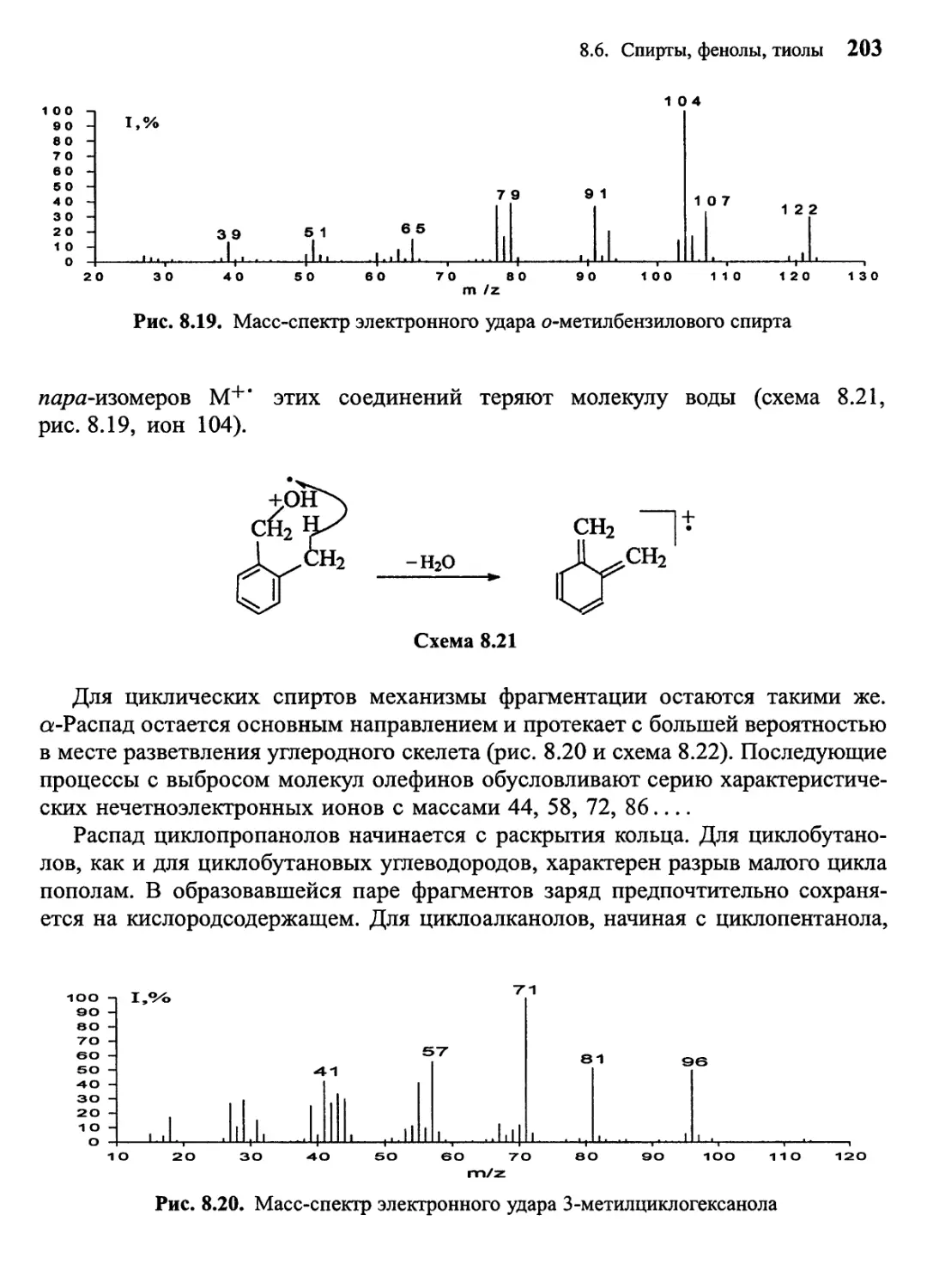
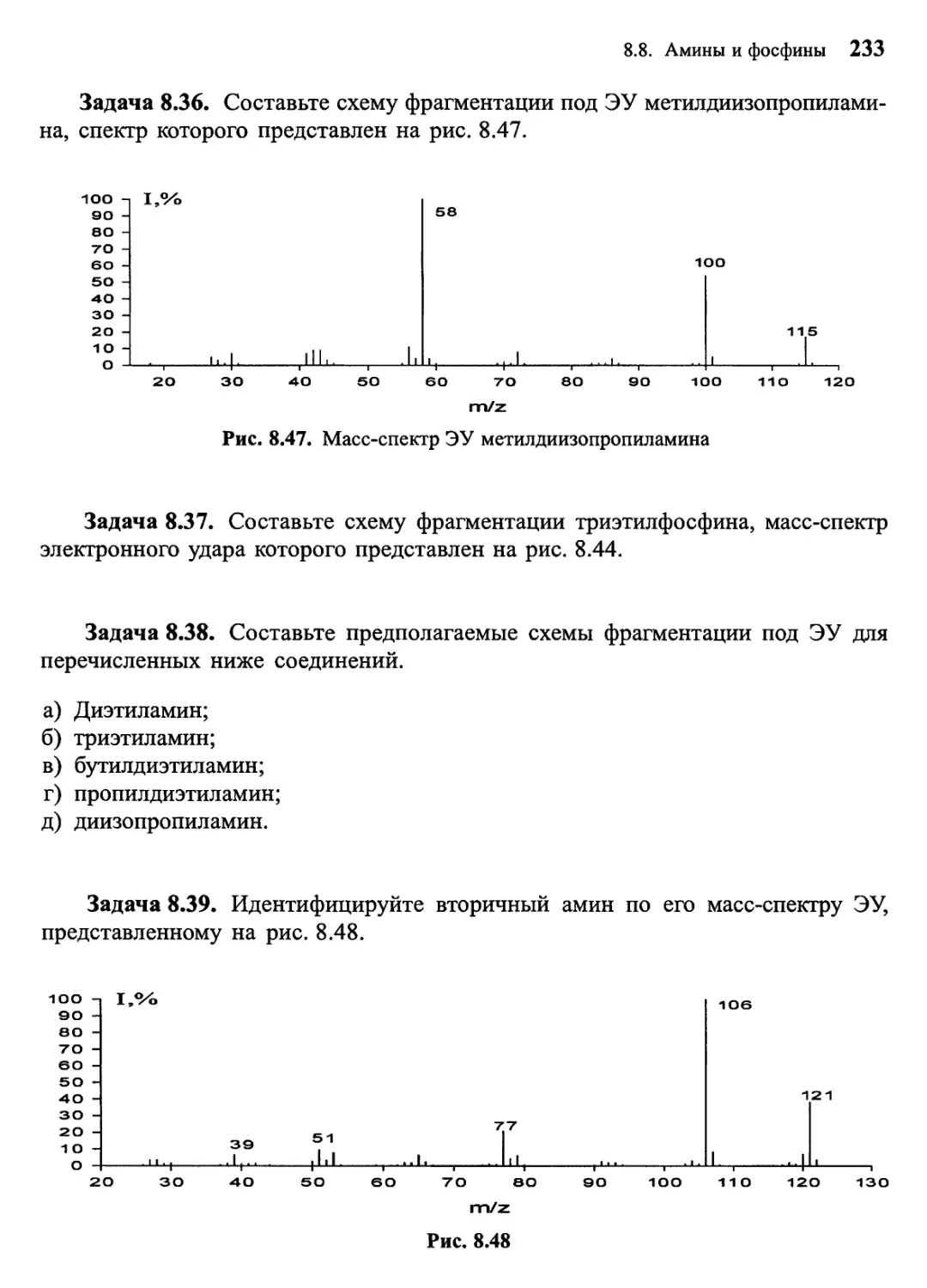
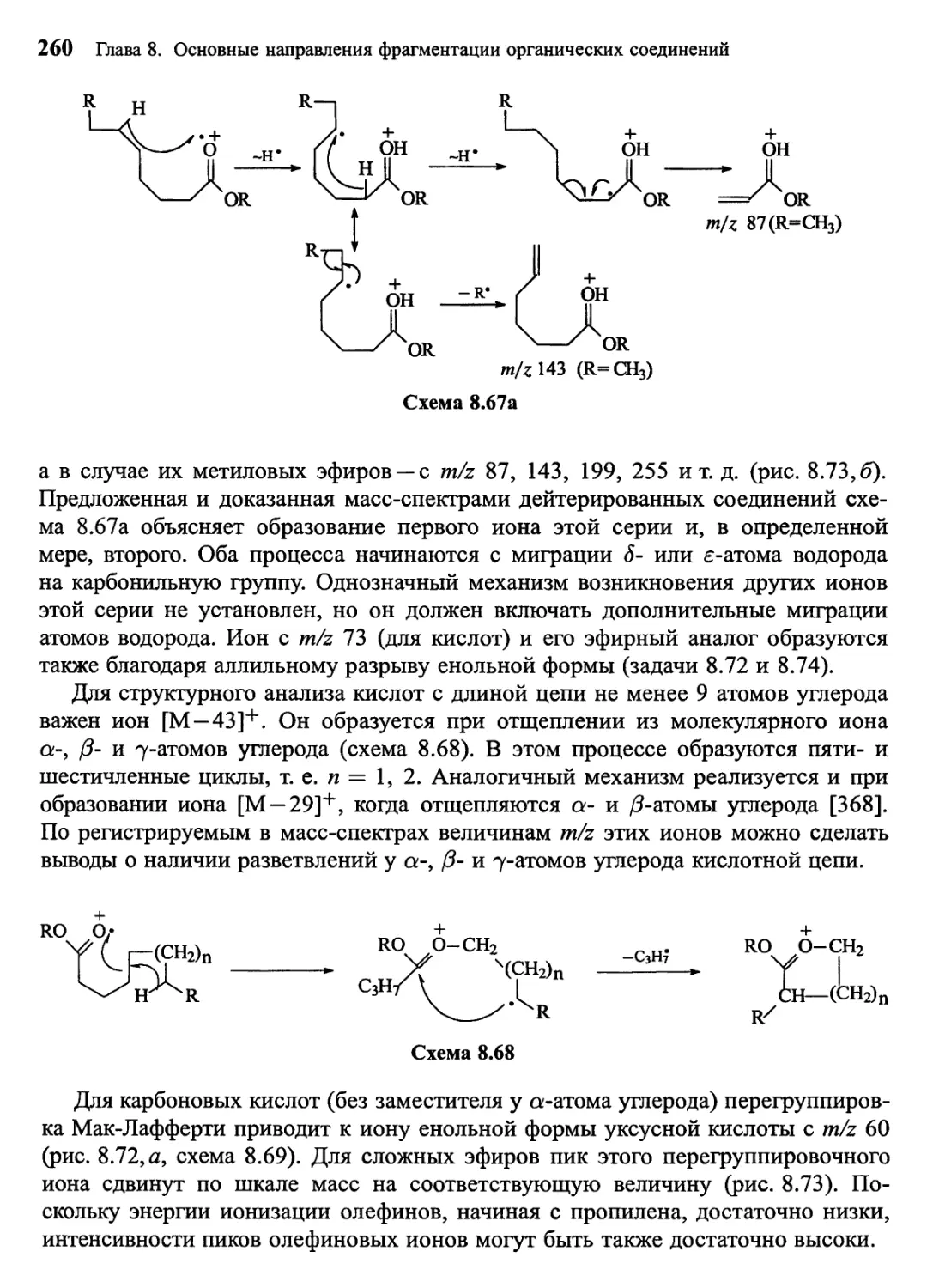
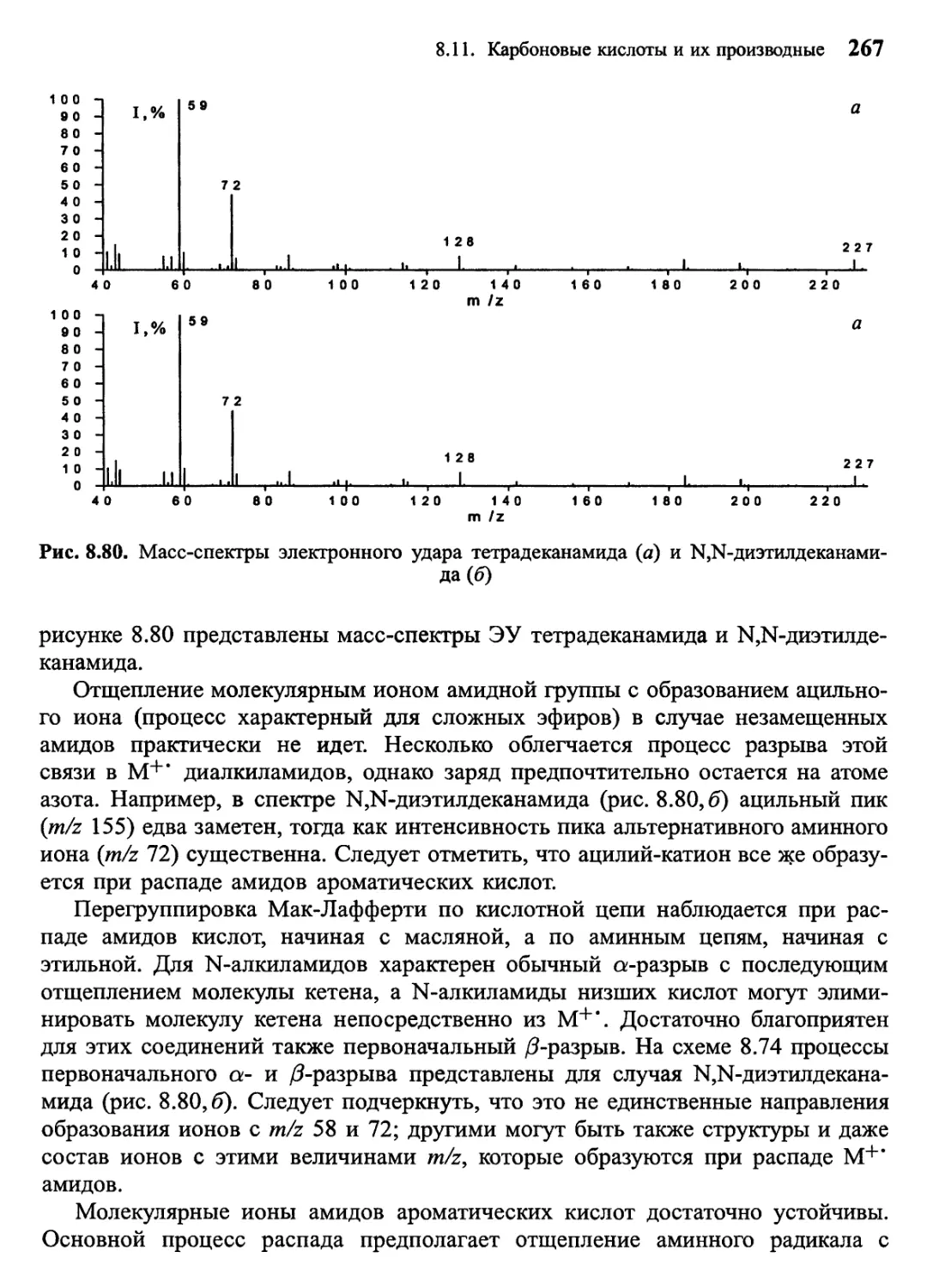
/




















































































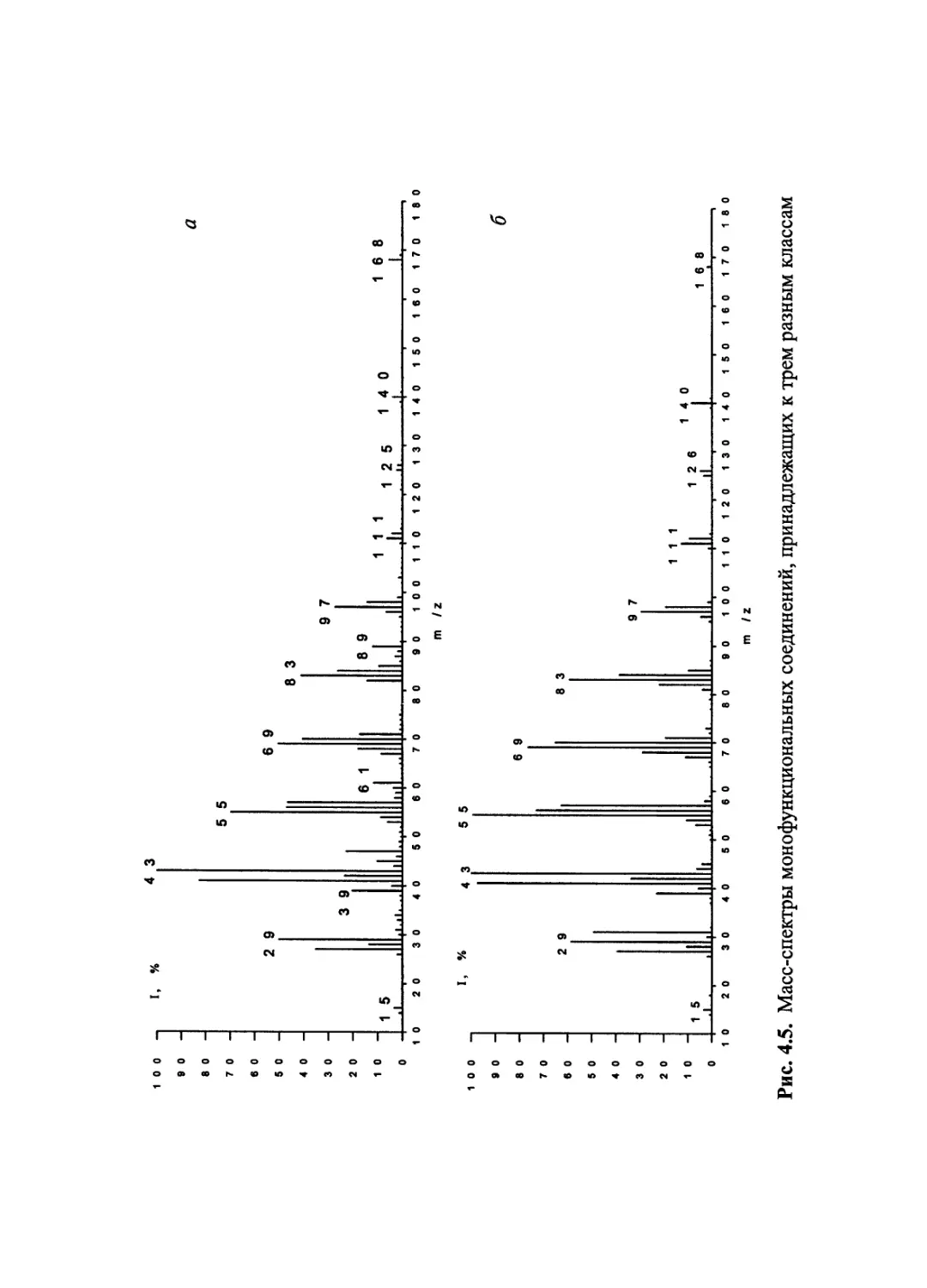



















































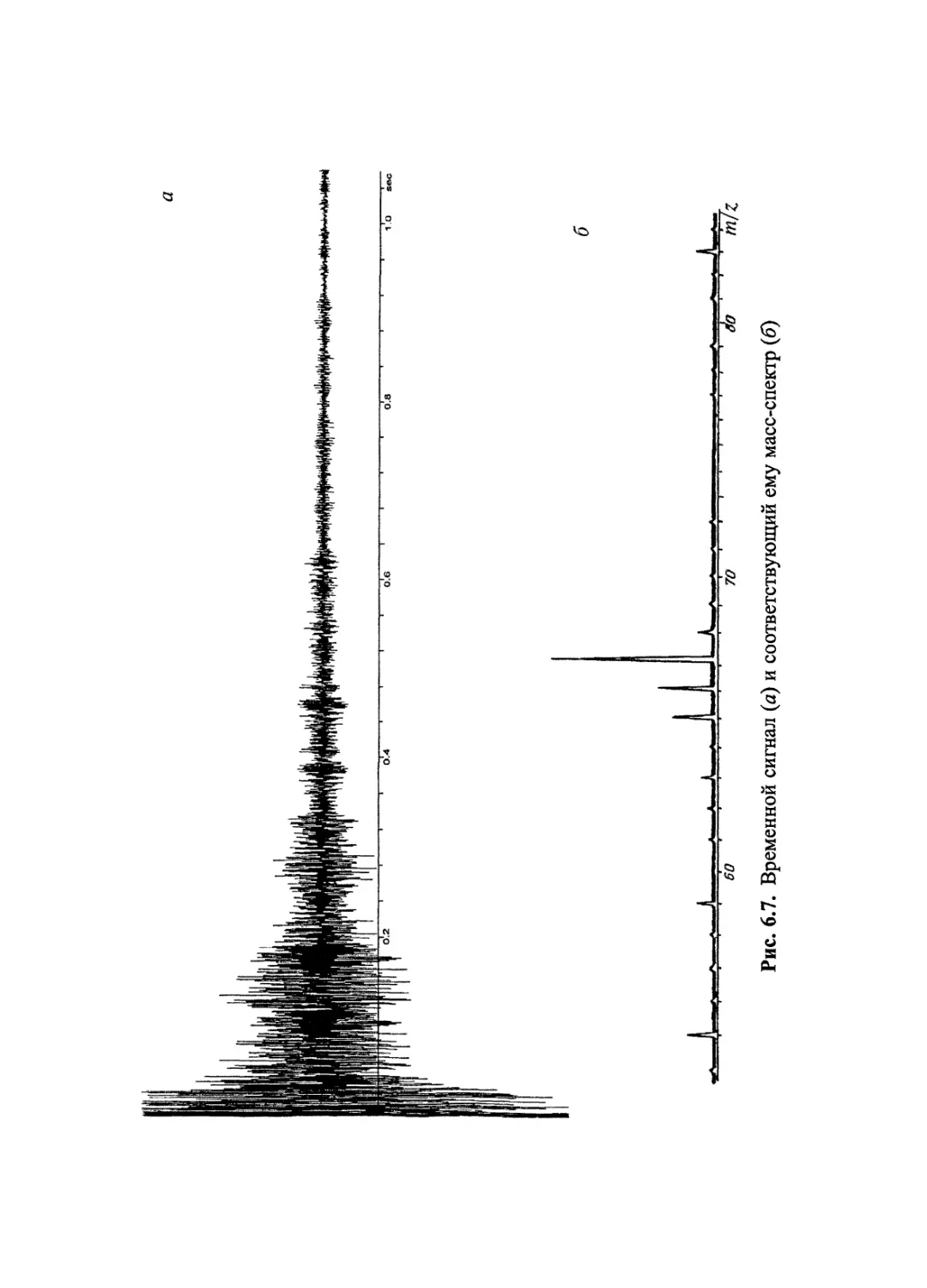




















































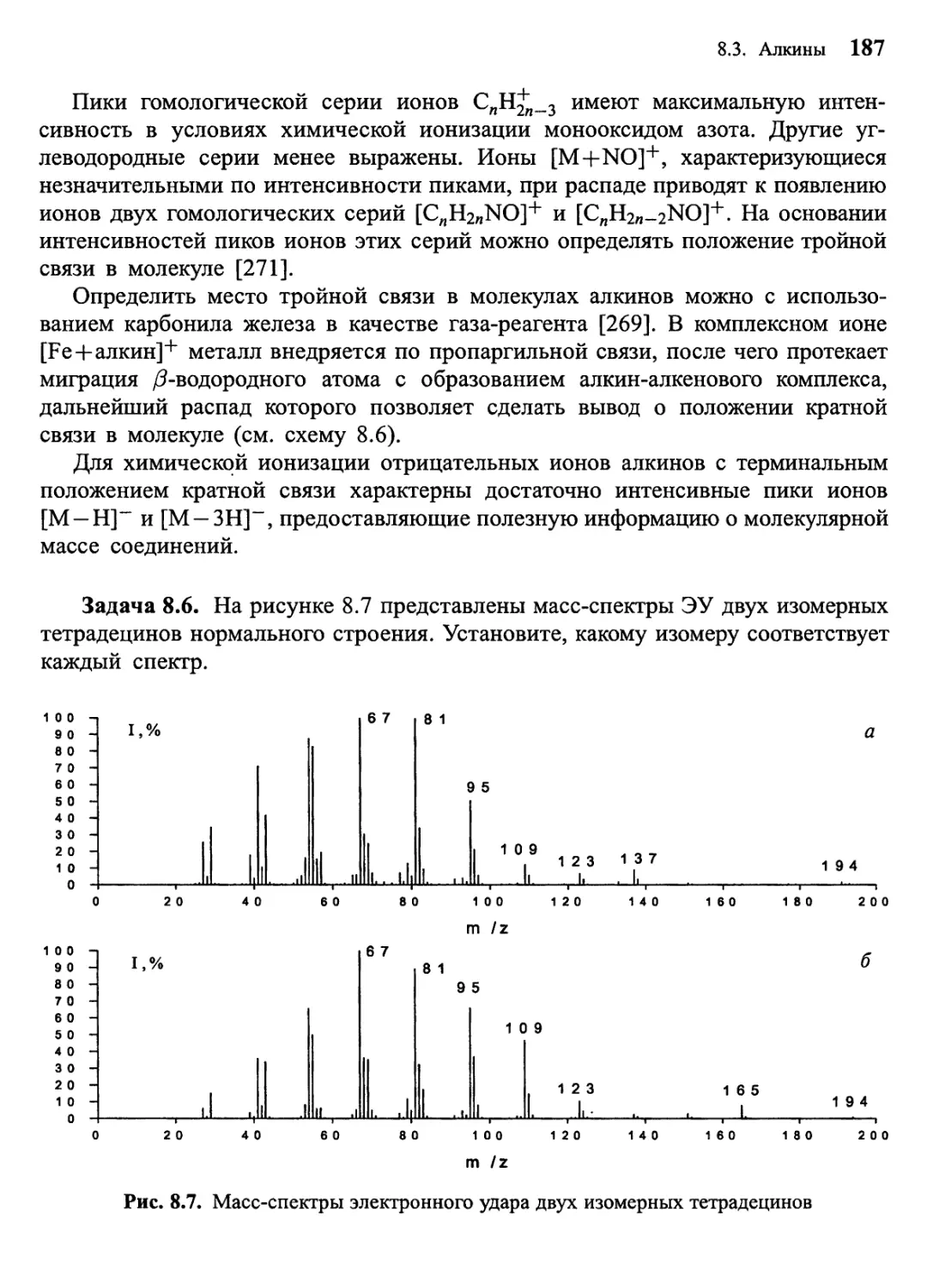






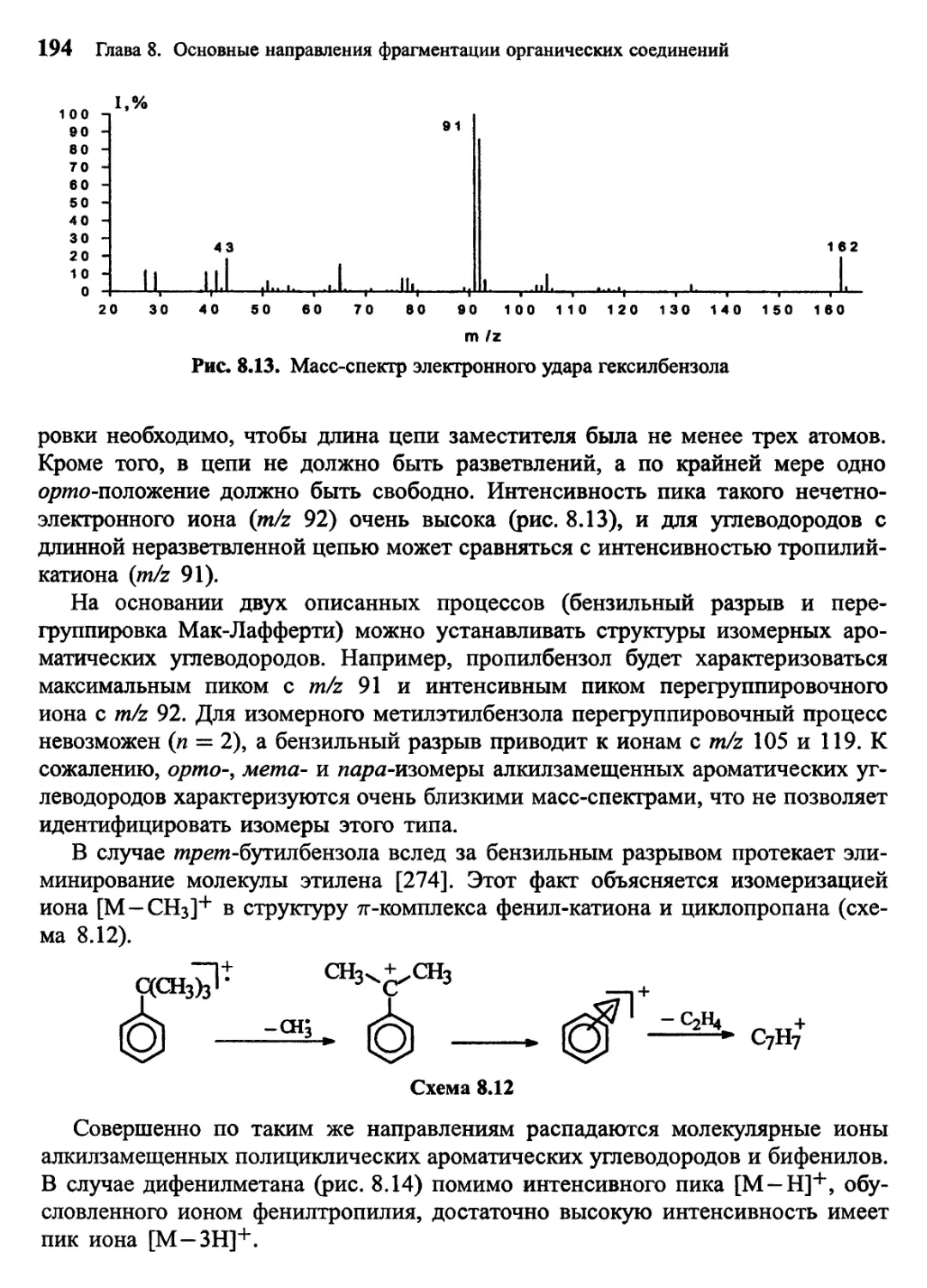


































































































































































































































































































Author: Лебедев А.Т.
Tags: другие физико-химические методы анализа (кроме оптических) органическая химия спектрометрия
ISBN: 5-94774-052-4
Year: 2003




Text
УДК 543.51+547
ББК 24.2
лзз
Рецензент:
доктор химических наук П. Б. Терентьев
Лебедев А. Т.
ЛЗЗ Масс-спектрометрия в органической химии / А. Т. Лебедев. —
М.: БИНОМ. Лаборатория знаний, 2003. —493 с, ил.— (Методы в
химии).
ISBN 5-94774-052-4
В учебном издании рассмотрены основы современной масс-спектрометрии
органических соединений: методы ионизации и разделения ионов, физико-химические
основы процесса масс-спектрометрического распада и направления фрагментации
важнейших классов органических соединений, а также области применения масс-
спектрометрии. Основное внимание уделено подходам при установлении строения
органических соединений по масс-спектрам; этот материал подкреплен большим
количеством задач, решение которых позволит получить практические навыки работы
со спектрами.
Для студентов старших курсов химических, биохимических,
химико-технологических и экологических специальностей, а также аспирантов, преподавателей и
научных сотрудников, работающих в области органической химии.
УДК 543.51+547
ББК 24.2
По вопросам приобретения обращаться:
в Москве
«БИНОМ. Лаборатория знаний» @95) 955-03-98, email: lbz@aha.ru
в Санкт-Петербурге
«Диалект» (812) 247-93-01, e-mail: dialect@sndlct.ioffe.rssi.ru
© Лебедев А. Т., 2003
ISBN 5-94774-052-4 © БИНОМ. Лаборатория знаний, 2003
Посвящается
Павлу Арутюновичу Шарбатяну
и Всеволоду Николаевичу Бочкареву,
замечательным людям
и отличным масс-спектрометристам,
открывшим для меня
удивительный мир этой науки
БЛАГОДАРНОСТИ
Автор выражает благодарность за помощь в работе над книгой
Джону Бови, Фреду Глиссону, Дмитрию Загоревскому, Ирине Московцевой,
Виктору Рыжову, Татьяне Самгиной, Йоргу фон Хельдену, Марии Хрущевой.
Автор выражает искреннюю благодарность за финансовую помощь компаниям
«Sakhalin Energy Investment Company»
и
«Ондео Налко Энерджи Сервис»
«Что такое „масса"? Благодаря чему она способна
оказывать влияние на духовную жизнь индивида?»
3. Фрейд
«Психология масс и анализ человеческого „Я"»
«Теория становится материальной силой, как
только она овладевает массами.»
К Маркс
«К критике гегелевской философии права»
ПРЕДИСЛОВИЕ
К концу XX века инструментальные физико-химические методы анализа стали
неотъемлемой частью экспериментальной работы исследователя, работающего в
области естественных наук. Наиболее мощными и многоцелевыми среди них,
безусловно, являются спектроскопия ядерного магнитного резонанса и масс-
спектрометрия. Оба метода активно используются в химии, биологии,
медицине, экологии, контроле технологических процессов, криминалистике и т. д.
Характеризуя вновь синтезированное вещество, исследователь обязан получить
и описать его ЯМР и масс-спектры.
Говоря о достоинствах масс-спектрометрии, следует прежде всего отметить
чувствительность, экспрессность, информативность и надежность метода. Для
получения достоверного масс-спектра индивидуального соединения даже на
рутинном масс-спектрометре достаточно 10-9-10~10 г вещества. При
необходимости простого детектирования конкретного соединения в смеси порог
обнаружения может быть легко снижен до 10~~12-10~14 г. Использование
современного оборудования и современных методов ионизации позволяет в некоторых
случаях увеличить чувствительность метода еще на несколько порядков. Таким
образом, не разрешимая без волшебства традиционная русская задача о «поиске
иголки в стоге сена» может быть легко решена масс-спектрометрически, причем
масс-спектрометрический эксперимент сравним с поиском иголки в нескольких
миллионах стогах сена.
Для получения обычного спектра электронного удара индивидуального
соединения необходимо затратить всего 1-2 мин, а время анализа сложной смеси
органических соединений в режиме хроматомасс-спектрометрии определяется
исключительно хроматографическим временем удерживания компонентов. При
этом следует учесть, что в памяти компьютера, являющегося неотъемлемой
частью современного масс-спектрометра, остаются информация о временах
удерживания, площадях пиков, а также масс-спектры всех компонентов смеси,
т. е., вводя в прибор 1 мкл сложнейшей смеси органических соединений, на
«выходе» можно получить информацию о ее качественном и количественном
составе. Ни один другой метод не сочетает в себе такой экспрессности и
6 Предисловие
информативности. Надежность масс-спектрометрического анализа также очень
высока, поскольку масс-спектр является реальной характеристикой конкретного
вещества, отражающей его структурные особенности.
История масс-спектрометрии насчитывает около 100 лет. Годом рождения
масс-спектрометрии можно считать 1901 г., когда немецкий физик В.
Кауфман создал первый прототип параболического масс-спектрографа для изучения
«катодных лучей», или 1913 г., когда сэр Дж. Томсон впервые спектрально
«увидел» изотопы неона, или 1918 г., когда А. Демпстер сконструировал
первый магнитный масс-спектрометр с источником для электронной и
термической ионизации. На протяжении следующих 50 лет «масс-спектральная наука»
оставалась прерогативой исключительно физиков и физикохимиков. Используя
простейшие соединения, масс-спектрометристы того времени успешно измеряли
характеристики элементов, атомов, молекул (энергии ионизации, прочцости
связей, точные массы элементов и природную распространенность их изотопов).
Органическая масс-спектрометрия характеризуется полувековой историей. Это
очень небольшой период времени, поэтому сейчас, в начале XXI века, можно
лично поговорить практически со всеми классиками, стоявшими у истоков
создания этой науки. После второй мировой войны возникло понимание, что
метод можно использовать не только в качестве прецезионного
физико-математического инструмента, но и в качестве удобного способа идентификации
достаточно сложных соединений по набору характеристических фрагментных
ионов. Знание классических химических реакций органических соединений и
привлечение теорий устойчивости органических ионов позволили приписать
фрагментам конкретные структуры, сделать обобщения по аналогиям
фрагментации внутри классов органических соединений. Переход на такой уровень
использования масс-спектрометрии резко увеличил число пользователей метода
и привел к взрывному развитию приборного парка. Особенно важную роль
сыграло то, что метод оказался применимым при решении нефтехимических
проблем. Финансовые вложения нефтяных компаний в значительной степени
способствовали быстрому инструментальному и теоретическому становлению
нового научного направления. Огромный фактический материал по истории
масс-спектрометрии содержит богато иллюстрированная фотографиями и
рисунками отличная книга под редакцией М. Грейсона [1], изданная в 2002 г. к
50-летию Масс-спектрометрического общества США.
Традиционно органическая масс-спектрометрия используется для решения
двух основных проблем: идентификация веществ и изучение фрагментации
ионизированных молекул органических соединений в газовой фазе в ионном
источнике [2]. С появлением хроматомасс-спектрометрии, ионно-циклотронного
резонанса [3], систем протока после разряда [4] возможности классического
метода значительно увеличились. Соединение масс-спектрометра с жидкостным
хроматографом еще более расширило круг изучаемых объектов. Новые методы
Предисловие 7
ионизации, в частности «электроспрей» [5] и МЛДИ [6], появившиеся к концу
XX века, позволили успешно работать со сложнейшими биоорганическими
молекулами, такими как полипептиды, белки, полисахариды, нуклеиновые кислоты,
молекулярные массы которых составляют миллионы дальтон [7]. Сегодня можно
проанализировать соединение практически любой сложности. В последние годы
широко ведутся исследования по анализу микроорганизмов [8], по
моделированию в газовой фазе химических реакций термолиза, фотолиза, превращений
катализируемых кислотами и основаниями [9], по изучению каталитических
процессов [10]. В этой связи уместно привести шутливое выражение одного
из «отцов» органической масс-спектрометрии Фреда Мак-Лафферти: «Если
химическую задачу нельзя решить с помощью масс-спектрометрии, ее вообще
нельзя решить» [11]. Признанием важности масс-спектрометрии для развития
современной науки стало присуждение в 2002 г. Нобелевской премии создателям
методов электроспрея и МЛДИ Джону Фенну и Коичи Танаке.
Наряду с очевидным использованием масс-спектрометрии в органической и
биоорганической химии для установления структур соединений хроматомасс-
спектрометрия стала сегодня основным методом качественного и
количественного определения органических загрязнений в объектах окружающей среды.
Современная химическая экология немыслима без этого метода. Изучение
метаболизма лекарственных средств и пестицидов в окружающей среде и живых
организмах также ведется с активным использованием масс-спектрометрии.
Метод незаменим в криминалистических исследованиях и при проведении допинг-
контроля на спортивных соревнованиях.
Масс-спектрометрия применяется для решения геохимических и космохи-
мических проблем, задач комбинаторной химии, иммунологии и медицины,
при идентификации микроорганизмов и т. д. Масс-спектрометрия имеет явное
преимущество перед другими физико-химическими методами, поскольку
оперирует с простейшими характеристиками вещества: массой молекулы и ее
основных фрагментов, а также с отношением количеств этих фрагментов. Это
позволяет достаточно легко усвоить основы метода и научиться работать с масс-
спектрами не только подготовленным студентам, но и школьникам старших
классов, изучающим органическую химию. Учитывая, что расшифровка масс-
спектра может быть сродни решению головоломки типа «puzzle», овладение
этим методом принеся несомненную пользу, как увеличение «багажа» знаний,
увлекательно своей простотой.
Тем не менее занимаясь масс-спектрометрией органических соединений уже
более 20 лет, я постоянно сталкиваюсь с ситуацией, когда даже опытные
химики-органики пасуют перед масс-спектром, предпочитая извлечь
необходимую информацию о структуре с помощью других, более знакомых им методов.
Объяснение этому я нахожу в отсутствии учебной литературы по интерпретации
масс-спектров и в отсутствии спецкурсов по этой дисциплине в вузах России.
8 Предисловие
Книги по масс-спектрометрии советских авторов, изданные в 70-80 годах
прошлого века, затрагивали отдельные аспекты органической масс-спектрометрии
[12-31]; среди них в качестве учебной литературы можно было бы выделить
лишь два пособия [15, 27]. Русскоязычный читатель не был знаком и с
большинством иностранных изданий; на русский язык были переведены лишь
фундаментальная пионерская монография Джона Бейнона [2] и еще три книги
[32-34], причем две из них [32, 33] достаточно давно. Следует лишь сожалеть,
что замечательный учебник Мак-Лафферти [35], претерпевший уже четыре
издания, так и не переведен на русский язык. Уже во время работы над этим
пособием вышла в свет книга группы авторов «Основы масс-спектрометрии
органических соединений» [36]. В ней хорошо изложены классические аспекты
органической масс-спектрометрии, подкрепленные представительным набором
задач.
Данное пособие направлено на решение двух основных задач: учебной и
познавательной. С материалом можно знакомиться последовательно или выборочно
по конкретным главам или разделам. Учебную функцию несут прежде всего
гл. 2-4, 8-10, в которых рассмотрены основные теории и законы фрагментации
органических соединений. Большое внимание уделено именно работе с масс-
спектром, подходам к его расшифровке с извлечением максимальной
структурной информации. Описание основных направлений фрагментации различных
классов органических соединений (от алканов до белков и нуклеиновых кислот)
сопровождается реальными масс-спектрометрическими задачами. Решая эти
задачи, читатель сможет закрепить теоретический материал и затем успешно
справляться с подобными проблемами в своей последующей научной работе.
Большое число вариантов типовых задач позволяет использовать книгу не только
для индивидуальной работы, но и для семинаров в студенческих группах. Глава
9 посвящена вопросам количественного масс-спектрометрического анализа.
Последнее десятилетие XX века ознаменовалось революционными
новациями в органической масс-спектрометрии. В русскоязычной литературе пока
отсутствуют описания современных методов ионизации, анализа и
расшифровки спектров сложных биологических соединений. В данной работе наряду с
описанием классических методов ввода и ионизации образца, а также
разделения и детектирования ионов значительное место уделено теоретическим и
практическим аспектам современной масс-спектрометрии: электрораспыление
и другие методы стыковки жидкостного хроматографа с масс-спектрометром,
матричная лазерная десорбционная ионизация (гл. 5), масс-спектрометрия с
преобразованиями Фурье, варианты времяпролетной масс-спектрометрии и т. д.
(гл. 6), а также тандемная масс-спектрометрия, спектры с переменой заряда,
нейтрализация—реионизация, спектрометрия ионной подвижности (гл. 7).
Очень важно, чтобы масс-спектрометрией как методом исследования владели
и его применяли в работе непосредственно химики, биологи, экологи, а не узкий
Предисловие 9
круг профессиональных масс-спектрометристов. Никто столь досконально не
знает своей задачи, как сам исследователь, работающий с веществом. Если он
сможет правильно поставить масс-спектрометрическую задачу и при этом
владеет техникой интерпретации масс-спектров, эффективность работы со
спектрами будет существенно выше, чем у профессионального масс-спектрометриста,
далекого от конкретной исследовательской проблемы. Для правильной
расшифровки масс-спектра может оказаться необходимой информация о производителе
исходного реактива, о побочных процессах изучаемой реакции, о примесях к
реагентам или даже о реакции, которая ставилась в этой посуде за неделю до
данного эксперимента.
ВВЕДЕНИЕ
Масс-спектрометрия является физико-химическим методом анализа,
заключающимся в переводе молекул образца в ионизированную форму с последующим
разделением и регистрацией образующихся при этом положительных или
отрицательных ионов. Масс-спектр позволяет сделать выводы о молекулярной массе
соединения, его составе и структуре. Масса самого тяжелого иона в спектре
равна молекулярной массе анализируемого соединения. Принято представлять
масс-спектр в виде графика или таблицы (рис. 1).
В случае графического изображения по оси абсцисс откладывается масса
ионов (точнее величина отношении массы иона к его заряду), а по оси ординат —
их интенсивности, т. е. относительное количество ионов данного вида. Принято
выражать интенсивность в процентах к полному ионному току (суммарной
интенсивности всех ионов в спектре) или к интенсивности максимального
иона (рис. 1). В качестве единицы размерности массы в масс-спектрометрии
используются термины: углеродные единицы (у. е.), атомные единицы массы
(а. е. м.), дальтоны (Да).
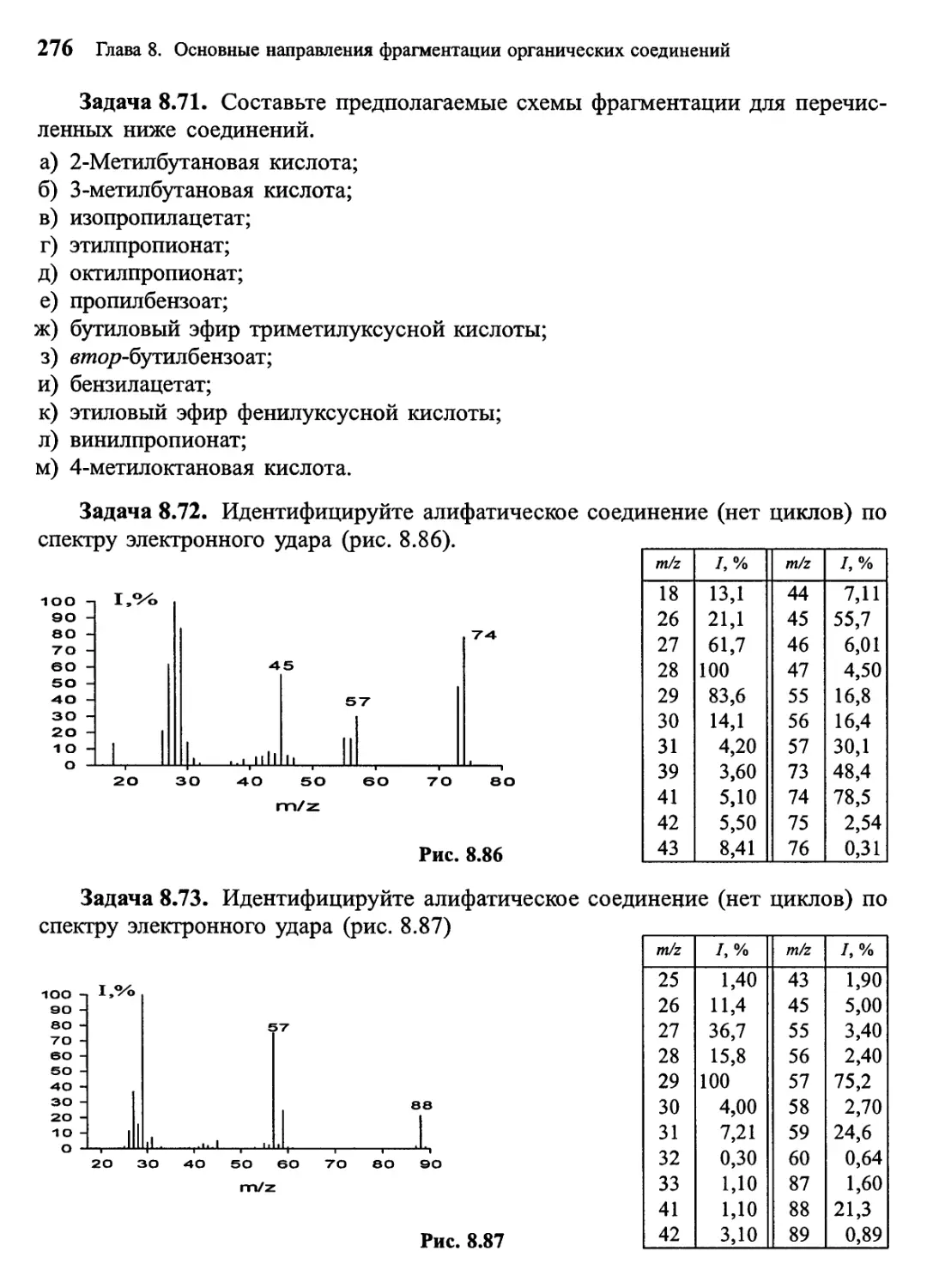
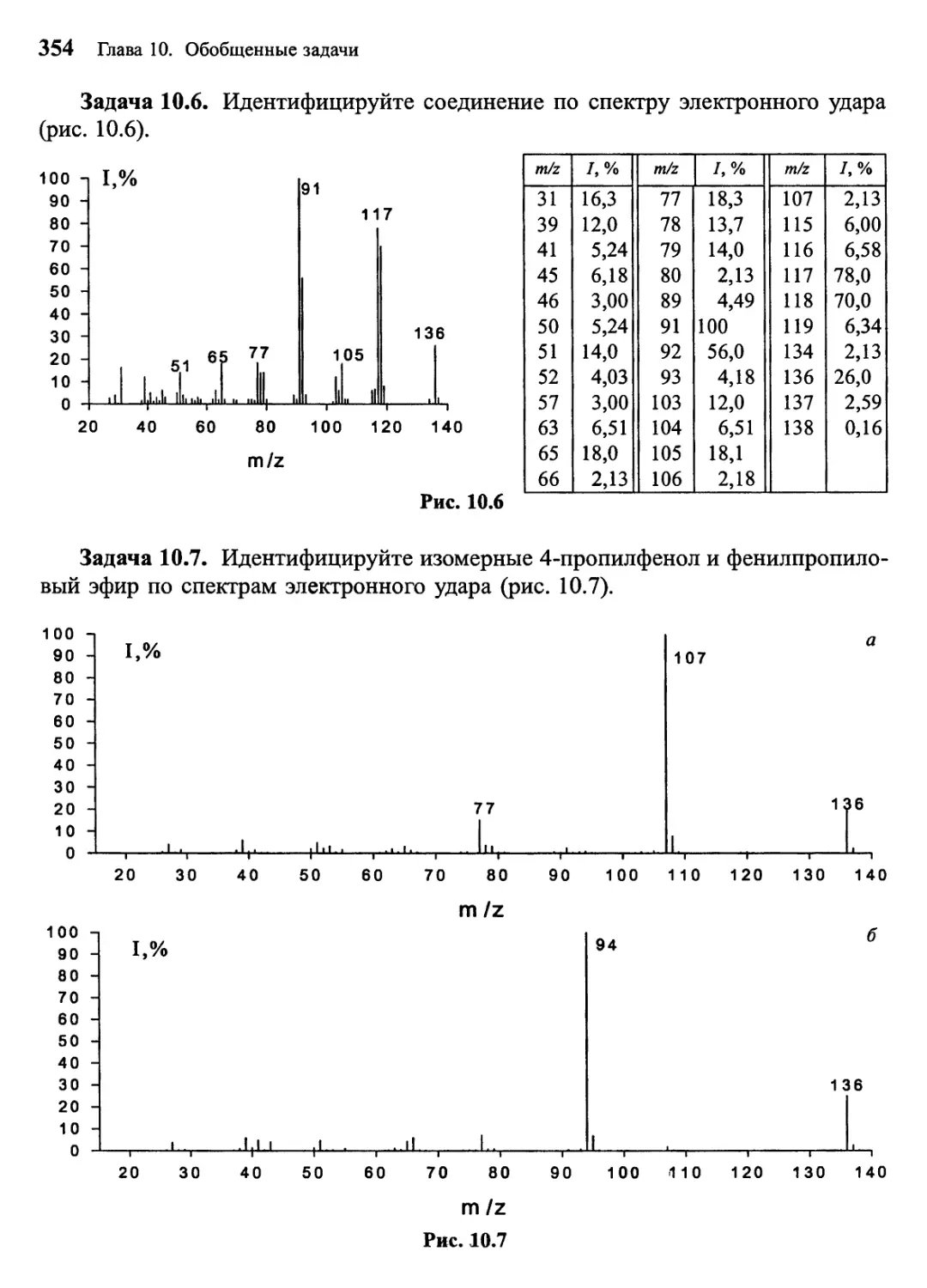
Задача 1, Попробуйте установить, какому распространенному
органическому соединению принадлежит масс-спектр, представленный на рис. 1. Масс-
спектрометрия высокого разрешения показала, что точная молекулярная масса
соединения 60,0211 Да, что соответствует составу С2Н4О2. Обращайте на данном
этапе внимание только на наиболее интенсивные пики в спектре, которые
обусловлены всей молекулой (ион с m/z 60) и ее наиболее значимыми
структурными фрагментами. В данном случае наиболее важными осколочными ионами
являются фрагменты с m/z 45, 43, 28 и 15.
Для получения масс-спектра прежде всего необходимо ввести образец в
источник ионов (гл. 1), затем перевести его молекулы в заряженную форму
(положительные или отрицательные ионы, гл. 2), разделить эти ионы по массам
и зарегистрировать их массы и количество (гл. 6). В современных приборах
управление всеми операциями, а также обработку результатов осуществляет
компьютер, являющийся неотъемлемой частью масс-спектрометра. На выходе
можно получить спектр в любом виде, провести вычитание или усреднение
спектров, провести сравнение с библиотекой масс-спектров. Принципиальная
блок-схема масс-спектрометра представлена на рис. 2.
Введение 11
ю
20
30
40
m/z
50
60
70
m/z
14
15
28
29
30
43
44
45
46
47
60
61
62
/,% |
0,7
33,0
56,0
0,65
0,11
100,0
2,2 '
26,0
0,31
0,10
62,0
1,48
0,26
Рис. 1. Масс-спектр соединения с брутто-формулой С2Н4О2 в графическом и табличном
виде
Система
ввода
->
Ионизация
->
Разделение
ионов
->
Регистрация
Компьютер
->
Обработка
информации
->
Спектр
Рис. 2. Блок-схема масс-спектрометра
Для решения более сложных задач (например, анализ неразделенных смесей)
схема процесса может быть значительно сложнее (гл. 7, тандемная масс-спектро-
метрия).
Масс-спектрометрическая фрагментация органических соединений
подчиняется определенным законам (гл. 2, 3), поэтому и при отсутствии спектра
анализируемого соединения в компьютерных библиотеках можно установить его
молекулярную массу, состав, наличие функциональных групп, а иногда и точную
структуру по характеру распада (гл. 4). В распаде родственных соединений есть
много общего (гл. 8). Этот факт помогает правильной интерпретации спектров
вручную, хотя требует определенной практики (гл. 10). Кроме того, масс-спек-
трометрия является наиболее чувствительным методом количественного анализа
органических соединений (гл. 9).
Глава 1
СИСТЕМА ВВОДА ОБРАЗЦА
Для того чтобы избежать нежелательных химических реакций в результате
взаимодействия молекул и ионов, в источнике масс-спектрометра, как правило,
поддерживается высокий вакуум A0~5-10_6 ммрт. ст.). Поэтому уже сам ввод
образца является самостоятельной технической задачей.
1.1. Баллон напуска
Одной из первых систем ввода, практически не используемой в настоящее
время, служил баллон напуска (рис. 1.1). Анализируемое вещество находилось
в газообразном состоянии (давление около Ю-2 ммрт. ст.) в баллоне объемом
50-2000 см3, внутренняя поверхность которого была остеклена для
предотвращения нежелательных химических реакций на стенках сосуда. В поздних вариантах
предусматривалась система нагрева пробы в баллоне. Этот резервуар соединялся
с источником ионов отверстием очень небольшого диаметра, через которое
осуществлялось молекулярное натекание образца в масс-спектрометр.
Альтернативными вариантами соединительного отверстия служили
капилляры или пористые диафрагмы. Эти устройства позволяли получить стабильную
концентрацию вещества в источнике и были необходимы на начальных этапах
развития органической масс-спектрометрии, когда применялись
малоэффективные вакуумные системы, а скорость развертки масс-спектра составляла минуты.
Очевидным недостатком данного ввода является необходимость переводить ве-
1.2. Прямой ввод 13
щество в газовую фазу в условиях неглубокого вакуума, что для многих образцов
весьма проблематично. Конструкции стыковки баллонов напуска с ионными
источниками подробно описаны в фундаментальном труде Джона Бейнона [2].
1.2. Прямой ввод
Развитие техники позволило в 60-х годах XX века перейти на более
совершенную систему ввода, которая является общепринятой и широко распространенной
в настоящее время. Это — прямой ввод вещества в область ионизации. Твердый
образец помещается в специальную микрокапсулу (стекло, кварц, керамика,
металл), которая штоком вводится непосредственно в ионный источник, т. е.
испарение осуществляется прямо в источнике ионов в условиях глубокого
вакуума. При необходимости образец может быть нагрет с помощью
программируемой печки до температуры 400-500°С, а в некоторых случаях, как, например,
в условиях десорбционной химической ионизации (разд. 5.3), —выше 1000°С.
Понятно, что в этих условиях количество соединений, доступных масс-спек-
трометрическому анализу, значительно возрастает. Полное испарение введенного
образца позволяет измерить количество соединения, т. е. прямой ввод позволяет
наряду с качественным проводить и количественный анализ веществ (гл. 9).
Важно обращать внимание не только на температуру прямого ввода, но и
на температуру источника ионов. Если вещество труднолетучее, а прямой ввод
перегрет, оно может конденсироваться на стенках источника, что ведет к его
быстрому загрязнению. Кроме того, последовательные процессы испарения и
конденсации приводят к увеличению роли деструкции вещества и,
соответственно, к искажению масс-спектра. Процессы деструкции имеют место и в случае
слишком высокой температуры источника. Принципиальная схема прямого ввода
представлена на рис. 1.2.
Программируемый нагрев образца в вакууме позволяет решить одновременно
три задачи: во-первых, перевести в газовую фазу широкий круг органических
Спираль нагрева Капилляр (ампула) с образцом
га
Вентиль,
J закрывающий вход
Источник ионов
Вакуумный насос Шток
Рис. 1.2. Система прямого ввода
14 Глава!. Система ввода образца
соединений; во-вторых, подобрать оптимальную температуру съемки, когда
воспроизводится качественный масс-спектр; в-третьих, анализировать смеси
соединений с разной степенью летучести. Такое фракционное испарение
позволяет в некоторых случаях провести достаточно быстрый качественный и
количественный анализ смеси без ее предварительного разделения.
Важной характеристикой прямого ввода является существенное уменьшение
количества образца, необходимого для получения масс-спектра. В данном случае
следует опасаться ввести слишком много вещества. Считается, что твердый
образец в капилляре на конце штока должен быть едва различим глазом. Избыточное
количество образца может привести к искажению масс-спектра из-за протекания
ионно-молекулярных реакций и вызвать быстрое загрязнение источника ионов.
Многие масс-спектрометристы предпочитают наносить вещество из раствора и
вводят шток в источник после испарения растворителя.
Еще один важный параметр съемки для наиболее распространенных спектров
электронного удара связан с положением образца относительно пучка
электронов. Если образец расположен достаточно далеко, его молекулы достигают пучка
электронов только после ряда последовательных столкновений со стенками
источника. При этом возрастает роль процессов термодеструкции. Однако если
поместить образец слишком близко к пучку электронов, наконечник штока может
приобрести отрицательный заряд и начать отталкивать электроны, что приводит
к расфокусировке пучка.
Система прямого ввода применяется в основном в сочетании с методами
электронного удара и химической ионизации, хотя его модификации используются в
случае бомбардировки быстрыми атомами (разд. 5.10) и для некоторых других
методов ионизации.
13. Мембранный ввод
(Membrane Inlet Mass Spectrometry, MIMS)
Достаточно перспективным является мембранный ввод пробы [37]. Мембрана
изготавливается из органического материала и пропускает в источник
масс-спектрометра соединения, растворимые или адсорбируемые материалом мембраны и
обладающие высоким коэффициентом диффузии в нем. Анализируемое
соединение должно быть достаточно летучим, чтобы испаряться на вакуумированной
стороне мембраны. В одном из приводимых ниже вариантов мембрана
размещается вне источника ионов (рис. 1.3) [38], в другом — капиллярная мембрана
вводится непосредственно в источник (рис. 1.4) [39].
Достоинством такого ввода является селективность в пропускании веществ
разной природы. Поскольку мембрана не пропускает воду, неорганические газы
и соли, она может использоваться для эффективного мониторинга загрязнения
воздуха или воды органическими соединениями, а также для контроля био-
1.4. Хроматомасс-спектрометрия 15
Источник ионов
Мембрана
? 1
Выход Вход
а со
8
Капиллярная
мембрана
Рис. 1.3. Размещение мембраны вне ис- Рис. 1.4. Размещение мембраны внутри источ-
точника ионов ника ионов
химических процессов. Например, в работе [40] показано, что предел
обнаружения в воде для широкого круга органических соединений с молекулярной
массой <300 Да может достигать нанограммов в литре. В литературе описаны
возможности непрерывного мониторинга органических соединений в сточных и
природных водах [38], в воздухе [41], мониторинга биологических реакций [42].
1.4. Хроматомасс-спектрометрия, ГХ-МС
(GC-MS)
На сегодняшний день хроматомасс-спектрометрия (хромасс, ГХ-МС) является
наиболее широко используемой разновидностью органической масс-спектромет-
рии, хотя, начиная с 90-х годов XX века, многочисленные варианты
комбинированного метода высокоэффективная жидкостная хроматография — масс-спек-
трометрия (разд. 1.5) используются все шире и в ближайшее время, вероятно,
займут лидирующее положение.
Стыковка газового хроматографа и масс-спектрометра была абсолютно
логичной, поскольку оба метода использовались для анализа смесей органических
соединений в газовой фазе и обладали приблизительно равной
чувствительностью. Единственной проблемой для объединения двух методов в едином приборе
являлось рабочее давление. Газовый хроматограф работает при атмосферном
давлении, а масс-спектрометр— в условиях глубокого вакуума. Основные
принципы стыковки были сформулированы и претворены в жизнь в 1957 году [43].
Первоначальные проблемы, связанные с недостаточной мощностью вакуумных
систем и набивными колонками с рабочими потоками газа-носителя 30 мл/мин,
решались установкой сепараторов различного типа [2]. Эти устройства
размещались между выходным концом колонки хроматографа и ионным источником
масс-спектрометра и предназначались для обогащения пробы анализируемым
веществом за счет избирательной откачки значительно более легкого газа-носителя
(водород, гелий). Появление более мощных вакуумных систем и капиллярных
16 Глава!. Система ввода образца
колонок с меньшими потоками @,5-2,0 мл/мин) значительно облегчило задачу, а
замена металла или стекла, из которых изготавливались колонки, на плавленый
кварц позволила ввести конец колонки непосредственно в ионный источник. Все
это сделало метод ГХ-МС простым и эффективным.
Не стоит забывать, однако, что стыковка хроматографа и масс-спектрометра
все-таки является компромиссом, поскольку давление в ионном источнике
оказывается несколько выше идеального, а выходное отверстие хроматографической
колонки функционирует в условиях вакуума, что несколько ухудшает
разрешающую способность метода. Подробнее об этом можно прочитать в книге Чепмена
[34]. Метод прежде всего предназначен для анализа смесей органических
соединений и заключается в их разделении на колонке хроматографа с
последовательным выходом компонентов из колонки в ионный источник масс-спектрометра,
где происходит их ионизация. Масс-спектрометр должен проводить запись
спектра (сканирование) с достаточной частотой, чтобы зарегистрировать масс-
спектр каждого соединения несколько раз. Это условие является важнейшим,
поскольку концентрация вещества очень быстро изменяется. В зависимости от
того, на восходящей или нисходящей стороне хроматографического пика будет
записан масс-спектр, будут дискриминированы или низкие, или высокие массы
(рис. 1.5).
Безусловно, лучшим будет спектр, зарегистрированный на вершине
хроматографического пика, где максимальна концентрация вещества и минимальны ее
изменения во времени. Желательно получить 5-10 спектров каждого компонента
смеси, а для улучшения качества спектра часто необходимо проводить
усреднения и вычитания фона. Продемонстрировать эффект вычитания фона можно
на примере, представленном на рис. 1.6а-1.6г. В данном случае спектр, снятый
у подножия хроматографического пика, вычитается из спектра, снятого на
вершине пика. Эту процедуру осуществляет компьютер. Результирующий спектр
очищен от наложения фона и позволяет значительно улучшить сходимость с
библиотечным спектром.
Очень важна скорость сканирования и для количественного анализа.
Недостаточное число сканирований приводит к значительному искажению площади
хроматографического пика (рис. 1.7). Современные приборы позволяют
сканировать спектр за 0,1-0,5 с, что удовлетворительно сочетается с шириной
хроматографического пика (как правило, несколько секунд). Если учесть, что
средний хроматомасс-спектрометрический анализ длится 30 мин, при скорости
сканирования 0,3 с в памяти компьютера должно остаться 6000 спектров. Этот
факт наглядно демонстрирует невозможность получения качественных
результатов без стыковки прибора с мощным компьютером. Кстати, времяпролетные
спектрометры (разд. 6.4) позволяют регистрировать 100 и более полных масс-
спектров в секунду. Эта особенность привела к созданию быстрой хроматомасс-
спектрометрии (Rapid GC-MS), позволившей сократить время анализа смесей в
5-10 раз [44].
1.4. Хроматомасс-спектрометрия 17
1 О О
9 О
8 О
7 О
6 О
5 О
4 О
3 О
2 О
1 О
О
1,4
2 0 0
1 2 3
1 0 5
7 7
9 5
5 1
10 0 ?
90 -
8 0-
7 0-
6 0-
5 0-
4 0-
30 -
??? 0 -
0 -
?,%
2 7
3 9
¦¦? .
5 1
?
J
L
1 0 5
7
.jj
I7 9 5
I...II. „I
4-
L_
m
/z
1 2 3
L , .
6
0
10 0 ?
9 0-
80 -
7 0-
6 0-
5 0-
4 0-
30 -
??? 0 -
0 -
I,%
*
5 0
5 1
., J . .
1 0 0
m /
1 0 5
77 9
¦ - ??
? d 1
b
*¦'!
1
1 5 0
?
1 2 3
I _
Ju
20 0
2 0 0
L_
2 5 0
в
10 0 15 0
m /z
Рис. 1.5. Масс-спектры л-фторбензофенона на восходящей стороне (а), на нисходящей
стороне (б) и вершине хроматографического пика (в)
Хромасс добавляет к масс-спектрометрической информации еще один очень
важный параметр — время удерживания. Именно благодаря этому параметру
появляется возможность во многих случаях проводить качественный и
количественный анализ изомеров, масс-спектры которых практически неразличимы.
Хромасс позволяет также детектировать ультрамикрокомпоненты на фоне
высоких концентраций других соединений. Для этой цели используется мониторинг
заданных ионов (разд. 9.2). Тем не менее следует отметить, что число
соединений, которые можно проанализировать методом ГХ-МС, значительно меньше,
чем при использовании масс-спектрометра с прямым вводом. Действительно, в
последнем случае можно получить масс-спектр соединения, обладающего хотя
бы слабой летучестью в условиях глубокого вакуума и температуре 300-400 °С,
не говоря уже об альтернативных методах ионизации (бомбардировка быстрыми
атомами, полевая десорбция, десорбционная химическая ионизация). В случае
же ГХ-МС необходимо, чтобы вещество прошло через колонку газового
хроматографа при атмосферном давлении и температуре 250-300 °С (в некоторых
случаях до 400 °С). Это приводит к невозможности анализа без предварительной
дериватизации труднолетучих, высокополярных, термолабильных соединений.
Рис. 1.6а. Масс-спектр на вершине хроматографического пика
Scon 834 <?2·7.00? mln): ISJEFIF^AI .D
Рис. 1.66. Масс-спектр у подножия хроматографического пика (спектр фона)
8-
s
1 ? ? ? ¦ ? ? ? ? ?
?
?
¦ ¦ < ? ¦ <
if
i
m
J
щ
_j
ч
Г ?
r ?-
g I
г ~]
8—J
1-Я
? 1
? ? I · ? ? ?
Д
О
К
к
я
«
?
Ч
о
о
й
К
?
<?
о
cd
?
CD
И
О
в
S
&
о
к
U
со
о
К
У
am
8
И
On
On
О
<L>
?
о
13
м
к
?.
и
ё
?
?
?
20 Глава!. Система ввода образца
Рис. 1.7. Вид и площадь наблюдаемого сигнала хроматографического пика в зависимости от
скорости сканирования, а — аналоговый сигнал; б — за время выхода компонента
зарегистрировано 13 масс-спектров; в — за время выхода компонента зарегистрировано 7 масс-спектров;
г —за время выхода компонента зарегистрировано 3 масс-спектра
В методе реакционной хроматомасс-спектрометрии перед колонкой
хроматографа или между колонкой и масс-спектрометром устанавливается
реакционная камера, в которой можно осуществлять заданные превращения
анализируемых соединений. В таком варианте дериватизация, разделение и анализ
компонентов пробы осуществляются в режиме «on-line». Реакционная хроматомасс-
спектрометрия может использоваться для улучшения разделения компонентов
смеси или для проведения структурных анализов. Например, дейтерирование
алкенов в реакционной камере позволяет установить положение двойной связи
в молекуле [21].
В 2001 г. в США вышла в свет книга [45], посвященная теории, практике и
последним достижениям хроматомасс-спектрометрии.
