/




























































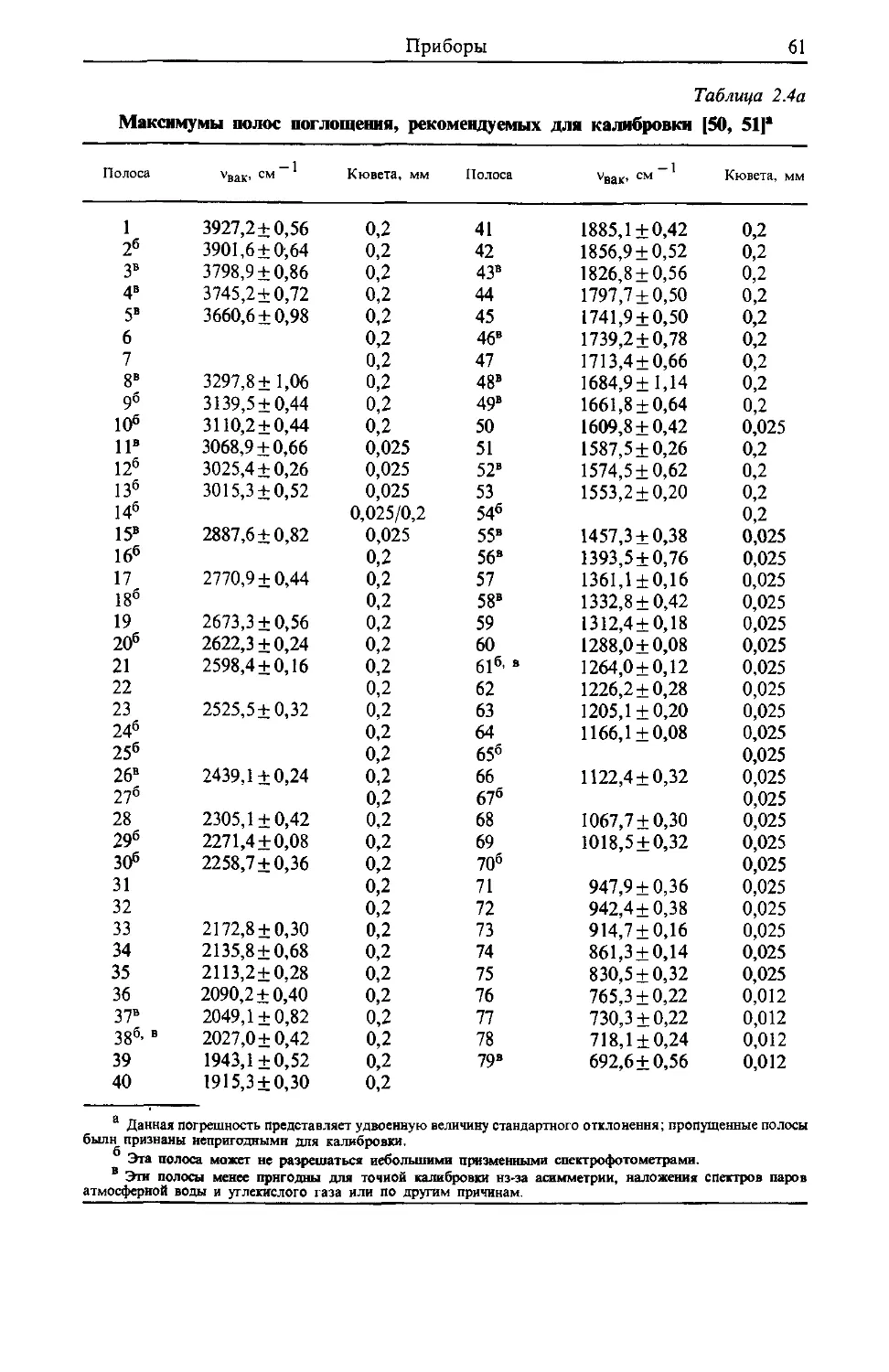





























































































































































































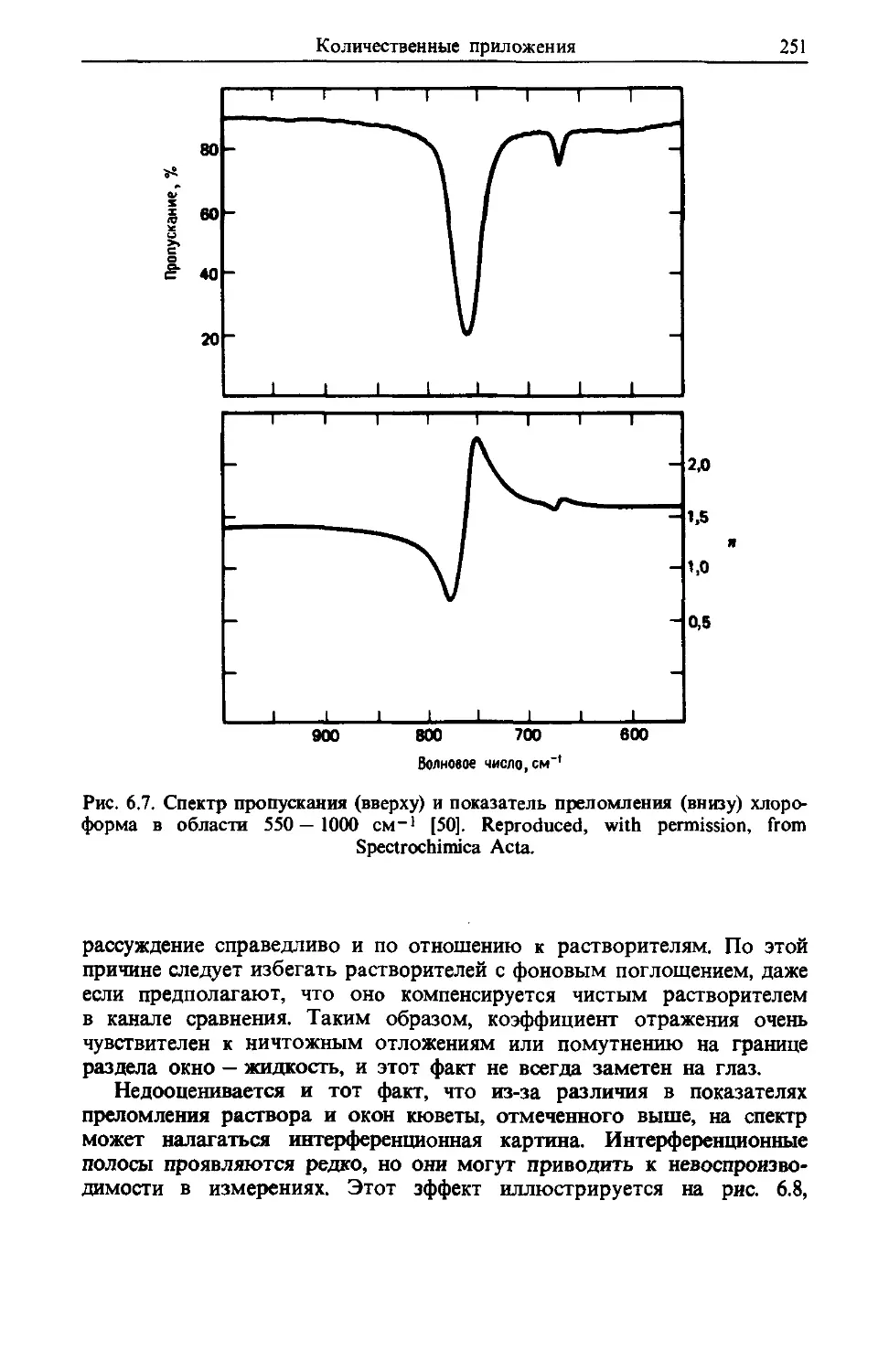

















































































Text
APPLIED
INFRARED SPECTROSCOPY
FUNDAMENTALS, TECHNIQUES,
AND ANALYTICAL PROBLEM-SOLVING
A.Lee Smith
Dow Corning Corporation
A WILEY-INTERSCIENCE PUBLICATION
JOHN WILEY AND SONS
NEW YORK ¦ CHICHESTER ¦ BRISBANE ¦ TORONTO
А.Смит
ПРИКЛАДНАЯ
UK-
СПЕКТРОСКОПИЯ
ОСНОВЫ, ТЕХНИКА, АНАЛИТИЧЕСКОЕ ПРИМЕНЕНИЕ
Перевод с английского
канд. хим. наук Б. Н. ТАРАСЕВИЧА
под редакцией
д-ра хим. наук, проф. А. А. МАЛЬЦЕВА
МОСКВА
МИР 1982
ББК 22.344
С50
УДК 543.422.4
Смит А.
Прикладная ИК-спектроскопия: Пер. с англ. — М.: Мир,
1982.-328 с, ил.
Книга американского ученого, представляющая собой подробное справочное
руководство по применению метода ИК-спектроскопии в аналитических целях -
для качественного и количественного анализов различных классов химических
соединений и их сложных смесей. Приводятся многочисленные примеры исполь-
использования метода в промышленных лабораториях, в частности при анализе
многокомпонентных систем, для контроля производственных процессов, при анализе
промышленных загрязнений.
Предназначена для научных работников, сотрудников лабораторий химиче-
химических предприятий, преподавателей, читающих курс экспериментальных методов
молекулярной спектроскопии.
1805000000-140
С 04Н01НЙ
Редакция литературы по химии
i 1979 by John Wiley and Sons, Inc. All rights reserved.
Authorized translation from English language edition
published by John Wiley and Sons, Inc.
i Перевод на русский нзык, «Мир», 1982
ПРЕДИСЛОВИЕ
РЕДАКТОРА ПЕРЕВОДА
Прошло 30 лет с тех пор, как появился первый серийный ИК-
спектрометр отечественного производства ИКС-1. За эти годы ИК-
спектроскопия стала рядовым методом быстрого структурно-группово-
структурно-группового анализа вещества, позволяющим также определять его строение
и количественный состав в различных агрегатных состояниях и широком
интервале температур. Почти в любой химической лаборатории
используются ИК-спектрофотометры или имеется потребность в них.
Сегодня каждый, кто связан с химией или изучает состав вещества,
обязан хорошо ориентироваться как в ИК-спектроскопии, так и в ряде
других физических методов. Чтобы полностью охарактеризовать любое
химическое соединение, необходимо получить его спектр ЯМР, ИК-
и масс-спектры, одновременно проводя элементный анализ. Следует
подчеркнуть, что эти методы не конкурируют между собой, а гармо-
гармонично дополняют друг друга. Поэтому неверно высказываемое иногда
мнение, что ИК-спектроскопия в химии отошла на второй план.
Активное развитие нового поколения автоматизированных с помощью
мини-ЭВМ ИК-спектрофотометров позволяет существенно повысить
точность, чувствительность и скорость количественных определений
и работать при очень больших оптических плотностях (например,
определять небольшие количества биологических веществ в водных
растворах). Очень перспективным оказался метод нарушенного полного
внутреннего отражения (НПВО).
Для подготовки кадров, владеющих методами ИК-спектроскопии,
в вузах ввели специальные лекционные курсы «Физические методы
в химии» и «Молекулярная спектроскопия». В учебной литературе
и лекциях по органической химии уже стало обязательным рассмотрение
ИК-спектроскопии. Многие осваивают ее самостоятельно.
На этом фоне особенно остро ощущается явный недостаток литера-
литературы на русском языке, где были бы в доступной форме обобщены
практика и аналитические приложения ИК-спектроскопии (вопросы
структурно-группового анализа освещены хорошо). Фактически имеются
только книга В. М. Чулановского «Введение в молекулярный спект-
спектральный анализ» (М.—Л.: ГИТТЛ, 1951) и переводная книга И. Кес-
слера «Методы инфракрасной спектроскопии в химическом анализе»
(М.: Мир, 1964), хотя принципы количественных измерений ИК-полос
поглощения в условиях инструментальных искажений начинали разраба-
Предисловие редактора перевода
тываться в нашей стране В. М. Чулановским, А. В. Иогансеном,
Г. Г. Петрашом, С. Г. Раутианом, В. И. Малышевым, А. Ф. Василь-
Васильевым и др.
Предлагаемая книга А. Смита весьма своевременна по следующим
причинам. Во-первых, она содержит очень ценные практические сведения,
необходимые в повседневной работе на спектральных приборах,
а также обширную, хорошо классифицированную библиографию по
ИК-спектроскопии. Во-вторых, она имеет подчеркнуто аналитическую
направленность. В-третьих, книга на конкретных примерах знакомит чи-
читателя с теми возможностями и перспективами исследования вещества,
которые связаны с появлением нового поколения спектральных при-
приборов, в том числе и лазерных спектрометров, серийно выпускаемых
зарубежными фирмами. И наконец, все разделы проникнуты идеей
оптимизации исследований с аргументированным обоснованием того,
что может ИК-спектроскопия и в чем состоит ее ограниченность.
Согласно этой идее, тщательный подбор режимов работы спектраль-
спектрального прибора и продуманный выбор методики позволяют даже на
спектрофотометрах среднего класса получать удовлетворительные
результаты.
Большим достоинством книги является то, что она, с одной стороны,
доступна широкому кругу исследователей, не имеющих специальной
подготовки, а с другой — даже опытные спектроскописты найдут в ней
много нового и смогут пользоваться ею как справочником.
Для перевода книга оказалась не из легких по целому ряду причин.
До сих пор в отечественной литературе нет установившейся терми-
терминологии по спектрофотометрии, тем более когда речь идет о переводе
с иностранного языка. Имеющийся ГОСТ противоречит международным
рекомендациям ШРАС и требованиям «Журнала аналитической химии».
Особую трудность вызвали перевод термина „absorbance" и его бук-
буквенное обозначение (А или D). В какой-то степени внесли осложнения
авторский жаргон и некоторая небрежность в определении ряда понятий
(волнового числа, аппаратной функции и т. д.). Если у читателей
возникнет неудовлетворенность краткостью изложения и отсутствием
более глубокого теоретического рассмотрения вопросов количественного
анализа, этот пробел можно будет восполнить, пользуясь рекоменду-
рекомендуемой литературой.
То обстоятельство, что в библиографии отсутствуют работы со-
советских авторов и лишь небольшое число цитированных в ней моно-
монографий имеется на русском языке, позволило в ряде глав привести
дополнительную литературу, ссылки на которую в тексте даются
курсивными цифрами.
Хочется надеяться, что книга А. Смита не только поможет в повсе-
повседневной практической работе, но и позволит правильно оценить
перспективу развития ИК-спектроскопии в недалеком будущем.
А. Мальцев
ПРЕДИСЛОВИЕ
После того как в течение 60-х годов метод ИК-спектроскопии
достиг кажущейся завершенности, наступил новый период его активного
развития. Возрождение интереса к этому методу стимулировалось вы-
выпуском сложной аппаратуры, позволившей поднять экспериментальную
технику и методы анализа до такого уровня, который был немыслим
всего несколько лет назад. В будущем возможны еще большие
достижения.
В основу настоящей книги положен расширенный вариант главы,
написанной для учебника Кольтгофа и Элвинга «Курс аналитической
химии». Я считал, что ее полезно опубликовать отдельно, поскольку
в ней содержится тот необходимый материал, которого нет в большин-
большинстве других монографий. Эта книга базируется на практическом мате-
материале, подчеркивающем основные идеи и ограниченность аналитической
ИК-спектроскопии для химиков. Для более детального ознакомления
с данным предметом в книге даются обширные библиографические
списки. Альтернативным подходом могло бы стать всестороннее рас-
рассмотрение ИК-спектроскопии в одной книге, но такое энциклопедическое
издание устарело бы, еще не будучи напечатанным.
Ввиду аналитической направленности книги теоретические вопросы
изложены в ней в такой степени, чтобы читатель только „почувствовал"
основы молекулярной динамики. Для получения из спектра максималь-
максимальной информации важно иметь хорошую технику. Однако даже приме-
применение ЭВМ не исключает случайностей и небрежностей как в ходе
приготовления образца, так и при работе на спектрофотометре, и этим
вопросам уделено очень большое внимание. Важно также, чтобы всякий,
кто имеет дело со спектральным прибором, понимал, как он работает;
с этой целью рассмотрены основные принципы конструкций существу-
существующих спектрофотометров. Представляются полезными списки ссылок
на каталоги спектров и обзоры, посвященные специальным вопросам.
Как мне кажется, количественный анализ методами ИК-спектроскопии
используется недостаточно широко и понимается не всегда правильно,
поэтому в книге ему отведено центральное место и для иллюстрации
многообразия его возможностей приведено несколько примеров. Рас-
Рассмотрены факторы, влияющие на групповые частоты, но групповые
частоты отдельных функциональных групп не обсуждаются — по сле-
следующим причинам: во-первых, имеются превосходные книги, посвящен-
Предисловие
ные этому вопросу, и, во-вторых, по моему убеждению, излишнее
доверие к таблицам и диаграммам и исключение из исследования
стандартных спектров являются ошибочными. Поскольку эта книга
предназначена для решения аналитических задач, в ней не затрагива
ется ряд интересных проблем, которые, по-видимому, больше принад
лежат области физической химии, хотя граница между физической
и аналитической химией, по общему мнению, довольно расплывчата.
Отсутствует рассмотрение таких вопросов, как хемосорбция, механизм
гетерогенного катализа, исследование структуры молекул, анализ коле-
колебательно-вращательных спектров, определение расстояний между энер-
энергетическими зонами в полупроводниках.
В книге особое внимание уделено основам методов и пределам
их возможностей, так что читатель получает солидную базу для
продолжения работы в соответствии с его собственными более
специальными интересами.
Здесь я хочу поблагодарить тех, кто внес вклад в настоящую книгу.
По счастливой случайности я имел возможность познакомиться и рабо-
работать с некоторыми из лучших в мире спектроскопистов-практиков,
которым я обязан многим — советами, руководством, поддержкой.
Я особенно благодарен за помощь К. Кравер, П. Гриффитсу,
Н. Харрику, Р. Ханна и Р. Найквисту, которые прочитали отдельные
части рукописи и высказали полезные замечания. Наконец, я весьма
признателен г-же Г. Биндер и г-же М. Фримен за их самоотверженные
усилия при перепечатке рукописи.
Мидленд, Мичиган А. Смит
январь 1979
Глава 1
ВВЕДЕНИЕ
Обзор приложений ИК-спектроскопии
Инфракрасная (ИК) спектроскопия используется в различных об-
областях науки, и в каждой из них придается этому термину различный
смысл. Для химика-аналитика это удобный метод решения таких задач,
как, например, определение пяти изомеров гексахлорциклогексана,
качества парафина, смолы, полимера, эмульгатора в змульсии для поли-
полировки, опознание страны, из которой вывезен контрабандный опиум.
Физику ИК-спектроскопия представляется методом исследования энер-
энергетических уровней в полупроводниках или определения межатомных
расстояний в молекулах. Она может быть также полезна и при изме-
измерении температуры пламени ракетного двигателя. Для химика-органика
это метод идентификации органических соединений, позволяющий вы-
выявлять функциональные группы в молекулах и следить за ходом хими-
химических реакций. Для биолога ИК-спектроскопия — перспективный метод
изучения транспорта биологически активных веществ в живой ткани,
ключ к структуре многих естественных антибиотиков и путь познания
строения клетки. Физикохимику метод позволяет приблизиться к пони-
пониманию механизма гетерогенного катализа и кинетики сложных реакций.
Он служит дополнительным источником информации при расшифровке
структуры кристаллов. В этих и многих других областях знания
ИК-спектроскопия служит исследователям мощным средством изучения
тайн вещества. Вероятно, справедливо будет сказать, что из всех инстру-
инструментальных методов ИК-спектроскопия наиболее универсальна.
История
На существование ИК-излучения впервые в научной литературе было
указано в 1800 г. Уильямом Гершелем [3], который для измерения
теплового эффекта солнечного света внутри и за пределами видимого
спектра использовал стеклянную призму и зачерненный термометр
в качестве детектора. Хотя Гершель первым пришел к выводу, что
ИК-излучение имеет ту же природу, что и видимый свет, он позже
отказался от этого, так как получил две различные кривые для „яр-
„яркости" и теплового эффекта. Однако главный интерес для Гершеля
представляла астрономия, и в дальнейшем он не исследовал „тепловой
спектр".
10 Глава 1
В течение последующих 80 лет интереса к этому явлению не было,
но в период с 1882 по 1900 г. исследователи, используя сравнительно
грубые методы, находившиеся в их распоряжении, совершили быстрое
вторжение в ИК-область. Абни и Фестинг [1], например, сфотографи-
сфотографировали спектры поглощения 52 соединений в области до 1,2 мкм
и нашли связь полос поглощения с присутствием в органической
молекуле некоторых групп. Джулиус [5], используя призму из каменной
соли (NaG) и болометр в качестве приемника излучения, исследовал
спектры 20 органических соединений. Он установил, что соединения,
содержащие метальную группу, поглощали при 3,45 мкм, но в более
длинноволновой области его измерения были ошибочны.
Вплоть до того времени, пока не была развита теория ИК-погло-
щения, не существовало способов определить, обусловлено ли оно
наличием отдельных атомов в молекуле, межмолекулярными эффектами
или внутримолекулярными движениями. Работа Джулиуса подтвердила
справедливость последнего, в частности, что именно группы атомов
в химическом соединении определяют картину поглощения.
Активность в этой области росла, но только В. Кобленцу своей
работой суждено было заложить настоящую основу ИК-спектроскопии.
В своих классических работах, начатых около 1903 г., он исследовал
ИК-спектры сотен веществ, как органических, так и неорганических [2],
с такой точностью и полнотой, что многие из его спектров, полученных
с призмой из Nad, остаются полезными и поныне.
Первые исследователи испытывали огромные экспериментальные
трудности. Им приходилось не только проектировать и собирать свои
собственные приборы, но и делать все остальное, в том числе шлифо-
шлифовать и полировать призму, серебрить зеркала, изготавливать радиометр.
Приборы градуировались по величинам показателей преломления ка-
каменной соли, измеренным либо ими самими, либо другими исследова-
исследователями. Спектрометры обычно устанавливались на фундаменте, а изме-
измерения зачастую проводились ночью, чтобы уменьшить влияние вибраций
на чувствительный радиометр или микрорадиометр. Так как каждая
точка в спектре должна измеряться отдельно, а на один микрометр
приходилось по крайней мере 10 точек, то измерение спектра пред-
представляло утомительную работу, занимавшую не менее 3—4 или более
часов [4].
В ранних работах было установлено, что каждое соединение имеет
свою собственную и неповторимую картину ИК-поглощения и что не-
некоторые группы даже в различных молекулах дают полосы поглощения
с приблизительно одинаковой длиной волны. Однако из-за трудоемкости
измерения спектров химики фактически не использовали этот метод
вплоть до 1940-х годов.
Вторая мировая война привела не только к повышенному спросу на
аналитическую аппаратуру, но и к быстрому развитию электроники.
Появилась возможность электронного усиления очень слабых сигналов,
получаемых от крошечного термоэлемента ИК-спектрометра, и регист-
Введение 11
рации их на ленте самописца. Из-за высокой инерционности термо-
термоэлемента и использования сигнала постоянного тока серьезной пробле-
проблемой был дрейф измеряемых величин. Тем не менее при везении
и осторожности можно было за 1-2 ч получить спектры довольно
хорошего качества.
Следующим значительным шагом было усовершенствование техники
изготовления термоэлектрических приемников (детекторов) с достаточно
малым временем отклика, чтобы модулировать (прерывать) излучение
с частотой от 5 до 10 Гц. Это новшество позволило устранить дрейф
в системе регистрации спектрометра и открыло путь к созданию
двухлучевых приборов *, шкалу которых стало возможным калибровать
в процентах пропускания в зависимости от линейной шкалы длин
волн или волновых чисел [7].
Эти усовершенствования вывели ИК-спектроскопию из царства
скуки, и, как только химики осознали ее возможности, популярность
метода быстро возросла. После периода интенсивных усилий, в тече-
течение которого были исследованы тысячи веществ и выявлены ограни-
ограничения, связанные с аппаратурой, у спектроскопистов появилась потреб-
потребность в лучшем разрешении и большей точности измерения волновых
чисел. Этим требованиям в какой-то мере отвечала дифракционная
решетка, а с появлением интерферометров возможность получения
точных контуров полос поглощения жидкостей и твердых тел стала
вполне реальной. Однако не следует слишком надеяться на скорую
возможность получения спектров, свободных от шумов, которые точно
передавали бы контуры, частоты и интенсивности поглощения молекул
и не были бы искажены самим спектрофотометром ** или артефактами
кюветы образца.
Возможности ИК-спектроскопии
ИК-спектр поглощения,—вероятно, уникальное в своем роде физи-
физическое свойство. Не существует двух соединений, за исключением опти-
оптических изомеров, с различающимися структурами, но одинаковыми
ИК-спектрами. В некоторых случаях, таких, как полимеры с близким
молекулярным весом, различия могут быть практически не заметны,
но они всегда есть. В большинстве случаев ИК-спектр является
„отпечатком пальцев" молекулы, который легко отличим от спектров
других молекул.
* Такой тип спектральных приборов общепринято называть спектрофото-
спектрофотометрами, хотя автор книги этого и не придерживается. Мы сочли полезным
при переводе чаще пользоваться последним названием. — Прим. ред.
** В этом вопросе автор противоречит сам себе, так как уже имеется целый
ряд конструкций серийных спектрофотометров, позволяющих регистрировать
собственные контуры полос поглощения газообразных, жидких и твердых
образцов (см. гл. 2). — Прим. ред.
12 Глава 1
Кроме того что поглощение характеристично для отдельных групп
атомов, его интенсивность прямо пропорциональна их концентрации.
Таким образом, измерение интенсивности поглощения дает после
простых вычислений количество данного компонента в образце.
По своим возможностям метод почти универсален. Образцы могут
быть жидкими, твердыми или газообразными. Они могут быть орга-
органическими или» неорганическими, хотя неорганические вещества иногда
не дают хорошо выраженных спектров. В обычных условиях для
ИК-излучения прозрачны только одноатомные газы и неполярные
молекулы (Ne, He, О2, N2, H2).
Другое ограничение заключается в том, что такой распространен-
распространенный растворитель, как вода, имеет в ИК-области очень сильное погло-
поглощение и, кроме того, растворяет окна кювет, в качестве которых
используют пластинки из кристаллов солей *, Метод ИК-спектроскопии
обычно не очень чувствителен к примесям, если они не превышают 1 %.
Это, конечно, может быть как благом, так и бедствием, все зависит
от точки зрения и решаемой проблемы. Подобным же образом может
огорчить и тот факт, что положения характеристических полос погло-
поглощения для многих групп различны при переходе от одной молекулы
к другой, но это подтверждает индивидуальность спектра поглощения
и дает больше для понимания структуры молекулы, чем если бы
полосы были неизменны.
Спектроскопия в ИК-области встречается с рядом специфических
трудностей. В силу того что излучение невидимо, юстировку оптических
деталей нельзя проводить визуально. Энергия, с которой приходится
иметь дело, крайне мала и уменьшается с увеличением длины волны.
Из-за низкой энергии сигнал приемника не намного выше уровня
шума, возникающего в результате хаотического теплового движения
электронов в контуре приемника. Более того, поскольку все части
спектрометра теплые (по сравнению с абсолютным нулем), они излучают
энергию в ИК-области и на детектор попадает значительное количество
паразитной энергии, которая должна быть отделена от полезного сиг-
сигнала. Кто-то сказал, что ИК-спектрометрию грубо можно сравнить
с фотографированием эмиссионного спектра на раскаленном добела
спектрографе.
Выбор аналитической методики
Как правило, ИК-спектроскопия должна использоваться везде, где
требуется знать специфические особенности. Температура плавления,
показатель преломления или удельный вес позволяет получить только
* Выпускаемые в последние годы новые оптические водонерастворимые
материалы для ИК-области спектра, например BaF2, кремний, германий и т. д.,
позволяют обойти это затруднение (см. (табл. 4.2). — Прим. ред.
Введение 13
отдельную точку, которую можно сравнивать с другими веществами.
В противоположность этому ИК-спектр дает почти бесконечное мно-
множество таких точек. Обычно он позволяет более надежно определять
функциональные группы, чем химический анализ.
Когда требуется провести совместно качественный и количественный
анализы, предпочитают метод ИК-спектроскопии. Приложения такого
типа включают контроль за органическими реакциями, особенно когда
их ход не достаточно хорошо известен и необходимо определить
конечные продукты.
Метод ИК-спектроскопии рекомендуется при количественном ана-
анализе сложных нелетучих смесей, таких, как полимеры, или в случае,
когда газохроматографическое разделение компонентов смеси за-
затруднено.
В то же время ИК-спектр не даст различий в составе или струк-
структуре, когда они соизмеримы с величиной случайных приборных ошибок,
и не покажет, действительно ли данный образец удовлетворяет техни-
техническим условиям, за исключением тех случаев, когда они связаны
с наличием или отсутствием некоторых полос поглощения. Спектр, как
и другие сложные измерения, может быть интерпретирован ошибочно,
если исследователи имеют лишь поверхностные знания в этой
области.
В некоторых случаях другие методы могут оказаться более экспресс-
экспрессными или более чувствительными. Например, ядерный магнитный резо-
резонанс (ЯМР) зачастую дает больше информации о строении молекул
некоторых классов растворимых органических веществ без спектров
сравнения или стандартов. Стандарты менее важны также в масс-
спектрометрии, где объем исследуемого образца может быть и меньше,
но вещество должно быть летучим, однако область применения метода
порой уже, чем в случае ИК-спектроскопии. Газовая хроматография,
масс-спектрометрия и ультрафиолетовая (УФ) спектроскопия имеют
превосходную чувствительность к следовым количествам (естественно,
в пределах их чувствительности). Кроме того, для некоторых веществ
эти три метода способны давать и превосходные количественные
результаты. Спектроскопия комбинационного рассеяния (КР) света
может быть использована в аналитических целях аналогично ИК-
спектроскопии, но чаще как дополняющий, а не конкурирующий
метод [б]. Таким образом, ясно, что аналитик должен сознавать
возможности и ограничения всех доступных методов.
Определения основных понятий
Каждая область науки имеет собственный язык, и здесь было бы
полезно дать определения некоторых терминов, использованных в этой
книге.
14 Глава 1
А В
1СИ
С D
Рис. 1.1. Связь единиц. Длина волны соответствует расстоянию АВ; вол-
волновое число — число волн, приходящееся на один сантиметр, CD; частота —
число волн, проходящих через фиксированную точку С в единицу времени.
Спектр можно рассматривать как последовательное расположение
электромагнитного излучения по длинам волн. Длины волн могут
изменяться от 102 мм до миллионов метров. Для удобства использу-
используются ангстрем (А), равный 10"8 см, и микрометр (мкм), ранее назы-
называвшийся микроном (ц) и равный 10 ~4 см. В ИК-спектроскопии часто
используется волновое число v, которое связано с длиной волны X
соотношением v (см) = A04Д), где X выражено в микрометрах*.
Для наглядности волновое число может быть представлено как число
целых длин волн электромагнитного излучения в одном сантиметре
(рис. 1.1). Волновое число прямо пропорционально энергии и частоте
колебаний структурного элемента исследуемого соединения:
где ст — волновое число в вакууме в см"; /— частота в секундах (для
обозначения частоты физики часто используют v, но мы будем исполь-
использовать f, чтобы избежать путаницы с обозначением волнового числа v);
Е — энергия в эргах; h — постоянная Планка; Хвак — длина волны
в сантиметрах, измеренная в вакууме (или приведенная к вакууму);
п — показатель преломления воздуха. С общетеоретической точки зрения
правильнее использовать частоту или волновое число в вакууме, так
как длина волны излучения зависит от показателя преломления среды,
в которой она измерена, а частота нет.
ИК-область обычно рассматривают, начиная с красного края види-
видимого спектра, примерно с 14000 см G000 А, или 0,7 мкм), где глаз
перестает воспринимать диспергированное излучение (таким образом,
„инфра" значит „ниже красного")- Так называемая фундаментальная
ИК-область начинается примерно с 3600 см, или 2,8 мкм. Аналити-
Аналитически полезная область распространяется от 3600 см примерно до
300 см, или 33 мкм. В настоящей книге рассматривается главным
образом средняя ИК-область, хотя бегло упоминается и „ближняя",
* Строго говоря, по принятому определению волнового числа X уже здесь
должно быть измерено ие на воздухе, а в вакууме. Тогда отпадает необхо-
необходимость вводить а (см. ниже). — Прим. ред.
Введение 15
расположенная между 14 000 и 3600 см 1. Нелегко определить „дальнюю"
ИК-область, но приближенно ее считают от 300 до 20 см *. Для ана-
аналитических целей она почти не используется. Излучение с частотами
ниже 20 см ~1 E00 мкм) относится к микроволновой или радиоволновой
области.
ЛИТЕРАТУРА
1. Abney W. de W., Festing E. R., Phil. Transact., 172, 887 A882).
2. Coblentz W. W., Investigations of Infrared Spectra, Carnegie Institute, Washington,
D. G, Publication No. 35, 1905 (reprinted 1962 by Coblentz Society and Perkin
Elmer Corporation).
3. Herschel W., Phil. Transact. Roy. Soc, 90, 284 A800).
4. Jones R. N., Appl. Opt., 2, 1090 A963).
5. Julius W. H., Verhandl, Konikl, Acad. Wetenschappen Amsterdam, 1, 1 A892).
6. Shane H. J., Appl. Spectrosc, 25, 430 A971).
7. W-ight N., Herscher L W., J. Opt. Soc. Am., 37, 211 A947).
* Все эти границы, действительно, весьма условны, но сейчас принято за
верхнюю границу дальней оптической ИК-области считать предел пропускания
окон из Csl, т.е. 200 см E0 мкм), а за нижнюю 10—30 см, которая
определяется коиструктивными возможностями серийных приборов. — Прим. ред.
Глава 2
ПРИБОРЫ
Быстрое увеличение числа различных типов спектральных приборов
создает затруднения даже для опытного спектроскописта. Тем не менее
общие принципы, заложенные б их конструкции, вполне доступны для
понимания. Кратко обсудим существующие в настоящее время системы
ИК-спектрометров, чтобы читатель при желании мог без больших
затруднений ориентироваться в более подробных описаниях. Для начала
было бы полезно приспособить схему Вайнфорднера, предложенную
для классификации приемников излучения [86], к классификации
спектрометров, как показано на рис. 2.1. Приборы, в которых инфор-
информация накапливается последовательно во времени, называют сканиру-
сканирующими. По мере сканирования каждого спектрального элемента инфор-
информация накапливается с помощью одноканального приемника. При-
Приборы с пространственным разделением, использующие многоканаль-
многоканальные приемники, в средней ИК-области практически не применяются;
примером такого прибора в видимой области служит спектрограф,
регистрирующий спектр на фотопластинку. Многоканальные спектро-
спектрометры — это такие приборы, в которых одноканальный приемник
одновременно получает много сигналов, соответствующих различным
элементам спектра. Эти сигналы проходят через один канал, но
расшифровываются таким образом, что дают информацию о каждом
отдельном спектральном элементе.
Физика ИК-излучения
Для понимания работы различных типов ИК-спектрометров и
спектрофотометров рассмотрим некоторые из физических принципов,
лежащих в основе их работы.
С точки зрения физика, конструкция, устройство и работа ИК-
спектрофотометров исчерпывающе рассмотрены Стюартом [79]. Как
отмечалось ранее, ИК-области присуща низкая энергия (так как она
обратно пропорциональна длине^олны). Поэтому конструирование
ИК-спектрофотометров направлено на максимальное увеличение энер-
энергии, проходящей через прибор. Точно так же, работая на спектрофото-
спектрофотометре, всегда нужно помнить об этом основном ограничении и вы-
выбирать режимы работы прибора такими, чтобы они отвечали требованиям
законов физики.
С последова-
последовательным _ одноканальный
сканированием детектор
Инфракрасные
спектрометры
С простран-
пространственным
разделением
- Диспергирую-
Диспергирующие
. Недиспергиру-
ющие
г Диспергирую-
I щие
. многоканальный __1
детектор " I „
L-Недиспергиру-
ющие
Ч Диспергирую-
Диспергирующие
Недиспергиру-
ющие
Сканирующие
Многощелевые
Растровые
Лазерные
полупроводниковые
С вращающимся
фильтром
Оптико-акусти-
Оптико-акустические
Детектор
типа„видикон"
Многоканальные
анализаторы
процессов
Преобразование
Адамара
Преобразование
Фурье
Рис. 2.1. Классификация ИК-спектрометров и спектрофотометров.
18
Глава 2
Все ИК-спектрофотометры независимо от того, просты они или
сложны, имеют определенные общие элементы: источник, оптическую
систему, приемник и усилитель.
Источник
Идеальным источником для ИК-спектроскопии был бы монохрома-
монохроматический излучатель высокой интенсивности, непрерывно перестраива-
перестраиваемый в широком интервале длин волн. Хотя лазеры с перестраиваемой
частотой отчасти приближаются к идеальному источнику (стр. 32), их
использование в обычных спектрофотометрах кажется довольно отдален-
отдаленным будущим. Обычно применяются источники, излучающие непре-
непрерывный спектр, приближающийся к излучению абсолютно черного тела.
Мощность излучения W абсолютно черного тела выражается через его
температуру Т и длину волны X законом Планка
¦&) "'Г'1
B.1)
где с1 и с2 — постоянные.
Наиболее распространенными источниками в области 100-4000 см
являются глобар, представляющий стержень из карбида кремния,
и штифт Нернста, состоящий из смеси оксидов циркония, тория
и иттрия. Источник с несколько пониженной излучательной способ-
способностью, но с гораздо большим временем жизни может быть изготовлен
бооо к
I
Рис.
г 4 6
Длина волны, мкм
2.2. Зависимость излучательной способности абсолютно черного тела
от длины волны [39]. Reproduced with permission of Book Company.
Приборы 19
из нихромовой проволоки, свернутой в плотную спираль. Из рис. 2.2,
на котором показан график зависимости излучательной способности
от длины волны и температуры, видно, что увеличение температуры
источника приводит к значительному росту интенсивности излучения
в коротковолновой области и лишь к слабому — в длинноволновой.
С повышением энергии коротковолнового излучения в приборе увеличи-
увеличивается уровень рассеянного света, поэтому не очень выгодно переходить
к высокотемпературным источникам света. В дальней ИК-области
(< 100 см) ртутная лампа высокого давления излучает лучше, чем
накаленные нити или стержни. В любом спектрофотометре размеры
источника должны быть такими, чтобы его изображение заполняло
апертуру прибора во всех областях спектра. Обзор источников ИК-
излучения сделан Канном [7].
Оптическая система
Назначение оптической системы — направлять излучение по нужному
пути. Использование отражательных, зеркал с наружным покрытием
предпочтительнее, чем линз, так как последние имеют хроматическую
аберрацию и преломляющая оптическая система должна постоянно
перестраиваться с изменением длины волны.
Отражающая оптика может иметь плоские, сферические либо более
сложные поверхности, такие, как тороиды, параболоиды или эллипсоиды
(рис. 2.3). Оптические системы с большей апертурой для уменьшения
аберрации и потери энергии обычно требуют использования асфери-
асферической оптики. В частности, зеркало коллиматора — обычно внеосевой
параболоид, а фокусирующее зеркало термоэлемента — эллипсоид.
Хотя зеркала такой формы и дают значительно лучшее изображение,
чем сферические, они оптически несовершенны даже теоретически,
за исключением случая точечного источника.
Как правило, чем больше апертура оптической системы (т. е. чем
меньше длина светового пучка относительно сечения оптических деталей),
тем выше требования к качеству и юстировке зеркал, щелей и других
элементов. Желательно иметь оптическую систему с большей апертурой,
так как в этом случае при данной спектральной ширине щели имеется
выигрыш в энергии в отличие от системы с малой апертурой. Для
детального рассмотрения конструкций спектрофотометров можно обра-
обратиться к работам Конна [17] и Стюарта [79] (см. также дополнитель-
дополнительную литературу [i-6]).
Приемники излучения
Приемники ИК-излучения делятся на две группы: тепловые и кван-
квантовые (или фотонные). Первая группа включает термоэлементы (спай
разнородных металлов, в котором при нагревании возникает электро-
электродвижущая сила — э. д. с, которая меняется с температурой), болометры
20
Глава 2
Рис. 2.3. Параболическое зеркало (а), которое фокусирует параллельный
пучок света в точке F; эллиптическое зеркало (б) с двумя фокусами Fj и F2;
тороидальное зеркало (в), которое имеет различающиеся фокусные расстояния
в горизонтальной и вертикальной плоскостях.
(сопротивления с большим температурным коэффициентом), а также
пневматические приемники. Ко второй группе относятся квантовые
приемники с внутренним фотоэффектом как с активацией примесями,
так и без нее. Энергия ИК-излучения слишком низка для того, чтобы
воздействовать на фотографические эмульсии или фотоэлектрические
приемники.
Термоэлемент по своей природе является тепловым устройством,
и потому он чрезвычайно малоэффективен (эффективность < 10~б) для
измерения очень малых изменений температуры в ИК-спектрофотометре,
Приборы 21
Время ответной реакции на изменение сигнала у термоэлемента больше,
чем у квантового приемника. Однако чувствительность термоэлемента
не зависит от длины волны, за исключением длинноволнового излучения
(более 30 мкм), где эффективность чернения* падает. Для дальней
ИК-области более эффективен пневматический приемник.
Абсолютные чувствительности термоэлементов и болометров к пуль-
пульсирующему сигналу обсуждены Вильямсом [84]. Грубо говоря, чувстви-
чувствительность термоэлемента обратно пропорциональна площади приемника
и его теплоемкости. Другие факторы, влияющие на чувствительность,
включают также термоэлектрический коэффициент и полное рассеяние
тепла. Болометры также обладают большей чувствительностью при
меньшем размере приемника и меньшей теплоемкости. Следовательно,
целесообразно иметь рабочий элемент по возможности меньших раз-
размеров и с минимальной теплоемкостью. Кроме того, элемент с низкой
теплоемкостью быстрее реагирует на периодический или прерывающийся
сигнал, и, как будет показано в дальнейшем, желательно, чтобы
детектор имел малое время ответа. Для повышения чувствительности
и уменьшения шумов болометр и термоэлементы обычно работают
в вакууме. Иначе говоря, чувствительность подобных термоэлементов
может меняться в широких пределах из-за различий в термоэлектри-
термоэлектрических коэффициентах.' Из доступных термоэлементов целесообразно
использовать наиболее чувствительные. Мощность излучения, попа-
попадающего на приемник спектрофотометра, имеет порядок 10 ~7 Вт
и вызывает изменение температуры элемента в несколько тысячных
долей градуса. В результате возникает сигнал, равный долям микро-
микровольта.
В пневматическом приемнике Голея, или оптико-акустическом при-
приемнике (ОАП) [31,9], используется тепловое расширение непогло-
щающего газа, находящегося в зачерненной приемной камере, задняя
стенка которой представляет собой гибкую пленку с зеркальным
покрытием с внешней стороны. Движение зеркала усиливается оптиче-
оптической системой и регистрируется фотоэлементом (рис. 2.4). Чувствитель-
Чувствительность такого приемника примерно того же порядка, что и термо-
термоэлектрических приемников, но требования к фокусировке на приемную
площадку не столь жесткие, а чернение может быть сделано более
чувствительным к длинноволновому излучению.
В квантовых приемниках отдельные фотоны возбуждают электроны.
В обычном фотоэлементе эти возбужденные электроны вырываются
с поверхности электрода, но в ИК-области энергия фотонов слишком
низка, чтобы таким путем вызвать фототок. Вместо этого исполь-
используются такие полупроводниковые устройства, в которых при поглощении
фотона электроны возбуждаются из непроводящей, валентной зоны
в метастабильное состояние или в зону проводимости. Энергия фотонов
* Имеется в ввду чернение приемной площадки термоэлемента. — Прим.
перев.
22
Глава 2
Рис. 2.4. Одна из схем приемника Голея.
7 — диафрагма; 2 — окно, прозрачное для ИК-излучения; 3 — линейчатый растр;
ный фотоэлемент [31]. Courtesy of Eppley Laboratory.
— светочувствитель-
светочувствитель101
Длина волны, мкм
Рис. 2.5. Спектральная чувствительность приемников с внутренним фото-
фотоэффектом [4]. Reproduced, with permission, from Infrared Physics.
Приборы 23
должна быть достаточной, чтобы „перебросить" электроны через энерге-
энергетическую щель Eg и сделать приемник проводником (т. е. v > EJH).
Высота энергетического барьера Et зависит от температуры и пони-
понижается при охлаждении (рис. 2.5).
Приемники с внутренним фотоэффектом, такие, как PbS, PbSe
и InSb, в 10-100 раз более чувствительны, чем термоэлектрические,
но они эффективны только в ограниченных областях длин волн
(рис. 2.5), поэтому их можно использовать только в специальных целях.
Чувствительность и спектральная область работы этих приемников
могут быть улучшены охлаждением до температуры жидкого азота.
Полупроводниковые (примесные или несобственные) детекторы можно
использовать вплоть до 250 см, но для реализации их уникальных
свойств требуется охлаждение до температуры жидкого азота, а в не-
некоторых случаях и жидкого гелия. Несмотря на этот недостаток,
их крайне малое время отклика делает эти приемники весьма привле-
привлекательными для ряда целей.
Пироэлектрические детекторы с использованием, например, три-
глицинсульфата (ТГС) используются в интерферометрах из-за их высокой
чувствительности в широкой области ИК-частот и интенсивностей.
Однако при высоких скоростях модуляции происходит некоторое
уменьшение их эффективности. Такие приемники являются сегнето-
электриками, которые ниже температуры Кюри обладают сильной
температурной зависимостью электрической поляризации. Это свойство
может быть использовано для детектирования очень малых изменений
температуры, вызванных излучением, прошедшим через спектрометр
[19]. Более детальные обзоры ИК-приемников даны в других
работах [57, 63, 70, 78, 3, 5-9].
Усиление
В ранних конструкциях ИК-спектрометров использовалось опти-
оптическое усиление сигнала приемника, получаемого с гальванометра
Или радиометра. В современных приборах почти исключительно
используется модуляция излучения с электронным усилением.
Для этого имеется несколько причин. Усилители переменного
тока более стабильны, чем постоянного, а настройкой усилителя только
на частоту модуляции можно избавиться от всех других частот, что
значительно понижает уровень шума. Кроме того, дрейф показаний
прибора может быть сведен к минимуму, так как полезный сигнал в
однолучевом спектрометре постоянно сравнивается с нулевым, когда
свет перекрыт. В случае же двухлучевых спектрофотометров полез-
полезный сигнал «опирается» ие на нуль, а на сигнал, проходящий через
второй канал прибора, в котором отсутствует изучаемый образец.
К недостаткам следует отнести то, что теряется по крайней мере
половина света, но эта потеря с избытком компенсируется ука-
указанными преимуществами.
24 Глава 2
Спектрометры
с последовательным сканирование
спектр*.
В этом разделе кратко обсуждаются принципы работы некоторых
из обычных оптических схем, используемых в ИК-спектрометрах и
спектрофотометрах. Для более детального ознакомления со спецификой
конкретных приборов можно обратиться к инструкциям и описаниям,
выпускаемым заводами-изготовителями. Поскольку спектрометры для
ближней ИК-области сходны с приборами для видимой области, они
в этом разделе не обсуждаются. Показанные здесь схемы не обязательно
реализуются в любых реально существующих приборах, но большинство
серийных спектрометров и спектрофотометров построено по одной
из них.
Диспергирующие приборы
Рассмотрим оптику осветителя и монохроматора раздельно. Схема
осветителя для простого однолучевого спектрометра изображена на
рис. 2.6, а. Осветитель состоит из источника ИК-излучения (S), моду-
модулятора (С) и зеркала (SM), фокусирующего изображение источника
на щель. Образец обычно помещают там, где сечение светового пучка
минимально. Преимущество расположения модулятора около источника
заключается в том, что можно проводить количественные измерения
спектров нагретых и охлажденных образцов, так как их собственное
излучение не модулируется и, следовательно, не регистрируется
спектрометром.
Типичная схема осветителя двухлучевого спектрофотометра с
оптическим нулем показана на рис. 2.6,6. Здесь оба луча «видят»
в основном (но не точно) один и тот же участок ИК-источника,
так что влияние температурных флюктуации минимально. После про-
прохождения через кюветы образца и сравнения пучки объединяются
зеркалом (СМ), которое часто представляет собой вращающийся
полудиск A80°-сектор). Оптический клин или гребенка (А) вводится
или выводится сервомеханизмом (следящей системой) из канала срав-
сравнения настолько, чтобы поглощение в этом канале было равно погло-
поглощению вещества в канале образца. Движение этого ослабителя (атте-
(аттенюатора) связано с пером самописца, которое осуществляет запись
спектра прямо в процентах пропускания.
На рис. 2.6, в показана оптическая схема осветителя другого двух-
двухлучевого спектрофотометра, в котором регистрируется так называемое
электрическое отношение (ratio recording), а оптический ослабитель не
используется. В этой схеме оба луча модулируются, но с разными
частотами, что позволяет сначала декодировать оптические сигналы от
двух пучков, а затем сравнивать их интенсивности через электрические
сигналы.
Рис. 2.6. Схемы блохов спектральных приборов.
о — олнолучевой освепгкяь спектрометра; б — двухлучевой осаетшель спеттрофотомсгра с опгичеогнм
нулем; в — двухлучевой остетитсль спектрофотометра, регистрирующего отношение электрнчетанх си ша-
шало»; г — монохроматор Ляттром; д~ монохроматор Литтрова двойного прохождения.
26 Глава 2
Наиболее распространенные моиохроматоры
Простой монохроматор Литтрова, оптическая схема которого пока-
показана на рис. 2.6, г, можно сочетать с любым осветителем и получить
одно- или двухлучевой спектрофотометр. После осветителя пучок света
проходит через входную щель (S^) и попадает сначала на зеркало
(С), которое превращает его в параллельный, а затем на призму (Р),
установленную в минимум отклонения. Для того чтобы просканировать
требуемый интервал длин волн, зеркало Литтрова (LM) поворачива-
поворачивается винтовым или кулачковым механизмом. В случае дифракционных
монохроматоров в положении LM находится дифракционная решетка,
а около St — подходящие светофильтры. Световой пучок дважды
проходит через призму и фокусируется в плоскости щели S2.
Пучок почти монохроматического света, проходя через выходную щель
S2, фокусируется зеркалом DM на приемнике D. Сигнал от него
усиливается, фильтруется и используется для приведения в движение
в канале сравнения аттенюатора, связанного с пером самописца (система
с оптическим нулем), или только пера самописца (однолучевой спектро-
спектрометр или система с регистрацией электрического отношения).
Входная и выходная щели моиохроматора расположены вертикаль-
вертикально. Для компенсации значительного изменения энергии с изменением
длины волны их ширина меняется во время сканирования, но между
собой они остаются равными.
Две щели необходимы по следующей причине. Входная щель огра-
ограничивает угол в горизонтальной плоскости, в пределах которого
распространяется свет, попадающий в монохроматор. Она выступает
в роли линейного источника, подобно щели эмиссионного спектро-
спектрографа. Хорошо известно, что для спектрографа, дающего стигматическое
изображение, спектральные линии имеют такую же форму, как и щель.
Уменьшение ширины щели приводит к уменьшению ширины спектраль-
спектральных линий (вплоть до дифракционного предела Рэлея).
В ИК-монохроматоре вместо фотопластинки, используемой в ка-
качестве приемника, устанавливается выходная щель, с помощью которой
в сущности осуществляется сканирование по спектру. Легко видеть, что
энергия, попадающая на приемник, состоит из пучка близких длин волн,
который приближается к монохроматическому по мере уменьшения
ширины щели. Ширина этой полосы энергии на половине высоты в
максимуме интенсивности известна как спектральная ширина щели.
Энергия, прошедшая через входную щель монохроматора, пропор-
пропорциональна ширине щели. Так как имеются две щели и каждая про-
пропускает некоторую долю света, то общая величина энергии выходя-
выходящего излучения пропорциональна произведению ширины двух щелей.
Если обе щели имеют одинаковую ширину, то энергия, пропускаемая
монохроматором, пропорциональна квадрату ширины щели. Об этом
соотношении важно помнить в процессе работы на спектрометре.
В двухлучевых и некоторых однолучевых приборах щели запро-
запрограммированы механическим кулачком или электрическим сигналом
Приборы 27
таким образом, чтобы энергия, достигающая детектора, оставалась
примерно постоянной при изменении длины волны. Изображение вход-
входной щели не должно превышать размеров приемной площадки детек-
детектора. Этим условием определяется максимально устанавливаемая
ширина щелей.
Излучение диспергируется либо призмой, либо решеткой. Исторически
именно призма была тем центром, вокруг которого развивалась ИК-
спектроскопия. В начальный период призмы изготавливали из природных
минералов, но такие материалы ограничены размерами, а их оптические
свойства далеко не совершенны. Развитие техники выращивания
больших монокристаллов галогенидов щелочных металлов и других
материалов сделало возможным широкое использование призм высокого
качества и приемлемой стоимости. Несомненно, что наиболее популяр-
популярным материалом для призм является каменная соль, так как ее про-
пропускание перекрывает значительную часть фундаментальной ИК-области
или области «отпечатка пальцев» при разумном компромиссе между
ценой, областью пропускания и разрешением.
Для того чтобы призма диспергировала излучение и возникал
спектр, показатель преломления материала призмы должен изменяться
с длиной волны. Чем «быстрее» это происходит, тем выше дисперсия.
Так как это условие выполняется вблизи полос поглощения, то
наилучшую дисперсию призма имеет как раз на границе пропускания.
Преимущество призменных монохроматоров заключается в их
большей простоте по сравнению с дифракционными. Недостатками
призменных монохроматоров являются ограниченное разрешение, его
зависимость от длины волны и чувствительность дисперсии к из-
изменению температуры. Призмы также искривляют изображение прямой
входной щели в фокальной плоскости выходной щели, причем кривизна
проекции щели в виде сегмента параболы зависит от длины волны.
Для компенсации этого эффекта, который может быть основной
причиной, ограничивающей реальное разрешение спектрофотометра,
одну из щелей (или обе) обычно искривляют* [79].
По существу дифракционные приборы превосходят призменные
благодаря лучшей разрешающей способности, а она сопровождается
более высокой точностью при качественных и количественных
измерениях спектров. Повышенное разрешение связано с более высоким
пропусканием энергии; как показывают расчеты, для эквивалентных
монохроматоров при оптимальных условиях яркость или световой
поток, проходящий через решетку, больше, чем через призму из
NaCl, в 6,7 раза при 625 см, в 25 раз при 1250 см, в 50 раз
при 2500 см и в 100 раз при 5000 см [47]. На практике эти
цифры несколько ниже [11]. Другим преимуществом решетки является
* Приборам с дифракционными решетками также свойственно искривление
изображения входной щели. — Прим. ред.
28 Глава 2
то, что разрешающая способность практически не меняется с
длиной волны.
Необходимо отметить, что для решеток характерна поляризация
ИК-излучения, причем степень поляризации зависит от направления
падающего луча к поверхности решетки [26, 87]. Поляризация
происходит и в призменных спектрометрах, но в меньшей степени*.
К недостаткам решеток относятся необходимость устранения неже-
нежелательных порядков спектров, больший уровень рассеянного света,
менее эффективное использование излучения и ограниченная спектраль-
спектральная область работы для одной решетки. Два первых недостатка
можно преодолеть при использовании призмы для предварительного
разложения в схеме двойного монохроматора или полосовых фильтров,
которые имеют очень низкое пропускание для нежелательных частот
и хорошее пропускание в нужной области спектра. Для того чтобы
перекрыть полный спектр, обычно применяют две или большее число
решеток и несколько фильтров. Требования к отрезающему фильтру
довольно жесткие, так как коротковолновое излучение, которое состав-
составляет большую часть рассеянного излучения, гораздо интенсивнее
(см. рис. 2.2). Эффективность решетки может быть повышена наре-
нарезанием канавок таким способом, чтобы увеличить интенсивность в
определенном порядке (угле блеска).
Двойной монохроматор, используемый в некоторых спектрометрах,
представляет собой, как это -ясно из названия, два последовательно
соединенных монохроматора. Преимущество такой конструкции состоит
в очень малом рассеянном свете и увеличении дисперсии почти в
два раза Недостатком являются повышенная сложность и стоимость
двух монохроматоров вместо одного.
Система двойного прохождения, которая в сущности превращает
одинарный монохроматор в двойной, показана на рис. 2.6, д. Она
обычно используется в однолучевых приборах с простым немодулируе-
мым источником, так как модулирование в точке С применяется для
разделения одно- и двукратно прошедшего излучения. Однако систему
двойного прохождения можно также комбинировать с двухлучевой
оптикой источника. Монохроматор двойного прохождения имеет те же
преимущества в дисперсии и рассеянной энергии, что и двойной
монохроматор, но, поскольку модулятор стоит после кюветного
отделения, прибор чувствителен к температуре образца. При количест-
количественном анализе нулевая линия может смещаться в ходе сканирования
из-за нагревания излучением образца или ножей щели.
Существуют и другие схемы монохроматоров, но они распрост-
распространены не так широко, как схема Литтрова. Сравнение некоторых
двухлучевых систем, которые могут быть использованы в ИК-спект-
рофотометрии, было проведено Голеем [32].
* Собственную поляризацию прибора необходимо учитывать при работе с
ориентированными образцами.— Прим. перев.
Приборы 29
Многощелевые спектрометры
Многощелевые спектрометры, предложенные в 1949 г. Голеем
[30], никогда не находились в серийном производстве. Многощелевая
схема основана на прямой пропорциональности мощности излучения,
пропущенного спектрометром, числу щелей. Множество узких щелей,
дающих высокое разрешение, можно объединить таким образом, что
энергия, прошедшая через монохроматор, увеличится.
Действие многощелевого спектрометра можно понять, если пред-
представить обычный монохроматор, в котором узкий интервал частот
проходит через выходную щель и попадает на детектор. Для просто-
простоты мы условно считаем, что излучение монохроматично и имеет
частоту v0. Поскольку моиохроматор стигматичен при v0, излучение,
проходящее через входную щель, будет попадать на соответствую-
соответствующую точку на выходной щели. Например, если нижняя часть входной
щели закрыта, то у выходной щели будет затемнена верхняя
часть. Представим вторую входную щель, также освещенную источ-
источником света,. находящуюся в плоскости первой щели и смещенную
в сторону на малое расстояние А. Пучок излучения из второй вход-
входной щели с той же частотой v0 будет попадать на вторую
выходную щель, также смещенную на расстояние d по отношению
к первой выходной щели. При этом энергия, достигающая детектора,
удваивается без потери в разрешении. Теперь возникает проблема:
излучение некоторой другой частоты (не v0) проходит через входную
щель 1 и выходную щель 2, а также через входную щель 2 и выход-
выходную щель 1. Каким же путем необходимо закодировать излуче-
излучение от каждой щели так, чтобы спектрометр реагировал только
на тот свет, который прошел через соответствующие входную и
выходную щели? Голей решил эту проблему, создав систему щелевых
вырезов во вращающихся дисках, которые действовали и как щель, и
как прерыватель. Прерыватель был сконструирован таким образом, что
частота прерывания нежелательного излучения (например, пропущенного
входной щелью 1 и выходной щелью 3) отличалась от частоты модуля-
модуляции полезного излучения (например, прошедшего через входную щель 2
и выходную щель 2) и выделялась соответствующим усилителем.
Спектр сканировался как обычно — вращением зеркала Литтрова в
монохроматоре. Голей демонстрировал 10-кратиое увеличение пропуска-
пропускающей способности против теоретического роста в 32 раза при 64 щелях.
Увеличение было меньше ожидаемого из-за частичного перекрывания
пучка осью прерывателя и других механических потерь света.
Растровые спектрометры
Развивая работы Голея, Жирар [27 — 29] модифицировал вход-
входную и выходную апертуры таким образом, что прозрачные и
непрозрачные элементы создавали регулярную картину (растр). Один из
30 Глава 2
Рис. 2.7. Гиперболический растр Жирара [28, 29].
растров показан на рис. 2.7. В данном случае цель остается преж-
прежней — повысить количество энергии без потери в разрешении. На
месте щелей расположены входной и выходной растры. Растры про-
пропускают 50% падающего света, и один из них находится в колебательном
движении (например, смещается вертикально), так что при этом
модулируются только соответствующие элементы входящего и выходя-
выходящего лучей. Для правильно рассчитанных растров добиваются более
чем 100-кратного увеличения светосилы.
Можно отметить ряд проблем, связанных с растровыми спектро-
спектрометрами. Во-первых, входной и выходной растры должны быть изго-
изготовлены с высокой точностью, но не совсем идентичными; в расчет
необходимо принять искажения в спектрометре. Во-вторых, некоторые
кюветы и приставки (например, многоходовые газовые кюветы или
микрокюветы) могут создавать неоднородность в световом пучке и
уширять аппаратную функцию, в результате чего происходит потеря
спектральной чистоты. В-третьих, из-за большого количества немодули-
рованного излучения, достигающего приемника, любые колебания, осо-
особенно в области частот колебаний растра, приводят к шумам.
Теория, преимущества, приложения и проблемы растровых спектро-
спектрометров обсуждены Морэ-Бэйли [62, 1, 2, W].
Приборы
Рис. 2.8. Характерные спектры пропускания различных типов интерференци-
интерференционных фильтров.
а — узкополосный фильтр; 6 - полосовой фильтр; в - отрезающий фильтр, пропускающий коротковолно-
коротковолновое излучение; г — отрезвющий фильтр, пропускающий длинноволновое излучение [79, с. 173]. Courtesy
of Marcel Dekker, Inc.
Недиспергирующие приборы
Спектрометры
с использованием даэлектрического
фильтра
Сложная вакуумная технология нанесения на прозрачную под-
подложку покрытий из чередующихся слоев диэлектриков с различаю-
различающимися показателями преломления сделала доступными полосовые,
отрезающие и узкополосные фильтры (рис. 2.8). Так как фильтры
изготовлены из диэлектрических материалов, то они не поглощают
падающего излучения, а только отражают и пропускают его. Узкопо-
Узкополосные фильтры можно изготовлять такими, что они будут иметь
совсем узкую полосу пропускания и прекрасно отражать остальные
частоты. Искусно изготовленный узкополосный фильтр на подложке
в виде диска, в котором частота пропускаемого излучения изме-
изменяется по окружности непрерывно и одинаково, можно использовать
в качестве монохроматора. В этом случае сканирование осущест-
32
Глава 2
Рис. 2.9. Схема спектрометра с диэлектрическим фильтром.
Излучение от источника 5 проходит через модулятор С, медленно вращающийся фильтр в виде
с переменным пропусканием, щель 5lf кюветное отделение 55 и попадает на приемник D.
вляется медленным вращением диска. При этом не требуется ни
призм, ни решеток. Таким образом, реализуется компактный, относи-
относительно простой ИК-спектрометр (рис. 2.9). Прибор достаточно портати-
портативен и может быть использован в полевых условиях. Спектрометр
для количественного анализа на основе диэлектрического монохроматора,
управляемый микро-ЭВМ, впервые описан в работе [81].
Перестраиваемые лазеры
Малая ширина полосы (в пределах от 1 до 10~6 см) и высокая
концентрация мощности перестраиваемых лазеров столь привлека-
привлекательны, что, без сомнения, в ближайшие несколько лет появятся
ИК-спектрометры нового поколения для специальных целей. Главными
системами, используемыми в настоящее время, являются лазеры на
полупроводниковых диодах (ПД), лазеры с переворотом спина с
использованием комбинационного рассеяния (ПСКР) и параметрические
генераторы (ПГ).
Полупроводниковые диодные лазеры (рис. 2.10) представляют
собой кристаллы, такие, как РЬ^^пДе, PbSi-^Se* и Pb^Cdi-^S.
Их стехиометрический состав и относительная концентрация составных
частей рассчитаны таким образом, чтобы получить излучение в полосах,
попадающих в среднюю ИК-область. Каждая полоса имеет ширину
около 40 см, но доступны не все частоты внутри этого интер-
интервала. Каждый непрерывный участок полосы может быть смещеи на
1 —2 см изменением тока диода. Кристаллик диода-полупроводиика
должен эксплуатироваться при низких температурах, и перестройка
номинальной длины волны излучения осуществляется изменением
температуры..
Лазеры с переворотом спина с использованием комбинационного
рассеяния (ПСКР) с накачкой лазером на СО2 [67] имеют довольно
Приборы
33
Рис. 2.10. Устройство полупроводникового лазера на р — и-переходе.
7— р-добавки: 2 —контакт; 5 - п-добавки; 4 - отполированная граяь; 5 —выход излучения [40]. Repro-
Reproduced with permission of Chapman and Hall, Ltd.
широкий интервал перестройки частот. Это происходит при варьировании
магнитного поля, в которое помещено все устройство и которое также
требует охлаждения до низких температур. Спектральные ширины
наблюдаемых полос менее 0,05 см. Спектрометр с лазером ПСКР,
смонтированный на аэростате, был использован для изучения содержа-
содержания NO в стратосфере при концентрациях порядка одной части на
миллиард [66].
В устройствах с параметрической генерацией используется лазерное
возбуждение и перестройка частоты происходит при изменении темпе-
температуры. Область перестройки частот этих лазеров и ширина полосы
обычно больше, чем для лазеров ПД и ПСКР [3].
Имеется ряд обзоров, в которых обсуждаются лазеры, перестраи-
перестраиваемые в ИК-области [1, 16, 41, 56, 65, 83, 11, 12}.
Оптико-акустические приборы
Оптико-акустическая спектроскопия является методом, родственным
с предыдущими в том отношении, что в качестве источника света
в анализаторе используется лазер с перестраиваемой частотой. Лазер-
Лазерный луч, промодулированный со звуковой частотой, направляют в
камеру образца, в одну из стенок которой вмонтирован чувствительный
емкостный микрофон. Когда частота модуляции излучения лазера
соответствует частоте полосы поглощения газа в кювете, газ, нагреваясь,
расширяется, при этом возникают колебания давления с частотой
модуляции. Эти колебания давления регистрируются емкостным микро-
микрофоном. Метод крайне чувствителен; он позволяет при подходящих
условиях обнаруживать концентрации порядка нескольких частей на
миллиард, а при удачных обстоятельствах и даже меньше [9, 22, 23,
54, 55].
3 - 586
34 Глава 2
Спектрометры
с пространственным
детектированием
Приборы с пространственным детектированием можно предста-
представить как ИК-спектрографы с множеством детекторов. (В спектрогра-
спектрографах излучение всех длин волн фокусируется на приемнике. В от-
отличие от них монохроматоры сконструированы так, что от аберраций
свободна только та часть излучения, которая проходит через выход-
выходную щель.) В одном из таких приборов, который был построен,
приемник представлял линейный ряд пироэлектрических детекторов,
а сканирование осуществлялось электронным лучом, аналогично тому
как это происходит в телевизионной камере. Из-за сложности устрой-
устройства прибор стоит дорого и сложен по конструкции, но он идеально
подходит для скоростной спектроскопии, так как не имеет подвиж-
подвижных элементов, за исключением электронного пучка.
Многоканальные спектрометры
Термин многоканальный (мультиплексный) заимствован из теории
связи, где он означает систему передачи многих потоков информации
одновременно по одному каналу. Многоканальные спектрометры привле-
привлекательны своей способностью использовать энергию ИК-излучения
гораздо эффективнее, чем более распространенные спектрометры с по-
последовательным (по времени) диспергированием. Выигрыш у многока-
многоканальных спектрометров или «выигрыш Фелжетта» [24] обусловлен тем,
что длины всех волн измеряются одновременно, т. е. отсутствует вы-
выходная щель (которая задерживает приблизительно 99,9 % излучения).
Преимущество многоканального спектрометра можно количественно
оценить и с другой точки зрения. В экспериментальном изме-
измерении, характеризующемся случайным шумом, отношение сигнал/шум
можно повысить, повторяя измерения N раз. Полезный сигнал будет
увеличиваться пропорционально числу измерений N, но шум, частично
усредняясь, возрастет только в yN раз. Таким образом, выигрыш
составляет в yN раз. Действительно, в многоканальной спектроско-
спектроскопии проводят многократные измерения интенсивности каждого раз-
разрешаемого элемента и при этом ожидают существенного роста
отношения сигнал/шум. Такое рассуждение справедливо для де-
детекторов, в которых шум не зависит от уровня сигнала, как, на-
например, в случае тепловых приемников, но не фотоэлементов [59].
Результирующий выигрыш энергии можно использовать для увеличения
скорости регистрации, уменьшения отношения сигнал/шум или при
анализе меньших образцов. Декодирование сигнала обычно является
сложной, но выполнимой посредством ЭВМ операцией.
Приборы
35
10 11
Рис. 2.11. Спектрометр с преобразованием Адамара с 2047 щелями (опти-
(оптическая схема Черни — Тернера).
1 — полевая диафрагма в вп и- шели G7,748 мм х 1,0 мм); 2 — 90е-уголковый зеркальный отражатель;
3 — входящий луч; 4 — выходящий луч; 5 - маска в виде матрицы с общим числом щелей 2047, на-
находящихся в фокальной плоскости; 6 — дифракционная решетка, 150 штрих/мм, угол блеска в области
2,0 мкм; 7 - спектрограф Черни— Тернера; * — ИК-приемиик PbS с активной площадью 1,0 мм2;
9 —к усилителю и записывающему устройству; 10 - регулируемая щель; // — оптический модулятор
Тунинга - Форка, частота 800 Гц [21]. Reproduced with permission of The American Chemical Society.
Диспергирующие спектрометры:
спектрометры
с преобразованием Адамара
Оптическая схема спектрометра, действие которого основано на
преобразовании Адамира, показана на рис. 2.11. Необходимо заметить,
что монохроматор имеет вполне обычный вид вплоть до щели,
ограничивающей поле спектра. В этом месте излучение разложено в
спектр (подобно невидимой радуге) вдоль плоскости диафрагмы маски.
Однако вместо ограниченного потока излучения, вырезаемого выходной
щелью, через щелевую полевую диафрагму и многощелевую маску,
изготовленную в точном соответствии с картиной, определяемой
матрицей преобразования Адамара, проходит весь интервал длин
волн. Каждый прозрачный или непрозрачный прямоугольный участок
маски соответствует элементу разрешения спектрометра. Упрощенная
схема маски Адамара и соответствующая матрица показаны на рис.
36
Глава 2
Спектральная
диафрагма
Даижение
'НТПН
О 1 О
1
1 1
О 1 1
О 1 1
1 1
о
О 1
О 1 1
10 111
Рис. 2.12. Простая кодирующая маска Адамара с семью элементами и
соответствующая ей матрица Адамара.
а - ширина элемента. Чтобы продемонстрировать связь с маской, матрица представлена в косой фор-
форме [53]. Reproduced, with permission, from Spectrochimica Acta.
2.12. Спектр сканируется перемещением маски вдоль горизонтальной
плоскости шаговым двигателем, так что каждый участок спектра или
беспрепятственно проходит, или блокируется «щелью» маски (рис. 2.12).
Далее, как обычно, излучение собирается на приемнике и сигнал
усиливается. В одной из конструкций излучение «дедиспергируется»
(дисперсия вычитается), проходя через монохроматор в обратном на-
направлении, и попадает на детектор, расположенный за входной щелью
(рис. 2.11). При отсутствии в кюветном отделении образца, а следо-
следовательно, и поглощения в процессе перемещения маски около вы-
выходного отверстия сигнал не изменяется. В случае поглощающего
образца, когда через прозрачную часть маски проходит участок длин
волн, соответствующий полосе поглощения, детектор «чувствует» из-
изменение в сигнале, а при попадании излучения на непрозрачную часть
маски изменения сигнала на детекторе не происходит. Выходящий с
детектора сигнал, который соответствует сумме всех длин волн, про-
прошедших через спектрометр, направляется в память ЭВМ, декодируется
и выдается в виде графика «длина волны — интенсивность» (т. е.
спектра) [64]. Система называется «спектрометром Адамара» из-за
того, что маска в выходной фокальной плоскости сконструирована в
соответствии с матрицей Адамара [77]. Если спектр содержит N
Приборы 37
разрешаемых элементов (например, 2000 единиц при разрешении в
1 см в интервале 2000 — 4000 см), то в результате сканиро-
сканирования возникает система N уравнений, последующее решение которых
определяет спектр.
Преимущества спектрометра с преобразованием Адамара (СПА)
включают достоинства мультиплекса, т. е. гораздо более высокое
пропускание по энергии, чем у дифракционного спектрометра (чис-
(численно в J/JV/2 раз), одновременную фиксацию всех длин волн, в том
числе и интересующих исследователя, и использование хорошо раз-
разработанной техники сканирующих спектрофотометров. Недостатками
СПА являются необходимость применения ЭВМ для декодирования
данных, ошибки в кодировании, возникающие из-за оптических аберра-
аберраций, и отсутствие контроля за температурой маски. Кроме того,
эффективность дифракционной решетки высока только в относительно
ограниченном интервале, поэтому охват полного спектра практически
невозможен.
Рассчитано, что в СПА с 255 щелями отношение сигнал/шум
увеличивается в 8 раз [20]. Был построен спектрометр с 2047 щелями
[21], но о его характеристиках не сообщалось.
Утверждалось, что СПА по своей природе превосходит интерферен-
интерференционный спектрометр, но Гиршфельд и Винтьес [44] считают, что
на самом деле верно обратное. Это заключение было проверено
Мибурном [61], который рассчитал относительные характеристики для
восьми типов спектрофотометров.
Однако при спектрофотометрировании отдельных узких спект-
спектральных интервалов СПА сохраняет преимущества многоканальных
спектрометров, избегая некоторых недостатков интерференционного
ИК-спектрометра.
Недиспергирующие спектрометры:
спектрометры на основе интерферометров
Спектрометр на основе интерферометра имеет ряд важных пре-
преимуществ перед диспергирующими спектрометрами. Некоторые из
них внутренне присущи самой конструкции, а другие обусловлены
тем, что данные помещаются в цифровом виде в память ЭВМ.
Принцип конструкции довольно прост (рис. 2.13). Интерферометр*
состоит из фиксированного и подвижного зеркал и светоделителя.
Источник ИК-излучения и приемник вместе с соответствующей оптикой
образуют спектрометр.
Работу прибора можно понять, обратившись к рис. 2.13 и 2.14
(более подробные объяснения даны в других работах [36, 37, 45, 58]).
* Здесь речь идет об интерферометре Майкельсона, однако существуют
и другие типы интерферометров. — Прим. перев.
38
Глава 2
Рис. 2.13. Оптическая схема ИК-иитерферометра.
/-предельные положения поддижного зеркала; 2 - неподвижное зеркало (М{1; 3 — подаижное зеркало
0М2); 4-приемник; S — светоделитель (Mr); tf-источник. Courtesy Block Engineering, Inc.
Л
ЛЛЛЛАЛЛЛЛ,
Рис. 2.14. Выходной сигнал с приемника интерферометра в зависимости
от смещения зеркала х для монохроматического источника (а) и для
идеального полихроматического источника (б). В положении 0 оба плеча
интерферометра имеют одинаковую длину [45]. Reproduced, with permission,
from Applied Spectroscopy.
Рассмотрим случай монохроматического источника. Половина излу-
излучения источника, отражаясь от полупрозрачного зеркала светоделителя,
направляется на зеркало М1( отражается и вновь через светоделитель
попадает на приемник. Другая часть излучения проходит сквозь све-
светоделитель к зеркалу М2, отражающему его обратно к светодели-
Приборы 39
телю, который частично отражает излучение на приемник. Когда
оптические пути обоих плеч интерферометра равны, два пучка интер-
интерферируют с усилением амплитуды. Если зеркало М2 смещено в каком-
либо направлении на расстояние Х/4, то путь излучения в этом
плече изменяется на 2А./4 или Х/2 и оба пучка, интерферируя, взаимно
ослабляются. По мере движения зеркала М2 на расстояние А./4 периоди-
периодически возникает интерференция с усилением или ослаблением излу-
излучения. Разница длин оптических путей двух плеч интерферометра на-
называется разностью хода. В ИК-интерферометрах максимальная
разность хода лежит обычно в пределах от 2 до 21 см. По мере
движения зеркала М2 приемник «видит» световой пучок, интен-
интенсивность которого меняется по косинусоидальному закону, как это
показано на рис. 2.14, а. Если / (х) - интенсивность света, попадаю-
попадающего на приемник, х — смещение зеркала в сантиметрах, В (v) -
интенсивность источника как функция частоты v в см, то уравнение
для сигнала имеет вид
I(x) = B(v)cosBnxv). B.2)
Если теперь добавить второй монохроматический источник, имею-
имеющий другую частоту, то результирующая зависимость в координатах
«положение зеркала — интенсивность» будет выглядеть так, как показано
на рис. 2.14, а, за исключением того, что пики будут касаться оси
абсцисс при других положениях. Приемник будет «видеть» сумму двух
косинусоидальных волн, или
1{х) = B(v1)cosBnxv1) + B(v2)cosBrocv2). B3)
Если добавлять третью, четвертую и т. д. до бесконечного числа
частот (т. е. рассматривать полихроматический источник), то в приемнике
возникает сигнал от суммы косинусоидальных волн:
+ х
1(х)= | B(y)cosBnxv)dv. B.4)
- оо
Этот сигнал будет в основном постоянным для большинства
положений подвижного зеркала, но, когда оба плеча интерферометра
имеют одинаковую длину оптического пути, все косинусоидальные
волны находятся в фазе и на интерферограмме будет наблюдаться
«центральный всплеск» (рис. 2.14,6). (На практике идеально полихрома-
полихроматического источника не существует, так как всегда имеется некоторое
атмосферное поглощение. Поэтому в области крыльев интерферограммы
будут наблюдаться затухающие колебания — «вигли» *.)
Чтобы понять, как возникает интерферограмма и как происходит
ее декодирование (расшифровка) при наличии полос поглощения, можно
использовать сходные рассуждения. Предположим, что теперь из поли-
* От английского "wiggles" — звон, колебание. - Прим. перев.
40
Глава 2
Рис. 2.15. а — интерферограмма излучения черного тела, прошедшего через
полистирол; 6 — ИК-спектр полистирола, полученный по однолучевой схеме
[46]. Reproduced with permission of American Chemical Society.
хроматического источника вычитается одна частота (т. е. вводится
полоса поглощения). Вдоль интерферограммы возникает обратная коси-
нусоидальная волна с этой частотой. Вычитание второй частоты
приведет к вычитанию второй косинусоидальной волны и т. Д.
Результирующая интерферограмма будет синтезирована из всех частот,
за исключением тех, что поглощены образцом. Типичная интерфе-
интерферограмма и результирующий спектр полистирола, полученный на
однолучевом приборе, показаны на рис. 2.15.
Уравнение B.4) является одним из двух уравнений преобразования
Фурье по косинусам. Второе уравнение имеет следующий вид:
B(v)= J I(x)cosBnxv)dx.
B.5)
Эти два уравнения определяют взаимосвязь между интерферограм-
мой и спектром. Преобразование интерферограммы в спектр произво-
производится на ЭВМ с использованием алгоритма преобразования Кули-
Тьюки [18, 25]. Для расчета спектра в интервале 400 — 3800 см с
разрешением 0,5 см требуется примерно 16000 машинных слов.
Поскольку на практике протяженность интерферограммы не может
быть от + оо до — оо, ее ограничивают, используя подходящую сгла-
сглаживающую математическую функцию. В случае прямоугольной функции,
представленной на рис. 2.16,6, получается после фурье-преобразования
косинусоидальная волна, а результирующая функция имеет вид, показан-
показанный на рис. 2.16,г. Боковые сигналы могут быть подавлены умножением
Приборы
41
лллАллллл/
v V v \J\\J v v v v v+j
-•**
¦*'
V
¦•*»
Л
4-,
Прямоугольная апоОизация
Рис. 2.16. Влияние прямоугольной сглаживающей функции.
а — бесконечная косинусоидальиая волна; б - сглаживающая функция; в — косинусоидальная волиа, полу-
чающаяся в результате перемножения функций а и б; г — изображение преобразования Фурье сгла-
сглаженной косинусоидальной волны [45]. Reproduced, with permission, from Applied Spectroscopy.
интерферограммы на аподизирующую* функцию, такую, как треуголь-
треугольная (рис. 2.17,6). Используются и другие аподизирующие функции, от
выбора которых зависят отчасти разрешение, контур полос и фото-
фотометрическая точность измерения поглощения [2, 14, 35, 36].
Приближенный расчет выигрыша энергии в интерферометре по срав-
сравнению с диспергирующим спектрометром дает превосходный резуль-
результат. Мы уже обсуждали выигрыш Фелжетта ]/N, где JV - число
разрешаемых элементов (N = 3400 и yN = 58 для интервала 3800 —
400 = 3400 см при разрешающей способности 1 см). Дополни-
Дополнительно интерферометр имеет более высокий геометрический фактор —
коэффициент, назьшаемый выигрышем Жакино (по имени французского
исследователя), и его появление можно объяснить тем, что входное
отверстие интерферометра имеет круглую, а не щелеобразную форму.
За счет этого от интерферометра ожидается выигрыш в энергии
в 80-200 раз.
Однако наблюдаемый в действительности выигрыш для отношения
сигнал/шум гораздо меньше — порядка 3-10 раз по сравнению с
дифракционным спектрометром. Причины такого расхождения были
систематически исследованы Гриффитсом, Слоаном и др. [38]. Они
* Аподизация — операция уменьшения побочных максимумов за счет выбора
подходящей функции выходного отверстия, на которую умножается интерфе-
рограмма. — Прим. перев.
42
Глава 2
-х /V V V V VIV V V V V \J+*
о
А А А
-X -X, ^ V V v ^*Х\
о
A
Рис. 2.17. Влияние треугольной аподизации.
а - косинусоидальная волна; б — функция аподюации; в - косикусоидальная волна, подвергшаяся аподшации
в результате умножения а на б; г — изображение преобразования Фурье косинусоидальной волны,
подвергшейся аподизацин [45]. Reproduced, with permission, from Applied Spectroscopy.
нашли, что этот выигрыш сложным образом зависит от разрешения,
длины волны, типа приемника и других факторов. Для современных
спектрометров главной причиной указанного несоответствия является
МЭШ (мощность, эквивалентная шуму) приемника на основе тригли-
цинсульфата (ТГС), которая в 20 раз ниже, чем у термоэлемента.
В этой же связи хорошо сконструированный разделитель пучков по
своим параметрам может быть лучше решетки, однако некоторые из
ранее. созданных разделителей менее эффективны. Ниже 600 см
разделитель пучков из Ge/KBr работает хуже, чем решетка. В цик-
циклическом режиме дифракционный прибор превосходит интерферометр.
Суммарный результат действия перечисленных факторов представлен
на рис. 2.18. Другие исследования приводят к аналогичным результа-
результатам [8, 74]. Несомненно, что устранение некоторых из недостатков
интерферометров потребует дальнейшего совершенствования и в ко-
конечном счете их реальное преимущество приблизится к теоретичес-
теоретическому. Считают, что дальнейшее улучшение работы интерферометров
связано с использованием процесса, известного под названием «чир-
пинг»*, т. е. введением преломляющих материалов в одно плечо
интерферометра [72, 73]. Центральный пик интерферограммы таким
* Английский термин "chirping" эквивалентен русскому термину «расщеп-
«расщепление». Чтобы в интерферометре ие происходило потери информации, все
полосы должны иметь близкую амплитуду, соответствующую возможностям
записывающего устройства. С этой целью в каждое из плеч интерферометра
вводят пластинки с различающейся дисперсией. Расщепление улучшает отношение
сигнал/шум, если шум. обусловлен ограниченностью динамического диапазона
записывающей системы [73]. - Прим. перев.
Приборы
43
3
е-
4000
Частота,см"
Рис. 2.18. Вычисленный общий выигрыш (в логарифмической шкале) при
работе на спектрофотометре с преобразованием Фурье (типа «Digilab FTS-14»)
по сравнению со спектрофотометром с дифракционной решеткой (типа «Beck-
man 4240»).
Спектры пропускания получены на двухлучевом приборе с разрешением 2 см (верхние кривые),
4 см" (средние кривые) и 8 см~' (нижние кривые) [38]. Reproduced, with permission, from Applied
Spectroscopy.
образом уширяется. При этом расчеты усложняются, но отношение
сигнал/шум улучшается.
В силу своих уникальных характеристик фурье-спектрометрия
применима к ряду проблем, которые было бы трудно или невозможно
решить на дифракционном спектрометре. Некоторые из этих приложений
описаны более полно ниже, и они включают такие проблемы, требующие
быстрого сканирования, как съемка спектров газовых хроматограмм,
сточных вод непосредственно в потоке или кинетические измерения в
газовой фазе.
