное производство 33
Глава IV. Переработка ископаемого твердого топлива 31
1. Коксование каменною угля 34
2. Конденсация и улавливание химических продуктов коксования . ЗУ
3. Применение коксового газа и его очистка 4?
4. Переработка химических продуктов коксования 44
5. Коксование сланпев 51
6. Полукоксование 52
7. Деструктивная гидрогенизация угля 53
Глава V. Газификация твердого топлива 55
1. Производство водяного газа 59
2. Газогенераторы 61
3. Подземная газификация углей 62
334
Стр.
Глава VI. Переработка нефти н горючих газон 63
1. Свойства моторных топлив 64
2. Переработка нефти 65
3. Прямая перегонка нефти 66
4. Кр.екинг нефтепродуктов 68
5. Риформинг-процесс 75
6. Нефтяные и природные газы и их переработка 77
7. Очистка нефтепродуктов 79
Часть вторая. ТЕХНОЛОГИЯ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
Глава VII. Производство технических газов 81
1. Производство водорода 81
2. Производство азота н кислорода 91
Глава VIII. Технология связанного азота 92
1. Проблема фиксации азота воздуха 92
2. Синтез аммиака 94
3. Производство азотно? кистсты , . 10О
4. Концентрирование азотной кислоты 109
Глава IX. Производство серной кислоты 114
1. Применение и сорта серной кислоты 114
2. Сырье 115
3. Получение сернистого газа и его очистка 118
4. Получение серной кислоты 127
Глава X. Производство содовых продуктов 143-
1. Сырье и методы его переработки 143
2. Аммиачный способ 145
3. Производство извести и углекислого газа 156.
4. Производство каустической Соды 159
5. Другие продукты содового производства . . 162
Глава XI. Технология химических продуктов для сельского хозяйства 163
I. Удобрения 164
1. Фосфорные удобрения 165
2. Калийные удобрения 178
3. Азотные удобрения 182
4. Жидкие удобрения 186-
5. Многосторонние удобрения 187
6. Микроудобрения 190
П. Ядохимикаты 192
Глава XII. Промышленный электролиз и электротермия 194
1. Основы электрохимии 194
2. Электролиз воды 197
3. Электролиз водных растворов хлористого натрия ....... 199
4. Переработка хлора 204
5. Электролиз расплавов 210
6. Электротермические процессы 213
335
Г л ава XIII. Химическая технология силикатов 222
1. Глинистое сырье н основы производства керамических изделий . 223
2. Химическая технология вяжущих материалов 236
3. Производство стеклянных изделий 247
Часть третья. Химическая технология органических веществ
Глава XIV. Основной органический синтез 255
1. Синтезы, на основе водорода и окиси углерода 255
2. Синтезы на основе двуокиси углерода. Производство мочевины
(карбамида) 259
3. Синтезы на основе предельных углеводородов 261
4. Синтезы на основе олефиновых углеводородов 267
5. Синтезы на основе ацетилена 273
Глава XV. Синтезы на основе ароматических углеводородов 280
1. Нитрование 280
2. Восстановление нитросоединений 281
3. Сульфирование 283
4. Получение фенолов, E-нафтола 284
5. Хлорирование ароматических соединений 285
6. Инсектициды. 4,4' — Дихлордифенилтрихлорметилметан (ДДТ).
Гексахлоран 287
7. Производство фенола из хлорбензола 288
8. Получение фенола совместно с ацетоном через изопропилбензол
(кумол) 289
9. Окисление с разрушением ядра 290
Глава XVI. Технология высокомолекулярных соединений 291
1. Каучук 294
2. Производство смол и пластмасс 302
3. Технология искусственного волокна 313
4. Технология синтетического волокла 317
Литература 325
Предметный указатель 31'9
Содержание 334