1.5. Жидкостная хроматография—масс-спектрометрия,
ЖХ-МС (LC-MS)
Решить проблему анализа тяжелых полярных термолабильных соединений
можно при замене газового хроматографа на жидкостный. При всей принципиальной
очевидности такого решения создать эффективный метод ЖХ-МС оказалось
не столь простой задачей. Наиболее сложным моментом оказывается стыковка
жидкостного хроматографа с масс-спектрометром. Масс-спектрометр обычно
работает в условиях глубокого вакуума A0~6 мм рт. ст.), а скорость потока
через стандартную колонку жидкостного хроматографа составляет примерно
1 мл/мин. Две наиболее часто используемые в современной хроматографии
жидкие фазы —метанол и вода. Испаряясь в источнике ионов, 1 мл метанола
образует 0,55л пара, а 1 мл воды—1,24л пара. Учитывая, что вакуумная
система масс-спектрометра может поддерживать требуемое для работы давление
только в том случае, если приток паров в ионный источник не превышает
10 мл/мин, понятно, что соединительный узел (интерфейс) системы ЖХ-МС
должен эффективно осуществлять обогащение анализируемым соединение^
примерно в 100 раз за счет удаления растворителя. При этом надо помнить, что
проводящие жидкости могут быть причиной возникновения электрической дуги
1.5. Жидкостная хроматография—масс-спектрометрия 21
в магнитных секторных приборах, работающих в условиях высокой разности
потенциалов, а нелетучие неорганические соли, используемые в качестве буфера
в хроматографии, приводят к проводящим отложениям на линзах источника
ионов, вызывая расфокусировку прибора.
К настоящему времени существует значительное число интерфейсов ЖХ-МС.
Часть из них уже практически не используется. Ниже представлены наиболее
важные варианты.
1.5.1. Ленточный транспортер
(Moving Belt)
Исторически первым интерфейсом можно считать ленточный транспортер [46],
появившийся в 1976 г. Принципиальная схема устройства представлена на
рис. 1.8.
Его основной конструкционной деталью является тонкая полиимидная
кольцевая лента. Элюат из колонки хроматографа попадает на ленту, движущуюся
по направлению к источнику ионов. На пути следования он проходит вдоль
инфракрасных испарителей, которые переводят в газовую фазу значительную
долю подвижной фазы. Далее лента проходит вакуумный замок, и в условиях
вакуума остатки растворителя откачиваются насосами. В месте перегиба ленты,
в ионном источнике, находится импульсный нагреватель, который переводит
в газовую фазу молекулы образца, подвергающиеся электронному удару или
химической ионизации. Двигаясь в обратном направлении, лента очищается
от избытка нанесенного образца благодаря дополнительным испарителям, а
иногда и растворителям, и возвращается за новой порцией элюата в исходном
(теоретически чистом) виде.
ИК-испаритель
Вакуумные прокладки
Изолятор
1L—ш—ш7^
Выход из ЖХ
я
Полиимидная лента
? ?
Нагреватель
образца
ПТ^^
0,1-0,5 5-50
мм рт.ст.
Промывочный
сосуд
Очищающий
нагреватель
Рис. 1.8. Принципиальная схема ленточного транспортера
22 Глава!. Система ввода образца
К достоинствам метода следует отнести:
1. Возможность использования стандартных библиотек масс-спектров
электронного удара для идентификации исследуемых веществ.
2. Возможность работы со стандартными колонками для жидкостной
хроматографии, поскольку метод использует скорости потока до 1.5 мл/мин. Такие
потоки приемлемы при распылении элюата на ленту.
3. Неорганические соли, используемые в качестве буфера при хроматографиро-
вании, не мешают анализу.
Тем не менее ленточные транспортеры не получили широкого признания,
поскольку лишь незначительно расширяли круг органических соединений,
доступных масс-спектрометрическому анализу. В ионном источнике все равно
требовался термический перевод вещества в газовую фазу. Лента транспортера
примерно через 10 прохождений оказывалась загрязненной, что приводило к
наложению масс-спектров друг на друга и мешало надежной идентификации.
Качество спектров и интенсивность характеристических ионов существенно
зависели от количества образца, т. е. воспроизводимость была плохой. В настоящее
время ленточные транспортеры не используются, хотя в 70-80 годах прошлого
века вышло в свет много работ, результаты которых базировались на применении
этого метода.
1.5.2. Прямой ввод жидкости
(Direct Liquid Introduction, DLI)
Данный метод тоже следует рассматривать скорее как историю масс-спектро-
метрии. Он более или менее активно использовался в 80-х годах XX века,
но уступил место более прогрессивной технике, основанной на аналогичном
принципе. Фактически все современные интерфейсы ЖХ-МС, за исключением
проточного варианта бомбардировки быстрыми атомами, могли бы быть названы
прямым вводом жидкости.
Для реализации прямого ввода жидкости на выходе из хроматографиче-
ской колонки устанавливается диафрагма с отверстием 2-5 мкм (рис. 1.9).
Возникающая струя мелких капель, движется с большой скоростью к источнику
Обогреваемый
участок удаления
? растворителя
«- гг-1 ksm I I
Ионный источник
Рис. 1.9. Принципиальная схема метода прямого ввода жидкости
1.5. Жидкостная хроматография—масс-спектрометрия 23
ионов. На пути следования капли проходят через камеру испарения, где большая
часть растворителя переходит в газовую фазу. Оказываясь в источнике
масс-спектрометра, молекулы анализируемого вещества подвергаются химической
ионизации (разд. 5.2), тогда как молекулы растворителя играют роль газа-реагента.
Основной недостаток метода— низкая скорость потока. Давление в источнике
становится слишком высоким при потоках более 50 мкл/мин. Поэтому требуется
либо разделять поток из стандартных хроматографических колонок, уменьшая
тем самым чувствительность метода примерно в 20 раз, либо использовать
узкие колонки. Кроме того, узкое отверстие диафрагмы постоянно забивается,
а добавки неорганических солей к подвижной фазе приводят к быстрому
ухудшению работы масс-спектрометра.
1.5.3· Поток частиц
(Particle Beam)
В этом варианте интерфейса поток из колонки хроматографа направляется через
капилляр в стеклянный распылитель, где элюат превращается в облако мелких
капель, разорванных концентрическим потоком гелия. Это облако
перемещается внутри стеклянной испарительной камеры, стенки которой подогреваются,
а давление поддерживается на уровне чуть ниже атмосферного. При этом
происходит частичное испарение растворителя, и размер капель уменьшается.
Испарительная камера заканчивается узким отверстием, за которым следует
сепаратор молекулярного пучка с вакуумной откачкой (рис. 1.10). Вылетая
из испарительной камеры, частицы приобретают высокую скорость благодаря
сужению потока. Поскольку более тяжелые молекулы анализируемого вещества
менее подвержены диффузии, чем атомы гелия или молекулы растворителя,
именно они и проходят в узкое входное отверстие сепаратора, а из него в масс-
спектрометр.
Испарительная
камера
?
К насосу
Капли образца
Капилляр
Рис. 1.10. Принципиальная схема интерфейса с потоком частиц
24 Глава 1. Система ввода образца
Частицы анализируемого вещества в ионном источнике подвергаются
химической или электронной ионизации. Получение спектров электронного удара
является преимуществом метода, поскольку позволяет использовать
компьютерные библиотеки масс-спектров (разд. 4.4). Такой интерфейс может работать
с потоками жидкости из хроматографа в диапазоне от 0,1 до 1,0 мл/мин.
Чувствительность метода для получения полного масс-спектра лежит на уровне
единиц нанограммов (Ю-9 г). Этот метод фактически заменил ленточный
транспортер, хотя и не устранил недостатков, связанных с невозможностью работы с
высокомолекулярными, нелетучими и термолабильными соединениями.
1.5.4. Термораспыление или термоспрей (Thermospray, TSP) и
плазмораспыление или плазмаспрей (Plasmaspray)
Принципиальная схема методов термораспыление и плазмораспыление
представлена на рис. 1.11. В этом случае поток из колонки жидкостного
хроматографа направляется в нагреваемый капилляр. Диаметр капилляра 0,1 мм, что
значительно больше по сравнению с отверстием для метода прямого ввода
жидкости. Это устраняет случаи закупорки — серьезный недостаток прямого
ввода жидкости. Температура капилляра поддерживается на уровне температуры
кипения растворителя. В результате из капилляра вырывается струя пара, которая
попадает в ионизационную камеру. В случае плазмаспрея подача напряжения на
разрядный электрод или просто пучок электронов создают условия химической
ионизации (разд. 5.2), причем роль газа-реагента играют пары растворителя.
Работая в режиме плазмораспыления, исследователи замечали, что часто
удается регистрировать ионы даже в случае выключенного электрода. Анализ
этого эффекта привел к возникновению техники термораспыления. Дело в том,
что некоторые анализируемые соединения уже в растворе находятся в виде
положительных или отрицательных ионов. Используемые в большинстве случаев в
качестве растворителей в жидкостной хроматографии вода и метанол с
добавками кислот и оснований могут протонировать или депротонировать субстрат.
Появлению заряда служат также добавки электролитов, например ацетата аммония.
Струя пара, выходящая из капилляра, представляет собой аэрозоль небольших
заряженных капель. Нагретый источник ионов и работающие вакуумные насосы
заставляют эти капли при движении уменьшаться в размере. В конечном итоге
градиент поля на каплях может достигать критических значений, что приводит к
устранению сольватной оболочки и появлению свободных ионов в газовой фазе.
Выталкивающий потенциал направляет эти ионы в узкое отверстие, ведущее к
анализатору, а избыток растворителя откачивается насосами (рис. 1.11).
Термоспрей и плазмаспрей применимы к потокам жидкости из хроматографа
в диапазоне от 0,5 до 2,0 мл/мин, что позволяет работать с обычными
аналитическими колонками. Следует отметить нежелательность использования в
качестве буфера нелетучих неорганических солей из-за возможного образования
1.6. Сверхкритическая флюидная хроматография—масс-спектрометрия 25
Разрядный
электрод
Выталкивающий
электрод
> К насосу
Рис. 1.11. Принципиальная схема термоспрея (плазмаспрея)
отложений в источнике ионов. В качестве недостатка метода можно отметить
его плохую воспроизводимость. Интенсивность спектра, а также наличие или
отсутствие фрагментных ионов существенно зависят от состава раствора,
наличия добавок, температуры капилляра и источника. В связи с этим важным
становится выбор оптимальных условий регистрации спектра для каждого
анализируемого вещества. Чувствительность метода лежит в очень широком
диапазоне, от единиц нанограммов до десятков микрограммов. Увеличение
доли пиков фрагментных ионов в спектре достигается увеличением потенциала
на выталкивающем электроде. Причиной тому становятся более эффективные
столкновения разогнанных ионов с молекулами, так как в этой части ионного
источника давление достаточно высоко.
Для ознакомления с другими существующими системами ввода образца
в масс-спектрометр целесообразно предварительно усвоить основные методы
ионизации веществ. Подробное описание проточного варианта бомбардировки
быстрыми атомами, химической ионизации при атмосферном давлении,
электрораспыления, ультразвукового распыления, а также метода пиролитической масс-
спектрометрии представлено в гл. 5. Для углубленного изучения принципов и
применения ЖХ-МС можно обратиться к книгам [47, 48].
1.6. Сверхкритическая флюидная
хроматография—масс-спектрометрия, СФХ-МС
(Supercritical Fluid Chromatography/Mass
Spectrometry, SFC/MS)
Помимо газа или жидкости подвижной фазой для хроматографического
разделения органических соединений может служить вещество, находящееся в
сверхкритическом состоянии. В сверхкритической флюидной хроматографии
(СФХ) используемое в качестве подвижной фазы вещество можно представить
26 Глава 1. Система ввода образца
как очень плотный газ. Наиболее часто используемый диоксид углерода в таком
состоянии имеет свойства промежуточные между жидкостью и газом, т. е. это
плотный газ с высокой сольватирующей способностью. Растворение образца
в таком материале можно рассматривать как процесс, родственный переводу
вещества в газовую фазу, но протекающий при достаточно низкой температуре.
Как уже отмечалось в разд. 1.5, посвященном ЖХ-МС, основной проблемой
стыковки жидкостного хроматографа и масс-спектрометра является большой
объем вещества (растворителя), поступающего в ионный источник, что трудно
совместимо с условиями высокого. вакуума в нем. В СФХ поток подвижной
фазы значительно меньше, т. е. комбинация СФХ-МС осуществима с меньшими
сложностями по сравнению с ЖХ-МС. Фактически метод СФХ-МС является
промежуточным между ЖХ-МС и ГХ-МС и в плане инструментальных
требований, и в плане свойств анализируемых соединений. Важно, что метод позволяет
анализировать более тяжелые и более термолабильные соединения по сравнению
с ГХ-МС.
В 80-х годах XX века казалось, что метод может существенно потеснить ЖХ-
МС благодаря большей разрешающей способности и уже существующим в то
время эффективным интерфейсам. Однако изобретение электроспрея (разд. 5.12)
и создание метода капиллярного электрофореза (разд. 1.7) отодвинули СФХ-МС
на вторые позиции. Тем не менее метод по-прежнему широко используется
для анализа пестицидов, силиконов, неионогенных ПАВ, природных соединений
и т. д. Еще одним преимуществом СФХ-МС над ГХ-МС является тот факт, что
в сверхкритическом состоянии вещество, используемое в качестве подвижной
фазы, является отличным растворителем и очень эффективно экстрагирует
органические соединения разных классов из образцов пищи, полимерных
материалов, биоты, объектов окружающей среды.
Существует огромное разнообразие интерфейсов для стыковки
сверхкритического флюидного хроматографа с масс-спектрометром [49]. Для метода
подходят практически все интерфейсы, используемые в ЖХ-МС, хотя они
могут быть слегка модифицированы. Результаты часто улучшаются при
увеличении полярности подвижной фазы, например при использовании в СФХ-МС
электроспрея (разд. 5.12) положительным образом сказывается добавление к
подвижной фазе метанола или другого полярного органического
растворителя [50]. Помимо электроспрея активно используется химическая ионизация
при атмосферном давлении (разд. 2.12). Применение ленточного транспортера
(разд. 1.5.1), потока частиц (разд. 1.5.3) или специальных капиллярных
ограничителей позволяет работать в режимах электронного удара (разд. 2.1) и химической
ионизации (разд. 5.2). Следует подчеркнуть, что в случае химической ионизации
газом-реагентом служит не СО2, а метан, изобутан или аммиак. Последний
часто оказывается предпочтительным, поскольку процесс ионизации аммиаком
подвержен менее значительному влиянию изменений парциального давления
СО2 в процессе анализа. Это связано прежде всего с очень низким сродством
1.7. Капиллярный электрофорез—масс-спектрометрия 27
к протону молекулы углекислого газа. Некоторое «раскрытие» ионизационной
камеры, хотя и ухудшает чувствительность, позволяет получать классические
спектры электронного удара. Ионизация молекул образца осуществляется как
напрямую, при их взаимодействии с электронами, так и в результате
перезарядки при взаимодействии с СО^"\ Такие спектры перезарядки (разд. 5.2)
оказываются практически аналогичными обычным спектрам электронного удара.
Их даже можно использовать для поиска по компьютерным библиотекам масс-
спектров [51].
Говоря об анализаторах, следует отметить, что большинство опубликованных
работ выполнено на квадрупольных приборах, для которых вакуумные
требования минимальны (разд. 6.5). Однако исследования велись также на магнитных
(разд. 6.1) и времяпролетных (разд. 6.7) масс-спектрометрах, а также ионных
ловушках (разд. 6.6).
1.7. Капиллярный электрофорез—масс-спектрометрия
(Capillary Electrophoresis/Mass Spectrometry,
СЕ/MS)
Создание капиллярного варианта электрофореза [52] существенно повысило
разрешающую способность разделения компонентов смесей (до 107 теоретических
тарелок), и, как следствие, эффективность анализа, а стыковка электрофорети-
ческого капилляра с масс-спектрометром подняла этот метод на новый уровень
[53, 54]. В данном случае можно с натяжкой говорить о принципиально другой
системе ввода образца в масс-спектрометр, потому что в качестве интерфейса
используются системы ЖХ-МС. Наилучших результатов удалось достигнуть при
использовании на выходе из колонки техники электрораспыления (разд. 5.12).
Возможно также использование техники ультразвукового распыления (разд. 5.13)
[55-57]. Тем не менее капиллярный электрофорез — масс-спектрометрия по
праву считается самостоятельным методом анализа прежде всего в случае
биологических молекул (белков, аддуктов с ДНК и т. д.). Поскольку наряду
с очень малым количеством пробы (менее 10~9 л) капиллярный электрофорез
позволяет проводить сверхбыстрое разделение компонентов смеси с шириной
пика в несколько миллисекунд [58], необходимо осуществлять такое же быстрое
сканирование спектра. Впечатляющие результаты получены при использовании
времяпролетного анализатора с ортогональным ускорением [59] и масс-спектро-
метрии с преобразованиями Фурье [60].
Глава 2
ФИЗИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА
МАСС-СПЕКТРОМЕТРИЧЕСКОГО
РАСПАДА
Поскольку масс-спектрометрия имеет дело с положительными или
отрицательными ионами, после ввода вещества в прибор необходимо провести
ионизацию молекул образца. На сегодняшний день существует несколько
десятков методов ионизации. Часть из них используется очень активно, часть —
только в единичных экспериментах. Популярность метода может достигать
максимума в какой-то момент времени, а затем с появлением новых, более
эффективных, сходить на нет.
2.1. Электронный удар или электронная ионизация, ЭУ
(Electron Impact, Electron Ionization, EI)
Среди различных методов ионизации особое положение занимает электронный
удар (ЭУ). Исторически это первый метод ионизации органических соединений.
Он же остается и наиболее распространенным на сегодняшний день. Основными
достоинствами метода являются надежность и универсальность. Кроме того,
в существующих компьютерных библиотеках масс-спектров (Wiley и NIST)
используются именно спектры электронного удара. Теории масс-спектрометри-
ческого распада и подходы к интерпретации спектров, о которых идет речь в
этой главе и последующих (гл. 3, 4), также базируются в основном на
первоначальном образовании молекулярного катион-радикала в результате электронной
ионизации.
Название метода —электронный удар — несколько не соответствует
действительности. Реального удара электронов по молекуле не происходит. Электрон,
пролетая вблизи молекулы, возбуждает ее электронную оболочку. Собственные
электроны молекулы перемещаются на более высокие орбитали и могут
покинуть молекулу. В связи с этим в последнее время термин «электронный удар»
(особенно в англоязычной литературе) заменяется на «электронную ионизацию».
Пучок электронов генерируется катодом (проволока или пластина из рения
или вольфрама) и ускоряется потенциалом 12-70 В по направлению к аноду
(рис. 2.1). Вещество в газовой фазе (давление 10~5-10~6 ммрт. ст.)
взаимодействует с электронами и ионизируется. Формально можно представить процесс
ионизации уравнением 2.1:
М + ё=М+'+2ё B.1)
2.1. Электронный удар или электронная ионизация 29
я
Катод
Ионы
А
Отверстие для
ввода образца
Анод
к ?_/? верите
JL ввода обра
Выталкивающий
электрод
Рис. 2.1. Принципиальная схема источника электронного удара
В результате образуется молекулярный ион (М+*). Это — нечетноэлектронный
ион, т. е. катион-радикал. Эффективность ионизации очень низка. Фактически
ионизируется примерно одна из десяти тысяч молекул образца. Более 99,99%
неионизованных молекул вещества откачивается из источника вакуумными
насосами. Этот факт, тем не менее, позволяет еще раз подчеркнуть высочайшую
чувствительность масс-спектрометрии, когда для получения спектра нужны
нано- и пикограммовые количества вещества. Вероятность ионизации меняется
от вещества к веществу. Эта характеристика соединения имеет количественный
показатель, называемый сечением ионизации.
Важным параметром является энергия ионизирующих электронов. График
зависимости ионного тока от энергии ионизирующих электронов представлен
на рис. 2.2. Ионный ток достигает максимума при энергиях электронов около
50 эВ. Стандартные масс-спектры ЭУ принято снимать, используя
ионизирующие электроны с энергией около 70 эВ.
Использование электронов с энергией около 70 эВ помимо высокой
эффективности ионизации объясняется и большей стабильностью спектра. Пучок
электронов не монохроматичен, причем разброс по энергиям, как правило, очень
велик и может достигать 5 эВ. Это приводит к тому, что условия взаимодей-
ЭИ 50 10° 150
Энергия электронов
Рис. 2.2. Зависимость величины ионного тока от энергии ионизирующих электронов
30 Глава 2. Физические основы процесса масс-спектрометрического распада
ствия вещества с низкоэнергетическими электронами (крутая часть графика
на рис. 2.2) не стабильны, а соответственно, не стабилен и масс-спектр. Чем
меньше угол наклона кривой эффективности ионизации в какой-либо точке, тем
выше стабильность масс-спектра, поэтому высокоэнергетическая часть кривой
предпочтительна.
Для того чтобы молекула образца потеряла свой электрон, необходима
энергия ионизирующих частиц выше какого-то определенного критического
значения. Эта энергия характеристична для каждого индивидуального соединения
и называется энергией ионизации (ЭИ). Поскольку каждая молекулярная или
атомная орбиталь имеет свою собственную энергию, молекула имеет несколько
значений энергии ионизации (см. метод фотоэлектронной спектроскопии [61]). В
масс-спектрометрии под энергией ионизации подразумевается первый потенциал
ионизации, т. е. энергия высшей занятой молекулярной орбитали. Величина
ЭИ для большинства органических соединений лежит в диапазоне 6-12 эВ.
Если энергия электронов, эмиттируемых катодом, ниже ЭИ, ионизация образца
невозможна, и масс-спектр не будет получен.
В процессе ионизации молекулярный ион получает избыточную внутреннюю
энергию в диапазоне 0-20 эВ. Эта избыточная энергия равномерно
распределяется по всем связям, причем превышение энергии какой-либо связи, ведет к ее
разрыву с отщеплением нейтрального фрагмента и образованием осколочного
иона. Минимальная энергия ионизирующих электронов, при которой в масс-
спектре помимо молекулярного будет регистрироваться осколочный ион,
называется энергией появления (ЭП) данного иона. Чем выше энергия ионизирующих
электронов, тем большее число направлений распада М+' реализуется, причем,
если избыточная энергия осколочного иона остается высокой, могут идти
вторичные процессы его распада. Принимая во внимание, что различия в ЭП
осколочных ионов очень незначительны, небольшие изменения энергии
ионизирующих электронов могут приводить к весьма существенным изменениям в
масс-спектре. Если учесть отсутствие монохроматичности пучка ионизирующих
электронов, понятно, что оптимальные условия получения спектра достигаются
при энергиях электронов 50-70 эВ, когда максимален выход ионного тока,
минимален наклон кривой эффективности ионного тока (рис. 2.2), а также
задействованы все возможные направления распада М+\
Наряду с однозарядными образуются двух и более зарядные ионы.
Количество многозарядных ионов существенно меньше, чем однозарядных, и зависит
прежде всего от структуры молекул образца, а также от условий эксперимента.
Например, повышенным выходом многозарядных ионов характеризуются
полициклические ароматические углеводороды (разд. 8.5).
В некоторых случаях, когда хотят увеличить интенсивность пика М+в,
снимают спектр, используя ионизирующие электроны с энергией 12-20 эВ. Следует
подчеркнуть, что в этом случае возрастает только относительная интенсивность
пика М+' и пиков перегруппировочных ионов (см.ниже) по отношению к интен-
2.1. Электронный удар или электронная ионизация 31
100 -| I, %
80
6 0
4 О
2 О
О
2 О
1 00
1,%
80
6 О ?
4 О
2 О ?
О
20
100 ?
8 0 -J
60 -J
40
20 ?
О
?,%
2 О
4 О
ilJ-
7 О Э В
1 О О
1 2 О
2 О Э В
m /z
1 4 э В
8 О
1 О О
1 2 О
6 0 8 0
m /z
1 О О
1 2 О
Рис. 2.3. Масс-спектр электронного удара этилпропионата (молекулярная масса 102 Да) при
энергиях ионизирующих электронов 70, 20 и 14 эВ.
сивности пиков осколочных ионов, тогда как абсолютная интенсивность всех
пиков в спектре падает. Кроме того, происходит потеря определенной информации,
так как многие направления фрагментации не реализуются в таких условиях.
Это положение проиллюстрировано на рис. 2.3. Видно, что при уменьшении
энергии ионизирующих электронов уменьшается число и интенсивность пиков
в спектре, хотя доля молекулярного иона возрастает. Важный вывод, который
следует помнить: если пик молекулярного иона отсутствует в масс-спектре,
полученном при энергии ионизирующих электронов 70 эВ, его не будет и при
меньшей энергии электронов. В этом случае можно говорить о нестабильности
молекулярного иона данного соединения. Кстати, значительное число
органических соединений характеризуется нестабильными молекулярными ионами в
условиях электронного удара. Это один из существенных недостатков данного
метода ионизации.
Наряду с положительными ионами в условиях электронного удара образуются
и отрицательные ионы (молекулярные анион-радикалы). К сожалению, качество
спектров отрицательных ионов в этом случае оставляет желать лучшего. В
частности, доминирующий пик в спектре зачастую обусловлен малоинформативным
ионом (Cl~, F~, CN~). Кроме того, выход отрицательных ионов значительно
меньше, чем положительных. Тем не менее информация, заключенная в масс-
32 Глава 2. Физические основы процесса масс-спектрометрического распада
спектрах отрицательных ионов, может служить хорошим дополнением к
полученной по спектрам положительных ионов. Детали фрагментации молекулярных
анион-радикалов основных классов органических соединений, полученных в
условиях электронного удара, подробно рассмотрены в работе [62].
Качество и информативность спектров отрицательных ионов можно повысить
при использовании электронов с малыми (тепловыми) энергиями. Метод
называется резонансный захват электрона и заключается в ассоциации электрона
одной из молекулярных орбиталей молекулы образца [26]. Как правило, ионы
образуются в нескольких резонансных областях с энергиями электронов 0-10 эВ.
Эти области могут характеризоваться разными выходами ионов и разными
направлениями фрагментации молекулярных анион-радикалов. Например,
молекулы 2-диазо-2-цианоацетамида и изомерного 4-циано-5-гидрокси-1,2,3-триазола
захватывают электроны в четырех резонансных областях энергий: 0,1, 1,5, 5,0
и 7,5 эВ, причем в трех последних ионные токи оказываются на 2-3 порядка
ниже [63]. Наличие нескольких резонансных областей придает масс-спектру
дополнительное измерение, поскольку химическое соединение характеризуется
несколькими масс-спектрами. Этот факт открывает хорошие перспективы по
масс-спектрометрической идентификации структурно близких соединений. К
сожалению, эти потенциальные возможности резонансного захвата электрона
реализованы на сегодняшний день далеко не в полной мере, хотя в последнее
время метод электронного захвата все шире и эффективнее используется для
анализа биологических молекул [64]. Другим примером использования
электронного захвата является метод селективного определения в нефти соединений,
содержащих гетероатомы. Метод основан на разности энергий электронов, которые
захватываются соединениями разных типов. Если большинство углеводородов
нефти захватывают электроны только с энергиями более 10 эВ, то гетероатом-
ные компоненты образуют анион-радикалы при взаимодействии с электронами,
энергия которых приближается к нулю [65].
Возвращаясь к классическому варианту спектров ЭУ, можно отметить, что
поскольку давление в источнике ионов в условиях ЭУ— 10~5-10~6 мм рт. ст.,
а образец можно нагревать до нескольких сотен градусов, в газовую фазу
переходят многие органические соединения. Тем не менее для анализа
термолабильных, высокомолекулярных, труднолетучих соединений метод не пригоден.
В спектрах ЭУ многих веществ пик М+' имеет низкую интенсивность либо
вообще отсутствует. Широкий разброс по энергиям ионизирующих электронов
не позволяет с достаточной точностью определять характеристики молекул и
ионов (энергии появления и ионизации). Это основные недостатки электронного
удара, работа над устранением которых привела к созданию представительного
ряда альтернативных методов ионизации (гл. 5).
2.2. Физические основы масс-спектрометрического распада 33
2.2. Физические основы
масс-спектрометрического распада
Существует ряд более или менее строгих подходов к описанию процессов
образования и распада молекулярных ионов. К сожалению, на сегодняшний день
ни один из них не может надежно предсказать массовые числа и интенсивности
пиков всех ионов в спектре даже достаточно простых органических соединений.
Во избежание чрезмерного углубления в физику и математику в данном разделе
будут отражены лишь основные подходы, преимущества и недостатки
существующих теорий.
Практически любой процесс генерирования заряженной частицы сопряжен
с передачей ей дополнительной внутренней энергии. В частности, в случае
наиболее распространенного (и наиболее жесткого) метода ионизации, каким
является электронный удар, образующиеся молекулярные ионы могут иметь
избыточную энергию в диапазоне 0-20 эВ. Учитывая, что 1 эВ = 96,48 кДж,
наиболее «горячие» ионы имеют энергию порядка 2000 кДж, т. е. значительно
выше энергии реакционных частиц, образующихся в растворах. Распределение
образовавшихся молекулярных ионов по энергиям (рис. 2.4, верхняя часть)
можно представить в виде вероятностной функции Р(Е). Эта кривая в определенной
мере напоминает фотоэлектронный спектр данной молекулы. В растворах в
результате столкновений происходит усреднение энергии частиц. В
ионизационной камере масс-спектрометра все определяется методом ионизации. В случае,
например, химической ионизации достаточно высокое давление в источнике
ионов приводит к многочисленным столкновениям ионов, радикалов и молекул,
Р(Е)
\gk(P)
6
5
/
эи
Рис. 2.4. Функции Р(Е) и к(Е) молекулярного иона органического соединения ABXY.
Диаграмма Вархафтига
2—1113
34 Глава 2. Физические основы процесса масс-спектрометрического распада
т. е. можно говорить об определенном усреднении их внутренней энергии.
Напротив, условия высокого вакуума при использовании электронного удара
препятствуют столкновениям частиц и судьба образовавшегося иона зависит
исключительно от той внутренней энергии, которую он приобрел в результате
ионизации. Именно эта энергия определяет то, что одна часть ионов достигнет
детектора без фрагментации, другая —в виде каких-либо перегруппировочных
ионов, третья —в виде каких-либо осколочных ионов, четвертая —в виде фраг-
ментных ионов второго поколения и т. д. Следует подчеркнуть, что, поскольку
речь идет о мономолекулярных реакциях изолированных частиц, живущих очень
короткое время, набор и количество продуктов реакции определяются прежде
всего кинетическими, а не термодинамическими факторами.
Здесь имеет смысл напомнить, что молекулярный ион образуется в
результате ионизации молекулы (потери электрона). Его величина m/z соответствует
молекулярной массе соединения. Фрагментные ионы образуются при распаде
молекулярного. Осколочные ионы образуются в результате простых разрывов
связей. Перегруппировочные ионы наряду с разрывами связей требуют
образования новых связей между атомами (группами), не связанными напрямую в
исходной молекуле.
В 1952 г. практически одновременно возникли две теории, направленные на
интерпретацию мономолекулярных реакций в газовой фазе в условиях глубокого
вакуума. Розеншток [66] предложил физическое описание процессов в
источнике масс-спектрометра. Эта теория была названа «квазиравновесной» (КРТ),
поскольку постулировала, что за 10~12 с избыточная энергия равномерно
распределяется по всем связям в молекуле, точнее по всем энергетическим состояниям
молекулярного иона, т. е. «квазиравновесие» между всеми этими состояниями
достигается до того, как начнется распад. Вероятности путей фрагментации
определяются только энергией и структурой каждого конкретного иона, а не
методом ионизации, механизмом образования или структурой предшественника
(для осколочных ионов). Исключения связаны, как правило, с простейшими
молекулами, случаями далеко отстоящих друг от друга возбужденных электронных
состояний и быстрыми (к > 10й) процессами распада. Близкая КРТ теория,
описывающая процессы в нейтральных молекулах, была названа по фамилиям
ее авторов Раиса, Рампсберга, Касселя и Маркуса — РРКМ [67].
Обе теории базируются на определенных допущениях:
1. Вращательные, колебательные, электронные и поступательные движения
частиц независимы друг от друга.
2. Движения ядер могут быть описаны уравнениями классической механики,
хотя и с учетом квантовохимических коррекций.
3. Все микросостояния частиц равновероятны, т. е. энергия равновероятно
распределяется по всем степеням свободы иона.
2.2. Физические основы масс-спектрометрического распада 35
4. Существует энергетическая граничная поверхность, разделяющая
родительские и дочерние ионы. Это поверхность может пересекаться только в одном
направлении, т. е. для любого родительского иона достижение этого
переходного состояния означает необратимый распад с образованием продуктов.
Основная задача теорий — расчет скоростей распада частиц с разрывом любой
химической связи в зависимости от избытка внутренней энергии. Рассмотрим
несколько подробнее ионизацию под электронным ударом. Сам процесс потери
электрона нейтральной молекулой протекает за Ю-16 с. По принципу
Франка—Кондона [68] расстояния между ядрами при этом не изменяются. Колебания
ядер могут начаться через 10~12-10-13 с. После ионизации молекулярные ионы
могут оказаться на любых уровнях всех возбужденных (вращательных,
колебательных, электронных) состояний. Важно, что ион в возбужденном электронном
состоянии живет не более 10~~8 с. За это время в результате излучательных или
безызлучательных процессов ион переходит в основное электронное состояние
либо распадается. Поскольку время присутствия ионов в источнике электронного
удара составляет 10_6 с, около 99% времени ион находится там в основном
электронном состоянии. Когда, представляя схему фрагментации, указывают
заряд иона на конкретном атоме, это не означает, что в результате ионизации
электрон удален именно из этого места. Речь идет об изображении иона в
основном электронном состоянии.
Вероятность какой-либо трансформации (изомеризация, распад)
молекулярного иона в зависимости от его внутренней энергии (рис. 2.4, нижняя часть)
связана с константой скорости к(Е) конкретной реакции. Молекулярные ионы,
энергия которых ниже критической энергии (Eq) для самой низкоэнергетической
реакции распада, регистрируются в виде М+\ Предположим, что молекулярный
ион соединения ABXY может распадаться или с разрывом центральной связи
В—X, или претерпевать перегруппировку с образованием химической связи
между ранее несвязанными А и ? (схема 2.1).
AY4" + В = X <- ????+' -> АВ+ + ??'
Схема 2.1
Вернемся к рис. 2.4. Если ?(М+#) < ??(??+#), ион стабилен и
регистрируется как М+\ Обозначим как ??/2(??+') такую энергию, при которой ровно
половина М+' распадается с образованием ??+#, а половина остается в виде
М+'. Аналогично ??/2(??+) соответствует энергии, при которой половина М+'
распадается с образованием ??+', а половина —с образованием АВ+, т. е. для
представленного на рис. 2.4 случая все молекулярные ионы с энергией выше
??/2(??+) будут распадаться с образованием АВ+, так как в этой точке скорость
образования АВ+ становится выше скорости образования ??+*. Формально
площади под кривыми на рис. 2.4 с обозначениями М+\ ??+', ??+ соот-
36 Глава 2. Физические основы процесса масс-спектрометрического распада
Рис. 2.5. Зависимость интенсивности пиков ионов в спектре от внутренней энергии М+*
ветствуют долям тока этих ионов в полном ионном токе. Однако необходимо
подчеркнуть, что эти доли являются начальными. Для вычисления реальной
интенсивности соответствующих пиков в масс-спектре необходимо учитывать
вторичные процессы распада этих первичных ионов.
При изменении распределения внутренней энергии М+" (функция Р(Е)) вид
масс-спектра может существенно меняться. Некоторые реакции частично или
полностью подавляются более эффективными для данной энергии. Изменением
распределения Р(Е) объясняется, в частности, изменение спектра электронного
удара при снижении энергии ионизирующих электронов (рис. 2.3).
Один из вариантов записи масс-спектрометрической информации в
графическом виде представлен на рис. 2.5. Пусть молекулярный ион ABXY+*
распадается (схема 2.1) с простым разрывом связи (ион АВ+) или с перегруппировкой
(ион AY+*). Изменение спектра с изменением внутренней энергии М+# наглядно
демонстрирует рис. 2.5. Вертикальное сечение позволяет получить масс-спектр
при конкретном значении энергии М4*".
Как видно из рис. 2.4 и 2.5, реакция с низшей ?? далеко не всегда приводит
к наиболее интенсивному фрагментному иону. Для рассматриваемой молекулы
???? реакция образования иона ??+" имеет предпочтительную энтальпию, в
то время как энтропийный фактор благоприятствует возникновению тона АВ+.
Действительно, в первом случае рвутся две связи (АВ и ??) и образуются
две связи (?? и ВХ), т. е. энергетические затраты невелики. Во втором случае
происходит разрыв одной связи (ВХ), но новых связей не образуется. Однако
стерические требования в первом случае значительно строже. Ион АВ+ может
образоваться, как только связь ВХ будет обладать достаточной для разрыва
связи колебательной энергией, а для возникновения иона ??+' необходим
подход А с искажением валентных углов к ? на достаточное для образования
2.2. Физические основы масс-спектрометрического распада 37
ABXY
Рис. 2.6. Изменение потенциальной энергии вдоль координаты реакции для простого
разрыва связи {слева) и перегруппировки (справа)
связи расстояние, что реализуется лишь для небольшой части многочисленных
конформаций М+', имеющих достаточную для такого процесса энергию. Этот
простой пример демонстрирует конкуренцию различных направлений распада и
объясняет сложность масс-спектров в случае многоатомных молекул.
Изменение потенциальной энергии вдоль координаты реакции для простого
разрыва связи и перегруппировки представлено на рис. 2.6. Для реакции
простого разрыва связей (рис. 2.6, слева) обратный процесс практически всегда не
требует энергии или протекает с минимальной энергией активации ЕГ9 а для
перегруппировок (рис. 2.6, справа) обратный процесс может иметь значительную
энергию активации.
Ранее были введены определения энергии ионизации (ЭИ) молекулы и
энергии появления (ЭП) иона. Если для определения 3?(??+') фиксировать
появление в спектре пика иона AY+*, будет измерена не Eq(AY+'), а ??/2(??+').
Разность Е\/2 — Eq называется кинетическим сдвигом. Обычно он невелик
@,01-0,1 эВ), но может достигать величин до 2 эВ. Для случая АВ+ измерение
2?о(АВ+) еще больше усложнено, поскольку дополнительно к кинетическому
сдвигу возникает так называемый «конкурентный сдвиг»; при ? < ??/2(??+)
М+' будет предпочтительно распадаться с образованием AY+\
Масс-спектрометрическое измерение ЭИ, ЭП и сродства к электрону в
сочетании с известными термохимическими характеристиками нейтральных частиц
позволяет вычислить теплоты образования ионов и энергии связей. Масс-спек-
трометрически можно изучать нестабильные радикалы и получать информацию
об энергиях связей, недостижимую при использовании других методов.
Измерение ЭП фрагментных ионов является важным методом установления
их структур. Пусть необходимо установить структуру иона АВ+, образующегося
при распаде ABXY+\ Если ионы такого же состава были изучены ранее
(например, при их образовании из ABCD+', AB2Y+* и т. д.) и для них были
38 Глава 2. Физические основы процесса масс-спектрометрического распада
установлены структуры и теплоты образования, необходимо измерить ЭП иона
АВ+, образующегося из ABXY+", и рассчитать теплоту его образования по
уравнению 2.2:
?#(??+) = ЭП + ?#(????) - ?#(??*) B.2)
Совпадение полученной величины АЯ(АВ+) с аналогичной величиной для
иона АВ+ известной структуры, образующегося в другом процессе, является
весомым свидетельством в пользу их одинаковой структуры. Понятно, что
если ?#(??+) значительно отличается от АН всех изученных ранее ионов
АВ+, можно утверждать, что исследуемый ион обладает другой структурой.
Развитие компьютерных расчетов с помощью эмпирических методов или аЪ
initio позволило проверять приписываемые ионам структуры при сравнении
экспериментальных и расчетных величин теплот их образования.
2.3. Метастабильные ионы
Кинетический сдвиг при определении ЭП обусловлен тем, что осколочный ион
появляется в масс-спектре только из тех М+*, энергия которых достаточна для
протекания соответствующей реакции с константой скорости не ниже 106.
Однако существует возможность регистрировать процессы с константой скорости
105. Дело в том, что ион покидает источник электронного удара за 10~6 с, но
достигает детектора за 10~5 с, т. е. обладая достаточной энергией, он может
распасться по пути следования.
Рассмотрим, как будет регистрироваться этот ион, называемый «метаста-
бильным», в магнитном секторном приборе. Пусть ион т\ покидает ионный
источник, ускоряется напряжением V и распадается с образованием иона т^
и нейтральной частицы т\ в бесполевом пространстве между источником и
магнитным анализатором.
mf —* т^ + т®
Пусть vn скорость иона с массой тп. Тогда в бесполевом пространстве
правомерны уравнения сохранения энергии и момента количества движения:
т\ v\ = nt2v\ + miv\ B.3)
ТП\ V\ = 7W2V2 + JW3V3 B.4)
Так как т\ =т2 + тз, оба уравнения могут быть справедливы только в случае
vi = V2 = V3, т. е. в первом приближении можно считать, что продукты распада
движутся в том же направлении и с теми же скоростями, что и родительский
ион. Магнитный сектор масс-спектрометра является анализатором моментов.
Поэтому метастабильный ион с реальной массой W2 и скоростью vi будет
регистрироваться вместе со стабильным ионом т*, обладающим скоростью ?*
2.3. Метастабильные ионы 39
(уравнение 2.5). Необходимо лишь, чтобы их моменты количества движения
были равны.
m*v* = W2Vi B.5)
Уравнение 2.6 справедливо для любого стабильного иона.
Ht^^lHin^eV B.6)
2 2 v J
Исходя из двух уравнений B.5 и 2.6) получаем уравнение 2.7 для расчета
регистрируемой массы метастабильного иона:
т* = ™* B.7)
Таким образом, метастабильный ион, образующийся в первом бесполевом
пространстве однофокусного магнитного масс-спектрометра, будет
регистрироваться в спектре с кажущейся массой, рассчитываемой по уравнению 2.7.
Метастабильные ионы проявляются в виде слабых уширенных пиков. Низкая
интенсивность обусловлена незначительной долей молекулярных ионов, энергия
которых благоприятствует их распаду вне источника. Даже наиболее
низкоэнергетический процесс (перегруппировка с образованием AY+", рис. 2.4) приводит
к слабому пику метастабильного иона. Для более высокоэнергетических
реакций образование стабильного иона в результате менее энергоемкого процесса
выгоднее образования альтернативных метастабильных ионов. Пики последних
могут вообще не наблюдаться в спектре. Уширение пика метастабильного иона
связано с высвобождением части внутренней энергии при распаде. Эта энергия
переходит в кинетическую, причем направление приращения может быть от
полностью совпадающего до прямо противоположного относительно скорости
движения распадающегося иона.
Регистрация в спектре метастабильного иона очень полезна, поскольку
позволяет доказать протекание конкретной реакции, связывающей родительский
и дочерний ионы. Схемы масс-спектрометрической фрагментации считаются
надежно установленными, если они подтверждены пиками метастабильных
ионов. В то же время пики метастабильных ионов ухудшают разрешение в
спектре. В зависимости от количества энергии, выделяющейся при фрагментации
в бесполевом пространстве, форма пиков метастабильных ионов может быть
разной (рис. 7.5). К сожалению, эти пики исчезают из спектра в результате его
компьютерной обработки.
Так как электростатический анализатор пропускает ионы в зависимости от их
энергии, метастабильные ионы, обладающие меньшей энергией по сравнению со
стабильными, через него не проходят. Тем не менее пики метастабильных ионов
проявляются в масс-спектре, полученном на двухфокусном приборе прямой
геометрии (разд. 6.2), если фрагментация идет во втором бесполевом пространстве
после прохождения электростатического анализатора.
40 Глава 2. Физические основы процесса масс-спектрометрического распада
N-
-N2
?"
I
Ph
ОН
d?
Схема 2.2
Многие масс-спектрометры позволяют работать в режиме получения
спектров исключительно метастабильных ионов с дефокусировкой стабильных. В
данном случае речь идет о варианте тандемной масс-спектрометрии (гл. 7), но
без активации соударением. Так можно получать все виды спектров
(родительских ионов, дочерних ионов, выбросов идентичных нейтральных частиц). Такие
спектры позволяют устанавливать направления фрагментации, решать
структурные задачи. Однако интенсивность сигналов и даже наличие или отсутствие
пиков конкретных ионов в спектрах зависит от энергии родительских ионов.
Поэтому, в отличие от спектров активации соударением, различия в спектрах
дочерних ионов двух родительских ионов не обязательно свидетельствуют об их
разной структуре.
Примером использования метастабильных ионов может служить масс-спек-
трометрическое исследование К-фенил-4-циано-5-гидрокси-1,2,3-триазола [69].
Ионы [?—?2]+' триазолов обычно циклизуются в азирины [70, 71] (схема 2.2).
Однако спектр метастабильных дочерних ионов свидетельствует о том, что
одним из направлений распада [?—?2]" >1-фенил-4-циано-5-гидрокси-1,2,3-
триазола является отщепление радикала NCO". Такой процесс невозможен
для азирина и заставляет предположить более сложную трансформацию иона
[M-N2]+\ например образование оксоиндола при взаимодействии радикального
центра с бензольным ядром (схема 2.2).
2.4. Полуколичественная теория
масс-спектрометрического распада
К сожалению, строгая математизированная теория (КРТ или РРКМ) не
применима к реальным органическим молекулам, поскольку за исключением
простейших молекул все другие случаи требуют слишком большого времени даже
с использованием современной компьютерной базы. Кроме того, значительные
ошибки могут быть вызваны незнанием реальных структур ни родительских
2.4. Полуколичественная теория масс-спектрометрического распада 41
ионов, ни дочерних ионов, ни переходного состояния. Распределение энергии по
связям также не очевидно. Фактически, самым большим успехом этих строгих
физических теорий можно считать расчеты спектров легких алканов (пропан,
бутан), т. е. простейших молекул с одним типом химических связей. Возможно, в
будущем с созданием следующих поколений компьютеров эти проблемы удастся
преодолеть.
Существует упрощенный подход в рамках полуколичественной КРТ для
описания процессов фрагментации более сложных молекул. Для расчета констант
скоростей реакций распада используется уравнение 2.8:
k = V[—E
где к — константа скорости, ? — частотный фактор, ? — избыток внутренней
энергии, 2?о — критическая энергии конкретной реакции, N — число осцилляторов.
Частотный фактор ? обратно пропорционален мере стерических затруднений
в активированном комплексе (переходном состоянии) и фактически является
численной характеристикой изменения энтропии в процессе; для простого разрыва
связи он принимается равным ее колебательной частоте (ИК-спектроскопия).
Для перегруппировочного процесса требуется конкретное расположение атомов
в пространстве, что приводит к уменьшению величины v. Число осцилляторов в
молекуле, содержащей ? атомов, рассчитывается по формуле: N = Ъп — 6. Однако
на практике оказывается, что не все осцилляторы равноценны, и величина к
завышается. Поэтому число осцилляторов обычно уменьшают в три раза.
В случае использования этой упрощенной теории для расчета интенсивностей
фрагментных ионов в спектре нельзя забывать про вторичные процессы распада
этих ионов при наличии у них достаточной внутренней энергии. Интенсивность
пика иона в спектре зависит и от скорости его образования, и от скорости его
распада.
B.8)
Глава 3
ОСНОВНЫЕ ПРАВИЛА И ПОДХОДЫ
К ИНТЕРПРЕТАЦИИ МАСС-СПЕКТРОВ
Накопление достаточного эмпирического материала по направлениям
фрагментации органических соединений позволило выявить определенные закономерности
и создать качественные теории масс-спектрометрического распада. Допущения,
сделанные в этих теориях, не искажают реального смысла и позволяют легко
понять суть процессов, а также запомнить и использовать для расшифровки
масс-спектров возможные механизмы фрагментации соединений в зависимости
от их структуры. Кстати, используя материал по масс-спектрометрическому
распаду органических соединений, К. Биман [72] еще в 1962 г. предложил
первую классификацию основных реакций фрагментации. За этим последовала
классификация Бенца [73]. В России вопросам классификации масс-спектромет-
рических реакции много внимания уделяет В. В. Тахистов [22, 23, 74].
Наиболее признанными среди качественных теорий
масс-спектрометрического распада являются концепция локализации заряда и неспаренного электрона,
а также оценка стабильности ионов и нейтральных частиц. Несмотря на
качественный характер эти подходы оказываются весьма полезными для реальной
работы со спектрами. В обоих случаях используется принцип минимальных
структурных изменений на каждой стадии распада, а структура молекулярного
иона принимается идентичной структуре исходной молекулы, если, конечно,
какие-либо процессы изомеризации М+' до распада не доказываются особо.
Бессмысленно противопоставлять эти две качественные теории друг другу.
Критикуя второй подход, можно утверждать, что электроны в молекуле
обобществлены и невозможно установить точное место локализации заряда и
неспаренного электрона. Совсем не обязательно предполагать, что для начала распада
требуется какой-либо пусковой механизм, за исключением накопления
достаточной энергии в конкретной химической связи. Однако обе теории позволяют
сделать лишь качественные выводы, и использование их в комплексе помогает
успешно интерпретировать спектры разнообразных органических соединений.
3.1. Стабильность ионов и нейтральных частиц
Рассмотрим вновь реакцию распада иона ABXY+* (схема 2.1):
AY*' + В=Х <- ABXY+' -> АВ+ + XY#
3.1. Стабильность ионов и нейтральных частиц 43
.ABXY
Рис. 3.1. Энергетическая диаграмма образования иона АВ+ из молекулы ABXY
Известно, что с минимальными допущениями в подавляющем большинстве
случаев можно считать, что энергия активации обратной реакции Ег отсутствует
или очень мала. Используя постулат Хэммонда [75], можно предположить, что
для эндотермической фрагментации переходное состояние будет значительно
ближе к продуктам реакции, чем к исходной частице (рис. 3.1). Следовательно,
стабильность этих продуктов в значительной мере определяет эффективность
процесса фрагментации. Важно учитывать стабильность не только дочернего
иона, но и отщепляющейся нейтральной частицы.
При оценке стабильности можно использовать обычные критерии, известные
из курса органической химии. Третичный алкильный карбокатион устойчивее
вторичного, а вторичный —первичного. Для карбанионов этого типа порядок
стабильности обратный. Бензильный и аллильный ионы устойчивы благодаря
резонансной стабилизации, причем для бензильного катиона помимо четырех
резонансных форм существует возможность энергетически выгодной в газовой
фазе трансформации в катион тропилия, который имеет уже семь резонансных
форм (схема 3.1).
[_0^сн2 ^^ 0=??2 ^^ +0=??2 *-* С^°ч
>—?-?-?-?-?
Схема 3.1
44 Глава 3. Основные правила и подходы к интерпретации масс-спектров
Напротив, ^р2-гибридизованные фенильный и винильный карбокатионы
менее устойчивы по сравнению с их д/?3-гибридизованными аналогами.
Резонансная стабилизация приводит к значительному увеличению стабильности в случае
возможного участия и-электронов гетероатомов (схема 3.2).
CH2~NH2 —- CH2=NH2 СН2-ОСНз «*—** СН2=ОСН3
Схема 3.2
Распад может осуществляться по определенному направлению благодаря
удобной делокализации электрона в нейтральном продукте фрагментации. В
этом случае вновь «работают» классические правила органической химии.
Делокализация электрона вследствие эффекта резонанса увеличивает
стабильность аллильного или бензильного радикалов, участие связей ?-СН приводит к
повышенной стабильности третичных алкильных радикалов и т. д. Тем не менее
следует помнить, что стабилизация заряда более существенна, чем стабилизация
неспаренного электрона. Если трет-С^^ стабильнее Н-С4Н9" на 1,5 эВ, то
трет-С^Щ стабильнее «-С4Н9 всего на 0,4 эВ.
Зачастую ионы распадаются с выбросом небольших нейтральных молекул:
водород, метан, вода, монооксид и диоксид углерода, монооксид азота,
хлористый водород, сероводород, метанол и т. д. Большая отрицательная величина
теплоты образования этих молекул создает преимущества для реализации
соответствующего направления распада. Альтернативные процессы,
сопровождающиеся выбросами менее стабильных частиц, проигрывают конкуренцию, что
может приводить к отсутствию соответствующих пиков в спектре. В результате
маскируется наличие в соединении каких-то важных функциональных групп.
3.1.1. Правило выброса максимального алкильного радикала
Важнейшим исключением, когда интенсивность пика ионов падает с
увеличением их термодинамической стабильности, является предпочтительное отщепление
большего алкильного радикала.
Этот эффект называется правилом выброса максимального алкильного
радикала. Правило «работает» всегда и часто помогает при интерпретации
спектров. В качестве примера можно привести фрагментацию молекулярных
ионов третичных аминов (схема 3.3). На первой стадии М+# теряет один из трех
углеводородных радикалов (R1", R2' или R3'). Среди трех альтернативных
фрагментов интенсивность пика ионов, образующихся при выбросе максимального
радикала, будет наивысшей, а при выбросе минимального радикала —низшей.
Аналогично среди первичных фрагментов, образующихся при распаде спиртов,
максимальную интенсивность будет иметь пик иона [М — R,-]+, возникающего
при отщеплении максимального радикала (схема 3.4). Кстати, интенсивный
3.1. Стабильность ионов и нейтральных частиц 45
?· 2"
CH2=N—СН2— R2 «^- R—СН2—N—СН2—R2 -^» R—СН2—Й=СН2
I I I
сн2 сн2 сн2
L· ' з r3
R R3 R
?-
R3*
R1— СН2— N — СН2—R2
II
сн2
Схема 3.3
пик иона [М—Н]+ означает, что атом углерода, от которого отщепляется атом
водорода, не связан с алкильными радикалами.
В некоторых случаях правило выброса максимального алкильного радикала
имеет не только качественный, но и количественный аспект. Загорский [76],
изучая фрагментацию аминов и спиртов, показал, что интенсивности пиков ионов,
обусловленных выбросами алкильных радикалов из молекулярных ионов, могут
быть рассчитаны, причем отношение интенсивностей пиков ионов [М—R,]+
обратно пропорционально отношению масс этих ионов.
Это правило применимо к отщеплению радикалов не меньше этильного,
а интенсивности пиков вторичных ионов должны суммироваться с
интенсивностью пика соответствующего первичного иона. В частности, распад М+*
4-этилоктанола-4 (схема 3.5) начинается с альтернативного отщепления трех
алкильных радикалов (этильного, пропильного или бутильного). При энергии
ионизирующих электронов 13 эВ, что позволяет минимизировать вторичные
+ +· +
ОН ОН ОН
R2 +^- R1—С—R2 -^C Rf—С
R3 R3 R3
-R
+
ОН
з·
R1 С R2
Схема 3.4
46 Глава 3. Основные правила и подходы к интерпретации масс-спектров
+ +· +
он он он
II -с2Н5 I -gjk II
с—с3н7 с2н5 — с—с3н7 И с2н5—с
г
C4H9 C4H9 C4H9
m/z\29 I m/z\\5
J -С4Н9
+
OH
с2н5—с—с3н7
m/z101
Схема 3.5
процессы распада, интенсивности пиков первичных фрагментных ионов с
массами 129, 115 и 101 составляют 78, 85 и 100% соответственно. Рассчитанные
интенсивности должны составлять 101/129 = 0,78 и 101/115 = 0,88. Как
видно, сходимость результатов достаточно хорошая. При количественной оценке
отщепления метильной группы предложенный расчет дает интенсивность пика,
завышенную в 2-3 раза.
Задача 3.1. Оцените интенсивности первичных фрагментных ионов,
образующихся молекулярным ионом перечисленных ниже соединений при потере
алкильных фрагментов.
а) Бутилметилэтиламин;
б) вшо^-пентилизобутилэтилавдин;
в) ewop-пентилизопропилпропиламин;
г) emop-бутил-трет-бутилпропиламин;
д) З-метилгептанол-3;
е) 3,4,5-триметилоктанол-4.
3.1.2. Правило Стивенсона (правило Стивенсона— Одье)
Разрыв простой связи в нечетноэлектронном ионе может привести к двум парам
ионов и радикалов (схема 3.6):
АВ+ +XY' <- ????+· -> АВ" +??+
Схема 3.6
Вопрос о том, какой фрагмент будет заряженным, впервые рассмотрен
Стивенсоном в 1951 г. [77]. Исходная формулировка правила звучала следующим
образом: если ЭИ(АВ) < ЭИ(ХУ), то ЭЩАВ+) = 3H(AB)+D(AB-XY); но если
3.1. Стабильность ионов и нейтральных частиц 47
Таблица 3.1
ЭИ фрагментов и интенсивности пиков комплементарных ионов
Соединение АВ—XY
HOCH2-CH2NH2
(СН3JСН-СН2ОН
(СН3JСН-СН(ОН)СН3
(СН3)зС-СН2ОН
(СН3)зС-СН(ОН)СН3
(CH3KC-CH,NH2
С1СН2-СН2ОН
ВгСН2-СН2ОН
ЭИ(АВ)
-7,6
7,55
7,55
6,93
6,93
6,93
9,3
8,6
Интенсивность
АВ+(% к макс.)
2,3
100
14,5
100
100
7,7
4,0
15,2
ЭИ(ХУ)
~6,2
~7,6
-6,9
-7,6
-6,9
-6,2
-7,6
-7,6
Интенсивность
ХУ+(%кмакс.)
100
67 |
100
7,4 !
79,2 |
100 !
100
100
ЭИ(АВ) > ЭИРСУ), то ЭЩАВ+) > 3H(AB)+D(AB-XY), т. е. во втором случае
образование иона АВ+ требует избытка энергии.
Современная трактовка правила более конкретна. Фрагмент с большей ЭИ
имеет большую склонность к удержанию неспаренного электрона.
Следовательно, вероятность образования иона с меньшей энергией ионизации будет
больше [78]. Поскольку такой ион, как правило, и более стабилен, его пик в
спектре будет интенсивнее, чем пик альтернативного иона. Результаты
экспериментального подтверждения правила Стивенсона (табл. 3.1) опубликованы в
работе [79].
Если разница в ЭИ альтернативных радикалов составляет более 0,3 эВ, пик
иона с меньшей ЭИ доминирует в спектре. Если различия в ЭИ составляют
менее 0,3 эВ, интенсивности пиков примерно равны.
Правило Стивенсона выполняется и для распада с перегруппировкой. В
этом случае в результате реакции образуются нейтральная молекула и катион-
радикал, т. е. за заряд конкурируют две молекулы. Катион-радикалом становится
та молекула, энергия ионизации которой ниже (схема 3.7).
AY*' + ВХ «- ????+" -> ?? + ВХ+·
Схема 3.7
Подтверждением этого постулата служат результаты, представленные в
табл. 3.2, полученные при изучении перегруппировки Мак-Лафферти [80] в М+"
альдегидов (схемы 3.8а и 3.86 [81]).
На первой стадии процесса (схема 3.8а) 1,5-сигматропный сдвиг атома
водорода приводит к интермедиату с разделенными радикальным и катионным
центрами. На второй стадии разрыв С—С-связи обусловливает образование пары
молекул (алкена и енола), одна из которых является нейтральной, а вторая —
катион-радикалом. Иногда перегруппировку представляют как согласованный
48 Глава 3. Основные правила и подходы к интерпретации масс-спектров
/??>+
\-:< О
сн
I II
сн2 с
"СН^ ЧСН3
chJ)
+
но
-RCHCH2
+
но
но
и*
CHf" ЧСН3
1
с
Ы{ ^снз
.с
СН/" NCH3
чсн НР
? г"*1
сн2' с
-Сз1^° , к-сн=о?'
Схема 3.8а
С
tH^ \СН3
R /??>+
" 1^ <г|!
сн2^ ?
он
сн2 \сн3
.с
сн2^ \сн3
Схема 3.86
процесс 1,5-сигматропного сдвига и разрыва С—С-связи (схема 3.86), однако
многочисленные исследования показали, что в большинстве случаев процесс
протекает постадийно [82].
Для бутаналя и пентаналя ЭИ енола ниже, чем ЭИ альтернативного алкена.
Как следствие, интенсивности пика енольного фрагмента выше в 5-10 раз.
Для последующих гомологов ЭИ образующихся терминальных олефинов лежат
в диапазоне 9,4-9,6 эВ, т. е. примерно равны ЭИ енола. В результате пики
соответствующих ионов в спектрах примерно равны. Метальный заместитель
в положении 2 понижает ЭИ енола, и интенсивность его пика становится
на порядок выше, чем у альтернативного алкена. Напротив, заместитель в
положении 3 приводит к образованию алкена с внутренней двойной связью и
ЭИ фрагментов и интенсивности их пиков в спектрах альдегидов [81]
Таблица 3.2
Соединение
Бутаналь
Пентаналь
Гексаналь
2-Метилпентаналь
3 -Метилпентанал ь
Фрагмент —
ЭИ (эВ)
С2Н4-10,5
С3Н6~ 9,8
С4Н8- 9,6
С3Н6- 9,8
С4Н8- 9,3
Интенсивность, %
(относительно ?27)
3,4
2,4
16,1
1,7
27,0
Фрагмент —
ЭИ (эВ)
С2Н40-9,5
С2Н40-9,5
С2Н40-9,5
С3Н6О-9,0
С2Н40-9,5
Интенсивность, %
(относительно ?27)
16,5
26,4
15,0
25,1
5,9
?27 — полный ионный ток, т. е. сумма интенсивностей пиков всех ионов в спектре в диапазоне
от m/z 27 до М+\
3.1. Стабильность ионов и нейтральных частиц 49
меньшей ЭИ (для бутена-2 9,3 эВ). В результате пик алкена становится в 4 раза
интенсивнее пика енола.
Необходимо подчеркнуть, что правило Стивенсона описывает реакции с
термодинамическим контролем. Если реакция определяется кинетическим
контролем, правило неприменимо.
3.1.3. Правила распада четноэлектронных ионов
Правило, сформулированное в 1980 г. Карни и Мандельбаумом [83], гласит, что
четноэлектронные ионы распадаются в основном с выбросом молекул, а не
радикалов, т. е. из катионов образуются прежде всего катионы, а не катион-
радикалы. Это связано с тем, что процесс разрыва связи с образованием двух
радикалов существенно более энергоемкий. Учитывая также, что стабильность
радикальных частиц меньше чем молекул или катионов, понятно, что такой
процесс не может конкурировать с альтернативным отщеплением молекул. Заряд
в результате фрагментации четноэлектронного иона может сохраняться на том же
атоме, что и у предшественника, или мигрировать. Правило, однако, не означает,
что эти реакции полностью запрещены. Они протекают, но образующиеся ионы,
как правило, малоинтенсивны; хотя и здесь имеются исключения. В частности,
можно привести в качестве примеров последовательное отщепление метальных
групп триметилсилильными производными аминов, спиртов или кислот, а также
последовательное отщепление атомов хлора или брома из соответствующих
полигалогенпроизводных.
Аналогом правила Стивенсона для четноэлектронных ионов являются
закономерности, отмеченные Филдом [84] и Бовеном [85]. Рассмотрим реакцию
распада четноэлектронного иона (схема 3.9):
АВ+->А++В
Схема 3.9
Выгодность обратной реакции, идущей практически без энергии активации,
зависит от сродства молекулы В к катиону А+. Чем выше это сродство, тем
активнее протекает реакция. Филд [84] сделал допущение, что сродство к
катиону должно изменяться параллельно сродству к протону, т. е. вероятность
того, что разные катионы АВ+ будут распадаться с образованием
идентичного иона А+, обратно пропорциональна величинам сродства к протону для
соответствующих молекул В. Подтверждением служит сравнение выгодности
отщепления молекул аммиака, воды и хлористого водорода протонированными
молекулярными ионами соответствующих аминов, спиртов и алкилхлоридов.
Действительно, аммиак (СП = 9,1 эВ) практически не отщепляется из МН+
аминов; пик, обусловленный выбросом молекулы воды (СП = 7,7 эВ) из МН+
спиртов весьма интенсивен в их спектрах ХИ; а отщепление НС1 (СП = 6,4 эВ)
из МН+ алкилхлорида является доминирующим процессом.
50 Глава 3. Основные правила и подходы к интерпретации масс-спектров
Бовен [85] также использовал для установления направления распада четно-
электронного иона величины сродства к протону (схема ЗЛО):
А + ВН+ <- Н(АВ)+ ->АН+ + В
Схема ЗЛО
Заряд будет сохраняться на фрагменте с большей величиной сродства к
протону. В частности, в спектре метастабильных ионов катиона С2Н50=СН^~
пик иона СН2ОН"*" (m/z 31) является максимальным, а альтернативный пик иона
СгН^" (m/z 29) отсутствует вовсе. Дело в том, что конкурентную борьбу за
протон между молекулами этилена (СП = 7,3 эВ) и формальдегида (СП = 7,9)
выигрывает формальдегид, обладающий большим сродством к протону Следует
подчеркнуть, что с увеличением энергии предшественников закономерности,
отмеченные Бовеном, прослеживаются не столь четко. Уже в спектре активации
соударением иона С2Н50=СН2~ интенсивность пика иона С2Н5" только в 6 раз
меньше интенсивности пика иона СН2ОН", а в обычном масс-спектре, где
задействованы самые разные механизмы образования ионов, интенсивности пиков
соответствующих фрагментов могут быть прямо противоположными
приведенному выше примеру. Как и правило Стивенсона, правила Филда и Бовена
применимы лишь к реакциям ионов с термодинамическим контролем. Реакции
кинетического контроля могут привести к нарушению описанных закономерностей.
ЗЛА Правило степеней свободы
Это правило сформулировано в 1975 г. в работе Бенте и др. [86]. Правило
названо эффектом степеней свободы и связывает интенсивность пика метаста-
бильного иона для вторичного распада фрагментного иона с размером
молекулярного иона. В работе [86] представлено сравнение результатов эксперимента
и расчета для распада представительного ряда молекулярных ионов алкан-2-
онов. В качестве первичного фрагмента рассматривали ион, образующийся в
результате перегруппировки Мак-Лафферти (схема 3.8). Этот ион далее
элиминировал метальный радикал (схема 3.11). Для гомологического ряда найдено,
что десятичный логарифм отношении интенсивности пика метастабильного иона
для последующего распада первичного фрагмента к интенсивности пика этого
НО
sai 9 -rchch2 I -сн3 +
*~н ^ ъ-~ едо
сн, с
s с ш^ \сн3
^CAf \CH3
Схема 3.11
3.1. Стабильность ионов и нейтральных частиц 51
первичного фрагмента линейно связан обратно пропорциональной зависимостью
с числом колебательных степеней свободы в исходном молекулярном ионе.
Суть эффекта заключается в том, что избыточная энергия молекулярного
иона распределяется пропорционально числу колебательных степеней свободы
в образующихся ионе и нейтральном фрагменте. Таким образом, чем больший
нейтральный фрагмент отщепился, тем меньшим будет разброс избыточной
энергии фрагментного иона. Именно эта энергия и обусловливает вторичные
процессы распада.
3.1.5. Прочность химических связей
Энергия образования фрагментного иона из нейтральной молекулы может быть
представлена в виде суммы энергии гомолитического разрыва конкретной связи
в молекуле и энергии ионизации соответствующего образовавшегося радикала.
Энергия связей атомов в органической молекуле лежит, как правило, в диапазоне
2-4 эВ, тогда как энергия ионизации равна 6-12 эВ. Следовательно, ЭИ является
значительно более важным фактором. Влияние ЭИ фрагментов на
интенсивность пиков ионов в масс-спектрах регламентируется правилом Стивенсона
(разд. 3.1.2). Однако в тех случаях, когда конкурирующие процессы приводят к
ионам близкой стабильности, прочность связи становится решающим фактором.
Например, изучение меченных дейтерием по метиленовым группам бензило-
вых эфиров фенилуксусной кислоты [87] показало, что ионы С7Н+ образуются
в основном при разрыве связи С—О, которая обладает меньшей энергией по
сравнению со связью С—С (схема 3.12).
1 2
С7Н* - — PhCH2|-C0b-j-CH2Ph ^—> С7Н7
Схема 3.12
При распаде М+* 1-хлор-З-бромпропана (рис. 3.2) процесс отщепления
одного из атомов галогенов приводит к двум альтернативным ионам На1(СН2)з~.
Энергии ионизации соответствующих радикалов должны быть крайне
близки, но поскольку связь С—Вг менее прочна, чем С—О, пик иона [М —Вг]+
(m/z 77 и 79) в 8 раз интенсивнее суммы пиков ионов [М — О]" и [М—НО]4"
(m/z 120-123). Аналогично, в спектрах а-бром-о>-иодалканов интенсивности
пиков ионов [М —1]+ всегда выше, чем пиков [М —Вг]+.
3.1.6. Структурные и стереохимические факторы
Принято считать, что высокие энергии ионов в масс-спектрометрии
нивелируют различия, вызываемые пространственными характеристиками молекулы в
классических реакциях органических соединений в растворах. Однако это
справедливо лишь отчасти. Максимальное влияние стерические факторы оказывают
52 Глава 3. Основные правила и подходы к интерпретации масс-спектров
1 00
90
80
70
60
50
40
30
20
1 0
1,%
4 1
1 58
?
JJ
-?
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170
m /z
Рис. 3.2. Масс-спектр электронного удара 1-бром-З-хлорпропана
на структуру активированного комплекса, а наиболее наглядно проявляются в
перегруппировочных процессах. Поэтому конкурентоспособность таких реакций
зачастую значительно ниже, чем это следует из величины их критической
энергии. В связи с этим увеличение внутренней энергии М+* приводит к
уменьшению количеств продуктов перегруппировок. Одним из следствий этого
является тот факт, что многие интенсивные пики в спектрах метастабильных
ионов очень малы в обычных масс-спектрах. Уменьшение энергии молекулярных
ионов (например, при использовании ионизирующих электронов с энергией
12-20 эВ), напротив, приводит к возрастанию относительных интенсивностей
пиков перегруппировочных ионов в ущерб пикам осколочных ионов. Сравнивая
масс-спектры соединения, полученные при нормальной и пониженной энергии
ионизирующих электронов, можно легко понять, какие пики обусловлены
осколочными, а какие — перегруппировочными ионами (рис. 2.3).
Хорошим примером влияния структурных факторов на интенсивность пиков
ионов является фрагментация хлорзамещенных и бромзамещенных алканов. В
отсутствие разветвлений в цепи аномально высокую интенсивность в
гомологической серии фрагментных ионов CwH2/jHal~^ имеют пики ионов C/^HgHal^",
что объясняется протеканием реакции циклизации с образованием пятичленных
структур (схема 3.13).