44 Глава 2
Явное преимущество фурье-спектрометрии обусловлено тем, что
спектральные данные вводятся в цифровом виде в память ЭВМ и с
ними легко обращаться; шум можно снизить повторным сканиро-
сканированием и усреднением сигнала, спектры можно умножить на коэффи-
коэффициент пересчета, разделить или вычесть из другого спектра и т. д.
Однако дифракционные спектрофотометры, оборудованные ЭВМ для
обработки данных, могут выполнять те же операции [10, 60]. Некоторые
из этих «трюков» требуют превосходной воспроизводимости по длинам
волн — порядка 10 см [42, 43].
Одним из недостатков фурье-спектрометрии является потребность
в очень точных, а поэтому дорогостоящих деталях интерферометров;
например, наклон подвижного зеркала в процессе сканирования не
должен изменяться больше чем на половину длины волны [34].
Для преобразования интерферограммы необходима также ЭВМ,
и трудности с обслуживанием в случае неисправности могут создавать
препятствия в работе для спектроскопистов, привыкших к дисперги-
диспергирующим спектрофотометрам. Спектральный интервал, хотя и достаточ-
достаточный, ограничен обычной областью D00 — 3800 см), и из-за понижения
эффективности светоделителя работа прибора ухудшается (т. е. увели-
увеличиваются шумы) вблизи пределов этого интервала. Различные спект-
спектральные области требуют различных светоделителей. Интерференцион-
Интерференционный спектрофотометр всегда сканирует полный спектр, и на каждую
длину волны затрачивается рдйнаковое время; в дифракционном спект-
спектрофотометре использование замедлителя скорости позволяет скани-
сканировать быстрее или пропускать те области спектра, которые не пред-
представляют интереса или где поглощение отсутствует. Ложный электри-
электрический сигнал или пропущенная точка может оказать заметное влияние
на спектр, что проявляется в виде искажения контуров полос или потери
разрешения. Если отсутствует необходимая оптическая или электрическая
фильтрация [46], то при интегральном преобразовании (свертке) может
возникнуть ложное спектральное поглощение (в английской терминоло-
терминологии "aliasing" или "folding"). В монографии Гриффитса [36] имеется
хорошее обсуждение ИК-спектроскопии с преобразованием Фурье (см.
также [/, 10, 11, 14, 15]).
Выбор спектрофотометра
Существуют разнообразные конструкции серийных приборов, и
выбор данного спектрофотометра определяется в первую очередь за-
задачами, которые будут на нем решаться*. Другие критерии включают
доступность, качество обслуживания и индивидуальный вкус спектро-
спектроскописта-исследователя.
Если, например, рассчитывают использовать прибор для общего
* В настоящее время в СССР производятся ИК-спектрофотометр ИКС-29,
спектрометры ИКС-31 и ИСМ-1.- Прим. перев.
Приборы 45
качественного и количественного анализов, а также для исследования
строения молекул, то можно выбрать двухлучевой спектрофотометр с
довольно высокой степенью сложности. Если прибор будет использо-
использоваться химиком-органиком для идентификации веществ, подобно аппа-
аппарату для определения температуры плавления, то требуется мини-
минимум сложности. Для рутинного количественного анализа существуют
дешевые «ИК-колориметры» с низким разрешением, шкалой ординат
высокой точности и встроенной микро-ЭВМ. Если предполагается
выполнение широкого круга сложных работ, то следует рассмотреть
возможность приобретения интерференционного спектрометра. Гриф-
Гриффите [34] проанализировал три ситуации, когда ИК-интерферометрия
значительно превосходит дисперсионную спектроскопию: 1) задача, в
которой требуется разрешение большого числа элементов (например,
спектр с высоким разрешением в широком интервале), 2) необ-
необходимость быстрого сканирования, 3) регистрация таких слабых сиг-
сигналов, что для записи спектра требуется неприемлемо длительное
время (например, сильно и слабо поглощающие образцы). Произво-
Производительность интерференционного спектрометра может быть заметно
выше, когда нужно проанализировать большое число образцов, требую-
требующих минимальной предварительной подготовки. Интерферометр не имеет
особого преимущества при простой регистрации спектров разнообраз-
разнообразных образцов, требующих различных методов подготовки. Он не имеет
также никаких преимуществ при рутинном анализе в ограниченных
спектральных областях. При таких условиях диспергирующие спектро-
спектрофотометры предпочтительнее [8].
Использование нового ИК-прибора обычно быстро выходит за
пределы тех задач, которые предполагалось решать вначале. По-
Поэтому цена не является решающим фактором. Обычно целесообразно
приобретать прибор, возможности которого шире потребностей се-
годняшего дня.
Принципы выбора
режима работы
спектрофотометра
Владеть хорошим ИК-спектрофотометром еще не значит иметь
хорошие спектры. Даже если допустить, что прибор находится в опти-
оптимальном режиме работы, определяющее значение для получения ре-
результатов имеет квалификация оператора. Можно с уверенностью
утверждать, что, по-видимому, 90% всех ИК-спектрофотометров (за
исключением простейших моделей) не эксплуатируются в своем наи-
наилучшем режиме работы. Перед обсуждением вопросов оптимизации
работы приборов определим некоторые специальные термины, которые
важны для понимания следующих разделов.
46
Глава 2
s/N = 500
250 40
Нуль
Рис. 2.19. Отношение сигнал/шум для нескольких уровней шума.
Определения
специальных терминов
Шум. Беспорядочные изменения сигнала создают шум. Шум изме-
измеряется величиной среднеквадратичного отклонения (СКО) пера самописца
от среднего положения за какой-то период времени. Уровень сред-
среднеквадратичного шума составляет около 25 % от величины максимальной
амплитуды шума (рис. 2.19). Шум является результатом ряда процес-
процессов: 1) джонсоновского шума, или теплового движения электронов в
приемном элементе; 2) статистических тепловых флюктуации в
элементе; 3) беспорядочного движения электронов в проводниках
и других компонентах цепи усиления; 4) ложных электрических сигна-
сигналов, возникающих в неисправных деталях усилителя или из-за плохой
его конструкции; 5) электрических сигналов, возникающих вне прибора.
Для хорошо сконструированного спектрофотометра при правильной
настройке шум в самописце будет в значительной степени джонсонов-
ским, который подчиняется соотношению
где к — постоянная Больцмана, Т— температура, R — сопротивление
детектора, Д/— ширина полосы пропускания частот (которая изменя-
изменяется обратно пропорционально времени отклика).
Разрешение. Это наименьший интервал частот или длин волн,
различимый при данных условиях. В диспергирующем спектрофотометре
Приборы
47
Рис. 2.20. Некоторые возможные аппаратные функции спектрофотометра.
Геометрическая ширина щели указана на половине высоты максимума энергии.
Рис. 2.21. Влияние конечной ширины щели монохроматора на контур по-
полосы.
а- истинный контур полосы; б —аппаратная функция; « — наблюдаемый контур полосы.
I A/2
С
S
Частота
Рис. 2.22. Ширина полосы поглощения, измеренная на половине высоты
оптической плотности.
48
Глава 2
оно зависит от разрешающей силы диспергирующего элемента, ши-
ширины щели, конструкции и юстировки оптической системы. В интерфе-
интерференционном спектрофотометре разрешение зависит от максимальной
разности хода, расходимости пучка, функции аподизации и, конечно,
качества оптики.
Постоянная времени. Для системы с критическим затуханием —
это время, необходимое для того, чтобы после мгновенного воздей-
воздействия постоянного по величине излучения сигнал на выходе достиг
1/е от своего предельного значения (где г — основание натурального
логарифма).
Время отклика. Время отклика пера самописца — это время, необхо-
необходимое для пробега пером всей шкалы при условиях слабого затуха-
затухания. Численно оно примерно равно учетверенной постоянной времени.
Отношение сигнал/шум. Это отношение отклонения на полную
шкалу к среднеквадратичной величине шума (рис. 2.19).
Ширина щели (геометрическая). Это величина раскрытия щелей,
измеренная в микро- или миллиметрах.
Ширина щели (спектральная). Это интервал частот, пропущенных
монохроматором через выходную щель, измеренный на половине
максимальной интенсивности. Спектральная ширина щели для диспергиру-
диспергирующего спектрофотометра может быть вычислена из геометрической [13]
при условии, что известны некоторые параметры, характеризующие
прибор.
Аппаратная функция. Это распределение интенсивности в зависимости
от частоты для излучения за выходной щелью, если входная щель
освещена монохроматическим светом. Некоторые из идеальных аппарат-
аппаратных функций показаны на рис. 2.20. Реальная аппаратная функция
спектрофотометра может быть определена в результате регистрации
узкой полосы поглощения с известным доплеровским контуром [48,
3, 16, 77]. Этот метод свободен от тех ошибок, которые возникают
при определении аппаратной функции путем сканирования излучения
такого когерентного источника, как лазер. Неправильно съюстированный
спектрофотометр может давать асимметричную аппаратную функцию
(рис. 2.20, в) или даже с несколькими максимумами. Наблюдаемый
контур полос является сверткой аппаратной функции и естественного
контура полосы (рис. 2.21).
Ширина полосы. Это ширина полосы в единицах волнового
числа или длины волны, измеренная на половине высоты макси-
максимальной оптической плотности (рис. 2.22). Иногда ее называют полу-
полушириной полосы.
Нуль. Положение пера, достигаемое при перекрывании энергии
в канале образца. Для каждой длины волны имеются истинная
нулевая точка и ложный нуль, который отличается от первой количеством
рассеянного излучения.
Рассеянное или паразитное излучение. Это излучение нежелательных
частот, попадающее на приемник. Наиболее общими причинами рас-
Приборы 49
Таблица 2.1а
Полосы поглощения, пригодные для измерения уровня рассеянного света [52]
Интервал волновых Интервал длин Вещество* Толщина поглощаю-
чнсел, см волн, мкм оещст,тво шего слоя, мм
3600-3620
3260 - 3335
3045-3075
2380-2460
1675-1760
1420-Г 590
1190-1240
1120-1125
880-940
740-800
645-700
а Все измерения,
2,76-2,78
3,00-3,07
3,25-3,28
4,07-4,20
5,68-5,97
6,30-7,05
8,07-8,42
8,90-8,93
10,6-11,4
12,5-13,5
14,3-15,5
Феиол
Фенилацетилен
Хлористый метилен
Хлороформ
Апетон
Сероуглерод
Хлороформ
Тетрахлорэтилен
»
Четыреххлористый углерод
Бензол
хроме фенола, выполнены для неразбавленных жидкостей.
20,0 @,05 М
раствор)
0,5
1,0
10
0,5
1,0
1,0
1,0
1,0
0,1
0,5
Таблица 2.16
Материалы окон для измерения уровня рассеянного света"
Интервал волновых Интервал длин волн, п Толщина поглощающего
чисел, см мкм вещество слоя мм
1000-2000 5,0-10,0 SiO2 (кварц) 7
700-1200 8,5-16,0 LiF 7
500-900 11,0-20,0 CaF2 7
250-500 20,0-40,0 NaCl 14
250-300 33,0-40,0 KBr 14
a Данные из инструкции по обслуживанию прибора «Perkin - Elmer 521».
сеянного излучения являются царапины, загрязнения, мельчайшие не-
несовершенства зеркал или диспергирующих элементов, спектры неже-
нежелательных порядков от решеток, отражения от поверхностей внутри
монохроматора, нагревание щелей или образца, нагревание или загрязне-
загрязнение модуляторов.
Приближенное измерение уровня рассеянного света может быть
выполнено несколькими способами. В одном из них используется такой
толстый слой образца, чтобы измеряемая полоса имела фактически
полное поглощение. Этот метод не точен, так как сам образец погло-
поглощает некоторую часть коротковолнового излучения, которое могло бы
внести вклад в рассеянную энергию. Могут быть также записаны
4-586
50 Глава 2
спектры некоторых веществ, имеющих очень интенсивную, резкую полосу
поглощения в области нужных частот (табл. 2.1а). В другом обще-
общепринятом методе используется окно из стекла, кварца, LiF, CaF2, NaQ
или КВг, которое пропускает высокочастотное излучение (источник
большей части рассеянного света), но отсекает частоты несколько
более высокие, чем частота исследуемой полосы (табл. 2.1 б). Если
зарегистрировать сигналы от такого окна, частично пропускающего
ИК-излучение, и от непрозрачной заслонки, то разница между ними
даст уровень рассеянного света в спектральном приборе.
Рассеянное излучение в серийных спектрофотометрах уменьшается
за счет тщательного конструирования и разумного использования
длинноволновых пропускающих диэлектрических фильтров. Ниже
600 см проблема усложняется, так как отношение нежелательного
излучения к полезному становится очень большим.
Оптимизация режима
работы спектрофотометра
Было бы полезно напомнить, что рабочий режим ИК-спектрофото-
метра ограничен энергией и установка режима работы сводится к дости-
достижению оптимального компромисса между величинами, влияющими
на условия записи спектров. Эти условия зависят от того, с какой
целью должен быть снят спектр, и от природы образца. Режимы
работы, которые являются наилучшими в одной ситуации, могут
оказаться непригодными при других обстоятельствах.
При накоплении и сопоставлении значительного числа спектров
необходимо задать набор стандартных режимов работы спектрофото-
спектрофотометра, чтобы все спектры были записаны при воспроизводимых усло-
условиях. Тщательная и воспроизводимая запись спектров рутинных образцов
может показаться напрасной тратой времени, но, как показывает опыт,
многие „рутинные" спектры неожиданно отсутствуют в каталоге эта-
эталонных спектров. Даже если это не так, из хорошего спектра может
быть получена более полная информация, чем из снятого небрежно.
„Обзорный" режим работы спектрофотометра можно использовать
только для контроля толщины образца; спектры на этой скорости
лучше не регистрировать. Последующее рассмотрение „правил работы"
применимо к диспергирующим спектрофотометрам. Для интерферен-
интерференционных приборов это будет сделано позже.
Исследователю, работающему с диспергирующим спектрофотомет-
спектрофотометром, важно понимать сущность трех основных параметров, определя-
определяющих работу прибора, и использовать их для оптимизации регистра-
регистрации спектров. Даже если имеются автоматические программы режимов
работы прибора, оператор все равно должен понимать, какие параметры
изменяются и как эти изменения будут влиять на спектр.
Оператору доступны три основные регулировки: разрешение (ширина
щели), уровень шума (усиление) и скорость сканирования (время
Приборы 51
отклика). Из этих параметров могут быть произвольно выбраны любые
два, тогда третий определяется этим выбором. (При этом полагается,
что искажения в спектре должны быть минимальны.) Хотя сказанное
относится к работе двухлучевых спектрометров с оптическим нулем,
в приборах, регистрирующих электрическое отношение (в которых нет
оптического клина), действуют те же ограничения в отношении
основных переменных.
Ранее (стр. 24-29) упоминалось, что в спектрофотометрах с опти-
оптическим нулем (наиболее распространенном типе приборов, по данным
настоящей книги) перо самописца приводится в движение электромеха-
электромеханической следящей системой. Она состоит из замкнутого контура,
включающего гребенку или оптический ослабитель в канале сравнения,
термоэлемент или другой приемник, усилитель и сервомотор (мотор
отработки), который вводит или выводит оптический ослабитель из
пучка сравнения. Регулировка усиления цепи этой следящей системы
оказывает определяющее влияние на работу спектрофотометра. Если
усиление слишком мало, то система будет реагировать медленно
и неполно, если слишком велико, то сервомотор ослабителя (и перо)
будет давать очень сильные выбросы или даже переходить в режим
автоколебаний. Демпфирование (успокоение) следящей системы можно
регулировать временем отклика, поэтому время отклика и усиление
должны быть подобраны таким образом, чтобы получить наилучший
отклик следящий системы. В свою очередь с временем отклика должна
согласовываться скорость сканирования. При сканировании с большей
скоростью, чем может реагировать перо, ничего не приобретается,
а теряется многое.
На энергию сервосистемы влияет как ширина щели, так и устанавли-
устанавливаемое усиление следящей системы. Эти два параметра должны быть
сбалансированы таким образом, чтобы энергия в следящей системе
поддерживалась на постоянном уровне *. Более высокое усиление исполь-
используется с узкой щелью, и наоборот. Естественно, что большее усиление
приводит к повышению уровня шумов в спектре, а узкая щель —
к лучшему разрешению.
Количественная связь между отношением сигнал/шум S/N, временем
сканирования t и шириной щели w выражается как
S/N = ctU2w2, B.7)
где с — постоянная.
Это уравнение получается примерно следующим образом. Уравнение
B.6) для джонсоновского шума записывают в виде
JV~(A/I/2 = (постоянная времени следящей системы)/2. B.8)
* Ширина щели не остается постоянной в процессе сканирования по спектру.
Закон, по которому автоматически раскрывается щель, называется щелевой про-
программой. Разные щелевые программы соответствуют различным уровням
энергии. — Прим. перев.
52 Глава 2
Так как сигнал пропорционален квадрату ширины щели, или S ~ w2,
то при объединении этих выражений получается уравнение B.7).
Из уравнения B.7) видно, что отношение сигнал/шум увеличивается
пропорционально квадрату ширины щели. Удвоение ширины щели
уменьшает шум в 4 раза (при условии, что усиление следящей системы
уменьшается, чтобы компенсировать большее количество энергии,
достигающей приемника).
Шум можно также уменьшить, увеличив время сканирования. За-
Запишем уравнение B.7) другим способом:
Разрешение ~ 1/w = c't1'* (S/Ы)'1/2. B.9)
Таким образом, чтобы удвоить разрешение, уменьшая ширину щели
в 2 раза, необходимо увеличить время сканирования в 16 раз.
Очевидно, если спектр сканируется настолько медленно, что серво-
сервосистема может следовать за сигналами, то значительное увеличение
разрешения может быть достигнуто только при практически неприем-
неприемлемом увеличении времени сканирования.
Поскольку удвоение разрешения удваивает число спектральных (раз-
(разрешаемых) элементов, то было высказано также соображение о том,
что время сканирования должно быть увеличено в 32, а не в 16 раз
[74]. Однако в зависимости от определения „элемента разрешения",
способа измерения и ширины измеряемого спектра поглощения любой
из двух коэффициентов может считаться правильным.
ИК-спектры, представленные в цифровом виде, могут быть сглажены
в процессе преобразования данных в ЭВМ с помощью усредняющего
полинома со скользящей точкой [85]. Если полезный сигнал скрыт
шумами, то для повышения отношения сигнал/шум можно использовать
метод повторного сканирования с последующим накоплением данных
(стр. 120).
Здесь уместно обсудить скорость сканирования в связи с постоянной
времени электронной системы и скоростью отклика следящей системы.
Назначение полосового фильтра (через который проходят сигналы
с частотой модуляции, а все остальные сигналы задерживаются)
в схеме усилителя состоит в том, чтобы уменьшить случайные
электрические сигналы (шум) и прохождение частоты 50 Гц из сети.
Чем на более узкую полосу частот настроен фильтр, тем ниже
уровень шума, но при этом вся система будет более инертной. Кроме
того, в некоторых схемах устанавливается дополнительный фильтр для
электронного усреднения шума. Обычно эти параметры являются
лимитирующими при определении скорости сканирования, так как
скорость отклика мотора пера самописца и связанной с ними электри-
электрической цепи обычно меньше, чем время отклика усилителя.
При сканировании могут возникать довольно большие ошибки как
в интенсивности, так и в положении максимумов особенно для
острых узких полос. Согласно Стюарту [80], „для регистрации интенсив-
интенсивности с ошибкой меньше 2 % требуется скорость сканирования менее 0,4
Приборы
53
40
30
25
20
15
10
П I I I I I I I I I I 1111 I I I I I Г
1 I
111
III
О 2 4 6 8 10 12 14 16 16 20 22 24
Ширина полосы, см~'
Рис. 2.23. Распределение ширин полос поглощения в спектрах 347 жидких
и твердых образцов, зарегистрированных на спектрофотометре со спектраль-
спектральной шириной щели 1 см-'.
наблюдаемой ширины полосы, отнесенной к постоянной времени. Это
эксперимент, который спектроскописты должны прочувствовать сами,
чтобы помнить о том, что очень быстрое сканирование может привести
к ошибкам". Ширины, наблюдаемые для ряда случайно выбранных
полос поглощения, показаны на рис. 2.23.
На основании этого теперь можно установить „генеральную линию"
поведения при работе на спектрофотометре. Бегло опишем методику,
а затем покажем, по какой системе следует изменять основные пара-
параметры, чтобы удовлетворить такие специальные требования, как высокое
разрешение, быстрое сканирование или высокая количественная точ-
точность. Более полное обсуждение этого процесса дано Поттсом
и Смитом [69,3, 16, 17].
Предполагается, что блоки спектрофотометра функционируют нор-
нормально, а сам прибор эксплуатируется согласно заводской инструкции.
Сначала произвольно выбирается щелевая программа. Она может
быть установлена согласно инструкции или лучше в 1,2 — 1,5 раза шире.
Если в приборе имеется замедлитель скорости, то можно выбрать
время сканирования 1 ч F0 см~х/мин) (позже это время можно сокра-
сократить), если нет, то подходящим временем является 0,5—0,6 ч. Для
54 Глава 2
уменьшения искажений время отклика следящей системы должно быть
согласовано с временем сканирования; за это время необходимо
сканировать не более 0,4 ширины полосы. Так как большинство полос
в спектрах жидкостей и твердых тел имеет ширину по крайней мере
3 см (рис. 2.23), то для времени отклика пера порядка 1 с (время
пробега всей шкалы) подходящим временем сканирования является 1 ч.
Величина усиления подбирается так, чтобы в систему отработки пера
поступало нужное количество энергии. При правильной регулировке
усиления перо „проскакивает" уровень сигнала на 2—4% при частичном
перекрывании и открывании канала образца (этот тест нужно проводить
в той области, где нет атмосферного поглощения, например около
1000 см). Далее проверяется уровень шума. Желателен невысокий
уровень шума (например, ± 0,2 %) как показатель того, что спектрометр
работает с максимальной эффективностью. Гладкий след пера указывает
на „вялость" следящей системы. Столь же нежелателен избыточный
шум, так как он мешает работе схемы автоматического замедления
скорости и искажает спектр.
Если уровень шума слишком высок или слишком низок, то регули-
регулируется щель и цикл повторяется до тех пор, пока результат не станет
удовлетворительным.
Дрейф пера или баланс устанавливается так, чтобы перо не дви-
двигалось при полностью перекрытых пучках (образца и сравнения). Затем
включается автоматическое замедление скорости и сканирование прово-
проводится с обоими закрытыми каналами. При этом замедление увеличи-
увеличивается до тех пор, пока сканирование не прекратится (работа прибора
замедляется уже на шумах), после чего замедление несколько умень-
уменьшают. Теперь время сканирования можно уменьшить в 2 — 3 раза,
и в процессе записи полосы поглощения спектрофотометр автомати-
автоматически будет понижать скорость по отношению к первоначально
выбранной (до 1 см~х/с в предыдущем примере). В дальнейшем для
достижения подходящей скорости записи спектра может оказаться необ-
необходимой некоторая регулировка замедления.
Следующий абсолютно необходимый шаг — проверка правильности
выбранных условий. Образец, спектр которого содержит набор узких и
широких полос, такой, как инден [12, 51], записывают сначала с нор-
нормальной скоростью, а затем со скоростью, составляющей 25% от
нормальной. Оба спектра должны быть в основном идентичны. Если
этого нет, то нужно провести тонкую регулировку, чтобы добиться
приемлемых условий работы.
Однажды установленные стандартные условия работы спектрофото-
спектрофотометра должны периодически проверяться (раз в день в лаборатории,
где работы ведутся интенсивно) записью спектра стандартного образца
(стр. 58). Время от времени может потребоваться некоторая незначи-
незначительная регулировка усиления и времени сканирования. Щелевая про-
программа, однажды установленная, должна поддерживаться строго
постоянной.
1
0,25
0,25
1
2
1
1
2
1
0,5
0,5
1/2
1
1
16
16
4
0,25
Приборы 55
Таблица 2.2
Изменение параметров работы спектрофотометра при постоянном
уровне энергии в системе отработки
Условие Шум Щель Вра",^^?тШ"
Норма
Низкий уровень шума
Низкий уровень шума
Высокое разрешение
Высокое разрешение
Быстрое сканирование
Используя уравнение B.7), можно вычислить значения переменных
параметров, которые встречаются при специальных режимах записи
спектров. Некоторые эквивалентные соотношения между переменными
параметрами приведены в табл. 2.2. В качестве примера представим,
что желательно просканировать часть спектра со шкалой ординат,
растянутой в 10 раз. Если это достигнуто только за счет усиления и
не проведены некоторые компенсирующие регулировки, то шум также
возрастет в 10 раз и никакого преимущества достигнуто не будет. Однако
можно увеличить щель в J/10 раз и отрегулировать усиление, чтобы
сохранился первоначальный уровень шума, а сканирование вести с обыч-
обычной скоростью. В то же время можно повысить время отклика и время
сканирования в A0J = 100 раз или использовать комбинацию регулиро-
регулировок. Можно, например, увеличив щель в 2 раза, увеличить время
сканирования в 4 раза и согласиться на несколько более высокий
по сравнению с нормальным уровень шума. Порядок установки рабочих
параметров для специальных условий обобщен в табл. 2.3.
Правила работы для интерференционных спектрофотометров были
сформулированы и экспериментально проверены Гриффитсом [33]. Для
спектрофотометра любого типа отношение сигнал/шум, измеренное при
данном разрешении, пропорционально квадратному корню из времени
измерения. В интерферометрах с быстрым сканированием время раз-
развертки увеличивается за счет использования дополнительных циклов
сканирования и усреднения сигнала либо непосредственно в интер-
ферограмме, либо в спектре (обычно в первом). В интерферометрах
с медленным сканированием, таких, которые обычно используются
для работы в дальней ИК-области, скорость движения зеркала может
быть понижена, но при этом требуется регулировка постоянной вре-
времени фильтра шумов [52]. Разрешение интерферометра изменяется
за счет изменения разности хода (расстояния, на которое перемещается
подвижное зеркало). Геометрический фактор интерферометра обычно
соответствует такой разности хода, чтобы при приемлемом разрешении
56 Глава 2
Таблица 2.3
Обобщение операции по установлению рабочих параметров регистрации
спектров для специальных условий* Л
Низкий уровень шума
Установить универсальные условия '->
Определить коэффициент понижения уровня шума
Раскрыть щели в (коэффициент) *'2 раз и уменьшить усиление в (коэффициент)
раз или увеличить время отклика и время сканирования в (коэффи-
(коэффициентJ раз
Записать контрольный спектр
Ограниченная энергия
Установить универсальные условия
Определить коэффициент ослабления энергии
Раскрыть щели в (коэффициентI'2 раз или увеличить усиление в (коэффи-
(коэффициент) раз и увеличить время отклика и время сканирования
в (коэффициентJ раз
Записать контрольный спектр
Высокое разрешение
Установить универсальные условия
Определить коэффициент сужения щелей
Восстановить энергию в сервосистеме, увеличив усиление
Увеличить время отклика и время сканирования до получения приемлемого
уровня шума
Записать контрольный спектр
Reprinted with permission from Applied Optics [69].
(например, 2 см *) сохранить оптимальные условия работы. На практике
при измерениях с низким разрешением максимальный теоретический
геометрический фактор не достигается из-за ограничений, налагаемых
размерами источника и приемника. В этих условиях геометрический
фактор не изменяется при изменении разрешения (случай постоянного
геометрического фактора). Для того чтобы извлечь пользу из повышен-
повышенной разности хода в некоторой точке по мере увеличения разрешения,
расходимость пучка должна быть уменьшена (случай переменного
геометрического фактора). Эта операция обычно выполняется измене-
изменением диаметра диафрагмы, помещенной в фокусе осветительной
системы. Эта диафрагма служит той же цели, что и входная щель
монохроматора.
В случае постоянного геометрического фактора (когда площадь
входного отверстия остается неизменной) увеличение разрешения за
счет увеличения разности хода в 2 раза приводит к увеличению
времени измерения также в 2 раза, а отношение сигнал/шум пони-
понижается в J/2 раз. Для восстановления прежней величины отношения
сигнал/шум время измерения должно быть вновь удвоено, таким обра-
Приборы 57
зом, в целом время измерения увеличивается в 4 раза (по сравнению
- диспергирующим спектрометром, в котором требуется 16-кратное
«еличение времени сканирования). Наоборот, уменьшение разрешения
2 раза в случае интерференционного спектрофотометра приводит
« уменьшению времени сканирования в 4 раза, а для диспергирующего
пектрофотометра — в 16 раз. Гриффите [33] проверил это соотношение,
использовав спектрофотометр FTS-14 с разрешением 2 и 4 см.
Ситуация меняется в случае переменного геометрического фактора,
когда при более высоком разрешении расходимость пучка должна быть
понижена. Чтобы увеличить разрешение в 2 раза, необходимо уменьшить
апертуру источника в ]/2 раз, что снижает геометрический фактор
в 2 раза. Восстановление „потерянной" энергии требует увеличения
времени измерения в 2 раза, что повышает уровень шума в у 2 раз,
а это в свою очередь вызывает увеличение времени измерения еще
в 2 раза, а в целом в 4 раза. Разность хода также удваивается, что
приводит к возрастанию времени измерения еще в 4 раза. Следовательно,
общее увеличение времени измерения, как и в случае диспергирующего
прибора, - в 16 раз.
На разрешение также влияет аподизация. Спектры, имеющие при-
примерно одинаковые отношение сигнал/шум и разрешение, можно изме-
измерить при одинаковом числе сканирований, либо используя данную
разность хода и треугольную аподизацию, либо половинную разность
хода и прямоугольную аподизацию. В последнем случае время измере-
измерения вдвое меньше, но у резких полос проявляются побочные макси-
максимумы.
Суммируя, можно сказать, что если интерференционный спектрофо-
спектрофотометр настроен на максимальное пропускание для регистрации спектров
с низким разрешением и сокращения времени сканирования, то усиление
должно быть меньше, чем в аналогичном случае для дифракционного
спектрометра, в 4 раза (другими словами, преимущество интерферо-
интерферометра перед дифракционным спектрометром меньше при низком раз-
разрешении). В то же время если для повышения разрешения интерферен-
интерференционного спектрофотометра нужно понизить геометрический фактор,
то порядок подбора условий точно такой же, как и для дифракцион-
дифракционного спектрофотометра.
Другие факторы,
влияющие на работу спектрофотометров
Мы видели, как фундаментальные физические законы ограничивают
работу ИК-спектрофотометра. Ограничения другого рода могут воз-
возникать из-за разъюстировки оптики прибора, неисправных деталей или
неподходящих окружающих условий.
Для руководства установкой и настройкой ИК-спектрофотометров
согласно заводским техническим условиям заводы-изготовители обычно
присылают инженера по обслуживанию. При этом достигается некото-
58 Глава 2
рое улучшение работы спектрофотометра, но настройка оптики не-
неопытным персоналом почти однозначно приводит к ухудшению работы.
Оптические элементы, если они хорошо установлены с самого начала,
больше не юстируют и в дальнейшем не требуют внимания.
Однако другие детали, особенно электронные лампы, если они
используются, иногда нуждаются в замене. Источники ИК-излучения
постепенно выходят из строя и также требуют периодической замены.
В большинстве инструкций по работе имеются указания на последо-
последовательность выявления неисправностей, которой нужно следовать при
возникновении неполадок. Порядок поиска отказавшей детали заключа-
заключается в последовательной изоляции систем (оптической, механической,
электронной), затем отдельных блоков (усилителя, пера самописца, серво-
сервомотора и т. д.) внутри этих систем и, наконец, неисправной детали.
Неправильная работа может быть вызвана импульсными электрическими
помехами, распространяющимися либо через воздух, либо через цепь
питания. Если окружающая температура относительно не постоянна,
то калибровка прибора по длинам волн, вероятно, может смещаться.
Многие исследователи, использующие двухлучевые спектрофотомет-
спектрофотометры, пренебрегают возможностью почти полного поглощения в некоторых
областях атмосферными парами воды и углекислым газом, особенно
при 2320 и 1400—1700 см, в результате чего перо может стано-
становиться инертным и малочувствительным к оптическим сигналам.
Быстрое сканирование в этих областях может дать спектр с явно
повышенным уровнем шума, так как скорость сканирования сравнима
со скоростью модуляции и два луча „видят" различные части атмосфер-
атмосферных полос поглощения. (Аналогичный эффект можно наблюдать, если
в монохроматоре пути обоих лучей не совсем одинаковы или коли-
количество рассеянного света в двух пучках различно, что возможно из-за
загрязнений зеркал осветителя.)
Для решения проблемы атмосферного поглощения спектрофотометр
продувают сухим воздухом или азотом. Для полного устранения помех
кюветное отделение необходимо заключать в кожух и продувать
быстрым потоком сухого газа. К счастью, такие тщательные меры
предосторожности необходимо применять редко.
В дальней ИК-области (ниже 400 см) проблема осложняется
сильными чисто вращательными полосами паров воды. В этой области
требуется особенно тщательная продувка, а при очень низких частотах
A0—200 см") рационально вакуумирование либо всего спектрометра,
либо его части.
Проверка работы
и калибровка спектрофотометра
Настоятельно рекомендуется для каждой серии спектров проводить
установку стандартных условий работы прибора, причем она должна
быть воспроизводимой и при последующих измерениях того же типа. Если
Приборы
59
11 12
11 12
Длина волны, икм
И 12 11 12
И 12
И 12
и л
и
l V
2.6 см
900800900800900800900 800 900800900800
Частота, см
Рис. 2.24. Влияние спектральной ширины щели на интенсивности полос
поглощения 861 и 903 см-' в циклогексане [75]. Reproduced, with permission,
from Applied Spectroscopy.
оператор выбирает условия сканирования произвольно, то могут про-
происходить невероятные ошибки в интенсивностях, контурах полос и часто-
частотах поглощения. Спектры, полученные при таких условиях, приводят
к дезинформации, что хуже, чем их отсутствие. Влияние ширины щели
на интенсивности полос средней резкости и средней ширины показано
на рис. 2.24.
По мере разработки спектрофотометров с более высоким разреше-
разрешением и универсальностью требования к предельным условиям ра-
работы узлов прибора становятся все более жесткими. Важно часто их
контролировать, чтобы застраховаться от непредвиденного ухудшения
качества спектральных измерений, так как даже наиболее надежные узлы
электроники спектрофотометров могут расстраиваться, источник может
искривляться, а оптические поверхности могут загрязняться и мутнеть.
По-видимому, многие практические операции по выбору режима
работы являются динамическими, т. е. такими, в ходе которых в стан-
стандартных условиях записывается спектр подходящего тестового образца.
При правильном выборе стандарта по одному спектру можно проводить
ежедневную проверку точности длины волны, отношения сигнал/шум,
разрешения, нуля и воспроизводимости поглощения. Достаточно часто
должно проводиться измерение рассеянного света и „гладкости"
линии /0. Другие второстепенные параметры, используемые для характе-
характеристики спектрофотометра, также могут нуждаться в периодической
проверке.
Стандартный образец для калибровки в идеале должен отвечать
следующим условиям: 1) быть стабильным; 2) иметь постоянную или
60
Глава 2
,W\4,-\/
4000
3400
2800
2200
1850
•0,200 MM
-0,025 мм
1800
1600
1400
1200
1000
71*
— 0,025мм 76
— 0,012 мм
900800 700 600500400300200
Рис. 2.25. Спектр индена (см. табл. 2.4). Courtesy Perkin — Elmer Corporation.
воспроизводимую толщину, 3) иметь много полос со значительно
различающимися поглощением и шириной, которые попадают во все
спектральные области, 4) быть легко доступным. Ни одно из веществ
не удовлетворяет всем этим критериям, но тем не менее используется
ряд достаточно подходящих соединений. Широко известным материалом
для калибровки в области 600 — 3400 см является пленка полистирола
(толщиной 0,07 мм), хотя в ее спектре и нет многих полос из тех,
Приборы
61
Таблица 2.4а
Максимумы полос поглощения, рекомендуемых для калибровки |50, 511"
Полоса
i
26
Зв
4В
5В
6
7
8В
9б
106
11"
12б
13б
14б
15В
166
17
18б
19
206
21
22
23
24б
256
26В
27б
28
29б
ЗО6
31
32
33
34
35
36
37В
38б'в
39
40
vBaK, см '
3927,2+0,56
3901,6 + 0,64
3798,9 + 0,86
3745,2+0,72
3660,6 + 0,98
3297,8 ±1,06
3139,5 + 0,44
3110,2 + 0,44
3068,9 + 0,66
3025,4 + 0,26
3015,3 + 0,52
2887,6 + 0,82
2770,9 ±0,44
2673,3 + 0,56
2622,3 + 0,24
2598,4 ±0,16
2525,5 + 0,32
2439,1 + 0,24
2305,1 ±0,42
2271,4 + 0,08
2258,7 ±0,36
2172,8 + 0,30
2135,8 + 0,68
2113,2±0,28
2090,2 + 0,40
2049,1 ±0,82
2027,0 ±0,42
1943,1+0,52
1915,3 + 0,30
Кювета, мм
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,025
0,025
0,025
0,025/0,2
0,025
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
Полоса
41
42
43В
44
45
46В
47
48В
49В
50
51
52В
53
546
55"
56"
57
58"
59
60
616в
62
63
64
65б
66
676
68
69
70б
71
72
73
74
75
76
77
78
79"
vBaK, см '
1885,1 ±0,42
1856,9 ±0,52
1826,8 + 0,56
1797,7 + 0,50
1741,9±0,50
1739,2 ±0,78
1713,4 + 0,66
1684,9+1,14
1661,8 + 0,64
1609,8 + 0,42
1587,5 + 0,26
1574,5 + 0,62
1553,2±0,20
1457,3 + 0,38
1393,5±0,76
1361,1 + 0,16
1332,8 ±0,42
1312,4+0,18
1288,0 ±0,08
1264,0 + 0,12
1226,2 + 0,28
1205,1 + 0,20
1166,1 ±0,08
1122,4 + 0,32
1067,7 + 0,30
1018,5±0,32
947,9 + 0,36
942,4+0,38
914,7 + 0,16
861,3 + 0,14
830,5 + 0,32
765,3 + 0,22
730,3 + 0,22
718,1 + 0,24
692,6+0,56
Кювета, мм
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,2
0,025
0,2
0,2
0,2
0,2
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,025
0,012
0,012
0,012
0,012
а Данная погрешность представляет удвоенную величину стандартного отклонения; пропущенные полосы
были признаны непригодными для калибровки.
Эта полоса может не разрешаться небольшими призменными спектрофотометрами.
в Эти полосы менее пригодны для точной калибровки нз-за асимметрии, наложения спектров паров
атмосферной воды и углекислого газа или по другим причинам.
62 Глава 2
Волновые числа
Номер полосы
1
2
3
4
а Если не указано ocof
Полосы 3 и 8 принадлежат
(в вакууме) в
камфоры
спектре смесн равных
п циклогексанона"
Волновое число, см Номер полосы
592,1
551,7
521,4
490,2+1
5
6
7
8
)о, то точность измерений составляет ±0,5 см
камфоре, полоса 4 — циклогексанону [50].
Таблица 2.46
количеств индепа,
Волновое число, см ~~
420,5
393,1
381,6
301,4
. Толщина кюветы 0,05 мм.
которые требуются. Используется газообразный аммиак B00 мм рт. ст.
в 5-сантиметровой кювете), но его давление в кювете со временем
меняется. Поттс [68] рекомендовал 0,03-миллиметровый слой 1,2-ди-
бромпропана. Было предложено также использовать инден (толщина
слоя 0,02 мм), 77 полос поглощения которого перекрывают интервал
690-4000 см [50, 51] и спектр которого представлен на рис. 2.25
(см. табл. 2.4 а). При правильном хранении чистый инден устойчив [50].
Дополнительные полезные полосы появляются при добавлении к индену
циклогексанона и камфоры (табл. 2.46). В спектре этой смеси имеется
ряд точек, удобных для проверки не только точности шкалы длин волн,
но и других ранее упоминавшихся параметров.
В сборнике спектров и частот Комиссии по молекулярной структуре
и спектроскопии при Международном союзе теоретической и прикладной
химии (ШРАС) [15] приведены данные по калибровке призменных
и дифракционных ИК-спектрофотометров, полностью перекрывающие
область 600—4300 см. Необходимо заметить, что положение любой
полосы, кроме изолированной и совершенно симметричной, будет зави-
зависеть от спектральной ширины щели. Таким образом, важно, чтобы
разрешение спектрофотометра, который должен быть откалиброван,
приблизительно соответствовало разрешению прибора, на котором был
измерен спектр, или использовались бы полосы, мало чувствительные
к спектральной ширине щели [15, 71, 82]. Влияние этих факторов
отмечено в уже упомянутых таблицах ШРАС.
Переносимость оптических плотностей
Хотя воспроизводимость оптической плотности в максимуме полос
при последовательных записях спектров на данном спектрофотометре
может быть очень хорошей (возможно, порядка 0,3 %), однако при
регистрации того же образца на другом приборе, даже того же типа
Приборы 63
и модели (или на том же приборе в другое время), совпадение
может оказаться не лучше чем 10—20%. Очевидно, если не приняты
меры для стандартизации условий каждого анализа на данном приборе,
то при определении пределов вероятных аналитических ошибок должна
быть проявлена значительная сдержанность.
Имеется несколько причин такого положения. Во-первых, если
ширина щели, усиление, время отклика и скорость сканирования не
согласованы между собой, то даже на одном и том же спектрофото-
спектрофотометре результаты будут ошибочными. Во-вторых, чтобы получить
количественное значение коэффициента поглощения [5, 3, 16, 17~\, эффек-
эффективная ширина щели должна быть менее 20% от ширины полосы, но
в то же время использование узких щелей не совместимо с низким
уровнем шума, необходимым для хорошей количественной точности.
Влияние спектральной ширины щели на величину оптической плотности
показано на рис. 2.24. Оптическая плотность чувствительна к аппаратной
функции спектрофотометра. Отрицательно может сказываться и рас-
рассеянное излучение. В-третьих, оптические ослабители, используемые в
двухлучевых спектрофотометрах с оптическим нулем, обладают нели-
нелинейностью, даже когда они изготовлены методом фотоцинкографии.
Эта нелинейность не обнаруживается проверкой закона Бера [76].
Если желательно получить переносимую величину оптической плот-
плотности, то необходимо работать на щелевых программах, дающих
спектральные ширины щелей не больше чем 0,2w, где w — ширина самой
узкой полосы в спектре. Точные контуры полос регистрируются при
соответствующем соотношении между скоростью сканирования, шири-
шириной щели и временем отклика. Важно также убедиться в линейности
фотометрического клина в пределах требуемой точности. Такую про-
проверку можно выполнить, используя ослабители в виде вращающихся
дисков с точно откалиброванными секторными прорезями [49,18~\.
Необходимо отметить, что первое требование справедливо только
в том случае, если требуются переносимые данные. В большинстве же
случаев количественного анализа для получения низкого уровня шума
целесообразно использовать более широкие щели, чем нормальные
(см. гл. 6).
Интегральные интенсивности умеренно изолированных полос, при
условии тщательной проверки спектрофотометра и использования под-
подходящего метода интегрирования, могут быть воспроизведены в раз-
различных лабораториях с точностью, лучшей чем ± 2 % [6].
ЛИТЕРАТУРА
1. Allkins J. R., Anal. Chem., 47, 752А A975).
2. Anderson R. J., Griffiths P. R., Anal. Chem., 47, 2339 A975).
3. Baldwin G. C, An Introduction to Non-Linear Optics, Plenum Press, New
York, 1969.
64 Глава 2
4. Bratt P., Engeler W., Levinstein H., MacRae A., Pehek J., Infrared Phys., 1,
27 A961).
5. Brodersen S, J. Opt. Soc. Am., 44, 22 A954).
6. Brownlee R. T. C, Cameron D. G., Ternai В., Topsom R. D., Appl. Spectrosc.,
25, 564 A971).
7. Cann M. W. P., Appl. Opt., 8, 1645 A969).
8. Chenery D. H., Sheppard N., Appl. Spectrosc, 32, 79 A978).
9. Claspy D. C, Infrared Optoacoustic Spectroscopy and Detection, in Y.-H. Rao,
Ed., Optoacoustic Spectroscopy and Detection, Academic Press, New Y ork, 1977.
10. Coates J. P., Am. Lab., 8 A1), 67 A976).
11. Coates V. J., Spectrochim. Acta, 15, 820 A959).
12. Coblentz Society Board of Managers, Anal. Chem., 38 (9), 27A A966); 47,
945A A975).
13. Coblentz Society Review Committee, Appl. Spectrosc, 11, 109 A957).
14. Codding E. G., Horlick G., Appl. Spectrosc, 27, 85 A973).
15. Cole A. R. H., Tables of Wavenumbers for the Calibration of Infrared Spectro-
Spectrometers, 2nd ed., Pergamon Press, Oxford, 1977.
16. Colles M. J., Pidgeon С R., Rep. Progr. Phys., 38, 329 A975).
17. Conn G. К. Т., Avery D. G., Infrared Methods, Academic Press, New York, 1960.
18. Cooky J. W., Tukey J. W., Math. Comput, 19, 297 A965).
19. Cooper J., Rev. Sri. Instrum., 33, 92 A962).
20. Decker J. A., Jr., Appl. Opt., 10, 510 A971).
21. Decker J. A., Jr., Anal. Chem., 44 B), 127A A972).
22. Dewey С F., Jr., Opt. Eng., 13, 483 A974).
23. Dewey С F., Jr., Kamm R. ?>., Hackett С. Е., Appl. Phys. Lett., 23, 633 A973).
24. Fellgett P., J. Phys. Radium, 19, 187, 237 A958).
25. Forman M. L, J. Opt. Soc. Am., 56, 978 A966).
26. George R. S., Appl. Spectrosc, 20, 101 A966).
27. Guard A., Opt. Acta, 7, 81 A960).
28. Girard A., Appl. Opt., 2, 79 A963).
29. Girard A., J. Phys., (Paris), 24, 139 A963).
30. Golay M. J. ?., J. Opt. Soc. Am., 39, 437 A949).
31. Golay M. J. ?., Rev. Sci. Instrum., 20, 816 A949).
32. Golay M. J. ?., J. Opt. Soc Am., 46, 422 A956).
33. Griffiths P. R., Anal. Chem., 44, 1909 A972).
34. Griffiths P. R., Anal. Chem., 46, 645A A974).
35. Griffiths P. R., Appl. Spectrosc, 29, 11 A975).
36. Griffiths P. R., Chemical Infrared Fourier Transform Spectroscopy, Wiley,
New York, 1975.
37. Griffiths P. R., Foskett С. Т., Curbelo R., Appl. Spectrosc. Rev., 6, 31 A972).
38. Griffiths P. R., Shane H. J., Hannah R. W., Appl. Spectrosc, 31, 485 A977).
39. Hackforth H. C, Infrared Radation, McGraw-Hill, New York, 1960.
40. Hinkley E. D., Opto-electronics, 4, 69 A972).
41. Hinkley E. D., Nill K. W, Blum F. A., Top. Appl. Phys., 2, 125 A976).
42. Hirschfeld Т., Appl. Spectrosc, 30, 549 A976).
43. Hirschfeld Т., Appl. Spectrosc, 30, 550 A976).
44. Hirschfeld Т., Wyntjes G., Appl. Opt., 12, 2876 A973).
45. Horlick G., Appl. Spectrosc, 22, 617 A968).
46. Horlick G., Malmstadt H. V., Anal. Chem., 42, 1361 C(>70).
47. Jacquinot P., J. Opt. Soc. Am., 44, 761 A954).
48. Jansson P. A., J. Opt. Soc. Am., 60, 184 A970).
Приборы 65
49. Jones R. N., Escolar D., Hawranek J. P., Neelakantan P., Young R. P.,
J. Molec. Struct., 19, 21 A973).
50. Jones R. N., Nadeau A., Can. J. Spectrosc, 20, 33 A975).
51. Jones R. N., Nadeau A., Spectrochim. Acta, 20, 1175 A964).
52. Kartha B. K, quoted by K. S. Seshadri and R. N. Jones, Spectrochim. Acta,
19, 1013 A963).
53. Keir M. J., Dawson J. В., Ellis D. J., Spectrochim. Acta, 32B, 59 A977).
54. Kreuzer L.B., J. Appl Phys., 42, 2934 A971).
55. Kreuzer L. В., Kenydh N. D., Patel С. К. N., Science, 177, 347 A972).
56. Kuhl J., Schmidt W., Appl. Phys., 3, 251 A974).
57. Levinstein H., Anal. Chem., 41 A4), 81A A969).
58. Low M. J. D., Coleman I., Spectrochim. Acta, 22, 369 A966).
59. Marshall A. G., Comisarow M. В., Anal. Chem.,. 47, 491A A975).
60. Mattson J. S., Anal. Chem., 49, 470 A977).
61. Meaburn J., Appl. Opt., 14, 2521 A975).
62. Moret-Bailly J., Grille Spectrometers, in K. N. Rao, Ed., Molecular Spectroscopy:
Modern Research, Academic Press, New York, 1972, p. 327.
63. Moss T. S., Infrared Phys., 16, 29 A976).
64. Nelson E. D., Fredman M. L, J. Opt. Soc. Am., 60, 1664 A970).
65. Nil! K. W., Proc. Soc. Photo-Opt. Instrum. Eng., 49, 56 A975).
66. Patel С. К. N., Burkhardt E. G., Lambert С A., Science, 184, 1173 A974).
67. Patel С. К. N., Shaw E. D., Phys. Rev. Lett., 24, 451 A970).
68. Potts W. J., Jr., Chemical Infrared Spectroscopy, Vol. 1, Techniques, Wiley,
New York, 1963.
69. Potts W. J., Jr., Smith A. L, Appl. Opt., 6, 257 A967).
70. Putley E. H., Phys. Technol., 4, 202 A973).
71. Rao K. N., Humphreys С X, Rank D. H., Wavelength Standards in the Infrared,
Academic Press, New York, 1966.
72. Sheahen T. P., Appl. Opt., 13, 2907 A975).
73. Sheahen T. P., Appl. Opt., 14, 1004 A975).
74. Sheppard N., Greenler R. G., Griffiths P. R., Appl. Spectrosc, 31, 448 A977).
75. Shane H. J., Appl. Spectrosc, 16, 5 A962).
76. Sloane H. J., Gallaway W. S., Appl. Spectrosc, 31, 25 A977).
77. Sloane N. J. A., Fine Т., Phillips P. G., Harwit M., Appl. Opt., 8, 2103
A969).
78. Smollett M., Infrared Phys., 8, 3 A968).
79. Stewart J. ?., Infraret Spectroscopy: Experimental Methods and Techniques,
Marcel Dekker, New York, 1970.
80. Stewart J. S., Paper No. 205, Pittsburgh Conference on Analytical Chemistry-
and Applied Spectroscopy, March 1961.
81. Telfair W. В., Gilby A. C, Syrjala R. J., Wilks P^A., Jr., Am. Lab., 8 A1),
91 A976).
82. Воробьев В. Г., Никитин В. А., Оптико-мех. промышленность, 38, 54
A971).
83. Whiffen D. H., Lasers in Infraed Spectroscopy, in A. R. West, Ed., Molecular
Spectroscopy, Heyden, London, 1977.
84. Williams V. Z., Rev. Sci. Instrum., 19, 135 A948).
85. Willson P. D., Edwards T. H., Appl. Spectrosc. Rev., 12, 1 A976).
86. Winefordner J. D., Fitzgerald J. J., Omenetto N., Appl. Spectrosc, 29, 369
A975).
87. Yamaguchi A., Ichishima I., Mizushima S., Spectrochim. Acta, 12, 294 A958).
5 - 586
66 Глава 2
Дополнительная литература
1. Пейсахсои И. В. Оптика спектральных приборов. — Л.: Машиностроение,
1975.
2. Тарасов К. И. Спектральные приборы.-Л.: Машиностроение, 1977.
3. Малышев В. И. Введение в экспериментальную спектроскопию.— М.: Наука,
1979.
4. Тарасов К. И., Блох А. А. и др. Проектирование спектральной аппарату-
аппаратуры/Под ред. К. И. Тарасова.—Л.: Машиностроение, 1980.
5. Прикладная инфракрасная спектроскопия. Пер. с англ./Под ред. Д. Кендал-
ла.-М.: Мир, 1970.
6. Лебедева В. В. Техника оптической спектроскопии.—М.: Изд. МГУ, 1977.
7. Марков М. Н. Приемники инфракрасного излучения. — М.: Наука, 1968.
8. Шоль Ж., Марфан И., Мюнш М., Торель П., Комбет П. Приемники инфра-
инфракрасного излучения. Пер. с франц.—М.: Мир, 1969.
9. Панкратов Н. А., Короткое В. П., Оптико-мех. пром-сть, № 2, 47 A974).
10. Толмачев Ю. А. Новые спектральные приборы.—Л.: Изд. ЛГУ, 1976.
11. Колебательная спектроскопия. Современные воззрения. Тенденции развития.
Пер. с англ./Под ред. А. Барнса, У. Орвилл-Томаса. — М.: Мир, 1981, с. 54.
12. Лазерная спектроскопия атомов и молекул. Пер. с англ./Под ред. Г. Вальте-
Вальтера.-М.: Мир, 1979.
13. Мерц Л. Интегральные преобразования в оптике. Пер. с англ.-М.: Мир,
1969.
14. Инфракрасная спектроскопия высокого разрешения. Сб. статей. Пер. с англ.-
М.: Мир, 1972.
25. Белл Р. Введение в фурье-спектроскопию. Пер. с англ.—М.: Мир, 1975.
16. Сешадри К., Джонс Р., Успехи физических наук, 85, 87 A965).
17. Петраш Г. Г. Исследование аппаратурных искажений и методы их учета
в ИК-спектроскопии. - В сб. Труды ФИАН, т. 27.-М.: Наука, 1964.
18. Воробьев В. Г., Круглякова М. А., Юдина С. Л., Изм. Техн., № 11, 25 A972).
Глава 3
СПЕКТРОСКОПИЧЕСКАЯ ЛИТЕРАТУРА
Литература по ИК-спектроскопии делится на две группы: научные
статьи и книги по некоторым аспектам ИК-спектроскопии или ее
применению и атласы ИК-спектров. Количество публикаций первого
рода растет так быстро (по данным Мак-Дональда [102], около
4000 статей в год в течение первой половины 70-х годов), что едва
ли кто-либо может их сосчитать, не говоря уже о том, чтобы
прочитать их. Часто появляются новые монографии, посвященные
общим и специальным вопросам ИК-спектроскопии, часть которых
приведена в табл. 3.1. Среди лучших источников информации о теку-
текущих исследованиях кроме «Chemical Abstracts» можно назвать двух-
двухгодичные обзоры, публикующиеся в журнале «Analytical Chemistry».
Фундаментальные обзоры, посвященные методам (например, ИК, масс-
спектрометрии, газовой хроматографии), появляются в апрельских
выпусках четных лет; в нечетные годы публикуются обзоры при-
прикладного характера (например, по полимерам, лекарственным пре-
препаратам, нефти, резине).
Эталонные ИК-спектры
Состояние дел По второй группе литературы нельзя считать удо-
удовлетворительным. Хотя в ИК-спектрах нет недостатка, их качество
и польза от них меняются в широких пределах. Кроме того, коли-
количественный перенос спектральной информации из этих спектров факти-
фактически невозможен по причинам, уже обсуждавшимся ранее (стр. 61).
Для идентификации вполне можно использовать качественные
спектры, имеющиеся в литературе. Списки некоторых из наиболее
легко доступных атласов и коллекций спектров приведены в табл. 3.2
и 3.3. По мнению автора, таблицы максимумов поглощения, хотя
и лучше, чем ничего, но гораздо менее полезны, чем собственно
спектры, так как в ширине и контурах полос содержится много
нужной информации. Вероятно, наиболее полезной картотекой для
любой лаборатории была бы созданная самим исследователем по
спектрам, получаемым на его собственном приборе. Организация
библиотеки спектров обсуждена более полно в следующем разделе.
Значение хорошей библиотеки эталонных ИК-спектров нельзя пере-
переоценить. Умелое использование корреляционных таблиц "и групповых
68 Глава 3
Таблица 3.1
Монографии d обзоры по ИК-спектросконип"
Тема, круг вопросов
Аналитическая ИК-спектроскопия
Приложения
Адсорбция
Биология
Хроматографические фракции
Покрытия, краски, лаки
Судебная экспертиза
Спектры газов и паров
Применение в промышленности
Неорганические объекты
Изотопзамещеииые соединения
Фосфорорганические соединения
Металлоорганические соединения
Лекарственные препараты
Полимеры и пластмассы
Почвы, минералы
Поверхности
Поверхностио-активиые вещества
Основы ИК-спектроскопии
Библиография по ИК-спектроскопии
Калибровка ИК-спектрометров
Применение ЭВМ в ИК-спектроскопии
Терминология
Дальняя ИК-область
Фурье-спектроскопия
Литература
5, 9, 72, 124, 50
96
81, 119, 140
36, 57, 1
24, 78
42, 2
142, 148, 58
87, 152
1, 52, 62, 82, 113, 115,
3, 4, 24, 51, 56, 58
123 ,
116, 141, 42
1, 62, 99, 114, 5, 6
10, 139
39, 47, 70, 73, 78, 79, 158,
7-10
50, 51, 58, 85, 145, //, 12
65, 69, 89
77
4, 30, 33, 34, 41, 72, 86,
124, 127, 144
31, 13
29, 128, 57
100, 101, 30
5, 44, 52
20, 55, 14
16, 53, 63, 101, 143, 15
Недиспергирующие газовые анализаторы 75
Групповые частоты
Теория групп
Водородная связь
Указатели к литературным спектрам
Спектрофотометры
Диспергирующие
Фурье-спектрометры
Интерпретация спектров
Лазеры в спектроскопии
Матричная изоляция
Молекулярная спектроскопия
Оптико-акустическая спектроскопия
Физика ИК-излучения
И, 17, 18, 30, 38, 45,
83, 112, 16, 17
37, 54, 18, 19
80, 122, 134
6, 7, 8, 62, 20
34, 137
63, 143
12, 76, 135, 147, 21-23
14, 107, 55
66, 54, 55
13, 15, 21, 74, 93, 153,
155, 25-30
26
64
Спектроскопическая литература 69
Продолжение табл. 3.1
Тема, круг вопросов Литература
Программированное обучение, интерпре- 12, 35, 76, 22, 31, 32
тация
Ежегодники по специальным вопросам 25, 46, 143
Методы
| Нарушенное полное внутреннее отра- 68, 2, 33 — 35, 56
жение
Лабораторная практика 5, 9, 47, 104, 124, 138
Отражение 149, 157
Теоретическая спектроскопия 21,93,136,153, 27, 36—38
Колебательно-вращательные спектры 3, 74, 136, 153
Отнесения колебаний 45, 133, 147
В качестве дополнительной можно рекомендовать литературу, ие вошедшую в пере-
перечисленные рубрики: вода [39, 40], органические растворители [41], ионообменные смолы
[43], ароматические и гетероциклические соединения [44], л-комплексы переходных элементов
[45]. — Прим. перев.
частот может направить идентификацию неизвестного вещества по
правильному пути, но достоверная идентификация (по ИК-спектрам)
может быть проведена только при сопоставлении с подлинным эта-
эталонным спектром. Эти спектры служат и другой полезной цели,
даже если точное соответствие не найдено; они указывают путь
к вероятным структурам (или помогают исключить постулированные
структуры) путем сравнения сходных молекул, содержащих некоторые
одинаковые группы. Для опытного спектроскописта библиотека эталон-
эталонных спектров так же важна, как и спектрофотометр.
Информационно-поисковые системы*
Одно дело иметь коллекцию спектров и совсем другое отыскать
в ней нужный спектр. Индексацию спектров можно проводить по
молекулярной формуле, химическому классу, виду поглощения или
путем комбинирования этих систем. В табл. 3.1 представлены некоторые
предметные и формульные указатели к спектрам, которые имеются
в научной литературе. Нетрудно составить формульный указатель
или к опубликованным спектрам, или к личной картотеке, и это
* В СССР информационно-поисковая система широкого назначения для
молекулярной спектроскопии разработана в Институте органической химии
н Вычислительном центре СО АН СССР под руководством академика В. А. Коп-
тюга [44, 46—49]. — Прим. перев.
70 Глава 3
Таблица 3.2
Атласы ИК-спектров
American Petroleum Institute (API) Research Project 44, Chemistry Dept., Texas
Agricultural and Mechanical University, College Station, Texas. Около 3500 спект-
спектров главным образом нефтяных углеводородов и родственных соединений.
Coblentz Society, Deskbook of Infrared Spectra, С D. Craver, Ed., P. O. Box
9952, Kirkwood, Mo. 63122. Около 900 рассчитанных ИК-спектров высшего
качества (большей частью в области 250-4000 см), иллюстрирующих
основные классы химических соединений.
Coblentz Society Spectra (issued through Sadtler Research Laboratories). Около
10000 спектров, среди них 5000 рассчитанных [27], по состоянию на 1977 г.
Colthup, Daly et al. [30]. Около 600 интерпретированных общих спектров
F00—4000 см) с уменьшенной ординатой.
Documentation for Molecular Spectroscopy, Working Atlas of Infrared Spectroscopy,
Butterworths, London, 1972. Содержит 800 спектров органических соединений,
представляющих главные классы F00—4000 см).
Infrared Data Committee of Japan, Sanyo Shuppan Boeki Co., Inc., Hoyu
Bldg., 8,2-Chome, Takara-cho, Chuo-ku, Tokyo. Около 14000 ИК-спектров,
большинство из которых снято на новых дифракционных приборах.
Меске R., Langenbucher F. (eds.), Infrared Spectra of Selected Chemical Compounds,
Heyden, London. Содержит 1879 спектров. Табулированы также данные по
волновым числам.
Pouchert С. J., The Aldrich Library of Infrared Spectra, 2nd ed., Aldrich
Chemical Co., Milwaukee, Wise., 1975. Около 11000 спектров, расположенных
по химическим классам.
Sadtler Research Laboratories, Standard Infrared Spectra, 3314 Spring Garden St.,
Philadelphia, Pa. 19104. Около 40000 спектров, снятых на дифракционных
приборах, и 50000—на призменных. Имеются специальные и фирменные
коллекции спектров, связанные с отдельными областями.
Schroder, Meier [132]. Содержит ИК-спектры и спектры КР 1014 соединений,
включающих углеводороды, N-гетероциклы, пестициды, лекарственные
препараты, полимеры и др. с линейной шкалой волновых чисел.
Thermodynamic Research Center Project (TRC), Chemistry Dept., Texas A and
M. University, College Station, Tex. 77843. Около 1180 спектров продуктов
нефтехимии и других важных промышленных химикатов.
Таблица 3.3
Специализированные коллекции спектров
Класс веществ спектров Область спектра, см ~~ Литература
Барбитураты 30 625-4000 92
9 650-4000 98
29 650-4000 23
Лекарственные препараты 817 625 —5000 B—16 мкм) 10
60 250-4000 67
175 666-5000 B-15 мкм) 71
Спектроскопическая литература
71
Класс веществ
Компоненты полировочной эмуль-
эмульсии
Эфирные масла
Взрывчатые вещества
Длинноволновые ИК-спектры раз-
различных веществ
Волокна
Ароматические и вкусовые ком-
компоненты
Газы, пары
Галогенсодержашие углеводороды
Неорганические вещества
Минералы
Наркотики
Природные вещества
Составляющие красок и лаков
(включая полимеры, пигменты,
растворители и добавки)
Пестициды
Пигменты
Пластификаторы
Полимеры, пластмассы
Дейтерированные растворители
Поверхностно-активные вещества
Терпеновые спирты
Терпены
Аппреты для текстиля
Очищенные триглицериды
Число
спектров
24
78
335
68
268
33
31
200
24
36
50
176
68
1566
33
99
66
616
303
238
875
G39)
9
23
400
740
2821
76
24
78
280
312
25
1454
24
466
141
72
84
13
Продолжение
Область спектра, см ~~
600-4000
600-4000
666-5000 B-15 мкм)
50-3700
666-5000 B-15 мкм)
666-5000 B-15 мкм)
625-5000 B-16 мкм)
625-4000
300-4000
300-4000
200-4000
666-5000 B-15 мкм)
666-5000 B-15 мкм)
303-714 A4-33 мкм)
666-4000 B,5-15 мкм)
300-4000
625-5000
300-4000
700-4000
400-4000
45-3800
Только таблицы
3200-3800 и 125-700
666-5000 B-15 мкм)
625-5000 B-16 мкм)
250-4000
666-4000 B,5-15 мкм)
250-4000
286-5000 B-35 мкм)
200-1500
400-4000
400-4000
666-3333 C-15 мкм)
556-5000 B-18 мкм)
600-4000
625-5000 B-16 мкм)
666-5000 B-15 мкм)
666-5000 B-15 мкм)
500-4000
250-4000 B,5-40 мкм)
табл. 3.3
Литература
90
91
129
132
139
146
111
19
150
151
22
125
126
20
ПО
109
121
142
148
28
115 ,
58,
145
97
156.
24
78
59
108
2
28
78
88
78 ,
103
77
105
106
117
118
72 Глава 3
может послужить полезным вспомогательным средством. Более слож-
сложной задачей является разработка схемы идентификации неизвестных
веществ по их спектрам поглощения.
В большинстве поисковых систем применяется двоичный код, когда
спектр разбивается на ряд пронумерованных интервалов одинаковой
ширины (например, 1 мкм, или 100 см). Присутствие кодируемой
полосы поглощения в интервале кодирования отмечается в ЭВМ
единицей, или прорезью на краю перфокарты, или некоторым другим
способом. Отсутствие поглощения кодируется «0» или отсутствием
прорези. Аналогичным образом можно закодировать и другую инфор-
информацию (в том числе и о химических группах).
Машинные поисковые системы часто используются для подбора
неизвестных .ИК-спектров. Некоторые из них описаны в литературе
[48, 49], существуют и коммерческие поисковые службы, основан-
основанные на этих принципах. Чтобы быть полезным, поисковые системы
должны иметь следующие признаки:
Легкость введения данных с использованием общепринятых и легко-
понимаемых терминов.
Не иметь жестких требований к точному 1:1-соответствию неиз-
неизвестного и эталонного спектров (большинство реальных образцов —
неочищенные вещества или смеси).
Использовать информацию об областях прозрачности. Отсутствие
полосы часто важнее, чем ее наличие.
Допускать возможность малых ошибок в определении длины волны
при кодировании.
Кодирование известных химических характеристик вещества. Жела-
Желательными признаками такой системы являются:
Быстрый поиск (по картотеке).
Взаимодействие ведущего поиск оператора с ЭВМ, чтобы иметь
возможность производить небольшие изменения во входных данных.
При понижении вероятности правильного ответа возможность пере-
перечисления «ближайших ошибок».
Эрли [49] опубликовал результаты количественной оценки для трех
связанных поисковых систем ИК-спектров, где определяются надеж-
надежность, точность, ложные поиски, время поиска, расходы и общий
критерий качества. 138 спектров произвольно выбрали из нескольких
картотек как «неизвестные» и провели поиск среди всех 92 000 спектров
Американского общества испытания материалов (ASTM). Самые слож-
сложные программы поиска дали почти одинаково хорошие результаты,
когда ими пользовались и новичок, и опытный эксперт.
В картотеке ASTM имеется значительное число дубликатов спектров;
это неизбежно, так как 3 — 5 % входных данных содержат ошибки
в виде избыточной информации [43], за исключением правильно зако-
закодированных спектров. Это предохраняет по крайней мере от равного
процента ошибки при кодировании полосы. Поэтому при любом
рутинном поиске необходима некоторая снисходительность.
Спектроскопическая литература 73
Во всех картотеках закодированных спектров, основанных на карто-
картотеке ASTM, используется двоичная система. Были предложены системы,
которые используют как интенсивности, так и контуры полос [56,
120]. Перекодирование больших существующих картотек является тяже-
тяжелой работой, и мало вероятно, чтобы она была предпринята в обоз-
обозримом будущем, хотя дополнительные данные могут обеспечить опре-
определенные преимущества.
Возникает интригующий вопрос: можно ли ЭВМ запрограммировать
так, чтобы она «думала», подобно спектроскописту, интерпретирующему
спектр? Некоторые скромные успехи были достигнуты на ограничен-
ограниченной основе с использованием узкого круга соединений и дополни-
дополнительных данных, таких, как протонный резонанс и масс-спектры
[60, 130]. Другой подход заключается в том, что при распознавании
спектра в ЭВМ используетср набор подлинных «обучающих» спектров
для установления вероятностных корреляций, которые применяются при
классификации ИК-спектров в соответствии с химическими классами
[32, 61, 94, 95, 154].
Оценка ИК-спектров
Критерии оценки эталонных ИК-спектров были опубликованы об-
обществом Кобленца [27]. Выделяются четыре класса спектров: спектры
I класса являются физическими константами материала независимо
от того, на каком спектрофотометре они получены; спектры II класса —
это эталонные спектры чистых материалов, снятые на исследователь-
исследовательском уровне с использованием хорошего дифракционного спектро-
спектрофотометра с оптимальными рабочими параметрами при режимах,
принятых в лабораторной практике; спектры III класса — аналитические
эталонные спектры определенных веществ, зарегистрированные с при-
применением хороших лабораторных методов и высококачественного приз-
менного или дифракционного спектрофотометра, которые не удовлет-
удовлетворяют критериям II класса, и, наконец, спектры, которые по той
или иной причине не попадают ни в один из классов. Рекомендации
для спектров, подготавливаемых к публикации в журналах, основанные
на приведенных критериях и развитые в других работах [40], обобщены
в табл. 3.4.
В любом случае на всех спектрах, для какой бы цели их ни
снимали, всегда должны быть указаны условия регистрации: щелевая
программа, скорость сканирования, спектрофотометр, способ приготов-
приготовления образца и другие важные параметры, относящиеся к делу.
Создание собственной картотеки
Со временем почти каждый исследователь, имеющий ИК-спектро-
фотометр, собирает картотеку эталонных спектров. Для того чтобы
ее успешно использовать, материал нужно как можно быстрее индек-
74
Глава 3
Таблица 3.4
Предлагаемые требования к ИК-спектрам для публикации а: специальные критерии
Чистота соединения:
Разрешение спектрофотометра:
Точность калибровки по длинам
волн:
Проверка калибровки:
Уровень шума:
Энергия:
Рассеянное излучение:
Постороннее поглощение:
Энергия сервосистемы:
Документация:
Интенсивность:
Формат:
Допускается минимальное число полос при-
примесей (не более трех) при условии, что они
отмечены
Как с призмой NaCl или лучше
±5 см в интервале 2000-4000 см";
±3 см ~' ниже 2000 см ~'
Запись спектра индена или другого вторич-
вторичного стандарта раз в неделю
Порядка 1 % от величины кйксимума
По крайней мере 50 % от нормального уровня
во всех областях (кроме участка 4,3 мкм, где
поглощает СО2)
Менее 5%
Поглощение воды в таблетках менее 0,1
единицы оптической плотности, поглощение
паров воды менее 2% Т
Нормальная; инертность и повышенная чув-
чувствительность неприемлемы
Указать тип и модель спектрофотометра;
призма или решетка; дать время сканиро-
сканирования и другие относящиеся к делу данные
наряду с физическим состоянием образца
(суспензия, растворитель, жидкая пленка)
По крайней мере одна полоса, имеющая
Т < 25 %, не более одной полосы с Т < 1 %,
базовая линия > 75 % Т, для таблеток
>40% Г при 2000 см
Оригинальный бланк, копирование вручную
не допускается
Стандартные ИК-спектры должны удовлетворять или превосходить спецификации общества Кобленца,
установленные для национальной системы эталонных данных [27], которая тоебует в основном спектры
чистых соединений с примесным поглощением в области 650-4000 см от иммерсионной среды
или растворителя (оно должно быть отмечено). Используются только оригинальные спектры или
фотокопии (перерисовка не допускается). По возможности исследователи должны использовать технические
условия II класса.
сировать и классифицировать. Вообще говоря, в картотеку должны
помещаться только спектры чистых соединений или веществ известного
или воспроизводимого состава. Общепринято сохранять оригинальный
спектр, а для работы использовать копию. В этой связи карточки
размером не более 20 х 60 см во многих случаях позволяют проводить
достаточно точное считывание и гораздо предпочтительнее больших
карточек, применяемых в ряде случаев. С карточками меньшего размера
легче обращаться, копировать, их можно хранить в ящике для картотек
и, что важнее всего, ряд таких карточек можно разместить на пульте
Спектроскопическая литература 75
прибора для сопоставления. Эталонные спектры можно пронумеровать
последовательно или разместить по группам (которые, однако, быстро
становятся громоздкими). Их можно шифровать через эмпирические
формулы, а полимеры, патентованные продукты и материалы неизвест-
неизвестного состава — специальными именными индексами. Важно, чтобы
спектры для картотеки эталонов были записаны стандартно и воспро-
воспроизводимо.
Для сотрудника маленькой или средней лаборатории, который хочет
составить собственную картотеку ИК-спектров, существует возможность
выбора. Если картотека не превышает 2000 карточек, то можно
использовать перфокарты с перфорацией по краю и ручную сортировку.
До 10 000 соединений можно нанести на 250 карточек системы
«peak-a-boo» [131]*. Некоторые коммерческие исследовательские служ-
службы, использующие ЭВМ, допускают подсоединение частной картотеки,
защищенной таким образом, что пользоваться ею может только
ее составитель.
При кодировании спектра на перфокарте учитываются не все полосы,
так как при избыточной или недостаточной перфорации пропадает
индивидуальность спектра. Критерий, используемый в картотеке ASTM,
заключается в том, что для кодирования выбирают полосы с погло-
поглощением, большим чем 1:10 по отношению к самой интенсивной
полосе в спектре. Другие исследователи используют отношение 1:5,
так что кодируется еще меньшее число полос. В последнем случае
для кодирования одного вещества требуется 5 — 8 полос.
ЛИТЕРАТУРА
1. Adams D. M., Metal-Ligand and Related Vibrations, St. Martin's Press,
New York, 1968.
2. Afremow 1. C, Vandeberg J. Т., J. Paint Technol., 38, 169 A966).
3. Allen H. C, Jr.. Cross P. C, Molecular Vib-Rotors, Wiley, New York,
1963. •
4. Alpert N. L., Reiser W. E., Szyrmnski H. A., IR; Theory and Practice
of Infrared Spectroscopy, 2nd ed., Plenum Press, New York, 1970.
5. American Society for Testing and Materials, 1977 Annual Book of ASTM
Standards, Part 42, ASTM, Philadelphia, 1977.
6. American Society for Testing and Materials, Alphabetical List of Compound
Names, Formulas, and Reference to Published Infrared Spectra; an Index
to 92,000 Published Infrared Spectra. ASTM, Philadelphia, 1969.
7. American Society for Testing and Materials, Molecular Formula List of
Compounds, Names, and References to Published Infrared Spectra, AND 31,
ASTM, Philadelphia, 1969.
8. American Society for Testing and Materials, ASTM Serial Number List
* «Peak-a-boo» — игра в прятки. Карты, на которых закодированы нужные
характеристики, складывают в стопу н рассматривают на просвет. Совпадающие
на всех картах отверстия указывают спектры, имеющие сразу все характе-
характеристики. — Прим. перев.
76 Глава 3
of Compound Names and References to Published Infrared Spectra, Vol. 3,
ASTM, Philadelphia, 1969.
9. American Society for Testing and Materials, Committee E-13, Manual on
Recommended Practices in Spectrophotometry, ASTM Philadelphia, 1966.
10. Association of Official Analytical Chemists, Infrared and Ultraviolet Spectra
of Some Compounds of Pharmaceutical Interest, revised ed., AOAC, Washington,
D.C., 1972.
11. Avram M., Mateescu G. D., Infrared Spectroscopy: Applications in Organic
Chemistry, Wiley Interscience, New York, 1972'
12 Baker A. J., Cairns Т., Eglinton G., Preston E. J., More Spectroscopic
Problems in Organic Chemistry, 2nd ed., Heyden, London, 193Й.
13. Banwell С N.. Fundamentals of Molecular Spectroscopy, 2nd ed., McGraw-
Hill, New York, 1973.
14. Колебательная спектроскопия. Современные воззрения. Тенденции развития.
Пер. с англ./Под ред. А. Барнса, У. Орвилла-Томаса. — М.: Мир, 1981.
15. Barrow С. М., Introduction to Molecular Spectroscopy, McGraw-Hill, New
York, 1962. ,
16. Белл Р. Введение в фурье-спектроскопию. Пер. с англ.—М.: Мир, 1975.
17. Беллами Л. Новые данные по ИК-спектрам сложных молекул Пер. с англ.—
М.: Мир, 1971.
18. Bellamy L. J., The Infrared Spectra of Complex Molecules, Vol. 1, .3rd ed.,
Wiley, New York, 1975. (Имеется перевод 1-го и 2-го изданий: Беллами Л.
Инфракрасные спектры сложных молекул. Пер. с англ.—М.: Мир, 1957,
1963.)
19. Bellanato J., Hidalgo A. #., Infrared Analysis of Essential Oils, Heyden,
London, 1971.
20. Bentley F. F., Smithson L. D., Rozek A. L., Infrared Spectra and Cha-
Characteristic Frequencies 700 — 300 cm, Interscience, New York, 1968.
21. Califano S., Vibrational States, Wiley, New York, 1976.
22. Chasan D. ?., Norwitz C, Microchem. J., 17, 31 A972).
23. Chatten 1. C, Levi L., Appl. Spectrosc, 11, 177 A957).
24. Chicago Society of Paint Technology, Infrared Spectroscopy: Its Use in the
Coatings Industry, Federat. Soc. Paint Technology, Philadelphia, 1969.
25. Clark R. J. #., Hester R. F., Advances in Infrared and Raman Spectro-
Spectroscopy, Vol. 1, Heyden, London, 1975; Vol. 2, 1976; Vol. 3, 1977; Vol. 4,
1978.
26. Claspy D. C, Infrared Optoacoustic Spectroscopy and Detection, in Y.-H.
Pao, Ed., Optoacoustic Spectroscopy and Detection, Academic Press, New
York, 1977.
27. Coblentz Society Board of Managers, Anal. Chem., 38, (9), 27A A966);
47, 945A A975).
28. Coblentz Society Inc., Special Collections: CSC-2, Halogenated Hydrocarbons;
CSC-3, Plastizers and Other Additives 1977.
29. Cole A. R. H., Tables of Wavenumbers for the Calibration of Infrared
Spectrometers, 2nd ed., Pergamon Press, Oxford, 1977.
30. Colthup N. В., Daly L. H., Wiberley S. E., Introduction to Infrared and
Raman Spectroscopy, 2nd ed., Academic Press, New York, 1975.
31. Comeford J. J., Rao С N. R., Dikshit S. K., Kudchadkev S. A., Gupta D. S.,
Bibliography of Infrared Spectroscopy Through 1960, National Bureau of
Standards, Institute of Applied Technology, Washington, D. C, 1976, Paper
No. NBS-SP-428, Parts 1-3.
Спектроскопическая литература 77
32. Comerford J. M., Anderson P. G., Snyder W. #., Kimmel H. S., Spectro-
chim. Acta, 33A, 651 A977).
33. Сопку R. Т., Infrared Spectroscopy, 2nd ed., Allyn and Bacon, Boston,
1972.
34. Conn G. К. Т., Avery D. G., Infrared Methods, Academic Press, New
York, 1960.
35. Cook B. W., Jones K., A Programmed Introduction to Infrared Spectroscopy,
Heyden, London, 1972.
36. Copier H., Infrared Analysis of Chromatographic Fractions, Druk. Elinkwijk,
Utrecht, The Netherlands, 1968.
37. Cotton F. A., Chemical Applications of Group Theory, Interscience, New
York, 1963.
38. Craver С D., Desk Book of Infrared Spectra, Coblentz Society, РОВ 9952,
Kirkwood, Mo., 1977.
39. Craver С D., Polymer Characterization: Interdisciplinary Approaches, Plenum
Press, New York, 1971.
40. Craver С D., Grasselli J. G., Smith A. L., Anal. Chem., 47, 2065 A975).
41. Кросс А. Введение в практическую инфракрасную спектроскопию. Пер. с
англ.-М.: ИЛ, 1961.
42. De Faubert Maunder M. J., Practical Hints on Infrared Spectrometry for
a Forensic Analyst, Hilger, London, 1971.
43. De Haseth J. A., Woodruff H. В., Isenhour T. L., Appl. Spectrosc, 31,
18 A977).
44. Denney R. C, A Dictionary of Spectroscopy, Halsted Press, New York, 1973.
45. Dolphin D., Wick A. E., Tabulation of Infrared Spectral Data, Wiley,
New York, 1977.
46. Durig J. R., Vibrational Spectra and Structure, A Series of Advances, Vol. 4,
Elsevier, Amsterdam, 1975; Vol. 5, 1976; Vol. 6, 1977.
47. Эллиот А. Инфракрасные спектры и структура полимеров. Пер. с англ.—
М.: Мир, 1972.
48. Erley D. S., Anal. Chem., 40, 894 A968).
49. Erley D. S., Appl. Spectrosc., 25, 200 A971).
50. Farmer V. C, The Infrared Spectra of Minerals, Mineralogical Society,
London, 1974.
51. Farmer V. C, Palmieri F., Characterization of Soil Minerals by IR Spectroscopy,
in J. E. Gieseking, Ed., Soil Components, Vol. 2, Springer, New York,
1975, p. 573.
52. Ferraro J. R., Low-Frequency Vibrations of Inorganic and Coordination
Compounds, Plenum Press, New York, 1971.
53. Ferraro J. R., Basile L. J., Fourier Transform Infrared Spectroscopy, Acade-
Academic Press, New York, 1977.
54. Ferraro J. R., Ziomek J. S., Introductory Group Theory and Its Application
to Molecular Structure, 2nd ed., Plenum Press, New York, 1975.
55. Финч А., Гейтс П. Н., Редклиф К., Бентли Ф. Применение длинноволновой
ИК-спектроскопии в химии. Пер. с англ. — М.: Мир, 1973.
56. Fox R. С, Anal. Chem., 48, 717 A976).
57. Freeman S. К., Gas Chromatography and Infrared and Raman Spectrometry,
in L. S. Ettre and W. H. McFadden, Eds., Ancillary Techniques of Gas.
Chromatography, Wiley, New York, 1969, pp. 227 - 267.
58. Gadsden J. A., Infrared Spectra of Minerals and Related Inorganic Compounds,
Butterworths, London, 1975.
78 Глава 3
59. Gore R. С, Hannah R. W., Pattacini S. С Porro T. J., J. Assoc.
Offic. Anal. Chem., 54, 1040 A971).
60. Gray N. А. В., Anal. Chem., 47, 2426 A975).
61. Gray N. А. В., Anal. Chem., 48, 2265 A976).
62. Greenwood N. N., Ross E. J. F., Straughan B. P., Index of Vibrational
Spectra of Inorganic and Organometallic Compounds, Vol. 1, Butterworth,
London, 1972, and succeeding volumes.
63. Griffiths P. R-. Chemical Infrared Fourier Transform Spectroscopy, Wiley,
New York, 1975.
64. Hackforth H. C, Infrared Radiation, McGraw-Hill, New York, 1960.
65. Hair M. L., Infrared Spectroscopy in Surface Chemistry,«*Marcel Dekker,
New York, 1967.
66. Hallam H. E., Vibrational Spectroscopy of Trapped Species, Wiley, New
York, 1973.
67. Hannah R. W., Pattacini S. C, The Identification of Drugs from Their
Infrared Spectra, Perkin-Elmer Applications Study No. 11, 1972.
68. Харрик Н. Спектроскопия внутреннего отражения. Пер. с англ.—М.: Мир,
1970.
69. Harrick N. J., Весктапп К. Н., in P. F. Kane, Ed., Characterization
of Solid Surfaces, Plenum Press, New York, 1974. pp. 215-245.
70. Хаслам Дж., Виллис Г. А. Идентификация и анализ полимеров. Пер.
с англ.—М.: Химия, 1971.
71. Hayden A. L., Sammul О. R., Selzer G. В., Carol J., J. Assoc. Offic.
Agric. chem., 45, 797 A962).
72. Hediger H., Infrared Spectroscopy: Principles, Uses, Interpretation, Vol. 11,
Methods of Analysis in Chemistry, Akad. Verlagsges, Frankfurt, 1971.
73. Henniker J. C, Infrared Speotrometry of Industrial Polymers, Academic
Press, New York, 1967.
74. Герцберг Г. Колебательные и вращательные спектры многоатомных моле-
молекул. Пер. с англ.-М.: ИЛ, 1949.
75. Hill D. W., Powell Т., Nondispersive Infrared Gas Analysis in Science,
Medicine and Industry, Hilger, London, 1968.
76. Hill R. R., Rendell A. ?., The Interpretation of Infrared Spectra, Heyden,
London, 1975.
77. Hummel D. O., Identification and Analysis of Surface-Active Agents, Wiley-
Interscience, New York, 1962.
78. Hummel D. O., Scholl F. K, Infrared Analysis of Polymers, Resins, and
Additives: an Atlas, Vol. 1, Plastics, Elastomers, Fibers, and Resins, Part 1:
Text; Part 2, Spectra, Tables, Index, Wiley-Interscience, New York, 1969;
Vol. 2, Additives and Processing Aids, 1973.
79. Ivin K. J., Structural Studies of Macromolecules by Spectroscopic Methods,
Wiley, New York, 1976.
80. Joesten M. D., Schaad L. J., Hydrogen Bonding, Marcel Dekker, New
York, 1974.
81. Jones D. W., Introduction to the Spectroscopy of Biological Polymers, Acade-
Academic Press, New York, 1976.
