/














































































Author: Дормидонтов Ю.П.
Tags: спектральные методы анализа оптические методы анализа органическая химия спектроскопия спектрометрия спектральный анализ методы спектроскопии
ISBN: 5-7944-0132-X
Year: 2001




Text
Дормидонтов Ю.П.
МЕТОДЫ УФ, ИК И ЯМР СПЕКТРОСКОПИИ И ИХ ПРИМЕНЕНИЕ В ОРГАНИЧЕСКОЙ ХИМИИ
Учебное пособие
МИНИСТЕРСТВО ОБРАЗОВАНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
Пермский государственный университет
Ю.П. Дормидонтов
МЕТОДЫ УФ, ИК И ЯМР СПЕКТРОСКОПИИ И ИХ ПРИМЕНЕНИЕ В ОРГАНИЧЕСКОЙ ХИМИИ
Учебное пособие по спецкурсу
Пермь 2001
ББК 24.2
Д68
УДК 543.4'547
Дормидонтов Ю.П.
Д68 Методы УФ, ИК и ЯМР спектроскопии и их применение в органической химии: Учебное пособие по спецкурсу // Пермский ун-т. Пермь, 2001.154 с.
ISBN 5 7944-0132-Х
Издание подготовлено в соответствии с программой спецкурса по физическим методам исследования для студентов государственных университетов специальности 2018- Химия.
Рассматриваются общие вопросы электромагнитного спектра, теоретические аспекты и практическое применение методов УФ, ИК и ПМР спектроскопии.
Предназначено студентам химического факультета, знакомящихся с основами физических методов исследования, а также может быть полезно студентам V курса и магистрам, специализирующимся по кафедре органической химии.
Табл. 16. Ил. 47. Библ. 20 назв.
Печатается по решению редакционно-издательского совета Пермского госуниверситета.
Рецензенты: кафедра физической и коллоидной химии Пермской государственной фармацевтической академии (зав. кафедрой доктор химических наук, профессор ВЛ. Гейн), доктор химических наук, профессор Пермской сельскохозяйственной академии A.11. Козлов.
ISBN 5-"1944-0 * 32-Х
© Ю.П. Дормитонтор. 2001
Введение
Современное состояние химии невозможно представить без применения физических методов исследования веществ, использующих отражение, поглощение или эмиссию электромагнитного излучения. Таких методов существует большое количество: от первых, появившихся на рубеже XIX-XX вв., методов дипольных моментов и рефрактометрии до современных типа РСА, ХПЯ, ЭПР и др. И хотя среди них нельзя, да и не имеет смысла выделять какой-то один или несколько методов как основные или всеобъемлющие, поскольку каждый метод решает свои задачи, тем не менее среди них необходимо отметить оптическую и радиоспектроскопию и масс-спектрометрию, как наиболее экспрессные и информативные.
Полный спектр электромагнитных колебаний занимает диапазон длин волн от десятков метров до долей нанометра, от радиоволн до жесткого у-излучения. Поэтому из спектров можно получить качественную и количественную информацию практически о всех видах движений, совершающихся на уровне молекул, атомов, ядер и электронов.
Однако не существует единого метода, при помощи которого можно было бы за один прием исследовать все виды и формы движения и таким образом исчерпывающе охарактеризовать химический объект. Для каждого диапазона частот или длин волн используется свой метод. И развитие физических методов определялось и определяется уровнем развития технической физики, наличием подходящих источников излучения, чувствительных детекторов, степенью разработки средств измерения и способов подавления помех. Физические методы рождались, развивались и существуют на стыке двух наук - физики и химии: первая разрабатывает и предоставляет методы и аппаратуру для исследования, а вторая - объекты для этого. Это приводит к постоянному развитию и физики, и химии.
В большинстве физических методов исследования происходит взаимодействие вещества с электромагнитным излучением (его электрической или магнитной компонентой) во всем диапазоне частот. При этом происходит изменение энергии молекул вещества в соответствии с соотношением Бора:
ДЕ = Е-Е0 =hv,
где ДЕ- изменение энергии системы, Е и £0 - энергия системы в возбужденном и стационарном состоянии, h - постоянная Планка, v- частота.
Если энергия возбужденного состояния больше энергии стационарного (£ > £0), т.е. ДЕ положительна, то наблюдается поглощение энергии; если £ £(), т.е. ДЕ отрицательна, то происходит излучение энергии.
В первом случае имеют дело со спектрами поглощения, во втором - с эмиссионными спектрами.
Электромагнитное излучение характеризуется волновыми и квантовыми параметрами. Волновой параметр выражается длиной волны Л (нм, см, м) или частотой тл(сек '), которые между собой связаны закономерностью:
с । и = — сек, 2
где с - скорость света.
Излучение может быть охарактеризовано не только частотой, но и волновым числом и, обратным длине волны (и = 1/2), иногда также называемой частотой, и измеряемым в см '. Оно показывает количество длин волн излучения, укладывающихся в 1 см. Истинная частота (сек ') показывает количество колебаний в 1 сек.
Квантовый параметр излучения - это энергия перехода между двумя (стационарным и возбужденным) энергетическими уровнями изучаемой частицы и измеряется в электроновольтах (эВ/молекула) или кило-калориях/моль (ккал/моль).
Связь между энергетическими и волновыми параметрами выражается следующими соотношениями:
ЛЕ - h v = h-c/A. = h-c- v ,
. 107 , 28591,2 , 1239,81 „
2 нм = ---- cm ' = -------- ккал/моль =-------- эВ,
v E E
IO7
v см ! = 349,758-£ ккал/моль = 8065,75-£ эВ =---- нм,
2
£ ккал/моль = 23,0609-£ эВ = 0,00285912- v см ' =
28591,2
2
нм,
123981
£ эВ = 0,000123981 v см~' = 0,0433634-£ ккал/моль =-нм.
2
Например, длине волны 400 нм соответствует волновое число 25000 см энергия 3,1 эВ/молекуле или 71,5 ккал/моль. Отсюда следует, что чем выше энергия излучения, тем меньше длина его волны (2), больше частота (и) или волновое число (v ).
Электромагнитный спектр простирается от /-лучей с длиной волны (2) 10 10 см до радиоволн (2 порядка I05 см), те. длины волн по всему диапазону изменяются на 15 порядков. Также изменяется и энергия от I О7 эВ и более для /-лучей до 10 8 эВ для радиоволн.
Высокоэнергетическая область спектра начинается /-лучами (£ -
4
|0н-10-8 см, Е~ 107 эВ), которые характеризуют изменения в энергетическом состоянии ядер, вызывая внутриядерные превращения (у-резо-нансная спектроскопия).
Рентгеновские лучи (/1 = 1О'-10 6 см, Е ® I О5 эВ) изменяют энергетическое состояние внутренних, прилегающих к ядру электронов, не затрагивая ядра. Изучение взаимодействия данных электронов и ядра дает возможность определить энергию связи электронов как с ядром, так и между собой (рентгеноскопия).
Ультрафиолетовое и видимое излучение, объединяемое в одну область электронных спектров (Л = 10 6-1О4 см, Е ® 10 эВ) соответствует изменению энергии внешних, валентных электронов (электронная, или УФ спектроскопия).
Инфракрасные лучи (Л = 1 (У4-10 2 см, Е ® I О"1 эВ) вызывают изменения энергии колебательных уровней молекулы, те. колебаний связей и функциональных групп (колебательная, или ИК спектроскопия).
Микроволновое поглощение (Л = 10-2-10 см, Е ® 10 3 эВ) связано с изменением энергии вращения атомов в молекуле и с колебаниями атомов в кристаллической решетке (микроволновая спектроскопия).
Радиоволны (Л > 10 см, Е < 10 5 эВ) соответствуют изменению энергетического состояния спинов ядер и электронов (радиоспектроскопия, подразделяемая на метод ядерного магнитного резонанса - ЯМР, ядерного квадрупольного резонанса-ЯКР, электронного парамагнитного резонанса - ЭТТР, электронного циклотронного резонанса-ЭЦР и др.).
Излучение любого источника состоит из лучей с различной длиной волны и называется полихроматическим. С помощью специальных устройств может быть получено излучение с определенной длиной волны и, соответственно, энергией. Такое излучение называется монохроматическим.
Другой характеристикой монохроматического излучения является его интенсивность, связанная с количеством квантов, проходящих через единицу площади в единицу времени. Она определяется законом Бу-гера-Ламберта-Бера:
lg (4Z/) = к п-
где /0 и / -интенсивности падающего (/0) и прошедшего J (/) света, п - число молей поглощающего вещества на —► пути светового потока, к- постоянная, определяемая природой поглощающего вещества.
Для растворов вещества в растворителе, прозрачном
1 в данном интервале частот (рис. I), то же уравнение имеет
вид:
5
D = lg- = £-cl,
/
где D - оптическая плотность вещества (определяется экспериментально); /-толщина поглощаюшего слоя или длина светового пути в растворе, см; с - концентрация вещества, моль/л; £ - мольный коэффициент поглощения, или коэффициент экстинкции (иногда - просто экстинкция), л/молысм.
Область интенсивного поглощения излучения называется полосой поглощения, а совокупность полос представляет собой спектр поглощения соединения.
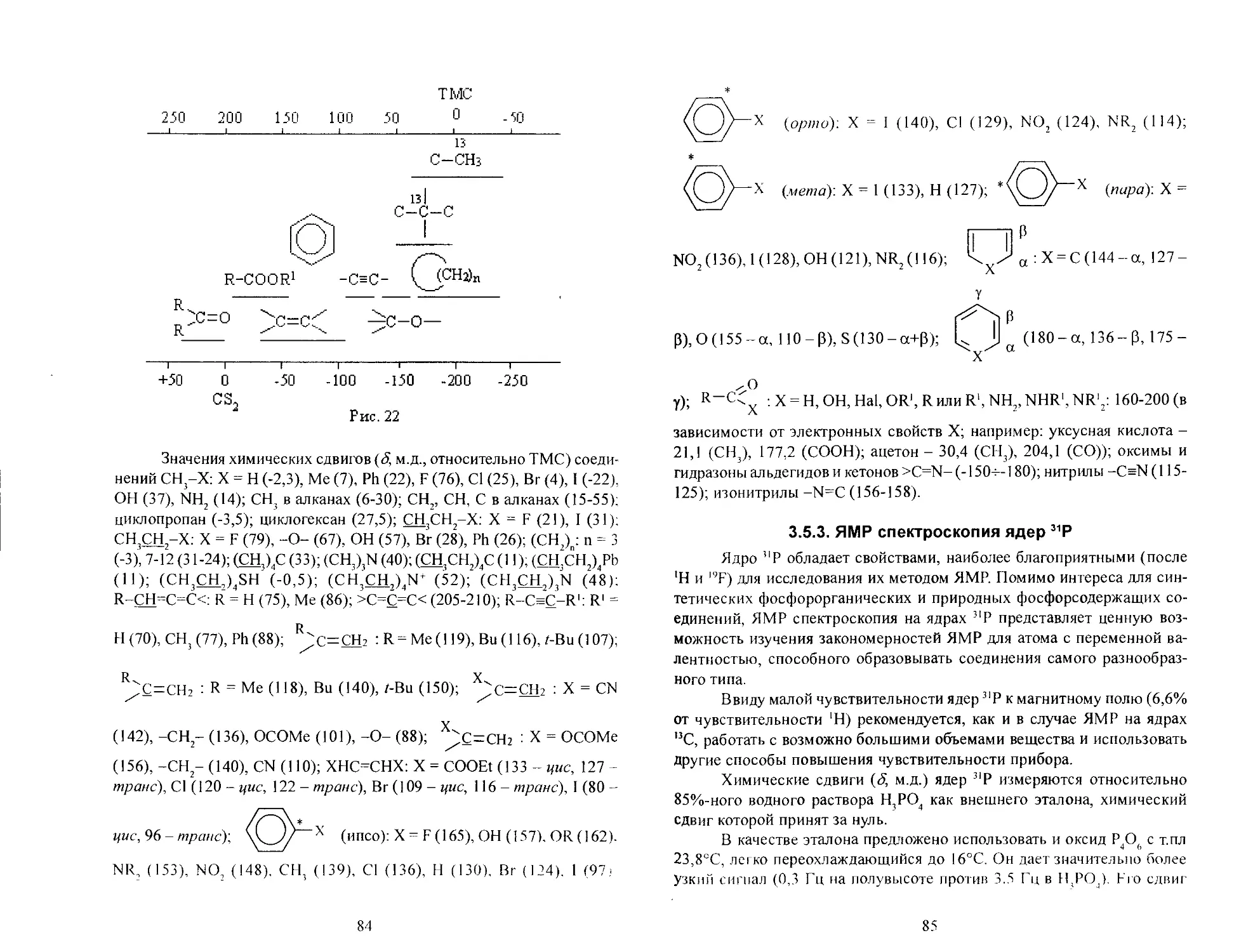
Электронный спектр соединения записывается как функция оптической плотности от длин волн: Df(X) или молярного коэффициента поглощения - экстинкции (гили ее логарифма) от длин волн: lg а ИК спектр - как функция процентного поглощения от волнового числа: % погл. Д v ). Электронный спектр может быть описан записью, например: Я (в гексане) 240 нм (г3400). В последнее время электронные спектры отображают как зависимость lg cfl v ), так как в этом случае энергия линейно изменяется по оси абсцисс.
1. Электронная (или УФ) спектроскопия
Весь диапазон электромагнитного спектра, изучаемый данным видом спектроскопии, подразделяется на 4 области.
1. Область 5-120 нм (область Лаймана). Исследуется при помощи дифракционных решеток, поскольку для нее не существует прозрачного материала. Эта область не имеет практического использования для исследования органических соединений.
2. Область 120-185 нм (область Шумана). Для изучения этой области используется флюоритовая оптика (из монокристалла CaF,, другие материалы имеют ограниченную прозрачность); источником излучения служат газоразрядные (водородные или гелиевые) трубки высокого напряжения, а приемником - специальные фотопластинки. Необходим глубокий вакуум (в этом интервале длин волн кислород прозрачен) или атмосфера азота, который прозрачен до 145 нм. В связи с другими техническими трудностями данная область имеет ограниченное применение для исследования.
Области Лаймана и Шумана образуют единую область дальнего ультрафиолета.
3. Область 185-400 нм (ближний УФ). Материал оптики - кварц, источник излучения - водородные лампы, приемники излучения - фото
6
элементы. Широко используется для изучения строения и свойств органических и неорганических соединений.
4. Область 400-800 нм (видимая область). Оптика - стеклянная или кварцевая, источники излучения - вольфрамовые лампы, а приемники излучения - фотоэлементы. Обычно измерение УФ спектров в пределах 185-800 нм проводят на одном приборе, снабженном кварцевой оптикой и сменными источниками и приемниками излучения, поэтому спектры в видимой области изучаются вместе с электронными спектрами. В интервале 200-800 нм воздух прозрачен, поэтому две последние области удобны для измерений. Доступность метода определяется и конструкционной простотой приборов.
Поглощение света в видимой и УФ областях обусловливается изменением в электронном состоянии молекулы, те. переходами валентных ст- и л-электронов со связывающих орбиталей или электронов неподе-ленных пар гетероатомов (w-электронов) с несвязывающих орбиталей на разрыхляющие о*- и я*-орбитали возбужденного состояния с повышенной энергией. Разные электроны в невозбужденном состоянии имеют различную энергию и поэтому возбуждаются излучением с различной длиной волны.
Последовательность энергетических уровней электронов в молекуле органического соединения представлена на рис. 2.
Разрыхляющая о*-орбиталь
Разрыхляющая л*-орбиталь
Несвязывающая и-орбиталь
Связывающая л-орбиталь
Связывающая ст-орбиталь
Рис. 2
Как видно из рисунка энергия связывающих л-орбиталей выше энергии связывающих ст-орбиталей, а для разрыхляющих о*- и л*-орбита-лей закономерность - обратная .
Трем типам электронов (ст-, л- и п-) соответствуют четыре типа электронных перехода: сто-ст*, л—>л*. п->а* и п>л* (первые два перехода образуют группу У->И-переходов, два вторых - N -->(?).
Таким образом, по энер| ии (Е) электронные переходы связаны следующей закономерностью: ст—>а* п-^а* > т—- п .
7
Вероятность перехода, определяющая интенсивность полосы поглощения. сильно зависит от изменения симметрии состояния молекулы при возбуждении, поэтому при более детальной классификации электронных переходов необходимо обратиться к анализу симметрии состояний, основанному на применении теории групп.
Но не вдаваясь в положения математического аппарата, констатируем, что любые л—>л*-переходы разрешены по симметрии, а п—^-переходы запрещены, поэтому интенсивность поглощения, связанного с тс—>л*-переходом высокая, а п—>л*-переходов - низкая. Аналогично, ст—><т*-переходы являются разрешенными, а п-^а* - запрещенными.
В электронной спектроскопии интенсивность полос поглощения обычно измеряется значением молярного коэффициента поглощения в максимуме (Л ) или минимуме (Л ) полосы (е или £ , или 1g £ или 1g fmin). Иногда поглощение может характеризоваться точкой перегиба. Полосы поглощения могут быть охарактеризованы также и интегральной интенсивностью А по формуле:
А = jf,, • ,
где г - молярный коэффициент поглощения при частоте г; ( и г,- частоты, ограничивающие рассматриваемую полосу поглощения.
Интегральная интенсивность определяется как площадь под кривой, вычерченной в координатах e-v.
Кроме энергетического фактора (энергия кванта должна быть равна энергии электронного перехода) на возможность поглощения накладывают ограничения также и правила, определяющие условия поглощения (правила отбора).
1. Правила отбора по спину запрещает переходы, при которых происходит изменение спина. Переходы, происходящие без изменения спина, называются синглет-синглетными (интенсивность полос высокая), а происходящие с изменением спина - синглет-триплетными (интенсивность полос низкая или они вообще отсутствуют в спектре).
2. Правило отбора по симметрии заключается в следующем: если симметрия основного и возбужденного состояний одинакова, то переход запрещен. Если хотя бы один из параметров перехода (смещение центра электрической тяжести, изменение значения или направления вектора электрического тока и др.) изменяется, то переход разрешен.
3. Правило отбора по локальной симметрии касается соединений, содержащих ненасыщенные группировки (ОО, OS. ON, N'- N, ON, N=O и др ), дающих поглощение, вызванное сииглет-синглезными /?->л*-пе-
8
реходами. Несмотря на синглет-синглетность переходов, интенсивность этих полос, как правило, невелика, поскольку w-электроны редко находятся на чистых р-орбиталях, а значит нет «чистого» п—>л*-перехода, и поэтому он оказывается запрещенным. В этом случае говорят, что переход запрещен по локальной (групповой) симметрии.
4. Общее правило отбора: если при поглощении энергии происходит возбуждение более чем одного электрона, то такие переходы также запрещены (этот запрет может частично сниматься другими факторами, данный вопрос рассматривается в специальной литературе).
Как видно из рис. 2, наиболее энергетически емкими переходами являются ст—>сг*- и п—>о*-переходы, поэтому соединения, не содержащие кратных связей (СпН2п+|Х, где X = Н, Hal, ОН, ЫН2 и др.) и для которых возможны только данные переходы, возбуждаются при воздействии на них лучей с высокой энергией (2 < 200 нм, область Шумана, что, как указывалось выше, связано с определенными трудностями в изучении спектров).
В то же время соединения, содержащие кратные С=С, С=С, С=О, C=N, C=N и др. связи, в которых возможны л—>л*- или п~> л*-переходы (а иногда и оба вместе) поглощают длинноволновое излучение (Я > 200 нм), что соответствует ближней ультрафиолетовой или видимой области.
1.1. Структура органических молекул и электронные спектры. Хромофоры и ауксохромы
Поглощение в ближнем ультрафиолете и видимой области обусловлено л—>тс*- и п—>л*-переходами, которые реализуются только в молекулах, содержащих кратные связи. Атомная группировка, включающая хотя бы одну кратную связь и придающая соединению способность к избирательному поглощению в данных областях, называется хромофором. Хромофоры подразделяются на изолированные и сопряженные. К первым относят группировки с одной кратной связью (С=С, С=О и N=N и др.), а ко вторым - структурные элементы сопряженных кратных связей.
Соединение, содержащее сопряженный хромофор, поглощает в более длинноволновой области и с большей интенсивностью, чем соединение с изолированными хромофорами. В этом случае спектр полихромофорного соединения трактуется просто как результат суммирования поглощения соответствующих изолированных хромофоров.
Некоторые из хромофоров (например, С=С или С=С-С=С) обеспечивают поглощение в ближнем ультрафиолете за счет только тс-^-тг*-переходов, другие (С=0-хромофор) - за счет «—>я*-перехода (крайне
9
редко за счет л—> л*-перехода), а третьи (например, сопряженный С=С-С=О-хромофор) вследствие реализации сопряжения - как так и и—>л*-переходов.
Атомную группировку, не содержащую связей и не имеющую максимума поглощения в ближнем ультрафиолете, но включение которой в систему хромофора приводит к увеличению длины волны я-*т?- или п-я*-переходов и повышению интенсивности поглощения, называют ауксохромом. Типичными ауксохромами являются группы ОН, OR, NH2, NR, и др., т.е. группы, содержащие гетероатом с неподеленной (свободной) электронной парой.
При выявлении взаимосвязи электронного спектра и структуры молекулы признается целесообразным наблюдение за изменениями в положении и интенсивности полосы поглощения при переходе от некоторого родоначального хромофора, «ответственного» за поглощение, к модифицированному путем введения в его систему дополнительной хромофорной или ауксохромной группы. Для характеристики спектральных изменений, вызванных модификацией структуры, используется специальная терминология:
батохромный сдвиг - смещение полосы поглощения в длинноволновую область спектра;
гипсохромный сдвиг - смещение полосы в коротковолновую область спектра;
гиперхромный эффект - увеличение интенсивности поглощения; гипохромный эффект - уменьшение интенсивности поглощения.
Эти же термины используются и для описания изменений в спектре, вызываемых заменой растворителя.
Полосы поглощения в УФ спектрах могут заметно различаться своими параметрами: положением, интенсивностью и формой: полосы со сходными признаками в определенной мере соответствуют родственным хромофорным группам. Это позволило классифицировать полосы и сформулировать эмпирические критерии для их отнесения.
При интерпретации полос при структурном анализе различают три вида полос:
1. Очень интенсивные полосы (g> 103) соответствуют я—>/^-переходам сопряженных систем (в литературе обозначаются как К-полосы; аналогичные по интенсивности полосы я—>л*-переходов в ароматических системах обозначают как Е-полосы: Е с е = 104-105 соответствует разрешенным переходам, а Е, с £= 2000-12000 - запрещенным.
2. Слабые полосы л?—» л*-переходов (е< 100), характерные для непредельных гетероаюмных функциональных групп и радикалов (R-no-
10
лосы).
3. Полосы средней интенсивности (г = 100-1000) соответствуют запрещенным я—>л*-переходам в ароматических структурах бензольного типа (В-полосы).
Дополнительными эмпирическими признаками для классификации полос, кроме их интенсивности, служат смещение максимума поглощения при переходе от неполярных растворителей к полярным и наличие тонкой колебательной структуры.
Замена неполярного растворителя полярным приводит к увеличению частоты и—> л*-перехода, т.е. к гипсохромному сдвигу R-полосы. При этом В-полосы, а также К-полосы соединений, подобных дивинилу или стиролу, обычно не испытывают смещения, но К-полосы соединений типа акролеина, нитробензола или анилина, отвечающие переходам с внутримолекулярным переносом заряда при аналогичной замене растворителя, испытывают батохромный сдвиг.
Тонкая колебательная структура встречается у полос всех трех типов и может быть надежным признаком определенного хромофора. В частности, тонкая структура характерна для К-полос полиинов, В-полос аренов, R-полос алкилнитритов и т.д.
1.2. Электронные спектры отдельных классов органических соединений
Насыщенные соединения: в алканах и циклоалканах возможны только сг->о*-переходы, лежащие в дальнем ультрафиолете (Л : метана 125 нм, этана 135 нм; в общем, чем длиннее и разветвленнее алкан, тем больше Л, но не выше 170-175 нм). В замещенных алканах (галогениды, спирты, эфиры, меркаптаны, сульфиды, дисульфиды, амины и др.) наряду с полосами ст—>о*-переходов присутствуют полосы и—>о*-переходов, лежащие при длинах волн на 30-40 нм больших (батохромный сдвиг), чем ст—>о*-переходов.
Ненасыщенные соединения. Алкены, циклоалкены или полиены с изолированными двойными связями поглощают (л-->л*-переход) в интервале 165-200 нм (2тах этилена 165 нм), и чем больше алкильных заместителей присоединено к двойной связи, тем наблюдается больший батохромный сдвиг. Одновременно наблюдается и гиперхромный эффект.
Поглощение алкинов с изолированной связью также обусловлено л->л*-переходом. Для них характерен батохромный сдвиг по сравнению с соответствующим алкеном (Лтах ацетилена 173 нм). Влияние алкильных заместителей хотя и заметно, но проявляется не так заметно, как для
11
алкенов.
Для сопряженных ди- или полиенов наблюдается батохромный сдвиг и гиперхромный эффект по сравнению с алкенами (Лтах 1,3-бута-диена217 нм, 1,3,5-гексатриена 256 нм).
Замена в сопряженной системе С=С-связи на ОС-связь практически не меняет положение полос поглощения, но вызывает уменьшение их интенсивности.
В спектрах ди- и полиинов наблюдается батохромный сдвиг по сравнению с алкинами (173-185 нм) до 210-250 и 340-390 нм соответственно.
Карбонильные соединения (альдегиды, кетоны, кислоты и их производные: эфиры, галогенангидриды, ангидриды, амиды). В этих соединениях возможны переходы п-^л* и п—>о* (последние - не анализируются). Наиболее характерны полосы и—>л*-переходов. Хотя их молярный коэффициент погашения мал (г< 100-1000) и не всегда может быть проанализирован, он наиболее изучен: с увеличением полярности растворителя его полоса сдвигается в коротковолновую область (гипсох-ромный сдвиг; почему?); в кислых средах она исчезает (почему?).
При переходе от формальдегида (Лтах 295, 185 и 155 нм, что соответствует я->л*- и о*-переходам) к ацетальдегиду или ацетону (и далее к насыщенным альдегидам или кетонам) наблюдается гип-сохромный сдвиг и—»л*-переходов и батохромный л->л*-переходов.
В дикарбонильных соединениях поглощение зависит от расположения С=О-групп: в спектрах а-дикетонов, а-кетоальдегидов, а-кетоэ-фиров, а-диэфиров имеет место небольшой гипсохромный сдвиг и гиперхромный эффект по сравнению с аналогичным монокарбонильным соединением.
Азометины (соединения со связью >C=N~), являющиеся изоэлек-тронными аналогами альдегидов и кетонов, поглощают в интервале 240-250 нм (батохромный сдвиг).
При переходе к спектрам тиокарбонильных соединений наблюдается значительный батохромный сдвиг по сравнению с аналогичными О-содержащими соединениями.
Ароматические соединения. Бензол имеет три полосы поглощения, связанные с л-->л*-переходами: хпик 180 (Ех или просто Е, s= 50000), 200 (Е2 или К, е= 7000) и 260 нм (В, £= 200). При введении алкильных заместителей и галогенов происходит незначительный сдвиг полос в длинноволновую область спектра с увеличением интенсивности. (Полоса при 180 нм исчезает, поскольку она в бензоле связана с сохранением кольцом симметрии правильного шестиугольника.) Введение заместите
12
лей, обладающих ±М-эффектами (ОН, NR2, СО, C=N и др.) приводит к батохромному сдвигу, который зависит от величины, но не направленности эффекта.
При переходе к спектрам конденсированных ароматических систем (нафталин, антрацен, фенантрен и др.) происходит (наряду с изменением характера происхождения полос) батохромный сдвиг поглощения.
Аналогичная картина (батохромный сдвиг) наблюдается и в спектрах гетероциклов, причем чем меньше электроотрицательность гетероатома, тем меньше сдвиг; чем больше гетероатомов в ядре, тем больше сдвиг; чем больше колец образуют гетероароматическую систему (например, пиррол-индол-карбазол), тем больше сдвиг.
1.3. Применение УФ спектроскопии
При интерпретации УФ спектров можно руководствоваться рядом эмпирических правил: I) полосы низкой интенсивности (lg е< 2) обусловлены наличием в молекуле карбонильной, тиокарбонильной, нитро-или нитрозогруппы; 2) полосы поглощения с Лтах= 250-300 нм (lg е= 2-3) связаны с бензольными соединениями; 3) полосы с 2тах > 200 нм (lg £> 4) характеризует соединения с сопряженными кратными связями.
По УФ спектрам легко отличаются соединения, содержащие сопряженные или несопряженные системы (для первых наблюдается значительный батохромный сдвиг).
По значению Ял устанавливается структура сопряженной системы в полиенах:
/тах = 217 + 5-А + ЗОВ + 5-С, где 2тах и 2I7 - длины максимумов поглощения в спектрах изучаемого сопряженного полиена и 1,3-бутадиена; А - число алкильных заместителей, присоединенных к сопряженной системе (могут содержать кратные связи, но не сопряженные с основной системой); В - число сопряженных двойных связей минус 2 (они содержатся в 1,3-бутадиене); С - число экзоциклических двойных связей (отходящих от 5-7-членных колец).
Для циклических шестичленных диенов за основу берется число 256 (вместо 217), а для пятичленных - 240. Остальные инкременты остаются прежними.
В случае непредельных стероидных углеводородов (!) начальное значение длины волны определяется взаимным расположением двойных связей: если они находятся в одном шестичленном цикле (кольце), то эта величина равна 253, если в разных, то 214. Если сопряженные двойные связи находятся и в одном и в разных шести- или пятичленных циклах, то используют значение 253.
13
Если в состав соединения входят шести- или пятичленные ненасыщенные кольца, то за основу берется значение 256 или 253.
СН2==СН-С=СН-СН=СН-СНз
Пример 1. '
Л (выч.) = 217 + 5-2 + 30-1 = 257 max'- '
Пример 2.
^\^СН-СН=С(СНз)2
Л (выч.) = 214 + 5-5 + 30-3 + 5-1 =334
Как правило, разница между экспериментально найденными и вычисленными значениями z.m,x не превышает ±2-3 нм, что позволяет делать выводы о строении сопряженной системы.
Структура непредельных альдегидов или кетонов может быть установлена по УФ спектрам их 2,4-динитрофенилгидразонов по формуле:
Л = 330 + 7-А + 17 + 9-В max
где Лтж и 330 - длины максимумов поглощения в спектрах 2,4-ДНФГ изучаемого карбонильного соединения и формальдегида, А - количество алкильных заместителей, присоединенных к C=N-rpynne или в конец (!) сопряженной системы; 17 - инкремент первой сопряженной двойной связи; В - число сопряженных двойных связей минус 2 (входит в фрагмент C=C-C=N).
/1 = 330 + 7-1 =337
O2N
Пример 5. СН3-СН=С—C=N-NH^O)—NO2
СН3 СН3 '--7
/1= 330 + 7-2 + 17 = 361
Нарушение сопряжения двух фрагментов вследствие стерических препятствий для сопряжения приводит к упрощению спектра, который становится похожим на спектр остатка одного из основных соединений.
14
По экспериментально найденной оптической плотности (D), зная концентрацию раствора (с) и толщину кюветы (/) по уравнению
D = £ с / можно рассчитать экстинкцию соединения, а для известного соединения (т.е. известно е) можно определить концентрацию раствора.
Если Лтах двух веществ, в том числе и таутомерных форм, имеют близкие значения, то, используя свойство аддитивности интенсивностей их поглощения, можно рассчитать состав смеси.
Пример 6.
СНз— СО-СН2— СО—СНз СНз—С(ОН)=СН-СО-СНз
Лтах275 нм (£= 100) Лтю 270 нм (е= 12000)
Мольный коэффициент поглощения данной смеси в гексане, этаноле и воде равен 11200, 9500 и 1800 соответственно. Следовательно, в гексане равновесие сдвинуто в сторону енольной формы на 92,6% (11200:12100), в этаноле - на 78,5%, в воде - на 15,7%.
УФ спектры можно использовать для контроля за превращениями в процессе реакций при наличии избирательного (различающегося) поглощения в конечном и исходном соединениях.
Пример 7.
I 2
—* СНзСОСОСНз—* Атах 275, 425 нм
А-тах 260, 310 нм
(оба п—>л*)
СН3СН2—С-СНз -II О
Атах 190 нм (л-^л*)
275 нм (п—>л*)
(оба п—>л*)
3 4
—»- СНзСН-С-СНз —* СН2=СН-С-СНз~*-I II II
Вг О о
Атах 180 нм (л->л*)
Атах 210 нм (я->я*)
320 нм (и—>л*)
285 нм (п—
Атах 280 нм (л—>л*) Атах 255 нм (л—>л*)
420 нм (н—>л*) 305 нм (и—>л*)
Реакцию 1 можно проконтролировать по исчезновению максимума поглощения при 190 нм с появлением в длинноволновой области нового максимума со значительно меньшей интенсивностью («--> л*-пере-ход) при 425 нм. Реакции 2 и 6 контролируются по исчезновению макси
15
мумов поглощения при 275,425, 280 и 420 нм. Реакция 3 контролируется исчезновением максимумов при 190 и 275 нм с появлением новых максимумов при 180 (гипсохромный сдвиг) и 285 нм (батохромный).
По УФ спектрам можно различить цис-транс- (син-анти-)-то-меры одного и того же соединения по интенсивностям полос я—>л*- или п—>л*-переходов (для транс- и инти-изомеров она значительно выше).
Наиболее надежные результаты применения УФ спектров для структурных исследований можно получить при сравнении спектра анализируемого образца со спектром ближайшего аналога (если таковой имеется), на основании чего можно сделать заключение об электронных взаимодействиях в молекуле, характере переходов и, тем самым, определенные выводы о строении образца.
1.4. Возможности и ограничения метода
Возможности метода: доказательство наличия в исследуемом веществе группировок-хромофоров сопряженной диеновой, полиеновой или ароматической систем или карбонильной группы или их отсутствия (по отсутствию максимумов поглощения при соответствующих длинах волн); в простейших случаях возможность определения типа хромофора, длины цепи сопряжения, количество алкильных групп, присоединенных к хромофору; количественный анализ смесей, включая регистрацию изменения концентраций растворов во времени.
Ограничения метода: ограниченность применения вследствие того, что некоторые соединения не имеют максимума поглощения в исследуемом диапазоне длин волн; сравнительно малые возможности при решении структурно-аналитических задач; сильное влияние (в ряде случаев) природы растворителя на характер спектра и возможность отклонений от закона Бугера-Ламберта-Бера; возможность фотохимической изомеризации в процессе съемки спектра (например, цис-транс-кзоме-ризация в ди- и полиеновых системах).
1.5. Задачи для самостоятельного решения
I. Рассчитать длину максимума поглощения для соединений:
а) (Сп3)2С=сн-сн-С(Сн-,)2 ; х-ги ;
НзС 1Н С(СНз)—СИ— СНз
ШСНз
СНз ’
16
б) (СНз)гС=С(СНз)—CH=N-NH-C6H3(NO2)2-2.4 ;
(СНз)гС—СН—СИ = СН—С(СгН5)—N—NH—C6H3(NO2)2-2,4 .
