/




















































































































































































































Author: Фролов Ю.Г. Гродский А.С.
Tags: химия практикум коллоидная химия лабораторные работы
Year: 1986




Text
Лабораторные
работы
и задачи
по коллоидной
химии
Под ред. проф. Ю. Г. ФРОЛОВА
и доц. А. С. ГРОДСКОГО
^Допущено Министерством высшего
и среднего специального образования, СССР
в качестве учебного пособия
для студентов химико-технологических
специальностей высших учебных заведений?»
МОСКВА
•ХИМИЯ*
VJ86
541
Л 12
УДК 541. 18 (076. 5)
Рецензенты: 1. Кафедра физикохимии полимеров Казан-
ского химико-технологического института. 2. Проф. И. А. Ту-
торский.
Авторы: Фролов Ю. Г., Гродский А. С., Назаров В. В.,
Моргунов А. Ф., Шабанова Н. А., Дворецков Г. А.,
Кривощепов А. Ф., Ким В. Е., Жигунова Л. К., Растегин Ю. И.
УДК 541.18 (076.5)
Лабораторные работы и задачи по коллоидной
химии. — Под ред. Ю. Г. Фролова и А. С. Грод-
ского. — М.: «Химия», 1986. — 216 с, ил.
Представлены лабораторные работы, контрольные вопросы и задачи
по основным разделам курса коллоидной химин: поверхностные явления
и поверхностное натяжение, адсорбционные равновесия, методы иссле-
дования дисперсных систем, образование и свойства лиофильных и лио-
фобных дисперсных систем. В каждом разделе приведены краткие теоре-
тические сведения и примеры решения задач по данной теме.
Предназначено для студентов хнмико-технологических специальностей
высших учебных заведений, изучающих курс коллоидной химии.
Табл. 47. Ил. S7.
л
1805000000—072
050(01)—86 72 8Ь
© Издательство «Химия», 1986 г.
СОДЕРЖАНИЕ
Пр едисловие..............................................................5
I. Поверхностные явления и поверхностное натяжение..............................7
Работа 1. Определение полной поверхностной энергии жидкостей . . . . . И
Работа 2. Исследование адсорбции неэлектролитов из бинарных растворов
•— —-4*. на твердых поверхностях.........................................18-
I 'Работа 3} Исследование влияния поверхностно-активных веществ на смачи-
-____вание и адгезию......................................................20
Работа 4. Определение критического натяжения смачивания неполярных по-
лимеров . . 25
Работа 5. Исследование влияния электрического потенциала на поверхност-
ное натяжение............................................................27
Контрольные вопросы и задачи..............................................30
II. Адсорбционные равновесия................................................ 37
. г
Работа 6.. Изучение адсорбции ПАВ из растворов на твердом адсорбенте 40
Работа -7. Исследование влияния строения молекул ПАВ на их поверхност-
ную активность. Определение параметров адсорбционного слоя 43
Работа 8. Определение удельной поверхности адсорбентов методом газовой
хроматографии . .........................................................46
Работа 9. Хроматографическое разделение смеси ионов с помощью ионооб-
менных смол............................•.............................51
Работа 10. Разделение смеси полимера и минеральной соли и определение
молекулярной массы полимеров методом гель-хроматографии . . 58
Контрольные вопросы и задачи .......................................... 63
III . Кинетические явления и кинетические методы исследования дисперсных систем 75
Работа И. Исследование молекулярно-кинетических свойств дисперсных си-
стем ............................................................... .... 79
Работа 12. Дисперсионный анализ низкодисперсных порошков методом седи-
ментации в гравитационном поле.......................................... 81
Работа 13. Дисперсионный анализ высокодисперсных порошков методом се-
диментации в центробежном поле.........................................89
i Работа 14"?ЧЭлектрофоретическое определение электрокинетического потенциа-
—™»Хла................................-............................... 93
Работа 15. Определение электрокинетического потенциала методом электро-
осмоса ................................................................97
Работа 16. Определение изоэлектрической точки золя гидроксида железа ме-
тодом электрофореза....................................................99
Контрольные вопросы ь задачи..............................................102
IV . Оптические свойства и оптические методы исследования дисперсных систем . .111
Работа 17. Определение размеров частиц дисперсных систем турбидиметриче-
ским методом . . .......................................113
Работа 18. Дисперсионный анализ микрогетерогенных систем методом оптв-
ческой микроскопии.....................................................117
Работа 19. Определение размеров частиц высокодисперсных систем методом
просвечивающей электронной микроскопии.................................123
Контрольные вопросы и задачи ......................................... 126
1* а
V. Образование и свойства лиофильных дисперсных систем.................129
Работа 20. Исследование мицеллообразования в растворах ПАВ..........132
Работа 21. Исследование солюбилизирующей способности растворов ПАВ . . 135
Работа 22. Исследование влияния длины углеводородной цепи молекул ПАВ
на термодинамические параметры адсорбции и мицеллообразова-
ния в водных растворах.............................................137
Работа 23. Адсорбционное титрование латексов........................143
Работа 24# Определение молекулярной массы полимеров и мицеллярной мас-
сы ПАВ нефелометрическим методом...................................146
Работа 25. Исследование растворов амфотерных полиэлектролитов .... 151
Контрольные вопросы и задачи........................................154
VI. Образование, устойчивость и свойства лиофобных дисперсных систем . . . 159
Работа 26. Синтез гидрозоля гидроксида железа, изучение его коагуляции и
стабилизации . ...............................................'**•*163
Работа 27. Исследование кинетики коагуляции латексов..............*.167
Работа 28. Получение эмульсий и изучение их свойств.............. . 171
Работа 29. Получение пен и изучение их устойчивости.................174
/ Контрольные вопросы и задачи.......................................178
V0. Структурно-механические свойства дисперсных систем..................185
Работа 30. Исследование вязкости структурированной жидкости с помощью
6И* капиллярного вискозиметра.............................188
Работа 31. Исследование реологических свойств неньютоновских жидкостей
• с помощью ротационного вискозиметра..................... 19t
Работа 32. Изучение зависимости вязкости растворов полимеров от их кон-'~-"-/
центрации с помощью реовискозиметра................................194
Работа 33. Изучение реологических свойств межфазных адсорбционных пле-
нок ..........................- ...................................198
Контрольные вопросы и задачи.................................... 204
Ответ ы к задачам...................................................... 210
Рекомендуемая литература................................................211
Приложения..............................................................211
ПРЕДИСЛОВИЕ
Коллоидная химия, как и химия вообще, является в большой степени
наукой экспериментальной. Для овладения ею недостаточно только тео-
ретической подготовки, знания идей и законов этой науки. Необходимо
также приобретение навыков экспериментального исследования, что
требует освоения современных приборов, умения обработать результаты
эксперимента и сделать соответствующие выводы и заключения. Задача
практикума по коллоидной химии состоит в обучении студентов экспе-
риментальным методам исследования поверхностных явлений, методам
получения и исследования свойств дисперсных систем.
В данном пособии представлены лабораторные работы, задачи и
контрольные вопросы по всем разделам курса коллоидной химии.
Для сознательного выполнения экспериментальной работы и реше-
ния задач в помощь студенту перед каждым разделом данного пособия
представлены краткие теоретические введения. Конкретные дополни-
тельные теоретические сведения даны также в начале описания работ.
Задачи, предлагаемые в конце каждого раздела, должны способствовать
более глубокому усвоению теоретического курса, помогать грамотно
обрабатывать экспериментальный материал, применять знания по кол-
лоидной химии в дальнейшей практической работе.
В связи с тем, что коллоидная химия завершает общехимическое
образование, нет необходимости, в данном пособии специально останав-
ливаться на определении погрешностей измерений, обработке опытных
данных и графических методах представления экспериментов. Все это
можно найти в подобных пособиях по дисциплинам, изучаемым ран^е
(на I и II курсах), например, по физической химии или физике.
Лабораторные работы и задачи по коллоидной химии, представлен-
ные в данном руководстве, отражают многолетний опыт всего коллек-
тива кафедры коллоидной химии Московского химико-технологического
института имени Д. И. Менделеева. Руководство написано в соответ-
ствии с действующей программой курса коллоидной химии для химико-
технологических специальностей высших учебных заведений, утвержден-
ной в 1979 г. учебно-методическим управлением Министерства высшего
и среднего специального образования СССР.
В экспериментальной отработке лабораторных работ и составлении
задач принимали участие практически все сотрудники кафедры. Теоре-
тические введения написаны проф. Ю. Г. Фроловым.
5
В написании лабораторных работ принимали участие следующие со-
трудники кафедры коллоидной химии: доц. А. С. Гродский (работы 1,
3, 4, 6, 7, 9, 10, 12, 17—19, 22—26, 32), асе. Г. А. Дворецкой (работы
12, 18), асе. Л. К. Жигунова (работа 1), асе. В. Е. Ким (работы 1, 4,
8, 22, 29), асе. А. Ф. Кривощепов (работы 13—15, 17, 24, 31), доц.
А. Ф. Моргунов (работы 5, 9—11, 14—16, 25, 27), доц. В. В. Назаров
(работы 1,8—10, 12, 17—19, 22, 29, 30, 33), асе. Ю. И. Растегин (ра-
бота 19), доц. Н. А. Шабанова (работы 2, 20, 21, 27, 28).
Контрольные вопросы составлены проф. Ю. Г. Фроловым, доц.
В. В. Назаровым и доц. А. С. Гродским. Подготовка задач к изданию
осуществлена доцентами Н. А. Шабановой и А. Ф. Моргуновым.
Подготовка рукописи к изданию в основном была выполнена доцен-
тами А. С. Гродским, В. В. Назаровым и А. Ф. Моргуновым.
Авторы выражают благодарность всем сотрудникам кафедры за по-
мощь в оформлении рукописи. Они также заранее благодарят читате-
лей за критические замечания и советы по улучшению пособия.
АВТОРЫ
I. ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ
И ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
Поверхностный, межфазный слой представляет собой область посте-
пенного (плавного) изменения свойств при переходе от одной фазы
системы к другой. Также постепенно меняется структура поверхностного
слоя — от структуры одной фазы до структуры другой фазы. Образо-
вание поверхностного слоя есть результат взаимодействия смежных
фаз. Изменения свойств и структуры поверхностного слоя обусловлены
действием поверхностной энергии.
При постоянных температуре и давлении поверхностная энергия
Гиббса определяется произведением поверхностного натяжения (фак-
тор интенсивности) о на площадь поверхности (фактор емкости) s:
Gs = as (1.1)
Площадь поверхности зависит от ее кривизны и дисперсности фаз.
Дисперсность D линейно связана с удельной поверхностью syA:
5Уд = -^=4- = ^ (1.2)
где V — объем дисперсной фазы; k — коэффициент формы частиц; d—диаметр ча-
стицы.
Для сферических частиц уравнение (L2) принимает вид
_ nd2 _________________________________6___
5уд~ (1/6) jtd3 — d
(1-3)
Кривизна поверхности Н в данной точке определяется производной
площади поверхности по объему:
или с помощью соотношения
(1-5)
где г1 и г2— радиусы окружностей, получаемых при пересечении поверхности, прохо-
дящими через нормаль к ней в данной точке двумя перпендикулярными плоскостями.
Например, кривизна поверхности сферических частиц радиусом г равна
ds 2
„ 1 2 1
Я = -х-—- ——, а
2 г г
(1.6)
Поверхностные явления выражаются в том, что состояние находя-
щихся в поверхностном слое молекул иное по сравнению с молекулами,
находящимися в объеме тела. Молекулы в объеме тела равномерно
окружены такими же молекулами, и поэтому их силовые поля пол-
ностью скомпенсированы. Молекулы поверхностного слоя взаимодей-
ствуют как с молекулами одной фазы, так и с молекулами другой
фазы, в результате чего равнодействующая молекулярных сил в по-
верхностном слое не равна нулю и направлена внутрь той фазы, с ко-
торой взаимодействие больше. Таким образом возникает поверхностное
натяжение о, стремящееся сократить поверхность.
Поверхностное натяжение можно представить и как энергию пере-
носа молекул из объема тела на поверхность или как работу образова-
ния единицы поверхности. Поверхностное натяжение можно выразить
частной производной от энергии Гиббса по величине межфазной по-
верхности при р и Т = const (при постоянных числах молей компо-
нентов) :
Г—1
V ds )р, т, tij
(1-7)
Отсюда следует, что для индивидуального вещества поверхностное на-
тяжение есть энергия Гиббса, приходящаяся на единицу поверхности.
Внутренняя (полная) энергия поверхностного слоя Us (в расчете
на единицу площади) связана с о уравнением Гиббса — Гельмгольца:
или
Us — о + qs (1.9)
где qs — теплота образования единицы поверхности (индекс г означает отнесение па-
раметра к единице площади поверхности); Т — температура.
Стремление системы к уменьшению поверхностной энергии Гиббса
выражается в самопроизвольном уменьшении межфазной поверхности
(изменение формы и кривизны, проявление процессов коагуляции, ко-
алесценции и др.) и уменьшении поверхностного натяжения (проявле-
ние процессов адсорбции, адгезии и смачивания, возникновение элек-
трического потенциала и др.).
Изменение кривизны поверхности (удельной поверхности) вызывает
изменение внутреннего давления в телах. Разность давлений Ар, на-
пример, внутри жидкого тела с кривизной поверхности и без нее, назы-
вается капиллярным (избыточным) давлением. Связь капиллярного
давления с кривизной поверхности описывается уравнением Лапласа:
Л/,1=0'~ДЭ • 10)
Это соотношение лежит в основе многих методов определения по-
верхностного натяжения. С изменением кривизны поверхности (удель-
ной поверхности) меняется также давление пара над веществом. Связь
между этими параметрами находит выражение в уравнении капилляр-
ной конденсации Кельвина (Томсона):
р oFM ds
lny;—Rf--iv (LI1>
где р и ps — давление насыщенного, пара над поверхностью, имеющей кривизну, и над
ровной поверхностью соответственно; ’/м— мольный объем вещества в конденсирован-
ном состоянии; R—универсальная газовая постоянная.
Если жидкость или твердое тело имеют поверхность раздела с дру-
гой жидкостью или твердым телом, то между ними проявляется адге-
зия (прилипание). Работа адгезии Wa, характеризующая взаимодей-
ствие фаз (она отнесена к единице площади поверхности), определя-
ется уравнением Дюпре:
И7а = ОД 1 + Оз, ] — ОД з (1.12)
где о2. 1 — поверхностное натяжение твердого тела или жидкости 2 на грдййЦе с га-
зом 1; Оз, 1—поверхностное натяжение жидкости или твердого тела 3 на границе с
газом 1; <т2, з — поверхностное (межфазное) натяжение на границе конденсированных
фаз.
Из уравнения Дюпре следует, что чем больше работа адгезии (взаи-
модействие между фазами), тем меньше межфазное натяжение о2, з-
Взаимодействие между жидкой и другой конденсированной фазой
можно оценить также с помощью краевого угла (угла смачивания) 0,
определяемого уравнением Юнга:
Сложение уравнений (1.12) и (1.13) приводит к уравнению Дюп-
ре— Юнга, позволяющему по экспериментально определенному cos 0
рассчитать работу адгезии:
1Га = <т2>1 (1 + cos0) . (1.14)
Уменьшение о может происходить также в результате самопроиз-
вольного концентрирования в поверхностном слое веществ с меньшим
поверхностным натяжением — адсорбции.
Величину адсорбции обычно выражают двумя способами. Согласно
одному способу ее определяют как количество вещества в поверхност-
ном слое А, приходящееся на единицу площади поверхности или еди-
ницу массы адсорбента (абсолютная величина адсорбции):
Д = = (1.15)
где cs — концентрация компонента в поверхностном слое, имеющем объем V. и тол-
щину h.
Й По другому способу величину адсорбции Г определяют как избыток
компонента в поверхностном слое (на единицу площади поверхности)
по сравнению с его количеством в равном объеме объемной фазы (из-
быточная адсорбция)
или что то же самре-;.
где cv — равновесная концентрация компонента в объеме; с0 — исходная концентрация
компонента в объеме; V — объем фазы.
id Сравнивая уравнения (I. 15) и (I. 16), получим связь между вели-
чинами адсорбции А и Г:
Г = Д — cvh (1.18)
lid Необходимо обратить внимание на то, что при экспериментальном
определении величину адсорбции обычно рассчитывают по разности
исходных и равновесных концентраций адсорбата (адсорбируемого
компонента), т. е. определяют величину Г (I. 17). При больших избыт-
ках, когда cs cv, равновесной концентрацией адсорбата в объеме
можно пренебречь, величины Л и Г приблизительно равны.
Рассмотрение баланса распределения компонентов при адсорбция
из бинарных жидких растворов, когда в поверхностных слоях проис-
ходит замена молекул одного компонента 1 на молекулы другого 2,
приводит к следующей зависимости между величинами адсорбции и
концентрациями компонентов (в мольных долях) при условии, что
Г1= — Г2 (увеличение концентрации одного компонента в поверхност-
ном слое происходит за счет уменьшения концентрации другого ком-
понента):
Г2 = ^2-^1 - Л1%2 (I-19)
где X) и хг — мольные доли компонентов в объеме раствора.
Величины адсорбции компонентов раствора Г/ и поверхностное на- !
тяжение связаны между собой фундаментальным адсорбционным урав-
нением Гиббса:
— do= £ (1.20) i
i j
где щ — химические потенциалы компонентов. j
При малых концентрациях с адсорбата в бинарном растворе соот-
ношение (1.20) переходит в следующее уравнение: J
В зависимости от знака производной ds/dc растворенные вещества
делят на поверхностно-активные (da/dc < 0) и поверхностно-инактив-
ные (da/dc > 0). Поверхностно-активные вещества (ПАВ) характери-
зуются поверхностной активностью g, которая является мерой способ-
ности вещества понижать поверхностное натяжение на границе раздела
фаз. Эта величина численно равна производной da/dc, взятой с обрат-
ным знаком, при стремлении концентрации ПАВ к нулю:
с Поверхностная активность ПАВ графически определяется как тан-
генс угла наклона ос касательной, проведенной к изотерме поверхност-
ного натяжения в точке ее пересечения с осью ординат, взятый со зна-
ком минус.
Поверхностное натяжение зависит также от электрического потен-
циала поверхности. Термодинамическими соотношениями между по-
верхностной и электрической составляющими энергии являются урав-
нения Липпмана:
do/dty = — qs — первое уравнение Липпмана (I. 23)
П2<т/Лр2 =— С — второе уравнение Липпмана (1-24)
где (р—-электрический потенциал; qs — плотность заряда; С — емкость двойного элек-
трического слоя на поверхности.
Графическую зависимость поверхностного натяжения от электриче-
ского потенциала называют электрокапиллярной кривой. Она имеет
вид параболы, максимум которой отвечает потенциалу нулевого заряда.
Экспериментальная электрокапиллярная кривая позволяет с помощью
дифференцирования получить значения плотности заряда (1.23) и ем-
кости двойного электрического слоя (1.24).
10
Работа 1. ОПРЕДЕЛЕНИЕ ПОЛНОЙ ПОВЕРХНОСТНОЙ
ЭНЕРГИИ ЖИДКОСТЕЙ
Цель работы: измерение поверхностного (межфазного) натяжения;
определение зависимости поверхностного натяжения чистых жидкостей
от температуры; расчет полной поверхностной энергии и ее состав-
ляющих.
В соответствии с уравнениями (1.8) и (1.9) полная поверхностная
энергия Us содержит две составляющие qs и о. С повышением темпе-
ратуры поверхностное натяжение о уменьшается, а теплота образова-
ния единицы поверхности qs увеличивается. Это объясняется тем, что
с повышением температуры расстояние между молекулами в жидких
телах увеличивается и соответственно равнодействующая межмолеку-
лярных сил (и, следовательно, поверхностное натяжение) уменьшается.
Вместе с тем с ростом разрыхленности поверхностного слоя увеличи-
вается его энтропия. При линейной зависимости поверхностного натя-
жения от температуры, что обычно наблюдается для большинства жид-
костей, поверхностное натяжение уменьшается на величину Tdo/dT,
тогда как энтропийная составляющая qs увеличивается. Таким обра-
зом, полная поверхностная энергия для таких систем является темпе-
ратурным инвариантом.
Для определения полной поверхностной энергии необходимо знать
поверхностное натяжение и его температурный коэффициент.
Наиболее доступными для экспериментального измерения поверх-
ностного натяжения являются системы жидкость — газ и жидкость —
жидкость. Существующие методы дают возможность измерять о при
неподвижной межфазной поверхности (статические) и при движущейся
поверхности раздела (динамические). Недостатком динамических ме-
тодов является сложность их аппаратурного оформления. Кроме того,
для надежного измерения поверхностного натяжения растворов, и, в
частности, растворов ПАВ, необходимо их выдерживать определенное
время для установления равновесия в поверхностном слое.
На практике наиболее часто используют статические или полуста-
тические методы, позволяющие измерять равновесные значения по-
верхностного натяжения жидкостей. К статическим относятся методы
капиллярного поднятия жидкости и висячей (лежащей) капли. Полу-
статическими являются методы максимального давления в капле (пу-
зырьке), отрыва кольца или пластины и сталагмометрический метод.
Метод капиллярного поднятия. В основе метода лежит
'зависимость высоты поднятия жидкости h в' узком капилляре от ее
поверхностного натяжения. В соответствии с уравнением Лапласа из-
быточное давление связано с высотой h жидкости в капилляре соотно-
шениями
. 2<Т , . . ~
Др =— 1 (1.25)
г м
Др = i\ogh (I. 26)
где г№— радиус кривизны мениска жидкости в капилляре; Др — разность плотностей
жидкости и газовой фазы; g — ускорение свободного падения.
Вводя так называемую капиллярную постоянную а
а2 = 2о/(Др§; = гм/г (I. 27)
и учитывая угол смачивания 0 жидкостью стенок капилляра радиу-
сом г, из уравнений (1.25) и (1.26) получаем:
i\r>grh a2 kpg
2 cos 0 2 cos 0
(1.28)
Последнее соотношение известно как уравнение Жюрена. Таким
образом для определения поверхностного натяжения жидкостей этим
методом экспериментально находят высоту поднятия h, радиус капил-
ляра г и угол смачивания 0. Метод капиллярного поднятия является
одним из наиболее точных (относительная погрешность менее 0,01 %).
£Метод максимального давления в пузырьке основан
на измерении давления, при котором происходит отрыв пузырька газа
(воздуха), выдуваемого в жидкость через капилляр.
При медленном продавливании пузырька из капилляра в жидкость
в нем возникает избыточное внутреннее давление Ар, которое согласно,
закону Лапласа определяется поверхностным натяжением ож-г и кри-
визной поверхности пузырька [см. уравнение (1.25)]. Радиус кривизны
/м изменяется по мере продавливания пузырька в жидкость. Из рис. 1
видно, что в начальный момент пузырек имеет большой радиус кри-
визны и поверхность его почти плоская (гм^>г). Со временем радиус
кривизны уменьшается, пузырек становится все более выпуклым и при.
гм=г избыточное давление внутри пузырька достигает максимального
значения Армакс. Это давление соответствует внешнему давлению в ка-
пилляре. Для дальнейшего увеличения размера пузырька не требуется'
повышение внешнего давления, поскольку с ростом пузырька внутрен-
нее давление в нем в соответствии с уравнением Лапласа уменьшается.
В результате воздух, находящийся в трубке, устремляется к сформиро-
вавшемуся пузырьку и приводит к его отрыву от капилляра. Таким
образом, определение поверхностного натяжения рассматриваемым ме-
тодом сводится к измерению внешнего давления, равного Армакс.
При определении поверхностного натяжения методом максимального
давления в пузырьке следует также учитывать гидростатическое дав-
ление слоя жидкости, находящейся над ним. Однако, если глубина по-
гружения капилляра в жидкость незначительна и радиус г мал, по-
правкой на это давление можно пренебречь.
Максимальное давление в пузырьке в простейшем варианте можно
измерить с помощью прибора Ребиндера (рис. 2). Прибор состоит из
измерительной ячейки 4 с капилляром 3, аспиратора 1, с помощью ко-
торого создают внешнее давление, и микроманометра.
Измерение АЛиакс проводят следующим образом. Исследуемую жид-
кость наливают в ячейку 4 до уров-
ня, при котором кончик капилляра 3
погружается в нее не более чем на
1 мм (избыток жидкости отбирают
с помощью капилляра). Ячейку сое-
диняют отводной трубкой 2 с аспи-
ратором 1 и краном 5 микромано-
метра.
— Рис. 1. Схема, иллюстрирующая формиро-
вание пузырька воздуха на выходе из ка-
пилляра.
12
2
Рис. 2. Схема установки для определения поверхностного иатяжеиия методом макси-
мального давления:
/ — аспиратор; 2 — соединительная трубка; 3— капилляр; 4 — ячейка; 5 — трехходовой край; 6—регуля-
тор уровня манометрической жидкости; 7 — кронштейн; 8 — манометрическая трубка.
Манометр устанавливают в горизонтальном положении с помощью
регулировочных ножек (установка производится по уровню микрома-
нометра) .
Вращая регулятор уровня 6 манометрической жидкости, устанав-
ливают мениск в манометрической трубке против нулевой отметки.
Кран 5 поворачивают по часовой стрелке, соединяя микроманометр
с системой разрежения. Открывают кран аспиратора 1. В установке
создается разрежение, в результате чего манометрическая жидкость
поднимается в трубке 8.
Сформировавшийся на конце капилляра 3 пузырек воздуха при до-
стижении Дрмакс, пробивая поверхностный слой, лопается. В этот мо-
мент давление в системе снижается и манометрическая жидкость начи-
нает опускаться, но затем в результате образования нового пузырька
она снова поднимается. Таким образом, уровень манометрической жид-
кости все время колеблется. Чтобы уменьшить пульсацию жидкости
в измерительной трубке, добиваются равномерного проскока пузырь-
ков, с интервалом 20—30 с. Время образования и отрыва пузырьков
воздуха регулируют путем изменения скорости вытекания воды из ас-
пиратора. Если показание манометра ДрмаКс в течение 2—3 мин не из-
меняется, то его считают установившимся и записывают в журнал.
Чтобы исключить трудоемкую операцию по измерению радиуса ка-
пилляра, для определения поверхностного натяжения используют
относительный метод. Для этого находят константу ячейки k, которую
рассчитывают по значениям максимального давления Дрст и поверх-
ностного натяжения <гст для стандартных жидкостей:
k = аст/Дрст (1.29)
Определив коэффициент k и измерив давление Др макс для исследуе-
мой жидкости, рассчитывают значение ож.г — k&pMaKc.
Метод максимального давления может быть использован и для из-
мерения межфазного натяжения на границе раздела жидкость —
13
Рис. 3. Схема установки для определения
межфазного натяжения:
а — схема ячейки для измерения межфазного натя-
жения; б—схема прибора УГАЗП-1 КТ; 1 — мано-
метрическая трубка; 2—приемник; 3—капилляр;
4 — резервуар; 5 — трехходовой кран; 6 — баллон;
7—электропечь.
жидкость аж.ж. Например, аж.ж можно определить с помощью универ-
сального газового прибора УГАЗП-1 КТ, предложенного П. П. Пугаче-
вичем. Упрощенная схема этого прибора приведена на рис. 3. При на-
гревании воздуха в баллоне 6 под действием возникающего избыточного
давления Др жидкость с плотностью pi выдавливается из капилляра 3
в жидкость с плотностью р2 (р2 > Р1).
В момент достижения максимального избыточного давления Дрмакс
условия механического равновесия на срезе капилляра 3 радиуса г
и на границе раздела фаз в манометрической трубке 1 могут быть вы-
ражены соответственно уравнениями (1.30) и (1.31):
2 ст _
Р + ЛРмакс + = р + gpi/l, + gp2/l2 -1--- (I. 30)
Р + Армакс + gPi (Н + h) — р + gpihx + gp2 (Л2 + h) (1-31)
где p, H, h, hi и hi — см. рис. 3, a.
Из совместного решения уравнений (1.30) и (1.31) получаем вы-
ражение для расчета межфазного натяжения:
стж-ж= W(Pz —Pi)Ar (1.32)
Таким образом, измерив максимальное смещение границы раздела
фаз h в манометрической трубке относительно среза капилляра, можно
рассчитать межфазное (и поверхностное — при продавливании воздуха)
натяжение. Важным преимуществом этого метода является то, что в
расчетное уравнение (1.32) не входит краевой угол.
Измерения проводят следующим образом. В тщательно промытую
и высушенную измерительную ячейку наливают необходимое количе-
ство жидкостей с известными плотностями р! и р2. Ячейку укрепляют
в воздушном термостате. Через некоторое время включают печь 7 и
нагревают воздух в баллоне 6. С помощью крана 5 соединяют баллон
с резервуаром 4. Под действием давления Др жидкость из манометри-
ческой трубки 1 перемещается в приемник 2. Одновременно на срезе
14
капилляра 5 формируется капля. Как только от капилляра 5 оторвется
первая капля жидкости, кран 5 поворачивают, отсоединяя измеритель-
ную систему от баллона 6. При достигнутом избыточном давлении про-
давливание капель продолжается. После отрыва 3-4 капель измеряют
при помощи катетометра расстояние между срезом капилляра и гра-
ницей раздела фаз в манометрической трубке 1. Измерения проводят
несколько раз и находят среднее значение h.
Межфазное натяжение рассчитывают по формуле (1.32). Радиус
капилляра определяют по значениям h, измеренных для жидкостей с
известным межфазным натяжением:
а, мДж/м2 а, мДж/м2
Вода — бензол 35,0 Вода — этилацетат 6,8
। Вода — н-гептан 50,2 Вода — бутанол 1,8
(I/ Метод отрыва кольца. Определение поверхностного натяже-
ния методом отрыва кольца сводится к измерению силы F, необходи-
мой для отрыва проволочного кольца от поверхности жидкости. Одним
из основных условий определения о рассматриваемым методом является
полное смачивание кольца исследуемой жидкостью. В этом случае при
отрывании кольца вместе с ним поднимается и столбик жидкости, сила
тяжести которого равна приложенной силе. Отрыву жидкости препят-
ствуют силы поверхностного натяжения. В момент равновесия, когда
внешнее усилие достигает значений сил поверхностного натяжения,
столбик жидкости разрушается, и кольцо отрывается от поверхности
жидкости.
Сила F определяется поверхностным натяжением раствора и раз-
мерами кольца:
F — 2лстж.г/?/ + 2лстж.г (R' + 2г) = 4ляж.г/? (I. 33)
1де R' — внутренний радиус кольца; R — средний радиус кольца; г — радиус прово-
локи, из которой изготовлено кольцо (г R).
Соотношение (1.33) справедливо только в том случае, если вместе
с кольцом поднимается столбик жидкости в виде полого цилиндра пра-
! вильной формы. В реальных условиях поверхность столбика жидкости
; имеет более сложную форму. Согласно исследованиям Гаркинса форма
этой поверхности зависит от отношения куба среднего радиуса кольца
: к объему поднимаемой жидкости в момент отрыва /?3/У и отношения
радиуса кольца к радиусу его сечения R/r. С учетом этого в уравне-
ние (I. 33) должен быть введен поправочный коэффициент р. Тогда
С-34)
Из последнего соотношения следует, что поверхностное натяжение жид-
кости пропорционально силе F, прикладываемой в момент отрыва
кольца:
<тж.г = kF (1.35)
Максимальное усилие F обычно измеряют с помощью прибора —
чувствительных торзионных весов. Схема прибора приведена на рис. 4.
Основной частью прибора является платиновое кольцо 7, подвешенное
к крючку 6 коромысла весов. Равномерное усилие для отрыва кольца
от поверхности исследуемой жидкости создают поворотом барабана 2.
Силу F фиксируют по шкале весов с помощью стрелки 3.
15
Рис. 4. Схема прибора для определения поверх-
ностного натяжения методом отрыва кольца:
1— винт подъема (опускания) столика; 2,5— барабаны; 3 —
стрелка; 4 — шкала; 6 — крючок; 7 — измерительное кольцо;
8 — чашка; 9 — подвижный столик; 10— арретир.
При выполнении измерения необходи-
мо соблюдать следующие условия:
1) плоскость кольца и поверхность жид-
кости должны быть строго параллельны
и горизонтальны; 2) кольцо должно пе-
ремещаться поступательно, медленно под-
нимаясь вверх.
Перед измерениями проверяют пра-
вильность установки весов по уровню.
Кольцо тщательно промывают в хромо-
вой смеси и дистиллированной воде и ос-
торожно высушивают фильтровальной бу-
магой. Подготовленное к работе кольцо
подвешивают на крючок торзионных ве-
сов. Исследуемую жидкость слоем тол-
щиной 5—7 мм наливают в чашку о Ф
чашку устанавливают на подвижный сто-
лик 9. При этом кольцо 7 не должно касаться жидкости, поэтому столик
предварительно опускают вниз до упора с помощью винта 1. Освободив
арретир 10 весов путем поворота его, вращением барабана 2 приводят
левую неподвижную стрелку 3 на нулевое деление шкалы. С помощью
винта 1 столик с чашкой поднимают вверх до касания кольца поверх-
ности жидкости. Затем вращением этого же винта подвижный столик
опускают вниз до тех пор, пока правая подвижная стрелка весов 4 не
достигнет красной черты на правой шкале, что соответствует установке
прибора в рабочее положение.
Поворачивают левый барабан 2 (от себя) и регистрируют положе-
ние стрелкй 3 в момент отрыва кольца от поверхности жидкости.
Для каждого раствора проводят три измерения и берут среднее
значение силы F. Поверхностное натяжение рассчитывают по формуле
(1.35). Коэффициент k находят по данным измерения силы отрыва
кольца для стандартной жидкости с известным поверхностным натя-
жением.
1/1 Сталагмометрический метод. Определение поверхностного
натяжения этим методом заключается в измерении объема или веса
капли жидкости, медленно отрывающейся от кончика капилляра в ниж-
нем конце сталагмометрической трубки. В основе метода лежит поло-
жение о том, что в момент отрыва сила тяжести капли q уравновеши-
вается силами поверхностного натяжения F. Силы поверхностного натя-
жения действуют вдоль окружности шейки капли, и препятствуют ее
отрыву. В момент отрыва можно считать, что
q = F «г 2ЛГ(ТЖ.Г (I. 36)
где г — внутренний радиус капилляра.
Обычно отрыв капель не происходит по линии внутреннего перимет-
ра капилляра сталагмометрической трубки радиусом г, а осуществля-
ется в шейке капли, имеющей меньший радиус. Поэтому для точного
16
определения значения <тж.г в выражении (1.36)' величину г следует
умножить на поправочный коэффициент |3', зависящий от радиуса и
объема капли:
<7 = F = 2л₽'гогж.г (1-37)
Вес капли чаще всего определяют следующим образом. Сталагмо-
метрическую трубку заполняют исследуемой жидкостью определенного
объема V и измеряют число капель п, вытекающих из данного объема.
Вес капли рассчитывают по уравнению
(1.38)
п \
где р — плотность жидкости. —
В связи со сложностью определения радиуса капилляра г и коэф-
фициента |3Z поверхностное натяжение находят путем сравнения дан-
ных по истечению из сталагмометрической трубки исследуемой жидко-
сти и жидкости с известным поверхностным натяжением. Значение <тж-г
рассчитывают по формуле
где аС1, рст и «ст — значения сг, р и п для стандартной жидкости.
Число капель п и пст можно регистрировать автоматически с по-
мощью счетчика, установленного на сталагмометре. Рассчитывают сред-
нее значение п числа капель из пяти измерений. Измерения проводят
в условиях медленного формирования капель (примерно 1—3 капли
в 1 мин), при этом скорость истечения жидкостей поддерживают по-
стоянной. Перед началом работы для удаления загрязнений из капил-
ляра сталагмометрическую трубку несколько раз промывают хромовой
смесью и водой.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
i Для проведения работы необходимы:
Прибор для определения поверхностного натяжения.
j Термостат.
[ Пипетки емкостью 1 и 10 мл.
Исследуемая и стандартная (вода) жидкости.
j Измеряют поверхностное натяжение исследуемой жидкости при не-
\ скольких значениях температуры (исследуемую жидкость, интервал
!температур и метод определения сж-г указывает преподаватель). Пе-
! ред измерениями проводят термостатирование жидкостей при каждом
! значении температуры.
• По данным измерений строят график зависимости сж-г = f(T) и по
[ тангенсу угла наклона полученной прямой находят значение темпера-
турного коэффициента ds/dT. Для каждой температуры по уравне-
ниям (1.8) и (1.9) рассчитывают qs и Us и делают вывод о влиянии
[температуры на термодинамические параметры поверхностного слоя
; ЖИДКОСТИ.
, Зак. 673
Таблица 1.1. Экспериментальные и расчетные данные для вычисления полной
поверхностной энергии жидкости
Температура, К Измеряемый параметр для расчета ^ж-г (F или Др) Поверхност- ное натяжение аж.г, Дж/м2 Температур- ный коэффициент dddT Теплота образования единицы поверхности qs, Дж/м2 Полная поверхностная энергия Us, Д,ж/ъл^
Результаты измерений оформляют в виде таблицы (см. табл. I. 1).
Работа 2. ИССЛЕДОВАНИЕ АДСОРБЦИИ НЕЭЛЕКТРОЛИТОВ
ИЗ БИНАРНЫХ РАСТВОРОВ НА ТВЕРДЫХ ПОВЕРХНОСТЯХ
Цель работы: определение гиббсовской (избыточной) адсорбции на
твердом адсорбенте из бинарных растворов; расчет гиббсовской адсорб-
ции компонентов раствора по изменению состава раствора при адсорб-
ции; построение изотерм избыточных величин адсорбции и их анализ.
При адсорбции из разбавленных растворов можно принять, что ак-
тивность растворителя не меняется. При адсорбции из концентрирован-
ных растворов следует учитывать адсорбцию и растворителя, и раство-
ренного вещества.
Об адсорбции из раствора обычно судят по изменению его состава,
который определяют до и после адсорбции одним из аналитических
методов. В концентрированных растворах неэлектролитов концентра-
цию компонентов удобнее выражать в мольных долях х.
Если твердый адсорбент массой m приводится в контакт с раство-
ром, содержащим п0 молей растворителя и растворенного вещества, то
в результате адсорбции А[ молей компонента 1 (растворитель) и Л2
молей компонента 2 (растворенное вещество) будут находиться на по-
верхности. Здесь А[ и А2 — моли, приходящиеся на единицу массы твер-
дого сорбента. При равновесии в жидкой фазе остается п\ и и2 молей
компонентов 1 и 2.
Тогда общее число молей в системе составит
п^ = п.\ п2 A\in А2т (I. 40)
Введем обозначения: xoi и л'О2—мольные доли компонентов 1 и 2
до адсорбции, которые составят
tii A- Aim
Toi =-------------------------,
По
и х2 — мольные доли компонентов 1
п2 + А2т
Хо2 ----------
По
и 2 после адсорбции, равные
(1-41)
П1 . «2
-----i-----> Х2 1 — Xi — -----------:----
Щ + tl2 til + п2
(I- 42)
Изменение мольной доли компонента 2 в растворе в результате
адсорбции Дх2 можно выразить следующим образом:
п2 + А2т
Л г — г _ г__________1 ____________«2 _ A2nitn - Ain2m
2 02 2 «1 + «2 + Aim + A2m nt + n2 («i + «2) «о
или
n0 Ax2 _
m
A2X1 —
(I. 44)
18
Левая часть уравнения (1.44) представляет выражение гиббсовской
адсорбции, и поэтому
Г2 = -^-^- = Л2Х! - А,х2 (1.45)
т
Изотермы, соответствующие уравнению (1.45), называют изотермами
состава. Для анализа уравнения (1.45) его удобно записать в виде
Г2 = Х|%2 (Л2/х2 — Л1/%1) (I. 46)
Если A2/x2>Ai/xi, то Гг > 0, т. е. гиббсовская, или избыточная,
адсорбция компонента 2 положительна и, наоборот, при А2/х2 < А\/х\
Гг < 0, т. е. гиббсовская адсорбция компонента 2 отрицательна. Так
как перед скобкой в уравнении (1.46) стоит произведение XiX2, то кри-
вая для гиббсовской адсорбции будет иметь максимум (Г2 > 0) или
минимум (Гг<0).
Если Л1/Х1~Лг/х2, то возможно, что гиббсовская адсорбция одного
из компонентов при малых концентрациях имеет один знак, а с ростом
концентрации может изменить знак на обратный. В точке пересечения
изотермы с осью состава будет соблюдаться условие
Л1/Х, == Л2/х2 или Г1 = Г2==0
которое отвечает адсорбционной азеотропии, т. е. отсутствию разделе-
ния веществ.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
1 Для проведения работы необходимы:
Прибор для определения концентрации растворов по показателю
преломления (интерферометр или рефрактометр).
Мерные колбы емкостью 50 мл.
Конические колбы с притертыми пробками емкостью 100 мл.
Бюретки емкостью 50 мл, пипетки емкостью 25 мл.
Адсорбент, например, активный уголь.
Взаимно смешивающиеся жидкости, например, толуол — этанол.
В мерных колбах готовят 10 растворов двух жидкостей, изменяя
соотношение компонентов от чистой жидкости 1 до чистой жидкости 2.
Из приготовленных растворов отбирают пипеткой по 25 мл, переносят
в конические колбы и прибавляют в каждую колбу определенную на-
веску сорбента, например, 3 г активного угля на 25 мл раствора. Колбы
закрывают притертыми пробками и оставляют на 1 ч, периодически
взбалтывая содержимое для установления адсорбционного равновесия.
В исходных растворах и в растворах после адсорбции определяют по-
казатели преломления и nf.
Порядок расчета величины гиббсовской адсорбции компонента 2
следующий. Рассчитывают число молей каждого компонента nOi и н02
в исходной смеси, общее число молей обоих компонентов и мольные
доли каждого компонента в исходной смеси до адсорбции хоь х02-
Р1Р1 Р2р2 I /т л'гх
п01 = — п°2 = ~м— п° ~ п°' + п°2 Р-
М 1 /И 2
Хо, =------- ХО2=--------------- (1.48)
«01 + П02 Пах + Пц2
где Vj, Г2 — объемные количества компонентов 1 или 2 в растворе; pi. рг — плотности
исходных жидкостей; Mi, М2 — молекулярные массы компонентов 1 и 2.
2* jg
Таблица I. 2. Данные для расчета гиббсовской адсорбции
Номер колбы И- мл мл d «0 d nl n0l П02 no X0l X02 X2 * 11 । , СЧ O') I Ц о < * 1 " 1 Г2 _ ДХ2П0
tn Моль/г
1 2
10
По полученным данным строят калибровочный график показателя
преломления бинарного раствора от его состава (мольной доли ком-
понента 2).
Из калибровочного графика определяют состав раствора после ад-
сорбции, т. е. мольные доли и х2 компонентов (xi -j- х2 — 1).
Рассчитывают разность между мольными долями каждого компо-
нента до адсорбции и после адсорбции:
Дх1 = х01 — xi
Дх2 = *02 — хг (I. 49)
Знак при значении Ах указывает на отрицательную или положитель-
ную величину гиббсовской адсорбции.
По формуле (1.45) рассчитывают гиббсовскую адсорбцию и строят
изотермы состава раствора при адсорбции в координатах Г2 = /(хг)
или Г1 = /(х1). Анализируют полученные данные. Экспериментальные
и расчетные данные записывают в таблицу (см. табл. 1.2).
Работа 3. ИССЛЕДОВАНИЕ ВЛИЯНИЯ ПОВЕРХНОСТНО-
АКТИВНЫХ ВЕЩЕСТВ НА СМАЧИВАНИЕ И АДГЕЗИЮ
Цель работы.: оценка работы смачивания и работы адгезии; изучение
влияния адсорбции ПАВ на смачивание твердых поверхностей мето-
дом измерения краевых углов.
Количественно адгезия оценивается работой адгезии IFa, которая
соответствует работе, затрачиваемой на разрыв межфазного соедине-
ния и приходящейся на единицу площади поверхности. Ее можно рас-
считать по уравнению (I. 12) или (I. 14).
Смачивание количественно характеЩ1зуют краевым углом 0 или
-cos 0. Краевой угол 0 определяется как угол между касательной, про-
веденной к поверхности смачивающей жидкости, и смачиваемой по-
верхностью твердого тела, при этом он всегда отсчитывается от каса-
тельной в сторону жидкой фазы. Касательную проводят через точку
соприкосновения трех фаз.
Различают равновесные и неравновесные (динамические и статиче-
ские) краевые углы. Равновесные краевые углы 0Р определяются только
значениями поверхностных натяжений на границах раздела всех трех
20
фаз. Равновесию отвечает минимум поверхностной энергии Гиббса си-
стемы, поэтому для каждой системы при данных внешних условиях 0Р
имеет только одно значение. Неравновесных краевых углов может быть
множество, так как они измеряются в условиях отклонения системы
от состояния термодинамического равновесия.
Равновесные краевые углы, рассчитанные на основе баланса сил,
действующих по периметру смачивания, определяются уравнением
Юнга (1.13). Если поверхностное натяжение на границе твердое те-
ло— газ (Тт-г больше, чем поверхностное натяжение на границе твердое
тело — жидкость <гт.ж, то краевой угол 0Р < 90°, поверхность твердого
тела является лиофильной (при смачивании водой — гидрофильной).
К материалам с гидрофильной поверхностью относятся, например,
кварц, стекло, оксиды металлов. Жидкость не смачивает поверхность,
если <Тт-г < От-ж и 0Р > 90°. В этом случае поверхность является лио-
фобной (гидрофобной). К материалам с гидрофобной поверхностью
относятся металлы, у которых поверхность не окислена, большинство
полимеров, а также все органические соединения, обладающие низкой
диэлектрической проницаемостью.
Явления адгезии и смачивания тесно связаны между собой. Влия-
ние смачивания на адгезионное взаимодействие отражает уравнение
(I. 14). Из этого уравнения видно, что чем лучше смачивание (меньше
0Р), тем больше работа адгезии. Максимальные значения могут
реализоваться лишь при достижении полного смачивания поверхности
твердого тела, когда cos 0Р = 1 и 1Еа = 2ож-г = 1ЕК, (где Wk— работа
когезии для смачивающей жидкости).
Таким образом, смачивающая способность жидкостей и адгезион-
ное взаимодействие их с твердыми телами в основном определяются
природой веществ, составляющих контактирующие фазы. Сопоставле-
ние уравнений (I. 12) и (I. 14) показывает, что высокая адгезия между
фазами может реализоваться лишь при определенном соотношении зна-
чений <гт.г и стж-г (щ-г > сГж-г)- Решающее значение при этом играет со-
стояние поверхности твердого тела и его поверхностное натяжение.
Природу поверхности твердого тела, а значит, и характер контакт-
ного взаимодействия его со смачивающей жидкостью можно изменить
путем модифицирования поверхности, например, проводя ее химиче-
скую обработку. Один из широко распространенных способов измене-
ния состава поверхностных слоев основан на адсорбции на них поверх-
ностно-активных веществ. Адсорбционное модифицирование твердых
поверхностей проводят либо из водных растворов, либо из органических
растворителей. В результате адсорбции ПАВ изменяется не только зна-
чение 0Р, но и скорость растекания смачивающей жидкости.
Влияние ПАВ на смачивание зависит от того, на какой поверхности
раздела фаз они адсорбируются, Молекулы ПАВ могут адсорбиро-
ваться как на твердой поверхности, так и на границе раздела жид-
кость— газ. Если поверхность твердого тела гидрофобная, то из вод-
ных растворов ПАВ адсорбируются и на твердой поверхности, и на
границе раствор — воздух. На межфазной поверхности молекулы ПАВ
располагаются в соответствии с правилом уравнивания полярностей
Ребиндера. В результате значения стт-ж и аж.г уменьшаются и согласно
уравнению (I. 13) поверхность твердого тела смачивается лучше. С уве-
личением адсорбции ПАВ твердая поверхность становится менее гидро-
фобной, происходит так называемая гидрофилизация поверхности.
21
а
с
Рис 5. Схема, поясняющая
определение краевых углов:
а — для углов 0<9О°; б — для
углов 0>9О°.
В результате адсорб-
ции ПАВ может вооб-
ще произойти переход
от несмачивания к сма-
чиванию, т. е. инверсия
смачивания. Если по-
гидрофильная, жидкость хорошо смачивает ее
верхность твердого тела
и адсорбция ПАВ на ней, как правило, не происходит. В этом случае
изменение значений 0Р связано в основном с уменьшением поверхност-
ного натяжения на границе жидкость — газ.
Об изменении характера взаимодействия твердой поверхности со
смачивающей жидкостью в результате адсорбции ПАВ можно судить
по работе смачивания Ц7СМ. Работа смачивания определяется как раз-
ность От-г — Ст-ж. Поскольку достаточно надежных методов измерения
поверхностного натяжения на границе с твердыми телами нет, для
расчета ТРСМ удобнее использовать следующее уравнение:
и7сМ = 0Гж-гС0зеР (1-50)
Нетрудно заметить, что работа смачивания в зависимости от знака
косинуса краевого угла может быть либо положительной, либо отрица-
тельной. Отсюда следует, что в зависимости от ориентации молекул
ПАВ на межфазной поверхности при адсорбции может измениться не
только абсолютная величина работы смачивания, но и ее знак.
Краевой угол, входящий в формулы для расчета работы адгезии
и смачивания, находят по основным размерам капель жидкости, нано-
симых на твердые поверхности: высоте h и диаметру основания d
(рис. 5). Значения cos 0 рассчитывают по формуле
cos9 = ..w2.)2..-.^ \/
(d/2)2 + h2
При d/2 < h можно использовать более простую формулу
cos9=1--S72
(1-51)
(1.52)
Параметры капли Ли d измеряют с помощью установки (рис. 6),
основными узлами которой являются катетометр (типа КМ.-6), измери-
тельная ячейка-кювета и осветительное устройство, обеспечивающее
контрастное изображение капли и исследуемой поверхности.
Измерения проводят следующим образом. Измерительную ячейку —
кювету 8 устанавливают на металлический держатель 9. На подстав-
ку 16 в ячейке помещают исследуемую пластинку и, повернув тумблер
трансформатора 13, включают лампу осветителя 12. Лампу следует
включать только на время измерения. Отворачивают винт 2 на задней
стороне каретки 4 катетометра и каретку устанавливают таким обра-
зом, чтобы объектив 6 находился примерно на уровне пластинки. За-
крепив измерительную каретку винта 2, с помощью микрометрического
винта 1 проводят более точную установку зрительной трубы 5 по вер-
22
тикали. После этого, перемещая фокусирующим винтом 7 препарато-
водителя вперед или назад держатель 9, а также перемещая его вправо
или влево винтом 10, добиваются резкого изображения профиля пла-
стинки в окуляр-микрометре 3.
Затем в кольцевое углубление кюветы наливают 1 мл исследуемой
жидкости, с помощью микрошприца или пипетки осторожно наносят
каплю этой жидкости на поверхность пластинки у самого ее края, об-
ращенного в сторону объектива, и кювету закрывают крышкой 15. Для
(получения воспроизводимых результатов необходимо наносить капли
жидкости примерно’одинаковых размеров, так чтобы диаметр их осно-
вания не превышал 2—3 мм.
При исследовании смачивания поверхностей водой измерения крае-
вых углов можно начинать через несколько минут после нанесения
капли воды. При исследовании смачивания растворами ПАВ капли
жидкости на пластинке следует выдерживать предварительно 15—•
20 мин (это связано с довольно медленным формированием адсорб-
ционных слоев ПАВ на межфазных границах раздела).
С помощью винтов 1, 7, 10 окончательно регулируют положение
держателя для получения наиболее резкого изображения контуров
капли и пластинки. При этом нужно учесть, что контрастность изобра-
жения зависит также и от степени освещенности ячейки, регулировку
которой осуществляют поворотом рукоятки трансформатора 14 (целе-
сообразно работать при небольшом накале лампы осветителя) и ири-
совой диафрагмы 11.
Параметры h и d определяют с помощью окуляр-микрометра 3.
В фокальной плоскости окуляра винтового окуляр-микрометра уста-
новлены: неподвижная окулярная шкала, разделенная на 8 делений,
и подвижная шкала с перекрестием и индексом в виде двух параллель-
ных штрихов, расположенных точно над перекрестием. Подвижная
шкала приводится в движение вращением барабана микрометрического
Рис. 6. Схема установки для измерения краевых углов (а) и ячейки-кюветы (б):
1,7,10 — регулировочные винты; 2 —винт подъема (опускания) каретки; 3 — окуляр-микрометр; 4— карет-
ка; 5 — зрительная труба; 6 — объектив; 8 — измерительная ячейка; 9— держатель; 11 — диафрагма
осветителя; 12 — осветитель; 13— тумблер включения трансформатора; 14 — рукоятка для рег\ли>ова-
ния напряжения; 15— крышка кюветы; 16—подставка.
23
винта. При повороте барабана на один оборот перекрестие и располо-
женные над ним штрихи перемещаются в поле зрения окуляра ровно
на одно деление неподвижной шкалы. Шкала барабана микровинта раз-
делена на 100 частей. Измеряемая величина h или d определяется по
делениям неподвижной шкалы и делениям шкалы барабана. Например,
двойной штрих в поле зрения расположен между делениями 1 и 2, а
индекс барабана находится против деления 18 шкалы барабана, сле-
довательно, показание окуляр-микрометра составляет 118 делений.
С помощью винтов /, 7 и 10 добиваются такого положения капли,
чтобы вся она располагалась в поле зрения окуляра между делениями
0 и 8. Для измерения диаметра основания капли вращением микромет-
рического винта катетометра 1 перемещают перекрестие окуляр-микро-
метра по вертикали и совмещают перекрестие с изображением пла-
стинки (с границей раздела капля-—поверхность пластинки). Вращая
барабан окуляр-микрометра 3, подводят центр перекрестия к левой
крайней точке капли (точка А на рис. 5) и по шкалам микрометра
отсчитывают число делений п\, отвечающих ее положению в окуляр-
микрометре. Затем, вращая барабан против часовой стрелки, совме-
щают перекрестие с правой крайней точкой капли (точка В, рис. 5),
фиксируя ее положение по шкалам микрометра (щ) . Вычисляют раз-
ность Дп==П2 —П1, которая определяет диаметр основания капли d.
При определении высоты капли перекрестие окуляра первоначально
совмещают с серединой основания капли путем перемещения его до |
положения барабана, соответствующего делению (nt + п2) /Ъ. Затем
окуляр-микрометр поворачивают вокруг тубуса на 90° и с помощью
барабана центр перекрестия подводят к вершине капли (точка С,
рис. 5). Записывают показания по шкалам микрометра. После этого '
вращением барабана перекрестие совмещают с изображением границы -
раздела поверхность пластинки — жидкость. Высоту h находят по
разности показаний (п2— «1).
Примечание. При исследовании смачивания большое значение имеет чистота ;
пластинок, а также посуды, с помощью которой готовят растворы. Даже небольшое :
загрязнение может существенно повлиять на результаты измерений. Поэтому пластин-
ки полимера следует брать аккуратно только за угол или ребро, а пипетки при смене
раствора необходимо каждый раз тщательно промывать.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Прибор для определения краевых углов.
Прибор для измерения поверхностного натяжения жидкости. ‘
Колбы емкостью 50 мл. !
Микрошприц или пипетка с капиллярным кончиком.
Пластинки полимера, например, фторопласта. .
Раствор ПАВ, например, олеата натрия.
Готовят 5—6 водных растворов ПАВ различной концентрации (по- .
верхностно-активное вещество, с которым проводят работу, и концен-
трации растворов указывает преподаватель). Измеряют поверхностное j
натяжение аж-г приготовленных растворов (методика определения <гж.г :
приведена в работе 1) и краевые углы 0 на границах раздела пла- '
стинка полимера — раствор ПАВ — воздух и пластинка полимера — ;
вода — воздух с помощью установки, описанной выше.
24
Таблица I. 3. Экспериментальные и расчетные данные определения работы
смачивания и адгезии
как среднее арифметическое. По формулам (1.14) и (1.50) рассчиты-
вают работу смачивания ИДМ и адгезии 1Га для каждого раствора.
Экспериментальные и расчетные данные записывают в таблицу (см.
табл. I. 3).
Строят изотерму смачивания cos 0 = f (сПАВ) и кривую зависимости
работы смачивания от концентрации, по ним определяют, при какой
концентрации ПАВ поверхность исследуемого полимера становится
гидрофильной, т. е. находят точку инверсии смачивания.
Работа 4. ОПРЕДЕЛЕНИЕ КРИТИЧЕСКОГО НАТЯЖЕНИЯ СМАЧИВАНИЯ
НЕПОЛЯРНЫХ ПОЛИМЕРОВ
Цель работы-, изучение влияния поверхностного натяжения жидкостей
на смачивание твердых тел; определение поверхностного натяжения
полимерных материалов и оценка для них работы адгезии.
Экспериментальное определение поверхностного натяжения жидко-
стей обычно не вызывает затруднений. Достаточно надежных методов
определения поверхностного натяжения на границе твердое тело — газ
оу.г и твердое тело — жидкость от.ж нет, поэтому для оценки их значе-
ний используют косвенные методы. Один из таких методов заключается
в определении критического поверхностного натяжения смачивания <ткр.
Критическое поверхностное натяжение <ткр находят по зависимости ко-
синуса краевого угла натекания cos0НТ (0НТ— статический краевой
угол, образующийся при натекании жидкости на твердую поверхность)
от поверхностного натяжения жидкости на границе с воздухом <тж.г.
Для многих твердых (низкоэнергетических) поверхностей при умень-
шении поверхностного натяжения смачивающей жидкости косинус
краевого угла cos 0НТ линейно увеличивается. Это позволяет экстрапо-
лированием прямой cos 0НТ = f (сж-г) до пересечения с прямой
cos0HT=l, параллельной оси абсцисс, находить значение <тж.г, при
котором достигается полное смачивание данной твердой поверхности.
Полученное значение ож.г является критическим поверхностным натя-
жением смачивания.
Величина <ткр практически не зависит от природы и свойств смачи-
вающих жидкостей, и в основном определяется свойствами смачивае-
мого твердого тела. Можно принять, что <ткр соответствует его поверх-
ностному натяжению, т. е. <тКР = ат.г. Это положение подтверждается
правилом Антонова, согласно которому межфазное натяжение на
25
границе двух песмешивающихся жидкостей равно разности поверхност-
ных натяжений взаимно насыщенных жидкостей на границе их с воз-
духом. Применительно к взаимодействию жидкости с твердой поверх-
ностью, когда она полностью смачивается, согласно этому правилу по-
лучаем:
М-ж = "ж-r - ат-1 d-53)
В то же время из уравнения Юнга (I. 13) при условии полного
смачивания твердой поверхности (cos 0 = 1) имеем:
°т-ж = °т-г — Н-г <L 54)
Следовательно, при полном смачивании твердого тела:
ат-г стж-г
и (1.55)
С<Р М-г
Следует отметить, что равенство (I. 55) соблюдается не всегда, по-
скольку для его выполнения необходимо, чтобы межфазное натяжение
оу-ж было равно нулю или хотя бы Стт-г и стж.г значительно превышали
значение стт-ж- Как правило, это условие реализуется лишь для низко-
энергетических поверхностей, а для высокоэнергетических поверхно-
стей не выполняется. Поэтому для них корреляции между значениями
поверхностных натяжений оц-г и огкр не наблюдается.
Определив критическое поверхностное натяжение смачивания о'кр,
можно также рассчитать работу адгезии W3. При линейной зависимо-
сти cos 0m- = f (ож-г) можно записать, что
cos 01И. — ! — Ь (<тж.г — о'кр) (I. 56)
где b — тангенс угла наклоны прямой cos Оцт = / (ож.г) к осн абсцисс.
Если твердая поверхность достаточно однородна и практически не
имеет шероховатостей, экспериментально измеряемые статические углы
0цТ близки к равновесным краевым углам 6Р. Поэтому в соответствии
с уравнениями (I. 14) и (1.56) имеем:
1Еа = <тж.г(2 + 6оК]))-6о^г (1-57)
Уравнение (1.57) для Wa представляет собой уравнение параболы,
вершина которой находится при стж.г = 1/& + 0,5стКр- Таким образом,
максимальная работа адгезии равна
+ (1.58)
Соотношения (1.57) и (1.58) хорошо согласуются с эксперименталь-
ными данными для многих систем.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Установка для измерения краевого угла.
Прибор для измерения поверхностного натяжения жидкости.
Конические колбы с притертыми пробками емкостью 50 мл.
Микрошприц или пипетка с капиллярным кончиком.
26
Пластинки полимера, например фторопласта, политрифторэтилена
или полиэтилена.
Жидкости с различным поверхностным натяжением.
Определяют краевые углы 0НТ при смачивании полимера чистыми
жидкостями с различным поверхностным натяжением. Подготовку ис-
следуемых полимерных пластинок и определение краевых углов про-
водят по методике, приведенной в работе 3. Краевой угол для каждой
жидкости измеряют три раза и значение 0НТ находят как среднее ариф-
метическое.
Затем измеряют поверхностное натяжение исследуемых жидкостей
одним из методов, приведенных в работе 1. Поверхностное натяжение
жидкостей также находят как среднее значение из результатов трех
параллельных измерений.
На основе полученных данных строят зависимость cos 0НТ = f (<тж-г
и экстраполяцией прямой до cos0HT=l определяют сгКр. По формуле
(1.57) рассчитывают значения работы адгезии Wa между полимером и
исследуемыми жидкостями и строят кривую Wa = f (аж-г).
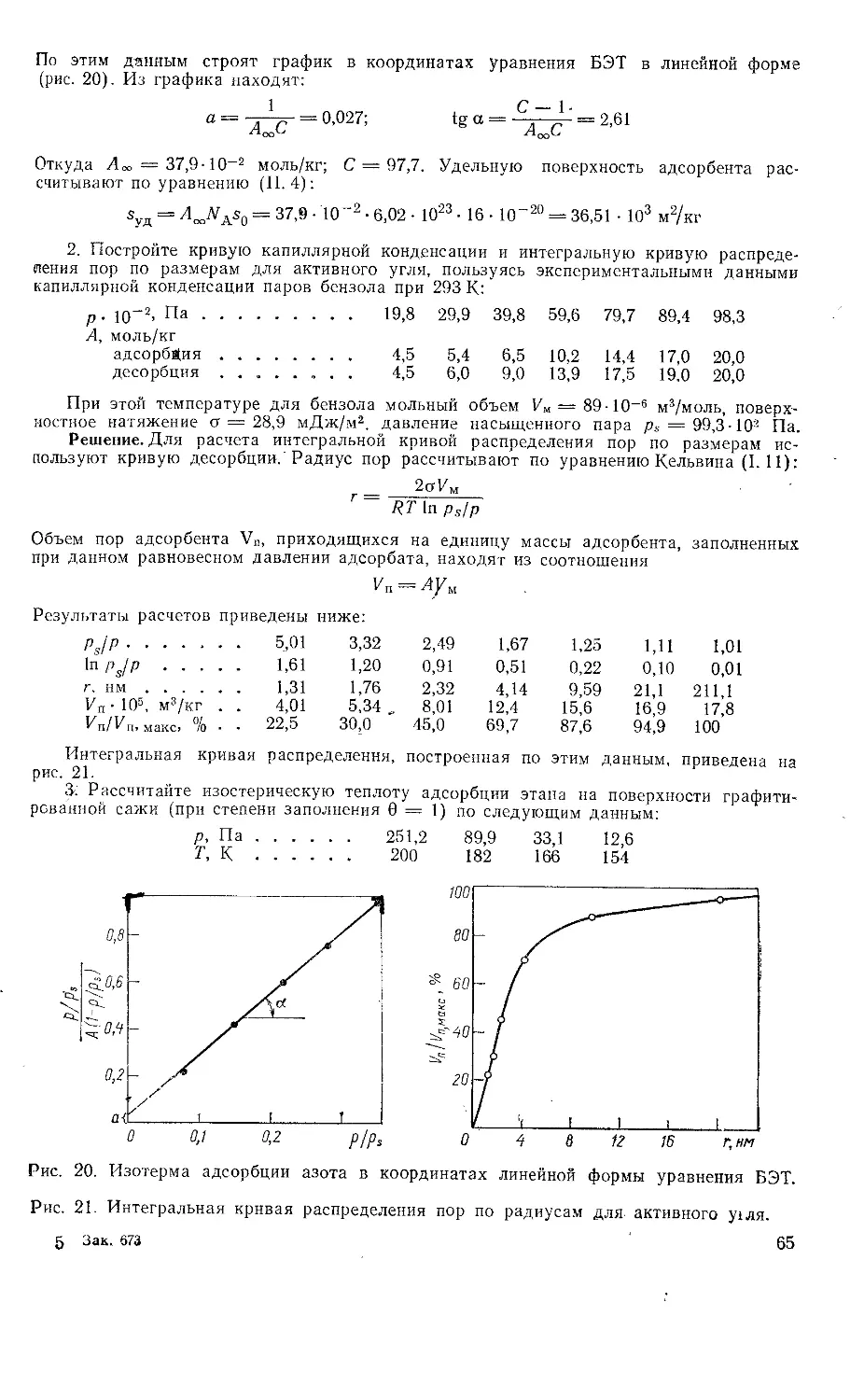
Работа 5. ИССЛЕДОВАНИЕ ВЛИЯНИЯ ЭЛЕКТРИЧЕСКОГО ПОТЕНЦИАЛА
НА ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
Цель работы: определение зависимости поверхностного натяжения на
границе ртуть — раствор и краевого угла от электрического потен-
циала; исследование влияния природы электролита на точку нулевого
заряда.
Связь между межфазным поверхностным натяжением и электриче-
ским потенциалом поверхности выражается уравнениями Липпмана
(1.23) и (1.24). Зависимость поверхностного натяжения от электриче-
ского потенциала называют электрокапиллярной кривой. Для межфаз-
ной границы ртуть — раствор электрокапиллярные кривые получают
обычно с помощью капиллярного электрометра. Используя уравнения
Липпмана, по электрокапиллярной кривой можно рассчитать плотность
заряда на поверхности ртути, дифференциальную емкость двойного
электрического слоя для определенного состава раствора и определить
точку нулевого заряда (т. н.з.), т. е. то значение потенциала, при кото-
ром плотность поверхностного заряда qs = 0, а о имеет максимальное
значение.
В присутствии поверхностно-активных веществ в растворе т. н. з.
может измениться. Например, ионы С1", Вг~, Г специфически адсорби-
руются на поверхности ртути, снижают о и приводят к смещению т. н. з.
в область более отрицательных значений электрического потенциала.
Характер влияния потенциала на поверхностное натяжение может
быть исследован по изменению краевого угла 0. Если на поверхность
ртути (или другого металла), находящейся в водном растворе элек-
тролита, нанести небольшую каплю органической жидкости, которая
нерастворима , в воде (рис. 7), то на трехфазной границе устанавлива-
ется равновесие сил поверхностного натяжения в соответствии с урав-
нением Юнга (I. 13). Если органическая жидкость неполярна и не яв-
ляется проводником, то значения ор, 3 и сч, 2 практически не зависят
от потенциала поверхности металла. Косинус краевого угла в этом
27
Рис. 7. Силы, действующие на границе раздела фаз: металл (3)—водный раствор
электролита (2) — органическая жидкость (7).
Рис. 8. Зависимость краевого угла 0 и cos 0 па поверхности ртути от электрического
потенциала.
случае является функцией только поверхностного натяжения на границе
металл — водный раствор о2,з:
Оо О - kl
cosO = —Ц----- (1.59)
«2
где kt и кг — константы.
Из уравнения (1.59) следует, что при потенциале нулевого заряда,
когда о2,з имеет наибольшее значение, cos 0 максимален и, следова-
тельно, краевой угол имеет наименьшее значение. При отклонении по-
тенциала от т. н. з. поверхностное натяжение на границе металл —
раствор электролита уменьшается и соответственно увеличивается 0.
Таким образом, зависимость краевого угла от потенциала проходит
через минимум (рис. 8).
Из уравнений (1.23) и (1.59) следует, что заряд единицы поверх-
ности qs — — du2,3/d(p = — k2d(cos д) /dtp положительный при потенциа-
лах электрода, более положительных по сравнению с <ри. 3 •—точкой
нулевого заряда. И наоборот, когда <р •< срн. 3, на поверхности имеется
отрицательный заряд.
Рис. 9. Схема установки для измерения краевого угла на поляризуемой поверхности
ртути:
1—ртуть; 2 —измерительная кювета; 3—вспомогательный электрод; 4—тумблер включения; 5—источ*
ник питания; о —реостат; 7—вольтметр; 8—анод; 9—катетометр; 10—осветитель.
28
Экспериментально определяемые значения краевых углов при раз-
личных потенциалах твердой металлической поверхности (платина,
серебро, цинк и др.), как правило, плохо воспроизводимы вследствие
энергетической неоднородности и шероховатости таких поверхностей.
Поверхность жидкого металла (ртуть, амальгамы, галлий) обычно од-
нородна, что позволяет провести исследование зависимости cos 0 от ф.