82. Jones L. H., Inorganic Vibrational Spectroscopy, Vol. 1, Marcel Dekker,
New York, 1971.
83. Jones R. N, Infrared Spectra of Organic Compounds: Summary Charts
of Principal Group Frequencies National Research Council of Canada, Ottawa,
1959.
Спектроскопическая литература 79
84. Jurs Р. С, Isenhour Т. L, Chemical Applications of Pattern Recognition,
Wiley-Interscience, New York,. 1975.
85. Karr C, Jr., Infrared and Raman Spectroscopyy of Lunar and Terrestrial
Minerals, Academic Press, New York, 1975.
86. Kemmner G., Infrared Spectroscopy: Principles, Applications, Methods, Vol. 5,
Chemical Monographs, Franckh, Stuttgart, 1969.
87. Прикладная инфракрасная спектроскопия. Пер. с англ./Под ред. Д. Кен-
далла.-М.: Мир, 1970.
88. Kendall D. N., Hampton R. R., Hausdorf Я., Pristera F., Appl. Spectrosc,
7, 179 A953).
89. Киселев А. В.,Лыгин В. И. Инфракрасные спектры поверхностных соеди-
соединений и адсорбированных веществ. — М.: Наука, 1972.
90. Lenzen С, Delcambe L., Inf. Bull., Int. Cent. Inf. Antibiot., 10, 78 A972).
91. Lenzen C, Delcambe L. Inf. Bull., Int. Cent. Inf. Antibiot., 11,
157 A973).
92. Levi L., НиЫеу С ?., Anal. Chem., 28, 1591 A956).
93. Levin I. N.. Molecular Spectroscopy, Wiley, New York, 1975.
94. Liddell R. W., Ill, Jurs P. C, Anal. Chem., 46, 2126 A974).
95. Liddell R. W, Jurs P. C, Appl. Spectrosc, 27, 371 A973).
96. Литтл Л. Инфракрасные спектры адсорбированных молекул. Пер. с англ. —
М.: Мир, 1969.
97. Manning J. J., Appl. Spectrosc, 10, 85 A956).
98. Manson J. M., Cloutier J. A. R., Appl. Spectrosc, 15, 77 A961).
99. Maslowsky E., Jr., Vibrational Spectra of Organometallic Compounds, Wiley,
New York, 1976.
100. Mattson J. S., Mark H. В., Jr., MacDonald H. C, Jr., Computers in
Chemistry and Instrumentation, Vol. 5, Laboratory Systems and Spectro-
Spectroscopy, Marcel Dekker, New York, 1977.
101. Mattson J. S., Mark H. В., Jr., MacDonald H. C, Jr., Computers in
Chemistry and Instrumentation, Vol. 7, Infrared, Correlation, and Fourier
Transform Spectroscopy, Marcel Dekker, New York, 1977.
102. McDonald R. S., Anal. Chem., 48, 196R A976).
103. McNiven N. L., Court R., Appl. Spectrosc, 24, 296 A970).
104. Miller R. G. J., Stace В. С, Laboratory Methods in Infrared Spectro-
Spectroscopy, 2nd ed., Heyden, London, 1972.
105. Mitzner B. M., Mancini V. J., Lemberg S., Theimer E. Т., Appl. Spectrosc,
22, 34 A968).
106. Mitzner B. M., Theimer E. Т., Freeman S. K, Appl. Spectrosc, 19,
169 A965).
107. Mooradian A., Jaeger Т., Stokseth P., Tunable Lasers and Applications,
Vol. 3, Springer Series in Optical Sciences, Springer-Verlag, Berlin, 1976.
108. Morris W. M., Jr., Haenni E. O., J. Assoc. Offic. Agric Chem., 46,
964 A963).
109. Morris W. M., J. Assoc. Offic. Anal. Chem.. 56, 1037 A973).
110. Morrison R. D., Am. Dyest. Rep., 52, 867 A963); ASTM Test D 276-72,
1976 Book of ASTM Standards, Part 33, Textiles - Fibers and Zippers;
High Modulus Fibers, American Society for Testing and Materials, Phila-
Philadelphia, 1976, p. 46.
111. Murphy J. E., Schwemer W. C, Anal. Chem., 30, 116 A958).
112. Nakanishi K., Solomon P. H., Infrared Absorption Spectroscopy, 2nd ed.,
Holden-Day, San Francisco, 1977.
80 Глава 3
113. Накамото К. Инфракрасные спектры неорганических и координационных
соединений. Пер. с англ.—М.: Мир, 1966.
114. Nakamoto К., McCarthy P. J., Spectroscopy and Structure of Metal Chelate
Compounds, Wiley, New York, 1968.
115. Nyquist R. A., Kagel R. O.. Infrared Spectra of Inorganic Compounds,
Academic Press, New York, 1971.
116. Nyquist R. A., Potts W. J., Jr., Vibrational Spectra of Phosphorum Com-
Compounds, in M. Halman, Ed., Analytical Chemistry of Phosphorus Compounds,
Interscience, New York, 1972.
117. O'Connor R. Т., McCall E. R., Morris N. M., Tripp J. W., U. S.
Agric. Res. Serv., South. Reg. [Rep.], report No. ARS-S-47, 1974.
118. Parkash S., Blanshard J. M. V., Spectrochim. Acta, 31A, 951 (W5).
119. Parker F. S., Applications of Infrared Spectroscopy in Biochemistry, Biology,
and Medicine, Plenum Press, New York, 1971.
120. Penski E. C, Padowski D. A., Bouck J. В., Anal. Chem., 46, 955 A974).
121. Pierson R. H., Fletcher A. N.. Gantz E. S., Aijal. Chem., 28, 1218 A956).
122. Пиментел Дж., Мак-Клеллан О. Водородная связь. Пер. с англ.— М.: Мир,
1964.
123. Pinchas S., Laulicht I., Infrared Spectra of Labeled Compounds, Academic
Press, New York, 1971.
124. Potts W. J., Jr., Chemical Infrared Spectroscopy, Vol. 1, Techniques, Wiley,
New York, 1963.
125. Pristera F., Fredericks W. E., U. S. Clearinghouse Fed. Sci. Technol.
Inform., AD 1969, No. 859846.
126. Pristera F., Halik N.. Castelli A., Fredericks W., Anal. Chem., 32, 495
A960).
127. Rao C. N. R., Chemical Applications of Infrared Spectroscopy, Academic
Press, New York, 1963.
128. Rao K. N., Humphreys С J., Rank D. #., Wavelength Standards in the
Infared, Academic Press, New York, 1966.
129. Sammul O. R., Brarmon W. L., Hayden A. L., J. Assoc. Offic. Agric. Chem.,
47, 918 A964).
130. Sasaki S., Abe #., Ouki Т., Sakamoto M., Ochiai S., Anal. Chem., 40,
2220 A968).
131. Schlichter N. E., Wallace E., Appl. Spectrosc., 17, 98 A963).
132. Schroder В., Meier W., Raman/IR Atlas, Vol. 1, Verlag Chemie, Weinheim,
Germany, 1974.
133. Shimanouchi Т., Tables of Molecular Vibrational Frequencies, Consolidated
Volume I, U. S., Government Printing Office, S. D. Cat. No. C13, 48 : 39,
1972.
134. Shuster P., Zundel G., Sandorfy C, The Hydrogen Bond: Recent Developments
in Theory and Experiments, Vol. 2, Structure and Spectroscopy, North-
Holland, Amsterdam, 1976.
135. Сильверстейн Р., Басслер Г., Моррил Т. Спектрометрическая идентифи-
идентификация органических соединений. Пер. с англ.—М.: Мир, 1977.
136. Steele D., Theory of Vibrational Spectroscopy, Saunders, Philadelphia, 1971.
137. Stewart J. E., Infrared Spectroscopy: Experimental Methods and Techniques,
Marcel Dekker, New York, 1970.
138. Stine К. Е., Modern Practices in Infrared Spectroscopy, Beckman Instru-
Instruments, Fullerton, Ca., 1970.
139. Sunshine I., Gerber S. R., Spectrophotometric Analysis of Drugs, Charles,
C. Thomas, Springfield, 111, 1963.
Спектроскопическая литература 81
140. Thomas G. J., Jr., Kyogoku Т., Biological Science, in E. G. Brame, Jr.,
and J. G. Grasselli, Eds., Practical Spectroscopy, Vol. 1, Infrared and
Raman Spectroscopy, Marcel Dekker, New York, 1977, Part C, p. 717.
141. Thomas L. C, Interpretation of the Infrared Spectra of Organophosphorus
Compounds, Heyden, London, 1974.
142. Thompson В., Hazardous Gases and Vapors: Infrared Spectra and Physical
Constants, Beckaman Instruments, Fullerton, Ca., technical report No. 595,
August 1974.
143. Vanasse G. A., Spectrometric Techniques. Vol. 1, Academic Press, New
York, 1977.
144. Van der Maas J. #., Basic Infrared Spectroscopy, 2nd ed., Heyden, London,
1972.
145. Van der Marel, H. W., Beutelspacher #., Atlas of Infrared Spectroscopy
of Clay Minerals and Their Admixtures, Elsevier, New York, 1976.
146. Wayland L., Weiss P. J., i. Assoc. Offic. Agric. Chem., 48, 965 A965).
147. Weitkamp #., Barth R., Infrared Structural Analysis: a Dualistic Interpreta-
Interpretation Scheme, Thieme, Stuttgart, Germany, 1972.
148. Welti D., Infrared Vapour Spectra, Heyden, ^London, 1970.
149. Wendlandt W. W., Hecht H. G., Reflectance Spectroscopy, Interscience,
New York, 1966.
150. Wenninger J. A., Yates R. L., J. Assoc. Offic. Anal. Chem., 53, 949
A970).
151. Wenninger J. A., Yates R. L., Dolinski M., J. Assoc. Offic. Anal. Chem.,
50, 1313 A967).
152. White R. C, Handbook of Industrial Infrared Analysis, Plenum Press,
New York, 1964.
153. Вильсон Е., Дешиус Дж., Кросс П. Теория колебательных спектров
молекул. Пер. с англ.— М.: ИЛ, 1960.
154. Woodruff Н. В., Ritter G. L., Lowry S. R., Isenhour Т. L., Appl.
Spectrosc., 30, 213 A976).
155. Woodward L. A., Introduction to the Theory of Molecular Vibrations and
Vibrational Spectroscopy, Oxford, U. P., London, 1972.
156. Yamaguchi K., Spectral Data of Natural Products, Vols. 1 and 2, Elsevier,
New York, 1970.
157. Young E.F., Hannah R. W., in W. W. Wendlandt, Ed., Modern Aspects
of Reflectance Spectroscopy, Plenum Press, New York, 1968.
158. Збинден Р. Инфракрасная спектроскопия высокополимеров. Пер. с англ.—
М.: Мир, 1966.
Дополнительная литература
/. Методы-спутники в газовой хроматографии. Пер. с англ./Под ред. Л. Эттра,
У. Мак-Фаддена. — М.: Мир, 1972.
2. Седова Т. А. Применение спектроскопии внутреннего отражения в судебной
экспертизе. - Л.: Изд. ЛГУ, 1978.
3. Давидович Р. Л., Кайдалова Т. А., Левчишина Т. Ф., Сергиенко В. И.
Атлас инфракрасных спектров поглощения и рентгенометрических данных
комплексных фторидов. — М.: Наука, 1972.
4. Харитонов Ю. Я., Давидович Р. Л., Костин В. И. Атлас длинноволновых
инфракрасных спектров поглощения комплексных фторидов металлов
III—V групп н уранила.— М.: Наука, 1977.
6 - 586
82 Глава 3
5. Чумаевский Н. А. Колебательные спектры элементоорганнческнх соединений
элементов ГУБ н VB групп. — М.: Наука, 1971.
6. Гар Т. К., Минаева Н. А., Миронов В. Ф., Чумаевский Н. А. Инфракрасные
спектры поглощения соединений германия. — М.: Наука, 1977.
7. Дехант И., Данц Р., Киммер В., Шмольке Р. Инфракрасная спектроскопия
полимеров. Пер. с нем.—М.: Химия, 1976.
8. Жбанков Р. Г. Инфракрасные спектры целлюлозы и ее производных.—
Минск: Наука и техника, 1964.
9. Инфракрасные спектры поглощения полимеров и вспомогательных ве-
веществ/Под ред. В. М. Чулановского — Л.: Химия, 196^,
10. Грибов Л. А. Теория инфракрасных спектров полимеров.— М.: Наука, 1977.
11. Болдырев А. И. Инфракрасные спектры минералов.—М.: Недра, 1976.
12. Плюснина И. И. Инфракрасные спектры силикатов.— М.: Изд. МГУ, 1967.
13. Справочная литература по молекулярной спектроскопии.— Новосибирск:
Изд. СО АН СССР, 1974.
14. Техника спектроскопии в дальней инфракрасной, субмиллиметровой и
миллиметровой областях спектра. Пер. с англ./Под ред. Д. X. Мартина — М.:
Мир, 1970.
15. Инфракрасная спектроскопия высокого разрешения. Сб. статей. Пер. с
англ.- М.: Мир, 1972.
16. Беллами Л. Инфракрасные спектры молекул. Пер. с англ. — М.: ИЛ, 1957.
17. Наканиси К. Инфракрасные спектры и строение органических соединений.
Пер. с англ.—М.: Мир, 1965.
18. Хохштрассер Р. Молекулярные аспекты симметрии. Пер. с англ.— М.: Мир,
1968.
19. Багавантам С, Венкатарайуду Т. Теория групп и ее применение к физи-
физическим проблемам. Пер. с англ. - М.: ИЛ, 1959.
20. Hershenson H. M., Infrared Absorption Spectra, Index to 1958-1962, New York,
London, Academic Press, 1964.
21. Бранд Дж., Эглинтон Г. Применение спектроскопии в органической химии.
Пер. с англ.—М.: Мир, 1967.
22. Казицына Л. А., Куплетская Н. Б. Применение УФ-, ИК-, ЯМР- и
масс-спектроскопии в органической химии.—М.: Изд. МГУ, 1979.
23. Дайер Д. П. Приложения абсорбционной спектроскопии органических
соединений. Пер. с англ.—М.: Химия, 1970.
24. Лоусон К. Инфракрасные спектры поглощения неорганических веществ.
Пер. с англ.— М.: Мир, 1964.
25. Маянц Л. С. Теория и расчет колебаний молекул.—М.: Изд. АН СССР,
1960.
26. Свердлов Л. М., Ковнер М. А., Крайнов Е. П. Колебательные спектры
многоатомных молекул. — М.: Наука, 1970.
27. Волькенштейн М. В., Грибов Л. А., Ельяшевич М. А., Степанов Б. И.
Колебания молекул. — М.: Наука, 1972.
28. Грибов Л. А. Введение в теорию и расчет колебательных спектров много-
многоатомных молекул.—Л.: Изд. ЛГУ, 1965.
29. Грибов Л. А. Введение в молекулярную спектроскопию. — М.: Наука, 1976.
30. Эляшберг М. Е., Грибов Л. А., Серов В. В. Молекулярный спектральный
анализ и ЭВМ.—М.: Наука, 1980.
31. Мальцев А. А. Молекулярная спектроскопия. — М.: Изд. МГУ, 1980.
32. Иоффе Б. В., Костиков Р. Р., Разин В. В. Физические методы определения
строения органических молекул.— Л.: Изд. ЛГУ, 1976.
Спектроскопическая литература 83
33. Применение спектроскопии нарушенного полного внутреннего отражения
(НПВО) в народном хозяйстве. — М.: ЦНИИТЭИ приборостроения, 1976.
34. Якутии В. И., Струков О. Г., Успехи химии, № 8, 1504 A972).
35. Золотарев В. М., Лыгин В. И., Тарасевич Б. Н., Успехи химии, № 1,
24 A981).
36. Сивин С. Колебания молекул и среднеквадратичные амплитуды. Пер.
с англ.- М.: Мир, 1971.
37. Бахшиев Н. Г. Спектроскопия межмолекулярных взаимодействий.—Л.:
Наука, 1972.
38. Коптев Г. С, Пентин Ю. А. Расчет колебаний молекул.—М.: Изд. МГУ,
1977.
39. Юхневич Г. В. Инфракрасная спектроскопия воды.—М.: Наука, 1973.
40. Молекулярная физика и биофизика водных систем. Вып. 1.— Л.: Изд. ЛГУ,
1973.
41. Атлас спектров. ИК-, УФ-, КР-, ПМР-спектры растворителей/Под ред.
В. А. Коптюга.-Новосибирск: Изд. СО АН СССР, 1975.
42. Шагидуллин Р. Р., Мухаметов Ф. С, Нигматуллина Р. Б., Виноградо-
Виноградова В. С, Чернова А. В. Атлас ИК-спектров фосфорорганических соедине-
соединений.- М.: Наука, 1977.
43. Семушин А. М., Яковлев В. А., Иванова Е. В. Инфракрасные спектры
поглощения ионообменных материалов.—Л.: Химия, 1980.
44. Атлас спектров ароматических и гетероциклических соединений/Под ред.
В. А. Коптюга. Вып. 1-16.- Новосибирск: Изд. СО АН СССР, 1972-1978.
45. Алексанян В. Т., Локшин Б. В. Строение молекул и химическая связь.
Т.5. Колебательные спектры тг-комплексов переходных элементов. Итоги
науки и техники.-М.: ВИНИТИ, 1976.
46. Дробышев Ю. П., Нигматуллин Р. С, Лобанов В. И., Коробейничева И. К.,
Бочкарев В. С, Коптюг В. А., Вестник АН СССР, № 8, 75 A970).
47. Бархаш В. А., Соколов С. П., Секерина Л. Ф., Дробышев Ю. П.,
Коптюг В. А., Изв. СО АН СССР, сер. хим., № 14, вып. 6, 111 A974).
48. Смирнов В. И., Нигматуллин Р. С, Коптюг В. А., Журн. прикл. спектр.,
22, № 3, 499 A975).
49. Нигматуллин Р. С, Смирнов В. И., Журн. прикл. спектр., 21, № 2 307 A974).
50. Кесслер И. Методы инфракрасной спектроскопии в химическом анализе.
Пер. с нем. — М.: Мир, 1964.
51. Лоусон К. Инфракрасные спектры поглощения неорганических веществ.
Пер. с англ.—М.: Мир, 1964.
52. ГОСТ 7601-78. Физическая оптика. Термины, буквенные обозначения и
определения основных величин.—М.: Госкомитет по стандартам, 1979.
53. Лазерная спектроскопия атомов и молекул. Пер. с англ./Под ред.
Г. Вальтера.-М.: Мир, 1979.
54. Крейдок С, Хинчклиф А. Матричная изоляция. Пер. с англ.— М.: Мир, 1978.
55. Криохимия. Пер. с англ./Под ред. М. Московица, Г. Озина.— М.: Мир, 1979.
56. Раков А. В. Спектрофотометрия тонкопленочных полупроводниковых струк-
структур.—М.: Советское радио, 1975.
57. Воробьев В. Г., Никитин В. А., Оптнко-мех. пром-сть, № 5, 60 A974).
55. Колебательные спектры в неорганической химии: Сб. статей. — М.: Наука,
1971.
Глава 4
МЕТОДЫ ПОДГОТОВКИ ОБРАЗЦОВ
Разнообразие методик подготовки образцов для съемки ИК-спектров
почти беспредельно, и исследователь должен выбрать одну из них,
наилучшим образом подходящую для решения соответствующей
проблемы. Здесь изложены некоторые наиболее полезные методы,
однако возможны и их многочисленные вариации. В ежегоднике Амери-
Американского общества испытания материалов (ASTM) [3] содержатся практи-
практические рекомендации по подготовке образцов для качественного и коли-
количественного анализов, а также для других целей.
Иногда природа образца подсказьшает метод подготовки, но обычно
существует возможность выбора. Для новичка спектроскописта имеется
большой соблазн записать спектры всех жидкостей между сжатыми
солевыми пластинками, а все твердые образцы — в таблетках с КВт.
Хотя эти методики действительно используются, они имеют серьезные
ограничения и не могут быть приняты как стандартные для всех
образцов.
В связи с тем что физическое состояние образца может сильно
влиять на ИК-спектр, целесообразно заранее определить иерархию
методов, которые будут использоваться в лаборатории. Последователь-
Последовательность применения методов определяется типами образцов, с которыми
приходится сталкиваться, и методами их приготовления, использован-
использованными при создании библиотеки эталонных спектров. Например, в
лаборатории, проводящей химические работы общего характера, для
жидкого образца можно избрать следующий порядок: 1) раствор,
2) неразбавленная жидкость в тонкой кювете, если вещество нераство-
нерастворимо, и 3) жидкость, сжатая между солевыми пластинками, так назы-
называемая «жидкая пленка». Для порошков и рыхлых твердых образцов
логична следующая последовательность: 1) раствор, 2) суспензия в вазе-
вазелиновом масле, 3) таблетки с КВг и 4) пиролизат. Такие методы,
как нарушенное полное внутреннее отражение (НПВО), обычно остав-
оставляют для исследования специальных случаев.
Растворы
Во многих случаях предпочитают приготовление образцов в виде
растворов. Хотя этот метод несколько сложнее, чем некоторые другие,
он имеет огромное преимущество, обусловленное высокой воспроизво-
Методы подготовки образцов 85
димостью. Можно сравнивать спектры, снятые с интервалом в несколько
лет, и при этом без труда заметить даже незначительные отличия.
Если известны концентрация и толщина кюветы (как это и должно
быть), то в этом случае легко можно выполнить полуколичественный
анализ. Кроме того, концентрация должна быть согласована с толщиной
кюветы таким образом, чтобы при регистрации спектра правильно
передавались контур и структура всех сильных полос. Это редко удается
сделать при записи спектров чистых жидкостей в кюветах фиксирован-
фиксированной толщины. (Исключениями из этого утверждения являются алифа-
алифатические углеводороды и алифатические растворители, которые имеют
более информативные спектры, если они не разбавлены.) Наконец,
в случае твердых образцов устраняются эффекты полиморфизма, ко-
которые могут оказывать серьезное влияние на спектр.
Растворители
Выбор растворителя всегда является компромиссом. Поскольку все
общепринятые растворители поглощают в ИК-области, исследователь
должен использовать тонкие слои и выбирать те из растворителей,
которые имеют окна прозрачности в интересующих областях спектра.
Не всегда легко найти такой прозрачный растворитель, в котором обра-
образец был бы достаточно растворим для получения требуемой концентра-
концентрации. (Однако иногда анализ смесей может быть значительно упрощен,
если использовать преимущество селективной экстракции ИК-раствори-
телем.)
Растворитель должен быть химически инертным по отношению к
образцу. Например, первичные и вторичные амины реагируют с CS2 с
образованием тиокарбаматов, и для них наилучшим способом является
приготовление жидких пленок. Алифатические амины также подверга-
подвергаются медленному фотохимическому взаимодействию с СС14 с образова-
образованием солянокислых аминов. Вещества, чувствительные к влаге, могут
реагировать с водой, остающейся в любом растворителе, особенно при
больших разбавлениях, до тех пор, пока растворитель не будет
полностью осушен.
Поначалу может создаться впечатление, что на двухлучевом спектро-
спектрофотометре можно работать с любым растворителем, так как поглоще-
поглощение в пучке образца компенсируется растворителем в пучке сравнения.
Однако в областях сильного поглощения растворителя энергия в спектро-
спектрофотометр не попадает, и говорят, что прибор «мертв».
Для обычной работы в области 625—4000 см B,5 — 16 мкм)
общепринято использовать СС14 или С2С14 при 1330—4000 см
B,5 — 7,5 мкм) и CS2 при 625—1330 см G,5—16 мкм). Не существует
растворителей, совершенно не влияющих на растворенное вещество, но
указанные неполярные растворители дают минимальный эффект из-за
их относительно однородного диэлектрического поля. Необходимо
отметить, что пары СС14 и CS2 крайне токсичны. Средняя максимально
86 Глава 4
допустимая концентрация в атмосфере, в которой работающий может
находиться без вреда для здоровья, составляет 10 частей на миллион
для СС14 и 20 частей на миллион для CS2*. Запах растворителя не
может быть надежным предостережением, так как порог чувствитель-
чувствительности обоняния для СС14 около 50 частей на миллион. Обонянием
можно обнаружить около 1 части на миллион CS2 в воздухе, но предел
чувствительности увеличивается со временем экспозиции. Кроме того,
CS2 горюч, поэтому работать с этими растворителями можно только
в хорошо вентилируемом вытяжном шкафу.
В качестве еще одной пары менее токсичных растворителей (но
с более высоким фоновым поглощением) можно использовать C2Cl4 в
области 1000-4000 см B,5-10 мкм) и н-гептан в области
250-1000 см A0-40 мкм).
Другими растворителями, применяемыми в более ограниченных
спектральных областях, являются НСС13, диоксан и диметилформамид.
Растворяющее действие таких веществ обусловлено их полярной при-
природой, что приводит к сильному ИК-поглощению и взаимодействию
с растворенным веществом. Дейтерированные растворители имеют окна
прозрачности, отличающиеся от их протонированных аналогов, и не
слишком дорого стоят в тех малых количествах, которые необходимы
для ИК-спектроскопии [83].
Для длинноволновой области выбор доступных растворителей шире.
В интервале 260 — 625 см A6 — 38 мкм) можно использовать СС14,
дихлорметан и гексан. Многие другие растворители имеют только
одну-две полосы и их можно применять в ограниченных спектральных
областях.
Для сведения к минимуму интерференционных полос, которые могут
возникать в виде фона (стр. 250—253), растворители и материалы окошек
должны быть близки по показателю преломления.
Чтобы скомпенсировать слабые флюктуации базовой линии, в канал
сравнения обычно устанавливается кювета подобранной толщины с
чистым растворителем. Однако всегда необходимо помнить, что там,
где пропускание растворителя падает ниже 30 %, сервосистема будет ра-
работать плохо и для получения правильных результатов необходима
коррекция усиления.
В специальных случаях в качестве растворителей можно применять
Н2О и (или) D2O. Используя очень тонкие кюветы (около 0,02 мм)
из KRS-5, BaF2, AgCl или Иртран-2 и регулируя щели спектрометра
так, чтобы скомпенсировать потерю энергии, можно получать вполне
приемлемые спектры водных растворов в области 830—1540 см
F,5—12 мкм) [20, 46, 100]. С D2O можно работать в другой спектраль-
спектральной области, но если этот растворитель используется с образцами,
* В СССР приняты следующие среднесуточные предельно допустимые
концентрации: 2,0 мг/мЗ для СС14 и 0,005 мг/мЗ для CS2.- Прим. перев.
Методы подготовки образцов
87
ю
1 1 1
1 1 '
л
[
)
i i i I! i
А
1
л
111
и
V
V
п
1400
1200
1000
1400
1200
Рис. 4.1. Влияние разбавления на спектр mpem-бутилового спирта.
а — 10%-ный раствор CS2 в кювете толщиной 0,1 мм; б — ]%-ный раствор в кювете толщиной 1 мм.
имеющими подвижный протон, то будет наблюдаться обмен. Водные
растворы можно исследовать и методом НПВО.
Влажность образцов влияет не только на их растворимость, но и
может приводить к помутнению окон кюветы. Влажный образец можно
высушить перед растворением, добавляя 2,2-диметоксипропан [30], кото-
который реагирует с водой в кислой среде с образованием метанола и
ацетона. Как реагент, так и продукты летучи и могут быть удалены
нагреванием.
Концентрация
Растворы обьино готовят, взвешивая определенное количество образ-
образца в мерной колбочке, а затем добавляя растворитель до метки.
Большинство органических веществ дает хорошие спектры при кон-
концентрациях 1 г/10 см3 в кювете толщиной 0,1 мм в интервале
625 — 4000 см B,5—16 мкм). Такие сильно поглощающие вещества,
как фтор- и кремнийорганические соединения, разбавляют до 0,2 г/10 см>
для области 625—1330 см G,5 — 16 мкм). Более высокие концентрации
и толщины могут понадобиться для работы ниже 600 см, за исклю-
исключением очень сильно поглощающих образцов.
88 Глава 4
Реакционноспособные или летучие вещества можно взвешивать в
колбочке с притертой пробкой, наполовину заполненной растворителем.
Аналогичным путем можно готовить образцы даже таких веществ,
которые кипят ниже комнатной температуры, при условии их охлажде-
охлаждения ниже температур кипения. Для работы с летучими, реакционно-
способными или радиоактивными растворами используют специальное
оборудование [87].
Очень важно стандартизировать растворители и условия разбавле-
разбавления, так как спектры могут резко изменяться при разбавлении или
изменении растворителей (рис. 4.1).
Толщина образца
На выбор толщины кюветы может влиять количество имеющегося
в распоряжении образца или его растворимость. Очень тонкие кюветы
(< 0,05 мм) не только трудно изготавливать, но и заполнять и опо-
опорожнять, в то время как кюветы толщиной 0,2 мм могут приводить
к чрезмерному поглощению растворителя. Удобным компромиссом
являются кюветы толщиной 0,1 мм. Для анализа следовых количеств в
областях высокой прозрачности растворителя могут использоваться кю-
кюветы с толщиной ггоглощающего слоя до 5 см. Однако перед тем, как
приготовлять образец с большой толщиной поглощающего слоя,
необходимо проверить, насколько удовлетворительно пропускание
растворителя в интересующей области спектра.
Проверка кюветы
Следует ожидать, что в конце концов окна кюветы станут мут-
мутными или покроются слоем адсорбирующихся загрязнений. О состоянии
окон не всегда можно судить по их внешнему виду, поэтому целесо-
целесообразно контролировать кювету, записывая скомпенсированный спектр
чистого растворителя. Ежедневная проверка спектра кюветы может
сберечь спектроскописту много бесплодных часов на попытки иденти-
идентифицировать ложные полосы.
Пленки
Одним из простейших методов приготовления образцов является
метод жидкой пленки. Он применим к нелетучим, нереакционноспо-
собным образцам и полезен в случае нерастворимых жидкостей или
для получения качественных обзорных спектров. Капля образца сжима-
сжимается между двумя солевыми пластинками или помещается на плоскую
стеклянную поверхность, а затем «вытирается» солевой пластинкой.
Желательно, чтобы в пределах сечения светового луча спектрометра
толщина образца была одинаковой. Очевидно, что спектры, полученные
таким путем, не очень воспроизводимы, и приходится прибегать к
Методы подготовки образцов 89
методу проб и ошибок, чтобы получить удовлетворительный результат.
Для исследования смол, лаков и других материалов, растворимых
в летучих растворителях, пленки можно получать испарением
растворителя. Тонкий слой раствора смолы, нанесенного на окно,
быстро высушивается под тепловой лампой, и полученные таким
способом спектры обычно свободны от мешающего влияния раство-
растворителя.
Образцы для съемки ИК-спектров водных эмульсий часто готовят,
высушивая несколько капель эмульсии на окне из AgCl. Непосредственно
эмульсии можно исследовать методом НПВО.
Растворы полимеров можно помещать на стекло, гладкую поверх-
поверхность пластика или ртуть, а после испарения растворителя регистри-
регистрировать спектры свободных пленок (без подложки). В спектрах пропуска-
пропускания таких пленок часто наблюдается интерференционная картина, ко-
которая усложняет спектр. Если это создает проблему, то от интерферен-
интерференции можно избавиться либо используя НПВО, либо располагая пленку
по отношению к лучу под углом Брюстера [58].
Суспензии
Сложности приготовления образцов твердых веществ, которые не-
нерастворимы в обычных растворителях для ИК-спектроскопии, чаще
всего возникают при их растирании до мелкодисперсных порошков,
образующих суспензии (взвеси) * в вазелиновом масле или КВг. В обоих
случаях цель состоит в создании однородного распределения частиц
в луче и в улучшении пропускания света взвешенными частицами в
среде, имеющей близкий с образцом показатель преломления.
Суспензии в вазелиновом масле
Вазелиновое масло** широко используется для приготовления
суспензий, но его недостатком является сильное поглощение в области
валентных и деформационных колебаний СН-связей ***. Это затрудне-
затруднение можно преодолеть путем использования хлорированных или фтори-
фторированных масел в области 1340—4000 см B,5 — 7,5 мкм).
Применяя подходящую технику, нетрудно приготовить хорошую
суспензию [14]. Важно, чтобы размер растертых частиц был меньше
длины волны ИК-излучения. Для этого малое количество вещества
(чем меньше, тем лучше, обычно не более 10 — 20 мг) растирают сначала
* По-английски — «mulls». — Прим. ред.
** Это высококипящая фракция нефти, содержащая насыщенные углеводо-
углеводороды. За рубежом его называют минеральным маслом. Имеется также фирменное
название «nujol» (нуйол) со средним составом C2S. — Прим. ред.
*** Это области 2800-3000 и 1350-1500 см.- Прим. ред.
2 -
я
с
о
с
IS
s
C
с
-е-
r-i
¦ч1
92
Глава 4
И 12 13
1000
Рис. 4.З. Эффект Христиансена в ацетате ртути (суспензия в вазелиновом
масле).
аккуратно в агатовой ступке, а затем превращают в тонкий порошок
энергичными движениями пестика. Далее порошок собирают на дне
ступки в плотную лепешечку, добавляют каплю вазелинового масла,
после чего продолжают растирание до получения однородной массы.
Полупрозрачная паста затем переносится на солевое окно с помощью
пластмассового шпателя и раздавливается вторым окном.
Секрет приготовления хорошей суспензии — в растирании. С малыми
образцами эта операция выполняется в течение 1 — 5 мин. Влияние на
спектр хорошего и плохого растирания показано на рис. 4.2. Когда
твердое вещество растерто не качественно, часть рабочего луча закрыта
непрозрачными частицами, часть — частицами подходящего размера, а
на некоторых участках они отсутствуют совсем. Поэтому прошедшее
излучение будет давать ложный нуль и в результате спектр получится
искаженным. Другое искажение спектра, показанное на рис. 4.3, возни-
возникает из-за эффекта Христиансена [101], который вызван большими
изменениями показателя преломления образца в окрестности полосы
поглощения (ср. с рис. 4.9). Так как количество рассеянного излучения
пропорционально квадрату разности показателей преломления образца
и матрицы, то образец хорошо пропускает по одну сторону полосы
поглощения, там где показатели преломления близки, и сильно
рассеивает по другую сторону. В результате такое асимметричное погло-
поглощение напоминает скорее дифференциальную кривую, чем спектр погло-
Методы подготовки образцов 93
щения. Эффект можно свести к минимуму, доведя размеры частиц до
величины, меньшей чем длина волны падающего излучения. Также
эффективно механическое размалывание в маленькой шаровой или
вибромельнице, но следить за этим процессом несколько сложнее, чем
при ручном растирании в ступке. Если нагревание или давление в ходе
механического размалывания влияет на кристаллическую структуру
образца (что бывает часто), то вид спектра будет зависеть от времени
растирания.
Таблетки с КВг
Метод взвесей в КВг, называемый еще методом прессования табле-
таблеток, впервые предложен в 1952 г. [105, 111]. Он заключается в тщательном
перемешивании тонкоизмельченного образца с порошком КВг (или
другим галогенидом щелочного металла) с последующим прессованием
смеси в пресс-форме, в результате чего получается прозрачная или
полупрозрачная таблетка. Наилучшие результаты достигаются при
откачке пресс-формы, что позволяет избавиться от включений воздуха
в таблетки. Преимущества метода прессования таблеток следующие:
1) отсутствие большинства мешающих полос поглощения, 2) возмож-
возможность контроля за концентрацией образца и 3) удобство хранения
образцов. Метод имеет также значительные недостатки, которые будут
обсуждены позже.
Для успешного приготовления таблеток с КВг могут использоваться
те же методы измельчения, что и в случае суспензий в вазелиновом
масле. Те же требования предъявляются и к размеру частиц. Наилучшие
результаты получают при тщательном растирании одного образца и при
последующем смешивании его (без растирания) с порошком КВг.
Растирание образца предпочтительнее проводить в агатовой ступке.
Иногда для облегчения растирания образца добавляют несколько капель
подходящего растворителя, такого, как хлористый метилен или гексан,
к смеси КВг — образец. Растворитель испаряется во время последующе-
последующего легкого растирания.
Оптимальное диспергирование растворимых образцов в матрице из
КВг достигается сушкой с охлаждением (лиофильной сушкой). Этот
метод наиболее пригоден для водорастворимых образцов, но может
быть использован и с веществами, растворимыми в органических
растворителях [101, 106]. Водный раствор образца и КВг заморажи-
замораживается в виде тонкой пленки льда на стенках вакуумируемого
стеклянного сосуда, охлаждаемого смесью сухого льда с ацетоном или
жидким азотом. Затем сосуд вакуумируется на несколько часов, в течение
которых вода сублимируется, оставляя сухой пушистый порошок.
Порошок прессуется в таблетку с использованием либо стандартной,
либо микротехники в зависимости от размеров образца. Вещества,
нерастворимые в воде, растворяют в таком органическом растворителе,
который сублимируется, подобно воде. Этот раствор вмерзает в корку
94 Глава 4
из замерзшего водного раствора КВг. Затем, как обычно, проводится
лиофилизация.
Как было показано, метод прессования таблеток особенно ценен при
исследовании волокон целлюлозы [90, 91] и шерсти [113], для которых
размалывание может привести к изменению степени кристалличности.
Показатели преломления КВг и этих волокон довольно близки, поэтому
относительно крупные частицы дают удовлетворительные спектры.
Остроумный метод анализа поверхностей красок, лаков, пластиков,
металлов или стекол с применением метода прессования таблеток с
КВг был описан Джонсоном [69, 70]. Порошок КВг используется для
абразивного истирания поверхности, при этом удаляется слой образца
толщиной 50—100 А. Затем из этого порошка прессуют таблетку и
получают вполне хорошие спектры. Если требуется, то обработку можно
проводить повторно и последовательно изучать различные слои. Для
того чтобы гарантировать воспроизводимость удаления слоев с поверх-
поверхности, можно использовать шлифовальный станок. Более быстрым исти-
истирающим действием обладает бромистый калий, смешанный с обрезками
стальной проволоки, которую потом удаляют магнитом. В качестве
примеров можно привести определение углеводородов на стекле, фтале-
вого эфира на нержавеющей стали и амидов на полиэтилене. Исследо-
Исследовались также причины адгезионного разрушения лакокрасочных покры-
покрытий. Для исследования распределения концентраций по толщине на
внутренних поверхностях артерий и вен они подвергались абразивному
действию струи порошкообразного КВг [71].
Значительные трудности встречаются при приготовлении образцов
упругих пленочных или каучуковых нерастворимых материалов. Сущест-
Существует элегантная методика для размола веществ в механической мельнице
после замораживания в жидком азоте [112, 117]. Стальной цилиндр
для размалывания, в котором находятся образцы и два шарика из
нержавеющей стали, выдерживается в жидком азоте до прекращения
кипения. Затем цилиндр помещают в механическую вибромельницу и
включают ее на короткое время. Такие материалы, как эпоксидная
смола, кора, табак, целлюлоза, бумага, ногти, превращаются в тонкий
порошок, который, как обычно, запрессовывается в таблетку с КВг.
Спектры материалов, подготовленных таким путем, имеют отличное
качество.
Метод прессования таблеток с КВг имеет некоторые существенные
недостатки, которые не всегда осознаются в полной мере. Вероятно,
наиболее серьезные проблемы связаны с изменением либо кристалли-
кристаллической структуры, либо состава образца. Спектры твердых веществ,
обладающих полиморфизмом, будут различаться в зависимости от
степени размола и величины давления (рис. 4.4) [7]. Изменения в спектрах
фенолов и органических кислот, очевидно, вызываются адсорбцией их
молекул на частицах галогенидов щелочных металлов [116]. В ходе
приготовления таблетки образцы могут реагировать с атмосферными
водой и углекислым газом, хотя имеются пути, позволяющие избежать
Методы подготовки образцов
95
Вазелиновое масло
МММ 1ТПЕ
* 7 в
Длина волны
7 U
Рис. 4.4. Полиморфизм в а-нафталинацетамиде, вызванный размалыванием.
Слева изображен спектр суспензии в вазелиновом масле, в середине и справа — спектры таблеток с
KI после размалывании на механическом вибраторе [7]. Reprinted with permission of American Chemical
Society.
этих трудностей [89, ПО]. Частичный или полный ионный обмен,
особенно для неорганических солей, солянокислых органических аминов
и других оснований, может приводить к таким спектральным измене-
изменениям, которые делают аналогичные исследования совершенно бесполез-
бесполезными. Эта проблема связана с адсорбированной водой, временем
и температурой, при которых происходит контакт соль — кислота, и
размером частиц КВг [18, 28]. В работе [85] сделан вывод о том, что
изменение свободной энергии было бы подходящим критерием для
предсказания ионной реакции обмена.
Полосы адсорбированной воды неизменно проявляются в областях
1640 и 3450 см, хотя вакуумная сушка и уменьшает их [7]. При
хранении таблетки часто мутнеют из-за образования в них микротре-
микротрещин [88].
Кажется наиболее целесообразным рекомендовать метод прессования
таблеток с КВг для образцов, которые 1) нерастворимы в обычных
ИК-растворителях, 2) аморфны или имеют устойчивую кристаллическую
структуру и 3) не содержат ионов, способных к обмену.
Нарушенное полное внутреннее отражение (НПВО)
Метод НПВО широко применяется для получения спектров «неу-
«неудобных» объектов, таких, как смолы, пищевые продукты или сырая
резина. Хотя явление полного внутреннего отражения впервые наблю-
наблюдалось еще Ньютоном, возможности его применения в ИК-спектроско-
пии были замечены только в 60-е годы [33, 34, 59]. В последние
годы происходили интенсивное развитие метода и быстрый рост
производства серийного оборудования, что сделало метод доступным
96
Глава 4
для спектроскопистов. Однако получение оптимальных результатов тре-
требует от исследователя понимания используемых в методе физических
принципов, которые здесь кратко изложены. Для более детального и
строгого знакомства с методом можно рекомендовать монографию Хар-
рика [59].
Основы
Рассмотрим сначала, что происходит, когда электромагнитное
излучение проходит через границу раздела между двумя прозрачными
веществами с различающимися показателями преломления. При нор-
нормальном падении луча на границу раздела (луч а на рис. 4.5)
излучение частично поглощается, а частично отражается. Доля отра-
отраженного излучения R выражается следующим образом:
R=(n2- naJ
(п2 + щJ '
где «! и п2 — показатели преломления сред у границы раздела *.
Далее рассмотрим луч Ь; луч Ь" — отраженный от границы раздела,
а луч Ь' — преломленный согласно закону Снеллиуса:
и! sin 9 = п2 sin ф. D.2)
Луч с ведет себя аналогичным образом. При увеличении угла 0,
до некоторой величины 9С угол ф возрастает до 90° и излучение
через границу раздела не проникает. Угол 6С называется критическим
Рис. 4.S. Поведение светового луча, выходящего из оптически более плотной
среды в менее плотную под различными углами к границе раздела.
* Необходимо отметить, что п2> п1у т. е. излучение падает на границу
раздела из оптически более плотной среды.— Прим. перев.
Методы подготовки образцов
97
—'), ж 1,33 -»<т,
.— л, =4 -<Tj - 1,33
10/20 30 40 50 80 70V80 90
Рис. 4.6. Зависимость отражения от угла падения для границы раздела
между средами с показателями преломления л, = 4 и я2 = 1,33 для перпен-
перпендикулярно (Л1) и параллельно (Л|0 поляризованного света по отношению
к плоскости падения в случаях внешнего отражения (сплошные линии) и внутрен-
внутреннего отражения (пунктирные лииии).
8С — критический угол; вц - угол Брюстера; 9р - главный угол (дополнительный к вц) [59]. Reproduced
with permission of Intencience Publishers.
Образец
Образец
5
Рис. 4.7. Схематическое представление нарушенного полного внутреннего
отражения.
а — призма однократного отражения; б —элемент многократного отражения.
7-586
98
,э
Волновое число,см"
1SOO 1400 1300 1200 «100 WOO
II I I I I I I I I I I I I I I I
в в 10
Длика волны, ики
Рис. 4.8. Сравнение спектров НПВО (верхний) и пропускания (нижний) клейкого вещества липкой ленты.
100 Глава 4
углом, и при 9 > 9С происходит полное внутреннее отражение.
Критический угол вычисляется по формуле
9С = arcsin —. D.3)
Можно отметить, что внутреннее отражение фактически совершенно
противоположно внешнему (от зеркала с наружным покрытием),
когда при каждом отражении теряется несколько процентов энергии
падающего излучения. При внутреннем отражении луч может претерпе-
претерпевать тысячи отражений без потери энергии, за исключением потерь
на поглощение средой. Для излучения, претерпевшего полное внутреннее
отражение, поведение его параллельно и перпендикулярно поляризо-
поляризованных компонент несколько отличается (рис. 4.6).
Использование спектроскопии НПВО основано на том факте, что,
хотя на границе раздела и происходит полное внутреннее отраже-
отражение, излучение на самом деле проникает на некоторую глубину в
оптически менее плотную среду (рис. 4.7, а). Это проникающее из-
излучение, называемое затухающей волной, может частично поглощаться
образцом при оптическом контакте с более плотной средой (роль
которой выполняет призма) в той точке, где происходит отражение.
Отраженное излучение дает спектр поглощения, который похож на спектр
пропускания образца (рис. 4.8). Однако это получается не так просто;
в действительности спектр НПВО зависит от нескольких параметров,
включающих показатели преломления призмы и образца, угол падения
излучения и площадь образца, число отражений, длину волны излучения
(а также от поляризации излучения. — Прим. перев.). Следующий раздел
посвящен тому, как оптимизировать эти переменные величины для
получения спектров, наиболее близких к спектрам пропускания.
Практика
Для получения спектра НПВО образец прижимается к рабочей по-
поверхности призмы или элемента многократного отражения (рис. 4.7,6),
через которую излучение посредством специальной оптической системы
направляется в спектрофотометр. Призма изготовляется из материала с
высоким показателем преломления, такого, как AgCl, KRS-5 или Ge
(табл. 4.1). Материал призмы должен быть прозрачным при толщине
до нескольких сантиметров, прочным, поддаваться полировке до
высокого класса и химически инертным.
Среди наиболее важных факторов, влияющих на спектр, можно
выделить показатели преломления призмы и образца. Показатель
преломления любого вещества резко изменяется в области полосы
поглощения, как это показано на рис. 4.9. Если лр и ns — показатели
преломления призмы и образца соответственно, a nsp = njnp (nip < 1), то
чем больше и,р (т. е. чем ближе друг к другу показатели преломле-
преломления призмы и образца), тем глубже внутрь образца проникает затухаю-
Методы подготовки образцов
101
Таблица 4.1
Характерные свойства оптических материалов, используемых для изготовления
элементов внутреннего отражения
Рабочая Средний Потери на
Матепиат область показатель Критический Твердость отражение Птммечани»
материал СПектраа, преломле- угол, град по Кнупу B поверхно- "Римечания
мкм ння сти), %
Хлорид серебра 0,4—20
Бромид серебра 0,5 — 30
K.RS-6, хлорид 0,4-32
таллия, бромид
таллия
Алмаз 1 — 3,8
5,9-100
KRS-5, бромид 0,6-40
таллия, иодид
таллия
Се лени д цинка 0,5—15
(Иртран-4)в
Теллурид кад- 1 — 23
мия (Иртран-6)
Селенид 1 — 12,5
мышьяка
Кремний 1,1 — 6,5
33-150
Германий 2—12
? Толщина слоя материала
° О типах алмаза см. [5).
В отечественной литературе
2,0
2,2
2,2
2,4
2,4
2,4
2,7
2,8
3,4
_
4,0
2 см, кроме
КО-4. - Прим,
30
27
27
25
24,6
24,6
22,2
20,9
15,6
14,5
алмаза,
перев.
9,5
7000
40,2
150
45
1150
600
толщина
20
26
26
31
31
31
38
39
52
59
которого
Светочувствите-
Светочувствителен, реагирует с
металлами
Тип П-аб
Хрупкий, под
действием кис-
кислот выделяет
H2Se
Хрупкий, неус-
неустойчив к дейст-
действию щелочи
составляет около 1 см.
щая волна и тем более контрастным получается спектр. Однако в
сильно поглощающих образцах могут быть области, в которых пока-
показатель преломления превосходит ту же величину для призмы и излуче-
излучение не будет больше испытывать полного внутреннего отражения
(а только зеркальное. — Прим. перев) (рис. 4.9). В результате спектр
будет искажен. Это не относится к случаю очень тонких образцов,
толщина которых меньше глубины проникновения затухающего го-
лучения.
Призмы НПВО могут иметь различную геометрию и рабочие
углы (как правило, 45 и 60°). В любом случае, для того чтобы происходи-
происходило полное внутреннее отражение, угол падения 8 должен быть
102
Глава 4
Показатель преломления образца
.Полоса поглощения образца
Показатели преломления элементов НПВО
Полос» поглощения,
обусловленные внутренним
отражением
—•-. ,-•- "з
Длина волны Л —»•
Рис. 4.9. Влияние показателя преломления призмы НПВО на полосу погло-
поглощения (пунктирная линия), имеющую показатель преломления я$ (сплошная
линия), близкий к показателям преломления призм л, (штрих-пунктириые
линии). Получающиеся в результате внутреннего отражения полосы поглоще-
поглощения приведены для п^<п3; ns~n2; па"> щ. Courtesy American Society for
Testing and Materials [2]. Copyright 1977.
больше критического угла 8С. Так как критический угол меняется
по полосе поглощения, то рабочий угол 8 должен быть заметно
больше, чем 8С (обычно на 4 - 5°. - Прим. перев.). Выбор очень большого
рабочего утла 9 нецелесообразен из-за уменьшения глубины проник-
проникновения с увеличением 8. В качестве эмпирического правила определения
8 Уилкс и Гиршфельд [127] предлагают при вычислений критического
угла прибавлять 0,2 к показателю преломления образца и затем 3°
к этому критическому углу для компенсации сходимости луча. Влияние
угла падения на спектр НПВО показано на рис. 4.10 и 4.11 и
обсуждено Уилксом [126]. Необходимо заметить, что «искаженные»
спектры, полученные с элементами из низкопреломляющих материалов
и с малыми углами падения, полезны при определении оптических
постоянных веществ [40, 64].
Другими факторами, определяющими качество спектра, являются
число отражений (которое увеличивается при уменьшении угла паде-
падения) и площадь контакта образца с элементом НПВО. Число отраже-
отражений* можно увеличить, используя более тонкий и длинный элемент
многократного отражения.
* Число отражений N связано с длиной элемента I, его толщиной t и
рабочим углом в соотношением Af = -ctgв.— Прим. перев.
Методы подготовки образпов
103
4000 3000
1 ^°
a.
20
n
1 1 '
-
-
1 1
2000
1500
л И
И*
г
1 1
CM"'
1 1 '
?[/
У v/
1000
I
\
V
V
1
900
ч
* л
\М
i
800
1 1
^ V
1
1
1
700
1 1
' 1 -
1/
-
1
100
80
60
40
20
0
6
12 13 14 15
8 9 10 11
Длина волны, мкм
Рис. 4.10. Спектры НПВО сорбцновой кислоты с элементом из KRS-5.
Верхний спектр получен при 8=45С, нижний — при 8 =60; [126]. Reproduced, with permission, from Applied
Spectroscopy.
Глубина проникновения dp имеет тот же порядок величины, что
и длина волны. Ее можно вычислить по формуле
D.4)
где Xj — длина волны излучения в призме, равная Х/пр. Влияние этого
фактора на спектр состоит в том, что полосы в длинноволновой
области более интенсивны, чем в коротковолновой, по сравнению со
спектром пропускания того же вещества (ср. с рис. 4.8).
Поскольку глубину проникновения варьируют, меняя либо материал
призмы НПВО, либо угол падения, либо то и другое (и особенно
если имеется возможность проводить вычитание спектров с использо-
использованием ЭВМ), можно в принципе получать профиль концентраций по
глубине от поверхности. Эта проблема обсуждалась Томпкинсом [118]
и Гиршфельдом [66], которые утверждают, что при благоприятных
условиях можно проникнуть на глубину до 2Х и разрешить отдель-
отдельные слои толщиной Х./10 — А/Ж
Однако не следует делать вывод о том, что проникающий луч
104
Глава 4
1200
1000
800
600
400
Рис. 4.11. Спектры НПВО NaClO3, показывающие влияние угла падения;
призма KRS-5.
0-9 = 60°; 6-45°; «-8 = 30°; г - спеггр пропускания суспензии в вазелиновом масле [16]. Repro-
Reproduced, with permission, from Spectrochimica Acta.
обрывается на определенной глубине в оптически менее плотной
среде. Величина его электрического поля падает экспоненциально,
согласно рис. 4.12, и dp определяется просто как глубина, на которой
поле убывает в е раз по сравнению с начальным значением.
В повседневной практике спектры НПВО большинства образцов
Методы подготовки образцов
105
Рис. 4.12. Амплитуды стоячей волны, возникающей вблизи поверхности, на
которой происходит полное внутреннее отражение.
Амплитуда электрического поля имеет синусоидальную форму в оптически более плотной среде 1 и убывает
экспоненциально в оптически менее плотной среде 2. Reproduced, with permission!, from Harrick [59].
получают, используя элемент многократного отражения из KRS-5 с
углом 45°. Контрастность спектра регулируется изменением площади
контакта. Когда размеры образца ограничены, гарантировать наи-
наилучшие условия получения спектров можно только при использовании
активней зоны элемента. Так как излучение, попадающее на диспергирую-
диспергирующий спектрофотометр, характеризует поверхность элемента НПВО
неравномерно, то на этой поверхности можно обнаружить небольшие
области максимальной чувствительности, или «горячие точки». Активные
области можно найти, последовательно закрывая узкими полосками
липкой ленты соседние участки элемента [92]. Образец должен по воз-
возможности целиком покрывать рабочую грань элемента. Если этого не
удается сделать, то положение нулевой линии искажается подобно
тому, как это происходит в случае пропускания пленки с отверстием
или жидкостной кюветы с пузырьком. Спектры наилучшего качества
получаются при использовании в канале сравнения подобранной под
106 Глава 4
пару приставки НПВО с элементом. Такая схема позволяет компенси-
компенсировать атмосферное поглощение и оптические потери. В связи с тем что
от 20 до 60 % излучения теряется на границе раздела элемент — воз-
воздух, для сохранения подвижности сервосистемы необходимо увеличить
щель (или усиление) (стр. 50-55).
Для контроля качества новых элементов внутреннего отражения
необходимо записать их спектры. В предполагаемом для использования
интервале длин волн запись спектра пропускания элемента не должна
иметь наклона или полос. В случае плохо отполированных элементов в
коротковолновой части спектра наблюдается рассеяние. Допуски
к плоскостности и углам элементов внутреннего отражения довольно
жесткие. По этой причине, а также из-за трудности изготовления
высококачественно отполированных поверхностей лучше не пытаться
переполировывать поцарапанный элемент, а вернуть его изготовителю
для переделки.
Подготовка образца
Зачастую при использовании метода НПВО наибольшие затруд-
затруднения вызывает получение воспроизводимого оптического контакта
между элементом внутреннего отражения и образцом. В случае
мягких образцов, таких, как эластомеры, каучуки или адгезивы,
проблем не возникает и с элементами многократного отражения
получаются достаточно интенсивные спектры. Волокна можно плотно
намотать на элемент. Для гибких пленок, волокон, бумаги, тканей
хороший оптический контакт обеспечивается с помощью резиновой
прокладки, которая одновременно предохраняет элемент от поврежде-
повреждений. Нужно только следить за тем, чтобы эта прокладка не
контактировала с поверхностью элемента, что может привести к по-
появлению дополнительных полос в спектре. Винтовые прижимные устрой-
устройства предохраняют образец от слишком сильного поджатия в держателе
во избежание деформации или разрушения элемента МНПВО.
В спектрах НПВО порошков отсутствуют коротковолновое рассея-
рассеяние и эффект Христиансена, которые встречаются в абсорбционных из-
измерениях [16, 60]. Порошки можно суспензировать в летучем раство-
растворителе, а затем, испарив его, осадить на поверхность элемента
(однако в случае анизотропных порошков может происходить их
самопроизвольная ориентация на поверхности, что сказывается на
спектрах при использовании поляризованного излучения). Порошки
можно наносить на липкую ленту, которая затем легко поджима-
поджимается к элементу. Лучшие результаты получаются для довольно тонко
растертых и однородных по размеру частиц порошков, и, конечно, липкий
слой должен быть полностью покрыт порошком и не проявляться
в спектре.
При подготовке твердых образцов могут возникнуть некоторые труд-
трудности. В случае хрупких материалов их лучше всего измельчать
и готовить к съемке, как обычные порошки. Если же нужно
Методы подготовки образцов 107
получить спектр плоской твердой поверхности, то оптический контакт
часто можно значительно улучшить, нанося на поверхность образца
тонкий слой вазелинового масла*. Если полосы вазелинового масла
перекрывают спектр образца или поверхность образца слишком груба,
чтобы дать спектр, то между элементом и образцом зажимают
пластинку AgQ толщиной 0,5 мм [63].
Жидкости дают хорошие спектры НПВО, и водные растворы успешно
исследовались этим методом [1]. Разработан специальный элемент
в виде вертикальной пластинки двойного прохождения, которая непосред-
непосредственно погружается в исследуемую жидкость [57]. Глубина погруже-
погружения определяет контрастность спектра.
В случае микрообразцов наилучшие результаты достигаются тогда,
когда образец в виде исключительного тонкого слоя наносится
на всю рабочую поверхность элемента [43]. При толщине образца
менее 1 мкм спектр не искажается даже вблизи критического угла на
границе раздела элемент НПВО — воздух. Если же образец нельзя
нанести в виде такого тонкого слоя, то для его более эффективного
применения можно либо воспользоваться комбинацией элемента од-
однократного отражения со световым конденсором, либо взять отража-
отражательные элементы специальной формы или уменьшенного размера.
Наибольшая чувствительность достигается при подборе таких условий,
которые дают максимальную глубину проникновения [62].
Очистка элементов НПВО должна проводиться осторожно и при
минимальном контакте с полированными поверхностями. Остатки
твердых или порошкообразных образцов удаляют, прижимая липкую
ленту к образцу, а затем снимая ее. Обычно достаточно споласкивать
элемент в ряде растворителей (ацетон, толуол, метилэтилкетон), держа
его вертикально, чтобы лучше высушить. В случае трудных образцов
может потребоваться 30-кратная обработка растворителем в ультразву-
ультразвуковой ванне. При сушке элемента НПВО последняя капля растворителя
на нижнем конце удаляется промоканием бумажной салфеткой. Элемент
можно брать только за нерабочие грани, используя резиновые напальч-
напальчники. Плазменная очистка применяется только для элементов из
кремния или германия, но не из KRS-5.
Практическое руководство по спектроскопии НПВО имеется в
ASTM (метод Е-573-76) [2, с. 435].
Газообразные образцы
Обычно особых трудностей с подготовкой газообразных образцов
нет, за исключением тех из них, которые реагируют с материалами
окон или корпуса кюветы. В этом случае для конструирования
* Вообще говоря, вазелиновое масло не всегда можно использовать таким
образом. Показатель преломления иммерсионной жидкости должен быть проме-
промежуточным между показателями преломления материала НПВО и образца. -
Прим. перев.
108 Глава 4
кюветы следует использовать специальные материалы. В качестве
окон можно брать полиэтиленовую пленку, но она не выдерживает
больших перепадов давлений*. Прокладки лучше использовать из
таких инертных материалов, как витон или тефлон, поскольку другие
материалы могут загрязнять образцы из-за адсорбции и десорб-
десорбции [52].
Для уменьшения влияния уширения полос за счет соударений
(стр. 180—182) давление в кюветах обычно доводят до атмосферного
одним из инертных газов, например сухим азотом. Такая процедура
увеличивает чувствительность к следовым количествам составных
частей (за исключением измерений, сделанных при высоком разреше-
разрешении), а также позволяет проводить количественные измерения. Це-
Целесообразно предусмотреть некоторые меры для перемешивания газов,
так как их взаимная диффузия замедленна. В качестве мешалки
может служить небольшой кусочек тефлона, который встряхивают
внутри кюветы.
В тех случаях, когда требуется высокая чувствительность, как,
например, при исследованиях загрязнений атмосферы, очень полезны
многоходовые газовые кюветы с большой длиной оптического пути,
предложенные Уайтом [124]. Промышленность выпускает оптические
кюветы с длиной оптического пути до 120 м, а в работе [97]
описана специальная кювета с общей длиной пути 1 км. Сообща-
Сообщалось [55], что для таких загрязнений атмосферы, как СО, N2O, NO,
NO2, HNO3, О3 и СгН4, может быть достигнута чувствительность
0,1-1 частей на миллиард.
Анализ следовых количеств в многоходовых газовых кюветах
значительно затрудняется селективной адсорбцией или десорбцией
веществ со стенок кюветы. Перед съемкой спектра кювета должна быть
«промыта», т. е. многократно заполнена и откачана. В некоторых
случаях для получения точных данных об истинном составе может
оказаться необходимым поддерживать непрерывный поток свежего
образца, протекающего через кювету.
Следовые количества вредных и ядовитых паров, содержащихся
в атмосфере, можно адсорбировать на древесном угле, помещенном
в трубочку, а затем элюировать растворителем для идентификации
по ИК-спектру. В обычную ловушку помещают 150 мг древесного
угля, через который со скоростью 1 л/мин прокачивают 10 л воздуха.
В одной из работ [26] для извлечения загрязнений из угла исполь-
использовали 0,5 мл CS2. При комнатной температуре экстракция проис-
происходит примерно на 50 %, но было показано, что охлаждение угля
вместе с CS2 до температуры жидкого азота повышает ее эф-
* Листовой полиэтилен низкого давления толщиной 1 мм хорошо вы-
выдерживает при ножевом уплотнении перепад давлений около 1 атм.—
Прим. ред.
Методы подготовки образцов 109
фективность до 80—100%. После 5—10-минутной экстракции CS2
переносится в жидкостную кювету толщиной 6 мм (объем 0,43 мл),
в канал сравнения помещается кювета с растворителем н произво-
производится запись спектра. Из десяти исследованных соединений девять
можно идентифицировать при концентрации 1 объемная часть на
миллион или меньше.
Специальные методы подготовки образцов
Отражение
Очень тонкие покрытия (около 0,01 мм) на гладких металли-
металлических подложках можно исследовать за счет отражения излучения
от металлической поверхности, помещая образец в обычную при-
приставку зеркального отражения. Пленки, толщина которых значительно
меньше, чем длина волны излучения, не дают спектров поглощения,
если излучение направлено перпендикулярно металлической поверхно-
поверхности; это обусловлено возникновением стоячей электромагнитной вол-
волны с узловой точкой вблизи отражающей поверхности. Молекулы в
узловой точке не взаимодействуют с излучением [48]. Используя
скользящее падение и многократное отражение, можно получать
ИК-спектры таких тонких слоев, как монослои [11, 119]. Рассматри-
Рассматриваемые системы применялись при исследованиях адсорбции СО на
свежих металлических поверхностях [119] и окисления металлов [98].
Диффузное отражение в ИК-области почти не использовалось,
вероятно, из-за экспериментальных трудностей, но на специально
сконструированном интерференционном спектрофотометре работа та-
такого рода стала возможной [128]. В ближней ИК-области спектро-
фотометрия диффузного отражения оказалась полезной при количе-
количественном анализе содержания белков, жиров н влаги в зерне и
других сельскохозяйственных продуктах [122].
Пиролиз
Когда все попытки получить спектр терпят неудачу, трудные об-
образцы можно подвергнуть пиролизу или сухой перегонке с последую-
последующим анализом ИК-спектров летучих продуктов [56]. Образец весом
1 г или меньше измельчают и помещают в пробирку из боросили-
катного стекла, расположенную горизонтально. Дно нагревается на
пламени горелки Бунзена, и образующиеся летучие продукты соби-
собираются в горловине. Они легко могут быть перенесены на солевое
окно для съемки ИК-спектра. Контролируемый пиролиз на серийном
оборудовании дает более воспроизводимые результаты и позволяет
также исследовать образцы в парообразном состоянии [120].
Как было найдено, в большинстве случаев спектры пиролизатов
ПО Глава 4
похожи на спектры исходных соединений. Даже тогда, когда этого
не наблюдается, спектры вполне воспроизводимы, поэтому их можно
сравнивать, если они подготовлены тем же способом. Например,
при пиролизе неизвестных полимеров были идентифицированы полиуре-
полиуретаны [72]; волокна, используемые при изготовлении ковров, дают
различающиеся продукты пиролиза [95].
Спектры при высоких
и низких температурах
Обычно нецелесообразно регистрировать рутинные спектры при
температурах, сильно отличающихся от комнатной, так как на
нагревание требуется время. Однако иногда возникают задачи, кото-
которые могут быть решены только при использовании высоко- или
низкотемпературных методик. Сейчас выпускаемые промышленностью
кюветы лучше, чем самодельные, но для решения специальных
задач в литературе имеется описание целого ряда конструкций.
Для температур, лишь ненамного превышающих комнатную, впол-
вполне пригодны термостатированные держатели кювет, нагреваемые цир-
циркулирующей жидкостью или электрическим током. Когда важно знать
температуру образца, в него вводят тонкую термопару, так как
измерения, выполненные в других частях кюветы, могут оказаться
в значительной степени ошибочными. Расплавы твердых веществ при
высоких температурах исследуются в специальной кювете, в которой
образец удерживается на платиновой сетке (силами поверхностного
натяжения) [13, 47].
В высокотемпературной ИК-спектроскопии кроме затруднений, свя-
связанных с достижением и поддержанием требуемой температуры,
имеются еще два. Горячая кювета с образцом может нагревать
спектрометр, но, изолировав держатель, этот эффект можно свести
к минимуму. Вторая, и более серьезная, проблема заключается в
том, что сам образец является источником ИК-излучения. Для устра-
устранения этого эффекта применяют либо однолучевой спектрометр, либо
регистрирующий электрическое отношение двухлучевой спектрофото-
спектрофотометр, в которых ИК-излучение модулируется до того, как оно
пройдет через образец (при этом приборы не реагируют на непромоду-
лированное излучение, испускаемое образцом)*.
Конструирование низкотемпературных кювет отчасти более сложное
дело, так как образец должен быть изолирован от атмосферной
влаги, которая может конденсироваться на нем. Это обычно дости-
достигается вакуумированием, что вызывает очередную трудность - необ-
необходимость разработки герметичной оболочки для кюветы. Эту пробле-
* Из отечественных приборов для этой цели подходят спектрометры
ИКС-21 и ИКС-31.- Прим. ред.
Методы подготовки образцов
111
Частота,см
Рис. 4.13. Участок спектра моногидрата лактозы (суспензия в вазелиновом
масле) при температурах 298 и 113 К (вверху) и 20 К (внизу). При более
низких температурах нужно меньше вещества [75]. Reproduced, with permission,
from Applied Spectroscopy Reviews.
му часто решают, предварительно охлаждая кювету и затем напыляя
образец на холодное окно, где он немедленно замораживается и
не создает заметного давления пара.
Метод, называемый матричной изоляцией [9, 27, 54, 84], широко
используется для изучения неустойчивых молекул и свободных ра-
радикалов. В нем образец испаряется (например, из маленькой печки)
и непрерывно намораживается на холодное окошко* вместе с го-
гораздо большим количеством инертного газа — азота или аргона, ко-
который образует матрицу. Концентрация молекул образца в заморо-
замороженной инертной матрице настолько низка, что они не взаимодейст-
* Как правило, в этом методе используется температура, близкая к
температуре жидкого гелия A0-20 К).-Прим. перев.
112 Глава 4
вуют друг с другом. После осаждения достаточного количества ве-
вещества производится запись спектра.
Сходные методики для качественного анализа сложных газовых
смесей названы псевдоматричной изоляцией [102, 103]. Здесь образец
напыляется импульсно за очень короткое время. Считается, что этот
метод довольно чувствителен к малым количествам примесей.
Описана 2-метровая многоходовая кювета для получения спектров
веществ, растворенных в жидком аргоне [68].
Низкотемпературная спектроскопия нашла применение при исследо-
исследовании некоторых типов веществ, имеющих при комнатной темпера-
температуре плохо разрешенные спектры (рис. 4.13), таких, как аминокислоты
и полипептиды [36], целлюлозные материалы [82], сополимеры по-
ливинилацетата и акрилата [53], углеводы [74, 75]. Метод можно
использовать для того, чтобы различать вещества, спектры которых
при комнатной температуре почти одинаковы (например, к-гексил-
бромид и к-гептилбромид) [17].
Калибровка температуры кювет может проводиться по известным
точкам плавления ряда веществ [38]. Германн [61] опубликовал ис-
исчерпывающий литературный обзор по низкотемпературной спектро-
спектроскопии вплоть до 1968 г.
Спектрофотометр воспринимает холодный образец как поглотитель
тепла, и, если световой пучок не модулируется до того, как он пройдет
через образец, спектр будет искажен (в том смысле, что действи-
действительное положение нуля значительно смещено). Этот эффект не имеет
особого значения при качественной регистрации спектров, но при
более точной работе его нужно учитывать.
Сочетание ИК-спектроскопии
с хроматографией
Фракции, выходящие из газового хроматографа, можно собрать,
а их спектры записать как от сконденсированных жидких образцов
[10], так и от паров по мере их выхода из хроматографа, используя
метод остановленного потока («stopped-flow») или проточный метод
(«on-the-fly») [6]. Улавливание фракций обычно требует умелого обра-
обращения с малыми количествами образца A0—100 мкг) и в этой
связи представляет меньший интерес, чем методики, позволяющие
проводить измерения в момент выделения. Однако преимущество
первого способа состоит в том, что можно получать спектры, со-
сопоставимые с эталонными. Низколетучие продукты улавливают на
порошкообразном КВг, помещенном в капилляр, из которого прес-
прессуют таблетки [93].
Спектры хроматографических фракций обычно записьюают в виде
паров при 180—200 °С. Методы улавливания паров или остановленной
струи можно применять с обычными ИК-спектрофотометрами [15,
23, 81]. Используя специальную хроматографическую приставку, в
00
I
ел
4000
a
is
XOQ
moo
%
r
-I
¦
m
2000,
1500
1 J J
J
I
1
1300 1200
1100
9
1000
1
V
900
I
1
1
1 1
MO
*
Ml
ем-i
1 "'I
z
э
1
¦ ;
1
700
.
|:
]
I1 ¦
4
¦
1
0
Длина волны, мкм
1200 1200 1100 1900 МО
1
2.T
Длина волны, мкм
14
Рис. 4.14. ИК-спеггры газохроматографической фракции — .ме/ив-метилстнрола.
а - однократное сканирование » течение 30 с; 6-16-«ратяое ожанирование F с на одно оанирование) с усреднением сигналов [41]. Reproduced, with permission,
from Applied Spectroscopy.
114 Глава 4
которой созданы оптимальные условия для ввода образца, можно
повысить чувствительность, обычно недостаточную для регистрации
малых количеств [107]. Применяется также «вкалывание» в хроматограф
повышенных объемов проб (< 100 мкл), а хорошее разделение фрак-
фракций достигается при увеличении давления во всей системе до 120 Н/см2.
Падение давления в колонке относительно мало, поэтому остановка
газового потока на время сканирования ИК-спектра не ухудшает
разрешения хроматографа.
Проточный метод требует использования либо специального ско-
скоростного диспергирующего спектрофотометра [73], либо интерферен-
интерференционного спектрометра. Наилучшая чувствительность достигается при
оптимальном соотношении объема кюветы, скорости потока и вре-
времени сканирования [50, 121].
В литературе имеются примеры анализов при совместном исполь-
использовании газового хроматографа и диспергирующего спектрофотометра
[41]. Спектры, показанные на рис. 4.14, получены от тазохроматогра-
фической фракции нефти. Методом хроматомасс-спектрометрии была
установлена молекулярная формула этой фракции — С10Н14, которой
отвечает структура либо индана, либо одного иэ изомеров метил-
стирола. Даже если качество этого спектра не сравнимо с качеством
спектра, полученного при более медленном сканировании и для
образца большего объема, и то с уверенностью можно сказать, что
эта фракция — .м-метилстирол. В других примерах, приведенных в
указанной статье, для идентификации выделенных микрообразцов
требуется применение таких дополнительных методов, как ЯМР и
спектроскопия комбинационного рассеяния света. Поскольку эти ме-
методы требуют 0,1 — 1 мкл вещества, они наиболее ценны, когда в
распоряжении имеется соответствующее количество образца. Кроме
того, они позволяют быстро разделять и характеризовать компо-
компоненты, не прибегая к фракционной перегонке.
Благодаря тому, что интерференционные спектрометры имеют пре-
преимущества перед остальными в условиях ограниченной энергии излу-
излучения, они дают лучшие результаты, особенно когда размеры образца
малы. В одном из исследований было показано, что всего 10 нг
изобутилметакрилата, проанализированные в потоке, после усреднения
16 сканирований и сглаживания дают вполне интерпретируемый спектр
с разрешением 8 см (рис. 4.15). Интерферометр был снабжен ох-
охлаждаемым детектором на основе теллурида ртути и кадмия.
Улавливание газохроматографической фракции в кювете повышает
чувствительность, улучшает отношение сигнал/шум и позволяет про-
проводить усреднение по большему числу сканирований (при условии,
что образец не разлагается при температуре кюветы). Обзор при-
приложений ИК-спектроскопии в газовой хроматографии представлен
Фриманом [39] и Литтлвудом [80], но, поскольку развитие методов
происходит быстро, лучшим источником информации является текущая
литература.
Методы подготовки образцов
115
CONDITIONS
SAMPLE:
DATE:
INJECTION:
OVEN:
INJECTION PORT
DETECTOR
LIGHT PIPE:
TRANSFER LINES:
COLLECTION VENT
CARRIER:
COLUMN:
CAN:
TIM:
HANOt.
ATTENUATION:
SFIITTEB RATIO:
SCAN MODE:
CHART:
MISC
ISOBUTYLMETHACRYLATE
2J17/77
0.5 0L t20 *G/»»L MECL9)
120 ISOTHERMAL
250
250 FID
120
200
200
HE. 30CC/MIN
10» OV-17 ON 00/100 MESH
CHROM WIHP) в' X l/Г SS
2
N.A.
N.A.
N.A.
N.A.
EV IEVERY MOOEI
1MIN/INIJMV F.S.I
10 NO TO IRON THE FLY
TOP: UNSMOOTHID
M.SS
10 КАМЮ «CAN RtF.
¦OTTOM: SMOOTHED
волновое число, см"
Рис. 4.15. Спектр 10 нг изобутилметакрилата, снятый в потоке на
ИК-фурье-спектрометре. Верхний спектр без сглаживания, нижний — со сглажи-
сглаживанием [121]. Reproduced, with permission, from Applied Spectroscopy.
Спектр газовой фазы при повышенных температурах заметно отли-
отличается от спектра конденсированного состояния при комнатной тем-
температуре (наибольшие отличия наблюдаются для легких молекул и
тех, которые в обычных условиях образуют водородные связи). Час-
Частоты полос также смещаются. Для идентификации могут понадо-
понадобиться эталонные спектры, зарегистрированные в близких условиях [123].
ИК-спектроскопию можно применять совместно с другими ви-
видами хроматографии. Из-за малого количества образца трудно исполь-
использовать ИК-спектроскопию для идентификации веществ, разделенных
методом тонкослойной хроматографии. В качестве такого приема
предложено переносить адсорбент с пятном с хроматограммы и
экстрагировать вещество, но толщина слоя адсорбента делает эту
операцию сомнительной - [25]. Подход in situ описан Гриффитсом и
сотр. [44, 94]. Они сообщили, что с помощью фурье-спектрометра
обнаружено 1 — 10 мкг адсорбированных пестицидов. Адсорбент (си-
ликагель или оксид алюминия) нанесли на подложку из AgCl тол-
толщиной 100 мкм. После хроматографирования образца пятна про-
проявили в парах иода. Затем пластинку поместили в спектрофотометр
и на область пятна направили свет от источника с помощью кон-
конденсора D х). Усреднение проводили по 400 сканированиям с раз-
116 Глава 4
решением 4 см~'. Спектры, полученные на однолучевом спектрометре,
сопоставляли со спектрами от соседних, свободных от образца участ-
участков пластинки. Так были отделены полосы адсорбента. Обработка
пластинок фторуглеродной смазкой или вазелиновым маслом заметно
снизила рассеяние коротковолнового излучения. Целенаправленное
развитие тонкослойной хроматографии по пути усложнения и исполь-
использования спектрометра, имеющего более чувствительный детектор, по-
позволило зарегистрировать приемлемый спектр 10 нг метиленового
синего [45].
ИК-спектры полимеров и других объектов, разделенных методом
гель-проникающей хроматографии, можно получить, удалив раствори-
растворитель испарением с выбранных участков хроматограммы и проведя
с остатком те же операции, что и с микрообразцом. Фракции
гель-хроматограммы можно детектировать, используя микрокювету
толщиной 1 мм и устанавливая спектрометр на фиксированное вол-
волновое число [24]. Если требуется провести детектирование при другой
длине волны, то образец следует заменить.
Работа с микрообразцами
При анализе микрограммовых количеств вещества [12] техника под-
подготовки образцов имеет особое значение. Ничто не может заменить
практический опыт, и время, потраченное на проведение предвари-
предварительных измерений образцов известного состава перед работой с не-
неизвестным объектом, не будет потеряно зря. Рекомендуемые мето-
методики ИК-микроанализа собраны в ASTM (метод Е-334-68) [2].
Предприятия, поставляющие приборы, производят также дополни-
дополнительные приспособления различных типов для работы с микрообраз-
микрообразцами: объективы-микроскопы, газовые кюветы малого объема, жид-
жидкостные микрокюветы и микропресс-формы для изготовления таблеток
с КВг. Удовлетворительное оборудование можно изготовить и в
лаборатории.
При работе с микрообразцами исследователь должен так оптими-
оптимизировать энергию пропускания, чтобы получить наилучший спектр.
Что лучше: сконцентрировать образец на небольшой площади и
уменьшить диаметр луча или нанести образец на большую площадь,
чтобы избежать виньетирования (частичного диафрагмирования) луча
я использовать растяжку по ординате для повышения интенсивности
спектра? Ответ на этот вопрос в некоторой степени зависит от
типа используемого спектрометра и возможности представлять данные
по поглощению в цифровом виде. Однако, вообще говоря, площадь
сечения пучка в месте расположения образца должна быть как
можно меньше, но в то же время соответствовать требованию
заполнения щелей светом при их максимальной ширине. Некоторая
потеря энергии допускается из-за виньетирования верхней и нижней
частей щелей. Эта потеря существенно не зависит от длины волны
Методы подготовки образцов 117
и может компенсироваться использованием ослабителя в канале срав-
сравнения и расширением щелей для поддержания работы сервосистемы.
Когда нельзя заполнить щели по всей ширине, потеря энергии
становится зависимой от длины волны, а это будет вызывать замедлен-
замедленное движение пера в некоторых областях и возможное отклонение
от линии фона (линии 100 %). При анализе малых образцов полезен
световой конденсор, уменьшающий без значительной потери энергии
сечение пучка в месте положения образца.
Для увеличения интенсивности спектра может использоваться рас-
растяжка по ординате, но, так как при этом шум возрастает во
столько же раз, что и интенсивность полосы, щели нужно увели-
увеличить в корень квадратный раз из коэффициента растяжки, а усиление
соответственно уменьшить. Увеличиваются также наклон линии 100 °/а
и другие артефакты, поэтому одновременно с растяжкой по ординате
редко удается записать спектр в широкой области. Однако, если
спектр представить в цифровом виде, можно точно скорректировать
линию 100 % и растяжка по ординате становится практически выгодной.
Этот процесс более полно будет описан в настоящем разделе позже.
Техника прессования таблеток с КВг широко применяется для при-
приготовления микроОбразцов нелетучих твердых веществ [5, 77]. Обычно
для диспергирования образца в КВг используется лиофилизация, так
как измельчение и количественный перенос нескольких микрограмм
вещества сопряжены с некоторыми трудностями. Кроме того, измель-
измельчение и другие манипуляции вносят загрязнения в количествах, зна-
значительно превышающих концентрацию образца. Необходимо еще раз
подчеркнуть, что работа с микрограммовыми количествами образца
независимо от методики должна выполняться только с исключительно
чистым оборудованием и с предельной осторожностью.
Разработана система концентрирования микрообразцов в матрице
из КВг [42]. Треугольный кусочек КВг из слегка спеченного порошка
(фирменное название «Wick-Stick») помещается в стеклянную ампулу,
куда вносятся образец и растворитель. Тот и другой мигрируют
в КВг, и растворитель испаряется с верхней части элемента из КВг,
Образец остается на кончике, который отламывают и прессуют в
микротаблетку. Спектры могут быть получены примерно от 50 мкг
вещества, а образец подвергается только минимальной обработке.
Приготовление микрообразцов даже слаболетучих веществ в виде
таблеток с КВг часто дает неудовлетворительные результаты из-за
потерь при испарении. Гриффите и Блок [51] испытывали ряд эфиров
фосфиновой кислоты, имеющих давление паров около 10 мм рт. ст.
при 20 °С. Они показали, что большая часть образца испарялась
при перенесении КВг и образца в микропресс и около 90 % мате-
материала рассеивалось за несколько минут. Аналогичный эксперимент
провел Кинг [76], исследуя потерю 2,6-диметоксифенола, летучесть
которого даже ниже, чем эфиров фосфиновых кислот. Он нашел,
что, когда ' качестве носителя образца используется растворитель
118
Глава 4
3400
2000
Рис. 4.16. Спектр 6 нг трифенилфосфата с диафрагмой 100 мкм; 1000 скани-
сканирований, разрешение 8 см~' [22]. Reproduced with permission of American
Chemical Society.
(CS2) с последующим испарением на порошке КВг, потери зависят
от разбавления вещества. Чем разбавленнее раствор, тем выше потери.
В любом случае, даже при использовании интерференционного спектро-
спектрофотометра с растяжкой по ординате, для получения спектра требует-
требуется больше 10 нг вещества. Практика показывает, что для слаболету-
слаболетучих веществ предел чувствительности выше для разбавленных раст-
растворов в кювете малого объема, чем для микротаблеток с КВг.
Как было показано, жидкостная микрокювета позволяет получать
без светового конденсора спектры растворов с содержанием вещества
10—100 мкг [114]. Если используется световой конденсор, то твердые
образцы, такие, как волокна или нелетучие жидкости, можно исследо-
исследовать без предварительной подготовки. В качестве держателя образца
предлагалось использовать часовые камни [4].
Энергетический выигрыш интерференционного спектрофотометра
позволяет использовать маленькие образцы. Апертурная диафрагма
диаметром в несколько десятых долей миллиметра в латунной про-
прокладке может быть использована для закрепления крошечных образцов.
Гиршфельд [65] определил оптимальный диаметр микрокюветы для
фурье-спектрометра.
Применение фурье-спектрометра с конденсором, уменьшающим раз-
размеры светового пучка в 8 раз, позволило исследовать микрообразцы
Методы подготовки образцов
119
3400
600
Рис. 4.17. Спектр 0,9 нг ацетата целлюлозы, содержащего 10% трифеиил-
фосфата; 96000 сканирований, разрешение 8 см [22]. Reproduced with per-
permission of American Chemical Society.
весом менее 1 нг [22] *. Подготовка образцов производится под
микроскопом, так как они часто не видны невооруженным глазом.
Образцы помещают на тонкую пластинку из NaCl и центруют
внутри отверстия диаметром 50—200 мкм, пробитого в тонкой ла-
латунной прокладке. Размер отверстия выбирают таким образом, чтобы
оно полностью заполнялось образцом. Оптимальная толщина образ-
образца — несколько микрометров, и в случае необходимости применяют
различные методы, чтобы ее уменьшить. Следует соблюдать крайнюю
осторожность и чистоту, так как одна частица порошка весит в
10—100 раз меньше всего исследуемого образца. На рис. 4.16
представлен спектр 6 нг трифенилфосфата, просканированный 1000 раз,
а на рис. 4.17 — спектр 0,9 нг ацетата целлюлозы с 90 пг трифенил-
трифенилфосфата, просканированный 96000 раз (накопление спектров прово-
проводилось в течение 40 ч). Ожидается, что дальнейшее совершенство-
совершенствование приборов и техники эксперимента позволит исследовать еще
меньшие образцы. Проводя работы такого рода, не следует забывать
о том, что ИК-спектры кристаллических частиц (как положение по-
* Необходимо учитывать, что в фокусе светового конденсора температура
может повышаться до 50 — 80 °С. — Прим. перев.
120
Глава 4
Применение ЭВМ Ш
растккки спектров
Рис. 4.18. Микроанализ при совместном использовании спектрофотометра и
микро-ЭВМ.
/«-линия 100% элемента НПВО,; S-\ мкг бирлана (Birlane), осажденного на элемент НПВО;
Л-результат вычитания Б-А, выполненного на ЭВМ; /"-спектр В, растянутый по ординате ¦
200 раз после сглаживай и»; Д - стандартный спектр НПВО бирлана [19). Reproduced, with permission,
from American Laboratory. Copyright International Scientific Communications.
A ¦
? ¦
1400 1200 1000
800
1400 1200 1000 800
Рис. 4.19. Иллюстрация накопления спектров при многократном сканировании.
.4 —спектр отражении от металлической поверхности; Б — та же поверхность с нанесенным слоем
силиконового масла толщиной 20 Л; В-результат 15-кратного сканирования спектра Б; Г—спектр В
после 10-кратлой растяжжн по ординате и сглаживания [19]. Reproduced, with permission, from American
Laboratory. Copyright International Scientific Communications.
