/





































































































































































































































































































































































































































































Text
PHYSICAL METHODS IN CHEMISTRY
Saunders Golden Sunburst Series
RUSSELL S. DRAGO
University of Illinois, Urbana
W.B. SAUNDERS COMPANY Philadelphia • London • Toronto
р. Драго
ФИЗИЧЕСКИЕ МЕТОДЫ В ХИМИИ
Перевод с английского
конд. хим. наук А.А. Соловьянова под редакцией академика О. А. Реутова
ИЗДАТЕЛЬСТВО «МИР» МОСКВА 1981
УДК 543.422
Книга представляет собой современное пособие, с помощью которого химики самых различных специальностей смогут овладеть основами практически всех физических методов исследования структуры химических соединений и их реакционной способности, а также научиться применять их для структурного и количественного анализа.
Во втором томе рассматривается теория таких важных современных спектроскопических методов исследования, как ЯМР, ЭПР, мёссбауэровская спектроскопия, и на примере большого числа соединений самых различных классов показывается, как проводят изучение их структуры и реакционной способности.
Предназначена для широкого круга научных работников, преподавателей и студентов химических и химико-технологических высших учебных заведений.
Редакция литературы по химии
2603040000
20506-198
041(01)-81
084-80
д
© 1977 by W. В. Saunders Company © Перевод на русский язык, «Мир», 1981
9. СПЕКТРОСКОПИЯ ЭЛЕКТРОННОГО ПАРАМАГНИТНОГО РЕЗОНАНСА *
ВВЕДЕНИЕ
9.1. ОСНОВНЫЕ ПРИНЦИПЫ МЕТОДА ЭПР
Электронный парамагнитный резонанс представляет собой явление поглощения излучения микроволновой частоты молекулами, ионами или атомами, обладающими электронами с неспаренными спинами. Называют это явление по-разному: «электронный парамагнитный резонанс» (ЭПР) **, «электронный спиновый резонанс» и «электронный магнитный резонанс». Все эти три термина эквивалентны и подчеркивают различные аспекты одного и того же явления. ЯМР и ЭПР характеризуются общими моментами, и это должно помочь понять суть метода ЭПР. В спектроскопии ЯМР два различных энергетических состояния (если I = 1/2) возникают из-за различного расположения магнитных моментов относительно приложенного поля, а переходы между ними происходят в результате поглощения радиочастотного излучения. В ЭПР различные энергетические состояния обусловлены взаимодействием спинового момента неспаренного электрона (характеризуемого ms = + г/2 для свободного электрона) с магнитным полем — так называемый электронный эффект Зеемана. Зеемановский гамильтониан, описывающий взаимодействие электрона с магнитным полем, дается выражением
H = g&HSz, (9.1)
где д для свободного электрона равен 2,0023193; 0—электронный магнетон Бора efi/2mec, который равен (9,274096 + 0,000050)-10“21 эрг-Э-1; Sz—оператор спина; Н—напряженность приложенного поля. Действие этого гамильтониана на электронные спиновые функции а и 0, соответствующие ms = + 1 /2 и ms = — 1/2, приводит к результату, показанному на рис. 9.1. Спиновое состояние 0 характеризуется магнитным моментом, направленным вдоль поля, в то время как в ЯМР низшее энергетическое состояние соответствует mi = + V2 (<М- Низшее энергетическое состояние в ЭПР соответствует ms = — 1/2, поскольку знак заряда электрона противоположен знаку заряда протона. Энергия перехода определяется выражением
АЕ = gfiH. (9.2)
* В списке общей литературы приведены обзоры работ по ЭПР. Мы особенно рекомендуем читателю книгу Вертца и Болтона.
** В русской литературе принято пользоваться именно этим термином.— Прим, перев.
6
Глава 9
Разность энергий спиновых состояний а и 0 в магнитных полях обычно используемой в методе ЭПР напряженности (несколько тысяч эрстед) соответствует частотам микроволнового диапазона. Для поля напряженностью в 10000 Э АЕ, согласно уравнению (9.2), составляет 28026 МГц,
Рис. 9.1. Снятие вырождения электронных спиновых состояний а и 0 под действием приложенного магнитного поля. (Обратите внимание на различие в основном состоянии ЭПР и ЯМР.)
тогда как в магнитном поле такой же напряженности энергия переходов протонного ядерного момента равна 42,58 МГц (т. е. излучение лежит в радиочастотном диапазоне). Магнитный момент электрона составляет — 9,2849-10 “21 эрг-Э-1, а протонный ядерный момент 1,4106-10“ 23 эрг-Э"1.
Спектр ЭПР снимают при фиксированной частоте, обычно около 9500 МГц, или 9,5 ГГц, если напряженность поля равна 3400 Э (X-диапазон), или 35 ГГц, если напряженность поля составляет около 12500 Э (Q-диапазон). Поскольку чувствительность прибора возрастает приблизительно пропорционально V2 и при этом увеличивается спектральное разрешение, обычно предпочитают работать при более высокой частоте. Однако при проведении измерений в Q-диапазоне необходимы образцы меньшего размера, поэтому чувствительность не столь высока, как следует из закона v2. Кроме того, достичь однородности (5Н/Н) поля более высокой напряженности, которое необходимо при более высокой частоте, значительно сложнее. Наконец, если исследуются водные растворы, то по мере увеличения частоты возрастает поглощение энергии растворителем, а это приводит к снижению чувствительности.
Для исследований ЭПР не следует выбирать такие растворители, как вода, спирты и т. д., так как они отличаются высокой диэлектрической проницаемостью и сильно поглощают энергию микроволнового излучения. Их можно использовать только в тех случаях, когда образец дает сильный сигнал и помещается в специальную ячейку (с очень небольшим диаметром ампулы для образца). Методом ЭПР можно изучать газы, растворы, порошки, монокристаллы и замороженные растворы. Проводить исследование замороженных растворов удобнее всего, когда
Спектроскопия электронного парамагнитного резонанса
7
Таблица 9.1
ОБЫЧНО ИСПОЛЬЗУЕМЫЕ СТЕКЛА2
Чистые вещества
З-Метилпентан Серная кислота Сахар
Метилциклопентан Фосфорная кислота Триэтаноламин
Нуйол (парафиновое мае-
л о) Этанол 2-Метилтетрагидрофуран
Изопентан Изопропанол Ди-н-пропиловый эфир
Метилциклогексан Пропанол-1 цис-транс- Декалин
Изооктан Бутанол-1 Триацетин (триацетат глицерина)
Борная кислота Глицерин Толуол
Смеси
Компоненты Соотношение
З-Метилпентан/изопентан 1/1
Изопентан/метилциклогексан 1/6
Метилциклопентан/метилциклогексан 1/1
ц ис-Пентен-2/шранс-пентен-2 Пропан/пропен 1/1
Изопропилбензол/пропан/пропен 2/9/9
Эганол/метанол 4/1, 5/2, 1/9
Изопропиловый спирт/изопентан 3/7
Этанол/изопентан/диэтиловый эфир 2/5/5
Альфанол 79'/смесь первичных спиртов Изопентан/н-бутиловый спирт 7/3
Изопентан/изопропиловый спирт 8/2
Изопентан/н-пропиловый спирт 8/2
Диэтиловый эфир/изооктан/изопропиловый спирт 3/3/1
Диэтиловый эфир/изооктан/этанол 3/3/1
Диэтиловый эфир/этанол 3/1
Изооктан/метилциклогексан/изопропиловый спирт 3/3/1
Диэтиловый эфир/толуол/этанол 2/1/1
Изопропиловый спирт/изопентан 2/5/5
Пропанол/диэтиловый эфир 2/5
Бутанол/диэтиловый эфир 2/5
Диэтиловый эфир/изопентан/диметилформамид/этанол 12/10/6/1
Вода/пропиленгликоль 1/1
Этиленгликоль/вода 2/1
Триметиламин/изопентан/диэтиловый эфир 2/5/5
Триэтиламин/изопентан/диэтиловый эфир 3/1/3
Метилгидразин/метиламин/триметиламин 2/4/4
8
Глава 9
Продолжение табл. 9.1
Смеси
Компоненты Соотношение
Днэтиловый эфир/изопентан/этанол/пиридин Ди-н-бутиловый эфир/диизопропиловый эфир/диметиловый эфир Дифениловый эфир/1,1-дифенилэтан/трифенилметан Диэтиловый эфир/изопентан Дипропиловый эфир/изопентан Дипропиловый эфир/метилциклогексан Диэтнловый эфир/t/ ис-пентен-2/шранс-пентен-2 Этилиодид/изопентан/диэтиловый эфир Этилбромид/метилциклогексан/изопентан/метилциклопентан Этанол/метанол/этилиодид Этанол /метанол /пропи лиодид Этанол/метанол/пропилхлорнд Этанол/метанол/пропилбромид Диэтиловый эфир/изопентан/этанол/1-хлорнафталин З-Метилпентан/изопентан Пропиловый спирт/пропан/пропен Диизопропил амин/пропан/пропен Дипропиловый эфир/пропан/пропен Толуол /мети ленх л ори д 12/10/6/1 3/5/12 3/3/1 1/1-1/2 3/1 3/1 2/1 1/2/2 1/4/7/7 16/4/1 16/4/1 16/4/1 16/4/1 8/6/2/2 1/2 2/9/9 2/9/9 2/4/4 1/1 или избыток то- луола
Толуол/ацетон 1/1 или избыток толуола
Толуол/метанол или этанол 1/1 или избыток то- луола
T олу ол/ацетонитрил 1/1 нли избыток толуола
T олу ол /хлороформ 1/1 нли избыток то- луола
2-Метилтетрагидрофуран/метанол 2-Метилтетрагидрофуран/пропионитрил 2-Метилтетрагидрофуран/метиленхлорид 2/1 2/1 2/1
а Приведенные данные заимствованы из работы Meyer В., Low Temperature Spectroscopy, American Elsevier Publishing Co., New York, 1971. Печатается с разрешения авторов.
замерзший растворитель образует стекло. Растворители, молекулы которых симметричны или склонны к образованию водородных связей, обычно хороших стекол не дают. Например, циклогексан не образует
Спектроскопия электронного парамагнитного резонанса
9
хорошее стекло, а метилциклогексан образует. Некоторые растворители и смеси растворителей, которые при замерзании дают хорошие стекла, приведены в табл. 9.1.
Большое значение имеет также материал, из которого изготовлена ампула для образца. Если отношение сигнала к шуму мало, предпочтительна ампула из кварца, поскольку пирекс в большей степени поглощает микроволновое излучение и сам способен дать сигнал ЭПР.
Известно много факторов, которые видоизменяют электронные энергетические состояния в магнитном поле. Мы рассмотрим эти факторы постепенно при обсуждении спектров ЭПР сложных систем.
Различие в энергиях переходов ЭПР для разных молекул определяется изменением величины ^-фактора в уравнении (9.2), тогда как в ЯМР принято фиксировать gN и вводить константу экранирования, чтобы описать различные резонансные энергии, т. е.
АЕ = - gN pN (1 - ст) Я Ат,.
(9-3)
Когда мы подойдем к более сложным системам, мы рассмотрим параметры, влияющие на 0-фактор
ЯДЕРНОЕ СВЕРХТОНКОЕ РАСЩЕПЛЕНИЕ
9.2. АТОМ ВОДОРОДА
Приступая к обсуждению энергии переходов ЭПР, прежде всего познакомимся с электрон-ядерным сверхтонким взаимодействием (СТВ). Атом водорода (в свободном пространстве) представляет собой достаточно простую систему ввиду его сферической симметрии и отсутствия анизотропных эффектов. Рассматривая явление ЭПР, мы будем использовать оператор Гамильтона, называемый эффективным спин-гамильто-нианом, который количественно описывает все наблюдаемые эффекты и позволяет осуществить полную интерпретацию спектра ЭПР.
Полный спин-гамильтониан для атома водорода (в свободном пространстве) имеет вид:
Н = gfUl 5 - gN $NH I + al S = 00 (HXSX + HySy + HZSZ) - Pn + HyIy + + HZIZ) + a!xSx + aiySy + alz Sz.
Для сферически симметричной системы в магнитном поле, направленном вдоль оси z, он упрощается* до
Я — g$HSz — gN HIz + al-S.
(9-4)
Первый член этого гамильтониана уже рассматривался [уравнение
* Нх и Ну равны нулю, Н = Hz. Эффекты Sx, Sy, 1Х и 1у не обяза j ельно нулевые.
10
Глава 9
(9.1)], он дает зависимость энергии от напряженности поля, представленную на рис. 9.1. О втором члене гамильтониана мы уже говорили при обсуждении ЯМР: он описывает взаимодействие ядерного момента атома водорода с магнитным полем. Второй член меньше первого и имеет противоположный знак (состояние с т/ = + 1 /2 является низшим). Совместное влияние первых двух членов уравнения (9.4) на энергии спиновых состояний атома водорода в магнитном поле показывает рис. 9.2Д. В приведенном примере напряженность магнитного поля фиксирована и штриховые линии показывают изменения энергии, вызываемые введением нового члена в гамильтониан. Для того чтобы определить энергию атома водорода в магнитном поле, мы используем для этого гамильтониана [уравнение (9.4)] базис из четырех возможных электронных и ядерных спиновых функций: <pt = |aeaN)>, <р2 = 1аеРмХ <р3 = |PeaN> и <p4 = |PepN>. Начнем с расчета энергий, обусловленных первыми двумя членами гамильтониана Но. Мы должны решить систему уравнений <<р„|Н0|<рт> — Е<<р„|<рт> = 0, где пит могут быть равны или не равны. Таким образом, секулярный детерминант 4 х 4 в этом базисе включает диагональные члены вида
<aepN|gPHSz ЛмРмН/2<хер^)> E<aepN|aepN>
<\аеРм|^РНЕг|ае)> • |pw)> <аеР1у|17мРм#Л:|Рм^ ’ |ае> — Е = 0.
Поскольку для этой задачи операторами являются fz и Sz, а и Р представляют собой собственные функции, т. е. Sz|ae>= 1/гОСе, Sz|Pe> = — - 72ре, fz|aN> = 72aN и 41 Pn> = Т VzPw- Кроме того, Sz не действует на ядерную спиновую функцию, а не действует на электронную спиновую функцию, что приводит к
<acPN|H0|aepN> - E<aepN|aepN> = у д$Н + у gN pN Н - Е = 0.
Теперь должно быть очевидно, что все не диагональные элементы, обусловленные этим гамильтонианом, равны нулю, поскольку все они имеют вид <<р„|Н0|<рт> — <<р„|<рт>, который отличен от нуля только при п = т. Поскольку матрица гамильтониана диагональна, детерминант уже разложен, и мы непосредственно получаем четыре значения энергии, что и показано выше для aepN. На рис. 9.2, В приведены эти четыре величины: Еь Е2, Е3 и Е4. Обычными правилами отбора для ЭПР являются Диц = 0 и Дт, = + 1. Следует отметить, что два перехода ЭПР (Ди1; = 0), показанные на рис. 9.2, В, имеют одну и ту же энергию. Если рассматривать только два первых члена гамильтониана, спектр ЭПР атома водорода должен быть таким же, как и спектр свободного электрона, т. е. при напряженности поля hv/gfi или д = 2,0023 должна наблюдаться одна линия.
Далее мы перейдем к члену al S гамильтониана. Этот член описывает взаимодействие электронного и ядерного спиновых моментов, которое с классической точки зрения соответствует скалярному про-
A 5
Свободный электрон н ~gftHSz в отсутствие магнитного поля
£<.=-{дрн+\дм^
Еъ = +г9Рн + 'г9нРнН В н=9РН5г~9мР^
£*
Дт = 2
'W, -----------------------4"
Ез-
~Z«
r4u
“4Й
Г
Н=дрН5г-днрмН1г
al
Щш+дцЦцН)
+0
a1
«®n+gNfiNn) Д Н=9^-9Лн1г +al‘S
Рис. 9.2. Влияние различных членов гамильтониана на энергию атома водорода в магнитном поле.
(З^ад/ соответствует т$ = — 1/2 и mj = +1/2: рер у соответствует ms = - 1/2 и mj = - 1/2; ае|3дг соответствует ms = + 1/2 и mj = = - 1/2; соответствует
ms = + 1/2 и mi = + 1/2.
12
Глава 9
изведению этих векторов I и S. Параметр а говорит о величине взаимодействия и имеет размерность энергии. Он носит название константы контактного ферми-взаимодействия, и его величина зависит от электронной плотности на ядре ф(20) в соответствии с уравнением 8л
а = —g₽gN₽N№(0)|2. (9.5)
Для атома водорода слейтеровская орбитальная функция имеет вид фь = (1/лао)ехр( - г/а0), где а0—радиус Бора, равный h2/me2 = = 0,52918 А. Подстановка в уравнение (9.5) величины ф для s-орбитали водорода при г = 0 дает a/h = 1422,75 МГц. Поскольку ядерное СТВ, о котором шла речь, включает скалярное произведение ядерного и спинового моментов, оно имеет х-, у- и z-компоненту, и поэтому
а! • $ = а (1Х • Sx + 1у Sy + /2 • Sz). (9.6)
Элементы, порожденные членом Iz-Sz, действующим на базис a.eaN и т. д., также являются только диагональными, поскольку <ф„ | alz • Sz | <pm> = 0, если т / п. Так, например, | alz | aN> = 0. Теперь диагональные элементы имеют следующий вид:
<aeaN \aSz Iz\a.e a.Ny=^-a
<aePN|aS2-4|aePN> = -^-a
<Peaw|aS2 • f2|PeaJV> = -
<pepw|aS2-f2|pepw>=^a
Чтобы получить значения энергии, показанные на рис. 9.2,Г, эти величины следует прибавить к энергиям, представленным на рис. 9.2,В. Вклады ± */4a в ^i, и Т-Д- члена aIzSz приведены внизу рис. 9.2,Г. Если мы теперь посмотрим на стрелки, показывающие изменение электронного спина, то увидим, что энергии двух переходов уже не равны. Один переход приводит к тому, что спектральная линия смещается на величину 1 /2 а в область более низкой энергии, чем та, которая соответствует g = 2,0023 (см. рис. 9.2,Г), а другая линия характеризуется более высокой (на 1/2а) энергией. Расстояние между этими двумя линиями равно а.
Чтобы закончить задачу, мы должны теперь учесть вклад IXSX и /у$у. Это лучше всего осуществить с помощью операторов сдвига, которые работают так же, как описанные ранее 1+ и I . (см. гл. 8, разд. 6). Для операторов электронного спина
S+ = S2-l- iSv, s_=sx-s...
Спектроскопия электронного парамагнитного резонанса 13
Таким образом,
S+L = (Sjx + $Х) + i(sX - sjy)
и
s_ ix = sjx + $X - i - sjy).
Решая совместно эти уравнения, получим
$Х + $Х= ^+L +
По аналогии с тем, что давали операторы 1+ и можно вывести следующие соотношения:
= 1<хД>, S_f+ |ctepjv)> = IPgCtjv)), S_f+laealV> =°
Все другие действия S_I+ или S+I_ на базис дадут нулевой результат. Таким образом, если мы рассматриваем представленную на рис. 9.3 матрицу 4x4, то единственными^ ненулевыми матричными элементами, получаемыми при действии S+I_ и S_I+, являются
<aePN|aS+f_|PeaN> =а,
<PeaJv|aS_f+|aePN> =а.
Отсюда можно получить матричные элементы
<aePN|aSX + aSXl₽e«N> = (V2)a и
<PeaN|aSX + aSX|ae₽iv> = (1/2) «•
Завершить этот раздел можно, построив полный детерминант, соответствующий исходному спин-гамильтониану [уравнение (9.4)], действуя на базис <р с тем, чтобы получить энергии <<рт|Н|<р„> = Е<фт|ф„>-Детерминант, показанный на рис. 9.3, равен нулю. Отметим, что он является блочно-диагонализованным, так что две величины энергии Et и Е4 получают непосредственно. Мы также видим, что IXSX и приводят к недиагональным элементам, которые смешивают <р2 и Фз- Ре-шая* с помощью теории возмущений результирующий детерминант 2x2, получаем (при втором порядке)
111 а2
Е2 = + —gN$NH - —а + — аи~~------——
2 2 4 4(gPH + gNpNH)
/1 1 \ 1 а2
+ —gNf>NH I —а .
\2 2 / 4 4(д$Н + gN$NH)
* Точное решение имеет вид Е =
± у Кз₽ + 9мМ2Я2 + а2]1'2.
l“A>
IA“w>
I AM
l«A>
I
I AM
- ЭЛЮ + |а - Е 0 0 0
0 + ЭЛН) - |а - Е 1- 0
0 + д^нН) - |а - Е 0
0 0 0 -UgHH - gw^) + la - Е £. 4
Рис. 9.3. Секулярный детерминант для атома водорода в отсутствие поля.
Спектроскопия электронного парамагнитного резонанса 15
Из рис. 9.2,Д, где продемонстрировано влияние этих недиагональных элементов на энергетические уровни, видно, что энергии обоих переходов возрастают на одну и ту же величину. Поскольку недиагональные элементы малы по сравнению с диагональными, эффекты, обусловленные этим членом гамильтониана, называются эффектами второго порядка. Таким образом, эффекты второго порядка не влияют на величину а, которую отсчитывают на спектре, но оказывают влияние на регистрируемую величину д. Более интересно то, что теперь из-за смешивания функций базиса первоначально запрещенный спектральный переход Е3 -> Е2 (одновременное изменение положения электронного и ядерного спинов) становится разрешенным *.
9.3 РЕГИСТРАЦИЯ СПЕКТРОВ ЭПР
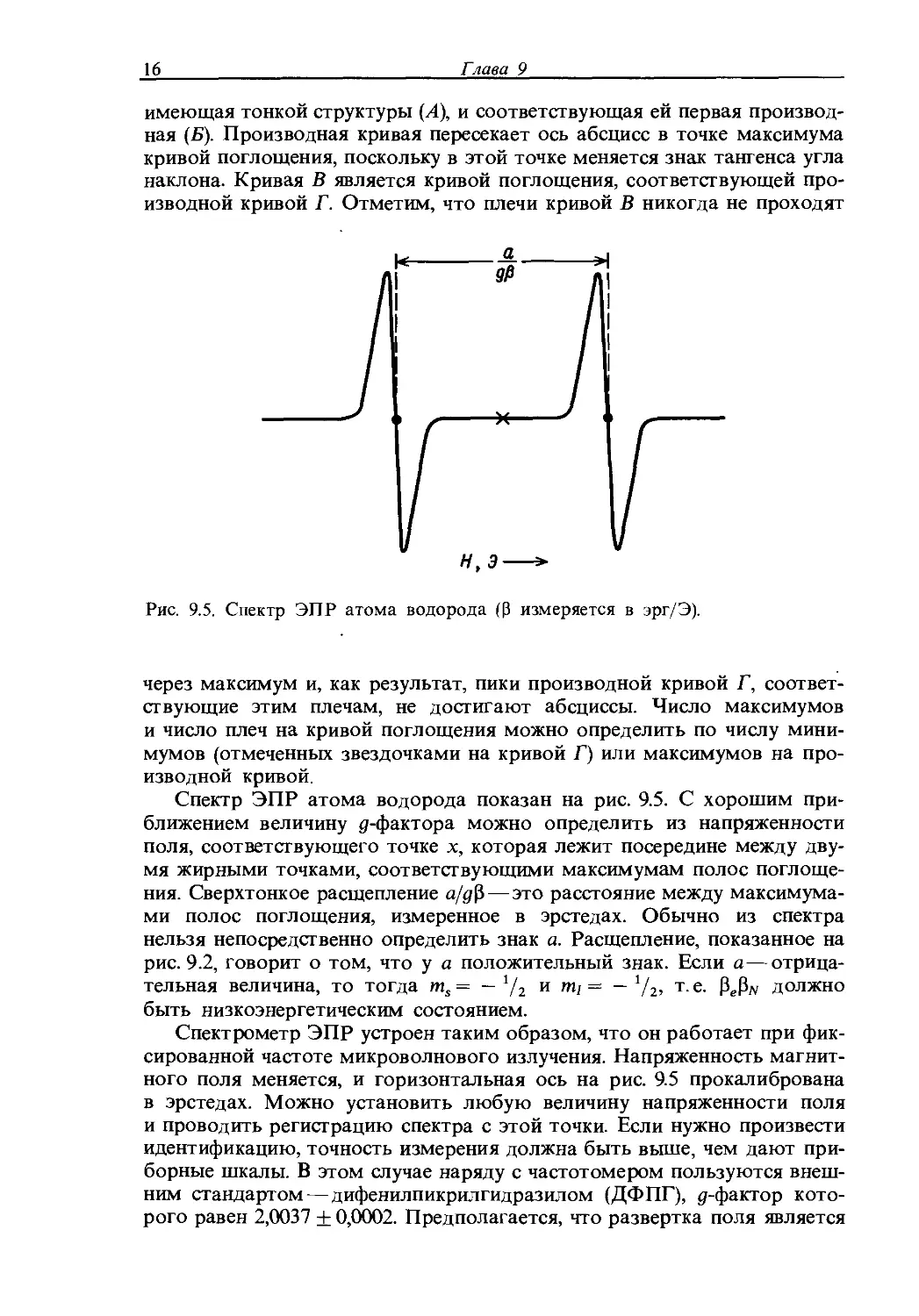
Спектр ЭПР, как и спектр ЯМР, можно представить в виде кривой зависимости интенсивности I от напряженности приложенного магнитного поля. Однако обычно спектры ЭПР представляют в виде первой
Рис. 9.4. Два способа представления спектров ЭПР: спектры поглощения (А, Б) и соответствующие производные кривые (В, Г).
Производной (или производной кривой), т. е. в виде зависимости первой производной (тангенса угла наклона) кривой поглощения от напряженности магнитного поля. Если линии поглощения широки, то первая производная дает лучшее представление о характере спектра. Один тип спектра легко перевести в другой. Соотношение между двумя видами спектров показано на рис. 9.4, где представлены одиночная линия, не
* Разрешенность этого перехода показана Керрингтоном и Мак-Лечланом. (Керрингтон А., Мак-Лечлан Э., Магнитный резонанс и его применение в химии.— М.: Мир, 1970.)
16
Глава 9
имеющая тонкой структуры (Л), и соответствующая ей первая производная (Б). Производная кривая пересекает ось абсцисс в точке максимума кривой поглощения, поскольку в этой точке меняется знак тангенса угла наклона. Кривая В является кривой поглощения, соответствующей производной кривой Г. Отметим, что плечи кривой В никогда не проходят
Рис. 9.5. Спектр ЭПР атома водорода (р измеряется в эрг/Э).
через максимум и, как результат, пики производной кривой Г, соответствующие этим плечам, не достигают абсциссы. Число максимумов и число плеч на кривой поглощения можно определить по числу минимумов (отмеченных звездочками на кривой Г) или максимумов на производной кривой.
Спектр ЭПР атома водорода показан на рис. 9.5. С хорошим приближением величину g-фактора можно определить из напряженности поля, соответствующего точке х, которая лежит посередине между двумя жирными точками, соответствующими максимумам полос поглощения. Сверхтонкое расщепление а/дР—это расстояние между максимумами полос поглощения, измеренное в эрстедах. Обычно из спектра нельзя непосредственно определить знак а. Расщепление, показанное на рис. 9.2, говорит о том, что у а положительный знак. Если а—отрицательная величина, то тогда ms = — 1/2 и = — 1/2, т. е. PeP)v должно быть низкоэнергетическим состоянием.
Спектрометр ЭПР устроен таким образом, что он работает при фиксированной частоте микроволнового излучения. Напряженность магнитного поля меняется, и горизонтальная ось на рис. 9.5 прокалибрована в эрстедах. Можно установить любую величину напряженности поля и проводить регистрацию спектра с этой точки. Если нужно произвести идентификацию, точность измерения должна быть выше, чем дают приборные шкалы. В этом случае наряду с частотомером пользуются внешним стандартом — дифенилпикрилгидразилом (ДФПГ), g-фактор которого равен 2,0037 + 0,0002. Предполагается, что развертка поля является
Спектроскопия электронного парамагнитного резонанса 17
линейной, поэтому 3-факторы для других линий можно отсчитывать от линии стандарта. Ось поля прокалибрована в эрстедах, а д считается безразмерной величиной, поскольку
д = /iv/pH,
где v — фиксированная зондирующая частота, а Н — переменная величина, которая определяется из спектра. Зондирующую частоту находят с помощью частотомера.
Параметр а иногда выражают в эрстедах (Э), мегагерцах (МГц) или см-1. Следует подчеркнуть, что расстояние между линиями в спектре в эрстедах находят с помощью соотношения а/дР, в котором а измеряется в эргах, а р — в эрг/Э. Если д / 2, некорректно приводить расстояние между линиями а в эрстедах. Чтобы получить а в эрстедах, необходимо умножить а, измеренное в эргах, на дР и разделить на дер (где де — ^-фактор свободного электрона, равный 2,0023193). Поскольку а характеризует энергию, лучше говорить о ней как об энергии. Для этого нужно умножить расстояние между линиями, выраженное в эрстедах, на зР. где р измеряется в см-1-Э-1. Эти единицы не зависят от 3-фактора. Значение а в МГц получают, умножая а (см-1) на с(3-1О10 см/с) и деля на 106.
9.4. СВЕРХТОНКОЕ РАСЩЕПЛЕНИЕ В ИЗОТРОПНЫХ СИСТЕМАХ, СОСТОЯЩИХ БОЛЕЕ ЧЕМ ИЗ ОДНОГО ЯДРА
Энергии первого порядка уровней атома водорода выражаются с помощью уравнения (9.7), которое не учитывает небольшое ядерное зеемановское взаимодействие:
Е = gPHms + amsmi. (9.Т)
Подстановка величин ms и тг в это уравнение позволяет воспроизвести энергии, приведенные на рис. 9.2,Г, Для ядра с произвольным ядерным спином проекция ядерного магнитного момента на направление эффективного поля на ядре может принимать любое значение 27 + 1, соответствующее квантовым числам — I, —1 + 1, 1—1, I. Эти ориентации
приводят к 27+1 различным ядерным энергетическим состояниям (одному для каждого значения т,), и если каждое из них взаимодействует с электронным моментом, в спектре ЭПР появляются 27 + 1 линий. Поскольку различия в энергиях малы, будем считать, что все уровни с одной и той же величиной ms заселены одинаково, а линии поглощения ЭПР имеют равную интенсивность и удалены друг от друга на одинаковое расстояние. Например, для неспаренного электрона 14N, где 7=1, ожидаются три полосы.
Далее мы рассмотрим, как изменится спектр, если электрон взаимодействует с несколькими ядрами, т. е. если он делокализован на нескольких ядрах. Чтобы не усложнять картину, предположим, что частицы быстро вращаются во всех направлениях и что 3-фактор незначительно отли
18
Глава 9
чается от ^-фактора свободного электрона. В качестве примера рассмотрим метильный радикал. Как показывает рис. 9.6, добавление квантовых чисел ядерного спинового углового момента индивидуальных протонов приводит к четырем различным величинам полного ядерного
m +|
т т ш +± 2
ш ш Ш -1
ш -4
Рис. 9.7. Четыре перехода в спектре ЭПР метильного радикала (спектр ЭПР изображен на рис. 9.8). (Как и для атома Н, состояние + т/ низшее для т = — 1/2, а состояние — т/ низшее для т$ = + 1/2, что следует из члена IS.)
Рис. 9.6. Схема возможного расположения ядерных спинов протонов в метильном радикале.
спинового момента М;, что в свою очередь приводит (рис. 9.7) к четырем переходам (АМ; = 0, Ams = ± 1. Поскольку полную величину М; = = + 1/2 или — 3/2 (см. рис. 9.6) можно получить тремя различными способами, a Mj = + 3/2 или — 3/2 —только одним, вероятность реализации первого варианта первой системы в три раза больше, чем последнего, и наблюдаемые интенсивности для соответствующих переходов (рис. 9.7) относятся как 1 :3 :3 :1.
Вообще, когда спектр поглощения расщепляется п эквивалентными ядрами с одинаковым спином Ц, число линий определяет выражение + 1. Если расщепление вызывается как набором п эквивалентных ядер со спином так и набором т эквивалентных ядер со спином Ij, число линий дается выражением (2н/; + 1)(2»;/(- + 1). Применение этих общих правил демонстрируют следующие примеры:
1. Если радикал содержит п неэквивалентных протонов, на которых делокализован электрон, спектр состоит из 2" линий.
2. Если неспаренный электрон делокализован на п эквивалентных протонах, в спектре наблюдается п + 1 линий (2н/ + 1). Это число меньше,
Спектроскопия электронного парамагнитного резонанса
19
чем число линий, ожидаемых для неэквивалентных протонов, т. е. 2", поскольку некоторые из возможных комбинаций ядерных спинов вырождены (см. рис. 9.6). Спектр метильного радикала, показанный на рис. 9.8, по этим причинам состоит из четырех линий.
Рис. 9.8. Производный спектр метильного радикала в СН4-матрице при 4,2 К.
Спектры, ожидаемые для различных чисел неэквивалентных протонов, можно легко предсказать, рассматривая поочередно расщепление, вызываемое каждым из протонов, как это показано на рис. 9.9. Если сигнал расщепляется двумя эквивалентными протонами, суммарное значение М, для трех уровней может быть равно = + 1, 0 и — 1. Поскольку суммарное Мг = 0 можно получить двумя способами (суммируя + 1/2 и — 1 /2, а также — х/2 и + Vi), центральный уровень является дважды вырожденным. В спектре наблюдаются три линии (AM, = 0, Ams = + 1), отношение интенсивностей которых составляет 1:2:1. Выше мы рассмотрели систему с тремя протонами (например, метильный радикал), аналогичные соображения применимы и для представленной на рис. 9.9 системы с большим числом протонов.
Относительные интенсивности полос даются коэффициентами биномиального разложения. Следует помнить, что эта формула применима лишь при эквивалентных протонах или других ядрах с I = 1/2.
3. Если неспаренный электрон делокализован на двух совокупностях неэквивалентных протонов, число линий, ожидаемое в спектре, определяется произведением чисел, ожидаемых для каждой совокупности: (Znlj + l)(2n/j•+ 1). Анион-радикал нафталина, который можно получить при переносе электрона от натрия к нафталину, содержит неспаренный электрон, делокализованный по всему нафталиновому циклу. Нафталин содержит две различные совокупности из четырех эквивалентных протонов, которые должны приводить к двадцати пяти линиям в спектре ЭПР его анион-радикала, что и обнаружено экспериментально.
4. Если электрон делокализован на ядрах, спин которых превышает ' Iг, то для расчета ожидаемого числа линий можно использовать процедуру, аналогичную применяемой для протонов. Если электрон делокализован на нескольких эквивалентных ядрах, спин которых превышает 1 /2,
20
Глава 9
число ожидаемых спектральных линий предсказывает формула 2nl + 1. Например, для электрона, делокализованного на двух эквивалентных атомах азота, ожидается пять линий. Группировка спинов, аналогичная изображенной на рис. 9.6, показывает, что интенсивности пяти линий будут относиться как 1 :2 :3 :2 :1.
+ 1 (1).---''
О (2) .
-1 (1)->
э +2(1)
+ 4(1) -------
------‘--..-2(1)
"Т(1)
-1 Я).--'--Г^?--.,-К4)
'------*'-< +2(1)
+ g (1)
+ 1 (5) + 1 (10) 4 (10)
(5)
-1(1)
-j (D -1(5) -1(10) + 1(10) + 1(5) + j(1)
0 1 2 3 4 5
Число протонов
Рис. 9.9. Сверхтонкие энергетические уровни, являющиеся следствием взаимодействия иеспареииого электрона с переменным числом эквивалентных протонов [2]. Числа в скобках показывают вырождение соответствующих уровней и, следовательно, относительные интеисивиости линий соответствующих переходов.
5. Если электрон делокализован на нескольких неэквивалентных атомах, общее число ожидаемых линий получают, умножая числа линий, ожидаемые для каждого атома. Схема, представленная на рис. 9.10 для электрона, делокализованного на двух неэквивалентных ядрах с / = 1, часто используется для того, чтобы показать возможное расщепление. Три линии в ряду А представляют расщепление линии ЭПР на ядре с I = 1 и константой СТВ а. Каждая из этих линий расщепляется на три компоненты в результате делокализации электрона на втором неэквивалентном ядре с I = 1 и константой СТВ а', что приводит к девяти линиям (ряд Б). В последующих разделах для интерпретации спектров используется схема, аналогичная приведенной на рис. 9.10. Форма спектра и расстояние между линиями в нем будут зависеть от резонансного поля, д-фактора и констант СТВ а и а'. Часто в наблюдаемом спектре не удается обнаружить всех ожидаемых линий, поскольку ширина линий велика по сравнению с а/д$ и две соседние линии могут не разрешаться. Например, спектр, приведенный на рис. 9.11. может быть обусловлен поглощением гипотетического радикала Н - Х+ <=s Н — X , где 1 = 1 для X.
Спектроскопия электронного парамагнитного резонанса 21
Две линии в ряду обусловлены протонным расщеплением; каждая линия ряда Б в свою очередь расщепляется на три компоненты в результате взаимодействия с ядром X. Таким образом, мы должны ожидать шесть линий равной интенсивности, однако можно зарегистрировать только пять линий, если две центральные компоненты не разрешаются;
а
(—1------1
I I I А
а'
6
III III III Б
а
Рис. 9.10. Три линии, которые можно ожидать в том случае, когда электрон находится у ядра с 1=1 (А), и девять линий, являющихся результатом расщепления на втором неэквивалентном ядре с I = 1 (Б).
A I I
Б I I II I I
Рис. 9.11. Гипотетический спектр поглощения радикала Н—X + (I = 1 для X).
в таком случае возникнет полоса, площадь которой вдвое превышает площадь других полос (рис. 9.11).
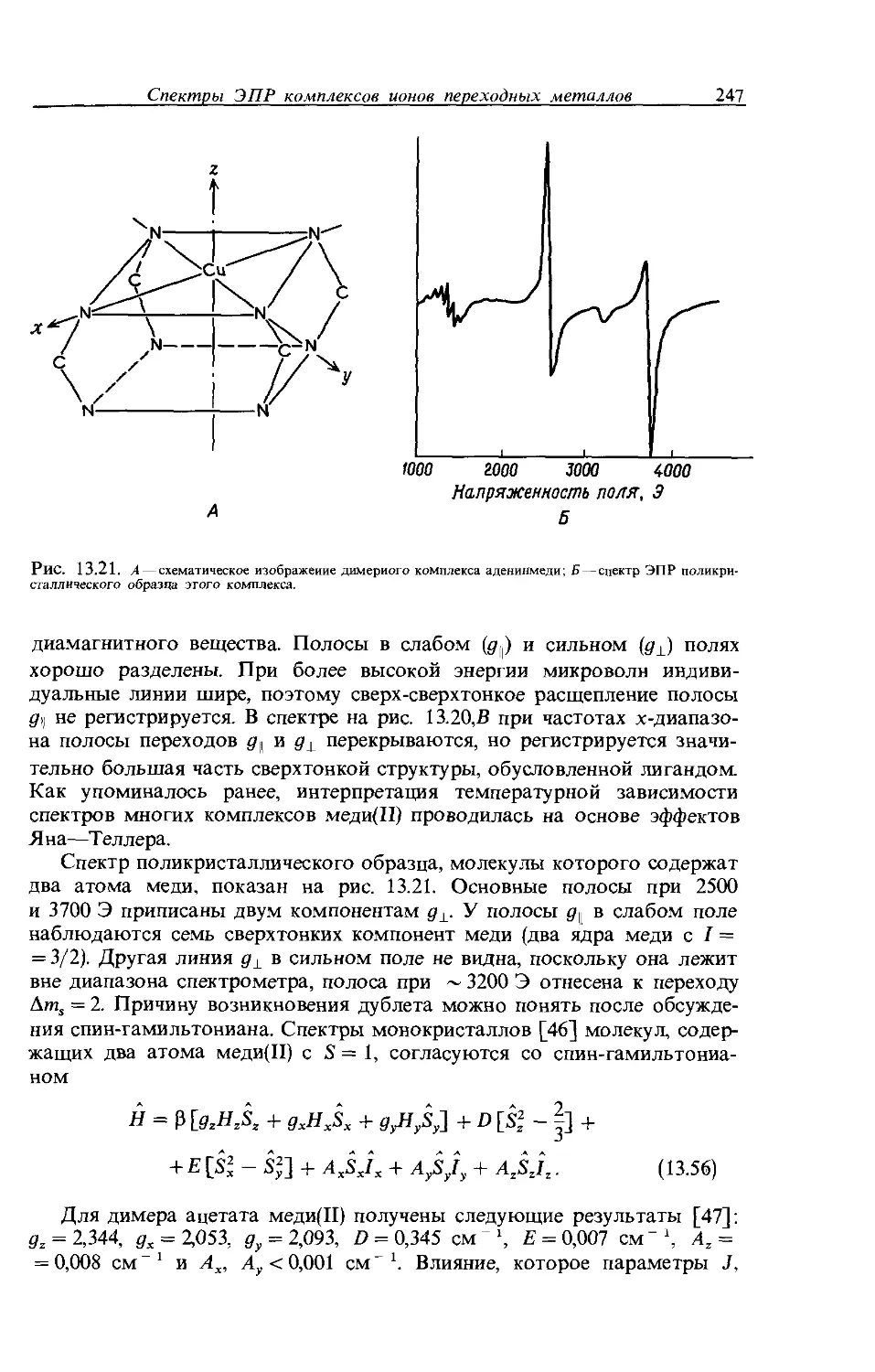
Спектр ЭПР [3] бмс-(салицилальдимин)меди(П) представляет собой интересный пример, который демонстрирует те свойства ядерного расщепления, которые мы обсуждали (рис. 9.12). Этот спектр получен для твердого вещества и не является изотропным; анизотропию спектра специально рассматривать не будем. Четыре основные группы линий
Рис. 9.12. Производный спектр ЭПР бнс-(салицилальдимин)меди(П) при изотопно чистой 63Си. Звездочкой обозначен калибровочный пик ДФПГ. Данные Maki А.Н., McGarvey B.R., J. Chem. Phys., 29, 35 (1958).
22
Глава 9
являются результатом расщепления на ядре 63Cu (I = 3/2). Сверхтонкая структура в каждой из четырех групп состоит из 11 линий относительной интенсивности 1:2:3:4:5:6:5:4:3:2:1. Эти линии являются результатом расщепления на двух эквивалентных атомах азота и атомах водорода, Н' на рис. 9.12. Общее число линий должно быть равно 15: (2nNIN + 1)(2нн/н + 1) = 5 х 3 = 15. В истинном спектре для каждой подгруппы обнаружено 11 линий, что вызвано перекрыванием некоторых из 15 линий (рис. 9.13). Линия электрона, не расщепляемая ядром, показана в ряду А, расщепление на двух эквивалентных ядрах азота — в ряду Б, а расщепление на двух эквивалентных протонах — в последнем ряду, В.
4d 4е 4f 4е 4d
В I I || I II | II I II I I
d, 2d, d + е, 2е, е + f,2f, f + е, 2е, е + d, 2d, d
Рис. 9.13. Интерпретация спектра ЭПР бис-(салицилальдимин)меди(П).
А—нерасщепленный переход; Б — расщепление на двух эквивалентных атомах азота (d, е н f—относительные интенсивности 1, 2 и 3); В — последующее расщепление на двух эквивалентных протонах.
Два азота расщепляют резонансную линию на пять с относительной интенсивностью 1 :2 :3 :2 :1. Эти величины обозначены в ряду Б как 4d, 4е и 4/, где d= 1, е = 2 и /= 3. Расщепление на двух эквивалентных протонах приводит к трем линиям для каждой линии ряда Б с отношением интенсивностей 1:2:1. Интенсивности указаны буквами под линиями ряда Б, их получают, суммируя ожидаемые интенсивности. Поскольку относительные интенсивности равны d = 1, е = 2 и /= 3, отношение интенсивностей линий в ряду есть 1:2:3:4:5:6:5:4:3:2:1. Экспериментальный спектр согласуется с такой интерпретацией. Приемлемость ее также подтверждают следующие данные:
1. Дейтерирование групп N — Н" (см. рис. 9.12) приводит к соединению, которое дает идентичный спектр.
* Б
Рис. 9.14. Основная молекулярная геометрия комплекса Co3(CO)9Se (Л) и спектр ЭПР (£) монокристалла FeCo2 (CO)9Se, в который внесено около 0,5% парамагнитного Co3(CO)9Se при 77 К.
В этом спектре 22 сверхтонкие компоненты; молекулярная ось третьего порядка параллельна направлению магнитного поля. (Печатается с разрешения Strouse С.Е., Dahl L.F., J. Amer. Chem. Soc., 93, 6032 (1971). © Am. Chem. Soc.)
Спектроскопия электронного парамагнитного резонанса 23
—--------------
2. Если заместить протоны Н' на метильные группы, то полученное соединение дает спектр ЭПР, состоящий из четырех основных групп линий каждая из которых в свою очередь включает пять линий, обусловленных только расщеплением на ядрах азота. Сверхтонкое расщепление на протонах N — Н" или протонах метильных групп либо очень мало, либо вообще отсутствует.
Этот спектр убедительно свидетельствует о делокализации неспаренного электрона комплекса на лиганде. Объяснить это можно только образованием ковалентных связей металл — лиганд, поскольку только при смешивании волновых функций иона металла и лиганда можно получить вклады лиганда в молекулярную орбиталь комплекса, которая содержит неспаренный электрон.
Другое интересное применение ЭПР [4] можно продемонстрировать на примере спектра ЭПР соединения Co3(CO)9Se, который наряду со структурой этого соединения показан на рис. 9.14. Наличие в спектре 22 линий говорит о том, что один неспаренный электрон в этой системе полностью делокализован на трех атомах кобальта (/с„ = 7/2)- В результате состояние окисления каждого атома кобальта соответствует + 2/3.
9.5. ВКЛАДЫ В КОНСТАНТУ СВЕРХТОНКОГО РАСЩЕПЛЕНИЯ В ИЗОТРОПНЫХ СИСТЕМАХ
Из уравнения (9.5) следует, что
а = ^-g$gN Pv I 12-
Сверхтонкое расщепление, обусловленное делокализацией плотности неспаренного электрона р(г.\-) на атоме водорода молекулы, выражается уравнением
а = ~^-gpgN pv Ph (rN), (9.8)
где p (гдг) можно приближенно рассматривать как разность в средних числах электронов у ядер, спиновые моменты которых характеризуются ms = + */2 и — */2. Если преобладает спиновая плотность + */2, т. е. доминируют электроны со спиновыми моментами, направленными против поля, то говорят, что это ядро находится под действием отрицательной спиновой плотности. Избыток электронной плотности с ms = — */г на' зывается положительной спиновой плотностью. Положительная спиновая плотность изображается стрелкой, направленной вдоль внешнего поля, а отрицательная плотность изображается стрелкой, направленной против поля. Некоторые авторы называют положительную спиновую плотность спином а, если даже волновая функция, применяемая для расчета матричных элементов для этого электрона, обозначается как pt„ Таким образом, принято придавать спиновой плотности в точности
24
Г.шва 9
противоположный знак, нежели электронным спиновым волновым функциям. В этой книге мы постараемся не обозначать спиновую плотность символами а и р и пользоваться ими только для обозначения спиновых волновых функций.
Следует также отметить, что плотность неспаренного спина на атоме в молекуле не согласуется непосредственно с вкладами атомов в молекулярную орбиталь, содержащую неспаренный электрон. Последний эффект мы назовем плотностью неспаренного электрона. Неспаренный электрон на орбитали одного атома в молекуле может поляризовать спаренные спины ортогональной ст-связи, так что один из электронов будет большее время проводить в окрестности одного атома, чем в окрестности другого. В результате на ядре атома появляется плотность неспаренного спина, если даже на этом атоме и отсутствует плотность неспаренного электрона. Эта мысль будет более понятна, если мы рассмотрим следующий пример.
Некоторые из первых попыток интерпретации СТВ были связаны с ароматическими радикалами, в которых неспаренный спин находится в тг-системе, как, например, в C6H5NO7- Расчет осуществлялся по методу Хюккеля, и для определения величины плотности неспаренного электрона у различных атомов углерода использовались квадраты /^-коэффициентов углерода в МО, на которой находится неспаренный электрон. Экспериментально наблюдаемое сверхтонкое расщепление обусловлено протонами цикла, которые ортогональны я-систсмс. Непосредственно на них плотность неспаренного электрона находиться не могла, но плотность неспаренного спина тем не менее на них ощущалась из-за так называемой спин-поляризации, или косвенного механизма. Мы попытаемся дать предельно простое представление этого эффекта, используя метод валентных схем. Рассмотрим две резонансные формы, представленные на рис. 9.15 для связи С-Н в такой системе, в которой неспаренный электрон находится на Рп-орбитали углерода. В отсутствие взаимодействия между я- и ст-системой (так называемое приближение идеального спаривания) мы можем записать волновые функции связывающей и разрыхляющей ст-орбиталей, используя метод валентных схем:
\[/° +\|/ц) И \|/*=—F-(Vl-^Ilb
|/2 |/2
где и фц—волновые функции структур I и II, изображенных на рис. 9.15, которые мы не будем пытаться детализировать в терминах АО и спинового базиса. Если учитывать взаимодействие ст- и я-систем, то можно обнаружить, что структура I более устойчива, чем структура II, и поэтому ее вклад в основное состояние превышает вклад II, что приводит к следующим функциям валентных схем:
ф° = яф, + (а > Ь) и
ф* = (а' < Ь').
Спектроскопия электронного парамагнитного резонанса
25
В результате электроны ст-связи С — Н поляризуются, и на атоме водорода появляется спиновая плотность, знак которой противоположен знаку плотности неспаренного электрона на рг-орбитали углерода. Таким образом, причиной большей стабилизации структуры I по сравнению со структурой II является электронное обменное взаимодействие.
I
II
Рис. 9.15. Резонансные формы фрагмента ст-связи С- Н с неспаренным электроном на углероде.
Это тот же самый эффект, который приводит к тому, что низшее возбужденное состояние гелия ls12s1 является триплетом, а не синглетом. Если мы пометим два электрона, которые для каждой структуры, изображенной на рис. 9.15, находятся в основном на углероде, мы увидим, что они могут обмениваться в структуре I:
яТ Ь]
СД| Н СД| н
h а
но обмен невозможен в структуре II. Таким образом, структура I стабилизуется в результате квантовомеханического взаимодействия, аналогичного резонансу.
Более полное описание этого эффекта в терминах молекулярных орбиталей дано в книге Дьюара [5]. Если рассматриваются взаимодействия e2/rtj в молекуле, один из полученных видов интеграла описывает отталкивательное взаимодействие:
е2
Jmn = Ж (О (0 (/’) 'Рп (/') Л,' Л/ (9-9)
ГЧ
Он характеризует кулоновское отталкивание между электронными плотностями [|ф*(г)фт(г)(/Т|—плотность в I] электронов i и j, находящихся на орбиталях тип, где т может быть равно или не равно п. Существуют и другие интегралы, которые мы обозначим как Ктп, которые имеют нулевое значение, если спины электронов спарены, и отличны от нуля, если спины параллельны. Интегралы Ктп имеют следующий вид: е2
ктп = ИМ/*(ОФП(О—ожожа- (9Л°)
ГЧ
Эти интегралы носят название обменных интегралов, поскольку они характеризуют обменное взаимодействие орбиталей, на которых находятся электроны i и j. Если спины двух электронов параллельны, квадраты
26
Глава 9
их волновых функций показывают, что вероятность их нахождения в непосредственной близости друг от друга исключительно низка по сравнению со случаем антипараллельных спинов. Таким образом, кулоновское отталкивание снижается, если спины параллельны, поскольку электроны находятся далеко друг от друга. Величина интеграла Ктп зависит от того, насколько сильно перекрываются две орбитали тип. Параметр 'l/m(/)'l/n(/)^Tj ранее был назван дифференциальным перекрыванием. Интеграл перекрывания дифференциального перекрывания двух ортогональных орбиталей равен нулю, т.е.
Ж(/)ОЖ = 0-
Однако если на интеграл дифференциального перекрывания в элементе объема (hj подействовать е2/гу, умноженным на фт(г)ф„(г), и проинтегрировать по элементам объема и d^, то полученный результат отличен от нуля, но это уже будет обменный интеграл. Обменные эффекты не только приводят к спиновой поляризации, описанной выше, но и к тому, что первое возбужденное триплетное состояние гелия лежит ниже, чем первое возбужденное синглетное состояние; их следствием являются закономерности, которые суммирует правило Гунда.
Спиновая плотность на атоме водорода связи С — Н, возникшая за счет того, что на л:-орбит ал и (2pz) находится плотность неспаренного электрона, дается выражением
ан = 2Рс, (9-11)
где рс — плотность неспаренного электрона на 2рг-орбитали углерода, a Q — величина пн, если электрон полностью связан с углеродом. Спиновая плотность на протоне отрицательна, поэтому ан также отрицательная величина и отрицательной должна быть и Q. В системах, где рс известна, Q можно рассчитать. Экспериментально Q варьирует от — 22 до — 27 Э. Приближенная величина — 23 Э, полученная с помощью метода Хюккеля для ароматических радикалов, приемлема для большинства задач, которыми мы будем заниматься. В расширенном методе Хюккеля перекрывание нулевым не считается, и коэффициенты молекулярных орбиталей нормируются таким образом, чтобы учесть это перекрывание. Поэтому величина Q зависит от метода расчета МО, т. е. от способа определения рс.
Если МО, на которой находится неспаренный электрон, имеет узел на одном из атомов углерода п-системы, возникают аналогичные обменные взаимодействия с низкоэнергетическими заполненными молекулярными п-орбиталями, приводящие к отрицательной спиновой плотности на указанном атоме углерода. (Более подробно мы обсудим этот вопрос при рассмотрении аллильного радикала, которое проводится ниже.) Результирующее обменное взаимодействие этого неспаренного спина с ст-связью С — Н приводит к появлению на водороде положительной спиновой плотности. В методе Хюккеля, расширенном методе Хюккеля и других ограниченных методах (т. е. в расчетах, при которых на каждую
Спектроскопия электронного парамагнитного резонанса 27
молекулярную орбиталь помещаются два электрона) обменные взаимодействия не учитываются. Результаты расчетов просто говорят о наличии на атоме углерода или на атоме водорода узла. Для устранения этого недостатка, например, применительно к атому водорода, непосредственно связанному с атомом углерода, на ортогональной С2р-орби-тали которого находится неспаренный электрон, используют уравнение (9.11). Часто проводят лишь качественное обсуждение и других поляризационных эффектов в молекуле, а при попытке количественной подгонки расчетных и экспериментальных констант взаимодействия в молекуле пренебрегают атомами углерода или водорода, где эти эффекты
Н (4,06) (13,93) Н Ь Н (13,93)
С1
I I
(14,98) Н Н (14,98)
Рис. 9.16. Протонное сверхтонкое расщепление в аллильном радикале.
доминируют. Это обсуждение можно сделать более конкретным, если рассмотреть [6] протонное сверхтонкое расщепление в аллильном радикале (рис. 9.16). В тг-системе радикала находятся три электрона, волновые функции которых даются следующими выражениями:
Ф1 — “2“(Ф1 + ]/2фг + фз) (связывающая орбиталь) (9.12)
, 1 / Фг= ~7^(ф1- Фз) 1/2 Фз= у(ф1- ]/2ф2+ Фз) (несвязывающая орбиталь) (9-13) (разрыхляющая орбиталь) (9-14)
Неспаренный электрон находится на орбитали ф2, поэтому можно предсказать, что плотность неспаренного электрона на концевом атоме углерода С1 равна pri = (1/]/2)2 = 0,5, где 1/]/2 — коэффициент С1 в МО, на которой находится неспаренный электрон. Как следует из уравнения (9.11), при Q = — 23 ан равна — 11,5. Более того, на центральном атоме углерода С2 не должно быть плотности неспаренного электрона, и в отсутствие какого-либо вида спиновой поляризации, затрагивающей тг-электронную плотность углерода, следует ожидать нулевую константу взаимодействия для этого атома водорода или для 13С в этом положении. Однако такого не наблюдается. Чтобы интерпретировать константы взаимодействия этого центрального атома водорода, необходимо рассмотреть поляризацию двух видов. При качественной интерпретации рассматривают заполненную орбиталь ф1 и записывают для нее две отдельные спиновые орбитали и ф1ь. На каждую спиновую орбиталь помещают только один электрон. Вол
28
Глава 9
новые функции выражают через приведенные выше волновые функции аллильного радикала в таком виде:
Фы = Ф1 + Из
фп>= Ф1 - Из (где Х«1)
Если спин направлен вдоль поля в низкоэнергетической ф1а и против поля в ф1(и на атомах 1 и 3 по сравнению с атомом 2 должно наблюдаться увеличение спиновой плотности, направленной вдоль поля. В ф1Ь при спиновой плотности, направленной против поля, на атоме 2 должна быть большая величина отрицательной спиновой плотности, чем на атомах 1 и 3. Таким образом, мы не переводим каких-либо неспаренных электронов на старую орбиталь фь а только влияем на распределение неспаренных спинов на трех атомах, что приводит к отрицательной (противоположной приложенному полю) спиновой плотности на С2. Эта отрицательная спиновая плотность затем спин-поляризуется под действием электронной пары связи С — Н [см. обсуждение уравнения (9.11)] так, что спиновая плотность оказывается на атоме водорода. Обменное взаимодействие неспаренного электрона, находящегося на ф2 (главным образом, на С1 и С3), с парой электронов, находящихся на ф1( снижает энергию ф1о по сравнению с ф1ь. Два атома водорода, связанные с концевым атомом углерода, неэквивалентны по симметрии, но до сих пор мы не говорили ни о каких эффектах, которые могли бы сделать их неэквивалентными с точки зрения распределения спиновой плотности. Такая неэквивалентность выявится с введением обменной поляризации, затрагивающей заполненные молекулярные ст-орбитали.
Итак, мы описали косвенные механизмы размещения плотности неспаренного спина на атоме водорода. Если свободный радикал является ст-радикалом, например винильным радикалом Н2С = С—Н, протонные АО молекулы дают вклад в молекулярные ст-орбитали, на которых находится неспаренный электрон. Таким образом, неспаренный электрон делокализован непосредственно на протоне, и ан пропорциональна ф2. Поскольку расчеты по методу Хюккеля не годятся для ст-си-стем такого вида, то вначале расчет МО проводится с помощью расширенного метода Хюккеля. Методика расчета ф2 на ядре водорода изложена в работе [7], вн рассчитывается как
ан,Э= 1887фн2 (9.15)
Спиновая поляризация в этих расчетах не учитывается. Часто большая часть плотности неспаренного электрона находится на данном атоме радикала. В таком случае спиновая поляризация больше влияет на протоны, непосредственно связанные с этим атомом. Для указанного атома можно ожидать плохого соответствия между экспериментом и расчетом. Как правило, при заметной прямой делокализации влияние спиновой поляризации для других протонов в молекуле (за некоторыми исключениями) относительно незначительно.
Описаны два полуэмпирических количественных подхода, в которых учитывается спиновая поляризация. В одном из них, описанном Мак-
Спектроскопия электронного пара магнитного резонанса 29
Дечланом [8], применяются волновые функции из расчетов по ограниченному методу, и к ним вводятся поправки на обменные эффекты. Результаты этого подхода оказались не слишком обнадеживающими. Во втором подходе использовались молекулярные орбитали, полученные при неограниченных расчетах, т. е. спиновые орбитали. Наиболее общим является так называемый метод ЧПДП [9], который был параметризован Поплом и др. для расчета спиновых плотностей. Для атомов, для которых доминирует спиновая поляризация, этот подход широко не проверялся, но в настоящее время его, конечно, следует использовать. Выходными данными являются одноэлектронные орбитали; чтобы получить чистую спиновую плотность на атоме, суммируют все положительные и отрицательные спиновые плотности на атомах во всех заполненных молекулярных орбиталях. Для радикала пиридина расчетные данные, полученные методом ЧПДП, плохо согласуются с экспериментальными [15].
Гораздо сложнее применить результаты, полученные при расчетах молекулярных орбиталей, к сверхтонкому расщеплению от взаимодействия с атомами, отличными от атома водорода. В отличие от протонов, для которых характерны только описанные выше прямой и косвенный механизмы СТВ, на сверхтонкое расщепление от взаимодействия с 13С влияют и другие факторы: 1) Неспаренные электроны на р(л)-орбитали могут поляризовать заполненные 2s- и 1 s-орбитали того же самого атома. 2) Может иметь место прямая делокализация электронной плотности на 25-орбиталь ст-радикала. 3) Спиновая плотность на соседнем атоме углерода за счет поляризации ст-связи С — С может вызывать появление спиновой плотности на 2s- и 2р-орбиталях углерода, резонанс которого поддается интерпретации. Расчеты [10—13] для сверхтонкого расщепления, вызываемого 14N, 33S и 17О, оказались более успешными, чем в случае 13С. Так, удалось интерпретировать спектры кремнийсодержащих радикалов [13]. Обнаружено, что влияние спиновых плотностей на соседних атомах для этих ядер имеет меньшее значение, чем для ядер 13С.
Результаты расчетов МО применяются для отнесения полос в спектре ЭПР органического радикала, а также для установления геометрии свободных радикалов. Например, можно установить, плоский ли радикал СН3- или лежит ли связь С — Н винильного радикала вдоль оси связи С — С. Если обнаруживается, что рассчитанные константы СТВ в значительной степени зависят от геометрии радикала (т. е. для различных структур проводится ряд МО-расчетов), соответствие рассчитанных и экспериментальных результатов может служить подтверждением правильности установления геометрии [14]. В некоторых примерах расчеты МО послужили доказательствами природы радикала, образовавшегося в эксперименте [Па, 14]. Например, у-облучение пиридина дает, как полагают, катион-радикал пиридина, т. е. частицу, у которой удален один из электронов неподеленной пары. Результаты, приведенные в табл. 9.2, показывают, что в действительности образуется 2-пиридильный радикал [Па].
30
Глава 9
Результаты расчетов, проведенных методом МО, и экспериментальные данные, полученные методом ЭПР, позволили установить, что в ст-системах имеет место очень большая степень делокализации электрона. Например, значительные количества плотности неспаренного
Таблица 9.2
Изотропные константы сверхтонкого расщепления (Э), рассчитанные расширенным методом Хюккеля и найденные экспериментально
Рассчитанные для Эксперимен-тальные
C5H5N + C5H4N (2-пиридил)
«N 52,5 33,8 29,7
ан 27,0(2,6Н) 5,3 )
9,7(3,5Н) 9,3 1
38,8 (4 Н) 7,6 [ 4,3
0,0 J
электрона обнаружены на протонах метильных групп CH3NH2 » C2H5NH2- Молекулярная орбиталь неподеленной пары представляет собой не локализованную орбиталь неподеленной пары азота, а делокализованную молекулярную орбиталь. Степень делокализации на протоны в различных ротамерах варьирует [16].
АНИЗОТРОПНЫЕ ЭФФЕКТЫ
9.6. АНИЗОТРОПИЯ ^-ФАКТОРА
Свойства 0-фактора радикала NO2, захваченного [17] в монокристалле KNO3, дают представление еще об одном аспекте спектроскопии ЭПР. Если на кристалл наложить поле, направленное вдоль оси NO2 (ось вращения второго порядка), то мы получим g = 2,006. Если поле направлено вдоль оси х или вдоль оси у (плоскость молекулы, включающая у), д = 1,996. В твердом состоянии молекула быстро вращается вокруг оси z, поэтому тот же самый результат получается для осей х и у, параллельных полю. Зависимость 0-фактора от ориентации еще более заметна, если исследуются комплексы ионов переходных металлов (см. далее) и комплексы лантаноидов и актиноидов.
До сих пор мы имели дело с так называемыми изотропными спектрами. Такие спектры дают радикалы сферической или кубической симметрии. В спектрах радикалов более низкой симметрии, находящихся в твердом состоянии, как 0-фактор, так и а анизотропны. Обычно для этих низкосимметричных систем спектры растворов имеют качественно изотропный вид, поскольку анизотропные эффекты усредняются до ну
Спектроскопия электронного парамагнитного резонанса 31
ля из-за быстрого вращения молекул. Сейчас мы рассмотрим, как возникают анизотропные эффекты и как их можно определить, а позднее (гл. 13) обсудим, как анизотропию а и ^-фактора можно использовать для получения информации об электронном основном состоянии комплексов переходных металлов.
Анизотропия д-фактора возникает в результате взаимодействия спинового углового момента с орбитальным угловым моментом. Спиновый угловой момент ориентируется в зависимости от направления поля, но орбитальный угловой момент, который связан с электронами, движущимися по молекулярным орбиталям, привязан к орбитальной волновой функции. Рассмотрим орбитальный вклад в момент электрона, находящегося на круговой молекулярной орбите, которая может прецессировать вокруг оси z молекулы. На рис. 9.17 схематически показаны две
А
Рис. 9.17. Взаимодействие проекций спина и орбитального углового момента для двух различных молекулярных ориентаций относительно приложенного поля.
различные ориентации этой орбитали в молекуле относительно направления поля. На рис. 9.17,Л вектор* орбитального магнитного момента Hl и вектор спинового магнитного момента ц, имеют одно и то же направление. На рис. 9.17,Б показана другая ориентация молекулы. Момент электрона характеризуется той же самой величиной, что и раньше, но теперь результатом является векторная сумма моментов, поскольку направления щ. и ц, не совпадают. Если бы орбитальный момент отсутствовал, тогда момент электрона должен был быть изотропным. Если влияние орбитального момента мало, то оно учитывается д-фактором, и в результате этот д-фактор становится анизотропным. Для описания g-фактора в этом случае необходим тензор, уравнение (9.16), который определяет эффективный спин, т.е. g-S = Seff. Для различных ориентаций д-тензор учитывает орбитальные эффекты, удлиняя и укорачивая Seff. Следует подчеркнуть, что если даже основное состояние
* Направление вектора орбитального углового момента определяется правилом правой руки, это направление противоположно направлению вектора магнитного момента электрона.
32
Глава 9
молекулы не имеет связанного с ним орбитального углового момента, то индуцированное полем примешивание к основному состоянию возбужденного состояния, которое обязано иметь орбитальный угловой момент, может привести к анизотропии «/-фактора. Информация, которую дает анизотропия g-тснзора об электронной структуре молекулы, обсуждается в гл. 13, посвященной ЭПР комплексов ионов переходных металлов.
Чтобы описать взаимодействие электронного спинового момента с магнитным полем и магнитным ядром [уравнение (9.4)] изотропных систем, запишем гамильтониан как
H = gfiHSz —gNPNHIz+ al-S,
где g-фактор и а—скалярные величины. Если исследуется анизотропный свободный радикал в твердой фазе, как д так и а должны быть заменены на тензоры или матрицы. Член g$HSz гамильтониана превращается в p.S’ g Н, который можно представить матрицей
Ж sv
gxx
Syx
Szx
Sxy
Syy
Szy
Sxz
Syz
Szz
Hx
(9.16)
На тензор заменяется также и а в члене al S. В данном случае х, у и z определяются в лабораторной системе координат, т. е. они являются осями кристалла. Недиагональный элемент gzx дает вклад в д-фактор вдоль оси z кристалла, когда поле приложено вдоль оси х. Эта матрица диагональна, если оси кристалла совпадают с молекулярной системой координат, которая диагонализует g. Если оси не совпадают, а кристалл зондируется вдоль своих осей х, у и z, то в матрицах, как это будет показано позднее, возникнут недиагональные элементы. Матрицу д можно привести к диагональному виду, выбрав соответствующим образом систему координат.
Если методом ЭПР исследуется монокристалл, то при наличии анизотропного д-фак гора измеряемая величина д является функцией ориентации кристалла относительно направления поля, поскольку мы определяем эффективный д-фактор, ориентированный вдоль поля. Если мы определим молекулярные оси X, Y и Z, которые приводят к диагональному виду д-тензор, и возьмем в качестве примера такую систему, где они совпадают с осями кристалла, эффективная величина д-фактора для произвольной ориентации кристалла выражается как
0eff = (g2)xx cos2 Qnx + (g2)yy cos2 0Hl + (g2)zz cos2 0Hz =
= (g2)xx12x + (g2)yyi2 + (02)zz£ (9.17)
где 0w,, 0Hl и 0H- — углы между направлением поля Н и осями А", У и Z соответственно. Косинусы этих углов, так называемые направляющие косинусы, часто обозначают 1Х, 1у и lz. Поскольку из тригонометрии из
Спектроскопия электронного парамагнитного резонанса
33
вестно, что /* + /j; + /2 = 1, то задать направление можно двумя параметрами. Приведенное выше уравнение можно записать в матричном виде
Xg2\x 0 0 4
?е«2 = [4441 0 (g2)yy 0 4 (9.18)
0 0 (Л 4
Поскольку Sx, Sy и Sz, как и Нх, Ну и Hz, определяются в терминах молекулярной системы координат х, у и z, то их можно заменить на те же самые направляющие косинусы. Молекулярная система координат, которая приводит к диагональному виду g-тензор, может не совпадать с произвольными осями, связанными с морфологией кристалла. Поскольку описываемый эксперимент осуществляется с использованием легко регистрируемых осей монокристалла, приведенное выше уравнение следует переписать в недиагональном виде
ge«2 = 14 4 4]
(g2)xx
(g2\x
(?% (g\v (g2)xv
(g2) Г/
\6 /sz ‘я
(j?2) I
'•5 >yz у
(g2Y- I, \o -'zzj Z _
(9.19)
Уравнение (9.19) пригодно для расчета всех компонент тензора. Матрица симметрична, т. е. (д2)ух = (д2)ху, поэтому достаточно рассчитать только шесть независимых компонент. Удобнее всего ориентировать кристалл в магнитном поле относительно наблюдаемых осей кристалла, поэтому оси х, у и z определяются через наблюдаемые оси монокристалла. Sx, Sy и Sz, как и Нх, Ну и Hz, определяются через эти оси.
Предположим, что кристалл установлен, как показано на рис. 9.18, т. е. ось у перпендикулярна направлению поля, так что кристалл можно поворачивать вокруг оси у, при этом в плоскости ху между Н и z образуются различные углы 0. В этом случае /2 равен cos0, а 1Х равен sin0, где 0 — угол между Н и осью z. Подставляя эти величины в уравнение (9.19) и умножая матрицы, получаем
б'ш = (92)xxSin20 + 2(g2)xxsin0cos0 + (g2)zzcos2Q. (9.20)
Если кристалл вращают в плоскости yz, то 1Х = 0, I = sin0 и lz = cos0. Подстановка в уравнение (9.19) и матричное умножение тогда дает
g2ft = (g2)wsin20 + 2(g2)vzsin0cos0 + (g2)zzcos20. (9.21)
Аналогичным образом вращение в плоскости ху приводит к
g2ff = (д2)хх cos2 0 + 2 (д2)ху sin 0 cos 0 + (д2)уу sin2 0. (9.22)
Таким образом, эти уравнения привязывают матрицу в уравнении (9.19) к экспериментально наблюдаемым д2{[. В описанном эксперименте определяется величина g-фактора, но не его знак, поскольку при ана-
3-574
34
Глава 9
лизе используется квадрат измеренного ^-фактора. Если кристалл вращается в одной из плоскостей, достаточно провести только три измерения g2ff (соответствующие трем различным величинам 0), чтобы определить три компоненты д2-тензора в соответствующих уравнениях.
Рис. 9.18. Установка кристалла для вращения в плоскости xz.
Для плоскости xz измеряют (g2)zz при 0 = 0 и (д2)хх при 0 = 90°. Зная эти величины и g2ff при 0 = 45°, можно определить (д2)хг. Таким 2
путем можно измерить шесть независимых компонент д -тензора. На практике проводится ряд измерений, а данные анализируются методом наименьших квадратов. После этого определяется матрица преобразования, которая вращает систему координат и приводит к диагональному виду д2-тензор. Эта операция дает молекулярную систему координат для диагонализации д2-тензора, а корни квадратные
2
из отдельных диагональных матричных элементов д дают дхх, дуу и д22 в этой специальной системе координат. Проводить описанную процедуру можно, только если все молекулы в монокристалле имеют одну и ту же ориентацию молекулярных осей относительно осей кристалла. Поэтому, снимая спектр ЭПР монокристалла, часто одновременно проводят его рентгеноструктурное исследование.
9.7. АНИЗОТРОПИЯ СВЕРХТОНКОГО ВЗАИМОДЕЙСТВИЯ
Мы начали предыдущий раздел с описания анизотропии ^-фактора, наблюдаемой при исследовании методом ЭПР ЫО2, захваченного монокристаллом KNO3, при различных ориентациях относительно поля. Величины а этой системы также очень анизотропны. Если молекулярная ось второго порядка параллельна приложенному полю, наблюдаемая
Спектроскопия электронного парамагнитного резонанса
35
константа СТВ азота составляет 176 МГц, тогда как при перпендикулярной полю ориентации она равна 139 МГц. В жестких системах взаимодействия электронных и ядерных диполей приводят к анизотропным компонентам в электрон-ядерном СТВ. Классическое выражение, описывающее взаимодействие двух диполей, рассматривается в гл. 8, те же самые основные соображения используются и в данной главе. Для взаимодейст вия электронного момента с ядерным моментом гамильтониан имеет следующий вид:
Hdipol = - 0МN\~ - 3(ST"4 (9-23i
г г3
Его знак противоположен тому, который мы получили для взаимодействия двух ядерных диполей; причины этого объясняются в разделах, посвященных жидким кристаллам и ЯМР в твердом теле. Подставляя S = Sx + Sy + Sz, I = Ix + Iy + Izur = х + у + z и расписывая эти векторы, получаем
(sxiy+ syix) -
(Sxiz+ szix) -
(9.24)
Если применить этот гамильтониан к электрону, находящемуся на орбитали, то приведенные в скобках величины заменяют на средние величины, угловые скобки мы используем для обозначения величины, усредненной по электронной волновой функции. В матричном виде мы тогда имеем
(9.25)
В сокращенном виде это выражение записывают как
Hdipol = hS-T-I, (9.26)
где Т—тензор дипольного взаимодействия (Гц), который служит мерой анизотропного ядерного СТВ. Гамильтониан теперь приобретает вид
Н = $SgH+gNpNHI+hSAi,
(9.27)
36
Глава 9
где первый член в правой части описывает электронное зеемановское взаимодействие, второй член—ядерное зеемановское взаимодействие, а третий член — СТВ. Параметр А в третьем члене включает как изотропную, так и анизотропную компоненты СТВ, т. е.
А = Т + al. (9.28)
Если гамильтониан, выраженный уравнением (9.27), используется применительно к свободным органическим радикалам, можно сделать несколько упрощающих допущений. Во-первых, энергия ядерного зеемановского взаимодействия д\ • / обычно мала по сравнению с другими членами. (Вспомните проведенное ранее обсуждение энергии переходов в ЭПР и ЯМР.) Во-вторых, анизотропия g-фактора мала,
Рис. 9.19. Ориентация спинового и ядерного моментов в приложенном поле.
и мы, рассматривая СТВ, примем, что g-фактор изотропен *. (Применительно к некоторым комплексам переходных металлов это допущение неприемлемо; см. далее.) Предполагается, что электронный зеемановский член является доминирующим энергетическим членом, поэтому S квантуется на направление Н, которое мы обозначим как ось z. Этот пример показывает (и в этом мы убедимся снова и снова), что часто удобно выбирать систему координат в соответствии с наибольшим энергетическим эффектом. Далее мы рассмотрим, как ориентируется ядерный момент относительно оси z. Наше обсуждение носит общий характер, но оно может помочь нам рассмотреть этильный радикал, ориентированный так, как это показано на рис. 9.19, где группа — СН3 не вращается. Отметив этот момент, сфокусируем внимание на ядре На, которое участвует в дипольном взаимодействии с электроном. Ядерный момент квантуется не на ось z, а на вектор эффективного поля Heff, который представляет собой сумму векторов внешнего поля Н (ядерный зеемановский эффект) и сверхтонкого поля, создаваемого находящимся рядом электроном. Если СТВ велико (~ 100 Э), сверхтонкое поле на ядре (т. е. поле, создаваемое электронным магнитным моментом на ядре атома водорода) составляет около 11700 Э. (Это поле
* Если анизотропия g-тензора сравнима с анизотропией СТВ, такого допущения делать нельзя. Указанная ситуация обсуждается в гл. 13 и в кн.: Abra-gam A., Bleany В., EPR of Transition Ions, р. 167, Clarendon Press, Oxford, 1970.
Спектроскопия электронного парамагнитного резонанса 37
значительно отличается от поля ~ 3000 Э, создаваемого магнитом, и поля ~ 18 Э, создаваемого на электроне значительно меньшим по величине ядерным моментом.) Таким образом, пренебрежение ядерным зеемановским членом I в уравнении (9.27) в какой-то степени оп-
равдано. В результате для большинства свободных органических радикалов, для которых ^-фактор изотропен, гамильтониан значительно упрощается по сравнению с тем, что дает уравнение (9.27), и мы имеем
Н = g$HSz + hSz (Ajx + AzyIy + AZZIZ). (9.29)
Второй член в правой части уравнения дает z-компоненту элекгрон-ядерного СТВ, учитывающую как вклады Т* и I так и вклад Iz, поскольку z-поле не квантует I, но квантует S. Если этот гамильтониан действует на |aeaN> и другие волновые функции, в секулярном детерминанте возникают недиагональные матричные элементы. Диагонализация этого детерминанта и определение энергии дает следующее: ^}Е-^Н±^А--+А-’+А" РА } £ -
Чпен. включающий квадратный корень, заменяет член 1/4а, полученный при расчете al S для атома водорода. Энергия СТВ выражается, таким образом, как
. z____________ (9.30)
AEhf= Az2y+Azz .
Величина А включает как изотропную (а), так и анизотропную (Т) компоненты СТВ. Поскольку в решении анизотропные компоненты усредняются до нуля, очень просто взять одну треть следа А и разложить А на Т и а. (Этот результат предполагает, что растворитель или твердая решетка не оказывают влияния на электронную структуру.)
Полученные выражения применимы к любой ориентации молекулы относительно приложенного поля. Если исследуется монокристалл, кристаллографические и молекулярные оси которого не совпадают, определить все компоненты тензора СТВ можно так же, как и при расчете g-тензора. Система координат, которая приводит к диагональному виду д-тен-зор, не обязательно совпадает с той системой координат, которая приводит к диагональному виду тензор А, и ни одна из этих систем координат может не быть молекулярной системой координат [176]. Если молекула характеризуется полной симметрией (т. е. в систему включаются все лиганды), так что у нее есть ось вращения м-порядка, то эта же ось будет диагональной для g и А и она должна совпадать с молекулярной осью z.
Угловую зависимость СТВ для такого случая, когда поле, обусловленное этим взаимодействием, велико, I = 1/2, и система имеет аксиальную симметрию, можно выразить путем подстановки rcos0 вместо z
38
Глава 9
и г sin 0 вместо х и у в уравнение (9.24). Фактически мы делим ядер-ный момент (рис. 9.19) на компоненты, параллельную и перпендикулярную полю. Гамильтониан, включающий электронный зеемановский член, принимает следующий вид:
Н = g&HSz + hSz {[а + В (3 cos20 - 1)] Iz + ЗВ cos 0 sin 0/х}. (9.31)
В результате действия этого гамильтониана на базис получаются энергии, выражаемые уравнением
hM
E = g$HMs± -y^[(a-B)2 + 3B(2a + B)cos20]1/2, (9.32)
где а — константа изотропного СТВ, В—константа анизотропного СТВ, а 0 — угол между осью z молекулы и направлением поля. Константа СТВ А, измеряемая экспериментально, представляет собой разность между энергиями соответствующих уровней; определяют ее (в см ') с помощью выражения
A = h [(а — В)2 + ЗВ(2а + В) cos 20]1/2.
Часто в литературе для описания анизотропии д и А в аксиальных системах применяют следующее уравнение:
ДЕ = hv = (±д1: + + ат, + | |(0||- 0±)ряо+ BmJ(3cos20-l),
v - / L- J (9.33)
где 0 — угол между осью z и направлением магнитного поля, а — константа изотропного СТВ, а В — константа анизотропного СТВ. Это уравнение получают из уравнения (9.32) путем учета анизотропии д-фак-тора и при допущении, что оба тензора, д и А, имеют диагональный вид в одной и той же системе координат.
Поскольку при анализе анизотропии СТВ имеют дело с А2 [см. уравнение (9.30)], обычно из эксперимента ЭПР не удается получить знак константы взаимодействия*. Однако для органических радикалов, которые имеют неспаренный электрон на р-орбитали, его можно легко предсказать (в этом случае отсутствует анизотропное СТВ сферической 2з-орбитали с плотностью неспаренного электрона). Вернемся теперь к уравнению (9.25). Допустим, что электрон находится на гипотетической орбитали, которая может быть представлена единичным вектором. Если эта гипотетическая орбиталь лежит вдоль оси z, то z = г, х = 0 и у -- 0, а все недиагональные члены имеют нулевое значение. При подстановке в уравнение (9.25) получаем, что Тхх = /с<1/г3>, Туу = k(i/r3> и Tzz = — /с<2/г3>* **. (Матричные элементы в том виде, как они записаны
Для некоторых систем можно определить знак константы взаимодействия, используя метод ЯМР, поскольку спин, направленный вдоль поля, вызывает сдвиг в слабое поле, а спин, направленный против поля, вызывает сдвиг в сильное поле.
** Константой пропорциональности является gfigx $N/h.
Спектроскопия электронного парамагнитного резонанса 39
в уравнении (9.25), имеют противоположный знак, но член, описывающий взаимодействие положительного ядерного момента и отрицательного электронного момента, отрицателен.) Отметим, что след равен нулю.
Для того чтобы задача стала более конкретной, мы рассмотрим электрон, находящийся на р2-орбитали. Для решения этой задачи удобно перейти к полярным координатам, заменив z, х и у на г cos 0, г sin 0 cos ф и г sin 0 sin ф соответственно. Для электрона в полярных координатах (с учетом отрицательного знака) получаем
Тхх = -y9P9NPN <(3 cos20—1)/г3>
Т„ = - ^-0P0nPn<(3cos20 - 1)/г3> уу 2
Tzz = 0P0N Pn<(3cos20 — l)/r3>.
Два последних матричных элемента легко определить, проведя подстановку вместо х и у в уравнении (9.25) и заменив <соз2ф> на 1/2 для аксиальной системы. Отметим, что след равен нулю. Если мы примем теперь, что электрон может быть локализован в каком-либо месте п-орбитапи. то нам придется провести интегрирование сначала по всем возможным углам для радиус-вектора электрона на этой орбитали, а затем по всем радиусам г. Сделав это, находим
4 rzZ = y6'P6'NPN<l/r3X (9-34)
где <1/г3>— средняя величина параметра 1/г3. Если обозначить Гг2 как 4/5Рр, то
Гхх=-|Рр и Tw=-jPp. (9.35)
Эти соображения могут помочь предсказать знак анизотропных компонент константы СТВ.
Интересно применить эти уравнения к тензору анизотропного СТВ для ядра 13С, который зависит главным образом от плотности неспаренного электрона на р-орбитали атома. Рассмотрим знаки Тхх, Туу и Tzz для этой системы. Три ориентации р-орбитали в молекуле относительно направления приложенного поля показаны на рис. 9.20. Штриховыми линиями указаны области, где функция 3cos20 — 1 равна нулю. Это позволяет учесть знаки для различных областей линий поля, создаваемого ядерным моментом. Поэтому, глядя на рис. 9.20, можно решить, каков знак Tzz [уравнение (9.34)]. Например, как следует из рис. 9.20,А, если рг-орбиталь направлена вдоль поля, почти полное усреднение дипольного взаимодействия ядерного момента по рг-орбитали происходит в положительной части конуса. Поэтому можно ожидать, что Tzz представляет собой большую положительную величину. Для ориентации
40
Глава 9
вдоль оси х, показанной на рис. 9.20,Б, Тхх будет отрицательным и большим по абсолютной величине; то же самое справедливо для Туу при ориентации, показанной на рис. 9.20,В. Анализ [18, 19] сверхтонкой структуры радикала обогащенной изотопом 13С малоновой кислоты Н13С(СООН)2 дает ас=92,6 МГц, Тхх=-50,4 МГц, Г„=-59,8 МГц
Рис. 9.20. Наглядное представление дипольного усреднения электронного и ядерного моментов.
А—р-орбиталь ориентирована вдоль поля; Б и В—р-орбиталь перпендикулярна полю.
и Tzz = + 120,1 МГц. После приведения к диагональному виду тензора СТВ становятся известными относительные знаки Тхх, Туу и Tzz (след должен быть нулевым), но не абсолютные, т. е. все те знаки, которые даны выше, могут быть изменены на противоположные. Однако выводы, которые можно сделать на основе рис. 9.20, говорят в пользу того, что приведенные знаки являются правильными.
Полезно заранее выяснить знаки компонент анизотропного протонного СТВ для радикала С — Н. Как и на рис. 9.20, три ориентации Рп-орбитали этого радикала, показанные на рис. 9.21, говорят о том, что Tzz мал, в то время как Туу положителен, а Тхх отрицателен. Визуальное усреднение р-орбитали по конусу магнитных линий ядерного момента также говорит о том, что Tzz мал. Обратите внимание, что конусы, изображающие линии магнитного поля, создаваемого ядерным моментом, изображены у ядра, чей момент вызывает расщепление посредством дипольного взаимодействия с электроном. Если оси х, у и z определены относительно фиксированных осей кристалла (которые совпадают с молекулярными осями), как на рис. 9.21, расчет [20] показывает, что неспаренный электрон, целиком находящийся на р-орбитали, должен приводить к тензору анизотропного СТВ:
тн
-38 0
0
о 4-43
0
О’ 0 -5
МГц
Спектроскопия электронного парамагнитного резонанса
41
Рис. 9.21. Наглядное представление дипольного усреднения ядерного момента атома водорода под действием электронного момента р-орбитали углерода.
А—Но параллельно оси z кристалла; Б — Но параллельно оси у кристалла; В — Но параллельно оси х кристалла, наблюдение осуществляется вдоль осн р-орбнтали сверху вниз.
Экспериментальный тензор протонного сверхтонкого взаимодействия для ос-ириюна радикала малиновой кисло ш, uouiauHo экспериментальным данным, определяется как
±91 О
О ±29
О О
О О ±58.
МГц
Поскольку константа изотропного СТВ а составляет одну треть следа А„, она равна + 59 МГц, и соответственно тензор анизотропного СТВ Т должен иметь следующий вид:
Тц
Г-32 О О
О +30 0 МГц
0 0+1.
+ 32 0
0 -30
0 0
МГц
о о
Аргументы, которыми мы пользовались при обсуждении рис. 9.21, и сравнение с теоретическим тензором говорят о том, что верным должен быть левый тензор. Его матрица возникает из а = — 59 МГц. Константа изотропного СТВ должна быть отрицательной, поскольку положительная а должна давать такие тензорные компоненты, которые соответствуют величинам, большим, чем для всего электрона.
Определение констант изотропного и анизотропного СТВ, проведенное для нескольких органических и неорганических радикалов, дало информацию относительно молекулярной орбитали, на которой находится неспаренный электрон. Величину В для одного электрона, находящегося на р-орбитали различных атомов, можно рассчитать с помощью ССП-
42
Глава 9
волновой функции из уравнения
2
в=уй Pjv<r 3>- (9.36)
Для 13С константы анизотропного СТВ выражаются как
-В
-В
+2В_
где В, рассчитанная из ССП-волновых функций, составляет 91 МГц. В случае НС(СООН)2 экспериментальное значение Tzz для 13С составляет + 120,1 МГц, тогда как для электрона, локализованного на С2р-орбитали, она равна 182 МГц. Поэтому делается вывод, что рс равна 0,66. При исследовании радикала, полученного из соединения, обогащенного изотопом 13С, установлено, что ас= +92,6 МГц. Изотропная константа электрона, полностью находящегося на s-орбитали, равна 3110 МГц. Измеренная ас соответствует СТВ С2,-спиновой плотности величиной 0,03. Поэтому можно предположить, что рассматриваемый радикал почти плоский.
Величина константы изотропного 13С-СТВ говорит в пользу плоской структуры [21] радикала СН3, ас = 38,5 Э. Для радикала CF3, как установлено, ас = 271,6 Э [21]; следовательно, у этого радикала пирамидальная структура, причем неспаренный электрон находится на s-орбитали.
Константа изотропного I4N-CTB в NO2 составляет 151 МГц, а максимальное значение константы анизотропного СТВ равно 12 МГц. Из величины СТВ 1540 МГц, ожидаемой для одного электрона на 2х-орби-тали азота, и величины СТВ 48 МГц, ожидаемой для электрона на 2р-орбитали, рассчитано, что ps составляет 0,10, и обнаружено, что рр составляет 0,25 при отношении 2p/2s = 2,5. Для хр2-орбитали это соотношение должно быть равно 2,0, поэтому полученный результат предполагает, что орбиталь, связывающая кислород, имеет больший р-характер, чем хр2-орбиталь, и что угол превышает 120°. Из микроволнового спектра NO2 в газовой фазе следует, что угол равен 134°.
9.8. ЭПР ТРИПЛЕТНЫХ СОСТОЯНИЙ
Теперь рассмотрим спектроскопию ЭПР молекул, в которых имеется более чем один неспаренный электрон. Примером может служить триплетное состояние нафталина, которое образуется при его УФ-облучении. Для регистрации спектра ЭПР использовали монокристалл дурола, в кристаллическую решетку которого были внесены указанные частицы. Внедрению нафталина в решетку дурола помогает похожая форма этих молекул, при этом сильно увеличивается время жизни триплетного состояния нафталина. Спектр состоит из трех линий, причем
Спектроскопия электронного парамагнитного резонанса 43
величина резонансного поля резко меняется при изменении ориентации кристалла. Происходящие изменения нельзя объяснить с помощью анизотропных д- и а-тензоров, которыми мы пользовались ранее. Анизотропия в этой системе обусловлена электрон-электронным спиновым взаимодействием и описывается спин-гамильтонианом, выражаемым уравнением (9.37). Этот гамильтониан очень похож на гамильтониан, описывающий дипольное взаимодействие электронного и ядерного спинов [уравнение (9.23)]:
И = gpg (Si + S2) + g2p2 J S1 '/2 - -3A'()(S2'r) I (9.37)
где r—вектор, соединяющий два электрона (1 и 2).
Влияние указанных эффектов на спектр ЭПР зависит от того, на какую область распространяется взаимодействие электронного и ядерного
..И 13аП0ЛНе SMOS
ms = 0 А
ms = -1
Рис. 9.22. Синглетное основное состояние (А) и компоненты nig = 1, 0 и — 1 состояния S = 1 (Б).
спинов. Ранее в этой главе мы рассматривали кулоновский (J) и обменный (К) интегралы. Если две молекулярные орбитали отличаются по энергии меньше, чем на разность энергий обмена и отталкивания, тогда возникает триплетное состояние. Основное состояние молекулы нафталина — синглет, и время жизни возбужденного триплетногр состояния значительно, поскольку триплет-синглетный переход запрещен. Для основного состояния S = 0 и ms = 0; электронная конфигурация основного состояния показана на рис. 9.22,Л. Электронные конфигурации триплетного состояния, для которого S= 1, а ш,= 1, 0 и — 1, даны на рис. 9.22, Б. Если в молекуле происходят только обменные и электростатические взаимодействия, то в отсутствие магнитного поля три конфигурации ms = 1, 0 и — 1 вырождены. Магнитное поле должно снимать вырождение (см. рис. 9.23,Л), и, как и при S' = 1/2, в этом случае должен наблюдаться лишь один переход. Однако магнитное диполь-дипольное взаимодействие между двумя неспаренными электронами снимает вырождение компонент ms при S = 1 даже в отсутствие поля (рис. 9.23, Б). Это устранение вырождения в отсутствие поля носит название расще
44
Глава 9
пления в нулевом поле. При наложении магнитного поля уровни расщепляются так, что можно зарегистрировать два перехода Ат, = + 1, как это показано на рис. 9.23^Б. Ранее мы упоминали, что в спектре ЭПР триплетного нафталина наблюдаются три линии. Две из них обусловлены переходами Ams = ± 1, показанными на рис. 9.23,Б, а третья — переходом Ams = ± 2 (между ms = +1 и ms = — 1), который становится
Рис. 9.23. Влияние расщепления в нулевом поле на ожидаемые переходы ЭПР.
А—расщепление в нулевом поле отсутствует. Б—умеренное расщепление в нулевом поле; штриховые стрелки демонстрируют результат, полученный при фиксированной частоте, а сплошные стрелки—результат, полученный при фиксированном поле. В —большие эффекты расщепления в нулевом поле; предполагается, что магнитное поле параллельно осн диполя молекулы.
разрешенным, если расщепление в нулевом поле мало по сравнению с частотой микроволнового излучения. В последнем случае не удается отнести точно величины ms= +1, Ои — 1 к существующим состояниям. Если расщепление в нулевом поле велико, как на рис. 9.23,В, ms становятся нормальными квантовыми числами, а энергии разрешенных переходов Am, = + 1—слишком большими, чтобы эти переходы можно было наблюдать в микроволновом диапазоне. В таком случае спектр ЭПР не регистрируется.
Поскольку электрон-электронное взаимодействие является дипольным [уравнение (9.37)], оно описывается симметричным тензором— так называемым тензором расщепления в нулевом поле D.
Hd = SDS. (9.38)
Элементы тензора D имеют тот же самый вид, что и элементы тензора Т в уравнении (9.25). Этот дипольный D-тензор объясняет большую анизотропию, наблюдаемую в спектр» бирадикала. Используя главные оси, которые приводят к диагональному виду тензор расщепления в нулевом поле, мы можем записать
HD = - XS2x - YS2 - ZS2,
Спектроскопия электронного парамагнитного резонанса 45
где X — элементы Dxx диагонализованного тензора расщепления в нулевом поле. Поскольку след тензора равен нулю, X + У + Z = 0, гамильтониан нулевого поля можно записать с помощью двух независимых констант D и Е:
Hd=D(S2-±SS) + E (S2 - S2y).
Поскольку для систем с аксиальной симметрией X = У, последний член становится равным нулю. Если молекула характеризуется кубической симметрией, расщепление в нулевом поле зарегистрировать не удается.
Действуя этим гамильтонианом на волновые функции триплетного состояния, можно рассчитать энергии как функции напряженности поля и ориентации. Результаты говорят о значительной анизотропии спектра. Спектр триплетного состояния нафталина описывается следующим образом: д (изотропный) = 2,0030, D/hc= +0,1012 см-1 и E/hc= — — 0,0141 см “ *. Величины D и Е зависят от того, насколько сильно взаимодействуют два спина. Эти параметры становятся пренебрежимо малыми, если два электрона локализуются в частях молекулы, достаточно далеко отстоящих друг от друга.
В жидкостях тензор D, след которого равен нулю, усредняется до нуля. Большие флуктуирующие поля, обусловленные большими спин-спиновыми взаимодействиями, меняющими свое направление, в молекуле с соответствующим расщеплением при нулевом поле вызывают эффективную релаксацию. Таким образом, линии в спектре обычно настолько широки, что их не удается зарегистрировать. Спектры ЭПР триплетных состояний (за некоторыми исключениями) в растворе наблюдать не удается, если только два спина не отстоят один от другого на большое расстояние (т. е. D и Е малы).
9.9. КВАДРУПОЛЬНОЕ ЯДЕРНОЕ ВЗАИМОДЕЙСТВИЕ
Ядро с ядерным спиновым квантовым числом I 1 также характеризуется электрическим моментом, и неспаренный электрон взаимодействует как с магнитным ядерным, так и с электрическим моментом. Градиент электрического поля на ядре может взаимодействовать с ква-друпольным моментом (такое взаимодействие изучается с помощью спектроскопии ядерного квадрупольного резонанса), и это взаимодействие влияет на энергии электронных спиновых состояний через ядерно-электронное магнитное взаимодействие как возмущение второго порядка. Влияние квадрупольного взаимодействия обычно носит сложный характер, поскольку этому взаимодействию сопутствует значительно большее магнитное СТВ. Ориентация ядерного момента квантуется как по отношению к градиенту электрического поля, так и по отношению к направлению магнитного поля. Если направление магнитного поля и оси кристалла параллельны, квадрупольное взаимодействие приводит только к небольшому смещению всех энергетических уровней на по
46
Глава 9
стоянную величину, что не вызывает никаких изменений в наблюдаемых переходах. Однако, если оси не параллельны, возникает конкуренция между электрическим и магнитным полями. В таком случае на расположение сверхтонких линий оказывается двоякое влияние: 1) все энергетические уровни смещаются на постоянную величину и 2) расстояние между энергетическими уровнями меняется, что приводит к увеличению разделения соседних линий в спектре ЭПР, особенно на концах спектра.
Квадрупольный эффект легко отличить от другого эффекта второго порядка, который приводит к постепенному увеличению или уменьшению протяженности спектра. Изменение в расположении спектра, обусловленное другим эффектом второго порядка, происходит в том случае, когда напряженность магнитного поля, создаваемого ядром, становится сравнима с напряженностью внешнего магнитного поля. В этом случае неравенство в разделении можно устранить, увеличив напряженность поля.
Конкуренция квадрупольног о электрического и магнитного полей приводит также к появлению дополнительных линий, которые обычно запрещены правилом отбора Дт7 = 0. Возможны также переходы Дш, = + 1 и Дш, = + 2 [22]. Константу ядерного квадрупольного взаимодействия дает анализ запрещенных линий. Для этого исследуют методом ЭПР монокристалл диамагнитного соединения, в решетку которого внесено изучаемое соединение. Спектр с такими переходами получен (рис. 9.24) для бпс-(2,4-пентандионата) меди(П) [б3Си(асас)2], внесенного в Pd(acac)2. Запрещенные переходы отмечены на рис. 9.24,Л стрелками, другие линии характеризуют четыре разрешенных перехода
Рис. 9.24. а -спектр («2-диапазон) Си(асас)2 при 0 = 87°; запрещенные линии указаны стрелками. Б-угло-вая зависимость расстояния между каждой парой запрещенных линий (Am/ = +1) в спектре А. Кривые рассчитаны с использованием величины Q' = 3,4-10'* см-1, значки относятся к экспериментальным данным. Буквы I т и /1 обозначают соответственно пары в слабом, среднем и сильном полях. (Печатается с разрешения из So Н., Belford R.L.. J. Amer. Chem. Soc., 91, 2392 (1969). © Am. Chem. Soc.)
Спектроскопия электронного парамагнитного резонанса 47
(7(б 3Си) = 3/2). Разделение линий в спектре и интенсивность запрещенных линий значительно зависят от угла 0. Изменение в разделении показано на рис. 9.24,Б. Путем матричной диагонализации спектры можно с помощью вычислительной машины подогнать к спин-гамильтониану:
н = РОцНА + <71 (ад + HySy)] + ASZIZ + B(SXIX + Syiy) +
+ Q'Ul - + 1)] - gNpNH-i,
член Q' [I2 —|1(1 +1)] отвечает за квадрупольные эффекты. Об остальных членах этого выражения мы уже говорили выше. Величина Q'/hc = (3,4 + 0,2)-10 “4 см “1 согласуется со всеми данными; если I = 3/2, 4Q'—величина, связанная с градиентом поля e2Qq (см. гл. 14) на ядре меди, т. е. Q' = 3e2qQ/4I(2I +1). При аналогичных исследованиях Cu(bzac)2, внесенного в Pd(bzac)2, и бис-(дитиокарбамата) меди(П) [Cu(dtc)2], внесенного в диамагнитный Ni(dtc)2, получены величины Q' соответственно (3,3 + 0,21)10 ” 4 см ” 1 и (0,7 ± 0,1)10 “ 4 см “ *. Для полости в </х2^у2-орбитали свободного иона меди Q’ приблизительно равна 1610”4 см”1, а ковалентность связывания должна снижать наблюдаемую величину (Q' является мерой градиента электрического поля в направлении оси симметрии). Распределение заряда в комплексе серы очень близко к симметричному: предполагается, что это может быть следствием значительной ковалентности связи медь—лиганд.
9.10. ШИРИНА ЛИНИИ ЭПР
В этом разделе мы вкратце рассмотрим ряд факторов, влияющих на ширину линии ЭПР. Многие из них аналогичны тем факторам, которые мы уже обсуждали в главе, посвященной ЯМР.
Уширение, обусловленное спин-решеточной релаксацией, возникает по причине взаимодействия парамагнитных ионов с термическими колебаниями решетки. Изменение во времени спин-решеточной релаксации в различных системах достаточно велико. Для некоторых соединений это время настолько велико, что их спектры удается наблюдать при комнатной температуре. Поскольку, как правило, время релаксации увеличивается с уменьшением температуры, хорошо разрешенные ЭПР-спектры многих солей переходных металлов можно получить лишь при температурах жидкого азота, водорода или гелия.
Спин-спиновое взаимодействие является следствием небольших магнитных полей, которые существуют на соседних парамагнитных ионах. Под действием этих полей общее поле у ионов слегка меняется и энергетические уровни сдвигаются. Возникает распределение энергий, которое вызывает уширение сигнала. Поскольку уширение есть функция (l/r3)(l — 3cos20), где г — расстояние между ионами, а 0 — угол между направлением поля и осью симметрии, то оно существенно зависит от направления приложенного поля. Этот эффект можно ослабить, если
48
Глава 9
увеличить расстояние между парамагнитными ионами, разбавив их изоморфным диамагнитным материалом; например, в кристалл диамагнитного ZnSO4 можно ввести небольшие количества CuSO4.
Быстрые химические процессы влияют на ширину спектральных линий и их существование, как и в ЯМР. Резонансные линии, разделенные
Рис. 9.25. Спектры ЭПР нитроокиси ди-трет-бутила в С2Н5ОН при 25° С.
Концентрация иитроокиси, М: А —10-4; Б—|0 2: В—10
в пределе прекратившегося обмена, будут уширяться с увеличением скорости процесса, а затем сливаться с образованием одной средневзвешенной линии. Если исходить из типичной для органического радикала полуширины линии на ее полувысоте ~ 0,1 Э, то значительное уширение линии будет происходить уже для процессов, константы скорости первого порядка которых составляют около 5-107 с-1.
Процессы электронного спинового обмена весьма распространены в свободнорадикальных системах, и они решающим образом влияют на ширину спектральных линий и характер спектра. В жидкофазных системах это чаще всего бимолекулярные столкновения, в ходе которых два радикала обмениваются электронами. Наблюдаемая при этом картина аналогична описанной при обсуждении спектров ЯМР. На рис. 9.25 показаны спектры ЭПР растворов с различной концентрацией нитроокиси ди-трет-бутила [(CH3)3C]2NO. В качестве растворителя использовался Ы,Ы-диметилформамид. По мере увеличения концентрации (А -» В) скорость бимолекулярного процесса увеличивается и линии уширяются. Константа скорости этого процесса составляет 7109 л/моль-с; это говорит о том, что скорость процесса лимитируется диффузией. По мере того как раствор, чей спектр показан на рис. 9.25^8, становится значитель
Спектроскопия электронного парамагнитного резонанса 49
но более концентрированным, единственная резонансная линия сужается, как это происходит в пределе быстрого обмена в ЯМР. Это сужение носит название обменного сужения.
Перенос электрона между радикалом и диамагнитной частицей также может происходить с такой скоростью, которая вызывает уширение спектральных линий. Одной из первых была исследована система, в которой происходил обмен электроном между нафталином и его анион-радикалом. Если растворителем служил ТГФ, константа скорости второго порядка переноса электрона составляет 6-107 л/моль с [25а]. Эта величина в сто раз меньше, чем для процесса, контролируемого диффузией. Полагают, что снижение скорости обусловлено тем, что наряду с переносом электрона происходит перенос положительного противоиона ионной пары анион-радикала.
Ширина линий в спектре может по ряду причин различаться. Мы упоминали ранее, что спиновая плотность на протонах группы СН3 эти-ламина зависит от конформации. Временная зависимость этого типа процесса может повлиять на ширину линий различных протонов в молекуле различным образом. Быстрый обмен между различными конфигурациями ионной пары с анион- или катион-радикалом также может привести к большему уширению одних линий по сравнению с другими [256, 26].
9.11. СПИН-ГАМИЛЬТОНИАН
Спин-гамильтониан действует только на спин-переменные и описывает различные взаимодействия в системах, содержащих неспаренные электроны. Его можно рассматривать как стенографический способ представления описанных выше взаимодействий. Спин-гамильтониан ЭПР для иона, находящегося в поле аксиальной симметрии (т. е. тетрагональном или тригональном), имеет следующий вид:
Н = - yS(S + 1)] + д\\ f}HzSz + д± ₽(HXSX + нД) +
+ AtSJz + A±(SJX + хД) + Q'[Iz - jl(l + 1)] -gN$NH0-I. (9.39)
Первый член описывает расщепление в нулевом поле, следующие два— влияние магнитного поля на спиновую вырожденность, остающуюся после расщепления в нулевом поле. Члены Ац и А± служат мерой сверхтонкого расщепления параллельно и перпендикулярно единственной в своем роде оси, a Q' характеризует изменения в спектре, обусловленные квадрупольным взаимодействием. Все эти эффекты рассматривались ранее. Последний член учитывает тот факт, что ядерный магнитный момент |1\ может взаимодействовать непосредственно с внешним полем \i\H0 = gNfiNH0I. Это взаимодействие может повлиять на парамагнитный резонанс лишь в том случае, когда неспа
4 - 574
50
Глава 9
ренные электроны взаимодействуют с ядром в ядерном сверхтонком или квадрупольном взаимодействиях. Если даже такое взаимодействие имеет место, этот эффект пренебрежимо мал по сравнению с описываемыми другими членами спин-гамильтониана.
В случае искажения более низкой симметрии главные (/-факторы переходят в дх, ду и дг; константами СТВ становятся А*, Ау и Az. В этой ситуации необходимо ввести еще два члена: E(S2 — S2), описывающий дополнительное расщепление в нулевом поле, и Q'(12х — /^), описывающий дальнейшее квадрупольное взаимодействие. При этом Q и Q" часто заменяют на Р и Р' соответственно.
Значение спин-гамильтониана состоит в том, что он дает стандартный феноменологический путь, с помощью которого спектр ЭПР можно описать через небольшое число констант. Определив экспериментально величины этих констант, можно далее использовать их в расчетах электронных конфигураций и энергетических состояний иона. Следует отметить, что для данной системы не все члены в уравнении (9.39) одинаково важны. Для ядра, не имеющего спина, все члены, содержащие I, равны нулю. Равен нулю и первый член, если расщепление в нулевом поле отсутствует.
9.12. ПРИМЕРЫ ПРИМЕНЕНИЯ
Спектр ЭПР CuSiF6-6H2O, введенного в соответствующую диамагнитную соль цинка, при 90 К состоит из одной полосы с частично разрешенной сверхтонкой структурой и характеризуется почти изотропным д-фактором [27]. В поле кубической симметрии основное состояние Си2 + орбитально дважды вырождено. Хотя симметрия Cu(H2O)6SiF6 ближе к тригональной, чем к кубической, орбитального вырождения это не снимает. В результате происходит ян-теллеровское искажение. Однако возможны три искажения той же самой энергии, которые снимают орбитальное вырождение: это три тригональных искажения со взаимно перпендикулярными осями (растяжение вдоль трех осей связей с транс-лигандами). В результате следует ждать трех переходов ЭПР, по одному для каждой частицы. Поскольку экспериментально обнаружен лишь один переход, было высказано предположение, что кристаллическое поле находится в резонансе между этими тремя искажениями [28]. При понижении температуры спектр становится анизотропным и состоит из трех совокупностей линий, соответствующих трем различным ионам меди, искаженным тремя различными тетрагональными искажениями [29]. Переходы наблюдаются в интервале температур 50—12 К; три перпендикулярные тетрагональные оси образуют ребра прямоугольной ячейки твердого вещества, а тригональная ось представляет собой диагональ остова. Другие смешанные соли меди, как установлено, характеризуются аналогичными переходами. У (Си, Mg)3La2(NO3)12-24D2O переход наблюдается при 33—45 К, у (Zn, Си) (ВгО3)2-6Н2О при температуре ниже 7 К, причем переход неполный. Авторы работы [30] иссле
Спектроскопия электронного парамагнитного резонанса 51
довали ЭПР-спектры CuSiF6-6H2O и получили следующие параметры:
90 К 20 К
д = 2,221 ± 0.005 9z = 2,46 ± 0,01
д[ = 2,230 ± 0,005 Эх = 2,10 + 0,01
А = 0.0021 ± 0,0005 см"1 <7? = 2,10 ± 0,01
В = 0,0028 ± 0,0005 см"1 0,0110 ± 0,0003 см1
< 0,0030 см “ 1
Квадрупольное взаимодействие разрешено не было. Аналогичное поведение, т. е. резонирующее кристаллическое поле при умеренных температурах, зарегистрировано в спектрах некоторых трис-комплексов ме-ди(П) с 2,2'-дипиридилом и 1,10-фенантролином [31].
Спектр ЭПР комплекса [(NH3)5Co — О — О — Co(NH3)s]5 + представляет собой интересный пример того, как из данных о спиновой плотности и сверхтонком расщеплении можно получить сведения о структуре соединения. В данном случае можно предложить четыре варианта структуры: 1) два атома кобальта(Ш) связаны между собой кислородным мостиком О2 ; 2) атомы кобальта(Ш) и кобалыа(ТУ) соединены между собой перекисным мостиком О| ; 3) два атома кобальта эквивалентны благодаря одинаковому взаимодействию одного неспаренного электрона с обоими атомами кобальта; 4) электрон взаимодействует с обоими атомами кобальта, но в большей степени с одним, чем с другим.
Структура 1 должна давать одну линию, в то время как структура 2 должна приводить к 8 линиям (I = 7/2 для Со). Структуру 3 должны характеризовать 15 линий, а структуру 4—64 линии. Полученный спектр [32] состоит из 15 линий, что устраняет маловероятные структуры 2 и 4 и говорит в пользу структур 1 и 3 или их смеси. О том, насколько вероятна структура 1, могут сказать данные по СТВ с ядром 17О.
Исследование [33] аддуктов комплексов кобальта(П) с кислородом состава 1:1 привело к внутренне согласующейся интерпретации констант изотропного и анизотропного взаимодействий с ядрами 17О и 59Со. В зависимости от того, какой лиганд соединен с атомом кобальта, аддукты описывались как связанные с О2 или с О/.
Одно из достоинств метода ЭПР—исключительно высокая чувствительность к небольшим количествам парамагнитного вещества. Например, при благоприятных условиях сигнал радикала дифенилпикрилги-дразила (ДФПГ) можно регистрировать, если в резонаторе спектрометра находится всего 10“12 г вещества. Эта высокая чувствительность метода используется для изучения радикалов серы. Если серу нагревать, диамагнитные циклы S8 расщепляются с образованием высокомолекулярных цепей Sx, на каждом конце которых находится по неспаренному
г
52
Глава 9
электрону. Цепи настолько длинны и концентрация радикалов настолько низка, что парамагнетизм нельзя обнаружить, используя метод Гойе, в то же время ЭПР-спектр снять удается [34]. Сравнивая площадь этого сигнала с площадью полосы, полученной в результате внесения известного количества ДФПГ, можно определить число неспаренных электронов (которое пропорционально площади под кривой поглощения). Таким образом было определено общее число радикалов в системе, а поскольку количество серы заведомо известно, то удалось рассчитать среднюю молекулярную массу частиц -SSXS-. Концентрация радикалов при 300° С равна 1,1-10'3 М и при 171° С цепь в среднем состоит из 1,5- 10б атомов. Установив зависимость концентрации радикалов от температуры, удалось определить теплоту диссоциации связи S —S; она оказалась равной 33,4 ккал/ моль в расчете на одну связь.
Медь(П) образует комплексы различных геометрических структур, электронные спектры которых похожи и магнитная восприимчивость примерно одинакова. Поэтому прийти к каким-то определенным выводам относительно структуры этих соединений можно, только если изучаются твердые соединения, а не их растворы и если можно воспользоваться результатами кристаллографических исследований. Недавние исследования пятикоординационных аддуктов, образуемых различными льюисовыми основаниями и гексафторацетилацетонатом меди (II), привели к созданию метода, позволяющего различить [35] апикальные и экваториальные изомеры тетрагональной пирамиды.
Поскольку метод ЭПР отличается высокой чувствительностью, он широко применяется на практике при изучении биологических систем [36]. Этим методом определены: состояния окисления металла, координационное число металла и виды лигандов во многих металлопроте-инах. Измерения, как правило, проводят в замороженных растворах. Интерпретация результатов трудна, и выводы базируются на аналогиях между спектрами этих соединений и спектрами модельных соединений. Примеры такого применения метода рассматриваются в главе, посвященной спектрам ЭПР ионов переходных металлов.
Опубликовано несколько работ, в которых методом ЭПР проводилась идентификация и установление структуры радикалов, образующихся под действием высокоэнергетического излучения [37]. Среди многих интересных радикалов, образующихся в таких условиях, обнаружены о;, сю, сю2, ро]“, SO3 и сю3.
В настоящее время разработаны два вида метода двойного резонанса: электрон-ядерный двойной резонанс (ЭЯДР) и электрон-электронный двойной резонанс (ЭЭДР). Основное назначение ЭЯ ДР — увеличение точности и разрешения при определении небольших сверхтонких расщеплений, определении того, какие ядра отвечают за расщепление, и установлении ядерных ^-факторов. ЭЭДР может помочь в отнесении перекрывающихся резонансных линий во многом тем же самым образом, что и двойной резонанс, используемый в ЯМР. С этими специальными методами читатель может познакомиться в книге Вертца и Болтона [38].
Спектроскопия электронного парамагнитного резонанса 53
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. Adrian F.J., et al., Adv. Chem. Ser., 36, 50 (1960).
2. Carrington A., Quart. Rev., 17, 67 (1963).
3. Maki A.H., McGarvey B.R., J. Chem. Phys., 29, 35 (1958).
4. Strouse C.E., Dahl L.F., J. Amer. Chem. Soc., 93, 6032 (1971).
5. Дьюар М.Дж.С. Теория молекулярных орбиталей в органической химии.— М.: Мир, 1972.
6. Fessenden R.W., Schuler R.H., J. Chem. Phys., 39, 2147 (1963).
7. Drago R.S., Petersen H., Jr., J. Amer. Chem. Soc., 89, 3978 (1967).
8. McLachlan A.D., Mol. Phys., 3, 233 (1960).
9. Pople J. A., et al., J. Amer. Chem. Soc., 90, 4201 (1968) и ссылки в этой работе; Pople J. A., Beveridge D.L., Approximate Molecular Orbital Theory, McGraw-Hill, Boock Co., New York, 1970.
10. Karplus M., Fraenkel G.K., J. Chem. Phys., 35, 1312 (1961).
11. a. Cramer R.E., Drago R.S., J. Amer. Chem. Soc., 90, 4790 (1968).
6. Sullivan P.D., J. Amer. Chem. Soc., 90, 3618 (1968).
12. Broze M., Luz Z., Silver B. L, J. Chem. Phys., 46, 4891 (1967).
13. Biddles I., Hudson A., Mol. Phys., 25, 707 (1973).
14. a. Drago R.S., Petersen H., Jr., J. Amer. Chem. Soc., 89, 5774 (1967).
6. Cramer R.E., Drago R.S., J. Chem. Phys., 51, 464 (1969).
15. Fessenden R. W., Neta P., Chem. Phys. Letters, 18, 14 (1973).
16. Fitzgerald R., Drago R. S., J. Amer. Chem. Soc., 90, 2523 (1968).
17. a. Livingston R., Zeldea H., J. Chem. Phys., 41, 4011 (1964).
6. Chien J.C.W., Dickerson L.C.. Proc. Natl. Acad. Sci., USA, 69, 2783 (1972).
18. McConnell H.M.. Fessenden R.W.. J. Chem. Phys., 31, 1688 (1959).
19. Cole T., Heller C., J. Chem. Phys., 34, 1085 (1961).
20. McConnell H.M., et al., J. Amer. Chem. Soc., 82, 766 (1960).
21. Fessenden R.W., Schuler R.H., J. Chem. Phys., 43, 2704 (1965).
22. Atkins P.W., Symons M.C.R., J. Chem. Soc., 1962, 4794.
23. Hirota N., Hutchison C.A., Jr., Palmer P., J. Chem. Phys., 40, 3717 (1964) и ссылки в этой работе.
24. So Н., Belford R.L, J. Amer. Chem. Soc., 91, 2392 (1969); Belford R.L., Huang D. T., So H., Chem. Phys. Letters, 14, 592 (1972) и ссылки в этой работе.