Измерение краевого угла 0 на поверхности ртути при различных
потенциалах проводят с помощью установки, схема которой приведена
на рис. 9. Установка состоит из измерительной ячейки 2, источника
постоянного тока 5, реостата 6, вольтметра 7, осветителя 10 и катето-
метра 9. Ячейка представляет собой стеклянную кювету с плоскопа-
раллельными стенками, на дне которой находится слой ртути 1 (5—
8 мм), являющейся катодом. В верхней части ячейки располагается
анод, изготовленный из платинированной платины. Катод соединяется
с источником тока через вспомогательный электрод (контактная стек-
лянная трубка 3, в нижней части которой имеется платиновый впай).
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Установка для измерения краевого угла на поверхности ртути.
Пипетка с тонким оттянутым концом.
Раствор индифферентного электролита, например KF.
Раствор ПАВ, например КВ масляная кислота (снижающие поверх-
ностное натяжение ртути).
Органическая жидкость, например толуол или декан.
В измерительную ячейку на слой ртути наливают раствор индиф-
ферентного электролита. Затем на поверхность ртути осторожно пи-
петкой наносят маленькую каплю органической жидкости (диаметр
основания капли не должен превышать 1 мм). Включают осветитель 10.
Изменяя положение столика, на котором закреплена кювета, а также
высоту расположения объектива катетометра 9, добиваются четко ви-
димой границы капли и поверхности ртути в поле зрения окуляра.
Предварительно для установления равновесия между окружающим
раствором и каплей проводят «тренировку» капли. Для этого включают
тумблер 4 и с помощью реостата 6 постепенно увеличивают напряже-
ние, подаваемое на электроды (катодная поляризация ртути). При этом
капля сначала несколько расплывается, а затем по мере увеличения
поляризации ртути начинает принимать форму, близкую к сферической
(не следует давать слишком большое напряжение, что может приве-
сти к отрыву капли от поверхности ртути). После этого с помощью
реостата постепенно уменьшают напряжение. «Тренировку» капли про-
водят 3—4 раза.
Измеряют высоту капли h и диаметр ее основания d в отсутствие
напряжения. Порядок измерения размеров капли указан в работе 3.
Затем подают напряжение на ячейку и измеряют размеры капли, уве-
личивая реостатом отрицательный потенциал на ртути на 0,1 В. После
каждого увеличения напряжения следует выждать 2 мин до установ-
ления равновесной формы капли. Напряжение увеличивают до тех пор,
пока капля не примет форму, близкую к сферической. Затем также
ступенчато уменьшают напряжение и измеряют размеры капли.
29
К электролиту, находящемуся в кювете, добавляют несколько мил
лилитров раствора ПАВ, аккуратно перемешивают стеклянной палоч-
кой водную фазу и измерения повторяют.
Для каждого результата измерения размеров капли по формуле
(1.51) рассчитывают косинус краевого угла и находят, значения угла 9,
Экспериментальные и расчетные данные записывают в таблицу (см.
табл. I. 4).
Таблица I. 4. Экспериментальные и расчетные данные исследования влияния
электрического потенциала на краевой угол
Электрический потенциал поверхности ртути ср, В Диаметр основания капли, дел. Высота капли. дел. CoS 6 а
«1 «2 Дгг «1 «2 Дп
Строят графики зависимости cos 9 и 9 от потенциала поверхности
ртути ер для раствора индифферентного электролита и раствора, содер-
жащего ПАВ. По графикам определяют изменение то'рси нулевого за-
ряда, вызванное ПАВ. Графическим дифференцирован'ием зависимости
cos 9 от <р определяют знак заряда поверхности ртути при потенциалах
более положительных и отрицательных по сравнению с точкой нулевого
заряда.
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАЧИ
КОНТРОЛЬНЫЕ ВОПРОСЫ
(р Что изучает коллоидная химия и каковы признаки ее объектов?
2. По каким признакам классифицируют объекты коллоидной хи-
мии? Приведите примеры дисперсных систем.
3. Какие поверхностные явления изучает коллоидная химия?
^^Что является мерой гетерогенности и степени раздробленности
дисдерсных систем?
Какими параметрами характеризуют степень раздробленности и
Какова связь между ними?
Что такое поверхностное натяжение и в каких единицах оно
измеряется?
Как зависит поверхностное натяжение от природы вещества, об-
разующего поверхность (межмолекулярного взаимодействия)?
8. Какие методы используются для определения поверхностного на-
тяжения жидкостей и твердых тел?
9. На чем основано измерение поверхностного натяжения жидкостей
методом капиллярного поднятия?
19. На чем основано измерение поверхностного натяжения жидко-
стей методом наибольшего давления пузырька воздуха? Положитель-
ным или отрицательным будет избыточное давление в жидкости на
границе с воздушным пузырьком?
11. На чем основано определение поверхностного натяжения мето-
дом отрыва кольца и сталагмометрическим методом?
30
12. Как и почему зависит поверхностное натяжение тел от темпе-
ратуры?
13. По какому уравнению можно рассчитать полную поверхностную
энергию? Какие данные необходимы для такого расчета?
14. Как влияет температура на теплоту и энтропию образования
единицы поверхности и на полную поверхностную энергию неассоции-
рованных жидкостей?
15. Что называется адсорбцией и как количественно ее характе-
ризуют?
16. Напишите фундаментальное адсорбционное уравнение Гиббса
и дайте определение избыточной адсорбции.
17. Каково соотношение между избыточной Г и абсолютной адсорб-
циями А? В каких случаях можно принять А ~ Г? Что такое отрица-
тельная гиббсовская адсорбция?
18. Что такое поверхностная активность? Какие вещества называ-
ются поверхностно-активными?
ту. Что называют адгезией и смачиванием? Какие параметры ис-
пользуют для их количественной характеристики?
gfo. Покажите взаимосвязь между адгезией и способностью жидко-
сти смачивать твердую поверхность. В чем состоит различие между яв-
лениями адгезии и смачивания?
72Ц Как влияет природа твердого тела и жидкости (межмолекуляр-
ноеЧазаимодействие в них) на смачивание и адгезию?
(22. Дайте характеристику и приведите примеры гидрофильных и
гидрофобных поверхностей. Как можно повлиять на смачивание по-
верхности?
(g3. Чем обусловлено улучшение смачивания водой гидрофобных по-
верхностей при введении в нее ПАВ?
Й) Что такое интегральная и дифференциальная теплоты смачи-
вания и какие существуют методы их определения?
Иб) Что такое углы натекания и оттекания и как по ним можно
найти равновесный краевой угол?
72(р Как влияет неоднородность и шероховатость твердых поверх-
ностей на их смачивание и адгезию?
(2J/ Что такое коэффициент Гаркинса? Каковы условия растекания
жидкостей? Рассмотрите особенности растекания жидкостей на твер-
дых поверхностях.
28. Как влияет кривизна поверхности и природа жидкости на ее
внутреннее давление? Каковы причины поднятия (опускания) жидко-
стей в капиллярах?
29. Чем обусловлена сферическая форма капель жидкости в усло-
виях невесомости?
30. Как влияет дисперсность вещества на его реакционную способ-
ность, давление пара, растворимость, константу равновесия химический
реакций?
31. Почему в капиллярах пар конденсируется при давдениях более
низких, чем на плоской поверхности?
32. Как влияет электрический потенциал на поверхностное натяже-
ние тел? Какими уравнениями выражается взаимосвязь между этими
параметрами?
33. Какие количественные характеристики двойного электрического
слоя можно определить по электрокапиллярным кривым?
31
34. Как влияют поверхностно-активные анионы и катионы на потен-
циал нулевого заряда?
35. Как изменяется вид электрокапиллярной кривой по мере увели-
чения концентрации индифферентного электролита и чем это обуслов-
лено?
ПРИМЕРЫ РЕШЕНИЯ ЗАДАЧ
1. Определите энергию Гиббса Gs поверхности капель водяного тумана массой
т = 4 г при 293 К, если поверхностное натяжение воды а — 72,7 мДж/м2, плотность
воды р — 0,998 г/см3, дисперсность частиц D = 50 мкм-1.
Решение. Энергия Гиббса поверхности определяется по уравнению (1.1):
Gs — os
Связь между удельной поверхностью «уд, поверхностью s, объемом V и дисперс-
ностью D выражается соотношением (I. 3):
Syn = у- = 67)
Отсюда поверхность капель тумана составляет:
s = 6 DV = 67) —
Р
и
т 4 • 10~3
= 67)——с = 6 • 5 • 107- ——з----0,0727 = 87,41 Дж
О УУо
2. Рассчитайте полную поверхностную энергию 5 г Эмульсии бензола в воде с
концентрацией 75 % (масс.) и дисперсностью D — 2 мкм-1 при температуре 313 К.
Плотность бензола при этой температуре р — 0,858 г/см3, поверхностное натяжение
о = 32,0 мДж/м2, температурный коэффициент' поверхностного натяжения бензола
da/dT = —0,13 мДж/(м2-К).
Решение. Полная поверхностная энергия Us рассчитывается по уравнению Гибб-
са— Гельмгольца (I 8):
Поверхность s капель бензола 75 %-ной эмульсии массой т — 5 г составляет:
s = 6 7)7 = 67)— =
Р
6-2 - 10s 6 4 • 5 • 0,75 • 10 3
858
= 52,4 м2
Отсюда
Us = (32,0 -10 3 + 0,13 • 10~3 • 313) • 52,4 = 3,81 Дж
3. Рассчитайте давление насыщенных паров р над каплями воды с дисперсностью
7) = 0,1 пм—1 при температуре 293 К. Давление паров воды над плоской поверхностью
при этой температуре ps = 2338' Па, плотность воды р = 0,998 г/см3, поверхностное
натяжение воды о — 72,7 мДж/м2.
Решение. Влияние кривизны поверхности их давление насыщенного пара выра-
жается уравнением Кельвина И 11):
р 2gVM ,2toV27) 2-0,0727g 18 10~3 -2- 108
П Ps ~ RTr Rf/p~ 998-8,31-293 . — 0,21
Отсюда
p/ps = 1,23 и р == 1,23 • 2338 = 2875 Па
4. Две вертикальные параллельные пластинки частично погружены в жидкость на
расстоянии d = 1 мм друг от друга. Угол смачивания (.1 пластинок жидкостью со-
ставляет 30°. Поверхностное натяжение жидкости о = 65 мДж/м2, разность плотностей
жидкости и воздуха Др = 1 г/см3.
32
Рассчитайте избыточное давление в жидкости и силу взаимного притяжения пла-
стинок, если их размеры составляют 5X5 см.
Решение. Капиллярное (избыточное) давление в жидкости между параллельными
пластинками рассчитывают по уравнению
. 2а cos 0 2 • 65 • 10~3 • 0,87 ,,О1ТТ
Др =-----------------------г,—--= 113,1 Па
d МО 3
При смачивании поверхности пластинок избыточное давление уменьшает внутреннее
давление в жидкости, что приводит к ее поднятию по сравнению с уровнем жидкости
в сосуде и появлению силы, прижимающей пластины:
р = kps = 113,1 • 0,05 • 0,05 = 0,28 Н
Такую силу надо приложить к каждой пластинке перпендикулярно ее поверхно-
сти, чтобы оторвать пластинки друг от друга.
5. В воздухе, содержащем пары воды, образуется туман при температуре 270,8 К
(коэффициент пересыщения у равен 4,21). Рассчитайте критический размер ядер кон-
денсации и число молекул, содержащихся в них. Поверхностное натяжение воды
о = 74 мДж/м2, мольный объем воды V„ — 18-10—е м3/моль.
Решение. Радиус г равновесного зародыша в зависимости от пересыщения систе-
мы определяется по уравнению Кельвина (I. 11):
Отсюда
.. 2аУм
RT In у
In
Р
Ps
In у
_ 2аУы
RTr
2-74-10 3-18-10~6 * * *
8,31 -270,8-In 4,21
= 8,24 • 10 10
м = 0,824 нм
Число молекул и в одном зародыше составит:
УяппаАД 4 - 3,14 (8,24 • Ю-10)3 • 6,02 • 1023
п = = - ядра—А ------------------------------= 78
Ум 3-18-10 6
6. Рассчитайте работу адгезии ртути к стеклу при 293 К, если известен крае-
вой угол 0 = 130°. Поверхностное натяжение ртути а = 475 мДж/м2. Найдите коэф-
фициент растекания f ртути по поверхности стекла.
Решение. Выражение для работы адгезии через краевой угол дается уравнением
Дюпре—Юнга (1.14):
Г а = аж.г (1 + cos 0) = 475 (1 + cos 130°) = 475 - (1 — 0,64) = 171 мДж/м2
, Коэффициент растекания рассчитывают по соотношению f = 1Уа—!УК, где
|>'к = 2аНй представляет работу когезии; f = 171 —2-475 = —779 мДж/м2, т. е. ра-
стекания нет.
ЗАДАЧИ
1. Определите энергию Гиббса поверхности 5 г тумана воды, если по-
верхностное натяжение капель жидкости составляет 71,96 мДж/м2,
а дисперсность частиц 60 мкм-1. Плотность воды примите равной
0,997 г/см3.
2. Аэрозоль ртути сконденсировался в виде большой капли объемом
3,5 см3. Определите, на сколько уменьшилась поверхностная энергия
ртути, если дисперсность аэрозоля составляла 10 мкм-1. Поверхност-
ное натяжение ртути примите равным 0,475 Дж/м2.
3. Определите поверхностное натяжение бензола при 293, 313 и
343 К. Примите, что полная поверхностная энергия не зависит от тем-
пературы и для бензола равна 61,9 мДж/м2. Температурный коэффи-
циент da/dT = — 0,13 мДж/ (м2 • К).
4. Рассчитайте полную поверхностную энергию 5 г эмульсии бен-
зола в воде с концентрацией 55 % (масс.) и дисперсностью 3 мкм-1
3 Зак. 673
33
при температуре 313 К. Плотность бензола 0,858 г/см3, межфазное
поверхностное натяжение 26,13 мДж/м2, температурный коэффициент
поверхностного натяжения бензола da/dT =— 0,13 мДж/(м2-К).
^/Рассчитайте избыточное давление внутри капель бензола, равно-
весных с паром, если удельная поверхность системы составляет
6-108 м“], а поверхностное натяжение бензола 28,87 мДж/м2 при 293 К.
6. Рассчитайте избыточное давление в капле воды (за счет кривиз-
ны) с удельной поверхностью 3-106 м-1 при температуре 313 К, если
поверхностное натяжение воды при 298 К составляет 71,96 мДж/м2,
а температурный коэффициент поверхностного натяжения воды
da/dT = - 0,16 мДж/(м2-К).
7. Рассчитайте капиллярное давление в капле ртути с дисперс-
ностью 1 мкм~', если поверхностное натяжение ртути составляет
0,475 Дж/м2.
8. Чтобы стряхнуть ртуть в медицинском термометре, нужно со-
здать ускорение, равное 10g. Рассчитайте диаметр перетяжки в капил-
ляре термометра, если поверхностное натяжение ртути 0,475 Дж/м2,
длина столбика ртути выше перетяжки 5 см, плотность ртути
13,54 г/см3.
9. Найдите поверхностное натяжение жидкости, если в капилляре
с диаметром 2 мм она поднимается на высоту 15 мм. Плотность жидко-
сти 0,998 г/см3, краевой угол мениска равен 0°. Сделайте предположе-
ние о природе жидкости.
10. Для определения поверхностного натяжения воды взвешивают
капли, отрывающиеся от капилляра и измеряют диаметр шейки капли
в момент ее отрыва. Оказалось, что масса 318 капель воды равна
5 г, а диаметр шейки капли — 0,7 мм. Рассчитайте поверхностное на-
тяжение воды.
11. Покажите, чему равна разность hi — h2 воды в двух сообщаю-
щихся капиллярах с диаметрами di и d2. Плотность и поверхностное
натяжение жидкости равны соответственно р, ст, краевые углы менисков
равны нулю.
12. Насколько изменится разность уровней воды в двух сообщаю-
щихся капиллярах диаметрами 0,1 и 0,3 мм при нагревании от 293 до
343 К, если поверхностное натяжение при этих температурах равно
соответственно 72,75 и 64,0 мДж/м2. Плотность воды при 293 и 343 К
составляет соответственно 0,998 г/см3 и 0,978 г/см3.
13. Поверхностное натяжение жидкости, смачивающей стекло' из-
меряют, определяя высоту между уровнями двух менисков в U-образ-
ной капиллярной трубке; диаметры капилляров колен трубки равны
1 и 10 мм. Рассчитайте поверхностное натяжение жидкости с плот-
ностью 0,998 г/см3, зная, что разность двух уровней менисков в капил-
лярах составляет 9 мм.
14. На какую высоту поднимается вода между двумя вертикаль-
ными стеклянными пластинами, частично погруженными в эту жид-
кость, если расстояние между ними 0,5 мм? Плотность и поверхностное
натяжение воды соответственно равны 0,997 г/см3 и 71,96 мДж/м2,
Краевой угол 0 примите равным 0°.
Н5) Вычислите поверхностное натяжение воды, определяемое мето-
домкапиллярного поднятия, если при 298 К вода поднялась в капил-
ляре на высоту 35,3 мм. Диаметр капилляра определен путем измере-
ния длины столбика и массы ртути, заполнившей капилляр под дав-
34
лением: длина столбика ртути составила 8,04 см, масса его 0,565 г.
Плотарсть ртути 13,54 г/см3, плотность воды 0,997 г/см3.
Между двумя параллельными пластинами находится слой воды
толщиной 0,5 мкм. Рассчитайте давление, сжимающее пластины, если
угол смачивания 0 — 0°, поверхностное натяжение воды равно
71,96 мДж/м2. Определите силу, которую необходимо приложить для
отрыва пластин друг от друга, если размер каждой 10 X Ю см.
17. Капля воды массой 0,1 г введена между двумя параллельными
стеклянными пластинами, причем краевой угол 0 — 0°. Какова сила
притяжения между пластинами, если они находятся друг от друга
на расстоянии 1 мкм. Поверхностное натяжение воды составляет
(71,96 мДж/м2, плотность воды 0,997 г/см3.
(18. С какой силой притягиваются две вертикальные и параллель-
ные стеклянные пластинки, частично погруженные в воду, если рас-
стояние между ними равно 1 мм? Ширина пластинок 15 см, поверхност-
ное натяжение воды 71,96 мДж/м2, угол смачивания 0°. Высота пла-
стинок такова, что поднявшаяся вода не доходит до их верхних краев.
Плотность воды примите равной 0,997 г/см3.
(ДУ? Определите, насколько давление паров над каплями воды диа-
метром 0,2 мкм больше, чем давление паров над плоской поверх-
ностью при температуре 298 К. Поверхностное натяжение воды
71,96 мДж/м2, мольный объем воды 18,05 см3/моль.
Как изменится это давление, если дисперсность капель увеличится
в 10 и 100 раз?
20. Определите, при каком пересыщении давление паров над кап-
лей бензола диаметром 2-10_2 мкм при 313 К соответствует равновес-
ному. Поверхностное натяжение бензола 26,13 мДж/м2, а плотность
0,858 г/см3. Что будет происходить, если пересыщение паров станет
больше или меньше рассчитанной величины.
21. Рассчитайте равновесное- давление паров над каплями воды с
дисперсностью 20 мкм-1 при температуре 333 К, если поверхностное на-
тяжение воды при температуре 293 К составляет 72, 75 мДж/м2, а тем-
пературный коэффициент поверхностного натяжения dv/dT —
= — 0,16 мДж/(м2-К). Давление насыщенных паров воды над плоской
поверхностью при 60°С равно 20,58-103 Па, а плотность воды
0/983.г/см3.
К <ц2^>. Определите равновесное давление паров над каплями воды и
четыреххлористого углерода с дисперсностью 0,1 нм-1 при температуре
293 К. Давление насыщенных паров над плоской поверхностью при этой
температуре для воды и четыреххлористого углерода составляет соот-
ветственно 23,38-102 и 13-103 Па; плотность соответственно равна 0,998
и 1,593 г/см3; поверхностное натяжение 72,75 и 25,68 мДж/м2. Обра-
тите внимание, как влияет природа жидкости на давление насыщен-
ных паров в дисперсной системе.
23. Рассчитайте равновесное давление паров над водой, находя-
щейся в капилляре радиусом 1 мкм при 293 К, предполагая, что угол
смачивания равен 0°. Выразите результат в процентах от давления
насыщенного пара воды. При 293 К плотность воды 0,998 г/см3, поверх-
ностное натяжение 72,75 мДж/м2, давление насыщенного пара 2338 Па.
24. Определите равновесное давление паров над каплями воды и
бензола радиусом 0,05 мкм при температуре 313 К. Примите, что давле-
ние насыщенных паров над плоской поверхностью при этой температуре
3*
35
для воды и бензола равно соответственно 77,6-102 и 24,08-103 Па, моль-
ные объемы 18,1 и 93,4 см3/моль, а поверхностные натяжения 69,55 и
26,13 мДж/м2.
25. Рассчитайте давление паров воды над вогнутым мениском в ка-
пиллярах радиусом 0,001 и. 10 мкм при 293 К. Угол смачивания при-
мите равным нулю. Плотность воды 0,998 г/см3, давление пара над
макцефазой 2338 Па, поверхностное натяжение воды 72,75 мДж/м2.
/25>Оцените размер частиц SrSO4, зная, что их растворимость на
3 %(масс.) больше растворимости крупных кристаллов. Межфазное
натяжение при 298 К примите равным 85 мДж/м2, плотность SrSO4
3$6-£/см3.
\,^27/ Рассчитайте межфазное натяжение в системе CaF2 — вода, зная,
что растворимость частиц CaF2 диаметро1м 0,3 мкм превышает раство-
римость крупных кристаллов (при 293 К) на 18 % (масс.). Плотность
CaFz примите равной 2,5 г/см3.
\j28. Определите поверхностную активность уксусноэтилового эфира
по приведенным ниже значениям поверхностного натяжения водных
растворов его при 298 К:
с, ммоль/л......
<т, мДж/м2 .......
7,8 15,6 31,2 62,5 125 250 500
69,6 68,0 65,1 61,5 56,2 49,7 41,5
Постройте изотерму гиббсовской адсорбции. Поверхностное натяже-
ние воды 71,96 мДж/м2.
29. Рассчитайте работу адгезии для воды, глицерина, трикрезил-
фосфата и бензола, смачивающих фторопласт. Поверхностное натяже-
ние (на границе с воздухом) воды, глицерина, трикрезилфосфата и бен-
зола соответственно равны 71,96; 63,2/40,9; 28,9 мДж/м2, а краевые
углы составляют 108, 100, 75 и 46°.
30. Рассчитайте работу адгезии в системе вода —графит, зная, что
краевой угол равен 90°, а поверхностное натяжение воды составляет
71,96 мДж/м2. Определите коэффициент растекания воды на графите.
31. Рассчитайте работу адгезии ртути к стеклу при 293 К, если
известен краевой угол 0 — 130°. Поверхностное натяжение ртути
475 мДж/м2. Найдите коэффициент растекания ртути по, поверхности
стекла.
32. Краевой угол воды на парафине равен 111° при 298 К- Для
0,1 М раствора бутиламина в воде поверхностное натяжение составляет
56,3 мДж/м2, краевой угол на парафине равен 92°. Рассчитайте по-
верхностное давление пленки бутиламина, адсорбированного на поверх-
ности раздела парафин — вода. ..Поверхностное натяжение воды
71,96 мДж/м2.
33. Смачивание поверхности стекла водой меняется при введении
катионного ПАВ, например додецилметиламмонийбромида. Постройте
изотерму смачивания [cos 0 = f (спав) 1 определите точку инверсии
смачивания (cos 0 = 0)' и рассчитайте работу адгезии, используя при-
веденные ниже данные:
СПАВ’ ММОЛЬ/л.....
<тж.г, мДж/м2.....
9, град ........
0 1 • 10-’ 1 • 10~2
72 71,5 70,1
0 47 85
1 2 5 10
63,2 56,2 50,9 41,0
92 82 57 0
34. Рассчитайте, как изменится в результате адсорбции катионного
ПАВ работа адгезии при смачивании стекла водным раствором доде-
36
цилметиламмонийбромида. Поверхность стекла покрыта слоем октаде-
кана. Постройте изотерму смачивания по следующим данным:
СПАВ’ ммоль/л ......
аж.г, мДж/м2.........
0. град......... . .
О 1 • 10-4 1 • 10~2 1 2 5 10
72 71,5 70,1 63,2 56,2 50,9 41,0
106 105 96 72 60 0 0
Сравните полученную изотерму с изотермой смачивания для стекла, не
обработанного октадеканом (см. задачу 33). Объясните, как ориенти-
руются молекулы ПАВ в поверхностном слое. Рассчитайте работу ад-
гезии.
35. Рассмотрите возможность растекания водного раствора вале-
риановой кислоты по поверхности ртути, исходя из значений поверх-
ностных и межфазных натяжений: ар.р_Во3дух = 25 мДж/м2, Снв-воздух =
= 475 мДж/м2, OHg-p-p = 329 мДж/м2. Если раствор будет растекаться
по поверхности ртути, то как при этом ориентируются полярные груп-
пы валериановой кислоты: к воде или к ртути? Объясните почему.
36. Экспериментально получено значение коэффициента растекания
гептанола по воде, равное 37 мН/м. Рассчитайте межфазное натяжение
на границе вода — гептанол, принимая значения поверхностных натя-
жений воды и гептанола соответственно 71,96 и 26,1 мН/м.
37. Капля бензола растекается по поверхности воды, а после взаим-
ного насыщения двух жидкостей образует линзу. Объясните это явле-
ние. Рассчитайте начальные и конечные значения коэффициентов рас-
текания бензола по воде, используя приведенные ниже значения по-
верхностных натяжений (в мН/м) для различных поверхностей раз-
дела при 293 К:
Вода — воздух..........................72,75
Бензол — воздух..........i.............28,87
Вода — бензол......................... 35,00
Вода, насыщенная бензолом, — воздух . . . 62,20
Бензол, насыщенный водой, — воздух . . . 28,80
Оцените значение межфазного натяжения на границе взаимонасыщен-
ных растворов воды и бензола.
38 Постройте изотерму гиббсовской адсорбции, используя значения
поверхностных натяжений водных растворов додецилсульфата натрия
при 20°С:
СПАВ> ммоль/л. .... 2,16 3,96 6,6 8,3 9,3 9,8 10,2 11,2
<тж.г, мДж/м2 ..... 62 54 47 43 42 41 41 41
Определите поверхностную активность этого ПАВ и критическую
концентрацию мицеллообразования. Поверхностное натяжение воды
72,75 мДж/м2.
II. АДСОРБЦИОННЫЕ РАВНОВЕСИЯ
Величина адсорбции зависит от природы поверхности адсорбента, при-
роды адсорбата и его концентрации (давления), температуры и др.
Графическая зависимость адсорбции от концентрации адсорбируемого
вещества в объемной фазе при данной температуре называется изотер-
мой адсорбции.
37
Адсорбция из предельно разбавленных растворов или смесей газов
подчиняется закону Генри:
А — кгс или А — к'гр (II- 1)
где /<г и /<г— константа Генри; с — концентрация адсорбата в объемной фазе;
р — давление пара адсорбата.
Аналитическим выражением изотермы мономолекулярной адсорб-
ции при более высоких концентрациях и на ровной поверхности явля-
ется уравнение изотермы Ленгмюра:
Л = Лсо-^4>- или 4 = А £'р (II. 2)
1 + ДС 1 + А р
где Л,», — предельная мономолекулярная адсорбция — емкость монослоя; К и К'—
константы адсорбционного равновесия, характеризующие энергию адсорбции.
Для высоких давлений пара изотерма адсорбции описывается об-
щим уравнением обобщенной теории Ленгмюра — уравнением полимо-
лекулярной адсорбции БЭТ (Брунауэра, Эммета и Теллера) :
_________A^Cp/ps__________
(1 — P/Ps) [1 + (С — 1) p/ps]
(II-3)
где С — константа, характеризующая энергию взаимодействия сконденсированного ад-
сорбата с поверхностью адсорбента; ps — давление насыщенного пара адсорбата.
Уравнение Ленгмюра (II. 2) и уравнение БЭТ (П.З) широко ис-
пользуются для определения удельной поверхности адсорбентов, ката-
лизаторов и других дисперсных систем. Удельная поверхность худ свя-
зана с емкостью монослоя Аоо соотношением
*уд = АЛа*0 (II. 4)
где N — число Авогадро; So— площадь, занимаемая одной молекулой адсорбата в
насыщенном адсорбционном слое.
Для описания адсорбции на пористых телах с переходными порами
(мезопористые адсорбенты) используют уравнение капиллярнрй кон-
денсации Кельвина (1-11), которое позволяет определить размеры пор.
Если тело в основном имеет микропоры, то применяют уравнение тео-
рии объемного заполнения микропор:
nnrn
In У = In [In (ps/p)K (II. 5)
где V — объем заполненных пор при данном давлении; — общий объем пор в адсор-
бенте; р — коэффициент аффинности, характеризующий природу адсорбата; Е— ха-
рактеристическая энергия адсорбции; п — показатель степени, выражаемый целыми
числами от 1 до 6.
Энергетические параметры адсорбции (изменение энтальпии, энтро-
пии, энергии Гиббса) обычно рассчитывают из температурных зависи-
мостей адсорбции. Например, из уравнений Клапейрона — Клаузиуса
или Бант-Гоффа можно получить следующую теоретическую изостеру
(А — const):
А// ,
In р =+ const (II.6)
где А// — изменение энтальпии при адсорбции.
По экспериментально построенной зависимости в координатах урав-
нения (II. 6) можно определить (по тангенсу угла наклона) дифферен-
38
циальные мольные (изостерические)' энтальпии адсорбции при данных
степенях заполнения (при данных А).
Стандартная энергия Гиббса AG° адсорбции связана с равновесной
константой адсорбции К соотношением
: Дб° = -/?ПпК (II. 7)
Зная стандартную энтальпию и энергию Гиббса, легко вычислить стан-
дартную энтропию адсорбции.
Поверхностно-активные вещества отличаются высокой адсорбцион-
ной способностью, и для них А ~ Г. Это позволяет применительно к
ПАВ совместно решить уравнения Гиббса (1.21) и Генри (II. 1).Со-
вместное решение дает линейную изотерму поверхностного натяжения
при малых концентрациях ПАВ в растворе: -
а = (То - KrRTc ' (II. 8)
где <То — поверхностное натяжение растворителя.
Принимая разность о0 — о за поверхностное (двухмерное) давле-
ние л, получим уравнение состояния идеального двухмерного газа:
nsa = RT (П-9)
где sM — поверхность, на которой распределен 1 моль ПАВ.
Совместное решение адсорбционного уравнения Гиббса (1.21)'
с уравнением Ленгмюра (II. 2) для ПАВ дает уравнение Шишковского,
связывающее изменение поверхностного натяжения раствора с концен-
трацией растворенного ПАВ в объеме:
(т = <т0-Ао<ЛГ1п(1 + Лс) (II. 10)
В этом случае уравнение состояния двухмерного газа имеет вид
n(sM — s,.t.0) = RT (II. 11)
где «м, о = 1/А„, т. е. поверхность, занимаемая 1 моль ПАВ в адсорбционно насыщен-
ном мономолекулярном слое. , ? - -
Уравнения Гиббса, Генри, Ленгмюра и Шишковского по экспери-
ментальным данным о поверхностном натяжении растворов позволяют
{рассчитать следующие величины и характеристики: адсорбцию ПАВ
:на межфазной границе раствор — воздух и раствор — твердый адсор-
'бент; толщину адсорбционного слоя; линейные размеры молекул ПАВ;
предельную адсорбцию поверхностного мономолекулярного слоя; удель- *
ную поверхность твердого адсорбента, катализатора, а также исследо-'
вать свойства поверхностных пленок.
В настоящее время широко распространена ионообменная адсорб-
ция с помощью ионитов, которые представляют собой твердую матрицу
с ионогенными функциональными группами. Если матрица несет отри-
цательный заряд (фиксирован анион), а обмениваются катионы, ад-
-• сорбент является катионитом. Если матрица заряжена положительно,
а подвижные противоионы несут отрицательный заряд, адсорбент явля-
* ется анионитом. Иониты со смешанными группами называют амфо-
литами.
Основными характеристиками ионообменных адсорбентов являются
их обменная емкость и константа ионного обмена, характеризующая
избирательную способность ионита. Обменная емкость — количество
ионов (в эквивалентах), поглощенных единицей массы сорбента или
39
единицей объема набухшего ионита. При ионообменной адсорбции про-
исходит стехиометрический обратимый обмен ионов Mj и М2 между
объемом раствора электролитов и адсорбентом:
г2М1 + ZiM2 < Д- ZiM2 + z2Mi (II. 12)
где М, и М2 — ионы, адсорбированные на ионите.
Равновесное состояние описывается уравнением константы ионного
обмена:
где а и а — термодинамические активности обменивающихся ионов соответственно в
ионите и растворе; Zi, Z2 — заряды ионов 1 и 2.
Возводя левую и правую часть полученного выражения в степень,
равную l/(ziz2), получаем уравнение Никольского для обмена поли-
валентных ионов:
оУгга!/г'
1^' - 1 /ТТ t
На практике обычно вместо активностей используют концентрации.
С помощью ионообменной адсорбции можно извлечь ионы или раз-
делить их, изменить кислотность раствора или сделать его нейтраль-
ным и т. д.
Адсорбция лежит в основе метода разделения компонентов смесей,
называемого хроматографией. Хроматографическое разделение проис-
ходит при движении подвижной фазы (раствор, газовая смесь) относи-
тельно другой неподвижной фазы (обычно адсорбент или инертный но-
ситель, пропитанный жидкостью) вследствие различного сродства раз-
деляемых веществ с фазами.
В зависимости от агрегатного состояния контактирующих фаз раз-
личают четыре вида хроматографии: газоадсорбционную, газожидкост-
ную, жидкостно-адсорбционную, жидкостно-жидкостную. По оформле-
нию процесса хроматографию делят на колоночную и плоскослойную
(тонкослойную и на бумаге). Существует три метода проведения хро-
матографии: фронтальный, вытеснительный и элюентный (проявитель-
ный). При первом методе разделяемую смесь непрерывно подают через
хроматографическую колонку. В вытеснительном и проявительном ме-
тодах после подачи разделяемой смеси в колонну вводят соответствен-
но или вытеснитель, который сорбируется лучше разделяемых веществ,
или чистый растворитель, слабо реагирующий с адсорбентом.
Работа 6. ИЗУЧЕНИЕ АДСОРБЦИИ ПАВ ИЗ РАСТВОРОВ
НА ТВЕРД ОМ АДСОРБЕНТЕ
Цель работы: получение изотерм поверхностного натяжения раство-
ров ПАВ на границе с воздухом; определение предельной адсорбции
ПАВ из водного раствора на угле; вычисление удельной поверхности
адсорбента.
I Типичные ПАВ имеют асимметрично построенные молекулы, состоя-
щие из двух частей: активной полярной группы, хорошо взаимодей-
ствующей с молекулами воды, типа —ОН, —СООН, —NH2, —NO2,
40
—SO3H, —SO3Na, —COONa и др., и неполярной гидрофобной груп-
пы — углеводородного радикала.
Такие дифильные молекулы, способные взаимодействовать одновре-
менно с полярными и неполярными средами, самопроизвольно накап-
ливаются на границах раздела фаз, понижая энергию Гиббса поверх-
ности и образуя адсорбционный слой определенной структуры. В ад-
сорбционных слоях молекулы ПАВ ориентируются полярными группами
в сторону полярной среды (воды), а гидрофобной неполярной частью —
в сторону менее полярной фазы (воздуха, углеводородной жидкости).
По мере заполнения поверхности раздела вода — воздух молекулами
ПАВ поверхностное натяжение на этой границе резко снижается. В раз-
реженных адсорбционных слоях молекулы ПАВ располагаются вдоль
поверхности. Такое, расположение ПАВ приводит к наибольшему экра-
нированию молекул воды и обеспечивает минимальное поверхностное
натяжение раствора. \
С ростом концентрации раствора число молекул ПАВ в адсорбцион-
ном слое увеличивается. При некоторой концентрации раствора может
образоваться предельно насыщенный адсорбционный слой, так назы-
ваемый «частокол Ленгмюра». В этом случае поверхностный слой от-
вечает конденсированной пленке и поверхность воды оказывается
сплошь покрытой углеводородными участками молекул ПАВ. Поверх-
ностное натяжение растворов при этих концентрациях приближается
к значению поверхностного натяжения самих поверхностно-активных
веществ на границе с воздухом.
При введении адсорбентов в водные растворы ПАВ молекулы ПАВ
адсорбируются на границе вода — твердая поверхность. Согласно пра-
вилу Ребиндера при адсорбции ПАВ разность полярностей между ад-
сорбентом и растворителем уменьшается. Все полярные гидрофильные
поверхности адсорбируют ПАВ из неполярных и слабополярных жидко-
стей. Неполярные сорбенты, такие, как уголь или некоторые полимер-
ные материалы, наоборот, хорошо адсорбируют ПАВ из полярных
жидкостей.
На адсорбцию ПАВ из растворов существенное влияние оказывает
и пористость сорбента. Влияние пористости определяется соотношением
размеров пор и молекул ПАВ. С уменьшением размеров пор адсорбция
небольших молекул ПАВ, как правило, возрастает. Однако это наблю-
дается, только если молекулы ПАВ имеют размеры, позволяющие про-
никнуть в поры адсорбента.
По истечении определенного времени в системе адсорбент — водный
раствор ПАВ устанавливается равновесие между количеством А моле-
кул ПАВ, перешедших на поверхность сорбента, и их объемной равно-
весной концентрацией с. Это равновесие может быть описано уравне-
нием Ленгмюра (II. 2), при этом емкость монослоя А™ отвечает пре-
дельной адсорбции. Для более точного определения величины А со пред-
почтительнее использовать уравнение Ленгмюра в линейной форме:
/ ’л ' <IL15>
Графически зависимость 1/А=/'(1/с) выражается прямой, пересе-
кающей ось ординат. Отрезок, отсекаемый от оси ординат, определяет
величину, обратную емкости монослоя. Тангенс угла наклона прямой
позволяет найти константу адсорбционного равновесия К»
41
Величину адсорбции А для ПАВ обычно рассчитывают, определяя
концентрацию раствора до и после адсорбции:
А ~ Г = v (11.16)
' т
где с», с — начальная и равновесная концентрации ПЛВ.в растворе; т — масса адсор-
бента; V — объем раствора ПАВ, в который введен адсорбент.
Разность концентраций поверхностно-активного вещества с0 — с
удобно определять по изменению поверхностного натяжения раствора.
Определив экспериментально емкость монослоя Лоо по формуле
(II. 4), можно рассчитать удельную поверхность адсорбента худ, т. е.
поверхность, приходящуюся на единицу массы адсорбента. Для этого
необходимо знать площадь s0, занимаемую одной молекулой адсорбата
в насыщенном адсорбционном слое на границе раздела фаз. Согласно
исследованиям Ленгмюра и Гаркинса площадь, занимаемая одной мо-
лекулой большинства одноосновных жирных кислот и спиртов, состав-
ляет 0,2—0,3 нм2.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Прибор для измерения поверхностного натяжения.
Весы с разновесами.
Конические колбы емкостью 50 мл.
Воронки и фильтровальная бумага.
Бюретки и пипетка емкостью 25 мл.
Активный уголь или другой неполярный сорбент.
Раствор ПАВ, например 0,6 М раствор бутилового спирта или 0,3 М
раствор изопентилового спирта.
Берут 6 навесок адсорбента (активного угля) по 1 г. Из исходного
раствора ПАВ готовят путем разбавления 6 водных растворов в соот-
ветствии с данными:
Номер колбы ................
Объем исходного раствора
ПАВ, мл.....................
Объем добавляемого раство-
рителя, мл.............. .
1 2 3 4 5 6
50 35 25х 15 10 5
0 15 25 35 40 45
Растворы ПАВ с помощью пипетки делят на две равные части (по
25 мл). Одну серию растворов используют для измерения поверхност-
ного натяжения и построения калибровочной изотермы а —/(спав)-
Метод измерения поверхностного натяжения выбирается по указанию
преподавателя (методики определения стж.г приведены в работе 1).
Измерения поверхностного натяжения начинают с растворов наимень-
шей концентрации.
В растворы второй серии вносят навески адсорбента (по одной
в каждую колбу) и оставляют на 1—1,5 ч, периодически взбалтывая,
для установления адсорбционного равновесия в системе. Затем рас-
творы ПАВ отделяют от адсорбента путем фильтрования и тем же ме-
тодом измеряют значения поверхностного натяжения.
По результатам измерений <тж-, растворов первой серии (до адсорб-
ции) строят калибровочную кривую о =/(спав) и по ней определяют
42
Таблица 11. 1. Результаты исследования адсорбции ПАВ на угле
Номер колбы До адсорбции После адсорбции А, моль/г 1/Д Ис
с0’ моль/л наибольшее давление пузырька Др или сила отрыва кольпа F °Ж-Г’ Дж/м2 наибольшее давление пузырька Др нли сила отрыва кольца F °Ж-Г‘ Дж/м2> С, моль/л
равновесные концентрации ПАВ после адсорбции. Для каждого рас-
твора по формуле (II. 16) рассчитывают значения А и строят изотерму
адсорбции в координатах А — с.
Полученные результаты записывают в таблицу (см. табл. II. 1).
Для определения величины А<х> строят график зависимости 1/А —
= f(l/c). По найденному значению Ах рассчитывают удельную поверх-
ность адсорбента зуд (для бутилового ^Гзрр0нтилового спиртов s_q при-
нимают равной 0,3 нм2).
Работа 7. ИССЛЕДОВАНИЕ ВЛИЯНИЯ СТРОЕНИЯ МОЛЕКУЛ ПАВ
НА ИХ ПОВЕРХНОСТНУЮ АКТИВНОСТЬ. ОПРЕДЕЛЕНИЕ ПАРАМЕТРОВ
АДСОРБЦИОННОГО СЛОЯ
Цель работы: получение изотерм поверхностного натяжения и адсорб-
ции для водных растворов алифатических спиртов; определение соот-
ношения поверхностных активностей ПАВ в их гомологическом ряду;
расчет толщины адсорбционного слоя и площади, занимаемой одной
молекулой ПАВ в насыщенном адсорбционном слое.
Поверхностно-активные свойства ПАВ зависят от числа метилено-
вых г]эупп в углеводородной цепи, природы и содержания полярных
группу Адсорбционная способность молекул ПАВ характеризуется по-
верхностной активностью g. Поверхностную активность можно найти
графически по экспериментальной изоте.рме поверхностного натяжения
о — f(c). На рис. 10 представлены изотермы поверхностного натяжения
для соседних членов гомологического ряда ПАВ. Приведенные кривые
показывают^что с удлинением углеводородного радикала гомолога по-
верхностная активность g повышается.
„Дюкло и Траубе установили экспериментальное правило, согласно
которому поверхностная активность жирных
кисл.от, спиртов, аминов и других веществ в
гомологических рядах на границе раствор —
воздух возрастает в 3,2 раза при увеличении
углеводородной цепи на каждую СН2-группу:
'?re-n/'7re = const ~3-2 Ш. 17)
где п — число метиленовых групп в углеводородном ра-
дикале.
Рис. 10. Изотермы поверхностного натяжения растворов
ПАВ с углеводородным радикалом, содержащим п,
в + 1 и п + 2 метилен.овъгх 'групп.
43
Это правило выполняется лишь для водных растворов ПАВ. Для
растворов ПАВ в неполярных растворителях поверхностная активность
при увеличении длины углеводородного радикала, наоборот, уменьща-
ется (обращение правила Дюкло — Траубе).
Ленгмюр дал теоретическое обоснование эмпирическому правилу Дюкло — Траубе.
Адсорбция и ориентация молекул ПАВ на границе фаз жидкость — воздух являются
самопроизвольно протекающими процессами, сопровождающимися уменьшением энер-
гии Гиббса системы. При введении ПАВ в полярную среду (воду) практически не-
гидратирующиеся углеводородные цепи ПАВ раздвигают молекулы воды, встраиваясь
в ее структуру. На осуществление этого требуется совершение работы против молеку-
лярных сил. Обратный процесс—выход молекул ПАВ на межфазную поверхность
(с ориентацией углеводородных цепей в сторону неполярной среды) идет самопроиз-
вольно с уменьшением энергии Гиббса системы, что соответствует работе адсорбции е.
Работа адсорбции зависит от длины углеводородной цепи ПАВ и. в расчете на 1 моль
молекул, состоящих из п лис л а СНг-звеиьев, составляет;
s = —AG = <pAA/i (II. 18)
где ср — работа адсорбции, отнесенная к одной СНг-группе.
Применительно к адсорбции из разбавленных растворов на основе уравнений
Генри (II. I) и изотермы Вант-Гоффа можно получить следующее выражение для
константы Генри /Ср:
А / AG \ / грААп \
Аг = —= ехр(^ или Ку = ехр I —— J (11.19)
Уравнение (II. 19) показывает, что значение константы Ку с увеличением числа
СНз-групп в молекуле ПАВ растет в кратное число раз. Для двух соседних членов
гомологического ряда ПАВ при условии постоянства концентрации и температуры
можно записать:
/<рААя\ - Г срАА (п + 1) 1
^Г(п) = ехР (—11 АГ(п+1) = ехр [--| (11.20)
Отсюда
А фАд А
Кг(„+1)/КГ (и) = ехР у-J ~ (И. 21)
Полученное выражение для соотношения Ку (n+ij/Ap (п) отражает правило Дюк-
ло —Траубе.
Коэффициент р равен 3,2 только при 20 °C. При повышении температуры его
значение уменьшается, приближаясь в пределе к единице. Уменьшение константы р об-
условлено возрастанием десорбции молекул ПАВ при повышении температуры и сни-
жением различия между поверхностной активностью гомологов.
; z Измерение поверхностной активности и адсорбции ПАВ позволяет
определить параметры адсорбционных слоев: площадь, занимаемую од-
ной молекулой, So и толщину поверхностного слоя 6. Величины sa и б
рассчитывают по экспериментально найденным значениям предельной
адсорбции Аоо. Предельную адсорбцию А» определяют по изотермам
адсорбции Г = f(c), для построения которых вычисляют несколько зна-
чений ds/dc. Для веществ с ярко выраженными поверхностно-актив-
ными свойствами величину адсорбции А можно принять равной гибб-
совской адсорбции Г (в молях на 1 м2 поверхности).
Площадь so, приходящуюся на одну молекулу в насыщенном адсорб-
ционном слое, вычисляют по уравнению
So==A~l!~ Ш-22)
лАлсо
44
Толщину адсорбционных слоев б рассчитывают по формуле
6 = . (11.23)
р
где М—молекулярная масса ПАВ; р— плотность ПАВ,
Сопоставление вычисленных значений толщины слоя б с длиной
ориентированных молекул дает возможность оценить тип поверхност-
ной пленки, определить ориентацию молекул ПАВ в адсорбционном
слое.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ .
Для проведения работы необходимы:
Установка для измерения поверхностного натяжения.
Бюретки. ,
Конические колбы емкостью 50 мл.
Фильтровальная бумага.
0,2 М растворы пропилового, бутилового и пентилового спиртов.
Готовят 5 водных растворов пропилового, бутилового и пентилового
спиртов разбавлением исходных растворов водой в соответствии с дан-
ными: > -V? ЧЛ. /
Объем исходного раствора Г
спирта, мл................. 25 19 12,5 6,3 3,2
Объем добавляемой воды, мл 0 6 12,5 18,7 21,8
Концентрация раствора с,
моль/л....................... 0,2 0,15 0,1 0,05 0,025
С помощью установки, указанной преподавателем, определяют по-
верхностное натяжение приготовленных растворов (методику измере-
ний см. в работе 1). Измерение поверхностного натяжения следует про-
водить быстро, так как может происходить испарение спирта с поверх-
ности раздела фаз.
По полученным данным для каждого спирта строят изотермы по-
верхностного натяжения и по ним находят поверхностную активность
g = — (d<j/de)c->o, а также значения da/de в нескольких точках изотерм.
По уравнению (1.21) вычисляют значения Г и строят изотермы адсорб-
ции Г — [(с). Данные расчета g, da/dc и Г для исследуемых спирточ
записывают в таблицу (см. табл. II. 2).
Таблица II. 2. Экспериментальные и расчетные данные по адсорбции
алифатических спиртов
Концентрация раствора спирта, моль/л Наибольшее давление в пузырьке Др или сила отрыва F Поверхностное натяжение аж-г’ Дж/м2 g=- \dcjc. >0 do de Г, . 2 моль/м
Затем рассчитывают соотношение поверхностных активностей для
соседних гомологов:
gC4H9OH gCsHtiOH
gC3H7OH £с4н9он
45
По изотермам Г = /(с) определяют значения ГмаКс и, приняв, что
для ПАВ Гмацс ~Аоо, по уравнениям (11.22) и (11.23) рассчитывают s0
и 6.
Полученные значения сравнивают с табличными и делают вывод об
ориентации молекул ПАВ в насыщенном адсорбционном слое.
Работа 8. ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ ПОВЕРХНОСТИ АДСОРБЕНТОВ
МЕТОДОМ ГАЗОВОЙ ХРОМАТОГРАФИИ
Цель работы: определение удельной поверхности адсорбента по удер-
живаемому объему; определение удельной поверхности адсорбента ме-
тодом тепловой десорбции (БЭТ).
Газовая хроматография используется для решения таких физико-
химических задач, как определение коэффициентов распределения и ак-
тивности, термодинамических функций распределения и адсорбции.
Этот метод применяется также для определения удельной поверхности
адсорбентов, катализаторов, наполнителей.
Наибольшее распространение для оценки 5УД получили следующие
хроматографические методы: метод, основанный на установлении изо-
термы адсорбции по параметрам хроматографического пика; фронталь-
ный метод, по которому величина адсорбции рассчитывается по пло-
щади, заключенной между концентрационными фронтами адсорбата и
несорбирующегося газа; метод тепловой десорбции, в котором количе-
ство сорбирующегося вещества находится по количеству адсорбата, по-
ступившему при нагревании из адсорбента в поток газа-носителя.
На рис. 11 приведена принципиальная схема газового хроматографа.
Газ-носитель из баллона / под давлением поступает в дозатор 2 (до-
затор-испаритель служит для ввода пробы в поток газа-носителя) и
последовательно проходит хроматографическую колонку 3 и детек-
тор 4. Сигнал детектора усиливается (блок 6) и подается на потенцио-
метр 7. Для испарения жидкой или, что реже, твердой пробы, в доза-
торе поддерживается необходимая температура.
Хроматографическая колонка представляет собой металлическую
или стеклянную трубку, заполненную насадкой (адсорбентом). Детек-
тор предназначен для определения содержания компонентов в потоке
газа-носителя. Работа детекторов основана на измерении одного из фи-
зических параметров компонента (теплопроводность, потенциал иони-
зации, плотность и др.).
Из детектора через уси-
литель сигнал поступает в
самопишущий потенциометр,
который выписывает хрома-
тограмму—графическое изо-
бражение зависимости вы-
Рис. 11. Блок-схема газового хро-
матографа:
1 — баллон с газом-носителем; 2 — доза-
тор-испаритель; 3—хроматографическая
колонка; 4 — детектор; 5 — термостат;
6—электронный усилитель; 7—самопи-
шущий потенциометр.
46
ходного сигнала детектора от времени или объема газа-носителя
(рис. 12).
На хроматограмме точка О соответствует моменту ввода пробы в
колонку. Вещество, не адсорбируемое в колонке, выходит через время
То (на хроматограмме — точка О'). Время удерживания такого компо-
нента называется «мертвым» временем удерживания. Оно пропорцио-
нально свободному объему хроматографической колонки Уо и обратно
пропорционально объемной скорости газа-носителя v:
t0 = V0/v (11.24)
Время, прошедшее от момента ввода пробы до появления максимума
пика, называется временем удерживания компонента тг. При постоян-
ной скорости движения диаграммной ленты самописца время удержи-
вания удобно выражать расстоянием I, пройденным пером самописца
от точки О до А. Объем газа-носителя, прошедшего через хроматогра-
фическую колонку за время тг, называется удерживаемым объемом Vr.
Поскольку удерживаемый объем зависит не только от природы адсор-
бента и адсорбата, но и от свободного объема колонки, то вводят по-
нятие приведенного удерживаемого объема У':
7; = 7г-70 = И(тг-т0) (11.25)
Скорость газа-носителя обычно экспериментально определяют при ком-
натной температуре и атмосферном давлении, поэтому в приведенный
объем вводят поправку, учитывающую условия работы хроматографи-
ческой колонки. Рассчитанный таким образом удерживаемый объем на-
зывается исправленным удерживаемым объемом Ум:
И„ = /< (П. 26)
где /’—поправочный коэффициент, учитывающий сжимаемость газа и отличие условий
хроматографирования от условий внешней среды.
Величину, полученную как отношение Vu к массе адсорбента т,
находящегося в хроматографической колонке, называют удельным
удерживаемым объемом:
Vm = Vu/m (II. 27)
В практической работе используется еще ряд характеристик, таких,
как относительный удерживаемый объем, коэффициенты разделения и
селективности, и т. д.
Для количественной обработки хроматограмм можно использовать
либо высоту пика, пропорциональную концентрации вещества, либо
площадь пика, пропорциональ-
ную абсолютному содержанию
компонента в газовой смеси. Ча-
ще определяют площадь пиков,
используя один из следующих ме-
тодов: планиметрию, вырезание и
взвешивание пиков, расчет пло-
щади как произведение основа-
ния пика а на половину высоты —
Рис. 12. Хроматограмма и ее основные
параметры.
47
или же с помощью электронных, аналоговых или механических ин-
теграторов. Количество анализируемых компонентов по площади пиков
можно определить, пользуясь калибровочной кривой (метод абсолют-
ной калибровки), или при помощи вводимого стандарта (метод внут-
ренней нормализации).
Наиболее простым из хроматографических методов определения
удельной поверхности является метод, основанный на измерении удер-
живаемых объемов. Для двух образцов 1 и 2 адсорбента одной и той же
природы, но с различной удельной поверхностью можно записать:
Vml = Bsya ! exp (Q/RT)- (II. 28)
V m2 ~ Bsyll2exp(Q/RT)’’ (11.29)
где Vmt и Vmz — удельные удерживаемые объемы, рассчитанные по уравнению (II. 27);
В — постоянная при данной температуре, зависящая от изменения энтропии адсор-
бата при адсорбции; syA t и syA 2 — удельная поверхность адсорбентов; Q — теплота
адсорбции.
Если удерживаемые объемы измерять при одной температуре, то
= или зуд 2 = (П. 30)
У ml $уд 1 Vml
Из уравнения (11.30) следует, что, вычислив Vmi и Vm2 по уравне-
ниям (11.25) и (11.27) для двух образцов адсорбента и зная удельную
поверхность одного из них дУД1, можно рассчитать удельную поверх-
ность второго сорбента.
Рассмотренный сравнительный метод применим для адсорбентов од-
ной природы и требует знания удельной поверхности для одного из
образцов. Этих недостатков лишен метод тепловой десорбции. Со-
гласно этому методу по изменению состава газового потока (гелий с
добавкой азота), проходящего через хроматографическую колонку с
исследуемым адсорбентом, определяют количество азота, адсорбиро-
ванного из газовой смеси при охлаждении адсорбента жидким азотом
и десорбированного с него при последующем нагревании его до ком-
натной температуры. Изменяя концентрацию азота в газовой смеси,
можно установить количество адсорбированного газа при различных
концентрациях азота в исходной газовой смеси и, следовательно, по-
строить изотерму адсорбции и вычислить по ней, используя линейную
форму уравнения БЭТ, предельную емкость монослоя и соответствую-
щую ей удельную поверхность адсорбента.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Вариант 1. Определение удельной поверхности адсорбентов
по удерживаемому объему
Для проведения работы необходимы:
Хроматограф.
Микрошприц.
Адсорбент, например силикагель.
Адсорбат, например бензол или гептан.
Для выполнения работы может быть использован любой газовый
хроматограф с катарометром в качестве детектора. Удобнее работать
48
Таблица II. 3. Экспериментальные и расчетные данные по определению
удельной поверхности адсорбента
Номер колонки V, мл/мин Z, мм V мин Vr- мл V'„ мл т, г Vm. ЫЛ'Г Дд.
1 2
с хроматографом, в котором проба вводится в одну из двух равноцен-
ных колонок, а вторая колонка является сравнительной (подается чис-
тый газ-носитель). Для того чтобы ячейки детектора работали в иден-
тичных условиях, т. е. чтобы электрическое сопротивление элементов
детектора определялось только составом газовых потоков, скорость
газа-носителя через них должна быть примерно одинаковой (допусти-
мое отклонение ±2 мл/мин).
Одну из колонок хроматографа (колонка 7) заполняют силикагелем
(масса гп\) с известной удельной поверхностью $уд1, а вторую — адсор-
бентом (масса тг), удельную поверхность которого необходимо опре-
делить. Условия хроматографического анализа: длина колонки 1 м,
внутренний диаметр 3 мм; температура термостата колонок 160 или
180°С для бензола или гептана соответственно; газ-носитель — азот,
его расход 30 мл/мин; скорость движения диаграммной ленты КСП—
12 мм/мин.
Работу выполняют в следующей последовательности. Включают хро-
матограф (под руководством преподавателя) и регулируют скорость
газа-носителя v в хроматографических колонках. Затем с помощью
микрошприца в хроматографическую колонку 1 вводят 5—7 раз по
2 мкл адсорбата (момент ввода пробы отмечается на диаграммной лен-
те. Каждую последующую пробу вводят только после полного проявле-
ния пика, соответствующего предыдущей пробе. Аналогичным образом
вводят адсорбат в колонку 2 с а/щорбентом, удельную поверхность ко-
торого определяют. *
Рассчитывают значения тг как отношения расстояний /, пройденных
пером самописца от момента ввода пробы (точка О на рис. 12) до се-
редины пиков (точка А), к скорости движения диаграммной ленты са-
мописца. Затем по уравнениям (11.24) — (11.27) рассчитывают вели-
чины т0, V' и Vm. По уравнению (II. 30) вычисляют удельную поверх-
ность адсорбента в колонке 2. Для расчета используют средние значе-
ния Vm, 1 и Vm, 2, найденные по результатам нескольких измерений. По-
лученные данные записывают в таблицу (см. табл. II. 3).
Вариант 2. Определение удельной поверхности адсорбентов методом
тепловой десорбции азота
Для проведения работы необходимы:
Хроматографическая установка.
Секундомер.
Адсорбент.
Азот, гелий.
4 Зак. 673
49
Рис. 13. Блок-схема хроматографической
установки для определения удельной по-
верхности методом тепловой десорбции:
1,2— баллоны стазами; 3 — смеситель; 4 — пенный
расходомер; 5 — потенциометр; 6,7— камеры детек-
тора; 8 — колонка.
Блок-схема хроматографической
установки, используемой для опре-
деления удельной поверхности ад-
сорбентов методом тепловой де-
сорбции, представлена на рис. 13.
Потоки гелия и азота из балло-
нов 1 и 2 подаются в определенном соотношении в смеситель 3, из ко-
торого газовая смесь поступает в сравнительную камеру детектора 6 и
далее в колонку 8 с исследуемым адсорбентом, в которой при охлажде-
нии происходит адсорбция азота. Из колонки газовая смесь поступает
в измерительную камеру детектора 7. Детектор фиксирует изменение
состава газовой смеси в результате адсорбции. Сигнал детектора посту-
пает на самопишущий потенциометр 5.
Объемная скорость газовой смеси должна быть постоянной при всех
значениях объемной доли азота в смеси. Скорость подачи смеси кон-
тролируют на выходе из детектора пенным расходомером 4.
Условия хроматографирования: длина колонки — 0,1 м; диаметр ко-
лонки— 3 мм; скорость газового потока через колонку — 30 мл/мин;
начальное содержание азота в газовой смеси составляет 5% (об.).
Включают хроматограф и с помощью редукторов на баллонах 1 и 2
устанавливают необходимую скорость подачи газовой смеси. При от-
сутствии охлаждения хроматографической колонки через сравнитель-
ную и измерительную камеры детектора проходит газовая смесь оди-
накового состава, поэтому вначале перо самопишущего потенциометра
записывает «нулевую» линию (рис. 14).
Хроматографическую колонку с исследуемым адсорбентом поме-
щают в сосуд Дьюара с жидким азотом, продолжая пропускать через
нее газовую смесь. При охлаждений' колонки в результате адсорбции
азота и изменения состава газовой смеси перо потенциометра начинает
отклоняться от «нулевой» линии. Смещение пера потенциометра проис-
ходит до тех пор, пока сорбент полностью не будет насыщен азотом
при данной концентрации азота в газовой смеси. Равновесие счи-
тается установленным, когда перо самописца снова отмечает «нуле
вую» линию. Затем сосуд с жидким азотом удаляют, и происходит пол
ная десорбция азота с поверхности адсорбента при комнатной темпе
ратуре. При этом самописец вычерчи-
вает кривую десорбции (см. рис. 14).
Операции адсорбции и десорбции при
данном составе газовой смеси повто-
ряют несколько раз (для оценки вос-
производимости результатов).
Подобные операции проводят, ме-
няя концентрацию азота в газовой
Рис. 14. Хроматограмма адсорбции и десорбции
азота.
50
Таблица II. 4. Экспериментальные и расчетные данные по определению
удельной поверхности адсорбента методом тепловой десорбции
X, % (об.) р, Па p/ps А P/Ps A(l-p/ps) Лоо
смеси (через 5%) в интервале 5—25 % (об.). Количество азота, адсор-
бированное при каждой исходной концентрации азота в газовой смеси,
определяют по площади пика на кривой десорбции, используя калибро-
вочный график.
Для получения калибровочного графика «объем азота (мл)’ — пло-
щадь пика» проводят хроматографический анализ модельных азотге-
лиевых смесей. Рассчитывают величину адсорбции А как отношение
сорбированного азота к массе сорбента в колонке.
Парциальное давление азота р, при котором проводилась адсорб-
ция, находят по формуле
где х — содержание азота в газовой смеси, % (об.); р0 — барометрическое давление.
Далее рассчитывают относительное давление азота p/ps (где ps —
давление насыщенного пара азота). Строят изотерму адсорбции А —
— p/ps. Представляют изотерму в координатах линейной формы урав-
нения БЭТ:
p/ps ,
ЛА—P/Ps) plps
Отрезок, отсекаемый на оси ординат, соответствует величине 1/ЛооС,
а тангенс угла наклона этой прямой равен (С— 1)/ЛооС. Из этих дан-
ных рассчитывают значение Лоо. Полученные результаты записывают
в таблицу (см. табл. II. 4).
Найдя значение Лоо, по уравнению (II. 4) вычисляют удельную по-
верхность исследуемого адсорбента, приняв для азота so == 0,162 нм2.
Работа 9. ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ СМЕСИ ИОНОВ
С ПОМОЩЬЮ ИОНООБМЕННЫХ СМОЛ
Цель работы: определение полной обменной емкости катионита и кон-
станты ионного обмена; разделение смеси катионов на ионообменной
хроматографической колонке.
Ионный обмен представляет собой обратимое стехиометрическое за-
мещение подвижного иона, связанного с ионогенной группой ионита,
на другой одноименно заряженный ион, находящийся в растворе. Ко-
личественной характеристикой ионита является полная обменная ем-
кость ПОЕ. Определение ПОЕ можно осуществить статическим или ди-
намическим методом, основанном обычно на реакциях, протекающих
4
61
в водных растворах:
RSO,HX + NaOH —> RSO~Na+ + Н2О
RNH+OH~+HC1 —> RNH3hCr+H2O
При статическом методе смолу, например катионит в Н+-форм<
титруют раствором щелочи.
При динамическом методе ПОЕ определяется с помощью хромате
графических колонок. Через колонку, заполненную ионообменной смс
лой, пропускают раствор электролита и регистрируют зависимость koi
центрации поглощаемого иона в выходящем растворе (элюате) от обт
ема прошедшего раствора (выходная кривая).
В работе ПОЕ сульфокатионита в Н+-форме определяют динамиче
ским методом по количеству кислоты в элюате, образующейся в ре
зультате вытеснения из смолы ионов Н+ ионами Na+:
RSO~H+ + Na+ + СГ —> RSO~Na+ + H+ + СГ
ПОЕ рассчитывают по формуле
ПОЕ=^2^ (II. 3!
т
где Уобщ — суммарный объем раствора, содержащий вытесненную из смолы кислот;
с—концентрация кислоты; т — масса ионообменной смолы в колонке.
Константу ионного обмена можно определить из данных о равне
веском распределении ионов в статических условиях (равновесное сс
стояние при ионном обмене описывается законом действия масс),
также динамическим методом по скорости перемещения зоны веществ
по слою смолы (элюентная хроматография). Если через колонку с ка
тионитом, в верхней части которой находится сорбированный ион М21
пропускается раствор кислоты, то в смоле происходит многократны
процесс обмена:
zRSO3'H++M2+ ч=±: (RS03“)2M2+ + zH+
В условии равновесия при распределении ионов между протекаю
щим раствором и слоями ионита (равновесная хроматография) отне
шение между концентрациями иона Мг+ в смоле с и растворе с (пр
малых значениях концентрации) равно
[н+1г ,ттг
с К''2 [н+]г
Скорость перемещения хроматографической зоны с постоянной кон
центрацией иона по высоте колонки равна
dr s(dc/dc) v 1
где v — объемная скорость пропускания раствора кислоты; s — площадь сечения кс
донки.
В реальных условиях равновесие не успевает полностью устано
виться вследствие медленной диффузии ионов в зернах смолы, что при
водит к размытию хроматографической зоны. Приведенное уравнени
(П. 34) хорошо описывает скорость движения зоны с максимально воз
52
можной концентрацией иона. Выражение (11.34) можно преобразовать
следующим образом:
dx .... h к — dx hv
j~- _ ’ *макс — yTMaKC, — (11.35)
а L тмакс UX Имакс
где ЦМакс и т„акс — объем протекающего через колонку раствора и время, отвечающие
максимуму выходной кривой; h — длина колонки.
При малых концентрациях иона Мг+ и постоянной концентрации
кислоты производную dc/dc можно заменить на отношение конечных
величин с/с и считать концентрацию ионов водорода в смоле [Н+] рав-
ной ПОЕ. Тогда
[ПОЕ]2
_ д12 (11.36)
de с [Н J
Из уравнений (11.34) — (11.36) следует:
к ___ Емакс [Н+]
Kl' J ~ ЛгЮЁрАГ ( 37)
Для двухвалентного катиона М2+, например кобальта или меди,
концентрационная константа равновесия
. [м2+] [н+]2
’2 [м2+][н+]2
рассчитывается по уравнению (11.37) при г — 2. Значение ЕмаКс нахо-
дят с учетом поправки на свободный объем колонки Vo-
^макс = ^акс-Е0 (11.38)
*
где Емакс — объем элюата, вышедшего из колонки от начала элюирования М2+ кис-
лотой до появления максимальной концентрации М2+ в элюате (максимум на выход-
ной кривой).
При хроматографическом разделении ионов широко используется
различная их склонность к образованию комплексных соединений. На-
пример, кобальт и медь могут быть разделены в колонке с сульфока-
тионитом при помощи десорбента — раствора цитрата калия.
Если пропустить через катионит в К+-форме раствор, содержащий
небольшое количество разделяемых элементов в отсутствие комплексо-
образователя, то ионы кобальта и меди поглощаются в верхнем слое
смолы.
Разделить кобальт и медь динамическим методом, промывая колон-
ку раствором, содержащим ионы К+, трудно, так как константы обмена
ионов Со 2+ и Си2+ на ион К+ отличаются не намного.
Если через колонку пропускать раствор цитрата калия (десорбент),
то при контакте его со смолой происходит частичная десорбция меди
и кобальта вследствие комплексообразования, например:
(RSO“)2M2+ + 2К+ + А3“ ч=> 2RSO3~K+ + [MA]~
где А3- — комплексообразующий ион лимонной кислоты.
С лимонной кислотой ионы кобальта и меди образуют несколько
различных комплексных соединений [МА]~, [МА2]~4 и др., которые не
адсорбируются на катионите.
53
Рис. 15. Выходные кривые хроматографиче-
ского разделения ионов меди и кобальта.
Общая концентрация ионов меди
или кобальта в растворе равна
сойщ=[м‘-’+] + f [MAZ] =
1
= [М2+] ( 1 + 2 [АГ' ) (П. 39)
где Р/ — константы устойчивости комплексных ионов; i — число комплексообразующих i
ионов в комплексном соединении. :
Уравнение (11.34) для скорости перемещения вещества по колонкеi
с учетом выражений (II. 36) и (II. 39) в присутствии комплексообразо- '
вателя имеет вид ;
/ ‘ \ \
4к+]2 1+У₽г[АГ I
dx __ v _______ \ 1 / <nJ
~dV - s dc/dco6^ sKk+ М2+ [ПОЕ]2 (IL4O)I
i
Из уравнений (11.39) и (11.40) следует, что в присутствии ком-
плексообразователя уменьшается концентрация катионов, способных
адсорбироваться на катионите, возрастает концентрация неадсорбиру-
ющихся комплексных соединений (при постоянном собш) и в результате
увеличивается скорость перемещения ионов данного вещества по ко-
лонке. При протекании вымывающего раствора через колонку происхо-
дит многократная десорбция и сорбция разделяемых ионов, причем ка-
тионы меди, образующие более устойчивые комплексные соединения,
перемещаются вдоль слоя смолы с большей скоростью, чем ионы ко-
бальта. В результате в колонке формируются различные по окраске
зоны — для меди голубая, для кобальта оранжевая; первым из колонки
выходит раствор, содержащий комплексные соединения меди, затем —
кобальта (рис. 15).