лос, так и их интенсивности) зависят от размера частиц (см.
стр. 264—266 настоящей книги, а также [104]).
Рост объема цифровой информации, получаемой от спектрофото-
спектрофотометров, регистрирующих электрическое отношение, или интерференцион-
интерференционных спектрофотометров, может привести к возникновению нового
Методы подготовки образцов 121
направления в практике ИК-микроанализа. Обычные спектрофотометры
с оптическим нулем, даже с цифровым выходом, менее приемлемы
из-за механических проблем, связанных с оптическим клином (инер-
(инерция, мертвый ход, потеря энергии при пропускании, близком к нулю)
ЭВМ выполняет с численными данными четыре основные операции:
растяжка шкалы оптической плотности, численное сглаживание, вы-
вычитание оптической плотности и накопление спектров [19].
Процесс в основном происходит следующим образом. Используя
стандартную или полумикротехнику, записывают спектр образца, пред-
представляют его в цифровом виде; производится также «холостая»
запись. Даже если оба этих спектра кажутся одинаковыми, все равно
спектр образца содержит информацию о нем. «Холостой» спектр
вычитается из спектра образца для внесения поправки на раствори-
растворитель, наклон линии 100 % примеси и т. д. Этот спектр растягивается
в нужное число раз, возможно до 100-200. Поскольку при этом уро-
уровень шума также увеличивается, для получения качественного спектра
проводят численное сглаживание. Дальнейшее уменьшение уровня
шума проводится накоплением спектров образца с их последующим
усреднением. В областях сильного поглощения растворителя спектр
образца, конечно, затемнен и не может быть выделен. Некоторые
примеры спектров микроОбразцов, записанных на диспергирующем
спектрофотометре, регистрирующем электрическое отношение и свя-
связанном с лабораторной мини-ЭВМ,, показаны на рис. 4.18 и 4.19.
Спектры при высоких давлениях
Некоторые исследователи спектроскопически изучали поведение
материалов при давлениях вплоть до 10000 атм. Такие исследования
выдвигают много специальных задач, особенно в отношении конструи-
конструирования кювет. Лучшим материалом для окон кювет высокого дав-
давления на сегодняшний день является алмаз и особенно так называе-
называемый алмаз типа Н-а, почти полностью прозрачный в ИК-области.
Оптическое отверстие такой кюветы, по понятным причинам, мало,
поэтому требуется оптический конденсор. Другая проблема связана
с калибровкой давления внутри кюветы. Эти и другие вопросы
рассмотрены в обзоре Ферраро и Безила [37], посвященном конструк-
конструкциям кювет, спектральным приборам, окнам, калибровке и включаю-
включающем обширную библиографию.
Спектры излучения
Проблема получения спектра образца, температура которого
только на несколько градусов выше комнатной, с использованием
его собственного излучения может показаться искусственной, но такой
эксперимент успешно выполнен на интерференционном спектрометре.
122
Глава 4
Жидкости и твердые вещества исследуются в виде тонких пленок,
осажденных на хорошо отражающую зеркальную подложку. Главная
трудность заключается в том, что образец должен быть не только
очень тонким, но и определенной толщины, в противном случае
внешние слои будут поглощать энергию, излучаемую внутренними.
Градиент температуры также создает проблемы. Для образца
подходящей толщины полосы поглощения будут наблюдаться и в
испускании; другими словами, спектр будет обращенным. Толстые
образцы дают контуры с интенсивными полосами, приближающиеся
к кривой излучения черного тела. В соответствующих условиях хорошие
спектры излучения дают газы [49]. Спектры излучения можно
получить и на обычных диспергирующих спектрометрах, но с большими
трудностями и худшими результатами [32].
Хитроумное оптическое устройство, использованное Лауером и
Петеркином [78, 79], позволило с помощью спектра излучения, полу-
полученного на интерференционном спектрометре, исследовать поведение
тонкой пленки смазки под нагрузкой.
Абсорбционная спектроскопия почти неизменно дает лучшие
результаты, чем эмиссионная, кроме, возможно, случая веществ с очень
слабым поглощением [67], и поэтому ей нужно отдавать пред-
предпочтение, когда это возможно.
Водные растворы
Вода, будучи сильно полярным соединением, является хорошим
растворителем для полярных веществ. Однако из-за очень сильного
поглощения ее применение в ИК-спектроскопии ограничено специаль-
специальными областями, такими, как биологические исследования [115]. Когда
используются небольшие толщины кювет порядка 10-25 мкм, можно
4000
3000
2000 1600 1200 1000 600
Рис. 4.20. Обычный спектр пропускания воды, снятый в кювете из AgBr
толщиной около 12 мкм [20]. Reproduced with permission of Heyden and
Son, Ltd.
Методы подготовки образцов
123
Рис. 4.21. Влияние температуры образца на спектр воды, полученный вы-
вычитанием с помощью ЭВМ.
Л — повета с образцом находится при температуре окружающей среды,- Б — кювета находится в тепловом
равновесен со спектрометром; В — разностный спектр Б —Л (с 10-кратной растяжкой по ординате);
Г — разность между двумя спектрами, зарегистрированными после того, как образец каждый раз
находился в тепловом равновесии в течение 20 мин [20]. Reproduced with permission of Heyden and
Son, Ltd.
проводить исследования в значительной части области «отпечатка
пальцев». Интенсивности полос около 3200 см и ниже 800 см
таковы, что невозможно использовать эти области, за исключением
очень тонких кювет порядка 2,5 мкм и менее, однако такие тол-
толщины трудно получить и почти невозможно воспроизвести. Поэтому
областью возможного использования является 800—3100 см"', при этом
вблизи 1640 см возникают некоторые трудности (рис. 4.20). Эту
область можно расширить, применяя тяжелую воду). Кроме того, в
таких тонких слоях поглощение образцов очень слабое, особенно в
разбавленных растворах.
Использование ЭВМ при регистрации таких спектров может сделать
анализ водных растворов практически доступным, если не рутинным
[20]. Техника подготовки остается той же, что и ранее описанная для
микрообразцов. Вначале из спектра образца вычитается фоновое погло-
поглощение чистой воды. При этом следует обратить внимание на
достижение теплового равновесия, так как спектр воды чувствителен
к изменению температуры (рис. 4.21). Необходимо также иметь в виду
124
Глава 4
Рис. 4.22. Спектр водного раствора.
А - раствор аспирина; Б — спектр А после вычитания спектра воды и 15-кратной растяжки [20]. Repro-
Reproduced with permission of Heyden and Son, Lid.
сильное взаимодействие воды с другими полярными молекулами, что
оказывает влияние на их спектры. Взаимодействие растворитель — раст-
растворенное вещество может приводить к неполной компенсации. Спектр
в цифровом виде переводится в шкалу оптической плотности, умножа-
умножается на подходящий коэффициент для растяжки по ординате, переводится
обратно в шкалу пропускания и, наконец, подвергается операции
сглаживания. На рис. 4.22 показан спектр водного раствора
лекарственного препарата. Наблюдаются полосы цитрата натрия, аце-
тилсалицилата натрия и растворенного СО2 B350 см). Эти спектры
зарегистрированы с использованием диспергирующего (регистирующе-
го электрическое отношение) спектрофотометра с цифровым выходом
и процессором.
Методы подготовки образцов 125
Конструкция кювет и уход за ними
Жидкостные кюветы постоянной толщины
Обычная ИК-кювета состоит из металлического корпуса с каналами
для заполнения и удаления жидкости, двух окон, прозрачных в ИК-области
(в одном из них имеются отверстия для введения в зазор раствора),
и прокладки или уплотнения, которое разделяет окна и определяет
толщину поглощающего слоя. Для обеспечения герметичности между
отверстием для заливки и просверленным окном используют прокладку
из свинца или полимера, устойчивого к растворителям. Корпус, как
правило, делают из латуни, нержавеющей стали или алюминия, а
уплотнения — обычно из свежеамальгамированного свинца, который
вскоре становится довольно твердым и хорошо соединяется с
поверхностью окон. Можно использовать другие амальгамированные
металлы (например, прокладки из тонкой меди или латуни), а также
листовые золото, серебро, индий или тефлон.
Производится множество типов жидкостных кювет, широко различа-
различающихся по своим основным качествам. При выборе конструкции
для работы в лаборатории нужно иметь в виду следующие факторы:
1) легко ли она заполняется, не попадает ли при этом раствор на
наружные поверхности окон; 2) удобно ли из нее удалять вещество
и промывать; 3) возможно ли разбирать кювету, чистить, перепо-
лировывать окна и вновь собирать ее в лабораторных условиях;
4) соответствует ли объем кюветы обычно используемым количе-
количествам образца; 5) соответствует ли стоимость кюветы тем задачам,
для которых ее предполагается использовать. Рекомендуется предвари-
предварительно опробовать несколько конструкций, прежде чем выбрать стан-
стандартную.
Для изготовления окон имеется целый ряд материалов, различаю-
различающихся такими характеристиками, как область прозрачности, твердость,
обрабатываемость, стоимость, показатель преломления. Значение пропус-
пропускания очевидно. Тонкое окно по сравнению с толстым будет про-
пропускать более длинноволновое излучение, давая возможность ис-
использовать кюветы в несколько более широкой области длин волн, чем
это позволяет делать призма из того же материала. Например, тон-
тонкая таблетка с КВг удовлетворительно пропускает до 250 см, в то
время как граница пропускания для призмы из КВг составляет около
400 см.
Твердость и обрабатываемость влияют на легкость, с которой окно
можно переполировать, если оно помутнело или поцарапано. Среди
кристаллов галогенядов щелочных металлов довольно хорошо поддается
полировке NaCl, что позволяет изготавливать плоские прозрачные
окна. В то же время более мягкий и менее хрупкий CsBr
труднее полируется.
Величина показателя преломления важна в том отношении, что
126 Глава 4
от нее зависят потери при отражении от поверхностей окна:
[П2 + Mi)
где «1 и п2 — показатели преломления двух граничащих сред. Для
NaCl сих 1,53 R = 0,04, т. е. около 4% на одну поверхность, гранича-
граничащую с воздухом. В то же время AgCl имеет л?2иК = 11%на одну
поверхность. Чтобы снизить потери при отражении, на оптические мате-
материалы с большими показателями преломления наносят иногда покрытия
(например, Ag2S на AgCl), которые обычно непрочны. Для съемки спектров
водных растворов описана германиевая кювета с просветляющим по-
покрытием [29]. В кюветах с высокопреломляющими окнами возникает
интерференционная картина, налагающаяся на спектр образца, даже
если кювета заполнена жидкостью (стр. 250—252).
Разборные кюветы, состоящие из держателя, двух окон и прокладки,
иногда используются для вязких жидкостей или других веществ, не
поддающихся съемке в обычной жидкостной кювете. Воспроизводимость
толщины обычно не очень хорошая, но достаточна для качественного
анализа.
Спектры образцов, реагирующих с солями, можно зарегистриро-
зарегистрировать в кюветах с окнами из инертных материалов. Если таких кювет
нет, то образцы могут быть предварительно насыщены размельченной
солью или помещены в полиэтиленовые пакетики, которые закрепляются
между солевыми пластинками.
В табл. 4.2 приведены свойства некоторых материалов, широко ис-
используемых для изготовления окон. Более точные и детальные
сведения имеются у Смита [108] (см. также [4]). Характеристики
пропускания материалов, используемых в длинноволновой ИК-области
30-500 см, даны в работе [35].
Жидкостная кювета должна быть сконструирована так, чтобы ее легко
можно было промыть чистым растворителем и высушить. Для удаления
вязких и клейких образцов может потребоваться неоднократное
промывание растворителем, протекающим в чередующихся направле-
направлениях. Остатки растворителя удаляются продувкой кюветы сжатым сухим
воздухом. Если воздух недостаточно сухой, то из-за охлаждения окон
при испарении растворителя внутри кюветы может сконденсироваться
влага и окна помутнеют.
Жидкостные кюветы переменной толщины
Жидкостные кюветы переменной толщины используются главным
образом для компенсации в дифференциальной спектроскопии. Толщина
кюветы образца редко бывает решающей, но для точной компенсации
растворителя в соответствии с толщиной слоя образца и его концент-
концентрацией толщина кюветы сравнения должна регулироваться очень тонко.
В литературе описано несколько типов кювет переменной толщины,
Методы подготовки образцов
127
Таблица 4.2
Свойства некоторых оптических материалов, используемых для изготовления окон
Материал окна
Химический
состав
Область Чувствительность Показа-
пропуска- к агрессивным тель пре-
прения, мкм средам ломлеиия
Примечания
Стекло
Кварц
LiF
Сапфир
Флюорит
Иртран 1а
Сервофракс б
BaF2
Каменная соль
Иртран IIа
Сильвин
Иртран IIIa
Иртран IVа
КВг
AgCl
Ge
Si
KRS-5
CsBr
Csl
Полиэтилен
—
SiO2
LiF
A12O3
CaF2
MgF2
As2S3
BaF2
NaCl
Zn2S
KC1
CaF2
Zn2Se
KBr
AgCl
Ge
Si
Tl2BrI
CsBr
Csl
0,35-2 HF, щелочь 1,
0,2-4 HF
0,2-7 Кислота
0,2-5,5
0,2-10 Соли Ntf4
2-8
1-12 Щелочь
0,2-13
0,2—16 Вода, глицерин
1-14
0,3 — 21 Вода, глицерин
0,2-11
1-21
0,2 — 27 Вода, спирт
0,6—25 Металлы, свет
2-20
1,5-? HF, щелочь
0,7-38 Спирт, HNO3
0,3—40 Вода, спирт
0,3 — 50 Вода, спирт
(СН2СН2)„ 20-200
i Торговое название «Eastman 1
ICodak Company».
Торговое название «Servo Corporation of America».
5-1,9
1,43
1,39
1,77
1,40
1,3
2,59
1,45
1,52
2,24
1,49
1,34
2,5
1,53
2,0
4,0
3,4
2,38
1,66
1,74
1,52
Высокая твердость,
нехрупкий
Нерастворим в воде
Поликристалличе-
Поликристаллический
Размягчается при
195 °С
Нерастворим в воде
Нерастворим в
большинстве раст-
растворителей
Поликристалличе-
Поликристаллический, нехрупкий
Поликристалличе-
Поликристаллический
Гигроскопический
Очень мягкий
Длинноволновая
граница зависит от
чистоты
Токсичный, мягкий
Гигроскопически й,
мягкий
Гигроскопический,
мягкий
Очень мягкий
а некоторые имеются в продаже. Самыми сложными считаются кюветы,
в которых одно окно неподвижно, а другое перемещается с помощью
кольца по винтовой резьбе [21, 125]. Такие кюветы должны отвечать
очень высоким требованиям к плоскостности окон, их плоскопарал-
лельности и точности механической системы.
Более простая кювета сравнения состоит из двух окон, разделенных
128 Глава 4
клиновидной прокладкой. Перемещая кювету поперек луча, эмпирически
можно подобрать такое положение, при котором будет происходить
компенсация. Другой прием заключается в создании клиновидной
полости в солевом блоке методом горячего прессования [8].
Газовые кюветы
Кюветы для газообразных образцов изготавливаются просто из
стеклянной трубки большого диаметра с припаянным краном для
откачки. Окна приклеивают разогретой смесью воска и канифоли,
глифталевым, эпоксидным или другим клеем. В одной из конструк-
конструкций уплотнение создается за счет давления, когда окно прижимается
к прокладке (типа "O-ring") при помощи кольца с резьбой и легко
снимается для чистки и полировки.
Уход за кюветами.
Переполировка оптических материалов
Неизбежно, рано или поздно, на окнах кюветы накапливаются за-
загрязнения, они мутнеют, покрываются царапинами и вытравливаются.
Загрязнения обнаруживаются при периодической записи рабочей и ком-
компенсирующей кювет, заполненных растворителем. Дело в том, что
видимые загрязнения могут не давать ИК-поглощения, а загрязнения,
поглощающие в ИК-области, могут быть не видимы глазом. Для уда-
удаления загрязнений кювету заполняют таким растворителем, как диоксан,
ацетон или диметилформамид, и оставляют на несколько часов.
Однако чаще кювету разбирают, а окна переполировывают.
Разборка кюветы обычно не представляет сложности, хотя для
отделения окон от амальгамированной прокладки может потребоваться
довольно кропотливая работа с острым лезвием бритвы (не нужно
пытаться сохранить прокладку, так как она не пригодна для вторич-
вторичного использования). Перед сборкой кюветы рекомендуется прочистить
отверстия для заполнения и корпус.
Для того чтобы научиться хорошо переполировывать окна, требуются
некоторые практические навыки, хотя техника этого дела не сложна и ее
может освоить каждый практик-спектроскопист. (Полировка призм
требует более высокой квалификацин, и ее лучше оставить оптику-
профессионалу.) В случае необходимости начальная шлифовка выпол-
выполняется на плоском стеклянном диске с использованием абразивного
порошка и воды или керосина в качестве смазки. Обычно же полируют
только мелким порошком E00 меш). Можно применять также аб-
абразивную бумагу, закрепленную на плоской стеклянной поверхности,
внимательно следя за тем, чтобы поверхность кристалла не становилась
выпуклой. После окончания шлифовки окно должно быть тщательно
Методы подготовки образцов 129
очищено от следов абразивного материала, так как присутствие любой
маленькой частицы сразу обнаруживается по появлению царапин во
время полировки.
Существует несколько методов переполировки кристаллов галогени-
дов щелочных металлов. Поскольку эти материалы мягкие (по сравнению
со стеклом), они поддаются полировке сравнительно быстро, и хотя
поверхности по качеству получаются хуже, чем для стеклянной оптики,
они вполне пригодны для использования в ИК-области. Метод полиров-
полировки на ткани — простейший для освоения, но он наименее пригоден
для получения плоской поверхности. Для того чтобы сделать полироваль-
полировальник, кусок прочной плотной ткани, такой, как тонкий шелк, плотно
натягивают на стеклянный диск или другую плоскую поверхность.
На ткань наносят небольшие пятна полирующего вещества, например
крокуса, барнсита, алоксита или шамвы с водой, этиленгликолем или
этанолом. Полировку ведут до тех пор, пока полируемая поверхность
не станет гладкой, и затем окно быстро перемещают на сухой участок
полировальника, где продолжают полировку. Таким способом можно
получить очень чистые поверхности, но они неизбежно будут несколько
скруглены.
В другом методе в качестве полировальника используется по-
поверхность стеклянной пластинки. Стекло слегка увлажняют, или поды-
подышав на него, или пульверизатором, и немедленно приступают к поли-
полировке. Получается вполне плоская поверхность, но ее чистота несколько
хуже, чем при обработке другими способами.
Третий метод, в котором применяют полировальник из смолы,
более трудоемок, чем два предыдущих, но он позволяет получать
плоские пластинки хорошего оптического качества. Полировальник
готовят из оптической смолы и воска [99]. Поверхность смоляной
подушки слегка увлажняют тонкой водной суспензией оптического
полирующего материала и круговыми движениями производят поли-
полировку кристалла. Через некоторое время кристалл со смолы переносят
на мягкую сухую ткань, где продолжают полировку, периодически
контролируя качество на оптически ровной поверхности с помощью
натриевой лампы, до тех пор, пока не будет достигнута требуемая
плоскостность и чистота. Работая на краях или в центре полироваль-
полировальника, можно увеличить вогнутость или выпуклость поверхности
пластинки. Интерференционная картина, наблюдаемая в монохромати-
монохроматическом свете, в действительности представляет собой «контурную карту»,
которая показывает степень успеха этой операции. Наиболее приемлемым
является минимальное число интерференционных колец C — 4). Большое
число интерференционных колец с неправильными контурами указывает
на неоднородность поверхности, которая возникает из-за переувлажнения
смолы. Обилие колец правильной формы является признаком кривиз-
кривизны (выпуклости или вогнутости). Секрет изготовления плоской поверхно-
поверхности заключается в плоскостности полировочной смолы. Полировальник
выравнивают, смачивая и помещая на него тяжелую стеклянную
9 - 586
130 Глава 4
плоскость на 1 — 2 ч. Эту операцию можно периодически повторять
в процессе полировки, поэтому в ходе обработки большого числа
пластинок обычно попеременно используют два полировальника.
Для предохранения полированных поверхностей от помутнения
нужно надевать тонкие резиновые напальчники (они продаются в магази-
магазинах медицинского оборудования), которые удобнее резиновых перчаток.
Следует еще раз напомнить, что полировальник нужно тщательно
предохранять от попадания твердых частиц после шлифовки.
Навык выполнения таких операций, как раскалывание, сверление и
резка кристаллов галогенидов щелочных металлов, приобретается опыт-
опытным путем.
Более твердые кристаллы (например, CaF2, BaF2 и LiF) полиру-
полируются теми же методами, что и описанные выше, только вместо воды
используется концентрированная НС1 [31]. С учетом ее свойств работа
должна проводиться под тягой. По общему мнению, хлористое серебро
полируется с добавкой тиосульфата натрия или бутиламина в этаноле
[86]. Работая с хлористым серебром, нужно помнить, что при
контакте оно легко реагирует с менее благородными металлами, чем
серебро, что отрицательно сказывается как на AgCl, так и на металле.
Сборку кюветы нужно проводить в определенной последователь-
последовательности, обращая особое внимание на полированные поверхности окон,
и помнить о следующем: 1) еще до монтажа прокладка тщательно
выравнивается, мелкие частицы и заусенцы удаляются; 2) в случае
свинцовой прокладки она амальгамируется ртутью, насыщенной свин-
свинцом; 3) необходимо позаботиться и убедиться в том, что внутренние
поверхности параллельны.
После затвердевания амальгамы C — 4 ч) кювету нужно заполнить
летучей жидкостью и проверить, не течет ли она. Протечки обычно
возникают из-за неплоскостности окон и (или) прокладки кюветы,
и их нельзя устранить, не перебрав кюветы.
Полированные поверхности галогенидов щелочных металлов крайне
неустойчивы, и их хранят либо в сухом эксикаторе, либо в камере,
температура которой поддерживается на несколько градусов выше
комнатной с помощью маленькой электрической лампочки.
Определение толщины кювет
Толщина кювет 0,03 — 0,6 мм наиболее точно определяется по
интерференционной картине [109]. Способ измерения заключается в
том, что в спектре пустой кюветы наблюдаются максимумы и минимумы,
возникающие в результате взаимного усиления и ослабления излуче-
излучения, отраженного от внутренних поверхностей окон кюветы (рис. 4.23).
Расстояние между окнами точно вычисляется по уравнению
. . и ХД2 . ч и 10
t (мкм) = — \ , или t (мм) = — , D.6)
Методы подготовки образцов
131
-sit
ifji
Ш
и
1
1
f- SI!»
i L"'l :-Л\
П \]r\ •!]
j !T:;i:r::
J *.•!;:•!
¦ :¦.!:¦;
t*40r
Щ
¦Ш
¦it
ri:
Hi!
Ж
17
1
И
MOO
7
Si
ЦА.З
.2
¦ - -. -1
iVu
\H:
Ж
I
№.
:-
0 10
1
7»»-
iui
Ml
щ
|
4)
wt*
¦¦he :H
-,-; i!
0,100
щ
1
!
ш
1400
Длина
'iFi?!
' HPW
Bill
-|..ЬШ1
r ffl?f
волны,
MKM
10
¦
a Lk
i ;i-
: . :
fi L:
|f
Ш ,__
%
\...
Ш
ft*
ii'i
iili
\i
ii
10
»
II
r1
¦to
V
\)
4
т
1
0
\}
15
u.
\l
T
1.
t
i
-40-
1Л
—f~
1
1
)
¦ l
«
17
1
0
20
1
39
1 '
»
0
Волновое число,см*
Рис. 4.23. Определение толщины кюветы по интерференционной картине.
Первый максимум нумеруется нулем.
где t — толщина кюветы, Xt и Х2 — длины волн двух максимумов,
vt и v2 - соответствующие частоты (см), а п — число максимумов
между \i и \2- В случае кювет толщиной более 0,6 мм зазор определя-
определяется по толщине прокладки, измеряемой с помощью микрометра.
Если интерференционную картину получить нельзя, то применяют
менее точный метод, который состоит в следующем. Стандартную
жидкость (например, ССЦ или С6Н12) помещают в кювету неизвестной
толщины и сравнивают значения оптической плотности одной из
удобных полос с тем, которое было получено для той же жидкости
в кювете известной толщины [96]. Эта полоса должна быть широкой и
иметь пропускание 30 — 50 %. Чтобы свести к минимуму ошибки,
обусловленные временным изменением параметров спектрофотометра,
обе кюветы нужно записать с небольшим интервалом во времени.
ЛИТЕРАТУРА
1. Ahlijah G. Е. В. К, Mooney E. F., Spectrochim. Acta, 25A, 619 A969).
2. American Society for Testing and Materials, 1977 Annual Book of ASTM
Standards, Part 42, ASTM, Philadelphia, 1977.
3. American Society for Testing and Materials, Committee E-13, Manual of
Recommended Practices in Spectrbphotometry, ASTM, Philadelphia, 1966.
4. Anderson D. R., Wilson T. ?., Anal. Chem., 47, 2482 A975).
5. Anderson D.H., Woodall N.B., Anal. Chem., 25, 1906 A953).
6. Azarraga L. V., McCall A. C, Environ. Prot. Technol. Ser., EPA-660-/2-73-034/
/273-034, 1974.
132 Глава 4
7. Baker A. W., J. Phys. Chem., 61, 450 A957).
8. Bams E. M., Hopkins R. R., Appl. Spectrosc, 15, 153 A961).
9. Barnes A. J., Rev. Anal. Chem., 1, 193 A972).
10. Blake B. H., Erley D. S., Beman P. L., Appl. Spectrosc., 18, 114 A964).
11. Blanke J. F., Vincent S. ?., Overend J., Spectrochim. Acta, 32A, 163 A976).
12. Blinn R. C, Adv. Chem. Ser., 104, 81 A971).
13. Boland P. G., Milne J. W., Spectrochim. Acta, 29A, 1214 A973).
14. Bradley К. В., Potts W.. A., Jr., Appl. Spectrosc, 12, 77 A958).
15. Brady R. F., Jr., Anal. Chem., 47, 1425 A975).
16. Brooker M. H., Irish D. E., Spectrochim. Acta, 28A, 701 A972).
17. Caspary R., Appl. Spectrosc, 22, 694 A968).
18. Cleverley В., Appl. Spectrosc, 30, 465 A976).
19. Coates J. P., Am. Lab., 8A1), 67 A976).
20. Coates J. P., Eur. Spectrosc. News A6), 25 A978).
21. Coates V. J., Rev. Sci. Instrum., 22, 853 A951).
22. Cournoyer R., Shearer J. C., Anderson D. H., Anal. Chem., 49, 2275 A977).
23. Crooks J. E., Gerrard D. L, Maddarn W. F., Anal. Chem., 45, 1823 A973).
24. Dawkins J. V., Hemming M., J. Appl. Polym. Sci., 19, 3107 A975).
25. De Leenheer A., J. Chromatogr., 74, 35 A972).
26. Diaz-Rueda J., Sloane H. J., Obremski R. J., Appl. Spectrosc., 31, 298 A977).
27. Downs A. J., Peake S. C, Molec. Spectrosc., 1, 523 A973).
28. Drew D. M., van Getnert J. I., Appl. Spectrosc., 25, 465 A971).
29. Edgell W. P., Appl. Spectrosc, 25, 276 A971).
30. Erley D. S., Anal. Chem., 29, 1564 A957).
31. Erley D. S., Blake В. Н., Long A. W., Appl. Spectrosc, 14, 25 A960).
32. Fabbri G., Baraldi P., Appl. Spectrosc, 26, 593 A972).
33. Fahrenfort J., Spectrochim. Acta, 17, 698 A961).
34. Fahrenfort J., Visser W. M., Spectrochim. Acta, 18, 1103 A962).
35. Fateley W. G., Witkowski R. E., Carbson G. L, Appl. Spectrosc, 20, 190
A966).
36. Feairheller W. R., Miller J. Т., Jr., Appl. Spectrosc, 25, 175 A971).
37. Ferraro J. R., Basile L. Jt, Appl. Spectrosc, 28, 505 A974).
38. Ford T. A., Seto P. F., Folk M., Spectrochim. Acta, 25A, 1650 A969).
39. Freeman S. K., Gas Chromatography and Infrared and Raman Spectrometry,
in L. S. Ettre and W. H. McFadden, Eds., Ancillary Techniques of Gas
Chromatography, Wiley, New York, 1969, pp. 227 - 267.
40. Fujiyama Т., Crawford В., Jr., J. Phys. Chem., 72, 2174 A968).
41. Gallaher K. L, Grasselli J. G., Appl. Spectrosc, 31, 456 A977).
42. Garner H. R., Packer H., Appl. Spectrosc, 22, 122 A968).
43. Gilby A.C., Cassels J., Wilks P. A., Jr., Appl. Spectrosc, 24, 539 A970).
44. Gomez-Taylor M. M., Kuehl D., Griffiths P. R., Appl. Spectrosc, 30, 447
A976).
45. Gomez-Taylor M. M., Griffiths P. R., Appl. Spectrosc., 31, 528 A977).
46. Coulden J. D. S., Spectrochim. Acta, 15, 657 A959).
47. Greenberg J., Hallgren L. J., Rev. Sci. Instrum., 31, 444 A960).
48. Greenler R. G., J. Chem. Phys., 44, 310 A966).
49. Griffiths P. R., Appl. Spectrosc, 26, 73 A972).
50. Griffiths P. R., Appl. Spectrosc, 31, 284 A977).
51. Griffiths P. R., Block F., Appl. Spectrosc, 27, 431 A973).
52. Cruenfeld M., Ginell R., Appl. Spectrosc, 24, 380 A970).
53. Haken J. K., Werner R. L., Appl. Spectrosc, 22, 345 A968).
Методы подготовки образцов 133
54. Hallam Н. ?., Vibrational Spectroscopy of Trapped Species, Wiley, New York, 1973.
55. Hanst P. L., Lefohn A. S., Gay B. W., Jr., Appl. Spectrosc, 27, 188 A973).
56. Harm D. L., Anal. Chem., 25, 1140 A953).
57. Harrick N. J., Anal. Chem., 43, 1533 A971).
58. Harrick N. J., Appl. Spectrosc, 31, 548 A977).
59. Харрик Н. Спектроскопия внутреннего отражения. Пер. с англ. — М.: Мир,
1970.
60. Harrick N. J., Riederman N. Я., Spectrochim., Acta, 21, 2135 A965).
61. Hermann Т. S., Harvey S. R., Honts С N., Appl. Spectrosc, 23, 435, 451,
461, 473 A969).
62. Hirschfeld Т., Appl. Spectrosc, 20, 336 A966).
63. Hirschfeld Т., Appl. Spectrosc, 21, 335 A967).
64. Hirschfeld Т., Appl. Spectrosc, 24, 277 A970).
65. Hirschfeld Т., Appl. Spectrosc, 30, 353 A976).
66. Hirschfeld Т., Appl. Spectrosc, 31, 289 A977).
67. Hordwik A., Appl. Opt., 16, 2827 A977).
68. Jeannotte A. C, II, Overend J., Spectrochim. Acta, 33A, 849 A977).
69. Johnson W. T. M., Offic. Dig. Federal. Soc. Paint TechnoL, 32, 1067 A960).
70. Johnson W. Т. М., Offic. Dig. Federat. Soc. Paint Technol., 33, 1489 A961).
71. Johnson W. Т. М., Permeys R., Appl. Spectrosc, 28, 328 A974).
72. Kaczaj J., Appl. Spectrosc, 21, 180 A967).
73. Katlafsky В., Dietrich M. W., Appl. Spectrosc, 29, 24 A975).
74. Katon J. ?., Develop. Appl. Spectrosc, 9, 3 A971).
75. Katon J. ?., Phillips D. В., Appl. Spectrosc Rev., 7, 1 A973).
76. King S. S. Т., J. Agric. Food Chem, 21, 526 A973).
77. Kirkland J. J., Anal. Chem., 29, 1127 A957).
78. Lauer J. L, Peterkin M. ?., J. Lubr. Technol., 97, 145 A975).
79. Lauer J. L, Peterkin M. E., J. Lubr. Technol., 98, 230 A976).
80. Littlewood В., Chromatographia, 1, 223 A968).
81. Louw С W., Richards J. F., Appl. Spectrosc, 29, 15 A975).
82. McCall E. R., Morris N. M., Tripp V. W., O'Connor R. Т., Appl.
Spectrosc, 25, 196 A971).
83. McNiven N. L, Coutr R., Appl. Spectrosc, 24, 296 A970).
84. Milligan D. E., Jacox M. ?., Matrix Spectra, in K. N. Rao and
С W. Mathews, Eds., Molecular Spectroscopy: Modern Research, Academic
Press, New York, 1972.
85. Milne J. W., Spectrochim. Acta, 32A, 1347 A976).
86. Mitzner B. M., J. Opt. Soc. Am., 47, 328 A957).
87. Nadeau A., Jones R. N., Spectrochim. Acta, 26A, 742 A970).
88. Norris W. P., Olsen A. L, Brophy R. G., Appl. Spectrosc, 26, 247 A972).
89. Nortia Т., Kontas E., Spectrochim. Acta, 29A, 1493 A973).
90. O'Connor R. Т., Cellulose and Fabrics in G. L. Clark, Ed., Encyclopedia of
Spectroscopy, Reinhold, New York, 1960. pp. 392 - 409.
91. O'Connor R. Т., Du Pre E. F., McCall E. R., Anal. Chem., 29, 998 A957).
92. Paralusz С. М., J. Colloid Interface Sci., 47, 719 A974).
93. Parliment Т. Н., Microchem. J., 20, 492 A975).
94. Percival С J., Griffiths P. R., Anal. Chem., 47, 154 A975).
95. Perenich T.A., Tuazon E.C., Appl. Spectrosc, 30, 196 A976).
96. Perry J. A., Appl. Spectrosc. Rev., 3, 229 A970).
97. Pitts J. N., Finlayson-Pitts B. J., Winer A. M., Environ. Sci. Technol., 11,
568 A977).
134 Глава 4
98. Poling G. W., J. Electrochem. Soc, 116, 958 A969).
99. Potts W. J., Jr., Chemical Infrared Spectroscopy, Vol. 1, Techniques, Wiley, New
York, 1963.
100. Potts W. J., Jr., Wright N., Anal. Chem., 28, 1255 A956).
101. Price W. C, Tetlow K. S., J. Chem. Phys., 16, 1157 A948).
102. Rochkind M. M., Anal. Chem., 40, 762 A968).
103. Rochkind M. M., Spectrochim. Acta, 27A. 547 A971).
104. Ruppin R., Englman R., Rep. Progr. Phys., 33, 149 A970).
105. Schiedt U., Reinwein H., Z. Naturforsch., 7b, 270 A952).
106. Schwarz H. P., Childs R. C, Dreisbach L, Mastrangelo S. V., Kleschick A.,
Appl. Spectrosc, 12, 35 A958).
107. Shops R. H., Varano A., Industr. Res., 19, B), 86 A977).
108. Smith A. L., Infrared Spectroscopy, in J. W. Robinson, Ed, Handbook of
Spectroscopy, Vol. II, CRC Press, Cleveland, 1974.
109. Smith D. C, Miller E. C, J. Opt. Soc. Am., 34, 130 A944).
110. Stoats P. A., Morgan H. W., Appl. Spectrosc, 22, 576 A968).
111. Stimson M. M., O'Donnell M. J., J. Am. Chem. Soc, 74, 1805 A952).
11Z Strait L. A., Hrenoff M. K., A Techniques for Preparing Solid Organic
Samples for Infrared Analysis, in Symposium on Spectroscopy, ASTM Spec.
Tech. Publ. No. 269, 190 A960).
113. Strasheim A., Buijs K., Spectrochim. Acta, 16, 1010 A960).
114. Sumas E. C, Williams J. F., Walker J., Kidd D., Appl. Spectrosc, 27, 486
A973).
115. Thomas G. J., Jr., Kyogoku Y., Biological Science, in E. G. Brame, Jr., and
Grasselli J. G., Eds., Practical Spectroscopy Vol. 1, Infrared and Raman
Spectroscopy, Part C, p. 717; Marcel Dekker, New York, 1977.
116. Tolk A., Spectrochim. Acta., 17, 511 A961).
117. Tompa A. S., Appl. Spectrosc., 22, 491 A968).
118. Tompkins H. G., Appl. Spectrosc, 28, 335 A974).
119. Tompkins H. C, Appl. Spectrosc., 30, 377 A966).
120. Truett W. L., Am. Lab, 9 F), 23 A977).
121. Wall D. L., Mantz A. W., Appl. Spectrosc, 31, 592 A977).
122. Watson С A., Anal. Chem., 49, 835A A977).
123. Welti D., Infrared Vapour Spectra. Heyden, London, 1970.
124. White J. U., J. Opt. Soc. Am., 32, 285 A942).
125. White J. U., Rev. Sci., Instrum., 21, 629 A950).
126. Wilks P. A., Jr., Appl. Spectrosc., 22, 782 A968).
127. Wilks P. A., Jr., Hirschfeld Т., Appl. Spectrosc., Rev., 1, 99 A967).
128. Willey R. R., Appl. Spectrosc., 30, 593 A976).
Дополнительная литература
1. Якушин В. И., Струков О. Г., Успехи химии, № 8, 1504 A972).
2. Золотарев В. М., Лыгин- В. И.. Тарасевич Б. Я., Успехи химии, № 1, 24
A981).
3. Гомон Г. О. Алмазы.—Л.: Машиностроение, 1966.
4. Воронкова Е. М., Гречушников Б. Н., Дистлер П И., Петров И. П.
Оптические материалы для инфракрасной техники. —Л.: Наука, 1965.
Глава 5
КАЧЕСТВЕННЫЙ АНАЛИЗ ПО ИК-СПЕКТРАМ
Введение
ИК-спектр органического соединения часто называют «отпечатком
пальцев», который неповторим, и поэтому ИК-спектроскопия явля-
является мощным методом идентификации органических соединений.
Метод крайне полезен при расшифровке неизвестных структур,
так как некоторые химические группы (метильная, карбонильная и др.)
имеют характеристические полосы поглощения, положение и интенсив-
интенсивность которых более или менее постоянны. Такие «групповые частоты»
можно переносить от одного соединения к другому. Более детально
групповые частоты будут обсуждены после краткого введения в теорию
ИК-спектров поглощения.
Теория ИК-спектров
В общем случае в молекуле могут происходить следующие типы
движений: 1) поступательное движение молекулы как целого, которое
может рассматриваться как движение центра масс; 2) вращение
молекулы вокруг центра масс; 3) колебания отдельных атомов, проис-
происходящие таким образом, что положение центра масс не изменяется
и молекула не вращается; 4) движение электронов в молекуле; 5) вращение
электронов и ядер атомов вокруг своих осей (спины электронов и
ядер). Случаи 1, 4, 5 прямо не связаны с аналитической ИК-
спектроскопией и в дальнейшем не обсуждаются.
Число степеней свободы (СС) частицы равно числу координат, не-
необходимых для определения ее положения в пространстве. Общее
число СС системы из N частиц равно 3JV. Если структуру молекулы
рассматривать жесткой, то ее положение в пространстве можно
задать тремя координатами центра масс. У молекулы имеются еще три
вращательные СС, так что необходимы еще три координаты для
фиксирования ее ориентации в пространстве. Поэтому число колебатель-
колебательных степеней свободы равно ЗАГ — 3 — 3, или 3N — 6. (Линейные
молекулы, такие, как НС1, О2, COj и С2Н2, имеют только две
вращательные СС, так как вращения в обычном смысле вокруг оси,
проходящей через все атомы, не происходит. Поэтому линейная
молекула имеет 3iV — 5 колебательных степеней свободы.) Это значит,
что у нелинейной молекулы из N атомов имеется 3N - 6 основных
типов колебаний.
Способность вещества поглощать энергию ИК-излучения зависит от
136 Глава 5
суммарного изменения дипольного момента молекулы при вращении
или колебании. Так или иначе, такие изменения происходят в зависимо-
зависимости от распределения электрических зарядов в молекуле. Например,
можно вообразить, что в молекуле оксида азота (NO) атомы азота и
кислорода связаны упругой связью. Атом кислорода окружен восемью
электронами, атом азота - семью. В процессе колебания происходит
изменение распределения зарядов; это означает, что поток излучения
«видит» осциллирующий заряд. Следовательно, в ИК-области спектра
оксид азота имеет полосу поглощения с частотой, соответствующей
частоте колебаний ядер. В то же время при колебаниях ядер в гомо-
ядерных молекулах, таких, как О2) Н2, N2 или С12, суммарного изме-
изменения дипольного момента молекулы не происходит, и эти газы не по-
поглощают ИК-излучение. Если немного подумать, то становится ясным,
что способность молекулы к поглощению излучения в процессе данного
колебания зависит только от распределения ее электрических зарядов.
Были разработаны математические методы предсказания ИК-активности
различных колебаний молекулы на основании анализа ее симметрии
[119, 1, 2].
Полная энергия молекулы (с хорошей степенью приближения)
представляется в виде суммы поступательной, вращательной, колебатель-
колебательной и электронной энергий:
Е = Епосг + ?вр + Екол + Е^. E.1)
Поступательная энергия мало влияет на молекулярные спектры и здесь
не рассматривается.
В 1913 г. Нильс Бор впервые предположил, что атомы и молекулы
не могут иметь произвольную энергию*, а должны находиться в ди-
дискретных энергетических состояниях. Следовательно, переходы между
энергетическими состояниями ведут к поглощению или излучению
вполне определенных и характерных порций энергии (квантов), которые
наблюдаются либо как эмиссионные линии от возбужденных молекул,
либо как полосы поглощения в ИК-, видимой и УФ-областях. На этой
важной концепции основана вся спектроскопия.
В процессе развития квантовая механика дала более точный метод
описания атомных явлений, чем это было доступно классической меха-
механике. Однако представленная ниже полуклассическая картина более
наглядна.
Вращательные спектры
Вращающиеся молекулы, находящиеся в газовой или паровой
фазе, принимают только определенные значения энергии, поэтому они
поглощают излучение только тех частот, которые соответствуют перехо-
* Здесь, конечно, идет речь о внутренней энергии, а не о поступательной.-
Прим. ред.
Качественный анализ по ИК-спектрам
137
.L
J
-8
* ¦ I I I I I 1 I l~
0 V
Рис. 5.1. Энергетические уровни и переходы для жесткой двухатомной мо-
молекулы (а) и получающийся вращательный спектр (б). Reproduced, with
permission of Van Nostrand — Reinhold, from G. Herzberg, Spectra of Diatomic
Molecules, Princeton, N. J., 1950.
дам между вращательными энергетическими уровнями. Так как эти
энергии низки, то вращательные линии поглощения обычно располо-
расположены в дальней ИК-области (ниже 500 см).
Молекулы можно классифицировать согласно симметрии их враще-
вращения (т. е. по относительным значениям главных моментов инерции).
Простейшими молекулами, имеющими вращательный спектр, являются
двухатомные и линейные многоатомные, у которых два момента
инерции одинаковы, а третий равен нулю. Следующим простейшим типом
молекул с точки зрения симметрии вращения являются сферические
волчки (с тремя равными моментами инерции), например метан. Сфе-
Сферические волчки не имеют вращательных ИК-спектров, поскольку в
процессе вращения молекулы дипольный момент не изменяется. Более
сложным типом волчков являются симметричные волчки (с двумя рав-
равными моментами инерции), к которым относятся молекулы NH3
и С6Н6. Самый сложный тип волчков - асимметричные волчки (с тремя
неравными моментами инерции). Это, например, молекулы Н2О,
С2Н4 и большинство многоатомных молекул.
138 Глава 5
Вращательная энергия Еър зависит от симметрии волчка и моментов
инерции рассматриваемой молекулы, а также от вращательного кванто-
квантового числа J, которое связано с угловым моментом молекулы
выражением
угловой момент =]/j{J + 1)-у-> E.2)
где J принимает целочисленные значения, как и большинство
других квантовых чисел. Для линейных молекул и сферических волчков
Etp = J(J+l)Bhc, E.3)
где В = h/Sn2tf; h - постоянная Планка, с — скорость света, / - момент
инерции относительно оси вращения. Для перехода J = 0 -»J = 1 из-
изменение энергии равно
А? = 2Bhc. E.4)
Уравнение E.3) показывает, что чем больше момент инерции моле-
молекулы, тем теснее располагаются вращательные линии. Типичная
диаграмма вращательных уровней и вращательный спектр двухатомной
молекулы показаны на рис. 5.1. Переходы в ИК-области подчиняются
правилу отбора AJ = + 1.
Колебательные спектры
Для понимания поведения колеблющейся молекулы рассмотрим
сначала простую колебательную систему, состоящую из груза иа
пружине. Кинетическая энергия К дается выражением
K = ±-mv2, E.5)
где т — масса, at)- скорость движения груза. Потенциальная энергия
сжатия и растяжения пружины V достигает максимума в крайних
(верхнем и нижнем) положениях груза. (Когда пружина растянута
или сжата, она способна производить работу и, следовательно,
обладает потенциальной энергией.) Сумма потенциальной и кинетиче-
кинетической энергий — величина постоянная. Если х — смещение груза из
положения равновесия под действием какой-то силы, то возвращаю-
возвращающая сила F пропорциональна смещению, но направлена в противопо-
противоположную сторону:
F = -кх, E.6)
где к — коэффициент пропорциональности или «силовая постоянная».
Тогда сила равна производной с отрицательным знаком от потен-
потенциальной энергии:
Качественный анализ по ИК-спектрам 139
Интегрируя, получаем
V=]kxdx=\kx2, E.8)
о 2
что является выражением для потенциальной энергии. Осциллятор с
потенциальной функцией, описываемой уравнением E.8), известен как
«простой гармонический осциллятор». Рис. 5.2 иллюстрирует связь
кинетической, потенциальной и полной энергий.
Для того чтобы из уравнения колебательного движения найти
частоту, используем второй закон движения Ньютона, согласно кото-
которому сила равна массе, умноженной на ускорение:
F = tm. E.9)
Однако ускорение — это вторая производная смещения (х) по времени,
поэтому
F=m-^r = mx. E.10)
Объединяя уравнения E.7) и E.10), получаем
t
х = х. E.11)
т
Решение этого уравнения, х, должно быть такой периодической
функцией, чтобы ее вторая производная была равна ей самой, умножен-
умноженной на (— к/т). Такой функцией является косинус, и мгновенное
смещение осциллятора в момент времени t равно
х = AcosBnft + b), E.12)
где/— частота колебания, А — максимальная амплитуда, а Ь — фазовая
постоянная, которая подбором граничных условий может быть обра-
обращена в нуль. Тогда
х = - 4it2/2/lcosBn#) = - 4я2/2х. E.13)
Теперь х = — (к/т) х, если
к/т = 4я2/2, E.14)
откуда частота колебаний осциллятора равна
Это уравнение приближенно выполняется и для молекулярных систем.
В случае двухатомной молекулы частота (колебания в секунду) выра-
140
Глава 5
V(x>
рЕ (полная)
¦Кинетическая
энергия
| Потенциаль-
I ная энергия
! 1
JC ш -Л IS О X = А
Рис. 5.2. Связь кинетической н потенциальной энергий гармонического осцил-
осциллятора с полной энергией Е.
i
\
\ '
4
• t
¦¦ /
/
1
1
-/
7
7
4
3
2
1
0
Рис. 5.3. Кривая потенциальной энергии для ангармонического осциллятора.
Показаны некоторые из возможных переходов.
Качественный анализ по ИК-спектрам 141
жается как
т + М
' E16)
где Мит- массы двух атомов, а ц - их приведенная масса.
Для многоатомных молекул это выражение становится гораздо более
сложным, так как в каждое колебание обычно вовлекается много
атомов, а тем самым и много силовых постоянных.
Другое осложнение возникает (даже в случае двухатомных молекул)
из-за того, что молекулярные колебания в действительности не явля-
являются «гармоническими». Это означает, что потенциальная энергия
может быть несимметричной функцией относительио положения равно-
равновесия колеблющихся масс и совсем не подчиняется простому выражению
типа E.8). Вместо него можно написать
V= ybc2 + Ах3 + Вх4 + Сх5 + ... . E.17)
Если ангармоничность мала, то смещение может быть представлено как
х = А1 cos BKft + fej) + А2 cosBjc2/t + b2) + Аъ cos Bn3ft + b3) + .... E.18)
Кривые потенциальной энергии для молекул похожи на кривую,
показанную на рис. 5.3.
Колебательная энергия, как и вращательная, квантована. Это озна-
означает, что осциллятор может находиться только в определенных
энергетических состояниях. На низшем уровне молекула не покоится,
а колеблется с некоторой частотой около положения равновесия.
В квантовой механике энергия гармонического осциллятора выражается
как
E.19)
где v — колебательное квантовое число, которое может принимать
только положительные целочисленные значения, v — частота основного
колебания в см. Правило отбора для гармонического осциллятора
Av = + 1, и его спектр состоит только из одной линии, так как
энергетические уровни расположены на равных расстояниях друг от
друга. Ангармоничность проявляется в следующем: 1) в «нарушении»
правила отбора, так что могут осуществляться переходы с Av =
= + 2, + 3, ... (их называют обертонами), и 2) в изменении расстоя-
расстояния между уровнями (обычно они располагаются плотнее друг к другу
по мере увеличения v). Хотя в реальной молекуле имеется как механи-
механическая, так и электрическая ангармоничность, только последняя обуслов-
обусловливает появление обертонов. Заселенность различных колебательных уров-
уровней молекулами определяется распределением Больцмана.
Некоторые колебания могут быть вырожденными, т. е. их частоты
совпадают. Вырождение колебания v2 иллюстрируется на примере
142
Глава 5
Рис. 5.4. Колебания СО2.
молекулы СО2 (рис. 5.4). В спектре этой молекулы будет проявляться
меньше частот, чем CJV — 6) или CJV — 5). Случайное вырождение про-
происходит тогда, когда два различных колебания имеют почти оди-
одинаковую энергию. В этом случае взаимодействие частот иногда при-
приводит к их взаимному изменению (одна растет, а другая убывает)
и перераспределению интенсивностей соответствующих полос [119].
Хотя чисто вращательные ИК-спектры большинства молекул наблю-
наблюдаются только в дальней ИК-области, вращательные энергетические
уровни сопровождают колебательные (рис. 5.5), так что в спектрах
газовой или паровой фазы на основное поглощение, обусловленное
колебательным переходом, неизменно налагаются вращательные
«крылья». Совокупность линий, расположенных со стороны низких
частот (переходы с AJ = — 1), называется Р-ветвъю, поглощение, совпа-
совпадающее с чисто колебательной частотой, — Q-ветвъю (AJ = 0), а линии
со стороны высоких частот (AJ = + 1) — R-ветвъю. В некоторых
случаях Q-ветвъ не наблюдается*.
* Для колебательно-вращательных спектров правила отбора по J могут
быть AJ=±1hAJ=0, ±1b зависимости от симметрии формы колебаний. —
Прим. ред.
Качественный анализ по ИК-спектрам
143
J'
I I
I I
?essesssss
«««««а: а: а; а: а:
I I I I
а. а.
S
а.
€ ? §
а. а. а.
J"
- 10
/ 4 /\
•» 9 I 7 6 5 4 3 2 1 0 12345» 7 I 9 10
XIJJ J i J
11 1 1 1 1 1
-
w J J J J
1 f f f IT
т W 9 I 7 6 S « 3 2 1 vo -1 -2 -3 -4 -5 -6 -7 -I -J -10
Рис. 5.5. Диаграмма энергетических уровней, поясняющая тонкую структуру
колебательно-вращательной полосы и образующегося спектра с учетом коле-
колебательно-вращательного взаимодействия (а) и без его учета (б). Коротковол-
Коротковолновая часть спектра слева. Reproduced, with permission of Van Nostrand —
Reinhold, from G. Herzberg, Spectra of Diatomic Molecules Princeton, N. J., 1950.
144 Глава 5
Рис. 5.6. Формы некоторых колебаний СН2СВг2 [229]. Reproduced, with
permission, from Journal of Chemical Physics.
Иногда вывод о происхождении данной колебательной полосы
можно сделать, рассматривая колебательно-вращательный контур (стр.
219). Взаимодействия между колебательными и вращательными энерге-
энергетическими уровнями, вызываемые центробежным растяжением и корио-
лисовым взаимодействием (особенно в случае вырожденных колебаний),
могут искажать контур полос.
При колебаниях многоатомных молекул все атомы движутся в
фазе, но с различными амплитудами. Строго говоря, не существует
колебания, локализованного на отдельной группе атомов, хотя может
быть и такая ситуация, когда колеблется почти одна связь. Для
иллюстрации рассмотрим некоторые колебания Н2С=СВг2, показанные
на рис. 5.6 [229]. Можно заметить, что в валентном колебании
СН C028 см~г) участвуют как валентное колебание О=С, так и
деформационное СН A372 см). Этот механический эффект в коле-
колебательных системах является одной из причин того, что групповые
частоты изменяются при переходе от одной молекулы к другой,
даже если силовые постоянные неизменны (стр. 157 — 161). Очевидно, что
обозначение нормальных колебаний как «валентное СН» или «деформа-
Качественный анализ по ИК-спектрам 145
ционное NH» в лучшем случае носит приближенный характер, а
действительные движения могут быть гораздо сложнее, чем это
подразумевается в их названии. Иногда колебания так сильно
перемешаны и делокализованы, что не существует простого термина
для достаточно полного описания этого движения.
Нормальные колебания
Каждое из 3JV — 6 колебаний многоатомной молекулы называют
нормальным (в том смысле, что оно не зависит от других колеба-
колебаний). Форма нормального колебания двухатомной молекулы ясна,
но для многоатомных молекул это сделать гораздо труднее, так как
требуется совместно решить 3N — 6 или более уравнений, включающих
массы и силовые постоянные. Кроме того, обычно силовых посто-
постоянных больше, чем частот. Тем не менее, используя дополнительную
информацию, такую, как свойства симметрии молекулы, силовые по-
постоянные, перенесенные из сходных молекул, частоты колебания изотопо-
замещенных молекул, а также вводя некоторые упрощающие предпо-
предположения, можно (при достаточном трудолюбии) пытаться решить эту
задачу*. Использование ЭВМ в значительной степени ускоряет такие
расчеты, и в результате этого в настоящее время можно проводить
расчет нормальных колебаний для весьма сложных молекул.
Важно отметить, что симметрия колебаний определяет их активность
в ИК-спектрах и спектрах КР. С использованием методов теории
групп [61, 88] были составлены таблицы, позволяющие предсказать
число и активность нормальных колебаний для каждого типа сим-
симметрии.
Классификация колебаний по симметрии и математический аппарат
расчета нормальных колебаний выходят за рамки содержания этой
главы. Более полное качественное описание дано Поттсом [215], а
строгое изложение — Герцбергом [119], а также Вильсоном, Дешиусом и
Кроссом [272],
Первый шаг при расчете частот и форм нормальных колебаний
состоит в построении функции потенциальной энергии. При записи
потенциальной функции обычно используют не декартовы, а внутренние
координаты. Внутренние координаты выражаются как изменения меж-
межатомных расстояний и углов. Их преимущество заключается в том,
* Здесь уместно отметить, что в теории колебаний многоатомных моле-
молекул принято различать два основных типа задач: 1) расчет частот и форм
нормальных колебаний по заданным параметрам молекулы (эта задача назы-
называется прямой колебательной задачей и решается однозначно); 2) определение
силовых постоянных по экспериментально найденным частотам колебаний
(эта задача называется обратной колебательной задачей и единственного
решения в общем случае не имеет; см., например, [1, с. 416-424]).-
Прим. перев.
10 - 586
146 Глава 5
что они дают более наглядную картину молекулярных колебаний и
облегчают алгебраические преобразования.
Численные значения силовых постоянных зависят от формы потен-
потенциальной функции, используемой для составления уравнений движения.
При сравнении силовых постоянных из разных работ нужно быть
уверенным, что они получены для одного и того же силового поля.
Наиболее полная потенциальная функция должна включать члены,
описывающие взаимодействие между всеми атомами в молекуле, неза-
независимо от того, связаны они или нет. Такая функция неудовлетвори-
неудовлетворительна по той причине, что, во-первых, малые силовые постоянные
вряд ли имеют физический смысл и, во-вторых, большое число не-
неизвестных членов делает решение уравнения невозможным, за исключе-
исключением случая простейших молекул.
Были предложены более приемлемые потенциальные функции.
Предполагается, что в валентно-силовом поле силы, участвующие в
молекулярных колебаниях, сосредоточены на химических связях, и
поэтому силовые постоянные выражаются через изменения длин связей
и углов. В приближении центрально-силового поля предпочтения
валентным связям не делается и силовые постоянные определяются
через взаимодействия между атомами независимо от того, связаны
они непосредственно или нет. Поскольку ни одно из этих полей
полностью не удовлетворяет исследователей, было предложено моди-
модифицированное силовое поле, известное как поле Юри — Бредли (иногда
также неудовлетворительное!). Поле Юри — Бредли, по существу, явля-
является комбинацией двух предыдущих. Предполагается, что главные силы
действуют вдоль химических связей, но дополнительно включаются
члены, описывающие взаимодействие между соседними несвязанными
атомами. Таким образом, в этом поле рассматривается притяжение
и отталкивание между соседними несвязанными атомами (эффекты
поля), которые, как найдено, влияют на формы, интенсивность и
частоту нормальных колебаний. Кроме того, если координаты выб-
выбраны удачно, то силовые постоянные имеют физический смысл и их
можно переносить из одной молекулы в другие со сходной струк-
структурой.
Когда определены силовые постоянные, можно получить форму
любого колебания. Для каждой частоты имеется одна нормальная
координата, которая описывает смещения всех атомов, участвующих в
этом колебании. Путем соответствующих преобразований нормальную
координату можно выразить через декартовы или внутренние
координаты, которые дают действительные смещения атомов в нагляд-
наглядной форме. Детали этих и других расчетов нормальных колебаний
представлены в [56, 78, 244, /].
Каждая из 3N — 6 нормальных координат может быть представ-
представлена своей собственной диаграммой потенциальной энергии, подобно
показанной на рис. 5.3. В этом случае абсциссой является нор-
нормальная координата, а не межатомное расстояние.
Качественный анализ по ИК-спектрам
147
Рис. 5.7. Примерные формы нормальных колебаний для некоторых распрост-
распространенных органических групп. Reproduced, with permission, from Smith [242].
Copyright The Chemical Rubber Company, CRC Press, Inc.
a — симметричное валентное СНз; б — асимметричное валентное СНз; в — симметричное деформационное
СН3 («зонтичное»—umbrella); г — асимметричное деформационное СН3; д — маятниковое СН3; « — кру-
крутильное СНз; ж — симметричное валентное СНз.
10*
148
Глава 5
Рис. 5.7. Продолжение.
з - асимметричное валентное СШ; и — симметричное деформационное СНг («ножничное»-scissors);
к - маятниковое СН2; л — крутильно-деформациоиное СН2 (twist); м - веерное СН2; н - веерное ви-
виниловой СНг; о — крутильне-деформационное виниловой С = С; л — симметричное валентное СОС; р — асим-
асимметричное валентное СОС.
Качественный анализ по ИК-спектрам
149
Рис. 5.7. Продолжение.
с - валентное ОН; т — плоскостное деформационное ОН; у — крутильное ОН; ф — валентное СО.
Здесь полезно дать краткое объяснение принятых обозначений
типов симметрии колебаний и нумерации основных (фундаментальных)
частот. В спектроскопической литературе колебания, симметричные по
отношению к оси симметрии, относят к типу А. Нижний индекс
или штрих вверху указывает на подтип внутри этого типа. Асиммет-
Асимметричные колебания относят к типу В, дважды вырожденные — к
типу Е, трижды вырожденные — к типу F. Отдельные колебания
нумеруются, начиная с самой высокочастотной полосы полносиммет-
полносимметричного типа колебания в порядке убывания частоты, затем переходят
к следующему наиболее симметричному типу колебаний и опять
проводят нумерацию в порядке убывания частот. Так, для CHQ3
валентное колебание СН (тип Аг), vt = 3033 см, симметричное валент-
валентное колебание СС13 (тип Ах), v2 =667 см, симметричное деформаци-
деформационное колебание СС13 (тип Аг), v3 = 364 см, деформационное коле-
колебание СН (тип Е), v4 = 1205 см, асимметричное валентное
колебание СС13 (тип Е), v5 = 760 см, и асимметричное деформа-
деформационное колебание СС13 (тип Е), v6 = 260 см (представленное
здесь описание носит приближенный характер). Дальнейшее обсуждение
и многочисленные примеры этих обозначений можно найти у Герцберга
[119, 1, 2].
В колебательной спектроскопии часто используются наглядные
термины (например, маятниковое СН2, крутильное СС13 или веерное CF2
колебания). Некоторые из этих форм движений изображены на
150 Глава 5
рис. 5.7; точная форма нормальных колебаний зависит от силовых
постоянных связей и масс атомов, участвующих в колебании. Для
сокращенного описания молекулярных движений часто используются
следующие символы: v (stretch — валентное), 8 (deformation - деформа-
деформационное), р (rocking — маятниковое), а> (wagging — веерное) и т (torsion —
крутильное) *.
Контуры полос
Уже отмечалось, что в случае колебательных спектров паров и
газов полосы поглощения имеют вращательную структуру, обра-
образующуюся в результате наложения вращательных энергетических уров-
уровней на колебательные. В жидком состоянии и растворе вращатель-
вращательная структура исчезает, так как вращение сильно затруднено. (Мо-
(Молекулы с малыми моментами инерции, находящиеся в неполярных
растворителях, должны, по-видимому, иметь неквантованное враще-
вращение [146].) По сравнению с узкими линиями** все полосы поглощения
имеют контуры, симметричные относительно центрального максимума
со слабыми крыльями в обе стороны. Факторами, оказывающими
влияние на распределение интенсивностей в газах [223], являются есте-
естественная ширина линии, возникающая из-за затухания излучения,
эффект Доплера, ударное уширение и специфические межмолекулярные
взаимодействия. В конденсированных фазах контуры полос обусловлены
главным образом столкновениями ближайших соседей и специфически-
специфическими взаимодействиями. Иногда важное значение приобретают также
изотопное расщепление, резонанс Ферми и «горячие» полосы (стр. 151).
Для эмпирического описания наблюдаемых контуров полос предло-
предложен ряд математических формул. Одна из наиболее удачных — функция
Лорентца, которая имеет вид
где (a/b2) = lg(/o/J)mai; 2b = Av1/2 — ширина полосы на половине высоты;
10 — интенсивность полосы в максимуме; v0 — частота в максимуме.
Функция Лорентца применяется при аналитическом разделении
* В литературе иногда встречаются обозначения и некоторых других типов
нормальных колебаний. В циклических соединениях происходят пульсацион-
ное (breathing — дыхание) колебание, соответствующее симметричному расшире-
расширению и сокращению цикла, и сморщивающее (ring puckering) деформационное
колебание, приводящее к выходу атомов кольца из его плоскости.— Прим.
перев.
** Отдельные линии вращательной структуры полос поглощения газов
также имеют контур, похожий иа контур полос, ио контуры линий можно
исследовать только при большом разрешении.— Прим. ред.
Качественный анализ по ИК-спектрам 151
контуров сложных полос поглощения жидкостей и при расчетах
интегральных интенсивностей. В обзоре Сешадри и Джонса [233]
рассмотрены влияние на контуры полос собственно природы вещества,
давления, температуры, регистрирующей аппаратуры и т. д., методы
приведения наблюдаемых контуров к идеальным и определение
интегральных интенсивностей (см. также [5]). В работе [145] описан
метод «псевдосвертки», с помощью которого исключаются инструмен-
инструментальные искажения наблюдаемых контуров полос.
Интенсивности полос
Вопрос об интенсивностях ИК-полос настолько тесно связан с
изменениями электронной конфигурации в процессе колебания, что в
явном виде может быть решен только путем расчета полной волновой
функции молекулы. Было сформулировано математическое описание
этого явления, представляющее некоторую ценность при расчете моментов
связей, но априорный расчет интенсивностей полос поглощения столь
груб, что почти не имеет практического значения. Однако эмпири-
эмпирические корреляции интенсивности со структурой иногда полезны.
Если av - коэффициент поглощения при частоте v, то основное со-
соотношение между наблюдаемыми интенсивностями и дипольным момен-
моментом связи |х (в первом приближении) записывается в виде
, , Nn[d\iT
где Q - нормальная координата колебания, JV - число молекул в 1 см3
образца, сия имеют обычные значения. Левая часть уравнения
E.21) может быть точно измерена для изолированных полос при
использовании очень узкой щели. Для более широких щелей необхо-
необходимо вводить поправку [143, 224]. Даже после того, как получено
точное значение интеграла (в координатах частота — оптическая плот-
плотность, но не пропускание), остается сомнение в знаке квадратного
корня. Интерпретация правой части этого уравнения рассмотрена
Коулсоном [62], который отметил проблемы, обусловленные пони-
пониманием дипольного момента связи и его изменения по нормальной
координате Q. Дальнейшее обсуждение интенсивностей полос можно
найти в других работах ([119, с. 260; 233; 272, с. 162; 5]).
Обертоны и составные полосы
Ангармоничность приводит к тому, что правило отбора для
гармонических осцилляторов становится не столь строгим, а это способ-
способствует возникновению обертонов и составных полос (рис. 5.3).
В общем случае поглощение обертонов колебаний попадает приблизи-
приблизительно в область Bvi) — b, где Ъ = 2 — 10 см (иногда и больше).
Некоторые полосы имеют отрицательную ангармоничность. Это озна-
152 Глава 5
чает, что обертон проявляется при частоте, большей, чем удвоенная
основная частота. Возможны случаи, когда основное колебание имеет
определенную симметрию, при которой первый обертон вовсе не про-
проявляется. Активность обертона можно предсказать, исходя из симметрии
колебания [119, с. 126]. Сходным образом составные (комбинацион-
(комбинационные) полосы наблюдаются при частотах, несколько меньших, чем сумма
основных частот. Иногда проявляются разностные полосы, образующи-
образующиеся при переходах из возбужденных колебательных состояний. В этом
случае соотношение v12 = (vj — v2) выполняется точно. Если в спектре
появляется разностная полоса, то должна возникать также соот-
соответствующая суммарная полоса. В другом типе разностных полос,
обычно известных как «горячие» или еще „upper-stage", переходы также
осуществляются между возбужденными колебательными состояниями.
Частота такого перехода v = (vj + vk) — v, или в частном случае при
переходе (v = 1) -* (v = 2), v = 2vt - vt будет отличаться от \к на
несколько см.
Горячие полосы обычно малоинтенсивны и редко наблюдаются, за
исключением спектроскопии высокого разрешения газов.
Интенсивность обертонов и составных полос для веществ в конден-
конденсированном состоянии обычно ниже, чем основных, в 10 — 100 раз.
Однако бывают и исключения, например интенсивность первого обер-
обертона деформационного колебания —NCS одинакова с основной
полосой.
Почти все полосы, наблюдаемые в ближней ИК-области @,7 -
2,5 мкм), за исключением нескольких электронных переходов, являются
либо обертонами или составными частотами валентных колебаний с
участием атома водорода, либо комбинациями частот валентных
колебаний атомов водорода с другими колебаниями. Это обусловлено
двумя причинами. Во-первых, в силу малости массы атома водорода
амплитуда его колебаний велика. Следовательно, движение в значи-
значительной степени ангармонично, что ведет к повышению интенсивности
полос обертонов. Во-вторых, большинство колебаний без участия атомов
водорода имеет более низкие частоты, так что в ближнюю ИК-область
спектра попадают только вторые и более высокие обертоны и составные
частоты более высоких порядков (которые гораздо менее интенсивны,
чем первые гармоники). Поэтому во многих работах, выполненных
в ближней ИК-области, используются многочисленные обертоны и
составные частоты колебаний атома водорода. В некоторых случаях
константы ангармоничности для различных колебаний столь сильно
различаются, что полосы, перекрывающиеся в основной ИК-области,
разрешаются в ближней ИК-области. В обзоре Кайе [154] кратко
обсуждается спектроскопия ближней ИК-области и представлена
библиография методов и их приложений до 1958 г. Может оказаться
полезным и обзор [266], охватывающий более поздние работы.
Были опубликованы также корреляционные диаграммы для ближней ИК-
области [56, 102].
Качественный анализ по ИК-спектрам 153
Групповые частоты:
их использование и ограничения
Общие положения
Как было экспериментально показано, некоторые химические группы,
такие, как СН3, С=О, Р=О и С=С, поглощают почти при
постоянных длинах волн независимо от молекулы, в которой они
содержатся. Такие полосы поглощения называют групповыми часто-
частотами*. Они часто позволяют быстро и однозначно подтвердить
наличие или отсутствие фрагмента, ответственного за поглощение.
В то же время редко, если это вообще возможно, можно найти по-
постоянную групповую частоту. Обычно положение частоты изменяется
внутри некоторого интервала, который и определяется как внутренними,
так и внешними факторами. В этом разделе будет кратко рассмот-
рассмотрено влияние внутренних факторов (электронных и механических) на
колеблющуюся группу; внешние влияния (окружение) обсуждаются в сле-
следующем разделе.
Напомним, что частота колебаний системы из двух тел равна
где /- частота колебаний, к - силовая постоянная связи, а |х - приве-
приведенная масса. Для двухатомной молекулы ц вычислить легко, но к
можно точно определить только экспериментально.
В ходе поиска эмпирического соотношения между силовыми по-
постоянными двухатомных молекул и другими физическими параметрами
были рассмотрены колебательные спектры большого числа молекул.
Широкое распространение получило выражение Бэджера [9]
где к0 — силовая постоянная (в дин/см), De ~ равновесное межатомное
расстояние, atj и Ьу — константы, зависящие от положения атомов i и j
в периодах периодической системы.
Улучшенное соотношение, предложенное Горди [106], имеет вид
к = 1,67 ЛП ^Цр- + 0,30, E.24)
где к — силовая постоянная, N — порядок связи, d — межъядерное рас-
расстояние, ХА и Хв — соответственно электроотрицательности атомов
А и В.
В связи с этими и другими сходными выражениями важно отме-
отметить следующее: 1) результаты носят только приближенный характер,
* Эти частоты называют также характеристическими. — Прим. перев.
154 Глава 5
так как силовая постоянная валентных колебаний зависит от контура
кривой потенциальной энергии, являющейся функцией как электронной
конфигурации, так и положения близлежащих электронных состояний;
2) строго говоря, эти формулы применимы только к двухатомным
молекулам, хотя, если колебание локализовано между двумя группами,
моделирующими двухатомную молекулу, их можно использовать и в
случае многоатомных молекул. Ограниченное применение этих функций
наводит на мысль о том, что они пригодны только для прибли-
приближенных расчетов частот и точное отнесение полос должно основывать-
основываться на более строгой теории.
Теперь рассмотрим типы колебаний, ответственных за появление
групповых частот в спектре. Еще раньше отмечалось, что моле-
молекулу можно смоделировать N массами, связанными пружинками (хими-
(химическими связями), и что такая система имеет 3N — 6 нормальных
колебаний. Строго говоря, в любом из этих нормальных колебаний
участвуют все JV атомов. В очень хорошем приближении неко-
некоторые из этих колебаний локализованы на отдельной группе и именно
они обусловливают групповые частоты.
Такая же ситуация наблюдается, когда имеется легкий атом водорода,
колеблющийся относительно более тяжелого атома углерода, как при
валентном колебании С—Н. Движение главным образом сосредото-
сосредоточено на более легком атоме и, следовательно, подвергается незна-
незначительному влиянию остальной части молекулы.
Возникновение второго типа групповых частот обусловлено тем,
что связанные атомы имеют близкие массы и колебания слабо взаи-
взаимодействуют с остальной частью молекулы. Примерами такого рода
являются частоты валентных колебаний кратных связей, как в группах
> С=О и C=N. Аналогично такие более или менее изолированные
группы, как фенильная, связанная с углеродом, или метильная — с
кремнием, находятся в слабом колебательном взаимодействии с
остальной частью молекулы, по крайней мере для некоторых нор-
нормальных колебаний.
Эти типы групповых частот почти всегда легко выявляются,
поскольку они попадают в относительно узкие интервалы длин волн.
Существует также класс групповых частот, в котором положение
полос изменяется в широких пределах, но его можно скоррелиро-
вать с массой, резонансом или электронными влияниями остальной
части молекулы. К настоящему времени причины некоторых из таких
корреляций до конца не поняты, и их нужно рассматривать в
значительной степени как эмпирические (но полезные) соотношения.
До сих пор мы обращали внимание на положение полос на шкале
частот и связывали его с молекулярной структурой. Эти измерения
можно выполнить точно, а данные, полученные на разных приборах,
легко сопоставить. Вероятно, столь же важными, но, конечно, гораздо
менее понятными являются изменения интенсивности, контура и
ширины полосы поглощения. Бессознательно эти параметры используются
Качественный анализ по ИК-спектрам 155
каждым спектроскопистом, но только в последнее время начались
экспериментальные исследования, направленные на решение проблемы с
помощью количественных измерений.
Недавний прогноз широкого использования интенсивностей для
качественного анализа пока не оправдался, отчасти из-за трудности
разделения перекрывающихся полос сложного спектра, отчасти из-за
того, что многие спектрофотометры не позволяют получать точные
значения интегральных интенсивностей, не прибегая к обычно трудоем-
трудоемкому интегрированию. Еще меньше внимания уделяется проблеме вы-
выявления количественных связей контуров полос с известными молеку-
молекулярными параметрами. Здесь же опять инструментальные ограничения
и перекрывание полос также сдерживают прогресс исследований.
Внутренние факторы,
влияющие на групповые частоты
Если бы химические связи всегда были независимы от соединения,
в котором они находятся, то проблема молекулярной динамики легко
решалась бы методами классической механики. Однако, как известно
каждому химику, множество неуловимых факторов влияет на длину,
полярность, направление, прочность связей. Факторами, прямо влияю-
влияющими на групповые частоты и интенсивности в молекулярном
спектре, являются изменения атомной массы, колебательное взаимодей-
взаимодействие, резонанс, индуктивный эффект и эффекты поля, сопряжение,
водородная связь, напряжение углов и связей [56]. Эти возмущающие
факторы обсуждаются в следующих разделах.
Изменения масс
Из уравнения E.16), связывающего массу и частоту, можно получить
частоту валентных колебаний между атомами А и В. При заме-
замещении атома В на С изменяется квадратный корень из приведен-
приведенной массы, т. е.
\ E.25)
Гас V »
при условии равенства других параметров. Однако, за исключением
случая изотопного замещения, другие параметры редко остаются без
изменения, так как при замещении атомов одного типа на другой
вместе с массой изменяются полярность, длина и прочность связи.
Например, если вычисляются частоты колебаний галогеноводородов
с использованием HI в качестве стандарта (т. е. в предположении,
что силовые постоянные для других галогеноводородов такие же, как и
для HI), то для НВг получается 2314 см (расчет) и 2564 см
(эксперимент), для НС1 — 2333 см (расчет) и 2886 см (эксперимент)
и для HF - 2360 см (расчет) и 2968 см (эксперимент). Очевидно,
156 Глава 5
что в этом ряду силовые постоянные изменяются вместе с массами.
Длина связи также изменяется с размером атома галогена, и частоты
в ИК-спектрах являются чувствительной мерой этого параметра [191,
193].
Если А, В или С — группа атомов, то простое уравнение E.25)
уже больше неприменимо, так как эффективная масса группы не явля-
является суммой атомных масс. Это происходит из-за того, что, хотя все
атомы группы колеблются в фазе, направление движения и ампли-
амплитуды их колебаний не всегда совпадают. Сопоставим, например,
валентные колебания С—С1 в СН3С1 и СН3СН2С1 (в которых элект-
электрические эффекты примерно одинаковы), полагая, что силовые постоян-
постоянные равны, а массы групп СН3 и С2Н5 составляют 15 и 29 а. е. м.
соответственно. Для частоты С—С1 в СН3СН2С1 вычисление дает
608 см по сравнению с величиной 657 см, наблюдаемой экспе-
экспериментально. Этот результат показывает, что эффективная масса этиль-
ной группы меньше суммы составляющих ее атомных масс, как мы
могли бы предположить интуитивно. Поэтому, когда имеют дело с груп-
группой атомов, даже если их электрические и стерические эффекты
сходны, изменения частоты уже нельзя связывать прямо с массой
без анализа нормальных координат.
Изотопное замещение приводит к сдвигу частоты валентного коле-
колебания, очень близкому к расчетному. Хорошо разработанной методикой
изучения колебаний, связанных с атомом водорода, является его заме-
замещение на дейтерий*. Для чисто валентного нормального колебания
атома водорода, связанного с бесконечной массой, при замещении на
дейтерий можно ожидать сдвига частоты в J/2 = 1,414 раз, но в реаль-
реальной молекуле величина смещения зависит от типа нормального ко-
колебания. Кримм [161, 162], например, показал, что при замещении
дейтерием в изолированной группе СН2 для различных типов нормаль-
нормальных колебаний частотный сдвиг происходит в следующих отно-
отношениях: валентное симметричное колебание СН2 в 1,379 раза, маятниковое
СН2 в 1,379 раза, валентное асимметричное СН2 в 1,349 раза, плоскост-
плоскостное деформационное СН2 в 1,349 раза, веерное СН2 в 1,323 раза,
крутильное СН2 в 1,414 раза. Там, где нормальные колебания различ-
различных типов взаимодействуют друг с другом, как в полиэтилене, эти числа
не получаются точно такими, но их относительные величины располо-
расположены в том же порядке.
Наблюдаемые частоты в изотопозамещенных образцах несколько
отличаются от рассчитанных и по причине небольшого, но заметного
изменения длины связи. Например, в Н35С1 г0 = 1,2837 А, но для
D35C1 r0 = 1,2813. Разница в 0,002 А существенна. В общем случае
замещение более тяжелым изотопом ведет к сокращению длины
связей. Это кажущееся несоответствие возникает из-за того, что
* Об изменении частот при изотопном замещении см. [1, 2, 7].— Прим.
перев.
Качественный анализ по ИК-спектрам 157
наблюдаемая длина связи является средней величиной, взятой между
крайними положениями атома в процессе колебания. Более тяжелые
изотопы колеблются с меньшей амплитудой, и поэтому средняя длина
связи кажется меньшей *. Равновесная длина связи ге, вычисленная для
неколеблющейся молекулы, совершенно не зависит от массы.
Геометрия
Заметное влияние на частоту и интенсивность колебательного
спектра поглощения оказывают также симметрия и геометрия молекулы.
Напомним, что колебание активно в ИК-области, если оно приводит
к изменению дипольного момента, а интенсивность полосы поглощения
зависит от изменения дипольного момента, которое падающее излучение
«видит» в процессе колебания. Ясно, что отсутствие характеристи-
характеристического поглощения в определенном месте спектра не должно обяза-
обязательно указывать на отсутствие данной группы. Хорошим примером яв-
является связь С=С. Ее валентное колебание не проявляется в спектрах
таких молекул, как Н2С=СН2, С12С=СС12, и транс-изомеров по той
X н
\ /
с=с
/ \
н х
причине, что колебание происходит около центра симметрии и не
вызывает изменения дипольного момента (однако это колебание весьма
активно в спектре КР). То же рассуждение применимо к метилацетилену
и ацетилену: в спектре первого наблюдается поглощение связи
—С=С—, а в спектре второго нет.
Могло бы показаться, что даже ^ис-изомеры
Н Н
\ /
с=с
/ \
Вг вг
будут проявлять только слабое поглощение, отвечающее валентному
колебанию —С=С—, так как через связь С=С можно провести
плоскость симметрии перпендикулярно плоскости молекулы. Однако,
наоборот, молекула имеет сильную полосу поглощения двойной связи
1589 см, поскольку в ней валентное колебание участвует на 80%.
В оставшихся 20% участвуют колебания связей СН и СВг, которые
вносят заметное изменение в дипольный момент [230]. Необ-
* Дело не только в амплитуде колебаний, а в наличии ангармоничности.—
Прим. ред.
158 Глава 5
холимо отметить, что, хотя для газовой фазы правила симметрии
выполняются точно, в жидкостях и твердых веществах молекулы воз-
возмущены их соседями и «запрещенное» поглощение может слабо про-
проявиться в спектре.
Групповые частоты также могут смещаться под действием механи-
механических факторов. Подходящим примером является валентное колебание
О=С в ряду циклоалкенов [56, 180]:
сн, сн, сн,
о о п v ~x v x
1646 1611 1566 1641 1641 1675
В случаях а, б, г колеблющаяся система должна дополнительно
работать против примыкающих простых связей С—С, при этом увели-
увеличивается силовая постоянная и частота повышается. В структурах д и е
колебание опять должно работать против связи =С—СН3 и частота
вновь повышается*. Изучение колеблющихся механических моделей
подтверждает указанные тенденции в изменении частот [54].
Колебательное взаимодействие
В молекуле колебания смежных групп, имеющих примерно одина-
одинаковые частоты, могут иногда взаимодействовать и давать смешан-
смешанное колебание, в котором участвуют обе группы. Такое колебатель-
колебательное взаимодействие носит довольно общий характер, но это не
мешает участвующим колебаниям оставаться групповыми. Например,
хорошо известная частота 1620 см в замещенных этиленах в дейст-
действительности на 60 % валентное колебание С=С и на 35 % деформа-
деформационное СН. Когда замещением водорода на дейтерий это взаимодей-
взаимодействие снимается, частота валентного колебания С=С падает до своего
нормального значения 1520 см [230]. Другим примером являются
полосы амид II и амид III с максимумами 1550 и 1290 см,
которые характерны для вторичных амидов с открытой цепью. Эти
полосы возникают как результат взаимодействия деформационного ко-
колебания NH и валентного колебания С—N [93]. Степень взаимодейст-
взаимодействия не изменяется существенно при переходе от одной структуры
к другой, и результирующие частоты достаточно постоянны.
Для того чтобы такое взаимодействие происходило, необходимо
выполнение следующих условий: 1) оба колебания должны иметь оди-
одинаковую симметрию; 2) оба колебания должны иметь примерно
* Колебания связей С=С в циклоалкенах рассмотрены в книге [8, с. 31].—
Прим. перев.
Качественный анализ по ИК-спектрам 159
одинаковую частоту; 3) взаимодействие между группами должно быть за-
заметным (это утверждение означает, что группы находятся в непосредст-
непосредственной близости и существует механизм переноса колебательной энергии
от одной группы к другой).
Особый случай наблюдается при взаимодействии обертона или сос-
составной частоты с основным колебанием. Это явление называется резо-
резонансом Ферми (по имени Э. Ферми, который впервые предложил
объяснение расщепления основной полосы 1330 см в СО2 [86]). В ре-
результате этого взаимодействия возникают две полосы там, где ожидается
одна (фундаментальная). Обертон или комбинационная полоса «заим-
«заимствует» интенсивность из фундаментальной, и обе полосы «отталкива-
«отталкивают» друг друга, так что ни одна из них не находится на том месте,
где ожидается согласно расчетам. Относительные интенсивности и
расстояния между полосами зависят от того, насколько близки невоз-
невозмущенные частоты. Их положение можно приближенно вычислить по
формуле
vo = 4(v- + vb) ± "f <v" - vb) ф-=-?-, E.26)
где две величины v0 относятся к невозмущенным частотам, va и
vb - наблюдаемые частоты, а 1а и 1Ь - их интенсивности [165, 246].
Другим примером резонанса Ферми является взаимодействие в
альдегидах валентного колебания СН (частота 2800 см) с первым
обертоном плоского деформационного колебания СН (частота 1400 см" ^
приводящее к характерному дублету в области 2700-2900 см.
Резонанс Ферми не всегда легко идентифицировать, но его можно
ожидать, когда полоса, одиночная при нормальных условиях, расщепля-
расщепляется. Иногда поведение пары полос в зависимости от физического
состояния и растворителя может подтвердить наличие взаимодействий
типа резонанса Ферми [30, 33, 262]. В метальных соединениях от
резонанса Ферми между обертоном деформационного колебания СН
и валентными колебаниями СН избавляются частичным дейтерированием
группы с образованием CHD2. В обзоре [190] представлены другие
способы определения резонанса Ферми.
Порядок связи
Согласно правилу Горди [уравнение E.24)], силовая постоянная
прямо пропорциональна порядку связи. Например, силовые постоян-
постоянные С—С, С=С и С^еС имеют значения около 4,6-10 , 10-Ю
и 15-10 дин/см соответственно.
Как будет видно в дальнейшем, колебательные частоты можно
скоррелировать с длиной связи, которая в свою очередь зависит от
ее порядка. Порядок связи берется не в виде интегральной величины,
а согласно предложенному Полингом [208, с. 239] уравнению, которое
160 Глава 5
связывает длину связи D(ri) и ее порядок:
D(ri) = Dl -0,71 lg n. E.27)
Порядок связи п и число связи п совпадают для п = 1, 2 или 3,
но для дробных п они не равны. В бензоле, например, п = 3/2, но
п = 5/3, что отражает избыточную энергию резонанса молекулы *.
Частота валентного колебания связи является чувствительной мерой
ее длины. Для связей С—О и С—N [169] их длина коррелирует
с частотой: по линейному закону для С—N и более сложным образом
для С—О в области 800—2400 см. Из-за колебательного взаимодей-
взаимодействия имеются отдельные отклонения от графиков, но в целом наблю-
наблюдается хорошая корреляция. Изучая валентные колебания X—Н
(Х=С, N, О), Бернштейн [32] показал, что длина связи может быть
определена из колебательных частот с такой же точностью, как и из
вращательных спектров, полученных с высоким разрешением. Здесь вновь
корреляционный график не линеен. Тенденции изменения длины связи
в зависимости от структуры были исследованы Мак-Кнном и др. [192].