25. a. Ward R.L., Weissman S.I., J. Amer. Chem. Soc., 79, 2086 (1957); Miller T.A., Adams R.N., J. Amer. Chem. Soc., 88, 5713 (1966).
6. Bolton J.R., Carrington A., Mol. Phys., 5, 161 (1962).
26. Freed J.H., Fraenkel G.K., J. Chem. Phys., 37, 1156 (1962).
27. Bleaney B., Ingram D.J.E., Proc. Phys. Soc., 63, 408 (1950).
28. Ahragam A., Pryce M.H.L, Proc. Phys. Soc., 63, 409 (1950).
29. Bleaney B., Bowers K.D., Proc. Phys. Soc., 65, 667 (1952).
30. Bleaney B., Bowers K.D., Trenam R.S., Proc. Roy. Soc. (London), A228, 157 (1955).
31. Allen H.C., Jr., Kokoszka G.E., Inskeep R.G., J. Amer. Chem. Soc., 86, 1023 (1964).
32. Eheworth E.A.V., Weil J. A., J. Phys. Chem., 63, 1890 (1959).
33. Tovrog B., Kitko D., Drago R. S., J. Amer. Chem. Soc., 98, 5144 (1976).
34. Gardner D.M., Fraenkel G.K., J. Amer. Chem. Soc., 78, 3279 (1956).
35. McMillin D., Drago R.S., Nusz J. A., J. Amer. Chem. Soc., 98, 3120 (1976).
36. См., например, Yang C.S., Heuennekens F.M., Biochemistry, 9, 2127 (1970).
37. Эткинс П., Саймонс M. Спектры ЭПР и строение неорганических радикалов.— М.: Мир, 1970.
38. Вертц Дж., Болтон Дж. Теория и практические приложения метода ЭПР.— М.: Мир, 1975, гл. 13.
54
Глава 9
ОБЩИЙ СПИСОК ЛИТЕРАТУРЫ*
Вертц Дж., Болтон Дж. Теория и практические приложения метода ЭПР.—М.: Мир, 1975.
Poole С.Р., Farach Н.А., The Theory of Magnetic Resonance, Wiley-Interscience, New York, 1972.
Керрингтон А., Мак-Лечлан Э. Магнитный резонанс и его применение в химии.— М.г Мир, 1970.
Пул Ч. Техника ЭПР-спектроскопии.— М.: Мир, 1970.
Goodman В. A., Raynor J.B., Adv. Inorg. Chem. Radiochem., 13, 135 (1970).
Bersohn R., Determination of Organic Structures by Physical Methods, Vol. 2. eds.
Nachod F.C., Phillips W. D., Academic Press, New York, 1962.
Carrington A., Quart. Revs., 17, 67 (1963).
Atherton N. M., Electron Spin Resonance, Halsted Press, 1973.
Эткинс П„ Саймонс M. Спектры ЭПР и строение неорганических радикалов.— М.: Мир, 1970.
Сликтер Ч.П. Основы теории магнитного резонанса с примерами из физики твердого тела.— М.: Мир, 1965.
Golding R.M., Applied Wave Mechanics, Van Nostrand-Reinhold, New York, 1964.
Абрагам А. Ядерный магнетизм.— M.: Мир, 1963.
Раке G. Е.. Estle Т. L., Paramagnetic Resonance, 2nd Ed., Benjamin W. A., Inc., New York, 1973.
Fischer H., Magnetic Properties of Free Radicals, Landolt-Bernstein Tables, New Series Group II, Vol. I. Springer-Verlag, Berlin, 1965.
Упражнения
1. Превратите производные кривые в кривые поглощения:
-vV +W
А Б
2. а. Сколько линий ci ерхтонкой структуры можно приписать делокализации неспаренного электрона по кольцам катиона дибензо лхрома?
б. Пользуясь методом группировки спинов, аналогичным представленному на рис. 9.6, объясните, сколько должно быть линий в спектре ЭПР этого соединения и каковы должны быть их относительные интенсивности?
3. а. Ацетат меди(П) представляет собой димер, в котором два атома меди сильно взаимодействуют. В его спектре ЭПР семь линий с относительной интенсивностью 1 :2:3:4:3:2:1. Для ядер меди 1 = 3/2; основное состояние ацетата меди синглетное, а возбужденное — триплетное. Исходя из этих данных, объясните, почему в спектре семь
* Работы, посвященные в основном ЭПР ионов переходных металлов, см. в гл. 13.
Спектроскопия электронного парамагнитного резонанса
55
линий и почему у них такая относительная интенсивность. [Ответ на этот вопрос можно найти в работе Bleaney В., Bowers K.D., Proc. Roy.Soc. (London), A214, 451 (1952).]
б. Как изменится интенсивность линий, если образец охладить? Объясните почему?
4. Как должен выглядеть спектр ЭПР (SO3)2NO2-?
5. Предположим, что мы получили анион
а. Сколько линий будет в его спектре и какой будет их относительная интенсивность?
б. Какими доводами вам нужно воспользоваться и какие эксперименты нужно провести, чтобы показать, что электрон делокализован на кислороде ?
в. Для этого аниона ан = 2,37 Э. Сравните спиновую плотность на атоме водорода в этом ионе со спиновой плотностью на свободном атоме водорода.
г. Как, зная знак константы протонного СТВ, определить, где находится неспаренный электрон — на с- или на л-молекулярной орбитали?
д. Примите, что ан = 2,37Э и что неспаренный электрон находится в л-системе, и рассчитайте спиновую плотность на ближайшем соседнем атоме углерода.
6. Константа СТВ с ядром 13С в метильном радикале составляет 41 Э, а константа протонного СТВ равна 23 Э. Изобразите спектр радикала 13СН,. [Ответ можно найти в работе Cole Т„ et al., Mol. Phys., 1, 406 (1958).]
7. Как будет выглядеть спектр анион-радикала хлорбензола, если предположить, что все сверхтонкие линии могут быть разрешены.
8. Для какого из бимолекулярных процессов — с константой скорости 107 или 1О10 л/моль-с — уширение линии должно быть больше? Все остальные параметры процессов адекватны.
9. Как много линий следует ожидать в спектре ЭПР (CN)5CoO2Co(NH3)5? Объясните почему.
10. Для радикала NH2 получен следующий спектр:
а. Переведите его в спектр поглощения.
б. Как можно определить, какое расщепление — большее или меньшее — обусловлено водородом?
56
Глава 9
в. Предположим, что большее расщепление обусловлено азотом. Чтобы объяснить спектр, постройте диаграмму, аналогичную приведенной на рис. 9.10.
11. а. Сколько линий должно быть в спектре ЭПР гипотетической молекулы SC13 (7 для S равна 0, а для С1 равна 3/2)?
б. Используя метод группировки спинов, аналогичный изображенному на рис. 9.6 и 9.7, объясните, чем обусловлено появление этих линий, и укажите стрелками переходы. Какова должна быть относительная интенсивность линий?
12. Спектр ЭПР радикала циклопентадиена С5Н5-, быстро вращающегося в монокристалле циклопентадиена, приведен ниже.
а. Запишите соответствующий спин-гамильтониан.
б. Интерпретируйте спектр.
13. Интерпретируйте приведенный ниже спектр СН2ОН.
1,153
Спектроскопия электронного парамагнитного резонанса
57
14. Ниже приведен спектр C6H5Ge(CH3)3. Примите, что все расщепления обусловлены протонами фенильного кольца, и интерпретируйте спектр. Рассчитайте а.
15. Запишите спин-гамильтониан, интерпретируйте спектр, рассчитайте а анион-радикал а.
16. а. Интерпретируйте спектр и для нитроокиси замещенного нитрозила рассчитайте а.
(СН3)2 /Nt С
I уС-С6Н5
(CH3)2^CxN<
о
58
Глава 9
Н-----10 Э------И
б. Что можно сказать о делокализации неспаренного электрона?
17. Интерпретируйте спектр калиевой соли анион-радикала бифенила и рассчитайте а.
Спектроскопия электронного парамагнитного резонанса
59
18. Интерпретируйте спектр пиразин-аниона и рассчитайте а.
10 Э -----Н
19. Ниже приведен спектр анион-радикала S2, который содержит 40% ядер S( / = 0) и 60% ядер 33S (I = 3/2). Интерпретируйте спектр и определите а для
—I___________I_________I_________I_________I____
1850 1900 1950 2000 2050 Э
20. Используя метод Хюккеля для расчета МО в сопряженных органических системах, можно с помощью соотношения Мак- Коннела приближенно определить для них константы протонного СТВ. Константа СТВ для i-го протона at выражается как at = Qpt, где р,- = CJ,; С,,— коэффициент различных атомных 2р-орбиталей углерода в молекулярной орбитали, на которой находится неспа-рениый электрон.
а.Атомные 2рг-орбитали углерода, составляющие тг-систему, ортогональны ст-связи sp2 С —Н. Почему в таком случае на протоне имеется плотность неспаренного электрона?
60
Глава 9
б. Схема МО бензола имеет следующий вид:
----Фв
----К Ф5
11 Фг> Фз
НН Ф1
В анион-радикале бензола неспаренный электрон может определяться 1
6 2
( ) либо \|/4, либо \|/5. Расчет МО по методу Хюккеля этих вол-
5 3
4
новых функций дает
Фа = Ж ~ Фз + Ч>5 - Фе),
1
Ф5 =----(2ф1 - Ч>2 - Фз + 2ф4 - ф5 - Фб)-
/12
В п-ксилоле вырожденность этих двух МО снимается, причем имеет более низкую энергию.
Используйте соотношение Мак-Коннела и рассчитайте константы протонного СТВ для анион-радикала »-ксилола. Изобразите спектр этого анион-радикала.
21. В результате исследования методом ЭПР анизотропного монокристалла при v = 9,520 ГГц удалось установить, как меняется g-фактор при вращении монокристалла в плоскостях xz (1), yz (2) и ху (3).
Спектроскопия электронного парамагнитного резонанса
61
Из известной величины напряженности поля в резонансе Н, используя соотношение ДЕ = hv и уравнение (9.2), можно рассчитать 3-фактор. Для микроволнового излучения частотой 9,520 ГГц
_ hv _ 6801,9 9eff = W = Н '
а. Интерпретируйте из графика и рассчитайте все элементы з2-тензора.
б. Что даст диагонализация з2-тензора?
в. Какие этапы включает диагонализация з2-тензора?
г. Какие нужно провести операции, чтобы получить матрицы направляющих косинусов?
д. Запишите спин-гамильтониан.
10. ЭЛЕКТРОННАЯ СТРУКТУРА И СПЕКТРЫ ИОНОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ
ВВЕДЕНИЕ
Предмет этой главы уже был темой нескольких монографий [1—12]. Здесь мы дадим обзор электронной структуры ионов переходных металлов и разовьем несколько важных идей, которые будут способствовать пониманию спектроскопии комплексов ионов переходных металлов— нашего основного объекта. Системы ионов переходных металлов рассматриваются и в последующих трех главах, поскольку в этих ионах имеются неспаренные электроны, что приводит к различным осложнениям. Как это часто бывает, эти осложняющие факторы, если их удается понять, дают много информации о соединениях, образуемых ионами переходных металлов. Осложнения возникают по причине электрон-электронных взаимодействий, спин-орбитального взаимодействия и влияния магнитного поля на системы, обладающие неспаренными электронами. Ранее мы уже обсуждали многие из этих тем, но, чтобы понять их до конца, лучше всего рассмотреть примеры, взятые из химии ионов переходных металлов.
ЭЛЕКТРОННЫЕ СОСТОЯНИЯ СВОБОДНЫХ ИОНОВ
10.1. ЭЛЕКТРОН-ЭЛЕКТРОННЫЕ ВЗАИМОДЕЙСТВИЯ И ОБОЗНАЧЕНИЯ ТЕРМОВ
Один или более электронов можно разместить на пяти а-орбиталях газообразного иона металла разнообразными способами. Мы можем указать разницу в энергиях, возникающую по причине различных межэлектронных отталкиваний и различных орбитальных угловых моментов, для этих различных конфигураций с помощью обозначений (или символов) термов. Любое обозначение терма группирует вместе все вырожденные размещения в газообразном ионе. Простейшим примером служит конфигурация d1. Разместить электрон с ms=+l/2 на пяти d-орбиталях можно пятью способами. Каждый из этих вариантов носит название конфигурации микросостояния.
Электронная структура и спектры ионов переходных металлов 63
В отсутствие внешнего электрического или магнитного поля пять микросостояний вырождены, кроме того, существуют еще пять вырожденных состояний, соответствующих ms = — 1/2. Эти десять микросостояний образуют десятикратную вырожденность так называемого терма 2D (см. далее).
Терм основного состояния для любой (/"-конфигурации можно установить, разместив электроны на d-орбиталях. При этом в первую очередь заполняются орбитали, имеющие большие величины mt, электроны размещаются по одному и не спариваются до тех пор, пока на каждой орбитали не будет находиться по одному электрону, т. е. все происходит согласно правилам Гунда. Величины mt для орбиталей, на которых находятся электроны, можно суммировать алгебраическим путем, чтобы получить величину L для каждого терма. В более законченной форме это звучит так: квантовое число mt индивидуального электрона связано с вектором, имеющим компоненту шг(й/2л), направленную вдоль приложенного поля. ML представляет собой сумму одноэлектронных величин mt. Правила сложения векторов требуют, чтобы ML принимало значения L, L— 1, ..., — L, поэтому максимальное значение ML дается величиной L. Для обозначения величин L используются буквы S, Р, D, F, G, Н, I, соответствующие L, равному 0, 1, 2, 3, 4, 5 и 6. Спиновую мультиплет -ность состояния определяют как 2S + 1 (S по аналогии с L представляет собой максимально возможное Ms, где Ms = Sms) Xms) и указывают с помощью индекса вверху слева от символа терма. Мультиплетность отвечает за число возможных проекций S на направление магнитного поля, т. е. если 5=1, мультиплетность три говорит о том, что Ms = 1, О, — 1 (т. е. z-компонента спинового углового момента может быть направлена вдоль поля, перпендикулярно полю и против поля). Полная вырожденность терма определяется как (2L + 1)(25 + 1). Расстановка 2L+ 1 относится к орбитальной вырожденности и описывается ML, принимающим значения L, L— 1, ..., — L. Как упоминалось ранее, символ описывает случай dl. Он десятикратно вырожден, причем пятикратная орбитальная вырожденность соответствует ML 2, 1, 0, — 1 и — 2. В dl-ионе только терм основного состояния 2D обусловлен ЗгУ-орбиталями. Значение квантового числа 5 для терма (или состояния) определяется максимальным значением Ms, которое равно сумме значений ms всех неспаренных электронов. Заполненные подоболочки не дают никакого вклада в L или S, поскольку суммы значений ms и mt равны нулю.
Рассмотрим далее (^-конфигурацию. В этом случае возможны 45 способов размещения двух электронов с ms = ±1/2 на пяти d-орбиталях. Используя описанную выше процедуру к микросостоянию
t t
получим L = 3 и 5=1, что приводит к терму 3F основного состояния, который 21 раз вырожден в отсутствие спин-орбитального взаимодействия. Другие 21 микросостояние составляют высокоэнергетические
64
Глава 10
(возбужденные) состояния, т.е. электрон-электронное отталкивание характеризуется большей величиной. Все термы «/"-конфигурации можно найти, построив таблицы, аналогичные табл. 10.1. Чтобы не нарушать
Таблица 10.1
Микросостояния «Р-иона с положительными значениями ML
последовательности изложения, начнем строить таблицу со строки ML — 4. Это значение ML можно получить только при наличии двух электронов на орбитали с mt = 2, т. е. только если спины спарены. Результирующее микросостояние обозначается как 2+ 2 “ и помещается в столбец табл. 10.1 под Ms = 0. Аналогичным образом для ML= 3, возможны микросостояния 2+1 + ,2+1-,2“1+ и 2 ~ 1 “, соответствующие значениям Ms + 1, 0, 0 и — 1. То же самое можно осуществить для ML, равного 2, 1 и 0. Микросостояния, соответствующие отрицательным значениям ML, в таблице не показаны. Их получают, умножая значения ML для положительных микросостояний ML на — 1; например, для ML= — 3 возмбжны следующие микросостояния: — 2+—1\ —2+ — — 1 “, — 2 — 1 + и — 2 — 1 ~ . Обратите внимание, что, например, микросостояния 2 + 1 “ и 1 ” 2+ эквивалентны, поскольку порядок, в котором мы записываем электроны, не имеет значения, однако эти микросостояния отличаются от микросостояния 2 “ 1 +, так как в последнем электрон с т( = 2 уже не имеет ms= + 1/2.
Высшему значению ML должен соответствовать терм или состояние 'G, и его компонентами ML являются 4, 3, 2, 1, 0, — 1, —2, —3, —4. Конфигурация с ML = 3 для терма 'G выбирается произвольно, поскольку мы только выполняем роль счетовода. Истинная волновая функция для этой компоненты терма 'G представляет собой линейную комбина
Электронная структура и спектры ионов переходных металлов 65
цию* двух микросостояний, указанных для ML = 3. Получить волновую функцию для данных значений L, ML, S и Ms можно с помощью [1] операторов сдвига. То же самое справедливо всякий раз, когда есть выбор микросостояний. Теперь перейдем к следующему высокому значению ML, а именно ML=3. Можно установить, что компонентам Ms — = + 1, 0 и — 1 должен соответствовать терм 3F. Этот терм вырожден 21 раз; 12 микросостояний с неотрицательными значениями ML. произвольно приписанными ему, заключены в табл. 10.1 в скобки. Далее перейдем к состоянию с L = 2, которое должно быть синглетом (т. е. S = 0). Микросостояния, произвольно отнесенные к терму 1D, обведены кружком. Далее мы заключаем в скобки те же микросостояния терма 3Р. Последний терм—это 'S. Каждый из этих термов представляет собой вырожденный набор состояний, и каждый терм отличается по энергии от любого другого **.
Энергии всех этих термов можно рассчитать [1] и выразить с помощью параметров Кондона — Шортли Fo, F2 и F4. Эти параметры являются символами различных интегралов электронного отталкивания в ионе. Выражение, определяющее зависимость энергии любого терма от этих параметров, не зависит от иона металла. В то же время величины этих параметров меняются с ионом металла. Например, E(3F) = = F0 — 8F2— 9F4, a E(3P) = FO + 7F2 — 84F4. Энергия перехода 3P— 3F представляет собой разность энергий этих двух термов, или 15F2 — — 75F4. Аналогичные выражения существуют для всех других переходов, относящихся к термам газообразного иона. Весь спектр можно воспроизвести с помощью параметров F2 и F4. Это справедливо для любого «12-иона.
В ионе V(III) переход 3F — 3P происходит при 13000 см1, а переход XF — 3D — при 10600 см-1. Решая систему уравнений
E(3P—3F) = 15F2-75F4= 13 000 см-1, (10.1)
Е(Ч) - 3F) = 5F2 + 45F4 = 10600 см “ \ (10.2)
получим F2 = 1310 см “ 1 и F4 = 90 см ” \
Рака переопределил эмпирические параметры Кондона — Шортли таким образом, что расстояние между состояниями с максимальной
* Каждое из микросостояний на самом деле обозначает волновую функцию в виде детерминанта, например
2Э-^-|2+(1)
1/2 2 + (2) Г (2)
** Индивидуальные собственные функции терма, как и весь терм, часто называют состояниями. Обычно различие очевидно. В тех случаях, когда различие выражено не столь явно, мы будем пользоваться для описания всей совокупности вырожденных состояний термином терм или уровень, а для описания индивидуальных состояний термином компонентные состояния.
5-574
66
Глава 10
мультиплетностью — функция только одного параметра В:
B = F2- 5F4. (10.3)
Второй параметр С необходим для того, чтобы выразить разность энергий между термами различной мультиплетности:
С = 35F4. (10.4)
После перегруппировки F2 = В + С/1 и F4 = С/35. Подставим эти величины в уравнения (10.1) и (10.2):
Е(3Р — 3F) = 15В,
E^D - 3F) = 5В + 2С.
Для V(III) мы имеем В = 866 см -1 и С/В = 3,6.
В заключение результаты электрон-электронных взаимодействий в «/2-ионе приводят к терму 3F основного состояния и возбужденным состояниям, обусловленным «/-орбиталями, показанными на рис. 10.1.
^’G
Электронные Электронные взаимодействия взаимодействия отсутствуют
(5 вырожденных d-орбиталей)
Рис. 10.1 Термы, обусловленные элек-трон-электронными взаимодействиями в газообразном 42-ионе. Цифра в скобках указывает вырождение каждого уровня (без учета спин-орби-тального взаимодействия).
Используя операции, аналогичные приведенным в табл. 10.1, можно определить термы, обусловленные различными «/"-ионами. Результаты для п от 1 до 9 представлены в табл. 10.2.
Во многих случаях конфигурацию d9 можно рассматривать как эквивалентную d1, если исходить из вырожденных состояний, обусловленных положительной дыркой, имеющейся в «/’-конфигурации. Можно представить d9 как di0 с позитроном, способным аннигилировать с любым из десяти электронов. Эта концепция носит название форма-
Электронная структура и спектры ионов переходных металлов
67
Таблица 10.2
Свободные термы для различных </"-ионов
п Термы
d' d9 2D
d2 ds 3F3P1G1D1S
d3 d1 4F^P 2H2G2F2D2D2P
d* d6 5D3H3G3F3F3D3P3P1I 1G1G1F1D1D1SlS d5 6S4G 4F 4О4Р21 2H2G2G2F2F2D2D2D2P2S
лизма дырок. По тем же самым соображениям возникают следующие эквиваленты:
d2 d3 d4
ds, d7, d6.
10.2. СПИН-ОРБИТАЛЬНОЕ ВЗАИМОДЕЙСТВИЕ
В СВОБОДНЫХ ИОНАХ
Как говорилось в гл. 9 (см. рис. 9.18), взаимодействие магнитного диполя электронного спинового момента с орбитальным моментом L • S представляет собой спин-орбитальное взаимодействие. Изменение величины спин-орбитального взаимодействия в различных электронных конфигурациях также приводит к расщеплению термов, о которых уже шла речь. При рассмотрении этого эффекта широко используются две схемы: так называемая схема взаимодействия Рассела — Саундерса, или схема L -S-взаимодействия, и схема j j-взаимодействия. Если электрон-электронные взаимодействия приводят к большим энергетическим расщеплениям термов по сравнению с расщеплениями, обусловленными спин-орбитальным взаимодействием, пользуются первой схемой. В этом случае мы по существу рассматриваем спин-орбитальное взаимодействие в качестве возмущения энергий отдельных термов.
Схема //-взаимодействия применяется в том случае, когда результатом спин-орбитального взаимодействия являются большие по величине расщепления, а электрон-электронные взаимодействия достаточно малы, чтобы их рассматривать как возмущение спин-орбитальных уровней. К //-схеме обычно прибегают при изучении редкоземельных элементов и ионов третьего ряда переходных металлов. Согласно //-схеме, спиновый угловой момент отдельного электрона взаимодействует с его орбитальным моментом с образованием суммарного вектора углового момента этого электрона j. Отдельные j суммируются и дают вектор J полного углового момента атома.
L S-схема взаимодействия применима к ионам большинства переходных элементов первого ряда, поэтому рассмотрим эту схему более
68
Глава 10
подробно. Выше мы уже говорили о том, что индивидуальные орбитальные угловые моменты электронов mt взаимодействуют с образованием суммарного углового момента, обозначаемого L. Взаимодействие спиновых моментов дает S. Результирующий угловой момент, включающий спин-орбитальное взаимодействие, обозначается J, а соответствующее квантовое число J может принимать все последовательные целочисленные значения в интервале абсолютных величин от L — S до L+ S. Для основного терма минимальное значение J относится к состоянию низшей энергии совокупности, если подоболочки, например d-орбитали, заполнены менее чем наполовину; максимальное значение J относится к состоянию низшей энергии, если подоболочка заполнена более чем наполовину. Если оболочка заполнена наполовину, существует только одно значение J, поскольку L = 0.
Чтобы все сказанное выше стало более понятным, разберем несколько примеров. Клеточная диаграмма основного состояния атома углерода имеет следующий вид:
Is
2s
n
+ 1 о -1
Квантовое число L, получаемое путем сложения т, всех электронов, находящихся на незаполненных орбиталях, для углерода равно единице: L = +1+0=1. Квантовое число S, сумма спиновых квантовых чисел (ms. = ±1/2) всех неспаренных электронов, для углерода также равно единице: S= 1/2+ 1/2= 1. Мультиплетность равна трем, и терм основного состояния обозначается как 3Р. Значения J, определяемые как |L — — S|, ..., |L+S|, соответственно равны |L—S|= 1—1 = 0, |L+S| = 1 + 1 = = 2, поэтому J = 0, 1 и 2 (единица — единственное целое число, необходимое для завершения серии). Рассматриваемая подоболочка заполнена менее чем наполовину, поэтому состояние с минимальным значением J имеет низшую энергию.
Основное состояние углерода записывается как 3Р0, где нижний индекс 0 относится к величине J.
Клеточная диаграмма основного состояния V3 + записывается как
+ 2+1 0 -1 — 2
т т
при обозначении терма 3F2 (L = 3, S = 1, J = 4, 3, 2). Возбужденное состояние для этой частицы демонстрирует следующая клеточная диаграмма :
п
Электронная структура и спектры ионов переходных металлов 69
это микросостояние входит в терм 1G4 (L = 4, 5 = 0, J = 4). Для азота с клеточной диаграммой
т т т
L = О, S = 3/2 и J = 3/2, терм 4S3/2- Отметим, что в этом случае имеется только одно значение J с L = 0, поскольку \L + S| = \L — S| = 3/2.
Практически каждый может установить следующие термы основных состояний элементов, которые приведены в скобках: 3Р2 (S), 2Р3/2 (С1), 3F2 (Ti), 7S3 (Cr), 3F4 (Ni), 3P0 (Si), 4S3/2 (As) и 4/9/2 (Pr).
Энергию спин-орбитального взаимодействия обычно описывают двумя параметрами: I; и X. Параметр 2, описывает энергии спин-орбитального взаимодействия единственного электрона. Он является мерой силы взаимодействия спинового и орбитального углового моментов единственного электрона в данном микросостоянии и, таким образом, характеризует свойство микросостояния, а не терма. Соответствующий взаимодействию оператор— это Д-S. Параметр £, определяется как
Zeff в2
Л 2 <?~3>, (Ю.5)
2т с
где 3>— средняя величина г-3, т — масса электрона, с—скорость
света, a Zeff—эффективный заряд ядра.
Параметр X используется для описания соответствующего свойства терма. Оператор теперь имеет вид XL • S. Величины Хид связаны соотношением
Х= ±£/2S. (10.6)
Параметр — в принципе положительная величина. Если оболочка заполнена менее чем наполовину, знак X положителен, если она заполнена более чем наполовину, знак X отрицателен. Он имеет смысл, если мы пользуемся положительными дырками, из-за которых для конфигураций с более чем наполовину заполненными оболочками необходимо изменить знак уравнения (10.5). Итак, для оболочки, которая заполнена менее чем наполовину, низшее значение J соответствует низшей энергии и положительному значению X.
Эквивалентной операторной формой L S является 1/2(J2 — L2 — S2), если состояния можно охарактеризовать квантовыми числами L, S и J. Вклад спин-орбитального взаимодействия в энергию любого уровня выражается тогда как
yX[J(J + 1) - L(L+ 1) - S(S + 1)]. (10.7а)
Разность энергий двух соседних состояний терма, возникших из-за спин-орбитального взаимодействия, определяется выражением
A£J>J + 1 = X(J 4- 1). (10.76)
Например, разность энергий состояний терма с7 = 3и7 = 4 равна 4Х.
70
Глава 10
Более того, в L-S-схеме спин-орбитальное взаимодействие происходит таким образом, что центр тяжести энергии терма сохраняется, т. е. средняя энергия остается той же самой. Основное состояние для 72-си-стемы, 3F, характеризуется значениями J, равными 4, 3 и 2, причем 2 относится к наименьшей энергии, поскольку оболочка заполнена менее чем наполовину. Полное обозначение терма основного состояния — 3F2.
____ j = o
____ J = 4
(5) A J = 2 зр W
(3) - A J=1
\ (D-2A J = o
____ J = 2
(9) ЗЛ J = 4 / /
3F(21)/ \\ w
\(5)~ 4X J = 2 // J
-------1
''-----2
Рис. 10.2. Расщепление (^-конфигурации под действием спин-орбитального взаимодействия. Справа показано расщепление состояния 3Р2 магнитным полем Но.
Для возбужденного состояния возможно лишь J, равное 2. Возбужденное состояние 3Р характеризуется J = 0, 1, 2, в то время как 3G характеризуется только J = 4, a lS—только J = 0.
Теперь, используя уравнение (10.7а), можно рассчитать вклад спин-орбитального взаимодействия в энергии всех состояний J. Для основного уровня 3F, где 7 = 2, мы получаем 1/2 л[2(2 + 1) — 3(3 + 1) — 1(1 + + 1)] = — 4л. Этот результат приведен на рис. 10.2 наряду с результатами аналогичных расчетов влияния 7.LS на все состояния 72-системы. Спин-орбитальное взаимодействие снимает не все вырождение, и оставшееся вырождение, соответствующее целочисленным значениям Mj от 7 до — 7, указано в скобках над каждым уровнем. Отметим, что уравнение (10.76) выполняется и центр тяжести сохраняется. Например, в терме 3Р умножение вырождения на изменение энергии дает 57.. — ЗА — — (1)(2)7. — 0. Вырождение индивидуальных состояний J устраняется магнитным полем. Расщепление на Mj-состояния показано только для основного терма с 7 = 2 (рис. 10.2).
Электронная структура и спектры ионов переходных металлов 71
КРИСТАЛЛИЧЕСКИЕ ПОЛЯ
10.3. ВЛИЯНИЕ ЛИГАНДОВ НА ЭНЕРГИИ d-ОРБИТАЛЕЙ
Обычно, когда проводится исследование ионов переходных металлов, мы имеем дело не с индивидуальными ионами, а ионами, входящими в состав комплексов. Для определения влияния лигандов, входящих в комплексы ионов переходных металлов, на энергии <7-орбиталей пользуются двумя приближениями кристаллического поля. Электроны иона металла в комплексе отталкиваются друг от друга, отталкиваются они и от электронной плотности основания Льюиса (лиганда). Если отталкивание между электронами металла и электронной плотностью лигандов мало по сравнению с межэлектронным отталкиванием, применяют так называемое приближение слабого поля. Если лиганды—сильные основания Льюиса, отталкивание между электронами металла и электронами лигандов превышает по величине межэлектронное отталкивание, в этом случае используется приближение сильного поля.
Базисом в этих задачах могут быть орбитали, выраженные комплексными волновыми функциями, угловые зависимости которых представлены сферическими гармониками
уо = (5/8)1/2(3cos20 - 1)-(27с)~ 1/2,
У±> = (15/4)1/2 sine cosе-(2тг)-1/2屓(’,
У2±г = (15/16)112 sin 20 • (2л)- 1/2е±21ч,
Кроме того, можно использовать и действительные тригонометрические волновые функции, которые представляют собой линейные комбинации комплексных орбиталей. Они выражаются как
dz~ = |0> (dZ2 в действительности rf(Z2-r2/3))
dy2 = (»7]/2)[|-1> + | + 1>] dx= (1//2)[|-1> - | + 1>] dxy= -07|/2)[|2>-|-2>] ^=(1//2)[|2> + |-2]
В приближении слабого поля в качестве базиса применяют собственные функции свободноионных термов (которые учитывают межэлектронное отталкивание в совокупности <7-уровней). Например, для терма 3F подходят волновые функции, соответствующие ML = + 3, + 2, +1 и 0. Они обозначаются как )3>, |2> и т. д. Гамильтониан выражается как
Н = Но+ V,
где Но—свободноионный гамильтониан, а V рассматривается как возмущение лигандом электронной плотности в Но. Возмущение V имеет
72
Глава 10
исключительно простой вид: оно включает только электростатическое отталкивание со стороны лигандов, которые рассматриваются как точечные заряды. Для октаэдрического комплекса возмущение определяется выражением
V^eZJr^ (10.8)
1
где е — заряд электрона, Z,— эффективный заряд на i-м лиганде, r,j—расстояние от d-электрона до i-ro заряда (это задача d1). Это выражение можно сравнить с полным гамильтонианом, приведенным в гл. 3. Использование упрощенного гамильтониана [уравнение (10.8)] приводит к теории кристаллического поля. Следует подчеркнуть, что такая постановка задачи просто описывает электростатическое отталкивание между d-электронами и электронной плотностью лигандов, и в этом случае мы можем говорить только об относительных энергиях d-орбиталей.
Для того чтобы рассчитать интегралы <ML\ V\ M'Ly, V записывают так, чтобы упростить интегрирование [2, 3]. По этой причине многие параметры, связанные с радиальной частью матричных элементов, в се-кулярном детерминанте имеют следующий вид: I/6 (Ze2r2 4« 5). где г24 соответствует средней четвертой степени радиуса d-электронов центрального атома, а—расстояние между металлом и лигандом, а размерность Ze та же самая, что и у е. Эта радиальная величина обозначается как lODty и имеет размерность энергии.
Запишем секулярный детерминант для октаэдрического комплекса с таким гамильтонианом, действующим на d1-кoнфигypaцию. Используя базис комплексных d-орбиталей, получаем
|2>
|2> Dq — Е
|1>
Р>
1~1>
|-2> 5Dq
|1> Р> |-1>
—4Dq - Е
6Dq — Е
—4Dq - Е
1~2>
5Dq
Dq - Е
Это дает корни
Е(|1» = — 4Dq E(|-l»= -4Dq Е(|0» = 6Dq
и уравнение в виде детерминанта |2>
2> Dq — Е
-2> 5Dq
|-2>
5Dq
Dq — Е
= 0
Электронная структура и спектры ионов переходных металлов 73
При решении этого детерминанта получаются две величины энергии: одна при — 4Dq и одна при 6Dq.
Как и в расчете по методу Хюккеля, подставляя найденные значения энергии, в секулярные уравнения, выведенные из секулярного детерминанта, можно получить волновые функции:
и
Ф4 = -i2-1/2(|2> - | - 2» ф5=212(|2> + | -2».
Данные волновые функции—это волновые функции dxy- и dx2 - /-орбиталей, последняя направлена на лиганды, а первая — между лигандами. Отметим, что октаэдрическое кристаллическое поле смешивает волновые
6Dq--
Рис. 10.3. Расщепление одноэлектронных d-орбиталей кристаллическим полем Oh.
-4Dq—
dyz dxv
функции \2У и | —2> и делает более удобным использование действительных dxy- и dx2 _у2-орбиталей для описания комплекса. Поскольку вырожденность '|1> и |-1> не снимается, волновые функции фь ф2, Фз можно рассматривать как действительную или мнимую комбинацию, что достаточно удобно. Эти результаты суммированы на рис. 10.3, из которого видно, что образуются два вырожденных набора орбиталей, разделенных 10Dq. Таким образом, для б/'-системы с энергией, соответствующей lODq. предполагается один d — d-электронный переход. В симметрии Oh три вырожденные dxz-, dyz- и d^-орбитали преобразуются как /2а и приводят к основному состоянию 2Т2а, в то время как dz2- и dx2_ ^-орбитали ведут к возбужденному состоянию 2£а. Эти символы сообщают следующую информацию: 1) символ Т указывает, что состояние орбиталь-но трижды вырождено, а Е говорит о двукратном вырождении; 2) верхний индекс 2 говорит о том, что спиновая мультиплетность равна двум, т. е. что один электрон не спарен; 3) g говорит о четном симметричном состоянии.
Рассмотрим далее слабое поле с октаэдрическим б/2-комплексом. Вначале займемся термом 3F, который описывается базисом |3/ ... |0> ... | —3>. Поскольку в базис для 3F входят все функции с Ms = 3/2, эта цифра при написании символа опускается и указывается только ML, т. е. полная запись должна иметь вид |3, 3/2>. Базисные волновые функции мы приводить не будем. Методы получения таких волновых функций с использованием операторов сдвига рассматриваются в моногра-
74
Глава 10
фии Больхаузена [1].
Запишем секулярный детерминант
|3>
<3| — 3Dq — Е
<2|
<ч
<01
<-1| 151/2Р?
<-2|
<-3|
|1>
— Dq — Е
|0> |-1> |-2>
151/2Р? SDq
— 6Dq — Е — Dq — Е
7Dq - Е
— 3Dq - Е
5О?
решениями которого являются
Ei = — 6Dq Е5= 2Dq
Е2 = — 6Dq Е6= 2Dq
Е3= — 6Dq Еп = 12Dq
Е4= 2Dq
Энергии * и волновые функции для соответствующих уровней приведены на рис. 10.4.
3А2 О)
3Г2 О)
3Г, (9)
12Dq [2"1/2 (|2> + |-2»]
[24-V2 (3|3> + 151/2 |-1»] [24-1/2 (3| — 3> + 151^2 |1»]
2Dq [2-’/2 (|2> - |-2»]
— 6Dq [24-1/2 (151/2 |-3> —3(1»]
[|0>]
[24“1/2 (151/2 |3> — 3| — 1»]
Рис. 10.4. Расщепление терма 3F 42-иона кристаллическим полем Ок.
При этом анализе ковалентность связи металл — лиганд совершенно не учитывается. В результате, если бы мы попытались рассчитать Dq, то полученная величина значительно отличалась бы от экспериментальной. В теории поля лигандов допускается существование ковалентности связи и Dq (и другие параметры, которые будут обсуждены вкратце) рассматривается как эмпирический параметр, который получается из электронного спектра. Формулировка задачи во всех других отношениях аналогична.
* Dq определяется для одноэлектронной системы. Для многоэлектронной системы для каждого состояния используют свою величину Dq (Ballhausen С. J., Gray Н. В., Chemistry of Coordination Compounds. Vol.l. A. E. Martell. Ed., Van Nostrand Reinhold, Princeton, N.J.). Однако это редко осуществляется на практике из-за весьма приближенного характера модели.
Электронная структура и спектры ионов переходных металлов
75
10.4. АСПЕКТЫ СИММЕТРИИ d-ОРБИТАЛИ, РАСЩЕПЛЯЕМОЙ ЛИГАНДОМ
Как это обычно бывает, когда используется сильно упрощенный гамильтониан, о корректности результатов говорит симметрия. Например, мы упоминали в гл. 2, что соответствующие комбинации двойных произведений векторов х, у и z дают неприводимые представления для <7-орбиталей и и(х вырожденностей. Применив уже рассмотренные принципы (гл. 2), можно показать, как получают все те состояния, которые обусловлены одноэлектронными уровнями. Этот подход можно распространить и на многоэлектронные системы различной геометрии.
Начнем с изучения влияния октаэдрического поля на полное представление, для которого базис образует совокупность <7-волновых функций. Чтобы получить это полное представление, необходимо найти элементы матриц, которые выражают результат действия каждой из операций симметрии группы на наш базис из б/-орбиталей. Характеры этих матриц содержат представление, которое мы ищем. Поскольку все <7-орби-тали четны, т. е. симметричны по отношению к операции инверсии, в результате операции инверсии никакой новой информации получить не удастся. Таким образом, мы можем иметь дело с более простой чисто вращательной подгруппой О, а не Oh. Если вы хотите убедиться в этом сами, то вспомните, что в любой группе, включающей i (например, £)4Л или Сзл), соответствующая группа вращений (например, £>4 или С3) имеет то же самое неприводимое представление для двойных произведений, за исключением нижних индексов и и g в первой группе. Напомним, что <7-волновые функции состоят из радиальной, спиновой и угловой (0 и <р) компонент. Радиальной компонентой мы пренебрегаем в силу ее ненаправленного характера, поскольку она не меняется при любых операциях симметрии. Кроме того, мы примем, что спиновая компонента не зависит от орбитальной и в данной ситуации пренебрежем первой. Угол 0 определяется относительно главной оси, например оси вращения, поэтому он не меняется при любом вращении и им также можно пренебречь. Меняется только <р; эта составляющая волновой функции выражается как е‘”1,<₽. (Для б/-орбиталей I = 2, a mt принимает значения 2, 1, 0, — 1, —2.) Для того чтобы определить влияние поворота на угол а на е‘”1,<₽, мы отметим, что такое вращение вызывает следующие изменения:
е2«<₽ е«<₽ е° e-i<p e~2i<?
^2<(ф+а) Поворот £«Ф+а) на угол а 0
£—i(g>+a)
£-2i(cp+a)
76
Глава 10
Матрица, которая, действует на наш базис из <7-орбиталей, записывается как
e2i“ 0 0 0 0
0 eia 0 0 0
0 0 е° 0 0
0 0 0 e~ia 0
.0 0 0 0 e-2ia
А матрица общего вида для вращения любого набора орбиталей выглядит как
0 .. 0 0
0 l)ia . • 0 0
0 0 ^(1— X)ia 0
0 0 • • 0 e-Ha
Если а = 0, каждый элемент равен, очевидно, единице. Мы дадим след этой последней матрицы * без вывода
sin U + 11 а
Х(а) =---------------, (10.9)
где а # 0. Из следа, определяемого этой формулой, мы получим характер представления для любой операции вращения. Подставляя а непосредственно в формулу (10.9), мы находим, что полный характер для вращения Сз пяти <7-орбиталей выражается как
Х(С3) =
Характеры для других вращений можно получить аналогичным путем. Если а = 0 или 2л, необходимо прежде всего установить предел неопределенности, т. е.
sin (I + | ](2л) \ Ч о
—> - sin л-----------------0 ’
Суммируемые величины образуют геометрическую прогрессию.
Электронная структура и спектры ионов переходных металлов 77
В соответствии с правилом Лопиталя
Х(0) = 2/+ 1
Х(7г) = 2/ + 1 для целого / или — (2/ + 1) для полуцелого /.
Таким образом, для тождественного преобразования а = 0 и Х(Е) равен 21 + 1. Используя таблицу характеров точечной группы О и приведенную выше формулу для определения характеров различных операций над пятью <7-орбиталями, получаем
Е 6С4 3C2(=Cj) 8С3 6С2
Хг 5 -1 1 -1 1
Из формулы разложения следует, что Хг = Е + Т. Поскольку rf-орби-тали четны, мы можем переписать этот результат как
ХГ = Eg + T2g .
Это и есть результат проведенного нами анализа кристаллического поля. Результаты аналогичного анализа симметрии даны в табл. 10.3.
Таблица 10.3
Представления различных орбиталей в точечной группе Oh
Тип орбита-
ли I Неприводимое представле-
ние3
s 0 а1я
Р 1 Ч»
d 2 eg+t2g
f 3 a2g + tlu + t2u
9 4 atg + eg + tlg + t2g
/1 5 e„ + 2tlu + t2u
' 6 atg + a2g + eg + tie + 2t2g
a Нижние индексы g и и определяются природой рассматриваемых атомных орбиталей. Если / четно (gerade), орбиталь обозначают нижним индексом д, если / нечетно (ungerade), то индексом и.
Таким же способом можно установить влияние других симметрий на одноэлектронные уровни. Можно также воспользоваться корреляционной таблицей, которая показывает, как представления группы Oh меняются или разлагаются на представления ее подгруппы, если меняется симметрия. В табл. 10.4 содержится такая информация для некоторых типов симметрии, с которыми обычно приходится сталкиваться в комплексах ионов переходных металлов.
Имея неприводимые представления (табл. 10.3) различных атомных орбиталей в точечной группе О-„ и корреляционную таблицу' (табл. 10.4),
Таблица 10.4
Корреляционная таблица для точечной группы Oh
Oh О Td Вм &2d Cm Cm D3d D3 c2h
A-ig -41 -41 -4i9 -41 -41 -41 Alg Ai -49
Aig А2 -42 В1Я Bi Bi -42 -42s a2 Bg
Е £ -41g + Blg 4i + Bi -41 + Bt Л1 4- Л2 E* E -4g + Bg
т\ Л A2g Eg Л2 4- E Л2 + E Л2 4~ El 4~ B2 -429 + Eg a2 + e Ag 4- 2Bff
т2я т2 т2 Bis + Eg b2 + e B2 + E Ai +Bt +B2 -419 + Eg Ai 4- E 2-49 + Bg
^lu А1 -42 ^lu Bl a2 a2 Aiu Ai Au
А2и л2 -41 B1„ -41 b2 Ai a2u a2 Bg
Еи Е Е -41a + Blu -4] + Bi A2 + Ai 4- A2 Eg E Au + Bu
т1и Л тг A2u + Eu b2 + e Ai+E Ai + + B2 A2u + Eu A2 4- E Au 4- 2BU
т2я т2 т\ B2a + Eu A2 + E Bi+E Л2 4- Bi 4- B2 ^iu + Eu Ai+E 2ЛН 4- Bu
Электронная структура и спектры ионов переходных металлов 79
можно установить неприводимые представления разных орбиталей в различных точечных группах. Результаты, полученные для одного электрона, находящегося на различных орбиталях, применимы также к термам многоэлектронных систем. Например, термы 3F, 3Р, lG, [D и ^-конфигурации можно рассмотреть как р-, g-, d- и s-орбитали. Нижние индексы д и и, приведенные в табл. 10.3, при этом не используются, но они зависят от природы д и и взятых атомных орбиталей. Таким образом, табл. 10.3 применима как к термам, так и к орбиталям. Например, терм D пятикратно вырожден подобно пяти d-орбиталям; он описывается волновой функцией для каждого из пяти значений М;. Эти волновые функции имеют Ф-составляющую, выражаемую как e'MLIf. Из табл. 10.3 и 10.4 можно видеть, что состояние D свободного иона расщепляется на состояния Ед + Т2д в октаэдрическом поле и на состояния А1д + Big + Ед + В2д в тетрагональном поле £)4Л. Аналогичным образом терм 3F приводит к А2д+Т1д+Т2д в октаэдрическом поле и к Bi+Л2-1-2Е + В2 в поле С4„.
Рассмотрим теперь состояния, обусловленные ^-конфигурацией, в том случае, когда кристаллическое поле превышает межэлектронные отталкивания *. Межэлектронные отталкивания рассматривают как возмущения d-электронных конфигураций сильным полем. Другими словами, идентифицируют различные состояния кристаллического поля, строят собственные функции, а e2/riJ используют как возмущение. Термы различных d"-KOH(j)nrypaunfi записать просто. 2Т2д и 2Ед—термы d1-конфигурации. Термы, обусловленные конфигурациями с дополнительным электроном, d" + 1, описывают прямым произведением симметрий dn-терма и добавленного электрона. Для d~ мы имеем конфигурации t2g, (2дёд и ед. Поэтому для t2g t2gx t2g, что ведет к Т 1а+ Т2д+ Ед+ А1п. В случае t2eg мы имеем t2g х ед, что ведет к Т1д+ Т2д, и для е2 ед х е8, что ведет к Ед + А1д + А2д. Далее определим мультиплетность приведенных термов и покажем связь этих термов сильного поля с термами газообразных ионов. Мультиплетности определяются методом снижения симметрии [1, 2]. Спиновую вырожденность еа-конфигура-ции можно определить, снижая симметрию от Oh до £>4Л с тем,
* Общая вырожденность, которая характеризует систему q электронов, заполняющих подоболочку с z-вырожденными орбиталями (каждая из которых занята двумя электронами с противоположно направленными спинами), выражается как
(2Д!
q\(2z - q)! '
Для /^-конфигурации
(2 х 3)!
—------— = 15,
2!(6 - 2)!
а для
Г 6f 1Г 4! 1
I---------- I ---------I = [151-Г4] = 60.
L 21(6 - 2)! L 11(4— 1)! J
80
Глава 10
чтобы устранить вырождение. Корреляционная таблица (табл. 10.4) показывает
----- а1д
----- Ь1д
При этом важно помнить, что состояния ведут себя таким же образом. В соответствии с принципом Паули а]д должна быть в D4h синглетом, что ведет к 1Alg(ajg дает а1д х а1д, откуда получается 1Alg)‘, bjg должно быть синглетом 1Alg (bjg ведет к Ь1д х Ь1д, что дает 1А1д), а a}gb}g ведет к Ь1д х а1д, что дает 1В1д или 3В1д. Эти состояния при
октаэдрической симметрии (Aig, Л2д и Еа) должны в точечной группе D4h переходить в следующие состояния (см. таблицу групповой корре-
ляции, табл. 10.4):
А1д
А2д
Ед
А1д
В1д
А1д
В'д
Мы определили мультиплетность состояний Й4Л; поскольку снижение симметрии не может изменить спиновую вырожденность, определить последнюю можно, вернувшись к более высокой симметрии состояний Oh. Состояние гА1д в D4h должно соответствовать синглету 1А1д в Oh. Другое состояние гА1д должно следовать из Ед, причем последним должно быть 1Е1д. Состояние В1д в Й4Л, проистекающее из 1Е1д, также должно быть синглетом. И наконец, 3В1д в Й4Л связано с А2д, которое должно быть 3Л2в.
Dih Oh
Alg(1Alg)
Звгд A2g(3A2g)
4, Eg(1£g)
1Big
Конфигурация t2g приводит к должны изучить корреляционную метрии Oh и найти более низкую в одномерные представления или
состояниям А1д + Ед+Т1д+Т2д. Мы таблицу для этих состояний в сим-симметрию, которая превращает их
ГЧ ГГТТГЧХ t V ТТТЧЛ ттТ» TT<aTTTTTT
wj 1*11*1 j VpillUlTY upv^vi aD.ltlinn,
Электронная структура и спектры ионов переходных металлов 81
Табл. 10.4 показывает, что этим требованиям удовлетворяют С2Й и С2г- Для С2А получаем
Он ^19 ^9 Т19 т2д С2> > Л ’ Ли+Вд » 49+В9 + В9 * Ад+ Ад+ Вд
Поскольку t2g в Oh приводит к орбиталям ад + ад + Ьд в С2А, возможны следующие конфигурации: aglag2, aglbg, ад2, ад2Ьд, Ьд. Как
и раньше.
«91 X «91 ведет к А
Я91 X «92 ведет к ^9
Я91 X Ь9 ведет к вд
«92 X «92 ведет к ^9
«92 X Ь9 ведет к вд
Ь9 X Ь9 ведет к ^9
Нижние индексы 1 и 2 при орбитали ад используются для того, чтобы различить два разных представления ад, которые вытекают из t2g в группе более низкой симметрии. Поскольку первое, четвертое и шестое двойные произведения, которые записаны выше, соответствуют двум электронам, занимающим одну и ту же орбиталь* (ад1, ад2 и Ьд соответственно), произведения должны быть синглетными состояниями 1А , 1 2Ад и гАд соответственно. Второе, третье и пятое двойные произведения соответствуют электронам на различных орбиталях и приводят к синглетному и триплетному состояниям: 'А + 3Ад и 21В^ + 23В?. Состояния в точечной группе C2h приведены в левом столбце табл. 10.5. В правом столбце даны коррелирующие состояния в Oh из табл. 10.4. Поскольку в С2А возможны три триплетных состояния симметрии Вд, Вд и Ад, они должны вытекать из состояния Все другие состояния, вытекающие из t2g, синглеты. Другие возможные корреляции отсутствуют. Таким же образом можно коррелировать состояния конфигураций t2geg.
Подводя итог описанной выше процедуре, следует отметить, что снижение симметрии проводилось следующим образом:
1. Орбитали в точечной группе более высокой симметрии были скоррелированы с орбиталями в точечной группе более низкой симметрии.
* Если мы пометим орбитали 1. 2 и 3, то получим 1 х 1,1 х 2, 1 х 3, 2 х 2,
2 х 3 и 3 х 3. Для 1x1, 2х2иЗхЗна одной и той же орбитали находятся два
электрона.
6-574
82
Глава 10
2. К этим орбиталям более низкой симметрии было добавлено необходимое число электронов.
3. Далее определены были электронные состояния, вытекающие из электронных конфигураций в точечной группе более низкой симметрии.
4. Электронные состояния более низкой симметрии с учетом спиновой мультиплетности были скоррелированы с электронными состояниями более высокой симметрии. (Это фактически и есть повышение симметрии.)
Далее следует показать, как термы, возникающие в сильном поле,
Таблица 10.5
Связь мультиплетностей в точечных группах С2ь и 0h
связаны с термами слабого поля, поскольку с изменением силы поля в ряду комплексов данных форм металла должен происходить непрерывный переход от одних термов к другим. Мы можем показать это на примере </2-иона, используя два принципа:
1. Все состояния слабого поля коррелируют с состояниями одной и той же симметрии и одинаковой мультиплетности в сильном поле.
2. С изменением силы поля лигандов состояния одной и той же симметрии и одной и той же спиновой вырожденности не могут пересекаться.
На рис. 10.5 слева представлены свободноионные состояния и соответствующие состояния в слабом поле, а справа—соответствующие состояния в сильном поле. Конфигурации в бесконечно сильном поле показаны в крайней правой позиции.
Если исходить из конфигурации е2 в бесконечно сильном поле, мы приходим к состоянию 1А. В конфигурацию t2 также входит состояние 1А1. Только если связи проведены так, как показано на рисунке, можно избежать пересечения. То же самое справедливо для 1£. Поскольку в слабом поле имеется только одно состояние 3/12, эта корреляция проста. И наконец, только такая корреляция не приводит к пересечениям состояний одинаковой симметрии и мультиплетности, входящих в конфигурации t2e и t2.
Результаты расчета энергий различных уровней при переходе от слабого поля к сильному представлены в графическом виде диаграммами Танабе—Сугано [13] для различных ^"-конфигураций (см. приложение
Электронная структура и спектры ионов переходных металлов 83
III). Энергии представлены как функции Е/ В от Dq/B, где В — параметр Рака. Использование величин £/ В и Dq/ В вызвано тем, что они позволяют удобным образом преобразовать уравнение для энергии для построения графика. Поскольку рассматриваемые состояния характеризуются различной мультиплетностью, то они являются функцией
поле
Рис. 10.5. Корреляция состояний </2-иона в сильном и слабом полях.
параметра Рака С в той же мере, что и параметра В. Таким образом, диаграмму можно построить только для данного отношения С/В. Для этих диаграмм в качестве уровня нулевой энергии берется низший терм.
Диаграммы Оргела используются для представления части той информации, которая полностью дана в диаграммах Танабе — Сугано. В диаграммы Оргела входят лишь те термы, мультиплетность которых совпадает с мультиплетностью основного состояния. С помощью диаграмм Оргела вполне можно интерпретировать электронные спектры разрешенных по мультиплетности переходов, поэтому в оставшейся части главы (например, рис. 10.9) мы часто будем прибегать к этим Диаграммам.
6*
84
Глава 10
10.5. ДВОЙНЫЕ ГРУППЫ
Ранее мы показали, как с помощью таблицы характеров можно найти характер представления, для которого р- и d-орбитали образуют базис в различных симметриях. В предыдущем разделе мы также показали, что характер %(а) любой операции симметрии, соответствующей повороту на угол а базисных орбитально волновой функции или волновой функции состояния с квантовым числом углового момента I или L выражается уравнением (10.9):
Х(а) =
Это уравнение можно применять для состояний, характеризующихся полным угловым моментом J (где J = L + S), путем простой замены J на I. Если электронов четное число и если J целочисленно, полное представление в любой симметрии можно разложить на неприводимые представления точечной группы, как это мы сделали в предыдущем разделе. Однако, если J имеет полуцелое значение (т. е. S нечетно), поворот на 2л (что представляет собой операцию тождественного преобразования) не дает тождественной величины характера:
Х(а + 2л) =
(ос + 2л) 2
Можно показать, что для проведения тождественного преобразования необходим поворот на 4л. Для того чтобы избежать этой сложности, поворот на 2л в этом примере рассматривается как операция симметрии, которую мы обозначим символом R. Обычная группа вращений расширяется таким образом, чтобы она включала произведение R на все существующие вращения. Новая группа называется двойной группой. Используя уравнение (10.9), можно получить характеры для вращений. Характеры Е и R (т. е. а — 0 и а = 2л) требуют раскрытия неопределенности вида
2
0’
что дает x(0) = 2J + 1 и %(2л) = 2J + 1 для целого J или — (2J + 1) для полуцелого J, как об этом говорилось ранее. Таблицы характеров для
Электронная структура и спектры ионов переходных металлов 85
двойных групп вращений Т>4 и О' приведены в приложении II; таблицы характеров для других групп имеются в литературе *. Наиболее распространены две системы обозначений неприводимых представлений; в одной из них используют символы с индексами Г;, а в другой—символы со штрихами, которые применяли и мы. Прямые произведения представлений двойных групп можно получить таким же образом, как и раньше, их можно представить в виде сумм неприводимых представлений.
Для большей ясности рассмотрим некоторые примеры. Если J — по-луцелое, для определения эффектов спин-орбитального взаимодействия необходимо воспользоваться двойными группами. Поскольку спин-ор-битальные эффекты обусловлены взаимодействием спинового и орбитального моментов электрона, мы занимаемся представлением прямого произведения этих двух эффектов. В качестве примера определим влияние октаэдрического поля и спин-орбитального взаимодействия на 4F-свободноионное состояние </7-иона. Как и в предыдущем разделе, мы можем получить полное представление в точечной группе О и разложить его:
хт(/=3)=я2+ Т. + Т2.
<Д-Ион в слабом поле 0h дает, как показано в диаграмме Танабе—Су-гано, основное состояние 47'1(! и возбужденное состояние 4Т2й и 4Я2з. В двойной группе О' эти состояния соответствуют Т'ДГД, Т'2(Г5) и Я2(Г2). Взяв S = 3/2 и подставляя вместо / в уравнение (10.9) S, мы порождаем в точечной группе О неприводимое представление ^(ГД, т. е. одно из новых неприводимых представлений двойной группы. Возьмем прямые произведения спиновой и орбитальной составляющих и разложим их, как и раньше, что даст
Г2хГ8 = Г8
Г4 х Г8 = Г6 + Г7 + gr8
Г5 х Г8 = Гб + Г7 + 2Г8
Как мы видим, спин-орбитальные эффекты не расщепляют Г2, но они расщепляют Г4 и Г5 на четыре состояния. Мы можем получить представления двойных групп, превратив L и S в J и использовав уравнение (10.9) для значений J, равных 9/2, 7/2, 5/2 и 3/2. Эта операция необходима, если спин-орбитальное расщепление сравнимо по величине или превышает расщепление в кристаллическом поле. В описанном выше подходе мы рассматривали сильное кристаллическое поле и дополнительное небольшое спин-орбитальное расщепление в нем. Результаты суммирует диаграмма, приведенная на рис. 10.6.
* Sugano S., Tanabe Y.. Kamimura H., Multiplets of Transition Metal Ions in Crystals, Academic Press, New York, 1970.
86
Глава 10
А 5 В Г Д Е Ж
Рис. 10.6. А — газообразный ион; Б — расщепление сильным полем 0h; В — последующее действие XL S меньшей величины; Г—свободный ион; Д—расщепление в результате сильного спии-орбитального взаимодействия; Е — последующее действие более слабого поля лигаидов; Ж—корреляция состояний в промежуточной области. Чтобы не усложнять картину, ни одно нз этих состояний не показано пересекающимся. Состояния различных двойных групп симметрии могут пересекаться. Состояния, относящиеся к одной и той же двойной группе симметрии, участвуют в конфигурационном взаимодействии.
Важно помнить, что, имея дело с эффектами спин-орбитального взаимодействия (с которыми нам часто придется сталкиваться в последующих главах) в системах с полуцелыми значениями J, необходимо применять двойную группу.
10.6. ЭФФЕКТ ЯНА—ТЕЛЛЕРА
Существует еще один эффект, который оказывает влияние на электронную структуру комплекса и который мы должны рассмотреть перед тем, как перейти к обсуждению электронных спектров. Рассмотрим молекулу с неспаренным электроном, находящимся на дважды вырожденной орбитали, например октаэдрический комплекс Cu(II). Вспомните, что в результате искажения геометрии молекулы от наиболее симметричной (Oh) до, например, D4h можно понизить энергию молекулы.
ео 44 -4- -4- Ч
-> 44 аю
‘2о 44 44 44 44 b2o
ео
Oh
Электронная структура и спектры ионов переходных металлов 87
Здесь конфигурация ед расщепляется на компоненты blg и а1д. Поскольку два электрона находятся на стабилизованной п19-орбитали и только один электрон занимает дестабилизованную Ь19-орбиталь, молекула как целое стабильна. Чем это обусловлено, легко понять, если обратиться к простой электростатической теории кристаллического поля; орбиталь, направленная на лиганд, дестабилизована, и чем ближе находится лиганд, тем выше энергия. Тетрагональное растяжение (удлинение двух связей М — L вдоль оси z и укорачивание четырех других связей вдоль осей х и у) дестабилизует d,> _ v,2 (Ь19)-орбиталь и стабилизует dz2(alg)-орбиталь. Точно так же тетрагональное сжатие должно поднимать dz2 и понижать dx2-y2- Ян и Теллер первыми отметили, что такое искажение нелинейной молекулы происходит в том случае, когда оно сопровождается понижением энергии. Таким образом предполагается, что ян-теллеровское искажение происходит всегда, если имеется орбитально вырожденное (Е или Т) состояние и если существует подходящее по симметрии колебание, позволяющее молекуле менять геометрию. Один неспаренный электрон на двукратно вырожденной паре е-орбиталей приводит к состоянию Е, а один или два неспаренных электрона на трехкратно вырожденных орбиталях t приводят к состоянию Т.
Отметим, что подобный вывод можно сделать относительно спин-орбитального взаимодействия. О существовании орбитального углового момента электрона говорит простая одноэлектронная схема. Для того чтобы у электрона был орбитальный угловой момент, он должен находиться на вырожденных орбиталях, что позволит ему свободно перемещаться с одной орбитали на другую и при этом вращаться вокруг оси. Рассмотрим, например, dxz- и </у2-орбитали металлоцена. Вырожденность этой пары орбиталей допускает вращение вокруг оси и существование углового момента. Все состояния Е и Т при этом характеризуются наличием спин-орбитального взаимодействия, если не считать состояний Е в точечных группах 0h и Td. В этих последних случаях состояния Е составлены из dx2-y2- и </г2-орбиталей, поэтому электрон не может вращаться вокруг оси.
Некоторые ученые, включая самого Теллера, утверждают, что эффект Яна—Теллера отсутствует, если состояние по какой-либо причине расщепляется. Другие считают, что сочетание ян-теллеровского искажения с другими факторами устраняет вырождение. В связи с последним утверждением возникает вопрос: если вырожденное состояние расщепляется, обусловлено ли это ян-теллеровским искажением, искажением, вызванным компонентами более низкой симметрии, или спин-орби-тальным взаимодействием? Поскольку часто величины перечисленных эффектов сравнимы (200—2000 см1), можно сказать, что расщепление обусловлено некоторой неопределенной комбинацией этих трех эффектов. Однако в состояниях Е ян-теллеровские искажения, как правило, больше, чем в состояниях Т, поэтому в последних обычно доминирует спин-орбитальное взаимодействие.
88
Глава 10
ПРИМЕНЕНИЕ
10.7. ОБЗОР ЭЛЕКТРОННЫХ СПЕКТРОВ
КОМПЛЕКСОВ «О„»
Электронные спектры комплексов переходных металлов можно интерпретировать с помощью теории кристаллического поля. При обсуждении комплексов «ОА» мы будем заниматься системами с локальной симметрией Oh, хотя симметрия всей молекулярной системы может быть и не такой. При описании типа расположения донорных атомов, непосредственно связанных с металлом, мы не будем строго придерживаться терминов симметрии и не будем учитывать остальные атомы лигандов. Естественно, такое допущение не всегда оправдано. В данном разделе мы рассмотрим, как интерпретировать и предсказывать электронные спектры и как оценить величины наблюдаемого (/-орбитального расщепления. Мы должны дать представление об эффективном методе координационной химии—использовании электронных спектров при решении структурных проблем. Все эти вопросы более подробно обсуждаются в ряде монографий, в которых ссылки на работы, содержащие спектры многих комплексов [1, 2, 4, 5, 9, 10, 12].
Напомним читателю, что правила отбора для электронных переходов обсуждаются в гл. 5. Здесь же мы применим эти правила к некоторым комплексам ионов переходных металлов. Прежде всего рассмотрим высокоспиновые октаэдрические комплексы Мп11, т. е. комплексы с ds -конфигурацией, где отсутствуют разрешенные по спину переходы d — d. Все эти переходы в данном случае запрещены как по мультиплет-ности, так и по правилу Лапорта. В отсутствие вибронного взаимодействия и переходов с переносом заряда комплексы Мп11 должны быть бесцветными. Ион гексааквамарганца(П) окрашен в бледно-розовый цвет, и все полосы поглощения в видимой области характеризуются очень низкой интенсивностью.
Тот факт, что переходы, разрешенные по мультиплетности, обычно дают широкие линии, в то время как переходы, запрещенные по мультиплетности,—узкие, может помочь отнесению полос в спектре. Разрешенные по мультиплетности переходы t2g -> ед приводят к возбужденному состоянию, в котором равновесное межъадерное расстояние между ионом металла и лигандом больше, чем в основном состоянии. При электронном переходе межъядерное расстояние меняться не должно (принцип Франка — Кондона), поэтому электронно возбужденные молекулы находится в колебательно возбужденных состояниях, в которых длины связей соответствуют основному состоянию. Взаимодействие возбужденного состояния с молекулами растворителя, находящимися не в первой координационной сфере, меняется, так как при образовании возбужденного состояния ближайшие молекулы растворителя удалены от иона металла на различные расстояния. Поскольку растворитель не может реорганизоваться за время перехода, данное возбужденное колебательное состояние различных молекул взаимодей
Электронная структура и спектры ионов переходных металлов
89
ствует с молекулами растворителя, находящимися на различных расстояниях от него. Изменение энергии сольватации влияет на энергию колебательно возбужденного состояния, и в результате появляются широкие полосы поглощения.
В некоторых переходах, запрещенных по спину, перестройка осуществляется на данном уровне. Например, в комплексах Сгш переходы происходят с основного состояния, в котором на /21/-орбитали находятся три неспаренных электрона, на возбужденное состояние, в котором на Г29-орбитали находятся два спаренных и один неспаренный электрон. В этих переходах, запрещенных по мультиплетности, различие в равновесных межъядерных расстояниях в основном и возбужденном электронных состояниях часто невелико. Результатом этих переходов на низкоэнергетический колебательный уровень возбужденного состояния, кривая потенциальной энергии которого аналогична по форме и по равновесному межъядерному расстоянию кривой потенциальной энергии основного состояния, являются узкие линии.
Как уже говорилось ранее, в тетраэдрической молекуле отсутствует центр симметрии, поэтому при d— d-переходах комплексы Td часто дают несколько более интенсивные линии (е = 100—1000), чем октаэдрические комплексы.
d1- и ^-комплексы
Проследить связь между окраской комплекса иона переходного металла, обусловленной d— d-переходом, и Dq проще всего на примере d1-комплекса, например комплекса Tiln в октаэдрическом поле. Основное состояние свободного иона описывается термом 2D, и, как указывалось ранее, вырожденные d-уровни расщепляются октаэдрическим полем на совокупность из трехкратно вырожденного 2Т29-состояния и двукратно вырожденного 2Е9-состояния. Расщепление составляет 10 Dq (рис. 10.7). С увеличением Dq возрастает и энергия ДЕ (а следовательно, и частота) перехода. Тангенс угла наклона линий Т2д и Ед составляет соответственно — 4Dq и + 6Dq. Величину Д (см-1) можно получить непосредственно из частоты полосы поглощения. Например, максимум полосы поглощения Ti(H2O)|+ лежит при 5000 А (20000 см ' ). Величина Д для воды, связанной с Ti3 + , составляет около 20000 см 1 (Dq равно 2000 см ’). Поскольку этот переход происходит с поглощением желто-зеленой компоненты видимого света, пропущенный свет пурпурный (голубой + + красный). При изменении лиганда меняется и окраска комплекса. Цвет раствора дополнителен к поглощенному (или поглощенным) цвету, поскольку окраску определяют линии пропускания. Визуально наблюдаемые линии следует относить достаточно осторожно, поскольку часто путают, например, фиолетовый и пурпурный цвета.
Диаграмму энергетических уровней для d’-комплекса в октаэдрическом поле получают обращением картины для d1-комплекса (см. рис. 10.8). Инверсия применима, поскольку основное состояние d’-конфигу-рации дважды вырождено [t2«eg может быть t$g (dx2_,.d2(d,i)1 или
90
Глава 10
t2g(dx? _ z)1 (dz2)2], а возбужденное состояние — трижды вырождено [1|яе4 может быть (dxy)2 (dyz)2 (dxz)1 (e9)4, или (dxy)2 (d^)1 (dXz)2 (e9)4, или (rfxy)1 (dyz)2 (dxz)2 (e9)4). Таким образом, мы имеем дело с переходом 2Е -> -> 2 Т2 В действительности электронный переход вызывает переход положительной дырки с уровня ед основного состояния на уровень t2g возбужденного состояния, и соответствующая энергетическая диаграмма
Рис. 10.7. Расщепление уровней dl-конфигурации в поле Ок. В скобках заключен вырожденный набор (/-орбита-лей.
получается в результате обращения диаграммы для d1-комплекса. Для того чтобы сохранить центр тяжести, тангенсами углов наклона линий на рис. 10.8 должны быть — 6Dq для Е и + 4Dq для Т2-
Результаты, представленные выше, часто суммируют с помощью диаграммы Оргела, как это показано на рис. 10.9. Для d1 -комплекса те
Рис. 10.8. Расщепление двух d-уровней в <19-комплексе полем Ок.
Рис. 10.9. Диаграмма Оргела для высокоспиновых d -, d4-, d6- и d9-KOMnneK-сов.
траэдрическое расщепление приводит к противоположному результату, чем октаэдрическое расщепление, поэтому тетраэдрические d1 и октаэдрические d9 комплексы характеризуются аналогичными диаграммами Оргела (рис. 10.9). Расщепление состояний в зависимости от Dq для октаэдрических комплексов с электронными конфигурациями d1 и d6
Электронная структура и спектры ионов переходных металлов
91
и для тетраэдрических комплексов с электронными конфигурациями d^ и d9 приведено в правой части рис. 10.9. Только одна полоса в спектре обусловлена d — d-переходами, ее относят к переходу 2T2t/ -> 2Е9. Левая сторона диаграммы Оргела применима как к октаэдрическим d4- и d9-, так и к тетраэдрическим d1- и d6-KOMn;ieKcaM. Единственный наблюдаемый d — d-переход отнесен к 2Е -» 2Т2.
d2-, d1-, d3- и d8-кoнфигypaции
Ранее было показано, что триплетные состояния газообразного d2-иона — это 3F и 3Р. Более того, мы показали, что октаэдрическое поле расщепляет термы 3F на триплетные состояния 3Т19, 3Т19 и 3Л29; состояние 3Л2а возникает из е2, состояния 3Tlt/— из t29 и (хотя ранее мы им не занимались) другое состояние 3Т19 — из t2geg. Для того чтобы понять электронный спектр, достаточно рассмотреть только триплетные состояния, поскольку основное состояние — триплет. Можно предложить следующее упрощенное описание с использованием одноэлектронной орбитали. Для основного состояния октаэдрического d2-KOMnneKca возможны следующие вырожденные конфигурации: dxy, dxz, d°z; dxy, dxz, dyz; dxy, d°z, dxy. Основное состояние орбитально трехкратно вырождено, и для его описания используют обозначение 3Tlt/ (F). (F) указывает на то, что состояние восходит к F-терму газообразного иона. В дополнение к основному состоянию 3Т19(Е) существует также возбужденное состояние, соответствующее конфигурации, для которой на уровне t2g спарены два электрона. Переходы на эти состояния запрещены по мультиплетности, но иногда наблюдаются слабые линии, приписываемые этим переходам.
Далее мы рассмотрим возбужденное триплетное состояние t2geg. Если электрон возбужден с dxz- или d^-орбитали, так что оставшийся электрон находится на d^-орбитали, возбужденный электрон испытывает меньшее электрон-электронное отталкивание со стороны электрона и d^-орбитали, если он размещен на dz2-op6HTann. Орбиталь dx2_y2 менее выгодна, поскольку занимающий ее электрон находится ближе к электрону, оставшемуся на d^-орбитали (конфигурации А на рис. 10.10). Аналогично, если электрон возбужден с d^-орбитали, его положение на dx2-^-орбитали более устойчиво. Оставшийся электрон может находиться либо на dxz-, либо на dyz-op&nTaswi, что дает конфигурации Бк В. Эта совокупность (рис. 10.10, А — В) приводит к состоянию 3T2s, которое орбитально трехкратно вырождено и имеет спиновую мультиплетность, равную трем. Конфигурации (dxy, dx2 . у-У, dxz, dz2; dyz, dz2) характеризуются более высокой энергией и также дают орбитально трехкратно вырожденное состояние 3Т19(Р). Другие возможные конфигурации, соответствующие t2selg, включают обращение одного из электронных спинов с образованием состояний с синглетной мультиплет-ностью. Переходы в эти состояния из основного триплетного состояния запрещены по мультиплетности. Наконец, двухэлектронный переход,
92
Глава 10
дающий возбужденное состояние е2 или d^, d*2_v2, приводит к однократно вырожденному состоянию 3Д29. Полезно указать, как эти состояния связаны с состояниями газообразного иона. В разделе, посвященном обозначениям термов (разд. 10.1), показано, что основное
Рис. 10.10. Возможные электронные конфигурации для
dx2-yi dzt
dxy dxz dyz В
состояние газообразного иона У1П(г/2-ион) — 3F. Поле лигандов в комплексе снимает семикратное орбитальное вырождение этого состояния (т. е. Ml = 3, 2, 1, 0, — 1, — 2, — 3) до двух трехкратно вырожденных состояний, 3Tlg(F) и 3Т29, и одного невырожденного состояния, 3Д29. Это показано на диаграмме Оргела (рис. 10.11) для ^-комплекса Oh.
Для нулевого Dq (газообразный ион) существуют только два триплетных состояния: 3F и 3Р. С увеличением Dq 3F расщепляется на 3T19(F), 3Т29 и Зл29. Вырожденность состояния 3Р полем лигандов не
Рис. 10.11. Диаграмма Оргела для высокоспиновых d2-, d3-, d1- и ds-комплексов.
снимается, и это состояние в октаэдрической симметрии превращается в триплетное состояние 3Т19(Р), где (Р) указывает, что соответствующее состояние возникает из состояния 3Р газообразного иона. Зависимость энергии указанных состояний от Dq дана как на диаграммах Оргела, так и Танабе — Сугано [13] (см. приложение IV). Использование диаграмм Оргела для прогнозирования спектров и отнесения линий мы покажем на примере комплексов V111 и Ni11.
В комплексе V111 могут происходить три перехода, затрагивающие показанные на рис. 10.11 состояния: 3T19(F)-> 3Т29, 3Tig(F) -> 3Т19(Р) и
Электронная структура и спектры ионов переходных металлов 93
Таблица 10.6
Максимумы пот лощении (см-1) октаэдрических комплексов Ni11
Лиганд
3Л29-»3Т29 3.12„ > ’Т,„(Г) \429-»3Т19(Р)
Н2О 8 500 15400 26000
NH3 10 500 17 500 28 200
(CH3)2so 7730 12970 24040
HC(O)N(CH3)2 8 500 13 605 (14900) 25 000
CH3C(O)N(CH3)2
7 575 12740 (14285) 23 810
3T19(F)-> 3Л29. Переход к 3А2д в V,n двухэлектронный; он относительно маловероятен, и поэтому интенсивность его низка. Экспериментально он не наблюдается. Спектр, полученный для октаэдрических комплексов V111, состоит из двух полос поглощения, отнесенных к 3T19(F) -> 3T29(F) и 3T19(F) -> 3Т19(Р). В V(H2O)g+ они расположены при 17000 и 24000 см 1 соответственно.
Для октаэдрических комплексов никеля (II) диаграмма Оргела (левая сторона рис. 10.11, ds) демонстрирует три ожидаемых перехода: 3Л2а -> -> 3Т2д, 3A2f) -> 3 T19(F) и 3Л29 -> 3Т19(Р). (Аналогичные результаты полу-
p. СМ'1
Рис. 10.12. Молярный коэффициент поглощения е для некоторых комплексов никеля(П) в растворе CH3NO2: Ni(NH3)6(СЮ4), (/). Ni[HC(O)N(CH3),]6(ClO4)2 (2) и .M[CH3C(<J)N(CH3)2J6(CKJ4)2 (J).
94
Глава 10
чены при использовании диаграммы Танабе — Сугано, см. приложение IV.) Экспериментальные максимумы поглощения, соответствующие этим переходам, приведены в табл. 10.6 для октаэдрических комплексов Ni11. (Числа в скобках соответствуют плечам у основной линии.) Спектры октаэдрических комплексов NH3, HC(O)N(CH3)2 и CH3C(O)N(CH3)2 приведены [14] на рис. 10.12. Эти комплексы окрашены соответственно в пурпурный, зеленый и желтый цвета.
10.8. РАСЧЕТ Dq и р ДЛЯ «Ой»-КОМПЛЕКСОВ Nin
Информация, содержащаяся в диаграммах Оргела, более точно выражается рядом уравнений (они выведены в разд. 10.3), которые связывают энергии различных состояний с величиной Dq лиганда. Для Ni11 в октаэдрическом поле энергии (Е) состояний относительно сферического поля выражаются уравнениями (10.10)—(10.12).
Для 3Т2д: е= — 2Dq (10.10)
Для 3А29: E=—\2Dq (10.11)
Для 3T19(F) и 3Г19(Р): [6Z)qp — 16(Z)q)2]+ [ —6Z)q — р]Е + Е2 = 0(10.12) где р — энергия состояния 3Р. Последнее уравнение имеет два корня, соответствующих энергиям состояний 3Tlg(F) и 3Т19(Р).
Из уравнений (10.10) и (10.11) видно, что энергии как 3Т2д, так и 3А29 — линейные функции Dq. Для любого лиганда, который образует с ионом никеля октаэдрический бесспиновый комплекс, разность энергий между состояниями 3Т2д и 3Л29 комплекса равна 10Z)q. Из диаграмм Оргела и Танабе — Сугано следует, что 3Л2<Г—► 3T2t/— переход низшей энергии. Поскольку этот переход служит непосредственной мерой различия в энергиях указанных состояний, А (или 10Z)q) можно приравнять к энергии перехода, т. е. к частоте соответствующей ему полосы (см”1).
Уравнение (10.12) можно решить для энергий других состояний. Однако приведенные выше уравнения выведены при допущении, что лиганды— это точечные заряды или точечные диполи и что связь металл— лиганд нековалентна. Если это допущение справедливо, то определенную таким образом величину Dq можно подставить в уравнение (10.12) и рассчитать, исходя из характеристик атомного спектра газообразного иона, энергию 3Р [10] и энергию других двух уровней в комплексе. Частоты ожидаемых спектральных переходов определяют из полос, соответствующих разностям между энергиями уровней 3Tig(F) — 3А29 и 3Т1д(Р) — 3А2д. Экспериментальные энергии, полученные из спектров, почти всегда ниже, чем величины, рассчитанные таким путем. Отклонение приписывают ковалентности.
Ковалентность снижает положительный заряд на ионе металла в результате индуктивного эффекта лигандов. При пониженном положительном заряде радиальная протяженность d-орбиталей возрастает, при этом электрон-электронные отталкивания ослабевают и энергия состояния ~Р снижается. В теории кристаллического поля ковалентность не
Электронная структура и спектры ионов переходных металлов 95
принимается во внимание, а в теорию поля лигандов, как мы увидим далее, она включается с помощью введения дополнительного параметра.
Разность энергий между состояниями 3Р и 3F в комплексе снижается относительно разности для газообразного иона под влиянием ковалентности, и в результате разность энергий для газообразного иона нельзя использовать в качестве р [в уравнении (10.12)]. Лучше оценивать р для каждого комплекса экспериментально. Уравнение (10.12) можно использовать при таких расчетах, взяв величину Dq для перехода 3А29 -> 3Т2д и экспериментальную энергию ДЕ для перехо'да 3Л2а -> 3Т19(Р). Единственная неизвестная величина, оставшаяся в уравнении (10.12),— это р. Снижение 3Р служит, помимо всего прочего, мерой ковалентности. Этот эффект носит название нефелауксетииеского, иногда его выражают с помощью параметра (3°, показывающего процент снижения энергии состояния 3Р в комплексе по сравнению с энергией состояния 3Р в свободном газообразном ионе [13]. Параметр 0° рассчитывают по уравнению
Р° = [(В-В')/В]100, (10.13)
где В—рассмотренный ранее параметр Рака для свободного газообразного иона, а В' — тот же самый параметр для комплекса. Следует отметить, что р в уравнении (10.12) пропорциональна В. Для комплекса никеля (II) энергию 3Р можно наряду с Dq подставить в уравнение (10.12) и рассчитать другой корень. Разность между этой рассчитанной энергией и энергией состояния 3Л29 дает частоту средней полосы [3А29 3E19(F)]. Со-
ответствие рассчитанной и экспериментальной величин для этой полосы — хороший довод в пользу симметрии Ой. Проведенное выше обсуждение станет более понятным, если обратиться к приложению V, где приведен расчет Dq, 0° и частоты перехода 3A2s -> 3Tlg(F) для Ni[(CH3)2SO]6 (С1О4)2.
Чаще всего 0° заменяют на 0, который определяется как
Р = Д/В. (10.14)
Если записать уравнение (10.13) как 0° = (1 — 0) • 100, то соотношение между этими параметрами становится явным.
Спектральные данные, полученные для многих других ионов, использовать для определения Dq и 0 не так просто, поскольку возникают различные осложнения, обусловленные спин-орбитальным взаимодействием. Влияние этого взаимодействия продемонстрировано на рис. 10.13 на примере ^-иона. Вследствие спин-орбитального взаимодействия (с. о.) трехкратно вырожденное состояние Т2д расщепляется, энергия основного состояния снижается и степень его снижения зависит от величины взаимодействия. Если энергия основного состояния снижается в результате спин-орбитального взаимодействия, энергии всех полос в спектре получают вклад, обусловленный этим снижением Ас 0 . Если вклад в полную энергию, обусловленный Дс 0 , нельзя определить, рас-
96
Глава 10
чет Л и Р неточен. Спин-орбитальное взаимодействие в возбужденном состоянии — проблема не столь сложная, поскольку переходы происходят часто к обоим расщепленным уровням и энергии можно усреднить. Если основное состояние расщепляется, то заселяется только более низкий уровень. Таким образом, точные значения Dq и Р без поправок на спин-орбитальное взаимодействие можно получить только для ионов
&ист ^набл
Рис. 10.13. Вклад спин-орбитального взаимодействия в А.
А—расщепление (/-уровней в отсутствие спин-орбитального взаимодействия; Б — расщепление уровня под действием спин-орбитального взаимодействия.
с основным состоянием А или Е (например, Ni2 + ). Ян-теллеровские искажения, влияющие на энергии уровней, также вызывают осложнения.
Ni11, Мп11 (слабое поле), Со111 (сильное поле) и Ст111 образуют ряд октаэдрических комплексов, спектры которых позволяют точно рассчитать Dq и Р без значительных осложнений, создаваемых спин-орби-тальным взаимодействием и ян-теллеровскими искажениями. В комплексах Tinl влияние этих эффектов невелико. В тетраэдрических комплексах величина расщепления под действием спин-орбитальных взаимодействий в большей степени сближается с величиной расщепления под действием кристаллического поля (Dq', расщепление в тетраэдрическом поле составляет около 4/9Dq). В результате спин-орбитальное взаимодействие дает заметный вклад в энергии наблюдаемых полос. В работе [14] описана процедура расчета Dq и Р для тетраэдрического комплекса Со11. Пример такого расчета дан в приложении V.
Как ст-, так и л-связывание лиганда с ионом металла дают вклад в величину Dq. При л-связывании затрагиваются 129-орбитали металла, поскольку они имеют необходимую симметрию и нужную направленность. Если в образовании л-связей участвуют незанятые орбитали лиганда (например, d-орбиталь (C2H5)2S или р-орбиталь CN“), Dq больше, чем' в отсутствие этого эффекта. Если л-связывание происходит между заполненными орбиталями лиганда и заполненными орбиталями t2g (как, например, у ОН- и Со111), конечным результатом этого взаимодействия является разрыхление и Dq снижается. Эти эффекты продемонстрированы на рис. 10.14. В первом случае (рис. 10.14,А) показано, что d-электроны tlg взаимодействуют со свободными орбиталями лиганда, в результате чего энергия t2 понижается и повышается энергия л-орби-
Электронная структура и спектры ионов переходных металлов 97
талей лиганда в комплексе. Свободная к *-орбиталь лиганда может участвовать в этом виде взаимодействия. Поскольку энергия t2g понижается, а энергия ед не меняется (е9-орбитали направлены на ст-электронные
Б Комплекс Комплекс л-Орбиталь в отсутствие с л-связыванием лиганда л-связывания
Рис. 10.14. Влияние л-связывания на энергию (^-орбитали и на Dq.
А— заполненные орбитали металла, свободные орбитали лигаида; Б — заполненные орбитали металла, заполненные орбитали лиганда.
пары лигандов), Dq возрастет. Во втором случае (рис. 10.14,Б) заполненные л-орбитали лиганда взаимодействуют с более высоко энергетическими заполненными л-орбиталями металла, понижая Dq и повышая энергию t2g.
Полезно связать энергии наблюдаемых d—d-переходов с энергетическими уровнями, используемыми при описании октаэдрических комплексов с помощью метода молекулярных орбиталей (МО). На рис. 10.15 показана диаграмма МО для комплекса Oh (л-связывание не учитывается). Разность энергий Т2д и Ед составляет 10Dq. По мере увеличения прочности ст-связи металл — лиганд Ед понижается, а Ед увеличивается на ту же самую величину, в то время как Dq возрастает. Если электроны металла Т2д образуют л-связи со свободными р- или d-орби-талями лиганда, энергия уровня Т2д в комплексе снижается, a Dq увеличивается. Электрон-электронные отталкивания электронов Т2д и несвязывающих электронов металла повышают энергию совокупности Т2д и понижают А. Изложенные выше соображения были использованы при интерпретации спектров ацетилацетонатов некоторых переходных металлов [15, 16].
7-574
98
Глава 10
Величина Dq определяется многими факторами: взаимодействиями, обусловленными электростатическим возмущением, отталкиванием электронов металла и лигандов, ст-связью металл — лиганд, тг-связью металла с лигандом и тг-связью лиганда с металлом. Более подробно все эти вопросы рассматриваются в монографиях [9, 10] и статье [18].
Рис. 10.15. Описание октаэдрического комплекса методом МО (л-связывание во внимание не принимается и л-электроны не учитываются).
Расчет значений Dq и 0 дает важные сведения о взаимодействии иона металла с лигандом. Так, например, установлено, что шестикоординационные комплексы никеля с амидами типа R(CON(R2)R3 характеризуются меньшими Dq и 0, если Rx и R2 — алкильные группы, а не атомы водорода. В то же время известно, что по отношению к фенолу и иоду донорная способность этих амидов увеличивается с ростом числа алкильных групп. Поэтому было высказано предположение, что между соседними координированными молекулами амида [14] в комплексах металлов возможны пространственные взаимодействия. Исследование комплексов никеля (II) некоторых первичных алкиламинов показало, что если даже вода замещает в комплексах амины, они взаимодействуют с никелем более сильно, чем вода, и почти так же сильно, как аммиак [19]. Авторы работы [20] сообщили также о высоких значениях Dq для никелевых комплексов этиленимина [20]. При объяснении причин неустойчивости алкиламинных комплексов в воде учитывалась энергия сольватации [19].
Электронная структура и спектры ионов переходных металлов 99
Величина 10Z><gr для различных ионов металлов обычно возрастает в ряду
Мп2 + < Ni2 + < Со2 + Fe2 + < V2 < Fe3 + < Сг3 + < V3 + < Со3 + < Мп4 + < Mo3 + < Rh3 + < Pd4 + < 1г3 + < Re4 + < Pt4 + .
Для типичных лигандов характерен так называемый спектрохимический ряд:
I “ < Вт “ < — SCN < F “ < мочевина < ОН “ < СН3СС>7 < СгО^- < Н2О < — NCS “ < глицин < C5H5N ~ NH3 < этилендиамин < SOj- < о-фенантролин < NO2 < CN ~
Йоргенсен [4, 5] сообщил о примечательных параметрах, которые позволяют предсказать величины 10Dq и Р для различных комплексов
Таблица 10.7
Эмпирические параметры для прогнозирования IOD4 и В с по-
мощью уравнения (10.15) и (10.16)а
Лиганд f h Ион металла g 103, cm 1 ' k
6F" 0,9 0,8 V(II) 12,3 0,08
6Н2О 1.00 1 0 Cr(III) 17.4 0,21
6 мочевина 0,91 1,2 Mn(II) 8.0 0.07
6NH, 1,25 1,4 Mn(lV) 23 0,5
Зеп 1.28 1.5 Fe(III) 14.0 0.24
Зох2 0,98 1,5 Co(III) 19,0 0,35
6СГ 0,80 2,0 Ni(II) 8,9 0,12
6CN 1,7 2,0 Mo(III) 24 0,15
6Вг 0.76 2,3 Rh(III) 27 0,30
3dtp 0,86 2,8 Re(IV) 35 0.2
C5H5N 1,25 — Ir(III) Pt(IV) 32 36 0,3 0,5
а Jorgensen С. К., Absorption Spectra and Chemical Bonding in Complexes, Per-gamon Press, New York. 1962.
ионов переходных металлов. Подставив в уравнения (10.15) и (10.16) эмпирические параметры, представленные в табл. 10.7, мы получим величины lODg и В для комплекса.
10Z)g =/д(см-1 • 10-3), (10.15)
B=B0(l-hk), (10.16)
Во — параметр межэлектронного отталкивания для свободного иона.
Т
100
Глава 10
10.9. ВЛИЯНИЕ ИСКАЖЕНИЙ НА ЭНЕРГЕТИЧЕСКИЕ
УРОВНИ d-ОРБИТАЛЕЙ
Поскольку октаэдрическое, квадратно-плоскостное и тетраэдрическое кристаллические поля вызывают различные расщепления пяти d-орбиталей, значительное влияние на d — d-переходы оказывает геометрия комплексов ионов металлов. Спектральные данные для этих переходов должны давать информацию о структуре комплексов. В этом разделе мы прежде всего обсудим вопрос о том, как структура влияет на энергии различных состояний иона металла, и затем используем эти данные для определения структур различных комплексов.
Е У
2О / ----\ «г \ рг d*y
dxz
Газообразный Кубическое Тетрагональное
Рис. 10.16. Орбитальное расщепление d1-комплексов в кубическом и тетрагональном полях.
поле поле
Структуру шестикоординационных комплексов можно классифицировать как кубическую, аксиальную или ромбическую, если эквивалентность лигандов вдоль осей х, у и z характеризуется соответственно соотношениями х = у = z, х = у / z и х / у Г z. Тетрагональное и тригональное искажения, т. е. растяжение или сжатие вдоль оси третьего порядка, приводят к обычным аксиальным структурам. На рис. 10.16 изображено расщепление термов ^-комплекса. Поскольку в таком комплексе d—d-электроные отталкивания отсутствуют, состояния можно скоррелировать с d-орбиталями, что и показано на рисунке. Чтобы в тетрагональном комплексе щранс-Т1А4В2+ расщепление происходило так, как это показано на рис. 10.16, лиганд А должен занимать более высокое положение в спектрохимическом ряду, чем лиганд В. Отталкивания между электронами лиганда и электронами металла меньше, если электроны находятся на орбиталях, направленных прямо на лиганды В, лежащие на оси z. В результате энергия dг2-opбитaли меньше, чем dx2_у2-орбитали, а энергия dxz и d^-орбитали меньше, чем d^-орбитали, и в спектре тетрагонального комплекса должно наблюдаться больше линий, чем в спектре октаэдрического комплекса.
На рис. 10.17 показаны энергии и расщепления для различных состояний спин-спаренного комплекса Сош. Поскольку в этом ионе более одного d-электрона, мы должны рассматривать состояния, а не орбитали. Возбужденное состояние 1Т1д октаэдрического комплекса расщепляется на состояния 1А2д и 1Ед в тетрагональном поле, в то время как 1Т2д расщепляется на 1Ед и 1В2д. На этом же рисунке показано также
Электронная структура и спектры ионов переходных металлов 101
расщепление в ромбическом поле. Гидратированный трис-(глицинато)-кобальт(Ш) существует в виде двух изомеров: фиолетового (а) и красного (Р). Один изомер должен быть кубическим (т. е. х = у = z), а другой— ромбическим. В спектре Р-изомера наблюдаются две полосы [18]. 7-Изомер также дает две полосы, одна из которых асимметрична и должна включать две или более неразрешенные полосы поглощения. Таким образом, а-изомер должен быть ромбическим изомером, а Р-изомер кубическим.
Рис. 10.17. Расщепление различных состояний комплекса кобальта(Ш) в кубическом, тетрагональном и ромбическом полях.
Afg Uy
Кубическое Тетрагональное Ромбическое поле поле поле
Как цис-, так и транс-СоА^В^-изомеры тетрагональны. Однако различие в энергиях между состояниями 1А2д и гЕд (ДЕ, на рис. 10.17) в транс-комплексах обычно вдвое больше [21, 22] [АЕ( (цис) = — — СДа—Ав) и АЕ( (транс) = 2С(Ал — Ав), где С — постоянная, обычно <1, а А^ и Ав—расщепление в кристаллическом поле лигандов А и В (т. е. положение этих лигандов в спектрохимическом ряду), а знак минус объясняет тот факт, что энергии 1 Ед и 1А2д меняются местами в цис- и транс-комплексах]. Обычно, если Ал и Ав заметно различаются, расщепление гА2д и гЕд приводит для 1А1д->' 1гГ1д к дублету в спектре транс-комплекса, в то время как в спектре цис-комплекса эта полоса просто уширяется [22]. Установлено также, что цис-изомеры часто характеризуются большей величиной коэффициента поглощения для d —d-переходов, чем транс-изомеры. Типичные спектры таких комплексов приведены на рис. 10.18. Если Ал и Ав имеют близкие величины, указанным критерием пользоваться нельзя. Ультрафиолетовую линию переноса заряда можно также использовать для того, чтобы различить цис-и транс-комплексы кобальта (III), поскольку частота полосы цис-комплекса обычно выше. Бензоилацетонаты Со1П и Сгш служат примером таких комплексов, в которых Ал и Ав почти равны, а транс-комплекс характеризуется большим £ [24].
Приняв некоторые допущения относительно расстояния между металлом и лигандом, дипольного момента лиганда и эффективного заряда на ядре никеля, можно рассчитать [25] изменение в энергии для различных состояний, происходящее по мере того, как расположение лигандов меняется от Oh до D4h. Это изменение соответствует удлине-
102
Глава 10
нию связи металл — лиганд вдоль dz2 до бесконечности. На рис. 10.19 показано изменение энергии для различных уровней в зависимости от искажения. На оси абсцисс отложены проценты, соответствующие постепенному ослаблению двух транс-связей металл — лиганд (т. е. 100% ослабления характеризует квадратно-плоскостной комплекс). Таким
в
А
Б
100
о
траке- Со(еп)гГг+
10000 15000 20000 25000 30000 35000
СМ'1
Рис. 10.18. Спектры водных растворов октаэдрического трис-(элилендиамин)ко-бальта(Ш) (Л), t/uc-дифторо-комплекса (Б) и транс-дифторо-комплекса (В).
образом, легко увидеть, почему спектр искаженного (тетрагонально или по типу плоского квадрата) комплекса никеля должен отличаться от спектра правильного октаэдрического комплекса.
Рис. 10.19 демонстрирует, как по мере увеличения степени тетрагонального искажения спектральные характеристики монотонно меняются и, наконец, спектры сильно искаженных тетрагональных комплексов напоминают спектры квадратно-плоскостных комплексов. При больших искажениях мультиплетность состояния низшей энергии для Ni(II) становится синглетной и образуется диамагнитный комплекс. Диамагнитные тетрагональные или квадратно-плоскостные комплексы дают спектры с высокоинтенсивными полосами поглощения (е = 100—350) с максимумами при 14000—18000 см-1. В спектре может быть одна, две или три полосы [26, 27] и отнесение полос становится затруднительным. Однако исходя из спектральных данных или данных по магнетизму, можно легко отличить квадратно-плоскостные или сильно искаженные тетрагональные комплексы никеля(П) от его почти октаэдрических или тетраэдрических комплексов.
Энергии и свойства различных уровней меняются как при искажении от симметрии Oh и D4fl, так и при искажении от 04(1 (плоский комплекс) до D2d и далее до Td (рис. 10.20).
Электронная структура и спектры ионов переходных металлов 103
На рис. 10.21 представлены спектры комплексов Oh, Dih, D2d и Td [25, 28, 29, 30]. Переходы 7]-комплекса никеля NiCI] отличаются большими величинами е, поскольку у комплекса отсутствует центр симметрии.
Рис. 10.19. Влияние тетрагонального искажения на энергетические уровни комплекса никеля (II) [Furlani С., Sartori G., J. Inorg. Nucl. Chem., 8, 126 (1958)].
В этом случае при описании комплекса методом МО d- и р-орбитали могут смешиваться. Вклад р-орбиталей основного и возбужденного состояний сообщает в некоторой степени разрешенный характер d -> -» р-переходу, и интенсивность его увеличивается. Смешивание в нецентросимметричных молекулярных орбиталях лигандов также приводит к увеличению интенсивности полос. Поэтому, как можно видеть из рис. 10.21, где представлена зависимость е/5 от X, для различных структур получаются различные спектры. Ожидается, что в спектре комплекса Td (см. рис. 10.20) будут наблюдаться три полосы vn v2 и v3, соответствующие трем спин-разрешенным переходам: 37](F) -> 3Т2, vt; 37](F) -> 3Л2, v2; 3T[(F) -» 37\(Р), v3 (см. рис. 10.21). Полоса Vj лежит в интервале между 3000 и 5000 см-1, часто ее маскирует полоса поглощения либо органической части молекулы, либо растворителя. Ее часто наблюдают для Ni2+ в силикатных стеклах и в NiCl^-. Полоса v2 лежит в интервале 6500—10000 см-1 и имеет заметный коэффициент поглощения (е = = 15 — 50). Полосы v3 обнаруживают в видимом диапазоне (12000—17000 см-1), они характеризуются заметным поглощением (е = = 100—200).
t
Qih---------> Pit! ------------------> Td
Рис. 10.20. Энергетические уровни для комплексов Td, Dih и D2d [Furlani C., Sartori G., J. Inorg. Nucl. Chem., 8, 126 (1958)].
A, MM
Рис. 10.21. Электронные спектры поглощения некоторых комплексов никеля. 1 -Ni(Ph3PO)4(ClO4)2 в CH3NO2 (D2j); 2-Ni[(CH3)2SO]6(ClO4)2 в (CH3)2SO (Ой); 3— NiCl4 в CH3NO2 (7^); 4 — №(П)(диметилглиоксимат)2 в СНС13 (D4h).
Кривая 3— зависимость е/5 от А..
Электронная структура и спектры ионов переходных металлов 105
Предполагается [28], что комплекс Ni[OP(C6H5)3]4(ClO4)2 имеет конфигурацию D2d. Полосы поглощения лежат при 24300, 14 800 и 13 100 см _ \ соответствующие величины е составляют приблизительно 24, 8 и 9.
10.10. ДАННЫЕ О СТРУКТУРЕ,
ПОЛУЧАЕМЫЕ ИЗ ЭЛЕКТРОННЫХ СПЕКТРОВ
Из электронных спектров часто можно без труда получить достоверную информацию о расположении лигандов в комплексах переходных металлов. Тетраэдрические комплексы обычно легко отличить от шестикоординационных уже по интенсивности полос. Спектры комплексов никеля(П) и кобальта(П) дают особенно много информации. Комплекс Ni{OP[N(CH3)2]3}4Cl2 может иметь тетраэдрическую D2d, квадратноплоскостную тетрагональную или другую искаженную октаэдрическую геометрию. Сходство электронных спектров этого комплекса и NiCl4-(см. рис. 10.21) говорит [30] о том, что №{ОР[М(СН3)2]3}4С12-первый синтезированный катионный тетраэдрический комплекс никеля(П). Вероятность такой структуры подтверждает также сходство дифракционных картин, наблюдаемых при отражении рентгеновских лучей от поликристаллических образцов комплексов никеля(П) и цинка(П). Предполагается, что комплекс цинка(П) с конфигурацией 3d10 тетраэдрический. Диаграммы Оргела или Танабе — Сугано для d8—комплекса Td выглядят так же, как и диаграмма для октаэдрического комплекса кобальта (II) (OfcJ7) при более низкой величине Dq. Поэтому высокоэнергетическая полоса в видимой области приписывается переходу 4T1(F) -»4Т1(Р), а низкоэнергетическая полоса — переходу 4T1(F) -> 4А2. Обычно наблюдаемое расщепление видимой полосы относят к спин-орбитальному взаимодействию, которое устраняет вырожденность состояния 4Т1(Р).
Авторы работы [31] показали, что электронный спектр никеля в другом комплексе — Ni(NO3)4“—характерен для шестикоординационного комплекса, а некоторые из нитратных групп могут быть бидентатными. Во многих случаях цвет комплекса иона переходного металла — плохой индикатор его структуры. Октаэдрические комплексы никеля(П) обычно дают три полосы поглощения в интервалах 8000—13 000, 15000—19 000 и 25000—29000 см-1. Точное положение полос зависит от параметров Аир. Коэффициенты поглощения, соответствующие этим полосам, обычно не превышают 20. Как указывалось в разделе, посвященном расчетам Dq, совпадение рассчитанной и экспериментальной найденной частот средней полосы рассматривалось как доказательство существования комплекса «Ол».
Бесспиновые тетрагональные комплексы никеля(П), в которых два лиганда, занимающие либо цис-, либо транс-положения, отличаются от четырех других, но имеют схожие величины Dq, дают спектры, очень похожие на спектры комплексов Oh. Например, если искажение в транскомплексе близко к нулю (см. рис. 10.17), ожидается типичный спектр
106
Глава 10
октаэдрического комплекса. Вообще коэффициенты поглощения у тетрагональных комплексов выше, чем у октаэдрических. Правило среднего окружения связывает положение максимумов поглощения в спектрах этих слегка искаженных тетрагональных комплексов с величинами Dq лигандов. Положение полосы определяется средней по всем лигандам окружения величиной Dq [4, 32].
Никель(П) образует большое число пятикоординационных комплексов [33]. Известны геометрические структуры, в основе которых лежат тригональная бипирамида и тетрагональная пирамида. Для многих комплексов характерно отклонение от указанной геометрии [34]. Циам-полини [35] подробно проанализировал электронные спектры этих комплексов, и читатель может обратиться к оригиналу. Часто, располагая лишь электронным спектром, трудно различить тетраэдрическую и некоторые пятикоординационные конфигурации.
Электронные спектры комплексов кобальта(П) во многих случаях могут дать ценную структурную информацию. Большинство шестикоординационных комплексов имеют высокоспиновую электронную конфигурацию. Диаграмма Оргела этих комплексов представлена на рис. 10.11. Основное их состояние — 4Tlg, и спин-орбитальное взаимодействие значительно. В комплексах этой группы теоретически допустимы три перехода; 4Tlg(F) -> 4T2g, 4Tig(F) -> 4Л2з и 4Tlg(F) -> 4Tlg(P). Переход 4T19(F) -> 4Л29 двухэлектронный и не неблюдается. Электронные спектры октаэдрического Co(H2O)g + и тетраэдрического СоС1]~ показаны на рис. 10.22. Полоса для октаэдрического комплекса при ~20 000 см-1 приписывается переходу 4F19(F) -> 4Т19(Р). Плечо появляется потому, что спин-орбитальное взаимодействие в возбужденном состоянии 4Т19(Р) снимает вырождение. Другая полоса поглощения— при 8350 см-1—приписывается переходу 4Tl^F) -> 4T2g.
Диаграмма энергетических уровней тетраэдрического комплекса Со(П) подобна аналогичной диаграмме Сг(1П). Все возможные комплексы должны быть высокоспиновыми (см. диаграммы Танабе — Сугано в приложении IV). Полоса поглощения при ~ 15000 см-1 приписана переходу 4Л2 -> 4Т1(Р'), а тонкая структура — спин-орбитальному взаимодействию состояния Т. Из-за существования спин-орбитального взаимодействия возникают также некоторые спин-переходы квартет—дублет. Другая показанная полоса отнесена к переходу 4Л2 -» 4F1(F). Предполагается, что ожидаемый переход 4Л2 -> 4Т2 характеризуется полосой в интервале 3000—4500 см-1; этот интервал не охватывается большинством спектрофотометров, работающих в видимой и УФ-областях, и часто перекрывается колебательными переходами лигандов (т. е. ИК-полосами). Синтезировано несколько пятикоординационных комплексов кобальта(П), их спектры опубликованы и интерпретированы [35а].
Комплексы [356] меди(П) характеризуются большим разнообразием геометрических структур, часто низкосимметричных. В спектрах их обычно наблюдается широкая полоса с максимумом поглощения при 15000 + 5000 см1, которая, как полагают, включает все ожидаемые
Электронная структура и спектры ионов переходных металлов 107
переходы. Возможно, однако, что переход высшей энергии происходит еще дальше в УФ-диапазоне и маскируется полосами переноса заряда. Таким образом, электронный спектр меди(П) мало ценен при установлении структуры. Положение полосы может грубо коррелировать с величиной поля лигандов связанных групп.
Рис. 10.22. Электронные спектры Со(Н2О)^ + (0,021 М Co(BF4)2 в Н2О) (Л) и СоС14“ (0,001 М СоС12 в ЮМ НС1) (Б). Штриховая линия дает разрешение двух полос, образующих наблюдаемый спектр.
Доступные спектральные данные позволяют сделать аналогичные выводы относительно структур других комплексов ионов переходных металлов. Левер [10] указывает на заметные различия между спектрами разных структур. Для расшифровки структур комплексов, наряду с электронными спектрами, используются ИК-спектры и данные магнитных методов [36]. Использование данных по магнетизму посвящена следующая глава.
Мы рассмотрели лишь несколько примеров, демонстрирующих применение спектроскопии в видимой, ультрафиолетовой и инфракрасной
108
Глава 10
областях с целью получения информации о структурах комплексов. При интерпретации спектров необходимо учитывать и число полос, и частоты, и коэффициенты поглощения. Чтобы быть уверенным в том, что изменения в спектре не обусловлены влиянием растворителя, необходимо сопоставлять спектры комплексов, снятых в растворе, со спектрами, снятыми для твердого вещества (в виде суспензии или спектра отражения). Подобные изменения могут вызывать перегруппировка лигандов, замена лиганда молекулами растворителя или увеличение координационного числа в результате сольвации.
Известны и другие примеры применения спектроскопии в видимой области для установления структуры. Так, величина Dq различна для групп — NO2 и — ONO. В результате различия в средней величине Dq комплекс [Co(NH3)5ONO2]2 + окрашен в красный цвет, а комплекс [Co(NH3)5NO2]2 +—в желтый. В статье [37] говорится об ограничениях, имеющихся при таком применении спектральной техники.
Использование электронных спектров для получения структурной информации прекрасно иллюстрируют результаты исследования электронной структуры иона ванадила [38]. При интерпретации спектра ва-надил-иона VO2 + полагают, что в связи V — О имеет место значительное л-связывание. Соединения, в которых, согласно данным рентгеноструктурного анализа, содержится группа VO2+, дают сходные электронные спектры переноса заряда и в твердом состоянии и в растворе. Поэтому можно предположить, что водные растворы этих комплексов содержат группы VO(H2O)2 + , а не V(H2O)g + . Протонирование VO2 + в принципе должно заметно влиять на спектр переноса заряда. Предполагается, что кислород не протонируется, поскольку его основность ослаблена из-за образования л-связи с ванадием. Полный расчет по методу МО для VO(H2O)2+ представлен в статье [38], там же дано отнесение полос в спектре водного раствора VOSO4-5H2O. Аналогичные исследования других окси-катионов также свидетельствуют о значительном тг-связывании металл — кислород [39] и помогают установлению электронной структуры этих частиц.
Электронный спектр приносит особую пользу при определении координационного числа и расположения лигандов в металлофермен-тах. Электронный спектр карбоангидразы, в которой цинк(П) заменен на кобальт(П), показывает, что ион металла находится в центре искаженного тетраэдра [40]. Однако, когда проводятся такие исследования, необходимо особенно тщательно убедиться, что структура фермента не меняется при замене металла. Если фермент при этом остается активным, то можно до некоторой степени быть уверенным, что структура его не изменилась. В указанном примере последующий рентгеноструктурный анализ подтвердил, что лиганды группируются вокруг цинка(Н) в виде искаженного тетраэдра. Проводя интерпертацию видимого спектра эритрокупреина—белка, содержащего медь(П), авторы работы [40] пришли к выводу, что в данной системе координируются по крайней мере четыре азотсодержащих донорных лиганда.
Голубой, содержащий медь(П), белок стеллацианин был переведен
Электронная структура и спектры ионов переходных металлов 109
в производное кобальта(П) [41]. Исследования показали, что медь(П) и кобальт(П) конкурируют за одно и то же место в белке. Поскольку спектры соединений, содержащих кобальт(П), интерпретировать легче, чем спектры производных меди(П). авторы смогли прийти к выводу: кобальт находится либо в центре искаженного тетраэдра, либо в пятикоординационном окружении. Интенсивная линия переноса заряда указывает на существование связи Со — SR. Отнесение всех линий спектра нативного медьсодержащего белка было проведено по аналогии. Существование порфириновых комплексов в ферментных системах можно установить по наличию в спектре характеристической полосы Соре в области 25000 см-1. Эта полоса обусловлена связанным с лигандом переходом п -> тг* типа перехода с переносом заряда (см. гл. 5). В электронных спектрах порфириновых комплексов обнаружены также две другие полосы низкой интенсивности. Существование этих полос и их сдвиги при введении заместителей в циклы можно понять, проведя расчеты по методу МО [42]. Положения этих полос использованы для классификации цитохромов.
ПАРАМЕТРЫ СВЯЗЫВАНИЯ, ПОЛУЧАЕМЫЕ ИЗ СПЕКТРОВ
10.11. ПАРАМЕТРЫ ст- И я-СВЯЗЫВАНИЯ, ПОЛУЧАЕМЫЕ ИЗ СПЕКТРОВ ТЕТРАГОНАЛЬНЫХ КОМПЛЕКСОВ
Как уже говорилось ранее, если под действием локального окружения симметрия комплекса снижается от Oh или Td, в спектре появляются дополнительные линии. Это, в частности, можно видеть при сравнении спектров Co(NH3)g+ и Co(NH3)5C12 + . Как следует из диаграммы Тана-бе — Сугано (приложение IV), для ^-комплекса сильного поля разрешены по спину переходы 1Alg —> 1Tlg и *Alg —> 1T2g, поэтому полосы относят именно так, как показано на рис. 10.23. Если одна из NH3-rpynn заменяется на другую группу, которую мы произвольно поместим на <1г2-орбиталь, то она уже взаимодействует с кобальтом иначе, чем исходная группа. Если проведено замещение на хлор, ст-взаимодействие должно быть более слабым, чем тг-взаимодействие. Влияние этих взаимодействий на относительные энергии dz2-, dxz- и ^„-орбиталей в Co(NH3)5C12 + и Co(NH3)g+ различно: более сильное тг-взаимодействие с С1 “ повысит энергию dxz- и J^-орбитали, а более слабое ст-взаимодействие понизит энергию ^-орбитали. Вырожденность триплетного состояния снимается, 1Т1 расщепляется на состояния 1А и и в результате мы получаем спектр, показанный на рис. 10.23,Б.
Как предсказывает теория [22], расщепление линии 1Т2 слишком мало, чтобы его можно было наблюдать экспериментально. Если в спектре появляются дополнительные полосы, для его описания необходимы дополнительные параметры, помимо iODq и В. Расчет этих па
по
Глава 10
раметров сильно зависит от отнесения наблюдаемых электронных переходов.
Герлох и Слейд [3] рассмотрели несколько низкосимметричных конфигураций, мы же вкратце обсудим лишь тетрагонально искаженные
Рис. 10.23. Спектры Co(NH3);'' (Л) и Co(NH3)5C12+ (5).
шестикоординационные комплексы. В данном случае полный потенциал имеет следующий вид:
^4h)=Coct+Ctetr. (10.17)
Если матричные элементы рассчитаны, мы получаем параметр, который раньше обозначали как Dq, и еще два других радиальных параметра, которые обусловлены Vletr, далее мы будем обозначать их Рх и Dt. Как показано в уравнениях (10.18) и (10.19), Ds включает члены с г-2, a Dt— члены с г'4.
Ds = lze2r-2( Ц-
7 \а3ху
Dt = -Ze2r-4|-4
7 \ а3
(10.18)
(10.19)
где а и b — различные расстояния между металлом и лигандом (а относится к лигандам ху, а b — к лигандам z). Поскольку Dt представляет собой функцию его можно связать с Dq. (Вспомните, что Dq—функция г-4.) Как показано в уравнении (10.20), Dt служит мерой разности в Dq между аксиальным и экваториальным центрами:
Dt = 4[Dq(xy) - Dq(z)].
(10.20)
Электронная структура и спектры ионов переходных металлов 111
Следует подчеркнуть, что ни Dq(xy), ни Dq(z) не адекватны аналогичным параметрам, используемым для описания лиганда в октаэдрическом комплексе [43].
Мак-Клюр [22] предположил следующее: симметрия комплекса такова, что ев- и Г2в-орбитали металла — это соответственно только ст-разрыхляющие и только л-разрыхляющие орбитали, и определил параметры Зст и 5л как
= стг-стху= — ~$-Ds -
(10.21)
3 5
Зст = л2 — пху = -^Ds + ^Dt.
(10.22)
Параметры ст и л можно рассматривать как косвенные индикаторы относительных ст- и л-разрыхляющих свойств лигандов. В работах [43—45] опубликованы величины этих параметров для различных лигандов и проведена их интерпретация. Согласно результатам спектральных исследований комплексов, при ст-связывании наблюдается следующий порядок взаимодействия:
NH3 > Н2О > F“ > СГ > Вг“ > Г
л-Отталкивание меняется в ряду
Г> Вг > СГ> F > NH3
10.12. МОДЕЛЬ УГЛОВОГО ПЕРЕКРЫВАНИЯ
Альтернативным подходом (имеющим несколько преимуществ) к параметризации спектров комплексов переходных металлов может служить модель углового перекрывания [3, 46]. Эта модель исходит из приближенного подхода к энергиям соединений переходных металлов в рамках метода МО. В первую очередь мы рассмотрим простой монокоорди-национный комплекс М—L. Если М—переходный металл, нас больше всего интересуют энергии ^/-орбиталей комплекса. Пять </-орби-талей комплекса симметрии Сжи охватывают ст-, л- и 5-представле-ния, т. е. d(z2)—это ст-представление, d(xn-) и d(yz)— л-представление, а d(xy) и d(x2 — у2) — S-представление. Рассматривая, например, ст-взаимо-действие, мы можем записать секулярные уравнения
Я а тга тле С<т г
М”
HaML-S°MLE НЧ-Е
= о
(10.23)
где Нм и HaL—энергии соответствующих орбиталей металла и лиганда; Нмг—обменный интеграл между орбиталями металла и лиганда, а Sml—интеграл перекрывания. Как правило, Нм » Н” и двухатомный интеграл перекрывания S^l мал, поэтому один из корней, скажем Ег,
112
Глава 10
достаточно близок к энергии а другой, Е2, достаточно близок к энергии HaL. При таком допущении мы можем записать два приближенных детерминанта:
Корни
тта г тта ест rja ПМ~ 1 П ML — d ML17 М tjct ест тта тта тта = o,
Щл ~ Н L #ML — Swift's, jja ест тта jja тт ^Mf^ML^L ** L~ ^2 = 0.
можно записать в явном виде:
£1 = (Hml)2 + (SaMLHaM)2 Н°м + tjct Tja “M “ L
р „„ (Н^)2-2Н^Н15^ + (5 ст rja \2 МЬЛ U
П2 tjct тта «М “ « L
(10.24)
(10.25)
(10.26)
Используя приближение Вольфсберга—Гельмгольца для H^l в виде
HaML=S^L(H^+№L), (10.27)
можно переписать уравнения (10.25) и (10.26) как
~ (Н°м + нп2
Е, - Я£, = = -Mg £ (S^L)2, (10.28)
“М “ L
(Н" + /Г)2
£2_ = Еа= - (S-J2. (10.29)
М L
*
Ео — положительная величина и, таким образом, выражает дестабилизующее влияние на энергии орбиталей металла, в то время как Е„ — отрицательная величина, и она выражает стабилизующее воздействие на орбитали лиганда. Как показывает уравнение (10.28), дестабилизующее влияние на данную орбиталь металла пропорционально (Sml)2- Этот эффект должен быть мал, если разность — Н° велика, а S^l мал. Е* можно в принципе рассчитать по уравнению (10.28), но на практике более целесообразно выразить ее параметрически. С этой целью интеграл перекрывания S^L можно представить в виде произведения радиальной и угловой составляющих
S°ML = SMLF1, (10.30)
Где SML — интеграл радиальных функций орбиталей металла и лиганда, a Eg—угловая составляющая. SML зависит от того, какие орбитали ме
Электронная структура и спектры ионов переходных металлов 113
талла и лиганда мы рассматриваем и каково расстояние между металлом и лигандом; F„ зависит только от геометрического расположения металла и лиганда. Если геометрия комплекса известна, рассчитать F^ легко.