2. Определить степень прохождения (%) реакции димеризации 5-этил-циклопентадиена (Лтах 247 нм, е= 3400) за I, 2 и 4 ч в растворе гексана, если в каждый момент времени г равна 985, 1530 и 2310 (димер при 247
нм полностью прозрачен).
3. Примените УФ спектроскопию для контроля за превращениями:
2. Инфракрасная (ИК) или колебательная спектроскопия
ИК спектроскопия является одним из самых важных и применяемых методов исследования веществ. Под действием ИК излучения (Л = 800-106нм, v = 12500-10 см ', Е= 0,2-0,01 эВ) происходят изменения в колебаниях атомов и функциональных групп в молекуле, таких как С-Н, С-С, С=С, ОС, С-О, С—О, C-N, C=N, C=N и др. Это приводит к появлению в спектре полос поглощения, по которым определяется наличие в молекулах тех или иных связей или групп.
Колебания связей и групп могут быть валентными (v) и деформационными (<5).
Вся ИК область (12500-10 см ') подразделяется на ближнюю (к видимой области) ИК (12500-5000 см '), фундаментальную (5000-250 см ') и дальнюю (250-10 см '). В каждой из этих областей решаются свои вопросы по идентификации, установлению строения веществ и обнаруже
ния его каких-либо специфических особенностей. В этом плане самой важной является фундаментальная ИК-область, в которой сосредоточены наиболее характерные для структурного анализа колебания функцио нальных групп и различных связей.
2.1. Теоретические основы ИК спектроскопии
ИК спектры обусловлены переходами между двумя энергетическими уровнями молекулы, находящейся в основном электронном состоянии, и связаны с тем, что кванты ИК излучения непосредственно возбуждают колебания связей и функциональных групп.
Для двухатомной молекулы (A-В) возможен только один тип колебательного движения, связанный с растяжением или сжатием связи (модель - пружина, рис. 3) между атомами А и В, имеющими массы т и да,.
т । т
к--------г----------1
Рис. 3
Если пружину растянуть, азатем освободить от действия приложенной силы, то происходит простое колебание атомов около положения оавновесия (:), причем частота колебаний (г) определяется по закону Гука:
где /ч приведенная масса тел А и В, т.е. гармоническое среднее отдель-
т, да. . .,
тых масс !. да,. =—: ' ), л - силовая постоянная или возвращающая да, + да.
< ила на единицу смещения, связанная с упругостью связи.
Согласно квантовой теории, энергия колебательного движения
может иметь .тишь значения, определяемые уравнением:
Е, =(V + \/2)-hv,
де Г- 0, I.2. 3, ...
Зависимость колебательной энергии приведена на рис. 4.
( of taciio посуулагу Бора, поглощаемая частота излучения опре-с.тяеж я гю уравнению.
ЛЕ £, Е,„
/
При ишлощении кванта энергии молекула переходит с (гулевого уровня (К, £0 = '/,/?о) на первый (Ко, £0 = ’/,/? г) и, следовательно, час гота поглощаемого излучения (о) равна характеристической колебательной частоте. Переход с уровня Го на уровень К, будет происходить с частотой 2 иит.д. Отсюда v- основная частота перехода, 2г- первый обертон, Зг-второй и т.д.
Поглощаемая колебательная энергия быстро распределяется равномерно меж-
ду колебательным, вращательным и поступательным движениями множества молекул. Тогда, согласно закону распределения Больцмана, относительное число молекул с энергией £ определяется по уравнению:
где к- постоянная (константа) Больцмана, равная 1,38...Ю~28 кДж/К.
Если принять относительное число молекул на уровне У равным 1 и учесть, что колебательные энергии имеют значения порядка 1000 см 1 (0,1 эВ), то при обычной температуре (300 К) относительное число молекул, находящихся на уровне составит 0,0083. Отсюда видно, что поскольку почти все молекулы находятся на самом низком колебательном уровне (Г), колебательные переходы происходят только с пего. Однако даже с этого уровня не все переходы разрешены. Поэтому правило отбора с учетом того, что действующее пока колебание является строго гармоническим, запрещает переходы, которые изменяют уровень перехода И более, чем на единицу. Следовательно, разрешенным является основной переход Г'о—> К, и в этом приближении колебательный спектр должен содержать только одну основную полосу.
Обычно молекулярные колебания существенно, но не строго, гармонические. Если ангармоничность достаточно велика, то в спектре обнаруживается слабое поглощение, отвечающее первому обертону с частотой 2 г и интенсивностью около 0,1 интенсивности основной полосы (интенсивность полосы второго обертона с частотой 3 г составляет 0,01 часть интенсивности основной полосы и практически никогда не видна в ИК спектрах). Ангармоничность ведет также к сближению энергетических уровней, так что полоса первого обертона появляется при частоте несколько меньшей, чем 2 г.
19
Поскольку световые волны, в том числе и ИК излучение, являются колеблющимися электрическими и магнитными полями, то для их возбуждения необходима осцилляция электрических зарядов. Наоборот, если световая волна поглощается, то вместо нее должен возникнуть осциллирующий заряд. Следовательно, для того чтобы непосредственно поглощать ИК излучение, молекула должна колебаться так, чтобы происходило смещение центра электрического заряда, т.е. чтобы дипольный момент (//) молекулы при колебании изменился. Так как //равен произведению величины заряда на расстояние между ними, то с изменением длины связи он должен изменяться. Среднее значение момента является постоянным дипольным моментом молекулы.
При колебаниях изменения длины связи (например, в молекуле НС1) являются (рис. 5) небольшими (пунктирные линии), в основном бесконечно малыми величинами, так что изменение ц можно представить наклоном (d/ddr). кривой зависимости //от г в области равновесной длины связи (г = 0).
Теория показывает, что интенсивность ИК поглощения пропорциональна (d/jJdf)j и квадрату напряженности электрической компоненты световой волны. Значение (dp/dr)0 не связано непосредственно со значением постоянного дипольного момента (//), а только с наклоном функции //=/(г) при г = 0. Если равновесная длина связи совпадает с длиной кривой, то
значение (d^dr) = 0, даже если значение // велико. И наоборот, если кривая резко меняет наклон в зависимости от г, то значение (d/jJdr)0 может быть довольно большим, даже если значение //мало.
Например, у молекулы СО //= 0,1 D (дебай), а интенсивность поглощения (молярный коэффициент поглощения) г = 530 см2/моль (л/моль-см). У молекулы НС1 //= 1,03 D, а г = 360 см2/моль.
Таким образом, в ИК спектрах проявляются, т.е. активны, колебания, которые сопровождаются изменением дипольного момента молекулы.
Для количественного описания колебаний больших молекул необходимо к представлению о сопротивлении связи в двухатомной молекуле растяжению или сжатию добавить положение, согласно которому углы между связями сопротивляются деформации.
0
Длина связи, т
Рис. 5
20
Молекула из «и» атомов должна иметь столько же степеней свободы, сколько их имеют атомы вместе взятые. Так как каждый атом имеет 3 степени свободы поступательного движения, то молекула должна иметь Зп степеней свободы. Из них по 3 степени требуются для описания поступательного и вращательного движения в целом, а оставшиеся (Зя-6) степени свободы относятся к колебательному движению. В линейных молекулах вращение вокруг межядерных осей происходят только совместно с определенными колебаниями и поэтому вращение у них имеет две степени и для колебательного движения таких молекул остается Зп-5 степеней свободы.
Так как колебания происходят при небольших отклонениях атомов от положения равновесия, т.е. с пренебрежением ангармоничностью, то в общем случае движение атомов описывается Зп-6 (Зп-5) нормальных колебаний, при которых все атомы в молекуле движутся с одинаковыми частотой и фазой, т.е. достигают положения максимальных смещений в одно и то же время с одновременным прохождением через положения равновесия. Однако амплитуды колебаний различных атомов весьма различны.
Если молекула обладает элементами симметрии, то не все Зи-6(5) нормальных колебаний спектрально активны, так как каждая форма нормального колебания определенным образом преобразуется при выполнении той или иной операции симметрии. Существует 3 случая поведения нормального колебания по отношению к любой операции симметрии:
1) нормальное колебание остается неизменным-симметричное колебание;
2) оно изменяет знак на обратный - антисимметричное колебание;
3) оно переходит в другую форму нормального колебания - вырожденное колебание по отношению к данной форме симметрии.
Вырождение колебания заключается в том, что различные колебания имеют одинаковые частоты, вследствие чего в спектре вместо нескольких полос появляется одна. Дважды вырожденные колебания встречаются в том случае, если молекула имеет ось симметрии третьего (и выше) порядка. Трижды вырожденные колебания бывают у молекул, имеющих более чем одну ось симметрии третьего порядка.
Так как в ИК спектрах активны колебания, сопровождающиеся изменением дипольного момента молекулы, а в спектрах КР (комбинационного рассеяния) активны колебания, связанные с изменением их поляризуемости, то существует правило альтернативного запрета: в случае центросимметричных молекул в ИК спектрах активны колебания,
21
антисимметричные относительно центра симметрии, а в КР спектрах -колебания, симметричные относительно этого центра.
В этом плане рассмотрим колебания воды и ацетилена.
В случае воды (Н2О, три атома) имеется три типа колебаний: Зи-6 = 3. Два колебания связаны с валентными колебаниями (у) ОН-груп-пы , а третье является деформационным (3) и отражает изменения угла между связями.
Валентные колебания ОН-группы будут симметричными (у), когда обе связи растягиваются или сжимаются одновременно и поэтому длины связей О-Н достигают наибольших или наименьших значений в одно и то же время. В антисимметричном колебании (у ) одна связь О-Н растягивается, в то время как другая связь укорачивается, т.е. их фазы колебаний противоположны (рис. 6).
Деформационные колебания (3) происходят при частотах, намного ниже частот валентных колебаний, так как для деформации угла требуется меньшая энергия, чем для растяжения связей.
Молекула ацетилена Н-С=С-Н линейная и имеет семь (3 -4-5 = 7) нормальных колебаний, из которых деформационные колебания дважды вырождены, так как осуществляются в двух взаимно перпендикулярных плоскостях с одинаковыми частотами (рис. 7).
Колебания с и и3, происходящие вдоль оси молекулы, связаны в основном с изменением длины связи С-Н, причем колебание с у, симметрично (у), а колебание с у3 - антисимметрично (у^). Колебания с у2 происходят также вдоль оси молекулы, но частота колебания определяется в основном силовой постоянной связи С=С (массы одинаковы).
Деформационные колебания и 32 симметричны и антисимметричны колебаниям связи С-Н. В ИК спектре активны только колебания с частотами у = 3287 см_| и = 729 см1, происходящие с изме-нением дипольного момента. В КР спектре про-являются только 2 интенсивные полосы с у = 3374 см-1 и и, - 1974 см'. Колебания с 3 оптически s 2
неактивны.
Таким образом, колебания молекулы ацетилена четко разделяются на валент ные и деформационные. Однако если хотя бы один из атомов водорода в этой молекуле заменить метильной группой, то такое строгое деление колебаний исчезает. Так, колебания связи С=С в метилаце-тилене (пропин, СН,С=СН, ус = 2142 см ') весьма существенно зависят от одинарной связи С-С и в малой
степени от величины углов Н-С-С. В то же время в симметричные деформационные колебания (<7) метильной группы небольшой вклад вносит растяжение связи С=С.
Экспериментальные исследования большого числа соединений, обладающих одними и теми же функциональными группами, показали, чго независимо от изменений в остальной части молекулы, эти одинаковые группы поглощают в сравнительно узком интервале частот. Такие частоты получили название характеристических, или групповых.
Например, все гидрокспсоединения (в частности, спирты и фенолы) поглощают около 3600 см-1, что характерно тля валентных колебаний ОН-группы.
К характеристическим частотам относятся колебания групп СН, СН„ СН„ ОН. NH, NH,, С- С, С^С, C=N, C=N и др
Для грубой оценки областей, в которых проявляются отдельные характеристические колебания, можно воспользоваться формулой:
Так. например, iруппа С-Н, рассматриваемая как двухатомная молекула, имеет приведенную массу т ,Н1 - 12/13 аг. ед. У изолированной С-С-связи те = 6. Так как частота меняетс я е щнообразно в зависимости от
III, , H./w |(( , При I/ . 1000 М и 1 г: v> г - с .и лк : ( 180
— . то. еч.тщ силовые постоянные неизменны, у (/1; (
см ', что попадает в пределы интервала, наблюдаемого в действительности.
Колебание будет тем более характеристичным, чем больше параметры колеблющейся группы атомов (К и те) отличаются от параметров остальной части молекулы.
Так, характеристичными будут колебания групп, содержащих легкий атом водорода (СН?, СН2, CH, NH,, NH, ОН и др.). В этом случае частоты настолько высоки, что их поглощение четко отделяется от всех других частот в спектре и они лежат в области высоких частот. Водород, присоединенный к ненасыщенному атому углерода, дает полосу валентного колебания около 3000 см-1.
Группы, содержащие кратные связи (С=С, C=N, С=О, С=С, ON и др.) имеют характеристическое поглощение, относящееся к колебаниям, в которых, в основном, происходит растяжение соответствующих связей. При этом группы, содержащие тройные связи, имеют более высокие характеристические частоты, чем группы с двойной связью, так как у первых групп возвращающие силы (силовые постоянные К) больше, чем у вторых.
Если же атомы близки по массам и соединены связями с соизмеримыми силовыми постоянными, то невозможно отделить колебания, относящиеся к индивидуальным связям (С-С, С-О, C-N), и полосы поглощения отвечают колебаниям большого числа углов и связей. Поэтому любое замещение, небольшое изменение геометрии молекулы приводит к сдвигам полос поглощения. Колебания, определяемые изменением углов (деформационные), лежат в области значительно меньших частот, чем колебания, связанные с изменением длин связей (валентные), так как возвращающие силы при деформации углов значительно меньше (рис. 8).
Обычно обертоны слабее основных полос в ~ 10 раз. Однако основные полосы имеют столь широкое распределение по интенсивностям, что обертон одной сильной основной полосы может быть более интенсивным, чем другая слабая основная полоса. В особых условиях, когда энергия обертона случайно совпадает с энергией другого основного колебания, наблюдается явление резонанса и обертон становится ненормально интенсивным. Резонанс приводит к появлению двух переходов близкой интенсивности, причем каждый из переходов имеет в значительной степени характер основного. Это явление называется резонансом Ферми и имеет широкое распространение.
В ИК области трудно получить точные данные по интенсивности, так как излучение, попадающее на детектор, никогда не является строго
Обертоны
V(£c-H) V(=c-H)
V(H-H) V(C-H)
____________1700
V(C=C=C) ^C-H) 7(C-H)
V(C®H) ^O-H) ^0-H)
^N-H)
О О о о о
О О **"> о
СП о 00 СП CS
CN CS
V(»C)
2 S
V(C-C, с-0. C-N) §
Е500
Функциональные группы
Рис. 8. Области поглощения некоторых структурных элементов v- валентные колебания, 8- деформационные плоскостные колебания, у- деформационные внеплоскостные колебания
монохроматическим, а сами полосы довольно узкие. Положение усугубляется трудностями, которые возникают при подготовке образца: малые объемы растворов и тонкая, легко разрущаемая кювета. Поэтому данные по коэффициенту экстинкции £пах невоспроизводимы и рассматриваются как кажущиеся f
При уменьшении разрешающей способности прибора ширина полосы увеличивается, а ее интенсивность уменьшается. Площадь полосы (А) меняется намного меньше (в зависимости от разрешающей способности прибора) и поэтому является более надежным критерием интенсивности, однако ее вычисления довольно трудоемки.
24
25
2.2. Основные параметры полос ИК поглощения. Положение полос в зависимости от условий записи спектра
Положение полосы поглощения в ИК спектре определяется как положение максимума поглощения (vmax, см'1), а ее ширина - как ширина на ее полувысоте (ЛЛ|/2) (рис. 9). Для расчета интегральных интенсивностей полос вычерчивают кривые в координатах е- и и считают площади, обрамляемые этими кривыми.
Для определения относительной интенсивности полос выбирают шкапа наиболее интенсивную
по шкале пропускания (или поглощения) и принимают ее интенсивность за 100%. Если интенсивность других полос составляет менее 30% от интенсивности наиболее интенсивной полосы, то такие полосы являются слабыми (сл.), если их интенсивности составляют 30-50%, то они являются средними {ср.), при интенсивности более 50-55% полосы -сильные (с.).
Когда видимый или ИК свет проходит через прозрачную среду, то часть его рассеивается и беспорядочно распространяется по отношению к направлению входящего луча. Если он монохроматический, то боль-
шая часть рассеяния (рассеяние Релея) состоит из света с неизменной частотой, а небольшая часть его имеет частоты, отличные от частот па
дающего света. Этот тип рассеяния, направленный перпендикулярно направлению падающего луча, называется комбинационным рассеянием (эффект Рамана), а спектры называются спектрами комбинационного рассеяния (СКР). Они дают информацию о частотах молекулярных колебаний.
В паро- или газообразном состояниях вещества, когда молекулы практически не ассоциированы, межмолекулярные взаимодействия отсутствуют или крайне незначительны. Поэтому истинный спектр веще
ства получается для его паров. Однако такие спектры не могут быть использованы для сравнения, так как это связано с трудностями испарения большинства органических (не говоря о неорганических) веществ, а также с изменением форм полос поглощения из-за появления элементов тонкой вращательной структуры.
В жидкости или расплаве молекулы находятся в окружении других молекул и под влиянием диэлектрических свойств среды, приводящих к ассоциации, или наличия межмолекулярных водородных связей (ММВС) полимерного типа происходит изменение частот колебаний. Эти специфические взаимодействия затрудняют сравнение характеристических частот групп различных веществ. В случае углеводородов и других относительно неполярных веществ взаимодействие между молекулами почти минимально в различных агрегатных состояниях. Тем не менее и в этих случаях обычно наблюдается понижение частот « на 1% при переходе от газообразного к жидкому состоянию, что объясняется значительным увеличением диэлектрической проницаемости.
Спектр твердого вещества изменен еще в большей степени, чем жидкого, не только благодаря усилению межмолекулярных взаимодействий, но и вследствие специфического влияния кристаллической решетки. Поэтому в спектре кристаллического состояния могут появляться дополнительные полосы, в особенности в области «отпечатков пальцев», или, наоборот, некоторые полосы исчезают. Появление дополнительных полос поглощения наблюдается, например, в спектрах высших жирных кислот, для которых в данной области имеется серия равномерно расположенных узких полос поглощения, обусловленных /иранс-расположе-нием метиленовых групп. В жидкой фазе в этой области имеется широкая полоса поглощения.
Под влиянием межмолекулярных взаимодействий, возникающих между атомами в элементарной ячейке, в спектре твердого соединения может происходить расщепление полос поглощения.
Исходя из вышеизложенного, можно заключить, что более надежные данные о частотах поглощения групп получаются при записях спектров растворенных веществ. Как правило, растворители, в особенности неполярные (гексан, циклогексан, СС14, CS2, С2С14 и др.), оказывают небольшое влияние на положение полос поглощения. Только в случае образования водородных связей происходит существенное снижение частот поглощения групп, участвующих в образовании водородных связей.
Так, и(шС в фенилацетилене снижается с 3337 см-1 в парах до 3250 см'1 в эфирном растворе за счет образования ММВС Ph-C^C-H-OEt, и, следовательно, удлинения связи =С-Н. То же самое наблюдается и для
26
27
фенола: ион 3654 см 1 в парах и 3344 см-1 в эфире. Классификация функциональных групп в зависимости от влияния растворителя представлена в табл. 1. Таблица 1
Группа в растворенном веществе Кислый растворитель (Н-СС13, H-OR и др.) Основный растворитель (R2O, R3N) и ароматические соединения (^--электроны)
S+ 8-Диполь X = Y (С=О, C=N, Р=О, S=O, N=O) s- s+ Диполь X- H (X = RO, RNH, RS или «кислый» С) Неполярная Х=Х (например С=С, N=N и др.) Сильная ассоциация: Ассоциация не проис- X=Y- X-Z (Z = СС13, OR ходит или она незна-и т.д.): Vx-y уменьшает- чительная; i\=Y прак-ся, полоса уширяется тически не изменяется Слабая ассоциация, i\_H Сильная ассоциация, может незначительно например -O-T?-OR2, уменьшаться, например, 15;_и уменьшается, по-H-O-HCCI3 лоса уширяется и ста- новится более интенсивной Очень незначительный сдвиг, если нет взаимодействия 1\=х с и полярной группы или если нет резонанса Ферми
Растворитель может оказать влияние также и на положение различных равновесий: таутомерных (например, ацетилацетон), конформерных (1,2-дибромэтан), ассоциативных (карбоновые кислоты). В твердом состоянии и в виде чистой жидкости карбоновые кислоты существуют, в основном, в виде димеров:
О — Н—О
R-c' zC-R о-н — о'
Разбавление неполярным растворителем до низкой концентрации приводит к равновесию мономер«->димер, которое еще сдвинуто далеко в сторону димера. В полярном растворителе, например CH3CN, при той же молярной концентрации равновесие сдвигается в сторону более полярного мономера (табл. 1).
За исключением соединений с сильными водородными связями, влияние межмолекулярных взаимодействий приводит к снижению частот поглощения до 25 см Наибольшее снижение в положении группо
вых частот наблюдается под влиянием внутримолекулярных взаимодействий, которые определяются влиянием масс, взаимодействием колебаний и резонансом Ферми, напряжением цикла и стерическим взаимодействием, индуктивным или мезомерным оттягиванием (или подачей) электронной плотности, диполь-дипольным взаимодействием и внутримолекулярной водородной связью (ВМВС).
2.3. Влияние массы атомов, взаимодействия колебаний и резонанса Ферми на характер спектров
Влияние массы атомов на частоту колебаний применяется в ИК спектроскопии для отнесения частот к соответствующим колебаниям, что можно наблюдать при изотопном обмене. Наиболее широко оно используется в случае группировок Х-Н при замене водорода дейтерием или тритием. При этом полосы поглощения, отвечающие колебаниям с участием водорода, смещаются в низкочастотную область в соответствии с измененной массой и практически неизменной силовой постоянной.
В качестве примера приведены частоты поглощения метанола и его дейтеропроизводных (табл. 2).
Таблица 2
Если колебания связей Х-Н или Х-D (как в случае метанола) не взаимодействуют с другими колебаниями, то отношение их частот равно V2 .
Группа | Колебание, см’1 | СНзОН | CH3OD | CD3OH | CD3OD
ОН Иэн 3687 2720 3690 2724
^он 1346 897 1297 776
СНз И» 2973 2965 2235 2228
Ц 2845 2840 2077 2080
<5as 1477 1500 1047 1060
1455 1458 1134 1135
С-О иС-0 1034 1041 968 983
Изотопный эффект используется в случае не только атомов водорода, но также и других изотопов, например, кислорода и азота. Соответствующие полосы при этом смещаются незначительно, но достаточно отчетливо для правильного их отнесения. Взаимодействие имеется во всех нормальных типах колебаний, но наиболее очевидно оно проявляется в том случае, когда группировки одного и того же типа связаны непосредственно. Так, для алленовой группировки отдельные колебания связей С=С можно было ожидать при 1650 см~', но в действительности все три
28
29
атома углерода участвуют в антисимметричном колебании (> С = С = С <), которое сильно поглощает при 1950 см*1, и в симметричном колебании (> С = С = С <), слабо поглощающем при 1050 см*1.
Резонанс Ферми обычно привлекается для объяснения расщепления полосы, одиночной при нормальных условиях. Так, например, расщепление полосы поглощения амидного карбонила некоторых лактамов даже в разбавленных растворах объясняется тем, что ее интенсивность поделена между колебанием группы O=C-N< и ближайшим обертоном или комбинационной полосой.
2.4. Влияние электронных эффектов на характер спектров
Электронное распределение в органических молекулах с учетом индуктивного (±7) и мезомерного (+М) эффектов дает простое и.удовлет-ворительное объяснение для большинства сдвигов частот групп. Разные значения частот для групп F-H, О-Н, N-H, С-Н и др. почти полностью определяются различными электроотрицательностями атомов F, О, N, С, ... и состоят в индуктивном оттягивании (-/) электронов от атома водорода. Следовательно, существует определенное соотношение между значением v и положением атома X в периодической системе элементов Д.И. Менделеева. Индуктивное действие очень быстро (по закону (*/3)", где п - количество атомов углерода между атомом X и рассматриваемым атомом водорода) затухает в системе простых связей, но четко проявляется в замещенных бензола и в системах, аналогичных ему по передаче индуктивного эффекта.
Во многих молекулярных группировках, особенно тех, которые имеют кратные связи, индуктивным эффектам противопоставлены эффекты мезомерной делокализации. Например:
О X Br OPh ОМе Н Me NMe, СбНз С . г'с-о, см
X (в гексане) 1760 1750 1735 1714 1697 1660
Ненасыщенный характер двойной связи С=О зависит от ее способности взаимодействовать по мезомерному механизму (+М) с группой (или атомом) X и от электроотрицательности (-/) группы X: кратность двойной С=О-связи увеличивается (г'( 0 повышается) при оттягивании от нее электронов за счет -/-эффекта группы X и уменьшается (vc=0 понижается) при подаче на нее электронов за счет +/- или +А/-эффекта.
Так при X = Вг превалирует -/-эффект атома брома, а при X =
NMe2 - мезомерный (+М) эффект атома азота.
-/-эффект 1г
+ Л/-эффект
н > m
о //
Ph -/-эффект
44 ,Ме
I N-Me
+Л/-эффект
|-/]<|+Л^
Сопряжение между тг-электронами сопряженной системы двойных связей является еще одним примером мезомерного влияния. Так, сопряжение С=О-группы с С=С-связью приводит к понижению обеих частот (vc=0 и vc=c) из-за образования предельной резонансной структуры: \S+/Q /15_ +
с=С-С=О С~С=С—О
"II ' Ъ a б
Снижение кратности С=О-связи в биполярной структуре б обусловливает понижение значения г-с=0, а увеличение полярности С=С-свя-зи приводит, как правило, к значительному увеличению интенсивности. Удлинение цепи сопряжения оказывает более слабое действие, чем влияние первой двойной С=С-связи. Так, в полиеновых альдегидах значение vc=0 лишь немногим меньше частоты поглощения в спектре кротонового альдегида (и = 1):
СН3-(СН=СН)„-СНО л = 0 ис=0 " 1724 см1
и=1 vc=0 1685 см*1
п = 2 ис=0 1677 см*1
л = 3 ис=0 1674 см*1
Значение и может резко снизиться, если возможна делокализация зарядов за счет взаимодействия +М- и -Л/-эффектов:
.Q Л)5+^ х-с=с-с=о
8-
В таких случаях бывает трудно сделать правильное отнесение из-за близости частот поглощения связей С=С и С=О, тем не менее колебания С=О-группы лежат, как правило, при больших частотах, чем колебания связей >С=С<.
Однако важным является то, что в обычной области карбонильно
30
31
го поглощения нет никаких полос, и поэтому для выделения карбонильной полосы, чувствительной к действию растворителя, применяется методика сдвига полосы поглощения в зависимости от характера растворителя.
Суммируя все вышесказанное по поглощению С=О-группы, необходимо сделать вывод, что чем больше дефицит электронной плотности на атоме углерода, тем выше частота поглощения этой группы.
2.5. Влияние дипольных взаимодействий на характер спектров
Две или более полярные группировки, находящиеся в одной молекуле и электронно изолированные друг от друга простыми связями, могут оказывать взаимное влияние, если расположены близко одна от другой. Такое дипольное взаимодействие не через химические связи, называемое эффектом поля, по происхождению является электростатическим. Оно особенно проявляется в а-галогенкетонах, где сильный электроотрицательный атом галогена может оказаться вблизи карбонильной группы. Так, в спектрах а-бромкетонов типа R-COCH2Br наблюдаются две полосы С=О-группы, обусловленные существованием двух вращательных изомеров, причем копланарно заслоненный конформер имеет более высокую частоту:
г;_0 1740 см-1 vc=0 1720 см 1
Подобные дипольные взаимодействия известны для сложных эфиров с атомами галогена в a-положении, лактонов и др., а также для соединений с другими гетероатомами: ОН, OR, OCOR, =О, NR2 и т.д.
Как правило, те конформации или конфигурации, в которых гетероатом оказывается в непосредственной близости от карбонильной группы, имеют более высокие значения гс=0, тогда как изомеры, где он наиболее удален от карбонила, поглощают при «нормальных» частотах, обусловленных электронными взаимодействиями.
2.6. Влияние внутримолекулярных водородных связей (ВМВС) на характер спектров
В органических соединениях водородная связь чаще всего наблюдается между группами ОН или NH и такими электроноакцепторными центрами как эфирный или карбонильный атомы кислорода, атомы азота или тт-электронные системы. ВМВС возникает в том случае, когда взаимодействующие группы, расположенные в одной молекуле, могут сблизиться на расстояние длины водородной связи, которая является индивидуальным свойством молекулы и поэтому не зависит от условий записи спектра (индивидуальное состояние вещества или его раствор) и кон
центрации раствора.
Метилсалицилат О-Ацетилпирокатехин
i'c=0: 1684 см 1 (-46 см 1736 см4 (-32 см ') 1778 см1 (+10 см1) и^н: 3210 см 1 (-400 см 1) 3410 см 1 (-200 см4) 3585 см ' (-25 см ')
Сравнение vc=i0 и и0 н метилсалицилата и О-ацетилпирокатехина сделано с vc o и и0 н метилбензоата (С6Н5СООСН3, ис_0 1730 см4), фенилацетата (СН3СООС6Н;, vc=0 1768 см4) и фенола (С6Н3ОН, vQ н 3610 см-1).
Из этих примеров видно, что наиболее прочная ВМВС (сдвиг в низкочастотную область) наблюдается в том случае, когда в результате ее образования возникает шестичленный цикл. Довольно прочная ВМВС наблюдается и в случае образования семичленного цикла. ВМВС с образованием пятичленного цикла, как правило, непрочны (сдвиги и и +0 н невелики или имеют противоположный знак). Аналогичные сдвиги наблюдаются и для связей N-H, если они (вместо связей О-Н) участвуют в образовании ВМВС.
Если соединение образует ММВС, то сдвиги полос поглощения С=О-, ОН- или NH-групп в низкочастотную область в спектрах индивидуальных веществ аналогичны вышеописанным, но при переходе к спектрам в растворах в инертных (неполярных) растворителях типа СС14 они сдвигаются в высокочастотную область до значений частот неассоциированных групп.
33
32
2.7. Применение ИК спектроскопии
Для обсуждения непосредственного практического применения ИК спектроскопии необходимо привести значения основных характеристических частот поглощения различных групп атомов: в табл. 3 приведены значения валентных (г и гД плоских деформационных (J и и вне-плоскостных деформационных (г) колебаний. Интенсивность полос оценивается качественно: с. - сильная, оч. с. - очень сильная, ср. - средняя, сл. - слабая, пер. - переменная.
Таблица 3
Группа ц см 1 Интенсивность Примечание
АЛКАНЫ
-СН3 2975-2950 С. Ks
2885-2860 с. Ц
1470-1435 ср.
1385-1370 с. %
-СН2- 2940-2915 с. Ks
2870-2845 с. Ц
1480-1440 ср. 3?н2
1250-1100 с. Скелетные колеба-
ния, ряд полос
АЛКЕНЫ
а) Колебания С-Н
rch=ch2 3095-3010 ср. vas СН2
2975 ср. цСН2
3040-3010 ср. 4н
1420-1410 ср. <Vh2
1300-1290 пер. 4н
995-985 с. Ген
915-905 с.
r'r2c=ch2 3095-3075 ср исн2
1850-1780 ср. обертон усц
1420-1410 ср- <$?н2
895-885 с. Гсн2
Продолжение табл. 3
Группа V, см 1 Интенсивность Примечание
RlCH=CHR2 1310-1290 С. <^н
980-960 с. усн (Е-изомер)
725-675 с. Ген (Z-изомер)
б) Колебания С =с
С=С несопряженная 1680-1620 пер.
rch=ch2 1645-1640 ср.
Диены 1650 ср.
Полиены 1650-1580 сл. широкая полоса
С=С, сопряженная с фе- «1625 оч. с.
нильным радикалом
С=С, сопряженная с С=О 1660-1580 оч. с.
АЛКИНЫ
r-c=ch 3310-3300 ср. 1/сн
2140-2100 сл.
700-600 сл. <%н
R’-CsC-R2 2260-2190 сл. гс=г; не проявляется
при высокой симметрии
АРЕНЫ
а) Колебания кольца
1625-1575 пер. Обычно ближе к 1600 см-1
1525-1475 пер. Обычно ближе к
1500 см-1
1590-1575 пер. Для сопряженных
систем. Как правило, интенсивна.
б) Валентные колебания С-Н
3080-3030 ср. Обычно несколько полос
34
35
Продолжение табл. 3
Группа ц см 1 Интенсивность Примечание
в) Внеплоскостные (И колебания С-Н
Монозамещение 770-730 710-690 С. с.
1,2-Дизамещение 770-735 с.
1,3-Дизамещение 900-860 ср.
810-750 с.
725-680 с.
1,4-Дизамещение 860-800 с.
1,2,3-Тризамещение 860-800 с.
720-685 ср.
1,2,4-Тризам еще ние 860-800 с.
900-860 с.
1,3,5-Тризамещение 900-860 ср.
865-810 с.
730-675 с.
1,2,3,4-Тетразамещение 860-800 с.
1,2,3,5-, 1,2,4,5-Тетра-и пентазамещение 900-860 ср.
СПИРТЫ И ФЕНОЛЫ а) Валентные колебания ОН
Неассоциированная ОН-труп па 3650-3590 пер. Чаще сильная, узкая полоса
ММВС: димеры 3550-3450 пер. Узкая полоса
полиассоциаты 3400-3200 с. Широкая полоса
ВМВС 3590-3420 пер. Узкая полоса
Хелаты 3200-2500 сл. Очень широкая
полоса
б) Колебания группы С-О-Н
Первичные спирты 1350-1260 С. Все полосы широ-
1075-1000 с. кие
Вторичные спирты 1350-1260 с.
1125-1030 с.
Третичные спирты 1410-1310 с.
1170-1100 с.
36
Продолжение табл. 3
Группа V, см 1 Интенсивность Примечание
Фенолы 1410-1310 С.
1230-1140 с.
ПРОСТЫЕ ЭФИРЫ
Алифатические 1150-1060 с.
-^с-о-с^
Ароматические и виниловые 1270-1230 с.
1075-1020 с.
( =С-О-С^- )
АЛЬДЕГИДЫ
(колебания группы >С=О)
Предельные алифатические а,р-Непредельные ^с=с-сно
1740-1720
1705-1685
Сопряженные полиеновые
1680-1660
( ^С=С-С=С-СНО )
Ароматические (Ar-С НО) 1715-1695
Группа-СНО 2880-2650
ср. гсн; может быть и обертон или составная частота
КЕТОНЫ
(колебания группы >С=О)
Алифатические (R'-CO-R2) 1725-1700
а,р-Непредельные 1695-1660
с.
с. Интенсивность полосы >С=С< повышена
37
Продолжение табл. 3
Группа ц см 1 Г1Н1СН-сивность Примечание
Арилалкилкетоны 1700-1680 с.
(Ar-CO-R)
Диарилкетоны 1670-1660 с.