Электронные эффекты
Распределение электронов в молекуле (которое определяет как хими-
химические свойства, так и силовые постоянные связей) зависит от многих
факторов: одни из них известны, другие нет. Предпринимались попытки
разделять электронные взаимодействия на индуктивные, резонансные
и эффекты поля для атомов (или групп) на основании химического
и физического поведения молекул, содержащих эти группы. Были
созданы различные шкалы реакционной способности, в которых заме-
замещающим группам или атомам приписывались некоторые относительные
величины. Эти величины полезны для предсказания скоростей и равно-
равновесий реакций новых молекул [171].
Большие усилия были затрачены на поиски корреляции ИК-группо-
вых частот с константами заместителей. Цель таких корреляций состоит
в том, чтобы дать возможность предсказывать сдвиги групповых
частот на основании свойств заместителей; такая информация неоценима
в том случае, когда подтверждается или отвергается постулированная
структура.
Индуктивный эффект. Способность атома в молекуле оттягивать
электроны на себя измеряется его электроотрицательностью. Относи-
Относительные величины электроотрицательности были получены для всех
элементов [107], и, хотя их точность ограничена одной-двумя знача-
значащими цифрами, эта концепция полезна. Происхождение индуктивного
эффекта связано с разностью электроотрицательностей атомов в одной
молекуле или группе.
Электроотрицательность полезно рассматривать скорее как свойство
• Об энергии резонанса см. [9, с. 85-88; 10, с. 65].- Прим. перев.
Качественный анализ по ИК-спектрам 161
связи, а не атома [219], так как величина, полученная для атома, ме-
меняется в зависимости от его химического состояния (например, 2,3 для
углерода в СН3 и 2,8 для углерода в C^N). Химические группы также
имеют относительные величины электроотрипательности, которые свя-
связаны с константами полярных заместителей [68].
Ряд частот валентных колебаний кратных связей был скоррелирован
с суммой электроотрицательностей заместителей, включая фосфо-
рильную (R3P=O) [19], карбонильную (R2C=O) [147] и другие группы
[65]. Шкала электроотрицательностей, основанная на силовых постоян-
постоянных связей А—Н, была предложена в работе [271].
Для молекул, содержащих а-связи, индуктивный эффект определя-
определяется как влияние заместителей X на достаточно удаленный реакцион-
реакционный центр Y (например, XCH2CH2CH2Y). Количественной мерой коэф-
коэффициента индукции в алифатических системах является а*, введенная
Тафтом [251].
Индуктивный эффект изменяет порядок связи и ее длину, влияя
тем самым на частоту колеблющейся группы. Например, карбонильные
группы подвергаются воздействию следующим образом:
R R
Заместители X, которые усиливают тенденцию кислорода притягивать
электроны (кислород более электроотрицателен, чем углерод), т. е. до-
доноры электронов, как в структуре б, понижают порядок связи и
уменьшают частоту валентного колебания карбонильной группы. За-
Заместители, которые оттягивают электроны и тем самым благоприят-
благоприятствуют структуре а, увеличивают частоту колебаний карбонильной
группы. Например, хлорангидриды кислот имеют полосу поглощения
около 1800 см, а кетоны — около 1715 см.
Однако не обязательно придерживаться того, что весь сдвиг возни-
возникает из-за изменения силовой постоянной связи С=О. Почти во всех
исследованиях, касающихся корреляции частоты и интенсивности карбо-
карбонильной группы, делаются следующие предположения: 1) колебательное
взаимодействие валентного колебания С=О с другими молекулярными
движениями пренебрежимо мало; 2) наблюдаемая ИК-частота колебания
карбонильной группы непосредственно связана с суммой различных
электрических эффектов, действующих на эту связь. Оверенд и Шерер
[205] эффектно показали, что такие допущения необязательно верны
и предположили, что существует невозмущенная силовая постоянная
связи, которая отражает ее реальную силу. Например, галогенпроизвод-
ные карбонильных соединений, наблюдаемые частоты которых лежат
И - 586
162 Глава 5
Силовые
Молекула
COF2
COBrF
COC1F
COBr2
COC12
постоянные галогенпронзводных
соединений
у(набл.), см ~
1928
1874
1868
1828
1827
V*
1861+9
1864+9
1848 + 9
1874+9
1856 + 9
Таблица 5.1
карбонильных
К(С=О), мдин/А
12,61
12,64
12,43
12,79
12,54
в интервале шириной 100 см, имеют почти одинаковые силовые
постоянные (табл. 5.1). Величина v* является расчетной частотой,
исправленной с учетом взаимодействий.
Причина ничтожного различия в электронных эффектах атомов
фтора и брома в этом удивительном примере неясна, и из этих данных
нельзя сделать вывода о том, что такие силы не влияют на частоту
колебаний карбонильной группы в галогенпроизводных карбонильных
соединениях. На основании расчетов, выполненных для молекул другого
типа, было сделано заключение о сильной зависимости v?o (и Ксо)
от электронных эффектов. Смещение частоты происходит в предсказан-
предсказанном направлении, но из-за полярных эффектов его величина отличается
от предполагаемой.
Приложено много усилий для изучения индуктивного влияния за-
заместителей на групповые частоты. Важно отметить, что индуктивный
эффект преобладает только в том случае, если заместитель находится
на заметном расстоянии от колеблющейся группы (например, отделен
по крайней мере одним углеродным атомом) [21].
Индуктивные эффекты были постулированы, чтобы объяснить пере-
передачу поляризации химических связей [31]. Дьюар [73] принял противо-
противоположную точку зрения. Он предположил, что в целом эффект воз-
возникает из-за электростатического взаимодействия через пространство
(т. е. эффект поля). Несмотря на то что имеются некоторые экспери-
экспериментальные данные в пользу этой точки зрения [245, 257], все же
общепринятым остается мнение, что взаимодействие в значительной
степени распространяется через связь, хотя, вероятно, этот механизм
не преобладает в передаче полярных эффектов.
Мезомерия, резонанс н сопряжение. Как „мезомерия", так и „резонанс"
относятся к квантовомеханическому понятию, которое заключается в
том, что молекулярная структура стабилизируется за счет вкладов
нескольких гипотетических стационарных состояний путем минимиза-
минимизации полной энергии системы [9]. Термин „мезомерия" был предло-
предложен Ингольдом [134] в 1933 г. для описания смещения электро-
Качественный анализ по ИК-спектрам 163
нов в системе с сопряженными связями (например, R2SP-c=C—С=Ь ).
Для того чтобы происходил резонанс такого типа, необходимы
следующие факторы: 1) предпочтительное энергетическое состояние для
резонансных форм; 2) наличие электронов для образования кратных свя-
связей (например, свободные электронные пары на атомах азота и кислоро-
кислорода); 3) копланарность атомов, участвующих в резонансе. Так, нельзя
ожидать заметного вклада от формы С1+=С—СО" в ацетилхлориде,
поскольку вычисленная разность энергий между этой и нормальной
формами, составляющая около ПО ккал, подтверждает наше интуи-
интуитивное заключение, основанное на относительной силе электронного
притяжения.
„Сопряжение" относится к случаю чередующихся кратных и простых
связей. Резонанс или перегибридизация приводят к тому, что кратные
связи передают часть их л-электронной плотности находящейся между
ними простой свяак. При этом частота колебаний простой связи повы-
повышается, а кратной связи уменьшается. В случае сопряженных связей
С=С наблюдается понижение частоты на 20—40 см. Возникает
также расщепление полосы поглощения, соответствующее валентным
колебаниям связей С=С, происходящим в фазе и не в фазе. Кроме
того, увеличивается интенсивность поглощения. Такие наблюдения не
обязательно означают изменение порядка связи, если иметь в виду зна-
значительное колебательное взаимодействие, происходящее между валент-
валентным С=С и деформационным СН колебаниями.
Влияние резонанса на полосу поглощения карбонильной группы
состоит в понижении частоты на 30 — 40 см (ср. 1715 см для ацетона
и 1685 см для ацетофенона). Постулируется, что при взаимодействии
с соседней л-связью электронная плотность на л-связи карбо-
карбонильной группы уменьшается:
о о
II I
с ^ с
/ \ / / \ +/
R C=C R С-С
/ \ / \
Индуктивное влияние атомов, участвующих в таком резонансе,
несомненно, также изменяется. Поскольку в результате изменения
индуктивного эффекта сопряжение усиливается или ослабляется, пред-
предсказать его величину не всегда легко. Эффекты, наблюдаемые в моле-
молекулах с сопряженными связями, более подробно обсуждаются в ра-
работах [22, 24].
Для корреляции структуры со скоростью реакции и данными по
равновесию для мета- и иара-замещенных производных бензола было
предложено уравнение Гаммета. Оно позволяет измерять электронное
влияние заместителя на реакционный центр в ароматических системах.
Уравнение, связывающее эффект заместителя а со скоростью реакции
164 Глава 5
(или константой равновесия), имеет вид
E.28)
где k0 относится к стандартному состоянию (незамещенное производное
бензола) и р - константа пропорциональности, которая отражает чув-
чувствительность реакции к полярным заместителям. Типичные корреляции
ИК-данных с величинами а Гаммета включают частоты колебаний
одного, двух или трех смежных атомов водорода [20], частоты коле-
колебаний карбонильной группы в ацетофенонах и бензофенонах [96, 144],
интенсивности полос поглощения валентных колебаний NH в аминах
[160], интенсивности полос поглощения групп C=N в ароматических
нитрилах [40].
Константы заместителей н ИК-частоты. Для корреляции скоростей
реакций, констант равновесия и других свойств различных семейств
производных бензола было предложено большое число констант за-
заместителей (например, некоторые из них: ст~, а*, стс, а*, а;, ст?).
Спектроскописты потратили больше времени на поиски корреляций
этих параметров и на хорошее соответствие данных, чем на логи-
логические соображения, связанные с химической структурой.
Свейн и Луптон [248] отметили, что быстрое увеличение числа
констант заместителей уже дошло до нелепости, и предложили любую
константу заместителя представлять в виде линейной комбинации
эффектов резонанса и поля (последний определяется так, чтобы включить
индуктивные эффекты):
g=/F + rR, E.29)
где F и R — константы поля и резонанса, характеризующие заместитель
(например, Н, ОМе, Вг, NH2), а / и г - весовые факторы, не зависящие
от заместителя, но различающиеся для каждого набора констант
(<зт, <зр, <т+, о1). До тех пор пока не была доказана правильность
этой концепции, использование только одного резонансного параметра
было сомнительным [42].
Позднее возникла тенденция определять полярные (или индуктив-
индуктивные) эффекты как результат действия несопряженных, пространственно
разделенных заместителей на равновесие или скорость процесса неза-
независимо от того, передается эффект через связи или через пространство.
Конечный результат измеряется в шкале os, которая основана на ста-
статистическом анализе данных по реакционной способности, равновесию
и физическим свойствам, взятых из многих источников [257]. Величины
Ст; образуют ряды, которые похожи, но не идентичны рядам а*,
описанным Тафтом. Шкалы индуктивности для алкильных заместителей
почти одинаковы независимо от модели, используемой для их полу-
получения [173].
Аналогичные шкалы были созданы для резонансных эффектов. Су-
Существуют четыре отдельные шкалы, каждая из которых применима
Качественный анализ по ИК-спектрам 165
NMe2
ОМе
F
С1
Me
Ph
СОМе
CO2R
NO2
Н
Et
г-Bu
OAc
0,06
0,27
0,50
0,46
-0,04
0,10
0,28
0,30
0,65
0,00
-0,05
-0,07
0,39
Константы заместителей D2]
°«
-0,34
-0,45
-0,45
-0,23
-0,11
0,04
0,47
0,34
0,46
0,00
-Л
-0,52
-0,45
-0,34
-0,23
-0,11
-0,11
0,16
0,14
0,15
0,00
-0,83
-0,61
-0,45
-0,23
-0,11
-0,11
0,16
0,14
0,15
0,00
Таблица 5.2
4
-1,75
-1,02
-0,57
-0,36
-0,25
-0,30
0,16
0,14
0,15
0,00
в различных ситуациях в зависимости от того, является ли бензольное
кольцо невозмущенным (с$), обедненным электронами (а^), обога-
обогащенным электронами (а«) или находится в резонансе с карбоксильной
кислотной группой (сгк) [257]. Константы для некоторых распростра-
распространенных заместителей представлены в табл. 5.2.
Полное влияние на молекулу описывается двухпараметровым урав-
уравнением
v - v0 = р,а, + ряа«, E.30)
где v — v0 — частотный сдвиг. Используя многие литературные данные,
Броунли и Топсом [42] рассмотрели связь частотных сдвигов в ИК-
спектре с параметрами реакционной способности (индуктивным и резо-
резонансным) для алифатических соединений [42], а Броунли и др. [41] -
для ароматических молекул. В большинстве случаев применение двухпа-
раметрового подхода дает лучшие корреляции. Эти авторы подчерк-
подчеркнули важность исследования широкого ряда заместителей для установ-
установления корреляций. Имеются обзоры по индуктивному [173, 245] и элект-
электронному эффектам заместителей [257]. Ниже представлены некоторые
примеры корреляций из имеющихся в литературе.
а. Асимметричное валентное колебание NO2 в алифатических нитро-
соединениях [181]. В соединениях типа R(CH3JCNO2 частота асиммет-
асимметричного валентного колебания NO2 связана с константой а* прибли-
приближенным уравнением
= 23,92а* + 1538,5 см E.31)
с коэффициентом корреляции 0,981.
166
Глава 5
920 930 940 9S0 960 970 980 990
V (крутильное колебание ОС в
G-CH-CHj), см
1000
Рис. 5.8. Зависимость частоты крутильного колебания С = С в G — СН = СН2
от величин рКа соответствующих кислот GCH2COOH [217]. Reproduced, with
permission, from Spectrochimica Acta.
1740 -
1739 -
"oo
1730 -
1725
-0,2
0,0
0,2
0,4.
0,6
0,8
Рис. 5.9. Зависимость vc
растворе ССЦ от величин ат
для .мета-замещенных метилбензоатов в
[69]. Reproduced, with permission, from Spectro-
Spectrochimica Acta.
Качественный анализ по ИК-спектрам
167
115-
110 -
105 -
-0,2
0,2
0,4
От
0,6
0,8
Рис. 5.10. Зависимость квадратного кория из интенсивности поглощения
л«7па-замещенных метилбензоатов в растворе ССЦ от величин ит; величины А
измерены в л/(мольсм2) [69]. Reproduced, with permission, from Spectrochi-
mica Acta.
б. Крутильно-деформационное колебание в G—Ctt=CH2 [217].
Исследование 69 винильных соединений выявило корреляцию частот
крутильного колебания в НС=СН2 с величинами рКа соответственно
замещенных производных уксусной кислоты (рис. 5.8). Точка для
C=N выпадает из кривой в силу гидролиза группы C^N, поэтому
точное значение рКа для циануксусной кислоты не получается. Точки
для метильной, этильной и изопропильной групп могут отклоняться
от корреляционной линии из-за колебательного взаимодействия. Хотя
причина изменения частоты крутильно-деформационного колебания
группы СН=СН2 в зависимости от индуктивной силы заместителей
неясна, это соотношение полезно.
в. Частота валентного колебания С=О в метилбензоатах [69].
На рис. 5.9 показана корреляция частот валентных колебаний группы
0=0 в ряду мета-замещенных метилбензоатов в разбавленных раство-
растворах ССЦ с простым параметром заместителя <зт Гаммета. Использо-
Использование двух констант заместителей дает двухпараметровое уравнение
vm - v0 = 12,79а/ + 5,63ал,
E.32)
которое описывает экспериментальные точки со среднеквадратичным
отклонением, примерно вдвое меньшим, чем при корреляции с <тш.
Заместители были выбраны таким образом, чтобы охватить широкую
область свойств, и включали NMe2, OMe, F, О, Вг, Me, H, СО2Ме
и NO2.
Аналогичные тенденции проявляются в отношении интенсивностей;
график однопараметровой зависимости от ат показан на рис. 5.10.
168 Глава 5
Двухпараметровое уравнение
= - 8,80а, -0,50<тк E.33)
дает несколько меньшее рассеяние.
Топсом и сотр. [41, 42, 152] подробно рассмотрели корреляции
ИК-частот и констант заместителей.
Выводы. Обсуждение электронных эффектов можно суммировать
следующим образом:
Индуктивные эффекты: 1) располагаются в порядке увеличения
констант заместителей <7/ или а* Тафта: чем больше величина константы,
тем больше эффект; 2) изменяют частоты валентных колебаний про-
пропорционально величине константы; 3) хорошо коррелируют с частот-
частотными сдвигами в тех случаях, когда заместитель замещает по крайней
мере один или два атома в колеблющейся группе.
Сопряжение кратных связей: 1) понижает частоту и 2) повышает
интенсивность валентного колебания кратной связи.
Резонанс: 1) зависит от способности электронов образовывать кратные
связи; 2) осуществляется с наибольшей вероятностью, когда участвующие
в нем атомы лежат в одной плоскости; 3) неизбежно изменяет
индуктивный эффект участвующих атомов; 4) оказывает большое
влияние на частоту; его величина и направление сдвига являются
функцией порядка связи и вторичного индуктивного эффекта.
Очевидно, что корреляции сдвигов частоты или интенсивности
с индуктивными или резонансными параметрами, хотя и полезны
с эмпирической точки зрения, не могут быть использованы для много-
многообещающих выводов о молекулярной структуре. Не только индуктивный
и резонансный эффекты и эффект массы создают сложную и часто
неясную картину, но и использование частот вместо силовых постоянных
может привести к ошибочным заключениям о действии этих эффектов.
В то же время нельзя ожидать от спектроскописта-аналитика расчета
колебательных частот для каждой молекулярной системы, с которой он
сталкивается. В аналитической работе эмпирическое приближение пока
является лучшим и дополнительные корреляции частот или интенсив-
ностей с другими физическими свойствами крайне полезны.
Влияние ассоциации
В широком смысле молекулы в любой конденсированной фазе нахо-
находятся в ассоциированном состоянии, и взаимодействие растворителя
с растворенным веществом или молекул индивидуального соединения
друг с другом будет вносить изменения в колебательный спектр. Этот
тип взаимодействия обсуждается далее. Здесь же ограничимся отдель-
отдельными взаимодействиями, которые осуществляются наряду с вандер-
ваальсовым притяжением.
Один из типов самоассоциации найден в соединениях с электронно-
ненасыщенными связями, таких, как А12 (СН3N и Вех (СНз)гх- Алкилли-
Качественный анализ по ИК-спектрам
169
3600
3200
2800 3600
Волновое число,см
3200
2800
Рис. 5.11. Поглощение водородной связи в некоторых типичных ассоцииро-
ассоциированных группах.
а — 2-этилгексановая кислота; 6 — н-пропиловый спирт; в — ацетамид; г — н-гексиламин. а и б ~ спектры
10 %-ных растворов в CG4, в — спектр суспензии в вазелиновом масле, г — спектр жидкой пленки.
170
Глава 5
\
\
\
^ 3
•-г
2,9 3,0
Длина волны, мкм
3,1
Рис. 5,12. Концентрационная зависимость поглощения в области валентных
колебаний водородных связей н-бутанола в СС14.
/-0,005 М в кювете толщиной 20 мм; 2-0,075 М в кювете толщиной 3 мм; J — 0,152 М в
кювете толщиной 1 мм; 4 - 1,7 М в кювете толщиной 0,025 мм [164]. Reproduced, with permission,
from Spectrochimica Acta.
тиевые соединения особенно склонны к образованию полимеров этого
типа [264], которые существуют и в газовой фазе [265]. Некоторые
алкоксиды металлов также образуют ассоциаты [15]. ИК-спектры поли-
полимеров довольно резко отличаются от спектров, ожидаемых для
мономеров.
Кислоты Льюиса образуют комплексы с карбонильными соедине-
соединениями. Сдвиг частоты валентных колебаний группы С=О в ацетоне
при комплексообразовании с BF3 составляет 70 см, а в ацетофеноне —
107 см [59, 247]. По-видимому, эти сдвиги могут коррелировать
с основностью группы С=О. Эфиры образуют комплексы с НС1 и HF
как в конденсированной, так и в газовой фазах [8].
Несомненно, что наиболее распространенным типом ассоциации
является водородная1 связь. Она возникает в результате взаимодей-
взаимодействия между протонодонорной (ХН) и протоноакцепторной молекулами.
Обычно донорами и акцепторами считаются электроотрицательные
атомы таких групп, как —ОН- -О—, —OH-N—, —NH-O— и
NH-N— (или в некоторых случаях л-электроны). Межмолекулярные
взаимодействия СН (как в растворах НСС13) также иногда рассматри-
рассматриваются как водородная связь.
Качественный анализ по ИК-спектрам 171
Влияние водородной связи на ИК-спектр состоит в смещении и уши-
рении полос валентных колебаний ХН (рис. 5.11). Интегральные
интенсивности также заметно повышаются. Вполне удовлетворительного
объяснения этих фактов еще нет, но ряд полезных наблюдений сделан,
и они здесь кратко рассматриваются.
Кэннон сформулировал критерии образования водородных связей
[47]: 1) связь X—Н должна иметь частично ионный характер (или быть
такой, что ионный характер может быть индуцирован поляризацией);
2) акцепторный атом должен иметь свободную электронную пару на
асимметричной орбитали; 3) чтобы взаимодействие было максимальным,
связь X—Н и орбиталь свободной электронной пары должны быть
коллинеарны (это не означает, что связи должны лежать на одной
прямой линии, т. е. —О—Н---0—). Можно добавить, что стерическое
влияние громоздких групп, окружающих донор протонов, будет пре-
препятствовать образованию водородной связи.
Теплота образования обычной межмолекулярной водородной связи
составляет 3 — 10 ккал/моль. Очень широкие полосы поглощения часто
состоят из нескольких перекрывающихся полос, соответствующих равно-
равновесным концентрациям димеров, тримеров и других полимеров
(рис. 5.12). Относительные количества различных ассоциатов зависят
от концентрации растворенного вещества, растворителя и температуры.
Только при разбавлениях 10 - 10 моль/л концентрация полимерных
частиц становится пренебрежимо малой. Из-за сильной зависимости
интенсивности от внешних факторов полосы поглощения ОН и NH нельзя
использовать для количественного анализа, кроме особых случаев.
Теплоты образования различных ассоциатов можно определить,
исследуя концентрационные зависимости от температуры. Это было
сделано для некоторых алифатических [174] и ароматических спиртов
[184]. Типичные результаты приведены в табл. 5.3.
Таблица 5.3
Энергии водородных связей спиртов в СС14,
измеревные методом ИК-спектроскопии
Спирт
Метанол
Этанол
трет-Ъутаноп
Фенол
л-Крезол
и-Хлорфенол
Тип ассоииата
Димер
Полимер
Димер
»
Полимер
Димер
»
»
Энергия, ккал/моль
9,2 + 2,5
4,7 + ?
7,2± 1,6
4,8+1,1
5,3 ±0,5
5,12+0,10
6,09 + 0,18
3,78 + 0,20
172 Глава 5
Некоторые молекулы даже в газовой фазе находятся в виде димеров
и полимеров, связанных водородной связью. Например, HF образует,
вероятно, ассоциаты (HFN. Энтальпии образования димеров уксусной
и масляной кислот равны 8,5 + 0,5 ккал/моль [220].
Сдвиг частоты поглощения при переходе от неассоциированной
к ассоциированной форме коррелирует с расстоянием от X до Y
во фрагменте X—Н- -Y [26, 27, 179, 232]. Чем меньше это расстояние,
тем сильнее смещение частоты.
Сдвиг частоты данного донора протонов в зависимости от различ-
различных оснований (акцепторов протона) может быть связан с теплотой
образования водородных связей [10, 108]. В качестве иллюстрации
можно привести энтальпии (в ккал) некоторых систем фенол —акцептор:
бензол A,6); этилацетат D,5); диэтиловый эфир E,4); метилэтилкетон
E,3) [218]. Также наблюдается корреляция ширин полос и интеграль-
интегральных интенсивностей с энтальпией, по крайней мере для некоторых
соединений [18].
Можно предположить, что протон, участвующий в водородной
связи, движется в потенциальной яме с двойным минимумом, соответ-
соответствующим положениям X—Н-Y и Х---Н—Y с максимумом в точке
Х-Н-Y. Барроу [16] установил экспериментальные критерии суще-
существования двойного минимума. В этом случае определенные энерге-
энергетические уровни расщепляются, и в результате в некоторых случаях
полосы основных частот и обертонов становятся дублетными.
Некоторые молекулы имеют такую структуру, что в них кроме
уже рассмотренных мелсмолекулярных водородных связей могут обра-
образовываться внутримолекулярные водородные связи между близлежа-
близлежащими группами [164]. В этот класс попадают некоторые орто-замещен-
ные ароматические соединения, такие, как о-хлорфенол:
Так как ассоциация стерически выгодна, то теплота образования может
быть более низкой (оценочно 1-4 ккал/моль). Сдвиги частот и ширины
полос меньше и не обязательно связаны с теплотой образования связи.
Кроме того, протоны могут ассоциироваться с более слабыми основа-
основаниями, такими, как я-электроны в аллильной группе 2-аллилфенола
[14], где при ассоциации наблюдался сдвиг на 63 см (рис. 5.13).
Внутримолекулярная водородная связь сохраняется даже при крайних
разбавлениях.
Несмотря на непостоянство поглощения ассоциированного водорода,
эта полоса иногда используется в качественном анализе. Например,
крайне широкое поглощение с многочисленными минимумами, наблю-
наблюдающееся в жирных кислотах (рис. 5.11), является, конечно, характе-
Качественный анализ по ИК-спектрам
173
/
2,70 2,80 2,90 3,00
2,70 2,80 2,90 3,00
длина волны, мкм
Рнс. 5.13. Спектры 2-аллилфенола при концентрациях 0,7, 0,07, 0,007 моль/л
в области валентного колебания ОН [14]. Reproduced, with permission, from
Journal of the American Chemical Society.
ристичным. Иногда, но не всегда можно различить самоассоцииро-
самоассоциированные амины и спирты; в несвязанном состоянии они различаются
легче (приложение 2). Вода, если она присутствует, мешает, но ее
часто можно определить по полосе деформационного колебания около
1620 см (рис. 5.14).
Водородная связь вызывает также другие изменения в спектрах. Если,
например, акцептором протона является карбонильная группа, то частота
ее колебаний понижается на 50 см, и в некоторых случаях ассо-
ассоциацию удобнее исследовать по этому эффекту.
Изменяются также внутренние колебания донорной молекулы. На-
Например, для спирта ожидается, что образование водородной связи
должно оказать большее или меньшее влияние на валентное колебание
С—О, деформационное колебание ОН и крутильное колебание СОН.
Эти нормальные колебания часто бывают смешанными, и поэтому
их трудно характеризовать. Некоторые детали колебательных спектров
ассоциированных донорных молекул детально обсуждены Пиментелом
и Мак-Клелланом [211], которые сделали следующие обобщения:
174
Глава 5
Длина волны, мкм
5 6
2000
1800 1600
Волновое число, см
Рис. 5.14. Полоса деформационного колебания воды в этаноле.
1) плоскостные деформационные колебания X—Н попадают в интервал
1000-1700 см и при образовании водородной связи смещаются
в область более высоких частот; 2) в ассоциированных жидких
и твердых веществах внеплоскостные крутильные колебания X—Н
лежат ниже 800 см; 3) валентные и деформационные колебания
X—H--Y имеют низкие частоты - порядка ^200 см; 4) ва-
валентное колебание С—О в спиртах попадает в область 1000-1070 см,
и при образовании водородной связи его частота понижается.
Для дальнейшего ознакомления с многочисленными эксперимен-
экспериментальными и теоретическими исследованиями водородной связи читатель
может обратиться к литературе [24, 111, 137, 159, 197, 210, 228, 238].
Влияние внешних воздействий
Термин внешние влияния носит весьма неопределенный характер,
и отделение их от внутренних эффектов довольно произвольно, но
в данном случае под внешними воздействиями подразумевают такие,
которые в какой-то степени подвергаются контролю исследователя.
Качественный анализ по ИК-спектрам 175
В эту категорию включают физическое состояние (газ, жидкость, твердое
вещество, раствор), растворитель, концентрацию и температуру. Каждый
из этих переменных параметров оказывает заметное влияние на поло-
положение групповой частоты, и для извлечения максимальной пользы
из корреляций групповых частот необходимо помнить о возможности
внешних возмущений.
Физическое состояние
Нигде влияние молекулярного окружения на картину ИК-поглощения
химических соединений не проявляется так резко, как при переходе
от газа или пара к конденсированному состоянию (рис. 5.15). В газо-
газовой фазе молекулы оказывают незначительное взаимное влияние на коле-
колебание и вращение друг друга. Как уже было показано (стр. 140—143),
результирующий спектр представляет собой ряд полос поглощения,
каждая из которых состоит из многих узких линий, соответствующих
отдельным колебательно-вращательным переходам, и перекрывает
широкую область длин волн. В жидкостях и растворах каждая молекула
ограничена «клеткой» из других молекул, так что они непрерывно
сталкиваются друг с другом и уже не могут совершать квантованного
вращательного движения. В результате тонкая вращательная структура
колебательной полосы исчезает и контур полосы поглощения становится
несколько похожим на вероятностную функцию. Причины, вызывающие
сильные локальные возмущения, включают дисперсионные силы, ди-
диэлектрические эффекты, диполь-дипольные и вандерваальсовы взаимо-
взаимодействия и такие специфические взаимодействия, как водородная связь.
Величина смещения полос поглощения при переходе от газа к жид-
жидкости весьма непостоянна. Она может достигать 100 см, но обычно
меньше 25 см. Например, частота валентного колебания С=О
этилацетата в газовой фазе имеет величину 1763 см, а в растворе
СС14 уменьшается до 1741 см. В ацетоне такое уменьшение при
переходе от пара к раствору CCL, составляет 18,5 см. Валентные
колебания С—Н в ацетилене довольно чувствительны к физическому
состоянию, вероятно, из-за ассоциации в растворах через мостиковые
водородные связи. В парах фенилацетилена наблюдается полоса погло-
поглощения 3340 см, а в растворе СС14—.3316 см. Для симметричных
пятиатомных молекул теоретическое толкование проблемы было дано
Хейкленом [116], который нашел, что длина связи увеличивается
при переходе от пара к жидкости.
В области 500 — 4000 см частоты почти всегда понижаются при
переходе к конденсированной фазе. Однако для некоторых низких
частот B00-340 см) было показано, что в жидкой фазе они выше,
чем в газовой [84].
В кристаллическом состоянии силовые поля, действующие на от-
отдельные молекулы, периодичны, что приводит к дальнейшим изменени-
изменениям в спектре (рис. 5.15). Полосы поглощения обычно становятся более
176
Глава 5
длина полны, мкм
5 6
7 ,
10 11 1213 IS
4000 ЗвОО 3200 2800 2400
2000 1800 1600
Волновое число, см^1
Рис. 5.15. Спектры л-дихлорбензола в парообразном состоянии (верхний), в
виде раствора (средний) и затвердевшего расплава (нижний).
узкими, часто расщепляются, могут появляться новые. Это происходит
из-за того, что каждая элементарная ячейка играет роль колеблющегося
элемента, но она обычно содержит больше одной молекулы. Поэтому
существует возможность движений в фазе и не в фазе, которые
могут иметь различающиеся частоты. Могут также происходить либрация
(ограниченное вращение) отдельных молекул и колебания решетки с низ-
низкой частотой. Эти энергетические состояния перекрываются с колеба-
колебательными и проявляются в спектрах поглощения. К тому же сильные
локальные силовые поля могут как изменять частоту колебательной
полосы, так и снимать запрет с обычно неактивных в ИК-области
нормальных колебаний.
Качественный анализ по ИК-спектрам 177
Такие молекулы, как жирные кислоты, при переходе в кристал-
кристаллическое состояние могут принимать одну предпочтительную конфор-
мацию, повышая интенсивность тех характеристических полос, которые
полезны для определения длины цепи [194]. В веществах, подобных
1,2-дихлорэтану, поворотная изомерия в кристаллическом состоянии
часто исчезает, поскольку все изомеры при затвердевании занимают
низшее энергетическое состояние, в результате чего спектр упрощается
[234]. Явление полиморфизма хорошо известно, и ИК-спектры раз-
различных кристаллических форм одного и того же вещества могут
часто заметно различаться. Переохлаждение некоторых жидкостей
приводит к образованию стекол. Обычно спектр стекла не очень
сильно отличается от спектра жидкости. Спектры монокристаллов
ряда веществ были исследованы и интерпретированы с точки зрения
структуры решетки. Для предсказания активности колебаний кристаллов
разработаны правила отбора [85]. Влияние изменения фазы и давления
на колебательные спектры рассмотрено Дэвисом [67].
Кроме тех случаев, когда исследуются эффекты твердого состояния,
спектроскописту желательно исключить как можно больше переменных
факторов, влияющих на спектр; поэтому всегда, когда это осуществимо,
твердые вещества лучше всего регистрировать в виде растворов.
Растворитель
Тому факту, что и положение и интенсивность полос, обусловлен-
обусловленных групповыми частотами, изменяются при переходе от одного
растворителя к другому, вероятно, не уделялось должного внимания.
Причины этих сдвигов начинают понимать только сейчас, и в этом
разделе будут рассмотрены главные факторы, влияющие на раство-
растворенный образец.
Исторически первую серьезную попытку объяснить уменьшение
частоты, обычно наблюдаемое в жидкостях, предпринял Кирквуд
[156] (на основе довольно ограниченных экспериментальных данных),
а позже Бауэр и Мага [17], взгляды которых впоследствии были
положены в основу теории, известной как теория КБМ. В этой теории
поглощающая частица рассматривается как простой двухатомный осцил-
осциллятор, находящийся в полости внутри растворителя с диэлектрической
постоянной е, а сдвиг полосы поглощения объясняется мгновенно
наведенной поляризацией среды растворителя колеблющимся диполем.
Выражение, известное как соотношение КБМ, связывает частотный
сдвиг с диэлектрической постоянной среды. Как будет показано позже,
это приближение является в значительной степени упрощенным по
сравнению с реальной ситуацией даже в случае неполярных раствори-
растворителей. Успех более совершенных выражении [44, 221] также остается
ограниченным.
Последующие проверки теории КБМ показывают ее непригодность.
Было исследовано влияние различных растворителей на отдельную
12 - 586
178 Глава 5
частоту, такую, как частота валентного колебания С=О в ацетоне
[256]. Эта частота, например, меняется от 1707 см в СН212 до
1722,5 см в к-гексане, интегральная интенсивность полосы изменяется
в 1,6 раза от к-гексана до пентахлорэтана, ширина полосы лежит
в пределах от 13 см в СН2С12 до 19,5 см в CS2. Однако частота
валентного колебания C=N и-метилбензонитрила в разных раст-
растворителях сдвигается незначительно, но интенсивность изменяется
в 2,3 раза, а ширина — в 1,6 раза [39]. Примеры такого типа иллюстри-
иллюстрируют непостоянство, которое следует ожидать от растворителей, но они
не очень помогают разобраться в различных факторах, вызывающих
эти изменения.
Было высказано предположение [30] о том, что частотный сдвиг
в значительной степени обусловлен ассоциацией с молекулами раст-
растворителя и что диэлектрические эффекты имеют относительно малое
значение.
При исследовании сдвигов, происходящих под действием раствори-
растворителей, целесообразнее использовать спектроскопию высокого разрешения
для изучения данного вещества в смеси двух растворителей, взятых
в разных соотношениях [25]. Если влияние растворителя — простая
функция свойств объема (например, диэлектрической постоянной),
то частота и интенсивность будут плавно изменяться по мере изме-
изменения состава растворителя. В то же время если имеют место специ-
специфические взаимодействия с молекулами растворителя, то должны наблю-
наблюдаться две полосы, интенсивности которых изменяются с составом
растворителя.
Систематические исследования такого типа были проведены Кага-
райсом [148] и Ветселом [267]. Используя циклогексан он и ацетон
как растворяемые вещества, а «-крезол (с которым ожидается ассо-
ассоциация) и циклогексан (который должен быть относительно инертным)
в качестве пары растворителей, они показали не только специфические
взаимодействия, происходящие с карбонильной группой кетонов
(рис. 5.16), но и то, что образуются комплексы 1:1 и 1:2, и вы-
вычислили константы их образования. Сдвиг частоты валентного колебания
С=О в циклогексаноне составил 16 см для комплекса 1:1 и допол-
дополнительно 5 см для комплекса 1 :2. Отмечены изменения интен-
сивностей и ширин полос.
Для менее активной пары растворителей — хлороформа и четырех-
хлористого углерода эти же авторы показали, что кетоны образуют
с хлороформом комплексы 1:1 и 1:2, в которых молекулы хлоро-
хлороформа, вероятно, связаны с карбонильными группами водородной
связью. Сдвиги частот карбонильного поглощения, обусловленные
ассоциацией, составили около 8 см для циклогексанона и 4 см
для ацетона. Наблюдались дополнительные небольшие смещения поряд-
порядка 3—4 см, которые были отнесены за счет неспецифических взаимо-
взаимодействий (включая объемные диэлектрические эффекты).
Исследование пары растворителей циклогексан — четыреххлористый
Качественный анализ по ИК-спектрам
179
0,6
0,4
0,2
I ' 1^1 ¦
Линия Крезол, М
10
'/ \ 2 0,0185
3 0,0609
4 0,0938
-№•
I
I
Линия Крезол, М
5 0,0938
6 0,211
7 0,549
8 1,413
13 • 0,0385м циклогексаноиа
5
' б
5\
1730 1720 1710
-W-
I
I
1700 1690 1730 1720
Волновое чисяо, см"'
1710 1700 1690
Рис. 5.16. Спектры циклогексанона в смесях л-крезола и циклогексана в
области валентных колебаний С=О [267]. Reproduced, with permission, from
Spectrochimica Acta.
углерод позволило сделать интересное заключение о том, что между
циклогексаноном (и предположительно другими полярными молекулами)
и четыреххлористым углеродом происходит образование слабой, но
отчетливо различимой координационной пары. Было показано, что
общий сдвиг частоты под действием растворителя составляет 6,8 см,
из которого около половины приписывается специфическому взаимодей-
взаимодействию.
Ветсел и Кагарайс пришли к выводу, что заметный вклад в смещение-
частоты карбонильного поглощения отдельного вещества в различных
растворителях вносят как специфические взаимодействия растворителя
с растворенным веществом, так и неспецифические, например объемные
диэлектрические эффекты и дисперсионные силы. Относительное значение
двух типов взаимодействий явно зависит от энергии ассоциированной
пары растворитель — растворенное вещество, имеющей большие диполь-
ные моменты и дающей максимальные специфические взаимодействия
и наибольшие сдвиги частот.
Величина сдвига зависит также от того, какая группа участвует
в ассоциации. Например, легко видеть, что ассоциация группы
X—Не растворителем, действующим как основание Льюиса, зависит
от кислотности протона, и, как было показано, частота валентных
колебаний N—Н в замещенных анилинах и нафтиламинах [43] линейно
12*
180 Глава 5
зависит от их рКа. Можно ожидать, что такие группы, как углерод —
галоген, будут проявлять специфические взаимодействия в несколько
меньшей степени; действительно, было найдено, что около половины
сдвига, возникающего в этих группах под действием растворителя,
обусловлено дипольными взаимодействиями, а оставшаяся часть — не-
неспецифическими [113]. Взаимодействия с растворителем вносят большой
вклад в изменения интенсивностей полос валентных колебаний групп
NH и ОН в дифениламине и а-нафтоле [201]; то же, по-видимому,
относится и к другим полярным группам.
Ясно, что изменение частот колебаний в молекулах зависит как
от растворителя, так и от взаимодействующей группы [112]. Это поло-
положение хорошо иллюстрируется исследованием крайнего случая — раст-
раствора п-диоксана в воде [95]. Ниже представлены некоторые из наблю-
наблюдаемых сдвигов и их направление при переходе от чистого п-диоксана
к разбавленному водному раствору:
vn (валентное BUCH) + 19 см-1,
v17 (валентное колебание кольца В„)-5 см,
v2j (валентное А„СН) + 12 см,
v26 (валентное колебание кольца А„)-1 см,
v16 (маятниковое Вц) + 4 см,
v27 (валентное колебание кольца Аи)-4 см.
Такие заметные изменения в поведении различных групповых частот
при замене растворителя положены в основу элегантного метода,
позволяющего охарактеризовать групповые частоты, который более
детально рассматривается позже.
Теперь ясно, что не существует совершенно инертного растворителя;
даже такой растворитель, как CCI4, участвует в специфическом вза-
взаимодействии с молекулами растворенного вещества. Исходя из этого,
можно было бы утверждать, что при записи спектров нужно совсем
отказаться от использования растворителей. Однако эта точка зрения
не учитывает того факта, что сильные межмолекулярные взаимодей-
взаимодействия происходят и тогда, когда исследуемая жидкость выступает
в качестве растворителя самой себя. Далее, так как загрязненные
или смешанные образцы, скорее, правило, чем исключение, то взаимодей-
взаимодействие с примесями, вероятно, приведет к гораздо большим изменениям
частот и интенсивностей, чем это наблюдается в относительно инерт-
инертных растворителях. По-видимому, наиболее постоянные и воспроизво-
воспроизводимые групповые частоты, интенсивности и контуры полос получаются
при использовании для повседневной работы пары растворителей
ССЦ—CS2, как это и было рекомендовано ранее (стр. 84—87).
' Концентрация
Газы. ИК-спектр поглощения газа при низком давлении состоит
из большого числа крайне узких, острых линий, соответствующих
переходам между отдельными колебательно-вращательными энергети-
Качественный анализ по ИК-спектрам
181
Рис. 5.17. Ударное уширение полосы 1310 см СН4.
о-50 мм рт. ст. СН4; 6 - 50 мм рт. ст. СН4 и 40 мм рт. ст. N2; в - 50 мм рт. ст. СН4 и
700 мм рт. ст. N2.
ческими уровнями. Поскольку большинство из этих линий лежит
за пределами разрешающей способности обычного ИК-спектрофото-
метра, в спектре поглощения обычно наблюдается некоторая усредненная
огибающая, а отдельные переходы видны только у молекул с враща-
вращательными уровнями, расположенными далеко друг от друга, как,
например, в НС1. По мере повышения общего давления (при добавле-
добавлении прозрачного постороннего газа) число столкновений с поглощаю-
поглощающими молекулами возрастает. Во время таких столкновений молекула
уже не может свободно вращаться, и если ее вращательная энергия
изменяется при поглощении излучения, то один или оба рассматри-
рассматриваемых энергетических уровня будут, вероятно, беспорядочно смещаться.
Следовательно, усредненная полоса поглощения уширится, и спектро-
спектрофотометр будет способен разрешать ее в большей степени, что приведет
к кажущемуся увеличению интенсивности. Это явление, известное
как ударное уширение, имеет важное значение в спектроскопии газов
и паров.
Влияние добавок азота при постоянном давлении газообразного
метана показано на рис. 5.17. Поглощение резко увеличивается во время
начального добавления азота и менее заметно при более высоких
давлениях. Увеличение давления инертного газа от 0 до 700 мм
может изменить интенсивность полосы поглощения в-5 или более раз.
Эффективность ударного уширения полос молекулами постороннего
газа зависит в некоторой степени от физического размера — чем
больше молекула, тем больше влияние. Однако правила, согласно
182
Глава 5
0,00
I
i I I
о полоса <3,7мкм
с Полоса 3,1 мкм
д полоса 7,4 мкм
О 100 200 300 400 500 600 700 800 900 1000
Парциальное давление С02,ммрт.ст.
Рис. 5.18. Ударное уширение некоторых полос ацетилена под действием
СО2 [5]. Reproduced, with permission, from Applied Spectroscopy.
которому молекулы можно расположить по их эффективности, нет.
Например, было показано, что полоса поглощения метана 1310 см
G,65 мкм) реагирует на посторонние газы в следующей последо-
последовательности: Не < Аг < Н2 = О2 < СО2 < С2Н6 < С3Н6 [53]. Однако для
валентных колебаний NO в N2O порядок другой: Ar<O2<N2<
< С2Н6 < СО2 = Н2. Кроме того, различные полосы поглощения одного
и того же газа могут по-разному реагировать на ударное уширение.
На рис. 5.18 показано поведение некоторых полос поглощения в ацетилене
в зависимости от парциального давления СО2 [5].
Имеющиеся в настоящее время сведения об ударном уширении
не могут нас удовлетворить полностью. С уверенностью можно
сказать лишь то, что усиление поглощения зависит от исследуемого
образца, особенностей наблюдаемой полосы поглощения, а также
от природы и давления газа, используемого для уширения. С прак-
практической точки зрения запись спектров всех газов, как «растворов»
в азоте при атмосферном давлении, имеет значительное преимущество.
При таком способе сводятся к минимуму изменения интенсивности
за счет давления, а- для сред со слабым поглощением улучшается
чувствительность. Это также позволяет избежать изменения интенсив-
ностей полос при натекании воздуха в кювету.
Жидкие и твердые вещества. В тех случаях, когда образуется водо-
водородная связь или возникают другие типы межмолекулярных ассоци-
атов, некоторые полосы поглощения могут быть довольно чувстви-
чувствительны к концентрации (см. рис. 4.1). Причины такого поведения
Качественный анализ по ИК-спектрам 183
обсуждались ранее. Рекомендуется для всех образцов данного типа
принять стандартные условия: растворитель, концентрацию и толщину
поглощающего слоя.
Температура
Влияние значительных изменений температуры на спектры хорошо
известно. Например, при 78 К, когда большинство веществ находится
в твердом состоянии, наблюдаются более узкие полосы поглощения,
изменяются интегральные интенсивности, могут появляться и новые по-
полосы. При температурах выше окружающей среды интенсивности погло-
поглощения в максимуме уменьшаются, а ширины полос увеличиваются. В га-
газах наблюдаются изменения контуров полос. Даже незначительные изме-
изменения температуры могут оказывать заметное влияние на спектры
ассоциированных молекул. Здесь обсуждаются некоторые из причин
такой температурной зависимости.
В газах распределение молекул по различным вращательным (и коле-
колебательным) энергетическим уровням подчиняется распределению Больц-
мана, которое зависит от температуры. По мере ее повышения доля
молекул в более высоких энергетических состояниях увеличивается
и контуры огибающих колебательно-вращательных полос изменяются
так, как показано на рис. 5.19. Более частые столкновения вызывают
уширение полос. Если возможны поворотные изомеры, то изменение
температуры может привести к смещению равновесия между ними,
что вызывает соответствующие изменения в спектрах. Такая ситуация
возникает в бутадиене-1,3 [225], в котором транс-форма преобладает
при комнатной температуре. При несколько более высокой температуре
A00 °С) количество цис-формы (или, возможно, гош-) становится значи-
значительным и изменения в некоторых полосах поглощения отражают
это перераспределение изомеров.
В спектрах жидкостей и растворов изменение температуры влияет
на положение полосы, ее интенсивность и ширину [129, 240]. Например,
частота валентного колебания СН в хлороформе [178] изменяется
на 3 см, а интегральная интенсивность — на 32% в интервале
от — 58 до + 60 °С. Однако самоассоциация молекул может влиять
как на эту полосу, так и в гораздо меньшей степени на интенсивность
других полос A7—25%). Падение интенсивности происходит линейно
с повышением температуры; такое влияние объясняется уменьшением
индуцированного дипольного взаимодействия с увеличением межмоле-
межмолекулярного расстояния. Похожие эффекты были замечены в алифати-
алифатических соединениях [74]. Как было показано, ширины полос в толуоле,
циклогексане и ацетонитриле изменяются в 1,5—4 раза в интервале
температур от — 60 до + 60 °С [223]. Уже отмечалась сильная темпе-
температурная зависимость в веществах с водородными связями. Подобным
же образом температура часто влияет на распределение поворотных
184
Глава 5
T* 1000 К
100 К
Га 300 К
+20+10 0-10-20 m
R
1000 К
40 +20
0 -20 -40
+ 10
+5
-5
-10 -15
Рис. 5.19. Зависимость от температуры распределения интенсивности линий
вращательной структуры в колебательно-вращательных полосах НС1 с вра-
вращательной постоянной В — 10,44 см (а) и гипотетической молекулы с
В =2 см (б). Reproduced, with permission of Van Nostrand — Reinhold, from
G. Herzberg, Spectra of Diatomic Molecules, Princeton, N. J., 1950.
изомеров, и на этой зависимости основан удобный метод исследования
разностей энергий между ними [234].
Изменение ширины полос Хигучи, Танака и др. [120] объяснили,
исходя из броуновского вращательного движения молекул, находящихся
в растворе. Полученные ими зависимости ширины полос от температуры
или вязкости растворителя согласуются с идеей о том, что более
частые столкновения молекул растворенного вещества с молекулами
растворителя приводят к увеличению ширины полос. Они вычислили
также барьер потенциальной энергии переориентации молекул несколь-
нескольких растворенных веществ. Как полагают, этот барьер возникает глав-
главным образом из-за специфического взаимодействия молекул с ближай-
ближайшими соседями и имеет высоту порядка 1,5 — 3 ккал/моль. Теоретические
аспекты релаксации переориентации изложены Гордоном [105] и Шимизу
[236]. В ходе экспериментов с полярными растворителями [121] было
показано, что другие, менее очевидные факторы также могут вносить
значительный вклад в уширение ИК-полосы в растворе.
Температурная зависимость спектров поглощения кристаллических
веществ не изучалась широко, но, пока не изменяется кристаллическая
форма, температура, по-видимому, оказывает относительно слабое
влияние на фундаментальные колебания. Комбинационные полосы
колебаний решетки и фундаментальных колебаний отвечают темпера-
температурной зависимости, соответствующей больцмановскому распределе-
распределению.
Качественный анализ по ИК-спектрам 185
Интерпретация спектров
Интерпретация ИК-спектров является для химика своего рода вы-
вызовом. Хотя наиболее очевидные спектральные особенности можно
сразу же связать с составом образца, спектр содержит такое огромное
количество информации, что даже умелый практик может воспользо-
воспользоваться только ее частью. Кроме того, спектры не отличаются «хорошим
поведением» в том отношении, что интенсивности полос не пропорци-
пропорциональны концентрациям соответствующих групп, с которыми они
связаны, а сдвиги групповых частот подвергаются влияниям, понятым
только отчасти. Тем не менее уже тот факт, что даже новичок,
сопоставляя неизвестный и известный спектры, уверенно может иден-
идентифицировать неизвестное вещество, приносит удовлетворение. По мере
приобретения опыта метод может быть использован для решения струк-
структурных проблем, которые трудно или невозможно решить другими
методами. Короче говоря, ИК-спектроскопия кое-что дает каждому.
Хотя идентификация неизвестных веществ и определение молекуляр-
молекулярной структуры обсуждаются раздельно, практически первая проблема
иногда решается сама по себе в ходе решения второй. Под неизвест-
неизвестными веществами подразумеваются обычные образцы, которые попадают
в спектроскопические лаборатории обслуживания. Они лежат в диапазоне
от дистилляционных фракций до коммерческих продуктов и сверх
того содержат загрязнения сомнительного характера и неясного про-
происхождения. Допустимо предположить, что все образцы этого типа -
смеси или в лучшем случае содержат примеси.
В то же время определение структуры проводится на тщательно
очищенных образцах (фактически невозможно получить структурную
информацию о неизвестной смеси). Важно также получить по воз-
возможности полные спектральные данные, включая ИК-спектры поглоще-
поглощения в широкой области спектра, спектры КР и данные по деполя-
деполяризации. В некоторых случаях оказываются полезными исследования
сдвигов частот под влиянием растворителей, контуров полос в газовой
фазе и другие измерения, обсуждаемые ниже.
Идентификация неизвестных веществ
Для того чтобы охарактеризовать неизвестные вещества, стандарт-
стандартного подхода нет. Можно только предложить несколько прибли-
приближенных вариантов, которые, судя по прошлому опыту, имеют некоторые
шансы на успех. Необходимо подчеркнуть, что не все неизвестные
вещества можно идентифицировать. Во многих случаях все, чего можно
достигнуть, — это только ориентировочно идентифицировать основные
функциональные группы.
Шансы на успешное решение задачи возрастают пропорционально
затраченным усилиям, и поэтому с ними всегда нужно соспоставлять
ценность полученных результатов. В любой проблеме наступает момент,
186 Глава 5
когда дальнейшая затрата времени и труда не приносит пользы.
Часто выявление основных групп является удовлетворительным реше-
решением проблемы. Действительно, иногда достаточно гарантировать, что
данная структура или группа отсутствует. В таких случаях очевидно,
что полная идентификация нерациональна. Дело в том, что важно
понять суть проблемы и представить себе путь ее решения до начала
работы над ней.
Если считается, что следует отдать предпочтение ИК-спектроскопии
(как это обычно и бывает), по крайней мере для классификации
неизвестного вещества, то значительная информация может быть полу-
получена еще до съемки спектра. Очевидно, важны его физическое сос-
состояние и свойства. Например, вещество будет лучше охарактеризовано
в случае бесцветных кристаллов, чем окрашенных смолистых или дег-
дегтеобразных масс. Полезную информацию могут дать испытания на
вязкость (для жидкостей) и растворимость, приблизительная температура
плавления, проверка вещества под микроскопом. Поведение малой
пробы при внесении в пламя обычно указывает, является ли материал
органическим или неорганическим и, если верно первое, присутствуют
ли в нем ароматические группы. Более совершенная методика иссле-
исследований в пламенах может выявить присутствие металлоорганического
соединения [243]. Для жидкостей или летучих твердых веществ сведения
об их чистоте дает газохроматографический анализ. Из-за того что пики
могут перекрываться или могут образовываться нелетучие остатки
чаще, чем предполагают многие химики, опасно считать, что оди-
одиночный пик на хроматограмме указывает на чистый образец.
Использование предыстории образца является важной частью любой
идентификации. Знание всех реагентов и условий реакций или обработки
вещества до его получения часто превращает трудную задачу в простую.
При записи спектров образцов, приготовленных другими исследо-
исследователями, спектроскопист должен иметь в виду возможность случайных
загрязнений за счет смазки кранов, растворителей, экстракции из
резиновых и пластиковых трубок и т. д. (Список полос поглощения
возможных примесей представлен в табл. 5.4, стр. 198.)
Здесь целесообразно определить, действительно ли образец достаточ-
достаточно чистый или содержит два или более главных компонента. В по-
последнем случае интерпретация спектра на основании групповых частот,
как описано ниже, достаточно затруднена.
Неизвестное вещество может быть или продуктом основной реакции,
или примесью в исходных веществах. Проверка методом ИК-спектро-
ИК-спектроскопии исходных веществ оправдана и должна составлять часть любой
серьезной попытки исследовать новую реакцию.
После записи спектра образца тем или иным методом, как обсуж-
обсуждалось в гл. 4, спектрограмма анализируется. Если образец был
нерастворим в органических растворителях, то можно подозревать,
что это неорганическое вещество. Удивительно, что не всегда легко
отличить неорганические вещества по их спектрам, особенно если
Качественный анализ по ИК-спектрам 187
они были записаны в виде суспензии в вазелиновом масле, которое
мешает наблюдению полос в области колебаний СН. Обычно неорга-
неорганические вещества имеют широкие полосы поглощения, но это бывает
не всегда.
Валентные колебания атомов водорода очень характерны и поз-
позволяют определять тип исследуемого соединения. Спектры образцов
записывают при стандартном разбавлении, как было рекомендовано
ранее, и по интенсивности и положению полос поглощения групп
СН выясняют, является ли молекула алифатической, ароматической
или той и другой одновременно. Кроме того, по спектру можно
оценить количество структур каждого типа.
Теперь рассмотрим спектр так, как это делает спектроскопист
(т. е. по областям длин волн), проиллюстрируем и обсудим некоторые
важные особенности каждой области. Это обсуждение не претендует
на полноту и дается скорее для того, чтобы указать на некоторые
типичные особенности ИК-спектра, а также проследить за ходом
рассуждений спектроскописта при анализе неизвестного спектра.
1. Область колебаний групп NH и ОН: 2500-3650 см B,75-
4,0 мкм). До тех пор пока нет сильных внутримолекулярных водо-
водородных связей, в разбавленных растворах инертных растворителей
обычно наблюдается поглощение, обусловленное валентными колебани-
колебаниями неассоциированных групп ОН и NH. На приборе среднего
класса можно отличить —ОН от —NH или —NH2 и даже найти
различие между первичными, вторичными и третичными спиртами
[260]. Слабая полоса в этой области может быть первым обертоном
поглощения С=О или может возникнуть из-за присутствия малого
количества воды (неассоциированная Н2О в ССЦ поглощает около
3700 и 3620 см). Органические кислоты легко отличить по широкому
несимметричному поглощению, распространяющемуся до 2000 см
и ниже.
2. Область валентных колебаний группы СН: 2800-3300 см
C-3,6 мкм). Самая высокая частота валентных колебаний СН,
а именно 3300 см C,03 мкм), принадлежит —С^еС—Н. Аромати-
Ароматические и ненасыщенные группы поглощают около 3100 см~' C,22—
3,34 мкм). В сильно напряженных циклах частоты валентных колебаний
СН также достигают этой области. Большинство алифатических соеди-
соединений имеет поглощение от 2800 до 3000 см C,34—3,6 мкм),
а молярный коэффициент поглощения пропорционален длине цепи
для неразветвленных алканов [92, 276]. Метокси-группа и группа
CH3N поглощают около 2780-2832 см C,53-3,60 мкм), а группа
—СН2О— часто проявляет слабое поглощение при более низких
частотах, чем обычные валентные колебания СН. Как ароматические,
так и алифатические альдегиды имеют характерное поглощение при
2700-2775 см C,6-3,7 мкм). Область валентных колебаний СН
подробнее рассмотрена в работах [56, 268].
3. Область прозрачности: 1850-2700 см C,7-5,4 мкм). Полосы
188 Глава 5
поглощения, лежащие в этой обычно прозрачной области, сразу
же становятся заметными, и их происхождение легко установить.
Они обусловлены хлористоводородными солями аминов, которые имеют
сложное поглощение от 2000 до 2800'см C,6 — 5 мкм), группами
SH около 2540-2590 см C,8-3,9 мкм), РН около 2275-2440 см
D,1-4,4 мкм), —feN около 2220-2260 см D,4-4,5 мкм), SiH
около 2090-2260 см D,4-4,8 мкм), — feC— около 2100-2260 см"
D,4-4,8 мкм), —N=C около 2110-2150 см D,6-4,7 мкм) и С=
=С=С около 1950 см E,1 мкм). Иногда резонанс Ферми может
привести к расщеплению полосы и в ряде случаев к усложнению
отнесения.
4. Область двойной связи: 1430 — 1950 см E,1—7 мкм). Самыми
распространенными и характеристичными группами с двойной связью
являются карбонильные. Вероятно, они наиболее изученный класс
групп, поглощающих в ИК-области. В то время как некоторые
структуры можно отличить просто по положению полосы валентного
колебания С=О, другие в силу совпадения частот однозначно можно
отнести, только прибегая к помощи других областей спектра. Как
уже отмечалось, органические кислоты и обычно альдегиды легко
идентифицируются по полосе поглощения карбонильной группы и по
поглощению групп ОН или СН. Сложные эфиры кроме полосы
валентных колебаний С=О имеют сильное поглощение С—О—R
около 1200 см. В кетонах также проявляются полосы средней
интенсивности около 1000—1370 см"'. Сильное поглощение в интервале
1540-1650 см F,1—6,5 мкм) может указывать на ионизированную
карбонильную группу (например, в металлосодержащих солях органи-
органических кислот), на плоскостные деформационные колебания NH в ами-
аминах, валентные колебания N=0 в нитратах или валентные колебания
С=О в амидах. Для определения природы поглощения здесь опять
необходимо рассмотреть другие спектральные области. Поглощение,
обусловленное валентными колебаниями С=С в алифатических соеди-
соединениях, находится в области 1630—1690 см E,9 — 6,1 мкм), если
только к одному или обоим атомам углерода не присоединен атом
фтора. В этом случае поглощение смещается в область более высоких
частот и число атомов фтора коррелирует с положением полосы.
Более тяжелые галогены понижают эту частоту, так как в валентном
колебании С;=С участвует также некоторая доля деформационного
колебания СН. Ценная структурная информация может быть получена
из положения этой полосы и полосы внеплоскостных деформационных
колебаний в области 800-1000 см A0-12,5 мкм) [217]. В аромати-
ароматических соединениях с малой степенью замещения наблюдаются три
(а при лучшем разрешении четыре) резкие полосы в области 1450 —
1650 см F—7 мкм). Этим полосам сопутствует более слабое поглоще-
поглощение около 1000 — 1200 см (8,3—10 мкм) и характеристические вне-
плоскостные деформационные колебания С—Н около 670—900 см
A1 — 15 мкм). Высокозамещенные ароматические соединения имеют
Качественный анализ по ИК-спектрам 189
поглощение переменной интенсивности около 1400 см~' G,1 мкм).
Число и положение заместителей обычно можно установить по погло-
поглощению в области 670 — 900 см или по обертонам и составным
частотам в интервале 1660 — 2000 см E — 6 мкм) [274]. Для наблюде-
наблюдения этих высокочастотных полос требуются концентрации в 10 раз
более высокие, чем обычные (см. рис. 1 в приложении 1).
5. Область «отпечатков пальцев»: ниже 1500 см~' F,7 мкм). В этой
области кроме характерного поглощения отдельных типов молекул
наблюдается ряд полезных групповых частот. Среди колебаний группы
С—Н можно назвать ножничное колебание метиленовой группы
в алканах вблизи 1467 см F,82 мкм). Асимметричное деформа-
деформационное колебание группы СН3 около 1460 см F,85 мкм) и симмет-
симметричное около 1380 см G,25 мкм) полезны для отнесения. Хорошо
известно, например, что геминальные диметильные группы дают в этом
положении дублет. Другие характеристические колебания С—Н отно-
относятся к внеплоскостным деформационным колебаниям атома водорода
в ненасыщенных и ароматических соединениях. Кроме того, в этой
области проявляются валентные колебания С—F около 1200 см
(8,3 мкм), валентные колебания С—О—С в прстых и сложных
эфирах около 1200 см, валентные колебания С—О и деформаци-
деформационные колебания ОН в спиртах в интервале 1000—1260 см G,9 —
10 мкм), валентные колебания Р=О около 1200 см (8,5 мкм),
валентные колебания SiO в области 1000-1100 см (9-10 мкм)
и валентные колебания СС1 около 700-800 см A2,5-14 мкм).
Каждый исследователь, работающий в области спектроскопии,
быстро осваивает эти и другие более специальные корреляции, свя-
связанные с его собственными интересами, и поэтому в течение не-
нескольких минут способен сформировать свое мнение о природе
неизвестного вещества. (Более полный набор корреляционных таблиц
дан в приложениях 1 и 2.)
Следующая стадия включает проверку картотеки спектров эталон-
эталонных веществ, строение которых доказано спектрами и другими ме-
методами. Если неизвестное вещество сразу не найдено, то можно
просмотреть библиотеку ИК-спектров, отдав предпочтение поисковой
системе на базе ЭВМ (стр. 69—73), которая выдает на печать под-
подходящие соединения в том порядке, в каком они удовлетворяют
решению задачи. В случае непригодности такого приближения клю-
ключом к идентификации неизвестного вещества часто является крити-
критическое рассмотрение спектров родственных структур, сопоставление
со спектрами достоверных образцов либо постулирование некоторой
приемлемой структуры, основанной на анализе спектров сходных
соединений [63]. В последнем случае должны быть использованы
независимые методы, подтверждающие структуру, такие, как химический
анализ, ЯМР, масс-спектрометрия и другие. Ценную информацию
может дать определение молекулярной массы. В некоторых случаях
может быть необходима такая химическая обработка, как омыление
190
Глава 5
Спектр
Нет
Да
Да
Провести поиск
по картотеке
Привлечь
дополнительную
информацию
Постулировать
структуру или
фрагменты
Проверить
картотеку
Аля соединений
\
Идентификация
окончена
Рис. 5.20. Последовательность операций, используемых спектроскопистом в
ходе идентификации неизвестного вещества [241]. Reproduced, with permission,
from American Laboratory. Copyright International Scientific Communications,
Inc.
I I
5 1
I
s
i
a.
5 58
192 Глава 5
сложных эфиров. Описываемый здесь процесс является критической
стадией в ходе идентификации — это тот момент, когда спектроскопист
реализует свою квалификацию, опыт и химическую интуицию для
построения приемлемой структуры, которая удовлетворяла бы имею-
имеющимся доказательствам. Это также тот момент, когда достигается
наибольший успех в ходе идентификации. Рассматриваемый процесс
показан в виде диаграммы на рис. 5.20.
Такой подход иллюстрируется на примере решения реальной задачи,
возникшей в лаборатории автора. На рис. 5.21 изображен спектр
белого кристаллического вещества (температура плавления 65 — 70 °С) —
продукта реакции стирола с металлическим литием и дихлордифенил-
силаном. Очевидно, оно принадлежит к классу ароматических соеди-
соединений и не содержит ни алифатических групп СН, ни NH, ни ОН.
Ароматическое кольцо является монозамещенным A600—2000 см)
и связано с электроотрицательным элементом второго периода перио-
периодической системы (полосы 688 и 755 см-1). Сразу же возникает
мысль о дифениле, но после сравнения со стандартным спектром
эту возможность приходится исключить. Полосы поглощения С—О—С
A250 см-1) отсутствуют, так что вещество не может быть фенило-
вым эфиром. Спектр га/кшс-стильбена похож, но не идентичен. Сле-
Следующее предположение — г^ис-стильбен, но это жидкость, как и 1,1-ди-
фенилэтилен. Тетрафенилэтилен плавится при 227 °С (слишком высоко).
Температура плавления трифенилэтилена составляет 72 °С, но спектр
не совпадает с имеющимся. Спектр дифенилацетилена (температура
плавления 62,5 °С) подходит, за исключением слабого поглощения
960 см-1, которое приписывается примеси транс-стильбена. В этом
случае задача решается не обращением к поисковой системе на базе
ЭВМ, а простым сравнением температур плавления и нескольких
спектров из картотеки эталонов.
Анализ смесей
Иногда спектры смесей расшифровываются с использованием поис-
поисковых систем либо на базе ЭВМ, либо на перфокартах, как было
описано ранее (стр. 69 — 73). Если имеется не более двух-трех компо-
компонентов и их спектры есть в картотеке эталонных спектров, то иденти-
идентификация возможна с помощью как полос, так и не спектральной
информации. Владельцы интерференционных и диспергирующих спектро-
спектрофотометров, в которых встроена своя ЭВМ, позволяющая запомнить
спектр, могут использовать прием, известный как вычитание опти-
оптической плотности или spectral stripping, при котором эталонные
спектры отдельных компонентов, умноженные на подходящий масштаб-
масштабный множитель, последовательно вычитаются из спектра смеси, оставляя
спектр других компонентов. Иногда для оптической компенсации
полос основного компонента смеси применяют двухлучевые спектро-
спектрометры. В канал образца помещают кювету с веществом, содержащим
Качественный анализ по ИК-спектрам
193
юо
SOOO 2000
1000 см"'
Коэффициент „разведения"
3000
2000
1000 см"
Рис. 5.22. а — спектр смеси толуола, циклогексана и гексана; б — отношение
оптических плотностей двух смесей после последовательного частичного ис-
испарения [125]. Reproduced, with permission, from Analytical Chemistry.
примесь, а в канал сравнения — кювету переменной толщины с одним
известным компонентом и, тщательно регулируя толщину, добиваются
компенсации поглощения этого компонента в образце [263]. Затем
записывают спектр, полагая теоретически, что это спектр только
оставшихся компонентов. На практике молекулярные взаимодействия,
влияние показателя преломления, отклонения от закона Бера при-
приводят к тому, что на спектре возникают аномальные сигналы, а
полосы имеют характерную форму [126]. Кроме того, когда исключае-
исключаемый компонент имеет некоторые полосы с пропусканием менее 25 %,
перо в этой области либо совсем не реагирует, либо дает очень
слабый сигнал, даже если другие полосы перекрываются этой полосой
поглощения (стр. 50 — 53). Компенсационные методы можно достаточно
успешно применять только в ограниченных спектральных областях, осо-
особенно там, где исключаемые компоненты имеют только слабые полосы
поглощения.
Наиболее удовлетворительные результаты можно получить, исполь-
используя такие физические методы разделения смеси на составные части,
как жидкостная или газовая хроматография, дистилляция или экстрак-
13 - 586
194
Глава 5
Толуол
юо
3000
2000
ЮОО СМ
Цикпогексан
300О
2000
1000 см"'
Гексан
3000 2000 ЮОО см
Рис. 5.23. Спектры компонентов смеси, вьщеленные с помощью ЭВМ [125].
Качественный анализ по ИК-спектрам 195
ция растворителем. Теория разделения и методы детально обсуждаются
Каргером и др. [150].
В случае смесей из нескольких компонентов для получения спектров
чистых составных частей (при отсутствии предварительной информации
об их числе или составе) можно применять частичное фракционирование
в совокупности с вычитанием спектров индивидуальных веществ [125].
В этом способе используется отношение спектров частично разделенных
компонентов. Процесс разделения может состоять из частичного испа-
испарения смеси, пропускания ее через твердый адсорбент либо из частичной
экстракции или осаждения одного компонента. Нет необходимости в
высокой степени разделения и в знании поведения отдельных веществ.
Первое, что нужно сделать в этом случае,— записать спектр смеси
(рис. 5.22, а), позволить ей частично испариться и вновь записать
спектр, с помощью ЭВМ получить отношение двух спектров, опреде-
определить число плоских областей различной высоты, равное числу компо-
компонентов (рис. 5.22, б), и, наконец, математически установить спектр
каждой из составных частей. Результат такого анализа смеси толуола,
циклогексана и гексана показан на рис. 5.23. Эту процедуру удобнее
всего проводить на интерференционном спектрофотометре с ЭВМ.
Общий метод определения спектров чистых компонентов из спектров
Смесей был описан и использован для анализа полимеров Кёнигом
и др. [158].
Использование корреляционных таблиц
В тех случаях, когда прямая идентификация не удается, необходим
последовательный анализ спектра по корреляционным таблицам. Назна-
Назначение корреляционных таблиц — подсказать возможные соединения и
структуры, которые можно использовать в дальнейших исследова-
исследованиях. Интерпретация спектров, основанная исключительно на корреля-
корреляциях положений полос по табличным данным, будет почти наверняка
ошибочной.
Корреляционные таблицы являются естественным продуктом су-
существования характеристических полос поглощения в ИК-области.
Они указывают наиболее вероятную область появления частоты погло-
поглощения (определенную эмпирически в результате изучения большого
числа известных структур), когда известно, что присутствует опреде-
определенная группа. Так как точное положение групповой частоты зависит
от многих факторов, то области поглощения могут быть широкими
и значительно перекрываться друг с другом.
Правильно используемые корреляционные таблицы могут быть
полезным дополнением для того, чтобы охарактеризовать структуру
вещества. Но если пределы их применения не осознаны, то они
могут вводить в заблуждение и давать ошибочные результаты. Важно
иметь в виду следующие ограничения:
13*
196 Глава 5
1. На основании корреляционных таблиц однозначные структурные
определения обычно невозможны. Исследователь, пытающийся про-
проводить интерпретацию спектра механически, используя только корреля-
корреляционную таблицу, почти наверняка обречен на неудачу из-за перекры-
перекрывания областей поглощения, возмущений, влияющих на положение
групповых частот, и из-за того, что в таблицах представлены только
наиболее характерные полосы.
2. Спектр исследуемого вещества должен быть зарегистрирован
примерно при таких же условиях, которые использовались при со-
составлении таблицы. В качестве наглядного примера можно отметить,
что большинство корреляционных таблиц не подходит для интерпре-
интерпретации спектров в газовой фазе, так как многие частоты не по-
попадают в области поглощения обычно используемого конденсированного
состояния.
3. При интерпретации следует рассматривать не отдельные полосы
поглощения, а спектр в целом. Например, карбонильные группы
могут давать слабый обертон, который ошибочно можно принять
за следы группы ОН. Сильные полосы поглощения в области
690—870 см-1 могут указывать на ароматические группы, но, если
им не сопутствуют резкие полосы около 1450—1600 и 1100 см-1,
они, вероятно, обусловлены некоторыми другими группами, такими,
как СС1.
4. Контуры и относительные интенсивности могут иметь такое же
или еще большее значение, чем положения полос. Ароматические полосы
обычно резкие, и их относительная интенсивность в области 1450—
1600 см-1 может указывать на тип замещения в фенильной группе.
Полоса поглощения связи О=С около 1650 см резкая, и ее невоз-
невозможно спутать с полосой амид I (полоса поглощения С=О), которая
попадает в это же место. Спектроскопист-практик без труда приведет
многочисленные примеры такого рода.
При правильном использовании корреляционные таблицы выступают
в роли путеводителя, позволяющего выявить возможности дальнейшего
исследования. Существует ряд превосходных таблиц групповых частот,
классифицированных по группам (что лучше, чем по областям длин
волн) [23, 24, 56, 199, 239, //]. Дальнейшее изучение этих моногра-
монографий, а также ссылки на соответствующие спектры в картотеках
и литературе быстро создадут такую возможность, когда неизвестная
полоса может быть отождествлена с определенной структурой или
по крайней мере с наличием конкретной группы в молекуле.
Корреляционная диаграмма и таблица групповых частот для не-
некоторых наиболее распространенных органических групп даны в прило-
приложениях 1 и 2. Более детальные корреляционные диаграммы опубли-
опубликованы в [56, 142, 242]. Были созданы обширные таблицы частот
поглощения для многих структур; обсуждалось влияние на групповые
частоты взаимодействия растворенного вещества с растворителем и
водородных связей [75].
Качественный анализ по ИК-спектрам 197
Если основные полосы поглощения отнесены, то, как правило,
не следует пытаться интерпретировать слабые полосы. Преувеличенное
значение, которое приписывают происхождению слабых полос погло-
поглощения, в аналитической работе пользы не приносит. Если же необхо-
необходимо отнести слабые полосы, то лучше сначала провести разделение
веществ, а потом идентифицировать их отдельно.
Аналитические методы идентификации узких классов соединений,
основанные на использовании последовательных схем и наличии или
отсутствии некоторых полос, полезны только для неспектроскопистов.
Достоверная идентификация должна основываться на прямом сравнении
неизвестного спектра с подлинным стандартным спектром, а не только
по корреляционной диаграмме, так как с ее помощью можно иденти-
идентифицировать только те вещества, которые использовались при ее со-
составлении. Образцы должны быть достаточно чистыми и подготовлены
так же, как и при съемке стандартных спектров. В рамках этих
ограничений такие схемы могут быть полезны и, возможно, позволят
исследователю сэкономить значительное время.
Для сотрудников, которые хотят в какой-то мере освоить интер-
интерпретацию спектров, опубликованы практические задачи [199, 239, И, 12].
Существуют также программированные обучающие системы [13, 58,123].
При тщательной работе с такими упражнениями можно глубоко
проникнуть в сущность интерпретации ИК-спектров.
Использование химических производных
Хотя ИК-спектр и является уникальной характеристикой вещества,
его «отпечатком пальцев», нередко при интерпретации спектра возни-
возникают неоднозначности. Спектры двух соединений, таких, как соседние
члены гомологического ряда, могут оказаться очень похожими, доступ-
доступный спектральный интервал — слишком ограниченным для того, чтобы
сделать выбор между сходными структурами; спектры бывают не-
нечеткими, как это случается для некоторых сильно полярных молекул;
интересующая составная часть смеси может оказаться неподдающейся
прямому определению, ее концентрация — слишком низкой, а ее полосы
поглощения могут фактически перекрываться со спектром матрицы.
В таких случаях часто бывает полезным синтезировать химическое
производное, которое имеет более характерный спектр, чем сходное
вещество. В качестве иллюстрации приведем несколько примеров.
Очень характерные спектры получаются для барбитуратов при их
реакции с п-нитробензилхлоридом' [50] или с водными растворами
сульфата меди и пиридина [172]. Анионные поверхностно-активные
вещества можно идентифи.циро,вать, осаждая их в виде бариевых солей
и регистрируя затем ЛК-спектры [136]. Аминокислоты можно охаракте-
охарактеризовать спектрами их производных — З-фенил-2-тиогидантоинов
[80]. 3,5-Динитробензоаты гомологических спиртов имеют своеобразные
спектры [76]. и их обычно можно различить, даже если этого
Таблица 5.4
Полосы поглощения распространенных примесей
Примерная^.
частота, см "~
Длина
волны, мкм
Соединение
или группа
Происхождение примеси
3700
3650
3450
2,70
2,74
2,9
2350 4,26
2330 4,3
2300 и 2150 4,35 и 4,65
1996 5,01
1400-2000
1820
1755
5-7
5,52
5,7
1700-1760 5,7-5,9
Н2О
Н2О
Н2О
СО2
СО2
CS2
во2
Н2О
СОС12
Фталевый
ангидрид
С=О
1720
1640
5,8
6,1
1520-1620 6,2-6,6
1520
1430
1360
1270
1110
1000-1110
980
935
907
837
823
794
788
720 и 730
728
667
6,6
7,0
7,38
7,9
9,0
9-10
10,2
10,7
11,02
11,95
12,15
12,6
12,7
13,7 и 13,9
13,75
14,98
NOf
SiCH3
4
SiOSi
K2SO4
(СН2О)Х
CC12F2
NaNO3
KNOj
CC14 (nap)
CCI4 (жид-
(жидкость)
Полиэтилен
Na2SiF6
CO2
Вода в растворителе (толстые слои)
Вода в некоторых кварцевых окнах
Вода, связанная водородными связями;
обычно в окнах из КВг
Атмосферное поглощение
Испаряющийся сухой лед
Подтекающие кюветы
Метабораты в окнах из галогенидов
щелочных металлов
Атмосферное поглощение
Продукт разложения в очищенном СНС13
Продукт разложения эфира фталевой кис-
кислоты или смолы
Продукты экстрации из прокладок в проб-
пробках
Из пластиковых трубок
Окклюдированная или кристаллизацион-
кристаллизационная вода
Продукты реакции органических кислот
с окнами из галогенидов щелочных метал-
металлов или таблетками нз КВг
Подтекающие кюветы
Загрязнения в окнах из галогенидов ще-
щелочных металлов
Загрязнения в окнах из галогенидов ще-
щелочных металлов
Силиконовое масло или смазка
Примесь в КВг для таблеток
Стекло, силиконы
Продукт разложения сульфатов в таб-
таблетках из КВг
Налет при осаждении газообразного форм-
формальдегида
Испарение фреона-12
(см. 1360 см)
Продукт разложения нитратов в таблетках
из КВг
Подтекающие кюветы
Плохая сушка кюветы или загрязнение
В любой об- Интерфе-
ласти
ренционные
полосы
4 из NaCl
Атмосфера
В пустых кюветах или в случае слишком
высоких показателей преломления мате-
материалов окон могут возникать интерферен-
интерференционные полосы
Качественный анализ по ИК-спектрам 199
нельзя сделать с помощью температуры плавления производных.
Из фруктовых приправ (fruit flavors) методом бумажной хроматогра-
хроматографии были выделены алифатические спирты и идентифицированы по
ИК-спектрам их эфиров с и-нитрофенилазобензойной кислотой [37]. Бы-
Были описаны спектры 2,4-динитрофенилгидразонов кетонов и альдегидов
[140]. Часто рентгенограммы изучаемых соединений имеют отличитель-
отличительные особенности, которые могут быть использованы для того, чтобы
подтвердить результат, полученный ИК-методом, или принять окон-
окончательное решение. Даже ионы металлов можно идентифицировать
по спектрам их 8-гидроксихинолятов [209] или других внутрикомплек-
сных соединений [122]. Как в качественном, так и в количественном
анализе методу приготовления химических производных химик-анали-
химик-аналитик должен уделять большее внимание.
Полосы поглощения примесей
В ходе идентификации неизвестных веществ часто обнаруживается
малое, а иногда и значительное содержание компонентов, которые,
без сомнения, не связаны с изучаемой задачей. Например, нет ничего
необычного в том, что даже в литературе можно найти спектры,
которые осложнены силиконовой смазкой, попадающей из шлифов
или кранов, фталевым эфиром (пластификатором) из пластико-
пластиковых трубок или полимерных прокладок крышек сосудов с образцом.
Каждая лаборатория, проводящая работы с ИК-спектрами, может
составить список полос поглощения примесей, возникающих из этих
и других источников, общих для подобных лабораторий. Такие полосы
были табулированы Лоунером [168] и частично представлены в табл. 5.4.
Мак-Карти [188] опубликовал спектры соединений, используемых для
полировки окон из галогенидоэ щелочных металлов.
Следы растворителя или других загрязнений в канале сравнения
будут давать обратный спектр, вычитаемый из нормального спектра
образца. Другие спектральные артефакты включают' эффект Христиан-
сена (стр. 89 и 92), перегруппировку энергетических уровней из-за
колебательных возмущений [81] и эффект «мертвого пера» в области
сильного поглощения растворителя (стр. 50—53 и 85).
Выводы: основные принципы
и порядок идентификации неизвестного вещества
Для оптимизации получения информации из спектра необходимо
свести к минимуму постороннее влияние растворителя и другие
взаимодействия. Спектры образцов нужно записывать согласно тща-
тщательно выбранным воспроизводимым методикам, предпочтительно
в растворах ССЦ и CS2. Нерастворимые вещества можно подготовить
к съемке, используя один из подходящих способов: пленки, суспензии
200 Глава 5
в вазелиновом масле, нарушенное полное внутреннее отражение или
таблетки с КВг. При подготовке образца важно избегать произвола
в выборе методики.
Идентификация неизвестного вещества может быть проведена сле-
следующим образом:
1. Определить, какая требуется информация об образце и как ее
предполагается использовать.
2. Отметить такие явные характеристики вещества, как форма, цвет,
запах, растворимость.
3. Получить, насколько это возможно, полные сведения о предысто-
предыстории образца.
4. Записать ИК-спектр.
5. Критически проанализировать спектр, используя знание групповых
частот и их изменений при изменении молекулярной структуры.
Если возможно, то привлечь данные по ЯМР, УФ-спектроскопии
и масс-спектрометрии.
6. Сравнить со спектрами структур, сходных с той, которая пред-
предполагается на основании предыдущего анализа.
7. Если соответствие не получено, то. обратиться к поисковой
системе и, используя положительную и отрицательную информации,
как было описано ранее (стр. 69), попытаться найти соответствие
с неизвестным веществом.
8. Если требуется, то провести разделение веществ и для каждого
все повторить, начиная с п. 4.
9. Используя корреляционные диаграммы, предположить для сравне-
сравнения дополнительные структуры.
10. Получить дополнительные сведения, такие, как элементный
анализ, спектры ЯМР, масс-спектры, спектры КР, молекулярные массы.
11. Постулировать приемлемую модель для соединения, согласую-
согласующуюся со спектральными данными, с его химическими и физическими
свойствами.
Приложения
Полимеры
ИК-спектроскопия сыграла большую роль в развитии промышлен-
промышленности полимеров. Известно, что метод широко используется на
практике для идентификации пластиков. ИК-спектроскопия имеет важное
значение в производстве красителей для качественной идентификации
связующих, пигментов и растворителей, для оценки качества сырье-
сырьевых материалов, дозировки компонент смол, в исследованиях по
стабилизации и окислению, в задачах по определению качества про-
продуктов. После некоторого физического разделения эластомеров на
компоненты их можно идентифицировать на главные и второстепен-
Качественный анализ по ИК-спектрам 201
ные. Метод ИК-спектроскопии часто применяют для характеристики
поверхности волокон и тканей, подвергнутых обработке с целью
очистки, придания огнестойкости и т.д. Кроме таких практических
приложений метод может дать ценную информацию о тактичности
(регулярности), кристалличности, пространственном расположении и
разветвленности полимерных цепей. В действительности области при-
применений метода столь широки, что можно лишь указать на наиболее
полезные из них и отметить некоторую литературу, относящуюся
к делу.
Зачастую метод приготовления образца в подходящем для анализа
виде представляет большую проблему, чем расшифровка самого спектра,
и это может явиться ключом к решению задачи. Некоторые из ме-
методов, применение которых может оказаться успешным, были де-
детально изложены в гл. 4 и в книге Хенникера [118].
Идентифшсацт
Доступ к хорошему каталогу эталонных спектров совершенно
необходим, и несколько из них указаны в табл. 3.3. Идентификация
и анализ полимеров обсуждаются в превосходной книге Хаслама
и Виллиса [115]. Очень полезны книги Хюммеля [131] по ИК-анализу
полимеров, смол и добавок; то же можно сказать и о содержании
и эталонных спектрах книги членов Чикагского общества технологии
красителей [51, 13].
Во многих случаях полимерный материал содержит два или более
компонентов, и результирующий спектр является приблизительно сум-
суммой спектров составляющих гомополимеров. За некоторым исключе-
исключением, невозможно с помощью одной ИК-спектроскопии обнаружить
разницу между сополимерами и смесью гомополимеров. Слоистые
полимерные пленки ведут себя в случае спектров пропускания как
однородные фазы, но если методом НПВО исследуются обе стороны
пленки по отдельности, то часто можно получить спектры отдельных
компонентов. В случае смесей сополимеров, мешающих определению
друг друга, спектр можно упростить посредством химического или
физического разделения составных частей [52, 114]. Например, поли-
полиуретаны можно подвергнуть щелочному гидролизу и провести экстрак-
экстракционное фракционирование для последующей идентификации [60]. Не-
отвержденные красители можно разделить экстракцией растворителем
и центрифугированием, а отдельные компоненты исследовать согласно
стандартным методикам.