Рассмотрим, как проводится разложение на составляющие на простом примере: лиганд L перекрывает р2-орбиталь металла М в фрагменте М — L. На рис. 10.24,А лиганд расположен на оси г, а связь М — L
Рис. 10.24. Перекрывание с /^-орбиталью во фрагментах М —L.
А
направлена вдоль этой оси. Если мы определим FJ (ст-лиганд перекрывается с р-орбиталью) как единицу, перекрывание в этом случае определяется величиной Sml. Если направление связи М — L, имеющей ту же самую длину, не совпадает с осью z, чистое перекрывание уменьшается. Радиальная составляющая остается той же самой, поэтому снижение сопровождается уменьшением угловой составляющей FJ. На рис. 10.24,5 связь М — L направлена вдоль оси х, ст-орбиталь лиганда ортогональна pz (перекрывание равно нулю), и поскольку радиальная составляющая не меняется, то угловой член F*J становится нулевым.
Подставляя уравнение (10.30) в уравнение (10.28), получаем
Обозначая
Е* =
(Н°м + HI)2 Я a rja
М — L
М)2.
запишем уравнение (10.31) как
Е* = еЖ)2.
(10.31)
(10.32)
(10.33)
{н°м + hi)2 с2
H°M + H°L ML’
Уравнение (10.33) показывает, что ст-разрыхляющее влияние на данную d-орбиталь можно выразить параметром еП и числом (Fda), которые имеются в стандартных таблицах (см. далее).
При л-связывании лиганд может взаимодействовать либо через низкоэнергетические заполненные орбитали, так что сохраняются те же
8 — 574
114
Глава 10
самые условия, что и при о-связывании, либо через высокоэнергетические свободные орбитали, так что « H"L, и поправки к энергиям орбиталей металла имеют связывающий характер. В обоих случаях, однако, ек можно определить с помощью уравнения (10.32), единственное различие заключается в знаке: ек положителен для разрыхляющего эффекта и отрицателен для связывающего.
Нетрудно видеть, что энергии d-орбиталей металла в монокоордина-ционном комплексе М — L определяются как
E(dz2) = ea,
E(dxz) = E(dyz) = еп,
(10.34)
E(dxy) = E(dxr-_.-) = e6
В комплексах ионов переходных металлов картина значительно усложняется, поскольку мы имеем дело с перекрыванием d-орбиталей и располагаем многими лигандами. Рассмотрим, например, октаэдрический комплекс. Система координат, показаннная на рис. 10.25,А, фиксирует положение истинных d-орбиталей этого комплекса. Можно использовать локальную (штрихованную) систему координат, связанную с каждым лигандом, так что связь металл — лиганд теперь станет осью z'. Ось х' находится в плоскости, образуемой z и z'. На рис. 10.25,Б показана локальная система координат для лиганда L2. С помощью полярных координат лиганда можно связать координаты точки в штрихованной системе координат с координатами в нештрихованной системе координат. Выразим теперь координаты d-орбитали, положение которой в нештрихованной системе координат известно, с помощью переменных в штрихованной системе координат. Полученные соотношения представлены в табл. 10.8, их можно распространить на комплексы любой геометрии.
Чтобы показать, как пользоваться табл. 10.8, определим для L2 со-
Рис. 10.25. Система координат (нештрихованная) для шестикоординационного комплекса (А), локальная (штрихованная) система координат (Б), определение 0 и ф, данных в табл. 10.8 (В).
Таблица 10.8
Соотношения между d-орбиталями, определенными в штрихованной и иештриховаииой системах координат (рис. 10.25)
z'2 уУ x'z' x'y' У2-у'2
Z2 -(1 + 3cos20) 4 0 |/з sin 20 2 0 j/з (1 — cos 20)
yz [/з sin ф sin 20 COS ф cos 0 sin ф cos 20 — cos ф sin 0 1 . . — - sin ф sin 20
XZ [/з —— cos ф sin 20 — 5Шф COS0 СО5ф cos 20 5Шф sin0 —-coscp sin 20
ху ]/2 . —sin 2ф(1 - cosQ) со52ф sin0 1 —sin 2ф sin 20 соэ2ф cos0 ^5т2ф(3 + cos 20)
х2 — у2 [/з —— со$2ф(1 — cos20) -5ш2ф sin0 ^со52ф sin20 — sin 2ф cos0 ^со52ф(3 + cos20)
116
Глава 10
отношение между d-орбиталями в штрихованном и нештрихованном базисах. Запишем с помощью обозначений, принятых на рис. 10.25, угловые полярные координаты лигандов в октаэдрическом комплексе:
Лиганд 1 2 3 4 5 6
0 0 90 90 90 90 180
ф 0 0 90 180 270 0
Для Ь2 0 = 90° и ф = 0. Подставляя эти величины в строку 1 табл. 10.8, находим, что комбинация d-орбиталей, которую мы должны иметь в штрихованной системе (записанной вверху), должна быть эквивалентна d22-op6HTann. Этот результат получают при подстановке координат вместо 0 и ф в первую строку:
1 , l/з
dz2 = —(1 + 3cos20)(z'2) + O(y'z') —v—sin 20(x'z') +
+ O(x'y') + ^^(1 — cos20)(x'2 — y'2) =
= -yz'2+Xl(x'2-y2). (10.35)
Энергии взаимодействия орбиталей z 2 и x2 — у2 с лигандом будут выражаться через и е5, где нижние индексы ст и 5 указывают, что взаимодействие лиганда с dz ^-орбиталью носит ст-характер, а взаимодействие лиганда с dx., _г2-орбиталью — 5-характер. Коэффициенты — — 1/2 и + |/з/2 можно рассматривать как характеристики перекрывания d^-орбитали с орбиталями dz,2 и dx,2_y,2. Поскольку энергия пропорциональна квадрату перекрывания [уравнение (10.32)], мы можем записать энергию взаимодействия лиганда с dz2-op6Hranbro как
1 3
E(dz2) = jea + |е5. (10-36)
Теперь мы должны рассчитать влияние Ь2 на энергии всех d-орбиталей, для чего подставим 0 = 90° и ф = 0 в выражения, приведенные в табл. 10.8. Полученные результаты можно выразить в виде матрицы.
-1 0 0 0
2
0 0 0 -1
0 0-1 0
0 10 0
— 0 0 0
2
0
о о
1
2
Электронная структура и спектры ионов переходных металлов 117
Поскольку коэффициенты, составляющие столбцы, выражены в штрихованных координатах, они соответствуют ст-, л-, л-, S- и 5-взаимодействию. Возводя в квадрат эти коэффициенты, определяем энергии:
1 3
E(d.y) = +
= е8
Жг) = ел
Жу) = ек
E(dx2_yl) = ^е„+ ^-е6 (10.37)
Подстановка нулевых значений 9 и <р лиганда L, в табл. 10.8 приводит к матрице
1 0 0 0 0’
0 10 0 0
0 0 10 0
0 0 0 1 0
0 0 0 0 1
Возводя коэффициенты в квадрат, находим энергию взаимодействия лиганда L] с каждой орбиталью, т. е. результат, полученный ранее из уравнения (10.34).
Повторим эту операцию для других лигандов:
Лиганд 1 Лиганд 2 Лиганд 3
E(dz2) E(dyz) e 71 1. . 3 4 ° 4 5 le +ae 4 4e5
E(dxz) e 71
E(dxy) es Л
E(dx2_y2) ei \e Iе.+
Лиганд 4 Лиганд 5 Лиганд 6
E(d.2) E(dyz) 4 c 4 о e5 -<?4 4 » 40 eK
E(dxz) e5 ел
E(dxy) e e5
E(dx2_y2) 4- -e 4 4 5 -f + 4 ° 4 6 4
Полученные результаты можно проверить, проведя суммирование в каждом столбце. В результате этой операции мы увидим, что каждый лиганд дает один тип ст-, два типа л- и два типа 5-взаимодействия. Суммируя вклады всех индивидуальных лигандов, мы в итоге получим
118
Глава 10
выражения для энергий d-орбиталей октаэдрических соединений:
£(dz2) = Зс„ + Зе§
£(dyz) = 4с„ + 2е§
Жг) = 4ея + 2е8 (10.38)
£«у) = 4е„ + 2е§ £(dx2_y2)=3ea + 3es
Естественно, энергия трех г2!)-°рбиталей одинакова; то же самое справедливо для двух е9-орбиталей. Если вкладом es пренебречь, разность между энергиями орбиталей ед и t2g составит Зс„ — 4ея, что в теории кристаллического поля соответствует А. В комплексе Td энергии е- и 12-орбиталей определяются как 8/Зея + 4/3es и 4/3es + 8/9ея + 16/9<, соответственно. Отметим, что при таких параметрах Аг; = 4/9Аоя. В комплексах более низкой симметрии добавляются величины энергий всех лигандов и рассчитываются энергии d-орбиталей. Численные значения параметров е„, еп и es определяют из энергий d-орбиталей октаэдрических комплексов. Значения еа для различных комплексов параметризуют в соответствии с интегралом перекрывания. Значение описанного подхода состоит в том, что совокупность параметров, полученную для данного лиганда и данного металла, можно использовать для объяснения спектров комплексов многих переходных металлов, если учесть геометрию комплекса и перекрывание. В работе [47] приведены соотношения между Dq, Ds, Dt, 3<т, Зя и еа и еп.
В заключение следует подчеркнуть, что если результаты расчетов, как в рассмотренном примере, зависят от отнесения полос, проведенное отнесение следует проверить, используя данные, полученные при изучении монокристалла поляризованным излучением (гл. 5).
НЕКОТОРЫЕ ВОПРОСЫ,
СВЯЗАННЫЕ С ЭЛЕКТРОННЫМИ ПЕРЕХОДАМИ
10.13. ОДНОВРЕМЕННЫЕ ПАРНЫЕ ЭЛЕКТРОННЫЕ ВОЗБУЖДЕНИЯ
Электронные спектры высокоспиновых октаэдрических и тетраэдрических комплексов железа(Ш) согласуются с диаграммами Танабе — Су-гано. В этих спектрах обнаружены три перехода: 6А1д -> 4Т2, 6А1д -> 4 7’, и 6А1д -» ЛА1д [если энергия 4£(£>) достаточно низка, наблюдается четыре перехода], и поскольку величина Dq для октаэдрических комплексов выше, чем для тетраэдрических, в первых переходы 4 7] и 4T2(G) характеризуются более высокими энергиями. Все d — d-переходы запрещены по мультиплетности, и интенсивность их мала. Однако исследование элек
Электронная структура и спектры ионов переходных металлов 119
тронного спектра шестикоординационного димера с кислородным мостиком (HEDTAFe)2O (где HEDTA — оксиэтилэтилендиаминтриацетат) [48, 49] приводит к поразительным результатам.
Рис. 10.26. Спектр поглощения 0,20М раствора (HEDTAFe)2O)-6H2O в воде при 296 К. Рассчитанные положения полос парного электронного переноса указаны стрелками.
Рассмотрим спектр этого соединения (рис. 10.26). Полосы а — d корректным образом можно отнести к четырем d— d-переходам октаэдрического комплекса: 6At -> 4Г15 6At -> 4E2(G), 6А1 -> 4А14Е1 и 6А1-> 4E(D). Интенсивности этих полос на два порядка превышают интенсивности таких же полос в типичном спектре комплекса железа(Ш).
Такое увеличение интенсивности запрещенных по спину переходов обычно для спин-связанных систем, поскольку взаимодействие частично смягчает правило отбора по спину.
Энергия четырех интенсивных (е — h) полос в ультрафиолетовом диапазоне слишком низка, и очень многое говорит о том, что они обусловлены переходами с переносом заряда. Интенсивность этих полос слишком высока для d — d-перехода в единственом ионе металла, и авторы работы [48] приписывают их одновременным d—d-переходам в двух центрах железа(Ш). Последние связаны, так что по спину разрешено парное возбуждение [50, 51]. Полоса при 29,2 • 103 см 1 отнесена
120
Глава 10
к одновременному переходу а в одном центре и b в другом (vo + v(, = = 29,4-103 см-1; полоса / при 32,5-103 см-1—к переходам а + с (32,3-• 103см полоса# при 36,8- 103см“ 1—кпереходамЬ + b и полоса h при 42,6-103 см 1—к переходам b + d. Это явление носит название одновременного парного электронного возбуждения, а полосы поглощения сокращенно обозначают символом SPE. Аналогичные эффекты наблюдаются и в других системах [52, 53], подробно они исследованы для (NH3)5CrOCr(NH3)| + . Результаты исследования показали, что увеличение интенсивности обусловлено вибронным взаимодействием, вызванным обменом электрических диполей.
10.14. ПОЛОСЫ ПЕРЕНОСА ЭЛЕКТРОНА
МЕЖДУ ЦЕНТРАМИ С РАЗЛИЧНОЙ СТЕПЕНЬЮ ОКИСЛЕНИЯ
Металломеры — это соединения, в молекулах которых содержится два или более атомов металла. Можно синтезировать металломеры с мостиковыми группами, которые позволяют атомам металла находиться в различной степени окисления, как, например, в [L5Mln—X — — МПЬ5]"+. Соединения «смешанной валентности» долгое время привлекали внимание исследователей в связи с тем, что все такие соединения интенсивно окрашены. Сравните, например, берлинскую лазурь KFe[Fe(CN)]6 с K3Fe(CN)6 и K4Fe(CN)6. В настоящее время установлено, что интенсивная окраска берлинской лазури обусловлена переносом электрона между двумя центрами с различной степенью окисления. Поясним это на примере димера пиразина рутения [54]:
(NH3)5Ru(II)—Ru(III)(NH3)55+
(NH3)5Ru(III)—Ru(II)(NH3)55+
Чтобы не усложнять картину, мы рассмотрим влияние только мостикового пиразина, а другие лиганды учитывать не будем. Пометим эти два атома рутения следующим образом:
Ruain— N
-Run
Построим кривую зависимости потенциальной энергии [55] этой молекулы от X (расстояния Ru'11— N) так, чтобы сумма расстояний Ruo — N и R(, — N оставалась постоянной. Кривую типа а (рис. 10.27,А) мы получим в том случае, если зададимся гармонической функцией потенциальной энергии, т. е. К=1/2/сХ2. Построим аналогичную кривую для Ru"—
Электронная структура и спектры ионов переходных металлов 121
— N (рис. 10.27,Б). Равновесное расстояние Ru'1 — N больше, чем равно-
Ruon—N,
-Ru/n
весное расстояние Ru'n— N. Обозначим разность Хо. Эти две кривые можно рассматривать как характеристику всей системы в целом. И исходя из этой характеристики, определим условие переходов электрона
X -
Р
Рис. 10.27. Кривые потенциальной энергии для различных классов соединений смешанной валентности. Системы варьируют от полностью локализованных (А) до соединений с промежуточной делокализацией (Б) и соединений с полной делокализацией (В). X—координата, вы-ражающая расстояние Ruoni— —N как сумму Ru„—N и Rub—N, поддерживается постоянной.
122
Глава 10
между двумя центрами. Eth— энергия активации перехода, ее приписывают молекуле, в которой длины связей Rua — N и Rub — N одинаковы. Тепловая энергия, необходимая для преодоления электроном барьера между металлами, Eth, определяется выражением [55]
£th= |k(|Xo)2 = = |е1Т. (10.39)
Согласно принципу Франка—Кондона, электронный переход не вызывает изменения молекулярных координат, поэтому энергия полосы переноса электрона между двумя центрами с различной степенью окисления в спектре поглощения (т. е. перехода с переносом заряда с одного атома Ru на другой) можно изобразить вертикальной линией Егг.
Реальная ситуация более сложна, поскольку координата X зависит от всех лигандов и электронов комплекса. В зависимости от того, насколько близко расположены металлические центры и насколько велико перекрывание орбиталей (по одной на каждом атоме рутения), те две орбитали, на которых может находиться неспаренный электрон, в состоянии смешиваться с образованием связывающей и разрыхляющей комбинаций. Это должно приводить к картине, изображенной на рис. 10.27,Б или В. Параметр, обозначенный (рис. 10.27,Е) как 2Hres, представляет собой удвоенный резонансный интеграл, т. е. недиагональный элемент в секулярном детерминанте между двумя d-орбиталями (по одной на каждом атоме рутения), на которых может находиться неспаренный электрон [56]. Из рис. 10.27,Б видно, что Ел < 1/4Егт. Согласно рис. 10.27,В, неспаренный электрон полностью делокализован в любой момент времени, а электронное поглощение нельзя по-настоящему назвать переносом электрона между двумя центрами с различной степенью окисления.
Робин и Дей [57] отметили, что на рис. 10.27,В один из энергетических минимумов соответствует электронной волновой функции, описывающей состояние, в котором неспаренный электрон находится в основном на одном атоме металла, но и в небольшой степени делокализован на другом атоме. Эти авторы предположили, что соединения смешанной валентности можно разделить на три класса (I — III) в зависимости от характера распределения электронной плотности на двух металлических центрах (делокализация отсутствует, небольшая делокализация, полная делокализация электронной плотности между двумя центрами).
Рассматриваемый комплекс рутения [55] дает интенсивную полосу поглощения в ближней ИК-области при 1570 нм Эту полосу приписывают электронному переходу, показанному на рис. 10.27,А, и обозначают Егт. Константа скорости электронного обмена, рассчитанная из энергии этого перехода для описанной выше приближенной модели с помощью уравнения (10.39), составляет 3109с-1. Электронный переход описывается как переход [2, 3] -> [3, 2].
Наличие высокоинтенсивных линий в области d — d-переходов в спектрах других комплексов было использовано как доказательство
Электронная структура и спектры ионов переходных металлов 123
существования соединений металлов смешанной валентности. Интерпретацию полосы как результата перехода в соединении смешанной валентности можно подтвердить зависимостью интенсивности перехода от степени окисления или восстановления [и, и]-окислительного состояния комплекса при образовании [и, т]-состояния. Максимум наблюдается в том случае, когда образуется [и, т], он исчезает при образовании [т, т].
10.15. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Идеи, изложенные в настоящей главе и гл. 5, имеют важное значение не только при решении структурных задач; с электронными переходами связаны явления флуоресценции и фосфоресценции. В фотохимических реакциях участвуют электронно возбужденные молекулы, и для того, чтобы разобраться с механизмами этих реакций, необходимо иметь представление о структуре и реакционной способности возбужденных частиц. В некоторых случаях синглет-триплетное возбуждение молекул приводит к образованию реакционноспособных радикалов. Часто молекулы, не способные к образованию комплекса, находясь в основном состоянии, приобретают такую способность, если одна из молекул возбуждена (такой комплекс называется эксиплексом). Таким образом, идеи, касающиеся электронных переходов, изложенные в настоящей главе л гл. 5, важны для многих областей.
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. Ballhausen C.J., Introduction to Ligand Field Theory, McGraw-Hill, New York, 1962.
2. Figgis B.N., Introduction to Ligand Fields, Interscience, New Vork, 1966.
3. Gerloch M., Slade R. C., Ligand Field Parametres, Cambridge University Press, New York, 1973.
4. a. Jorgensen C.K., Absorption Spectra and Chemical Bonding in Complexes, Pergamon Press, New York, 1962.
6. Jorgensen С. K., Modern Aspects of Ligand Field Theory, North Holland Publishing Co., Amsterdam, 1971.
5. Schliifer H. L., Gliemann G., Basic Principles of Ligand Field Theory, Interscience, New York, 1969.
6. Moore С. E., Atomic Energy Levels Circular 467, Vol. 2, National Bureau of Standarts, 1952. Тома 1 и 3 содержат аналогичные данные относительно других газообразных ионов.
7. Condon Е. U., Shortley G. Н., Theory of Atomic Spectra, Cambridge University Press, New York, 1957.
8. Griffith J. S., The Theory of Transition Metal Ions, Cambridge University Press, 1961.
9. McClure D.S., Electronic Spectra of Molecules and Ions in Crystals, Academic Press, New York, 1959.
10. Lever A.B.P., Inorganic Electronic Spectroscopy, American Elsevier, New York, 1968.
11. Spectroscopic Properties of Inorganic and Organometallic Compounds, Vols. I — IV, The Chemical Society, London, 1968 —1971.
124
Глава 10
12. Dunn Т. М., Modern Coordination Chemistry, ed. Lewis J. and Wilkins R. G., Interscience, New York, 1960.
13. Tanabe Y., Sugano S., J. Phys. Soc. Japan, 9, 753, 766 (1954).
14. Drago R.S., Meek D.W., Joesten M.D., LaRoche L., Inorg. Chem., 2, 124 (1963).
15. Cotton F.A., Goodgame M., J. Amer. Chem. Soc., 83, 1777 (1961).
16. Barnum D. W., J. Inorg Nucl. Chem., 21, 221 (1961).
17. Piper T.S., Carlin R.L., Chem. Phys., 36, 3330 (1962).
18. Liehr A.D., Ballhausen C.J., Phys. Rev., 106, 1161 (1957).
19. Drago R.S.,-Meek D.W., Longhi R., Joesten M.D., Inorg. Chem., 2, 1056 (1963).
20. Kiser R.W., Lapp T.W., Inorg Chem., 1, 401 (1962).
21. Basolo F., Ballhausen C.J., Bjerrum J., Acta Chem. Scand., 9, 810 (1955).
22. McClure D. S., Advances in the Chemistry of Coordination Compounds, ed. Kirshner S., p. 498, Macmillan, New York, 1961, и ссылки в этой работе.
23. Basolo F., J. Amer. Chem. Soc., 72, 4393 (1950); Shimura Y.J., J. Amer. Chem. Soc., 73, 5079 (1951); Nakamoto K., Fujita J., Kobayashi R., Tsuchida R., J. Chem. Phys., 27, 439 (1957) и ссылки в этой работе.
24. Fay R.C., Piper T.S., J. Amer. Chem. Soc., 84, 2303 (1962).
25. Furlani C., Sartori G., J. Inorg. Nucl. Chem., 8, 126 (1958).
26. Manch W., Fernelius W., J. Chem. Educ., 38, 192 (1961).
27. Maki G., J. Chem. Phys., 29, 162 (1958); ibid., 29, 1129 (1958).
28. Cotton F.A., Bannister E., J. Chem. Soc., 1960, 1873.
29. Gill N.S., Nyholm R.S., J. Chem. Soc., 1959, 3997.
30. Donoghue J.T., Drago R.S., Inorg. Chem., 1, 866 (1962).
31. Straub D.K., Drago R.S., Donoghue J.T., Inorg. Chem., 1, 848 (1962).
32. Jorgensen С. K., Energy Levels of Complexes and Gaseous Ions. Gjellerups, Copenhagen, 1957.
33. Sacconi L, Pure Appl. Chem., 17, 95 (1968); Transition Metal Chem., 4, 199 (1968); Morassi R., Bertini L, Sacconi L, Coord. Chem. Rev., 11, 343 (1973).
34. Furlani C., Coord. Chem. Rev., 3, 141 (1968).
35. a. Ciampolini M., Struct Bonding, 6, 52 (1969).
6. Hathaway B.J., Billing D.E., Coord Chem. Rev., 5, 143 (1970).
36. Figgis B.N., Lewis J., Modern Coodination Chemistry, ed. Lewis J. and Wilkins R. G., Interscience, New York, 1960.
37. Linhard M., Siebert H., Weigel M., Z. Anorg u. Allgem. Chem., 278, 287 (1955).
38. Ballhausen C.J., Gray H.B., Inorg. Chem., 1, 111 (1962).
39. Gray H.H., Hare C.R., Inorg. Chem., 1, 363 (1962); Hare C.R., Bernal I., Gray-H.B., Inorg. Chem., 1, 831 (1962).
40. Brill A. S., Martin B. R., Williams R. J. P., Electronic Aspects of Biochemistry, ed. Pullman B., Academic Press, New York, 1964.
41. McMillin D.R., Holwerda R.A., Gray H.B., Proc. Nat. Acad. Sci., 71, 1339 (1974).
42. Cauhgey W.S., et al., J. Mol. Spect., 16, 451 (1965) и ссылки в этой работе.
43. Rowley D.A., Drago R.S., Inorg. Chem., 7, 795 (1968); ibid., 6, 1092 (1967).
44. a. Lever A.B.P., Coord. Chem. Rev., 3, 119 (1968).
6. Chiang R.L., Drago R.S., Inorg. Chem., 10, 453 (1971).
45. Burbridge C.D., Goodgame D.M.L., Goodgame M., J. Chem. Soc., A, 1967, 349.
46. a. Schaffer C.E., Jorgensen C.K., MoL Phys., 9, 401 (1965).
6. Larsen E., LaMar G.N., J. Chem. Educ., 51, 633 (1974).
47. Bertini L, Gatteschi D., Scozzafara A., Inorg. Chem., 15, 203 (1976).
48. Schugar H.J., Rossman G.R., Thibeault J., Gray H.B., Chem. Phys. Letters, 6, 26 (1970).
49. Gray H. B., Bioinorganic Chemistry, Amer. Chem. Soc. Monograph, 100, 365 (1971).
50. Lohr L, Coord. Chem. Rev., 8, 241 (1972).
51. Dexter D.L., Phys. Rev., 126, 1962 (1962).
52. Hansen A.E., Ballhausen C.J., Trans. Faraday Soc., 61, 631 (1965).
53. Giidel H.U., Dubicki L, Chem. Phys., 6, 272 (1974) и ссылки в этой работе.
54. Creutz С., Taube Н., J. Amer. Chem. Soc., 91, 3988 (1969).
55. Hush N.S., Progr. Inorg. Chem., 8, 391 (1967) и ссылки в этой работе.
Электронная структура и спектры ионов переходных металлов
125
56. Mayoh В., Day Р., J. Chem. Soc., Dalton, 1974, 846.
57. Robin M.B., Day P., Adv. Inorg. Chem. Radiochem., 10, 248 (1967).
УПРАЖНЕНИЯ
1. Определите с помощью диаграмм Танабе — Сугано (приложение IV), сколько линий должно быть в спектре поглощения октаэдрического комплекса Сг1П, если для лиганда Dq/B =1? Пометьте эти переходы и выпишите их в порядке увеличения длины волны.
2. В спектрах октаэдрических комплексов Со2 + с лигандами слабого поля (Dq/B = 0,7) наблюдаются три хорошо разрешенные полосы. Проведите качественное отнесение этих полос, используя диаграммы Танабе — Сугано, и выпишите их в порядке снижения частот. Каким будет спектр октаэдрического комплекса Со2 + с лигандами сильного поля?
3. Полосы спектра поглощения комплекса NiR^Clj соответствуют е « 150. Атомы R и С1 занимают сходные положения в спектрохимическом ряду. Координируются ли атомы хлора?
4. Выделены два изомера Co(NH3)4(SCN)2 . Как определить, в обоих ли изомерах группы SCN связаны через атом серы? Если в обоих изомерах координация осуществляется через серу, как установить, у какого из изомеров цис-конфигурация, у какого — транс-конфигурация? (Вспомните, что в спектрохимическом ряду — SCN и СГ располагаются рядом, в то время как — NCS-создает более сильное поле; Co(NH3)4C12 синтезировать легко.)
5. Используя диаграммы Танабе — Сугано, проведите отнесение линий в показанных ниже спектрах шестикоординационных акво-комплексов (за исключением А).
А
co
Cr*V4) 0,10Me 0,75MH;,SO4
5000 fOOOO 20000 30000 35000
5000 10000 20000 30000 35000
V.CM'1 E
5000 /0000 20000 30000 35000
V, CM'1
и
6. Рассчитайте" lODq и В для Н2О в комплексе, спектр которого приведен в задаче 5Ж.
7. Известно, что для <Р-конфигурапии [см., например. Lever А. В. Р., J. Chem. Ed., 45, 711 (1968)] энергия перехода ’iA2s^iT2д равна 10D<j и энергия перехода 4Л2,-»4Т 19(F) равна 7.5В' + 15D<j - 1/2(225В'2 + 100D<j2 - 18B'Bq)1/2. Рассчитайте по этим данным величины 10D<j и Р (В'/В, где В — 1030 см ') для а. Н2О, используя спектр задачи 5Б;
б. С2ОГ, используя следующий спектр:
/50
/00
50-
0,0050 М К3Сг(ох)3
оГ । । । । I । । । । I । । 1 1 I । । । । I । 1 । । I । I. 1 1 I
5000 10000 15000 20000 25000 30000 35000
V, СНГ1
в. для Н2О в комплексе Ni2+ величина 10Bq составляет 8500 см-1, а Р = = 0,88. Сравните результаты, полученные при решении задачи За с этими величинами и объясните причины различия. Сравните величины 101)г/ и Р, полученные для воды и оксалата в комплексе Сг(Ш);
г. исходя из того, что энергия перехода 4Л2й-> 4Т19(Р) определяется выражением 7,5В' + 151><7 + 1/2(225В'2 + 1001)г/2 — 18ОВ'£>6/)12, рассчитайте частоту этой полосы в спектре комплекса Сг(С2О4)з-.
128
Глава 10
8. Ион [Ni (пиридин)4(Н2О)2]г + дает полосы d—d-переходов при 27 000, 16 500 и 10150 см-1. Низкосимметричное расщепление не регистрируется. Рассматривая указанный комплекс как октаэдрический, определите 10£>г/. Сравните полученную величину со средним (правило усредненного окружения) величин Dq для двух шестикоординационных комплексов (их можно найти в табл. 10.4).
9. Почему октаэдрические комплексы (слабого поля) Мп2+ окрашены значительно менее интенсивно, чем такие же комплексы Сг3 + ?
10. В литературе опубликован электронный спектр трис(оксалат)хрома(Ш), внесенного в решетку NaMgAKCjO^), • 9Н2О. Если предположить, что это октаэдрический комплекс, то основное его состояние — а низшие возбужденные
состояния (не молекулярные орбитали) — 2Ед, 2Т1д, 2Т2д и 4Т2в. Наблюдаемые частоты полос и коэффициент поглощения приведены ниже:
V, см 1 е
17 500 40
23 700 67
14 500 2,6
15 300 2,0
20 700 0,3
а. Указанные выше переходы относятся к числу электронно разрешенных. Почему наблюдаемый спектр не согласуется с октаэдрической симметрией комплекса? (Интенсивность переходов, запрещенных по мульти-плетности, низкая; е обычно не превышает 5.)
б. Полученный спектр говорит о том, что истинная симметрия молекулы—D3. Понижение симметрии приводит к следующей корреляции:
21
г:
^2
Л2 + Е
+ Е Е
Если регистрация спектра проводилась с использованием света, поляризованного в плоскости, перпендикулярной и параллельной тригональной оси, то в первом случае наблюдаются все полосы, кроме полосы при 17 500 см-1, а во втором—только полосы при 17 500 и 15 300 см1. Исходя из того, что расщепления дублетных октаэдрических состояний в симметрии D3 не разрешаются и что энергия состояния 2Ед ниже, чем 2Т1д, отнесите переходы, используя D3-возбужденные состояния, и обоснуйте проведенное вами отнесение. (Помните, что некоторые дублетные линии представляют собой неразрешенные мультиплеты и поэтому соответствуют более чем одному переходу.)
11. В Re2Cl| (см. задачу 6 гл. 3) переход электрона с орбитали Ь2д на орбиталь Ь1и представляет собой d—d-переход в молекуле с центром инверсии. Разрешен ли он и почему?
12. Ниже показано расщепление энергетических состояний высокоспинового 3d2-Hona в поле Oh:
<Т
2 ig
Электронная структура и спектры ионов переходных металлов 129
а. Определите обозначение свободноионного основного терма (*). (Вспомните, что спиновая мультиплетность состояний Т и А та же, что и у свободного иона.)
б. Как будет выглядеть приведенная выше диаграмма, если учитывать спин-орбитальное взаимодействие (XL-S)? (Рассматривайте только состояние Т .) Приведите диаграмму расщепления и укажите величину J и вырождение для каждого уровня.
в. Приведите диаграмму расщепления для всех приведенных выше состояний J (/2-иона и обозначьте эти состояния соответствующими значениями Mj. Покажите, как влияет на уровни магнитное поле.
13. а. Из состояний, обусловленных наличием в ионе 2р-электронов, только одно является триплетом. Каково обозначение терма этого триплета и как вы это установили?
б. К каким состояниям J приводит этот триплет при учете спин-орбитального взаимодействия и какое из них имеет низшую энергию?
14. а. Определите неприводимые представления для термов, возникающих при расщеплении “‘/•'-состояния газообразного иона октаэдрическим полем лигандов.
б. Используя метод снижения симметрии, определите спиновую мультиплетность состояний в пункте а.
15. Определите неприводимые представления состояний, на которые при спин-орбитальном взаимодействии расщепляется уровень 2Т2. иона FetCN)^.
16. а. Выпишите операции двойной группы D'2, которая получается путем комбинирования операций В3 с операцией R. Убедитесь в том, что выписанные вами операции имеют свойства группы.
б. Как много классов входит в В3? Какие операции входят в каждый класс? Отвечая на этот вопрос, не забудьте, что эквивалентные операции симметрии относятся к одному и тому же классу.
11. МАГНЕТИЗМ
11.1. ВВЕДЕНИЕ
В этой главе мы рассмотрим некоторые аспекты магнетизма, которые имеют решающее значение для понимания спектров ЯМР и ЭПР комплексов ионов переходных металлов. Магнитные эффекты обусловлены электронами молекул, поскольку магнитный момент электрона в 103 раз превышает магнитный момент протона. В главе, посвященной ЯМР, мы рассматривали циркуляции спаренных электронов, которые вызывают диамагнитные эффекты. Неспаренные электроны также приводят к магнитным эффектам, которые зависят от числа неспаренных электронов и их размещения на орбиталях. Магнетизм исследуют, измеряя (см. далее) магнитную поляризацию соединения в магнитном поле. Различные типы поведения вещества в магнитном поле показаны на рис. 11.1. Чтобы описать поведение веществ в магнитном поле, удобно определить параметр, называемый магнитной индукцией В:
В = Но + 4лМ, (11.1)
где Но — напряженность приложенного магнитного поля, а М—намагниченность, а точнее, величина намагниченности, отнесенная к единице объема. Если разделить* обе части уравнения (11.1) на Но, получим
”^Г= 1 +4я7Т7= 1 +4лХ”' (11.2)
Отношение М'/Но обозначают символом х и называют магнитной восприимчивостью единицы объема. Объемная восприимчивость (безразмерная величина) связана с намагниченностью соотношением
Ъ = М’/Н0.
В/Но представляет собой проницаемость среды, это магнитный аналог диэлектрической проницаемости. Разделив х„ на плотность вещества d, мы получаем восприимчивость, отнесенную к единице массы, или
* Мы предполагаем, что в изотропной системе направления Но и М совпадают. Таким образом, хотя мы не имеем права делить вектор на вектор, мы можем исключить свойство направленности и осуществить деление, и в результате получим уравнение, содержащее только скалярные величины.
Магнетизм
131
удельную восприимчивость х9
Xv/d = Х9(см3/г).
(П-З)
Умножение %9 на молекулярную массу дает молярную восприимчивость %:
Х9 • Мол. масса = х (см3/моль).
(11-4)
Величина % отрицательна для диамагнитных веществ и положительна для парамагнитных. В упорядоченном кристалле восприимчивость мо
Рис. 11.1. Линии магнитного поля (т. е. линии, соответствующие постоянной напряженности поля) в вакууме (А), в парамагнитном веществе, внесенном в поле (Б), и в диамагнитном веществе, внесенном в поле (В).
жет быть анизотропной, т. е. ее можно представить тензором с несколькими компонентами. Мы рассмотрим четыре типа магнетизма: диамагнетизм, парамагнетизм, ферромагнетизм и антиферромагнетизм. Особенности этих типов магнетизма указаны в табл. 11.1. Последние два типа магнетизма можно выявить, изучая зависимость % от напряженности магнитного поля. Зависимость восприимчивости ферромагнетиков
Таблица 11.1
Различные виды магнетизма
Вид Знак Величина, сгсэ Зависимость X от поля . Источник
Диамагнетизм - 10-6 Не зависит Вызванная полем циркуляция спаренных электронов
Парамагнетизм + о- кг4 » Угловой момент электрона
Ферромагнетизм + 10'4—кг2 Зависит Выстраивание спинов под действием дипольного взаимодействия моментов со-
седних атомов
Антиферромагне-
тизм + 0—10~4 » Спаривание спинов U вследствие диполь-дипольных взаимодействий
9*
132
Глава II
и антиферромагнетиков от температуры также очень характерна (рис. 11.2).
Температура, при которой на кривой температурной зависимости X для антиферромагнетика появляется максимум, называется температурой Нееля. Температура, при которой на кривой температурной зависимости % ферромагнетика появляется излом, называется температурой
Рис. 11.2. Температурная зависимость магнитной восприимчивости ферромагнетика (/), парамагнетика (2) и антиферромагнетика (.?).
Кюри. Многие вещества, которые в твердом состоянии при температурах, близких к комнатной, ведут себя как парамагнетики, при температурах ниже температуры жидкого гелия (4,2 К) проявляют слабые ферромагнитные или антиферромагнитные свойства.
11.2. ВИДЫ МАГНЕТИЗМА
Диамагнетизм
Диамагнетизм обусловлен индуцированной полем циркуляцией спаренных электронов, которая приводит к возникновению магнитного поля противоположного направления. Таким образом, все молекулы испытывают воздействие диамагнитных эффектов. Диамагнитная восприимчивость атома пропорциональна числу электронов п и сумме квадратов средних радиусов орбит электронов гл
У-л=- £п^£Г‘ = -2>83 • 1010fr?. (11.5)
Магнетизм
133
Таблица 11.2
Константы Паскаля
Аюм X, 10 6 . СМ3/ моль Атом X• 10 ”. см3/моль
Н -2,93 F -6,3
С -6,00 С1 -20,1
С (ароматический) -6,24 Вг -30,6
N -5,57 I -44,6
N (ароматический) -4,61 Mg2 + -5
N (моноамид) -1,54 Zn2 + - 15
N (диамид, имид) -2,11 Pb2 + -32,0
О -4,61 Са2 + -10,4
О2 (карбоксильный) -7,95 Fe2+- -12,8
S -15,0 Cu2 + -12,8
Р -26,3 Co2 + -12,8
Ni2 + -12,8
Связь XB-10 6, см3/моль
UUZZZOo II III II III II II II uuuuZZu + 5,5 + 0,8 + 8,2 + 0,8 + 1,8 + 1,7 + 6,3
Атомы большего размера с большим числом электронов характеризуются большими диамагнитными восприимчивостями, чем атомы меньшего размера с меньшим числом электронов. Молярную восприимчивость молекулы или сложного иона можно получить с хорошим приближением, суммируя диамагнитные вклады всех атомов Хл и всех связей в функциональных группах Хв.
Х=Ехл+1хв- (11.6)
i ' 1 1
Параметры Ха и /в носят название констант Паскаля; наиболее распространенные константы Паскаля приведены в табл. 11.2. На примерах пиридина и ацетона мы далее покажем, как проводится расчет /. C5H5N. Сумма вкладов в %( х К) ь см3/моль):
5 х С (цикла) = — 31,2,
5 х Н = - 14,6,
1 х N (цикла) = — 4,6.
X = Е ХЛ( + £Ч = - 50,4'10 6 см3/моль-
134
Глава 11
Для описания функциональных групп используют величины констант для атомов углерода и азота, входящих в цикл, поэтому £ равна нулю. J
(СН3)2С = О. Сумма атомных вкладов (х 10”6, см3/моль):
3 х С = - 18,0,
6 х Н = - 17,6,
1 х О = - 4,6.
Сумма вкладов связей ( х 10“5, см3/моль)
1 х)с=(>= +6,3
2*4 + 2хв = “33>9 X 10-6 СМ3-МОЛЬ-’
Для комплекса переходного металла можно измерить лишь суммарную магнитную восприимчивость XMEAS> которая представляет собой сумму парамагнитного (х„._.) и диамагнитного вкладов.
1 АКА 1Д1А
^PARA = ^MEAS ~ ^DIA- (П-7)
Таким образом, для получения парамагнитной восприимчивости надо из суммарной восприимчивости вычесть диамагнитную, что можно осуществить: 1) используя для оценки XDIA константы Паскаля, приведенные в табл. 11.2; 2) измеряя диамагнитную восприимчивость лиганда и прибавляя ее к восприимчивости металла (табл. 11.2) и 3) используя восприимчивость аналогичного диамагнитного комплекса металла.
Парамагнетизм в простых системах, где S= 1/2
Парамагнитный вклад в восприимчивость обусловлен спиновым и орбитальным угловыми моментами, взаимодействующими с полем. В первую очередь мы рассмотрим систему, имеющую сферическую симметрию, с одним электроном и в отсутствие орбитального вклада в момент. Магнитный момент такой системы — векторная величина, выражаемая уравнением (11.8):
p=-00S, (11.8)
где S — оператор спинового углового момента, g — электронный (/-фактор (см. гл. 9), Р — магнетон Бора электрона, р = 0,93 • 1О-20 эрг/Э (см. гл. 9).
Гамильтониан Н, описывающий взаимодействие этого момента с наложенным полем, выглядит следующим образом:
Н - р-Н = 0PS H.
(11-9)
Магнетизм
135
Этот гамильтониан, действующий на спиновые волновые функции, имеет два собственных значения энергии (см. рис. 9.1):
Е = msg$H при ms= +|, (11.10)
а разность между ними определяется выражением
ДЕ = д$Н.
(11-11)
Если Н составляет приблизительно 25 кЭ, то ДЕ для свободного электрона с д = 2,0023 равна около 2,3 см-1, что достаточно мало по сравнению с кТ(205 см-1 при комнатной температуре). Оба состояния заселены при комнатной температуре, причем в основном состоянии имеется небольшой избыток заселенности.
Величина проекции на направление поля магнитного момента электрона ц„ в квантовом состоянии п выражается частной производной энергии этого состояния Е„ по полю Н, что и демонстрирует уравнение (11.12):
Ц„= -дЕ„/dH = -msg$. (11.12)
Для того чтобы определить магнитный момент объема образца любого вещества, мы должны взять сумму индивидуальных моментов состояний, взвешенную по их болъцмановским заселенностям.
Фактор Больцмана для расчета вероятности Рп заселенности дискретных состояний с энергией Е„ при тепловом равновесии выражается уравнением (11.13)
--И/)ехр—. (11.13)
кТ) L-! \ кТ J
В этом выражении N„ относится к заселенности состояния п, a N — к суммарной заселенности всех существующих состояний. У нас есть волновая функция для каждого состояния, и мы используем термин «уровень» для обозначения всех состояний, имеющих одну и ту же энергию. Сумма магнитных моментов, взвешенная по заселенности всех индивидуальных состояний, носит название «макроскопический магнитный момент М». Для моля вещества М определяется уравнением (11.14):
(11-14) m,
где JV — число Авогадро. Подставляя уравнение (11.13) для Р„ в уравнение (11.14), получаем уравнение (11.15) для системы с 5=1/2
+ 1/2
N Е Ц„ехр
М= -------------
136
Глава 11
Подстановка уравнения (11.12) для ц„ и уравнения (11.10) для Е„ и суммирование по всем ms= +1/2 приводит к
Если (д$Н/кТ)« 1 (<70Н составляет около 1 см-1 для <7 = 2,0 и полей напряженностью 5000—10000 Э, а кТ при комнатной температуре равно 205 см1), то можно сделать следующее приближение:
Подстановка уравнения (11.17) в уравнение (11.16) и упрощение дает
М = Ng2$2H/UT.
(11.18)
Поскольку молярная восприимчивость связана с моментом соотношением
Х = М/Н,
можно записать, что
X =ЖР2/4/сТ.
(11-19)
(11.20)
Уравнение (11.20) представляет собой так называемый закон Кюри, который предсказывает линейную связь между восприимчивостью и величиной, обратной температуре. Из него следует, что отрезок, отсекаемый на оси ординат, равен нулю, т. е. % -> 0 при Т -> со. Экспериментально такой вид зависимости обычно не наблюдают. Линейные графики зависимости подобного вида обнаруживают для многих систем, но отсечение отлично от нуля:
х = с/(т-0).
(11-21)
Уравнение (11.21), где С = 1Уа2р2/4/с. а 0 дает поправку на ненулевую величину отсекаемого отрезка, представляет собой математическое выражение закона Кюри—Вейса. Для систем, которые магнитно не разбавлены (т. е. для чистых твердых парамагнитных соединений), обычна ненулевая величина отсечения. В этих системах межионные или межмо
Магнетизм
137
лекулярные взаимодействия [1] приводят к группировке соседних магнитных моментов и в результате отсекаемый отрезок отличается от нуля.
Для молекул, не имеющих орбитального углового момента, уравнение (11.20) записывается как
Nq2$2
х = ( +1)' (11'22)
При 5=1/2 оно приводится к виду (11.20) и объясняет так называемые спиновые магнитные восприимчивости комплексов с любым числом неспаренных электронов.
Как х, так и М—макроскопические величины. При описании магнитных свойств комплексов переходных металлов обычно используют микроскопический параметр, называемый эффективным магнитным моментом peff. Измеряется он в магнетонах Бора и определяется следующим образом:
/ А1/2
Ueff = Vw) (*Т)1/2=2>828(*Л1/2- (ii-23)
В этом выражении магнитная восприимчивость — это XPARA- Уравнение (11.23) можно получить, заменив g2S(S + 1) в уравнении (11.22) на p2ff и решив его относительно peff. Таким образом, любые эффекты, под влиянием которых S становится плохим квантовым числом, включаются в ^-фактор. (Вспомните о переменности 0-фактора в ЭПР, гл. 9.) Уравнение (11.24) можно использовать для расчета чисто спиновых магнитных моментов для различных значений S:
peff (чисто спиновый) = 0[S(S + 1)]1/2, (11.24)
где 0 равен 2,0 для электрона, не имеющего орбитального углового момента; peff измеряется в магнетонах Бора.
В табл. 11.3 представлены величины чисто спинового магнитного момента для различных чисел неспаренных электронов. Для многих
Таблица 11.3
Чисто спиновые магнитные моменты для различного числа неспаренных электронов
Число неспа-
ренных элек- S pcff, магнетон Бора тронов
1 1/2 1,73
2 1 2,83
3 3/2 3,87
4 2 4,90
5 5/2 5,92
6 3 6,93
7 7/2 7,94
138
Глава 11
комплексов ионов переходных металлов наблюдаемые величины магнитного момента близки к рассчитанным по уравнению, учитывающему только спин [1—9]. Однако для многих других комплексов наблюдаемые магнитные моменты и температурная зависимость восприимчивости отличаются от рассчитанных. В этих случаях проявляется действие и других эффектов и поэтому необходим более полный анализ.
11.3. УРАВНЕНИЕ ВАН-ФЛЕКА
Теоретические предпосылки
В этом разделе мы рассмотрим орбитальные вклады, а также анизотропию магнитной восприимчивости для молекул низкой симметрии. Взяв в качестве главной молекулярной оси ось z, запишем необходимую часть гамильтониана, учитывающую эти дополнительные эффекты:
Н = XL-S + $(L+geS)-H, (11.25)
где операторы Ln S имеют х-, у- и z-компоненты, а д„—g-фактор свободного электрона. Первый член правой части уравнения (11.25) описывает спин-орбитальное взаимодействие (Л.—константа спин-орбитального взаимодействия) и, как нетрудно видеть, от поля он не зависит. Другие члены суммируют спиновые и орбитальные вклады в момент электрона. [Отметим, что уравнение (11.25) очень напоминает уравнение (11.9).] Используя этот гамильтониан, надо подумать о выборе базиса. Для свободного бД-иона, когда взаимодействием L • S пренебрегают, хорошим базисом служат комплексные функции | + 2), | +1 > и т. д., поскольку они сами по себе являются собственными функциями гамильтониана Н. Поэтому при расчете полной матрицы с элементами <<р„ |LZ + g„Sz । недиагональные элементы отсутствуют.
В бД-комплексе с лигандами, определяющими оси х, у и z, удобный базис составляют действительные орбитали. Так, например,
dxy= — (d+2 — d_2)
1|/2 и
2= ~ (d+2 + d_2).
[/2
В этом базисе не диагональные элементы отличны от нуля, например:
(dxy | Lz | dx2 _yi') — — (d+2 | Lz | d+2) +
+ <J+2 | Lz | d-2) — (d-2 | Lz | d+2) — (d-2 | L2 | d-2) =
= 2+ 0 + 0-(-2) = — 2i.
Магнетизм
139
Вклад Sz в этот матричный элемент равен нулю, поэтому
<б/+ху | Sz | d^2_y2 > = | dx2_y2 > < + |SZ | + > = (0) = 0.
Ненулевые недиагональные элементы ответственны за искажение волновой функции основного состояния под действием наложенного поля (ранее мы получили матричныеА элементы для Lz и Sz, но полный гамильтониан определяется как 0 (L + geS) Н. Это искажение осуществляется в результате примешивания подходящих возбужденных состояний. Диагональные элементы называются зеемановскими членами первого порядка, а не диагональные—зеемановскими членами второго порядка. Если недиагональные члены отсутствуют, все диагональные матричные элементы должны иметь первый порядок по Н и результирующие энергии также должны зависеть от Н в первой степени.
Недиагональные элементы, связывающие состояния очень различных энергий, обычно малы по сравнению с разностью энергий, поэтому этой задачей обычно занимается теория возмущений. При обсуждении уравнения Рамзея (гл. 8) мы видели, что это приближение приводит к членам общего вида
у <Ф„ | ОР | Фт>2
А Ет — Еп
т^п
Л
где Ор — неопределенный оператор. Вызванное полем смешение возбужденных состояний используется для интерпретации парамагнитного вклада в химический сдвиг. В данном случае мы с помощью теории возмущений получаем член аналогичного вида:
v [<ф; | Lz + geSz | ф,.> 0Н]2
/, Е.-Е. (П-26)
j*i
Если конфигурация иона отличается от d’-конфигурации, то преимущества теории возмущений становятся очевидными, поскольку полная матрица должна быть большой.
В базис, предназначенный для расчета полной матрицы комплекса слабого поля, должны входить волновые функции, учитывающие элек-трон-электронное отталкивание в приближении кристаллического поля. Для комплекса сильного поля хорошим базисом будут действительные d-орбитали. Таким образом, при нахождении наилучшего базиса большое значение имеют относительные величины факторов, влияющих на энергию d-орбиталей. Приведем приблизительные величины некоторых эффектов.
А) слабое поле, первый ряд переходных металлов:
е2/г0. > КП 104см’’ 104см-1
возмущение низкосимметричного КП > XL • S
102—103 см-1 102 см’
140
Глава И
Б) сильное поле, первый ряд переходных металлов:
КП > е2/гу ^возмущение КП > XL-S 5 104см-1 3-103см-1 103см-1 102см-1
В) третий ряд переходных металлов:
КП > XLS > е2/г0-104 см-1 103 см-1 103 см-1
Г) лантаноиды:
XLS > е2/г,; > КП
5-103см-1 5-103см-1 103см-1.
Эффект магнитного поля составляет около 1 см”
До сих пор мы не принимали во внимание спин-орбитальное взаимодействие (член Л L • S). Для ионов первого ряда переходных металлов его можно учесть, добавив энергию взаимодействия X L • S к энергиям уровней в качестве возмущения их величины. Такой подход вполне приемлем, если только X L • S мало по сравнению с электрон-электронными отталкиваниями и влиянием кристаллического поля. Диагональные матричные элементы L • S рассчитываются в базисе из действительных орбиталей и добавляются к энергиям как поправки. Если спин-орбитальное взаимодействие велико, подход, основанный на возмущении, неприемлем. Например, и (знак относится к значениям ms электрона) имеют одно и то же значение щ_, = 3/2 и смешиваются под действием L • S.
Вывод уравнения Ван-Флека
В этом разделе мы вкратце рассмотрим, как проводят расчет эффектов кристаллического поля в интересующих нас молекулах или ионах с помощью гамильтониана уравнения (11.25). Прежде всего вернемся к обсуждению влияния различных факторов на магнитный момент. Если мы выпишем вклады в энергию данного состояния п зависящих от поля эффектов, рассмотренных в предыдущем разделе, то получим уравнение (11.27):
£„ = Ет„ + Н£<’’ XL’S Член, описывающий эффект Зеемана первого порядка (диагональные элементы) <- Н2Е„т (11.27) Член, описывающий эффект Зеемана второго порядка (недиагональные элементы)
Напоминаем, что проекция магнитного момента на направление поля определяется производной —дЕп/8Н [уравнение (11.12)]. Мы видим, что
М агнетизм
141
первый член, Етп, не дает никакого вклада в магнитный момент данного состояния, вклад второго члена не зависит от напряженности магнитного поля и только вклад третьего члена зависит от поля. Член Е{Г,п уравнения (11.27) мы получили при выводе уравнения Кюри, только теперь в него вошел и орбитальный момент. Вклад члена второго порядка зависит от £; — Ej. Эта разность может быть очень велика, если электронное возбужденное состояние близко по энергии к основному состоянию и имеет корректную симметрию.
Для того чтобы определить влияние указанных эффектов на восприимчивость, вернемся к выводу закона Кюри и перепишем уравнение (11.15), заменяя ехр( —£„//сТ) на
/ -Етп-НЕ(1}п-Н2Е{2}„+ ...\ / НЕ^ \ /-£<°>\
ехр(------------гг------------) “ (‘ - - 111'28)
и принимая также, что
Ц„= = -£(1>- 2Н£<2’. (11.29)
дН
В результате уравнение (11.15) приводит к
(11.30)
Согласно уравнению (11.30), для парамагнитных соединений М = 0 при Н = 0, если только
(11.31)
Расписывая числитель и пренебрегая в уравнении (11.30) членами более высокого порядка, чем £<2], а также произведением £<2]£(1], мы из уравнений (11.30) и (11.31) при условии, что % = MJH [уравнение (11.19)],
получаем
(11.32)
142
Глава 11
где Етп— вклад взаимодействия /.L-S и т. д. Для основного уровня член Е*0] всегда равен нулю; для состояния с более высокой энергией величина, приводящая к этому энергетическому члену в отсутствие поля, заменяется на Е(„0). Е(„1) включает msg$H и другие вклады первого порядка, в Е{2}„ входят вклады зеемановского эффекта второго порядка.
Таким образом, восприимчивость определяется средневзвешенными по заселенностям восприимчивостями уровней. Следовательно, г-кратно вырожденный уровень имеет г компонентных состояний, каждое из которых должно быть включено в сумму в уравнении (11.32). Как используется это уравнение, станет более понятным после рассмотрения некоторых примеров.
Применение уравнения Ван-Флека
Мы продемонстрируем применение уравнения Ван-Флека [уравнение (11.32)] на примере основного состояния свободного иона металла с квантовым числом J (реализуется взаимодействие Рассела — Саундерса). Во всех примерах, которые рассматриваются в этом разделе, берется средневзвешенное по заселенностям индивидуальных моментов уровней. Вырожденность 2J +1 снимается магнитным полем, и относительные энергии результирующих уровней выражаются как гп,д$Н. Мы рассматриваем только основной уровень Е*0^ и Е<2„\ которые принимаются за нуль. (При анализе больцмановских заселенностей выбор нулевого уровня энергии произволен, для удобства мы полагаем энергию основного уровня в отсутствие поля Н, т. е. Е*0^, равной нулю.) Уравнение (11.32) принимает вид
nW
kT(2J + l)
JY^P2 /J2 + (J-l)2+...+ (-J + l)2 +(-J)2 kT \ 2J + 1
JVf/P /J(J + 1)(2J+1) \ У<?2Р2
------ ---------------- =---------J (J + 1).
кТ \ 3(27 + 1)' J ЗкТ ’
(11.33)
Вводя Цец для свободного иона получаем
Цегг = в [J(J + 1)]1/2 (магнетона Бора) (11.34)
Согласно схеме Рассела — Саундерса, для свободного иона д-фактор определяется следующим образом (это выражение приводится без вывода [2]):
а = 1 _|_ S(S + 1) ~ Е(Е+1) + J(J + 1) 2J (J + 1)
(11.35)
Магнетизм
143
Из этого уравнения следует, что в свободном ионе вклады в p.eff обусловлены как спиновым, так и орбитальным угловым моментом. Более того, если L=0, то J = S. При этом <7 = 2,00 и уравнение (11.34) сводится к уравнению (11.24) для чисто спинового магнитного момента.
Рассмотрим еще один пример. Продемонстрируем применение уравнения (11.32) на примере ^’-комплекса Ti3+ [3]. На рис. 11.3 показано расщепление термов газообразного иона кристаллическим
Г8
2^/
(10) \
10DQ
4 Энергия
л + ^н + 1~
х вн -г 4
-Л/2
X 4 /Нг
2. 3 Л
Газообразный Поле 0h AL-S Н
ион
Рис. 11.3. Расщепление состояния 2D газообразного иона полем Oh, z.L S и магнитным полем. Вырождение уровней указано в скобках, а энергии приведены справа.
полем, спин-орбитальным взаимодействием и магнитным полем. Выражения для энергий, приведенных на рисунке, получают из волновых функций, вытекающих из приближения слабого кристаллического поля, при действии на эти волновые функции операторов XL-S и Р (L + geS) • Н уравнения (11.25). Поскольку параметр ЮЛ? в октаэдрическом комплексе, как правило, довольно большая величина, проводя расчет восприимчивости с помощью уравнения (11.32), можно пренебречь состоянием 2Е. Рассмотрим эту задачу в целом, начиная с основного уровня 2Т2; пронумеруем состояния по мере возрастания их энергий. Запишем термы Е(„о>, Б*,1’ и Е(„2> для состояния 1—4: —X/2, —X/2, Л и л (см. гл. 10); 0, 0, — pH и +РН; — 4/3(Р2Н2/Х), 0, +4/3(Р2Н2/Х) и +4/3(Р2Н2/Х). Подстановка ука
144
Глава 11
занных величин в уравнение (11.32) и умножение каждого терма на вырождение соответствующего уровня дает
+ 2 (] — (2)(0)ехр( | +
\кТ J У и\2к1у_
+ еХР(2И7) + еХр1
+ 2
Поскольку X = (Яр2/3/сТ)Иегг
Heff =
- , / ЗХ
2 + ехр\~2kT
где р — магнетон Бора. Обратите внимание, что в соответствии с проведенным нами анализом закон Кюри не должен выполняться. По мере приближения Т к бесконечности p2ff стремится к нулю. Если Т мало, то мал и p2ff, если Т приближается к нулю, то это уравнение перестает выполняться, так как gfiH«кТ. Если X стремится к нулю, то Цеп стремится к 3. И наконец, когда Т стремится к нулю, мы получаем интересный результат, а именно что в этом случае система с неспаренным электроном имеет нулевую восприимчивость. Такой результат обусловлен сокращением спинового и орбитального вкладов. Справедливость этого вывода подтверждается экспериментально.
Рассматривая эту задачу, мы пренебрегали какими-либо вкладами возбужденного уровня 2Ед. Однако сделанное выше приближение справедливо для многих систем. Проводя исследование методом ЭПР, чувствительность которого выше, можно определить вклад возбужденного состояния в величину ^-фактора (см. гл. 12). При реализации спин-орбитального взаимодействия основной уровень (Г8) и возбужденный уровень Г8 состояния 2Е8(Г3 х Г6 = Г8) могут смешиваться [4], g-фактор при этом меняется от 40Н/ЗХ до 4Х/А + 40Н/ЗХ, где А — это lODg. Зеемановский член второго порядка смешивает основной уровень с возбужденным, а степень смешивания зависит от А.
Далее мы рассмотрим [9] комплекс хрома(Ш) (d3). Октаэдрическое поле приводит к основному состоянию */12 и возбужденным уровням 4Т2 и 4ТИ что показано для квартетных состояний в диаграммах Танабе—Сугано. Энергия 4Т2 на ~ 18000 см-1 выше, чем энергия 4Л2, по-
Магнетизм
145
этому вкладом последнего в восприимчивость можно пренебречь. Поскольку основное состояние — орбитальный синглет (Л), то вклада в магнитную восприимчивость он не дает (см. далее). Магнетизм определяет уравнение, выведенное для чисто спинового магнитного момента и S = 3/2. Рассмотрим далее влияние тетрагонального искажения на комплекс хрома(Ш). Как показано на рис. 11.4, это искажение снимает
Рис. 11.4. Расщепление состояния 4Л2 тетрагональным полем D. (D — параметр тетрагонального расщепления или расщепления в нулевом поле).
вырожденность состояний с ms = + г/2 и ms = + 3/2. Расщепление тетрагональной компонентой описывается параметром D. Поскольку такое расщепление существует и' в отсутствие поля, то оно является одним из многих эффектов, которые носят название расщепления в нулевом поле. При аксиальном расщеплении в нулевом поле его можно представить с помощью гамильтониана DS,?. Если приложенное поле параллельно главной молекулярной оси, восприимчивость этой системы получают путем введения величин для Е', которые для двух уровней, показанных на рис. 11.4, равны соответственно х/2 gz0 и 3/2gz0. Для более низкий энергетический уровень принимается за нуль, а для более высокого уровня при величине D мы имеем
/ [гехр(О) + 2ех₽(-^г)]
Таким образом, при D/kT « 1 (это справедливо для очень небольших искажений или для очень высоких температур) выражение для Xz сводится к (5/4) N д2$2/кТ, в то время как формулу (11.20) для S= 1 /2 получают при условии, что Т стремится к нулю или D велика *. Для того чтобы рассчитать среднюю восприимчивость порошкаЛ необходимо определить и используя компоненты х и у, L и S в уравнении (11.25) с добавлением члена DSi2- Таким путем можно рассчитать анизотропию %.
* Экспериментально найденное значение D для псведооктаэдрического Сг3 + составляет приблизительно 0,1 см “ 1 из-за спин-орбитального примешивания возбужденных состояний.
10-574
146
Глава 11
В качестве последнего примера рассмотрим комплекс никеля(П) с небольшим тетрагональным искажением. Расщепление для этого случая показано на рис. 11.5.
(0)exp(-0)+ (-fzP)2-exp
Хп кТ
<Лехр
кТ и
JV 1 + 2ехр
( о\ 2Ng2fi2 еХр( кт)
= ~кТ
1 + 2 ехр
Если D « кТ, то получается следующее выражение:
Для (NH4)2Ni(SO4)2-6Н2О экспериментально найденное [4] значение gz составляет 2,25, a D= —2,24 см'1, что приводит к величине Хц=4260-•1(Г6 см3/моль. Эксперимент дает 4230-10“ 6 см3/моль, тогда как, согласно уравнению (11.20), в котором учитывается только чисто спиновое взаимодействие, Хц должна быть равна 3359 • 10“6 см3/моль.
В этом разделе необходимо указать еще на один момент. Если электроны делокализованы на лигандах из-за ковалентности связи металла с лигандом, матричные элементы, соответствующие орбитальному угловому моменту, снижаются ниже величины, рассчитанной с исполь-
Энергия +0HZ + D
-рнг + D
О
(1)
Газообразный Кубическое Тетра- Н ион поле зональное поле
Рис. 11.5. Расщепление состояния иона никеля(П) в тетрагональном поле.
Магнетизм
147
зованием металлоцентрированного оператора L, примененного к волновым функциям орбиталей металла. Для компенсации такого снижения можно использовать kL, (где к—коэффициент орбитальной редукции; k < 1), чтобы ввести поправку на делокализацию электронной плотности на лиганды, снижающую орбитальный угловой момент.
11.4. ИСПОЛЬЗОВАНИЕ
ДАННЫХ ИЗМЕРЕНИЯ ВОСПРИИМЧИВОСТИ
Спин-орбитальное взаимодействие
Если уравнение (11.34) применять к комплексам ионов редкоземельных металлов, то получается прекрасное соответствие между рассчитанными и экспериментальными значениями восприимчивости (данные для некоторых трехзарядных ионов представлены в табл. 11.4)
Такое прекрасное соответствие обусловлено тем, что кристаллическое поле лигандов неэффективно гасит орбитальный угловой момент
Таблица 11.4
Рассчитанные и экспериментально найденные величины магнитного момента для ионов некоторых трехвалентных редкоземельных металлов
Элемент Конфигурация Терм peff, магнетон Бора
расчетный экспериментальный
Се 4/5s5p6 2F5/2 2,54 2,4
Рг 3H4
3,58 3,5
Nd 4/s ^9/2
3,62 3,5
Pm 4/
2,68
Sm 4/з 6Я5/2 1,5
0,84
Eu 4/ 7f г 0
0 3,4
Gd 4/ ”$7/2 7,94
8,0
Tb 4/s
9,72 9,5
Dy 4/ 6 M n 1 5/2
Ho 4/° sl8 10,63
Er 4/ri 4^15/2 10,60 10,4
Tm 4/2 X 9,59 9,5
Yb 4/13 2F 7/2 7,57 7,3
4.54 4,5
ю’
148
Глава 11
Таблица 11.5 Рассчитанные и экспериментально найденные величины магнитных моментов для комплексов 3</-иоиов (расчет проводился по уравнению (11.34) и формуле для магнитной восприимчивости, обусловленной чисто спиновым магнитным моментом)
V [он Конфигура- g[J(J + I)]'2 2[S(S + 1)] Heff (эксп.)
ЦИЯ Терм
Ti3+, V4+ 3d1 2d3/2 1,55 1,73 1,7—1,8
V3 + 3d2 3f2
1,63 2,83 2,6—2,8
Cr3 + , V2+ 3d3 ^3/2
0,77 3,87 — 3,8
Mn3 + , Cr2+ 3d4 5П
0 4,90 ~ 4,9
Fe3+, Mn2+ 3d5 6S5,2
5,92 5,92 ~ 5.9
Fe2 + 3d6 X
6,70 4,90 5,1—5,5
Co2 + 3d7 ^9/2 3,87
6,63 4,1—5,2
Ni2 + 3d8 X
5,59 2,83 2,8—4,0
Cu2 + 3d9 2d5/2
3,55 1,73 1,7—2,2
электронов, находящихся на внутренних Ф/’-орбиталях. Совершенно другие результаты получаются для ряда З^-ионов переходных металлов. В этом случае уравнение для чисто спиновой магнитной восприимчивости дает лучшее соответствие с экспериментальными данными (табл. 11.5).
Таким образом, во многих комплексах орбитальный вклад в значительной степени гасится кристаллическим полем. Известна очень прЪс-тая модель, которая позволяет предсказать, в каком случае полного гашения орбитального момента не происходит. Если электрон может занимать вырожденные орбитали и, следовательно, вращаться вокру|г оси, то он будет характеризоваться орбитальным угловым моментом. На орбитали, на которую перемещается электрон, не должно быть электрона с таким же самым спином.
Например, в октаэдрическом d1 -комплексе электрон может занимать при вращении вокруг оси z орбитали dxz и dyz, и в результате комплекс характеризуется орбитальным угловым моментом. В октаэдрическом высокоспиновом ^-комплексе как на dxz-, так и на ф^-орбитали находятся электроны с тем же самым спиновым квантовым числом, поэтому здесь орбитальный угловой момент отсутствует. Используя эту весьма приближенную модель, можно предсказать, что следующие октаэдрические комплексы должны характеризоваться эффективным гашением всего орбитального вклада в момент.
Магнетизм
149
Высокоспиновые системы
♦3 4.3 л1 4.3 ^2 4.6 ^2 4.6 „3
f2g> ^2gegi ^2gegi ^2дед9 ^2дед •
Низкоспиновые системы
f29 и t29e9.
Один электрон в е9-конфигурации может занимать только d,i- и 4У2_),2-орбиталь, поэтому вращаться вокруг оси он не будет. Если в состоянии Е электрон находится в подходящем поле лигандов на dxy- и dx2_ ^-орбиталях, тогда вклад орбитального момента возможен. Орбитальный момент гасится в е1-, е2-, e2t2-, e3t3-, и е4г2-тетра-эдрических высокоспиновых комплексах.
Орбитальные моменты многих молекул с основными состояниями Л2й и Ед отличаются по величине от значений, которые дает формула, учитывающая чисто спиновые магнитные моменты. Такое различие обусловлено двумя причинами: 1) примешиванием возбужденных состояний вследствие спин-орбитального взаимодействия и 2) зеемановскими эффектами второго порядка (парамагнетизм, не зависящий от температуры). Например,
(4effC429) = р (чисто спиновый) А - (И.36)
Последний член описывает не зависящий от температуры парамагнетизм, наведенный полем. Член 4Х/10£)</ обусловлен примешиванием возбужденных состояний в результате спин-орбитального взаимодействия. В комплексах более низкой симметрии момент, обусловленный примешиванием возбужденного состояния, выражается как
/ 2Х \ 4А'В2
9) = Ц (чисто спиновый) ^1 - (11.
Как можно видеть из проведенного выше обсуждения, магнитные моменты комплексов переходных металлов часто достаточно характерны для основного электронного состояния и структуры комплекса. В литературе известно много примеров использования такого рода данных. В качестве иллюстрации рассмотрим несколько комплексов никеля(П) и кобальта(П).
В октаэдрическом поле комплекс никеля(П) имеет орбитально невырожденное основное состояние 3242(t2ee9), поэтому никакого вклада от спин-орбитального взаимодействия ожидать не следует. Измеренные величины моментов варьируют в интервале 2,8 — 3,3 магнетона Бора, что очень близко к 2,83 магнетона Бора, которое получается, если учитывать чисто спиновый магнитный момент. Величины моментов октаэдрических комплексов несколько превышают значения моментов, имеющих чисто спиновый характер, из-за небольшого смешивания с мультиплетным возбужденным состоянием, в котором заметную роль играет
150
Глава 11
спин-орбитальное взаимодействие. Основное состояние тетраэдрического комплекса никеля(П) — 3ТЬ т. е. по существу (e4f2e), и возможен большой вклад орбитального момента. В результате, если даже в ок1аэд-рическом и тетраэдрическом комплексах никеля(П) имеются два неспаренных электрона, магнитные моменты тетраэдрических комплексов составляют около 4, а октаэдрических комплексов 3,3 магнетона Бора, или ниже. Экспериментально установлено [8, 10], что магнитные моменты NiClJ-, Ni(FMOTA)4+ [10а] (ГМФТА — гексаметилфосфотриамид) и NiX2-2(C6H5)3AsO (X — галоген) [106] превышают 4 магнетона Бора. Нихольм [7] предположил, что между величиной момента и искажением тетраэдрической симметрии комплексов никеля(П) существует обратная зависимость. Комплекс NiX2-2(С6Н5)3Р, как известно, сильно искажен, и его магнитный момент составляет примерно 3 магнетона Бора. Структуры №Х2-2ГМФТА и СоХ2-2ГМФТА (где X—СГ, Вг“, I-и NO3) установлены после проведения комбинированных магнитных и спектральных исследований [12]. Эта работа служит хорошим примером, демонстрирующим применение указанных методов.
Основное состояние октаэдрических комплексов кобальта(П)—4Т1я, и орбитальный вклад в момент предположительно должен быть большим. Примешивание возбужденного состояния несколько снижает момент, но тем не менее он обычно превышает 5 магнетонов Бора (чисто спиновый магнитный момент равен 3,87). Основное состояние тетраэдрических комплексов кобальта(П)—4Л2, и магнитный момент может быть небольшим, вероятно, может приближаться к значению, обусловленному вкладом чисто спинового магнитного момента. Однако возбужденное магнитное состояние в тетраэдрических комплексах сравнительно мало по энергии и может смешиваться с основным состоянием. Рассчитанные [13] и экспериментально установленные магнитные моменты лежат в интервале 4—5 магнетонов Бора. В соответствии с уравнением (11.36) для тетраэдрических комплексов кобальта(П) [13] обнаружена обратная зависимость между величиной момента комплекса и величиной Dq.
11.5. ВНУТРИМОЛЕКУЛЯРНЫЕ ЭФФЕКТЫ
Выше предполагали, что в твердом веществе электронные спины отдельных ионов металла не взаимодействуют. Теперь обратимся к молекулам, содержащим более одного иона металла с неспаренным спином, например
Магнетизм
151
Как правило, взаимодействие между металлоцентрированными орбиталями металла, если оно существует, можно записать для пары i, j гамильтонианом
H=-2JSrS;, (11.38)
где J—константа обменного взаимодействия, или параметр обмена, выраженный в единицах энергии. Его следует отличать от квантового
Рис. 11.6. Структура Си2(СН3СО2)4-2НЛО. Расстояние Си —Си составляет 2,6 А.
числа J. При таком гамильтониане параметр обмена J антиферромагнитного взаимодействия — величина отрицательная (что приводит к рпа-риванию электронов), a J ферромагнитного взаимодействия — величина положительная. (Единой формы записи гамильтониана (11.38) пока не выработано: — 2J часто заменяют на J, 2J и — J.)
Классическим примером системы с двумя металлическими центрами может служить дигидрат димера ацетата меди(П). Структура этой молекулы показана на рис. 11.6, где в качестве оси z взята ось связи металл-металл. Ионы меди(П) имеют ^-конфигурацию. Установлено, что при низких температурах данное соединение диамагнитно, а при близких к комнатной парамагнитно. Молекулу этого комплекса можно рассматривать как систему с двумя молекулярными орбиталями, представляющими собой по существу орбитали металла dxi _у2 (со значительным вкладом мостиковой ацетатной группы). На рис. 11.7 дано упрощенное представление этой части диаграммы МО*. (Напоминаем, что триплетное состояние и обменные взаимодействия рас-рассматриваются в главе, посвященной ЭПР.) Если Д < К, где Д — разность энергий МО между связывающей и разрыхляющей МО, а К — энергия спаривания спинов, то, следовательно, рассматриваемая система
* Его называют путем сверхобмена с участием мостиковых ацетатных групп. Оценки величины вклада прямого обменного взаимодействия, которое включает непосредственное перекрывание двух орбиталей атомов меди, весьма противоречивы.
152
Глава 11
ферромагнитна. Величина J в гамильтониане уравнения (11.38) для двух d1- или й9-ионов металла в димере выражается как
1 = (К-Д)/2. (11.39)
В основном состоянии Си2(СН3СО2)4 -2Н2О диамагнитна, но возбужденное триплетное состояние близко к нему по энергии и при умеренных температурах заселено. Установлено, что —21 = 284 см-1 [14]. Путем добавления гамильтониана уравнения (11.38) к g[iSzHz мы можем оце-
zz \ Рис. 11.7. Упрощенное представле-
d^-y2 / I Ч dx2_vz ние взаимодействия двух dx2_ 2-ор-। биталей металла в Си2(СН3СО)2-
\ I -2Н2О, приводящего к двум невы-
х' рожденным уровням.
МО
нить энергии различных уровней в этой системе и, используя уравнение (11.32), рассчитать изменение восприимчивости с температурой. Определим полное спиновое квантовое число системы S как
S = S1+S2.
Соответственно
s2= s?+ S2 + 2SrS2
и при перегруппировке Sx • S2 выражается как
Si -s2 = |(S2- S?- S2). (11.40)
Поскольку 52ф = 5 (S + 1) ф, то **
-2151-52ф = - J[S(S + IJ-SJSi + 1)-S2(S2 + 1)]\|/. (11.41)
Для дигидрата димера ацетата меди(П)
1
5, = S2 = — и S = 0 или 5=1.
2
Если 5 = 0, то -Л в то время как для 5 =1 - м о 1 1 4 ' -£ I LU II 1 11
** Комбинация — + 1) — S2(S2 + 1) является константой для каждого
уровня димера, и ее часто опускают, т. е. нулевой уровень энергии меняется.
Магнетизм
153
-lj + + дрНг
~iJ \~2J
-{j - 9fiHz,-2J - gf!Hz
0
Энергия разделения
Рис. 11.8. Энергии уровней в димерной ^’-системе.
Гамильтониан H = gfiSzH—2JSt-S2 приводит к результатам, представленным на рис. 11.8. Подставляя эти величины в уравнение (11.32), получаем
2(ИР)2 / J \ 2д2р2
X кТ еХР\2/сТ/ _ кт _
N ~ , / J \ / -3J\ ~ / _2Z\ ’
3 IkT) + ™\~2kT ) 3 + ехР [~ТГ;
Перегруппировка этого выражения дает
_ 2Л/УР2 1
Х ЗкТ . ехр(—2J//cT) ’ + 3
Поскольку Np2/3/c составляет около 1/8, мы при д = 2 получаем
1 1
X --------------------(11.42)
Т t ! ехр( —2J//cT) V
Из этого выражения следует, что х стремится к нулю, если Т стремится к бесконечности, и х становится малым, если мало Т. Поэтому х должна характеризоваться максимумом, который можно определить, приравняв нулю 8 In X/ 8 Т. Отсюда мы находим, что
4
J/kTc= (11.43)
т. е. х достигает максимального значения при
154
Глава 11
Если/ « /с Г или Т » Тс, восприимчивость подчиняется закону Кюри — Вейса, т. е.
3 1
4 Г+0’
(11.44)
где 0 = — J)1к.
Кроме того, если — J -> + оо, % стремится к нулю.
Величина J, обнаруженная для димерных карбоксилатов меди [15, 16], сильно зависит от мостиковой карбоксильной группы; например, 2J равно — 284 см “ 1 для дигидрата апетата и — 339 см “ 1 для дигидрата бутирата. При замене воды на другие основания Льюиса наблюдаются резкие изменения J.
В работе [17] описаны димерные системы железа(Ш) с мостиковыми гидрокси- и оксигруппами
Н
Н
Величина J первого комплекса составляет около — 8 см \ в то время как для второго* комплекса она равна —90 см-1.
11.6. РАВНОВЕСИЕ ВЫСОКИЙ СПИН—
НИЗКИЙ СПИН
Если рассмотреть диаграммы Танабе—Сугано для октаэдрических /4-, d5-, d6- и /’-комплексов, то можно увидеть, что для некоторых величин Dq/B основное состояние меняется от высокоспинового до низкоспинового. В /4-, /5-, d6- и /’-комплексах, таким образом, меняются соответственно состояния 5Ед и 3Т1д, 6А1д и 2Т2д, 5Т2д и 1А1д, 4Т1д и 2Ед. Если поле лигандов таково, что два состояния близки по энергии, возбужденное состояние заселено термически и система состоит из равновесной смеси двух форм. В работе [18а] описаны многочисленные исследования этого явления. Мы же рассмотрим комплексы железа(П) с лигандами типа оснований Шиффа, которые образуются при конденсации tren[N(CH2CH2NH2)3] с 2-пиридинкарбоксальдегидом, поскольку эти системы типичны и исследованы [186] как в твердом состоянии, так и в растворах. Комплекс такого типа показан на рис. 11.9, где, чтобы упростить картину, приведен только один из пиридиновых альдиминов. Синтезирован ряд таких соединений, в которых R, R' и R"—Н или СН3.
* Эта величина спорна, поскольку в комплексе содержится примесь желе-за(П1) [Moss Т., et al., J. Chem. Soc., Chem. Comm., 1972, 263].
Магнетизм
155
Обозначим эти комплексы следующим образом. I—[Fe(Py)3 tren]2+, если R = R' = R" = Н; II - [Fe(6-CH3Py) (Py)2tren]2+, если R = R' = = Н и R" = CH3; III— [Fe(6-CH3Py)2 (Ру) tren]2 +, если R = H и R' = = R" = CH3; IV — [Fe(6-CH3Py)3 tren]2 +, если R = R'= R" = СН3.
Рис. 11.9. Структурная формула трмс-[4-[(6-К)-2-пиридил]-3-аза-3-бутенил}аминожелеза(П).
Для некоторых из этих комплексов и в жидкой, и в твердой фазах обнаружено существование равновесия между низкоспиновым lAig(tfg) и высокоспиновым 5F2g(t2ge2) состояниями. Комплекс I низкоспиновый и при комнатной, и при более низких температурах, тогда как для комплексов II и III характерно состояние спинового равновесия как в твердом состоянии, так и в растворе. Комплекс IV при температурах, превышающих 180 К, является существенно высокоспиновым. В твердом состоянии спиновое равновесие в очень большой степени зависит от аниона. Термодинамические параметры такого взаимного превращения можно определить из температурной зависимости восприимчивости; так, установлено, что для комплексов II и III в растворе АН составляет соответственно + 4,6 и + 2,8 ккал/моль. Рентгеноструктурный анализ кристаллов показывает, что метильные группы — заместители в пиридиновом цикле — взаимодействуют с циклом. Таким образом, поле лигандов в комплексе IV ослаблено в такой степени, что этот комплекс представляет собой высокоспиновое соединение, тогда как комплекс I низкоспиновый. В молекуле IV, как обнаружено, среднее расстояние между азотом и металлом меньше приблизительно на 0,12 А, что обусловлено переходом из высокоспинового состояния в низкоспиновое.
11.7. ИЗМЕРЕНИЕ МАГНИТНОЙ ВОСПРИИМЧИВОСТИ
В этом разделе мы кратко рассмотрим методы измерения объемной магнитной восприимчивости и по ходу изложения укажем, в каких работах эти методы рассматриваются достаточно подробно. При измерении объемной магнитной восприимчивости по методу Гойе [20а] длинную стеклянную трубку постоянного сечения заполняют твердым веществом или раствором и подвешивают в однородном магнитном поле. Образец взвешивают при наложении поля и в отсутствие поля, разность результатов взвешивания соотносится с восприимчивостью и напряженностью поля. Если используется эталон с известной во
156
Глава 11
сприимчивостью, определять напряженность поля нет необходимости. Эванс [206] сообщил об остроумном и недорогом устройстве для рутинных измерений магнитной восприимчивости методом Гойе.
По методу Фарадея [19] в неоднородном магнитном поле подвешивают небольшое количество образца, так чтобы во всем объеме последнего Н(дН/8Х) была постоянной. Чувствительность метода очень высока, поэтому для измерений достаточно небольших количеств вещества; этот метод позволяет также измерять восприимчивость раствора.
Определять восприимчивость в широком диапазоне температур, вплоть до температуры жидкого гелия удобно также с помощью магнетометра [21]. Изменение индуктивности катушки при введении в нее образца можно связать с восприимчивостью последнего. В работах [22, 23] описано определение восприимчивости с использованием обычного моста индуктивности. Описан также исключительно чувствительный сверхпроводящий квантовый магнетометр с элементом Джозефсона [24].
Исследуя восприимчивость монокристаллов, можно определить величину ее анизотропии [25—28]. Как мы увидим в главах, посвященных ЭПР и ЯМР комплексов ионов переходных металлов, эти данные применяются в нескольких важных областях. Анизотропию магнитной восприимчивости обычно определяют методом Кришнана, устанавливая критический момент вращения. В статье [31] рассматривается использование метода ЯМР для измерения магнитной восприимчивости веществ в растворе. Раствор парамагнитного комплекса, содержащий внутренний стандарт, вводят в объем между двумя концентрическими трубками. Раствор того же самого инертного стандарта в том же самом растворителе, в котором растворен комплекс, вводят во внешнюю часть конструкции. В этом случае наблюдаются две линии стандарта, причем линия вещества, введенного в раствор парамагнитного комплекса, соответствует более высокой частоте. Сдвиг линии внутреннего стандарта? в парамагнитном растворе относительно диамагнитного раствора ДН/Н связывают с разностью объемной восприимчивости АХ„ двух жидкостей:
АН _ 2л Av Н ~ 3
Удельная восприимчивость растворенного вещества Хд выражается как
Y 3Av , Xo(4-<U
2тыт т
где Av—частотное разделение двух линий, выраженное в герцах; v— зондирующая частота; т—масса вещества в миллилитре раствора; Хо — удельная восприимчивость растворителя; d0 — плотность растворителя, a ds — плотность раствора. Следует отметить, что, определяя зависимость восприимчивости от температуры, необходимо вводить поправки на изменение плотности с температурой [32].
Магнетизм
157
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. Ginsberg А. Р„ Lines М. Е., Inorg. Chem., 11, 2289(1972).
2. Herzberg G., Atomic Spectra and Atomic Structure, 2nd ed., Dover Publications, New York, 1944, p. 109.
3. Kotani M., J. Phsy. Soc. Japan., 4, 293(1949).
4. Bleaney B., Stevens K. W. H., Rep. Progr. Phys., 16, 108(1953).
5. Mabbs F. E., Machin D. J., Magnetism and Transition Metal Complexes John Wiley, New York, 1973.
6. a. Figgis B. N., Lewis J., The Magnetic Properties of Transition Metal Complexes, in Progress in Inorganic Chemistry, Vol. 6, ed. by Cotton F. A., Interscience, New York, 1960.
6. Figgis B. N., Lewis J., The Magnetochemistry of Complex Compounds, in Modern Coordination Chemistry, ed. by Lewis J. and Wilkins R. G., Interscience, New York, 1960.
7. Nyholm R. S„ J. Inorg. Nuclear Chem., 8, 401(1958).
8. Gill N. S„ Nyholm R. S., J. Chem. Soc., 1959, 3997.
9. Carlin R. L, J. Chem. Educ., 43, 521(1966).
10. a. Donoghue J. T., Drago R. S„ Inorg. Chem., 1, 866(1962) и ссылки в этой работе.
б. Cotton F. A., et al., J. Amer. Chem. Soc., 83, 4161(1961) и ссылки в этой работе.
И. Venanzi L. М., J. Chem. Soc., 1958, 719.
12. Donoghue J. T., Drago R. S., Inorg. Chem., 2, 572(1963).
13. Cotton F. A., et al., J. Chem. Soc., 1960, 1873.
14. Kokoszka G. F., Gordon G„ Trans. Metal Chem., 5, 181(1969).
15. Figgis B. N„ Martin R. L„ J. Chem. Soc., 1956, 3837.
16. Kokot E., Martin R. L., Inorg. Chem., 3, 1306(1964) и ссылки в этой работе.
17. Schugar Н. J., Rossman G. R„ Gray H. B„ J. Amer. Chem. Soc., 91, 4564(1969).
18. a. Martin R.L., White A.H., Trans. Metal Chem., 4, 113(1968).
6. Hoselton M. A., Wilson L. J., Drago R. S., J. Amer. Chem. Soc., 97, 1722(1975).
19. См., например, Mulay L. N., Mulay I. L„ Anal. Chem., 44, 324R(1972).
20. a. Figgis B., Lewis J., Techniques of Inorganic Chemistry, ed. by Jonassen H., Weissberger A., Vol. IV, p. 137, Interscience, New York, 1965.
6. Evans D. F., J. Physics E; Sci. Instr., 7, 247(1974).
21. Foner S„ Rev. Sci. Instr., 30, 548(1959).
22. Pillinger W.L., Jastram P.S., Daunt J.G., Rev. Sci. Instr., 29, 159(1958).
23. McElearney J. N., Shankle G. E., Schwartz R. W„ Carlin R. L„ J. Chem. Phys., 56, 3755(1972).
24. Dawson J. W., et al., Biochem., 11, 461(1972) и ссылки в этой работе.
25. Mitra S., Trans. Metal. Chem., 7, 183(1972).
26. Gerloch M., et al., J. Chem. Soc., Dalton, 1972, 1559; ibid., 1972, 980 и ссылки в этой работе.
27. Horrocks W. D., Jr., Sipe J. P„ III, Science, 177, 944(1972).
28. Figgis B. N„ Wadley L. G. B., Gerloch M„ J. Chem. Soc., Dalton, 1973, 238.
29. Krishnan K. S„ et al., Phil. Trans. Roy. Soc., A, 1933, 231, 235.
30. Gordon D. A., Rev. Sci. Instr, 29, 929(1958).
31. Evans D. F„ J. Chem. Soc, 1959, 2005.
32. Ostfeld D., Cohen I. A., J. Chem. Educ, 49, 829(1972).
158
Глава 11
Упражнения
1. Рассмотрите следующую систему энергетических уровней, которая должна была бы появиться при расщеплении в нулевом поле состояния с S = 1:
£(|0» = - ^D - ^2И2 ,
4 ’ 3 3D
e(|-i» = 1d-9ph+
j bD
£(|l»=lD + gPH+ gW2.
3 OD
Здесь D—параметр расщепления в нулевом поле, другие символы имеют свой обычный смысл. Определите молярную парамагнитную восприимчивость этой системы.
2. Рассмотрим октаэдрический комплекс Fe(II)
R"
Для многих систем низкоспиновое состояние 1А1д лежит ниже высокоспинового состояния 5Т2д менее чем на 1000 см-1, так что при комнатной температуре оба состояния в заметной степени заселены. Состояние ‘А невырожденно, а состояние 5Т 15-кратно вырождено (почему?). Математически состояние Т в октаэдрическом поле эквивалентно состоянию Р в свободном ионе, т.е. можно считать, что L= 1 и ML= — 1, 0 и +1. Положим, что состояние ‘А не сдвигается при всех возмущениях.
Магнетизм
159
а. Запишите 15 базисных функций для состояния 5 Г, используя скобочные обозначения Дирака и эквивалентность Т и Р. (Вспомните, что волновая функция с ,,Ml‘ = 1 и Ms = — 2 записывается как |1, — 2>. При такой формулировке „АД“ и Ms часто называют «хорошими квантовыми числами».)
б. Предположим теперь, что комплекс тригонально искажен; при этом ось третьего порядка октаэдра сохраняется. Феноменологически влияние такого искажения описывается гамильтонианом
- /2 - \
Я = 8 --L? , V )
где 5—.параметр тригонального искажения или расщепления в нулевом поле, a Lz— z-компонента эквивалентного орбитального углового момента. Как это часто бывает, химические факторы, влияющие на величину 8, неясны, и полученные в эксперименте значения могут помочь установить эти факторы. Зеемановский гамильтониан при магнитном поле, ориентированном вдоль оси z, записывается как
Н = f,Hz(Lz + geSz).
Здесь мы рассматриваем только z-направление (параллельное оси третьего порядка), поэтому в итоге мы должны получить выражение для Хл. Ниже демонстрируется использование гамильтониана и волновых функций:
— 2> = (j- Ml J| 1, — 2> =(д- 1JI 1, -2> =
= -||1, ~2>SZ|1, -2> = MS|1, — 2> = — 2| 1, — 2>.
Примените два члена рассмотренного выше гамильтониана к волновым функциям состояния 5 Г, выведенным в пункте а. Покажите, что все недиагональные элементы матрицы 15 х 15 должны быть нулевыми. (Это означает, что все члены Ет уравнения Ван-Флека должны быть равны нулю.) Далее определите энергии 15 волновых функций. Подтвердите, что при построении полного гамильтониана центр тяжести уровней сохраняет постоянное положение, т. е. что Н = 8LZ2, хотя при корректном расщеплении центр тяжести сохраняться не должен.
в. Допустим, что разность в энергиях между 1А и 5Т параметризована с помощью Е. Энергетическая диаграмма при этом должна быть похожа на приведенную ниже
5А
5Т Т
lA 1 Ч__________________
oh Da Магнитное поле
Используя уравнение Ван-Флека, определите Хц как функцию Е, 6 и Г.
В настоящем эксперименте проявляется и спин-орбитальное взаимодействие, которое дает недиагональные элементы, осложняющие анализ. Чтобы при расчетах на ЭВМ можно было получить наилучшие величины X, Е, 8, осуществлена подгонка экспериментально полученной зависимости X—Т к теоретической. Интересно отметить, что соответствие кривых
160
Глава 11
было плохим, пока не было сделано допущение, что Е зависит от температуры.
3. Основное состояние р1 -конфигурации свободного иона 2Р. Рассмотрите этот ион в магнитном поле, направленном вдоль оси z.
а. Какова вырожденность этого состояния?
б. Запишите для этого состояния волновые функции, используя скобочные обозначения Дирака, т.е. \ML, Ms/-
в. Основным возмущением в этом состоянии является спин-орбитальное взаимодействие:
Н = 1L-S-
Рассмотрите основной вклад в сильном магнитном z-поле
Я да XLZSZ.
Приводит ли он к диагональным элементам, недиагональным элементам или к тем и другим в базисе пункта б? Используя Я, рассчитайте энергии этих волновых функций.
г. Зеемановский гамильтониан имеет следующий вид:
Я = PH(L + 2S) = ₽Я2(12 + 2SZ).
Рассчитайте энергии волновых функций пункта б, используя этот гамильтониан.
д. Используя энергии, полученные в пунктах в и г, и уравнение ВанФлека, рассчитайте X-
4. Как оказалось, при 300 К молярная магнитная восприимчивость комплекса составляет —186 -10“5 6 см3/моль. Рассчитайте молярную парамагнитную восприимчивость, введя поправку на диамагнетизм комплекса. Определите цеП и объясните, почему он имеет именно такое значение.
Н3С н h3cJ__Ьн
. 7\ уН
Cu(hfac)2—О—N X,
н3С"д рн
Н3С Н
5. а. Магнитный момент Co(N2H4)2C12 составляет 3,9 магнетона Бора. Является ли гидразин бидентатным лигандом? Предложите структуру комплекса.
б. Каким образом можно использовать электронную спектроскопию для подтверждения вывода, сделанного в пункте а?
6. В комплексах каких из перечисленных ионов переходных металлов, имеющих тетраэдрическую структуру, следует ожидать вклад спин-орбитального взаимодействия V3+, Cr3+, Cu2+, Со2+, Fe2+, Мп2 + ?
Магнетизм
161
7. В каких из следующих низкоспиновых квадратно-плоскостных комплексов следует ожидать орбитальных вкладов: d2, d3, <r, d5, d6?
8. Почему Fe2(CO)9 (с тремя мостиковыми и шестью концевыми карбонильными группами) диамагнитен?
9. Объясните, почему примешивание компоненты D4h к основному состоянию Td понижает магнитные моменты комплексов никеля(П)?
10. Каким должен быть магнитный момент комплекса Ег3 + ?
11. Ниже приведена зависимость эффективного магнитного момента от температуры для двух похожих трис-бидентатных комплексов Fe(III)
A: R = Н-прОПи/1 Б: R = изопропил
Температура, К
а. Исходя из электронной структуры комплекса Fe(III), объясните изменения в jreff в данном диапазоне температур для каждого из двух комплексов. Другими словами, почему приведенные на рисунке кривые так сильно различаются, хотя эти два комплекса кажутся такими похожими?
11-574
162
Глава 11
б. Точки на графиках соответствуют экспериментальным данным, а кривые представляют собой полученное с помощью метода наименьших квадратов иаилучшее приближение к теоретическому выражению. С помощью диаграммы энергетических уровней опишите, какими параметрами можно воспользоваться при таком теоретическом рассмотрении.
12. СПЕКТРЫ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА ПАРАМАГНИТНЫХ КОМПЛЕКСОВ ИОНОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ
12.1. ВВЕДЕНИЕ
На раннем этапе развития метода ЯМР было широко распространено мнение, что снять спектр ЯМР парамагнитного комплекса практически невозможно, поскольку электронный спиновый момент настолько велик, что он должен вызывать быструю релаксацию возбужденного ядерного состояния, а это даст малое 7] и широкую линию. Подобная ситуация действительно наблюдалась для некоторых парамагнитных комплексов, в частности комплексов Мп(П), однако во многих других случаях это предположение не подтвердилось. Например, на рис. 12.1 представлен рассчитанный [1] спектр ЯМР парамагнитного комплекса Ni(CH3NH2)g + , там же для сравнения дан спектр ЯМР CH3NH2. В связи со сказанным возникает несколько вопросов:
1. Почему же мы все-таки видим спектр парамагнитного комплекса?
2. Почему наблюдаемые сдвиги линий комплекса относительно ТМС столь велики по сравнению со сдвигами линий некоординированного лиганда? Обычно протонные сдвиги для большинства органических соединений лежат в интервале 10 м.д., а сдвиги комплекса значительно выходят из этого интервала.
Рис. 12.1. Рассчитанные протонные спектры ЯМР растворов CH3NH, (А) и Ni(CH,NH2)^ + (Б) при 60 МГц (масштабы графиков А и Б отличаются).
н
164
Глава 12
3. Почему сигнал протона NH лежит в сильном поле, тогда как сигнал протонов группы СН3 сдвинут в слабое поле?
На все перечисленные вопросы мы и ответим в этой главе.
12.2. ПРОЦЕССЫ РЕЛАКСАЦИИ
Чтобы ответить на первый из поставленных выше вопросов, необходимо провести более полный анализ процессов релаксации в парамагнитных системах. Для электрона в магнитном поле существуют два разрешенных спиновых состояния с ms = + ‘/г- Если электрон делокализован на протоне, в спектре ЯМР должны наблюдаться две исключительно широкие линии, обусловленные взаимодействием ядерного спина с двумя спиновыми состояниями электрона. Линии эти должны далеко отстоять друг от друга, поскольку а — константа сверхтонкого взаимодействия (СТВ), о которой говорилось в главе, посвященной ЭПР, в данной системе велика. Интенсивность линии, обусловленной электронами, магнитные моменты которых направлены вдоль поля, должна быть несколько больше, поскольку заселенность состояния с низшей энергией выше. Резонансная линия должна быть настолько широкой, что ее не удается наблюдать, поскольку движение молекулы с большим парамагнитным моментом должно приводить к очень большому флуктуирующему магнитному полю и небольшому ТР В терминах спектральной плотности (гл. 7) диапазон частот должен быть большим, а частотные компоненты очень интенсивными, что дает низкую величину Т\. Итак, приведенный на рис. 12.1 спектр октаэдрического комплекса Ni(II) остается необъясненным, поскольку то, что мы наблюдаем для данного вида протона г,— это одиночная линия, сдвинутая значительно меньше, чем на величину Я; (см. рис. 12.1). Константы взаимодействия электронного спина с ядерным спином в ЯМР мы обозначим At.
Электрон в приведенном выше примере меняет свое спиновое состояние со временем. Если по временной шкале ЯМР изменение происходит слишком быстро, чистым эффектом является усреднение до нуля осциллирующего поля на протоне, которое связано с электроном. В результате снижается эффективность релаксирующей способности электрона по отношению к протону. Очень быстрый межмолекулярный электронный обмен или обмен лиганда должны оказывать то же самое влияние, поскольку за счет этих эффектов у протона оказываются электроны с различными значениями ms. Эта картина очень напоминает явление усреднения, рассмотренное ранее в связи с ядерным спин-спи-новым расщеплением. Первый эффект похож на развязку протона в ядерной спин-спиновой системе, а последний похож на обмен протона группы О — Н этанола.
Электроны, релаксирующие в точности с той же самой частотой, что и А;, в наибольшей степени уширяют спектральные линии ЯМР. Обычное значение А{ составляет примерно 108 Гц. Скорость вращения молекулы в растворе характеризуется временем корреляции тс, которое составляет 10“11 с. Время жизни электронного спинового состояния
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 165
связано с те — временем электронной релаксации или Т1е — временем обмена лигандом (которое всегда меньше); те определяет время, необходимое для переноса энергии на другие степени свободы, а Т1е—время, которое проводит лиганд у данного металлического центра. Если 1/Т1е»А,- или 1/тс»/!,, на ядро будет оказывать влияние только средневзвешенное по заселенности двух электронных спиновых состояний; ядро будет взаимодействовать с электронной спиновой системой в незначительной степени и, таким образом, значительно слабее релаксировать при этих условиях. Таким образом, для наблюдения спектра ЯМР необходимо, чтобы время жизни электронного спинового состояния составляло 10 ~ 9—10“ 13 с. Напомним еще раз: быстрая ядерная релаксация приводит к широким линиям в спектре ЯМР, но быстрая электронная релаксация снижает эффективность механизма ядерной релаксации, увеличивает 7] для протона и вызывает сужение линии. Наблюдаемые линии не столь узки, как в спектрах диамагнитных молекул. Для регистрации спектров парамагнитных ионов используется радиочастотное излучение высокой мощности, поскольку низкие величины Т[ говорят о снижении опасности насыщения.
Комплексы Мп(П) представляют собой примеры систем с медленно релаксирующими электронами (1/те« Л;). Если сигнал ЯМР вообще наблюдается, то он имеет вид одиночной очень широкой линии. Однако в спектрах ЭПР, где наблюдаются переходы электронных спинов, эта медленная релаксация гарантирует длительное время жизни возбужденного состояния и, таким образом, получение узкой спектральной линии. Итак, линии спектров ЭПР систем с медленной релаксацией узкие, а линии спектров ЭПР систем с быстрой релаксацией широкие. Было бы удивительно, если бы удалось осуществить и эксперимент ЭПР, и эксперимент ЯМР с одним и тем же соединением и при одной и той же температуре. Эти методы дополняют друг друга.
Некоторые общие наблюдения, касающиеся времен жизни спиновых состояний электрона в различных комплексах ионов переходных металлов первого ряда, помогут определить, в каких случаях следует ожидать узких спектральных линий ЭПР или ЯМР. Время жизни электронных спиновых состояний комплексов с трехкратно вырожденными (Т) основными состояниями часто невелико, и соответственно в их спектрах ЯМР наблюдаются узкие линии, а в спектрах ЭПР — широкие. К такого рода комплексам относятся октаэдрические комплексы: dl, d2, низкоспиновый (/4, низкоспиновый ds, высокоспиновый d6 и высокоспиновый d7. Если октаэдрическая симметрия комплекса сильно искажена, состояние Т расщепляется, спектр ЯМР при этом часто уширяется, а спектр ЭПР сужается. Особенно широкие линии в спектре ЯМР получают для низкосимметричных комплексов конфигурации d2, где имеется только один электрон и вызывающее релаксацию расщепление в нулевом поле отсутствует. Узкие линии в спектрах октаэдрических комплексов Ni2 + (основное состояние А) обусловлены расщеплением в нулевом поле.
Тетраэдрические комплексы (основное состояние Т) также дают уз
166
Глава 12
кие линии в спектре ЯМР, типичным примером могут служить тетраэдрические комплексы Со2 + . К системам с особенно широкими линиями в спектрах ЯМР и узкими линиями в спектрах ЭПР относятся комплексы с октаэдрической d3-, высокоспиновой ds- и ^-конфигурациями.
12.3. СРЕДНЯЯ ПОЛЯРИЗАЦИЯ ЭЛЕКТРОННЫХ СПИНОВ
Обсудим теперь причину больших наблюдаемых сдвигов в парамагнитных системах. Различие в сдвигах между парамагнитным и аналогичным диамагнитным комплексами носит название изотропного сдвига. Чем обусловлен изотропный сдвиг, можно понять, если вспомнить, что в помещенном в магнитное поле комплексе с неспаренным электроном два спиновых состояния ms заселены неодинаково. Если мы примем, что для комплексов с ms = + и ms = — величины сдвигов должны различаться, то нам станет понятным, почему при быстром обмене спинов или при быстрой релаксации наблюдается средневзвешенный по мольным долям химический сдвиг. Назовем средневзвешенную величину ms для системы в магнитном поле средней поляризацией электронных спинов—Sg. Напомним, что в предыдущей главе эта разность в заселенности использовалась для расчета парамагнитной восприимчивости. Теперь для расчета влияния разности в заселенности на сдвиг ЯМР мы воспользуемся очень похожим подходом. Все факторы, влияющие на парамагнетизм, оказывают влияние и на наблюдаемый сдвиг, поскольку сдвиг можно рассматривать как результат действия дополнительного поля, создаваемого электронными моментами в непосредственной близости от ядра [2, 3]. Величина этого дополнительного поля на ядре зависит от типа электрон-ядерного взаимодействия (скалярного или дипольного), который в свою очередь определяется характером связей и геометрией молекулы. Скалярные и дипольные механизмы, ответственные за передачу магнитных эффектов электронов на ядра в молекуле, подробно обсуждаются в последних разделах главы. Поскольку среднюю поляризацию электронного спина Sq можно связать с наблюдаемым сдвигом, рассмотрим, какими причинами она обусловлена и как ее можно рассчитать.
В предыдущей главе мы видели, что
U=-0pS, (12.1)
а гамильтониан для взаимодействия электронного магнитного момента с полем записывается как
Н = -u-H = 0PSH. (12.2)
Используя распределение Больцмана применительно к системе с S = 1/2, получим
NjNp = ехр( —\E/kT) = exp ( — д$Н/кТ). (12.3)
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 167
Следовательно, полное число электронов в состоянии р выражается как Np = Naexp(^H/fcT).
Используя закон Кюри и тот факт, что g$H/kT « 1, мы показали в предыдущей главе, что
ехр(±0рН//сТ) яй 1 ±(0рн//сТ). (12.4)
Решая совместно уравнение (12.4) и уравнение, определяющее 1V₽, получаем
= (IV — AQ(1 +
где N = Ny + IVp. Теперь можно выразить Л'р следующим образом:
1
gJ3H kT
2 + ^ kT J
кТ
2кТ
Числитель в скобках имеет вид 1+пх, где п = 2, а х = д$Н/2кТ. При х«1 справедливо приближение (1 +.v)" г 1 + пх, и, следовательно,
fi+^y
N \ 2кТ J N / д$Н\ 0рн< = + 2кТ /
V + 2кТ )
Таким же образом получаем
IV/ _^рну
* 2 \ 2кТ)
Разность между Na и Л!р дает избыток неспаренных спинов, направленных вдоль поля, и ее можно рассматривать как суммарный вектор спиновых моментов, направленных вдоль и против поля. Эта разность представляет собой суммарный магнитный момент индуцированный в системе неспаренных электронов внешним полем. Среднее значение ms определяется средней поляризацией электронных спинов Sq, т. е. параметром, который интересует нас в эксперименте ЯМР. В нашем примере мы берем средневзвешенное по заселенностям состояний + 1/2 и — 1 /2 в виде
1\ / 1\
н— 1ЛУ. "ЬI — I-Wr
2/ \ 2 J р д$Н
N ~ 4кТ ’
(12.5)
168
Глава 12
Между Sg и объемным магнитным моментом М, с которым мы имели дело в предыдущей главе, существует прямая зависимость. Мы установили, что число избыточных спинов на низшем уровне в два раза превышает среднюю поляризацию электронных спинов Sg умноженную на полное число электронов N. Чтобы определить средний магнитный момент, число избыточных спинов следует умножить на магнитный момент одного спина в соответствующем состоянии, направленном вдоль поля, 1/20&, т. е.
M = ^0p(2NS,) = W02p2H/4/cT (12.6)
Это есть закон Кюри, о котором мы говорили в предыдущей главе и к которому пришли, определяя заселенности различных уровней. _
В более общем случае, когда число спинов превышает единицу, Sg определяется как
5
X Sexp(-Es/fcT)
S, = . (12.7)
Z exp(-Es//<T) s,= -s
Здесь ESg— энергия состояния с квантовым числом S. Уравнение (12.7) согласуется с выведенным ранее выражением для одноэлектронного спина (Sg = ±1/г). Из выражения (12.7) следует, что состояние с данным значением спинового магнитного квантового числа Sg взвешено в соответствии с его равновесной заселенностью. Взвешенные величины просуммированы по всем энергетическим уровням и разделены на общее число уровней, что дает среднюю поляризацию электронных спинов. Если к уравнению (12.7) применить приближение типа ДЕ « кТ, то можно представить экспоненту в виде степенного ряда, в котором мы рассмотрим только два первых члена. После алгебраического преобразования получается уравнение
- = 0^+11, (12.8)
где </av— средняя величина g-фактора. Уравнение (12.7) строго выполняется для зеемановского мультиплета, где известна ESf. Уравнение (12.8) выполняется, только если ESt= g$HSg и AES> « fcT. В главе, посвященной магнетизму, мы обсуждали эффекты (орбитальный угловой момент, расщепление в нулевом поле, эффекты Зеемана второго порядка), которые приводят к отклонениям от закона Кюри. Теперь, поскольку у нас есть выражение для S мы попытаемся, исходя из уравнения (12.8), получить выражение, позволяющее количественно оценить контактный сдвиг ЯМР. Если указанные условия не выполняются, можно вывести другие выражения, используя другие уравнения для Sg.
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 169
12.4. СКАЛЯРНЫЙ, ИЛИ КОНТАКТНЫЙ,
СДВИГ В СИСТЕМЕ С ИЗОТРОПНЫМ ^-ТЕНЗОРОМ
Скалярный, или «контактный», сдвиг описывается гамильтонианом
а 8л
н=—0e0~₽₽~I-S5(r). (12.9)
Это взаимодействие электронного и ядерного спинов рассматривалось в гл. 9 в разделе, посвященном контактному взаимодействию Ферми, там же дается объяснение всем принятым обозначениям. Этот эффект связан с влиянием плотности неспаренного спина, который делокализован непосредственно на ядре, исследуемом методом ЯМР. Подставляя среднюю поляризацию электронных спинов в уравнение (12.9), получаем
а 071
H = —gegNWN
0avpHS(S + l)l
•15 (г),
3kT
(12.10)
где gay учитывает любой орбитальный эффект, который может сделать S плохим квантовым числом. Символ Кронекера 8 ограничивает рассмотрение эффекта только этим ядром. Сравнивая уравнение (12.10) с уравнением, полученным для протона в диамагнитном окружении,
Н— — (12.11)
находим, что они аналогичны, если принять, что в уравнении (12.11) Н — эффективное магнитное поле Heff, обусловленное электронной плотностью на этом ядре |ф(0)|2. Обозначая интеграл вида ]ф;8(г)ф;(/т как ф2(0) (возведенная в квадрат величина волновой функции при г = 0), мы имеем
8л S(S + 1)
Heff= ~ ТgeРgavPH 3fcT 1Ф(0)12- (12.12)
Полное поле на ядре в эксперименте ЯМР представляет собой сумму Heff И Но магнита. Сдвиг, обусловленный парамагнетизмом, который мы пытались установить, вызван Heff. Он носит название скалярного сдвига и определяется из выражения для Heff [уравнение (12.12)] в эрстедах (Э). Если число электронов превышает единицу, для расчета сдвига (в предположении ge = gay) в конечное выражение необходимо ввести нормировочный член 1/2S, обусловленный принципом исключения Паули.
81г 1 ,, 5(5+1)
АН = —gfvp2H \ 1 |ф(0)|2. (12.13)
э Ж1
По ряду причин данные измерений контактного сдвига часто выражаются через А — константу взаимодействия электронного спина
170
Глава 12
с ядерным. Это объясняется в первую очередь тем, что сдвиг в резонансе зависит от температуры, а константа А (в рамках допущений, сделанных в связи с введением Sq) не зависит. Гамильтониан взаимодействия можно записать как
H = AIS, (12.14)
где А— константа изотропного сверхтонкого взаимодействия, по сути дела, представляет собой константу а, рассмотренную в главе, посвященной ЭПР. Заглавная буква используется в данном случае для того, чтобы показать, что в молекуле или ионе может находиться более одного неспаренного электрона. Мы определим А как
8л ,
А = —0e0jvPPjv|\|/(O)|2. (12.15)
Как говорилось ранее, при наличии более чем одного неспаренного электронного спина необходимо ввести нормировочный коэффициент 1/2S и для исследуемого комплекса заменить де на gav. При этом
А = |^0аУР^|ф(О)|2, (12.16)
где А выражена в эргах (эрг). Чтобы перевести А в другие единицы, можно воспользоваться соотношениями
Л(эрг) =/1Л (Гц), (12.17)
А(эрг) = даурА (Э). (12.18)
Выражение для контактного сдвига обычно записывают не в виде уравнения (12.13), а как функцию константы сверхтонкого взаимодействия А. Если мы подставим уравнение (12.15) в уравнение (12.13), то получим для изотропного сдвига следующее выражение:
АН _ Av A;gavpS(S + 1)
-JT--V 11219)
Теперь можно видеть, что очень небольшая величина А может привести к огромному изотропному сдвигу. Читателю следует перевести величины изотропных сдвигов на рис. 12.1, выраженные в единицах частоты (при комнатной температуре), в единицы напряженности поля,«<т.-е. в эрстеды. Тождество AH/H = Av/v, где v — фиксированная зондирующая частота, следует непосредственно из того, что для ядерных спинов hv = = д$Н. Зависимость Av от 1/Т должна иметь вид прямой линии, тангенс угла наклона которой для систем, подчиняющихся закону Кюри, пропорционален А;. Для систем с орбитально вырожденным основным состоянием, таких, как октаэдрические комплексы никеля(П) и тетраэдрические комплексы кобальта(П), уравнения (12.7) и (12.8) справедливы.
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 171
В условиях, когда приведенные для Sq выражения не справедливы, уравнения (12.13) и (12.19) также неприменимы. Для этих систем мы можем записать
АН Av At
= — =-----— <s2> Н---------------V Пд^Н
и задачей становится поиск корректного выражения для <SZ>.
12.5. ПСЕВДОКОНТАКТНЫЙ СДВИГ
Молекулы, для которых g-тензор неизотропен, удобно разбить на две группы: молекулы, в которых вклады эффектов Зеемана второго порядка значительны, и молекулы, в которых эти вклады невелики. Рассмотрим вначале последний случай. Зависимость изотропного сдвига от температуры можно выразить с помощью уравнения (12.19) со средней величиной g-фактора для любого орбитального углового момента. Если это сделано, результирующая величина А из кривой зависимости Av от 1/Твключает вклады не только скалярного, или контактного, члена, т.е. уравнение (12.15) больше не выполняется. Наблюдаемый изотропный сдвиг Av выражается как
(изотропная) Д^(скалярная) Т ДУ(псевдоконтактная) • (12.21)
Обсудим теперь вопрос об источнике этого псевдоконтактного, или дипольного, вклада.
Если молекула обладает неспаренным электроном, дипольный эффект передается через пространство и ощущается исследуемым ядром. Когда g-фактор изотропен, дипольные эффекты усредняются до нуля вследствие быстрого вращения молекулы в поле. Это явление рассматривалось в главе, посвященной ЭПР, где было показано, что этот же самый эффект приводит к дипольному вкладу в сверхтонкое взаимодействие, который усредняется до нуля в растворе. В тех случаях, когда g-фактор анизотропен, величина дипольного вклада в магнитное поле на интересующем нас ядре, обусловленная плотностью неспаренного электрона на металле, зависит от ориентации молекулы относительно поля. Поскольку для разных ориентаций g-фактор имеет различные значения, этот пространственный вклад не должен усредняться до нуля в результате быстрого вращения молекулы. Таким образом, те же самые эффекты, которые приводят к анизотропии g-фактора, дают и псевдокон-тактный вклад. Этот псевдоконтактный эффект, связанный с влиянием через пространство, можно сопоставить с анизотропным вкладом соседнего атома, рассмотренным в гл. 8. который, как было показано, зависит от разности в XDIA для различных ориентаций. То же самое справедливо для XPARA. Применяя уравнение (12.8), мы рассматриваем систему, в которой А/ меняется симбатно Ag [2]. Часть гамильтониана, описывающая псевдоконтактный вклад, аналогична гамильтониану дипольного взаимодействия, рассмотренному в гл. 9.
172
Глава 12
Авторы [2, 3] вывели уравнения для расчета вклада дипольного эффекта в изотропный сдвиг для ряда систем. Если Т1е « тс и 1/тс « (Етах — — Emin)/h (где Т1е—время жизни электронного спинового состояния, тс—время корреляции вращения молекулы, а Етах и Emin—максимальное и минимальное значения зеемановской энергии в зависимости от ориентации, т. е. АЕ = — цтахН, где ц — эффективный магнитный момент,
Рис. 12.2. Определение параметров уравнения (12.22).
а Н — напряженность магнитного поля), мы можем записать общее уравнение для псевдоконтактного вклада в сдвиг Av; (рс) i-го ядра как
1 sin20, cos2q>;
^(Хх-Ху)--------3----
(12.22)
В этом уравнении N — число Авогадро, %х, %у и xz—компоненты восприимчивости, а 0 и <р—углы, определенные на рис. 12.2. На этом рисунке N — ядро, исследуемое методом ЯМР, М—металл, а г—расстояние между этими двумя центрами. Уравнение (12.22) справедливо для всех систем, поскольку в него введены истинные восприимчивости, В тех случаях, когда применимо уравнение (12.8), оба уравнения можно записать несколько по-другому, вводя в них g-факторы, поскольку анизотропия g-тензора определяется проще, чем анизотропия /.
Переписывая уравнение (12.22) для аксиально симметричной системы при тех же самых требованиях, налагаемых на время жизни, мы получаем следующее уравнение для псевдоконтактного вклада:
Avj(pc) P2S'(S'+ 1) / 1 - 3cos20; \ , — sH—з—я ’•
(12.23)
где S' — эффективный спин, экспериментально наблюдаемый для исследуемой системы. Входящие в уравнение (12.23) параметры определены для показанной на рис. 12.3 молекулы с локальной аксиальной симметрией. Поскольку уравнение (12.23) проще, чем (12.22), во многих рабо-
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 173
тах исследуются аксиально симметричные системы. Если д-факторы не обеспечивают приемлемое описание восприимчивости, т. е. X/ / Ng^AS(S + 1)//сТ, уравнения (12.19) и (12.23) не выполняются. Как было показано, уравнения, базирующиеся на g-факторах, не пригодны для описания тригонально искаженного шестикоординационного комплекса пиразоилбората кобальта (П). Основное состояние 4Ед расщепляется на четыре дублета (см. гл. 13), каждый из которых характеризуется своим собственным g-фактором. Поскольку некоторые из уровней заселены
Рис. 12.3. Определение параметров уравнения (12.23). Показаны параллельная и перпендикулярная оси. Радиус-вектор от металла до исследуемого ядра обозначен г;, а угол, который он образует с осью вращения высшего порядка, 0(.
при комнатной температуре и не заселены при температуре жидкого гелия, при которой снимают спектр ЭПР, усредненный g-тензор обеспечивает лишь плохое описание восприимчивости при комнатной температуре. В работах [4—6] приведены уравнения для таких систем, для которых неприменимо уравнение (12.23).