Скорость перемещения вещества по колонке обратно пропорциональ-
на Умакс- Относительная скорость перемещения по колонке разделяе-
мых ионов может быть рассчитана по уравнению
(dx/dT)Cu2+ tC акс (Со) - ц0
(rfx/dT)Co2+ умакс (Си) - цо
1 + £ ₽z [А]
I
(П.41)
Поскольку константы ионного обмена К+ на Со2+ и Си2+ прибли-
зительно одинаковы (Дк+ Со2+ = АГК+ Си2+), относительная скорость чис-
ленно равна параметру, характеризующему относительную степень
комплексообразования разделяемых ионов.
54
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Часть 1. Определение ПОЕ смолы КУ-2
Для проведения работы необходимы:
Хроматографическая колонка, заполненная 5 г смолы КУ-2.
pH-метр типа pH-340.
Градуированные пробирки.
Мерный цилиндр емкостью 250 мл.
Стакан емкостью 50 мл.
Бюретка.
3 М раствор НС1.
1,5 М раствор NaCl.
0,2 М раствор КОН.
Фенолфталеин.
В работе используется хроматографическая колонка, устройство
которой изображено на рис. 16. Рабочий объем колонки 5, представ-
ляющей трубку с внутренним диаметром 10 мм и высотой 200 мм, за-
полнен сульфокатионитом КУ-2 с размером зерен 0,4—0,6 мм. Ем-
кость 2 для элюента соединена с рабочим объемом колонки трубкой 4,
благодаря которой создается избыточное гидростатическое давление,
необходимое для прохождения раствора через слой ионита. Тонкий
стеклянный стержень 3 способствует заполнению трубки раствором.
Скорость пропускания раствора регулируется краном 6.
Предварительно переводят ионогенные группы ка-
тионита в Н+-форму, пропуская через колонку 40 мл
3 М раствора НС1. Для этого 20 мл кислоты нали-
вают в емкость 2 и через несколько секунд (после пол-
ного выхода пузырьков воздуха из трубки 4) откры-
вают кран 6 и устанавливают необходимую скорость
вытекания раствора (1 капля в 1 с). Когда уровень
раствора опустится до нижней части трубки 4, за-
крывают кран 6 и наливают в емкость 2 новую пор-
цию кислоты. Снова устанавливают оптимальную ско-
рость элюирования.
Затем смолу промывают водой до тех пор, пока
pH в вытекающем растворе не будет иметь значение
3,5—4,0. Для этого потребуется около 50 мл воды.
Рекомендуется измерять pH после того, как из ко-
лонки выйдет 25—30 мл раствора. Отбирают алик-
вотные части по ~5 мл и анализируют их с помощью
рН-метра.
Через подготовленную таким образом колонку про-
пускают 1,5 М раствор NaCl. Элюат собирают порция-
ми по 3—4 мл в градуированные пробирки. В каждой
пробирке определяют точный объем пробы и измеряют
pH. Процесс вытеснения ионов Н+ из катионита
Рис. 16. Хроматографическая колонка:
1 — отводная трубка; 2 — емкость; 3— стеклянный стержень; 4— трубка; 5 —
колонка с ионитом; 6 — кран.
55
Таблица II. 5. Экспериментальные данные по определению ПОЕ смолы КУ-2.
Номер пробы Объем пробы, мл pH пробы Суммарный объем элюата V, мл
проводят до тех пор, пока pH элюата не будет иметь значение 2,7—3,0
Полученные результаты записывают в таблицу (см. табл. II. 5) и строят
выходную кривую, т. е. зависимость pH от объема вышедшего элюата
(суммарный объем проб).
Все растворы, содержащие вытесненную из смолы кислоту (остатки
элюата в пробирках, растворы из кюветы pH-метра, вода после про-
мывки пробирок и кюветы pH-метра), объединяют, сливают в мерный
цилиндр и измеряют объем УОбщ. Из полученного раствора отбирают
пипеткой аликвотную часть 10 мл и титрованием 0,2 М раствором КОН
в присутствии фенолфталеина определяют концентрацию кислоты в
объединенном растворе. Полную обменную емкость катионита КУ-2
рассчитывают по формуле (11.32).
Часть 2. Определение константы ионного обмена
Для проведения работы необходимы:
Хроматографическая колонка со смолой КУ-2.
pH-метр типа pH-340.
Фотоэлектрический колориметр (ФЭК-56 М).
Градуированные пробирки.
Стакан емкостью 50 мл.
3 М и 0,6 М растворы НС1.
Раствор соли кобальта(П) или меди(П) концентрацией 30 мг/мл.
Предварительно катионит переводят в Н+-форму, пропуская через
него 40 мл 3 М НС1 (см. определение ПОЕ смолы КУ-2). Затем смолу
промывают водой до тех пор, пока pH выходящего из колонки раствора
не будет равно ~3. После этого в свободную емкость 2 (см. рис. 16)
колонки вносят 1 мл 0,6 М раствора НС1, открывают кран 6 и дают
впитаться кислоте в верхний слой смолы. Затем в емкость 2 наливают
воду и собирают вытекающий раствор в пробирки порциями по — 1,5 мл
(объем уточняют по градуировке пробирок) и измеряют значение pH
в каждой пробе.
Строят график зависимости pH от объема вышедшего элюата и по
минимальному значению pH определяют свободный объем колонки Уо-
Через колонку пропускают 4 мл
раствора соли меди или 2,5 мл раство-
ра соли кобальта. Затем через колон-
ку пропускают 3 М НС1 со скоростью
1 капля за 2 с для элюирования ад-
сорбированных в верхней части слоя
смолы ионов М2+. Порции элюата по
Рис. 17. Зависимость оптической плотности от
объема элюата.
56
Таблица II. 6. Экспериментальные данные по определению константы
ионного обмена
Номер пробы D пробы Суммарный объем проб элюата V, мл
4 мл собирают в пробирки и с помощью- электрофотоколориметра опре-
деляют их оптическую плотность D, используя для растворов меди
светофильтр с максимумом светопропускания при 620 нм, а для раство-
ров кобальта — 480 нм. Раствором сравнения служит вода. Методика
определения оптической плотности приведена в работе 17. Полученные
результаты записывают в таблицу (см. табл. II. 6).
Строят график зависимости D от V (рис. 17) и по максимальному
значению оптической плотности находят объем элюата Умакс’ вышеД'
шего из колонки от начала элюирования до появления максимума на
выходной кривой, и по уравнению (11.38) рассчитывают значение УМакс.
Константу ионного обмена рассчитывают по уравнению (11.37), при
этом за концентрацию [Н+] принимают концентрацию НС1 в элюате;
значение ПОЕ определяют в первой части работы.
Часть 3. Разделение смеси ионов Си2+ и Со2+
Для проведения работы необходимы:
Хроматографическая колонка с ионообменной смолой КУ-2.
Фотоэлектрический колориметр (ФЭК 56М).
Градуированные пробирки.
Стакан емкостью 50 мл.
Раствор соли кобальта(II) и соли меди(II).
3 М раствор НС1.
1,5 М раствор КС1.
Раствор, содержащий 0,15 моль/л однозамещенного цитрата калия
и 0,15 моль/л двузамещенного цитрата калия.
Катионит предварительно переводят в К+-форму (см. определение
ПОЕ смолы КУ-2). Для этого через колонку со скоростью 2 капли
в 1 с пропускают последовательно растворы: 20 мл 3 М НС1, 40 мл
1,5 М КС1, 20 мл Н2О. Затем около 4 мл разделяемой смеси ионов меди
и кобальта пропускают через смолу и колонку промывают небольшим
количеством (10—15 мл) воды. При этой операции ионы Си2+ и Со2+
поглощаются в верхней части катионита (этот слой приобретает бурую
окраску). В емкость 2 (см. рис. 16) наливают раствор цитрата калия,
открывают кран колонки и собирают элюат в пробирки по 4,0—4,5 мл.
Скорость элюирования должна соответствовать 1 капле в 1 с.
Периодически добавляют в емкость 2 десорбент и проводят элюи-
рование до полного выхода кобальта из колонки, о чем свидетельствует
исчезновение оранжевой окраски элюата. С помощью фотоэлектроко-
лориметра измеряют оптическую плотность D каждой порции элюата
со светофильтром 620 нм (для определения содержания меди) и затем
со светофильтром 480 нм (для определения содержания кобальта).
Методика определения оптической плотности приведена в работе 17.
В качестве раствора сравнения используют воду. По калибровочным
57
графикам, построенным с применением стандартных растворов солей
меди и кобальта, находят концентрации Си2+ и Со2+ в каждой пробе.
Полученные результаты записывают в таблицу (см. табл. II. 7) и
строят выходную кривую хроматографического разделения Си2+ и Со2+
(см. рис. 15).
Таблица II. 7. Экспериментальные данные по хроматографическому разделению
Си2+ и Со2+
Номер пробы Объем пробы, мл D при 620 нм [Си2+], г/л D при 480 нм [Со2 + ], г/л Объем элюата V, мл
По уравнению (II. 40) рассчитывают параметр относительной сте-
пени комплексообразования ионов меди и кобальта; Го находят по дан-
ным определения константы ионного обмена.
Работа 10. РАЗДЕЛЕНИЕ СМЕСИ ПОЛИМЕРА И МИНЕРАЛЬНОЙ СОЛИ
И ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ МАССЫ ПОЛИМЕРОВ
МЕТОДОМ ГЕЛЬ-ХРОМАТОГРАФИИ
Цель работы: разделение смеси полимера и минеральной соли (NaCl);
определение молекулярной массы полимеров.
Гель-хроматография (гель-фильтрационная, гель-проникающая, мо-
лекулярно-ситовая хроматография) применяется для разделения и ана-
лиза высокомолекулярных соединений, а также для отделения их от
низкомолекулярных веществ. Этим методом можно определить молеку-
лярную массу полимеров, рассчитать кривую распределения макромо-
лекул по молекулярным массам.
Гель-хроматография является одним из видов жидкостной хромато-
графии. При анализе этим методом растворенное вещество распреде-
ляется между свободным растворителем и растворителем, находящимся
во внутренних полостях пористых частиц наполнителя. Свободный рас-
творитель является подвижной фазой, а пористые частицы, содержащие
растворитель, образуют неподвижную фазу.
Разделительная способность зависит от размеров молекул веществ
и размеров внутренних полостей частиц наполнителя. Большие моле-
кулы, эффективные размеры которых превышают размеры пор напол-
нителя, вместе с протекающим в колонке растворителем передвигаются
в зазорах между частицами и выходят из колонки первыми со свобод-
ным объемом Го (где Го— объем растворителя в колонке между час-
тицами наполнителя без учета суммарного объема пор Г5). Очень ма-
ленькие молекулы, способные проникать в поры, выходят из колонки
последними с общим объемом растворителя (элюента), равным сумме
(Vo+Vs). Молекулы промежуточного размера, которые частично мо-
гут проникать в поры, выходят с промежуточным объемом раствори-
теля, называемым удерживаемым объемом Vr:
Vr^V„ + KVs (11.42)
Коэффициент К в этом уравнении называется коэффициентом рас-
пределения, он характеризует меру доступности объема пор наполни-
58
теля колонки для данного компонента. Если К — 0, то Vr = Vo, т. е.
макромолекулы не проникают в поры наполнителя. Если К = 1, тс
Vr = Vo + Vз, т. е. молекулы свободно проникают во все поры. При
О < К < 1 молекулы проникают в часть объема пор.
В качестве наполнителя колонок в гель-хроматографии часто ис-
пользуют сшитые полимеры с порами различного диаметра. Для ана-
лиза водных растворов применяют гидрофильные вещества, хорошо
набухающие в воде: гели декстрана (сефадексы, молселекты), представ-
ляющие собой трехмерную структуру, агарозные гели, полиакриламид-
ные гели. Эти наполнители относятся к категории мягких, они сильно
сжимаются в колонке при относительно небольших давлениях. Для
органических растворов используют полужесткие наполнители: макро-
пористые полистирольные гели, а также полиметилметакрилат, поли-
винилацетатные гели (меркогель) и др. К жестким наполнителям, ко-
торые применяются для высокоэффективной жидкостной хроматогра-
фии при высоких давлениях, относятся силикагели и макропористые
стекла. Недостатком этих наполнителей является высокая адсорбцион-
ная активность силикатной поверхности (К > 1).
В гель-хроматографии применяются наполнители различных марок,
отличающиеся размером пор. При использовании в качестве наполни-
теля, например, сефадекса марки G-50 для разделения фракций поли-
этиленгликоля Н(—СНОН—СН2—)„ОН первыми будут выходить из
колонки фракции с относительной молекулярной массой больше 10 000
(7< = 0). Последними будут выходить фракции полимера с молекуляр-
ной массой, меньше 500, и в их числе часто присутствующие минераль-
ные соли (NaCl и др.), для которых Д =1. Таким образом, с помощью
сефадекса G-50 могут быть разделены фракции полиэтиленгликоля с
молекулярной массой в области 500—10 000.
В интервале фракционирования по молекулярной массе М опреде-
ленного гомологического ряда полимеров значения К в основном ли-
нейно зависят от lgМ:
K = ci—c2lgJM (П.43)
где Ci и сг — константы.
Разделение двух соседних компонентов 1 и 2 (фракций) характе-
ризуется коэффициентом разделения а, который определяется по урав-
нению
Vr(l)-Vr (2)
(A У, + ДУ2)/2
(II. 44)
где Ег(1) и Vr(2)—удерживаемые объемы компонентов; AVi и ДЦ2— ширина пиков
у основания, соответствующих компонентам 1 и 2.
При постоянной скорости потока растворителя, проходящего через
колонку, удерживаемые объемы пропорциональны времени появления
максимумов от начала хроматографирования (времени удерживания
тг). Значения ДЕ также могут быть выражены, в единицах времени. То-
гда уравнение (11.44) можно записать в виде
2 [тг (2) — тг (1)]
ДТ1 + Дт2
(11.45)
Таким образом, коэффициент разделения в основном определяется
шириной пиков и расстоянием между ними.
59
rtf
15
Ю
15
Рис. 18. Схема установки для гель-хро-
матографического разделения и анализа
полиэтиленгликоля и хлорида натрия:
1,2— сосуды; 3,6— краны; 4 — пробка на шлифе;
5— колонка; 7 — кондуктометрическая ячейка;
8 — щит; 9— милливольтметр; 10— самопишущий
потенциометр; 11 — рукоятка включения фото-
электроколориметра; 12 — рукоятка грубого
регулирования чувствительности; 13 — кювета;
14 — рукоятка точного регулирования чувстви-
тельности; 15— капилляр; 16— зажим.
Схема установки для проведения гель-хроматографии приведена
на рис. 18. Анализируемый раствор вносится в верхнюю часть колон-
ки 5 и проходит по колонке с растворителем (водой), подаваемым из
сосуда 2. Выходящий из колонки раствор для регистрации фракций
полимера (полиэтиленгликоля) смешивается с реагентом (0,01 М рас-
твор иода), который поступает из емкости 1. При смешивании образу-
ется окрашенное комплексное соединение. Смесь проходит через про-
точную кювету 13 фотоэлектроколориметра KF*, с помощью которого
измеряют светопоглощение раствора. Сигнал от фотоколориметра че-
рез щит подается на самопишущий потенциометр 10 (КСП-4).
Содержание низкомолекулярного соединения (NaCl) в выходящем
из колонки растворе определяется по электропроводности в измери-
тельной ячейке 7. Сигнал от ячейки через щит подается на милливольт-
метр 9 и самопишущий потенциометр 10. (Вначале потенциометр запи-
сывает сигнал от фотоколориметра, а затем после выхода из колонки
фракций полиэтиленгликоля переключается вручную на запись электро-
проводности.)
* Фотоэлектроколориметр типа KF позволяет быстро регистрировать изменения опти-
ческой плотности с одновременной записью сигнала на самопишущем потенциометре.
60
Расход элюента (растворителя, подаваемого в колонку) и расход
реагента постоянны, что обеспечивается применением сосудов Мари-
отта 1 и 2. Расход элюента зависит от высоты, на которой находится
сосуд 2, и гидродинамического сопротивления наполнителя колонки.
Расход реагента определяется длиной и диаметром капилляра, встав-
ленного в резиновый шланг, а также высотой размещения сосуда 1.
Эта высота выбирается такой, чтобы на 1 мл раствора, выходящего
из колонки, приходилось 5 мл реагента.
Так как расход элюента постоянный, то объем вышедшего раствора
пропорционален времени элюирования и перемещению диаграммной
ленты самопишущего потенциометра.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Установка для проведения гель-хроматографии (наполнитель, на-
пример, сефадекс G-50).
Пипетка емкостью 1 мл, 2 шт.
0,0IM раствор иода.
Анализируемый водный раствор, содержащий несколько олигомеров
полиэтиленгликоля, в их числе олигомер с молекулярной массой 20000
(содержание каждого олигомера 0,2 г/л) и NaCl (0,01 моль/л).
Заполняют сосуд 2 (см. рис. 18) дистиллированной водой до от-
метки и плотно закрывают пробкой с трубкой. В сосуд 1 наливают
0,5 л реагента (раствор иода) до отметки и плотно закрывают пробкой
с трубкой. Включают фотоэлектроколориметр и щит 8 в сеть. Тумблер
на щите переводят в положение «фотоэлектроколориметр». Рукоятку 11
фотоэлектроколориметра устанавливают в положение «~». При закры-
тых кранах 3 и 6 отсоединяют пробку 4 со шлангом от колонки и пол-
ностью открывают кран 6. При этом вода медленно вытекает из ко-
лонки и верхний ее уровень В — В понижается. Как только уровень
воды совпадает с уровнем А — А (граница слоя сефадекса), кран 6
закрывают. Затем в верхнюю часть колонки пипеткой вносят 1 мл ана-
лизируемого раствора. Касаясь кончиком пипетки внутренней стенки
колонки, раствор приливают осторожно и так медленно, чтобы не про-
исходило взмучивания частиц сефадекса. Открывают кран 6 и дают
впитаться раствору в сефадекс. Когда уровень раствора совпадет с
уровнем А — А, кран 6 закрывают.
Пипеткой также осторожно и медленно вносят в верхнюю часть ко-
лонки 1 мл воды. Вставляют пробку 4 в колонку, закрепляют ее и
включают тумблеры «прибор» и «диаграмма» потенциометра. С по-
мощью рукояток грубого 12 и точного 14 регулирования чувствитель-
ности фотоэлектроколориметра устанавливают перо самописца между
значениями 0,8 и 0,9 мВ по верхней шкале прибора. Открывают пол-
ностью кран 3 и подают элюент. Снимают зажим 16 и подают реагент
из емкости 1. Полностью открывают- кран 6 и одновременно делают
отметку на диаграммной ленте потенциометра о начале хроматографи-
рования. Через 6—8 мин проверяют положение пера самопишущего
потенциометра и при отклонении его от ранее установленного с по-
мощью рукоятки 14 фотоэлектроколориметра вновь устанавливают
61
Рис. 19. Выходная кривая при гель-хроматографировании смеси полиэтиленгликолей и
хлорида натрия.
перо между значениями 0,8—0,9 мВ (в дальнейшем регулировка при-
бора не производится).
Через 30—40 мин элюирования фракции полиэтиленгликоля выхо-
дят из колонки. За началом выхода NaCl наблюдают по показанию
милливольтметра 9. Когда милливольтметр покажет 20 мВ, тумблер
на щите 8 переключают в положение «электропроводность». При этом
перо потенциометра переходит влево и выходная кривая для хлорида
натрия вычерчивается в перевернутом виде по отношению к кривой
для полиэтиленгликоля (рис. 19).
После переключения тумблера в положение «электропроводность»
выключают фотоэлектроколориметр, переводя рукоятку 11 в положе-
ние «0», и зажимом 16 перекрывают подачу реагента. По окончании
элюирования хлорида натрия закрывают краны 3 и 6 и выключают
тумблеры потенциометра.
По хроматограмме с учетом скорости перемещения диаграммной
ленты находят значения параметров: время удерживания фракции по-
лиэтиленгликоля с молекулярной массой 20 000 т0, времена удержива-
ния остальных фракций полимера тг и хлорида натрия тыась а также
значения Ат0, Ат,, Атыась Рассчитывают коэффициенты распределения
для всех фракций полимера по формуле
= (11.46)
TNaCl ~ Т0
По калибровочному графику в координатах Ki = f(lgM), построен-
ному с использованием фракций полиэтиленгликоля с известной моле-
кулярной массой, определяют молекулярную массу фракций полимера
в исследуемом растворе. По уравнению (11.45) вычисляют коэффициент
разделения а для двух фракций полиэтиленгликоля.
62
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАЧИ
КОНТРОЛЬНЫЕ ВОПРОСЫ
у. Дайте определение понятия адсорбции. Что такое изотерма, изо-
стера и изопикна адсорбции?
(2. При каких условиях соблюдается при адсорбции закон Генри?
Каков физический смысл константы Генри?
Напишите уравнение изотермы адсорбции теории мономолеку-
тярной адсорбции Ленгмюра. Объясните физический смысл входящих
з него величин. При каких условиях это уравнение применимо?
Чем отличаются константы адсорбции в уравнениях Ленгмюра
и Генри и какова взаимосвязь между ними?
(Ь. Как определяют константы уравнения Ленгмюра? Какие термо-
динамические и геаме-т-р-ическц.е характеристики можно рассчитать,
зная эти к,онстант4т?
(о. Объясните физический смысл констант уравнения БЭТ. При ка-
кихдсловиях это уравнение выполняется?
(£. Как определяют константы уравнения БЭТ? Для чего применяют
это уравнение?
(8. Какие адсорбаты используют при определении удельной поверх-
ности адсорбентов методом БЭТ и при каких условиях проводят из-
мерения?
9. Применительно к каким адсорбентам адсорбция описывается тео-
рией капиллярной конденсации? Каковы исходные положения этой
теории? 4
10. Как рассчитываются кривые распределения пор по размерам
из данных капиллярной конденсации и каково их назначение?
11. Каковы особенности характеристической кривой адсорбента?
Что означает аффинность характеристических кривых?
12. Каковы особенности адсорбции на микропористых адсорбентах
и какая теория используется для описания адсорбции на этих сор-
бентах?
13. Как рассчитать общий объем пор у микропористого адсорбента?
14. Дайте определение интегральной и дифференциальной теплоты
адсорбции. Как находят изостерическую и «чистую» теплоты адсорбции?
15. Объясните, как влияет степень заполнения поверхности на теп-
лоту адсорбции и адсорбционный потенциал?
16. Чем отличается адсорбция из растворов от адсорбции газов и
паров? Какие уравнения используют для описания изотермы обменной
молекулярной адсорбции из растворов?
*"17. Какие уравнения описывают зависимость поверхностного натя-
жения растворов ПАВ от их концентрации? При каких условиях они
применимы?
18. Какие факторы влияют на агрегатное состояние адсорбционных
слоев молекул ПАВ?
19. Какие уравнения состояния используют для газообразных ад-
сорбционных пленок? Как в них учитываются собственные размеры
молекул и взаимодействие между ними?
«ц_20. Как рассчитать толщину адсорбционного слоя и «посадочную»
площадку молекул ПАВ, зная зависимость поверхностного натяжения
от состава раствора?
63
Расскажите об ориентации молекул алифатических спиртов
^или кислот) при адсорбции их из водных растворов на активном угле.
Чем определяется площадь молекулы в адсорбционном слое?
**22. Сформулируйте правило Дюкло — Траубе и поясните его физи-
ческий смысл. При каком строении поверхностных пленок соблюдается
это правило? В чем заключается обратимость этого правила?
я>23. Покажите взаимосвязь между константой адсорбции Генри и
поверхностной активностью.
•" 24. Что такое хроматография? Рассмотрите физико-химические
принципы хроматографического разделения.
Как классифицируют хроматографические методы по механизму
процесса разделения? Какие параметры количественно определяют сте-
певдигродства разделяемых веществ к фазам?
(26,- Как классифицируют хроматографические методы по агрегат-
ному. состоянию неподвижной и подвижной фаз?
. (27. Какие способы используют для проведения хроматографиче-
ского процесса и каковы их основы?
28. Как связана форма хроматографического пика с видом изо-
термы адсорбции (равновесная хроматография)?
29. Что собой представляют ионообменные адсорбенты? Укажите
их классификацию по основности, методы получения.
30. Что называют полной и динамической обменной емкостью иони-
та и как их определяют?
31. Чем обусловлено медленное установление равновесного состоя-
ния при обмене ионов между ионообменной смолой и раствором?
32. Рассмотрите процесс обессоливания воды с помощью катиони-
тов и анионитов на примере удаления-солей MgSO4 и Са(НСО3)2.
33. В чем заключаются особенности метода гель-хроматографии?
34. От каких факторов зависит эффективность работы хроматогра-
фических колонок?
ПРИМЕРЫ РЕШЕНИЯ ЗАДАЧ
1. Ниже приведены экспериментальные данные по адсорбции азота на ТЮг (рутиле)
при 75 К:
р-10-2, Па. . 60,94 116,41 169,84 218,65 272,25
А, моль/кг . . 0,367 0,417 0,467 0,512 0,567
Постройте график, соответствующий линейному уравнению БЭТ. Найдите кон-
станты А» и С. Рассчитайте удельную поверхность адсорбента. Давление насыщен-
ного пара азота при указанной температуре ps = 78,3-103 Па, площадь, занимаемая
одной молекулой азота, so = 0,16 нм2.
Решение. В линейной форме уравнение БЭТ (II. 3) имеет вид
pips _ 1 . С — 1 _ р
A(l—p/ps} А^С АХС ps
Вначале рассчитывают значения p/ps и : :
А (1 р!ps)
p/ps.................... 0,078 0,149 0,217 0.279 0.348
, ,,PlPs.— , кг/моль 0,21-9 0,420 0,593 0,756 0,941
-p/ps)
64
По этим данным строят график в координатах уравнения БЭТ в линейной форме
(рис. 20). Из графика находят:
а = -Дт- = 0,027;
^а = ^г = 2’61
Откуда А „о = 37,9 10~2 моль/кг; С = 97,7. Удельную поверхность адсорбента рас-
считывают по уравнению (II. 4):
s = A N кЗп = 37,9 • Ю'2 • 6,02 • 1023 • 16 • Ю’20 = 36,51 • 103 м2/кг
V) lL оо л. и
2. Постройте кривую капиллярной конденсации и интегральную кривую распреде-
ления пор по размерам для активного угля, пользуясь экспериментальными данными
капиллярной конденсации паров бензола при 293 К:
р. 10-2, Па................. 19,8 29,9 39,8 59,6 79,7 89,4 98,3
А, моль/кг
адсорбция................... 4,5 5,4 6,5 10,2 14,4 17,0 20,0
десорбция........... . . 4,5 6,0 9,0 13,9 17,5 19,0 20,0
При этой температуре для бензола мольный объем Км = 89-10-6 м3/моль, поверх-
ностное натяжение 0 = 28,9 мДж/м2, давление насыщенного пара ps = 99,3-102 Па.
Решение. Для расчета интегральной кривой распределения пор по размерам ис-
пользуют кривую десорбции.'Радиус пор рассчитывают по уравнению Кельвина (I. 11):
2оКм
RT In Ps/р
Объем пор адсорбента Vn, приходящихся на единицу массы адсорбента, заполненных
при данном равновесном давлении адсорбата, находят из соотношения
V п — Ау м
Результаты расчетов приведены ниже:
pjp 5,01 3,32 2,49 1,67 1.25 1,11 1,01
ln pjp 1,61 1,20 0,91 0,51 0,22 0,10 0,01
г, нм 1,31 1,76 2,32 4,14 9,59 21,1 211,1
Кп • Ю5, м3/кг . . 4,01 5,34 , 8,01 12,4 15,6 16,9 17,8
макс, % • • 22,5 30,0 45,0 69,7 87,6 94,9 100
Интегральная кривая распределения, построенная по этим данным, приведена на
рис. 21.
3. Рассчитайте изостерическую теплоту адсорбции этапа на поверхности графити-
рованной сажи (при степени заполнения 0 = 1) по следующим данным:
р, Па............... 251,2 89,9 33,1 12,6
Рис. 21. Интегральная кривая распределения пор по радиусам для активного у1ля.
Зак. 673 65
Решение. Для расчета дифференциальной (изостерической) теплоты адсорбции ис-
пользуем уравнение
In Р —---+ Ci
где q—изостерическая теплота адсорбции; С] — постоянная интегрирования.
Рассчитываем значения In р и 1/Т:
In р................... 5,52 4,50 3,50 2,53
1/Т-103, К-1........... 5,00 5,49 6,02 6,49
Строим график зависимости In р от 1/Т (рис. 22) и определяем тангенс угла на-
клона прямой:
tga = -JL
Таким образом, q = —tg aR = 2,01 • 103-8,31 = 16,7-103 Дж/моль.
4. Адсорбция растворенного в воде ПАВ на поверхности ртуть — вода подчиняет-
ся уравнению Ленгмюра. При концентрации ПАВ 0,2 моль/л степень заполнения по-
верхности 0 — 0,5. Рассчитайте поверхностное натяжение ртути на границе с раство-
ром при 298 К и концентрации ПАВ в растворе 0,1 моль/л. Предельное значение пло-
щади, занимаемой молекулой ПАВ на поверхности, s0 = 0,20 нм2, поверхностное на-
тяжение ртути на границе с водой равно 0,373 Дж/м2.
Решение. Зависимость поверхностного натяжения от концентрации ПАВ выража-
ется уравнением Шишковского (II. 10):
а = а0 ~ AxRT In (1 + Кс)
Константу равновесия К определяем из уравнения Ленгмюра (II. 2):
Кс
\ + Кс
0
Откуда
0,5 ' * .
= 5 л/моль
. 9 . __________
л с (1—0) 0,2-0,5
Емкость монослоя Аоо находим из соотношения (11.22):
Аоо = —-——------------УГ---------п = 8.3 • 10~6 моль/м2
s0A'a 20 • 10~2° • 6,02 • 1023
Зная К и Лоо, по уравнению (11.10) рассчитываем поверхностное натяжение ртути на
границе с раствором:
а = 0,373 — 8,3 • 10~6 • 8,31 • 298 • In (1 + 5 • 0,1) =
= 0,373 — 2,05 • 10~2 • 1п 1,5 = 0,365 Дж/м2
5. Ниже приведены значения поверхностного давления л валериановой кислоты
при 292 К при различной площади поверхности, приходящейся на 1 моль кислоты:
л-102, Н/м..................... 0,35 0,53 0,78 1,10 1,49 1,92 2,36 2,85 3,36
sM-10’5 м2/моль................ 6,92 4,66 3,49 2,77 2.44 2,27 2,11 2,03 1,89
Известно, что поверхностное давление раствора валериановой кислоты с концен-
трацией 4 ммоль/л равно 0,002 Н/м. Рассчитайте константы уравнения Шишковского.
Решение. Строим график зависимости л = /(.s,.,) (рис. 23). Точка перегиба на
кривой отвечает формированию плотного слоя молекул валериановой кислоты па по-
верхности водного раствора. Из графика находим площадь поверхности, приходящуюся
на I моль кислоты в плотном монослое:
«м, о = 2,5 • 105 м2/моль
Затем рассчитываем емкость монослоя:
= = 2ДЛ= 4 ’ 10-6 М0ЛЬ/м2
1
66
Рис. 22. К определению изостерической теплоты адсорбции этана иа поверхности гра-
фитированной сажи.
л-10г,н1м
Рис. 23. Зависимость поверхностного давления пленки от площади, приходящейся на
1 моль валериановой кислоты.
Принимая во внимание, что Со — о = л, из уравнения Шишковского (II. 10)
о = Со — A^RT In (1 + Кс)
определяем константу К:
|п<1 + *с)=оГ
In (1 + 0,0047<) =
0,002
4-10 6 -8,31 -293
К = 56,9 л/моль
6? Полистирольный сульфокатионит в Н+-форме в количестве т = 1 г внесли в
раствор КС1 с исходной концентрацией с0 = 100 экв/м3 объемом К = 50 мл н смесь
выдержали до равновесного состояния. Рассчитайте равновесную концентрацию калия
в ионите, если константа ионообменного равновесия К.к+ /рг+ = 2,5, а полная обмен-
ная емкость катионита ПОЕ — 5 экв/кг. к '
Решение. Для реакции ионного обмена у
Н+ + К+ =₽=* К+ + Н+
константа равновесия равна
Кк+/н + [k+][h+J
где [К+], [Н+] — концентрация ионов в ионите; [К+], [Н+]—концентрация ионов в
растворе.
Обозначим искомую концентрацию [К+] через х. В смоле ионы Н+ обмениваются
на эквивалентное количество ионов К+, а суммарное содержание ионов равно ПОЕ.
Поэтому
[Н+] = ПОЕ - [К+] = 5 - х
Равновесная концентрация ионов Н+ в растворе, появляющихся в результате вы-
теснения их из ионита, равна
[H+] = -&L=x.^_==£12£2 = 20x
V V 5 • 10-5
&*
67
Равновесная концентрация ионов К+ в растворе составляет:
[К+] = с0 - [Н+] = 100 - 20х
С учетом полученных выражений для равновесных концентраций ионов уравнение
константы ионного обмена можно записать так:
9 5 = __ * (20х>
(100 — 20х)(5 — х)
После преобразования получаем:
Зх2 — 50х +125 = 0
Решение квадратного уравнения дает
50 + д/502 — 4 • 3 • 125
Х' =----------2^3----------= 13’6
И
50 - д/502 -4-3-125 „ „
Х2 =-------v___------------= 3,06
Значение Xi намного больше ПОЕ, что неприемлемо. Следовательно, [К+] =
= 3,06 экв/кг.
ЗАДАЧИ
1. Удельная поверхность силикагеля, найденная методом низкотемпе-
ратурной адсорбции азота, составляет 4,1-10s м2/кг. Плотность сили-
кагеля 2,2 г/см3. Рассчитайте средний диаметр частиц силикагеля.
2. Ниже приведены данные об адсорбции паров воды макропорис-
тым силикагелем при комнатной температуре:
р-10-2,Па.......... 3,04 4,68 7,72 11,69 14,03 17,77
А, моль/кг......... 4.44 6,28 9,22 11,67 13,22 14,89
Пользуясь уравнением Ленгмюра, определите предельную емкость
силикагеля.
3. При измерении адсорбции газообразного азота на активном угле
при 194,4 К были получены следующие данные:
р-10”, Па.............. 1,86 6,12 17,96 33,65 68,89
А • 103, м3/кг......... 5,06 14,27 23,61 32,56 40,83
Значения А даны для азота при нормальных условиях.
Рассчитайте постоянные в уравнении Ленгмюра и удельную поверх-
ность активного угля, принимая плотность газообразного азота равной
1,25 кг/м3, а площадь, занимаемую одной молекулой азота на поверх-
ности адсорбента, равной 0,16 нм2.
4. Удельная поверхность непористой сажи равна 73,7-103 м2/кг.
Рассчитайте площадь, занимаемую молекулой бензола в плотном моно-
слое, исходя из данных об адсорбции бензола на этом адсорбенте при
293 К:
р, Па................... 1,03 1,29 1,74 2,50 6,67
А • 102, моль/кг........ 1,57 1,94 2,55 3,51 7,58
Предполагается, что изотерма адсорбции описывается уравнением
Лецкмюра.
Qx) Определите константы эмпирического уравнения Фрейндлиха,
используя следующие данные об адсорбции диоксида углерода на ак-
тивном угле при 293 К:
68
р- 10-3, Па .
А • 102, кг/кг
1,00 4,48 10,0 14,4 25,0 45,2
3,23 6,67 9,62 11,72 14,5 17,7
6. Используя уравнение БЭТ, рассчитайте удельнук) поверхность
адсорбента по изотерме адсорбции бензола (варианты I—IV):
I p/ps . . . . 0,04 0,08 0,16 0,22 0,27 0,36 0,46
А, моль/кг .... . . . . 0,348 0,483 0,624 0,724 0,805 0,928 0,13
II P/Ps . . . . 0,05 0,12 0,19 0,26 0,34 0,44 0,50
А, моль/кг .... . . . . 0,31 0,593 0,795 0,99 1,21 1,525 1,77
III P/Ps . . . . 0,03 0,07 0,12 0,17 0,24 0,31 0,38
А, моль/кг .... . . . . 0,196 0,301 0,373^ 0,423 0,488 0,520 0,625
IV P/Ps . . . . 0,02 0,05 0,11 0,19 0,25 0,30 0,36
А. моль/кг .... . . . . 0,104 0,196 0,298 0,387 0,443 0,488 0,550
Площадь, занимаемую молекулой бензола, примите равной 0,49 нм2.
7. Используя уравнение БЭТ, рассчитайте удельную поверхность ад-
сорбента по данным об адсорбции азота:
p/ps.....................
А 103, м3/кг...........
0,1 0,2 0,3 0,4
0,71 0,31 0,93 1,09
Площадь, занимаемая молекулой азота в плотном монослое, равна
0,16 нм2, плотность азота 1,25 кг/м3.
8. При обработке данных по адсорбции азота на графитированной
саже при 77 К с помощью графика, соответствующего линейному урав-
нению БЭТ, найдено, что тангенс угла наклона прямой составляет
1,5-103, а отрезок, отсекаемый на оси ординат, равен 5 единицам (ад-
сорбция выражена в м3 азота на 1 кг адсорбента при нормальных
условиях). Рассчитайте удельную поверхность адсорбента, предпола-
гая, что площадь, занимаемая одной молекулой азота, равна 0,16 нм2.
9. Ниже приведены результаты измерения адсорбции газообразного
криптона (при 77,5 К) на катализаторе:
р, Па........................ 13,22 23,99 49,13 75,70 91,22
А 103, м3/кг................ 1,27 1,5 1,76 1,9 1,98
Значения А даны для криптона при нормальных условиях. Определите
константы уравнения БЭТ и удельную поверхность катализатора, при-
нимая, что один атом криптона занимает площадь 0,195 нм2, ps =
— 342,6 Па, плотность криптона равна 3,74 кг/м3.
10. Используя уравнение БЭТ, рассчитайте удельную поверхность
адсорбента по изотерме адсорбции азота:
p/ps........... 0,0288 0,050
А, моль/кг . . 2,16 2,39
0,110 0,136 0,175 0,200
2,86 3,02 3,22 3,33
Площадь, занимаемая одной молекулой азота в адсорбционном слое,
равна 0,16 нм2.
11. Какую долю (в процентах) составляет давление паров воды
в капиллярах радиусом 10 ч и 10~2 мкм от нормального давления на-
сыщенного пара при 298 К? При расчетах примите, что краевой угол
равен нулю, а стн,о = 71,96 мДж/м2.
/T2j'Рассчитайте эффективный диаметр пор силикагеля по экспе-
римёТгг^льным данным адсорбции паров бензола на этом адсорбенте
при 293 К.
69
Адсорбция p/ps 0,554 0,634 0,763 0,782 0,826 0,882 0,999
А, моль/кг . , 1,62 1,88 3,95 4,35 4,90 17,2 19,2
Десорбция p/ps ..... 0,948 0,880 0,852 0,810 0,770 0,686 0,610 0,585
А, моль/кг . , 18,98 18,78 18,70 14,69 7,14 2,67 1,91 1,77
Поверхностное натяжение бензола 28,87 мДж/м2, мольный объем
88,79 см3/моль. Постройте интегральную кривую распределения объема
пор. но их радиусам.
ЦЗ/ Постройте изотермы адсорбции и десорбции, пользуясь экспери-
ментальными данными капиллярной конденсации паров воды в порах
активного угля при 293 К (варианты I—III):
I р- 10~2, Па А, моль/кг . . . 5,32 10,0 11,3 12,5 14,7 17,3 20,0 23,3
адсорбция ... 0,5 2,3 4,0 5,0 10,0 16,0 20,0 28,5
десорбция ... 0,5 2,5 5,0 7,5 15,0 23,0 27,6 28,5
II р- 10~2, Па А, моль/кг . . . 4,65 9,3 14,0 18,7 20,9 23,3
адсорбция . . . . 0,5 1,5 3,5 20,0 24,0 28,5
III') десорбция ... 0,5 1,5 13,0 27,0 28,0 28,5
p!ps . ... . 0,1 0,2 0,4 0,6 0,8 0,9 1,0
/ А, моль/кг
адсорбция ... 6,5 9,0 11,5 14,0 22,5 26,6 30,0
десорбция ... 7,0 10,3 13,5 16,5 25,0 27,6 30,0
Рассчитайте и постройте интегральную кривую распределения объе-
ма пор по размерам. Мольный объем воды 18 см3/моль, давление ps =
= 2338 Па, поверхностное натяжение воды 71,96 мДж/м2.
14. Постройте петлю гистерезиса и интегральную кривую распре-
деления объема пор адсорбента по размерам, используя эксперимен-
тальные данные капиллярной конденсации метанола на силикагеле
при 293 К (варианты I—II):
I р • 10 2, Па . . . . . . 16 32 64 79 96 НО 128
А, моль/кг адсорбция . 2,5 3,5 4,8 6,3 13,0 19,0 22,5
десорбция . . . • • 2,5 3,5 4,8 6,5 17,5 21,2 22,5
II p!ps 0,2 0,4 0,6 0,8 0,9 1.0
А, моль/кг адсорбция . десорбция . 0,8 0,8 1,3 1,3 1,6 1,7 2,2 3,0 4,0 3,7 3,87 3,87
При 293 К мольный объем метанола 40,6 см3/моль, поверхностное на-
тяжение 22,6 мДж/м2, давление насыщенного пара 128-Ю2 Па.
Рассчитайте характеристическую кривую и убедитесь в ее тем-
пературной инвариантности по данным об адсорбции бензола на мик-
ропористом активном угле при двух значениях температуры:
Т = 293 К
\ /₽> Па . . . 0,13 0,51 1,30 3,33 16,7 37,3 95,6 319,8
V А, моль/кг . . . . . . 1,13 1,69 2,25 2,82 3,94 4,50 5,25 5,77
Т = 323 К
р, Па . . . 0,042 0,30 1,87 14,70 49,5 102,9 272,5 721,4
А, моль/кг . . . . . . 0,41 0,68 1,36 2,38 3,26 3,80 4,61 5,09
Плотность жидкого бензола при 293 и 323 К соответственно равна 0,879 *
и 0,846 г/см3, давление насыщенного пара составляет 10 470 и 35 480 Па.
70
16. Рассчитайте и постройте характеристические кривые для бен-
зола и хлороформа, а также изотерму адсорбции хлороформа при
293 К по данным адсорбции бензола на микропористом активном угле
при 293 К:
р, Па......................... 0,13 0,51 1,30 3,33 16,7 37,3 95,6 319,8
А, моль/кг.................... 1,13 1,69 2,25 2,82 3,94 4,50 5,25 5,77
Коэффициент аффинности для хлороформа равен 0,87 (стандартное
вещество — бензол). Давление насыщенного пара бензола и хлоро-
форма при этой температуре соответственно равно 10,47-103 Па и
23,99-103 Па, плотность жидкого бензола 0,879 г/см3 и хлороформа
1,480 г/см3.
Q7) Рассчитайте и постройте интегральную кривую распределения
пор по размерам (варианты I—II) по данным капиллярной конденса-
ции паров метилового спирта на активном угле при 293 К:
CD р • 10 2, Па . . . . А, моль/кг • • .12,8 25,6 38,4 51,2 64,0 76,8 90 102
адсорбция . . . . . 7,5 8,0 8,3 8,6 9,4 10,2 11,4 13
десорбция . . . . . 7,5 8,3 9,0 9,6 10,0 11,0 11,7 13
п р- 10-2, Па ... . А, моль/кг . . 64,0 76,8 90,0 102,5 114,5 128
адсорбция . . . . . 24,0 28,3 31,0 36,0 46,0 55,0
десорбция . . . . . 24,0 30,0 37,5 44,0 50,0 55,0
Мольный объем спирта 40,6 см3/моль, давление насыщенного пара
128-102 Па, поверхностное натяжение 22,6 мДж/м2.
18. Рассчитайте и постройте характеристические кривые для бен-
зола и гексана, а также изотерму адсорбции гексана при 323 К по
данным адсорбции бензола на активном угле при 323 К:
р Па...................... 0,042 0,30 1,87 14,7 49,5 102,9 272,5 721,4
А, моль/кг................. 0,41 0.68 1,36 2,38 3,26 3.80 4,61 5,09
Коэффициент аффинности для гексана равен 1,46 (стандартное веще-
ство— бензол). Давление насыщенного пара бензола и гексана при
этой температуре соответственно равно 35,48-103 и 70,75-103 Па, а
плотность жидких бензола и гексана 0,846 и 0,631 г/см3 соответственно.
19. По уравнению Дубинина — Радушкевича рассчитайте объем пор
цеолита, используя данные по адсорбции этана (298 К):
р-10-3, Па............ 10 15 20 30
А, моль/кг............ 2,37 2,53 2,63 2,77
Давление насыщенного пара этана 37-10s Па, мольный объем этана
64 см3/моль.
2d По уравнению Дубинина — Радушкевича рассчитайте объем пор
сажи на основе данных об адсорбции паров бензола:
p/ps................ 0,3 0,4 0,5 0,6
А, моль/кг.......... 1,10 1,38 1,60 1,90
Молъцый объем бензола равен 88,8 см3/моль.
21. Постройте изотерму адсорбции и определите общую пористость
сажи (по уравнению Дубинина — Радушкевича), используя экспери-
ментальные данные адсорбции бензола на саже (варианты I—III):
71
p/ps.......... 0,1 0,2 0,3 0,4
А, моль/кг '' " <
I.............. 1,75 2,15 2,42 2,66
II............. 0,4 0,59 0,65 0,70
III............ 1,20 1,35 1,50 1,60
Мольный объем бензола равен 88,8 см3/моль.
22. Изотермы адсорбции газов А и В на некотором твердом теле
описываются уравнением Ленгмюра. При температуре 77 К степень за-
полнения поверхности 0 = 0,01 чистым газом А достигается при рА =
= 133-102 Па, а чистым газом В — при рв = 1330 Па. Рассчитайте раз-
ность теплот адсорбции газов А и В.
23. Измерена адсорбция азота на низкодисперсном непористом по-
рошке. Найдено, что при 77 и 90 К степень заполнения поверхности 0,
равная 0,5, достигается при p/ps соответственно 0,02 и 0,2. Пользуясь
уравнением БЭТ, рассчитайте изостерическую теплоту адсорбции, а
также дифференциальные изменения энтропии и энергии Гиббса ад-
сорбции при 77 К. Теплота испарения жидкого азота при 77 К состав-
ляет 5,66 кДж/моль.
24. Рассчитайте значения изостерической теплоты адсорбции и по-
стройте график ее зависимости от степени заполнения поверхности по
данным об адсорбции метана на графитированной саже при разных
температурах:
Л. моль/кг р, Па
113 К 123 К 133 К 143 К
0,0175 37,2 119,7 289,9 684,9
0,01 S 70,5 211,5 542,6 1243,6
0,03 115,7 356,4 957,6 2307,6
0,05 158,3 532,0 1516,2 3830,4
0,08 283,3 1050,7 3245,2 8551,9
0,10 731,5 2713,2 8645,0 22610,0
0,12 6716,5 16957,5 43225,0 85785,0
Емкость монослоя принять равной 0,12 моль/кг.
25. Величина адсорбции красителя (ПАВ) из раствора может быть
использована для оценки удельной поверхности порошков. При введе-
нии 1 г активного угля в 100 мл водного раствора метиленового голу-
бого концентрация красителя изменяется от начальной 1 • 10-4 моль/л до
конечной равновесной 6- 10—6 моль/л, а при добавлении 2 г угля к та-
кому же исходному раствору равновесная концентрация составила
4-10"5 моль/л. Считая, что адсорбция описывается уравнением Ленг-
мюра, рассчитайте sy;i угля. Площадь, занимаемую молекулой краси-
теля на поверхности, примите равной 0,65 нм2.
26. Растворенное в воде ПАВ адсорбируется на поверхности
ртуть — вода согласно уравнению Ленгмюра. При концентрации ПАВ
0,3 моль/л степень заполнения поверхности 0 составляет 0,6. Рассчи-
тайте поверхностное натяжение на границе раздела ртуть — раствор
при 298 К, если концентрация ПАВ равна 0,2 моль/л. Предельное зна-
чение площади, занимаемой молекулой ПАВ на поверхности, примите
равной 0,20 нм2, поверхностное натяжение на границе раздела ртуть —
вода 0,373 Дж/м2.
27. Найдите площадь, приходящуюся на одну молекулу в насы-
щенном адсорбционном слое анилина на поверхности его водного рас-
твора с воздухом, если предельная адсорбция анилина составляет
6-10-6 моль/м2.
72
28. Зная поверхностное давление «-гексилового спирта при 285 К
в зависимости от его концентрации в водном растворе:
с-104, моль/л...................... 6,2 8,1 12,5 17,2 25 34,3 49 68,6 98
п-103, Н/м......................... 2,3 2,5 3,9 5,7 7,9 9,4 13,4 16,3 19
рассчитайте величину адсорбции, площадь, приходящуюся на 1 моль,
Хм и площадь, занимаемую одной молекулой спирта в плотном моно-
слое s0, используя уравнение Гиббса.
29. При исследовании адсорбции стеариновой кислоты из ее рас-
творов в «-гексане различных концентраций с на порошке стали полу-
чены результаты: ,
с-105, моль/л..................... 1 2 4 7 10 15 20 25
Л-103, кг/кг...................... 0,786 0,864 1,00 1,17 1,30 1,47 1,60 1,70
Рассчитайте удельную поверхность порошка стали, принимая йлощадь
1 молекулы стеариновой кислоты в насыщенном монослое 0,20 нм2.
30. ! Рассчитайте по уравнению Ленгмюра адсорбцию пропионовой
кислоты из раствора с концентрацией 0,5 моль/л на поверхности раз-
дела раствор — воздух при 298 К, если поверхностное натяжение этого
раствора 55,6 мДж/м2, поверхностное натяжение воды 71,96 мДж/м2
и константа уравнения Ленгмюра К = 7,73 л/моль.
31, Ниже приведены данные об адсорбции жирных кислот на сили-
кагеле из толуольных растворов и на неполярном углеродном сорбенте
из водных растворов:
Силикагель — толуол Углеродный с, ммоль/л сорбент—вода А, моль/кг
с, ммоль/л А, моль/кг
Уксусная кислота 8 0,648 1,0 0,80
10 0,810 1,5 1,20
Пропионовая кислота 10 0,276 0,5 1,24
15 0,430 0,8 1,98
Масляная кислота 10 0,092 0,2 1,50
15 0,140 0,25 1,86
Считая, что изотерма адсорбции при указанных концентрациях опи-
сывается уравнением Генри, покажите применимость правила Траубе
и его обратимость при адсорбции жирных кислот на твердых адсорбен-
тах. Объясните влияние среды на константу адсорбции, используя пра-
вило уравнивания полярностей Ребиндера.
32,- Рассчитайте константы уравнения Шишковского, если поверх-
ностное давление «-валериановой кислоты при 292 К в зависимости от
площади поверхности, приходящейся на 1 моль этого вещества, со-
ставляет:
л- 103, Н/м ... 3,5 5,3 7,8 11,0 14,9 19,2 23,6 28,5 33,6
• ИГ5. м7моль 6,92 4,66 3,49 2,77 2,44 2,27 2,11 2,03 1,89
Раствор «-валериановой кислоты с концентрацией 3 ммоль/л имеет
поверхностное давление 4,2-10~3 Н/м.
1 33. При исследовании адсорбции уксусной кислоты на древесном
угле из водных растворов объемом 200 мл получены результаты:
Масса угля, г............... 3,96 3,94 4,00 4,12 4,04 4,00
Концентрация кислоты,
ммоль/л
до введения угля........ 503,0 252,2 126,0 62,8 31,4 15,7
равновесная в растворе. . 434,0 202,0 89,9 34,7 11.3 3,33
Покажите, что эти данные удовлетворяют изотерме адсорбции Фрейн-
длиха. Рассчитайте константы этого уравнения.
73
34. При изучении поверхностных пленок на весах Ленгмюра на по-
верхность воды наносили бензольный раствор пальмитиновой кислоты
CisHsiCOOH концентрацией 0,4 % (масс.). Взвешиванием определили,
что масса 100 капель раствора равна 0,33 г. На поверхность воды меж-
ду двумя перегородками нанесли 5 капель раствора. После испарения
бензола на поверхности осталась нерастворимая пленка кислоты. При
передвижении подвижной перегородки по направлению к неподвижной
зарегистрировано резкое увеличение поверхностного давления, когда
расстояние между перегородками составило 23 см в длину и 14 см
в ширину. Определите площадь, занимаемую одной молекулой, при об-
разовании монослоя на поверхности. Рассчитайте длину, приходящуюся
на одну группу СН2. Плотность пальмитиновой кислоты в жидком со-
стоянии и в виде пленки равна 0,850 г/см3.
35. В 200 мл 0,12 н. раствора NaOH ввели 5 г воздушно-сухого
сильнокислотного катионита в Н+-форме. После установления равно-
весия отфильтровали 100 мл раствора, для нейтрализации которого
потребовалось 20 мл 0,12 н. раствора НС1. Определите полную обмен-
ную емкость катионита.
36. В 150 мл раствора H2SO4 с концентрацией 0,110 моль/л ввели
3 г сильноосновного анионита в ОН~-форме. После установления рав-
новесия ионного обмена отобрали 50 мл раствора, для нейтрализации
которого потребовалось 22 мл раствора КОН концентрацией
0,05 моль/л. Рассчитайте полную обменную емкость анионита.
37. Полная обменная емкость сухого сульфокатионита КУ-2-8 в
Na+’форме равна 4,8 моль экв/кг. Определите предельно возможное
количество (в г) кобальта(II) и бария(II), которое может сорбировать
из соответствующих растворов 1 кг исходного ионита.
38. Полная обменная емкость анионита АВ-17-8 в С1_-форме равна
4,2 моль экв/кг. Рассчитайте предельно возможное количество (в -г)
кобальта(П) и золота(Ш), которое может сорбировать 1 кг исходного
ионита из растворов хлороводородной кислоты, если указанные эле-
менты находятся в виде комплексных анионов [СоС14]2~ и [АиС14]~.
Рассчитайте количество сульфокатионита в Н+-форме и анио-
нита в ОН~-форме, необходимое для очистки 1000 м3 природной воды,
содержащей 0,025 г/л NaCl, 0,04 г/л MgSO4, 0,12 г/л Са(НСО3)2. Пол-
ная обменная емкость катионита 4,2 экв/кг, анионита — 3,5 экв/кй.
40. Определите, какое количество (в кг) морской воды можно обес-
солить с помощью хроматографических колонок, содержащих 1 кг ка-
тионита и 1 кг анионита, если динамическая обменная емкость каж-
дого ионита равна 3,5 экв/кг. Концентрация солей, преобладающих
в воде, в % (масс.): NaCl — 2,74, MgCl2— 0,33, MgSO4 — 0,23.
41. Рассчитайте среднее значение константы ионообменного равно-
весия, описываемого уравнением Никольского, используя данные по
замещению ионов кальция из почвы на ионы натрия из раствора нат-
риевой соли:
Равновесная концентрация в
растворе, ммоль/л
Na............................ 3,26
Са . ...................... 37,84
Адсорбция А-102 на почве,
моль/кг
Na........................... 0,28
Са.......................... 39,72
6,60 13,80 21,25
36,72 34,62 31,87
0,60 1,20 1,89
39,56 39,40 38,68
74
42. Ввели 3 г полистирольного сульфокатионита в Н+-форме, пол-
ная обменная емкость которого 5,12 экв/кг, в 0,2 л водного раствора
CsCl исходной концентрации 8-10~2 моль/л. Определить равновесные
концентрации ионов Н+ и Cs+ в растворе и в ионите, если константа
ионообменного равновесия равна Хм/ц — 2,7.
В раствор, содержащий 0,028 моль/л RbCl, ввели 5 г феноло-
формальдегидного сульфокатионита в Na+’форме и смесь выдержали
до достижения равновесия ионного обмена. Рассчитайте, какая часть
рубидия будет адсорбироваться, если константа равновесия Ккь/^а —4,3,
полная обменная емкость 3,5 экв/кг (Ка+-форма ионита), объем рас-
твора 0,21 л.
III. КИНЕТИЧЕСКИЕ ЯВЛЕНИЯ И КИНЕТИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ ДИСПЕРСНЫХ СИСТЕМ
В микрогетерогенных системах (суспензиях, эмульсиях, газовых эмуль-
сиях, аэрозолях), частицы которых благодаря большой массе не могут
принимать участия в тепловом (броуновском) движении, происходит
седиментация — осаждение или обратный процесс — всплывание час-
тиц. Если движение потока частиц ламинарное и может быть описано
уравнением Стокса, то скорость оседания (всплывания) в гравитацион-
ном поле и связана с их размером следующим соотношением:
,, vg (р — Ро)
В
где v—объем частицы; g— ускорение свободного падения; р и ро — соответственно
плотность частицы и дисперсионной среды; В — коэффициент трения.
Для сферических частиц (В — 6лт]г) это уравнение принимает вид
.. . 2gr2 (Р — Ро)
9т]
где г — радиус частицы; ц — вязкость дисперсионной среды.
Способность системы к седиментации характеризуют константой се-
диментации:
(Ш. 1)
(III. 2)
тот _ v (р — ро)
г>еед — —g- ~ §
(III. 3)
где тот — относительная масса частицы (с учетом плотности среды; тот = т — аро).
Для сферических частиц уравнение (III. 3) принимает вид
5сед = —"(р~-ро)- (III. 4)
э,|
В центробежном поле расстояние х-, пройденное частицами при се-
диментации, увеличивается со временем т по экспоненте (при постоян-
ном числе оборотов центрифуги).
75
а если частицы сферические, то
х 2г2 (р — р0) со2т
х0 ~ 9ц
/ 9ц In x/xq
Л' 2 (р — р0) со2т
(III. 6)
где хо — начальное расстояние частиц от центра вращения; со — угловая скорость вра-
щения ротора центрифуги.
Соотношения (III. 1) — (III. 6) используются в седиментационном
анализе дисперсности различных материалов.
Седиментационный анализ дисперсности состоит в получении кри-
вой седиментации, т. е. зависимости массы осадка т дисперсной фазы
(осевшей к данному времени) от времени осаждения т. Очевидно, что
для монодисперсной (с одинаковыми частицами) системы такая зави-
симость является линейной:
m = (III-7)
п
где Q — общая масса дисперсной фазы; Н — первоначальная высота столба суспензии.
Все реальные дисперсные системы полидисперсны (частицы дис-
персной фазы имеют разные размеры), и поэтому скорости осаждения
частиц различных фракций разные: крупные частицы осаждаются бы-
стрее, мелкие — медленнее. По этой причине кривая седиментации вы-
пукла к оси ординат. Тангенсы угла наклона касательных в данных
точках кривой седиментации определяют скорости седиментации соот-
ветствующих фракций частиц. Зная скорости осаждения частиц отдель-
ных фракций, по уравнению (III. 2) можно рассчитать их размеры (ра-
диусы). Построением интегральной, а затем дифференциальной кри-
вых распределения частиц полидисперсной системы по радиусам (раз-
мерам) заканчивается седиментационный анализ.
Ультрамикрогетерогенные системы (золи) отличаются тем, что их
частицы принимают участие в тепловом движении, следуют всем моле-
кулярно-кинетическим законам, которые позволяют по соответствую-
щим экспериментальным зависимостям определить концентрацию, мас-
су и размер частиц дисперсной фазы.
К таким законам относится закон Эйнштейна — Смолуховского, ко-
торый устанавливает связь между средним сдвигом частиц Д и коэф-
фициентом диффузии D:
К2 = 2 Dr (III. 8)
где т — время между моментами измерения расстояний движущейся частицы.
К золям применимо также уравнение Эйнштейна для коэффициента
диффузии D. Если частицы сферические, то это уравнение имеет вид
Применительно к лиозолям справедлив осмотический закон Вант-
Гоффа (относящийся обычно к растворам):
л = RT = vkT (III. 10)
2V А
где я — осмотическое давление; v— число частиц в единице объема золя, т. е. частич-
ная концентрация.
76
Для аэрозолей можно использовать газовый закон Клапейрона —
Менделеева в следующей форме:
pV = -^~RT = nkT (Ш.11)
где п—число частиц в аэрозоле.
При осаждении частиц ультрамикрогетерогенных систем создается
градиент концентраций, который является движущей силой диффузии
частиц в направлении, обратном седиментации. При равенстве диффу-
зионного и седиментационного потоков устанавливается так называе-
мое диффузионно-седиментационное равновесие, характеризующее тер-
модинамическую седиментационную устойчивость таких систем. Час-
тичная концентрация на высоте h равна д--'
г vg (р — ро) J1 ,'ттт
vfc = v„exp----r I (III. 12)
где Vo — концентрация частиц на высоте h = 0.
Мерой термодинамической устойчивости к седиментации является
высота he, на протяжении которой концентрация дисперсной фазы из-
меняется в е раз:
h kT kT
е ~~ m0Tg ~ vg (р — ро)
(III. 13)
Чем больше высота he, тем система термодинамически более устойчива
к седиментации. Устойчивость повышается с ростом температуры,
уменьшением размера частиц и разности плотностей частицы и среды.
Мерой кинетической устойчивости к седиментации является вели-
чина, обратная константе седиментации:
•^сед тот 2г2 (р — р0)
Как видно, кинетическую устойчивость к седиментации можно ре-
гулировать путем изменения вязкости и плотности среды, плотности и
размеров частиц.
Относительное движение фаз дисперсных систем можно наблюдать
также под действием электрического поля, что обусловлено наличием
на межфазных поверхностях двойного электрического слоя, возникаю-
щего вследствие межфазного взаимодействия.
Различают три механизма образования двойных электрических
слоев: 1) поверхностная диссоциация функциональных групп, 2) ад-
сорбция ионов электролитов и 3) ориентирование полярных молекул
на межфазной границе. В результате указанных взаимодействий по-
верхность со стороны одной фазы заряжается положительно, а со сто-
роны другой — отрицательно.
Двойной электрический слой (ДЭС) состоит из заряженной поверх-
ности с потенциалом <р0 и противоположно заряженной части слоя,
в которой находятся противоионы (рис. 24). Одна часть противоионов
примыкает непосредственно к поверхности, образуя плотный (адсорб-
ционный) слой — слой Гельмгольца. Другая часть противоионов под
действием теплового движения распространяется в глубь фазы, обра-
зуя так называемый диффузионный слой, или слой Гуи.
77
Рис. 24. Двойной электрический слой и изме-
нение в нем потенциала.
Толщина плотного слоя Гельмголь-
ца принимается равной диаметру про-
тивоиона. Эту часть ДЭС можно рас-
сматривать как плоский конденсатор,
потенциал которого с увеличением
расстояния от поверхности снижается
линейно. По теории Гун — Чепмена
противоионы диффузной части ДЭС
распределяются в поле поверхностно-
го потенциала в соответствии с зако-
ном Больцмана. Теория показывает,
что потенциал в диффузной части слоя
снижается с расстоянием по экспо-
ненте. При малом значении потен-
циала эта зависимость выражается
уравнением
<p = <pee-xx ' (Ш. 15)
где <рб — потенциал диффузного слоя; х— расстояние от начала диффузной части
ДЭС; х — величина, обратная толщине диффузной части слоя.
Из уравнения (III. 15) следует, что за толщину диффузной части
слоя принято расстояние, на котором потенциал диффузной части слоя
<рб уменьшается в е раз.
В соответствии с той же теорией толщина диффузной части слоя
равна
(III. 16)
где So — электрическая постоянная; s — относительная диэлектрическая проницаемость
среды; F— постоянная Фарадея; I — ионная сила раствора; с01- — концентрация иона
в растворе; z, — заряд иона электролита. ,
Из уравнения (III. 16) следует, что X уменьшается с ростом кон-
центрации электролита, заряда его ионов и с понижением температуры.
При движении одной фазы относительно другой на плоскости сколь-
жения происходит разрыв ДЭС (как правило, в диффузной части) и
возникновение электрокинетического («дзета») g-потенциала (см.
рис. 24).
Под действием электрического поля могут двигаться дисперсная
фаза относительно дисперсионной среды (электрофорез) или диспер-
сионная среда относительно дисперсной фазы (электроосмос). Механи-
ческое движение дисперсной фазы относительно дисперсионной среды
(седиментация) вызывает возникновение электрической разности по-
тенциалов (потенциал седиментации). Если электрическая разность
потенциалов возникает при движении дисперсионной среды относи-
тельно дисперсной фазы, то такое явление называют потенциалом про-
текания.
Наибольшее распространение из электрокинетических явлений по-
лучили электрофорез и электроосмос. Значение электрокинетического
потенциала, возникающего при электрофорезе или электроосмосе, мож-
73
но рассчитать по уравнению Гельмгольца — Смолуховского:
ИЛИ
jw (III. 18)
ее0/
где т| — вязкость среды; Uo — линейная скорость движения фаз; Е — напряженность
электрического поля; % — удельная электропроводность среды; о — объемная скорость
движения среды; / — сила тока.
Электрокинетический потенциал зависит от тех же факторов, что
и толщина диффузной части ДЭС.
Работа 11. ИССЛЕДОВАНИЕ МОЛЕКУЛЯРНО-КИНЕТИЧЕСКИХ СВОЙСТВ
ДИСПЕРСНЫХ СИСТЕМ
Цель работы-, исследование распределения частиц дисперсной фазы по
высоте при диффузионно-седиментационном равновесии; определение
числа Авогадро.
Участие частиц дисперсной фазы в броуновском движении может
отражаться на седиментации. При оседании частиц в гравитационном
поле увеличивается их концентрация в нижних слоях, в результате
чего возникает диффузионный поток, направленный противоположно
потоку седиментации. Через определенное время может наступить диф-
фузионно-седиментационное равновесие. Распределение частиц при рав-
новесии в монодисперсной системе описывается гипсометрическим за-
коном, который для частиц сферической формы радиусом г имеет вид
г 4nr3g (р — р0) N. т
% = vfcexp[_------\RT °’ А (й, - й2)J (III. 19)
где v/ii и V/12 — частичная концентрация соответственно на высоте /г, и /г2; р и ро—•
соответственно плотность дисперсной фазы и дисперсионной среды.
Таким образом, определив концентрацию частиц на высоте h\ и h2,
радиус частиц г и зная плотность их и дисперсионной среды, можно
рассчитать число Авогадро Ад.
Для исследования броуновского движения в ультрамикрогетероген-
ных системах применяют ультрамикроскоп и микроскоп с сильным
увеличением (X 1200—1800), позволяющим наблюдать частицы разме-
ром 0,3—1 мкм.
Концентрацию частиц, находящихся на определенной высоте в дис-
персной системе после установления диффузионно-седиментационного
равновесия, можно найти методом мгновенного фотографирования че-
рез микроскоп. На фотографии подсчитывают число частиц (число за-
фиксированных частиц должно быть достаточно большим).
Фотографирование можно заменить визуальными наблюдениями.
Для этого следует уменьшить поле зрения микроскопа с помощью диа-
фрагмы, так чтобы видно было лишь несколько появляющихся и затем
исчезающих частиц, которые можно быстро сосчитать. Если сделать
такие подсчеты много раз (через определенные промежутки времени)
и суммировать найденные значения, то получается такой же результат,
как и при мгновенном фотографировании.
79
При больших увеличениях глубина резкости микроскопа очень мала,
поэтому найденные суммарные значения чисел частиц пропорциональ-
ны концентрации их на соответствующих высотах в золе.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Микроскоп МИН-8 с осветителем.
Стеклянная кювета (предметное стекло с выемкой глубиной
~0,17 мм).
Предметные и покровные стекла.
Пипетки емкостью 1 и 5 мл.
Колба емкостью 50 мл.
Воронка для фильтрования.
Бумажный фильтр.
Секундомер.
2 %-ный раствор канифоли в этиловом спирте.
Парафин.
В качестве исследуемой дисперсной системы используют золь ка-
нифоли в воде. Его получают методом физической конденсации при за-
мене растворителя, для этого приливают 5 мл раствора канифоли в
спирте по каплям к 25 мл воды при энергичном перемешивании. Обра-
зуется полидисперсный опалесцирующий золь с отрицательно заряжен-
ными частицами, имеющими сферическую форму. Грубодисперсные1
частицы отделяют, пропуская золь через бумажный фильтр, смочен-
ный водой.
В работе можно использовать также золи канифоли, близкие к мо-
нодисперсным, которые получают фракционированием с помощью по-
следовательного центрифугирования.
В кювету вносят пипеткой емкостью 1 мл несколько капель золя
канифоли и закрывают сверху покровным стеклом. Необходимо сле-
дить за тем, чтобы в кювете не было пузырьков воздуха. Для предот-
вращения высыхания золя на края покровного стекла наносят тонкий
слой теплого парафина, предварительно удалив выдавленные капли!
золя фильтровальной бумагой.
В микроскоп устанавливают объектив с увеличением X 90 и оку-
ляр X 8. На столик микроскопа помещают кювету с золем и включают
осветитель. На поверхность покровного стекла кюветы наносят каплю
иммерсионной жидкости, в которую погружают линзу объектива. Осто-
рожно фокусируют до появления в поле зрения микроскопа частиц
канифоли. Заменяют окуляр на X 15 и наблюдают броуновское дви-
жение частиц.
Устанавливают рабочий окуляр X 20, в котором имеется непрозрач-
ная диафрагма из тонкой фольги. В центре диафрагмы сделано отвер-
стие небольшого диаметра (прокол тонкой иглой для уменьшения поля
зрения).