Анализ затвердевших красителей и смол представляет собой более
трудную проблему; методы НПВО или пиролиза часто могут дать
предварительные сведения о составе этих материалов. Покрытия, под-
подвергнутые атмосферному старению, идентифицируются размалыванием
образца с последующим прессованием таблетки с КВг [77, 155]. В ходе
судебной экспертизы кювета высокого давления с алмазными окнами
202 ¦ Глава 5
использовалась для приготовления микрообразцов покрытий и плас-
пластиков [259].
Так как покрытия часто являются сложными смесями многих
компонентов (некоторые из них могут иметь сильное поглощение,
а другие — слабое), то такие грубые, усредненные анализы могут
ввести в заблуждение. Кравер [64] представила в качестве примера
две различные фталевые алкидные смолы. В их спектрах преобладал
фрагмент фталата, доля которого составляла только 26 %. Более полный
анализ таких композиций требует тщательного разделения на компо-
компоненты, которые затем необходимо охарактеризовать. Каппельмайер [149]
предложил последовательность операций для анализа покрытий на
основе смол, которые включают методы местных проб, омыление
и другие химические операции, а также экстракцию растворителем.
В качестве примера на рис. 5.24 показана схема анализа полиэфиров.
Пигменты часто можно идентифицировать по их ИК-спектрам
[2, 131] или рентгенографически, комбинируя, может быть, эти методы
с анализом на металлы.
Анализ покрытий часто осложнен тем, что некоторые ингредиенты
сами являются сложными смесями, такими, как шеллак, канифоль,
парафин, натуральные смолы. При идеальном разделении эти смеси
должны быть отделены как целое от других сходных сложных компо-
компонентов. При идентификации таких материалов стандартные спектры
полезны, но часто и неоднозначны. Крайне полезно знание сходных
рецептур и их ожидаемых компонентов. Их можно найти в нескольких
работах [189, 198, 249].
Неопытного, торопливого и неосторожного аналитика ожидает много
ловушек как в процессе подготовки образцов, так и при интерпрета-
интерпретации спектра. На некоторые из них указала Кравер [64], чью главу
об анализе покрытий должен прочитать каждый, кто проводит такого
рода работу. Практические приложения ИК-спектроскопии и газовой
хроматографии в производстве красителей обсуждены Хельменом [117],
который представил и иллюстративные примеры.
Эластомеры обычно являются сложными смесями, содержащими
один или более основных полимеров, пигменты и наполнители, пласти-
пластификаторы, катализаторы полимеризации, антиоксиданты, стабилизаторы
смазки, антистатики и т. д. Идентификация методом ИК-спектроскопии
всех этих компонентов в высокомолекулярном эластомере маловероятна.
Фактически эластомер, наполненный сажей, может быть настолько
непрозрачным, что совсем не будет давать ИК-спектра; в этом слу-
случае необходимо разделение. В различных публикациях рассмотрены
анализы конкретных полимерных систем; некоторые из них включены
во всеобъемлющий обзор приложений ИК-спектроскопии в резиновой
промышленности [114]. Для разделения компонентов в ходе подго-
подготовки к ИК-анализу часто применяют экстракцию растворителем
и методы хроматографии, включая тонкослойную, гель-проникающую,
колоночную и газовую.
Качественный анализ по ИК-спектрам
203
% №он,
вода
Нерастворимая часть
Полимерные спирты ,
полистирол и т.9.
(спектр)
Растворимая часть
Перегонка с
водяным паром
Остаток
Подкислить
на Jo рН 5,5,
экстрагировать
эфиром
Спирты
(спектр)
Остаток
Одноосновные
кислоты
(спектр)
Подкислить НС1
ворН (-2
ОсаОок
Двухосновные
кислоты
(спектр)
Фильтрат
Гликояи
(спектр)
Рис. 5.24. Схема химического и физического анализов полиэфира [64, 149].
Reproduced with permission of Wiley-Interscience.
Спектры волокон и тканей обычно снимают, используя метод
НПВО* [132], хотя они могут быть также размолоты и спрессо-
спрессованы в таблетки с КВг [187], подвергнуты пиролизу [49] или раз-
раздроблены и спрессованы в тонкие листки без подложки [214]. Волокна,
которые нельзя размолоть или приготовить в виде пленки, можно
закрепить параллельно друг другу в микродержателе; это позволяет
* При съемке ИК-спектров НПВО таких дисперсных объектов, как волокна,
нужно учитывать возможность появления ложных полос. — Прим. перев.
204 Глава 5
получить приемлемые спектры четырех-шести волокон [48]. Химически
модифицированные волокна хлопка были исследованы методом разност-
разностной спектроскопии [187, 203]. Если ткань обработана гидрофобной
или олеофобной аппретурой или антипиреновым покрытием, то в
спектрах НПВО и пропускания можно наблюдать полосы как во-
волокна, так и этих покрытий. Часто покрытия можно отделить и
идентифицировать простой экстракцией хлороформом, тетрахлорэти-
леном или метанолом [170]. Растворитель можно испарить с пластинки
многократного НПВО, и требуется ничтожное количество экстракта
для получения спектра, поддающегося идентификации (стр. 98 — 106).
Когда на волокне имеются антипирены, смазочные вещества и пласти-
пластификаторы, требуются более сложные процедуры экстрагирования. Ка-
Каталог спектров для идентификации реагентов, используемых для хи-
химической отделки, был составлен О'Коннором и др. [204, 13].
Структура
Значительные усилия, во многих случаях небезуспешные, были за-
затрачены на изучение структуры полимеров методом ИК-спектроскопии.
ИК-спектры полимеров с кристаллической и аморфной структурами
обычно различаются. Этим методом можно исследовать расположение
мономерных единиц в полимере и особенности их конфигурации,
упаковку и разветвленность цепей. Иногда, например, возможно отли-
отличить блок-сополимеры от статистических, если одна из мономерных
единиц содержит ассоциативные группы, а другая нет. В этом случае
количество ассоциативных групп дает меру неупорядоченности в
полимере. В некоторых случаях одна из мономерных единиц чувстви-
чувствительна к окружению, и в сополимере происходит изменение частоты
по сравнению с гомополимером. В качестве примера можно привести
[216] систему винилхлорид — винилиденхлорид, в спектре которой по-
полоса чистого поливинилхлорида 1250 см (8 мкм) при сополимери-
зации сдвигается к 1203 см (8,3 мкм). Эта полоса обусловлена
колебаниями изолированного фрагмента (—СН2СНС1—) в цепочках
поливинилиденхлорида.
Кристалличность обычно изучается при использовании поляризо-
поляризованного ИК-излучения [35]. В некоторых полимерах полосы «кристал-
«кристалличности» и «аморфности» имеют различающиеся частоты. Например,
в полиэтилентерефталате полоса 1343 см-i отнесена к кристалли-
кристаллической структуре, а полоса 1370 см-i обусловлена наличием областей
аморфности. Из этих полос поглощения можно вычислить соотноше-
соотношение между двумя формами [79]. Кримм [163], используя метод
ИК-спектроскопин, исследовал расположение полимерных цепей в крис-
кристаллическом полиэтилене.
Спектры изотактического и атактического полистиролов представлены
на рис. 5.25. В целом различия выражены не явно, но вполне за-
Качественный анализ по ИК-спектрам
205
I60O ИОО 1200 ЮОО 800
Частота, ем
600
400
Рис. 5.25. ИК-спектры изотактического и атактического полистиролов [182].
Reproduced, with permission, from Applied Spectroscopy.
2000
1600
1200 800
Частота, см
400
Рис. 5.26. ИК-спектры трех полиморфных форм изотактического полибу-
тена-1 [182]. Reproduced, with permission, from Applied Spectroscopy.
206
Глава 5
MOO
WOO MOO MOO
Частота, cm
boo юоо
aoo
eoo
400
Рис. 5.27. a — верхний спектр: естественная смесь аморфного и кристал-
кристаллического 1,4-/ирв«с-полихлоропренов при комнатной температуре; нижний
спектр: только 1,4-/и/м«с-полихлоропреи в аморфном состоянии при — 80°С;
б — разностный спектр, полученный с помощью ЭВМ для кристаллического
1,4-шрвнс-полнхлоропрена. Courtesy Perkin — Elmer Corporation.
метны в области 1060 см-i. Рис. 5.26 иллюстрирует полиморфизм
в полибутене-1, где модификация I представляет трехкратную спи-
спиральную конформацию, модификация II — четырехкратную, а модифи-
модификация III имеет ромбическую структуру [182]. Методом ИК-спектро-
скопии была также определена последовательность чередования в
этилен-пропиленовых сополимерах [258].
Для получения спектра кристаллического 1,4-юранс-полихлоропрена
из спектров частично кристаллического вещества и аморфного поли-
полимера было использовано вычитание оптической плотности (стр. 185 —
195), выполненное на интерференционном спектрофотометре [157].
Аналогичные результаты были получены на диспергирующем спектро-
спектрофотометре, соединенном с ЭВМ (рис. 5.27).
Качественный анализ по ИК-спектрам 207
Реакции
Для исследования поверхностного окисления полибутадиена при
30 °С Кёниг [157] использовал вычитание оптической плотности.
Его результаты показаны на рис. 5.28. Изменение соотношения цис-
и /пранс-ненасыщенности зафиксировано только через 10 ч C000 и
975 см. Частичное окисление (образование С—О) подтверждается по-
полосой 1065 см. В процессе более длительной обработки окисление
приводит к появлению групп ОН C300 см) и С=О A700, 1720 и
1770 см). Аналогично исследовалось радиационное разрушение
полиэтилена [250]. Старение тройного сополимера из акрилонитрила
бутадиена и стирола под действием подобных условий также иссле-
исследовали методом ИК-спектроскопии [66]. Метод НПВО был применен
для изучения разложения поликарбоната под действием УФ-излучения;
распределение продуктов реакции по глубине устанавливали последо-
последовательным удалением слоев полимера [99]. Тот же метод использо-
использовался и при исследовании деструкции эластомеров под действием
озона [7] *.
Методы, применяемые для получения структурной информации
из колебательных спектров, выходят за рамки этой главы; для полу-
получения более подробной информации читатель может обратиться к
специальным монографиям [135, 275], к другой литературе [36, 127]
или к прикладным обзорам по полимерам, которые появляются в
нечетные годы в апрельских обзорных выпусках журнала "Analytical
Chemistry".
Поверхностно-активные вещества
При выделении и идентификации поверхностно-активных веществ
(ПАВ) могут возникать очень интересные аналитические проблемы.
В композициях эти вещества часто содержатся в незначительных
количествах. Обычно они используются в смесях и даже в «чистом»
виде, как правило, содержат сходные по строению вещества, исходные
компоненты, продукты побочных реакций и добавки, не являющиеся
поверхностно-активными. Тогда первая стадия будет состоять в отде-
отделении ПАВ от других веществ, после чего можно попытаться начать
идентифицировать их по отдельности.
Систематическое разделение и анализ ПАВ детально рассмотрены
Розеном и Гольдшмитом [227], которые рекомендуют подходящие
химические и физические методы анализа, включая ИК-спектроскопию,
для характеристики продуктов такого разделения. В монографии Хюм-
меля [130] содержится ряд ИК-спектров ПАВ.
* Метод НПВО был использован также для изучения градиентов кон-
концентраций [14, IS].— Прим. перев.
208
Глава 5
10 ч
3007
1065 C-Os
975
3500
2700 2000
740 цис-С-Нь
800 см"
Рис. 5.28. Разностные спектры для полибутадиена, окисленного при 30°С в
течение 10, 23S и 640 ч [157].
Качественный анализ по ИК-спектрам 209
Биологические системы
ИК-спектроскопия не получила столь широкого распространения
среди биологов, как среди химиков, по той причине, что биологи-
биологические объекты часто не дают хорошо разрешенных спектров (по
крайней мере при комнатной температуре). Тем не менее сообщается
о некоторых интересных приложениях. В одном из них метод НПВО
применяется для получения спектра поверхности живой кожи. Удалось
проследить во времени за поглощением в организме инертных и
биологически активных веществ [90, 222]. Сообщается о проведении
методом ИК-спектроскопии таких стандартных измерений, как анализ
крови на содержание липидов [94, 231] и распознавание типов живых
тканей [34, 70]. Имеются обзоры по приложениям метода ИК-спектро-
ИК-спектроскопии к биологическим системам [139, 206, 207, 252, 16].
Неорганические,
металл оорганическне
и координационные
соединения
Несмотря на то что еще в 1906 — 1908 гг. Кобленц исследовал
ИК-спектры пропускания большого числа неорганических веществ,
возможности метода для качественного, количественного и структур-
структурного анализов этих веществ в значительной степени не учитывались
вплоть до 1950-х годов. В это десятилетие появилось несколько
сборников спектров, которые показали полезность ИК-спектроскопии
для идентификации, особенно в совокупности с рентгеноструктурным
и эмиссионным анализами. Кроме таких традиционных неорганических
веществ, как UF6, SiCl4, BF3 и NH3, в последние годы широко
изучаются координационные соединения. Спектроскопические данные
и эталонные спектры, относящиеся к этим двум классам веществ,
можно найти в нескольких монографиях [87, 109, 186, 200]. Существует
и другая родственная литература, включающая книги по колебательным
спектрам неорганических веществ [141], ИК-спектрам и спектрам КР
лунных и земных минералов [151]. Ферраро [87] рассмотрел низко-
низкочастотные колебания (в дальней ИК и КР) неорганических и коорди-
координационных соединений. Почвы и их составные части [82, 83, 89, 138,
261], а также минералы, используемые в производстве цемента [101],
были охарактеризованы по ИК-спектрам.
Колебательная спектроскопия успешно применяется при исследовании
классов таких гибридных молекул, как кремнийорганические [6] и
фосфорорганические [202, 253]. Имеется обзор по колебательным спект-
спектрам соединений переходных элементов [104]. Исследования по метал-
лоорганическим соединениям обобщены Адамсом [1] и Гольдштейном
[104] (см. также дополнительную литературу к табл. 3.3. — Прим.
перев.).
14-586
210 Глава 5
Проблемы окружающей среды
Естественно, что метод, который позволяет достоверно .проводить
идентификацию и количественно измерять промышленные загрязне-
загрязнения, должен найти широкое применение для контроля за состоянием
окружающей среды. Растущий объем литературы по применению
ИК-спектроскопии к проблемам окружающей среды подтверждает эту
мысль.
Анализ воздуха в полевых условиях может эыполняться на простых
портативных спектрофотометрах [103, 269, 270]. Применение много-
многоходовых газовых кювет с большой длиной оптического пути A0—20 м)
позволяет обнаружить многие промышленные загрязнения с чувстви-
чувствительностью несколько частей на миллион. Компромисс между ме-
мешающим поглощением и максимальной чувствительностью может быть
достигнут при использовании спектрометров с высоким разрешением.
Томпсон [254] собрал спектры и физические свойства 600 опасных
газов и паров. Количественный анализ газообразных загрязнений об-
обсуждается позже (стр. 272—274).
Метод ИК-спектроскопии применялся для изучения реакций в ат-
атмосфере (например, фотолиза и окисления). Содержимое специальной
многоходовой газовой кюветы с большой длиной оптического пути
облучалось светом, имитирующим солнечный, и проводилась непре-
непрерывная регистрация спектров поглощения реагентов и продуктов [98,
Таблица 5.5
Пределы обнаружения атмосферных загрязнений в кювете
с длиной оптического пути 1 км [213]
Соединение
Аммиак
Формальдегид
Муравьиная кислота
Азотная кислота
Азотистая кислота (цис)
Надазотная кислота
Пентаоксид азота
Озон
Пероксиацетилнитрат
Пероксид водорода
Метанол
Метилнитрат
Частота измерения,
931
967,5
2779,0
2781,5
1105,3
896,0
853,0
803,0
740,0
1248,0
1055,0
793,0
1162,0
1250,0
1033,0
853,0
Предел обнаружения,
часть на миллиард
2
2
4
4
4
4
4
2
2
10
4
¦х
j
8
2
2
i
il
'I!H
'I/iir!:
' Ш\\
<: ill
•и.
!'! P
In"
lirf
й
u
•in
I"!
ttri
i
i 1
ИХЛ
i'i'
• ,'
11
ii
. H
орэти
лен \
¦ > i
; t ;
iiii
m
ш
I'iilii:5! "Jyi
i?PrP
w
Ш
V.
¦ I j
•
¦::-
•*™
• •!
Mil
и
ill
ft 1
Il-J
ill
Щ1
ii
т n
L i:H HI Г
!!'j li; -
Ш
11 ill
i Jl •!:¦
: |i! !;:
i '•;
7TS
¦:.
•
•¦;
!:
' i
•¦•;
3000
Рис. S.29. Имитация атмосферного фотолиза.
Верхний спектр: 1,2-дихлорэтален E частей яа миллион) н NO2 (I часть на миллион) до облучении; нижний спепр: после облучении. Длина оптического пути 500 м.
Шкала абсцисс имеет обратный поршок по сраикнню с обычным [98]. Reproduced, with permission, from Environmental Science and Technology.
212
Глава 5
20
40
60
120
140
60 ЮО
Время, мин
Рис. 5.30. Фотоокисление 1,2-дихлорэтилена и NO2 [98].
160
213]. Типичные спектры, полученные до и после облучения в те-
течение 100 мин, показаны на рис. 5.29. Зависимость концентраций от
времени облучения представлена на рис. 5.30. В табл. 5.5 даны
пределы обнаружения для ряда компонентов смога, исследованных
в кювете с длиной оптического пути 1 км.
Методом ИК-спектроскопии с преобразованием Фурье [185] изуча-
изучались реакции гербицида «Линурон» (Linuron) в различных грунтовых
системах. Торфяные почвы требуют гораздо больших количеств герби-
гербицида. Для обнаружения химической реакции «Линурона», адсорбирован-
Качественный анализ по ИК-спектрам
213
I 1 1 1 1 1—I
2000
1500 КХЮ
Частота, см
Рис. 5.31. ИК-спектры сырой нефти (из Эквадора), не подвергшейся (а)
и подвергшейся (б) воздействию погодных условий. Разностный спектр (в)
получен, когда в канале образца находился образец нефти, подвергшейся
погодному старению, а в канале сравнения был исходный образец [38].
Reproduced, with permission, from American Laboratory. Copyright International
Scientific Communications.
ного на торфе (но не на глине), использовали вычитание спектров
(их оптических плотностей).
Чтобы идентифицировать источники нефтяных пятен, было предпри-
предпринято много попыток обнаружить «отпечатки пальцев» нефтей из раз-
различных месторождений. Главная проблема состоит в том, что про-
происходит испарение более легких фракций, а остающаяся фракция
подвергается погодному старению, так что спектр поглощения нефти
зависит от времени (рис. 5.31). Кроме того, спектр может быть
усложнен и такими загрязнениями, как животные и растительные
масла. В наиболее успешном методе характеристики нефтей, подверг-
214
Глава 5
шихся погодному старению, измеряется оптическая плотность при
ряде длин волн, выбранных статистически с максимальной чувстви-
чувствительностью к источнику нефти и минимальной к погодным воздейст-
воздействиям [3, 38, 153, 183; 17, с. 354]. Образцы сырой нефти можно
подвергнуть искусственному погодному старению и тем самым по-
получить соответствие нефтям из пятен; по-видимому, температура воды
оказывает доминирующее влияние на скорость процессов, свой вклад
вносят также освещенность и перемешивание [3, 12].
Анализ удаленных атмосфер и объектов
При получении ИК-спектров удаленных земных и небесных объектов
и атмосфер возникает несколько трудных проблем. Первым и главным
затруднением является сложность получения полезного сигнала из-за
обсуждавшегося ранее ограничения по энергии. В качестве источника
может служить солнце, но его недостатки очевидны. Использование
удаленных контролируемых источников ИК-излучения или обратных
отражателей возможно только в отдельных случаях. Спектры излучения
нагретых газов, таких, которые выходят из вытяжной трубы, можно
наблюдать методом корреляционной спектроскопии (стр. 274).
Возможно, что перестраиваемые лазеры представляют собой наиболее
перспективный источник для контроля состояния воздуха на расстоя-
расстоянии [124].
Вторая Проблема связана с интенсивным поглощением излучения
парами Н2О и СО2 атмосферы воздуха, особенно при большой длине
оптического пути (> 1 км), что требует работы в окнах прозрачности
между полосами (рис. 5.32).
Несомненно, на пределе возможностей дистанционной ИК-спектро-
скопии выполнена работа Ларсона и Финка [166], которые объединили
фурье-спектроскопию с телескопами с большой апертурой для опреде-
3000
5000
6000
7000
Рис. 5.32. ИК-спектр пропускания земной атмосферы в области 1—10 мкм.
Основные окна прозрачности расположены в областях 1000, 2000, 2800, 4500,
6200 и 8200 см. Adapted from Ridgway et al. [226] and reproduced, with
permission, from Applied Spectroscopy [166].
4000
eooo
6000
cm-
Рис. 5.33. ИК-спехтры отражения инея, образующегося из некоторых газов,
встречающихся в лабораторных условиях [166]. Reproduced, with permission,
from Applied Spectroscopy.
a - замороженные пары HjO, 90 К, R-38 см; 6 - замороженный СО,, 90 К, R=2,2 см'; «-за-
«-замороженный СН4, 60 К, Я=И см; г-замороженный NH,, 107 К, Я=-И см '; д-заморожш-
ный НА 91 К, R = 2,2 см-1; е - замороженный NH4HS, 87 К, R-2,2 см '.
г.о
8000
9000
Волновое число
Рис. 5.34. Спектры центральных областей планеты Марс, полярной у
шапки и их отношение. I
Треугольниками отмечены особенности, характерные для полярной шапки и не противоре- •*!
чашие присутствию «льда» СО2 [167]. Reproduced, with permission, from Astrophys. J. Lett. °
Рис. 5.35. Спектры трех основных породообразующих минералов.
Видны все наиболее интенсивные полосы, обусловленные ионами
железа. Adapted from Gaffey [97] and reproduced, with permission, from п o
Applied Spectroscopy [166].
i i i i I i i i i I i i i i I i i i i I i i i i I i i i
I I I I I
Полевой шпат
2,5
Качественный анализ по ИК-спектрам ^^ 217
ления поверхностного состава таких малых планет Солнечной системы,
как астероиды и спутники. Они наблюдали спектры отраженного
солнечного света. Оптическая схема была построена таким образом,
что сигнал от выбранного участка объекта, рассматриваемого через
телескоп, интерферометрически сравнивался с сигналом, получаемым
от соседнего участка неба. При этом уменьшалось влияние высокого
уровня фонового излучения и слабый сигнал от цели детектировался
с последующим усреднением по многим тысячам сканировании.
Как можно было предположить, различные соединения в заморожен-
замороженном состоянии будут давать разные ИК-спектры (рис. 5.33 и 5.34).
Кроме того, в спектре льда наблюдается полоса 6056 см-', интен-
интенсивность которой зависит от температуры. Таким образом, из спектров
отражения можно оценить температуру удаленных объектов, покрытых
льдом. Многие минералы имеют характеристические спектры отражения,
что позволяет, по крайней мере частично, идентифицировать состав
астероидов и планет (рис. 5.35).
Отнесения частот
Хотя строгая корреляция положений полос с наличием в моле-
молекуле некоторых групп атомов, основанная на представлениях клас-
классической механики, и может иметь определенную ценность, примени
ние такого прямого подхода ограниченно. Когда формы движения
атомов в группе определены (валентные, деформационные, маятни-
маятниковые), групповые частоты можно использовать не только для ха-
характеристики отдельных групп, но и более тонко — для предсказания
возможных сдвигов или аномальных эффектов, возникающих в соседних
группах. Отнесения частот важны, конечно, и для термодинамических
расчетов, основанных на спектроскопических данных. Хотя и не всегда
легко отнести групповую частоту, особенно при смешанных колебаниях,
существует ряд методов, которые можно использовать для решения
этой проблемы. Они кратко обсуждаются ниже.
Сравнение с родственными структурами
Несомненно, что большинство корреляций группа — частота прове-
проведено путем сравнения большого числа родственных (и предпочтитель-
предпочтительно простых) структур. Замена одного атома или группы сразу же
вызывает соответствующие изменения в спектре.
Успешное применение этого метода зависит от доступности под-
подходящих для такого сравнения образцов. Спектры высокомолекулярных
соединений можно интерпретировать, сравнивая спектры мономерных
единиц или модельных соединений, содержащих некоторые из тех
структур, которые предполагаются в полимере. Так, например, структура
поливинилхлорида [161] была определена с помощью таких модельных
соединений, как СН3СН2СН2С1, СН3СН2СНОСН3 и СН3СН2С(СН3JС1.
218 Глава 5
Была показана возможность отнесения довольно ограниченных спект-
спектральных интервалов к поглощению С—С1 в зависимости от того,
находится ли атом С1 в ю/юкс-положении относительно атомов Н или С,
имеющих общую связь С—С [237]. Эти данные вместе с результатами
по изотопному замещению и анализом симметрии элементарной
ячейки подтвердили конфигурацию полимера, постулированную на
основе исследований методом рентгенографии. Полезные сведения
о модельных структурах можно найти в таблицах частот и их от-
отнесений, составленных Шиманучи [235].
корреляционный метод был развит Колтупом, который, используя
метод МО в варианте ППДП/2, рассчитал электронные плотности
в замещенных этиленах [57] и ароматических соединениях [55]. Он
нашел ряд простых соотношений между различными Частотами и элек-
электронной плотностью.
Измерения интенсивностей
Очевидным развитием концепции групповых частот является пред-
представление о групповых интенсивностях. Этот параметр изучался неко-
некоторыми исследователями, но, как известно, в «спектроскопии интенсив-
интенсивностей» больше экспериментальных трудностей, чем в «спектроскопии
частот». Тем не менее был найден ряд интересных и полезных
соотношений. Липперт и Мек [177] предложили двумерную корре-
корреляционную диаграмму, на которой частоты откладываются по абсциссе,
а интенсивности — по ординате. График такого типа был использован
[255] для того, чтобы различить поглощение разных карбонильных
групп. Методом прессования таблеток с КВг [91] исследованы интен-
интенсивности полос поглощения в соединениях, содержащих группы
\ /
—C=N, С=С , —N=O и —S=O. Для успешного использования
шкалы интенсивностей, очевидно, требуется спектрофотометр с высоким
разрешением, а также накопление значительного объема первичной
информации по большому числу соединений.
Этот метод неприменим к некоторым группам, интенсивность
поглощения которых изменяется в широких пределах (например, из-за
мезомерного эффекта). Например, при переходе от соединения к соеди-
соединению интенсивность поглощения группы — C=N изменяется в 10 или
большее число раз. К счастью, частота колебаний этой группы
попадает в такую область спектра, где ее лишь иногда можно
спутать с поглощением других групп.
Данные, полученные-методом комбинационного рассеяния
и другими спектроскопическими методами
В работах [56, 18—20} обсуждены принципы и применение спектро-
спектроскопии КР. Для распознавания симметричных и несимметричных
колебаний в молекулах с высокой симметрией такие параметры, как
Качественный анализ по ИК-спектрам 219
активность спектров КР, интенсивность, степень деполяризации, могут
иметь огромное значение. Применение спектроскопии КР в качестве
дополнительного метода при определении молекулярной симметрии
хорошо известно, и ни одно серьезное исследование структуры молекул
не может быть выполнено без данных по КР.
Ценные сведения об отнесении частот в колебательном спектре
можно получить из других спектроскопических методов, таких, как
УФ-спектроскопия, которая в принципе используется при исследовании
ароматических, ненасыщенных и других соединений, содержащих
тс-электроны или неподеленные электронные пары, или микроволновая
спектроскопия, которая на основе детального изучения вращательных
спектров позволяет определять точные размеры молекул.
Колебательно-вращательная структура полос
Еще в 1933 г. было показано, что контур линий вращательной
структуры спектров паров и расстояние между ними определяются
направлением изменения дипольного момента в процессе колебания
и моментами инерции молекулы [100]. Результаты этой работы,
которая первоначально была выполнена на симметричных молекулах,
впоследствии были распространены и на несимметричные [11, 128].
Зная конфигурацию молекулы, можно зачастую предсказать контур
огибающей вращательной структуры для отдельного колебания и вычис-
вычислить расстояние между максимумами Р- и Д-ветвей. Однако данные
по молекулярной симметрии редко можно получить только из контуров
полос. Более детальную информацию дает исследование с высоким
разрешением вращательной структуры.
i
Расчет силовых постоянных
Силовые постоянные можно рассчитать, значительно усложнив
первоначальное предположение о том, что колебания в группе атомов
происходят так же, как и в двухатомной молекуле; полное определение
всех силовых постоянных включает учет взаимодействия несвязанных
атомов. Хотя первый метод и прост, но он слишком груб, чтобы
давать достаточно ценные результаты; последний слишком сложен для
решения большинства практических задач. Как уже обсуждалось ранее,
для получения силовых постоянных сложных молекул используются си-
силовые поля более простых молекул. Сейчас в этой области достигнут
значительный прогресс. Интересно представляют те силовые постоянные,
которые можно переносить с одной молекулы на другую, что позволяет
вычислять частоты со значительной точностью. Еще одно преимущество
расчета колебаний состоит в том, что нормальные координаты и рас-
распределение потенциальной энергии дают действительную форму сме-
смещений атомов и ясно указывают на смешанные колебания.
Для более детального ознакомления с этими методами читатель
может обратиться к литературе [46, 56, 78, 88, 272, 273, 1, 2].
220 Глава 5
Изотопное замещение
Как уже было показано (стр. 155—157), при замещении водорода
на дейтерий соответствующие полосы поглощения сдвигаются в 1,3 —
1,5 раза (примерно в у2 раз), причем смещения мало зависят от типов
колебаний. Замещение на дейтерий часто происходит сравнительно
легко и широко используется как метод изучения колебаний, проис-
происходящих с участием атома водорода.
Другие изотопы также дают полезную информацию. В некоторых
случаях естественное содержание изотопов вполне достаточно для вы-
выполнения законченного исследования. Например, в ряду карбонатов
щелочноземельных металлов Дешиус [72] обнаружил поглощение
A3СОзJ~ при естественном содержании 13С, равном 1,1 %. В спектрах
KNO3 наблюдались полосы A5NO3)~ [71]. Изучая спектры 6LiCH3
и 71лСНз, Вест и Глейз [265] показали, что эти соединения являются
полимерами даже в газовой фазе, и отнесли полосы в области
340—570 см~1 к колебаниям связи Li—С. В качестве дополнительного
метода отнесения частот Хатчинсон и др. [133] рассмотрели изотопное
замещение металлов.
Наконец, в качестве примера использования изотопного замещения
для отнесения частот приведем работу Линтона и Никсона [175,
176] по силилцианиду. Предшествующие исследователи, основываясь
на химических свойствах и аналогии, предположили, что продуктом
реакции SiH3l с AgCN является изоцианид. Линтон и Никсон, пред-
предполагая, что в этой реакции образуется нормальный цианид, синтези-
синтезировали образцы, обогащенные 13С и 'SN. Для валентного колебания
вцианиде вычисленные сдвиги составили — 51 см~' для i^CN и —31 см~1
для C15N. Для изоцианида вычисленные сдвиги были — 43 см
для N13C и — 38 см для 15NC. В эксперименте наблюдались сдвиги
— 51 см~' для образца, обогащенного 13С, и —30 см для образца,
содержащего 15N, что явилось решающим доказательством в пользу
нормального строения этого соединения.
Количественная связь частот в парах изотопозамещенных молекул
дается правилом Теллера—Редлиха и различными правилами сумм
[272]. Многочисленные данные по ИК-спектрам изотопозамещенных
соединений собраны Пинхасом и Лаулихтом [212].
Исследования в поляризованном свете
ориентированных образцов
Исследования в поляризованном свете особенно ценны для ориен-
ориентированных полимеров и монокристаллов. В ориентированном образце
полоса поглощения имеет максимальную величину, если момент
перехода параллелен электричесокму вектору излучения, и поглоще-
поглощение отсутствует, если эти векторы взаимно перпендикулярны. Однако
Качественный анализ по ИК-спектрам 221
необходимо проявлять осторожность и помнить о различии между
моментом перехода и направлением связи, так как их направле-
направления не обязательно совпадают.
Можно выделить два направления использования результатов изме-
измерений в поляризованном свете: 1) поиск связи моментов перехода
с молекулярной ориентацией в кристаллах с известной структурой;
2) определение структуры кристаллов или полимеров на основании
дихроичных отношений. В одной из таких работ методом НПВО
в поляризованном свете было проведено отнесение частот в ИК-спектрах
монокристаллов стеариновой кислоты и ее дейтерированного (d35)
аналога [196]. Для дальнейшего ознакомления с поляризационным
методом можно обратиться к литературе [35, 161].
Сдвиги частот под влиянием растворителя
Было показано (стр. 177—180), что различные нормальные колебания
по-разному реагируют на изменение растворителя. На этом эффекте
основан изящный метод, позволяющий охарактеризовать групповые
частоты. В нем сдвиг неизвестных полос в различных растворителях
сопоставляется со сдвигом аналогичной известной полосы в другой
молекуле. Полосы одного типа (например, карбонильных групп) прояв-
проявляют сходные сдвиги в широком интервале растворителей. Этот
эффект был использован для того, чтобы отличить полосу поглощения
группы С==О от полосы двойной связи С=С в пиридонах [29].
В результате такого исследования получается график типа представ-
представленного на рис. 5.36. Наименьшие сдвиги наблюдаются в относительно
инертных растворителях (гептан и циклогексан), а наибольшие —
в растворителях, способных образовывать водородные связи (бромо-
форм и йодистый метилен). Другим примером такого рода является зави-
зависимость сдвига полосы поглощения C==S от сдвига полосы С=О
в ацетофеноне [28]. В ходе этой работы была выявлена аномалия
в отнесении групповой частоты C=S.
Такой метод неприменим к неполярным связям, а также в тех
случаях, когда растворитель преимущественно ассоциируется с другой
частью молекулы, вследствие чего ожидаемый сдвиг исследуемой полосы
не наблюдается. Однако, несмотря на эти ограничения, метод смещения
частот под влиянием растворителей имеет большие возможности
для идентификации групповых частот и установления структуры.
В гл. 1 отмечалось, что оптические изомеры нельзя различать
методом ИК-спектроскопии. Это не совсем так. Было показано, что,
применяя оптически активный растворитель и интерференционный
спектрофотометр, можно наблюдать небольшие, но измеряемые изме-
изменения в положениях полос и интенсивностях, которые различаются
для двух изомеров и могут быть зафиксированы при вычитании
оптических плотностей [ПО]. Для иллюстрации можно привести пример
оптически активной яблочной кислоты (Я) в энантиоморфном 2-окта-
222
Глава 5
20
10
У
/
5 10 15 20
Дг/r х 103СО N-метил -2-пиридоиа
25
Рис. 5.36. Сдвиг полосы поглощения 1700 см в М-метил-2-пиридоне [29].
Reproduced, with permission, from Spectrochimica Acta.
ноле (О). Неожиданно обнаруживается, что спектры +Я в —О
полностью отличаются от спектров — Я в +О; спектры —Я в —О
и +Я в +О отличаются друг от друга и от спектров двух других
пар. Это явление можно объяснить слабой остаточной эллиптической
поляризацией интерферометра Майкельсона, которая снимает спектро-
спектроскопическое вырождение стереоизомеров.
ЛИТЕРАТУРА
1. Adams D. M., Spectrosc. Prop. Inorg. Organomet. Сотр., 5, 239 A972).
2. Afremow L. С, Vandeberg J. Т., J. Paint Technology, 38, 169 A966).
3. Ahmadjian M. Baer C. D., Lynch P. F., Brown С W., Environ. Sci. Technoe.,
10, 777 A976).
4. Allen H. C, Cross P. C, Molecular Vib-Rotors, Wiley, New York, 1963.
5. Altshuller A. P., Wartburg A. F., Appl. Spectrosc., 15, 67 A961).
6. Anderson D. R., Infrared, Raman and Ultraviolet Spectroscopy, in A. L. Smith,
Ed., Analysis of Silicones, Wiley, New York, 1974, Chap. 10, pp. 247-286.
7. Andries J. C, Diem H. E., J. Polym. Sci., Polym. Lett. Ed., 12, 281
A974).
8. Arnold J., Bertie J. E., Millen D. J., Proc. Chem. Soc. (London), 1961,
121.
9. Badger R. M., J. Chem. Phys., 2, 128 A934); 3, 710 A935).
10. Badger R. M., Bauer S. H., J. Chem. Phys., 5, 839 A937).
11. Badger H. M., Zumwalt L. R., J. Chem. Phys., 6, 711 A938).
12. Baer С D., Brown С W., Appl. Spectrosc., 31, 524 A977).
13. Baker A. J., Cairns Т., Eglinton G., Preston F. J., More Spectroscopic
Problems in Organic Chemistry, 2nd ed., Heyden, London, 1975.
Качественный анализ по ИК-спектрам 223
14. Baker A. W., Shulgin А. Т., 3. Am. Chem. Soc., 80, 5358 A958).
15. Barrachough С. G., Bradley D. C, Lewis J., Thomas I. M., J. Chem. Soc.,
1961, 2601.
16. Barrow G. M., Spectrochim. Acta, 16, 799 A960).
17. Bauer E., Magat M., J. Phys. Radium, 9, 319 A938).
18. Becker E. D., Spectrochim. Acta, 17, 436 A961).
19. Bell J. V., Heisler J., Tannenbaum H., Goldenson J., J. Am. Chem. Soc,
76, 5185 A954).
20. Bellamy L. J., J. Chem. Soc, 1955, 2818.
21. Bellamy L. J., Appl. Spectroscopy, 16, 61 A962).
22. Bellamy L. J., The Origins of Group Frequency Shifts, in M. J. Wells,
Ed., Spectroscopy, The Institute of Petroleum London, 1962, pp. 205-221.
23. Беллами Л. Новые данные по ИК-спектрам сложных молекул. Пер. с англ.—
М.: Мир, 1971.
24. Bellamy L. J., The Infrared Spectra of Complex Molecules, Vol. 1, 3rd ed.,
Wiley, New York, 1975. (См. [18] в гл. 3.)
25. Bellamy L. J., Hallam H. E., Trans. Faraday Soc, 55, 220 A959).
26. Bellamy L. J., Owen A. J., Spectrochim. Acta, 25A, 329 A969).
27. Bellamy L. J., Pace R. J., Spectrochim. Acta, 25A, 319 A969).
28. Bellamy L. J., Rogasch P. E., J. Chem. Soc, 1960, 2218.
29. Bellamy L. J., Rogasch P. E., Spectrochim. Acta, 16, 30 A960).
30. Bellamy L. J., Williams R. L.. Trans. Faraday Soc, 55, 14 A959).
31. Bent H. A., Chem. Rev., 61, 275 A961).
32. Bernstein H. J., Spectrochim. Acta, 18, 161 A962).
33. Bertran J. F., Ballester L., Dobrihalova L., Sanchez N.. Arrieta R., Spectrochim.
Acta, 24A, 1765 A968).
34. Bessette F., Can. J. Spectrosc, 20, 126 A975).
35. Bluhm A. L., Polarized Infrared Spectroscopy, in G. L. Clark, Ed., Encyclopedia
of Spectroscopy, Reinhold, New York, 1960, pp. 522-531.
36. Boerio F. J., Koenig J. L., J. Macromolec. Sci., Rev. Macromolec Chem.,
C7, 209 A972).
37. Bosvik R., Knutsen K, V., von Sydow С F. E., Anal. Chem., 33, 1162
A961).
38. Brown С W., Lynch P. F., Ahmadjian M., Baer С D., Am Lab., 7
A2), 59 A975).
39. Brown T. L., Spectrochim. Acta, 10, 149 A957).
40. Brown T. L., Chem. Rev., 58, 581 A958).
41. Brownlee R. T. C, Di Stefano J., Topsom R. D., Spectrochim. Acta, 31A,
1685 A975).
42. Brownlee R. T. C, Topsom R. D., Spectrochim. Acta, 31A, 1677 A975).
43. Bryson A., J. Am. Chem. Soc, 82, 4858, 4862 A960).
44. Buckingham A. D., Proc Roy. Soc. (London), A248, 169 A958); A255,
32 A960).
45. Buckingham A. D., Trans. Faraday Soc, 56, 753 A960).
46. Califano S., Vibrational States, Wiley, New York, 1976.
47. Cannon С G., Spectrochim. Acta, 10, 341 A958).
48. Carlsson D. J., Suprunchuk Т., Wiles D. M., Text. Res. J., 47, 456 A977).
49. Cassels J. W., Appl. Spectrosc, 22, 477 A968).
50. Chatten L. G., Levi L., Appl. Spectrosc, 11, 177 A957).
51. Chicago Society for Paint Technology, Infrared Spectroscopy: Its Use in the
Coatings Industry, Federat. Soc. Paint Technol., Philadelphia, 1969.
224 Глава 5
52. Cobler J. G., Plastics, in F. J. Welcher, Ed., Standard Methods of Chemical
Analysis, Vol. IIB, 6th ed., Van Nostrand, New York, 1963, pp. 2034-2104.
53. Coggeshall N. D., Saier E. L., J. Chem. Phys., 15, 65 A947).
54. Colthup N. В., J. Chem. Educ, 38, 394 A961).
55. Colthup N. В., Appl. Spectrosc., 30, 589 A976).
56. Colthup N. В., Daly L. H., Wiberley S. E., Introduction to Infrared and
Raman Spectroscopy, 2nd ed., Academic Press, New York, 1975.
57. Colthup N. В., Orloff M. K., Spectrochim. Acta, 27A, 1299 A971).
58. Cook B. W., Jones K., A Programmed Introduction to Infrared Spectroscopy,
Heyden, London, 1972.
59. Cook D., in G. Olah, Ed., Friedel-Crafts and Related Reactions, Vol. 1,
Interscience, New York, 1963, p. 767.
60. Corish P. J., Anal. Chem., 31, 1298 A959).
61. Cotton F. A., Chemical Applications of Group Theory, Interscience, New
York, 1963.
62. Coulson С A., Spectrochim. Acta, 14, 161 A959).
63. Craver С D., Desk Book of Infrared Spectra, Coblentz Society, РОВ
9952, Kirkwood, Mo., 1977.
64. Craver, Clara D., Surfaces, in E. G. Brame, Jr., and J. G., Grasselli,
Eds., Practical Spectroscopy, Vol. 1, Part C, Infrared and Raman Spectroscopy,
Marcel Dekker, New York, 1977, p. 933.
65. Daasch L. W., Spectrochim. Acta, 13, 257 A958).
66. Davis A., Gordon D., J. Appl. Polym. Sci., 18, 1159 A974).
67. Davis J. E. D.. J. Molec. Struct., 10, 1 A971).
68. Davis M. A., J. Org. Chem., 32, 1161 A967).
69. Deady L. W., Harrison P. M., Topson R. D., Spectrochim. Acta, 31A,
1671 A975).
70. Deb К. К., Spectrosc. Lett., 8, 185 A975).
71. Decius J. C, J. Chem. Phys., 22, 1941 A954).
72. Decius J. C, J. Chem. Phys., 22, 1946 A954).
73. Dewar M. J. S., Grisdale P. J., J. Am. Chem. Soc, 84, 3539, 3548 A962).
74. Dijkstra G., Spectrochim. Acta, 11, 618 A957).
75. Dolphin D., Wick A. E., Tabulation of Infrared Spectral Data, Wiley,
New York, 1977.
76. Douthit R. C, Garska K. J., Yarborough V. A.. Appl. Spectrosc., 17,
85 A963).
77. Drisko R. W., Crilly J. В., Mat. Prot. Perform., 11 A2), 49 A972).
78. Duncan J. L, Force Constant Calculations in Molecules, in R. F. Barrow,
D. A. Long, and D. J. Millea Eds., Molecular Spectroscopy, Vol. 3,
The Chemical Society, London, 1975, Chapter 2.
79. Edelmann K., Wyden #., Kaut. Gummi, Kunstst., 25, 353 A972).
80. Epp A., Anal. Chem., 29, 1283 A957).
81. Evens J. C, Spectrochim. Acta, 17, 129 A961).
82. Farmer V. C, Infrared Spectroscopy in Mineral Chemistry, in A. W. Nicol. Ed.,
Physicochem. Methods of Mineral Analysis, Plenum Press, New York, 1975.
83. Farmer V. C, Palmieri F., in J. E. Gieseking, Ed., Soil Components,
Vol. 2, Springer, New York, 1975, p. 573.
84. Fateley W. G., Matsubara I., Witkowski R. E., Spectrochim. Acta, 20,
1461 A964).
85. Fateley W. G., McDevitt N. Т., Bentley F. F., Appl. Spectrosc., 25, 155
A971).
Качественный анализ по ИК-спектрам 225
86. Fermi ?., Z. Physik, 71, 250 A931).
87. Ferraro J. R., Low-Frequency Vibrations of Inorganic and Coordination
Compounds, Plenum Press, New York, 1971.
88. Ferraro J. R., Ziomek J. S., Introductory Group Theory and its Appli-
Application to Molecular Structure, 2nd ed., Plenum Press, New York, 1975.
89. Fieldes M., Furkert R. J., Wells N.. N. Z. J. Sci., 15, 615 A972).
90. Fischmeister /., Hellgren L, Vincent J., Arch. Dermatol. Res., 253, 63
A975).
91. Flett M. St. C, Spectrochim. Acta, 18, 1537 A962).
92. Francis S. A.. J. Chem., 18, 861 A950).
93. Fraser R. D. В.. Price W. C, Nature, 170, 490 A952).
94. Freeman N. K.. Analysis of Blood Lipids by Infrared Spectroscopy, in
G. J. Nelson, Ed., Blood Lipids and Lipoproteins: Quantity, Composition,
and Metabolism, Wiley, New York, 1972, pp. 113-179.
95. Fritzsche H., Spectrochim. Acta. 17, 352 A961).
96. Fuson N.. Josien M. L., Shelton E. M., J. Am. Chem. Soc, 76, 2526
A954).
97. Gaffey M. J., J. Geophys. Res., 81, 905 A976).
98. Gav B. W.. Jr., Hanst P. L.. Bufalini J, J.. Noonan R. C, Environ.
Sci. Technol., 10, 58 A976).
99. Gedemer T. J., Appl. Spectrosc, 19, 141 A965).
100. Gerhard S. L., Dennison D. M., Phys. Rev., 43, 197 A933).
101. Ghosh S. N.. Chatterjee A. K.. J. Mat. Sci., 9, 1577 A974).
102. Goddu R. F., Delker D. A.. Anal. Chem., 32, 140 A960).
103. Golding K., Process Eng. 70, 1 A974).
104. Goldstein M.. Spectrosc. Prop. Inorg. Organomet. Сотр., 5, 333 A972).
105. Gordon R G., J. Chem. Phys., 43. 1307 A965).
106. Gordy IV., J. Chem. Phys., 14, 305 A946)!
107. Gordy W., Thomas W. J. O., J. Chem. Phys., 24, 439 A956).
108. Gramstad Т., Spectrochim. Acta, 19, 497 A963).
109. Greenwood N. N.. Ross E. J. P., Straughan B. P., Index of Vibrational
Spectra of Inorganic and Organometallic Compounds, Vol. 1, Butterworth,
London, 1972, and succeeding volumes.
110. Grieble D. L., Griffiths P. R., Hirschfeld Т., Anal. Chem., 50, 415 A978).
111. Hadii D., Chimia, 26, 7 A972).
112. Hallam H. E., Solvent Effects on Infrared Group Frequencies, in M. J. Wells,
Ed., Spectroscopy, Institute of Petroleum, London, 1962, pp. 245 — 264.
113. Hallam H. E., Ray Т. С, Nature, 189, 915 A961).
114. Hampton R. R., Rub. Chem. Technol., 45, 546 A972).
115. Хаслам Дж., Виллис Г. А. Идентификация и анализ полимеров. Пер.
с англ.—М.: Химия, 1971.
116. Heicklen J., Spectrochim. Acta, 17, 82 A961).
117. Helmen Т., Farbe Lack, 80, 715 A974).
118. Hennlker J. C, Infrared Spectrometry of Industrial Polymers, Academic
Press, New York, 1967.
119. Герцберг Г. Колебательные и вращательные спектры многоатомных молекул
Пер. с англ.- М.: ИЛ, 1949.
120. Higuchi S., Tanaka S., Kamada И., Spectrochim. Acta, 28A, 1721 A972).
121. Higuchi S., Tsuyama H.. Tanaka S., Kamada H., Spectrochim.. Acta, 31A,
1011 A975).
122. Hill A. G., Curran C, J. Phys. Chem., 64, 1519 A960).
15 - 586
226 Глава 5
123. Hill R. R., Rendell D. A.' ?., The Interpretation of Infrared Spectra,
Heyden, London, 1975.
124. Hinkley E. D., Environ. Sci. Technol., 11, 564 A977).
125. Hirschfeld Т., Anal. Chem., 48, 721 A976).
126. Hirschfeld Т., Kizer K., Appl. Spectrosc., 29, 345 A975).
127. Hblland-Moritz K., Siesler H. W., Appl. Spectrosc. Rev., 11, 1 A976).
128. Hollas J. M., Spectrochim. Acta, 22, 81 A966).
129. Hughes R. H., Martin R. J., Coggeshall N. .?>., J. Chem. Phys., 24,
489 A956).
130. Hummel D. O., Identification and Analysis of Surface-Active Agents, Wiley-
Interscience, New York, 1962.
131. Hummel D. O.,\Scholl F. K., Infrared Analysis of Polymers, Resins and
Additives: an Atlas, Vol. 1, Plastics, Elastomers, Fibers, and Resins, Part
l-.Text; Part 2, Spectra, Tables, Index, 1969; Vol. 2, Additives and Pro-
Processing Aids, 1973, Wiley-Interscience, New York.
132. Hummel D. O., Siesler H., Zoschke E., Vierling I., Morlock U., Stadtlaender Т.,
Melliand Textilber, Int., 54, 1340 A973).
133. Hutchinson В., Eversdyk D., Olbrkht S., Spectrochim. Acta, 30A, 1605
A974).
134. Ingold С. К., Structure and Mechanism in Organic Chemistry, Cornell U.
Pr., Ithaca, New York, 1953.
135. Ivin K. J., Structural Studies Of Macromolecules by Spectroscopic Methods,
Wiley, New York, 1976.
136. Jenkins J. W., Kellenbach К. О., Anal. Chem., 31, 1056 A959).
137. Joesten M. D., Schaad L. J., Hydrogen Bonding, Marcel Dekker, New
York, 1974.
138. Jonas K., Solymar K., Orban M., Acta Chim. Acad. Sci. Hung., 81, 443
A974).
139. Jones D. W., Introduction to the Spectroscopy of Biological Polymers,
Academic Press, New York, 1976.
140. Jones L. A., Holmes J. C. Seligman R. В., Anal. Chem., 28, 191 A956).
141. Jones L. H., Inorganic Vibrational Spectroscopy, Vol. 1, Marcel Dekker,
New York, 1971.
142. Jones R. N., Infrared Spectra of Organic Compounds: Summary Charts
of Principal Group Frequencies, National Research Council of Canada,
Ottawa, 1959.
143. Jones R. N., Escolar D., Hawranek J. P., Neelakantan P., Young R. P.,
J. Molec. Struct., 19, 21 A973).
144. Jones R. N., Forbes W. E., Mueller W. A., Can. J. Chem., 35, 504 A957).
145. Jones R. N.. Venkataraghavan R., Hopkins J. W., Spectrochim. Acta, 23A,
925 A967).
146. Jones W. J., Sheppard N., Rotation in Solution, in M. J. Wells, Ed.,
Spectroscopy, The Institute of Petroleum, London, 1962, pp. 181-204;
147. Kagarise R. E., J. Am. Chem. Soc, 77, 1377 A955).
148. Kagarise R. E., Whetsel К. В., Spectrochim. Acta, 18, 341 A962).
149. Kappelmeier C. P. A., Chemical Analysis of Resin-Based Coating Materials,
Wiley-Interscience, New York, 1959.
150. Karger B. L., Snyder L. R., Horvalh C, An Introduction to Separation
Science, Wiley, New York, 1973.
151. Karr C, Jr., Infrared and Raman Spectroscopy of Lunar and Terrestrial
Minerals, Academic Press, New York, 1975.
Качественный анализ по ИК-спектрам 227
152. Katritzky A. R., Topsom R. D., Linear Free Energy Relationships and
Optical Spectroscopy, in N. B. Chapman and J. Shorter, Eds., Advances
in Free Energy Relationships, Plenum Press, London, 1972.
153. Kawahara P. K., Santner J. F., Julian E. C, Anal. Chem., 46, 266 A974).
154. Kaye W., Near Infrared Spectroscopy, in G. L. Clark, Ed., Encyclopedia
of Spectroscopy, Reinhold, New York, 1960, pp. 494-505.
155. Kirkland J. J., Anal. Chem., 29, 1127 A957).
156. Kirkwood J. G., in W. West and R. T. Edwards, J. Chem. Phys., 5,
14 A937).
157. Koenig J. L., Appl. Spectrosc., 29, 293 A975).
158. Koenig J. L., D'Esposito L., Antoon M. K, Appl. Spectrosc., 31, 292 A977).
159. Kollman P. A., Allen L. C, Chem. Rev., 72, 283 A972).
160. Krueger R. J., Thompson H. W., Proc. Roy. Soc, (London), A243, 143
A957); A250, 22 A959).
161. Krimm S., Fortschr. Hochpolym.-Forsch., 2, 51 A960).
162. Krimm S., Polymers: Theory and Interpretation of Spectra, in G. L. Clark,
Ed., Encyclopedia of Spectroscopy, Reinhold, New York, 1960, pp. 535-544.
163. Krimm S., Infrared Studies of Chain Organization in Crystalline Polyethylene
in E. B. Mano, Ed., Proceedings of International Symposium on Macromo-
lecules, 1974, Elsevier Amsterdam, 1975, p. 107.
164. Kuhn I. P., Bowman R. E., Spectrochim. Acta, 17, 650 A961).
165. Langseth A., Lord R. С Kgl. Danske Videnskab. Selskab, Mat-fys. Medd.,
16, 6 A948).
166. Larson H. P., Fink U., Appl. Spectrosc., 31, 386 A977).
167. Larson H. P., Fink U., Astrophys., J. Lett., 171, L91 A971).
168. Launer P. J., Perkin-Elmer Instrum. News., 13, C), 10 A962).
169. Layton E. M., Jr., Kross R. D., Fassel V. A., J. Chem. Phys., 25,
135 A956).
170. LeBlanc R. В., Trimmer Т. Т, Text. Chem. Color, 5, 279 A973).
171. Leffler J. E., Grunwald E., Rates and Equilibria of Organic Reactions,
Wiley, New York, 1963.
172. Levi L., Hubley С. Е., Anal. Chem., 28, 1591 A956).
173. Levitt L. S.. Widing H. F., The Alkyl Inductive Effect, in R.. W. Taft,
Ed., Progress in Physical Organic Chemistry, Wiley, New York, Vol. 12,
1976, p. 119.
174. Liddel U., Ill, Becker E. D., Spectrochim. Acta, 10, 70 A957).
175. Linton H. R., Nixon E. R., J. Chem. Phys., 28, 990 A958).
176. Linton H. R., Nixon E. R., Spectrochim. Acta, 10, 299 A958).
177. Lippert E., Mecke R., Z. Elektrochem., 55, 366 A951).
178. Лисица М. П., Цященко Ю. П., Оптика и спектроскопия, 9, 438 A960).
179. Lord R. С, Merrifield R. ?., J. Chem. Phys, 21, 166 A953).
180. Lord R. C, Walker R. W., J. Am. Chem. Soc, 76, 2518 A954).
181. Lunn W. H., Spectrochim. Acta, 16, 1088 A960).
182. Luongo J. P., Appl. Spectrosc., 25, 76 A971).
183. Lynch P. F.. Tang S., Brown С W., Anal. Chem., 47, 1696 A975).
184. Maguire M. M., West R., Spectrochim. Acta, 17, 369 A961).
185. Mantz A. W.. Morita H. K., Appl. Spectrosc, 30, 587 A976).
186. Maslowsky E., Jr., Vibrational Spectra of Organometallic Compounds, Wiley,
New York, 1976.
187. McCall E. R., Miles S. H.. Tripp V. W., O'Connor R. Т., Appl. Spectrosc.,
18, 81 A964).
15*
228 Глава 5
188. McCarthy D. E., Appl. Spectrosc, 22, 66 A968).
189. McGraw-Hill, Inc., Modern Plastics Encyclopedia, McGraw-Hill, New York,
Annual.
190. McKean D. C, Spectrochim. Acta, 29A, 1559 A973).
191. McKean D. C, Spectrochim. Acta, 31A, 1167 A975).
192. McKean D. C, Biedermann S., Burger #., Spectrochim. Acta, 30A, 845
A974).
193. McKean D. C, Saur O., Travert L., Lavalley 'J. C, Spectrochim. Acta,
31A, 1713 A975).
194. Meiklejohn R. A., Meyer R. J., Aronovic S. M., Schuette H. A., Meloch V. W.,
Anal. Chem., 29, 329 A957).
195. Morris N. M., McCall E. R., Tripp V. W., Text, Chem. Color, 4, 283
A972).
196. Munch W., Fringeli U., Giinthard Hs. #., Spectrochim, Acta, 33A, 95
A977).
197. Murthy A. S. M., Rao С N. S.. Appl. Spectrosc. Rev., 2, 69 A968).
198. Myers R. R., Long J. S., Treatise on Coatings Film-Forming Compositions,
Part I, Marcel Dekker, New York, 1967.
199. Nakanishi K., Solomon P. H., Infrared Absorption Spectroscopy, 2nd ed.,
Holden-Day, San Francisco, 1977.
200. Накамото К. Инфракрасные спектры неорганических и координационных
соединений. Пер. с англ.—М.: Мир, 1966.
201. Nasser M. /., Appl. Spectrosc, 28, 545 A974).
202. Nyquist R. A., Potts W. J., Jr., Vibrational Spectra of Phosphorus Com-
Compounds, in M. Halman, Ed., Analytical Chemistry of Phosphorus Compounds,
Interscience, New York, 1972.
203. O'Connor R. Т., High Polym., 5, 51 A971).
204. O'Connor R. Т., McCall E. R.. Morris N. M., Tripp V. W., U. S.
Agric. Res. Serv., South. (Rep.), 1974, ARS-S-47 A974).
205. Overend J., Scherer J. R., Spectrochim. Acta, 16, 773 A960).
206. Parker F. S., Applications of Infrared Spectroscopy in Biochemistry, Biology,
and Medicine, Plenum Press, New York, 1971.
207. Parker F. S., Appl. Spectrosc, 29, 129 A975).
208. Pauling L., Nature of the Chemical Bond., 3rd ed., Cornell U. P., Ithaca,
New York, 1960.
209. Phillips J. P., Deye J. F., Anal. Chim. Acta, 17, 231 A957).
210. Pimentel G. C, McClellan A. L., Annu. Rev. Phys. Chem., 22, 347 A971).
211. Пиментел Дж., Мак-Клеллан О. Водородная связь. Пер. с англ.— М.: Мир,
1964.
212. Pinchas S., Laulicht I., Infrared Spectra of Labeled Compounds, Academic
Press, New York, 1971.
213. Pitts J. N.. Finlayson-Pitts B. J., Winer A. M., Environ. Sci., Technol.,
11, 568 A977).
214. Polcin У., Appl. Spectrosc, 28, 588 A974).
215. Potts W. J., Jr., Chemical Infrared Spectroscopy, Vol. 1, Techniques, Wiley,
New York, 1963.
216. Potts W. J., Jr., The Use of Infrared Spectroscopy in Characterization
of Polymer Structure, in Symposium on Plastics Testing and Standardiza-
Standardization, ASTM Special Technical Publication, No. 247, 1958, p. 225.
217. Potts W. J., Jr., Nyquist R. A., Spectrochim. Acta, 15, 679 A959).
218. Powell D. L., West R., Spectrochim. Acta, 20, 983 A964).
Качественный анализ по ИК-спектрам 229
219. Pritchard H. О., Skinner H. A., Chem. Rev., 55, 745 A955).
220. Pros A. W., van Zeggeren F., Spectrochim. Acta, 16, 563 A960).
221. Pullin A. D. В., Proc. Roy. Soc. (London), A255, 39 A960).
222. Puiinam N. A.. J. Soc. Cosmet. Chem., 23, 209 A972).
223. Раков А. В., Оптика и спектроскопия, 13, 369 A962).
224. Ramsay D. A., J. Am. Chem. Soc, 74, 72 A952).
225. Резникова Е. Б., Тюлин В. И., Татевский В. М., Оптика и спектроскопия,
13, 364 A962).
226. Ridgway S., Larson H. P., Fink U., The Infrared Spectrum of Jupiter,
in T. Gehrels, Ed., Jupiter, Arizona U. P., Tucson, 1976.
227. Rosen M. J., Goldsmith H. A., Systematic Analysis of Surface-Active Agents,
2nd ed., Wiley-Interscience, New York, 1972.
228. Sandorfy С Can. J. Spectrosc, 17, 24 A972).
229. Scherer J. R., Over end J., J. Chem. Phys., 32, 1720 A960).
230. Scherer J. R., Overend J., J. Chem. Phys., 33, 1681 A960).
231. Schmid P., Physiol. Chem. Phys., 7, 335, 349, 357 A975).
232. Schwarzmann E., Naturwissenschaft., 49, 103 A962).
233. Сешадри К., Джонс Р., УФН, 85, № 1, 87 A965).
234. Sheppard N., Rotational Isomerism about С — С Bonds in Saturated Mole-
Molecules Studies by Vibrational Spectroscopy, in H. W. Thompson, Ed., Advances
in Spectroscopy, Vol. I, Interscience, New York, 1959, pp. 288-353.
235. Shimanouchi Т., Tables of Molecular Vibrational Frequencies, Consolidated
Volume I, U. S. Government Printing Office, S. D. Cat. No. C13, 48 :39, 1972.
236. Shimizu #., Bull. Chem. Soc. Jap., 39, 2385 A966).
237. Shipman J. J., Folt V. L., Krimm S., Spectrochim. Acta, 18, 1603 A962).
238. Shuster P., Zundel G., Sandorfy C, The Hydrogen Bond: Recent Developments
in Theory and Experiments, Vol. 2, Structure and Spectroscopy, Elsevier
North-Holland, Amsterdam, 1976.
239. Сильверстейн Р., Басслер Г., Моррил Т. Спектрометрическая идентификация
органических соединений. Пер. с англ.—М.: Мир, 1977.
240. Slowinski E. J., Jr., Claver G. С, J. Opt. Soc. Am., 45, 396 A955).
241. Smith A. L., Am. Lab., 2 A0), 27 A970).
242. Smith A. L., Infrared Spectroscopy, in J. W. Robinson, Ed., Handbook
of Spectroscopy, Vol. II, CRC Press, Cleveland, 1974.
243. Spialter D., Ballester M., Anal. Chem., 34, 1183 A962).
244. Steele D., Theory of Vibrational Spectroscopy, Saunders, Philadelphia, 1971.
245. Stock, Leon M., J. Chem. Educ, 49, 400 A972).
246. Стрижевекий В. Л., Оптика и спектроскопия, 8, 165 A960).
247. Susz В. P., Lachavanne A., Helv. Chim. Acta, 41, 634 A958).
248. Swain С. G., Lupton E. C, Jr., J. Am. Chem. Soc, 90, 4328 A968).
249. Sward G. G., Gardner H. A., Paint Testing Manual: Physical and Chemical
Examination of Paints, Varnishes, Lacquers, and Colors, 13th ed., American
Society for Testing and Materials, Philadelphia, 1972.
250. Tabb D. L., Sevcik J. J., Koenig J. L., J. Polym. Sci., Polym. Phys.
Ed., 13, 815 A975).
251. Taft R. W., Jr., Separation of Polar, Steric, and Resonance Effects in
Reactivity, in M. S. Newman, Ed., Steric Effects in Organic Chemistry,
Wiley, New York, 1956, pp. 556-675.
252. Thomas G. J., Jr., Kyogoku Y., Biological Science, in E. G. Brame, Jr.,
and J. G. Grasselli, Eds., Practical Spectroscopy, Vol. 1, Infrared and
Raman Spectroscopy, Part C, Marcel Dekker, New York, 1977, p. 717.
230 Глава 5
253. Thomas L. С, Interpretation of the Infrared Spectra of Organophosphorus
Compounds, Heyden, London, 1974.
254. Thompson В., Hazardous Gases and Vapors: Infrared Spectra and Physical
Constants, Beckman Instruments, Fullerton, Ca., Technical Report No. 595,
August 1974.
255. Thompson H. W., Jameson D. A., Spectrochim. Acta, 13, 236 A958).
256. Thompson H. W., Jewell D. J.. Spectrochim. Acta, 13, 254 A958).
257. Topsom R. D.. The Nature and Analysis of Substituent Electronic Effects,
in R. W. Taft, Ed., Progress in Physical Organic Chemistry, Vol. 12,
Wiley, New York, 1976.
258. Tosi C, Ciampelli F., Fortschr. Hochpolym.-Forsch., 12, 87 A973).
259. Tweed F. T, Cameron R., Deak J. S., Rogers P. G., Forens. Sci., 4, 211
A974).
260. Van der Maas J. H., Lutz E. T. G.. Spectrochim. Acta, 30A, 2005 A974).
261. Van der Mdrel H. W., Beutelspacher H., Atlas of Infrared Spectroscopy
of Clay Minerals and Their Admixtures, Elsevier, New York, 1976.
262. Венкатесварлу К., Джагатхезан С, Оптика и спектроскопия, 13, 775
A962).
263. Washbum W. H., Appl. Spectrosc., И, 46 A975).
264. Weiner M., Vogel G., West R., Inorg. Chem., 1, 654 A962).
265. West R., Glaze W., J. Am. Chem. Soc, 83, 3580 A961).
266. Whetsel К. В., Appl. Spectrosc. Rev., 2, 1 A968).
267. Whetsel К. В., Kagarise R. E., Spectrochim. Acta, 18, 315, 329 A962).
268. Wiberley S. E., Bunce S. C, Bauer W. H., Anal. Chem., 32, 217 A960).
269. Wilkins P. ?., Air Qual. Instrum., 2, 246 A974).
270. Wilks P. A., Jr., Environ. Sci. Technol., 10, 1204 A976).
271. Wilmshurst J. K., J. Chem. Phys., 28, 733 A958).
272. Вильсон Е., Дешиус Дж., Кросс П. Теория колебательных спектров
молекул. Цер. с англ.-М.: ИЛ, 1960.
273. Woodward L. A., Introduction to the Theory of Molecular Vibrations
and Vibmional Spectroscopy, Oxford U. P., London, 1972.
274. Young С W., Duvall R. В., Wright N.. Anal. Chem., 23, 709 A951).
275. Збинден Р. Инфракрасная спектроскопия высокополимеров. Пер. с англ.—
М.: Мир, 1966.
276. Zenker W.. Anal. Chem., 44, 1235 A972).
Дополнительная литература
1. Волькенштейн М. В., Грибов Л. А., Ельяшевич М. А., Степанов Б. И.
Колебания молекул.-М.: Наука, 1972.
2. Коптев Г. С, Пентин Ю. А. Расчет колебаний молекул. - М.: Изд. МГУ, 1977.
3. Петраш Г. Г., Исследование аппаратурных искажений и методы их учета в
ИК-спектроскопии. В сб. Труды ФИАН. Т. 27.-М.: Наука, 1964, с. 3.
4. Маянц Л. С, Авербух Б. С. Теория и расчет интенсивностей в колеба-
колебательных спектрах молекул.-М.: Наука, 1971.
5. Грибов Л. А. Теория интенсивностей в инфракрасных спектрах многоатомных
молекул.-М.: Изд. АН СССР, 1963.
6. Бахшиев Н. Г. Спектроскопия межмолекулярных взаимодействий.—Л.:
Наука, 197Z
Качественный анализ по ИК-спектрам 231
7. Свердлов Л. М., Ковнер М. А., Крайнев Е. П., Колебательные спектры
многоатомных молекул.— М,: Наука, 1970.
8. Беллами Л. Новые данные по ПК-спектрам сложных молекул. Пер. с англ.—
М.: Мир, 1971.
9. Ингольд К. Теоретические основы органической химии. Пер. с англ. —
М: Мир, 1973.
10. Беккер Г. Введение в электронную теорию органических реакций. Пер. с
нем.-М.: Мир, 1977.
11. Наканиси К. Инфракрасные спектры и строение органических соединений.
Пер. с англ. - М.: Мир, 1965.
12. Казицына Л. А., Куплетская Н. Б. Применение УФ-, ИК-, ЯМР- и масс-
спектроскопии в органической химии.-М.: Изд. МГУ, 1979.
13. Инфракрасные спектры поглощения полимеров и вспомогательных ве-
веществ/Под, ред. В. М. Чулановского. - Л.: Химия, 1969.
14. Пентин Ю. А., Тарасевич Б. Н., Эльцефон Б. С, Вестник МГУ, сер. хим.,
№ 1, 13 A973).
15. Попов В. Я., Тарасевич Б. Н., Момотенко В. А., Вестник МГУ, сер. хим.,
№ 6, 721 A976).
16. Якутии В. И., Струков О. Г., Успехи химии, № $, 1504 A972).
П. Руководство по химическому анализу поверхностных вод суши/Под ред.
А. Д. Семенова. — Л.: Гидрометеоиздат, 1977.
/8. Гилсон Т., Хендра А. Лазерная спектроскопия КР в химии. Пер. с англ.—
М.: Мир, 1973.
19. Применение спектров комбинационного рассеяния. Пер. с англ./под ред.
А. Андерсона.— М.: Мир, 1977.
20. Сущинский М. М. Комбинационное рассеяние света и строение вещества.—
М.: Наука, 1981.
Глава 6
КОЛИЧЕСТВЕННЫЕ ПРИЛОЖЕНИЯ
Применение ИК-спектроскопии
в количественном анализе
Одно из наиболее значительных преимуществ ИК-спектра заклю-
заключается в его индивидуальности. Эта особенность имеет также большое
значение и при количественном анализе, поскольку часто для каж-
каждого компонента смеси можно найти отдельные полосы, на которые
другие составляющие либо совсем не влияют, либо их влияние
незначительно. Если же влияние обнаруживается, то обычно можно
с помощью стандартных методов провести коррекцию.
Другое преимущество состоит в том, что из качественного спектра
можно получать полуколичественные результаты. Этот факт не учитыва-
учитывается многими исследователями, которые используют аналитические
методы от случая к случаю и для качественного и полу количествен-
количественного анализов привлекают, порой без особой необходимости, одно-
одновременно ИК-спектры и газовую хроматографию.
Количественную ИК-спектроскопию можно использовать для иссле-
исследования крайне широкого круга объектов: она не ограничивается
летучими, кристаллическими или растворимыми веществами. Во многих
случаях нет необходимости в трудоемком приготовлении образца.
Метод применяется для решения самых разнообразных задач, таких,
как определение ненасыщенности полиэтилена [93], частиц кварца
в воздухе [107], D2O в Н,О [36], СО, в вине [24], СО в крови
и сердечной мышце [76], а также жиров, белков и сахара в мо-
молоке [1].
К недостаткам метода относится: опасное случайное перекрывание
полос, прозрачность гомоядерных молекул, время, которое приходится
затрачивать на приготовление образца, необходимость в первичных
стандартах, отсутствие возможности переноса калибровочных данных с
одного спектрофотометра на другой и (в некоторых случаях) недо-
недостаток чувствительности. Как количественный метод анализа многих
газов и летучих жидкостей газовая хроматография заменила ИК-
спектроскопию, что обусловлено ее удобством, точностью, быстротой
и чувствительностью. Спектроскопия ЯМР, так же как и масс-спектроме-
трия, дает уникальные возможности для исследования некоторых типов
жидких и растворимых веществ. Какой из этих методов выбрать
в каждом отдельном случае, зависит от анализируемой системы,
Количественные приложения 233
доступности приборов, требуемой точности и количества вещества,
необходимого для приготовления образца.
При планировании количественного ИК-анализа нужно задать
вопрос о необходимой точности определения. Обычный ответ — «как
можно точнее» - является совершенно бессмысленным утверждением.
Небольшое предварительное тестирование обычно выявляет реальные
пределы ошибок определения, а когда это сделано, можно переходить
к дальнейшему анализу. Эта точка зрения подчеркивается из-за
того, что время, сложность и стоимость анализа примерно про-
пропорциональны желаемой точности, и было бы неразумно выдавать
результаты, точность которых выше, чем это требуется. Полуколичествен-
Полуколичественный анализ, отвечающий относительной точности около + 10%, про-
провести не сложно, и для этого требуются только отдельные стандарты
чистых компонентов (а в некоторых особых случаях стандарты не тре-
требуются вовсе). Для проведения количественного анализа с точностью
порядка ± 1 % необходимо соблюдать ряд мер предосторожности, обыч-
обычно включающих тщательное приготовление стандартных смесей. При
благоприятных условиях можно добиться последующего увеличения точ-
точности, но при этом требуется исключительное внимание к деталям
анализа и максимальная тщательность в работе.
В дальнейшем кратко будут рассмотрены стандартные методы,
общие для ИК-анализа, а затем более полно — некоторые специальные,
такие, как определения с высокой точностью, анализ следов и струк-
структурно-групповой анализ.
Это вступление будет неполным, если не вспомнить часто
повторяемую истину о том, что анализ не может быть лучше
образца. Рассмотрение методов отбора образцов из больших количеств
веществ выходит за рамки этой главы и часто находится вне сферы
деятельности аналитика, но за правильное перемешивание и отбор пробы
из образца, представленного для ИК-анализа, несет ответственность
спектроскопист. Не следует предполагать, что жидкости, газы и порошки
однородны, и поэтому при подготовке пробы необходимо тщательное
перемешивание. Когда имеется достаточное количество вещества, то
нужно взвесить по крайней мере 1 г для приготовления раствора,
даже если будет использована только малая часть этого количества.
Подготовка образцов твердых веществ и полимеров представляет
собой более трудную задачу, и часто требуется большое искусство
для получения представительной (характеризующей вещество в целом)
пробы. Валтон и Гоффман [112] собрали библиографию, относящуюся
к методам подготовки образцов различных веществ.
234 Глава 6
Законы поглощения
Физические законы, которым подчиняется поглощение излучения,
детально обсуждаются в литературе [79], и поэтому здесь ограничимся
только определением терминов*.
Закон Бугера
Закон, первоначально установленный Бугером [18], а позже незави-
независимо от него Ламбертом, связывает поглощение образца с его толщиной.
Согласно ему, каждый слой равной толщины поглощает постоянную
часть энергии излучения, проходящего через него. Этот закон точен для
однородного образца и математически записывается как
-?-<А F-1)
где / — интенсивность или энергия излучения, Ь — единица толщины.
Множитель а, — константа пропорциональности, связанная с поглоща-
тельной способностью образца. В интегральной форме это соотноше-
соотношение имеет вид
1пА = а,Ь, F.2)
где /0 — интенсивность падающего, а /- интенсивность прошедшего
излучения.
Закон Бера
Соотношение между поглощением монохроматического света веще-
веществом и его концентрацией в гомогенной среде было сформулировано
Бером [14]. Он установил, что поглощение света частицами вещества
пропорционально их концентрации с:
или
ln-j- = асс. F.4)
* При переводе английских терминов, рекомендуемых ГОРАС [1], возникает
целый ряд трудностей, чем в какой-то степени объясняется отсутствие
единой терминологии в отечественной литературе. Имеющийся же ГОСТ
[2] на термины и определения по физической оптике часто противоречит
международным рекомендациям [1]. В переводе все дано в терминах и обозна-
обозначениях автора, за исключением слова «absorbance», переведенного наиболее
распространенным в нашей стране термином «оптическая плотность».—
Прим. ред.
Количественные приложения 235
Закон Бугера — Бера *
Комбинируя соотношения, можно написать
-\g— = \g— = abcc' = A. F.5)
Для удобства в дальнейшем концентрацию представим в виде произ-
произведения двух коэффициентов: с — доли исследуемой составной части
в образце и с' — концентрации образца в аналитическом растворе;
lg(/0//) обычно называют оптической плотностью А, константу а —
коэффициентом поглощения. Пропускание определяют как отношение
1/10, а процент пропускания — как 100///0- Обычно используются деся-
десятичные логарифмы, а коэффициент 2,3 включают в константу а.
Уравнение F.5) является основным в количественной спектрофото-
метрии. Для однородной смеси или раствора оно устанавливает прямо-
прямолинейную графическую зависимость оптической плотности от кон-
концентрации или толщины данного образца. Кроме того, из него следует,
что при одинаковой длине волны оптические плотности полос погло-
поглощения различных частиц образца суммируются (закон аддитивности).
Отклонения от законов поглощения
Часто происходят отклонения от уравнения F.5), но необходимо,
чтобы они не влияли на точность количественного анализа, если
их происхождение известно. Причины этих отклонений могут быть
внешними (несовершенство спектрофотометра и кювет) и внутренними
(взаимодействия в изучаемой системе).
Вероятно, наиболее важным фактором, вызывающим отклонение от
закона Бугера — Бера, является конечная ширина щелей, которые, естест-
естественно, имеются во всех спектрофотометрах. Как было показано ранее
(гл. 2), совместное действие ширины щели и аппаратной функции
спектрометра заключается в том, что на приемник попадает не монохро-
монохроматическое излучение, а скорее некоторый интервал длин волн. Кроме
того, этот интервал расширяется, если для снижения уровня шума раскры-
раскрывают щели. Так как закон [уравнение F.5)] справедлив только для
монохроматического излучения, то при ширине щели, большей, чем
ширина полосы, возникают ошибки. Рамсэй [90] составил таблицы
отношений истинных оптических плотностей в максимуме к наблюда-
наблюдаемым при различных оптических плотностях и ширинах щелей для
лорентцевых контуров полос. Например, при оптической плотности 1 и
отношении ширина щели/ширина полосы S:Avi/2=0,2 А (истин.):
:/4(набл.) = 1,03; для S:Avlj2 = 0,5 А (истин.): А (набл.) = 1,24. Ясно, что
* Как принято в- основном в отечественной литературе, правильнее говорить
о законе Бугера — Ламберта — Бера. — Прим. перев.
236 Глава 6
даже небольшие изменения ширины щели (или аппаратной функции)
спектрофотометра, на котором проводится количественный анализ, будут
влиять на наклон и линейность графика зависимости оптической плот-
плотности от концентрации.
Другими факторами, связанными с прибором и вносящими вклад
в отклонения от закона Бугера — Бера, являются потери на отражение
и рассеяние в образце, эффекты отражения луча и ошибки в установке
нулевой линии спектрофотометра. Позже эти факторы будут обсуждаться
подробнее.
Систематические ошибки возникают в том случае, если поглощаю-
поглощающие частицы не сохраняют свою идентичность в гомогенном растворе.
Обычно этот эффект включает межмолекулярную ассоциацию, особенно
водородную связь и мицеллообразование. Количественный анализ
ассоциатов будет обсуждаться позже.
Некачественное растирание твердых образцов, спектры которых за-
записывают в виде порошков, может привести к "значительным
отклонениям от закона Бера, так как «острота» полос сильно зависит
от дисперсности порошка (стр. 264 — 266).
Практика количественного анализа
В течение 1957 - 1960 гг. в журнале "Analytical Chemistry" было
опубликовано 150 различных методик количественного анализа, разра-
разработанных при финансовой поддержке общества Кобленца. Последую-
Последующие работы такого рода публикуются в журнале "Applied Spectroscopy".
Не следует проводить разработку новых методик количественного
анализа без предварительного ознакомления с указателем методов в
"Applied Spectroscopy" [103].
Количественный анализ методом ИК-спектроскопии выполняется
прямым или косвенным сравнением оптической плотности неизвестного
вещества при данной длине волны (часто в максимуме интенсивной
полосы поглощения) с оптической плотностью того же вещества
известной стандартной концентрации. Для расчетов наиболее полезным
параметром является оптическая плотность в максимуме, так как она
легко измеряется и прямо связана с концентрацией. Необходимо избе-
избегать измерений оптической плотности на краях полос, так как даже очень
маленькие ошибки в воспроизводимости длин волн приводят к большим
изменениям поглощения. Для анализа можно использовать любую
полосу (сильную или слабую) при условии, что концентрация раствора
и толщина кюветы выбраны таким образом, что оптическая плотность
попадает в оптимальный интервал. Целесообразно выбирать полосы,
минимально перекрывающиеся с другими полосами в спектре.
Согласно уравнению F.5), оптическая плотность пропорциональна
логарифму пропускания (с обратным знаком. — Прим. ред.) — функции,
неудобной для использования в серийных измерениях. При расчете опти-
оптической плотности с бланка может возникнуть желание непосредственно
Количественные приложения 237
Рис. 6.1. Эквивалентные уровни шума в линейной шкале для следующих
оптических плотностей: 0 fa), 1 F) к 2 (в).
зарегистрировать ее в линейном масштабе почти так же, как это
делается в УФ-спектрофотометре, что упрощает получение этих данных.
Кроме того, в силу аддитивности оптической плотности ее полная
величина при данной длине волны является просто суммой отдельных
оптических плотностей. Однако существует несколько причин, по
которым в ИК-спектрофотометрах данные представляются в виде
процента пропускания. Прежде всего электрический сигнал, который
подвергается усилению для регистрации, линеен по отношению к
проценту пропускания. Вполне возможно так видоизменить выход
усилителя посредством механического или электрического кулачка, что
перо будет непосредственно регистрировать оптическую плотность.
Обычно этого не делается, поскольку в ИК-области, где энергия и так
ограничена (рис. 6.1), при больших оптических плотностях будет сильно
увеличиваться уровень шума. Другая причина использования линей-
линейной шкалы процентов пропускания заключается в том, что в этом слу-
случае интенсивность более слабых полос увеличивается, а сильных -
уменьшается, и даже самые интенсивные полосы не достигают конца
шкалы.
Оптические плотности можно измерять, используя обычную милли-
миллиметровую шкалу и беря логарифм отношения 10/1 (рис. 6.2). В более
простом методе применяется прозрачная масштабная линейка, откалиб-
рованная в оптических плотностях. Точка А = оо совмещается с
действительной точкой 0% пропускания бланка, а величины /0 и /
считываются по шкале. Разность между этими числами и есть
оптическая плотность полосы. Тот же принцип реализуется на бланках
спектрофотометров с нанесенными по ординате оптическими плотно-
плотностями (рис. 6.2).
Измерения интегральных интенсивностей, которые определяются
как площадь, заключенная под контуром полосы, редко используются
для количественного анализа по той причине, что оптические плотности
в максимуме можно измерить гораздо точнее и воспроизводимо,
238
Глава 6
0.530
Рис. 6.2. Измерение оптической плотности с использованием миллиметровой
шкалы (а) и определение оптической плотности непосредственно на бланке (б).
по крайней мере на одном спектрофотометре. Кроме того, точные
измерения интегральных интенсивностей требуют либо линейной орди-
ординаты оптической плотности, либо утомительного ручного пересчета
величин процента пропускания в оптические плотности.
Для количественного анализа точка 0% пропускания должна быть
установлена по возможности наиболее точно еще до проведения из-
измерений пропускания. Ее нужно совместить с линией А = оо (или
0% пропускания) на бланке (однако вместо 100% пропускания лучше
устанавливать 90 — 95 %). На двухлучевых спектрофотометрах с оптиче-
оптическим нулем точно установить нуль трудно. Когда канал образца
перекрыт, оптический клин движется к 0 % пропускания и, следовательно,
для активации сервосистемы либо энергии недостаточно, либо ее совсем
нет. Движение пера в этой области затруднено, и поэтому нуль не
может быть установлен с большой точностью. В спектрофотометрах,
регистрирующих отношение электрических сигналов, эта проблема,
конечно, отсутствует. В качестве первого шага при установлении
нуля нужно убедиться в том, что дрейф нуля пренебрежимо мал; это
значит, что, когда перо находится в среднем положении и оба
канала полностью перекрыты, оно должно совершать движения только
в пределах обычного шума. Затем обе заслонки удаляются и в канал
образца вновь очень медленно вводится заслонка, чтобы по мере
приближения пера к нулю не проскочить его из-за инерционности
Количественные приложения 239
сервосистемы. Заслонку лучше изготовить из такого материала, который
непрозрачен для исследуемой области спектра, но, как указывалось
ранее (стр. 47 — 49), пропускает более высокочастотное излучение.
В этих же целях можно использовать толстый слой образца (в
10 — 100 раз толще, чем обычно) или другое вещество с сильным
поглощением в области данных длин волн (табл. 2.1), чтобы записать
полосу с фактически бесконечной оптической плотностью. Такие полосы
нужно сканировать с очень маленькими скоростями для уменьшения
ошибки из-за проскакивания пера. Другой метод, дающий, как
считается, более высокую точность количественных измерений, рассмат-
рассматривается ниже.
Полуколичественный анализ
Одним из преимуществ использования растворов в качестве образ-
образцов является возможность полуколичественной обработки спектров.
При этом спектрофотометр должен быть хорошо отрегулирован и отъ-
отъюстирован. Загрязненные вещества или компоненты смесей обычно
можно анализировать с относительной точностью ±10% или лучшей,
если для сравнения использовать чистые вещества.