12.6. КАЧЕСТВЕННАЯ ИНТЕРПРЕТАЦИЯ СПЕКТРОВ ЯМР ПАРАМАГНИТНЫХ МОЛЕКУЛ
Разделение контактного и дипольного вкладов
Константа изотропного сверхтонкого взаимодействия А, полученная в эксперименте ЯМР, обусловлена теми же самыми эффектами, которые дают константу сверхтонкого взаимодействия а, получаемую из спектра ЭПР. Если одну и ту же систему можно исследовать обоими методами, получаемые изотропные величины а или А должны быть идентичными. Метод ЯМР значительно более чувствителен, и большие протонные сдвиги (например, 50 Гц) позволяют рассчитать протонные константы СТВ, которые нельзя определить из спектров ЭПР. Кроме того, методом ЯМР можно по направлению сдвига определить знак константы взаимодействия, в то же время характер спектра ЭПР от знака константы не зависит. Поскольку природа эффекта в обоих случаях одинакова, все сказанное об А можно распространить и на а.
Прежде чем пытаться понять причины возникновения изотропного сдвига сигнала ядра в эксперименте ЯМР, необходимо разделить сдвиг данного ядра на скалярный (контактный) и дипольный (псевдокон-тактный) [см. уравнение (12.21)]. Можно подобрать системы, в которых дипольный вклад пренебрежимо мал по сравнению с контактным вкла
174
Глава 12
дом. Например, как следует из уравнения (12.23), псевдоконтактный вклад равен нулю, когда = д:. Если окружение атома металла имеет строго кубическую симметрию (например, тетраэдрическую или октаэдрическую), наблюдается только контактный сдвиг. Мы выделяем курсивом слово «строго», поскольку тонкие эффекты могут вызывать отклонение от кубической симметрии и могут привести к значительному псевдоконтактному вкладу (см. далее раздел, посвященный образованию ионных пар).
В системах, имеющих одновременно и псевдоконтактный, и контактный вклады, пользуются тремя традиционными подходами. Если геометрия молекулы известна из рентгеноструктурного исследования монокристалла и если структура ее одна и та же в растворе и в твердом состоянии, а также если известна анизотропия восприимчивости *, можно рассчитать [7] псевдоконтактный вклад. Рассчитав величину псевдоконтактного вклада, можно из уравнения (12.21), используя измеренную величину изотропного сдвига, определить контактный вклад. Рентгеноструктурные исследования и измерения восприимчивости монокристалла требуют и много времени, и больших материальных затрат. Эквивалентность структур в твердом состоянии и в растворе доказать очень трудно, и часто она лишь допускается. В благоприятных ситуациях значительные изменения в структуре можно установить с помощью спектральных методов.
Второй подход состоит в определении анизотропии g-фактора в тех системах, где электронная структура позволяет пользоваться уравнениями, базирующимися на g-факторах [например, уравнением (12.23)], т. е. если д2 ос /. К сожалению, время жизни электронных спиновых состояний, приводящее к хорошо разрешенному спектру ЭПР, обусловливает плохо разрешенные спектры ЯМР, и наоборот. В статье [8] описаны такие комплексы железа(Ш), для которых можно снять и спектр ЭПР, и спектр ЯМР. Результаты сопоставления измеренных величин восприимчивости с рассчитанными из (/-факторов и линейность кривой зависимости Av от 1/Т позволяют предположить, что (/-факторы приемлемы для оценки псевдоконтактного сдвига в этой системе.
Третий подход, называемый методом отношения, был предложен [9] для разделения изотропного сдвига в октаэдрических комплексах ко-бальта(П) на скалярную и дипольную составляющие. Было сделано допущение, что отношения скалярных сдвигов аналогичных протонов в комплексе кобальта(П) те же самые, что и для протонов аналогичного комплекса никеля(П). Зная величины как этих отношений, так и отношений геометрических факторов, для нескольких протонов удается рассчитать анизотропный член, например в уравнении (12.23), и затем рассчитать Av (дипольный) для каждого протона. Вообще некорректно предполагать, что октаэдрические комплексы никеля(П) и кобальта(П) характеризуются аналогичными картинами делокализации [10]. У высокоспинового комплекса кобальта(П) неспаренные электроны находятся
См. список литературы к гл. И.
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 175
как на t2g-, так и на ^-совокупности, которые могут перекрываться одновременно и я-, и ст-орбиталями лиганда, делокализуя электроны на орбиталях обоих типов непосредственно. Как происходит смешивание орбиталей металла и лиганда при образовании молекулярных орбита-лей октаэдрического комплекса, можно понять, рассмотрев упрощенный
ЭГь*
Я
X
'ct2g+^L*
d-Орбитали металла в поле 0h
Ье9
1-Н-л-ь------
Молекулярные орбитали
------Н-Яь
Молекулярные орбитали лиганда.
Рис. 12.4. Смешивание орбиталей металла 0h и лиганда при образовании комплекса Ni — L2 + . (L может быть, например, пиридином.) Симметрия октаэдрического комплекса такова, что расстояние между а- и л-системой в лиганде L поддерживается постоянным. Чтобы картина чрезмерно не усложнилась, на рисунке показан лишь фрагмент М —L. Отметим, что а » b, с » d и Ь'»а, тогда как d’»c'. Разрешенное по ci гметрии смешивание t2g и л не показано.
пример смешивания орбиталей одного лиганда и совокупности орбита-лей металла, находящихся под воздействием октаэдрического поля лиганда. В октаэдрическом комплексе мы должны использовать приведенные по симметрии 0h линейные комбинации а- и я-орбиталей лиганда. Рис. 12.4 демонстрирует смешивание орбиталей октаэдрического поля иона никеля(П) с а-, я *-орбиталями лиганда. Высокоспиновый комплекс кобальта(П) должен иметь на Г29-совокупности молекулярных орбиталей, представляющих собой главным образом МО металла, на один электрон меньше. Непосредственная делокализация неспаренных электронов как на а-, так и на я *-орбиталях пиридина возможна в комплексе кобальта(П) и невозможна в комплексе никеля(П). Сдвиги, наблюдаемые в спектрах некоторых комплексов бипиридила, показывают [10] непригодность метода отношения. Если орбитали лиганда могут взаимодействовать только с е9-орбиталями металла, механизмы смешивания для комплексов кобальта и никеля окажутся схожими. Кроме того, если неспаренные электроны делокализованы непосред
176
Глава 12
ственно в я-системе лиганда в комплексе кобальта(П), но если в этом лиганде имеются протоны, в контактный сдвиг которых я-делокализа-ция вклада не дает, отношение этих протонов в комплексах кобальта(П) и никеля(П) снова должно быть аналогичным [И]. При образовании тетраэдрических комплексов кобальта(П) и никеля(П) неспаренные электроны могут делокализоваться и на л-, и на ст-орбиталях лиганда, и эти комплексы также могут демонстрировать аналогичные отношения скалярных сдвигов. Тем не менее метод этот малопригоден [10] не потому, что никто никогда не найдет такой системы, к которой он был бы применим; дело в том, что необходимо заранее достаточно достоверно предсказать, какие МО лиганда участвуют в делокализации электрона при образовании комплекса или какие протоны занимают доминирующее положение в некоторых механизмах. Поскольку чаще всего комплексы, в которых должен быть отделен псевдоконтактный вклад, исследуют экспериментально для определения делокализации спина, этот метод большого практического значения не имеет.
Сложность в предсказании доминирующего механизма не столь очевидна при экспериментальном изучении изотропных сдвигов, поскольку она маскируется трудностями, возникающими из-за одновременного существования контактного и дипольного сдвигов. Например, после исследований методом ЭПР трис-(бипиридил)железа(1П) и определения псевдоконтактного сдвига [8] первоначальный вывод [12] о том, что контактные сдвиги комплексов железа(Ш) (/|9) и никеля(П) (е2) близки, оказался неверным.
Именно по этой причине большинство работ в области ЯМР парамагнитных комплексов посвящено исследованию систем, в которых доминирует один из вкладов — контактный или псевдоконтактный. Мы же уделим основное внимание системам с доминирующим контактным вкладом. В литературе обсуждался тот факт, что у молекул с почти изотропными (/-факторами псевдоконтактный вклад отсутствует. Комплексы общей формулы MLg + , где L—моно дентатный лиганд, не имеют псевдоконтактного вклада [13]. Если комплекс MLg+ характеризуется ян-теллеровским искажением, следует ожидать, что в шкале времени ЯМР в растворе оно будет динамическим. Если даже реализуется весьма маловероятная ситуация с нединамическим искажением, тогда быстрый обмен лигандов должен усреднять сдвиг до нуля, поскольку для двух лигандов, находящихся на оси z, функция 3cos2 0—1 вдвое больше, чем для четырех лигандов, находящихся на осях х и у, и имеет противоположный знак. Таким образом, средний псевдоконтактный вклад для всех шести лигандов равен нулю. Образование ионных пар может фиксировать искажение.
Скалярные сдвиги и ковалентность
Скалярный, или контактный, вклад в связывание лиганда в комплексе возможен только при ковалентном характере связи металл — лиганд. Поэтому неоднократно предпринимались попытки определить степень
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 177
ковалентности связывания в ряде комплексов, а также установить, исходя из величин таких сдвигов, возможно ли в комплексах обратное п-связывание металл — лиганд. При описании комплекса по методу МО орбитали лиганда и иона металла смешиваются с образованием молекулярных орбиталей. Плотность неспаренных электронов делокализована на лиганды, по-видимому, это обусловлено вкладом орбиталей лиганда в результирующие несвязывающие молекулярные орбитали главным образом металла (см. рис. 12.4). Эта плотность неспаренных электронов взаимодействует с ядром, спектр ЯМР которого исследуется, по такому же принципу, что и механизм прямой делокализации, рассмотренный в посвященной методу ЭПР гл. 9, где мы имели дело с влиянием этого взаимодействия на энергетические уровни электрона. Интерпретация данных по контактному сдвигу в терминах связывания затруднена по двум причинам. Методами ЭПР и ЯМР регистрируют плотность неспаренного спина. В дополнение к прямой делокализации плотности неспаренных электронов спин делокализуют косвенные механизмы поляризации спинов. Например (см. разд. 9.5), в метильном радикале отсутствует чистая плотность неспаренного электрона в а-струк-туре, но в результате поляризации спина неспаренный спин ощущается на протоне (см. рис. 9.15 и соответствующий текст). Используя молекулярные орбитали электронных пар, важно отличать в комплексе делокализацию неспаренного электрона (прямые механизмы) от делокализации спина (косвенный механизм) *. Если и прямой, и косвенный механизмы дают на атоме спиновую плотность одного и того же знака, эти два механизма нельзя различить (см. далее). Далее, исходя из контактных сдвигов, можно получить информацию только о несвязывающих орбиталях, по существу орбиталях металла. Степень взаимодействия этих орбиталей совсем не обязательно отражает взаимодействие связывающих молекулярных орбиталей [14]. Об этом, в частности, говорят результаты исследования контактных сдвигов в Ni(stien)2(RCO2)2 [где stien — стильбендиамин (1,2-дифенилэтилендиамин) и R — СН3, СН2С1, СНС12 и СС13]. По мере ухудшения координационных свойств анионных лигандов контактные сдвиги уменьшаются, вместо того чтобы расти. Увеличения сдвига следует ожидать при усилении ковалентного характера связи никель — stien, если исходить из механизма прямой делокализации и принять, что разрыхляющие орбитали отражают возросшую ковалентность, которая, как предполагается, должна быть следствием увеличения формального заряда на металле. Однако, поскольку сдвиг не увеличился, предложены возможные объяснения для наблюдаемого расхождения [14].
* Представление о том, что делокализация электрона зависит от делокализации спина, является только артефактом. Потребность в нем возникает, если в расчетах систем с неспаренным спином вместо неограниченных методов МО используются ограниченные.
12-574
178
Глава 12
Скалярные сдвиги в координированном пиридине
Принципы, используемые при качественной интерпретации контактных (скалярных) сдвигов, можно проиллюстрировать, объясняя снижение величины сдвига в ряду Н (2) > Н (3) > Н (4) при координации пиридина с комплексом никеля (П). Более того, если с металлом координируется 4-метилпиридин, то сдвиги сигналов протонов метильной группы происходят в направлении, противоположном сдвигам Н (4) пиридина. Область контактных сдвигов, наблюдаемых для молекулы лиганда, характеризует молекулярные орбитали лиганда, которые участвуют в делокализации спина (т. е. волновые функции для вкладов aL, и т. д. на рис. 12.4). Вклад в молекулярную орбиталь «неподеленной пары азота» в пиридине снижается в ряду Н (2) > Н (3) > Н (4). Таким образом, характерная картина а-делокализации обусловлена смешиванием е9-орбитали и о-МО пиридина. Это приводит к большому сдвигу для Н(2), меньшему сдвигу для Н(3) и самому маленькому сдвигу для Н (4). В то же время высшая заполненная или низшая свободная л-орби-таль водородных коэффициентов не имеет, но характеризуется большими углеродными коэффициентами для всех атомов углерода, поэтому в результате поляризации спина получаются грубо сравнимые сдвиги Н(2) и Н(4). Для октаэдрического комплекса, неспаренные электроны которого находятся только на орбиталях е9, часто получают доводы в пользу существования сдвига, обусловленного л-делокализацией, если даже л-орбитали лиганда ортогональны е9-орбиталям металла, на которых находятся неспаренные электроны. Это происходит потому, что плотность неспаренного электрона в ст-системе может спин-поляризо-вать заполненную орбиталь, представляющую собой по существу л-мо-лекулярную орбиталь (это тот же самый механизм поляризации спина, который рассматривается в главе, посвященной ЭПР), и тем самым приводит к отличной от нуля спиновой плотности в л-системе некоторых атомов. Это эквивалентно тому, что неспаренный электрон на МО IV (рис. 12.4) спин-поляризует пару на МО I. Больший спин остается на металле, что дает меньший спин на лиганде. Указанный эффект аналогичен обсуждавшемуся для аллильного радикала (рис. 9.16).
Возможно также, что в комплексе неспаренный электрон, находящийся на МО IV, спин-поляризует МО III (в которую некоторый вклад дает л-орбиталь лиганда) — заполненную МО, представляющую собой по существу /2я-орбиталь металла. Электрон с тем же самым спином, что и на орбитали е9, находится главным образом на металле, а электрон с противоположно направленным спином находится главным образом на части л*-МО, которая в основном является МО лиганда. Неспаренный спин в результате этих двух косвенных взаимодействий делокализован в л-системе лиганда, но на t2g- (в основном орбитали металла) и на л1-молекулярной орбитали (в основном орбитали лиганда) комплекса плотность неспаренного электрона отсутствует. Далее мы будем использовать термин спиновая плотность для обозначения неспаренного спина, обусловленного либо прямым, либо косвенным взаимо-
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 179 действиями, и термин плотность неспаренного электрона для обозначения прямой делокализации.
Основным доводом в пользу нахождения неспаренного спина в я-си-стеме ароматического лиганда типа пиридина или фенильной группы является результат замещения атома водорода цикла на группу СН3. Если наблюдаемый сдвиг протона СН3 меняет знак по сравнению со знаком сдвига протона, находящегося в том же самом положении в кольце незамещенного соединения, то спиновая плотность находится в я-системе. Это происходит потому, что спиновая плотность в я-систе-ме — преимущественно углеродной системе — делокализована непосредственно на метильные протоны, т. е. связанные в этими протонами орбитали атомов водорода характеризуются небольшими коэффициентами в я-молекулярной орбитали. В незамещенном ароматическом соединении 1 s-орбиталь водорода ортогональна я-системе, и я-спиновая плотность должна поляризовать о-связь С — Н, чтобы повлиять на протоны. В результате знак спиновой плотности на Н противоположен знаку спиновой плотности в я-системе.
Из проведенного выше обсуждения видно, что метод МО можно непосредственно использовать для интерпретации механизмов делокализации спинов в комплексе и для качественного описания этим же методом лиганда. В последнем случае мы будем интерпретировать спектр, исходя из молекулярных орбиталей лиганда и электронной конфигурации металла для соответствующей симметрии комплекса, и будем пытаться установить, какие молекулярные орбитали лигандов смешиваются с молекулярными орбиталями металла или какие из первых спин-по-ляризованы.
Делокализация спина
Если неспаренный спин делокализован на высшей заполненной я-ор-битали или низшей свободной я*-орбитали соединения (С6Н5)„Х (п = 1, 2 или 3, а X — основная функциональная группа), возникает характерная картина сдвигов, которая обусловлена образованием узла на мета-угле-роде в высшей заполненной я- или низшей свободной я*-молекулярной орбитали фрагмента С6Н5. В результате спин, направленный вдоль поля в углеродной я-системе, вызывает сдвиг в сильное поле сигналов орто- и иара-протонов и сдвиг в слабое поле сигналов .мета-протонов. Контактные сдвиги фенильных протонов тетраэдрических комплексов никеля (II) с (С6Н5)3Р дают картину, характерную для я-делокализации, со сдвигами сигналов орто- и пара-протонов в сильное поле (т. е. плотность неспаренного электрона делокализована непосредственно в я-си-стеме). Указывают ли эти результаты на то, что существует обратное я-связывание металл — лиганд, затрагивающее d-орбиталь фосфора и е9-набор никеля? Очевидно, что такая интерпретация неверна. я-Систе-ма фенильной группы не ортогональна неподеленной паре электронов фосфора в лиганде. Таким образом, если образуется «о»-связь металл — фосфор с акцепторной орбиталью металла, содержащей неспа
12:
180
Глава 12
ренный электрон, то этот неспаренный электрон может непосредственно делокализоваться в фенильной я-системе. В результате я-делокализация не будет сопровождаться обратным я-связыванием. Именно такая картина наблюдалась [15] для Ni(C6H5CH2NH2)6 + . В этой системе возможна прямая я-делокализация для протонов фенильной группы, если даже атом азота не имеет низкоэнергетических свободных орбиталей для обратного я-связывания, а вся плотность неспаренного электрона находится на е,(-орбиталях октаэдрического комплекса. При рассмотрении симметрии лиганда видно, что «я-орбитали цикла» неортогональны неподеленной паре азота, т. е. у молекулы как целого дискретные о- и я-системы отсутствуют.
Остается ответить на вопрос; почему сигналы протонов N — Н в (CH3NH2)6Ni2+ сдвигаются при комплексообразовании в сильном поле? Сигналы протонов метильной группы сдвигаются в слабое поле из-за прямой делокализации плотности неспаренного электрона. Большая часть плотности неспаренного электрона, делокализованной на лиганде, находится на азоте, а меньшая часть делокализована непосредственно на протоне N — Н. Поэтому значительная спин-поляризация связи N — — Н определяет сдвиг этого протона и приводит к отрицательной А (т. е. к сдвигу в сильное поле). Такая ситуация характерна, вероятно, для всех протонов N — Н, если азот непосредственно связан с никелем.
Из предыдущего обсуждения должны стать довольно понятными два момента. Изотропные сдвиги имеют важное значение для понимания электронной конфигурации металла в комплексе, но они мало используются для получения информации относительно деталей связывания металл — лиганд. При интерпретации протонных контактных сдвигов не обойтись без использования молекулярных орбиталей комплекса или, при приближенном подходе, орбиталей лиганда.
12.7. ПОЛУКОЛИЧЕСГВЕННАЯ ИНТЕРПРЕТАЦИЯ КОНТАКТНЫХ СДВИГОВ
Возможность качественной интерпретации с применением МО спектров ЯМР парамагнитных комплексов подтверждает достаточно хорошее соответствие результатов приближенных МО-расчетов и экспериментальных данных. Первоначально широкое распространение получил ограниченный расширенный метод Хюккеля [1,16—20]. В настоящее время разработаны программы расчета с применением неограниченного метода ЧПДП. Результаты, полученные этими двумя методами, обычно согласуются (см. далее).
Расчет контактного сдвига аналогичен расчету методом МО изотропных констант СТВ ЭПР, обсуждавшемуся в гл. 9. В идеальном случае весь комплекс должен рассчитываться по неограниченному методу МО, а спиновые плотности на индивидуальных атомах должны быть определены и превращены в А, как это описано для а в гл. 9. Как уже говорилось выше, первоначально для интерпретации протонных контактных сдвигов целого ряда металлоценов использовался расши-
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 181
ренный метод Хюккеля [18]. Во всех изученных соединениях протонные контактные сдвиги хорошо согласуются с результатами расчета методом МО всего комплекса. Более того, эти расчеты дают корректное основное состояние.
Основное состояние предсказывается при добавлении некоторого числа электронов металла к молекулярным орбиталям, показанным на рис. 12.5. В первичном механизме спиновой делокализации для ванадо-цена и хромоцена участвуют, как установлено, молекулярные орбитали,
------
-4—
-4- -4- e2y(dty.
Рис. 12.5. Энергии молекулярных орбиталей (по существу d-орбиталей) металлоцена. (К ванадоцену добавлены электроны. Главный компонент металла показан в скобках.)
которые представляют собой ст-орбитали плоского циклопентадиена, в то время как в кобальто- и никелецене основной вклад дают л-орби-тали свободного циклопентадиена. Обширное смешивание «ст-орбита-лей» цикла с орбиталями металла при образовании металлоцена — важный вывод, вытекающий из этой работы. В прошлом при описании связывания циклопентадиена с ионом металла ст-системой обычно пренебрегали. Однако изучение протонных сдвигов для ряда парамагнитных бкс-ареновых комплексов [20] показало, что вклад ст-орбиталей необходимо учитывать. Авторы работы [21] исследовали контактные сдвиги ядер 13С нескольких металлоценов. Оказалось, что полученные данные качественно согласуются с выводами, сделанными в результате изучения протонного ЯМР, тогда как данные по ЯМР на 13С не могут быть адекватно поставлены в соответствие с волновыми функциями, полученными с помощью расширенного метода Хюккеля. Такое несоответствие неудивительно, и, чтобы вспомнить, чем оно обусловлено, следует вернуться к гл. 9, где обсуждается расчет констант СТВ ядер 13С органических свободных радикалов.
Подгонка протонных контактных сдвигов должна давать некоторую уверенность в волновых функциях, полученных в расчетах по методу МО. Если это условие выполняется, то исходя из результатов определения контактных сдвигов можно сделать некоторые выводы [20] относительно связывания, например: 1) расстояние между молекулярными орбиталями е2у и а1у в комплексах бмс-бензолов больше, чем в комплексах бмс-циклопентадиена, что говорит о большем обратном связывании в первом случае; 2) степень электронной делокализации по МО не обязательно связана с рассчитанными порядками связей, поэтому по величине контактного сдвига нельзя судить о «стабильности»; 3) ст-МО цикла играют важную роль в связывании в обоих типах комплексов, 4) 4s-и 4р-орбитали металла характеризуются значительными порядками свя
182
Глава 12
зи с атомами цикла. Исследование методом ЯМР парамагнитных металлоорганических соединений подробно рассматривается в специальной монографии [22].
К сожалению, в большинстве парамагнитных комплексов ионов переходных металлов число атомов настолько велико, что расчет методом МО всего комплекса практически невозможен. Кроме того, даже если число атомов приемлемо, встает вопрос, может ли расчет, проведенный по расширенному методу Хюккеля или по методу ЧПДП, дать разумные волновые функции для соединений с такой большой разницей в величинах зарядов, какая сущес. вует между ионом металла и лигандом. При рассмотрении таких систем предполагается, что ион металла дает по крайней мере меньшее возмущение к вкладу протона в молекулярную орбиталь, представляющую собой главным образом МО непо-деленной пары, и в другие молекулярные орбитали свободного лиганда, участвующие в связывании. Это допущение разумно для большинства комплексов, в которых прочность связи металл — лиганд составляет 10—20 ккал/моль. С учетом этого приближения проводится расчет по методу МО свободного лиганда и анализ электронной плотности ф2 с использованием волновых функций нейтрального лиганда (см. гл. 3). Последний позволяет определить, какими должны быть величины А, если на каждой из орбиталей, которые, как ожидается, смешиваются с орбиталями металла при образовании комплекса, находится по одному электрону. Результаты таких расчетов для различных замещенных пиридинов представлены в табл. 12.1.
Определив константы взаимодействия для одного неспаренного электрона на каждой из нескольких орбиталей лиганда, можно рассчитать неспаренный спин, делокализованный на каждой из этих орбиталей в комплексе. Для этого мы составляем совместные уравнения с учетом наблюдаемых констант взаимодействия для орто- и пара-протонов комплекса 4-метилпиридина. В качестве примера можно взять уравнения для комбинаций о-донорной и л-разрыхляющей орбитали, необходимых для объяснения скалярных сдвигов в шестикоординационных комплексах никеля(П).
орто- +0,3272= + 7,23х + 2,92у (12.24)
и-Метил:
-0,0302= +0,11х-4,31у (12.25)
Здесь первое число, записанное в каждом уравнении,— наблюдаемый скалярный сдвиг для комплекса, х—доля неспаренного электрона на о-орбитали, а у—доля неспаренного электрона на л-разрыхляющей орбитали. Решения этой системы уравнений дает х = 4,20-10“2 и у = = 8,08-Ю-3. Ион металла рассматривается просто как зонд для внесения спина на молекулярные орбитали металла. Мы снова подчеркиваем, что, если механизм л-спинового вклада включает л-поляризацию, на молекулярной орбитали комплекса, составляющей главным образом эту л-систему, плотность неспаренного электрона отсутствует. Величина у говорит о вкладе л-орбитали в результирующую спиновую делокали-
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 183
Таблица 12.1
Константы взаимодействия для одного неспареиного электрона, находящегося на различных орбиталях лигандов типа пиридина __
Лиганд Положение А, Э
ль СУ л*
4-Метилпиридин О + 1,75 + 7,23 + 2,92
м + 2,33 + 2,85 + 1,42
п-СН3 -6,80 + 0,11 -4,31
Пиридин О + 1,51 + 7,21 + 2,05
м + 1,60 + 2,75 + 0,87
п + 6,20 + 7,30 + 5,58
З-Метилпиридин О + 3,19 + 6,85 + 2,08
м + 2,70 + 2,50 + 1,07
п + 3,26 + 7,02 + 5,47
л<-СН3 -4,17 + 0,63 -0,49
4-Винилпиридин О + 0,74 + 7,31 + 1,54
м + 1,76 + 2,90 + 0,95
н3 + 1,49 + 0,13 + 0,70
Н2 -1,96 + 0.16 -1,43
Hi + 1,06 + 0,70 + 2,44
зацию, наблюдаемую для комплекса. Аналогичную систему уравнений можно решить для ст-донорной и л-связывающей молекулярных орбиталей пиридина, что дает х = 4,40-10~2 и = 5,20-10 3 (где z—вклад л-связывающей молекулярной орбитали). Комбинации, включающие другие пары орбиталей, ведут к неприемлемым результатам. В этом примере для получения двух неизвестных—х и у или х и z— использовались две величины сдвига. В качестве контрольного оставалось только .мета-положение. Однако, если пользоваться этим методом применительно ко всем замещенным пиридинам, перечисленным в табл. 12.1, число протонов, подходящих для проверки в этом подходе, возрастает. Можно показать, что наблюдаемые сдвиги так же хорошо получаются с помощью комбинации ст + л или ст + л* либо смеси этих двух комбинаций. Таким образом, спектр ЯМР координированного пиридина при контактном сдвиге обусловлен главным образом механизмом ст-спино-вой делокализации с небольшим л-вкладом. ст-Вклад обусловлен прямой делокализацией плотности неспаренного электрона на лиганде. л-Вклад должен быть обусловлен спиновой поляризацией, поскольку л-орбитали пиридина ортогональны е9-орбиталям металла, а на 12я-орбиталях металла находятся только спаренные электроны. Нельзя определить, обусловлен ли л-вклад в комплексе заполненной л-орби-талью или свободной л*-орбиталью пиридина либо и той и другой вместе. Заполненная л-орбиталь может оказывать влияние вследствие спин-поляризации заполненной молекулярной орбитали комплекса, представляющей по существу МО пиридина (см. МО I на рис. 12.4). Свободная л *-орбиталь может принимать участие в результате спин-поляризации в комплексе заполненной молекулярной орбитали, пред
184
Глава 12
ставляющей собой по существу 12я-орбиталь металла, к которой примешана л *-орбиталь пиридина (см. МО III на рис. 12.4). Итак, метод МО позволяет нам понять, чем обусловлены спектральные сдвиги, но считать его исчерпывающим нельзя.
В большинстве ранних работ, посвященных этому вопросу, расчет МО проводился по методу ЧПДП, в котором непосредственно учитывается спин-поляризация. Однако в этом случае с лигандом надо обращаться, как со свободным радикалом. Обычно возмущение ионом металла не может дать ничего похожего, чтобы реализовалась такая ситуация. Если лигандом является пиридин, для согласования с экспериментальными данными используются характеристики фенильного радикала, поскольку характеристики радикала пиридина дают худшие результаты. Несмотря на упомянутые выше дефекты, эта модель дает хорошее соответствие для протонных и контактных сдвигов 13С 31 из 35 ядер в ряде координированных пиридинов [23, 24]. Интерпретация механизмов спиновой делокализации в комплексе вполне согласуется с результатами более ранних работ, полученных для протонов, и с результатами расчетов с помощью расширенного метода Хюккеля [19]. Метод ЧПДП также дает много полезной информации. Если рассматривать конечную цель этой работы — полуколичественную интерпретацию наблюдаемых сдвигов,— и расширенный метод Хюккеля, и метод ЧПДП обеспечивают требуемую точность.
Необходимо вновь подчеркнуть один важный момент. При использовании ограниченных методов расчета следует вводить поправку на косвенные спиновые вклады, обусловленные поляризацией, как это было сделано ранее для небольших л-вкладов в сдвиг пиридина *. При неограниченном методе расчета ЧПДП спин-поляризация учитывается [19, 25, 26]. Справедливость критерия существования вкладов л-спино-вой делокализации, который состоит в изменении знака протонного сдвига заместителей Н и СН3 в ароматическом кольце, становится сомнительной [25] при сравнении результатов расчетов, проведенных по методу ЧПДП и расширенному методу Хюккеля. Как упоминалось ранее, метильные протоны в комплексе 4-метилпиридина характеризуются константой, знак которой противоположен знаку константы 4-протона в комплексе пиридина. При расчете методом ЧПДП [25] аналога ст-ра-дикала 4-метилпиридина для метильного протона получается отрицательная константа взаимодействия. Этот результат не говорит о том, что механизм о-делокализации может приводить к отрицательной константе взаимодействия СН3. Таким образом, нельзя отвергать [25] давно принятый критерий изменения знака константы для СН3 и Н за счет л-спиновой делокализации. Необходимо помнить, что поляризация заполненной л-системы заложена в расчет методом ЧПДП свободного радикала. Детальный анализ результатов расчетов методом ЧПДП показал [26], что ст-механизм определяет 2- и 3-водородные сдвиги, но что
* Концепция спиновой поляризации вводится из-за недостатков ограниченных расчетов.
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 185
ЧПДП-аналог спиновой поляризации заполненной л-орбитали в расчете по методу Хюккеля дает главный вклад в отрицательную константу взаимодействия метильной группы. Таков результат анализа этой системы с помощью расширенного метода Хюккеля [19]. В оригинальной литературе [19, 26], посвященной этому предмету, термины «электронная делокализация» и «спиновая делокализация» используются произвольно, но если вы обратитесь к рис. 12.4, то сразу вспомните, что означают эти термины.
В заключение, чтобы показать, насколько важны приближенные волновые функции при интерпретации контактных сдвигов, мы рассмотрим сдвиги в спектрах некоторых комплексов N-окиси 4-метилпириди-на [27]. Картина наблюдаемых протонных контактных сдвигов напоминает механизм ^-делокализации со спином, направленным в л-системе вдоль поля. Исходя из этих сдвигов, можно сделать вывод, что при координации N-окись 4-метилпиридина должна вращаться таким образом, чтобы л-молекулярная орбиталь, которая представляет собой главным образом р2-орбиталь кислорода (ось z перпендикулярна плоскости цикла), смешивалась с ст-связывающей е9-совокупностью нике-ля(П). Это приводит к возможности прямой делокализации неспаренного спина по «орбитали цикла». Такой тип координации с вращением донора обнаружен в твердом аддукте этого донора. Расчет по методу МО указывает, что некоторые из высокоэнергетических молекулярных орбиталей донора представляют собой главным образом АО кислорода с очень небольшими коэффициентами АО водорода. Таким образом, если даже эти молекулярные орбитали участвуют в связывании с нике-лем(П), они должны давать по крайней мере небольшой непосредственный вклад в протонные контактные сдвиги.
В некоторых ситуациях (см. далее) необходимы системы с исключительно псевдоконтактным вкладом в изотропный сдвиг. N-окись пиридина относится к числу именно таких лигандов, которые обеспечивают псевдоконтактный вклад в сдвиг [28]. Если в комплексе определяющую роль играет о-делокализация и контактная и псевдоконтактная части сдвигов меняются одинаковым образом, то отсутствие контактных вкладов установить трудно. Однако при использовании такого лиганда, как N-окись пиридина, альтернирование протонных сдвигов, обусловленное узлами в л-системе, свидетельствует об определяющей роли контактного сдвига.
12.8. ПРИМЕНЕНИЯ ИЗОТРОПНЫХ СДВИГОВ
Определение структуры
Предпочтительное применение метода ЯМР для изучения парамагнитных комплексов часто обусловлено увеличением разделения сигналов, причина которого — неэквивалентность входящих в молекулу атомов. Продемонстрировать чувствительность этого метода можно на примере пятикоординационного комплекса основания Шиффа с нике-
186
Глава 12
лем(П), который может иметь либо тетрагонально-пирамидальную, либо тригонально-бипирамидальную структуру (рис. 12.6). Протонный спектр ЯМР [29] демонстрирует две линии для каждой а- и Р-СН2-группы и две серии сигналов для каждой совокупности ароматических протонов. Для группы СН3 наблюдается единственная линия, что говорит
Рис. 12.6. Тетрагонально-пирамидальная (а) и тригонально-бипирамидальная (б) структуры пентадентатных комплексов оснований Шиффа.
о присутствии в растворе только одного изомера, хотя в твердом состоянии их два [ЗОа]. Если бы в растворе присутствовали оба изомера, то для каждого положения кольца наблюдались бы четыре сигнала. Структура комплекса должна быть диссимметричной и должна включать неэквивалентные триметиленовые группировки и фенильные кольца. Авторы работы [29] считают, что эти данные исключают тетрагональнопирамидальную и подтверждают тригонально-бипирамидальную структуру комплекса. Однако этот вывод неверен, поскольку даже в тетрагональной пирамиде R-группа должна давать наблюдаемую неэквивалентность [306]. Неэквивалентность колец наблюдается в отдельных изомерах в твердом состоянии. В аналогичных диамагнитных комплексах цинка(П) можно наблюдать лишь очень незначительную неэквивалентность сигналов протонов группы СН2. Быстрая перегруппировка через промежуточную тетрагональную пирамиду должна усреднять сигналы. Такие процессы при комнатной температуре быстро не протекают (в шкале времени ЯМР), и эта молекула представляет собой один из немногих пятикоординационных жестких комплексов. Если заменить N — СН3 на серу, внутримолекулярная перегруппировка приводит к наблюдаемым эквивалентным циклам.
В спектре протонного ЯМР октаэдрического комплекса Co{[4,6-(CH3)2phen]3}2+ (см. рис. 12.7, где дана формула (CH3)2phen) из-за существования цис- и транс-изомеров возможны 32 линии. В полученном спектре этого комплекса наблюдается 31 линия.
Как упоминалось в гл. 8, спектры ЯМР энантиомеров в ахиральных растворителях идентичны. Однако чувствительность метода ЯМР при
Рис. 12.7. Протонный спектр ЯМР {Co[4,6-(CH3)2phen]3}2 + в растворе метанола при —20° С. [Печатается с разрешения La Mar G.N., Van Heck G.R., Inorg. Chem., 9, 1546 (1970). ©Am. Chem. Soc.]
188
Глава 12
исследовании парамагнитных комплексов часто достаточна для того, чтобы различить сигналы диастереомеров, образующихся в том случае, когда оптически активные энантиомеры координируются с оптически активными комплексами ионов переходных металлов. В монографии [31] приведено много других примеров определения структуры методом ЯМР.
Исследование образования ионных пар
Авторы [32—34] использовали вклад в сдвиг протонов алкиламмо-ниевой группы ионной пары R4N + MX3L~ для оценки расстояния между анионом и катионом (г) в ионной паре и для изучения эффектов сольватации. В первом случае задавались геометрией ионной пары. В спектре ионной пары с R — н-бутил наблюдаются четыре протонных сигнала. Этот спектр можно попытаться согласовать с уравнением (12.23) (или другой, более удобной формулой) путем варьирования расстояния в так называемом геометрическом факторе [(1 — 3cos20j)/r?]. Для удобства мы запишем уравнение для псевдоконтактного сдвига как
Av(pc)/v= -K/^GF, (12.26)
где К — составная константа,/(я) — некоторая функция д- или а-тензора, a GF—геометрический фактор. Отношение изотропных сдвигов двух бутильных протонов определяется следующим выражением:
Av;(pc)/ДуДрс) = (GFV(GF),. (12.27)
Геометрический фактор рассчитывается для заданной геометрии [33], а затем усредняется по всем ориентациям, которые может иметь динамическая ионная пара при свободном вращении в растворе. Поскольку те ядра комплекса, которые эквивалентны в свободном катионе, дают одну линию, группа R4N+ быстро вращается в шкале времени ЯМР. Отношение Av;/Av;- для различных пар протонов рассчитывают для разных расстояний между анионом и катионом, и расчет этот повторяют для различных возможных ориентаций аниона и катиона в ионной паре. Для оценки межионного расстояния используют ту структуру, которая лучше всего согласуется с данными по сдвигу. Для (С6Н5)3РС4Нд (С6Н5)3РСоВг^ расстояние между фосфором в катионе и металлом в анионе, по данным работы [35], составляет 8 А. Такая точность оценки вызывает подозрение, поскольку в растворе ионные пары многих систем обычно образуют довольно большие кластеры, особенно если диэлектрическая проницаемость растворителя низкая. Таким образом, сдвиг сигнала катиона представляет собой величину, усредненную по всему окружению катиона в кластере. Более того, убедительно показано [36], что для алкиламмониевых протонов ионной пары может иметь место контактный сдвиг, что делает сомнительным анализ многих систем, основанный только на псевдоконтактной части. О ковалентности связывания в ионной паре говорит [36] наблюдав-
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 189
мый контактный вклад в изотропный сдвиг сигнала 14N в [(h-C4H9)4N] [(С6Н5)3РСо!3]. Сдвиг происходит в направлении, противоположном предсказываемому знаком \д, который следует из направления протонного сдвига. Существование этого контактного члена демонстрирует исключительную чувствительность изотропных сдвигов, поскольку он обнаруживает очень небольшую величину ковалентности в связывании катиона с анионом в ионной паре. Авторы [36] предложили механизмы ковалентного взаимодействия в ионной паре.
Влияние растворителя на степень образования ионных пар проявляется [37] в величине наблюдаемого изотропного сдвига. Данные по сдвигу для системы [(h-C4H9)4N] [(С6Н5)3РСоВг3] можно привести в соответствие [38] с выражением для константы равновесия следующей реакции:
[(C4H9)4N + ] [(С6Н5)3 РСоВг3 ] (C4H9)4N+ +(С6Н5)3РСоВг3
(12.28)
Исследования проводились с растворителями с диэлектрической проницаемостью в интервале от 4,6 до 64, концентрация раствора электролита составляла 0,02—0,5 М. Сдвиг, обусловленный ионной парой «катион — анион», можно определить одновременно с величиной К из данных по сдвигу. Полученные результаты показали [38], что межионное расстояние в растворителях с высокой диэлектрической проницаемостью выше, чем в растворителях с низкой диэлектрической проницаемостью.
Как оказалось [39], тетраэдрические металлсодержащие анионы, которые сами по себе не анизотропны, могут давать псевдоконтактный вклад в неметаллическом катионе. Причину и величину ^-анизотропии можно в первом приближении объяснить электростатическим возмущением кристаллического поля сферического аниона, вызываемым катионом. Предполагается также, что за время жизни ионной пары тетраэдрическая структура аниона под действием катиона несколько искажается. Поскольку катион лежит на единственной в своем роде оси, он будет подвержен влиянию дипольного сдвига, обусловленного индуцированной в ионной паре анизотропией. Существует много эквивалентных путей, по которым катион может приблизиться к тетраэдрическому или октаэдрическому аниону для образования ионной пары, и все эти пути характеризуются сопоставимыми псевдоконтактными вкладами катиона. Таким образом, динамический процесс подобного вида не усредняет до нуля псевдоконтактный сдвиг катиона. В то же время Динамический процесс такого вида усредняет до нуля влияние псевдоконтактного сдвига на положение сигналов атомов тетраэдрического или октаэдрического аниона.
Природа второй координационной сферы в комплексах
Изотропный сдвиг ЯМР позволяет эффективно исследовать природу координационной сферы вокруг комплекса иона переход
190
Глава 12
ного металла в растворе [40]. Изучая [40] спектр ЯМР [(C4H9)4N] [СоР(С6Н5)313], растворенного в СНС13, можно увидеть, что сигнал протона хлороформа сдвинут в сильное поле. Сдвиг той же величины и также в сильное поле наблюдается и для никелевого (II) аналога. Поскольку знак \д для комплексов никеля (II) и кобальта (II) различается, то сдвиг в сильное поле для обоих комплексов нельзя объяснить псевдоконтактным членом [см. уравнение (12.23)]. На самом деле сходство сдвигов для комплексов никеля и кобальта указывает на отсутствие ожидаемого псевдоконтактного вклада. Последнее возможно при условии, что протоны хлороформа связаны с атомами иода, координирующимися с кобальтом (см. рис. 12.3) под углом 0 [уравнение (12.23)], равным 55°. В таком случае разность 1 — 3cos20 равна нулю, и псевдоконтактный вклад отсутствует. Взаимодействие протонов с иодом предполагается интуитивно, и правильность этого предположения подтверждают [40] результаты исследования, проведенного методом ИК-спектроскопии: изменения в частоте валентного колебания С — Н хлороформа наблюдаются в присутствии этих комплексов кобальта и никеля и не наблюдаются в присутствии свободного (С6Н5)3Р. При образовании водородных связей с фенильными группами координированного фосфина можно было бы ожидать образования водородных связей и с фенильными группами свободного (С6Н5)3Р. В среднем во взаимодействии с комплексом участвует около четырех молекул хлороформа. Таким образом, контактный вклад убедительно подтверждает существование заметного, но небольшого ковалентного взаимодействия при образовании водородной связи даже при таком слабом доноре протонов, как хлороформ.
В развитие [41, 42] этой идеи была исследована сольватация поли-(1-пиразолил) бората кобальта(П) анилином и пиридином. В этом комплексе (/-тензор очень анизотропен (д№ = 8,48, </ L = 0,96). Линии сдвигаются в сильное поле, следовательно, предпочтительное направление подхода анилина и пиридина перпендикулярно оси симметрии комплекса.
Рис. 12.8. Структура поли(1-пиразолил)бората кобальта(П), где X = В, Y = H (или заместитель). Известны родственные комплексы, в которых X = С и Y = Н (или заместитель). Симметрией всего исходного комплекса является D3tj.
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 191
В спектре анилина наибольшим сдвигом характеризуется сигнал орто-протонов, в то время как в спектре пиридина больше всего сдвинут сигнал 4-протонов. Если водородной связью связан анилин, ближе всего к комплексу должен подходить фрагмент анилина, содержащий азот. Положительный полюс диполя пиридина—фрагмент с 4-Н, и величина и направление сдвига говорят о том, что именно он ближе всего подходит к металлу. Эти предпочтительные ориентации наиболее вероятны среди распределения выгодных ориентаций.
Исследование кинетики и равновесий
Методом ЯМР исследуется кинетика процессов, протекающих с участием парамагнитных комплексов. Больший интервал сдвигов дает возможность более точно определять константы равновесий и исследовать более быстрые процессы химического обмена. Возможность такого применения метода ЯМР рассматривается в гл. 8.
Сдвигающие реагенты
mpuc-Хелатные комплексы редкоземельных металлов и р-дикетонов представляют собой кислоты Льюиса и вызывают большие псевдокон-тактные сдвиги в основаниях Льюиса, которые вводят в растворы, содержащие эти комплексы. Результирующие сигналы для некоторых из этих ионов (например, комплексов европия и празеодима) очень узки даже по сравнению с комплексами никеля(П). На рис. 12.9 показаны [44] протонные спектры ЯМР (частота 100 МГц) растворов цис-4-бу-тилциклогексанола в CDC13, содержащих Eu(dpm)3 [dpm — дипивалоил-метанато, (СН3)3СС(О) — СНС(О)С(СН3)3]. Спектр чистого спирта приведен на рис. 12.9,Л, спектр первого порядка с хорошо разделенными линиями показан на рис. 12.9,Б. Обратите внимание, что спин-спиновые взаимодействия все еще не затронуты.
Комплексы редкоземельных металлов, вызывающие псевдокон-тактные сдвиги, были названы [43] сдвигающими реагентами (СР). Между комплексом СР и избытком основания Льюиса быстро устанавливается равновесие. Комплексы лантана с такими фторсодержащими хелатообразующими соединениями, как 1.1.1,2.2,3.3-гептафтор-7,7-диме-тилоктандион-4,6 (CH3)3CC(O)CHC(O)CF2CF2CF3 (сокращенно названным fod), относятся к числу сильных кислот Льюиса и приводят к очень большим сдвигам. Вопросам использования сдвигающих реагентов посвящено несколько сот работ, в том числе исчерпывающий обзор [46], поэтому мы лишь рассмотрим, где применяются СР и какие осложнения при этом возможны.
Сдвигающие реагенты применяются, в частности, для упрощения спектров второго порядка (т. е. при A^J). При комплексообразовании сигналы различных протонов в молекуле сдвигаются по-разному, и с увеличением концентрации СР А становится больше, чем J (рис. 12.9). В системах, содержащих СР, образующих комплексы 1:1, средний сдвиг i-ro
192
Глава 12
протона, полученный в результате быстрого обмена, определяется как
8,.ave = ^8/ + ^5c, (12.29)
где 8f— химический сдвиг ядра в некоординированной молекуле, а 8С— химический сдвиг ядра в координированной молекуле, N— мольная доля. Для комплекса 1.1
Nf = 1 - Nc.
Решая совместно эти два уравнения, получаем
3,AV, = (1 - Nc)8f + NC8C = 8, + ЛЦ8, - 8Д. (12.30)
Если комплексообразование проходит полностью, зависимость 8i AEV от Nc имеет вид прямой, тангенс угла наклона которой равен 8С — 5f. Отсекаемый этой прямой отрезок 8 у представляет собой химический сдвиг, который наблюдается в спектре ЯМР свободного соединения, если используется спиновая развязка или если спектр второго порядка анализируется с помощью вычислительной машины.
Обычно СР участвуют в более сложной совокупности равновесий, чем просто в образовании аддукта 1:1. Однако при небольшом относительно субстрата содержании реагента зависимость отсчитываемого от
К.0 15,0 52,0 11,0 10,0 9,0 8,0 7,0 6,0 5,0 4,0 3,0 2,0 1,0 0,0
Рис. 12.9. Протонный спектр (100 МГц) i/uc-4-трет-бутилциклогексанола (20 мг, 1,28-Ю-4 моль) в CDC13 (0,4 мл) в присутствии следующих количеств Eu(dpm)3:
А — 0,0 мг; Б—10,3 мг;В—16,0 мг; Г—33,1 мг; Д— 60,2 мг. [Печатается с разрешения из Demarco P.V., et al., J. Am. Chem. Soc., 92, 5734 (1970).] © Am. Chem. Soc.
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 193
сигнала ТМС химического сдвига от молярного отношения закомплексованного субстрата имеет вид прямой [44]. Экстраполяция к нулевой мольной доле закомплексованного субстрата дает желаемый «развязанный» химический сдвиг исследуемого протона. Если же зависимость отклоняется от прямой или если ее надо экстраполировать на большое расстояние, значения химического сдвига получают с большой ошибкой. Комплексы ионов переходных металлов первого ряда были первыми системами, изученными таким образом. Если при изучении какой-либо системы возникают указанные выше осложнения, то следует попытаться найти комплекс иона переходного металла первого ряда, образующий аддукт 1:1, и провести расчет по уравнению (12.30). Можно также использовать комплекс, для которого константа равновесия второй стадии К2 намного превышает константу первой стадии Кг, и взять уравнение (12.30), видоизмененное применительно к комплексу состава 2:1.
С помощью сдвигающих реагентов в принципе можно определять геометрию молекул в растворе [40]. Этот эксперимент обычно проводится в диапазоне быстрого обмена. Предполагают, что спектральные сдвиги протонного ЯМР, обусловленные СР, имеют по своей природе дипольный характер. В идеальном случае можно задаться структурой молекулы и рассчитать по уравнению (12.22) дипольные сдвиги для большого числа различных ядер исследуемой молекулы. Чтобы добиться соответствия расчетных и экспериментальных данных по сдвигу, меняют задаваемую структуру молекулы. Поскольку структура исследуемой молекулы и структура комплекса в растворе, как и величина и положение магнитного диполя металлического центра в комплексе, неизвестны, то в общей сложности система имеет восемь неизвестных. Что это за неизвестные, можно увидеть из рис. 12.10, где показан такой жесткий лиганд, как пиридин, связанный в комплекс с СР. Для определения ориентации молекулы относительно СР нужны четыре параметра: 1) г—расстояние между металлом и донором; 2) а—угол между связями металл — донорный атом и азот — ордао-углерод; 3) Р—угол между плоскостью лиганда и магнитной плоскостью х, у металла; 4) у— угол, характеризующий поворот плоскости молекулы лиганда относительно оси азот — пара-углерод. Кроме того, нужны два угла для определения ориентации магнитной оси относительно связи металл — — донор. Для объяснения анизотропии х нужны еще два неизвестных. Чтобы получить строгое решение этой задачи, необходимо установить возможность существования в растворе сопоставимых количеств комплексов состава 1:1 и 2:1. (Большинство СР обладают двумя кислотными центрами.) Если в растворе комплексы 1:1 или 2:1 не доминируют наряду с Ki и К2, должны быть известны две совокупности неизвестных, о которых мы упоминали ранее, по одной для каждого комплекса. Однако, чтобы не усложнять картину, можно прекратить обмен между лигандами в комплексе и свободными лигандами. Из всего сказанного выше следует, что строго геометрии молекул в растворе определить нельзя. Таким образом, чтобы получить информацию относи
13-574
194
Глава 12
тельно геометрических структур в растворе, необходимо сделать некоторые упрощающие допущения.
Если допустить существование оси симметрии третьего порядка и наличие доминирующего типа комплекса, задачу можно решить. Для комплекса с аксиальной симметрией расчет можно проводить по уравнению (12.23), а не (12.22). Если взять отношение всех наблюдаемых сдвигов ЯМР к наибольшему наблюдаемому сдвигу Av7, то из уравнения (12.23) можно вывести соотношение
Av,. Avj
1 — 3cos2 0,-
3cos2 0; \
(12.31)
Таким образом, мы можем не рассматривать анизотропию х комплекса. (Это возможно, если исключить из рассмотрения протоны хелатирующего лиганда и заниматься только координированным основанием Льюиса.) Если допустить существование аксиальной симметрии, но считать, что направление связи металл—донорный атом отклоняется от магнитной оси z, то останется пять неизвестных [46]. Для определения ориентации молекулы относительно металла необходимы еще четыре неизвестных, и одна нужна для определения ориентации магнитной оси z относительно связи металл — донорный атом. Если считать,
Рис. 12.10. Определение переменных для задачи установления молекулярной структуры с использованием сдвигающего реагента.
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 195 что магнитная ось z совпадает со связью металл — донорный атом, то останутся только четыре неизвестные, необходимые для определения ориентации молекулы относительно атома металла. В последнем случае системы относятся к числу наипростейших, и их чаще всего и исследуют. Чтобы добиться наилучшего соответствия этих параметров с экспериментальными данными, созданы программы расчета на вычислительных машинах, однако параметры, получаемые в результате подгонки, несмотря на большое количество данных, часто имеют высокие коэффициенты корреляции.
Хотя замена уравнения (12.22) на (12.23) позволяет получить информацию о молекулярной структуре в растворе, ни одна из структур, изучаемая в растворе, не имеет в твердом состоянии ось симметрии третьего или более высокого порядка. Высшей осью симметрии является ось симметрии второго порядка комплекса Ho(dpm)3(4-pic)2. Результаты измерения магнитной анизотропии монокристалла этого комплекса показали [50], что для 4-метильных протонов 15% наблюдаемого сдвига ЯМР обеспечивает дополнительный член, ответственный за не-аксиальность симметрии [уравнение (12.22)]. Авторы работ [51—53] исследовали Eu[(dpm)3py2] и аналогичные комплексы Ей с такими лигандами, как 3- и 4-пиколин и 3,5-лутидин при комнатной и пониженных температурах. Как оказалось, используя уравнение (2.12), можно добиться соответствия расчетных данных и экспериментальных данных, полученных при комнатной температуре, и только с помощью этого уравнения можно достигнуть соответствия в том случае, когда экспериментальные данные получены при пониженных температурах. Были выдвинуты предположения о динамических процессах, которые могут объяснить причину успеха предположения о существовании аксиальной симметрии при комнатной температуре. К аксиальной симметрии может привести вращение субстрата относительно связи металл — — донорный атом [54] или же быстрое взаимное превращенн' различных геометрических изомеров через диссоциацию и ассоциацию субстрата [551. По мнению авторов работы [49], эффективная аксиальная симметрия возникает в СР потому, что уравнение (12.22) содержит тригонометрические функции <р, азимутального угла, только в нечетных степенях. Это как раз те тригонометрические функции, которые усредняются до нуля. При изучении релаксации аксиальной симметрией задаваться нельзя, поскольку уравнение релаксации включает тригонометрические функции ф только в четных степенях.
Величина изотропного сдвига зависит от иона редкоземельного металла, входящего в состав хелатообразующего лиганда. На рис. 12.11 показаны сдвиги для 2-протона хелатного бис-аддукта 4-пиколина с шрис-(дипивалоилметанато)лантаноидом, Ln(dpm)3 [50]. Аналогичные тенденции обнаружены и для других субстратов. Изменение природы металла не дает новой информации о молекулярных структурах в растворе, если исходить из изменения при этом величин сдвигов, поскольку появляются новые неизвестные.
Влияние СР на сигналы ЯМР на ядрах 13С дает дополнительную информацию, позволяющую проводить определение структур в раство-
13'
196
Глава 12
ре. Однако, согласно данным [46], в этом случае на сдвиг может оказывать влияние контактное взаимодействие Ферми, поэтому пользоваться полученными результатами нужно крайне осторожно.
Помимо комплексов лантаноидов, вызывающих сдвиг сигналов, применяются комплексы лантаноидов, способные уширять сигналы
Рис. 12.11. Наблюдаемые изотропные сдвиги сигнала 2-протона в 4-пиколине в комплексах 4-пиколин — Ln(dpm)3 состава 2:1. [LaMar G.N., Metz Е.А., J. Amer. Chem. Soc., 96, 5611 (1974).]
[56] . Поскольку эффекты сдвига и уширения имеют различную зависимость от г(г-3 и г-6 соответственно), две совокупности результатов дают дополнительную информацию к определению структуры. Ширина сигнала 2-метильных протонов аддуктов 2-пиколина с Ln(dpm)3 [50] приведена в табл. 12.2.
Однако определение структуры молекул в растворе с помощью СР осложнено многими явлениями, что ограничивает применение и точность метода. К такого рода осложняющим факторам относятся следующие:
1. Стехиометрию образующихся аддуктов необходимо определять, но часто ее предполагают.
2. Рассмотрение предполагает, что в растворе присутствует только один геометрический изомер аддукта. Однако на самом деле их может
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 197 быть много. Более того, лантаноиды имеют высокое сродство к воде, которая, если она присутствует в растворе, может конкурировать с субстратом за кислотные центры, что увеличивает число комплексов в системе.
Таблица 12.2
Ширина иа полувысоте [Av(l/2)] сигналов 2-метильных протонов 2-пиколина в аддуктах Ln(dpm)3
Ln(III) Av(1/2), Гц Ln(III) Av(1/2), Гц
Рг 5,6 Dy 200
Nd 4,0 Ho 50
Sm 4,4 Er 50
Eu 5,0 Tm 65
Tb 96 Yb 12
3. Если субстрат участвует в динамическом внутримолекулярном процессе, то определяется средняя конформация молекулы, а она может быть относительно неустойчивой.
4. Обычно предполагается, что комплекс аксиально симметричен, так что дипольные сдвиги можно рассчитать с помощью члена <(1 — — 3cos2 0)/г3). Системы, изученные к настоящему времени, аксиальной симметрией не обладают (см., однако, работу [49]).
5. Часто неизвестно положение главной магнитной оси в комплексе относительно лиганда.
6. Если субстрат сложен, необходимо допускать существование только одного связывающего центра.
Несмотря на эти осложнения, метод, основанный на использовании СР, позволяет различать основные структурные характеристики многих изомерных соединений. Например, можно различить как соединения [44]
так и соединения [57]
198
Глава 12
СР использовали для дифференциации внешнего и внутреннего фосфолипидных слоев в мембранах [58]. Они применялись также в конформационных исследованиях нуклеотидов в растворах [59] и в других биохимических системах [60]. Необходимо всегда учитывать возможность того, что СР способны влиять на конформацию биомолекулы.
Другим интересным применением метода является использование оптически активных СР для определения оптической чистоты. Идея аналогична той, которая обсуждалась в гл. 8, где описывалось применение оптически активных растворителей. В данном случае образование различных диастереоизомерных аддуктов характеризуется различными константами устойчивости, что дает для энантиомерных оснований различные сдвиги, усредненные по мольным долям. В работах [61—63] сообщается об использовании для этой цели различных оптически активных комплексов редкоземельных металлов.
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. Fitzgerald R.J., Drago R.S., J. Amer. Chem. Soc., 90, 2523 (1968).
2. См., например, Drago R.S., Zink J.I., Richman R.M., Perry W.D., J. Chem. Educ., 51, 371, 464 (1974); Keller H.J., Schwazhaus K.E., Angew. Chem. Int. Ed., 9, 196 (1970).
3. В качестве более строгого и математического рассмотрения этого и следующих разделов см. Jesson J.P., in NMR of Paramagnetic Molecules, ed. by LaMar G.N., Horrocks W.D., Jr., Holm R. H., Academic Press, New York, 1973.
4. Kurland R.J., McGarvey B.R., J. Magn. Res., 2, 286 (1970).
5. Golding R.M., Mol. Phys., 8, 561 (1965).
6. LaMar G.N., Jesson J.P., Meakin P., J. Amer. Chem. Soc., 93, 1286 (1971).
7. a. Horrocks W.D., Jr., Hall D.D., Inorg. Chem., 10, 2368 (1971).
b. Benelli C., Bertini I., Gatteschi D.G., J. Chem. Soc., Dalton, 1972, 661.
8. DeSimone R.E., Drago R.S., J. Amer. Chem. Soc., 92, 2343 (1970).
9. Kluiber R.W., Horrocks W.D., Jr., Inorg. Chem., 6, 430 (1967).
10. Wicholas M.L., Drago R.S., J. Amer. Chem. Soc., 90, 2196 (1968).
11. Horrocks W.D., Jr., Inorg. Chem., 9, 690 (1970).
12. LaMar G.N., Van Hecke G.R., J. Amer. Chem. Soc., 91, 3442 (1969).
13. Wicholas M.L., Drago R.S., J. Amer. Chem. Soc., 91, 5963 (1969).
14. Zink J.I., Drago R.S., J. Amer. Chem. Soc., 92, 5339 (1970).
15. Fitzgerald R.J., Drago R.S., J. Amer. Chem. Soc., 89, 2879 (1967).
16. Fitzgerald R.J., Drago R.S., J. Amer. Chem. Soc., 90, 2523 (1968).
17. Drago R.S., Petersen H., Jr., J. Amer. Chem. Soc., 89, 3978 (1967); 89, 5774 (1967).
18. Rettig M.F., Drago R.S., J. Amer. Chem. Soc., 91, 1361 (1969); 91, 3442 (1969).
19. Cramer R.E., Drago R.S., J. Amer. Chem. Soc., 92, 66 (1970).
20. Anderson S.E., Jr., Drago R.S., J. Amer. Chem. Soc., 92, 4244 (1970); 91, 3656 (1969); Inorg. Chem., 1'1, 1564 (1972).
21. Anderson S.E., Jr., Matwiyoff N.A., Chem. Phys. Lett., 13, 150 (1972).
22. Rettig M.F., NMR of Paramagnetic Molecules, ed. LaMar G.N., Horrocs W. D. and Holm R. H., Academic Press, New York, 1973.
23. Doddrell D., Roberts J.D., J. Amer. Chem. Soc., 92, 6839 (1970).
24. Morishima L, Yonezawa T, Goto K., J. Amer. Chem. Soc., 92, 6651 (1970).
25. Horrocs W.D., Johnson D.L., Inorg. Chem., 10, 1835 (1971).
26. Horrocs W.D., Jr., Inorg. Chem., 12, 1211 (1973).
27. Perry W.D., et al., Inorg. Chem., 10, 1087 (1971).
28. Johnson B.F.G., Lewis J., McArdle P., Norton J.R., J. Chem. Soc., Dalton, 1974, 1253.
29. LaMar G.N., Sacconi L„ J. Amer. Chem. Soc., 89, 2282 (1967).
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 199
30. a. Orioli P.L., DiVaira М., Sacconi L., Inorg. Chem., 10, 553 (1971).
6. Bertini I., частное сообщение.
31. a. Holm R.H., Hawkins C.J., NMR of Paramagnetic Molecules, ed. LaMar G.N., Horrocs W. D„ Jr., Holm К. H., Academic Press, New York, 1973. 6. LaMar G.N., Van Heck G.R., Inorg. Chem., 9, 1546 (1970).
32. LaMar G.N., J. Chem. Phys., 41, 2992 (1964); 43, 235 (1965).
33. Walker I.M., Rosethal L., Quereshi M.S., Inorg. Chem., 10, 2463 (1971).
34. Larsen D. W.. J. Amer. Chem. Soc.. 91. 2920 (1969); Inorg. Chem.. 5. 1109 (1966).
35. Fischer R.H., Horrocs W.D., Ir., Inorg. Chem., 7, 2659 (1968).
36. Brown D.G., Drago R.S., J. Amer. Chem. Soc., 92, 1871 (1970).
37. Fanning J.C., Drago R.S., J. Amer. Chem. Soc., 90, 3987 (1968).
38. Lim Y.Y., Drago R.S., J. Amer. Chem. Soc., 94, 84 (1972).
39. Walker I.M., Drago R.S., J. Amer. Chem. Soc., 90, 6951 (1968).
40. Rettig M.F., Drago R.S., J. Amer. Chem. Soc., 88, 2966 (1966).
41. Eaton D.R., Can. J. Chem., 47, 2645 (1969).
42. Eaton D.R., et al.. Can. J. Chem., 49. 1218 (1971).
43. Hinckley C.C., J. Amer. Chem. Soc., 91, 5160 (1969).
44. Demarco P. V., et al., J. Amer. Chem. Soc., 92, 5734 (1970).
45. Rondeau R.E., Sievers R.E., J. Amer. Chem. Soc., 93, 1522 (1971).
46. Hawkes G. E., et al., Nuclear Magnetic Shift Reagents, ed. Sievers, Academic Press, New York. 1973.
47. Davis R.E., Willcott M. R., Ill, Nuclear Magnetic Shift Reagents, ed. Sievers R.E., Academic Press, New York, 1973.
48. Davis R. E., Willcott M.R., III, J. Amer. Chem. Soc., 94, 1744 (1972); Gansow 0., et al., J. Amer. Soc., 95, 3389 (1973).
49. LaMar G.N., Metz E.A., J. Amer. Chem. Soc., 96, 5611 (1974).
50. Horrocs W.D., Jr., et al., Science, 177, 994 (1972); J. Amer. Chem. Soc., 93, 6800 (1971).
51. Cramer R.E., Dubois R., J. Amer. Chem. Soc., 95, 3801 (1973).
52. Cramer R.E., Dubois R., Seff K., J. Amer. Chem. Soc., 96, 4125 (1974).
53. Cramer R.E., Dubois R„ Furuike C.K.. Inorg. Chem.. 14. 1005 (1975).
54. Briggs J. P., et al., Chem. Comm., 1972, 1180.
55. Horrocs W.D., Jr., J. Chem. Soc., 96, 3022 (1974).
56. Marzin C., et al., Proc. Nat. Acad. Sci. (USA), 70, 562 (1973).
57. Akutani T, et al., Tetrahedron Lett., 1971, 1115.
58. Andrews S.B., et al., Proc. Nat. Acad. Sci. (USA), 70, 1814 (1973) и ссылки в этой работе.
59. Barry C.D., et al., Biochem. Biophys. Acta, 262, 101 (1972) и ссылки в этой работе.
60. Dwek R. A., et al.. J. Biochem. 21. 204 (1971).
61. Whitesides G.M., Lewis D.W., J. Amer. Chem. Soc., 92, 6979 (1970); 93, 5914 (1971).
62. Goering H.L., et al., J. Amer. Chem. Soc., 93, 5913 (1971).
63. Fraser R.R., et al., Chem. Commun, 1971, 1450.
Упражнения
1. Близок ли к нулю контактный вклад в изотропный сдвиг в протонном ЯМР для каждой из приведенных ниже структур? Равен ли нулю псевдоконтактный вклад? Объясните свои ответы.
а.
(тетраэдрическое расположение вокруг Со).
200
Глава 12
б. Аддукт н-бутанола с Eu(dpm)3, где dpm—это
?! О io
(СН3)3С 'ХГ С(СН3)3 н
2. Снят спектр ЯМР на ядрах 17О водных растворов парамагнитных ионов. Сигнал 17О воды, входящей в координационную сферу парамагнитного иона, сдвинут в сильное поле по сравнению с сигналом некоординированной воды. Водный раствор, содержащий парамагнитные ионы Dy3 + , дает только один 17О-сигнал. Водные растворы диамагнитного иона А13+ характеризуются двумя 17О-линиями. Рассмотрите диаграмму спектров.
а. Кратко объясните, почему в спектрах водных растворов комплексов Dy3+ наблюдается только одна линия, тогда как в спектрах растворов комплексов А13+ их две.
б. В спектре ЯМР на ядрах 17О раствора 0,1 М по Dy3+ и 10,0 М по воде наблюдается одна линия, сдвинутая в сильное поле по сравнению с сигналом некоординированной воды. Объясните, почему добавление 0,2 моль безводного комплекса А13+ приводит к сдвигу этой линии до 216 м.д.
в. Рассчитайте число молекул воды в координационной сфере А13 + .
Dy3+, Н2О ______________________|___________
АР+, Н2О ____________L.
А13+, Н2О, Dy3*______Ц.
0
гоо'м.д.
3. Для Ni(C6H5CH2NH2)^ + величины Av (изо) составляют: NH2 +6313; СН2 —2083; фенильные протоны: орто +78, мета —95 и пара +83 Гц.
а. Интерпретируйте эти сдвиги, исходя из механизма делокализации.
б. Каким образом при интерпретации сдвигов протонов фенильной группы вы можете исключить спин из существенно ст-МО-поляризации тс-МО?
4. а. Сколько линий должно быть в протонном спектре ЯМР указанного соединения и какие из них будут сдвинуты в большей степени при введении СР.
Н сн3 - i - он
б. Будут ли результаты различными, если добавленный СР оптически активен, и почему?
5. Объясните, как с помощью метода ЯМР можно определить, координиро-
4—N—Н
ван ли Ц \ кислотами Льюса через азот, амино- или имино-группы.
6. Показанный ниже стабильный радикал при комнатной температуре в растворе в бензоле при концентрации 10 “4 М дает спектр ЭПР, состоящий из
Спектры ЯМР парамагнитных комплексов ионов переходных металлов 201
трех узких линий. По мере увеличения концентрации радикала сигнал ЭПР уширяется, и 1 М раствор дает едва различимый сигнал, хотя в аналогичных условиях при 60 МГц регистрируется следующий сигнал ЯМР:
а. Объясните, чем обусловлены сигналы ЭПР при низкой и при высокой концентрации радикала.
б. Почему с ростом концентрации сигнал ЭПР исчезает, а сигнал ЯМР появляется?
в. Каким образом можно провести предварительный расчет величины А, наблюдаемой в приведенном выше спектре ЯМР? Объясните, какие данные нужно использовать в таком расчете?
7. Изотропные константы СТВ в анионе пиридина равны ан = 6,28, аИ11) = = 3,55, аН(3) = 0,82 и ан(4) = 9,70. Атомы в системе пронумерованы следующим образом:
(4)
а. Объясните, на какой МО — с' или л — находится неспаренный электрон.
б. Какой(ие) фактор(ы) следует принимать в расчет при интерпретации aN данного аниона.
в. Какой дополнительный фактор следует учитывать при интерпретации aN C5H5N+ (т. е. пиридина с удаленным одним электроном из неподеленной пары)?
г. Основываясь на приведенных выше данных, объясните, почему при координации шести молекул пиридина с образованием октаэдрического комплекса никеля(П) сигналы протонов 2, 3 и 4 сдвинуты в слабое поле соответственно на —3820, —1420 и —445 Гц [J. Amer. Chem. Soc., 92, 66 (1970)].
д. Что вы можете сказать о псевдоконтактном сдвиге в этом комплексе?
е. Объясните, почему сдвиг составляет +442 Гц, если метильную группу, находящуюся в положении 4. заменить на протон?
8. Четырехкоординационные комплексы никеля(Н) могут быть: 1) низкоспиновыми (S = 0) квадратно-плоскостными, 2) высокоспиновыми (S = 1) тетраэдрическими, 3) с промежуточной геометрией D2d при близких по энергии триплетном и синглетном состояниях; 4) возможно также равновесие между комплексами 1 и 2 типа, константа скорости взаимного перехода в данном случае равна КГ7 с.
202
Глава 12
а. Какой окажется температурная зависимость магнетизма для каждого из указанных комплексов (т. е. выполняется или не выполняется закон Кюри— Вейса). Объясните почему.
б. Можно ли различить комплексы типа 3 и 4, используя спектроскопию ЯМР, ЭПР или ИК-спектроскопию? (Укажите, что вы ожидаете получить при проведении исследования указанными методами.)
в. Опишите, что вы ожидаете получить при изучении спектра ЯМР и его температурной зависимости для каждого из указанных выше четырех типов комплексов.
9. Подвергните критическому рассмотрению следующие планы исследований. Если этот план можно осуществить при некоторых разумных условиях, укажите, что это за условия. Если план исследования превосходен, объясните, почему вы так думаете.
а. Смешивание Га9-уровней ионов переходных металлов первого ряда и лигандов необходимо исследовать с помощью контактных сдвигов ЯМР лигандов л-акцепторов в октаэдрических комплексах Ni2 + (4/'a) и Mn2 + (t^e2).
б. Существование ковалентности в комплексах редкоземельных металлов необходимо исследовать путем измерения изотропных протонных сдвигов.
в. Для установления структуры фермента в растворе следует использовать СР и реагенты, уширяющие сигналы.
13. СПЕКТРЫ ЭЛЕКТРОННОГО ПАРАМАГНИТНОГО РЕЗОНАНСА КОМПЛЕКСОВ ИОНОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ
13.1. ВВЕДЕНИЕ
Данная глава является продолжением гл. 9, и прежде, чем приступить к чтению изложенного здесь материала, следует хорошо понять принципы, рассмотренные в гл. 9—11. Однако в случае комплексов ионов переходных металлов основой интерпретации спектров ЭПР служит теория поля лигандов, и по этой причине изложение материала в настоящей главе связано с его изложением в гл. 9 лишь косвенно.
Спектры ЭПР комплексов ионов переходных металлов дают быструю информацию об электронных структурах этих комплексов. Дополнительная информация и осложнения, характерные для систем ионов переходных металлов, обусловлены возможным вырождением d-орбиталей и тем, что многие молекулы содержат более одного неспаренного электрона. Эти свойства приводят к орбитальным вкладам и эффектам нулевого поля. В результате существования заметных орбитальных угловых моментов ^-факторы комплексов многих металлов очень анизотропны. Спин-орбитальное взаимодействие также приводит к большим расщеплениям в нулевом поле (от К) см ' и больше) за счет смешивания основного и возбужденного состояний.
Теорема Крамерса [1] суммирует свойства многоэлектронных систем. Согласно этой теореме, у иона с нечетным числом электронов в отсутствие магнитного поля каждый уровень должен оставаться по меньшей мере дважды вырожденным. При нечетном числе электронов квантовое число т, должно иметь значение от +1/2 до +J. Таким образом, низшим уровнем любого иона с нечетным числом электронов должен быть по крайней мере дублет, называемый дублетом Крамерса. Это вырождение можно устранить магнитным полем, поэтому должен возникать регистрируемый спектр ЭПР. В то же время для системы с четным числом электронов т, = 0, +1, ..., ±J. Вырождение можно полностью снять кристаллическим полем низкой симметрии; в этом случае остаются только синглетные уровни, которые могут отличаться по энергии настолько сильно, что в микроволновом диапазоне спектр ЭПР не наблюдается. Это иллюстрируется расщеплением энергетических уровней, показанным на рис. 13.1. Для систем с четным числом электронов основное состояние невырожденно и энергия перехода между состояниями с J = 1 и 7 = 0 достаточно часто лежит вне диапазона энергий микроволн.
При исследовании комплексов ионов переходных металлов необходимо сделать несколько дополнительных замечаний о методике экспе-
204
Глава 13
Газообразный Кристалла- Слон- Магнитное Газа- Кристалла- Спин- Магнитное ион ческое орбитальное поле образный ческое орбитальное поле поле Oh взаимодействие Н I ион поле Of, взаимодействие Н (в нулевом пале) ' <в нулевом поле)
А б
Рис. 13.1. Устранение вырождения состояний газообразных ионов кобальта(П) (Л) и ванадия(1П) (Б) кристаллическим полем, спин-орбитальным взаимодействием и магнитным полем.
Считается, что состояние Т имеет эффективное Ц называемое L' и равное 1; для состояния А значение L' равно 0. Тогда J' = L' + S,...,L’ — S. Для основного состояния показаны только расщепления в магнитном и нулевом полях.
римента (отличающейся от описанной в гл. 9). На ширину линий в спектре ЭПР влияет ряд факторов, помимо тех, которые связаны непосредственно с прибором. Как и в случае ЯМР, важное значение имеют спин-решеточные, спин-спиновые и обменные взаимодействия.
Уширение, обусловленное спин-решеточной релаксацией, возникает в результате взаимодействия парамагнитных ионов с тепловыми колебаниями решетки. Пределы изменения времени спин-решеточной релаксации для различных систем велики. Время жизни отдельных соединений настолько велико, что позволяет наблюдать спектр при комнатной температуре, тогда как в случае других систем это невозможно. Поскольку время релаксации обычно растет с понижением температуры, для получения хорошо разрешенного спектра многие соединения переходных металлов необходимо охладить до температуры жидкого азота или гелия.
Спин-спиновое взаимодействие возникает из-за наложения магнитных полей от окружающих парамагнитных ионов. В результате поле на каждом ионе несколько изменяется, и энергетические уровни сдвигаются. Возникает распределение энергий, которое приводит к уширению сигнала. Поскольку этот эффект изменяется как функция вида (1/г3)-(1 — 3cos29), где г—расстояние между ионами и 0 — угол между
Спектры ЭПР комплексов ионов переходных металлов 205
направлением поля и осью симметрии, обусловленное им уширение демонстрирует заметную зависимость от направления поля. Величина спин-спинового взаимодействия снижается с увеличением расстояния между парамагнитными ионами, поэтому часто системы ионов переходных металлов удобно изучать при внесении их в изоморфное диамагнитное вещество. Например, комплекс меди можно изучать либо в виде порошка или монокристалла, разведя его в аналогичном комплексе цинка, либо в виде замороженного раствора. Разведение твердого вещества изолирует электронный спин данного комплекса от электронных спинов других парамагнитных молекул, и время жизни спинового состояния увеличивается. (См. обсуждение спектров, приведенных на рис. 9.25.) Замороженные растворы должны образовывать хорошие стекла. В противном случае образуются парамагнитные агрегаты, которые дают спектр с уширенными линиями. Часто необходимо удалять кислород из растворителя, поскольку он может вызывать уширение сигнала. Даже при образовании хороших стекол обычно не удается зарегистрировать сверхтонкие расщепления ниже чем 3 или 4 Э.
Химические обменные процессы заметно изменяют ширину линий. Этот эффект также можно ослабить путем разведения. Если в обмене участвуют эквивалентные парамагнитные частицы, то линии уширяются у основания и становятся уже у центра. Если в обмене участвуют различные ионы, то отдельные линии сливаются и дают один сигнал, который может быть широким или узким в зависимости от скорости обмена. Такой эффект наблюдается для CuSO4-5H2O, в элементарной ячейке которого имеются два различных центра меди [2].
Анизотропию параметров ЭПР можно наблюдать в монокристаллах. Информацию об анизотропии в системе можно также получить при исследовании порошков и стекол, поскольку результирующие спектры принадлежат системам, в которых нет усредняющего движения. Теперь посмотрим, почему информация об анизотропии получается из спектра, даже если молекулы в порошкообразном или стеклянном образце имеют исключительно большой набор ориентаций относительно направления приложенного поля. Рассмотрим молекулу с осью симметрии третьего или более высокого порядка, которую можно описать с помощью д№ и д±. Как видно из рис. 13.2, в молекуле много осей, которые можно пометить как д±. Поэтому в объеме образца со многими ориентациями ансамбля кристаллитов имеется больше возможных ориентаций для оси д±, расположенной по полю, чем для оси 0ц • (/-Фактор для любой ориентации выражается как
02=0±2sin20 + ^cos29, (13.1)
где 9 — угол между направлением главной оси (т.е. оси д$) и направлением поля. Поскольку все ориентации кристаллитов в твердом состоянии равновероятны, поглощение будет иметь место при всех направлениях поля от д^ до дъ а так как кристаллитов с дъ направленным по полю, больше, чем кристаллитов с направленным по полю д%, то наибольшая величина поглощения будет соответствовать q±. Если рас-
206
Глава 13
Рис. 13.2. Ось д^ и некоторые оси д± в кристалле с осью симметрии третьего или более высокого порядка.
сматривать вероятности различных ориентаций и вероятность перехода, соответствующую каждой из них, то можно предсказать появление спектра, изображенного на рис. 13.3,Л, первая производная которого показана на рис. 13.3,5. Этот пример идеализирован, и часто перекрывание характеристик, обусловленных дц и д±, сильно затрудняет определение их величин.
Если система ромбическая и дх> ду> gz, то спектр, полученный для поликристаллического образца при 7 = 0, напоминает спектр, представленный на рис. 13.4,Л. Если I = 1/2 и система имеет почти изотропные ^-факторы, но А? > Ау > Ах, то следует ожидать спектр, показанный на рис. 13.4,5. Спектр комплекса с аксиальной симметрией и I = 1/2, для которого д± > д\\ и Лц > А±, представлен на рис. 13.4,5. Другие системы довольно сложны, и вероятность неправильного отнесения стано
Рис. 13.3. Идеализированные спектры поглощения (Л) и его первой производной (Б) для неориентированной системы с 5=1/2, аксиальной симметрией и без сверхтонкого взаимодействия (д± > д^).
Спектры ЭПР комплексов ионов переходных металлов 207
вится очень большой. Значения g и А можно определить с достаточной надежностью только в относительно простых случаях. Имеются программы расчета на ЭВМ для получения спектров ЭПР поликристалли-ческих образцов простых систем, таких, так комплексы Cu(II).
Для ориентации молекул при исследованиях методом ЭПР можно также использовать [3] нематические фазы жидких кристаллов. В этом случае изучают молекулы растворенного вещества, которые не могут принимать сферическую форму; примером может служить молекула
Рис. 13.4. Спектры ЭПР порошкообразных образцов систем с 5=1/2.
А — ромбическая система с I = 0; Б — изотропный ^-фактор, I = 1/2 и Az > Ау > Ах ; В—аксиальная симметрия с I = 1/2, 0_i_ > и > Ах- в последних случаях можно получить надежное значение g и А только при использовании ЭВМ.
z
Рис. 13.5. Спектры ЭПР Co(meacacen) при 77 К [4].
А—структурная формула; Б — ориентация молекулы в замороженном ориентированном жидком кристалле; В—неориентированный замороженный раствор; Г — замороженный жидкий кристалл, ориентированный как на Б; Д — замороженный жидкий кристалл, ориентация которого изменена на 90° по сравнению с Б. Фаза в этом спектре обратна обычно используемой. [Reprinted with permission from Hoffmann B.M., Baso-lo F., Dimente D.L., J. Am. Chem. Soc., 95, 6497 (1973). Copyright by American Chemical Society.]
Спектры ЭПР комплексов ионов переходных металлов 209
Со(теасасеп), изображенная на рис. 13.5,А. Жидкокристаллический раствор этого низкоспинового комплекса Со(П) помещают в магнитное поле, чтобы дать возможность молекулам сориентироваться (как молекулам жидкого кристалла, так и молекулам растворенного вещества), а затем его охлаждают. Эта операция схематически показана на рис. 13.5,Б. Спектр ЭПР на рис. 13.5,Г [4а] характеризует образец, ориентированный относительно магнитного поля, как изображено на рис. 13.5,Б, в то время как спектр на рис. 13.5,Д характеризует образец, повернутый на 90° вокруг оси z (т. е. ось у параллельна полю) относительно приложенного поля. При повороте интенсивность части спектра, соответствующей д2. увеличивается, но участок спектра, соответствующий glt остается без изменения. Можно легко ошибиться, предположив, что мы имеем аксиальную систему с дъ соответствующим оси z (т. е. оси glf, перпендикулярной плоскости), и д2 и Цз, соответствующими дъ где дх и ду одинаковы. Однако для молекулярной системы координат, определенной на рис. 13.5, A, gz должен быть отнесен к д3, дх—к д, и ду—к д2. Эти отнесения в дальнейшем были подтверждены результатами исследования спектров ЭПР монокристалла [46]. При изучении жидкокристаллических веществ могут возникнуть сложности, если не показано, что молекулы жидкого кристалла не координируются с исследуемым комплексом.
13.2. ИНТЕРПРЕТАЦИЯ ^-ФАКТОРОВ
Введение
В противоположность g-факторам органических свободных радикалов {/-факторы ионов переходных металлов могут заметно отличаться от значения {/-фактора свободного электрона, равного 2,0023. Такие отклонения дают много информации об электронной структуре комплексов. Различие обусловлено тем, что спин-орбитальное взаимодействие в комплексах многих ионов переходных металлов по величине значительно превышает соответствующее взаимодействие в органических свободных радикалах (см. ниже). Таким образом, для понимания явления ЭПР существенное значение приобретают спин-орбитальные эффекты.
{/-Фактор неспаренного электрона в газообразном атоме или ионе, для которого применима схема взаимодействия Рассела—Саундерса, выражается с помощью приведенного ранее уравнения
J(J+1) + S(S+1)-L(L+1)
д = 1 4----------------------. (13.2)
2J(J+1) V
В конденсированной фазе у комплексов ионов переходных металлов первого ряда значения {/-фактора не только не отвечают этому выражению, но часто отклоняются также от чисто спиновых значений. В конденсированной фазе орбитальное движение электрона сильно возмуще-
14-574
210
Глава 13
но, и орбитальное вырождение, если оно существовало до погружения в химическое окружение, частично снимается или «гасится». Если у электрона есть орбитальный угловой момент, то имеется тенденция к его сохранению за счет слабого взаимодействия со спином. Поэтому эффект гашения в окружении лигандов, т.е. в «кристаллическом поле»,
Газообразный 0h D<h ион
Рис. 13.6. Расщепление состояния 2D полями Ок и D4h (сжатие вдоль оси z в £>«)•
и эффект сохранения за счет спин-орбитального взаимодействия конкурируют. Если бы не спин-орбитальное взаимодействие, то мы должны были бы всегда наблюдать изотропный g-фактор, равный 2,0023. Указанные эффекты можно продемонстрировать, рассматривая влияние кристаллического поля на (/'-ион. как это показано на рис. 13.6 для Oh и Р4Л (сжатие оси г). Уравнение (13.2) должно описывать gi-фактор газообразного 2D-иона. Октаэдрическое кристаллическое поле расщепляет состояние 2D на состояния 2Т2а и 2Еа. Вырожденное состояние Т2д может далее расщепляться за счет искажения (например, ян-теллеровского) или под действием тетрагонального поля лигандов до уровней Е и В2. В то же время спин-орбитальное взаимодействие стремится сохранить какую-то небольшую величину орбитального углового момента, поэтому в тетрагональном комплексе орбитальный угловой момент полностью не гасится.
Обычно это называют смешиванием близлежащего возбужденного состояния за счет спин-орбитального взаимодействия. Если величина спин-орбитального взаимодействия мала по сравнению с тетрагональным искажением (т. е. в случае больших искажений), то смешивание можно трактовать с помощью теории возмущений. В октаэдрическом комплексе спин-орбитальное взаимодействие существует в основном состоянии 2Т2д; рассмотрение этой ситуации с помощью теории возмущений неадекватно и не обеспечивает точности, необходимой для понимания спектра ЭПР. (Напоминаем, что такой подход был использован в гл. 11, посвященной магнетизму.)
Спектры ЭПР комплексов ионов переходных металлов 211
Системы с 5=1/2 с орбитально не вырожденными основными состояниями
Полный гамильтониан для систем со спин-орбитальным взаимодействием в магнитном поле выражается как
Н = Н (зеемановский) + Н (SO) = PH (L+ geS) + KL-S. (13.3)
Один из эффектов спин-орбитального взаимодействия должен видоизменять простые одноэлектронные d-орбитальные волновые функции. Он описывается членом XL-S гамильтониана. Например, спиновая волновая функция основного состояния 2В2а d'-иона в тетрагональном комплексе изменяется в результате спин-орбитального взаимодействия л L-S. Исходя из теории возмущений первого порядка, волновую функцию для дублета Крамерса | + >, учитывающую спин-орбитальные эффекты, можно записать в виде
|±>-N|0>+ (!-№)-- <13-4)
MLMS
Терм |0> представляет собой основное состояние 2В1в без учета спин-ор-битальных эффектов (т. е. для d'-иона с тетрагональным сжатием это один электрон на d^-орбитали), в то время как суммирование дает вклад, обусловленный спин-орбитальным подмешиванием возбужденных состояний. В этом примере член АЕ в знаменателе указывает на то, что Состояние 2Е будет давать наибольший вклад из всех подмешиваемых состояний. Из уравнения (13.4) видно, что если к основному состоянию не подмешивается орбитальный угловой момент, то | ± > = = |0>. Расчет матричных элементов в уравнении (13.4) дает коэффициенты, необходимые для записи соответствующих волновых функций. Эти функции затем используются с зеемановским гамильтонианом в уравнении (13.3), т. е.
H' = PLH + fiU3S-H,
чтобы получить матрицу 2x2, включающую | + > и | — >. Отметим, что мы оперировали полным гамильтонианом в уравнении (13.3), используя обе его части по отдельности. Член 7.L-S видоизменяет волновую функцию, на которую мы подействуем зеемановским гамильтонианом. Задача определения собственных значений решается путем использования операторов сдвига. Получающиеся энергии выражаются как g$Hms, где g — эффективный ty-фактор в направлении компоненты поля (г = х, у, z). Таким путем, используя Ег, Sz и зеемановский гамильтониан, мы имеем
gz = 2,0023 + 2Х
/<0|LJn> <n|LJ0>\
\ Е(0) - Е(и) J
(13.5)
14*
212
Глава 13
где |0>—волновая функция основного состояния, а |п>—волновые функции возбужденных состояний. Поскольку основное и возбужденные состояния, соответствующие переходам в комплексе с 5=1/2 с небольшим спин-орбитальным взаимодействием, адекватно описываются истинными заселенностями орбиталей, обозначения состояний и орбита-лей можно использовать взаимозаменяемо. Таким образом, видно, что ^-фактор сильно зависит от подмешивания возбужденного состояния за счет спин-орбитального взаимодействия.
Матричные элементы <0|Lz|n/> отличаются от нуля только в том случае, когда величина mi10/ равна величине mt функции |п/>. В базисе действительных орбиталей
И
dxy
(|+2> + | —2»
мы будем иметь ненулевые матричные элементы для (dx2_yi |£.| d.,,.'). (Это следует из тройного произведения Гх2_л2 х х Гху.) Для dl-комплекса, для которого можно использовать одноэлектронные орбитали вместо состояний, расчет всех матричных элементов приводит к
gz = 2,0023 +
8^
Е(0) —Е(и)’
(13.6)
причем Х= +^/25 для менее чем наполовину заполненной оболочки. Символ обозначает одноэлектронное спин-орбитальное взаимодействие. Если оболочка заполнена более чем наполовину, то мы обращаемся к формализму положительных вакансий, образование которых сопровождается электронными переходами. Знак второго члена 8с/[Е(0) — Е(и)] меняется за счет изменения знаменателя на Е(и) — Е(0). Если оболочка заполнена менее чем наполовину, то спин-орбитальные эффекты снижают значение 0-фактора по сравнению с 2,0023, но если оболочка заполнена более чем наполовину, то спин-орбитальные эффекты приводят к увеличению 0-фактора по сравнению с де. Величины
Е(0) и Е(и) представляют собой энергии соответственно основного и возбужденных состояний.
Для тетрагонального комплекса
0Х = 2,0023 +
2Х^ <0|Ех|и><и|Ех|0>
Е(0) — Е(и)
(13.7)
где 0Х = ду. Эти матричные элементы рассчитывают путем использования операторов сдвига, поэтому они отличаются от нуля лишь в том случае, когда значения для основного и возбужденного состояний
Спектры ЭПР комплексов ионов переходных металлов 213
различаются на +1. Для тетрагонального «^-комплекса (рис. 13.6)
2с
= 2,0023 + %—. (13.8)
Таким образом, результаты расчета матричных элементов можно представить в обобщенном виде, записав выражение для ^-факторов системы с S = 1/2:
g = 2,0023 +
Е(0)-Е(и)’
(13-9)
Значения п получают из так называемого «магического пятиугольника», показанного на рис. 13.7 который суммирует результаты расчета матричных элементов <0|Е|и>. Обратимся вновь к задаче определения
т(
Рис. 13.7. «Магический пятиугольник» для расчета значений п уравнения (13.9).
^-фактора в тетрагональном <Е-поле, используя этот пятиугольник. Прежде всего мы видим, что для электрона на ху-орбитали (т. е. в основном ху-состоянии) только электронная циркуляция на орбитали х2 — у2 может дать орбитальный угловой момент вдоль оси z. Тогда gz получает вклады только орбиталей ху и х2 — у2, которые, как видно из рис. 13.7, имеют величину п = 8. В этом случае уравнение (13.9) приобретает вид
8с
gz = 2,0023 + -----1-----. (13.10)
Еху-Ех2_у1
Поскольку дх является ненулевым матричным элементом только в том случае, когда |0/ и отличаются на величину Am(= 1, получаем
2с
дх = 2,0023 + \ (13.11)
Еху ~ Exz
И
gy = 2,0023 + \. (13.12)
7 /* _ р
^ху ^yz
214
Глава 13
Из этих формул фактически следует, что орбитали позволяют электронам циркулировать вокруг соответствующих осей, т. е. орбитали х2 — у2 и ху—вокруг оси z, орбитали ху и xz—вокруг оси х и орбитали ху и yz—вокруг оси у. «Магический пятиугольник» построить легко. Три ряда представляют орбитали, обладающие различными значениями mt: верхний ряд соответствует mt = Q, второй ряд—т,= ±1 и третий ряд—т, = +2. Следует подчеркнуть, что весь этот подход основан на идее возмущения и справедлив, если только величина пс/[Е(0) — £(п)] мала по сравнению с диагональными зеемановскими элементами. Если больше подходит теория возмущений второго порядка, то добавляется член, пропорциональный ^2/А£2.
Покажем, как можно использовать значения 0-фактора. На рис. 13.8 показаны расщепления d-уровней тетрагональных комплексов меди(П) под действием кристаллического поля с сжатием (Л) и удлинением (Б) расстояния металл — лиганд по оси четвертого порядка (z). В случае А при одном неспаренном электроне на орбитали dzi и m, = Q нет другой орбитали с компонентой т1 = 0, поэтому gz = 2,0023. Величина дх =
= ду выражается как
= 2,0023 + - 6-^ . (13.13)
~ ^z2
Отметим, что мы переставили порядок энергий, поскольку оболочка заполнена более чем наполовину. В то же время для тетрагонального растяжения (рис. 13.8, Б) при одном неспаренном электроне на >2-°рбитали
gz = 2,0023 + ---------. (13.14)
Величина дх = ду определяется выражением
дх = 2,0023 +~~---------• (13.15)
-£^2,2
Таким образом, 0-факгор можно использовать для того, чтобы различить эти две структуры. Комплексы с тетрагональным сжатием встречаются очень редко, зато комплексы с тетрагональным растяжением распространены. Порфириновые комплексы меди(П) характеризуются ^-факторами gz = 2,70 и дх = 2,04 [5].
Отметим интересную особенность 0-факторов некоторых шестикоординационных комплексов меди(П). Как упоминалось в гл. 9, если снимать при 90 К спектр монокристалла [Cu(H2O)6] SiF6, разведенного соответствующим диамагнитным комплексом цинка, то наблюдается одна полоса с частично разрешенной сверхтонкой структурой и почти изотропным 0-фактором [6J. Следует ожидать ян-теллеровского искажения, но реализуются три искажения одной и той же энергии, которые разрешают орбитальное вырождение. Они представляют собой три
Спектры ЭПР комплексов ионов переходных металлов
215
взаимно перпендикулярных тетрагональных искажения (растяжение или сжатие вдоль трех осей, связывающих транс-лиганды). В результате ожидаются три различных спектра ЭПР, по одному для каждого варианта. Поскольку обнаружен только один переход, было высказано
Рис. 13.8. Расщепления «/-уровней тетрагонально сжатого (А) и тетрагонально растянутого (В) комплексов.
предположение, что кристаллическое поле резонирует между тремя искажениями (так называемое динамическое искажение Яна—Теллера). С понижением температуры спектр становится анизотропным, он состоит из трех совокупностей линий, соответствующих трем различным окружениям иона меди, искаженным тремя различными тетрагональными искажениями [8]. Были обнаружены аналогичные переходы в других смешанных солях меди: (Си, Mg)3 La2(NO3)12-24D2O между 33 и 45 К; (Zn, Cu)(BrO3)2-6H2O (неполный переход) ниже 7 К. Сообщалось [9] о следующих параметрах для CuSiF6-6H2O:
90 К
31 = 2,221 ± 0,005
3±= 2,230 ± 0,005
А = 0,0021 ± 0,0005 см “ 1
В = 0,0028 ± 0,0005 см “ 1
20 К
gz = 2,46 + 0,01
gx = 2,10 + 0,01
gy = 2,10 + 0,01
Az = 0,0110 + 0,0003 см “J
, > < 0,0030 CM"1 j
Аналогичное поведение (т. е. резонирующее кристаллическое поле при умеренных температурах) было зафиксировано, например [10], в спектрах некоторых трис-комплексов меди(П) с 2,2'-дипиридином и 1,10-фенантролином [И], а также с трис-октаметилфосфорамидом [12]. Последний лиганд [(CH3)2N]2 Р(О)ОР(О) [N(CH3)2]2—бидентатное хелатирующее соединение, в котором координируются фосфорильные атомы кислорода. Указанные комплексы можно исследовать, не разводя их в диамагнитном веществе. Они подробно исследовались [12] в виде мокристалла с помощью спектроскопии ЭПР и рентгеноструктурного анализа. Из спектра при 90 К следует, что в комплексе имеется по крайней мере три магнитных центра, каждый из которых описывается р-тензо-
216
Глава 13
ром. Анализ спектра показывает, что эти центры относятся соответственно к искажениям вдоль осей х, у и z, локализуясь фактически в различных пределах динамических ян-теллеровских колебаний, происходящих при комнатной температуре. Обсуждалась [12] также интересная проблема определения хиральности молекулы при анализе спектра ЭПР монокристалла.
При исследовании методом ЭПР монокристаллов комплекса ионов переходных металлов обычно обнаруживают [13—15] комплексы, в которых в очевидной системе координат кристаллического поля д- и А-тензоры не диагональны. Ось, которая перпендикулярна зеркальной плоскости или совпадает с осью вращения, должна быть одной из трех главных осей молекулы. g-Тензор молекулы и А-тензор для любого атома, лежащего на этой оси, должны иметь главные значения вдоль этой координаты. Если в молекуле есть только одна ось, которая удовлетворяет приведенным выше требованиям, две другие оси, используемые в качестве базиса при анализе в кристаллическом поле, не обязательно будут главными осями соответствующих д- и А-тензоров, т. е. выбор этих осей не обязательно приведет к диагональному тензору. Например, бис-(диселенокарбамат) меди(П) имеет симметрию C2h [13, 14]. Ось вращения второго порядка является одной из осей, приводящих соответствующие компоненты д- и А-тензоров к диагональному виду, но две другие компоненты не диагональны в системе координат, соответствующей осям кристаллического поля. Если молекула обладает симметрией D2h, то три оси вращения второго порядка этой точечной группы должны быть главными осями как для g-тензора, так и для А-тензора. Таким образом, результаты исследования методом ЭПР могут дать информацию относительно симметрии молекулы. Для несимметричной молекулы совсем не обязательно, чтобы молекулярные оси совпадали с осями, которые приводят g-тензор или А-тензор* к диагональному виду. На самом деле система координат, приводящая А-тензор к диагональному виду, может и не диагонализировать g-тензор. Например, в витамине Bj 2 угол между системой главных осей х, у, которая приводит А-тензор к диагональному виду, и системой осей, которая приводит g-тензор к диагональному виду, составляет 50 [15].
Системы с сильным спин-орбитальным взаимодействием
Если спин-орбитальное взаимодействие велико, то для получения подходящих волновых функций нельзя пользоваться теорией возмущений, т. е. уравнение (13.4) неприменимо. Октаэдрический -комплекс с основным состоянием 2Т1д может служить примером комплекса, в котором спин-орбитальное взаимодействие велико. Если оператор спин-орбитального взаимодействия qL-S действует на шестикратно вырожденный ба
* Для низкосимметричных молекул д- и Л-тензоры могут быть действительно асимметричными (т. е. аху # аух). д -Тензор будет всегда симметричным.
Спектры ЭПР комплексов ионов переходных металлов 217
зис действительных орбиталей
Фг =(1/|/2)(|2, 1/2> — | — 2, 1/2», ф2 = (l/j/2)(|2, -1/2>-[-2, -1/2», Фз =И, 1/2» ф4 = |1, -1/2» ф5 = | - 1, 1/2» ф6 = |-1, -1/2»
(волновые функции выражены в виде \ML, то результатом будет новый базис из орбиталей, подходящих для ^-системы с большой величиной спин-орбитального взаимодействия. Эти орбитали получают на основе энергий из детерминанта 6x6 с матричными элементами
| • S | v|/j» Подстановка энергий снова в уравнения секулярного типа
дает собственные функции, приведенные ниже:
< Р1 = (1//3) (|/2ф! + ф6),
< Р2 = (1/0) ( - /Ж + Фз),
< Рз = Ф4,
<Р4=Ф5,
< р5 = (1/]/3) (ф2 + /2ф3), ф6 = (l/j/З) (Ф1 - |/2ф6).
Соответствующие величины энергии таковы: = — ^/2, Е2 = — ^/2, Е3 = — ^/2, Е4 = — ^/2, Е5 = и Е6 = Из этого анализа видно, что спин-орбитальное взаимодействие снимает шестикратное вырождение состояния Т, приводя к совокупности из двух уровней и совокупности из четырех уровней более низкой энергии, соответствующих Г7 и Г8 в двойной группе О' *. Далее нам необходимо определить влияние магнитного поля. Поскольку система с симметрией Oh магнитно изотропна (х, у и z), необходимо рассчитать только влияние Hz. Оператором Гамильтона Н (параллельным z) является Р(Ег + geSz)Hz. Результирующие энергии получены в гл. 11 и представлены на рис. 13.9. Расщепление в низкоэнергетической совокупности невелико при втором порядке по Н (т.е. Н2). Решение для g(&E = g$H) приводит к д$Н = 4(32Н2/3£; или д = 4рН/Зс « 0 для наинизшего уровня **. При заметном разделении Г8 и Г7 (например, = 154 см “ 1 для Ti3 +) сигнал ЭПР не будет регистрироваться, несмотря на то что состояние Г7 заселено. Решая это уравнение
* Состояния Г7 и Г8 соответствуют 2=1/2 и 2 = 3/2 для свободного иона. Эти обозначения получены путем разложения произведения 7/ (орбитальная часть) и Е’2 (Г6 для спина равно 1/2) в двойной группе О'.
** В некоторых системах зарегистрированы переходы с очень небольшими значениями д-фактора.
218
Глава 13
для д$Н для приведенных выше уровней, получаем значение (/-фактора, равное 2,0. Это состояние будет заселено при комнатной температуре, но большая величина спин-орбитального взаимодействия приводит
Поле 0h Спин- н
орбитальное взаимодействие
E=(-/3HZ +
Е = -i/2
Е = -(i/2) - (|/8гН//^
Рис. 13.9. Влияние спин-орбитального взаимодействия и приложенного магнитного поля на состояние Т.
к небольшим те, и спектры не наблюдаются. Для увеличения значения те необходима температура жидкого гелия, но тогда только состояния Г8 будут заселены. Таким образом, невозможность регистрировать спектр ЭПР этой системы прямо следует из теории кристаллического поля.
13.3. СВЕРХТОНКОЕ РАСЩЕПЛЕНИЕ И РАСЩЕПЛЕНИЕ В НУЛЕВОМ ПОЛЕ
Проявление в спектрах эффектов сверхтонкого расщепления в нулевом поле
Сверхтонкое расщепление на металле и расщепление в нулевом поле дают много информации о комплексах переходных металлов. Рис. 9.14 демонстрирует СТВ с кобальтом в случае Co3(CO)9Se. Прежде чем продолжать знакомство с этой темой, читателю полезно заново просмотреть раздел, посвященный анизотропии СТВ в гл. 9, и раздел, посвященный ЭПР триплетных состояний. Спин-гамильтониан для одного ядра со спином I и одного эффективного электронного спина S может быть записан с учетом сверхтонкого расщепления на металле и расщепления в нулевом поле
H = PH-g-S + S-D-S + /iS-A-7-fifNpNH-f. (13.16)
В предыдущем разделе мы обсуждали член pH • g • S и осложнения, обусловленные орбитальными эффектами. Следующий за ним член учитывает эффекты расщепления в нулевом поле, описанные ранее с по
Спектры ЭПР комплексов ионов переходных металлов
219
мощью дипольного тензора D, след которого равен нулю. Последние два члена появляются в том случае, когда I # 0.
В гл. 9 были рассмотрены эффекты расщепления в нулевом поле, обусловленные дипольным взаимодействием двух или более электронных спиновых моментов. В комплексах ионов переходных металлов член S-D-S используют для описания любого эффекта, который снимает спиновое вырождение, включая дипольные взаимодействия и спин-орбитальное расщепление. Низкосимметричное кристаллическое поле часто приводит к большим эффектам нулевого поля.
В аксиально-симметричном поле (т. е. тетрагональном или тригональном) спин-гамильтониан ЭПР, который можно использовать для получения соответствия с наблюдаемыми спектрами эффективных спиновых систем, более низких, чем квартетные, принимает вид
= П Sz2-|s(S+ 1) + g^HzSz + g^(HxSx + HySy) +
+ + л± (Sxix + Syiy) + q i2z - у (I + 1) - ypN h0 • i.
Первый член описывает расщепление в нулевом поле, следующие два члена—влияние магнитного поля на спиновую мультиплетность, остающуюся после расщепления в нулевом поле; члены с Лц и Л± являются мерой сверхтонкого расщепления параллельно и перпендикулярно главной оси, a Q'—мерой небольших изменений в спектре, вызванных ядерным квадрупольным взаимодействием. Все эти эффекты обсуждались в гл. 9. Последний член учитывает тот факт, что ядерный магнитный момент pN может непосредственно взаимодействовать с внешним полем pN Но = yPN Но • I, где у — гиромагнитное отношение ядра, a Pv—ядерный магнетон Бора. Он описывает ядерный эффект Зеемана, который вызывает переходы в ЯМР. Зеемановское ядерное взаимодействие может влиять на спектр парамагнитного резонанса только в том случае, когда неспаренные электроны взаимодействуют с ядром в ядерном сверхтонком или квадрупольном взаимодействиях. Если даже такое взаимодействие и реализуется, то его величина пренебрежимо мала по сравнению с величинами других эффектов.
В случае искажения более низкой симметрии имеются три различные компоненты дх, ду и gz и три различные константы сверхтонкого взаимодействия — Ах, АУли Поэтому необходимо включить два дополнительных члена: E(S2 — S2) — дополнительное расщепление в нулевом поле и Q"(I% — Iy)—дополнительное квадрупольное взаимодействие. Соответственно символы Р и Р' часто используют вместо символов Q и Q".
Значение спин-гамильтониана состоит в том, что он дает стандартный феноменологический способ описания спектра ЭПР посредством небольшого числа констант. Если величины этих констант определены экспериментально, то часто можно провести расчеты, связываю
220
Глава 13
щие эти параметры с электронными конфигурациями и энергетическими состояниями ионов.
Расщепление состояния 6S октаэдрического комплекса марганца(П) показано на рис. 13.10,Л- Это интересный случай расщепления нулевым полем основного состояния 6AIg комплекса Мп2 + с симметрией Oh. Спин-орбитальное взаимодействие подмешивает к основному состоянию возбужденные состояния 4Т2, которые расщепляются кристаллическим полем, и это смешивание приводит к небольшому расщеплению в нулевом поле уровней комплекса Мп2 +. Дипольное взаимодействие электронных спинов дает меньший эффект по сравнению с подмешиванием более высоко лежащих состояний комплекса. В этом примере очень интересны орбитальные эффекты, поскольку основным состоянием является 68, и поэтому возбужденное состояние 4Т2 может подмешиваться только за счет спин-орбитальных эффектов второго порядка. Таким образом, расщепление в нулевом поле относительно невелико, например порядка 0,5 см1 в некоторых порфириновых комплексах
Уровни Приложенное Ядерное Переходы
нулевого поле расщепление тонкой
поля А структуры
500 Э
Рис. 13.10. А — расщепление уровней в спектре октаэдрического комплекса Мп(П); Б — спектр монокристалла комплекса Мп2+, внесенного в MgV2Oe, демонстрирующий пять разрешенных переходов (тонкая структура), каждый из которых расщеплен на ядре марганца (I = 5/2) (сверхтонкая структура). При 300 К дх = 2,0042 + 0,0005, ду = 2,0092 ± 0,001 и д2 = 2,0005 ± 0,0005; Ах = Ау = Аг = - 78 ± 5 Э, Dx = 218 ± 5 Э, Dy = = — 87+5 Э и Dz = - 306 ± 20 Э. [Modified from Ng Н. N., Calvo C., Can J. Chem., 50, 3619 (1972). (Reproduced by permission of the National Research Council of Canada.)]
Спектры ЭПР комплексов ионов переходных металлов
221
О Увеличение
напряженности поля
напряженности поля
А
Рис. 13.11. Диаграмма энергетических уровней и переходы для молекулы или -иона с S = 1.
В отсутствие (Л) и в присутствии (Б) расщепления в нулевом поле. Система Б направлена вдоль оси z, поскольку эффект расщепления в нулевом поле очень анизотропен.
марганца(П) [16а]. Как показано на рис. 13.10, расщепление в нулевом поле приводит к трем дважды вырожденным спиновым состояниям: Ms = ±5/2, ±3/2, ±1/2 (крамерсово вырождение). Каждое из них расщепляется приложенным полем на два синглета, приводя в общей сложности к шести уровням. В результате этого расщепления можно ожидать пять переходов: ( — 5/2-> — 3/2, — 3/2—> — 1/2, — 1/2-> 1/2, 1/2—> —> 3/2, 3/2-> 5/2). Спектр расщепляется далее за счет ядерного сверхтонкого взаимодействия с ядром марганца (/ = 5/2). Это должно привести к появлению тридцати спектральных линий.
В противоположность термину сверхтонкое расщепление термин тонкое расщепление используют в том случае, когда полоса поглощения расщепляется из-за снятия вырождения в результате расщепления в нулевом поле. Компоненты тонкого расщепления имеют различные интенсивности: интенсивность центральных линий наибольшая, в то время как для боковых линий она наименьшая. В простых случаях разделение линий изменяется как функция 3cos20— 1, где 0—угол между молекулярной осью z и направлением магнитного поля.
На рис. 13.11 показано влияние расщепления в нулевом поле на систему с S = 1 при фиксированной молекулярной ориентации. (Напомним, что крамерсово вырождение отсутствует.) В отсутствие эффектов нулевого поля (рис. 13.11, А) два перехода с |Ams| = 1 вырождены, и наблюдается только одна полоса.
Для расщепления, показанного на рис. 13.11, Б, в спектре должны наблюдаться две линии. Конкретным примером систем такого типа служит основное состояние 3Л2й комплекса никеля(П) в поле Oh. Спин-орбитальное взаимодействие подмешивает возбужденные состояния, которые расщепляют конфигурацию 3Л2э. Напомним, что расщепление в нулевом поле очень анизотропно и обеспечивает механизм релаксации для электронного спинового состояния. Поэтому спектр ЭПР комплексов никеля(П) с симметрией Oh трудно регистрировать, и при исследовании, как правило, необходимо их замораживать до температуры жид
222
Глава 13
кого гелия или азота. При комнатной температуре можно наблюдать спектр ЯМР. В некоторых системах в спектре ЭПР можно наблюдать узкий двухквантовый переход с |Ams| = 2 [166].
Как упоминалось в гл. 9, расщепление в нулевом поле может быть настолько велико, что переходы с Ams = ± 1 не регистрируются. Например, переходы с Ams = + 1 не наблюдаются в комплексах V3 + (d2), но можно обнаружить, что под действием ядерного спина 51V (1 = 7/2) слабый переход с Ams = 2( — 1 -» +1) расщепляется на восемь линий. Механизм, за счет которого становится разрешенным переход Ams = 2, обсуждается для триплетных состояний органических соединений в гл. 9.
Далее мы рассмотрим эффективный спин S'. Мы уже пользовались этой концепцией, но теперь дадим ему формальное определение, чтобы описать, как некоторые из уже рассмотренных эффектов учитываются спин-гамильтонианом. Если кубическое кристаллическое поле оставляет основное состояние (например, состояние Т) орбитально вырожденным, то поля более низкой симметрии и спин-орбитальное взаимодействие будут снимать как орбитальное, так и спиновое вырождение. В случае нечетного числа неспаренных электронов крамерсово вырождение оставляет низшее спиновое состояние дважды вырожденным. Если расщепление велико, то этот дублет хорошо отделяется от дублетов, лежащих выше, и переходы наблюдаются только в низшем дублете, который ведет себя как более простая система с S = 1/2. Тогда мы говорим, что система имеет эффективный спин S', равный только 1/2 (S' = 1/2). Примером может служить комплекс Со2 +. В кубическом поле основным состоянием является 4F; под действием полей более низкой симметрии и спин-орбитального взаимодействия это состояние расщепляется на шесть дублетов. Если низший дублет отделен от других значительно больше, чем на кТ, то эффективный спин имеет величину 1/2 (S'= 1/2) вместо 3/2. Если эффективный спин S' отличается от спина S, то спин-гамильтониан может быть записан через S', а не через S.
Совершенно очевидно, что все описанные выше эффекты должны оказывать значительное влияние на характер спектра. Таким образом, качественная интерпретация при визуальном исследовании спектров ЭПР ионов переходных металлов чрезвычайно сложна. Необходимый опыт приобретается в ходе работы со многими спектрами и при нахождении аналогий между известными и неизвестными системами. Практическая сторона вопроса изложена в последнем разделе на примере характерных спектров различных г/”-систем. Кроме того, читатель может приобрести известные навыки, решая упражнения, которые приведены в конце главы.
Вклады в А
Сверхтонкое взаимодействие объединяет в себе контактное взаимодействие Ферми, дипольное взаимодействие ядерного спина с электронным и взаимодействие ядерного спина с орбитальным моментом
Спектры ЭПР комплексов ионов переходных металлов
223
электрона. Эти эффекты рассматривались в гл. 9. Для трактовки дипольного вклада читатель должен обратиться к уравнению (9.25) и к последующему его обсуждению. След тензора в уравнении (9.25) равен нулю, поэтому информация о дипольном взаимодействии может быть получена только из упорядоченных или частично упорядоченных систем.
Таблица 13.1
Вклады электронов, находящихся на d-орбнталях, в дипольное сверхтонкое взаимодействие
Орбиталь TJPd) T (PJ XX' Cl' Tyy(Pd)
4/7 -2/7 -2/7
d^f- -4/7 2/7 2/7
dyz 2/7 -4/7 2/7
2/7 2/7 -4/7
d*y -4/7 2/7 2/7
Как упоминалось в уравнениях (9.34) и (9.35), где компоненты тензора А с нулевым следом обозначены через Т. вклад в дипольное сверхтонкое взаимодействие для электрона, находящегося на р2-орбитали, выражается как
Для электрона, находящегося на рх-орбитали,
(13.17)
Т
ZZ
Рр
т
УУ
Аналогичные выражения могут быть получены для электрона, находящегося на одной из d-орбиталей. Для определения вклада дипольного взаимодействия параметры в табл. 13.1 следует умножить на
(13-18)
Знаки и величины этих параметров запомнить легко. Орбиталь «накладывается» на график (3cos20— 1 (-зависимости силового поля, создаваемого ядерным моментом, как это показано на рис. 13.12 для d^-орбитали. Для получения небольшого положительного значения Tzz на этом рисунке ось z молекулы направлена вдоль оси z поля.
224
Глава 13
Для получения небольшого положительного значения Тхх повернем далее орбиталь на 90° против часовой стрелки без вращения конуса, так чтобы ось х молекулы стала параллельной направлению поля (которое все еще направлено вдоль оси z, как это показано на рис. 13.12). Затем исходя из рис. 13.12, повернем молекулу таким образом, чтобы ось
Рис. 13.12. Ориентация «/„-орбитали относительно конуса 3cos20 — 1.
z молекулы стала перпендикулярной плоскости страницы, а ось у—параллельной направлению поля (т.е. оси z на рис. 13.12). Теперь лопасти «/„-орбитали находятся в отрицательной части конуса, и следует ожидать большого отрицательного значения Туу. Эти повороты соответствуют трем взаимно перпендикулярным ориентациям молекулы относительно направления поля. Зная знаки и величины компонент сверхтонкого тензора, можно получить информацию об атомных орбиталях комплекса, дающих главный вклад в молекулярные орбитали, на которых находится неспаренный электрон.
Контактное взаимодействие Ферми подробно рассматривалось в гл. 9 и 12. Плотность неспаренного спина «ощущается» на исследуемом ядре за счет прямого подмешивания 5-орбиталей к МО, на которой находится неспаренный электрон, и за счет спин-поляризации заполненных внутренних з-орбиталей под действием плотности неспаренных электронов на d-орбиталях. Если 45-орбиталь металла свободна, то она может подмешиваться к d-разрыхляющей орбитали, представляющей собой главным образом орбиталь металла; если на этой d-орбитали находится неспаренный электрон, то он частично занимает Дз-орбиталь металла.
Спин-поляризация может приводить к двум результатам. Если внутренние 15- или 25-орбитали спин-поляризованы а-спином, находящимся на 3d-op6HTaHH, на ядре появится избыток р-спина. Если заполненные Зз-орбитали спин-поляризованы электроном с а-спином, находящимся на 3d-op6m^H, на ядре появится а-спин. Эффект прямой делокализации на 4з-орбиталь может быть продемонстрирован путем сравнения
Спектры ЭПР комплексов ионов переходных металлов 225 констант контактного СТВ Ферми (ЛР Г), определенных для различных комплексов кобальта(П). Было обнаружено [17а], что AF с при координационном числе 6 лежит в интервале от —30 до —45 Э, при координационном числе 5 — в интервале от — 5 до — 25 Э и для квадратноплоскостной структуры с координационным числом 4 составляет 0 Э. Можно записать уравнение, представляющее AF с в виде суммы вкладов:
Лгс = х(А45) + (1-х)(Л3Д (13.19)
где /14,— константа прямого СТВ одного неспаренного электрона, находящегося на 45-орбитали (1320 Э-г/Р); A3d—константа СТВ, обусловленного спин-поляризацией заполненных «5»-орбиталей под действием неспаренного электрона, находящегося на Зй-орбитали (— 90 Э-gp). Применив это уравнение к полученным ранее результатам, можно получить х = 3 — 4%, х = 4,5—6% и х = 6,5% для подмешивания 45-орбитали в комплексах кобальта соответственно при шести-, пяти- и четырехкратной координации с ионом кобальта(П).