При постоянной высоте /г', которую фиксируют по делениям на ба-
рабане микрометрического винта, подсчитывают число частиц, находя-
щихся в поле зрения микроскопа в данный момент времени наблюде-
ния через каждые 5 с. Интервал времени между подсчетами фиксиру-
ется по секундомеру. Такие подсчеты при данной высоте проводят не
80
Таблица III. 1. Результаты определения числа частиц в дисперсной системе
Высота. МКМ Число частиц в поле зрения Суммарное число частиц
h'l h2 2, 1, 4, 1, 0, 2, 3, 0 . . . 4, 3, 1, 6, 4, 3, 1, 5 . . .
менее 150 раз. Затем с помощью микрометрического винта изменяют
наблюдаемую высоту на 40—60 мкм. При новой высоте h'2 проводят
такое же число подсчетов частиц через выбранный ранее интервал вре-
мени. Результаты измерений записывают в таблицу (см. табл. III. 1).
Для каждой высоты рассчитывают суммарное число частиц, заре-
гистрированных во всех подсчетах.
Расстояние (hi — h2) в дисперсной системе определяют с учетом по-
правки на иммерсионный слой:
- h2 = - /г') (III. 20)
где «1 — показатель преломления дисперсионной среды; Пг — показатель преломления
иммерсионной жидкости; (/Zj — Л2) — разность высот в делениях по барабану микро-
скопа.
Для определения среднего размера частиц используют окуляр с из-
мерительной шкалой. На предметное стекло наносят каплю золя и
дают ей подсохнуть на воздухе; при этом на поверхности стекла обра-
зуются участки, плотно прикрытые частицами. Полученный препарат
накрывают покровным стеклом и с помощью микроскопа подсчитывают
число частиц, имеющихся на отрезке между двумя соседними деле-
ниями шкалы окуляра. Такие измерения проводят 10 раз в разных
участках препарата. По известному значению цены деления измери-
тельной шкалы рассчитывают средний радиус частиц.
Зная плотность канифоли, дисперсионной среды и температуру золя,
по формуле (III. 19) рассчитывают число Авогадро.
Работа 12. ДИСПЕРСИОННЫЙ АНАЛИЗ НИЗКОДИСПЕРСНЫХ ПОРОШКОВ
МЕТОДОМ СЕДИМЕНТАЦИИ В ГРАВИТАЦИОННОМ ПОЛЕ
Цель работы: получение кривой седиментации для низкодисперсного
порошка; построение интегральной и дифференциальной кривых рас-
пределения; определение гранулометрического состава порошка.
В основе седиментационного метода анализа дисперсных систем
в гравитационном поле лежит зависимость скорости осаждения частиц
дисперсной фазы от их размеров под действием силы тяжести (уравне-
ние III. 2). Это уравнение справедливо только для условий, при кото-
рых выполняется закон Стокса (частицы имеют сферическую форму,
движутся л^минарно и независимо друг от друга с постоянной ско-
ростью, трение является внутренним для дисперсионной среды). По-
этому. описываемый метод дисперсионного анализа применяется для
суспензий, эмульсий, порошков с размерами частиц 10-5 4- 10-2 см.
При высокой скорости оседания частиц большего размера развивается /
6 3ак.673 g£
Рис. 25. Кривая седиментации бидисперсной системы.
турбулентный режим, а если сила тяжести превышает силу внутрен-
него трения, движение становится равноускоренным. Неприменимость
уравнения (III. 2) к очень малым частицам обусловлена участием их
в молекулярно-кинетическом движении и как следствие появлением
диффузионного потока частиц, направленного противоположно седи-
ментационному потоку. На движении таких частиц сильно сказываются
конвекционные тепловые потоки.
1 Для седиментационного анализа следует применять разбавленные
системы, для которых можно пренебречь изменением скорости движе-
ния частиц в результате их столкновения. Поскольку большинство ре-
альных систем (суспензии, порошки) имеют частицы неправильной
формы, по уравнению (III. 2) можно рассчитать так называемый экви-
валентный радиус, т. е. радиус частиц сферической формы, оседающих
с такой же скоростью. На практике дисперсную систему характеризуют
распределением частиц по размерам и фракционным составом системы
(содержание дисперсной фазы в заданных интервалах радиусов час-
тиц). Эти характеристики получают, анализируя кинетические кривые
осаждения (кривые седиментации), обычно представляющие собой за-
висимость массы осевшего вещества от времени осаждения.
Кинетика седиментации частиц монодисперсной системы описыва-
ется уравнением (III. 7). Кривая седиментация в этом случае пред-
ставляет собой прямую линию, выходящую из начала координат, тан-
генс угла наклона которой характеризует скорость накопления осадка:
tg ср = dm/dx — Qu)H = k — const (Ш. 21)
По достижении времени х = Н/и весь порошок переходит в осадок
,(m = Q). Г~"\
В бидисперсной системе, состоящей цз двух монодисперсных фрак-
ций, частицы разного размера оседают одновременно. Суммарная кри-
82
вая седиментации ОАВС (рис. 25) является суперпозицией кривых се-
диментации ОЕ и OD отдельных фракций 1 и 2. Она представляет
собой ломаную линию, состоящую из трех участков. Участок ОА соот-
ветствует совместному осаждению обеих фракций “со" скоростью
Am/dxtg Z_ АОА' = k, равной сумме скоростей осаждения фракций
1 и 2: k = k\ -ф k2 = tg /LA'OE -ф ig/LB'OD. К моменту времени п —
= Н/и\ более крупная фракция полностью осела. Ее масса moi соот-
ветствует отрезку А'Е. В то же время общая масса осевших частиц
к моменту времени п есть mi=AA'. Разность mi — mol = АЕ прихо-
дится на частицы фракции 2.
Участок АВ соответствует осаждению частиц только второй фрак-
ции. Все частицы этой фракции с массой то2, соответствующей отрез-
ку-В*П, осядут к моменту времени т2 = Н1и2. При т > т2 масса осевших
частиц не меняется, что характеризуется горизонтальным участком ВС.
Суммарная масса частиц, осевших ко времени т2, представляет собой
массу всех частиц системы: т2 = mOi + т02 = тмакс и характеризуется
отрезком ВВ'. Если продолжить участки АВ и ВС до пересечения с осью
ординат (точки G и Е), то из равенства треугольников OB'D и BLG
следует, что GL = B'D = mQ2, OG = OL — GL = m2 — т02 = т01. Таким
образом, продолжение участка АВ отсекает на оси ординат отрезок,
соответствующий массе фракции 1, а отрезок, отсекаемый про-
должениями участков АВ и ВС, соответствует массе частиц второй
фракции.
Размеры частиц этих фракций могут быть вычислены по временам
их полного осаждения тц и т2, если известна высота столба суспензии Н.
Поскольку линейная скорость осаждения и = Н/х, то из уравнения
(III. 2) следует, что радиус частиц равен
г =-\ГкИ7х (Ш.22)
где К = 9т|/2^(р— ро)—постоянная, зависящая от свойств частиц и дисперсионной
среды.
Прежде чем перейти к более сложной полидисперсной системе, вве-
дем некоторую функцию Ол_Лредставляющую собой по физическому
смыслу массу частиц во фракциях, нацело выпавших (ушедших из
всего столба суспензии Я) к моменту времени т.
В соответствии с этим определением при т = тт Qi==m01; при т = т2
Q2 = moi-ф т02. Для произвольных промежуточных точек ха, х-о, хс:
Qa — 0’, Qb = moi; = mOi + т02. При этом масса частиц, осевших к
моменту времени ть, может быть определена следующим образом:
mb = m^ + bbt=Qb+Gb'tgZ BGF = Qb + xb(^-^ _ (Ш.23)
В ..„„ддисперсной системе частицы различных радиусов оседают
одновременно, но с разными скоростями. Кривая седиментации такой
системы представлена на рис. 26. Предполагая независимость осажде-
ния частиц, можно представить эту кривую как наложение бесконеч-
ного числа кривых седиментации монодисперсных систем и применить
к ней те же рассуждения, что и к бидисперсной системе. В качестве
прямолинейных участков при этом можно рассматривать бесконечно
малые отрезки касательных, проведенных к кривой седиментации в той
или иной точке. Общее количество порошка, осевшего к произвольному
6* 83.
моменту времени ть в соответствии с (III. 23) выразится уравнением
(III. 24)
Величина Qi определяется отрезком, отсекаемым на оси ординат
продолжением касательной к кривой в точке ть и характеризует массу
частиц во фракциях, нацело выпавших к моменту времени ть По-
скольку радиус частиц, прошедших за время ti всю высоту суспензии
О = VKHlxi [см. (III. 22)], то Qi — это масса частиц системы с г rj.
Член (dtn/dx)x. Т[ характеризует массу частиц с г < и, осевших к мо-
менту Т1-
Аналогично, Q2 есть количество порошка с г г2 = 'у/КН/х, , и т. д.
Как правило, определяют относительную массу осевшего порошка
(в % от общего содержания дисперсной фазы в системе). В этом слу-
чае тмаКс=100%, а величины Qi, Q2, Qs представляют собой про-
центное содержание фракций с радиусами соответственно г rlt г г2,
Г > г3.
На основании сказанного выше можно графически построить инте-
гральную кривую распределения частиц по размерам — зависимость
величины Q (процентного содержания фракции частиц с радиусами
от максимального до г) от г. Общий вид такой кривой для полидис-
персной системы представлен на рис. 27, а. Интегральная кривая по-
зволяет определить процентное содержание фракций. Например, для
фракции, содержащей частицы размерами от г\ до г2, оно равно: AQi =
— Q2 — Qi-
Более наглядное представление о распределении частиц в системе
по размерам дает дифференциальная кривая распределения, представ-
ляющая собой зависимость массовой функции распределения F —
= |AQ/Ar|, в пределе \dQ/dr\ от радиуса частиц, (рис. 27,6).
Рис 26. Кривая седиментации полидисперсной системы.
§4
Рис. 27. Интегральная (а) и дифференциальная (б) кривые распределения частиц по
радиусам.
Для построения графика функции Е(г) можно использовать инте-
гральную кривую, определяя приращения AQ для серии фракций Дг.
При этом полученное значение У7(г) относят к среднему для данной
фракции радиусу. Дифференциальную кривую можно построить и не-
посредственно из кривой седиментации (см. рис. 26), определяя AQ
как отрезки, отсекаемые соседними касательными на оси ординат, на-
пример: AQi = Q2 — Qi. Для нахождения \г\ = г2-—и необходимо
определить радиусы частиц, осевших к моментам п и т2.
Очевидно, что на рис. 27,6 процентное содержание фракции частиц
с размерами от Г\ до г2 характеризуется площадью участка под кри-
вой, а площадь под всей кривой равна массе всех частиц системы
(100%). На кривой можно выделить три наиболее характерных для
системы размера частиц: минимальный (наименьший) гМИн, наивероят-
нейший гн, отвечающий максимальному значению функции, и макси-
мальный Гмакс-
Описанный выше способ построения кривых распределения называ-
ется «методом касательных». Более точные результаты можно полу-
чить с помощью ряда аналитических методов.
Одним из наиболее простых аналитических методов является ме-
тод, предложенный Н. Н. Цюрупой для медленно оседающих суспен-
зий. Согласно этому методу кривая седиментации описывается урав-
нением
m = Q,„ —(III. 25)
Т + TQ
где Qm и то — некоторые постоянные, имеющие соответственно размерности тит.
Физический смысл константы Qm становится ясным, если предпо-
ложить, что т->оо. При этом т/(т + то)--*-1 и m^Qm. Таким образом,
Qm характеризует количество порошка, которое оседает за бесконечно
большой интервал времени. Если за 100 % принять количество порош-
ка, осевшее за конкретный конечный промежуток времени, то Qm долж-
но быть больше 100 %.
При т = т0 т== Qm/2, поэтому то иногда называют «половинным
временем седиментации».
8&
Согласно (III. 24) общее количество порошка, осевшее к любому
моменту времени т:
m = Q+(4c)T (IIL26)
ИЛИ
~ / dm \ ,ттт
Q = m — - I т (III. 27)
\ ат /
Подставляя в (III. 27) m и dm/dx, в соответствии с уравнением (III. 25)’
получим:
/ Т \2
Q = Qm[T + T-J = Qm«2 (HI. 28)
Величина а может быть выражена через размеры частиц, определяе-
мые из уравнения (III. 22):
т
а (III. 29)
_____ * + П '« + ''
где г0 = УкЯ/т0.
Таким образом
( 'о V
Q==Qma2 = Qm -ж~—г (III. 30)
\ 'о + г /
Уравнение (III. 30) представляет собой аналитическое выражение
интегральной кривой распределения. Уравнение дифференциальной
кривой распределения может быть получено дифференцированием урав-
нения (III. 30) по г:
dQ
dr
—~ -у/а (1 — а) а2
С
го
(III. 31)
Значения а2 и в в зависимости от г/г0 приведены в табл. III. 2.
По уравнениям для интегральной и дифференциальной функций
распределения можно определить значения трех основных радиусов,
характеризующих полидисперсную систему. Минимальный радиус мож-
но получить из уравнения (Ш.ЗО) при Q= 100 %:
Гмин = Го (о, 1 VQm 0 (III. 32)
Дифференцируя уравнение (III. 31) по г и приравнивая производ-
ную нулю (для максимального значения функции), можно получить
значение наивероятнейшего радиуса:
гн = г0/2,24 (III. 33)
Таблица III. 2. Параметры а2 и е. пои разных соотношениях г/г0
г; г о а2 е г/го а2 8 Г/Го аз е
0,1 0,980 0,097 0,6 0,541 0,239 1,4 0,114 0,054
0,2 0,925 0,177 0,7 0,451 0,209 1,6 0,079 0,036
0,3 0,842 0,232 0,8 0,372 0,182 1,8 0,056 0,023
0,4 0,743 0,255 0,9 0,305 0,155 2,0 0,040 0,016
0,45 0,692 0,260 1,0 0,250 0,125 2,5 0,019 0,007
0,5 0,640 0,256 1 1,2 1 0,168 0,083 . 3,0 0,010 0,003
За максимальное значение радиуса принимают
гмакс 3r0 (III. 34)
при котором значение функции распределения F составляет ~0,01 от
ее максимального значения.
Если степень полидисперсности системы П определять отношением
максимального радиуса к минимальному, то в соответствии с (III. 34)
и (III. 32):
И = Гмд- =------JL---- (Ш. 35)
Гм,ш (0.1VQm-l)Z
Таким образом, степень полидисперсности зависит только от Qm,
а значение наивероятнейшего радиуса, характеризующего общую дис-
персность системы, — только от г0. Это позволяет рассматривать г0 как
коэффициент, характеризующий дисперсность, a Qm— как коэффици-
ент, характеризующий полидисперсность.
Из выражения (III. 30) следует, что процентное содержание любой
заданной фракции равно
AQ] = Q2 Q| = Q/na2 Qmal = Qm ~~ ®l) (III. 36)
где индексы 1 и 2 относятся к частицам с радиусами г± и г±.
Таким образом, в методе Н. Н. Цюрупы седиментационный анализ
сводится к определению двух коэффициентов: г0 и Qm-
Для нахождения этих величин уравнение седиментации (III. 25)
записывают в виде
т/m = r0/Qm + т/Qm (Ш.37)
В координатах х/т — т это уравнение прямой линии. Котангенс угла
наклона прямой к оси х равен Qm,
нат, To/QOT.
Постоянная го определяется с
т — т0. При этом для вычисления
а отрезок, отсекаемый на оси орди-
помощью уравнения (III. 22) при
постоянной К необходимы данные
О ВЯЗКОСТИ Т] и ПЛОТНОСТИ Ро жид-
кой фазы и плотности вещества по-
рошка р. Получив численные значе-
ния констант Qm и г0 и задавшись
рядом значений радиусов, с помо-
щью уравнений (III. 30) и (III. 31)
и табл. III. 2 строят интегральную
и дифференциальную кривые рас-
пределения.
Для получения кривой седимен-
тации определяют с помощью тор-
зионных весов (рис. 28) изменение
массы частиц, оседающих на ча-
шечку 8, помещенную в стеклянный
цилиндр 7 с суспензией исследуе-
мого порошка.
Рис. 28. Схема торзионных весов:
1 — арретир; 2— стрелка весов; 3 — риска; 4 — рычаг
уравновешивания; 5 — стрелка циферблата; 6 — крю-
чок для чашк’Ч 7— стеклянный цилиндр; 8 — чашка.
87
Диаметр цилиндра должен быть подобран таким образом, чтобы расстояние ме-
жду его стенками и чашечкой составляло ~3 мм. При меньших зазорах на осажде-
ние частиц будут сказываться пристеночные эффекты. Расстояние от чашечки до дна
цилиндра должно составлять — 2—3 см (при больших расстояниях могут быть не
учтены самые крупные частицы), от дна чашечки до поверхности суспензии (высо-
та Н) -15 20 см.
Чашечка подвешивается на крючок 6, которым заканчивается ко-
ромысло весов. При этом необходимо проследить, чтобы цилиндр был
установлен соосно с чашечкой.
Для проведения измерений арретир весов 1 перемещают в крайнее
правое положение (при этом стрелка циферблата 5 должна находиться
против нуля шкалы). Стрелка весов 2 отклоняется влево от риски 3.
Поворотом рычага 4 против часовой стрелки совмещают стрелку 2
с риской 3. Отсчитывают показания (в мг) по положению стрелки 5
Торзионные весы, как правило, рассчитаны на измерение навесок в пределах
0—500 мг. Учитывая, что масса чашечки в дисперсионной среде может быть больше
верхнего предела шкалы, торзионные весы тарируют с помощью груза, установленного
на рычаге противовеса внутри весов. Массу груза подбирают такой, чтобы показания
весов, отвечающие массе пустой чашечки в дисперсионной среде, находились в интер-
вале 0—100 мг. Оставшаяся до 500 мг часть шкалы определяет максимальную массу
порошка, которая может быть измерена при осаждении его иа чашечке.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Торзионные весы или микровесы другого типа.
Чашечка для взвешивания.
Стеклянный цилиндр емкостью 500 мл.
Мешалка (диск с отверстиями, закрепленный на стержне).
Секундомер.
Дисперсная система, например порошок молотого кварца или суль-
фата бария.
Определяют показание торзионных весов т0, отвечающее массе пус-
той чашечки в чистой дисперсионной среде (дистиллированной воде).
Воду наливают в цилиндр до метки (соответствующей уровню иссле-
дуемой суспензии). Чашечку опускают в цилиндр, несколько раз резко
поворачивают вокруг оси, чтобы избавиться от находящихся на ней
пузырьков воздуха, затем проводят взвешивание.
Приготавливают суспензию, для чего навеску 1,0—1,5 г исследуе-
мого порошка (из расчета получения —0,5 %-ной суспензии) вносят
в цилиндр с дистиллированной водой, погружают в жидкость мешалку
и плавными движениями вверх и вниз перемешивают суспензию. Пере-
мешивают до тех пор, пока весь порошок не распределится равномерно
по всему объему воды. Затем в суспензию быстро вносят измеритель-
ную чашечку и подвешивают ее к коромыслу весов, одновременно
включают секундомер. При этом, как и при взвешивании в чистой
дисперсионной среде, надо проследить, чтобы на чашечке не было пу-
зырьков воздуха и чтобы она не соприкасалась со стенками цилиндра,
располагаясь соосно с ним.
Отмечают показания весов тИзм с интервалами времени в 30 с, за-
тем интервалы увеличивают до 1 мин, 3 мин, 5 мин по мере того, как
за предыдущий интервал времени изменение массы становится незна-
88
Таблица III. 3. Экспериментальные результаты седиментационного анализа
Время т, с Показания весов отизм- мг т=отИзм-т»> мг т, % ат
чительным. Заканчивают измерения, когда за ~ 10 мин изменение
массы составит не более 2—3 мг.
Для каждого измерения определяют массу осадка m — таз« — т0
и процентное содержание частиц в осадке т = m/mMaKc‘ 100[%], где
Шмакс — максимальное значение т. Результаты записывают в таблицу
(см. табл. III. 3) и строят кривую седиментации m(%) = f(x).
Для построения интегральной и дифференциальной кривых распре-
деления ^используют «метод касательных»/или аналитический метод
Н. Н. Цю'рутг-^ттсгуКазани^^
Результаты обработки кривой седиментации по «методу касатель-
ных» записывают в таблицу (см. табл. III. 4).
Таблица III. 4. Результаты обработки данных седиментационного
анализа по «методу касательных»
Время т, с частиц гУмкм Q- % &ri~ri ri + l &0~i' °/а
При использовании метода Н. Н. Цюрупы кривую седиментации
строят в координатах т/т — г. Результаты расчетов записывают в таб-
лицу (см. табл. III. 5).
Таблица III. 5. Результаты обработки данных седиментационного анализа
аналитическим методом
Г, мкм г/го а2 Q, % е F
Значения радиусов в табл. III. 5 задают в пределах гмии < г < Гмакс!
1" мин) Г макс» а также гн вычисляют по уравнениям (III. 32) — (III. 34).
При обработке данных «методом касательных» указанные величины
определяют из дифференциальной кривой распределения.
По уравнению (III.35) находят степень полидисперсности П.
Работа 13. ДИСПЕРСИОННЫЙ АНАЛИЗ ВЫСОКОДИСПЕРСНЫХ ПОРОШКОВ
МЕТОДОМ СЕДИМЕНТАЦИИ В ЦЕНТРОБЕЖНОМ ПОЛЕ
Цель работы: определение гранулометрического состава высокодис-
персного порошка и построение дифференциальной кривой распреде-
ления частиц по размерам.
При дисперсионном анализе высокодисперсных систем или систем
с малой разностью плотностей частиц дисперсной фазы и дисперсион-
89
Рис. 29. Схема ротора седиментометра СВ-3:
1,6—магниты; 2—площадка; 3— корпус ротора, 4 — втулка; 5,7— противовесы уравновешивания коро-
мысла; И — противовес; 9—коромысло; 10— призма.
Рис. 30. Схема термостата седиментометра СВ-3:
I — датчик контроля и регулирования температуры; 2,5 — электронагреватели; 3—корпус ротора; 4-
датчики для регистрации угла поворота коромысла.
ной среды седиментацию проводят в центробежном поле с использова-
нием центрифуг различной конструкции.
Для определения гранулометрического состава высокодисперсных
порошков в работе используется седиментометр типа СВ-3. Принцип
работы этого прибора основан на осаждении частиц в центробежном
поле с непрерывным их взвешиванием и записью кинетического хода
процесса. Седиментометр СВ-3 состоит из центрифуги, узла управления
и вторичного прибора — потенциометра КСП-4, смонтированных в од-
ном корпусе.
Центрифуга прибора включает ротор с измерительным коромыслом,
электрический привод и термостат. Ротор (рис. 29) представляет со-
бой полый диск, верхняя часть которого закрывается крышкой, фикси-
руемой гайкой. Гайка навинчивается на центральную втулку 4, через
которую в ротор наливается исследуемая суспензия. Внутри ротора на
призме 10 установлено измерительное коромысло 9, предназначенное
для взвешивания оседающих частиц. Оно представляет собой горизон-
тальные маятниковые весы, на одном конце которых расположена пло-
щадка 2 для приема оседающих частиц, на другом — противовес 8.
Для измерения угла поворота коромысла в зависимости от массы
осевших частиц установлен постоянный магнит 1, второй, неподвиж-
ный магнит 6 закреплен на дне ротора. При установке в роторе изме-
рительное коромысло уравновешивается противовесами 5, 7. Привод
центрифуги обеспечивает фиксированные скорости вращения ротора:
2000, 4000, 7000 об/мин.
Для исключения влияния температуры окружающей среды на про-
цесс осаждения частиц центрифуга установлена в термостате
(рис. 30). В верхней крышке термостата имеется отверстие для нали-
вания суспензии в ротор. На дне термостата смонтированы электрона-
греватели 2, 5, датчики 4 для регистрации углов поворота коромысла
и датчики 1 для измерения и регулирования температуры. При враще-
90
нии ротора магниты индуцируют в датчиках 4 импульсные сигналы.
Отклонение измерительного коромысла от положения равновесия при
оседании частиц приводит к изменению временного интервала между
импульсами. Величина этого интервала преобразуется в пропорцио-
нальный электрический сигнал, который записывается на потенцио-
метре КСП-4.
На рис. 31 представлена кривая кинетического хода процесса седи-
ментации. Для определения гранулометрического состава порошка по
точке выхода кривой на плато (с учетом скорости движения диаграмм-
ной ленты) определяют время осаждения частиц минимального раз-
мера тМакс, а по точке начала подъема седиментационной кривой —
время осаждения частиц максимального размера тмин. По этим данным
рассчитывают минимальный гмин и максимальный гмакс радиусы частиц,
пользуясь формулой
г = 17’35 .106 а /- (III. 38)
N V (Р — Ро) т
где г — радиус частиц, мкм; N — скорость вращения ротора, об/мин; ц — вязкость дис-
персионной среды, Па-с; р, ро — плотность дисперсной фазы и дисперсионной среды,
кг/м3; т — время осаждения, с.
В интервале от гмин до гмаКс задаются значениями радиусов частиц,
для которых по формуле (III. 38) рассчитывают время осаждения. Ра-
диусы выбирают таким образом, чтобы при постоянном интервале
Аг = п+1 — г,- между Гмин и гмакс укладывалось 10—15 их значений.
Найденные значения времени осаждения (с учетом скорости движения
диаграммной ленты) наносят на временную ось кривой седиментации.
Из точек Ti, т2, ... проводят линии, параллельные оси ординат, до пе-
ресечения с седиментационной кривой (рис. 31). Из полученных точек
проводят параллели до пересечения с осью ординат. Эти параллели
отсекают на оси ординат отрезки APoz, пропорциональные содержанию
частиц данной фракции, т. е. с радиусами в интервале г;- — r<+i. Отно-
Рис. 31. Седиментационная кривая, полученная на приборе СВ-3.
91
сительное содержание частиц каждой фракции рассчитывают по фор-
муле
рАР-..-юо [%] (Ш.39)
* о, макс
где Ро, макс — предельная ордината седиментационной кривой.
Найденные значения AQOi используются при построении дифферен-
циальной кривой распределения частиц по размерам.
ПОРЯДОК. ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Седиментометр СВ-3.
Секундомер.
Мерный цилиндр емкостью 500 мл.
Высокодисперсная система, например высокодисперсный порошок
диоксида кремния.
Работу выполняют с помощью седиментометра следующим образом.
Открывают верхнюю крышку термостата. Снимают ротор с вала при-
вода, отвинчивают ключом крепежную гайку и снимают крышку ро-
тора. Затем проверяют правильность установки измерительного коро-
мысла и наливают в ротор 450 мл дистиллированной воды. Ротор
закрывают крышкой, так чтобы совпали риски на крышке и корпусе ро-
тора и с помощью ключа плотно заворачивают крепежную гайку.
Устанавливают ротор на валу привода (при этом паз во втулке
ротора должен совпадать со штифтом вала), а датчики для регистра-
ции угла поворота коромысла продвигают к центру ротора по направ-
ляющим до упора. Закрывают крышку термостата, зафиксировав ее
зажимом.
После этого переключатель масштаба устанавливают против циф-
ры выбранной скорости вращения ротора (по указанию преподавателя)
и включают клавиши «сеть», «ротор», «термостат» (в положение
«25°C»), клавишу «запись» ставят в положение «/°C». На потенцио-
метре КСП-4 включают тумблер «прибор».
Смешивают в стеклянном стакане навеску исследуемого порошка
(0,34-0,5 г) с 10 мл воды. Через 15 мин после включения прибора
переключают клавишу «запись» из положения «/°C» в положение «а».
Включают тумблер «диаграмма-» потенциометра КСП-4. Ручкой
«нуль» выводят перо самописца потенциометра на нулевую отметку
диаграммы или на 1—2 начальных деления (скорость движения диа-
граммы устанавливается по указанию преподавателя).
В отверстие на верхней крышке термостата вставляют стеклянную
воронку и суспензию исследуемого порошка наливают через воронку
в ротор центрифуги. Одновременно включают секундомер. Через ~15с
на диаграммной ленте делают временную отметку, которую принимают
за точку начала процесса осаждения. Работу заканчивают после вы-
хода седиментационной кривой, фиксируемой на диаграммной ленте,
на плато. После этого двигатель центрифуги останавливают переводом
клавиши «ротор» в положение «откл.» и все клавиши прибора перево-
дят в исходное положение.
Для всех фракций (выбранных интервалов Агг) рассчитывают зна-
чения Ft — |AQ0</Ar;|. Полученные экспериментальные и расчетные
92
Таблица III. 6. Экспериментальные и расчетные результаты седиментационного анализа
суспензий в центробежном поле
Радиусы частиц г., мкм Время седимента- ции т, с АР0 ^01 гг, ср, мкм
данные записывают в таблицу (см. табл. III. 6) и по ним строят диф-
ференциальную кривую распределения частиц по размерам Р(гц ср),
принимая, что средний радиус частиц каждой фракции Гц СР равен ft 4-
Работа 14. ЭЛЕКТРОФОРЕТИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
ЭЛЕКТРОКИНЕТИЧЕСКОГО ПОТЕНЦИАЛА
Цель работы: определение электрокинетического потенциала дисперс-
ных систем электрофоретическим методом: исследование влияния со-
става дисперсионной среды на ^-потенциал.
Для изучения электрофореза удобно использовать гидрозоль гидр-
оксида железа или синтетические латексы.
Частицы золя гидроксида железа могут перемещаться к аноду или катоду. При
синтезе золя методом гидролиза хлорида железа (III) в кипящей воде на поверхности
частиц избирательно адсорбируются ионы Fe3+ или Fe(OH)2+ (потенциалобразующие
ионы), которые содержатся в полученном слабокислом растворе в значительном из-
бытке по сравнению с содержанием ионов ОН-. В результате образуется устойчивый
золь, частицы которого заряжены положительно.
В том случае, когда в растворе имеется избыток щелочи, например КОН, поверх-
ность частиц гидрозоля железа заряжается отрицательно.
Синтетические латексы представляют собой дисперсии частиц полимера в воде,
получаемые полимеризацией мономера, например стирола, в присутствии ПАВ. Моле-
кулы ПАВ адсорбируются на поверхности частиц, образующихся из полимерных мо-
лекул. Если молекулы ПАВ способны диссоциировать на ионы, как, например, олеат
натрия CnHssCOONa, то частицы латекса получают заряд и приобретают электрофо-
ретическую подвижность.
Электрофоретическая подвижность частиц дисперсной фазы опре-
деляется величиной ^-потенциала. В соответствии с уравнением (III. 16)
толщина диффузного слоя, а отсюда и ^-потенциал уменьшаются с рос-
том концентрации электролита (при постоянной концентрации потен-
циалобразующих ионов и постоянных значениях температуры и ди-
электрической проницаемости).
Для определения ^-потенциала экспериментально находят скорость
перемещения заряженных частиц дисперсной фазы в электрическом
поле. При микроэлектрофорезе измеряют скорость перемещения инди-
видуальной частицы дисперсной фазы под микроскопом.При макроэлек-
трофорезе (или просто электрофорезе) определяют скорость перемеще-
ния границы раздела золь-—контактная жидкость, в качестве которой
используется либо дисперсионная среда золя, либо раствор электро-
лита, электропроводность которого равна электропроводности золя.
Определив сдвиг а границы раздела золь — контактная жидкость
УЗ
Рис. 32. Схема прибора для проведения электро-
фореза:
1 — электрофоретическая трубка; 2 — краны; 3 — электроды;
4 — гайки крепления: 5 — сосуд; 6 —зажим; 7 — резиновый
шланг.
за время т, рассчитывают электрофорети-
ческую подвижность:
a aL
и = —7Т
эФ тД г V
(III. 40)
где Е — напряженность электрического поля;
L — расстояние между электродами, V—прило-
женная разность потенциалов.
Из уравнений (III. 17) и (III. 40) сле-
дует, что значение ^-потенциала пропор-
ционально электрофоретической подвиж-
ности:
•П«эф
ее0
ku .
эф
(III. 41)
Электрофорез проводят в U-образной градуированной стеклянной
трубке 1 (рис. 32), снабженной двумя кранами 2, диаметры каналов
которых равны диаметру трубки. Резиновым шлангом 7 трубка соеди-
нена с сосудом 5, в который наливают исследуемый золь. Внешняя раз-
ность потенциалов подается к металлическим электродам 3, изготов-
ленным из платины или титана.
На одном стенде монтируются четыре электрофоретические трубки,
которые параллельно подключают к сети постоянного тока, что позво-
ляет исследовать зависимость ^-потенциала частиц золя от природы
и концентрации вводимого электролита.
Порядок работы с прибором следующий. Трубку тщательно промы-
вают дистиллированной водой, открывают краны 2 (см. рис. 32) и за-
жим 6, отвинчивают две крепежные гайки 4 и снимают электрофорети-
ческую трубку со стенда вместе с металлическими планками. <: по-
мощью которых трубка монтируется к стенду.
Поднимают электрофоретическую трубку, так чтобы верхняя ее
часть была приблизительно на одном уровне с сосудом 5. В сосуд 5
наливают 50 мл золя, при этом золь заполняет резиновый шланг и под-
нимается на некоторую высоту в трубке 1. Чтобы получить четкую
границу раздела золь — контактная жидкость, необходимо удалить пу-
зырьки воздуха, которые остаются в соединительных шлангах и около
кранов. Для этого сначала продавливают резиновый шланг 7 в несколь-
ких местах, затем опускают электрофоретическую трубку настолько,
чтобы в нее почти полностью перешел золь из сосуда 5. После этого
трубку приподнимают, так чтобы после обратного перетекания части
золя в сосуд 5 в коленах трубки осталась жидкость, уровень которой
был бы на 1—2 см выше крапов 2. Операцию подъема и опускания
трубки повторяют 3—4 раза. Краны поворачивают на 180° и анало-
гично несколько раз перемещают трубку вверх и вниз до полного уда-
ления пузырьков воздуха.
Выбрав такое положение трубки, чтобы над кранами находилось
немного золя (1—2 см), закрывают краны 2 и зажим 6. Золь, остав-
шийся над кранами, выливают в колбу, тщательно промывают оба
94
колена водой и соответствующей контактной жидкостью. Укрепляют
электрофоретическую трубку на стенде гайками 4.
В оба колена трубки наливают такое количество контактной жидко-
сти, чтобы она заполнила каждую трубку над кранами 2 на ~3/4 ее
высоты. Добавляя пипеткой некоторое количество контактной жидко-
сти, добиваются равного уровня ее в коленах трубки.
Устанавливают электроды. Открывают медленно и поочередно оба
крана 2, затем осторожно приоткрывают зажим 6 настолько, чтобы
уровни жидкости медленно поднимались в электрофоретической труб-
ке. После того, как электроды погрузятся в контактную жидкость, за-
крывают зажим 6. Граница между золем и контактной жидкостью
должна быть четкой, без размывов.
Отмечают положение границ раздела золь — контактная жидкость
в обоих коленах трубки и подключают ячейку к сети постоянного тока
(включают тумблер стенда).
Для определения напряженности электрического поля Е измеряют
«сдаваемое напряжение и расстояние от одного электрода до другого
вдоль трубки.
Электрофорез проводят в течение определенного времени, после
чего отмечают изменившееся положение границы раздела золь — кон-
тактная жидкость.
[ЮРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Вариант 1. Определение электрокинетического потенциала
полистирольного латекса
Для проведения работы необходимы:
Стенд с четырьмя электрофоретическими трубками.
Вольтметр.
Конические колбы емкостью 100 мл.
Бюретки и пипетка емкостью 5 мл.
Полистирольный латекс.
0,5 М. раствор NaCl.
Контактная жидкость (разбавленный раствор NaCl, электропровод-
ность которого равна электропроводности исходного латекса).
В колбы /—4, соответствующие номерам электрофоретических тру-
бок, наливают по 40 мл латекса. В колбу 2 вводят 1 мл раствора NaCl,
в колбу 3 — 2 мл,в колбу 4 — 4 мл и доводят общий объем раствора
водой во всех колбах до 50 мл. В колбу 1 электролит не вводят (опре-
деляют исходное значение g-потенциала латекса). Заполняют приготов-
ленными растворами электрофоретические трубки и проводят электро-
форез, как описано выше, в течение 60 мин.
Рассчитывают иЭф и ^-потенциал по формулам (III.40) и (111.41),
значения г) и е дисперсионной среды принимают равными соответ-
ственно 10-3 Па-с и 81. Экспериментальные и расчетные данные запи-
сывают в таблицу (см. табл. III. 7).
Строят график зависимости ^-потенциала от концентрации электро-
лита в латексе и анализируют полученные результаты.
у5
Таблица III. 7. Экспериментальные и расчетные результаты
электрофоретических измерений
1
2
3
4
Положение границы золь — контактная жидкость, см Сдвиг границы в колене трубки, см
колено ! трубки начальное конечное
левое правое левое правое левое правое левое правое
Вариант 2. Опеделение электрокинетического потенциала
золя гидроксида железа
Для проведения работы необходимы:
Стенд с четырьмя электрофоретическими трубками.
Вольтметр.
Конические колбы емкостью 100 и 250 мл.
Пипетки емкостью 1 и 5 мл.
Мерный цилиндр емкостью 50 мл.
Колонка с анионитом.
Золь гидроксида железа очищенный, 200 мл.
0,2 М раствор НС1.
0,2 М раствор КОН.
2 %-ный (масс.) раствор FeCl3.
0,01 М раствор Na2SO4.
0,002 М раствор СаС12.
Синтезируют золь гидроксида железа методом гидролиза FeCl3.
Для этого к 170 мл дистиллированной воды, нагретой до кипения, мед-
ленно приливают 30 мл 2%-ного раствора FeCl3, смесь доводят до ки-
пения и затем охлаждают при комнатной температуре. Для удаления
образовавшейся хлороводородной кислоты золь пропускают через ко-
лонку, заполненную слабоосновной смолой в ОН-форме.
В колбы 1—4, соответствующие номерам электрофоретических тру-
бок, наливают из мерного цилиндра по 50 мл очищенного золя и сле-
дующее количество электролита:
Колба 1 1 мл 0,2 М раствора НС1
Колба 2 1 мл 0,2 М раствора НС1 + 1 мл 0,01 М рас-
твора NaaSCh
Колба 3 0,4 мл 0,2 М раствора КОН
Колба 4 0,4 мл 0,2 М раствора КОН + 1 мл 0,002 М
раствора СаСЬ
В других четырех колбах (5—8) аналогично готовят контактные
жидкости, добавляя те же количества электролита к 50 мл воды вме-
сто золя. (Природа и концентрация электролита в задания могут быть
изменены.)
96
Приготовленными растворами заполняют электрофоретические труб-
ки (в первой используют золь 1 и раствор 5, во второй — золь 2 и
раствор 6 и т. д.). Электрофорез проводят в течение 40 мин. Опреде-
ляют знак заряда коллоидных частиц и рассчитывают ^-потенциал по
формулам (III. 40) и (III. 41). Результаты записывают в таблицу (см.
табл. III. 7). Объясняют полученные результаты.
Работа 15. ОПРЕДЕЛЕНИЕ ЭЛЕКТРОКИНЕТИЧЕСКОГО ПОТЕНЦИАЛА
МЕТОДОМ ЭЛЕКТРООСМОСА
Цель работы: определение ^-потенциала по скорости электроосмотиче-
ского переноса жидкости через капиллярную систему, эксперименталь-
ное определение зависимости ^-потенциала от концентрации электро-
лита в среде.
В приборах, предназначенных для исследования электроосмоса, дис-
персная фаза представляет собой пористую диафрагму из керамиче-
ских или других материалов. Дисперсионной средой является раствор
электролита. Под действием внешней разности потенциалов, приложен-
ной к электродам, ионы диффузного слоя смещаются к соответствую-
щему электроду и увлекают за собой в результате трения дисперсион-
ную среду.
С помощью электроосмоса можно определить знак заряда поверх-
ности дисперсной фазы и электрокинетический потенциал. Например,
если жидкость перемещается к катоду, то это свидетельствует об от-
рицательном знаке заряда твердой поверхности на границе с рас-
твором.
Если радиусы капилляров диафрагмы значительно больше толщины
диффузного слоя и перемещение жидкости не вызывает изменения ги-
дростатического давления, электрокинетический потенциал можно рас-
считать по уравнению Гельмгольца — Смолуховского (III. 18). При ра-
диусах капилляров пористой диафрагмы, сопоставимых с толщиной
диффузного слоя, в уравнение вводится поправка
^ + ^1 (ш.42)
ее0/
где х» — удельная электропроводность в объеме раствора (за пределами двойного
электрического слоя); xs — поверхностная проводимость, представляющая собой уве-
личение проводимости раствора в капилляре вследствие повышенной концентрации
ионов в двойном электрическом слое.
Поправка на поверхностную проводимость может быть найдена пу-
тем измерения электрического сопротивления диафрагмы, заполненной
дисперсионной средой и стандартным концентрированным раствором
электролита.
Определение электрокинетического потенциала методом электроос-
моса проводят при помощи установки, состоящей из источника посто-
янного тока, электроосмотической ячейки, миллиамперметра и пере-
ключателя полярности тока. Электроосмотическая ячейка (рис. 33) со-
стоит из разъемного корпуса /, две половины которого соединяются
между собой при помощи накидной гайки 4. Подвод электрического
тока осуществляется через неполяризующиеся электроды 8, представ-
ляющие собой вмонтированные в корпус трубки, которые на 2/3 запол-
нены студнем агара, содержащим электролит (КС1). В верхней части
7 Зак. 673
97
Рис. 33. Схема прибора для прове-
дения электроосмоса:
1 — корпус; 2 — штуцеры; 3 — капилляры; 4—
накидная гайка; 5 — мембрана; 6 — втулка;
7— прокладки; 8 — электроды, подсоединя-
емые к источнику тока; 9 — агаровые элект-
родные мостики; 10— стаканы с раствором
сульфата меди.
ячейки имеются штуцеры 2, в
которых устанавливаются гра-
дуированные капилляры 3.
При определении (;-потен-
циала порошкообразных мате-
риалов мембрану из исследуемого порошка помещают во втулку 6. При
использовании жестких мембран они устанавливаются в ячейке вместо
втулки 6. При работе с мембраной, приготавливаемой из порошка, втул-
ку 6 устанавливают на сухую гипсовую пластину и при помощи шпателя
набивают втулку влажным осадком (из данного порошка). Мембрана
должна быть плотной, без раковин. Втулку с мембраной закрывают
с обеих сторон прокладками из фильтровальной бумаги 7 и устанав-
ливают в одной из половин ячейки. Затем обе части корпуса ячейки
плотно соединяют при помощи накидной гайки 4.
Собранную ячейку устанавливают в держателях прибора таким об-
разом, чтобы агаровые электроды 8 были опущены в стаканы 10 с рас-
твором C11SO4. Обе половины ячейки при помощи пипетки через верх-
ние штуцеры 2 полностью заполняют раствором электролита, равно-
весным с использованным осадком (в ячейке не должно быть воздуш-
ных пузырей).
Далее устанавливают градуированные капилляры и проверяют гер-
метичность ячейки. Если положение менисков жидкости в капиллярах
не изменяется в течение 3—5 мин, это показывает, что прибор герме-
тичен. Прибор подключают к источнику постоянного тока, включают
тумблер «сеть» и по секундомеру измеряют время прохождения ме-
ниска жидкости между делениями капилляра. По направлению движе-
ния жидкости через мембрану к тому или иному электроду определяют
знак заряда частиц. Кроме того, по миллиамперметру фиксируют зна-
чение силы тока. Затем тумблер «сеть» выключают, изменяют поляр-
ность электродов переключателем полярности и снова проводят изме-
рение.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Установка для определения электрокинетического потенциала.
Секундомер.
Шпатель.
Гипсовая пластина.
Градуированные пипетки.
Мерные колбы емкостью 50 мл.
Стаканы емкостью 100 мл.
Порошок А12О3 (размеры частиц 10—100 мкм)’ или керамически
мембраны на основе А12О3.
0,015 М раствор хлорида алюминия.
98
Таблица III. 8. Экспериментальные и расчетные результаты
электроосмотических измерений
Концентрация электролита, моль/л Удельная электропровод- ность и, См-м“1 Сила тока I, А Время т, с Объемная скорость V, мЗ/с ^-Потен- циал, В
Из исходного 0,015 М раствора хлорида алюминия в мерных кол-
бах емкостью 50 мл готовят 6 растворов следующих концентраций:
0,3-10-3, 0,8-10-з, 1,2-10-з, 1,5-10-з, 2-Ю-3, 3-10-3Л1. Навески порошка
А12О3 (по 10 г) или керамические мембраны вносят в стаканы, прили-
вают приготовленные растворы, перемешивают и выдерживают 30 мин
для достижения адсорбционного равновесия. Затем осадки отделяют
от растворов декантацией или фильтрованием. При работе с жесткими
мембранами извлекают их из растворов пинцетом.
Определяют удельную электропроводность каждого фильтрата (ме-
тодика измерения приведена в работе 20).
Из выделенных осадков AI2O3 поочередно готовят мембраны. Мем-
брану устанавливают в электроосмотическую. ячейку, наливают в нее
соответствующий ей равновесный раствор А1С13 (фильтрат) и измеряют
время т переноса жидкости в капилляре, как описано выше. Для дан-
ной системы выполняют 5—6 измерений и находят среднее время тср
прохождения мениска между отсчетными делениями капилляра. Пос-
ле этого ячейку разбирают и промывают дистиллированной водой.
Таким образом проводят измерения для всех приготовленных мем-
бран.
Рассчитывают объемную скорость электроосмотического переноса
дисперсионной среды:
а = у/тСр (III. 43)
где V — объем дисперсионной среды.
Используя найденные значения V, по уравнению (III. 18) рассчи-
тывают электрокинетический потенциал для исследуемых систем (при
данных концентрациях электролита поправкой на поверхностную про-
водимость можно пренебречь). Экспериментальные и расчетные дан-
ные записывают в таблицу (см. табл. III. 8).
По полученным данным строят график зависимости ^-потенциала
(с учетом его знака) от концентрации электролита и объясняют ее.
Работа 16. ОПРЕДЕЛЕНИЕ ИЗОЭЛЕКТРИЧЕСКОЙ ТОЧКИ ЗОЛЯ
ГИДРОКСИДА ЖЕЛЕЗА МЕТОДОМ ЭЛЕКТРОФОРЕЗА
Цель работы-, определение изоэлектрической точки золя гидроксида же-
леза методом электрофореза; исследование влияния высокомолекуляр-
ных соединений на изоэлектрическую точку.
Частицы дисперсной фазы, находящиеся в жидкой дисперсионной
среде, в зависимости от состава раствора могут иметь на своей по-
верхности положительный или отрицательный электрический заряд.
7*
99
С изменением концентрации потенциалобразующих ионов меняется ве-
личина заряда поверхности и значение электрокинетического потен-
циала. При определенном составе раствора может быть достигнуто изо-
электрическое состояние, при котором отсутствует заряд на поверхности
и электрокинетический потенциал равен нулю.
В дисперсных системах, где потенциалобразующими ионами явля-
ются ионы Н+ и ОН, изоэлектрическому состоянию соответствует
определенное значение pH среды, которое называется изоэлектрической
точкой. Изоэлектрическая точка рНиэт зависит от кислотно-основных
свойств вещества дисперсной фазы. Для большинства гидрозолей гидр-
оксидов (кремния, титана, железа, алюминия и др.) рНиэт определяется
соотношением констант равновесия реакций отщепления и присоедине-
ния протона Н+:
^1 ,
МОН МО +н+
МОН + Н+ мон+
[МО"] [Н+]
[МОН]
[МОН+]
[МОН] [н+]
МОН — группа поверхностного слоя гидроксида, например sStOH.
Поверхность дисперсной фазы электронейтральна, если [МО-] =
= [МОН2+].Из приведенных уравнений для изоэлектрической точки сле-
дует соотношение рНИЭт = 72(pKi — р/О). Следовательно, чем менее
кислыми свойствами обладает вещество и соответственно поверхност-
ный слой в каком-либо растворителе, т_ем больше значение его изоэлек-
трической точки. Для гидрозоля диоксида кремния рНиэт « 2, а для
золя гидроксида алюминия 9,4.
Частицы гидрозоля диоксида кремния, имеющие положительный за-
ряд при pH <' 2, перемещаются в процессе электрофореза к катоду, а
при pH >2 — к аноду. Таким образом, изоэлектрическая точка может
быть найдена по экспериментальной зависимости электрофоретической
скорости частиц золя от pH среды. Изоэлектрической точке соответ-
ствует то значение pH, при котором электрофоретическая скорость и
электрокинетический потенциал равны нулю.
Изоэлектрическая точка золя может быть изменена в результате
адсорбции на поверхности частиц полиамфолитов (ПАВ или высоко-
молекулярных соединений). Поскольку при значениях pH среды, близ-
ких к изоэлектрической точке, золи, как правило, становятся неустой-
чивыми, адсорбционное модифицирование поверхности частиц часто
применяют для защиты их от коагуляции. При такой стабилизации
поверхность частиц приобретает свойства адсорбата. При этом заряд
частиц и изоэлектрическая точка зависят не только от природы ста-
билизатора, но и концентрации электролитов.
При использовании в качестве модификаторов поверхности белков
частицы золя в кислой среде вследствие диссоциации основных групп
белка (диссоциация кислотных групп подавлена) приобретают поло-
жительный заряд. В щелочной среде, когда диссоциируют преимуще-
ственно карбоксильные группы белка, частицы золя заряжены отрица-
тельно. При значениях pH, отвечающих изоэлектрической точке белка,
электрофоретическая подвижность золя равна нулю.
100
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Стенд с четырьмя электрофоретическими трубками.
pH-метр типа pH-340
Колонка с анионитом.
Конические колбы емкостью 100 и 500 мл.
Мерный цилиндр емкостью 50 мл.
Пипетки емкостью 1 и 5 мл.
2%-ный (масс.) раствор FeCl3.
0,02 и 0,2 М растворы NaOH.
0,02 М раствор НС1.
0,05 %-ный (масс.) раствор желатины.
Синтезируют и очищают 400 мл золя гидроксида железа (см. опи-
сание работы 14).
В колбы 1—4, соответствующие номерам электрофоретических тру-
бок емкостью 100 мл, наливают из мерного цилиндра по 50 мл очищен-
ного золя и при перемешивании добавляют раствор щелочи:
Колба 1 50 мл золя
Колба 2 50 мл золя + 0,4 мл 0,02 М раствора NaOH
Колба 3 50 мл золя + 3,5 мл 0,02 Л1 раствора NaOH
Колба 4 50 мл золя + 1 мл 0,2 М раствора NaOH
С помощью прибора pH-340 измеряют pH приготовленных раство-
ров. В других четырех колбах емкостью 100 мл готовят контактные
жидкости, добавляя указанное выше количество щелочи к 50 мл воды.
Приготовленными растворами заполняют электрофоретические трубки
(см. порядок выполнения работы 14) и проводят электрофорез в тече-
ние 30 мин. Результаты измерений и расчетов записывают в таблицу
(см. табл. III. 7 в работе 14). По полученным данным строят график
зависимости ^-потенциала от pH и определяют рНИЭт-
Аналогичным образом определяют изоэлектрическую точку золя
в присутствии желатины. Для приготовления растворов в 50 мл золя
вначале вводят раствор желатины, смесь тщательно перемешивают и
затем добавляют раствор щелочи или кислоты:
Колба 1 50 мл золя + 3 мл раствора желатины 4-5 мл 0,02 М НС1
Колба 2 50 мл золя + 3 мл раствора желатины + 1 мл 0,02 М НС1
Колба 3 50 мл золя + 3 мл раствора желатины
Колба 4 50 мл золя 4-3 мл раствора желатины -|- 1 мл 0,02 М раствора NaOH
Перед заполнением электрофоретические трубки тщательно промы-
вают водой.
Экспериментальные и расчетные данные записывают в таблицу,
строят график (; = f (pH) и определяют рНиэт.
Сопоставляют и объясняют результаты проведенных опытов.
101
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАЧИ
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Чем обусловлено броуновское движение частиц дисперсных систем?
В каких системах возможно броуновское движение? Приведите при-
меры.
2. Какова количественная взаимосвязь между броуновским движе-
нием частиц и тепловым движением молекул среды. Как можно рас-
считать число Авогадро, используя это соотношение?
3. Какие выводы можно сделать из теории броуновского движения,
обобщающей свойства истинных растворов и золей?
4. Как можно определить размеры дисперсных частиц или концен-
трацию их в лиозолях по осмотическому давлению?
5. Какие известны Вам методы дисперсионного анализа? Укажите
области их применения. Для каких дисперсных систем применяется
седиментационный анализ в гравитационном поле и в центробежном
поле?
- 6. Напишите уравнение Стокса для скорости седиментации в грави-
тационном поле. Каков физический смысл входящих в него величин?
Изменением каких параметров системы можно изменять скорость осаж-
дения частиц?
7. Каковы условия соблюдения закона Стокса при седиментации?
Какие отклонения наблюдаются при несоблюдении этих условий?
8. Что такое константа седиментации и что она характеризует? На-
пишите выражение для константы седиментации сферических частиц,
если осаждение их подчиняется закону Стокса.
9. Какие системы называют мрнодисперсными и полидисперсными?
Что служит характеристикой полидисперсности системы?
10. Каково назначение интегральных и дифференциальных кривых-
распределения частиц по размерам? Как изменяется вид кривых рас-
пределения по мере приближения полидисперсной системы к монодис-
персной?
И. Как определить содержание частиц для данного интервала раз-
меров по интегральной и дифференциальной кривым распределения?
12. Для каких систем применяется седиментационный анализ в цен-
тробежном поле? Как изменяется скорость оседания частиц в центро-
бежном поле в процессе седиментации? Напишите выражение для кон-
станты седиментации в центробежном поле.
13. Что такое диффузионно-седиментационное равновесие? Чем ха-
рактеризуются кинетическая и термодинамическая седиментационнах
устойчивость системы? Как определяют размеры частиц в условиях
диффузионно-седиментационного равновесия?
V 14. Каковы возможные причины возникновения двойного электри
ческого слоя на межфазной поверхности? Приведите примеры механиз
мов образования двойного электрического слоя в различных дисперс
ных системах.
V15. Дайте характеристику строения двойного электрического ело:
на поверхности раздела фаз. Как изменяется потенциал с расстояние?
от поверхности?
16. Расскажите об основных положениях теории строения двойног
электрического слоя. Какое соотношение лежит в основе этой теории
102
17. Что понимают под толщиной диффузной части двойного элек-
трического слоя? Чем определяется толщина плотной и диффузной час-
тей двойного электрического слоя?
18. Перечислите электрокинетические явления и объясните, чем они
обусловлены.
19. Что называют электрокинетическим потенциалом? Какие фак-
торы влияют на ^-потенциал? Как изменяется ^-потенциал отрицательно
заряженных частиц при введении в золь нитратов калия, бария и лан-
тана?
20. При каких условиях применимо уравнение Гельмгольца-—Смо-
луховского для скорсти электрофореза? Какими свойствами должна
обладать контактная жидкость?
21. Что собой представляют релаксационный эффект, электрофоре-
тическое торможение и поверхностная проводимость? В каких случаях
их необходимо учитывать при расчете ^-потенциала?
ПРИМЕРЫ РЕШЕНИЯ ЗАДАЧ
1. Определите коэффициент диффузии D и среднеквадратичный сдвиг Д частицы
гидрозоля за время т = 10 с, если радиус частицы г = 50 нм, температура опыта
293 К, вязкость среды г] = 10~3 Па-с. _
Решение. Среднеквадратичный сдвиг частицы Д за промежуток времени т опреде-
ляется по закону Эйнштейна — Смолуховского (III. 8):
А2 = 2 Dr
Коэффициент диффузии D рассчитывается по уравнению Эйнштейна (III. 9):
D = = 4,29.10^ м7с
6-3,14- 10 <50-10 9
Тогда средеквадратичный сдвиг частицы составит:
Д = У2Дт = д/2-4,29- 10“12- 10 =9,26- 10~е м = 9,26 мкм
2. Определите радиус частиц гидрозоля золота, если после установления диффу-
зионно-седиментационного равновесия при 293 К на высоте h = 8,56 см концентрация
частиц изменяется в е раз. Плотность золота р = 19,3 г/см3, плотность воды Ро —
== 1,0 г/см3.
Решение. Распределение частиц по высоте при установлении диффузионно-седи-
ментационного равновесия описывается гипсометрическим уравнением (III. 12):
In > Ц?(Р-РО)А
Vo kT
где v — объем частицы, равный для сферических частиц 4/зЯТ3.
Согласно условию задачи Vh = v0/e и lnvh/vo = —1. С учетом этого выражение
для радиуса частиц принимает вид
— 3/ 3kT 3 /___________3- 1,38- 10~~23-293__________
Г ~ V 4ngh (р — Ро) ~ V 4 • 3,14 • 9,81 • 8,56 • 10-2 - (19,3 — 1) • 103
= 3,98- 10-9 м = 3,98 нм
3. Рассчитайте осмотическое давление 30 %-ного (масс.) гидрозоля SiO2 при 293 К,
если удельная поверхность частиц зуд = 2,7-105 м2/кг. Плотность . частиц гидрозоля
р = 2,2 г/см3, плотность среды р0 — 1,15 г/см3. F
103
Решение. Осмотическое давление в дисперсных системах рассчитывается по урав-
нению Вант-Гоффа (III. 10);
л = vkT = kT
т
тле- с — массовая концентрация частиц; т — масса одной частицы.
Рассчитываем массовую концентрацию дисперсной фазы:
__________________0,3____________________0,3_________= 402 7 / 3
С“ 0,3/р + 0,7/ро — [0,3/2,2 • 103 + 0,7/1,15 • 103 ’ КГ/М
Для сферических частиц
. т — '/gad3^-
6 6 '
Поскольку 5уд = — , то d =----------. Тогда
ар 5удР
36л 36-3,14 , 1П ]n-2i
т = -д—5- = -г------7----------=1,19-10 кг
«Здр2 (2,7- 105)3 -(2,2-103)2
Рассчитываем осмотическое давление:
л =-----402,7 = -1,38 • 10“23 • 293 = 1,37 • 103 Н/м2
1,19- 10 21
4. Частицы бентонита дисперсностью D = 0,8 мкм-1 оседают в водной среде под
действием силы тяжести. Определите время оседания п на расстояние h = 0,1 м, если
плотность бентонита р = 2,1 г/см3, плотность среды р0 = 1,1 г/см3, вязкость среды
т) = 2-10“3 Па-с. Во сколько раз быстрее осядут частииы на то же расстояние в
центробежном поле, если начальное расстояние от оси вращения ха = 0,15 м, а ско-
рость вращения центрифуги п — 600 об/с.
Решение. Скорость оседания частиц с радиусом с в сплошной среде при соблюде-
нии закона Стокса (III. 2) выражается уравнением
h _ 2 (р = ро) gr2
Ti 9т]
откуда
где
___ 9r)/i4£>2
Т‘ “ 2 (р — р0) g
= 9 - 2 - 10~~3 - 0,1 - 4 - (8 - 105)2 _
1 2 • (2,1 - 1,1) • 103 • 9,81 °
Для частиц, оседающих в центробежном поле, справедливо соотношение (III. 6):
х _ 2г2 (р — ро) <й2 т2
ха 9ц
где х = %о + h\ и = 2лп — угловая скорость вращения центрифуги.
Время оседания в центробежном поле составит:
_ 9ц In (х0 + h)fxQ • 4D2 _
T2 2 (р — Ро) 4л2га2
__ 9 • 2 • 10~3 • In (0,15 + 0,1Ю>/0,15 - 4 • (8 • 105)2
2 • (2,1 — 1,1) • 103 • 4 • (3,14)2 • 6002 — 0,83 С
Искомое соотношение равно
5. Рассчитайте толщину диффузного ионного слоя % на поверхности частиц суль-
фата бария, находящихся в водном растворе NaCl концентрацией 25 мг/л. Относи-
тельная диэлектрическая проницаемость раствора при 288 К равна е = ’82,2.
104
Решение.
(Ill. 16):
Толщина диффузного ионного слоя рассчитывается по уравнению
ee0RT
2F21
где электрическая постоянная so =• 8,85-10-12 Ф/м,
= 96 500 Кл/моль, ионная сила раствора I = — >
постоянная Фарадея F ==
(с — концентрация в моль/м3).
Для электролита NaCl ионная сила равна:
/ = у(с-12 + с-12)=с
Выразим концентрацию раствора NaCl в моль/м3:
с =
25 _ 25
М ~ 58,44
= 0,428
Следовательно, I — 0,428. Рассчитываем толщину %:
./82,2 • 8,85 • 10 12 - 8,31-288
V 2-96 5002 • 0,428 “
1,477- 10 8 м== 14,77 нм
6. Рассчитайте электрокинетический потенциал на границе водный раствор — по-
ристая стеклянная мембрана по данным электроосмоса: сила тока / = 3-10-3 А, за
время 60 с переносится 0,63 мл раствора, вязкость дисперсионной среды г, = 10“3
Па-с, относительная диэлектрическая проницаемость среды е = 80,1. Электрическое
сопротивление мембраны с дисперсионной средой Ri = 4500 Ом, а сопротивление
мембраны, заполненной 0,1 М раствором К.С1, составляет R2 = 52 Ом. Удельная
электропроводность 0,1 М раствора КС1 равна xKci = 1,167 См-м~’.
Решение. Электрокинетический потенциал рассчитывается по формуле (III. 18):
~ /ее0
Объемная скорость переноса среды v при электроосмосе составит:
6,3-I0-7 1Л_ ,.-8 з/
о=------г?;-= 1,05-10 м/с
ои
Удельная электропроводность раствора в порах мембраны складывается и/ объ-
емной электропроводности X/ и поверхностной проводимости zs, т. е. х = Хи + х».
Электрическое сопротивление мембраны с дисперсионной средой равно
7?Г =---£--- =-
X., + X X
V s
где k — постоянная мембраны.
Если пористая мембрана заполнена 0,1 М раствором КС1, то поверхностной про-
водимостью можно пренебречь и сопротивление мембраны будет равно
Я2 = —
xv ХКС1
Следовательно
= k = /?2ХКС1
Отсюда
х = хКС1-^-= 1-1671,35- 10-2 См-м"1
и электрокинетический потенциал равен
г 1,05 • 10“8-10“3-1,35-10-2
ь =------- -------------- — = о 066 в
3- 10 3 - 80,1 -8,85- 10 12
10&
7. Рассчитайте электрокинетический потенциал на границе керамический фильтр —
водный раствор КС1, если при протекании раствора под давлением р — 3,1 • 104 Па по-
тенциал течения составил U — 1,2- 10 ’- В. Свойства дисперсионной среды при 298 К:
удельная электропроводность х = 0,141 См-м"1 (0,01 М раствор КС1), вязкость
г] = 8,94-Ю~4 Па-с, относительная диэлектрическая проницаемость а = 78,5.
Решение. Зависимость потенциала течения от приложенного давления и свойств
дисперсной системы выражается соотношением
Л
Т]Х J
Г Uip 1,2- 0,141 R
Q — _19 л — U,и/ й
ееор 78,5-8,85-10 12-3,1-104
ЗАДАЧИ
1. Среднеквадратичное значение проекции сдвига частицы гидрозоля
SiO2 за 3 с составляет 8 мкм. Определите радиус частицы, если вяз-
кость дисперсионной среды равна 1-10-3 Па-с при 293 К.
2. Определите проекцию среднего сдвига Л для частиц гидрозоля
за время 10 с, если радиус частиц 0,05 мкм, температура опыта 293 К,
вязкость среды 1 • 10_3 Па-с.
3. Удельная поверхность сферических частиц гидрозоля кремнезема
составляет: а) 1,1-104 м2/кг; б) 1,1-10® м2/кг; в) 1,1-106 м2/кг. Плот-
ность кремнезема 2,7 г/см3, вязкость дисперсионной среды 1-10-3 Па-с,
температура 293 К. Определите проекции среднего сдвига частиц золя
за время 4 с.
4. По данным Сведберга, коэффициент диффузии коллоидных час-
тиц золота в воде при 298 К равен 2,7-10~6 м2/сут. Определите дисперс-
ность частиц гидрозоля золота. Вязкость воды при 298 К равна
8,94- IO3 Па-с.
5. Ниже приведены результаты измерения среднеквадратичного
сдвига частиц суспензии гуммигута в воде, полученные Перреном:
Время сдвига, с . . 30 60 90 120
Сдвиг, мкм .... 7,09 10,65 11,31 12,00
На основании этих результатов вычислите среднее значение числа
- Авогадро. Радиус частиц суспензии 0,212 мкм, температура опыта
290 К, вязкость среды 1,1-10-3 Па-с.
,\ 6. Определите частичную концентрацию золя А12О3, исходя из сле-
у дующих данных: массовая концентрация 0,3 г/л, коэффициент диффу-
** зии сферических частиц золя 2-J.Q-6 м2/сут, плотность А12О3 4 г/см3,
вязкость среды 1-10-3 Па-с, температура 293 К.
7. Результаты экспериментов Сведберга по определению среднего
сдвига А частиц золя платины в разных средах при 293 К следующие:
П-Ш'1, Па-с -г, с Д, мкм
Ацетон 3,2 1,60 6,2
Вода 10,0 0,65 2,1
Пропиловый спирт 22,6 0,45 1,3
На основании этих данных определите дисперсность частиц золя,
а также коэффициенты диффузии частиц в указанных средах.
8. Для гидрозоля А12О3 рассчитайте высоту, на которой концентра-
ция частиц уменьшается в 2,7 раза. Форма частиц сферическая, удель-
ная поверхность дисперсной фазы гидрозоля: а) 109 м~'; б) 0,5-109 м-1;
106
в) 108 м-1. Плотность AI2O3 4 г/см3, плотность дисперсионной среды
1 г/см3, температура 293 К.
9. Ниже приведены результаты изучения равновесного распределе-
ния частиц гидрозоля селена по высоте под действием силы тяжести
(при 293 К).:
h, мкм ............. 50 850 1050 1250
Число частиц в еди-
нице объема .... 595 271 165 90
Используя эти данные, рассчитайте коэффициент диффузии частиц
селена в воде. Плотность селена примите равной 4,81 г/см3, плотность
воды 1 г/см3, вязкость воды 1 -10~3 Па-с.
10. Определите высоту, на которой после установления диффузион-
но-седиментационного равновесия концентрация частиц гидрозоля SiO2
уменьшится вдвое. Частицы золя сферические, дисперсность частиц:
а) 0,2 нм-1; б) 0,1 нм-1; в) 0,01 нм-1. Плотность SiO2 2,7 г/см3, плот-
ность воды 1 г/см3, температура 298 К.
11. В опытах Вестгрена было получено следующее установившееся
под действием силы тяжести распределение частиц гидрозоля золота
по высоте:
h, мкм ............. 0 50 100 200 300 400 500
Число частиц в еди-
нице объема .... 1431 1053 779 408 254 148 93
Определите средний размер частиц гидрозоля, если плотность дис-
персной фазы равна 19,6 г/см3, температура 292 К.
12. Осмотическое давление гидрозоля золота (форма частиц сфе-
рическая) с концентрацией 2 г/л при 293 К равно 3,74 Па. Рассчитайте
коэффициент диффузии частиц гидрозоля при тех же условиях, если
плотность золота 19,3 г/см3, а вязкость дисперсионной среды
1-10 '3 Па-с.
13. Рассчитайте отношение осмотических давлений двух гидрозолей
(форма частиц сферическая) при условии: одинаковая массовая кон-
центрация, но различная дисперсность частиц — Di = 40 мкм-1 и D2 —
= 20 мкм-1; 2) одинаковая дисперсность, но различная массовая кон-
центрация — С[ = 7 г/л и с2 = 3,5 г/л.
V14. Рассчитайте размер частиц диоксида кремния, если известно, что
время их оседания на расстояние 1 см составляет: а) 30 с; б) 60 мин;
в) 100 ч. Плотность дисперсной фазы и дисперсионной среды состав-
ляет соответственно 2,7 и 1,1 г/см3, вязкость дисперсионной среды
1,5-10-3 Па-с.
,4-5. Рассчитайте время, за которое сферические частицы стекла
в воде~Ъседают на расстояние 1 см, если дисперсность частиц состав-
ляет: а) 0,1 мкм-1; б) 1 мкм-1; в) 10 мкм-1. Плотность дисперсной
фазы и дисперсионной среды соответственно 2,4 и 1,0 г/см3. Вязкость
дисперсионной среды 1-10-3 Па-с.
16. Определите удельную поверхность порошка сульфата бария
(в расчете на единицу массы), если частицы его оседают в водной
среде на высоту 0,226 м за 1350 с (предполагая, что частицы имеют
сферическую форму). Плотность сульфата бария и воды соответственно
4,5 и 1 г/см3, вязкость воды 1-10-3 Па-с.
17. Рассчитайте, за какое время сферические частицы А12О3, рас-
пределенные в среде с вязкостью 1,5-10-3 Па-с, оседают на высоту
107
1 см, если удельная поверхность частиц составляет: a) 104 м-1;
б) 10s м“!; в) 10б м-1. Плотности дисперсной фазы и дисперсионной
среды равны соответственно 4 и 1 г/см3.
18. Граница между гидрозолем золота и дисперсионной средой в
центробежном поле ультрацентрифуги через 1 ч после начала опыта
находилась на расстоянии 3,70 см от оси вращения, а через 1,5 ч —
на расстоянии 3,78 см. Определите размер и удельную поверхность (в
расчете на единицу массы) сферических частиц гидрозоля, если ско-
рость вращения ротора центрифуги 8700 об/мин, плотность золота
19,3 г/см3, плотность воды 1 г/см3, вязкость воды !• 10_3 Па-с.