а-Дикетоны 1730-1710 с.
р-Дикетоны:
а) кетонная форма -1720 с.
(-СО-СН2-СО)
б) енольная форма 1640-1535 с. Имеется широкая
1 полоса v0H в об-
(—со-сн=с-он ) ласти 3200-2700
см”1
КАРБОНОВЫЕ КИСЛОТЫ
а) Колебания карбонильной группы (>С=О)
Предельные алифатические:
а) мономеры
б) димеры —1760 с.
1725-1700 с.
а,р-Непредельные
( ^С=С-СООН ):
а) мономеры «] 720 с.
б) димеры 1715-1680 с.
Ароматические (Аг-СООН):
а) мономеры
б) димеры ® 1720 с.
1700-1680 с.
Кислоты с ВМВС 1680-1650
б) Колебания группы О-Н
Неассоциированная группа 3550-3500 с. уон ОН
Ассоциированная группа ОН 3300-2500 с. Любая группа ОН 955-890 пер Широкая полоса уон, широкая полоса
38
Продолжение табл. 3
Группа V, см 1 Интенсивность Примечание
в) Колебания С-О-Н
1300-1250 ср.
1150-1050 ср.
г) Соли кислот
/9 1610-1550 с. и,
—С/Э ” С\'° 1420-1300 ср. ц
О О"
СЛОЖНЫЕ ЭФИРЫ
а) Колебания карбонильной группы >С=О
Предельные алифатические ( -^C-COOR ) 1750-1735 С.
а,р-Непредельные 1730-1715 с.
( ^С=С-COOR )
Ароматические (Ar-COOR) 1730-1715 с.
Виниловые 1800-1770 с.
( —СОО-С=С^ ) и фени
ловые (-СОО-Аг)
а-Кетоноэфиры и а-ди-эфиры Р-Кетоноэфиры: 1755-1740 с.
а) кетонная форма —1735 с (-CO-CH2-COOR) б) енольная форма 1655-1635 с (~C(OH)=CH-COOR) ’ Р-Кетоногруппа поглощает около «1750 см-1 Широкая, оч.с.; ис=с при 1630 см
39
Продолжение табл. 3
Группа V, см 1 Интенсивность Примечание
б) Колебания с участием связи C-OR
Алифатические кислоты 1200-1170 С.
а,р-Непредельные кислоты 1310-1250 с.
1180-1130 с.
Ароматические кислоты 1300-1250 с.
1150-1100 с.
АМИДЫ КИСЛОТ
а) Валентные колебания группы N-H
Первичные амиды:
а) неассоциированная группа 3540-3480 С. VasNH2
NH2 3420-3330 с. VSNH2
б) ассоциированная группа 3360-3320 ср- KsNH,
nh2 3220-3180 ср. ц NH2
Вторичные амиды: а) неассоциированная группа 3440-3420 с. vnii
NH (цис) б) неассоциированная группа 3460-3440 с. 'Ун
NH (транс) в) ассоциированная группа 3180-3140 ср. Ичн
NH (цис) г) ассоциированная группа 3330-3270 ср. 1/nh
NH (транс)
б) Полоса «Амид-1»
(полоса поглощения группы >С=О)
Первичные амиды:
а) для индивидуальных состояний — 1650 С.
б) для разбавленных раство- «1690 с.
ров Вторичные амиды: а) для индивидуальных состояний 1680-1630 с.
б) для разбавленных раство- 1700-1665 с.
ров
40
Продолжение табл. 3
Группа V, см 1 Интенсивность Примечание
Третичные амиды: 1670-1630 с.
р-Лактамы 1760-1730 с.
у-Лактамы «1700 с.
в) Полоса «Амид-П»
(составные частоты деформационных колебаний связей N-H и С- N)
Первичные амиды:
а) для индивидуальных состояний 1650-1620 С.
б) для разбавленных раство- 1620-1590 с.
ров Вторичные амиды: а) для индивидуальных состояний 1570-1515 с.
б) для разбавленных раство- 15501510 с.
ров
г) Полоса «Амид-Ш»
Первичные амиды 1420-1400 ср.
Вторичные амиды 1305-1200 ср. д) Другие полосы
Первичные амиды 770-620 ср Д|Н
Вторичные амиды 630-530 с. ди
АМИНЫ. ИМИНЫ И ИХ СОЛИ
а) Валентные колебания связей N-H
Первичные амины 3500-3300 ф NH, и ц NH2
Вторичные амины 3500-3300 пер. (ц = 346 + 0,876 vas)
Имины (>С=МН) 3400-3200 пер Имеется полоса
Ассоциированная группа NH 3400-3100 Ф группы >C=N-при 1660-1640 см~
41
Продолжение табл. 3
Группа ц см 1 Интенсивность Примечание
б) Деформационные колебания связей N-H
Первичные амины 1650-1580 ср., с. И
900-650 ср. Вторичные амины 1650-1550 сл. в) Колебания группы -^С— Алифатические амины 1220-1020 ср. Ароматические амины 1350-1250 с. г) Соли аминов /as И /5
+ —NH3 : а) для индивидуальных со- 3350-3150 ср. vas + ц, широкая
стояний б) для разбавленных раство- -3380 ср. полоса vas
ров =3280 ср- ц
= 1600 = 1300 2700-2250 ср. ср-c. vas + ц, широкая
^NH2 -^NH 1620-1560 2700-2250 ср. ср- полоса 7n+h2 широкая полоса
НЕПРЕДЕЛЬНЫЕ АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ
а) Азометины, оксимы, оксазины и др. (v>c_N_)
Связь >C=N- в открытой 1690-1635 пер.
цепи
Связи >C=N- в открытой цепи а,р-непредельных соединений 1665-1630 пер.
42
Продолжение табл. 3
Группа ц см 1 Интенсивность Примечание
Связи >C=N-в а,р-непре- 1660-1480 пер. дельных циклах
б) Нитрилы (связь -C=N) и др, соединения
Предельные нитрилы ( -^C-C=N ) а,р-Непредельные нитрилы 2260-2240 2235-2215 ф- с.
( ^С=С-C=N )
Арилнитрилы (Ar C=N) 2240-2220 с.
Изонитрилы (-N=C) 2185-2120 с. Ич-с
Изоцианаты (-N=C=O) 2275-2240 с. V-N=C=O
Карбодиимиды (-N=C=N-) 2155-2130 с. V_n.c=N-
Азосоединения (-N=N-) 1600-1400 пер. Hm=n
Азиды ( R—N=N=N ) 2160-2120 1350-1180 с. с:/. Чч'з 4:i
Соли арилдиазония + Ar—N=N-X~ 2300-2230 с. Xn+=n
N-Окиси алифатических 970-950 с. vn->o
аминов ( -^N—►() )
N-Окиси пиридинов и пири- 1300-1250 пер. VN->O
мидинов ( ^N—►() )
Азоксисоединения ( — N=N—►() ) 1310-1250 с. v'n>0
43
Продолжение табл. 3
Группа ц см 1 Интенсивность Примечание
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ КОВАЛЕНТНЫЕ СВЯЗИ N-O
а) Нитросоединения (колебания группы NO2)
Первичные и вторичные 1565-1545 С. 4is
1385-1360 с. И
Третичные 1545-1530 с. н
а,р-Непредельные 1530-1510 с.
1360-1335 с. к
Ароматические 1550-1510 с. 4>s
1365-1335 с. н
Все нитросоединения 860-840 с. <%5
«750 с.
б) Нитраты (R-ONO,, колебания группы NO2)
1655-1610 С.
1300-1255 с. ц
в) Нитрамины (R^N-NO?, колебания группы NO2)
Предельные 1585-1530 с.
Все нитрамины 1300-1260 с. И,
г) Нитрозосоединения (R-NO, колебания группы NO)
Алифатические 1600-1500 С.
Ароматические «1500 с.
д) Нитриты (R-O-N=O)
Тр<7ис-форма 1680-1650 04. с. Гко
815-750 с. колебания с участием NO
Цис-форма 1625-1610 04. с.
850-810 с. колебания с участием NO
Все нитриты 3360-3320 ср. Обертон vsj0
44
Продолжение табл. 3
Группа V, см 1 Интенсивность Примечание
СЕРУСОДЕРЖАШИЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
а) Колебания связей S-H и C' S
Колебания S-H (меркаптаны 2590-2550 сл. И;н
и тиофенолы)
Колебания >C=S:
тиоальдегиды и тиокетоны 1200-1050 с. H>s
тиоамиды (-C(=S)-N<) 1550-1460 с. H>s
1300-1100 с. 4_-s
б) Колебания связи S=O
Сульфоны предельные и 1350-1300 04. с. Vas
непредельные (R2SO2) 1160-1120 04. с. Ц
Сульфонаты (R -SO2- OR) 1420-1330 с. ^as
1200-1145 с. К
Сульфаты (ROSO2OR) 1440-1350 с.
1230-1150 с. Vs
Сульфокислоты (RSO3H) 1260-1150 с. 14s
1080-1010 с. к
ЕАЛОГЕНОПРОИЗВОДНЫЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
C-F:
монозамещенные 1110-1100 с. Одна полоса
дизамещенные 1250-1050 04. С. Две полосы
поли(или перемещенные 1400-1100 04. С. Много полос
С-С1:
монозамещенные 750-700 с.
ди- или полизамещенные 800-700 04. С.
С-Вг =650 с.
С-1 600-500 с.
ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Связи: >Р-Н 2440-2350 ср. Узкая полоса
>P-Ph 1450-1435 ср
^Р->О 1350-1175 с.
45
Продолжение табл. 3
Продолжение табл. 3
Группа ц см 1 Интенсивность Примечание Группа V, см 1 Интенсивность Примечание
>Р-О-Аг 1240-1180 С. внг 2400-2200 c. Одна или несколь-
>Р-О-А1к 1050-990 оч. с. 1130-1040 c. КО полос
>Р-ОН 2700-2560 сл. И>н BF/ «1060 ОЧ. c.
«1030 ОЧ. c.
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ВгО3~ 810-790 ОЧ. c.
Фуран и бензофуран 3165-3125 с. Ч:-н СОз2- 1450-1410 880-800 04. C.
«1565 с. Колебания кольца cp.
«1500 с. Колебания кольца НСО3 1420-1400 c.
1000-990 c.
1030-1015 885-870 с. с. И’-о Наиболее характе- 840-830 705-795 c. c.
800-740 3125-3050 ристичная С10з“ 980-930 04.C.
Тиофен и бензотиофен с. с. <5с-Н И:-н С1О4‘ СгО42’ 1140-1060 950-800 04. C. c. Широкая полоса Несколько полос
«1520 с. Колебания кольца 950-900
«1040 с. Сг2О72- c.
750-690 с. <JC_H. Самая сильная из полос yN-H (в растворах) Колебания кольца CN“, CNO , CNS" СО: 2200-2000 c.
Пиррол и индол 3440-3400 «1565 ср. пер. нормальные карбонилы мостиковые карбонилы 2100-2000 «1830 c. c.
«1500 пер. Колебания кольца HF2 «1450 c.
Пиридин, хинолин и изохинолин (* ТОЛЬКО для пириди- 3070-3020 «1200* с. с. |/с-н Юз’ МпО4 «1230 920-890 850-840 c. 04. C. Несколько полос
нов) 1100-1000* с. C.
900-670 с. " <А-н NH4+ 3335-3030 04. C.
«710* с. . 1485-1390 C.
1650-1580 ср. N3 2170-2080 C.
1580-1550 сл. - Колебания кольца NO2’ 1375-1175 СЛ.
1510-1480 ср. . 1400-1300 C. У комплексных
Пиримидины и пурины 3060-3010 ср. И?-н нитрилов две по-
1000-960 ср. 1250-1230 840-800 1410-1340 лосы
825-775 1580-1520 ср. ср- ^с-н Колебания кольца NO3' 04. C. СЛ. 04. C.
860-800 СЛ.
НЕОРГАНИЧЕСКИЕ ИОНЫ no2+ 1410-1370 c.
AsO43~ «800 с. NO+ 2370-2230 c.
AsF6’ 705-690 оч.с.
46
47
Окончание табл. 3
Группа V, CM 1 Интенсивность Примечание
NO‘ (координационные со- 1940-1630 c.
единения)
NO” (координационные со- 1170-1045 c.
единения)
NO~ (нитрозилгалогениды) 1850-1790 c.
PF6 850-840 ОЧ. c.
РО?’, НРО42-, Н2РО4“ 1100-950 c.
s2o32- 1660-1620 СЛ.
1000-990 c.
SO42- 1130-1080 ОЧ.c.
680-610 ср-
HSO4 1180-1160 c.
1080-1000 c.
SO,2' 880-840 c.
SeO42' «1100 nep.
«830 c.
SiF62' «725 c.
SiO32“ 1100-900 c.
UO22+ 940-900 c.
По сравнению с другими физико-химическими методами исследований ИК спектры дают наиболее полную информацию о функциональных группах, имеющихся в соединении. Для удобства интерпретации ИК спектров приведены сравнительно узкие интервалы частот поглощения с указанием групп, которые могут поглощать в этом интервале (см1): 3690-3300 - валентные колебания неассоциированной группы О-Н (пары или разбавленные растворы одно- или двухатомных спиртов), ассоциированной группы О-Н (димеры спиртов, ВМВС в диолах, разбавленные растворы карбоновых кислот, кетоноспирты, гидроксиэфиры), связей N-H (первичные и вторичные N-ариламиды, первичные и вторичные амины в спектрах их разбавленных растворов) или обертон полосы >С=О; 3300-3150 - валентные колебания связей С-Н в ацетиленах и в HCN, колебания NH, в первичных или NH во вторичных амидах, ОН - в димерах карбоновых кислот; 3150-3000 - колебания связей =С-Н в алкенах, аренах, гетаренах, виниловых, простых и сложных эфирах, ассоциированной группы ОН, участвующей в образовании прочной ВМВС. колебания группы N'Hj и-NH в монозамещенных амидах, анти
симметричные колебания связей С-Н в соединениях СН3На1, СН2На12 и циклопропаноне; 3000-2800 - симметричные колебания связей С-Н в СН3На1 и СН,На12, антисимметричные и симметричные колебания связей С-Н в алканах и алкильных радикалах, присоединенных к кратной связи, ареновому или гетареновому кольцу; 2800-2600 - валентные колебания групп -N+H3, >N+H2 и =N+H; 2700-2550 - поглощение фосфорных гидроксикислот; 2650-2350 - колебания связей О-D, S-H, Р-Н; 2350-2200 - колебания связей Si—Н в алкилсиланах; 2200-2000 - колебания групп -С=С-, -C=N, -N}, -N=C или С=О в оксиде углерода, кетене или карбонилах металлов общей формулы Мх(СО)у; 1980-1650 - колебания алленового фрагмента (>С=С=С<) и связей >С=О в карбонилах металлов, карбонильных соединениях (альдегиды, кетоны, кислоты и их производные), >C=N- в азометинах, >С=С< в алкенах.
Ниже 1600 см идут специфические колебания связей и групп атомов, интерпретацию которых целесообразнее проводить на основании данных, приведенных в табл. 3.
Практическое использование ИК спектроскопии заключается в применении ее для подтверждения или установления строения вновь синтезированного соединения; обнаружения, установления характера и качественной оценки прочности водородной связи; контроля за протеканием реакции и распознавания соединений по их ИК спектрам.
1) После получения данных о качественном и количественном анализах, данных ЯМР ('Н, |3С, l9F и др.) и масс-спектрометрии с помощью ИК спектроскопии можно подтвердить в анализируемом соединении наличие функциональных групп (>С=О, NH2, NH, ОН и др.) или кратных связей (>С=С<, -С=С-, >C=N-, -C=N и др.), цис-транс- (или син-анти) изомерию, трео- или эритро-конформации и решить другие вопросы структурного характера.
В этом случае прибегают к «описанию» ИК спектра синтезированного соединения.
Пример 8. Описать ИК спектр метилового эфира г/ис-п-ацетилфе-нилакриловой кислоты
Н 3С- со С= С- СООСНз
'---' н н
Наиболее характерными полосами в его ИК спектре, которые необходимо привести, являются полосы кетонного и сложноэфирного карбонилов, 1,4-дизамещение бензольного кольца и г/ис-конфигурация эти-лиденового фрагмента.
48
49
Пример 9. При описании ИК спектра ацетилацетона, находящегося также и в енольной форме
НзС-^/^ХНз
снз—С-СН2— с-снз Т П
II II (К ..о
о о н*
обязательно указать условия записи спектра: индивидуальное состояние или раствор, вид растворителя, концентрация раствора и температура записи спектра, поскольку от этих факторов в значительной степени зависит характер спектра. Далее необходимо указать полосы поглощения С=О-групп в дикетоне и еноле, поглощение ассоциированной прочной ВМВС ОН-группы и С=С-связи в еноле. Можно (но не обязательно) также привести полосы поглощения различных С-Н-связей.
11ример 10. Описывая ИК спектр бис-триметилстанниловог,о эфира бутиндиовой кислоты (Me.SnOCO-C^C-COOSnMej,, обязательно привести полосы поглощения связей С^С, С=О и C-O-Sn. Можно привести поглощение (г и vj связей С-Н.
2) В случае не очень сложных соединений по ИК спектрам возможно установить (после получения данных качественного и количественного составов) строение полученного соединения или предложить его возможную структуру.
Пример И. Соединение имеет состав С5Н10О2. В его ИК спектре среди прочих полос поглощения имеются полосы при 1710 и3150 см-1 (в спектре индивидуального состояния), не изменяющиеся при переходе к спектру в растворе СС14. Кроме того, присутствует широкая полоса в интервале 2970-2850 см
Наличие в спектре двух первых полос свидетельствует о присутствии в соединении С=О- и О-Н-групп. (Отсутствие азота в брутто-фор-муле говорит об отсутствии аминогрупп, а отсутствие поглощения около 1600 см 1 или в интервале 2100-2250 см 1 - об отсутствии С=С или С=С-связей. Брутто-формула отвергает также наличие в молекуле бензольного кольца.) Неизменяемые значения частот С=О- и ОН-групп при переходе от спектра индивидуального вещества к спектру в растворе СС14 свидетельствуют о наличии в соединении прочной ВМВС, которая возможна при наличии в молекуле шестичленного цикла. Отсюда вырисовывается контур молекулы:
50
Методом «отсечения» (С5Н10О2 - С3НО2) получается остаток молекулы (С2Н9), который необходимо распределить по оставшимся пяти свободным связям у атомов углерода. Поскольку колебания связей С-Н прописаны в виде широкой полосы поглощения и не представляется возможным выделить отдельные колебания связей С-Н в СН3-, СН2- или СН-группах, то однозначно решить вопрос о строении данного соединения не представляется возможным. Можно лишь предложить одну из возможных структур:
н н
СНз\ Лк /СНз с с;
II I н о. „о
••Н
Н Н
С2Н5\ 'с^ „н с с'
II I н о. 'О
-н
н н
Ik .<k /С2Н5 с с;
II I н
о. Д)
••Н
Н СПз сн3\ kk и 'с с Н I II о. .0 н-‘
н н
СНЗ\ Л1
,с с
СНз^1 II
о. .0 н-’
Н СНЗ
,СХ ,СНз
'С с
Н I II
(к .о
н-‘
НзС СНз \ /
о... .о
Н-"
И С2Н5 \ / bk _н к с
H I 11
Ck .0
Пример 12. В ИК спектре соединения состава С Н наиболее характерными полосами поглощения являются полосы при 3255,3060,2235, 1655 и 835 см
Полоса с ц равной 3255 см может принадлежать только концевой =С-Н-группе (отсутствие в брутто-формуле атомов кислорода или азота, что могло бы означать наличие в соединении связей О-Н или N-H), что подтверждается наличием полосы с v = 2235 см 1 (-С=С— связь). Наличие полос поглощения с г= 3060 и 1655 см 1 свидетельствует о наличии в молекуле фрагмента -С=С-Н, а сильная полоса при 835
51
см - об 1,4-дизамещении бензола. Отсюда, искомым соединением является 1,4-диэтинилбензол:
Н~С=С—С=С-Н
Пример 13. Установить или предложить структурную формулу вещества состава C4H9NO, если в его ИК спектре (для индивидуального состояния) имеются полосы поглощения при 3300,3160,2970-2850,1655, 1540 и 1470-1430 см-1. При переходе к спектру в растворе СС14 низкочастотные полосы (1470-1430 см1) остаются на месте, высокочастотные (3300 и 3160 см-1) сливаются в одну широкую полосу около 3440 см-1, а полосы при 1655 и 1540 см*1 сдвигаются до 1680 и 1530 см1.
Наличие полосы поглощения при 1655 см’1 свидетельствует о наличии в соединении карбонильной группы, а повышение ее до 1680 см'' о включении ее в ВМВС. Полосы с г-* 3300 и 3160 емтй-могут говорить о наличии в соединении группы NH2 (и. и и) или NH, если она входит в состав вторичного амида (и и и ). Присутствие в соединении первичной аминогруппы отвергается наличием одной, а не двух полос поглощения связей N-H в растворе СС14. Гидроксильная группа в соединении присутствовать не может, так как в молекуле содержится один атом кислорода. Широкие полосы в интервале 2970-2850 и 1470-1430 см 1 соответствуют валентным и деформационным колебаниям связей С-Н. Отсюда на основании ИК спектра данному соединению можно придать формулу метилпропиоамида (1), этилацетамида (2), пропил- (3) и изопропилформамида (4):
CH3CH2CONHCH, (1), CH3CONHCH,CH3 (2),
H-CO-NHCH,CH2CH3 (3), H-CO-NHCH(CH3)2 (4)
Две последние формулы исключаются, поскольку формамиды поглощают при более высоких значениях частот (I 700- 1710 см '). поэтому строение данного соединения отвечает одной из двух формул (I) или (2).
3) Поскольку при образовании водородной связи значения частот поглощения ОН-, NH,-, NH, SH-групп (и некоторых других) понижаются, по наличию и величине этого сдвига в низкочастотную область от значений неассоциированных групп (см. табл. 3) можно судить о наличии и прочности водородной связи. Вместе со сдвигом полосы уменьшается ее интенсивность и увеличивается полуширина, причем чем прочнее водородная связь, тем больше сдвиг, меньше интенсивность и больше полуширина.
Для выяснения водородной связи (внутри- или межмолекулярная, ММВС) необходимо записать спектр в растворе инертного растворите
ля (гексан, циклогексан, СС14 и др.) и по структуре полученного спектра можно сделать вывод о характере водородной связи: при наличии в соединении ММВС последние в растворе разрываются и частоты групп, участвующих в образовании водородной связи, принимают значения, близкие к значениям неассоциированных групп; если водородные связи внутримолекулярные, то значения частот и характер полосы остаются практически теми же, что и в спектре конденсированной фазы соединения; если полоса ассоциированной группы в спектре раствора остается практически на месте с одновременным появлением полосы неассоциированной группы, следует вывод о наличии в соединении как внутри-, так и межмолекулярных водородных связей: при растворении первые (ВМВС) не разрываются с сохранением полосы на прежнем месте, а вторые (ММВС) разрываются с появлением полосы неассоциированной группы.
4) При проведении каких-либо реакций происходи г изменение функциональных групп, исчезновение одних или появление других функций, изменение углеродного скелета молекулы и т.д. Это позволяет вести контроль за протеканием реакций, который осуществляется по исчезновению полос, характерных для колебаний какой-либо группы в исходном соединении.
Пример 14.
Первую стадию можно контролировать по исчезновению симметричных и антисимметричных колебаний связей С-Н метильной группы, вторую по исчезновению полосы поглощения ОН-группы, третью и четвертую - по исчезновению полос поглощения сложноэфирного карбонила и эфирной связи -C(=O)-O-Et (появляется полоса Амид-1, но она лежит значительно ниже, чем полоса поглощения сложноэфирного карбонила) и пятую стадию - по исчезновению полосы Амид-1.
52
53
Пример 15.
нс=сн
1
г-*- СбНб
2 3
---► СНзСНО —* СНзСН(ОС2Н5)2
4 5 6
—► ch2=ch-cn —*• сн2=сн-соон —*•
7 / \
—* СН2=СН-СООСНз —*/—сн2—сн— 1 \ СООСНз/п
Первое, второе и четвертое превращения можно контролировать по исчезновению колебаний =С-Н- и С^С-связей (в случае четвертой реакции появляется поглощение связи C=N, но оно лежит несколько ниже, чем поглощение связи С=С). Третья реакция контролируется исчезновением из спектра полосы колебаний С=О-группы, а пятое - исчезновением колебаний связи C=N. За протеканием шестой реакции можно наблюдать по исчезновению полосы -О-Н-группы. Полное использование метилакрилата в результате реакции 7 можно проконтролировать по отсутствию полосы поглощения С=С-связи в полимере.
Пример 16.
1 2 3
—► СбНзСОСООН —* CbHsCOCOOEt —►
С6Н5СОСНз —
Первую реакцию можно контролировать по исчезновению полос поглощения (v и н) алифатических С-Н-связей (2975-2860 см1), вторую - по исчезновению поглощения ОН-группы, а третью - по исчезновению поглощения кетонной и сложноэфирной групп. Реакция 4 контро
лируется исчезновением полосы С=О-группы исходного ацетофенона (появляющаяся полоса С=О-группы м-нитроацетофенона лежит значительно выше). Аналогично можно проконтролировать реакцию 5 (по понижению частоты поглощения в м-аминоацетофеноне по сравнению с м-нитроацетофеноном). Реакция 6 контролируется переходом двух полос поглощения группы NH2 (^ и v) в одну или две (v к и vм), но лежащие значительно ниже, чем полосы поглощения связей NH2 в м-аминоацетофеноне.
5) Можно ли с помощью ИК спектров распознать соединения? Пример 17. Ацетон, ацетофенон, бензофенон.
СНз—С-СНз С6Н5—С-СНз СбН5—С-СбН5
II II II
О о о
Распознать данные соединения по их ИК спектрам можно по положению полос поглощения карбонильных групп (v ). Поскольку частота зависит от величины дефицита электронной плотности (8+) на ато-ме углерода (чем она больше, тем выше частота), а 8+ зави-
сит от характера радикалов, связанных с С=О-группой, то донорные заместители, обладающие +1- или +Л/-эффектом погашают 8+, понижая тем самым vc=Q. Электроноакцепторные группы действуют противоположно. Учитывая -(-/-эффект СН3-групп и +Л/-эффект фенильных радикалов (+М> +Г), 8+ на атоме углерода в ацетоне будет максимальным, а в бензофеноне - минимальным. Следовательно, значения частот нс=0 будут связаны следующей закономерностью: и, > и,_, . >
'с-О (бе ф )'
Пример 18. М,Ы-Диметиланилин, бензойная кислота, этилбензоат, бензамид, N-метилбензамид и КЙМ-диметилбензамид.
C6H5N(CH3)2 (1), C6HSCOOH (2), C6H5COOEt (3), C6H5CONH2 (4), C6H5CONHCH, (5), C6HSCON(CH3), (6)
В ПК спектре соединения 1 отсутствует поглощение С=О-группы, а соединения 6 - поглощение 0-11- или N-H-связей, поэтому их спектры будут отличаться от спектра соединений 2-5 и друг от друга (в спектре соединения 1 отсутствует поглощение С=О-группы, которое есть в спектре соединения 6). В спектре соединения 3 также отсутствует поглощение О-Н- или N-H-связей, но в его ИК спектре поглощение С=О-группы лежит выше, чем в спектре соединения 6 (К=01ф]1р > нс ч)ам11д). В спектрах соединений 4 и 5 полосы амид-1 будут иметь близкие значения, но в спектре соединения 4 будут наблюдаться две полосы (г и v ) поглощения
54
55
связей N-H, в то время как в спектре соединения 4 - только одна полоса (или две, и и см. пример 16). Различить соединения 2 и 5 можно по положению как полос поглощения vc=0 (см. выше), так и по поглощению выше 3300 см 1 (vNH < vQH).
Пример 19. Диэтиловые эфиры цис-1,2- (1), транс-1,2- (2) и 1,1-циклопропандикарбоновых (3) кислот.
НДН нДСООЕТ y\COOEt
I (1), Н (2), (3)
EtOOC COOEt EtOOC H COOEt
Соединение 3 от соединений 1 и 2 можно отличить по положению полосы поглощения С=О-групп, поскольку соединение 3 является эфиром Р-дикарбоновой кислоты, а соединения 1 и 2 - эфирами у-дикето-кислот (в них частота ниже). Соединение 1 является гщоизомером, поэтому в его спектре будет присутствовать полоса около 970 см ', а в спектре соединения 2 (/ираис-изомер) - полоса около 700 см-1.
2.8. Техника фундаментальной И К спектроскопии
Область фундаментальной ИК спектроскопии - диапазон от 5000 до 250 см4 (20-400 нм). Она исследуется при помощи спектральных приборов: UR-20, Specord и др., принципиальная схема которых приведена на рис. 10.
.кЬ—ы-!
* кт 6 T л ГП
1 2 4LJ_________L4! 7 8
3
Рис. 10.
1 - источник излучения - силитовые (керамические) стержни; 2 - кювета с образцом - солевая, обычно из монокристалла NaCl, КВг, LiF, CaF, пли AgCl; 3 - монохроматор; 4 - входная и выходная щели; 5 - фокусирующая оптика; 6 - диспергирующая оптика - солевые сменные призмы из LiF (от 5000 до 1700 см '), CaF2 (до 1100 см '), NaCl (до 700 см1), КВг (до 400 см1), Csl (до 250 см ’); 7 - приемник излучения - термоэлементы, болометры; 8 - регистрирующее устройство.
Для работы с растворителями, подобными воде, которые могут растворить солевую оптику, ячейки изготовляют из специального оптического материала - иртрана-2.
Шкала волн прибора обычно меняется при его работе, поэтом} ес необходимо регулярно калибровать. Для калибровки в диапазоне 5000-650 см в качестве стандарта используют полистирол.
Методом ИК спектроскопии можно исследовать газы, жидкости, твердые вещества или растворы:
а) при изучении газообразных веществ необходимы специальные, так называемые газовые, кюветы с большой длиной светового пути;
б) жидкие соединения наносят в виде пленки на пластины из материала, прозрачного в исследуемой области, обычно из NaCl;
в) твердые вещества чаще всего изучают в виде суспензии (пасты) с вазелиновым маслом (нуйолом) или гексахлорбутадиеном. В последнее время широкое распространение получил метод записи ИК спектров в пасте с перфторвазелиновым маслом, особенно в тех случаях, когда необходимо проанализировать поглощение различных С-Н-связей.
Для приготовления суспензии образец измельчают в агатовой ступке агатовым же пестиком и добавляют 1-2 капли суспендирующего вещества и смешивают (растирают) его с образцом до получения однородной пасты.
Эту пасту тонким слоем наносят между пластинами из NaCl или КВг, которые помещают на пути светового луча. Качество получаемого спектра сильно зависит от способа приготовления суспензии: чем она тоньше, тем качественнее спектр.
Нужно иметь в виду, что само вазелиновое масло имеет поглощение в области валентных и деформационных колебаний связей С-С и С-Н, поэтому для исследования поглощения вещества в этих областях (3000-2850 и 1470-1350 см1) целесообразно заменить вазелиновое масло гексахлорбутадиеном или перфторвазелиновым маслом.
Твердые вещества можно исследовать и в виде таблеток с КВг, Для чего исследуемое вещество и КВг (сухой!) тщательно перемешивают и измельчают в агатовой ступке и прессуют в таблетку толщиной 1-1,5 мм (давление около 20 атм/см2), которую укрепляют в приборе и просвечивают. При этом нужно иметь в виду, что спектр соединения может быть искажен из-за взаимодействия вещества с бромидом калия при высоком давлении;
г) для исследования растворов наиболее удобны двухлучевые приборы, позволяющие вычитать из поглощения раствора (растворитель + образец) поглощение растворителя. Для этого раствор образца в кювете помещают на пути одного луча света, а другую оптически идентичную Кювету с растворителем - на пути второго луча (луча сравнения). При этом измеряется поглощение, соответствующее разности noi лощения
56
57
двумя кюветами (с раствором и растворителем), что приводит к компен сации полос растворителя. Растворитель должен быть по возможности инертным и неполярным (СС14, C2CI4, CS,, циклогексан и др.), не образовывать с веществом сильных водородных связей или межмолекулярных ассоциатов, не иметь собственных интенсивных полос поглощения в исследуемой области и давать возможность готовить достаточно концентрированные растворы (порядка 0,1 М; при проведении тонких исследований, например водородных связей, возможно приготовление менее концентрированных растворов).
В ИК области трудно получить точные данные по интенсивность' полос поглощения, так как излучение, попадающее на детектор, никогда не является истинно монохроматическим, а сами полосы - довольно узкими. Положение усугубляется трудностями, которые возникают при подготовке образца: малые объемы довольно концентрированных растворов и тонкая, легко разрушаемая кювета, которая сильно «боится»-влаги. Поэтому данные по интенсивности полос поглощения зачастую трудно воспроизводимы и приходится пользоваться их качественным описанием (см. стр. 26). В спектрах некоторых соединений две или более полос могут сливаться в одну и тогда после указания значения частоты поглощения добавляется буква ш (широкая), что свидетельствует, как правило, о включении групп, отвечающих за происхождение этой полосы (например, О-Н- или N-H-связи), во внутри- или межмолекулярные водородные связи или о составном характере этой полосы, что часто наблю-да-ется в спектрах соединений, содержащих много разных, но похожих связей - алканы, циклоалканы и др., в спектрах которых проявляются колебания (валентные и деформационные, симметричные и антисимметричные) связей С-Н и С-С.
2.9. Возможности и ограничения метода
Возможности метода: доказательство присутствия в веществе группировок, обладающих характеристическими частотами колебаний: доказательство тождественности образцов; качественный и количественный анализ смесей при известных спектрах компонентов; текущий контроль за ходом реакций; анализ двухкомпонентных простых или равновесных смесей.
Ограничения метода: практически полное отсутствие информации об углеродном скелет исследуемого вещества.
58
2.10. Задачи для самостоятельного решения
I. Можно ли применить ИК спектроскопию для контроля за сле-
дующими превращениями?
а) СгНб С2Н4 С2Н2
(~с=с-\
г-»- СНзСООН —► СН3СООС2Н5
CH3CH2OH -
L-*- СНзСНО —
—*- СНзСН(ОН)— сшсно —СНзСН=СН-СНО
59
2. Предложить возможную структурную формулу или установить строение соединения состава:
a) C8HgNO. Данные ИК спектра (v, см1): 750, 1610, 1665, 2850-2950, 3050,3220,3400. При переходе к спектру в растворе СС14 полоса с v = 3400 см-1 повышается до 3520 см-1, а остальные полосы остаются практически на месте.
б) С9Н18О2. Данные ИК спектра (v, см1): 1180, 1740, 2870, 2970. При переходе к спектру в растворе в СС14 все полосы остаются на месте.
в) C8H10O3S. Данные ИК спектра (v, см-1): 1540, 1735, 2860-2960, 3070 (ш), 3120, 3420 (ш). При переходе к спектру в растворе в СС14 полосы при 1735 и 3420 см-1 повышаются до 1745 и 3560 см-1, а остальные остаются практически на месте.
3. Можно ли по ИК спектрам различить:
а) соединения общей формулы: х COOEt ; Где X = Н, Me,
NH2, MeNH, Me2N, O2N.
б) бензофенон, флуоренон, антрахинон.