Вклад взаимодействия ядерного спина с электронной орбиталью в константу взаимодействия связан с псевдоконтактным вкладом, рассмотренным в гл. 12. Гамильтониан имеет вид
H = ^[-(L’S)(L.I)-(L.I)(L.S)]. (13.20)
Здесь ядерный момент взаимодействует не только со спиновым моментом (что рассмотрено ранее), но и с орбитальным моментом. Ядер-ное взаимодействие со спиновым моментом характеризуется тензором с нулевым следом, но след тензора взаимодействия с орбитальным моментом отличается от нуля. Поэтому для спектров ЯМР в растворе наблюдается псевдоконтактный вклад.
13.4. ИНТЕРПРЕТАЦИЯ д- И 4-ТЕНЗОРОВ ДЛЯ СИСТЕМ С S' = 1/2 В РАМКАХ ТЕОРИИ ПОЛЯ ЛИГАНДОВ
В этом разделе при анализе спектры ЭПР интерпретируются с использованием в качестве базиса d-орбиталей комплекса. Ковалентность связывания учитывается путем снижения параметра спин-орбитального взаимодействия § и значения <г “ % свободного иона. Базисные действительные орбитали смешиваются за счет спин-орбитального взаимодействия при использовании теории возмущений^ первого порядка и гамильтониана спин-орбитального взаимодействия I • s. Приводятся результаты для нескольких d-электронных конфигураций и в дальнейшем обсуждаются на отдельных примерах. Выражение для расчета компонент ^-тензора уже обсуждалось.
15-574
226
Глава 13
d9 -Конфигурации
Используем формализм дырок, чтобы параметр £, был отрицателен, и рассмотрим случай основного состояния х2 — у2. Крамерсов дублет занимает низшее положение; обозначим его символом ф*, фф, где знаки + и — относятся к величинам ms. На этом примере продемонстрируем применение уравнения (13.4) и определим теперь Г через подходящие волновые функции ф+, фф. Волновые функции, которые смешиваются за счет спин-орбитального взаимодействия, имеют вид
Фа = Na\d^_y2y + iak\dxyy — ~a2\dyzy - |a3|dxz>,
Фа Na | dx2 — у2У I dxy У + 2^21 3“ 2^з I Xz У •
(13.21)
(13.22)
Здесь Na—нормировочный коэффициент; величины а—это а{ = fyEia, где Е1а, например, выражено через Е (| dx? _ » — Е (| dxy У), а Е2а—через E(|dx2_>r2» — E(|dxz>). Величины а малы, поэтому Na близко к единице. Полагая Na равным 1, получаем (как обсуждалось выше)
* 2с
вхх = 2<фа+ £ lxk + gesxk\^a у=де + ---= де + 2а3, (13.23)
к &х2-у2 bxz
а 2£
дуу = 21<фа+£ 1ук + де ^1Фа У = де + = де + 2«2, (13.24)
к Ьх2-У2 Eyz
g?z = 2<фа+£L + geszk\y\i;y = де + —-— F- = де + 8аР (13.25)
к Ьх2-У2 Ьху
(Отметим, что те же результаты получаются при использовании «магического» пятиугольника.)
Для того чтобы рассчитать А, рассмотрим снова все матричные элементы фа+ и ф“ с х-, у- и z-компонентами оператора
£ (4 ~ ^"sk + -ak) = А,
к '
где dk = 4.sk — (lk • sk)lk — lk (lk • 5k). Эта форма оператора обсуждалась в работе [17]. Читатель, которого интересует расчет этих матричных элементов, должен знать проблему фаз, связанную с выбором волновых функций. Полное обсуждение имеется в работах [17—19]. Здесь просто приводим результаты.
Спектры ЭПР комплексов ионов переходных металлов
227
Ахх = 2Р<К | Ах 11|/„ > =
= Р I 2 3
7 7Ex2_y2-Eyz
2 3
= Pj 2а 3 — .X" + — — —а2 ,
Ауу = 2iP < фa IАIК > =
= Р I 2 3
“l_Ex2_y2-Eyz 7 7Ex2_y2-Exz
= Pd 2а2 — Jf + - — ^а3 ,
Azz = 2Р<i|/+ | Az|К > =
__________________ 4 3 (_____________________ _________£_______ Exi _ y2 — Exy 7 7\ Exi _ y2 — Eyz Ex2 _ y2 — Exz
4 3
8a3 — JC — — + — (a2 + a3)
(13.26)
(13.27)
(13.28)
Суммирование k производится по всем электронным дыркам (в этой системе одна), а Pd = 0e0Jvppjv<r-3>. Символом JfPd обозначается вклад контактного взаимодействия Ферми; члены ±(2/7)Pd и ±(4/7)Pd описывают дипольный вклад, а другие члены — взаимодействие ядерного спина с орбитальным угловым моментом электрона. В случае раствора должен получаться изотропный /1-тензор, в котором
^iso о (^хх 4" ^уу 4” Azz
(13.29)
Член в скобках — аналог псевдоконтактного вклада в ЯМР.
Низкоспиновые комплексы кобальта(П) имеют две дырки в dx2_y2 и одну в <7.2-орбиталях. Используя «магический» пятиугольник, можно получить gzz, gxx и gyy в следующем виде:
gzz = 2,0023,
gxx = 2,0023 +------= 2,0023 + 6Ь,,
Ухх е.2-е„ 1
(13.30)
(13.31)
228
Глава 13
где bt = £/(£> - Eyz), И
6E
gyy = 2,0023 +--------= 2,0023 + 6b2, (13.32)
Ez2 ~ ^xy
где b2 = y(Ez2— Exy). Сверхтонкие компоненты выражаются как
^хх — ?d (у 3 \ — - — Ж + 6bj + -b2 ) (13.33)
Луу d /2 4 \ — - — + 6b2 + 7 bi , (13.34)
A-zz + у - JC - + b2) (13.35)
При наличии соответствующих выражений для (/-факторов и величин § для свободного иона можно рассчитать ^-факторы для нескольких комплексов и сравнить их с экспериментальными значениями. Результаты расчета совпадают с предсказаниями теории кристаллического поля. Они суммированы [20] в табл. 13.2.
Таблица 13.2
Сопоставление экспериментальных н теоретических значений д-факторов [20]
Комплекс 0эксп 0теор X (комплекс) X (ион)
VO[(CH3CO)2CH]2 VO2+ X (ион) = 170 cm'1 1,948 (0=0j) 1,921 0,67
V(H2O)i + V2 + X (ион) = 56 см'1 1,965 (g=0L=0ii) 1,964 0,99
Cr(H2O)3 + Cr3 + X (ион) = 91 см'1 1,977 (0 = 01= 0ц) 1,961 0,61
Cr(NH3)| + 1,986 1,969 0,49
Cr[(CH3CO)2CH]3 1,981 1,961 0,51
Сг(еп)Г 1,987 1,969 0,45
Cr(CN)63' 1,992 1,975 0,37
Ni(H2O)i + Ni2+ X (ион) = —324 см“1 2,25 (0 = 01 =0ц) 2,30 0,83
Ni(NH3)2 + 2,16 2,24 0,66
Cu(H2O)i + Си2 + X (ион) = — 828 см “1 2,46 (0 = 0ц) 2,593 0,77
Cu(NH3)i + 2,223 2,468 0,47
Cu [(CH3CO)2CH]2 2,266 2,443 0,60
Cu[NHCHC6H5O]2 2,200 2,408 0,49
Cu[S2CN(C2H3)2]2 2,098 2,347 0,28
Спектры ЭПР комплексов ионов переходных металлов
229
Можно отметить, что экспериментальные результаты неизменно ближе к величине 2,0023, чем величины, предсказываемые теорией кристаллического поля. Расхождение может быть устранено путем придания эмпирического эффективного значения параметрам § или X, для того чтобы согласовать рассчитанную величину g с экспериментальной. Тогда степень отклонения результатов простой модели кристаллического поля определяется отношением X (комплекс)/! (газообразный ион). Увеличение ковалентности связывания в комплексе должно вызывать уменьшение этого отношения. В теории поля лигандов берут £, (или л.) и Р из величин для свободного иона. Пониженные величины часто интерпретируют в терминах ковалентных эффектов (см. ниже).
Использование приведенных выше уравнений и информации, которую можно с их помощью извлечь, можно продемонстрировать на примере спектра ЭПР бкс-(малеонитрилдитионато)меди(П).
Этот комплекс был разведен в диамагнитном комплексе никеля(П), после чего методом ЭПР исследовали монокристалл полученного соединения [17]. Системы координат, в которых А- и 0-тензоры диагональны, совпадают, что приводит к величинам дхх = 2,026 + 0,001, дуу = 2,023 ± + 0,001 и 0.. = 2,086 + 0,001. Главные значения А-гензора равны соответственно (39 ± 1) -10-4, (39 + 1)-10-4 и (162 + 2)-10-4 см-1. Величина Aiso, полученная из спектра растворов, составляет 76 • 10“4 см-1. Из ^-факторов и уравнений (13.23)—(13.25) для основного состояния dx2_y2 можно определить значения аг « а2 ~ а3 « 0,01. Подставляя эти значения в уравнения (13.26)—(13.28) и решая их относительно А (основное состояние dx2_y2), получаем
= Pd (-0,48 - X],
А± = Pd ( + 0,30-
(13.36)
(13.37)
Так как Aiso составляет 76-10 4 см-1, то Ац и Aj_ должны иметь одинаковый знак, т. е. (39 + 39 + 162): 3 = 76. Поскольку |л J > |А±|, величина ,Ж' должна быть положительной, что обычно и наблюдают на практике. Для получения большего абсолютного значения АЙ, чем А±, нужно, чтобы знак Aj был отрицательным [это следует из уравнений (13.36) и (13.37)]. Теперь, зная знаки и величины Ав и А±, из уравнений (13.36) и (13.37) можно определить Pd и .#): Pd 1,6 • 10“2 см 1 (по сравнению с 3,5 • 10“2 см-1 для свободного иона) и Ж' к, 0,55. Контактное СТВ Ферми характеризуется величиной А =(0,55) (1,6- 10-2 см-*). Такой анализ нужен для подтверждения часто базирующегося на интуиции отнесения основного состояния,
230
Глава 13
поскольку при неправильном отнесении могут получиться нереальные значения Ж и Pd.
Снижение величины Pd комплекса по сравнению с величиной для свободного иона интерпретируют ковалентным взаимодействием с лигандами. Однако при расчете значений Р из величин А, знаки которых неизвестны, не обойтись без химической интуиции. Например, при исследовании [21] K3Co(CN)6, облученного рентгеновскими лучами, были обнаружены парамагнитные частицы с ^ = 2,010, <?±=2,120, Ац/дР = = 83,5 Э и A/gfi = 26,9 Э. Установлено, что значение Pd комплекса составляет 0,0088 см-1 по сравнению со значением Pd « 0,023 см-1 для свободного иона, откуда следует, что орбиталь, на которой находится неспаренный электрон, имеет d-характер только на 38%. Если бы знаки Аи и были противоположными, то значение Pd составляло бы 0,0147, что значительно более похоже на истину.
Чтобы сделать вывод о различии в электронной структуре неразумно также сравнивать только ограниченные участки спектров ЭПР ряда соединений, например только gfl или А±. Так, для многих комплексов Со(П) [22] с координационным числом 5 величина Ай лежит в интервале от 90-10-4 до 100-10-4 см-1, а величина Pd—часто в интервале от ~ 0,017 до 0,020 см-1. Водные растворы Co(CH3NC)5 + в присутствии избытка CH3NC [23] имеют величину =61 • 10-4 см-1, что свидетельствует на первый взгляд о большей ковалентности, чем обычно. Однако рассчитанное значение Pd составляет 0,0180 в соответствии со многими данными для других изученных соединений.
При интерпретации контактного взаимодействия Ферми в терминах ковалентности связывания можно «попасть в ловушку». Если симметрия 45-орбитали подходит для ее смешивания с d-орбиталью, на которой находится неспаренный электрон, то появится прямой вклад в . Исследование такого рода рассматривалось в связи с уравнением (13.18).
Предыдущее обсуждение строилось на использовании параметров Pd и значения которых в комплексе ниже по сравнению со значениями в свободных ионах. В другом аналогичном подходе [20, 24] используются коэффициенты молекулярных d-орбиталей из орбиталей металла и лиганда, например, для симметрии Й4Л; если пренебречь точной формой орбиталей лиганда, можно получить следующие одноэлектронные орбитали:
а1э = Sj (dzi) — &2 (лиганд), Ь29 = «1 (dxy) — а2 (лиганд), big = Pi (dxi _ yi)— p2 (лиганд), e9(i) = 7i (%) (лиганд), ^2) = 71 (dyz) — 72 (лиганд).
Можно вывести следующие уравнения для g-фактора и А неспаренного
Спектры ЭПР комплексов нлнов переходных металлов
231
электрона, находящегося на Ь29-орбитали:
д.. = 2,0023 -II ’
A£(b19 - b2g) ’
= 2,0023 -
ЬЕ(ев-Ь2д)’
Л - Pd
— а] + Э&А + 0|| — 2) + -01 — 2)
A± = Pd
_а^_| + ^ + П(01_2)
В этих уравнениях использованы Е, и Pd для свободных ионов; таким образом, «1Р1 = Е, (комплекс)/!; (ион).
Сообщалось о многих исследованиях, в которых комплекс меди(П) анализировали так же, как это описывается выше. Однако не все результирующие тенденции в изменении значений а2 имеют смысл для комплексов меди(П). Были предложены [25, 26] две несовместимые интерпретации этих тенденций исходя из изменений ковалентности связывания металл—лиганд.
13.5. СВЕРХТОНКОЕ РАСЩЕПЛЕНИЕ, ОБУСЛОВЛЕННОЕ ВЗАИМОДЕЙСТВИЕМ ЭЛЕКТРОНОВ С МАГНИТНЫМИ МОМЕНТАМИ ЯДЕР ЛИГАНДА
Сверхтонкое расщепление на ядрах лиганда зависит от контактного взаимодействия Ферми (F. С.), дипольного взаимодействия с ионом металла (DIP), дипольных эффектов, обусловленных электронной плотностью на р-орбитали лиганда (LDP), и псевдоконтактного вклада иона металла (LPC), возникающего за счет взаимодействия орбитального углового момента неспаренного электрона с ядерным спином лиганда. Если сверхтонкая структура, обусловленная лигандом, разрешена, то последний член обычно мал по сравнению с другими. При наличии интенсивного спин-орбитального взаимодействия следует ожидать большого псевдоконтактного вклада, но релаксационные эффекты осложняют наблюдение спектра ЭПР и. следовательно, сверхтонкого расщепления на лиганде. Значения Ай и А± выражают с помощью уравнений (13.38) и (13.39):
А|| = Ар с + 2 (XDIP + Aldp), (13.38)
= Af с - 2 (Aqip + Aldp). (13.39)
Эти уравнения относятся к параллельной и перпендикулярной ком
232
Глава 13
понентам тензора сверхтонкого взаимодействия лиганда. Трудности возникают, если при измерении экспериментатором сверхтонкого взаимодействия с лигандом (Л±), исходя из обусловленной лигандом тонкой структуры сверхтонкой линии металла (например, азотная сверх-сверх-тонкая структура сверхтонкой линии кобальта), два тензора сверхтонкого взаимодействия недиагональны в одной и той же системе координат. Для того чтобы полностью понять систему, необходимо исследовать монокристалл одновременно методом ЭПР и с помощью дифракции рентгеновских лучей. Если спектр раствора характеризуется разрешенной сверхтонкой структурой, обусловленной лигандом, то Лг.с. можно измерить непосредственно. Уравнения (13.38) и (13.39) нельзя использовать для определения .4dip и ,4ldp. Однако, если известна структура, можно получить разумные значения XDjp из уравнения
Лор =------;--(см"1), (13.40)
аЛ
где а — расстояние между лигандом и металлом. Уравнение (13.40) выведено для условия, когда расстояние между металлом и лигандом велико по сравнению с расстоянием между ядром атома металла и электроном. Если такое приближение не справедливо, используются другие уравнения [27]. Величины XF C. и XLDp связаны с s-орбитальным вкладом as и р-орбитальным вкладом ар в молекулярную орбиталь следующими уравнениями:
Л.с. = ^|^?РР1у1Ф(0)|Ч2, (13.41)
^LDp=|yPPn<7T>X- (13.42)
Сообщалось [28] о величинах А^с. и А^пр для одного электрона, находящегося на 5- и р-орбиталях, из которых можно рассчитать процент s- и р-характера. Из уравнений (13.41) и (13.42) можно рассчитать отношение %$/ %р, которое указывает на «гибридизацию лиганда» в комплексе. Этот подход предполагает, что делокализация происходит главным образом по прямому механизму, т.е. он не учитывает спин-поляриза-ции.
Использование этих идей мы продемонстрируем на некоторых примерах, не вдаваясь в подробности. Спектр ЭПР [29] комплекса дает
Спектры ЭПР комплексов ионов переходных металлов 233
A||(14N)/g0 = 15,6 Э, X±(14N)/gP = 12,7 Э и .4is0 = 13,7 Э. Значения А для электрона, целиком находящегося на s- и р-орбиталях, как сообщалось, составляют соответственно Л°/рР = 34,1 Э и А°/д$ = 550 Э. Это приводит к отношению °/op/°/os, равному ~ 1, что указывает на sp-гибридиза-цию и линейную структуру Fe — N — О. В то же время отношение p/s для нитрозильного атома азота [30] Fe(CN)5NO3- составляет 1,6, что согласуется с изогнутой металл-нитрозильной структурой (хр2-гибрид должен иметь отношение 2).
Недавно удалось наблюдать [31] сверхтонкое расщепление на атомах фосфора в спектрах ЭПР аддуктов (С6Н5)3Р и PF3 тетрафенилпор-фиринкобальта(П). Как сообщалось, отношение p/s в первом случае составляло 2,7, а во втором — 0,47.
13.6. ОБЗОР СПЕКТРОВ ЭПР КОМПЛЕКСОВ ИОНОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ ПЕРВОГО БОЛЬШОГО ПЕРИОДА
В этом разделе дается краткий обзор некоторых результатов, полученных при исследовании различных «/"-комплексов методом ЭПР. Более полное обсуждение читатель может найти в работах [19, 20]. Прежде чем приступить к рассмотрению результатов, следует упомянуть, что спин-орбитальное взаимодействие—главный фактор, определяющий электронную релаксацию в этих системах. При ознакомлении с этим разделом читатель может столкнуться с Такими утверждениями, как «расщепление в нулевом поле вызывает быструю релаксацию» или «анизотропия g-факгора ведет к небольшим временам жизни электронного спинового состояния» и т.д. Все эти выражения говорят об очевидных эффектах спин-орбитального взаимодействия в молекуле. Ранее уже обсуждалась связь спин-орбитального взаимодействия с релаксационными эффектами. Комплексы ионов переходных металлов второго и третьего периодов значительно более сложны для исследования методом ЭПР, поскольку в этом случае значения констант спин-орбитального взаимодействия много больше.
(f-Конфигурация
Результаты исследований ионов с электронной конфигурацией d1 можно согласовать со спин-гамильтонианом
Н = Р [gxHxSx + gyHySy + gzHzSz~] + AXSX1X + AySyIy + AZSZIZ.
(13.43)
В октаэдрическом поле лигандов основным состоянием является 2Т2д, и в этом основном состоянии имеет место значительное спин-орбитальное взаимодействие. Все крамерсовы дублеты, обусловленные 2Т2д, близки по энергии и интенсивно смешиваются под действием спин-орбитального взаимодействия. Это приводит к небольшим значениям те.
234
Глава 13
В тетраэдрическом поле лигандов возникает основное состояние 2£(х2— у2, z2), в котором спин-орбитальное взаимодействие первого порядка отсутствует. При такой геометрии подмешивание расположенных поблизости возбужденных состояний 2Т2д к основному состоянию за счет спин-орбитального взаимодействия второго порядка * приводит к низким временам спиновой релаксации для электрона и широким полосам поглощения. Комплексы обычно должны быть исследованы при температурах, близких к температуре жидкого гелия. Возбужденное состояние 2 Г расщепляется под действием спин-орбитального взаимодействия. Если поле лигандов искажено (например, как в VO2 + ), то основное состояние становится орбитальным синглетом, а возбужденные состояния не подмешиваются. При более высоких температурах наблюдаются узкие спектральные линии ЭПР.
Были выведены [20] уравнения с использованием -волновых функций и соответствующего спин-гамильтониана, которые связывают д-фак-тор в тригонально искаженном комплексе с величиной искажения. Искажение выражается через 5 (см-1)— расщепление состояния 2Т. В шрис-(ацетилацетонато)титане(Ш) было обнаружено большое искажение с 5 = 2000—4000 см-1. В результате такого расщепления время жизни электронного спинового состояния увеличивается, и можно зарегистрировать спектр ЭПР при комнатной температуре.
d2 -Конфигурация
Известно очень мало примеров спектров ЭПР октаэдрических комплексов <72-ионов из-за сильного спин-орбитального взаимодействия в основном состоянии 3Т1. Основным состоянием тетраэдрических комплексов является 3Л2, поэтому следует ожидать больших времен релаксации и большей легкости в регистрации спектров ЭПР. Спектры этих систем можно согласовать с S = 1 и спин-гамильтонианом
+ Е [S2 - S2] + A SZIZ + В [_SXIX + SJJ. (13.44)
Комплекс V3 + в октаэдрическом окружении [32] в А12О3 дает д№ = 1,92, д = 1,63, D= +7,85 и А = 102. Примером изученного комплекса Td [33] может служить комплекс V3 - в решетке CdS. При температуре жидкого азота наблюдаются значения g-фактора и D, равные соответственно 1,93 и 1 см-1.
* В теории возмущений второго порядка Е-компонента этого расщепленного состояния подмешивается к основному состоянию.
Спектры ЭПР комплексов ионов переходных металлов 235
^-Конфигурация
Основным состоянием октаэдрических (^-комплексов является 4А2, которое должно обладать крамерсовым дублетом низшей энергии. Если расщепление в нулевом поле мало, как это показано на рис. 13.13, А, то иногда удается зарегистрировать три перехода и из двух наблюдаемых переходов можно получить параметр расщепления в нулевом поле. Если расщепление в нулевом поле велико по сравнению с частотой спектрометра, то наблюдается только одна полоса (рис. 13.13, Б). Вообще спектры можно согласовать со спин-гамильтонианом
+ Е [Sx - Sy] + X„SJZ + Д± [Sxfx + S/J. (13.45)
В некоторых системах [33] трудно распознать «типичные участки» для Сг3 +. Спектр на рис. 13.13,Г рассчитан с помощью ЭВМ с использова-
Рис. 13.13. А—небольшие расщепления основного состояния 442 «^-комплекса в нулевом и магнитном полях (поле направлено вдоль оси z) и результирующий спектр; Б—в виде стекла в ДМФ Н2О н СН3ОН [33] при 9,3 ГГц. £>>0,4 см £<0,01; В — mpaHc-ECrfCsHsN^ChP в виде стекла в ДМФ , Н2О, СН3ОН [33] при 9,211 ГГп; Г—рассчитанный на ЭВМ [33] спектр для случая В при gii -ff± = 1,99, £>= 0,164 см-1 и £ = 0. [Reprinted with permission from Pedersen E., Toftlund ££., Inorg. Chem., 13,1603 (19^4). Copyright by American Chemical Society.]
236
Глава 13
нием изотропного g-фактора, D = 0,164 см-1 и Е = 0 см-1. В работе [33] имеется много спектров тетрагональных комплексов Сг(Ш) и проведен их детальный анализ.
«/’-Системы интенсивно изучали, особенно комплексы Сг3 + . В октаэдрических комплексах электроны металла находятся на орбиталях г2д, поэтому сверхтонкое взаимодействие с лигандом обычно мало. д-Фак-тор для этой системы определяется, согласно теории кристаллического поля, выражением
g = 2,0023
8Х
АЕ(4Т29-4А29) '
(13.46)
В основном состоянии 4A2ff отсутствует спин-орбитальное взаимодействие, и состояние 4’Т2д подмешивается к 4A2g только в небольшой степени. Уравнение (13.46) отличается от приведенных ранее в двух отношениях. Спин-орбитальное взаимодействие описывается X (который может быть и положительным, и отрицательным) и характеризует состояние. При наличии более чем одного неспаренного электрона разности энергий также могут быть выражены через разности энергий соответствующих электронных состояний. Расчет в случае комплекса V(H2O)’ + с использованием АЕ = 11 800 см “ 1 и X = 56 см1 дает значение д-факто-ра, равное 1,964, которое близко к наблюдаемому значению 1,972 [34]. Для комплекса Cr(H2O)g+ АЕ = 17400 см-1,Х = 91см-1и предсказываемое значение g ниже экспериментального, равного 1,977 [35]. В случае комплекса Мп4+ расхождение даже больше: рассчитанное значение равно 1,955, а измеренное 1,994. Этот результат можно объяснить меньшей точностью приближения кристаллического поля и увеличением ковалентности связывания по мере роста заряда на центральном атоме.
^-Конфигурация
Известно очень немного спектров ЭПР для «/4-электронной конфигурации. Основное состояние 5Е этой системы в слабом кристаллическом поле Oh не имеет орбитального углового момента, поэтому S—хорошее квантовое число. Расщепление в нулевом поле уровней +2, + 1 и 0 приводит к четырем переходам, если расщепление мало, как это показано на рис. 13.14, и ни к одному, если расщепление велико. Ожидаемые ян-теллеровские искажения и сопровождающие их большие расщепления в нулевом поле часто делают невозможной регистрацию спектров.
Для низкоспиновых «/4-ко индексов информацию можно получить, обратившись к разделу, посвященному «/’-системам (напоминаем о формализме дырок).
Низкоспиновая «^-конфигурация, 5 = 1/2
В сильном поле лигандов октаэдрической симметрии основным состоянием является 2Т2. Спин-орбитальное взаимодействие расщепляет
Спектры ЭПР комплексов ионов переходных металлов
237
Рис. 13.14. Расщепления основного состояния 5Е ^-комплекса в нулевом и магнитном полях (поле направлено вдоль оси z).
этот терм на три близко лежащих крамерсовых дублета, однако из-за большого спин-орбитального взаимодействия спектр ЭПР можно наблюдать только при температурах, близких к температуре жидкого гелия. Поскольку в этой системе имеется пять d-электронов, ситуация аналогична случаю d1-комплекса, если не считать положительной дырки. Ян-теллеровские силы стремятся исказить такие системы, как МХ£“, по-
Рис. 13.15. Диаграмма энергетических уровней низкоспиновой </5-системы в полях Oh и D3, которые расщеплены спин-орбитальным взаимодействием и магнитным полем. При учете спин-орбитальных эффектов используются представления двойной группы D3 (штрихованные значения).
238
Глава 13
этому значения g-факторов, предсказываемые уравнением (13.9), наблюдают редко. Расщепление дублетного состояния свободного иона полем 0h за счет искажения D3 спин-орбитальным взаимодействием и магнитным полем показано на рис. 13.15. Поскольку спин полуцелый, при рассмотрении спин-орбитального взаимодействия используются представления двойной группы и штрихованные символы для представлений. Если определить параметр искажения 5, как на рис. 13.15, то можно вывести выражения для д-фактора:
3(^ + 28)
011 К + 23)2 + 852]1/2 ’
25 - 3^ л = ------------5------
[(£ + 25)2 + 8V]1'2
(13.47)
(13.48)
Отметим, что при 8->0 в соответствии с уравнением (13.9) и д^, и д± стремятся к нулю. Например, Fe3+ в K3Co(CN)6 демонстрирует [37] спектр ЭПР с д( = 0,915 и д± як 2,2. Подстановка значения gf в уравнение (13.47) и использование ^=—103 см-1 для свободного иона дает значение 8 як 200 см-1.
Большие отклонения от октаэдрической симметрии приводят к тому, что орбитальный синглет имеет наименьшую энергию, при этом он хорошо отделяется от орбитально не вырожденных возбужденных состояний. Времена электронной релаксации увеличиваются, и спектр ЭПР
Рис. 13.16. Схематическая структура гемоглобина.
можно наблюдать при более высоких температурах. Примерами таких систем могут служить низкоспиновые производные гемоглобина Fe(III) [38] (рис. 13.16), которые характеризуются большим тетрагональным искажением из-за плоской структуры гема. Примерами оснований, которые создают низкоспиновое окружение, являются N3, CN- и ОН-. Экспериментальные значения g-фактора для составляют дх — 1,72, ду = 2,22 и дг = 2,80. Таким образом, большая анизотропия дх- и ду-факторов обусловлена взаимодействием отдельной d'-орбитали железа с л-орбиталью азота координированной гистидиновой группы, которая связывает глобин в гем. Исследования методом ЭПР [39] комплек
Спектры ЭПР комплексов ионов переходных металлов 239
сов бипиридила и фенантролина с железом(Ш), рутением(Ш) и осми-ем(Ш) были проанализированы с помощью энергетической диаграммы, аналогичной изображенной на рис. 13.15, на которой показано искажение, приводящее к состоянию 2 А более низкой энергии, чем состояние 2£ (т. е. 5 отрицателен). Спин-делокализация на лиганд исследована как функция металла этого ряда.
Высокоспиновая «^-конфигурация
Эта d-электронная конфигурация исследовалась очень тщательно. Высокоспиновые комплексы имеют основные состояния sS, а другие секстетные состояния отсутствуют. Другим ближайшим термом является 4Т1, и для его подмешивания необходимы спин-орбитальные взаимодействия второго порядка, поэтому его вклад мал. Таким образом, время жизни электронного спина велико, и спектры ЭПР можно легко регистрировать при комнатной температуре и в кристаллических полях любой симметрии. Более того, при нечетном числе электронов крамерсово вырождение наблюдается даже при большом расщеплении в нулевом поле. Энергетические уровни комплекса Мп(П) изображены на рис. 13.10. Результаты, полученные для высокоспиновых комплексов, можно согласовать с гамильтонианом
А А А ОС А
H = «/pHS + D S2-|j + £[S;
;2-s2] + xs-i +
+ |п S2 + S4 + S4-^ +i|q£ 35S4-^S2 +
(13.49)
Члены с более высокими степенями S появляются потому, что оператор октаэдрического кристаллического поля связывает состояния со значениями Ms, отличающимися на + 4; они приводят к более сложному базису и большему числу ненулевых недиагональных матричных элементов. На рис. 13.17 показаны расщепление энергетических уровней и спектр, ожидаемый для неискаженного октаэдрического комплекса железа) III).
В комплексах железа(Ш) с небольшим тетрагональным искажением D«hv и £ = 0. Энергетические уровни и ожидаемый спектр показаны на рис. 13.18, Л. Наблюдаемые «/-факторы очень близки к 2,00 из-за исключительно малой величины спин-орбитального взаимодействия. Поэтому можно легко наблюдать спектры ЭПР при комнатной температуре. Если D»hv, то возникнет ситуация, изображенная на рис. 13.18,Б, и наблюдаются лишь переходы между | + 1/2> и | — 1/2>. Если даже более высоко лежащие уровни и заселены, то AMS А 1 для возможных переходов и ни одна спектральная линия не наблюдается. Можно рассчитать «/-факторы, используя в качестве базиса только |5/2, 1 /2> и 15/2, — 1/2> и зеемановский гамильтониан Н = д'ц рЯ& + д\р {HXSX + HySy). Если
240
Глава 13
Рис. 13.17. Расщепление энергетических уровней (Л) и спектр (Б), ожидаемый для октаэдрического комплекса железа(Ш) (направление поля Н параллельно главной оси октаэдра).
Спектры ЭПР комплексов ионов переходных металлов
241
Рис. 13.18. Энергетические уровни и ожидаемый спектр d’-иона в слабом (А) и сильном (Б) тетрагональных полях (направление поля Н параллельно тетрагональной оси). [Kokoszka G.F., Duerst R. W., Coord. Chem. Rev., 5, 209 (1970).]
16-574
242
Глава 13
направление поля Н параллельно оси z, то имеем
(13.50)
Решение уравнения (13.50) приводит к ЛЕ = gef>H. и дц = де.
Если направление поля Н параллельно оси х, то после использования = Sx ± iSy получим
1_5 Л
12’2/ 0
_5 _ А
2’ 2/
о
(13.51)
Диагонализация уравнения (13.51) приводит к ЛЕ = Зде$Нх и д± = = Зде « 6,0.
Такую ситуацию хорошо демонстрирует рис. 13.16, где В—лиганд слабого поля, например F " или Н2О, который вызывает образование высокоспинового комплекса. Параметр расщепления в нулевом поле D был измерен для нескольких систем такого типа путем изучения спектра в дальней инфракрасной области в магнитном поле. Для различных комплексов были получены значения в интервале 5 — 20 см-1 [40].
Рис. 13.19. Крамерсовы дублеты в ромбической симметрии (D и Е не равны нулю) высокоспинового комплекса железа(Ш). Три главные компоненты показаны в скобках.
Спектры ЭПР комплексов ионов переходных металлов 243
И наконец, следует рассмотреть случай геометрического искажения комплекса с устранением аксиальной симметрии. При этом параметры расщепления в нулевом поле D и Е отличаются от нуля. Как показано на рис. 13.19, гамильтониан снова приводит к трюм крамерсовым дублетам. Решая матрицу этого гамильтониана с использованием диагонализирующих ее волновых функций, находим, что три крамерсовых дублета являются линейными комбинациями (5/2, ±5/2), (5/2, ±3/2) и |5/2, ± 1/2). Таким образом, переходы в пределах каждого крамерсова дублета разрешены; соответствующие значения g-фактора показаны на рис. 13.19; расстояние между крамерсовыми дублетами достаточно велико, чтобы между ними не происходили переходы, но при обычных температурах все они в значительной степени заселены и наблюдается много линий. Примером такого комплекса [41] может служить Na[Fe(edta)]-4Н2О (где edta—этилендиаминтетраацетат), разведенный в монокристалле аналогичного комплекса Co(III). Спектр демонстрирует один почти изотропный переход при g = 4,27 и два очень анизотропных перехода с главными значениями g-фактора, равными соответственно 9,64 и 1,10.
(^-Конфигурация
Эту систему тщательно не исследовали. Низкоспиновые комплексы диамагнитны, а высокоспиновые комплексы с симметрией Oh напоминают d4-комплексы. Высокоспиновый комплекс железа(П) при 4,2 К характеризуется д-фактором 3,49 и шириной спектральной линии 500 Э. Спин-орбитальное взаимодействие в основном состоянии велико, имеются в комплексе и близко лежащие возбужденные состояния, которые могут к нему подмешиваться. Если эффекты нулевого поля малы, то в основном состоянии с J = 1 должны наблюдаться два перехода. В искаженном октаэдрическом поле эффекты нулевого поля велики, и спектр ЭПР комплекса не регистрируется. Примером такой системы может служить дезоксигемоглобин.
(^-Конфигурация
Основное состояние для высокоспинового (/’-комплекса с симметрией Oh представляет собой 4Т1Я (F). При интенсивном спин-орбиталь-ном взаимодействии измерения ЭПР возможны лишь при низких температурах. При S = 3/2 и трех орбитальных компонентах в Т получается в общем 12 низко лежащих спиновых состояний. При низких температурах, необходимых для регистрации спектра из-за проблем спиновой релаксации, заселен только низко лежащий дублет, что дает лишь одну линию при эффективном S' = 1/2 с д-фактором 4,33. Имеется обзор, посвященный исследованию таких систем [42].
Тетраэдрические комплексы кобальта)!!) с основными состояниями ±42 аналогичны (/’-комплексам, если не считать того, что в первом случае возбужденное состояние 4Т2 ближе по энергии к состоянию 4Л2.
244
Глава 13
При большей величине спин-орбитального взаимодействия для комплексов кобальта(П) обнаружены более широкие линии.
В сильных кристаллических полях дублет 5=1/2 состояния 2Е имеет низшую энергию. Поскольку в состоянии 2Е отсутствует спин-орбитальное взаимодействие и поскольку вблизи него нет дублетных состояний, время жизни электронного спина велико, что часто позволяет регистрировать спектры ЭПР с узкими линиями при температуре жидкого азота и комнатной температуре.
Спин-гамильтониан для низкоспиновых d1 -комплексов обычно выражается в виде
Н - Р[3х^х^х + ву^у^у + dz^z^zl + 4А4 + AySyly + Az$zlz-
В пяти- и шестикоординационных тетрагональных d1 -комплексах неспаренный электрон находится на (/,2-орбитали. Для этой электронной конфигурации в предположении аксиальной симметрии
0В= 2,0023, (13.53)
91 = 2,0023 - -----г - (13.54)
ДЕ (z — xz, yz)
Этот факт был использован [32] для изучения образования аддуктов координационно ненасыщенных комплексов кобальта с различными аксиально координирующимися основаниями В. Хорошее перекрывание между неподеленной парой донора, координирующегося через атомы азота или фосфора, и (/,2-орбиталью приводит к легко наблюдаемой сверхтонкой структуре. Вейланд использовал большое гиромагнитное отношение (и, следовательно, большое сверхтонкое взаимодействие) 31Р, чтобы получить отношения гибридизации для различных доноров РХ3, образующих комплексы с Со(тетрафенилпорфирин) [31] и Co(sa-len) [43]. При исследовании [44а] 2:1-аддуктов основания BF2 с бис-(ди-фенилглиоксим)Со(11) было обнаружено, что значения Р [см. обсуждение уравнений (13.36) и (13.37)] для кислородсодержащих доноров выше, чем для азотсодержащих доноров. Для ряда из десяти азотсодержащих доноров было также найдено, что Р варьирует от 0,0216, если В—хину-клидин, и до 0,0147, если В — N-метилимидазол.
В низкоспиновых «/'-комплексах dzz- и 4л-орбитали принадлежат к одному и тому же неприводимому представлению и могут смешиваться [см уравнение (13.18)]. Поэтому для получения информации о коэффициентах молекулярных орбиталей и ковалентности в этих системах нельзя использовать контактный член Ферми УС, выведенный из уравнений (13.30) — (13.35).
Сообщалось [446] о регистрации спектров ЭПР аддуктов О2 с различными комплексами кобальта(П). Неспаренный электрон в этой системе находится главным образом на О2. Несмотря на это, наблюдается заметное сверхтонкое взаимодействие. Взаимодействие с металлом вызвано спин-поляризацией заполненной молекулярной орбитали ад
Спектры ЭПР комплексов ионов переходных металлов
245
дукта О2, обусловленной спариванием неспаренного электрона металла, находящегося на (/.г-орбитали исходного пятикоординационного комплекса кобальта(П), с одним из разрыхляющих электронов молекулы О2. Степень переноса электрона с кобальта(П) на координированную молекулу О2 оценивалась по величине константы анизотропного сверхтонкого взаимодействия кобальта. При некорректном анализе данных ЭПР была получена для многих систем степень переноса электрона значительно ниже 90% или выше [44в].
Высокоспиновая ds-конфигурация
Основным состоянием газообразного иона является 3F, причем низшее положение в октаэдрическом поле занимает орбитальный синглет. rf-Оболочка заполнена более чем наполовину, поэтому спин-орбитальное взаимодействие ведет к значению ^-фактора, превышающему значение для свободного электрона. Расщепление в нулевом поле делает трудной регистрацию спектров ЭПР, если только не использовать низкие температуры. Найденные значения ^-фактора обычно близки к изотропным.
(^-Конфигурация
(/’-Конфигурацию исследовали очень тщательно. В октаэдрическом поле основным состоянием является 2Ед. Ожидается большой ян-телле-ровский эффект, позволяющий регистрировать спектр ЭПР при комнатной температуре. В тетрагональных комплексах основным состоянием является dx2_y2 (оси х и у направлены на лиганды) и наблюдаются узкие линии. Отметим, что в этом эксперименте можно обнаружить квадрупольное взаимодействие спина с ядром меди (см. гл. 9). Данные исследования методом ЭПР согласуются со спин-гамильтонианом
Н = P[gz Hz Sz + дх Нх Sx + ду Ну %] + Az Sz Iz + Ах Sx Ix +
+ AySy Iy + Q' [I2 - +l)-]-gNf,NH-i. (13.55)
Несколько типичных спектров ЭПР комплексов меди(П) показано на рис. 13.20. На рис. 13.20,Л изображен изотропный спектр раствора. У полосы в сильном поле наблюдаются как протонная, так и азотная сверхтонкие структуры, обусловленные взаимодействием с лигандом, чего нет у полос в слабом поле. Этот результат приписывают разности во временах релаксации для этого перехода, который зависит от связанного с ним значения т: [45]. Растворитель влияет на молекулярное время корреляции, которое в свою очередь влияет на вид спектра [45].
На рис. 13.20,Б изображен анизотропный спектр при частотах Q-диапазона. Такие спектры наблюдают для стекло- или порошкообразных образцов комплексов меди, разведенных в кристаллической решетке
6
Рис. 13.20. «Типичные» спектры ЭПР комплексов меди(П).
А —типичный спектр раствора для квадратно-плоскостного лиганда — основания Шиффа [Hasty £., Colburn T.J., Hendrickson D.N., Inorg. Chem., 12, 2414 [1973)]; Б —спектр стекло- или порошкообразного образца аксиального комплекса при частотах Q- диапазона; В—спектр комплекса в виде стеклообразного или порошкообразного образца для случая А при частотах Х-диапазоиа.
2650 2850 3050 3250 3450
Напряженность поля, Э
В
Спектры ЭПР комплексов ионов переходных металлов
247
Б
Рис. 13.21. А— схематическое изображение димерного комплекса аденинмеди; Б — спектр ЭПР поликри-сталлического образца этого комплекса.
диамагнитного вещества. Полосы в слабом (д^) и сильном (д±) полях хорошо разделены. При более высокой энергии микроволн индивидуальные линии шире, поэтому сверх-сверхтонкое расщепление полосы gill не регистрируется. В спектре на рис. 13.20,В при частотах х-диапазо-на полосы переходов д^ и gL перекрываются, но регистрируется значительно большая часть сверхтонкой структуры, обусловленной лигандом Как упоминалось ранее, интерпретация температурной зависимости спектров многих комплексов меди(П) проводилась на основе эффектов Яна—Теллера.
Спектр поликристаллического образца, молекулы которого содержат два атома меди, показан на рис. 13.21. Основные полосы при 2500 и 3700 Э приписаны двум компонентам д±. У полосы д^ в слабом поле наблюдаются семь сверхтонких компонент меди (два ядра меди с I = = 3/2). Другая линия д± в сильном поле не видна, поскольку она лежит вне диапазона спектрометра, полоса при ~ 3200 Э отнесена к переходу Ams = 2. Причину возникновения дублета можно понять после обсуждения спин-гамильтониана. Спектры монокристаллов [46] молекул, содержащих два атома меди(П) с 8 = 1, согласуются со спин-гамильтониа-ном
Н = ₽ [дгНX + gxHxSx + 0Д/Д] + D [Sz2 -1] +
+ Е [S2 - S2] + AXSXIX + А// + AZSJZ. (13.56)
Для димера ацетата меди(П) получены следующие результаты [47]: = 2,344, дх = 2,053, ду = 2,093, D = 0,345 см ’ \ Е = 0,007 см “ \ Az = = 0,008 см “1 и Ах, Ау < 0,001 см_ \ Влияние, которое параметры J,
248
Глава 13
D и Е и оказывают на спектр ЭПР, показано на рис. 13.22. При |j\ = = 260 см “ 1 в Си2(ОАс)4 в спектре ЭПР не наблюдаются переходы между состояниями с S = 0 и S = 1. Обменное взаимодействие приводит к низкоэнергетическому состоянию S = 0, поэтому с падением температуры снижается интенсивность сигналов. Эта температурная зависимость приводит к значению J, равному — 260 см “ 1, что соответствует разделению состояний с S = 0 и S = 1 величиной 2J, или — 520 см 1. В рассмотренном ранее спектре порошкообразного образца расщепление полос gtl и д± обусловлено двумя переходами с ДМ5 = + 1, усредненными по ориентациям. В относительно редкой ситуации, когда параметр обмена J меньше, чем доступная энергия микроволнового
Рис. 13.22. Влияние взаимодействий J и эффектов нулевого поля на спектр ЭПР монокристалла одной ориентации и энергетические уровни молекулы, содержащей два <19-иона меди.
Спектры ЭПР комплексов ионов переходных металлов 249
излучения в эксперименте ЭПР, можно наблюдать переходы ЭПР между состояниями S = 1 и S = 0 димерного комплекса меди. Сообщалось [48] о «внешнесферных» димерах в [Cu2(tren)2X2](BPh4)2, где X = = NCO- и NCS-, a tren — 2,2',2"-триаминотриэтиламин. «Внешнесфер-ный» димер образуется из двух тригональных бипирамид комплекса Си(П) за счет образования водородной связи между некоординированным концом O(S) группы OCN “ (SCN “), связанной через азот с одним атомом меди, и протоном N — Н координированного tren. Как показано на рис. 13.22, при частотах Х-диапазона из синглет-триплетных переходов могут быть измерены значения J в интервале от ~ 0,15 до ~ 0,05 см“ 1.
13.7. МЕТОД ДВОЙНОГО РЕЗОНАНСА
Два метода двойного резонанса—двойной электрон-ядерный резонанс (ДЭЯР) и двойной электрон-электронный резонанс (ДЭЭР) — имеют относительно ограниченное применение в исследованиях ЭПР. В методе ДЭЯР переход ЭПР наблюдают в системе, в которой насыщен переход ядерного спина, а в методе ДЭЭР измерения проводят при насыщении другого перехода электронного спина. Как и в методе двойного резонанса ЯМР, в результате эффекта Оверхаузера наблюдается увеличение интенсивности. Во многих случаях [49 — 51] можно достичь преимуществ, аналогичных тем, что рассматривались для аналогичных экспериментов ЯМР
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. См. Tinkham М., Group Theory and Quantum Mechanics, p. 143, McGraw-Hill Book Co., New York, 1964.
2. Bugguley D.M.S., Griffits J.H.E., Nature, 162, 538(1948).
3. Fackler J.P., Smith J. A., J. Amer. Chem. Soc., 92, 5787(1970);
Fackler J.P., Levy J.D., Smith J. A., ibid., 94, 2436(1972).
4. a. Hoffman B.M,, Basolo F., Diemente D.L., J. Amer. Chem. Soc., 95, 6497(1973).
6. Cariati F., Morazzoni F., Busetto C., Del Piero D., Zazzetta A., J. Chem. Soc,, Dalton, 1975, 556.
5. Manohanon P. T., Rogers M. T., in Electron Spin Resonance of Metal Complexes, ed. Teh Fu Yen, Plenum Press, New York, 1969.
6. Bleaney B., Ingram D. J., Proc. Phys. Soc., 63, 408(1950).
7. Abragam A., Pryce M.H.L., Proc. Phys. Soc., 63, 409(1950).
8. Bleaney B., Bowers K. D., Proc. Phys. Soc., 65, 667(1952).
9. Bleaney B., Bowers K.D., Trenam R.S., Proc. Roy. Soc. (London), A, 228, 157(1955).
10. Allen H. C., Jr., Kokoszka G. F., Inskeep R. G., J. Amer. Chem. Soc. 86, 1023(1964).
И. В качестве других примеров см. Ham F.S., im Electronic Paramagnetic Resonance, ed. by Geschwind S., Plenum Press, New York, 1972.
12. Koch R.C., Joesten M.D., Venable J.H., Jr., J. Chem. Phys., 59, 6312(1973).
13. Kirmse R., et al., I. Chem. Phys., 56, 5273(1972).
14. Buluggiu E., Vera A., J. Chem. Phys., 59, 2886 (1973).
15. Pilbrow J.R., Winfield M.E., Mol Phys., 25, 1073(1973).
16. a. Yonetoni, et al., J. Biol. Chem., 245, 2998(1970).
6. Orton J. W., et al., Proc. Phys. Soc., 78, 554(1961).
17. a. Symons M.C.R., Wilkerson J.G., J. Chem. Soc., A, 1971, 2069.
250
Глава 13
б. Maki А. Н., Edelstein N., Davidson A., Holm R. Н., J. Amer. Chem. Soc., 86, 4580(1964).
18. Griffith J. S., The Theory of Transotion — Metal Ions, Cambridge University Press, New York, 1961.
19. Golding R. M., Applied Wave Mechanics, Van Nostrand, New York, 1969.
20. McGarvey B.R., Electron Spin Resonance of Transition Metal Complexes, in Transition Metal Chemistry, Vol. 3, pp. 89 — 201, ed. by Carlin R. L., Marcel Dekker, New York, 1967.
21. Lin W.C., McDowell C.A., Ward D.J., J. Chem. Phys., 49, 2883 (1968).
22. Wayland B.B., et al., J. Amer. Chem. Soc., 96, 2795(1974).
23. Kimball M.E., Pratt D.W., Kaska W.C., Inorg. Chem., 7, 2006(1968).
24. a. Maki A.H., McGarvey B.R., J. Chem. Phys., 29, 31(1958).
6. Kivelson D., Nieman R., J. Chem. Phys., 35, 149(1961).
25. Zink J. I., Drago R.S., J. Amer. Chem. Soc., 94, 4550(1972).
26. Kuska H.A., Rodgers M.T., J. Chem. Phys., 43, 1744(1965); Kuska H.A., Rodgers M.T., Drullinger R.E., J. Phys. Chem., 71, 109(1967).
27. Goodman B. A., Raynor J. B., Adv. Inorg. Chem. Radiochem., 13, 135(1970).
28. Эткинс П., Саймонс M., Спектры ЭПР и строение неорганических радикалов.— М.: Мир., 1970.
29. Sweeney W.V., Coffman R.E., J. Phys. Chem., 76, 49(1972).
30. McNeil D.A.C., Raynor J.B., Symons M.C.R., Proc. Chem. Soc. 1964, 364.
31. Wayland B.B., Abd-Elmageed M.E., J. Amer. Chem. Soc., 96, 4809 (1974).
32. Foner S., Low W„ Phys. Rev., 120, 1585(1960).
33. Pedersen E., Toflund H., Inorg. Chem., 13, 1603(1974).
34. Borcherts R.H.. Kikuchi C.. J. Chem Pin- 40. 2270(1964).
35. Jorgensen С. K., Acta Chem. Scand., 8, 1686(1957).
36. Керрингтон А., Мак-Лечлан Э„ Магнитный резонанс и его применение в химии.— М.: Мир, 1970.
37. Lin W.C., McDowell С. A., Ward D.J., J. Chem, Phys., 49, 2883 (1968).
38. Ingram D. J. E., Biological and Boochemical Applications of Electron Spin Resonance, Plenum Press. New York, 1969.
39. DeSimone R. E., Drago R.S., J. Amer. Chem. Soc., 92, 2343(1970).
40. Brackett G.C., Richards P.L., Caughey W.C., J. Chem. Phys., 54, 4383(1971).
41. Aaasa R., Vanngard T., Arkiv Kemi, 24, 331(1965).
42. Kenedy F.S., et al., Biochem. Biophys. Res. Comm., 48, 1533(1972) в печати.
43. Wayland В. В., Abd-Elmageed M.E., Mehne L.F.,
44. a. Tovrog B., Drago R.S., в печати.
6. Tovrog В., Kitko D.J., Drago R.S., J. Amer. Chem. Soc., 98, 5144(1976).
c. Hoffman B.M., Diemente D.L., Basolo F., J. Amer. Chem. Soc., 92, 61(1970); Crumbliss A.L., Basolo F., J. Amer. Chem. Soc. 92, 55(1970).
45. Lewis W. B., Morgan L. O., Transition Metal Chemistry, Vol. 4, p. 33, ed. Carlin R. L., Marcel Dekker, New York, 1968.
46. Kokoszka G.F., Duerst R.W., Coord. Chem. Rev., 5, 209(1970).
47. Bleaney B., Bowers K.D., Proc. Roy. Soc. (London), A, 214, 451(1952).
48. Duggan D.M., Hendrickson D.N., Inorg. Chem., 13, 2929(1974).
49. Вертц Дж., Болтон Дж., Теория и практические приложения метода ЭПР.— М.: Мир, 1975.
50. Hyde J. 8., Electron-Electron Double Resonance, Varian Associates Reprint No, 256.
51. Hyde J. S„ Sneed R.C., RistG.H., J. Chem. Phys., 51, 1404(1969).
РАБОТЫ ОБЩЕГО ХАРАКТЕРА
1. Вертц Дж., Болтон Дж., Теория и практические приложения метода ЭПР.— М.: Мир, 1975.
2. Abragam A., Bleaney В., EPR of Transition Ions, Clarendon Press, Oxford, England, 1970.
Спектры ЭПР комплексов ионов переходных металлов 251
3. Orton J. IV., Electron Paramagnetic Resonance, Gordon and Breach Publishers, New York, 1968.
4. Golding R.M., Applied Wave Mechanics, Van Nostrand, New York, 1969.
5. Griffith J. S., The Theory of Transition-Metal Ions, Cambridge University Press, New York, 1961.
6. Керрингтон А., Мак-Лечлан Э., Магнитный резонанс и его применение в химии.— М.: Мир, 1970.
7. Пул Ч., Техника ЭПР-спектроскопии.— М.: Мир, 1970.
8. McGarvey В. R., Electron Spin Resonance of Transition Metal Complexes, in Transition Metal Chemistry, Vol. 3, pp. 89 — 201, ed. Carlin R. L., Marcel Dekker, New York, 1967.
9. Kokoszka G„ Gordon G., Technique of Inorganic Chemistry, Vol. 7, pp. 151—271, ed. Jonasse H. B., Weissberger A., Interscience, New York, 1968.
10. Goodman B.A., Raynor J.B., Adv. Inorg. Chem. Radiochem., 13, 135(1970).
11. Bersohn R., Determination of Organic Structures by Physical Methodes, Vol. 2, ed. NachodF. C., Phillips W. D., Academic Press, New York, 1961.
12. Carrington A., Quart. Rev., 17, 67 (1963).
13. Carrington A., Longuet-Higgins H. C., Quart. Rev., 14, 427 (1960).
Упражнения
1. Ниже представлен типичный спектр раствора комплекса Си2 + , который можно охарактеризовать g = 1/3 рц + 2/3 д± и А = 1/3 Лц + 2/3А±.
а. Определите д и А.
б. Этот спектр снят при частоте X-диапазона (9,4-109 Гц). Почему А необходимо выражать в герцах?
252
Глава 13
2. Представленный ниже спектр ЭПР раствора типичен для комплексов ванадила (для V I = 7/2). Объясните характер расщепления.
3. Студенты часто с трудом понимают, почему спектр ЭПР замороженного или порошкообразного образна должен давать (например, для аксиальносимметричных соединений) рц, д., Ац и Л±. Они говорят: «Вы видите суперпозицию всех возможных ориентаций. При промежуточных ориентациях у вас есть средние д и А, например д2 =zgl2cosz0-t-gi2sinz0. Таким образом, спектр должен быть очень широким с некоторыми особенностями». Этот аргумент корректен постольку поскольку, но при этом студенты забывают, что мы наблюдаем первую производную. Например, допустим, что соединение характеризуется gt = 2,0 и д: = 2,1. Если вспомнить, что здесь значительно больше перпендикулярных направлений, чем параллельных, то окажется, что спектр поглощения в идеальном случае должен выглядеть как на А :
Поскольку линия уширяется, на самом деле кривая поглощения больше похожа на Б. Тогда первая производная будет иметь вид
Далее ситуация несколько осложняется за счет сверхтонкого взаимодействия, но существо дела не меняется.
Сколько будет наблюдаться линий в изображенном ниже спектре ЭПР порошкообразного образца диэтилдитиофосфината меди (для 63Cu I = 3/2)? Указание: Л и (Си) « 5Л±(Си). Какие другие ядра в состоянии дать сверхтонкую структуру?
Спектры ЭПР комплексов ионов переходных металлов
253
4. Ниже представлены спектр замороженного раствора (вверху) и спектр при комнатной температуре (внизу) комплекса ванадила d, /-тартрата (в наличии только один тип частиц). Что вы можете сказать о структуре этого комплекса? Винная кислота имеет формулу
Н Н
I I
ноо-с-с-соон
I I
он он
254
Глава 13
5. Дифенилпикрилгидразил (ДФПГ) относится к ненных стандартов в спектроскопии ЭПР. Он
числу наиболее распростра-имеет структуру
При концентрации 10 3 М в растворе ксилола спектр ДФПГ имеет вид
Спектры ЭПР комплексов ионов переходных металлов
255
Чем обусловлено наблюдаемое в спектре расщепление? Почему его наблюдают только в разбавленном растворе?
6. Ниже приведен спектр фталоцианина кобальта (для Со I = 7/2), 04/1-порфирин Со (II) аналогичен по структуре порфирину меди в предыдущей задаче.
1400 /700 2000 2300 2600 2900 3200 3500 3800 4/00
Спектр ЭПР фталоцианина ft-кобальта, магнитно разведенного в порошке фталоцианина, свободного от ^-металла прн 77 К.
а. Каковы значения Зц, з_, Лц и А±? Каково происхождение наблюдаемой сверхтонкой структуры?
б. Растворение фталоцианина кобальта в 4-метилпиридине приводит к образованию аддукта состава 1 :1. Спектр замороженного образца показан ниже. Используя диаграмму расщепления D4>, для низкоспинового комплекса Со(П), укажите, какую орбиталь занимает неспаренный электрон? Почему каждая из восьми компонент в сильном поле расщепляется на три линии?
Спектр ЭПР фталоцианина кобальта в растворе 4-метилпиридина (77 К).
в. Почему вы видите сверх-сверхгонкое взаимодействие с атомом азота пи-ридина, но не с четырьмя атомами азота—донорами фталоцианина?
7. Теграфенилпорфирин меди (II) (СпТФП, см. структуру Г ниже) имеет квадратно-плоскостную структуру D4h, где атом меди лежит в плоскости четырех эквивалентных атомов азота порфириновой группы. Спектр ЭПР образца Си1Ф11, внесенною в свободный лиганд, показан ниже. Этот образец содержит 100% 63Сп. Естественное содержание меди таково: 69% 63Сп и 31% 65Сп; для обоих изотопов I = 3/2.
а. Спектр А представляет собой спектр порошкообразного образца, полученный при частоте 9,4 ГГц (А'-диапазон), в то время как спектр Б полу-
256
Глава 13
чен при 35 ГГц (Q-диапазон). Почему Зц и лучше разделяются на Б, чем на Л?
б. Объясните источник большого числа полос, наблюдаемых в области gt спектра Б, а также характер расщепления, наблюдаемого для каждой полосы (см. спектр В).
в. Какова диаграмма расщепления кристаллическим полем для квадратноплоскостного комплекса? Как характер расщепления на спектре В согласуется с занятостью орбиталей, предсказываемой для комплекса Си (II) вашей диаграммой расщепления кристаллическим полем?
г. Если ваше соединение меди не имеет оси симметрии третьего или более высокого порядка, то на что должен быть похож спектр ЭПР порошкообразного образца?
д. Если использовать соединение меди с естественным содержанием изотопов, то как это должно сказаться на характере спектра В?
Спектры ЭПР комплексов ионов переходных металлов
257
Отметим, что усиление сигнала в низком поле приводит к сигналу В.
8. Предскажите число спектральных линий для
а. Со(Н2О)|+,
б. Сг(Н2О)| + .
Укажите, как расщепление в нулевом поле и крамерсово вырождение находят применение в этих примерах.
9. Следующие данные исследования методом ЭПР ряда высокоспиновых октаэдрических комплексов гексафторидов металлов взяты из работы Ргос. Roy. Soc. (London), 236, 535 (1956):
Комплекс g ^металл 'Ю4, CM 1 ^F104, CM ^металл Температура, К
MnF*" gx=gy=gz=2,<x> 96 17 55Мп = 5/2 300
CoFf gx=2,6 Лх = 43 Лх = 20 59Со = 7/2 20
gy=6,Q5 Лу = 217 Л, = 32
0г = 4,1 Л2 = 67 Л2 = 21
CrF3" 0 = 2,00 Л = 16,2 Л = 3 53Сг = 3/2 300
3 = 1,98 Л = 16,9 Л = 1
а. Почему комплексы CrF|“ и MnFg- дают узкие линии ЭПР при комнатной температуре, тогда как для CoF^- такие линии наблюдаются только при 20 К? Каким эффектом (или эффектами) обусловлена необходимость регистрировать спектр ЭПР при таких разных условиях? Какой из этих комплексов лучше всего исследовать методом ЯМР при комнатной температуре?
б. Почему з-факторы для CrF^ и MnF* изотропны и близки к 2,0, в то время как для CoF*- они анизотропны и отличаются от 2,0?
в. Почему CoFg- и MnF£_ имеют большую Лг, чем CrF^-?
10. В ноябрьском выпуске 1973 г. журнала “Inorganic Chemistry” имеются спектры ЭПР жидкого и замороженного растворов, некоторых комплексов
17-574
258
Глава 13
ванадилтиофосфината при концентрации 10 3 М.
О
а. Объясните спектр раствора вещества, для которого R = CH3. (Нижний спектр рассчитан ЭВМ.) Какие параметры нужно, чтобы охарактеризовать его? Спины V, Р и Н равны соответственно 7/2, 1/2 и 1/2.
б. Объясните спектр замороженного раствора, для которого И = фенил. Какие параметры нужны, чтобы охарактеризовать его?
в. Каким должно быть математическое соотношение между параметрами спектра в п. «а» и параметрами в п. «б».
11. Ниже представлен спектр аксиально-симметричного комплекса Сп2+ в замороженном растворе, который представляет собой ^-систему. Предполагается, что все ядра меди относятся к изотопу 63Сп (/ = 3/2). Могут понадобиться следующие константы: р = 9,27-10~ 23 эрг/Э, h = 6,67-10 “ 27 эрг • с и v = 9,12 109 Гц (частота, при которой снят спектр).
а. Сколько параметров нужно для объяснения спектра? Из каких характеристик это следует?
б. Почему половина спектра в сильном поле (область J.) имеет большую интенсивность ?
в. Каково абсолютное значение Зц? Обоснуйте использование вами членов взятого уравнения.
12. Предполагается, что квадратно-пирамидальный комплекс Cu(hfac)2P(C6H5)3 (для 31Р I = 1/2; для 63Cu I = 3/2) может существовать в одной из следую
Спектры ЭПР комплексов ионов переходных металлов_____________259
щих изомерных форм [Inorg. Chem., 13, 2517 (1974)]:
Pfc6H5)3 о-_
О I °х I О
(wnf) (;си(п/
О О ОХ Р(С6Н5)5
аксиальный базальный.
Какой из указанных ниже спектров может вам помочь различить их? а. Электронный спектр?
б. Инфракрасный спектр или спектр комбинационного рассеяния (отвечайте обычным образом, т. е. спектр не дает полного представления)?
в. Спектр ЭПР?
г. Спектр ЭПР соединения, меченного 17О (для 17О 7 = 5/2)?
д. Спектр ЯМР?
14. СПЕКТРОСКОПИЯ ЯДЕРНОГО КВАДРУПОЛЬНОГО РЕЗОНАНСА (ЯКР) *
14.1. ВВЕДЕНИЕ
Если ядро с квадрупольным электрическим моментом (ядерный спин 7 > 1; см. разд. 7.2 и рис. 7.1) находится в неоднородном электрическом поле, являющемся следствием асимметрии электронного распределения, то может возникнуть градиент электрического поля (см. ниже). Квадру-полъное ядро будет взаимодействовать с этим градиентом электрического поля в различной степени в зависимости от различных возможных ориентаций эллиптического квадрупольного ядра. Поскольку квадру-польный момент возникает в результате несимметричного распределения электрического заряда в ядре, нас будет больше интересовать электрический квадрупольный момент, нежели магнитный момент. Число разрешенных ядерных ориентаций определяется ядерным магнитным квантовым числом т, которое принимает значения от +1 до — 7 (всего 27 + 1). Низший по энергии уровень квадруполя соответствует ориентации, для которой наибольшая величина положительного ядерного заряда располагается ближе всего к наибольшей плотности отрицательного заряда в электронном окружении. Разности энергий различных ориентаций не очень велики, и при комнатной температуре в группе молекул существует распределение ориентаций. Если электронное окружение ядра является сферическим (как в С1 “), то все ядерные ориентации эквивалентны и соответствующие энергетические состояния квадруполя вырождены. Если сферическим является ядро (7 = 0 или 1/2), то энергетических состояний квадруполя не существует. В спектроскопии ЯКР мы изучаем разности энергий невырожденных ядерных ориентаций. Эти разности энергии обычно соответствуют радиочастотному диапазону спектра, т.е. от ~ 0,1 до 700 МГц.
Для того чтобы определить некоторые термины, которые имеют важное значение для ЯКР (и мёссбауэровской спектроскопии, гл. 15), полезно рассмотреть взаимодействие зарядов, диполей и квадруполей с плотностью отрицательного заряда. На рис. 14.1, Л показано взаимодействие положительного заряда, находящегося на оси z, с отрицательной электронной плотностью. Энергия выражается как — е2/г или — eV, где V( = —е/г)—электрический потенциал в точке г, в которой находится положительный заряд. На рис. 14.1,Б показан диполь, находящийся в по-
* Некоторые из принципов, описанных в разд. 14.1—14.4, имеют общее значение как для спектроскопии ЯКР, так и для мёссбауэровской спектроскопии; поэтому эти разделы нужно прочитать перед тем, как приступить к гл. 15.
Спектроскопия ядерного квадруполъного резонанса
261
ле, создаваемом электроном. Теперь энергия, связанная с ориентацией диполя, зависит от изменения потенциальной энергии вдоль диполя. Таким образом, нас интересует, как меняется вдоль диполя электростатический потенциал, или производная dV/dz, называемая также z-komho-нентой электрического поля, £z. На рис. 14.1, В показано взаимодействие электрического поля, создаваемого электроном, с квадруполем,
>г
Рис. 14.1. Взаимодействие положительного заряда (А), диполя (Б) и квадруполя (В) с z-компонентой электрического поля, создаваемого единичным отрицательным зарядом.
который можно представить в виде двух диполей с фиксированными одна относительно другой конфигурациями. Энергия такого взаимодействия зависит от скорости изменения (или градиента) электрического поля вдоль квадруполя. Таким образом, мы имеем дело с «изменением изменения» потенциала электрона, или второй производной V но z, т. е. d2V/dz2. Этот параметр, который в то же время представляет собой изменение компоненты электрического поля dEJdz, называется градиентом электрического поля.
В рассматриваемой молекуле ядро находится в облаке электронной плотности. Электрический градиент определяется через усредненный по времени электрический потенциал, создаваемый электроном. Кроме того, градиент электрического поля описывается симметричным тензором V 3 х 3, след которого равен нулю. Ядерный квадрупольный момент также описывается тензором Q 3 х 3. Энергия взаимодействия ядерного квадруполя Eq выражается как
£e = |QuVM,
262
Глава 14
где Qjj—тензор ядерного квадрупольного момента, a Vy—тензор градиента электрического поля. Произведение зависит от взаимной ориентации осей двух систем координат. Для Q удобно выбирать систему координат, оси которой совпадают с осями координат спиновой системы. При таком выборе цилиндрическая симметрия ядра позволяет определить тензор через один параметр — ядерный квадрупольный момент Q. Для того чтобы определить знак градиента электрического поля, необходимо знать знак ядерного квадрупольного момента. Можно также выбрать систему координат, в которой тензор градиента электрического поля диагоналей. Это так называемая система главных осей тензора градиента электрического поля, и его единственными ненулевыми элементами являются диагональные элементы, величины которых дают тензор со следом, равным нулю:
д2у д2у а2 к =0
дх2 ду2 dz2
При сравнении данного атома в различных молекулах необходимо знать ориентацию системы главных осей градиента электрического поля в молекулярной системе координат, для чего нужны три угла Эйлера (а, Р и у).
Асимметрию молекулы и направление оси z градиента поля qzz относительно осей кристалла можно исследовать, рассматривая спектр ЯКР монокристалла, находящегося в магнитном поле. Зеемановское расщепление зависит от ориентации, и детальный анализ спектров для различных ориентаций монокристалла дает возможность определить направление оси z градиента поля qzz. Эту ось можно сравнить с осями кристалла.
Если главные оси системы координат молекулы совпадают с главными осями тензора градиента электрического поля, то потенциальная энергия Eq взаимодействия квадрупольного момента с электрическим полем на ядре выражается как
е
Eq =—(VXXQXX+VyyQyy+V2zQzz)- (14.1)
Мы определяем градиент электрического поля Vzz как eqzz, где е — заряд электрона (4,8 10 ~ 10 эл.-ст. ед.). Поскольку след тензора градиента электрического поля равен нулю, для получения градиента поля необходимо определить еще только один параметр, что осуществлено в уравнении
V - у
(14.2)
'ZZ
Параметр ц называется параметром асимметрии. Величины Ухх, Ууу и Vzz часто записывают как eqxx, eqyy и eqzz. По условию, |q„| > |qxx| > 1^1, поэтому т| меняется от 0 до 1. Если след тензора равен нулю, то градиент поля полностью определяется eq и г|.
Спектроскопия ядерного квадрупольного резонанса
263
Подстановка выражений qzz и р в уравнение (14.1) и использование того факта, что qzz + qxx + qyy = 0, дают
г- _ Й2<? 1
|(П - 1)6хх - ^(П + VQyy + Q:
(14.3)
При аксиальной симметрии р равен нулю, и Eq становится равной (1/4) х х e2qQzz. (Отметим, что Qzz + Qxx + Qyy = 0.)
Рассмотренный выше классический подход можно перевести на квантовомеханический язык с помощью оператора
(14-4)
где суммирование производится по всем компонентам ядерного квадрупольного момента Q;j и градиента электрического поля qif В системе главных осей при р, определенном выше, наиболее распространенной формой гамильтониана является
Не =
4/(2/— 1)
3/2-/(/+ 1)-^-(/2+ +?_)
(14.5)
Произведение e2Qq или e2Q<y/7i (часто записываемое как eQqzz или eQ//zz/7i) называют константой квадрупольного взаимодействия. Оператор Hq действует на ядерные волновые функции. Если р = 0, то член, включающий операторы сдвига, опускается. Мы не будем заниматься точным расчетом матричных элементов; интересующийся этим вопросом читатель может обратиться к работам [1—3]. Достаточно сказать, что для получения энергий ядерных спиновых состояний в градиенте электрического поля, обусловленном распределением электронной плотности в молекуле, можно записать ряд секулярных уравнений и решить их.
14.2. ЭНЕРГИИ КВАДРУПОЛЬНЫХ ПЕРЕХОДОВ
В аксиально-симметричном поле (р = 0) энергии различных состояний ядерного квадруполя можно выразить уравнением
e2Qq [Зт2 -1(1 + 1)] = -----------------
т 4/(2/-1)
где I — ядерное спиновое квантовое число, а т — ядерное магнитное квантовое число. Для спина ядра 1 = 3/2 ядерное магнитное квантовое число т может принимать значения 3/2, 1/2, — 1/2 и — 3/2. Для т = 3/2 подстановка в уравнение (14.6) приводит к Е3/2 = + e2Qq/4. Поскольку т возводится в квадрат, величина энергии при т = — 3/2 идентична величине энергии при т = 3/2, результатом чего является дважды выро
264
Глава 14
жденный набор энергетических состояний квадруполя. Аналогично состояние с т = ±1/2 также двукратно вырождено. Энергия перехода АЕ, указанного на рис. 14.2 стрелкой, соответствует e2Qq/4 — (— e2Qc//4) = = e2Qq/2. Таким образом, для ядра со спином I = 3/2 в аксиально-симметричном поле следует ожидать одного перехода, а величина e2Qq, выраженная в единицах энергии, может быть рассчитана непосредственно
Е = +esQq/4
Е = -ezQq/4
Рис. 14.2. Энергетические уровни квадруполя в сферическом поле (А) и в аксиально-симметричном поле (Б).
из частоты поглощения: e2Qq = 2АЕ = 2/iv. Величина e2Qq часто выражается в единицах частоты (в МГц), хотя, строго говоря, для этого ее надо разделить на h. Для приведенного выше случая величина e2Qq должна вдвое превышать частоту перехода ЯКР.
Таким же образом, используя уравнение (14.6), можно рассчитать число переходов и частоту перехода (в единицах е22<?) для других ядер с различными значениями 1 в аксиально-симметричном поле. При I = = 7/2 будет четыре энергетических уровня (E+i/г, Е±3/2, Е±5/2 и Е±7д) и три перехода. Правило отбора для этих переходов суть Лт = + 1, поэтому наблюдаются переходы E±i/2 ->Е±з/2, Е±з/2->Е±5/2 и Е ±5/2 ->Е ±7/2. (Напоминаем, что при обычных условиях все уровни заселены.) При подстановке I и т в уравнение (14.6) получается, что энергия перехода Е ±з/2 ->Е ±5/2 вдвое превышает энергию перехода Е±1/2->Е±з/2. Энергии указанных уровней и влияние на них параметра асимметрии г| показаны на рис. 14.3. Отклонения от предсказанных частот в наблюдаемом спектре, когда т] = 0, приписываются отклонениям от аксиальной симметрии в образце, и, как это будет видно в дальнейшем, их можно использовать в качестве меры асимметрии.
В эксперименте ядерного квадруполъного резонанса (ЯКР) * для воздействия на переходы между различными ориентациями квадрупольно-го ядра в несферическом поле используют излучение радиочастотного диапазона. Эксперимент обычно проводят с использованием порошко-
* Обзоры по ЯКР имеются в общем списке литературы.
Спектроскопия ядерного квадрупольного резонанса
265
образного образца. Различные ориентации маленьких кристаллов относительно направления радиочастотного излучения влияют только на интенсивности переходов, но не на их энергии. Информацию о структуре соединения можно получить, рассматривая, как различные структурные
Рис. 14.3. Диаграмма энергетических уровней ядерного квадруполя для I = 7/2.
и электронные эффекты влияют на асимметрию электронного окружения. Для каждого химически или кристаллографически неэквивалентного квадрупольного ядра ожидается один набор сигналов. Кристаллографические расщепления обычно малы по сравнению с расщеплениями, обусловленными химической неэквивалентностью.
В ЯКР в качестве генераторов-детекторов обычно применяют регенеративные и сверхрегенеративные схемы. Сверхрегенеративная схема наиболее распространена, поскольку позволяет в поисках сигнала сканировать широкие линии и не осложняет работу. Ее недостаток, являющийся одновременно характеристикой ее действия, состоит в возникновении на месте резонансного сигнала мультиплета линий (рис. 14.4,Л). Истинная резонансная частота соответствует центральной линии мультиплета. Регенеративная схема в каждом случае дает единственную полосу, но она требует постоянной настройки, и работать в ней утомительно.
На рис. 14.5 показан спектр ЯКР на ядрах 35С1 соединения С13ВОРС13. В спектре обнаружены сигналы трех неэквивалентных ядер хлора. Два центра резонансных сигналов при 30880 и 31 280 МГц отмечены крестиком. Центр третьего сигнала в области 30950 МГц трудно установить с достаточной точностью из-за перекрывания с сигналом, имеющим центр при 31280 МГц.
Дополнительно к этим прямым измерениям разности энергий уровней квадруполя за счет поглощения энергии радиочастотного излучения та же информация может быть получена из тонкой структуры в чистом вращательном (микроволновом) спектре газа. Различные ядерные ориентации дают несколько различающиеся по величине моменты инерции, что приводит к тонкой структуре в микроволновом спектре. Могут быть проведены непосредственные измерения поглощения энергии ра-
266
Глава 14
диочастотного излучения в твердом теле. Столкновения и колебания в жидкости или даже в некоторых твердых веществах (особенно вблизи точки плавления) модулируют градиент электрического поля до такой степени, что время жизни квадрупольного состояния становится очень небольшим. Это приводит к бесконечному уширению сигнала, и линию часто не удается обнаружить. Подробное описание аппаратурного оформления метода можно найти в литературных источниках [4].
Рис. 14.4. а —мультиплет линий для единственного сигнала при сверхрегенеративиой схеме; Б—единственная линия при регенеративной схеме для того же сигнала, что и в Л.
Разность энергий между различными уровнями и, следовательно, частота перехода зависят как от градиента поля q, создаваемого валентными электронами, так и от квадрупольного момента ядра. Ква-друпольный момент eQ является мерой отклонения распределения электрического заряда ядра от сферически симметричного. Для данного изотопа величина eQ постоянна, и для многих изотопов она может быть получена из различных источников [5, 6]. Величина eQ может быть измерена в экспериментах с атомными пучками. Размерностью eQ является заряд, умноженный на квадрат расстояния, но чаще квадру-польный момент выражают через Q * в см2. Например, квадрупольный момент Q ядра 35С1 с ядерным спином 1 = составляет —0,08-Ю-24 2 ~
см ; отрицательный знак указывает на то, что распределение заря-да сжато относительно оси спина (см. рис. 7.1).
Вторым фактором, определяющим степень расщепления энергетических уровней квадруполя, является градиент поля q на ядре, вызванный электронным распределением в молекуле. Расщепление уровня квадруполя связано с произведением e2Qq. Для молекулы с аксиальной симметрией q часто лежит вдоль оси симметрии высшего порядка, и если известна величина eQ, то можно определить значение q. В несимметричном окружении энергии различных уровней квадруполя уже не выражаются уравнением (14.6), поскольку необходимо использовать полный гамильтониан уравнения (14.5). В случае 7 = 3/2 для энергий двух состояний можно вывести [2] следующие уравнения:
3e2e<?j/l +П2/3
£±3/2= ^(2/^1)^ (14'7)
Q часто измеряют в барнах; 1 барн = 10 24 см2.— Прим, перев.
Рис. 14.5. Спектр ЯКР на ядрах 35С1 соединения С13ВОРС13 при 77 К с маркировкой через 25 кГц.
268
Глава 14
и
_ — 3eЕ 2Qg|/T+г|2/3
±1/2 4/(2/-1)
(14.8)
Из разности уравнений (14.7) и (14.8) с учетом Е = hv видно, что для I = = 3/2 ожидается один переход с частотой v = (e2Qq/2h}]/ \ + г\2/3. Поскольку имеются два неизвестных г] и q, из измеренной частоты нельзя определить величину e2Qq. Как будет показано, эту задачу можно решить, используя результаты исследования методом ЯКР в слабом магнитном поле.
Известны уравнения, с помощью которых можно рассчитать энергию уровней ядер, чей спин отличается от 3/2. Во многих случаях, когда в спектре наблюдается больше чем одна линия, из него можно получить и параметр асимметрии г], и e2Qq. Для I = 1 такими уравнениями являются
- 2e2Qq ° 47(27—1)
и
Е±1 =
е26</(1 +п) 47(27—1)
(14.10)
Соответствующие энергетические уровни изображены на рис. 14.6,6, где К обозначает e2Qq/4I(2I — 1). На рис. 14.6,В показано возмущение этих уровней под действием приложенного магнитного поля. Этот
т = ±1
Е = КО -71)
Е = Kft + jj)
т = О
Е = —2К
Рис. 14.6. Энергетические уровни для квадрупольного ядра с I = 1 при различных условиях.
Д) Г] = 0 и приложено магнитное поле Но = 0; Б) г] /0 и Яо = О; В) д/0 и Яо/О, но постоянно.
Спектроскопия ядерного квадрупольного резонанса 269
эффект будет рассмотрен в следующем разделе. Как видно из рис. 14.6,Б, для 1=1 и г| + 0 имеются три перехода (помеченных как v+, v_ и v0), поэтому из спектра можно определить два неизвестных е2(Х? и т|. Энергии уровней на рис. 14.6 описываются уравнениями (14.9) и (14.10). Энергии v+ и т. д. находят по разности энергий уровней. Для любых двух переходов результирующие уравнения могут быть решены относительно двух неизвестных e2Qq и т|. Интересно отметить, что для ядра с I = 1 в аксиально-симметричном поле в спектре следует ожидать появления только одной линии (см. рис. 14.6, Л).
Энергии уровней квадруполя как функция т| были рассчитаны [2] для случаев, когда I отличается от 1 или 3/2. Составлены [3] таблицы, с помощью которых можно рассчитать ц и e2Qq из спектральных данных для ядер с 7 = 5/2, 7/2 и 9/2. Если величина ц значительна, то правило отбора Ат = + 1 нарушается и в спектре часто можно наблюдать полосы с Ат = + 2.
14.3. ВЛИЯНИЕ МАГНИТНОГО ПОЛЯ НА СПЕКТРЫ
Если проводить эксперимент ЯКР с образцом, помещенным в постоянное магнитное поле, то к гамильтониану ЯКР необходимо добавить гамильтониан Нм, описывающий взаимодействие магнитного поля с ядерным магнитным дипольным моментом:
Нм= — g,vP,vH-1. (14.11)
Слабое магнитное поле (gN$NH « e2Qq) выступает как возмущение HQ. В общем влияние этого поля на энергии заключается в сдвиге невырожденного уровня квадруполя и в расщеплении двукратно вырожденного уровня [т. е. имеющего m + 0, см. уравнение (14.6)]. Это изменение энергии невырожденных уровней квадруполя показано на рис. 14.6, Б и В для ядра, в котором 1=1 и ц 0.
Для ядра с I = 1 квадрупольный спектр с двумя линиями может возникнуть в случае ц^О, как отмечалось выше (рис. 14.6,Б), или для ядер с г] = 0, находящихся в двух неэквивалентных центрах решетки. Исследование спектра образца, находящегося во внешнем магнитном поле, позволяет различить эти два варианта. В первом случае (ц + 0) снова должны наблюдаться две линии, но с энергиями, отличающимися от тех, что наблюдались в отсутствие поля; во втором случае каждый двукратно вырожденный уровень должен расщепляться, приводя к спектру с четырьмя линиями.
Как упоминалось ранее, для ядра с 1 = 3/2 каждый из уровней Е±1/2 и Е±з/2 двукратно вырожден в несимметричном поле. Как следствие этого, e2Qq и ц нельзя определить непосредственно. Это вырождение устраняется магнитным полем, в результате чего появляются четыре уровня. В спектре наблюдаются четыре перехода: +1/2->+3/2, + 1/2->—3/2, —1/2->+3/2 и —1/2->—3/2 (А|т| = + 1). Разности энергий, соответствующие этим переходам, зависят от Н, e2Qq и ц [как показывают уравнения (14.7) и (14.8)], поэтому для этой системы можно
270
Глава 14
рассчитать q и г] из спектров образца в отсутствие и при наличии приложенного поля [7].
14.4. СВЯЗЬ МЕЖДУ ГРАДИЕНТОМ ЭЛЕКТРИЧЕСКОГО ПОЛЯ И МОЛЕКУЛЯРНОЙ СТРУКТУРОЙ
Рассмотрим теперь, как можно получить информацию об электронной структуре молекулы из величин q и г]. Градиент поля на атоме А в молекуле и электронная волновая функция связаны уравнением
4moiA = ?{ X [ZB(3cos20AB - 1)/ К’в] - f Ф*Х[(Зсо820а, - 1)/^J y\idt}.
В / А п
(14.12)
Как видно из этого уравнения, градиент поля в молекуле gmol является чувствительной мерой плотности электронного заряда в непосредственной близости от ядра, поскольку уравнение (14.12) включает величину ожидания <1/г3>. В первом члене суммирование проводится по всем ядрам, окружающим квадрупольное ядро, а во втором члене — по всем электронам. При известной молекулярной структуре первый член рассчитать легко. ZB обозначает заряд ядра любого атома в молекуле, отличающегося от ядра А, градиент поля на котором исследуется; 0АВ— угол между осью связи или осью вращения высшего порядка для А и радиус-вектором КАВ, связывающим А с В. Второй член представляет собой градиент поля в молекуле, создаваемый электронной плотностью, и называется градиентом электрического поля qci. Наконец, ф—волновая функция основного состояния и 0А|> — угол между связью или главной осью и радиус-вектором гАп для и-го электрона. Этот интеграл взять трудно. В приближении ЛКАО можно написать
осс
= - 2е X ££ Ciu /г, (14.13)
« i J
где <?А = (3cos20 — 1)/г3, и—верхний индекс молекулярной орбитали, а i и j—индексы атомных орбиталей. Ciu и CJu обозначают коэффициенты ЛКАО атомных орбиталей ф, и ф7 в молекулярной п-орбитали. Интеграл может быть одно-, двух- и трехцентровым. Очевидно, для расчета нужны хорошие волновые функции, а для интерпретации q необходимо обратиться к расчетам с помощью уравнения (14.13). Для некоторых атомов трехцентровые вклады малы (например, для N и С1), и ими можно пренебречь. Разделяя одно- и двухцентровые члены в уравнении (14.13) и обозначая интеграл j ф^дф;4т как q‘£ и т. д., можно переписать уравнение (14.13) в виде уравнения
А АВ/А ВА
^Ае1= + X ади^ + 2Е
и ' i j и j (14.14)
Спектроскопия ядерного квадрупольного резонанса
271
Существует несколько полуэмпирических методов расчета градиента электрического поля. Коттон и Харрис [8] предположили, что ядерная часть уравнения (14.12) (т. е. первый член) может сокращаться с частью обусловленной суммарной атомной заселенностью на соседнем атоме В, т. е.
е у ZB(3cos20AB- 1) - ^АВ
В#А А В#А
2Z I с?^+2ХХ Z ciucjtJrt и j и i j
’(14.15)
е
Это приводит к уравнению А А В^А
<1™.= -е[2££С?ц<й + 2££ X CiuCjuq‘£]. и i и i j
(14.16)
Далее, предполагая, что двухцентровый интеграл может быть составлен в таком виде, что он пропорционален интегралу перекрывания
(14.17)
можно написать
А А А
<?moi= -e£<?A(2£C,2„ + 2£ X С;„С;Лц)- (14.18)
1 и и j
Видно, что градиент поля получается путем умножения q‘r( на суммарную атомную заселенность Р [уравнение (3.31)], или
А
<1то1 = еР У, Ф;
3cos2 0—1
Фг
(14.19)
Для атома А с валентными s- и р-орбиталями суммирование проводится по четырем орбиталям, для которых qA в случае s-орбитали равно нулю и
/ 1 1 \
« = -е«а,|Рг-
где q3t — градиент поля, создаваемый одним электроном, находящимся на р-орбитали. Величина в скобках в уравнении (14.18) представляет собой в точности суммарную атомную заселенность Р атомной орбитали ф; (см. гл. 3), выраженную через молекулярные орбитали. В зависимости от относительных заселенностей рл-, ру- и рг-орбиталей величина q может быть положительной или отрицательной. Если константа квадрупольного взаимодействия выражена в мегагерцах, то имеем
e2Qq = _{ е26<?а1 \ (р _ 1р _ 1Р \ h \ h 1 2 х 2’’
(14.20)
где e2Qq/h — константа квадрупольного взаимодействия, а e22<?at//» — константа взаимодействия с одним электроном, находящимся
272
Глава 14
на р-орбитали. Для того чтобы можно было применить эти уравнения с большей точностью, рг-орбиталь должна совпадать с q.z.
Уравнение (14.20) представляет собой МО-аналог выражения валентных схем Таунса и Дейли [9], о котором сообщалось ранее. Их подход основывается на следующих аргументах. Поскольку s-орбиталь сферически симметрична, электронная плотность на этой орбитали не создает градиента поля, а поскольку электроотрицательность исследуемого атома не является минимальной по сравнению с другими атомами в молекуле, максимальный градиент поля на этом атоме <?то1 представляет собой атомный градиент поля qM, создаваемый одним электроном, находящимся на рг-орбитали изолированного атома. Если исследуемый атом более электроотрицателен, чем атом, с которым он связан, атом с квадрупольным ядром окружен в молекуле большей электронной плотностью, чем изолированный атом. Электронная «занятость» р-орбиталей атома с квадрупольным ядром в молекуле e2QqM и параметр е22</то1 (который определяется из спектра ЯКР рассматриваемой молекулы) связаны соотношением
е2е<?т01 = [1 - s + d - i (1 - s - </)] e2Q9at, (14.21)
где e2Qqat—константа квадрупольного взаимодействия, если р-орбиталь занята одним электроном, s — доля «-характера атома с квадрупольным ядром с соседним атомом, d—доля ^-характера этой связи, I—доля ионности связи (для молекулы А — В = сфд + «/фв и i = с2 — d2). Если исследуемый атом электроположителен, то i меняет знак. Для некоторых атомов величины e2QqM и qat имеются в таблицах [4, 5, 10, 11]. Если возможно л-связывание, то этот эффект также необходимо учесть. Используется также другой вариант уравнения (14.21) [12]:
e22<?moi = (1 - « + d - i - л)е22^а1, (14.22)
где п — степень л-связывания, а другие символы имеют тот же самый смысл, что и ранее.
С увеличением ионности связи электронное окружение приобретает сферическую симметрию (где qmol = 0) и величина e22qmol уменьшается. Как показывает уравнение (14.22), гибридизация р-орбитали с s-орби-талью приводит также к уменьшению е22«?то1. Смешивание s-орбитали с р-орбиталью снижает градиент поля, поскольку s-орбиталь сферически симметрична. В ковалентной молекуле увеличение градиента поля вызвано вкладом «/-орбиталей в связывание.
Существует несколько проблем, связанных с этими подходами к интерпретации градиентов поля. Во-первых, если даже указанный выше подход корректен в случае интересующей нас системы, то в уравнение (14.22) входят четыре неизвестных, а мы имеем только одну измеряемую величину, е22«?то1. Исследователи, пытающиеся интерпретировать эту величину, вынуждены, таким образом, предполагать ответ и давать разумное объяснение данным, полученным на основе предложенных моделей. Во-вторых, О’Конски и Ха [13] показали, что допущение Котто
Спектроскопия ядерного квадрупольного резонанса
ТП
на и Харриса [8], введенное в уравнение (14.15), в общем случае не является правильным. Если это уравнение неприменимо, то уравнения (14.20)—(14.22) также неверны.
Сообщалось о полуэмпирическом подходе к интерпретации градиента поля [14], в котором не делается описанных выше сомнительных приближений. Этот метод подробно обсуждается в оригинальной литературе.
Еще одним фактором, осложняющим квантовомеханические расчеты и интерпретацию параметров мёссбауэровской спектроскопии (см. ниже) и спектроскопии ЯКР, является эффект Штернгеймера [15]. Этот эффект состоит в поляризации первоначально сферически симметричной внутренней оболочки электронов валентными электронами. Если электронная оболочка теряет сферическую симметрию, то электроны этой оболочки дают вклад в градиент поля на ядре. Подобно спин-поляризации (обсуждается в гл. 12), этот эффект является артефактом, обусловленным тем, что расчет всего кристалла по методу молекулярных орбиталей неполный. Существуют два вклада в экранирование Штернгеймера, и их можно продемонстрировать, если рассматривать лиганды или ионы, окружающие электронную плотность атома, в виде точечных зарядов. Сферическое расширение электронной плотности оболочки показано для положительных точечных зарядов на рис. 14.7, А и Б, а возникающая асимметрия—на рис. 14.7, В. Мы искусственно нарушаем поляризацию, чтобы продемонстрировать эти два вклада в экранирование Штернгеймера.
Экранирование для систем с заполненной оболочкой обычно выражается с помощью формального уравнения
е22<?оьз = (1 - y^e2Qq0,
где e2Qq0— градиент поля, рассчитанный в отсутствие каких-либо эф-
Рис. 14.7. Схематическая иллюстрация эффекта Штернгеймера.
А.—сферическая оболочка электронной плотности; Б—радиальное расширение, обусловленное электрическими зарядами, находящимися вне оболочки; В—эллиптическая поляризация под действием двух положительных зарядов.
18-574
274
Глава 14
фектов Штернгеймера, и угг— параметр экранирования i-ro заряда, находящегося на расстоянии г от ядра. Знак ун зависит от расстояния. Для зарядов, расположенных вне электронной плотности оболочки центрального атома, угг обычно отрицателен; в этом случае используют термин антиэкранирование. Для зарядов, находящихся внутри орбита-лей валентных электронов центрального атома, знак yrj обычно положителен, и в этом случае говорят об экранировании. Константы экранирования были рассчитаны для зарядов, находящихся вне оболочек большого числа ионов; для их обозначения используют символ у^. Величину yrj, фигурирующую в расчетах эффекта Штернгеймера в молекулах, определить трудно, но, вероятно, она значительно ниже у^. Многие исследователи, работающие в области ЯКР, предполагают, что величина эффекта Штернгеймера постоянна в ряду аналогичных комплексов.
14.5. ПРИМЕНЕНИЯ
Известны спектры ЯКР многих молекул, в которые входят следующие ядра: 27А1, 75As, 197Au, 10В, 1 ’В, 135Ва, 137Ва, 209Bi, 79Вг, 81Вг, 43Са, 35С1, 37С1, 59Со, 63Cu, 69Ga, 71Ga, 2Н, 201Hg, 127I, 115In, 25Mg, 14N, 55Mn, 23Na, 93Nb, 17O, 185Re, 187Re, 33S, 121Sb, 123Sb и 181Ta.
Интерпретация данных no e2Qq
Как упоминалось ранее, нельзя строго интерпретировать величины e2Qq через ионность связи, л-связывание, s- и ^-гибридизацию. Как показано в табл. 14.1, этот эффект можно четко различить для соедине-
Таблица 14.1
Величины e2Q</moi Для некоторых двухатомных галогенидов
Молекула * e2£>4mol Молекула e22«moi
FC1 -146,0 BrCl 876,8
ВгС1 -103,6 LiBr 37,2
га -82,5 NaBr 58
тга -15,8 DBr 533
ка 0,04 FBr 1089
RbCl 0,774 DI -1827
CsCl _ 3 Nal -259,87
а Величина e12Qqmei для 35С1, 79Вг и 1271 равна соответственно — 109,74, 769,76 и —2292,84; e2Qqmni относится к подчеркнутым атомам.
ний, ионность связи в которых значительно различается. Более положительные величины e22<?moi наблюдаются для более ионных соединений.
Спектроскопия ядерного квадрупольного резонанса
275
Были сделаны попытки «объяснить» данные по e2Qq для большого числа галогенсодержащих соединений в предположении, что d-гибридизация не имеет значения, и путем оценки ионности связи или «-гибридизации [12, 16].
Если различие в ионности связи достаточно велико, чтобы его можно было предсказать, то величины е22дто! меняются в соответствии с изменением ионности. Однако для систем, в которых различие в ионности связи не столь очевидно, если исходить из электроотрицательности и других соображений, интерпретация различия в e2Qq с точки зрения относительной важности эффектов, учитываемых уравнением (14.22), сомнительна. Одно из наиболее успешных исследований было проведено в ряду замещенных хлорбензолов. Найдено линейное соотношение между константой Гаммета ст и частотой квадрупольного резонанса [17]. Результаты укладываются в соотношение у(МГц) = 34,826 + + 1,024ст. Электроноакцепторные заместители характеризуются отрицательными значениями ст и приводят к меньшим величинам e2Qq, поскольку возросшая ионность связи С — С1 увеличивает отрицательный заряд на атоме хлора.
Общую проблему ковалентности связывания металл—лиганд в ряду комплексов изучали с помощью непосредственного измерения квадрупольного резонанса [18]. Путем сравнения характера изменения силовых постоянных с изменением констант взаимодействия хлорного квадрупольного резонанса была предложена [19] разумная интерпретация данных для целого ряда соединений MCI" Л Строгой интерпретации данных (особенно когда речь идет о небольших изменениях) снова препятствует недостаток информации о четырех переменных в уравнении (14.22).
В спектре ЯКР на ядрах 55Мп (7 = 5/2) наблюдается два перехода, поэтому можно определить как qZ2, так и т). Известно об исследованиях методом ЯКР ряда металлоорганических карбонилов марганца [20J. Замещение метильной группы в ряду (СН3)„С6Н6..пМп(Со)з приводит только к небольшим изменениям параметра асимметрии. Имеется обзор [21], посвященный исследованиям ЯКР на ядрах 59Со. Из величины градиента поля можно сделать вывод о цис- и транс-изомерии октаэдрических комплексов: для [цис-Со (еп)2С12]С1 величина e2Qq составляет 33,71, тогда как для [транс-Со(еп)2С12]С1 — 60,63. Рациональное объяснение этой разницы будет дано в следующей главе, в разделе, посвященном параметрам парциального градиента поля.
Термохимически энергии связей были оценены и сопоставлены с результатами исследования методом ЯКР на ядрах 35С1 соединений GeClg-, SnClg- и PbCig-. Авторы работ [22] высказываются в пользу сделанного ранее вывода [23], что полученные результаты не согласуются с более высокой электроотрицательностью свинца по сравнению с германием и оловом.
Этот обзор результатов исследования методом ЯКР посвящен установлению ясности в ковалентности химических связей и других их характеристиках и ни в коей мере не является полным. Поскольку интер
18'
276
Глава 14
претация результатов сомнительна, изложение было кратким. Для того чтобы выделить переменные в уравнении (14.22), необходимо располагать большей информацией, чем только данными о градиенте поля.
Влииние кристаллической решетки на величину e2Qq
Осложнения и ограничения, связанные с применением спектроскопии ЯКР, обусловлены тем, что непосредственное измерение переходов ядерного квадрупольного резонанса может осуществляться лишь в твердых веществах. Из-за сложности микроволнового спектра больших молекул эти измерения—единственный источник информации о ядерном квадрупольном резонансе. Измерения в твердых веществах осложнены эффектами решетки. В тех случаях, когда молекулы исследовали обоими методами (путем непосредственных измерений и с помощью микроволновой спектроскопии), было обнаружено, что величина e2Qq обычно на 10—15% ниже в твердом веществе. Высказано предположение [5], что это уменьшение вызвано увеличением ионности связи в твердом состоянии. Известно несколько примеров молекул, значительно ассоциированных в твердом состоянии, но не в газовой фазе или в растворе (например, 12 и CNC1). В этих случаях необходимо с большой осторожностью делать выводы относительно молекулярных характеристик на основании величин e2Qq. Величины e2Qq для кобальта в некоторых его комплексах могут проиллюстрировать эту проблему [21]. Величина e2Qq для mpaHC-[Co(NH3)4Cl2]Q и траис-[Со(еп)2С12]С1 равна соответственно 59,23 и 60,63 МГц, т. е. различие составляет 1,40 МГц. В то же время для траис-[Со(еп)2С12]МО3 величина е22<?(Со) равна 62,78 МГц. Изменение аниона меняет e2Qq на 2,15 МГц. Величина е22<?(С1) для K2SnCl6 и [(CH3)4N]2SnCl6 составляет соответственно 15,063 и 16,674 МГц. Высказаны предположения [22в, 22с] относительно возможных причин такого различия. В следующем разделе будет рассмотрено несколько примеров, в которых кристаллическая решетка влияет на число линий, наблюдаемых в спектре.
Структурная информация, получаемая из спектров ЯКР
Поскольку градиент поля на неэквивалентных ядрах в молекуле различается, следует ожидать для каждого из этих ядер свою спектральную линию (или набор линий в зависимости от 7). Вообще окружение атома, установленное методом ЯКР, согласуется с результатами, полученными с помощью рентгеноструктурного анализа. В спектре ЯКР на ядрах хлора для каждого из следующих соединений: K2SeCl6, Cs2SeBr6, (NH4)2TeCl, (NH4)2SnBr6 и K2PtCl6 — обнаружена только одна линия [18].
Как упоминалось ранее, появление мультиплетов в спектрах ЯКР может быть обусловлено следующими эффектами:
1. Химической неэквивалентностью атомов в молекуле.
2. Химически эквивалентными атомами в молекуле, занимающими
Спектроскопия ядерного квадрупольного резонанса 277
неэквивалентные положения в кристаллической решетке твердого вещества.
3. Расщеплением вырождения уровней энергии квадруполя за счет асимметрии градиента поля. Расщепления квадрупольных уровней другими магнитными ядрами в молекуле, аналогичные спин-спиновому расщеплению в ЯМР, в спектрах ЯКР часто не наблюдают, но их можно обнаружить при наличии более совершенной аппаратуры. Обычно величина этих расщеплений меньше или того же порядка, что и ширина линий. Сообщалось [24] о таком расщеплении, наблюдаемом в спектре НЮ3, где сигнал ядра 1271 расщепляется на протоне.
Неэквивалентность положений в решет ке демонстрирует спектр ЯКР на ядрах брома соединения K2SeBr6, в котором при комнатной температуре наблюдается одна линия, а при температуре сухого льда—две. Это различие объясняется изменением в кристаллической фазе. Метод ЯКР — один из наиболее мощных методов определения фазовых изменений и получения структурной информации о фазовых переходах. Хотя соединения K2PtI6, K2SnBr6 и К2ТеВг6 включают «октаэдрические» анионы, в кристаллической решетке галогены неэквивалентны, и все спектры на ядрах галогенов состоят из трех линий, указывая на существование по крайней мере трех различных окружений галогенов.
В спектрах ЯКР на ядрах хлора соединений TiCl4 [25а], SiCl4 [25b] и SnCl4 [25b] обнаружены четыре линии в каждом. На основании этого был сделан вывод, что кристаллические структуры этих веществ похожи.
Проблема отличия неэквивалентных положений в решетке от химической неэквивалентности молекулярной конфигурации (которая связана со структурой молекулы в газовой фазе или в некоординирующемся растворителе) в некоторых случаях трудна. Как правило, различие в частотах линий, обусловленное неэквивалентностью положений в решетке, мало по сравнению с различиями в частотах, связанными с химической неэквивалентностью ядер в молекуле. Если наблюдаются лишь небольшие разделения спектральных линий, то с помощью одного метода трудно установить, с каким из двух эффектов мы имеем дело. Другой тип трудностей, с которыми приходится сталкиваться в методе ЯКР, демонстрирует спектр ЯКР на ядрах хлора соединений М2С110 [26]. Если даже в структуре молекулы Nb2Cl10 содержатся и концевые, и мостиковые атомы хлора, то в спектре наблюдается единственный хлорный сигнал, приписываемый мостиковому атому [26]. В спектрах Re2Cl10 и W2C11O наблюдается большое число линий [26]. Следовательно, аргументы, основанные на отнесении числа различных типов ядер в молекулярной структуре к числу резонансных линий, которые наблюдаются в спектре, могут иметь сомнительную ценность.
Исследования ЯКР на ядрах 14N (/ = 1) трудны, но могут дать очень интересную информацию. Поскольку I = 1, и e2Qq, и параметр асимметрии г] могут быть рассчитаны из спектров ЯКР. В спектре ЯКР на ядрах 14N соединения BrCN сигнал представляет собой дублет, что может быть обусловлено двумя неэквивалентными атомами азота в кри
278
Глава 14
сталлической решетке или расщеплением азотного сигнала из-за асимметрии градиента поля. Первое объяснение устраняется рентгеноструктурным анализом монокристалла [27]. Как было установлено, структура твердого BrCN согласуется с линейными цепями типа
- Br—C=N ••• Br—C=N ••• Br—C=N
Здесь азот находится в аксиально-симметричном окружении, и следует ожидать только одну линию. Однако предполагается, что взаимодействия между цепями снижают симметрию окружения азота, и это приводит к двум спектральным линиям. Для BrCN можно записать различные резонансные формы, а величины e2Qq указывают, что на броме находится формальный положительный заряд. Заметное увеличение e2Qq для брома наблюдается в твердом веществе по сравнению с газовой фазой, что может быть обусловлено увеличением вкладов структуры Br + CN _ в основное состояние в твердом веществе из-за стабилизации Вг+ за счет координации. Если связь N-Br—С описывается pd-гибридом, то e2Qq будет также увеличиваться за счет возросшего вклада d-орбиталей в связь углерода с бромом.
14№Квадрупольные переходы в пиридине и различных его координационных соединениях характеризуются [28] большими изменениями энергии. Из обсуждения рис. 14.6 следует, что при г] / О ожидаются три перехода (v+, v0 и v_); из этих данных можно определить e2Qq и ц. Типичные результаты представлены в табл. 14.2.
Таблица 14.2
Параметры 14№квадрупольных переходов н градиента поля в пиридине н его координационных соединениях (все частоты даны в кГц, температура 77 К). (Кристаллографическая неэквивалентность, если она имеет место, разрешена.)
Соединение v + v_ Vo e2Qq h Л
Пиридин (Ру) 3892 2984 908 4584 0,396
Пиридинийнитрат 1000 580 420 1053 0,798
Py2ZnCl2 2387 2332 2078 2038 309 294 2977 2913 0,207 0,202
PyZn(NO3)2 2124 1884 240 2672 0,180
2097 1877 220 2649 0,166
Py2CdCl2 2850 2298 552 3432 0,320
—
Иодный спектр ЯКР твердого иода свидетельствует о большом параметре асимметрии ц [29]. Поскольку атом иода в молекуле иода аксиально-симметричен, значительная асимметрия рассматривается как результат межмолекулярного связывания в твердом состоянии. Большая величина ц в иодном спектре ЯКР молекулы НЮ3 говорит в пользу структуры Ю2(ОН), а не НЮ3. Структура НЮ3 имеет ось С3, поэтому qxx = qyy. В работах [1, 2] имеется много дополнительных примеров исследований такого рода.
Спектроскопия ядерного квадрупольного резонанса
279
Из параметра асимметрии г) может быть получена информация о л-связывании. Обсуждались методы расчета г) для различных ядер. Одинарная сг-связь с галогеном должна приводить к аксиально-симметричному градиенту поля. Двойные связи ведут к асимметрии, и г) зависит от степени л-связывания. Был сделан вывод, что в винилхлориде, винил-бромиде и винилиодиде существует заметное (~ 5%) л-связывание углерод — галоген [30]. Берсон [31], исходя из т), провел подробное исследование проблемы качественной оценки степени л-связывания углерод — галоген.
Величины г], полученные из спектров ЯКР соединений Sil4, Gel4 и Snl4, были интерпретированы с точки зрения очень небольшой степени ( ~ 1%) двоесвязанности [32] в связи галогена с центральным атомом, что может быть обусловлено эффектами твердого состояния.
Спектры ЯКР на ядрах 75As, 121Sb, 123Sb, 209Bi, 35C1, 37Ci, 79Br и 81Br соединений с общей формулой R3MX2 исследовались [33] при R = CH3, CH2C6H5 и С6Н5, X = Cl, F и Вг и М = As, Sb и Bi. Были обнаружены очень небольшие параметры асимметрии, свидетельствующие о существовании во всех этих соединениях оси третьего порядка. Из этих данных следует, что большинство соединений имеет структуру тригональной бипирамиды. Однако в случае соединения мышьяка [(CH3)3AsBr + ]Вг ~ это не так, хотя, вероятно, катион имеет симметрию С3г,-
Спектры ЯКР на ядрах 37Ci, 35С1, 121Sb, 1271 соединений 21С1-А1С1з и 2ICl-SbCi5 показывают [34], что эти вещества должны иметь формулы 1С12 А1С14 и IQJSbClg . Спектры ЯКР V-образных катионов IC1J, 13 и 12С1+ были исследованы в нескольких различных соединениях. Были исследованы [35] также спектры ЯКР на ядрах 35С1 хлоралюминиевой группы в ряду соединений М„(А1С14)т. Из энергий переходов ЯКР можно судить о том, свободен ли анион А1С14, или он сильно координирован с катионом. «Ионные» переходы А1С14 были обнаружены в диапазоне от 10,6 до 11,3 МГц. Сильная координация А1С14 приводит к удлинению мостиковых связей Al — С1 и заметному увеличению диапазона и средней частоты хлорных переходов.
В следующей главе, посвященной мёссбауэровской спектроскопии, рассмотрена модель парциального градиента поля (ПГЦ) для корреляции градиентов поля на центральном атоме. Она оказывается полезной для установления молекулярных структур на основании данных мёссбауэровской спектроскопии. Эту модель можно также использовать для структурного анализа в случае ЯКР. Поскольку большинство данных, по которым была построена и проверена модель ПГЦ, получено с помощью мёссбауэровской спектроскопии, этот вопрос обсуждается в следующем разделе.
14.6. МЕТОДЫ ДВОЙНОГО РЕЗОНАНСА
Сообщалось [36—38] об использовании методов ядерного двойного резонанса для регистрации квадрупольных переходов. Эти методы зна
280
Глава 14
чительно увеличивают чувствительность по сравнению с предыдущими методами, а также позволяют наблюдать переходы с очень низкой частотой, например, в спектрах на ядрах дейтерия переходы наблюдаются в интервале 100—160 кГц [39].
Метод двойного резонанса с адиабатическим размагничиванием является новым методом в этой области. Рассмотрим образец с квадру-польным ядром в молекуле, в которой имеется несколько протонов. Если образец помещен в магнитное поле и мы ждем достаточно долго, чтобы наступило равновесие, то, как это обсуждалось в главе, посвященной ЯМР, будет существовать избыток протонных ядерных моментов, расположенных вдоль поля, которые участвуют в ларморовой прецессии и дают вклад в суммарную намагниченность. Если образец удалить из поля, то суммарная намагниченность упадет до нуля, поскольку индивидуальные моменты располагаются в соответствии со своими собственными локальными полями. Беспорядочная ориентация этих локальных полей в отсутствие внешнего поля приводит к нулевой суммарной намагниченности. Эта ситуация изображена на рис. 14.8 слева, в той части, которая помечена как «образец удален из поля».
Если 7] больше, чем время, необходимое для удаления образца из поля, то избыточная заселенность спинов +1/2 сохранится, но они будут прецессировать вокруг направления суммарного локального поля на ядре, возникающего за счет спин-орбитального взаимодействия с соседними протонами. По всему образцу намагниченность равна нулю, но если этот образец вновь поместить в магнитное поле, то в образце одновременно возникает намагниченность, причем не придется ждать столько же времени, сколько необходимо для процесса 7]. Эта ситуация изображена на рис. 14.8 в той части, которая помечена как «образец повторно внесен в поле». Интенсивность намагниченности можно измерить немедленно после повторного внесения образца в магнитное поле, используя 90°-ный импульс и измеряя кривую СИС (рис. 14.8). Если период времени между удалением образца из поля и повторным его внесением туда достаточно велик по сравнению с 7], то намагниченность будет падать по мере рандомизации спинов.
Теперь рассмотрим эксперимент, в котором образец облучается радиочастотным излучением, соответствующим энергии квадрупольного перехода ядра В, после удаления образца из поля. Кроме того, предположим, что время между удалением образца из поля и повторным его внесением туда мало по сравнению с 7] протонов. Эффект этого радиочастотного излучения заключается в рандомизации ядер В за счет индуцированных им квадрупольных переходов в спиновой системе В. При выполнении соответствующих условий относительно амплитуды приложенного радиочастотного излучения, отвечающих наличию локального поля на протоне, рандомизация спиновой системы В влияет на рандомизацию спиновой протонной системы. Это происходит следующим образом. Если образец удален из поля, то разность энергий между состояниями m = + 1/2 и m = — 1/2 (т. е. энергия перехода ядра водорода) снижается до нуля. В этом процессе наступит момент, когда разность
Спектроскопия ядерного квадрупольного резонанса
281
из поля внесен в поле
Рис. 14.8. График зависимости намагниченности. Образец удален из поля, повторно внесен в поле, подвергнут воздействию 90°-ного импульса после чего происходит спад свободной индукции.
энергий для ядер водорода совпадет с разностью энергий квадру-польных состояний ядра В и произойдет резонансный обмен энергией, вызывающий рандомизацию протонов. Процесс рандомизации часто называют увеличением спиновой температуры системы. В результате рандомизации протонной спиновой системы из-за переходов в ядрах В намагниченность, восстанавливающаяся при повторном внесении образца в магнитное поле, имеет меньшую величину, чем в противном случае. В результате наблюдается и меньший СИС. Если частота, вызывающая квадрупольные переходы, не подходит для резонанса, то ядро В не рандомизуется, и большая часть намагниченности восстанавливается. Путем систематического увеличения частоты излучателя В «спектры» квадрупольных переходов системы В наносятся на схему в зависимости от влияния на СИС протонной спиновой системы.
Путем использования метода двойного резонанса спин-эха [39—41] можно устранить большую часть неоднородного дипольного уширения (дефекты кристалла и т. д.), что приводит к очень широким линиям в непосредственном эксперименте ЯКР. С помощью этого метода снят спектр ЯКР соединения А12Вг6 [40].
Если имеются два квадрупольных ядра, окруженных ядрами с I = 0 [например, как в случае D — Мп(СО)5], то можно наблюдать дипольное взаимодействие ядер марганца и дейтерия [42]. Как обсуждалось в главах, посвященных ЯМР, из величины дипольного взаимодействия можно определить длину связи. Исходя из спектра ЯКР на ядрах 55Мп, для длины связи Мп — D получим величину 1,61 А, что превосходно согласуется с данными по рассеянию нейтронов [43].
282
Глава 14
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. Сликпгер Ч.П., Основы теории магнитного резонанса с примерами из физики твердого тела, разд. 6.3.—М.: ИЛ, 1965.
2. Bersohn R., J. Chem. Phys., 20, 1505 (1952).
3. Livingston R., Zeldes H., Tables of Eigenvalues for Pure Quadrupole Spectra, Oak. Ridge Natl. Lab. Rept. ORNL—1913 (1955).
4. a. Das T.P., Hahn E.L., Nuclear Quadrupole Resonance Spectroscopy, Academic Press, New York, 1958.
b. Lucken E. A. C., Nuclear Quadrupole Coupling Constants, Academic Press, New York, 1969.
5. O’Konsk: С. T, Determination of Organic Structures by Physical Methodes, Vol.
2, ed. Nachod F. C, Phillips W. D., Academic Press, 1962.
6. Попл Дж., Шнейдер В., Бернстейн А., Спектры ядерного магнитного резонанса высокого разрешения.—М.: ИЛ, 1962.
7. Dean С., Phys. Rev., 86, 607 А (1952).
8. Cotton F.A., Harris С.В., Proc. Nat. Acad. Sci. (USA), 56, 12 (1966); Inorg. Chem., 6, 376 (1967).
9. Townes C.H., Dailey B.P., J. Chem. Phys., 17, 782 (1949).
10. Cohen M.H., Reif F., Solid State Physics, Vol. 5, ed Seitz F., Turnbull D., Academic Press, New York, 1957.
11. Orville-Thomas W.J.. Quart. Rev, 11, 162 (1957).
12. Dailey B.P.. J. Chem. Phvs.. 33, 1641 (1960).
13. О Konski C.T., На T.K., J. Chem. Phys., 49, 5354 (1968).
14. White W.D., Drago R.S., J. Chem. Phys, 52, 4717 (1970).
15. Sterheimer R.M., Phys. Rev, 130, 1423 (1963); ibid, 146, 140 (1966) и ссылки в этой работе.
16. Townes С.Н., Dailey В.Р., J. Chem. Phys, 23, 118 (1955); Gordy W., Disc. Faraday Soc, 19, 14 (1955); Whitehead M.A., Jaffe H.H., Trans. Faraday Soc, 57, 1854 (1961).
17. MealH.C., J. Amer. Chem. Soc, 74, 6121 (952); Bray P. J., Barnes R.G., J. Chem. Phys, 27, 551 (1957) и ссылки в этой работе.
18. Nakamura D.. Ito К.. Kubo M.. Inorg. Chem, 1, 592 (1962); ibid, 2, 61 (1963); J. Amer. Chem. Soc, 82, 5783 (1960); ibid, 83, 4526 (1961); ibid, 84, 163 (1962); Ku-bo M., Najamura D., Adv. Inorg. Chem. Radiochem, 8, 257 (1966).
19. Brown T.L., McDugle W. G., Kent L.C., J. Amer. Chem. Soc, 92, 3645 (1970).
20. Brill T.B., Koltar A. J., Inorg. Chem, 13, 470 (1974).
21. Brown T.L.. Accts. Chem. Res, 7, 408 (1974) и ссылки в этой работе.
22. a. Welsh W.A., Brill ТВ., et al., Inorg. Chem, 13, 1797 (1974).
b. Brill T.B., Gearhart R.C., Welsh W.A., J. Mag. Res, 13, 27 (1974).
c. Brill T.B., J. Chem. Phys, 61, 424 (1974).
23. Drago R. S.. Matwiyoff N.A., J. Organometal. Chem, 3, 62 (1965).
24. Livingston K., Zeldes H., J. Chem. Phys, 26, 351 (1957).
25. a. Reddoch A.H., J. Chem. Phys, 35, 1085 (1961).
b. Schawlow A.L., J. Chem. Phys, 22, 1211 (1954).
26. Edwards P.A., McCarley R.E., Inorg. Chem, 12, 900 (1973).
27. Geller S., Schawlow A.L., J. Chem. Phys, 23, 779 (1955).
28. Hsieh Y.N., Rubenacker G.R., Cheng C.P., Brown T.L., J. Amer. Chem. Soc, 1977, в печати.
29. Dehmelt H.G., Naturwiss, 37, 398 (1950).
30. Howe J. A., Goldenstein J.H., J. Chem. Phys, 26, 7 (1957); 27, 831 (1957) и ссылки в этой работе.
31. Bersohn R., J. Chem. Phys, 22, 2078 (1954).
32. Robinson H., Dehmelt H.G., Gordy W., J. Chem. Phys, 22, 511 (1954); Kojima S., et al, J. Phvs. Soc. Japan, 9, 805 (1954).
33. Brill T.B., Long G.G., Inorg. Chem, 9, 1980 (1970).
34. Merryman D.J., Corbett J.D., Inorg. Chem, 13, 1258 (1974).
35. Merryman D.J., Edwards P.A., Corbett J.D., McCarley R.E., Inorg. Chem, 13, 1471 (1974).
36. Hartmann S. R., HahnE.L., Phys. Rev, 128, 2042 (1962).
Спектроскопия ядерного квадрупольного резонанса
283
37. Slusher R.E., Hahn E.L., Phys. Rev., 166, 332 (1968).
38. Edmonds D. T, et al., Advances in Quadrupole Resonance, Vol. 1, p. 154, Heyden, London, 1974; Rev. Pure Appl. Chem., 40, 193 (1974).