19. Рассчитайте толщину диффузного ионного слоя X на поверхно-
сти твердой пластинки, помещенной в водные растворы с содержанием
индифферентного электролита КС1: а) 1-10~5; б) 1 • 10~3; в) IX
X Ю-1 моль/л. Относительную диэлектрическую проницаемость рас-
творов при 298 К примите равной 78,5. Постройте график зависимости
<р/<рб от расстояния, которое изменяется от Z до 5Z.
20. Рассчитайте толщину диффузного ионного слоя X на поверхно-
сти пластинки при 300 К в водном растворе, 1 л которого содержит
0,05 г NaCl и 0,01 г Ba(NO3)2 (индифферентные электролиты). Отно-
сительная диэлектрическая проницаемость раствора равна 76,5. Во
сколько раз изменится X, если раствор разбавить чистой водой в
4 раза?
21. Рассчитайте толщину диффузного ионного слоя X частиц дис-
персной фазы при 293 К в водных растворах, содержащих 1-10~4 моль/л
одного из следующих электролитов: NaCl, СаС12, MgSO4. Считая, что
относительная диэлектрическая проницаемость растворов линейно из-
меняется от 87,8 до 69,7 при повышении температуры от 273 до 323 К,
постройте зависимость Л. от Т для раствора NaCl.
22. Используя теорию Гуи — Чепмена для слабозаряженной поверх-
ностТГГ"оцените значение потенциала ф и объемной плотности заряда р
на расстоянии х = 15 нм от поверхности. Окружающей средой явля-
ется водный раствор NaCl с концентрацией 5-10-4 моль/л, температура
293 К, относительная диэлектрическая проницаемость среды 80,1, по-
тенциал поверхности фо ~ Фв = 0,02 В. Определите, во сколько раз из-
менятся ф и р, если х увеличить в три раза.
23. Рассчитайте объемную плотность заряда на границе диффузного
слоя рв дисперсной фазы по следующим данным: фв=.0,03 В, окружа-
ющей средой является водный раствор КС1 с концентрацией
3-10~4 моль/л, относительная диэлектрическая проницаемость среды
80,1, температура 293 К- Чему равна плотность поверхностного заряда,
обусдлвленного диффузным слоем?
*4 $ Рассчитайте емкость диффузного слоя дисперсной фазы. Дис-
персионной средой является водный раствор СаС12 концентрации
2-Ю-4 моль/л при 283 К с относительной диэлектрической проницае-
мостью 83,8. Определите, во сколько раз изменится емкость, если к рас-
твору СаС12 добавить равный объем водного раствора NaCl такой же
мольной концентрации?
25. Рассчитайте электрокинетический потенциал поверхности квар-
ца по данным, полученным при исследовании электроосмотического
переноса жидкости через кварцевую мембрану: сила тока 2-Ю-3 А,
объемная скорость раствора КС1, переносимого через мембрану,
0,02 мл/с, удельная электропроводность раствора 1,2-10~2 См-м-1,
108
вязкость 1-10“3 Па-с, относительная диэлектрическая проницаемость
80,1.
26. Рассчитайте электрокинетический потенциал на границе раз-
дела фаз кварц — водный раствор КС1 по следующим данным электро-
осмоса: сила тока 2-10~3 А, время переноса 0,1 мл раствора ПО с,
удельная электропроводность раствора 6,2-10'2 См-м~', вязкость
10~3 Па-с, относительная диэлектрическая проницаемость 80,1.
27. Постройте график зависимости электрокинетического потенциа-
ла поверхности дисперсной фазы (электрокорунд, а-А12Оз) от концен-
трации электролита сЭл (гексаметафосфат натрия) по эксперименталь-
ным данным электроосмоса;
сэл> %••••*• v - 103, мл/с . . . / - 103, А z0 • 102, См • м-1 . • 102, См • м~! . . . . 0,02 . . . 1,85 . . . 1,70 . . . 2,40 . . . 7,76 0,05 1,25 1,70 3,91 6,42 0,10 2,01 2,15 6,10 0,49 0,20 1,28 3,30 10,20 0,20
и — соответственно удельная электропроводность дне-
персионнои среды и поверхностная проводимость.
Относительная диэлектрическая проницаемость растворов 80,1, вяз-
кость 1 • 10-3 Па-с. Определите знак заряда поверхности мембраны,
если растворы под действием тока перемещаются к катоду.
28. Рассчитайте объем раствора, перенесенный через мембрану из
корунда за 1 ч в результате электроосмоса слабого раствора электро-
лита под действием э.д. с. 100 В. Электрокинетический потенциал по-
верхности корунда 0,08 В, относительная диэлектрическая проницае-
мость среды 80,1, вязкость 1-Ю-3 Па-с, электрическое сопротивление
мембраны с этим раствором = 3900 Ом.
Для определения удельной электропроводности раствора, включаю-
щей поправку на поверхностную проводимость, было измерено элек-
трическое сопротивление мембраны в 0,1 н. растворе КО, которое ока-
залось равным /?2 = 41,0 Ом. Удельная электропроводность 0,1 н. рас-
твора КО равна 1,167 См-м-1. Рассчитайте расход электроэнергии, не-
обходимый для переноса 1 м3 раствора.
' 29/ Рассчитайте электрофоретическую скорость передвижения час-
тиц золя трисульфида мышьяка по следующим данным: (/потенциал
частиц — 42,3 мВ, расстояние между электродами 0,4 м, внешняя раз-
ность потенциала 149 В, вязкость среды 1-10~3 Па-с, относительная
диэлектрическая проницаемость 80,1.
30. Определите (/потенциал корундовой мембраны по результатам
электроосмоса растворов КС1:
cKCi> моль/л . . . . . . 0,01 0,02 0,10 0,50
См • м~1 . . . . . . 0,128 0,250 1,167 5,55
R, Ом . . 565 520 340 71,6
1 103, А . . 10 26 зо- 50
v • 103, мл/с . . . . . . 0,780 0,628 0,730 0,185
7? — сопротивление мембраны, заполненной соответствующим
раствором КС1.
Относительная диэлектрическая проницаемость растворов 80,1, вяз-
кость 1 • 10“3 Па-с, температура 293 К.
109
Для всех растворов рассчитайте толщину диффузного ионного слоя %
и сопоставьте характер изменения ^-потенциала и X при увеличении
концентрации КС1.
31. Рассчитайте ^-потенциал поверхности частиц бентонитовой гли-
ны по результатам электрофореза при следующих условиях: расстоя-
ние между электродами 25 см, напряжение 100 В, за 15 мин частицы
перемещаются на 6 мм к аноду, относительная диэлектрическая про-
ницаемость среды 78,2 (при 298 К), вязкость 8,94-10~4 Па-с.
32. Частицы аэросила SiO2 в водной среде при pH = 6,2 имеют
электрокинетический потенциал, равный —34,7 • 10 3 В. На какое рас-
стояние и к какому электроду сместятся частицы за 30 мин, если на-
пряжение в приборе для электрофореза 110 В, расстояние между элек-
тродами 25 см, относительная диэлектрическая проницаемость среды
80,1, вязкость 1-10~3 Па-с.
33. Рассчитайте электрокинетический потенциал частиц корунда
в водном растворе по следующим данным: скорость электроосмоса че-
рез корундовую мембрану 0,02 мл/с, удельная электропроводность рас-
твора 1,2-10“2 См-м”1, поверхностная проводимость 2-Ю"2 См-м-1,
вязкость раствора 1 • 103 Па-с, сила тока при осмосе 1,5-10—2 А, отно-
сительная диэлектрическая проницаемость раствора 80,1.
34. Рассчитайте электрокинетический потенциал частиц кварцевого
стекла, а также толщину диффузного ионного слоя, если скорости пе-
редвижения этих частиц в водных растворах NaCl концентрацией
5-10“4 и 67-10”3 моль/л равны- соответственно 2,2 и 0,4 мкм/с при по-
стоянной напряженности электрического поля 100 В/м. Вязкость рас-
творов 1,14-10—3 Па-с, относительная диэлектрическая проницаемость
82, температура 288 К.
35. Рассчитайте электрокинетический потенциал по эксперименталь-
ным данным электрофореза золя гидроксида кремния в растворах
Cd(NO3)2:
cCd(NOa)2 ’ ммоль/л....... о 1 3,6 15,0
Электрофоретическая подвиж-
ность и0 109, м2/(с • В) . ... 25 19 11 6,5
Относительная диэлектрическая проницаемость среды 80,1, вязкость
1-10~3 Па-с, дисперсная фаза перемещается к аноду. Постройте гра-
фическую зависимоссть ^-потенциала от концентрации Cd(NO3)2. Объ-
ясните полученную зависимость.
36. Рассчитайте электрокинетический потенциал частиц золя
Fe(OH)3 по данным электрофореза: внешняя э.д. с. 170 В, расстояние
между электродами 0,45 м, смещение границы золя к катоду составило
12 мм за 30 мин. При температуре опыта, равной 298 К, вязкость дис-
персионной (водной) среды 8,94-10—* Па-с и относительная диэлектри-
ческая проницаемость 78,2.
37. Определите электрокинетический потенциал на границе раздела
фаз керамический фильтр — водный раствор КС1, если при протекании
раствора под давлением 2-Ю4 Па потенциал течения равен 6,5-10-3 В.
Удельная электропроводность среды 1,3-10-2 См • м-1, вязкость 10-3Па-с,
относительная диэлектрическая проницаемость 80,1.
38. Рассчитайте потенциал течения, возникающий при продавлива-
нии этилового спирта через мембрану из карбоната бария под давле-
нием 9,81-103 Па, если ^-потенциал равен 54-10~3 В, удельная электро-
не
проводность среды 1,1-10—4 См-уН, вязкость 1,2-10~3 Па-с, относи-
тельная диэлектрическая проницаемость 25.
39. Водный раствор NaCl под давлением 4,9-104 Па проходит через
кварцевую мембрану. Вычислите потенциал течения на границе мем-
брана— раствор, если ^-потенциал равен 0,04 В, удельная электропро-
водность среды 1-Ю-2 См-м-1, вязкость 1-10-3 Па-с, относительная ди-
электрическая проницаемость 80,1.
40. Рассчитайте потенциал, возникающий при течении водного рас-
твора через мембрану из полистирола под действием давления 2-104 Па,
если известно, что при электроосмосе в этой дисперсной системе объ-
емная скорость раствора равна 8-10—4 мл/с при силе тока 4-Ю-4 А.
41. Постройте графическую зависимость ^-потенциала на границе
раздела стекло — водный раствор электролитов Ca(NO3)2, A1(NO3)3 и
Th(NO3)4 от их концентрации:
Гдл, моль/л ... ^-потенциал, мВ, 3- 10”7 3- 10~6 1 • 10~4 1 • 10”2
для Ca(NOs) . . . — 145 — 120 —80 —33
A1(NO3)3 . . . — 115 — 15 +55 +30
Th(NO3)4 . . . —90 +45 + 140 +83
Объясните влияние заряда катионов на изменение ^-потенциала.
IV. ОПТИЧЕСКИЕ СВОЙСТВА И ОПТИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ ДИСПЕРСНЫХ СИСТЕМ
Из оптических методов исследования в коллоидной химии применя-
ются те методы, с помощью которых можно проводить дисперсионный
анализ, т. е. определять размер и форму частиц, удельную поверхность,
концентрацию дисперсной фазы. К таким методам относятся световая
и электронная микроскопия, методы, основанные на рассеянии лучей,
двойном лучепреломлении и др.
Наиболее информативными, и поэтому широко используемыми ме-
тодами определения дисперсности и формы частиц являются световая
и электронная микроскопия. С помощью этих методов можно непосред-
ственно наблюдать частицы и измерять их размеры. Нижний предел
световой микроскопии составляет до 100 нм, электронной микроско-
пии— до 2—5 нм. Следует иметь в виду, что электронная микроскопия
имеет существенный недостаток, а именно: она применима только для
исследования сухих образцов и не может быть исйользонана для на-
блюдения их, например, в жидких средах.
Указанный недостаток отсутствует у оптических методов, основан-
ных на рассеянии света (опалесценции). Они не уступают электронной
микроскопии и по чувствительности.
Светорассеяние, или опалесценция, принадлежит к дифракционным
явлениям, обусловленным неоднородностями, размеры которых меньше
длины волны падающего света. Такие неоднородности рассеивают свет
во всех направлениях. Теория светорассеяния (опалесценции) впервые-
была развита Рэлеем. В ее основе лежит уравнение для интенсивности
света /р, рассеянного единицей объема дисперсной системы со сфери-
ческими диэлектрическими частицами, значительно меньшими длины
Ш
волны падающего света:
[*vw^ Т
F W-0 +cos20)J
(IV. 1)
где /о — интенсивность падающего света; F — функция показателей преломления;
v — концентрация частиц в единице объема системы; v — объем чарицы; X — длина
волны падающего света; R — расстояние частицы от источника света; 0 — угол между
направлениями распространения рассеянного света и падающего света.
Функция F определяется соотношением
F = 24л3
га1 - гао у
п-1 + 2«g J
(IV. 2)
где rii и По — соответственно показатель преломления вещества дисперсной фазы и
дисперсионной среды.
Уравнение Рэлея лежит в основе оптических методов определения
размеров частиц и концентрации дисперсной фазы: ультрамикроскопии,
нефелометрии и турбидиметрии.
Ультрамикроскопия от обычной микроскопии отличается тем, что
объект (дисперсная система) освещается сбоку, а наблюдают рассеян-
ный свет. Вследствие этого частицы кажутся светящимися точками на
темном фоне, и разрешающая сила микроскопа резко возрастает, что
позволяет наблюдать частицы с диаметром до 2—3 нм.
Нефелометрия — метод исследования, при котором измеряют интен-
сивность рассеянного света, падающего на кювету с дисперсной систе-
мой. Обычно объемная концентрация с дисперсной фазы известна или
легко определяется. Поэтому соотношение (IV. 1) при данной длине
волны удобно записать в виде (0 — const)
/р = I0kw2 — Iokcv (IV. 3)
где k — константа; с — w — объемная концентрация дисперсной фазы.
Из уравнения (IV. 3) следует, что, зная концентрацию или раз-
мер частиц в стандартной системе, можно рассчитать соответственно
размер частиц или их концентрацию в исследуемой дисперсной
системе.
Турбидиметрия основана на измерении интенсивности проходящего
через дисперсную систему света. Рассеянный свет можно считать фик-
тивно поглощенным, и поэтому есть все основания принять, что зако-
номерности рассеяния света подчиняются уравнению Бугера — Лам-
берта—Бера:
In /0//п = 2,3D = т1 (IV. 4)
где Ifr— интенсивность света, прошедшего через систему; D = 1g /0//„ — оптическая
плотность; г — мутность системы; I — толщина слоя системы.
Мутность т = /р//(ь а отсюда и оптическая плотность D в соответ-
ствии с уравнением (IV. 1) пропорциональны концентрации и квадрату
объема частиц. Это позволяет определять размеры частиц и их концен-
трацию по оптической плотности системы методом сравнения со стан-
дартными системами с помощью фотоэлектроколориметра.
Ы2
Работа 17. ОПРЕДЕЛЕНИЕ РАЗМЕРОВ ЧАСТИЦ ДИСПЕРСНЫХ СИСТЕМ
ТУРБИДИМЕТРИЧЕСКИМ МЕТОДОМ
Цель работы: экспериментальное определение размеров частиц высоко-
дисперсных систем.
Уравнение Рэлея может быть использовано для определения раз-
меров частиц сферической формы, если их радиус г не превышает
1/20 длины волны X падающего света. При 0 = 90° согласно уравне-
нию (IV. 1) радиус таких частиц равен
Необходимым условием использования уравнений (IV. 1) и (IV. 5)'
является отсутствие поглощения света, а также вторичного светорас-
сеяния. Поэтому уравнение Рэлея применимо только для так называе-
мых «белых золей», т. е. не поглощающих свет дисперсных систем, и
при очень малых концентрациях дисперсной фазы.
С увеличением размеров частиц закон Рэлея перестает соблюдаться
и интенсивность рассеянного света становится обратнс^ропорциональ-
ной длине волны в степени, меньшей чем четвертая. В этом случае поль-
зуются либо уравнениями, вытекающими из общей теории светорассея-
ния, либо эмпирическими соотношениями. В_ частности, если размер
(диаметр) частиц составляет от 1/10 до 1/3 длины световой волны и
показатели преломления частиц и среды не сильно различаются, для
описания светорассеяния в системе можно воспользоваться следую-
щими эмпирическими уравнениями, предложенными Геллером:
£> = /гХ~га и x = k'‘k~n (IV. 6)
где k и k' — константы, не зависящие от длины волны.
Зависимости lg D (или 1g т) от 1g X в соответствии с уравнениями
(IV. 6) представляют собой прямую линию,_ тангенс угла наклона ко-
торой равен показателю степени п с минусом. Значение показателя
степени п в этих уравнениях зависит от соотношения между размером
частицы и длиной волны падающего света, характеризуемого пара-
метром Z:
Z = 8лг/Х (IV. 7)
С увеличением Z значение п уменьшается, стремясь в пределе к 2
для частиц, радиус -которых больше длины волны. При малых значе-
ниях Z соблюдается уравнение Рэлея и п — 4. Значения п для Z от 2
до 8 приведены в табл. IV. 1.
Показатель степени п в уравнении (IV. 6) находят на основе тур-
бидиметрических данных. Для этого экспериментально измеряют опти-
ческую плотность системы при различных длинах волн (в достаточно
узком интервале X) и строят график в координатах lg D — lg X. Пока-
затель п определяют по тангенсу угла наклона полученной прямой.
По значению п находят соответствующее значение параметра Z (см.
табл. IV. 1), а затем но формуле (IV. 7) рассчитывают средний радиус
частиц исследуемой дисперсной системы.
Следует отметить, что этот метод, как и уравнение Рэлея, применим
только для «белых» золей, т. е. для неокрашенных дисперсных систем
(метод базируется только на светорассеянии),
8 Зак. 673 14-3
Таблица IV. 1. Показатель степени п в уравнении Геллера
в зависимости от параметра Z
п Z п Z
3,812 2,0 2,807 5,5
3,686 2,5 2,657 6,0
3,573 3,0 2,533 6,5
3,436 3,5 2,457 7,0
3,284 4,0 2,379 7,5
3,121 4,5 2,329 8,0
3,060 5,0 — —•
Оптическую плотность золя определяют с помощью прибора
ФЭК-56М, оптическая схема которого приведена на рис. 34.
Световой луч от источника света 5, пройдя через светофильтр 6.
попадает на призму 7, которая делит световой луч на два потока. От-
разившись от зеркал 4 и 8, параллельные световые лучи проходят че-
рез кюветы 3 и 9, падают на зеркала 1 и 11 и затем поступают на фо
тоэлементы 12. По ходу правого светового луча можно устанавливать
последовательно две кюветы (одна с золем, другая с дисперсионно?
средой).
Раздвижная диафрагма 10, расположенная по ходу правого луча
света, при вращении связанного с ней барабана меняют свою площадь
и тем самым меняет интенсивность светового потока, падающего на
правый фотоэлемент. Раздвижная диафрагма 2, расположенная по ход)
левого луча, служит для ослабления интенсивности светового потока.
Рис. 34. Оптическая схема прибора ФЭК-56М:
1,4,8,11— отклоняющие- зеркала; 2.10— раздвижные диафрагмы; 3,9— кюветы: 5 — источник света- 6-
светофильтр; 7—призма; 12--фотоэлементы.
114
падающего на левый фотоэлемент. При равенстве интенсивностей обоих
световых потоков стрелка регистрирующего микроамперметра нахо-
дится в нулевом положении.
Правый световой лун является измерительным, левый — компенса-
ционным.
Порядок работы на приборе ФЭК-56М следующий. Включают фо-
тоэлектроколориметр и «прогревают» его в течение ~30 мин. Устанав-
ливают электрический нуль прибора, для чего рукояткой на верхней
панели прибора световые лучи перекрывают шторкой (рукоятка в пра-
вом положении) и рукоятками «нуль» на левой панели устанавливают
стрелку микроамперметра на «О».
На пути левого светового луча устанавливают кювету, заполненную
дисперсионной средой. В правый кюветодержатель помещают две кю-
веты: одну с дисперсионной средой, другую — с исследуемой системой
(золем) и вращением рукоятки на правой панели прибора на пути пра-
вого светового луча устанавливают кювету с золем. Индексы правого
и левого барабанов устанавливают на «О» по шкале оптической плот-
ности (нанесена красными цифрами). Затем шторку, перекрывающую
световые лучи, переводят в положение «открыто». Вследствие поглоще-
ния или рассеяния света исследуемой системой (в данном случае —
рассеяния) на правый фотоэлемент будет падать световой поток мень-
шей интенсивности, чем на левый фотоэлемент, и стрелка микроампер-
метра будет отклоняться от нулевого положения. Вращая барабан ле-
вой раздвижной диафрагмы, стрелку микроамперметра возвращают на
«О» (уравнивают интенсивности обоих световых потоков). Затем пово-
ротом рукоятки на правой панели прибора по ходу правого луча уста-
навливают кювету с дисперсионной средой. При этом стрелка микро-
амперметра, установленная на «О», смещается, так как фотометриче-
ское равновесие снова нарушается (дисперсионная среда прозрачнее, ,/
и интенсивность светового потока, падающего на правый фотоэлемент,
увеличивается). Вращением правого барабана добиваются первона-
чального нулевого положения стрелки и отсчитывают по шкале пра-
вого барабана значение оптической плотности исследуемой системы,
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Фотоэлектроколориметр ФЭК-56М.
Мерные колбы емкостью 100 мл.
Пипетки емкостью 1 и 10 мл.
Высокодисперсный золь, например полистирольный латекс.
Водный 1 %-ный раствор аммиака.
Вариант 1. Определение размеров частиц дисперсных систем,
подчиняющихся уравнению Рэлея
Из исследуемого золя (латекса) с известным содержанием дисперсной
фазы готовят четыре пробы с кратностью разбавления 1 :2000, 1 :3000,
1 :5000, 1 : 10 000. Вначале готовят пробу латекса с кратностью разбав-
ления 1 : 10Q. Для этого пипеткой вносят 1 мл исходного латекса в мер-
ную колбу, содержащую около 10 мл 1 %-ного водного раствора ам-
миака и тем же раствором доводят объем жидкости в колбе до 100 мл.
8* 115
Таблица IV. 2. Экспериментальные данные для расчета размеров частиц
по уравнению Рэлея
Объемная концентрация латекса с, см3/см3 Оптическая плотность D °ср Мутность — 1 Т, СМ т/с
Из полученного латекса аналогичным способом готовят остальные
пробы.
Измеряют оптическую плотность латексов, при этом используют
кюветы длиной 5 см и светофильтр № 6 (Хвак = 540 нм). Для каждого
образца латекса измерения проводят три раза и определяют среднее
значение D. По формуле (IV. 4) рассчитывают мутность т. Полученные
значения записывают в таблицу (см. табл. IV. 2).
В эту же таблицу записывают значения объемной с концентрации
латекса, которую рассчитывают на основе данных о плотности частиц
полимера (плотность частиц полистирольного латекса составляет
1,06 г/см3).
Строят график в координатах т/с — с. По этому графику экстрапо-
ляцией находят величину т/с при с — 0. Полученные значения т/с при
с = 0 используют для расчета радиуса г частицы латекса по формуле
(IV. 5). При расчете функции F принимают, что показатель преломле-
ния дисперсионной среды (воды) п0 = 1,333, а частиц полистирола tix —
— 1,593. Длину волны в данной среде находят как Х = ХВаК/по-
Вариант 2. Определение размеров частиц дисперсных систем,
не подчиняющихся уравнению Рэлея
Сначала измеряют оптическую плотность золя (латекса) с помощью
фотоэлектроколориметра, используя светофильтр № 3. Значение опти-
ческой плотности латекса должно находиться в пределах 0,70—0,95.
Если значение D образца меньше или больше указанных, следует со-
ответственно увеличить или уменьшить концентрацию дисперсной фазы
в латексе. Затем определяют оптическую плотность образца латекса
при различных длинах волн падающего света (светофильтры № 3—9).
При каждой длине волны оптическую плотность измеряют три раза и
определяют среднее значение D. Значения длин волн, соответствующие
светофильтрам прибора ФЭК-56М, составляют:
Номер светофиль-
тра ............. 3 4 5 6 7 8 9
%вак, нм....... 400 440 483 540 582 620 625
Далее находят значения lgXBaK и lg£>cp, строят график в координа-
тах IgDcp — 1g Хвак и определяют показатель степени п в уравнении
(IV. 6). По данным табл. (IV. 1) (предварительно строят график в ко-
ординатах Z— п) находят значение параметра Z, соответствующее ра-
нее определенному п. Полученные результаты записывают в таблицу
(см. табл. IV. 3).
По уравнению (IV. 7) рассчитывают радиус частиц латекса. Сле-
дует учесть, что при расчете г в уравнение (IV. 7) нужно подставить
116
Таблица TV. 3. Экспериментальные данные для расчета размеров частиц
дисперсных систем, не подчиняющихся уравнению Рэлея
Номер свето- фильтра ?'вак’ нм 1g к D ^Dcp Л , Z
среднее значение длин волн %Ср в том интервале, в котором опреде-
лялся показатель степени п. Величину Хср находят по соотношению
^вак. макс 4* ^вак. мин
,ср _ _
(IV. 8)
Найденное значение г соответствует среднему радиусу частиц ла-
текса.
Работа 18. ДИСПЕРСИОННЫЙ АНАЛИЗ МИКРОГЕТЕРОГЕННЫХ СИСТЕМ
МЕТОДОМ ОПТИЧЕСКОЙ МИКРОСКОПИИ
Цель работы-, определение основных гранулометрических характери-
стик микрогетерогенной системы (среднего размера частиц, интеграль-
ной и дифференциальной кривой распределения частиц по размерам)
с помощью микроскопа.
Изучение гранулометрического состава дисперсной системы с по-
мощью оптического микроскопа заключается в визуальном определении
размеров, числа и формы частиц непосредственно в поле зрения ми-
кроскопа или по микрофотографиям.
При дисперсионном анализе, выполняемом с помощью микроскопа,
чаще всего определяют линейный размер частицы, причем однозначно
линейный размер можно измерить только у частиц правильной формы,
например, сферических частиц эмульсии. Частицы порошка, суспензии
обычно не имеют правильной геометрической формы, и их характери-
зуют эквивалентным размером. Как правило, в качестве эквивалент-
ного размера используют диаметр или радиус сферической частицы,
эквивалентной данной частице по какому-либо свойству (объему, по-
верхности, скорости оседания и др.).
Эквивалентный размер можно определить по результатам измере-
ния размеров частицы по разным направлениям (длина, ширина, вы-
сота), с учетом ее формы. Однако определение даже двух размеров
всех частиц систем по их проекции на плоскость — очень трудоемкая
задача. Поэтому чаще всего методом микроскопии определяют стати-
стический диаметр (или радиус), характеризующий один линейный раз-
мер частицы.
Есть несколько видов статистического диаметра, например, длина
проекции частицы на произвольно выбранное направление (диаметр
Фере), длины различным образом заданных хорд проекции частицы
на плоскость вдоль заданного направления. Статистическими диамет-
рами можно охарактеризовать систему только при исследовании боль-
шого числа частиц, случайным образом ориентированных в препарате.
Реальные системы, являющиеся, как правило, полидисперсными,
117
характеризуют средним диаметром или радиусом, условно заменяя ре-
альную полидисперсную смесь системой частиц правильной формы и
одинакового размера. Возможны различные способы усреднения в за-
висимости от того, какие параметры полидисперсной и заменяющей
ее монодисперсной системы предполагаются одинаковыми. Наиболее
часто используют следующие виды усреднения (запишем их для сред-
него радиуса частиц):
среднечисленный радиус (одинаково число частиц)
~rn='Lniril'£lni (IV-9)
где я,- — число частиц с радиусом г.', п. — суммарное число частиц в системе,
среднеповерхностный (одинакова суммарная поверхность)
Е V?/E (IV-10)
среднемассовый или среднеобъемный (одинаковы общая масса или
объем частиц)
Эти три средних радиуса равны друг другу для монодисперсной систе-
мы; для полидисперсной системы они различны: гт > rs > гп.
Отношение
— г /г/г т (IV. 12)
характеризует полидисперсность системы и называется коэффициентом
полидисперсности.
Для монодисперсных систем k = 1, для полидисперсных систем
k < 1, причем чем меньше значение k, тем больше разброс частиц по
размерам.
Для удобства обработки результатов дисперсионного анализа час-
тицы системы группируют по фракциям, которые соответствуют опре-
деленным интервалам размеров. При расчете по формулам (IV. 9) —
(IV. И) число частиц щ в интервале радиусов Аг; (от г, до п + Ап)
относят к среднему для этой фракции радиусу п, ср = п+ Аг,/2 (в даль-
нейшем изложении под п подразумевается г,. ср).
Результаты микроскопического дисперсионного анализа чаще всего
оформляют графически в виде гистограмм, интегральных и дифферен-
циальных кривых распределения. При построении гистограммы по оси
ординат откладывают значения содержания частиц в принятых интер-
валах радиусов (в % от общего числа учтенных частиц):
а по оси абсцисс — радиус частиц г.
Такая гистограмма дает наглядное представление о распределении
частиц по размерам при условии, что интервалы радиусов во фракциях
одинаковы (Ari = Аг2 = Агз ==..,). Чаще по оси ординат откладывают
плотность распределения F = \Qn/\r (рис. 35), которая не зависит от
величины интервалов Аг. Кривая, проведенная через точки, соответ-
ствующие серединам интервалов гистограммы Аг, построенной в коор-
динатах AQ„/Ar — г с учетом разброса этих точек, является дифферен-
циальной кривой распределения частиц по размерам. ,
на
Рис. 35. Гистограмма дисперсной
системы и дифференциальная кри-
вая численного распределения ча-
стиц по размерам.
При построении инте-
гральной кривой распределе-
ния частиц по размерам для
каждой фракции опреде-
ляют величину Qnl = У, AQra/
i
(где AQn( — число частиц
во фракциях с радиусами
г/^г,). Значения Qn (в %
от общего числа учтенных частиц) откладывают по оси ординат, зна-
чения радиусов — по оси абсцисс. Такая интегральная кривая характе-
ризует численное распределение частиц по размерам Qn = f{r).
По данным микроскопического анализа могут быть построены так-
же интегральные кривые массового (объемного) и поверхностного рас-
пределения частиц по размерам, соответственно Qm — f(r) и Qs = f(r).
Наиболее просто это сделать для систем с частицами правильной фор-
мы, знание радиуса которых позволяет рассчитать массу, объем и по-
верхность частиц. Для построения кривых распределения Qm — f(r) и
Qs — f(r) рассчитывают AQm« и AQSi:
ДО . =
(IV. 14)
AQsi
(IV. 15)
Отношения AQmt/Ап и AQSi/Ап используют для построения диффе-
ренциальных кривых массового и поверхностного распределения частиц
по размерам.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Оптический микроскоп с микрометрической сеткой.
Предметные и покровные стекла.
Шпатель, ступка с пестиком, измерительный цилиндр.
Исследуемый порошок, например, кварц, корунд, сажа, и дисперси-
онная среда, например парафиновое масло, глицерин, дистиллирован-
ная вода.
Приготовление препаратов. Для микроскопического ана-
лиза используют эмульсию (приготовление см. в работе 28) или суспен-
зию порошка.
При использовании масел или глицерина в качестве дисперсионной
среды для приготовления суспензии небольшое количество порошка
(на кончике шпателя) вносят в каплю жидкости, помещенную на пред-
метное стекло и смесь тщательно перетирают, затем накрывают покров-
ным стеклом. Стекла должны быть предварительно тщательно вымыты
хромовой смесью.
119
носят на предметное стекло и
Рис. 36. Препарат эмульсии в поле
зрения окуляра микроскопа с микро-
метрической сеткой.
Если в качестве дисперсион-
ной среды используют воду, то
навеску порошка перетирают
в ступке с небольшим коли-
чеством воды, после чего
смесь переносят в цилиндр и
разбавляют водой из расчета
получения ~С),5 %-ной (масс.)
суспензии. Для предотвраще-
ния агрегации частиц к сус-
пензии добавляют несколько
капель жидкого стекла или
раствора дифосфата натрия
Na4P2O7. Одну-две капли
эмульсии или суспензии на-
накрывают покровным стеклом.
При анализе высокодисперсных частиц в водной среде следует увеличить вязкость
дисперсионной среды, для чего одну-две капли приготовленной водной эмульсии
(суспензии) вносят при перемешивании в 1 мл нагретого раствора желатины (3 г же-
латины на 1 л воды).
Методика микроскопического анализа. Для анализа
используют оптический микроскоп любого типа, предназначенный для
лабораторных исследований, в окуляр которого вставляют микрометри-
ческую сетку. Шкала микрометрической сетки разбивает поле зрения
микроскопа на 100 квадратов, сторона каждого из которых составляет
5 делений сетки (рис. 36). Цена деления сетки зависит от кратности
увеличения окуляра и объектива и определяется с помощью окуляр-
микрометра. Для некоторых сочетаний окуляров и объективов цена де-
ления микрометрической сетки приведена в табл. IV. 4.
Предметное стекло с образцом эмульсии или порошка помещают
под объектив микроскопа и, перемещая тубус микроскопа по верти-
кали, добиваются наилучшей резкости изображения частиц.
Чтобы получить наилучшее для подсчета частиц увеличение, рас-
сматривают образец с разными окулярами и выбирают такой, чтобы
в пределах микрометрической сетки находилось не более 30—40 и не
менее 15 частиц.
Таблица I V. 4. Цена деления шкалы микрометрической сетки
в зависимости от увеличения объектива и окуляра
Увеличение окуляра Цена деления шкалы, мкм, при увеличении объектива
Х8 | Х4О
X 7 21,7 4,3
X И) 17,0 3,3
X 15 14,0 3,0
120
Для суспензий в качестве статистического диаметра частиц прини-
мают наибольший размер проекции частицы на плоскость наблюдения
вдоль одной из сторон микрометрической сетки. В одну фракцию вклю-
чают все частицы, диаметр которых содержит одно и то же целое число
делений сетки. Если число малых частиц в пределах микрометриче-
ской сетки значительно превышает число крупных частиц, целесооб-
разно мелкие частицы считать не по всей сетке, а только в какой-то
ее части, пересчитывая затем содержание таких частиц на всю сетку.
Например, частицы диаметром <1 деления (первая фракция) считают
в 16 центральных квадратах, 1—2 и 2—3 деления (вторая и третья
фракции) — в 36 центральных квадратах. Для определения числа час-
тиц, приходящихся на всю сетку, число частиц первой фракции умно-
жают на 100/16, а второй и третьей — на 100/36.
Для получения достоверных результатов подсчет частиц по фрак-
циям следует проводить не менее 6 раз в разных местах данного пре-
парата.
Выбор нового поля зрения осуществляют произвольным перемеще-
нием образца под микроскопом с помощью микрометрических винтов
предметного столика. Результаты подсчета частиц записывают в таб-
лицу (см. табл. IV. 5).
Для каждой фракции по данным табл. IV. 5 рассчитывают средний ра-
диус частиц по формуле
* 2 \ 2 /
где х — цена деления микрометрической сетки; т — целое число делений, укладываю-
щееся в диаметре частиц данной фракции.
Дальнейшую обработку данных проводят в соответствии с табл. IV. 6.
По данным этой таблицы по уравнениям (IV.9) — (IV. 12) рассчи-
тывают средние радиусы частиц г«, Д, гт и коэффициент полидисперс-
ности системы k.
Затем рассчитывают интегральные Qra = f(r), Qm = f(r) и диффе-
ренциальные \Qn/Xr — f(r), AQm/Ar — f(r) кривые распределения час-
тиц по числу и по массе; исходные данные записывают в таблицы
(см. табл. IV. 7 и табл. IV. 8). При расчетах принимают, что интервал
радиусов для каждой фракции одинаков и составляет половину цены
деления сетки (Аг — х/2).
Таблица IV. 5. Результаты подсчета частиц (капель) микроскопическим анализом
Номер фракции Подсчитанное число частиц (капель)
поле 1 поле 2 поле 3 поле 4 поле 5 поле 6 всего ni
1 (d < 1 дел.) 2 (1 < d < 2 дел.) 3 (2 < d < 3 дел.) 4 (3 < d < 4 дел.)
121
Таблица IV. 6. Результаты дисперсионного анализа
Средний радиус частиц фракции г-,мкм Число частиц фракции ni nirl n.r2 I I n .r3. 1 I n.r* I i
Гг гз
Xnir* X,nir“i
Таблица IV. 7. Исходные данные для построения кривых
численного распределения частиц по размерам
Г-, MKM = 100- % ^nl Содержание частиц с r>ri Д0„/ЛЛ %/мкм
rl Д(4. ДР„! + д<2„2 + Д(4з + +Д^ Д(?П1/ДГ
Г2 Д(42 Д(2п2 + Д(4з+ + ДРК2/Д^
r3 Д(4з ДР„з + ... +AQ,t/e AQrt3/Ar
rk Д(^ *Qnk/^
100
Таблица IV. 8. Исходные данные для построения кривых
массового распределения частиц по размерам
Г-, мкм n’rk ЛОт“ЕМ'10°'% Относительная масса частиц с r^ri Qm, % AQm^r, %/mkm
Г\ дет1 AQ , 4- AQ <> 4- AQ q 4~ ... 4“ AQ b 1 ^m2 1 ^-шЗ ^-mk дРт1/дг
о . 4~ AQ q “4- ... “4- AQ . 1 1 1 ^-tnk
гз Д(Зт3 о 4~ • • 4' AQ m3 1 ‘ mk ^m3/^r
rk ^Qmk AQ ^mk
100
122
По результатам проведенных расчетов строят интегральные и диф-
ференциальные кривые распределения частиц по размерам.
Работа 19. ОПРЕДЕЛЕНИЕ РАЗМЕРОВ ЧАСТИЦ
ВЫСОКОДИСПЕРСНЫХ СИСТЕМ МЕТОДОМ ПРОСВЕЧИВАЮЩЕЙ
ЭЛЕТРОННОЙ МИКРОСКОПИИ
Цель работы: получение электронных микрофотографий частиц дис-
персных систем и определение по ним размеров и гранулометрического
состава.
Принцип действия и устройство электронного мик-
роскопа. Принцип электронно-микроскопического метода заключа-
ется во взаимодействии узкого электронного пучка с достаточно тон-
ким объектом, слабо поглощающим электроны. Длина волны де Бройля
для электронов, разогнанных до высоких скоростей в вакууме, состав-
ляет ~ 0,005 нм, что значительно меньше межатомных расстояний в
конденсированном веществе. Поэтому основными явлениями, возникаю-
щими при взаимодействии электронного пучка с веществом, являются
рассеяние и интерференция.
Проходя через объект, электроны сталкиваются с ядрами атомов,
в результате чего часть из них рассеивается под определенным углом,
причем число рассеянных электронов (и угол рассеяния) определяется
числом столкновений, которое в свою очередь зависит от плотности
объекта, его толщины и скорости электронов. Формирование контраст-
ного изображения объекта на флюоресцентном экране микроскопа свя-
зано с разной степенью рассеяния электронов различными участками
объекта. Пучок электронов, прошедший через наиболее толстую часть
объекта и имеющий наибольший угол рассеяния, доходит до флюорес-
центного экрана значительно ослабленным, в результате интенсивность
свечения соответствующего участка экрана мала. При прохождении
через более тонкие участки объекта электронный пучок рассеивается
меньше и вызывает в соответствующих местах более интенсивное све-
чение экрана. Так упрощенно можно представить формирование кон-
трастного изображения объекта на экране электронного микроскопа.
Электронные микроскопы состоят из трех основных систем.
1. Электронно-оптическая система (колонна микроскопа) служит
для формирования электронного, а затем и светового изображения
исследуемого объекта. 2. Вакуумная система служит для создания раз-
режения в колонне микроскопа (10-2—10-3 Па), чтобы обеспечить боль-
шой (~ 1,5 м) свободный пробег электронов. 3. Система электрического
питания предназначена для снабжения различных узлов микроскопа
постоянным электрическим током и высоким напряжением (50—•
150 кВ), необходимым для ускорения потока электронов.
Колонна микроскопа, являющаяся основной частью прибора, вклю-
чает следующие узлы.
1) Осветительная система предназначена для получения электро-
нов и формирования электронного пучка. Она состоит из электронной
пушки, в которой нагретая до высокой температуры вольфрамовая нить
испускает электроны, ускоряемые электрическим полем, и конденсорной
линзы (электромагнитного или электростатического типа), которая с
помощью магнитного или электрического поля фокусирует электрон-
ный пучок на исследуемый образец,
123
2) В камере объектов размещаются исследуемые образцы, которые
вводят через специальное шлюзовое устройство и устанавливают на
специальный столик, способный перемещаться в горизонтальной плоско-
сти, что позволяет рассматривать их различные участки.
3) Пройдя через образец, электронный пучок попадает в фокуси-
рующую систему, служащую для увеличения изображения.
4) В фотографической камере сформированное электронно-микро-
скопическое изображение исследуемого объекта проецируется на флюо-
ресцентный экран и при необходимости фиксируется на фотопленке
или фотопластинке. Изображение на экране можно наблюдать через
специальные окна камеры. Кассеты с фотопластинками заменяют
через вакуумный шлюз, не нарушая герметичности колонны микро-
скопа.
Приготовление препаратов. Наиболее важным этапом в
электронно-микроскопическом анализе является приготовление образца
для исследования, поскольку от него во многом зависит качество и
информативность полученных изображений. Методика приготовления
образцов определяется видом объекта и методом исследования.
При прямом методе исследования в микроскопе наблюдают непо-
средственно изучаемый объект, например высокодисперсные частицы
или ультратонкие срезы материалов. При косвенном методе в микро-
скопе исследуют отпечаток (реплику), полученный с объекта, напри-
мер, с поверхности композиционных материалов или замороженной
жидкости. Наиболее часто прямым методом исследуют свободнодис-
персные коллоидные системы типа твердое тело — жидкость (золи, сус-
пензии) и твердое тело — газ (порошки, аэрозоли), поскольку твердые
частицы не меняют своей формы и размеров при высушивании и ваку-
умировании. Образцы наносят на пленку-подложку, прозрачную для
электронов (толщиной 10—20 нм). Пленка такой толщины имеет не-
большую механическую прочность, поэтому ее предварительно поме-
щают на специальную поддерживающую металлическую сетку (обычно
медную) с размером ячеек не более 0,1 мм. В качестве материалов
пленок-подложек используют полимеры (нитроцеллюлозу, поливинил-
формальдегид), углерод, кварц и др.
Полимерные пленки получают при испарении тонких слоев раство-
ров полимеров, нанесенных на поверхность воды или стекла. Углерод-
ные и кварцевые пленки получают распылением материалов в электри-
ческой дуге в специальных вакуумных установках. Пары углерода и
кварца осаждают на чистые стеклянные пластинки, покрытые слоем
полимера, на поверхность слюды или монокристаллов хлорида натрия.
Затем пленки отделяют от поверхности и переносят на поддерживаю-
щие сетки. Такие пленки в отличие от полимерных устойчивее к дей-
ствию электронного луча и химически инертны. К недостаткам угле-
родных пленок следует отнести их гидрофобность.
При препарировании систем типа твердое тело — жидкость их раз-
бавляют до содержания твердой фазы 0,01—0,05 % жидкостью, в ко-
торой дисперсная фаза нерастворима. Затем каплю образца с помощью
микропипетки наносят на пленку-подложку. Для более равномерного
распределения образца на подложке часто применяют распыление кап-
ли с помощью ультразвука. При высушивании образцов лиофобных
золей может происходить агрегация частиц, поэтому в них предвари-
тельно добавляют стабилизатор, например желатину.
124
Прямым методом электронно-микроскопического анализа изучают
также структуру различных композиционных материалов, полимеров
и т. д., делая с них срезы. Ультратонкие срезы толщиной не более
200 нм получают с помощью специальных приборов — ультрамикрото-
мов. Образец закрепляют на конце обогреваемого стержня, совершаю-
щего колебательные или вращательные движения. При движении об-
разец подходит к ножу микротома, который снимает с него тонкий срез.
За промежуток времени между двумя последовательными колебания-
ми стержень с образцом нагревается и расширяется. Регулируя ско-
рость нагревания, можно получить срезы различной толщины. Обра-
зующиеся срезы падают на поверхность жидкости, откуда их переносят
на сетки с пленкой-подложкой.
Косвенный метод (метод реплик) применяется в основном для ис-
следования структуры поверхностей. Часто эти поверхности получают
искусственно, производя сколы исследуемых систем (полимеров, ком-
позиционных материалов, замороженных золей, эмульсий и растворов
полимеров). Таким образом, исследуя реплику, получают информацию
и о внутреннем строении системы.
По способу получения реплики можно разделить на одноступенча-
тые (негативные) и двухступенчатые (позитивные). При получении од-
ноступенчатой реплики на поверхность исследуемого объекта предва-
рительно наносят тонкую пленку вещества (полимер, углерод и др.),
воспроизводящую рельеф поверхности, а затем ее отделяют либо ме-
ханически, либо растворением образца.
Для приготовления двухступенчатой реплики сначала получают
пластиковый отпечаток, представляющий собой массивный слепок, вос-
производящий структуру исследуемого объекта. На этот первичный
отпечаток наносят тонкую пленку и получают вторичный отпечаток,
который отделяют от первичного обычно путем растворения послед-
него. Полученные реплики переносят на сетки.
В качестве материалов для получения одноступенчатых реплик
используют те же вещества, что и для получения пленок-подложек.
Контрастность изображения в репликах обусловливается различной
толщиной отдельных участков пленки. Наилучшую контрастность обес-
печивают углеродные и кварцевые реплики.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Электронный микроскоп.
Световой микроскоп.
Отсчетный микроскоп (МИР-12).
Предметные сетки с подложками.
Мнкропипетки.
Пинцет.
Чашки Петри.
Синтетический латекс, например, полистирольный.
Для приготовления препарата исходный латекс с известным содер-
жанием дисперсной фазы разбавляют дистиллированной водой, добав-
ляя небольшое количество стабилизатора (в качестве стабилизатора
используют эмульгатор данного латекса). Микропипеткой отбирают
125
небольшое количество приготовленного латекса и наносят на пленку-
подложку каплю размером 1—2 мм. Готовят 3—4 препарата. Образцы
сушат в закрытой чашке Петри при комнатной температуре.
Приготовленные препараты предварительно ' просматривают в све-
товом микроскопе при увеличении 30—100 раз и выбирают сетки с наи-
более чистой, ровной и нерастрескавшейся пленкой. Затем препараты
исследуют с помощью электронного микроскопа. (Работу на электрон-
ном микроскопе проводят под руководством преподавателя.)
Сетку с препаратом вводят через шлюзовое устройство в камеру
объектов колонны микроскопа. Сначала пленку-подложку освещакхг
электронным лучом небольшой интенсивности (тренировка). Происхо-
дящая при этом карбонизация поверхности полимерной пленки-под-
ложки значительно повышает ее устойчивость к действию электронного
пучка большей интенсивности. Затем увеличивают интенсивность пучка
и просматривают весь препарат при небольшом увеличении (5000—
10 000 раз), выбирая участок, наиболее подходящий для съемки. После
этого устанавливают необходимое рабочее увеличение, наводят на рез-
кость и фотографируют. Данную операцию повторяют 2—4 раза, ис-
следуя разные участки пленки. При этом общее число отснятых частиц
должно быть не менее тысячи. (Операции проявления фотопла-
стинок и получения фотоотпечатков проводят под руководством лабо-
ранта.)
Для фотографий определяют общий коэффициент увеличения, кото-
рый равен произведению увеличений микроскопа и фотоувеличителя.
Затем с помощью отсчетного микроскопа измеряют диаметры 700—
800 частиц, разбивают их на фракции по интервалам размеров и опре-
деляют долю частиц каждой фракции -по отношению к общему числу
частиц (методику микроскопического анализа см. в работе 18). Строят
гистограмму распределения частиц по размерам и по формулам (IV. 9),
(IV. 11), (IV.12) вычисляют среднечисленный гп и среднемассовый гт
радиусы частиц и коэффициент полидисперсности k.
Рассчитывают и строят интегральные Qn — f(r), Qm=f(r) и диф-
ференциальные AQn/Ar = f(г), AQm/Ar = f(г) кривые распределения
частиц по размерам (см. работу 18).
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАЧИ
КОНТРОЛЬНЫЕ ВОПРОСЫ
jj Какие оптические явления наблюдаются при падении луча света
на дисперсную систему? Какие методы исследования дисперсных сис-
тем основаны на этих явлениях?
Какие оптические методы используются для определения разме-
ров частиц дисперрных систем? Укажите границы применимости (по
дисперсности) этих методов.
3. Как рассчитать по данным микроскопического анализа средне-
численный, среднеповерхностный и среднемассовый радиусы частиц?
По какому параметру можно судить о полидисперсности системы?
4. Какую информацию о дисперсной системе дают интегральная и
дифференциальная кривые распределения частиц по размерам?
5. Каковы преимущества и недостатки электронной микроскопии,
применяемой для определения размеров частиц дисперсных систем?
126
Чем обусловлено светорассеяние в дисперсных системах и истин-
ных растворах? Какими параметрами количественно характеризуют
рассеяние света в системе?
Какие золи называют «белыми»? Какова связь между оптической
плотностью и мутностью белых золей? Для каких дисперсных систем
применимо уравнение Рэлея?
Как влияют размеры частиц на зависимость оптической плотно-
сти^<белых» золей от длины волны падающего света?
- С$ Чем различаются методы нефелометрии и турбидиметрии? Какие
уравнения используются для определения характеристик рассеяния
св^ж?
'-’'ЦО/Для каких дисперсных систем применимо уравнение Дебая?
Кадие параметры дисперсных систем определяют по методу Дебая?
-0I.JB чем заключаются особенности метода уЛьтрамикроскопии?
Длй“йаких дисперсных систем применим этот метод? Какие характери-
стики дисперсных систем могут быть определены этим методом?
ПРИМЕРЫ РЕШЕНИЯ ЗАДАЧ
1. Поток света с длиной волны % = 528 нм, проходя через эмульсию СС14 в воде
толщиной слоя I — 5 см, ослабляется в результате светорассеяния в два раза. Рас-
считайте радиус частиц дисперсной фазы, если ее объемное содержание cv = 0,8 %,
показатель преломления ССЦ Я1 = 1,460, воды Яо = 1,333. Свет рассеивается в соот-
ветствии с уравнением Рэлея и ослабляется по закону Бугера — Ламберта — Бера.
Решение. Уравнение Рэлея (IV. 1) для интенсивности света, рассеиваемого едини-
цей объема дисперсной системы во всех направлениях, имеет
24л3 / я? — я^ \2 24л3 / я2 — я2 \2
1 -— _____ I _______ | -VD2 / = ---- I -------- 1 Г
р Я,4 \ п\ + 2я2/ 0 Я,4 \ п2} + 2я2 J v
Интенсивность света при прохождении через белый золь уменьшается в соответ-
ствии с уравнением Бугера — Ламберта — Бера (IV. 7):
следующий вид:
4
•уяг3/0 = т/0
По условию задачи Ц/1а = 2. Тогда
2,30 2,31g 2
г = щГ = 13’85 м~‘
Подставляя полученное значение т в уравнение Рэлея, находим радиус капель
эмульсии:
13,85-(5,28-ДО 7)4
32-3,144-8- 10~3
С 1,4602 + 2-1,3332 \2
V 1,4602 — 1,3332 7
= 2,23- 10-8
м = 22,3 нм
I
ЗАДАЧИ
1. Определите диаметр частиц аэрозоля, используя результат исследо-
вания методом поточной ультрамикроскопии: в объеме 2,2-10~2 мм3
подсчитано 87 частиц аэрозоля (дыма мартеновских печей). Концен-
трация аэрозоля 1 • 10_4 кг/м3, плотность дисперсной фазы 2 г/см3,
форма частиц сферическая.
127
2. Используя уравнение Рэлея, сравните интенсивности света, рас-
сеянного двумя эмульсиями с равными радиусами частиц и концентра-
циями: бензола (ли = 1,501) в воде и н-пентана (/и = 1,357) в воде.
Показатель преломления воды п0 = 1,333.
3. Покажите, в каком случае и во сколько раз интенсивность рас-
сеянного дисперсной системой света больше: при освещении синим све-
том (Xi = 410 нм) или красным светом (7,2 = 630 нм). Светорассеяние
происходит в соответствии с уравнением Рэлея, и интенсивности па-
дающих монохроматических пучков света равны.
4. С помощью нефелометра сравнивают светорассеяние стандарт-
ного и исследуемого гидрозолей мастики равных концентраций. Интен-
сивности рассеянных световых потоков одинаковы при высоте освещен-
ной части исследуемого золя hi — 5 мм и высоте стандартного золя
h2 = 21 мм. Средний радиус частиц стандартного золя 120 нм. Рассчи-
тайте радиус частиц исследуемого золя.
J 5, При изучении ослабления синего света золями мастики получены
следующие данные:
Концентрация золя, % (масс.) 0,60 0,20 0,08 0,04
Толщина слоя, мм . . .<fN. • 2,5 2,5 20 20
Доля прошедшего света, % 3,1 29,4 2,6 15,9
Покажите применимость уравнения Бугера_—Ля-мберта---Бара Для
этой дисперсной системы. Рассчитайте, какая додя света будет рас-
сеяна 0,25 %-ным золем при толщине поглощающего слоя равной
10 мм.
6. Рассмотрите возможность применения уравнения Бугера — Лам-
берта — Бера для гидрозолей гидроксида железа, используя данные по
ослаблению монохроматического света (7, = 500 нм) этими дисперс-
ными системами:
Концентрация золя,
% (масс.) 0,20 0,10 0,08 0,04 0,02
Толщина слоя, мм . . . Доля прошедшего све- 2,5 2,5 2,5 5,0 5,0
та, % 1,7 11,8 18,6 18,2 43,0
Определите, какая доля света будет рассеяна 0,02 %-ным золем,
находящимся в кювете длиной 30 мм.
7. Два пучка монохроматического света равной начальной интен-
сивности с 7,1 = 440 нм (синий свет) и Т,2 = 630 нм (красный свет) про-
ходят через эмульсию бензола в воде. Рассчитайте отношение интен-
сивностей прошедшего света, если толщина слоя эмульсии равна:
а) 1 см; б) 5 см; в) 10 см. Содержание дисперсной фазы 0,10 % (об.),
средний радиус частиц эмульсии 40 нм, показатель преломления бен-
зола и воды соответственно ni = 1,501 и п0 — 1,333. При расчете при-
мите, что ослабление света происходит только в результате светорас-
сеяния и показатели преломления не зависят от длины волны света.
г-&-Свет с длиной волны 540 нм и начальной интенсивностью 10 про-
ходит через слой эмульсии тетралина в воде толщиной: а) 5 сМ;
б) 1-0 см; в) 15 см; г) 20 см. Рассчитайте долю прошедшего света
1п/1о и постройте график зависимости ее от радиуса частиц дисперсной
фазы, изменяющегося в результате коалесценции от 10 до 50 нм. Со-
держание дисперсной фазы 0,05 % (масс.)., показатель преломления
тетралина и воды п\ — 1,540, п0 = 1,333.
128
9. Используя закономерности светорассеяния в соответствии с тео-
рией Рэлея и ослабления светового потока в соответствии с законом
Бугера — Ламберта — Бера, рассчитайте радиус частиц дивинилсти-
рольного латекса (варианты I—IV)
ской плотности D в кювете длиной
“Концентрация латекса, г/л . .
Л, нм...................
D.......................
по результатам измерения оптиче-
5,01 см при длине волны света
I II III IV
0,2 0,5 0,4 0,8
400 440 490 540
0,347 0,402 0,552 0,203
Плотность и показатель преломления дисперсной фазы равны
0,945 г/см3 и 1,653, показатель преломления воды 1,333.
10.' Рассчитайте радиус частиц полистирольного латекса (варианты
I — IV) по зависимости оптической плотности D от длины волны света к:
к, нм I D IV
II III
400 0,562 0,900 0,795 —.
440 0,414 0,704 0,566 —•
490 0,289 0,518 0,382 0,336
540 0,207 0,387 0,267 0,266
582 0,159 0,306 0,202 0,221
630 0,120 0,237 0,150 0,180
При расчете используйте уравнение Геллера.
V. ОБРАЗОВАНИЕ И СВОЙСТВА ЛИОФИЛЬНЫХ
ДИСПЕРСНЫХ СИСТЕМ
Дисперсные системы делят на лиофильные, обладающие термодинами-
ческой агрегативной устойчивостью, и лиофобные, которые термодина-
мически неустойчивы к агрегации, но могут быть устойчивы кинети-
чески.
Термодинамическая устойчивость лиофильных дисперсных систем
означает, что они равновесны (состояние отвечает минимуму энергии
Гиббса), обратимы и образуются самопроизвольно как из макрофаз,
так и из истинного раствора.
Процесс образования лиофильных дисперсных систем аналогичен
процессу растворения и может быть представлен термодинамическим
соотношением:
AG = А/7 — Г AS (V. 1)
Самопроизвольному образованию лиофильной дисперсной системы от-
вечает условие
AG < 0 или А/7 — Г AS < 0
Поскольку образуется гетерогенная система, поверхностная энер-
гия должна быть скомпенсирована энтропийной составляющей, т. е.
частицы дисперсной системы должны участвовать в молекулярно-кине-
тическом (тепловом) движении. Отсюда следует, что лиофильные си-
стемы могут быть только ультрамикрогетерогенными, а поверхностное
натяжение на границе частица — среда должно быть очень малым. Зна-
чение поверхностного натяжения, при котором обеспечивается термо-
динамическая устойчивость дисперсных систем, определяется соотноше-
9 Зак. 673 1 29
нием Ребиндера—-Щукина:
ПМакс< ykTla7 (V.2)
где у — безразмерный коэффициент; k — константа Больцмана; а — средний размер
частиц.
Расчеты показывают, что межфазное поверхностное натяжение
в лиофильных дисперсных системах в зависимости от размера частиц
может иметь значения в пределах от 1,4- 1О4 до 1,4 мДж/м2.
Опыт показывает, что такие малые межфазные поверхностные на-
тяжения на границе жидкость — жидкость в широком температурном
и концентрационном интервале характерны при резко выраженной лио-
фильности молекул компонента, образующего дисперсную фазу, т. е.
одна часть молекулы должна быть гидрофильной, другая — олео-
фильной.
Типичными представителями лиофильных дисперсных систем явля-
ются растворы коллоидных ПАВ (ассоциативные коллоиды) и растворы
полимеров (молекулярные коллоиды). В растворах коллоидных ПАВ
мицеллы (частицы) образуются вследствие ассоциации дифильных мо-
лекул. При ассоциации лиофильные части молекул ПАВ (имеющие
большее сродство к растворителю) располагаются на периферии ми-
целлы, внутри ее находятся лиофобные части молекул. Так, в водных
растворах неполярные углеводородные радикалы молекул ПАВ обра-
зуют ядро мицеллы, а полярные группы обращены к воде. В неполяр-
ных средах образуются обратные мицеллы, т. е. внутри мицеллы рас-
полагаются полярные группы.
По способности ПАВ диссоциировать в водных средах, поверхност-
но-активные вещества классифицируют на ионогенные и неионогенные.
Ионогенные ПАВ диссоциируют в водных растворах, и их в свою оче-
редь подразделяют на анионные (углеводородный радикал в составе
аниона), катионные (с органическим катионом) и амфолитные (амфо-
терные). Соответствующее строение имеют двойные электрические
слои мицелл, образованных разными ПАВ. Например, анионные ПАВ
в воде образуют мицеллы с отрицательно заряженным ядром и поло-
жительно заряженными противоионами.
Мицеллообразование в растворах коллоидных ПАВ является наибо-
лее термодинамически выгодным процессом по сравнению с процессами
образования истинного раствора или разделения фаз. Это обусловлено
переходом углеводородной или полярной части дифильных молекул
ПАВ в подобную им по полярности фазу. Например, полярные группы
молекул ПАВ обращаются к воде, поскольку они гидратированы, а
углеводородные радикалы выталкиваются из водной фазы. Оба эти про-
цесса сопровождаются выделением теплоты, что способствует уменьше-
нию энергии Гиббса системы.
Способность ПАВ к образованию мицелл в существенной степени
зависит от длины углеводородного радикала. ПАВ с небольшими угле-
водородными радикалами, например низшие спирты, кислоты и их
соли, находятся в растворе в молекулярно-дисперсном состоянии при
любых концентрациях. Такие ПАВ называют истинно растворимыми.
Их применяют в качестве смачивателей, диспергаторов, вспенивателей.
ПАВ, способные к мицеллообр-азованию в жидких средах, называются
коллоидными. Они имеют большие углеводородные радикалы, от их
размеров зависит значение концентрации, выше которого образуются
130
мицеллы (критическая концентрация мицеллообразования, ККМ). Чем
больше размер углеводородного радикала у ПАВ, тем меньше раство-
римость ПАВ в воде и тем ниже ККМ.
Между мицеллами и молекулярно-растворенными молекулами ПАВ
существует динамическое равновесие
истинный раствор золь
При концентрациях, незначительно превышающих ККМ, мицеллы
имеют сферическую форму. С ростом концентрации мицеллы взаимо-
действуют (или это происходит через стадию молекулярного раство-
ра), образуя более крупные мицеллы цилиндрической, дискообразной,
палочкообразной, пластинчатой формы. Затем могут формироваться
жидкокристаллические структуры.
Коллоидные растворы ПАВ способны к солюбилизации, т. е. в них
резко увеличивается растворимость веществ, плохо растворимых в дан-
ном растворителе. Растворение происходит в результате внедрения мо-
лекул растворяемого вещества внутрь мицелл.
Значение ККМ уменьшается также с уменьшением гидратации (ги-
дрофильности) противоионов. Введение электролитов (индифферент-
ных) снижает ККМ у ионогенных ПАВ и слабо влияет на ККМ неионо-
генных ПАВ. Введение неэлектролитов (органических растворителей)'
при наличии солюбилизации приводит к повышению устойчивости ми-
целл, т. е. к уменьшению ККМ в водных растворах ПАВ. При отсут-
ствии солюбилизации, как правило, ККМ увеличивается за счет уси-
ления растворяющей способности среды.
Имеется много методов определения ККМ. Все они основаны на
резком изменении физико-химических свойств растворов ПАВ при пе-
реходе от молекулярного раствора к мицеллярному. На рис. 37 пока-
заны графики зависимости свойство — состав раствора. Концентрации
ПАВ, при которых наблюдаются перегибы на графиках «свойство —
состав», соответствуют ККМ.
К лиофильным дисперсным цистемам относятся также растворы по-
лимеров. Молекулы полимеров имеют размеры, при которых они про-
являют свойства отдельной фазы (микрофазы). В то же время рас-
творы полимеров имеют свойства истинных растворов, так как моле-
кулы полимеров участвуют в молекулярно-кинетическом движении. Та-
ким образом, растворы полимеров, как
и лиозоли вообще, относятся к системам
переходным между истинными гетеро-
генными системами и истинными раство-
рами.
Взаимодействие полимера с раствори-
телем обычно начинается с набухания,
т. е. с поглощения растворителя полиме-
ром, объем и масса которого при этом
увеличивается. Степень набухания опре-
деляется массой жидкости, поглощенной
Рис. 37. Зависимости мутности т, эквивалентной
электропроводности X, поверхностного натяже-
ния о, показателя преломления п, осмотического
давления л раствора ионогенного ПАВ в воде от
его концентрации.
9
131
единицей массы вещества:
где mi и т2 — масса полимера до и после набухания.
Набухание может быть ограниченным и неограниченным. При огра-
ниченном набухании степень набухания достигает определенного пре-
дельного значения и далее не увеличивается — система переходит в со-
стояние геля (студня). При неограниченном набухании система пере-
ходит в конечном итоге в раствор, т. е. набухание переходит в раство-
рение. На стадии набухания происходит диффузия растворителя в по-
лимер, и только после достаточного увеличения расстояния между
макромолекулами последние способны диффундировать в растворитель
(стадия растворения).
Кинетика набухания описывается обычно следующим уравнением:
ах = ар (1 — е“йх) (V.4)
где ат и ар — соответственно степень набухания ко времени т и после достижения
равновесия; k—константа скорости набухания.
В разбавленных растворах полимеров макромолекулы могут быть
как в расправленном (вытянутом) состоянии (в хороших растворите-
лях), так и свернуты в клубок. При этом ориентирование полярных
и неполярных частей макромолекулы подобно ориентированию при об-
разовании мицелл из ПАВ. В макромолекулах так же, как и в мицел-
лах, может происходить солюбилизация. С повышением концентрации
растворов полимеров макромолекулы ассоциируют, образуя надмоле-
кулярные структуры — ассоциаты.
О свойствах растворов полимеров часто судят по зависимости осмо-
тического давления л от концентрации с. Эту зависимость представ-
ляют в виде вириального ряда степеней концентраций:
л/c^RT (А + А2с + Азс2+ ...) ' (V.5)
где А- — 1/М, Аг, А3— соответственно первый, второй, третий вириальные коэффи-
циенты; М — молекулярная масса полимера.
При небольших концентрациях соблюдается линейная зависимость
л/с от с, т. е. можно ограничиться вторым вириальным коэффициен-
том, который характеризует степень отклонения раствора от идеаль-
ного, обусловленную взаимодействием макромолекул с растворителем.
Работа 20. ИССЛЕДОВАНИЕ МИЦЕЛЛООБРАЗОВАНИЯ
В РАСТВОРАХ ПАВ
Цель работы: определение критической концентрации мицеллообразо-
вания ПАВ различными методами.
В водных растворах коллоидных ПАВ при очень низких концентра-
циях, соответствующих ККМ, образуются сферические мицеллы, кото-
рые содержат от 20 до 100 молекул и характеризуются узким распре-
делением частиц по размерам. При увеличении концентрации ПАВ про-
исходит переход мицеллы из одной формы в другую (цилиндрическую,
дискообразную и т. д.) при соответствующей критической концентра-
ции — ККМ2, ККМ3 и т. д.
432
_В__работе для определения ККМ используется кондуктюметрячв-^
ский~~метод и метод, основанный на измерении поверхностного натя-<
/жёния. ~——— — ' —.
Кондуктометрическое определение ККМ основано на измерении
концентрационной зависимости электропроводности растворов ионоген-
ных~~ТТАВ. В о6ласТй~кбнцентраций до ККМ~зависимостйГУЕёльнои д
эквивалентной электропроводности от концентрации ПАВ. соответствуют
аналогичныАА'ЗВтпп1'ИОстята"'71ЛЯ рДстворов'" средних по силе электроли-
тов. При концентрации, соответствующей ККМ, на графиках зависимо-
стей наблюдается/излом, обусловленный образованием сферических
ионных мицелл. ‘‘Подвижность ионных мицелл меньше подвижности
ионов и, кроме того$ значительная часть противоионовунаходится в
плотном слое Гельмгольца, что существенно уменьшает электропровод-
ность раствора ПАВ. Поэтому при увеличении концентрации ПАВ
больше ККМ эквивалентная электропроводность более резко уменьша-
ется, а возрастание удельной электропроводности значительно ослабля-
ется. По изменению удельной электропроводности х можно также опре-
делить ККМ2 (рис. 38);-
Поверхностное- натяжение водных растворов ПАВ уменьшается с
ростом ^концентрации вплоть до ККМ. Изотерма о = f (1п сПА’в
(рис. 39) в области низких концентраций ПАВ имеет криволинейный
участок, на котором в соответствии с уравнением Гиббса адсорбция
Г на межфазной границе возрастает с ростом концентрации. При опре-
деленной концентрации ст криволинейный участок изотермы переходит
в прямую с постоянным значением do/dlnc, т. е. адсорбция достигает
постоянного и максимального значения. В этой области на межфазной
границе формируется насыщенный мономолекулярный адсорбционный
слой. При дальнейшем увеличении концентрации ПАВ (сплв>ККМ)
в объеме раствора образуются мицеллы и поверхностное^ натяжение
практически не изменяется. ККМ определяется по излому изотермы
при выходе ее на участок, параллельной оси In с.
Измерение поверхностного натяжения позволяет определять ККМ
как ионогенных, так и неионных ПАВ. Исследуемые ПАВ необходимо
Рис. 38. Зависимость удельной электропроводности х раствора ионогенного ПАВ от
его концентрации.
Рис. 39. К определению ККМ по изменению поверхностного натяжения раствора ПАВ.
133
тщательно очищать от примесей, поскольку их присутствие может
явиться причиной появления минимума на изотерме при концентрациях,
близких к ККМ.
ПОРЯДОК. ВЫПОЛНЕНИЯ РАБОТЫ
Вариант 1. Определение ККМ в растворе ПАВ
кондуктометрическим методом
Для проведения работы необходимы:
Прибор для измерения электропроводности водных растворов, на-
пример мост переменного тока Р-577.
Кондуктометрическая ячейка с платиновыми электродами.
Мерные колбы емкостью 100 мл.
Пипетки емкостью 25 мл.
Раствор ионогенного ПАВ, например олеата натрия.
0,02 М раствор КС1 (раствор готовят из дважды перекристаллизо-
ванной соли).
Предварительно определяют константу кондуктометрической ячейки.