Как было показано, измерение оптической плотности включает
измерение не только пропускания (/) в максимуме полосы поглощения,
но и интенсивности фона A0). Эту точку измерить точно бывает трудно,
но обычно можно удовлетворительно провести «базовую» линию
[51, 88, 121]. Этот метод определения /0 — в лучшем случае только
приближение, но он легко осуществляется, воспроизводим и широко
применяется. Аналитическая полоса регистрируется полностью, и через
ее края, где нет Поглощения, проводится прямая базовая линия
(рис. 6.3, а). Однако по-настоящему преимущество метода проявляется
в том случае, когда измеряемая полоса попадает на плечо другой
полосы поглощения (рис. 6.3,6) и базовая линия проводится в виде
касательной к зарегистрированной кривой, как показано на рисунке.
Обычно в методе базовой линии труднее всего определять точки
краев. Базовую линию нужно проводить как можно ближе к вообра-
воображаемому следу пера, если бы полоса отсутствовала. Иногда это
приближение может быть очень плохим (как для полосы b на
рис. 6.3, в). Когда проводится определение с использованием серии
стандартов и строится калибровочная кривая, действительное положение
точек краев базовой линии не имеет большого значения, если
только они используются одинаковым образом и при условии, что
не возникает новых мешающих полос. Любая систематическая ошибка
автоматически компенсируется калибровочной кривой. Методы про-
проведения базовой линии обсуждаются Поттсом [88].
Если полуколичественный анализ проводится от случая к случаю
с использованием спектров чистых веществ в качестве стандартов,
то из-за неоднозначного проведения базовой линии могут возникать
240
Глава 6
il
р
il
Ч
¦й W
¦f - - vf ТТ
ml
H
г
x~
11
¦f
rrq-
§?!
1
ц
r.
r
1
к.
4г
.if!
: i :
W
и
I ¦.;:
Г
f
Л#:-;:
й
f::'
Рис. 6.3. а — проведение базовой линии для изолированной полосы; б — проведе-
проведение базовой линии в случае перекрывающихся полос; в — базовая линия,
плохо аппроксимирующая фон.
довольно большие ошибки. При этом можно поддаться соблазну и
нарисовать фон, который предположительно соответствует действитель-
действительности. Эту процедуру можно сделать несколько менее субъективной,
предположив, что перекрывающиеся полосы имеют лорентцевы или по
крайней мере симметричные контуры. Тогда серию полос можно пере-
перевести на кальку так, что при их суммировании получается наблюдае-
наблюдаемый контур. Однако такие оценки не могут быть точнее, чем
метод базовой линии, и, конечно, менее воспроизводимы.
Количественный анализ средней точности
Как отмечалось, одна из принципиальных причин отклонения от
закона Бера обусловлена конечной шириной щели ИК-спектрофотометра.
Тем не менее оптимальную точность при количественном анализе
получают, раскрывая щели по крайней мере в 2, а предпочтительнее
в 3 раза. Причина этого, если сказать кратко, заключается в том,
что можно справиться с отклонениями от закона Бера, но не с шумами
спектрофотометра. Лучший способ повышения отношения сигнал/шум
Количественные приложения
241
929
Установка разрешения
970 980
1000
10 20 30 40 50
Ширина щели (в мкм при 3650см)
60
Рис. 6.4. Зависимость ошибки оптической плотности от ширины шели и
ошибка установки точки 0% пропускания [83].
/ - ошибки установки нуля пропускания учтены; 2 - ошибки установки нуля пропускания исключены.
состоит в расширении щелей [см. уравнение B.7) в гл. 2: S/N ~ w2],
при этом приходится мириться с некоторым ухудшением контура
полос, возникающим в результате увеличения щелей [83, 88]. Факти-
Фактически при этом для хорошего спектрофотометра с решеткой потеря в
разрешении будет совсем небольшой, так как обычная ширина
щели меньше, чем ширина полос большинства жидкостей.
Для точной количественной работы важна воспроизводимая (но не
обязательно точная) установка нуля. Для проведения этой операции
на двухлучевом спектрофотометре с оптическим нулем Перри [83]
рекомендовал использовать сетку с пропусканием около 15%.
Сетка тщательно очищается и помещается в держатель так, чтобы ее
можно было воспроизводимо вводить в канал образца на довольно
Т6 - 586
242
Глава 6
2 1,3 1 08 0,6 О/
0.2
I I il
Т 1 1
16
Рис. 6.5. Зависимость относительной ошибки концентрационного отношения
х от пропускания интенсивной полосы Г для х =0,1, 0,2, 0,5 и 1 (АГ=0,01).
Пуштирная лши1- хривм Твимаяа-Лотиана дл» относительной ошибки определения концектрацни по
изолированной полосе [77]. Reproduced, with permUsion, from Applied Spectroscopy.
большом расстоянии от фокуса луча. С сеткой обращаются так же
тщательно, как с оптическим элементом, т. е. не прикасаясь к опти-
оптической части. Ее пропускание определяется приблизительно; с этого
момента цифра считается точной и, согласно ей, устанавливается нуль
Количественные приложения 243
спектрофотометра. Любое различие между точками истинного и
предполагаемого 0% пропускания автоматически учитывается в калиб-
калибровочной кривой. Если спектрофотометр был настроен для данной
серии измерений, то регулировка не должна изменяться до окончания
измерений. Резкое влияние ширины щели и установки нуля на ошибку
оптической плотности показано на рис. 6.4.
Линию фона или Jo можно установить, применяя уже обсуждав-
обсуждавшийся метод базовой линии, наполняя кювету образца чистым раст-
растворителем (либо используя аналогичную кювету сравнения с раствори-
растворителем) или, если возможно, выбирая участок спектра, где нет погло-
поглощения. И вновь любая разность между измеренным и истинным /0
будет компенсироваться калибровочной кривой.
В частном случае двухкомпонентной смеси или в тех случаях,
когда концентрация одного компонента постоянна, можно использовать
отношение оптических плотностей двух полос вместо рабочих кривых,
построенных по исправленным оптическим плотностям. Отпадает необ-
необходимость измерения толщины кюветы и концентрации, так как эти
величины сокращаются. Такой способ применим к полимерным плен-
пленкам, а также к спектрам НПВО жидкостей и растворов.
Из-за быстрого изменения показателя преломления жидкости вблизи
полосы поглощения количественный анализ нужно проводить в разбав-
разбавленном растворе предпочтительно с концентрацией 2 вес.% на объем
или менее [42]. Образцы взвешивают в мерных колбочках (стр.
84—88) с соответствующей точностью и немедленно доводят раство-
растворителем до метки. Аналитические полосы должны быть выбраны пре-
преимущественно изолированными, т. е. они не должны перекрываться по-
полосами других компонентов или растворителя. Это не должны быть
самые сильные полосы в спектре. Для получения минимальной
ошибки отсчета оптической плотности толщину кюветы нужно по-
подобрать таким образом, чтобы оптическая плотность имела величину
около 0,43 (или 37 % пропускания); на практике вполне удовлетворитель-
удовлетворительным является интервал ОД —0,7 B0 — 60% пропускания). Если исполь-
используется метод измерения относительной оптической плотности, то
оптимальная величина зависит от относительных интенсивностей двух
полос, как показано на рис. 6.5. Материалы окон кюветы нужно
выбирать так, чтобы их показатель преломления соответствовал по-
показателю преломления растворителя. Толщину кюветы можно определить
либо по интерференционной картине, либо сравнивая с оптической
плотностью полосы в спектре доступной неразбавленной жидкости,
измеренной в кювете, прокалиброванной интерференционным методом.
Тогда оптическая плотность А данной полосы будет пропорциональна
толщине кюветы t, т. е.
А2 t2
где индексы 1 и 2 относятся к двум кюветам. Естественно, что
16*
244 Глава 6
при этом все параметры прибора должны находиться в оптимальном
режиме, как и в случае количественных измерений.
При повседневном количественном анализе, когда известен состав
всех компонентов, любая из двух систем может быть использована
для получения значений оптических плотностей полос (которые нужно
измерить). Перед началом сканирования спектрофотометр обычно уста-
устанавливают на ту начальную длину волны, при которой поглощение
отсутствует, и проводят медленную запись всей полосы поглощения.
При очень большой скорости сканирования перо будет воспроизводить
контур полосы не точно и оптическая плотность окажется слишком
низкой. Преимущество сканирования состоит в том, что можно зафикси-
зафиксировать неожиданные сдвиги максимума поглощения или присутствие
постороннего поглощения, которое может влиять на измерение.
В методе измерения по одной точке устанавливается длина волны
максимума полосы и значения оптической плотности считываются со
шкалы ординат. Одним из преимуществ такого способа является
электрическая фильтрация с большой постоянной времени и усреднение
флюктуации шума по гораздо более длительному периоду времени,
чем это возможно при сканировании. Кроме того, полное время,
необходимое для выполнения такого измерения, может быть не-
несколько меньше. Очевидно, усилия, затраченные на эти измерения,
оправдывают использование метода только для рутинного анализа.
Время от времени нужно записывать спектры образцов, измеряемых
этим методом, для того, чтобы обнаружить любые неожиданные из-
изменения в их составе.
Измерения по точке легко проводить на однолучевых спектрофото-
спектрофотометрах, и, по-видимому, точность таких измерений несколько лучше, чем
в случае двухлучевых спектрофотометров.
Выводы
Предлагаемая ниже последовательность действий может служить
руководством по количественным ИК-измерениям. Естественно, что не
существует универсальной методики, и, чтобы видоизменить или модифи-
модифицировать метод в нужном направлении, необходимы опыт и мастерство
спектроскописта. Предполагается также, что спектроскопист уже рассмот-
рассмотрел различные аналитические возможности для решения его задачи и
решил, что ИК-спектроскопия — наиболее полезный метод. Развитие
метода может быть основано на следующей схеме:
Формулировка задачи. Сколько компонентов присутствует? Какой
из них нужно определить? С какой точностью? Какие концентра-
концентрационные пределы предполагаются для каждого компонента? Эта стадия
включает запись ряда предварительных спектров для получения
полуколичественной картины системы.
Выбор оптимальных условий. Записать спектры каждой составной
части. Выбрать аналитические полосы с минимальным перекрыванием.
Количественные приложения 245
Подобрать такую комбинацию толщины кюветы и концентрации(ий),
чтобы поглощение растворителя было пренебрежимо мало, а пропускание
аналитических полос составных частей при средних концентрациях
попадало бы в область 20 — 60%. Подобрать несколько лучшее отноше-
отношение сигнал/шум, чем требуется, чтобы удовлетворить точности определе-
определения; установить ширину щели или программу (обычно в 2 — 3 раза
большую, чем нормальная), отрегулировать чувствительность спектрофо-
спектрофотометра и для нахождения оптимальной проверить несколько скоростей
сканирования.
Приготовление стандартов. Для каждого компонента требуются по
крайней мере четыре стандарта. Проверить линейность графика зависи-
зависимости оптической плотности от концентрации. Если нужно определить
несколько компонентов, то следует приготовить смешанные стандарты.
Проверить закон аддитивности. Выбрать наилучший метод расчета
концентраций. Если необходимо, то определить поправку на перекры-
перекрывание полос.
Запись спектров образцов. Использовать такой режим работы прибора
и подготовки образца, как и при работе со стандартами. Необходимо
часто проверять точность по стандартам или известным образцам. Ста-
Статистически оценить результаты.
Количественный анализ высокой точности
Развитие методов, описанных выше, может привести к повышению
точности. Уже отмечалось значение правильного приготовления образ-
образцов, но важно помнить о следующем: чтобы добиться, скажем,
точности анализа 0,5%, образец должен быть представительным и од-
однородным по крайней мере в такой же степени, даже если пределы
других ошибок не даны. При стремлении к большей точности необ-
необходимо иметь наиболее представительный образец, и во многих слу-
случаях точность анализа ограничена потерей однородности образца.
В ходе очень точных работ могут проявляться ошибки, обуслов-
обусловленные испарением. Это на самом деле так, если растворенное
вещество имеет высокое давление пара при комнатной температуре, в
результате чего концентрация может быстро изменяться в зависимости
от времени пребывания на воздухе. Если с образцами и стандартами
обращаться аналогичным образом, то ошибки, связанные с подготовкой
пробы и испарением, будут оказывать примерно одинаковое влияние.
Сходные проблемы возникают при работе с веществами, чувстви-
чувствительными к влаге и кислороду. В зависимости от условий может
потребоваться работа в сухой камере и даже в инертной атмосфере.
При работе с образцами, реагирующими с водой, могут потребовать-
потребоваться специальные меры предосторожности, включая использование герме-
герметичных и хорошо осушиваемых стеклянных сосудов и кювет. В любом
случае такие вещества должны находиться в минимальном контакте
с атмосферой. Целесообразно также применять среднеразбавленные
246 Глава 6
растворы с концентрацией порядка 2 %, а все сосуды должны быть по
возможности заполнены и тщательно закрыты. Как будет показано
в следующем разделе, посвященном ошибкам, оптическая плотность
чувствительна к температуре образца.
Ранее отмечалось, что раскрытие щели спектрометра в w .раз
делает возможным понижение уровня шума в w2 раз. Дальнейшего
повышения точности анализа можно добиться, используя разностную
спектроскопию в сочетании с растяжкой по ординате.
Разностная, или двухлучевая, спектроскопия с компенсацией стандарт-
стандартным образцом (иногда называемая дифференциальной*) применялась в
УФ- и видимой областях в течение многих лет, но в ИК-области не
имела широкого распространения. Такое положение сложилось по двум
причинам: до недавнего времени возможности спектральных прибо-
приборов были сильно ограничены, а очень маленькие толщины образцов,
используемые в ИК-области, делают точную компенсацию трудной.
Тем не менее для анализов с высокой точностью или для некоторых
типов анализов следовых количеств техника разностной спектроскопии
является непревзойденной.
Здесь отдается предпочтение термину «разностные спектры», так
как «дифференциальные» относятся к бесконечно малым разностям и
поэтому не точно отражают положение вещей. Кроме того, его можно
спутать со спектроскопией производных, которая имеет дело с
первой или второй производной кривой поглощения [28].
Из дополнительных приспособлений к ИК-спектрофотометру, исполь-
используемому в сочетании с разностной спектроскопией, особенно полезно
устройство для растяжки по ординате. Оно имеется в продаже или его
можно сконструировать в лаборатории [113]. Это приспособление
производит электрическое умножение выходного сигнала, так что ампли-
амплитуда отклонения пера увеличивается в 5, 10 или 20 раз и т. д. по
желанию. Увеличение амплитуды сигнала полезно только при условии
сохранения прежнего незначительного уровня шумов. Для этого необ-
необходимо увеличить ширину щели в корень квадратный раз из
коэффициента растяжки и понизить усиление, чтобы ограничить уро-
уровень шума. Растяжка по ординате наиболее эффективно применяется
в коротких интервалах длин волн, так как крайне трудно настолько
хорошо отрегулировать кюветы, чтобы проводить сканирование в
широкой области.
Теперь предлагается сам метод точного количественного анализа.
Предположим, что нужно проанализировать примерно 40% А в В.
* В отечественной литературе нет устоявшейся точки зрения на эти тер-
термины. На этот вопрос обратили внимание Бернштейн И. Я. и Камин-
Каминский Ю. Л. [3]. За последние годы существенно развилась еще и двухволновая
спектрофотометрия в видимой н УФ-областях спектра. Здесь сохранена термино-
терминология оригинала. — Прим. перев.
Количественные приложения
247
Рис. 6.6. Определение [H(CH3)SiO] в [(CH3JSi0]x методом разностной спектро-
спектроскопии при S-кратной растяжке.
а —разностный метол. Кривая / — образец в сопоставления с 2,82% стандартом; крива» 2 — образец
в сопоставлении с 2,98% стандартом; кривая 3 — образец в сопоставлении с 3,19% стандартом; 6 —метод
перекомпенсацин; S означает, что в канале сравнения находится стандарт, а в канале образца - ис-
исследуемый раствор. Для кривых, не отмеченных буквой S, места стандарта и раствора изменены.
Готовятся стандарты, содержащие 38, 39, 40, 41 и 42% А. Устанав-
Устанавливая анализируемый образец в канал образца и каждый из стандартов
последовательно в канал сравнения, получаем серию спектральных
кривых, показывающих только разницу поглощения исследуемого об-
образца и стандартов. Используя растяжку по ординате, можно так уве-
увеличить эту разность, что маленькие различия в составе будут
отражаться в виде больших различий в спектре (рис. 6.6, а). Если,
например, используется 10-кратная растяжка и одновременно понижен
нормальный уровень шума в 5 раз, то нужно увеличить геометриче-
геометрическую ширину щели в у 10¦ ]/5 = ~ 7 раз (и соответственно уменьшить
усиление). Предельная точность фактически достигается тогда, когда
ширина щели увеличена до такой величины, что изображение
выходной щели монохроматора полностью покрывает приемную площад-
248 Глава 6
ку детектора и достигает ее края [88, гл. 7]. На калибровочный
график обычно наносят расстояние между максимумами кривых, так как
при растяжке по ординате шкала оптических плотностей теряет смысл.
Точность можно повысить в два раза и за счет другого фактора,
применяя метод перекомпенсации [92]. Первый раз сканирование
проводят, как было описано выше, а во время повторной записи
образцы меняют местами. Кюветы остаются в прежнем положении.
Разность при этом удваивается по сравнению с одним сканированием
(рис. 6.6,0). Имеющиеся расхождения в пропускании кювет при этой
процедуре автоматически компенсируются. В разностной спектроскопии
можно достичь точности порядка 0,1 % и выше. Еще в 1953 г. разност-
разностный метод был использован для определения циклогексана (содер-
(содержание 85 %) в нефтяных концентратах с воспроизводимостью и
точностью 0,1 % [46].
Вместо приготовления стандартных растворов Перри [83] предложил
применять такие нейтральные светофильтры, как сетки. Подходят так-
также вращающиеся калиброванные секторы [66]. При работе с сетками
важно помнить следующее:
1. Сетки должны иметь оптическую плотность 0,3-0,4 D0-50%
пропускания). Более высокие оптические плотности получают комбина-
комбинацией этих сеток.
2. Обращаться с сетками нужно как с оптическими элементами,
т. е. предохранять их от пыли и отпечатков пальцев.
3. Установка сеток должна быть воспроизводимой, и они должны
размещаться в канале сравнения достаточно далеко от фокуса.
4. Быть уверенным в том, что сетки не повернуты ни относительно
друг друга, ни относительно щели.
Оптическая плотность каждой сетки тщательно измеряется несколько
раз при оптимальных условиях, выбранных для количественного анализа,
а затем берется усредненное значение. Любая неучтенная ошибка
компенсируется калибровочной кривой. Сетки используются как в ком-
комбинации друг с другом, так и поодиночке.
Несмотря на то что компенсация в канале сравнения проведена,
спектры стандартных образцов должны регистрироваться при проведе-
проведении каждого анализа, если производится растяжка по ординате.
Во время определения параметры прибора должны оставаться не-
неизменными.
Для проведения анализа методом разностной спектроскопии пред-
предполагается следующий общий порядок:
1. Записать спектры образца и искомых компонентов.
2. Выбрать аналитическую длину волны, на которой влияние различ-
различных мешающих факторов минимально.
3. Установить толщину и концентрацию раствора такими, чтобы
пропускание аналитической полосы было 30-40%. Обоснование такого
выбора дано Поттсом [88, гл. 7].
4. Подобрать параметры прибора и особенно ширину щели, чтобы
Количественные приложения 249
получить требуемую воспроизводимость (precision)* оптической плот-
плотности.
5. Приготовить стандарты, включая ожидаемую концентрацию
образца.
6. Записать спектр в пределах аналитической полосы, поместив
кювету с анализируемым веществом в канал образца, а соответствующие
стандарты в канал сравнения.
7. Если разница между спектрами слишком мала, то определить,
в какой степени нужно растянуть ординату, чтобы получить требуемую
воспроизводимость.
8. Повторить п. 5, используя растяжку по ординате. Увеличить
ширину щели в корень квадратный раз из коэффициента растяжки
(и понизить усиление, добившись нормальной чувствительности пера).
9. Построить калибровочную кривую и определить неизвестную
концентрацию.
Источники ошибок
в количественном анализе
Цель настоящего раздела — обратить внимание на некоторые из
возможных источников ошибок в количественном ИК-анализе и пред-
предложить методы их минимизации. В некоторых случаях, по крайней
мере в простом случае однокомпонентных определений, можно оценить
величину каждого из источников в общую ошибку. Труднее оценить
ожидаемую воспроизводимость при анализе многокомпонентных смесей.
В дальнейшем предполагается, что явных ошибок удалось избежать
и что погрешности взвешивания и калибровки мерной колбы пре-
пренебрежимо малы.
Толщина кюветы определяется достаточно точно из интерференци-
интерференционной картины (стр. 130), но некоторая погрешность может возникнуть
либо случайно, либо из-за неточности в калибровке спектрофотометра.
Представим, например, что кювета толщиной 0,100 мм дает 20 макси-
максимумов в интервале 835 — 1835 см. На абсциссе с линейной шкалой час-
частот сдвиг бумаги или постоянная ошибка в калибровке, скажем на 5 см,
не будет влиять на вычисленные толщины. Однако нелинейная ошибка
в калибровке на +5 см при 1835 см и на —5 см при 835 см даст
расчетную толщину 0,101 мм, или ошибку в 1 %. В то же время
при использовании линейной шкалы длин волн смещение диаграммной
бумаги на 0,025 мкм приведет к ошибке в толщине кюветы в 0,7%
* См. [4], где приведены рекомендуемые Научным советом по аналитиче-
аналитической химии АН СССР термины, определения и обозначения метрологических
характеристик анализа вещества. В данной книге „precision" переводилось и как
„точность", н как „воспроизводимость" в зависимости от смысла. — Прим. ред.
250 Глава 6
и нелинейная ошибка в калибровке на +0,025 мкм при 5,45 мкм
и на —0,025 мкм при 11,98 мкм даст ошибку в 1 %.
Иногда кажущаяся толщина кюветы меняется от одного спектрофо-
спектрофотометра к другому. Этот эффект наблюдается на кюветах с неплоскими
и непараллельными окнами в результате того, что луч спектрофото-
спектрофотометра будет пересекать различные рабочие участки окон. При этом
длина оптического пути для чечевицеобразной или клиновидной полости
будет различаться по мере перемещения от центра к краю окна.
На хороших кюветах с отклонением окон от плоскостности на три-
четыре длины волны света натриевой лампы такого эффекта не
наблюдается.
Крайне желательно работать в тех областях спектра, где поглоще-
поглощение растворителей пренебрежимо мало. Если полос поглощения раство-
растворителя избежать нельзя, то иэ-за их неточной компенсации могут воз-
возникнуть ошибки. Этот эффект становится более заметным для кон-
концентрированных растворов и областей сильного поглощения раство-
растворителей [42].
Даже если растворитель и не поглощает, пропускание кювет образца
и сравнения, вероятно, должно различаться по уже обсуждавшимся
причинам. Коррекция часто выполняется, особенно в методе „cell-in-
cell-out" (кювета вводится и выводится из светового пучка), следующим
образом. При каждой аналитической длине волны измеряется разница
в оптической плотности двух кювет, когда они заполнены раствори-
растворителем. После подсчета разности в пропускании кюветы образца, когда
она заполнена чистым растворителем и исследуемым раствором при
некоторой длине волны, где нет поглощения, вносится соответствующая
поправка в величину оптической плотности образца. Эти наблюдения
были выполнены автором, однако даже такая процедура не дает полной
коррекции в случае загрязнения окон кюветы, и единственным средством
получения приемлемых данных является замена кюветы образца.
Мартин [75] оценил величину ошибки, возникающей из-за разности
пропускания кювет, в зависимости от самого пропускания.
До тех пор пока показатель преломления растворителя точно
не согласован с показателем преломления материала окон, будет иметь
место некоторая потеря света из-за отражения на границе раствор -
поверхность окна. Постоянная потеря энергии на отражение не столь
существенна, важнее быстро изменяющиеся потери, которые могут
происходить по нескольким причинам.
Хорошо известно, что ..показатель преломления жидкости резко
меняется вблизи сильной полосы поглощения. Когда записывают
спектры неразбавленных жидкостей или концентрированных растворов,
в некоторых областях спектра почти наверняка должны быть значи-
значительные изменения в отражении. Рис. 6.7 иллюстрирует изменение
показателя преломления хлороформа около полосы 760 см [50] [ср.
уравнение D.1)]. Влияние показателя преломления на спектр уменьшается,
если используются растворы с концентрацией 2 % и менее [42]. То же
Количественные приложения
251
80 -
I «О
40 -
20 "
1
¦—¦"«¦-»•¦¦¦•¦1
1
1
1
1
¦¦^¦¦-¦^
1
1
1
1
1
|
N
\
I
1
/
1
г
/
1
1
г
1
1
г*—.
1
1
———
1
1
-
2,0
1,5
-0,5
900
800 700
Волновое число, см
в 00
Рис. 6.7. Спектр пропускания (вверху) и показатель преломления (внизу) хлоро-
хлороформа в области 550—1000 см-1 [50]. Reproduced, with permission, from
Spectrochimica Acta.
рассуждение справедливо и по отношению к растворителям. По этой
причине следует избегать растворителей с фоновым поглощением, даже
если предполагают, что оно компенсируется чистым растворителем
в канале сравнения. Таким образом, коэффициент отражения очень
чувствителен к ничтожным отложениям или помутнению на границе
раздела окно — жидкость, и этот факт не всегда заметен на глаз.
Недооценивается и тот факт, что из-за различия в показателях
преломления раствора и окон кюветы, отмеченного выше, на спектр
может налагаться интерференционная картина. Интерференционные
полосы проявляются редко, но они могут приводить к невоспроизво-
невоспроизводимости в измерениях. Этот эффект иллюстрируется на рис. 6.8,
252
Глава 6
0,3
illllllif! iifllliiHlilliiili mmm
==5====:~:5=======EE=r3S5SS-3=.
liliiii! I illiililSelPiiiiil i 1^§Ш1Ш
Рис. 6.8. Полоса поглощения в обычных условиях (а) и та же полоса при
наложении на нее интерференционной картины (усиленной для ясности) (б).
Амплитуды полос
подбора
"окна
1,5
1,5
1,5
интерференции, возникающих из-за
материала
"раство;
1,427 или
1,397 или
1,357 или
кюветы и растворителя
>а
1,577
1,610
1,659
Отражение
0,00062
0,00125
0,0025
Таблица 6.1
неправильного
|Н5]
МП. °,
0,025
0,50
1,0
на котором интенсивность полосы поглощения прямо зависит от того,
соответствует ли ее максимум максимуму или минимуму интерферен-
интерференционных полос. Точки, между которыми проведена базовая линия, будут
оказывать аналогичное влияние, так что суммарный эффект может
быть довольно значительным. Этот эффект был оценен Уайтом
и Вардом [115]; некоторые из их данных представлены в табл. 6.1.
Очевидно, чем больше разница в показателях преломления, тем большее
значение приобретает проблема интерференции. Показатели преломле-
преломления для некоторых широко распространенных материалов окон
и растворителей приведены в табл. 6.2, на основании которой можно
оценить вклад интерференции для некоторых сочетаний раствори-
растворитель — окно.
От интерференционных эффектов можно избавиться, если подобрать
Количественные приложения
253
Длина
волны,
мкм
2
3
4
5
6
7
8
9
10
И
12
13
14
15
16
18
20
25
30
35
Показатели преломления
NaCl
1,526
1,525
1,522
1,519
1,515
1,511
1,507
1,501
1,495
1,488
1,480
1,471
1,461
1,451
1,440
—
_
_
_
—
КС1
1,475
1,474
1,472
1,470
1,468
1,465
1,463
1,460
1,456
1,452
1,447
1,442
1,437
1,431
1,425
1,414
1,390
_
_
—
КВг
1,538
1,536
1,535
1,534
1,533
1,532
1,530
1,528
1,526
1,524
1,522
1,519
1,516
1,513
1,510
1,500
1,492
1,463
_
—
материалов окон
CsBr
1,670
—
—
1,666
—
-
—
1,662
-
—
1,656
1,655
1,653
1,648
1,642
1,625
1,608
1,596
со,
,447
,442
,438
,438
,431
,428
1,422
1,406
,384
1,339
,169
,7
1,60
1,559
,533
1,525
1,497
1,486
1,494
Таблица 6.2
и растворителей |102]
cs2 c,ci.
,582 1
,575
,566
,548
,450
,783
,657
,636
,628
,622
,618
1,615
1,614
1,612
,610
,48
,48
,48
,47
,47
1,47
1,46
1,45
,42
—
,50
—
1,54
НСС13
1,43
1,43
1,43
1,42
1,42
1,41
1,37
1,41
1,40
1,36
1,26
-
с6н,2
1,41
1,40
1,42
1,41
1,41
соответствующим образом показатели преломления растворителя и ма-
материалов окон, нанести специальные покрытия на них или использовать
кюветы такой большой толщины, что интерференция не возникнет.
Другой, более тонкий, эффект, связанный с неправильным подбором
показателей преломления, состоит в небольшом сдвиге максимума
поглощения (< 1 см). Этот сдвиг происходит из-за того, что отражение
окна кюветы изменяется с изменением показателя преломления
раствора, и, если окно и раствор имеют различные показатели пре-
преломления, отражательная способность (а следовательно, и пропускание)
будет различаться по обе стороны полосы поглощения [123].
Сходимость пучка образца обычно не оказывает влияния на длину
оптического пути, но если используется световой конденсор или при-
приставка с микроскопом, то эффект может стать заметным. Блоут и др.
[17] рассмотрели этот случай количественно и вывели поправочные
коэффициенты, зависящие от угла сходимости, показателя преломления
и измеряемого поглощения, с помощью которых можно скорректировать
наблюдаемое поглощение.
Похожий эффект, который состоит в изменении фокуса сходящегося
луча, может возникнуть на толстых окнах или образцах. Его влияние
254 Глава 6
на спектрофотометр неопределенно, но, как предполагают, на интенсив-
интенсивность, аппаратную функцию и рассеянное излучение он оказывает
воздействие. Рекомендуется применение пар кювет с приблизительно
равными толщинами окон.
Для предохранения от воздействия влаги воздуха на растворимые
солевые оптические детали многие современные ИК спектрофотометры
термостатируются при температуре выше комнатной. Кюветы, уста-
установленные в кюветное отделение, также подвергаются тепловому
воздействию; кроме того, на них попадает достаточно интенсивное
тепловое излучение. В результате этого температура раствора может
изменяться на несколько градусов в зависимости от длительности
пребывания кюветы в спектрофотометре. Температурные зависимости
полос поглощения различаются, но они обычно гораздо сильнее, чем
те, которые возникают из-за ожидаемых изменений плотности. Для
деформационных колебаний группы СН3 в метиловых эфирах это
изменение составляет около 0,2 % оптической плотности при изменении
температуры на один градус [33]. Для полос ассоциированных молекул
могут наблюдаться очень сильные температурные зависимости. В одном
исследовании [54] было показано, что при увеличении температуры
на один градус оптическая плотность полосы гидроксидной группы воз-
возрастает на 0,01 единицы, или на 40% при повышении температуры
кюветы от 24,5 до 37 °С, когда ее помещают в кюветное отделение
серийного спектрофотометра. Толщина кюветы также может меняться
с температурой. Количественный анализ высокой точности, очевидно,
требует хорошего термостатирования.
Было широко изучено влияние некоторых хорошо известных источ-
источников ошибок на количественные измерения ИК-спектров и проведена
оценка величины результирующих ошибок. Эти факторы включают
погрешности в пропускании, положении нулевой линии и /0, обусловлен-
обусловленные шумами и рассеянным светом, ошибки записи на самописце,
возникающие из-за конечного времени отклика сервосистемы, и огра-
ограниченное разрешение спектрофотометра.
Проблема шумов была в какой-то степени решена в современных
диспергирующих спектрофотометрах путем применения более эффектив-
эффективных оптических систем, усилителей и приемников, но ее нужно прини-
принимать во внимание при получении точных величин пропускания. Как
отмечалось ранее (стр. 50—56), шум может быть уменьшен либо
увеличением постоянной времени сервосистемы, либо расширением
щелей для того, чтобы повысить энергию, проходящую через спектро-
спектрофотометр. Последний путь более практичен, но он приводит к тому,
что из-за потери разрешения интенсивность в максимуме понижается.
Однако при количественном анализе воспроизводимость важнее, чем
абсолютная точность оптической плотности, так что вообще стараются
увеличить ширину щелей (при соответствующем ослаблении усиления)
до тех пор, пока не будет достигнут приемлемый уровень шума
Количественные приложения
255
20
16
Г f I
*- Пропускание
Линия 100%
го
40 60
Пропускание, %
80
100
Рис. 6.9. Процент относительной ошибки в определении концентрации при
ошибке пропускания в 1% (верхняя кривая) или ошибка в 1% в положе-
положении линии 100% (нижняя кривая) [91]. Reproduced, with permission, from
Analytical Chemistry.
[85; 88, гл. 6]. Для лучших количественных измерений этот уровень
может быть ±0,1 % или меньше.
Погрешность в определении нуля независимо от шума или рассеян-
рассеянного света будет влиять на точность изменений оптической плотности.
Аналогично ошибки в оценке 10 отражаются на значениях оптической
плотности. Эти факторы рассмотрены Робинсоном [91], из статьи
которого взят рис. 6.9, и другими исследователями [62, 75]. Непосто-
Непостоянство источника излучения или системы усиления также может давать
вклад в ошибку величины пропускания [75]; результирующий эффект
будет таким же, как и под влиянием шума.
На основании этих и подобных исследований в общем было
принято, что для снижения ошибок необходимо работать с величинами
пропускания 20—60%. Из рис. 6.5 и 6.9 следует, что вероятность
ошибки сильно возрастает ниже 10 и выше 80% пропускания,
и вблизи пределов шкалы пропускания нельзя проводить даже грубый
полуколичественный анализ. При высокой оптической плотности образца
значительный вклад в ошибку, вероятно, будет вносить его собственное
излучение [70].
256 Глава 6
Двухлучевые спектрофотометры, в которых используется система
оптического нуля, подвержены ошибкам, возникающим в результате
нелинейности оптического клина [97]. Несмотря на это, спектрофо-
спектрофотометры хорошего качества имеют ошибку в пропускании менее 0,4%.
При проверке закона Бера этот эффект не обнаруживается и не важен,
если анализы проводятся на одном спектрофотометре, особенно при
использовании одного и того же участка оптического клина. В двухлу-
чевых спектрофотометрах, регистрирующих отношение электрических
сигналов, важна линейность усилителя.
Понятие об „ошибке слежения" или „динамической точности" отно-
относится к способности сервосистемы или пера самописца реагировать
на сигнал приемника излучения. Так как все спектрофотометры имеют
определенную постоянную времени, то реальная спектрограмма является
только приближенным отражением сигнала спектрофотометра [89].
Ошибку слежения можно уменьшить за счет регулировки спектрофото-
спектрофотометра и малых скоростей сканирования. При проведении анализа целе-
целесообразно сканировать аналитические полосы при нескольких скоростях.
Оптическая плотность данной полосы будет зависеть от времени сканиро-
сканирования при больших скоростях, пока не будет найдена такая скорость,
при которой оптическая плотность максимальна. Скорость, выбранная
для анализа, должна быть еще меньше этой скорости.
Иногда проявляются другие инструментальные факторы, такие, как
сдвиги длин волн, возникающие при изменении температуры окружа-
окружающей среды, или атмосферное поглощение. Заключительная часть этого
раздела посвящена факторам, которые осознаются исследователями
в меньшей степени, но тем не менее" оказывают значительное влияние
на точность спектрофотометра.
Вообще известно, что сильная разъюстировка оптики спектрофото-
спектрофотометра ведет к худшему разрешению из-за потери оптической чистоты
сигнала и снижения отношения сигнал/шум. Кроме того, к ощутимой
потере разрешения могут приводить более тонкие изменения, например
искривление источника излучения или слабое помутнение зеркал, но они
могут оказывать заметное влияние только на долговременную воспро-
воспроизводимость спектрофотометра. Такое изменение возникает из-за того,
что лучи образца и сравнения следуют через спектрофотометр по
несколько различающимся оптическим путям; это оказывает влияние
на аппаратную функцию, рассеянный свет, линию 10 и на соответствие
спектрального состава лучей образца и сравнения. После юстировки
нужно проводить проверку калибровочных кривых, а источники ИК-
излучения, особенно штифты Нернста, периодически проверять на
искривление.
Любое измерение на двухлучевом спектрофотометре в областях
поглощения атмосферы или растворителя требует более длительного
времени сканирования или более широких щелей, чем обычно, из-за
потерь энергии в обоих лучах. В области поглощения паров воды
на спектре часто возникают шумы из-за движения воздуха в кюветном
Количественные приложения 257
Таблица 6.3
Влияние отдельных видом однопроцентных ошибок на ошибку измерения
оптической плотности 175]
Измеренные значения пропускания, %
Вид ошибок
20 30 40 50 60 70 80 90
Измерение /0 A % шу- 0,43 0,62 0,83 1,09 1,44 1,96 2,80 4,48 9,49
ма)
Измерение / A % шу- 4,34 3,11 2,77 2,73 2,89 3,26 4,01 5,60 10,55
ма)
Рассеянный свет 4,14 2,56 1,98 1,67 1,47 1,33 1,22 1,13 1,07
Неравенство кювет 0,43 0,62 0,83 1,09 1,44 1,96 2,80 4,48 9,49
Максимальная полная 9,34 6,91 6,41 6,58 7,24 8,51 10,83 15,69 30,60
ошибка
отделении и недостаточного раскрытия щелей для устранения этого
шума. Единственное средство — закрыть кюветное отделение и дождать-
дождаться равновесия до регистрации спектра.
Среди других общих источников ошибок можно назвать тепловое
излучение и отражение оптическим клином, модулятором, ножами щелей
и образцом или веществом сравнения. Эти факторы вносят погрешность
порядка 0,1—0,2% в положение нулевой линии в двухлучевом приборе
[106] и 1-2%-в однолучевом.
Ошибка измерения оптической плотности в процентах, возникающая
при накоплении однопроцентных ошибок из всех перечисленных, источ-
источников, была подсчитана Мартином [75] и представлена в табл. 6.3.
На практике, конечно, некоторые ошибки сокращаются, а их величины
отличаются от 1 %. Тем не менее эта таблица полезна для иллюстра-
иллюстрации неточности результатов измерений в методе ИК-спектроскопии.
Большинства ошибок, перечисленных в этом разделе, можно избежать
или свести к минимуму при условии, что: 1) спектрофотометр нахо-
находится в хорошем состоянии, а его режим работы оптимален; 2) строятся
рабочие кривые для искусственных смесей; 3) прибор периодически
проверяется на образцах известного состава; 4) запись спектров как
стандартов, так и образцов проводится в постоянных воспроизводимых
условиях. Короче говоря, для количественных измерений ИК-спектров
важна хорошая техника анализа.
Расчеты
Однокомпонентные системы
В простейшем случае один или большее число компонентов образца
имеют не перекрывающиеся полосы и для количественных расчетов
можно воспользоваться уравнением F.5). Сначала из спектра отдельного
17-586
258
Глава 6
ДС
Концентрация
Рис. 6.10. А — график, изображающий закон Бера для монохроматического
излучения; Б — экспериментальная кривая, полученная со щелью конечной
ширины; В — предполагаемая кривая, полученная при использовании единствен-
единственной стандартной точки с концентрацией 100%. Ошибка в этом случае
соответствует величине ДС.
вещества или его раствора с известной концентрацией определяют
коэффициент поглощения а и используют его для вычисления концентра-
концентраций составных частей в образцах. Например, если оптическая плотность
полосы для чистого вещества равна 0,312 в кювете толщиной 0,101 мм
и с концентрацией раствора 2,17%, то
а =
0.312
ccb
¦ = 0,01424.
F.7)
B,17) A00) @,101)
В образце с оптической плотностью 0,305 в кювете толщиной 0,115 мм
и с концентрацией раствора 2,05% содержание этого вещества будет
А 0,305
с =
= 9Г
F.8)
аЬс @,01424) B,05) @,115)
При анализах этого типа, которые проводятся периодически с исполь-
Количественные приложения 259
зованием одного и того же стандарта, принято объединять коэффициенты
(bed'/А) для стандарта, поэтому а' = (Ьсс'/А), где а' — обратная величина
по отношению к коэффициенту поглощения а. Тогда имеем с = Аа'/Ьс',
что облегчает вычисления.
При условии выполнения закона Бера точность результатов этих
вычислений ограничена только ошибками измерения. Если закон Бера
не выполняется (общий случай), то будут получаться только прибли-
приближенные концентрации, но калибровочная кривая (или поправочная
кривая), построенная по известным стандартам, гарантирует макси-
максимальную точность (рис. 6.10). Если закон Бера выполняется, то состав
многокомпонентных смесей можно вычислить точным решением систем
уравнений, если же нет, то можно использовать приближенные или
графические методы.
Анализ многокомпонентных систем
в условиях выполнения закона Бугера — Бера
Когда имеется несколько компонентов, существует вероятность того,
что некоторые аналитические полосы будут перекрыты. Однако довольно
редко случается, чтобы полосы всех веществ, образующих смесь, взаимно
перекрывались при всех длинах волн, выбранных в качестве аналити-
аналитических. Конечно, целесообразно выбирать полосы с минимальным
перекрыванием. В случае перекрывающихся полос, когда ассоциация
в растворе отсутствует, оптическая плотность при длине волиы X
является суммой оптических плотностей поглощения каждого из z-x ком-
компонентов при этой длине волны:
+ a2bc2 + ... + dibCi, F.9)
где обозначения те же, что и ранее, кроме ct — ее'. Тогда для
п компонентов и N длин волн имеем
А а = aiAbci.+ a2Abc1 + ... + anAbcm
Ав = a1Bbc1 + a2Bbc2 + ...
F.10)
Если для анализа выбирают сильные полосы с незначительным или
пренебрежимо малым перекрыванием, то диагональные члены аиЬсь
а2вЬс2... будут большими, а другие — небольшими или равными нулю.
Значения а определяют из уравнения F.5), измеряя оптическую плотность
каждого компонента при всех длинах волн XN. Недиагональные коэффи-
коэффициенты отличаются некоторой неопределенностью, так как эти частоты
17*
i + a2Nbc2 + ... + anNbcn.
260 Глава 6
обычно не попадают в максимум поглощения. Если одна или несколько
из них лежат на крыле резкой полосы, то коэффициенты особенно
чувствительны к ошибкам в измерении частот. Когда число компонентов
мало, система уравнений F.10) решается просто. В случае п = 2
_ Л.Аа2в — Ава2А .
ABaiA-AAaiS
Очевидно, что решение системы i уравнений значительно услож-
усложняется при i = 3, 4, 5 ... .
Ряд коэффициентов си сг ... ct в уравнениях F.10) образует матрицу,
решение которой по отношению к концентрациям дает обратную
матрицу:
с, = кпАл + кпАв + ... + klnAN,
с2 = к21Ал + к22Ав + ... + k2nAN, F.13)
с„ = кп1АА + клАв + ... + kmAN.
Для двухкомпонентной системы уравнения F.11) и F.12) образуют
аналогичную матрицу. При х ^ 3 эти уравнения легче решать для
коэффициентов ки в численном виде, чем в неявной форме. Такие
решения можно выполнить, используя стандартные методы матричной
алгебры; специальные области применения и примеры даны в литера-
литературе [4, с. 86; 13, с. 403—412]. Для систематических анализов двух-
и трехкомпонентных систем расчеты можно сильно упростить, если
построить номограммы, что не требует больших усилий [101]. Для
оценки недиагональных коэффициентов методом скорейшего спуска
Перри [83, 86] предложил использовать одну-две смеси известного
состава.
Уравнения F.10) можно решить с помощью подходящих прибли-
приближений. Если недиагональные члены малы (малое перекрывание), то
решения сходятся быстро. В качестве первого приближения предполага-
предполагается, что перекрывание мало (т. е. коэффициенты aiB, a2A, aiB ... anN,
пф N равны нулю), и определяются концентрации сь сг ... ct. Начальные
значения с,- затем подставляются в уравнения F.10), откуда и полу-
получается новое значение сг. Этот процесс продолжается до тех пор, пока
не будет достигнута требуемая степень точности [4, метод Е-168-67].
На практике аналитические полосы часто можно выбрать так, чтобы
перекрывание для одного или большего числа компонентов было или
пренебрежимо малым, или относительно постоянным. Тогда анализ
в значительной степени упрощается, поскольку концентрация этой
Количественные приложения 261
составной части вычисляется в первую очередь, а поправки находятся
из графиков или таблиц, которые относятся к другим оптическим
плотностям. Таким образом, выполняя вычисления в нужной последо-
последовательности, можно провести полный расчет посредством только
вычитания поправочных членов, избегая утомительных алгебраических
и приближенных вычислений.
Анализ многокомпонентных систем
в условиях отклонений от закона Бугера — Бера
Если закон Бугера — Бера не выполняется, но аддитивность сохра-
сохраняется, то вычисления можно провести одним из нескольких способов.
При концентрациях, меняющихся не в очень широких пределах,
искривленные графики закона Бера могут считаться приблизительно
прямыми линиями, если пользоваться ограниченной областью кон-
концентраций. В этих пределах используются линейные уравнения, обсуж-
обсуждавшиеся в предыдущем разделе. Для более широких интервалов
концентраций вводятся члены, отражающие взаимодействия. Затем
коэффициенты вычисляются по методу наименьших квадратов [10].
Во многих случаях, особенно когда имеется широкий интервал
концентраций, удовлетворительные результаты получаются при исполь-
использовании метода графических последовательных приближений [30]. Для
п компонентов строят п2 графиков зависимостей оптических плотностей
от концентраций при п длинах волн. Для каждого компонента можно
быстро найти неисправленные концентрации и из этих результатов,
применяя поправочные кривые, последовательно определить лучшие
значения концентраций. Если взаимное перекрывание полос не слишком
велико, то значения концентраций должны быстро сойтись в пределах
экспериментальной ошибки.
В литературе имеется указание на совместное использование алгеб-
алгебраических и графических методов [13, с. 414—417; 87]. Сначала концент-
концентрации вычисляются в предположении выполнимости закона Бера, а затем
расчет повторяется с учетом поправочных коэффициентов (нанесенных
на график в зависимости от концентрации).
Анализ без стандартов;
вторичные стандарты; процедура нормировки
Анализ без чистых стандартных образцов возможен при следующих
условиях: 1) число компонентов п известно; 2) имеются аналитические
полосы с незначительным или пренебрежимо малым перекрыванием;
3) можно приготовить л смесей состава, меняющегося по возможности
в широких пределах. Такие условия часто осуществляются в случае
последовательного ряда дистилляционных фракций. Если имеются две
смеси хну компонентов 1 и 2, то для двух частот А и В можно,
262 Глава 6
как и ранее, написать
ААх = афс1х, ААу y
АВх = a2bc2x, ABy = a2bc2y, F.14)
а также
clx + c2x=l (или 100%),
с1у + с2у = 1. F.15)
Таким образом, имеются шесть простых уравнений с шестью неизвест-
неизвестными (аь а2, с1х, с2х, с1у, с2у), которые относительно легко решаются.
По мере увеличения числа компонентов системы уравнений F.14)
и F.15) также усложняются. Поскольку при этом составы смесей
сближаются, точность определений понижается.
Если компоненты взаимно мешают друг другу, то остается возмож-
возможным составить систему уравнений для определения состава бинарных
смесей, но в этом случае потребуются данные о дополнительных смесях,
так как появляются два новых неизвестных коэффициента поглощения
[88, гл. 6].
Если для определения коэффициентов поглощения чистые стандарты
недоступны, то для расчета значений а применяется процедура,
описанная выше. Можно использовать смеси или даже образцы,
в которых концентрация каждой составной части независимо опреде-
определена некоторым другим методом. В этом случае точность ИК-анализа
полностью определяется точностью независимого метода. Кроме того,
возможность существования неизвестных и неидентифицированных
(и возможно, мешающих) компонентов делает использование вторичных
стандартов непривлекательным.
Результаты, полученные для многокомпонентных смесей, иногда
нормируют к 100 %, но это не стандартная операция. Она имеет смысл,
если только все компоненты в равной степени подвергаются некоторому
влиянию неизвестной величины, такой, как ошибка разбавления или
погрешность в определении толщины кюветы, и если установлено, что
никаких дополнительных компонентов в смеси нет. Сумма концентраций
всех компонентов является хорошей проверкой как на точность анализа,
так и на наличие неизвестных составных частей*.
Статистическая оценка результатов
Каждая стандартная аналитическая процедура должна быть подверг-
подвергнута статистическому анализу. В результате этого можно не только
определить вероятные пределы ошибок, но и найти их источники.
* Теоретические основы современных методов количественного анализа
многокомпонентных систем по спектрам поглощения даны в [5]. - Прим. ред.
Количественные приложения
263
Подробная информация имеется в литературе [3, 6, 7], поэтому здесь
кратко обсуждаются только элементарные сведения.
Сначала установим различие между точностью или воспроизводи-
воспроизводимостью измерения н абсолютной точностью. Оценки стандартного
отклонения, основанные на различии в результатах повторных анализов,
относят к воспроизводимости. Разность между предполагаемой и най-
найденной для синтетических стандартов величинами дает меру точности.
Полезны оба вида оценок. Хорошо известно, что 67 % величин повтор-
повторных анализов попадают внутрь стандартного отклонения 1 % или + о,
95 % в интервал + 2о и 99 % в интервал + За. Истинное среднеква-
среднеквадратичное отклонение о отличается от стандартного отклоне-
отклонения s. Для п измерений, если х — наблюдаемая величина 1-го измере-
Таблща 6.4
Повторные измерения концентраций
а. Измерения единичного твердого
образца
Среднее =
s =
Коэффициент вариации =
б. Измерения образца в растворах
Среднее =
s =
Коэффициент вариации =
Оператор № 1
0,640
0,652
0,635
0,643
0,638
0,638
0,645
0,648
0,627
0,643
0,641
0,0070
1,09%
85,3
84,3
81^5
80,3
80,3
85,0
83,5
84,1
80,5
84,6
82,94
2,06
2,48 %
Оператор № 2
0,651
0,640
0,636
0,632
0,627
0,629
0,633
0,638
0,630
0,633
0,635
0,0069
1,09%
81,2
82,2
81,5
80,5
78,2
82,2
84,5
84,8
85,8
83,4
82,43
2,64
3,20%
264 Глава 6
ния,
Щ ()
П— 1
или, если все наборы продублированы, a d — разность между парами
измерений.
-№-¦
Если s меняется приблизительно пропорционально числу измерений,
то общепринятым параметром сравнения является коэффициент ва-
вариации, или относительное стандартное отклонение, равное «/(истинное
значение). Для иллюстрации в табл. 6.4 (а) представлена статистическая
обработка результатов измерений полосы поглощения твердого веще-
вещества двумя операторами в двухмесячный период. Из этих результатов
можно оценить вероятные ошибки измерения, в том числе и инстру-
инструментальные, каждого из операторов и сравнить их (другой проверкой),
чтобы увидеть, действительно ли имеется значительная разница.
Повторяя измерения с растворенными образцами, можно добавить ста-
стадии взвешивания и растворения, как в табл. 6.4F). Аналогично обраба-
обрабатываются другие аналитические измерения. Отсюда можно сделать
заключения относительно погрешности каждой стадии анализа.
Вероятно, наиболее осмысленные данные получаются при использо-
использовании „слепых" образцов, т. е. одно и то же вещество представляется
на анализ под кодовыми номерами. Использование кодированных
образцов помогает устранить сознательное или бессознательное пре-
предубеждение при сборе данных.
Специальные задачи и методы
Количественные измерения спектров газов
В большинстве случаев количественный анализ газовых смесей легче
проводить методами газожидкостной хроматографии или масс-спектро-
метрии. Однако с успехом может быть использована и ИК-спектро-
скопия при условии, что обращается внимание на соответствующую
стандартизацию подготовки образца и ударное уширение. Последний
эффект можно уменьшить, доводя давление всех газообразных образцов
до 760 мм рт. ст. таким инертным газом, как азот. Сорбция паров
некоторых соединений внутри кюветы может быть уменьшена предвари-
предварительной обработкой кюветы веществом образца.
Количественные измерения ИК-спектров паров обсуждены Фриделем
и Квейзером [41], которые также описали принятые методики запол-
заполнения кювет летучими жидкостями. С веществами, газообразными
при комнатной температуре, работают в обычных вакуумных системах.
Очень разбавленные газовые смеси можно приготовить диффузионным
методом [78].
Количественные приложения 265
Анализ твердых веществ
Среди ряда исследователей в области ИК-спектроскопии, а к ним
принадлежит и автор, существует мнение, что нужно избегать коли-
количественных измерений твердых веществ как таковых. Тем не менее иногда
нет возможности выбора, и, если принять меры предосторожности,
методом прессования таблеток с КВг зачастую можно получить
удовлетворительные результаты.
Трудность анализа порошков обусловлена зависимостью оптической
плотности от однородности образца. Джонс [65] показал, что, если
в образце 10% составляют прозрачные включения и имеется полоса
с истинной оптической плотностью 1, наблюдаемая величина равна
0,775. В этой же работе приведены ошибки и для других отношений
площади прозрачной части образца к площади поглощающей. Отмеча-
Отмечается также, что эффект быстро возрастает по мере увеличения опти-
оптической плотности. Этот эффект назван „мозаичным", и его величина
зависит от размера частиц, их формы и распределения в образце.
По мере роста концентрации частиц область прозрачности (и величина
этой ошибки) уменьшается [63]. Другим, часто не учитываемым
фактором является зависимость интенсивности полосы кристаллических
веществ от размера частиц. Исследование кристаллического твердого
хлоранила показало, что при изменении размера частиц от 12 до
160 мкм коэффициент поглощения некоторых полос (в матрице из КВг)
может уменьшиться в 4 раза (рис. 6.11). Аналогичный эффект наблю-
наблюдался на кварце [111]. Наряду с изменением интенсивности может
происходить также сдвиг по частоте. Причина этого явления заклю-
заключается в том, что наблюдаются главным образом поверхностные,
а не объемные колебания, и именно они чувствительны к диэлектри-
диэлектрической постоянной окружающей среды [94]. Отсюда следует, что
неравномерное распределение поглощающих частиц в канале образца
из-за их слишком большого размера или изменение распределения
частиц по размерам от одного образца к другому приведет
к аномальным интенсивностям полос. Обычно рекомендуется, чтобы
диаметр частиц был меньше самых коротких длин волн используемого
излучения (в большинстве случаев 2 мкм). Если спектры раствора
получить не удается, то для проведения продуманных количественных
измерений с таблетками из КВг или суспензиями нужно быть уверенным
в том, что образец подходящим образом измельчен до требуемой
степени дисперсности.
Опубликован ряд количественных методов, в которых используются
матрицы из КВг. Успешное применение этих методов, очевидно, зависит
от воспроизводимости размалывания и диспергирования образцов.
Количественный анализ суспензий по ИК-спектрам можно выполнить,
вводя внутренний стандарт. На практике это означает смешивание
известного количества не мешающего вещества, имеющего легко
измеряемую полосу поглощения, с навеской исследуемого образца
266
Глава 6
200
WO
i
0
300
200
Ю0
0
800
500
400
300
200
100
0
i i i i i i
i i i i i i
***
1-МИ3
• 1 1 1 1 1
1260 cm-'
1 1 1
740 cm1
1 1 1
1690 см
1 t 1
1 1 1 1 1 1 i 1 1
BSOcm .
1 1 1 1 1 1 1 1 1
1560 cm'1-
11 iiiiiii
- C52 1110cm-' -
1 1 1 1 1 1 1 | |
20 40 BO 80 Ю0 120 1401 BO 180 0 20 «О вО 60 ЮО 120 МО 160 160
Средний размер частиц, мки
Рис. 6.11. Зависимость молярного коэффициента поглощения хлоранила от раз-
размера частиц для различных полос поглощения [98]. Reproduced, with permis-
permission, from Applied Spectroscopy.
и сравнение относительных оптических плотностей полос. Такое отно-
отношение не зависит от толщины образца. Некоторыми из веществ,
предлагаемыми в качестве внутренних стандартов, являются карбонат
кальция, азид натрия, d-аланин, роданид свинца, нафталин и роданид
калия. Из этого ряда простейшие спектры наблюдаются у неоргани-
неорганических веществ, а роданида имеют сильную полосу поглощения около
4,8 мкм, которая обычно не перекрывается с полосой поглощения
образца.
Среди применений внутреннего стандарта можно назвать анализ
поливинилацетатхлорида на содержание ацетата [116], определение
количества свободной кислоты в алюминиевых мылах методом прессо-
прессования с КВг [116] и анализ изомерных фталиевых кислот, спектры
которых записывались в виде суспензий в вазелиновом масле [19].
Количественный анализ полимерных материалов
Количественный анализ полимеров представляет собой несколько
специальную проблему, и во многих случаях этот метод нужно приспо-
приспосабливать к конкретной задаче. Большинство трудностей возникает
Количественные приложения 267
из-за нерастворимости полимерных материалов в обычных раствори-
растворителях, прозрачных в ИК-области, и присущих им погрешностей при
подготовке стандартов. Здесь кратко изложены некоторые изящные
решения этих задач.
В отдельных случаях с помощью обычных методов сополимери-
зации можно получить удовлетворительные стандарты, но окончатель-
окончательный состав таких композиций зачастую неизвестен с точностью,
достаточной для их использования в качестве стандартов. Для того
чтобы охарактеризовать полимерную композицию, полезны методы
химического анализа, если они доступны. Третий метод стандарти-
стандартизации основан на применении меченых атомов. В одном из примеров
этилен, меченный С, использовали для приготовления этиленпропи-
леновых сополимеров и их составы определяли из удельных актив-
активностей [34]. В другом случае стандарты для ИК-анализа тройного
сополимера метилнзопропенилкетона, бутадиена и акрилонитрила были
приготовлены с использованием метилизопропенилкетона, меченного
14С. Содержание акрилонитрила определяли по содержанию азота
методом Дюма [105]. Для определения состава стандартов применяют и
метод ЯМР. Методы стандартизации этиленпропиленовых сополимеров
были рассмотрены в обзоре Тоси и Чиампелли [109], а Хэмптон
[47] дал таблицу из 39 литературных ссылок, относящихся к ме-
методам количественного анализа полимеров.
Исследование полимера методом ИК-спектроскопии можно провести
несколькими путями. Наиболее распространенный метод — отливка
тонкой пленки полимерного материала из растворителя. Хотя в этом
случае толщина неодинакова и точно неизвестна, она может быть
исключена из расчетов, если использовать отношение оптических плот-
плотностей двух полос — по одной от каждого компонента. Метод при-
применим к сополимерам любого числа мономеров при условии, что
каждый из них имеет отдельную полосу поглощения. Величины про-
пропускания в максимуме полосы должны быть оптимальны, как показа-
показано на рис. 6.5.
Полимеры, содержащие наполнители и пластификаторы, часто гото-
готовят к съемке экстрацией растворителем [47]. Пластификаторы могут
оказаться растворимыми в мягких растворителях, таких, как CS2 или
этиловый эфир, и их экстрагируют из измельченного полимера в ап-
аппарате Сокслета. Экстракт в CS2 можно прямо перенести в ИК-
спектрофотометр. От наполнителя полимер отделяется более жестким
растворителем, например о-дихлорбензолом. В этом случае из раство-
раствора можно отлить пленку полимера, а спектр наполнителя получить
методом прессования с КВг или методом суспензии в вазелиновом
масле. Примером такого рода является количественный анализ соста-
состава поливинилхлорида [21].
Подготовка образцов для анализа методом НПВО обсуждается
позже (стр. 270).
К другому методу, применимому как для нерастворимых, так и для
268 Глава 6
сильнонаполненных полимеров, относится пиролиз [15]. В данном при-
примере сополимеры бутадиеннитрилаг и фенола нагревали в вакууме
до 540—580 °С, а пиролизат собирали в ловушку, охлаждаемую су-
сухим льдом. На основании анализа конденсата для фенольного ком-
компонента получены результаты с воспроизводимостью ±2% в преде-
пределах от 0 до 33 %. Было также оценено количество негорючего
наполнителя. Более полный анализ конкретных полимеров обсужден
в других работах [47, ПО], там же имеются ссылки на методы
количественного анализа различных типов полимеров по ИК-спектрам.
Интересный подход к анализу полимерных пленок без обслуживаю-
обслуживающего персонала осуществили Джонсон и др. [64], которые соединили
стандартный спектрофотометр с ЭВМ для автоматического анализа
72 последовательных образцов.
Количественный анализ ассоциированных образцов
Как было показано, межмолекулярное взаимодействие сильно за-
зависит от концентрации. Поэтому колебательные частоты и интенсив-
интенсивности поглощения молекул, участвующих в ассоциации, также обычно
изменяются с концентрацией. Отдельные компоненты смеси могут
вступать в самоассоциацию, взаимодействовать с растворителем или
другими компонентами в различных отношениях в зависимости от
их относительных концентраций. Такая ассоциация наблюдается, когда
участвующие молекулы имеют фрагменты с кислыми и основными
(в смысле Льюиса) свойствами.
Первичные ароматические амины являются хорошим примером та-
такого межмолекулярного взаимодействия. В ходе исследования пер-
первого обертона валентного колебания NH было найдено [114], что
молярный коэффициент поглощения этой полосы меняется от 1,87
в циклогексане до 0,61 в ацетонитриле. В других растворителях
получаются промежуточные значения. Этот эффект в данном случае
обратен тому, что наблюдалось для соответствующей основной по-
полосы. Ясно, что изменение растворителя в такой системе требует
перестройки заново рабочей кривой.
Другим хорошо известным примером является концентрационная
зависимость полосы гидроксидной группы. Часто бывает необходимо
измерять спектроскопическими методами концентрацию либо самой
гидроксидной группы, либо молекул, содержащих эту группу. Иногда
это можно сделать, используя один из методов, обсуждаемых ниже.
Однако из-за тенденции гидроксидных групп к образованию водород-
водородной связи с кислородом и другими электроотрицательными атомами
(стр. 168—174) ИК-спектроскопия в данном случае не столь полезна
для количественного анализа, как в других. Если растворитель или дру-
другие находящиеся в растворе вещества, достаточно инертны, то от-
относительные интенсивности полос ассоциированных и неассоциирован-
ных групп ОН определяются концентрацией и температурой раствора;
Количественные приложения 269
при этом разбавление раствора благоприятствует диссоциации. За-
Загрязнения (например, вода) или другие протонодонорные или прото-
ноакцепторные вещества будут изменять равновесие диссоциации и от-
относительные интенсивности поглощения двух полос. Конечно, при
этом ни одна из них не будет следовать закону Бугера — Бера, и по-
попытки количественного анализа окажутся безуспешными. Кроме того,
подразумевается, что любая другая группа, участвующая в ассоциации,
в большей или меньшей степени может проявлять сходные "эффекты.
Если существует такая возможность, то необходимо соблюдать не-
некоторую осторожность при выборе полос для количественного ана-
анализа. Хорошее знание групповых частот и их природы неоценимо
при таком выборе.
Для того чтобы обойти эти трудности, в некоторых случаях ис-
используют специальные методы. Вероятно, наиболее общим из них яв-
является разбавление образца инертным растворителем по крайней мере
до 0,01 моль/л, когда концентрация ассоциатов становится пренебре-
пренебрежимо малой. В этом случае наряду с высокопрозрачным раствори-
растворителем необходимо применять кюветы с большей, чем обычно, тол-
толщиной. В коротковолновой ИК-области спектра этому критерию
удовлетворяет СС14.
Во втором методе пренебрегают эффектами ассоциации. Этот под-
подход иногда приводит к успеху, но только при условии, что состав
системы относительно постоянен и мешающие примеси либо отсутст-
отсутствуют, либо на них можно сделать поправку. Так, гидроксидное
число алкидных смол определялось из оптической плотности полос
ассоциированной группы ОН в растворе СН2С12. Результат был исправ-
исправлен на кислотное число смолы и на содержание в ней воды [80].
Очевидно, такой эмпирический метод требует различных калибровочных
кривых для каждого типа смол и частых проверок точности определе-
определения.
В третьем методе анализа ассоциированных объектов их спектры
записывают при условии полной ассоциации. Это можно сделать,
используя в качестве растворителя либо основание Льюиса (или
кислоту в зависимости от условий), либо само исследуемое вещество.
Например, полипропиленгликоли можно проанализировать на гидрок-
сидные группы in situ [22], так как группы ОН образуют внутри-,
молекулярную водородную связь с кислородом простого эфира, и
возникающая в результате этого полоса поглощения достаточно точно
подчиняется закону Бугера — Бера. Для коррекции величины опти-
оптической плотности группы ОН может оказаться необходимым не-
независимое определение воды. В ближней ИК-области в качестве ас-
ассоциирующего растворителя для связывания гидроксидных групп и га-
гарантии воспроизводимости анализа часто используется хлороформ.
Простые и сложные полиэфиры анализировались с целью определения
гидроксидного числа в области 2—3,2 мкм, при этом в качестве
растворителя применялся ССЦ, содержащий 10% СНС13 [54]. Смеси
270 Глава 6
Н2О с D2O можно анализировать с помощью отношения оптических
плотностей ^25бо/^35зо [36]- Полосы поглощения ассоциатов особенно
чувствительны к флюктуациям температуры, и в случае их исполь-
использования для анализа температура образца должна тщательно кон-
контролироваться.
Суммируя, можно сказать, что лучше избегать количественных
измерений на частотах тех групп, которые участвуют в ассоциации.
Если же такие измерения необходимы, то условия следует поддержи-
поддерживать по возможности неизменными и достаточно часто проверять
калибровку.
Групповой анализ
Интересным примером применения групповых частот в количест-
количественном анализе является прямое определение таких функциональных
групп, как альдегидные, кислотные, спиртовые, при этом структура
конкретных молекул не учитывается. Такое определение концентра-
концентраций групп представляет интерес для нефтяной и химической про-
промышленности. В одном из исследований [95, 96] спирты, кислоты,
альдегиды, кетоны, сложные и простые эфиры были определены из-
измерением их поглощения при 3635 см B,75 мкм), 3550 см"
B,82 мкм), 2720 см C,68 мкм), 1720 см E,8 мкм), 1140-1300 см
G,7-8,8 мкм) и 1060-1220 см-1 (8,2-9,4 мкм) соответственно.
В другой работе [61] проводился контроль методом ИК-спектроско-
пии содержания транс-ненасыщенности в жирах, нефти и сложных
эфирах.
Точность этих анализов зависит от постоянства коэффициента
поглощения для каждой функциональной группы при переходе от
одной структуры к другой и в меньшей степени от взаимодействия
между членами разных групп. В общем случае ошибка группового
анализа больше, чем ошибка анализа многокомпонентных смесей
отдельных веществ, и составляет величину порядка 10—20%. В то
же время родственные группы, такие, как метиленовые фрагменты
в нормальных парафиновых углеводородах, имеют одинаковые коэффи-
коэффициенты поглощения, независимые от длины цепи [124], которые,
как предполагается, можно определять со значительной точностью.
Нарушенное полное внутреннее отражение
Количественный анализ с использованием метода НПВО можно
применять в том случае, если образец содержит такой внутренний
стандарт, интенсивность поглощения которого не изменяется в за-
зависимости от состава образца. Можно использовать и внешние
стандарты, но ряд мешающих факторов осложняет этот метод [118].
Во-первых, положение образца и эффективность его контакта с по-
Количественные приложения 271
верхностью элемента отражения должны быть воспроизводимы с вы-
высокой степенью точности. Во-вторых, воспроизводимыми должны быть
и другие параметры (угол падения и расположение оптических эле-
элементов). Эти условия чаще выполняются для жидкостей, чем для
твердых веществ, но и здесь, особенно в случае тех образцов, ко-
которые имеют тенденцию к расслаиванию, может возникнуть другая
трудность, обусловленная, вероятно, градиентом электрического поля
у поверхности элемента отражения. По этой причине излучение «ви-
«видит» ту часть образца, которая не представляет собой основную
массу (а только поверхность толщиной 0,1Л, — 10Х.. — Прим. перев.).
Иногда один из компонентов раствора адсорбируется на поверхно-
поверхности элемента, в результате чего ставится под сомнение возможность
количественных измерений.
Использование относительных интенсивностей полос дает более
точный метод количественного анализа твердых образцов. В бинарной
системе для каждого компонента выбирают по одной полосе погло-
поглощения. Для получения наилучших результатов полосы ие должны
перекрываться и должны находиться не слишком далеко друг от
друга (по длинам волн). Записываются спектры серии стандартов
и строятся калибровочные кривые, которые используются для анализа
неизвестных образцов. Воспроизводимость положения образца и пло-
площади контакта не оказывает слишком большого влияния на ре-
результаты, так как обе полосы должны подвергаться этим воздей-
воздействиям в равной степени (хотя было бы хорошо проверить такое
утверждение). В одной из работ анализировались смеси нейлонового
и хлопчатобумажного волокон в области 20—80% с использованием
элемента МНПВО из K.RS-5 толщиной 1 мм с углом падения 60°
[118]. Содержание винилацетата в этиленвинилацетатных сополимерах
контролировалось посредством отношения оптических плотностей по-
полосы валентного колебания карбонильной группы A735 см-1) и полосы
деформационного колебания СН A465 см) [38].
Независимо от способа проведения анализа спектрофотометр дол-
должен быть так отрегулирован, чтобы сервосистема получала доста-
достаточно энергии, в противном случае будут получаться ошибочные
Анализ следовых примесей
Хотя обычно не считается, что метод ИК-спектроскопии чувст-
чувствителен к следовым примесям, существует ряд методик, повышающих
его чувствительность до нескольких частей на миллион. Содержание
этого раздела ограничено определением известных компонентов малой
* Некоторые аспекты количественного анализа методом НПВО освещены
в обзоре [8]. — Прим. перев.
272 Глава 6
концентрации в больших образцах в отличие от анализа микрообраз-
цов, которые действительно представляют проблему в методе ИК-
спектроскопии. Однако некоторые приемы, описываемые ниже, были
успешно использованы для анализа следов этим методом.
Образцы с большой толщиной поглощающего слоя
В пробных спектрах поглощения матричного вещества с толщиной
образца в 10—1000 раз большей, чем обычно, иногда обнаруживают-
обнаруживаются «окна», в которых можно найти полосы примесей. При решении
такого рода задач часто важна область валентных колебаний С—Н.
Когда этот метод пригоден, он быстр, чувствителен, точен и дол-
должен быть первым приближением при любом анализе примесей. Этот
метод применялся для определения влажности хладагента дихлорфтор-
метана в количестве от 0 до 100 частей на миллион [32]. Много-
Многоходовые кюветы с длиной оптического пути до 50 м дают чувстви-
чувствительность порядка нескольких частей на миллион для многих обыч-
обычных газов, загрязняющих атмосферу (табл. 6.5) [59].
Если в распоряжении имеется подходящее вещество для сравне-
сравнения, то для исследования толстых образцов в качестве альтерна-
альтернативного метода можно использовать разностную спектроскопию в
сочетании с растяжкой по ординате. В таком случае чувствитель-
чувствительность диспергирующих спектрофотометров ограничена только шири-
шириной рабочего элемента приемника, так как величина растяжки по
ординате и толщина образца могут увеличиваться до тех пор, пока
Таблица 6.5
Чувствительность к обычным загрязнеппям атмосферы при
использовании 40-метровой газовой кюветы [59]
(соединение ~
СН2=СНСНО
H2S
НС1
NO,
со
С2Н4
so2
сн4
о3
Минимальная концентрация^ ¦
см
1720
1290
2920
1630
2160
946
1370
1300
1050
определению
мг/м3
0,05
70,0
12,0
0,03
1,0
0,12
0,09
0,06
0,11
поддающаяся
объемная часть
на миллион
0,02
50,0
7,0
0,014
0,8
0,10
0,03
0,085
0,05
а Минимальные количества, поддающиеся определению в атмосфере, иногда могут
быть выше из-за мешающего поглощения других составных частей.
Количественные приложения 273
соответствующее расширение шели не перестанет обеспечивать приемле-
приемлемое отношение сигнал/шум.
Отметим некоторые не очевидные на первый взгляд ограничения
рассматриваемого метода. Во-первых, спектрофотометр должен быть
хорошо отъюстирован, а кюветы должны иметь хорошее пропускание,
в противном случае будет наблюдаться сильный наклон линии фона.
Вторая трудность возникает в областях атмосферного поглощения
из-за случайных потоков воздуха. Это влияние можно уменьшить,
если закрыть кюветное отделение и продуть его и спектрофотометр
сухим газом. В-третьих, нельзя полагать, что если, например, 10
частей на миллион некоторого вещества обнаруживаются при 5-кратной
растяжке по ординате, то при 20-кратной растяжке можно будет
обнаружить 2,5 частей да миллион. Такая пропорциональность очень
часто не соблюдается из-за потери разрешения при расширении щели,
увеличенного наклона фона и повышенной трудности компенсации
основных составных частей исследуемого образца.
Разностная спектроскопия была применена для определения при-
примесей в моющих средствах [16], для анализа в газовой смеси шести
компонент, содержащих С4 [84], и для исследования разветвленности
полиэтилена [119, 120].
В интерференционных спектрофотометрах при растяжке по орди-
ординате используется многократное сканирование и специальное сглажи-
сглаживающее устройство для улучшения отношения сигнал/шум. Компен-
Компенсация полос колебаний решетки в образце полупроводникового крем-
кремния толщиной 2 см с помощью специально очищенного образца
сравнения позволила обнаружить в кремниевом стержне всего лишь 7
частей кислорода на миллиард [67].
Метод ИК-спектроскопии применялся для решения многих сложных
задач, связанных с промышленными загрязнениями и защитой ок-
окружающей среды. Например, с помощью интерференционного спектро-
спектрофотометра был выполнен анализ дымовых газов с очень низким со-
содержанием примесей [57]. Высокотоксичный карбонил никеля был опре-
определен в количестве, меньшем, чем 1 часть на миллиард, в присутст-
присутствии 106-кратного избытка мешающего СО [74]. Бейкер и Карлсон [7]
применили интересный метод анализа гексафторацетона (ГФА)
в воздухе. Они добились высокой аналитической чувствительности,
введя ГФА в реакцию с К2СО3 и Н2О, в результате которой по-
получается HCF3; последний обнаруживается при гораздо меньшей кон-
концентрации, чем ГФА. Пределы обнаружения многих загрязнений можно
понизить до 0,05 — 1 части на миллион осушкой (со специальным
осушителем) и повышением давления воздуха в кювете. Описан спектр,
зарегистрированный в 20-метровой кювете при давлении 10 атм [6].
Для определения CC12F2 в атмосфере (ПО частей на триллион, что
эквивалентно содержанию у поверхности земли) был проанализирован
с помощью моделирования на ЭВМ [81] солнечный спектр в области
800-1250 см.
18 - 586
274 Глава 6
ИК-лазеры нашли применение для анализа малых содержаний
диоксида азота [40], винилхлорида [39] и различных других газов
[45]. Были выбраны наилучшие частоты для анализа О3, N2O, CO,
СН4, Н2О и СО2 посредством таких узкополосных ИК-источников,
как лазеры [44]. Чувствительности порядка нескольких частей на
миллиард были достигнуты при использовании перестраиваемых ла-
лазеров с собирающими отражателями [55, 56].
Обзоры Ханста [48, 49] посвящены спектроскопическим методам
анализа загрязнений воздуха.
Корреляционная спектроскопия
С помощью аналитической методики, называемой корреляционной
спектроскопией* [117], чувствительность газового анализа может быть
повышена до нескольких частей на миллиард. В этой методике спектр
входящего в прибор излучения сопоставляется со спектром эталонной
газовой смеси (target gas), содержащейся в кювете спектрометра.
В сущности спектр образца усиливается спектром эталонной газовой
смеси и результат интегрируется. Области спектра, в которых происхо-
происходит взаимное перекрывание полос, гасятся. Метод позволяет наблюдать
спектры как испускания, так и поглощения.
Описаны три системы корреляционной спектроскопии. В первой
выходная щель дифракционного монохроматора заменена маской из
серии щелей, которые соответствуют спектру поглощения эталонной
газовой смеси. Вторая маска покрывает ту же спектральную область,
но она сконструирована так, чтобы пропускать только излучение
фона спектральных интервалов, смежных с полосами поглощения.
Маски коррелятора попеременно вводятся перед выходной щелью, и
промодулированный сигнал, возникающий на приемнике, соответствует
интенсивности спектра.
Вторая система сходна с недиспергирующим анализатором с га-
газовым фильтром (стр. 284—288), в котором эталонная смесь исполь-
используется в качестве сенсибилизатора. Применение такого устройства
было описано для контроля содержания СО в окружающем воздухе
[26].
В третьем варианте корреляционным спектрометром является ин-
интерферометр. Спектр эталонной смеси хранится в памяти прибора
в виде интерферограммы. Обработка данных легко проводится на
ЭВМ, которой снабжен спектрометр. Описано применение этого метода
* В отечественной литературе термин «корреляционная спектроскопия»
(correlation spectroscopy) еще не установился. Широко известна только одна
из систем корреляционной спектроскопии — промышленные автоматические
ИК-газоаналнзаторы. Изложение автором принципов корреляционной спектро-
спектроскопии оказалось мало понятным и для лучшего понимания следует обра-
обратиться к книге [117].- Прим. ред.
Количественные приложения 275
Таблица 6.6
Дистанционное обнаружение загрязнений
атмосферы по спектрам испускания8
Соединение
СО
NH3
СН4
NO2
4УВСТГм„Ь"™аТрдЧаСТЬ
5
10
0,6
2
а Длина оптического пути 5 м, температура источ-
источника 200 °С [581.
для обнаружения NO, CO, SO2, HC1 и HF в дымовых газах (по
спектрам поглощения) [52]. Пределы обнаружения обычных газов
по спектрам испускания показаны в табл. 6.6 [58].
Методы концентрирования
Физическое или химическое концентрирование искомой примеси
является мощным методом определения следов. Существует много
очень точных методов анализа, которые можно использовать для
исследования исходного образца, но при этом имеется риск непол-
неполного определения примеси.
Разработано так много способов выделения малых составных
частей, что здесь будут упомянуты только некоторые. Для выделе-
выделения следовых примесей используются методы хроматографии, экстрак-
экстракции растворителем, кристаллизации и дистилляции. Ранее обсуждалась
комбинация ИК-спектроскопии и хроматографии. Бумажная хрома-
хроматография применяется для разделения биологических веществ; для
получения ИК-спектра достаточно всего нескольких микрограммов ве-
вещества [108]. Широко распространен простой и эффективный метод
экстракции растворителем. Например, экстракция из отработанной воды
четыреххлористым углеродом позволяет определять 0,1 часть нефти
на миллион и 10 частей фенола на миллиард [100]. Силиконовые
жидкости, используемые в качестве антивспенивающих добавок в пи-
пищевых продуктах, были определены в количестве нескольких частей
на миллион экстракцией CS2 или другим растворителем [60]. Дроб-
Дробная кристаллизация в сочетании с разностной спектроскопией приме-
применялась для определения катехина и сходных примесей в гидрохиноне
[9]. Для повседневного контроля за содержанием добавок к полиэти-
полиэтилену проводят их выделение растворителями из измельченного поли-
полимера с последующей экстракцией СС14 и CS2 [104].
18*
276 Глава 6
Косвенные методы
Незначительные добавки, содержащиеся в веществе, могут хи-
химически реагировать с образованием производных или «уникальных»
соединений, к которым чувствителен метод ИК-спектроскопии. Вода
в неорганических жидкостях определялась в ходе реакции с карби-
карбидом кальция, в которой образуется ацетилен; количество последнего
измерялось методом ИК-спектроскопии [37]. При этом достижимы
чувствительности ниже 1 части на миллион, но в некоторых слу-
случаях необходимо специальное оборудование. В несколько больших
количествах воду можно определить обработкой 2,2-диметоксипропа-
ном в кислой среде с последующим измерением полосы поглощения
карбонильной группы образовавшегося ацетона [29]. Еще одним при-
примером косвенного метода является определение следов кислорода
в металлическом натрии. Натрий превращается в NaCl по реакции
Вюрца с к-амилхлоридом, оксид натрия реагирует с СО2, образуя
Na^Oj. Матрица из NaCl прессуется в таблетку, и определяется
количество карбоната. Этим методом можно определить 20—300 частей
кислорода на миллион [31].
Комбинация методов
Часто бьшает целесообразным совместное использование ИК-спект-
ИК-спектроскопии с УФ-спектроскопией, масс-спектрометрией, газожидкостной
или жидкостной хроматографией. Таким путем были успешно решены
многие неразрешимые другими методами задачи, например идентифи-
идентификация продуктов окисления о-ксилола [20]. Для рутинных анализов
эти методы менее распространены, но нельзя не учитывать такого
случая, когда методами титрования, полярографии или УФ-спектро-
скопии можно определить один или большее число компонентов,
к которым метод ИК-спектроскопии не чувствителен. Важным сообра-
соображением является относительная легкость, быстрота, точность совмест-
совместного использования методов по сравнению с ИК-спектроскопией.
Примеры
Для иллюстрации возможностей применения ИК-спектроскопии при
решении широкого круга задач из литературы выбран ряд примеров
количественных определений. Условия проведения этих анализов не
обязательно оптимальны; при использовании приемов, ранее обсуждав-
обсуждавшихся в этой главе, в некоторых случаях могут быть получены лучшие
результаты. Многие примеры количественного анализа методом ИК-
спектроскопии никогда не были опубликованы, так как они связаны
с запатентованными методами и соединениями.
а. Определение концентрации 0,1 —0,8 % 4-метил- 2,6-ди-тре»1-бутил-
Количественные приложения
277
О.2
I «.4
0,6-
1.0
1
: 1
: е,9
1
[\
1
з эа
мкм
Рис. 6.12. Спектры вазелинового масла с ингибитором и без него.
.масло, масло с ингибитором,
оба масла [5]. Reproduced, with permission, from
Applied Spectroscopy.
фенола в вазелиновом масле [5]. Фенол использовался в качестве
ингибитора окисления, причем был необходим быстрый и точный
метод контроля. Так как гидроксидная группа защищена разветвлен-
разветвленными mpew-бутильными группами, то при низких концентрациях
водородные связи отсутствуют и соответствующую полосу можно
использовать для анализа. Для такого анализа был применен также
метод отношения оптических плотностей [27].
Образец: вазелиновое масло.
Форма образца: жидкость в кювете толщиной 1 мм.
Измеряемая полоса: ОН, 3650 см B,74 мкм). Спектр сканиро-
сканировался в интервале 2,65 — 2,85 мкм. Нулевая оптическая плотность изме-
измерялась при 2,82 мкм (рис. 6.12).
Расчеты: графические; строился график зависимости А = Ащ — Аг&
от концентрации.
Воспроизводимость: s = 0,006% в интервале 0,2 — 0,8%.
Точность: s = 0,01 %
Необходимое время: 5 мин.
б. Анализ пяти ароматических соединений С10 [82]. Хотя состав-
составные части этой смеси летучи и, следовательно, поддаются анализу
методом газожидкостной хроматографии, этот метод в равной степени
применим и к нелетучим веществам. Спектры образцов записаны без
разбавления, толщины кювет нормированы (этого нельзя было бы
сделать при наличии других, неизмеряемых компонентов). Необходимое
время около 45 мин.
278
Глава 6
Образец, аналитические длины волн в мкм (длины волн стандартов)
относительные значения коэффициента поглощения для каждой аналитической
длины волны:
Вещество
8,56
(8,79)
10,0
A0,53)
10,65
A0,53)
11,54
A1,40)
8,92
(8,79)
Изобутнлбензол
в/яо/>-Бутилбензол
о-Диэтнлбензол
,/И-Днэтил бензол
и-Диэтилбензол
0,870
0,031
0,002
0,215
0,063
-0,068
0,529
-0,083
0,053
0,003
-0,074
-0,142
0,395
-0,021
-0,013
-0,079
0,004
0,056
0,591
0,064
0,244
0,023
0,071
0,020
0,760
Рис. 6.13. Зависимости оптической плотности от концентрации для смесей
нзопропанол — вода, определенные по спектрам НПВО [72]. Reproduced, with
permission, from Spectrochimica Acta,
Форма образца: неразбавленная жидкость в кювете толщиной 0,1 мм.
Стандарты: чистые углеводороды (99 %).
Измеряемая полоса: оптическая плотность при длине волны стан-
стандарта вычиталась из оптической плотности при аналитической длине
волны.
Расчеты: алгебраические.
Количественные приложения
279
1320
1714
Рис. 6.14. Проведение базовой линии и измерение поглощения в максимумах
полос полиэфира и шерсти [12]. Reproduced, with permission, from Applied
Spectroscopy.
Воспроизводимость: s не вычислялась; средняя абсолютная ошиб-
ошибка 0,5 %, средняя относительная ошибка 2,7 %.
в. Анализ бинарных смесей спирт — вода [72]. В этой работе опи-
описан анализ смесей метанол — вода, этанол — вода, к-пропанол — вода
и изопропанол — вода в интервале концентраций 0—100%, но экспери-
экспериментальные данные приведены только для последней смеси. Чтобы
избежать работы с тонкими кюветами, использован метод МНПВО
с 45°-м германиевым элементом. Несколько удивляет выполнимость
закона Бугера — Бера для групп с водородными связями.
Образец: спирт—вода.
Форма образца: жидкость в кювете МНПВО.
Измеряемая полоса: изопропанол, 950 см; вода, 3350 см (ис-
(исправленная на поглощение спиртовой группы ОН).
Расчеты: графические (рис. 6.13).
Воспроизводимость: s < 4 % из усреднения по семи измерениям.