39. Ragle J.L., Sherk K.L., J. Chem. Phys., 50, 3553 (1969).
40. Emshwiller M., Hahn E.L., Kaplan D., Phys. Rev., 118, 414 (1960).
41. Ragle J.L., et al., J. Chem. Phys., 61, 429, 3184 (1974).
42. a. Ireland P.S., Olson L.W., Brown T.L., J. Amer. Chem. Soc., 97, 3548 (1975). b. Ireland P.S.. Brown T. I... J. Mag. Res.. 20, 300 (1975).
43. LaPlacaS.J., et al., Inorg. Chem., 8, 1928 (1969).
РАБОТЫ ОБЩЕГО ХАРАКТЕРА
1. Cohen M.H., Reif F., Solid State Physics, Vol. 5, ed. Seitz F., Turnbull D., Academic Press, New York. 1957.
2. Das T. P., Hahn E. L., Nuclear Quadrupole Resonance Spectroscopy, Academic Press, New York, 1958.
3. Lucken E. A. C., Nuclear Quadrupole Coupling Constants, Academic Press, New York, 1969.
4. Smith J. A. S., ed., Advances in Nuclear Quadrupole Resonance, Heyden and Sons, Ltd., London, Vol. 1, 1974; Vol. 2, 1975; Vol. 3, 1977.
Упражнения
1. а. Рассчитайте энергии различных состояний квадрупольного ядра с 1 = 2 и выразите их через е Qq.
б. Как много переходов следует ожидать и каково соотношение между энергией переходов и e2Qq‘l
2. а. Используя уравнения, приведенные в настоящей главе для расчета энергий уровней 0 и + 1 ядра с I = 1 в асимметричном поле, рассчитайте частоту через e2Qq и т| для переходов 0-» +1 и 0-> —1.
б. Выразите разность энергий между двумя переходами в п. «а» через т| и e2Qq.
в. Каким образом из этих данных можно определить г| и д?
3. Опишите эксперимент ЯКР, который должен дать информацию о степени л-евязывания в связях фосфор — сера в PSC13 и (C6H5)3PS. Можете ли вы определить из эксперимента ЯКР, образует ли сера sp2-гибрид и использует ли р-орбитали в связывании, или рх- и ру-орбитали серы в равной степени участвуют в связывании? (Заметим, для 33S I = 3/2.)
4. Сообщалось о том, что сигнал ЯКР на ядрах 1271 соединения Asl3 представляет собой синглет, но характеризуется очень большим параметром асимметрии [28]. Рентгеноструктурный анализ монокристалла показывает, что As находится в октаэдрическом окружении. Объясните большую величину параметра асимметрии.
5. Укажите число резонансных линий для следующих ядер при указанных условиях:
а. 1271 (1 = 5/2), т| =0, Н0 = 0.
б. ,4N (1 = 1), т] = 0. Н0 = 0.
в. 75As (I = 3/2), т] = 0, Но = 0.
г. ,2‘Sb (1 = 5/2), т| = 1, Но = 0.
д. 14N (1 = 1), т| = 1, Н0=0.
е. 14N (1=1), п = 1, Но^0.
6. Квадрупольная энергия ядра выражается как
_ e2Q<?[3m2 - Ц1 + 1)]
41(21 - 1)
Ядро 59Со имеет I = 7/2, содержание его в природе составляет 100%.
284
Глава 14
а. Как много переходов ЯКР кобальта будет наблюдаться в спектре K3Co(CN)6? Какова энергия этих переходов, если её выразить через e2Qq?
б. Повторите то же самое для K3Co(CN)5Br.
7. Рассмотрите ЯКР на ядрах азота для каждой из следующих систем. Как много линий следует ожидать при наличии и в отсутствие магнитного поля? (Для 14N I = 1.)
a. NH3.
б. NHJ.
в Пиридин
8. Частоты в спектре ЯКР на ядрах 59Со (для 59Со 7 = 7/2) Cl3SnCo(CO)4 составляют 35,02 (±5/2-» L7/2), 23,37 (±3/2^+5/2) и 11,68 МГц (±1/2-> -» ±3/2). Рассчитайте г] и e~Qq. (Указание: каково отношение частот, если г] = = 0?)
9. Известно соединение с формулой СН31п12. Полагают, что оно построено из ионов, т. е. (CH3)2In + Inl4. Как много резонансных линий следует ожидать в спектре ЯКР на ядрах 1151п и 1271 для этого соединения при условии, что нет кристаллографической неэквивалентности аниона и катиона? Для катиона г] = = 0,05. О какой структуре катиона говорит такая небольшая величина?
10. В спектре на ядрах 14N твердого пиридина (C5H5N) при 77 К линии лежат при 3,90 и 2,95 МГц. Каковы e2Qq/h и г] для азота в пиридине?
11. В спектре ЯКР на ядрах 35С1 соединения HgCl2 линии лежат при 22,05 и 22,25 МГц при 300 К.
I I 22,05 22,25
В спектре ЯКР на ядрах 35С1 комплекса HgCl2 • диоксан единственная линия при 300 К лежит при 20,50 МГц. В этом комплексе имеет место взаимодействие Hg <- О донорно-акцепторного характера.
а. Каков вероятный источник расщепления линий в чистом HgCl2?
б. Электрические квадрупольные моменты ядер 35С1 и 37С1 составляют соответственно 0,079-10 “ 24 и 0,062-10 “ 24 см2. При какой частоте должны лежать линии в спектре ЯКР на ядрах 37С1 соединения HgCl2?
в. Объясните уменьшение резонансной частоты в спектре ЯКР на ядрах 35С1 комплекса сулемы с диоксаном по сравнению с чистым HgCl2, исходя из />-орбитальной заселенности в рамках уравнения (14.13).
15. МЁССБАУЭРОВСКАЯ СПЕКТРОСКОПИЯ
15.1. ВВЕДЕНИЕ
Мёссбауэровская спектроскопия [1], которая в тексте сокращенно называется МБ-спектроскопией, регистрирует переходы, обусловленные поглощением у-лучей веществом. Эти переходы характеризуются изменением ядерного спинового квантового числа I. Условия поглощения зависят от электронной плотности вокруг ядра, а число наблюдаемых спектральных полос связано с симметрией соединения. В результате этого можно получить структурную информапию. Многие из идей и символов, используемых в данной главе, были описаны в гл. 14.
Для того чтобы понять принцип действия этого метода, рассмотрим вначале газообразную систему, состоящую из радиоактивного источника у-лучей и образца, который может поглощать у-лучи. Ядро источника, испускающее у-квант, переходит в основное состояние. Энергии испущенных у-лучей (£у) лежат в диапазоне 10—100 кэВ и выражаются с помощью уравнения
Ey = E, + D-R, (15.1)
где Ег—разность энергий между возбужденным и основным состояниями ядра источника, D—доплеровский сдвиг, обусловленный поступательным движением ядра, и R— энергия отдачи ядра. Энергия отдачи, аналогичная той, которая высвобождается, когда пуля покидает ствол винтовки, обычно составляет от 10 “ 2—10 3 эВ и выражается уравнением
R = Еу/2тс2, (15.2)
где т—масса ядра, а с — скорость света. Доплеровский сдвиг объясняет тот факт, что энергия у-кванта, испущенного ядром газообразной молекулы, движущейся в том же направлении, что и квант, отличается от энергии у-кванта, испущенного ядром молекулы, движущейся в противоположном направлении. Распределение энергий, обусловленное поступательным движением ядер источника в различных направлениях, носит название доплеровского уширения. Левая кривая на рис. 15.1 демонстрирует энергетическое распределение испущенных у-лучей, вызванное доплеровским уширением. Ширина кривой есть результат доплеровского уширения. Пунктирная линия на рис. 15.1 соответствует Ег—разности энергий между основным и возбужденным состояниями ядер источника. Разность энергий R между пунктирной линией и макси-
286
Глава 15
мумом левой кривой суть энергия отдачи, полученная ядром источника при испускании у-кванта.
В МБ-спектроскопии энергия у-кванта, вызывающего переход в образце, выражается как
Ey = E, + D + R. (15.3)
В этом случае R добавляется потому, что возбуждающий у-квант должен иметь достаточную энергию, чтобы вызвать переход и эффект отдачи поглощающего ядра. Величина D имеет тот же смысл, что и раньше, а Ег по предположению одна и та же для источника и образца.
Энергия
Рис. 15.1. Распределение энергий испущенных и поглощенных '/-квантов.
Правая кривая на рис. 15.1 демонстрирует энергетическое распределение у-лучей, необходимое для поглощения. Связь между энергиями образца и источника видна из всего рисунка. Как показывает площадь заштрихованного участка рисунка, вероятность того, что энергия у-кванта источника будет поглощаться образцом, невелика. Поскольку ядерные энергетические уровни квантованы, вероятность поглощения у-кванта источника образцом, в результате которого произойдет переход в образце, очень мала. Основной причиной несогласования энергий у-квантов является энергия отдачи, так как центр энергетического распределения испущенного излучения лежит при Er — R, в то время как центр энергетического распределения излучения, необходимого для поглощения, лежит при Er + R. Величина R для газообразных молекул (~ 10“1 эВ) значительно превышает типичную величину доплеровской энергии. Для того чтобы кривые энергетического распределения источника и образца перекрывались, доплеровская энергия должна быть достаточно большой, т. е. источник должен двигаться со скоростью 2 • 104 см/с, чего достичь нелегко. Однако, если величину R можно уменьшить или если можно найти условия для перехода, не сопровождающегося отда
Мёссбауэровская спектроскопия 287
чей ядра, вероятность поглощения образцом у-квантов источника должна быть большой. Как показывает уравнение (15.2), R можно понизить за счет увеличения массы т, что осуществляется путем помещения ядер источника и образца в кристалл, так что эффективной массой становится масса кристалла. По этой причине мёссбауэровские спектры почти всегда регистрируют для вещества в твердом состоянии и при использовании твердого источника.
Поместив источник и образец в твердые кристаллические решетки, мы не оказали воздействия на переходы без отдачи для всех ядер, но увеличили вероятность перехода без отдачи. Причина этого заключается в том, что энергия у-лучей может привести к возбуждению колебаний решетки. Эта энергия влияет тем же самым образом, что и энергия отдачи в газе, т. е. она приводит к снижению энергии излучающей частицы и увеличению энергии поглощающей частицы. Некоторые характеристики кристалла и условия эксперимента для излучения и поглощения не меняют исходного колебательного состояния решетки, т. е. будут удовлетворять условиям перехода без отдачи. Следует подчеркнуть, что эти условия определяют просто интенсивность наблюдаемых линий, поскольку этим эффектом задается только число частиц с подходящей энергией. Нас не интересует абсолютная интенсивность полос, поэтому здесь не обсуждается этот аспект МБ-спектроскопии. Однако упомянем, что для некоторых веществ (обычно твердых молекулярных веществ) решеточные и молекулярные колебания возбуждаются до такой степени, что при комнатной температуре происходит только небольшое число переходов без отдачи и спектр не наблюдается. Часто спектр регистрируют путем значительного понижения температуры образца.
Переходя к твердому состоянию, мы в значительной степени уменьшаем ширину резонансных линий по сравнению с тем, что показано на рис. 15.1. В твердом состоянии доплеровское уширение становится пренебрежимо малым и R имеет величину около 10“4 эВ для у-квантов с энергией 100 кэВ и излучателей с массовым числом 100. Полная ширина линии на ее полувысоте дается с помощью принципа неопределенности Гейзенберга как ДЕ = h/т = 4,5610 ~ 16/0,977-10 “7 = 4,67-10 ~ 9 эВ, или 0,097 мм/с (для 57Fe). Ширина линии—величина бесконечно малого порядка по сравнению с энергией источника 1,4-104 эВ. Времена жизни возбужденных состояний мессбауэровских ядер лежат в интервале от ~10-5 до 10“10 с,-что ведет к ширине линий большинства ядер от 10'11 до 10 ~6 эВ. Этот вопрос обсуждается в работах [1—5], в которых более подробно рассматривается МБ-спектроскопия.
Основное внимание мы уделим факторам, влияющим на энергию, необходимую для поглощения у-квантов образцом. Существуют три типа взаимодействий ядер с химическим окружением, которые приводят к небольшим изменениям энергии, необходимой для поглощения: 1) сдвиги резонансных линий за счет изменения в электронном окружении, 2) квадрупольные взаимодействия и 3) магнитные взаимодействия. Эти эффекты дают информацию, имеющую значение с химической точки зрения, и будут рассмотрены в первую очередь.
Прежде чем приступить к обсуждению указанных факторов, целесо-
288
Глава 15
образно описать процедуру получения спектров и показать типичный МБ-спектр. Электронное окружение ядра влияет на энергию у-лучей, необходимую для того, чтобы вызвать ядерный переход от основного состояния к возбужденному, т. е. Ег. Энергия у-лучей источника может меняться в диапазоне разностей энергий, обусловленных электронными окружениями в различных образцах, за счет движения источника относительно образца. Чем выше скорость такого движения, тем выше средняя энергия излученных у-лучей (за счет эффекта Доплера), и наоборот. Изменение энергии AES кванта источника, движущегося относительно образца, выражается как
ДЕ5 = — Ecos0, (15.4)
с
где Е— стационарная энергия кванта, v0 — скорость движения источника и 0 — угол между вектором скорости образца и линией, соединяющей образец и источник. Если источник движется прямо на образец, то cos 0=1. Для регистрации МБ-спектра источник перемещают относительно образца и определяют скорость движения источника, при которой наблюдается максимум поглощения у-лучей.
В качестве наглядного примера рассмотрим МБ-спектр Fe3 + FeIn(CN)6, где Fe3+ и Fe111 обозначают железо(Ш) в слабом и сильном кристаллическом полях соответственно. Это вещество содержит железо в двух различных химических окружениях, и, чтобы вызвать переходы в различных ядрах, требуются у-лучи двух различных энергий. Для получения МБ-спектра источник передвигают относительно фиксированного образца и строят график зависимости поглощения у-лучей от скорости движения источника (рис. 15.2). Полосы соответствуют скоростям движения источника, при которых поглощение у-лучей источником максимально. Отрицательные относительные скорости соответствуют движению источника от образца, а положительные относительные скорости—движению источника к образцу. Относительные скорости движения источника наносят на ось абсцисс рис. 15.2 и эту величину связывают с энергией у-лучей. Для. источника 57Fe, испускающего у-лучи с энергией 14,4 кэВ, энергия меняется на 4,8-10“8 эВ, или на 0,0011 кал/моль на каждый 1 мм/с скорости движения источника. Этот результат может быть рассчитан из уравнения
ДЕ, =-----' ----14 4.10з эВ = 4 8.10- 8 эВ (15.4)
s 3,00-1011 мм/с '
Полученная энергия эквивалентна частоте 11,6 МГц (v = E/h, где h = = 4,14-10 15 эВ-с). Для других ядер с энергией у-лучей Е., (в кэВ)
£
1 мм/с = 11,6-“[447 МГц.
Обращаясь снова к абсциссе рис. 15.2, можно видеть, что разность энергий между ядерными переходами Fe3+ и FeniB FeFe(CN)6 мала,
Мёссбауэровская спектроскопия 289
она составляет около 2-10“8 эВ. Путем сравнения спектра на рис. 15.2 со спектрами большого числа цианидных комплексов железа линию при 0,03 мм/с приписали [6] Fe111, а линию при 0,53 мм/с—катиону
Рис. 15.2. МБ-спектр FeFe(CN)6.
___I______I________1_
-1,0 0 1,0
мм/с
Fe3 +. Различные положения линий, обусловленные разными химическими окружениями, характеризуются величинами скорости движения источника в см/с или мм/с и носят названия изомерных сдвигов, центровых сдвигов или химических сдвигов.
Теперь мы проанализируем информацию, которая содержится в параметрах, полученных из спектра.
15.2. ИНТЕРПРЕТАЦИЯ ИЗОМЕРНЫХ СДВИГОВ
Две различные линии на рис. 15.2 обусловлены разностями изомерных сдвигов двух различных атомов железа в октаэдрических центрах. Изомерный сдвиг—результат электростатического взаимодействия распределения заряда в ядре с электронной плотностью, вероятность существования которой на ядре конечна. Конечную вероятность перекрывания с плотностью ядерного заряда имеют только s-электроны, поэтому изомерный сдвиг можно рассчитать, рассматривая это взаимодействие. Следует помнить, что р-, d- и другие электронные плотности могут оказывать влияние на s-электронную плотность путем экранирования s-электронной плотности от заряда ядра. Предполагая, что ядро представляет собой однородно заряженную сферу радиуса R, а s-электронная плотность вокруг ядра постоянна и задается функцией ф/ (0), разность между электростатическим взаимодействием сферически распределенной электронной плотности с точечным ядром и той же самой электронной плотности с ядром радиуса R выражается как
5Е = К[ф/ (0)]Я2, (15.5)
где К—ядерная постоянная. Поскольку R имеет различные значения для основного и возбужденного состояний, электронная плотность вокруг ядра взаимодействует с двумя этими состояниями различным образом и, следовательно, влияет на энергию перехода, т. е.
8Ег - 8Е9 = К[ф/ (0)](Е2 - К2), (15.6а)
19-574
290
Глава 15
где нижние индексы «е» и «д» относятся соответственно к возбужденному и основному состояниям. Влияние х|/2 (0) на энергию перехода показано на рис. 15.3 для ядра 57Fe, которое имеет I = 1/2 для основного состояния и I = 3/2 для возбужденного. Энергии этих двух состояний в разной степени подвержены влиянию (0), и поэтому энергия перехода меняется.
(е)
(е>
(9)
V<0)
Рис. 15.3. Изменение энергий МБ-переходов для различных величин v|/s2(0).
Этот рисунок представляет собой графическую иллюстрацию уравнения (15.6а) для двух различных величин фз(0) и ядра 57Fe. Для того чтобы можно было пользоваться этой диаграммой, различия в ф/(0) должны быть обусловлены кубическим или сферическим распределением связанных атомов.
Для данного ядра величина R постоянна, но х|/2 (0) меняется от соединения к соединению. Центровой сдвиг в МБ-спектре представляет собой разность между энергией этого перехода в образце (или поглотителе) и энергией того же перехода в источнике. Она выражается через разность в уравнениях вида (15.6а) для источника и образца, или
центровой сдвиг = K(R2 - R92){[»|// (0)]а - [i[/s2 (0)]ь}, (15.66)
где нижние индексы «а» и «Ь» относятся соответственно к поглотителю и источнику. Обычно в работе используют стандартные источники (например, 57Со в Pd для мёссбауэровского спектра железа или BaSnO3 для спектра олова). Ядро 57Со переходит в ядро 57Fe в возбужденном состоянии за счет электронного захвата. В результате излучения у-кван-та возбужденное ядро 57Fe переходит в стабильное ядро 57Fe. При использовании стандартного источника (0)]6 заменяется на постоянную С. Кроме того, изменение радиус-вектора Re — Rg мало, что приводит к следующему обычно используемому выражению для центрового сдвига:
8R , центровой сдвиг = К'----[х|/2 (0)а — CJ, (15.6в)
R
где 8R = Re — Rg, С — постоянная, характеризующая источник, и К' = = 2KR2. Для данного ядра К', 5R/R— константы, поэтому центровой
Мёссбауэровская спектроскопия
291
сдвиг прямо пропорционален s-электронной плотности на ядре образца. Термин центровой сдвиг используют для экспериментально определяемого центра линии; термин изомерный сдвиг используют в том случае, если в центровой сдвиг внесена поправка на небольшой доплеровский вклад от теплового движения мёссбауэровских ядер. Знак 87? зависит от разности между эффективными зарядовыми радиусами ядра R возбужденного и основного состояний: R^ — R^. Радиус возбужденного ядра 57Fe меньше радиуса ядра в основном состоянии, и увеличение s-электронной плотности приводит к отрицательному сдвигу. Для олова знак 8R положителен, поэтому наблюдается противоположная тенденция в изменении сдвига с s-электронной плотностью.
Как упоминалось ранее, электронная плотность на р- или d-орбиталях может экранировать s-электронную плотность от заряда ядра за счет того, что электронная плотность на р- и d-орбиталях пронизывает s-орбиталь. Расчеты по методу Хартри—Фока показывают [6, 7], что уменьшение числа d-электронов вызывает заметное увеличение полной s-электронной плотности на ядре железа. Поэтому при одинаковых лигандах и при отрицательном 8R/R катион Fe2 + имеет заметно больший центровой сдвиг, чем Fe3 + . Если исследовать эти ионы в ряду соединений, то интерпретация затрудняется, поскольку d-, s- и р-элек-тронные плотности видоизменяются за счет ковалентного связывания. Например, для 57Fe увеличение 45-электронной плотности приводит к снижению центрового сдвига, а увеличение 3d-3neKrpoHHoft плотности—к его росту. На основании этого были интерпретированы МБ-спектры ряда высокоспиновых комплексов железа [7]. В случае 119Sn центровой сдвиг растет с увеличением s-электронной плотности и снижается с увеличением р-электронной плотности.
15.3. КВАДРУПОЛЬНЫЕ ВЗАИМОДЕЙСТВИЯ
Обсуждение центрового сдвига, проведенное в предыдущем разделе, применимо к системам со сферическим или кубическим распределением электронной плотности. Как отмечалось в гл. 14, вырождение ядерных энергетических уровней для ядер с I > 1/2 устраняется некубическим распределением электронов или лигандов. Для нецелых спинов расщепление не снимает (+)- или (— (-вырождения уровней с т,, но мы наблюдаем свой уровень для каждого + иц. Таким образом, градиент электрического поля может привести к I + 1/2 различным уровням для полуцелых значений (например, 2 для 7 = 3/2, что соответствует ±1/2 и + 3/2). Для целых значений I получаем 27 + 1 уровней (например, 5 для 7 = 2, что соответствует 2, 1, 0, —1 и —2). На рис. 15.4 показано влияние этого расщепления на ядерные энергетические уровни и характер спектра 57Fe. Основное состояние не расщепляется, но возбужденное состояние расщепляется, что приводит к двум линиям в спектре. Центровой сдвиг определяется из центра двух результирующих линий. Если и основное, и возбужденное состояния имеют большие значения I, то получается сложный МБ-спектр.
292
Глава 15
Квадрупольное расщепление
2 2
/ Центровой Квадрупольное сдвиг расщепление
мм/с
—4,0 0 4,0
ММ/С
Рис. 15.4. Влияние некубического электрического окружения на ядерные энергетические уровни 57Fe (Л) и МБ-спектр (Б), МБ-спектр на ядрах железа соединения Fe(CO)5 при температуре жидкого азота (В) [9J.
т
Гамильтониан для квадрупольного взаимодействия имеет тот же вид, что и в случае ЯКР:
= 47 ff- 1) [(31‘ - -О + + М + Л •
Для 1 = 3/2 (57Fe и 119Sn) квадрупольное расщепление выражается как
квадрупольное расщепление =+ т]2/3)1/2. (15.7)
Мёссбауэровская спектроскопия 293
Все символы были определены в гл. 14, посвященной ЯКР. Для ядра 57Fe нельзя определить величины из квадрупольного расщепления q и т|. Не менее важен знак константы квадрупольного взаимодействия. Если уровень пу = + 3/2 соответствует высокой энергии, то знак положителен; знак отрицателен, если при = +1/2 при квантовом числе I = 3/2 энергия максимальна. Если судить по спектру порошкообразного образца, то переходы между уровнями с nq = ±1/2 и + 3/2 имеют одинаковые интенсивности, поэтому знак константы квадрупольного расщепления определить трудно. Однако его можно установить из спектра упорядоченной системы или при исследовании поликристаллическо-го образца в магнитном поле (см. ниже). Для систем, в которых значения I для основного и возбужденного состояний превышают значение I для железа, спектры имеют более сложный вид и содержат больше информации. Расщепление возбужденного состояния не происходит в сферически симметричном или кубическом поле, но оно имеет место только при наличии градиента поля на ядре, вызванного асимметричным распределением р- или d-электронной плотности в молекуле. Градиент поля существует в тригонально-бипирамидальной молекуле пентакарбонила железа, поэтому ожидается расщепление ядерного возбужденного состояния, приводящее к появлению дублета в спектре (рис. 15.4,В).
Если совокупности t2g- и е9-орбиталей в октаэдрических комплексах ионов переходных металлов имеют равные заселенности в компонентных орбиталях, то квадрупольное расщепление равно нулю. Низкоспиновые комплексы железа(П) (12я) не дают квадрупольного расщепления, если только не снимается вырождение, и эти орбитали могут взаимодействовать различным образом с молекулярными орбиталями лиганда. В то же время высокоспиновый комплекс железа(П) (t2geg) не имеет разбаланса в совокупности Г2я-орбиталей, и часто наблюдают большое квадрупольное расщепление. Если расположение лигандов вокруг железа(П) в точности октаэдрическое, то dxy-, dyz- и б/Х2-орбигали должны быть вырожденными и никакого расщепления наблюдаться не будет. Однако эта система подвержена ян-теллеровскому искажению, которое может привести к большому градиенту поля. Если энергия разделения Г2я-орбиталей от ян-теллеровского искажения имеет порядок величины kТ, то наблюдается квадрупольное расщепление, сильно зависящее от температуры. Основное состояние в искаженном комплексе можно установить, если известен знак q. Знак можно определить из МБ-спектра ориентированной системы или при исследованиях в сильном магнитном поле. Аналогичные соображения применимы к высоко-и низкоспиновым соединениям железа(Ш).
Факторы, влияющие на величину градиента поля, рассмотрены в гл. 14. Было показано, что для получения дополнительной информации о типах связи они имеют ограниченное применение.
294
Глава 15
15.4. МАГНИТНЫЕ ВЗАИМОДЕЙСТВИЯ
В магнитном поле вырождение ядерных спиновых состояний с т, = = ±1/2 и т. д. снимается. Для 57Fe правила отбора Ат,= 0, ±1 приводят к симметричному спектру из шести линий. Спектр нулевого поля диамагнитного соединения, состоящий из двух линий, расщепляется на дублет и триплет при малых т]. Дублет обусловлен переходами ±1/2-» -» + 3/2 и — 1/2—► +3/2. Если дублет лежит в направлении положительной скорости, то знаки константы квадрупольного расщепления и q положительны. Подробная интерпретация часто затруднительна, но знак т| можно установить [10]. Измерения градиента поля в ферроцене позволили установить, что он положителен [11]. Очень интересный результат состоит в том, что знак q для комплекса бутадиена с трикарбо-
А
___________I_________
О +
Скорость, мм/с Б
Рис. 15.5. Магнитное и квадрупольное расщепление в ферромагнитном соединении 57Fe.
А — диаграмма энергетических уровней; Б — ожидаемый МБ-спектр. [Copyright © 1973 McGraw-Hill Book Со. (UK) Limited. From Bancroft G. M., Mossbauer Spectroscopy. Reproduced by permission.]
Мёссбауэровская спектроскопия 295
нилом железа противоположен знаку q для комплекса циклобутадиена с трикарбонилом железа.
В образцах ферромагнитных веществ существует внутреннее магнитное поле, которое полностью снимает вырождение ядерных энергетических уровней. Такая система показана на рис. 15.5. На спектр влияют e2Qq, Н1оса1 и ориентация Н1оса1 относительно главных осей градиента электрического поля. В общем случае задача сложнее, и более подробные сведения об этом читатель может найти в работах, приведенных в общем списке литературы.
Если проводить мёссбауэровские эксперименты с парамагнитными соединениями, находящимися в магнитном поле, то можно получить много информации. Большую часть имеющихся в настоящее время результатов можно описать с помощью следующего спин-гамильтониана:
H=£>[S22-|S(S+ 1)] + E(S2-S2) + pS-0-H + S-A-i- (15.8)
— 0N P.V I • H + (2^— 1) - / (/ + 1) + В (f2 - f2)}EFG •
Таким образом, из этих экспериментов дополнительно к обычной информации, доступной из мёссбауэровского эксперимента, можно определить как параметры нулевого поля D и Е, так и компоненты константы сверхтонкого расщепления. Нижний индекс EFG в последнем члене означает, что выражение записано в системе координат, которая приводит градиент поля к диагональному виду (она может не совпадать с А). Поле, действующее на ядро, Heff, может, как полагают, представлять собой сумму приложенного поля и внутреннего поля Hint, обусловленного парамагнетизмом неспаренного электрона:
Heff = Hint + H. (15.9)
Если Hint # 0, то сверхтонкое взаимодействие приводит к эффективному полю, которое расщепляет основное и возбужденное состояния. Эти вопросы подробно обсуждались в литературе [12 -14].
15.5. МЁССБАУЭРОВСКАЯ ЭМИССИОННАЯ СПЕКТРОСКОПИЯ
Ядро 57Со за счет электронного захвата переходит в ядро 57 Fe (Т1/2 для ядра 57Fe составляет 0,1 мкс), при этом заселяется возбужденное состояние ядер железа. Для того чтобы исследовать энергетические уровни ядер 57Fe, образующихся при распаде ядер источника, испущенные у-лучи могут поглощаться стандартным поглотителем, настроенным на одну энергию. В качестве источника готовится и используется кобальтовый-57 аналог исследуемого соединения. Из этого эксперимента получают [15] информацию о короткоживущих ком
296
Глава 15
плексах железа. Необходимо быть уверенным в том, что интересующий нас комплекс железа останется нетронутым, когда в процессе распада кобальта образуются высокоэнергетические атомы железа. В табл. 15.1 представлены результаты, найденные для некоторых оксигенированных комплексов диметилового эфира протопорфирина IX с 57Со в зависимости от основания, занимающего аксиальное положение. Их следует сравнить с величинами и 8 (соответственно 2,23 и 0,27) для оксигенированного гемоглобина, полученными в абсорбционных экспериментах.
На рис. 15.6, А показан эмиссионный спектр пятикоординационного комплекса 1-метилимидазола перед оксигенированием, а на рис. 15.6, Б — спектр того же комплекса после оксигенирования.
Скорость поглотителя, мм/с
Рис. 15.6. Эмиссионные МБ-спектры 1-метилимидазольного аддукта кобальтового комплекса диметилового эфира протопорфирина IX (Л) и аддукта с О2 (Б).
Мёссбауэровская спектроскопия
297
Таблица 15.1
Результаты исследования с помощью мёссбауэровской эмиссионной спектроскопии [12] квадрупольных расщеплений и изомерных сдвигов для оксигенированных 57Со-комплексов диметилового эфира протопор-фирниа IX. В первой колонке приведены лиганды, координированные в транс-положении относительно О2.
Лиганд AEg, мм/с SFe, мм/с
1-Метилимидазол 2,17 0,29
1, 2-Диметилимидазол 2,32 0,30
Пиридин 2,28 0,27
Пиперидин 2,25 0,30
Этилметил сульфид 2,27 0,30
Метод мёссбауэровской эмиссионной спектроскопии имеет особенно важное значение в тех случаях, когда исходное соединение железа трудно синтезировать и выделить.
15.6. ПРИМЕНЕНИЯ
Для того чтобы продемонстрировать, какого типа информацию может дать МБ-спектроскопия, рассмотрим несколько применений этого метода к решению химических задач. В табл. 15.2 приведены данные, которые необходимы для исследования указанных изотопов методом МБ-спектроскопии.
Факсимиле спектров некоторых комплексов железа показаны на рис. 15.7. Как упоминалось ранее, для высокоспиновых комплексов железа с шестью эквивалентными лигандами практически сферическое электрическое поле ожидается на ядре Fe3+(d5)(t39e9), но не на ядре Fe2+(d6)(t29e9). В результате наличия градиентов поля на ядре нужно ожидать квадрупольного расщепления в спектрах высокоспиновых комплексов железа(П), но не высокоспиновых комплексов железа(111). Это следует из спектров А и Б комплексов, показанных на рис. 15.7. Для низкоспиновых комплексов железо(П) имеет конфигурацию t®9, а желе-зо(Ш) — ?29- Поэтому в комплексах сильного поля квадрупольное расщепление теперь ожидается для железа(Ш), но не для железа(П). Этот вывод согласуется с экспериментальными данными, полученными для ферро- и феррицианид-ионов. Если расположение шести лигандов в комплексе железа(П) сильного поля не приводит к их эквивалентности, как, например, в [Fe(CN)5NH3]3-, то результатом будет квадрупольное расщепление в комплексе железа(П) сильного поля. Квадрупольное расщепление грубо связано с различием в заселенностях ^/-орбиталей (см. гл. 3) выражением
<1 valence = Kd L^Nd,2 + Ndx2_y2 + Ndxy ~ dxz + N dr)] ’
Таблица 15,2'
Мёссбауэровские изотопы, представляющие химический интерес
Изотоп Энергия у-кванта, кэВ Период полураспада предшественника Полуширина, мм/с Спин 2", барны Сто-Ю’19. см2в Содержание в природе, % 104^-R
57Fe 14,4 270 сут 0,19 1/2-> 3/2 + 0,3 23,5 2,19 - 18 ±4
61Ni ‘ 67,4 1,7 ч, 3,3 ч 0,78 .3/2 - 5/2 + 0,13 7,21 1,19
67Zn 93,3 60 ч, 78 ч 3,13-Ю-4 5/2 - 3/2 + 0,17 1,18 4,11
73Ge 67 76 сут 2,19 9/2 -> 7/2 -0,26 3,54 7,76
83Kr 9,3 83 сут, 2,4 ч 0,20 9/2 -> 7/2 + 0,44 18,9 11,55 + 4 + 2
99 Ru 90 16,1 сут 0,15 5/2->3/2 >0,15 1,42 12,72
i°7Ag 93,1 6,6 ч 6,68 IO’11 1/2->7/2 - 0,54 51,35
119Sn 23,9 245 сут 0,62 1/2->3/2 -0,07 13.8 8,58 + 3,3 + 1
121Sb 37,2 76 г. 2,10 5/2->7/2 -0.29 2,04 57,25 - 8,5 ± 3
125Te 35,5 58 сут 4,94 1/2—> 3/2 + 0,19 2,72 6,99 + 1
129j 27,8 33 сут. 70 мин 0,63 7/2-> 5/2 -0,55 3,97 0 + 3
129Xe . 39,6 1,6 • 107 г. 6,84 1/2->3/2 -0,41 1,95 26,44 0,3
133Cs 81,0 7,2 г. 0,54 7/2 -> 5/2 - 0,003 1,02 100
177Hf 113,0 6,7 сут, 56 ч 4,66 7/2->9/2 + 3 1,20 18,50
18’Ta 6,25 140 сут, 45 сут 6,48-IO'3 7/2 -> 9/2 + 4,2 17,2 99,99
182W 100,1 115 сут 2,00 0->2 - 1,87 2,46 26.41 + 1,3
187Re 134,2 23,8 ч 203,8 5/2 -> 7/2 + 2,6 0,54 62,93
186Os 137,2 90 ч 2,37 0->2 + 1,54 2,89 1,64
193Ir 73,1 32 ч 0,59 3/2 ->1/2 + 1,5 0,30 62,7 + 0,6
195Pt 98,7 183 сут 17.30 1/2->3/2 - 0,63 33,8
197Au 77,3 65 ч, 20 ч 1,85 3/2->1/2 + 0,58 0,44 100 ±3
5 Copyright © 1973 McGraw-Hill Book Со. (UK) Limited. From Bancroft C.M., Mossbauer Spectroscopy. Reproduced by permission. в Квадрупольиый момент основного состояния, где и основное, н возбужденное состояния имеют I > 1/2.
Поперечное сечение поглощения мёссбауэровских у-кваитов.
Мёссбауэровская спектроскопия
299
-0,2 0 +0,2 +0,4
А
—I___I___I I____
-о,/ 0 +0,2 Б
-0,2.-0,1 0 +0,1+0,2 Г
Рис. 15.7. МБ-спектры некоторых комплексов железа(П) и железа (III).
А — бесспииовое железо(И)— FeSO4-7H2O; Б — бесспииовое железо(111) — FeCl3; В — железо(И) со спаренными спинами—K4Fe(CN)6-3H2O; Г — железо(Ш) со спаренными спинами — K3Fe(CN)6. [Brady P.R., Wigley-P.P.F., Duncan J.F., Rev. Pure Appl. Chem., 12, 181 (1962).]
где qvalence— вклад q валентных электронов, находящихся на d-орбиталях. Для р-электронов имеем
^valence Кр Е Nрг 3" P, + *
Измеренные при комнатной температуре величины и 8 для большого числа комплексов железа [1] представлены в табл. 15.3. Для комплексов железа изомерные сдвиги в положительном направлении соответствуют снижению электронной плотности вблизи ядра. Для высокоспиновых комплексов существует корреляция между изомерным сдвигом и s-электронной плотностью. Увеличение 8 на 0,2 мм/с эквивалентно снижению s-электронной плотности заряда на ядре на 8% [8]. Отрицательные величины, полученные для низкоспиновых феррицианидных комплексов по сравнению с высокоспиновыми комплексами железа(Ш), свидетельствуют о большей электронной плотности на ядре феррициа-нид-иона. Этот результат объясняли интенсивным тг-связыванием в феррицианидах, которое удаляет d-электронную плотность от иона металла, что в свою очередь снижает экранирование s-электронов. Указанный эффект приводит к увеличению s-электронной плотности на ядре и уменьшению 8. Как сильные ст-доноры, так и сильные тг-акцепторы снижают 8.
МБ-спектр вещества, приготовленного из сульфата железа(Ш) и
300
Глава 15
Таблица 15.3
Квадрупольное расщепление АЕ„ и изомерный сдви! § для некоторых соединений железа (3 и АЕ^ даны в мм/с)
Соединение ДЕе 8 Соединение AEe 8
Высокоспиновое Fe(II) Низкоспиновое Fe(II)
FeSO4-7H2O 3,2 1,19 K4 [Fe(CN)6] • 3H2O — -0,13
3,15 1,3 — -0.16
<0.1 + 0.05
FeSO4 (безводный) 2.7 1.2
Fe(NH4)2(SO4)26H2O 1,75 1,19 Na4 Fe(CN)J 10H2O <0,2 -1,01
1,75 1,3 Na3 Fe(CN)5NH3] 0.6 -0,05
FeCl2-4H2O 3,00 1,35 K2 [Fe(CN)5NO] 1,85 -0,27
FeC4H4O6 2,6 1,25 1,76 -0,28
FeF2 2,68 — Zn [Fe(CN)5NO] 1,90 -0,27
FeC2O4 2H2O 1,7 1,25
Высокоспиновое Fe(III) Низкоспиновое Fe(III)
FeCl3 • 6H2O 0,2 0,85 K3 [Fe(CN)6] — -0,12
— -0,17
FeCl3 (безводный) 0.2 0,5 0,26 -0,15
FeCl3- 2NH4C1 H2O 0,3 0,45 Na3[Fe(CN)6] 0,60 -0,17
Fe(NO3)3-9H2O 0,4 0,4
Fe2(C2O4)3 0,5 0,45
Fe2(C4H4O6)3 0,77 0,43
Fe2O3 0,12 0,47
K4Fe(CN)16, идентичен спектрам соединений, синтезированных либо из сульфата железа(П) и K3Fe(CN)6, либо путем окисления атмосферным кислородом соединения, полученного из сульфата железа(П) и K4Fe(CN)6. Спектры этих соединений говорят о том, что катион в них — высокоспиновое железо(Ш), тогда как анион—низкоспиновое железо(П).
Был исследован [9] МБ-спектр нитропруссида натрия Na2Fe(CN)5NO. Поскольку этот комплекс диамагнитен, его рассматривали ранее как содержащий железо(П) и NO+. МБ-спектр представляет собой дублет с A Eg и S, равными соответственно 1,76 и — 0,165 мм/с. Сопоставление последней величины с опубликованными результатами [8] для ряда комплексов железа позволило заключить, что она близка к величине 8 железа(1У). МБ-спектр и магнетизм согласуются со структурой, в которой имеет место интенсивное тг-связывание неспаренного электрона на совокупности г2я-орбиталей железа с неспаренным электроном азота, как это показано на рис. 15.8. Для возникновения железа (IV) в заполненную связывающую тг-орбиталь должна давать большой вклад атомная орбиталь азота, а в вакантную разрыхляющую тг-орбиталь—атомная орбиталь железа. Поскольку экранирование s-электронов d-электронами снижается, на азоте должна локализоваться большая тг-электронная плотность, а величина 8 железа должна приближаться к величине 8 железа (IV). Так как электронная плотность находится там, где ранее была разрыхляющая тг-орбиталь окиси азота, наблюдается снижение частоты валентного колебания N — О в инфракрас
Мёссбауэровская спектроскопия
301
ном спектре. Очень большое квадрупольное расщепление согласуется с очень интенсивным л-связыванием в цепочке Fe — N — О.
МБ-спектры часто оказываются полезными для определения состояния окисления атомов. Было показано, что спектры, вначале приписанные хелатам салицилальдоксима и некоторым другим хелатам высокоспинового железа (II), на самом деле являются спектрами продуктов
Рис. 15.8. Орбитали, участвующие в л-связывании группы Fe — N с гоуппой NO в Fe(CN)5NO^.
окисления железа (III). Окисленные соединения давали центровой сдвиг (относительно Na2Fe(CN)5NO-2H2O) около 0,6 мм/с и небольшое квадрупольное расщепление. Это расщепление, меньшее, чем ожидалось для железа (II), объясняли обратным л-связыванием. Аутентичное соединение, приготовленное в условиях, в которых полностью исключался контакт с кислородом, давало обычный центровой сдвиг железа(II) и большое квадрупольное расщепление (~1,4). В табл. 15.4 представлены величины центрового сдвига различных состояний окисления высокоспиновых соединений железа.
Таблица 15.4
Изомерные сдвнгн для высокоспиновых соединений железа (сдвиги относительно Na2Fe(CN)5NO-2H2O)
Состояние окисления
Изомерный сдвиг
~ +2,2
~ +0,7
+ 4
~ +0,2
+ 6 -----0,6
+ 2
~ + 1,4
Дрикамер и др. [16] исследовали влияние давления на МБ-спектры широкого ряда производных железа. Под действием давления (~ 165 кбар) на трпс-(ацетилацетонато) железо(Ш) образуется новое соединение, приписываемое производному железа (II). Наблюдаемое изменение обратимо.
Определение состояний окисления соединений олова из МБ-спектров не столь строго, как в случае соединений железа. Величины 5 ниже 2,65 мм/с часто обусловлены оловоm(IV), а большие величины — оло-вом(П). Известны и исключения. Изомерные сдвиги некоторых четырех-и шестикоординационных соединений олова (IV) значительно меняются а зависимости от средней электроотрицательности по Полингу Хр-групп, присоединенных к атому металла. Известно [17] о существовании следующих корреляций:
изомерный сдвиг (мм/с, SnX4) = 4,82 — 1,27 %
изомерный сдвиг (мм/с, SnX^-) = 4,27 — 1,16хр.
302
Глава 15
Если исключить фтор, то последнее уравнение приобретает вид изомерный сдвиг (мм/с, SnXg-) = 4,96 — 1,40 хр.
Сообщалось [18, 19] о МБ-спекграх карбонильных соединений железа Fe(CO)5, Fe2(CO)9 и Fe3(CO)12. В случае Fe(CO)5 и Fe2(CO)9 они имеют такой вид, как следует ожидать из их известных структур. Структура Fe3(CO)12, установленная на основании МБ-спектра, не согласуется с данными ИК-спектроскопии и предварительными результатами рентгеноструктурных исследований. Как видно из рис. 15.9, А, МБ-спектр
Рис. 15.9. МБ-спектр (Л) и структура (Б) Fe3(CO)12.
указывает на существование более чем одного типа железа. Внешние две полосы приписаны одному типу железа, а две внутренние—другому. Вообще площади полос грубо пропорциональны количеству присутствующего типа железа. Окончательные исследования кристаллического соединения подтвердили структуру, изображенную на рис. 15.9, Б, что согласуется и с результатами МБ-спектроскопии. Спектры Fe (II) Х2 (СО)2Р2 (где X = С1, Вг и I, а Р = фосфины или фосфиты) интерпретировали с точки зрения существования пяти различных изомеров.
С помощью МБ-спектроскопии были исследованы некоторые системы со спиновым равновесием между высоко- и низкоспиновыми комплексами железа(П). Типичными являются результаты [20], полученные для гексадентатного лиганда {4-[(6-Р)-2-пиридил]-3-азабутенил] 3амина. Спектры соединений с двумя или тремя метильными группами R характеризуют при 77 К низкоспиновое железо (II) (1Я1), тогда как при 294 К большой изомерный сдвиг и большое квадрупольное расщепление характерны для высокоспинового железа (II) (5Т2). При промежуточных температурах в спектре наблюдаются обе формы. Эти данные говорят
Мёссбауэровская спектроскопия
303
о существовании истинного равновесия (5T2i+1A1) и о том, что время жизни этих состояний по шкале времени МБ-спектроскопии велико, т. е. времена жизни должны быть не ниже 10 ” 9 с.
Известны [5, 12, 21, 22] интересные применения МБ-спектроскопии к биологическим системам. Пероксидаза хрена является гемопротеином
Рис. 15.10. Подобная кубану структура оболочки Fe4S4(S — — R — )4 некоторых ферредоксинов.
железа(III). Ее можно окислить в две одноэлектронные стадии: на первой стадии образуется соединение красного цвета, а на второй — зеленого. Изменения в МБ-спектре [23] при окислении до любой из форм интерпретировали с точки зрения образования железа (IV). Удаление второго электрона, в результате чего образуется зеленая форма, происходит, как полагают, с белка или порфирина.
МБ-спектроскопия находит широкое применение при изучении ре-докс-центров, существующих в некоторых классах железосерусодержа-щих белков. Исследована кристаллическая структура фотосинтетического (высокого потенциала) белка ферредоксина HPred из Chromatium, и показано, что он содержит оболочку из Fe4S4(SCys)4 (SCys — цистеин), имеющую подобную кубану структуру, изображенную на рис. 15.10. Аналогичные единицы присутствуют и в других белках. Соединение HPred легко окисляется до формы НРОХ. Кристаллическая структура не дает информации о состояниях окисления железа или заряде кластера. МБ-спектры нескольких белков, содержащих эту единицу, характеризуются дублетом, обусловленным квадрупольным расщеплением [24]. Путем сравнения изомерного сдвига HPred с изомерными сдвигами других соединений железа был сделан вывод [24], что оболочка состоит в среднем из двух ионов Fe3 + и двух ионов Fe2 +. Атомы железа антиферромагнитно взаимодействуют, что приводит к диамагнитному веществу, а электроны металла в этой системе делокализованы, так что существует только один тип атомов железа. Чувствительность изомерного сдвига к состоянию окисления показана в табл. 15.5.
Синтезировано [25], например, такое модельное со-
единение: [(C2H5)4N]2[Fe4S4 (SCH2C6H5)4], Известно, что заряд на его анионе равен —2, поэтому это должна быть система 2Fe2++2Fe3+. МБ-спектр [26], показанный на рис. 15.11, и кристаллическая структура идентичны спектру и структуре HPred или окисленного ферредоксина с эквивалентными атомами железа и изомерным сдвигом 0,36 мм/с, что подтверждает правильность отнесения соответствующих ферредоксинов к системам 2Fe2 + + 2Fe3 + [24].
304
Глава 15
Таблица 15.5
Центровые сдвиги для различных железосерусодержащих соединений
Соединениеа
S
Fe3+ (рубредоксин) 0,25
3Fe3 + + lFe2+ (Chromatium НРОХ) 0,32
2Fe3+ +2Fe2+ (Chromatium HPred 0,42
и окисленный ферредоксин)
lFe3+ + 3Fe2+ (восстановленный ферредоксин) 0,57
Fe2+ (рубредоксин) 0,65
2 Символ, подобный 3Fe3 + + lFe2 + , просто подразумевает среднее состояние окисления для атомов железа, соответствующее этой комбинации.
МБ-спектр [(C2H5)4N]2[Fe4S4(SCH2C6H5)4], изображенный на рис. 15.11, А, представляет собой квадрупольно расщепленный дублет, обусловленный низкой симметрией окружения железа. Линии в дальнейшем расщепляются магнитным полем, как показано на рис. 15.11, Б. Сплошная линия на рис. 15.11, Я—результат подгонки с помощью метода наименьших квадратов к лоренцевой форме линии. Сплошная ли-
Рнс. 15.11. МБ-спектр [(C2H5)4N]2[Fe4S4(SCH2C6H4)4],
А — при 1,5 К и Но = 0; Б—при 4,7 К и Но = 80 кЭ. [Holm R.H. et al., J. Amer. Chem. Soc., 96, 2644 (1974). Copyright by American Chemical Society.]
Мёссбауэровская спектроскопия 305
ния на рис. 15.11, Б — рассчитанный на ЭВМ спектр для Н = 80 кЭ, р = 0 и АЕе = 1,26 мм/с. Исходя из соответствия со спектральными данными установлено, что знак главной компоненты градиента поля положителен.
Рис. 15.12. Геометрия и координаты для транс- (Л) и i/uc-MB4A2 (5).
Как уже не раз отмечалось, градиент поля трудно интерпретировать. Однако было найдено возможным параметризовать ионы и группы, присоединенные к нейтральному иону металла, и использовать эти параметры, называемые аддитивными парциальными квадрупольными расщеплениями, для прогнозирования квадрупольного взаимодействия. Основной является модель точечного заряда. В системе координат, в которой градиент электрического поля диагоналей, вклады заряда Z в Vxx, Vyy и Vzz выражаются как
Vxx = Zer “ 3(3sin 2 0 cos2 <р — 1),
Vyy = Zer - 3(3sin2 0 sin2 <p — 1),
VZ2 = Zer ~ 3(3cos2 0 — 1),
где 0 и <p определены, как обычно для полярной системы координат. Например, для тетрагонального комплекса (рис. 15.12) мы должны взять каждый лиганд (точечный заряд) и просуммировать индивидуальные вклады в Vxx, Ууу и В табл. 15.6 приведены результаты этих расчетов для цис- и транс-комплексов, изображенных на рис. 15.12.
Суммируя вклады в табл. 15.6, для транс-комплекса получаем
Vxx = Vyy = ( - 2[А] + 2[В])е,
Кг = (4[А]-4[В])е,
20-574
306
Глава 15
Таблица 15.6"
Индивидуальные вклады точечных зарядов в тензор градиента электрического поля в транс- и 1/ис-МА2В4. Параметр [А] равен Z^e/r\
трвис-МА2В4
Лиганд (рис. 15.12) е <Р sin 0 cos 0 sin ф COS ф KJe KJe
Аз 0 0 0 1 0 1 -[А] -[А] + 2[А]
а2 180 0 0 -1 0 1 -[А] -[А] + 2[А]
Вз 90 0 1 0 0 1 + 2[В] -[В] -И
В2 90 90 1 0 1 0 -[в] + 2[В] -[в]
В3 90 180 1 0 0 -1 + 2[В] -И -И
В4 90 270 1 0 -1 0 -[в] + 2[В] -[В]
г/ис-МА2В4
Лиганд 0 Ф sin 0 cos 0 sin ф COS ф Vxx/e
Аз 90 0 1 0 0 1 + 2[A] -[A] -[A]
а2 90 90 1 0 1 0 -[A] + 2[A] -[A]
Вз 0 0 0 1 0 J -И -[B] + 2[B]
в2 180 0 0 -1 0 1 -[B] -[B] + 2[B]
В3 90 180 1 0 0 -1 + 2И -[B] -[B]
В4 90 270 1 0 -1 0 -[B] + 2[B] -[B]
сору 1 Copyright © 1973 McGraw-Hill . Reproduced by permission. Book Co, (UK) Limited. From Bancroft G. M., Mossbauer Spectros-
где [А] и [В]—неопределенные вклады лигандов А и В. Для цис-комплекса имеем
Vxx= ^ = ([А] - [В])е, Ии = (-2[А] + 2[В])е.
Отношение квадрупольных расщеплений транс- и ц нс-комплексов равно
(4[А]~ 4[В])е _
(— 2[А] + 2[В])е
Если возможны оба изомера, то с помощью МБ-спектроскопии их просто различить, поскольку изомеры характеризуются приблизительно таким отношением. В приближении парциального квадрупольного взаимодействия величины ПКР эмпирически относят к [А] и [В] и предсказывают градиент поля. С помощью приведенных выше уравнений легко определяется цис- или транс-изомерия. Для другой геоме
Мёссбауэровская спектроскопия
307
трии необходимо вывести подходящие уравнения. Соединение Fe(CO)2(PMe3)2I2 может существовать в пяти изомерных формах, рассчитанные квадрупольные расщепления которых значительно отличаются. Можно синтезировать два изомера с наблюдаемыми квадрупольны-ми расщеплениями —0,90 и +1,31 мм/с. Они легко идентифицируются как изомер с транс-Р, цис-I, цис-СО и изомер с транс-расположением всех соответствующих групп [27]. Параметры парциального квадрупольного расщепления (ПКР) для различных лигандов [27], связанных с железом(П), приведены в табл. 15.7.
Таблица 15.7а
Параметры [27] парциального квадрз iki.ii.iioi о расщепления (ПКР) лигандов для желе1а(П)
Лиганд ПКР Лиганд ПКР
NO + 6 + 0,01 P(OPh)3 -0,55
Х“ -0,30 CO -0,55
N2 -0,37 PPh2Et -0,58
N.7 -0,38 PPh2Me -0,58
CH3CN -0,43 depb/2 -0,59
SnCh -0.43 P(OEt)3 -0,63
H2O® -0,45 depe/2 -0,65
SbPh3 -0,50 P(OMe)3 -0,65
NCS“ -0,51 PMe3 -0,66
AsPh3 -0,51 dmpe/2 -0,70
NH® -0,52 ArNC -0,70
NCO -0,52 CN-6 -0,84
PPh3 -0,53 H -1,04
3 Copyright © 1973 McGraw-Hill Book Со. (UK) Limited. From Bancroft Mdssbauer Spectroscopy. Reproduced by permission.
б Величины ПКР выведены из даииых, полученных при комнатной температуре.
Если использовать величины ПКР для лигандов, представленные в табл. 15.7, наряду с соответствующими уравнениями для комплексов в табл. 15.8, то получатся предсказанные величины (см. табл. 15.8) [27].
Сообщалось [27] о других параметрах, которые можно использовать в случае соединений олова. Этот подход с большим успехом был использован для многих других систем [27, 28]. Очевидно, в модели точечного заряда имеются сомнительные места [29], но требуется провести больше работ, чтобы можно было сказать, когда ее нельзя использовать эмпирически.
Знак и величина градиента поля могут быть использованы для получения информации об электронном основном состоянии комплекса иона переходного металла. Приближенную величину градиента поля для раз-
308
Глава 15
Таблица 15.8“
Предсказанные н наблюдаемые [27, 28] квадрупольные расщепления (в мм с) прн 295 К
Комплексы Наблюдаемые Предсказанные
mpaHc-FeCl2(ArNC)4 + 1,55
t/uc-FeCl2(ArNC)4 -0,78 -0,78
[FeCl(ArNC)5] С1О4 0,73 + 0,78
mpaHc-Fe(SnCl3)2(ArNC)4 + 1,05
t/uc-Fe(SnCl3)2(ArNC)4 0,50 -0,52
t/uc-FcCISnCI,(ArNC)4 0,61 -0,69 (т]=0,60)
mpmtc-FcH2(dcpb)2 -1,84
mpaHc-FeHCl(depe)2 <0,12 -0,20
mpaHc-Fe(EtNC)4(CN)2 -0,60
uuc-Fc(EtNC)4(CN)2 0,29 + 0,30
mpaHc-[FeH(ArNC)(depe)2] BPh4 -1,14 -0,98
mpaHc-[FeH(CO) (depe)2] BPh4 1,00 -0,46
а Copyright © 1973 McGraw-Hill Book Со. (UK) Limited. From Bancroft G. M., Mossbauer Spect-roscopy. Reproduced by permission.
личных основных состояний можно оценить путем добавления вкладов заселенных d-орбиталей, приведенных в табл. 15.9.
Параметр “ 3> представляет собой величину ожидания 1/г3 для со-
Таблица 15.9
Значение н г] для различных атомных орбиталей
Орбиталь q n
Pz 0
Рх +|«3> -3
Ру + |<r-> + 3
dx2-y2 + *<r-3> 0
dz2 ->3> 0
dxy 0
dxz -?«> + 3
dyz -3
Мёссбауэровская спектроскопия
309
ответствующей орбитальной функции. Эта таблица построена путем использования различных орбитальных функций для расчета матричных элементов :
, 3cos2 0 — 1 q^ = q = -<^l-----з----М>,
г
., , 3sin2 0cos2tp । , х riq = <Ф1------р--— 1Ф>-
В типичном случае прогнозируется, что основное состояние ферроцена a^/dy), -/) должно иметь значение qZ2, равное 2( —4/7<г-3» +
+ 4(4/7)<г~3» = 8/7<г~3}. Наблюдается большой положительный градиент поля. При образовании катиона феррицения квадрупольное расщепление уменьшается, что согласуется с потерей е2<гэлектРона [30].
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. Debrunner Р. G., Frauenfelder Н., in Introduction to Mdssbauer Spectroscopy, ed. May L., Plenum Press., New York, 1971.
2. Bancroft G. M., Mdssbauer Spectroscopy — An Introduction for Inorganic Chemists and Geochemists, McGraw-Hill Book Co. (UK). Limited, 1973.
3. Weissbluth M„ biiuctuie and Bonding, 2, 1 (1967).
4. Bearden A. J., Dunham W.R., Structure and Bonding, 8, 1 (1970).
5. Gibb T.C., Greenwood N.N., Mdssbauer Spectroscopy, Chapman and Hall, 1971.
6. Duncan J. F., Wigley P.W.R., J. Chem. Soc., 1963, 1120.
7. Watson R.E., Freeman A. J., Phys. Rev., 120, 1125 (1960).
8. Walker L.R., Wertheim G. K., Jaccarino V, Phys. Rev. Letters, 6, 98 (1961).
9. Epstein L. M., J. Chem. Phys., 36, 2731 (1962).
10. Collins R.L., Travis J. C„ Mdssbauer Effect Methodology, 3, 123 (1967).
11. Collins R.L., J. Chem. Phys., 42, 1072 (1965).
12. Miinck E„ in The Porphirins, Dolphin D.H., ed., Academic Press, New York, 1975.
13. Oosterhuis W.T., Structure and Bonding, 20, 59 (1974).
14. Lang G., Quart. Rev. Biophys., 3, 1 (1970).
15. Marchant L., Sharrock M., Hoffman B.M., Miinck E., Proc. Nat. Acad. Sci. USA, 69, 2396 (1972).
16. Champion A.R., Vaughan R.W., Drickamer H.G., J. Chem. Phys., 47, 2583 (1967) и ссылки в этой работе.
17. Parish R.V., Prog. Inorg. Chem., 15, 101 (1972).
18. Herber R.H., Kingston W. R„ Wertheim G.K., Inorg. Chem., 2, 153 (1963).
19. Herber R.H., King R.B., Wertheim G.K., Inorg. Chem., 3, 101 (1964).
20. Hoselton M. A.. Wilson L. J.. Drago R.S.. J. Amer. Chem. Soc.. 97. 1722 (1975).
21. Marshall W., Lang G., Proc. Phys. Soc. (London), 87, 3 (1966).
22. Moss Т.Н., Ehrenberg A., Bearden A.J., Biochemistry, 8, 4159 (1969).
23. Debrunner P„ in Spectroscopi Approaches to Biomolecular Conformation, ed. Urry D. W., American Medical Association Press, Chicago, 1969.
24. Jhompson C.L., et al., J. Biochem., 139, 97 (1974) и ссылки в этой работе.
25. HolmR.H., Endeavour, 1975, 38 и ссылки в этой работе.
26. Holm R.H., et al., J. Amer. Chem. Soc., 96, 2644 (1974) и ссылки в этой работе.
27. Bancroft G. М., Coord. Chem. Rev., 11, 247 (1973) и ссылки в этой работе.
28. Bancroft G.M., Butler К. D., Inorg. Chim. Acta, 15, 57 (1975).
29. Marks A.P., Drago R.S., Herber R.H., Potasek M.J., Inorg. Chem., 15, 259 (1976).
30. Morrison W.H., Jr., Hendrickson D.N., Inorg. Chem., 13, 2279 (1974).
310
Глава 15
ПЕРИОДИЧЕСКИЕ ИЗДАНИЯ
1. Mossbauer Effect Methodology, Vols. 1—9, Plenum Press, New York, 1965.
Упражнения
1. Какой эффект, увеличивающий электронную плотность на ядре, влияет на относительные энергии основного и возбужденного состояний 57Fe и 119Sn? Объясните ожидаемый изомерный сдвиг, исходя из радиусов эффективных ядерных зарядов этих состояний.
2. Предположим, что вы прочитали статью, в которой автор утверждает: в МБ-спектре низкоспинового комплекса железа(Ш) две линии являются результатом ян-теллеровского искажения. Подвергните критике этот вывод.
3. Нарисуйте структуру SnF4 и объясните, почему квадрупольное расщепление наблюдается в этом соединении, а не в SnCl4.
4. Предположим, что вас интересует, имеет ли место л-связывание в Sn — — О или Sn — S и где оно больше: в соединении (C6H5)3SnOCH3 или (C6H5)3SnSCH3. Опишите эксперимент с использованием МБ-спектроскопии, который мог бы объяснить эту проблему.
5. В каком из соединений (SnCl2Br2, SnCl3Br или SnF3I) величина AEg выше и почему?
6. Проанализируйте изображенный ниже МБ-спектр продукта реакции сульфата железа с феррицианидом калия.
7. Ниже представлен МБ-спектр Fe(CO)5 при температуре жидкого азота, а. Почему необычные единицы оси х эквивалентны энергии?
мм/с
б. Что обусловливает появление наблюдаемого дублета? Покажите (и обозначьте), из каких энергетических уровней он возникает.
Мёссбауэровская спектроскопия
311
8. Используя выражение для точечного заряда, укажите три диагональные компоненты градиента электрического поля для двух изомеров МА3В3. Предскажите знак квадрупольного расщепления в этих изомерах.
9. Соединение (CH3)2SnCl2 представляет собой полимер, в котором мостиковые атомы хлора имеют октаэдрическую координацию вокруг атома олова. Метильные группы занимают транс-положения. Рассматривая связь СН3 — Sn в качестве оси z градиента поля, предскажите знаки q и e2Qq.
10. Какое из приведенных ниже соединений имеет наибольшее квадрупольное расщепление для меченого атома:
а. цис- цли транс- * Fe(CO)4Cl2? t
б. (CH3)3SnCl(C3„) или полимерный (CH3)3SnCl (D3h) с мостиковыми атомами хлора вокруг атома олова)?
в. Высокоспиновый цис- или транс-* Fe(NH3)4Cl2?
11. Фермент путидаредоксин имеет два центра с двумя атомами железа. Окисленная форма характеризуется приведенным ниже спектром.
___I_________I_________I_________I_________I_________I
-0,1 0 +0,1
Скорость, см/с
g-Тензоры анизотропны. Одинаковы или различны центры фермента, содержащие атомы железа?
12. Какой из приведенных ниже МБ-спектров принадлежит K3Fe(CN)6 и K4Fe(CN)6 и почему?
13. В статье [J. Amer. Chem. Soc.. 97, 6714 (1975)] исследуют ряд квадратно-пирамидальных комплексов железа(Ш) с набором N4S донорных атомов (сера — аксиальный донор). Электронная струк1ура этих комплексов сильно
312
Глава 15
зависит от точной природы макроциклического лиганда N4 и аксиального лиганда, о чем свидетельствуют результаты различных спектральных исследований.
а. Используя диаграммы одноэлектронных энергетических уровней, покажите, какие спиновые состояния можно в действительности ожидать для комплексов железа(Ш), имеющих структуру квадратной пирамиды. Вспомните, что относительные энергии d-орбитали могут слегка меняться в зависимости от набора доноров.
б. Используя представленные ниже спектры, отнесите возможные спиновые состояния к комплексам А, Б, и В. Объясните, как характеристики МБ-спект-ров повлияли на ваше решение и почему.
Скорость, мм/с
16. ИОНИЗАЦИОННЫЕ МЕТОДЫ: МАСС-СПЕКТРОМЕТРИЯ, ИОННЫЙ ЦИКЛОТРОННЫЙ
РЕЗОНАНС И ФОТОЭЛЕКТРОННАЯ
СПЕКТРОСКОПИЯ*
МАСС-СПЕКТРОМЕТРИЯ
16.1. ОПИСАНИЕ ПРИБОРА И ХАРАКТЕРИСТИКА СПЕКТРОВ
Существует много различных типов масс-спектрометров. Детали конструкции и относительные достоинства различных типов приборов описаны в литературе [1—7]. Большинство основных принципов масс-спектрометрии можно продемонстрировать, описав принцип действия простого масс-спектрометра, изображенного на рис. 16.1. Образец, находящийся в емкости, вводится через отверстие, входит в ионный источник а и проходит через электронный пучок в точке в, пучок обозначен штриховой линией. При взаимодействии образца с электронами, имеющими достаточную энергию, образуются положительные ионы, движущиеся по направлению к ускоряющим пластинам гид, поскольку между задней стенкой (напускной щелью) и передней стенкой этого устройства существует небольшая разность потенциалов. Отрицательные ионы притягиваются задней стенкой, которая заряжена положительно относительно передней стенки, и разряжаются на ней. Положительные ионы проходят через пластины гид, ускоряются под действием большой разности потенциалов (несколько тысяч вольт) между этими пластинами и покидают ионный источник через отверстие б. Заряженные ионы движутся по круговой орбите под влиянием магнитного поля. Полуокружность, помеченная е, есть траектория движения ускоренного иона в магнитном поле напряженности Н. Радиус полуокружности г зависит от следующих параметров: 1) ускоряющего потенциала V (т. е. от разности потенциалов между ускоряющими пластинами г и б), 2) массы иона т, 3) заряда иона е и 4) напряженности магнитного поля Н. Связь между этими параметрами выражается уравнением**
* Раздел, посвященный фотоэлектронной спектроскопии, написан проф. Д. Хендриксоном из Иллинойского университета.
** Это уравнение выводится просто: потенциальная энергия иона eV превращается в кинетическую энергию -mv2 (где т — масса, a v — скорость). При полном ускорении
eV = ^mv2. (16.2)
В магнитном поле центробежная сила mv2/r уравновешивается центростремительной силой Hev. Определяя г из Hev = mv2/r, получаем выражение г = mv/eH. Если с его помощью исключить v из уравнения (16.2), то получится уравнение (16.1).
314
Глава 16
т Н2г2
~ё~ 2V
(16.1)
Если ионы проходят через щель ж в детектор, то появляется сигнал. Для того чтобы обеспечить свободное движение ионам, источник
Напускная система
Рис. 16.1. Схема масс-спектрометра с 180°-ным отклонением.
и путь, который проходят ионы, должны находиться в условиях разрежения при давлении около 10 “ 7 мм рт. ст. Давление пара образца у напускной щели должно быть по крайней мере 10 ~2 мм рт. ст. при температуре напускной системы, хотя специальные методы позволяют работать и при более низких давлениях. Как правило, для эксперимента достаточно 1 или 2 мкмолей вещества.
Поскольку параметры Н, V и г представляют собой измеряемые величины, можно определить отношение m/е. Отметим, что двузарядные положительные ионы с массой 54 дают такую же линию, что и однозарядный ион с массой 27. При обычных условиях работы масс-спектрометра большинство ионов несет один заряд. Двузарядные ионы встречаются гораздо-^реже, а ионы, несущие большее число зарядов, в значительных концентрациях не образуются.
Регистрация масс-спектра заключается в определении отношения т/е для всех фрагментов, образующихся в процессе бомбардировки молекулы пучком высокоэнергетических электронов. Для того чтобы осуществить это, можно двигать щель детектора и измерять величину г для всех частиц, непрерывно генерируемых при электронной бомбарбировке в ионном источнике. Экспериментально такой вариант неосуществим. Значительно проще непрерывно варьировать Н или V [см. уравнение
Ионизационные методы
315
(16.1)], так чтобы все частицы в конечном счете двигались по полуокружности фиксированного радиуса. Интенсивность сигнала, непосредственно связанная с числом ионов, попадающих в детектор, может быть построена как функция Н или V в зависимости от того, какой из этих параметров изменяется. На практике проще всего измерять меняющийся потенциал, поэтому напряженность поля часто поддерживают постоянной. Если Н и г постоянны, то V обратно пропорциональна m/е [в
Рис. 16.2. Масс-спектр фрагмента в диапазоне т/е от 40 до 48. Интенсивность пиков автоматически снижена, и число горизонтальных линий над каждым пиком указывает коэффициент снижения.
соответствии с уравнением (16.1)], и, чтобы получить обычный масс-спектр, отношение т/е можно построить как функцию интенсивности сигнала. На рис. 16.2 изображена узкая область типичного масс-спектра.
В приборах с варьируемым потенциалом результаты легко представить через величины т/е. Как можно видеть из сравнения различия в разности потенциалов, соответствующих величинам т/е 400 и 401, с различием для т/е 20 и 21 [рассчитанных по уравнению (16.1)], точность меняется с массой. В современных приборах низкого разрешения можно добиться точности в интервале (±0,4— ±1) т/е до 1000 единиц массы. При высоком разрешении точность составляет 2—3 м. д. при очень больших массах (> 3000), если имеются стандартные соединения, пики которых отличаются на 10—12% от пиков неизвестных масс. Сполна фторированный керосин—одно из таких распространенных стандартных соединений, положение линий которого известно вплоть до 900 единиц массы.
В различных приборах возможны некоторые изменения в принципе действия масс-спектрометра, описанного выше; о некоторых из них мы коротко расскажем. Один тип масс-спектрометров аналогичен описанному выше, если не считать того, что он регистрирует спектр отрицательно заряженных частиц. В другом, времяпролетном приборе вместо траектории-полуокружности реализуется прямолинейная траектория (в
316
Глава 16
отсутствие магнитного поля). Ионы получают не непрерывно, а импульсами, и им позволяют диффундировать к детектору. Тяжелые ионы движутся медленнее, чем легкие. Спектр представляет собой график зависимости интенсивности от времени пролета частицы. Имеются масс-спектрометры с простой и двойной фокусировкой. Главное различие между ними заключается в наличии в последнем электростатического сектора, который приводит к фокусировке ионов по энергии до того, как они
Рис. 16.3. Представление масс-спектра этанола.
подвергнутся анализу магнитным полем. В результате этого значительно улучшается разрешение на коллекторе. В лучших приборах с двойной фокусировкой достигается точность в 1 м. д.
В масс-спектрометрах для бомбардировки образца обычно используются электроны с энергией 70 эВ, хотя напряжение можно варьировать в широких пределах. В спектрометрах с ионизацией полем [8], чтобы добиться эффекта ионизации, используют электрическое поле напряженностью 107—108 В/см. В этом методе молекула получает значительно меньшее количество энергии, и ионизационный процесс называется «мягким». Электрон при этом удаляется за счет квантовомеханического туннельного эффекта. В последующих разделах обсуждаются некоторые достоинства различных ионизационных методов.
Масс-спектр, регистрируемый прибором, может быть протяженным и неудобным для записи. Поэтому данные часто представляют в виде нового графика, где по ординате откладывают интенсивность в виде отрезков прямой (относительное содержание каждого фрагмента), а по абсциссе — отношение т/е. Содержание фрагмента выражают в процентах интенсивности его пика относительно наиболее интенсивного пика в спектре. Часто полезно интенсивность выразить через единицы полной ионизации S. В этом случае ее выражают в процентах вклада, который данный пик дает в полную ионизацию; он получается путем деления интенсивности данного пика на сумму интенсивностей всех пиков. Если масс-спектр не регистрируют во всем диапазоне от 1 до молекулярной массы вещества, то нижний предел отмечают на ординате с помощью
Ионизационные методы
317
нижнего индекса у S (например, S12 означает, что спектр регистрировали, начиная с т/е 12 до молекулярной массы вещества). Типичный график представлен на рис. 16.3. Для сопоставления интенсивностей различных пиков в одном и том же спектре можно вполне ограничиться относительным содержанием. Для сопоставления интенсивностей пиков в различных спектрах необходимо использовать единицы полной ионизации. Как правило, пики с интенсивностью, равной 1% S, регистрируются легко.
Определим теперь разрешающую способность прибора. В большинстве исследований органических и неорганических соединений необходимо знать отношение т/е с точностью в одну единицу (т. е. это 249 или 250). Разрешение прибора иногда выражают как m/Am, если два пика, т и т + Ат, разделены и минимальная интенсивность между двумя пиками составляет только 2% полной т. Например, разрешение 250 означает, что два пика с т/е 250 и 251 разделены и что в минимуме между ними перо возвращается в положение, соответствующее не более 2% полного ионного тока (являющегося характеристикой интенсивности на рис. 16.2) относительно базисной линии. В приборах с худшим разрешением это невозможно для пиков с большими массами, и значение отношения т/е, для которого пики разрешаются, служит критерием определения разрешения.
16.2. ПРОЦЕССЫ, ПРОИСХОДЯЩИЕ
ПРИ СТОЛКНОВЕНИИ
МОЛЕКУЛЫ С ВЫСОКОЭНЕРГЕТИЧЕСКИМИ ЭЛЕКТРОНАМИ
Важно подчеркнуть достаточно очевидный момент, что детектор анализирует только те частицы, которые попадают в него. Поэтому мы должны рассматривать не только частицы, получающиеся в ионизационных процессах, но и реакции, в которых могут участвовать эти частицы в течение 10“ 5 с, необходимых для их движения от ускоряющих пластин до детектора. Если молекула А бомбардируется электронами средней энергии, то начальные процессы, в которых она участвует, можно выразить с помощью уравнений
А + е - А + +2е, (16.3)
А + е -> А"+ 1 + (и + 1)е, (16.4)
А + е -> А-. (16.5)
Процесс, описываемый уравнением (16.3), наиболее распространен и наиболее важен в масс-спектрометрии. Он реализуется в том случае, если энергия бомбардирующего электрона равна или выше энергии ионизации молекулы (7—15 эВ). Если энергия бомбардирующего электрона равна потенциалу ионизации, то вся она должна быть передана молекуле, чтобы удалить из нее электрон. Вероятность такого события мала.
318
Глава 16
По мере увеличения энергии электронного пучка вероятность ионизации при столкновении возрастает и возникают пики с большей интенсивностью. При дальнейшем росте энергии электронов большая ее часть передается образующемуся молекулярному иону. Она может быть настолько большой, что в ионе рвутся связи, и происходит фрагментация частицы. Ускоряющий потенциал бомбардирующего электрона, которого только-только хватает для начала фрагментации, называется потенциалом возникновения фрагментарного иона. Если энергия электрона достаточно высока, то в молекуле может происходить разрыв более чем одной связи. Следующая последовательность реакций описывает процессы с участием гипотетической молекулы В — С — D — Е, когда она бомбардируется электронами:
а) Процесс ионизации:
BCDE+e -> BCDE++2e (16.6)
б) Фрагментация положительного иона:
BCDE+ -> В+ + CDE- (16.7)
BCDE+ -> ВС+ + DE- (16.8)
ВС+ -> В+ + с- (16.9)
или ВС+ -> В- + С+ (16.10)
BCDE+ -> DE+ + ВС- (16.11)
DE+ -> D+ + Е- (16.12)
или DE+ -> D- + Е+ (16.13)
BCDE+ -> ВЕ+ + CD - и т. д. (16.14)
BCDE+ -> CD+ + BE- и т. д. (16.15)
в) Образование пар:
BCDE+ + е -> ВС+ +DE“ +е (16.16)
г) Резонансный захват:
BCDE + e -> BCDE (16.17)
Возможны и другие типы расщепления иона, но только положительные ионы могут двигаться к детектору и приводить к появлению пиков в масс-спектре. При реализации приведенной выше схемы, если массы В, С, D и Е различны, в масс-спектре появятся пики, соответствующие фрагментам В +, ВС +, С +, DE +, D +, Е +, BE + и CD +. Для того чтобы ион BCDE+ расщепился на В+, С - и DE- [уравнение (16.9)], а не на ВС++ DE- [уравнение (16.8)], в молекулу BCDE необходимо ввести больше энергии.
Ионизационные методы
319
В уравнении (16.14) ион в процессе диссоциации перегруппировывается, что приводит к образованию фрагментов со связями, которых нет в исходной молекуле BCDE. Эти процессы перегруппировки осложняют интерпретацию масс-спектра, и, чтобы предсказать, когда будет происходить подобный процесс, необходимо знать много примеров. Как правило, к этому прибегают только в том случае, когда нужно объяснить появление пиков неожиданных масс или неожиданной интенсивности.
Ион-молекулярные реакции могут приводить к появлению в масс-спектре пиков, которые соответствуют массам, большим, чем молекулярная масса образца. Этот процесс можно представить уравнением
BCDE+ + CDE- -> BCDEC+ + DE- (16.18)
В ходе реакции (16.18) происходит столкновение молекулярного иона с нейтральной молекулой; эта реакция—процесс 2-го порядка, и скорость ее пропорциональна произведению концентраций реагентов. Интенсивность пиков, получающихся благодаря этому процессу, будет зависеть от произведения парциальных давлений BCDE+ и CDE. Изучая масс-спектр при различных давлениях, можно видеть, что относительные интенсивности этих пиков меняются, и поэтому такой процесс можно легко идентифицировать.
Все рассмотренные выше реакции представляют собой мономолеку-лярные процессы распада. Генерация ионов в ходе электронной бомбардировки часто приводит к потере наименее прочно удерживаемого электрона, и ионы часто образуются в колебательно возбужденных состояниях с избытком внутренней энергии. В некоторых молекулах образца происходит потеря низкоэнергетического электрона, что приводит к иону в электронно возбужденном состоянии. Ион в возбужденном состоянии может подвергаться внутренней конверсии энергии, в результате чего он переходит в основное электронное состояние с избытком колебательной энергии. Молекула может диссоциировать в любое из возбужденных состояний, участвующих во внутренних конверсиях с безызлучательным переносом энергии. В этом случае ион фрагментирует, как только он начинает колебаться. Таким образом, в данном образце получаются ионы с широким энергетическим распределением, и фрагментация может происходить по различным механизмам. Полезно рассмотреть временные шкалы для некоторых обсужденных процессов. Время одного валентного колебания составляет ~ 10“ 13 с, максимальное время жизни возбужденного состояния—около 10“8 с и время, которое ион проводит в ионизационной камере масс-спектрометра, равно 10" 5—10" 6 с. Следовательно, для перехода иона с избыточной электронной энергией в более низкое электронно возбужденное состояние с избытком колебательной энергии времени вполне хватает. Поэтому мы наблюдаем процессы в ионизационной камере через регистрируемые молекулярные ионы в различных энергетических состояниях, которые подвергаются быстрой внутренней конверсии энергии, образуя индивидуальные ионы с различным количеством избыточной энергии. Фрагментация протекает по первому порядку с различными
320
Глава 16
скоростями в зависимости от электронного состояния и избытка колебательной энергии индивидуального иона. Вот почему могут происходить все различные процессы, характеризующие фрагментацию иона BCDE+, и все они могут найти свое отражение в масс-спектре.
16.3. ПРИМЕНЕНИЕ МАСС-СПЕКТРОМЕТРИИ
ДЛЯ ИДЕНТИФИКАЦИИ
Применение масс-спектрометрии для идентификации очевидно. Чтобы получить воспроизводимый спектр, обычно используют электронный пучок с энергией 40 — 80 эВ, поскольку этот ускоряющий потенциал выше потенциала возникновения большинства фрагментов. Как показывают уравнения (16.6) — (16.16), может происходить много различных процессов фрагментации, приводящих к большому числу пиков в спектрах простых молекул. На рис. 16.3 изображены пики достаточной интенсивности, обнаруженные в масс-спектре этанола. Учитывая очень слабые пики, которые на этом рисунке не показаны, в общей сложности в масс-спектре этанола наблюдается около 30 пиков. Эти пики низкой интенсивности представляют большую ценность для идентификации, но обычно при интерпретации спектра (т. е. при отнесении процессов фрагментации, приводящих к этим пикам) их не рассматривают. Полезная сводка литературных источников по масс-спектрам многих соединений (в основном органических) приведена в списке литературы в конце главы. Интересный пример идентификации продемонстрирован на рис. 16.4, где показаны масс-спектры трех изомеров этилпиридина. Спектры этих трех очень сложных соединений заметно различаются, что представляет ценность для идентификации. Оптические антиподы и рацематы дают идентичные спектры. Проблему при идентификации создают примеси, поскольку основные фрагменты этих примесей приводят к появлению в масс-спектре нескольких пиков низкой интенсивности. Если одно и то же вещество приготовить в двух различных растворителях, то спектры могут достаточно различаться при условии, что весь растворитель не удален из вещества. Загрязнение углеводородной смазкой также может привести ко многим линиям.
16.4. ИНТЕРПРЕТАЦИЯ МАСС-СПЕКТРОВ
Интерпретация масс-спектра представляет собой отнесение каждого основного пика в спектре к определенному фрагменту. Интенсивный пик соответствует высокой вероятности образования иона в процессе фрагментации. В отсутствие перегруппировки [уравнение (16.14)] расположение атомов в молекуле часто может быть установлено на основании масс образующихся фрагментов. Например, интенсивный пик при т/е 30 в спектре метилгидроксиламина говорит в пользу структуры CH3NHOH по сравнению со структурой H2NOCH3, поскольку этот пик может возникнуть только при разрыве связи О — N в первом соединении, но не может быть результатом какого-либо простого механизма
Ионизационные методы
321
т/е
Рис. 16.4. Масс-спектры трех изомеров этилпиридина. (© 1960 McGraw-Hill
Book Company. From Biemann К., Mass Spectrometry. Reproduced by permission.)
фрагментации в последнем соединении. Для определения структуры важнее фрагменты с большей массой, нежели с небольшой.
Часто отнесению пиков в масс-спектре помогают соображения о вероятных продуктах фрагментации различных молекулярных структур.
21 -574
322
Глава 16
Энергия, необходимая для получения фрагмента из молекулярного иона, зависит от энергии активации расщепления связи, которая часто связана с прочностью разрываемой связи. Распределение регистрируемых ионов зависит не только от этого, но и от стабильности образующегося положительного иона. В большинстве случаев обнаружено, что стабильность положительного иона имеет наибольшее значение, а она в свою очередь связана с эффективностью, с которой результирующий фрагмент может делокализовать положительный заряд. Фрагментация НОСН2—CHjNHj происходит с образованием -CHjOH и CHjNHj (m/> = 30) или и СН2ОН+ (т/е = 31). Поскольку атом азота не
обладает такой электроотрицательностью, как атом кислорода, резонансная форма СН2 = NHj делает этот ион более устойчивым, чем аналогичная форма СН2 = ОН +. Поэтому заряд более эффективно делокализован по частице CH2NH^, чем по частице СН2ОН + , и пик т/е = 30 в десять раз интенсивнее, чем пик т/е = 31. Сера стабилизирует заряд не так эффективно, как кислород, поскольку л-связывание сера — углерод не столь эффективно, как л-связывание кислород — углерод. Поэтому пик т/е = 31, обусловленный фрагментом СН2ОН + из HSCH2CH2OH, имеет вдвое большую интенсивность, чем пик т/е = 47, обусловленный CH2SH +.
Если при этом образуются более устойчивые частицы, то положи-
тельные ионы перегруппировываются. Например, ион —СН2+
перегруппировывается в ион Г |1 •
Интенсивность пика этого фрагмента будет зна-
чительно выше по сравнению с тем случаем, если эту перегруппировку не учитывать.
Появление большого числа различных фрагментов часто помогает установить структуру молекулы. Однако даже в этом случае необходимо соблюдать осторожность. Ион, образующийся в ионизационной камере, подвергается многим колебательным процессам; эти процессы могут сопровождаться перегруппировками с образованием связей, которых нет в исходном соединении [см., например, уравнение (16.14)]. Образование новых ионов затрудняет установление химических процессов, которые приводят к появлению в масс-спектре различных пиков. Это в свою очередь создает трудности для выяснения влияния прочности связи или других свойств молекулы на относительные количества образующихся ионных фрагментов. Была предпринята попытка количественно рассмотреть масс-спектрометрическую фрагментацию на основании так называемой квазиравновесной теории [10]. Внутреннюю энергию распределяют по всем возможным осцилляторам и ротаторам молекулы и рассчитывают скорости распада по различным направлениям. Каждому колебательному уровню приписывается весовой фактор или частотный фактор (т. е. энтропийный член). Для молекулы реального размера полный анализ сложен. Вводятся приближения, приводящие
Ионизационные методы
323
к высокопараметризованному подходу [10]. В настоящее время квази-равновесная теория—в большей степени область исследования, нежели ценный метод спектрального анализа.
Масс-спектрометрическое правило сдвига [11] широко используется при установлении структуры алкалоидов и иллюстрирует основную идею общей применимости. Часто можно определить местонахождение заместителя в молекуле, если существуют низкоэнергетические направления распада сложной молекулы и если на этот распад влияет заместитель. Для этого находят фрагмент, молекулярная масса которого увеличилась на массу, соответствующую массе заместителя или характеристического фрагмента последнего.
Разработан метод использования масс-спектрометрии для определения последовательности чередования пептидов с короткой цепью. Читателя, интересующегося этим вопросом, мы отсылаем к обзорной статье [12].
Низкая летучесть многих веществ затрудняет их анализ с помощью масс-спектрометрии. Летучесть часто можно увеличить путем преобразования полярных групп в молекуле, например, карбоксильную группу можно превратить в метиловый эфир или триметилсилильный эфир. При исследовании низколетучих веществ очень удобен метод ионизации полем (см. ниже).
Сочетание масс-спектрометрии с газожидкостной хроматографией дает превосходный метод анализа смесей. В этом случае требуются очень небольшие количества вещества. Масс-спектрометр используется в качестве детектора в газожидкостной хроматографии, и многочисленные масс-спектры регистрируются по мере поступления компонентов из колонки. Частично разрешенные пики в хроматограмме легко идентифицируют по изменению во времени масс-спектра вещества, соответствующего этому пику.
Многочисленные примеры и подробное рассмотрение факторов, приводящих к образованию стабильных ионов органических соединений, можно найти в монографиях, цитированных в списке литературы, и в работе [9]. В литературе обсуждаются и общие принципы прогнозирования перегруппировок образующихся ионных фрагментов. Если исходя из данной структуры можно объяснить появление в спектре пиков основных фрагментов и отнести эти пики к фрагментам, основываясь на разумных процессах фрагментации, то такое отнесение послужит важным доводом в пользу существования именно этой структуры.
16.5. ВЛИЯНИЕ ИЗОТОПОВ НА ХАРАКТЕР
МАСС-СПЕКТРА
Если исследовать вещество, в котором элемент имеет более чем один стабильный изотоп, достаточно распространенный в природе, то для каждого фрагмента, содержащего этот элемент, наблюдается более чем один пик. В спектре СН3Вг наблюдаются два пика почти равной интенсивности при т/е = 94 и 96, что соответствует в основном (СН3—
21*
324
Глава 16
79Вг)+ и (СН3 81Вг)+. Изотопы 79Вг и 81 Вг содержатся в природе почти в равных количествах (50,54 и 49,46%), поэтому два пика фрагментов, содержащих бром, имеют почти равную интенсивность и разделены двумя единицами т/е. Если во фрагмент входят два эквивалентных атома брома, то различные комбинации изотопов дают сигнал в виде триплета с отношением интенсивностей 1:2:1. Кроме этих пиков, в спектре появятся также «пички», обусловленные небольшим природным содержанием D и 13С, что соответствует всем комбинациям масс 12С, 13С, D, Н, 79Вг и 81Вг. Результирующая группа пиков данного фрагмента имеет важное значение при их отнесении к фрагменту. Относительные интенсивности пиков зависят от относительного содержания в природе изотопов атомов фрагмента, например, СО+ может включать фрагменты с массовыми числами 28, 29, 30 и 31. Их относительные количества можно рассчитать с помощью простой теории вероятности [13, 14]. Чтобы провести такие расчеты, созданы программы для ЭВМ [15, 16]. Эти характеристические участки спектра весьма полезны для его отнесения в случае молекул, в которых атом имеет более чем один распространенный изотоп. Молекулы, содержащие ионы переходных металлов, часто дают такие характеристические участки спектра. Использование пиков 13С позволяет определить число атомов углерода во фрагменте.
Преимущества масс-спектрометрии высокого разрешения можно продемонстрировать [17] на примере некорректного отнесения пика с т/е = 56 в спектре низкого разрешения Fe[(C2H5)2NCS2]3 к атому железа. В спектре высокого разрешения пик должен находиться при т/е = = 55,9500, но он не был обнаружен. Вместо этого появился пик при т/е = 56,0350, который приписали фрагменту C3H6N.
Другое важное применение масс-спектрометрии, основанное на использовании изотопов, состоит в исследовании обменных реакций с участием соединений, содержащих нерадиоактивные изотопы. Для определения скорости обмена изучают во времени содержание изотопа в продукте превращения меченого исходного вещества. Продукт или исходное соединение можно разложить до газообразного вещества, содержащего метку, и из масс-спектра получить изотопное отношение. Эти вещества можно также исследовать непосредственно, и из анализа изменений в спектре различных фрагментов можно установить местонахождение и количество метки. Определяя, какие пики в спектре изменяются при внедрении изотопа, можно выявить части молекулы, участвующие в обмене. С помощью метки и масс-спекгрального анализа было показано, что эфирный кислород в продукте реакции метанола с бензойной кислотой принадлежит метанолу:
С6Н5С(О)-ОН + СН318ОН -> С6Н5С(О)-18ОСН3 + Н2О.
Еще одно интересное применение можно продемонстрировать на примере следующих обменных реакций:
BF3 + BX3 -> BX2F + BF2X,
3RBX2 + 2BF3 -> 3RBF2 + 2BX3 (а также BF„Xm),
3R2BX + BF3 -> 3R2BF + BX3 (а также BF„Xm),
Ионизационные методы
325
где R—алкил или винил, а X—хлор или бром. Были получены фрагменты, соответствующие продуктам, однако при попытке разделения последних выделили только исходные вещества [18]. Для процесса обмена предполагалось существование четырехцентрового промежуточно-
R zcl zF
го продукта типа Для того чтобы определить, обмени-
сь 4F
ваются или нет алкильные группы в ходе реакции
3RBX2 + 2BF3 -> 3RBF2 + 2ВХ3 (и BXmF„), трехфтористый бор был обогащен изотопом 10В. Отсутствие повышенного содержания 10В во фрагментах, содержащих алкильные или ви-нильные группы, позволило сделать вывод, что ни алкильные, ни ви-нильные группы не обмениваются при условиях, когда частица RBX2 стабильна.
Следует указать, что в рассмотренных выше применениях нет необходимости метить все молекулы соединения, поскольку вполне достаточно небольшого обогащения.
16.6. ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНЫХ МАСС;
МЕТОД ИОНИЗАЦИИ ПОЛЕМ
Если молекулярный ион, который образовался в процессе, аналогичном процессу (16.6), стабилен, то молекулярную массу вещества можно определить непосредственно из пика с наибольшим массовым числом, интенсивность которого не зависит от давления. Например, молекулярную массу Fe(CO)4.(CF2 — CF2 — СН2 — CF2) нельзя определить [19] обычными способами, однако из пика молекулярного иона в масс-спектре следует, что это вещество представляет собой мономер с молекулярной массой 368. Для многих соединений молекулярный ион достаточно стабилен, чтобы пользоваться указанным методом определения молекулярной массы, но часто это не так. Главная проблема состоит в определении пика молекулярного иона.
Многие молекулы либо не обладают достаточной летучестью, либо недостаточно устойчивы по отношению к электронной бомбардировке, чтобы можно было определить молекулярную массу с помощью масс-спектрометрии, если только не применять метод ионизации полем. Если молекулярные ионы нельзя зарегистрировать при температуре испарения вещества и бомбардировке электронами с энергией 70 эВ, то они обычно не наблюдаются и при более низкой энергии электронов. Хотя снижение энергии электронов приводит к увеличению интенсивности пика молекулярного иона по сравнению с пиками фрагментов, абсолютная интенсивность пика молекулярного иона снижается. В методе ионизации полем в зазоре между двумя металлическими электродами создается электрическое поле напряженностью ~ 5-107 В/см. Как только газообразная молекула попадает в такое поле, она ионизуется. Этот процесс носит название ионизации полем. На силу тока образующихся
326
Глава 16
ионов влияет скорость поступления нейтральных молекул в область поля. Органические молекулы могут также адсорбироваться на поверхности электрода и затем ионизоваться высоким электрическим полем в процессе, называемом десорбцией полем. Одно из преимуществ метода десорбции полем — это возможность исследования молекул вещества, которое не обладает достаточной летучестью или разрушается при сублимации. Электроды покрываются раствором вещества и растворитель испаряется, оставляя пленку вещества на электроде. Затем для ионизации вещества прикладывается электрическое поле.
Кроме исследования менее летучих веществ, другим преимуществом методов ионизации полем («мягких» ионизационных методов) является то, что они позволяют наблюдать пик молекулярного иона высокой интенсивности для многих веществ, для которых этот пик трудно или невозможно зарегистрировать при использовании методов с электронной
он он
Молекулярная масса 150
т/е —-
РИС. 16.5. А — формула при ионизации полем.
D-рибозы; Б — масс-спектр при электронной бомбардировке; В — масс-спектр
Ионизационные методы
327
бомбардировкой. Рис. 16.5 демонстрирует разницу между спектрами D-рибозы, полученными при электронной бомбардировке и методом ионизации полем. В спектре, соответствующем электронной бомбардировке, пик молекулярного иона обнаружить практически нельзя и его не удается надежно отличить от пика примеси. Спектр в случае метода ионизации полем дает интенсивный пик молекулярного иона исходного вещества. Наиболее интенсивная линия в спектре соответствует молекулярному иону исходного вещества, молекулярная масса которого увеличена на одну единицу. Такой результат часто наблюдают в случае сильно абсорбирующихся полярных молекул, особенно содержащих гидроксильные группы; пик фрагмента, масса которого на единицу превышает массу молекулярного иона исходного вещества, возникает за счет отщепления протона от второй молекулы на поверхности электрода после, ионизации первой молекулы. При использовании эмиттеров в виде проволоки интенсивность этой линии значительно снижается [8].
В методе десорбции полем молекулы могут поступать в область испарения за счет абсорбции из газовой фазы и диффузии вдоль твердого электрода. Во многих случаях при использовании газообразных образцов трудно провести различие между процессами ионизации и десорбции полем.
16.7. РАСЧЕТ ТЕПЛОТ СУБЛИМАЦИИ;
ЧАСТИЦЫ В ПАРЕ НАД ТВЕРДЫМИ ВЕЩЕСТВАМИ С ВЫСОКОЙ ТЕМПЕРАТУРОЙ ПЛАВЛЕНИЯ
Расчет теплоты сублимации основан на том факте, что интенсивность пиков в спектре прямо пропорциональна давлению пара образца в ионном источнике. Образец помещают в емкость с отверстием очень небольшого диаметра (ячейка Кнудсена), соединяющим ее с ионным источником, поэтому вещество может попасть в источник только за счет диффузии через это отверстие. Если ячейка термостатирована и в ней имеется достаточное количество образца, так что часть его всегда находится в твердом виде, то теплоту сублимации образца можно определить, исследуя изменения интенсивности пика (которая связана с давлением пара) в зависимости от температуры образца. Небольшое количество образца, диффундирующее в ионный источник, не оказывает заметного влияния на равновесие. При таких исследованиях были получены интересные результаты относительно природы частиц, присутствующих в паре над некоторыми твердыми веществами, имеющими высокие температуры плавления. В паре над хлоридом лития были обнаружены мономеры, димеры и тримеры, а в паре над хлоридами натрия, калия и цезия—мономеры и димеры [20].
В паре над твердым Сг2О3 найдены частицы Сг, СгО, СгО2, О и О2. Сообщалось о потенциалах возникновения этих частиц и об энергиях разрыва связей [21]. Пар над МоО3, как оказалось, содержит тримеры, тетрамеры и пентамеры. Рассчитаны давления паров, изменения свободной энергии и энтальпии сублимации [22].
328
Глава 16
16.8. ПОТЕНЦИАЛЫ ВОЗНИКНОВЕНИЯ
И ПОТЕНЦИАЛЫ ИОНИЗАЦИИ
Как упоминалось ранее, молекулярный ион возникает каждый раз, когда происходит столкновение молекулы вещества с электроном, энергия которого равна энергии ионизации молекулы или превышает ее. Типичная зависимость, связывающая энергию электрона с числом ионных фрагментов данного типа, образующихся при бомбардировке (т. е. с относительной интенсивностью данного пика), изображена на рис. 16.6.
Рис. 16.6. Кривая эффективности ионизации.
Она называется кривой эффективности ионизации. Если энергия электронов заметно ниже энергии ионизации, то никаких ионов не возникает. Если энергия электронов равна энергии ионизации, то появляется пик очень низкой интенсивности, поскольку для ионизации в этом случае необходимо, чтобы при столкновении вся энергия электрона передавалась молекуле, вероятность чего не очень высока. По мере увеличения энергии электронов вероятность передачи ими энергии, достаточной для ионизации молекулы, увеличивается. При этом интенсивность пика растет, пока кривая не достигнет насыщения. Хвост кривой при низких энергиях возникает потому, что энергии электронов в пучке различны. Таким образом, для определения энергии ионизации необходимо проэк-страполировать кривую (пунктирная линия на рис. 16.6). В литературе [21] имеется подробное описание различных способов экстраполяции кривой и возникающих при этом ошибок. Если наблюдаемый пик представляет собой пик молекулярного иона (е + RX -+ RX + + 2е), то энергию ионизации молекулы можно определить путем экстраполяции кривой эффективности ионизации. Если пик принадлежит фрагменту, то экстраполяция кривой эффективности ионизации дает потенциал возникновения этого фрагмента. Например, если исследуемый пик является пиком фрагмента R+ молекулы RX, то потенциал его возникновения Ar+ получается путем экстраполяции кривой эффективности ионизации для этого пика. Потенциал возникновения связан со следующими пара-
Ионизационные методы
329
метрами:
^r+ = ^r-x + 1r + + Ее, (16.19)
где DR_X—газофазная энергия разрыва связи R —X, 1К—потенциал ионизации R, Ек— кинетическая энергия образующихся частиц и Ее — энергия возбуждения фрагментов (т. е. электронная, колебательная и вращательная энергии, если фрагменты образуются в возбужденных состояниях). Обычно Ек и Ее малы, и уравнение (16.19) адекватно аппроксимируется уравнением
Лк+ = Dr_x + 1r- (16.20)
Если известна DR _ х, то из данных по потенциалу возникновения можно рассчитать 1R. Часто, зная 1R, можно вычислить Dr-x- Чтобы применять уравнение (16.20), значение /к должно быть ниже значения 1Х; иными словами, X диссоциирует или электронно возбуждается. Эксперименты такого типа представляют собой лучший метод расчета энергий разрыва связей, но в этом случае потенциалы ионизации получаются с меньшей точностью, чем при использовании других методов.
Статья [22], посвященная масс-спектрометрическому исследованию фосфина и дифосфина, превосходно демонстрирует, какую информацию можно получить при таких исследованиях. В ней сообщается о величинах энергии диссоциации и потенциала возникновения основных положительных ионов, а также рассмотрена энергетика процессов фрагментации и предложен их механизм.
ИОННЫЙ ЦИКЛОТРОННЫЙ РЕЗОНАНС (ИЦР)
16.9. ОСНОВНЫЕ ПРИНЦИПЫ ИЦР
Спектрометр ИЦР, выпускаемый в настоящее время промышленностью, представляет собой по существу масс-спектрометр, в котором используется метод регистрации сигнала спектрометров магнитного резонанса. Как и в масс-спектроскопии, в этом методе генерируется положительный ион с массой т и зарядом е. В однородном магнитном поле Н этот ион ускоряется и движется по круговой орбите, плоскость которой перпендикулярна направлению магнитного поля. Движение иона по этой орбите описывается циклотронной частотой а>с, выражаемой как
сос = еН/тс, (16.21)
где с—скорость света. Поскольку сос не зависит от скорости v иона, все ионы с данным значением т/е характеризуются данным значением а>с. Однако распределение скоростей приводит к распределению радиусов орбит:
сос = v/r. (16.22)
Этот вращающийся на орбите ион может поглощать энергию переменного электрического поля EJt) частоты со1; если coj = сос.
330
Глава 16
В эксперименте две параллельные пластины представляют собой часть резонансной цепи чувствительного регенеративного детектора-генератора, который является источником поля E^t). Путем развертки CDj регенеративного детектора-генератора во всем диапазоне частот со£. ионов образца при постоянном Н регистрируются поглощение энергии и масс-спектр. Более распространена регистрация масс-спектра при изменении Н при ПОСТОЯННОЙ йр
Важность метода ИЦР заключается не в использовании его в качестве другого вида масс-спектроскопии, а в результатах, которые можно получить из эксперимента двойного резонанса. В этом эксперименте исследуют влияние поступательной энергии данного иона на интенсивность сигнала другого иона, который может взаимодействовать с данным ионом в ион-молекулярной реакции. Например, в ходе наблюдения за сигналом А + накладывается электрическое поле вспомогательного генератора, частота которого сос соответствует В + . Спектр А+ меняется, если А + и В + взаимодействуют в химической реакции. Обычно при проведении эксперимента вторую частоту варьируют в диапазоне частот, характеризующих все другие ионы, находящиеся в таком состоянии, что и В т .
Для большинства экзотермических реакций константа скорости к уменьшается с увеличением кинетической энергии Е реагирующего иона, т. е. dk/dE отрицательна. Знак dk/dE можно определить из эксперимента ИЦР, поскольку увеличение или уменьшение интенсивности иона-продукта при облучении реагирующего иона указывает соответственно на положительный или отрицательный знак dk/dE. Если интенсивность не изменяется, то облучаемый ион, вероятнее всего не участвует в образовании продукта. Альтернативной, но маловероятной возможностью является равенство dk/dE нулю *. Экзотермическая реакция должна протекать в отсутствие облучения, но в случае эндотермической реакции это не так.
Существуют две основные области применения ИЦР: а) определение абсолютных и относительных термохимических параметров и б) исследование возможных ион-молекулярных реакций и их механизмов, а также измерение констант скорости. Чтобы проиллюстрировать сказанное выше, обсудим некоторые результаты исследования реакций переноса протона в системах, содержащих Н2О, СН3ОН и С2Н5ОН. Спектр простого резонанса говорит о присутствии ионов ОН “, СН3О “ и С2Н5О “ с т/е = 17, 31 и 45. При проведении эксперимента двойного резонанса получаются следующие результаты:
1) Облучение пиков с т/е — 17 и 31 приводит к снижению интенсивности пика с т/е = 45.
2) Облучение пика с т/е = 17 приводит к снижению интенсивности пика с т/е = 31.
* Если при облучении реагирующие ионы удаляются из реакционной камеры, то сигнал двойного резонанса может быть получен для реакции, константа скорости которой не зависит от кинетической энергии реагирующего иона.
Ионизационные методы
331
3) Интенсивность пика с т/е = 17 не меняется, если ускорять ионы с т/е = 31 и 45.
Эти результаты свидетельствуют о том, что в ячейке ИЦР протекают следующие необратимые реакции:
НО - + СН3ОН -> СН3О - + Н2О,
НО" +С2Н5ОН -> С2Н5О- +Н2О,
СН3О +С2Н5ОН -> С2Н5О“ +сн3он.
Напомним, что воздействие на один ион может увеличить концентрацию другого иона только в том случае, если в отсутствие возмущения оба иона химически реагируют. Полученные результаты приводят к следующему порядку реакционной способности при переносе протона:
С2Н5ОН > СН3ОН > Н2О.
Для подробного знакомства с применениями этого метода отсылаем читателя к работам [25, 26].
ФОТОЭЛЕКТРОННАЯ СПЕКТРОСКОПИЯ*
16.10. ВВЕДЕНИЕ
Столкновение фотонов с атомами или молекулами может привести к испусканию фотоэлектронов. В течение последних двух десятилетий фотоэлектронная спектроскопия развилась в многообещающую область химии. Фотоэлектронная спектроскопия отличается от описанных ранее спектроскопических методов, в которых измеряются характеристики поглощенного, испущенного или рассеянного электромагнитного излучения. В этом методе предмет изучения—кинетическая энергия испущенных при ионизации электронов.
Частично из-за потребности в монохроматическом излучении возникли два раздела фотоэлектронной спектроскопии. Рентгеновская фотоэлектронная спектроскопия, сокращенно обозначаемая как РФС или ЭСХА (электронная спектроскопия для химического анализа), использующая рентгеновские лучи в качестве источника ионизирующего излучения, изучает в основном электроны оболочки (т.е. невалентные электроны). Создание этого метода приписывают Сигбану и сотр. [27]. В ультрафиолетовой фотоэлектронной спектроскопии (УФС) используют ультрафиолетовое излучение, имеющее более низкую энергию, и, таким образом, исследуют энергии связи валентных электронов. Обязанная своим развитием главным образом Тернеру и его сотрудникам [28], УФС предназначалась не только для измерения энергий связывания валентных электронов, но и для наблюдения за возбужденными колебательными состояниями молекулярного иона, образующегося в процессе фотоионизации.
* Этот раздел написан Д. Хендриксоном.
332
Глава 16
16.11. ТЕОРЕТИЧЕСКИЙ ПОДХОД
Энергетика
При использовании вакуума в качестве точки отсчета энергия Еъ, необходимая для освобождения электрона из системы, может быть рассчитана из закона сохранения энергии:
Eb — ^source — ^kin — Ег. (16.23)
Здесь £source— энергия ионизирующей радиации, £kin—кинетическая энергия фотоэлектрона и £г—энергия отдачи атома или молекулы. Можно показать, что энергия отдачи атома или молекулы при испускании фотоэлектрона пренебрежимо мала, за исключением случая рентгеновской фотоионизации атома водорода [27]. Фотоэлектронный спектр представляет собой график зависимости числа фотоэлектронов, падающих на соответствующее счетное устройство, от кинетической энергии фотоэлектрона. Пики (т. е. максимумы в скорости счета) соответствуют фотоионизации электронов с различных уровней (т. е. молекулярных орбиталей) образца. Типичный спектр РФС изображен на рис. 16.7. В этом случае спектрометр РФС настроен таким образом, что регистрируются фотоэлектроны, возникающие за счет ионизации ls-электронов атомов азота в mpaHC-[Co(NH2CH2CH2NH2)2(NO2)2](NO3). В фотоэлектронном спектре наблюдаются три максимума, и они отнесены к ионизации трех различных типов атомов азота. Таким образом, некоторая (небольшая) часть молекул твердого вещества теряет Nls-электрон с атома азота аниона NO3, а другие молекулы—N^-электроны с атома азота либо NO2, либо этилендиамина. В энергиях связи электронов оболочки наблюдаются химические сдвиги во много электрон-вольт (1 эВ = = 8067 см “1 = 23,06 ккал/моль); мы вернемся к этому позднее. Следует подчеркнуть, что из данной молекулы испускается только один электрон, либо Nls-электрон, либо валентный электрон со связывающей орбитали. Вероятность многократной ионизации мала.
В случае УФС, как мы увидим, разрешение таково, что можно легко регистрировать колебательную структуру, связанную с электронным состоянием ионизуемой молекулы. Аналогия с электронной абсорбционной спектроскопией очевидна. В эксперименте УФС фотоионизации с испусканием электрона сопутствует электронный переход из основного состояния исходной молекулы в основное электронное состояние (иногда в возбужденное состояние, см. ниже) ионизованной молекулы. В электронной абсорбционной спектроскопии колебательная структура наблюдается для возбужденного электронного состояния, а в УФС — для электронного состояния ионизованной молекулы. Тогда явная форма уравнения (16.23) для энергии, необходимой для освобождения электрона из молекулы, выглядит как
£ь = 7 + £vlb + £rot = £source — £kln, (16.24)
Ионизационные методы
333
где I—адиабатическая энергия удаления электрона, a £vlb и £rot— квантованные энергии колебания и вращения различных электронных состояний ионов, которые являются следствием фотоионизации. Энергия адиабатической ионизации представляет собой энергию перехода между основным состоянием молекулы и основным состоянием иона. Энергия вертикальной ионизации измеряется в переходе, для
Рис. 16.7. Is-фотоэлектронный спектр атома азота в транс-[Co(NH2CH2CH2NH2)2 (NO2)2] • NO3 [51].
Энергия связи, эВ
которого межъядерные расстояния в ионе не отличаются от межъядерных расстояний в исходной молекуле. Таким образом, энергия вертикальной ионизации равна энергии адиабатической ионизации или превышает ее. Во многих случаях из спектра УФС можно определить обе энергии (см. ниже).
В эксперименте УФС в качестве ионизирующего излучения используют вакуумный ультрафиолет; обычно источником такого излучения является гелиевая [однократно ионизованный гелий, обозначаемый как Не(1)] резонансная лампа с энергией 21,21 эВ. Однако можно применять и другие разрядные лампы, например лампу Ar (I) или лампу с двукратно ионизованным гелием, Не(П). Энергия этих ламп ограничивает УФС исследованиями валентных электронов; как правило, измерения проводят с использованием газообразных образцов. Известно несколько работ, посвященных исследованию растворов [29] и твердых веществ [30].
334
Глава 16
Поскольку составной частью прибора РФС является источник рентгеновского излучения, который ионизует образец, этим методом можно определять энергии связывания как валентных электронов, так и электронов оболочки. Обычно используют рентгеновское излучение Ка Mg и Al с энергией соответственно 1253,6 и 1486,6 эВ. Методом РФС исследовали твердые вещества, газы, жидкости, растворы и замороженные растворы. В случае твердых веществ и замороженных растворов рассчитанные энергии связывания электронов относят к энергии уровня Ферми твердого вещества. Уровень Ферми соответствует высшему заполненному уровню электронного слоя структуры твердого вещества при О К. Уравнение сохранения энергии (16.23) преобразуется к виду
Eb = Дючгсе — ^kin — Фкрес (16.25)
В этом уравнении опущена незначительная энергия отдачи и введена работа выхода cpspec (~ 4 эВ) внутренних металлических поверхностей спектрометра РФС. Работа выхода материала спектрометра — это энергия, необходимая для удаления электрона с поверхности спектрометра. Работа выхода образца отличается от работы выхода материала спектрометра. Образец в спектрометре РФС находится в электрическом контакте со спектрометром, и, если имеется достаточное число носителей заряда (многие образцы представляют собой диэлектрики и носители заряда образуются в ходе облучения), уровни Ферми для образца и спектрометра будут одни и те же. Уравнение (16.25) можно понять, рассмотрев эксперимент РФС. При фотоионизации электрон образца получает некоторую кинетическую энергию £,. Для того чтобы попасть в спектрометр, электрон должен пройти через входную щель. Поскольку рабочие потенциалы спектрометра и образца различны, кинетическая энергия электрона изменяется до £kin, что обусловлено либо ускорением, либо замедлением фотоионизованного электрона входной щелью. В камере спектрометра электрон имеет кинетическую энергию Ekin, и эта энергия измеряется прибором. Таким образом, для соотнесения энергии связывания с уровнем Ферми в выражение вводится cpspec. К счастью, нет необходимости знать работу выхода каждого образца.
Разрешение
Теоретическое разрешение, возможное в эксперименте УФС, где определяются энергии связывания валентных электронов, обсуждалось Тернером [31]. Напомним, что измерения проводятся в газовой фазе. Разрешение в спектре УФС ограничивается скоростью движения молекулы-мишени в сочетании со скоростью движения фотоэлектрона (фактически это явление аналогично доплеровскому уширению) величиной ~10-3 эВ. Если вместо камеры, заполненной газообразным веществом, использовать пучок молекул-мишеней, то можно достичь разрешения 10“ 4 эВ. В случае пучка распределение молекулярных скоростей относительно источника более однородно. Вклад в ширину спектральных линий УФС за счет времени жизни возбужденного состояния
Ионизационные методы
335
иона, вызванного фотоионизацией, невелик ( ~ 10 “ 7 эВ). Однако в некоторых случаях возникший при фотоионизации ион распадается, что приводит к уширению наблюдаемых линий. Если же ион устойчив и если рассматривать только перечисленные выше факторы, то следует ожидать теоретического разрешения ~ 10-3 эВ. Экспериментально такого разрешения достичь не удалось. Ограничения накладывают как вращательная тонкая структура ( ~ кТ) колебательного пика, так и спин-орбитальное расщепление. Экспериментально было получено энергетическое разрешение 0,010 эВ (см., например, работу [32]). Ухудшение разрешения, связанное с прибором и источником ионизирующего излучения, было сведено до минимума.
В случае РФС наблюдаемая полуширина линии (полная ширина на половине высоты) значительно выше, чем в случае УФС. Фотоионизация электронов оболочки приводит к возбужденным состояниям, время жизни которых значительно короче, чем в случае УФС, поскольку время жизни пропорционально Е~3—энергии фотоионизационного перехода. Данные по поглощению и испусканию рентгеновских лучей [33] показывают, что присущая внутренним атомным уровням ширина линий снижается с уменьшением атомного номера и может быть порядка 0,1 эВ или менее для элементов с низким атомным номером. Тогда можно ожидать, что теоретический предел полуширины спектральных линий РФС будет иметь порядок 0,1 эВ при фотоионизации ls-электро-нов углерода, 1 s-электронов азота, 2р-электронов фосфора и других аналогичных электронов.
Однако в противоположность УФС естественная ширина линий обычных источников рентгеновских лучей РФС довольно значительна и играет большую роль в определении полуширины экспериментально наблюдаемых спектральных линий [27]. В РФС обычно используют рентгеновский дублет а это рентгеновское излучение образуется
в том случае, когда электроны «падают» из оболочек Ln и Ещ (спин-орбитальное расщепление 2р-атомных уровней) в дырку оболочки К (1s-атомный уровень). Естественная ширина линий, связанная либо с переходом Цх -> К, либо с переходом Еш -> К, составляет 0,7 эВ для рентгеновского излучения А1; в этом случае дублеты перекрываются, приводя к эффективной ширине ~ 1,0 эВ. Магниевое рентгеновское излучение Ка^ состоит из дублета шириной ~ 0,8 эВ. Источники рентгеновских лучей с большими энергиями (например, Сг, Си или Мо) характеризуются шириной дублетной компоненты, превышающей 1,0 эВ. Таким образом, эффективный предел ширины линий РФС устанавливается «естественной» шириной источника рентгеновского излучения, несколько модифицированной естественной шириной, связанной с уровнем, с которого происходит фотоионизация. Некоторые вклады обусловлены также недостатками приборов. При изучении твердых веществ экспериментально наблюдаемая полуширина спектральных линий РФС для пиков Cls, Nls, Р2р, S2j, и подобных им составляет ~ 1,5 эВ. Эксперименты РФС с газообразными веществами дают значительно более узкие линии. Например, полуширина линии Nels для газообразного неона составляет 0,8 эВ [27]. Разница в полуширине линий для газообраз
336
Глава 16
ного и твердого веществ приписывается заряду поверхности твердого вещества (обычно диэлектрика) и различию в кристаллических полях, действующих на атомы на различных глубинах твердого вещества. Как было показано в работе [34], существует возможность исключить влияние ширины рентгеновской линии на спектр РФС с помощью развертки.
Средняя глубина, с которой уходят электроны из твердого вещества, зависит от кинетической энергии фотоэлектронов. Изучение [35] металлов показало, что при кинетической энергии 1000 эВ средняя глубина может достигать ~ 100 А, тогда как при энергии 10 эВ она должна быть 10 А. Большое значение для РФС имеет чистота поверхности образца. С помощью этого метода можно исследовать химию поверхностей, как, например, в гетерогенном катализе.
Теорема Купманса
Как УФС, так и РФС могут быть использованы для исследования валентных электронов в молекулах, и нас как раз интересует та информация, которую можно получить об этих электронах из фотоэлектронного спектра. На рис. 16.8 в качестве примера изображен спектр УФС газообразного азота. В случае источника Не(1), устанавливающего предел ионизации в 21,21 эВ, можно наблюдать три колебательно-структурированных фотоионизационных процесса (~ 15,6, ~ 17,0 и ~ 18,18 эВ). Их можно приписать ионизации с трех высших заполненных молекулярных орбиталей N2(2ou-, ли- и Зо9-орбиталей). Отнесение пиков основано на наблюдаемой колебательной структуре. Следует отметить, что в спектре РФС имеются те же три линии (колебательная структура не видна из-за худшего разрешения) в дополнение к пику при 37,3 эВ для ионизации с 2о9-уровня и единственному пику при 409,9 эВ для 1о9- и 1ои-уровней [27].
Эти данные подводят нас к теореме Купманса, согласно которой энергия вертикальной ионизации для удаления электрона с молекулярной орбитали равна собственному значению с обратным знаком, полученному при расчетах молекулярных орбиталей с помощью метода самосогласованного поля (ССП МО) Хартри—Фока [36] (стабильная орбиталь имеет отрицательное собственное значение). Основное допущение этой теоремы состоит в том, что молекулярные орбитали, соответствующие исходной молекуле, будут теми же, что и для ионизованной молекулы. При наличии электронной релаксации (т. е. при изменении молекулярных орбиталей в ионизованной молекуле, обусловленном изменением энергии электронного отталкивания) или при заметном изменении энергий корреляции (член, не включенный в расчет по методу МО; он учитывает зависимость координат каждого электрона от координат всех других электронов) теорема Купманса не выполняется.
Энергии вертикальной ионизации легко определить для рассмотренного образца N2 (рис. 16.8). В табл. 16.1 приведены энергии вертикальной ионизации N2 и собственные значения, полученные при нескольких
Ионизационные методы
337
Oq Оц Is Is—ft-----------------H----------tt-/S
6
Рис. 16.8. УФС-спектр газообразного азота [39].