в) метиламин, диметиламин, триметиламин, анилин, N-метиланилин, Ы,Ы-диметиланилин?
4. Описать ИК спектры соединений:
a) C6F -CO-C(=NOH)-C6F5; б) (C6Hs)2C(OH)-CH2CH2COOEt;
в) H2N—6Q/— CON(CH3)2
3. Спектроскопия ядерного магнитного резонанса (ЯМР спектроскопия)
Метод ЯМР применяется в органической химии с конца 1950-х гг. Однако благодаря своей эффективности он быстро совершенствовался и в настоящее время является одним из важнейших методов исследования органических соединений (наряду с ИК спектроскопией и масс-спектрометрией). Но он не заменяет данные методы. Они взаимно дополняют друг друга.
Поскольку теория ЯМР наиболее подробно разработана применительно к ядрам водорода (протонам), то рассмотрение основных положений проведем применительно к методу протонного магнитного резонанса (ПМР) с кратким упоминанием положений, относящихся к другим магнитным ядрам, отмечая сходства и различия в поведении в магнитном поле протонов и данных ядер.
3.1. Магнитные свойства и резонанс ядер. Ширина спектральной линии
Как и в УФ или ИК спектроскопии, в основе метода ЯМР лежит соотношение Бора: ЛЕ = hv, но изменение энергии в этом случае связано с магнитными свойствами ядер.
Ядра некоторых элементов или их изотопов обладают магнитным моментов (//), величина которого зависит от ядерного спинового числа I. Оно может принимать значения: 0, ±'/2, ±1, ±3/2,... в зависимости от массового числа атома (количества протонов и нейтронов в нем, |/?1 + ‘п ) и номера элемента ('/? ) в периодической системе элементов.
Массовое число:
Атомный номер:
Спиновое число:
нечетное четный или нечетный
'/ 3/ 5/
'2, '2,
четное четное
четный нечетный
0 1,2,3,...
-----<4----Орбита
f J прецессии
/Ось в ращения \ / ядра
/СЛ Ядро, обладающее спином
Н Н апряжение 0 магнитного поля
Рис. И
C4N7,2Н,) имеют спин / = 1. При Мере спин является полуцелым MSi14;/=3/2 для "Bs, 35С1р, 79Вг!
Поскольку ядро имеет электрический заряд, то при вращении ядра со спином 7^0 возникает магнитное поле, направление которого совпадает с осью вращения ядра. Такое ядро можно рассматривать как крошечный магнит с магнитным моментом д
Протон и нейтрон имеют спин 1= ±‘/2. Ядра |2С6, |8О8 и 32S|6, содержащие равные количества протонов и нейтронов, имеют I = 0 и поэтому немагнитны. Ядра с четным массовым числом и нечетным номером нечетном массовом числе и любом но-числом (/ = '/2 для 'Нр l9Fg, |3С6, 3IP|S, ; /= % для |7О8, '%).
Ядро со спином / может находиться в магнитном поле в 2/+ 1-со-
стояниях. При / - 0 ядра имеют одно энергетическое состояние (2 0+1),
60
I
61
для них не наблюдаются переходы с одного энергетического состояния па другое и поэтому они не являются объектами исследования методом ЯМР.
Ядра с I > I кроме магнитного резонанса (/г) обладают электрическим квадрупольным моментом и поэтому исследуются методом ядер-ного квадрупольного резонанса (ЯКР).
Для ядер с 7 = ±7 во внешнем магнитном поле напряженностью Н возможны две ориентации с энергетическими уровнями, равными ±А^о-
...-- %ДЯ=+/гЯ0(>||Я^
S'o - энергия ядра в отсутствие магнитного поля Яо
•АЕ= 2/rH0
Рис. 12
Из рисунка видно, что разность энергий между возбужденными уровнями ЛЕ = 2р77
ЛЕ можно выразить не только через //, но и через гиромагнитную постоянную у, включающую в себя // и 7: y = 2n/nlh-J (при I = У/л = уОтсюда:
h-v ЛЕ = -!--Но.
2л
С учетом соотношения Бора (ЛЕ = h- и) получаем формулу:
2л
Это соотношение является основным соотношением ЯМР.
Внешнее магнитное поле ориентирует магнитный момент ядра параллельно (//1| Я) или антипараллельно (/Ц Я) своему направлению. Однако из-за наличия у ядра механического момента вращения (щ) при взаимодействии последнего с полем ось вращения ядра отклоняется от направления поля и описывает коническую поверхность вокруг оси поля (рис. 12). Движение такого типа называется прецессией (на этом явлении основано устройство навигационных приборов - гироскопов; простейшим примером прецессирующего движения является вращение обыкновенного волчка).
Частота прецессии равна частоте электромагнитного излучения, необходимого для перевода ядра из одного спинового состояния в дру-
62
гое: + '/, ~'/г
Таким образом, для наблюдения ЯМР необходимо поместить образец в сильное однородное магнитное поле напряженностью Н и подействовать на него излучением с частотами и, к. и ит.д. При совпадении одной из них с ц определяемой из основного уравнения ЯМР, ядра будут переходить с одного ядерного магнитного уровня на другой. Такие переходы впервые в 1946 г. наблюдали две группы американских ученых под руководством Э.М. Перселя (на протонах парафина, СН ) и Блоха (на протонах воды), т.е. было открыто явление протонного магнитного резонанса (метод ПМР).
Вероятности переходов на верхний и нижний уровни одинаковы, однако заселенности уровней различны: на нижнем уровне она выше, чем на верхнем, поскольку система стремится занять состояние с более низкой энергией (принцип Ле-Шателье). Соотношение заселенностей (в соответствии с распределением Больцмана) при комнатной температуре не превышает 10"3 от общего числа магнитных ядер. Именно небольшой, хотя и конечный избыток ядер на нижнем уровне, обусловливает явление ЯМР, т.е. поглощение разночастотного излучения с частотой ив соответствии с основным уравнением ЯМР.
При длительном поглощении излучения существовавший вначале на низшем уровне избыток ядер может уменьшаться, что приведет к уменьшению интенсивности сигнала поглощения, который при определенных условиях может исчезнуть. Такое явление называется насыщением. При этом заселенности обоих спиновых уровней выравниваются.
Возвращение с верхнего уровня на нижний, не сопровождающееся излучением, называется релаксацией. Известны два ее типа: спин-ре-шеточная, заключающаяся во взаимодействии изучаемого ядра со всей системой вещества, т.е. его кристаллической решеткой; ее время (Г,) в твердых телах или вязких жидкостях велико и достигает нескольких часов, а в жидкостях и газах мало (10 2-102 сек); и спин-спиновая, которая характеризует обмен спиновыми состояниями между соседними ядрами и определяется временем Т.
От величин Т и 73, зависит естественная ширина полосы: она обратно пропорциональна среднему времени жизни возбужденного состояния. В случае долгоживущих состояний наблюдаются узкие линии, а короткоживущих - широкие.
Времена релаксаций большинства невязких органических жидкостей или растворов имеют величину порядка I сек, что соответствует Ширине спектральной линии около 1 Гц.
Суммарное магнитное ноле, взаимодействующее с прецессирую-
63
щим ядром, является суммой постоянного внешнего поля и локальных полей решетки. У твердых веществ и вязких жидкостей движения молекул ограничены, взаимодействия их с магнитным полем невелики (большие времена спин-решеточных релаксаций) и резонансные сигналы имеют очень большую ширину и малую интенсивность. В невязких жидкостях или растворах величина локального магнитного поля очень маленькая (быстрое движение молекул), время спин-спиновой релаксации (время возбужденного состояния) велико, поэтому сигналы высокоинтенсивные и имеют небольшую ширину.
Кроме времени спин-спиновой и спин-решеточной релаксаций на ширину спектральной линии влияют еще два фактора: присутствие в образце парамагнитных молекул (например, растворенного кислорода) или ионов, а также ядер, обладающих электрическим квадрупольным момен том. При тепловом движении молекулы эффективно взаимодействуют с хаотически меняющимися во времени электростатическими полями, ядро быстро отдает спиновую энергию решетке (малое время спин-решеточной релаксации) и это приводит к уширению сигнала.
В заключение необходимо привести магнитные свойства ядер, наиболее важных для спектроскопии ЯМР органических соединений (табл. 4).
Таблица 4
Ядро Спиновое квантовое число,! Магнитный момент,// Гиромагнитное отношение, у Резонансная частота ц0 в поле 1 Т (тесла) =10000э Относительная чувствительность при постоянном поле Но Природное содержание, % Квадрупольный момент, Q
н Ч 2,79277 2,675 42,577 1,000 99,98 —
'Н2 1 0,85735 0,411 6,536 0,009 0,0156 0,003
10в 3 1,8007 0,288 4,575 0,02 18,83 0,111
"в 7, 2,6880 0,858 13,660 0,165 81,17 0,036
|3С 7, 0,70216 0,673 10,705 0,016 1,108 —
l4N 1 0,40369 0,193 3,076 0,001 99,635 0,02
I5n 7, -0,28298 -0,271 4,315 0,001 0,365 —
|7О 7, -1,8930 -0,363 5,772 0,029 0,037 -0,004
l4F 7? 2,6273 2,517 40,055 0,834 100,0 —
29s, -0.55492 -0,531 8.460 0.079 4,70 -
Окончание табл. 4
Ядро Спиновое квантовое число, / Магнитный момент, // Гиромагнитное отношение, у Резонансная частота ц0 в поле 1 Т (тесла) = 10000 э Относительная чувствительность при постоянном поле Но Природное содержание, % Квадрупольный момент, Q
31р 33S 35С1 37С1 79Вг 81Вг |271 свободный электрон Ч ч ч Ч Ч Ч Ч 1,1316 0,64274 0,82089 0,68329 2,099 2,2626 2,7939 -1836,0 1,083 17,235 3,265 4,172 3,472 10,667 11,498 8,519 39449,0 0,066 100,0 0,74 75,4 24,6 50,57 49,43 100,0 -0,064 -0,08 -0,062 0,33 0,28 -0,75
3.2. Магнитное экранирование и химический сдвиг протонов
Ядра водорода, а также другие магнитные ядра в органических молекулах окружены электронами, вращение которых вокруг ядра создает свое поле. Она препятствует взаимодействию внешнего магнитного поля с ядром. Иными словами, электроны заслоняют (экранируют) ядро, поэтому напряженность поля вблизи ядра отличается от напряженности внешнего поля В результате экранирования изменяется частота, при которой наблюдается явление резонанса. Это изменение называется химическим сдвигом. Магнитное экранирование и, следовательно, химический сдвиг определяются положением данного протона в молекуле. Для эквивалентных протонов значения химических сдвигов одинаковы и они дают один резонансный сигнал, а различающиеся окружением в молекуле протоны обладают различным экранированием, имеют различные химические сдвиги и дают раздельные сигналы, что позволяет определять положения протонов в молекуле.
Положение резонансного сигнала зависит от напряженности поля так как она определяет силу, ориентирующую ось вращения протона, т.е. угол прецессии (у(рис. 11). Следовательно, для выражения химического сдвига необходима величина, не зависящая от Н В качестве тако-
64
65
ной принято положение сигнала протонов в гетраметилсилане ('|МС. Me Si). Выбор его в качестве эталона обусловлен тем, что он содержи: двенадцать структурно эквивалентных сильно экранированных протонов, дающих вследствие этого легко опознаваемый узкий сигнал в са мом сильном поле. Преимуществом ТМС является его хорошая растворимость в обычных органических растворителях и химическая инертность (недостатки: низкая температура кипения и нерастворимость в воде).
Вследствие инертности и растворимости ТМС его можно добав лять практически к любому веществу в любом органическом растворителе, но низкая температура кипения (27°С) ограничивает его примене ние, поэтому более употребительным эталоном является гексдметилди-силоксан (ГМДС, Me3Si-O-SiMe3; т.кип. 10ГС). Он, как и ТМС, химически инертен и хорошо растворим в основных органических раствори телях. С учетом распределения электронной плотности в ТМС и ГМДС г последнем протоны менее экранированы (более деэкранированы-т.е. на них легче воздействовать радиочастотным излучением) и их сигнал находится в более слабых полях (на 0,055 м.д.) относительно ТМС.
ТМС, ГМДС или другое вещество может использоваться как внутренний, так и внешний эталон. В первом случае он в очень небольших количествах добавляется в ампулу с образцом, а во втором помещается в капилляр, находящийся в ампуле. В этом случае в величину химического сдвига вводится поправка на магнитную восприимчивость материала, из которого изготовлен капилляр.
При работе с водными растворами в качестве внешнего эталона используют 3-триметилсилилпропансульфонат натрия (Me3Si-CH,CH2CH,--SO3Na) или его дейтерированный по метиленовым группам аналог, в котором сигнал триметилсилильной группы смещен в слабые поля от сигнала ТМС на 0,015 м.д.
Если /7 - напряженность магнитного поля, при которой наблюдается резонанс протонов эталона (в частности, ТМС), И - напряженность поля, при которой наблюдается резонанс протонов образца, то (// - Н)т)/Н0 и есть величина, не зависящая от Нд. Она является безразмерной и характеризует экранирование данного протона, т.е. его химический сдвиг.
Поскольку измерение напряженности магнитного поля значительно более сложное и менее точное; и поскольку она пропорциональна резонансной частоте, химический сдвиг также равен отношению (щ гv)/i;i, где и и i'q - резонансные частоты протонов образца п эталона; г - рабочая частота прибора.
Поскольку /-/. и //* (в килоэрстедах, кЭ) или iy2[q и rq (в МГн)
66
хотя и ра сличаются, но незначительно, и в !0J раз меньше //(| и и, ученые-,химики условились: чтобы иметь значащие цифры сразу после запятой в величинах химических сдвигов, получаемые в результате деления, умпожа1ь по I06. обозначая полученные значения как миллионные доли (м.д.). Если принять г т.е. г = 0, если обозначить химический сдвиг через Й, то
е Vo6p(rU) ,
8 =—1-------10 (м.д.).
И (МГц)
Согласно основному уравнению ЯМР, резонансный сигнал любого протона должен иметь одну характеристическую частоту для заданной Н. Однако, в действительности сигналы различных протонов имеют разные частоты в зависимости оттого, в каком химическом окружении они находятся. Поэтому в уравнение вводится множитель - безразмерное число о: называемое константой экранирования.
г = ^-/70(1-ст).
Разность (I - ст) называется электронным деэкранированием ядра и чем оно больше, т.е. чем меньше ст, тем менее экранировано ядро и тем в более слабых магнитных нолях (требуется излучение с меньшей энергией) находится резонансный сигнал протона (э го положение относится и к другим магнитным ядрам).
Для измерения химических сдвигов прогонов кроме шкалы Л в которой сигнал ТМС принят за нуль, и значения величин й, находящиеся слева от него, имеют положительные значения (чем левее сигнал, тем больше значения 8:: 1,2, 3,... м.д., тем в более слабых полях он находится!), существуют шкала г, в которой сигнал ТМС принят за 10 (химические сдвиги имеют значения 9, 8, 7, ...). Соотношение между шкалами: й+ т= 10.
Как видно из табл. 5 и 6, на величину химического сдвига, опредя-емую магнитным экранированием ядра, влияет множество факторов, в связи с чем зависимость между положением ядра в молекуле и его химическим сдвигом носит эмпирический характер. Основным из таких факторов является электронная плотность вокруг данного протона: чем опа выше, тем большее сопротивление она оказывает внешнему разночастотному излучению для возбуждения им ядра и, следовательно, для этого необходима большая энергия. Резонансный сигнал проявляется в более сильном поле. В соответствии с этим протоны, обладающие кислыми свойствами (с минимальной электронной плотностью), резонируют в ^слабых нолях. Па электронную плотность вблизи ядра существенно
67
Таблица 5
Химические сдвиги (£+0,1, м.д.) группировок -СН3, ^СНг , ^;СН
-СН3 -СН2- \1/ о I
Положение в молекуле, м.д.
СН3-СН2- 0,9 -сн2-сн2- 1,3 \|/ о 1 ж 1,5
СН3-СН< 1,7 -сн2-сн< 1,9 ^сн-с^ 2,1
сн3-с6н5 2,3 —сн2—С6Н5 2,6 >СН-С6Н5 2,9
СН3-С(=О)- 2,1 -СН2-С(=О)- 2,4 >СН-С(=О)- 2,5
СН3-О- 3,3 -сн2-о- 3,5 >сн-о- 3,7
СН3-ОСО- 3,7 -сн2-осо- 4,2 >сн-осо- 4,3
сн3-соон 2,3 -сн2-соон 2,2 >сн-соон 2,4
CH3-COOR 2,3 -CH2-COOR 2,2 >CH-COOR 2,4
СН3-С1 3,1 -СН2-С1 3,5 >СН-С1 4,1
СН3-Вг 2,7 -СН2-Вг 3,4 >СН-Вг 4,2
СН3-1 2,2 -СН2-1 3,2 >СН-1 4,2
CH3-S- 3,1 -CH2-S- 3,3 >CH-S- 3,5
CH3-N< 2,2 -ch2-n< 2,5 >CH-N< 2,9
Таблица 6
Химические сдвиги других протонов
Протоны и их положение в молекуле £, м.д. Протоны и их положение в молекуле 8, м.д.
R-CH, 0,9-1,0 R-CH2-Br 3,4-3,6
ЦИКЛО-С6Н|2 1,42 CH3O-R 3,3-3,7
r-ch2-r 1,3-1,5 rch2-o- 3,4-4,0
СН3СН2С1 1,5 СН3-О-С6Н5 3,7-3,9
R-CH2-CH2C1 1,7 RCH2C1 3,4-3,8
r2ch-ch,ci 1,6-1,9 r2chor‘ 3,7-4,1
R2C=C(R)-CH3 1,7-2,0 rcooch2r' 3,6-4,5
R-CsC-СНз 2,0 R2CHCI (Br, 1) 4,0-4,6
R3CH 1,5-2,5 RCH2NO2 4,2-4,5
R-CHj-COOR1 2,0-3,0 огненна 4,5
R2C=C(R)-CH2-R' 1,9-2,4 rnh2 1,0-5,0
R-C=CH 2,0-3,0 r2c=ch2 4,6-5,5
R-CH->-CO-R' 2,0-2,8 R-OH 1,0-5,5
C6H5CO-CH3 2,6 r2c=ch-r' 5,5-5,9
68
Окончание табл. 6
Протоны и их положение в молекуле <5, м.д. Протоны и их положение в молекуле <5, м.д.
R-CH2-C6H5 2,5-3,0 R-CHC1-, 5,6-5,9
\ ——\
СНз 2,9 СНз 6,42
/
н
rch2-s- 3,3 с6н5-н 7,26
R2C=C(R)-CHR2 2,9-5,3 С6Н5-ОН 4,0-12,0
r-ch2-i 3,0-3,4 R-C(=O)H 8,5-11,0
R-COOH 10,0-13,0
R-CH=CH-OH 15,0-18,0
влияет индукционный эффект заместителя и присутствие соседних непредельных группировок. Поэтому атомы водорода, находящиеся в
XC=CZ /
a-положении к двойной связи ( > 8 1,7-2,1 м.д.), аромати
ческому кольцу ( Аг—С^Н )или>С=О-группе ( ^СН—С^ ,52,1-2,6
М.д.) или во фрагменте Н—С—X = Hal, О, S, N, 5 3,3-4,2 м.д.), легко идентифицируемы. Еще большая разница в магнитном экранировании наблюдается, если водород присоединен к углероду в sp2- или sp-гибридизации (Д м.д.): 4,5-6,0 (=СН), 2,3-2,6 (=СН), 6,5-8,0 (С-Н), 8,5-11,0 (Н-С=О). В данном случае помимо деэкранирования существенное влияние на величину химического сдвига оказывают так называемые кольцевые токи электронов, создаваемые непредельными группировками, особенно ароматической системой, тройной связью и карбонильной группой. Они могут как деэкранировать в ароматических структурах, так и экранировать протон при 5р-гибридизованном атоме углерода.
Для ароматических протонов существует определенная зависимость между химическим сдвигом и характером, а также взаимным расположением заместителей. Протоны бензола резонируют при 7,26 м.д. Яри наличии в кольце электроноакцепторного заместителя (-СООН, ~NO, и др.) сигналы смещаются в слабые поля на величину до 1 м.д. Если заместитель имеет неподеленную пару электронов (-ОН, -NH, и
69
др.), сигнал смещается в сильные ноля до 0,5-1,0 м.д. Алкильные заместители, обладающие -t /-эффектом, влияют на величину химического сдвига в значительно меньшей степени, чем заместители, обладающие -А/-’)ффектом.
Наиболее сильное влияние заместитель оказывает на орио-про-тон, несколько меньшее на ия/эя-протон и еще меньшее нащеотя-протон. В случае алкильных заместителей химический сдвиг сигналов ароматических протонов не зависит от положения заместителей и сигналы про являются в виде одного, как правило, широкого сигнала.
Полициклические арены дают сложный сигнал в интервале 7,0-9,0 м.д.
В основных гетероциклических ароматических системах химические сдвиги протонов попадают, в принципе, в ту же область, что и у бен зола. Но различное положение протонов к гетероатому (а- и 0-положе-ния в фуране, пирроле и тиофене; а-, 0- и у-положения в пиридине) довольно велико и поэтому сигналы данных протонов легко идентифицируемы, причем а-прозоны резонируют в самом слабом поле (индукционное действие гетероатома).
Для групп ОН в спиртах и фенолах, NH и NH, в аминах или амн дах положения сигналов протонов нехарактеристичны и в значительной степени зависит от участия данных групп в образовании водородных связей. Однако определить, что сигнал принадлежит именно этим группам, экспериментально относительно несложно, так как его положение зави ент от концентрации раствора и температуры, при которой записываете-спектр: при разведении сигнал сдвигается в сильное поле, а при пониже нии температуры - в слабое. Кроме того, эти сигналы исчезают при ра створении образца в 1),0 (если возможно) за счет водородного обмена.
Положение сигнала протона СООН-группы в карбоновых кислотах (i) 10-13 м.д.) довольно постоянно и меньше изменяется даже при сильном разбавлении, поэтому оно более характеристично.
Если на магнитное экранирование протона действуют одновременно две или более группировки, то смещение его сигнала в область слабых или сильных полей зависит от характера и величины электронодонорного или электроноакцепторного действия заместителей.
Таким образом, по величине химического сдвига может быть определено положение протона в органической молекуле. Однако сигналы от протонов, незначительно отличающихся по магнитному экранированию, могут частично или полностью перекрываться. Так, сигналы метиленовых групп в ациклических или циклических системах часто слип, юзея в один широкий сложный сигнал.
70
Протоны, связанные с неуглеродными атомами в бинарных соединениях, также могут быть исследованы методом ПМР. Химические сдвиги (£ м.д.): Н, (4,43), NH. (0.18), И,О (0.83). Sill. (3,23). PH. (1,71), H,S (0,31), HF (2,73), НС1 (-6,22), HBr(-4,12). HI (-13.02).
3.3. Полуэмпирические соотношения между химическим сдвигом и строением молекулы
Согласно теоретическим представлениям различают 4 типа вкладов в химический сдвиг.
1. Электронное влияние заместителей.
2. Молекулярные магнитные поля за счет удаленных связей.
3. Молекулярные электрические поля за счет постоянных диполей.
4. Взаимодействие электронов через пространство.
Если электронное влияние заместителей на химический сдвиг определяется только внутри одной молекулы, то явления, обозначенные в пп. 2-4, могут происходить и межмолекулярно (раздел 3.4).
3.3.1. Электронное влияние заместителей
Введение заместителей вызывает изменение состояния электронов через соседние и даже несколько связей и может распространяться на всю молекулу. Чаще всего оно передается через 1 -3 связи. Через большее число связей, как правило, оно становится незначительным и им можно пренебречь.
Заместители с +1- или +Л7-эффектами подают электроны, экранируя изучаемый протон и сдвигая его сигнал в область сильных полей, заместители с или -М-эффектами действуют в противоположном направлении. Чем больше по модулю I- или Л7-эффекты, тем больше сдвиги в соответствующую область спектра. Отсюда, по величине эффекта можно прогнозировать положение сигнала в спектре ЯМР, а по величине химического сдвига - оценить эффект заместителей, а также рассчитать «т-константы Гаммета или о*-константы Тафта.
3.3.2. Молекулярные магнитные поля за счет удаленных связей
Под влиянием внешнего магнитного поля /7 в системе электронов индуцируются токи, которые, в свою очередь, генерируют магнитное поле, экранирующее ядро собственной электронной оболочкой. Но оно может воздействовать и на те протоны, которые не связаны непосредственно с данными циркулирующими элск!ронами.
71
Подвижные электроны /г-связей особенно легко индуцируют магнитные поля, которые оказывают значительное влияние на химические сдвиги протонов.
Например, для молекулы бензола, помещенной в магнитное поле, наблюдается следующая картина (рис. 13).
Рис. 13 Рис. 14
Циркулирующие электроны тт-связи генерируют магнитное поле Н’, которое не просто противоположно по направлению полю Нд, но и является анизотропным, т.е. различным по направлениям в системе координат (х,у, z). Если внутри бензольного кольца направления полей Н' и Н противоположны, то за его пределами имеются замкнутые направленные силовые линии, параллельные полю Н,., поэтому ароматические протоны за пределами кольца попадают в сферу действия параллельных полей И'. В результате для возбуждения этих протонов требуется не такое сильное внешнее магнитное поле, как в отсутствие кольцевых токов, и их сигналы обнаруживаются в более слабых (<5= 7-9 м.д.) полях по сравнению с сигналами многих других протонов.
С помощью «конуса анизотропии» (рис. 14) можно получить наглядное качественное представление о том, в каких пространственных областях происходят сдвиги сигналов в более сильные или, соответственно, слабые поля.
В общем случае можно сказать, что влияние молекулярного магнитного поля на химический сдвиг протона зависит от расстояния и (вследствие анизотропии этого магнитного поля) ориентации исследуемого протона относительно системы связей, индуцирующих это поле.
Имеются соединения, в которых протоны пространственно располагаются лишь в положительной части «конуса анизотропии». Тогда индуцированное поле Н' суммируется с Нд и вследствие этого сигналы протонов сдвигаются в более сильные поля, часто даже за сигнал ТМС,
72
т.е. наблюдаются отрицательные значения химических сдвигов, как например в ПМР спектрах бнцикло[4,4,1 ]ундека-1,3,5,7,9-пентаена (1,6-ме-тано[10]анулен) (А), транс-15,16-диметнл-15,16-днгидропирана (В) и [18]анулена (С), а также порфиринов.
'>11 кольца - 8,14-8,67 м.д.
<5Н (внешн.) - 9,28 м.д.
Вместе с бензолом эти соединения составляют группу ануленов с 4л+2л-электронами (и = 1, 2, 3, 4), которые в соответствии с правилом Хюккеля обладают ароматическим характером. Поэтому делокализацию л"-электронов в цикле в основном состоянии этих систем можно устано-. вить с помощью ПМР спектроскопии по эффекту кольцевого тока, который является качественным критерием ароматичности. Такие молекулы называются диатропными, а молекулы в которых совсем нет кольцевого Тока, - атропными.
Среди кратных связей особенно сильной магнитной анизотропией обладают двойные С=С-, С=О-, C=N- и N- O-связи, а также тройные связи С=С и C=N (см. рис. 15).
*
j t
71
I
Рис. 15
Таким образом, магнитная анизотропия может быть положительной (в межконусном пространстве дефицит электронной плотности, +) или отрицательной, поэтому сигналы протонов, лежащих в конусе электронной плотности, сдвигаются в слабое поле, если в нем отрицательный заряд, и в сильные поля при наличии в конусе положительного заряда по сравнению с сигналами соединений, в которых отсутствует маг-
нитная анизотропия.
Последнее подтверждается сравнением химических сдвигов протонов групп СН3 в толуоле (8 = 2,32 м.д.) и пропине (8= 1,80 м.д.), свидетельствующем о повышении экранирования протонов вблизи оси связи С=С. Напротив, в областях, удаленных от оси тройной связи, ожидается деэкранирующий эффект. Экспериментальное подтверждение этого
эффекта найдено при изучении ПМР спектра 4-этинилфенантрена, в котором сигнал протона Н сдвинут на 1,71 м.д. в слабое поле по сравнению с его положением в спектре самого фенантрена.
Вклад цилиндрических или химических связей в химический сдвиг протона (Л8) за счет индуцированного магнитного поля рассчитывают
по уравнению
Л8 = (зсо?^-1),
3R' х ’
где Л% разность диамагнитных восприимчивостей связей в двух на-
74
правлениях в пространстве (величина анизотропии, табл. 7), R - длина вектора, соединяющею протон с центром святи, <р - уюл между осью связи и вектором (рис. 16).
Связь I .. н R С’Н И? с“с С Л ' С с с D 1 А ОС Рис. 16 C=N Таблица 7 !%_( 10 111 см7молекула) -11,00 -13,98 -20,55 -27,5 -57,0
Для напряженных (3- или 4-членных) циклических систем приведенные значения Л % неприменимы.
Различие в химических сдвигах аксиальных и экваториальных прогонов в циклогексане (Дкв - J ~ 0,5 м.д.) объясняется, главным образом, анизотропией простой связи С-С (рис. 17, 18).
3.3.3. Молекулярные электрические поля за счет постоянных диполей
Если в молекуле имеются группы, создающие постоянные электрические диполи (С=О, С—X, C^N, N=O и др.), то они образуют электрические поля, которые действуют непосредственно через пространство. Это ведет к смещению электронов вдоль оси связи С-Н, изменяя тем самым экранирование протона и его химический сдвиг.
Данное явление в большинстве случаев нельзя отделить от индукционного эффекта группы, однако существуют соединения, в которых дипольсоздающие группы настолько удалены от рассматриваемого протона, что действие этого эффекта через сдвиги практически не имеет значения. В то же время протон может располагаться в пространстве так близко к дипольной группе, что электрическое поле будет оказывать существенное влияние на химический сдвиг.
75
3.3.4. Взаимодействие электронов через пространство
Если протоны расположены пространственно так близко, что их электронные оболочки вступают в обменное (ван-дер-ваальсово) взаимодействие, то происходит смещение (АЗ, м.д.) сигналов в слабое поле по сравнению с соединениями, где такое взаимодействие отсутствует.
Таблица 8
Соединение
^Н-Н, нм
Соединение
*н-н, нм
М м.д.
Если расстояние между протонами 7? превышает 0,21 нм, то с"зигом за счет взаимодействия через пространство можно пренебречь (табл. 8).
3.4. Влияние внешних воздействий на химический сдвиг
Температура. Химические сдвиги протонов связей С-Н в основном очень мало меняются с изменением температуры, но положение сигналов протонов в группах О-Н, N-H и S-Н сильно зависят от температуры, поскольку последние склонны к образованию водородных связей. Эти протоны в ассоциатах имеют значения химических сдвигов иные, чем в изолированных молекулах.
76
,.IL ..I-L ,-fk
() O’ O'
I I I
R R R
Изменение температуры влияет на образование ассоциатов, а тем самым на химический сдвиг: чем прочнее водородные связи, тем в более слабых полях находятся сигналы данных протонов. С понижением температуры водородные связи становятся более прочными. Наиболее прочные внутримолекулярные водородные связи наблюдаются для соединений, в которых возможно образование 6-членного кольца, в частности хелатного типа (<5= 15-20 м.д.). Влияние температуры на прочность ВМВС, а следовательно, и на химический сдвиг протонов в ОН-, NH-или SH-группах весьма незначительно.
Концентрация. Химические сдвиги протонов связей С-Н также мало зависят от концентрации. Для протонов, связанных с гетероатомами и образующих ММВС, увеличение концентрации повышает прочность водородных связей, сдвигая сигналы протонов в группах ОН, NH или SH в слабые поля. В случае ВМВС концентрация также мало влияет на химические сдвиги данных протонов.
Растворитель. Сравнивать химические сдвиги протонов различных соединений можно только для спектров, записанных в одном и том же растворителе. Поскольку на практике это не всегда возможно, стараются подобрать неполярные растворители (СС14, CS2, С2С14, цикло-С6Н|2 и др.).
В ароматических растворителях (бензол, толуол, пиридин и др.) значения химических сдвигов изменяются; это объясняется их сильным анизотропным эффектом (наличие кольцевых токов, см. раздел 3.3.2).
Химические сдвиги, измеренные в полярных растворителях, как правило, отличаются от значений, измеренных в неполярных растворителях. Поскольку большинство из них содержат атомы водорода, что усложняет спектр образца, то используются их дейтерированные производные: СО3ПО2(нитрометан-Д), CD3CN (ацетонитрил-Д), CD3-CO-CD3 (ацетон-Д, дейтероацетон - ДАЦ), CD3OH (метанол-сЦ, H-CON(CD3)2 или D-CON(CD3)2 (диметилформамид-Д или -d?, ДМФА-Д или -d?), (CD3)2S-»O (диметилсульфоксид-d^ ДМСО-б6), CF3COOH (трифторуксусная кислота, ТФУК) и др.
С помощью этих растворителей (при записи спектров разбавленных растворов) можно определить константы спин-спинового взаимодействия (см. с. 89) и таким образом различить первичные, вторичные и : третичные спирты, экваториальные и аксиальные ОН-группы в цикло-
77
алканолах и т.д. Решшь ли вопросы при работе с неполярными растворителями нельзя из-за быстрого внутри- или межмолекулярного протонного обмена (см. раздел 3.6).
Разность химических сдвигов одного и того же протона в различных растворителях называется сдвигом за счет растворителя. Часто он позволяет получить дополнительную информацию о молекулярной структуре.
Хиральные растворители. Если какой-либо рацемат (7? + S) растворить в хиральном растворителе, то R- и S-энантиомеры по разному взаимодействуют с ним, что выразится в различии химических сдвигов про-
*
тонов, связанных с хиральным атомом углерода ( —С—И ). в качестве таких растворителей наиболее часто используют 1-фенилтрифторэтанол
а*
СН—СНз
Nib
).
На основе различия в химических сдвигах в хиральных раствори телях для ряда аналогичных соединений, из которых хотя бы один имеет известную конфигурацию, можно установить абсолютную конфигурацию остальных соединений.
Парамагнитные сдвиги. Если к раствору образца добавить немного парамагнитного комплексного соединения (сдвигающего, или шифт-реагента, shift-reagenfy. трис(2,2,6,6-тетраметилгептан-3,5-дионато- или дипивалоилметанато, dpm)- или трис( 1,1,1,2,2,3,3 -гептафтор-7,7-димети-локтан-4,6-дионато- или фтороктандионато, (офевропия (3+) или -празеодима (3+)
М(<1рт)з
М = Ей3* или Рг’',
78
то наблюдается изменение ширины линии и :химических сдвигов прозонов, находящихся у атомов углерода в диамагнитной молекуле образца, которые особенно легко присоединяют ион металла.
В спектрах молекул лигандов (спирты, кетоны, амины и тиолы; кислоты, как и вода, разрушают шифт-реагенты) сигналы протонов смещаются в слабые -Eu(dpm)3 или сильные -Pr(dpm)3 поля, если комплекс имееточень короткое время спин-решезочиой релаксации (Т < 10" сек).
Сильнее всего изменяются химические сдвиги протонов, наиболее близко расположенные к иону металла, но для них же наблюдается и
максимальное уширение сигналов.
Тем не менее, таким образом удается различить сигналы а-, Р-, у-и §-СН2-групп алифатических спиртов, цис- и /ир<7нс-1,2-циклогександи-олов (для первых - сдвиг в слабые поля по сравнению со вторыми) и т.д.