В ячейку наливают такой объем раствора КС1 точно известной кон-
центрации, чтобы электроды были полностью погружены в него. Ячей-
ку помещают в термостат, термостатируют 4—5 мин, подключают элек-
троды к клеммам кондуктометра и измеряют сопротивление Ro рас-
твора КС1 между электродами. Константу К рассчитывают по формуле
К —
(V. 6)
где Хо — удельная электропроводность раствора ' КС1 при температуре опыта.
Значения удельной электропроводности 0,01М раствора КС1 при
различных температурах:
t. °C х, См*м t, °C х, См*м
16 0,1173 21 0,1305
17 0,1199 22 0,1332
18 0,1225 23 0,1359
19 0,1251 24 0,1386
20 0,1278 25 0,1413
После измерения электропроводности раствора КС1 ячейку много-
кратно промывают дистиллированной водой.
Из исходного раствора ПАВ путем последовательного разбавления
вдвое готовят 10 растворов.
Для этого в мерную колбу вносят 50 мл исходного раствора ПАВ
известной концентрации и доводят его объем до 100 мл дистиллирован-
ной водой. Из приготовленного раствора отбирают 50 мл и переносят
в другую мерную колбу с последующим доведением объема до метки
дистиллированной водой. Растворы готовят непосредственно перед из-
мерением электропроводности (для предотвращения гидролиза ПАВ).
Измеряют сопротивление каждого раствора по методике, приведенной
выше для раствора КС1. Удельную % и эквивалентную X электропровод-
ности растворов рассчитывают по формулам
и = K/R, X = х/с (V. 7)
где К — константа кондуктометрической ячейки; с — концентрация раствора ПАВ.
131
Результаты записывают в таблицу (см. табл. V. 1)’.
Таблица V. 1. Результаты, кондуктометрических измерений для растворов ПАВ
СПАВ’ экв/л Vе ПАВ R, Ом , к, См-м 1 К, См*м2/экв
Строят графики зависим'остей Л = f (VcnAB ), и = /(сПав)и находят
ККМ исследуемого ПАВ.
Вариант 2. Определение ККМ по изменению
поверхностного натяжения растворов ПАВ
Для проведения работы необходимы:
Прибор для определения поверхностного натяжения.
Мерные колбы емкостью 100 мл.
Пипетки емкостью 25 мл.
Раствор ПАВ, например олеата натрия.
Готовят растворы ПАВ последовательным разбавлением исходного
раствора известной концентрации, как описано в варианте 1 работы.
Для предотвращения гидролиза растворы готовят непосредственно
перед измерением поверхностного натяжения. Рекомендуется исполь-
зовать растворы со значениями pH 10—11.
Поверхностное натяжение измеряют одним из методов, описанных
в работе 1.
По полученным данным строят график зависимости о = /(1пс) и по
излому кривой находят ККМ.
Работа 21. ИССЛЕДОВАНИЕ СОЛЮБИЛИЗИРУЮЩЕЙ СПОСОБНОСТИ
РАСТВОРОВ ПАВ
Цель работы: определение зависимости мольной солюбилизации масло-
растворимого красителя от концентрации ПАВ; определение ККМ ме-
тодом солюбилизации.
Солюбилизация нерастворимых или малорастворимых в воде ве-
ществ, например углеводородов, спиртов, фенолов, красителей, заклю-
чается в растворении их во внутренней части (углеводородных ядрах)
мицелл мыла. В результате солюбилизации вещество равновесно рас-
пределяется между мицеллами и водной фазой. Процесс солюбилиза-
ции в растворах ПАВ включает стадии растворения солюбилизата в
оде, диффузии его молекул к поверхности мицелл и проникновения
нутрь мицелл.
В зависимости от строения молекул ПАВ и солюбилизата возможен различный
характер их включения в мицеллу. Неполярные вещества (углеводороды) растворя-
ются во внутренней углеводородной части мицелл (рис. 40, а). Полярные вещества,
например спирты, располагаются между молекулами ПАВ в мицелле, как бы раздви-
гая их (рис. 40,6). При солюбилизации в растворах неионогенных ПАВ молекулы
солюбилизата могут не проникать внутрь мицелл, а располагаться на их поверхности
среди беспорядочно изогнутых оксиэтиленовых цепей (рис. 40, в).
135
Рис. 40. Солюбилизация в мицеллах ПАВ неполярных углеводородов (а), полярных
веществ (б), углеводородов в мицеллах неиоиогениых ПАВ (и).
Молекулы ионогенного ПАВ (1), неионогенного ПАВ (2), неполярного вещества (3), полярного ве-
щества (4).
Солюбилизация приводит к набуханию мицелл и соответственно
к увеличению их размеров. Процесс солюбилизации является медлен-
ным, равновесие может устанавливаться в течение нескольких суток.
Перемешивание и повышение температуры ускоряет наступление рав-
новесия. С ростом концентрации ПАВ солюбилизация растет, отражая
перестройку мицелл в растворах, причем их насыщение солюбилизатом
может не достигаться.
Контроль за солюбилизацией можно осуществлять различными ме-
тодами. Рефрактометрический метод исследования солюбилизации ос-
нован на измерении показателя преломления растворов ПАВ в зави-
симости от содержания солюбилизируемого вещества в растворе. Пока-
затель преломления увеличивается и достигает постоянного значения
для раствора, насыщенного солюбилизатом. Появление избыточных ко-
личеств углеводорода в водном растворе приводит к образованию
эмульсии, поэтому резко возрастает мутность дисперсной системы. Это
дает возможность контролировать солюбилизацию также турбидимет-
рическим методом.
Солюбилизирующую способность ПАВ часто оценивают с помощью
олеофильных красителей (например, судан III, оранжевый ОТ). Такие
красители, практически нерастворимые в воде, растворяются в гидро-
фобной части мицелл, окрашивая раствор. Интенсивность окраски
раствора тем выше, чем больше количество коллоидно-растворенного
красителя. Содержание солюбилизированного красителя определяют,
измеряя оптическую плотность раствора. По оптической плотности с по-
мощью калибровочного графика определяют количество солюбилизиро-
ванного красителя в единице объема раствора S. Мольную солюбили-
зирующую способность 5м данного раствора ПАВ рассчитывают как
отношение полученного значения S к концентрации ПАВ:
SM = S/c (V. 8)
кде с — концентрация ПАВ в растворе, моль/л.
По концентрационной зависимости оптической плотности солюби-
лизированных растворов ПАВ можно определить ККМ, экстра-
полируя начальный участок кривой до пересечения с осью концен-
трации.
136
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Фотоэлектроколориметр, например ФЭК-56М.
Конические колбы с пробками емкостью 50 мл.
Мерная колба емкостью 50 мл.
Воронки для фильтрования.
Бюретки или градуированные пипетки.
Фильтровальная бумага.
Раствор ПАВ, например 0,2 А! раствор олеата натрия.
Краситель, например судан III, пинацианолхлорид.
В конических колбах готовят 6—8 растворов ПАВ последователь-
ным разбавлением исходного раствора. В каждый приготовленный рас-
твор ПАВ вносят 5—10 мг красителя (количество на кончике скальпе-
ля). Колбы закрывают пробками, смеси перемешивают интенсивным
встряхиванием, выдерживают 40—60 мин и фильтруют через бумаж-
ный фильтр.
С помощью фотоэлектроколориметра измеряют оптическую плот-
ность D каждого фильтрата, начиная с раствора минимальной концен-
трации, при определенном светофильтре (например, для раствора с Су-
даном III используют светофильтр № 6). Методика определения опти-
ческой плотности приведена в работе 17.
По калибровочной кривой [D = f(c) для раствора красителя] на-
ходят мольную солюбилизирующую способность SM растворов ПАВ.
Результаты исследования записывают в таблицу (см. табл. V. 2).
Для построения калибровочной кривой измеряют оптическую плот-
ность растворов красителя (без ПАВ) в бензоле. Растворы готовят
следующим образом. Точную навеску красителя (~10 мг) вносят в
мерную колбу, приливают некоторое количество бензола, перемеши-
вают и доводят объем раствора до метки бензолом. Из исходного рас-
твора последовательным разбавлением готовят 5—6 растворов.
Таблица V. 2. Результаты исследования солюбилизации красителя
в зависимости от концентрации ПАВ
Концентрация раствора ПАВ Оптическая плотность D Dcp SM
г/л моль/л
Строят графики зависимости D и SM от концентрации ПАВ. Опре-
деляют ККМ путем экстраполяции начальных участков зависимостей
на ось концентраций.
Работа 22. ИССЛЕДОВАНИЕ ВЛИЯНИЯ ДЛИНЫ
УГЛЕВОДОРОДНОЙ ЦЕПИ МОЛЕКУЛ ПАВ
НА ТЕРМОДИНАМИЧЕСКИЕ ПАРАМЕТРЫ АДСОРБЦИИ
И МИЦЕЛЛООБРАЗОВАНИЯ В ВОДНЫХ РАСТВОРАХ
Цель работы: установление взаимосвязи между поверхностными и объ-
емными свойствами водных растворов солей карбоновых кислот; опре-
деление зависимости поверхностной активности и ККМ от длины угле-
137
водородного радикала ПАВ; расчет работы адсорбции и мицеллообра-
зования.
Свойства растворов коллоидных ПАВ, в частности, адсорбционная
способность и способность образовывать мицеллы зависят от числа ме-
тиленовых групп в углеводородной цепи ПАВ, природы и содержания
полярных групп. Так, при увеличении длины углеводородного радикала
на одну СН2-группу поверхностная активность ПАВ возрастает в
3,2 раза (правило Траубе).
Поверхностная активность связана с параметрами адсорбции, в
частности с константой Генри Кг-
g = g° ~ £. = = к (V. 9)
с с 1
или
In g = In Дет — In с (V. 10)
где а» и о — поверхностное натяжение соответственно растворителя и раствора ПАВ.
Для растворов коллоидных ПАВ поверхностное натяжение раство-
ров линейно уменьшается в области малых концентраций вплоть до
ККМ (до о = оккм), оставаясь далее постоянным. Поэтому для них
уравнение (V. 9) принимает вид
сто — стккм _ const ... ...
8 ККМ ~ ККМ (V' П)
In ККМ = const — In g (V. 12)
Соотношение (V. 12) указывает на наличие однозначной взаимо-
связи между объемными и поверхностными свойствами растворов кол-
лоидных ПАВ.
При малых концентрациях ПАВ в соответствии с уравнением (II. 7)
изменение адсорбционного потенциала (е = — А6°) при увеличении
длины углеводородного радикала ПАВ на одну метиленовую группу
равно
Де = - (до’ , - ДвА = RT In = %т In (V. 13)
V п+' п) *Г(«) Sn
где п — число метиленовых групп в углеводородном радикале.
Согласно теории Ленгмюра зависимость поверхностной активности от
числа метиленовых групп в углеводородной цепи ПАВ описывается
уравнением
, Iе , Деге ...
lng- = a +— = а + -^ (V. 14)
ИЛИ
lng = a +
МАфя
RT
где а — константа для данного гомологического ряда ПАВ; ср = Де/Уд— вклад од-
ного метиленового звена в изотермическую работу адсорбции ПАВ.
Из уравнения (V. 14) следует, что для водных растворов (е > 0)
с ростом длины углеводородного радикала молекул ПАВ работа ад-
сорбции увеличивается и соответственно повышается их поверхностная
138
активность. Объединяя уравнения (V. 10) и (V. 14), получаем:
ЛДт
1п с = 1п Да — а-п (V. 15)
1\1
При одинаковой депрессии поверхностного натяжения (До = const)
для растворов ПАВ — гомологов уравнение (V. 15) принимает вид
ЛДт
1п сда = “---ну « (V-16)
К1
где а' = In Да — а^= const.
Из уравнения (V. 16) видно, что чем больше п, тем меньше сда, т. е.
чем больше метиленовых групп в углеводородной цепи ПАВ, тем мень-
ше концентрация ПАВ, необходимая для снижения поверхностного на-
тяжения на одну и ту же величину (До).
Образование мицелл в растворах коллоидных ПАВ, как и адсорб-
ция молекул ПАВ в поверхностном слое, протекает самопроизвольно.
При концентрации ПАВ ниже ККМ энергия Гиббса системы уменьша-
ется за счет адсорбции ПАВ на границе раствор — воздух. При этом
углеводородный радикал молекулы ПАВ выталкивается из воды в га-
зовую фазу. Так происходит вплоть до достижения предельной емкости
адсорбционного слоя. При дальнейшем увеличении концентрации ПАВ
в растворе снижение энергии Гиббса системы может достигаться только
за счет структурных изменений в объеме раствора, т. е. путем образо-
вания мицелл в растворе.
Длина углеводородной цепи оказывает решающее влияние на ми-
целлообразование ПАВ в водных средах. Чем длиннее цепь, тем больше
оказывается выигрыш энергии в результате когезии углеводородных
радикалов и, следовательно, меньше необходимая концентрация ПАВ
в растворе для образования мицелл. Критическая концентрация ми-
целлообразования зависит также от сил электростатического отталки-
вания между ионизированными гидрофильными группами, поскольку
сближение этих групп в процессе мицеллообразования требует опреде-
ленной затраты энергии на преодоление сил кулоновского отталки-
вания.
В равновесном мицеллярном растворе химические потенциалы ПАВ
в объеме раствора и в мицелле равны. При наличии двойного элек-
трического слоя у мицеллы ПАВ это условие можно выразить следу-
ющим образом:
ц° + RT In а = ц° + RT In а + Е = jT + RT In а + z/<p0 (V. 17)
где |Г и ц° — химический потенциал ПАВ соответственно в объемной фазе и в ми-
целле при стандартных условиях; а и а — активность ПАВ соответственно в растворе
и в мицеллах; Е — работа заряжения, т. е. работа, необходимая для перемещения
1 моль ионов из объема водного раствора в мицеллу; zi — заряд иона; F — число
Фарадея; <р0 — электрический потенциал на поверхности мицеллы.
Для мицеллярных растворов неионогенных ПАВ Е 0. В раство-
рах ионогенных ПАВ, например анионного, находящегося в растворе
одно-одновалентного электролита, поверхность мицеллы сильно заря-
жена. В этом случае <ро может быть найдено с помощью уравнения Пу-
ассона — Больцмана и соотношения, связывающего объемный и поверх-
ностный заряды. В конечном виде взаимосвязь ф0 с концентрацией про-
139
тивоионов (катионов) в объеме раствора с0< выражается уравнением
_ RT г ( \ , -1
фо F [ П V2ee0J?7-J П М
(V. 18)
где q—поверхностная плотность заряда мицеллы.
Уравнение (V. 18) позволяет рассчитать потенциал <р0, а следова-
тельно и Е, при условии, если все противоионы находятся в диффуз-
ном слое. Поскольку часть противоионов в реальной дисперсной си-
стеме находится в плотном слое (слое Гельмгольца), то для учета
этого при расчете работы заряжения Е следует ввести поправочный
коэффициент р (как правило, [3 < 1):
Е = Pz/Фо (V. 19)
Тогда согласно уравнению (V. 18) имеем:
(V. 20)
Подставляя полученное значение Е в уравнение (V. 17), находим
для мицеллярного раствора ПАВ (а = ККМ; а — 1):
In ККМ = + ₽ In (-2ggo7?7. ) - ₽ In сог
(V.21)
Следовательно, зависимость ККМ от концентрации электролита для
данного ПАВ приближенно можно выразить следующим уравнением:
In ККМ = - ₽ In с0. + К (V. 22)
где К — постоянная для мицеллярного раствора данного ПАВ
В отсутствие электролита cOi = ККМ. Поэтому .
1пКММ = -^-(-т-Ьг') +Г (V.23)
\ 1 + р /
где е' — — и° — работа мицеллообразования, равная работе по переносу 1 моль угле-
водородных радикалов молекул ПАВ из мицеллы в объем раствора; К' — постоянная.
Чтобы рассчитать работу мицеллообразования, необходимо знать
поправочный коэффициент 0. Для этого исследуют влияние противо-
ионов на ККМ.
С учетом зависимости s' от длины углеводородного радикала урав-
нение (V. 23) принимает вид
<EnN д / 1 \
in ККМ =-----(д-qrp-) + к' <v- 24)
где <р' — вклад одной СН2-группы в работу мицеллообразования ПАВ.
Таким образом, для гомологического ряда ПАВ можно записать:
Последнее соотношение указывает на то, что значения работ ад-
сорбции и мицеллообразования близки между собой. Это объясняется
тем, что оба процесса (адсорбция и мицеллообразование) связаны с
переходом углеводородных цепей молекул ПАВ из раствора в близкую
к ним по полярности фазу.
140
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Прибор для измерения поверхностного натяжения.
Конические колбы емкостью 50 и 250 мл.
Мерные колбы емкостью 100 мл.
ПАВ: лаурат натрия СцН23СООЫа, пальмитат натрия Ci5H3iCOONa,
миристат натрия Ci3H27COONa.
0,025М раствор карбоната натрия.
0,25 М раствор хлорида натрия.
Часть 1. Определение поверхностной активности
и работы адсорбции для гомологического ряда ПАВ
Готовят исходные растворы ПАВ (по 100 мл) следующих концентра-
ций: лаурат натрия — 0,05 М; миристат натрия — 0,006ТИ; пальмитат
натрия — 0,002 ЛГ. Для этого навески ПАВ вносят в конические колбы
емкостью 250 мл, наливают по 20 мл 0,025 М раствора Na2CO3 и 80 мл
воды. Смеси нагревают (не доводя до кипения) для растворения ПАВ
и затем охлаждают до комнатной температуры. Переливают в мерные
колбы емкостью 100 мл и доводят объем растворов до метки.
В шесть колб емкостью 50 мл наливают указанные ниже объемы
приготовленных растворов ПАВ и общий объем проб доводят водой
до 20 мл (за исключением пробы в шестой колбе):
Номер колбы ................
Объем исходного раствора, мл
лаурата натрия .............
миристата натрия . . . .
пальмитата натрия . . . .
1 2 3 4 5 6
1 2 5 9 14 20
0,5 1 3 6 12 20
1 2 4 7 12 20
Измеряют поверхностное натяжение всех приготовленных раство-
ров ПАВ (методику определения о см. в работе 1).
По полученным данным строят графики зависимости о = f (In сПАВ)
для каждого гомолога и по ним находят ККМ и о-ккм (см. рис. 39).
По формуле (V. 11) рассчитывают поверхностную активность исследо-
ванных ПАВ. Полученные данные записывают в таблицу (см.
табл. V. 3).
Рассчитывают соотношения между поверхностными активностями
и ККМ в данном гомологическом ряду согласно формуле (V. 25).
Строят график в координатах Ing — п. По тангенсу угла наклона
полученной прямой рассчитывают значения Ае и <р [см. уравнение
(V. 14)] и вычисляют работу адсорбции для каждого ПАВ (е = Аеп).
Таблица V. 3. Результаты исследования коллоидно-химических свойств
растворов ПАВ
ПАВ Число метиленовых груди In ККМ ККМ, моль/л аккм> Дж/м2
Лаурат натрия Миристат натрия Пальмитат натрия
141
Часть 2. Определение работы мицеллообразования
для гомологического ряда ПАВ
Готовят 50 мл исходного раствора лаурата натрия концентрации
4-10-2 моль/л (для растворения ПАВ колбу нагревают до 60—80°С).
В двенадцать колб емкостью 50 мл отбирают указанные ниже объемы
приготовленного раствора ПАВ:
Номер колбы . ...............
Объем исходного раствора
лаурата натрия, мл
I серия ................
II серия.................
1 2 3 4 5 6
0,2 0,5 1,0 1,5 2,5 5,0
0,2 0,5 0,7 1,0 2,0 4,0
Затем в колбы вводят такое количество NaCl, чтобы во всех рас-
творах суммарная концентрация Со< ионов Na+ оставалась постоянной:
для I серии растворов Со; = 0,1 моль/л, для II серии растворов с0» =
= 0,2 моль/л. Объем всех проб доводят водой до 20 мл.
Объем вводимого раствора хлорида натрия рассчитывают по фор-
муле
^NaC!
__ 20 (С0г ~ сПАв)
cNaCI
(V. 26)
где Со; — суммарная концентрация ионов натрия в растворе ПАВ; смас1 — концентрация
исходного раствора NaCl.
Измеряют поверхностное натяжение всех приготовленных растворов
(см. описание работы 1). Строят графики зависимости о = /(1пспдв)
и находят ККМ при различных концентрациях противоионов Na+. Ре-
зультаты записывают в таблицу (см. табл. V. 4). Поскольку в отсут-
ствие посторонних электролитов с0« = ККМ в таблицу включают также
данные определения ККМ, полученные без введения хлорида натрия
(часть 1 работы).
Таблица V. 4. Результаты исследования влияния концентрации противоионов на ККМ
Концентрация Na+, моль/л 1псо£ In ККМ ККМ, моль/л
Без электролита 0,1 0,2
По данным табл. (V. 4) строят график зависимости In ККМ =
= /(lncoi)- По тангенсу угла наклона полученной прямой рассчитывают
поправочный коэффициент р [см. уравнение (V. 22)]. По результатам,
полученным в части 1 (см. табл. V. 3) строят график в координатах
In ККМ — п. В соответствии с уравнением (V. 24) тангенс угла наклона
этой зависимости равен
tgy RT
По тангенсу угла наклона tgy находят значения <р' и работы ми-
целлообразования (е7 == ф'пАа) . Сравнивают полученные значения ра-
бот адсорбции е и мицеллообразования е7,
142
Работа 23. АДСОРБЦИОННОЕ ТИТРОВАНИЕ
ЛАТЕКСОВ
Цель работы: определение степени насыщенности поверхности латекс-
ных частиц ПАВ, площади, занимаемой молекулой ПАВ в насыщенном
адсорбционном слое, и удельной поверхности дисперсной системы.
Способность коллоидных ПАВ образовывать мицеллы при малом
содержании их в растворе лежит в основе широко применяемого на
практике метода адсорбционного титрования латексов. Этот метод ис-
пользуется для определения таких важнейших характеристик латексов,
как степень адсорбционной насыщенности поверхности частиц 0,, их
размеров, площади, занимаемой одной молекулой ПАВ при образова-
нии насыщенного адсорбционного слоя s0, а также удельной поверх-
ности системы SyA.
Метод адсорбционного титрования латексов состоит в следующем.
В латекс, на поверхности частиц которого уже содержится определен-
ное количество стабилизатора (ПАВ), дополнительно вводят ПАВ. Мо-
лекулы ПАВ адсорбируются в основном на поверхности полимерных
частиц. Концентрация же свободного стабилизатора в водной фазе
вследствие его высокой поверхностной активности ничтожно мала и
значительно меньше концентрации, при которой образуются мицеллы.
После завершения адсорбции ПАВ, которая, как правило, носит моно-
молекулярный характер, дальнейшее добавление его приводит к ассо-
циации молекул или ионов в водной фазе латекса. Этому отвечает пе-
региб на кривых адсорбционного титрования, например, кривых зави-
симостей поверхностного натяжения или удельной электропроводности
от объема вводимого в латекс раствора ПАВ (рис. 41).
Степень адсорбционной насыщенности латексов характеризует сред-
нюю плотность упаковки молекул ПАВ в адсорбционных слоях. От
нее во многом зависит устойчивость латексов к коагуляции, поведение
их при хранении и в процессах'переработки в изделия. Степень адсорб-
ционной насыщенности поверхности частиц латекса определяется как
л __ Aj _______Aj
1 A- Al + Аад
(V.27)
где Ai — количество (моль) ПАВ в исходном латексе, приходящееся на единицу мас-
сы (г) полимера; Л»— общее количество ПАВ (моль), необходимое для образования
насыщенного адсорбционного слоя на поверхности частиц, отнесенное к единице массы
(г) полимера; Аад — количество (моль) стабилизатора, адсорбированное единицей мас-
сы (г) дисперсной фазы в процессе титрования.
Следовательно, для нахождения б,х
степени адсорбционной насыщенности
латексов необходимо знать две вели-
чины: Ai и Аад, которые определяются
экспериментально. Значение А< опре-
деляют колориметрическим или кон-
дуктометрическим методом, Аад нахо-
дят путем адсорбционного титрования
латексов. Необходимо учитывать, что
Рис. 41. Зависимость поверхностного натяже-
ния о и удельной электропроводности х ла-
текса от объема вводимого раствора ПАВ.
143-
при титровании на поверхности латексных частиц адсорбируется не все
количество введенного ПАВ, определенная часть ПАВ остается в водной
фазе латекса, и в конечной точке титрования (соответствует точке пере-
гиба на кривой адсорбционного титрования) содержание его достигает
ККМ. Поэтому при определении значения Аад из общего израсходо-
ванного объема стабилизатора следует вычесть количество ПАВ, до-
бавленное в водную фазу латекса до достижения ККМ.
Зная общее количество ПАВ, необходимое для образования насы-
щенного мономолекулярного слоя на поверхности полимерных частиц,
т. е. (А; + Аад), можно рассчитать также удельную поверхность ла-
текса Худ. Для этого независимым методом находят величину s0. Зна-
чение Худ вычисляют по формуле
худ = (Аг + Аад)5^А (V. 28)
Поскольку для систем, содержащих частицы сферической формы,
удельная поверхность худ равна
худ=-^- (V.29)
asp
где ds — среднеповерхностный диаметр частиц (см. работу 18); р — плотность веще-
ства, составляющего дисперсные частицы.
то, зная 5УД и so, можно рассчитать средний диаметр латексных частиц:
+ (V’30)
Если средний диаметр полимерных частиц ds найден независимым
методом, например, турбидиметрическим методом, то уравнение (V. 30)
позволяет рассчитать х0 по данным адсорбционного титрования.
Обычно при определении величины Аад для учета ПАВ, введенного
при титровании, но оставшегося в водной фазе, проводят титрование
нескольких образцов латексов с различной концентрацией полимера.
По точкам перегиба на кривых адсорбционного титрования находят
объем Уккм (см. рис. 41). По значениям Уккм определяют количество
ПАВ, адсорбировавшегося на частицах латекса Аад. Для этого исполь-
зуют уравнение материального баланса перераспределения стабилиза-
тора при смешении исходного раствора ПАВ с образцом латекса:
^ККМСПАВ = ^слат + ^ККМ
ИЛИ (V- 31)
^ККМСПАВ
V
= ^аЛаТ + ККМ
где спдв—концентрация ПАВ в растворе, используемом при титровании; слат — кон-
центрация полимера в данной серии образцов латекса; V — объем образцов латекса.-
Согласно уравнению (V.31) величина Аад определяется как тангенс
угла наклона прямой, построенной в координатах УккмСпав/Е—слат,
а отрезок, отсекаемый этой прямой на оси ординат, соответствует зна-
чению ККМ данного ПАВ. Последнее справедливо лишь тогда, когда
исходный латекс не содержит свободного стабилизатора. В остальных
случаях приведенный способ определения ККМ не является достаточно
строгим.
,144
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Установка для измерения поверхностного натяжения или мост пе-
ременного тока с кондуктометрической ячейкой для измерения электро-
проводности.
Термостат.
Фотоэлектроколориметр ФЭК-56М.
Конические колбы емкостью 50 и 200 мл.
Пипетки емкостью 1 и 10 мл.
Полистирольный латекс.
Раствор ПАВ, например 0,1 М раствор олеата натрия.
0,1 М раствор гидроксида натрия.
Предварительно определяют диаметр частиц латекса. Для этого
измеряют оптическую плотность исходного латекса при различных дли-
нах волны падающего света с помощью фотоэлектроколориметра (ме-
тодику измерения и расчета диаметра частиц см. в работе 17).
В колбах емкостью 200 мл готовят три образца латекса с концен-
трацией дисперсной фазы 5, 10 и 15 % (масс.) в объеме по 120 мл.
Доводят pH приготовленных образцов до значений 9—10, для чего
в них вводят по каплям 0,1 М раствор NaOH (pH контролируют с по-
мощью рН-метра).
Из полученных образцов готовят три серии латексов с различным
содержанием ПАВ. Для этого отбирают 30 колб емкостью 50 мл (по
10 для каждой серии), нумеруют
пипетки вносят по 10 мл латекса,
дующих объемах Упав:
Номер колбы.................... 1 2
Объем раствора ПАВ, добав-
ляемый в латекс, мл
I серия образцов (5%-ный
латекс) .................... 0 0,25
II серия образцов (10%-
ный латекс) ................ 0 0,5
III серия образцов (15%-
ный латекс)................. 0 1,0
их и в каждую колбу с помощью
затем вводят раствор ПАВ в сле-
3 4 5 6 7 8 9 10
0,50 1,0 1,5 2,0 2,5 3,0 3,5 4,0
1,0 1,5 2,0 3,0 4,0 5,0 6,0 7,0
2,0 3,0 4,0 5,0 6,0 7,0 8,0 9,0
Объем золей во всех колбах доводят дистиллированной водой до
постоянного значения V = 20 мл и выдерживают их 30—40 мин для
установления адсорбционного равновесия. После этого измеряют по-
верхностное натяжение <т или удельную электропроводность к латек-
сов. Если в качестве стабилизатора латексных частиц применены ионо-
генные ПАВ, предпочтительнее кондуктометрический метод, поскольку
он позволяет более точно определять точку излома на кривых титро-
вания.
Методика измерения о приведена в работе 1, методика кондуктомет-
рических измерений — в работе 20. Для каждого образца латекса из-
мерение величины <т или % проводят не менее трех раз и рассчитывают
среднее значение параметра. Полученные данные записывают в таб-
лицу (см. табл. V. 5).
Для каждой серии латексов строят кривые титрования %=^(Еп\в)'
или о = И^пав) и по ним находят значения Еккм при различных кон-
центрациях полимера в латексе слат, Затем строят график в координа-
10 Зак. 673
145
Таблица V. 5. Результаты адсорбционного титрования латексов
I серия образцов
(5%-ный латекс)
II серия образцов
(10%-ный латекс)
Ш серия образцов
(15%-ный латекс)
Номер
колбы
среднее
значение
О ИЛИ х
о.
Дж/м2
среднее
значение
о или х
о, х,
Дж/м2 См-м”1
среднее
значение
о или х
тах УккмСпав/У — Слат и определяют Аад (при построении графика слат
выражают в г/л, а спав в моль/л). По формулам (V.27)— (V.30) рас-
считывают степень адсорбционной насыщенности латексных частиц 0,,
площадь so, приходящуюся на одну молекулу стабилизатора в насы-
щенном адсорбционном слое, и удельную поверхность системы 5УД.
Необходимые для расчета значения А> и р получают от преподавателя.
Работа 24. ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ МАССЫ ПОЛИМЕРОВ
И МИЦЕЛЛЯРНОЙ МАССЫ ПАВ НЕФЕЛОМЕТРИЧЕСКИМ МЕТОДОМ
Цель работы: определение молекулярной массы полимера и второго
вириального коэффициента; определение мицеллярной массы ПАВ.
Дебай, исходя из флуктуационной теории светорассеяния и исполь-
зуя уравнение (V. 5) для осмотического давления, получил соотношение
между мутностью т раствора полимера, его массовой концентрацией с
и молекулярной массой полимера Л4:
Нс 1
“=^- + 2^ (V.32)
где Аг — второй вириальный коэффициент в уравнении состояния растворов полиме-
ров (V. 5); Н — константа для данной системы полимер — растворитель.
Для неполяризованного (естественного) падающего света констан-
та Н равна
где п и по — показатель преломления соответственно раствора и растворителя; Хвак —
длина волны света в вакууме.
Согласно уравнению (V. 32) зависимость Нс/т, от концентрации
раствора в области малых значений с графически выражается прямой
линией. Отрезок, отсекаемый этой прямой на оси ординат, соответствует
величине, обратной молекулярной массе полимера М. Тангенс угла на-
клона прямой определяет термодинамическое сродство между раство-
ренным веществом и растворителем, которое в данном случае харак-
теризуется вторым вириальным коэффициентом А2.
Таким образом, для нахождения молекулярной массы полимеров
методом Дебая достаточно получить концентрационную зависимость
мутности т раствора ПАВ и найти значение константы ТУ, для чего до-
полнительно требуется измерить инкремент показателя преломления
(п— п0)/с. Для определения этой величины лучше всего использовать
146
интерферометр или дифференциальный рефрактометр. Значения пока-
зателя преломления п0 и инкремента показателя преломления (и—по)/с
для некоторых систем полимер — растворитель приведены в табл. V. 6.
Необходимо отметить, что уравнение Дебая применимо только для
сильно разбавленных растворов полимеров, когда с <0,1 %. При этом
оно выполняется только в том случае, если размеры макромолекуляр-
ных клубков не превышают 40—50 нм, т. е. меньше 1/10Х. При боль-
ших размерах рассеивающих частиц в них возникает внутримолекуляр-
ная интерференция и суммарная интенсивность светорассеяния си-
стемой уменьшается. В результате при расчете по уравнению (V. 32)
получаются заниженные значения молекулярной массы. Для определе-
ния истинных значений М в таких системах необходимо учесть зависи-
мость интенсивности рассеянного света от угла 9 [см. уравнение
(IV. 1)] и в уравнение Дебая ввести соответствующую поправку.
При определении молекулярных масс полимеров методом Дебая
следует также учесть, что параметр т отражает светорассеяние, обус-
ловленное только рассеивающими частицами, и не связан с рассеянием
света растворителем, т. е. является избыточной величиной:
т = Гобш — то (V. 34)
где Тобщ — суммарное значение мутности растворенного вещества и растворителя;,
то — мутность чистого растворителя
Метод Дебая может быть применен также к растворам ПАВ, содер-
жащим мицеллы, если они являются рэлеевскими частицами и рас-
твор достаточно разбавленный. В этом случае уравнение Дебая при-
нимает следующий вид:
И (с - ККМ) = 1 + 2Л2 _ ККМ) (V 35)
г —ГККМ Мм
где т и тккм — мутность раствора и мутность его при ККМ соответственно; Л4М — йи-
целлярная масса ПАВ.
С помощью уравнения (V. 35) можно найти такие важнейшие
характеристики ПАВ, как мицеллярная масса Мм и вириальный коэффи-
циент А2, позволяющий оценить взаимодействие мицелл с растворите-
лем. Значение Мм согласно уравнению (V.35) определяется, как вели-
чина, обратная отрезку, отсекаемому на оси ординат на графике зави-
симости Н (с—ККМ)/(т — Тккм) = f(с — ККМ). Зная молекулярную
массу М ПАВ, можно рассчитать также число агрегации п — Мы/М.
Таблица V.6. Показатель преломления некоторых растворителей п0 и (п — пв)1с
для систем полимер — растворитель при — 546 нм
Система полимер — растворитель гао (п-п0)/«
Полистирол — бензол 1,498 0,106
Полистирол — толуол 1,494 0,109
Полиметилметакрилат — ацетон 1,357 0,134
Поливинилацетат — ацетон 1,357 0,104
Поливиниловый спирт — вода 1,330 0,170 л/а
10 *
147
Рис. 42. Оптическая схема нефелометра НФМ:
1— источник света; 2— рассеиватели; 3,10— отсчетные барабаны; 4,9— объективы; 5 — светофильтр;
6 — окуляр; 7,11,16— линзы; 8 — призмы; 12— светоловушка; 13— камера; 14— кювета; 15 — конденсор;
17—стеклянная пластинка.
При определении молекулярных характеристик методом светорас-
сеяния следует обращать особое внимание на очистку исследуемых
растворов. Поскольку интенсивность рассеянного света пропорциональ-
на квадрату объема частиц, наличие примесей, имеющих большие раз-
меры, чем рассеивающие свет частицы, может вызвать значительные
погрешности при обработке экспериментальных данных. Обычно очист-
ку растворов проводят фильтрованием через стеклянные пористые или
бактериальные фильтры, используют также метод центрифугирования.
Мутность исследуемых растворов определяют с помощью нефело-
метра типа НФМ (рис. 42). Световой поток от монохроматического
источника света 1 падает на прозрачную пластинку 17 и, пройдя через
нее, а также через линзу 16 и конденсор 15, попадает в кюветный
блок 13, в котором помещается кювета 14 с исследуемой системой.
Световой поток, прошедший через кювету, гасится в светоловуш-
ке 12. Свет, рассеянный под углом 135° к падающему потоку, проходит
через линзу 11, попадает в фотометрическую головку и вызывает осве-
щение одной половины поля зрения в окуляре 6. Часть светового по-
тока, отраженного от пластинки 17, попадает на рассеиватель 2, а за-
тем в фотометрическую головку, что создает определенную яркость
освещения второй половины поля зрения.
Световые потоки, попавшие в фотометрическую головку, проходят
соответствующие измерительные диафрагмы, каждая из которых свя-
зана со своим отсчетным барабаном 3 и 10. Оба световых потока,
пройдя объективы 4 и 9, призмы 8, фокусирующую линзу 7 сходятся
к оптической оси окуляра 6. В результате наблюдатель видит поле
зрения в форме круга, разделенного пополам вертикальной линией. При
этом яркость левой половины поля зрения определяется световым по-
током, прошедшим диафрагму, связанную с барабаном 3, а яркость
правой половины — потоком, прошедшим другую диафрагму.
Определение мутности исследуемой системы основано на уравнива-
нии световых потоков, рассеянных раствором в кювете 14 и рассеива-
телем 2, путем изменения степени раскрытия измерительных диафрагм.
Пои исследовании сильно разбавленных растворов уравнивание световых
148
потоков проводят с помощью диафрагмы, установленной на пути све-
тового потока от рассеивателя. Для этого диафрагму на пути светового
потока от раствора полностью открывают, устанавливая барабан 10
на отметке «100» (по черной шкале), отсчетный барабан 3 также уста-
навливают на отметке «100» и затем, постепенно вращая его, добива-
ются одинаковой освещенности обеих половин поля зрения в окуляре.
По черной шкале барабана 3 отсчитывают число делений, показываю-
щих отношение площади диафрагмы при данном раскрытии к площади
се максимального раскрытия (в %).
При равенстве световых потоков выполняется соотношение
т-100 = ТрШр (V. 36)
где т — мутность исследуемой системы; тр — мутность рассеивателя; тр— число деле-
ний, отсчитанных по черной шкале барабана 3.
Для определения мутности растворов в абсолютных единицах рас-
сеиватель предварительно градуируют по эталонной призме с извест-
ной мутностью тЭт. Равенство световых потоков, идущих от эталонной
призмы и от рассеивателя, выражается следующим уравнением:
тэт • 100 = тртр (эт) (V.37)
где тр(ЭТ) — степень открытия диафрагмы, находящейся на пути светового потока от
рассеивателя (в единицах шкалы барабана 3).
Объединение уравнений (V. 36) и (V. 37) дает уравнение для рас-
чета мутности исследуемой жидкости в абсолютных единицах:
т_
т = гэт--Е— (V.38)
тр (эт)
Мутность растворов обычно выражают в см~’.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Часть 1. Определение молекулярной массы полимеров
Для проведения работы необходимы:
Нефелометр НФМ.
Пипетки емкостью 5 и 10 мл.
Колбы Бунзена со стеклянными фильтрами.
Водоструйный насос.
Раствор полимера, например полиметилметакрилата в ацетоне или
поливинилового спирта в воде.
Растворитель.
Предварительно растворитель и исходный раствор полимера тща-
тельно очищают от возможных механических примесей, для чего их
фильтруют через стеклянные фильтры (типа G = 4, G = 5, или
ПОР-16).
Камеру 13 нефелометра заполняют водой, обеспыленной методом
ультрафильтрации, и помещают в нее эталонную призму. Устанавли-
вают пригодные для исследуемых систем светофильтр и рассеиватель
(по указанию преподавателя), включают источник света и определяют
значение тР(ЭТ).
149
Таблица V. 7. Экспериментальные и расчетные данные для определения
молекулярной массы полимера и второго вириального коэффициента
нефелометрическим методом
Концентрация раствора с, г/100 см3 Число делений по шкале барабана m СР Мутность раствора тобщ’ см“‘ г“тобщ Т0’ см“1 Яс/т
После этого в измерительную кювету наливают 20 мл растворителя,
кювету закрывают стеклянной крышкой и устанавливают ее вместо
эталонной призмы в центре камеры 13. При том же рассеивателе и
светофильтре определяют значение тр. По формуле (V. 38) рассчиты-
вают мутность растворителя т0-
Аналогичным образом измеряют тР и рассчитывают тОбщ исходного
раствора полимера. Затем из кюветы пипеткой отбирают 5 мл раствора
и добавляют в таком же объеме обеспыленный растворитель (для от-
бора раствора и внесения растворителя используют отдельные пипет-
ки). Раствор перемешивают путем осторожного покачивания кюветы
и после термостатирования определяют его мутность. Таким образом
проводят 7-8 операций по разбавлению раствора и измерению его мут-
ности. Измерения тР для каждого исследуемого раствора проводят не
менее трех раз.
Для определения значений М и А2 вычисляют избыточную мутность
растворов, которую находят как разность мутности раствора тОбщ и
мутности растворителя то [см. уравнение (V. 34)], затем по уравнению
(V. 33) рассчитывают константу Н.
Для каждой концентрации раствора полимера рассчитывают вели-
чину Нс/т. Экспериментальные и расчетные данные записывают в таб-
лицу (см. табл. V. 7).
Строят график зависимости Яс/т = f(c) и по нему находят моле-
кулярную массу полимера как величину, обратную отрезку, отсекае-
мому на оси ординат, и второй вириальный коэффициент по тангенсу
угла наклона прямой.
Часть 2. Определение мицеллярной массы ПАВ
Для проведения работы необходимы:
Нефелометр.
Пипетки емкостью 5 мл.
Водный раствор ПАВ, например додецилсульфата натрия или пре-
парата ОП-10.
Обеспыленная вода или бидистиллят.
При проведении работы и для приготовления исходных растворов
ПАВ используют воду, обеспыленную методом ультрафильтрации, или
бидистиллят. Определяют мутность растворителя и растворов ПАВ в
соответствии с указаниями, данными в части 1 работы. Значения ККМ
и тккм указываются преподавателем или определяются эксперимен-
тально.
150
Таблица V. 8. Экспериментальные и расчетные данные для определения
мицеллярной массы ПАВ нефелометрическим методом
Концентрация раствора ПАВ, с, г/100 см3 Число делений по шкале барабана пг mp' ср Мутность раствора тобщ' см~‘ т“тобщ Т0’ см”"1 TKKM’ см-1 Я (с —ККМ)
По формуле (V. 33) вычисляют константу Н (значения и0 и
(п— по)с определяются экспериментально при помощи интерферомет-
ра). Экспериментальные и расчетные данные записывают в таблицу
(см. табл. V. 8).
По данным таблицы строят график зависимости Н(с — ККМ)/т—
— тккм = f(с — ККМ); экстраполируя прямую до пересечения с осью
ординат, находят мицеллярную массу ПАВ.
Работа 25. ИССЛЕДОВАНИЕ РАСТВОРОВ
АМФОТЕРНЫХ ПОЛИЭЛЕКТРОЛИТОВ
Цель работы: определение изоэлектрической точки раствора желатины
по зависимости вязкости и мутности от pH среды.
Полиэлектролиты благодаря наличию ионогенных групп диссоции-
руют в воде и других полярных растворителях. Степень диссоциации
групп амфотерных полиэлектролитов зависит от их природы, pH и
ионного состава раствора.
При увеличении степени диссоциации возрастает электростатиче-
ское отталкивание одноименно заряженных групп макромолекул, что
приводит к существенному изменению их конформации в растворе, а
именно: цепи, свернутые в клубок, распрямляются и стремятся при-
нять форму, приближающуюся к линейной. В результате этого увели-
чивается эффективный размер молекул и существенно изменяются фи-
зико-химические свойства растворов, например, возрастает вязкость,
изменяется интенсивность светорассеяния. При уменьшении степени
диссоциации макромолекулы, наоборот, сворачиваются, приобретая
конформации с наибольшим значением энтропии в системе. Если pH
раствора поддерживают постоянным, то в результате электростатиче-
ского'-вта’ймодействия ионизированной части полярных групп и тепло-
вого движения устанавливаются определенные конформации молекул.
СбСТояние равновесия зависит от величины заряда полииона, состава
раствора, температуры.
Исследуемая в данной работе желатина представляет собой про-
дукт переработки коллагена — распространенного в природе белкового
вещества. В молекулах желатины содержатся как кислотные (карб-
оксильные), так и основные (амино) группы. Поэтому в водных рас-
творах желатина проявляет свойства, присущие амфотерным полиэлек-
тролитам, т. е. происходит ионизация кислотных и основных групп:
—СООН —СОО_ + Н+
—NH2 + H+ —NH+
151
Степень ионизации каждой группы зависит от pH среды и ионной
силы раствора. Для полиамфолитов характерным является такое со-
стояние, когда число ионизированных кислотных групп равно, числу
ионизированных основных, т. е. суммарный заряд макромолекул равен
нулю. Это наблюдается при определенной концентрации ионов водо-
рода, отвечающей изоэлектрической точке. В изоэлектрическом состоя-
нии макромолекула стремится свернуться в наиболее плотный клубок.
При изоэлектрической точке не происходит передвижения молекул
под действием внешнего электрического поля, наблюдается минималь-
ная вязкость раствора, максимальное светорассеяние и набухание, наи-
большее осмотическое давление.
~ В кислой среде (относительно изоэлектрической точки), например
в присутствии НС1, диссоциация карбоксильных групп подавлена и
макромолекулы содержат в основном положительно заряженные груп-
пы — RNH^.
В слабокислой среде с увеличением содержания в растворе НС1
степень диссоциации аминогрупп повышается. В результате электро-
статическое отталкивание групп — RNHJ возрастает и происходит раз-
вертывание молекулярных клубков полиамфолита. Это сопровождается
увеличением вязкости и уменьшением мутности раствора. При значи-
тельном содержании НС1 (большое количество хлорид-ионов) степень
диссоциации основных групп понижается в результате образования
солевой формы RNH3C1, а эффективные размеры молекулы снова
уменьшаются.
При возрастании pH раствора относительно изоэлектрической точки
(изменяется от нейтральной до слабощелочной) преобладает процесс
диссоциации кислотных групп, что приводит к постепенному разверты-
ванию молекулярных клубков. При значительном повышении щелоч-
ности, так же как и при увеличении кислотности, количество заряжен-
ных групп уменьшается в результате образования RCOONa и макро-
молекулы снова сворачиваются в плотные клубки. В соответствии
с таким поведением макромолекул желатины на кривой зависимости
свойства раствора полиамфолита от pH среды имеется несколько экс-
тремумов.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Вариант 1. Определение изоэлектрической точки раствора
желатины по зависимости вязкости от pH среды
Для проведения работы необходимы:
Установка для измерения вязкости.
Секундомер.
pH-метр типа pH-340.
Конические колбы емкостью 50 мл.
Пипетки емкостью 10 мл.
1,5%-ный (масс.) раствор желатины.
0,05М хлороводородная кислота.
0,01 М раствор гидроксида калия.
В пронумерованные колбы (2—8) вносят пипеткой по 10 мл охлаж-
денного до комнатной температуры и отфильтрованного раствора же-
152
Таблица V. 9. Результаты измерения pH и вязкости растворов желатины
для определения изоэлектрической точки
Номер колбы pH раствора Время истечения т, с Относительная вязкость Т|
-
латины. Затем в колбы добавляют растворы хлороводородной кислоты,
щелочи и воды в следующих объемах (в колбу 1 наливают 20 мл
воды):
Номер колбы ..... 1 * 2 3 4 5 6 7 8
Объем НС1, мл ... — 10 6 3 1 — — —
Объем раствора КОН,
мл................. — — — — — — 2 5
Объем воды, мл .... 20 — 4 7 9 10 8 5
* Без желатины.
Измеряют pH всех приготовленных растворов. Для этого в кювету
pH-метра наливают 1—1,5 мл раствора и осторожно соединяют ее со
стеклянным электродом; значение pH определяют сначала по шкале
прибора с интервалом от —1 до 14, а затем более точно фиксируют
по шкале с интервалом в три единицы pH. После каждого измерения
кювету и электрод следует тщательно промыть водой.
Затем определяют вязкость растворов и растворителя (воды) с по-
мощью капиллярного вискозиметра Уббелоде (методика измерения
приведена в работе 30). Измерение проводят при постоянном давлении
(давление выбирается по указанию преподавателя). Относительную
вязкость растворов желатины рассчитывают по формуле
Пота = п/Ло = т/т0 (V .39)
где т] и т]о — вязкость раствора и растворителя (воды); т и т,->— время истечения
раствора и растворителя.
Экспериментальные и расчетные данные записывают в таблицу
(см. табл. V. 9). По данным таблицы строят график зависимости отно-
сительной вязкости г]отн от pH раствора и по минимуму на кривой
определяют изоэлектрическую точку желатины.
Вариант 2. Определение изоэлектрической точки раствора
желатины по зависимости мутности от pH среды
Для проведения работы необходимы:
Фотоэлектроколориметр, например ФЭК-56М, с кюветами длиной
3 см.
pH-метр типа pH-340.
Конические колбы емкостью 50 мл.
Пипетки емкостью 10 мл.
1,5 %-ный (масс.) раствор желатины.
Концентрированная и 0,05 М хлороводородная кислота.
0,01 М раствор гидроксида калия,
153-
Таблица V. 10. Результаты измерения pH и мутности раствора желатины
для определения изоэлектрической точки
Номер колбы pH раствора Оптическая плотность D Мутность раствора т, см"1
В пронумерованные колбы (1—9) вносят по 10 мл отфильтрован-
ного раствора желатины и затем добавляют растворы хлороводородной
кислоты, щелочи и воду в следующих объемах:
Номер колбы 1 2 3 4 5 6 7 8
Объем НС1, мл 10 4 1 0,5 — — — — —
Объем раствора КОН, мл . . — — — — — 1 3 6 10
Объем воды, мл . — 6 9 9,5 10 9 7 4 —-
Измеряют pH всех приготовленных растворов, как указано в ва-
рианте 1 работы. Затем определяют оптическую плотность D раство-
ров с помощью прибора ФЭК-56М со светофильтром № 2 (А. = 364 им).
Методика измерения оптической плотности приведена в работе 17. Рас-
творы из кювет после измерений выливают обратно в колбы. По окон-
чании измерения увеличивают кислотность растворов в колбах 1 и 2,
для чего в колбу 1 добавляют одну каплю конц. ИС1, а в колбу 2 —
две капли кислоты и снова измеряют значения pH и D этих растворов.
Мутность растворов желатины вычисляют по формуле (IV. 7). Экс-
периментальные и расчетные данные записывают в таблицу (см.
табл. V. 10). По данным таблицы строят график зависимости мутности
раствора от pH и по максимуму находят изоэлектрическую точку же-
латины.
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАЧИ
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. По какому признаку дисперсные системы делят на лиофобные и
лиофильные? Чем объяснить самопроизвольное возрастание межфаз-
ной поверхности при образовании лиофильных дисперсных систем?
2. Какой параметр используется в качестве критерия лиофильности
дисперсной системы? Какие виды энергии сопоставляются в уравнении
Ребиндера — Щукина?
3. Какие дисперсные системы относят к лиофильным? Приведите
примеры таких систем. Как происходит формирование частиц дисперс-
ной фазы в лиофильных системах?
4. Как классифицируют поверхностно-активные вещества?
5. Чем отличаются коллоидные ПАВ от истинно растворимых? Что
называют критической концентрацией мицеллообразования?
6. Какие существуют методы определения ККМ? Почему при кон-
центрациях, превышающих ККМ, поверхностное натяжение растворов
ПАВ практически не изменяется?
7. Какие факторы влияют на ККМ? Как и почему влияет длина
углеводородного радикала на ККМ в разных по полярности раствори-
телях?
154
8. Каким образом ориентируются молекулы ПАВ в мицеллах, обра-
зующихся в полярной и неполярной средах? От чего зависит форма
мицелл в растворах коллоидных ПАВ?
9. Как влияет на ККМ. природа полярной группы молекул ПАВ?
Каково влияние добавок индифферентного электролита на ККМ
ионогенных и неионогенных ПАВ?
10. В чем проявляется взаимосвязь поверхностных и объемных
свойств растворов коллоидных ПАВ?
11. Какое явление называют солюбилизацией? Чем обусловлено это
явление? Каково практическое значение этого явления?
12. Расскажите о практическом применении ПАВ. На чем осно-
вано использование ПАВ в качестве стабилизаторов дисперсных
систем? В чем заключается механизм моющего действия растворов
ПАВ?
13. Каковы особенности растворения полимеров? Какой процесс на-
зывается набуханием? В каких случаях происходит ограниченное и
неограниченное набухание полимера?
14. Укажите характеристики набухания полимеров в низкомолеку-
лярных жидкостях. Что такое степень набухания и как она определя-
ется?
15. Что характеризует второй вириальный коэффициент в уравне-
нии состояния растворов полимеров? По изменению каких свойств рас-
творов полимеров можно определить этот параметр?
16. Как влияет pH раствора на форму молекул полиамфолитов?
Что такое изоэлектрическая точка полиэлектролитов?
17. Как влияют индифферентные электролиты на заряд и форму
молекул полиэлектролитов?
18. Как влияет изменение конформаций макромолекул на вязкость
и светорассеяние растворов полиамфолитов?
19. Каково практическое применение растворов полимеров? Рас-
смотрите факторы, обеспечивающие агрегативную устойчивость дис-
персных систем при стабилизации их полимерами.
ПРИМЕРЫ РЕШЕНИЯ ЗАДАЧ
1. Критическая концентрация мицеллообразования (ККМ) додецилсульфата натрия
при 20, 40 и 60°C составляет соответственно 1,51-10-3; 1,62-10-3 и 1,87-10~3 моль/л.
Рассчитайте стандартную теплоту, энергию Гиббса и энтропию мицеллообразования
при 20 “С.
Решение. Теплоту мицеллообразования q определяем по температурной зависимо-
сти ККМ:
A//., q
In ККМ = + const = - -7- + const
Рассчитываем значения In ККМ и 1/7:
In ККМ................. —6,50 —6,42 —6,28
1/7-103, К"1 . . . . 3,41 3,19 3,00
Строим график зависимости In ККМ от 1/7 (рис. 43). Теплоту мицеллообразования
определяем по тангенсу угла tg а наклона прямой:
tg« = -4-=-537
А
155
/
Энтропию мицеллообразования
Рис. 43. К определению теплоты мицеллообразо-
вания додецилсульфата натрия.
Отсюда
q = 8,31 • 537 = 4,46 кДж/моль
Энергию Гиббса рассчитываем по соотноше-
нию
AG° = RT In ККМ = 8,31 • 293 • In 1,51 10~3 =
= — 15,83 кДж/моль
находим, пользуясь термодиначеским уравнением
AG° = Д77° - Т ^SOU
ММ м
-------т
— 4,46 — (—15,83)
= 0,039 кДж/(моль • К)
293
2. Постройте кривую кинетики набухания каучука в четыреххлористом углероде
в координатах а — т по следующим экспериментальным данным:
Время набухания т, мин 5 30 90 150 210 240 270 300
Степень набухания ат 0,33 1,15 2,33 2,91 3,25 3,41 3,58 3,58
Определите графическим способом константу скорости набухания К-
Решение. При ограниченном набухании полимеров степень набухания изменяется
во времени в соответствии с уравнением
</ат ,, г
— К (амакс ат)
Интегрируя это уравнение, получаем:
1п ---=
амакс ат
Зависимость In (aMaKC/(aMaKC - аД] от т является линейной. Согласно исходным
данным амакс = 3,58. мости: т, мин Рассчитываем / .... 5 ханные 30 для построения графика 210 этой зависи- 240
90 150
амакс .... 1,1 1,47 2,86 5,34 10,85 21,06
амакс
। амакс амакс — .... 0,097 0,387 1,052 1,676 2,38 3,047
Из графика (см. рис. 44) находим:
К. = tg 0 = 0,012 мин-1
3. Постройте график зависимости приведенного осмотического давления от кон-
центрации раствора сополимера стирола и метакриловой кислоты в толуоле (7’ =
= 300 К) по следующим данным:
Концентрация раствора
с-10~3, г/м3 ............... 1,1 2,8 5,4 7,6
Осмотическое давление л, Па 9,8 37,3 106,4 187,4
По графической зависимости л/с = / (с) определите относительную молекулярную
массу М полимера и значение второго вириальиого коэффициента А.
Решение. Приведенное осмотическое давление растворов полимеров описывается
уравнением (V. 6) ;
т“ет(т+л=»+М
156
Рис. 45. Зависимость приведенного осмотического давления от концентрации раствора
полимера.
Рис. 44. К определению константы скорости набухания каучука в четыреххлорисгом
углероде.
При малых концентрациях полимера в растворе коэффициент А3 равен нулю.
Рассчитываем значения л/с и строим их зависимость от с (рис. 45);
с-10-3, г/м3 ........... 1,1 2,8 5,4 7,6
(л/с) 103, Па • м3/г . . , 8,91 13,32 19,70 24,66
Экстраполируя зависимость на значение с = 0, находим:
RT ч 8,31 300
----= 6 7 10 3 М —----------т = 372 000
М ’ 6,7 10-3
Вириальный коэффициент А2 определяется по тангенсу }гла наклона полученной
прямой:
= ПТТ = ЯЧ1 = i’003• 10~9 м3• М0ЛЬТ2
i\l o,ol • oUU
Положительное значение второго вириального коэффициента указывает на хорошее
термодинамическое сродство между полимером и данным растворителем.
ЗАДАЧИ
1. Напишите формулу мицеллы олеата натрия в водном растворе, если
число агрегации равняется 60. Как изменится строение мицеллы при
введении в раствор больших количеств NaCl?
2. Оцените поверхностную активность лаурилсульфата на границе
раздела его водного раствора с воздухом, если известно, что при ККМ,
равной 0,015 моль/л, поверхностное натяжение составляет 30 мДж/м2.
Поверхностное натяжение воды примите равным 71,96 мДж/м2.
3. Рассчитайте теплоту мицеллообразования,..а также, стандартную
энергию Гиббса и энтропию процесса при 293 К, используя следующие
значения ККМ для додецилсульфата натрия в растворах NaCl:
Т, К . .................... 293 311
ККМ, ммоль/л
' в 0,01 М 'растворе NaCl . . -5,13 -5,37
н 0,2 М растворе NaCl . . 0,76 0,87
Проанализируйте изменение термодинамических
образования и влияние электролита.
333
6,17
1,45
функций мицелло-
157
4. Полистирол с молекулярной массой 300000 адсорбируется из
толуольного раствора на углеродном адсорбенте, имеющем удельную
поверхность 0,12 м2/г. Величина предельной адсорбции при образова-
нии монослоя равна 0,033 г/кг. Рассчитайте площадь, приходящуюся
на одну молекулу полистирола в плотном монослое, и число молекул
на поверхности 1 кг адсорбента.
5. Используя уравнение Дебая, рассчитайте мицеллярную массу
ПАВ и радиус мицелл в воде по следующим данным:
Концентрация раствора с, г/л 0,5 1,0 1,5 2,0 4,0
Мутность раствора т-109, см-1 1,70 2,68 3,54 3,85 5,09
Константу в уравнении Дебая примите равной Н = 3-10~и, плот-
ность ПАВ 1140 г/см3, ККМ 0,1 г/л.
6. Рассчитайте радиус мицелл ПАВ в водной среде, считая их сфе-
рическими, по следующим данным: коэффициент диффузии мицелл при
313 К равен 0,69- 10~п м2/с, вязкость среды 8-10~4 Па-с.
7. Постройте изотерму поверхностного натяжения о = f (In с) по ре-
зультатам измерения поверхностного натяжения водных растворов
додецилсульфата натрия C^IksOSOsNa на границе с воздухом при
293 К:
С, о-ю3. Дж/м2 С, ммоль/л 0-103, Дж/м2 с, ммоль/л о-ю3. Дж/м2
0,2 67,3 1,2 51,8 5,0 36,8
0,3 65,0 1,4 49,9 6,0 36,5
0,5 61,0 1,6 48,0 7,0 36,4
0,6 59,4 1,8 46,7 10,0 36,3
0,8 56,4 2,0 45,1 15,0 36,3
1,0 54,0 3,0 40,6
Объясните, какие процессы, происходящие на поверхности системы
и в ее объеме, обусловливают появление точек перегиба на изотерме.
Рассчитайте площадь, занимаемую одной молекулой ПАВ в монослое
и ККМ.
8. Постройте кинетическую кривую набухания каучука в четырех-*
хлористом углероде по следующим -экспериментальным данным:
Время набухания т, мин 6 30 90 150 210 240 270 330
Степень набухания аг 0,33 1,15 2,33 2,91 3,25 3,41 3.58 3,58
Определите графическим способом константу скорости набухания К.
• 9. Постройте график зависимости приведенного осмотического дав-
ления л от концентрации с раствора сополимера стирола и метакрило-
вой кислоты в толуоле (Т — 300 К) по следующим данным:
с, г/л...... 1,1 2,8 5,4 7,6 9,4 8,5
л-10'2, Па. . 0,098 0,373 1,064 1,874 2,717 2,330
Рассчитайте молекулярную массу сополимера и второй вириальный
коэффициент.
10. По уравнению Дебая рассчитайте молекулярную массу поли-
стирола и второй вириальный коэффициент, используя следующие дан-
ные по измерению светорассеяния растворов полистирола в толуоле:
Концентрация раствора, г/л 1,11 1,45 1,88 2,43 2,87
Мутность раствора т • 108, м~1 3,68 4,47 5,55 6,50 7,13
Постоянную Н примите равной 1,17-10-13,
158
11. При эмульсионной полимеризации стирола в присутствии лау-
рата калия получен полистирольный латекс со следующими парамет-
рами: концентрация полимера 45 г/л, удельная поверхность 3-107 м2/м3,
плотность частиц 1,05 г/см3. Для определения степени адсорбционной
насыщенности поверхности полимера молекулами ПАВ проведено ад-
сорбционное титрование, при котором в 50 мл латекса введено
1,1 • 10—3 моль лаурата калия.
Рассчитайте степень адсорбционной насыщенности исходного ла-
текса, зная, что площадь одной молекулы лаурата калия в насыщен-
ном адсорбционном слое составляет 0,3 нм2. Примите во внимание, что
при образовании насыщенного монослоя лаурата калия на поверхно-
сти частиц полимера в дисперсной среде достигается критическая кон-
центрация мицеллообразования 16,0 ммоль/л.
VI. ОБРАЗОВАНИЕ, УСТОЙЧИВОСТЬ И СВОЙСТВА
ЛИОФОБНЫХ ДИСПЕРСНЫХ СИСТЕМ
Синтез лиофобных дисперсных систем (суспензий, золей, в том числе
аэрозолей, эмульсий) осуществляют методами диспергирования и кон-
денсации. Диспергирование твердых и жидких веществ в выбранных
средах проводят в шаровых и коллоидных мельницах, мельницах виб-
ропомола, ультразвуковых установках и др. Эффект диспергирования
усиливается при введении в среду ПАВ (эффект Ребиндера). Конден-
сационные методы основаны на физической или химической конденса-
ции атомов или молекул с последующим образованием новой фазы
в виде дисперсных частиц, распределенных в объеме среды (газооб-
разной, жидкой или твердой).
При конденсации необходимо пересыщение, так как возникающие зародыши имеют
большее равновесное давление пара (для жидкости) или большую растворимость (для
твердых частиц) благодаря большой кривизне поверхности (малому радиусу частиц).
Зависимость радиуса зародышей от пересыщения выражается уравнением Кельвина
(1.11). При образовании зародыша в случае лиофобных систем требуется затрата ра-
боты на создание новой поверхности. Учет этой работы и работы пересыщения дает
следующее выражение для работы образования зародыша в таких системах:
А = — AG = — ‘/3as
где s — поверхность зародыша.
Из этого уравнения следует, что затрата работы тем меньше, чем меньше поверх-
ностное натяжение и размеры зародыша. Окончательные размеры частиц дисперсной
фазы зависят от соотношения между скоростью образования зародышей и скоростью,
их роста, который самопроизвольно происходит в пересыщенных системах.
’т,
^Методом физической конденсации получают золи, дымы, дисперсные-
металлы. При химической конденсации частицы новой фазы образу-,
ются в результате протекания в системе химической реакции с образо-.
ванием малорастворимых соединений.
/.Лиофобные дисперсные системы термодиначески неустойчивы, так
как частицы дисперсной фазы склонны к агрегации. Их термодинами-
ческая агрегативная неустойчивость обусловлена избытком поверхност-
нойЛЯГергии. Межфазное поверхностное натяжение в лиофобных систе-
мах больше рассчитанного по соотношению Ребиндера — Щукина
(V. 2). Поэтому они не могут быть получены самопроизвольным дис-
159
пергированием как лиофильные системы; для их образования должна
быть затрачена внешняя энергия.
Укрупнение частиц дисперсной фазы при потере агрегативной устой-
чивости достигается в результате изотермической перегонки (раство-
рение мелких и рост крупных частиц в соответствии с уравнением Кель-
вина) или за счет слипания (слияния) частиц — коагуляции. Наиболее
распространен процесс коагуляции. В зависимости от природы системы
и концентрации дисперсной фазы этот процесс может заканчиваться
или осаждением частиц, или структурообразованием.
^Лиофобные дисперсные системы характеризуются кинетической аг-
регативной устойчивостью, определяемой скоростью процесса коагуля-
ции. Кинетика коагуляции описывается уравнением Смолуховского:
1 + 7<Vot или ^=T+ve <VLl)
где Vv — суммарное число частиц дисперсной фазы ко времени т; vo — первоначаль-
ное число частиц; 9 = l/(/Cv0)—время половинной коагуляции; К — константа ско-
рости коагуляции.
Константа К определяется соотношением
Е = Еб/>ехр^ или К р ехР (VI. 2)
где Кб — константа скорости быстрой коагуляции; Р — стерический множитель, учиты-
вающий благоприятные пространственные расположения частиц при столкновении;
— энергия взаимодействия частиц, или потенциальный барьер; k — константа Больц-
мана; г| — вязкость дисперсионной среды.
Константа скорости коагуляции К (константа скорости медленной
коагуляции) является мерой кинетической агрегативной устойчивости.
Если ДЕ = 0 и Р = 1, то эта константа равна константе скорости быст-
рой коагуляции, зависящей от вязкости -среды и температуры системы.
Если ДЕ ф 0 и Р 1, то не все соударения частиц эффективны, и про-
исходит медленная коагуляция. Замедление коагуляции, обусловлен-
ное потенциальным барьером, характеризуется фактором устойчиво-
сти, или коэффициентом стабильности:
IV — — р exp J (VI. 3)
При значительном потенциальном барьере может наступить такое
состояние системы, когда скорость агрегации частиц будет равна ско-
рости дезагрегации и система окажется термодинамически устойчивой
к .коагуляции.
Таким образом, агрегативная устойчивость коллоидных систем обус-
ловливается термодинамическими и кинетическими факторами. Термо-
динамические факторы, действие которых направлено на снижение
поверхностного натяжения и увеличение энтропии, уменьшают вероят-
ность эффективных соударений между частицами, создают потенциаль-
ные барьеры. Кинетические факторы снижают скорость столкновения
частиц и связаны в основном с гидродинамическими свойствами си-
стемы.
Различают следующие факторы устойчивости (стабилизации) дис-
персных систем.
1. Электростатический фактор (термодинамический); заключается
в уменьшении поверхностного натяжения вследствие возникновения
двойного электрического слоя на поверхности частиц в соответствии
160
с уравнением Липпмана. 2. Адсорбционно-сольватный фактор (термо-
динамический) ; состоит в уменьшении поверхностного натяжения в ре-
зультате взаимодействия частиц с дисперсионной средой (уравнение
Дюпре) или благодаря адсорбции стабилизаторов (адсорбционное
уравнение Гиббса). 3. Энтропийный фактор (термодинамический); про-
является в стремлении дисперсной фазы к равномерному распределе-
нию по объему системы под действием теплового движения. 4. Струк-
турно-механический фактор (кинетический); связан с тем, что на раз-
рушение пленок, образующихся на поверхности частиц и обладающих
упругостью и механической прочностью, требуется энергия и время.
5. Гидродинамический фактор (кинетический); заключается в сниже-
нии скорости движения частиц при изменении вязкости и плотности
дисперсионной среды. 6. Смешанные факторы наиболее характерны для
реальных систем; агрегативная устойчивость обеспечивается действием
нескольких факторов одновременно.
Каждому фактору устойчивости соответствует специфический метод
его нейтрализации. Например, электростатический фактор очень чув-
ствителен к введению' электролитов. Действие структурно-механиче-
ского фактора можно предотвратить с помощью веществ, разжижаю-
щих упругие структурированные слои на поверхности частиц, а также
механическим, термическим способами и др.
Устойчивость дисперсных систем определяется балансом энергии
притяжения и энергии отталкивания частиц. Энергия притяжения обус-
ловлена межмолекулярными силами, главным образом силами Ван-
дер-Ваальса. В первом приближении эта энергия обратно пропорцио-
нальна квадрату расстояния между частицами^Ьо теории ДЛФО (Де-
рягина, Ландау, Фервея, Овербека), учитывающей только электроста-
тическую составляющую расклинивающего давления (давления оттал-
кивания), энергия отталкивания убывает с расстоянием по экспонен-
циальному закону.
Для области малых электрических потенциалов суммарная энергия
взаимодействия частиц (пластин) равна
\ и (й) = 2г0^1е-^ - -^2 (VI. 4)
.----
где <р6—электрический потенциал диффузного слоя; х—величина, обратная тол-
щине диффузного слоя; А* — константа Гамакера; й — расстояние между частицами
(пластинами); е — диэлектрическая проницаемость дисперсионной среды; g0 — электри-
ческая постоянная.