расчетах по методу МО. (Энергии даны в эВ.) Между значениями, полученными методами РФС и УФС, наблюдается хорошее соответствие, однако собственные значения, полученные методом самосогласованного поля Хартри—Фока, не согласуются с наблюдаемыми результатами. На самом деле порядок <т9 2р и л„2р обращен, что может служить указа-
22 -574
338
Глава 16
Таблица 16.1
Данные фотоэлектронной спектроскопии для газообразного азота (энергии выражены в эВ)
Орбиталь Конечное состояние РФСа УФС6 Собственные значения, рассчитанные для N2 методом ССП МО Хартри — РасчетЕ для N2‘
Фока0
ст92Р %+ пи2р 2п„ 21„+ 15,5 16,8 18,6 15.57 16,96 18,75 17,28 16,75 21,17 15,99 15,34 19,74
2К 37,3 40,10
o9/o„ls 2Г9„ 409,9 /426,61\
(426,71 )
а См. работу [27]. 6 См. работу [39]. В Cade Р. Е., et al.. J. Chem. Phys .. 44, 1973 (1966).
нием на различие в энергиях релаксации. Если даже расчеты видоизменить путем учета энергий различных состояний N2 и N2 и различия в полных электронных энергиях, то наблюдается только удовлетворительное соответствие. И снова происходит обращение <Уд2р и л„2р. В этом примере теорема Купманса дает лишь качественное соответствие, что справедливо и в общем случае. Другой пример приведен в табл. 16.2. Некоторые типы полуэмпирических расчетов по методу МО дают, видимо, полуколичественное соответствие с наблюдаемыми
Таблица 16.2 Данные фотоэлектронной спектроскопии для моноокиси углерода
Орбиталь РФСа УФС6 Расчет по методу Хартри — Фока °
Зст 14,5 13,98 15,08
lit 17,2 16,58 17,35
2 а 20,1 19,67 21,85
1а 38,3 41,40
с15 295,9 309,13
O1S 542,1 562,21
б См. работу [27].
в Al-Joboury M.I., et al., J. Chem. Soc., 1965, 616.
Neumann D.B., Moskowitz J.W., J. Chem. Phys., 50, 2216 (1969).
Ионизационные методы
339
100
эв
Рис. 16.9. УФС-спектр газообразного аммиака [28].
энергиями вертикальной ионизации. В литературе [37] продолжается обсуждение нарушения теоремы Купманса.
Если даже теорема Купманса строго и не выполняется, то все-таки полезно знать, какие пики в фотоэлектронном спектре могут быть связаны с различными молекулярными орбиталями в исходной молекуле. Например, в гл. 3 рассматривались симметрия и строение молекулярных орбиталей NH3. Было установлено, что семь атомных орбита-лей в симметрии С3„ образуют представление, которое сводится к трем неприводимым представлениям и двум неприводимым представлениям е. Восемь валентных электронов NH3 заполняют две из at- и одну из е-молекулярных орбиталей, образуя конфигурацию основного состояния
...(2а1)2(1е)4(3а1)2.
Только другая заполненная орбиталь, 1^-орбиталь, представляет собой по существу атомную 15-орбиталь азота. Спектр Не(1) (21,21 эВ) показан на рис. 16.9, где наблюдаются вертикальные ионизации с уровня За{ при 10,88 эВ и с уровня 1е при 16,0 эВ (первый максимум). Это отнесение выполнено с помощью результатов различных расчетов по методу МО. Источник Не(П) (42,42 эВ) с более высокой энергией был использован для регистрации вертикальной ионизации с уровня 2аг при 27,0 эВ [38]. Интересно отметить, что пик 1е при ~ 16 эВ имеет характер дублета, т.е. расщепляется. Это расщепление отнесено за счет ян-телле-
22*
340
Глава 16
ровского искажения в ионе, что обусловлено конфигурацией (2а1)2(1е)3(За1)2. Расщепление составляет 0,78 эВ. Известны и другие примеры ян-теллеровского расщепления [28].
Поперечные сечения и угловые распределения
Сравнительно недавно [27] были получены спектры РФС газообразных веществ, ранее исследуемых методом УФС. Полученные интересные результаты основаны на относительных поперечных сечениях фотоионизации валентных электронов в зависимости от энергии источника. Например, для рентгеновского излучения с большей энергией электроны на молекулярной орбитали, составленной главным образом из атомных s-орбиталей, имеют более высокое относительное поперечное сечение (и, следовательно, большую интенсивность спектральной линии), чем электроны на молекулярной орбитали, составленной в основном из атомных 2р-орбиталей. Сопоставление спектров РФС и УФС указывает на различные относительные интенсивности соответствующих пиков. Пик, обусловленный электронами на молекулярных орбиталях, составленных главным образом из атомных орбиталей s-типа, имеет большую относительную интенсивность в спектре РФС, чем в спектре УФС.
16.12. ФОТОЭЛЕКТРОННЫЕ СПЕКТРЫ
Спектры УФС
Вид колебательной структуры полос УФС дает некоторую информацию о характеристиках связывания фотоионизованных электронов. Ионизация несвязывающих электронов приводит к иону с тем же межъядерным расстоянием, что и в исходной молекуле. Поэтому в спектре будет преобладать колебательный пик низшей энергии (г = 0, г' = 0) (рис. 16.10). Возвращаясь к спектру N2 на рис. 16.8, мы видим, что ионизация электронов либо с уровня Зст9 при 15,6 эВ, либо с уровня 2<т„ при 18,8 эВ в каждом случае дает по существу один интенсивный пик. Это обусловлено тем, что данные уровни имеют соответственно слабо разрыхляющий и слабо связывающий характеры.
Как показано на рис. 16.10, фотоионизация либо сильно связывающих, либо сильно разрыхляющих электронов приводит к изменениям равновесных длин связей. В этих случаях наиболее интенсивный пик (т. е. соответствующий вертикальной ионизации) будет характеризоваться более высокой энергией, чем пик, соответствующий адиабатической ионизации (см., например, пик л„ для N2). На самом деле относительные интенсивности (называемые коэффициентами Франка — Кондона) соответствующих колебательных пиков могут быть с достаточным успехом [39] рассчитаны теоретически. Для получения характеристик молекулярной орбитали, с которой удален электрон, оказываются также полезными колебательные частоты, наблюдаемые для состояния иони-
Ионизационные методы
341
Рис. 16.10. Колебательные состояния исходной молекулы М и ионизованной молекулы М + .
А—в этом случае электрон уходит с несвязывающей орбитали и два электронных потенциала лежат один над другим. Наиболее интенсивен переход г' = 0ч_г=0 (указан стрелкой); Б — ямы смещаются (изменились размеры молекул), если электрон уходит либо со связывающей, либо с разрыхляющей орбитали. В этом случае переход v’ = 3 v = 0 приводит к полосе с наибольшей интенсивностью.
зованной молекулы. Частота валентных колебаний основного состояния N2 составляет 2345 см-1, и ее необходимо сопоставить с частотами различных пиков. В табл. 16.3 приведены частоты валентных колебаний, наблюдаемых для различных ионизованных состояний N2 ; сопоставление каждой из этих величин с частотой 2345 см-1 N2 говорит о характерах МО (см. таблицу).
Таблица 16.3
Колебательная структура в УФС-спектре
Пик v(N—N), см 1 Подразумеваемый характер МО
<Т9 (2р) 2150 Слабо связывающий
(2р) 1810 Сильно связывающий
(2s) 2390 Слабо разрыхляющий
Кроме колебательной структуры, в спектрах УФС наблюдается и другая тонкая структура. Первый пик (т. е. с низшей энергией связывания) в спектрах СН3С1, СН3Вг и СН31, очевидно, обусловлен высшей занятой молекулярной орбиталью; она локализована в основном на атоме галогена (некоторый С—Н-связывающий характер и X—Н-разрых-
342
Глава 16
ляющий характер) [40]. Этот пик указывает на спин-орбитальное взаимодействие. Разделение в приведенном ниже ряду соединений меняется следующим образом: СН31+, 0,62 эВ; СН3Вг+, 0,31 эВ; СН3С1+, ~ 0 эВ [28]. Величина этих спин-орбитальных взаимодействий согласуется с тем, что предсказывал Малликен: 0,625, 0,32 и 0,08 эВ соответственно [41]. Были проведены и другие исследования спин-орби-тальных взаимодействий. Фактически недавняя работа [32], в которой достигнуто высокое разрешение в 10 мэВ, позволила зарегистрировать спин-орбитальные расщепления в NO, равные 0,012 — 0,016 эВ.
К настоящему времени методами УФС и РФС исследовано значительное число небольших газообразных молекул. Изучены также некоторые радикалы и частицы в возбужденных состояниях. Были зарегистрированы [42] три ожидаемых состояния О+ (2Р, 4S и 2D) [42]. Для получения возбужденных частиц О2 использовали безэлектродный микроволновый разряд; их фотоэлектронный спектр демонстрирует колебательно-структурированный пик, обусловленный образованием О2 (2ПЯ). Сопоставление с пиком, соответствующим ионизации основного состояния О2 (2Е“) до того же ионного состояния, дает величину потенциала адиабатической ионизации О2 (1Дв), равную 11,09 + 0,005 эВ [43].
1000
0
Энергия электронов, эВ
Рис. 16.11. УФС-спектр кислорода. (НН1Н.А.О., Day Р., Physical Methods in Advanced Inorganic Chemistry, Interscience, N. Y., 1968, p. 88.)
Ионизационные методы
343
Изучены и некоторые другие частицы такого типа (например, SO и NF2).
По сравнению с диамагнитными соединениями парамагнитные соединения характеризуются более сложными спектрами УФС и РФС. Молекула кислорода имеет два неспаренных л9-электрона. Спектр УФС кислорода приведен на рис. 16.11. Фотоионизация электрона с частично заполненной разрыхляющей молекулярной ля(2р)-орбитали характеризуется первым пиком в спектре УФС, реализуется только одно ионное состояние. В то же время фотоионизация электрона с одной из других заполненных молекулярных орбиталей приводит в каждом случае к двум электронным состояниям иона О2 . Таким образом, если электрон удаляется с заполненной связывающей л„-орбитали, то на ней остается неспаренный электрон, спин которого может быть параллелен или антипараллелен спинам двух неспаренных электронов, находящихся на разрыхляющей л9-орбитали. Если спин оставшегося электрона параллелен спинам электронов на л9-орбитали, то мы будем иметь три неспаренных электрона, полный спин S = 3/2 и электронное состояние 4Пи для молекулы О2 . При другом направлении спина электронным состоянием молекулы О2 будет 2ПИ. Состояния 4П„ и 2П„ молекулы О2 имеют различные энергии, и, таким образом, ионизационный пик ^„-орбитали расщепляется. В табл. 16.4 приведены наблюдаемые характеристики молекулы О2, полученные из спектров УФС и РФС.
Таблица 16.4
Данные по вертикальной ионизации О2 [27]
Одноэлектрон- Электронное ная молекуляр- состояние ная орбиталь иона РФС, эВ УФС, эВ
л„2р Ч 13,1 12,10
V? [Ч 17,0 16,26
14 ? ?
п82р (% 18,8 18,18
21,1 20 31
a„2s f4L„ 25,3 24,5 [с Не (II)]
14 27,9
4s 39,6
) 2У ^9 41,6
lsO2 4 543,1
544,2
Для сравнения на рис. 16.12 показан спектр РФС газообразного О2. Видно, что пик 1 s-кислорода также расщепляется, в этом случае из 1,1 эВ. Это расщепление связано не с различием в энергиях молекулярных орбиталей l.sou и 1sct9 кислорода, которое, согласно расчетам, мало. Напомним, что в случае N2 расщепление такого рода не наблюдалось. Более того, если исходить из отношения Iso^/lsn^ отношение
344
Глава 16
интенсивностей двух полос Ols составляет 2:1, а не 1:1, Снова, как и в случае валентных электронов, неспаренный электрон, находящийся на ls-орбитали О2, взаимодействует с двумя неспаренными электронами, находящимися на разрыхляющей молекулярной л9-орбитали О2 . Ионизация даже этих электронов оболочки кислорода приводит к двум состояниям 2Е и О2, которые значительно различаются по энергии.
Наблюдать такое расщепление очень интересно. Одноцентровые обменные интегралы определяют разность энергий между квартетным и дублетным состояниями. Таким образом, межэлектронные отталкива-
Скорость счета, импульс/200с
Скорость счета, импульс/200с
Рис. 16.12. РФС-спектр электронов оболочки и валентных электронов кислорода, возбужденного рентгеновским излучением KotMg.
Два пика наблюдаются для каждой полностью занятой орбитали, включая ls-орбнталь электронов оболочки кислорода [27]. Пики А, В, С и пики в области больших энергий связи, чем энергия пика Ou, являются пиками-сателлитами (см. ниже).
Ионизационные методы
345
Рис. 16.13. А — фотоэлектронный спектр NO, полученный прн использовании линии Не при 304 А; три небольших пика, помеченных буквой а. обусловлены линией Не прн 320 А; время регистрации 75 ч; Б—фотоэлектронный спектр В'1 £ +-состояния NO, полученный при использовании линии Не при 304 А; B-фотоэлектронный спектр NO, полученный при использовании линии 584 А. Разрешение ЮмэВ. Развертка пиков указана пунктирными линиями [32].
346
Глава 16
ния (e2/r,j, где гу—расстояние между i-м и j'-м электронами, а е—заряд электрона) обменного типа даже между валентными неспаренными электронами и электроном оболочки кислорода велики. Как обнаружено, такие межэлектронные спиновые поляризации имеют важное значение для объяснения больших сдвигов линий в спектрах ЯМР парамагнитных молекул (см. гл. 12). Как будет описано в последнем разделе, расщепление за счет электронного обмена наблюдается также для электронов оболочки парамагнитных комплексов ионов переходных металлов.
Из проведенного выше обсуждения очевидно, что УФС-спектры относительно больших молекул содержат довольно много информации о потенциалах ионизации, энергиях колебаний ионизованной молекулы, спин-орбитальных взаимодействиях, ян-теллеровских расщеплениях и электронных обменных взаимодействиях. К сожалению, полосы часто перекрываются и появляются широкие линии с неразрешенной колебательной структурой. Примером небольшой молекулы, в спектре которой наблюдается большое число линий, служит газообразная NO. На рис. 16.13 показаны спектры этой молекулы, полученные Асбринком и сотр. [32] при разрешении 10 мэВ и источнике Не(1) и при разрешении 25 мэВ и источнике Не (II). С процедурой отнесения линий читатель может познакомиться в цитированной работе, однако даже внимательное рассмотрение рис. 16.13 показывает, что в спектре разрешены как обменное, так и спин-орбитальное расщепления.
Наконец, разработаны общие методы изучения паров веществ, которые по своей природе не могут быть внесены в виде газа. К таким веществам относятся очень реакционноспособные свободные радикалы, свободные атомы, молекулы в возбужденных состояниях или вещества, имеющие очень низкую упругость паров. Берковиц и сотр. [44] генерировали такие частицы в молекулярном пучке, который затем взаимодействовал с пучком света: два этих пучка входили в спектрометр под прямым углом. Для коллектирования фотоэлектронов использовали анализатор в виде цилиндрического зеркала. Возникала необходимость улучшения отношения сигнала к шуму. Указанный метод использовали для изучения Т1С1, TIBr, Til, InCi, InBr и Ini в газовой фазе. Эти молекулы довольно нелетучи, поэтому их невозможно исследовать обычными методами.
Химические сдвиги в РФС
В 1957 г. Сигбан и сотр. [45] зарегистрировали «химический сдвиг» в энергиях связи ls-электронов меди. Было найдено, что сдвиг в Еь (1s) между Си и СиО составляет 4,4 эВ (по сравнению с 8979 эВ), но в то время не было дано объяснение сдвигу. В начале 60-х годов группа Сиг-бана обнаружила сдвиги в энергиях связи 2s- и 2р-электронов серы [46]. Было установлено, что энергии связи электронов оболочки можно скоррелировать с состоянием окисления серы. С тех пор были проведены многочисленные исследования в том же направлении. Например, на
Ионизационные методы
347
Состояние окисления ксенона,
Рис. 16.14. График зависимости относительных энергий связи 345/2-электронов оболочки ксенона от состояния его окисления. (Данные взяты из работы [55].)
рис. 16.14 изображены графики зависимости энергий связи З^-электро-нов ксенона от состояния его окисления в ряду фторидов ксенона. Наблюдается монотонный и, по-видимому, линейный рост. Обычно обнаруживают, что энергия связи электронов оболочки зависит от химического окружения и что она возрастает с увеличением состояния окисления атома. При этом сдвиги велики. Весьма удивительно то, что по величине они сравнимы с энергиями химических реакций!
При качественной интерпретации соотношения между химическими сдвигами энергий связи электронов оболочки и распределением заряда в молекулах возникло много фальсификаций. В гл. 3 упоминалось, что с помощью метода молекулярных орбиталей можно рассчитать формальный заряд (5) на атоме в молекуле. Напомним, что формальный заряд определяется как электронная плотность на атоме в молекуле минус электронная плотность на свободном атоме. Из рис. 16.15 следует, что можно коррелировать формальный заряд на атоме азота в молекуле (полученный с помощью итерационных расчетов по расширенному методу Хюккеля) с наблюдаемыми энергиями связи ls-электронов азота для ряда азотсодержащих соединений. Отметим, что для корреляции со сдвигом в энергиях фотоионизационных переходов электронов оболочки используют заряд основного состояния атома, который определяют произвольным образом. Наблюдаемый успех либо случаен, либо обусловлен тем, что члены, такие, как энергии электронной релаксации, сохраняют постоянное значение.
Неизменно верят в то, что сдвиг в энергиях связи электронов оболочки обусловлен как зарядом на исследуемом атоме, так и зарядами на других атомах в молекуле. В этой «модели потенциала» сдвиг
348
Глава 16
в энергиях связи электронов оболочки ДЕ можно связать с формальным зарядом на атоме А(3Д) в молекуле [27]:
ДЕ = k&A + VA+l, (16.26)
где к и /—эмпирические параметры, используемые для подгонки с имеющимися данными по энергиям связи электронов оболочки. Член VA представляет собой кулоновский потенциал, обусловленный зарядами соседних атомов, т. е. формальные заряды на соседних атомах qB суммируют, считая их точечными:
7А = Е qB/RAB, (16.27)
В^А
где Едв— расстояние между ядрами. О влиянии плотности зарядов на соседних атомах на энергию связи электронов оболочки данного атома говорят данные, полученные в диапазоне энергий связи (~ 1,7 эВ) 2р-электронов калия для ряда калиевых солей [47]. Эта «модель потенциала» применялась ко многим элементам. Было выполнено значительное число возможных эмпирических, полуэмпирических и неэмпирических МО-расчетов. Стаки и сотр. [48] сопоставили распределения
Рассчитанный заряд на атоме азота,
Рис. 16.15. График зависимости энергий связи ls-электронов азота от зарядов на атомах азота, рассчитанных с помощью расширенного метода Хюккеля [51].
Ионизационные методы
349
заряда, полученные из модели потенциала применительно к результатам исследования методом РФС ряда небольших молекул, с распределениями заряда, вытекающими из некоторых теоретических расчетов, квадрупольных моментов и результатов экспериментов по дифракции рентгеновских лучей и нейтронов.
Обсуждалась теоретическая основа уравнения (16.26). Можно показать, что, если приближение нулевого дифференциального перекрывания допустимо и если выполняется теорема Купманса (или при постоянном расхождении), для химических сдвигов энергий связей электронов оболочки [1, 49] получается уравнение, аналогичное уравнению (16.26). Уравнение (16.26) было также модифицировано различными способами. В одном случае для объяснения различных плотностей валентных электронов фактически был разложен член fc6A. Таким образом, для атома второго периода должен быть один член для 2х-электронной плотности, к (2s) Зд (2s), и один для 2р-электронной плотности, к (2р) Зд (2р). Нет необходимости говорить, что модель с большим числом параметров дает лучшее совпадение.
Независимо от подходов, основанных на модели потенциала, для аналитических целей и для прогнозов можно использовать различные корреляции (как и любую другую хорошую корреляцию). В 1896 г. Анжели [50] сообщил о синтезе соединения Na2N2O3. В 1969 г. Хендриксон и сотр. [51] обнаружили в спектре Na2N2O3 два пика Nls при энергиях связи 403,9 и 400,9 эВ. Наличие двух пиков Nls приблизительно равной интенсивности исключает из рассмотрения структуру II (из трех наиболее вероятных структур N2O|“, изображенных ниже), в которой из резонансных форм следует эквивалентность двух атомов азота.
6|г-
C)=N—N [()=N—О—N—О]2~ [О—N=N—О—О]2~
хо|
I II III
Расчеты N2O3“ по методу МО (как итерационные по расширенному методу Хюккеля, так и с помощью метода ППДП) дали заряды на атомах азота в структуре I, которые согласуются с корреляциями заряд— атомный заряд. Структура монокристалла, установленная с помощью рентгеноструктурного анализа в 1973 г. [52], говорит в пользу того, что N2O|“ существует в виде I.
Отметим, что для соединений олова и железа удалось обнаружить линейные корреляции между энергиями связи электронов оболочки и мёссбауэровскими изомерными сдвигами [53]. Была также установлена корреляция связей электронов оболочки хлора с частотами ядерного квадрупольного резонанса [54].
Интересной вариацией темы коррелирования энергий связи электронов оболочки с другими молекулярными характеристиками является тонкая концепция «эквивалентных оболочек» [55]. Этот подход возник, вероятно, из желания построить термохимический цикл (подобный ци
350
Глава 16
клу Борна — Габера), поскольку кажется, что атомные оболочки с одним и тем же зарядом можно рассматривать как химически эквивалентные. Другими словами, известно, что некоторые термодинамические параметры (теплоты образования) могут характеризовать изменение энергии связи электронов оболочки. Эта идея может стать более понятной, если рассмотреть следующий пример. Фотоионизация электрона Nls из газообразной молекулы азота может быть представлена уравнением
hv
N2 (газ) —NN * + (газ) + е (вакуум), (16.28)
Еь
в котором звездочка указывает на то, что ls-электрон удален с одного атома азота. Очевидно, нельзя использовать термодинамические данные для расчета изменения энергии (ДЕ) в процессе (16.28), так как теплота образования газообразного NN * + неизвестна. Однако, чтобы сделать это уравнение более приемлемым для такого подхода, эквивалентные оболочки можно заменить. В этой операции есть одна тонкость. Поскольку речь идет о валентных электронах газообразного NN * +, оболочка fN* + ) точно так же может быть оболочкой кислорода, где заряд ядра кислорода, будучи больше заряда ядра атома азота на единицу, заменяет дырку в ls-оболочке. Формальный процесс замещения может быть записан в виде уравнения
NN*++O6+-NO++N*6+ (16.29)
Если эти две оболочки эквивалентны, то изменение энергии в ходе процесса (16.29) равно нулю (ДЕ = 0). Как оказывается, необходимо только, чтобы то же изменение энергий ДЕ наблюдалось для того же самого типа замещения в ряду ls-ионизованных молекул азота. Складывая уравнения (16.28) и (16.29), получаем
N2+O6 + -> NO + + N*6 + +е. (16.30)
Изменение энергии в ходе процесса (16.30) равно либо Еь для ls-элек-трона в N2, либо этой Еь плюс (предположительно) член с постоянной энергией. Энергия связи ls-электронов азота была измерена для N2 и многих азотсодержащих молекул. Для других молекул (все они считаются газообразными) процессы замещения эквивалентных оболочек можно записать в виде следующих уравнений:
NH3 + О6 + -> ОН3+ + N *6 + + е,
(CH3)2NH + O6+ -> (СН3)2ОН+ + N*6+ + е,
HCN + О6 + -> НСО + + N *6 + + е,
NO + О6+ -> О2+ + N*6+ + е,
NO2 + О6+ -> О3+ + N*6+ + е, (16'31)
CH3NH2 + О6 + -> СН3ОН2+ + N *6 + + е,
NNO + О6 + -> NO2 + N *6 + + е.
Ионизационные методы
351
Рис. 16.16. График зависимости относительных энергий связи ls-электронов азота от относительных термодинамических энергий. Данные взяты из работы [55].
В приведенных выше уравнениях известны теплоты образования молекулярных частиц, и для каждого процесса могут быть получены относительные термодинамические энергии (ЕТ). Например, Ет для уравнения с NH3 определяется как теплота образования ОН3 минус теплота образования NH3. График зависимости Ет от энергий связи ls-электронов азота (Еь) демонстрирует исключительно хорошую корреляцию (рис. 16.16). Такой тип замещения эквивалентных оболочек дает хорошие корреляции и для данных по энергиям связи электронов в других элементах, например в углероде (1s) и ксеноне (3d5/2) [55]. Этот вид корреляций полезен, поскольку дает возможность из некоторых измеренных энергий связи электронов оболочки и известных термодинамических параметров предсказать различные, еще не определенные термодинамические величины. Изучение приведенных выше уравнений показывает, что их можно использовать для определения сродства к протону. По некоторым непонятным причинам сродство к протону (РА) молекулы В берется как положительное число и приравнивается изменению энергии процесса (16.32) с отрицательным знаком.
-РА
В + Н+ -> ВН + . (16.32)
В табл. 16.5 приведены некоторые значения РА, определенные с помощью приближения эквивалентных оболочек, а также известные значения РА.
Еще не известные теплоты образования также можно определить
352
Глава 16
для различных молекул с помощью этого приближения. Например, AHJ для газообразной NON можно оценить как 120 ккал/моль, и это дает АН = — 100 ккал/моль для процесса
NON (газ) -> NNO (газ) АН = — 100 ккал/моль. (16.33)
Прежде чем закончить обсуждение химических сдвигов в энергиях связи электронов оболочки, вкратце рассмотрим данные РФС по соединениям со «смешанной валентностью», которые обсуждались в гл. 10. Старейшим из известных координационных комплексов является
Таблица 16.5
Сродство к протону
Основание РА, ккал/моль Основание РА, ккал/ моль
c5h5n 247 С6Н,ОН 192
ch3nh2 247 NC13 190
NH3 214а Н2О 169а
CHjNO, 209 NF, 133
NH,OH 208
a ~ - Эти два значения были определены в других экспериментах и использованы в качестве базиса для корреляции.
берлинская лазурь. Состав этого интенсивно окрашенного в голубой цвет соединения выражается формулой KFe[Fe(CN)6]. Интенсивный цвет связан с так называемой полосой переноса заряда между центрами различной валентности. Спектр РФС Fe(2p3/2) берлинской лазури состоит из относительно узкого пика и широкого пика при 4,4 эВ для более высокой энергии связи [57]. Оба пика приписаны различным ионам железа. Рентгеноструктурный анализ [57] этого соединения показывает, что существуют три различных типа атомов железа. Два из них относятся к высокоспиновым атомам железа(Ш): один находится в окружении шести атомов азота, а другой связан с четырьмя атомами азота и двумя атомами кислорода. Низкоспиновые атомы железа(П) координируются с атомами углерода. Отметим, что такая интерпретация спектра РФС была подвергнута критике [58].
Обменное расщепление или другая тонкая структура (см. ниже) в спектре РФС должна быть проанализирована самым тщательным образом. К соединениям со смешанной валентностью относится также [(NH3)5Ru (пиразин) Ru(NH3)5]5 + , описанный в гл. 10. Это соединение было также исследовано методом РФС [59]. Как сообщалось, в спектре РФС наблюдаются два ионизационных пика, обусловленные двумя неэквивалентными ионами переходного металла. Следует отметить, что пики ls-углерода находятся в том же спектральном диапазоне, что и ионизационные пики металла, и вывод сделан исходя из результата вычитания пиков углерода из спектра. Метод РФС характеризуется шкалой
Ионизационные методы
353
очень малых времен, иногда порядка 10'17 с. Таким образом, он может быть использован для регистрации некоторых скоростей более быстрого переноса заряда между центрами различной валентности в соединениях со смешанной валентностью.
Структура пиков РФС
Из проведенного ранее обсуждения химических сдвигов ионизационных пиков РФС электронов оболочки можно сделать вывод, что для электронов оболочки всегда наблюдаются простые спектры, например, для каждого заметно различающегося окружения атома азота наблюдается один пик для ls-электронов азота. К счастью, это не всегда так [27]. Мы уже видели, что парамагнитные частицы, такие, как О2, вызывают обменные расщепления линий электронов оболочки. Такие же расщепления, обусловленные обменными процессами, обнаружены и в спектрах РФС парамагнитных комплексов ионов переходных металлов. Кларк и Адамс [60] сообщили о Зз-обменном расщеплении хрома величиной около 4,5 эВ в Cr(hfa)3 и 3,1 эВ в Cr(h5-C5H5)2. Может возникнуть вопрос, должен ли анализ такого расщепления способствовать пониманию деталей контактных сдвигов Ферми в ЯМР, наблюдаемых для парамагнитных частиц.
Возможно даже, что более много обещающей и интригующей тонкой структурой в спектрах РФС является структура, связанная с процессом «встряхивания». Было бы очень удивительно, если бы все, что происходило при столкновении у-кванта очень высокой энергии с молекулой, ограничивалось бы фотоионизацией одного валентного электрона или электрона оболочки. Одновременно с фотоионизацией электрона может происходить возбуждение одного из оставшихся электронов до первоначально свободной орбитали. Это явление называется «встряхиванием». Таким образом, структура пиков РФС с более высокой энергией и низкой интенсивностью, обусловленных электронами оболочки, может быть использована для изучения различных электронных переходов, происходящих одновременно с фотоионизацией. Эти пики-сателлиты обнаружены в диапазоне энергий, превышающих на величину до 50 эВ энергии связей, характеризующие основные пики. Очевидно, электронное поглощение при 50 эВ (= 404000 см ' 1 = 25 нм) представляет собой поглощение с очень высокой энергией в области вакуумного ультрафиолета.
Пример такой структуры с пиками-сателлитами в спектре РФС изображен на рис. 16.12, где наблюдаются широкие пики, лежащие при более высоких энергиях связи, чем два пика Ols для молекулы кислорода. Пики, помеченные буквами А, В и С, представляют собой полосы, характеризующие процесс «встряхивания»; они возникают как сателлиты у пиков, обусловленных фотоионизацией валентных электронов. Аналогично рис. 16.17 демонстрирует структуру «встряхивания» для пика Nls молекулы азота. Пики, помеченные символами а', а3, а4, а5 и а6, обусловлены немонохроматичностью рентгеновского излучения Ко. Mg.
23-574
354
Глава 16
<000
<500
500
430 420 4<0 400 390
Энергия связи,, эВ
Рис. 16.17. Фотоэлектронный спектр ls-электронов азота, возбужденного рентгеновским излучением К7.Mg. Основной пик при энергии связи 410 эВ представляет собой линию Nls, возбужденную излучением Ка.1 2 Mg. Спектр демонстрирует также линии, возбужденные сателлитами рентгеновского излучения, и линии, соответствующие «встряхиванию» и неупругому рассеянию на молекуле N2 [27].
О
Многие из пиков, помеченных буквами А, В, С и D, приписаны возбуждениям «встряхивания» [27]. Вероятно, пики «встряхивания» могут быть идентифицированы для многих молекулярных частиц и, возможно, окажутся полезными для идентификации электронных структур многих систем. Следует отметить, что ярко выраженные сателлиты «встряхивания» были обнаружены в фотоэлектронных спектрах Ni(CO)4, Fe(CO)5, Cr(CO)6, W(CO)6 и (CO)5CrX (X = NH3, PPh3 и т.д.) [61].
Ионизационные методы
355
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. Biemann К., Mass Spectrometry, McGraw-Hill, New York, 1962.
2. Джонстон P., Руководство по масс-спектрометрии для химиков-органиков.—М.: Мир. 1975.
3. Benyon R.A. W.. Saunders R.A.. Williams А.Е.. The Mass Spectra of Organic Molecules, Elsevier, Amsterdam, 1968.
4. Litzow M. R., Spalding T. R., Mass Spectrometry of Inorganic and Organometallic Compounds, Elsevier, New York, 1973; Lewis J., Johnson B.F. G., Acc. Chem. Res., 1, 245 (1968); Bruce M.I., Adv. Organomet. Chem., 6, 273 (1968).
5. Budzikiewicz H„ Djerassi C., Williams D.H., Mass Spectrometry of Organic Compounds, Holden-Day San Francisco, 1967.
6. Shrader S.R., Introductory Mass Spectrometry, Allyn and Bacon, Inc. Boston, 1971.
7. Me Lafferty F.W., Interpretation of Mass Spectra, Benjamin W. A., New York, 1967.
8. Beckey H.D., Angew. Chemie, Int. Ed., 8, 623 (1969).
9. Williams D., in Determination of Organic Structures by Physical Methodes, Vol.
3, eds. Nachod F. C., Zuckerman J. J., Academic Press, New York, 1971.
10. Rosenstock H.M., Krauss M., in Mass Spectrometry of Organic Ions ed. McLaf-ferty F. W., Academic Press, New York, 1963.
11. Biemann K., Spiteller G., J. Amer. Chem. Soc., 84, 4578 (1962).
12. Jones J.H., Quart Rev. Chem. Soc., 22, 302 (1968).
13. Hugentobler E., Loliger J., J. Chem. Ed., 49, 610 (1972).
14. Lederberg J., J. Chem. Educ., 49, 613 (1972).
15. Maurer D„ et al., J. Chem. Educ., 51, 463 (1974).
16. Bell H.M., J. Chem. Educ., 51, 548 (1974).
17. Collard J., Ph. D. thesis, U. of Illinois, Urbana, 1975.
18. Brinckman F.E., Stone F.G.A., J. Amer. Chem. Soc.. 82, 6135 (1960).
19. Hoehn H. A., Pratt L., Watterson K., J. Chem. Soc., 1961, 2738.
20. Milne T.A., Klein H.M., J. Chem. Phys., 33, 1628 (I960).
21. Grimley R.T., Burns R.P., Ingram M.G., J. Chem. Phys., 34, 664 (1961).
22. Berkowitz J., Ingram. M. G., Chupka W. A.. J. Chem. Phys., 26, 842 (1957).
23. Барнард Дж., Современная масс-спекгрометрия.—М.: Мир, 1957.
24. Wada Y., Kiser R. W, Inorg. Chem., 3, 174 (1964).
25. Baldeschwieler J.D., Science, 159, 263 (1968).
26. a. Baldeschwieler J.D., Woodgate S.S., Acct. Chem. Res., 4, 114 (1971).
b. Beauchamp J.L. Ann. Rev. Phys. Chem., 22. 527 (1971)
c. Brauman J. L. Blair L. K.. in Determination of Organic Structures by Physical Methodes, Vol. 5, eds. Nachod F. C., Zuckerman J. J., Academic Press, New York, 1973.
27. Siegbahn K., Nordling C.. Johansson G„ Hedman J., Heden P.F., Hamrin K., Ge-lius U., Bergmark T„ Werme L. 0., Manne R., Baer ESCA Applied to Free Molecules, North-Holland/American Elsevier, New York, 1969.
28. Turner D. W., Baker C., Baker A.D., Brundle C.R., Molecular Photoelectron Spectroscopy, Wiley-Interscience, New York, 1970.
29. Aulich H„ Baron B., Delahay P., J. Chem. Phys., 58, 603 (1973).
30. Eastman D.E., Photoemission Spectroscopy of Metals, in Techniques in Metals Research VI, ed. Passaglia E., Interscience, New York, 1971; Fischer T.E., Surface Sci., 13, 30 (1969); Brundle C. R., in Surface and Defect Propertise of Solids, Vol. 1, eds. Roberts M. W., Thomas J. M., Specialist Periodical Reports, The Chemical Society, London, 1972.
31. Turner D. W„ Nature, 213, 795 (1967); Turner D. W„ Adv. Mass Spec., 4, 755 (1968).
32. Edquist 0., Asbrink L., Lindholm E., Z. Naturforsch., 26a, 1407 (1971).
33. Parratt LG., Rev. Mod. Phys., 31, 616 (1959).
34. Rendina J.F., larson P.E., Bulletin from GCA/McPherson Instrument Co., 1975.
35. Carlson T. A., in Electron Spectroscopy, ed. Shirley D. A., North-Holland, Amsterdam, 1972.
23*
356
Г.шва 16
36. Coopmans Т., Physica, 104 (1934); Roothaan С. С. J., Rev. Mod. Phys., 23, 61 (1951).
37. Gederbaum L.S., Hohlneicher G., von Niessen W., Chem. Phys. Let. 18, 503 (1973); Davis D. W., Hollander J.M., Shirley D.A., Thomas T.D., J. Chem. Phys., 52, 3295 (1970); Hillier I.H., Saunders V.R., Wood M.H., Chem. Phys. Lett., 7, 323 (1970); Richards W. G„ Int J. Mass Spec. Ion Phys., 2, 419 (1969).
38. Potts A.W., Price W.C., Proc. Roy, Soc. Lond., A, 326, 181 (1972).
39. Turner D. W., May D.P., J. Chem. Phys., 45, 471 (1966).
40. Kato H., Morokuma K., Yonezawa T, Fukui K., Bull. Chem. Soc. Japan, 38, 1749 (1965).
41. Mulliken R.S., Phys. Rev., 47, 413 (1935).
42. Jonathan N„ Morris A., Okuda M., Smith D.J., Ross K.J., in Electron Spectroscopy, ed. Shirley D. A., North-Holland, Amsterdam, 1972.
43. Jonathan N., Smith D.J., Ross K.J., J. Chem. Phys., 53, 3758 (1970).
44. Berkowitz J., Dehmer J.L., J. Chem. Phys., 57, 3149 (1972) и ссылки в этой работе.
45. Siegbahn К., et al., Phys. Rev., 110, 776 (1958).
46. Fahlman et al., Nature, 210, 4 (1966).
47. Moddeman W.E., Blackburn J.R., Kumar G., Morgan K.A., Jones M.M., Albrid-ge R. G.. in Electron Spectroscopy, ed. Shirley D. A., North-Holland, Amsterdam, 1972.
48. Stucky G.D., Matthews D.A., Hedman J., Klasson M., Nordling C., J. Amer. Chem. Soc., 94, 8009 (1972).
49. Ellison F.O., Larcorn L., Chem. Phys. Lett., 10, 580 (1971).
50. Angeli A., Gazz. Chim. ItaL. 26, 17 (1896).
51. Hendrikson D.N., Hollander J. M., Jolly W.L., Inorg. Chem., 8, 2642 (1969).
52. Hope H.. Sequeira M.R.. Inorg. Chem.. 12, 286 (1973).
53. Adams I., Thomas J. M., Bancroft G. M., Butler K. D. Barber M., Chem. Commun., 1972, 751; Swartz W. E„ Watts P. H., Lippincott E. R„ Watts J. C., HuheeyJ. E.. Inorg. Chem., 11, 2632 (1972); Barber M., Swift P., Cunningham D., Frazer M.J., Chem. Commun., 1970, 1338.
54. Clark. D.T, Briggs D., Adams D.B., J. Chem. Soc.. Dalton. 1973, 169.
55. Jolly W.L. Hendrickson D.N.. J. Amer. Chem. Soc., 92, 1863 (1970); Hollander J. M., Jolly W. L., Acct. Chem. Res., 3, 193 (1970).
56. Wertheim G.K., Rosencwaig A., J. Chem. Phys., 54, 3225 (1971); Leibfritz D., Bremser W., Chem. Ztg., 94, 982 (1970).
57. Buser N.J., Ludi A., Petter W., Schwarzenbach., Chem. Commun., 1972 1299.
58. Jolly W.L., Coord. Chem. Rev., 13, 47 (1974).
59. Citrin P.H., J. Amer. Chem. Soc.,, 95, 6472 (1973).
60. Clark D.T, Adams D.B., Chem. Commun., 1971, 740.
61. Barber M„ Connor J. A., Hillier I.H., Chem. Phys. Lett., 9, 570 (1971).
Упражнения
1. Если ускоряющий потенциал в масс-спектрометре снижается в ходе эксперимента, то будет ли пик с большим или меньшим m/е зарегистрирован в спектре первым?
2. л-Связывание сера—углерод не столь эффективно, как л-связывание азот — углерод. Какой из пиков в спектре HSCH2CH2NH2 будет иметь большую интенсивность: с т/е =30 или 47?
3. Обратитесь к рис. 16.4 и вспомните обсуждение соотношения между делокализацией заряда и стабильностью положительного иона. Объясните, почему
Qch2+
имеет наиболь
шую интенсивность для изомера этилпиридина с этильной группой в положении 3.
Ионизационные методы
357
4. а. Для того чтобы определить потенциал ионизации метильного радикала, пик с каким отношением т/е в масс-спектре СН4 необходимо исследовать в зависимости от ускоряющего потенциала?
б. Запишите уравнение для потенциала возникновения фрагмента в п. «а» через потенциал ионизации и энергию диссоциации.
в. Можно ли при расчете энергии диссоциации в п. «б» из термохимических данных пользоваться одной четвертой величины теплоты образования (из газообразных углерода и водорода) СН4?
5. Спектры УФС даже относительно небольших молекул, подобных бутадиену, сложны (см. рисунки). Используя расчет бутадиена по методу Хюккеля, отнесите пики в спектре этого соединения (пики при 8,6 и 10,95 эВ—примесные пики). Три колебательные прогрессии (1520, 1180 и 500 см-1) были качественно идентифицированы в первой полосе при 9,08 эВ. Обсудите колебательную структуру этой полосы (и форму, и величину) в соответствии со сделанным вами отнесением. (Полосы наблюдаются в ИК-спектре основного состояния бутадиена при 1643, 1205 и 513 см-1.)
6. Эффекты координации небольших молекул с переходными металлами можно исследовать методом фотоэлектронной спектроскопии. Спектры приве-
358
Глава 16
дены ниже для CO и W(CO)6. Качественно объясните, что наблюдается, если исходить из связывания в этом комплексе. Примите во внимание соображения симметрии и энергетические соображения, если они подходят для ваших целей.
600
Импульс/с
w(co>,
. VV ( vV|g
4*"v***z I 1 I I I I I I I I I
21 20 19 18
16 15 14 13 12
ЭВ
1 10 9 8
7. Ниже приведены УФС-спектр и МО-диаграмма аммиака. Указаны связывающие (ь), несвязывающий (") и разрыхляющие (*) уровни.
2p—
\W
\ е*\
V
// и
Ионизационные методы
359
а. Какие явления приводят к возникновению двух полос при 11 и 16 эВ?
б. Что вызывает появление тонкой структуры, наблюдаемой для каждой полосы? (Объясните одной—двумя фразами.)
в. Следует ли тонкая структура, которую вы предсказали, из данной МО-диаграммы и почему? Как вы можете объяснить аномалии? Указание: рассмотрите формы частиц «до» и «после».
17. РЕНТГЕНОВСКАЯ КРИСТАЛЛОГРАФИЯ *
17.1. ВВЕДЕНИЕ
С помощью рентгеновской кристаллографии можно в общем случае определить точный состав и расположение атомов почти в любой молекуле. Однако на сделанное выше заявление накладываются некоторые ограничения. Во-первых, молекула должна находиться в кристаллическом твердом состоянии, что приводит к геометрическим искажениям, возникающим при упаковке ее с соседними молекулами. Во-вторых, система не должна подвергаться фотохимическому разложению при облучении ее рентгеновским излучением в течение дня [1]. В-третьих, интересующая нас система должна образовывать подходящие для кристаллографического исследования кристаллы, исключающие две проблемы, наиболее распространенные при решении структурных задач: двойникование и разупорядочивание [2]. В-четвертых, число атомов, положения которых следует определить, не должно быть слишком большим.
Уменьшение усилий, необходимых для установления кристаллической структуры, позволяет все чаще использовать в исследованиях рентгеновскую кристаллографию. В настоящее время при благоприятных условиях можно провести исследования от кристаллизации до воспроизведения структуры в течение двух — десяти дней. При правильной организации работы сроки получения результатов, их обработки и доведения структуры до деталей можно сократить. Вероятно, каждому ученому в ходе своих исследований приходится обращаться к рентгеновской кристаллографии. Цель настоящей главы—дать достаточно информации, чтобы читатель мог получить представление о рентгеноструктурном исследовании кристаллов.
Много полезной информации часто можно получить, используя рентгеноструктурные методы, не предназначенные для исследования монокристаллов. Например, симметрия элементарной ячейки может быть установлена с помощью методов, исследующих пленки вещества, а для идентификации соединений могут быть использованы результаты, полученные при изучении порошкообразных веществ или веществ, нанесенных на поверхность путем разбрызгивания или осаждения. Химик-экспериментатор должен разбираться по крайней мере в этих менее сложных методах.
В данной главе мы ограничимся, хотя и не полным, но наглядным описанием ряда примеров наиболее важных аспектов рентгеновской
* Эта глава написана М. Даггеном, сотрудником Калифорнийского университета. С благодарностью были восприняты полезные замечания Дж. Стаки и Р. Райана.
Рентгеновская кристаллография
361
кристаллографии. Вопросы, которые требуют привлечения подробного математического аппарата и не могут быть использованы новичком в этой области немедленно, здесь не рассматриваются. Методы генерирования рентгеновских лучей и их регистрации описаны в литературе [3]—они представляют собой умозрительную схему кристаллографии, которой мы и займемся.
17.2. СИММЕТРИЯ ТВЕРДОГО СОСТОЯНИЯ
С точки зрения рентгеновской кристаллографии монокристалл состоит из повторяющихся трехмерных участков электронной плотности [4] (при дифракции рентгеновских лучей ядра не регистрируются). Расположение электронов внутри этой кристаллической решетки как раз и определяет направления и интенсивности пучков рассеянных ими рентгеновских лучей. Естественно, электронная плотность определяется структурой молекулярных единиц и порядком их упаковки в кристалле. Упаковка молекул в кристалле характеризует симметрию распределения электронной плотности и размер наименьшего повторяющегося в одном направлении трехмерного фрагмента кристалла, называемого элементарной ячейкой. Используя простую фотографическую технику, часто можно определить размер и симметрию элементарной ячейки, а зная число молекул в ячейке, часто можно получить информацию об элементах симметрии интересующей нас молекулярной частицы. Этой информации может быть вполне достаточно для удовлетворительного определения молекулярной структуры, хотя для получения и интерпретации фотографических данных требуется только несколько часов работы в вечернее время. Разделы 17.2—17.5 посвящены соотношениям симметрия — интенсивность, которые необходимо понять для проведения исследований подобного типа. Ниже мы рассмотрим различные аспекты симметрии элементарной ячейки.
а. Элементарная ячейка. Элементарная ячейка представляет собой наименьший объем кристалла, который можно выбрать в качестве единицы, включающей всю информацию о структуре и симметрии кристалла. Между ее тремя определяющими ребрами и тремя углами возможны следующие соотношения:
а = Ь = с и а=Р = у = 90°, а^Ь^с и 90°,
а также другие промежуточные варианты. Первое соотношение — необходимое, но не достаточное условие для кубической решетки, тогда как второе описывает триклинную решетку. Чаще всего встречаются следующие типы решеток: моноклинная (а = у = 90°, Р =£ 90° и с) и ромбическая (а = Р = у = 90° и а Эти элементарные ячейки по-
казаны на рис. 17.1. Сделанных определений достаточно для непосредственного обсуждения. Более полное описание семи различных кристаллических систем дано в приложении VII, которое, как предполагается, будет прочитано после знакомства с разд. 17.2.
Если размедэ элементарной ячейки известен (скажем, а = Р = у = 90°, а = 10 А, Ь = 15 А и с = 20 А), то рассчитать плотность кристалла р из объема
362
Глава 17
Рис. 17.1. Общие формы и определяющие ребро и угол для моноклинной (Л) и ромбической (Б) элементарных ячеек.
V можно с помощью следующей формулы:
р = и1И/И4, (17.1)
где М—молекулярная масса, А — число Авогадро и п— число формульных (или молекулярных) масс в элементарной ячейке. Если измерена р кристалла (обычно путем флотации) с известным элементным составом молекулярных частиц, то можно рассчитать п. Величина п имеет большое значение, поскольку, как это будет показано позднее, она может дать информацию о молекулярной симметрии исходя из свойств симметрии решетки.
б. Операторы. Для определения пространственной группы используется больше операторов симметрии, чем для определения точечной группы. К этим операторам относится оператор трансляции, поскольку теперь мы передвигаем молекулу в трех направлениях. Два новых оператора носят название оператора скольжения и винтового оператора. Соответствующими им элементами симметрии являются винтовая ось и плоскость скольжения. Операция, соответствующая винтовой оси второго порядка, поворачивает содержимое ячейки на 180° (2п/п, п = 2) и затем сдвигает его вдоль оси на половину длины параллельного ребра элементарной ячейки (1/и длины в ходе одной операции при правом вращении). Операция скольжения отражает содержимое от плоскости и затем сдвигает его на половину элементарной ячейки в некотором направлении в той же плоскости. Эти операции показаны на рис. 17.2 и более подробно будут рассмотрены позднее.
Винтовая ось второго порядка на рис. 17.2 лежит вдоль ребра а при у= 1/2 и z= 1/2. Она обозначается символом 2Р Общим обозначением операции, соответствующей винтовой оси, является Nm, где N — порядок собственной оси вращения, а т (которое может принимать целочисленные значения 1, 2, ..., N — 1) указывает на трансляцию на m/N периода элементарной ячейки параллельно оси вращения. Плоскость скольжения на рис. 17.2 называется а-скольжением перпендикулярно b при
Рентгеновская кристаллография
363
Рис. 17.2. Свойства преобразований формы под действием винтовой оси второго порядка (на рисунке обозначена как винтовая ось) параллельной а (Л), и плоскости скольжения, перпендикулярной Ь, которая движется вдоль а (Б).
2- элементарной ячейки
у = —1/2. Все дробные обозначения относятся к частям длины элементарной ячейки в этом направлении (т. е. измерение х, у и z проводится в направлениях а, b и с соответственно).
Теперь приступим к определению некоторых операций пространственной группы и дадим их символические обозначения. Эта информация будет неполной, но наглядной.
1. Собственная ось второго порядка. Рис. 17.3,Л демонстрирует собственную ось второго порядка, параллельную b при х=1/4 и z = 0, месторасположение которой обычно обозначают символом (1/4, 0, 0)*. В случае решеток все операции симметрии описываются произведением операций точечной группы по отношению к осям элементарной ячейки а, Ь, с и операции трансляции. Например, операция симметрии второго порядка над точкой s (рис. 17.3,Л) описывается символом 2ь[1/2, 0, 0], где 2Ь подразумевает операцию вращения второго порядка вокруг оси Ь, а квадратные скобки обозначают трансляцию в направлениях а, b и с соответственно. Операторы второго порядка, если поворот осуществляется вокруг осей, параллельных а и с, будут обозначаться символами 2а и 2с. Дробные обозначения координат даны в круглых скобках.
В матричной записи операция 2Ь [1/2, 0, 0] имеет вид
Матрица 3x3 характеризует операцию вращения, проводимую относительно начала координат. Как и при более раннем обсуждении то
* На всех диаграммах отсчет ведется относительно начала координат, находящегося в верхнем левом углу. Точка, к которой смещается ось от начала координат, находится на грани элементарной ячейки, перпендикулярной проходящей через нее оси.
364
Глава 17
чечных групп, матрица 3x3 превращает х в — х, у в — у и z в — z. Символ [1/2, 0, 0] представляет собой операцию трансляции на половину длины элементарной ячейки в направлении а. Математически операция трансляции точки на t1; t2, t3 определяется как
[tp t2, t3] (x, у, z) = (tj+x, t2+y, t3 + z).
Операция 2b [1/2, 0, 0] над точкой (x, у, z) включает сначала операцию 2Ь (матричное умножение), а затем векторное сложение [1/2, 0, 0] (или наоборот, поскольку эти операции коммутируют). Комбинация этих двух операций—вращения вокруг оси b и трансляции [1/2, 0, 0] — эквивалентна вращению второго порядка относительно точки (1/4, 0, 0). Для проверки этого утверждения рассмотрим точку (0, 1/2, 0). Видно, что элемент, изображенный на рис. 17.3,Л, должен переводить эту точку в точку (1/2, 1/2, 0). Рассчитывая результат другого вращения второго порядка относительно получившейся точки, имеем
т. е. исходная точка восстанавливается.
В примере, изображенном на рис. 17.3,Б, мы имеем 2С при (1/4, 1/4, 0) или 2С [1/2, 1/2, 0].
2. Винтовые оси. Как упоминалось ранее, винтовые оси возникают в том случае, когда трансляция осуществляется в направлении оси вращения, например, 2а [1/2, 0, 0] обозначают как 2,. Если винтовая ось находится при у = 1/4 и z = 1/4, то операция записывается как 2а [1/2, 1/2,
Рис. 17.3. Преобразование точки под действием собственной оси второго порядка («-►) (Л), параллельной Ь, и собственной оси второго порядка, параллельной с (Б).
Рентгеновская кристаллография
365
Вид едоку
Рис. 17.4. Альтернативные изображения, демонстрирующие винтовую ось второго порядка, параллельную а и находящуюся на A-грани при у = 1/4, z= 1/4.
На левом рисунке знак 1/4 отмечает положение элемента 1/4 длины элементарной ячейки за плоскостью бумаги. Правый рисунок представляет собой вид слева левого рисунка.
1/2]. При осуществлении этой операции над любой точкой мы производим сначала вращение второго порядка 2„ вокруг оси а, т. е. относительно начала координат (у = 0, z = 0), а затем производим трансляцию (в квадратных скобках) результата операции 2Д. Эта винтовая ось показана на рис. 17.4.
В точечных группах наличие двух операторов (таких, как две перпендикулярные оси второго порядка) приводит к существованию и третьег го оператора. То же справедливо в некоторых пространственных группах. Например, собственная ось второго порядка вдоль а при (0, 0, 1/4) и собственная ось второго порядка вдоль b при (1/4, 0, 0) порождают винтовую ось второго порядка 2; вдоль с при х= 1/4 и у = 0 [при (1/4, 0, 0)]. Это можно изобразить с помощью выражения
2
а
1
о, о,—
2
•2Ь |,0,0
= 2,
ri,0,r|
2 2
Такой тип умножения следует осуществлять путем простого добавления компонент оператора трансляции и проведения матричного умножения операторов вращения (как и в операциях точечных групп):
трансляции
(1 0 0\ /-1 О 0\ /-1 о о
0 -1 0 1-1 0 1 °) = ( 0-10
О О -1/ \ О 0 -1/ \ О 01
2а 26 2С
366
Глава 17
^а
b
Рис. 17.5. Два обозначения а-скольжения.
Различие между тире и точками указывает на трансляцию соответственно в плоскости бумаги и перпендикулярно ей.
3. Зеркальные плоскости. Зеркальная плоскость, перпендикулярная b и пересекающая b при у — 1/4, может быть представлена в виде опера-
/1 ° °\ w6=|0 -1 ol
\0 0 1/
тора отражения в сочетании с трансляцией перпендикулярно плоскости, в этом случае [0, 1/2, 0].
4. Плоскости скольжения. Плоскость скольжения вдоль а перпендикулярно b при у= 1/4 обозначают как ть [1/2, 1/2, 0] (рис. 17.5). Здесь ть представляет собой элемент отражения перпендикулярно оси b при b = = 0, а скобки характеризуют трансляцию, как и в случае оператора, соответствующего винтовой оси. Описанный элемент должен называться а-скольжением, если трансляция осуществляется в направлении а. Если плоскость по-прежнему перпендикулярна Ь, то указанная операция может быть с-скольжением при трансляции вдоль с или п-скольжением при трансляции одновременно наполовину длины элементарной ячейки вдоль b и наполовину вдоль с.
5. Центрированные решетки. Другим оператором пространственных групп, не имеющим аналога в точечных группах, является центрирующий оператор. Этот оператор приводит к трем общим типам кристаллических решеток, которые называют гранецентрированными (обозначаются F), бокоцентрированными (А, В или С *) и объемноцентрированны-ми (7). Симметрию этих решеток можно описать только операциями трансляции, которые включают трансляции только наполовину длины ребра ячейки. Например, в Д-центрированной решетке для каждой точки (х, у, z) должна существовать эквивалентная точка (х, 1/2 + у, 1/2 + z).
* Различаются только три грани, принимая во внимание идентичность операторов [0, 0, 0] и [1, 1, 1].
Рентгеновская кристаллография
367
Таким образом, /I-центрирующий оператор представляет собой [0, 1/2, 1/2]. Аналогично гранецентрирующий оператор (т. е. F-центрирующий) требует, чтобы для каждой точки (х, у, z) существовали три дополнительные точки (1/2 + х, 1/2 + у, z), (х, 1/2 + у, 1/2 + z) и (1/2 + х, у, 1/2 + + z), так что F-центрирующими операторами являются [1/2, 1/2, 0]. [0, 1/2, 1/2] и [1/2, 0, 1/2] (в дополнение к операции тождественного преобразования [0, 0, 0]). Объемноцентрирующим оператором служит [1/2,1/2,1/2]. Эти типы решеток показаны на рис. VII. 1 (в приложении VII). Отметим, что если атом или молекула находится в начале координат ячейки, то другая такая же частица находится в центре грани А (перпендикулярно а) при /1-центрировании; при F-центрировании еще три частицы находятся в центрах граней А, В и С; аналогичный особый случай можно указать для объемного центрирования.
На практике чаще встречаются нецентрированные элементарные ячейки, которые называют примитивными решетками. Следует подчеркнуть, что определение операции центрирования требует, чтобы группы находились в центре (например, в центре грани) только в том случае, если другая группа находится в начале координат.
в. Пространственные группы. Символы пространственных групп записывают таким образом, чтобы их можно было однозначно отождествить с кратчайшей возможной записью каждой из 230 различных групп, которые могут быть образованы из рассмотренных выше различных операций. Мы проиллюстрируем метод записи, а также продемонстрируем совокупности операторов, составляющих группы. Рассмотрим ромбическую систему, которая даст также несколько еще не обсужденных операторов.
Из символа пространственной группы Рпта (читается как «Р—п—т—а») следует, что решетка этого типа относится к примитивной решетке; элементами симметрии этой группы являются «-скольжение, перпендикулярное оси а, зеркальная плоскость, перпендикулярная оси Ь, и а-скольжение, перпендикулярное оси с. Условия, используемые при записи символов такого вида, и вытекающая из них информация сведены в табл. 17.1. В первом столбце приведены семь различных кристаллических систем наряду с симметриями точечных групп элементарной ячейки (т. е. симметрией, которой они обладали бы, если бы не было трансляции). В столбце «характеристическая симметрия» приведены те существенные элементы симметрии, которые делают кристалл единственным в своем роде по отношению к приведенным точечным группам. В столбце «положение в символе точечной группы» описаны условия записи этого символа и указан порядок (первичный, вторичный, третичный), в котором элементы симметрии перечислены в символе. В приведенном выше примере Рпта Р—символ решетки, а п, т и а соответственно первичный, вторичный и третичный символы.
Какие же другие элементы симметрии порождаются тремя элементами, перечисленными в символе Рпта, и где эти последние должны находиться в кристалле? На эти вопросы можно ответить следующим образом.
Таблица 17.1
Стандартные элементарные ячейки, их симметрия и символы
Кристаллографическая система и точечные группы Соотношения между осями и углами3 Характеристическая симметрия I Число решеток Бравэ j системе Символы решеток Положение в символе точечной группы Пример
первичный вторичный третичный
Триклинная 1, 1 а b с Симметрия 1 Р Только один символ, который обозначает все Р1,
а Р Y 90° только первого порядка (тождественное преоб- направления в кристалле Р1
разование или инверсия)ж
Моноклинная 1-й вариант Ось второго по- 2 1-й ва- \р Символ дает природу единственной оси второго Р21/С
2, т, 2/т3 а b с а = Р = 90‘ / у 2-й вариант рядка (вращения или инверсии) только в одном направлении; берется как ось z риант 1В 2-й ва- J Р риант 1С порядка4 (вращения и(или) инверсии) 1-й вариант: единственная в своем роде ось ze 2-й вариант: единственная в своем роде ось у С2/с
а Ь с а = у = 90° 1= р в первом варианте и как ось у во втором варианте
Ромбическая а # b # с Оси второго по- 4 Р 2-й порядок (вра- 2-й порядок (вра- 2-й порядок (вра- Рпта,
222, mm2, ттт п R „ 00° рядка (вращения С6 щение и(или) ин- щение и(или) ин- шение и (или) ин- Р2,2Л
или инверсии) в I версия) вдоль версия) вдоль версия) вдоль
трех взаимно перпендикулярных направлениях F оси X ОСИ у ОСИ Z
Тетрагональная а = Ь с Ось четвертого 2 Р* 4-й порядок (вра- 2-й порядок (вра- 2-й порядок (вра- Р4Ьт.
4. 4. 4/mJ; 422, а = р = у = 90° порядка (враще- I щение и(или) ин- щение и(или) ин- щение и(или) ин- 14тт
4тт, 42т, ния или инверсии) вдоль оси Z версия) вдоль версия) вдоль версия) вдоль оси z осей х и у других направле-
4/ ттт3 НИЙи
24-574
Тригональная (Ромбоэдри-и гексагональ- ческие оси) нпя —h —
3. 3: 32, Зт, Зт; а ~ ~ С 6, 6/т; 622, 6mm, а = р = у < 6т2 <120° ^90°
(Гексагональные оси)
а = Ь # с а = р = 90°, у = 120° Кубическая _ а = b = с 23, m3; 432, 43m, тЗт а = р = у = 90
Ось третьего по- 1 рядка (вращения или инверсии) вдоль диагонали кристалла с использованием ромбоэдрических осей или ...
Вдоль направле- 1 ния оси z с использованием гексагональных осей
Четыре оси 3 третьего порядка, каждая из которых отклоняется на 54° 44' от кристалл ографи-ческих осей
Rr
3-й или 6-й порядок (вращение и(или) инверсия) вдоль оси z
2-й или 4-й порядок (вращение и(или) инверсия) вдоль особых плоскостей u
2-й порядок (вращение и(или) инверсия) вдоль осей х, у и и (х. у и и в плоскости на 120°)
2-й порядок (вра щение и(или) инверсия) перпендикулярно осям х, у и и в особой плоскости11
Р3221, КЗс, Гб, 22, Рбсс
3-й порядок (вращение и(или) инверсия) вдоль диагонали кристалла
3-й порядок (вращение и(или) инверсия) вдоль особых направлений и
РД Fm3, 143т
р
р I
F
q Знак подразумевает неравенство, что обусловлено симметрией; естественно, может иметь место случайное совпадение.
Если рассматривать решетки сами по себе, то бокоцеитрироваииую ромбическую решетку принято обозначать символом С. В пространственных группах точечной группы mm2 условие «единственная в своем роде ось z» требует, чтобы бокоцеитрироваииую решетку иногда обозначали С, а иногда А (или В).
Тетрагональные решетки Р и I можно также обозначать как С и F, но только в том случае, если векторы а и Ь, перпендикулярные вектору с, не являются самыми короткими.
Решетка R описывается здесь исходя из ромбоэдрических осей, но ее можно также привязать и к гексагональным осям. Если эти варианты необходимо различать, то в первом случае используются символы 7?obv или Rrev, а во втором — символ 7?hex.
Элементами моноклинной симметрии являются ось вращения второго порядка и(или) зеркальная плоскость (перпендикулярная этой оси, если имеются оба элемента).
При кристаллографическом исследовании обычно встречается второй вариант.
Ось инверсии во всех случаях подразумевает наличие зеркальной плоскости, перпендикулярной оси вращения (несобственная ось в точечных группах). Как видно для ортогональных систем, пространственная группа часто получает наименование, исходя из обозначения плоскости.
Косая черта указывает на плоскость скольжения или зеркальную плоскость, перпендикулярную обозначенной оси вращения.
В каждом случае подробные сведения можно найти в работе [3]—для наших целей оии не имеют особого значения.
370
Глава 17
Нам известны четыре операции группы [включая оператор тождественного преобразования 7-(0, 0, 0)]. Запишем их, оставляя незаполненными места, где информацией мы пока не располагаем:
1,0,0\
0,1,0 1- [0, 0, 0], .0,0, 1/
тождественное преобразование
и-Скольжение * та [ — , 1/2, 1/2], диагональная трансляция, положение на оси х не известно. (Положение на оси х не известно потому, что конечная величина зависит от расположения «-скольжения плоскости на оси х. По определению плоскости и-скольжения у и z будут транслироваться наполовину периода элементарной ячейки.)
Зеркальная плоскость ть [0, —, 0], не известно положение на оси у.
а-Скольжение тс [1/2, 0, — ], не известно положение на оси z. Эти три последние операции (та, ть и тс) могут быть попарно перемножены:
Из теории точечных групп мы знаем, что тс 2С порождает центр инверсии 1; определим его в качестве начала координат ячейки
тс
- 0 - -2 2’ и’ 2 '
0, | = 1 [0, 0, 0].
2 2 L
Это справедливо, поскольку тождественное преобразование в повторяющейся системе
1 1
— + — =1=0.
2 2
В результате можно заполнить пропущенные места во всех приве-
та обозначает зеркальную плоскость, перпендикулярную оси а.
Рентгеновская кристаллография
371
денных выше уравнениях, что приводит к
та 1 1 1 • Wh о, —, 0 = 2С 1 1 —, о, -
7’ 7’ 7 и 2 2 ’ 2
ть
тс
0, —, 0 • т.
2
1 1“
7’ °’ 7
1
7’
11
°’2 =2‘
1 1 1
т“ 7’7’7
= 2Ь
1 1 1
7.2
Таким образом, операциями для Рпта являются
1 (0, 0, 0)
1 (0, 0, 0) ’1 1 г „ Г1 1 f
т„ -. -
|_2 2 2_ а[2’ 2’ 2_
«ь 0, |, 0 2„ 0, 1 0
1 о, 2 ’ 2
тс
2С 0, с 2 2
Представляем читателю возможность показать, что эти восемь операций составляют математическую группу; мы будем называть ее пространственной группой.
Рассмотрим свойства преобразований точки (х, у, z) в Рпта, соответствующих последовательно перечисленным выше восьми элементам симметрии:
х, У, 2
х, У, 2
[1/2 — х, 1/2 +у, 1/2+ z] [1/2 +х, 1/2 — у, 1/2 —z]
[х, 1/2 —у, z] [х, 1/2 +у, z]
[1/2+ х, у, 1/2 — z] [1/2 —х, у, 1/2+ z]
(Отметим, что х, у и z в кристаллографической записи означают соответственно — х, — у и — z.) Помещение атома в любую точку (х, у, z) в элементарной ячейке приводит к появлению атомов в семи других перечисленных точках. Аналогично для любой молекулы в элементарной ячейке существует семь других молекул, которые симметрично расположены по отношению к этим семи точкам. Для определения полного содержимого ячейки необходимо только перечислить одну восьмую часть точек.
24*
372
Глава 17
Рассмотрим плоскость, где у = 1/4 (а х и z имеют произвольные значения). Видно, что для восьми операций Рпта в этой плоскости находятся только четыре связанные по симметрии точки. Если бы существовали лишь четыре молекулы в каждой элементарной ячейке симметрии Рпта, то они могли бы иметь следующие координаты:
(х, 1/4, z), (1/2 -х, 3/4, 1/2+ z), (1/2 +х, 1/4, 1/2 - z), (х, 3/4, z).
(Координаты этих точек получаются путем подстановки у = 1/4 в общие уравнения, приведенные выше.) Эти точки носят название особых при симметрии т. Они могут существовать (см. разд. 17.7в), если только молекулярная точечная группа включает в качестве элемента симметрии зеркальную плоскость и если последняя совпадает с такой же плоскостью элементарной ячейки.
Общие и особые точки и их точечная симметрия перечислены для всех пространственных групп в первом томе «Интернациональных таблиц для рентгеновской кристаллографии» [7]. При наличии данных относительно элементарной ячейки можно непосредственно установить возможные молекулярные симметрии (в том и только в том случае, если молекулы находятся в особых точках, а пространственная группа определена однозначно).
В качестве примера рассмотрим молекулу титаноцена (C5H5)2Ti. Было много споров относительно предложенной геометрической структуры этого соединения, поскольку теоретические соображения говорят в пользу изогнутой структуры, тогда как вполне возможна структура, аналогичная структуре ферроцена. Обнаружено, что (C5H5)2Ti существует только в димерной форме, и, таким образом, этот вопрос имеет смысл только для недавно синтезированной молекулы (C5Me5)2Ti, в которой все атомы водорода замещены на метильные группы. Это соединение в растворе представляет собой мономер; если его выделить в виде твердого кристалла, то в элементарной ячейке симметрии Р21/с содержатся две молекулы [5]. В этой группе общая точка порождает четыре молекулы на элементарную ячейку, в то время как особых точек всего две с симметрией 1. Очевидно, для того чтобы молекула (C5Me5)2Ti находилась в центре симметрии 1, ее структура должна иметь центр инверсии, и поэтому одно циклопентадиенильное кольцо будет порождать другое, параллельное первому. Поскольку при воздействии рентгеновских лучей кристаллы этого вещества при комнатной температуре медленно разлагаются, точные данные по интенсивности рентгеновского излучения получить трудно; однако ограниченный набор данных согласуется со сделанным предположением о наличии только центровой симметрии.
Мы продолжим обсуждение общих свойств симметрии в случае пространственной группы P2-J с (читается как «Р-два-один-на-с» или «Р-два-один в нижнем индексе-на-с»). Точечная группа, соответствующая этой пространственной группе, получается путем превращения элементов симметрии пространственной группы в их эквиваленты в точечной группе. Например, в этом случае 2t выводится из оси вращения второго
Рентгеновская кристаллография
373
порядка, ас— из зеркальной плоскости. Таким образом, точечная группа, соответствующая пространственной, есть 2/т. Из первого столбца табл. 17.1 видно, что мы имеем дело с моноклинной системой, которая относится к числу наиболее распространенных пространственных групп. Ее операции содержатся в наименовании, и табл. 17.1 позволяет установить, что это за операции. Как отмечалось ранее, примитивная решетка имеет винтовую ось второго порядка с перпендикулярной ей плоскостью с-скольжения. Выполняется следующее соотношение:
2ь[-,1/2, -] т„[0, -,1/2]=1[0,0,0].
Взяв центр инверсии в качестве начала координат, получаем
2Ь [0, 1/2, 1/2] -ть [0, 1/2, 1/2] = I [0, 0, 0].
Для моноклинных систем в качестве единственной оси рассматривается ось Ь, которая представляет собой ось второго порядка (собственную или несобственную).'
В триклинной кристаллической системе, где а/Ь/с и а / р / у, единственными операторами являются оператор тождественного преобразования и, возможно, центр инверсии, а две единственные триклинные пространственные группы — Р1 и Р1 (читаются как «Р-один» и «P-один с черточкой»); последняя группа обладает центром инверсии.
г. Диаграммы элементарных ячеек. На рис. 17.6 в виде диаграммы показаны элементы симметрии пространственной группы Р2г/с. Жирные точки—это центры инверсии. Центры инверсии, которые находятся не в углах элементарной ячейки, возникают по той причине, что комбинация операции инверсии в начале координат и операции трансляции от одной точки решетки до другой приводит к центру инверсии на половине пути между этими двумя точками решетки. Обозначенные специальным значком (рис. 17.6) оси 2; перпендикулярны плоскости бумаги, а оси 215 обозначенные стрелками, лежат в этой плоскости; 1/4 указывает на плоскость скольжения, параллельную плоскости бумаги, но отстоящую от нее на 1/4 размера ячейки вдоль перпендику-
Рис. 17.6. Символические обозначения элементов симметрии, содержащихся в пространственной группе P2Jc.
374
Глава 17
лярной оси (Ь), и транслирующую в направлении стрелки. Штриховые линии обозначают плоскости скольжения, перпендикулярные плоскости бумаги и транслирующие параллельно штиховым линиям. Эти символы даны в «Интернациональных таблицах для рентгеновской кристаллогра-
-----
Рис. 17.7. Символическое обозначение элементов симметрии Рпта.
фии». Из-за трансляции элементарной ячейки [1,0,0], [0,0,1], [0,1,0] и т. д. в ней находится более чем один элемент каждого из четырех типов элементов. Например, 2Ь [0, 1/2, 1/2]-[1, 0, 0] равно 2Ь [1, 1/2, 1/2], что соответствует 2j при х = 1/2 и z = 1/4.
Для пространственной группы Рпта можно построить диаграмму симметрии из набора приведенных ранее операторов. Наряду с тремя зеркальными плоскостями и тремя осями второго порядка имеется еще и центр инверсии. К трем операторам, включающим зеркальные плоскости, относятся н-скольжение при х = 1/4, зеркальная плоскость при у = 1/4 и а-скольжение при z = 1/4. Осями являются 2; вдоль а при у = = z = 1/4, 2j вдоль b в начале координат и 2] вдоль с при х = 1/4 и у = = 0. Рис. 17.7 демонстрирует все элементы симметрии элементарной ячейки, порожденные данными восемью операторами. Штрихпунктир-ная линия изображает плоскость н-скольжения, движущуюся по диагонали (направление делит пополам угол между осями b и с), а все центры инверсии проецируются на переднюю грань ячейки, хотя можно видеть, что один центр, возникающий при (1/2, 0, 0), связан с центром инверсии, находящимся в начале координат, винтовой осью (при х = 1/4, b = 0) и поэтому находится при z = 1/2.
17.3. НАПРАВЛЕНИЕ ДИФРАКЦИИ РЕНТГЕНОВСКИХ ЛУЧЕЙ
Получив некоторое представление о свойствах симметрии внутренней структуры кристалла, займемся теперь анализом взаимодействия рентгеновских лучей с этим кристаллом. Для этого используем соотно
Рентгеновская кристаллография
375
шение, обычно встречающееся в вводных курсах химии и называемое законом Брэгга*:
nA. /1
апв-— 7
(17.2)
Из этого математического выражения следует, что если электромагнитная волна с длиной X попадает под углом G на две параллельные плоскости, расстояние между которыми составляет d (рис. 17.8), интерференция лучей, исходящих из точек О и С, происходит лишь в том
Рис. 17.8. Отражающие плоскости и направления падающей и отраженной волн, демонстрирующие геометрические соотношения закона Брэгга.
случае, когда угол G удовлетворяет закону Брэгга (и— целое число). Волны с одной фазой возникают при «отражении» под углом G.
Эта концепция применима к дифракции в кристалле, поскольку кристаллическая решетка может быть описана с помощью набора параллельных плоскостей с различными расстояниями d между ними. Если пучок рентгеновских лучей падает на любой набор плоскостей под углом, для которого выполняется соотношение Брэгга, то из кристалла будет исходить единственный вторичный пучок. И на самом деле, когда на монокристалл вещества действует пучок интенсивного рентгеновского излучения, из него в различных направлениях испускаются многие тысячи более слабых пучков или отражений, как это показано на рис. 17.9. Угол между каждым отраженным пучком и падающим пучком излучения определяется расстоянием между рассеивающими плоскостями.
Как описать этот набор плоскостей, используя параметры элементарной ячейки? Можно рассматривать их как определяемые точками равной электронной плотности в ячейке; в этом случае решетка задается симметрией распределения электронной плотности, аналогичным образом плоскости могут задаваться решеткой. Рассмотрим двумерные решетки и наборы плоскостей, показанных на рис. 17.10. Все возможные наборы плоскостей могут быть заданы с помощью так называемых ин-
376
Глава 17
Падающий пучок рентгеновских кучей
=-]
Поглотитель пучка
Рис. 17.9. Иллюстрация явления рассеяния кристаллом.
дексов Миллера, которые определяют их однозначно в зависимости от числа частей, на которые эти плоскости делят ребра элементарной ячейки. На рис. 17.10, А плоскости делят а на две части и с—на одну. В нашей двумерной схеме этот набор плоскостей обозначается как (2, 1). На рис. 17.10,Б показаны плоскости (3, 1), а на рис. 17.10,В — плоскости (1, 2). Трехмерная элементарная ячейка на рис. 17.11 демонстрирует сегменты первых плоскостей (h, k. l) = (2, 1, 2) параллельного бесконечного набора, где h, к и / соответствуют осям а, b и с. Этот набор плоскостей приведет к одному дифрагированному пучку, если падающий пучок составляет с этими плоскостями такой угол 0, что sin0 = nX/2 (1/^212)- В то же время отметим, что, хотя рентгеновские лучи рассеиваются электронами, которые распределены в ячейке, а не только в определяемых нами плоскостях, мы в дальнейшем увидим, что рассеяние от содержимого ячейки всегда будет выражаться через рассеяние от наборов плоскостей, задаваемых формой ячейки. Распределение электронов в ячейке определяет только относительные интенсивности отраженных пучков.
Тот факт, что в эксперименте по рассеянию рентгеновских лучей наблюдается лишь один пучок, характеризующий целый ряд параллельных плоскостей, в сочетании с неудобным обратным соотношением между 0 и dhkl, вызывает желание описать решетку таким образом, чтобы 0 был прямо связан с расстоянием и каждый ряд плоскостей в реальной решетке представлялся бы точкой в новой обратной решетке (о.р.).
Рис. 17.10. Наборы плоскостей, проведенных через узлы решетки вдоль оси b кристалла.
Рентгеновская кристаллография
377
Обращаясь к закону Брэгга, мы видим, что sinG, характеризующий отклонение между падающим и отраженным пучками, обратно пропорционален расстоянию d между плоскостями в кристаллической решетке. Структуры с большим d будут иметь сжатую дифракционную картину, а структуры, в которых d мало — растянутую. Если бы обратное соотношение между sin G и d можно было заменить на прямое, то интерпретация дифракционной картины упростилась бы. Это достигается конструированием обратной решетки.
Точка, выбранная в качестве начала координат в о. р., совпадает с началом координат в реальной решетке. Точку в о.р., соответствующую набору плоскостей в реальной решетке, можно найти, исходя из начала координат и вычерчивая нормали к ближайшим Миллеровским
Рис. 17.11. Участки плоскостей (2, 1, 2) в произвольной элементарной ячейке.
плоскостям hkl, не проходящим через начало координат. Нормаль оканчивается на расстоянии l/d^ от начала координат. Проводя эту операцию со всеми наборами плоскостей в реальной решетке, получаем полную обратную решетку. (Отметим, что нахождение точек в о. р. определяется исключительно размером и формой реальной элементарной ячейки, но не ее молекулярным содержимым.) Этот процесс схематически изображен на рис. 17.12 для нескольких плоскостей реальной решетки.
Наша следующая задача—распространение анализа рис. 17.12 на случай трех измерений. Если это сделать, то получится ряд плоскостей (они показаны точками на рис. 17.13), соответствующих о.р.
Реальная решетка имеет индексы Миллера (0, к, Г), (1, к, Г), (2, к, Г) и т. д. Вместо того чтобы брать в качестве начала координат точку на ребре реальной решетки, как мы это делали на рис. 17.12, поместим ее в центр. Выберем точку А в реальной решетке на оси а. Существует целый набор возможных плоскостей, параллельных линии ОА с h, равным нулю [т. е. набор плоскостей (0, к, /)]. Перпендикуляры, опущенные из О на плоскости этого набора, напоминают спицы колеса, центром которого является точка О. Все обратные решетки, построенные на основе нормалей к этим плоскостям, будут лежать в плоскости 0, к, I обратной решетки, показанной на рис. 17.13. Все плоскости
378
Глава 17
реальной решетки, пересекающие ось а в точке А, будут давать точки (по одной на реальную плоскость) в плоскости 1, I, к обратной решетки (этот факт объясняется в работе [3]). Аналогичные плоскости возникают для других значений h.
Рис. 17.12. Связь между некоторыми точками реальной и обратной решеток.
А и В—точки реальной решетки, которые вместе с началом координат О определяют элементарную ячейку. Они лежат в плоскости при с = 0. А — точки реальной решетки со стрелками от начала координат до Миллеровских плоскостей; Б—обратная решетка, которая представляет собой график обратных величин расстояний, указанных стрелками на рис. А. Плоскость 200 не проходит через точки решетки и соответствует дифракции Брэгга второго порядка, т.е. 2/dioo определяется как иррациональная плоскость 200, a n/dl0o— как пропорциональная плоскость иОО.
Рентгеновская кристаллография
379
Рис. 17.13. Схематическая конструкция обратной решетки в трех измерениях.
Ребра обратной решетки, показанные на рис. 17.14, обозначаются буквами со звездочкой: а*, Ь* и с*. Звездочкой помечены и соответствующие углы. Для ромбической ячейки направления а и а*, b и Ь* и с и с* совпадают, но длины их различаются. Для моноклинной ячейки соотношения между а и а* и с и с * зависят от р, в то время как соотношение между b и b * с углом никак не связано:
а* = —г- Ь* = с* = —Ц-, (17.3)
а sin Р b с sin р
а = у = а* = у* = 90°, Р* = 180° - р.
Рис. 17.14. Соотношение между реальной и обратной элементарными ячейками моноклинной системы.
380
Глава 17
Объем реальной ячейки выражается следующим образом:
V = = abcsin Р, (17.4)
где И* — объем обратной ячейки. Отметим, что каждая ось о. р. перпендикулярна грани исходной ячейки; например, а * перпендикулярна плоскости Ьс, а а — нормаль к плоскости Ь*с*. Таким образом, каждая ось реальной решетки перпендикулярна набору плоскостей обратной решетки, например, а — нормаль к плоскостям Okl, ikl, 2kl и т.д. В векторном виде можно, например, записать а* = (Ь х с)/И (Отметим, что а = (Ь* х х с*)/И)
Как можно описать условия для дифракции, исходя из параметров обратной решетки, показано на рис. 17.15, где изображена сетка hOl моноклинной обратной решетки вместе с падающим пучком рентгеновского излучения, проходящим через начало координат (помеченное точкой О). Термин сетка часто используют для описания расположения точек о. р. в плоскости. Центр окружности радиуса 1 /А. лежит в точке D. Если кристалл поворачивать вокруг точки О таким образом, чтобы точка Р все время находилась на построенной окружности, то из геометриче-
Рис. 17.15. Двумерное представление сферы отражения и ее связь с законом Брэгга.
Рентгеновская кристаллография
381
ских соображений следует, что
• Л ОР
s,n9'W
(17-5)
Из построения обратной решетки видно, что величина отрезка ОР равна l/d№l, поэтому
X
sin 0 = —-— или 2dhlcl sin 0 = X 2“hkl
(17.6)
и закон Брэгга выполняется. Набор реальных плоскостей, представленных точкой Р, перпендикулярен ОР, и легко показать, что дифрагированный пучок имеет направление, указанное сегментом DP.
Распространяя это обсуждение на случай трех измерений, можно сказать, что любая точка обратной решетки, лежащая на сфере отражения (определяемой длиной волны, направлением падающего пучка и началом координат элементарной ячейки), в принципе приводит к дифрагированному пучку, выходящему из кристалла в направлении, определяемом центром сферы и точкой пересечения о. р. со сферой. Отсюда немедленно следует, что по мере уменьшения X (т. е. по мере увеличения энергии рентгеновских лучей) размер сферы растет и при пересечении сферы обратной решеткой наблюдается больше отражений. Отметим, что о. р. вращается вместе с кристаллом вокруг начала координат, которое находится на поверхности сферы отражения, а не в центре ее. Таким образом, для данной кристаллической системы можно получить больше информации. В действительности оказывается, что число возможных отражений N выражается как
„ (4/3).(2ДР V*
(17-7)
17.4. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ—ПЕРВЫЙ ЭТАП
а. Фотография монокристалла. Существует несколько методов, которые могут быть использованы для получения фотографических данных о монокристалле либо по отдельности, либо вместе. Сложность этих методов увеличивается (но при этом они чаще применяются) с конструированием фотокамер, которые сортируют тысячи дифрагированных кристаллом пучков на более определенные совокупности. Для того чтобы продемонстрировать, зачем нужна обратная решетка, схема построения которой дана в последнем разделе *, опишем прецессионный метод. Геометрия прецессионной камеры показана на рис. 17.16. Плоскость сетки обратной решетки касается сферы отражения в точке О, которая является началом координат и, макроскопически, местом нахождения кристалла. Кристалл укрепляется на оси, перпендикулярной
Применение фотографического метода обосновано в работе [3].
382
Глава 17
падающему пучку (обычно приклеивается к стеклянной нити). Поскольку сетка о. р. тангенциальна сфере, прямая ось может совпадать с падающим пучком. Другие плоскости о. р. убудут параллельны показанной плоскости; если ось а совпадает с пучком, то плоскость Okl обратной решетки касается сферы только в точке О, тогда как плоскости Ikl, Ikl, ... пересекают сферу по окружностям. Там, где эти плоскости касаются сферы, возникают пучки рентгеновского излучения, направленные от центра сферы. Нулевая плоскость даст отражения только в том случае, когда кристалл вращается вокруг оси подвеса (рис. 17.16,Б). Камера может быть установлена таким образом, чтобы круговое пересечение нулевой плоскости со сферой отражения двигалось вместе с камерой вокруг падающего пучка, сохраняя постоянным угол между сеткой о. р. и падающим пучком. Следовательно, «окружность отражения» движется вокруг начала координат о. р., подставляя в положение для отражения каждую точку о. р. (ограничиваемую только углом прецессии ц). Если
Рис. 17.16. Геометрические соотношения в прецессионной камере.
А —нулевая плоскость сферы отражения (р = 0°); Б — нулевая плоскость наклонена по отношению к сфере отражения; В — расстояние на точках нулевой плоскости о.р. и рентгеновские лучи, падающие на пленку.
Рентгеновская кристаллография
383
пленка установлена так, что она всегда параллельна сетке о. р. во время движения камеры, пленка регистрирует расположение точек, являющихся неискаженным представлением сетки о. р., которая обусловливает появление этих точек.
Следует упомянуть еще и о том, что конус отраженных пучков возникает за счет каждой из плоскостей Oik, Ilk, Ilk, ..., но поскольку размер конуса в каждом случае различен, конусы могут быть разделены экраном, помещенным между кристаллом и пленкой. Этот экран представляет собой металлическую пластинку с отверстием диаметром 2—5 мм, который рассчитан таким образом, чтобы пропускать только отраженные пучки, вызванные окружностью отражения корректного размера. Размер (радиус) отверстия можно подобрать так, чтобы наблюдались отражения только от необходимой плоскости nkl. На практике и составляет 0, 1 или 2 (редко больше), а центр экрана фиксируется с помощью целлофановой ленты.
Предполагая, что интересующий нас кристалл подвешен на обратной оси, скажем b *, моноклинной решетки, какие операции мы должны осуществить, чтобы определить параметры элементарной ячейки и пространственной группы? Сначала мы поворачиваем кристалл до тех пор, пока ось, скажем а *, не станет перпендикулярной пучку и, таким образом, плоскости hkn не займут отражательного положения для п (в зависимости от выбранного экрана). При использовании обычных методов можно достичь такого совпадения оси с пучком с точностью + 0,05°. Теперь, отметив угол, при котором обнаружена зона hkO, вращаем кристалл до обнаружения сетки Ofc/. Угол между этими двумя направлениями оси, если он меньше 90°, составляет Р * (символом Р пользуются, если угол превышает 90°). Если же плоскости hkO и Ofc/ регистрируются на пленке в виде точек, расположенных в форме прямоугольника, то разделение вдоль осей фотографий дает возможность рассчитать а *, b * и с * (с помощью формулы, характеризующей геометрию камеры). При обоих положениях ось Ъ * перпендикулярна пленке, тогда как вертикальными осями для плоскостей hkO и Ofc/ являются соответственно а * и с *. Эти расчеты дают все данные, необходимые для определения объема и формы элементарной ячейки. На рис. 17.17 изображена сетка hkO соединения [Cu2 (tren)2 (CN)2] (BPh4)2, полученная при прецессионном фотографировании (tren — 2, 2', 2"-триаминотриэтиламин) [6].
Используя программы автоиндексирования, можно определить оси элементарной ячейки и углы, не имея полного набора фотографий, на которых видна каждая сетка нулевой плоскости (Ofc/, /10/, /ifcO). В таких программах в качестве входных данных служит большое число беспорядочных отражений (зарегистрированных либо на пленке, либо с использованием дифрактометра). С их помощью находят все такие реальные оси, для которых индексы всех отражений относительно данной оси — целые числа. Получаются углы между всеми парами возможных осей и их длины в ангстремах. Углы, которые отличаются от 90°, или оси аналогичной длины могут быть сгруппированы по три, и эти группы наилучшим образом описывают элементарную ячейку.
384
Глава 17
Очевидно, что положение отраженных пучков при прецессионном фотографировании характеризует только форму и размер элементарной ячейки. Параметры ячейки определяют расстояние между точками обратной решетки и, таким образом, через геометрическое устройство
Рис. 17.17. Прецессионная фотография, изображающая сетку hkO соединения [Cu2(tren)2(CN2](BPh4)2. Ось а* горизонтальна (ось подвеса кристалла), а ось Ь* вертикальна.
камеры—расстояние между пятнами на фотографии. В следующем разделе будет дан количественный анализ факторов, приводящих к изменению интенсивностей зарегистрированных пятен. Мы уже сталкивались с тем, что интенсивность зависит от точного распределения электронной плотности в элементарной ячейке. Поскольку элементы симметрии, характеризующие элементарную ячейку, требуют, чтобы ее содержимое было организовано определенным образом, картина интенсивности, наблюдаемая при прецессионном фотографировании, характеризует эти элементы симметрии. Это утверждение будет обосновано позднее. Теперь же мы будем говорить, что для каждого элемента симметрии в элементарной ячейке, включающего трансляцию (или центрирование), на фотографии будет возникать соответствующая картина систематических погасаний. Например, для ячейки симметрии Р 2Jc полностью отсутствует каждое нечетное отражение оси О/сО. Это указывает на наличие винтовой оси второго порядка, параллельной Ъ. Аналогично в зоне Okl чередующиеся ряды отражений свидетельствуют об отсутствии перпендикулярности оси 00/, что характеризует плоскость с-скольжения.
Рентгеновская кристаллография
385
Таблица 17.2
Некоторые наиболее распространенные элементы симметрии н результаты погасания
Элемент симметрии Отражение от: Отсутствуют, если:
Винтовая ось второго Вдоль а MX) /i = 2n +1
порядка Вдоль b OkO k=2n +1
Вдоль с 00/ l=2n +1
Скольжение ±а />-Скольжение Ok/ k = 2n + 1
с-Скольжение Ok/ l=2n+l
и-Скольжение Ok/ k+l = 2n+ 1
Скольжение Lb а-Скольжение hOl h = 2n+ 1
с-Скольжение hOl l=2n+l
и-Скольжение hOl h+l = 2n+ 1
Скольжение ±с а-Скольжение hkO h = 2n+ 1
Ь- Скольжение hkO k = 2n+ 1
и-Скольжение hkO h+k = 2n+ 1
С-Центрирование hkl h + k = 2n+ 1
В-Центрирование hkl h + l = 2n+ 1
Л-Центрирование hkl k + l=2n+ 1
В табл. 17.2 приведены некоторые наиболее распространенные элементы симметрии и характеризующие их систематические погасания.
На практике очевидны три момента: 1) если только картины запечатлелись в памяти, то погасания и соответствующие элементы симметрии быстро распознаются на серии фотографий; 2) необходимо искать как сетку Okl, так и сетку 1к/, чтобы определить, перпендикулярна ли плоскость с-скольжения оси а, поскольку потеря чередующихся рядов в Ок/ похожа на большее разделение в о. р.; присутствие всех рядов в Ikl дает точное разделение в о. р. и говорит о погасаниях в 0k/; 3) погасания, вызванные наличием одного типа элементов симметрии, могут скрывать погасания, которые в противном случае должны быть обусловлены другим типом элементов симметрии. Это одна из причин, по которой не удается установить пространственную группу, к которой относится кристалл (т. е. на основании полученных данных можно отнести кристалл к двум или более пространственным группам). Кроме того, важно знать, какие прецессионные фотографии будут демонстрировать какую-либо симметрию в обратной решетке, включая зеркальные плоскости и оси второго порядка. Например, если существует зеркальная плоскость, перпендикулярная оси а, то интенсивность отражений* hkl и hkl одна и та же; таким образом, одна сторона зоны hkO (или /10/) будет зеркальным отражением интенсивностей другой ее стороны. Для того чтобы определить пространственную группу, важно сохранить след этих наблюдаемых зеркальных плоскостей. В прецессионных фотогра
* h означает отрицательную величину.
25-574
386
Глава 17
фиях нулевой плоскости всегда имеется центр инверсии, не зависящий от решетки.
Удобно, что в одной из таблиц «Интернациональных таблиц для рентгеновской кристаллографии» [7] перечислены все возможные пространственные группы для данного набора элементов симметрии, обнаруженных фотографическим путем (стр. 349 2-го издания). Как правило, корректная схема разметки осей о.р. не известна до тех пор, пока не определена пространственная группа. Затем оси метятся в соответствии с их связью с найденными элементами симметрии и в согласии с принятыми условиями.
б. Дифракционные картины порошкообразных образцов. Эти дифракционные картины регистрируются одним из двух методов. Первый и все еще наиболее распространенный метод состоит в том, что небольшое количество образца помещается в тонкий капилляр, находящийся в центре цилиндрической камеры. Отверстие в боковой части цилиндра предназначено для входа пучка рентгеновского излучения. Как только пучок падает на капилляр, последний начинает вращаться вокруг своей оси, и на пленке, которой обложена внутренняя поверхность цилиндра, регистрируется картина рассеянных лучей. На рис. 17.18 изображена конструкция камеры.
В другом методе может быть использован счетчик, чувствительный к рентгеновскому излучению. Этот прибор сконструирован так, как показано на рис. 17.19. Счетчик движется по дуге, регистрируя изменения в интенсивности рассеянных рентгеновских лучей. Этот метод проще и быстрее, а также характеризуется значительно лучшим разрешением, чем можно достичь с помощью пленки; поэтому в дальнейшем мы будем обсуждать использование дифрактометра, предназначенного для исследования порошкообразных образцов.
Прежде всего необходимо выяснить, на что похожа в этом случае дифракционная картина и как она возникает? Представим себе, что порошок— это набор многих небольших кристаллов. Каждый кристалл даст отраженные пучки, если только точки о. р. касаются сферы отражения. В принципе, для данной точки о.р. это может случиться для многих различных ориентаций кристалла при одном главном условии: угол между отраженным и падающим пучками должен быть всегда одинаковым, т. е. любое отражение может возникнуть только при одной величине 0 (см. рис. 17.15). Как показано на рис. 17.20, для этого необходимо, чтобы любое отражение было усреднено по всем ориентациям кристалла в конусе рассеянного рентгеновского излучения, т.е. в принципе может быть зарегистрирован один такой конус для каждого возможного отражения, хотя обычно отражением является наиболее интенсивное 100 или подобное ему. Как только счетчик сдвинется на некоторый небольшой угол, скажем на 3°, от падающего пучка по направлению к 90°, щель пересечет каждый конус и зарегистрирует его как пик в дифракционной картине порошкообразного образца.
На рис. 17.21 изображено несколько дифракционных картин порошкообразных образцов, полученных с помощью дифрактометра, для родственных химических систем [8]. В левой части представлены дифрак-
Рис. 17.18. Конструкция камеры, предназначенной для исследования порошкообразных образцов, находящихся в капилляре, с помощью регистрации на пленке.
Рис. 17.19. Геометрия дифрактометра, предназначенного для исследования порошкообразных образцов, в котором образец нанесен на стеклянную пластинку, а в качестве детектора используется счетчик.
Рис. 17.20. Конус отраженных рентгеновских лучей одного и того же индекса, возникших за счет рассеяния от беспорядочно распределенных кристаллов в порошкообразном образце.
25*
388
Глава 17
ционные картины для [Ni2 (tren)2X] (BPh4)2, где X — С2О4 , (N3)2 или (OCN“)2, a tren — 2,2',2"-триаминотриэтиламин— лиганд-треножник:
Из электронных спектров следует, что в каждом случае Ni2 + октаэдрически координирован. Измерения магнитной восприимчивости указывают, что во всех трех случаях пары ионов никеля магнитно взаимодействуют. Инфракрасный спектр говорит о том, что азид-ионы связаны эквивалентно с каждым концом. ц-Оксалато-системы распространены относительно широко, а ренгтеноструктурные исследования монокристалла указывают на димерную структуру типа
Как видно из дифракционных картин Б и В (рис. 17.21) порошкообразных азидо- и цианато-систем, положения и интенсивности пиков почти в точности совпадают с этими же параметрами на рис. 17.21, А. Сходство в положениях пиков означает, что во всех трех случаях размеры элементарной ячейки одинаковы. В этом есть определенная польза, но важно содержимое ячейки, поэтому следует подробно изучить интенсивности. Имеет место такое хорошее соответствие, что было бы удивительным, если бы две из этих структур сильно отличались одна от другой. Каждый катион почти определенно является димером, и для того чтобы размер димера во всех случаях оставался почти одним и тем же, системы с X = (N3)2 и (OCN“)2 можно изобразить следующим образом:
Рентгеновская кристаллография
389
Рис. 17.21. Экспериментальные дифракционные картины порошкообразных образцов [Ni2(tren)2X] (BPh4)2, где X—С2О4“ (Л), (N2)2 (Б), (OCN“)2 (В), осажденный из водного раствора (OCN“)2 (Г), перекристаллизованный из растворителя (SCN“)2 (Д) и (SeCN“)2 (Е).
Многие ученые в прошлом ошибались, основываясь в своих выводах относительно структуры на ограниченном наборе данных и на соответствии только положений линий. Необходимо тщательно рассчитать вес данных и представить вероятность корректного вывода. Был проведен полный рентгеноструктурный анализ монокристаллических образцов описанных выше азидо- и цианато-систем, и было обнаружено, что они действительно изоструктурны и их углы различаются лишь незначительно.
Еще одной иллюстрацией применимости метода исследования порошкообразных образцов служат результаты сопоставления дифракционных картин В и Г, где В получена при изучении осажденного порошка цианато-комплекса, а Г—при изучении выращенного монокристалла обычным кристаллографическим методом. Отметим, что картины отчетливо различаются. Однако из измерений магнитной восприимчивости следует, что и в том и в другом случае геометрия димерного катиона одна и та же. Упаковка кристаллической решетки в двух указанных случаях различна, хотя с электронной и химической точек зрения это одно и то же вещество. Кристаллизация, которая приводит более чем к одной пространственной группе, встречается не ча
390
Глава 17
сто. Она иногда приводит к различным молекулярным возмущениям, которые огорчают экспериментатора до тех пор, пока дифракционные картины для порошкообразных образцов не помогут выяснить возможную причину наблюдаемых эффектов.
Дифракционные картины Д и Е исключительно похожи во многих отношениях и имеют много общего с картиной Г относительно наиболее интенсивных линий. Следует предположить, что при замене SCN и SeCN на OCN, если даже геометрии систем похожи, большие электронные плотности на атомах S и Se должны приводить к заметному изменению интенсивностей линий при рассеянии рентгеновских лучей. Поэтому можно сказать, что дифракционные картины Д и Е также похожи, как и следовало бы ожидать, если бы все три структуры были похожи. Результаты согласуются с аналогичными структурами. Особенно заметно увеличение интенсивности пиков при больших углах с увеличением массы мостиковой группы; оно станет понятным после обсуждения, проведенного в разд. 17.5.
17.5. ДИФРАКЦИЯ РЕНТГЕНОВСКИХ ЛУЧЕЙ— ИНТЕНСИВНОСТИ
В этом разделе описаны некоторые факторы, влияющие на измеряемую интенсивность отраженных пучков, а также математическая процедура, используемая для расчета интенсивности любого отражения на основе данных о содержимом элементарной ячейки. Мы увидим, что измерение интенсивности само по себе не может дать достаточной информации для прямого расчета положения атомов, и поэтому должен использоваться итерационный метод, в котором сравниваются измеренные и рассчитанные интенсивности и применяемая атомная модель улучшается до тех пор, пока не будет достигнуто адекватное соответствие двух наборов величин.
Для того чтобы получить данные о группировке содержимого в элементарной ячейке, необходимо измерять интенсивности рассеянных пучков рентгеновского излучения. И в методе с пленкой, и в методе со счетчиком кристалл движется во время измерений так, что точки о. р. пересекают сферу отражения с одной стороны до другой. Поскольку точки о.р. растягиваются по сфере отражения, интегральная интенсивность зависит частично от угла между направлением движения и поверхностью сферы при пересечении. Время, необходимое для пересечения точкой о.р. сферы, увеличивается по мере того, как угол приближается к нулю. Необходимо также объяснить различие в «отражаемости» рентгеновских лучей, электрический вектор которых перпендикулярен и параллелен плоскости отражения. Лорентцева и поляризационная поправки соответственно могут быть использованы для исправления наблюдаемой интенсивности (Ihkl) отражения hkl следующим образом:
|Гш1= (17-8)
Рентгеновская кристаллография
391
где лорентцева поправка равна L= 1/sin 20, поляризационная поправка равна р = (1 + cos220)/2, а К — постоянная, зависящая от свойств системы, используемой для получения данных. Результирующая Fhkl называется амплитудой наблюдаемого структурного фактора. Если говорить об измерении, то Fhkl представляет собой абсолютное значение структурного фактора Fobs.
При определении кристаллической структуры значения Fobs сопоставляются со значениями Fcalc, рассчитанными из предполагаемого расположения атомов в элементарной ячейке. Для того чтобы рассчитать
Рис. 17.22. Кривая фактора рассеяния для атома углерода.
sin в/Л
структурные факторы из данной атомной модели, производится суммирование рассеяния по всем атомам в элементарной ячейке. Следует рассмотреть два ключевых момента. Первый состоит в том, что рассеяние на электронном распределении каждого атома j описывается с некоторым приближением фактором сферического рассеяния fj. Этот фактор выражает рассеивающую способность атома и пропорционален числу электронов в атоме и углу, под которым происходит рассеяние (sin 9,7. или 1/2 dhkl — расстояние, обратное расстоянию точки о.р. от начала координат). Типичная кривая фактора рассеяния (в данном случае для атома углерода) в предположении, что атомы имеют сферическую форму, изображена на рис. 17.22. Снижение рассеивающей способности в зависимости от угла обусловлено тем, что комбинированное рассеяние на всем электронном окружении атома, которое дает свой вклад, как если бы оно было связано с положением ядра, имеет тенденцию к уменьшению благодаря увеличению разности фаз между волнами, возникающими в широкой области пространства. Программы для ЭВМ, предназначенные для расчета структурных факторов, обычно требуют в качестве входных данных таблицу, в которой фактор рассеяния представлен в зависимости от значений sin 0//_. Тогда для расчета вклада атома в данный Fhkl вычислительная машина может интерполировать таблицу к значению sin0//., соответствующему индексу hkl.
Второй момент состоит в том, что структурные факторы рассчитываются, как если бы все рассеяние в элементарной ячейке происходило на электронной плотности в начале координат. Для этого рассеивающие способности атомов, дающих вклад в Fcalc. должны быть сложены
392
Глава 17
как векторы, чтобы можно было учесть различие в фазах между волной, возникающей в начале координат, и волноц, возникающей • в месте нахождения атома. Другими словами, у структурного фактора Рш есть и величина, и фаза—его величиной является число электронов, которые при рассеянии в фазе (скажем, все содержимое находится в начале координат) должны показывать ту же дифрагирующую способность, что и истинное атомное содержимое элементарной ячейки; Рш имеет результирующую фазу всех атомов в элементарной ячейке, которые дают свой вклад в этот фактор.
Расчет разности фаз для любого отражения hkl между началом координат и некоторой точкой (х, у, z) в ячейке прост. (Здесь х, у и z выражаются в относительных координатах, т. е. А/а, kJb, А./с.) Отражение hkl обусловлено набором плоскостей, расположенных таким образом, что разность фаз между любой парой соседних плоскостей составляет 2п рад. Это означает, что для отражения hkl ребра элементарной ячейки характеризуются разностью фаз 2ith, 2nk и 2л/ рад, причем каждое ребро делится плоскостями отражения на h, к и I частей. Тогда разность фаз между точками (х, у, z) и (0, 0, 0) для отражения hkl представляет собой сумму координатных фаз, т. е.
= 2 л (hx + ку + Iz). (17-9)
Здесь мы должны суммировать вклады структурных факторов всех атомов ячейки. В то же время важно ввести комплексное представление структурных факторов, поскольку это значительно облегчает расчет фазовых соотношений. Напомним, что сложение двух волн гг и г2, имеющих свою амплитуду и фазу, можно рассматривать как сложение двух векторов на комплексной плоскости. Это проиллюстрировано на рис. 17.23, где справедливы следующие соотношения:
А = а1 + а2 В = Ьг+Ь2
|К| = 1/а2 + В2 и а = tg - 11-V I
\А/
Теперь R как векторную величину (с величиной и фазой) можно выразить любым из следующих способов:
R = А + iB,
R = |Я| (cos ot + i sin ot) или
R = |Я|?в.
Предполагая, что структурный фактор Fhkl следует рассчитывать путем суммирования по всем атомным вкладам, его амплитуду получаем в виде
FM = V&fjay + Sjjbf = /£/,cos8/ + £//Sin8/. (17.10) ill i
Рентгеновская кристаллография
393
Используя уравнение (17.9), это выражение можно переписать как = V^hkl +
где
Аш = cos 2л (hx} + kyj + /z7)
J
и
Вш = Xfj sin 2л (hXj + ky} + Izj). j
Фазовый угол результирующего структурного фактора равен
(17.11)
Лш
Все эти выражения обычно применяются для расчета структурных амплитуд при сопоставлении с Fobs.
Приведенные выше уравнения для Fhkl можно записать в более компактной форме:
(17.12) j
используя математическую эквивалентность е2,х и cos 2х + i sin 2х. Рассматривая это выражение для FhU, можно получить некоторое представление о важнейшей проблеме, с которой приходится сталкиваться при установлении структуры с помощью рентгеновской кристаллографии. Рш имеет величину и фазу; оба параметра определяются суммой по всем атомным положениям в решетке. Экспериментально можно измерить только величину каждого Fhkl, поэтому на первый взгляд проблема кажется неразрешимой, поскольку в задаче определения положения всех атомов не известно столько фаз, сколько имеется наблюдаемых парамет-
Рис. 17.23. Векторная диаграмма, показывающая сумму двух комплексных переменных на комплексной плоскости.
394
Глава 17
ров. В следующем разделе будут описаны два метода, с помощью которых можно обойти фазовую проблему, что делает практически возможным установление структуры.
В настоящий момент, исходя из выражения для Fhkl как функции атомных координат, мы в состоянии предсказать влияние симметрии на наблюдаемые интенсивности. Ниже будет показано, что систематические погасания, перечисленные в разд. 17.4, могут быть выведены из этих соображений.
В уравнении (17.12) проводится суммирование по всем атомам в элементарной ячейке, даже по тем, которые связаны по симметрии. Не исключено, что связанные по симметрии атомы могут быть описаны более определенным аналитическим выражением относительно их полного вклада в Fhkl.
Элементарная ячейка содержит только п единственных в своем роде атомов, что обычно меньше общего числа атомов. Эти п атомов определяют асимметрическую ячейку; для общего числа атомов п х т в элементарной ячейке имеется т асимметрических ячеек. В случае примитивной центрированной ячейки и в зависимости от симметрии величина т составляет 2, 4 или 8 для триклинной, моноклинной и ромбической решеток. Теперь Fhkl можно переписать в виде
= ^f„(.Xe2ni(hXm-"+kym'" + lz"'-''\ (17.13)
п т
где сумма в скобках часто обозначается как Тт. Значение этого выражения состоит в том, что вклад атомов в Fhkl может быть рассчитан для т атомов за один раз. На самом деле часто оказывается, что в том случае, когда т атомов связаны по симметрии, математические выражения упрощаются.
Особенно прост и важен случай, когда т = 2 и две асимметрические ячейки связаны центром инверсии, т. е. х, у, z = х, у, z. Вклад в структурный фактор каждой пары атомов равен
/т- \ _ v 27u(/ixm>n+ kym „+ lzm„_
11 hkl/n — L,e ~
m
= cos 2л (hx + ky + lz) + i sin 2л (hx + ky + lz) + cos 2л ( — hx — ky — lz) +
+ i sin 2л (— hx — ky — lz) = 2cos 2л (hx + ky + lz).
Таким образом, для центрированной ячейки, когда в элементарной ячейке имеется центр инверсии,
Fhki = 2£./„cos 2л (hx* + кУп + к") • п
Видно, что в этом случае структурный фактор действителен (т. е. фазовый угол равен либо нулю, либо л); это часто оказывает большую помощь для успешного определения структуры.
Рентгеновская кристаллография
395
Рассмотрим более сложный пример, когда в элементарной ячейке имеется винтовая ось второго порядка, такая, как в пространственной группе P2JC. Здесь координаты положения (х, у, z) преобразуются в координаты (х, 1/2 + у, 1/2 — z) за счет операции, соответствующей этому элементу симметрии. Можно записать (в этом случае используются только косинусы, поскольку известно, что Р2г/с— центрированная пространственная группа, т. е. xyz->xyz):
k I
Тт = 2 [cos 2л (hx + ку + lz) + cos 2л ( — hx + ку + — + — — lz)].
При использовании тригонометрического тождества для косинуса суммы углов это выражение можно переписать в виде
Thkl= 2 cos2n(hx + lz) cos 2яку — sin2n(/ix + /z) sin 2 л/су +
/ k+l
+ cos 2л (— hx — lz) cos 2л ( ку 4-—
I k+l
— sin 2л (— hx — lz) sin 2л1 ку 4--—
или, группируя члены,
/ i
Thkl = 2 < cos 2л (hx + lz) cos 2nky + cos 2л ( ку 4-
Г • ( k+/\T)
— sin2n(hx + lz) sin 2nky — sin 2л I ку 4--—I >
Формулы для тригонометрических функций суммы углов могут быть вновь использованы для раскрытия cos 2л [/су + (/с + 1)/2] и sin 2л [/су + + (/с +1)/2], и так как
/ р ч- / \
cos 2л ( —-— j = ( — l)k + '
и sin 2л --------------------------------- = О,
\ 2 7
то
к + / cos 2л (/су ч--—) = ( — l)k +' cos 2л/су
и
sin2л f ку + j = (— l)k+ 'sin 2 л/су.
396
Глава 17
Теперь можно записать конечное выражение
Тш = 2 {cos 2л (hx + Iz) [cos Inky + (— I)*1 + 1 cos 2n/cy] —
— sin 2л (hx + Iz) [sin 2л/су — (— l)k + ' sin 2л/су]}.
Если к + l четно, то
= 4cos 2л (hx + Iz) cos 2л/су,
а если к + I нечетно, то
Thkl = — 4sin 2 л (hx + Iz) sin 2л/су.
Для оси 0/с0 обратной решетки, если к нечетно, к + I нечетно и Тш = 0; аналогично для hQl, если I нечетно, к + I нечетно и Тш = 0. Таким образом, мы показали, что систематически погасания как для винтовой оси, так и для плоскости скольжения возникают в том случае, когда для расчета структурного фактора используются условия существования оси 2, в центрированной структуре. Интересно также отметить, что Т hk, = T-hki. Это означает, что дифракционные картины имеют центр симметрии.
17.6. ПРАКТИЧЕСКИЕ ПРИМЕНЕНИЯ—ВТОРОЙ ЭТАП
а. Получение и обработка данных по интенсивности. К настоящему моменту многие детали экспериментальных операций, относящихся к определению структуры с помощью рентгеновской кристаллографии, в общих чертах описаны. Теперь нас интересует измерение интенсивностей. В методах, в которых используется пленка, интенсивности измеряют лишь изредка, поэтому мы сконцентрируем наше внимание на использовании дифрактометра, управляемого ЭВМ*.
Имея подходящий кристалл и изучив свойства его элементарной ячейки с помощью пленочных методов, можно затем измерить интенсивности, укрепив кристалл в дифрактометре таким образом, чтобы была известна ориентация обратной решетки относительно геометрии дифрактометра. В этом случае ЭВМ будет знать, где находится каждое отражение hkl. На схеме типичного гониометра дифрактометра (рис. 17.24) показан диапазон движений при координировании дифрактометра и кристалла. На самом деле не слишком трудно установить кристалл вручную. Установку можно также осуществить, направляя беспорядочные отражения на дифрактометр и используя автоиндексирующую программу (описанную ранее). Этот метод находит все большее применение. Как только в счетчике зарегистрированы три отражения за счет подгонки углов х, ср, со и 20 при условии, что они корректно индексированы (— h # h и т. д.), ЭВМ может рассчитать матрицу ориентаций (и постоянные ячейки); из этих данных ЭВМ может определить углы дифрактометра, соответствующие любым значениям h, к и I. Естествен
* Такой прибор называют автоматическим рентгеновским дифрактометром.— Прим, перев.
Рентгеновская кристаллография
397
но, точность этой матрицы зависит от точности измерения отражений. Все необходимые данные можно получить, если матрица адекватна для нахождения (с точностью в пределах 0,02° для каждой дуги) любого желаемого отражения в обратной решетке. Может возникнуть обратная задача измерения положения, скажем 15 или 20 отражений. Тогда ЭВМ
Рис. 17.24. Углы гониометра дифрактометра.
использует программу для определения наилучшей матрицы путем получения соответствия со всеми отражениями методом наименьших квадратов. В конечном счете можно воспользоваться этим методом, поскольку он наиболее удобен для определения параметров элементарной ячейки и их погрешностей.
В этом случае электронно-вычислительной машине необходимо знать интервал значений h, к и /, в котором она должна измерять интенсивности; например, для моноклинной системы может потребоваться лишь единственный набор данных (h = — 20, 20, к = 0, 20, I = 0, 20). В то же время для получения лучших результатов два (или все) набора данных могут быть подвергнуты совместному усреднению. Выбор метода зависит от времени и значимости структуры. Число (единственных в своем роде) данных, необходимых для системы, зависит от числа определяемых параметров. Если таких параметров 200, то необходимо получить по крайней мере 1200 отражений, а еще лучше 2000. К счастью, элементарные ячейки кристаллов больших молекул имеют значительные размеры; при меньших V* число наблюдаемых параметров возрастает. Обычно дифрактометр (с излучением Мо) используется для получения всех данных от 20 = х до 20 = у, где г » 3° и не столь мал, как при приближении к падающему пучку, а у равен ~ 45° или углу, при котором интенсивности отличаются от уровня шумов.
Если ЭВМ направляет гониометр на поиск пика для измерения его интенсивности, то углы регулируются в пределах почти одного градуса по 20 пика, и затем гониометр медленно сканирует по пику до тех пор, пока счетчик не просуммирует полную интегральную интенсивность и ЭВМ не зарегистрирует время, необходимое для сканирования. Затем
398
Глава 17
регистрируется фоновое излучение по обе стороны пика обычно в течение 10 с. Автоматический дифрактометр печатает на бумажной ленте, карточках, магнитной ленте или каком-нибудь другом регистрационном материале такую информацию, как х, со, 20, ср, фон, время, необходимое для измерения фона, интенсивность пика и время, необходимое для измерения каждого отражения.
Как только вся эта информация получена, программа приведения данных трансформирует данные по интенсивности в величины Fobs, делая поправки на лорентцевый и поляризационный члены, фон и т. д. Кроме того, рассчитывается статистический вес каждого отражения (т. е. величина, по которой можно судить об ожидаемой точности измерения отражения). Как правило, с наименьшей точностью определяются наиболее интенсивное (из-за экспериментальных проблем) и наименее интенсивное (из-за небольшого числа импульсов за время счета) огра: жения, и они определяют величины cshkl. Позднее будет показано, что при уточнении атомных параметров в модели элементарной ячейки, приводящем к максимальному соответствию между Fobs и Fcaic, при определении ошибки становится важным взвешивание индивидуальных величин F. Желательно располагать зависимостью погрешности Fcalc и Fobs, которая является функцией и |Fobs|, и sin9/z..
Однако вначале мы займемся только методом, дающим величины Fobs и «пустые» элементарные ячейки. Используя ряд Фурье, электронную плотность в любой точке ячейки можно выразить через наблюдаемые структурные факторы следующим образом (см. работу [3], стр. 222):
р (x,y,z) = -^-XXXFhkie~2ni<kx+ky+lz>.
V h k I
Если и наблюдаемые амплитуды, и наблюдаемые фазы подставить в это выражение, то получится трехмерное распределение электронной плотности в элементарной ячейке, которое позволяет довольно просто определять местонахождение интересующих нас атомов и молекул, поскольку каждый пик электронной плотности соответствует положению атома. Как уже неоднократно говорилось, именно неопределенность фаз приводит к трудностям, которые в течение многих лет мешают эффективно использовать рентгеновскую кристаллографию.
Кристаллография систем, содержащих атом тяжелого металла, часто упрощается за счет использования при определении структуры метода тяжелого атома.