-R
СИ?
Схема 3.13
СН2-
I
сн2
+
?
сн,
~сп{
Любые разветвления в углеродной цепи резко снижают
конкурентоспособность данного процесса. На рисунке 3.3 представлены масс-спектры 1-
хлороктана и 2-хлороктана. Видно, что максимальный пик (m/z 91) в спектре
3.1. Стабильность ионов и нейтральных частиц 53
1 00
90
80
70
во ?
50
40
30 Ч
20 А
1 0 -|
о
10
1 00 ?
90 А
80 А
70 А
60 А
50 А
40 А
30 А
20 ?
1 0 -|
О 4—
20
!,%
91
1 05
I.
20 30 40 50 60 70 80 90 100 110 120 130 140 150
l,%
m /z
^ii
1 05
m /?
Рис. 3.3. Масс-спектры ЭУ 1-хлороктана (а) и 2-хлороктана (б)
первого соединения полностью отсутствует в спектре второго. Интенсивности
пика иона с m/z 105 (аналог циклического иона для 2-хлороктана) составляет
уже не 100%, а лишь 6% (рис. 3.3,5). Аналогичные процессы характерны для
алкилмеркаптанов (образование пятичленных циклов) и аминов (образование
шестичленных циклов).
Важную роль играет размер цикла в переходном состоянии при отщеплении
из М"' монофункциональных алкилпроизводных простейших молекул (HHal,
Н2О, H2S). Для элиминирования из М+" алкилгалогенидов НС1 и НВг
предпочтительным является пятичленное переходное состояние A,4-сигматропный сдвиг
атома водорода). Для тиолов этот процесс обусловливает 40% элиминируемого
сероводорода, тогда как основным F0%) является 1,4-элиминирование A,5-сиг-
матропный сдвиг). Для спиртов 1,4-элиминирование —доминирующий процесс
[88].
В качестве еще одного примера пространственных эффектов можно привести
перегруппировочный процесс по типу перегруппировки Мак-Лафферти,
приводящий к образованию интенсивных пиков ионов в масс-спектрах алкилбензолов
(схема 3.14). При наличии разветвлений в алкильной цепи или заместителей в
обоих opmo-положениях этот процесс практически полностью подавляется.
С каждым годом увеличивается число работ, посвященных использованию
масс-спектрометрии для решения стереохимических проблем. Масс-спектры
электронного удара стереоизомеров, как правило, практически неразличимы.
Однако использование низкоэнергетических методов ионизации (ХИ, ПИ и т. д.)
54 Глава 3. Основные правила и подходы к интерпретации масс-спектров
r^VJ^1 ^ ^У OP1 -RCHCH2, ^/^
Схема 3.14
в сочетании с тандемной масс-спектрометрией позволяет делать надежные
выводы о пространственной структуре молекул.
Иногда и спектры ЭУ оказываются информативными. Исследования
фрагментации изотопно-меченных соединений показали [89], что 1,4-элиминирование
НС1 и воды из М+ * хлорциклогексана и циклогексанола соответственно
протекает стереоспецифично из конформации ванна, когда гетероатом и элиминируемый
атом водорода оказываются на расстоянии 1,7 А. Стереоспецифично протекает и
1,3-элиминирование НС1 (диаксиальное расположение атомов хлора и водорода).
Однако выбросы Н2О и HOD абсолютно равновероятны при элиминировании
молекулы воды из М+# циклогексанола меченного дейтерием в положение 3.
Минимальное расстояние между аксиальными заместителями в положениях 1
и 3 составляет 2,3 А. Это расстояние оказывается непреодолимым в случае
циклогексанола, и М+' до протекания элиминирования трансформируется с
раскрытием цикла. Более длинная (на 0,4 А) связь С—О позволяет реакции
1,3-элиминирования идти непосредственно в неизомеризованном М+\
Соотношение интенсивностей пиков ионов [М — Вг]+/М+* для цис-1,2-
дибромциклогексана составляет 1,55, а для транс-изомера —64,5. Очевидно,
существует анхимерное влияние со стороны второго атома брома (схема 3.15)
в диаксиальной конформации [90].
Вг:^
Схема 3.15
Важные стереохимические результаты можно получать при использовании
химической ионизации с применением хиральных газов-реагентов. Например,
энантиомерные ?-аминокислоты можно различать химической ионизацией
оптически чистым 2-метилбутанолом [91].
Следует подчеркнуть, что стереохимические аспекты масс-спектрометрии
многогранны и не могут быть изложены в рамках учебного пособия.
Интересующимся этим направлением можно порекомендовать обратиться к обзорам
3.1. Стабильность ионов и нейтральных частиц 55
М. Грина [92], А. Мандельбаума [93, 94] и монографии Дж. Сплиттера и Ф. Ту-
речека [95].
3.1.7. орто-Эффект
Одним из наиболее очевидных проявлений структурных различий является
opmo-эффект. Это явление хорошо известно и активно используется для
синтетических целей в органической химии. Спектры подавляющего большинства
орто-замещенных ароматических соединений существенно отличаются от их
мета-и лорд-изомеров. В качестве примера масс-спектрометрического орто-эф-
фекта обычно приводят фрагментацию молекулярных ионов алкилсалицила-
тов. В спектрах ЭУ этих соединений наблюдаются интенсивные пики ионов
[М —RDH]+', отсутствующие в спектрах их мета-и ия/?я-изомеров (схема 3.16).
~~lt
II *0
о
Схема 3.16
Несколько примеров орто-эффекта рассмотрены в гл. 8 при обсуждении
основных процессов фрагментации различных классов органических
соединений. В частности, на схеме 8.21 представлено элиминирование молекулы
воды М+* о-метилбензилового спирта, на схеме 8.24 —аналогичный процесс
для М+Ф о-крезола. В спектре о-толуиловой кислоты пик иона [М — И20]+\
образующегося в результате opmo-эффекта, является максимальным в спектре
(рис. 8.75). Характеристический ион с m/z 149, пик которого доминирует в
спектрах диалкилфталатов, также обусловлен qpmo-эффектом (схема 8.73).
Сложный перегруппировочный процесс, протекающий в орто-замещенных алкил- и
циклопропилнитробензолах, представлен на схеме 8.92. Образование индолов
по Фишеру (схема 8.94) в источнике масс-спектрометра тоже может считаться
проявлением opmo-эффекта. Задачи 8.21 и 10.2 требуют выявления процессов
распада на основе орто-эффекта.
Масс-спектрометрическому наблюдению орто-эффекта посвящен
представительный обзор X. Шварца [96]. В нем дана классификация процессов, связанных
с opmo-эффектом, даны примеры необычных реакций элиминирования, реакций
внутримолекулярной циклизации, обменных и восстановительных процессов.
Определенные аналогии в протекании реакций, обусловленных орто-эф-
фектом, в растворах и в ионизационной камере масс-спектрометра позволяют
делать прогнозы о возможном синтезе тех или иных соединений на основании
изучения масс-спектра их предшественников. В частности, молекулярные ионы
56 Глава 3. Основные правила и подходы к интерпретации масс-спектров
f*!* - ЛП+ _ ^
си,
И х 'x=o,nh ULC xhx=o,nh* kX_x
II
о
Схема 3.17
О
О
'NH-СГ ^ ^^NH-
O О
Схема 3.18
о-карбокси- и о-карбоксамидофенилциклопропанов [97] распадаются с
предварительной изомеризацией в пяти- и шестичленные гетероциклические структуры
(схема 3.17).
Аналогичная реакция протекает в серной кислоте, причем за 2-3 месяца
устанавливается термодинамическое равновесие между пяти- и шестичленны-
ми гетероциклами. Однако соотношение этих продуктов легко предсказать за
несколько минут, измерив соотношение интенсивностей пиков ионов [М—СНз]+
и [М—С2Н4]4"' в масс-спектре соответствующего исходного фенилциклопропа-
на [97].
На основании данных тандемной масс-спектрометрии показано [98], что
М+* замещенных К-(о-циклопропилфенил)бензамидов могут изомеризовать-
ся до распада с образованием 3-арил-1-этил-1Н-бензоксазинов и 5-этил-2-
оксодибензоазепинов (схема 3.18). Метильная группа в К-[о-A-метилциклопро-
пил)фенил]бензамидах препятствует последнему процессу, оставляя образование
бензоксазинов единственно возможной реакцией циклизации. Химический
эксперимент подтвердил прогнозы, сделанные на основании масс-спектров [98].
3.2. Концепция локализации заряда
и неспаренного электрона
Краеугольным камнем этого подхода к интерпретации процессов распада в масс-
спектрометрии, впервые предложенного К. Джерасси [88] и развитого Ф. Мак-
Лафферти [35], служит постулат о том, что в сложной молекуле распад
инициируется зарядом или неспаренным электроном, локализованными в наиболее
благоприятном месте молекулы. Это в лучшем случае приближение, однако такой
3.2. Концепция локализации заряда и неспаренного электрона 57
подход удобен для запоминания большого числа реакций самых разнообразных
по структуре частиц.
Если речь идет о молекулярном ионе, необходимо выделить для него
основное (невозбужденное) электронное состояние, т. е. удалить электрон с высшей
занятой молекулярной орбитали. Таким образом, наиболее благоприятные для
неспаренного электрона и заряда места в молекулярном ионе связаны с потерей
электрона с минимальной энергией ионизации. Энергетические требования в
этом случае аналогичны известным в УФ-спектроскопии для электронных
переходов: ? < ? < п.
При использовании для ионизации электронов с энергией 70 эВ
собственный электрон образца может быть удален с любой молекулярной орбитали
с энергией меньшей этого значения. Однако в результате излучательных и
безызлучательных процессов исходный электронно-возбужденный ион должен
перейти в основное электронное состояние. Обычно время жизни возбужденных
электронных состояний не превышает 10~~8 с. Поскольку время пребывания
ионов в источнике электронного удара 10~6 с, можно говорить о том, что
подавляющую часть времени ион находится в источнике в невозбужденном
состоянии. Концепция локализации заряда и электрона становится тем более
справедливой, чем уже разброс избыточной энергии в М+' (функция Р(Е)). В
этом случае увеличивается доля низкоэнергетических реакций с lg k(E) < 8.
Радикальный центр и центр, несущий заряд, могут располагаться как на
одном атоме (например, удаление «-электрона), так и на разных атомах
(например, удаление электрона ?-связи). Во фрагментных ионах эти центры могут
оказаться на значительном расстоянии друг от друга [99]. Например, выброс
молекулы СО молекулярным ионом циклогексанона приводит к возникновению
линейной цепочки из пяти метиленовых групп, причем неспаренный электрон и
положительный заряд находятся на противоположных концах цепи (схема 3.19).
Безусловно, такой ион далее может изомеризоваться в более стабильную
структуру.
о4) о\
Схема 3.19
Иногда такие ионы оказываются стабильнее своих изомеров с расположением
заряда и неспаренного электрона на одном атоме. Например, ион СНзОН+"
значительно менее стабилен, чем ион ??^??^, генерируемый при распаде
этиленгликоля. Расчеты и эксперименты показали, что эти два иона находятся
на дне колодцев потенциальной энергии, что не позволяет им изомеризоваться
друг в друга [100].
58 Глава 3. Основные правила и подходы к интерпретации масс-спектров
?^??-^?—^ А' + В=Х-? (СН3-СН2-ОН - аН3' + СН2=5н)
A^Lx *> ?' +???
4C=(J *· R"+RCeO
А^-ВГ-Х ** А + В=Х
R ?-
N+ >R-N-CHo-CHi ** R—?? + СН2=СН2
l2- LJ12
СН2-СН2
Схема 3.20
Инициирование фрагментации радикальным центром (?-распад) вызвано
тенденцией электрона к спариванию. Неспаренный электрон участвует в
образовании новой связи с соседним атомом. При этом рвется другая связь этого а-
атома (отсюда ?-распад). Три представленных в общем виде варианта ?-распада
(схема 3.20) проиллюстрированы реальными примерами (в скобках).
Тенденция к инициированию распада радикальным центром параллельна
донорным свойствам этого центра. Наиболее активно эти процессы будут
проходить, когда удаляется «-электрон атома азота. Напротив, галогены менее
активны в этих реакциях. Эти утверждения наглядно демонстрируются основными
направлениями распада основных классов органических соединений (гл. 8).
Движущей силой инициирования распада центром, несущим заряд, является
притяжение электронной пары. Тенденция к инициированию распада
заряженным центром для гетероатомов противоположна отмеченной для радикального
центра. Наиболее активны галогены, менее — сера и кислород, замыкает ряд азот.
Можно говорить об увеличении выгодности этого процесса с увеличением
электроотрицательности атома. Три представленных в общем виде варианта распада
(схема 3.21), инициированного центром, несущим заряд, проиллюстрированы
реальными примерами (в скобках).
Вторые реакции в схемах 3.20 и 3.21 для вариантов распадов,
инициируемых радикальным центром и центром, несущим заряд, связаны с одинаковым
исходным ионом и одинаковыми продуктами. Различие заключается лишь в том,
какой из двух комплиментарных продуктов будет катионом, а какой радикалом.
Для определения интенсивностей соответствующих пиков следует использовать
А-В-Х -А++В-Х (С2Н5-0-С2Н5 С2н?+0-С2Н5)
А-В=Х —¦> А-В-Х - А++В-Х Ъ=0 - R++RC=0
АСё-Х —^ А+ + в-х ic2H5-o-c2H5 —- с2н^ + но-с2н5)
Схема 3.21
3.2. Концепция локализации заряда и неспаренного электрона 59
правило Стивенсона (разд. 3.1.2). Для выявления механизма (в рамках концепции
локализации) необходимо иметь в виду, что стабилизация заряда более
существенна, чем стабилизация неспаренного электрона (см.выше). Поскольку
реакции, инициируемые заряженным центром, подразумевают миграцию заряда, они,
как правило, менее благоприятны по сравнению с реакциями, инициируемыми
неспаренным электроном.
Задача 3.2. Какие реакции фрагментации, инициированные центрами,
несущими заряд и неспаренный электрон, должны протекать при распаде следующих
соединений:
а) Диэтилсульфид (удален и-электрон S);
б) метилэтилкетон (удален и-электрон О);
в) бутанол-2 (удален «-электрон О);
г) гексен-3 (ионизирована 7г-связь);
д) изобутилхлорид (удален «-электрон С1);
е) триэтиламин (удален «-электрон N).
Перегруппировочные процессы также могут быть разделены на два
указанных типа. Наиболее типичным примером перегруппировки, инициируемой
радикальным центром, является перегруппировка Мак-Лафферти (схема 3.22):
* /Н <t R\·^ HA но ноЛ1"
СН V СН) Н° -RCHCH: " I
°W\<* ^Ааь аьА* «чАя,
Схема 3.22
Атом водорода через шестичленное переходное состояние A,5-сигматропный
сдвиг) мигрирует к атому кислорода (тенденция к спариванию электронов). В
образовавшемся интермедиате реакцию распада инициирует радикальный центр,
расположенный на 7-атоме углерода.
В качестве примера перегруппировки, инициируемой центром, несущим
заряд, можно привести вторичный процесс фрагментации аминов, являющийся
по сути 1,3-сигматропным сдвигом (схема 3.23):
ЛР1 -RCHCH2 +
Ofe=N-LCH2 CH2=NH
R R
Схема 3.23
60 Глава 3. Основные правила и подходы к интерпретации масс-спектров
Если теория Вудворда—Гофмана предсказывает значительную критическую
энергию для 1,3-сигматропных сдвигов в нейтральных молекулах, теоретические
расчеты [101] и экспериментальные факты показывают, что этот постулат не
распространяется на заряженные частицы. В данном случае гидрид-ион со своей
парой электронов мигрирует через четырехчленное переходное состояние на
атом азота с одновременным гетероциклическим разрывом связи С—N и
отщеплением молекулы алкена. Заряд сохраняется на атоме азота. Образовавшийся
ион аналогичен исходному и при соответствующем R и достаточной энергии
может отщепить еще одну олефиновую молекулу по такому же механизму.
Подобные процессы характеризуются низкой энергией активации, а для их
протекания необходима длина углеводородной цепи не менее двух атомов. В
связи с этим интенсивность вторичных и последующих процессов распада
аминов, обусловленных такими перегруппировками, может быть очень высокой,
а интенсивность пиков первичных фрагментных ионов, рассчитанная на основе
функций к(Е) и Р(Е) для М4", существенно ниже.
Задача 3.3. Какие ионы образуются в результате перегруппировки Мак-
Лафферти для перечисленных ниже соединений? Оцените с учетом правила
Стивенсона интенсивности пиков олефинового и енольного ионов.
а) Пентаналь;
б) пентанон-2;
в) деканон-4;
г) З-метилпентанон-2;
д) З-пропилгексанон-2;
е) 3-фенилбутаналь;
ж) 5-фенилпентанон-2.
Задача 3.4. Почему перегруппировка Мак-Лафферти подавлена или
невозможна в случае перечисленных ниже соединений?
а) Бутанон-2;
б) 1-фенилпентанон-З;
в) гептен-5-он-2;
г) гексен-4-он-2;
д) октен-4-он-З.
3.2. Концепция локализации заряда и неспаренного электрона 61
3.2.1. Фрагментация, удаленная от места локализации заряда
Отмечены случаи, когда определенные процессы распада идут без видимого
участия со стороны центра, несущего заряд [102-106]. Наиболее хорошо изученным
примером является фрагментация анионов жирных кислот (схема 3.24).
/Н/Н\ -Но
шнсн^чзг! С сн-ссн^-соо" ^ ,„ ч *т »CT2=CT-(ai2b-cocr
ЧоДо1 -Ш3(СН2)„-СН=СН2 т
Схема 3.24
Реакция идет именно по указанному на схеме механизму [102], что было
доказано спектрами дейтерированных аналогов, а также регистрацией нейтральных
частиц методом нейтрализации—реонизации (разд. 7.4). Отщепляются именно
молекулы водорода и алкена. Эксперимент с холестериновым производным
показал, что скручивание молекулы с приближением в пространстве разрывающихся
связей к заряженному центру в данном случае роли не играет, т. е. идет именно
удаленная от заряда фрагментация. Для протекания такого процесса необходима
цепочка из не менее чем четырех атомов углерода, причем интенсивность
процессов возрастает с увеличением длины алкильной цепи.
Аналогичные процессы зарегистрированы при распаде анионов алкилсульфа-
тов, алкилсульфонатов, N-ацилированных аминокислот, четвертичных алкилам-
мониевых и алкилфосфониевых ионов, протонированных молекулярных ионов
алкиламинов [102].
Глава 4
ПРАКТИЧЕСКИЕ ОСНОВЫ
ИНТЕРПРЕТАЦИИ МАСС-СПЕКТРОВ
Итак, вы имеете масс-спектр органического соединения. С чего начинать процесс
расшифрозки? Во-первых, желательно иметь исчерпывающую информацию об
истории появления данного образца. Здесь важны многие данные (метод синтеза
и выделения, природа исходных реагентов и растворителей, наличие примесей
и т. д.). Во-вторых, имеет смысл прежде всего свериться с компьютерными
библиотеками спектров (разд. 4.4). В идеале задача может быть решена уже
на этой стадии. В любом случае полученная при сравнении нового спектра со
спектрами известных соединений информация может помочь отнести образец
к конкретному классу соединений, сделать выводы о наличии каких-либо
функциональных групп.
Если библиотека спектров не предоставляет однозначного ответа, следует
обратить внимание на общий вид спектра: отметить параметры съемки,
обратить внимание на наиболее интенсивные пики, на характерные группы пиков.
Если, например, спектр характеризуется большим числом фрагментов, пики
которых имеют все большую интенсивность при движении вниз по шкале масс,
скорее всего образец является алифатическим соединением. Напротив, редкие
интенсивные пики характерны для ароматических структур. Однако начинать
основную работу со спектром следует с установления пика молекулярного иона.
4.1. Молекулярный ион
Информация, которую можно извлечь, анализируя область молекулярного
иона, поистине огромна. Масса М+* —это молекулярная масса анализируемого
соединения. Соотношение изотопных пиков (см. ниже) позволяет установить
примерный элементный состав, а измерение точной массы М+' с помощью масс-
спектрометрии высокого разрешения —точный элементный состав соединения.
Относительная интенсивность пика М+# позволяет сделать определенные
предположения о его структуре, принадлежности анализируемого соединения к тому
или иному классу. Например, для углеводородов относительная интенсивность
М+' в полном ионном токе увеличивается по мере увеличения степени
ненасыщенности соединения. Доказательством этого положения может служить ряд
интенсивностей (в полном ионном токе) пика молекулярного иона углеводородов
с десятью атомами углерода в молекуле: декан 1,0%, децен-1 1,1%, бутилцикло-
4.1. Молекулярный ион 63
гексан 4,2%, декадиен-4,6 4,7%, 2,6-диметилоктатриен-2,4,6 7,5%, бутилбензол
10,2%, 1-фенилбутен-2 11,0%, 1 -метил- 1Н-инден 19,9%, нафталин 58,7%.
К сожалению, многие соединения под ЭУ не дают пика молекулярного иона;
он не стабилен. В связи с этим необходимо научиться правильно определять
пик М+#в спектре. Ион должен удовлетворять четырем необходимым, но не
достаточным условиям, для того, чтобы считать пик молекулярным:
1) иметь самую большую массу в спектре (разд. 4.2);
2) быть нечетноэлектронным;
3) быть способным образовать важнейшие ионы с большой массой за счет
выброса реальных нейтральных частиц;
4) включать все элементы, наличие которых в образце можно увидеть по
фрагментным ионам.
Если хотя бы одно из этих условий не выполняется, ион не молекулярный;
если выполняются все четыре условия, ион может быть молекулярным.
Рассмотрим подробнее представленные условия. Первое положение
очевидно, поскольку масса целой молекулы заведомо больше массы любого ее
фрагмента.
Количество электронов в ионе можно проверить, рассчитав степень его
ненасыщенности:
R = x--y+-z+l D.1)
где R — степень ненасыщенности (число кратных связей и циклов в ионе); х, у и
? — индексы в брутто-формуле иона Cx?LyNzOn. Понятно, что степень
ненасыщенности можно определить только после установления элементного состава иона.
Если в состав входят другие элементы, индексы х, у, ?, ? будут суммами атомов
соответствующих валентностей (для Си Si 4—?, ???3— ?, OhS2 — ?, Ни Hal
1 —у). Необходимо отметить, что если перечисленные элементы присутствуют
в других степенях окисления, формула D.1) может привести к неправильному
результату. Например, используя стандартные значения валентностей для серы
и фосфора B и 3 соответственно) в группах SO2 или РО^-, по стандартной
формуле мы придем к неправильному результату.
Определив /?, мы устанавливаем не только степень ненасыщенности иона, но
и количество электронов в нем. Если R — целое число, ион нечетноэлектронный
и, следовательно, может быть молекулярным; если R — дробь, ион четноэлек-
тронный и молекулярным быть не может.
В качестве примера вычислим степень ненасыщенности ионов состава
C5H9N302ClBr и Ci2SiHnPSBr3. В первом случае х = 5, у = 9+1 + 1 = 11,
? = 3, ? = 2. Тогда R = 5 — 11/2 + 3/2 + 1 = 2, т. е. ион содержит две двойные
связи, или одну тройную связь, или два цикла, или цикл и двойную связь. Кроме
того, поскольку R —- целое число, такой ион, будучи нечетноэлектронным, может
быть молекулярным.
64 Глава 4. Практические основы интерпретации масс-спектров
Во втором случае ? = 12+1 = 13, у = 11 + 3 = 14, ? = 1, ? = 1, т. е.
Л=13 —7+1/2+1=7,5. Следовательно, степень ненасыщенности иона равна 7,
а дробная величина указывает на то, что ион является четноэлектронным и не
может быть молекулярным.
Альтернативным вариантом расчета степени ненасыщенности может служить
способ замены гетероатомов углеводородными фрагментами. Все одновалентные
элементы (за исключением водорода) заменяются группами СНз,
двухвалентные— СН2, трехвалентные — СН, четырехвалентные (за исключением самого
углерода) — С. Полученная формула сравнивается с формулой алкана с таким же
содержанием атомов углерода. Разность между числом атомов водорода в алкане
Bи + 2) и в образце, деленная на два, дает величину R. Для разобранных выше
примеров получаем:
- C5H9N302ClBr = С5Н9 + ЗСН + 2СН2 + 2СН3 = Ci2H22. Брутто-формула
соответствующего алкана Ci2H26 (додекан). R — B6 — 22)/2 = 2.
- Ci2SiHnPSBr3 = C12H11 + С + СН + СН2 + ЗСН3 = Ci8H23. Брутто-формула
соответствующего алкана CisH38 (октадекан). R = C8 — 23)/2 = 7,5.
Третье необходимое условие позволяет проверить правильность выбора М+#
на основании первичных фрагментных ионов. Обычно М+# легко отщепляет
молекулы СО, С02, Н20, С2Н4, HHal; радикалы Alk", H#, Hal", ОН' и т. д.
Следует помнить, что потери из М+* от 5 до 14 или от 21 до 25 а. е. м.,
приводящие к возникновению интенсивных пиков ионов, крайне
маловероятны. Если в спектре такие пики все же присутствуют, пик М+#, по-видимому,
выбран неверно; или в образце имеются примеси. Например, если в масс-спектре
чистого соединения самый тяжелый ион имеет массу 120, а следующий за ним —
112, ион 120 —не молекулярный, а фрагментный, т. е. в данном случае М+"
нестабилен и не регистрируется в спектре.
Четвертое условие также достаточно очевидно. Проиллюстрировать его
можно следующим примером. Если где-нибудь в средней части спектра чистого
соединения (например, в области m/z 100-200) отчетливо проявляются пики с
изотопным соотношением, характерным для брома, хлора и т. д. (разд. 4.2), а
кандидат в М+* в области m/z 300-400 явно не содержит таких атомов, это не
молекулярный ион.
Задача 4.1. Может ли ион с максимальной массой быть молекулярным и
обусловить образование следующей серии фрагментов:
а) 130, 129, 115, 113, 102, 101, 97...;
б) 128, 127, 120, ИЗ, 100...;
в) 100, 82, 78, 77, 66, 65, 57...;
г) 162, 161, 147, 134, 115, 105...-
д) 178, 177, 176, 152, 151...;
е) 154, 153, 152, 151, 150, 127, 126...;
ж) 143, 142, 128, 125, 119, 115...;
4.2. Определение элементного состава ионов на основании изотопных пиков 65
з) 124, 123, 111, 109, 107, 106, 96, 95...;
и) 220, 219, 218, 217, 201, 205, 185, 163...;
к) 179, 178, 161, 150, 136...;
л) 100, 85, 71, 70, 69, 57...;
м) 58, 57, 43, 29, 28, 27, 15...;
н) 100, 99, 85, 81, 57...?
Задача 4.2. Может ли ион с максимальной массой быть молекулярным и
обусловить образование следующей серии фрагментов:
а) CioHi4, C10H13, СюНц, С9Н11, C9H9, CgH9, C7H7...;
б) С12Н15О2, СюНюО, С9Н12О2, CgHioO, C10H13...;
в) C8Hi2, CgHn, C7H9, С6Н12, СбН7...;
г) C13H15N02, C13H14N02, C12H12N02, C9H6N02, C13H15NO, С13Н150,
C12H15...;
Д) CioHg, C10H7, СюНб, С10Н5, CgHo, CsHs...;
e) C10H10O45 C9H803, C9H703, C8H702 ...;
ж) C10H12N, C10HnN, C9H9N, C8H7N, C9Hn ...;
3) С7Н7СЮ, С7Н6СЮ, С6Н6С1, С7Н7О, C7H60...;
и) C10H7CI, СюНбС1, C10H5CI, C10H7, СюНб, С9Н7 ...;
?) 04?8, С4Нб, Q3H5, C3H4, C2H5, С2Н4...;
л) C8H13N302, C8H12N302, C7H10N3O25 C8H13N20, C7H10NO3, C7H13N2... ?
4.2. Определение элементного состава ионов
на основании изотопных пиков
Как уже отмечалось выше, масс-спектрометрия высокого разрешения способна
определить точный элементный состав ионов. Однако благодаря тому, что
большинство химических элементов имеет несколько стабильных изотопов,
элементный состав ионов можно с определенной степенью надежности установить
по обычным (низкого разрешения) спектрам. Проявление присутствия изотопов
можно наглядно продемонстрировать спектрами сероуглерода (рис. 4.1, а), этил-
хлорида (рис. 4.1,6) и этилбромида (рис. 4.1, в).
Вывод о том, что ионы с массами 76 (рис. 4.1, а), 64 (рис. 4.1,6) и 108
(рис. 4.1, в) связаны с потерей двух атомов водорода из молекулярного иона,
неверен. На самом деле именно эти три иона являются молекулярными, а более
тяжелые (+2 Да) ионы обусловлены более тяжелыми природными изотопами
атомов серы C4S), хлора C7С1) и брома (81Вг) соответственно.
В таблице 4.1 представлены данные по природной распространенности
изотопов важнейших (для органических соединений) элементов. Эта таблица является
небольшим фрагментом сводной табл. 1 (см. приложения). В дальнейшем мы
будем называть элементы А, А+1,А+2в зависимости от того, какой изотоп они
имеют помимо основного.
3—1113
66 Глава 4. Практические основы интерпретации масс-спектров
100
90
во
70
60
50
40
30
20
10
1,%
76
78
20
100
90
80
70
60
50
40
30
20
10
30
40
50
60
70
80
90
1 i(%)
_1 1 1 1 1
-| .
m /?
ь
64
66
б
1
30
40
50
m /z
60
70
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
?,%
? . _
1 08
-?—? . .—U
?
1 1 0
1
?
75
80
85
90
95
m /?
100
105
1 10
115
Рис. 4.1. Масс-спектры сероуглерода (?), этилхлорида (б) и этилбромида (в)
Определение элементного состава соединения следует начинать с пика ?+2.
Необходимо учитывать, что в случае присутствия в молекуле нескольких атомов
(А+2)-элементов в спектре могут появиться интенсивные пики ?+4, М+6
и т. д. Существует простое правило для проверки наличия в составе основных
(А+2)-элементов. Если интенсивность пика ?+2 составляет менее 3% от
интенсивности пика М, соединение не содержит атомов хлора, брома, серы
и кремния. Правило применимо и для осколочных ионов, а его справедливость
подтверждается данными табл. 4.1.
Хлор, бром, сера и кремний легко детектируются масс-спектрометрически
благодаря характерной для каждого элемента мультиплетности сигнала. На
рисунке 4.1,6 и в представлены спектры соединений с одним атомом хлора
4.2. Определение элементного состава ионов на основании изотопных пиков 67
Таблица 4.1
Природная распространенность изотопов химических элементов
Элемент
Водород
Углерод
Азот
Кислород
| Фтор
Кремний
Фосфор
Сера
Хлор
Бром
Иод
Изотоп
?
2D
I2C
13с
14N
15N
160
170
180
19F
28Si
29Si
30Si
31p
32s
33 s
34S
36S
35C1
37C1
79Br
81 Br
127I
Тип
изотопа
A
A+l
A
A+l
A
A+l
A
A+l
A+2
A
A
A+l
A+2
A
A
A+l
A+2
A+4
A
A+2
A
A+2
A
Интенсивность, % !
(относительно
$2 изотопов)
99,985
0,015
98,89
?,??
99,64
0,36
99,76
0,04
0,20
100,00
92,18
4,71
3,12
100,00
95,02
0,75
4,21
0,11
75,40
24,60
50,57
49,43
100,00
Интенсивность, % |
(относительно
наиболее
распространенного
изотопа)
100,00
0,02
100,00
1,12
100,00
0,37
100,00
0,04
0,20
100,00
100,00
5,11
3,38
100,00
100,00
0,79
4,44
0,11
100,00
32,63
100,00
97,75
100,00
Тип
элемента
А*
А+1
А+1
А+2
А
А+2
А
А+2*
А+2
А+2 !
А
* Несмотря на присутствие изотопов А+1 у водорода и А+4 у серы эти элементы считаются
А и А+2 соответственно, так как распространенность указанных изотопов очень низка и может
проявляться только при наличии очень большого числа атомов ? или S в молекуле образца
(разд. 5.12).
68 Глава 4. Практические основы интерпретации масс-спектров
и брома соответственно. Приближенно можно считать, что в случае хлора
отношение интенсивностей пиков А и А+2 составляет 3 : 1, а в случае брома —
1:1. Наличие двух и более атомов (А+2)-элемента в составе иона вновь дает
уникальное соотношение изотопных пиков в мультиплете, благодаря которому
природа и количество (А+2)-элементов может быть легко установлены.
Ион, содержащий ? атомов (А+2)-элемента, будет характеризоваться ? + 1
пиками, отстоящими друг от друга на 2 а. е. м. Интенсивность этих пиков можно
рассчитать по формуле биноминального распределения (уравнение 4.2), где а
и Ъ — природное соотношение изотопов соответствующего элемента (для О 1
и 0,325; для Вг 1 и 0,98; для Sin 0,044), а « — число таких атомов в ионе.
Например, если ион содержит 4 атома хлора, интенсивность пика А+4 (третьего
в мультиплете) по отношению к А составит 4 ? 3 ? ?2 ? 0,3252/2 = 0,635, т. е.
63,5%.
„_ь , п{п-\)ап-2Ъ2 х п(п-\){п-2)ап-ъЪъ
(а + Ь)п-=ап + пап-'Ь+— ^ +
л
(„ _ 1 Vh - ?.Vti - Ч W~4/>4
+ ... D.2)
2! 3!
п(п-\){п-2)(п-Ъ)ап-*ЪА
4!
Для упрощения вычислений удобно пользоваться округленными значениями
распространенности изотопов в природе (для О 3:1, для Вг 1:1, для S
25:1). Например, для соединения с тремя атомами хлора будут наблюдаться
4 пика ионов М, М+2, М+4 и М+6 с относительными интенсивностями
C + 1K = 27+27 + 9 + 1.
На рисунке 4.2 представлены частичные спектры (область высоких значений
т/т) тетрахлорбифенила и тетрабромбифенила. На спектрах отчетливо видны
кластеры изотопных ионов с содержанием 4, 3 и 2 атомов хлора (брома) в
молекуле.
Если в ионе имеется два разных (А+2)-элемента, расчет интенсивностей
сигналов в мультиплете осуществляется с помощью матричного перемножения.
Например, для дибромдихлорбензола СбНгВг^СЬ в мультиплете (без учета
изотопов углерода) будет 5 сигналов, отстоящих друг от друга на 2 а. е. м. Расчет
интенсивностей внутри этого мультиплета можно провести следующим образом:
Два атома хлора C + IJ дадут триплет с соотношением 9:6: 1.
Два атома брома A + IJ дадут триплет с соотношением 1:2:1.
Перемножая матрично одно на другое, получаем:
(9
6
1)х
A
:2
9:
9:
:1) =
18 :
6 :
24 :
9
12
1
22
6:
2
8:
1
1
Таким образом, относительные интенсивности пиков в мультиплете будут
9 : 24 : 22 : 8 : 1, или, нормируя на 100%,-37,5 : 100 : 92 : 33 : 4. Как
4.2. Определение элементного состава ионов на основании изотопных пиков 69
1 О О
9 0 4
80 -|
70
6 О
5 О ?
4 О
ЗОН
20 -^
1 О ?
О
1,%
li- Д.Ill «... .Willi
?
jib
2 0 0 2 2 0
m /?
1 00
90
8 0 Ч
70 -|
60
50
40 -J
30 A
2 0 4
1 0 -I
0
1 80
I,%
i
ли-, , mi, . , , ,
200 220 240 260 280 300 320 340 360 380 400 420 440 460 480 500
Рис. 4.2. Частичный спектр (область высоких значений m/z)\ тетрахлорбифенила (а) и
тетрабромбифенила (б)
видно из данного примера, интенсивность пика ? может оказаться меньше
интенсивностей изотопных пиков.
Задача 4.3. Определите интенсивность пика М+8 по отношению к ? в
спектре соединения, содержащего:
а) 5 атомов хлора;
б) 6 атомов брома;
в) 5 атомов серы (без учета вклада 33S и 36S).
Задача 4.4. Определите интенсивность пика М+6 по отношению к ? в
спектре соединения, содержащего:
а) 4 атома хлора;
б) 7 атомов брома;
в) 6 атомов серы (без учета вклада 33S и 36S);
г) 2 атома хлора и 3 атома брома;
д) 3 атома брома и 2 атома хлора;
е) 4 атома брома и 2 атома серы (без учета вклада 33S и 36S);
ж) 3 атома хлора и 3 атома серы (без учета вклада 33S и 36S);
з) 3 атома хлора и 3 атома брома;
и) 2 атома хлора и 4 атома брома.
70 Глава 4. Практические основы интерпретации масс-спектров
Кислород формально также считается (А+2)-элементом, однако природная
распространенность изотопа 180 составляет всего 0,2% от основного изотопа
160. В связи с этим надежно определить точное число атомов кислорода по
интенсивности изотопных пиков в реальном спектре практически невозможно.
Тем не менее сделать определенные предположения о числе атомов кислорода в
молекуле вполне правомерно. Например, если интенсивность пика иона М+2 в
спектре образца с небольшим числом атомов углерода в молекуле больше 0,5%
от интенсивности М+#, можно предположить, что в состав соединения входит
один или более атом кислорода.
После того как удалось установить присутствие в анализируемом образце
(А+2)-элементов, необходимо перейти к определению (А+1)-элементов. Для
этого следует рассмотреть интенсивность пика М+1 (Ф+1 для фрагментных
ионов). (А + 1)-элементы — углерод, водород и азот, причем на практике водород
можно исключить, так как природное содержание дейтерия очень мало и
начинает проявляться только в спектрах соединений с очень высоким содержанием
этого элемента. В случае фрагментных ионов необходимо учесть, что ион Ф+1
может быть составным. Помимо изотопного иона такую же величину m/z может
иметь фрагмент с другим элементным составом.
Углерод является важнейшим элементом органических соединений. В
зависимости от источника анализируемого образца содержание изотопа 13С может
колебаться в диапазоне 1,08-1,12% от 12С. Хотя существуют специальные масс-
спектрометрические подходы для определения точного содержания 13С в образце
и, как следствие, определения происхождения данного образца (разд. 4.2.2), при
интерпретации масс-спектров обычно используется величина отношения 13С к
12С 1,1%.
Наличие одного атома углерода в молекуле метана приводит к появлению
наряду с пиком М+'с m/z 16 пика иона с m/z 17 и интенсивностью 1,1% от М+*
за счет молекул 13СН4, количество которых в природе составляет 11 на каждые
1000 молекул 12СН4. Увеличение числа атомов углерода в молекуле приводит к
увеличению интенсивности пика ионов М+1 до величины 1,1 и%, где « — число
атомов углерода в молекуле. Для определения числа атомов углерода в молекуле
по масс-спектру необходимо разделить интенсивность пика М+1 в процентах от
? на 1,1. Полученное частное означает максимально возможное число атомов
углерода в молекуле. Надо помнить, что расчет может оказаться сложнее (см.
ниже) при наличии пика иона [М —Н]+.
Большое число атомов углерода приводит к увеличению вероятности
одновременного присутствия в молекуле двух и большего числа атомов 13С, что
вызывает рост интенсивностей пиков изотопных ионов [М+2], [М+3] и т. д.
(табл. 4.2). Для каждого атома другого элемента, присутствующего в ионе,
необходимо увеличить интенсивность пика А+1 на 0,37% (для азота), на 0,04%)
(для кислорода), на 5,1% (для кремния), на 0,8% (для серы); а пика А+2 —на
4.2. Определение элементного состава ионов на основании изотопных пиков 71
Таблица 4.2
Вклад изотопов углерода в интенсивности пиков изотопных ионов.
Интенсивность основного пика (А) принята за 100%
Число
атомов углерода
в молекуле
? ?
2
3
4
5
6
7
1 8
9
10
11
12
13
14
15
16
17
18
19
20
30
40
50
60
70
80
90
100
А+1
?~~
2,2
3,3
4,4
5,5
6,6
7,7
8,8
9,9
11,0
12,1
13,2
14,3
15,4
16,5
17,6
18,7
19,8
20,9
22,0
33,0
44,0
55,0
66,0
77,0
88,0
99,0
ПО
А+2
0
0,01
0,03
0,07
0,12
0,18
0,25
0,34
0,44
0,54
0,67
0,80
0,94
1,10
1,27
1,45
1,65
1,86
2,07
2,30
5,26
9,44
14,8
21,4
29,2
38,2
48,5
59,9
А+3
0
0
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
0,01
0,02
0,02
0,03
0,04
0,05
0,06
0,07
0,09
0,11
0,13
0,15
0,54
1,32
2,54
4,55
7,29
10,9
15,6
21,5
А+4
0
0
0
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
0,04
0,13
0,32
0,71
1,34
2,32
! 3,74
5,74
А+5
о
0
0
0
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
<0,01
0,01
0,03
0,09
0,19
0,39
0,68
1,21
0,2% (для кислорода), на 3,4% (для кремния), на 4,4% (для серы), на 32,5% (для
хлора), на 98,0% (для брома).
Величину изотопного пика за счет присутствия в молекуле нескольких
изотопов углерода 13С легко рассчитать самостоятельно. Интенсивность пика А+2
(в % к пику А) для соединения, содержащего ? атомов углерода, определяется
по формуле:
/(%)=100<^@,011J D.3)
72 Глава 4. Практические основы интерпретации масс-спектров
m/z
Ъ1
38
39
40
47
49
50
51
53
55
62
63
Л%
8,3
14,6
51,6
20,3
6,5
2,1
9,7
8,3
5,5
13,2
5,4
9,4 |
m/z
64
65
66
67
74
75
76
77
79
94
95
96
Л%
4,4
40,5
53,3
2,35
2,8
1,5
1,2
1,6
1,2
100
6,6
0,4
m/z
Рис. 4.3. Масс-спектр соединения с молекулярной массой 94 Да
Интенсивность пика А+3 (в % к пику А) для соединения, содержащего ?
атомов углерода, определяется по формуле:
7(%)=100С*@,011K D.4)
Интенсивность пика А+4 (в % к пику А) для соединения, содержащего ?
атомов углерода, определяется по формуле:
/(%)= 100(^@, ОНL D.5)
В общем виде интенсивность пика А+т (в % к пику А) для соединения,
содержащего ? атомов углерода, определяется по формуле:
/(%)=100С™@,011Г D.6)
На этой стадии расшифровки спектра можно не учитывать вклад атомов
азота в сигнале М+1, поскольку соответствующие коррективы будут внесены
в дальнейшем. После подсчета числа атомов углерода по пику М+1 необходимо
внести коррективы в интенсивность пика ?+2, в частности, точнее оценить
число атомов кислорода. В качестве примера рассмотрим масс-спектр соединения в
графическом и табличном виде, представленный на рис. 4.3.
Молекулярный ион (m/z 94) имеет максимальную интенсивность в спектре.
Относительная интенсивность иона ?+ 2 (m/z 96) составляет 0,4% от М+#.
Следовательно, вещество не содержит в составе хлора, брома, серы и кремния,
а число атомов кислорода может составлять от 0 до 2. Относительная
интенсивность пика М+1, равная 6,6% позволяет сделать вывод о наличии 6 атомов
углерода в молекуле F,6/1,1 = 6). По таблице 7.2 устанавливаем вклад изотопов
100-
90-
80 Ч
70 4
60-|
50-
40-
30-
20-
10-
0-
30
1,%
In.
70
100
4.2. Определение элементного состава ионов на основании изотопных пиков 73
13С в суммарную интенсивность пика ?+2. Для шести атомов С этот вклад
составляет 0,18%. Таким образом, можно сделать вывод, что в молекуле может
быть не более одного атома кислорода @,4 — 0,18 = 0,22). Масса 6 атомов
углерода 72 Да; масса 1 атома кислорода 16 Да; суммарно 88. Оставшиеся 6 а. е. м.
(94 — 88 = 6) могут быть отнесены только к атомам водорода. Следовательно,
состав соединения СбНбО. Другие варианты химически реальных составов (с
атомами фтора или азота) не проходят либо по массе, либо по интенсивности
изотопных пиков. На рисунке 4.3 представлен масс-спектр фенола.
4.2.1. Азотное правило
Для большинства элементов, входящих в состав органических соединений,
имеется случайное, но очень удобное для масс-спектрометристов, соответствие
между валентностью и массой наиболее распространенного изотопа: либо оба
числа четные, либо — нечетные. Важнейшим исключением является азот. Так
появилось азотное правило, кажущееся, на первый взгляд, весьма странным:
если соединение содержит четное число атомов азота или азота в составе
нет, его молекулярная масса четная; если число атомов азота нечетное,
молекулярная масса тоже нечетная. Правило применимо не только к молекуле
и молекулярному иону, но и к фрагментным ионам. В этом случае оно
формулируется следующим образом: нечетноэлектронный ион имеет четную массу,
если содержит четное число атомов азота; четноэлектронный ион имеет
четную массу, если содержит нечетное число атомов азота.
Азот является (А+ 1)-элементом, причем природная распространенность
изотопа 15? составляет примерно 0,4% от 14?. Поскольку помимо азота только
углерод вносит значимый вклад в интенсивность пика А+1, пользуясь различиями
в интенсивностях изотопов 13С и 15? и азотным правилом, можно оценить число
атомов азота в молекуле.
Для расчета интенсивностей изотопных пиков, обусловленных присутствием
в молекуле атомов азота, можно использовать формулы, аналогичные
представленным для случая углерода. В общем виде интенсивность пика А+т (в % к
пику А) для соединения содержащего ? атомов азота определяется по формуле 4.7:
/(%) = 100С™@,0037)" D.7)
Когда установлена природа и количество (А+2)- и (А+1)-элементов в
молекуле, несложно сделать аналогичные заключения и о моноизотопных элементах.
Эти элементы имеют специфические массы (табл. 4.1), поэтому их определение
обычно не вызывает затруднений.
Следует помнить, что установление элементного состава по изотопным пикам
может оказаться проблематичным, если, например, в образце были примеси с
массами ионов в области кластера молекулярного иона исследуемого
соединения. Для веществ с основными функциями (например, аминов) даже в спектрах
74 Глава 4. Практические основы интерпретации масс-спектров
ЭУ зачастую присутствует пик иона [М+Н]+, искажающий изотопную картину.
Необходимо также учитывать, что интенсивности пиков в реальных условиях
подвержены незначительным колебаниям от спектра к спектру. Для установления
точного соотношения изотопных пиков лучше всего записать несколько спектров
и усреднить результаты.
Задача 4.5. Установите примерное число атомов углерода в соединениях по
изотопным пикам М+\ В большинстве случаев спектр представлен в форме:
величина m/z (интенсивность к максимальному пику в спектре в %). Пик
молекулярного иона указан в ряду первым (в скобках везде, как показано для
в), приведено отношение m/z).
а) М+- 100%, М+1 7,8%, ?+2 0,2%;
б) М+- 100%, М+1 0%, М+2 0%;
в) М+· (m/z 128) 100%, М+1 (m/z 129) 11,2%, М+2 (m/z 130) 0,5%;
г) 134 A00), 135 F,6), 136 C3), 137 B,1);
д) 133 A00), 134 (9,3), 135 @,3);
е) 156 A00), 157 B,3), 158 @);
ж) 120 A00), 121 (8,6), 122 D,7);
з) 180 A00), 181 D,6), 182 (98), 183 D,5);
и) 159 A00), 160 A0,4), 161 C3), 162 C,4);
к) 77 A00), 78 B,6), 79 @);
л) 184 E0), 185 A,1), 186 A00), 187 B,2), 188 E0), 189 A,1);
м) 107 C3), 108 B,7), 109 @,08).
Задача 4.6. Определите элементный состав молекулярных ионов по
интенсивности изотопных пиков. В большинстве случаев спектр представлен в
форме: величина m/z (интенсивность к максимальному пику в спектре в %). Пик
молекулярного иона указан в ряду первым (в скобках везде, как показано для а),
приведено отношение m/z).
а) M+,(m/z 108) 100%, М+1 (m/z 109) 7,7%, М+2 (m/z ПО) 0,4%.
б) 79 A00), 80 E,9), 81 @,1);
в) 128 A00), 129 A1,0), 130 @,5);
г) 98 A00), 99 F,3), 100 D,5);
д) 98 A00), 99 B,2), 100 F6,0), 101 A,4), 102 A1,0), 103 @,2);
е) 208 (80), 209 A2,4), 201 A,2);
ж) 204 A00), 205 G,7), 206 @,2);
з) 85 G5), 86 C,4), 87 C,3);
и) 158 A00), 159 G,4), 160 E,2), 161 @,2);
к) 206 (80), 207 (8,8), 208 G9), 209 (8,8), 210 @,4);
л) 170 A00), 171 C,4), 172 @,04).
4.2. Определение элементного состава ионов на основании изотопных пиков 75
Задача 4.7. Идентифицируйте соединения по изотопным пикам
молекулярного иона. Спектр представлен в форме: величина ??/? (интенсивность к
максимальному пику в спектре в %). Пик молекулярного иона указан в ряду
первым.
а) 94 A00), 95 A,1), 96 (98), 97 A,1);
б) 64 A00), 65 B,2), 66 C3), 67 @,7);
в) 64 A00), 65 @,9), 66 D,8);
г) 67 A00), 68 D,8), 69 @,1);
д) 142 (80), 143 (8,8), 144 @,4);
е) 46 E0), 47 A,1), 48 @,1);
ж) 84 G5), 85 C,9), 86 C,3);
з) 88 A00), 89 A,1), 90 @);
и) 59 E0), 60 A,3), 61 @,1);
к) 156 E0), 157 A,1), 158 @).
Задача 4.8. Рассчитайте интенсивности изотопных пиков молекулярного
иона для перечисленных ниже соединений.
а) Дибромметан;
б) хлороформ;
в) четыреххлористый углерод;
г) сероуглерод;
д) дихлорэтан;
е) тетрабромбензол;
ж) бромхлорметан;
з) бромоформ;
и) дибромдихлорметан.
Задача 4.9. Установите молекулярный ион и определите его элементный
состав в следующих сериях. Спектр представлен в форме: величина ??/?
(интенсивность к максимальному пику в спектре в %).
а) 94 A,9), 95 G,1), 96 A00), 97 F,5), 98 @,2);
б) 160 @,8), 161A,0), 162 A00), 163 A0,8), 164 C2,9), 165 C,6), 166 @,2);
в) 93 A,1), 94 A00), 95 C,8), 96 (8,8), 97 @,26);
г) 134 B,1), 135 @,4), 136 B6,0), 137 B,6), 138 @,16);
д) 126 F,0), 127 (9,8), 128 A00), 129 A1,0), 130 @,5);
е) 260 A,3), 261 A2,2), 262 A00), 263 A9,5), 264 A,9);
ж) 127 A,8), 128 A6,0), 129 A00), 130 A0,0), 131 @,5);
з) 121 (89,0), 122 A00), 123 G,9), 124 @,7);
и) 328 C3,0), 329 B,2), 330 A00), 331 F,6), 332 A00), 333 F,6), 334 C3,0),
335 B,2).
76 Глава 4. Практические основы интерпретации масс-спектров
4.2.2. Определение содержания изотопа 13С в природных образцах
Круговорот углерода в природе осуществляется с участием углеродсодержащих
соединений атмосферы, гидросферы, литосферы и биосферы. Существует
определенная разница в содержании изотопа углерода 13С между соединениями. Для
оценки отклонения содержания изотопа 13С от среднего значения используется
шкала ?(%?). Отклонение рассчитывается по уравнению D.8):
513С =
A3C/12C)Q
(l3C/UQs
? 1000 D.8)
где индексы о и s означают соотношение изотопов углерода в образце и
в стандарте. В качестве нулевой точки был выбран карбонатный материал
литосферы. Наиболее близкую к этому значению величину A,95%о) имеет
стандартный образец NBS-19. Существует ряд других стандартных образцов.
Например, NBS-22 нефть-(-29,74%о), NBS-18 карбонат кальция-(-5,01%о).
Обычно величины 513С для растений лежат в диапазоне (— 15%о) — (—30%о), для
нефти — (—20%о)-(—36%о). Наиболее обеднен изотопом 13С атмосферный метан:
513С^ - 47%о.
Для точного определения 513С образец сжигают до СО2 и замеряют
интенсивности пиков ионов с m/z 44, 45, 46, после чего проводят корректировку для
устранения вклада изотопа 170. При использовании магнитного анализатора и
трех детекторов для каждой массы D4, 45, 46) можно получить результаты с
относительным стандартным отклонением до 0,001%. Точность измерения и
использование шкалы SnC позволяют получать достаточно интересные результаты.
В частности анализ останков дает возможность различить древних европейцев и
американцев. Дело в том, что в Европе основным пищевым продуктом была
пшеница, а в Америке — кукуруза. Различие в изотопном составе этих растений
составляет несколько единиц по шкале ?13 С [107].
4.2.3. Расчет изотопной чистоты соединений
Масс-спектрометрия предоставляет отличную возможность оценить число
тяжелых изотопов в молекуле. Химик-органик сталкивается с такой задачей довольно
часто, например, изучая механизмы реакций. Обычно синтезируют образцы,
обогащенные изотопами 2Н, 13С, 14С, 15?, 180, хотя используют изотопы и
других элементов.
Определять содержание меченых молекул в образце лучше всего по спектрам
электронного удара, поскольку в этом случае не идут ионно-молекулярные
реакции. Для расчетов можно использовать наиболее интенсивные пики в
спектре, обусловленные ионами с максимальными величинами m/z. Оптимальным
кандидатом является молекулярный ион. Однако если в спектре немеченого
соединения присутствуют значимые пики ионов [М+Н]+ или [М —Н]+, расчет
4.2. Определение элементного состава ионов на основании изотопных пиков 77
Таблица 43
Расчет интенсивностей изотопных пиков в масс-спектре дейтерированного ацетофенона
Изотопомер
1 *
d2
d3
10% d0
? 10% dx
20% d2
60% d2
Udo — d$
Нормализованная Y,do—d-$ (%)
m/z 120
100,0
10,0
10,0
16,2
m/z 121
8,8
100,0
0,88
10,0
10,88
17,6
m/z 122
0,54
8,8
100,0
0,05
0,88
20,0
20,93
33,9
m/z 123
0,54
8,8
100,0
0,05
1,76
60,0
61,81
100,0
m/z 124
0,54
8,8
0,11
5,28
5,39
8,7
m/z 125
0,54
0,32
0,32
0,5
будет затруднен. Иногда это препятствие можно обойти. Для устранения пиков
ионов [М+Н]+ необходимо постараться записать спектр с минимально
возможным напуском образца. Для учета вклада ионов [М—Н]+ необходимо знать,
из какого положения молекулы отщепляется атом водорода и может ли в этом
положении оказаться атом дейтерия. При проблематичном ответе на эти вопросы
можно попытаться поработать с ионом точно известного состава, например
[М —СНз]+, [М — СО]"', [М —С1]+, при этом важно, чтобы рядом с выбранным
ионом, не было иона другого состава с массой на 1-2 Да легче или тяжелее.
Рассчитаем интенсивность кластера молекулярного иона образца
ацетофенона, 60% молекул которого содержат три атома дейтерия, 20% — 2 атома дейтерия,
10%— 1 атом дейтерия, а 10% молекул изотопно незамещены. Составим таблицу
(табл. 4.3), учитывая, что кластер молекулярного иона чистого немеченного
ацетофенона имеет следующий вид: 120 A00%), 121(8,8%), 122@,54%). Последняя
строка табл. 4.3 представляет собой искомый вид спектра.
Обычно приходится решать обратную задачу, когда по спектральной картине
необходимо рассчитать процентное содержание соединений с разным
количеством тяжелых изотопов в молекуле. Пусть для образца меченого
ацетофенона получен следующий масс-спектр: 120 B0,0%), 121 D2,0%), 122 A00%),
123 A4,0%), 124 A,0%), 125 (<0,03%). Решать задачу удобно с помощью
таблицы (табл. 4.4).
Ион с m/z 120 может быть обусловлен только немеченным ацетофеноном.
Поскольку интенсивность пика этого иона 20%, будем считать, что в смеси 20
частей немеченного соединения. Исходя из этого рассчитаем интенсивности
пиков ионов с m/z 121 и 122, обусловленных изотопными ионами немеченного
соединения. Если 100 частей этого соединения дают спектр 120 A00%), 121 (8,8%),
122 @,54%), то 20 частей дадут- 120 B0,0%), 121 A,76%), 122 @,11%). Именно
эти цифры представлены во второй строке табл. 4.4. В третьей строке (остаток 1)
представлен спектр образца после вычета вклада немеченного соединения. Сле-
78 Глава 4. Практические основы интерпретации масс-спектров
Таблица 4.4
Расчет доли молекул ацетофенона с различным содержанием атомов дейтерия
Изотопомер
Спектр
d0
Остаток 1
d\
Остаток 2
*
Остаток 3
d3
Остаток 4
m/z 120
20,0
20,0
-
-
-
-
-
-
-
m/z 121
42,0
1,76
40,24
40,24
-
-
-
-
-
m/z 122
100,0
0,11
99,89
3,54
96,35
96,35
-
-
-
m/z 123
14,0
-
14,0
0,2
13,8
8,5
5,3
5,3
-
m/z 124
По
-
?,?
-
?,?
0,5
0,5
0,47
<0,03
m/z 125
0ДJ
~
0,02
-
0,02
-
0,02
0,02
-
дующим шагом необходимо оценить долю монодейтерированного ацетофенона.
Интенсивность 40,24 пика с m/z 121 обусловлена М+" этого соединения.
Рассчитаем интенсивности изотопных пиков для такой доли этого продукта. Получим
следующий ряд D-я строка) 121 D0,24%), 122 C,54%), 123 @,2%). Вновь
проводим вычитание этих интенсивностей из спектра (остаток 1) и получаем
остаток 2, т. е. спектр, обусловленный только соединениями с двумя и тремя
атомами дейтерия в молекуле. Повторяем эту процедуру еще дважды и получаем
остаток 4. Здесь присутствует только пик иона 124 с интенсивностью 0,03%, т. е.
это уровень ошибки измерения. Таким образом, мы получили следующий ряд:
do 20 частей, d\ 40,24 части, аг 96,36 части, d^ 5,3 части. Принимая сумму изото-
померов за 100%, получаем доли каждого: 12,3% do, 24,9% d\9 59,5% J2, 3,3% ?/3·
Задача 4.10. Рассчитайте процентное содержание изотопомеров в спектре
образца ацетофенона, характеризующегося следующим масс-спектром:
а) 120 A5,0%), 121 C2,0%), 122 E0,0%), 123 A00%), 124 (8,7%), 125 @,5%);
б) 120 D0,0%), 121 D0,0%), 122 A00%), 123 E0%), 124 C,5%), 125 @,25%).
Задача 4.11. Рассчитайте процентное содержание изотопомеров в спектре
образца и-метоксифенола, характеризующегося следующим масс-спектром:
а) 123 @,21%), 124 C0,0%), 125 F0,0%), 126 (80,0%), 127 A00%), 128 G,7%),
129 @,6%);
б) 124 @,2%), 125 A,2%), 126 A00%), 127 B8,0%), 128 A,8%), 129 @,05%).
Задача 4.12. Рассчитайте процентное содержание изотопомеров в спектре
образца 4-фтор-2-метоксианизола, характеризующегося следующим
масс-спектром:
а) 155 @,12%), 156 B0,0%), 157 A00%), 158 A2,0%), 159 @,8%), 160 @,02%);
б) 155 @.06%), 156 A0,0%), 157 B3,0%), 158 D2,0%), 159 E6,0%), 160 A00%),
161 E9,2%), 162 C,7%), 163 @,37%).
4.3. Фрагментные ионы 79
Задача 4.13. Рассчитайте вид спектра в области молекулярного иона для
спектра образца, содержащего 10% немеченных, 20% d\, 30% di и 40% d$
меченых молекул 4,7-дифторнафтилметилкетона.
4.3. Фрагментные ионы
Когда получена исчерпывающая информация о молекулярном ионе, следует
перейти к рассмотрению фрагментных ионов. Как уже отмечалось выше,
фрагментные ионы можно разделять на перегруппировочные и осколочные, однако
для интерпретации масс-спектра полезно пользоваться также другой
классификацией. Все важнейшие фрагментные ионы можно разделить на три вида.
1) Наиболее тяжелые ионы, образующиеся из М+' в результате выброса
простейших частиц, т. е. без существенной перестройки структуры исходной
молекулы.
2) Ионы, характеризующиеся наиболее интенсивными пиками в спектре.
3) Характерные серии ионов, различающихся на гомологическую разность, т. е.
на 14 а. е. м.
Необходимо учитывать, что интенсивности пиков фрагментных ионов
первого и третьего типа могут быть незначительными. Очень важно научиться не
пропускать их.
4.3Л. Гомологические серии ионов
Если индивидуальные пики ионов с большими массами важны для
установления начальных характеристических путей фрагментации, лабильных групп, то
гомологические серии ионов с низкой массой несут информацию о наличии
гетероатомов, степени ненасыщенности, и, в конечном итоге, принадлежности
соединения к тому или иному классу. Для таких ионов (как правило, чет-
ноэлектронных) значительно проще установить элементный состав, чем для
более тяжелых, для которых могут существовать сотни изобарных составов.
В таблице 4.5 представлены гомологические серии ионов, характерные для
наиболее распространенных классов органических соединений.
Следует подчеркнуть, что в спектре, например, алифатического спирта
наблюдаются и алкановая B9, 43, 57...), и алкеновая B7, 41, 55), и спиртовая C1,
45, 59...) серии. Алкановая серия присутствует в масс-спектре любого
соединения, содержащего алкильную группу. В случае изобарных серий (например, ал-
каны и кетоны) следует обращать внимание на интенсивность изотопных пиков.
Так, для изобарных ионов с массой 43 (составы СНзСО и С3Н7) интенсивность
изотопного пика с массой 44 составит 2,2% и 3,3% соответственно. Еще проще
обстоит дело с (А+2)-элементами. В этом случае в спектре проявляются две
серии характеристических ионов за счет изотопов А и А+2.
80 Глава 4. Практические основы интерпретации масс-спектров
Таблица 4.5
Гомологические серии ионов некоторых классов органических соединений
Класс соединения
Алканы
Алкены, нафтены
Алкины, диены
Спирты, простые эфиры
Альдегиды, кетоны
Кислоты, сложные эфиры
Тиолы, сульфиды
Амины
Алкилхлориды
Алкилфториды
Алкилбромиды
Алкилиодиды
Нитрилы
Алкилбензолы
Формула
С"Щп+\
спи2п_1
СпН2п_3
СяН2л+10+
0,?2?_!?+
СлН2л_!0+
CwH2n+iS+
CwH2n+2N+
СлН2лС1+
C„H27IF+
СлН2иВг+
C„H2J+
0,?2„_2?+
m/z
15,29,43,57,71,85...
27,41,55,69,83...
25,39,53,67,81...
31,45,59,73,87...
29,43,57,71,85...
45,59,73,87, 101...
47,61,75,89, 103... (no 32S)
30,44,58,72,86, 100...
35, 49, 63, 77, 91, 105 ... (по 35С1)
19,33,47,61,75...
79,93, 107, 121... (по 79Вг)
127, 141, 155, 169...
40, 54, 68, 82, 96...
38, 39, 50-52, 63-65, 75-78, 91, 105,
119...
Если степень ненасыщенности молекулы или количество функциональных
групп в ней велико, значимые серии ионов с низкой массой, отстоящих друг от
друга на СН2-группу, в спектре отсутствуют. Хорошим примером могут служить
алкилбензолы. В области низких масс гомологическая серия отсутствует, хотя
характеристические ионы имеются. Ароматическая серия все же наблюдается,
хотя она сдвинута в область более высоких масс (табл. 4.5; ионы 91, 105, 119).
Если алкильная группа связана с заместителем с высокой ЭИ, в спектре
возникает, иногда достаточно интенсивная, серия нечетноэлектронных ионов
общей формулы СЛН^.
Задача 4.14. К какому классу принадлежат монофункциональные
соединения, спектры которых представлены на рис. 4.4?
Безусловно, анализ гомологических серий имеет свои ограничения. Низкие
интенсивности пиков в некоторых сериях требуют внимания и опыта
исследователя, чтобы не пропустить их. Зачастую в спектрах самых разнообразных
соединений доминируют пики алкановой серии. Распад с участием одной
функциональной группы может полностью подавить или существенно
замаскировать другие направления фрагментации полифункциональных соединений. В
последнем случае полезно бывает использовать при интерпретации масс-спектра
данные ИК-спектроскопии.
Задача 4.15. На рисунке 4.5 представлены масс-спектры трех сдединений,
характеризующихся близким набором фрагментных ионов. Пики молекулярных
ионов в двух случаях отсутствуют. Постарайтесь определить класс соединений
vo
? ?
- ?
?
? ? ? ? ? ? ? ? ? ?
???????????
00>(OS(OU)«C)(Mr
?
? ? ? ? ? ? ? ? ? ?
???????????
00>«N(DU)*nCMr-
? *"*
- ?
- ?
? ? ? ? 1 ? ? ? 1 ?
???????????
?
?
?
?
S
?
?
?
?
?
•?*
?
?
?
1
•?·
?
?
?
2
?
?
?
?
?
s
?—?—?—?—?—?—?—?—г
?—?—?—?—?—?—?—?—?—г
2
о
о
?
к
s
?.
ts
s
я
35
IS
9
о
о
??
к
о
S
Hi
w
I
о
S3
о
2
<L>
и
о
I
о
о
cd
я
? ? ? ? ? ?
о о о о
84 Глава 4. Практические основы интерпретации масс-спектров
на основании гомологических серий ионов. Учтите, что интенсивности пиков в
некоторых сериях могут быть низкими.
4.3.2. Выбросы простейших нейтральных частиц
Самые простые, но важные заключения можно сделать на основании выбросов
нейтральных фрагментов из М+\ В результате этих процессов возникают ионы
с большой массой: [М—1]+, [М—15]+, [М—18]+', [М — 20]+* и т. д. Например,
интенсивный сигнал иона [М —Н]+ означает не только наличие лабильного
атома водорода, но и отсутствие других лабильных групп в этом положении
(правило выброса максимального алкила, разд. 3.1.1). В области высоких масс
важны практически все пики, даже если их интенсивность ниже 1%. Эти ионы
очень важны и для подтверждения правильного определения молекулярного иона
(разд. 4.1).
4.3.3. Наиболее интенсивные пики в спектре
Извлечение информации из наиболее интенсивных в спектре пиков иногда
оказывается достаточно сложным делом. Регистрация пика иона с конкретной
целочисленной величиной ??/? не дает однозначного определения его состава.
Даже использование масс-спектрометрии высокого разрешения (разд. 6.3) для
определения элементного состава иона не является методом установления его
структуры, поскольку он может образоваться прямо из М+" с минимальным
искажением исходной структуры или в результате ряда перегруппировочных
процессов. Например, интенсивный пик иона с m/z 30 не дает права однозначно
приписать вещество к первичным аминам, так как этот ион легко образуется
и из вторичных, и из третичных аминов при достаточной длине их алкильных
радикалов (разд. 8.7 ).
Однако если незначительные по интенсивности пики можно проигнорировать
без особого ущерба для результата процесса интерпретации, состав ионов,
характеризующихся максимальными пиками, желательно установить однозначно.
Они, как правило, приводятся в схеме фрагментации, отражающей основные
направления распада анализируемого соединения. Эти ионы могут
характеризоваться большой массой и теоретически иметь много возможных изобарных
составов. Поэтому для установления их состава может понадобиться масс-
спектрометрия высокого разрешения, а также получение спектров родительских
и дочерних ионов. Если исследователь работает долгое время с определенным
классом соединений, то состав и структура основных ионов, как правило, уже
известны и проводить отнесение фрагментных пиков в новых спектрах легко.
Задача оказывается более сложной, если речь идет о неизвестных образцах,
принадлежащих к самым разнообразным классам органических соединений.
86 Глава 4. Практические основы интерпретации масс-спектров
стоинством электронного удара являются стандартные условия регистрации
спектра. Благодаря этому спектры одних и тех же соединений, полученные на
разных приборах, оказываются похожими и доступны сравнению. Программное
обеспечение этих библиотек позволяет проводить не только рутинное сравнение
спектра нового образца с библиотечными, но и делать выборки с учетом
элементного состава, интенсивности конкретных пиков, молекулярной массы и т. д.
Работая с этими выборками, оператор может проявить свои знания, использовать
дополнительную информацию, проигнорировать пики, про которые известно,
что они обусловлены примесями. В этом случае надежность правильной
идентификации соединения значительно повышается.
Прежде чем проводить сравнение полученного спектра с библиотечными,
имеет смысл оценить его качество и сделать возможные процедуры его
улучшения. В частности, в разд. 1.4 описан метод устранения фона в режиме
ГХ-МС. Обработанный спектр (рис. 1.6) отлично сходится с библиотечным,
хотя для исходного спектра компьютер не предлагал этого варианта структуры.
В главе 9 описана процедура компьютерной обработки хроматографического
пика, обусловленного двумя соединениями с близкими временами выхода.
Использование компьютерных библиотек, безусловно, существенно облегчает
процесс идентификации соединения по его масс-спектру, но в целом машина не
может пока полностью заменить квалифицированного специалиста. Даже если
спектр соединения имеется в библиотеке, не всегда очевидно, что компьютер
его правильно определит, точнее, поставит на первое место среди выбранных
им вариантов. На этой стадии последующее «ручное» сравнение предложенных
вариантов со спектром образца очень полезно. Если спектр анализируемого
вещества отсутствует в библиотеке, компьютер все равно предложит
наиболее подходящие, с его «точки зрения», варианты. Эта информация наверняка
облегчит задачу оператора, поскольку может дать необходимый толчок для
отнесения вещества к тому или иному классу соединений. В настоящее время
создаются алгоритмы для элементов машинной расшифровки масс-спектров. Эти
программы требуют творческого подхода высококвалифицированного оператора
и позволяют получать впечатляющие результаты, например предложить наиболее
вероятную структуру соединения, спектр которого отсутствует в библиотеке.
4.5. Использование для интерпретации
дополнительной масс-спектральной информации
Зачастую оказывается, что обычного спектра электронного удара недостаточно
для установления структуры вещества. В этом случае необходимо использовать
другие возможности масс-спектрометрии. Например, отсутствие пика
молекулярного иона в спектре электронного удара может потребовать получение
4.4. Библиотеки масс-спектров 85
Не следует также слепо использовать таблицы, приводимые в масс-спек-
трометрической литературе, связывающие величину m/z регистрируемого пика
со структурным фрагментом молекулы (табл. 3 приложения). Такие таблицы
могут быть полезными, но пользоваться ими надо с осторожностью. Например,
регистрация иона С2Н3О4" часто интерпретируется в таблицах как ацетильный
фрагмент (СНзСО), при этом вещество относится к метилкетонам. Однако фраг-
ментный ион СНзСО+ возникает в процессе фрагментации (перегруппировка
Мак-Лафферти) многих кетонов с достаточной длиной алкильной цепи.