г. Анализ композиций полиэфир — шерсть, содержащих 25 —75 % по-
полиэфира [12]. Был нужен быстрый и точный метод для характерис-
характеристики изделий из волокон полиэфира и шерсти. Для съемки спектров ис-
использовался метод МНПВО, а так как это бинарная система, можно
было бы еще привлечь метод отношения оптических плотностей.
Воспроизводимость была бы лучше, если расширить щели и умень-
уменьшить усиление (чтобы понизить уровень шумов на спектре).
Образец: волокна из полиэфира и шерсти.
Форма образца: ткани, плотно прижатые к обеим сторонам эле-
элемента МНПВО из KRS-5 с углом 45°.
280
Глава 6
'I 2,0
1,0
20 40 60
Содержание полиэфира,/'.
Рис. 6.15. Калибровочный график зависимости А (полиэфир)//! (шерсть)
от процентного содержания полиэфира [12]. Reproduced, with permission,
from Applied Spectroscopy.
Выбор полосы: шерсть, 1520 см 1 (перекрывание с полосой по-
полиэфира компенсируется при использовании отношения полос); по-
полиэфир, 1714 см-1 (рис. 6.14).
Приготовление стандарта: необработанные шерстяная и полиэфир-
полиэфирная ткани размельчаются по отдельности в мельнице Уайли, уравни-
уравнивается влажность, проводится взвешивание, приготовляется водная
суспензия, которая высушивается.
Расчеты: графические (рис. 6.15).
Воспроизводимость: s не вычислялась, но 9-кратное повторение
для образцов усредненного состава дало результаты с отклонением
в пределах 10 %.
д. Определение HF, HC1, SiF4 и HCF3 в газообразном WF6 [25].
Ю00 4000 ЭООО 2600 2000
Волновое число
1М0 1400 1300 1200 1100
1000
7 • I 10
Длина волны, мкм
12
Рис. 6.16. Спектр поглощения WF6 при <~ 1,3 атм и 25°С в кювете толщиной 10 см. Показаны также полосы
поглощения примесей [25]. Reproduced, with permission, from Applied Spectroscopy.
282 Глава 6
Важным методом получения металлического вольфрама высокой чисто-
чистоты является восстановление газообразного WF6. Примеси в этом газе
оказывают большое влияние на свойства металла, и их содержание
должно тщательно контролироваться. Иногда из-за исключительной
реакционной способности WF6 анализ осложняется; например, он реаги-
реагирует с влагой, образуя HF. Использована специальная система под-
подготовки образцов, которая позволила промывать ИК-кюветы сухим
и очищенным азотом.
Образец: газообразный WF6 при атмосферном давлении.
Кювета: 10-сантиметровая, изготовленная из нержавеющей стали
типа 304 с окнами из BaF2.
Выбор полосы (рис. 6.16):
_. Чувствительность,
Соединение Частота, см ~ весовая часть на миллион
HF 3850 B,60 мкм) 100
НС1 2860 C,50 мкм) 100
SiF4 1030 (9,73 мкм) 10
HCFj 1150 (8,71 мкм) 10
Приготовление стандарта: введение стандартных добавок примесных
газов в кювету производится газовым шприцом (для SiF4 исполь-
используется тефлоновый шприц).
Расчеты: алгебраические (нелинейные для HF и НС1).
Воспроизводимость: s не вычислялась, но «лучшая воспроизводи-
воспроизводимость, достигаемая в последовательных рутинных анализах при ис-
использовании сдвоенных образцов, составляла ± 6 %». Большая часть
этой ошибки обусловлена приготовлением образца; при повторном
сканировании одного и того же образца воспроизводимость состав-
составляет около ± 1 %.
е. Количественный анализ содержания Me3Si0j,2 и SiOH йа по-
поверхности обработанной SiO2 [2]. Поверхность силикагеля и аэросила
SiO2 образована силанольными группами (SiOH) и адсорбированной
водой. Поверхность можно модифицировать, проведя следующую
реакцию с (CH3KSiCl:
(CH3KSiCl ->=SiOSi(CH3K + HC1.
В ходе изучения адсорбции интересно знать степень обработки. Этот
анализ бьш разработан с целью получения такой информации. Посколь-
Поскольку точное количество порошка в канале образца определить трудно,
использовалось отношение оптических плотностей полос обрабатываю-
обрабатывающего реагента (и SiOH) и субстрата. Калибровка для (CH3KSi01/2
была выполнена по результатам анализа на углерод. Было принято,
Количественные приложения 283
что на необработанной поверхности содержание гидроксидных групп
составляет 8 мкмоль/м?. Уменьшение оптической плотности полосы
SiOH прямо пропорционально увеличению оптической плотности по-
полосы (CHjKSi01/2.
Образец: порошок SiO2 (силикагель или аэросил).
Форма образца: порошок, помещенный между пластинками NaCl,
имеющий пропускание 15 — 40% при 800 cm~i.
Выбор полосы: (CH3KSiOi,2, 2965 см-1 C,37 мкм); SiOH, 940 см-1
A0,6 мкм), субстрат SiO2, 800 см-1 A2,5 мкм).
Приготовление стандарта: SiO2 был обработан (CH3KSiCl, промыт
и высушен при 110°С в течение ночи. Для построения калибро-
калибровочной кривой стандарты подвергались анализу на содержание уг-
углерода.
Расчеты: графические.
Воспроизводимость: j = 0,0039 для ACH/Asi = 0,1131, или 3,5 отн.%.
Кинетические исследования
Прямое использование методов ИК-спектроскопии в исследованиях
по химической кинетике возможно при условии, что реакция явля-
является достаточно медленной и время отклика спектрофотометра не ог-
ограничивает точность измерений. Существует несколько методов изу-
изучения зависимости концентрации от времени. Простейший из них
состоит в том, что реакция в небольшой аликвотной4 пробе реакцион-
реакционной смеси останавливается или разбавлением, или введением де-
дезактивирующего катализатора, или понижением температуры, а образ-
образцы сканируются в подходящих условиях. В другом методе прово-
проводится реакция непосредственно в кювете ИК-спектрофотометра при
многократной записи спектра (или его части). Если интерес пред-
представляет только одна составная часть реакционной смеси, то спек-
спектрофотометр устанавливается на фиксированную частоту полосы погло-
поглощения и оптическая плотность наблюдается как функция времени;
примером такого рода является исследование термического разложения
оксида этилена [99]. Во многих случаях, когда нельзя использовать
растворитель, удовлетворительные результаты можно получить, ра-
работая с толстыми образцами в области обертонов. Конечно, при этом
должна тщательно контролироваться температура образца.
Камерон и др. [23] описали остроумную кювету и систему сме-
смешивания, которые позволяют использовать более медленное сканиро-
сканирование при исследовании реакций, протекающих с не слишком боль-
большими скоростями @,1 — 1 с), на обычных спектрофотометрах (рис. 6.17
и 6.18).
На интерференционных спектрофотометрах можно достигнуть бо-
более быстрого сканирования. Сообщалось [69] о наблюдении за газо-
газофазным нитрованием бутадиена смесью NO2 — N2O4 при односекунд-
ном сканировании с интервалами 6,5 с. Специальный метод, в ко-
284
Глава 6
9
Рис. 6.17. Устройство для введения реагентов в реактор. Время реакции
изменяется при изменении скорости потока и(или) расстояния иглы плунжера
от ИК-кюветы.
/ - давление азота: 2 - PhSiClj, эфир; 3 — вода, эфир; 4 — расходомеры; 5 — реактор; б - выход; 7 — ИК-юо-
вета; 8 — иасос; 9 — термостат [23]. Reproduced, with permission, from Industrial Engineering Chemistry
Fundamentals. Copyright American Chemical Society.
тором в системе непрерывного сканирования с преобразованием
Фурье объединено спектральное и временное модулирование несущей
частоты, позволяет регистрировать короткоживущие частицы с разре-
разрешением по времени лучше 2 мкс [73].
Непрерывно действующие
автоматические ИК-анализаторы
Естественно, что такой полезный лабораторный метод, как ИК-
спектроскопия, был применен на предприятиях химической и нефтя-
нефтяной промышленности для непрерывного анализа состава органических
смесей. От анализа до контроля какого-либо химического или фи-
физического процесса только один шаг, и поэтому многие из них
управляются сигналами, поступающими с ИК-контролирующего уст-
устройства. Хотя проточные ИК-анализаторы вытесняются другими ти-
типами устройств, особенно газожидкостными хроматографами, во мно-
Количественные приложения
285
oPhSLClj(OH)
QPhSUOH),
0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9
-Среднее время г, с
Рис. 6.18. Зависимость концентрации силанов при гидролизе PhSiCl3 от
среднего времени реакции [23].
Msi = 0,073
Обшее содержание силана в молях
Литры раствора
п,.» Моли добавленной Н,0
Ми п = 0,215 -—,
П2и Литры раствора
Моли добавленной НО
«на
Литры раствора
гих случаях ИК-спектроскопия остается лучшим аналитическим ме-
методом.
Так как условия работы на промышленных предприятиях обычно
отличаются повышенными загрязненностью, агрессивностью, уровнем
вибрации, температурами, то должны применяться" специально скон-
сконструированные прочные, надежные, закрытые приборы. В литературе
описаны приборы различного уровня сложности от простейших фото-
фотометров до многоточечных диспергирующих анализаторов [9, iff].
В этом разделе будут рассмотрены некоторые из основных конструк-
конструкций и коротко обсуждены их преимущества и недостатки.
286 Глава 6
Недиспергирующие анализаторы |35]
Простейший анализатор недиспергирующего типа, состоящий из
источника, кюветного отделения, приемника излучения и регистрирую-
регистрирующего устройства, показан на рис. 6.19. Он очень чувствителен, но
любой газ, поглощающий ИК-излучение, будет вызывать сигнал.
Прибор можно сделать частично селективным, используя фильтр либо
в виде отдельной детали, либо как специальное покрытие приемника.
Например, органическая смола, нанесенная вместо черни на рабочую
поверхность приемника, делает прибор чувствительным в области
частот валентных колебаний С—Н и устраняет мешающее влияние,
скажем, углекислого газа. Та же смола, используемая в виде све-
светофильтра в пучке, будет вызьшать противоположный эффект.
Простой фотометр является однолучевым прибором и поэтому
чувствителен к флюктуациям излучения источника, изменениям чувст-
чувствительности приемника и загрязнениям в потоке исследуемого газа.
Если добавить второй приемник и кювету (ячейку), как показано
на рис. 6.20, то эти эффекты можно устранить, а прибор сделать
более селективным. Одна из кювет F\, служащая фильтром, заполнена
газом, который должен определяться как примесь, поэтому изменения
концентрации в кювете образца SC не будут сказываться на излу-
излучении, проходящем через этот канал. Другая кювета F2 может быть
пустой или содержать мешающий газ при подходящем давлении, если
таковой присутствует в исследуемом потоке. Приемники Dl и D2
включены навстречу друг другу, и сигнал, выходящий из анализа-
анализатора, равен разности между двумя большими сигналами. Такую
систему принято называть анализатором с негативной фильтрацией
(negative filter analyzer). Если два пучка подобраны точно, то анали-
анализатор чувствителен к определяемому компоненту и не чувствителен
к другим помехам. Такой анализатор использовался для анализа бу-
тенов в газообразном бутадиене и этилбензола в жидком мономере
стирола [122].
Принцип позитивной фильтрации можно реализовать на приборе
сходной конструкции, используя селективный приемник (positive filter
analyzer). В наиболее общем виде такой приемник имеет форму
камеры, содержащей анализируемый газ (или газ с полосами погло-
поглощения, совпадающими с полосами жидкого образца, который анали-
анализируется в потоке). Одна из стенок камеры является тонкой ме-
металлической диафрагмой, которая совместно с решеткой действует как
конденсорный микрофон [71]. Если излучение модулируется, то нагре-
нагревание и охлаждение газа в приемнике вызывают изменения давления,
которые приводят к возникновению переменного сигнала на выходе.
Этот сигнал усиливается и используется как мера концентрации газа
в кювете образца. Одна из таких конструкций показана на рис. 6.21.
Недиспергирующие газовые анализаторы подробнее рассматрива-
рассматривались в работе [53]. Систематические операции для повышения чув-
Количественные приложения
287
S
Рис. 6.19. Простой фотометрический анализатор.
S - источник ИК-излучения; SC-кювета образца; D — приемник; М — регистрирующее устройство.
к
S SC
Рис. 6.20. Недиспергирующий анализатор с негативной фильтрацией.
S О
JL
SC
L
См
Рис. 6.21. Недиспергирующий анализатор с позитивной фильтрацией.
5-источник излучения; SC и RC - кюветы образца и сравнения соответственно; F — кювета фильтра;
/>! и D2 — камеры приемника; М — регистрирующее устройство.
ствительности недиспергирующих двухлучевых анализаторов и мини-
минимизации помех описаны в работах [8, 68].
Если присутствует большое количество поглощающих примесей, то
ни анализатор с негативной фильтрацией, ни анализатор с позитив-
позитивной фильтрацией не дает полностью удовлетворительных результатов,
хотя и сообщалось, что прибор с негативной фильтрацией позво-
позволяет добиваться лучшей компенсации. Анализаторы обоих типов наи-
288 Глава 6
более эффективны в том случае, когда аналитические полосы находятся
вблизи максимума кривой излучения, так как при этом на приемник
попадает большая энергия. Для устранения некоторых из недостат-
недостатков недиспергирующих анализаторов были созданы прочные миниа-
миниатюрные спектрометры, используемые на предприятиях.
Подобно диспергирующим анализаторам для использования в про-
промышленности были также приспособлены спектрометры с диэлектри-
диэлектрическим фильтром (стр. 31). Проточные устройства позволяют
отбирать пробы в различных точках, как, например, при контроле
состояния воздухд на химических производствах [43].
Диспергирующие анализаторы
Сложные задачи, включающие анализ мешающих компонентов,
часто можно решить с использованием диспергирующих анализаторов.
Фактически почти любой анализ, проводимый в лаборатории, может
быть выполнен таким прибором в автоматическом режиме. Описано
[11] несколько типов анализаторов, соответствующих одно- и двух-
лучевым лабораторным спектрометрам и спектрофотометрам. Такие
приборы можно приспособить для анализа многокомпонентных систем
последовательным выведением длин волн, на которых проводится
анализ. Для коррекции нулевых концентраций в случае помех можно
подключить небольшую ЭВМ.
Достоинствами двухлучевых диспергирующих анализаторов явля-
являются их более высокая селективность и стабильность. Непрерывное
сравнение двух длин волн в таком приборе позволяет эффективно
исключать помехи. Баланс пучков осуществляется либо по методу
оптического нуля, либо регистрацией отношения электрических сиг-
сигналов.
В целом диспергирующие анализаторы легче приспособить к ре-
решению различных задач, а их регулировка требует меньше времени.
Такие характеристики, как стабильность, надежность и чувствитель-
чувствительность, довольно высоки, а иногда и превосходят соответствующие
характеристики недиспергирующих анализаторов; помехи со стороны
других примесей устраняются легче. Эти приборы лучше подходят
для работы в энергетически ограниченной области спектра ниже
1000 см-1 A0 мкм).
При работе с ИК-анализаторами возникают обычные проблемы
подготовки образца и запаздывания показаний прибора. Потоки образ-
образца через тонкие жидкостные кюветы могут оказаться столь медлен-
медленными, что потребуется обходной трубопровод для основных потоков
анализируемого вещества с целью «шунтирования» ИК-кюветы. При
использовании элемента НПВО толщина кюветы несущественна и через
кюветное отделение может направляться сильный поток. В любом
случае «мертвое время» должно быть минимальным. Для удаления
загрязнений может потребоваться фильтрование потока образца, а для
Количественные приложения 289
предохранения от затвердевания образца - повышенная температура.
Почти в каждом случае возникают свои собственные проблемы и за-
зачастую большие усилия требуются для решения задач, связанных
с подготовкой образца, чем для приспособления анализатора к ана-
аналитической системе.
Большинство анализаторов снабжено периодически действующим
автоматическим или ручным устройством для калибровки по образцу
известного состава. Частая и тщательная проверка калибровки также
важна в проточных анализаторах, как и при проведении лаборатор-
лабораторных измерений. При правильной эксплуатации ИК-анализаторы редко
простаивают [11].
ЛИТЕРАТУРА
1. Adda J., Blanc-Patin Е., Jeunet R., Grappin R., Mocquot G., Pousardieu B.,.
Ricordeau G., Lait, 48, 145, 293 A968).
2. Ahmed A., Gallei E., Appl. Spectrosc., 28, 430 A974).
3. American Society for Testing and Materials, 1976 Annual Book pf ASTM
Standards, Part 30, ASTM, Philadelphia, Method E 180-67, 1976, p. 538.
4. American Society for Testing and Materials, 1977 Annual Book of ASTM
Standards, Part 42, ASTM, Philadelphia, 1977.
5. Bain G. #., Appl. Spectrosc, 10, 193 A956).
6. Baker В. В., J. Am. Ind. Hyg. Assoc, 35, 735 A974).
7. Baker В. В., Jr., Carlson D. P., Anal. Chem., 46, 1583 A974).
8. Baker W. J., Anal. Chem., 28, 1391 A956).
9. Bard С. С, Porro Т. J., Rees H. L., Anal Chem., 27, 12 A955).
10. Barnett H. A., Bartoli A., Anal. Chem., 32, 1153 A960).
11. Bam A. M., Ruhl H. D., Chem. Eng. Progr., 64, (8), 45 A968).
12. Basch A., Tepper E., Appl. Spectrosc., 27, 268 A973).
13. Bauman R. P., Absorption Spectroscopy, Wiley, New York, 1962.
14. Beer A., Ann. Physik. Chem. [2], 86, 78 A852).
15. Ветку F. F., Rappaport G., Charles J. Cleary Awards for Papers on
Materials Sciences, Library of Congress, Washington, 1962, pp. 57—70.
16. Blakeway J. M., Putinam N. A., Anal. Chem., 35, 630 A963).
17. Blout E. R., Parrish M., Jr., Bird G. R., Abbate M. J., J. Opt. Soc. Am.,
42, 966 A952).
18. Bouguer P., Essai d'Optique sur la Gradation de la Lumiere, 1729.
19. Bradley К. В., Potts W. J., Jr., Appl. Spectrosc, 12, 77 A958).
20. Brown R. A., Quiram E. R., Appl. Spectrosc, 17, 33 A963).
21. Burley R. A., Bennett W. J., Appl. Spectrosc, 14, 32 A960).
22. Burns E. A., Muraca R. F., Anal. Chem., 31, 397 A959).
23. Cameron J. H., Kleinhenz T. A., Hawley M. C, Ind. Eng. Chem., Fund.,
14, 328 A975).
24. Caputi A., Jr., Udea M., Am. J. Enol. Viticult., 24, 116 A973).
25. Chaney С L., Chin J., Appl. Spectrosc, 28, 139 A974).
26. Chaney L. W., McClenny W. A., Environ. Sci. Technol., 11, 1186 A977).
27. Cleverley В., Dolby R., Appl. Spectrosc, 32, 187 A978).
28. Collier G. L., Panting А. С. М., Spectrochim. Acta, 14, 104 A959).
29. Critchfield F. E., Bishop E. Т., Anal. Chem., 33, 1034 A961).
19 - 586
290 Глава б
30. Daasch L. W., Anal. Chem., 19, 779 A947).
31. deBruin H. J., Anal. Chem., 32, 360 A960).
32. Diamond W. J., Appl. Spectrosc., 13, 77 A959).
33. Dijkstra G., Spectrochim. Acta, 11, 618 A957).
34. Drushel H. V., Iddings F. A., Anal. Chem., 35, 28 A963).
35. Fastie W. G., Pfund A. H., J. Opt. Soc. Am., 37, 762 A947).
36. Fochler H. S., Appl. Spectrosc., 17, 105 A963).
37. Forbes J. V., Anal. Chem., 34, 1125 A962).
38. French A. R., Benham J. V, Pullukat T. J., Appl. Spectrosc., 28, 477 A974).
39. Freund S. M., Sweger D. M., Anal. Chem., 47, 930 A975).
40. Freund S. M., Sweger D. M., Travis J. C, Anal. Chem., 48, 1944 A976).
41. Friedel R. A., Queiser J. A., Anal. Chem., 29, 1362 A957).
42. Fujiyama Г., Herrin #., Crawford B. L., Jr., Appl. Spectrosc., 24, 9 A970).
43. Gearhart H. L., Cook R. L., Whitney R. W., Hefner R. D., Anal. Chem.,
49, 893 A977).
44. Golden B. M., Yeung E. S., Anal. Chem., 47, 2132 A975).
45. Green B. D., Steinfeld J. /., Environ. Sci. Technol., 10, 1134 A976).
46. Hammer С F., Roe H. R., Anal. Chem., 25, 668 A953).
47. Hampton R. R., Rub. Chem. Technol., 45, 546 A972).
48. Hanst P. L., Adv. Environ. Sci. Technol., 2, 91 A971).
49. Hanst P. L., Appl. Spectrosc, 24, 161 A970).
50. Hawranek J. P., Jones R. N., Spectrochim. Acta, 32A, 111 A976).
51. Heigl J. J., Bell M. F,, White J. U., Anal. Chem., 19, 293 A947).
52. Herget W. F., Jahnke J. A., Burch D. E., Gryvnak D. A., Appl. Opt.,
15, 1222 A976).
53. Hill D. W., Powell Г., Nondispersive Infrared Gas Analysis in Science,
Medicine and Industry, Hilger, London, 1968.
54. Hilton С L., Anal. Chem., 31, 1610 A959).
55. Hinkley E. D., Environ. Sci. Technol., 11, 564 A977).
56. Hinkley E. D., Opto-electronics, 4, 69 A972).
57. Hirschfeld Т., Opt. Eng., 13, 15 A974).
58. Hirschfeld Т., Res. Devel., 27 G), 20 A976).
59. Hollingdale-Smith P. A., Can. J. Spectrosc., 11, 107 A966).
60. Homer H. J., Weiler J. E., Angelotti N. C, Anal. Chem., 32, 858 A960).
61. Huang A., Firestone D., J. Assoc. Offic. Anal. Chem., 54, 1288 A971).
62. Hughes H. K., Appl. Opt., 2, 937 A963).
63. Jakes J., Slabina M., Schneider В., Appl. Spectrosc., 26, 389 A972).
64. Johnson D. R., Cassels J. W., Brame E. G., Westneat D. F., Anal. Chem.,
34, 1610 A962).
65. Jones R. N., J. Am. Chem. Soc, 74, 2681 A952).
66. Jones R. N., Escolar D., Hawranek J. P., Neelakantan P., Young R. P.,
J. Molec. Struct., 19, 21 A973).
67. Kagel R. O., Baker J. A., Appl. Spectrosc, 28, 65 A974).
68. Koppius O. G., Anal. Chem., 23, 554 A951).
69. Lephardt J. O., Vilcins G., Appl. Spectrosc, 29, 221 A975).
70. Low M. J. D., Abrams L., Appl. Spectrosc., 20, 416 A966).
71. Luft K. F., Z. Tech. Physik, 24, 97 A943).
72. Malone С. Р., Flournoy P. A., Spectrochim. Acta, 21, 1361 A965).
73. Mantz A. W, Appl. Spectrosc., 30, 459 A976).
74. Mantz A. W., Appl. Spectrosc, 30, 539 A976).
75. Martin A. ?., Trans. Faraday Soc., 47, 1182 A951).
Количественные приложения 291
76. Maxwell J. С, Barlow С. Н., Spallholz J. E., Caughey W. S., Biochem.
Biophys. Res. Commun., 61, 230 A974).
77. McDowell R. S., Appl. Spectrosc, 26, 405 A972).
78. McKelvey J. M., Hoelscher H. ?., Anal. Chem., 29, 123 A957).
79. Meehan E. J., Fundamentals of Spectrophotometry, in I. M. Kolthoff and
P. J. Elving, Eds., Treatise on Analytical Chemistry, Part I, Section D-3,
Vol. 5, p. 2753, Interscience, New York, 1964.
80. Murphy J. F., Appl. Spectrosc., 16, 139 A962).
.81. Nordstrom R. J., Shaw J. #., Skinner W. R., Chan W. #., Calvert J. G.,
Uselman W. M., Appl. Spectrosc, 31, 224 A977).
82. Perry J. A., Anal. Chem., 23, 495 A951).
83. Perry J. A., Appl. Spectrosc. Rev., 3, 229 A970).
84. Perry J. A., Bain G. H., Anal. Chem., 29, 1123 A957).
85. Perry J. A., Bain G. H., Trover W. В., Appl. Spectrosc., 10, 191 A956).
86. Perry J. A., Sutherland R. G., Hodden N., Anal. Chem., 22, 1122 A950).
87. Pillion E., Rogers M. R., Kaplan A. M., Anal. Chem., 33, 1715 A961).
88. Potts W. J., Jr., Chemical Infrared Spectroscopy, Vol. 1, Techniques, Wiley,
New York, 1963.
89. Potts W. J., Jr., Smith A. L., Appl. Opt., 6, 257 A967).
90. Ramsay D. A., J. Am. Chem. Soc, 74, 72 A952).
91. Robinson D. Z., Anal. Chem., 23, 273 A951).
92. Robinson D. Z.; Anal. Chem., 24, 619 A952).
93. Rueda D. R., Balta Calleja F. J., Hidalgo A., Spectrochim. Acta, 30A,
1545 A974).
94. Ruppin R., Englman R., Rep. Prog. Phys., 33, 149 A970).
95. Saier E. L, Cousins L. R., Basila M. R., Anal. Chem., 34, 824 A962).
96. Saier E. L, Hughes R. H., Anal. Chem., 30, 513 A958).
97. Schnurmann R., Kendrick E., Anal. Chem., 26, 1263 A954).
98. Sharpless N. E., Gregory D. A., Appl. Spectrosc., 17, 47 A963).
99. Simard G. L., Steger J., Mariner Т., Sqlley D. J., Williams V. Z., J.
Chem. Phys., 16, 836 A948).
100. Simard R. G., Hasegawa I., Bandaruk W., Headington С. Е., Anal. Chem.,
23, 1384 A951).
101. Smith A. L., Appl. Spectrosc, 12, 153 A958).
102. Smith A. L., Infrared Spectroscopy, in J. W. Robinson, Handbook of
Spectroscopy, Vol. II, CRC Press, Cleveland, 1974.
103. Smith A. L, Kiley L. В., Appl. Spectrosc, 18, 38 A964).
Г04. Spell H. L., Eddy R. D., Anal. Chem., 32, 1811 (I960).
105. Sterling G. В., Cobler J. G., Erley D. S., Blanchard F. A., Anal. Chem.,
31, 1612 A959).
106. Stewart J. ?., Appl. Opt., 1, 75 A962).
107. Toma S. Z, Goldberg S. A., Anal. Chem., 44, 431 A972).
108. Toribara T. Y., Di Stefano V, Anal. Chem, 26, 1519 A954).
109. Tosi C, Ciampelli F., Fortschr. Hochpolym. Forsch., 12, 87 A973).
110. Try on M., Horowitz E., Infrared Spectrophotometry, in G. M. Kline, Ed.,
Analytical Chemistry of Polymers, Part II, Interscience, New York, 1962,
pp. 291-334.
111. Tuddenham W. M., Lyon R. J. P., Anal. Chem., 32, 1630 A960).
112. Walton W. W., Hoffman J. I., Principles and Methods of Sampling in
I. M. Kolthoff and P. J. Elving, Eds., Treatise on Analytical Chemistry,
Part I, Section A, Vol. 1, p. 67, Interscience, New York, 1959.
292 Глава 6
113. Westneat D. F, Anal. Chem., 33, 812 A961).
114. Whetsel К. В., Robertson W. E., Krell M. W., Anal. Chem., 32, 1281 A960).
115. White J. U., Ward W. M., Anal. Chem., 37, 268 A965).
116. Wiberley S. E., Sprague J. W., Campbell J. E., Anal. Chem., 29, 210 A957).
117. Wiens R. #., Zwick H. #., Trace Gas Detection by Correlation Spectroscopy,
in J. S. Mattson, H. B. Mark, Jr., and H. G. MacDonald, Jr., Eds.,
Infrared, Correlation, and Fourier Transform Spectroscopy, Marcel Dekker,
New York, 1977.
118. Wilks P. A., Jr., Appl. Spectrosc, 23, 63 A969).
119. Willbourn A. #., J. Polym. Sci., 34, 569 A959).
120. Wood D. L., Luongo J. P., Mod. Plast., 38, 132 (March, 1961).
121. Wright N., Ind. Eng. Chem., Anal. Ed., 13, 1 A941).
122. Wright N., Hersher L. W., J. Opt. Soc. Am., 36, 195 A946).
123. Young E. F., Hannah R. W., in W. W. Wendlandt, Ed., Modern Aspects of
Reflectance Spectroscopy, Plenum Press, New York, 1968.
124. Zenker W., Anal. Chem., 44, 1235 A972).
Дополнительная литература
1. Номенклатурные правила ИЮПАК по химии. Т. 1. Неорганическая химия,
физическая химия, полутом 2.-М.: ВИНИТИ, 1979, с. 308.
2. ГОСТ 7601-78. Физическая оптика. Термины, буквенные обозначения и оп-
определения основных величии. — М.: Госкомитет по стандартам, 1979.
3. Бернштейн И. Я., Каминский Ю. Л. Спектрофотометрический анализ
в органической химии.—Л.: Химия, 1975.
4. Ж. аналит. химии, 30, 2058 A975).
5. Васильев А. Ф. Теоретические основы современных методов количествен-
количественного анализа многокомпонентных систем по спектрам поглощения. Докт.
дисс- М.: 1976.
6. Зайдель А. Н. Ошибки измерений физических величин. - Л.: Наука, 1974.
7. Доерфель К. Статистика в аналитической химии.—М.: Мир, 1969.
8. Золотарев В. М., Лыгин В. И., Тарасевич Б. #., Успехи химии, № 1,
24 A981).
9. Прикладная инфракрасная спектроскопия. Пер. с англ./Под ред. Д. Кендал-
ла.- М.: Мир, 1970.
10. Центр, ин-т научн.-техн. электротехнич. пром-сти и приборостроения. Ав-
Автоматические газоанализаторы: Сб. статей.—М.: 1961.
Приложение 1
КОРРЕЛЯЦИОННЫЕ ДИАГРАММЫ
см'*
3600
3500
3400
3300
3200
3100
3000
2900
2800
Спярг
joeqmm
I c-o
Амив
Первич-
Первичный амин
(рюпор)
Com
Спирты сн-гаиья
Вторичный
Этжсив
сив
Борнин
I Парппмай
Оисни
«миг
оси
'I
ен.
lCHs
-2,8
-2,9
-3,0
-3,1
-3,2
-3,3
-3,4
-3,5
-3,6
294
Приложение 1
ом '
2700
2600
2500
2400
2300
2200
2100 -
2000 -
1900 L
soh
1 Нитрилы
1 ДЛИфВТИЧ
I ароматиигсчив
SIH
CIC
Аллах
МКМ
-- 3,7
- 3,8
-- 4,0
- 4,2
- 4,4
- 4,6
"- *,*
-¦ 5,0
- S.2
Корреляционные диаграммы
295
см
1900 Ь
цмс/нмсшй
8й
1800
lu
Галогемнгиври|
фткой
«ислотв I
Га/мгаиаигиВр«е|
«ро««г«««««<
Циклический
•вгивриВ
|/1тоапп(шй
екги'риа
мрбомгг
Алиштачсиий
1700
№00
Обертон
сн=сн.
5-члеиный
пактом
|ИМК|
• Cm
тыпл
Нитрит
Вторичный
Тргичнш)
ЗИИО
lawn I C=C
Нитрат
MKM
- 5,3
- 5,4
- 5,5
- 5,4
- 5,7
- 5,8
- 5,9
- 6,0
- 6,1
- 6,2
- 6,3
296
Приложение 1
см*
1600 -
1500
1400
1300
Ароматически
кольцо
вторичный
аимо
Спи
кислот
Амино-
Аминокислоты
Сель
первич-
первичного
амина
I Вторичный
[карванат
Ароматическое
Соль
первич-
первичного
амина
кольцо
Метил
снэо
CH}S
Сулырат
.Геииналь-
ГииНиие-
Метю тмима»
|/Ру"М
Нетилкетон
Лактам
Соли
кислот
Иитимувкат
со»
аммони
Сулыроиамио
Лактам
,снэ
Сом
вторичного
амииа
Иочаеина
Нитоо
**
g
сн2
—
Карболовая
кислота
1 Винил
цис-i
СН=СН
Нитро ['слоеные
яриры
борной
1 КИСЛОТЫ
Сулырон
MKI
- 6.3
- 6,4
- 6,5
- 6.6
- 6.7
- 6,8
- 6,9
- 7,0
- 7,1
- 7,2
- 7.3
- 7.4
- 7.5
- 7.6
- 7.7
- 7.8
Корреляционные диаграммы
297
см
1300
Кароомпая
кислота
Нитрат
Оюжнмй эфир
уксусной кисло-
TW
1200
1100
Вторичный
ароматический
амин
Алифатичес-
Алифатический карбонат
Ароматический
кзроонат
Ароматический
простой эци
рулыроним норий
Алифатичес-
Цикли-
чвжий
простой
эфир
СульфОками!
•Сн„
э - 7,6
Феноа
(таервый)
-- 8,0
Бторичиьш
спирт
Алифатический
простой >фир
Сульоюн
1000
спирт
¦ Аигиорио
Сулмриномя
кислота
СулмомЫ)
Аромати-
Ароматический
простой
- 8,2
-8,4
-8,6
- 8,8
- 9,0
-9,2
- 9,4
- 9,6
-9,8
10,0
298
Приложение 1
см"
1000 -
900
800
700
|Виим
|с=сн.
1,1.-
3«мещеии« |
ЧвТОртИЧНМ
1MMQI
СМИ
ITpam-
см=сн
Й 1НГИ1
I
РО-
Ароиети-
I, I, 4 -
IatMtmenii
ироматх-
ивскои
iHtttptr
1,2-
I Замшами •
I ароматическом
¦ кольце
1,3, S-
1, 3-
Замвще
аромятнчвском
кольце
1™Т
ком колеи»
мкм
-. 10
- и
Злокмви
I, I, ¦-
Замещение I ароя»
тицеском кольце
Нитрит
I, г, s-
ое •рона-
я
Г'3"
чкккй ¦ эроштм-
изотие- ческам
цпшат квкы»
СН=СН
|НИТ|ЯТ
1,2,3-
Замешеии: ¦ аро
Нитргг
1 Замещение в ар»
магическом кольц
...I
CCI,
600
(мстежетшм
- 12
- 13
- 14
- 15
- 16
Корреляционные диаграммы
299
2000 f900 f800 T7CXJ 2000 1900 Г800 1700 20С0 1900 18001700
2000 1900 1800 1700 2000 1900 1800 1700 2000 1900 1800 170O
CM"'
Рис. 1. Характеристичность спектра в области обертонов и составных полос
для различных типов замещения в бензольном кольце.
Полосы малоинтенсивны, и поэтому спектры получены при толщине поглощающего слоя ~0,1 мм. Вид
спектра более информативен, чем волновые числа полос поглощения. Данные взяты из работы Young С. W.',
Du Vail R.B., Wright N.. Anal. Chem., 23. 709 A951). Reproduced with permission of American
Chemical Society.
Приложение 2
ХАРАКТЕРИСТИЧЕСКИЕ ЧАСТОТЫ ПОГЛОЩЕНИЯ
В приведенной ниже таблице представлены характеристические ча-
частоты некоторых из наиболее часто встречающихся химических групп.
Таблица не претендует на исчерпывающую полноту и ее цель — показать
возможные пути для дальнейшего исследования структуры. Чистое
вещество можно охарактеризовать с помощью ИК-спектров только
при продуманном использовании стандартных спектров и нескольких
сборников из предлагаемых в гл. 3. Для дальнейшего изучения ре-
рекомендуются монографии, в которых даны более полные обзоры и об-
обсуждения корреляций [1, 2], и сборник модельных спектров [3].
В этой таблице все частоты поглощения даны в волновых числах
(в некотором интервале) для растворов в инертных растворителях,
если не оговорено особо. Часто положение полосы внутри интервала
связано с некоторой структурной особенностью молекулы. Интервалы
имеют приблизительный характер; существует вероятность того, что
некоторые структуры поглощают вне данного интервала. Обозначения
типов нормальных колебаний также носят приближенный характер.
Например, в валентном колебании С—О—С в простых эфирах участвуют
и некоторые другие скелетные колебания. Использованы следующие обо-
обозначения: v* — асимметричное валентное колебание, 5s — симметричное
деформационное колебание, со - веерное колебание, р - маятниковое ко-
колебание, R - алифатическая группа, Аг - арильная (ароматическая)
группа, X - галоген, А" - анион, М+ - катион, S - сильная (интенсив-
(интенсивная полоса), М - средняя, W - слабая, V - переменная интенсивность,
ip - плоскостное колебание, оор — внеплоскостное колебание, комб.—
комбинационная полоса.
1. Карбоаовые кислоты При комнатной температуре, кроме
(дамершя форма) случаев крайне разбавленных раство-
растворов, карбоновые кислоты находятся
в виде димеров
О"*Н—О
• \
R—С С—R
\ •
О—Н"О
Характеристические частоты поглощения
301
vqh 2500-3100 S
vc=o 1700- 1720 S
5он(ф) 1395-1440 W
vc_0 1280-1315 S
5он(ооР) 875-960 M
шсн 1180-1345 W
Карбоновые кислоты
(мономерная форма)
3500-3580 W
vc=o 1740-1800 S
5ОН 1280-1380 М
vc_o 1075-1190 S
2. Галогенангндрнды кислот
О vc==o 1795-1810 S
II
RCX
920-965 М
~1200 S
850-890 V
3. Соли кшслот
О'
R—С
О
М+
=о 1540-1650 S
fcrzro 1360-1450 М
Широкая (см. рис. 5.11). Узкие слабые
полосы часто появляются около
2500-2700 см
Электроотрицательные группы в а-пс-
ложении повышают частоту (на 15 —
20 см для Q или F и 6 см для Вг).
Сопряжение с группой С=С или
арильной группой понижает частоту
до ~1690 см
Широкая
В твердом состоянии наблюдается
прогрессия полос. Вели N — число по-
полос, то число углеродных атомов в
кислоте равно 2N (четное число) или
2N - 1 (нечетное число)
Наблюдается в паровой фазе при по-
повышенных температурах, в газохрома-
тографических фракциях или в очень
разбавленных растворах
Резкая
Сопряжение с двойной связью или
фенильным кольцом понижает часто-
частоту на 25—30 см'1. Хлорангидриды
ароматических кислот могут иметь
вторую, более слабую полосу при
1735-1750 см (дублет резонанса
Ферми)
Хлорангидриды ароматических кислот
Частоты, зависящие от кристалличе-
кристаллической структуры и поэтому трудно
поддающиеся предсказанию
Формиаты: -2390, 1600,
1360, 775
Ацетаты: 1550-1600,
1400-1450,
~ 1050, 1020,
925
302
Приложение 2
Оксалаты: vc=
~ 1620, 1320,
770, 515
4. Спирты
а. Одноатомные:
Ароматические (см. Фенолы)
Первичные: vOH ~364О М
RCH2OH 3200 - 3400 S
vco 1000-1075 S
Вторичные: vOH ~3630 M
RR'CHOH 3200 - 3500 S
vco Ю75-1150
Третичные: vOH ~362Q M
RR'R"COH
vco
б. Многоатомные
voh '
в. Углеводы:
voh '
vc-o
5. Альдегиды
3200-3520 S
1100-1210 S
спирты:
~3600 М
3350-3550 S
~ЗЗОО S
1000-1125 S
О vCH 2800 - 2900 М
II 25сн
ИГН Vr-—rs
2695-2775 М
1720-1740 S
8СН 1380-1410
Аг(С=О)Н vc=0 1685-1710 S
1260-1310 W
1160-1210 М
6. Амиды
а. Первичные: vNH ~335O, 3180
О
RCNH,
vc=o ~ 1665 s
5nh2 ~ 1630 S
vCN ~1400 M
Разбавленный раствор в ССЦ
Широкая. ОН, связанная водородной
связью (см. рис. 5.11)
Раствор в CQ4
Широкая
Насыщенные спирты с неразветвлен-
ной цепью, ~ 1100 см; разветвление
понижает частоту на 10—15 см
Раствор в ССЦ
Широкая
Если К = н-алкил, то vco~ 1140; раз-
разветвление, ненасыщенность или ариль-
ные группы понижают частоту
Разбавленный раствор в ССЦ
Твердое вещество или жидкость
Широкая
Много полос
Дублет резонанса Ферми (иногда вид-
видна только одна полоса)
Электроотрицательный заместитель на
а-углеродном атоме увеличивает час-
частоту (на 15-20 см для С1 или F,
6 см-1 для Вг и 10 см-1 для OR).
Сопряжение понижает частоту. Обра-
Образование водородной связи понижает
частоту на 5 — 10 см
Групповые частоты для амидов не-
непостоянны, так как спектры обычно
получают для твердых веществ.
Возмущения в твердом состоянии,
включая водородную связь, велики и
часто подавляют другие эффекты
В разбавленном растворе, 3520 и
3100 см
В разбавленном растворе
Амид I 1 эти полосы попадают в об-
Амид П J ласти 1670-1690 и 1615-
1620 см
Характеристические частоты поглощения
303
PNH:
,(ip) -1150 W
, 600-750 W
б, N-Замещенные
(транс):
0
II
R—С—N—R'
1
H
в. N-Замещеив
(цис): см. .
vNH ~330O S
2vcn ~ 3100 W
vc=o 1630 - 1680 S
^CN ~^" °NH "^ 1550 S
vCNH -1250 V
<oNH(oop) ~700 W
Лактамы
г. N-Двузамещенные:
7. Соли аминов
а. Первичных:
(R-NH3)+A-
vc_o 1630-1680 S
vNH. 3200-2800 S
Проявляется не всегда
Широкая
В разбавленном растворе, 3400 —
3470 см
Амид I. В разбавленном растворе,
1670-1700 см
Амид II. Очень характеристична.
Раствор, 1510-1540 см
Амид III
Широкая
Соли ароматических аминов имеют
Комб. 2800-2000 W
Комб. ~2000 V
5» 1560-1625 S
S4NHj 1505-1550 S
б. Вторичных:
(RjNH2)+A- vNH 2700-3000 S
Комб. 2300-2700 W
5цНа 1560-1620 М
в. Третичных:
(R3NH)+A" vNH 2330-2700 S
г. Четвертичные
аммониевые
соединении:
RN(CH3K+A" 900-980
8. Аманы
а. Перннчиые \ftHj 3330-3350 М
алифатические:
RCH2NH2 vsNH2 3250-3450 М
vSich, 2770-2830 V
5nh2 1590-1650 М
<oNHl 750-850 S
б. Перничные
ароматические:
ArNH2 vjS,H 3420-3520 М
v'nh 3325-3420 М
vfS,H3 3390-3500 М
fc 3330-3420 М
более низкую частоту
Обычно несколько полос
Много полос
Много полос
Нет характеристических полос погло-
поглощения кроме —N(CH3K"
Много полос. Обычно имеет полосы
около 3020, 1485 и 1410 см
Vs = 0,98^ для эквивалентных связей
NH
Кроме того, часто имеется плечо око-
около 3200 см
Много широких полос
Раствор
Раствор
Жидкость
Жидкость. Имеется более слабая по-
полоса при 3170-3250 см
304
Приложение 2
в. Вторичные
алифатические: vNH - 3300 W
RNHR' ~3250 W
(%н 700-750 М
г. Вторичные
ароматические:
ArNHR vNH ~3400 М
5chn -1510 S
vCn 1250-1340 S
vnr 1180-1280 M
д. Третичные:
9. Аминокислоты
(твердые)
+H,N О"
\ //
НС—С i
/ \\
R О.
vNH+ 2600-
3100 S
Комб.
-2100 W
v?o 1555-
1605 S
vfco 1390-
1430 S
10. Соли аммония
(твердые)
(NH4)+A" vNH* 3100-3330 S
5nhj 1390-1485 S
11. Ангидриды
а. Алифатические
с открытой цепью:
О О f~1820 S
II II Vc=o1~1750M
С С 1
/ \ / \ vco 1040-1050 S
R О R'
Раствор
Жидкость
Ароматическая полоса около 1500 см"
Лучший путь для идентификации тре-
третичных аминов заключается в их об-
обработке минеральной кислотой (на-
(например, 2 капли образца + 1 капля
смеси концентрированной НС1 с эта-
этанолом) и записи спектра производного
в области 2200-3000 см. Погло-
Поглощение около 2600 см указывает на
солянокислую соль третичного амина
(см. Соли аминов)
Много полос до 2200 см~
Широкая
Механическое взаимодействие приво-
приводит к образованию дублета. Электро-
Электроотрицательные заместители в ot-по-
ложенни увеличивают частоту
б. Сопряженные
с открытой цепью:
-1775 S
1720 М
Характеристические частоты поглощения
305
в. Циклические,
несопряженные
пятичленные циклы:
О
\ /
/ \
\
С
l~1860 W
Vc=o j~1780 VS
vco 1000-1130 S
vco 895-955 S
~1860 W
г. Циклические,
сопряженные
иятичлениые циклы:
V&*> t-1760 S
vco 895-955 S
д. Циклические,
шестичленные циклы:
-1800 М
12. Соединения бора
а. Борные эфиры:
•{:
~138О S
VS
B(ORK
RB(ORJ
RjBOR
б. Соединения,
содержащие связи ВО
~900-1000 S
в. Бораиы: vBh 2350-2640 S
RBH2 vBhb 1540-2220 S
г. ВОН (твердые): vOH 3200 - 3300 S
д. Боразнны: vNH 3425-3505 М
vBN 1330-1470 S
8BN 680-700
е. ВСНз ЬЪн> 1405-1460
8СНз 1280-1330
1430-1440 S
13.
ж. В-фенил:
Карбаматы
а. Первичные:
О
H,NCOR
Конденсированные
vNH 3250-3340
Vc=o -1705
Удвоение полосы из-за присутствия
изотопов 10В и "В
Если атом азота находится в коорди-
координации с атомом бора, то полоса vbo
не наблюдается
Нормальные бораны
Водородные мостиковые связи в выс-
высших боранах
Широкая
Дублет
Колебания кольца
Раствор (СНС13)
3390-3480
~ 1730, если R — ароматическое коль-
кольцо
306
Приложение 2
б. Вторичные: vNH 3250-3340 3390-3480
HR-NCOOR vc^ 1715-1735 1705-1725
1530-1540 1510-1530
в. Третичные: v^^ 1683-1691
R'R'NCOOR
14. Карбонаты
О
||
ArOCOAr
~ 1800 S
1205-1220
О
ArOCOR
1720 S
1210-1250 S
О
II
ROCOR
o ~ I740 s
voco 1240-1280 S
15. Эпокснды
О
/\
R—С—С—R
/ \
H H
16. Сложные эфиры
О 2*0=0
II
RCOR'
vCH 2990-3050 M
1230-1280 W
815-950 V
805-880 V
775-850 М
750-775 W
3450 W
1735-1750 S
1720-1725 S
1715-1740 S
1160-1210 S
1230-1260 S
1280 S
17. Простые эфары
ROR' .VcH2 2835-2878 V
Колебания с раширеннем и сужением
цикла («дыхательные»)
Однозамещенное кольцо
Двузамешенное кольцо A,1 и 1,2)
Тризамещениое кольцо
Часто ошибочно приписывается сле-
следовым количествам ОН
Сопряжение понижает частоту на
10—20 см, а внутримолекулярные
водородные связи — на 40 см. Элект-
Электроотрицательная группа в ос-положе-
нии увеличивает частоту и . приводит
к расщеплению карбонильной полосы
в дублет
Эфиры муравьиной кислоты
R-Ненасыщенная или ароматическая
группа
Насыщенные сложные эфиры, кроме
ацетатов
Только ацетаты
Ароматические (фталаты, бензоаты и
т.д.)
Для алифатической группы, следую-
следующей за кислородом
Характеристические частоты поглощения
307
VCH3OA1 ~2835 М
vCoc 1085-1140 S
1210-1310 S
1010-1050 М
vCoc П70-1250 S
18. Фторированные углеводороды
а. Монофтор:
CF vCf 1000-1400 М
б. Днфтор:
CF2
в. Трнфтор: v 1120-1350 S
CF3 680-780 М
19. Галогеннрованные
соединения
а. Хлор: vca 560-830 W
°>снс1 1240-1300 S
Vccu 700-830 S
б. Бром: vCBr 515-680 М
в>сн2вг -1230 S
в. Иод: vCI 485-610 М
2j ~1170 S
vCF 1120-1280 S
20. Гетероциклические
соединения
а. Фураны,
2-замещенные:
1570-1605
1475-1510
1380-1400
820-885
740-800
Метоксигруппа, связанная с аромати-
ароматическим кольцом
Алифатические простые эфиры. Обыч-
Обычно ~П25 см
Ароматические простые эфиры с алк-
оксигруппой
Ароматические простые эфиры с алк-
оксигруппой
Циклические соединения (например,
ди океан)
Не очень характерная
Не очень полезная область
О
б. Пнрролы: -3490
~1565
-1500
N
Н
Если есть водородные связи, то
3100-3400 см
в. Тнофены, 1514-1534
2-замещенные: 1430—1454
1347-1361
О
г. Тиофены, -1530
3-замещенные: ~ 1410
~137О
308 Приложение 2
Д. Пяркдины, 1585-1615
2-ммешеыиые: 1568—1576
1465-1477
1428-1438
е. Пирнднны, 1590-1600
3-замешенные: 1572-1582
1465-1485
1417-1425
ж. Пнридины, 1600- 1610
4-замещенные: 1552-1570
1480-1520
1410-1420
з. N-Оксиды
пиридина: 1200-1320
845-880
21. Углеводороды
а. Насыщенные
алифатические:
Метальные
группы v?H 2952-2972 S
RCH3 v?H' 2862-2882 М
S?H3 1450-1475 М
5b/ 1377-1383 М
ArCHj v?H 2920-2930 М
\?СН1 2860-2870 М
RCH(CH3J 8s<^.„- j. Дублет гем-диметильной группы
-1170 М
1130-1150 V
RCH(CH3JR' 1151-1159 М
{
1235-1255 М
О 1160-1210 М
|| vCHj 2900-3000 W
RCCHS бен, 1405-1440 М
«о, 1350-1375 S
RNCH3 v 2780-2805 S
ArNCHj v 2810-2820
ROCH3 V 2955-2992 S
v8 2865-2897 S
28 2815-2840 V
8 1440-1470 M
RSCHj 5" 1415-1440 M
5' 1290-1330 W
Характеристические частоты поглощения
309
Метмлевовые группы
RCH2R' vf;H2 2916-2936 S
vfcHj 2843-2863 M
5sCh2 1450-1475 M
R(CH2)B>3 рСн2 772-730 W
О
RCH.CR }
RCH.CssNl
RCH,NO, j
RCH2OR
RCH2OH
RCH2NH
RCH2NHR )
RCH2NR'2 [
iv 2900-3000 M
'6 1405-1445 S
Ш
v» 2922-2955 S
V 2835-2880 S
5 1445-1475 M
V 2920-2960 S
vs 2760-2820 S
1445-1475 M
RCH2SR' 5 1415-1440 M
o> 1220-1270 S
RCH2C1 5 1430-1460 M
a> 1240-1300 S
Изолированные СН
R3CH 289°
1340
б. Алкены: Vs 3077-3092 M
Вшшл: vs 3012-3025 M
RCH=CH2 2<o 1805-1840 W
vc=c 1638-1648 M
5CH 1412-1420 M
ш 985-995 S
@ 905-910 S
транс v 3000-3050 M
RCH=CHR' vc-c 1668-1678 IV
ш 965-980 S
цис vc-c 1630-1660 M
RCH=CHR' рсн 1440-1430 M
ш 650-730 M-S
Вштлнден:
R' v" 3077-3100 M
\ 2o> 1775-1790 M
C=CH, vc-^ 1640-1660 M
885-895 S
В твердом состоянии может прояв-
проявляться в виде дублета
He имеет практического значения
Меняется под влиянием индуктивного
эффекта группы R (см. рис. 5.8)
Меняется в зависимости от резонанс-
резонансных свойств заместителей
Меняется под действием суммарного
индуктивного эффекта заместителей.
Уменьшается на 40 см на каждый
атом С1, О и т. д.
Отрицательная ангармоничность
Меняется в зависимости от резонанс-
резонансных свойств заместителей
310
Приложение 2
Тризамешенные:
RR'C=CHR" v 2990-3050 W
vc-c 1667-1692 W
a> 790-840 M
в. Алкмны:
RfeCH vCH 3270-3340 S
v^; 2100-2140 W
5Сн 610-700 M
г. Аллены:
RC=C=CR vc-^ 1940-1950
5CHj -850
д. Ароматические:
vCH 3010-3110 M
1660-2000 W
vcc ~1600 V
~158O V
vcc
~ 1490 VI
~1450 V
5CH 730-885
Однозамещенные
1, 2-Дизамещенные (орто)
1, З-Дизамещенные (мета)
1, 4-Дизамешенные (пара)
1, 2, З-Тризамещенные
1, 2, 4-Тризамещенные
1, 3, 5-Тризамещенные
1, 2, 3, 4-Четырехзамещенные
1, 2, 3, 5-Четырехзамещенные
1, 2, 4, 5-Четырехзамещенные
1, 2, 3, 2, 5-Пятизамещеяные
22. Гндроперокснды
ROOH vqh ~356О
3390-3435
~815
RR'R"COOH 830-845
Если R — электроотрицательная груп-
группа, то может наблюдаться дублет
Три или четыре пика
Характеристическая картина поглоще-
поглощения обертонов (см. рис. 1 в прило-
приложении 1)
Нормальные колебания кольца
Запрещены, если у молекулы есть
центр симметрии. Слабые для непо-
ляриых заместителей, более сильные —
для полярных; полоса 1580 см"' осо-
особенно интенсивна для О- или N-заме-
щенных колец
Нормальные колебания кольца. Для
электронодонорных групп интенсив-
интенсивность полосы 1490 см*1 повышена.
В пара-замещенных молекулах эти по-
полосы попадают в область 1500 и
1370 см
Частоты более чувствительны к поло-
положению заместителей, чем к их при-
природе. Однако корреляция становится
ненадежной для сильно полярных за-
заместителей, таких, как NO2 или СООН
735-765; 685-710
740-760
770-790; 680-705
800-830
760-780; 705-745
870-885; 805-825
830-850; 680-700
~82О
~865
~865
~865 '
Раствор в СС14 (свободные ОН)
Связанные водородной связью
Две полосы, если алкильная группа
имеет четное число атомов углерода
Характеристические частоты поглощения
311
23. Имнды
О О
II II
RCNCR'
I
н
о
vNH -3280
vc=o 1733-1737 S
1503-1505
1167-1236
732-739
Твердое состояние. Нециклические
соединения
Твердое состояние. Нециклические
соединения
\
NH
24.
25.
R
\
О
Изоцнанаты
vc=o 1735-1790 M
1680-1745 S
Vs 2263-2275 S
Изошмнураты Vc_o A680-1700 S
0
II R
A/
N N
1 1
vc=o 1710-1715 S
О
О
Имеется также плечо около 1755 см '
Ароматическое замещение. Плечо око-
около 1780 см
26.
27.
28.
0
Изоннтрнлы (нзоцнаниды)
RNC
ArNC
Изотноцшшаты
RNCS
RCH2NCS
ArNCS
Кетоны
v 2134-2146 S
v 2110-2130 S
Vs 2050-2150 S
v' 650-700
5CH2 1318-1347 S
v» 925-945
v~_n 1705-1725 ;
RCR'
Заместители на а-углеродном атоме
увеличивают частоту на ~15—20 см
(F или Q), на ~6 см (Вг), на
~10 см (OR). Вращательные изо-
изомеры могут приводить к удвоению
полос. Сопряжение с одной группой
понижает частоту на 30—40 см,
с двумя — на ~60 см. Образование
водородной связи понижает частоту
на 5-10 см
312
Приложение 2
о
II
RCCH,
"сн,
5С=О
1350-1370 S
~595
1715-1822 S
Из-за механического взаимодействия
с валентными колебаниями С—С час-
частота зависит от размера цикла
29.
N-
I
С
Лактамы (фк-Н-замешевные
яы)
[—Н vNH ~32OO S
Комб. ~3100 М
=О vc^ ~1650
1700-1560
1730-1760
1440-1490
1310-1350
-800
6NH
vCN
30. Лактоны
О
II
(у
1735-1750 S
1760-1795 S
Для шести- и семичленных циклов
Для пятичлеиных циклов
Для четырехчленных циклов
Широкая
Шестичленные циклы
Пятичленные циклы. Если в кольце
имеется ненасыщенность, то в карбо-
карбонильной области наблюдаются две
полосы
31. Меркаптаны
RSH vSH 2540-2590 W
vcs 550-700 W
32. Нитраты (ковалентно-связанные)
RONO2 v?,o 1625-1660 S
v^o' 1270-1285 S
vNO2 840-870 S
8 745-760 S
6 690-710 S
33. Нитрилы
R—C=N VcN 2240-2260 M
ArC=N
2220-2240 V
34. Нитриты (ковалентно-связанные)
R—О—N=O vno 1648-1681 S
(транс)
1605-1625 М
(цис)
vNO 750-814 S
35. Нитросоедннешм
RCH2NO2 v^Oj 1545-1556 S
v^,21368-1390 M
RCHXNO2 v» 1556-1580 S
v» 1340-1368 M
При сопряжении понижается до 2215—
2235 см
Частота и интенсивность зависят от
заместителей в ароматическом кольце
На частоту влияют электроотрица-
электроотрицательные заместители [см. уравнение
E.31)]
Характеристические частоты поглощения
313
ArNO2 v* 1500-1530 S
vs 1330-1370 М
36. Нитрозоамины
R2N—N=O vN0 1425-1460
vNN 1030-1150
ArRN—N=O vN0 1450-1500
vCN 1160-1200
vNN 925-1025
37. Ннтрозосоединения
О О vNO 1330-1420 S
N=N
\
\
N=N
vN0 1323-1344 S
v 1390-1397 S
v -1409 S
v 1176-1290 S
v 1253-1300 S
О
\
38. Окснмы
R2—C=N—OH v0H 3150-3300
vCN 1620-1690
vNO -930
Пероксиды
RCH2OOCH2R' voo 830-1000 W
39. Фенолы
АЮН Vqh 3593-3617 M
v0H 3200-3250 V
8OH 1330-1390 M
vco 1180-1260 S
40. Соединения фосфора
а. Фосфнны:
R.PH3_n vPH 2275-2440 M
5pH: 1080-1090 M
coPH! 910-940
8Ch2p 1405-1440
б. Оксиды фосфнноа:
R3P-»O vpo 1140-1300 S
vP0 ~1150 S
~1190 S
в. Фосфорные кислоты:
Rn(HOK_nPO voh 2550-2700 M
2100-2350 M
Алифатические. Обычно определяются
в виде димеров
Алифатические, в твердом состоянии
Ароматические
Ароматические
Алифатические
Алифатические
При идентификации не используются
Разбавленный раствор (свободная ОН)
Связанная ОН
Твердое состояние. Эти частоты по-
понижаются в растворе
Твердое состояние
Многие полосы наблюдаются в виде
дублетов из-за поворотной изомерии
в соединениях фосфора
Резкая полоса
Частота меняется в зависимости от
суммы электроотрицательностей за-
заместителей
Алифатические
Ароматические
Широкая
21 - 586
314
Приложение 2
г. Эфиры
фосфорной
кислоты:
(ROKP -»
(АгО)зР-
О vP0
VcOP
> О vco
VPO
д. Фосфиты:
(ROKp
е. Другие
PvF
PCI
PBr
POP
po|-
POi-
VPO
~1280 S
970-1050 S
740-830 S
1160-1260 S
914-994 S
855-875
корреляции:
815-890
440-580
400-485
870-1000
970-1030
1000-1100
Только для метокси- и этоксипроиз-
водных
POCH3 ~2960; 1460; 1170-П90
POC2H5 -2990; 1485; 1450; 1395;
1375; 1155-1165
POC6H5 1160-1240; 855-994
PCH2 1405-1440
PCH3 1280-1310; 860-960
P-*S 580-750
P—N 930-1110
P=N (циклические) 1100-1320
41. Креминнорганнческие соединения
а. Снлаиы:
RnSiH,t_n vsiH 2100-2250 S
8siH 800-840 S
8siH2 840-900 S
920-940 S
8siH 910-930 S
930-945 S
б. Снлоксаны: vsiOsi 1000—1100 S
vSl0Si ~1020 S
vsiosi ~ 1080 S
() vsi0Si ~ 1080 S
и. Другие корреляции:
SiCH3 5CHj 1250-1280 S
3 760-860 S
SiC2H5 1220-1250 M
1000- 1020 M
945-970 M
1430 M
1100-1125 S
~73O M
690-700 M
Точное положение зависит от индук-
индуктивных эффектов заместителей
Полимеры с длинной цепью имеют
две сильные широкие полосы около
-1080 и 1020 см
SiC6H5
Характеристические частоты поглощения
315
SiOCHj 2840 S
1190 M
1090 S
800-850 M
SiOR 1075-1100 S
945-990 M
SiOAr 920-970 M
SiOH vOH -3960 W
3200-3400 M
бон 830-950 M
SiCH=CH2 vcc 1590- 1610 M
-1400 M
990-1020 M
940-980 M
SiCH2Si 1040-1080 S
SiNHSi vNH 3390-3420 M
~1170 S
910-950 S
SiNH2 vNH 3840; 3400 M
1540 M
SiOCOCH3 v^-o 1700-1770 S
1190-1250 S
1000-1050 S
925-970 S
SiCl v 460-650 S
SiClj v» 535-600 S
v* 460-540 M
SiCl3 v» 570-620 S
Vs 450-535 M
42. Сульфаты
О v* 1390-1415 S
|| Vs 1187-1200 S
ROSOR'
О
Группа, чувствительная к гидролизу
Разбавленный раствор
Группа, связанная водородной связью
Группа, чувствительная к гидролизу
Группа, чувствительная к гидролизу
Группа, чувствительная к гидролизу
43. Сульфиды
RSR'
RSSR'
КО hi 2 S
CH3S—
ArS—
V
V
570-705 W
-500 W
v» 2922-2948
V8
8
@
6a
5s
P
2846-2878
1410-1435
1220-1270 S
1415-1440
1290-1330
960-1030
-1090
Используется редко. Лучше идентифи-
идентифицировать методом спектроскопии КР
Дисульфиды. Используется редко
316
Приложение 2
44. Сульфиновые кислоты
О vqh 2340-2790
II vs^o 990-1090
RSOH vso 810-870
45. Сульфиты
О
II
ROSOR'
1198-1220
46. Сульфонамиды
О v|o 1310-1380 S
|| / v|o 1140-1180 S
RSN vNH 3300, 3250
|| \ 6 ~1570
О vSN 900-910
vNH 3270-3330
47. Сульфоиы
О v|o 1290-1340 S
|| v|o 1120-1165 S
RSR' Зад, 500-610 M
|| cos02 460-525 M
О
48. Сульфоновая кислота
О vqh ~29OO S
|| -2400 М
RSOH v^ 1342-1352
|| v|=o П50-1165
О vso 895-910
49. Соли сульфоновой
кислоты
О '
II
RSO
Твердое состояние
Первичные сульфонамиды
Вторичные сульфонамиды
Часто и растворе ССЦ проявляется
в виде триплета
Широкая. Эти соединения очень легко
гидролизуются с образованием суль-
сульфатов оксония, спектры которых от-
отличаются от спектров солей сульфо-
ната
1120-1230 S
1025-1080 W
(ArSO3)"M+ -1230, 1190, ИЗО,
1040
50. Сулъфонилхлорнды
О v|o 1360-1390 S
|| v|o 1168-1185 S
RSCI
О
51. Сульфоксццы
О
||
RSR'
1030-1080 S
980-1020
При наличии водородной связи
Характеристические частоты поглощения
317
52. Тнокарбонильиые
соединения
S Vc=s 740-1420 V
Не так полезна, как полоса валентных
колебаний С=О. Валентные колеба-
колебания часто смешиваются с другими
колебаниями
а. Тритиокарбонаты:
S vc_s -1070
II
RSCSR'
б. Сложные дитноэфнры:
S 1170-1225
II
RCSR'
в. Ксантогеиаты:
S 1200-1250 S
|| 1110-1140 M
ROCSX 1020-1070 S
г. Тиоамиды:
S vNHl -3380
|| -3180
RCN 6 -1630
Г
Твердое состояние
1550
53. Тноцнанаты
R—SCN vSCN 2160-2175 M
54. Тнолоные эфиры
S vc_o 1680-1700
|| vcs 930-1030
RCSR'
RCSAr
1690-1710
920-1020
О
II
ArCSR
1640-1680
940-905
О
II
ArCSAr'
1650-1700
895-920
318 Приложение 2
о
II
HCSR
~1675
~755
55. Пронзнодные мочевины
О
II
RNCNR'
Н Н
vnh
vc=c
~ЗЗОО
, 1635-1660
1575-1613
Спектры мочевины похожи на спектры
амидов
ЛИТЕРАТУРА
1. Bellamy L. J., The Infrared Spectra of Complex Molecules, Vol. 1, 3rd ed.,
Wiley, New York, 1975. (См. [18] в гл. 3.)
2. Colthup N. В., Daly L Я., Wiberley S. G., Introduction to Infrared and
Raman Spectroscopy, 2nd ed., Academic Press, New York, 1975.
3. Craver C. D., Desk Book of Infrared Spectra, Coblentz Society, Kirkwood,
Mo., 1977.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсолютно черное тело 18
2-Аллилфенол 172
Алмазные окна 121
Альдегиды 199
Амид II, III 158
Аминокислоты 112, 197
Амины 85
Аммиак 61, 209
Анализ
воздуха 288
групповой 270
количественный 232, 245
источники ошибок 108, 249
полимерных материалов 266
порошков 265
следовых примесей 272
Аналитические полосы 243
Ангстрем 14
Ангармоничность 141, 151
Анилины 179
Апертура 19
Ароматические соединения 188
Ассоциация 168, 268
количественный анализ 268
Атмосферное поглощение 58, 106, 214, 273
Аттенюатор 24 см. Оптический клии, Осла-
Ослабитель
Ацетилен 175, 182, 276
Ацетон 178
Базовая линия 86, 239
Баланс 54
Биологические системы 209
Ближняя ИК-область 152
Болометр. 21
Бумага 94, 106
Бутадиен-1,3 183, 286
нитрование 283
Бутены 286
Вазелиновое масло 89
Ветви Я, Q, Р 142, 219
„Вигли" 39
Вииилхлорид 274
Виньетирование 116
Влажность 87
Внутренний стандарт 265
Внутрикомплексные соединения 199
Вода 86, 122, 173, 187, 214, 232, 270, 273
анализ смеси спирт — вода 279
ассоциированная 95
Водные растворы 122
Водородная связь 170, 174
внутримолекулярная 172
межмолекулярная 171
Волновое число 14
Волокна 106, 203, 279
нейлоновые 271
Волчки
асимметричные 137
симметричные 137
сферические 137
Вращательные спектры 136
правила отбора 138
Время
отклика 48
сканирования 51
Выигрыш
Жакино 41
Фелжетта 34
Вырождение колебаний 141
Высокотемпературная спектроскопия ПО
Газовые кюветы 128
многоходовые 108, 210, 272
Газы 180, 264
Галогеноводороды 155
Гармонический осциллятор 139
правило отбора 141
энергия 141
н-Гексан 178, 195
Гексафторацетон 273
Гексафторид вольфрама 280
урана 209
к-Гексилбромид 112
к-Гептилбромид 112
Геометрический фактор 56, 57
к-Гептан 85
Гидроксидная группа 170, 270, 268
Глобар 18
Глубина проникновения 100, 103
Горячие полосы 152
Групповые частоты 153, 187
Дальняя ИК-область 15, 58
Двойная связь 157, 159, 188
Двойное прохождение 28
Декодирование иитерферограммы 39
1,2-Дибромпропан 61
2,2-Диметоксипропаи 87, 276
2,6-Диметоксифенол 117
Динамическая точность 256
320
Предметный указатель
2,4-Динитрофенилгидразоны 199
Диоксан 86, 180
Диоксид азота 274
Диоксид кремния 282
Дипольный момент связи 151
Дисперсия 27
Дифениламин 180
Дифенилацетилен 192
Дифракционная решетка 27
поляризация света 27
рассеянный свет 28
Дифференциальная спектроскопия 246, 272
см. Разностная спектроскопия
Дихлорфторметан 272
Длина волиы 14
связи 156, 160
Дымовые газы 273
Жидкостные кюветы 125
Жирные кислоты 177
Загрязнения атмосферы 108
Закон
Бугера 234
Бугера — Бера 235
отклонения 235, 261, 269
Планка 18
Снеллиуса 96
Затухающая волна 100
Зеркало
внеосевой параболоид 19
коллиматора 19
Литтрова 26
параболическое 19
торическое 19
эллиптическое 19
Идентификация 185, 199
Изотопное замещение 156, 220
ИК-анализаторы 284
диспергирующие 288
недиспергирующие 286
ИК-область, границы 14, 15
Индеи 61
Интенсивность 151, 183, 218
влияние размера частиц 265
интегральная 237
точность измерения 52
Интерференция 86, 131, 251
Иитерферограмма 42
Интерферометр 37
Информационно-поисковые системы 68
Иртраи-2 86
Источники ИК-излучения 18
Калибровка спектрофотометра 58
Калибровочная кривая 243, 256, 271
Камфора 61
Карбонил никеля 273
Карбонильная группа 153, 161, 164, 167, 170,
175, 178, 188, 207
Кинетика 283
Кислоты Льюиса 170, 269
Классификация ИК-спектров 73
Колебательно-вращательная структура по-
полос 219
Колебательное взаимодействие 158
Колебательные спектры 138
правила отбора 141
Комбинационное рассеяние света 13, 114,
145, 157, 200, 218
Конденсор 118, 253
Константы заместителей 164
Контуры полос 150, 183
Концентрация образца 87, 180, 243, 269
Концентрирование 275
Координационные соединения 209
Корреляционная спектроскопия 274
Корреляционные таблицы 195
Коэффициент
вариации 264
поглощения 62, 151, 235, 258
и-Крезол 178
Критический угол 96
KRS-5 86, 270
о-Ксилол 276
Кюветы 88, 272
толщина, определение 130
Лазеры с перестройкой частоты 32, 214, 274
полупроводниковые 32
с параметрической генерацией 33
с переворотом спииа 32
Лиофильная сушка 93
Липиды 209
Литература по ИК-спектроскопии 68
Масс-спектрометрия 13, 189, 200, 276
Матричная изоляция 111
Метилбензоаты 167
л-Метилбензоиитрил 178
4-Метил-2,6-ди-трет-бутилфенол 276
Метиленовый синий 116
.м-Метилстирол 114
Метод
наименьших квадратов 261
скорейшего спуска 260
Микроволновая спектроскопия 219
Микрометр 14
Микрообразцы 107, 116
Многокомпонентные системы 259
Мозаичный эффект 265
Монохроматор 24
двойной 28
Литтрова 26
Мощность излучения 18
Нарушенное полное внутреннее отражение
95, 201, 204, 270
анализ в потоке 288
внутренний стандарт 270
многократное 201, 204, 279
поляризованное излучение 220
Негативная фильтрация 286
Неорганические соединения 209
Нефть 213, 270, 275
Низкотемпературная спектроскопия 112
п-Нитробензилхлорид 197
Нормальные колебания 145
формы 146
Предметный указатель
321
Нормальные координаты 146, 151
Нормировка 262
Нулевая линия 48, 238, 254
Обертоны 151
Область „отпечатков пальцев"' 189
Однокомпонентные системы 257
Озон 108, 207
Оксид углерода 232, 273
Оксиды азота 108, 283
Оптическая плотность 235, 258
аддитивность 235
воспроизводимость 61
вычитание 192, 207
измерение 237, 258
точность 62, 257
Оптические изомеры 221
Оптические материалы 253
полировка 128
свойства 127
Оптический клин 24 см. Ослабитель, Ат-
Аттенюатор
Осветитель 24
Ослабитель 24, 62, 117
Основания Льюиса 179, 269
Отношение сигиал/шум 34, 37, 51
Отражение 109, 126, 236
Ошибки в количественном анализе 264
Пептиды 112
Пиролиз 109, 201, 268
Поверхностно-активные вещества 197, 207
Поворотные изомеры 183
Поглощение атмосферы 58, 256
Подготовка образцов 84
для съемки спектров НПВО 106
Позитивная фильтрация 286
Показатель преломления 92, 96, 100, 250
Полибутен-1 206
Поливиннлхлорид 217
Поливинилиденхлорид 204
Полимеры 200
аморфность 204
количественный анализ 266
кристалличность 204
полиморфизм 204
реакции 207
Полиморфизм 94, 177, 206
Полистирол 60, 204, 206
Полиуретаны ПО, 201
1,4-трпнс-Полихлоропрен 206
Полиэтилен 232, 273, 275
Полиэтилентерефталат 204
Полиэфиры 202
Полуколичественный анализ 239
Поляризованный свет 202
Порядок связи 159
Постоянная времени 48
Правило Теллера — Редлиха 220
Преобразование
Кули-Тъюки 40
Фурье 40
Приемники ИК-излучения 19
То лея 21
квантовые 21
оптико-акустические 21
пироэлектрические 23
полупроводниковые 23
с внутренним фотоэффектом 23
тепловые 19
Приведенная масса 141
Приготовление образцов
выбор метода 84
газы 107
пленки 88
порошки 106
растворы 84
концентрация 87
суспензии 89
в вазелиновом масле 89
таблетки с КВг 93
эмульсии 89
Примеси 198
Пропускание 235, 255
Псевдоматричная изоляция 112
Псевдосвертка 151
Разностная спектроскопия 246, 273, 275
Разностные полосы 152
Разность хода 39, 56
Разрешение 46, 52, 55
Рассеяние света" 236
Рассеянный свет 19, 48, 59
Растворители лля ИК-спектроскопии 85,
177, 221
дейтерированные 86
Растр 29
Растяжка спектра по ординате 118, 121,
246, 247, 272, 273
Резонанс Ферми 150, 159, 188
Ртутная лампа 19
Светоделитель 38
Светофильтр
диэлектрический 31, 50
нейтральный 248
отрезающий 28, 31
полосовой 31
узкополосный 31
я-Связь 163
Сглаживающая функция 40
Сероуглерод 85, 108, 118, 178, 267, 275
Силикагель 283
Силовая постоянная 138, 145, 156, 162, 219
Силовое поле
валентно-силовое 146
центрально-силовое 146
Юри — Бредли 146
Сканирование 31
быстрое 55, 283
многократное 116, 119, 217, 273
скорость 52, 244, 256
оптимальные условия 54
Соотношение
Бэджера 153
Горди 153, 159
Кирке уда — Бауэра — Мага 177
Сопряжение 162, 168
Составные полосы 151
Спектрометры
322
Предметный указатель
диспергирующие 24, 35
интерференционные 35, 55
многоканальные 16, 34
миогошелевые 29
недиспергирующие 31, 37
с диэлектрическим фильтром 31, 288
оптико-акустические 33
с перестраиваемым лазером 32
растровые 29
сканирующие 16, 24
с преобразованием Адамара 35
с пространственным разделением 16, 34
Спектроскопия производных 246
Спектрофотометры
двухлучевые 24, 58, 85, ПО, 288
дифракционные 27
интерференционные 45, 212, 283
калибровка 58
настройка 58
оптимальные условия регистрации 54
с оптическим нулем 24, 26, 288
призмеииые 27
с регистрацией электрического отношения
24, 26, 124, 238, 288.
термостатирование 254
Спектры излучения 121
Спирты 187
алифатические 171
ароматические 171
водородная связь 171, 173
Средняя ИК-область 15
Стеклообразное состояние 177
Стирол 286
Схема Вайнфорднера 16
Таблетки с КВг 93, 117, 201
прессование 265
Температура ПО, 183, 254
Кюри 23
Теория групп 145
Термоэлемент 20
Толуол 195
Требования к ИК-спектрам 74
Трифеиилфосфат 119
Тройная связь 157, 159
Тяжелая вода 86, 232, 270
Фенол 275
Фосфиновых кислот эфиры 117
Фосфорильная группа 161
Фотометрический клин 62
Фталевые кислоты 266
Функция
аподизируюшая 41, 57
аппаратная 48, 62, 235
Лорентца 150
потенпиальной энергии 145
Фурье-спектроскопия 43, 118, 212, 214
Химические производные 197
Хлоранил 265
Хлорид серебра 89, 107, 115
Хлороформ 86, 149, 183
Хроматография 112
бумажная 275
газовая 13, 112, 193, 202, 284
гель-проникающая 116, 202
жидкостная 193, 276
тонкослойная 116, 202
Целлюлоза 94, 112
Циануксусная кислота 167
Циклоалкены 158
Циклогексан 131, 178, 195
Циклогексанон 61, 178
Частичное фракционирование 195
Четыреххлористый углерод 85, 131,
178, 180, 269, 275
„Чирпинг" 42
Число степеней свободы 135
Ширина полосы 48
линии, естественная 150
Штифт Нернста 18, 256
Шум 46, 254
джонсоновский 46
статистический 46
уровень 54
175,
Углеводы 112
Углекислый газ 108, 124, 142, 182, 214,
232, 276
Угловой момент 138
Ударное уширение 181
Ультрафиолетовая спектроскопия 13, 219,
276
Уравнение
Гам мета 163
Полита 159
Усиление 23, 52, 54, 86
Феиилацетилен 175
З-Фенил-2-тиогидантоин 197
Щелевая программа 52, 53
Щель 26
кривизна 27
ширина 51, 62, 235, 241, 246
геометрическая 48
спектральная 26, 48, 62
Электронный эффект 160
индуктивный 160
мезомерный 162
Электроотрицательность 153, 160
Энергия молекулы 136
вращательная 138
кинетическая 138
Предметный указатель 323
Энергия Эффект
колебательная 141 Христиансена 92, 106, 199
потенциальная 139 Эффективность термоэлемента 20
Эпоксидная смола 94
Этилацетат 175
Этилбензол 286
Этилвинилацетат 271 Яблочная кислота 221
Эффект Ядерный магнитный резонанс 13, 114, 189,
Доплера 150 200, 267
СОДЕРЖАНИЕ
Предисловие редактора перевода 5
Предисловие 7
Глава 1. Введение 9
Обзор приложений ИК-спектроскопии 9
История 9
Возможности ИК-спектроскопии 11
Выбор аналитической методики 12
Определения основных понятий 13
Литература '. 15
Глава 2. Приборы 16
Физика ИК-излучения 16
Источник 18
Оптическая система 19
Приемники излучения % 19
Усиление 23
Спектрометры с последовательным сканированием спектра . . . 24
Диспергирующие приборы 24
Наиболее распространенные монохроматоры 26
Многощелевые спектрометры 29
Растровые спектрометры 29
Недиспергирующие приборы 31
Спектрометры с использованием диэлектрического фильтра 31
Перестраиваемые лазеры 32
Оптико-акустические приборы 33
Спектрометры с пространственным детектированием 34
Многоканальные спектрометры 34
Диспергирующие спектрометры: спектрометры с преобразо-
преобразованием Адамара 35
Недиспергирующие спектрометры: спектрометры на основе
интерферометров 37
Выбор спектрофотометра 44
Принципы выбора режима работы спектрофотометра 45
Определения специальных терминов 46
Оптимизация режима работы спектрофотометра 50
Другие факторы, влияющие на работу спектрофотометров 57
Содержание 325
Проверка работы и калибровка спектрофотометра .... 58
Переносимость оптических плотностей 62
Литература 63
Глава 3. Спектроскопическая литература 67
Эталонные ИК-спектры 67
Информационно-поисковые системы 69
Оценка ИК-спектров 73
Создание собственной картотеки 73
Литература 75
Глава 4. Методы подготовки образцов 84
Растворы 84
Растворители 85
Концентрация 87
Толщина образца 88
Проверка кюветы 88
Пленки 88
Суспензии 89
Суспензии в вазелиновом масле 89
Таблетки с КВг 93
Нарушенное полное внутреннее отражение 95
Основы 96
Практика 100
Подготовка образца 106
Газообразные образцы 107
Специальные методы подготовки образцов 109
Отражение 109
Пиролиз 109
Спектры при высоких и низких температурах ПО
Сочетание ИК-спектроскопии с хроматографией 112
Работа с микрообразцами 116
Спектры при высоких давлениях 121
Спектры излучения 121
Водные растворы 122
Конструкция кювет и уход за ними 125
Жидкостные кюветы постоянной толщины 125
Жидкостные кюветы переменной толщины 126
Газовые кюветы 128
Уход за кюветами. Переполировка оптических материалов 128
Определение толщины кювет 130
Литература 131
Глава 5. Качественный анализ по ИК-спектрам 135
Введение 135
Теория ИК-спектров 135
Вращательные спектры 136
326 Содержание
Колебательные спектры 138
Нормальные колебания 145
Контуры полос . , 150
Интенсивности полос 151
Обертоны и составные полосы 151
Групповые частоты: их использование и ограничения .... 153
Общие положения 153
Внутренние факторы, влияющие на групповые частоты. ... 155
Изменения масс 155
Геометрия 157
Колебательное взаимодействие 158
Порядок связи 159
Электронные эффекты 160
Влияние ассоциации 168
Влияние внешних воздействий 174
Физическое состояние 175
Растворитель 177
Концентрация 180
Температура 183
Интерпретация спектров 185
Идентификация неизвестных веществ 185
Анализ смесей 192
Использование корреляционных таблиц ,. ¦ , 195 .
Использование химических производных 197
Полосы поглощения примесей 199
Выводы: основные принципы и порядок идентификации неиз-
неизвестного вещества 199
Приложения 200
Полимеры 200
Идентификация 201
Структура 204
Реакции .* 207
Поверхностно-активные вещества 207
Биологические системы 209
Неорганические, металлоорганические и координационные
соединения 209
Проблемы окружающей среды 210
Анализ удаленных атмосфер и объектов 214
Отнесения частот 217
Сравнение с родственными структурами 217
Измерения интенсивностей 218
Данные, полученные методом комбинационного рассеяния
и другими спектроскопическими методами 218
Колебательно-вращательная структура полос 219
Расчет силовых постоянных 219
Изотопное замещение 220
Исследования в поляризованном свете ориентированных
образцов 220
Сдвиги частот под влиянием растворителя 221
Литература 222
Содержание 327
Глава 6. Количественные приложения 232
Применение ИК-спектроскопии в количественном анализе . . . 232
Законы поглощения 234
Закон Бугера 234
Закон Бера 234
Закон Бугера—Бера 235
Отклонения от законов поглощения 235
Практика количественного анализа 236
Полуколичественный анализ 239
Количественный анализ средней точности 240
Выводы 244
Количественный анализ высокой точности 245
Источники ошибок в количественном анализе 249
Расчеты 257
Однокомпонентные системы 257
Анализ многокомпонентных систем в условиях выполнения
закона Бугера—Бера 259
Анализ многокомпонентных систем в условиях отклонений
от закона Бугера—Бера . .' 261
Анализ без стандартов; вторичные стандарты; процедура
нормировки 261
Статистическая оценка результатов 262
Специальные задачи и методы 264
Количественные измерения спектров газов 264
Анализ твердых веществ 265
Количественый анализ полимерных материалов ..... 266
Количественный анализ ассоциированных образцов .... 268
Групповой анализ 270
Нарушенное полное внутреннее отражение 270
Анализ следовых примесей 271
Образцы с большой толщиной поглощающего слоя . . . 272
Корреляционная спектроскопия 274
Методы концентрирования 275
Косвенные методы 276
Комбинация методов 276
Примеры 276
Кинетические исследования 283
Непрерывно действующие автоматические И К-анали заторы 284
Недиспергирующие анализаторы 286
Диспергирующие анализаторы 288
Литература 289
Приложение 1. Корреляционные диаграммы 293
Приложение 2. Характеристические частоты поглощения .... 300
Литература 318
Предметный указатель 319
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги,
ее оформлении, качестве перевода и другие
просим присылать по адресу: 129820, Моск-
Москва, И-110, ГСП, 1-й Рижский пер., д. 2,
издательство «Мир».
А. Ли Смит
ПРИКЛАДНАЯ ИК-СПЕКТРОСКОПИЯ
Основы, техника, аналитическое применение
Ст. научн. редактор С. К. Оганесян
Мл. научн. редактор И. И. Землячева
Художник М. Н. Кузьмина
Технические редакторы Л. П. Бирюкова, Е. В. Яшук
Корректор Т. П. Пашковская *
ИБ № 2841
Сдаио в набор 11.09.81.
Подписано к печати 23.03.82.
Формат 60 х 906.
Бумага офсетная
Гарнитура тайме. Печать офсетная. Объем
10,25 бум. л.
Усл. печ. л. 20,50. Усл. кр.-отт. 20,50.
Уч.-изд. л. 20,12. Изд. № 3/1585.
Тираж 5000 экз. Зак. -*-а6. Цена 1 р. 30 к.
Издательство «Мир»
129820, Москва, И-110, ГСП, 1-й Рижский пер., 2
Ордена Октябрьской Революции, ордена Трудового
Красного Знамени Ленинградское производственно-
техническое объединение «Печатный Двор» имеш;
А. М. Горького Союзполиграфпрома при Госу-
Государственном комитете СССР по делам издательств,
полиграфии и книжной торговли. 197136, Ленин-
Ленинград, П-136, Чкаловский пр., 15.