Для определения энантиомерности наиболее подходящим реагентом является трис[3-(трифторметилгидроксиметилен)-б-камфарат]евро-
пия, Eu(tfc)3 (
У
асимметричного атома углерода в аминах ( )>а также и ДРУ-
гих протонов радикалов R1 и R2 в спектрах 7?-энантиомеров в значительно большей степени, чем в спектрах 5-энантиомеров.
3.5. Спектроскопия магнитного резонанса других ядер
В последние годы ЯМР спектроскопия других магнитных ядер приобретает все большее значение и определяется, главным образом, совершенствованием спектрометров. Существенное преимущество данного метода перед ПМР заключается в том, что области химических сдвигов в этом случае значительно больше (для протонов 0-20 м.д.) и поэтому сигналы ядер разделены и хорошо идентифицируемы.
Однако следует учитывать и некоторые недостатки такого же широкого, как и метод ПМР, применения методов ЯМР на других магнитных ядрах:
79
I) меньшею чувствительность ядер к магнитному полю;
2) меньшее природное содержание (кроме l9F) некоторых ядер (ПС |SN,, 29Si|s и др.);
3) квадрупольное уширение линий у ядер со спиновым квантовы',1 числом /> '/, (2Dp "В„ i4N7, ,7Os и др.);
4) очень большое время спин-решеточной релаксации (|3С6 и др.).
3.5.1. ЯМР спектроскопия ядер 19F
ЯМР на ядрах l9F является наиболее интенсивно развивающей^ , областью этого метода, что связано с наивысшей магнитной восприимчивостью (после протонов) ядер ”F, 100%-ным содержанием данного изотопа в природном фторе и важностью фторорганических соединений в теоретической и практической химии.
Теоретический анализ влияния р-электронов фтора на экранирование его ядра показал, что наиболее существенный вклад связан с характером связи Э-F: для полностью ионизированного атома фтора (фто рид-иона, F ), обладающего сферическим симметричным электронным облаком, вследствие чего ядро максимально экранировано, сигнал наблюдается в самом сильном магнитном поле (<5= 0 м.д.). В ковалентно построенной молекуле фтора (F2) облака р-электронов наиболее отклоняются от сферической, т.е. они наиболее деэкранированы и их сигнал смещается в самые слабые поля на 625 м.д. В данный интервал химических сдвигов укладываются сигналы всех ядер l9F в органических соединениях. Отсюда следует, что чем более поляризована в сторону фтора связь Э-F, тем в более сильных полях находится сигнал ЯМР |9Е
Для некоторых неорганических соединений сигналы ядер l9F лежат в более слабых полях, чем сигнал молекулы F2 (430 м.д. относительно CFCL). Например, сигналы l9F для нитрозилфторида (FNO) и дифтордиоксида (F2O2) лежат при 476 и 865 м.д. соответственно.
По химическим сдвигам можно обнаружить минимальные различия в химическом окружении ядер фтора: различить энантиомерные спирты или амины, содержащие, например, CF3-rpynnw у асимметрического атома углерода, обнаружить фторкарбениевые ионы и т.д.
В качестве внутреннего эталона и одновременно растворителя могут использоваться вещества, содержащие один или несколько магнитно эквивалентных атомов фтора: фтор- и гексафторбензолы, трифторуксусная кислота (ТФУК, для соединений, не содержащих подвижных атомов водорода), бензотрифторид (C6HSCF ), тетрафторметан (CF для । азообразных веществ) и др. Но единым внутренним стандартом выбран
80
фтортрихлорметан (CFC1,), сигнал которого принят за нуль.
Существенным отличием 5-шкал ЯМР 'Н и l9F является то, что химические сдвиги протонов всех соединений (за исключением двух-трех) имеют положительные значения относительно ТМС, а сдвиги ядер ,9F могут иметь (относительно CFCL) и положительные (влево от нуля, в область слабых полей) и отрицательные значения (это касается и сигналов других эталонных веществ, например CF3COOH).
+430 cf4 cf3cooh -64,0 -76,54 -76;3S Лм.д
0 -67,75 -113,12 -162,92 -195
CFCI3 F"
+76,54 +8,79 -36,58 -118,46
Рис. 19
Следовательно, для пересчета величин химических сдвигов ядер l9F от одного эталонного вещества к другому необходимо пользоваться следующими соотношениями:
^crcij = ^сг4 _64,0 = 5CsHjCCIj -67,75 = 5CF C00H -76,54 =
= 5СбН1Р-113,12 = 5са-162,92.
Значения химических сдвигов ядер l9F лежат в следующих интервалах м.д. (рис. 20).
Рис. 20
Ниже приведены более точные значения химических сдвигов ядер l9F в различного типа соединениях (5 м.д. относительно ТФУК): -SF. | (160-95), -SO,F (140-115), -C(=S)F (125-105), -C(-O)F (110-105-), CFX3
81
(70-0), CF3S-и CF3N-(30-20),-CFJ (=20),-CF2C1 (5-15), СГзС^- (5-
20), CF3C(=S)- (=5), CFsCF^ (2-3), -CF2O- (O-r-lO), CF3CH2- (=0), CF.CF - (04-10), =CF2 (-54-50), -CF2Br (=-20), =CF- или Ar-F (-25--120), —CFzCF^ (=-30), -CF2-C(=O)~ (-35--55), CF3CF - (-454-50), -
CF2CF - (-504-55), -CF2CH - (==-50), -C£2CF2H (=-55), -CF2CF2H (-60--
(-404-45),
(-454-60), x'C-C''F (-504-
55), CF3CE\ (-554-90).
Растворители и концентрация растворов могут оказывать влияние на химические сдвиги ядер |9Е Так сигнал фтора в замещенных фтор-бензолах смещается при бесконечном разбавлении в гептане на 1,4-1,6 м.д. в слабое поле. Однако этот сдвиг относительно невелик по сравнению с ПМР, учитывая протяженность спектрального интервала в ЯМР l9F (625 м.д.) и ПМР (=20 м.д.). Влияние растворителя компенсируется при проведении исследований в разбавленных растворах с применением
внутренних эталонов.
с 1
Рис. 21
Для получения значений 3, не зависящих от концентрации, экстраполируют 8 к. бесконечному разбавлению раствора, для чего записывают спектры растворов при их последовательном двойном разбавлении, получая зависимость 8 от с-1 (рис. 21).
3.5.2. ЯМР спектроскопия ядер 13С
Незначительное природное содержание ядер |3С (1,08%) в природном углероде и их малая чувствительность к магнитному полю (в 62,5 раза меньше, чем для протонов) предъявляют особую требовательность к ЯМР спектрометрам на этих ядрах: необходимо работать с образцами максимального объема и применять другие методы повышения чувствительности прибора, например импульсную ЯМР спектроскопию с Фурье-преобразованием в сочетании с накопителем. Используется и метод шумовой модуляции, когда на магнитное поле накладывается насы
82
щающее радиоизлучение с таким широким диапазоном частот, что подавляется взаимодействие ядер |3С сразу со всеми протонами. При этом проявляется эффект Оверхаузера, который вызывает дополнительное усиление сигналов ядер |3С.
Поскольку атомы углерода входят в состав всех органических соединений (sp3-, sp1- и sp-гибридизация в алифатических соединениях, C ip, в связях ^С=О и ^С—О—, —C=N , ^C=N— и -^С— и т.д.), то благодаря малой концентрации ядер |3С в природном углероде удается получить хорошо разрешенные спектры.
Химические сдвиги ядер |3С, как и протонов, крайне чувствительны к малейшим изменениям электронной конфигурации атома углерода, особенно к изменениям конформации молекул. Например, в спектрах производных циклогексана (z/mktzo-C6H||OR, где R = Н, Aik, Аг) сигнал атома углерода, связанного аксиальной связью с RO-группой, сдвинут на 4 м.д. в сильные поля по сравнению с сигналом того же атома, но связанного с экваториальной RO-группой. Объясняются такие сдвиги пространственными факторами: если протон или один из заместителей, связанных с изучаемым атомом углерода, вступает в пространственные взаимодействия с какой-либо электронодефицитной группой, то электронная плотность вокруг этого атома углерода повышается (увеличивается ' его экранирование) и сигнал смещается в область сильных полей.
Увеличением экранирования объясняется смещение сигнала |3С в область сильных полей (на 10-13 м.д.) в спектрах сопряженных карбонилсодержащих соединений по сравнению с несопряженными:
__ /С=С-С(=О)— X о 13
х | ’ /N—С(=о>— , —О—С(=О)— и
Т.д.
Значения химических сдвигов ядер |3С (интервал около 400 м.д.) измеряются относительно ТМС (его сигнал принят за нуль), ГМДС или сероуглерода (CS2), используемых в качестве внутренних эталонов. Они Имеют положительные значения при нахождении их в слабых полях по ! сравнению с сигналами эталона.
| Соотношение химических сдвигов (<5) относительно ТМС и CS2 Имеет зависимость:
^тмс = ^cs, +192,8 .
Интервалы химических сдвигов (А м.д.) ядер |3С в различного типа оединениях лежат в следующих диапазонах (рис. 22).
83
250 200
тмс о
150 100 50
~1------1-------Г.........I-------1-------1-----1—
+50 0 -50 -100 -150 -200 -250
CS, Рис. 22
Значения химических сдвигов (<S м.д., относительно ТМС) соединений CH-X: X = Н (-2,3), Me (7), Ph (22), F (76), Cl (25), Br (4), I (-22), OH (37), NH2 (14); CH3 в алканах (6-30); CH,, CH, С в алканах (15-55); циклопропан (-3,5); циклогексан (27,5); СН3СН2-Х; X = F (21), I (31); СН3СН-Х: X = F (79), -О- (67), ОН (57), Вг (28), Ph (26); (CH2)n; п = 3 (-3), 7-12 (31-24); (СН3)4С (33); (CH3),N (40); (СН,СН2)4С (11); (СН3СН2)4РЬ (Н); (CH3CH,)4SH (-0,5); (CH3CH,)4N+ (52)1 (CH3CH,)3N (48); R-CH=C=C<: R = H (75), Me (86); >C=C=C< (205-210); R-C=C-R': R1 = H (70), CH. (77), Ph (88); R"c=CH2 : R= Me(119), Bu (116), Z-Bu (107);
^С=СН2 : R = Me (118), Bu (140), Z-Bu (150); ^c=CH? : X = CN (142), -CH - (136), OCOMe (101), -O- (88); ^С^СНг : X = OCOMe (156), -CH - (140), CN (ПО); XHC=CHX: X = COOEt (133 - цис, 127 -транс), Cl (120 - цис, 122 - транс), Br (109 - цис, 116 - транс), I (80 -
цис, 96 - транс)', (ипсо): X = F (165), ОН (157), OR (162).
NR, (153), NO, (148). CH. (139), Cl (136), H (130). Br (124). 1 (97/
84
х (орто): X = I (140), Cl (129), NO, (124), NR, (114);
(мета): X - 1 (133), H (127);
(пара): X =
NO2 (13 6), 1 (128), OH (121), NR2 (116);
P), О (155-a, 110-P), S(130-a+P);
:X = C(144-a, 127-
(180-a, 136-P, 175-
y); r-C^x : X = H, OH, Hal, OR1, R или R1, NH,, NHR1, NR1,: 160-200 (в зависимости от электронных свойств X; например: уксусная кислота -21,1 (СН3), 177,2 (СООН); ацетон - 30,4 (СН3), 204,1 (СО)); оксимы и гидразоны альдегидов и кетонов >C=N- (-150^-180); нитрилы -C=N (115-125); изонитрилы -N=C (156-158).
3.5.3. ЯМР спектроскопия ядер 31Р
Ядро 3|Р обладает свойствами, наиболее благоприятными (после 'Н и l9F) для исследования их методом ЯМР. Помимо интереса для синтетических фосфорорганических и природных фосфорсодержащих соединений, ЯМР спектроскопия на ядрах 3|Р представляет ценную возможность изучения закономерностей ЯМР для атома с переменной валентностью, способного образовывать соединения самого разнообразного типа.
Ввиду малой чувствительности ядер 3|Р к магнитному полю (6,6% от чувствительности 'Н) рекомендуется, как и в случае ЯМР на ядрах |3С, работать с возможно большими объемами вещества и использовать другие способы повышения чувствительности прибора.
Химические сдвиги (8, м.д.) ядер 31Р измеряются относительно 85%-ного водного раствора Н3РО4 как внешнего эталона, химический сдвиг которой принят за нуль.
В качестве эталона предложено использовать и оксид Р4О6 с т.пл 23,8°С, легко переохлаждающийся до 16°С. Он дает значительно более узкий сигнал (0,3 Гц на полувысоте против 3.5 Гц в Н,РО4). Pro сдвиг
85
относительно Н РО составляет 112±0,1 м.д. в слабое поле. В качестве внутреннего эталона используют триметилфосфат (Ме3РО4), сигнал которого смещен на 2,4 м.д. в сильное поле относительно Н3РО4.
Общая шкала химических сдвигов ядер 3|Р составляет 500 м.д.. если не принимать во внимание резонанс элементного фосфора, дающего широкий пик в области -450 м.д.
Химические сдвиги ядер 3,Р (<5, м.д., относительно 85%-ной Н3РО4): РН3 (-215); МеРН2 (-130); Ме2РН (-95); Ме3Р (-60); (PhP)n (0 при п =2 или + 10 при п = 4); в интервале -60^+80 м.д. резонируют соединения R„PF5 „ (п = 0-5), (RO) Р(ОН)3 п (п = 0-2), Р(О)Х3 (X = F, ОН, R, OR, NR2 в различных сочетаниях); в интервале 90±5 м.д. - P(S)X3 (X = F, Cl, R, RO, NRj; PF3 при 100; P(NR2)3 около 110; (RO)3P и R,PF от 125 до 140; RPF2 и RPClj 135-200; PBr3 и PC13 при 220 и 230 соответственно.
3.5.4. ЯМР спектроскопия ядер 14N
Если спектры ЯМР на ядрах l5N записывают только на образцах, обогащенных данными ядрами, то резонансные сигналы ядер l4N наблюдаются и для обычных соединений. Химические сдвиги ядер l4N, непосредственно связанных с протонами (связи N-H), можно определить из спектров ПМР с помощью гетероядерного двойного резонанса, для чего варьируют частоту дополнительного радиоизлучения и одновременно наблюдают спектр ПМР. Как только с помощью данного приема достигается резонансная частота ядер l4N, обнаруживается исчезновение тонкой структуры или сужение сигнала протона в NH-rpynne.
Область химических сдвигов ядер l4N и l5N составляет около 1000 м.д.
В качестве внутренних стандартов в данном методе на ядрах l4N используют нитрат-ион (для водных растворов), сигнал которого принят за нуль, или нитрометан (для органических растворителей), связанных соотношением
^CH.NO, = ^NO,
Ниже приведены химические сдвиги сигналов ядер l4N в некоторых классах органических соединений (<5, м.д., относительно CH3NO2): RNOj от 45 до 0; O2N-R-NO2 от +5 до -10; Ar-NO2* (0-40); R(NO2), (20-50); хинолин (70-115); R-C(S)-NHR (210±5); R-C(S)-NH2 (225±5); R-C(O)-NHR' (220-250); R-C(O)-NH2 (230-270); RNH2, R2NH, R,N (305-355); NH/(325-350).
* Здесь и далее величины химических сдвигов отрицагсльны.
86
3.5.5. ЯМР спектроскопия ядер 11В
Оба встречающиеся в природе изотопа бора (|0В5 и "В5) имеют магнитные моменты, но чаще применяется резонанс на ядрах "В вследствие их большего природного содержания (81,2%) и большей величины магнитного момента. Поскольку ядро "В (/= 3/2) обладает также квадру-полъным моментом, сигналы ЯМР уширены по сравнению с сигналами 'Н и |9Е
В качестве внешнего или внутреннего эталона применяется триф-торборэфират (F3B OEt2, его сигнал принят за нуль). Как и в спектрах на других магнитных ядрах сигналы, расположенные слева от сигнала эталона (в слабых полях) имеют положительные значения химических сдвигов, справа - отрицательные.
Химические сдвиги ядер "В (А м.д., относительно F3B OEt2):
R4 R'\ Ar'\
R2/B-R3 (92-85); R2/B“X или Ar,/B“X (87-42);
„X Ri\ ^R3
К(илиАг)—(62-25); R2/N~B''R4 (60-30 - мономер или 25-10-
Rx .--IK /Н димер); R'-B-N-R2 или X-B-N-R тримеры (51-35);
X^
(50-32 (0,20-10 (2)); Y^B-Z (50--5); MBR4, MBAr,, MB(OR)4 (15-
-40), где X, Y, Z = Hal, OR, NR2, SR; M = Li, Na, K, Al.
3.5.6. ЯМР спектроскопия некоторых других магнитных ядер
Интенсивное развитие техники ЯМР способствует изучению резонанса изотопов с небольшим магнитным моментом и низким природным содержанием, и поэтому дающих слабые сигналы ЯМР.
Область химических сдвигов магнитного ядра 17О, как и для l4N, составляет около 1000 м.д., а для его аналогов (Se и Те) - значительно больше.
Основной вклад в химический сдвиг этих ядер вносит парамагнитная составляющая атомного экранирования (в отличие от протонов, химический сдвиг которых в основном зависит от диамагнитной составляющей), значения которой изменяются в широких пределах.
87
Благодаря большим сдвигам снижаются требования к точности определения, что позволяет использовать в ЯМР спектроскопии ядра, обладающие квадрупольным моментом и дающие поэтому широкие сиг налы.
Химические сдвиги кислорода, часто выступающего центром координации при образовании комплексов и сольватации кислородсодержащих соединений, в значительной степени зависят от природы растворителя (например, при переходе от раствора ацетона с изотопом 17О в циклогексане к раствору в муравьиной кислоте сигнал ядра |7О смещается в слабые поля на 48 м.д.).
Наиболее изучен резонанс на ядрах 17О в карбонилсодержащих соединениях (альдегиды, кетоны, кислоты, эфиры, амиды и т.д.), а также в соединениях, содержащих фрагмент С-О-С. Для первых сигналы лежат в интервале 240-610 м.д. (внешний эталон - Н2|7О), а для вторых -120-260 м.д. Для ядер 17О, как и для других магнитных ядер, выполняется правило: чем более деэкранировано ядро, тем в более слабых полях находится его сигнал в спектре ЯМР.
В отличие от ядер 17О для ядер 77Se и |27Те нельзя с полной уверенностью сказать, что весь диапазон их химических сдвигов исследован, хотя он и составляет порядка 4000-5000 м.д. Это связано с их более развитым электронным облаком, а также проявлением ими различных степеней окисления, что также сказывается на экранировании ядер.
Химические сдвиги ядер 29Si, ||5' ll9Sn и 207РЬ, помимо самостоятельной ценности, интересны для сравнения с химическими сдвигами |3С. Установлено, что порядок изменения химических сдвигов в аналогичных соединениях углерода и кремния противоположны. Это объясняется наличием у последнего свободных J-орбиталей, что приводит к возможной регибридизации кремния.
В случае олова, в основном повторяется та же картина, что и для кремния, но закономерность имеет скорее промежуточный между кремнием и углеродом характер, что может быть связано с уменьшением участия высших орбиталей в образовании связей. В этом плане свинец при нахождении в магнитном поле в большей степени напоминает углерод, чем кремний или олово.
Резонанс магнитных ядер типичных металлов (7Li, 23Na, 27А1), необходимость излучения которого обусловлена их широким применением в металлокомплексном катализе, наблюдается в сравнительно узких диапазонах частот, причем химические сдвиги данных ядер определяются в основном факторами среды, а не строением соединений, т.е. электронным экранированием.
88
3.6. Непрямое спин-спиновое взаимодействие
В спектрах ЯМР высокого разрешения помимо расщепления сигналов магнитных ядер, обусловленного их различным экранированием, наблюдается дополнительное расщепление, вызванное непрямым спин-спиновым взаимодействием.
Принцип взаимодействия магнитных ядер на примере взаимодействия протонов, поскольку взаимодействие других ядер по сути аналогично, следующий.
3.6.1. Взаимодействие 1Н-1Н
Спектр ПМР «нулевого» порядка 1,1,2-трихлорэтана состоит из двух сигналов (рис. 23, верхний спектр) относящихся к различно экра-
нированным протонам: | Н
/Н сьс-с-нз
С1
ТМС
Н
РР+Н2
i ТМС
Рис. 23
^Н'+Н1
Протон Н связан с атомом углерода, имеющим два электроотрицательных атома хлора, и, следовательно, его сигнал находится в более слабом поле по сравнению с сигналом протонов Н1 и Н2, имеющими одинаковое химическое окружение, а значит и одинаковый химический сдвиг.
Взаимодействие протонов, а точнее их спинов, друг с другом состоит в следующем.
Любой протон, например Н, сам является слабым магнитом и поэтому создает собственное магнит
ное поле, которое воздействует на протоны Н1 и Н2 как через пространство, так и через связи между ядрами.
Взаимодействие ядер через пространство называется прямым, через связи - непрямым. В жидких и газообразных веществах прямое взаимодействие пронаблюдать нельзя, так как вследствие быстрого молекулярного движения составляющие данного взаимодействия взаимно уничтожаются. В твердых телах прямое спин-спиновое взаимодействие приводит к такому уширению сигналов, что измерить химические сдвиги не
89
удается (наблюдается так называемый «широкополосный» ЯМР). Прямые спин-спиновые взаимодействия можно обнаружить в спектрах ПМР жидких кристаллов.
Непрямое спин-спиновое взаимодействие можно наблюдать в спектрах жидких и газообразных веществ.
В 1,1,2-трихлорэтане спины ядер водорода расположены хаотично, но в магнитном поле они выстраиваются вдоль силовых линий и имеют две разрешенные ориентации: параллельную направлению поля и анти-параллельную. Это значит, что в месте расположения протонов Н' или Н2 из-за действия магнитного поля протона Н возникают два дополнительных поля - усиливающее и ослабляющее. (Будет ли дополнительное поле усиливающим или ослабляющим при параллельной ориентации спина ядра Н внешнему полю, зависит от числа связей, разделяющих взаимодействующие ядра, и указывается знаком константы спин-спино-вого взаимодействия, см. далее рис. 27). Следовательно, в месте расположения протонов Н1 или Н2 наблюдаются две линии. Поскольку они относятся к одному виду ядер, то являются не отдельными сигналами, а двумя линиями (дублет) одного сигнала (рис. 23, нижний спектр).
Ядра Н1 и Н2 независимо друг от друга ориентируются параллельно или антипараллельно внешнему магнитному полю (рис. 24). Они так-p|i ________________ же индуцируют дополнительные магнит-
--------------------- ные поля в месте расположения протона ---------------------с с >_>Но Н. Если направления спинов параллельны Дополнительное ДруГ другу, то действие их либо усилива-поле равно нулю ющее (спины параллельны приложенно-Рис. 24____________________му полю), либо ослабляющее (спины ан-
типараллельны полю). Если спины ядер Н1 и Н2 антипараллельны друг другу, то их дополнительные поля взаимно уничтожаются и результирующее действие равно нулю. Вероятность того, что результирующее поле будет равно нулю, вдвое больше, чем вероятности параллельных ориентаций спинов. Следовательно, сигнал протона Н состоит из трех линий (триплет), причем интенсивность средней линии (дополнительное поле равно нулю) вдвое больше интенсивностей крайних линий (рис. 23, нижний спектр).
Расстояние между линиями дублета или триплета (и вообще мультиплета) называется константой непрямого спин-спинового взаимодей-с гвия (КССВ). Она обозначается J и измеряется в Гц (J ).
Поскольку взаимодействие всегда двустороннее, то Jb обоих мультиплетах одинакова.
Прогон Н1 индуцирует дополнительное магнитное поле в месте
90
расположения протона Н2 и наоборот. Но такое взаимодействие экспериментально пронаблюдать нельзя, так как оба ядра, будучи магнитно эквивалентными, имеют одинаковый химический сдвиг и их сигналы накладываются друг на друга.
Если сигнал содержит нечетное число линий, то химический сдвиг измеряется от сигнала эталона до центральной линии мультиплета, если же четное - то до его середины.
Во фторметане (F-CH'H2H3) протоны эквивалентны, т.е. имеют одинаковые химические сдвиги. Независимо друг от друга они могут принимать параллельную или антипараллельную ориентацию в магнитном поле (рис. 25).
н* -----
н2 ----► ◄------► <~
н3 ◄-----------------------------
---------к ;..—...................> Но +72...................................+'/2 -72 -3/2
Рис. 25
При суммировании составляющих спина по направлению Но получаются два усиливающих (+3/2 и +'/) и два ослабляющих (~3/2 и ~'/2) дополнительных магнитных поля, т.е. сигнал ядра l9F расщепится в четыре линии (квартет). Соотношение интенсивностей линий в сигнале составляет как 3:9:9:3, т.е. 1:3:3:1.
Ядро l9F имеет спин J = '/2 и, следовательно, две возможные ориентации в магнитном поле (у^), что приведет к расщеплению сигнала протонов в дублет (см. расщепление сигнала протонов Н1 и Н2 в спектре ЯМР 1,1,2-трихлорэтана, рис. 23 и спектр ЯМР 'Н и l4F имеет следующий вид (рис. 26).
<-J-+ Константа взаимодей-
ствия JH F (Гц) имеет одинако-|__ | вую величину для обоих муль-
типлетов.
Спектр ЯМР F Спектр ЯМР Н Мультиплетностьсигнала
Рис. 26 Wрассчитывается по формуле
N = 2n-J + 1, где п - число соседствующих магнитно-эквивалентных ядер, J - спин ядра.
Если взаимодействуют ядра с J = то формула расчета муаыи-плегностп упрощается
f N п t I.
Q1
Однако эта формула правильна лишь в том случае, когда разность химических сдвигов (<5) взаимодействующих ядер сильно превышает (минимум на один порядок) константу взаимодействия (J) или когда взаимодействуют разные магнитные ядра.
В общем случае мультиплетность (количество линий) сигнала в спектре ЯМР какого-либо магнитного ядра, взаимодействующего через 1 -3 связи с другими магнитными ядрами со спином J=, рассчитывается по формуле:
N = (и + 1 )(т + 1 )(р + 1 )(<? + 1 )(г + 1), где п, т, p,q,r- количество магнитных ядер одного типа.
Н
П. I
„ „ /Р-С-СН2-СООН
Например, сигнал протона Нв соединении ।
F
содержит 48 линий: N = 3-2-2-2-2 = 48, где 3 - количество линий в сигнале от взаимодействия с протонами СН2-группы, 2, 2, 2, 2 - количество линий от взаимодействия с ядрами 'Н, 3|Р и двумя неэквивалентными ядрами l9F.
Передача влияния магнитного поля ядра через несколько связей на другое ядро (взаимодействие магнитных ядер) представляется следующим образом (рис. 27):
t ; t ;
спин спин спин спин
ядра электрона электрона ядра
Рис. 27
Ориентация спина ядра в магнитном поле /7 (по принципу Хунда они ориентированы параллельно друг другу) требует антипараллельной
ориентации спина электрона, участвующего в образовании связи с соседним атомом. В электронной паре, образующей химическую связь, спины электронов антипарал-лельны (принцип Паули). Если второй электрон связи также принадлежит магнитному ядру, то спин этого ядра опять-таки (по принцип)
92
Хунда) ориентируется анти параллельно спину «своего» электрона.
Векторная модель непрямого спин-спинового взаимодействия (по Дираку и Ван-Флеку) выглядит следующим образом (рис. 28).
Как видно из рисунка, константа непрямого спин-спинового взаимодействия (КССВ) имеет положительный знак, когда спины взаимодействующих ядер и электронов антипараллельны, т.е. имеет меньшую энергию. Следовательно, константы взаимодействия протонов через две или четыре связи отрицательны (-V и -V), а через одну (например, в H-F), три или пять связей (+V и +\7) положительны.
Свойства КССВ
1. В отличие от химического сдвига константа не зависит от рабочей частоты прибора.
2. Константу взаимодействия магнитно-эквивалентных ядер нельзя наблюдать непосредственно, т.е. определить из спектра, но можно (при необходимости) рассчитать. Для этого заменяют один из протонов дейтероном, практически не меняя электронной конфигурации соединения, т.е. не изменяя химический сдвиг протона.
CH2XY -> CHDXY
Эта операция приводит к расщеплению синглетного, т.е. содержащего одну линию, сигнала протонов в спектре исходного соединения (при условии, что X и Y - немагнитные ядра) в дублет вследствие взаимодействия ядер 'Н и 2D, --------------ц которое можно измерить из спектра (рис. 29).
Н Поскольку константы взаимодействия про-
Лз-d порциональны гиромагнитным отношениям ________у ядер, то
р Азо Л-Уц-у,,, ./н н - Я-ун ун,
гис' где А - коэффициент пропорциональности, ун
и yD - гиромагнитные отношения протона и дейтерона.
Отсюда ^н~н = — = 6,55.
Лз-d /о
Следовательно, константа J в данного типа соединениях в 6,55 раза больше найденной из спектра константы J в дейтерированном соединении. Константа JH D газообразного Н-D равна 43 Гц, для структуры H-C-D - около 5 Гц, для структуры H-C-C-D - менее 1 Гц.
3. Абсолютная величина J в принципе тем меньше, чем больше вязей между взаимодействующими ядрами.
4. Константа Jзависит от свойств и гсоме!рии молекул (см. ниже).
93
3.6.1.1. Геминальные константы взаимодействия (JreM)
Геминальные константы J во фрагменте Н-С-Н имеют абсолютные значения в интервале 0-20 Гц. Исключение составляет константа взаимодействия протонов в формальдегиде, равная -43 Гц.
ВеличинаJ зависит от гибридизации атома углерода, с которым связаны протоны, что ведет к изменению диэдральных углов между свя-
\ Н
зями : при ср = 0° (этилен, протоны находятся в одной плоско-
сти) JH_н = +2 Гц, <р = 73° (циклопропанон) ./н н = -4,5 Гц, (р = 109°28' (С/,з)^н = -12,4Гц.
Влияние сверхсопряжения на геминальное взаимодействие выражается в зависимости Угем от диэдрального угла между плоскостью л-связи и связью С-Н (рис. 30). Под влиянием двойной связи Угем возрастает или уменьшается на AJ:
= Д, + ,
где - константа взаимодействия протонов у 5р3-гиб-ридизованного атома углерода (-12,4 Гц).
Величина A.J макси-
♦-у? мальна (по абсолютной величине), когда воображаемая линия, соединяющая протоны Н и Н', параллельна плоскости л-связи (^=30°). При повороте обеих связей С-И против часовой стрелки возникающие углы равны 60°, 90°, 120°, 150°, 180°. Как видно из рисунка двойная связь понижает Уем в соседней СН,-группе на 1,9 Гц; введение еще одной двойной связи понижает J еще на 1,9 Гц.
При введении в P-положение к метиленовой группе электроотрицательных заместителей Уем изменяется в зависимости от их пространственной ориентации.
94
BzO
H
н
OBz
Н'
Bz - бензоил (PhCO).
Константа J ем изменяется в сторону положительных значений, если в проекционной формуле Ньюмена электроотрицательный p-заместитель оказывается расположенным между протонами метиленовой группы.
Неподеленные пары электронов гетероатома, находящегося в a-положении к СН2-группе, также влия-
ют на7ем. Если одна из неподеленных пар атомов О или М, находящихся в a-положении к СН2-группе (-О-СН2- или >N-CH2-), параллельна связи С-Н, то kJ этой группы прибавляется около 1,8 Гц.
Константа Уем для СН2-групп, расположенных рядом с гетероато-» мом в пятичленных циклах более положительны, чем в шестичленных, i поскольку неподеленные пары электронов гетероатомов полнее перекры-5 ваются со связями С-Н в пятичленных циклах, чем в шестичленных, I вследствие почти заслоненного положения СН2-групп в пятичленном I цикле.
I 3.6.1.2. Вицинальные константы взаимодействия (7внц)
I' Под вицинальным взаимодействием подразумевают взаимодей-I ствие через три связи, например между протонами при двух соседних | атомах углерода.
1Ц
„с-сС
| Константы J всегда положительны и имеют значения в пределах 0-20 Гц.
Зависимость J через два хр’-гибридизованных атома углерода (три a-связи) зависит от диэдрального угла ср (рис. 31).
95
'' н н \
\ / \
— с—С-—'
Л,™ = Ai cos1(p+в (0° - 90°)’ /™ = А2 • С05>+ В (90° < (Р< 180°),
Рис. 31
где А( и А2 - константы, зависящие от вида фрагмента С-С; они сильно уменьшаются в присутствии электроотрицательных заместителей; константа В очень мала и на практике в расчет не принимается. При <р- 0° 10 Гц, ^=30°-6 Гц, <р= 60° -2 Гц, 90° - 0 Гц, ср= 120°-4 Гц, <р = 150°- 12 Гц, <р= 180°- 16 Гц.
Поскольку величина диэдрального угла характеризует, главным образом, циклоалканы, наиболее изучено вицинальное взаимодействие для находящегося в форме кресла циклогексана, для которого получены следующие значения констант взаимодействия:
= 8-14 Гц
= 0-6 Гц
= 0-6 Гц
Значения ./виц зависят от взаимного расположения взаимодействующих ядер, что отражено в следующих эмпирических правилах:
а) электроотрицательные заместители только в том случае уменьшают вицинальные константы взаимодействия (J ) гош-протонов (т.е. протонов связей С-Н, образующих диэдральный угол <р = 60°С), если они находятся в транс-положении относительно хотя бы одно из взаимодействующих протонов;
б) вицинальные константы взаимодействия J (угол tp = 180°), напротив, всегда уменьшаются в присутствии электроотрицательных заместителей.
С помощью этих правил можно объяснить, например, различные значения J между аксиальными и экваториальными протонами в циклогексаноле.
96
Вицинальные константы взаимодействия через одинл/Л и ,у/?2-гиб-
ридизованный атом углерода (I Н ) также зависят от диэдраль-н
ного угла аналогично Уиц через двалуЛгибридизованных атома. Только в этом случае необходимо использовать другие значения А, и А2 в уравнениях, связывающих J и эти параметры (см. с. 96).
Вицинальное взаимодействие через один ур3-гибридизованный атом углерода и один гетероатом (О или N) также зависит от диэдрально-го угла <р. В этом случае взаимодействие максимально при ср= 180°.
/нс-он = 4,5 Гц ^НС-ОН = 3 Гц
Наблюдать эти константы в спектре можно лишь при условии быстрого подавления пространственной переориентации протона при гетероатоме, что достигается сильным разбавлением растворов образца в диметилсульфоксиде или другом растворителе, образующем прочные ММВС с протоном, находящимся у гетероатома.
В соответствии с жесткой плоской структурой двойной связи имеется две возможности для вицинального взаимодействия (J рассмотрена ранее).
При взаимодействии протонов, находящихся в цис- или транс-положепиях, их константы лежат в интервалах: J =5-16 Гц,./ =13-
г цис ’ транс
21 Гц, т.е../ всегда больше./ . ipani цис
97
Помимо электронного влияния заместителей (чем выше его электроотрицательность, тем меньше J), величина константы зависит от угла между взаимодействующими ядрами, что наглядно отражается в их значениях для олефиновых протонов в циклоалкенах формулы:
(Гц): n = 1, J= 0.5-2,0; п = 2, 7= 2,5-4,0; п = 3, J= 5,1-7,0; п = 4, J= 8,8-10,5.