Тем не менее некоторые специфические ионы, характеризующиеся
интенсивными пиками в спектрах, сразу наводят на мысль об определенной
структуре: m/z 77 —фенил, m/z 91—тропилий С7Н7 (бензил), m/z 30 —аминогруппа
CH2NH2, m/z 105 —бензоил PhCO. Доминирующий в спектре пик иона с m/z 149
практически наверняка означает принадлежность исследуемого соединения к
диалкилфталатам. Кстати, эти соединения, широко используемые во всем мире
в качестве пластификаторов при производстве пластмасс, оказались достаточно
стабильными и в настоящее время являются приоритетными загрязняющими
веществами в окружающей среде. Фталаты, таким образом, очень часто
оказываются примесями, порой весьма неожиданными, в самых различных образцах.
Работая в режиме хроматомасс-спектрометрии, оператор часто сталкивается
с хроматографическими пиками, характеризующимися спектрами, в которых
доминируют пики ионов с массами 73, 147, 207, 281, 355 и т. д. Этот набор
обусловлен выбросами фрагментов наиболее распространенных полидиметил-
силиконовых фаз хроматографической колонки в источник масс-спектрометра.
В таблице 3 приложений представлены наиболее распространенные варианты
элементных составов ионов с целочисленными массами.
После извлечения всей возможной информации из масс-спектра надо
попытаться скомпоновать наиболее вероятную структуру. Если есть основания
заранее предполагать какую-либо структуру, необходимо попытаться нарисовать
теоретически возможные пути ее распада и сопоставить с полученным спектром.
4.4. Библиотеки масс-спектров
В настоящее время издано большое число атласов масс-спектров (в
подавляющем большинстве спектры электронного удара), однако значительно удобнее
пользоваться компьютерными библиотеками. На сегодняшний день существуют
две основные коммерческие библиотеки масс-спектров: Wiley и NIST.
Библиотеки содержат именно спектры электронного удара, поскольку максимальное
число образцов анализировалось и анализируется в настоящее время именно
этим методом. Интенсивная фрагментация позволяет проводить более надежное
сравнение спектров, тогда как спектры химической ионизации или полевой
ионизации характеризуются всего лишь несколькими пиками. Еще одним до-
4.5. Использование для интерпретации дополнительной масс-спектральной информации 87
спектра каким-либо «мягким» методом (химическая ионизация, полевая
ионизация). Проблема пиков примесей в спектре соединения, снятого с помощью
прямого ввода, может быть преодолена с помощью хроматомасс-спектромет-
рии. Достаточно очевидна важность масс-спектрометрии высокого разрешения
(разд. 6.3), поскольку информация о точном элементном составе молекулярного
и фрагментных ионов имеет первостепенное значение.
Менее ясна, на первый взгляд, необходимость использования спектров ме-
тастабильных ионов и спектров активации соударением. Эти две техники
позволяют прежде всего получить чистый спектр вещества, лишенный накладок
со стороны различных примесей. Кроме того, они дают возможность точно
установить направления распада, связывая конкретные родительские и дочерние
ионы. Эти виды спектров могут предоставить определенную информацию и о
структуре ионов (см. гл. 7).
Например, тот факт, что в спектрах сульфонов (схема 4.1) пики ионов с
массами [90+R]+ и [104+R]+, образующихся непосредственно из
молекулярного иона, однозначно свидетельствуют о его трансформации до распада с
образованием новой С—С-связи между атомами углерода малого цикла и второго
ароматического ядра [108]. В этом случае циклопропановая группа,
обладающая наименьшей ЭИ, и, следовательно, несущая в электронно-невозбужденном
состоянии заряд и неспаренный электрон, атакует по нуклеофильному или
радикальному механизму второе ароматическое ядро.
[104 + RJ+ ^ \=/ [90 + R]+
Схема 4.1
Иногда никакие варианты съемки масс-спектра не позволяют однозначно
установить структуру интересующего иона. В этом случае реальную помощь
могут принести масс-спектры изотопных аналогов анализируемого соединения.
Этот подход успешно используется и в других физико-химических методах, а
также в классической органической химии. В учебнике по масс-спектрометрии
[36] синтезу этих соединений для масс-спектрометрических целей посвящен
достаточно большой раздел.
+/СНЧ
О О О С=0 О 1+
CH30-C-CRr CR2-C-CHN2 ,Сч ,CR2 CH30-C-CR2-CR2-CH=C=0''
СН30 CR2
4-1 4-2 4-3
88 Глава 4. Практические основы интерпретации масс-спектров
В качестве примера использования изотопных аналогов для решения масс-
спектрометрических вопросов можно привести следующий эксперимент [109].
Ионы [?—?2]+? диазокетонов 4-1 распадаются по ряду направлений, которые
требуют их предварительной изомеризации в другие структуры, прежде всего
пирановую 4-2 или кетеновую 4-3.
Для выбора одной из двух альтернативных структур пришлось синтезировать
дейтерированный аналог (три атома водорода метильного радикала сложноэфир-
ной группы были заменены на атомы дейтерия). Дело в том, что одним из
путей распада иона [?—?2]4" является выброс метильной группы. Поскольку
в составе диазокетонов 4-1 радикалы R также представляли собой метальные
группы, необходимо было установить, какая именно группа отщепляется из
[?—?2]4". Спектр дейтерированного аналога показал, что элиминируется
именно метильная группа сложноэфирной функции. На этом основании предпочтение
было отдано пирановой структуре. Алкильная группа не может отщепиться из
этого положения в кетеновой структуре 4-3, тогда как для гетероциклической
структуры 4-2 такой процесс, напротив, весьма благоприятен (схема 4.2).
E3L. 5*™с*°
? ?
^С CR2
°* W
Схема 4.2
4.6. Схема фрагментации
Заканчивается процедура работы со спектрами составлением схемы
фрагментации. Эта схема должна отражать основные характеристические направления
распада молекулярного иона, а также состав фрагментных ионов, по возможности
их структуру, взаимосвязь этих ионов друг с другом, а иногда интенсивности их
пиков (схемы 4.3 и 4.4).
Невозможно разместить на схеме все ионы, пики которых наблюдаются в
спектре. Многие из них не несут никакой принципиальной химической
информации, а в многообразии представленных реакций можно потерять действительно
важные характеристики данного соединения. Нет смысла приводить также
тривиальные фрагменты. Например, в спектре любого сложного ароматического
соединения будут присутствовать пики с m/z 77 или 76, обусловленные бензольным
ядром. Пики этих ионов могут иметь высокую интенсивность, однако они (ионы)
не раскрывают своеобразие исходной молекулы и могут образоваться
практически из любого фрагмента с большей массой. Поэтому необходимо включать в
схему важные для групповой или индивидуальной характеристики фрагментные
+ СН
о' ^с=о
и ?
С CR2
снз<У Чж/
. сн
о* ^с=о
СН3-0 CR2
4.6. Схема фрагментации 89
^/к.0+ * *
~стз ^?^?^3 -снзсно ^ш2
ЧА
со
о о
147 @,6%) Mf 162 A4,8%) 118 C8,9%)
-СО
НСО* -СО
С8НтО+ С8Н50? С9Н90+ ОД*'
119 C,6%) 133 @,64%) 133 @,16%) 90 A7,8%)
Схема 4.3
ионы. Очень важно не забыть ионы, пики которых наиболее интенсивны в
спектре, однако важную информацию могут нести первичные осколочные и
перегруппировочные ионы, пики которых имеют низкую интенсивность.
Для того чтобы схема была достоверной, элементные составы всех
представленных ионов должны быть однозначно доказаны, а все направления
фрагментации подтверждены спектрами метастабильных ионов или активации
соударением. Иногда это отмечается непосредственно на схеме звездочкой над
стрелкой, связывающей два иона (схемы 4.3 и 4.4).
Изображение на схеме структур ионов в большинстве случаев не совсем
корректно. Обычно пользуются принципом наименьших структурных изменений
и стараются, насколько это возможно, сохранить структуру исходной молекулы,
последовательно отрывая от нее конкретные фрагменты. Однако более корректно
ограничиться брутто-формулой. Иногда авторы в своем исследовании пытаются
установить именно структуры ионов, которые, кстати, могут существенно
отличаться от структуры исходной молекулы. В этом случае приведенные структуры
обсуждаются в тексте особо, приводятся аргументы и, возможно, результаты
дополнительных экспериментов, на основании которых сделан выбор в пользу
приводимой структуры.
Если речь идет о схеме фрагментации индивидуального соединения, удобно
прямо на схеме рядом со значением m/z привести интенсивность пика иона
в процентах от максимального пика в спектре или от полного ионного тока
(схема 4.3). Однако часто схема фрагментации отражает характерные
направления распада для группы родственных соединений. В этом случае оптимальным
вариантом является буквенная маркировка иона на схеме непосредственно под
брутто-формулой или структурой иона, а количественные результаты
(относительные интенсивности характеристических фрагментных ионов) приводятся в
таблице, которая сопровождает схему (схема 4.4, табл. 4.6). Для обозначения
90 Глава 4. Практические основы интерпретации масс-спектров
w сн2 * х=/ чсн2 *
М А С
r^Q.^
в
CH2=N=CH2
D
Схема 4.4
-NCH2
RC7h?
?
Таблица 4.6
Относительные интенсивности характеристических фрагмент-
ных ионов в масс-спектрах ЭУ N-арилсульфонилазетидин-З-онов
R
?
СН3
ОСН3
С1
?02
м
-
-
0,3
-
-
Интенсивности пиков ионов, %
(относительно полного ионного тока)
А
0,5
0,4
0,6
0,5
0,1
В
0,3
0,2
0,9
0,4
0,1
С
0,1
0,1
0,8
-
0,1
D
75,0
77,5
73,7
77,7
65,7
?
1,1
0,7
0,7
0,2 1
0,5
фрагментных ионов используют прописные (А, В, С...) или строчные (а, Ь,
с...) латинские буквы. Можно встретить также обозначения Fb F2, F3..., а в
русскоязычной литературе Фь Фг> Фз · · · (от слова «фрагмент, fragment»).
В качестве обобщения материала, изложенного в данной главе, можно
привести последовательность действий для интерпретации масс-спектра,
предложенную Мак-Лафферти [35].
1) Изучить всю имеющуюся информацию о веществе (спектральную,
химическую, историю появления данного образца). Отметить все параметры съемки
масс-спектра. Проверить значения m/z.
2) Используя изотопные пики, вывести элементный состав всех фрагментов, для
которых это возможно; подсчитать их степень ненасыщенности.
3) Провести тест на молекулярный ион (максимальное значение m/zy нечетно-
электронность, выбросы нейтральных частиц).
4) Отметить все важнейшие нечетноэлектронные ионы, обусловленные пере-
группировочными процессами.
4.6. Схема фрагментации 91
5) Изучить общий вид спектра (стабильность молекулярного иона, лабильность
связей и т. д.).
6) Приписать возможные структуры сериям ионов с низкой массой, важнейшим
первичным нейтральным фрагментам, а также вторичным (по спектрам
метастабильных ионов и активации соударением), важнейшим
характеристическим ионам.
7) Постулировать структуру молекулы (тест по реальному спектру
идентифицированного вещества, по спектрам близким структур, по спектру,
предсказанному теоретически).
Глава 5
АЛЬТЕРНАТИВНЫЕ МЕТОДЫ
ИОНИЗАЦИИ ОБРАЗЦА
Наряду с электронным ударом (разд. 2.1) для проведения масс-спектрометри-
ческих экспериментов применяются десятки других методов ионизации. Часть
из них используется очень активно, часть — только в единичных экспериментах.
Популярность метода может достигать максимума в какой-то момент времени,
а затем с появлением новых, более эффективных, сходить на нет. Иногда, для
характеристики вещества полезно использовать несколько методов ионизации.
5.1. Ионизация фотонами
(Photoionization)
Устранить разброс ионизирующих частиц по энергиям позволяет метод
фотоионизации. Получить монохроматический пучок фотонов значительно проще,
чем пучок электронов. Энергия фотонов обратно пропорциональна длине их
волны. Энергии 1 эВ соответствует длина волны 12395 А. В коммерческих
приборах, первый из которых был построен в 50-х годах [ПО], используются
монохроматические пучки фотонов с разбросом энергий 0,01-0,02 эВ. Инертные
газы являются основными источниками фотонов для масс-спектральных
исследований. Газоразрядная трубка располагается вне источника ионов, куда фотоны
проникают через окошко из фторида лития. Энергии фотонов, излучаемых
инертными газами в условиях тлеющего разряда, лежат в диапазоне 10-40 эВ,
что позволяет ионизировать любые органические соединения. Например,
Тип Не I: Не^Не* -+Не + /и/ ? = 21,22эВ, ? = 584,3 А
ТипАг1: Ar-+Ar*-^Ar + Az/ ?=11,62эВ, ? =1066,7 А
Другим достоинством ионизации фотонами является полная передача энергии
при взаимодействии излучения с молекулой вещества. Метод чрезвычайно
полезен для установления энергетических характеристик (потенциалы ионизации
и появления) молекул, радикалов, ионов. Самостоятельный
физико-химический метод — ультрафиолетовая фотоэлектронная спектроскопия [61] —основан
на ионизации вещества фотонами.
В настоящее время доля работ с использованием классической
фотоионизации (за исключением процессов с применением лазера) невелика. Это связано с
экспериментальными сложностями, незначительной фрагментацией молекуляр-
5.2. Химическая ионизация 93
ных ионов, зависимостью фрагментации от энергии фотонов, а также
необходимостью переводить анализируемый образец в газовую фазу, как и в случае ЭУ.
5.2. Химическая ионизация, ХИ
(Chemical Ionization, CI)
В 1966 г. Мансон и Филд [111] описали новую технику получения
масс-спектра — химическую ионизацию (ХИ). Это «мягкий» метод ионизации. Избыточная
энергия молекулярных ионов в этом случае не превышает 5 эВ. Как следствие,
фрагментация оказывается весьма незначительной. Иногда спектр представляет
собой только пик молекулярного иона. Таким образом, основное достоинство
метода заключается в его способности предоставлять информацию о молекулярной
массе анализируемых соединений в тех случаях, когда ЭУ не в состоянии этого
сделать. Наиболее полным изданием, посвященным ХИ, является монография
А. Гаррисона [112].
Процесс ионизации осуществляется в основном в результате ионно-
молекулярных реакций. Главным отличием ХИ от ЭУ является существенно
более высокое давление в источнике ионов (до 1 мм рт. ст.). Это давление
создается за счет газа-реагента. Таким газом может служить практически
любое летучее вещество: гелий, вода, метан, аммиак, сероуглерод, бензол,
этилендиамин, изобутан и т. д. Желательно, но не строго обязательно, чтобы это
вещество образовывало один или несколько B-3) высокореакционноспособных
ионов при взаимодействии с электронами или в результате ионно-молекулярных
реакций. Само анализируемое соединение вносится, как и в случае ЭУ, в
микроколичествах с парциальным давлением на уровне Ю-5 мм рт. ст. Необходимые
для ионизации образца ионы газа-реагента образуются при взаимодействии
его молекул с разогнанными электронами с энергиями 200-500 эВ, аналогично
тому, как это происходит под ЭУ. Однако если в условиях ЭУ образующиеся
ионы не претерпевают столкновений с молекулами образца в связи с высоким
вакуумом в источнике, в случае ХИ такие столкновения неизбежны. В результате
в источнике создается плазма с преобладанием каких-либо ионов, которые
называются ионами-реагентами. Например, в случае метана в ионном источнике
протекают следующие основные реакции:
СН4 —> СНл , СНл , СН2
сн|' + са, -* сн^" + сн;
сн++сн4^с2н+ + н2
сн+· + сн4 -*· с2н+ + н2+н·
С2Н+ + СН4 -> С3Н+ + Н2 и т. д.
94 Глава 5. Альтернативные методы ионизации образца
В образовавшейся ионной плазме максимальную концентрацию имеют ме-
тонийкатион СН^" D4%) и этилкатион С2Н5" C0%). Именно эти частицы и
являются основными ионами-реагентами химической ионизации метаном.
Кстати, образование иона CHf в масс-спектрометре было впервые зафиксировано
В. Л. Тальрозе еще в 1952 г. [113]. Для изобутана ионом-реагентом является
mpem-бутилкатион, образующийся при отщеплении атома водорода от
молекулярного иона. Среди других газов-реагентов можно выделить часто
используемые воду и аммиак. В первом случае ионом-реагентом является НзО+, а во
втором—NH^". Достаточно часто в качестве газа-реагента используется водород,
причем образование иона-реагента Н3" было впервые описано Демпстером в
1916 г. [114].
В достаточно представительном обзоре Дж. Бродбельт [115] описаны
современные перспективные газы-реагенты. В частности, хорошие результаты
получены при использовании диметилового эфира [116]. Основным ионом-
реагентом является катион СНзОСН^, при взаимодействии которого с молекулой
анализируемого вещества идет образование трех продуктов: [М+СНзОСН2]+,
[М+СН3ОСН2-СН3ОН]+ и [М + СНзОСН2-СН20]+. Интенсивности пиков
этих ионов зависят от структуры соединений, включая тонкие стереохимические
различия. Химическая ионизация диметиловым эфиром позволяет различать
дизамещенные изомерные бензолы [117], определять длину цепочки полиэти-
ленгликолей [118].
Триметилборат в качестве газа-реагента приводит к возникновению реакци-
онноспособного иона-реагента (СН30JВ+ [119], с помощью которого удается
различать изомерные диолы и полиолы, получать структурную информацию об
амидах [120].
Полезный ион-реагент СН3ОСН2СН2ОСН2" образуется при использовании в
качестве газа-реагента 2-метоксиэтанола. Хорошие результаты получены при его
использовании для анализа нуклеозидных антибиотиков [121]. Ионом-реагентом
при использовании ацетофенона или его замещенных аналогов служит ароил-
катион. Его можно с успехом использовать для анализа гидроксилсодержащих
соединений [122].
Реакции между ионами-реагентами и молекулами образца можно разделить
на четыре типа.
1. Протонирование
М + ВН+-+МН+ + В
Способность или неспособность иона реагента ВН+ протонировать молекулу
образца легко установить при сравнении сродства к протону (СП) соединений
В и М. Если СП(М) > СП(В), идет реакция протонирования М. Чем больше
разность СП(М) и СП(В), тем большей избыточной энергией будет обладать
протонированный молекулярный ион МН+ (уравнение 5.1). Величины СП наи-
5.2. Химическая ионизация 95
более часто используемых газов-реагентов лежат в диапазоне от 423 кДж/моль
для водорода до 936 кДж/моль для этилендиамина.
? = СП(М) - СП(В) E.1)
Понятно, что при использовании водорода уровень фрагментации будет
максимальным, а при использовании этилендиамина—минимальным [123, 124].
Следует подчеркнуть, что в результате реакций этого типа образуются четно-
электронные ионы.
2. Перезарядка
М + Х+" ->М+#+Х
Для газов-реагентов, не содержащих доступных атомов водорода, важнейшей
альтернативой переносу протона становится перенос электрона, т. е. процесс
перезарядки. Избыточная внутренняя энергия образующегося молекулярного
иона рассчитывается по уравнению 5.2:
? = ЭР(Х+·) - ЭИ(М) E.2)
где ЭР — энергия рекомбинации, а ЭИ — энергия ионизации.
Вновь, как и в случае протонирования, чем меньше разность между двумя
величинами, тем меньшей избыточной энергией будет обладать молекулярный
ион. Учитывая, что энергии ионизации большинства органических молекул
лежат в диапазоне 6-12 эВ, а энергии рекомбинации типичных газов-реагентов — в
диапазоне от 9,2 эВ (бензол) до 24,6 эВ (гелий), можно подбирать газ-реагент
таким образом, чтобы образующийся молекулярный ион обладал необходимой для
каждого конкретного случая энергией. Таким образом, перезарядка позволяет
получать нечетноэлектронные молекулярные ионы, аналогичные образующимся
при ЭУ, но с заданной внутренней энергией [125-127].
3. Электрофильное присоединение
М + Х+-+МХ+
Наиболее наглядный пример этой реакции — образование интенсивных пиков
ионов [M+NKU]* широким кругом органических соединений при
использовании в качестве газа-реагента аммиака. Если молекула имеет достаточно высокую
величину сродства к протону, она протонируется ионом аммония, если же она
является слабым основанием с низким СП, в результате присоединения иона
аммония возникает стабильный комплекс, причем электроны для его
образования предоставляются гетероатомами или центрами ненасыщенности в молекуле.
Аналогичные аддукты образуются и при использовании других газов-реагентов
[124, 127, 128]: для ионизации метаном характерны ионы [М+С2Нз]+ и
[М+СзН5]+, для ионизации монооксидом азота —ионы [?+??]+.
96 Глава 5. Альтернативные методы ионизации образца
4. Отрыв аниона
АВ + Х+ ^АХ + В+
Этот процесс характерен для взаимодействия молекул, обладающих низкой
величиной СП, с протонирующими реагентами или для реагентов перезарядки
с образцами, для которых перенос заряда является эндотермическим процессом.
Примером является образование ионов [М —Н]+ при ионизации метаном [129]
алифатических углеводородов (разд. 8.1).
Большинство ионов реагентов способно участвовать более чем в одной из
указанных реакций. Выше уже упоминался ион аммония. Аналогично ион NO4"
может вести себя и как реагент перезарядки [127], и как реагент отрыва аниона
[130], и как электрофильная частица [127].
Иногда желаемый эффект достигается при использовании смеси газов-
реагентов. Так, применение влажного аргона приводит к образованию ионов
реагентов Аг+* и НзО+. Первый вызывает перезарядку с последующей интенсивной
фрагментацией, а второй действует как мягкий протонирующий агент, генерируя
стабильные ионы МН+ [131].
Химическая ионизация обычно используется для получения интенсивных
пиков молекулярных ионов. Для получения структурной информации в условиях
химической ионизации очень удобно использовать тандемную масс-спектромет-
рию с активацией соударением (гл. 7). Недостатком метода является, как и в
случае электронного удара, необходимость перевода образца в газовую фазу, что
не позволяет анализировать малолетучие и термолабильные соединения.
Еще одним направлением использования химической ионизации является
изучение H/D-обмена [132]. Таким образом можно сосчитать число активных
атомов водорода в молекулах анализируемых образцов. В качестве
газов-реагентов используются CH3OD, D2O, ND3. Помимо подсчета активных атомов
водорода метод иногда позволяет различать изомеры, а также генерировать в
газовой фазе изотопные аналоги, меченные по определенным положениям.
Очень эффективными ионами-реагентами оказались катионы металлов.
Впервые об использовании для масс-спектрометрического анализа катионов
щелочных металлов было заявлено в 1976 г. [133]. Можно упомянуть использование
ионов железа Fe+ для установления положения кратных связей в алкенах,
алкинах и кислотах [134], а также катионов меди Си+ для анализа карбонил-
содержащих соединений [135].
5.3· Химическая ионизация отрицательных ионов
(Negative Ion Chemical Ionization, NICI)
В стандартных условиях химической ионизации в ионизационной камере
присутствуют низкоэнергетичные тепловые электроны, а из молекулы
газа-реагента помимо положительных ионов образуются отрицательные ионы. Моле-
5.3. Химическая ионизация отрицательных ионов 97
кулы образца могут захватывать электроны с образованием анион-радикалов
(резонансный захват электронов, разд. 2.1) или вступать во взаимодействие с
ионами-реагентами. В последнем случае протекают ионно-молекулярные
реакции нескольких типов.
1. Депротонирование
А~+ВН^АН + В-
Этот процесс, как и в случае положительных ионов, контролируется
величинами сродства к протону. Если величина СП иона реагента А~ выше, чем иона
В~, реакция оказывается экзотермичной, и процесс идет. Величины СП наиболее
часто используемых ионов-реагентов лежат в диапазоне от 1689 кДж/моль для
NH^~ до 1395 кДж/моль для С1~. Понятно, что сильное основание NH^~ будет
депротонировать практически любое органическое соединение [136], тогда как
мягкий ион С1~ реагирует с весьма ограниченным кругом субстратов [137].
2. Перезарядка
В + А—->В—+А
Если величина сродства к электрону анализируемой молекулы выше чем
молекулы газа-реагента, может идти процесс перезарядки. Например, молекула
кислорода характеризуется низкой величиной сродства к электрону. В результате,
реагируя с органическими субстратами, ион О^' приводит к образованию
в результате перезарядки молекулярных анион-радикалов. Перезарядка может
вести и к потере характеристических ионов. В частности, если в ионном
источнике в результате какого-либо процесса образуются атомы фтора, обладающие
очень высоким сродством к электрону, они могут, реагируя с анион-радикалами
образца, превращать последние в нейтральные частицы [138].
3. Нуклеофильное присоединение
В + А--+АВ"
Анионы-реагенты с низкой величиной СП могут реагировать с молекулами
субстрата с образованием стабильных аддуктов. Одним из наиболее активных
в этом отношении ионов-реагентов является С1~. В результате нуклеофильного
присоединения он образует комплексы с широким кругом органических молекул
[139].
4. Нуклеофильное замещение
АВ + С-^ВС + А"
Любое основание является одновременно нуклеофилом, т. е. наряду с атакой
на атом водорода возможна атака на атом углерода. Наиболее активно вступают
4—1113
98 Глава 5. Альтернативные методы ионизации образца
в реакцию нуклефильного замещения ионы ОН" и 0^'[34]. Последний является
столь активным нуклеофилом, что может вытеснять атом водорода из
ароматических соединений.
Метод химической ионизации отрицательных ионов активно используется в
настоящее время и в фундаментальной масс-спектрометрии, и в прикладных
исследованиях. В обзорах Дж. Бови [136, 140] рассмотрена фрагментация де-
протонированных молекулярных ионов различных классов органических
соединений. В обзоре [141] представлены результаты использования данного метода в
экологических исследованиях.
5.4. Пульсирующая химическая ионизация
(Pulsed Positive, Negative Ion Chemical Ionization
Mass Spectrometry)
Этот метод, предложенный Хантом и др. [142], позволяет одновременно
получать масс-спектры химической ионизации положительных и отрицательных
ионов. Два вида спектров удачно дополняют друг друга, что важно и для целей
структурного определения, и для количественных расчетов, поскольку пределы
обнаружения для разных соединений зависят от заряда ионов. Метод успешно
используется на квадрупольных приборах (разд. 6.5), поскольку анализатор этого
типа легко пропускает и катионы, и анионы. Для магнитных секторных приборов
такой вариант неприемлем.
В данном режиме полярность вытягивающего потенциала переключается
со скоростью примерно 10000 раз в секунду, поэтому ионы сгруппированы в
«пакеты» с чередованием положительных и отрицательных. После прохождения
анализатора положительные ионы направляются на один детектор, а
отрицательные—на другой. Происходит регистрация сразу двух типов спектров. Важным
моментом является природа газа-реагента, который должен «работать» в обоих
режимах. Например, хорошим вариантом является смесь метана с монооксидом
азота, в которой роль основных ионов реагентов выполняют CHf и ОН~.
5.5. Десорбционная (прямая)
химическая ионизация, ДХИ
(Direct (Desorption) Chemical Ionization, DCI)
Скорости испарения и деструкции органических веществ имеют разные
температурные зависимости. При низких температурах предпочтительна деструкция, а
при высоких — испарение (рис. 5.1). Испарение труднолетучего вещества можно
осуществить при его быстром нагреве до температуры, при которой испарение
наиболее эффективно. Метод, использующий этот эффект в источнике химиче-
5.5. Десорбционная (прямая) химическая ионизация 99
lg*
Деструкция
Рис. 5.1. Зависимость скоростей деструкции и испарения от температуры образца
ской ионизации, был впервые описан в работе Балдвина и Мак-Лафферти [143].
Следует отметить, что аналогичный подход был использован и для получения
спектров электронного удара [144]. Этот метод называется электронный удар
в пучке. Название обусловлено тем, что образец на штоке прямого ввода
помещается как можно ближе к пучку электронов, что уменьшает вероятность
термодеструкции на внутренней поверхности источника.
В настоящее время для прямой химической ионизации используют
обогреваемые электрические эмиттеры. Очень хорошо зарекомендовали себя эмиттеры
полевой ионизации (разд. 5.7), но поскольку они сложны в изготовлении, обычно
используется вольфрамовая, рениевая или платиновая проволока диаметром
50-200 мкм. Для увеличения выхода молекулярных ионов проволока
покрывается нанесенным с помощью электрофореза полиимидным слоем. После снятия
спектра для повторного использования эмиттера желательна его прожарка. Как
правило, эмиттеры с полиимидным слоем годятся не более чем для 10 образцов.
Метод используется для анализа термолабильных и нелетучих соединений:
Сахаров, пептидов, солей, кислот. Спектр представляет собой интенсивный пик
молекулярного иона и ряд пиков фрагментных ионов, часть из которых может
быть обусловлена не фрагментацией молекулярного иона, а термодеструкцией
исходной молекулы. Зачастую ДХИ используют для увеличения выхода
молекулярных ионов. В этом случае полезно несколько повысить давление в
ионизационной камере за счет газа-реагента. Некоторое снижение абсолютной
чувствительности компенсируется определенным подавлением фрагментации.
Очень важную роль играет газ-реагент. Так фрагментация аскорбиновой кислоты
при использовании метана интенсивнее, чем при использовании аммиака (ионы
[M+NH4]"). Еще лучшие результаты получаются при применении анионов С1~.
В этом случае спектр представлен исключительно пиками иона [М+С1]-. Таким
образом, для снижения фрагментации предпочтительно использовать «мягкие»
ионы-реагенты ( NH^", Cl~, ОН").
100 Глава 5. Альтернативные методы ионизации образца
Для каждого вещества существует оптимальная температура съемки спектра.
Анализируя неизвестное соединение, его нагревают со скоростью 10 град./мин,
и находят оптимум. При повторной съемке резко нагревают вещество до этой
температуры. Существенным недостатком метода является быстрое ухудшение
качества спектра во времени. Это связано с процессами термодеструкции,
которые все-таки идут с заметной скоростью. Некоторого улучшения можно
добиться при быстрой развертке спектра [145].
5.6. Полевая ионизация (Field Ionization, FI)
Еще одним методом получения интенсивного пика молекулярного иона
является полевая ионизация. Автором данного подхода к анализу органических
молекул является Беккей [146]. Ключевую роль играет проволочный эмиттер,
поверхность которого покрыта микроиглами. В простейшем случае эмиттером
может служить острие иглы или лезвие бритвы. На эмиттер подается высокое
напряжение (8-10 кВ). Благодаря малому радиусу кривизны на концах игл
создается высокая напряженность электрического поля (до 1 В/А). Молекула
образца в газовой фазе, оказываясь в непосредственной близости от
эмиттера, подвергается воздействию этого поля. Основным механизмом ионизации
является туннелирование электрона из молекулы органического соединения на
эмиттер. Энергия, переносимая в результате такого процесса, составляет доли
электронвольта, т. е. избыточная энергия молекулярного иона М+" значительно
ниже, чем даже в условиях ХИ. Кроме того, другие электроны ионизирующейся
молекулы не возбуждаются, и М+# оказывается в основном (невозбужденном)
электронном состоянии. Высокий положительный потенциал эмиттера резко
выталкивает образовавшийся катион из источника. Среднее время присутствия
М+Ф в источнике полевой ионизации составляет 10-12 с, тогда как в случае ЭУ—
10~6 с. В результате даже возможные низкоэнергетические процессы
фрагментации не успевают реализоваться в полной мере, и спектр зачастую представляет
собой единственный пик, принадлежащий молекулярному иону [147].
Полевая ионизация, являясь наиболее мягким методом, в то же время не
устраняет недостатка ХИ и ЭУ, связанного с необходимостью перевода образца
в газовую фазу. Для его преодоления была предложена модификация метода,
получившая широкое признание и названная полевой десорбцией.
5.7. Полевая десорбция (Field Desorption, FD)
Аппаратурное оформление, а также механизм полевой десорбции, не отличаются
от описанных выше для полевой ионизации. Единственным, но принципиальным
отличием является нанесение раствора образца непосредственно на эмиттер.
5.8. Плазменная десорбционная масс-спектрометрия 101
После испарения растворителя эмиттер помещается в источник ионов. При
подаче на него высокого напряжения, в результате туннелирования электрона из
молекулы образца, ион образуется непосредственно в конденсированной фазе.
Поскольку энергия десорбции иона с поверхности одноименно заряженного
эмиттера значительно ниже, чем энергия десорбции исходной нейтральной
молекулы, ион переходит в газовую фазу. В отличие от полевой ионизации здесь
образуются в основном четноэлектронные протонированные молекулярные ионы
МН+. Добавка к анализируемому образцу кислот или неорганических солей
облегчает процесс ионизации с образованием ионов МН+ и [М+катион]+. В
этом случае, поскольку ионы присутствуют уже в конденсированной фазе, для
их извлечения требуется меньший потенциал (около 10~2 В/А). Метод хорошо
зарекомендовал себя для анализа Сахаров, пептидов, нуклеотидов, солей, кислот.
Отличные результаты получены также при анализе синтетических неполярных
полимеров [148, 149].
Определенные проблемы полевой десорбции (как и полевой ионизации)
связаны с изготовлением эмиттеров и нанесением на них образца. Тонкие
углеродные иглы обычно наносят на эмиттер пиролизом бензонитрила в
специальной камере. Длинные иглы требуют тщательного изготовления, а короткие
затрудняют нанесение образца. Спектры часто получаются неинтенсивными.
Для преодоления этого недостатка используется многократное сканирование
внутри узкого интервала масс с последующим интегрированием. В результате
устраняется нестабильность спектра и улучшается отношение сигнал/шум.
5.8. Плазменная десорбционная масс-спектрометрия
(Plasma Desorption Mass Spectrometry, PDMS)
Первое сообщение об использовании продуктов распада 252Cf для ионизации
органических веществ опубликовано в 1976 г. Макфарлейном [150].
Многочисленные продукты деления изотопа 252Cf обладают энергиями в сотни
мегаэлектронвольт. Когда они проходят через металлическую фольгу, на которую нанесен
анализируемый образец, создается быстрый локальный разогрев (до 10000 К).
В результате наблюдается процесс, аналогичный десорбционной химической
ионизации (разд. 5.5), при котором скорость десорбции (испарения)
органических молекул оказывается выше скорости их разложения. Метод характеризуется
очень высокой чувствительностью и позволяет получать спектры полярных
соединений [151]. Однако в связи с низкими ионными токами он может
использоваться только с времяпролетным анализатором в импульсном режиме.
Еще одним недостатком является необходимость работы с радиоактивными
продуктами распада. Эти ограничения не позволили плазменной десорбции
успешно конкурировать с другими методами анализа подобных соединений.
102 Глава 5. Альтернативные методы ионизации образца
5.9. Лазерная десорбционная масс-спектрометрия
(Laser Desorption Mass Spectrometry, LDMS)
Еще одним методом анализа нелетучих органических соединений является
лазерная десорбция [152]. Метод заключается в переводе молекул образца в
газовую фазу при его облучении лазером. Механизм ионизации во многом
сродни десорбционной химической ионизации и плазменной десорбции и связан
с испарением и ионизацией вещества в результате быстрого локального нагрева,
когда скорость десорбции превосходит скорость деструкции. Основным
недостатком, как и в случае плазменной десорбции, является необходимость
использования времяпролетных приборов, поскольку масс-спектрометры других типов
плохо сочетаются с импульсным лазером. Тем не менее следует отметить, что
модификация этого метода (см. МЛДИ, разд. 5.14) является на сегодняшний день
важнейшим методом анализа высокомолекулярных органических соединений.
Кроме того, метод успешно применяется в неорганической масс-спектрометрии.
5.10. Бомбардировка быстрыми атомами, ББА
(Fast Atom Bombardment, FAB);
вторичноионная масс-спектрометрия, ВИМС
(Secondary Ion Mass Spectrometry, SIMS)
Метод ионизации, позволивший в начале 80-х годов XX века вывести масс-
спектрометрию на новый качественный уровень, также использует принцип
бомбардировки образца энергетическими частицами. Принципиальная схема
ионизации бомбардировкой быстрыми атомами [153] представлена на рис. 5.2.
Art
Шток
Образец
на наконечнике
штока Выделение и
фокусировка
Ионы
Рис. 5.2. Принципиальная схема ионизации бомбардировкой быстрыми атомами
5.10. Бомбардировка быстрыми атомами 103
Пучок ускоренных атомов (обычно инертные газы) направляется на раствор
вещества, нанесенный на металлическую подложку, расположенную на конце
штока прямого ввода. Взаимодействуя с веществом, атомы создают интенсивный
локальный разогрев, в результате чего молекулы поверхностных слоев
отрываются в виде плотного газа, содержащего положительные и отрицательные
ионы, а также нейтральные частицы, которые также могут ионизироваться
непосредственно над поверхностью образца.
Для получения пучка атомов используется «атомная пушка». Сначала
получают ионы инертного газа, которые далее ускоряются потенциалом 6-8 кВ.
Разогнанные ионы взаимодействуют с атомами газа путем переноса заряда. Этот
процесс протекает без заметного изменения кинетической энергии. Непрореаги-
ровавшие ионы отклоняются специальной линзой, а образовавшиеся в результате
перезарядки ускоренные атомы с энергией 6-8 кэВ направляются под углом
60-70° на поверхность образца.
Реакция рекомбинации используется в альтернативном варианте «атомной
пушки». В этом случае пучок ионов ускоряется по направлению к образцу и
пересекает пучок электронов, движущихся в перпендикулярном направлении.
Захватывая электроны, ионы превращаются в атомы. При этом ни энергия, ни
направление движения разогнанных частиц не меняются. На выход ионов
анализируемого образца существенно влияет масса разогнанных частиц первичного
пучка. Например, пучок атомов Хе эффективнее, чем Аг и тем более Ne.
Если на шток нанесено сухое вещество, то через короткий промежуток
времени наблюдается резкое падение интенсивности пучка генерируемых ионов, так
как разрушается поверхностный слой кристаллической решетки вещества.
Поэтому используют растворенный образец. Диффузия в жидкой матрице протекает
достаточно быстро для того, чтобы обеспечить постоянную концентрацию
молекул образца на поверхности. В этом случае пучки ионов образца достаточной
интенсивности могут генерироваться в течение длительного времени B0 и более
минут). В качестве растворителя (матрицы) может использоваться широкий
круг нелетучих соединений. Наиболее распространен глицерин. Температурный
диапазон работы с глицерином составляет от —20 до +40 °С при оптимальной
температуре 25 °С. Вторым соединением по частоте использования в качестве
матрицы является ;и-нитробензиловый спирт.
Важно, чтобы образец был именно растворен в матрице. Если равномерного
растворения нет, качество спектров значительно ухудшается. Помочь может
добавление солюбилизаторов. Учитывая важность растворимости вещества,
неудивительно, что количественный анализ смесей с помощью ББА приводит
к непредсказуемым результатам в связи с большими различиями в
чувствительности к разным компонентам.
Метод ББА — модификация вторичноионной масс-спек грометрии
(Secondary Ion Mass Spectrometry, SIMS), которая давно используется в
неорганической масс-спектрометрии [154] и заключается в бомбардировке образца
104 Глава 5. Альтернативные методы ионизации образца
разогнанными ионами, например Cs+. Однако попытки анализировать
органические соединения [155, 156], хотя и позволили получить спектры ряда
труднолетучих веществ, не привели к серьезным успехам. Причина заключалась
в том, что исследователи анализировали сухое вещество, а быстрое
деформирование поверхности его кристаллической решетки приводило к получению
некачественных масс-спектров. Когда после успешного применения ББА
провели эксперименты по бомбардировке растворенного образца ионами, спектры
оказались не хуже, чем при бомбардировке атомами. В частности, ионы Cs" с
энергией 6 кэВ и атомы Хе с энергией 8 кэВ позволяют получать идентичные
спектры. Кстати, Беннингховена считают основоположником метода именно
благодаря работе [155], опубликованной в 1976 г. Тем не менее Г. Д. Танцырев
опубликовал свои результаты исследований с применением SIMS в 1971 г. [157],
а с использованием ББА —в 1973 г. [158].
Хотя важнейшим достоинством ББА является возможность получать масс-
спектры соединений с массами в несколько тысяч дальтон, это не мягкий метод
ионизации типа полевой десорбции. Бомбардировка образца быстрыми атомами
обеспечивает передачу молекулам энергии в широком диапазоне. В результате
наряду с пиками молекулярных ионов в спектре наблюдаются интенсивные пики
осколочных ионов, что позволяет в ряде случаев не прибегать к активации
соударением (гл. 7) для изучения фрагментации и структуры анализируемых
молекул.
Следует отметить принципиальное сходство спектров ББА и ХИ. Оба вида
характеризуются четноэлектронными ионами типа МН+ и [М —Н]~, но не М+"
и М-". Отчасти это является результатом ионно-молекулярныхреакций,
протекающих в плазме над поверхностью образца, отчасти перенос протона идет еще в
жидкой матрице. Использование в качестве растворителя материалов с большей
основностью приводит к образованию ионов [М+матрица]4". Качество спектров
можно повышать добавками, вызывающими образование ионов непосредственно
в жидкой фазе. Добавки кислот приводят к увеличению интенсивности пиков в
спектрах положительных ионов соединений с центрами основности, а добавки
щелочей используются для увеличения выхода отрицательных ионов в спектрах
соединений с кислотными группами. В частности, полипептиды с избытком
основных аминокислот [159] характеризуются спектрами положительных ионов
с интенсивными пиками МН+. В спектрах полипептидов с избытком кислотных
групп интенсивность этого пика мала, тогда как пики молекулярных депрото-
нированных ионов, напротив, весьма интенсивны. Иногда полезно добавлять к
образцу неорганические соли. Например, добавка NaCl к образцам олигосахари-
дов или полипептидов в глицериновой матрице [153] приводит к образованию
интенсивных пиков ионов [M+Na]+.
Бомбардировка быстрыми атомами успешно используется для анализа самых
разнообразных соединений: Сахаров, нуклеотидов, витаминов, солей,
антибиотиков, металлоорганических соединений, простагландинов, стероидов, порфиринов
5.10. Бомбардировка быстрыми атомами 105
и т. д. [34, 160, 161]. К сожалению, неудовлетворительные результаты
получаются при анализе неполярных полимеров (например, полистирола). Наиболее
впечатляющих успехов, однако, удалось достичь в анализе пептидов. Наличие
интенсивных пиков молекулярных и фрагментных ионов в сочетании с
диапазоном регистрируемых масс выше 10000 Да, а также ограниченный набор
природных аминокислот позволили достаточно легко устанавливать последовательность
аминокислотных звеньев. В спектре присутствуют пики, обусловленные отрывом
1, 2, 3, и т. д. аминокислот от молекулярного иона. Поскольку эти отрывы
идут по обоим концам исходной молекулы, можно составлять цепочку с обоих
концов (разд. 8.15). За прошедшие с момента появления метода 20 лет удалось
расшифровать последовательность аминокислот для тысяч и тысяч пептидов.
Модификацией ББА является метод бомбардировки тяжелыми
кластерами (Massive cluster impact, MCI). В этом случае в качестве энергетических
частиц используются кластеры ионов аммония с глицерином. В результате их
взаимодействия с образцом образуются многозарядные ионы, т. е. появляется
возможность получать спектры более тяжелых, чем в случае классической ББА,
биополимеров. Использование кластеров увеличивает также чувствительность
анализа [160].
5.10.1. Проточный (динамический) вариант
бомбардировки быстрыми атомами
(Continuous Flow or Dynamic Fast Atom Bombardment)
Принципиальная схема метода бомбардировки быстрыми атомами (ББА) в
проточном варианте [162] представлена на рис. 5.3.
Поток из колонки хроматографа направляется в капилляр штока ввода в
источник. Капилляр заканчивается сеткой или медной пластинкой, используемой
в качестве подложки в статическом варианте метода, в которой высверливается
Рис. 5.3. Принципиальная схема метода бомбардировки быстрыми атомами в проточном
варианте
106 Глава 5. Альтернативные методы ионизации образца
отверстие для поступления образца. Раствор подается в источник со скоростью
не более 10 мкл/мин. Температура подложки поддерживается на уровне 40-60 °С,
что позволяет избежать замораживания образца в результате активного
испарения растворителя в условиях высокого вакуума. Низкие приемлемые скорости
потока заставляют проводить его деление на выходе из колонки хроматографа,
что, безусловно, ухудшает чувствительность метода, которая, тем не менее, в
благоприятных случаях может достигать уровня пикомолей [163].
Еще одной конструктивной особенностью метода является подача в поток
глицерина в количестве 1-5%. Глицерин необходим для эффективной ионизации
(разд. 5.10), служа матрицей. Его можно добавлять непосредственно в жидкую
фазу перед хроматографированием. Однако в этом случае может наблюдаться
ухудшение разделения компонентов. Поэтому чаще врезают подвод глицерина
на выходе из колонки хроматографа [163].
5.11. Химическая ионизация
при атмосферном давлении
(Atmospheric Pressure Chemical Ionization, APCI)
Простейшая теоретическая стыковка жидкостного хроматографа с
масс-спектрометром может осуществляться, если источник ионов работает под атмосферным
давлением. Такой вариант прибора достаточно широко используется, начиная с
70-х годов XX века [164].
Принципиальная схема прибора представлена на рис. 5.4. Поток из обычной
колонки жидкостного хроматографа направляется в распылитель, где он
превращается в мелкодисперсный аэрозоль, смешиваясь с большим количеством
нагретого газа (обычно азот или воздух). Капельки аэрозоля в окружении
газового потока перемещаются в область испарения, где в газовую фазу переходит
Сепаратор
МСГатм
IP
!
t
Разрядная область
Газ-распылитель ^ насосу
Рис. 5.4. Принципиальная схема источника ионов, работающего при атмосферном давлении
5.12. Электрораспыление, электроспрей 107
большая часть молекул растворителя. Далее на пути потока следует область
ионизации. Так как в источнике поддерживается атмосферное давление,
ионизация осуществляется либо за счет коронного разряда, либо при взаимодействии
молекул с электронами, эмиттируемыми /^-излучателями (например, фольга из
63Ni). Поскольку количество молекул растворителя существенно превышает
количество молекул анализируемого вещества, создаются условия химической
ионизации (разд. 5.2). На выходе из источника ионов расположен ряд
последовательных сепараторов с узкими входными отверстиями. Здесь происходит
откачка легких молекул для снижения избыточного давления. В результате в
анализатор, работающий в условиях глубокого вакуума, поступают в основном
ионы анализируемого вещества. Метод хорошо зарекомендовал себя для анализа
небольших, как полярных, так и неполярных, молекул (< 1200 Да). Он пригоден
для пестицидов, стероидов, лекарственных препаратов и их метаболитов.
5.12. Электрораспыление, электроспрей
(Electrospray Ionization, ESI)
Электроспрей произвел революцию в масс-спектрометрии, выведя ее в 90-х
годах прошлого века на новый уровень. Метод позволил говорить не только об
органической масс-спектрометрии, но и о биоорганической, и даже
биологической масс-спектрометрии.
Впервые электрораспыление для ввода жидкой пробы в масс-спектрометр
было предложено Джоном Фенном и др. [165] на основе более ранних работ
Доуля [166]. За это изобретение в 2002 г. Фенну присуждена Нобелевская
премия.
2
?
Капилляр
\
Противоэлектрод Сепараторы
/
&/
mm
\
1И
N2 К насосу
о,
g
Рис. 5.5. Принципиальная схема метода электрораспыления
108 Глава 5. Альтернативные методы ионизации образца
Поток из жидкостного хроматографа (рис. 5.5) направляется в иглу диаметром
ОД мм, на которую подается высокое напряжение порядка 6 кВ. На выходе из
иглы в источнике ионов образуется аэрозоль из заряженных капель с высоким
поверхностным зарядом. Эти капли движутся к противоэлектроду, имеющему
потенциал Земли. В этом же направлении уменьшается и давление, хотя в целом
в этой части ионного источника (до противоэлектрода) давление поддерживается
на уровне атмосферного. По мере движения к входному отверстию первого
сепаратора капли уменьшаются в размере за счет испарения растворителя. Достигая
критического размера, при котором силы поверхностного натяжения далее не
могут противостоять силам кулоновского отталкивания (предел Релея), капля
«взрывается» с образованием более мелких капелек. Этот процесс повторяется.
В итоге возникают микрокапли, содержащие всего одну заряженную частицу,
которая может оказаться в газовой фазе после испарения остаточных молекул
растворителя. Альтернативный механизм предусматривает выброс заряженной
молекулы с поверхности одноименно заряженной капли. В обоих случаях в
газовой фазе оказываются несольватированные молекулы анализируемого вещества,
которые проходят через сепаратор и оказываются в анализаторе. Механизмы
ионизации продолжают изучаться и в настоящее время [5, 167-173].
Поток газа (как правило, азота), подающийся между противоэлектродом и
первым сепаратором, а также коаксиально капилляру (газ-распылитель) приводит
к улучшению распыления потока жидкости и лучшим условиям для десоль-
ватирования ионов. Такая модификация метода часто называется ионоспреем.
Этот вариант позволяет также увеличивать скорость потока жидкости в прибор
до 200 мкл/мин, тогда как в обычном варианте скорость потока составляет
1-40 мкл/мин. Дополнительные модификации интерфейса [174] позволяют
работать с потоками из обычных хроматографических колонок (до 2 мл/мин).
Наряду с попытками увеличения потока велись работы по уменьшению
потока жидкости. В результате был создан метод, названный «наноспреем» [175].
Эта модификация электроспрея оперирует потоками со скоростями в несколько
нл/мин и позволяет повысить эффективность образования ионов анализируемого
вещества почти на два порядка.
Тем не менее чувствительность метода пока остается непревзойденной при
использовании обычных потоков (до 20 мкл/мин). Качественный масс-спектр
можно получить при вводе в прибор менее одного аттомоля (Ю-18 М) вещества.
В последних модификациях коммерческих масс-спектрометров используется
Z-форма движения пучка ионов (рис. 5.6). Такой вариант благодаря
дополнительным линзам позволил направить в анализатор лишь заряженные частицы, а не
весь поток раствора образца. Это улучшило качество спектров и значительно
снизило загрязнение приборов.
Помимо высочайшей чувствительности и возможности работы с
термолабильными и нелетучими соединениями электроспрей предоставил
исследователям возможность анализировать высокомолекулярные соединения с молеку-
5.12. Электрораспыление, электроспрей 109
ГЛЗ
Рис. 5.6. Z-форма прибора для реализации метода электрораспыления
лярными массами до миллиона дальтон и выше [176]. Зарегистрированный
рекорд составляет 110000000 Да [7]. В этой работе [7] в масс-спектрометре с
преобразованиями Фурье удалось измерить молекулярную массу ДНК
бактериофага Т4, зарегистрировав многозарядные молекулярные ионы B8 000-35 000).
Особенных успехов удалось достичь при использовании электроспрея для
установления структур полипептидов, белков, нуклеиновых кислот [160]. Дело
в том, что анализатор масс-спектрометра делит ионы не по массам, а по
отношению массы к заряду (гл. 6). В результате ион с массой 100000 Да и
зарядом 100, регистрируется как однозарядный ион с массой 1000. Условия
электрораспыления приводят к возникновению именно многозарядных ионов. В
частности, анализируя полипептиды, легко получить ионы протонированные или
депротонированные по многим основным или кислотным участкам молекулы.
Поскольку ионы, образующиеся в условиях электроспрея, обладают значительно
меньшей внутренней энергией, чем, например, в случае бомбардировки
быстрыми атомами, их фрагментация практически отсутствует, а масс-спектр зачастую
представляет собой набор пиков, обусловленных молекулярными ионами с
различными зарядами (точнее [М+лН]л+). Для инициирования фрагментации этих
110 Глава 5. Альтернативные методы ионизации образца
ионов необходимо использовать метод тандемной масс-спектрометрии (гл. 7).
Типичный спектр полипептида представлен на рис. 5.7.
Измерив массу этих многозарядных ионов, легко вычислить молекулярную
массу соединения. В современных приборах это делает компьютер, но не
представляет труда провести подобные вычисления вручную. В отличие от
традиционной масс-спектрометрии (гл. 4) в этом методе для работы со спектрами
используют усредненные массы элементов: С 12,011, ? 1,00794, N 14,00674,
О 15,994, S 32,066. Это объясняется тем, что высокомолекулярные соединения
имеют очень сложную изотопную картину в области молекулярного иона,
особенно многозарядного молекулярного иона. Разрешающей способности метода
оказывается недостаточно, и пики получаются широкими, с неразрешенной
изотопной картиной. Максимум такого пика определяется именно усредненной
массой элементов.
Пусть М — масса молекулы, // — масса протона, т\ и т2 — массы двух
соседних пиков в масс-спектре с зарядами ? + 1 и ? соответственно. Поскольку
масс-спектрометр измеряет отношение массы к заряду, индивидуальные пики
в масс-спектре (рис. 5.7) регистрируются на шкале масс со значением, которое
определяется по уравнению:
М + пН
т = E.3)
?
Следовательно, масса молекулы может быть вычислена по уравнению:
М = п(т-Н) E.4)
Неизвестную величину ? можно легко определить на основе регистрируемых
масс двух соседних пиков, обусловленных ионами, отличающимися на один
заряд. В этом случае можно исходить из двух уравнений типа 5.3:
М + пН М + (п+\)Н
т2 = и ??? = —
? n + l
Решая эту систему уравнений относительно п, получаем
п = E.5)
ГП2 — Ш\
Результат необходимо округлить до ближайшего целого числа. Таким
образом, можно рассчитать молекулярную массу по каждой паре соседних пиков и
усреднить полученное значение.
Электроспрей может эффективно использоваться для анализа соединений с
массой до ~ 150 000 Да. В случае более тяжелых молекул спектры становятся
слишком сложными для расшифровки, так как очень большое число пиков
многозарядных ионов располагается в сравнительно небольшом диапазоне (от
m/z 500 до m/z 3000).
Задача 5.1. Рассчитайте молекулярную массу полипептида, спектр которого
представлен на рис. 5.7.
112 Глава 5. Альтернативные методы ионизации образца
5.13. Ультразвуковое распыление
(Sonic Spray)
Небольшое число работ выполнено с использованием ультразвукового
распыления [55, 56]. Этот метод позволяет получить спектры нелетучих термолабильных
полярных молекул при стыковке масс-спектрометра с жидкостным
хроматографом или системой для капиллярного электрофореза. В этом случае капилляр,
соединяющий хроматографическую систему с источником масс-спектрометра,
не обогревается и на него не подается высокий электрический потенциал.
Ключевую роль играет поток азота, который подается в систему с очень высокой
скоростью коаксиально капилляру с пробой (рис. 5.8). Начиная с определенной
скорости потока, прибор регистрирует ионы. Именно скорость потока имеет
первостепенную важность. Механизм ионизации (точнее десольватирования
имеющихся в растворе ионов) пока не изучен.
Сепараторы
Рис. 5.8. Принципиальная схема метода ультразвукового распыления
Метод хорошо зарекомендовал себя для анализа лекарственных препаратов и
их метаболитов. Его чувствительность иногда на 2 порядка превосходит
чувствительность традиционно используемой для этих целей химической ионизации при
атмосферном давлении [57].
5.14. Матричная лазерная
десорбционная ионизация, МЛДИ
(Matrix-Assisted Laser Desorption/Ionization, MALDI)
Настоящий прорыв к анализу сложнейших биоорганических молекул произошел
с появлением матричной лазерной десорбционной ионизации. Впервые метод
описан во второй половине 80-х годов XX века [177-179]. В 1988 г. были
опубликованы результаты анализа белков с массами до 70 000 Да [180], а в
1990 г. —уже до 116000 Да [181]. Рекордные для масс-спектрометрии массы
однозарядных ионов в несколько миллионов дальтон [182] были зарегистрированы
именно с помощью матричной лазерной десорбционной ионизации. Одному
из первооткрывателей метода Кончи Танаке в 2002 г. присуждена Нобелевская
премия в области химии.
5.14. Матричная лазерная десорбционная ионизация 113
Метод заключается в облучении короткими лазерными импульсами образца,
представляющего собой твердый раствор анализируемого соединения в
органической матрице. Матрица выбирается таким образом, чтобы ее молекулы активно
поглощали фотоны, эмиттируемые УФ- или ИК-лазером. Над поверхностью
образца создается плотная высокотемпературная плазма, в которой наряду с
молекулами и ионами матрицы оказываются и молекулы анализируемого
соединения. Ионизация последних путем поглощения энергии фотонов или в
результате ионно-молекулярных реакций приводит к образованию положительных и
отрицательных ионов, которые вытягиваются высоким потенциалом из области
ионизации и направляются в анализатор. Метод характеризуется интенсивными
пиками молекулярных ионов разного типа и низкой фрагментацией. Наиболее
термолабильные, труднолетучие, высокомолекулярные соединения стали
доступны масс-спектрометрическому анализу. К настоящему времени методом МЛДИ
успешно анализируются полипептиды, белки, нуклеотиды, полисахариды,
синтетические полимеры, гуминовые кислоты, фуллерены, органические комплексные
соединения и т. д. (рис. 5.9). Количество переменных в МЛДИ значительно
больше, чем в других методах, поэтому анализ нового соединения является
самостоятельной задачей, причем выбор условий эксперимента может привести
как к успеху, так и полному провалу. Важнейшими параметрами метода
являются природа матрицы, количественное соотношение матрица : анализируемое
соединение, длина волны, долгота импульса и мощность лазерного излучения,
способ пробоподготовки и т. д.
Матрица. Как и в методе бомбардировки быстрыми атомами, в МЛДИ
важнейшую роль играет матрица, однако ее роль в этом случае значительно отличается.
Правильный выбор материала является ключевым моментом для успешной
регистрации масс-спектра. Количество самых разнообразных органических
соединений, использованных в качестве матрицы, очень велико. Основные
требования к материалу матрицы заключаются в его высокой способности поглощать
лазерное излучение; кристаллизоваться с включением в структуру молекул
анализируемого вещества; инертности по отношению к анализируемому веществу;
хорошей растворимости в растворителях, используемых для пробоподготовки;
низкой летучести в условиях вакуума. Наиболее широкое применение в качестве
матриц нашли коричная кислота, З-амино-4-гидроксибензойная кислота,
никотиновая кислота, се-циано-4-гидроксикоричная кислота, 2,5-дигидроксибензой-
ная кислота, 6,7-дигидроксикумарин, 2-D-гидроксифенилазо)бензойная кислота,
3-гидроксипиколиновая кислота, 2,4,6-тригидроксиацетофенон и многие другие.
Обычно раствор матрицы в подходящем растворителе (концентрация ~10 мг/мл)
готовится ежедневно, поскольку он светочувствителен и подвержен
фоторазложению. В качестве растворителей наиболее часто используются вода, этанол,
ацетон, ацетонитрил, тетрагидрофуран, хлороформ и т. д. Источники МЛДИ с
повышенным давлением позволяют использовать в качестве матрицы воду и ряд
<
?
к
*
<L>
§
а
?
?
О.
?
?—?
??
ЧО
^
?
w
?
?
??
iJU
W
?
IS!
акс
S
cd
|
о
P<
<?
i
IT!
<D
§
О
Ц
&
??
о
•-?
m
??
^r
<?
СП
s^
?
S
X
Д «
S
5,
vE>
uQ
§
?
<?
3
ю
К
Л
1
*
w
н
l=J
О
w
&
о
5.14. Матричная лазерная десорбционная ионизация 115
других соединений. В этих условиях эффективность работы ИК-лазеров выше,
чем УФ-лазеров [182].
Лазеры. В идеале лазер должен эффективно передавать образцу
контролируемое количество энергии, причем во избежание термического распада эта энергия
должна переноситься в кратчайший период времени. Два наиболее активно
используемых типа лазера в коммерческих приборах — азотный и углекислотный.
Первый является ультрафиолетовым и характеризуется длиной волны 337 нм,
энергией фотонов 3,68 эВ. Долгота его импульса составляет обычно от 0,1 до
нескольких наносекунд. Второй является инфракрасным и характеризуется
длиной волны 10,6 мкм, энергией фотонов 0,12 эВ. Долгота его импульса составляет
от 100 не до 1 мкс. Помимо этих лазеров используются неодимовые, эрбиевые, а
также эксимерные (XeCl, ArF, KrF).
Пробоподготовка. Раствор анализируемого вещества в растворителе, который
хорошо смешивается с растворителем матрицы, в концентрации 5-10 мг/мл
добавляется к рабочему раствору матрицы. Молярное соотношение
матрица/анализируемое вещество должно составлять от 100:1 до 50000:1 и
оптимизироваться для каждого нового образца. После получения гомогенного
раствора 0,5-2 мкл наносится на специальную плашку из нержавеющей стали
и высушивается сначала при атмосферном давлении на воздухе, а затем при
нагревании струей теплого воздуха в слабом вакууме. Природа
взаимодействия матрицы с образцом пока недостаточно изучена. Во время высушивания
происходит кристаллизация матрицы с включением молекул анализируемого
вещества в кристаллическую решетку или же молекулы образца размещаются по
поверхности быстро образующихся кристаллов матрицы. Образование и размер
кристаллов в значительной мере зависят от природы матрицы и растворителя
и в меньшей степени от природы образца, поскольку количество последнего
значительно меньше.
Помимо соотношения матрица/анализируемое вещество на качество спектра
сильно влияет наличие примесей и добавок. При работе с биологическими
соединениями часто используются буферные растворы, соли, поверхностно-
активные вещества. Эти соединения оказываются в растворе анализируемого
образца и могут повлиять на качество спектров. Существенным преимуществом
МЛДИ по сравнению с электрораспылением является толерантность метода по
отношению к примесям и добавкам, хотя в некоторых случаях эти побочные
соединения могут приводить к уменьшению интенсивности основного сигнала
по разным причинам. Качество спектров, как правило, не ухудшается, а иногда
чувствительность может возрасти за счет образования ионов [М+катион]+.
Поэтому, если количество примесей не является чрезмерным, образцы можно
анализировать без предварительной очистки.
116 Глава 5. Альтернативные методы ионизации образца
Исследуемое
вещество
Матрица
Рис. 5.10. Взаимодействие лазерного импульса с образцом МЛДИ:
а — до лазерного импульса, б — после лазерного импульса
Проведение анализа. Ионизация. Короткий световой импульс лазера
поглощается молекулами матрицы и разрушает ее кристаллическую структуру.
Часть молекул отрывается от поверхности и образует высокотемпературный
суперплотный газ (зона шлейфа), в котором помимо молекул, ионов и ассоциатов
матрицы присутствуют молекулы анализируемого соединения (рис. 5.10).
Поскольку энергия лазера поглощается матрицей, молекулы образца оказываются в
зоне шлейфа, в газовой фазе, практически без приращения внутренней энергии,
т. е. с сохранением исходной структуры.
Механизмы ионизации образца в условиях МЛДИ пока недостаточно изучены
[183-185]. В масс-спектрах наблюдаются в основном пики однозарядных
молекулярных ионов, причем катионы и анионы могут регистрироваться при простой
смене полярности выталкивающего электрода. Помимо них часто наблюдаются
пики однозарядных мультимеров (типа М„Н+). Спектры образца оказываются
очень похожими даже при использовании столь разных лазеров, как УФ и ИК.
Этот факт указывает на общность механизмов ионизации для обоих вариантов.
Обычно разделяют процессы на первичные и вторичные. Первичная
ионизация включает процессы возникновения заряда у молекулы образца в
момент ее перехода из конденсированной в газовую фазу. Вторичная ионизация
обусловлена взаимодействием молекул образца с ионами матрицы в газовой
фазе. Поскольку доля образца по сравнению с долей матрицы очень мала,
взаимодействия молекул и ионов образца друг с другом маловероятны и не
имеют большой значимости.
Среди предложенных механизмов первичной ионизации можно выделить
взаимодействие молекулы образца А с кластером молекул возбужденной матрицы
М* и протонирование молекулы образца возбужденной молекулой матрицы:
м:+А-+М„+А+ + е-
М*+А^АН+ + [М-Н]-
5.14. Матричная лазерная десорбционная ионизация 117
Одновременно с переходом молекулы образца в газовую фазу может
осуществляться ее взаимодействие с катионом соли, присутствующей в
пробе случайно или добавленной намеренно. В этом случае возникают ионы
[А+катион]4".
Значительно больше процессов ионизации предложено для молекул самой
матрицы. Помимо указанных для молекул образца можно упомянуть двух- и
полифотонную ионизацию, термическую ионизацию и диспропорционирование.
В зоне шлейфа образуется газовая плазма и протекают процессы вторичной
ионизации. Механизмы этих реакций аналогичны описанным в разд. 5.2 и
включают протонирование и депротонирование, перезарядку и присоединение
аниона или катиона. Образовавшиеся молекулярные катионы и анионы образца
разных типов (М+\ МН+, [М — Н]~, [М+катион]+ и т. д.) обладают невысокой
внутренней энергией, т. е. их фрагментация незначительна.
Расширение образовавшейся газовой плазмы за область шлейфа приводит
к последующим взаимодействиям молекул и ионов образца с молекулами и
ионами плазмы. Возникают новые ионы, а внутренняя энергия ионов образца
усредняется в результате многочисленных столкновений с молекулами матрицы.
Интересные предположения о механизме ионизации сделаны Карасом и др.
[184]. Авторы приводят достаточно убедительные доводы в пользу того, что
исходные ионы образца (например, пептиды, осажденные из раствора при
рН = 2) являются многозарядными, а в источнике протекает их последовательная
нейтрализация. Для положительных ионов уменьшение заряда обусловлено
захватом тепловых электронов, образующихся в процессе фотоионизации молекул
матрицы и присутствующих в достаточном количестве в области шлейфа. Для
отрицательных ионов ключевую роль играют взаимодействия с протониро-
ванными молекулами матрицы. Результатом этих процессов является полная
нейтрализация первичных ионов. Немногочисленные оставшиеся однозарядные
ионы, однако, успевают покинуть реакционную зону и достичь детектора.
Нейтральные молекулы и частицы плазмы откачиваются насосами, а
образовавшиеся ионы вытягиваются и ускоряются высоким потенциалом (~25 кВ)
по направлению к анализатору. Практически во всех коммерческих приборах
используется времяпролетный анализатор, поскольку он характеризуется
неограниченным диапазоном масс (разд. 6.7), а основной задачей МЛДИ является как
раз получение информации о молекулярной массе высокомолекулярных
соединений. Кроме того, именно времяпролетные приборы лучше других работают с
импульсными методами ионизации и обладают высочайшей чувствительностью,
так как все образовавшиеся в результате ионизации ионы достигают детектора.
После того как детектора достигнет наиболее тяжелый ион, производится новый
лазерный импульс, и весь процесс ионизации и регистрации ионов повторяется.
Спектры после каждого лазерного импульса могут суммироваться до получения
качественной информации о молекулярной массе соединения. Обычно для
приемлемого результата оказывается достаточно 30-50 лазерных импульсов.
118 Глава 5. Альтернативные методы ионизации образца
Важным моментом является возможность направлять каждый последующий
лазерный импульс в новую точку образца. В случае равномерного
распределения анализируемого соединения в твердом растворе матрицы спектры будут
идентичными. Однако поскольку идеальной равномерности достичь практически
невозможно, концентрация молекул образца в каждом месте может несколько
отличаться, и перемещением луча можно найти оптимальный участок поверхности.
Для калибровки шкалы масс используются самые разнообразные соединения
с точно известной массой. Среди наиболее широко используемых можно назвать
грамицидин С (m/z 1141,5), бычий инсулин (m/z 5733,5), сыворотку бычьего
альбумина (m/z 66430). Используют варианты как внешней, так и внутренней
калибровки. В первом случае калибровочный стандарт помещают на отдельной
мишени, во втором — вместе с анализируемым соединением. Последний вариант
приводит к увеличению точности определения массы примерно на порядок.
Очень часто используют калибровку по двум точкам, когда в качестве реперов
выбираются пик протонированного (депротонированного) молекулярного иона
стандарта и пик его димера или двухзарядного иона. Используется также смесь
двух стандартов. Важно, чтобы пик стандарта с известной массой располагался
достаточно близко к пику молекулярного иона исследуемого соединения, но
не накладывался на него. Хотя разрешающая способность времяпролетных
анализаторов, как правило, не превышает 10-12 тысяч, точность измерения масс
достаточно высока и достигает 0,01%.
Метод МЛДИ используется прежде всего для установления молекулярной
массы соединения. Поскольку полностью избежать фрагментации не удается,
в спектре часто наблюдаются малоинтенсивные пики фрагментных ионов, не
имеющие значимости для структурных исследований. Если же возникает вопрос
об установлении структурных фрагментов соединения, необходимо использовать
определенные модификации обычного варианта анализа. Для этого используют
спектры метастабильных ионов (post source decay), полученные с помощью ре-
флектрона [186], или спектры активации соударением с использованием техники
ортогонального ускорения (разд. 6.7).
Вид спектра тяжелых молекул (рис. 5.9) значительно отличается от
классического вида спектра ЭУ. Когда речь идет о молекуле массой в несколько
сотен тысяч дальтон, разрешающей способности любого масс-спектрометра
оказывается недостаточно для разрешения всей изотопной картины в области
молекулярного иона. Обычные правила работы со спектром ЭУ, описанные
в гл. 4, неприемлемы для соединений с молекулярной массой более 2000 Да.
Прежде всего это касается извлечения информации на основе интенсивностей
изотопных пиков (разд. 4.2). Например, наличие в молекуле 100 атомов углерода
приведет к интенсивности пика М+1 за счет присутствия одного атома 13С 110%
от интенсивности пика М+*, обусловленного только атомами 12С. Пики ?+2
и М+3, обусловленные двумя и тремя атомами 13С в молекуле будут иметь
интенсивности 60 и 22% соответственно. Даже водород, который учитывается
при расшифровке спектра ЭУ как ?-элемент (разд. 4.2), поскольку природное
5.14. Матричная лазерная десорбционная ионизация 119
3
Рис. 5.11. Область молекулярного
иона соединения C146H212N30O56S4.