При больших потенциалах и расстояниях между частицами (пла-
стинами) эта энергия ^определяется уравнением
(VI. 5>
где со — концентрация противоионов в дисперсионной среде; у — постоянная, опреде-
ляемая величиной <рв-
Суммарная потенциальная энергия взаимодействия частиц отри-
цательна на близких и далеких расстояниях (преобладает энергия
притяжения). Она может быть положительна на средних расстояниях
(преобладает энергия отталкивания). Максимум потенциальной кривой
72Ц Зак. 673 161
или замораживании системы.
Рис. 46. Зависимость энергии взаимодействия
U двух частиц от расстояния между ними h.
(рис. 46) отвечает потенциальному
барьеру Д£. Первый минимум / соот-
ветствует непосредственному сопри-
косновению частиц, а второй II-—при-
тяжению частиц, между которыми
имеются прослойки среды.
Коагуляция лиофобных дисперс-
ных систем может происходить в ре-
зультате различных внешних воздей-
ствий, например при механическом
воздействии (ультразвука), действии
электрического поля, при нагревании
Коагуляция лиофобных золей может
быть вызвана также их сильным разбавлением или концентрирова-
нием. Наиболее часто коагуляция дисперсных систем происходит при
добавлении электролитов. Различают два типа электролитной ко-
агуляции коллоидных систем: 1) нейтрализационную, происходящую
в результате снижения поверхностного потенциала частиц; 2) концен-
трационную, протекающую вследствие сжатия диффузной части двой-
ного электрического слоя (потенциал поверхности в этом случае не
изменяется).
Нейтрализационная электролитная коагуляция характерна для кол-
лоидных систем, содержащих слабо заряженные частицы. Концентра-
ционная коагуляция обычно наблюдается в сильно заряженных дис-
персных системах.
Введение электролитов снижает высоту потенциального барьера
(см. рис. 46), но при небольших концентрациях электролита энергети-
ческий барьер остается достаточно велик и коагуляции частиц не про-
исходит. Агрегация наступает при введении определенного для данной
системы количества электролита, соответствующего порогу коагуляции.
Порог быстрой коагуляции ск определяет количество электролита, не-
обходимое для коагуляции единицы объема коллоидной системы при
полном исчезновении потенциального барьера А£. При сохранении не-
большого потенциального барьера в системе протекает медленная ко-
агуляция.
При электролитной коагуляции по концентрационному механизму
(для сильно заряженных частиц) порог коагуляции ск в соответствии
с правилом Дерягина — Ландау (обоснование эмпирического правила
Шульце — Гарди) обратно пропорционален заряду z противоионов в
шестой степени, т. е.
При нейтрализационной коагуляции (при малых потенциалах по-
верхности сро частиц) показатель степени при z в уравнении (VI. 6)
уменьшается до двух (правило Эйлерса — Корфа).
162
Работа 26. СИНТЕЗ ГИДРОЗОЛЯ ГИДРОКСИДА ЖЕЛЕЗА,
ИЗУЧЕНИЕ ЕГО КОАГУЛЯЦИИ И СТАБИЛИЗАЦИИ
Цель работы: синтез гидрозоля гидроксида железа конденсационным
методом; определение порога электролитной коагуляции золя и изуче-
ние зависимости его от заряда коагулирующего иона; определение за-
щитного числа стабилизатора (высокомолекулярного соединения).
Гидрозоль гидроксида железа синтезируют методом конденсации
путем проведения реакции гидролиза хлорида железа при 100 °C:
FeCl3 + 3H2O —> Fe(OH)3 + ЗНС1
Реакция гидролиза FeCl3 идет интенсивно с образованием высоко-
дисперсных нерастворимых в воде частиц Fe(OH)3.
Агрегативная устойчивость золя гидроксида железа обеспечивается
прежде всего наличием на поверхности дисперсных частиц двойных
электрических слоев. Элементарная частица такого золя называется
мицеллой. В основе мицеллы лежит нерастворимый в данной диспер-
сионной среде агрегат, состоящий из множества молекул (атомов):
[Fe(OH)3]ra, где п — число молекул (атомов), входящих в агрегат.
Поверхность агрегата может заряжаться благодаря избирательной
адсорбции ионов из дисперсионной среды или диссоциации молекул
в поверхностном слое агрегата. В соответствии с правилом Пескова —
Фаянса адсорбируются преимущественно ионы, входящие в состав аг-
регата, либо специфически взаимодействующие с ним. Ионы, сообщаю-
щие агрегату поверхностный заряд, называются потенциалопределяю-
щими. Заряженный агрегат составляет ядро мицеллы. При данном ме-
тоде получения золя гидроксида железа ядро [Fe(OH)3]„-mFe3+ имеет
положительный поверхностный заряд за счет адсорбции ионов Fe3+ из
среды (т — число адсорбированных ионов). Заряд ядра компенсиру-
ется эквивалентным зарядом противоположно заряженных ионов —
противоионов, расположенных в объеме среды. Противоионы, находя-
щиеся непосредственно у поверхности ядра (на расстояниях, близких
к диаметрам ионов), помимо электростатических сил испытывают силы
адсорбционного притяжения поверхности. Поэтому они особо прочно
связаны с ядром мицеллы и носят название противоионов адсорбцион-
ного слоя (их число т — х). Остальные противоионы составляют диф-
фузно построенную ионную оболочку и называются противоионами
диффузного слоя (их число соответствует х).
Мицелла гидрофобного золя является электронейтральной. Формулу
мицеллы ионостабилизированного золя гидроксида железа можно запи-
сать следующим образом:
f { [Fe(OH)3]„ mFe3+ • 3 (т - х) СГ} ЗхСГ
/ агрегат потенциал- противоионы ионы диф-
/ определяющие плотного фузного слоя
ионы слоя
ядро мицеллы
коллоидная частица
мицелла
В формуле мицеллы границы коллоидной частицы обозначены фи-
гурными скобками. Толщина адсорбционного слоя 6 мала (<1 нм)’
‘АН*
163
и постоянна. Толщина диффузного слоя X существенно больше, (может
быть >10 нм)~п сильно зависит от концентрации электролитов в си-
стеме [см. уравнение (III. 16)]. При высоких концентрациях электро-
литов или при введении многозарядных ионов ионный фактор х ста-
новится большим, а толщина Х= 1/х мала и стремится к нулю. В пре-
деле X = 0 и частица незаряжена. В этом случае противоионы адсорб-
ционного слоя полностью компенсируют поверхностный заряд ядра.
В результате достигается так называемое изоэлектрическое состояние,
часто сопровождающееся потерей агрегативной устойчивости системы.
Написанная выше формула мицеллы является только качественной,
она позволяет судить о структуре поверхностных ионных слоев, но не-
пригодна для количественной характеристики состава коллоидных
частиц.
В процессе коагуляции высокодисперсного золя гидроксида железа’
образуются сравнительно небольшие по размерам седиментационно
устойчивые агрегаты. Поэтому исследование коагуляции частиц Fe(OII)3
удобнее всего проводить с помощью турбидиметрического метода (см.
работу 17). Применимость этого метода основывается на сильной зави-
симости интенсивности светорассеяния от размеров частиц, itplr'коагу-
ляции частиц она повышается, соответственно увеличивается оптиче-
ская плотность золя. Поскольку при прохождении светового потока
через окрашенные золи часть света рассеивается, а часть поглощается,
то при изучении коагуляции в таких системах методом турбидиметрии
необходимо исключить поглощение света. Для золя Fe(OH)3 этого
можно достичь, проводя измерения при красном светофильтре, т. е. при
длине волны падающего света X = 620—625 нм.
Порог быстрой коагуляции находят по пороговому объему электро-
лита VK (мл), при котором оптическая плотность золя достигает мак-
симального значения, а при дальнейшем добавлении электролита не
изменяется (рис. 47). Значение ск рассчитывают по формуле
где Сэл — концентрация введенного электролита, моль/л; V — объем золя, мл.
Для предотвращения агрегации частиц и защиты гидрозолей и гид-
росуспензий от коагулирующего действия электролитов применяют
высокомолекулярные соединения и коллоидные ПАВ, растворимые в
воде, например белки, мыла, крахмал, декстрин. Их стабилизирующее
действие основано на образовании на поверхности частиц дисперсной
фазы адсорбционных гелеобразных пленок и связано как с уменьше-
нием межфазного натяжения, так и со структурно-механическими свой-
ствами поверхностных слоев.
Защитная способность полимеров или ПАВ относительно выбран-
ного золя характеризуется защитным числом S — количеством веще-
ства, требуемого для стабилизации единицы объема золя. Защитное
число S, как и порог коагуляции ск, определяют методом турбидимет-
рии. Защитное число S (г/л золя) вычисляют по уравнению
5 = _Ссфащ. (VI ,8)
где с(г—концентрация раствора стабилизатора, г/л; V3au>объем раствора стабили-
затора. необходимый для предотвращения коагуляции золя, мл.
164
Рис. 47. Зависимость оптической плотности D золя от объема электролита-коагуля-
тора V,.,.
Рис. 48. Зависимость оптической .плотности D золя от объема раствора стабилиза-
тора Уст-
Значение Узащ соответствует объему стабилизатора в золе, содер-
жащем пороговый объем VR электролита, при котором на кривой за-
висимости D = f(VCT) появляется нижний горизонтальный участок
(рис. 48).
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
ФотоэлектрОколориметр типа ФЭК-56М.
Электрическая плитка.
Коническая колба емкостью 250 мл.
Пробирки емкостью 20 мл.
Бюретки емкостью 25 мл и градуированные пипетки.
2 %-ный (масс.) раствор хлорида железа (III).
0,00125 М раствор сульфата натрия.
0,5М раствор ацетата натрия.
0,01 %-ный (масс.) раствор желатины.
Для получения гидрозоля Fe(OH)3 в колбу с 250 мл кипящей ди-
стиллированной воды наливают 10 мл раствора хлорида железа. Обра-
зовавшийся золь, красно-коричневого цвета, охлаждают до комнатной
температуры.
Далее исследуют коагуляцию золя гидрбкеида железа при введении
в него растворов сульфата натрия или ацетата натрия путем измерения
оптической плотности полученных систем.
В 10 пробирок наливают по 10 мл золя, воду и электролит (раствор
Na2SO4 или CH3COONa) в следующих объемах:
Номер пробирки . . 1
Объем воды, мл . . . 10,0
Объем электролита
Уэл, МЛ ...... 0
2 3 4 5
9,0 8.5 8,0 7,5
1,0 1,5 2,0 2,5
6 7 8 9 10
7,0 6,5 6,0 5,5 5,0
3,0 3,5 4,0 4,5 • 5.0
165
Таблица VI. 1. Результаты исследования коагуляции золя
гидроксида железа оптическим методом
Объем электролита
Уэл, мл
Оптическая плотность золя D
электролит NaCHsCOO
электролит Na2$O4
Электролит вводят в каждую пробу золя за 2—4 мин непосред-
ственно перед измерением ее оптической плотности.
Измеряют оптическую плотность золя в каждой колбе с помощью
фотоэлектроколориметра с применением светофильтра № 8 или № 9.
Методика измерения оптической плотности приведена в работе 17.
Полученные данные записывают в таблицу (см. табл. VI. 1).
Строят график зависимости £> = ДУЭл) для Na2SO4 и CH3COONa,
по нему находят пороговые объемы электролита VK, вызывающие
быструю коагуляцию золя, и по формуле (VI. 7) рассчитывают значе-
ния ск. Сравнивают найденные значения ск для Na2SO4 и CH3COONa
и объясняют их в соответствии с правилом Шульце — Гарди [законом
Дерягина — Ландау, см. уравнение (VI. 6)].
Затем определяют защитное число полимера — желатины относи-
тельно золя Fe(OH)3. Для этого готовят 10 проб, наливая в пробирки
золь и растворы в следующем объеме и последовательности:
Номер пробирки . . . .
Объем золя, мл ....
Объем воды, мЛ . . . .
Объем раствора жела-
тины Уст, мл............
Объем электролита, мл
1 2 3 4 5 6 7 8
10 10 10^ 10 10 10 10 10
^)До 2ф мл^читывД! , z
/ *' \ и sJ^Jpo^jjjst'a х
9
10
10
10
я обърмкб зойя, рд^твора. желдти-ны
и эЯекгоолп-Га х '
5,0 4,0 3,5 3,0 2,5 2,0 1,5 1,0 0,5 0
в объеме, соответствующем Гк
Как и при исследовании коагуляции, общий объем проб должен
быть одинаковым и составлять 20 мл. Электролит-коагулятор добав-
ляют через 10—15 мин после введения желатины (для адсорбции же-
латины на частицах золя). Оптическую плотность золя измеряют че-
рез 3—5 мин после введения электролита. Значения D записывают
в таблицу (см. табл. VI. 2).
Строят график зависимости £> = /(Гст). Находят объем раствора
желатины Узащ, необходимый для предотвращения коагуляции золя, и
по формуле (VI. 8) рассчитывают защитное число S.
Таблица VI. 2. Экспериментальные данные для определения
защитного числа желатины
№ пробирки
Объем раствора желатины
VcT> мл
Оптическая плотность золя D
166
Работа 27. ИССЛЕДОВАНИЕ КИНЕТИКИ КОАГУЛЯЦИИ ЛАТЕКСОВ
Цель работы: изучение кинетики коагуляции латексов электролитами
с одно- и двухвалентными катионами; определение порога быстрой
и медленной коагуляции; расчет фактора стабильности и энергетиче-
ского барьера отталкивания; расчет константы скорости быстрой ко-
агуляции и сравнение ее значения с теоретической величиной.
Кинетику электролитной коагуляции латексов, являющихся «белы-
ми» золями, удобно исследовать оптическим методом.
Оптическую плотность латекса, в котором протекает электролитная
коагуляция, можно выразить уравнением
I 24л3 / га? — га? \2 v-л
---------I —!-----) v V
2,3 X4 \n2 + 2n2J m ‘
4 I и ' т=\
(VI. &)
где Z — длина слоя латекса, рассеивающего свет; Л — длина волны падающего света;
/ц п0 — показатель преломления соответственно дисперсной фазы и дисперсионной
среды; vm, Vm — численная концентрация и объем m-мерных частиц.
Учитывая, что по теории кинетики коагуляции
vo (r/9)m~i 1
=--------- и у —--------------
(1 + т/е)т+1 Kv0
получаем следующее выражение для оптической
Vm
(VI. 10)
из уравнения (VI. 9)
плотности латекса:
E> = Av0Vf(l + Kvot)
(VI. И)
где vo — численная концентрация исходного латекса (до коагуляции); Vi = c/(pv0)—
объем частиц в исходном латексе (одномерных); с — массовая концентрация золя;
р — плотность вещества дисперсной фазы; А—константа, равная
/ 24л3 / п2 — п2 \2
2,3 л4 V + 2гао /
По мере развития процесса коагуляции латекса размеры частиц
возрастают, поэтому рассеяние света будет меньше, чем это следует
из уравнения (VI. И). Однако оптическая плотность латекса в началь-
ный период коагуляции линейно зависит от времени процесса т.
Дифференцируя по времени уравнение (VI. 11), получаем:
= (VI. 12)
В этом уравнении К — константа медленной коагуляции.
Отсюда можно получить уравнение для линейного участка кинети-
ческой кривой коагуляции:
которое позволяет найти константу коагуляции по зависимости опти-
ческой плотности от времени коагуляции.
Теоретическую константу быстрой коагуляции можно рассчитать по
уравнению
ЛЬТ
^б = -о— (VI. 14)
d4
167
Рис. 49. Зависимость (dD/dT)x_>0 от концентра-
ции электролита с:л в золе.
Фактор стабильности латекса нахо-
дят как отношение W — Кб/К.
Найденное таким образом значение
фактора стабильности дает возможность
оценить потенциальный барьер \Е (сте-
рический множитель принимается рав-
ным Р « 1):
Д5 = _ ЦТ In Г (VI. 15)
С ростом концентрации электролита в золе потенциальный барьер
уменьшается, скорость коагуляции частиц возрастает, соответ-
ственно этому увеличивается значение (djD/dr)t->o- Это позволяет по
графической зависимости (clD/dr^^ от концентрации электролита сЭл
определять порог быстрой коагуляции ск золя при условии, если W — 1
и К = Кб (рис. 49).
Для изучения кинетики электролитной коагуляции латексов исполь-
зуют оптический метод, определяя оптическую плотность серии проб
латексов после введения в них электролита. Оптическую плотность из-
меряют с помощью установки, состоящей из фотоэлектроколориметра
типа KF (см. примечание в работе 10), усилителя измерительной схе-
мы и автоматического самопишущего потенциометра КСП-4.
С помощью этой установки можно измерить оптическую плотность
двумя способами: в автоматическом непрерывном режиме фиксирова-
ния измеряемой величины или путем периодического измерения ее
(через определенные интервалы времени).
Порядок работы с прибором KF следующий. При втором способе
измерений вилку 1 (рис. 50) устанавливают в положение, обозначен-
ное «нормаль». Переключатель 2 устанавливают в положение «0» и
прибор включают в сеть. В гнездо 6 вставляют светофильтр.
В находящийся под крышкой 4 кюветодержатель помещают две
кюветы: слева'—кювету с растворителем (водой), справа — кювету с
исследуемым образцом латекса. С помощью рукоятки 7 кюветодержа-
тель устанавливают в положение, при котором поток света проходит
через кювету, запол-
ненную водой. Пере-
ключатель 2 переводят
в положение
После этого стрелку
измерительного при-
Рис. 50. Общий вид фото-
электроколориметра типа
KF:
1 — вилка; 2 — переключатель ;
3 — рукоятка грубого регулиро-
вания чувствительности; 4—
крышка отсека; 5 — рукоятка точ-
ного регулирования чувствитель-
ности; 6—гнездо светофильтра;
7—рукоятки кюветоде.ржателя;
8 — шкала измерительного прибо-
ра.
168
бора 8 устанавливают на нулевое значение (по шкале оптической плот-
ности) . Для этого рукоятку точной регулировки чувствительности 5 вы-
водят в крайнее правое положение (путем поворота ее против часовой
стрелки), а переключатель грубого регулирования 3 устанавливают в
таком положении, при котором стрелка измерительного прибора уходит
вправо от нулевого значения шкалы. Затем, вращая рукоятку 5 по ча-
совой стрелке, устанавливают стрелку измерительного прибора 8 на
нулевое деление.
Для измерений оптической плотности образца рукоятку 7 переме-
щают в левое положение до упора и тем самым на пути светового по-
тока устанавливают правую кювету с исследуемым раствором. Значе-
ния оптической плотности отсчитывают по показаниям стрелки измери-
тельного прибора 8.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Установка для измерения и регистрации оптической плотности.
Кюветы емкостью 10 мл, 2 шт.
Секундомер.
Пипетки емкостью 1 и 5 мл.
Шприц емкостью 1 мл.
Полистирольный латекс, 0,1 %-ный (масс.).
Электролиты: 5Л1 раствор NaCl и 0,5М раствор ВаСЬ.
Часть 1. Исследование влияния электролитов с одно- и двухзарядными
коагулирующими катионами на кинетику коагуляции латекса
Исследуют две серии проб, содержащих 5 мл латекса, воду и электро-
лит (в одной серии — хлорид натрия, в другой — хлорид бария):
Номер пробы........... ~ 1
Объем воды, мл ... . 4,5
Объем электролита, мл 0,5
2 3 4 5 6
4,4 4,3 4,2 4,1 4,0
0,6 0,7 0,8 0,9 1,0
Концентрация латекса во всех пробах равна 0,05 % (масс.), объем
каждой пробы 10 мл. Способ измерения оптической плотности указы-
вается преподавателем. Измерение D проводят, используя светофильтр
с длиной волны Хвак = 510—520 нм.
Сначала измеряют оптическую плотность латекса без добавления
электролита. Для этого в правую кювету фотоэлектроколориметра на-
ливают 5 мл исходного латекса (с = 0,1 %) и 5 мл воды и проводят
измерение, как указано выше.
Затем измеряют оптическую плотность латекса в присутствии элек-
тролитов. Для этого в кювету вводят латекс исходной концентрации
объемом 5 мл и воду от 4,0 до 4,7 мл (в зависимости от объема элек-
тролита, добавляемого затем в латекс). Кювету устанавливают в кюве-
тодержатель и с помощью рукоятки 7 помещают ее в световой поток.
Затем, оставляя переключатель 2 в положении «~-», в кювету с по-
мощью шприца или пипетки быстро вводят точно отмеренный объем
электролита. При работе по второму способу включают секундомер и
регистрируют D латекса через каждые 15 с в течение 3—4 мин.
Результаты, полученные при исследовании коагуляции латекса под
действием электролита, записывают в таблицу (см. табл. VI. 3).
11 Зак. 673
169
Таблица VI. 3. Данные по исследованию кинетики коагуляции латекса
оптическим методом
Номер пробы Электролит Концентрация электролита, моль/л Время после введения электролита т, с Оптическая плотность D в момент % \ du
По полученным результатам строят графики зависимости D — f(r)'
при разных концентрациях электролитов. На первых линейных участ-
ках определяют (dDldx)x^a (при всех значениях концентраций электро-
литов). По найденным значениям (dD/dx)x^a строят график зависимо-
сти (dD/dx)x^o — f (сэл) для каждого электролита и по нему определяют
пороги быстрой коагуляции латекса электролитами с одно- и двухза-
рядными коагулирующими катионами. Рассчитывают отношение най-
денных значений порогов быстрой коагуляции cKi/cK2-
Рассчитанное отношение сопоставляют с отношением порогов быст-
рой коагуляции, которое следует из правила Дерягина — Ландау
(правила Шульце — Гарди).
По значениям (с/Р/й'г)т >0 при cKj и ск2 с помощью уравнения
(VI. 13) рассчитывают константы скорости коагуляции К латекса ис-
следуемыми электролитами. Экспериментально найденные значения К.
сравнивают с теоретическими значениями, рассчитанными по уравне-
нию (VI. 14).
Часть 2. Изучение влияния электролитов
на кинетические параметры коагуляции латекса
Экспериментально исследуют зависимость оптической плотности ла-
текса при разных концентрациях хлорида натрия (меньше порогов
быстрой коагуляции). Методику работы см. в части 1. Результаты запи-
сывают в таблицу (см. табл. VI. 3).
По уравнениям (VI. 3), (VI. 13) — (VI. 15) рассчитывают константу
скорости коагуляции К, фактор стабильности IV и потенциальный
барьер Строят зависимость \Е — }(сэл). Объясняют полученные
результаты.
Часть 3. Изучение влияния степени адсорбционной насыщенности
частиц молекулами ПАВ на кинетику коагуляции латекса
Для выполнения этой части работы определяют пороги быстрой коагу-
ляции латексов с различной степенью адсорбционной насыщенности
ПАВ (содержащих различные количества ПАВ на поверхности час-
тиц). Используют латексы со степенью адсорбционной насыщенности
0,- — 0,2; 0,5 и 0,8. В качестве электролита-коагулятора используют
5 М раствор хлорида натрия. Для каждого образца латекса (с опреде-
ленным значением 0г) исследуют серию проб, содержащих 5 мл исход-
ного латекса, воду и электролит:
Номер пробы ..... 1 2 3 4 5 б 7 8
Объем воды, мл ... . 4,7 4,6 4,5 4,4 4,3 4,2 4,1 4,0
Объем электролита, мл 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
170
Измерения оптической плотности проводят по методике, описанной
в первой части работы. Результаты опытов записывают в таблицу,
аналогичную табл. VI. 3.
По полученным кинетическим данным D =/(сэл) для каждого об-
разца латекса определяют порог быстрой коагуляции ск и константу
скорости процесса Ц. Методика определения ск и К описана в первой
части работы.
По экспериментально найденному значению константы скорости
коагуляции К и теоретически рассчитанной константе скорости быст-
рой коагуляции Кб латекса вычисляют значения факторов замедле-
ния W и анализируют полученные экспериментальные и расчетные ре-
зультаты.
Работа 28. ПОЛУЧЕНИЕ ЭМУЛЬСИЙ И ИЗУЧЕНИЕ ИХ СВОЙСТВ
Цель работы, получение эмульсии, определение ее типа и изучение ее
устойчивости; получение обратной эмульсии.
Различают два основных типа эмульсий — дисперсии масла в воде
(м/в) и дисперсии воды в масле (в/м). Первые относятся к эмульсиям
прямого типа, вторые — к эмульсиям обратного типа.
В зависимости от содержания дисперсной фазы эмульсии классифи-
цируют на разбавленные [содержание дисперсной фазы ф менее 1 %
(об.)], концентрированные [ф до 74 % (об.)] и высококонцентрирован-
ные [ф свыше 74 % (об.)].
Эмульсии — типично лиофобные дисперсные системы (за исключе-
нием самопроизвольно возникающих «критических» эмульсий). Потеря
их агрегативной устойчивости может быть обусловлена процессами
изотермической перегонки или коагуляции (коалесценции капель) и
обычно сопровождается потерей седиментационной устойчивости (рас-
слоение системы). В качестве меры устойчивости эмульсии можно при-
нять время существования определенного объема эмульсии до полного
ее расслоения.
Устойчивость эмульсии повышают введением в систему стабилиза-
тора (эмульгатора), в качестве которого можно использовать электро-
литы, ПАВ и высокомолекулярные соединения. Агрегативная устойчи-
вость эмульсий определяется теми же факторами, которые обусловли-
вают устойчивость к коагуляции других лиофобных дисперсных си-
стем.
Разбавленные эмульсии могут быть достаточно устойчивы в присут-
ствии таких слабых эмульгаторов, как электролиты. Устойчивость
таких эмульсий связана в основном с наличием двойного электриче-
ского слоя на частицах дисперсной фазы. Устойчивость концентриро-
ванных и высококонцентрированных эмульсий в большинстве случаев
определяется действием структурно-механического барьера при образо-
вании адсорбционных слоев эмульгатора. Характерно, что образую-
щиеся межфазные адсорбционные слои обусловливают плавное изме-
нение свойств переходной зоны на границе раздела двух жидких фаз,
увеличивая лиофильность частиц дисперсной фазы. Наиболее сильное
стабилизирующее действие оказывают высокомолекулярные соедине-
ния и коллоидные ПАВ (мыла, неионогенные ПАВ), адсорбционные
слои которых имеют гелеобразную структуру и сильно гидратированы.
11»
171
Тип эмульсии, образующейся при механическом диспергировании,
в значительной мере зависит от соотношения объемов фаз. Жидкость,
содержащаяся в большем объеме, обычно становится дисперсионной
средой. При равном объемном содержании двух жидкостей при дис-
пергировании возникают эмульсии обоих типов, но «выживает» из них
та, которая имеет более высокую агрегативную устойчивость и опре-
деляется природой эмульгатора. Способность эмульгатора обеспечи-
вать устойчивость эмульсии того или иного типа определяется энерге-
тикой взаимодействия его с полярной и неполярной средами, которая
может быть охарактеризована при помощи полуэмпирической харак-
теристики— числа гидрофильно-липофильного баланса (ГЛБ) поверх-
ностно-активных веществ. ПАВ, имеющие низкие значения ГЛБ
(2—6), лучше растворимы в органических средах и стабилизируют
эмульсии в/м, тогда как при ГЛБ = 12—18 ПАВ лучше растворяются
в воде и стабилизируют эмульсии м/в.
Щелочные соли жирных кислот средней молекулярной массы всегда
дают эмульсии типа м/в, а соли двухвалентных металлов, например
магния, — эмульсии в/м. При постепенном увеличении концентрации
двухвалентных ионов в эмульсии м/в, стабилизированной мылом с ка-
тионом однозарядного металла, происходит обращение эмульсии и ее
переход в эмульсию типа в/м.
Особый случай представляет стабилизация эмульсий высокодис-
персными порошками. Такая стабилизация возможна при ограничен-
ном избирательном смачивании порошков (при краевом угле 0 больше
0°, но меньше 180°). При этом порошки лучше стабилизируют ту фазу,
которая хуже смачивается. Так, гидрофильный мел «бронирует» мас-
ляную фазу и не позволяет коалесцировать каплям масла в водной
дисперсионной среде. Таким образом, краевой угол, характеризующий
избирательное смачивание, при объяснении стабилизации эмульсий
тонкодисперсными порошками является аналогом ГЛБ молекул ПАВ.
На практике тип эмульсий определяют следующими методами. По
методу разбавления каплю эмульсии вносят в пробирку с водой. Если
капля равномерно распределяется в воде, — это эмульсия м/в. Капля
эмульсии в/м диспергироваться в воде не будет. Согласно методу окра-
шивания непрерывной фазы несколько кристаллов водорастворимого
красителя, например, метилового оранжевого или метиленового синего
окрашивают эмульсию м/в равномерно по всему объему. Эмульсия в/м
равномерно окрашивается по всему объему жирорастворимым краси-
телем. Тип эмульсий можно определить также по ее электропроводно-
сти (метод электропроводности). Высокие значения электропроводно-
сти указывают на то, что дисперсионной средой является полярная
жидкость, а эмульсия относится к типу м/в. Малые значения электро-
проводности показывают на образование обратной эмульсии (типа
в/м).
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Микроизмельчитель с регулятором напряжения.
Цилиндры емкостью 250 мл.
Бюретка емкостью 50 мл.
Пипетки емкостью 10 мл.
172
Стаканы емкостью 50 мл.
Конические колбы емкостью 50 мл.
Пробирки.
Вазелиновое масло, техническое.
Раствор ПАВ, например 0,1 М раствор олеата натрия или додецил-
сульфата натрия.
0,5М раствор MgCl2 или Mg(NO3)2.
Красители, например судан I или метиленовый синий.
Вариант 1. Получение эмульсии м/в и определение ее стабильности
Готовят растворы ПАВ объемом 40 мл путем разбавления исходного
раствора согласно следующим данным:
Номер колбы ..... 1 2 3 4
Объем раствора ПАВ,
мл 10 20 30 40
Объем воды, мл ... . 30 20 10 —
Затем получают эмульсию с помощью микроизмельчителя (рис. 51).
Цилиндр 2 емкостью 250 мл устанавливают в держатель 1. Поднимают
держатель с цилиндром до упора ограничителя, чтобы мешалка 3 во-
шла в цилиндр. Передвигая держатель с цилиндром, устанавливают
мешалку по центру цилиндра. Бюретку 4 закрепляют в штативе и на-
ливают в нее вазелиновое масло. В цилиндр 2 наливают 12,5 мл рас-
твора ПАВ из колбы 1.
Микроизмельчитель подключают к регулятору напряжения 6. Тумб-
лер 5, расположенный на основании микроизмельчителя, устанавливают
в положение «5000 об/мин».
Регулятор напряжения включают в сеть и устанавливают напря-
жение 130 В. В цилиндр 2 из бюретки подают 37,5 мл масла в течение
10 мин. После введения всего-масла продолжают перемешивать еще
10 мин.
Таким же образом готовят эмульсии с другими растворами ПАВ
(колбы 2—4).
Сразу после получения эмульсии
10 мл ее переливают из цилиндра
в пробирку и определяют время рас-
слоения ее на две макрофазы.
Остальную часть эмульсии пере-
носят в стакан и определяют ее тип
следующим образом.
1. Каплю эмульсии и каплю во-
ды помещают на предметное стекло
и стекло наклоняют так, чтобы кап-
ли пришли в соприкосновение. Если
капли сольются, то дисперсной фа-
зой является вода, если не сольют-
ся — масло. 2. На фильтровальную
Рис. 51. Установка для получения эмуль-
сий:
1 — держатель; 2 — цилиндр; 3 — мешалка; 4—
бюретка; 5 — тумблер; 6— регулятор напряже-
ния.
173
Таблица VI. 4. Экспериментальные результаты определения
типа эмульсии и ее устойчивости
Концентрация ПАВ, моль/л Концентрация эмульсии, % (об.) Время расслоения, мин Тнп эмульсии
бумагу наносят каплю эмульсии. Если средой является вода, то капля
сразу всасывается бумагой, на которой остается жирное пятно. Капля
эмульсии в/м не всасывается. 3. Эмульсию разливают в две пробирки.
В одну пробирку добавляют несколько капель красителя метиленового
синего, растворимого в воде, в другую несколько крупинок Судана I,
растворимого в масле. Эмульсия м/в окрашивается в синий цвет (су-
дан I в ней не растворяется). Эмульсия в/м окрашивается в красный
цвет (метиленовый синий в ней не растворяется).
Полученные данные записывают в таблицу (см. табл. VI. 4).
Вариант 2. Получение эмульсии обратного типа
Используя исходный 0,144 раствор ПАВ, получают эмульсию вазели-
нового масла в воде в соответствии с методикой, указанной в вари-
анте 1 работы.
Часть эмульсии (15 мл) переносят в стакан для определения ее
типа (методика определения типа эмульсии приведена в варианте 1).
В оставшуюся часть эмульсии при перемешивании вносят 5 мл 0,5 М
раствора MgCl2 или Mg(NO3)2. По окончании введения всего раствора
электролита перемешивание продолжают еще 5 мин.
Переносят 10 мл эмульсии в пробирку и измеряют время расслое-
ния ее на две фазы. Оставшуюся часть эмульсии используют для опре-
деления ее типа.
Полученные результаты записывают в таблицу, аналогичную
табл. VI. 4 (указывают тип эмульсии до и после введения в нее рас-
твора электролита). Анализируют полученные данные.
Работа 29. ПОЛУЧЕНИЕ ПЕН И ИЗУЧЕНИЕ ИХ УСТОЙЧИВОСТИ
Цель работы: получение пены и изучение, влияния ПАВ, высокомолеку-
лярных соединений и электролитов на ее устойчивость.
Как и все дисперсные системы, пены получают методами дисперги-
рования и конденсации. Методом диспергирования пены получают
посредством перемешивания или барботирования газов в жидкость.
Конденсационный метод основан на изменении физического состояния
раствора (при повышении температуры раствора или уменьшении внеш-
него давления), приводящем к пересыщению его газом.
Первой стадией процесса пенообразования является образование газовой эмуль-
сии (эмульсии газ — раствор ПАВ). На межфазной поверхности пузырьков (рис. 52, а)
образуется адсорбционный слой ПАВ. При флокуляции пузырьков на поверхности
раствора формируется пленочный каркас пены, характеризующийся тем, что прослойки
жидкости между адсорбционными слоями ПАВ на пузырьках пены взаимосвязаны,
благодаря чему образуется единая структура.
174
Структура пены определяется соотношением объемов газовой и жидкой фаз, и в
зависимости от этого соотношения пены могут иметь сферическую форму ячейки (ша-
ровая пена), полиэдрическую или переходную ячеистую.
Шаровая пена образуется, если объем жидкой фазы Гж превышает объем газовой
фазы Vr более чем в 10—15 раз. Пленки пузырьков этой пены имеют относительно
большую толщину. Чем меньше отношение Гж/Гг, тем меньше толщина пленки. По
мере старения пены пленки утончаются и шаровая пена превращается в полиэдриче-
скую.
Структура полиэдрических пен описывается геометрическими правилами Плато.
Три пузырька, стенки которых встречаются под углом 120°, образуют механически
устойчивую систему. При их соединении пленки, разделяющие их, образуют трехгран-
ный столбик жидкости, называемый каналом Плато—Гиббса, который играет важную
роль в механизме утончения пленок. Большая кривизна поверхности в области кон-
такта трех пузырьков приводит к значительному перепаду давлений между газовой
и жидкой фазами, в результате жидкость выдавливается из пленки в канал Плато—
Гиббса. Поскольку стенки всех пузырьков должны быть одинаковыми, то в одной
точке (узле) сходятся четыре канала Плато—Гиббса, образуя между собой углы
109° 28'.
Используя геометрические правила Плато, можно предсказать наиболее вероятную
форму ячейки пены. Она представляет собой пентагональный додекаэдр (рис. 52,6)—
фигуру, ограниченную 12 пятиугольными гранями. Характерно, что поверхность пен-
ных пленок в этом случае не имеет кривизны. Состояние такой пены близко к равно-
весному, и поэтому она наиболее устойчива.
Для шаровых пен различают седиментационную и агрегативную устойчивость. По
мере превращения шаровой пены в связную, полиэдрическую, понятие седиментацион-
ной устойчивости применительно к ней теряет обычный смысл.
Агрегативная устойчивость пен характеризуется скоростью укруп-
нения частиц дисперсной фазы за счет коалесценции и изотермической
перегонки. Стабилизация пен достигается с помощью ПАВ. В зависи-
мости от природы ПАВ и свойств образуемых ими адсорбционных
слоев, устойчивость пен обусловливается действием общих для дис-
персных систем факторов стабилизации (ионно-электростатический,
структурно-механический барьер и др.) и специфическим для пен и
эмульсий эффектом Гиббса — Марангони.
Эффект Гиббса — Марангони заключается в следующем. Тонкие
пленки, содержащие ПАВ, способны реагировать на локальные изме-
Узлы
а в
Рис. 52. Элемент шаровой пены из трех пузырьков (а) и элементарная ячейка поли-
эдрической пены (6).
175
нения толщины пленки. Течение жидкости в поверхностных слоях
пленки приводит к уносу ПАВ и, следовательно, к увеличению поверх-
ностного натяжения. Это вызывает возникновение двухмерного давле-
ния, направленного в сторону, обратную течению (эффект Марангони).
Вместе с тем увеличение ст повышает упругость пленки, препятствую-
щую механической деформации пленки (эффект Гиббса). Поэтому в
присутствии ПАВ утончение пленок происходит только по механизму
вытекания жидкости между адсорбционными слоями ПАВ. При малой
толщине зазора этот процесс идет со сравнительно малой скоростью.
В пенах (особенно в шаровых) кривизна отдельных участков не-
одинакова. Следовательно, жидкость в пленках и каналах находится
под различным давлением. В результате происходит разрушение пены.
Разрушение пены происходит по трем механизмам: а) вытекание жид-
кости из пены (синерезис), обусловливающее утончение пленок без
изменения объема пены; б) укрупнение больших ячеек пены и исчез-
новение маленьких из-за диффузии газа через пленки; в) разрыв пле-
нок, приводящий к разрушению пены. Преобладание одного или дру-
гого механизма зависит от многих факторов.
Свойства пен обычно характеризуют следующими параметрами:
1) кратностью — отношением объема пены к объему раствора, пошед-
шего на образование пены; 2) стабильностью — временем существова-
ния элемента пены (пузырька, пленки) или определенного объема пе-
ны; 3) дисперсностью — распределением пузырьков по размерам, или
средним размером пузырьков.
Важнейшим свойством пены является пониженное (по сравнению
с атмосферным) равновесное капиллярное давление в каналах Плато —
Гиббса ра, определяемое высотой столба пены h:
Pa = PL + PSh (VI. 16)
где pi — давление в нижнем слое пены; р — плотность раствора.
Капиллярное давление измеряют с помощью прибора, изображен-
ного на рис. 53. Установка состоит из U-образного водяного мано-
метра 1, цилиндра с поршнем 2 (шприца) и капиллярной трубки 3 с прива-
ренной пористой мембраной 4. На
трубке 3 на высоте 10 см от мембраны
нанесена метка 5.
Измерение проводят следующим
образом. Трубку 3 опускают в стакан
с раствором ПАВ на глубину 0,2—
0,5 см и при помощи поршня 2 уро-
вень мениска раствора в капиллярной
трубке 3 совмещают с меткой. Значе-
ние пониженного давления, регистри-
руемое манометром 1, является нача-
лом отсчета р0. Затем стакан с рас-
твором ПАВ заменяют стаканом с
Рис. 53. Установка для измерения капилляр-
ного давления в каналах Плато — Гиббса:
1 — манометр; 2— поршень шприца; 3 —капиллярная
трубка; 4— пористая мембрана; 5 — мет^а.
176
пеной и трубку 3 опускают в пену на глубину I см. При сопри-
косновении с пеной из-за пониженного давления в пенных кана-
лах жидкость стремится перейти из трубки 3 в пену, и следовательно,
мениск жидкости в трубке 3 опускается. Перемещая поршень 2, со-
здают дополнительное разрежение над мениском, добиваясь, чтобы
жидкость из капилляра не вытекала в пену, но при этом и не отсасы-
валась из нее. Это давление р регистрируют по манометру 1.
Кинетические измерения давления в пенных каналах проводят до
получения постоянного значения р, соответствующего максимуму утон-
чения пленки, поскольку в процессе старения пен в результате их утон-
чения кривизна пенных каналов увеличивается, а давление в каналах
Плато — Гиббса уменьшается. Равновесное капиллярное давление ра
находят как разность давлений р и р0.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Прибор для определения поверхностного натяжения.
Установка для измерения капиллярного давления в пене.
Пеногенератор, например сосуд с барботером.
Стеклянная кювета.
Колбы емкостью 100 мл.
Стаканы емкостью 150 мл.
Пипетки емкостью 10 мл.
1 %-ный (об.) раствор пенообразователя, например САМПО — смесй
вторичных алкилсульфатов и высших жирных спиртов.
1,5 %-ный (масс.) раствор полимера, например желатины.
0,01 М раствор электролита, например хлорида калия.
Часть 1. Исследование влияния концентрации ПАВ на давление
в пенных каналах
Из исходного раствора пенообразователя готовят растворы (по 20 мл)
концентрации [в % (об.)]: 0,5, 0,25, 0,125, 0,0625 и определяют их
поверхностное натяжение (методика измерения приведена в работе 1).
Из приготовленных растворов ПАВ получают пены. Для этого часть
раствора ПАВ наливают в пеногенератор, который подсоединяют
к водоструйному насосу, и барботируют через раствор воздух. Полу-
ченные пены помещают поочередно в стеклянную кювету и определяют
среднее значение диаметра ячеек пен d (как отношение длины кюветы
к числу ячеек пены, укладывающихся по длине кюветы).
Из растворов ПАВ снова готовят пены и для каждой из них изме-
ряют равновесное давление в каналах Плато — Гиббса ра по методике,
приведенной выше.
Таблица VI. 5. Экспериментальные и расчетные данные исследования пен
Концентрация пенообразователя, СПАВ’ % (°б-> Поверхностное натяжение о, мДж/м2 d, м Давление, Па R.t м
Ро р ро
177
Экспериментально определенное давление в пенных каналах ра
можно представить как разность избыточного давления в каналах
Плато — Гиббса и в пузырьке:
= (VI-17)
1\ и
где R — радиус кривизны пенных каналов.
Зная ра и d, рассчитывают радиус кривизны каналов Плато —
Гиббса R. Полученные данные записывают в таблицу (см. табл. VI. 5).
Строят графики зависимостей <г = /(спав) и р0 — /(спав) и анализируют
полученные результаты.
Часть 2. Исследование влияния электролита на давление
в пенных каналах
Готовят растворы пенообразователя (по 20 мл), как указано в части
1 работы, и добавляют к ним по 2 мл 0,01 М раствора КС1. Определяют
поверхностное натяжение полученных растворов.
Находят среднее значение диаметра ячейки пены d и давление
в пенных каналах ра для всех приготовленных растворов (методика
измерений описана выше). По уравнению (VI. 17) рассчитывают ра-
диус пенных каналов R. Экспериментальные и расчетные данные запи-
сывают в таблицу, аналогичную табл. (VI. 5). Строят графики зависи-
мостей о =/(спав) и =/(спав) и анализируют полученные резуль-
таты.
Часть 3. Исследование влияния высокомолекулярных веществ
на давление в пенных каналах
Готовят растворы пенообразователя (по 20 мл), как указано выше,
и в них добавляют по 0,2 мл 1,5 %-ного (масс.) раствора желатины.
Растворы тщательно перемешивают и определяют их поверхностное
натяжение.
Находят средний диаметр ячеек пены d и давление в пенных кана-
лах ра по уравнению (VI. 17). Рассчитывают радиус каналов Плато —
Гиббса R. Экспериментальные и расчетные данные записывают в таб-
лицу, аналогичную табл. VI. 5. Строят графики зависимостей о —
= /(спав) и ра = /(спав) и анализируют полученные результаты.
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАЧИ
КОНТРОЛЬНЫЕ ВОПРОСЫ
jj Назовите виды устойчивости дисперсных систем в соответствии
с классификацией Пескова. В чем заключается различие между лио-
фильными и лиофобными коллоидными системами?
2. Чем обусловлена агрегативная неустойчивость лиофобных дис-
персных систем? Какие процессы самопроизвольно происходят в этих
системах?
3. Какими методами получают лиофобные дисперсные системы?
Приведите примеры.
178
4. На что затрачивается работа при дроблении и измельчении ма-
териалов? Каким образом можно уменьшить работу измельчения и по-
высжь дисперсность измельчаемого материала?
5 Чем отличаются процессы гомогенной и гетерогенной конденса-
ций" и каковы причины возникновения метастабильного состояния в пе-
ресшценных системах?
(ддЧем определяется критический радиус зародыша новой фазы?
Как можно регулировать размеры частиц лиофобных дисперсных сис-
тем, получаемых методом конденсации?
7. Какой процесс называют коагуляцией? Чем завершается процесс
коагуляции? Какими способами можно вызвать коагуляцию лиофобной
коллоидной системы?
8. Что называют быстрой и медленной коагуляцией? Какова взаи-
мосвязь между скоростью коагуляции и видом потенциальной кривой
взаимодействия частиц?
9. Какие параметры дисперсной системы влияют на скорость коагу-
ляции частиц в соответствии с теорией Смолуховского? Чем отличаются
константы скорости быстрой и медленной коагуляции?
10. Каково различие между нейтрализационной и концентрацион-
ной коагуляцией лиофобных золей электролитами? Как влияет заряд
коагулирующего иона на порог быстрой коагуляции?
11. Действием каких факторов обеспечивается агрегативная устой-
чивость лиофобных дисперсных систем? Какие вещества используют
в к^естве стабилизаторов этих систем?
W Что такое расклинивающее давление и каковы причины его
возникновения? Назовите составляющие расклинивающего давления.
13. Какие составляющие расклинивающего давления рассматривает
теория устойчивости ДЛФО? Приведите примеры потенциальных кри-
вых взаимодействия между частицами для дисперсных систем с раз-
личной степенью устойчивости. Каковы особенности коагуляции частиц
в первом и вторичном энергетических минимумах в соответствии с тео-
рией ДЛФО?
14. В чем заключается сходство и различие суспензий и лиозолей?
15. Как классифицируют эмульсии? Какие вещества используют
в качестве стабилизаторов прямых и обратных эмульсий?
16. В чем заключаются особенности стабилизации пен? Какими па-
раметрами характеризуют устойчивость пен?
17. Как классифицируют аэрозоли? В чем причины принципиальной
агрегативной неустойчивости этих систем?
18. ‘Приведите примеры практического использования суспензий,
лиозолей, эмульсий, пен и аэрозолей.
ПРИМЕРЫ РЕШЕНИЯ ЗАДАЧ
1. Экспериментально получена зависимость общего числа частиц гидрозоля золота
в 1 м3 Vv от времени коагуляции т, вызванной электролитом NaCl:
т, с................... 0 125 250 375 425
v2-10-14, част./м3 .... 20,2 8,08 5,05 3,67 3,31
Вязкость среды т| = 1 -103 Па-с, температура 293 К.
Покажите применимость уравнения Смолуховского для описания кинетики коагу-
ляции данного золя. Рассчитайте время половинной коагуляции 0 и константу ско-
рости быстрой коагуляции Kq. Сравните значение константы скорости быстрой коагу-
ляции, рассчитанной теоретически, с экспериментальной величиной.
179
Рис. 54. К определению времени половинной коагуляции 0 золя.
Рис. 55. Зависимость энергии электростатического отталкивания U3, энергии молеку-
лярного притяжения Ua и суммарной энергии U взаимодействия частиц от расстояния
между ними.
Решение. Кинетика изменения общего числа частиц в теории Смолуховского
представлена уравнением (VI. 2):
_ 'Vo
1 + т/0
1 4kT
где время половинной коагуляции 0=—гг-, Kjeop = ——
Уравнение Смолуховского можно представить в виде
Рассчитываем значения при различном времени коагуляции:
т, с.................... О 125 250 375 425
v0/v2................... 1 2,5 4,0 5,5 6,1
Строим график зависимости от т (рис. 54). Как следует из графика, экспе-
риментальные данные соответствуют линейной зависимости, что указывает иа приме-
нимость уравнения Смолуховского. Из графика находим:
375
0 = ctg а — -=-=--— = 83,33 с
5,5 — 1
Тогда
С = ^0" = 20,2 • 10'4 • 83,33 = 5,94 ’ 10 М
= 4-1,38-10^-293 = 539.10-18 м3/с
° Зт) 3 • 10 3
Значения констант Кдксп и Кдео₽ близки, следовательно, коагуляция гидрозоля золота
является быстрой.
180
2. Рассчитайте и постройте потенциальную кривую взаимодействия сферических
частиц диаметром 200 нм в водном растворе NaCl, если потенциал <₽о = 20 мВ, кон-
станта Гамакера А* = 0,5-10~19 Дж, параметр х — 1-Ю8 м~* и температура 293 К.
Значения суммарной энергии взаимодействия частиц определите при расстояниях ме-
жду их поверхностями h = 2, 5, 10, 20 и 40 нм.
_ Решение, Согласно теории устойчивости ДЛФО для слабозаряженных поверхно-
стей и малых расстояний h (h 50 нм) суммарная энергия взаимодействия между
двумя частицами радиусом г рассчитываетсгупо уравнению
и = иэ + ии = 2ле0е/ф|11п (1 + e~Kh) -
где U3 — энергия электростатического отталкивания частиц; U..,— энергия их молеку-
лярного притяжения; 8о — электрическая постоянная; 8 — относительная диэлектриче-
ская проницаемость среды.
Находим значение U при h = 2 нм:
= 2 • 3,14 • 8,85 • 10“12 (817'1 • 10~7 • (2 • 10-2)2 • In (1 + е“012) = 10,77 • 1О~20 Дж
U ‘ = - ^•10~19-1-;°~7 = - 20,83 • IO"20 Дж
12-2- 10 '
U = 10,77 -10 20 — 20,83 -10 20 = — 10,06 - 10 20 Дж
Аналогично рассчитываем энергию взаимодействия частиц при др у-
гих расстояниях h:
Л, нм хД e-’t'i In (1 + е M,t) Уэ-1020, Дж УМ.1О20. Дж У-1О20. Дж
5 0,5 0,606 0,474 8,54 —8,33 0,21
10 1,0 0,368 0,313 5,64 —4,16 1,48
20 2,0 0,135 0,126 2,28 —2,08 0,20
40 4,0 0,018 0,018 0,32 — 1,04 —0,72
По данным приведенных расчетов строят график зависимостей U3,
UM, U от h (рис. 55).
ЗАДАЧИ 7'4
I. Золь Agl получен при добавлении 8 мл водного раствора KI кон-
центрацией 0,05 моль/л к 10 мл водного раствора AgNO3 концентра-
цией 0,02 моль/л. Напишите формулу мицеллы образовавшегося золя.
Как заражена частица золя? Каким методом можно определить этот
заряд?
2. Золь гидроксида железа(III) получен при добавлении к 85 мл
кипящейДдистиллированной воды 15 мл 2 %-ного раствора хлорида
железа(III). Напишите формулу мицелл золя Fe(OH)3, учитывая, что
при образовании частиц гидроксида железа(III) в растворе присут-
ствуют следующие ионы Fe3+, СП. Как заряжены частицы золя?
3. В воздухе, содержащем пары воды, образуется туман при темпе-
ратуре 269 К, когда коэффициент пересыщения становится равным
3,71. Рассчитайте критический размер ядер конденсации и число моле-
кул, содержащихся в них. Поверхностное натяжение воды 76,4 мДж/м2,
плотность воды 1 г/см3.
4. Рассчитайте концентрацию силиката натрия, находящегося в рас-
творенном состоянии в равновесии с аморфными частицами (удельная
поверхность частиц 3-105 м2/кг) гидрозоля кремнезема при 298 К- Рас-
творимость кремнезема в равновесии с макрофазой при этой же тем-
пературе составляет 0,015 % (масс.). Плотность частиц SiO2 равна
181
2,2 г/см3, поверхностное натяжение на границе кремнезема с раство-
ромг45 мДж/м2.
Рассчитайте энергию Гиббса образования зародыша критического
размера в пересыщенном растворе кремниевой кислоты,' полученной из
водного, раствора силиката натрия с помощью иоцного обмена. Поверх-
ностное натяжение на границе кремнезема с водой примите равным
45 мДж/м2. Коэффициент пересыщения раствора равен 3. Плотность
частиц 1,8 г/см3.
6. Гидрозоль кремнезема, полученный из силиката натрия путем
ионного обмена, после термической обработки содержит аморфные
частицы плотностью 2,2 г/см3. Оцените поверхностное натяжение крем-
незема на границе с водным раствором, если растворимость при 298 К
макрофазы составляет 0,015 % (масс.), а частиц с удельной поверх-
ностью 8-Ю4 м2/кг равна 0,016 % (масс.).
7. Время половинной коагуляции тумана минерального масла с
удельной поверхностью 1,5-107 м-1, концентрацией 25 мг/л составляет
240 с. Рассчитайте и постройте кривую изменения суммарного числа
частиц при коагуляции для следующих интервалов времени: 60, 120,
240, 480 и 600 с. Плотность масла 0,970 г/см12 3.
8. При изучении оптическим методом кинетики электролитной ко-
агуляции гидрозоля Agl, стабилизированного ПАВ, получено значение
константы скорости быстрой коагуляции, равное 3,2-10~!8 м3/с (при
293 К). Вязкость среды 1-10-3 Па-с. Сравните эту константу с констан-
той, даваемой теорией Смолуховского. Объясните влияние ПАВ на ха-
рактер коагуляции.
> 9. По экспериментальным данным время половинной коагуляции
гидрозоля составляет 340 с при исходной частичной концентрации час-
тиц 2,52-1014 част./м3, вязкости дисперсионной среды 1 • 10~3 Па-с и
температуре 293 К. Сделайте вывод, быстрой или медленной является
коагуляция. Как изменится скорость коагуляции, если вязкость среды
увеличить в 3 раза?
10. Во сколько раз уменьшится суммарное число частиц vs дыма
мартеновских печей через 1, 10, 100 с после начала коагуляции? Сред-
ний радиус частиц 20 нм, концентрация 1•10_3 кг/м3, плотность частиц
2,2 г/см3. Константа быстрой коагуляции, по Смолуховскому, равна
3-10Д6 м3/с.
(Гр Рассчитайте константу быстрой коагуляции золя серы под дей-
ствием хлорида алюминия, используя следующие экспериментальные
данные:
т, с.................. 0 2 4 10
10~17, част./м3 . . . 16,0 0,99 0,50 0,20
12. Рассчитайте время половинной коагуляции аэрозоля с дисперс-
ностью 0,25 нм-1 и концентрацией 1,5-10~3 кг/м3, если константа быст-
рой коагуляции, по Смолуховскому, равна 3-10-16 м3/с. Плотность час-
тиц аэрозоля примите равной 2,2 г/см3.
13. Рассчитайте время половиной коагуляции, используя экспери-
ментальные данные по изменению общего числа частиц при коагуляции
лиофобной дисперсной системы в воде:
т. с.................... О 7,0 15,0 20,2 28,0
• 10"15, част./м3 ... 32,2 24,2 19,9 16,7 14,2
182
Рассчитайте и постройте кривые изменения числа первичных и
Двойньтх частиц во времени.
Проверьте применимость теории Смолуховского к коагуляции
золя селена раствором хлорида калия, используя следующие экспери-
ментальные данные:
т, с................ О 0;66 4,25 19,00 43,00
v2-10“14, част./м3. . . 29,70 20,90 19,10 14,40 10,70
15. Рассчитайте число первичных частиц гидрозоля золота при ко-
агуляции электролитом к моменту времени т=150 с, если первона-
чальное число частиц в 1 м3 составляет vo = 1,93-1014, а константа ско-
рости быстрой коагуляции равна 0,2 • 10-17 м3/с.
16. Рассчитайте константу скорости быстрой коагуляции суспензии
каолина в воде по данным кинетики коагуляции, полученным с по-
мощью ультрамикроскопа (при 293 К):
т, с................ 0 100 175 250 400 500
Vv-10'“, част./м3. . . 5,00 3,78 3,23 2,86 2,22 1,95
17. При изучении коагуляции суспензии бентонитовой глины в воде
методом счета частиц в ультрамикроскопе получены следующие
данные:
т, с...................... 335 510 600 800
v2-10~14, част./м3. . . 2,52 1,92 1,75 1,49
Исходное число частиц в золе vo = 5-lO14 част./м3. Проверьте приме-
нимость уравнения Смолуховского для описания данных по кинетике
коагуляции. Рассчитайте время половинной коагуляции и число частиц
2-, 3-, 4-го порядка к моменту времени т = 800 с.
18. Пользуясь уравнением Смолуховского, рассчитайте и постройте
кривую изменения общего числа частиц при коагуляции гидрозоля
серы. Дисперсность исходного золя 0,05 им-', концентрация 6,5 мг/л,
плотность серы 0,9 г/см3. Вязкость дисперсионной среды при 295 К со-
ставляет 10-3 Па-с. Интервалы времени возьмите равными 1, 2, 4, 10
и 20 с.
19. Рассчитайте и постройте графическую зависимость энергии при-
тяжения для плоскопараллельных пластин в водной среде от расстоя-
ния между ними, изменяющегося от 5 до 100 нм. Константу молекуляр-
жх сил Гамакера примите равной 2-10-20 Дж.
• ,20. Рассчитайте энергию притяжения двух толстых параллельных
пластин в водной среде, если расстояния между поверхностями равны 5,
10, 15, 20 и 50 нм и постройте график зависимости энергии от рас- .
стояния. Константу молекулярных сил' Гамакера примите равной
2-10-20 Дж.
21. Рассчитайте и постройте графическую зависимость энергии при-
тяжения сферических частиц полистирола, находящихся в водной сре-
де, от расстояния между поверхностями частиц, которое изменяется
от 2 до 20 нм. Радиус частиц равен 50 нм, константа Гамакера А* —
= 0,5-10-20 Дж.
22. Рассчитайте и постройте графическую зависимость энергии
электростатического отталкивания двух толстых плоскопараллельных
183
пластин в водном растворе KCI от расстояния между поверхностями, из-
меняющегося от 5 до 150 нм. При расчете используйте следующие
данные: температура 300 К, фл = 0,02 В, концентрация электролита
/ 2-10_3 моль/л, диэлектрическая проницаемость среды 77,8.
23. Рассчитайте энергию электростатического отталкивания U3 двух
. плоскопараллельных пластин в водном растворе КД при расстояниях
/ между поверхностями 5, 10, 20, 30 и 50 нм и постройте графическую
/ зависимость U3 — f(h). Потенциал диффузного слоя <p6 = 3-10~2 В,
I концентрация раствора электролита с0 = 1 ммоль/л, температура рас-
• твора 293 К, диэлектрическая проницаемость среды 80,1.
24. Рассчитайте и постройте графическую зависимость энергии
электростатического отталкивания двух плоскопараллельных пластин
от концентрации NaCl в водном растворе, изменяющейся от 1 до
10 ммоль/л. При расчете примите расстояние между пластинами 20 нм;
потенциал диффузного слоя <р6 = 3-10_2 В; температура 298 К, диэлек-
трическая проницаемость среды 78,2.
25. Рассчитайте энергию отталкивания Ua сферических частиц ди-
оксида кремния диаметром 20 нм в водных растворах NaCl при рас-
стояниях между поверхностями частиц 1, 2, 5, 10 и 15 нм. Постройте
график зависимости U3 = f(h) при концентрациях электролита в рас-
творе Ci ^=5-10—4 и с2 = 5'10-2 моль/л. Примите равными потенциал
диффузного слоя <р6 = 4-10~2 В, температуру раствора 293 К, диэлек-
трическую проницаемость 80,1.
26. Постройте график энергии взаимодействия плоскопараллельных
пластин большой толщины в водном растворе одно-одновалентного
электролита по следующим данным: потенциал диффузного слоя <рв =
= 1 • 10”2 В, Со = 1 ммоль/л, константа Гамакера А* = 1,25-10~20 Дж,
диэлектрическая проницаемость среды 80,1. Значения энергии взаимо-
действия рассчитайте для расстояний между поверхностями пластин 5,
10, 20, 30 и 50 нм при 293 К.
27. Постройте потенциальную кривую взаимодействия двух плоско-
параллельных пластин, находящихся в водном растворе одно-однова-
,5 лентного электролита, при значении потенциала диффузного слоя <р6 =
= 0,02 В. Рассчитайте суммарную энергию взаимодействия U = U3 +
+ UM при расстояниях между поверхностями пластин, равных 5, 10, 20,
30 и 50 нм, используя следующие данные: константа Гамакера А* ~
= 5-10~20 Дж, концентрация электролита 9-10_4 моль/л, температура
273 К, диэлектрическая проницаемость среды 87,8.
‘ 28. Рассчитайте суммарную энергию взаимодействия (U - LL, 4-
+ U„) двух пластин, находящихся в водном растворе NaCl концентра-
ции 4 ммоль/л, при расстояниях между их поверхностями 5, 10, 15, 30
и 60 нм. Используйте следующие данные: <р6 = 0,03 В, константа Га-
макера А* = 3,5-10~20 Дж, температура 300 К, диэлектрическая прони-
цаемость 77,8. По полученным данным постройте график зависимости
29. Рассчитайте и постройте потенциальную кривую взаимодействия
сферических частиц диаметром 200 нм в водном растворе NaCl по сле-
дующим данным: потенциал <рв = 20 мВ, константа Гамакера А* =
= 0,5-10-19 Дж, параметр х = 1-108 м-1 и температура 293 К. Значения
суммарной энергии взаимодействия частиц определите при расстояниях
между поверхностями 2, 5, 10, 20 и 40 нм.
184
30, При исследовании коагуляции полистирольного латекса полу-
чены следующие значения порогов коагуляции:
Электролит . . . NaCl СаС12 А1С13
Порог коагуляции,
моль/л......... 0,4'7 8,8 •10“3 6-10-4
Рассчитайте соотношение порогов коагуляции и сопоставьте его с со-
отношением, получаемым в соответствии с правилом Дерягина —•
Ландау.
Порог коагуляции отрицательно заряженного гидрозоля As2S3
под действием КС1 равен 4,9-10-2 моль/л. С помощью правил Шуль-
це— Гарди и Дерягина — Ландау для этого золя рассчитайте пороги
коагуляции, вызываемой следующими электролитами: K2SO4, MgCl2,
MgSO4, AICI3 и A12(SO4)3.
32. Порог коагуляции положительно заряженного гидрозоля
Fe(QH)3 под действием электролита NaCl равен 9,25 ммоль/л. С по-
мощью правил Шульце—Гарди и Дерягина — Ландау для этого золя
рассчитайте пороги коагуляции, вызываемой следующими электроли-
тами: KNO3, ВаС12, K2SO4, MgSO4, КгСг2О7.
33. Порог коагуляции гидрозоля металлического золота, вызывае-
мой NaCl, равен 24 ммоль/л, а Кг8О4—11,5 ммоль/л. Используя пра-
вила Шульце — Гарди и Дерягина — Ландау, определите знак заряда
золя и рассчитайте порог коагуляции для следующих электролитов:
СаС12, MgSO4, A12(SO4)3, А1С13, Th(NO3)4.
VII. СТРУКТУРНО-МЕХАНИЧЕСКИЕ СВОЙСТВА
ДИСПЕРСНЫХ СИСТЕМ
Разбавленные агрегативно устойчивые дисперсные системы не обра-
зуют пространственной сетки из частиц дисперсной фазы (структуры),
и поэтому их реологические свойства близки или подобны свойствам
дисперсионной среды. Зависимость вязкости таких систем от концен-
трации дисперсной фазы описывается уравнением Эйнштейна:
Г) — По ( 1 + «<₽) ИЛИ Цуд = — — = аф (VII. 1)
По
где Т]о — вязкость дисперсионной среды; <р — объемная доля дисперсной фазы; а — ко-
эффициент формы частиц (для сферических частиц а = 2,5); т]уд — удельная вязкость.
Уравнение Эйнштейна соблюдается для дисперсных систем, течение
которых подчиняется закону Ньютона (ньютоновские жидкости):
Р = т)-^- = т)у (VII. 2)
где Р — напряжение сдвига; у и у — соответственно деформация и скорость деформа-
ции (течения).
С увеличением концентрации дисперсной фазы возрастает взаимо-
действие между частицами и обнаруживаются все более сильные от-
клонения от уравнения Эйнштейна. Вязкость концентрированных си-
стем растет с концентрацией почти по экспоненте. Одновременно
12 Зак. 673
185
Рис. 56. Кривые течения ньютоновской жидкости
(7) и псевдопластической структурированной
жидкообразной системы (2).
наблюдается зависимость вязкости от на-
пряжения сдвига (нагрузки), и закон
Ньютона перестает выполняться. Эти от-
клонения обычно обусловлены взаимо-
действием частиц и образованием струк-
туры, в которой частицы определенным
образом ориентированы относительно
друг друга. Такие системы называют не-
ньютоновскими жидкостями.
Наиболее общим уравнением, описы-
вающим течение неньютоновских жидкостей, является эмпирическое
уравнение Оствальда — Вейля:
Р = kyn ИЛИ = 1
(VII. 3)
где k и п—постоянные, характеризующие данную жидкообразную систему.
Если п=1, жидкость является ньютоновской и константа k совпа-
дает с ньютоновской вязкостью ц. Таким образом, отклонение п от
единицы характеризует степень отклонения свойств неньютоновских
жидкостей от свойств ньютоновских жидкостей. При п < 1 ньютонов-
ская вязкость уменьшается с увеличением скорости сдвига и напряже-
ния. Соответственно этому жидкости называют псевдопластическими.
При п > 1 ньютоновская вязкость растет с увеличением скорости
сдвига и напряжения. Соответственно эти жидкости называют дила-
тантными.
На рис. 56 представлена кривая течения структурированной жидко-
образной системы — реальной псевдопластической жидкости (кривая 2).
Для сравнения приведена зависимость у=Д(Р) для ньютоновской жид-
кости (прямая 1). На кривой течения структурированной псевдопла-
стической жидкости имеется три характерных участка. На участке ОА
эти система ведет себя подобно ньютоновской жидкости с большой вяз-
костью гамаке = etg ой. Такое поведение системы объясняется тем, что
при малых скоростях течения структура, разрушаемая приложенной
нагрузкой, успевает восстанавливаться. Медленное течение с постоян-
ной вязкостью без прогрессирующего разрушения структуры называется
ползучестью.
Для слабо структурированных систем начальный прямолинейный
участок кривой обычно небольшой, и его практически невозможно об-
наружить. Для сильно структурированных систем область значений Р,
при которых наблюдается ползучесть, может быть весьма значительной.
Напряжение Рк соответствует началу разрушения структуры.
При дальнейшем увеличении напряжения (участок АВ) зависи-
мость y = f(P) теряет линейный характер, при этом вязкость (ньюто-
новская) уменьшается. Переменные значения вязкости являются след-
ствием разрушения структуры. В точке В кривой течения структура
системы практически полностью разрушена. Напряжение, отвечающее
этой точке, называется предельным напряжением на сдвиг Рт. При
186
напряжениях Р > Рт, когда структура разрушена, система течет по-
добно ньютоновской жидкости, имеющей вязкость i]MHH = ctga2.
Рост взаимодействия между частицами приводит к упрочнению про-
странственной структуры в дисперсных системах. Жидкообразное тело
переходит в твердообразное. Образование структуры обычно связывают
с появлением у системы предела текучести РТ — минимальной нагрузки,
при которой тело начинает течь. Чем прочнее структура, тем выше
предел текучести.
Для идеального пластического твердого тела Бингам предложил
следующее уравнение:
Р — Рт = ЦплУ ИЛИ р = Рт + 'ПплУ (VII. 4)
где т|пл — пластическая вязкость, характеризующая способность структуры к разру-
шению при изменении нагрузки.
Различия между ньютоновской и пластической вязкостью видны из
соотношения между ними:
Реальные пластические- тела могут отличаться по реологическим
свойствам от идеальных бингамовских тел. Общим для них является
следующее эмпирическое соотношение:
р = + (VII. 6)
В соответствии с уравнением (VII. 6) при п = 1 система представ-
ляет собой бингамовское тело; п > 1 отвечает пластическому дила-
тантному телу и п < 1 — псевдопластическому твердообразному телу.
Тип и свойства структур, образующихся в коллоидных системах,
зависят от характера сил взаимодействия между частицами. Согласно
теории структурообразования все структуры в коллоидных системах
разделяются на два типа: коагуляционные и конденсационно-кристал-
лизационные. В основу этой классификации положена потенциальная
кривая взаимодействия частиц, вытекающая из теории ДЛФО (см.
рис. 46).
Коагуляционные структуры возникают за счет ван-дер-ваальсовых
сил притяжения частиц и образуются в результате коагуляции их на
расстояниях, отвечающих вторичному минимуму на потенциальной
кривой, когда между частицами дисперсной фазы имеются прослойки
среды. Наличие таких прослоек в местах контакта между частицами
обусловливает относительно небольшую прочность и ярко выраженные
пластические свойства структур. Для коагуляционных структур харак-
терны такие специфические свойства, как тиксотропия и реопексия.
Тиксотропия — способность структурированной системы восстанавли-
вать во, времени свои прочностные свойства после ее механического
разрушения." Реопексия — явление, обратное тиксотропйш^~в.озникнове~
ние и упрочнение структуры в результате механического воздействия.
Образованию коагуляционно-тиксотропных структур благоприят-
ствует наличие на поверхности частиц лиофобных участков (мозаич-
ность поверхности), по которым они контактируют. Анизометрия час-
тиц также способствует структурообразованию, так как на концах вы-
тянутых частиц из-за большой кривизны поверхности двойные электри-
ческие и сольватные слои менее развиты.