3.6.1.3. Константы дальнего взаимодействия
Под дальним взаимодействием понимают взаимодействие через четыре и более связей. Как известно, КССВ уменьшаются с ростом числа связей между взаимодействующими ядрами и составляют 0-4 Гц, причем через четыре связи они, как правило, отрицательны, а через пять - в основном положительны.
Величина аллильного взаимодействия (Н-С-С=С-Н) зависит от диэдрального угла <р между плоскостью л-связи и связью С-Н протона при насыщенном атоме углерода: J максимальны при их параллельности (рис. 32) вследствие максимального л-ст-перекрывания. Если взаимодействующие протоны находятся в г/мс-положении, то в этом случае , как правило, несколько меньше 7^“.
Рис. 32
Взаимодействие через четыре g-связи проявляется только в тех случаях, когда взаимодействующие протоны находятся в системе, обра-
Н С Н
зующей плоскую зигзагообразную структуру (в форме W): .
ио имеет место в производных циклогексана и выражается в ширине линий (В) на их полувысоге. В конкретных случаях возможность суще-с кования Л'-образных ыруктур целесообразнее проверить на шаро-иг-,, лвых ii.'iii Гцоарта-Бр.тюба моделях.
н
В = 0,6-0,9 Гц
Константы взаимодействия в системе Н-С-С(=О)-С-Н наблюдаются в спектрах производных циклогексанона или циклопентанона и имеют максимальное значение (2 Гц), когда оба протона находятся в экваториальном или квазиэкваториальном положениях.
Взаимодействие через пять связей наблюдается, главным образом, для ненасыщенных или ароматических соединений (Н-С=С-С=С-Н или Н-С-С=С-С-Н). Взаимодействие в последней системе называется го-моаллильным и проявляется в том случае (J = 1,6 Гц), когда орбитали СН-связей также расположены параллельно л-орбиталям (максимальное л-а-перекрывание).
Константы взаимодействия Н-Н Г и)
Геминальные: о'х'С''н > -124-15; N=C-CH2-C=N, -20,4; Ph-CH,-C=N, -18,5; CH3C=N, -16,9; СН3СООН, -14,5; Ph-CH3. -14,4; CH3NO2, -13,2; CH4, -12,4; CH3C1, -10,8; СН3ОН, -10,8; СН3Вг, -10,2; CH3F, Lh
-9,6; СН,1, -9,2; CH,CL, -7,5; ~СО-СХ , -164-20; Р<„ ,-0,54-9,5;
3 2 н н
Q9
—N-c^„ , ±74-±16,5. н
Вицинальные: СН3СН2Х (X = OR, NR,, Hal, металл) 6-9 (7, наиболее часто встречающееся значение); С12СН-СНС12 (2,0 - гош, 16,35 -Н
I
трансу, CLCH-CHF, (2,0 - гош, 10,25 - транс)', V 3,5-12 (5);
Н
Н । Н .
II । I
С N Н 2 С S Н (3)’ В—С К(илиС=О)_Н 5-9'
н н I
н
С(--О) Н | II г О //-у I I /-
I 1-5 (2-3), н-С-С(=О)-Н 5-8 (-6-)’ Н-С=С-Н 6'
н
с I
Н I С С III
12(10); 10-18(17); "" 9-13(10); Н-С-С-Н
—с—с—и неси
4-11,5 (7);
5-14 (8-12, а-а'), 0-7 (2-6, а-е'), 0-5 (2-4, е-е');
Н——Н 7,3-12,5 (цис), 3,5-8,0 (транс)-, п / \ п 2,5-5,2 (цис),
100
3,0 (е-а);
Н
2,7-3,8; /*\ 6,3 (цис), 3,8 (транс)',
Н—' л Н
0-2 (п = 1), 2-4 (п = 2), 5-7 (п = 3), 8-11 (п = 4), 10-13
(п = 5). ।
Дальние: Н-С-С-С-Н 0.4 (2,0); Н-С-Х-С-Н О-ь-З (-1,0);
Н-С-С=С-Н -0,5^-3,0 (-1,5); Н_С-У_С-Н -14-5 (-3,0);
I 1111
н-с-с=с-н .и.3 (.15); н-с—с—с-с-н 0-з (15).
н—с—с=с—с—Н 2-3-
9 (орто), 3 (мета), 0,5
0,5 (0);
5 (2-3), 8 (3-4), 1,5 (2-4 и 3-5),
4
0,5 (2-5 и 2,6); 5
3 4
2 1,8 (2-3), 3,6 (3-4), 0 (2-4), 1,5 (2-5); 5
и
3
2
5,4 (2-3), 4,0 (3-4), 1,5 (2-4), 3,5 (2-5);
н
2,5 (1-2, 1-3,2-3), 3,5 (3-
4
4), 1,5 (2-4), 2,0 (2-5); 5
1^2 3,5 (4-5), 1,5 (2-5), 0 (2-4).
101
3.6.2. Взаимодействие других магнитных ядер
3.6.2.1. Взаимодействие 'Н-13С
Значения У1н_>.с непосредственно связанных между собой ядер определяются, в основном, контактным вкладом во взаимодействие и поэтому линейно зависят от характера гибридизации .s-орбиталей атома углерода в направлении С-Н-связи, что видно из табл. 9, причем за исходную взята константа '</ в метане, для которого характерна наиболее «чистая» 5/?3-гибридизация:
Хс (выч.) = 125- ^-хаРа^теР(%) = 500.ан = £ + .
= 125 + рх+ ру + p:+g
где ан - доля S-характера, £ и р - аддитивные вклады заместителей в шкалах Малиновского или Дугласа (табл. 10), g - поправка на парное взаимодействие (табл. 11).
Таблица 9
Соединение Состояние гибридизации S-характер, %(«н) J, Гц (найд.) J, Гц (выч.)
Метан sp3 25 125 125
Бензол 33,3 159 167
Ацетилен -'Р 50 248 250
Таблица 10
Заместитель (X) (CH3X) 4 P Заместитель (X) '•7h-c (CH3X) P
н 125 41,7 0 NO, 147 63,6 22
F 149 65,6 24 CN 136 52,6 11
CI 150 66,6 25 C=CH 132 48,6 7
Вг 152 68,6 27 COOH 130 46,6 5
I 151 67,6 26 CHO 127 43,6 2
OR, ОН 143 59,6 18 COCH3 126 42,6 1
S(=O)CH3, SCH3, SH 138 54,6 13 C6H5 126 42,6 1
nh2 133 49,6 8 CH3 126 42,6 1
nh’ch3 132 48,6 7 CCI, 134 50,6 9
N(CH3)2 131 47,6 6 C(CH3)3 124 40.7 -I
нт
Таблица 11
Заместители X в СН2Х2 g, Гц Заместители X в СН,Х, g, Гц
F F 13 С1 СООН 2
F С1 9 OR OR 6
F CN 4 nr2 NR, 0
CI CI 3 NO, no2 0
Для расчета констант взаимодействия ' J в алкенах или альдегидах получены следующие эмпирические уравнения:
17н.с(сН2=13СНх)=157 + |/Л; ijH с(х=|3СН0)= 172,4 +4,01-рх, где £ и р - те же инкременты, что и для метана; 157 - '</ в этилене, 172,4 - значение, полученное по методу наименьших квадратов для алифатических альдегидов и близкое по значению '</ в формальдегиде (172 Гц).
Константы взаимодействия непосредственно связанных ядер С^и-с) в Различного типа соединениях приведены в табл. 12.
Таблица 12
Тип соединения Jh-C, Гц Тип соединения 41-с, Гц
Алканы, циклоалканы 120-210 Арены, алкены 160-170
Производные циклопропана 160-190 Альдегиды 170-270
Производные циклобутана 140-180 Алкины 240-270
Таким образом, изучение констант '</ служит надежным методом исследования тонкого электронного строения молекул. Однако этот подход не совсем пригоден для соединений с необычной структурой: карбоионов, гетаренов с большим числом гетероатомов, напряженных колец, поскольку экспериментально наблюдаемые константы в них часто плохо согласуются с вероятной гибридизацией орбиталей, что, возможно, связано с ролью иных вкладов, кроме контактного, в величину константы.
Значения ,/н с для ядер 'Н и 13С, разделенных двумя (V ) или более связями (VH с и др.), значительно меньше, чем . Для системы ядер 11С!’С она зависит от угла между связями, а для системы Н-С-С-13С - от диэдрального угла аналогично связям Н-С-Н или Н-С-С-Н, причем между этими величинами выполняется соотношение:
' ' <н = о,75±0,05 . Они составляют для данных систем 3,6-6,4 Г и
С С П
iuзависимо от гибридизации ядра НС (s/A sp- пли sp-).
103
Знаки констант 7 подчиняются в целом тем же закономерно с-
3 2 I
тям, что и констант J Так, в метилацетилене Н, С- С s С- Н JH ,,
J„ J н положительны, 7, , и 7„ _2 отрицательны.
3.6.2.2. Взаимодействие 'H-I9Fh l3C-I9F
Константы J играют очень большую роль в установлении строения фторорганических соединений. Абсолютные значения констант 7HF в различных системах ядер приведены в табл. 13.
Таблица 13
Структура системы Тип соединения 7н-т,Гц
H-F HF 615
H-C-F (гем.) -CF2H 52-57
-chf2 45-47
ji =сС 72-85,-160-372'
F
H-C-C-F (виц.) Фторалканы (7H_F усреднена 3-25, 10-22'
по конформациям) фторалкены: цис 1-20
12-52
транс 6-10, 22-103'
o-H-F в производных бензола
H-C-C-C-F м-H-F в производных бензола 6-8
H-C-C-C-C-F л-H-F в производных бензола 2-3
H-P-F 120, 500-14402
H-C-P-F CH3P(O)F2 6,6
J4 =сС P(O)F2 0
Н-С(-С), 2-P-F 0-1
B-F 10-110
Примечание. '7„ ;2 Константа 7„р .„ (в PF3 она равна 1400 Гц).
Как видно из таблицы, константы 7 F, как правило, больше по абсолютному значению и изменяются в более широких пределах, чем kohci ан гы 7 В целом обнаруживается сходство зависимости соответ-с ’ вхчощих констант 7 , ! и 71( н от характера заместителей, однако их знаки часто отличаются.
104
Так, во фторэтанах константы 2./н г и -'7Н г одного знака, а в триф-торэтене 37ц F и 27н F ,ся имеют одинаковые знаки и противоположны по знаку 37н F .. Полный анализ спектра винилфторида показал, что все три константы 7, , одного знака с 7, „ и ./ ,, . т.е. положительны.
Анализ констант 7 в производных бензола показывает, что либо все три константы (37 „ V ,, ,, 57 u „) положительны, либо одна из них ( '7n |[ F) отрицательна, а две другие положительны. Экспериментальная проверка знаков констант в данном случае затруднена из-за малого значения последней (2-3 Гц, табл. 14).
Константы ‘J отрицательны (i60-372 Гц, табл. 13). Для |3Саром-19Р ''4-f = 240-255 Гц, константы 27с F и 37C_F в о- или .м-дифтор-бензолах близки и положительны (12,5 и 11,8 Гц), a 47с_F в л-дифторбен-золах невелики (1-2,6 Гц) и могут быть разного знака.
3.6.2.3. Взаимодействие I9F-I9F
; Данное взаимодействие является более сложным, чем Н-Н или Н-F, и отличается от них как по значениям, так и по знакам, что объясня-j ется возможным взаимным перекрыванием р-орбиталей атомов фтора. Это приводит к одно- или двухэлектронному прямому (через простран-; ство) взаимодействию спинов этих магнитных ядер.
На практике при анализе F-F-взаимодействий используют эмпи-, рические закономерности изменения значений и знаков констант J и . их корреляции с параметрами заместителей (табл. 14).
105
Окончание табл. 14
Структура системы
Л-1-
0-19
115-
125
15-
35
7-10
го-
31
Структура системы
Структура системы
Значения констант F лежат в интервале 0-400 Гц, причем часто изменяются в широких пределах для одного и того же типа соединений при замене заместителей. При этом области JFF для различных структур нередко перекрываются, что затрудняет интерпретацию спектров и возможность использования значений JFF для корреляции со структурой молекулы.
Знаки констант J основаны на сравнении их с соединениями, в которых наряду с Н-F- и F-F-взаимодействиями наблюдается и Н-Н-взаимодействие, для которого знак известен.
%—С С+1 Fx. ^-F Fx ^F ^F
F'' 1 ’ , C-C (-) , C-C-C (+) , C=C (+) •
C=C'EH - r'C=C<*’ • F'-C=C|-’ - F"C'C=C-'FW
В зависимости от числа связей (п) между ядрами Р и X константы
106
F 06> ФГг 6-
F > T ’ T ’
‘ F
Г F
Ji Ji
OL ,+>. nJ w
Как видно из табл. 14, значения констант J u изменяются в очень широких пределах (142-343 Гц), причем резко различаются для алканов 1\ /
F—С—С—F и алкенов (5,0-9,0 Гц). Для соединений ' \ , где R - замести-
к С1
i тель, содержащий гетероатом, присоединенный к атому углерода, обна-ружена обратная зависимость между электроотрицательностью гетероатома (Е, по шкале Полинга) и константой взаимодействия геминальных |ядер F: .7vu = 540/Е.
- F1
Для констант ,/е(( в фторалкевах ( ~~ 1 ?7 ) отмечена их линейная зависимость от суммы химических сдвигов ядер l9F относительно СО/;
® Дг1я разных групп соединений, содержащих концевую = СД-грун-
? пу, получены несколько различающихся значений констант к и h (к -- 1,4 | и b = 217; £ = 0,938 и6 = 130,4).
| Однако для вицинальных ядер l9F подобные корреляции не обна-
| ружены или они имеют грубый характер.
) 3.6.2.4. Взаимодействие с участием ядер 31Р
Значения и знаки констант Jp х, где X - другое магнитное ядро, ' определяются валентным состоянием и гибридизацией взаимодейству-• ющих ядер, числом связей между ними и пространственной структурой молекулы. Наряду с прямым резонансом на ядрах 3|Р константы J могут быть получены при наблюдении резонанса на ядрах 'Н, l9F, |3С. ; . Они заключены в широкий интервал: от -1100 Гц в фторофосфонатах до : +1100 Гц в гидридах фосфора.
107
"./р имеют следующие знаки:
11 1 п = 2 п = 3 п = 4
Х= 'Н 4- + и - + 4- и -
X- нс + и - + И — -Г отсут.
Х = l9F - + + 4-
Х = ”Р 4* И - + отсут. отсут.
Константа 'J изменяется в пределах от 138 Гц в фосфин-анионе ( РН2) до 1092 Гц в тетрафторгидриде фосфора (HPFJ.
Изменение констант '</ в пределах одной группы соединений с одинаковой координацией фосфора связаны с изменением ^-характера связи Р-Н под влиянием заместителей и эффективного объема последних, приводящим к изменению валентных углов и регибридизации атома фосфора.
Значения геминальных констант в системе Н-С-Р V н также изменяются, хотя и меньше, чем 'Jp и могут быть положительными и отрицательными: от 59,0 Гц в i/wc-CH3CH=CH-P(O)Cl2 (2Jpll во фрагменте всегда положительна) до -21,5±0,5 Гц в соединениях RCH2P(O)C12.
Значения вицинальных констант V в еще большей степени зависят от взаимною расположения атомов в системе Н-С-С-Р, причем наблюдается полная аналогия (в знаках, но не в значениях) с вицинальными J н: максимальное значение при двугранном угле 180° (~25 Гц) и близкое к нулю при 90°. Это позволяет различать цис- и тран с-мзомеры непредельных фосфорорганических соединений.
В аллильных, алленовых и ацетиленовых фосфорсодержащих системах во многих случаях наблюдается дальнее взаимодействие ("/) через 4-8 связей (2-4 Гц), что обусловлено механизмом передачи спин-спинового взаимодействия через систему л-связей и согласуется с наблюдаемым чередованием знаков констант. В системах, содержащих гетероатом, эти константы значительно ниже по абсолютному значению.
Изменение констант ( подчиняется в целом тем же закономерностям: './р^ и V положительны, V отрицательна и обычно меньше V с. Константы V в системах |3С-С=С-Р больше при транс-распопо-жении взаимодействующих ядер, чем при г/мс-расположении. Наблюдается закономерное возрастание констант 'J при переходе от sp3- к sp2-и далее ^-гибридизации атома углерода и при введении электроотрицательных заместителей:
P(S)Me, CICH2P(O)C12 CICH,P(O)F.
Гц 56,1 ’ 116,0 166.0
CI.C-CH-P(OXOMe), CI,OC(Cl)-P(0)(()Me), CIOC- Р(())(ОМе),
108
27рс, Гц 189,7 209,1 302,4
Наличие в молекуле двух атомов фосфора позволяет, как правило, наблюдать взаимодействие 3|Р3|Р. Во многих случаях константы '7 могут быть найдены при анализе сложных мультиплетных сигналов в спектрах ПМР.
Интервал констант ‘7р р занимает около 700 Гц: от 583 Гц в тетра-этоксидифосфиноксосульфиде до -108 Гц в тетрагидродифосфине:
(EtO),P(O)-P(S)(OEt)4 [О2Р(О)-Р(О)НО]3+ F,P-PF, '7рр, Гц 583,0 465,5 227,4
Me2P(S)-P(S)Me2 Н2Р-РН2
'7рр, Гц 187,0 -108
Электроотрицательные заместители в меньшей степени взаимодействуют с Зх-электронами фосфора для образования связей, оставляя больше х-характера для образования Р-Р-связи, что приводит к смещению '7р р в сторону положительных значений.
Значительные константы2J наблюдаются в системах Р-Х-Р, где X ~ О, N, S, что позволяет эффективно исследовать изомеризацию этих соединений.
Константы 37р р в алкеновых (Р-С= С Р) или алкиновых (Р—С=С—Р) системах составляют 5,8-13,5 Гц, а 1 Jp р в алленовой системе (Р-С- С“С-Р) равна 3,2 Гц.
3.6.2.5. Взаимодействие с участием других ядер
Совершенствование техники ЯМР позволило в последнее время значительно расширить изучение спин-спиновой связи ядер 13С с различными магнитными ядрами. Для |3С13С-взаимодействия характерны в целом те же закономерности, что и для 'Н-'Н-взаимодействия. Константы ( - положительны, 2J( (. - отрицательны и по абсолютному значению ( 2 Гн) меньше констант 37с с (15-20 Гц). Константы '7 (Гц) коррелируют с х-характером орбиталей взаимодействующих С-атомов:
Сч/ -С,с 7=35-40; С/? -Ср. : 7=40-45; -С;>, : 7= 45-65;
С,д -С;>! :7= 67-76; С*,, -С„:7= 80-125; Cv,-Cjp:7= 155-175.
Исследование аммиака, обогащенного изотопом l5N (/= '/), и его дсйтероироизводных указывает на значительную спин-спиновую связь между ядрами 'Н и |SN: '.7,н ,-N = 61±1 Гц, которая в 1,335 раз больше, чем в РНр что соответствует более эффективному перекрыванию атомных орбиталей в молекуле аммиака.
109
В спектре 1 IMP неооогашенного аммиака также наблюдается спин спиновая связь ядер Н и ‘4Н, но в спектрах аминов данное взаимодействие проявляется нить в уширении сигнала протонов >N-H.
В перфтора минах (R .-NF,) константы N ,,F достигают 117 Г ц
Связь между ядрами 'Ни HN через две и более связи не обнаруживается, но в изонитцнлах ( CH-N=C=O) и четвертичных аммониевых ^C-N^
основаниях (1ц ) наблюдается заметная спин-спиновая связь
(до 1,8 Гц). Нарушение симметрии в тетраалкиламмониевых солях, особенно содержащих х/г-гибридизованный атом углерода, уменьшает константу J,H.uN.
Выявить взаимодействие с участием ядра |7О трудно из-за его очень низкого содержания в природном кислороде и значительного уширения сигналов. Константа ./,н , (82-85 Гц) в спиртах определена косвенные
путем Константы ~.J ч , в формильных соединениях (
лежат в интервале 4-14 Гн го соизмеримо с \71н_,,с. в этиленах. В фосфорорганических соединениях отчетливо наблюдается взаимодействие ядер |7О и 3,Р с константами р =- 145-180 Гц.
Наконец в формамиде найдена константа J„ ,, , равная -5,2 Гн.
3.7. Интегрирование сигналов
ЯМР спектр, кроме поюження в нем сигналов и их мультппле!-ностп, характеризуется еще одним параметром - площадью под сигнала ми. или их интенсивностью. Площадь можно измерять планиметром, как средневзвешенную величину (что делалось ранее), или с помощью встроенного в спектрометр интегратора, как делается в настоящее время, в результате чего над (или под) спектральной линией прописываются ступенчатые кривые различной высоты, показывающие количество магнитно-эквивалентных ядер, соответствующих данному сигналу.
Высоты кривых называются интегральными интенсивностями сигналов. Они могут дать ценную, часто - решающую, дополнительную информацию о строении соединения.
НО
-СПз
определить лишь относительное число протонов в молекуле. Так, если бы не различия в химических сдвигах, то этилформиат и диэтилмалонат дали бы (по интенсивностям сигналов) одинаковые спектры.
Н-СООСШСНз
1 2 3
Нх .СООСН2СН3
Н СООСШСНз 2 46 (1:2:3)
Из интеграла можно получить следующую информацию:
а) количественный элементный анализ содержания в молекуле элементов с магнитными ядрами ('Н, |3С, l9F, 31Р и др.);
111
t>) количество магнитно-эквивалентных ядер в молекуле. Так можно, например, определять длину углеродной цепи алифатических карбоновых кислот или соотношение различных фрагментов в сополимерах;
в) количественный изотопный анализ, т.е. определять степень изотопного замещения, например дейтерирования, в различных положениях молекулы;
г) количественный анализ простых или равновесных смесей. Преимущество такого метода состоит в том, что в процессе съемки положение равновесия не затрагивается (например, кето-енольное равновесие).
д) энантиомерность (оптическая чистота) - из различия сигналов энантиомеров в хиральных растворителях или при добавлении хираль-ного парамагнитного шифт-реагента, например Eu(tfs)3 (см. с. 79).
3.8. Анализ ЯМР спектров высокого разрешения
3.8.1. Определение типа спектра
Спиновой системой называют систему из двух или более ядер, между которыми осуществляется спин-спиновое взаимодействие. Ядра в системе обозначают латинскими буквами: А, В, С, ..., X, Y.
Ядра одного типа, имеющие одинаковые химические сдвиги, называют химически эквивалентными и обозначают одними и теми же буквами.
Вид спектра очень сильно зависит от того, насколько велика разница между химическими сдвигами и константами взаимодействия. Чтобы можно было сравнивать оба параметра, их следует привести к общей размерности, т.е. выразить в герцах.
Если разность химических сдвигов двух групп ядер того же порядка или меньше, что и КССВ, то для буквенного обозначения спиновой системы используют соседние буквы алфавита: АВ, А2В, АВС и т.д.
Если указанная разность достаточно велика, то, напротив, пользуются буквами, отстоящими в алфавите далеко друг от друга, например
Jv>АХ-система
Л V- J; АВ-система
Рис. 35,а
! I2
ГЛв к
J-----pj-----L-!----1_ ----------------------
Va z4v= 0; А2-система
Л v < J; АВ-система
Рис. 35,6
На рис. 35 J№ - константа взаимодействия ядер А и В; Av= vA-vB - разность их химических сдвигов.
3.8.2. Спектры первого порядка
З.8.2.1. Условия образования и признаки спектров первого порядка
Под спектром первого порядка подразумевают спектр, который можно анализировать достаточно точно с помощью расчета, если выполняется условие
Вблизи границы этого соотношения часто уже наблюдаются отклонения от реального вида спектра, однако параметры можно все-таки рассчитывать по первому порядку.
Следующее условие гласит: каждое ядро из одной группы химически эквивалентных ядер одинаково взаимодействует с любым ядром из другой группы также химически эквивалентных ядер. Ядра, не удовлетворяющие этим двум условиям, называют магнитно-неэквивалентными, и их спектры нецелесообразно причислять к спектрам первого порядка.
Н , I Н —С—F
В дифторметане । оба протона химически эквивалент-F1
ны (vH = vH,) и магнитно-эквивалентны (JH_F = JH, F, )•
Поскольку химические сдвиги ядер 'Н и l9F сильно различаются вследствие различных гиромагнитных отношений, эта система называется А,X,-системой.
Fk _ ^F
В 1,1-дифторэтене н1х-С— протоны химически эквивалентны (vH = vH,), но магнитно неэквивалентны (JH F JH _F,), поскольку
ИЗ
JH F отражает взаимодействие i/wc-ядер, a J , - транс-яаер. Магнитная неэквивалентность отмечается с помощью штрихов при буквенном обозначении системы и тогда данная система записывается как АА'ХХ', который нельзя причислить к спектрам первого порядка.
В спектрах первого порядка наблюдаются отдельные мультиплеты, причем линии мультиплета находятся на одинаковом расстоянии друг от друга, которое равно константе взаимодействия J. Соотношение интенсивностей линий в мультиплете соответствует коэффициентам разложения бинома (а + by (треугольник Паскаля):
п = 0 1 Синглет
п= 1 1 1 Дублет
п = 2 1 2 1 Триплет
п = 3 1 3 3 1 Квартет
п = 4 1 4 6 4 1 Квинтет (пентет)
п = 5 1 5 10 10 5 1 Секстет
п = 61 6 15 20 15 6 1 Септет
Если отношение Av/J* лишь немногим больше 6, вышеуказанное соотношение линий в мультиплете не соблюдается точно (рис. 36).
При переходе от АХ- к । । JiZ/»6(A2X) АВ-спектру в каждом муль-
типлете линии, ближайшие к
..- -.... A/AR'i другому мультиплету, увели-
"I J J Г - ( 2 ) чиваются по интенсивности,
а линии, наиболее удален-
Рис‘ Ш ные, уменьшаются.
В общем случае взаимосвязанные мультиплеты изменяют соотношение интенсивностей таким образом, что линии, ближайшие к общему центру тяжести двух мультиплетов, возрастают, а соответствующие периферийные линии - уменьшаются. Этот так называемый «эффект крыши» может оказать существенную помощь при анализе спектра, когда в нем присутствуют несколько спиновых систем.
3.8.2.2. Анализ спектров первого порядка
При анализе мультиплетов исходят из того, что каждый из них соответствует одному ядру или группе магнитно-эквивалентных ядер.
Сначала из спектра определяют константу взаимодействия - расстояние между двумя соседними линиями мультиплета, а затем отыскивают другой мультиплет с тем же самым расстоянием между линиями.
114
Если имеется полный ЯМР спектр одного вида ядер ('Н, 13С, 19F и др.), то в спектре обязательно должен быть как минимум еще один мультиплет, так как взаимодействие всегда двустороннее. Следовательно, расщепление во взаимосвязанных мультиплетах, т.е. расстояние между линиями, также должно быть равновеликим. После идентификации взаимосвязанных мультиплетов определяют химические сдвиги сигналов.
Расстояние от центра мультиплета до сигнала ТМС есть химический сдвиг сигнала того ядра (или группы магнитно-эквивалентных ядер), которым обусловлен этот мультиплет.
Расстояние между центрами мультиплетов (Л v. = v. - и) в Гц должно быть больше как минимум в 6 раз КССВ (J, Гц). 6 этом случае можно применять правило мультиплетности (треугольник Паскаля).
По мультиплетности сигналов можно определить количество эквивалентных ядер, участвующих во взаимодействии (рис. 37).
CH3CH2NO2
(система А2Х3 или А3Х2)
СН2
n + 1 = 4 "—— n + 1 = 3 два эквива- три эквивалентных ядра лентных ядра
Рис. 37
По мультиплетности одного сигнала всегда можно установить количество ядер, относящихся к другому сигналу, но следует учитывать тот факт, что в спектрах вышеуказанных типов (от двух различных видов ядер - АпХп) мультиплеты могут быть смешанными из-за взаимодействия с другими магнитными ядрами.
Спектры трех различных видов магнитных ядер (AmMcXn) могут полностью различаться в зависимости от изменений химических сдвигов (иА, им, их) и констант взаимодействия (J^, J^,
Ниже приведены примеры спектров первого порядка (рис. 38, рабочая частота прибора 100 МГц).
115
AX (CHCI2CHO)
AX2 (CHCI2CH2CI) ill
АМХ (тиазол)
AMX2 (O2NCH=CHCH2Br)
ill, ill____I. It .11 I__.
AX3 (CH3SH) ilL
A2X2(CH2F2)
A2X3 (CH3CH2NO2)
AMX3 (O2NCH=CHCH3)
lb .11 .
AXs [CICH(CH3)2]
nllhi____________________
Рис. 38
Анализ более сложных спиновых систем можно найти в специальной литературе (например: [16, 17]).
3.9. Применение ЯМР спектроскопии
Основное применение ЯМР спектроскопии заключается в установлении строения соединений (см. раздел 3.13) или подтверждении строения, установленного или предложенного на основании функционального и элементного анализов, других методов исследования (УФ- и ИК-спектроскопия, масс-спектрометрия), а также определения молекулярной массы. Он используется и для контроля за протеканием реакции.
Однако данный метод очень широко используется и для изучения динамических эффектов в органических соединениях.
3.9.1. Исследование быстрых перегруппировок
Временная частота, с которой осуществляется переориентация спинов ядер в молекуле, относительно невелика, но в ряде молекул за это время могут успевать протекать изомерные превращения или обратимые обменные взаимодействия, приводящие к равновесному состоянию. Последние легко происходят тогда, когда протон связан с молекулой непрочно и могут быть внутри- или межмолекулярными. Быстрые перегруппировки и обменные взаимодействия на характере спектров ПМР и при
116
малом времени полуреакции (10-2-10-6 с) могут приводить к усреднению сигналов спектра.
Для упрощенного рассмотрения перегруппировок методом ЯМР предположим, что существует только два энергетически равноценных состояния, между которыми происходит обмен протоном.
При медленном обмене протон долго находится в каждом из двух положений и дает два отдельных сигнала, обусловленных различным химическим окружением (рис. 39).
медленный обмен
промежуточный обмен
Рис. 39
Л
^анв
быстрый обмен
При ускорении обмена происходит слияние линий. Увеличение скорости обмена протонов в зависимости от типа обменного процесса может быть достигнуто с помощью повышения температуры, изменения концентрации, замены растворителя или добавления катализатора.
Условие для слияния линий:
к = тс- Av/ -/1,
где к- константа скорости обмена, A v= иАН- ив н - разность химических сдвигов (в Гц) протона в различных состояниях в отсутствие обмена.
Эта формула справедлива для медленных или, в некоторых случаях, промежуточных обменных процессов. Для быстрых процессов она имеет несколько другой вид:
к = тс • (j v)2 /2Д., где Вг - ширина сигнала (на его полувысоте, Гц) после слияния сигналов от протонов А и В.
Химический сдвиг сигнала после слияния определяется по формуле
у = (Ул Рл+Ув-Р»)/2’ где РАп Рв~ мольные доли форм А и В.
Классификацию процессов на «медленные, промежуточные и быстрые» следует производить только с учетом условия слияния линий. Если
117
слияния не происходит, то наблюдается таутомерное равновесие двух форм, причем одна из них не исчезает ни при каких внешних воздействиях. Обменный процесс называется медленным (в шкале времени ЯМР), если два различных положения протона можно обнаружить в спектре. Однако этот процесс может оказаться все еще слишком быстрым для того, чтобы, например, удалось химически разделить два изомера, которые превращаются друг в друга. Быстрым называется процесс, при котором различные положения протона уже нельзя различить в ЯМР спектре. Несмотря на это, различные положения протона при быстрых процессах еще могут быть обнаружены с помощью УФ- или ИК-спектров.
Изменение положения протонов происходит при инверсии кольца в циклических соединениях (например, 3-метиленбенз-1,5-дитиепин, рис. 40).
Рис. 40
При низких температурах время жизни форм А и В достаточно велико и протон Н1 (или Н2) дает два отдельных сигнала от положений и (аналогично Н2 и Н2). Поскольку протоны и Н2 или Н2 и эквивалентны, спектр относится к классической спиновой системе АВ. (Дальше взаимодействие Н1 или Н2 с алкеновыми протонами, приводящее к дополнительному расщеплению сигналов, в данном случае устранено путем облучения образца дополнительным излучением с резонансной частотой алкеновых протонов).
118
При повышении температуры скорость инверсии возрастает, сигналы от протонов Н^. и , как и Н2 и На, сливаются. Обе пары имеют одну точку слияния, поскольку время пребывания в двух зеркально-симметричных формах А и В одинаково.
Таким образом, благодаря быстрой инверсии протоны Н1 и Н2 становятся эквивалентными и между ними уже нельзя (при температурах выше -50°С) пронаблюдать взаимодействие.
Аналогично в спектре циклогексана при -100°С наблюдаются два сигнала от На и Не, которые при повышении температуры сближаются и при комнатной температуре в спектре присутствует один синглетный сигнал с <5= 1,42 м.д., так как при этой температуре скорость инверсии (106 превращений в секунду) намного превышает частоту резонансных переходов с одного энергетического уровня на другой.
Примером проявления внутримолекулярных обменных взаимодействий может служить и спектр ПМР 1,5-диацетилциклопентадиена, который енолизован практически полностью.
Две возможные енольные структуры постоянно быстро переходят друг в друга за счет перемещения протона от одного атома кислорода к другому, что приводит к усреднению сигнала протона разных метильных групп (5= 2,45 м.д., 6Н; в индивидуальной структуре метильные группы неэквивалентны). Аналогично усредняются сигналы от протонов в положениях 2 и 4, давая один дублет (расщепление от протона в положении 3) с 5= 7,15 м.д. (2Н). Сигнал протона ОН-группы лежит при 18 м.д. (с, 1Н).
В качестве примера межмолекулярного обмена можно сравнить спектры ПМР безводного этанола (рис. 446, система АМ2Х3: <£сн = 1,34 м.д., т, ЗН; £сн = 3,75 м.д., д.к, 2Н; £он = 5,6 м.д., т, 1Н) и 96%-ного этанола, в котором наблюдается быстрый обмен и протон ОН-группы не участвует в спин-спиновом взаимодействии. Поэтому его спектр ПМР
имеет следующие параметры: система А2Х3: <5сн = 1,36 м.д., т, ЗН; <5СН = 3,62 м.д., к, 2Н; <50н = 4,8 м.д., с, 1Н.
При растворении карбоновых кислот в воде имеет место высокоскоростной обмен между протонами воды и карбоксильной группы и в
119
спектре не проявляются их отдельные сигналы, а присутствует одиночный пик, соответствующий среднему магнитному экранированию того или иного протона. С понижением температуры произойдет раздвоение сигнала и в спектре будут присутствовать два синглетных сигнала протонов воды и СООН-группы.
3.9.2. Исследование заторможенного внутреннего вращения
В диметилформамиде связь -C(=O)-N имеет значительную долю двоесвязаиности вследствие взаимодействия свободной электронной пары атома азота и карбонильной группой, поэтому для него можно написать следующую предельную структуру (суперпозицию):
нз<\Д\О нзс\+ Р~
N-С N=C
Н3С2 Н Н3С2 Н
нзС\^ ,-р
N-C
Н3С2 Н
Поэтому СН3-группы оказываются неэквивалентными и дают два синглетных сигнала с 8= 2,95 (2СН3) и 2,80 ('СН3) м.д. (Сигнал протонов 'СН3-группы лежит в более сильных полях, вследствие их некоторого внешнего экранированияр-электронами атома кислорода. Сигнал альдегидного протона лежит при 8,06 м.д.).