1 — номинальная масса C408); 2 —
моноизотопная масса C409,3546);
3 — наиболее распространенная
масса C411,3613); 4 —средняя
масса C411,721)
содержание дейтерия составляет 0,015% от содержания протия, приведет к
появлению значимого изотопного пика М+1, если число атомов водорода в
молекуле более 100. Другие элементы, практически всегда присутствующие в
составе биологических молекул (N, О, S), усложняют изотопную картину еще
значительнее. В результате огромное число изотопных пиков проявляется в масс-
спектре в виде широкого сигнала, ширина которого может составлять сотни и
тысячи дальтон (рис. 5.9).
Кроме того, для высокомолекулярных соединений кумулятивный эффект
дефектов масс атомов (разд. 6.3) становится значительным и ведет к
существенным отклонениям номинальной массы, рассчитанной на основе целочисленных
атомных масс элементов, от регистрируемой массы.
В связи с этим для спектров тяжелых соединений приходится вводить еще
несколько определений молекулярной массы (рис. 5.11).
Номинальная масса — масса, рассчитанная суммированием целочисленных
атомных масс наиболее распространенных (легких) изотопов элементов.
Моноизотопная масса — масса, рассчитанная суммированием точных атомных
масс (с учетом дефекта массы) наиболее распространенных (легких) изотопов
элементов.
Наиболее распространенная масса — масса иона, пик которого наиболее
интенсивен в кластере молекулярного иона.
Средняя масса — масса, рассчитанная суммированием усредненных атомных
масс элементов.
На рисунке 5.11 отчетливо видно различие между этими вариантами
величин молекулярной массы соединения. Пик молекулярного иона с номинальной
массой 3408 Да в спектре отсутствует, поскольку суммарный дефект массы
оказывается больше 1 Дальтона. Минимальную массу в спектре имеет пик с
моноизотопной массой. Ее величина 3409,3546 Да. Интенсивность этого пика
уступает интенсивности последующих трех пиков в связи с высокой
вероятностью присутствия в молекуле 1-3 более тяжелых изотопов.
В частности, следующий за пиком с моноизотопной массой пик состоит
из пяти индивидуальных неразрешенных пиков, обусловленных присутствием
изотопов 2Н, 13С, 15N, 170, 33S. Рассчитаем массы и относительные интенсив-
w146' ^^"ЗО^бб^
3408
3410
3412
3414
3416
3420
120 Глава 5. Альтернативные методы ионизации образца
ности этих пиков. Присутствие одного атома дейтерия в молекуле приведет к
появлению пика иона с массой 3410,3580 Да и относительной интенсивностью
(в сумме интенсивностей этих пяти пиков) 1,7%. Присутствие одного атома 13С
в молекуле приведет к появлению пика с массой 3410,3609 Да и относительной
интенсивностью 89,2%. Присутствие одного атома 15N в молекуле приведет к
появлению пика с массой 3410,3517 Да и относительной интенсивностью 6,2%.
Присутствие одного атома пО в молекуле приведет к появлению пика с массой
3410,3588 Да и относительной интенсивностью 1,2%. Наконец присутствие
одного атома 33S в молекуле приведет к появлению пика с массой 3410,3540 Да и
относительной интенсивностью 1,7%. Чтобы увидеть эти пять пиков раздельно,
нужен прибор с разрешающей способностью (разд. 6.3) более 4000000.
Следующий пик на рис. 5.11 с целочисленной массой 3411 (наиболее
распространенная масса) состоит из 19 индивидуальных пиков, обусловленных
одновременным присутствием в молекуле двух изотопов А+1 или одного
изотопа А+2 A80 или 34S). Поскольку основной вклад, как и в предыдущем
случае, вносит изотоп 13С, центр масс этого составного пика будет
находиться около 3411,3613 Да. Кстати, средняя молекулярная масса, рассчитанная из
усредненных величин атомной массы элементов (разд. 5.12), находится рядом с
этим пиком и составляет 3411,7210 Да. Видно, что пик с такой массой в спектре
отсутствует, однако при получении спектров еще более тяжелых соединений, с
массами в десятки тысяч дальтон, разрешающей способности любого прибора
оказывается недостаточно для получения разрешенного кластера молекулярного
иона. Пики ионов представляют собой огибающую шириной в сотни и тысячи
дальтон, причем вершина этой кривой соответствует средней молекулярной
массе соединения (рис. 5.9).
Вариантом МЛДИ является метод поверхностной матричной лазерной
десорбционной ионизации (Surface Enhanced Laser Desorption Ionization,
SELDI), впервые описанный Хатченсом [187, 188]. Метод позволяет резко
повысить селективность анализа пептидов и белков благодаря модификации матрицы.
К обычному материалу матрицы для МЛДИ добавляется соединение, которое
может образовать химические связи с конкретным участком сложной молекулы.
Если на этот подготовленный микрочип нанести раствор смеси биомолекул, то
только те из них, которые имеют в своей структуре подходящие места
связывания, образуют химические связи с молекулами модифицированной матрицы.
Последующая промывка микрочипа удаляет все несвязанные молекулы. После
высушивания образец подвергается МЛДИ анализу [189, 190].
Метод SELDI показал превосходные результаты в решении биохимических и
медицинских проблем: установление структур белков, идентификация
биомаркеров, установление мест связывания антигенов, клинические исследования по
выявлению заболеваний (туберкулез, диабет, рак, панкреатит, болезнь Альцгей-
мера и т. д.), и многие другие [191-193].
5.15. Пиролитическая масс-спектрометрия 121
5.15. Пиролитическая масс-спектрометрия
(Pyrolysis/Mass Spectrometry, Py/MS)
В классической масс-спектрометрии большое внимание уделяется сохранению
исходной структуры анализируемого соединения. Поэтому спектры
термолабильных образцов снимают при минимально возможной температуре или с
помощью другого метода ионизации. Например, вместо ГХ-МС используют
ЖХ-МС. Основной задачей пиролитической масс-спектрометрии является
быстрое и эффективное термическое разложение веществ на более простые, которые
подвергаются последующему анализу. Сейчас, когда методы электрораспыления
или МЛДИ позволяют получать масс-спектры соединений с молекулярной
массой в сотни тысяч и миллионы дальтон, метод пиролитической
масс-спектрометрии используется в основном для анализа неразделимых смесей соединений:
горючие сланцы, синтетические полимеры, гуминовые кислоты,
микроорганизмы, другие биологические и геологические материалы. Основам метода и его
применению для анализа высокомолекулярных соединений посвящена книга
Хмельницкого, Лукашенко, Бродского [194].
Пиролиз можно осуществить в самостоятельном приборе, а пиролизат затем
перенести (например, в ловушке, охлаждаемой жидким азотом) в
масс-спектрометр. Можно проводить пиролиз перед колонкой хроматографа или даже
непосредственно в ионном источнике масс-спектрометра. В последнем случае
получается только интегральный спектр, являющийся суперпозицией спектров
всех соединений, образовавшихся в результате температурного разложения
образца. Во избежание появления продуктов окисления, маскирующих реальную
картину, пиролиз принято вести в среде инертного газа или в вакууме. В качестве
метода ионизации наиболее часто используют ЭУ, ХИ, полевую ионизацию,
ионизацию лазером. Пиролитическая масс-спектрометрия, как правило,
направлена на решение одной из двух задач:
1) Установление структурных фрагментов очень сложных соединений.
2) Сравнение образцов друг с другом (доказательство идентичности).
В первом случае желательно использовать комплексный метод, типа ГХ-МС
или ЖХ-МС, а во втором — предпочтительнее получать интегральный спектр.
Такой подход отлично зарекомендовал себя, в частности, для сравнения
микроорганизмов (разд. 8.17). В этом случае структуры отдельных продуктов распада
неважны, тогда как интегральный спектр можно рассматривать как «отпечатки
пальцев» данного вида бактерий, вирусов, грибов и т. д. Идентичность двух
образцов микроорганизмов друг другу устанавливают таким образом очень
быстро и эффективно. Важно лишь во избежание ошибок строго контролировать
условия проведения эксперимента. Они должны быть абсолютно идентичны по
скорости нагрева, типу разогрева образца и температуре пиролиза. Для целей де-
122 Глава 5. Альтернативные методы ионизации образца
тального сравнения интегральных масс-спектров предложен ряд математических
подходов [195, 196].
Использование масс-спектрометра как термогравиметрического детектора
позволяет надежно доказать, какое именно соединение выделяется образцом в
каждом из зарегистрированных по уменьшению массы процессов. Температуру
в этом случае медленно поднимают до нескольких сотен градусов.
Несколько большими скоростями нагрева характеризуется метод катодного
разогрева (с помощью сопротивления). Для проведения такого анализа образец
наносится непосредственно на проволоку из благородного металла. Проволока
с образцом электрически программируемо нагревается до нужной температуры
с нужной скоростью. Иногда для увеличения объема пробы ее помещают не
непосредственно на проволоку, а в нагреваемую от нее кварцевую трубку. Такой
подход хорошо зарекомендовал себя для анализа полимерных материалов.
Для получения спектров типа «отпечатков пальцев» наилучшие результаты
получены при использовании индуктивного нагрева в точке Кюри (Curie-point
inductive heating). Как уже отмечалось выше, для такого анализа
первостепенное значение имеет воспроизводимость условий эксперимента. Точкой Кюри
называется температура, при которой ферромагнетики теряют свои магнитные
свойства. Например, для железа это происходит при температуре 770 °С, для
никеля 358 °С, для кобальта 1120°С. Когда проволока (фольга), изготовленная
из ферромагнитного материала, помещается в радиочастотное электромагнитное
поле, происходит ее быстрый разогрев со скоростью до 104 К/с. Точка Кюри
достигается за доли секунды. В этот момент материал теряет свои магнитные
свойства, и нагрев прекращается. Однако как только температура упадет на долю
градуса, нагрев возобновляется. Если на проволоку (фольгу) нанести образец
анализируемого материала, его пиролиз будет проходить при точно известной
температуре.
Несколько пиролитических способов основано на лазерном нагреве образца.
Использование постоянного лазера, работающего в инфракрасном или видимом
диапазоне, позволяет проводить нагрев со скоростями, аналогичными
достигаемым пиролизом в точке Кюри. Пятно лазера обычно имеет диаметр 150-200 мкм,
а перемещая лазерный луч по поверхности образца, можно определять
неоднородности его состава.
Максимальными скоростями нагрева характеризуется метод лазерной
абляции, которую можно рассматривать как взрывной пиролиз. Пятно импульсного
ультрафиолетового лазера диаметром 10-25 мкм вызывает локальный разогрев
образца со скоростью до 106 К/с. Образующаяся над поверхностью плазма
состоит не только из молекул. В ней присутствуют радикалы, положительные и
отрицательные ионы. При этом можно получить масс-спектр без дополнительной
ионизации. Тем не менее в пиролитической масс-спектрометрии для получения
полной картины используют дополнительный метод ионизации. Перемещение
луча по поверхности позволяет получить информацию о поверхностной неодно-
5.15. Пиролитическая масс-спектрометрия 123
родности материала. Поскольку в результате абляции молекулы поверхностного
слоя образца переходят в газовую фазу, повторный импульс лазера,
направленный в то же место, захватит молекулы второго слоя, затем третьего и т. д.
Таким образом, можно получить информацию об изменениях состава образца от
поверхности в глубину Два этих подхода (поверхностное и глубинное
сканирование) эффективно применяются для решения криминалистических задач.
Например, метод широко используется для доказательства подлинности
картин или для правильного проведения реставрационных работ [197]. Поскольку
добавки к краскам хранились живописцами средних веков в глубокой тайне,
установление присутствия на холсте тех или других соединений позволяет
установить принадлежность полотна к той или иной школе. В частности,
венецианские художники эпохи Возрождения использовали в качестве матрицы
для красок белок яиц, белое вино и чесночный сок. При исследовании соскобов
с их картин можно найти соответствующие полисахариды, глицериды, белки.
Фламандские живописцы первыми стали использовать льняное масло.
Соответственно в спектре наблюдаются жирные кислоты. Характерные спектры дают
добавки казеина, смол, восков и т. д. Проведение масс-спектрометрического
эксперимента позволяет не только установить, какие добавки использовались
живописцем, но и оценить результат эффектов старения.
Глава 6
РАЗДЕЛЕНИЕ И РЕГИСТРАЦИЯ ИОНОВ
После того как проведена ионизация образца, необходимо разделить ионы,
образовавшиеся в источнике в результате самых разнообразных процессов. Для
этой цели используется несколько типов анализаторов. Каждый тип имеет свои
преимущества и недостатки. В связи с этим, приобретая дорогостоящий масс-
спектрометр, необходимо четко представлять круг задач, которые планируется
решать с его помощью.
6.1. Магнитный секторный масс-спектрометр
Хронологически первым анализатором масс был магнитный. Принципиальная
схема его устройства представлена на рис. 6.1.
Магнит
Ускоряющий электрод
Источник\ \ \ / / Детектор
Рис. 6.1. Принципиальная схема масс-спектрометра с магнитным анализатором
Ион, образовавшийся в источнике, выводится оттуда благодаря системе
электродов и ускоряется потенциалом V B-8 кВ) по направлению к анализатору.
Пусть т — масса иона, е — единичный заряд, ? —число таких зарядов у иона,
? —скорость иона. Тогда кинетическая энергия иона, на входе в магнитный
анализатор, выражается следующим уравнением:
zeV = mv2/2 F.1)
Попадая в магнитное поле напряженностью В перпендикулярно магнитным
силовым линиям, этот ион будет двигаться по окружности R, причем сила
Лоренца уравновешивается центробежной силой:
Bzev = mv2/R F.2)
6.1. Магнитный секторный масс-спектрометр 125
2БП
Электростатический
анализатор
Магнитный
анализатор
1БП
Рис. 6.2. Принципиальная схема магнитного секторного прибора двойной фокусировки
прямой геометрии. БП — бесполевое пространство
Уравнение 6.2 может быть представлено в форме BzeR = ???, которая
демонстрирует, что магнит является анализатором именно моментов, а не масс.
Комбинация уравнений 6.1 и 6.2 приводит к основному уравнению разделения
ионов в магнитном анализаторе:
m/ze = B2R2/2V
F.3)
Для получения масс-спектра можно сканировать напряженность магнитного
поля либо ускоряющее напряжение1. Однако изменение величины ускоряющего
напряжения всего в два раза вызывает некоторую дефокусировку ионного пучка
и потерю чувствительности. При этом изменяется и разрешающая способность
(см. ниже). Поэтому масс-спектры принято получать сканированием
напряженности магнитного поля.
Поскольку секторный магнит является анализатором моментов, а не масс,
ионы с одной массой, но различной кинетической энергией, не собираются
в точечном фокусе однофокусного магнитного прибора. Разброс кинетической
энергии ионов лимитирует достижимое разрешение. Для преодоления этого
недостатка на пути следования ионов ставят дополнительный
электростатический сектор (рис. 6.2).
1В масс-спектрографах осуществляется разделение ионов по радиусам их траекторий в постоянном
магнитном поле. В этом случае все ионы одновременно достигают фотопластинки, расположенной в фокальной
плоскости, и засвечивают ее. Исходя из расположения сигналов на пластинке и интенсивности засветки, можно
устанавливать массы ионов и их количество, т. е. получать масс-спектр.
126 Глава 6. Разделение и регистрация ионов
6.2. Электростатический анализатор.
Двухфокусный секторный масс-спектрометр
Электростатический анализатор может располагаться до или после магнитного.
В первом случае принято говорить о прямой геометрии прибора, во втором —
об обратной. Для большинства масс-спектрометрических задач расположение
анализаторов не принципиально, однако при работе в некоторых режимах тан-
демной масс-спектрометрии (MIKES, разд. 7.2.2.1) необходим прибор обратной
геометрии.
Попадая в электростатический сектор, ион движется по круговой орбите с
радиусом R таким образом, что сила электрического поля уравновешивается
центробежной силой:
mv2/R = zeE F.4)
Учитывая кинетическую энергию ионов (zeV = mv2/2), можно получить
следующее выражение:
R = 2V/E F.5)
Следовательно, через анализатор пройдут ионы с одинаковой энергией вне
зависимости от их массы.
Приборы с двойной фокусировкой характеризуются большей величиной столь
важного параметра, каким является разрешающая способность. Под
разрешением (R) понимается способность получать на данном приборе раздельное
изображение двух ионов (рис. 6.3) с массами т и (т + ?/и). Величина R
формально соответствует отношению т к Am.
Идеальная форма пиков ионов — прямоугольная, реальная — Гауссова. В
зависимости от глубины ложбины между двумя соседними пиками принято говорить
о разрешении на уровне 10% от высоты пиков для магнитных приборов и 50% —
для квадрупольных (разд. 6.5). Если Am = 1, то R теоретически является макси-
Ж
т т + Ат
Рис. 6.3. Определение разрешающей способности прибора
6.3. Масс-спектрометрия высокого разрешения, МСВР 127
мальной массой, регистрируемой прибором, когда пики остаются разделенными.
Разрешение 80000 не означает, что на приборе можно получить разрешенный
пик однозарядных ионов с такой массой. Регистрируемая масса
пропорциональна B2/V, но безграничное увеличение В сопряжено с техническими сложностями
и нецелесообразно, а уменьшение V понижает и разрешающую способность.
Для расчета разрешающей способности можно использовать один пик. В этом
случае в качестве Am используется полуширина пика с массой т.
Следует подчеркнуть, что секторные приборы характеризуются диапазоном
величин m/z до 10000, наивысшей точностью измерения масс (<5 м. д.) и
непревзойденным динамическим диапазоном A07). Давление в анализаторе секторного
прибора не должно превышать 10~6 мм рт. ст.
6.3. Масс-спектрометрия высокого разрешения, МСВР
Что же означают тогда величины разрешающей способности масс-спектрометров
в десятки и сотни тысяч? Дело в том, что каждый изотоп любого элемента имеет
уникальный характеристический дефект массы. Поскольку за стандарт принят
основной изотоп углерода 12С A2,000000), массы всех остальных изотопов
элементов не целые числа. Так масса основного изотопа водорода !Н 1,00782506,
азота 14N 14,00307407, кислорода 160 15,99491475 и т. д. Точные массы всех
изотопов представлены в табл. 2 приложения.
Измерение точной массы иона D-6 знаков после запятой) однозначно
определяет его элементный и изотопный состав. Проведение измерений с такой
точностью называется масс-спектрометрией высокого разрешения. Измерения
проводят вручную или с помощью компьютера, совмещая положение пика
неизвестного иона на шкале масс с ближайшим пиком иона известного состава.
В качестве реперов масс можно использовать любое соединение,
характеризующееся интенсивным пиком с величиной m/z, близкой измеряемой. Наиболее
распространенными для этой цели являются перфторкеросин, перфтортрибутил-
амин, другие полностью фторированные соединения. Использование перфтори-
рованных соединений объясняется их летучестью, а также тем, что фтор
является моноизотопным элементом. В спектрах таких соединений регистрируются
интенсивные сигналы фрагментных ионов, равномерно перекрывающие весь
диапазон от m/z 19 до М+".
Простейшим примером использования масс-спектрометрии высокого
разрешения может служить разделение мультиплета с целочисленной массой 28 Да.
Три соединения с такой массой практически всегда присутствуют на уровне
фона при съемке масс-спектров. Это —азот, монооксид углерода и этилен.
На однофокусном приборе с разрешением 500 молекулярные ионы всех трех
соединений проявятся в виде единого пика (рис. 6.4, а). Повышение разрешения
меняет картину (рис. 6.4, б, в, г).
128 Глава 6. Разделение и регистрация ионов
'?'?'?'?
Д = 250 R = 600 Я =1500 R = 5000
Рис. 6.4. Зависимость формы пика фоновых ионов с целочисленной массой 28 Да от
разрешающей способности масс-спектрометра
Действительно, для разделения пиков ионов СО и С2Н4 с массами 27,994915
и 28,03300 Да необходимо разрешение R = 28/B8,03300 - 27,994915) = 770,
тогда как для разделения пиков ионов СО и N2 с массой 28,006148 Да —уже 2500.
Если учесть, что число ионов с одинаковой целочисленной массой резко
возрастает с увеличением массы, то важность масс-спектрометрии высокого
разрешения и, естественно, увеличения разрешающей способности приборов
становится очевидной. Решение задачи установления элементного состава
соединения, включающего самые разные элементы, за несколько минут выгодно
отличается от трудоемкого последовательного определения с помощью
классического элементного анализа. Следует, правда, отметить, что элементный анализ
дает информацию об образце в целом (с учетом примесей), а масс-спектрометр
«видит» только «летучие» компоненты. Так что для точного установления
состава целевого соединения лучше использовать масс-спектрометрию, а для
проверки его чистоты — элементный анализ.
Масс-спектрометрия высокого разрешения очень важна также в анализе
биомолекул методами электроспрей и МЛДИ. В этом случае для правильного
определения молекулярной массы сложнейших соединений необходима очень
высокая точность измерения.
Современные секторные приборы обладают разрешающей способностью
60000-80000, в некоторых случаях до 150000. Сверхвысокое разрешение
достижимо при использовании масс-спектрометрии с преобразованиями Фурье
(ионно-циклотронный резонанс, разд. 6.4).
Неправильно считать, что увеличение разрешающей способности однозначно
ведет к увеличению точности измерения массы. Как только разрешения
оказывается достаточно для получения индивидуальных пиков с одной целочисленной
массой, которые принадлежат образцу и стандарту, бессмысленно повышать
разрешение далее, поскольку при этом падают и точность измерения, и
чувствительность анализа.
Основным вариантом измерения точной массы иона является совмещение
пика этого иона на экране осциллографа с пиком иона с известной массой.
Оба пика попеременно сканируются на экране за счет переключения величины
ускоряющего напряжения. Совмещение осуществляется поворотом тумблеров,
6.4. Масс-спектрометрия с преобразованиями Фурье 129
Таблица 6Л
Результаты измерения точной массы иона методом МСВР
Измеренная
масса иона
(Мизм)
163,9497
Вычисленная
масса иона
(Мвыч)
163,9503
163,9488
163,9521
163,9537
163,9447
Разность (м. д.)
Мизм - МВыч
0,6
-0,9
2,4
4,0
-5,0
Элементный
состав иона
C6N2S2
CgHSCl
C5H5S2C1
C3H4N2S3
C3HN202SC1
имеющих разную чувствительность по отношению к изменению ускоряющего
напряжения (V). Когда достигнуто совмещение пиков, коэффициент пересчета
можно прочитать на панели прибора по значениям выставленным на тумблерах.
Для расчета точной массы иона можно воспользоваться простейшей формулой:
Vs
т = kms = — ms F.6)
где индекс s относится к стандарту. Ручное измерение массы занимает несколько
минут и не может использоваться в режиме хроматомасс-спектрометрии, когда
вещество присутствует в источнике не более 10 с. Существует возможность
компьютерного измерения масс [198]. В этом варианте точность измерения ниже,
однако увеличение скорости измерения позволяет проводить измерения в режиме
гх-мс.
Результаты анализа часто приводятся в виде таблицы, в которой отмечается
измеренная масса иона, а также элементный состав и рассчитанная масса иона.
В таблице могут быть представлены ионы с разными составами, рассчитанная
масса которых наиболее близка измеренной экспериментально. В таблице 6.1
представлен пример установления элементного состава иона с помощью масс-
спектрометрии высокого разрешения. Наиболее близкие массы к измеренной
имеют 5 ионов. Поскольку известно, что молекула исследуемого соединения не
содержит атомов хлора и водорода, состав определяемого иона C6N2S2.
6.4. Масс-спектрометрия с преобразованиями Фурье
(Fourier Transform Mass Spectrometry, FT-MS)
Масс-спектрометрия с преобразованиями Фурье основана на принципе ионно-
циклотронного резонанса (ИЦР). Физические основы этого метода были
разработаны Лоуренсом [199] в 30-х годах XX века, первый масс-спектрометр этого
типа построен позже, в 50-х годах, Соммером [200]. Приложение преобразований
Фурье к ИЦР впервые было осуществлено Комисаровым и Маршалом в 1978 г.
5—1113
130 Глава 6. Разделение и регистрация ионов
В настоящее время метод является одним из наиболее популярных в масс-
спектрометрии [201].
Основным элементом прибора является ячейка, размещенная в однородном
магнитном поле. Ячейка может иметь разнообразные формы, простейшей из
которых является куб. Для создания магнитного поля применяют постоянные
магниты и электромагниты. Однако поскольку характеристики прибора
улучшаются при увеличении напряженности поля, наилучших результатов удалось
достичь при использовании суперпроводящих магнитов силой 3-9 Т.
Ионы образца, генерированные любым известным методом, направляются в
ячейку, в которой протекают процессы активации и детектирования ионов.
Входное отверстие в ячейку расположено на пластине, перпендикулярной силовым
линиям магнитного поля. Можно генерировать ионы и непосредственно в ячейке.
Оказываясь в ячейке, ион, обладающий определенной скоростью теплового
движения, испытывает на себе силу Лоренца. Эта сила перпендикулярна
направлению магнитного поля и скорости иона. Она уравновешивается центробежной
силой. При большой величине напряженности магнитного поля ион описывает
окружность (рис. 6.5):
F = та = mv2/r = zevB, F.7)
где т — масса иона, ? — компонента скорости иона в направлении
перпендикулярном магнитному полю, г —радиус траектории кругового движения иона,
е — единичный заряд, ? —число единичных зарядов у иона, В — напряженность
магнитного поля.
Если ?? угловая (циклотронная) частота ионов в радианах в секунду, то
уравнение 6.7 можно представить следующим образом:
mv/r = тис = zeB или шс = zeB/m F.8)
Обычно используется выражение для циклотронной частоты иона в герцах:
ус — ?</2? = zeB/2-кт или m/ze = B/2nvc F.9)
В частности, циклотронная частота иона N^~# в магнитном поле
напряженностью 1,0 ? составляет:
1,6х1(Г19Клх1,0Т
6,28 ? 28 Да ? 1,66 ? 107 кг/Да К Ц'
т. е. 5,7 ? 105 циклов в секунду.
Магнитное поле в масс-спектрометрах с преобразованиями Фурье, как
правило, не изменяется, а масса иона определяется при измерении его циклотронной
частоты. Следует подчеркнуть, что циклотронная частота не зависит от скорости,
а следовательно, кинетической энергии иона. Поэтому появляется возможность
достигать сверхвысокого разрешения (до сотен миллионов [202]),
недостижимого при работе на магнитных приборах. Однако разрешающая способность
6.4. Масс-спектрометрия с преобразованиями Фурье 131
уменьшается с увеличением массы иона и для достаточно тяжелых ионов она
становится равной или даже уступает разрешающей способности магнитных
анализаторов.
Удерживание иона в ячейке осуществляется при подаче на задерживающие
пластины, перпендикулярные силовым линиям магнитного поля, небольшого
равного напряжения. Например, если исследуются положительные ионы, на
пластины подается напряжение +3 В, если отрицательные —3 В. В результате
ионы в ячейке претерпевают гармонические колебания между
задерживающими пластинами. Комбинация магнитного и электрического полей создает
трехмерную ловушку, в которой ионы могут существовать часами. Столь
длительное время жизни ионов существенно отличает ИЦР от других масс-
спектрометрических методов, когда ионы живут милли- и микросекунды. Здесь,
правда, следует отметить, что масс-спектрометрия с преобразованиями Фурье
имеет значительно более строгие вакуумные ограничения. Для нормальной
работы прибора в условиях высокого разрешения требуется давление на уровне
10~9-10-10 мм рт. ст. Такое давление достигается при использовании мощных
криогенных или турбомолекулярных насосов.
Суперпозиция циклотронного движения и колебаний между
задерживающими пластинами, формально независимых друг от друга, приводит к
возникновению магнетронного движения ионов. Магнетронная частота зависит от
формы ячейки, расстояния между пластинами, напряженностей магнитного и
электрического полей и не зависит от массы ионов. Эта частота имеет величину
порядка 1-100 Гц, т. е. существенно ниже частоты циклотронного движения,
которая лежит в кило- и мегагерцевом диапазоне. Магнетронный радиус иона
определяется также начальным положением иона относительно основной оси
ячейки. Для достижения максимального разрешения и максимальной точности
определения массы магнетронный радиус должен быть минимизирован. Это
достигается генерированием или вводом ионов в ячейку вдоль основной оси
параллельно магнитным силовым линиям.
Простейшая последовательность событий для получения масс-спектра может
быть представлена следующим образом:
очистка!
возбуждение
детектирование]
-> [ионизация] ->[ н'онов j-[ ион*-ов j
Для очистки ячейки от ионов к задерживающим пластинам прикладывается
асимметричное напряжение (например, +10 и —10 В). В результате все ионы,
находившиеся в ячейке, покидают ее за несколько миллисекунд. Процесс
ионизации может включать образование ионов непосредственно в ячейке (электронный
удар), рядом с ячейкой в условиях глубокого вакуума (МЛДИ), вне магнитного
поля. В последнем случае образовавшиеся ионы направляются в ячейку с
помощью системы электродов. Для возбуждения и детектирования ионов их
облучают приложением радиочастотного поля к паре противолежащих пластин,
132 Глава 6. Разделение и регистрация ионов
?
Рис. 6.5. Циклотронное движение ионов до возбуждения (а); возбуждение ионов
резонансным радиочастотным импульсом (б); циклотронное движение ионов после возбуждения (в)
расположенных параллельно линиям магнитного поля. Когда собственная
частота иона оказывается в резонансе с частотой возбуждающего импульса, радиус
траектории иона увеличивается по спирали (рис. 6.5,6). Поскольку скорость и
радиус движения иона увеличиваются синхронно, угловая частота не изменяется
и ион остается в резонансе. Ионы с другими массами продолжают при этом
двигаться по своим нерезонансным орбитам. Если возбуждение продолжается
в течение длительного времени, ускоренные ионы достигают боковых
пластин и гибнут. Этот эффект, кстати, можно использовать для удаления ионов
определенной массы из ячейки, например, работая в режиме тандемной масс-
спектрометрии (разд. 7.4). Если же вовремя прекратить подачу радиочастотной
энергии, ионы будут продолжать вращаться по орбите с большим радиусом
(рис. 6.5, в). В идеальных вакуумных условиях при отсутствии столкновений
ионы теоретически должны оставаться на этих орбитах неограниченное время.
Процесс детектирования ионов представлен на рис. 6.6. Циклотронное
когерентное движение пучка разогнанных положительных ионов между
детектирующими пластинами, имеющими потенциал Земли и соединенными друг с другом
через сопротивление, обусловливает следующее. Когда положительные ионы
движутся от левой к правой пластине, их электрическое поле заставляет
электроны перемещаться в том же направлении по внешней цепи через сопротивление.
Во второй части циклотронного движения ионов электроны перемещаются в
обратном направлении. Это движение электронов называют наведенным током.
Это переменный ток с частотой, равной разности частот циклотронного и
магнетронного движения, а его амплитуда пропорциональна числу ионов данной
^
""
(
?
\
\
\
1
^ч
,1
.1
Усилитель
>-
R
Рис. 6.6. Детектирование резонансно ускоренных ионов
6.4. Масс-спектрометрия с преобразованиями Фурье 133
массы в ячейке. Наведенный ток создает небольшое переменное напряжение на
сопротивлении, которое может быть усилено и измерено. Таким образом, можно
регистрировать ионы, не удаляя их из ячейки.
В классическом варианте ионно-циклотронного резонанса для получения
спектра необходимо было линейно сканировать частоту или напряженность
магнитного поля. Это требовало значительного времени и большого количества
образца. Сущность масс-спектрометрии с преобразованиями Фурье заключается
в возбуждении сразу всех ионов в результате приложения широкого
радиочастотного импульса. Существующие синтезаторы частот способны активировать
ионы с собственными частотами от 10 кГц до 1 МГц за 1 мс.
Регистрируемый наведенный ток является комплексом синусоид с различной частотой
и амплитудой. Пример такого временного сигнала представлен на рис. 6.7, а.
Математические операции, основанные на преобразованиях Фурье, позволяют
перейти от временного сигнала к масс-спектру в обычном виде (рис. 6.7, б).
Хотя вся необходимая информация о массовых числах ионов содержится
в любом коротком сегменте временного сигнала, полезно записать сигнал
полностью. Дело в том, что разрешающая способность прямо пропорциональна
продолжительности зарегистрированного временного сигнала. Если обозначить
это время Г, то разрешение в спектре можно рассчитать по уравнению 6.10:
R = Tvc/2 F.10)
К сожалению, ? имеет ограничения. Амплитуда временного сигнала
уменьшается во времени, поскольку когерентность ионного пучка нарушается в
результате столкновений ионов с нейтральными частицами. Понятно, что
использование более глубокого вакуума будет способствовать увеличению
разрешающей способности. В частности, временной сигнал до 60 с получают в
вакууме 10~10 мм рт. ст.
Впечатляющие результаты получены при использовании масс-спектрометрии
с преобразованиями Фурье для анализа соединений большой массы (особенно
в сочетании с МЛДИ), для установления направления газофазных ионных и
фотохимических реакций. Особенно ценно сверхвысокое разрешение метода,
позволяющее устанавливать точные массы ионов. Отличные результаты
получены и при работе в режиме тандемной масс-спектрометрии (гл. 7). Причем
для целей тандемной масс-спектрометрии не требуется никаких дополнительных
конструкционных добавлений, существенно удорожающих соответствующие
приборы с магнитными, квадрупольными или времяпролетными анализаторами.
В начале 80-х годов XX века казалось, что масс-спектрометрия с
преобразованиями Фурье позволит анализировать ионы любой массы, однако существуют
не только практические, но и теоретические пределы. Прежде всего следует
отметить, что, начиная с определенной массы, сила радиального электрического
поля начинает превосходить силу Лоренца. Кроме того, радиус циклотронной
орбиты ионов, движущихся со скоростями теплового движения, увеличивается
?-s e
о
В
о
?
О
о
2
К
2
!
?
О
о
«
о
к
X
<?
<?
PQ
?©
о
К
Рч
6.5. Квадрупольный анализатор 135
с увеличением их массы, т. е. теоретический предел масс для каждой ячейки
определяется ее размерами. Первый фактор является более важным, чем второй.
В ячейке размером 2,5 см с задерживающим напряжением 1 В в магнитном поле
3 ? радиальное электрическое поле не позволяет анализировать однозарядные
ионы тяжелее 40 000 Да. Термический радиус анализируемого иона играет
значительно меньшую роль. Для указанных параметров анализа предел по этому
показателю составит 10000000 Да [203].
Методу масс-спектрометрии с преобразованиями Фурье посвящен ряд
недавно опубликованных обзоров [204-207].
6.5. Квадрупольный анализатор
Квадрупольный анализатор, часто называемый квадрупольным фильтром масс,
состоит из четырех параллельных стержней круглого или гиперболического
сечения (рис. 6.8).
Противоположные стержни электрически соединены и находятся под
напряжением, складывающимся из компоненты постоянного тока U и радиочастотной
компоненты Vocosujt. Вторая пара стержней имеет равную по величине, но
противоположную по знаку компоненту постоянного тока, а фаза радиочастотной
компоненты сдвинута на 180°. Ионы, вводимые в анализатор небольшим
ускоряющим напряжением A0-20 В), под действием электрического поля колеблются
относительно осей ? и у. Так как каждый ион имеет свою собственную частоту,
зависящую от массы, через квадруполь пролетают лишь те частицы,
частота которых находится в резонансе с радиочастотой квадруполя. Масс-спектр
генерируется сканированием U и Vq при сохранении постоянной величины
U/Vo. Регистрируемая масса т пропорциональна Vo, т. е. линейное изменение
Vo предоставляет калибруемую линейную шкалу масс [208].
Квадруполь легко управляется компьютером, имеет хороший динамический
диапазон A05), стыкуется со всеми системами ввода, способен без
модифицирования разделять и положительные, и отрицательные ионы. Его
достоинствами являются также быстрота сканирования, небольшие размеры, дешевизна и
+(t/4-KC0SG)i)
Рис. 6.8. Квадрупольный анализатор
136 Глава 6. Разделение и регистрация ионов
возможность работы при повышенном (до 5 ? 10~5 мм рт. ст.) давлении. Эти
факторы обусловили широчайшее распространение квадрупольных приборов
начиная с 70-х годов XX века, особенно для серийных стандартных анализов,
съемки рутинных спектров электронного удара или химической ионизации. Еще
один импульс для использования приборов этого типа связан с созданием метода
электрораспыления (разд. 5.12). Современные квадруполи работают в диапазоне
величин m/z до 3000-4000, что делает их чрезвычайно удобными для анализа
белков, пептидов, других биомолекул с применением электрораспыления,
когда образование многозарядных ионов происходит в условиях атмосферного
давления. Квадрупольные анализаторы отлично зарекомендовали себя также в
приборах гибридной геометрии для тандемной масс-спектрометрии (гл. 7). Для
этих же целей широко используются системы из трех или пяти квадруполей.
К недостаткам анализаторов этого типа можно отнести не самый широкий
диапазон масс (до m/z 3000-4000), низкую разрешающую способность и плохие
результаты при работе в режиме МЛДИ.
6.6. Ионная ловушка
Ионная ловушка была впервые описана одновременно с квадрупольным
анализатором, однако первый коммерческий прибор появился на рынке только в
1983 г. [209]. Тем не менее к настоящему времени ионная ловушка
превратилась из простого детектора к газовому хроматографу в высокоэффективный
хроматомасс-спектрометр с диапазоном регистрируемых масс в десятки тысяч
дальтон, разрешающей способностью до 25 000, возможностью работы в режиме
тандемной масс-спектрометрии с регистрацией до 10 поколений фрагментных
ионов и высочайшей чувствительностью.
Основой прибора являются три электрода (рис. 6.9): два концевых
(полюсных) гиперболической формы, обычно имеющие потенциал Земли, и один
кольцевой электрод между ними, на который подается радиочастотное напряжение,
обычно мегагерцового диапазона. Эта система электродов создает некое
подобие ячейки, используемой в методе ионно-циклотронного резонанса (разд. 6.4).
Ловушка может удерживать ионы достаточно долгое время. Для ионизации
образца используют электронный удар или химическую ионизацию. Работа
ведется в импульсном режиме. Например, импульсная подача электронов в
ловушку @,1-10 мс) вызывает ионизацию необходимого числа молекул образца.
Образовавшиеся ионы какое-то время удерживаются полем центрального
электрода. В классическом варианте нестабильного масс-селективного сканирования
импульсное изменение амплитуды радиочастотного напряжения, приложенного
к кольцевому электроду, заставляет ионы с определенным значением m/z
переходить на нестабильные траектории и покидать ловушку, попадая на электронный
умножитель. В результате генерируется масс-спектр.
6.6. Ионная ловушка 137
Ввод образца
ш
Катод
— Фокусирующая линза
Запирающий электрод
Полюсный электрод
Центральный кольцевой
электрод
N. \Умножитель
Полюсный электрод
Рис. 6.9. Принципиальная схема ионной ловушки
Вариантом этого метода является резонансное извлечение. В обычных
условиях работы ионы в зависимости от величины m/z имеют характеристические
частоты движения. Эти частоты зависят от многих параметров. Можно создать
условия резонанса для конкретных ионов благодаря дополнительному
радиочастотному сигналу, приложенному к концевым электродам. В условиях резонанса
ионы поглощают достаточную энергию для того, чтобы покинуть ловушку, после
чего они регистрируются внешним детектором. В этом случае ионы можно
извлечь из ловушки, приложив более низкое напряжение, чем в классическом
варианте. Основным результатом является возможность расширения диапазона
регистрируемых масс. Например, прибор с верхним диапазоном масс 650 Да
при работе в обычном режиме может регистрировать соединения с массой до
70 000 Да в режиме резонансного извлечения ионов.
Возможность удаления мешающих ионов на разных стадиях ускорения и
замедления движения ионов, индуцирование фрагментации ионов при их
столкновениях с атомами гелия позволяют успешно использовать ионные ловушки
для работы в режиме тандемной масс-спектрометрии, причем можно получить
138 Глава 6. Разделение и регистрация ионов
информацию о нескольких последовательных поколениях фрагментных ионов.
Такая техника зачастую обозначается в литературе (MS)W. Показана также
возможность достижения разрешающей способности 25 000 [209]. Результатам
исследований с использованием ионных ловушек посвящены два номера
журнала масс-спектрометрического общества США за 2002 г. [210, 211].
К достоинствам ионных ловушек следует добавить также небольшие размеры
и самую низкую среди масс-спектрометров стоимость прибора. К недостаткам
можно отнести протекание в ловушке ионно-молекулярных реакций, что
приводит к искажениям стандартного масс-спектра. В результате использование
стандартных компьютерных библиотек NIST и Wiley для идентификации соединений
по масс-спектрам оказывается менее эффективным, чем в случае магнитных или
квадрупольных анализаторов.
6.7. Времяпролетный анализатор
Этот тип анализатора масс [212] основан на простейшем принципе: скорость
разогнанных ионов обратно пропорциональна их массам (уравнение 6.11).
eV = mv2/2 или m = 2eV/v2 F.11)
где К —ускоряющее напряжение
Если ионы движутся в полой трубе, то места регистрации они достигают в
порядке увеличения своей массы. Анализатор такого типа крайне прост и дешев,
а его диапазон масс практически не лимитирован. Однако широкому
использованию времяпролетных приборов мешала низкая разрешающая способность и
невозможность их применения с непрерывными методами ионизации.
Время достижения ионом детектора можно рассчитать по формуле 6.12:
'="Ш (б12>
Для разности потенциалов 3 кВ и длины трубы дрейфа 0,5 м ион с m/z 500
достигает детектора за 15 мкс, а ион с m/z 50 —за 4,6 мкс, т.е. масс-спектр
в диапазоне от 50 до 500 Да можно зарегистрировать за время около 10 мкс.
Понятно, что время можно увеличивать, уменьшая разность потенциалов или
увеличивая длину дрейфа.
Основные проблемы времяпролетных приборов — это разброс ионов с одной
массой по времени, по скорости (энергии) и в пространстве. На рисунке 6.10
представлена схема линейного варианта времяпролетного анализатора,
предложенная в пионерской работе Мак-Л арена и Уайли [212]. Ионы, образовавшиеся
в левой части источника, ускоряются разностью потенциалов между стенкой
источника и сеткой по направлению к детектору. В области дрейфа происходит
6.7. Времяпролетный анализатор 139
V
К
*
о
1 h
х
л
8
о
Is
О
о
о
О
о
о
о
о
О
Область дрейфа
О
о
о
о
Оо
О °
о
?
?
*
?
s
Рис. 6,10. Схема линейного времяпролетнога масс-спектрометра
разделение ионов по скоростям (следовательно, по массам). Детектор
регистрирует интенсивность пучков изобарных ионов. В результате генерируется масс-
спектр. Следует отметить, что на детектор поступают все ионы, что существенно
улучшает чувствительность метода по сравнению со сканирующими
анализаторами, в которых детектора достигает фактически менее одного процента ионов.
Разброс по энергиям связан с тем, что ионы уже изначально (до ускорения)
имеют различную кинетическую энергию ??/. В результате полная энергия
движения ионов в области дрейфа будет (eV + AU).
Пространственный разброс связан с тем, что ионы в источнике находятся не
на одной линии, а занимают конкретный объем. Часть их оказывается ближе
к сетке, часть — дальше. Если обозначить это отклонение в пространстве от
среднего значения As, и, учитывая, что V = Es, получается, что энергия ионов в
области дрейфа равна eE(s ± As). Таким образом, происходит уширение сигнала
и, соответственно, потеря разрешения.
Прибор, представленный на рис. 6.10, не приспособлен для работы в
сочетании с классическими непрерывными методами ионизации органических
молекул (ЭУ, ХИ, полевая ионизация, электрораспыление), поскольку в этом
случае разброс во времени появления ионов бесконечен, что означает нулевую
разрешающую способность. Тем не менее использование импульсных методов
генерирования ионов (лазерная десорбционная масс-спектрометрия, масс-спек-
трометрия вторичных ионов и др.) позволяет достаточно эффективно работать
на линейных времяпролетных приборах. В этом случае время генерирования
ионного пучка ограничивается временем импульса, т. е. разброс ионов по
времени имеет конечное значение, причем, чем короче импульс, тем меньше разброс.
Указанные методы ионизации, а также МЛДИ, связаны с генерированием ионов
с поверхности. В результате помимо разброса образования ионов во времени
одновременно устраняется и пространственный разброс, наблюдающийся при
использовании методов ионизации молекул в газовой фазе. Для устранения
разброса по энергиям и повышения разрешающей способности линейных
анализаторов было предложено использование двух сеток, а также метод задержки
извлечения ионов из источника импульсной подачей напряжения [212]. Однако
140 Глава 6. Разделение и регистрация ионов
Источник
Область дрейф;
I I I I I I I I
Детектор
eV+U0
Рефлектрон
I I I I I I I
Рис, 6.11. Схема времяпролетного масс-спектрометра с рефлектроном
революционным решением стало использование отражателя (рефлектрон,
ионное зеркало), предложенное в 1973 г. Б. А. Мамыриным [213].
Схема времяпролетного масс-спектрометра с рефлектроном представлена на
рис. 6.11. Из группы ионов с одной массой частицы с большей кинетической
энергией проникают глубже внутрь отражающего поля рефлектрона, тем самым
затрачивая больше времени на замедление и последующее ускорение. Первыми
достигая ионного зеркала, они последними покидают его и догоняют более
медленные ионы в фокальной плоскости, в которой располагается детектор.
Такой принцип позволил проводить корректировку разброса по энергиям и
повысить разрешающую способность времяпролетных приборов до 10000.
Еще одним замечательным нововведением в масс-спектрометрию было
использование принципа ортогонального ускорения (рис. 6.12). Благодаря этому
методу удалось состыковать времяпролетный анализатор с непрерывными
источниками ионов. Фактически одновременно А. Ф. Додоновым в России [214]
и М. Гилхаусом в Австралии [215] были сконструированы масс-спектрометры с
ортогональным ускорением в сочетании с электроспреем и электронным ударом
Выталкивающий электрод
|.° ?·« 8'ае'аЛу·.* о·.· а'« 0»·?
Детектор
б
Выталкивающий электрод
Детектор
Рис. 6.12. Принципиальная схема метода ортогонального ускорения: а — выталкивающий
электрод выключен, б — выталкивающий электрод включен
6.8. Детектирование ионов 141
соответственно. В приборах этого типа импульсное приложение потенциала к
линзе, расположенной параллельно движению ионов от источника к детектору,
ускоряет сегмент коллимированного пучка ионов в направлении
перпендикулярном их движению. В итоге ионы достигают второго детектора в соответствии
со своей массой. После того как самые тяжелые ионы достигнут детектора,
ортогональный импульс может быть повторен. В этом случае разброс в скоростях
в ортогональном направлении отсутствует, а пространственный разброс невелик
и определяется только шириной пучка коллимированных ионов.
Важным достоинством времяпролетного анализатора является значительно
более высокая чувствительность по сравнению со сканирующими приборами.
Все ионы, образовавшиеся в результате импульса ионизации или оказавшиеся в
зоне действия импульса ортогонального ускорения, достигают детектора.
Следующий импульс можно осуществить, как только самый тяжелый ион достигнет
детектора. В реальной ситуации, например в экспериментах с ортогональным
ускорением, детектируется один из семи ионов каждой массы, образовавшихся
в источнике.
Для сканирующих приборов, напротив, характерно одномоментное
пропускание только ионов с конкретной массой. Например, если за одну секунду
сканируется диапазон масс от 10 до 1000 Да, то ионы с конкретной величиной
m/z достигают детектора лишь в течение 0,001 с, т. е. детектируется 0,1%
образовавшихся в источнике ионов. Таким образом, выигрыш в чувствительности при
переходе от сканирующего к времяпролетному анализатору составляет примерно
два порядка.
Резюмируя вышеизложенное, следует отметить следующие достоинства масс-
спектрометра с времяпролетным анализатором:
- более высокая чувствительность по сравнению со сканирующими приборами;
- очень высокая скорость записи спектра (до нескольких сотен спектров в
секунду);
- практически неограниченный диапазон масс;
- разрешающая способность более 10000;
- самые разнообразные источники ионов;
- идеальный второй анализатор для работы в режиме тандемной масс-спектро-
метрии (разд. 7.6);
- небольшие размеры.
6.8. Детектирование ионов
Последним звеном получения масс-спектра является процесс детектирования
ионов. Токи, создаваемые незначительным количеством ионов с одним
значением m/z, слишком малы A0_9-10~17 А) для непосредственного измерения.
Поэтому после прохождения анализатора пучок ионов направляется на специальное
142 Глава 6. Разделение и регистрация ионов
Ионы
140Г ф#°
Электроны
Рис, 6.13. Принципиальная схема электронного умножителя
устройство (умножитель), которое должно перевести поток ионов в адекватный
усиленный поток фотонов или электронов. Наиболее популярный электронный
умножитель представляет собой систему бериллиево-медных динодов, причем
потенциал каждого последующего несколько выше, чем предыдущего (рис. 6.13).
Попадая на первый динод, ионы выбивают с его поверхности эквивалентное
количество электронов. Эти электроны под действием приложенных к динодам
увеличивающихся потенциалов ускоряются для последующих столкновений с
поверхностью на каждой ступени динодной цепи, приводя к лавинообразному
возрастанию эмиссии электронов. Последний динод соединен с предусилите-
лем, который преобразует ток в напряжение в форме, удобной для цифровой
компьютерной записи. Коэффициент усиления такой системы может превышать
106, а срок службы составляет 1-2 года. Небольшой срок службы связан с
загрязнением поверхности в результате взаимодействия с ионами, а также с
работой в условиях невысокого вакуума. В некоторых приборах пучок ионов
ускоряется перед первым динодом электростатическим полем. Увеличение
энергии ионов приводит к увеличению выхода вторичных электронов, следовательно,
к увеличению чувствительности.
Фотоумножитель основан на принципе регистрации фотонов (рис. 6.14).
Поток ионов направляется на конверсионный динод и вызывает эмиссию вто-
Конверсионный динод
Экран
шФотоны
Ионы
? Фотоумножитель
Рис. 6.14. Принципиальная схема фотоумножителя
6.8. Детектирование ионов 143
Ионы
Детектор
mt ч-
Рис. 6.15. Схема решеточного детектора
ричных электронов. Электроны направляются на сцинтилляционный экран, где
происходит эмиссия фотонов, которые далее попадают на каскадный
умножитель, аналогичный описанной выше динодной системе. Усиленный поток
фотонов регистрируется. Обладая таким же коэффициентом усиления, что и
электронный умножитель, фотоумножитель имеет срок службы до 5 лет, так как
фотоны в отличие от электронов проходят через стекло, т. е. умножитель может
функционировать в запаянной трубке в условиях глубокого вакуума.
Масс-спектрографы исторически предшествовали масс-спектрометрам. В их
конструкции для регистрации ионов использовали фотографическую пластинку.
Количество ионов каждого типа устанавливали по интенсивности засветки. К
сожалению, и разрешение такой системы оставляло желать много лучшего, и
интенсивность засветки была не прямо пропорциональна количеству ионов.
Решеточный детектор представляет собой пластинку с просверленными в
ней цилиндрическими отверстиями диаметром 15-20 мкм (рис. 6.15). Расстояние
между центрами отверстий составляет 10-30 мкм. Сторона входа пластинки
имеет отрицательный потенциал (~1 кВ) относительно стороны выхода. Внутренняя
поверхность каналов покрыта полупроводниковым материалом, благодаря
которому осуществляется эмиссия вторичных электронов. Коэффициент умножения
такого устройства равен 105. При использовании 2-3 пластин можно повысить
его до 108. Выходя из канала, поток вторичных электронов попадает на анод, и
сигнал передается на процессор. Такое устройство напоминает фотографическую
пластинку. Ионы с разной величиной m/z при сканировании параметров
анализатора направляются на разные каналы и могут детектироваться одновременно.
Решеточный детектор, безусловно, более чувствителен, однако и стоимость
его выше. В связи с этим до настоящего времени электронный и
фотоумножители используются чаще.
Глава 7
ТАНДЕМНАЯ МАСС-СПЕКТРОМЕТРИЯ,
МС/МС
Одним из недостатков прямого ввода образца в источник является искажение
спектра вещества за счет примесей, присутствующих в образце. Возникают
лишние пики, порой весьма интенсивные, особенно если летучесть примеси
больше, чем летучесть основного компонента. Недостатками хроматомасс-
спектрометрии являются длительное время анализа и невозможность
прохождения через колонку некоторых компонентов. Мягкие методы ионизации (ХИ,
ПИ, электрораспыление) характеризуются интенсивными пиками молекулярных
ионов, однако отсутствие фрагментации затрудняет установление структуры
соединений. Одним из вариантов решения всех этих проблем стало использование
тандемной масс-спектрометрии. В настоящее время эта техника применяется
со всеми описанными выше анализаторами и используется для анализа самых
разных классов соединений. Тандемной масс-спектрометрии посвящено много
обзоров и монографий. Наиболее полно метод представлен в книге [216].
Современный масс-спектрометр может работать по алгоритму обычной
химической аналитической лаборатории, когда смесь химических соединений
сначала разделяется на компоненты, а затем устанавливаются структуры этих
компонентов (рис. 7.1).
? Смесь молекул
Г Источник ионов
? Смесь ионов
| Анализатор I. Выбор иона |
Камера соударений.
Фрагментация иона Mi*
I Смесь дочерних ионов
? Fq ,F/2 ...
I Анализатор II. Разделение ионов |
I
Детектор
? ,
Масс-спектр
Рис. 7.1. Принципиальная схема метода тандемной масс-спектрометрии
7.1. Активация ионов 145
Смесь ионизируется мягким методом. Образовавшиеся молекулярные ионы
по очереди проходят через первый анализатор. В бесполевом пространстве их
внутреннюю энергию повышают определенным образом, что вызывает
фрагментацию. Второй анализатор позволяет получить масс-спектр индивидуального
соединения. Практически все стадии этой процедуры очевидны. Исключение
составляет инициирование процесса фрагментации.
7.1. Активация ионов
Тот факт, что не все ионы, покинувшие источник, достигают детектора, известен
с момента «рождения» масс-спектрометрии. Речь не идет о неизбежной гибели
ионов на стенках прибора по пути следования. Дело в том, что внутренняя
энергия части ионов оказывается недостаточной для того, чтобы распасться в
источнике, но достаточной, чтобы начать фрагментировать во время движения
от источника к детектору. Такие ионы называются метастабильными. Они были
подробно рассмотрены в разд. 2.3. Внутренняя энергия иона может увеличиться
в результате столкновения с атомами атмосферных газов, обусловливающих
остаточное давление в приборе, или при столкновениях с металлическими
поверхностями. Пики таких ионов были зарегистрированы Астоном еще в 1919 г.
[217]. Долгое время эти пики рассматривались исключительно как помеха. Они
искажали реальный спектр и ухудшали разрешение. Однако позже [218] было
показано, что пики метастабильных ионов, а также пики, обусловленные ионами,
образовавшимися вне источника ионов в результате активации их
предшественников соударением, могут успешно использоваться для проведения структурных
исследований.
Первой ступенью тандемной масс-спектрометрии является повышение
внутренней энергии ионов, направленных из источника в анализатор. Для этой цели
используется ряд подходов.
7.1.1. Активация соударением или диссоциация,
индуцированная столкновениями
(Collisional Activation, CA; Collision-Induced Dissociation, CID)
Активация соударением, впервые целенаправленно осуществленная на
секторных магнитных приборах, основана на том, что разогнанные ускоряющим
напряжением ионы имеют высокую кинетическую энергию F-8 кэВ). В результате
их столкновений с атомами инертного газа в специальной камере
соударений, помещенной в бесполевом пространстве, небольшая часть кинетической
энергии переходит во внутреннюю. Во избежание потерь камера соударений
располагается в фокальной плоскости. Время столкновения иона с молекулой
длится 10~14-10-16 с. Приобретенной ионом внутренней энергии может ока-
146 Глава 7. Тандемная масс-спектрометрия, МС/МС
заться достаточно для его распада по одному или нескольким направлениям.
Число возможных процессов, инициируемых соударениями, весьма велико. На
схеме 7.1 представлено несколько наиболее вероятных из них.
т\ —> т? + т®
т~\ —> т^+ + е~
т\ —> т\ + 2е~
т\ —> т\ + т® + 2е~
Схема 7.1
Все эти процессы, а также ряд других, действительно протекают в камере
соударений. Вероятность того или иного процесса зависит от многих параметров,
включая кинетическую энергию ионов т+, природу газа в камере, давление
в камере, сечение реакции. Наиболее распространенной является первая из
представленных реакций. Ее сечение значительно выше по сравнению с другими
процессами.
Уравнение 7.1 связывает величину приобретаемой ионом в процессе
соударения энергии с характеристиками эксперимента:
Еы = EkinTz—г-г}—, G.1)
Mion+Mgas
где Ein — приобретаемая ионом внутренняя энергия, Е^п — энергия
поступательного движения иона, Mgas — молекулярная масса газа, используемого в камере
соударений, М{0п — масса иона.
Важнейшим параметром является кинетическая энергия родительского иона.
Подавляющее большинство работ выполнено на ионах с высокой энергией
F-8 кэВ) на секторных приборах или с низкой энергией A0-100 эВ) на квад-
рупольных приборах. Значительно реже используются ионы с промежуточными
величинами энергий. Как видно из уравнения 7.1, с уменьшением кинетической
энергии уменьшается и приобретенная ионом внутренняя энергия. Спектры
одного и того же предшественника с разными энергиями могут существенно
отличаться. Как и в случае уменьшения энергии ионизирующих электронов
(разд. 2.1), уменьшение энергии предшественника в экспериментах МС/МС
приводит к тому, что ряд возможных направлений распада не осуществляется.
Доминируют же в спектре пики, обусловленные наиболее энергетически
выгодными процессами. Таким образом, спектры высоких и низких энергий могут
дополнять друг друга при выполнении структурных исследований.
Тем не менее следует отметить, что эффективность соударений в случае
системы трех квадруполей (Еып < 100 эВ) оказывается значительно выше, чем
можно было ожидать (уравнение 7.1). Это связано с высокой фокусирующей
7.1. Активация ионов 147
способностью квадруполя, работающего в радиочастотном режиме, по
сравнению с камерой соударений в секторных приборах, и большей длиной квадруполя,
приводящей не только к единичным, но и многоразовым столкновениям.
Природа газа для соударений также существенно влияет на процесс
фрагментации (уравнение 7.1). Показано [219], что использование более тяжелого газа
приводит к увеличению эффективной энергии столкновений, т. е. чем тяжелее
газ, тем большую структурную информацию можно получить. Особенно это
важно для квадрупольных приборов и при исследовании органических молекул
с массой более 1000 Да.
Значительное влияние на процессы активации соударением оказывает
давление в камере. Обычно используется давление около Ю-2 мм рт. ст., но возможны
любые другие значения в зависимости от задачи эксперимента. Понятно, что
увеличение давления ведет к увеличению доли фрагментных ионов в результате
увеличения вероятности хотя бы одного столкновения родительского иона с
молекулой газа. Кроме того, увеличивается вероятность двух и более
столкновений. Классическая камера соударений имеет длину ~1 см, причем выходными
отверстиями для газа служат отверстия для входа и выхода ионов. Такая
конструкция, хотя и решает основную задачу, делает область соударений размытой,
что затрудняет однозначное определение параметров столкновений. В двух
модификациях [220, 221] использован поток инертного газа в перпендикулярном
направлении к пути следования ионов. Такой подход привел к улучшению
результатов эксперимента.
Инициировать фрагментацию иона можно по-другому. На сегодняшний день
помимо активации соударением используется фотодиссоциация, активация
электронами и поверхностно-индуцированная диссоциация. Эти методы
используются реже, однако их возможности еще недостаточно изучены, и, может быть, в
будущем они окажутся более востребованными.
7.1.2. Фотодиссоциация (Photodissociation)
Фотоны — удобные частицы для повышения внутренней энергии иона и
инициирования его фрагментации [222]. Преимуществами фотодиссоциации над
активацией соударением являются простота варьирования энергии фотонов при
использовании источников с разной длиной волны и полная передача энергии
фотона иону. Кроме того, можно использовать двухфотонное и мультифотонное
взаимодействие. Однако сечения реакций взаимодействия ионов с фотонами на
2-4 порядка ниже, чем при столкновениях ионов с молекулами газа. Поэтому для
эффективного применения этого метода необходимо либо использовать мощные
потоки излучения (лазеры), либо увеличивать время взаимодействия (масс-
спектрометрия с преобразованиями Фурье, ионная ловушка). В случае
квадрупольных или магнитных приборов техническое обеспечение взаимодействия
ионов с фотонами приводит к определенным инструментальным сложностям.
148 Глава 7. Тандемная масс-спектрометрия, МС/МС
Активация электронами (Electron Excitation) имеет низкие сечения реакции
и к тому же лишена преимуществ фотонов. В связи с этим применение
электронов весьма ограничено [223].
7.1.3. Поверхностно-индуцированная диссоциация
(Surface-Induced Dissociation, SID)
В этом случае поток разогнанных ионов направляется на металлическую
поверхность [224]. В результате неупругого столкновения часть ионов
нейтрализуется, часть отражается, а часть в результате повышения внутренней энергии
распадается по ряду возможных направлений. Отраженные родительские и
образовавшиеся фрагментные ионы движутся под прямым углом к исходному
пучку ионов и могут быть зарегистрированы. Преимуществами метода являются
отсутствие дифференцированной откачки газа и инструментальных сложностей,
связанных с использованием активации фотонами или электронами. Сечение
реакции достаточно высоко, хотя и уступает активации соударением. Этот метод
активации распада является наиболее перспективным.
7.2. Анализаторы ионов
в тандемной масс-спектрометрии
Практически все виды анализаторов, описанные в гл. 6, используются для
экспериментов тандемной масс-спектрометрии.
7.2.1. Система трех квадруполей
Наиболее наглядно продемонстрировать возможности МС/МС можно на примере
прибора с системой трех квадруполей. Принципиальная схема такого масс-
спектрометра представлена на рис. 7.2, а, а варианты работы на таком приборе —
на рис. 7.2, б-г.
Предположим, что в ионный источник этого прибора посредством прямого
ввода ввели смесь нескольких соединений с разными молекулярными массами
и ионизировали их каким-либо мягким методом, например химической
ионизацией. В результате в ионном источнике образуются устойчивые ионы М/Н+.
Если сканировать напряжение на одном из квадруполей, а другие использовать в
режиме пропускания всех ионов, получится классический масс-спектр, в котором
присутствуют только пики ионов МгН+.
Настроим первый квадруполь на пропускание ионов с массой М]Н. Все ионы
с другими массами погибают на стенках этого квадруполя. Второй квадруполь
будем использовать в качестве камеры соударений. Этот квадруполь настроен
на пропускание всех ионов, причем в него подается инертный газ, чаще всего
7.2. Анализаторы ионов в тандемной масс-спектрометрии 149
Источник
ионов
>
Qi
Q2
Камера
соударений
ь
Q3
ь
Детектор
Q1
Выбор
иона
А|
?
Q2
Камера
соударений
А+ А +
?
Q3
Разделение
Qi
Сканирование
W
Q2
Камера
соударений
?
Q3
Пропускание
ионов At
Qi
Сканирование
^
Q2
Камера
соударений
ъ
Q3 1
Сканирование с
ПОСТОЯННОЙ
разшщей в m/z
otQI I
Спектр дочерних
ионов для ??
Спектр
> родительских
ионов для At
Спектр ионов,
отщепляющих
> идентичные
нейтральные
частицы
Рис. 7.2. а — принципиальная схема тройного квадрупольного масс-спектрометра, б — схема
эксперимента для получения спектра дочерних ионов, в —схема эксперимента для
получения спектра родительских ионов, г — схема эксперимента для получения спектра ионов,
распадающихся с выбросом идентичных нейтральных частиц
гелий или аргон, для создания давления ~10~2 ммрт. ст. Прошедшие через
первый квадруполь ионы ???+ сталкиваются во втором с молекулами газа,
и за счет перехода части кинетической энергии приобретают дополнительную
внутреннюю энергию. Этой энергии может оказаться достаточно для
инициирования процессов фрагментации, в результате чего из второго квадруполя помимо
исходных ионов выходят и осколочные ионы, образовавшиеся при распаде части
ионов ???+. Обычное сканирование третьего квадруполя позволяет получить
спектр первого соединения, точнее спектр его молекулярного иона, содержащий
информацию и о молекулярной массе, и о массе основных структурных
фрагментов. Далее через первый квадруполь пропускают молекулярные ионы второго
компонента (М2Н4") и получают на выходе из третьего квадруполя его спектр.
Эта процедура повторяется с молекулярными ионами каждого из соединений
смеси. Этот тип спектра (рис. 7.2, б) называется спектром дочерних ионов.
Теперь рассмотрим аналогичную смесь органических соединений, но
несколько видоизменим задачу. Предположим, что среди 30-40 соединений,
образовавших в результате ХИ протонированные молекулярные ионы МгН+,
необходимо определить только фталаты. Наиболее интенсивный пик в спектрах
этих соединений обусловлен ионом с массой 149. Пусть первый квадруполь
работает в обычном режиме сканирования, второй, по-прежнему, выступает в
качестве камеры соударений, а третий пропускает только ионы с m/z 149. Когда
соударениям подвергается молекулярный ион соединения, не образующего при
распаде ион 149, на выходе из третьего квадруполя никакого сигнала нет. Если
150 Глава 7. Тандемная масс-спектрометрия, МС/МС
же в конфетный момент времени через первый квадруполь проходит МН+
фталата, в камере соударений образуется ион 149 и на выходе из третьего квад-
руполя появляется сигнал. Таким образом можно селективно зарегистрировать
только пики протонированных молекулярных ионов фталатов. Этот тип спектра
(рис. 7.2, в) называется спектром предшественников или спектром
родительских ионов.
Другой важной задачей, для решения которой используются спектры
родительских ионов, является установление путей образования конкретного
дочернего иона при проведении структурных исследований.
Третий вид спектров (рис. 7.2, г) связан с потерями нейтральных частиц
с одинаковыми массами. Предположим, необходимо определить карбоновые
кислоты в смеси органических соединений. Используя химическую ионизацию
отрицательных ионов, получают депротонированные молекулярные ионы
компонентов смеси. Второй квадруполь работает в качестве камеры соударений, а
первый и третий действуют в сканирующем режиме, причем в момент, когда
первый пропускает ион с массой А, третий пропускает ион с массой [А—44].
Сигналы в спектре дадут только те ионы, которые распадаются в камере
соударений с выбросом 44 а. е. м. В случае эксперимента с кислотами 44 а. е. м.
соответствуют потере молекулы СОг- Этот процесс характерен для распада
анионов карбоновых кислот и маловероятен для других распространенных
классов соединений.
Спектры ионов, отщепляющих идентичные нейтральные частицы, также
используется в структурных исследованиях.
Безусловно, наиболее распространенным вариантом является запись спектра
дочерних ионов, поскольку в этом случае речь идет о получении чистого
спектра без наложения каких-либо примесей, которые почти неизбежно
ухудшают качество обычного масс-спектра. Помимо очевидного анализа смесей
спектры дочерних ионов широко используются в структурных исследованиях.
Дело в том, что совпадение таких спектров для исследуемого иона и иона
известной структуры является наиболее убедительным, хотя и не абсолютным,
доказательством идентичности структур этих ионов.
Следует ещё раз подчеркнуть, что ускоряющее напряжение в квадрупольных
приборах составляет несколько десятков вольт, т. е. кинетические энергии
родительских ионов невелики, соударения являются низкоэнергетическими, и не все
возможные механизмы фрагментации могут быть задействованы.
7.2.2· Магнитные секторные приборы
Для работы в режиме тандемной масс-спектрометрии необходимо иметь прибор,
по крайней мере, с двумя анализаторами: одним магнитным и одним
электростатическим.
7.2. Анализаторы ионов в тандемной масс-спектрометрии 151
7.2.2.1. Спектр кинетических энергий ионов,
проанализированных по массе (MIKES)
Наиболее популярным вариантом является техника съемки спектров дочерних
ионов MIKES — Mass Analyzed Ion Kinetic Energy Spectrum, т. е. спектр
кинетических энергий иона, проанализированного по массе. Для работы необходим
прибор обратной геометрии (рис. 7.3).
в
?
Камера
соударений
???,?^,???...
w
Источник
ионов
м+,м;,м;...
w
Рис. 7.3. Принципиальная схема работы в режиме MIKES
Рассмотрим снова случай смеси органических соединений, помещенной в
ионный источник и подвергнутой химической ионизации. Магнитный
анализатор последовательно настраивается на пропускание протонированных
молекулярных ионов МгН+. Последовательность анализа можно проследить на примере
первого родительского иона с массой ???. При столкновении с атомами газа
в камере соударений во втором бесполевом пространстве между магнитным
и электростатическим анализатором разогнанные потенциалом в 7-8 кВ ионы
???+ теряют небольшую часть своей кинетической энергии за счет ее перехода
во внутреннюю. Этот процесс инициирует распад ионов по всем возможным
направлениям. Чем выше давление в камере, тем большее число исходных ионов
М/Н+ распадается. Обычно давление составляет ~10~2 мм рт. ст. В этом случае
ток пучка выбранных первичных ионов уменьшается на 10-20%, а пики фраг-
ментных ионов достаточно интенсивны для получения качественного спектра.
Покидая камеру соударений, ионы ???+, а также фрагментные ионы попадают
в электростатический анализатор. Как отмечалось выше, электростатический
анализатор является анализатором энергий. При получении обычного масс-
спектра напряжение на нем жестко связано с ускоряющим напряжением V = ??·
Такой режим позволяет пропускать через анализатор только ионы с кинетической
энергией eV. Ионы с меньшей или большей энергией в стандартных условиях
через электростатический анализатор не проходят.
Кинетическая энергия ионов ?]?+, покинувших источник, составляет:
e^ = (MiH)v?/2 D.2)
В результате соударений идут процессы распада (схема 7.2):
М!Н+-*т+ + т5;
MiH+—> m~l + т°5 и т. д.
Схема 7.2
спектр
>
152 Глава 7. Тандемная масс-спектрометрия, МС/МС
Интенсивность
1804
41
3886
88 91.2
5783
131
6360
144
-^Ч-
7<Ш
159
?, Д
m/z
Рис. 7.4. Спектр MIKE депротонированного молекулярного иона а-диазо-л-метилацетофе-
нона. Число, выделенное курсивом, означает величину напряженности электростатического
поля в вольтах при пропускании данного иона, а число, выделенное жирным шрифтом,
означает массу данного иона (точнее, величину m/z). Двухканальный самописец позволяет
видеть и наиболее интенсивные, и минорные пики в спектре
Образующиеся фрагментные ионы т^, т^ и др., в первом приближении,
движутся в том же направлении и с той же скоростью, что и исходный ион М]Н+.