б. Метод тяжелого атома. Этот метод, открытый Паттерсоном [9], основан на том факте, что, хотя и нельзя рассчитать р(х, у, z), можно рассчитать аналогичный ряд, в котором используются амплитуды, но не фазы Елн. Этот ряд, называемый функцией Паттерсона, записывается как
Р (х, у, z) = -^XLL |F^,|2 cos 2л (hx + ky + lz), V h k I
Рентгеновская кристаллография
399
где х, у и z—координаты положения на трехмерной карте Паттерсона. Паттерсон показал, что эта функция имеет максимальное значение в конце каждого межатомного вектора в решетке, хвост которого направлен в начало координат. Высота паттерсоновского пика пропорциональна nZ{Zj, когда пик соответствует вектору между i-м и у-м атомами (с атомным номером Z), а п пиков совпадают из-за требований по симметрии решетки. Если один из атомов в асимметрической ячейке имеет особенно большую массу (скажем, атом меди в ячейке, в которой, помимо этого атома, содержатся только атомы С, N и Н), то вектор Си — Си будет несомненно соответствовать наибольшему пику на карте Паттерсона. Исходя из положения этого пика на карте, можно определить координаты х, у и z атома меди в элементарной ячейке. После этого удается рассчитать карту электронной плотности, используя наблюдаемые величины Fhkl, которые определяются фазами, установленными за счет присутствия в известном месте наиболее сильного рассеивающего центра решетки. Поскольку только некоторые из фаз корректны (в центрированной ячейке) или близки к корректным (в нецентрированной ячейке), карта плотности в целом не будет точной, но, вероятно, когда Z2 (тяжелый атом) ~
^Z2 (легкие атомы)
некоторые участки карты будут четко обрисовывать элементы структуры. Если эти элементы структуры входят вместе с атомом металла в фазовую модель, то можно уточнить фазы. Путем неоднократного повторения этой процедуры можно в конце концов установить положение всех атомов.
Рассмотрим теперь более подробно, как из карты Паттерсона можно установить местонахождение атома. В пространственной группе Pljc стандартными координатами положения являются (х, у, z), (х, у, z), (х, 1/2 + у, 1/2 —z) и (х, 1/2 —у, 1/2 + z). Векторы между этими положениями могут быть рассчитаны путем вычитания, как показано в табл. 17.3. Из 16 векторов только девять будут независимыми:
Векторы Паттерсона Вырождение
О, 0, 0 4
2х, 2у, 2z 1
2х, 2у, 2z 1
2х, 2у, 2z 1
2х, 2у, 2z 1
2х, 1/2, 1/2+ 2z 2
О, 1/2+ 2у, 1/2 2
О, 1/2-2у, 1/2 2
2х, 1/2, 1/2 -Iz 2
Линия в пространстве Паттерсона, обозначенная 0, г, 1/2 (где v — относительная координата), называется линией Харкера и, как можно видеть, возникает за счет требований по симметрии, налагаемых на пло-
400
Глава 17
Таблица 17.3
Стандартные межатомные векторы
X, У, Z X, у, Z х, - + у, — — Z 2 У 2 Х’ЬУ’Ьг
X, У, Z 0, 0, 0 2х, 2у, 2z 2х, i --2z 1 2 0, ~ — 2у, - 2 2
У, Z 2х, 2у, 2z 0, 0, 0 o.l + 2,.l 2х, i + 2z 2 2
- 1 X, 2 + У’ 1 Z 2 2х, -, - + 22 2 2 ,1,1 0, 2у, - 2 2 0, 0, 0 2х, 2у, 2z
Х,|-У, 1+‘ o.l+2,.i ,,-11, 2х, — 2z 2 2 2х, 2у, 2z о, 0, 0
скость скольжения. На эту линию будет ложиться вектор с весом 2 для каждого связанного по симметрии набора атомов. Наибольший из векторов с фактором Z2 (металл)//2 (азот) (если азот является вторым по массе элементом решетки) связан с атомом металла, обычно его обнаружить легко*. Значение v составляет 1/2 ± у (в зависимости от того, какой конец вектора виден), и, таким образом, можно определить у. В плоскости и, 1/2, w должны быть еще два пика приблизительно той же интенсивности, что и интенсивность первых двух. Положение этих пиков позволяет рассчитать х и z атома металла. И наконец, эти значения могут быть проверены путем помещения вектора 2х, 2у, 2z в то место, где он предположительно находится.
Атом металла—главный атом в модели элементарной ячейки, теперь же мы займемся доведением последней. Для этого требуются всего лишь две программы для ЭВМ: программа Фурье, которую можно использовать для расчета функции Паттерсона, карт электронной плотности Fobs или карт плотности |Fobs| — |Fcalc|, и программа доведения по методу наименьших квадратов, которая, если модель завершена, но не точна, варьирует все неизвестные параметры таким путем, чтобы получить наилучшее соответствие между величинами Fobs и Fcalc (найденной из этой модели). Вторая программа также отвечает за расчет структурных факторов, используемых в программе Фурье.
в. Фурье-синтезы. Фурье-синтез при создании модели может сводиться к стандартной обработке данных, а может быть тонким искусством в полном смысле этого слова. Напомним процедуру, завершающую построение модели. Из положения тяжелого атома рассчитывается ряд структурных факторов. Последние вместе с рассчитанными фазами закладываются в программу Фурье; если же выбран другой способ действий, то фазы величин Fcalc используются в Fobs. Используя приведен
* Величину вектора металл — металл обычно рассчитывают, исходя из высоты исходного пика, который включает «собственные векторы» всех атомов.
Рентгеновская кристаллография 401
ное выше уравнение, рассчитывают фурье-карту Fo электронной плотности. Если фазы атома металла корректны только для тех отражений, чьи интенсивности определяются исключительно рассеянием на атоме металла, рассчитанная фурье-карта будет показывать атом металла и ничего более, и расчет оказывается неудачным. Это необычный результат, но следует хорошо представлять себе, что при определении карты электронной плотности в описанной процедуре основной вклад дают наибольшие Fo; для корректности фаз важнее всего те Fo, в которые дают большие вклады легкие атомы структуры. Если для расчета карты используются лишь те отражения, для которых Fealc (металл)
0,3Fobs, то, вероятно, на карте будут видны плотности более легких атомов.
Разностная фурье-функция рассчитывается из выражения
Ар = - |ес|)Л2-^+^+,2>,
Г h k I
где <хс—фаза Fc. Самые важные свойства разностной карты состоят в том, что отражения, для которых Fo х Fc, дают небольшие вклады; это, таким образом, не приводит к простому воспроизведению модели. Члены, для которых |FC| » |F0|, дают большие вклады в карту AF, хотя их вклад в карту Fo пренебрежимо мал. Это важно, поскольку наличие подобных членов приводит к большим ошибкам в модели. По этой причине полезно включить в AF-синтез данные по всем слабым и ненаблюдаемым точкам (ненаблюдаемая точка — это отражение, чья интенсивность в 1—2 раза ниже, чем статистическая ошибка измерения этого отражения).
Если структура завершена, то карта AF в любой области элементарной ячейки не имеет пиков или провалов. Если даже положения всех атомов определены, часто обнаруживают, что вокруг атомов, чьи электронные плотности нельзя хорошо согласовать с моделью стационарного атома, возникают странной формы области положительной и отрицательной плотностей. Теперь мы подошли к моменту, требующему введения концепции температурного фактора. Этот фактор отвечает за колебания молекул, вследствие чего атомы следует рассматривать, исходя из их усредненных по времени положений. Атомы можно рассматривать как колеблющиеся либо изотропно (в сферически симметричной форме), либо анизотропно (в форме эллипсоида). Различие состоит в том, что в первом случае для описания движения необходим только один параметр, а во втором случае—шесть. Смысл математического подхода заключается в простой корректировке фактора рассеяния на тепловое движение исходя из того, что «размазывание» электронной плотности вызывает более быстрое чем обычно уменьшение f в зависимости от sin0/Z. Для изотропного и анизотропного случаев соответственно можно записать
__ / — В(з1п20)Д2
J = J0e и
f — i-Qe-(^)B(Blih2a*2 + B22k2b*2 + &3/2c*2 + 2Bl2hka*b* + 2Bl3hla*c* + 2Вг3Л/6*с*)
29-574
402
Глава 17
Получить хорошие значения В из карт AF обычно трудно, особенно для анизотропного движения, поэтому необходимо обратиться к более мощному методу.
г. Доведение по методу наименьших квадратов. Допустим, что путем последовательных изменений модели, расчетов структурных факторов и фурье-расчетов мы обнаружили все атомы в ячейке с точностью ±0,1 А во всех направлениях. Теперь мы должны изучить метод дальнейшего улучшения модели. Прежде всего возникает вопрос, как много параметров следует определить? В случае анизотропной модели для каждого атома имеется девять параметров плюс еще один, масштабный коэффициент. Для сопоставления Fca]c и Fohs необходимо, чтобы средний Fc был бы тем же, что и средний Fo. Для этого Fo просто умножается на некоторый коэффициент, величина которого обычно близка к 1,0. Обычные ограничения, налагаемые на применение метода наименьших квадратов, состоят в том, что число переменных параметров не должно превышать некоторого определенного значения, скажем 270 (предел ЭВМ), число отражений должно по крайней мере в 5 раз (желательно в 10 раз) превышать число параметров и исходная пробная структура должна быть относительно точной (поскольку доведение нелинейно).
Каждый раз, когда запускается программа обработки данных по методу наименьших квадратов, она рассчитывает из данной модели структурные факторы (плюс таблица факторов рассеяния, информация о симметрии и т.д.) и, используя матричный метод, который имеет слишком много тонкостей, чтобы его здесь обсуждать, рассчитывает изменение каждого параметра, которое приводит к снижению величины функции
hkl
где W№1—весовые факторы, рассчитываемые как 1/о^;.
Качество модели в любой точке определяется коэффициентом R, который можно записать в виде
р _ E||F0| - |FJ|
2|F0|
или
EPFIIFol - IFJ!
ix. W — ------------
EPF|F0|
co взвешенными отклонениями. Как правило, при завершении доводки R и Rw должны иметь почти одно и то же значение—вполне приемлемо 0,05 или ниже.
После того как программа наименьших квадратов отработала несколько раз, используя в качестве входных данных результаты предыдущего расчета, улучшение должно достигнуть того момента, когда R не меняется, а параметры почти йе отклоняются от своих значений от цикла к циклу. Теперь доводку можно считать законченной.
Выходные данные расчетов по методу наименьших квадратов включают относительные координаты каждого атома и его тепловые пара
Рентгеновская кристаллография 403
метры, в том числе оценку стандартного отклонения для каждого параметра. Эти отклонения обычно недооценены, но являются хорошим относительным показателем погрешности. Для того чтобы расположить группы атомов в плоскости и иметь возможность представить пространственную картину в отдельных областях элементарной ячейки, можно теперь рассчитать длины связей и валентные углы. Оценка погрешностей может быть проведена с помощью относительных координат и отнесена к молекулярным величинам.
Иногда удается определить положения атомов водорода, когда структура в других отношениях хорошо доведена. Если известно, где они могут находиться, то их можно обнаружить на карте AF; если же это не так, то часто можно добавлять атомы водорода к структуре путем геометрического расчета их положений, основываясь на sp2- или хр3-гибридизации (естественно, метильная группа таким путем не локализуется из-за ее низкого барьера вращения). Введение атомов водорода в структуру может оказать определенную пользу, поскольку, например, расстояние металл — азот, равное 2,20 А, в координационном комплексе первичного амина может измениться до 2,10 А из-за заполнения электронной плотности более удаленного электрона атомами водорода.
д. Эффективные прямые методы. Это другой метод определения фаз, который ранее не был описан; его применимость для определения неорганических структур до недавнего времени была более ограниченной, чем применимость метода тяжелого атома. При совместных усилиях английских и бельгийских ученых [10] прямые методы стали столь эффективными, что все другие Методы могут вскоре показаться устаревшими. Программа их прямых методов, носящая название MULTAN 74, позволила во многих случаях определить структуры из Fobs и положения всех атомов без помощи исследователя. Выходные данные могут включать даже проекции молекулы.
Прямой метод описан в гл. 13 работы [3], и с ним необходимо познакомиться. Здесь же мы только укажем, что программа расчета прямых методов включает математическое соотношение, которое позволяет производить отнесение к сильным отражениям, основываясь на приближенных соотношениях между фазами групп отражений. Можно также оценить точность отнесения. Фазы можно приписать некоторым отражениям, а другие отражения получат фазы исходя из первоначального их набора. Если эту процедуру осуществить до того уровня, при котором фазы получают восемь или десять отражений одного независимого атома, то можно получить карту электронной плотности, показывающую содержимое ячейки. Как правило, процесс фазирования может требовать отнесения к некоторым точкам гипотетических значений, так что иногда находят до восьми возможных фазовых схем. Программа MULTAN 74 способна выбрать среди них наиболее вероятную. Она также включает алгоритм обработки данных, который учитывает предположительное число, тип и даже группировку атомов в элементарной ячейке (не их положения или ориентации, которые, естественно, неизвестны). Кроме того, MULTAN 74 облегчает поиск Е-карты для атомов в положении связывания, что приводит к согласованию предпола
26*
404
Глава 17
гаемых фрагментов или молекул. Если карта разумно согласуется с ожидаемым содержимым ячейки, то координаты атомов печатаются наряду с длинами связей, валентными углами и наиболее информативными проекциями молекулы.
После этого могут быть либо рассчитаны дополнительные фурье-карты для отделения нечетких областей, либо непосредственно проведены расчеты по методу наименьших квадратов.
17.7. ОСЛОЖНЕНИЯ И ПОПРАВКИ
Еще не были рассмотрены некоторые проблемы, с которыми приходится сталкиваться в ходе доводки структуры. К ним относятся двойникование, поглощение, разупорядочивание и либрационное движение.
а. Двойникование. В этом случае, если кристалл не подходит для получения данных, его просто не используют. По существу, эта проблема возникает за счет наложения двух кристаллических областей кристалла с небольшим нарушением порядка расположения между ними, приводящим к удвоению всех отражений (при фотографическом наблюдении). Некоторые кристаллы одного и того же вещества могут быть свободны от этого дефекта, однако их поиск длителен и часто безуспешен. Отдельные программы автоматических дифрактометров приспособлены для получения данных даже при исследовании кристаллов, имеющих такой дефект, но подобные исследования сопряжены с большими трудностями. Иногда нарушения в порядке расположения этих двух кристаллических двойников значительны. В некоторых случаях было обнаружено двойникование на 90°, и фотография имела нормальный вид, если не считать того, что систематические погасания не согласовались ни с какой пространственной группой. В таких случаях можно получить результаты и подогнать их к модели, в которой величины Fcalc просуммированы по двум- повернутым одна относительной другой ячейкам, каждая из которых характеризуется одним и тем же расположением молекул.
б. Поглощение. Поглощение становится проблемой, если кристалл имеет форму, значительно отклоняющуюся от сферической, или если рентгеновские лучи сильно поглощаются. Поскольку длина пути рентгеновских лучей через кристалл различна для различных значений hkl, величины Fobs, для которых пучок прошел большой путь в кристалле, будут искусственно занижаться. Если известны как расстояние каждой грани кристалла от его центра, так и углы дифрактометра, необходимые для приведения каждой грани в отражательное положение, то можно воспользоваться программой ЭВМ, чтобы ввести поправки в Fobs для данного изучаемого кристалла. Эти поправки необходимо вводить только в том случае, когда размеры кристалла значительно различаются, скажем в 6 или 7 раз, или когда линейный коэффициент поглощения цг превышает 30 для большинства нормальных кристаллов. Этот
Рентгеновская кристаллография
405
коэффициент можно рассчитать, зная содержимое ячейки:
р — плотность кристалла, суммирование проводится по всем атомам, р„—процентное содержание атома п и (ц/р); можно найти для атома каждого типа в «Интернациональных таблицах для рентгеновской кристаллографии». Интенсивность выходящих рентгеновских лучей I может быть выражена через интенсивность падающего излучения и толщину кристалла в месте прохождения пучка т:
/ = /ое"игт
Для различных отражений изменения 1/10 могут достигать 50% из-за эффектов поглощения. При изменении на 15% низшей наблюдаемой величиной R может быть 10%, и структура не будет точной.
Отметим, что даже для сферических кристаллов могут потребоваться поправки на поглощение, если вещество характеризуется сильным поглощением, хотя в этом случае такую поправку ввести легко. Предлагаем читателю показать, что длина пути все еще зависит от 0 даже в сферических системах.
в. Разупорядочивание и либрационное движение. Разупорядочивание, если оно обнаружено, представляет собой очень сложную проблему. Трудность возникает в том случае, когда определенная молекулярная группа в элементарной ячейке может находиться в любом из двух (или более) эквивалентных положений (связанных между собой элементами симметрии) и действительно наблюдается в обоих (или во всех) этих положениях в одном и том же кристалле. В некоторых элементарных ячейках группа находится в одном положении, которое отличается от положения в других элементарных ячейках. В программе расчета по методу наименьших квадратов должна учитываться возможность существования фракционных атомов; если группа находится в данном положении половину времени, то ее можно рассматривать как половину группы. Иногда случается, что атомы могут занимать более или менее непрерывный интервал между двумя крайними положениями; таким образом, электронная плотность растянута так, что удовлетворительного согласия можно достичь только при использовании специально построенной программы. Для таких разупорядоченных групп невозможно получить обычные точные параметры, например длины связей и валентные углы. То же справедливо для групп, которые подвергаются ли-брационному движению, т. е. вращению и колебанию одновременно. Такое движение нельзя учесть с помощью стандартных программ.
Полезные советы. Если вы собираетесь выращивать кристаллы для определения структуры, то пользуйтесь следующими характеристиками: 1) кристаллы должны иметь, насколько это возможно, одинаковые размеры во всех направлениях: порядка 0,2—0,4 мм; 2) если вы будете исследовать ионные системы и вас интересует катион, то старайтесь избе
406
Глава 17
гать сферических анионов, особенно С1О4. Этот анион почти всегда может занимать несколько положений, и невозможность точного определения его координат будет влиять неблагоприятным образом на установление оставшейся части структуры. Анион BPh4 велик (много атомов), но обычно хорошо упаковывается, а его кольца легко обнаружить даже на плохих фурье-картах. Поэтому его можно использовать для установления большей части исходной модели фаз.
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
1. Nolte M.J., Singleton Е„ Laing М., J. Amer. Chem. Soc., 97, 6396 (1975).
2. Wei C.H., Dajl L.F., J. Amer. Chem. Soc., 91, 1351 (1969) и ссылки в этой работе; Cotton F.A., Troup J.M., J. Amer. Chem. Soc., 96, 4155 (1974).
3. Stout G.H., Jensen L.H., X-ray Structure Determination, McMillan, New York, 1968.
4. В качестве вводного обсуждения геометрии кристаллов см. Livingston R. L., J. Chem. Educ., 44, 376 (1967).
5. Thick J. A., Bercaw J. А., неопубликованные результаты.
6. Duggan D.M., Hendrickson D.N., Inorg. Chem., 13, 1911 (1974).
7. International Tables for X-ray Crystallography, Vol. 1,, eds. Henry N. F. M., Longsdale K., Kynoch Press, Birmingham, England, 1965.
8. Duggan D.M., Hendrickson D.N., Inorg. Chem., 12, 2422 (1973); ibid., 13, 2929 (1974).
9. Patterson A.L., Z. Krist., A, 90, 517 (1935).
10. Main P., Woolfson M.M., Lessinger L., University of York, York, England, and Gemain G. and Declerq J.-P., Place Louis Pasteur, 1348, Louvain-La-Neuve, Belgium.
Упражнения
1. Кристалл [Cu2(tren)2(CN)2](BPh4)2 характеризуется нейтральной плавучестью в смеси толуола с п-бромтолуолом; 5 мл этой смеси весят 6,35 г. Рентгеновские фотографии говорят о том, что элементарная ячейка должна быть моноклинной с а = 13,792, b — 10,338, с — 20,316 и р = 94,27°. Сколько молекул этого соединения содержится в элементарной ячейке?
2. Установлено, что пространственной группой комплекса меди в первой задаче является Р TJc. Если молекулы действительно представляют собой димеры, как это записано, то какой элемент симметрии связан с этими двумя фрагментами? Если вы не располагаете никакими спектральными данными, то можно ли с помощью указанных результатов отличить мономерную форму этой системы от димерной?
3. Покажите, что наличие оси второго порядка вдоль с при х = 1/4, у = 1/4 и плоскости fe-скольжения, перпендикулярной с при z = 1/4, требует одновременного существования центра инверсии при (1/4, 0, 1/4).
4, Постройте таблицу умножения, чтобы показать, что четыре операции Р 2j/c составляют группу.
5. Изобразите плоскости ab и Ьс в диаграммах элементарной ячейки для пространственной группы С2/с.
6. Какое относительное число отражений о можно получить для [Cu2(tre£)2(CN)J(BPh4)2, используя медный (X = 1,54 А) и молибденовый (7. = = 0,712 А) источники рентгеновских лучей? (Для получения более подробной информации см. задачу 1.)
7. Какова пространственная группа системы, для которой систематическими погасаниями являются 0к/, / = 2п+1; /10/, /=2п + 1; hkO, h = 2n+l и прецессионная фотография которой показывает зеркальную симметрию относительно всех осей?
Рентгеновская кристаллография
407
8. Какие из приведенных ниже пар систем могут быть с успехом изучены при использовании порошкообразных образцов? Обоснуйте свой вывод.
a. [Ni(tren)N3](BPh4) и [Ni(tren)N3]PF6.
б. [Ni(tren)N3](BPh4) и [Cu(tren)N3] (BPh4).
в. Rh(CN mpem-Bu)4 BF4 и Rh(CNPh)4BF4 .
г. Rh(CNPh)4Cl и Rh(n-C1C6H4NC)4C1.
9. Плоскость с-скольжения, перпендикулярная а в начале координат, связывает х, у, z с х, у, 1/2 + z. Используя выражение для расчета структурного фактора, покажите, что условием для систематических погасаний, обусловленных этим элементом, является Ofc/, Z = 2n+1.
10. Паттерсоновская карта, рассчитанная с помощью данных для [Cu2(tren)2(CN)2](BPh4)2, демонстрирует пик при относительных координатах (0, 0,348, 0,500) и другой пик равной интенсивности при (0,330, 0,500, 0,702); эти пики характеризуются наибольшей интенсивностью на карте по сравнению с пиком, находящимся в начале координат. Каковы относительные координаты атома меди?
11. Каков вид области карты AF, демонстрирующей истинное положение атома, отстоящего, скажем, на ~ 0,5 А от атома в модели, где оба атома сферические (т. е. имеют изотропные температурные коэффициенты)? Если положение атома в модели правильно, а истинное тепловое движение скорее анизотропно, чем изотропно, как в модели, то какого типа различия в электронных плотностях следует ожидать?
12. Каково влияние максимального поглощения на относительные интенсивности отражений от кристалла [Cu2(tren)2(CN)2](BPh4)2, который напоминает пластинку толщиной 0,1 мм и шириной 0,8 мм. Нужно ли вводить поправки?
13. Покажите, что операция вращения второго порядка начала координат в сочетании с перпендикулярной трансляцией Т эквивалентна вращению второго порядка относительно оси Т/2. Означает ли это, что решеточная трансляция [1, 0, 0] требует, чтобы оператору 2Ь при (1/4, 0, 0) сопутствовал другой оператор при (3/4, 0, 0)?
ПРИЛОЖЕНИЯ
ПРИЛОЖЕНИЕ I
ТАБЛИЦЫ ХАРАКТЕРОВ ДЛЯ ХИМИЧЕСКИ ВАЖНЫХ ГРУПП СИММЕТРИИ
1. Неаксиальные группы
А 1
х* 2, у2 z2, ху
yz, XZ
х2, у2, z2 ху, XZ, yz
2. ГРУППЫ С„
z, Rz
x, y, Rx, R
Z, Rz
(x, y)(Rx, Ry)
x2, y2, z2, xy yz, xz
Е = ехр(2л</3)
2 .,2 ,2
(х2 — у2, xy)(yz, xz)
z, R.
2 + У2, Z2 x2 — y2, xy
y)(.Rx, kJ (yz, xz)
Приложения
409
С5 Е С5 С25 С35 CJ е = ехр(2га'/5)
А 11111 2, R, -V2 + у2, Z2
Ei fl Е Е2 Е2* Е* 1 11 Е* е2* е2 е f (х, y)(Rx, Ry) (yz, xz)
|1 Е2 Е* Е е2*1
г2 ) 1 Е2* Е Е* е2 [ (х2 - у2, ху)
С6 Е С6 с3 с2 с23 С56 е = ехр(2тп/6)
А 1 1 1 1 1 1 z, Rz Х2 + у2, Z2
В 1 -1 1 -1 1 -1
Ег fl 11 £ Е* — Е* — Е -1 -1 — £ -£* Е*') £ ( (X, у) (Кх, Ry) (xz, yz)
Г1 - Е* — Е 1 -в* — £ 1
е2 и — £ — Е* 1 — £ “П (х2 - у2, ху)
С2 г? С С2 Л V 7 V- 7 V 7 V- 7 V 7 V- 7 е = ехр(2л!/7)
А 1111111 2, Rz Х2+ у2, Z2
Е3 fl Е Е2 Е3 Е3* 82* £*1 11 Е* Е2* Е3* Е3 Е2 Е ( (X, у) (К„ Ry) (xz, yz)
Г1 £z Е3* Е* Е Е3 Е2*)
е2 1 Е2* Е3 Е Е* Е3* Е2 j” (х2 - у2, ху)
J1 Е3 Е* Е2 Е2* £ Е3*|
Е3 11 Е3* Е Е2* Е2 Е* Е3 j
С8 E Cs C4 C2 C3 C| Cf Cl e = exp(2ni/8)
tn tn Сч ь. 111 11111 1-11 11-1-1-1 fl e i —1 — i — e* — e e*1 11 E* -1 -1 i —E —E* E f J1 i — 1 1 — 1 — i i — Л 11 — i — 1 1 — 1 i —i If |1 — e i —1 — i e* e — e*1 11 — E* — i — 1 1 E E* — E f 2, Rz (X, y) (R*. Ry) x2+ y2, z2 (xz, yz) (x2 - y2, xy)
410
Приложения
3. ГРУППЫ Dn
»2 Е С,(7) С2(у) С2(х)
А 1 1 1 1 X2, у2, z2
1 1 -1 -1 Rz ху
в2 1 -1 1 -1 у. Ry xz
вз 1 -1 -1 1 х, Rx
Di Е 2С3 зс.
А1 1 1 1 X2 + у2, z2
А2 1 1 — 1 Z R,
Е 2 -1 о (x,y)(Rx,Ry) (x2-y2, xy)(xz, yz)
D4 E 2C4 C2( = C2) 2C2 2Q
Ar 11 1 11 X2 + y2, z2
a2 11 1-1-1 z, R
1-1 1 1-1 X2-y2
й2 1-1 1-11 xy
E 20-2 00 (x, y)(Rx, R) (xz, yz)
Ds E 2C5 2C| 5C2
Ar 1 1 1 1 x2 + y2, z2
a2 1 1 1 -1 z, Rz
Er 2 2 cos 72° 2 cos 144° 0 (x, у) Ry) (xz, yz)
e2 2 2 cos 144° 2 cos 72° 0 (x2~y2, xy)
D6 E 2C6 2C3 C2 3C2 3G
Ar 1 1 1 1 1 1 x2 + y2, z2
a2 1 1 1 1 -1 -1 z, Rz
Br 1 -1 1 -1 1 -1
B2 1 -1 1 -1 -1 1
Er 2 1 1 —2 0 0 (x, y) (Rx, Ry) (xz, yz)
e2 2 -1 1 2 0 0 (X2-/, xy)
Приложения
411
4. ГРУППЫ Ст
С2* Е С2 a„(xz)
Ai 1 1 1 1 z 2 2 2 X , у , Z
Л2 1 1 -1 -1 К; ху
В1 1 -1 1 -1 X, Ру XZ
в2 1 -1 -1 1 У, Рх yz
Для плоской молекулы ось х перпендикулярна ее плоскости.
C3v Е 2С3 За„
Ai 1 1 1 Z х2+ у2, Z2
Аг 1 1 -1 Rz
Е 2 -1 0 (х, y)(Rx, Ry) (х2 — у2, ху) (xz, yz)
ct E 2C4 c2 2a„ 2ad
Aj 1 1 1 1 1 z X2 + y2, z2
A2 1 1 1 -1 -1 Rz
Bi 1 -1 1 1 -1 x2 — y2
в2 1 -1 1 -1 1 xy
E 2 0 — 2 0 0 (x, y) (Rx, Ry) (xz, yz)
* Если в молекуле симметрии Civ атомы расположены в виде квадрата, плоскости должны проходить через большее число атомов такой группировки или пересекать наибольшее число связей.
E 2C5 2C2 5o„
Al 1 1 1 1 z x2+ y2, z2
a2 1 1 1 -1 Rz
Ei 2 2 cos 72° 2 cos 144° 0 (x, y) (R„ Rr) (xz, yz)
e2 2 2 cos 144° 2 cos 72° 0 (x2— y2, xy)
Се» E 2C6 2C3 C2 Зсту
Ai 1 1 1 1 1 1 z x2+ y2, z2
a2 1 1 1 1 1 -1 Rz
Bl 1 -1 1 -1 1 -1
B2 1 -1 1 -1 1 1
Ei 2 1 -1 — 2 0 0 (X, y) (Rx, Ry) (xz, yz)
e2 2 -1 -1 2 0 0 (x2 - y2, xy)
412
Приложения
5. ГРУППЫ C„„
^2h Е С2 i <3*
Л 1 1 1 1 R; x2, y2 z2 xy
вв 1 -1 1 -1 Rx, R у xz, yz
Л 1 1 -1 -1 z
в„ 1 -1 -1 1 X, у
C?>h Е с3 С23 S3 S53 £ = exp(27ti/3)
А' 1 1 1 1 1 1 Rz x2 + J 2, Z2
Е' р р £ £* £* £ 1 1 £ £* £*| £ [ (x, y) (x2- y2, xy)
А" Ч 1 1 -1 -1 z
Е" р 11 £ £* £* £ -1 -1 — £ -£* -£*) — £ j (Rx, Ry) (xz, yz)
^4ft Е С4 с2 cl i s4
А, 1 1 1 1 1 1 1 i R2 X2+ y2, z2
Вв 1 -1 1 -1 1 -1 1 -i x2 — y2, xy
р -1 — i 1 -1
Ея р — z -1 i 1 — i -1 z [ (R4, Ry) (xz, yz)
Аи Ч 1 1 1 -1 -1 -1 -r z
Ви 1 -1 1 -1 -1 1 -1 1
р -1 — i -1 1
Еи (x, У)
^5ft Е с5 с 1 С2 S5 S? 5 1 £ = exp (2rr;/5)
А'
Е'1
£'2
А”
Е'{
E'i
1 1 1
Il £ £2
) 1 £* £2*
J 1 £2 £*
Il £2* £
Ч 1 1
J1 £ £2
1 1 £* £2*
р е2 8*
р £2* £
1 £2* £2 1 £* £ 1 1 1 £ 1 E* 1 £2 E2* 1 £2* £2 1 £*) £ ( Rz (x, У) x2+ y2, z2
£ £* £2* £2 1 £2 1 £2* £* £ £ £* £2*) £2 j (x2 - y2, xy)
1 1 -1 -1 -1 -1 z
fi2* £* — 1 —£ — £2 -£2* -£*)
£2 £ -1 — £* — £2* — £2 — £ ( {Rx, Ry) (xz, yz)
£ £2* — 1 —£2 -£* — £ — E2p
£* £2 -1 -£2* — £ -£* -E2 f
Приложения 413
Е С6 С3 С2 С3 С* i S3 Si а„ S6 S3 8 = ехр(2тп76)
Ад Вд Е2д Аи ви Е1и Е2и 6. п ^2ft 111 111 111 1-1 1-1 1-1 1-1 1 fl s — e* —1 —s s* 1 e — s* 'll s* — s — 1 — s* 8 1 e* — e JI — s*—e 1 -s*-e 1 — s*—e Il — 8 —8* 1—8 —8* 1—8 —8* 11 1 11 1 -1 -1 -1 1-1 1-1 1-1-1 1-1 P 8 -8* -1 —S 8* -1 -8 8* p e*-s -1 -8* e -1 -8* 8 Jl — 8*—8 1—8*—8 —1 8* 8 11—8 8* 1 — 8 — e* — 1 S 8* »УППЫ Dnh E C2(z) C2(y) C2(x) i a(x 1 1 1 -1 1-1 — l—s e*l -1-8* 8 j 1 -8*-8 | 1 -8 — 8*1 -1 -1 -1 J 1 -1 1 1 S -8*1 1 e* —s j -1 8* 8 | -1 8 S*j y) ct(xz) a(j R. (R z u, z) „ Ry) y) x2 (x (X + У2, z2 z, yz] 2-)’2, xy)
Ад Вгв Bie Взд Аи Biu в2и в2и Е>зн 1 1111 1 1-1-11 1-1 1-1 1 - 1-1-1 1 1 - 1 111—1— 1 1 -1 -1 -1 - 1-1 1-1 -1 1-1-1 1 -1 E 2C3 3C2 2S3 3a„ 1 -1 1 -1 -1 1 -1 1 1 1 1 1 1 1 1 1 R. Ry R, z У X X2 , У2, z2 xy xz yz
А\ А'2 Е' А'{ A'i Е" 111111 1 1-1 1 1-1 2-1 0 2-1 0 1 1 1 -1 -1 -1 1 1 -1 -1 -1 1 2-1 0-2 10 E 2C4 C2 2C2 2C'i i 2S4 R, (X, y) (Rx, Ry) 2a„ 2 X2 (X2 (xz °d 4-У2- -y2- yz) A xy)
к к> — к> — <е к, — ы — sees <е ® <е <е 1111111 1 1 1-1-1 1 1 1-1 1 1-1 1-1 1-1 1-1 1 1-1 2 0 -2 0 0 2 0 1 1 1 1 1-1-1 1 1 1 -1 -1 -1 -1 1-1 1 1-1-1 1 1-1 1-1 1-1 1 2 0 -2 0 0 -2 0 1 1 I -1 - 1 1 - 1 -1 — 2 0 -1 -1 - -1 1 -1 -1 -1 1 - 2 0 1 1 1 1 0 r 1 1 1 0 R, (R„ z (X, y) Ry) x2+y2, z2 x2-y2 xy (xz, yz)
<rv проходит через атомы, a ad делит пополам валентные углы.
414
Приложения
В5Л Е 2С5 2Cj 5C2 2S5 2S| 5o„
A'i 1 1 1 1 1 1 1 1 x2+ y2, z2
Л'2 1 1 1 -1 1 1 1 -1
Е'1 2 2 cos 72° 2 cos 144° 0 2 2 cos 72° 2 cos 144° 0 (x, y)
Е'2 2 2 cos 144° 2 cos 72° 0 2 2 cos 144° 2 cos 72° 0 (x2 - y2, xy)
1 1 1 1 -1 -1 -1 -1
А"г 1 1 1 -1 -1 -1 -1 1 z
Е'[ 2 2 cos 72° 2 cos 144° 0 — 2 -2 cos 72° -2 cos 144° 0 (Rx, Ry) (xz, yz)
Е'1 2 2 cos 144° 2 cos 72° 0 — 2 — 2 cos 144° -2 cos 72° 0 2
E 2C6 2C3 C2 3C'2 3Q i 2S3 2S6 3ad 3o„
^10 1 1 1 1 1 1 1 1 1 1 1 1 x2 + y2, z2
A 2g 1 1 1 1 -1 -1 1 1 1 1 -1 -1 R.
Blg 1 -1 1 -1 1 -1 1 -1 1 -1 1 -1
1 -1 1 -1 -1 1 1 -1 1 -1 -1 1
El9 2 1 -1 — 2 0 0 2 1 -1 — 2 0 0 (R„ Ry) (xz, yz)
Ei9 2 -1 -1 2 0 0 2 -1 -1 2 0 0 (x2 - y2, xy)
lu 1 1 1 1 1 1 -1 -1 -1 -1 -1 -1
^2u 1 1 1 1 -1 -1 -1 -1 -1 -1 1 1 z
B1U 1 -1 1 -1 1 -1 -1 1 -1 1 -1 1
B2u 1 -1 1 -1 -1 1 -1 1 -1 1 1 -1
Elu 2 1 -1 — 2 0 0 — 2 -1 1 2 0 0 (*, y)
E2u 2 -1 -1 2 0 0 — 2 1 1 — 2 0 0
^8ft E 2C8 2CI 2C4 C2 4C2 4C5 i 2S8 2S| 2S4 <з„ 4о44о„
1 1 1 1 1 1 1 1 1 1 1111 X2+ y2, z2
7129 1 1 1 1 1 -1 -1 1 1 1 1 1-1-1 R,
Bia 1 -1 -1 1 1 1 -1 1 -1 -1 1 1 1-1
fi2a 1 -1 -1 1 1 -1 1 1 -1 -1 1 1-1 1
Bia 2 ^2 -^2 0 — 2 0 0 2 ^2 -^2 0-2 0 0 (Rx, Ry) (xz, yz)
E2g 2 0 0 — 2 2 0 0 2 0 0 -2 2 0 0 (x2 — v2, xy)
e39 2 -^2 ^2 0 — 2 0 0 2 -^2 ^2 0-2 0 0
1 1 1 1 1 1 1 -1 -1 -1 -1-1-1-1
A2u 1 1 1 1 1 -1 -1-1 -1 -1 -1-1 1 1 z
Bu, 1 -1 -1 1 1 1 -1-1 1 1 -1-1-1 1
B2u 1 -1 -1 1 1 -1 1 -1 1 1 -1-1 1-1
Eiu 2 ^2 -^2 0 — 2 0 0-2 -^2 ^2 0 2 0 0 (x,y)
E2u 2 0 0 — 2 2 0 0-2 0 0 2-2 0 0
Eju 2 -^2 ^2 0 — 2 0 0 —2 ^2 -^2 0 2 0 0
Приложения
415
7. ГРУППЫ Dnd
&2<1 E 2.S4 c2 2C2 2<rd
Al 1 1 1 1 1 x2+ y2, z2
^2 1 1 1 -1 -1 R,
в. 1 -1 1 1 -1 x2-y2
в2 1 -1 1 -1 1 z xy
E 2 0 — 2 0 0 (x, Ji), (R„ Ry) (xz, yz)
D3d E 2C3 3C2 > 2S6 3ad
Alg 1 1 1 1 1 1 x2 + y2, z2
A2g 1 1 -1 1 1 -1 R<
Eg 2 -1 0 2 -1 0 (Rx, Ry) (x2- y2, xy),
^1U Aiu 1 1 1 1 1 -1 -1 -1 -1 -1 -1 1 z (xz, yz)
Eg 2 -1 0 — 2 1 0 (x, y)
D^d E 2S8 2Q 2S1 C2 4C2 4<^
Ai 1 1 1 1 1 1 1 x2+y2, z2
Л2 1 1 1 1 1 -1 -1 R;
Bl 1 -1 1 -1 1 1 -1
B2 1 -1 1 -1 1 -1 1 Z
Ei 2 0 -У2 — 2 0 0 (x, У)
e2 2 0 — 2 0 2 0 0 (X2- У2, xy)
E3 2 -У2 0 /2 — 2 0 0 (R„ Ry) (xz, yz)
DSd E 2C5 2C2s 5C2 i 2Sil 2Si0 5ad
11 1111 11 x2+y2, z2
11 1-111 1-1 R;
Elg 2 2 cos 72° 2 cos 144° 0. 2 2 cos 72° 2 cos 144° 0 (R„ Ry) (xz, yz)
E2g 2 2cosl44° 2cos72° 0 2 2cosl44° 2cos72° 0 (x2- y2, xy)
^1U 11 11-1-1 -1-1
^2u 11 1 -1 -1 -1 -1 1 z
Eiu 2 2cos72° 2cosl44 0 —2 —2cos72° — 2cosl44° 0 y)
^2u 2 2cosl44° 2cos72° 0 -2-2 cos 144° -2 cos 72° 0
416
Приложения
D6d Е 2S12 2С6 2S4 2С3 25125 с2 6С'2 6ad
1 1 1 1 1 1 1 1 1 х2+ у2, Z2
Л 2 1 1 1 1 1 1 1 -1 -1
В, 1 -1 1 -1 1 -1 1 1 -1
в2 1 -1 1 -1 1 -1 1 -1 1
Е, 2 Уз 1 0 -1 -/з — 2 0 0 (х, У)
е2 2 1 -1 — 2 -1 1 2 0 0 (х2 - у2, ху)
Е3 2 0 — 2 0 2 0 — 2 0 0
е4 2 -1 -1 2 -1 -1 2 0 0
е5 2 -уз 1 0 -1 уз — 2 0 0 (R,, Ry) (xz, yz)
Ss Е S8 C4 d8 C2 ^8 ci Sl s = exp (2 га/8)
А 1 1 1 1 1 1 i 1 x2+ y2, Z2
В 1 -1 1 -1 1 -1 i -1 z
р И £ i -s* -1 — £ — i s*3 (x, y)
31 £* — i — £ -1 -s* i £ ( R„)
е2 j 1 31 i — i -1 -1 — i i 1 1 i — i -1 -1 ~4 4 (x2 - y2, xy)
Г1 -s* -1 s*
Е3 {1 — £ i s* -1 £ — i — 6*1 (xz, yz)
Приложения
417
9. КУБИЧЕСКИЕ ГРУППЫ
т Е 4С3 4С3 зс2 £ = exp (2тп/3)
А 1 1 1 1 x2 + y2+ z2
[1 £ £* п (2z2 — x2— y2,
31 £* £ If x2- y2)
Т з 0 0 -1 (Rx, Ry, Rz); (x, у, z) (xy, xz, yz)
T„ E 4C3 4C23 3C2 i 4S6 4S65 3aft £ = exp(2iti/3)
A, 1 1 1 1 1 1 1 1 x2 + y2+ z2
Au 1 1 1 1 -1 -1 -1 -1
E Г1 £ £* 1 1 £ £* If (2z2 — x2 — y2,
^9 31 £* £ 1 1 £* £ 1J x2 ~ У2)
P fl £ £* 1 -1 — £ -£* -1з
Пи 3 1 £* £ 1 -1 -£* — £ -If
T9 3 0 0 -1 1 0 0 (Rx, Ry, RJ (xz, yz, xy)
Tu 3 0 0 -1 -1 0 0 1 (x, У, z)
Ta E 8C3 3C2 6S4 6ad
Л1 1 1 1 1 1 x2 + y2 + z2
Л2 1 1 1 -1 -1
E 2 -1 2 0 0 (2z2 — x2— y2, Х2~У2)
Л 3 0 -1 1 -1 (Rx, Ry, Rz)
T2 3 0 -1 - 1 1 (X, У, z) (xy, xz, yz)
0 E 6C4 3C2(=Ci)»C3 6C2
Ar 1 1 1 1 1 X2+ y2 + Z2
Л2 E 1 2 -1 1 1 0 2-1 -1 0 (2z2 — x2— y2,
3 1-1 0 -1 (Rx, Ry, Rz); x2-y2)
T2 3 -1 -1 0 1 (x, y, z) (xy, xz, yz)
27—574
418
Приложения
о„ Е 8С3 6С2 6С4 зс2(=с|)1 6S4 8S6 3aft 6ad
Alg 1 1 1 1 1 1 1 1 1 1 x2+ y2+ z2
^2д 1 1 -1 -1 1 1 -1 1 1 -1
2 -1 0 0 2 2 0 -1 2 0 (2z2 — x2 — y2, x2 - y2)
3 0 -1 1 -1 3 1 0 -1 -1 (Ex, RJ
?2в 3 0 1 -1 -1 3 -1 0 -1 1 (xz, yz, xy)
^1и 1 1 1 1 1 -1 -1 -1 -1 -1
^2и 1 1 -1 -1 1 -1 1 -1 -1 0
Еи 2 -1 0 0 2 —2 0 1 — 2 0
т1и 3 0 -1 1 -1 -3 -1 0 1 1 (x, y, Z)
Т2и 3 0 1 -1 -1 -3 1 0 1 -1
10. ГРУППЫ Q, И D^h ДЛЯ ЛИНЕЙНЫХ МОЛЕКУЛ
C«v E 2C* ... ooar
Ai =E+ 1 1 ... 1 z x2+.y2, z2
a2=z 1 1 ... -1 R,
E, = П 2 2 cos ф... 0 (x, y); (Kx, Ry) (xz, yz)
e2 = A 2 2cos 2ф... 0 (x2-y2, xy)
Е3=Ф 2 2 cos Зф... 0
E 2C*W ... oo i 2S*, ... ooC2
4+ 1 1 1 1 1 1 x2 + y2, z2
1 1 -1 1 1 -1 R,
n9 2 2 cos ф 0 2 — 2 cos ф ... 0 (R„ Ry) (xz, yz)
2 2 cos 2ф 0 2 2 cos 2ф 0 (x2- y2, xy)
К 1 1 1 -1 -1 -1 z
1 1 -1 -1 -1 1
n„ 2 2 cos ф 0 — 2 2 cos ф ... 0 (x, y)
2 2 cos 2ф 0 — 2 — 2cos2<i> .. . 0
11. ИКОСАЭДРИЧЕСКИЕ ГРУППЫ*
1„ Е 12С5 12С25 20С3 15С2 i 12S10 12Sto 20S6 15c
Л 1 1 1 1 1 1 1 1 1 1 x2 + y2 + z2
3 1/2(1 +1/5) 1/2(1 -1/5) 0 -1 3 1/2 (1-1/5) 1/2(1 +1/5) 0 -1 (Rx, Ry, Rz)
3 1/2(1 —]/5) 1/2(14-1/5) 0 -1 3 1/2(1+ 1/5) 1/2(1 —1/5) 0 -1
G„ 4 -1 -1 1 0 4 -1 -1 1 0
нв 5 0 0 -1 1 5 0 0 -1 1 (2z2-x2-y2,
x2 — y2,
xy, yz, zx)
А. 1 1 1 1 1 -1 -1 -1 -1 -1
т1в 3 1/2(14-1/5) 1/2(1—]/5) 0 -1 -3 -1/2(1 -1/5) -1/2(1 + 1/5) 0 1 (x, У, г)
т2и 3 1/2(1 -1/5) 1/2(1+]/5) 0 -1 -3 -1/2(1 +1/5) -1/2(1 — 1/5) 0 1
Gu 4 -1 -1 1 0 — 4 1 1 -1 0
ни 5 0 0 -1 1 -5 0 0 1 -1
* Для чисто вращательной группы I таблицей характеров является выделенный в левом верхнем углу фрагмент; нижние индексы д должны быть, естественно опущены, а (х, у, z) приписано представление Tv
ПРИЛОЖЕНИЕ II
ТАБЛИЦЫ ХАРАКТЕРОВ ДЛЯ НЕКОТОРЫХ ДВОЙНЫХ ГРУПП
ГРУППЫ О3 И C'3v
D'3 С3 С23 Е R ЗС2 3C2R c3r C3R D3
C3V Сз С2 Е R За„ 3avR C23R C3R С'зе
А\ Г, А'г Г2 г3 111 1 1 1 111 1-1-1 ,22-1-1 0 0 Lz Xf У) ^x-> Ly z Lz x, y, Ex, Ly
’t «л U U U СТС СТ} 3, 'S ц, ГЧ ГЧ го — Ьв Ьв 1-1-1 1 i 1-1-1 1 -i i 2-21 -10 0
Приложения
421
группы с; и 54
с; Е R С4 С4К ci ClR ci CiR ci
Е R s4 S4K c2r c2 si SiR Si
А' Гс 1 1 1 1 1 1 i 1 2 Lz
В' г2 1 1 -1 -1 1 1 -i -1 Lz z
Е’ F3 1 1 i i -1 -1 — i — i } x + iy 1 x ± iy
|Г4 1 1 — i — i -1 -1 i i Lx + iLy J + iLy
^1/2 ГГ5 1 -1 со — со i — i co3 -co3
кв 1 -1 — со3 со3 — i i — co co
Е'312 Г7 1 -1 — со со i — i — co3 co3
1Г8 1 -1 со3 -со3 — i i co — co
со = exp (от/4)
ГРУППА Т'
T E R 3C2 3C2R 4C3 4C3K 4C2 4C1K
A' Г1 1 1 1 1 1 1 1
E' fb jr3 1 1 1 1 1 1 CO co2 CO co2 co2 co "s з
T r4 3 3 -1 0 0 0 0 x, y, z, Lx, Ly, Lz
L'l/2 r5 2 — 2 0 1 -1 -1 -1
^3/2 fr6 2 — 2 0 co — co co2 -co2
}Г7 2 — 2 0 co2 -co2 CO — co
со = ехр (2л1/3)
422
Приложения
ГРУППЫ О' И Td
О' 4C3 4Ci 3C| 3C4 3Ci 3C'2 E R 4C3R 4C3R 3CjR 3CjK 3C4R 3C'2R O'
rd 4C3 4C23 3Cj 3S4 3SJ 6a4 £ R 4C23R 4C3R 3CjR 3SdR 3S4R 6<3dR rd
-ч ГЧ in u u u u u X X 4 11111111 111 1 1 -1 -1 -1 22-1-1 2 0 0 0 3 3 0 0 -1 1 1 -1 3 3 0 0 -1 -1 -1 1 x, y, z, L„ Ly, Lz kJ [ Ss N • к »<
C> tfl tfl tu - - C tfl tfl w- Ui - >— ' M N N T T oo 'j a> 2-2 1 -1 0 ]/2 -]/2 0 2-2 1 -1 0 —]/2 ]/2 0 4-4-11 0 0 0 0
группа /4
D'4 E R c4 CiR Cl c4r C2 c2r 2C'2 2C'2R 2C5 2C^R
Tj 1 1 1 1 1 1 1
Л2 r2 1 1 1 1 1 -1 -1
Bi r3 1 1 -1 -1 1 1 -1
B'2 r4 1 1 -1 -1 1 -1 1
£i r5 2 2 0 0 — 2 0 0
£'2 r6 2 ^2 -1/2 0 0 0
Е'з Г7 2 — 2 -]/2 1/2 0 0 0
ПРИЛОЖЕНИЕ III
НОРМАЛЬНЫЕ КОЛЕБАНИЯ
ДЛЯ НАИБОЛЕЕ РАСПРОСТРАНЕННЫХ СТРУКТУР
Пирамидальные молекулы XY3
Плоские молекулы XY
424
Приложения
Молекулы ZXY3 с точечной группой симметрии С3„
Октаэдрические молекулы XY6
Приложения
425
Квадратно-плоскостные молекулы XY4
28-574
Плоские молекулы ZXY
426
Приложения
Тригонально-бипирамидальные молекулы XY5
»гМ19)
w
Молекулы X2Y6 типа этана
Приложения
427
»s(XY)
Плоскостные (Civ) А,(р)
Внеплоскостные (Сг) А(р)
А\(р) А(р)
и(ХХ)
вг(вр)
B(dp)
pt(w
Аг( dp) А(р)
Bt(dp)
В (dp)
Нелинейные молекулы X2Y2 (р— поляризованное, dp
деполяризованное)
ПРИЛОЖЕНИЕ IV
ДИАГРАММЫ ТАНАБЕ—СУГАНО ДЛЯ ПОЛЕЙ О*
Энергетическая диаграмма для конфигурации d2
Энергетическая диаграмма для конфигурации J3
* Эти диаграммы представляют собой полные энергетические диаграммы для указанных конфигураций, воспроизведенные из работы: Tanabe Y., Suga-по S., J. Phys. Soc. Japan., 9, 753, 766 (1964).
На диаграммах у обозначает отношение параметров Рака С/В. Сплошные линии, перпендикулярные оси Dq/B на диаграммах для конфигураций J4, d5, d6 и J7, указывают на переходы от слабого к сильному полю. Расчет и заложенные в нем допущения имеются в оригинальной работе. Диаграммы неприменимы к любому данному комплексу, но дают качественное представление об энергиях различных состояний в зависимости от Dq/B.
Приложения
429
о«/в
Энергетическая диаграмма для конфигурации J4
Энергетическая диаграмма для конфигурации d5
Энергетическая диаграмма для конфи- Энергетическая диаграмма для конфигурации гурации J7
430
Приложения
Энергетическая диаграмма для конфигурации d'
ПРИЛОЖЕНИЕ V
РАСЧЕТ А (1(Ю$) И 0 ДЛЯ О;,-К()М11ЛЕКСОВ Nin И ^-КОМПЛЕКСОВ Со11
РАСЧЕТ А и Р ДЛЯ ОКТАЭДРИЧЕСКИХ КОМПЛЕКСОВ Ni2+
Данные, приведенные в табл. 10.6 для Ni[(CH3)2SO]6(ClO4.)2, будут использованы для иллюстрации расчетов А, ₽ и частоты полосы 3А2д -> 3Tlg (F). Величина А, или 10 Dq, получается непосредственно из перехода низшей энергии ЭА29 -> Т2д, который лежит при 7728 см ~ \ Уравнение (10.12)
[6Dqp — 16(Dq)2] + [ — 6Dq — р]Е + Е2 = 0
используется доя расчета экспериментальной величины энергии ЭВ, т. е. р в уравнении (10.12). Параметр р равен 15В для комплекса никеля (II), где В—параметр Рака. Параметры Рака указывают на величину межэлектронного отталкивания между различными уровнями в газообразном ионе. Величина В постоянна, что дает возможность выразить разность энергий между уровнями высшей спиновой мультиплетности через некоторое целое число п, умноженное на В, т. е. пВ. Для различных ионов как п, так и В различны; в случае иона Ni2 + разность энергий между 3F и 3Р составляет 15В. В случае комплекса тот же член равен 15В'. Для использования уравнения (10.12) необходимо взять величину энергии состояния 3Т1д (В), которая представляет собой энергию, наблюдаемую для перехода 3Т1д (В) (24038 см ” '), плюс энергия уровня 3А2д, поскольку
Энергия, наблюдаемая для перехода (24038 см” ‘) = энергия 3Т1д(Р) — — энергия 3А2д,
поэтому
Энергия 3Т19 (В) = Е для перехода + Е (3А2д).
Таким образом, комбинируя это выражение с уравнением (10.11) [*(%,) = — 12Dq; отметим, что Dq = 7728/10 773], получим
Энергия 3Т1д (В) = 24038 см ” 1 — 12Dq = 14 762 см “ !.
*
Величина Е = 14762 см” 1 используется в уравнении (10.12) наряду с Dq = 773, чтобы получить величину
р = 13 818 см”1 = 15 В'.
Величина Е(3В) для газообразного иона Ni2+ составляет 15 В=
432
Приложения
= 15 840 см *, а Р равна [уравнение (10.14) или (10.13)]
13 818 15В'
В = -------=--------= 0,872
Р 15 840 15В
или
15 840 - 13 818
15 840
х 100 = 12,8%.
Для того чтобы рассчитать энергии состояний 3Т19 (F) и 3Т19 (Р), величины р = 13 818 см“ 1 и Dq = 773 см”1 подставляются в уравнение (10.12), и оно решается относительно Е. Получаются два корня: Е — = 14762 и 3694 см”1. Поскольку переходом является 3Л29 -> 3Tlg (F) или 3Т1д (Р), полосы поглощения соответствуют разностям
В[3Т19 (Р)]-В[3А29] и В[3Т19(Р)]-В[3Л29]
или Л А
% (F)] = 3694 - [ - 12(773)] = 12970 см ” 1
и
[*А2д -► 3Tlff(P)] = 1476212(773)] = 24038 см”1.
Соответствие рассчитанных и экспериментальных величин для полосы при 12970 см” 1 свидетельствует в пользу приведенных выше значений Р и Dq.
РАСЧЕТ ДИр ДЛЯ ^-КОМПЛЕКСОВ Со2 +
В поле тетраэдрической симметрии основное состояние 4Р комплекса Со2+ расщепляется на состояние 4 Л 2, 4Т2и4'Т1 (F). Переходы 4Л2 ->4Т2, 4А2 -> 4 7] (Р) и 4Л2 -» 4Т, (Р) обозначаются соответственно как vb v2 и v3. Для расчета Дир используются следующие соотношения
V!=4 (1)
v2 = 1,5Д + 7,5В' - Q, (2)
v3 = 1,5Д + 7,5В' + Q, (3)
Q = у [(0,6Д - 15В')2 + 0,64Д2]1 /2, (4)
где В' — эффективная величина члена межэлектронного отталкивания Рака в комплексе. Запишем вновь уравнения (10.14) и (10.13):
В' (комплекс)
В (свободный ион) или
о В (свободный ион) — В' (комплекс)
Р В (свободный ион) (
Для того чтобы продемонстрировать расчет, рассмотрим спектр тетраэ
Приложения
433
дрического комплекса Со(ТМГЦ + (где ТМГ — тетраметилгуанидин) Г1]. Полоса, отнесенная к v,. представляет собой дублет с максимумами при 530 нм (18 867 см1), £ = 204 и 590 нм (16949 см1), е = 269. Спектр в ближней инфракрасной области приводит к v2 в виде триплета: 1204 нм (8306 см1), 6 = 91,5; 1320 нм (7576 см-1), е = 85,0; 1540 нм (6494 см ~ *), е = 23,5. Степень расщепления состояний Т, за
счет спин-орбитального взаимодействия такова [2]: — и
Энергия перехода 4Л 2 ->4 7) (F) получается путем усреднения трех линий полосы v2 с использованием приведенных выше весовых коэффициен
тов:
9
--(6494) = 14612
6
^-(7576) = 11 364
-^-(8306) = 31 148
Общее — 57 124
4
30
Таким образом, средняя энергия v2 составляет 57124: — = 7617 см Е
Энергия перехода 4Л2^-47’1 (Р) (т. е. v3) получается путем усреднения двух линий и приводит к 17908 см-1. Для получения Дир решается ряд уравнений (1)—(4). Сложение уравнений (2) и (3) дает
д = v2 + уз - 15д' 3
а подстановка v2 и v3 для Со(ТМГ)4+ приводит к А = 5(1702 см”1 — — В'). Вычитая уравнение (2) из уравнения (3), получим
Q = y(v3 - V2) = 5146 см-1.
Возводя обе части уравнения (4) в квадрат и делая перестановку, получим
4g2 = А2 - 18В'А + 225 (В')2.
Подстановка 2 = 5146 см-1 и А = 5(1702 см-1—В') в уравнение (6) приводит к уравнению, которое может быть решено относительно В'. Один корень равен 821 см-1, а другой отрицателен. Если положительный корень подставить в А = 5(1702 см ~ 1 — В'), то получим величину А = 4405 см-1; р рассчитывается из уравнения (5).
1. Drago R.S., Lonhgi R.L., Inorg. Chem., 4, 11 (1965).
2. Stahl-Broda R„ Low W., Phys. Rev., 113, 775 (1959).
ПРИЛОЖЕНИЕ VI
ПРЕОБРАЗОВАНИЕ ДАННЫХ
ПО ХИМИЧЕСКИМ СДВИГАМ
Химические сдвиги 5 (в м. д.) некоторых соединений, часто используемых в качестве внешних стандартов, относительно 5 = 0 для тетраме-тилсилана таковы: циклогексан —1,6; диоксан —3,8; Н2О —5,2; СН2С12 -5,8; С6Н6 -6,9; СНС13 -7,7; H2SO4 (плотность 1,857) -— 11,6. (Большие по абсолютной величине отрицательные сдвиги указывают на меньшее экранирование.) Эти сдвиги получаются в чистых жидкостях относительно внешнего стандарта. В результате они могут быть использованы для преобразования данных и сопоставления результатов, полученных с различными веществами в качестве внешних стандартов. Для превращения 5, полученного относительно С6Н6 в качестве стандарта, в 5 относительно Si(CH3)4 (внешний стандарт) вычитаем 6,9 м. д. из величины 5 для С6Н6. Необходимо произвести проверку, чтобы убедиться в том, что знак 5 соответствует условию, приведенному в разд. 7.10 для протонов (5S — 6Л).
Превращение результатов, полученных относительно внешнего стандарта, в данные относительно внутреннего стандарта довольно не просто. Если величины химического сдвига при бесконечном разведении в СС14 превратить в величины относительно Si(CH3)4, используя приведенные выше данные, то получатся не значения т. Эта операция превращает химические сдвиги в величины 5 относительно чистого Si(CH,)4. Различие в 5 для Si(CH3)4 в чистой жидкости и при бесконечном разведении в СС14 составляет около 0,4 м. д. Чистая жидкость экранирована в большей степени.
Для сдвигов фторсодержащих соединений обычно в качестве стандарта с нулевым сдвигом берется F2. Сдвиги для других жидкостей относительно F2 составляют 375,6 для SF6, 414,3 для CFC13, 454,2 для CF3C1, 491,0 для CF4, 507,6 для CF3COOH, 543,2 для C6H5F, 598,9 для SiF4 и 625 для HF, где положительные значения говорят о фторе, экранированном в большей степени.
Многие сдвиги фосфорсодержащих соединений берутся относительно 85%-ной Н3РО4 в качестве стандарта.
ПРИЛОЖЕНИЕ VII
КРИСТАЛЛИЧЕСКИЕ СИСТЕМЫ И СИММЕТРИЯ
Соотношения между углами и ребрами, приведенные в разд. 17.2а для различных кристаллических систем, полезны, но не являются единственными в своем роде. Системы определяются на основе элементов симметрии, присутствующих в элементарной ячейке. На рис. VII. 1 показаны четырнадцать решеток Бравэ, которые возникают за счет введения оператора центрирования. Как описывалось для триклинной, моноклинной и ромбической решеток, эта операция симметрии обычно наклады-
а
Триклинная Р
Рис. VII. 1. Четырнадцать решеток Бравэ, распределенных среди семи кристаллических систем.
Примитивная решетка (Р) имеет одну эквивалентную точку в каждой элементарной ячейке при (ООО), объем-ноцентрированная решетка (/)—две эквивалентные точки в каждой элементарной ячейке при (ООО; 1/2 1/2 1/2); базоцентрированная решетка (С) — две эквивалентные точки в каждой элементарной ячейке при (ООО; 1/2 1/2 0), граиецентрированная решетка — четыре эквивалентные точки в каждой элементарной ячейке при (ООО; 0 1/2 1/2; 1/2 0 1/2; 1/2 1/2 0). Все эквивалентные точки решетки связаны между собой только трансляциями и имеют идентичные окружения.
436
Приложения
вает ограничения на углы и ребра групп, но может случиться, что триклинная ячейка в пределах ошибки эксперимента будет иметь углы в 90°. Если симметрия решетки не превышает 1 (оператор центра инверсии), то ячейка триклинная, и углы в 90° рассматриваются как случайные. Аналогично типами симметрии, определяющими соноклинную и ромбическую решетки, являются соответственно 2/ш и ттт. Они означают, что для моноклинных решеток можно обнаружить ось второго порядка, перпендикулярную зеркальной плоскости, и что для ромбических решеток существуют три взаимно перпендикулярные зеркальные плоскости. Для моноклинной системы эти ограничения по симметрии будут приводить к условию а = у = 90°. Как правило, р#90°, но иногда очень близок к этому значению. Суть заключается в том, что идентификация соответствующей кристаллической системы должна основываться на знании наблюдаемой симметрии, а не на размерах ячейки.
Отметим, что имеются еще четыре кристаллические системы, которые мы подробно не описывали и которые включают элементы вращения третьего, четвертого и шестого порядков. К ним относятся гексагональная, ромбоэдрическая, тетрагональная и кубическая системы. Они включены в табл. 17.1.
ПРИЛОЖЕНИЕ VIII
ХАРАКТЕРИСТИКИ ОТДЕЛЬНЫХ ЯДЕР
Изотопа Частота ЯМР для поля в 10 кЭ, МГц Содержание Относительная чувстви- Магнитный момент pNa в единицах ядерного магнетона (eh/4nmc) Спин I в единицах h/2n Электричес- Анизотроп- Изотропное
в природе, °/ /о тельность для одинакового числа ядер кий квадруполь ный момент Q в единицах 10“24 см2 ное сверхтонкое взаимодействие В, МГц6 сверхтонкое взаимодействие Ло, МГц0
при посто- при постоян-янном поле ной частоте
?пг 29,165 — 0,322 0,685 -1,9130 1/2
,'Н 42,577 99,9844 1,000 1,000 2,79270 1/2 — — 1420
2Н 6,536 1,56 10"2 9,64-10 3 0,409 0,85738 1 2,77 • 10 3 — 218
,6Li 6,265 7,43 8,51 10 “3 0,392 0,82191 1 4,6 10“4 — 152д
,7Li 16,547 92,57 0,294 1,94 3,2560 3/2 -4,2-10'2 — 402д
.’Be 5,983 100 1,39-10“2 0,703 -1,1774 3/2 2-10~2 — -358
юв 4,575 18,83 1,99-10“2 1,72 1,8006 3 0,111 17,8 672
UB 13,660 81,17 0,165 1,60 2,6880 3/2 3,55 10“2 53,1 2020
,13c 10,705 1,108 1,59-10“2 0,251 0,70216 1/2 — 90,8 3110
,14N 3,076 99,635 1,01-10“3 0,193 0,40357 1 2 10'2 47,8 1540
,15N 4,315 0,365 1,04 • 10 “3 0,101 -0,28304 1/2 — -67,1 -2160
,17O 5,772 3.7-10 2 2,91 • 10 2 1,58 -1,8930 5/2 — 4 10“3 -144 -4628
40,055 100 0,834 0,941 2,6273 1/2 — 1515 47910
,23Na 11,262 100 9,27-10“2 1.32 2,2161 3/2 0,1 — 886д
,25Mg 2,606 10,05 2,68 -10 2 0,714 -0,85471 5/2 — — —
,27A1 11,094 100 0,207 3,04 3,6385 5/2 0,149 59 2746
,2’Si 8,460 4,70 7,85-10“2 0,199 -0,55477 1/2 — -86,6 -3381
31p 17,235 100 6,64-10 2 0,405 1,1305 1/2 — 287 10178
Изотопа Частота ЯМР для поля в 10 кЭ, МГц Содержание Относительная чувстви- Магнитный момент В единицах ядерного магнетона Спин I в единицах /1/2л Электричес- Анизотроп- Изотропное
в природе, % тельность для одинакового числа ядер при посто- при постоян- кий квадру-польный момент Q в единицах 10“24 см2 ное сверхтонкое взаимодействие В, МГц6 сверхтонкое взаимодействие Ло, МГц6
янном поле ной частоте
,33S 3,266 0,74 2,26-10“3 0,384 0,64274 3/2 — 6,410“2 78 2715
35С1 4,172 75,4 4,71-10“1 0,490 0,82089 3/2 — 797•10“2 137 4664
,37С1 3,472 24,6 2,72-Ю-3 0,408 0,68329 3/2 -6,21 10“2 117 3880
,3’К 1,987 93,08 5,08-10“4 0,233 0,39094 3/2 — — 23 Р
,43Са 2,865 0,13 6,39-10“2 1,41 -1,3153 7/2 — — —
,45Sc 10,343 100 0,301 5,10 4,7491 7/2 — — 1833
,47Ti 2,400 7,75 2,10-10“3 0,659 -0,78712 5/2 — — -492
,4’Ti 2,401 5,51 3,76-10“3 1,19 -1,1023 7/2 — — -492
,51V 11,193 — 100 0,383 5,53 5,1392 7/2 0,3 — 2613
,53Cr 2,406 9,54 1,0-10 4 0,29 -0,4735 3/2 — — -630
,55Mn 10,553 100 0,178 2,89 3,4610 5/2 0,5 X — 3063
57Fe — 2,245 — — <0,05 — — 450
57Cor 10,0 — 0,274 4,95 4,6 7/2 — — —
58Cor 13,3 — 0,25 2,5 3,5 2 — — —
,5’Co 10,103 100 0,281 4,83 4,6388 7/2 0,5 3666
,60Cor 4,6 — 5-10“2 4,3 з,о 5? — — —
61Ni — 1,25 — — <0,25 — — — 1512
,63Cu 11,285 69,09 9,38 • 10 2 1,33 2,2206 3/2 -0,15 4952
,65Cu 12,090 30,91 0,116 1,42 2,3790 3/2 -0,14 5305
,67Zn 2,635 4,12 2,86-10“3 0,730 0,8735 5/2 — — 1251
,69Ga 10,218 60,2 6,93-10“2 1,201 2,0108 3/2 0,2318 — —
,71Ga 12,984 39,8 0,142 1,525 2,5549 3/2 0,1461 — —
,73Ge 1,485 7,61 1,40-10“3 1,15 -0,8768 9/2 -0,2 — —
,75As 7,292 100 2,51-10“2 0,856 1,4349 3/2 0,3 255 9582
438____________________Приложения
77Se 8,131 7,50 6,97-10“3 0,191 0,5333 1/2 — 376 13468
79Br 10,667 50,57 7,86-10“2 1,26 2,0990 3/2 0,33 646 21738
81Br 11,498 49,43 9,84- 10 2 1,35 2,2626 3/2 0,28 696 23432
85Rb 4,111 72,8 1,05 -10 2 1,13 1,3483 5/2 0,31 — 1012'
87Rb 13,932 27,2 0,177 1,64 2,7415 3/2 0,15 34171
87Sr 1,845 7,02 2,69 10 “3 1,43 -1,0893 9/2 —- —
89y 2,086 100 1,17 10'4 4,90-10“2 -0,1368 1/2 — — —
91Zz 4,0 11,23 9,4-103 1,04 -1,3 5/2 — —
93Nb 10,407 100 0,482 8,06 6,1435 9/2 — 0,4+ 0,3 — —
95Mo 2,774 15,78 3,22-IO'3 0,761 0,9099 5/2 — — -3528
97Mo 2,833 9,60 3,42-IO'3 0,776 -0,9290 5/2 — — -3601
"Tcr 9,583 — 0,376 7,43 5,6572 9/2 0,3 —
"Ru — 12,81 — — — 6/2 —
101Ru — 16,98 — — — 5/2 — —
103Rh 1,340 100 3,12-10“5 3,15-10“2 -0,0879 1/2 —
105Pd 1,74 22,23 7,79-10“ 4 0,47 -0,57 5/2 — —
107Ag 1,722 51,35 6,69-10 5 4,03-10“2 -0,1130 1/2 — — -3520
109Ag 1,981 48,65 1,01-IO'4 4,66-10“2 -0,1299 1/2 — -4044
11'Cd 9,028 12,86 9,54-10'3 0,212 -0,5922 1/2 —
"3Cd 9,444 12,34 1,09 10“2 0,222 -0,6195 1/2 —
115In' 9,329 95,84 0,348 7,23 5,5072 9/2 1,161 —
"7Sn 15,77 7,67 4,53-10“2 0,356 — 0,9949 1/2 —
"9Sn 15,87 8,68 5,18-10“2 0,373 - 1,0409 1/2 — —
121Sb 10,19 57,25 0,160 2,79 3,3417 5/2 -0,8
123Sb 5,518 42,75 4,57 10“2 2,72 2,5334 7/2 -i,o
125Te 13,45 7,03 3,16-10“2 0,316 -0,8824 1/2 —
127I 8,519 100 9,35-10“2 2,33 2,7939 5/2 -0,75 — —
1 29jr 5,669 — 4,96-10“2 2,80 2,6030 7/2 -0,43
177Hf — 18,39 — — — 1/2 или 3/2 — —
179Hf — 13,78 — — — 1/2 или 3/2 — — —
18'Ta 4,6 100 2,60-10“2 2,26 2,1 7/2 6,5 — —
Приложения____________________439
Изотопa Частота ЯМР для поля в 10 кЭ, МГц Содержание Относительная чувстви-fl природе, гельность для одинако- Магнитный момент в единицах ядерного магнетона (e/i/4irwc) Спин / в единицах h/2n Электричес- Анизотроп-кий квадру- ное сверхпольный мо-тонкое взаи- Изотропное сверхтонкое взаимодействие Ло, МГц”
7„ вого числа ядер при посто- при постоян-янном поле ной частоте
мент Q в единицах 10“24 ми2 модействие В, МГц6
183^ 1J5 14,28 6,98 -10"5 4,12 0,115 1/2 —
,185Re 9,586 37,07 0,133 2,63 3,1437 5/2 2,8 —
,187Re 9,684 62,93 0,137 2,65 3,1760 5/2 2,6 —
,189Os 3,307 16,1 2,24- I0 3 0,385 0,6507 3/2 2,0 —
‘“k 0,81 38,5 3,5-10“5 9,5- ИГ 2 0,16 3/2 -1,2 — —
0,86 61,5 4,2-10“5 0,104 0 17 3/2 -1,0
9,153 33,7 9,94 • 10 3 0,215 0,6004 1/2 — — —
197Au 0,691 100 2,14-ИГ5 8,1-ю;2 0,136 3/2 0,56
• 199Hg 7,612 16,86 5.72-103 0,179 0,4993 1/2 — —
• 201Hg 3,08 13,24 1,90 10"3 0,362 -0,607 3/2 0,5 —
203 24,33 29,52 0,187 0,571 1,5960 1/2 — — —
205y| 24,57 70,48 0,192 0,577 1,6114 1/2 —
,207Pb 8,899 21,11 9,13 10-3 0,209 0,5837 1/2 — —
,209Bi 6,842 100 0,137 5,30 4,0389 9/2 -0,4 —
Свободный электрон g = 2,00 27,994 — 2,85- Ю8 658 -1836 1/2 — — —
а Точка показывает, что магнитный момент измерен методом ЯМР
Гиромагнитное отношение можно определить из удг = цдг/Л/
Анизотропное сверхтонкое взаимодействие определяется как В— |/i 1$wPjV0₽<r-3>, где рассчитывается для валентных р-электронов из функций самосогласованного поля. Взаимодействия таковы, что главные тензора с нулевым следом составляют соответственно -1, -1 и +2, умноженные на приведенные числа
Изотропное сверхтонкое взаимодействие выражается как Ло = ~h~ | Фа(0) |2, где ф,(0)—величина S-волновой функции валентной оболочки в самосогласован-
ном поле у ядра нейтрального атома Д ГТ
Изотоп радиоактивен
е Экспериментально сверхтонкое взаимодействие изменяется с использованием метода пучков. [См. Kusch. Р., Taub H.f Phys. Rev., 75, 1477 (1949)]
Приложения
441
Некоюрые постоянные
Величина
Обоз- Постоянная
начение в единицах CGS в единицах СИ
к постоянная Больцмана 1,3807-10“16 >pi-K 1 1,3807-10-23 Дж-К-1
h постоянная Планка 6,6262-10 27 эрг-с 6,6262-10“34 Дж-с
с скорость света в вакууме 2,9979-10* 1 * ° см-с1 2,9979-108 м с-1
N число Авогадро 6,0220-1023 моль-1 1,6022-10“20 6,0220-10 3 моль
е заряд электрона ИЛИ 4,8032-10^10 1,6022-10“19 Кл
аем атомная единица массы 1,6606-10“ 24 г 1,6606-10“27 КГ
р магнетон Борг 9,2741-10“ 21 эрг-Э-1 9,2741-10“24 Дж-Т-1
Ра ядерный магнетон 5,0508-10“ 24 эрг-Э-1 5,0508-10“27 Дж-Т-1
те масса покоя электрона 9,1095 -10 28 г 9,1095-10“31 КГ
тр масса покоя протона 1,6726-10“24 г 1,6726-10“27 кг
магнитный момент электрона 9,2848-10“21 эрг-Э-1 9,2848-10“24 Дж-Т-1
л 3,14159 26535 89793 23846 26433 83279 50288
1 Т обозначает Тесла (кг с 2-А Кл обозначает кулон; 1 эрг — = 1 г см2-с-2.
Переводные коэффициенты
1 кал = 4,184 Дж (Джоулей)
1 эВ = 1,6022-10"19 Дж
1 см-1 = 1,9865 10 “ Дж
1 Гц = 6,6262 10“31 Дж
1 Кл = 1,3807 10“27 Дж
1 кВт = 3,6 • 106 Дж
1 эрг = 1 • 10“7 Дж
предметный указатель
Абсорбция связи в ЯМР 1, 261
Автокорреляционная функция 1, 310
Амбидентатные анионы 1, 235
Ангармонические колебания 1, 176
Анизотропный вклад
в химический сдвиг 1, 291
соседнего атома 1, 381
Анизотропный эффект 2, 30-42
Анизотропия
сверхтонкого взаимодействия 2, 34
соседних групп 1, 292
^-фактора 2, 30
Антистоксова линия 1, 195
Антиферромагнетизм 2, 131
Асимметрии параметр 1, 300
Асимметрическая ячейка 2, 394
Асимметричное колебание 1, 183
Асимметричный волчок 1, 226
Ассоциативный закон 1, 32
Атомная заселенность 1, 93
суммарная 1, 93
Базис векторов 1, 50
Биномиальное разложение 1, 283
Блоха уравнение 1, 250, 257
Больцмана распределение 1, 257
Борна-Оппенгеймера приближение 1, 112
Броуновское движение 1, 310
Брэгга соотношение 2, 375
Валентное колебание 1, НО
симметричное 1, 183
CN 1, 234
Вант-Гоффа уравнение 1, 117
Ван-Флека уравнение 1, 138, 2, 142
Вариационная энергия 1, 71
Вариационный принцип 1, 71
Вектор
намагниченности 1, 253
среднего расстояния 1, 143
электрического дипольного момента
1, 143
Векторное произведение 1, 40, 251
Векторы 1, 39
умножение 1, 40
Вероятность переходов 1, 266
«Ветвистый спектр» 1, 224, 282
Взаимодействие
виброиное 1, 147
виртуальное 1, 296
зарядов и квадруполей 2, 260
через пространство 1, 286
L S 2, 67
Винтовая ось 1, 23; 2, 362, 364
Вклад в сверхтонкое взаимодействие
2, 222
Внешний контроль 1, 272
Внешний стандарт 1, 274
Внутренний контроль 1, 272
Внутренний стандарт 1, 274
Возбуждения, одновременные парные
электронные 2, 118
Возбужденные состояния 1, 163
Волновое число 1, 108
Волновые функции
определение 1, 69
основа для неприводимых представ-
лений 1, 75
произведения 1, 102
свойства, выведенные из 1, 90
симметризованные, 1, 103
средняя величина 1, 69
Вольфсберга—Гельмгольца формула 1,
92
Вращательные переходы 1, 210
Вращательные состояния 1, 112
Вращательные спектры КР 1, 213
Вращающаяся система координат 1,
252
Вращающийся остов 1, 252
Временная зависимость 1, 263
Время жизни 1, 268, 335
электронного спинового состояния
2, 164
Время корреляции 1, 310
Время релаксации
винилацетата 1, 312
Предметный указатель
443
измерение (фурье-спектроскопия
ЯМР) 1, 307
определение 1, 255, 311
поперечной 1, 255, 313
продольной 1, 255
электронной 2, 165
«Встряхивания» процесс 2, 353
Вторая координационная сфера 2, 189
Вырожденность терма 2, 63
Вырожденные представления 1, 54
Гармоники сферические 2, 71
Гармонические колебания 1, 176
Гармоническое движение 1, 176
Геометрические преобразования ем.
Представления
Гибридизация лиганда 2, 232
Гипсохромный сдвиг см. Сдвиг в синюю область
Гиромагнитное отношение 1, 250
Гойе метод 2, 155
Градиент
поля 1, 299
электрического поля 2, 263, 270
Групповые колебания 1, 190
Дипольное взаимодействие 1, 389; 2, 171
гамильтониан 1, 390
определение 1, 311
Дипольные моменты 1, 26
Дипольный или псевдоконтактный вклад 2, 171
разделение 2, 173
Дирака дельта-функция 1, 391
Дисперсия оптического вращения
(ДОВ) 1, 163
кривая 1, 164
Диссимметрия 1, 25
Дистанционные эффекты 1, 381
Дифракция в кристалле 2, 375, 386
Дифференциальное перекрывание 1, 99
Диэдрические плоскости 1, 15
Длина волны 1, 108
Донорные свойства
аминов 1, 160
карбонильных соединений 1, 159
сульфоксидов, сульфонов и сульфатов 1, 159
Доплеровский сдвиг 2, 285
Доплеровское уширение 2, 285
Двойникование 2, 404
Двойной резонанс 1, 365
электрон-электронный 2, 249
электрон-ядерный (ДЭЯР) 2, 249
Двойные группы 2, 84
Делокализация
в сигма-системе 1, 96
неспаренного электрона 2, 177
спина 2, 179
Деполяризованные линии в спектре
КР 1, 201
Десорбция полем 2, 326
Деформационные колебания 1, ПО,
183
Диагонализация 2, 32, 37
Диагонализованная матрица 1, 41
Диагональные элементы 1, 41
Диаграммы симметрии 2, 374
Диамагнетизм 2, 131, 132
Диамагнитная восприимчивость 1,
274; 2, 132
Диапазон
медленного обмена 1, 335
почти быстрого обмена 1, 335
прекратившегося обмена 1, 335
промежуточного обмена 1, 335
Q 2, 6
X 2, 6
Диастереотопные ядра 1, 285
Димер пиразина рутения 2, 120
Единицы полной ионизации 2, 316
Единичная матрица 1, 41
Жоба метод 1, 125
Запрещенные переходы 1, ИЗ
Запрещенные по спину переходы 2, 89
Заселенность перекрывания 1, 93
Зеемана эксперимент 1, 267
Зеемановские члены второго порядка
2, 139
Зеркальная плоскость 1, 13; 2, 366
Зеркальное отражение 1, 25
Зеркально-поворотная ось 1, 15
Зеркальные плоскости 2, 366
Идентификации метод 1, 117, 125, 154
Излучение 1, 107
Изобестические точки 1, 121
Изомерный сдвиг
интерпретация 2, 289
описание 2, 289
р-электронная плотность 2, 291
s-электронная плотность 2, 291
Изотопный сдвиг 2, 166
Изохронные ядра 1, 285, 363
ИК-активные колебания в SF4 1, 208
29'
444
Предметный указатель
ИК-спектр 1гСОС1[Р(С6Н5)3Ъ-О2 1, 236
IrH2CO[P(C6H5)3J2 1, 236
IrN2Cl[P(C6H5)3]2 1, 236
М(СО)5Х 1, 238
ИК-спектроскопии метод 1, 213 использование симметрии 1, 204 определение структуры 1, 227 применение 1, 213 пропускание растворителей 1, 216
Импульсная развязка 1, 404
Импульсный ЯМР 1, 307
Инверсии центр 1, 10
Интеграл
момента перехода 1, 142
перекрывания 1, 71
Интегральная интенсивность 1, 142
Иода комплексы молекулярные 1, 157
Ионизации полем метод 2, 325
спектрометр 2, 316
Ион-молекулярные реакции 2, 319
Ионные пары
образование 2, 176
эффект сольватации 2, 188
ЯМР 2, 188
Ионный циклотронный резонанс
(ИЦР) 2, 329
спектрометр 2, 329
Исследование монокристалла PtClJ" поляризованным светом 1, 154
Карбонильные соединения, УФ-спектр 1, 155
Каша формула 1, 92
Квадратная матрица 1, 41
Квадратноволновая функция 1, 302
Квадрупольное ядерное взаимодействие 2, 45
Квадрупольное ядро 2, 260
Квадрупольный момент 1, 248, 299
Квадрупольный электрический момент 2, 260
Квазиравновесная теория 2, 322
Квантовомеханическое рассмотрение расщепления 1, 354
Кинетическое выражение для полной формы линии 1, 333, 338
Класс 1, 24, 37
Кнудсена ячейка 2, 327
Ковалентность 2, 176
Колебания
деформационные см. Деформационные колебания
решетки 1, 217
Колебательные энергетические состоя-
ния 1, 110
Комбинационного рассеяния спектроскопия (КР) 1, 193 поляризованные линии 1, 199 правило отбора 1, 197 применение 1, 213 резонансного 1, 201 эксперимент 1, 193
Коммутация 1, 24
d1 и «/’-Комплексы 2, 89
Кондона—Шортли параметры 2, 65 Константа
взаимодействия в NO2 2, 42
-электронного спина с ядерным 2, 168
-изотопного сверхтонкого в NO2 2, 42
-обменного 2, 151
- спин-спинового 1, 280-295
равновесия из спектральных данных 1, 119
сверхтонкого расщепления в изотропных средах 2, 23
экранирования 1, 261
Контактное ферми-взаимодействие см. Ферми-взаимодействие контактное
Контактный сдвиг интерпретация 2, 180 расчет 2, 180
Конфигурации d2, d3, d\ d8 2, 91 Конфигурационное взаимодействие 1, 139
Конфигурация микросостояния 2, 62 Корреляционная функция 1, 310 Косвенный механизм 2, 24
Коттона—-Крейнханцеля приближение 1, 240
Коэффициент
орбитальной редукции 2, 147 экстинкции 1, 117
Крамерса дублет 2, 203 теорема 2, 203
Кривая эффективности ионизации 2, 328
Кристаллического поля теория 2, 71, 72. См. также Резонирующее кристаллическое поле
Кришнана метод, критический момент вращения 2, 156
Круговой дихроизм (КД) 1, 163, 164 спектр Ni(Ph)3+ 1, 166
Кубическая решетка 2, 361 Кулоновские интегралы 1, 71 Купманса теорема 2, 336 Кюри-Вейса закон 2, 136 Кюри закон 2, 136 Кюри температура 2, 132
Предметный указатель
445
Ламберта-Бера закон 1, 117
Ларморова частота 1, 252, 258
ЛКАО-МО метод см. Метод ЛКАО-
МО
Локальная симметрия 1, 147
Лорентцева функция 1, 261
Магический пятиугольник 2, 213
Магнетизм 2, 130
Магнитогирическое отношение 1, 250
Магнетометр 2, 156
Магнитная неэквивалентность 1, 363
Магнитная поляризация 2, 130
Магнитного диполя оператор 1, 166
правила отбора 1, 166
переход 1, 151
Магнитной восприимчивости измерение 2, 155
Магнитный круговой дихроизм 1, 163,
167
Магнитный момент 2, 134
ионов редкоземельных металлов 2, 147
с учетом только спина 2, 138
Мак-Клюра определение параметров
5- и 5-связывания 2, ПО
Масс-спектров интерпретация 2, 320
группы изотопов 2, 324
ионизация полем 2, 323
потенциалы ионизации 2, 328
применение для идентификации 2,
320
процессы ионизации 2, 318
процессы перегруппировки 2, 319
теплоты сублимации 2, 327
фрагментация 2, 318
Fe[(C2H5)2NCS2]3 2, 324
Масс-спектрометр
принцип действия 2, 313
разрешение 2, 315
Масс-спектрометрическое правило
сдвига 2, 323
Масс-спектрометрия высокого разрешения 2, 324
в сочетании с газожидкостной хроматографией 2, 323
Матрица
обмена 1, 348
ориентации 2, 396
Матрицы 1, 41
ортогональные 1, 48
сопряженные 1, 43
Матричная изоляция 1, 218
Матричное умножение 1, 43
Матричные элементы 1, 186, 266
Межатомные кольцевые токи 1, 383
Мёссбауэровская спектроскопия
в биологии 2, 303
вклады точечных зарядов 2, 306 гамильтониан для квадрупольного взаимодействия 2, 291
градиент поля 2, 293
железокарбонильных соединений 2 302
железосерусодержащих соединений 2, 304
железосодержащих соединений 2, 299
интерпретация изомерных сдвигов 2, 289
комплексов диметилового эфира протопорфирина с 57Со 2, 296 магнитные взаимодействия 2, 294 оловянных соединений 2, 301 описание 2, 285
относительные скорости 2, 288
пероксидаз хрена 2, 303
применения 2, 297
ферредоксина 2, 303
ферромагнитных веществ 2, 295
ферроцена 2, 309
эмиссионная 2, 295
Na,Fe(CN)5NO 2, 300
Fc3 + Fein(CN)& 2, 288
[Fe(CN)5NH3]3- 2, 297
Fe(II)X2(CO)2P2 2, 303
Металломеры 2, 120
Метка с помощью насыщения спина 1, 369
Метод
ab initio 1, 72
внешнего стандарта 1, 274
внутреннего стандарта 1, 274
двойного резонанса 2, 279
ИЦР 2, 330
ЛКАО-МО, 1, 70
отношения 2, 175
побочных линий 1, 273
снижения симметрии 2, 79
спин-эха 2, 281
ССП 1, 97
ССП-ЧПДП 1, 96
тяжелого атома 2, 398
ЧПДП 2, 29
Микроволновая спектроскопия 1, 210
Микросостояния 2, 63
Миллера
индексы 2, 377
плоскости 2, 377
Модель
углового перекрывания 2, 111
электрического поля 1, 386
Молярная эллиптичность 1, 164
Молярный коэффициент экстинкции 1, 117, 140
446
Предметный указатель
Моноклинная решетка 2, 361
Монохроматическое излучение 1, 107
Морзе кривая 1, 128
Мультиплетность 1, 133
Накопление сигнала (САТ-экспери-мент) 1, 301
Направляющие косинусы 2, 32
Насыщение 1, 261
Недиагональные представления 1, 51, 77
Недиагональные элементы 1, 41, 77
Нееля температура 2, 132
Несобственное вращение 1, 15, 48
Неспарениый электрон см. Делокализация
Нефелауксетический эффект 2, 95
«Нечетный» переход 1, 145
Неэквивалентные протоны 1, 284
Нормально-координатный анализ 1, 185
Нормальные колебания 1, 110, 181
Нормальные координаты 1, 111
Нормальные моды 1, 110
Нормирование 1, 79
Нулевая частота 1, 269
Нулевая частотная компонента 1, 316
Обмена лигандов реакция 1, 341
Обменное сужение 2, 49
Образование пар 2, 318
Обратная решетка 2, 376
Обратный элемент 1, 32
Оверхаузера эффект 1, 372
ядерный 1, 372
Одноэлектронные молекулярные ор-
„ битали 1, 71
12-Оператор 1, 256, 265
12-Оператор 1, 265
Операторы 1, 68
винтовой 2, 362
Гамильтона 1, 70
диполя перехода 1, 206
коммутации правило 1, 265
магнитного диполя 1, 166
проекционные 1, 78
свойства 1, 264
сдвига 2, 12
скольжения 2, 362
трансляции 2, 362
углового момента 1, 69, 250
центрирующий 2, 366
Оптическая активность 1, 25
Оптической чистоты определение 2,
198
d- и р-Орбиталей смешивание 1, 150 действительные волновые функции 2, 71
«/-Орбиталей энергия 2, 71
Орбитально-вырожденное состояние 2, 91
Оргела диаграммы 2, 83
Основная частота 1, 178
Ось вращения 1, 11
Отдача энергии см. Энергия отдачи
Относительные координаты 2, 392
Отношение деполяризации 1, 201
Отпечатков пальцев метод см. Идентификации метод
Парамагнетизм 2, 131, 134
Парамагнитное экранирование 1, 375
Парамагнитный член 1, 376
Параметр
асимметрии 2, 262
межэлектронного отталкивания 2, 99 обмена 2, 151 связывания 2, 109
Z 1, 161
Парциальное квадрупольное расщепление 2, 305
Паскаля константы 2, 133
Паскаля треугольник 1, 283
Паттерсона функция 2, 398
Перенос электрона между двумя центрами с различной степенью окисления 2, 120
Переходы
нечетный 1, 145
разрешенные 1, 113
узкий двухквантовый 2, 222
п->л* 1, 133
п -> ст* 1, 133
л -* Л* 1, 133
ст —* ст* 1, 133 0 -> 0 1, 131
Планка постоянная 1, 107
Плоскополяризованный свет 1, 163
Плоскость скольжения 2, 362, 366
Плотность неспаренного электрона 2, 24, 26
Податливости постоянная 1, 189
Подгруппы 1, 35
Н j-Поле 1, 266
Полихроматическое излучение 1, 107
Полное колебательное состояние 1, 111
Полное представление определение 1, 49 получение 1, 61 разложение 1,61
Поляризованное излучение 1, 107, 199
Предметный указатель
447
Порядок групп 1, 33 связи 1, 91 спектра 1, 298, 352, 362 условное обозначение 1, 363
Полярность растворителей 1, 161
Потенциал возникновения фрагмен-
тарного иона 2, 318, 328
Потенциальной энергии кривая 1, 128
Правило отбора
в ИК-спектроскопии 1, 178
-спектроскопии КР 1, 195
коммутации 1, 265
общее 1, 113, 145, 166
правой руки 1, 250
применение для неорганических сое-
динений 1. 227
Предел крайнего сужения 1, 311
Представления
геометрические преобразования 1, 44
— недиагональные 1, 51
— неприводимые 1, 49
получение и разложение 1, 61
Преобразование подобия 1, 36, 44, 52
Приближение
гармонического осциллятора 1, 184
замыкания 1, 379
сильного поля 2, 71
слабого поля 2, 71
Приведенная масса 1, 181
Примитивные решетки 2, 367
Принцип неопределенности 1, 114, 268
Программы
автоиндексирования 2, 383
MULTAN 74 2, 403
Проекции молекулярных орбиталей 1,
76
Проекционные операторы 1, 78
Произведения
волновых функций 1, 102
операций симметрии 1, 23
прямые 1, 64
Производная кривой спектра ЭПР 2,
15
Пространственная группа 1, 23
Пространственная симметрия 1, 23
Протонный резонанс органических со-
единений 1, 262
Протоны неэквивалентные 1, 284
Прочность связи 1, 230
Прямая делокализации 2, 177
спинового комплекса кобальта(П) 2, 174
Прямое дипольное взаимодействие 1, 392
Прямые методы 2, 403
Псевдоконтактный сдвиг 2, 171
Работа выхода 2, 334
Равновесие высокий спин-низкий
спин 2, 154
Радикал СН3 2, 42
Радикал CF3 2, 42
Развязка 1, 365
неполная 1, 403
Разложения формула 1, 61
Размер цикла, влияние на донорные свойства 1, 160
Разностная полоса 1, 183
Разрешенные переходы см. Переходы
Рака параметры 2, 65
Рассела-Саундерса взаимодействие 2, 67
Рассеяния фактор 2, 391
Расчет Dg 2, 94
величина 2, 98
влияние спин-орбитального взаимодействия 2, 95
определение 2, 72
Расчеты
аллильного радикала 1, 87 молекулярных орбиталей 1, 87
Расщепление. См. также Квантовомеханическое рассмотрение расщепления
в нулевом поле 2, 43
сверхтонкое см. Сверхтонкое расщепление
спин-спиновое см. Спин-спиновое расщепление
тонкое см. Тонкое расщепление
Регенеративные схемы 2, 265
Резонансного комбинационного рассеяния спектроскопия 1, 201
Резонансные интегралы 1, 71
Резонансный захват 2, 318
Резонирующее кристаллическое поле 2, 215
Релаксационные процессы 1, 253, 268, 315
для парамагнитных систем 2, 164
Релаксация 1, 268
межмолекулярная, агент 1, 403
механизм 1, 268, 269
нулевая частота 1, 269
поперечная 1, 269
-время 1, 255
продольная, время 1, 255
Релеевское рассеяние света 1, 195
Рентгеновская кристаллография 2, 360 программа приведения 2, 398
Рентгеновская фотоионизация 2, 332
Рентгеновская фотоэлектронная спектроскопия (РФС) 2, 331
448
Предметный указатель
спектр 2, 332, 353
химические сдвиги 2, 346
Рентгеновской дифракции межа-
томный вектор 2, 399
Ромбическая решетка 2, 361
Ромбическая структура комплекса 2,
100
CAT-эксперимент см. Накопление сигнала
Сверхрегенеративная схема 2, 265
Сверхтонкое расщепление 2, 218
аллильного радикала 2, 27
в изотропных системах 2, 23 электрона на d-орбиталях 2, 223 — /^-орбиталях 2, 223
Свободноионный гамильтониан 2, 71
Сдвигающие реагенты, изомерные со-
единения 2, 197
нуклеотиды 2, 198
определение 2, 191
фосфолипидные слои 2, 198
Сдвиг в синюю область (гипсох-ромный сдвиг) 1, 140
Секулярный детерминант 1, 72, 266
Сила
диполя 1, 142
осциллятора 1, 142
Силовая постоянная 1, 176, 178
взаимодействия 1, 186
Символ SPE 2, 120
Символы решеток 2, 368
Симметризованная волновая функция см. Волновая функция
Симметричный волчок 1, 226
Симметрия
в квантовой механике 1, 75 молекулярных орбиталей 1, 204 операции 1, 9, 23 определение 1, 9
особые точки, название 2, 372
положения 1, 217
твердого состояния 2, 361
элементы 1, 9, 10
СИС-интерферограмма 1, 305
Система АВ 1, 356
матрица элементов 1, 358
Система АХ 1, 353
Систематические погасания 2, 384
Скалярный сдвиг 2, 176
Слабое и сильное поле, корреляция 2,
82
След матрицы 1, 41
Слейтера детерминант 1, 103
Смешивание
более высоколежащих состояний 1, 138
d- и р-орбиталей 1, 150
Собственная ось вращения 1, 11
Собственная функция 1, 69
Собственные значения 1, 69
Соединения смешанной валентности 2, 121
Соотношения Е и С 1, 232
Сопряженные матрицы см. Матрицы
Сопряженные элементы 1, 36
Соре-полоса 2, 109
Составные полосы 1, 183
Спада свободной индукции кривая 1, 263
АВ-спектр 1, 361
Спектральная плотность 1, 315
Спектрохимический ряд 2, 99
Спектры
второго порядка, примеры 1, 298, 352, 362
газов 1, 224
поглощения поляризованного излучения 1, 152
Спина делокализация в пиридине 2, 179
механизм 2, 179
примеры 2, 180
Спин-гамильтониан 2, 9, 49 атома водорода 2, 9
Спиновая плотность 2, 23
Спиновая температура 2, 281
Спиновое эхо, эксперимент 1, 313. См. также Метод спин-эха
Спин-орбитальное взаимодействие 1, 138, 147
в свободных ионах 2, 67
энергия 2, 65
Спин-орбитальные эффекты 1, 389
Спин-поляризация 2, 24
Спин-решеточная релаксация 1, 254, 268. См. также Релаксация
Спин-спиновая релаксация 1, 254. См. также Релаксация
Спин-спиновое взаимодействие 2, 47 влияние числа и природы связей 1, 285
квантовомеханическое рассмотрение 1, 354
константа 1, 280
механизм 1, 288, 389
определение знака констант 1, 368
-структуры 1, 289
Спин-спиновое расщепление 1, 280
Спин-спиновый обмен 1, 269
Средняя поляризация электронных спинов 2, 166
ССП-метод см. Метод
ССП — ЧПДП-метод см. Метод
Стационарный эксперимент 1, 258
Предметный указатель
449
Структурный фактор 2, 393
Сульфат-ион 1, 237
Сфера отражения 2, 381
Сферический волчок 1, 226
Таблицы умножения для групп 1, 33
Танабе-Сугано диаграммы 2, 82
Таунса-Дейли выражение 2, 272
Температурный фактор 2, 401
Тензор
А 2, 37
анизотропного сверхтонкого
взаимодействия для ядра 13С 2, 39
градиента поля 2, 262
градиента электрического поля 2, 263
поляризуемости 1, 195
расщепления в нулевом поле 2, 44
экранирования, расчет 1. 395
^-Тензора диагонализация 2, 32
определение 2, 5, 37
Теория
возмущений 1, 73
групп 1, 31
молекулярных орбиталей 1, 68
поля лигандов 2, 74
Тепловое движение 2, 401
Терм
символы 2, 62
А, кривая 1, 168
В, кривая 1, 168
С, кривая 1, 168
2D 2, 63
3Р 2, 65
Термотропные жидкие кристаллы 1, 396
Термы свободного иона 2, 67
Тетрагональное искажение 2, 100
Тетрагональное растяжение, сжатие 2, 87
Тиклинг-резонанс 1, 365
Тождественное преобразование 1, 11, 32
Тонкая структура 1, 130
Тонкое расщепление 2, 221
Точечное произведение 1, 40
Точечные группы в классификации мо-
лекул 1, 21
определение 1, 18
Тригональное искажение 2, 100
Триклинная решетка 2, 361
Угловой момент 1, 249, 250
Удельная эллиптичность 1, 164
Удельное вращение 1, 164
Узкий двухквантовый переход см.
Переходы
Ультрафиолетовая фотоэлектронная спектроскопия (УФС) 2, 331
спектры 2, 340
Ультрафиолетовый спектр, требование по симметрии 1, 132
3-Факторы
интерпретация 2, 209
системы с S = 1/2 2, 211
тетрагональный комплекс 2, 213
шестикоординационные комплексы
меди(П) 2, 214
Фарадея метод 2, 156
Ферми-взаимодействие, контактное 1,
390; 2, 12
гамильтониан 1, 391
механизм 1, 288
Ферми-резонаис 1, 184
Ферми-уровень 2, 334
Ферромагнетизм 2, 131
Ферроцитохром, УФ-спектр 1, 203
Фоновые колебания 1, 217. См. также
Колебания
Формальный заряд 1, 91, 93
Фотоионизация электронов оболочки
2, 335
Фотоны 1, 107
Фотохимические реакции 2, 123
Фотоэлектронная спектроскопия
газообразного азота 2, 338
моноокиси углерода 2, 338
прибор 2, 334
сродство к протону 2, 351
теплоты образования 2, 351
химический сдвиг 2, 332
Фотоэлектронный спектр 2, 332
колебательная структура 2, 340
образование О/ 2, 343
«смешанная валентность» соедине-
ния 2, 352
УФ 2, 333, 340
структура NjOj- 2, 349
Франка-Кондона принцип 1, 130
Функция
двугранного угла 1, 293
формы линии 1, 266
Фурье-карта 2, 101
Фурье ряды 1, 302; 2, 398
Фурье-спектроскопия ЯМР 1, 264
принципы 1, 301
Характеров таблицы 1, 50, 57
Харкера линия 2, 399
Химический сдвиг 1, 270
интерпретация 1, 384
450
Предметный указатель
локальные вклады 1, 373 оптические изомеры 1, 388 применение 1, 276 электроотрицательность 1, 293
ХПЯ 1, 373
Хюккеля метод 1, 86
Хюккеля расчеты молекулярных орбиталей 1, 86
Хюккеля расширенный метод для металлоценов и комплексов бнс-аре-нов 1, 92, 96
Центрированные решетки 2, 366
Центровые сдвиги 2, 289
Центр симметрии 1, 10
Циклическая группа 1, 34
Частота основного колебания 1, 177
Частотная зависимость 1, 263
Частотные компоненты 1, 316
«Четный» переход 1, 145
Ширина спектральной линии 1, 268,
269
естественная 1, 114
Шкала т 1, 273
Шредингера уравнение 1, 70
Штернгеймера антиэкранирование 2,
274
Штернгеймера эффект 2, 273
Эквивалентная ориентация 1, И
Эквивалентные атомы 1, 24
«Эквивалентные оболочки» 2, 349
Эквивалентный набор 1, 24
Экранирование
влияние растворителей 1, 274
локальный диамагнитный вклад в 1, 375
Электронная абсорбционная спектро-
скопия 1, 128
Электронная корреляция 1, 98
Электронная плотность 1, 90
Электронная спектроскопия, примене-
ние 1, 154
Электронное обменное взаимодей-
ствие 2, 25
Электронное состояние 1, 110
Электронные переходы
влияние растворителей на 1, 161
интенсивность 1, 142
симметрия 1, 135
Электронные состояния свободных ио-
нов 2, 62
Электронные спиновые функции 2, 5
Электронный спектр 2, 88
влияние тетрагонального искажения 2, 103
данные о структуре 2, 105
иона ванадила 2, 108
Электрон-электронные взаимодействия 2, 62
Электрон-электронный двойной резонанс 2, 52
Электрон-ядерный двойной резонанс 2, 52
кобальтаЦП) 2, 101
комплексов никеля 2, 93, 104
металлоферментов 2, 108
октаэдрического трпс-(этилендиа-
мин)кобальта(Ш) 2, 102
шестикоординационного димера
с кислородным мостиком 2, 119
Элементарная ячейка 2, 361
диаграммы 2, 273
Электронный парамагнитный резонанс (ЭПР)
анализ систем меди(П) 2, 231
трис-(бипиридил)железа(ПГ) 2, 176
влияние ковалентности в комплексе
2, 229
-электронного спинового обмена 2, 48
внешний стандарт 2, 16
Зеемана гамильтониан 2, 5
квадрупольное взаимодействие 2, 45
константы скорости 2, 48
применения 2, 50
спектр биологических систем 2, 52
-в жидких кристаллах 2, 207
-О2-аддуктов с комплексами Со(И)
2, 244
-витамина В12 2, 216
-димера меди(11) 2, 247
- бпс-(диселенокарбамата)меди(П) 2, 216
-d1 -комплексов 2, 233
-d2-комплексов 2, 234
-d3-комплексов 2, 235
-«(“-комплексов 2, 236
-«(’-высокоспиновых комплексов 2, 239
-d6 -низкоспиновых комплексов 2, 236
-d6-комплексов 2, 243
-«(’-комплексов 2, 243
-ds-высокоспиновых комплексов 2, 245
-«(’-комплексов 2, 245
-комплексов железа(Ш) 2, 239
— меди(П) 2, 52
— хромаЦП) 2, 236
Предметный указатель
451
-трис-(комплексов)меди(П) 2, 215
-низкоспиновых d’-систем 2, 244
-нитратных комплексов 2, 244
-октаэдрического Мп(П) 2, 220
- поликристаллических образцов 2, 207
-порошков 2, 205
-порфириновых комплексов меди(П)
2, 214
-радикала малоновой кислоты 2, 41
-регистрация 2, 15
- бис-(салицилальдимин)меди(П) 2, 21
-систем с S = 1/2 2, 211
-систем с сильным спин-орби-тальным взаимодействием 2, 216
-фосфорных лигандов 2, 233
стекла 2, 7
теория поля лиганда, интерпретация g и 4-тензоров 2, 225
триплетные состояния 2, 42
ЧПДП метод см. Метод ЧПДП
ширина линии 2, 47
эксперимент 2, 6
Энергетические уровни квадруполя
в аксиально-симметричном поле 2, 264
с I = 1 2, 268
Энергия
вертикальной ионизации 2, 333
квадрупольных переходов 2, 263
отдачи 2, 285
перехода 1, 161
связи электронов 2, 349
Энтальпия
ассоциации 1, 159
образование комплексов донор -12
1, 160
Эрмитова операция 1, 68
Эффект реакционного поля 1, 387
Эффективный спин-гамильтониан 2, 9
Эффективный спин S' 2, 222
Ядерная квадрупольная релаксация 1, 288, 300
Ядерное сверхтонкое взаимодействие 2, 9, 12
Ядерное сверхтонкое расщепление 2, 9
Ядерное спиновое квантовое число 1, 248
Ядерный квадрупольный момент 2, 261
Ядерный квадрупольный резонанс (ЯКР)
адиабатическое размагничивание 2, 280
двойного резонанса методы 2, 279
спектры, влияние магнитного поля 2, 269
-BrCN 2, 278
-двухатомных галогенидов 2, 274
-иода твердого 2, 278
-комплексов кобальта 2, 276
-карбонилов марганца 2, 275
-НЮ3 2, 277
-М.(А/С/4)„ 2, 279
-М2С110 2, 277
-МСТ6 2, 275
-R3MX2 2, 279
эксперимент 2, 264
Ядерный магнитный момент 1, 248
Ядерный магнитный резонанс (ЯМР) биномиальное разложение 1, 283 влияние внутримолекулярного обмена 1, 298
-квадрупольных ядер 1, 299
-химических реакций 1, 297
внутримолекулярные перегруппировки 1, 345
идентификация соединений 1, 278
импульсный 1, 307
кинетика 1, 333
константы скорости и энтальпии активации 1, 333
на ядрах фосфора 1, 248, 276, 279
— фтора 1, 279
несимметричное слияние линий в спектре 1, 345
определение порядков реакции 1, 339
Паскаля треугольник 1, 283
переход 1, 255
правило расщепления 1, 283
применение в кинетических исследованиях 1, 341
растворов жидких кристаллов 1, 395
расчет термодинамических параметров 1, 332
сдвиги линий 2, 163
спектр диастереомеров 2, 188
-0-дикетонов 2, 191
-квадрупольных ядер 1, 314
-парамагнитных комплексов 2, 176
-парамагнитных металлоценов 2, 180
- поли(1-пиразолил)бораткобаль-та(П) 2, 190
-энантиомеров 2, 188
-этанола 1, 280
-(Ви^)(СоРРк313) 2, 190
~(CH3NH2)6 Nii+ 2, 180
-Со {[4,6-(СЯ3)2 С6Я4]3}2 +
-FPO(OH)2 1, 289
-F2PO(OH) 1, 289
2, 186
-HPF2 1, 287
452 Предметный указатель
-НРО(ОН}2 1, 289
-HRh(CN)s 1, 278
-P4S3 1, 289
твердых тел 1, 393
усредненная обменом линия 1, 297
ширина полосы 1, 270
элементарные аспекты 1, 248
3С ЯМР
времена релаксации 1, 311
корреляционная диаграмма 1, 402
магнитный резонанс 1, 401
применение 1, 311, 401
сверхтонкого взаимодействия константа 2, 41
экранирования локальный парамагнитный член 1, 376
ЯМР-гамильтониан 1, 255
ЯМР-эксперимент 1, 259
квантовомеханическое описание 1, 264
Ядерный g-фактор 1, 255
Яна-Теллера искажение 2, 50, 215
Яна-Теллера эффект 2, 86
ОГЛАВЛЕНИЕ
ГЛАВА 9. СПЕКТРОСКОПИЯ ЭЛЕКТРОННОГО ПАРАМАГНИТНО-
ГО РЕЗОНАНСА...................................................... 5
Введение ......................................................... 5
9.1. Основные принципы метода ЭПР........................ 5
Ядерное сверхтонкое расщепление................................... 9
9.2. Атом водорода....................................... 9
9.3. Регистрация спектров ЭПР.......................... 15
9.4. Сверхтонкое расщепление в изотропных системах, состоящих более чем из одного ядра............................ 17
9.5. Вклады в константу сверхтонкого расщепления в изотропных системах........................................ 23
Анизотропные эффекты............................................. 30
9.6. Анизотропия g-фактора.............................. 30
9.7. Анизотропия сверхтонкого взаимодействия............ 34
9.8. ЭПР триплетных состояний.......................... 42
9.9. Квадрупольное ядерное взаимодействие .'............ 45
9.10. Ширина линии ЭПР.................................. 47
9.11. Спин-гамильтониан................................. 49
9.12. Примеры применения................................ 50
Список цитируемой литературы..................................... 53
Упражнения....................................................... 54
ГЛАВА 10. ЭЛЕКТРОННАЯ СТРУКТУРА И СПЕКТРЫ ИОНОВ ПЕ-
РЕХОДНЫХ МЕТАЛЛОВ...................................... 62
Введение......................................................... 62
Электронные состояния свободных ионов............................ 62
10.1. Электрон-электронные взаимодействия и обозначения термов ..................................................... 62
10.2. Спин-орбитальное взаимодействие в свободных ионах 67
Кристаллические поля............................................. 71
10.3. Влияние лигандов на энергии d-орбиталей............ 71
10.4. Аспекты симметрии d-орбитали, расщепляемой лигандом 75
10.5. Двойные группы..................................... 84
10.6. Эффект Яна — Теллера............................... 86
Применение....................................................... 88
10.7. Обзор электронных спектров комплексов .... 88
10.8. Расчет Dq и 0 для „(^’’-комплексов Ni11............ 94
454
Оглавление
10.9. Влияние искажений на энергетические уровни d-орбиталей 100
10.10. Данные о структуре, получаемые из электронных спектров 105
Параметры связывания, получаемые из спектров...................... 109
10.11. Параметры а- и л-связывания, получаемые из спектров тетрагональных комплексов.............................. 109
10.12. Модель углового перекрывания....................... 111
Некоторые вопросы, связанные с электронными переходами.............118
10.13. Одновременные парные электронные возбуждения ... 118
10.14. Полосы переноса электрона между центрами с различной степенью окисления..................................... 120
10.15. Фотохимические реакции............................. 123
Список цитируемой литературы...................................... 123
Упражнения........................................................ 125
Глава 11. Магнетизм............................................... 130
11.1. Введение........................................... 130
11.2. Виды магнетизма.................................... 132
11.3. Уравнение Ван-Флека................................ 138
11.4. Использование данных измерения восприимчивости . 147
11.5. Внутримолекулярные эффекты......................... 150
11.6. Равновесие высокий спин — низкий спин.............. 154
11.7. Измерение магнитной восприимчивости................ 155
Список цитируемой литературы...................................... 157
Упражнения........................................................ 158
ГЛАВА 12. СПЕКТРЫ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА ПАРАМАГНИТНЫХ КОМПЛЕКСОВ ИОНОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ........................................................ 163
12.1. Введение..................................... 163
12.2. Процессы релаксации.......................... 164
12.3. Средняя поляризация электронных спинов....... 166
12.4. Скалярный, или контактный, сдвиг в системе с изотропным
g-тензором.................................... 169
12.5. Псевдоконтактный сдвиг....................... 171
12.6. Качественная интерпретация спектров ЯМР парамагнитных молекул 173
12.7. Полуколичественная интерпретация контактных сдвигов 180
12.8. Применения изотропных сдвигов................ 185
Список цитируемой литературы...................................... 198
Упражнения........................................................ 199
ГЛАВА 13. СПЕКТРЫ ЭЛЕКТРОННОГО ПАРАМАГНИТНОГО РЕЗОНАНСА КОМПЛЕКСОВ ИОНОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ ..........................................................203
13.1. Введение............................................203
13.2 Интерпретация g-факторов............................209
13.3. Сверхтонкое расщепление и расщепление в нулевом поле 218
Оглавление
455
13.4. Интерпретация g- и Л-тензоров для систем с в
рамках теории поля лигандов..........................225
13.5. Сверхтонкое расщепление, обусловленное взаимодействием электронов с магнитными моментами ядер лиганда 231
13.6. Обзор спектров ЭПР комплексов ионов переходных металлов первого большого периода......................233
13.7. Метод двойного резонанса.............................249
Список цитируемой литературы......................................249
Упражнения........................................................ 251
ГЛАВА 14. СПЕКТРОСКОПИЯ ЯДЕРНОГО КВАДРУПОЛЬНОГО РЕ-
ЗОНАНСА (ЯКР)........................................... 260
14.1. Введение . . ....................................... 260
14.2. Энергии квадрупольных переходов . . .... 263
14.3. Влияние магнитного поля на спектры . .... 269
14.4. Связь между градиентом электрическою ж. ,я п молекулярной структурой 270
14.5. Применения.......................................... 274
14.6. Методы двойного резонанса......................... 279
Список цитируемой литературы...................................... 282
Упражнения........................................................ 283
ГЛАВА 15. МЁССБАУЭРОВСКАЯ СПЕКТРОСКОПИЯ ...... 285
15.1. Введение............................................ 285
15.2. Интерпретация изомерных сдвигов..................... 289
15.3. Квадрупольные взаимодействия........................ 291
15.4. Магнитные взаимодействия............................ 294
15.5. Мёссбауэровская эмиссионная спектроскопия............295
15.6. Применения.......................................... 297
Список цитируемой литературы...................................... 309
Упражнения........................................................ 310
ГЛАВА 16. ИОНИЗАЦИОННЫЕ МЕТОДЫ: МАСС-СПЕКТРОМЕТРИЯ, ИОННЫЙ ЦИКЛОТРОННЫЙ РЕЗОНАНС И ФО-
ТОЭЛЕКТРОННАЯ СПЕКТРОСКОПИЯ..............................313
Масс-спектрометрня.................................................313
16.1. Описание прибора и характеристика спектров...........313
16.2. Процессы, происходящие при столкновении молекулы с высокоэнергетическими электронами.................317
16.3. Применение масс-спектрометрии для идентификации. . . 320
16.4. Интерпретация масс-спектров.....................320
16.5. Влияние изотопов на характер масс-спектра..........’ 323
16.6. Определение молекулярных масс; метод ионизации полем 325
16.7. Расчет теплот сублимации; частицы в паре над твердыми веществами с высокой температурой плавления .... 327
16.8. Потенциалы возникновения и потенциалы ионизации 328
Ионный циклотронный резонанс (ИЦР).........................
16.9. Основные принципы ИЦР.......................
Фотоэлектронная спектроскопия..............................
16.10. Введение................................ . •
16.11. Теоретический подход..........................-
329
331
331
332
456
Оглавление
16.12. Фотоэлектронные спектры........................ 340
Список цитируемой литературы....................................355
Упражнения......................................................356
ГЛАВА 17. РЕНТГЕНОВСКАЯ КРИСТАЛЛОГРАФИЯ...................... 360
17.1. Введение........................................ 360
17.2. Симметрия твердого состояния.................... 361
17.3. Направление дифракции рентгеновских лучей........374
17.4. Практическое применение — первый этап............381
17.5. Дифракция рентгеновских лучей-интенсивности .... 390
17.6. Практические применения — второй этап........... 396
17.7. Осложнения и поправки............................404
Список цитируемой литературы....................................406
Упражнения .....................................................406
ПРИЛОЖЕНИЯ..................................................... 408
Предметный указателе............................................442
Р. Драго
ФИЗИЧЕСКИЕ МЕТОД1>1 В ХИМИИ
Т. 2
Ст. научный редактор С?К. Оганесян
Научный редактор Р. И. Краснова
Младший научный редактор Ю. В. Сергиевская
Художник М. В. Кузьмина
Художественный редактор Г. В. Шотина
Технический редактор Г. Б. Алюлина
Корректор К. Л. Водяницкая
ИБ № 1768
Сдано в набор 01.11.79.
Подписано к печати 27.01.81.
Формат 60 х 901/76
Бумага офсетная № 1.
Гарнитура тайме. Печать офсетная.
Объем 14,25 бум. л. Усл. печ.л. 28,50.
Уч.-изд. л. 28,79. Изд. № 3/0209.
Тираж 10000 экз. Зак. 574. Цена 2 руб. 30 коп.
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2.
Можайский полнграфкомбннат Союзполнграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
г. Можайск, ул. Мира, 93.
Отсканировал Семенсченко Владимир
chem_vova@mail.univ.kiev.ua; vova2002@mail.ru
Р. Драго ФИЗИЧЕСКИЕ МЕТОДЫ <Г В ХИМИИ