Внутреннее вращение вокруг связи N-СО должно привести к внутримолекулярному обмену положений метильных групп. Однако этот об-
N-C
Н3С2' Н
НЛ
N-C
HjCl' н
мен при комнатной температуре протекает с низкой скоростью, поскольку его энергетический барьер высок (около 88 кДж/моль или 21 ккал/моль). Время их пребывания в
цис- или транс-положениях по отношению к С=О-группе достаточно
велико, что и приводит к появлению двух раздельных сигналов.
Если повышать температуру, то эти сигналы уширяются и, наконец, при температуре выше 120°С сливаются в один пик и поэтому исчезает возможность различить СН3-группы в цис- и транс-положениях.
Заторможенное внутреннее вращение вокруг связей D—и
, где D - электронодонорная группа (атом азота или ароматическая л-система), А - акцептор электронов (атом кислорода или серы), наблюдается в тиоамидах, карбаматах, бензальдегиде и фурфуроле, про-
120
тонированных кетонах, нитрозаминах и нитритах (в последних эфирный кислород служит донором электронов).
Ме\ Мб\
,N-C< /N-C<
Me R’ Me OR ’
R-0
"n=o
В некоторых системах динамический процесс вызывает обмен между системами АВ и А2 для двух протонов. Например, в молекуле 1Ч-метил-2,4,6-тринитроанилина несимметричное замещение в аминогруппе ведет к тому, что при низкой температуре для двух ароматических протонов наблюдается система АВ. При повышении температуры вращение по связи ^С—NHMe ускоряется, эти протоны становятся эквивалентными и спектр вырождается в систему А2 (рис. 41).
-60,7°С
Рис. 41
121
В ряде случаев обнаружена заторможенность вращения трет.-бутильной группы, так что при низких температурах метильные группы становятся неэквивалентными. Например, для протонов mpem.-бу-тильной группы mpem.-бутилциклоаклканов (л > 2) наблюдаются два сигнала с отношениями интенсивностей 2:1 (2СН3:1СНД что объясняется их трансо
идной конформацией, в которой эквивалентны только две метильные груп-
пы. Барьер вращения при температуре коалесценции (-126°С) зависит от величины цикла и достигает максимума (32,6 кДж/моль) в mpem.-бутил-
циклооктане (л > 4).
В спектре l-m/7em.-бутил-5,6,7,8-тетрафтор-нафталина даже при комнатной температуре содержатся два сигнала неэквивалентных СН3-групп с отношениями интенсивностей 2(СН3):1(СН3). Наблюдается и тонкая структура прямого взаимодействия F-Н через пространство. При 134°С сигналы сливаются. Барьер вращения равен 77,4 кДж/моль (18,5 ккал/моль).
3.9.3. Исследование инверсии конфигурации
С помощью динамического ЯМР можно изучать не только заторможенное внутреннее вращение, но и инверсию конфигурации аммиака
и замещенных аминов,
О
.N
Н нн-н о
впервые установленную с помощью микроволновой спектроскопии. Ее также называют инверсией пирамидального атома. Она имеет место в дибензилметиламине, что показано с помощью ПМР. Можно было ожидать проявления
Ph— СН2
Ph-CH2
в его спектре метиленовых протонов в виде си-^N-Me стемы АВ (т.е. двух дублетов), поскольку они находятся в диастереотопном окружении. Однако вследствие быстрой инверсии при комнатной температуре сигналы вырождаются в синглет. Но если измерения проводить в кислой среде (pH < 2,0), то амин существует в протонированной форме и процесс ин
версии резко замедляется.
122
(PhCH2)2NH-Me
(PhCH2)2NH-Me
(PhCH2)2N—Me + | инверсия
(PhCH2)2N—Me +
H+
H+
Протоны СН2-групп в соли диастереотопны и дают спектр системы АВ. Из схемы видно, что инверсия азота происходит по стадии свободного амина и ее скорость зависит от концентрации амина и pH раствора. На стадии протонирование-депротонирование конфигурация амина не изменяется. Экспериментальным подтверждением данного вывода служит то, что спин-спиновое взаимодействие протонов в системе H-N-CHj исчезает при более низких концентрациях кислоты (pH = 2,0) до того, как метиленовые протоны станут энантиотопными. Дальнейшее повышение pH приводит затем к тому, что метиленовые протоны становятся магнитно-эквивалентными. Диастереотопия метиленовых протонов в данном соединении методом ПМР наблюдается при -155°С.
В циклических аминах также обнаруживается инверсия азота.
Система АА'ХХ' кольцевых протонов N-этилазиридина сливается в синглет при 108±5°С (барьер процесса равен 81 кДж/моль). В случае N-хлоразиридина температура коалесценции лежит выше температуры разложения вещества (>180°С), поэтому барьер инверсии должен быть очень высок. Это увеличение барьера, обусловленное влиянием хлора-заместителя, приводит к тому, что 7-хлор-7-азабицикло[4.1.0]гептан удалось разделить на диастереомерные инвертомеры.
Для циклических диазинов, например 2,3-диметил-2,3-диазабицик-ло[2.2.1]гептена-5, характерна последовательная инверсия обоих атомов азота с барьером в 60,7 кДж/моль при -9°С, в то время как для инверсии 1,2-диэтилдиазиридина /N-C2H5
С2Н5 требуется более 96
кДж/моль, что связано с размером кольца в нем, поскольку требуемое для инверсии плоское переходное состояние имеет более высокую энергию, чем большие циклы, что видно из уменьшения величин энергетических барьеров инверсии N-замещенных азиридина, азетидина и азоли
123
дина (пирролидина): [^N—R > '\^N—R > R (R = Me или Cl), вследствие уменьшения энергии дестабилизации переходного состояния (при R. = COOAlk ряд имеет обращенный вид).
Инверсионный барьер уменьшается, если заместитель у азота способствует выплощению пирамидальной конфигурации азота (sp3 -» sp2) /-и за счет электронных взаимодействий с не-
,-3' '^'З // поделенной парой электронов азота, как,
[>-N=N—С например, в N-п-нитрофенилазоазириди-
не, для которого барьер инверсии составляет 49,8 кДж/моль (11,9 ккал/моль).
Триалкилоксониевые соли изоэлектронны с аминами и для них также найдена инверсия конфигурации атома кислорода. Так, изопро-
пил-оксониевый ион оксирана ( C3H7-W ) имеет в спектре при 40°С синглет метиленовых протонов, который при -70°С превращается в два дублета системы АА'ВВ'. Подобные процессы инверсии наблюдались и в аналогичных соединениях фосфора и мышьяка.
3.9.4. Исследование инверсии циклов
Классическим примером данного процесса является инверсия циклогексанового кольца, характеризующаяся переходом конформации «кресло» (а) через конформации «твист» (б) или «ванны» (в) в эквивалентную кресловидную конформацию а'. Переходы между конформациями а и б или в невысоки по энергии (около 45,2 кДж/моль; переход а —> в требует чуть меньшей энергии) и происходят без угловых деформаций - просто путем вращения вокруг С-С-связей. Такие низкоэнергетические конформационные переходы называются псевдовращением.
Изучение спектра ПМР циклогексана-йц при разных температурах показало, что еще при -60°С протоны На и На дают один широкий сигнал, и только при -89°С наблюдаются два сигнала (рис. 42).
124
Из спектров определены константы скоростей ка. и км, оказавшиеся в два раза выше константы к Данные спектры и величины констант получены после подавления спин-спинового взаимодействия методом двойного ядерного резонанса.
В замещенных циклогексанах X также наблюдается инверсия цикла, протекающая около 80°С. Эта темпера-тура для всех систем соответствует об
ласти медленного обмена. Константы рассчитаны как отношение интегральных интенсивностей резонансных сигналов протонов ДиН (табл. 15), где А - разность свободных энергий конформеров или конформационная энергия заместителя, кДж/моль).
Таблица 15
X 1 А । Температура, °C к X А Температура, °C к
CN 0.24 -79 1.88 OCD3 0,547 -82 4,23
F 0,276 -86 2,10 SCD, 1,07 -79 16,1
1 0,468 -80 2,39 сн, 1,7
Br 0,476 -81 3,48 ;-С3Н, 2,1
Cl 0,528 -81 3,99 Z-C4H9 5,0
В циклогексене инверсия проявляется в том, что протоны Н и Н становятся эквивалентными. Энергия данного взаимопревращения (22,6 кДж/моль) почти такая же, как и для пик.чогептатриена (25 кДж/моль), который имеет форму «ванны». При -150°С обнаруживается неэквива-
125
лентность протонов Ht и Н в обоих соединениях, а при комнатной температуре они эквивалентны.
а
б
Сложные конформационные превращения и изомеризация связей обнаружена для [16]анулена, спектр ПМР которого при -30°С содержит один синглет при 6,74 м.д. (а). Детальный анализ изменения форм линий при пониженных (до -130°С) температурах показал, что: 1) [16]анулен существует в конформациях а и б, находящихся в быстром равновесии; при -60°С константа равновесия к = (р/р^ = 2,9;
2) каждая конформация обладает такой внутренней подвижностью, что за счет одновременного сдвига двойных связей все протоны последовательно занимают все четыре для а или шестнадцать для б магнитно различающихся положений для этих структур. Таким образом, конформер а проходит через 8, а б - через 32 идентичные конформации, которые отличаются только обменом положения некоторых протонов. Существование конформации б было установлено только после того, как нашли, что экспериментальные спектры не согласуются с полученными при расчете формами линий, если исключить вклад конформера б в равновесие.
3.9.5. Исследование валентной таутомерии
Быструю обратимую валентную таутомерию или валентную изомеризацию впервые с помощью ПМР спектроскопии наблюдали для 3,4-гомотропилидена. Она является вырожденной перегруппировкой Коупа, при которой циклопропановая связь (С3-С4) мигрирует в положение С'-С6 за счет [3,3]-сигматропного сдвига. Структуры а на' идентичны
126
2 по физическим и химическим свой-
ствам, поэтому такие перегруппиров-8<^Т с? / [С7 ки называются скрытыми реакциями
или Реакциями идентичности, а рав-
5 6 новесие д<-»д' - изодинамическим. В
а а’ ходе перегруппировки атом С7 ал-
лильного положения переходит в циклопропановое кольцо. Аналогично и другие атомы, кроме С2 и С5, также изменяют свои окружения. А поскольку эти изменения протекают при комнатной температуре, то го-мотропилиден был назван Флуктуирующей молекулой.
Переход проявляется в том, что протоны в положениях 7, 8, 1,3,4,6 становятся эквивалентными. Спектр «заторможенной» структуры наблюдается в области медленного обмена при температурах ниже -ЗО°С. При более высоких температурах наблюдается уширение линий.
Большой интерес представляет спектр ПМР бульвалена (рис. 43). Серия валентных изомеризаций вышеуказанного типа в нем приводит к эквивалентности всех протонов при 80°С и спектр содержит только один синглет (5= 4,35 м.д.). Понижение температуры ведет к уширению сигнала, затем его раздвоению и при -80°С спектр соответствует «заторможенной» структуре. В ней сигналы метиново-го протона и циклопропанового кольца перекрываются и дают один широкий сигнал при 2,10 м.д. В области слабых полей (3 = 5,55 и 5,65 м.д.) лежат сигналы неэквивалентных алкеновых
протонов, образующих спиновую систему АВ (Av< J, рис. 35). Энергия активации процесса, ответственного за усреднение химических сдвигов всех протонов найдена путем анализа температурной зависимости форм
127
линий и составляет 49 кДж/моль.
Но до сих пор с помощью ПМР не решен однозначно вопрос, участвует ли циклогептатриен в быстром и обратимом равновесии со своим валентным таутомером норкарадиеном, хотя для его производных -7-циано-7-трифторметил- и 7,7-ди(метоксикарбонил)гептатриена - наличие такой таутомерии установлено.
Так, в спектре последнего соединения, записанного при -139°С, имеется уширенный синглет (5= 2,86 м.д.) протонов Нй, дублет протонов Но (5= 5,72 м.д., JH н 9,5 Гц, система АВ) и три синглетных сигнала протонов неэквивалентных СН3-групп при 3,50 (эндо) и 3,74 (экзо) для б, 3,64 м.д. для а. Сигналы алкеновых протонов лежат в интервале 6,1-6,6 м.д. (м). Константа равновесия (ка/б), найденная из спектра, составляет 2,8-3,0. При комнатной температуре наблюдается усредненный спектр.
3.10. Ядерный магнитный двойной резонанс (ЯМДР)
Явление ЯМДР весьма сходно с явлением отсутствия спин-спинового взаимодействия в результате быстрого химического обмена (раздел 3.9.1).
Суть ЯМДР заключается в воздействии на образец сильного стационарного радиочастотного излучения с частотой, близкой к резонансной частоте изучаемого протона. При этом происходит его насыщение, в результате чего сигнал исчезает, и протон не участвует в спин-спиновом взаимодействии. Поскольку протон облучается двумя излучениями (от разных источников) с равными частотами, то метод называют методом двойного резонанса или подавления спин-спинового взаимодействия.
На рис. 44 приведен спектр ПМР (шкала S, внутренний эталон -ТМС) подкисленного (а) и чистого этанола (б), при насыщении (подавление резонанса) метильной группы (в), метиленовой группы (г) п
128
ТМС
ОН
СН3
СН2
Рис. 44
3, м.д.
давая соответственно квартет и триплет.
гидроксильной группы (д).
Протоны метиленовой группы подкисленного этанола (а) дают квартет (взаимодействие с протонами СН3-группы). Они не взаимодействуют с протоном ОН-группы из-за быстрого межмолекулярного обмена с ними, поэтому его сигнал прописывается в виде широкого синглета в очень слабых полях. В данном случае взаимодействуют только протоны метиленовой и метильной групп,
В безводном этаноле (б), когда протонный обмен практически отсутствует, протоны ОН- и СН2-групп участвуют во взаимодействии, вследствие чего сигнал протона ОН-группы прописывается в виде триплета, а СН2-группы - двойного квартета.
При насыщении протонов СН3-группы (в) они исключаются из взаимодействия с протонами СН2-группы и последние прописываются в виде дублета (расщепление от протона ОН-группы).
При насыщении протонов СН2-группы (г) она исключается из взаимодействия с протонами ОН- и СН3-групп, сигналы которых прописываются в виде синглетов.
После насыщения протона ОН-группы (б) спектр протонов алкильного радикала идентичен (без сигнала протона ОН-группы) спектру подкисленного этанола (а).
На рис. 45 приведен теоретический спектр ЯМР |3С алифатической части диэтилового эфира бензилмалоновой кислоты Ph-CH2-CH(COOCH2CH3)2 с учетом взаимодействия какого-либо ядра |3С только со «своими» протонами (а, взаимодействие с соседними протонами подавлено) и после неполного (б) и полного (в) подавления взаи-
129
модействия даже со «своими» протонами.
Для спектров, полученных в условиях двойного (тройного) резонанса, установлены специальные обозначения. Если обычный ЯМР спектр |3С обозначают как «ЯМР 13С», то спектр в условиях развязки от протонов обозначают «ЯМР |3С {'Н}>>. Иногда применяют и более подробные обозначения. Так, «ЯМР 13С {3|Рс 1 Нп}» означает спектр ЯМР 13С в условиях тройного резонанса, где применена селективная (с), т.е. неполная (см. рис. 44) развязка от атомов фосфора и полная (п) - от протонов.
Применение двойного резонанса изменяет не только мультиплет-ность, но и интенсивность сигналов, повышая ее (так называемый коллапс мультиплетов).
Важнейшей областью применения двойного резонанса является интерпретация с его помощью спектров ЯМР, когда получается более простой спектр. В сложных спектрах удается найти мультиплеты, обусловленные спин-спиновым взаимодействием. С его помощью могут быть проанализированы и спектральные характеристики. Например, из спектра типа АВХ часто бывает трудно определить КССВ, но из более простого спектра типа АВ, полученного наложением дополнительного поля, они легко определяются.
Метод тройного резонанса также может быть использован для интерпретации спектров. В этом случае на выбранные участки спектра накладывают два дополнительных стабильных разночастотных поля.
3.11. Возможности и ограничения метода
Несмотря на широкое применение в химии ЯМР на многих магнитных ядрах ("В, |3С, l4N, l4F, 3|Р, 3,S. 2<>Si и др ), метод ПМР все же более распространен и доступен. О возможностях данных методов вкратце упоминалось в тех разделах, в которых они были рассмотрены. Поэтому укажем возможности и ограничения метода ПМР.
I30
Возможности метода: определение числа типов протонов в молекуле; определение числа протонов каждого данного типа; определение положения протона в молекуле относительно непредельных группировок и гетероатомов; в простейших случаях определение числа протонов при соседних углеродных атомах; количественный анализ смесей, включая кинетические измерения; исследование быстрых перегруппировок и реакций обмена.
Ограничения метода: возможность перекрывания сигналов от близких по типу протонов; относительно низкая чувствительность.
3.12. Аппаратурное оформление и техника эксперимента
3.12.1. Блок-схема ЯМР спектрометра и его характеристики
В твердых и жидких телах магнитные ядра распределены по энергетическим состояниям, заселенность которых между основным (NJ и возбужденным (Na) состоянием соответствует закону Больцмана:
где ДЕ - разность энергий между состояниями Np nNa, k- постоянная Больцмана, Т- абсолютная температура.
Поскольку ДЕ в рассматриваемом случае очень мала, то число ядер в возбужденном (N^ состоянии лишь очень ненамного превышает их число в невозбужденном (NJ состоянии (Np> Na). При напряженности магнитного поля 1 Т (Тесла) и комнатной температуре ДЕ для протона составляет около 0,021 Дж/моль. Тогда избыток протонов на верхнем уровне, который определяет вероятность перехода, а, следовательно, и интенсивность регистрируемого сигнала, составляет только 0,001%.
В опытах по ядерному магнитному резонансу помещают ампулы (05 мм) с образцом в датчик (ячейку) спектрометра между полюсными наконечниками магнита (рис. 46).
На образец действуют электромагнитным излучением, частоту которого можно менять, и когда частота излучения совпадает с резонансной частотой ядра, оно возбуждается. Источником возбуждающего излучения является радиочастотный генератор (передатчик), а поглощенная образцом энергия измеряется радиочастотным мостом, после которого сигнал усиливается и записывается самописцем. Так получается спектр, по которому методом калибровки можно определить резонансую частоту. Таким образом, ЯМР спектрометр включает все элементы, кото-
131
передатчик
усыгитель
juttttHvm сатописву
Рис. 46
рые есть в оптических спектрофотометрах: источник излучения, ячейка для образца и детектор. Излучение, генерируемое катушкой L, практически монохроматично для любой отдельной частоты, поэтому приспособление для монохроматического излучения не нужно. Образец необходимо помещать в магнитное поле Яо до того, как он сможет поглощать энергию, т.е. до его облучения.
Наблюдается огромная разница между спектрами твердых тел и жидкостей или растворов. Для первых помимо взаимодействия ядер-ных магнитных моментов с внешним магнитным полем наблюдается и взаимодействие друг с другом (дипольное взаимодействие), поэтому в спектрах наблюдаются сигналы с большой шириной (до нескольких килогерц, так называемый широкополосный ЯМР, который является объектом изучения физики твердого тела).
В жидких телах направления магнитных моментов расположены хаотично и поэтому диполь-дипольное взаимодействие между ядрами исчезает, т.е. в сумме становится равным нулю и поэтому ширина линий в сигналах меньше 1 Гц. В этом случае говорят о спектроскопии ЯМР высокого разрешения.
Обычно в ЯМР спектрометрах используют электромагниты с напряженностью поля 1,4 или 2,3 Т, поэтому резонансная частота для протонов лежит в радиодиапазоне (З-Ю^-З Ю 9 Гц, Л = 100 м - 10 см).
Резонанс (в соответствии с формулой v = / • Н0/2л) экспериментально можно осуществить двумя путями: изменяя частоту облучения, вызывающего переходы при постоянном поле И,. (частотная разверт
132
ка), или изменяя напряженность поля при постоянной частоте облучения (полевая развертка), непрерывно меняя ее с помощью катушек развертки, укрепленных на полюсных наконечниках магнита. На практике используют оба метода.
При полевой развертке спектры записывают так, что напряженность поля в спектре возрастает слева направо, при частотной развертке при движении слева направо частота понижается, поэтому при обычном движении пера самописца слева направо первыми записывают сигналы ядер с большими резонансными частотами.
Кроме упоминавшихся электромагнитов в ЯМР спектрометрах используются и постоянные магниты, недостатком которых является большая чувствительность к изменениям температуры и внешних магнитных полей. Но они не требуют специальной аппаратуры для охлаждения воды, отводящей тепло от электромагнитов.
В последние годы сконструированы ЯМР спектрометры, в которых магнитное поле создается сверхпроводящими соленоидами, создающими поля значительно большей напряженности (до 10 Т), чем электромагниты или постоянные магниты, что резко увеличивает чувствительность метода (см. ниже). Однако стоимость и затраты на эксплуатацию таких приборов значительно выше, чем обычных спектрометров, поскольку соленоид должен постоянно охлаждаться жидким гелием.
Такие высокие напряженности полей соответствуют рабочим частотам для протонов в интервале 100-500 (и выше) МГц.
Каждый ЯМР спектрометр характеризуется его разрешающей способностью (Реп.), под которой понимают возможность различить в спектре линии, расположенные очень близко одна к другой. Она определяется по формуле
Р. сп. = Л v (Гц)/ и0 (Гц), где Av- ширина сигнала на его полувысоте, v - рабочая частота спектрометра.
Реп. современных приборов имеет величину 10ч-10~12 (при диаметре ампулы 5 мм). Таким образом, чем выше разрешение спектрометра, тем меньше разность частот двух резонансных сигналов, которые могут быть записаны раздельно, т.е. разрешены.
Тестом на Реп. обычно являются линии спектра ПМР ацетальдегида (одна из внешних линий квартета) или о-дихлорбензола, в спектре которого между второй парой линий должно быть 0,45 Гц (при v0 = 60 МГц).
Мерой чувствительности прибора является отношение сигнал/шум (S/R) в полученном спектре, определяемое по формуле
133
S/R = 2,5 А/В, где А - высота самого высокого пика от его вершины до середины «шумовой» линии, В - высота «шумовой» линии (от самого нижнего уровня «шума» до самого высокого).
Отношение S/R в современных приборах для ампул в 5 мм находится в интервале 20-50.
3.12.2. Дополнительные приспособления ЯМР спектрометров
Современные приборы высокого разрешения снабжаются накопителем, представляющим собой компьютер, в котором спектры могут когерентно накапливаться, причем накапливаются и шумы, но не когерентно. Это приводит к тому, что интенсивности полезных сигналов после каждого прохождения спектра (и) складываются, увеличиваясь в Jn раз, а интенсивность шумов увеличивается в значительно меньшей степени вследствие их некогерентности, суммируясь не после каждого прохождения. Таким образом, интенсивность полезных сигналов при многократном прохождении спектра возрастает значительно быстрее, чем случайных шумов.
Необходимым условием для применения накопителя является точное совпадение резонансных шумов при каждом прохождении спектра, т.е. высокая стабильность магнитного поля и рабочей частоты. Спектрометры, снабженные приспособлением для стабилизации Но и г0, позволяют производить накопление в течение нескольких часов и даже дней, так что можно накопить тысячи спектров. По окончании накопления записывают усиленный спектр.
Накопитель незаменим для записи спектров веществ, труднорастворимых или содержащих небольшие количества магнитного изотопа в природном элементе (например, |3С, l5N, ”S и др.), а также веществ, количества которых измеряются долями миллиграммов.
При регистрации спектра при частотной развертке частота в каждое данное мгновение времени соответствует данной точке спектра и позволяет получить информацию только об этой точке. Если бы можно было ввести сразу все частоты спектра, то за очень короткий промежуток времени получили бы гораздо больше информации.
Это достигается применением импульсной спектроскопии с фу-рье-преобразованием, в результате чего получают фурье-преобразован-ный спектр, соответствующий обычному ЯМР спектру. Время, необходимое для регистрации такого спектра, на два порядка величины мень
134
ше обычного.
При сочетании данного метода и накопителя удается за один и тот же интервал времени улучшить отношение S/R в 100 раз. Расшифровка накопленного фурье-преобразованного спектра производится с помощью компьютера.
В зависимости от области применения спектрометры ЯМР высокого разрешения могут быть снабжены другими приборами:
- температурной головкой для записи спектров при низких или высоких температурах и для термостатирования образца;
- оборудованием для записи ЯМР спектров различных магнитных ядер; - стабилизатором условий резонанса по иным магнитным ядрам (не тем, по которым записывается спектр);
- приспособлением для наблюдения двойного, тройного и т.д. резонанса (гомо- и гетероядерного);
- приборами для шумового подавления спин-спинового взаимодействия; - ампулами различного диаметра и вида (микроампулы, сферические, с вкладышами и т.д.).
3.12.3. Требования к образцу и подготовка к записи спектра
Первое необходимое условие для получения ЯМР спектра - присутствие в образце магнитных ядер. Наиболее часто встречающиеся в органической химии магнитные ядра и их характеристики приведены в табл. 5.
Чтобы в спектре получился заметный сигнал, образец должен содержать достаточное количество магнитных ядер.
Для ЯМР спектроскопии требуется значительно больше вещества, чем для УФ- и ИК-спектроскопии или масс-спектрометрии. Необходимое количество вещества определяется
- видом ядер, по которым необходимо записать спектр (их относительная чувствительность к магнитному полю приведена в табл. 4; чем меньше чувствительность, тем больше вещества необходимо для записи спектра);
- природным (см. табл. 4) или искусственно повышенным содержанием соответствующих ядер в молекуле (для записи ЯМР спектров ацетона на ядрах ,3С требуется больше вещества, чем на ’Н);
- количеством исследуемых ядер в молекуле (хлороформа необходимо больше, чем хлорметана);
- напряженностью магнитного поля, рабочей частотой и чувствительностью прибора (чем они выше, тем меньше вещества необходимо).
135
Для записи спектра ПМР с н0 = 100 МГц при однократной съемке требуется 1-20 мг вещества (при работе с накопителем требуется значительно меньше).
Для записи спектров ЯМР высокого разрешения образец должен находиться в газообразном или жидком с малой вязкостью состоянии. Твердые и подавляющее большинство жидких веществ должны быть растворены в таком растворителе, который по возможности не закрывал бы спектр растворенного вещества.
Приготовление раствора образца для измерения спектров ПМР довольно просто. Предпочтительнее использовать растворители, не содержащие протонов, однако это ограничение не очень существенно, так как сравнительно легко доступны дейтерированные растворители. Для полярных или слабополярных соединений наиболее подходящим растворителем является СС14, а для полярных - СНС13. Во избежание наложения резонансного сигнала протона хлороформа используется дейтерированный хлороформ (CDC13 с содержанием дейтерия 99,8%). Эти два растворителя используются в 90% случаев. Для слаборастворимых в них образцов используются другие дейтерированные растворители: диметил-сульфоксид-с16, ацетон-d^ ацетонитрил-б3, бензол-б6, D2O. Свойства наиболее часто используемых растворителей приведены в табл. 16.
Таблица 16
Растворитель Т.кип., °C Т.пл., °C Хим. сдвиг 8, м.д.
Название | Формула
Тетрахлорид углерода ссц 76,8 -22,8 —
Хлороформ Хлороформ-d СНС13 CDC13 61,3 -111,5 7,27
Диметилсульфоксид Диметилсульфоксид^6 (CH3)2SO (CD3)2SO 189,0 18,4 2,58
Ацетонитрил Ацетонитрил^ CH3CN CD3CN 82,0 -45,7 1,95
Ацетон Ацетон-с1б (СН3)2СО (CD3),CO 56,5 -95 2,05
Тетрахлорэтилен С2С14 187 возг.
Метиленхлорид СН2С12 40,1 -95,1 5,35
1,4-Диоксан l,4-flHOKcaH-d8 С4Н8О2 c4d8o, 101,5 12,0 3,68
Циклогексан С6Н12 81,4 6,5 1,43
Бензол Бензол^ С6Н6 C6D6 80,1 5,5 7,27
136
Окончание табл. 16
Растворитель Т.кип., °C Т.пл., °C Хим. сдвиг, S, м.д.
Название | Формула
Пиридин C5H5N 115,3 -42,0 6,9-8,5
Вода Н2О 100,0 0,0 4,8
Тяжелая вода D2O
Трифторуксусная кислота CF3COOH 72,4 -15,3 11,6
Сероуглерод cs2 46,3 -1115 -
Диоксид серы so2 -10,0 -75,5 -
В некоторых дейтерированных растворителях присутствуют небольшие (не более 0,5%) количества недейтерированного вещества, дающие, как правило, слабые сигналы с теми же химическими сдвигами, что и сигналы недейтерированных веществ.
Интенсивность сигнала можно повысить, работая при высоких температурах, когда растворимость существенно увеличивается. Но при более высокой температуре, согласно закону распределения Больцмана, ядерные уровни сближаются в большей мере, чем при низкой. В результате этого чувствительность метода при 150°С составляет приблизительно половину от чувствительности при 40°С, а при -100°С почти вдвое превышает ее.
При выборе растворителя следует учитывать и тот факт, что иногда исследуемое вещество получают в ЯМР-ампуле путем реакции исходного вещества с растворителем или вторым компонентом. Так, протонированные кетоны получаются в трифторуксусной кислоте, а простые карбокатионы впервые непосредственно наблюдались при взаимодействии арилфторидов с SbF5 с образованием соответствующих алкилгексафторантимонатов: R-F+SbF5—>R+[SbF6]~. Аналогичные результаты наблюдаются при использовании и других суперкислот, в том числе и «магической» кислоты - HOSO2F SbF5. В этих случаях выбор растворителя - это не просто стандартная процедура, поскольку в подобных примерах ЯМР-ампулы используются в качестве «реакционного сосуда».
Сигналы труднорастворимых веществ часто лишь едва заметны, так как в образце содержится недостаточное количество магнитных ядер. В таких случаях полезно увеличить объем образца, т.е. использовать ампулы большего, чем 5 мм диаметра; если допустима небольшая в таких случаях потеря разрешения, работать с расплавами или использовать накопитель.
Чтобы приготовить образец для съемки, 20-30 мг или около 50 мкл вещества помещают в ампулу с внешним диаметром 5 мм, растворя
137
ют это вещество в «0,5 мл растворителя и добавляют эталонное вещество, обычно ТМС или ГМДС. (Поскольку ТМС имеет низкую температуру кипения (27°С) и высокое давление паров, с ним трудно работать в малых количествах. Поэтому нужно иметь заранее приготовленные 5%(об.)-ные растворы ТМС в наиболее часто используемых растворителях). Высота заполнения ампулы - около 4 см.
Выбор эталонного вещества, как и растворителя, определяется исследуемой системой и результатами, ожидаемыми от спектра. Так, использование ТМС иили ГМДС для производных циклопропанона, а также соединений, содержащих триметилсилильные группы, может привести к перекрыванию резонансных сигналов образца и эталона. Поэтому в этих случаях в качестве растворителя и эталона используют циклогексан, метиленхлорид, бензол и другие соединения с точно известными хи-мическими сдвигами протонов. В водных растворителях как эталон можно использовать 1,4-диоксан или трет.-бутиловый спирт, но особенно удобно использовать частично дейтерированный 3-метилсилилпропионат натрия (CH3)3Si-CD2-CD2-COONa, сигнал которого в шкала б в водных или мета-нольных растворах находится точно при 0 м.д., как и у ТМС.
Рис. 47 Типы ампул, используемые в ЯМР для различных целей (для работы с внутренним эталоном - а, г, д (микроампула), е; для работы с внешним эталоном - б, в, ж; ампула с вкладышем - г; ампу-лы-«реакторы» - е. ж)
138
3.12.4. Измерение спектров при различных температурах
По ряду причин бывает необходимо регистрировать спектры ЯМР при различных температурах. Основные приложения этих экспериментов состоят в изучении зависимости формы линий от температуры, в результате чего получают информацию о константах скоростей внутри- и межмолекулярных перегруппировок (раздел 3.9) и детектируют стабильные интермедиаты. С повышением температуры можно улучшить растворимость плохо растворяющихся веществ (см. раздел 3.12.3).
Современные спектрометры, как правило, оборудованы датчиком с возможностью варьирования температуры от -170 до +220°С.
Для проведения этих измерений образец, охлажденный в проточной камере, охлаждаемой жидким азотом или нагретый с помощью электрической печи, подается в датчик. Температура потока газа проверяется с помощью термопары и регулируется автоматически. Другая система охлаждения для низкотемпературных измерений основана на использовании газового потока испаряющегося жидкого азота, охлаждающего образец, и температура измеряется путем варьирования потока газа. Предложена и методика, основанная на эффекте Джоуля-Томпсона, когда охлаждение осуществляется за счет внезапного расширения газа, находящегося под давлением. Теплообменник, где газ предварительно (до расширения) охлаждается газом, уходящим из камеры, допускает возможность понижения температуры до -110°С.
В идеальном случае измерение температуры можно было бы проводить в самой ампуле, но из-за технических трудностей это не всегда возможно. На практике температуру измеряют термопарой, находящейся в потоке азота у нижнего конца ампулы, или с помощью регистрации спектров стандартных образцов метанола или этиленгликоля до и после записи спектра образца. Для этих соединений различия в химических сдвигах /1и(в Гц) сигналов протонов СН3- или СН2-групп и ОН-групп являются температурно-зависимыми. Точные измерения привели к соотношениям, справедливым для спектрометров с и0 = 60 МГц.
Низкие температуры, метанол:
175-225 К: Т = 537,4 - 2,380/1v, 220-270 К: Т = 498,4 - 2,038-Лц
265-313 К: Т = 468.1 - 1.810-/1 и
Высокие температуры, этиленгликоль:
310-410 К: Т = 466,0 - 1,694 /1 и
Эти уравнения дают ошибку, не превышающую ±0,5°С для чистых образцов обоих веществ, в которых расщепления, обусловленные
139
спин-спиновым взаимодействием, исключались путем добавления следов (0,03%) концентрированной соляной кислоты.
Если сигналы протонов метанола или этиленгликоля не перекрываются с сигналами протонов образца, то наиболее точные температурные измерения в области от -110 до +140°С проводятся с помощью ампулы в (рис. 47), когда капилляр содержит метанол или этиленгликоль. При этом температура измеряется одновременно с записью спектра.
Выбор растворителя при проведении температурных измерений представляет собой специальную проблему. Для высоких температур используют диметилсульфоксид (т.кип. 189°С), гексахлорбутадиен-1,3 (т.кип. 214-216°С), декалин (декагидронафталин, т.кип. 195-196°С), нитробензол (т.кип. 210-211 °C). В этих экспериментах тетраметилсилан как эталонное вещество, заменяют другим эталоном, например 1,3,5-гексакис (тридейтерометил)-1,3,5-трисилациклогексаном (т.кип. 208°С, 8 = 0,327 м.д., с). При низких температурах в качестве растворителей используют ацетон-с!6 или сероуглерод (возможно в смеси с хлороформом), что позволяет получать температуры до -100°С. Ниже -100°С применяют бромтрифторметан (т.кип. -58-^-57°С) или дифтордихлорметан (т. кип. -28°С), диметиловый эфир (т.кип. -138,5°С) или сероокись углерода (т.кип. -138°С). Часто удовлетворительных результатов можно добиться только с помощью смеси нескольких растворителей.