Кинетическая энергия этих фрагментных ионов равна rri2v\/2> Шау\/2 и т. д. В
стандартных условиях съемки эти ионы не пройдут через электростатический
анализатор. Для того чтобы зарегистрировать эти ионы, необходимо, чтобы
напряжение на этом анализаторе было адекватно их кинетической энергии,
поскольку R = 2V/E0. Поэтому напряжение на анализаторе сканируется от
исходного значения Ео, равного ускоряющему напряжению V, до нуля. Когда
напряжение уменьшается до величины гп2Ео/(М\Н), через анализатор пройдут
ионы т^, образовавшиеся в камере соударений. При напряжении m4?o/(MiH)
будут регистрироваться ионы т^. Таким образом записывается полный спектр
MIKES. По оси абсцисс откладывается напряжение на электростатическом
7.2. Анализаторы ионов в тандемной масс-спектрометрии 153
анализаторе. Поскольку (???)/?? = ГП2/Е2 = ГП4/Е4 = Щ&и зная величины (???),
Ео и Е(9 можно легко вычислить значения Ш2, т^ и т. д. Пример такого спектра
представлен на рис. 7.4.
Разрешение в спектре не превышает 500, поскольку ширина пиков
достаточно велика. Выше говорилось о том, что скорость фрагментных ионов равна
скорости родительского иона. На самом деле это не совсем верно, и фраг-
ментные ионы получают при образовании определенное приращение скорости
(разд. 7.2.2.2).
Разрешение невелико не только по дочерним, но и по родительским ионам,
так как для выделения предшественника используется один магнитный
анализатор. Это положение можно исправить при использовании приборов продленной
или гибридной геометрии (разд. 7.2.5 и 7.2.6).
Понятно, что на приборе прямой геометрии, когда магнитный анализатор
расположен за электростатическим, MIKES невозможен. Тем не менее
существует метод записи спектров дочерних ионов и на таком приборе (разд. 7.2.2.3 —
связанные сканирования).
7.2.2.2. Высвобождение кинетической энергии при распаде
Хотя в использованном математическом описании процесса MIKES было
сделано допущение, что скорости поступательного движения родительского иона,
дочернего иона и нейтрального продукта равны исходной скорости
родительского иона, это лишь упрощение реальной ситуации. Это положение справедливо
только в том случае, когда обратная ионно-молекулярная реакция протекает без
энергии активации.
Рис. 7.5. Формы пиков в зависимости от количества высвобождающейся энергии.
Количество энергии изменяется в ряду а < б < в < г
Обычно в момент распада часть внутренней энергии родительского иона
переходит в кинетическую, причем вектор приращения может быть направлен
в любую сторону, от совпадающей с направлением скорости поступательного
движения распадающегося иона до полностью противоположного. В результате
форма пиков оказывается различной (рис. 7.5) и зависимой от
высвобождающейся энергии. Чем больше энергии выделяется, тем шире пик в спектре.
Для реакции
M+->F++N
154 Глава 7. Тандемная масс-спектрометрия, МС/МС
4140 4160
4180
4200 4220
4240 4260 4280 4300
4320
?,В
Рис. 7.6. Форма регистрации пика для расчета высвобождения кинетической энергии
при отщеплении частицы HN3 депротонированным молекулярным ионом 2-диазо-2-
цианоацетамида [225]
по спектрам MIKES можно вычислить количество высвобождающейся энергии
(уравнение 7.3):
K^eVmFs2 ^
16w/v
где s = /г/2 — полуширина пика в спектре. Измерить полуширину пика
достаточно просто даже вручную. Достаточно несколько раз записать его в узком
диапазоне и усреднить измеренные значения (рис. 7.6). Точность измерения
очень высока.
Величина К (уравнение 7.3) важна не только в качестве самостоятельного
параметра для установления внутренней энергии иона и параметров реакции.
Она может эффективно использоваться в структурных исследованиях. Например,
спектры ЭУ изомерных о-, л*-, л-бромтолуолов и бензилбромида практически
неразличимы. Однако полуширина пика, обусловленного потерей атома брома
молекулярным ионом этих соединений, в спектре метастабильных ионов
составляет 73, 271, 162 и 1,7 мэВ соответственно, т. е. различить эти изомеры не
составляет труда [226].
Если два иона с одной брутто-формулой элиминируют идентичную частицу,
причем полуширина пика, соответствующего этому процессу, одна и та же,
родительские ионы, по-видимому, имеют идентичные структуры. В частности,
идентичная ширина пика, соответствующая образованию иона m/z 66 из де-
протонированных молекулярных ионов 2-диазо-2-цианоацетамида и 4-циано-5-
гидрокси-1,2,3-триазола послужила дополнительным доказательством
изомеризации депротонированного молекулярного иона диазоамида в триазол [225].
7.2. Анализаторы ионов в тандемной масс-спектрометрии 155
7.2.2.3. Связанные сканирования
Техника связанных сканирований включает одновременное изменение двух из
трех основных параметров прибора: напряженности магнитного поля 5,
напряженности электростатического поля ? и ускоряющего напряжения V [216]. Тем
не менее поскольку с изменением ускоряющего напряжения V изменяется и
разрешение, наиболее популярными являются три метода, аналогичные
описанным выше для случая тройного квадруполя и основанные на одновременном
изменении В и Е, связанных определенной зависимостью.
Рассмотрим вариант записи спектра дочерних ионов с использованием
техники связанных сканирований. Камера соударений, в которой протекают процессы
фрагментации исследуемого иона, расположена в этом случае в первом беспо-
левом пространстве, т. е. между источником и первым анализатором. Порядок
расположения анализаторов может быть любым. Этот вид тандемной
масс-спектрометрии помимо структурных исследований очень широко используется в
сочетании с хроматомасс-спектрометрией для получения селективной информации
о компонентах смесей. Дефокусировка прибора относительно стабильных ионов
дает возможность увидеть на хроматограмме только интересующие пики. Такой
вариант эксперимента очень полезен при необходимости установить присутствие
конкретных соединений (даже в виде микропримесей) в сложнейших смесях.
7.2.2.3.1. Спектры дочерних ионов. Предположим, что необходимо записать
спектр дочерних ионов молекулярного иона mf. При столкновении разогнанных
ионов mf с атомами инертного газа в камере соударений будут идти процессы
его распада (схема 7.3):
mf —» mf + т%
mf —> mf + rrP5 и т. д.
Схема 7.3
Настроим прибор на пропускание стабильных ионов mf. В этот момент
электростатический анализатор пропускает все ионы, обладающие полной
кинетической энергией eV, а магнитный анализатор ионы mf, определяемые
уравнением 7.4: ^ ^
m]=B2R2/2V G.4)
Теперь будем уменьшать одновременно В и ? таким образом, чтобы В/Е =
const. Как только величины В и ? будут отличаться от исходных значений Во
и Ео, электростатический анализатор перестанет пропускать любые стабильные
ионы, образовавшиеся в источнике и ускоренные полным напряжением V. Какие
ионы (тх) будет пропускать магнитный анализатор (уравнение 7.5), когда эти
величины уменьшатся в т^/т\ раза? Он будет пропускать ионы mf,
образовавшиеся в камере соударений из mf. Действительно, на основании закона
сохранения энергии кинетическая энергия фрагментных ионов mf составляет
156 Глава 7. Тандемная масс-спектрометрия, МС/МС
em^V/mx, что равносильно тому, если бы эти ионы образовались в источнике
и были ускорены напряжением Vrri2/m\. В этом случае правомерно уменьшить
знаменатель дроби уравнения 7.5 в тг/тх раза.
^B2R2 ^в2*2
m*=^If = -2V- G'5)
ТП\
Разделив уравнение 7.4 на 7.5, получим:
ш\/ш2 = m\/mx G.6)
Таким образом, тх = Ш2, т. е. при изменении 5 в m2/mi раза, через магнитный
анализатор пройдут именно ионы т^, образовавшиеся в камере соударений из
т*. Ионы т^ в этот момент времени проходят и через электростатический
анализатор, так как их энергия равна eVm2/mi, a ? изменилось синхронно с В
в гп2/т\ раза. _
2F mi
?-~??^ GЛ)
mi
В результате фрагментные ионы mj будут регистрироваться прибором. При
уменьшении В и ? в т^/т\ раза будут регистрироваться ионы т^9
образовавшиеся из rnl в камере соударений. Просканировав, таким образом, В и ? от
исходных значений Eq и #о ДО 0, можно получить полный спектр дочерних
ионов, образовавшихся в камере соударений из т^~. На рисунке 7.7 представлен
пример спектра дочерних ионов, полученный методом связанных сканирований.
Видно, что, как и в случае MIKES, разрешение невелико.
7.2.2.3,2. Спектры родительских ионов. Для записи спектра родительских
ионов с помощью метода связанных сканирований необходимо изменять
одновременно В и ? при условии, что В2/Е = const. Пусть требуется установить
все родительские ионы, при распаде которых в камере соударений в первом
бесполевом пространстве образуются фрагменты mf, т. е. зарегистрировать все
реакции распада, представленные на схеме 7.4:
A+-»m+ + mij
Г+ —> т^2 + т® и т. д.
Схема 7.4
Исходно прибор настраивается на пропускание стабильных ионов т^~,
образовавшихся в источнике и ускоренных полным напряжением V. Далее величины
7.2. Анализаторы ионов в тандемной масс-спектрометрии 157
Интенси в ность
6325
155 \7016
172
?,В
mlz
Рис. 7.7. Связанные сканирования. Спектр дочерних ионов (В/Е = const) молекулярного
иона фениламиносульфонамида PhNHSC^NKb. Число, выделенное курсивом, означает
величину напряженности электростатического поля в вольтах при пропускании данного иона,
а число, выделенное жирным шрифтом, означает массу данного иона (точнее величину
m/z). Двухканальный самописец с разной чувствительностью позволяет видеть и наиболее
интенсивные, и минорные пики
В и Е, жестко связанные зависимостью В2/Е = const, синхронно уменьшаются
до нуля. Как только величины В и ? будут отличаться от исходных значений Во
и Eq, прибор перестанет пропускать любые стабильные ионы, образовавшиеся
в источнике и ускоренные полным напряжением V. Когда величины В2 и ?
изменятся в /W2/A раз, магнитный анализатор будет пропускать ионы тх с
кинетической энергией eVmi/A, аналогичные ионам из источника, ускоренным
напряжением Гтг/А.
^2
т*= т2 =т*2 G·8)
2—V
А
158 Глава 7. Тандемная масс-спектрометрия, МС/МС
Как видно из уравнения 7.8, это будут вновь ионы Ш2, но уже образовавшиеся
в камере соударений. Эти же ионы т\ в этот момент времени проходят и
через электростатический анализатор (уравнение 7.9), так как их энергия равна
eVmi/A, a E изменилось синхронно с В2 в W2/A раза.
*-т-»Ь G9)
А
Таким образом, ион /nj, образовавшийся в камере соударений из А, будет
зарегистрирован прибором. При изменении В2 и ? в тг/Ъ раза, будет
зарегистрирован ион т\, образовавшийся в камере соударений из своего предшественника
Б. Просканировав В и ? от исходных значений Eq и Во до нуля, можно получить
полный спектр родительских ионов, образующих в камере соударений ионы
W2. Масс-спектрометр все время регистрирует ионы W2, но образовавшиеся
из разных предшественников, а следовательно, имеющие разные кинетические
энергии.
На рисунке 7.8 представлен пример спектра родительских ионов, полученный
методом связанных сканирований. Разрешение в спектре не превышает
нескольких сотен. Следует, кроме того, отметить, что в спектре часто присутствуют
ложные пики (артефакты). Для большей надежности при установлении
протекания конкретной реакции распада А+ —> mf + m® желательно зарегистрировать
соответствующие пики и в спектре дочерних, и в спектре родительских ионов.
7.2.2.3.3. Спектры выбросов идентичных нейтральных частиц. Спектры
ионов, отщепляющих идентичные нейтральные частицы, используются
значительно реже. Предположим, необходимо установить ионы, которые распадаются"
в камере соударений с выбросом одинаковых частиц. Это может быть
эксперимент по изучению процессов распада молекулярного иона конкретного вещества.
Например, можно установить все фрагментные ионы, способные элиминировать
молекулу СО. Это может быть эксперимент в режиме ГХ-МС, когда в сложной
смеси соединений необходимо установить присутствие веществ определенного
типа. Например, по выбросам 44 Да в спектрах химической ионизации
отрицательных ионов можно зарегистрировать среди многих компонентов смеси только
карбоновые кислоты. По выбросам 35 и 70 Да в спектрах электронного удара
можно селективно установить присутствие полихлорированных соединений, а
по выбросам 18 Да —спирты.
Рассмотрим реакцию распада молекулярного иона М+ с выбросом
нейтральной частицы N и образованием фрагментного иона F+ в первом бесполевом
пространстве.
M+->F+ +N
Электростатический анализатор должен пропускать дочерние ионы.
Учитывая, что в первом приближении vm = vp, получаем, что для пропускания иона
7.2. Анализаторы ионов в тандемной масс-спектрометрии 159
Интенсивность
5878
174
7019
146
ЕВ
m/z
Рис. 7.8. Связанные сканирования. Спектр родительских ионов (В2/Е = const) иона
Ph—NH— N=C=C=0+ (m/z 146), образующегося при распаде N-фениламиносульфонилкарб-
этоксидиазоацетамида С2Н5О—CO—CN2"~~CO—NH—SO2— NHPh. Число, выделенное
курсивом, означает величину напряженности электростатического поля в вольтах при пропускании
данного иона, а число, выделенное жирным шрифтом, означает массу данного иона (точнее
величину m/z)
F+ напряжение на анализаторе должно понизиться до величины ?? (уравнения
7.10 и 7.11).
mUVM
Тогда
е?о =
R
R R
? ?? /им /wn
— = — = = 1 или тм
&0 ?Lq ??? Шм
mN
-?
G.10)
G.11)
G.12)
Магнитный анализатор должен пропускать дочерние ионы т* с кажущейся
массой (см. метастабильные ионы, разд. 2.3), которую можно вычислить по
160 Глава 7. Тандемная масс-спектрометрия, МС/МС
уравнению 7.13.
т =—^ = wF— = — (mM-mN) G.13)
Подставляя для wm величину из уравнения 7.12, получаем:
-?
Е2т^
^(¦-1
G.14)
Подставляя величину кажущейся массы в основное уравнение пропускания
ионов магнитным анализатором (разд. 6.1), получаем:
т* B2R2 Е2тк
Eiz
G.15)
*(-5
Переместив все постоянные величины в правую часть уравнения, получаем:
\ E0J IVrrm
—&— = eW2 = GЛ6)
Осуществляя одновременное сканирование В и ? от J?o и ^о до 0 в такой
зависимости, можно регистрировать только те ионы, которые тяжелее своего
фрагмента на массу N.
Часто используют [216] другое математическое выражение для сканирования
этого типа:
— = const G.17)
[д-г-сад-1]"
2
7.2.2.3.4. Получение карты метастабильных переходов. Этот вид масс-спек-
трометрического эксперимента используется еще реже. Исследователя, как
правило, интересует доказательство протекания конкретной реакции распада или
установление присутствия в смеси определенного соединения. Метод метаста-
бильного картирования подразумевает получение информации о всех реакциях,
протекающих при масс-спектрометрической фрагментации соединения. Карта
представляется в координатах ? — m/z и строится сканированием напряженности
магнитного анализатора от величины Во до нуля для каждого дискретного
значения Е. Часто ее представляют в трехмерном формате. Соответствующим
сечением по карте можно получить спектр любого родительского иона,
дочернего иона или выбросов любых идентичных нейтральных частиц.
Понятно, что метастабильное картирование является весьма трудоемким
занятием, а получаемая информация в большинстве случаев оказывается
избыточной.
7.2. Анализаторы ионов в тандемной масс-спектрометрии 161
7.2.2.3.5. Сканирование ускоряющего напряжения. Метод дефокусировки*
Этот вариант масс-спектрометрического эксперимента для получения спектров
родительских ионов возник одним из первых, однако в настоящее время он
практически не используется, поскольку уступает по эффективности описанным
выше методам. Для регистрации спектра сканируется величина ускоряющего
напряжения. Электростатический анализатор при этом имеет все время
постоянную напряженность, равную исходному ускоряющему напряжению: Ео = V$.
Если фрагментация происходит вне источника, кинетическая энергия
фрагмента оказывается ниже стандартного значения, и он не проходит через
электростатический анализатор. Однако если повысить ускоряющее напряжение,
энергии родительского иона и дочернего иона, образующегося в первом бес-
полевом пространстве, будут больше, и в определенный момент заряженный
фрагмент пройдет через электростатический анализатор. Рассмотрим реакцию,
протекающую в бесполевом пространстве:
Кинетическая энергия любого стабильного иона (образовавшегося в
источнике) равна:
? = ??0 = ^ = ^ G.18)
Для того, чтобы зарегистрировать ион F+, образовавшийся в бесполевом
пространстве, можно, не изменяя напряженность электростатического сектора,
увеличить величину ускоряющего напряжения, т. е. повысить энергию
предшественника. Необходимо, чтобы его (предшественника) скорость выросла до
величины vF согласно уравнению:
eV=^ G.19)
При делении уравнения 7.19 на уравнение 7.18 получаем:
У тм т?У
— = -51 или тм = -?- G·20)
Для получения спектра родительских ионов фрагментного иона F+ прибор
предварительно настраивается на пропускание стабильных ионов F+,
образовавшихся в источнике. Далее увеличивают ускоряющее напряжение. Как только V
будет отличаться от Ко, произойдет дефокусировка стабильных ионов. Любые
стабильные ионы, в том числе и F+, не будут проходить через
электростатический анализатор. Сигнал на детекторе исчезнет и появится вновь, когда
величина V будет соответствовать требуемой по уравнению 7.18 для образования
дочернего иона F+ с необходимой энергией. К сожалению, в отличие от других
техник тандемной масс-спектрометрии, описанных выше, требуется не понижать
разность потенциалов от исходной величины до нуля, а увеличивать ее. Этот
6—1113
162 Глава 7. Тандемная масс-спектрометрия, МС/МС
процесс имеет ограничения. Поэтому полного спектра при большой разнице в
массах ионов М+ и F4" записать нельзя. Кроме того, с изменением V изменяется
и разрешение.
7.2.3. Ионная ловушка
Этот вид анализатора позволяет переходить от обычной масс-спектрометрии к
тандемной без использования дополнительных инструментальных усложнений.
Для получения спектра дочерних ионов необходимо провести три
последовательных операции: выделение родительского иона, инициирование его фрагментации
и регистрацию дочерних ионов. Первая стадия легко выполнима методом масс-
селективной стабильности, когда приложением суммы постоянного и
радиочастотного поля из ловушки удаляются все ионы с массами больше и меньше, чем
у выбранного для анализа иона. Для этой цели можно использовать и метод
резонансного удаления. После того как в ловушке остался только исследуемый ион,
необходимо повысить его внутреннюю энергию, индуцировав тем самым его
фрагментацию. Кинетическая энергия ионов повышается благодаря
резонансному возбуждению, которое осуществляется приложением небольшого (до 5 В)
переменного напряжения к концевым электродам. Частота напряжения должна
соответствовать собственной частоте ионов. При этом амплитуда колебаний
ионов поддерживается на незначительном уровне, чтобы избежать резонансного
удаления ионов. Как и в обычной камере соударений, внутренняя энергия
иона повышается в результате перехода части его кинетической энергии во
внутреннюю в результате столкновений. Это достигается при столкновении иона
во время его движения внутри ловушки с атомами гелия, который подается туда
намеренно во всех режимах работы. Эффективность трансформации исходного
иона может достигать 100%. Образовавшиеся фрагментные ионы
детектируются обычным образом в режиме масс-селективной нестабильности. Следует
отметить, что ионная ловушка позволяет изучать только низкоэнергетические
столкновения.
Достоинством ионной ловушки является возможность проводить не только
МС/МС-эксперименты, но и многостадийные исследования. В этом случае после
генерирования фрагментных ионов один из них выбирается для дальнейшего
изучения, а остальные выводятся из ловушки. С этим ионом проводятся те же
операции по ускорению, инициированию столкновений и записи спектра теперь
уже внучатых ионов изначального предшественника. Эта последовательность
выделения иона и его фрагментации может проводиться многократно. Такая
техника называется (МС)Л. Опубликованы результаты по записи масс-спектров
фрагментов до десятого поколения исходного родительского иона [209]. Следует,
однако, отметить, что чувствительность анализа уменьшается при переходе к
каждому следующему поколению ионов, поскольку из всего набора фрагментов
выбирается лишь один.
7.2. Анализаторы ионов в тандемной масс-спектрометрии 163
7.2.4. Масс-спектрометрия с преобразованиями Фурье
Аналогично ионной ловушке метод масс-спектрометрии с преобразованиями
Фурье позволяет работать в режиме тандемной масс-спектрометрии не за счет
модификации самого прибора, а лишь при изменении последовательности
действий. Выше было описано, что последовательность событий для получения
обычного масс-спектра включает очистку ячейки, ионизацию, возбуждение и
детектирование ионов. Для случая двойной масс-спектрометрии с активацией
соударением возможна следующая последовательность получения спектра
дочерних ионов:
Г отбор родительского иона ]
[ионизация] —> (удаление ионов —>
I с другими значениями т/щ
["очистка
[ячейки
увеличение
кинетической энергии
выбранного иона
Г активация
[соударением
возбуждение
и детектирование
фрагментных ионов
Первые два процесса аналогичны используемым для получения обычного
масс-спектра. Чтобы оставить в ячейке только заданный родительский ион,
необходимо приложить к возбуждающим пластинам радиочастотные импульсы
с определенной частотой и амплитудой для удаления из ячейки всех ионов с
большими и меньшими массами. Существует много вариантов осуществления
такого процесса. В частности, широко используется метод сохраненной формы
волны с обратными преобразованиями Фурье (SWIFT, Stored Waveform Inverse
Fourier Transform), предложенный в середине 80-х XX века Маршаллом [227].
В этом случае параметры возбуждающих импульсов рассчитываются с помощью
обратных преобразований Фурье. В результате приложения таких импульсов все
ионы с более высокими и более низкими собственными частотами удаляются из
ячейки, а ионы с заданной массой получают приращение кинетической энергии
и переходят на большую циклотронную орбиту, т. е. оказываются
подготовленными для следующей процедуры — фрагментации, активированной соударением.
Таким образом, две стадии последовательности выполняются одновременно.
Следующим этапом является инициирование столкновений разогнанных
родительских ионов с молекулами инертного газа. Газ подается в ячейку
импульсным краном в нужный момент времени. Количество газа должно быть
оптимальным для осуществления достаточного числа столкновений, но не привести к
потере родительских ионов в результате радиальной диффузии, которая
заключается в нарушении направления циклотронного движения и может вызвать гибель
ионов, направив их на боковые пластины.
Возбуждение и детектирование фрагментных ионов осуществляется обычным
образом. Определенным недостатком описанного эксперимента является
образование дочерних ионов не в центре ячейки, что ухудшает разрешение в спектре.
Для его преодоления используют различные приемы. Например, хорошие ре-
164 Глава 7. Тандемная масс-спектрометрия, МС/МС
зультаты получены методом поддерживающего офф-резонансного возбуждения
(SORI, Sustained Off-Resonance Excitation). В этом случае [228] возбуждающая
частота находится то в резонансе, то не в резонансе с собственной частотой
родительского иона, т. е. циклотронная орбита иона то расширяется, то сужается.
При этом амплитуда возбуждающего импульса невысока и ион удаляется от
центра ячейки на незначительное расстояние. Напуск в ячейку инертного газа
приводит к большому числу низкоэнергетических столкновений, в результате
которых внутренняя энергия иона повышается, пока не достигает предела, за
которым начинается его распад. Фрагментные ионы образуются практически в
центре ячейки, что повышает эффективность их последующей регистрации.
Как и ионная ловушка, масс-спектрометр с преобразованиями Фурье
позволяет получать спектры многих поколений ионов. Следует, однако, помнить, что
чувствительность метода для каждого следующего поколения ионов оказывается
более низкой, так как на каждой стадии фрагментации происходит удаление
подавляющего числа образовавшихся частиц. Кроме того, удается изучать только
низкоэнергетические столкновения.
7.2.5. Приборы продленной геометрии
Иногда возникает необходимость зарегистрировать не только дочерние ионы
определенного предшественника, но и ионы последующих поколений. Такие
задачи возникают, например, при доказательстве структур ионов, при
составлении схем фрагментации органических соединений. Эти схемы оказываются
полезными для последующей идентификации структурно близких соединений
или их метаболитов. Как было показано выше, эта задача легко выполнима при
использовании ионных ловушек или масс-спектрометров с преобразованиями
Фурье (разд. 7.2.3 и 7.2.4). Однако подобные задачи можно решить на
модифицированных секторных или квадрупольных приборах.
Примером прибора продленной геометрии может служить масс-спектрометр с
пятью квадрупольными анализаторами (рис. 7.9). Первый квадруполь
настраивается на пропускание выбранного иона с массой ??. Второй квадруполь работает
в качестве камеры соударений, где происходит образование фрагментных ионов
Q1
??4
Q2
Ml+,M2+,M3-*
Q3
М2+
Q4
М21+,М22+
Q5
Источник
Детектор
Рис. 7.9. Запись спектра внучатых ионов выбранных предшественников на приборе с
системой пяти квадрупольных анализаторов
7.2. Анализаторы ионов в тандемной масс-спектрометрии 165
Источник
Р01
?
Е1
Р02
I
I I
в
РОЗ
I
I
Е2
Детектор
Рис. 7.10. Принципиальная схема прибора в конфигурации ЕВЕ. РО„ — реакционные области
(камеры соударений)
первого поколения. Третий квадруполь пропускает только тот фрагментный
ион, дальнейший распад которого необходимо проанализировать, например
М^. Четвертый квадруполь вновь выполняет функции камеры соударений, где
происходит образование фрагментных ионов второго поколения, образующихся
исключительно из М^. Работающий в обычном сканирующем режиме пятый
анализатор позволяет записать полный спектр фрагментных ионов,
образовавшихся из MJjj". Такой режим позволяет изучить последовательность превращений:
М+
М+
М++М++М+ + ..
Увеличение числа секторов в магнитных масс-спектрометрах тоже позволяет
проводить более сложные эксперименты. Уже добавление электростатического
анализатора к обычному двухфокусному прибору (система BEE или ЕВЕ,
рис. 7.10) позволяет помимо записи спектров внучатых ионов, как в случае
системы пяти квадруполей, проводить селекцию родительских ионов для записи
спектров дочерних ионов в режимах MIKES или связанных сканирований с
высоким разрешением.
В этом случае используется третья камера соударений (третья реакционная
область, РОЗ). Высокое разрешение по родительским ионам весьма полезно,
когда в источнике образуется большое число ионов, в том числе с одинаковой
целочисленной массой.
Безусловно, наиболее мощным секторным прибором с тремя анализаторами
является масс-спектрометр с конфигурацией ВЕВ (рис. 7.11). Используя третью
камеру соударений (РОЗ), как и в случае описанных выше конфигураций, ЕВЕ
или BEE, можно выделять родительский ион с высоким разрешением, а, исполь-
Источник
Р01
?
В1
Р02
I
I I
РОЗ
I
I
В2
Детектор
Рис. 7.11. Принципиальная схема прибора в конфигурации ВЕВ. РО„ — реакционные
области (камеры соударений)
166 Глава 7. Тандемная масс-спектрометрия, МС/МС
зуя вторую камеру соударений (Р02), можно достигать высокого разрешения по
дочерним ионам.
Существующие коммерческие приборы с четырьмя анализаторами
(конфигурации ЕВЕВ или ВЕЕВ) позволяют получать спектры четвертого поколения
фрагментных ионов, а также иметь высокое разрешение и для родительских, и
для дочерних ионов в случае МС/МС. Теоретически можно наращивать число
секторов неограниченно, но необходимо отметить, что добавление каждого
нового анализатора увеличивает размеры прибора и заметно его удорожает. Кроме
того, чувствительность анализа уменьшается с каждой дополнительной
стадией, поскольку каждый последующий магнитный анализатор пропускает только
один из всех фрагментных ионов, а каждый последующий электростатический
анализатор пропускает только ионы в узком диапазоне кинетических энергий.
Поскольку разброс энергий у фрагментных ионов, образовавшихся в камере
соударений, довольно велик, только часть их проходит через электростатический
анализатор с жестко заданной величиной напряженности поля.
7.2.6. Приборы гибридной геометрии
Как уже отмечалось выше, в экспериментах с активацией соударениями обычно
используется два диапазона кинетических энергий ионов. Секторные приборы
работают в диапазоне 6-8 кВ, а квадрупольные приборы —в диапазоне 50-100 В.
Возможность изучать и высокоэнергетические, и низкоэнергетические
столкновения, а также снизить стоимость приборов достигается при использовании
масс-спектрометров гибридной геометрии. Простейшим представителем этого
класса является прибор, в котором за магнитным анализатором следует квад-
руполь (рис. 7.12).
Использование магнита позволяет получать разрешение до 5000 по
родительским ионам, а замедляющего потенциала—изучать низкоэнергетические или
высокоэнергетические реакции распада в зависимости от того, где происходит
активация. Если задействована реакционная область 1 или 2, изучаются
высокоэнергетические столкновения, а если роль камеры соударений выполняет первый
Источник
Р01
I
в
Р02
I
Замедляющий
потенциал
II |
Q1
Q2
Детектор
Рис. 7.12. Принципиальная схема гибридного масс-спектрометра BQQ. РО„ — реакционные
области (камеры соударений)
7.2. Анализаторы ионов в тандемной масс-спектрометрии 167
Источник
Р01
?
Ускоряющее
напряжение
Р02
?
РОЗ
I
I |li
в
Детектор
Рис. 7.13. Принципиальная схема гибридного масс-спектрометра QEB
квадруполь (реакционная область 3), изучаются низкоэнергетические процессы.
Для записи спектров родительских ионов второй квадруполь настраивают на
пропускание выбранных дочерних ионов и сканируют напряженность
магнитного поля, используя для активации соударением третью реакционную область.
Для записи спектров ионов, выбрасывающих идентичные нейтральные частицы,
одновременно сканируют В и Q, при условии B2/Q = const.
Достаточно интересна конфигурация гибридного прибора QEB (рис. 7.13).
Первым анализатором является квадруполь, и есть возможность изучать и
высокоэнергетические, и низкоэнергетические процессы (реакционные области 1
и 2), а также получать высокое разрешение по дочерним ионам. Прибор может
работать и в режиме МС/МС/МС.
Задача 7.1. Какие варианты спектров можно получить на приборе BEQQ,
принципиальная схема которого представлена на рис. 7.14.
Отличные результаты получены при стыковке квадрупольных или секторных
приборов с времяпролетным анализатором. Варианты конфигурации QTOF,
BTOF, EBTOF, EBETOF (TOF — time of flight, времяпролетный анализатор)
позволяют существенно увеличить чувствительность метода для регистрации
дочерних ионов. Эффект увеличения чувствительности на времяпролетных приборах
по сравнению со сканирующими обсуждался выше. Принципиально ничего
не меняется и в режиме тандемной масс-спектрометрии. Камера соударений
Источник
Р01
?
Р02
I
Замедляющий
PQ3 потенциал
I I
— В ?
?^?-? ?
Q2
Детектор
Рис. 7.14. Принципиальная схема гибридного масс-спектрометра BEQQ. РО„ — реакционные
области (камеры соударений)
168 Глава 7. Тандемная масс-спектрометрия, МС/МС
располагается в этом случае перед времяпролетным анализатором, который
может быть укомплектован системой ортогонального ускорения и рефлектроном.
Выше было описано, как ионные ловушки и приборы ионно-циклотронного
резонанса (масс-спектрометры с преобразованиями Фурье) могут работать в
режиме тандемной масс-спектрометрии с регистрацией нескольких поколений
ионов без дополнительных инструментальных усложнений. Приборы этого типа
могут составлять часть гибридного масс-спектрометра. Как правило,
выделенные предшествующим анализатором родительские ионы направляются в ионную
ловушку или ячейку ионно-циклотронного резонанса, где к ним прикладывается
необходимая последовательность действий по активации, распаду и регистрации
фрагментных ионов второго или последующих поколений.
7.3. Инверсия заряда (Charge Inversion)
Еще один процесс, имеющий место при проведении экспериментов с тандемной
масс-спектрометрией, связан с потерей в результате активации соударением двух
электронов и превращением отрицательных ионов в положительные (схема 7.5).
Первое сообщение о наблюдении такого процесса появилось в 1975 г. [229].
Авторы зарегистрировали появление положительных ионов в результате активации
соударением анион-радикалов ряда ароматических соединений.
ABXY" + ? -»????+ + N + 2е"
Схема 7,5
Энергия, необходимая для такого процесса (схема 7.5), равна сумме энергий
сродства к электрону и ионизации молекулы (уравнение 7.21):
? = СЭ(АВХУ) + ЭИ(АВХУ) G.21)
В зависимости от приобретенной в результате соударения внутренней энергии
образовавшиеся положительные ионы могут достичь детектора без распада или
фрагментировать. Спектры инверсии заряда во многом напоминают спектры
активации соударением соответствующих ионов, полученных в результате
электронного удара. Существенным отличием является значительное уменьшение
роли перегруппировочных процессов. Это связано с крайне коротким временем
жизни таких инвертированных ионов (~10~~15 с). В этих условиях константы
скоростей реакций простого разрыва связей существенно превосходят константы
скоростей перегруппировочных процессов.
Метод инверсии заряда используется для решения трех задач:
1. Интенсификация процессов фрагментации для получения структурной
информации. В качестве примера на рис. 7.15 представлены спектры активации
7.3. Инверсия заряда 169
Рис. 7.15. Спектры активации соударением (а) и инверсии заряда (б) отрицательных
ионов с m/z 66, образовавшихся из депротонированного молекулярного иона 2-диазо-2-
цианоацетамида
соударением и инверсии заряда отрицательных ионов с m/z 66,
образовавшихся из депротонированного молекулярного иона 2-диазо-2-цианоацетамида.
Очевидно, что во втором случае спектр значительно информативнее [225].
2. Генерирование катионов с необычными структурами. Например, инверсия
заряда енолят-аниона ацетальдегида приводит к соответствующему катиону,
который в обычных условиях нестабилен и перегруппировывается без
энергии активации в ацетил-катион [230].
3. Генерирование и установление структур нейтральных частиц, образующихся
в результате потери анионом первого электрона. Фактически речь идет о
нейтрализации-реионизации (разд. 7.4), однако нейтрализуется
отрицательный ион, а в результате реионизации образуется положительный ион [231].
170 Глава 7. Тандемная масс-спектрометрия, МС/МС
7.4. Нейтрализация—реионизация
(Neutralization—Reionization Mass Spectrometry, NRMS)
Если на пути следования пучка ускоренных ионов поместить камеру соударений,
помимо процессов фрагментации протекают и процессы перезарядки (см. выше).
В частности, положительные ионы можно превратить в нейтральные частицы.
Для активации такого процесса в камере соударений применяются атомы ксенона
или пары металлов (Na, Hg). Использование металлов с очень низкой энергией
ионизации, хотя и увеличивает выход нейтральных частиц, однако их внутренняя
энергия оказывается слишком высокой и вызывает распад. Кроме того, пары
металлов быстро загрязняют прибор. В настоящее время для осуществления
нейтрализации используют самые разнообразные вещества. Чем ближе энергии
ионизации этих веществ к энергии ионизации образующейся частицы, тем
выше вероятность того, что эта частица «выживет» в течение эксперимента.
Хороших результатов удалось добиться при использовании диметилдисульфида,
аминов, монооксида азота. Для нейтрализации отрицательных ионов чаще всего
используется кислород [232].
В результате нейтрализации в камере образуются нейтральные молекулы
и радикалы. Считается, что нейтрализация является вертикальным по
Франку—Кондону процессом, т. е. образующиеся нейтральные частицы сохраняют
структуры исходных ионов. На выходе непрореагировавшие ионы отклоняются
специальным электродом, а нейтральные частицы продолжают следовать по
начальной траектории и оказываются во второй камере соударений (рис. 7.16),
где при столкновении с молекулами газа (например, смесь Не/Ог) вновь
происходит их ионизация путем перезарядки. В результате этого процесса наряду
с ионизацией происходит и фрагментация. Образующиеся ионы анализируются
обычным образом. Этим методом генерируются и исследуются нейтральные
частицы, живущие определенный промежуток времени [233-238]. Надо отметить,
что зачастую они находятся в возбужденных состояниях. Обычно время между
процессами перезарядки составляет около 10_6 с. Многие ионы в газовой фазе
имеют нетривиальные структуры, и таким образом можно получать нейтральные
частицы с такими же структурами. Если нестабильная в других
экспериментальных условиях или неизвестная ранее частица получена в масс-спектрометре, она
может образовываться (быть интермедиатом) и в других условиях: при
протекании газофазных (межзвездные процессы) реакций и реакций в конденсированной
фазе (каталитические процессы).
Метод нейтрализации—реионизации обычно осуществляется на секторных
приборах в условиях высокоэнергетических столкновений. В случае квадруполь-
ных приборов исходные ионы должны дополнительно ускоряться, а конечные
ионы — замедляться перед их разделением. Первичный пучок ионов должен
быть достаточно мощным, поскольку общий выход процесса в целом составляет
менее 0,1%.
7.4. Нейтрализация—реионизация 171
М,+
Камера соударений 1
Хе
Камера соударений 2
Не/02
мл м12+ м13+ м1п+
АНАЛИЗАТОР
Рис. 7.16. Принципиальная схема эксперимента нейтрализации—реионизации
Спектры нейтрализации—реионизации оказываются сложнее спектров
активации соударением. Это вызвано тем, что фрагментируют и нейтральная частица,
и образовавшийся в результате реионизации ион. Для дифференцирования этих
процессов приходится использовать специальные приемы. В частности метод
различий в распаде нейтральной частицы и иона (NIDD — Neutral and Ion
Decomposition Difference) был предложен для экспериментов по генерированию
нейтральных частиц из отрицательных ионов [239]. В этом варианте
последовательно в идентичных условиях снимают спектры нейтрализации—реионизации и
инверсии заряда. Во втором случае отрицательный ион в условиях эксперимента
теряет два электрона и превращается в положительный, минуя стадию
нейтрального фрагмента. Вычитая второй спектр из первого, получают спектр продуктов
диссоциации нейтральной частицы.
Ключевым моментом эксперимента является доказательство того, что
нейтральная частица сохранила структуру исходного иона. Для этой цели
сравнивают спектры активации соударением исходных ионов и ионов с той же массой,
образовавшихся после реионизации. Идентичность этих спектров означает, что
нейтральная частица сохраняет структуру своего предшественника.
Метод можно использовать и для анализа нейтральных частиц,
образующихся при фрагментации ионов в камерах соударений. В таком варианте метод
называется «диссоциативная ионизация, индуцированная столкновениями
(Collision Induced Dissociative Ionization, CIDI)» [240] или «реионизация
нейтрального фрагмента (Neutral Fragment Reionization, NpR)» [241]. В первом
172 Глава 7. Тандемная масс-спектрометрия, МС/МС
случае камера соударений не заполняется газом (реакции распада метастабиль-
ных ионов, разд. 2.3), во втором —в качестве газа для активации соударением
в первой камере, как праЬило, используют гелий, который не вызывает
нейтрализации ионов. Это важное преимущество метода нейтрализации—реионизации
по сравнению с обычной активацией соударением. В отличие от классического
варианта МС/МС здесь можно установить структуру не только заряженного, но и
нейтрального продукта реакции фрагментации. Благодаря такой технике удалось
доказать, что потеря 27 а. е. м. из молекулярного иона анилина обусловлена
отщеплением не HCN, a HNC [242], что метилацетат на 80% отщепляет СН3О, a
на 20% отщепляет СН2ОН [243]. Были генерированы в газовой фазе нейтральные
молекулы, не существующие в растворах: Y—СС—X (где X и ? обозначают
ОН или NHb) и т. д. [244]. Зарегистрированы металлоорганические соединения
с незаполненными лигандами валентностями [245], гипервалентные частицы
Нз [246], D30" [247] и др., ван-дер-ваальсовы комплексы Не2 [248]. Метод с
успехом использован для изучения полиатомных радикалов, соответствующих
интермедиатам в процессах радиационных и химических повреждений ДНК
[249]. Важных результатов удалось добиться в области металлоорганической и
координационной химии [232, 250, 251].
7.5. Спектрометрия ионной подвижности
(Ion Mobility Spectrometry)
Метод спектрометрии ионной подвижности, иначе называемый «плазменной
хроматографией», был впервые описан Коэном и Карасеком [252] в 1970 г., хотя
несколько раньше эксперименты с трубой дрейфа и внешним источником ионов
были проведены в группе Хастеда [253]. Метод можно рассматривать в качестве
тандемного, хотя на первой стадии разделение ионов идет не по их массам, а по
их структурам (формам).
Метод позволил получить интересные результаты при изучении электронных
состояний ионов, геометрии и структуры металлических и углеродных
кластеров, ионов органических молекул, включая большие молекулы (цитохром С,
апомиоглобин) и т. д. Информативность спектрометрии ионной подвижности
уступает современным масс-спектрометрическим методам, однако результаты
могут быть весьма полезными как сами по себе, так и в сочетании с
полученными другими методами.
Под подвижностью иона в газовой фазе подразумевается, насколько быстро
он движется под действием электрического поля через буферный газ.
Электрическое поле ускоряет ион, а столкновения с молекулами газа замедляют
его движение. В результате этих двух процессов создаются условия для
равномерного поступательного движения иона, которое характеризуется скоростью
7.5. Спектрометрия ионной подвижности 173
вход ионов
Рис· 7.17. Принципиальная схема прибора для измерения ионной подвижности
дрейфа V/). Мобильность (подвижность) иона —это отношение скорости дрейфа
к напряженности приложенного электрического поля.
K = vD/E G.22)
Параметром, определяющим скорость иона в трубе дрейфа, является отношение
напряженности электрического поля к плотности буферного газа (?/?). При
малых значениях ?/?, когда скорость дрейфа невелика по сравнению со скоростью
теплового движения ионов, их подвижность не зависит от напряженности поля.
Это явление называется слабопольным пределом (low field limit). Если скорость
дрейфа значительно превышает скорость теплового движения (сильнопольный
предел, high-field limit), подвижность зависит от величины E/N, и ионы могут,
в определенной степени, выравниваться в трубе дрейфа. Так как измерение
подвижности ионов преследует цель получить о них определенную
структурную информацию, эксперименты обычно проводят в слабопольном режиме. В
качестве буферного газа используют гелий, азот, воздух и т. д.
Принципиальное устройство прибора для измерения ионной подвижности
включает источник ионов, трубу дрейфа, анализатор и детектор (рис. 7.17).
Источник ионов может быть как внешним, так и внутренним. Описаны
эксперименты с использованием практически всех известных методов ионизации.
Для измерения времени дрейфа важно, чтобы ионы вводились в трубу узкими
группами. Поэтому хорошие результаты получаются, например, при
использовании матричной лазерной десорбционной ионизации (разд. 5.14), когда ионы
образуются под действием короткого лазерного импульса.
В варианте низкого разрешения A0-20) используют низкое давление
буферного газа (<10 мм рт. ст.) и напряженность электрического поля, равную ~10 В/см.
Длина трубы дрейфа в этом случае составляет несколько десятков сантиметров.
В варианте высокого разрешения B00-400) используют давление буферного газа
~500 мм рт. ст., напряженность электрического поля ~500 В/см и трубы дрейфа
длиной до нескольких метров. К сожалению, метод характеризуется низкой
разрешающей способностью, которая ограничена за счет диффузии ионного пучка
174 Глава 7. Тандемная масс-спектрометрия, МС/МС
при его движении по трубе дрейфа. Уравнение для определения разрешения
метода было предложено в 1975 г. [254]:
t / LEze
R=At = \Jl6KBnn2> G·23)
где L — длина трубы дрейфа, ? — напряженность электрического поля, ze~- заряд
иона, Г —температура, Кв — константа Больцмана.
Для повышения разрешающей способности можно увеличивать длину трубы
и напряженность поля или уменьшать температуру проведения эксперимента.
Уменьшение температуры действительно приводит к улучшению разрешения,
но эффективность этого подхода невелика. Например, понижение температуры
с 298 до 77 К приводит к повышению разрешения всего в два раза. Удлинение
трубы также имеет ограничения, поскольку при движении ионный пучок
расширяется в результате диффузии. При определенной длине дрейфа ионы начинают
гибнуть на стенках. Не является панацеей и напряженность поля. Для того
чтобы работать в режиме слабопольного предела, необходимо одновременно с
увеличением напряженности увеличивать и давление буферного газа. Высокое
давление заставляет использовать высокий потенциал для ввода ионов в трубу
дрейфа. Соударения ионов, обладающих высокой кинетической энергией, с
атомами газа ведут к переходу части энергии движения во внутреннюю, в
результате чего инициируется фрагментация.
Эффект активации соударением может сыграть и положительную роль.
Поскольку «горячие ионы» образуются на входе в трубу дрейфа, именно здесь
возможны процессы их изомеризации или распада. При дальнейшем движении
по трубе избыточная энергия быстро уменьшается в результате новых
многочисленных столкновений с молекулами газа. Поэтому длина всей трубы может
быть использована для разделения изомеризованных и фрагментных ионов.
Варьирование потенциала ввода позволяет получать информацию об энергии
активации процессов изомеризации и диссоциации. Аналогичные энергетические
параметры можно получить и при варьировании температуры эксперимента.
Поскольку время движения иона в трубе дрейфа определяется усредненным
значением сечения ионно-молекулярных взаимодействий, важнейшим
параметром оказывается геометрия иона. Чем сильнее скручен ион, тем быстрее он
достигает детектора. Именно на этом принципе основано изучение структуры
и геометрии ионов. Например, четыре пика в спектре иона С^ [255]
свидетельствуют об образовании в источнике четырех типов этих ионов, которым можно
приписать структуры фуллерена, графитовой плоскости, моноциклического и
бициклического колец. Увеличение потенциала ввода, увеличивая внутреннюю
энергию ионов, ведет к изменению в соотношении форм ионов С^. В частности,
бициклическая структура изомеризуется в моноциклическую благодаря
меньшему напряжению последней. Однако для изомеризации в фуллерен повышения
потенциала ввода оказывается недостаточно. Для реализации такого процесса
7.5. Спектрометрия ионной подвижности 175
необходимы более тяжелые кластеры, например С^ [256] или металлофулле-
рены [257]. Кстати, практически идентичные времена дрейфа для Cf6 и LaC^
означают, что металл расположен внутри сферы.
Изучение геометрии ионов белков в газовой фазе позволяет изучать
внутримолекулярные взаимодействия [258]. Например, исследование геометрии ионов
цитохрома С, полученных методом термораспыления (разд. 1.5.3), в зависимости
от величины заряда, температуры и потенциала ввода показало следующее. Для
ионов с небольшим зарядом (от +4 до —7) при комнатной температуре и малой
величине потенциала ввода характерна более свернутая форма по сравнению
с существующей в водных растворах. Это объясняется тем, что растворитель
препятствует плотной упаковке глобулярных белков, так как полярные боковые
цепи направлены в растворитель для увеличения взаимодействия, а молекулы
воды могут проникать в полости глобул. В отсутствие растворителя боковые
цепи располагаются на поверхности глобул, и белок существует в более плотной
форме. С увеличением заряда иона (от +4 до +20) происходит увеличение его
размера, т. е. его форма становится все менее компактной, ион раскрывается. В
этом случае кулоновское отталкивание успешно конкурирует с
внутримолекулярными взаимодействиями. Аналогичные процессы раскрытия глобул происходят
при увеличении температуры эксперимента или потенциала ввода.
Спектрометрия ионной подвижности позволяет также изучать реакции ионов
при добавлении к буферному газу реакционноспособных частиц. Например, ввод
в трубу дрейфа паров D2O позволяет изучать эффективность Н—D обмена для
ионов белков и пептидов конкретной геометрии.
В работах [259] и [260] продемонстрированы возможности метода для
детектирования взрывчатых веществ, лекарственных препаратов, компонентов
химического оружия, органических загрязнений окружающей среды. Хаген [261]
показал, что метод может использоваться для разделения ионов изомерных
полициклических ароматических углеводородов.
Тридцатилетняя история тандемной масс-спектрометрии свидетельствует о
возрастающем значении этого метода и связанных с ним комплексных методов
(ГХ-МС-МС, ЖХ-МС-МС, спектрометрия ионной подвижности) в научных
исследованиях. Эффективные структурные исследования сложных молекул
(пептиды, белки, полисахариды, нуклеиновые кислоты) не могут быть реализованы
без тандемной масс-спектрометрии. Преимущества метода для анализа смесей,
структурных исследований, фундаментальной науки, для устранения проблем,
связанных с искажением спектра из-за термических и ионно-молекулярных
процессов, а также наличия примесей в образце очевидны. Фактически только
высокая стоимость приборов препятствует повсеместному переходу от обычной
к тандемной масс-спектрометрии.
Глава 8
ОСНОВНЫЕ НАПРАВЛЕНИЯ
ФРАГМЕНТАЦИИ ВАЖНЕЙШИХ
КЛАССОВ ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
В данной главе рассматриваются процессы фрагментации молекулярных катион-
радикалов (ЭУ), протонированных (ХИ) и депротонированных (ХИОИ)
молекулярных ионов основных классов органических соединений от алканов до
белков и нуклеиновых кислот. Речь идет прежде всего о монофункциональных
производных, а в некоторых случаях о бифункциональных. Разобраны
важнейшие доказанные реакции распада. Для более углубленного изучения масс-
спектрометрического поведения органических соединений необходимо
обращаться к специализированным обзорам и статьям. Много полезной информации
по фрагментации под электронным ударом самых разнообразных соединений
содержится в книге [16].
В этой главе не рассматриваются направления фрагментации
гетероциклических соединений. Законы их распада остаются такими же, что и для других
органических соединений, однако многообразие структур и связанное с этим
многообразие реакций требуют специального обсуждения, что выходит за рамки
данного пособия. Для занимающихся масс-спектрометрией гетероциклов можно
посоветовать обратиться к книгам [15, 16, 262].
Если основные процессы фрагментации органических соединений под ЭУ в
той или иной мере описаны во многих учебных пособиях, то спектры
химической ионизации остаются в основном предметом оригинальных статей и обзоров.
Это связано со значительно меньшей универсальностью этого метода ионизации
и большим числом переменных. Поэтому во избежание неоправданного
увеличения объема данного пособия процессы в условиях химической ионизации
описаны здесь только в общих чертах, но имеются весьма многочисленные
ссылки на оригинальную литературу.
8.1. Алканы
Электронный удар. На рисунке 8.1 представлены спектры изомерных пен-
тадеканов общей формулы С15Н32. Все четыре соединения характеризуются
одинаковым набором пиков фрагментных ионов, отстоящих друг от друга на
14 а. е. м., однако интенсивности этих пиков существенно различаются. Пики
молекулярных ионов алканов малоинтенсивны (обычно менее 1% в полном
ионном токе); причем их интенсивность уменьшается с увеличением длины цепи
или числа разветвлений в молекуле. В спектрах сильно разветвленных алканов
8.1. Алканы 177
пик М+' может отсутствовать вовсе. Энергии ионизации алканов довольно
высоки, так как ионизируются электроны С—Н- или С—С-а-связей. Основную
гомологическую серию ионов можно изобразить общей формулой C„H2n+i
(m/z 29, 43, 57, 71...). Условимся называть эту серию алкановой.
Ионы алкановой серии образуются непосредственно из М+* при
разрывах любой из С—С-связей, или в результате отщепления молекулы олефина
из первичного алкильного иона. По правилу Стивенсона заряд в результате
фрагментации остается на том фрагменте, энергия ионизации которого ниже.
Максимальными в спектрах, практически всегда, являются пики ионов с m/z 43
(С3К7"), 57 (С4Н9"), 71 (CsH^), поскольку энергии ионизации соответствующих
алкильных радикалов минимальны и составляют 7,55, 6,93 и 6,94 эВ
соответственно. Следует отметить, что даже в случае алканов нормального строения
указанные фрагменты представляют собой изомеризованные структуры, так как
вторичные и третичные карбокатионы значительно стабильнее, а, следовательно,
процессы изомеризации термодинамически выгодны.
В общем случае процессы рандомизации и скрамблинга, т. е. полного
смешения атомов водорода и углерода, очень характерны для парафинов. В
связи с этим установить структуру сильно разветвленных алканов по их масс-
спектрам электронного удара практически невозможно. Для нормальных алканов
(рис. 8.1а) характерно экспоненциальное уменьшение интенсивностей пиков
ионов алкановой серии, что позволяет однозначно подтверждать структуру этих
соединений. При этом пик [М —СНз]+ либо полностью отсутствует, либо имеет
крайне низкую интенсивность (рис. 8.1а). Задача структурного анализа
разрешима и для алканов с одним-двумя разветвлениями. Для этих соединений разрыв
С—С-связей в месте разветвления более благоприятен по сравнению с разрывом
соседних связей, поскольку образуется вторичный, т. е. более устойчивый, ион.
Интенсивность соответствующих фрагментов оказывается аномально высокой
и нарушает экспоненциальное падение интенсивностей пиков ионов алкановой
серии, характерное для нормальных парафинов. Так, для 3-метилтетрадекана
(рис. 8.1в) появляется пик иона [М — СНз]+, а интенсивность пика [М — С2Н5]4"
выше, чем даже иона с m/z 99. В этих процессах четко выполняется
правило выброса максимального алкила (разд. 6.1.1). В случае 2-метилтетрадекана
(рис. 8.16) помимо интенсивного пика [М —СНз]+ очень высокую интенсивность
имеет пик иона [М —СзН7]+ с m/z 169. Такой процесс отщепления изопро-
пильной группы является характеристикой алканов с метальным заместителем
в положении 2. В случае 4,8-диметилтридекана (рис. 8 Л г) отчетливо видны
повышенные интенсивности пиков ионов с m/z 197, 169 и 141. Первый обусловлен
отщеплением метильного радикала, второй — пропильного, третий — амильного.
Обратите внимание на четкое выполнение правила выброса максимального
алкила. Понятно, что при наличии в цепи четвертичного атома углерода
процессы разрыва связей с образованием третичного катиона становятся особенно
благоприятными. В частности, пик иона с m/z 57 часто доминирует в спектрах
соединений с mpem-бутильной группой.
178 Глава 8. Основные направления фрагментации органических соединений
? оо
90
80
70
60
50
4 0
30
20
1 0
о
1 00
90
8 О
70
6 О
5 О
4 О
3 О
2 О
1 О
О
1 О О
90
80
70
60
5 О
4 О
30
20
1 О
О
1 О О
90
80
7 О
60
5 О
40
3 О
2 О
1 О
О
Рис.
1,%
^Ш ulllL
lib dl»_
2 1 2
l·
4 О
I,%
80
1 О О
1 2 О
m /z
16 0 18 0 2 0 0
T^iL и!,
„I. . ..?.
I,%
6 0
5 7
1 20
m /z
18 0 2 0 0
JL Л!
-J. X
1 8 3
6 0
5 7
1 0 0
1 2 0
m /z
1 4 1
1 69
I
2 1 2
40 60 80 100 120 140 160 180 200
m /z
8.1. Масс-спектры электронного удара пентадекана (?), 2-метилтетрадекана (б), 3-ме-
тилтетрадекана (в) и 4,8-диметилтридекана (г)
8.1. Алканы 179
Наряду с алкановой серией в спектрах можно выделить еще две серии ионов,
пики которых достаточно интенсивны. Ионы алкеновой серии с общей формулой
С„Н2Л_1 (m/z 27, 41, 55, 69...) образуются в основном при неселективном
отщеплении молекулы водорода ионами алкановой серии. В спектрах
разветвленных изомеров значительно увеличивается интенсивность пиков нечетноэлек-
тронных фрагментов серии С„Н^' с четной массой (схема 8.1).
С„Н2„+1— CR2— CH2R ~H » ROT3 + CnH2n-^CR2
Схема 8.1
Движущей силой этих процессов является выгодность образования
термодинамически стабильных замещенных алкенов. Интенсивность пика нечетноэлек-
тронного иона оказывается выше при разрыве связи в месте разветвления, т. е.
процессы подчиняются закономерностям, описанным для алкановой серии.
Химическая ионизация. Молекулы алканов обладают очень низкой величиной
сродства к протону. Они могут протонироваться по связям С—С или С—?
[263], однако ионы оказываются нестабильными и распадаются с образованием
алкильных катионов. Поэтому даже при использовании в качестве газа-реагента
метана (высокая протонирующая способность) наибольшую интенсивность в
спектре имеет ион [М —Н]+. Его образование можно рассматривать как потерю
молекулы водорода протонированным молекулярным ионом МН+ или как
реакцию отрыва аниона (разд. 2.3). Реакцию отрыва аниона можно считать также
основным механизмом образования алкильных ионов (схема 8.2). Безусловно,
фрагментные алкильные ионы могут возникать и при отщеплении молекулы
алкена первичными ионами [М —Н]+ (схема 8.2).
CioH22 + CH5" *¦ С8Нп + С2Нб + СРЦ
СюН2+1 * С8Нп + С2Н4
Схема 8.2
Интенсивность пиков ионов [М —Н]+ очень высока и может достигать 90% в
полном ионном токе. Этот факт выгодно отличает спектры ХИ от спектров ЭУ.
С увеличением числа разветвлений в молекуле интенсивность пика [М—Н]+
заметно уменьшается. Для повышения его интенсивности эффективным оказалось
использование в качестве газа-реагента монооксида азота [264].
Химическая ионизация отрицательных ионов. Крайне низкие величины
кислотности алканов приводят к тому, что они не депротонируются даже наиболее
сильным из доступных оснований, ионом NH^*. Кроме того, величины сродства
180 Глава 8. Основные направления фрагментации органических соединений
к электрону для подавляющего большинства алкильных групп являются
отрицательными, т. е. алкильные анионы оказываются нестабильными относительно
соответствующих радикалов. Получить эти анионы тем не менее можно при
фрагментации анионов карбоновых кислот.
Задача 8.1. Интенсивности каких пиков в спектрах ЭУ будут аномально
высоки по сравнению с пиком изомера нормального строения?
а) 4,10-Диметилгексадекан;
б) 3 Д 2-диметилгексадекан;
в) 3,9,14-триметилгептадекан;
г) 2,2-диметилдекан;
д) 5,10,15-триметилоктадекан.
Задача 8.2. Какие различия будут наблюдаться в спектрах электронного
удара?
а) 2-Метилоктан и 3-метилоктан;
б) 4-метилдекан и 5-метилдекан;
в) 3,3-диметилнонан и 3-метилдекан;
г) 5,5-диметилнонан и 3,6-диметилнонан;
д) 2,2-диметилоктан и 3,4-диметилоктан;
е) 3,3,7-триметилдодекан и 3,7-диметилтридекан.
Задача 8.3. Какие нечетноэлектронные фрагменты будут интенсивны при
распаде М+"?
а) 3,3-Диметилоктан;
б) 3,4-диметилоктан;
в) 3,5-диметилоктан.
Задача 8.4. Идентифицируйте метилалкан по спектру ЭУ (рис. 8.2).
юо
90
80
70
60
SO
ЛО
ЗО
20
Ю
I/^q
120
m/z
Рис. 8.2
8.2. Алкены и диены 181
Задача 8.5. Идентифицируйте диметилалкан по спектру ЭУ (рис. 8.3).
юо
90
80
70
во
50
40
зо
20
лЬ
О
1,<*?>
120 140
т/г:
Рис. 8.3
8.2. Алкены и диены
Электронный удар. Молекулярные ионы алкенов чуть более интенсивны, чем
в случае алканов. На рисунке 8.4 представлены масс-спектры нескольких
соединений этого класса. Спектры характеризуется аналогичными гомологическими
сериями ионов: алкановой (C^Hj^) и алкеновой (C^H^j). Однако в отличие
от алканов интенсивности пиков второй серии алкенов выше, чем первой.
Образование алкеновой серии обусловлено аллильным распадом (схема 8.3),
приводящим к образованию устойчивых благодаря резонансной стабилизации
аллильных катионов.
R-CH2-CH-CH-R
R
? ?
+ CH2^C№^CH-R
Схема 8.3
Перегруппировки, приводящие к возникновению нечетноэлектронных
фрагментов (СЛН^*), как правило, не селективны. Однако заметную роль играет
процесс (схема 8.4), протекающий через шестичленное переходное состояние
(вариант перегруппировки Мак-Лафферти).
я^Г
-Н*
R
'Ж
R
?
R
Схема 8.4
СНз
?
R
Первый потенциал ионизации молекулы алкена обусловлен удалением
электрона 7г-связи, т. е. можно считать, что в электронно-невозбужденном состоянии
заряд и неспаренный электрон расположены на соседних атомах углерода,
которые были соединены двойной связью. Однако в результате аллильных
перегруппировок (схема 8.5) и неселективных процессов рандомизации, которые
182
Глава 8. Основные направления фрагментации органических соединений
1 00
90
8 0
7 О
6 О
50
4 О
30
20
1 О
1,%
55
6 9 8 3
7 0
I L
100 15 0
m /?
10 0 -? ?, %
90 4
80 A
7 О А
6 0 4
5 0-^
40 J
30 4
2 0 4
1 0 4
4 3
70 8 3
1 1 1
_1 JL.
100 150
m /?
100 ? ?,%
9 0 4
80 ?
70 J
60 -|
5 ?
4 ?
30
2 ?
1 ?
29
4
4 1
..
3
.
5
...
5
6
...??
9
8 3
??
9 7
? ]
200
250
100 15 0
m /?
2 5 0
00 ?
90 -
? ? ?
? ? ?
50 -
4 0-
30 -
20 -
1 0 -
0 -
?,%
>J_
4 1
2
J
9
??
43
L
5
г^
6
5 7
6 9
iLn
и
L
8 3
j
JlL
9 7
? 111
ШЦ nil, ..!¦¦ ? ¦ ,..
2 24
1 ' ' 1 '
г
1
m /z
Рис. 8.4. Масс-спектры электронного удара гексадецена-1 (?), гексадецена-3 (б), гексадеце-
на-7 (в) и 2-метилпентадецена-1 (г)
8.2. Алкены и диены 183
протекают с большой скоростью (\gk « 10), заряд и неспаренный электрон де-
локализуются по углеродной цепи. Установить по масс-спектру первоначальное
положение двойной связи практически невозможно. Единственным исключением
являются тетразамещенные алкены.
R-CH-CH-CH2-CH2~R ^ R-CH2-CH-CH-CH2-Ri=^R-CH2-CH2-CH-CH-R
Схема 8.5
Не поддается установлению также принадлежность соединения к цис- или
транс-изомерам, если, правда, нет спектров обоих изомеров. Дело в том, что
молекулярный ион, как правило, более интенсивен в спектре mpawc-изомера.
Из-за неселективности масс-спектров для анализа алкенов используют
методы дериватизации. Распространено эпоксидирование с последующей
трансформацией в кетоны, диолы или аминоспирты [265]. Метод реакционной хро-
матомасс-спектрометрии (разд. 1.4) используется для анализа алкенов после их
каталитического дейтерирования в алканы [21]. Поскольку микрореактор может
быть установлен до или после колонки хроматографа, работу можно вести в двух
вариантах, хроматографируя или алканы, или алкены. Использование помимо
масс-спектров информации о временах удерживания повышает эффективность
этого подхода.
Установить положение кратной связи в молекуле алкена можно по спектрам
полевой ионизации (разд. 2.7). Благодаря минимальному времени пребывания
молекулярного иона в источнике A0~~12 с) многостадийные аллильные
перегруппировки (схема 8.5) не успевают значительно исказить исходную структуру, а
аллильные разрывы (схема 8.3) позволяют идентифицировать изомеры [266].
Для диенов характерна серия гомологических ионов С„Н^_3. Интенсивность
пика М+' невысока (рис. 8.5). Процессы распада протекают по механизмам,
аналогичным описанным для алкенов. Задача различия изомеров по масс-
спектрам очень сложна и может быть решена только в особых случаях при
использовании широкого набора стандартов. Полезно также использовать другие
физико-химические методы.
Химическая ионизация. В спектрах химической ионизации метаном, как и в
спектрах ЭУ, доминируют две серии ионов (алкановая и алкеновая). Пики прото-
нированных молекулярных ионов МН+ обладают достаточной интенсивностью
(до 20% в полном ионном токе) и позволяют установить молекулярную массу
соединения. Дополнительную помощь в идентификации оказывают пики ионов
[? —?]4*. Эти ионы образуются в основном при отщеплении атома водорода
из аллильного положения исходной молекулы, поэтому интенсивность их пиков
падает с увеличением числа алкильных групп вокруг кратной связи [267].
К сожалению, ни положение двойной связи, ни различия между цис- и
/иранс-изомерами установить по спектрам ХИ метаном нельзя. Определенных
184 Глава 8. Основные направления фрагментации органических соединений
юо ? ?,?/?
80
60 -?
40 А
20 А
53
h , .1
67
8 1
95
,1 09
12 3 13 7
20
ЮО ? ?,%
80
60
40
20
60
53
80 1 00
m /?
95
1 20
140
67
_ц
81
I ¦ ¦ Ll.
1 09
,12 3
[L- J
1 37
1 60
1 51
20
40
60
80 100
m /z
1 20
1 40
1 60
Рис. 8.5. Масс-спектры ЭУ додекадиена-5,7 (?) и 3,4,5,6-тетраметилоктадиена-2,5 (б)
успехов удалось добиться при замене метана изобутаном [268]. Вид спектра
оказывается идентичным наблюдаемому для метана, однако появляется новый пик,
обусловленный присоединением к молекуле образца mpem-бутильного катиона.
Различия в соотношении интенсивностей пиков ионов [М+С4Н9]+/[М—Н]+
(~2 для 1/мс-изомеров и <1 для транс-изомеров) оказываются достаточными
для отнесения образца к тому или иному изомеру. На основании анализа
интенсивностей фрагментных ионов в спектрах ?/ис-изомеров можно также
установить положение двойной связи в исходной молекуле. Однако этот подход
неприменим для транс-изомеров.
Такой же результат можно получить при использовании в качестве
газа-реагента монооксида азота. Отношение интенсивностей пиков ионов
[?+??]+/[? —Н]+ составляет ~2 для г/мс-изомеров и <1 для транс-изомеров
[268]. В спектрах ХИ алкенов окисью азота помимо максимальных пиков
ионов [М —Н]+ и [?+??]+ присутствуют достаточно интенсивные пики М+\
Интенсивность последних увеличивается с увеличением замещенности при
двойной связи, так как дополнительные алкильные группы уменьшают энергию
ионизации олефина, и, начиная с определенного значения, реакция перезарядки
(см. разд. 2.3) становится экзотермичной.
Определить место двойной связи в молекулах олефинов, а также диенов,
алкинов и ненасыщенных карбоновых кислот и их эфиров можно с использо-
8.3. Алкины 185
ванием карбонила железа в качестве газа-реагента [269]. В комплексном ионе
[Fe+алкен]4" металл внедряется по аллильной связи, после чего /3-водород-
ный атом мигрирует с образованием промежуточного комплекса (схема 8.6).
В условиях активации соударением диссоциация этого комплекса приводит к
элиминированию меньшего олефинового лиганда.
Схема 8.6
Для диенов характерны пики протонированных молекулярных ионов МН+
и связанная с ними гомологическая серия ионов C^H^j. Процессы распада
аналогичны протекающим для олефинов.
Химическая ионизация отрицательных ионов. Олефины достаточно легко
депротонируются в газовой фазе, однако аналитическая ценность спектров
этого типа невелика. В частности, максимальной интенсивностью в спектрах
ХИОИ олефинов влажной закисью азота (ион-реагент ОН~) обладают пики
ионов [?—?+?2?]"" и [?—?+?2?—ЬЬО]-. Депротонированные
молекулярные ионы [М—Н]~, а также ионы [М—ЗН]~, напротив, характеризуются низкой
интенсивностью.
Для химической ионизации отрицательных ионов диенов характерны
достаточно интенсивные пики ионов [М —Н]~ и [М—ЗН]~, предоставляющие
полезную информацию о молекулярной массе соединений. К сожалению значительной
структурной информации спектры этого типа не несут [136].
8.3. Алкины
Электронный удар. Ацетиленовые углеводороды характеризуются низкой
стабильностью молекулярных ионов. Пик М+" терминальных алкинов может
отсутствовать в спектре. По мере перемещения тройной связи к центру молекулы
стабильность М+* увеличивается. На рисунке 8.6 представлены масс-спектры
электронного удара изомерных додецинов с терминальной и внутренней тройной
связью.
Характерной для алкинов является гомологическая серия ионов общей
формулы СлН2^_з· Основной механизм образования этих ионов аналогичен аллиль-
ному разрыву связи в М+* алкенов. В данном случае образуются пропаргильные
ионы, стабилизированные, хотя и в меньшей степени, чем аллильные, эффектом
резонанса. В отличие от алканов и алкенов перегруппировочный процесс
(схема 8.7), сопровождающийся образованием шестичленного циклического иона,
186 Глава 8. Основные направления фрагментации органических соединений
1 0 0
8 0
6 0
4 0
2 0
0
-
-
I,
%
1
4
1
J
||
¦ ??
6 7
1ш_
л
8 1
H±J I-1-.
9 5
1^-
10 9
«
1 0 О
/?
1.4 О
10 0 ?
80 -
6 0-
40 -
2 0-
0 -
1,%
и 1.
J J
??, , „ll
6 7
La
8 1
I. ,,
9 5
1 0 9
ll
1 2 3
—r-b
1 3 7
I. t
6
1 6 6
m /z
Рис. 8.6. Масс-спектры электронного удара додецина-1 (а) и додецина-5 (б)
позволяет делать выводы о положении кратной связи в исходной молекуле. Пики
ионов, образование которых протекает по этому механизму, имеют повышенную
интенсивность в ряду экспоненциально уменьшающихся интенсивностей пиков
ионов алкиновой серии. В частности, в масс-спектре додецина-1 максимальную
интенсивность имеет пик иона с m/z 81 (рис. 8.6, а), а в спектре додецина-5
повышенную интенсивность имеет пик иона с m/z 137 (рис. 8.6,6). Эффективность
этого процесса значительно снижается при наличии разветвлений в молекуле
алкина [270].