12*
187
Коагуляция в первичном минимуме приводит к образованию кон-
денсационно-кристаллизационной структуры, в которой частицы дис-
персной фазы связаны химическими связями. В таких структурах час-
тицы не могут двигаться относительно друг друга, и поэтому для них
характерно разрушение без обратимого восстановления. Конденсаци-
онно-кристаллизационные структуры, как правило, обладают высокой
прочностью и проявляют упругие свойства. Пластические свойства их
выражены значительно слабее и связаны, главным образом, с приро-
дой вещества частиц.
Обычно в коллоидных системах образуются структуры смешанного
типа, характеризующиеся преобладанием в первый период коагуляци-
онных контактов и дальнейшим упрочнением связей между частицами
с переходом к структурам конденсационно-кристаллизационного типа.
Такой переход от одних структур к другим часто наблюдается в про-
цессах, сопровождающихся увеличением концентрации системы, на-
пример, при высушивании суспензий или спекании материалов с коагу-
ляционной структурой.
Переход к конденсационным структурам обычно сопровождается
уменьшением пластических свойств и ростом прочности. Большинство
реальных твердых тел неорганической природы имеют кристаллизаци-
онно-конденсационные структуры.
Для типичных твердых тел реологические кривые строят в коорди-
натах напряжение — деформация. При малых напряжениях у них про-
исходят обратимые упругие деформации, за пределом упругости — пла-
стические деформации и затем твердое тело разрушается. Хрупкие тела
(керамика, бетоны, стекло и др.) разрушаются при нагрузках, меньших
предела текучести (предела упругости).
Работа 30. ИССЛЕДОВАНИЕ ВЯЗКОСТИ
СТРУКТУРИРОВАННОЙ ЖИДКОСТИ С ПОМОЩЬЮ
КАПИЛЛЯРНОГО ВИСКОЗИМЕТРА
Цель работы', получение кривых течения для ньютоновской жидкости
(воды) и неньютоновской жидкости (раствора полимера при двух кон-
центрациях); определение предельного напряжения сдвига и вязкости
растворов полимера; построение графиков зависимости вязкости от
нагрузки.
Реологические методы и приборы подразделяют на интегральные и
дифференциальные с учетом «однородности» и «неоднородности» полей
напряжений и деформаций.
Дифференциальные методы дают возможность исследовать непо-
средственно изменение деформации во времени в каждой точке системы
при ее течении, т. е. установить поле деформаций и скоростей дефор-
маций, а иногда и поле напряжений. К таким методам относятся микро-
киносъемка процессов течения, рентгеновское просвечивание и др. Ин-
тегральные методы позволяют наблюдать суммарный эффект течения.
К наиболее применяемым интегральным методам, при которых прояв-
ляется неоднородность полей напряжений и деформаций, относится ка-
пиллярная вискозиметрия, метод внедрения конуса, метод падения
шарика.
Капиллярные вискозиметры используют для исследования реологи-
ческих свойств относительно невязких жидкостей. Разработано боль-
188
Д
Рис. 57. Вискозиметр Уббелоде.
шое число конструкций капиллярных вискозиметров. Наи-
<4 / Л более пригодным для получения реологических кривых
\)\/ является вискозиметр Уббелоде (рис. 57).
'Г - /-3 Вискозиметр Уббелоде представляет собой U-образную
трубку с расположенными на одном уровне небольшими
резервуарами (шариками) одинакового объема. В одно из
колен впаян капилляр, через который исследуемая
жидкость под давлением перетекает из одного шарика в
другой.
Измерение с помощью капиллярного вискозиметра сво-
дится к определению времени вытекания жидкости через капилляр с из-
вестными геометрическими параметрами. При стационарном ламинар-
ном течении жидкости вязкость рассчитывают по уравнению Пуазейля
(получающемуся путем интегрирования уравнения Ньютона):
— = пг* (VII. 7)
т 8г|/
где V — объем жидкости, вытекающей из капилляра радиусом г и длиной I за вре-
мя т; Др — разность давлений на концах капилляра; Г| — вязкость (динамическая)
жидкости.
При работе с вискозиметром Уббелоде объем вытекающей жидко-
сти (равный объему шарика) постоянен. Поэтому при расчетах можно
использовать уравнение Пуазейля в более простом виде:
— = (VII. 8)
Т Г|
где k — константа, характерная для данного вискозиметра.
Измерение вязкости методом капиллярной вискозиметрии проводят
с помощью установки, схема которой приведена на рис. 58. Порядок
работы на ней следующий. В тщательно вымытый вискозиметр с по-
мощью воронки, вставленной в левое колено, наливают исследуемую
жидкость. Жидкость наливают в таком количестве, чтобы ее уровень
доходил примерно до середины шариков (см. рис. 57). Жидкость в пра-
вый шарик прокачивают с помощью резиновой груши. После этого во-
ронку вынимают и вискозиметр помещают в термостат.
После термостатирования на левое колено вискозиметра надевают
резиновую трубку и соединяют его с системой разрежения, как это
показано на рис. 58. В системе создают требуемое разрежение, для
чего используют вакуумный насос. Предварительно проверяют поло-
Рис. 58. Схема установки
для измерения вязкости с
помощью капиллярного ви-
скозиметра:
1,3— двухходовые краны; 2 —
ресивер; 4—моностат; 5—мано-
метр; 6 — трехходовой кран; 7 —
вискозиметр; 8 — термостат.
189
жение кранов установки: краны 1 и 3 должны быть закрыты, а кран
6— в положении «вакуум» (вискозиметр соединен с атмосферой).
Включают насос (на рис. 58 не показан), открывают кран /ив сис-
теме создают требуемое давление, которое контролируют по мано-
метру 5 (давление регулируют с помощью крана 3). Давление можно
снизить, поворачивая трехходовой кран 6 в положение «атмосфера».
Установив нужное давление, краны 1 и 3 закрывают, а насос выклю-
чают.
С помощью резиновой груши засасывают жидкость в вискозиметре
выше отметки а в правом колене (см. рис. 57), а трубку, надетую на
это колено, зажимают пальцами. Переводят кран 6 в положение «вис-
козиметр» (при этом вискозиметр соединяется с системой вакуума) и
тут же разжимают резиновую трубку. Жидкость при этом перетекает
из правого колена вискозиметра в левое. По секундомеру фиксируют
время, в течение которого жидкость проходит расстояние от верхней
отметки а до нижней отметки в, т. е. определяют время истечения по-
стоянного объема жидкости V через капилляр вискозиметра.
После прохождения уровня жидкости через отметку в кран 6 пере-
водят в положение «вакуум», в противном случае жидкость из виско-
зиметра попадает в вакуумную систему.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Установка для измерения вязкости.
Секундомер.
Воронка.
Резиновая груша.
Растворы полимера двух концентраций, например желатины или
карбоксиметилцеллюлозы.
Вискозиметр заполняют дистиллированной водой и определяют вре-
мя истечения т при давлении 50—60 см столба манометрической жидко-
сти. Далее проводят измерения при других давлениях, уменьшая его
каждый раз примерно на 5 см столба жидкости (всего проводят
7—8 измерений).
Аналогичные измерения проводят для растворов полимера, только
для них измерения следует начинать с наименьших давлений (~10—
15 см столба манометрической жидкости).
После окончания работы вискозиметр промывают дистиллирован»
ной водой с помощью водоструйного насоса. Полученные результаты
записывают в таблицу (см. табл. VII. 1).
Таблица VII. 1. Результаты измерения времени истечения
с помощью капиллярного вискозиметра
Вода Раствор полимера концентрации
е1 С2
Др, Па т, с 1/т. с-1 Др, Па т. с 1/т, с—1 т), Па-с Др, Па т, с 1/т. с-1 П. Па-с
190
По данным табл. VII. I строят кривые течения для воды и растворов
полимеров в координатах 1/т — Др и находят для растворов полимеров
предельные напряжения сдвига Рт, Рк и Рт (в Н/м2). Используя спра-
вочные данные о вязкости воды (см. работу 32), по тангенсу угла на-
клона кривой течения для нее находят константу k в уравнении (VII. 8)
и по этому уравнению рассчитывают значения ц растворов полимеров
как функцию Ар. Для растворов полимеров строят графики в коорди-
натах т| — Ар.
Работа 31. ИССЛЕДОВАНИЕ РЕОЛОГИЧЕСКИХ СВОЙСТВ
НЕНЬЮТОНОВСКИХ ЖИДКОСТЕЙ С ПОМОЩЬЮ
РОТАЦИОННОГО ВИСКОЗИМЕТРА
Цель работы: Получение реологических кривых течения и эффектив-
ной вязкости суспензий пылевидного кварца, определение зависи-
мостей предела текучести от концентрации твердой фазы в суспен-
зиях и эффективной вязкости суспензий от вязкости дисперсионной
среды.
Для оценки реологических характеристик дисперсных систем наи-
большее распространение нашли ротационные вискозиметры, которые
характеризуются широкими пределами измерений и высокой воспроиз-
водимостью результатов. Рабочий узел таких вискозиметров чаще
всего представляет собой два коаксиальных цилиндра (кроме комби-
нации цилиндр — цилиндр могут применяться конус — конус, полусфе-
ра— полусфера и т. д.), в зазор между которыми наливается исследуе-
мая жидкость. Один из цилиндров неподвижен, другой приводится во
вращение. -У некоторых типов приборов вращается наружный цилиндр,
а у других — внутренний.
Ротационные вискозиметры работают в режиме либо постоянной
скорости деформации (у = const), либо постоянного напряжения сдви-
га (Р = const). В приборах, работающих в режиме постоянной скоро-
сти деформации, один из цилиндров вискозиметра вращается с посто-
янной скоростью, увлекая за собой исследуемую жидкость, которая,
в свою очередь, приводит во вращение второй (измерительный) ци-
линдр), связанный с динамометрическим устройством. При этом реги-
стрируется изменение крутящих моментов или пропорциональных им
напряжений сдвига.
В приборах с постоянным напряжением сдвига к одному из цилин-
дров прикладывается постоянный крутящий момент, второй цилиндр
при этом неподвижен. Регистрируется скорость вращения подвижного
цилиндра, пропорциональная скорости деформации исследуемой систе-
мы. Ряд конструкций ротационных вискозиметров, работающих в ре-
жиме постоянного напряжения, разработан М. П. Воларовичем. В этих
вискозиметрах вращающийся внутренний цилиндр приводится в движе-
ние через блоки падающими грузами. Скорость деформации и вязкость
определяют в установившемся режиме течения, так как для коллоид-
ных систем ламинарный поток устанавливается не мгновенно, как в
ньютоновских жидкостях, а во времени, что связано с наличием в них
определенной структуры.
В данной работе предлагается использовать ротационный вискози-
метр типа РВ-8М. (рис. 59).
191
Рис. 59. Схема ротационного вискозиметра:
1—лимб; 2 — блок; 3 — стопор; 4— внутренний цилиндр; 5 — внеш
ний цилиндр в термостате; 6 — подъемная площадка; 7 — винт
S —маховик; 9—подъемный механизм; 10 — стопорный винт.
Рабочий узел этого вискозиметра представ-
ляет собой два коаксиальных цилиндра с риф-
леными поверхностями — внутренний 4 и внеш-
ний 5. Внутренний цилиндр (ротор) закреп-
ляется при помощи резьбового соединения на
оси, установленной на подшипниках. На этой
же оси закреплен лимб 1, по кото >ому ведут
отсчет оборотов ротора. К лимбу прикреплена
капроновая нить, пропущенная через блок 2.
На конце нити находится чашечка для груза.
Лимб зафиксирован стопором 3, при опу-
скании которого груз начинает перемещаться,
приводя в движение ротор с отсчетным
лимбом.
Исследуемую суспензию (объемом 10 мл) наливают во внешней
цилиндр вискозиметра, который устанавливают на подъемной площад-
ке 6 и фиксируют винтом 7. Площадку с цилиндром поднимают вверх
до упора вращением маховика 8 подъемного механизма 9 и фиксируют
стопорным винтом 10. При этом торец ротора должен находиться в од-
ной плоскости с торцом внешнего цилиндра, а суспензия должна пол-
ностью заполнять зазор между цилиндрами.
Для расчета вязкости системы определяют скорость вращения ро-
тора путем регистрации времени, необходимого для совершения двух
оборотов лимба в установившемся режиме течения. Стационарный ре-
жим течения устанавливается после 1 —1,5 оборотов ротора (лимба).
Эффективную вязкость суспензии рассчитывают по формуле
Р Pt
г1 = Т = й1П7-
(VII. 9)
где ki — константа прибора (определяется путем калибровки по эталонным жидкостям
с известной вязкостью); N— число оборотов лимба за время т.
Напряжение сдвига находят так:
P = k?F (VII. 10)
где ki — постоянная, зависящая От геометрических размеров внутреннего и внешнего
цилиндров; для используемого вискозиметра ki = 9,25-10~3 Н/(м2-кг); F— масса
груза.
Перед измерением вязкости при другой нагрузке груз поднимают
путем вращения верхней части лимба по часовой стрелке при закры-
том стопоре (ротор при этом не вращается).
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Ротационный вискозиметр и разновесы.
Секундомер.
Стаканы и градуированная пипетка емкостью 10 мл.
192
Металлический шпатель.
Пылевидный кварц.
Раствор водорастворимого полимера, например 1 %-ный раствор
натриевой соли карбоксиметилцеллюлозы.
Часть 1. Исследование влияния концентрации дисперсной фазы
на реологические свойства суспензий
Готовят четыре суспензии пылевидного кварца с различной концентра-
цией твердой фазы:
Номер образца . . . .
Навеска кварца, г . •
Объем раствора поли-
мера, мл..............
Концентрация дисперс-
ной фазы, % (масс.) . .
12 3 4
11 13 14 15
7 7 6 6
60 65 70 72
Суспензии тщательно перемешивают шпателем. Исследуемую сус-
пензию наливают во внешний цилиндр вискозиметра и проводят изме-
рение вязкости, как указано выше, при последовательно увеличиваю-
щихся нагрузках. Нагрузки увеличивают до тех пор, пока продолжи-
тельность двух оборотов лимба вискозиметра не будет составлять
3—4 с (этот предел обусловлен переходом к турбулентному режиму
течения). Для каждого образца суспензии проводят не менее 7—8 из-
мерений.
Рассчитывают скорость деформации y = N/(ki%) и по уравнению
(VII. 9) эффективную вязкость. Полученные результаты записывают
в таблицу (см. табл. VII. 2).
По расчетным данным строят реологические кривые течения у =
= f(P) и эффективной вязкости T) = f(P). По кривым течения опреде-
ляют предел текучести Рт для каждого образца суспензии, строят гра-
фик зависимости Рт от концентрации дисперсной фазы и анализируют
полученные результаты.
Часть 2. Исследование влияния вязкости дисперсионной среды
на реологические свойства суспензий
Из исходного раствора водорастворимого полимера готовят водные рас-
творы по 10 мл с концентрациями 0,4, 0,6, 0,8 % (масс.). Навески квар-
ца по 14 г помещают в 4 фарфоровых стакана и с помощью пипетки
наливают в них по 5 мл исходного и приготовленных растворов поли-
мера. Суспензии тщательно перемешивают шпателем.
Таблица VII. 2. Результаты измерения скорости деформации
и эффективной вязкости суспензий с различной концентрацией
дисперсной фазы
Номер образца Концентрация дисперсной фазы, % (масс.) Р. г Р. Н1м2 т, с N, об. Y, об/с ц, Н-с/м2
193
Таблица VII. 3. Результаты измерения скорости деформации
и эффективной вязкости суспензий с различной вязкостью
дисперсионной среды
помер образца Концентрация полимера, % (масс.) F, г Р, Н/м2 X, с АГ. об- V. об/с ц. Н»с/м2
Исследуемую суспензию наливают во внешний цилиндр вискози-
метра и измеряют вязкости при последовательно увеличивающихся
нагрузках.
По полученным экспериментальным данным рассчитывают скорость
деформаций у и эффективную вязкость г] (порядок расчета указан в
первой части работы). Полученные результаты записывают в таблицу
(см. табл. VII. 3).
По расчетным данным строят реологические зависимости y = f(P)
и T] = f(P). По кривым эффективных вязкостей x\ = f(P) определяют
значения вязкостей Цмакс и цМ1ш, соответствующих неразрушенным и
предельно разрушенным структурам для каждого образца суспензии,
и строят графики их зависимости от вязкости дисперсионной
среды. Вязкость растворов натриевой соли карбоксиметилцеллюлозы
(ЫаКМЦ):
Концентрация
NaKMII, % (масс.) 0,4 0,6 0,8 1,0
Вязкость, Н-с/м2 . . 0,45 0,85 2,0 4.0
Анализируя полученные экспериментальные данные, делают заклю-
чение о влиянии концентрации дисперсной фазы и вязкости дисперси-
онной среды суспензий на процессы структурообразования в системе.
Работа 32. ИЗУЧЕНИЕ ЗАВИСИМОСТИ ВЯЗКОСТИ
РАСТВОРОВ ПОЛИМЕРОВ ОТ ИХ КОНЦЕНТРАЦИИ С ПОМОЩЬЮ
РЕОВИСКОЗИМЕТРА
Цель работы: получение реологических кривых для растворов полиме-
ров при различных концентрациях; определение предельных напряже-
ний сдвига и построение зависимости вязкости раствора от концентра-
ции полимера.
Экспериментально наблюдаемые концентрационные зависимости
удельной вязкости растворов полимеров показывают, что их вязкость,
как правило, не подчиняется закону Эйнштейна (уравнение (VII. 1).
С повышением концентрации растворов т]уд растет более резко по кри-
вой, обращенной выпуклой частью к оси концентрации (рис. 60).
Аномалия вязкости растворов полимеров обусловливается особен-
ностями макромолекул, а также образованием структур в растворе при
увеличении концентрации полимера. Находящиеся в растворе сверну-
тые в клубки макромолекулы всегда удерживают внутри себя некото-
рое количество растворителя. Наличие связанного растворителя при-
водит к увеличению размеров полимерных клубков-частиц и суще-
ственно влияет на вязкость системы.
194
На течение растворов полимеров и их вязкость большое влияние
может оказывать также изменение формы макромолекул. При наложе-
нии внешнего давления возможно распрямление полимерных клубков
и ориентация их по направлению течения. В результате ориентации
макромолекул гидродинамическое сопротивление потоку и вязкость
раствора уменьшаются. При относительно больших концентрациях рас-
творов распрямление и ориентация полимерных молекул затруднены.
Поэтому при повышении концентрации растворов гибкоцепных макро-
молекул вязкость увеличивается более резко, чем предсказывает урав-
нение Эйнштейна.
Концентрационная аномалия вязкости для растворов высокомоле-
кулярных соединений может быть обусловлена и проявлением межмо-
лекулярных взаимодействий в системах полимера с растворителем и
макромолекул друг с другом. Эти взаимодействия можно учесть, если
в выражение для удельной вязкости раствора ввести члены, пропор-
циональные квадрату, кубу и т. д. концентрации растворенного веще-
ства. После замены концентрации раствора с степенным рядом урав-
нение для т]уД принимает вид
ЧУД == Ас + Вс2 + Сс3 + ...
или (VII. И)
Чуд/с = А + Вс + Сс2 + ...
где Т]уД/с — приведенная вязкость раствора; А, В, С — коэффициенты разложения в ряд
по с.
Хаггинс предложил следующее уравнение для приведенной вязко-
сти растворов полимеров, в котором коэффициенты В и С, обусловлен-
ные межмолекулярными взаимодействиями в системе, выражаются че-
рез характеристическую вязкость [т]] и константы К.' и К" соответ-
ственно:
= [т1] (1 + ТС h] с + X" hl2 е2 + ...) (VII. 12)
Характеристическая вязкость [г|] определяет гидродинамическое
сопротивление макромолекул потоку жидкости в предельно разбавлен-
ных растворах, когда полимерные молекулы находятся на больших
расстояниях друг от друга и практически не взаимодействуют. Значе-
ния характеристической вязкости находят путем экстраполяции вели-
чины г)уд/с к нулевой концентрации раствора:
[4] = lim (Чуд/С) (VII. 13)
с->0
Постоянная К' характеризует взаимодействие макромолекул с рас-
творителем. Ее называют вискозиметрической константой Хаггинса.
Значение константы К' позволяет оценить
степень сродства между полимером и рас-
творителем. Чем больше компоненты рас-
твора различаются по природе, тем больше
коэффициент К'. Увеличение константы
Хаггинса при ухудшении «качества» рас-
творителя обусловливается возрастанием
числа случайных контактов макромолекул.
Рис. 60. Зависимость удельной вязкости раствора
полимера от концентрации.
195
Вискозиметрическая константа К" определяет взаимодействие между
макромолекулами при парных столкновениях.
Выражение (VII. 12) является общим уравнением, связывающим
вязкость раствора полимера с его концентрацией. При небольших зна-
чениях с, когда взаимодействие между полимерными клубками не про-
является, оно переходит в соотношение
Пуд/с = hi + К' h]2 с (VII. 14)
Соотношение (VII. 14) является уравнением прямой, тангенс угла
наклона которой определяется вискозиметрической константой Хаггин-
са К'.
Уравнение Хаггинса применимо для растворов макромолекул, при-
нимающих форму плотных, «непротекаемых» для растворителя сфери-
ческих частиц, и достаточно хорошо соблюдается только для растворов
с относительно небольшой концентрацией. При увеличении содержания
полимера в растворе взаимодействие между макромолекулами приво-
дит к их ассоциации. При определенных для данного раствора концен-
трациях ассоциация завершается образованием пространственной
структуры. В результате приведенная вязкость раствора резко возрас-
тает. В этом случае уравнение Хаггинса не соблюдается и раствор ха-
рактеризуется так называемой структурной вязкостью г]стр.
Для структурированных растворов полимеров зависимость вязкости
от напряжения сдвига выражается полной кривой течения (см. рис. 56),
имеющей участки наибольшей г)макС и наименьшей Г|МИн ньютоновской
вязкости, между которыми находится область структурной г)СтР вяз-
кости.
Прочность пространственной структуры характеризуется критиче-
ским напряжением сдвига Рк. С ростом концентрации растворов поли-
меров взаимодействие между макромолекулами повышается и образу-
ются более прочные структуры. Поэтому при увеличении концентрации
растворов значения Рк и т)макс повышаются. Таким образом, концентра-
ционная зависимость вязкости растворов полимеров дает ценную ин-
формацию о структурных особенностях исследуемых систем и тем са-
мым позволяет оценить влияние межмолекулярных взаимодействий на
их реологические параметры.
Для измерения вязкости в данной работе используют прибор типа
реовискозиметра Хепплера. Реовискозиметры относятся к приборам
с рычажным нагружением, они позволяют использовать грузы разной
массы, и поэтому с их помощью можно проводить измерения в довольно
широком интервале напряжений сдвига. С помощью реовискозиметра
можно измерять вязкость как ньютоновских жидкостей, так и струк-
турированных систем.
Схема реовискозиметра Хепплера приведена на рис. 61. Основными
элементами его являются индикатор 1, арретир 2, коромысло 4, на
котором через шарнир 5 с маятником 6 крепится стержень с шариком 9,
термостат 8 и измерительная пробирка 10. Постоянная нагрузка Р на
исследуемую систему, которая помещается в цилиндрическую пробир-
ку, создается с помощью груза, устанавливаемого на чашечке 3. Под
действием приложенной нагрузки шарик вдавливается в систему и де-
формация у регистрируется по индикатору часового типа. Скорость
деформации у определяется по времени т прохождения стрелкой инди-
196
катора определенного числа делений на его шкале,Время т фиксируют
по секундомеру.
Динамическую вязкость системы находят как
T] = P/y = kPi; (VII. 15)
где k — константа, определяемая геометрическими характеристиками пробирки и ша-
рика.
Константу k находят путем калибровки по стандартной жидкости
с известной вязкостью. В качестве стандартной жидкости может быть
использована дистиллированная вода.
Значения динамической вязкости воды при различных температурах
следующие:
i, °C Ч-Ю3, 1Ьс/м2 t, °C Ч-Ю3, Н-с/м2 t, °C Ч-Ю3. Н-с/м2
19 1,023 23 0,936 27 0,854
20 1,002 24 0,914 28 0,836
21 0,981 25 0,894 29 0,818
22 0,958'1 26 0,874 30 0,801
Измерения выполняют 3 раза и по ним рассчитывают среднее значение
времени опускания шарика тст. Константу k рассчитывают из соотно-
шения
k = ^~ (VII. 16)
/•"С ст
Измерение с помощью реовискозиметра проводят следующим обра-
зом. В измерительную пробирку наливают исследуемую жидкость в та-
ком объеме, чтобы ее уровень находился примерно на 1 см выше узкой
цилиндрической части, и с помощью прижимного приспособления фик-
сируют ее в термостате. Арретиром (в виде эксцентрика) устанавли-
вают коромысло в верхнее положение и опускают шарик, соединенный
со стержнем, в пробирку на небольшую глубину, так чтобы он был
погружен только в ее измерительной части (с постоянным по высоте
внутренним диаметром). Стержень с шариком вставляют в маятник 6
и укрепляют с помощью зажима 7. Устанавливают на чашечке 3 тре-
буемый груз и измеряют скорость опускания шарика в исследуемую
жидкость. Для этого прибор переводят в рабочее положение, освободив
арретир. При прохож-
дении стрелки индика-
тора через нулевую от-
метку включают секун-
домер. При опускании
шарика в измеритель-
ной части пробирки
на 30 мм, чему соот-
ветствует положение
стрелки индикатора на
Рис. 61. Схема реовискози-
метра Хепплера:
1— индикатор; 2 — арретир; 3 —
чашечка для груза; 4 — коромыс-
ло; 5 — шарнир; 6 — маятник; 7—
зажим; 8 — термостат; 9— шарик,
укрепленный на стержне; 10 —
пробирка.
197
Таблица VII. 4. Экспериментальные данные по определению вязкости
растворов полимера с помощью реовискозиметра
Концентрация раствора с, г/л Нагрузка Р Время опускания шарика Т, с Вязкость раствора Я» Па-с
г/см2 Н/м2
делении 30, секундомер выключают. По окончании измерения пово-
ротом арретира шарик возвращают в исходное верхнее положение.
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Реовискозиметр Хепплера.
Секундомер.
Колбы емкостью 50 мл.
Пипетки емкостью 10 мл.
Водный раствор полимера, например раствор натриевой соли карб-
оксиметилцеллюлозы.
Готовят четыре раствора полимера (по 25 мл) концентрации 2, 5, 8
и 10 г/л путем разбавления исходного раствора. По описанной выше
методике находят вязкость приготовленных растворов.
Для растворов полимеров концентраций 2 г/л рекомендуются сле-
дующие нагрузки: 10, 20, 30, 40 и 50 г/см2, для растворов концентрации
5 и 8 г/л измерения следует проводить при нагрузках 10, 20, 30, 40, 60,
80 и 100 г/см2, для раствора полимера концентрации 10 г/л используют
нагрузки 20, 30, 40, 50, 60, 80, 100 и 120 г/см2.
При расчете вязкости растворов по формуле (VII. 15) нагрузку Р выражают в
Н/м2, поэтому указанные выше значения Р в г/см2 необходимо умножить на пересчет-
ный коэффициент 98,1.
Полученные результаты за-писывают в таблицу (см. табл. VII. 4)'.
По данным таблицы для всех растворов строят графики зависимо-
сти вязкости т] от нагрузки Р и определяют значения Рк и т]мин.
По значениям т]МИн рассчитывают относительную и удельную вяз-
кость растворов. По полученным данным строят кривую т]уд = f(c)'
и анализируют найденную зависимость.
Работа 33. ИЗУЧЕНИЕ РЕОЛОГИЧЕСКИХ СВОЙСТВ МЕЖФАЗНЫХ
АДСОРБЦИОННЫХ ПЛЕНОК
Цель работы-, определение реологических свойств адсорбционного слоя
молекул высокомолекулярного соединения на границе раздела жид-
кость — жидкость.
Существуют два основных способа исследования реологических
свойств структур. В соответствии с первым способом нагрузка, дей-
ствующая на систему, поддерживается постоянной (Р = const), а из-
меряется зависимость деформации у от времени действия нагрузки т.
198
Рис. 62. Модель Максвелла (а) и деформационная кривая этой модели (б).
При втором способе измеряется деформация при разных нагрузках Р.
По полученным зависимостям 7 = ИТ) или y = f(P) могут быть рас-
считаны параметры, характеризующие структурно-механические свой-
ства дисперсной системы.
Структурно-механические свойства реальных тел моделируются с помощью ком-
бинаций из простейших идеальных реологических моделей: модели Гука, модели Нью-
тона и модели Сен-Венана — Кулона. Эти три модели иллюстрируют соответственно
идеально упругое тело, идеально вязкую'жидкость и идеально пластичное тело. Соеди-
няя последовательно и (или) параллельно эти простейшие модели, можно получить
составную модель, параметры который будут близки к свойствам реального тела.
Так, последовательное сочетание упругого и вязкого элементов дают модель
Максвелла, иллюстрирующую свойства упруго-вязкого тела, учитывающего упругие
свойства жидкости. Схема модели приведена на рис. 62, а, а на рис. 62, б представлена
зависимость деформации для этой модели от времени действия нагрузки.
При наложении постоянной нагрузки Р к этой модели вначале деформируется
элемент Гука (мгновенная обратимая деформация уо), а затем начинается вязкое
течение (необратимая деформация yt), обусловленное деформацией элемента Ньютона.
По окончании действия нагрузки (р = 0) упругая деформация исчезает, а модель со-
храняет необратимую деформацию, обусловленную вязким течением. По величине у0
может быть рассчитан модуль упругости Ei.
Е\—Р/Уо (VII. 17)
а по необратимой деформации yt можно найти динамическую вязкость (при dy/dr =
= const):
Т]1= — т (VII. 18)
Yi
Математическая запись модели Максвелла имеет следующий вид
dy 1 dP Р
du Ei du "Г г]!
(VII. 19)
(VII. 20)
199
Рис. 63. Кривая релаксации напряжения.
При y = const и dy/dt — 0 решение последнего
уравнения дает:
Р = Л,ехр(—т/Z) (VII. 21)
где X = "ni/jEj — время релаксации напряжения.
Уравнение (VII. 21) описывает изменение на-
пряжения в модели со временем (релаксацию на-
пряжения) при постоянной деформации с началь-
ным напряжением Ро (рис. 63).
Поведение вязкоупругих тел, обладющих
эластичностью (упругим последействием), иллю-
стрирует модель Кельвина — Фойгта, состоящая
из параллельно соединенных элементов Гука и Ньютона (см. рис. 64, а). Кривая де-
формации этой модели представлена на рис. 64,6.
Приложение постоянной нагрузки (р = const) к этой модели приводит в дви-
жение поршень элемента Ньютона, но скорость движения его с течением времени
уменьшается, так как на пружину (элемент Гука) приходится все большая часть
усилия. Деформация модели постепенно приближается к предельной упругой дефор-
мации. При снятии напряжения (Р = 0) система возвращается в исходное состояние,
при этом скорость деформации уменьшается. Этот процесс замедленной деформации
называется упругим последствием, и оно обусловливает эластичность системы. По
деформации уэ, максимально достижимой при данной нагрузке, рассчитывается модуль
эластической деформации.
Е, = Р/уэ (VII. 22)
Математически модель Кельвина — Фойгта записывается следующим образом:
Решением этого уравнения при Р = const является соотношение
У = -£-(1 — е'~г/0) (VII. 24)
С2
где 0 = rja/jEa — время релаксации деформации.
Уравнение (VII. 24) описывает изменение деформации во времени (релаксацию
деформации).
Известны и более общие модели. Например, последовательное соединение моделей
Максвелла и Кельвина — Фойгта (рис. 65, а) позволяет смоделировать систему, обла-
дающую упругой деформацией, упругим последействием, а также способностью к ре-
Рис. 64. Модель Кельвина — Фойгта
(а) и деформационная кривая этой модели (б).
200
р
Рис. 65. Составная модель упруго-пласти-
ческого тела (а) и деформационная кри-
вая этой модели (б).
лаксации напряжений. Деформационная кривая составной модели приведена на
рис. 65, б.
Математически эта модель описывается следующим уравнением:
У = -Х-+ т +-^-(1 — е-т/е) (VII. 25)
Многие межфазные адсорбционные пленки имеют деформационную
кривую, аналогичную приведенной на рис. 65,6. На основании такой
кривой можно рассчитать ряд параметров, характеризующих структур-
но-механические свойства пленки, а именно:
1) модуль упругости Ег [см. уравнение (VII. 17)];
2) модуль медленной эластической деформации Е2 [см. уравнение
(VII. 22)];
3) равновесный модуль эластичности
Еэ = —у (VII. 26)
Уо + Уэ
4) степень эластичности
а = т~Д7;;--’100== F Sp "10°™ (V1L27)
Уо + Уэ г,2
Структурно-механические характеристики адсорбционных пленок
определяют методом плавающей магнитной пластинки с помощью при-
бора, схема которого приведена на рис. 66.
На предметном столике 1 микроскопа (МИР-12) помещаются вспо-
могательный магнит 2, рабочая кювета 5 и основной электромагнит 6.
Ферромагнитная пластинка 4 помещается на границе раздела фаз в ра-
бочей кювете 5, и ее положение регулируется с помощью вспомогатель-
ного магнита 2, На основной электромагнит подается напряжение от
13 Зак. 673 201
Рис. 66. Схема установки для определения реологических характеристик адсорбцион-
ных пленок:
1 — предметный столик микроскопа; 2 — вспомогательный магнит; 3 — микроскоп; 4 — ферромагнитная
пластинка; 5—измерительная кювета; 6 — основной электромагнит; 7 — источник постоянного тока;
$—вольтметр.
источника постоянного тока 7, измеряемое вольтметром 8. Смещение
пластинки, вызываемое действием электромагнита 6, фиксируется по
шкале измерительного микроскопа 3. Предварительно прибор калиб-
руют с использованием торзионных весов, определяя зависимость между
силой F, действующей на пластинку, и напряжением U, подаваемым
на основной электромагнит.
Работу на установке выполняют в следующем порядке. В кювету 5
наливают раствор полимера (уровень раствора должен находиться
примерно на середине высоты магнита 6). Кювету устанавливают в дер-
жателе на предметном столике микроскопа и с помощью пластмассо-
вого пинцета на поверхность раствора аккуратно помещают пластинку.
Вращая вспомогательный магнит, устанавливают пластинку таким об-
разом, чтобы она располагалась параллельно боковым стенкам кю-
веты. Правильность установки пластинки на поверхности раствора кон-
тролируют путем наблюдения в окуляре микроскопа.
Передвигая электромагнит 6, устанавливают его на определенном
расстоянии I между торцевой плоскостью его сердечника и ближайшей
к ней стороной пластины (рекомендуется I ш 30 мм). Значение I фик-
сируют по шкале предметного столика микроскопа. Затем сверху
по стенке кюветы аккуратно вводят несколько миллилитров (3—
5 мл) неполярного растворителя. Фиксируют время введения раство-
рителя.
Если при введении растворителя пластинка погрузится на дно кюветы, допу-
скается извлечение ее намагниченным пинцетом и последующее опускание на границу
раздела полярная жидкость — неполярная жидкость.
Снова проверяют правильность расположения пластинки и расстоя-
ния I. Поворотом лимба микроскопа совмещают вертикальную линию
перекрестия шкалы окуляра с концом пластинки, ближайшим к элек-
тромагниту 6. Через определенное время после введения неполярной
жидкости на электромагнит накладывают рабочее напряжение U и од-
новременно включают секундомер. Через определенные промежутки
времени т (~10 с) по шкале микроскопа измеряют смещение пластин-
ки А/ до тех пор, пока оно не превысит 1 мм. Отключают питание
электромагнита (время выключения отмечают по секундомеру) и снова
через те же промежутки времени измеряют по шкале микроскопа изме-
нение остаточной деформации.
202
ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для проведения работы необходимы:
Установка для измерения реологических характеристик межфазных
пленок.
Секундомер.
Пинцеты, пластмассовый и стальной, намагниченный.
Пипетка емкостью 5 мл.
Водный раствор полимера, например 1,5 %-ный (масс.) раствор же-
латины.
Неполярный растворитель, например октан.
Измеряют смещение магнитной пластинки на границе раздела вод-
ный раствор полимера — неполярная жидкость по методике, указанной
выше. Вначале проводят измерения через 25—30 мин, а затем через
1 ч после введения в кювету неполярного растворителя (для вновь при-
готовленной системы). По полученным данным рассчитывают деформа-
цию у = Д//П (П — периметр пластинки). Результаты измерений запи-
сывают в таблицу (см. табл. VII. 5).
Таблица VII. 5. Экспериментальные результаты определения деформации
адсорбционной пленки полимера
Время после приложения и снятия внешнего поля %, с Смещение пластинки, Д/, мм Деформация у
10
20
...
По данным таблицы строят деформационные кривые Y==f(x). Счи-
тая, что при незначительных смещениях пластинки (Д/ 1 мм), дей-
ствующее на нее со стороны электромагнита усилие практически по-
стоянно, по калибровочному графику F = f(U) определяют F, соответ-
ствующее рабочему напряжению. По значению F рассчитывают напря-
жение Рп= F /П.
По графикам Y==f(x) с помощью уравнений (VII. 17), (VII.22),
(VII. 26) и (VII. 27) рассчитывают параметры, характеризующие
структурно-механические свойства пленки: модуль упругости £ь мо-
дуль эластической деформации Е2, равновесный модуль эластичности
Еэ и степень эластичности а. Результаты сводят в таблицу (см.
табл. VII. 6) и анализируют изменение реологических параметров меж-
фазной пленки во времени.
Таблица VII. 6. Результаты расчета структурно-механических свойств
адсорбционной пленки
Время с момента контакта жидких фаз, мин Ер Н/м Е2, Н/м Еэ, Н/м а, %
13=»
203
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАЧИ
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какими основными структурно-механическими свойствами характе-
ризуются дисперсные системы? Каким методом они выявляются?
2. Каковы причины возникновения структур в дисперсных си-
стемах?
3. Назовите простейшие идеальные реологические модели (элемен-
ты). Как зависят деформации этих моделей от приложенной нагрузки?
4. Какая реологическая модель иллюстрирует упруго-вязкие свой-
ства систем? Что собой представляет время релаксации напряжения?
Какова взаимосвязь (качественная) между временем релаксации и
агрегатным состоянием тел?
5. Какая реологическая модель иллюстрирует эластичность (упру-
гое последействие)? Как изменяется во времени деформация вязко-
упругого тела?
6. Какая реологическая модель иллюстрирует пластические свой-
ства дисперсных систем? Какими параметрами характеризуют проч-
ность структур?
7. Назовите два основных типа структур дисперсных систем (клас-
сификация Ребиндера). Как они образуются (проиллюстрируйте по-
тенциальной кривой взаимодействия частиц) и чем отличаются их рео-
логические характеристики? Приведите примеры реальных структур
различных типов.
• 8. Как классифицируют дисперсные системы по их реологическим
свойствам? Приведите типичные кривые течения для них.
9. Какие жидкости называются ньютоновскими? Напишите уравне-
ние Ньютона для течения жидкостей. Объясните физический смысл
входящих в него параметров. Нарисуйте кривые течения и вязкости
тля ньютоновских систем.
10. Нарисуйте кривые течения и эффективной вязкости для струк-
турированных систем. Покажите на графиках предельное статическое
напряжение сдвига Рк и предельное напряжение сдвига Рт, а также
вязкости, соответствующие неразрушенной и полностью разрушенной
структурам.
11. Какие изменения происходят в системах с коагуляционной
структурой при напряжениях Р < Рк, Рк < Р < Рт и Р > ~Рт? Объяс-
ните явление ползучести.
12. . Что представляют собой явления тиксотропии и реопексии? Чем
обусловлены эти явления и для каких структурированных систем они
характерны? Приведите примеры таких структурированных дисперс-
ных систем.
13. Какое уравнение выражает зависимость вязкости жидких агре-
гативно устойчивых дисперсных систем от концентрации дисперсной
фазы? При каких условиях оно применимо?
ч 14. Как зависит вязкость растворов полимеров от их молекулярной
массы, формы макромолекул и их термодинамического сродства к рас-
творителю? Напишите уравнения Марка — Хаувинка и Хаггинса и объ-
ясните, при каких условиях они выполняются.
15. Что называют относительной, удельной и характеристической
вязкостью? Как их определяют?
•204
* 16. Объясните принцип действия капиллярного вискозиметра. На-
пишите уравнение Пуазейля для объемной скорости движения жидко-
сти.в капилляре. Как калибруются капиллярные вискозиметры?
17. Объясните принцип действия ротационных вискозиметров. Для
каких систем используются приборы этого типа?
< 18. Как осуществляется переход от коагуляционно-тиксотропных
структур к конденсационным (кристаллизационным) и наоборот? Воз-
действием каких факторов можно вызвать эти переходы?
19. Каким образом размеры частиц и взаимодействие между ними
влияют на структурно-механические свойства дисперсных систем?
20. В чем заключается эффект Ребиндера? Какие вещества высту-
пают в качестве понизителей твердости материалов? Приведите при-
меры использования этого эффекта.
ПРИМЕРЫ РЕШЕНИЯ ЗАДАЧ
1. Реологические свойства 20%-ной суспензии бентонитовой глины в исследуемом
интервале нагрузок описываются реологической моделью, состоящей из последователь-
но соединенных элемента Гука и модели Кельвина — Фойгта со следующими парамет-
рами: модуль упругости элемента Гука Е = 1,5-103 Па; модуль эластичности
Еэ — 1,3-103 Па; вязкость элемента Ньютона г] = 1,2-105 Па-с. Рассчитайте дефор-
мацию, развивающуюся в системе за 100 с при напряжении сдвига Р = 10 Па.
Решение. Полная деформация модели при последовательном соединении элемен-
тов складывается из деформаций элемента Гука и модели Кельвина — Фойгта:
10
1,5- 103
/ -1,3-Ю3 ч
Ю I, ,TnF'100) 1п-з
1,3- 103 У — 11,/5 - 10
2. При изучении структурно-механических свойств графитовых паст глубина по-
гружения конуса пластометра Ребиндера составляла h = 2 мм при нагрузке F — 3 Н.
Константа конуса Kl = 0,4. Рассчитайте предельное напряжение сдвига графитовой
пасты.
Решение. Предельное напряжение сдвига вычисляется по формуле
Р =КГ — =0,4-----------—з-тг = 3- 105 Па
т L й2 (2 • 10-3)2
3. При изучении структурно-механических свойств суспензий пылевидного кварца
С помощью прибора Вейлера — Ребиидера определено усилие = 1,5-10—2 Н, не-
обходимое для смещения рабочей пластины прибора. Длина пластины 1 см, ширина
0,5 см. Рассчитайте предельное напряжение сдвига Рт суспензии.
Решение. Предельное напряжение сдвига, характеризующее прочность структуры
системы, вычисляется по уравнению
п ____ Т'макс
где s — боковая поверхность пластины.
р 1,5-10~2
т 2-0,5- 10 2 • 10 2
= 150 Па
4. Рассчитайте толщину гидратных оболочек 6 золя АЬОз, если реологическими
измерениями установлено, что при концентрации 12 % (масс.) золь является ньюто-
новской жидкостью с вязкостью г)= 1,18-10—3 Па-с. Радиус частиц золя г равен 10 нм.
Плотность частиц дисперсной фазы р — 4 г/см3, дисперсионной среды ро = 1-г/ем3 '
Вязкость дисперсионной среды т)0 — 1 • Ю-3 Па-с. Коэффициент формы частиц <<£== 2,ЭД
Решение. Вязкость данного золя может быть выражена уравнением Эйнштейна? ‘
11 = Йо (1 +^Рг)
где фг — объемная доля дисперсной фазы с учетом гидратации.
205
Объемная доля дисперсной фазы с гидратными оболочками составит:
фг = = 0>072
Пой 1 10 3 2,5
Объемная доля дисперсной фазы без гидратных оболочек равна
= сА12О3/Р = 12/4 = 0
сА120з/Р + сН2о/Ро 12/4 + 88/1
Если п — число частиц в' единице объема, то
<рг __ п • 4/Зл (г + б)3 _f 1 । AV
<р п 4/Злг3 \ г )
Откуда
•4(f)1’-]“ '» 1О"[(Ж)'В- ‘J-2'87-'0"’ ”-2’97""
5. При измерении вязкости растворов полистирола в толуоле с помощью капил-
лярного вискозиметра (типа вискозиметра Оствальда) получены следующие данные:
Концентрация раствора
с, г/л............. 0 1,70 2,12 2,52 2,95 3,40
Время истечения рас-
твора т, с............ 97,6 115,6 120,2 124,5 129,8 134,9
Рассчитайте значения относительной, удельной, приведенной вязкости растворов
полимеров и постройте график зависимости 1)уд/с = /(с). Определите характеристи-
ческую вязкость (т|| и значение вискозиметрической константы Хаггинса К7.
Решение. Относительная вязкость определяется как отношение вязкости раствора г]
к вязкости чистого растворителя г]»:
Потн — ‘П/'По
Удельная вязкость раствора представляет собой приращение вязкости за счет рас-
творенного вещества, отнесенное к вязкости растворителя:
П ~ 'По ,
Иуд — ~ Нота *
По
Согласно уравнению Пуазейля (VII. 7) можем записать:
где р и ро — соответственно плотность раствора и растворителя; т и т0 — время исте-
чения раствора и растворителя.
Приведенная вязкость г)пр есть отношение удельной вязкости к концентрации рас-
твора с
Пир = Иуд/С
Результаты расчета приведены ниже:
Концентрация раствора
с, г/л.................. 1,70
Поти....................... 1,184
Иуд........................ 0,184
т)уД/с, л/г................ 0,108
2,12 2,52 2,95
1,231 1,276 1,330
0,231 0,276 0,330
0,109 0,110 0,111
3,40
1,382
0,382
0,112
Строят график в координатах г]уд/с—с (Рис- 67). Экстраполируя зависимость на
нулевую концентрацию, определяем значение характеристической вязкости:
Н] = (lim = 0,104 л/г
X с Л->о
206
Рис. 67. Зависимость приведенной вязкости раствора
полистирола в толуоле от концентрации.
Согласно Хаггинсу [см. уравнение (VII. 14)] при ма-
лых концентрациях раствора
= [1)1 + К' [Т)12 с
Вискозиметрическую константу Хаггинса К' находим по тангенсу угла наклона пря-
мой:
tg а = К' [т]]2 = 0,0023
К'
_ 0,0023
— 0.1042
= 0,216
ЗАДАЧИ
1. Используя уравнение Эйнштейна, определите вязкость золя AgCl,
если концентрация дисперсной фазы составляет: а) 10 % (масс.),
6)^10% (об,). Частицы золя- имеют сферическую форму. Плотность
AgCl равна 5,56 г/см3. Дисперсионная среда имеет вязкость
1 • 10“3 Па-с и плотность 1 г/см3
J2. Определите вязкость золя А12О3, если концентрация дисперсной
фазы золя составляет: а) 8% (масс.); б) 8 % (об.). Частицы имеют
сферическую форму, плотность А12О3 — 4,0 г/см3. Вязкость и плотность
дисперсионной среды соответственно 1-10-3 Па-с и 1 г/см3. Найти от-
носительное увеличение вязкости.
3. Рассчитайте массовую концентрацию гидрозоля диоксида крем-
ния, если известно, что его вязкость на 10 % больше вязкости диспер-
сионной среды. Частицы SiO2 имеют сферическую форму, плотность их
равна 2,7 г/см3, плотность дисперсионной среды 1 г/см3.
4. Рассчитайте толщину гидратных оболочек частиц золя диоксида
кремния, если экспериментальными методами установлено, что вязкость
15 %-ного (масс.) золя составляет 1,3-10-3 Па-с, а диаметр частиц
равен 16 нм. Плотности частиц дисперсной фазы золя и дисперсионной
среды соответственно 2,7 и 1 г/см3. Вязкость дисперсионной среды
1-10~3 Па-с. Коэффициент формы частиц а = 2,5.
5. Сопоставьте графически теоретические значения вязкости гидро-
золей диоксида кремния, рассчитанные по уравнению Эйнштейна, с экс-
периментально найденными значениями:
Ств, % (масс.).................
т) - 103, Па-с, при диаметре
частиц
5,9 нм......................
21,0 нм.....................
10 15 20 30 40
1,10 1,17 1,30 1,83 2,72
1,10 1,15 1,24 1,48 1,82
Определите концентрации, при которых начинается процесс структу-
рообразования. Сделайте выводы о влиянии размера частиц золя на
этот процесс. Плотности дисперсной фазы золя и дисперсионной среды
соответственно 2,7 и 1 г/см3. Вязкость дисперсионной среды 1-10~3Па-с,
Коэффициент формы частиц а = 2,5.
207
РУ Рассчитайте вязкость 50 %-ного водного раствора глицерина,
если при приложении к нему напряжения 18 Н/м2 скорость развития
деформации составляет 3-103 с-1.
7. Определите относительную деформацию образца полиуретанового
каучука при различных температурах, если к нему приложено посто-
янное напряжение 5-10® Н/м2. Используйте следующие значения мо-
дуля упругой деформации:
Г, К............... . 233 263 293
£-10~6, Н/м2............ 167,0 20,0 11,1
8. По экспериментальным данным, полученным при помощи капил-
лярного вискозиметра:
Р, Н/м2 .............. 10 15 20 25
Скорость деформации
Y - 103, с-1........ 1,18 1,76 2,36 2,90
Постройте кривую течения для исследуемой жидкости и рассчитайте
ее вязкость.
9. Определите диаметр сечения капиллярного вискозиметра длиной
5 см, если 3 мл ньютоновской жидкости с вязкостью 1 • 10~3 Па-с про-
текают через него под давлением в 100 Н/м2 за 61 с.
10. Определите вязкость машинного масла, если через капилляр
длиной 6 см и диаметром 1 мм оно протекает со скоростью 2,04 X
X Ю~3 см3/с под давлением в 100 Па.
И. Течение 12 %-ной суспензии бентонитовой глины в исследуемом
интервале нагрузок описывается уравнением Бингама для вязкопла-
стичного тела. Постройте кривую течения суспензии, рассчитайте пре-
дельное напряжение сдвига и пластическую вязкость ее по следующим
экспериментальным данным:
Р, Н/м2 ................. 20 25 30 35 40
Скорость деформации V- с~1 250 480 710. 940 1100
12. В дисперсной системе, представляющей собой упруговязкое
тело Максвелла, под действием нагрузки мгновенно развивается упру-
гая относительная деформация, равная 400 %. Рассчитайте начальное
напряжение в системе и промежуток времени, за которое оно умень-
шится в 100 раз. Модуль упругости и коэффициент ньютоновской вяз-
кости системы составляют соответственно 500 Н/м2 и 50 Па-с.
13. При помощи ротационного вискозиметра, работающего в ре-
жиме постоянной скорости деформации, были получены реологические
характеристики 20%-ных водных суспензий графита при различных
значениях pH дисперсионной среды:
Скорость деформации V» с"”1 Напряжение А Н/м2, при pH
13 И 7 4 2
50 1,8 8,2 14,2 9,6 1,о
100 2,4 9,0 15,0 10,2 1,8
200 3,0 10,0 16,0 11,2 2,2
400 4,4 10,8 16,6 12,0 3,0
800 7,0 12,8 17,6 13,8 4,6
1200 9,6 15,0 18,8 ' 16,0 6,6
208
Постройте кривые течения, рассчитайте предел текучести и построй-
те график зависимости его от pH среды суспензий.
14. При помощи ротационного вискозиметра исследованы 76 %-ные
водные суспензии электрокорунда, в дисперсионную среду которых
введен электролит (КС1), вызывающий агрегацию частиц дисперсной
фазы:
Р, Н/м2...............
Чэфф, Па-с, при кон-
центрации КС1
без КС1.............
5 • 10~4 моль/л , . .
0,1 моль/л..........
10 20 40 60
0,5 1,6 3,0 3,7
4,4 4,5 4,5 4,5
9,0 7,5 6,3 5,8
80 100 120 160
3,9 4,0 4,0 3,9
4,5 4,5 4,5 4,4
5,2 4,9 4,8 4,7
Постройте графики зависимости эффективной вязкости от напря-
жения и определите типы структур в суспензиях (структурные измене-
ния при деформировании имеют обратимый характер).
15. При помощи прибора Вейлера — Ребиндера изучены структурно-
механические свойства 25 %-ных олеосуспензий железного сурика в
зависимости от концентрации ПАВ (олеиновой кислоты):
спав> % (масс.)........................... 0 0,5 2,0 6,0 20,0
Наибольшее усилие, FMaKC • Ю2, И 3,4 2,0 1,2 0,5 0,2
Рассчитайте предельные напряжения сдвига и постройте график их
зависимости от концентрации ПАВ. Длина рабочей пластинки прибора
8 мм, ширина 6 мм.
16. Рассчитайте модули упругости натурального каучука при раз-
личных температурах, если под действием постоянного напряжения
1 • 10s Н/м2 относительная деформация составляла:
Г, К.................................. 223 283 313
у, %.................................... 2 40 80
17. Рассчитайте по уравнению Марка — Хаувинка молекулярную
массу натурального каучука, если характеристическая вязкость его
раствора в бензоле [р] = 0,126 м3/кг, константа 7( = 5-1СН5, параметр
а = 0,67.
18. При измерении вязкости растворов полистирола в толуоле с по-
мощью капиллярного вискозиметра получены следующие данные:
с, г/л....................... о 1,70 2,12 2,52 2,95 3,40
Время истечения рас-
твора т, с.......... 97,6 115,1 120,2 124,5 129.9 134,9
Рассчитайте значения относительной, удельной, приведенной вязко-
сти растворов полимеров и постройте график зависимости r\ya/c = f(c).
Определите характеристическую вязкость [р] и вискозиметрическую
константу Хаггинса К'.
19. При измерении вязкости растворов 1,4цис-полиизопрена в то-
луоле получены с помощью капиллярного вискозиметра следующие
данные:
с, г/л ........ . о 1,41 1,94 2.59 3,24 3,89
Время истечения рас-
твора т, с........ 171,5 216,1 234,0 257,3 282,6 308,1
209
Рассчитайте значения относительной, удельной и приведенной вяз-
кости растворов. Постройте график зависимости т]уд/с — f(c) и опреде-
лите характеристическую вязкость раствора [г|] и вискозиметрическую
константу Хаггинса К',
ОТВЕТЫ К ЗАДАЧАМ
Приведены ответы к наиболее сложным задачам.-
I. 1. 129,9 Дж. 2. 99,7 Дж. 4. 3,85 Дж. 5. 1,15-107 Па. 6. 1,39-106 Па.
(7. 1,9-10е Па. 8. 28,6 мкм. 9. 73 мДж/м2. 10. 70 мДж/м2. 12. 20,3 нм.
13. 24,5 мДж/м2. 14. 29,4 мм. 15. 70ЛмДж/м2. 16. 2,88-105 Па, 2880 Н.
17. 1,44-104 Н^0. 1,2. 21. 21 300 Па<2£> 2,9-103 и 1,9-103 Па. 23. 2335 Па.
26. 0,214 мкм@>0,968 Дж/м2. 30. 71,96 и —71,96 мДж/м2. 32. 23,8 йН/м.
36. 8,86 мН/м.
II. 1. 6,5 нм. 2. 30,3 моль/кг. 3. 5-10-2 м3/кг, 0,6 Па-1, 220 м2/г.
4. 0,49 нм2. 5. К = 1,45-10-3, п = 2,22. 6. 177, 295, 118 и 115 м2/г.
7. 2,9 м2/г. 8. 2,86 м2/г. 9. 1,5-10~3 м3/кг, 133, 7,8 м2/г. <То> 270 м2/г.
11. 98,95 и 90,03 %. 19. 0,24 дм3/кг. 20. 0,19 дм3/кг. 21. О',262, 0,070 и
0,151 дм3/кг. 22. 1,47 кДж/моль. 23. 18,2 кДж/моль, —0,204 кДж/
,'(моль-К),—2,5 кДж/моль. 25. 4,69 м2/г. 26. 0,359 Дж/м2. 29. 0,69 м2/кг.
30. 3,31 • 10-6 моль/м2. 32. 4-10—6 моль/м2, 0,18 л/ммоль. 34. 0,208 нм2,
0,15 нм. 35. 3,84 экв/кг. 36. 9,9 экв/кг. 37. 141,4 г Со, 329,6 г Ва.
38. 124 г Со, 827 г Аи.@ 612 и 735 кг. 40. 6,07 кг. 41. 0,263 (л/кг)1'2.
42. Концентрация цезия в смоле 3,25 экв/кг, в растворе 31,28 экв/м3,
43. 91 %. '
П1.(21\9,26 мкм. 3. 4,12, 13,0 и 41,2 мкм. 5. АД (средн.) = 7,3 X
ХЮ23 моль-1. 6. 2,25-1019 м-3'. 8. 1,21 эд, 0,15 м, 1,21 мм. 9. 6,71 X
ХЮ-12 м2/с. @ 2,61 м, 32,6 см, 0,326 мм. 11. 62,8 нм. 12. 7,17Х
X Ю-11 м2/с. 15. 131 с, 3,64 ч, 364 ч. 16. 142 м2/кг. 17. 0,025, 2,55 и 255 с.
18. 1,87 нм, 8,28-104 м2/кг. 19. 96, 9,6 и 0,96 нм. 20. 9,49 нм, увеличится
в 2 раза. 22. 6,6 мВ, 2,49-104 Кл/м3 <р и р уменьшатся в 9,04 раза.
23. 6,88-104 Кл/м3, 1,21-10~3 Кл/м2.724; 5,94-10-2 Ф/м2, емкость умень-
шится в 1,225 раза. 25. 0,169 В. 26. 39,7 мВ. 28. 427 см3, 6,0 (кВт-ч)/м3.
29. 10,5 мкм/с. 31. —21,5 мВ. 32. 1,95 см. 33. 0,06 В. 34. 34,5 и 6,3 мВ,
13,6 и 1,18 нм. 36. 22,8 мВ. '37. 59,6 мВ. 38. 0,888 В. 39. 13,9 мВ.
40. 40 мВ.
IV. 1. 0,282 мкм. 4. 193,6 нм. 5. 0,997. 6. 0,9935. 7. 1,3, 3,9, 15,2.
9. 29,9, 26,3, 36,3 и 23,5 нм.
V. 2. 2,79-10-3 (Дж-м)/моль. 4.. 1,81-103 нм2, 6,6-1016 . 5. 1,49-104,
1,7 нм. 7. 0,37 нм2, 5 ммоль/л. 11. 0,16.
ЛУ1. 4. 0,13 % (масс.). 5-3.-J0-19 Дж. 6. 46 мДж/м2. 10. В 5 и 409 раз.
@4,5-J0-18 м3/<2. 12. 1,7-10-'4 с. 13. 20 с. 15. 1,7-1014 част/м3. 16. 6Х
X ю~18 м3/с. ’ /Д дд
VII. 2. 10,5-10-3 Па-с, 1,2-10~3 Па-с.^Д 10,1 %.(<4.) 2 нм.-8. 8,5Х
X 10-3 Па-с. 9. 1-10-3 м. 10. 2-Ю-2 Па-с. 12. 2-10~3 Па, 0,46 с. 18. h] =
— 0,101 м3/кг, /с = 0,35.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
1. Фролов Ю. Г. Курс коллоидной химии. Поверхностные явления и дисперсные си-
стемы. М., Химия, 1982. 400 с.
2. Фридрихсберг Д. А. Курс коллоидной химии. Л., Химия, 1984, 368 с.
3. Щукин Е. Д., Перцов А. В., Амелина Е. А. Коллоидная химия. М., изд-во МГУ,
1982. 348 с.
4. Адамсон А. Физическая химия поверхностей. Пер. с англ./Под ред. 3. М. Зорина,
В. М. Муллера. М., Мир, 1979. 588 с.
5. Грег С., Синг К. Адсорбция, удельная поверхность, пористость. Пер. с англ. 2-е
изд. М., Мир, 1984. 306 с.
6. Шинода Д. и др. Коллоидные поверхностно-активные вещества. Пер. с англ./Под
ред. А. Б. Таубмана, 3. Н. Маркиной. М., Мир, 1966. 320 с.
7. Зонтаг Г., Штренге Д. Коагуляция и устойчивость дисперсных систем. Пер. с нем./
/Под ред. О. Г. Усьярова. Л., Химия, 1973. 152 с.
8. Практикум по коллоидной химии./Под ред. И. С. Лаврова. М., Высшая школа,
1983. 215 с.
9. Практикум по коллоидной химии и электронной микроскопии./Под ред. С. С. Воюц-
кого, Р. М. Панич. М., Химия, 1974. 224 с.
приложения
ОСНОВНЫЕ ФИЗИКО-ХИМИЧЕСКИЕ ВЕЛИЧИНЫ, ИСПОЛЬЗУЕМЫЕ
В КУРСЕ КОЛЛОИДНОЙ ХИМИИ
А — абсолютная адсорбция, моль/кг, моль/м2; константа Гамакера, Дж.
а — определяющий размер, м; активность, моль/л.
В - коэффициент трения, п-с/м.
с — концентрация, моль/л, г/л.
D — дисперсность, м-1; оптическая плотность; коэффициент диффузии, м2/с.
Е — напряженность электрического поля, В/м; потенциальный барьер, Дж; модуль
упругости, Н/м2.
F — сила, Н.
f — коэффициент растекания, Дж/м2.
G — энергия Гиббса, Дж.
g — поверхностная активность, Дж-м/моль.
Н — энтальпия, Дж; кривизна поверхности, м“'; постоянная Дебая.
I — сила тока, А; ионная сила, моль/л.
К — константа адсорбционного равновесия.
k — постоянная Больцмана, Дж/К.
М — относительная молекулярная масса.
/VА — число Авогадро, моль-1.
Р — напряжение сдвига, Н/м2.
р — давление, Па.
q — теплота, Дж; поверхностный заряд, Кл.
7? — универсальная газовая постоянная, Дж/(моль-К); сопротивление, Ом.
г — радиус, м.
S — энтропия, Дж/К; константа седиментации.
211
s — площадь поверхности, м2.
Т — температура, К.
U — разность потенциалов, В; энергия, Дж.
и — линейная скорость, м/с.
— мольный объем, м3/моль.
v — объемная скорость, м3/с.
х — мольная доля компонента.
z — заряд иона.
а — коэффициент разделения.
р — коэффициент аффинности; константа устойчивости комплексных соединений.
Г — гиббсовская адсорбция, моль/м2.
у — относительная деформация.
у — скорость деформации, с-1.
А — смещение (сдвиг) по выбранному направлению, м.
б — толщина адсорбционного слоя, м.
е — относительная диэлектрическая проницаемость.
е0 — электрическая постоянная, Ф/м.
t. — электрокинетический (дзета) потенциал, В.
Т| — вязкость, Па-с.
6 — краевой угол, время половинной коагуляции, с.
и — величина, "Обратная толщине диффузной части двойного электрического слоя,
м~’; удельная электропроводность, См/м.
Л — толщина диффузной части двойного электрического слоя, м; эквивалентная
электропроводность, См • м2/моль.
ц — химический потенциал, Дж/моль.
v — число частиц в единице объема.
— поверхностное давление, Н/м; осмотическое давление, Па.
р — плотность, кг/м3.
а — поверхностное натяжение, Дж/м2, Н/м.
т — время, с; мутность, см~’.
<р — потенциал, В; объемная доля.
Таблица 1. Поверхностное натяжение воды при различных температурах
t. °C °ж-г- мДж/м2 А °C °Ж-Г» мДж/м2 । t, °с аж-г» мДж/м2
10 74,22 17 73,19 24 72,13
и 74,07 18 73,05 25 71,97 -
12 73,93 19 72,90 26 71,82
13 73,78 20 72,75 27 71,66
14 73,64 21 ‘ ' 72,59 28 71,50
15 73,49 22 72,44 29 71,35
16 73,34 23 72,28 30 71,18
212
Таблица 2. Физико-химические свойства некоторых органических жидкостей
Вещество Молеку- лярная масса Плот- ность (20 °C), г/см3 Поверхностное натяжение, мДж/м^ при температуре (в °C)
20 30 40 50
Этанол 46,07 0,789 22,4 21,6 20,7 19,9
Пропанол-1 60,10 0,804 23,7 22,9 22,2 21,4
Пропанол-2 60,10 0,785 21,3 20,5 19,7 19,0
Бутанол-1 74,12 0,810 25,4 24,5 23,6 22,7
Пентанол-1 88,15 0,814 25,8 24,9 24,0 23,2
Пентанол-2 88.15 0,810 24,0 23,0 22,0 21,0
Гексанол-1 102,18 0,819 26,2 25,4 24,6 23,8
Гексан 86,18 0,659 18,4 17,4 16,3 15,3
Толуол 92,14 0,867 28,5 27,3 26,1 24,9
Хлороформ 119,38 1,498 27,3 26,0 24,7 23,4
(15 °C)
Четыреххлорнстый углерод 153,82 1,595 27,0 25,8 24,6 23,4
Уксусная кислота 60,05 1,049 27,6 26,6 25,6 24,6
Хлорбензол 112,56 1,107 33,3 32,3 31,1 29,9
Нитробензол 123,11 1,203 43,9 42,7 41,5 40,2
Анилин 93,14 1,022 43,3 42,2 41,2 40,1
Ацетон 58,08 0,791 24,0 22,9 21,8 20,7
Таблица 3. Площадь, занимаемая молекулами ПАВ
в предельно насыщенном адсорбционном слое
ПАВ Формула 50 HO20’ M2
Лаурат натрия CH3(CH2)ioCOONa 41
Миристат натрия CH3(CH2)i2COONa 34
Пальмитат натрия CH3(CH2)14COONa 25
Стеарат натрия CH3(CH2)i6COONa 23
Олеат натрия C8Hj7CH=CHC7H14COONa 28
и-Додецил бензол сульфон ат натрия Ci2H25C6H4SO3Na 33
Додецилсульфат натрия C12H25SO4Na 33
Октнлэтиламмонийхлорид C8H17(C2H5)NH2Cr 34
Цетил пиридиний хлорид C i вЫз з N С 5H5 C1 46
Октанол-1 C8Hi7OH 30
Октиловый эфир этиленгликоля CsHiyOCHgCHgOH 32
л-Октилфениловый эфир октаэтилен- гликоля CSH17C6H4O(C2H4O)8H 53
п-грег-Октнлфениловый эфир окта- этиленгликоля C8H17C6H4O(C2H4O)8H 58
я-трет-Октилфеннловый эфир декаэти- ленгликоля C8Hi7C6H4O(C2H4O)I0H 74
n-трет-Октилфениловый эфир гексаде- каэтиленгликоля 80
213
Таблица 4. Критические концентрации мицеллообразования ПАВ
в водных растворах
ПАВ Формула ККМ, моль/л Метод определения
Анионные ПАВ
Деканоат натрия C9H)9COONa 9,4 • Ю-2 Удельная электропро-
9,5- 10-2 водность Поверхностное натя- жение
Додеканоат натрия CnHzaCOONa 2,5- 10-2 2,3- 10-2 2,4- 10~2 Удельная электропро- водность Поверхностное натя- жение Солюбилизация кра- сителя
Миристат натрия Ci3H27COONa 6,9- 10~3 Удельная электропро- водность
Олеат натрия С 17Нзз COON а 1,1 • io-3 2,1 10 То же Поверхностное натя- жение
Додецилсульфат натрия Ci2H25SO4Na 8,1 • 10~3 8,3- 10-3 Удельная электропро- водность Поверхностное натя- жение
Тетрадецилсульфат натрия Ci4H29SO4Na 2,1 • 10-3 Удельная электропро- водность
Додецилсульфонат натрия Ci2H25SO3Na 9,8 • 10-3 Поверхностное натя- жение
и-Додецилбензолсульфонат Ci2H25C6H4SO3Na 1,2 • 10-3 Удельная электропро-
натрия Катионные ПАВ водность
Додецил аммонийхлорид CJ2H25NH3Cr 1,5-Ю-2 Удельная электропро- водность
Тетрад ециламмонийхлорид С14Н29ЙН3СГ 2.8-10-3 То же
Додецилметил аммонийхло- C12H26(CH3)NH2C1- 1,5- Ю-2 »
рид J. 2
Додецилдиметил аммоний- хлорид CI2H25(CH3)2NHCr 1,6- 10 »
Додецилтриметил аммоний- хлорид Cl2H26(CH3)3NCr + 2,0 • Ю-2 »
Додецилпиридииийхлорид C12H25NC5H6C1- Неионогенные П 1,5- Ю-2 AB Поверхностное натя- жение
Додециловый эфир тетра- этиленгликоля Ci2H25O(C2H4O)4H 4-10-5 По верхностиое н атя- жение
Додециловый эфир гекса- этиленгликоля С12Н25О(С2Н4О)6Н 8,7 • IO-5 То же
Додециловый эфир окта- этиленгликоля C12H25O(C2H40)8H 8,3- 10-5 Солюбилизация кра- сителя
Додециловый эфир додека- этиленгликоля C12H25O(C2H4O)I2H 1,4 - 10—4 Поверхностное натя- жение
п-трет-Октил фениловый эфир триэтилеигликоля C3Hi7C6H4O(C2H4O)3H 1,1 • 10-4 То же
w-трет-Октил фениловый эфир пентаэтиленгликоля C3H17C6H4O(C2H4O)5H 1,3- 10 »
w-трет-Октил фениловый эфир гептаэтиленгликоля С8Н17С6Н4О(С2Н4О)7Н 1,8- 10-4 »
214