3.13. Примеры задач, решаемых с помощью ЯМР спектроскопии
3.13.1. Задачи с решениями
Пример 20. Сделать отнесение сигналов в спектре ПМР 2-нитро-бутана, имеющих химические сдвиги (8, ТМС) при 0,95; 1,50; 1,95 и 4,70 м.д. и рассчитать их мультиплетность (здесь и далее учитывается взаимодействие через две или три связи).
deb a
В 2-нитробутане - СН3—СН2—CH(NO2)—СН3 в самых сильных полях (0,95 м.д.) резонируют протоны d, сигнал которых расщеплен в триплет от протонов соседней СН2-группы (N = n + 1). Протоны другой метильной группы (а), чуть более деэкранированные, чем протоны d, в спектре дают дублет (расщепление от СН-группы) при 1,50 м.д. Протоны СН2-группы резонируют при 1,95 м.д. и их сигнал прописывается в виде мультиплета, содержащего 8 линий [(3+1 )х( 1 + 1)]. Наиболее деэк-ранированным протоном (4,70 м.д.) является протон b (действие электроноакцепторной нитрогруппы), который взаимодействует с двумя по-
140
чти эквивалентными СН3- и СНо-группами и поэтому вместо 12 линий в сигнале [(3+1)х(2+1)] последний прописывается в виде секстета с интенсивностями линий в нем 1:4:7:7:4:1 (происходит наложение линий друг на друга такого типа:
13 3 1
13 3 1
13 3 1
1 4 7 7 4 1
Пример 21. Рассчитать мультиплетность и распределение интенсивностей линий в сигнале соединений:
а б а б а б
1) СН3ОН ;2) (СН3)3С-СН2Вг ; 3) CHD2C1 ;
а б в г
4) (СН3)2СН-О-СН2СН3
1) а - дублет (1:1), б- квартет (1:3:3:1); 2) при J= 0 а и б - синглеты, при J> 0 а - триплет (1:2:1), б - мультиплет из 10 линий в соответствии с коэффициентами разложения бинома (а+Ь)": 1:9:36:84:126:126:84:36:9:1; 3) а - триплет (1:2:1); б - дублет (1:1); 4) а -дублет (1:1), б - септет (1:6:15:20:15:6:1), в - квартет, г - триплет.
Пример 22. Сделать отнесение сигналов (8= 7,36,7,82,8,16 и 8,38 м.д.) и рассчитать их мультиплетность в спектре м-бромнитробензола.
Наиболее деэкранированным является протон а-его
Вг сигнал (с) имеет химический сдвиг 8= 8,38 м.д. Сле-
pj^ дующим по деэкранированию является протон b (8 Т/"-J = 8,16 м.д., д). Дублетный сигнал (8= 7,82 м.д.) соот-..-X—XX ветствует протону d. Наименее деэкранирован про-
J NO2 тон с (5= 7,36 м.д.), взаимодействующий с протона-
Нр, ми b и d и поэтому прописывается в виде дублета
дублетов (д.д.).
Пример 23. Рассчитать мольный состав, предварительно сделав отнесение сигналов протонов, смеси тетраметилсилана, циклогексана, 1,4-диоксана, mpem.-бутилхлорида, уксусного ангидрида, дихлорметана и хлороформа, если в ее спектре ПМР сигналы протонов каждого вещества имеют одинаковые интенсивности.
Соединения содержат 12, 12, 8, 9, 6, 2 и 1 протон. Приняв интенсивность каждого сигнала за 1, находим вклад одного протона соединения в интенсивность:1 /|2:'/|2:'/'g:'/'9:'/6:'/2:1 (общийзнаменатель-72). Следовательно, мольное соотношение тетраметилсилана: циклогексана: ди-оксана:тре/и.-бутилхлорида:уксусного ангидрида:дихлорметана:хлоро-форма = 6:6:9:8:12:36:72.
141
Пример 24. Определить мольный состав смеси бензола, толуола, бензальдегида, нитробензола и гексаметилбензола, если в ее спектре ПМР содержатся сигналы (8. м.д., в скобках - интегральная интенсивность): 1,8с (36Н), 2,3с (9Н), 7,10-7,40м (60Н), 7,85д (4Н), 8,20д (1 ОН), 9,7с (2Н).
Распределение электронной плотности в соединениях показывает, что в самом сильном поле (1,8 м.д.) лежит сигнал протонов метильных групп в гексаметилбензоле, в котором интенсивность одного протона (а) равна 2 ед. (36:18). Сигнал с 8= 2,3 м.д. принадлежит протонам СН3-группы толуола: Ь = 3 ед. (9:3); следовательно, в интенсивность сигнала ароматических протонов (60) толуол вносит 15 ед. (3x5). Сигнал с 8= 9,7 м.д. принадлежит сигналу альдегидного протона бензальдегида (его интенсивность с = 2), следовательно из двух дублетных сигналов о/иио-протонов (7,85 и 8,20 м.д.) первый принадлежит бензальдегиду (2x2 = 4), а второй - нитробензолу, в котором интенсивность одного протона (d) равна 5 ед.
Бензальдегид и нитробензол вносят в интенсивность сигнала ароматических протонов би 15 ед. (2x3 и 5x3), следовательно, на долю бензола остается 24 ед. (60 - 15 - 6 - 15). Значит, интенсивность одного протона в бензоле (е) = 4.
Если х - мольная доля в смеси каждого соединения, то ах + b-х + с-х + d-x + е-х = 100, т.е. 2х + Зх + 2х + 5х + 4х = 100. Следовательно, х
= 6,25.
Отсюда, бензола в смеси 25% (мол.), толуола- 18,75%, бензальдегида и гексаметилбензола - по 12,5% и нитробензола - 31,25%.
Пример 25. Применить ПМР спектроскопию для контроля за превращениями (здесь и далее - без учета возможных побочных реакций):
СНО СООН
142
8
9
—► o2n—GJ)-—ch2cn----------* 02N——ch2ch2nh2
Ct. 1 - протоны чистых бензола и толуола резонируют при 7,27 и 7,10 м.д., но в спектре последнего имеется синглетный сигнал с £=2,3 м.д. протонов СН3-группы. По мере протекания реакции интенсивность последнего сигнала будет увеличиваться. Ароматические протоны, давая в начале реакции широкий синглет или мультиплет, будут смещать центр (синглета или мультиплета) в сторону сильных полей (к 7,1 м.д.). На первых порах интенсивности сигналов протонов СН3-группы и Нар не будут соответствовать 3:5, характерному для толуола, поскольку в последнее значение будут вносить вклад протоны бензольного кольца. Как только соотношение интенсивностей сигналов будет строго соответствовать 3:5, можно считать, что превращение бензола в толуол закончилось.
Ст. 2 - по исчезновению сигнала протонов СН3-группы.
Ст. 3 - по исчезновению сигнала альдегидного протона.
Ст. 4 - по исчезновению сигнала протона ОН-группы, но данное решение некорректно, поскольку с уменьшением в реакционной массе кислоты, интенсивность данного сигнала будет уменьшаться и в итоге будет затруднительно обнаружить его в шумах прибора. Поэтому более надежно воспользоваться соотношением интенсивностей сигналов протонов ароматических, СН2- и СН3-групп (5:2:3, при незакончившейся реакции соотношение будет другим - см. ст. 1).
Ст. 5 - по исчезновению сигналов алифатических протонов.
Ст. б ио исчезновению синглетов при 2,30 и 7,10 м.д. и обнаружению синглета около 2,50-2,60 м.д. и двух дублетов в более слабых (чем даже 7,27 м.д.) полях с интенсивностями 3:2:2.
Ст. 7 - сигнал протонов СН2-группы будет прописываться в более слабых полях, чем СН3-группы, с одновременным уменьшением интенсивности и, в итоге, исчезновением его и появлением в более слабом поле сигнала СН2-группы с интенсивностью (относительно интенсивности сигналов ароматических протонов) 1:1:1.
Ст. 8 - сигнал протонов СН2-группы сдвинется в слабые поля по сравнению с сигналом протонов в спектре бромида.
Ст. 9 - протоны -СН2-СН2-группы будут резонировать, давая два триплета в более сильных полях, чем протоны СН2-группы в нитриле (поэтому об окончании реакции можно судить по исчезновению сигнала протонов последней, а также по интенсивностям сигналов ароматических и алифатических протонов (1:1:1:1).
143
Пример 26. Сделать отнесение сигналов ядер 13С в спектре цикло-гептатриена (<5^ = 31, 125, 131 и 136 м.д.) и рассчитать их мультиплет-4 з ность, учитывая взаимодействие ядер |3С-'Н (без учета вза-Оимодействия |3С-13С).
2 Наиболее экранированным ядром является С7, поэто-
I му его сигнал лежит при 31 м.д. и прописан в виде трипле-та триплетов (триплет от взаимодействия с протонами при С, и С6, расщепляющийся в триплет при взаимодействии со «своими» протонами). Самыми деэкранированными являются ядра С3 и С4 (136 м.д., дублет дублетов, д.д - от взаимодействия с протонами при С2 и С5 и «своими» протонами). Сигнал при 131 м.д. принадлежит ядрам С2,и С; (д.д), а сигналы ядер С, и С6 имеют химический сдвиг 8= 125 м.д. (дублет триплетов, д.т - триплет от взаимодействия с протонами при С7, расщепляющийся в дублет после взаимодействия со «своим» протоном).
Пример 27. Сделать соотнесение сигналов ядер ”F (8= -116,2 и
-107,9 м.д. относительно CFC13) к • 1- или 2-фторнафталину и рассчитать их мультиплетность.
Поскольку величины химических сдвигов отрицательны, т.е. лежат вправо от сигнала CFC13, сиг
нал с 8= -107,9 м.д. лежит в более слабом поле. С учетом распределения электронной плотности фтор в 1-фторнафталине более деэкранирован, поэтому его сигнал и имеет это значение 8. Он прописан в виде дублета; сигнал l9F в спектре 2-фторнафталина - дублет дублетов. (Расщепление
от протонов в 1 и 3 положениях).
В примерах 28-34 необходимо по составу и спектру ПМР установить строение соединения (химические сдвиги измерены относительно ТМС; в скобках приведены интегральные интенсивности сигналов).
Пример 28. С9Н,2. £н (м.д.): 2,15с (3), 6,8с (1).
Судя по составу, соединение имеет высокую степень ненасыщенности (до алкана, С9Н2О, недостает 8 атомов водорода, степень ненасыщенности 4). Оно, безусловно, имеет бензольное ядро (имеется сигнал с 8= 6,8 м.д.) и три СН3-группы, поскольку только они могут дать синглетные сигналы. Эти группы в кольце находятся в 1,3,5-положениях, чтобы ароматические протоны дали синглет в спектре. Значит анализируемое соединение -- 1,3,5-триметилбензол (мезитилен). В спектре 1,2,3- или 1,2,4-триметилбензолов ароматические протоны будут давать более слож
ные по мультиплетности резонансные сигналы.
144
Пример 29. С|3Н12. с>н (м.д.): 3,95с (1), 7,1с (5).
Поскольку сумма интенсивностей в два раза меньше количества атомов водорода в соединении, истинное соотношение интенсивностей равно 2.10. Синглетный сигнал с интенсивностью 2 может дать только СН2-группа. Оставшееся количество атомов углерода и водорода (С|2Н|о) соответствует двум фенильным радикалам и соединение является дифе-нилметаном.
Пример 30. С3Н5С1О2, £н (м.д.): 3,85с (3), 4,1с (2).
Мультиплетность и интенсивность сигналов свидетельствует о наличии в соединении СН3- и СН2-групп, не взаимодействующих между собой. Остаток -СС1О2 состоит из СО2-группы и атома хлора. Отсюда возможны две структуры соединения: СН3СООСН2С1 (1) или С1СН2СООСН3 (2). Если бы соединение имело структуру 1, то протоны СН3-группы резонировали бы около 2,5 м.д., а такого сигнала нет. Следовательно, спектр соответствует соединению 2 - метиловому эфиру хлоруксусной кислоты, что подтверждается положением сигнала с интенсивностью 3 и 8= 3,85 м.д., что согласуется с табличными данными.
Пример 31. С9Н10О. £н (м.д.): 2,0с (3), 5,1с (2), 7,4с (5).
Мультиплетность и интенсивности сигналов свидетельствуют о наличии в соединении СН3- и СН2-групп и фенильного радикала (остаток -СО2 = СОО). Из нескольких структурных формул, содержащих
идентифицированные фрагменты:
СООСН2СН3
СООСН3
СН3
ряет только последняя - бензилацетат.
Пример 32. С6Н10О4. с>н (м.д.): 1,15т (3), 4,15к (2).
Судя по интенсивностям (3+2) и мультиплета остям (т+к) сигналов и количеству протонов в соединении (10), оно содержит две эквивалентные СН3СН,-группы. Остаток - две COO-группы. Следовательно, анализируемое соединение - диэтилоксалат, С2Н;ООС-СООС2Н5.
Альтернативная структура дипропионилпероксида (СН3СН2СОО-)2 отвергается, т.к. протоны СН2-групп резонировали бы около 2,5 м.д.
Пример 33. С6Н|4О2. Ан (м.д.): 1,1т (6), 1,15д (3), 3,50к (4), 4,65к(1).
Интегральные интенсивности сигналов свидетельствуют, что
145
в соединении содержится три СН3-группы, причем две из них (6 = 1,1 м.д ) - эквивалентны и им соответствуют два квартета (3,50 м.д.). Два
оставшихся сигнала соответствуют фрагменту СН3 j--н . Следовательно, данное соединение является 4-метил-3,5-диоксагептаном СН3СН2ОСН(СН3)ОСН2СН3 (1,1 -диэтоксиэтан).
Другие соединения с данной брутто-формулой либо не могут существовать [(С2Н5)2С(ОН)ОСН3], либо их строение не соответствует условиям спектра ПМР [С2Н5О-СН(ОСН3)-С2Н5].
Пример 34. C|0H,0F4O. <5н (м.д.): 2,15с (3), 4,25т (2), 5,85т.т. (1), 7,15с (1), 7,20-7,30м (3).
Первый сигнал относится к протонам СН3-группы, не взаимодействующим с другими магнитными ядрами ('Н или ”F). Наличие сигналов , их количество, мультиплетность и интегральные соотношения свидетельствуют о ме/иа-замещении в бензольном ядре. Интенсивности сигналов (2:1) протонов алифатической части (C3H3F4) свидетельствуют о наличии в ней СН2- и СН-групп. С учетом атомов фтора фрагмент C3H3F4 имеет строение -CH2CF2CF2H (другое взаимное расположение атомов водорода и фтора даст совершенно другую мультиплетность сигналов ’Н). Следовательно, анализируемое соединение имеет одну из двух фор-
13 структуре I протоны СН.О- и СН2-групп будут резонировать около 3,8 и 2,5 м.д. соответственно, но таких сигналов в спектре нет, поэтому структурой соединения, имеющего вышеприведенные ЯМР-спектраль-ные параметры, является структура II.
3.13.2. Задачи для самостоятельного решения
1. Сделать отнесение сигналов 'Н и рассчитать их мультиплетность в спектре ПМР: а) и-этоксиацетанилида (Д м.д.): 1,4; 2,1; 4,0; 6,8; 7,4 и 7,9; б) 5-бром-2-пропионилтиофена (<5, м.д.); 1,3; 2,2; 7,2 и 8,0; в) 2,4,4-триметилпентена-1 (<5, м.д.): 1,0; 1,25; 1,95; 5,6 и 5,8.
2. Сделать отнесение сигналов |3С{'Н} (£, м.д.) и рассчитать мультиплетность сигналов при их неполной развязке и без развязки в спектре: а) 2,2-диметилбутана - 8,8; 28,9; 30,4 и 36,5; б) 1,4-нафтохина- 126,9;
146
131,8; 133,7; 138,5 и 184,7; в) N,N-диметил-!,3-пропандиамина - 32,2; 40,8; 45,4 и 57,8.
3. Сделать отнесение сигналов l9F (<5, м.д., относительно CFC13) и рассчитать их мультиплетность с учетом взаимодействий ’Н-l9F и l9F-19F в спектре: а) 1,1,3-трихлорперфторпропана-(-64,8), (-71,0) и (-114,0); б) трифторвинилбензола - (-102,0), (-133,0) и (-193,0); в) перфторпири-дина - (-87,8), (-13 3,4) и (-161,2).
4. Рассчитать мультиплетность сигналов магнитных ядер (без 13С) в спектрах: а) диметил-1,1-дифторацетонилфосфина; б) комплекса трифторид бора-триметиламин; в) 1,1-диметил-2,2-бис(трифторметил)гид-разина.
5. Рассчитать мольное содержание смесей по их спектрам ПМР (приведены величины химических сдвигов, 8, м.д., сигналов, их мультиплетности и интегральные интенсивности): а) метанола, этанола, 1- и 2-пропанолов, 2-метилпропанола-2 и 2,2-диметилпропанола-1 -0,9с (9), 1,0т (3), 1,1с (2), 1,15с (18), 1,2т (3), 1,25д (12), 1,5м (2), 3,25септ (2), 3,4т (2), 3,6к (2), 3,75с (6); б) mpem.-бутилбензола, анизола, Ы,Ы-диметилани-лина, ацетофенона и пропиофенона - 1,2т (6), 1,3с (9), 2,6с (24), 2,75с (9), 3,3к (4), 3,8с (9), 6,8-7,5м (65); в) кетонной и енольной форм дибензоил-метана- 3,3с (8), 5,5с (1), 7,2-7,8м(50).
6. Применить ПМР спектроскопию для контроля за превращениями:
147
7. Описать ПМР и (для двух последних соединений) ЯМР спектры (без развязки, с частичной и полной развязкой от протонов): диэтилового эфира 4,4'-дифеновой кислоты, ди(1 -нафтил)гидроксиэтанола, и-фтор-бензил-2-Н-перфторпропионата, диметилфторфосфина и дифенилена.
8. При окислении двух изомерных углеводородов (А и В) состава СнН16 перманганатом калия, в спектрах ПМР, имеющих сигналы (Д м.д.) при 0,8с, 2,4с и 7,2с (интегралы 9:2:5) и 0,8т, 1,3с, 1,8к и 7,2с (3:6:2:5) соответственно получена одна кислота (С). Определить структуру соединений А-С.
9. При пинаколиновой перегруппировке алкандиола (А; 8, м.д. = 0,9с, 3,5с; отношение интенсивностей 6:1) получен кетон (В; 8, м.д. = 0,9с, 2,1с; интенсивности - 3:1). Определить структуру А и В.
10. Изобутилен обработан бромоводородом. В спектре ПМР продукта присоединения имеются сигналы (8, м.д.): 1,0д, 2,1м, 3,2д. Соотношение интенсивностей 6:1:2. Установить строение вещества и указать, произошло ли присоединение по правилу Марковникова.
11. Установить строение соединений по ЯМР 13С{Нс} (8, м.д., муль-типлетность): а) С;Н|2, 13,7к, 22,7т, 34,6т; б) С|4Н10, 2,3д, 5,35д, 5,45д, 51с, 70с; в) C9HlgO, 22,6к, 24,4д, 52,3т, 210,0с.
12. В нижеследующих задачах по составу соединений и данным ИК и ПМР спектров установить их строение (приводятся данные ИК спектра, ц и спектра ПМР - в скобках интегральная интенсивность сигнала; при необходимости приводятся другие спектральные параметры): a) CgH|gO: v= 2920-2860, 1400, 1100 см1; £(м.д.) = 1с; б) С7Н16О3: v = 2970-2880, 1180см1; £(м.д.) = 1,15т (9), 4,4к (6), 5,0с (1); в) CgH|4: v= 2900-3000, 1620 см1; £(м.д.) = 1,8с (6), 6,0с (1); г) С|6Н|4: v= 3050, 1600 см1; ^(м.д.) = 2,2с (3), 7,1д (2), 7,3д (2); д) C|4H14S2: v= 3050, 1600 см ‘; 5(м.д.) = 3,6с(2), 7,2с (5); e)C|2H|g: v= 2900-2850,1700,1450 см1; 8
148
(М.д.) = 1,05с (1), 1,55с (2); ж) CgH|2: 3000-2850, 1600 см1; <У(м.д.) = 0,95д(6), 2,40м (1), 2,85м (1), 6,Од (2), 6,35д(2); з) С|4Н|0: v= 3050, 1600 см ';^(м.д.) = 7,1с(1), 7,2д(2), 7,Зд(2);и)С4Н2О3: v= 1760, 1600 см1; £(м.д.) = 6,5с; к) C|2H|g: v= 2950-2870,1610см1; £(м.д.) = 2,Ос; л) С,Н12О2: v= 3450, 1600 см1; £(м.д.) = 2,8с (3), 2,4т (2), 2,6т (2), 6,9д (2), 7,1д (2); м) CgHgO2: v= 3125, 1695 см-1 (не изменяются в растворе СС14, объяснить причину); £(м.д.) = 3,8с (3), 6,8-7,5м (4), 10,5с (1); н) С)0Н|0О: v= 3050, 1680, 1610,920см"1; £(м.д.) = 2,20с(3), 5,85д(1, JH_H 15Гц), 6,10д (1, JH н 15 Гц), 7,45м (3), 7,60д(2); о) С1(.Н14О3: v= 3050, 1690, 1250 см~ '; ^(м.д.) = 2,5с(3),7,6д(2),8,Зд(2);п)С|0Н|6О4: и= 2950-2890,1160
см1; <5 (м.д.) = 2,35с (1), 3,55с (1); р) C?H5F7O3: v= 1770, 1730 см"1; 8 (м.д.) = 1,25т (3), 4,40к (2) (в спектрах ЯМР ”F содержатся два сигнала); с) C24H22FgO2: v= 3050,1700 см"1; £(м.д.) = 2,20с (3), 2,30д (6), 6,8с (2); т) C22H22FgO2: v= 2950-2880, 3450 см~‘ (в растворе СС14 сдвигается до 3630 см1, объяснить причину); £(м.д., в растворе СС14) = 2,2с (6), 4,35с (1), 6,8с (1), 7,0-7,1 м (2) (Что произойдет при записи спектра ПМР в растворе ДМСО-с16?); у) C|6H4F10O2: v= 1800, 1655 см '; £(м.д.) = 2,60т (2), 3,05т(2);ф)С|0Н9Р9О3: v= 1760, 1740см-1; £(м.д.)= 1,3с(вспектреЯМР l9F содержится один сигнал); х) C6H13NO2: v = 2970-2880, 1680, 1600 см1; £(м.д.) = 2,1т(2), 3,45т(2), 3,3с (9); ц) С9Н|4О4: v= 1740, 1590 см1; 8(м.д.) = 1,15т (6), 1,9д (3), 4,2д.к. (4), 6,8к (1); ч) C|5H24Og: v = 2940-2860,1740 см-'; 5(м.д.) = 1,1 т(6), 1,6т (1), 4,Ок (4), 4,5т(1); ш) C12H|2FeO:
3050, 1690, 1600 см1; £(м.д.) = 2,2с (3), 3,80с (5), 4,25д (2), 4,80д (2); щ) С,311,3FSO3: и-= 3380, 1720 (не изменяются в растворе СС14, объяснить причину), 1650 см-1; 8 (м.д.) = 1,15с (6), 1,25т (3), 4,20к (2), 5,1 Од (1), 10,8д (1); э) С7Н„О4Р: и = 1710 см-'; 8 (м.д.) = 0,95д.к (2), 1,1д.т (3), 1,25с (6).
3.13.3. Ответы к задачам
УФ спектры:
1.а)237; 295; 308. б) 370; 386.
2. 29; 45; 68.
ИК спектры:
2. а)
, б) /-Bu-COOBu-r, в)
ОН
149
Спектры ПМР:
1. а) 1,4т(СН3), 2,1с (СН3СО), 4,0к (СН2), 6,8д (Н ) 7,9ш.с. (NH); б) 1,3т (СН3), 2,2к (СН2), 7,2д (4-Н^), 8,Од (3-Н^); в”) 1,0с [(СН3)3С], 1,25д (СН2), 1,95д (СН3), 5,6д.д. и 5,8д.д. (=СН2).
2. а) 8,8 (С4, к, т.к), 28,9 С, к, -), 30,4 (С2, с, м-30 линий), 36,5 (С3, т, к.т); б) 126,9 (С7, д, д.д), 131,8 (С8, д, д.д), 133,7 (С2, д, д), 138,5 (С9, с, м - 8 линий), 184,7 (С1, с, д.д); в) 32,2 (С, т, т.т), 40,8 (С3, т, т.т), 45,4 (С2, т, м -27 линий), 57,8 (СН3, к, т.к).
3. а) -64,8 (CF23, т), -71,0 (CF, т), -114,0 (CF22, д.т); б) F'(Z)F2C=C(F3)-Ph: -102,0 (F1, д.д), -133,0 (F2, д.д), -193,0 (F3, д.д); в) -87,8 (2-F, д), -133,4 (4-F, д.д),-161,2 (д.д).
4. а) СН3 (д), СН3СО (с), CF2 (д), Р (т. септ.); б) СН3 (д.д), BF3 (д.д), BF3 (м - 80 линий), В(-С)3 (м - 80 линий); в) СН3 (д.д), CF3 (д.д), N'F3 (м - 42 линии), N2(-C)2 (м - 98 линий).
5. а) 22,2:11,1:11,1:22,2:11,1; б) 7,7:23,1:30,8:23,1:15,4; в) 4:1.
8. Неопентил- и трет.-пентилбензоаты (А, В), бензойная кислота (С). гт+
9. (СН3)2С(ОН)-С(ОН)(СН3)2 —► (СН3)3С-СО-СН3
10. Нет (1-бром-2-метилпропан).
11. а) Пентан; б) дифенилацетилен; в) 2,6-диметилгептанон-4.
12. a) Jty-mpem.-бутиловый эфир; б) ортомуравьиный эфир; в) 2,5-диме-тилгексадиен-2,4; г) 9,10-диметилантрацен; д) дибензилсульфид; е) гек-саметилбицикло[2.2.0]гексадиен-2,5; ж) 5-изопропилциклопентадиен-1,3; з) фенантрен; и) малеиновый ангидрид; к) триизопропилиденциклопро-пан; л) 2-и-метоксифенилэтанол; м) о-гидроксиацетофенон; н) транс-4-фенил-3-бутенон; о) 4,4'-диацетилдифенилоксид; п) диэтиленкеталь 1,4-циклогександиона; р) этиловый эфир перфторизопропилглиоксиловой кислоты; с) 1,6-димезитилперфторгександион-1,6; т) 1,6-ди-н-ксилил-1,6-дигидрокси-1,6-дигидроперфторгексан (все сигналы сдвигаются в слабые поля до 6,45, 7,05 и 7,3 м.д., сигнал при 6,45 м.д. расщепится в дублет и появится сигнал (д) протона ОН-группы около 11 м.д.); у) 5,5-ди(пентафторфенил)бутиролактон; ф) трет.-бутиловый эфир пер-фтор-mpem.-бутилглиоксиловой кислоты; х) триметил-2-карбоксилато-этиламмоний, (CH3)3N+-CH2CH2COO“; ц) диэтиловый эфир циклопропан-1,1-дикарбоновой кислоты; ч) тетраэтиловый эфир 1,1,3,3-пропан-карбоновой кислоты; ш) ацетилферроцен; щ) этиловый эфир 2,2-диме-тил-З-гидрокси-З-пентафторфенилгликолевой кислоты; э) 5,5-диметил-2-этилфосфа-1,3-диоксандион-4,6.
150
Библиографический список
1. Штерн Э., Тимонс К. Электронная абсорбционная спектроскопия в органической химии. М.: Мир, 1974. 290 с.
2. Бернштейн И.Я., Каминский Ю.Л. Спектрофотометрический анализ в органической химии. Л.: Химия, 1975. 230 с.
3. Беллами Л. Инфракрасные спектры сложных молекул. М.: Мир, 1963. 592 с.
4. Беллами Л. Новые данные по ИК-спектрам сложных молекул. М.: Мир, 1971. 320 с.
5. Наканиси К. Инфракрасные спектры и строение органических соединений. М.: Мир, 1965. 216 с.
6. Дайер Джон Р. Применение абсорбционной спектроскопии органических соединений. М.: Химия, 1970. 164 с.
7. Смит А. Прикладная ИК-спектроскопия. М.: Мир, 1982. 328 с.
8. Драго Р. Физические методы в химии. М.: Мир, 1981. Т. 1. 424 с.
9. Казицина Л.А., Куплетская Н.Б. Применение УФ-, ИК- и ЯМР-спектроскопии в органической химии. М.: Высшая школа, 1971. 264 с.
10. Казицина Л.А., Куплетская Н.Б. Применение УФ-, ИК- иЯМР-и масс-спектроскопии в органической химии. М.: Изд-во Московского ун-та, 1979. 240 с.
11. Иоффе Б.В., Костиков Р.Р., Разин В.В. Физические методы определения строения органических соединений. М.: Высшая школа, 1984. 336 с.
12. Бенуэл К. Основы молекулярной спектроскопии. М.: Мир, 1985. 384 с.
13. Вилков Л.В., Пентин Ю.А. Физические методы исследования в химии. М.: Высшая школа, 1987. 368 с.
14. Сергеев Н.М. Спектроскопия ЯМР (для химиков-органиков). М.: Изд-во МГУ, 1981.279 с.
15. Жунке А. Ядерный магнитный резонанс в органической химии. М.: Мир, 1974. 176 с.
16. Ионин Б.И., Ершов Б.А., Кольцов А.И. ЯМР-спектроскопия в органической химии. Л.: Химия, 1983. 270 с.
17. Гюнтер X. Введение в курс спектроскопии ЯМР. М.: Мир, 1984. 480 с.
18. Дерроун Э. Современные методы ЯМР для химических исследований. М.: Мир, 1992. 403 с.
19. Миронов В.А., Янковский С.А. Спектроскопия в органической химии. Сборник задач. М.: Химия, 1985. 232 с.
20. Гордон А., Форд Р. Спутник химика. М.: Мир, 1976. С. 181-245, 271-350.
151
ОГЛАВЛЕНИЕ
Введение....................................................3
1. Электронная (или УФ) спектроскопия.......................6
1.1. Структура органических молекул и электронные спектры. Хромофоры и ауксохромы...................................9
1.2. Электронные спектры отдельных классов органических соединений..............................................11
1.3. Применение УФ спектроскопии........................13
1.4. Возможности и ограничения метода...................16
1.5. Задачи для самостоятельного решения................16
2. Инфракрасная (ИК) или колебательная спектроскопия......17
2.1. Теоретические основы ИК спектроскопии..............18
2.2. Основные параметры полос ИК поглощения. Положение полос в зависимости от условий записи спектра............26
2.3. Влияние массы атомов, взаимодействия колебаний и резонанса Ферми на характер спектров....................29
2.4. Влияние электронных эффектов на характер спектров...30
2.5. Влияние дипольных взаимодействий на характер спектров ...32
2.6. Влияние внутримолекулярных водородных связей (ВМВС) на характер спектров....................................33
2.7. Применение ИК спектроскопии........................34
2.8. Техника фундаментальной ИК спектроскопии...........56
2.9. Возможности и ограничения метода...................58
2.10. Задачи для самостоятельного решения...............59
3. Спектроскопия ядерного магнитного резонанса (ЯМР спектроскопия)..............................................60
3.1. Магнитные свойства и резонанс ядер. Ширина спектральной линии.......................................61
3.2. Магнитное экранирование и химический сдвиг протонов.65
3.3. Полуэмпирические соотношения между химическим сдвигом и строением молекулы............................71
3.3.1. Электронное влияние заместителей...............71
3.3.2. Молекулярные магнитные поля за счет удаленных’ связей...............................................71
3.3.3. Молекулярные электрические поля за счет постоянных диполей..............................................75
3.3.4. Взаимодействие электронов через пространство..76
3.4. Влияние внешних воздействий на химический сдвиг....76
3.5. Спектроскопия магнитного резонанса других ядер.....79
3.5.1. ЯМР спектроскопия ядер 19F....................80
3.5.2. ЯМР спектроскопия ядер |3С....................82
152
3.5.3. ЯМР спектроскопия ядер 3|Р ....................85
3.5.4. ЯМР спектроскопия ядер l4N.....................86
3.5.5. ЯМР спектроскопия ядер "В......................87
3.5.6. ЯМР спектроскопия некоторых других магнитных ядер . 87
3.6. Непрямое спин-спиновое взаимодействие...............89
3.6.1. Взаимодействие 'Н-'Н...........................89
3.6.1.1. Геминальные константы взаимодействия (JreM).94
3.6.1.2. Вицинальные константы взаимодействия (J ).95
3.6.1.3. Константы дальнего взаимодействия.........98
3.6.2. Взаимодействие других магнитных ядер..........102
3.6.2.1. Взаимодействие 'Н-|3С....................102
3.6.2.2. Взаимодействие 'H-I9F и l3C-l9F..........104
3.6.2.3. Взаимодействие l9F-l9F...................105
3.6.2.4. Взаимодействие с участием ядер 31Р.......107
3.6.2.5. Взаимодействие с участием других ядер.......109
3.7. Интегрирование сигналов............................110
3.8. Анализ ЯМР спектров высокого разрешения............112
3.8.1. Определение типа спектра......................112
3.8.2. Спектры первого порядка........................ИЗ
3.8.2.1. Условия образования и признаки спектров первого порядка.....................................ИЗ
3.8.2.2. Анализ спектров первого порядка..........114
3.9. Применение ЯМР спектроскопии.......................116
3.9.1. Исследование быстрых перегруппировок..........116
3.9.2. Исследование заторможенного внутреннего вращения.. 120
3.9.3. Исследование инверсии конфигурации............122
3.9.4. Исследование инверсии циклов .................124
3.9.5. Исследование валентной таутомерии.............126
3.10. Ядерный магнитный двойной резонанс (ЯМДР).........128
3.11. Возможности и ограничения метода..................130
3.12. Аппаратурное оформление и техника эксперимента.......131
3.12.1. Блок-схема ЯМР спектрометра и его характеристики ..131
3.12.2. Дополнительные приспособления ЯМР спектрометров........................................134
3.12.3. Требования к образцу и подготовка к записи спектра .. 135
3.12.4. Измерение спектров при различных температурах...139
3.13. Примеры задач, решаемых с помощью ЯМР спектроскопии...........................................140
3.13.1. Задачи с решениями ..........................140
3.13.2. Задачи для самостоятельного решения..........146
3.13.3. Ответы к задачам.............................149
Библиографический список...................................151
153
Методы УФ, ИК и ЯМР спектроскопии и их применение в органической химии
Учебное пособие по спецкурсу
Юрий Павлович Дормидонтов
Редактор ГА. Гусман
Технический редактор ГА. Ковальчук
Корректор Л-. Я. Бобкова
Компьтерный набор С. Б. Лавриков
ИБ № 263
Лицензия ЛР № 020409 от 12.02.97.
Подписано в печать 7.10.99
Формат 60х84'/|6. Бум. тип. № 1. Печать офсетная. Усл. печ. л. 9,0.
Уч.-изд. л. 7,56. Тираж 100 экз. Заказ 47
Редакционно-издательский отдел Пермского университета.
614600. Пермь, ул. Букирева, 15.
Типография Пермского университета.
614600. Пермь, ул. Букирева, 15.