RO
+ RCH2
Схема 8.7
Химическая ионизация· В спектрах химической ионизации алкинов метаном
и изобутаном пики МН+ и [М —Н]+достаточно интенсивны, что позволяет
установить молекулярную массу соединений. Среди осколочных ионов выделяются
гомологические серии ионов C/iH^J_1 и С„Н^_3. В условиях ХИ изобутаном
высокую интенсивность имеет пик продукта присоединения [М+С4Н9]4", а по
интенсивностям пиков алкеновой серии C/JH^J_1 можно определять положение
тройной связи в молекуле [271].
8.3. Алкины 187
Пики гомологической серии ионов СЛН^__3 имеют максимальную
интенсивность в условиях химической ионизации монооксидом азота. Другие
углеводородные серии менее выражены. Ионы [?+??]+, характеризующиеся
незначительными по интенсивности пиками, при распаде приводят к появлению
ионов двух гомологических серий [CnH2„NO]+ и [C„H2«-2NO]+. На основании
интенсивностей пиков ионов этих серий можно определять положение тройной
связи в молекуле [271].
Определить место тройной связи в молекулах алкинов можно с
использованием карбонила железа в качестве газа-реагента [269]. В комплексном ионе
[Fe + алкин]" металл внедряется по пропаргильной связи, после чего протекает
миграция /3-водородного атома с образованием алкин-алкенового комплекса,
дальнейший распад которого позволяет сделать вывод о положении кратной
связи в молекуле (см. схему 8.6).
Для химической ионизации отрицательных ионов алкинов с терминальным
положением кратной связи характерны достаточно интенсивные пики ионов
[М — Н]~ и [М —ЗН]~, предоставляющие полезную информацию о молекулярной
массе соединений.
Задача 8.6. На рисунке 8.7 представлены масс-спектры ЭУ двух изомерных
тетрадецинов нормального строения. Установите, какому изомеру соответствует
каждый спектр.
10 0 ?
90 -
8 0-
70 -
60 -
50 -
40 -
30 -
20 -
1 0 -
0 -
1,%
, ¦¦'!
N
Li
И
67 |
Lb
8 1
?
ll. ...
) 5
? 10 9
ll, ll
1 2 3
-rJ·—
1 3 7
-? —r-
a
1 94
1 ' ' ?
2 0
100 -?
9 0 -| I>%
80
70 4
60 4
50 -J
4 0 -|
30
20
1 0
40 60 80 100 120 140 160 180 200
m /z
|67 ^
8 1
95
1 0 9
1 2 3
1 6 5
1 94
20
80 100 120 140 160 180 200
m /z
Рис. 8.7. Масс-спектры электронного удара двух изомерных тетрадецинов
188 Глава 8. Основные направления фрагментации органических соединений
8.4. Алициклические углеводороды
Электронный удар. Основная характеристическая серия гомологических
ионов для моноциклических насыщенных углеводородов (рис. 8.8) совпадает с
алкеновой (C^H^j). Соответственно, для бициклических — характерна алкино-
вая серия (СлН^_з) и т. д. С увеличением длины алкильных заместителей
значительную интенсивность приобретают пики ионов алкановой серии (C^Hj^).
Масс-спектрометрии малых циклов посвящена монография [17].
Молекулярные ионы малых циклов менее устойчивы по сравнению с циклогексанами и
циклопентанами. Раскрытие циклопропанового кольца в молекулярном ионе
может протекать до начала фрагментации, поэтому спектры похожи на спектры
изомерных алкенов. Для циклобутановых углеводородов важное направление масс-
спектрометрического распада связано с разрывами противолежащих С—С-связей
цикла. Из двух родственных нечетноэлектронных ионов более высокую
интенсивность будет иметь пик фрагмента с меньшей энергией ионизации (правило
Стивенсона, разд. 7.1.2). В частности, на рис. 8.8,6 интенсивность пика иона с
m/z 84 превосходит интенсивность пика иона с m/z 28.
ън<1*
(У «~C2Hs' Q-C^l^ ^J-c2h5 -с2Н4> ^ + с2н5_^ +г
/иД83
m/z 55
/и/г 112
-С2Нб
chST -^
m/z 82
С5Н7
m/z 67
C2H5
&-
C3H?
m/z 41
Схема 8.8
m/z 84
C2H5
m/z 56
—4 trC2Hs I^ <**
m/z 83
-C3H7
¦^j-CiHs
m/z 69
Пи1си молеьсулярных ионов алкилциклогексанов более интенсивны, чем
соответствующие пики алкилциклопентанов. На схеме 8.8 представлены основные
направления распада молекулярного иона этилциклогексана, спектр которого
изображен на рис. 8.8, г. Отщепление экзоциклического заместителя протекает
легко, особенно с увеличением длины алкильного заместителя. Он может
отщепляться в виде радикала или с миграцией атома водорода в виде ал-
кана или алкена. В результате в спектрах монозамещенных
алкилциклогексанов высокой интенсивностью обладают пики ионов с m/z 84, 83 и 82, а
8.4. Алициклические углеводороды 189
1 О О
9 О
8 О
7 О
? О
S О
4 О
3 О
2 О
1 О Ч
О 4-
2 О
10 0 -?
9 0 -I
8 0 Ч
7 0 -]
6 0 -\
5 0 -А
4 0 ?
ЗОН
2 0 -I
1 О -I
1,%
2 О
10 0 -?
9 0 4
8 0 -I
7 0 Ч
6 О -I
5 О
4 О
3 О
2 0 Ч
1 О
2 О
1.%
4 0 5 0
7 О
m /?
4 0 5 0
7 О
m /?
40 50 60 70 80 90
m /z
m /?
1 1 2
*--, r-J ,
10 0 110 12 0
10 0 110 12 0
10 0 110 12 0
0 0 -?
9 0 -
8 0-
7 0 -
6 0 -
5 0-
4 0 -
30 -
2 0 -
1 0 -
0 -
?,%
_u J
5 ?
4 1
jj ^JL
5 5
I U J
18 3 г
112
L ?
10 0 110 12 0
Рис. 8.8. Массчзпектры электронного удара алициклических углеводородов с брутто-
формулой CgH^: а — амилциклопропана, б — бутилциклобутана в — пропилциклопентана, г —
этилциклогексана
в спектрах монозамещенных алкилциклопентанов—пики ионов с m/z 68, 69,
70. Установление изомеров нафтенов с помощью масс-спектрометрии весьма
затруднительно.
Процессы фрагментации молекулярных ионов полициклических нафтенов, а
также спиранов не отличаются высокой селективностью и в основном совпадают
с изложенными для моноциклических углеводородов. Поскольку стабильность
молекулярного иона возрастает с увеличением степени ненасыщенности (в
данном случае числа циклов), интенсивность пика М+' в спектрах полициклических
190 Глава 8. Основные направления фрагментации органических соединений
нафтенов достаточно высока и позволяет надежно устанавливать молекулярную
массу соединения.
Химическая ионизация. Масс-спектры этого типа могут с успехом
использоваться для установления молекулярной массы алициклических углеводородов.
Максимальной интенсивностью в спектре (до 75% в полном ионном токе)
обладает пик иона [М — Н]+. Пики ионов МН+ менее интенсивны, хотя
отчетливо наблюдаются в спектрах. Основные фрагментные ионы принадлежат
гомологическим алкановой и алкеновой сериям [272]. Спектры ХИ не имеют
ценности для структурного анализа, хотя надо отметить, что информации об
исследованиях нафтенов с помощью химической ионизации недостаточно.
Использование для анализа алициклов ХИ отрицательных ионов невозможно по
причинам, аналогичным описанным для алканов (разд. 8.1).
Задача 8.7. Составьте схему фрагментации метилциклопентана, спектр ЭУ
которого представлен на рис. 8.9.
-100 ?
эо -|
во 4
70 А
?? -\
50 -4
ло -1
зо А
20 А
1 О -J
1,«М»
Рис. 8.9. Масс-спектр электронного удара метилциклопентана
8.4.1. Циклоалкены
Электронный удар. Пики молекулярных ионов циклоалкенов достаточно
устойчивы. Характеристической серией ионов оказывается алкиновая (СИН^_3)>
хотя интенсивности пиков ионов алкеновой и алкановой серии могут быть
достаточно высоки. Аллильные перегруппировки могут приводить к весьма
существенным трансформациям исходной структуры.
Очень характерен для циклоалкенов ретродиеновый тип распада,
заключающийся в распаде цикла с образованием алкенового и диенового фрагментов
(схема 8.9). Поскольку энергия ионизация диена, как правило, ниже, заряд чаще
сохраняется на нем (m/z 54 на рис. 8.10). Алкильные заместители в положениях
1, 2, 3 и 6 приводят к соответствующему сдвигу по шкале масс диенового
иона, а заместители в положениях 4 и 5 элиминируются как часть молекулы
алкена. По этому признаку можно делать определенные выводы о структуре
соединения. Введение электронодонорных заместителей в положение 4 или 5
8.4. Алициклические углеводороды 191
1+
m/z 54
-^-са
-сн3"
V
m/z67
Схема 8.9
циклогексенового цикла может уменьшить ЭИ соответствующего алкена. В
результате пик такого иона будет более интенсивен, чем пик альтернативного
диена.
Еще один доказанный процесс, приводящий к иону с максимальной
интенсивностью в спектре (m/z 67) связан с отщеплением метильной группы (рис. 8.10).
Методом изотопных меток доказано, что отщепляется в основном атом углерода
из положения 4 кольца (схема 8.9). Ион [М — Н]+ образуется при
отщеплении аллильного атома водорода из М+\ Этот резонансно стабилизированный
фрагмент отщепляет последовательно две молекулы водорода с образованием
ароматического фенилкатиона (m/z 77, рис. 8.10).
юо
80 ?
во
40
20 -|
1,%
йЛ
_1ЛД
m/z
Рис. 8.10. Масс-спектр электронного удара циклогексена
Химическая ионизация. Информация по этому виду спектров циклоалкенов
минимальна. Максимальную интенсивность в спектрах химической ионизации
метаном имеют пики [М — Н]+. Этот факт позволяет надежно устанавливать
молекулярную массу циклоалкенов. В условиях химической ионизации
отрицательных ионов молекулы этих углеводородов легко депротонируются, однако
спектры малоинформативны [136].
192 Глава 8. Основные направления фрагментации органических соединений
Задача 8.8. На рисунке 8.11 представлены масс-спектры ЭУ двух изомерных
метилциклогексенов. Что можно сказать об их структурах?
Рис. 8.11
8.5. Ароматические углеводороды
Электронный удар. Молекулярные ионы незамещенного бензола и его
полициклических аналогов характеризуются максимальными по интенсивностям
пиками в спектре. С увеличением числа конденсированных колец доля М+' в
полном ионном токе возрастает. Например, для бенз[а]пирена интенсивность
пика М+* составляет более 50% в полном ионном токе (рис. 8.12), причем второй
и третий по интенсивности пики в спектре принадлежат двухзарядным ионам
М+* и [М—Н]+. Другие интенсивные пики обусловлены однозарядными ионами
[М-Н2]+\ [М—С2Н2]4", [M—Н2,—С2Н2]+\ а также двухзарядными ионами
10О ?
90 -
во -
70 -
60 -
во -
40 -
ЗО -
20 -
1 О -
?.^?.
-? 1 з I
, .il II
126
.
... ?·(?
I I "",l I ' ,ml
252
I,
Рис. 8.12. Спектр электронного удара бенз[а]пирена
8.5. Ароматические углеводороды 193
такого же состава. Аналогичные процессы характерны для любых незамещенных
полициклических ароматических углеводородов.
Важность качественного и количественного определения полициклических
ароматических углеводородов (ПАУ) как приоритетных загрязняющих веществ
определила интенсивный поиск их селективного анализа. В частности,
существенные различия величины сечения ионизации в процессе резонансного
захвата электрона отмечены в зависимости от структуры изомерного ПАУ [273].
Молекулярные ионы характеризуются интенсивными пиками в спектрах ЭУ
и в случае замещенных ароматических углеводородов (рис. 8.13). Для этих
соединений доминирующим процессом становится бензильный разрыв с
образованием резонансно стабилизированного иона (схема 8.10). При распаде мо-
нозамещенного бензола происходит образование полностью рандомизованного
тропилий-катиона (m/z 91), который далее последовательно теряет две молекулы
ацетилена (схема 8.10). Этот катион можно считать первым в гомологической
серии характеристических для алкилбензолов ионов с общей формулой Ph(CH2)+.
Если есть возможность альтернативного отщепления нескольких алкильных
радикалов, интенсивность пиков соответствующих фрагментных ионов
определяется правилом выброса максимального алкильного радикала (разд. 6.1.1).
?$^* ^ ?/**— (©) =S&. «** -3*. с**
m/z 91 m/z 65 m/z 39
Схема 8.10
Процесс отщепления заместителя с образованием фенильного катиона идет,
но значительно менее эффективно, поскольку прочность фенильной связи С—С
A10,8 ккал/моль) значительно выше, чем бензильной G4,1 ккал/моль), а
стабильность фенил-катиона значительно ниже, чем бензил-катиона или
тропилий-катиона. Тем не менее пики фрагментных ионов, образующихся при разрыве связи
Alk—Ph, становятся весьма интенсивными при распаде полиалкилзамещенных
бензолов.
Для интерпретации масс-спектра очень важен перегруппировочный процесс,
идущий по механизму перегруппировки Мак-Лафферти. Это направление
распада для случая монозамещенного бензола проиллюстрировано схемой 8.11. В
результате образуется молекулярный ион толуола. Для протекания перегруппи-
UCI> — U^ ULiH2 J- кХен,
Схема 8.11
7—1113
194 Глава 8. Основные направления фрагментации органических соединений
1,%
I 1 1 t I I I I I I 1
91 I
43
_Д ?,?,? ,? ,1, ,. и,, J
162
J] .J, ^ I Д_
? ¦ ·? "i"" ? 1 ? ? —i"" ? '¦ ? """I ¦ ¦¦ ?—- 1 1 r—
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160
m /z
Рис. 8.13. Масс-спектр электронного удара гексилбензола
ровки необходимо, чтобы длина цепи заместителя была не менее трех атомов.
Кроме того, в цепи не должно быть разветвлений, а по крайней мере одно
opmo-положение должно быть свободно. Интенсивность пика такого нечетно-
электронного иона (m/z 92) очень высока (рис. 8.13), и для углеводородов с
длинной неразветвленной цепью может сравняться с интенсивностью тропилий-
катиона (m/z 91).
На основании двух описанных процессов (бензильный разрыв и
перегруппировка Мак-Лафферти) можно устанавливать структуры изомерных
ароматических углеводородов. Например, пропилбензол будет характеризоваться
максимальным пиком с m/z 91 и интенсивным пиком перегруппировочного
иона с m/z 92. Для изомерного метилэтилбензола перегруппировочный процесс
невозможен (п = 2), а бензильный разрыв приводит к ионам с m/z 105 и 119. К
сожалению, орто-, мета- и ияря-изомеры алкилзамещенных ароматических
углеводородов характеризуются очень близкими масс-спектрами, что не позволяет
идентифицировать изомеры этого типа.
В случае mpem-бутилбензола вслед за бензильным разрывом протекает
элиминирование молекулы этилена [274]. Этот факт объясняется изомеризацией
иона [М-СНз]+ в структуру ?-комплекса фенил-катиона и циклопропана
(схема 8.12).
6 —==*- 6 —
Схема 8.12
Совершенно по таким же направлениям распадаются молекулярные ионы
алкилзамещенных полициклических ароматических углеводородов и бифенилов.
В случае дифенилметана (рис. 8.14) помимо интенсивного пика [М—Н]+,
обусловленного ионом фенилтропилия, достаточно высокую интенсивность имеет
пик иона [М—ЗН]+.
С2Ч4 +
с7н7
8.5. Ароматические углеводороды 195
100 -.
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
¦ , ,1,,
65
.'¦ Л ,¦
, ,,???
91
?_^
115
—, л ,
1ь52 I
,„ ? "Л
? 168
L
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180
m/z
Рис. 8.14. Масс-спектр электронного удара дифенилметана
Этот эффект связан с образованием флуоренил-катиона в результате нуклео-
фильной атаки одного бензольного ядра по катионному центру в орто-положе-
нии другого (схема 8.13). Следует отметить, что даже если в орто-положениях
находятся заместители, происходит их элиминирование с образованием
флуоренил-катиона.
о-*-о—?
???
Схема 8.13
Ароматические углеводороды являются хорошими моделями для изучения
скрамблинга в масс-спектрометрии. В частности, исследование механизма
отщепления молекулы ацетилена из М+* бензола показало, что до распада
происходит полная рандомизация атомов углерода и водорода. Например,
молекулярный ион 1,3,5-тридейтеробензола теряет молекулы С2Н2, C2HD и C2D2 в
отношении 1:3:1. Изучение распада соединений, меченных изотопами 13С
и D, показало, что все С—?-связи в молекулярном ионе бензола разрываются и
статистически реорганизуются до или во время фрагментации, т. е. скрамблинг
атомов водорода осуществляется в результате миграций, а скрамблинг атомов
углерода —в результате валентной изомерии.
Аналогичные процессы характерны и для более сложных ароматических
систем. Например, М+" пентадейтерофенилбензола СбН5—Сб05 теряет молекулы
С2Н2, C2HD и C2D2 после полной рандомизации. Отщепление метильного
радикала молекулярным ионом стильбена протекает только после полной
рандомизации атомов углерода и водорода. Для протекания этого процесса необходимо
более 10000 разрывов и образований новых связей. Это событие кажется
невероятным, но действительно возможно, поскольку константы скоростей этих
реакций имеют порядок 1012-1013, а время жизни иона в источнике масс-
спектрометра в условиях ЭУ составляют 10~6 с.
196 Глава 8. Основные направления фрагментации органических соединений
Для алифатических соединений, особенно в случае наличия функциональных
групп, при использовании энергии электронов 70 эВ скрамблинг не столь
характерен, однако при понижении энергии ионизирующих электронов этот процесс
становится заметным. Заметим, что в общем случае, чем быстрее идут реакции
фрагментации, тем меньше вероятность рандомизации.
Химическая ионизация. Для незамещенных ароматических углеводородов
при ХИ, как и в случае электронного удара, фрагментация незначительна.
Максимальную интенсивность имеет пик протонированного молекулярного иона
МН+. Наряду с ним в спектрах проявляются лишь пики ионов, обусловленных
реакцией электрофильного присоединения ([М+С2г15]+ и [М+С3Н5]4* для
химической ионизации метаном). Сечение ионизации в случае ХИ метаном
меньше, чем в условиях ЭУ. При использовании химической ионизации водородом
в спектрах присутствуют только пики ионов М+" и МН+. Сечение ионизации
при этом сравнимо с наблюдаемым в условиях ЭУ. Доля МН+ увеличивается с
увеличением энергии ионизации молекулы. Соотношение интенсивностей пиков
ионов МН+ и М+* может использоваться для идентификации изомерных ПАУ
[275]. Для этой цели используют в качестве газа-реагента смесь метана и аргона.
Интенсивность пиков МН+ остается высокой и для алкилзамещенных
ароматических углеводородов. С увеличением длины и разветвленности алкильного
заместителя возрастает интенсивность пиков гомологической серии
ароматических ионов (m/z 91, 105, 119...). Образование этих ионов может протекать как
при фрагментации протонированного молекулярного иона, так и в результате
ионизации по механизму отрыва аниона (схема 8.14).
СН3 +
ш5+ + R—^^—СН-С3Н7 R-^^— CH-CH3 + C3Hg + CH4
Схема 8.14
Начиная с бутильного заместителя, в спектре появляются пики ионов
алкановой серии. Наличие в молекуле wpem-бутильной группы приводит к
максимальному по интенсивности в спектре пику иона с m/z 57. Алкановые
ионы образуются при отщеплении ароматического фрагмента из МН+. Еще
одним процессом распада МН+ является элиминирование молекулы олефина
(схема 8.15).
С5Нц+ + R-<0> - R—<?^-сн-с3н7 - R-®> + °5???
Схема 8.15
8.5. Ароматические углеводороды 197
Процессы, представленные на схеме 8.15, по-видимому, протекают через
промежуточное образование комплексов VIII-1 [276]. Водородные сдвиги в
алкильном катионе комплекса VIII-1 могут приводить к образованию более
стабильного катиона.
R~"@K CsHl1 :S==?: R~© -+сн-С3н7
комплекс ???-l
Химическая ионизация позволяет установить молекулярную массу
ароматического углеводорода и различить структурные изомеры. Однако, как и в
случае ЭУ, различить орто-, мета- и лара-изомеры по спектрам ХИ не удается.
Количество алкильных заместителей в бензольном ядре можно установить при
использовании в качестве газа-реагента дейтерированного метанола CH3OD
[277]. Этот реагент позволяет провести замещение всех ароматических атомов
водорода на дейтерий.
При использовании монооксида азота в спектрах алкилбензолов доминируют
пики М+'и [?+??]+. Соотношение пиков этих ионов зависит от энергии
ионизации молекулы [278].
Химическая ионизация отрицательных ионов (ХИОИ). Незамещенный
бензол не реагирует с гидроксил-анионом в связи с отсутствием кислых атомов
водорода. Однако в спектрах незамещенных полициклических ароматических
углеводородов наблюдаются пики ионов [? —?]~, [?+??]~, [?—?+??]~,
[?—?+?2?]~. Сечения ионизации ПАУ выше, чем в условиях ЭУ, что
позволяет использовать ХИОИ для детектирования этих соединений в следовых
количествах [279]. Для алкилбензолов образование ионов [М — Н]~ при
отщеплении бензильного атома водорода является доминирующим процессом. Депро-
тонированные молекулярные ионы очень устойчивы. Они могут присоединять
молекулу ?2?. Образовавшиеся ионы могут в свою очередь терять молекулу
воды. Для химической ионизации 0~# характерно образование ионов [М —Н]~~,
[М-2Н]-, [М-Н+ОГ, [M-R+0]-.
Инициирование фрагментации депротонированных молекулярных ионов
алкилбензолов требует активации соударением [280]. Наиболее характеристичным
является элиминирование молекулы алифатического углеводорода с захватом
атома водорода из opwo-положения (схема 8.16).
R1 R1
¦ (СбЩТ^КНг + R2H
Схема 8.16
об — об
198 Глава 8. Основные направления фрагментации органических соединений
Элиминирование молекулы С4Н4 является доминирующим направлением
распада иона [М—Н]~дифенилметана [281]. Этот процесс доказан изучением
спектров изотопных аналогов и протекает селективно, вероятно, через
промежуточную валентную изомеризацию в структуру бензола Дьюара (схема 8.17).
фГ-
·* PhCH-G=CH + C4H4
Схема 8.17
Задача 8.9. Какие отличия во фрагментации позволяют различить
перечисленные ниже соединения по их масс-спектрам электронного удара?
а) Пропилбензол и изопропилбензол;
б) бутилбензол, метилпропилбензол и диэтилбензол;
в) 2,4-диметилбутилбензол и 2,6-диметилбутилбензол;
г) бутилбензол, изобутилбензол, emop-бутилбензол и mpem-бутилбензол;
д) 1-пропилнафталин и 1,4,7-триметилнафталин;
е) 2-бутилантрацен, 5-метил-2-пропилантрацен и 2,3,4,7-тетраметилантрацен.
Задача 8.10. Какие интенсивные пики первичных фрагментов будут
наблюдаться в спектрах электронного удара перечисленных ниже соединений?
а) emqp-Амилбензол;
б) орто-метилизобутилбензол;
в) 1-метилнафталин;
г) 2-этилнафталин;
д) 2-пропилнафталин;
е) 2-этилантрацен;
ж) 1-бутилантрацен;
з) 1-пропилбенз[а]антрацен;
и) 2-трет-бутилнафталин.
Задача 8.11. Идентифицируйте соединение, спектр электронного удара
которого представлен в графическом и табличном виде (рис. 8.15).
-lOO
эо
80
70
60
50
40
ЗО
20
Ю
О
1,%
91
40 вО 80 ЮО 120 140
m/z
160
m/z
27
29
39
41
51
57
63
65
/,% J
2,61
3,61
3,28
7,11
4,14
1,97
1,36
8,23
m/z
77
78
79
90
91
92
93
I 104
/,% 1
3,03
5,40
2,56
1,20
100
72,9
5,03
1,15 |
m/z
105
115
117
119
133
148
149
[ 150
?%
1,73
1,25
1,26
2,54
4,83
20,7 !
2,50
0,14
Рис. 8.15
8.6, Спирты, фенолы, тиолы 199
Задача 8.12. Объясните образование устойчивого иона с m/z 165,
образующегося при распаде трифенилметана под электронным ударом.
Задача 8.13. Идентифицируйте соединение по его спектру электронного
удара (рис. 8.16).
100
90
80
70
60
50
40
30 -|
20 -|
10
о
1,%
178
76 89
151
1
X
40 60 80
100 120 140
m/z
160 180
m/z
| 39
50
51
63
74
75
76
88
I 89
/,% ?
1,58
1,60
1,61
3,41
2,17
3,86
9,51
7,18
12,2
m/z
150
151
152
175
176
177
178
179
| 180
/,% |
3,93
5,96
6,03
2,89
15,0
7,22
100
15,8
из
Рис. 8.16
8.6. Спирты, фенолы, тиолы
8.6.1. Спирты
Электронный удар. Введение в молекулу углеводорода гидроксильной
группы приводит к снижению энергии ионизации соединения. Однако при этом
уменьшается и интенсивность пика М+\ В спектрах третичных, а иногда
и вторичных спиртов пик М+# отсутствует вовсе (рис. 8.17,6 и в). Первый
потенциал ионизации молекулы спирта обусловлен удалением w-электрона атома
кислорода. Это означает, что в электронно-невозбужденном состоянии заряд и
неспаренный электрон локализованы на атоме кислорода. Нестабильность М4" *
вызвана благоприятными процессами фрагментации, инициируемыми
ионизированной гидроксильной группой.
Необходимо отметить, что возможность образования водородной связи между
молекулами спиртов иногда приводит к появлению в спектре пиков ионов
МН+. Интенсивность таких пиков не постоянна и зависит от давления паров
образца в источнике. Снижение напуска образца приводит к уменьшению их
интенсивности или полному исчезновению.
Определенные искажения в масс-спектр вносит также термическая
деструкция молекул. В результате возникают ионы [? —2?]+#, [?—?2?]4"',
[М—Нг,—НгО]*'. Эти же ионы могут образовываться и за счет обычных
200 Глава 8. Основные направления фрагментации органических соединений
1 00
90
80
70
60
50
40
30
20
1 0
о
1 00
90
80
70
60
50
40
30
20
1 0
0
1 00
90
80
70
60
50
40
30
20
1 0
О
Рис.
1,%
JJ
55
69
83
97
LJ
1 1 1
1 25
1 4 О
1 68
1 86
20
1,%
60
55
80
1 00
m /z
1 20
140
JLJj ?,?????
69
II, .?
83
97
1 1 5
¦ Nijl. ...I.li
1 60
1 80
1 68
20
?,%
40
60
10 100
m /z
1 20
JLL
57
69
87
???.,.,?,?
140
1 29
160 180
1 43
О 20 40 60 80 100 120 140 160 180
m /z
8.17. Масс-спектры электронного удара додеканола-1 (а), 2-метилундеканола-5 (б) и
2,6-диметил-4-пропилгептанола-4 (в)
процессов масс-спектрометрической фрагментации М+# (доказано спектрами ме-
тастабильных ионов).
Наиболее характерный процесс фрагментации молекулярных ионов спиртов
связан с ?-распадом и последующими перегруппировочными процессами с
выбросами молекул олефинов. Интенсивности пиков первичных ионов
определяются правилом выброса максимального алкила (разд. 6.1.1, схема 8.18,
рис. 8.17, в). Ионы характеристической «спиртовой» серии имеют значения
m/z 31, 45, 59, 73 и т. д. Пики ионов этой серии в масс-спектрах могут быть
как очень интенсивными, так и едва заметными (рис. 8.17). Схема 8.18 отражает
8.6. Спирты, фенолы, тиолы 201
+
ОН
/-С4Н9—С
-С4Н8
-1-С4Н9
+·
ОН
1-С4Н9—С—/-С4Н9
С3Н7
m/z 186
-С3Н7
+
ОН
II
/-С4Н9— С—/-С4Н9
Схема 8.18
основные направления фрагментации 2,6-диметил-4-пропилгептанола-4, спектр
которого приведен на рис. 8.17, е.
Ионы с m/z 1Ъ, 87, 31 (рис. 8.17, в) образуются в результате вторичных
перегруппировочных процессов, протекающих через четырехчленное переходное
состояние. Образование иона с m/z 45 можно объяснить протеканием перегруп-
пировочного процесса через шестичленное переходное состояние (схема 8.19).
сн2 он
сн3-сн) Сен
тД87
-СзН6
Схема 8.19
сн2=сн-он2
m/z 45
Наряду с пиками спиртовой серии высокую интенсивность будут иметь пики
ионов алкановой и алкеновой серий. Особенно это проявляется при распаде
неразветвленных соединений. Эти углеводородные фрагменты могут
обусловливать в случае тяжелых спиртов практически весь ионный ток (рис. 8.17, а).
Важным направлением распада спиртов является элиминирование молекулы
воды. Атом водорода, отщепляющийся вместе с гидроксильной группой, может
захватываться из самых разных положений, хотя наиболее вероятными являются
202 Глава 8. Основные направления фрагментации органических соединений
положения 2, 3 и 4. В результате этого процесса возникают олефиновые или
циклановые ионы. Выброс молекулы воды может сопровождаться
элиминированием молекулы этилена. Показано, что при этом отщепляются а- и /?-атомы
углерода. Этот процесс (образование иона [М—46]+#) протекает по механизму,
представленному на схеме 8.20. По значению m/z этого иона можно судить о
наличии разветвлений у а- и /?-атомов углерода, поскольку в этом случае вместе
с молекулой воды отщепляется не С2Н4, a C2H3R.
/ОН
сн2( н
СЩ R2
-R!CHCH2
-Н20
Схема 8.20
CHf^R2
Для бензиловых спиртов (рис. 8.18) характерно элиминирование атома
водорода, за которым следует отщепление молекулы СО. Далее этот фрагмент с
выбросом молекулы водорода превращается в фенил-катион. Следует отметить,
что отрыв атома водорода может идти от любого из атомов углерода, что
подразумевает трансформацию М+* до распада [32].
Отщепление от М+" гидроксильной группы протекает в случае бензиловых
спиртов, поскольку в результате образуется стабильный бензильный (тропилие-
вый) катион. Фрагмент с m/z 105 (рис. 8.18) образуется в результате
окислительного процесса с отщеплением молекулы водорода и образованием молекулярного
иона бензальдегида, который в дальнейшем теряет атом водорода [282].
Для алкильных производных бензилового спирта характерны процессы,
описанные выше для алкилбензолов. Проявление оряю-эффекта можно
наблюдать при фрагментации о-алкилбензилового спирта. В отличие от мета- или
ЮО
??
80
70 -I
во -J
50 -I
40
зо
ZO -\
ло
19°Л>
lLl
lLL
1.Ml
108
50
TO
mf-z.
100
Рис. 8.18. Масс-спектр электронного удара бензилового спирта
8.6. Спирты, фенолы, тиолы 203
1 00
90
80
70
60
50
40
30
20
1 О
104
-J 1,%
79 9 1
107
1 22
39
5 1
65
70 80
m /z
Рис. 8,19. Масс-спектр электронного удара о-метилбензилового спирта
лара-изомеров М+# этих соединений теряют молекулу воды (схема 8.21,
рис. 8.19, ион 104).
-н20
Схема 8.21
Для циклических спиртов механизмы фрагментации остаются такими же.
?-Распад остается основным направлением и протекает с большей вероятностью
в месте разветвления углеродного скелета (рис. 8.20 и схема 8.22). Последующие
процессы с выбросом молекул олефинов обусловливают серию
характеристических нечетноэлектронных ионов с массами 44, 58, 72, 86
Распад циклопропанолов начинается с раскрытия кольца. Для циклобутано-
лов, как и для циклобутановых углеводородов, характерен разрыв малого цикла
пополам. В образовавшейся паре фрагментов заряд предпочтительно
сохраняется на кислородсодержащем. Для циклоалканолов, начиная с циклопентанола,
-то
90
80
70
60
SO
40
зо
20
ю
1,%
¦ ¦¦ '?
вО 70
m/z
ЭО ЮО НО 120
Рис. 8.20. Масс-спектр электронного удара 3-метилциклогексанола
204 Глава 8. Основные направления фрагментации органических соединений
важную роль в масс-спектрометрическом распаде играют перегруппировочные
процессы. В частности, максимальную интенсивность в спектре имеют пики
ионов, образовавшихся в результате миграции атома водорода и отщепления
алкильного радикала (m/z 57 и 71 на схеме 8.22). Миграция атома водорода
протекает селективно, как это показано на схеме. Механизм процесса подтвержден
спектрами дейтерированных аналогов [32].
m/z6%
+
он
m/z 57
-с2щ - +.
- СН2=СН-ОН
m/z 44
Схема 8.22
На основании молекулярной массы соединения и массы перегруппировоч-
ных ионов (m/z 57, 71, 85 и т. д.) можно делать выводы о месте алкильного
радикала в молекуле. Если алкильный заместитель расположен в положении
2 циклогексанового кольца, из двух альтернативных фрагментных ионов этого
типа чуть более интенсивным будет незамещенный (m/z 57). Объяснить этот
факт можно большей легкостью разрыва С—С-связи в месте разветвления. Для
циклогексанолов с алкильным заместителем в положении 3 предпочтительным
оказывается образование более тяжелого из пары альтернативных фрагментов.
По-видимому, решающим оказывается факт образования замещенного олефина.
Если заместитель находится в положении 4, он будет элиминирован в составе
алкильного радикала вне зависимости от первоначального разрыва связей С\ —Св
или С]—С2. Заместитель в положении 1, напротив, всегда остается в составе за-
8.6. Спирты, фенолы, тиолы 205
???
90
80
70 -|
во
50
+о
ЗО -j
20 -|
10
\°л>
31
<45
so во
m/z
90 ???
ЮО
90
80
70
во
50
40
ЗО
20
Ю
1,%
..|||,Щ
148 162
20 ЗО -40 SO 60 70 80 90 ЮО НО 120 130 140 1SO 160 170
m/z
ЮО ?
эо -
во -
70 -
во -
50 -
40 -
ЗО -
20 -
Ю -
о -I
1/2
/о
31
у
J
«43
J
,
Щ
57
.
? ^
и
? ?
J jjl
73
III
86
,|
jU
юз
^lll , ,, ,, ,, ,.,.
в
163
ЗО 40 50 60 70 80 ЭО ЮО 11 О 120 130 140 150 160 170
m/z
Рис. 8.21. Масс-спектры электронного удара 1,2-бутандиола (а), глюкозы (б) и сахарозы (в)
ряженного фрагмента этого типа. Однако метальный радикал может отщепляться
из М+' (?-распад), приводя к интенсивным пикам фрагмента [М—СНз]+.
Элиминирование воды может протекать с захватом атома водорода в
основном из положения 4, а также из положений 3 и 5 цикла [89]. Дальнейший распад
иона [? — ?2?]+' обусловливает образование ряда углеводородных фрагментов,
характеризующихся интенсивными пиками в спектре (схема 8.22).
Гликоли и полиолы распадаются по тем же механизмам, что и
монофункциональные спирты. Пики их молекулярных ионов имеют очень низкую
интенсивность или вообще отсутствуют. Наиболее благоприятны ?-разрывы при
атомах углерода, связанных с гидроксильными группами (m/z 31, 59, 61 для
1,2-бутандиола, рис. 8.21, а).
Низкая летучесть и низкая термическая устойчивость Сахаров приводит
к появлению в спектрах электронного удара большого числа пиков ионов,
обусловленных продуктами термической деструкции исходной молекулы. На
рисунке 8.21,5 представлен масс-спектр глюкозы. Пик молекулярного иона
отсутствует. В области высоких значений m/z можно отметить последовательные
выбросы молекул воды и метилового спирта, однако интенсивности пиков этих
206 Глава 8. Основные направления фрагментации органических соединений
ионов малы. Доминируют в спектре пики с m/z 18 (?^?"), 31 (СНгОН"),
42 (СН2СО+·), 59 (С2Н30+), 60 (С2Н40^), 72 (С3Н4О+·), которые
образуются как в результате масс-спектрометрического распада, так и в результате
термической деструкции. Спектры дисахаридов характеризуются полным
доминированием ионов с низкими массами (рис. 8.21, в). С увеличением степени
полимеризации возможности электронного удара значительно уменьшаются по
причине термолабильности и крайне низкой летучести этих соединений. Для
увеличения термической стабильности молекулы используют метилированные
или ацилированные производные Сахаров. Хотя молекулярные ионы отсутствуют
даже для моносахаридов, и в этих случаях масс-спектры содержат меньше
пиков, обусловленных продуктами термического распада. Основные направления
распада можно классифицировать, допустив, что в электронно-невозбужденном
состоянии заряд и неспаренный электрон локализованы на гетероциклическом
атоме кислорода [15]. Не вдаваясь в подробности, следует отметить, что в
настоящее время накоплен весомый материал по фрагментации Сахаров, обобщенный
в обзорах О. С. Чижова [283, 284].
Химическая ионизация несколько улучшает ситуацию, но только для
низкомолекулярных Сахаров. Причина заключается в необходимости переводить
вещество в газовую фазу. Пики молекулярных ионов низкой интенсивности
появляются в масс-спектрах ХИ метаном [285] и изобутаном [286]. По этим
спектрам можно делать и определенные структурные выводы. Напротив,
спектры ХИ ионом аммония [287] и хлорид-анионом [288] позволяют легко
устанавливать молекулярные массы Сахаров. Спектры характеризуются интенсивными
пиками продуктов присоединения [?+??4]4" и [М+С1]~. Поскольку в спектрах
олигосахаридов [289] в результате разрыва гликозидных связей наблюдаются
интенсивный пик молекулярного иона и пики фрагментных ионов (альтернативно
эти спектры можно получить с использованием тандемной масс-спектрометрии),
стало возможно масс-спектрометрическое секвенирование Сахаров.
Использование десорбционной химической ионизации (разд. 2.3) позволило,
в некоторой степени, преодолеть проблемы с летучестью образцов [290]. Как
и в случае классической химической ионизации, лучших результатов удается
добиваться при использовании мягких ионов-реагентов (??^ и С1~).
Основными современными методами ионизации являются МЛДИ (разд. 2.15)
и электрораспыление (разд. 5.12), которые позволяют установить не только
молекулярную массу, но также природу и последовательность моносахаридов,
места их связывания [291-295].
Химическая ионизация. Протонированные молекулярные ионы МН+
алифатических, алициклических, аллиловых и бензиловых спиртов не стабильны и
распадаются в момент образования с выбросом молекулы воды. В условиях
ионизации метаном и более мягким изобутаном пик МН+ в спектре отсутствует.
Ион [??—Н20]+, напротив, характеризуется максимальным пиком в спектре.
8.6. Спирты, фенолы, тиолы 207
Установить молекулярную массу спирта можно также по иону [М —Н]+, пик
которого весьма интенсивен. Этот ион образуется по механизму отрыва аниона
(гидрид-иона) в момент ионизации (разд. 2.2). Отщепление происходит от а-ато-
ма углерода. Поэтому в спектрах третичных спиртов пик этого иона не
наблюдается. Ион [М—Н]+ далее элиминирует молекулу воды с образованием алкеново-
го иона. Дальнейший распад первичных ионов ([МН—НгО]* и [М — Н,—Н^О]4")
связан с выбросами молекул олефинов. Интенсивность пиков соответствующих
ионов алкановой и алкеновой серий зависит от избыточной энергии первичных
ионов. При ионизации изобутаном внутренняя энергии иона меньше, и, как
следствие, интенсивности пиков вторичных фрагментов значительно ниже.
Пик протонированного молекулярного иона спирта можно увидеть в спектре
полифункциональных соединений с благоприятным пространственным
расположением функциональных групп. В частности, пик МН+ максимален по
интенсивности в спектрах гексен-4-ола-1 и гексен-5-ола-1 [296]. В спектрах
изомерных 1,4-циклогександиолов (рис. 8.22) интенсивность пика МН+ крайне
низка для транс-изомера и очень высока для t/ис-изомера. Причина заключается
в образовании внутримолекулярной водородной связи в t/wc-изомере.
нрон нон +
н-^^—н н-^_^-ок
Эффективный масс-спектрометрический метод структурного анализа
изомерных диолов предложен Эйхманом [297]. Метод основан на измерении
соотношения интенсивностей пиков и определения путей распада ионов [М+Н]+ и
[М+СН3ОСН2, — СНзОН]4" в спектрах ХИ диметиловым эфиром. Триметил-
борат в качестве газа-реагента позволяет различать стереоизомерные диолы и
полиолы [298].
По спектрам химической ионизации монооксидом азота можно различить
изомерные первичные, вторичные и третичные спирты [299]. Третичные
спирты характеризуются только ионом [М —ОН]+, вторичные — ионами [М—Н]+,
[М—ОН]4" и [? —2?, +NO]+, а первичные — наряду с указанными еще ионом
[М — ЗН]+. Ключевым моментом является реакция окисления молекулы спирта,
сопровождающаяся потерей двух атомов водорода и образованием
карбонильного соединения. Возникающие из первичных спиртов альдегиды реагируют с
ионом-реагентом NO+ или по механизму электрофильного присоединения, или
по механизму отрыва аниона (гидрид-иона) с образованием иона [М—ЗН]+.
Образующиеся из вторичных спиртов кетоны могут только присоединять NO".
Перегруппировка 1,2-диолов, протекающая в ионном источнике
масс-спектрометра в условиях химической ионизации [300], совершенно аналогична
классической пинаколиновой перегруппировке этих диолов в растворе, впервые
208 Глава 8. Основные направления фрагментации органических соединений
описанной Р. Фиттигом еще в 1849 г. На схеме 8.23 этот процесс представлен на
примере 2,3-диметилбутандиола-2,3.
HOvOH (ЮН СН3
ЫЛ/СНз ~?2? сн3>|/СНз снзч1
' сн3 сн3/ч^"сн3 сн3/ ??3
Схема 8.23
При последующем более детальном изучении пинаколиновой
перегруппировки в условиях химической ионизации была подтверждена идентичность ее
механизма для целого ряда алифатических, алициклических и ароматических
соединений [301]. В процессе перегруппировки арильная группа мигрирует
предпочтительнее алкильной. Это наблюдение совпадает с известными ранее
данными о миграционной способности радикалов в кислотно-катализируемой
пинаколиновой перегруппировке в растворах [302].
Химическая ионизация отрицательных ионов. В результате депротониро-
вания молекула спирта теряет протон гидроксильной группы. Образующийся
алкоголят-анион обусловливает максимальный пик в спектре. Вторым
характеристическим ионом является [М —ЗН]~, образующийся в результате двух-
стадийного процесса элиминирования молекулы водорода депротонированным
молекулярным ионом (схема 8.24). Расчеты аЪ initio и измерение изотопных
эффектов при фрагментации дейтерированных аналогов подтвердили протекание
1,2-элиминирования через промежуточное образование комплекса альдегида с
гидрид-анионом [303].
RCH2CH2O" * [H"(RCH2CHO) ] ·* [ H2(RCHCHO)~] ~**2 ш RCHCHO
Схема 8.24
Более интенсивная фрагментация наблюдается при использовании в качестве
иона-реагента О-'. В работе [304] предложен механизм образования ряда фраг-
ментных ионов. В частности, последовательность превращений, изображенная
на схеме 8.25, позволяет делать выводы о заместителях у ?-атома углерода.
н-'°Н
-н2о ?
? «- ch=ch-r'
ch=ch-r'
Схема 8.25
Если величина сродства к электрону углеводородного радикала положительна
(фенил, бензил и др.), алкоголят-анионы могут распадаться с выбросом моле-
8.6. Спирты, фенолы, тиолы 209
кулы формальдегида и образованием соответствующего аниона. Анион PhCH2
образуется также при распаде депротонированных молекулярных ионов спиртов
Ph(CH2)„OH, где ? = 3-5 [305].
Сигматропные [1,3]-сдвиги достаточно редки в химии растворов [306], однако
и практика масс-спектрометрии, и теоретические расчеты [101] свидетельствуют
о выгодности этих процессов для заряженных частиц. В частности, такой
процесс протекает весьма активно в случае снятия стерического напряжения.
Например, депротонированный 1-винилциклобутанол изомеризуется до распада
в циклогексанон (схема 8.26). Поскольку никаких иных пиков в спектрах
активации соударением иона [М—Н]~ циклогексанона не наблюдается, авторы
[307] заключили, что перегруппировка протекает для 100% исходных молекул
винилциклобутанола.
Схема 8.26
Инициирование пинаколиновой перегруппировки щелочами менее известно.
Такая перегруппировка протекает в стерически напряженных системах [308]
или в а-хлоргидринах [309]. В работе [310] методом МС/МС исследованы
два основных механизма этой перегруппировки в условиях ХИ ионами ??^~.
Если потенциально мигрирующими группами являются алкильные радикалы и
нет стерических ограничений для того, чтобы атомы кислорода могли принять
сшяш-конформацию, реакция идет через образование эпоксидного интермедиата
(схема 8.27а). В случае миграции ароматических радикалов [311] либо в
пространственно затрудненных системах (^wc-2-метоксициклогексанол) протекает
именно пинаколиновая перегруппировка (схема 8.276).
сн30
<??3??
л
сн3
D
с—с
U
/:?3
СН30"Н-СН2<
сн3
,сн3 -сн3он сн2 сн3
/ч/-сн3 сн/ |Чяз
Схема 8.27а
nf~
^^^ \г
ОСНз
сн3о
- СН3ОН
>
Схема 8.276
210 Глава 8. Основные направления фрагментации органических соединений
8.6.2. Фенолы
Фенолы характеризуются интенсивным пиком молекулярного иона.
Специфический «фенольный» распад заключается в элиминировании из М+# частиц
СО и НСО' (схема 8.28). Вероятно, в газовой фазе часть молекулярных ионов
благодаря кетоенольной таутомерии имеет форму кетона.
Наличие алкильного заместителя в молекуле фенола приводит к реализации
бензильного распада и перегруппировочного процесса по механизму
Мак-Лафферти, которые описаны для случая ароматических углеводородов (разд. 8.5).
Эти процессы доминируют и могут в существенцой мере маскировать
«фенольный распад». Например, интенсивность пика [М — Н]+ в спектрах метилфенолов
практически равна интенсивности пика М+". Следует подчеркнуть, что для о-и
/?-алкилфенолов с достаточной для протекания перегруппировки Мак-Лафферти
длиной заместителя, предпочтительным оказывается бензильный разрыв, а для
.метя-изомеров — перегруппировочный процесс. Это объясняется возможностью
дополнительной стабилизации бензильного катиона благодаря участию
заместителя. Энергия образования катиона уменьшается, и процесс простого разрыва
связи успешно конкурирует с перегруппировкой Мак-Лафферти. Для
мета-изомера, такое участие заместителя невозможно [312].
он ?
,СН2 _н2о
&
Схема 8.29
Для opmo-замещенных фенолов можно также отметить процесс,
обусловленный opwo-эффектом. Он связан с элиминированием молекулы воды (схема 8.29).
Интенсивность пика [М—НгО]-1-' невелика, но позволяет отличить о-алкилфенол
от его мета- и ляра-изомеров (рис. 8.23). Для мета- и ляря-замещенных
алкилфенолов, как и для самого фенола, элиминирование молекулы воды не
характерно.
Химическая ионизация. Пики протонированных молекулярных ионов
фенолов доминируют в спектрах химической ионизации. Однако отсутствие
фрагментации делает спектры этого типа менее информативными, чем спектры
электронного удара. Учитывая, что информацию о молекулярной массе фенола
8.6. Спирты, фенолы, тиолы 211
-too
??
80
то
во
so
ДО
зо
20
Ю
19°уЬ
Ah
39
jliL
JL·
юо
??
во
то
во
SO
40
ЗО
20
Ю
1.<И»
39
????
60 70
m/z
Рис. 8.23. Масс-спектр электронного удара о- (а) и л*-метилфенола (б)
можно легко получить по спектру ЭУ, серьезных исследований по ХИ фенолов
не проводилось.
Для фенолят-аниона (условия химической ионизации отрицательных ионов)
характерны выбросы молекулы СО и радикала НСО' [313], т. е. те же процессы,
которые описаны для ионизации электронным ударом.
8.6.3. Тиолы
Энергии ионизации тиолов примерно на 1 эВ ниже, чем у аналогичных спиртов.
Молекулярные ионы достаточно стабильны. Их пики имеют интенсивность до
10% в полном ионном токе и отчетливо проявляются в спектрах даже для
достаточно «тяжелых» соединений (рис. 8.24). Процессы фрагментации тиолов
под ЭУ аналогичны, описанным для спиртов. В спектрах прослеживаются пики
ионов алкановой, алкеновой и тиольной (m/z 47, 61, 75...) серий. Присутствие
серы в составе иона легко детектируется по пику изотопного иона А+2
(разд. 4.2). М+# тиолов элиминируют молекулу H2S, однако альтернативно
может отщепляться радикал SH*. Элиминирование молекулы сероводорода может
сопровождаться выбросом молекулы этилена.
Для меркаптанов с неразветвленной углеводородной цепью аномально
высокую интенсивность в тиольной серии имеет пик иона с m/z 89 (рис. 8.24).
Ион имеет циклическую структуру и образуется по механизму внутреннего
замещения, представленному на схеме 8.30.
R^\
SH
-R
Схема 8.30
$н
m/z 89
212 Глава 8. Основные направления фрагментации органических соединений
ЮО ?
во -
70 -
во -
SO -
ло -
зо -
20 -
ю -
\9<У6
А-Л
.
J il
I
ш „
? 5?
??
il11,
84
1 .
а
118
|
ЮО
GO
ао
70
во
50
40
ЗО
20
Ю
ЗО ЛО 50 вО ГО вО ©? 100110120
m/z
55
юз
К ,??,
202
J
ЮО 120
nVz
Рис· 8.24. Масс-спектры электронного удара гексантиола (а), додекантиола (б)
Циклоалкилтиолы распадаются аналогично вторичным тиолам. Основной
для алициклических спиртов процесс, приводящий к максимальным пикам в
спектрах (m/z 71 на схеме 8.22 и рис. 8.20), значительно менее характерен для
серосодержащих аналогов. В спектрах циклоалкилтиолов интенсивность пика
иона с m/z 73 (для замещенных в положения 2, 3, 5, 6 m/z 72 + R) не превышает
10% (рис. 8.25).
Особенности распада тиофенолов позволяют предположить, что для
молекулярных ионов этих соединений кетоформа более выгодна, чем енольная. Эта
форма может легко объяснить наличие в спектрах интенсивных пиков ионов
[М — CS]4", [М—СгНг]*'. Помимо этого, в отличие от реакций распада фенолов,
интенсивно идут процессы с элиминированием радикала SH' и атома серы
(рис. 8.26).
ЮО
90 ?
80
70
во
SO -\
40
ЗО
20
Ю
?,? , ?
??,? „.II
20 ЗО 40 50 вО 7? вО ?? ЮО НО 120 130 140
m/z
Рис. 8.25. Масс-спектр электронного удара 2-метилциклогексантиола
ЮО ?
ЭО -
во -
70 -
во -
so -
«to -
зо -
20 -
Ю -
О -
\°si>
39
... ¦!¦ I
51
-4\
вв
, II. 1,
84 ?
? ? , , 1
но
L ,
вО 7?
m/z
ЭО ЮО НО 120
Рис. 8.26. Масс-спектр электронного удара тиофенола
8.6. Спирты, фенолы, тиолы 213
Задача 8,14 Пик какого первичного фрагмента будет наиболее интенсивным
в спектрах перечисленных ниже соединений?
а) Метилэтилпропилкарбинол;
б) изоамилвторбутилэтилкарбинол;
в) октанол-3;
г) З-метилдеканол-4;
д) 3,3-Диметил-4-этилоктанол-4;
е) гексадеканол-1;
ж) гептадеканол-5.
Задача 8.15. Как с помощью масс-спектрометрии можно различить
перечисленные ниже изомерные соединения?
а) Бутанол-1 и бутанол-2;
б) пентанол-1, пентанол-2 и пентанол-3;
в) гексанол-1 и 2-метилпентанол-1;
г) 2-метилоктанол-1 и З-метилоктанол-1;
д) диметилэтилкарбинол и диэтилкарбинол;
е) 4,4,4-трифенилбутанол-2 и 3,4,4-трифенилбутанол-2;
ж) бутантиол-1 и бутандиол-1,4.
Задача 8.16. Идентифицируйте соединение по его спектру электронного
удара (рис. 8.27).
ЮО ?
ЭО -
80 -
70 -
во -
SO -
АО -
ЗО -
20 -
10 -
о -I
19<У*
III
I , lUI+J
31
L-
42
, ?
-4UjUa r
59
I,
-Ц
m/z
19
26
27
28
29
j 30
/,%
1,09
4,50
18,9
6,85
17,5
2,55
m/z
31
32
33
39
41
1 42
/, % 1
100
2,64
1,24
3,18
6,96
9,38
m/z
43
45
57
59
60
1 61
7, %
3,47
1,81
1,50
10,5
7,03
0,22
m/z
Рис. 8.27
Задача 8.17. Какие различия будут наблюдаться в спектрах электронного
удара перечисленных ниже соединений?
а) 2-Метилциклогексанол и 3-метилциклогексанол;
б) 2-метилциклогексанол и 4-метилциклогексанол;
в) 2-метилциклопентанол и циклогексанол;
г) 2,4,6-триметилфенол и 4-пропилфенол;
д) 2-фенилэтанол и 1-фенилэтанол;
е) декантиол-1 и З-метилнонантиол-1;
ж) гидрохинон и тиофенол.
214 Глава 8. Основные направления фрагментации органических соединений
Задача 8.18. Какое из перечисленных
представленным на рис. 8.28 масс-спектром?
а) Ундекан;
б) деканол-2;
в) 3,4-диметилоктанол-2;
г) деканол-3;
д) деканол-1;
е) З-метилнонанол-3;
ж) 3,4-диэтилциклогексанол;
з) 2,7-диметилоктанол-4;
и) деканаль;
к) 7-метилнонанол-1;
л) децен-1.
ниже соединении характеризуется
Дайте аргументированный ответ.
ЮО ?
90 -
80 -
70 -
во -
50 -
АО -
ЗО -
20 -
Ю -
О -
т,%
<*
jL|
1 I
и
[59
6?
и
. 83
1 ,,к
111
-О-
12?
20 АО GO 80 ЮО 120 140 160
m/z
Рис. 8.28
m/z
27
28
29
31
39
41
42
43
1 44
45
55
56
57
[ 58
/,% 1
10,5
3,61
18,1
20,4
5,25
31,0
7,42
26,9
2,35
1,53
37,3
11,2
17,8
9,84
m/z
59
60
69
70
71
72
83
84
97
111
112
129
130
I 140
/,%
100
3,42
70,3
9,10
2,08 j
2,55
6,09
2,78
1,94
15,0
1,82
12,4
1,07
2,38
Задача 8.19. Идентифицируйте соединение по его спектру электронного
удара (рис. 8.29).
ЮО
90
80
70
во
50
40
ЗО
20
ю
о
-
-
-
-
-
\J°A>
31
III I.
—,,| l,| , ,—^??
?5
?-. ,
61
?-1 Г ! 1 Г \
20 2S ЗО 3S 40 45 SO 55 60 65 70 75 ??
m/2
Рис. 8.29
m/z
26
! 27
29
30
31
33
39
41
42
43
/,% I
3,07
16,7
18,1
1,86
18,3
1,90
1,80
1,89
2,97
17,2 1
m/z
44
45
46
57
61
62
63
75
76
1 77
/,%
6,18 i
100 i
2,46
1,82
4,71
0,11
0,02
0,20
0,58
0,02
Задача 8.20. Составьте предположительные схемы фрагментации для
распада перечисленных ниже соединений под электронным ударом.
а) З-Этилгексанол-3;
б) диэтилбутилкарбинол;
8.6. Спирты, фенолы, тиолы 215
в) 5-этилдеканол-З;
г) метилэтилпропилкарбинол;
д) триэтилкарбинол;
е) триизопропилкарбинол;
ж) 4-метилфенол;
з) 3-этилфенол;
и) 3-метилциклопентанол.
Задача 8.21. На рисунке 8.30 представлены масс-спектры электронного
удара 2-бромфенола и 3-бромфенола. Сделайте отнесение.
0 о
9 О
8 О
7 0
6 О
5 О
4 0
3 0
2 О
1 0
1 0 О
9 О
8 О
7 О
6 О
5 О
4 О
3 О
2 О
1 О
О
I , %
6 5
9 3
? ?, %
¦и.. .il
J Jk-
4 0 6 0
8 0 10 0 12 0
m /?
Рис. 8.30
40 60 80 100 120 140 180 180
m /?
1 7 2 || б
14 0 16 0 18 0
Задача 8.22. Идентифицируйте соединение по его спектру электронного
удара (рис. 8.31).
100-,
QO
вон
70
60
50
40
ЗО
20
Ю
1,%
64
39 I
ill ..1.1 .1, .In
80 100 120
m/z
m/z
28
37
38
39
46
49
50
51
53
60
/, % ?
2,99
6,90
9,79
12,8
2,25
3,38
5,12
2,30
6,27
1,90
m/z
64
62
63
64
65
66
72
73
74
L75
/,% 1
6,12
10,7
33,0
49,8
21,0
2,52
2,09
9,45
4,13
4,_3lJ
m/z
92
93
99
100
101
102
128
129
130
1 131
/,%
20,9
2,70
6,48 |
8,85
2,67
3,09
100
6,86 !
33,9
2,21
Рис. 8.31
216 Глава 8. Основные направления фрагментации органических соединений
Задача 8.23. Какие различия будут наблюдаться в спектрах химической
ионизации водородом, метаном и изобутаном деканола-1?
Задача 8.24. На рисунке 8.32 представлен масс-спектр электронного удара
этилциклогексанола. Определите положение заместителя.
юо -.
so J 1,%
80 А
ГО А
вО А
so A
АО А
зо А
20 А
ю А
О -| 1
АЛ
5?
Q7
81
Л ЛО
ilii
95
*±>Ч-
20 ЗО 40 50 вО 70 вО ЭО ЮО 110 120 130
m/z
Рис. 8.32
Задача 8.25. Идентифицируйте монофункциональное соединение с
молекулярной массой 132 Да по спектру ЭУ (рис. 8.33).
юо
90
80
70
60
50
40
30
20
10
о
л^Л\
56
70
N.. ..ШИ.ц!.. .Ilji
132
98
30 40
50
60
70
80
m/z
Рис. 8.33
90
100
110 120 130
Задача 8.26. Идентифицируйте соединение по спектру электронного удара
(рис. 8.34).
ЮО ?
?? -
во -
70 -
во -
50 -
АО -
ЗО -
20 -
Ю -
1,%
1, ... .
АО
63
?..1,.??.?
во
89
J-llu., .ill ,?
во юо
m/z
115
-J
120
ЛАА
L
???
Рис. 8.34
! m/z
39
50
51
57
58
62
63
65
72
/,% ?
5,89
3,86
4,10
5,58
7,61
4,78
9,83
3,84
2,55
m/z
74
75
86
87
88
89
90
101
1 из
/,% 1
3,19
2,95
2,02
2,79
2,67
8,83
1,78
2,32
2,61
m/z
114
115
116
117
143
144
145
146
/.%
8,23
73,7
36,6
3,31
1,99
100
10,9
0,75
8.7. Простые эфиры и сульфиды 217
8.7. Простые эфиры и сульфиды
Электронный удар* Пики молекулярных ионов простых эфиров, за
исключением низших гомологов имеют незначительную интенсивность, однако эти ионы
все же более стабильны, чем М+" спиртов. Заряд и неспаренный электрон в
электронно-невозбужденном состоянии локализованы на атоме кислорода. За
счет протекания ионно-молекулярных реакций, как и в случае спиртов, возможно
образование ионов МН+. Для устранения этого эффекта необходимо понизить
давление паров образца в источнике. В целом масс-спектры эфиров
характеризуются теми же гомологическими сериями ионов, что и спектры спиртов:
алкановой, алкеновой, спиртовой (рис. 8.35).
На схеме 8.31 в качестве иллюстрации типичных процессов распада
представлены основные направления фрагментации изобутилпропилового эфира.
Первичные реакции инициируются радикальным центром (атом кислорода). В
результате ?-разрывов возникают ионы спиртовой серии, пики которых, как
правило, достаточно интенсивны. Положительный заряд в М+' локализован
на атоме кислорода, который достаточно сильно оттягивает на себя электроны
обобществленных с атомами углерода пар. Это активизирует реакции,
инициируемые зарядом, с образованием алкильных катионов. Эфиры с большими алкиль-
ными радикалами характеризуются интенсивными пиками нечетноэлектронных
фрагментов серии С„Н^#, которые образуются в результате элиминирования
молекулы ROH. Процесс миграции атома водорода в этой реакции неоднозначен,
как и в случае отщепления молекулы воды в спиртах. Следует отметить, что
молекула воды может отщепляться и молекулярными ионами простых эфиров
(m/z 70 на рис. 8.35, а), т.е. этот процесс не может служить критерием для
отнесения неизвестного образца к спиртам или эфирам.
, ? ? ? ? ,
¦i-r-
55 60
lil.i,
Рис. 8.35. Масс-спектр электронного удара изобутилметилового эфира (я) и
изобутилпропилового эфира (б)
218 Глава 8. Основные направления фрагментации органических соединений
сн2=он
m/Z3\
-СзнЛ
С4Н9
m/z 57
С3Н7
m/Z43
С4Н8
m/z56
сн2=он
/и/г 31
t t /\ -t
+. -C3H7· +· ' -C2H5* ' +
сн2=о-сн2-С2Н5 CH3-CH-CH2-0-CH2-C2H5 сн3-сн-а^-о=сн2
? ДО 116) / т/*Ю
-C3Hs|
1
???5? QH^"
m/z29 m/? 42
Схема 8.31
CH2=0—CH3
/иД45
Вторичные перегруппировочные процессы (ионы с m/z 31 на схеме 8.31)
инициированы зарядом и протекают в основном как 1,3-сигматропный сдвиг
через четырехчленное переходное состояние с миграцией гидрид-иона, однако
возможен и 1,5-сигматропный сдвиг через шестичленное (схема 8.32). В обоих
случаях к атому, несущему положительный заряд, мигрирует гидрид-ион.
CHi
WH
+ -CiHg
сн2=он «*-
m/z3\
\ 4п
сн3>
СН2ГН
сн^-о=сн2
0Л+.
СН2^0>-СН2-
С3Н6
??/? 87
Схема 8.32
»сн2=о-сн3
m/z 45
На рисунке 8.36 представлен масс-спектр циклогексилэтилового эфира.
Максимальную интенсивность имеет пик иона с m/z 85, образующегося по
механизму, описанному для циклогексанола. Второй по интенсивности пик в спектре
принадлежит иону с m/z 57, который образуется при элиминировании молекулы
00 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
??.
57
41
J..I. , ..U
?. L-
J JL
85
i-, Ц
128
_ . L—
20 30 40 50 60 70 80 90 ЮО HO 120 130
m/z
Рис. 8.36. Масс-спектр электронного удара циклогексилэтилового эфира
8.7. Простые эфиры и сульфиды 219
+·
едо н
m/z 128 rn/z 85
Схема 8.33
этилена ионом с m/z 85 (схема 8.33). Элиминирование из М+* молекулы
алифатического спирта (ион с m/z 82 на рис. 8.36) аналогично отщеплению
молекулы воды из М+' алициклических спиртов (схема 8.22).
Арилалкиловые эфиры характеризуются очень интенсивными пиками
молекулярных ионов. Для арилметиловых эфиров активно идет отщепление
молекулы формальдегида или последовательное отщепление метильной группы и
молекулы СО (аналог «фенольного» распада, представленного на схеме 8.23).
Если в ароматическом ядре такого эфира присутствуют длинные алкильные
заместители, то потере молекулы формальдегида предшествует бензильный разрыв
(схема 8.10). Еще одной реакцией распада М+' алкилариловых эфиров является
отщепление молекулы олефина (схема 8.34а). Образующиеся молекулярные
катион-радикалы фенолов характеризуются интенсивными пиками и очень
полезны для установления принадлежности образца к изомерным алкилфенолам
или фенилалкиловым эфирам. Для полиалкоксибензолов возможны
последовательные процессы отщепления молекул олефинов согласно схеме 8.34а.
Аг-<>-СН2— СН2—R * Аг-ОН + К-СН=СН2
Схема 8.34а
Дифениловый эфир распадается под ЭУ с выбросами атома водорода,
молекулы СО или радикала НСО\ Пик молекулярного иона максимален в спектре,
причем распаду может предшествовать изомеризация (схема 8.346).
*** - ссо - ???
Схема 8.346
? ?
:JC *>
~со >
-нсо
Ацетали и кетали имеют большую склонность к ?-разрыву, так как в
результате образуется резонансно стабилизированный ион (схема 8.35). Как
следствие, М+" этих соединений не стабильны и в спектрах ЭУ часто не
220 Глава 8. Основные направления фрагментации органических соединений
регистрируются. Тем не менее молекулярные массы этих соединений можно
установить, поскольку наряду с пиками ионов [М—R!]+ в спектрах наблюдаются
также интенсивные пики ионов [М—OR]+ и [М—OR2]+.
R1
RO-CH-OR2
-R1
- RO=CH-OR2
Схема 8.35
—* RO-CH=OR2
Диалкилсульфиды характеризуются интенсивным пиком молекулярного иона.
Повышенная интенсивность пика М+2 является важным указанием на наличие
в составе образца атома серы (разд. 4.2). Отсутствие пиков ионов [М —SH]+ и
[М—H2S]+# позволяет легко отличать сульфиды от изомерных тиолов.
Основные процессы фрагментации, как и в случае простых эфиров,
инициированы центром, несущим неспаренный электрон, и связаны с выбросом алкиль-
ных радикалов с разрывом Са—С^-связи. Интенсивность образующихся фраг-
ментных ионов определяется правилом выброса максимального алкила. В
частности, при распаде М+* гексилэтилсульфида (рис. 8.37) пик иона [М—CsHu]"
максимален в спектре, тогда как пик альтернативного иона [М —СНз]+ едва
заметен.
В отличие от простых эфиров разрывы связей алкильных заместителей с
гетероатомом протекают достаточно интенсивно, причем образуются не только
алкильные ионы, но и серосодержащие фрагменты с сохранением заряда на
гетероатоме (m/z 117 и m/z 61 на рис. 8.37). Этот процесс может сопровождаться
миграцией атома водорода (m/z 62 на рис. 8.37). Следует отметить, что ионы
с m/z 117 и m/z 61 могут образоваться и по другому механизму, связанному
с первоначальной реакцией замещения (схема 8.30 для тиолов). В этом случае
этильный радикал отщепляется от гексильной группы с образованием пятичлен-
ного циклического иона (схема 8.36), дальнейший распад которого объясняет
возникновение ионов с m/z 47, 56, 57, 61, 89. Несколько механизмов
задействовано и в образовании алкильных ионов (m/z 41, 43, 55, 56, 69). На схеме 8.36
представлены лишь некоторые из них. Этот пример демонстрирует богатство
ЮО ?
ЭО -
80 -
70 -
©О -
50 -
40 -
ЗО -
20 -
ю -
о -
I
,%
,—L
л·
д!
1
1л
S
1 ¦ ? "'
6
? 62
Li
I 1
1LJJ
75
8«4
??. , 11. ?, ,
117
? ,
¦l±t r-
146
¦ ? ' *
¦ь-,
?? ЭО
m/z
Рис. 8.37. Масс-спектр электронного удара гексилэтилсульфида
8.7. Простые эфиры и сульфиды 221
ОН?
ml
-н*
m/z 55
ЗДз-s
тД117
l··· +
С4Нв S-C^H5
m/z 56 m/z 61
~C2H4
-C2H5
CH21:CH2
s
?
m/z 61
4
+. -СНз +
hs-c2h5 * hs=ch2
m/z 62 m/z 47
c*hS'
m/z 84
-C3H5 / V-C3H7
+ -СзНб
CoHis-s-qhs
w/z 146
-c2H4
-C2H4
Q
0№
-C5H" тДП7
-СзНб
-SH* +.
-C4H8
m/z 56
C3H7 C3H5 -
/и/г 43 m/z 41
QsHn
m/z 83
m/z%9
-H
У
+ -C2H4
CH2=S-C2H5 * CH2=SH G1H7
m/z 75 m/z 47 m/z 55
Схема 8.36
и поливариантность процессов, протекающих при фрагментации органических
соединений под электронным ударом.
Среди вторичных процессов фрагментации можно выделить перегруппи-
ровочный процесс в ионах, обусловленных ?-распадом. Он протекает, как и
в случае спиртов или эфиров, через четырех- или шестичленное переходное
состояние и сопровождается элиминированием молекулы алкена.
Фрагментация сульфидов с алициклическим заместителем в целом
аналогична описанной для родственных циклоалкилтиолов и алициклических простых
эфиров. Фрагментация арилсульфидов (рис. 8.38) аналогична описанной выше
00 @ О
0 0 0
? ? ?
70 -
ео -
50 -
АО -
зо -
20 -
Ю -
О -
1,%
А
ш
5
Lj_
, II 1, . ?„, , , , 1
no
il
л.
T-J
23
Ji—,
15
—? г-'
2
L
©О 100
m/z
ПО 12О 130 ??? ISO
Рис. 8.38. Масс-спектр электронного удара фенилпропилсульфида
222 Глава 8. Основные направления фрагментации органических соединений
для соответствующих простых эфиров. Спектры малолинейчаты и обусловлены
?-разрывами (m/z 123) и перегруппировками Мак-Лафферти по алкильной цепи
(m/z ПО).
Химическая ионизация. В отличие от спиртов в условиях химической
ионизации протонирующими газами-реагентами простые эфиры характеризуются
максимальными по интенсивности пиками протонированных молекулярных ионов
МН+. Это позволяет однозначно определять молекулярные массы этих
соединений. Среди фрагментных ионов можно выделить [М—Н]+, RO=CH^, R+ и
ROH^, которые могут образовываться как непосредственно во время процесса
ионизации, так и в результате распада МН+ или [М — Н]+. Химическая
ионизация монооксидом азота приводит к образованию ионов [М — Н]+ в качестве
единственного продукта.
Первое сообщение о протекании перегруппировки Кляйзена в ионизационной
камере масс-спектрометра в условиях химической ионизации появилось в работе
[314]. Позже этот процесс был детально изучен на большой серии аллилфе-
ниловых эфиров с применением тандемной масс-спектрометрии [315]. Было
достаточно убедительно показано, что механизм (схема 8.37) перегруппировки,
происходящей при масс-спектральном распаде этих соединений, совершенно
аналогичен наблюдаемому в растворе при его инициировании трихлоридом бора
[316] или трифторуксусной кислотой [317, 318]. Аналогичная перегруппировка
протекает и для пропаргилфениловых эфиров [315].
г+
^ он ^ он + - сн, ^ .он
се—со- ^ ?
сн2
m/z 135 Схема 8.37
Химическая ионизация отрицательных ионов. В условиях химической
ионизации отрицательных ионов характеристическим является разрыв С—О-связи
(схема 8.38). Для диалкиловых эфиров этот процесс протекает столь интенсивно,
что пики депротонированных молекулярных ионов в спектрах не наблюдаются.
НО" + ROR -+ RO" + [R-H] + Н20
Схема 8.38
Для эфиров, содержащих двойную связь, характерно депротонирование по
аллильному положению. Ионы [М — Н]~ претерпевают перегруппировку по
8.7. Простые эфиры и сульфиды 223
механизму Е1сь с образованием интермедиата (схема 8.39), который далее
распадается до алкоголят-аниона [319].
-с2н4
Схема 8.39
Для ряда депротонированных молекулярных ионов эфиров с
ненасыщенными заместителями зарегистрированы перегруппировочные процессы, хорошо
известные в классической органической химии. В частности, ион [М—Н]~~
претерпевает перегруппировку Виттига по механизму 1,2- или 3,2-сигматропного
сдвига [320], за которой следует перегруппировка Коупа [321] в окси-варианте
(схема 8.40).
ОЮ
^°^-* д^ —"О
PhCHOR
Схема 8.40
Перегруппировка Виттига протекает также при распаде депротонированных
ионов алкилбензиловых эфиров (схема 8.41). Реакция идет через интермедиат,
который может быть комплексом радикала с анион-радикалом или аниона с
нейтральной молекулой [322].
[ R'iPhCHCO]
[ R-(PhCHO)]
Схема 8.41
Задача 8.27. Оцените интенсивности пиков фрагментных ионов,
образующихся по механизмам, инициированным центрами, несущими положительный
заряд и неспаренный электрон, в случае перечисленных ниже соединений.
Используйте материал этого раздела, а также материал и примеры, изложенные
в разд. 3.1.2 (правило Стивенсона).
а) Метилэтиловый эфир;
б) изопропилэтиловый эфир;
в) mpew-бутилметиловый эфир;
г) диизопропиловый эфир;
224 Глава 8. Основные направления фрагментации органических соединений
д) бензилэтиловый эфир;
е) пропилфениловый эфир;
ж) аллилбутиловый эфир.
Задача 8.28· Объясните образование фрагментных ионов (m/z 123, ПО,
109, 77, 65, 45), пики которых имеют значительную интенсивность в спектре
ЭУ пропилфенилсульфида (рис. 8.38). Составьте схему фрагментации для этого
соединения
Задача 8.29. Какие различия будут наблюдаться в масс-спектрах
перечисленных ниже изомерных соединений?
а) Диэтиловый эфир и метилпропиловый эфир;
б) метилэтиловый эфир и пропанол-1;
в) изопропилметиловый эфир и пропилметиловый эфир;
г) пропилфениловый эфир и этилбензиловый эфир;
д) третбутилэтиловый эфир и неопентилметиловый эфир;
е) лдра-толилэтиловый эфир и фенилпропиловый эфир;
ж) фенилэтиловый эфир и 4-этилфенол;
з) гексилтиол-1 и дипропилсульфид.
Задача 8.30. Составьте схему фрагментации для вторбутилпропилового
эфира, спектр ЭУ которого представлен на рис. 8.39.
ЮО -?
90 -I
80 А
70 А
60 А
SO А
АО А
зо А
20 А
Ю -1
1,%
J-L
ЮО НО
m/z
Рис. 8.39. Масс-спектр электронного удара e/wop-бутилпропилового эфира
Задача 8.31. Составьте предполагаемые схемы фрагментации под ЭУ для
перечисленных ниже соединений.
а) Диизопропиловый эфир;
б) бензилэтиловый эфир;
в) втор'бутип-трет-бутиловый эфир;
г) дибензиловый эфир;
д) метилгексиловый эфир.
8.8. Амины и фосфины 225
Задача 8.32. Идентифицируйте соединение по спектру ЭУ (рис. 8.40).
100
90
80
70
60
50
40
30
20
10
о
? ?>%
51
65
77 92
. I. J ¦..,., I.
-?
80
152
.I..
186
40
60
100
120
m/z
140
160
m/z
45
50
51
52
58
62
63
65
66
69
71
л % 1
3,80
9,16
29,8
1,73
2,20
1,46
3,68
13,9
2,81
8,92
1,18
m/z
74
75
76
77
79
80
82
83
84
92
1 93
/,% 1
2,66
2,02
2,20
10,0
1,75
2,01
2,85
1,30
2,04
7,56
3,09
m/z
102
108
109
110
115
121
139
141
142
151
1 152
/, % 1
1,99
2,87
6,53
1,90
2,26
1,16
2,69
2,64
2,17
1,70
8,13
m/z
153
154
160
171
183
184
185
186
187
188
1 189
/,%
4,26
2,06
1Д9
5,70
3,70
26,1
65,1 I
100
15,8
4,69
0,61
Рис. 8.40
180
8.8. Амины и фосфины
Электронный удар. Аминогруппа обладает более сильными электронодонор-
ными свойствами по сравнению с гидроксильной. Положительный мезомерный
эффект атома азота больше, а отрицательный индуктивный эффект меньше,
чем у атома кислорода. Поэтому процессы распада спиртов, инициируемые
радикальным центром, еще в большей степени характерны для аминов.
На рисунке 8.41 представлены масс-спектры электронного удара изомерных
аминов. Амины обладают низкой энергией ионизации (9,4 эВ для этиламина).
В электронно-невозбужденном состоянии заряд и неспаренный электрон будут
локализованы на атоме азота. К сожалению, интенсивность пиков молекулярных
ионов аминов очень низка из-за высокой интенсивности процессов распада. Тем
не менее азотное правило (разд. 7.2.1) позволяет установить принадлежность
вещества к классу аминов.
8—1113
vo
о
о
(? *
о
' CM
?
О ^
? ?
?
?
о -"
•? ?
I I I I I I I 1 I I
???????????
? ? ? ? ? » ? ? ? 1
???????????
OaCN«IOt(9Nr
i ? ? ? ? ? ? ? ?
???????????
O0)«S(DI0tnN<>
ccj
s
се
8
03
ее
?
8.
С
?
о
о
oe
S
8.8. Амины и фосфины 227
Склонность аминогруппы к ионизации и инициированию распада хорошо
видна на примере этаноламина (схема 8.42). ЭИ фрагмента CH2NH2 составляет
6,2 эВ, а фрагмента СН2ОН — 7,6 эВ (табл. 3.2). В результате интенсивность пика
иона CH2NH2" в спектре составляет 100%, а интенсивность пика альтернативного
иона СН2<ЭН+ — 2,3%. В отличие от спиртов, в спектрах которых пики ионов
[?—?2?]+? могут быть весьма интенсивными, ионы [? —??^]" не
образуются.
НО=СН2 «« HO-CT2~Oi2--NH2 «·—·> ??-(?2-??2-??2 * CH2=NH2
Схема 8.42
Повышенное давление паров образца в ионном источнике приводит, как и в
случае спиртов, к появлению ионов МН+. Эти ионы имеют четную массу и
могут маскировать наличие атома азота в молекуле. Ионно-молекулярную природу
образования этих ионов легко распознать варьированием напуска образца.
Основные фрагментные ионы обусловлены ?-разрывами и последующими
перегруппировочными процессами с отщеплением молекул олефинов. Эти
реакции аналогичны описанным ранее для спиртов и простых эфиров (разд. 8.6 и
8.7) и приводят к характеристической гомологической серии «аминных» ионов
(m/z 30, 44, 58, 72...). Для первичных аминов в спектре доминирует пик иона
CH2NH2" с m/z 30 (рис. 8.41, а). С увеличением длины углеводородного
радикала возрастает доля /?, 7> S...-разрывов. Вероятность разрывов С—С-связей
уменьшается с удалением от аминогруппы, однако в спектрах неразветвлен-
ных первичных аминов пик иона с m/z 86 имеет повышенную интенсивность
(рис. 8.41, а). Он образуется в результате реакции представленной на схеме 8.43.
Любые разветвления в цепи приводят к резкому торможению этого процесса.
Следует отметить, что в отличие от тиолов и алкилгалогенидов образуется
именно шестичленный цикл. Правда, в случае аминов этот процесс выражен
менее ярко.
н н
XJ — О
Схема 8.43
Следующей стадией распада первичных ионов [М —Alk]+ является
отщепление молекулы олефина с миграцией гидрид-иона к центру, несущему заряд.
Обычно водород мигрирует через четырехчленное переходное состояние, но
возможна и реализация шестичленного. На схеме 8.44 эти процессы представлены
для случая дигексиламина (рис. 8.41,6). Заряд сохраняется на атоме азота во
всех этих последовательных процессах.
Распад циклоалкиламинов обусловлен теми же движущими силами, что и
распад алкиламинов. На рисунке 8.42 представлен масс-спектр N-этил-циклогек-
228 Глава 8. Основные направления фрагментации органических соединений
юо
90
80
70
60
SO ?
4-0
ЗО
20
ю ?
1,%
56
.1 II.?
50
ТО 80
m/z
ЮО НО 120
Рис. 8.42. Масс-спектр электронного удара N-этилциклогексиламина
силамина. Первой стадией трансформации молекулярного иона является разрыв
связей алицикла, инициированный радикальным центром. Именно в результате
этого процесса образуются фрагментные ионы, характеризующиеся наиболее
интенсивными пиками в спектре.
С4Н9 /Н
+ -C6Hi2
CH2=NH2 -
m/z 30
C3H7
^СБ-Н
chL ™Л (
|А^+ \^/>Ч+ -СзНю +
CH2^NH=CH2 «*—*> NCH2-NH-CH2 -CH2=NH-CH3
тДП4 /иД44
Схема 8.44
На схеме 8.45 представлены основные направления фрагментации М+"
циклогексилэтиламина. Схема не претендует на всю полноту описания реакций
распада, но дает представление об основных процессах, характерных для
распада циклоалкиламинов. Практически аналогично распадаются и производные
циклопентана [32].
СН9—/Н
ед-йн ед-йн Qh5-nh ?* (^
Q? CH3 -^ CH2-NH
CH2=NH
m/z 112
m/z84
—C2H4
CH2=CH-NH-C2H5
iw/*7l
Схема 8.45
+
NH2
I
m/z 56
8.8. Амины и фосфины 229
юо
©о
SO
7?
©О
SO
-*о
ЗО
20
1 О
о
I I
?,°/?
I—J-ь Ць-
aiu p-i
77
?.
j, Ш«
¦
Ю6
?
? ^
13S
?
L-
80
m/2
ЮО 120 140
Рис. 8.43. Масс-спектр электронного удара 4-пропиланилина
Фрагментами ион с m/z 84 на рис. 8.42 (m/z 56 для незамещенных по
атому азота алкилциклогексиламинов) аналогичен иону с m/z 57, образующемуся
при распаде алициклических спиртов. Его масса позволяет делать выводы о
наличии алкильных заместителей в различных положениях циклогексанового
кольца. В случае 2-алкилзамещенных из двух альтернативных пиков с массами
56 и 55 + R для незамещенных по атому азота алкилциклогексиламинов более
интенсивным будет первый, для 3-алкилзамещенных—второй. Для 4-алкилзаме-
щенных заместитель отщепляется в любом случае, и в спектре незамещенных
по атому азота алкилциклогексиламинов присутствует только ион с m/z 56.
Фрагментный ион с m/z 71 может образоваться при разрыве циклогексанового
кольца с одновременным отщеплением двух молекул этилена (схема 8.45). Для
1-алкилциклогексиламинов интенсивный пик часто обусловлен ионом,
образующимся при отщеплении заместителя.
Анилины характеризуются максимальным в спектре пиком молекулярного
иона. Достаточно высокую интенсивность имеет пик иона [? —?]4", имеющий
азатропилиевую структуру. Основной процесс фрагментации связан с
последовательным элиминированием из М+# молекулы HNC и атома водорода, что
аналогично потере молекулы СО и атома водорода из М+" фенолов, т. е. по
крайней мере, часть молекулярных ионов анилина существует в иминоформе
(схема 8.46).
Схема 8.46
Для алкилзамещенных анилинов основное направление распада связано с
отщеплением алкильного радикала (бензильный разрыв) и образованием бензиль-
ного (аминотропилиевого) катиона (рис. 8.43), который далее последовательно
элиминирует молекулы HNC и Е^- Для алкилариламинов доминирует ?-распад с
разрывом алифатической С—С-связи.
230 Глава 8. Основные направления фрагментации органических соединений
Ион [М—3]+ образуется при распаде молекулярных ионов диариламинов
аналогично тому, как это было описано выше для случая дифенилметана, и,
вероятно, имеет структуру карбазолил-катиона (схема 8.47).
Схема 8.47
Для фосфинов характерны те же направления распада, что и для аминов.
Однако более активно протекают процессы, инициируемые центром, несущим
заряд. В частности, помимо ?-разрывов С—С-связей активно идут перегруп-
пировочные процессы с отщеплением молекулы олефина и миграцией атома
водорода к фосфору. Ионы с m/z 90 и 62 при распаде триэтилфосфина (рис. 8.44)
принадлежат диэтилфосфину и моноэтилфосфину соответственно.
Достаточно интенсивные пики в спектрах алкилфосфинов могут
принадлежать ионам, образующимся в результате /^-разрывов (схема 8.48).
R2P-CH2-CH2—R
Схема 8.48
R2pJ
сн2
Следует подчеркнуть, что в отличие от аминов, ?-разрывы становятся менее
благоприятны для фосфинов и соответствующих соединений других элементов
V группы. Причина кроется в более высоком энергетическом барьере
трансформации атомов этих элементов из тетрагональной в тригональную форму.
Например, разница между энергией ионизации молекулы (С2Н5)зЭ и энергией
появления иона (С2Н5JЭ=СН2~ (где Э = N или Р) для фосфина на 1,17 эВ
больше, чем для амина.
Дифенилфосфин (рис. 8.45) характеризуется интенсивным пиком
молекулярного иона. Ион [М — 3]+ образуется по механизму, изложенному для случая
дифенилметана и дифениламина. Максимальный пик в спектре (m/z 108)
обусловлен отщеплением молекулы бензола из М+\
юо
90
80
7?
60
50
40
ЗО
20
Ю
л 1,%
¦ ? ,3*.!..!., ?.
-С2НЧ
75 _С2Нд
-С2Н4
70 80
nrVz
Рис. 8.44. Масс-спектр электронного удара триэтилфосфина
8.8. Амины и фосфины 231
юо
??
во
70
во
SO
40
зо
20
Ю
О
1,*&
о1_^
ЮО 120
m/z
Рис. 8.45· Масс-спектр электронного удара дифенилфосфина
Химическая ионизация. Амины легко протонируются в газовой фазе. В
спектрах химической ионизации водородом, метаном и изобутаном пики протониро-
ванных молекулярных ионов МН+ аминов имеют максимальную или близкую
к ней интенсивность. Два других важных характеристических фрагментных
иона—[М—Н]+ и [М—R]+. Последнему приписывается иминиевая структура,
т. е. структура основных первичных фрагментов, образующихся под ЭУ. Расчеты
показывают, что наиболее вероятным механизмом образования ионов этого типа
является отрыв карбаниона в процессе ионизации. Альтернативный процесс
отщепления молекулы алкана из МН+ менее вероятен, поскольку
характеризуется высоким энергетическим барьером [323]. Элиминирование молекулы NH3
из МН+ значительно менее выгодно, чем отщепление молекулы Н2О из МН+
спиртов (разд. ЗЛ.З).
Химическая ионизация отрицательных ионов. Два основных направления
распада депротонированных молекулярных ионов аминов связаны с
элиминированием молекулы водорода или молекулы алкана (схема 8.49). Последний
процесс может привести к альтернативному образованию карбаниона [324].
Представленные на схеме направления распада подтверждены спектрами
изотопно-меченных соединений. В спектрах активации соударениями
интенсивности пиков ионов, образующихся при отщеплении молекул водорода и алкана
примерно равны.
Депротонированный анилин распадается аналогично фенолят-аниону. В
данном случае отщепляется молекула HNC с образованием циклопентадиенил-
аниона.
R2CH-NH »
Т -RH ?-
[R-(Rra=NH)] ——\_
? - RCH=NH
R"
[H~(R2Q*NH)] -?*?_
—R2ON-
-^RfOT-C*=NH
RCH=N~
-> R'CH-CH=]SIH
Схема 8.49
232 Глава 8. Основные направления фрагментации органических соединений
В работе [325] масс-спектрометрически на примере трансформации бензил-
дифениламина в бензгидриланилин была изучена перегруппировка Стивенса,
которая может считаться азотным аналогом перегруппировки Виттига (схема 8.50).
Перегруппировка является доминирующим направлением реакции, о чем
свидетельствуют высокие выходы продуктов как в растворах при действии бутиллития
[326], так и в условиях химической ионизации отрицательных ионов [327]. Для
доказательства механизма протекания этого процесса использовали изотопно-
меченные соединения и тандемную масс-спектрометрию, однако установить,
идет этот процесс согласованно или постадийно, не удалось.
Ph2NCHPh
PhNCHPh2
PhN CHPh
Схема 8.50
Задача 8.33. Какие первичные ионы образуются при распаде под ЭУ
перечисленных ниже соединений? Оцените относительные интенсивности их пиков.
а) Бутилпропилэтиламин;
б) диметилэтиламин;
в) mpem-бутилизопропиламин;
г) mpem-бутилизопропилэтиламин;
д) emop-бутилпропилэтиламин;
е) изобутил-вто/?-бутил-т/?ет-бутиламин.
Задача 8.34. Для соединений, перечисленных в задаче 8.33, приведите
схемы образования вторичных перегруппировочных фрагментных ионов аминной
серии, которые могут образоваться при распаде первичного фрагментного иона,
характеризующегося максимальным пиком в спектре.
Задача 8.35. Составьте схему фрагментации под ЭУ бутилизопропиламина,
спектр которого представлен на рис. 8.46.
юо
90
80 -\
70
60
50
40
30
20
Ю
О
1,%
_4HJ
LiL
70 80
m/z
90 ЮО НО 120
Рис. 8.46. Масс-спектр ЭУ бутилизопропиламина
8.8. Амины и фосфины 233
Задача 8.36. Составьте схему фрагментации под ЭУ метилдиизопропилами-
на, спектр которого представлен на рис. 8.47.
юо
90
во
7?
во -I
50 А
АО А
зо А
20 -J
ю А
1,°Л>
л^
ill,
90 ЮО НО 120
m/z
Рис. 8.47. Масс-спектр ЭУ метилдиизопропиламина
Задача 8.37. Составьте схему фрагментации триэтилфосфина, масс-спектр
электронного удара которого представлен на рис. 8.44.
Задача 8.38. Составьте предполагаемые схемы фрагментации под ЭУ для
перечисленных ниже соединений.
а) Диэтиламин;
б) триэтиламин;
в) бутилдиэтиламин;
г) пропилдиэтиламин;
д) диизопропиламин.
Задача 8.39. Идентифицируйте вторичный амин по его масс-спектру ЭУ,
представленному на рис. 8.48.
юо ? ?,«?>
90
во
??
во
50 А
АО А
зо -|
20
?? ?
О
39
51
¦ II
4L_
70 80
m/z
©О ЮО НО 120 130
Рис. 8.48
234 Глава 8. Основные направления фрагментации органических соединений
Задача 8,40. Идентифицируйте соединение по его масс-спектру ЭУ,
представленному на рис. 8.49.
1,%
юо
??
80
70
60
50
40
30
20
10
43
76
63
118
30 40 50 60
70 80
??/?
90 ЮО 1Ю120
Рис. 8.49
m/z
33
34
39
: 41
? 42
I 43
44
45
/,%
8Д4
9,15
6,22
34,4
6,50
100
3,36
7,26 |
m/z
46
47
48
57
59
60
61
1 63
/,% 1
4,71
8,52
7,50
5,14
6,03
3,50
5,29
9,871
m/z
74
75
76
77
103
118
119
I 120
/,%
9,24
9,13
49,0
7,62
5,47
19,0
1,25
0,04
Задача 8.41. Как можно различить перечисленные ниже изомерные
соединения по спектрам ЭУ?
а) Метилэтиламин, пропиламин и изопропиламин;
б) дибутиламин и emop-бутилбутиламин;
в) дипентиламин и изопентилпентиламин;
г) дипропиламин и диизопропиламин;
д) 1-метилциклопентиламин и 2-метилциклопентиламин;
е) циклогексиламин и 2-метилциклопентиламин;
ж) циклогексиламин и N-метилциклопентиламин;
з) 2-, 3-, и 4-этилциклогексиламины;
и) дифениламин и 4-аминобифенил.
8.9. Алкилгалогениды, арилгалогениды
Электронный удар. Молекулы алкилгалогенидов легче теряют один из
электронов неподеленных пар гетероатома. Энергии ионизации невысоки, а заряд и
неспаренный электрон в электронно-невозбужденном состоянии М+#
локализованы на атоме галогена, которые, однако, не склонны к удерживанию заряда.
Поэтому интенсивность пика молекулярного иона невысока, а пики фрагментов,
содержащих атом галогена, также малоинтенсивны. Сравнивая производные
фтора, хлора, брома и иода, можно отметить, что интенсивность пика М+"
возрастает при уменьшении электроотрицательности гетероатома (рис. 8.50).
Для фторидов характерна гомологическая серия ионов с m/z 33, 47, 61, 75
и т. д.; для хлоридов (по 35С1) —серия ионов с m/z 49, 63, 77, 91 и т. д.; для
бромидов (по 79Вг) —серия ионов с m/z 93, 107, 121, 135 и т. д.; для иодидов —
8.9. Алкилгалогениды, арилгалогениды 235
? оо
9 0
80
70
60
50
40
30
20
1 О
о
1 00
9 О
80
70
60
5 О
4 О
30
20
1 О
О
1 00
90
80
70
6 О
50
4 О
3 О
20
1 О
О
1 00
90
8 О
7 О
6 О
50
40
3 О
20
1 О
О
Рис.
1,% |43
3 3
Jjjlj
69
83 .97
„? „?,? L, У.1,1
20 40 60 80 100 120 140 160 180 200 220 240 260
m /?
9 1
Li
1 05
., I,.. .
1 76
20 40 60 80 100 120 140 160 180 200 220 240 260
m /?
?, % |4 з в
5 7
1 35
? ''' '¦ г '—
20 40 60 80 100 120 140 160 180 200 220 240 260
m /?
4 3
85
14 1 155
"? ? ' ? " ' ? ? 1 "~
20 40 60 80 100 120 140 160 180 200 220 240 260
m /?
8.50. Масс-спектры электронного удара l-фтордекана (?), 1-хлордекана (б),—1-бром-
декана (в) и— 1-иоддекана (г)
236 Глава 8. Основные направления фрагментации органических соединений
серия ионов с m/z 141, 155, 169 и т. д. В случае хлора и брома, естественно,
наблюдается еще одна серия ионов с массами на две единицы больше за счет
изотопов 37С1 и 81Вг. Кстати, наличие изотопов позволяет очень просто
детектировать присутствие в молекуле атомов хлора и брома, а также устанавливать
их количество (разд. 4.2). Присутствие иода также легко обнаружить благодаря
большой атомной массе и тому факту, что иод имеет всего один природный
изотоп. Интенсивности пиков ионов характеристических серий могут быть как
весьма высоки (рис. 8.50,6 и в), так и крайне низки. Для алкилхлоридов и
алкилбромидов характерна еще одна особенность. В ряду пиков
гомологической серии аномально высокую интенсивность имеют пики ионов С4Н8СГ**
и С4Н8Вг+. Они образуются по механизму внутримолекулярной циклизации
(схема 8.51), описанному выше для алкилтиолов и алкиламинов. Образуются
именно пятичленные циклы [328]. Разветвления в углеродной цепи подавляют
этот процесс. Для фтора и иода он вообще не характерен (рис. 8.50, ? и г).
R< {+, -R· ? Hal
\J —'?
Схема 8.51
Поскольку положительный заряд лучше стабилизируется «мягкими» атомами,
можно было бы предположить большую склонность иода к локализации заряда
во фрагментных ионах. Для ионов На1+ это именно так. В спектрах многих
иодпроизводных интенсивности пиков ионов 1+ и Н1+# очень высоки. Они могут
быть максимальными в спектре. Однако для ионов СН2=На1+ наблюдается
обратная последовательность. Объяснить этот факт можно более значительным
мезомерным эффектом атома фтора, электроны которого расположены на той
же 2р-орбитали, что и электроны углерода. Увеличение радиуса атома галогена
уменьшает возможность подачи электронов для стабилизации заряда. В
результате интенсивность пика ионов СН2=На1+ уменьшается.
Для иодидов, бромидов, а также третичных и реже вторичных хлоридов
характерна реакция элиминирования из М+' атома галогена. В условиях
ионизации электронным ударом может протекать и гетероциклический разрыв связи в
нейтральной молекуле (схема 8.52). Молекулярные ионы фторидов и первичных
хлоридов отщепляют галоген в виде HHal, причем атом водорода мигрирует из
положения 2 или 4 исходной молекулы (четырех- или шестичленное переходное
состояние).
RHal->R++Cr
Схема 8.52
8.9. Алкилгалогениды, арилгалогениды 237
юо
90
80 -I
?? А
во А
50 ?
40
зо
20
10
1/М>
ЭО
Jul
20 ЗО 40 50 вО 70 80 ЭО 100 110 120 130 140 150 160 170 180 190 200 210
m/z
Рис. 8.51· Масс-спектр электронного удара 4-бромпропилбензола
Бензилгалогениды теряют под ЭУ атом галогена с образованием устойчивого
углеводородного иона, вероятно тропилиевой структуры, пик которого, как
правило, максимален в спектре. Выброс атома галогена оказывается более
предпочтительным, чем альтернативный выброс метильной группы в
аналогичном положении. Например, в спектре 2,4,6-триизопропилбензилхлорида [329]
интенсивность пика иона [М —С1]+ в 5 раз выше интенсивности пика иона
[М-СН3]+.
Атомы галогена (за исключением фтора) легко отщепляются из М+*
ароматических галогенпроизводных. Интенсивность пика соответствующего иона
может быть очень высока, вероятно, в результате трансформации в более
устойчивую структуру. Определить положение атома галогена в ароматическом
ядре практически невозможно. Например, в разд. 4.2.2.2 уже упоминалось, что
спектры ЭУ о-, м-, л-бромтолуолов и бензилбромида практически неразличимы
ни по массовым числам ионов, ни по интенсивностям их пиков.
Бензильный разрыв предпочтителен для алкилзамещенных арилгалогенидов.
Еще одной особенностью этой группы соединений является элиминирование
молекулы HHal первичным ионом [М—R]+ замещенной тропилиевой структуры
с образованием иона с m/z 89 (рис. 8.51, схема 8.53).
Hal~\i>
R*
Hal^0CH2-
Схема 8.53
Hal
-HHal
С7Н5
m/z 89
Химическая ионизация* Протонированные молекулярные ионы МН+ ал-
килгалогенидов, циклоалкилгалогенидов и бензилгалогенидов оказываются
неустойчивыми и легко отщепляют молекулу HHal, причем интенсивность
последующего распада углеводородного фрагмента зависит прежде всего от
238 Глава 8. Основные направления фрагментации органических соединений
энергетики реакции протонирования (разд. 5.2). Максимальный, а иногда и
единственный пик в спектре принадлежит иону [МН—НХ]+ [330].
Арилгалогениды, напротив, характеризуются интенсивным пиком МН+.
Исключением является иодбензол, для которого характерен ион М+" (ХИ
водородом). Ионы МН+ распадаются с выбросом атома галогена или молекулы
галогеноводорода. Протекание одной или другой реакции в зависимости от
природы галогена полностью совпадает с тем, как это описано для случая
ЭУ. Кстати, в первой реакции происходит образование нечетноэлектронного
иона, что совершенно не типично вообще для масс-спектрометрического
распада (разд. 3.1.3), а для химической ионизации в особенности. Фенил-катион,
образовавшийся при отщеплении молекулы HHal, далее присоединяет молекулу
водорода (газ-реагент) с образованием протонированного молекулярного иона
бензола [331].
Ион СбН^" образуется также в условиях ХИ водородом полигалогенпроизвод-
ных. В этом случае идут последовательные реакции элиминирования молекул
HHal и присоединения молекулы водорода [332]. На основании интенсивности
пика этого иона можно делать важные структурные выводы, различать
изомеры. В частности, его интенсивность очень высока в спектре 2-хлорбифенила,
умеренно высока в спектре 4-хлорбифенила и крайне низка в спектре 3-хлор-
бифенила. Аналогичные наблюдения сделаны и относительно дихлорбифени-
лов [333].
В условиях химической ионизации метаном интенсивность процессов
фрагментации ниже, поскольку величина сродства к протону метана выше, чем
водорода (разд. 5.2). В спектрах арилгалогенидов, помимо максимальных по
интенсивности пиков МН+, присутствуют достаточно интенсивные пики ионов
[М+С2Н5]+ и [М+СзН5]+. Фенил-катион, образующийся при отщеплении
молекулы HHal, реагирует с молекулой газа-реагента (метан) с образованием
иона [МН—ННа1+СН4]+, интенсивность пика которого может быть достаточно
высока [331, 334].
Процесс образования ионов [МН —ННа1+Н2]+ наблюдается и в условиях
ХИ арилгалогенидов изобутаном. Пики этих ионов имеют значительную
интенсивность даже в спектрах достаточно сложных галогенпроизводных, например,
N-галогенфенилсульфонилазетидин-З-онов [335] и N-галогенфенилсульфонил-
пирролидин-3-онов [336]. В данном случае реакцию его образования можно
представить как замещение атома галогена атомом водорода в протонированном
молекулярном ионе (схема 8.54).
МН+ + С4Н10 -> [??-Hal + Н]+ + НаГ + С4Н; или
МН+ + С4Ню -* [??-Hal + Н]+ + HHal + С4Н8
Схема 8.54
8.9. Алкилгалогениды, арилгалогениды 239
Достаточное внимание было уделено химической ионизации полигалоген
производных, многие из которых являются приоритетными загрязняющими
веществами окружающей среды. Неароматические полихлорированные
соединения в условиях ХИ метаном характеризуются очень низкой интенсивностью
пика МН+ [337]. Максимальную массу и интенсивность пиков имеют
ионы [МН—НС1]+ и [МН—2НС1]+. Аналогичная ситуация наблюдается и в
случае ароматических производных, содержащих массивные алифатические
заместители.
Для полихлорированных бензолов, бифенилов и других полициклических
ароматических соединений ион МН+ характеризуется максимальным по
интенсивности пиком в спектре [333, 338, 339]. Однако следует отметить, что ХИ не
дает дополнительных преимуществ по сравнению с электронным ударом ни для
качественного, ни для количественного определения этих соединений.
В работе [340] показано, что изомерные гексахлорбифенилы различимы с
использованием тандемной масс-спектрометрии, если во втором квадруполе
осуществить взаимодействие М+" этих соединений с аммиаком. Интенсивности
пиков ионов [M+NH3—НС1]+ значительно разнятся от изомера к изомеру.
Очень много внимания уделяется количественному определению
полихлорированных дибензодиоксинов и дибензофуранов. Будучи суперэкотоксикантами,
эти соединения опасны для окружающей среды, присутствуя в водоемах даже в
концентрации ниже 10~13 г/л. Безусловно, разработка наиболее чувствительных
методов их надежного и селективного определения очень важна. Различные
подходы к определению этих соединений, в том числе методами химической
ионизации, представлены в [25, 341].
Химическая ионизация отрицательных ионов. Взаимодействие алкилгалоге-
нидов с сильными основаниями в газовой фазе приводит к реакциям замещения
(Sn2) или элиминирования (Е2). Сродство к электрону атома галогена столь
высоко, что в спектрах ХИОИ этих соединений зачастую можно увидеть только
пик иона Hal". Детектировать депротонированные молекулярные ионы, как
правило, не удается. Исключением являются фториды. В частности,
взаимодействие этилфторида с анионом СБзО~ [342] приводит наряду с реакцией
элиминирования к образованию /?-фторэтиланиона (схема 8.55).
———- F~+ CD3OH + С2Н4
34% » -CH2CH2F + СЮзОН
—— * [F™-HOCE>3] + C2H4
Схема 8.55
240 Глава 8. Основные направления фрагментации органических соединений
Достаточно много внимания было уделено резонансному захвату электрона
молекулами полигалогенсодержащих соединений [343-345]. Отмечается, что в
условиях эксперимента (химическая ионизация метаном) моногалогенпроизвод-
ные не дают пика молекулярного иона, тогда как максимальную интенсивность
имеет пик иона На1~. При увеличении числа атомов галогена в молекуле
интенсивность пика М~# увеличивается. Для пента- или гексахлорфенилпроиз-
водных, а также пента- или гексабромфенилпроизводных пик иона М""" может
быть максимальным в спектре [346]. Предел обнаружения полихлорированных
дибензофуранов и дибензодиоксинов в условиях резонансного захвата электрона
можно улучшить на несколько порядков по сравнению с условиями электронного
удара [346]. Предел обнаружения можно улучшить дополнительно при
добавлении к метану кислорода. В этом случае происходит замещение атома хлора
на атом кислорода (схема 8.56). В случае полихлорированных дибензодиоксинов
[347] реакция может идти и с разрывом С—О-связей (схема 8.56), т. е. появляется
возможность делать выводы о степени замещения бензольных колец.
J6C—ЖЧ-^-?
Схема 8.56
Использование самой разнообразной масс-спектрометрической техники для
анализа полихлорированных бифенилов описано в обзоре [348],
полихлорированных дибензодиоксинов и дибензофуранов — в обзоре [341], ДЦТ и
родственных соединений—в обзоре [349], легких полигалогенированных
углеводородов — в обзоре [350], полихлорированных полициклических пестицидов — в
обзоре [351]. Эти вопросы освещаются в книге Е. С. Бродского и Р. А. Хмельницкого
[25].
Задача 8.42· Как можно по спектрам электронного удара различить
перечисленные ниже изомерные соединения?
а) 1-Бромгексан и 3-бромгексан;
б) 1-хлоргептан и 1-хлор-2-метилгексан;
в) 3-пропилбромбензол и 1-фенил-З-бромпропан;
г) (о-хлорметил)фенол и (м-хлорметил)фенол.
8.9. Алкилгалогениды, арилгалогениды 241
Задача 8.43. Идентифицируйте соединение по масс-спектру электронного
удара, представленному на рис. 8.52.
ЮО ?
ЭО -
80 -
70 -
60 -
50 -
40 -
ЗО -
20 -
10 -
О -
1,%
¦¦!.,
с
ц.
1
-L_^
77
, ..??
1ц—^—| 1 jj
112
jL,
ЗО 40
50 60 70 80
m/z
ЭО ЮО НО 120
Рис. 8.52
m/z
I 37
38
39
49
50
51
52
15б
д% |
3,28
8,57
1,80
2,40
13,7
16,3
1,65
5,18
m/z
57
61
62
63
73
74
75
17б
/,% I
1,69
1,03
0,92
0,75
2,86
5,63
5,67
4,05
m/z
11
78
85
86
112
113
114
1 115
/,%
48,2
3,23
1,05
1,05
100
6,84
32,1
2,24
Задача 8.44. Идентифицируйте соединение по масс-спектру электронного
удара, представленному на рис. 8.53.
ЮО ?
ЭО -
80 -
70 -
60 -
50 -
40 -
ЗО -
20 -
1 О -
о -I
1,%
93|
79
—? '
1
У
1 17-4
L—,—,—J
20 40 60 80 ЮО 120 140 160 180
m/z
Рис. 8.53
m/z
79
80
81
82
87
91
92
/,% 1
27,4
4,18
26,6
4,09
1,40
14,8
5,89
m/z
93
94
95
96
158
160
I 162
/, % ?
100
6,66
84,1
0,93
1,50
2,98
1,46
m/z
172
173
174
175
176
177
/,%
45,8
0,50
90,2
1,01
44,8
0,48
Задача 8.45. Идентифицируйте соединение по масс-спектру электронного
удара, представленному на рис. 8.54.
ЮО ?
ЭО -
80 -
70 -
60 -
50 -
40 -
зо -
20 -
Ю -
О -
19°А>
ь_
6
ul
971
1
I ^ J
117
li LL_
20
40
60
80
m/z
ЮО
120
m/z
26
27
35
36
37
47
49
60
161
/,% I
19,5
11,7
6,49
2,42
2,11
3,23
1,25
7,74
49,0
m/z
62
63
82
84
97
98
99
100
| 101
/,% 1
5,57
16,0
2,07
1,42
100
3,27
63,0
1,57
11,0
m/z
102
117
118
119
120
121
122
123
/,%
0,24
14,0
0,31
13,9
0,30
4,59
0,10
0,51
Рис. 8.54
242 Глава 8. Основные направления фрагментации органических соединений
Задача 8.46. Идентифицируйте соединение по масс-спектру электронного
удара, представленному на рис. 8.55. Что представляют собой ионы с m/z 129—
132?
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
?,%
L,
47
Jll ?
7 1 V 96
71
, ??,.,?
l\
1 18
JiJ
?] d
141
j, J
155
II
190 I
? li
Ji-.—Д
225
llil.l. , 1
262
L
20 40 60 80 100 120 140 160 180 200 220 240 260
m /z
m/z
35
36
37
47
48
49
59
71
73
76
/,% 1
3,09
3,38
1,04
18,4
4,62
6,24
3,10
10,6
3,45
1,10
m/z
77
82
83
84
85
94
95
96
97
[_98
/,% 1
1,08
4,57
19,4
4,01
7,09
15,3
8,79
10,1
0,99
1,04
m/z
106
108
110
117
118
119
120
121
122
129
Д% |
10,8
7,20
1,25
2,98
43,5
4,41
29,6
2,06
4,88
2,05
m/z
130
131
132
141
143
145
147
153
155
157
/, % 1
4,10
3,42
1,52
28,1
27,8
9,37
1,02
15,9
16,1
5,24
m/z
164
166
188
189
190
191
192
193
194
1 196
/,% 1
1,36
1,75
36,7
1,61
48,4
2,15
24,5
1,07
5,44
0,45
m/z
223
224
225
226
227
228
229
230
231
1 233
/,% 1
60,1
2,52
100
4,40
66,7
2,93
22,2
0,98
3,70
0,95
m/z
258
260
261
262
263
264
266
268
/,%
17,4
34,8
1,53
29,0
1,28
12,9
3,22
0,43!
Рис. 8.55
Задача 8.47.
Идентифицируйте соединение по <
на рис. 8.56.
100 ?
90 -
во -
70 -
во -
50 -
40 -
ЗО -
20 -
ю -
1,%
31
I Т°
69
119
20 АО 60 вО ЮО 120 ???
m/z
m/z
I 12
19
31
32
50
69
70
100
119
120
/,%
1,50
1,20
18,3
0,20
10,1
100
1,09
0,60
41,6
0,88
Рис. 8.56
8.10. Карбонильные соединения 243
Задача 8.48. Идентифицируйте соединение по масс-спеюру электронного
удара, представленному на рис. 8.57. Какова природа иона с m/z 81,5? Пик
молекулярного иона отсутствует, но его легко вычислить.
? ?,%
?
J 81
1 35
1 I.
,5
128
[.. il. I
?"? ? ¦ ¦ 1 1
40 80 120 16
m /z
163
207
1 '
0 200
m/z
35
37
79
80,5
81
81,5
82,5
83,5
91
93
/,% |
7,61
2,52
9,51
12,5
9,47
20,8
9,7
1,39
1,12
1,09!
m/z
126
127
128
129
130
161
162
163
164
1 165
/,% 1
8,81
0,10
11,7
0,12
2,94
60,2
0,66
100
1,09
46,8
m/z
166
167
205
206
207
208
209
210
211
1 212
/,% |
0,51
6,69
6,90
0,08
16,1
0,18
11,5 !
0,12l
2,28
0,03
100
90
80
70
60
50
40
30
20
10
0
Рис. 8.57
Задача 8.49. Составьте предполагаемую схему фрагментации 4-бромфенола.
Задача 8.50. Идентифицируйте соединение по его спектру ЭУ (рис. 8.58).
Используйте информацию об интенсивностях следующих пиков: 95 (90%),
96 F,0%), 97 @,20%).
100 -? ?,%
90 -I
80
70 -I
60 -I
50
40
30
20
10
50
Jl.
75
95
JLi
174
20
40
60
80
100
m/z
Рис. 8.58
120
140
160
180
8.10· Карбонильные соединения
Электронный удар. Энергии ионизации альдегидов и кетонов лежат в
диапазоне 9,4-9,8 эВ. Интенсивность пика молекулярного иона, как правило,
достаточна для его надежной регистрации. При переходе к соединениям с длинной
цепью пик М4" может не наблюдаться в спектрах альдегидов. В электронно-
244 Глава 8. Основные направления фрагментации органических соединений
100
90
80
70
60
50
40
30
20
10
0
100
90
80
70
60
50
40
30
20
10
0
41
1,%
57
70
82
1 12
1,%
40
43
60
80
72
m /z
100
120
140
160
57
jj.
mill., .. ?. 111
хЮ
40 60 80 100 120 140 160
m /?
Рис. 8.59. Масс-спектры электронного удара деканаля (а) и 2-этилгексаналя (б)
невозбужденном состоянии заряд и неспаренный электрон локализованы на
атоме кислорода. Однако благодаря эффекту резонанса (схема 8.57) заряд
может находиться и на атоме углерода карбонильной группы. Кстати, механизм
подавляющего большинства реакций карбонильных соединений в растворах
предусматривает именно такую резонансную форму.
+·
О
II
Rr-C—R
О
I
R^C—R
+
Схема 8.57
При фрагментации М+' карбонильных соединений значительную роль
играют перегруппировочные процессы, хотя большинство важнейших фрагментов
обусловлено простыми разрывами связей. На рисунке 8.59, ? и б представлены
спектры двух альдегидов алифатического ряда.
Для альдегидов ?-распад менее характерен, чем для кетонов. Пик иона НСО+
наблюдается только в спектрах низших гомологов, а также альдегидов с
сильными электроноакцепторными группами (например, C3F7CHO). Пик иона [М —Н]+
имеет заметную интенсивность только в случае дополнительной стабилизации
8.10. Карбонильные соединения 245
заряда (например, ароматические альдегиды). На схеме 8.58 представлены
основные направления фрагментации 2-этилгексаналя (рис. 8.59,6).
+·
С4Н9-сн~с^н
с2н5
m/z 128
ОН
сн2=сн-сС
m/z 57
?
у
он
С4Н9—CH=CV
mlz 100
?
+·
yOH
1 ^н
/иД 44
Схема 8.58
-С4Н»
-С2Н4
mlz 72
При переходе от пропионового к масляному альдегиду появляется новое
направление распада — перегруппировка Мак-Лафферти (схема 8.59). Благодаря
этому процессу для альдегидов без заместителей у ?-атома углерода характерен
ион с m/z 44 (рис. 8.59, а).
\^Р -
СН2 ?
xch/nh
CH2R1
R-CH=CH2, *
+
НО
Я?) С
4 / Ч
ОТ ХН
- RCHCH2
Схема 8.59
Если в ?-положении находится алкильный заместитель R, значение m/z этого
перегруппировочного иона увеличивается на соответствующую величину (ион
с m/z 72 в спектре 2-этилгексаналя на рис. 8.59,6). Кроме того, становится
246 Глава 8. Основные направления фрагментации органических соединений
юо
90
во
то
ео
50
ДО
зо
го
1 О
1,*^&
il
Э-4
I
60 70
Рис. 8.60. Масс-спектр электронного удара циклогексанкарбоксальдегида
возможна вторичная перегруппировка с элиминированием молекулы олефина
(ион с m/z 44 в спектре 2-этилгексаналя на рис. 8.59,6). Разветвление у ?-атома
углерода дает возможность протекать перегруппировке Мак-Лафферти по обеим
алкильным цепям. Надо лишь, чтобы длина цепочки была не меньше двух
атомов углерода. Хотя образующиеся ионы обусловлены выбросами молекул, а
не радикалов, интенсивности их пиков могут быть предсказаны на качественном
уровне по правилу выброса максимального алкила (ионы с m/z 72 и 100 в спектре
2-этилгексаналя на рис. 8.59,6). В спектрах ?-алкилзамещенных альдегидов
высокую интенсивность имеют также пики четноэлектронных ионов, возникающих
при отщеплении радикала R1 из енольного иона, образующегося в результате
перегруппировки Мак-Лафферти (схема 8.59, и ионы с m/z 72 и 57 на схеме 8.58).
Как уже отмечалось выше (разд. 3.1.2), заряд в результате перегруппировки
может оставаться на прежнем месте или мигрировать. В случае альдегидов
миграция заряда приводит к возникновению олефинового иона (схема 8.59).
Для незамещенного в ?-положении альдегида в спектре наблюдается пик иона
[М—44]+. Его интенсивность может быть очень высокой.
Достаточно стабильный молекулярный ион циклогексанкарбоксальдегида
распадается с выбросом молекулы воды или радикала НСО". Образующиеся
углеводородные ионы определяют всю последующую картину фрагментации
(рис. 8.60).
Для ароматических альдегидов очень характерен последовательный или
одновременный выброс атома водорода и молекулы СО (пики ионов с m/z 123
и 95 на рис. 8.61). Пик молекулярного иона, как правило, весьма интенсивен.
Важным отличием от алифатических альдегидов является интенсивный пик иона
[М —Н]+, представляющего собой устойчивый резонансно стабилизированный
бензоилкатион АгСО+.
На рисунке 8.62 а, б представлены масс-спектры ЭУ двух алифатических
изомерных кетонов. Разрывы С—С-связей с двух сторон от карбонильной группы
приводят к образованию двух пар алкильных и ацильных ионов. Из двух ациль-
ных катионов более интенсивный пик имеет образовавшийся при отщеплении
более крупного алкила, а из двух алкильных —более стабильный (вторичный,
третичный, аллильный и т. д.). Например, в случае децил-трет-бутилкетона
интенсивность пика иона С4Н9СО4" будет выше, чем С10Н21СО4", а интенсивность
8.10. Карбонильные соединения 247
юо -,
90
80 -I
70 ?
во
50
ЛО -\
зо -I
20 -|
Ю
1.%
iiUL
ЮО 1?? 120 130
Рис. 8.61. Масс-спектр электронного удара л-фторбензальдегида
пика С4Н9" выше, чем СюН^. Эти первичные ионы могут обладать
внутренней энергией достаточной для протекания вторичных процессов распада. На
схеме 8.60 отражены основные направления фрагментации под ЭУ деканона-5
(рис. 8.62,6).
ОЙ
m/z7l
С5НПС=0
m/z 99
+·
О
CsHn-C-QHo
+·
ОН
С4Н9С=0
m/z 85
+·
ОН
I
С5НЦ—С=СН2 ~С4И* СН2=С-СНз "СзНб O^C-QHo
m/z 114 /иД58 «Д100
Схема 8.60
Молекулярные ионы кетонов могут претерпевать перегруппировку Мак-Лаф-
ферти с участием любого из алкильных радикалов при длине цепочки не менее
трех атомов углерода и наличии 7-атома водорода. Возможны последовательные
перегруппировки с образованием конечного иона со структурой енольной формы
ацетона (схема 8.61). Перегруппировка не идет, если 7-атом углерода имеет sp2-
или .ур-гибридизацию,а также в случае а,/?-непредельных кетонов и альдегидов.
?
+
но
но'^^
R
-RCHCH2
СН3
но
сн2
Схема 8.61
о4»
I I I I I I I I I I
vo
о
X
5
».
\
?> —-
DO
N.
tf4
1 1 i
—
1 1 1 1 I 1 1
-
-
ooooooooooo
Ofl)(OSOIO<iDNr
ooooooooooo
8.10. Карбонильные соединения 249
юо
90 -I
80 -J
70
во
SO
АО -\
ЗО
20 -]
10 -J
1,%
^11
69
98
60 7?
m/z
Рис. 8.63. Масс-спектр электронного удара циклогексанона
Распад алициклических кетонов изучен достаточно хорошо и был подробно
описан еще в 60-х годах прошлого века [32, 352-354]. Молекулярные ионы
ненапряженных алициклических кетонов достаточно устойчивы. Их пики имеют
высокую интенсивность в спектрах ЭУ.
На рисунке 8.63 представлен масс-спектр электронного удара
циклогексанона. Схема 8.62 отражает основные направления распада М+" циклогексанона.
Интенсивность пика молекулярного иона составляет 30% от интенсивности
максимального пика в спектре (рис. 8.63). Отщепление из М+" молекулы воды
(ион с m/z 80 на рис. 8.63) идет в результате последовательных, не всегда
селективных, перегруппировочных процессов. Еще более сложные структурные
изменения исходной молекулы приводят к элиминированию этильного радикала,
причем на 80% отщепляются атомы углерода из положений 3 и 5 исходного
цикла. На примере циклогексанона можно еще раз подчеркнуть, что даже для
столь простого и хорошо изученного соединения и даже при использовании
современной масс-спектрометрической техники и меченых соединений, крайне
затруднительно получить абсолютно исчерпывающую информацию по всем
процессам, протекающим при распаде молекулярного иона.
?4"
-н2о
?,??
m/z S0
Ч-с2н5
+
с4н5о
m/z 69
~?·
—С2Щ
-со
m/z 42
Схема 8.62
+
О
оъ -СзН?
-СН3
+
* с=о
m/zS3
m/z 55
250 Глава 8. Основные направления фрагментации органических соединений
В частности, в работе [355] показано, что фрагментация циклогексанона
значительно сложнее, чем это принято представлять в учебной масс-спектро-
метрической литературе. Например, ион с m/z 55, характеризующийся
максимальным пиком в спектре, является составным (80% С3Н3О и 20% С4Н7).
Кислородсодержащий фрагмент образуется не только из М+* (схема 8.62), но
и из иона [М — С2Н4]+' в результате потери метального радикала, а также
из иона [М-СзНб]"*" в результате потери атома водорода. Углеводородный
фрагмент может появиться в результате пяти реакций фрагментации (схема 8.63).
Составными являются и большинство других ионов в спектре. Отметим тот факт,
что элиминирование молекулы этилена из М+* может протекать по нескольким
механизмам с вовлечением атомов углерода С2, СЗ, С4 и значительным скрам-
блингом атомов водорода [32, 353].
м+-->с4н++с2н3о·
?" -* [М-Н]+ -+ С4Н+ + СН2СО
м+· -+ [м-сн3]+ -> с4н|+со
м+· -> [м-со]+в -+ с4н++сн;
М+# -> [М-СН2СО]+' -> С4Н^ + Ы
Схема 8.63
Тем не менее установленные пути фрагментации позволяют делать
структурные выводы на основании анализа масс-спектров. Например, важное
значение имеет максимальный ион в спектре циклогексанонов (m/z 55 на схеме
8.62). Алкильный заместитель в положении 3 приводит к увеличению доли
разрыва связи Ci — Сб по сравнению с Ci~С2 примерно в 4-5 раз, поскольку
в результате фрагментации образуется более термодинамически стабильный ион
с внутренней кратной связью (m/z 69, схема 8.64). Алкильный заместитель в
положении 2, с одной стороны, облегчает разрыв связи Ci— C2 (более
замещенной по сравнению с Ci~Сб), но с другой стороны, облегчается миграция
атома водорода из положения 2 при разрыве связи Ci— Сб. Результатом является
приблизительное равенство интенсивностей двух пиков альтернативных ионов.
При фрагментации 4-алкилзамещенных заместитель элиминируется при любом
первичном разрыве С—С-связей у ?-атома углерода. Кстати, ион с m/z 55
характеризуется максимальным или близким к максимальному пиком в спектрах
не только циклогексанона, но и циклопентанона и циклоалканонов с большим
размером цикла.
Алкиларил- и диарилкетоны характеризуются очень интенсивными пиками
М+". Фрагментация прежде всего определяется ?-разрывами, причем в случае
алкиларилкетонов в спектре доминирует пик более устойчивого ароматического
8.10. Карбонильные соединения 251
+
О
-С4Н9
сн3
t
m/z 55
+
О
СНз СНз
Схема 8.64
бензоильного иона (m/z 105 на рис. 8.64). Пик альтернативного ацильного
иона часто едва заметен (m/z 71 на рис. 8.64). Наличие алкильной цепочки
достаточной длины приводит к реализации перегруппировки Мак-Лафферти
(m/z 120 на рис. 8.64). М+" диарилкетонов может элиминировать молекулу СО с
последующими выбросами 1-3 атомов водорода. Пики этих фрагментов не очень
интенсивны, но отчетливо видны в малолинейчатых спектрах этих соединений.
Химическая ионизация. Пик протонированного молекулярного иона МН+
кетонов доминирует в спектре химической ионизации протонирующими газами-
реагентами. Кроме него могут наблюдаться лишь незначительные пики ацильных
ионов, образующихся в результате ?-разрывов, и кластерных ионов [М+А1к]+,
соответствующих используемым газам-реагентам (разд. 2.3). Для
инициирования фрагментации необходимо использовать реагенты, для которых характерна
максимальная экзотермичность процесса ионизации. Протонированные
молекулярные ионы альдегидов значительно менее устойчивы. Максимальную
интенсивность имеет, как правило, пик иона [МН —НгО]". Могут наблюдаться также
пики более легких углеводородных ионов. Процесс отщепления воды протекает
неоднозначно [356, 357]. В частности, в состав элиминируемой молекулы воды
не всегда входит атом водорода, который протонировал молекулу альдегида.
ОО ?
ЭО -
во -
ТО -
во -
SO -
4.О -
ЗО -
20 -
Ю -
О -
I... .,
U iLj—
??
±^
-СО
-? · '¦¦г '
Ю5
1—,
-С31Н/
-с2н„
| -"* ¦ -, ^__
, 14в
—К—
Рис. 8.64. Масс-спектр электронного удара бутирофенона
252 Глава 8. Основные направления фрагментации органических соединений
Для спектров химической ионизации монооксидом азота кетонов характерно
протекание процессов перезарядки и электрофильного присоединения [358]. В
первом случае образуется М+', а во втором [M+NO]+. Соотношение интенсив-
ностей их пиков определяется величиной энергии ионизации кетона. Для
протекания перезарядки энергия ионизации должна быть ниже 8,8 эВ. В спектрах
альдегидов [359], помимо пиков указанных ионов, достаточно интенсивны пики
[М —Н]+. Эти ионы образуются по механизму отрыва аниона (разд. 5.2).
Изомеризация протонированного молекулярного иона пивалоинового
альдегида в протонированный ион метилизопропилкетона (схема 8.65) протекает
по механизму [301], близкому классической пинаколиновой перегруппировке.
Эта реакция представляет собой частный случай изомеризации альдегидов в
более термодинамически устойчивые кетоны. Подобный процесс, протекающий
в растворах в условиях кислотного катализа, был ранее описан в работе [360].
снзч| /.он сн3. V/Он сн3 | хт
с—с *" с—с " с—с
сн3' ^н сщ-х^^сщ сщ' ^?3
?
Схема 8.65
Химическая ионизация отрицательных ионов. Депротонирующие агенты
эффективно взаимодействуют с карбонильными соединениями в газовой фазе с
образованием енолят-анионов [М —Н]~, пики которых имеют максимальную
интенсивность в спектрах. Меньшую интенсивность имеют пики ионов [М —ЗН]~.
Для инициирования процессов распада енолятов необходимо использовать
метод активации соударениями. Работы в этом направлении проводились
многими группами исследователей [361-366]. Енолят ацетона выбрасывает атом
водорода и молекулу метана. Схема 8.66 отражает основные направления
фрагментации енолята З-этилпентан-2-она [364, 365], связанные с элиминированием
молекулы метана или с последовательным отщеплением двух молекул этилена.
Результаты исследований подтверждены спектрами дейтерированных аналогов.
СНз СНз Схема 8.66 "
8.10. Карбонильные соединения 253
Для депротонированных циклогексанонов (схема 8.67) характерны
отщепления молекулы водорода с возникновением двух изомерных ионов и реакция
разрыва цикла с отщеплением молекулы этилена [362, 365]. Последний процесс
можно рассматривать как деструкцию енолят-аниона по механизму ретрореак-
ции Дильса—Альдера. Аналогичным образом распадаются и алкилзамещенные
циклогексаноны [362].
-с2н4
Схема 8.67
Достаточно насыщенными оказываются масс-спектры химической ионизации
карбонильных соединений ионами 0~" [367]. Ионизация осуществляется в
результате нуклеофильной атаки О-' на атом углерода карбонильной группы и
ведет к разрывам связей С—? и С—С и образованию ионов [М—Н]~, [М—2Н]~',
RCOO- и R^COO-. Потеря молекулы водорода осуществляется с участием
атомов водорода одного или двух ?-атомов углерода, причем эти фрагментные
ионы могут распадаться далее с выбросами углеводородных радикалов и
образованием различных енолят и алкоголят-анионов. Таким образом, на основании
этих спектров можно устанавливать структуры карбонильных соединений. В
особо сложных случаях показано [367], что существенную помощь могут оказать
спектры тандемной масс-спектрометрии с инверсией заряда (разд. 7.3).
Задача 8.51. Какие ионы возникают в результате перегруппировки Мак-
Лафферти при распаде перечисленных ниже соединений под электронным
ударом,
а
б;
в
г
д
е
ж
3
и
к
З-Метилоктанон-4;
3-метил-5-этилоктанон-4;
3,5-диэтилоктанон-4;
3 -этил октанон-2;
4-метилдеканон-5;
нонанон-4;
4-пропилдеканон-5;
4,5 - диэтилу ндеканон-5;
децен-З-он-5;
6-метил-8-этилтетрадецен-5-он-7.
254 Глава 8. Основные направления фрагментации органических соединений
Задача 8.52. Какие перегруппировочные процессы протекают при распаде
перечисленных ниже соединений под электронным ударом?
а) Фенилпропилкетон;
б) бензилметилкетон;
в) бензилбутилкетон;
г) /?-фенилэтилэтилкетон.
Задача 8.53. Какие различия наблюдаются в масс-спектрах электронного
удара перечисленных ниже изомерных соединений?
а) Пентаналь и 2-метилбутаналь;
б) бутаналь и 2-метилпропаналь;
в) 2-метилбутаналь и 3-метилбутаналь;
г) 3,5-диметилоктаналь и 4,4-диметилоктаналь;
д) гексаналь и гексанон-2;
е) гексен-2-аль и гексен-5-аль;
ж) 4,6-диметилдеканон-5 и 4-этилдеканон-5.
з) бутилфенилкетон и mpem-бутилфенилкетон;
и) пропилфенилкетон и 4-пропилбензальдегид;
к) бензилбутилкетон, бензил-втор-бутилкетон и /3-фенилэтилпропилкетон;
л) 3-метилбензальдегид и ацетофенон;
м) циклогексилметилкетон и циклогексилуксусный альдегид.
Задача 8.54. На рисунке 8.65, а и б представлены спектры изомерных
3,3-диметилгексаналя и 4,4-диметилгексаналя. Сделайте отнесение.
100-1
00 Ч
8 0-1
70 А
60 А
50 А
40 А
30 А
20 А
1 О А
1,%
20
100 -j I ,%
90 -I
80 4
70 4
60 А
50 А
40 А
30 А
10 ? ? Г
m/z
55
20
60
m/z
Рис. 8.65
8.10. Карбонильные соединения 255
Задача 8.55. Объясните, почему интенсивность пика с m/z 104 столь высока
в спектре 4-фенилбутаналя (рис. 8.66).
ОО ?
ЭО -
80 -
70 -
60 -
50 -
40 -
ЗО -
20 -
Ю -
О -
I
j—
,<Уо
-J r-J
91
.1
J ,-J
104
|
II ,
148
, L
«40 60 ?? 100 120 140
m/z
Рис. 8.66
Задача 8.56. Какие ионы будут иметь максимальные пики в спектрах ЭУ
перечисленных ниже соединений?
а) Изопропилбутилкетон;
б) метилэтилкетон;
в) ewop-бутилизопропилкетон;
г) бензилэтилкетон;
д) изопропилметилкетон.
Задача 8.57. Составьте схему фрагментации бутирофенона, спектр которого
представлен на рис. 8.63.
Задача 8.58. Составьте предполагаемые схемы фрагментации под ЭУ
перечисленных ниже соединений.
а) Пентанон-2;
б) пентанон-3;
в) З-метилпентанон-2;
г) пропилэтилкетон;
д) изопропилпропилкетон;
е) дибутилкетон;
ж) mpem-бутилпропилкетон.
256 Глава 8. Основные направления фрагментации органических соединений
Задача 8.59. Идентифицируйте соединение, масс-спектр электронного удара
которого представлен на рис. 8.67.
юо
90
80
70
60
SO
40
зо
20
1 О
о
Х*/ъ
58
40
-45
50
55
60
m/z
m/z
26
27
28
! 29
j 30
31
38
39
40
/,% ?
16,8
70,8
100
97,4
6,51
3,67
0,76
3,38
1,45
m/z
41
42
43
53
55
56
57
58
1 59
/, %
1Д4
3,07
1,56
0,42
2,08
0,45
19,3
66,5
2,29
Рис. 8.67
Задача 8.60. Идентифицируйте соединение, масс-спектр электронного удара
которого представлен на рис. 8.68.
юо
90
80
70
60
SO -I
40 А
ЗО -4
20 -|
ю
19°Л>
72
20 ЗО 40 SO 60 70
m/z
m/z
15
26
27
28
29
39
41
/,% ?
4,31
1,12
8,34
3,25
17,6
2,78
1,031
m/z
42
43
44
57
71
72
j 73
/,%
3,12
100
2,23
8,87
1,29
25,0
1,11
Рис. 8.68
Задача 8.61. Составьте предполагаемые схемы фрагментации под
электронным ударом и укажите различия, с помощью которых можно отличить
следующие изомерные соединения по их масс-спектрам.
а) 2-Метилциклогексанон и 3-метилциклогексанон;
б) 2,3-диметилциклогексанон и 2,4-диметилциклогексанон;
в) 2,5-диметилциклогексанон и 2,6-диметилциклогексанон;
г) 2,4-диметилциклогексанон и 3-этилциклогексанон;
д) 3-винилциклогексанон и 4-винилциклогексанон;
е) циклогексанон и 2-метилциклопентанон;
ж) 2-метил-З-этилциклогексанон и 2-этил-З-метилциклогексанон.
8.10. Карбонильные соединения 257
Задача 8.62. Идентифицируйте соединение, масс-спектр электронного удара
которого представлен на рис. 8.69.
100 -?
90 -|
80
70
60
50
40
30
20
10
О
1,%
44
56
-Н^
72
?, 1Д ,!¦
82
30 40
50 60 70
m/z
-т
80 90 100
Рис. 8.69
m/z
27
28
29
30
39
40
41
42
43
44
45
/,% |
39,3
6,02
39,4
2,17
25,7
3,34
71,5
12,8
53,9
100
19,8
m/z
46
53
54
55
56
57
58
59
62
67
68
/,% I
1,03
3,45
3,54
14,9
80,7
51,6
7,32
1,75
1,57
9,87
1,12|
m/z
69
70
71
72
73
81
82
83
98
100
/,% ?
1,21
1,25
8,39
17,4
1,23
1,18
13,3
1,02
1,13
1,33
Задача 8.63. Идентифицируйте соединение, масс-спектр электронного удара
которого представлен на рис. 8.70.
юо
90
80
70
60
SO
40
ЗО
20
Ю
О
1,°/о
43
58
85
ЮО
—? a*lr »*t
20 ЗО 40 50 SO ТО 80 ЭО ЮО
m/z
15
18
27
28
29
39
40
41
/,% ?
4,03
0,89
6,41
1,13
10,1
7,11
1,13
17,3
m/z
42
43
44
45
55
56
57
1 58
/,% 1
4,29
100
2,98
1,60
1,42
1,77
21,9
42,0
m/z
59
67
69
85
86
100
101
1 102
/,%
1,60
1,60
0,60
16,5
0,89
20,0
1,32
0,08
Рис. 8.70
Задача 8.64. Идентифицируйте соединение, масс-спектр электронного удара
которого представлен на рис. 8.71.
ОО ?
ЭО -
80 -
70 -
во -
50 -
40 -
ЗО -
20 -
Ю -
г» -
1,%
- --Ь, .
95
75
„ .1, J, , , J
L .. ,. . ,
123
?
21
j. ,, I
8
?
40
80
120
m/z
160
200
Рис. 8.71
m/z
50
51
69
74
75
76
81
94
95
/,% ?
2,77
1,84
3,51
3,09
19,4
1,32
0,51
3,82
46,8
m/z
96
123
124
125
188
217
218
219
I22O
1,% |
3,13
100
7,86
0,44
2,06
3,35
28,4
4,12
0,32
9—1ИЗ
258 Глава 8. Основные направления фрагментации органических соединений
8.11· Карбоновые кислоты и их производные
Электронный удар. В электронно-невозбужденном состоянии заряд и неспа-
ренный электрон в молекулярных ионах карбоновых кислот и сложных эфиров
локализован на атомах кислорода. На рисунке 8.72 представлены масс-спектры
электронного удара додекановой и 2-пропилнонановой кислот. Масс-спектры
изомерных сложных эфиров представлены на рис. 8.73.
Пики молекулярных ионов интенсивны в спектрах даже достаточно тяжелых
соединений этих классов. Однако при увеличении длины алкильных радикалов
(как кислотной, так и спиртовой части), а также числа разветвлений в цепи
молекулярные ионы становятся менее устойчивы, а их пики можно не увидеть
в спектре (рис. 8.73, а и в). Основные направления фрагментации М+" связаны
с разрывами связей рядом с карбонильной группой. Ионы, образовавшиеся при
разрыве связи С—О, как правило, более интенсивны. Важнейшим исключением
является ион ОН+, интенсивность пика которого в спектрах кислот крайне мала.
Напротив, пики ионов OR+ могут иметь значительную интенсивность в спектрах
сложных эфиров.
Ацилий-катион устойчив для легких гомологов, но с увеличением длины цепи
интенсивность его пика заметно снижается E3% в спектре масляной кислоты,
7% в спектре нонановой кислоты, 4% в спектре пальмитиновой кислоты).
1 29
oiULL Jl
1 57
?.|? I,
1 71
200
100 120 140
m /?
102
160
1 80
1 15
1 29
1 71
200
200
? u г-1 "П ¦ 1 1
20 40 60 80 100 120 140 160 180 200
m /?
Рис. 8.72. Масс-спектры электронного удара додекановой (а) и 2-пропилнонановой
кислот (б)
8.11. Карбоновые кислоты и их производные 259
100 ?
90 -|
80
70
60
50 ?
40
30
20
10 ?
I (% )
43
55 70
61
83
Ll
97
112
j__ li
-?
90
20 30 40 50
60
70 80
m /Z
100 110 120 130 140
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
n -
I,%
,l ,, ,|
55
J ill ,l„,
74
87
I. . ..In
I. „Ii, I. ...
143
.. I,
157 169
I. L
б
200
I
20
40
100
90
80
70 ?
60
50 -j
40 A
30 -]
20 A
10 A
I,%
60 80 100 120 140
m /z
43
160 180 200
56
h и
69
84
,» „,???,?,?
9.9
117
JiL.
1 ¦'""» "? ? t-
20 30 40 50 60 70 80 90 100 110 120 130 140
m /z
Рис. 8.73. Масс-спектры электронного удара децилацетата (?), метилундеканоата (б) и
гексилгексаноата (в)
Следует отметить, что неселективная потеря атома водорода молекулярными
ионами многих карбоновых кислот приводит к образованию ионов [М—Н]+.
Разрывы С—С-связей в алкильной цепочке кислот и сложных эфиров
приводят к появлению ионов с m/z 59, 73, 87, 101... характеристической
гомологической серии с общей формулой [(CH2)«COOR]+. Через каждые 4 метиленовых
звена наблюдается повышенная интенсивность пика соответствующего иона в
серии. В случае карбоновых кислот нормального строения повышенные
интенсивности будут иметь пики ионов с m/z 73, 129, 185, 241 и т. д. (рис. 8.72, а),
260 Глава 8. Основные направления фрагментации органических соединений
+
ОН
OR
m/z 87(R=CH3)
OR
OR
m/z 143 (R=CH3)
Схема 8.67a
а в случае их метиловых эфиров —с m/z 87, 143, 199, 255 и т. д. (рис. 8.73,6).
Предложенная и доказанная масс-спектрами дейтерированных соединений
схема 8.67а объясняет образование первого иона этой серии и, в определенной
мере, второго. Оба процесса начинаются с миграции <5- или ?-атома водорода
на карбонильную группу. Однозначный механизм возникновения других ионов
этой серии не установлен, но он должен включать дополнительные миграции
атомов водорода. Ион с m/z 73 (для кислот) и его эфирный аналог образуются
также благодаря аллильному разрыву енольной формы (задачи 8.72 и 8.74).
Для структурного анализа кислот с длиной цепи не менее 9 атомов углерода
важен ион [М—43]+. Он образуется при отщеплении из молекулярного иона
а-, /?- и 7-атомов углерода (схема 8.68). В этом процессе образуются пяти- и
шестичленные циклы, т. е. ? = 1, 2. Аналогичный механизм реализуется и при
образовании иона [М —29]+, когда отщепляются а- и /?-атомы углерода [368].
По регистрируемым в масс-спектрах величинам m/z этих ионов можно сделать
выводы о наличии разветвлений у а-, /?- и 7-атомов углерода кислотной цепи.
+
RO О/ + +
W ,_(сН9)п RO 0-СН2 RO 0-СН2
kAXR с3нА ^ <Ьн-(Ьн2)п
V_^4R r/
Схема 8.68
Для карбоновых кислот (без заместителя у ?-атома углерода)
перегруппировка Мак-Лафферти приводит к иону енольной формы уксусной кислоты с m/z 60
(рис. 8.72, а, схема 8.69). Для сложных эфиров пик этого перегруппировочного
иона сдвинут по шкале масс на соответствующую величину (рис. 8.73).
Поскольку энергии ионизации олефинов, начиная с пропилена, достаточно низки,
интенсивности пиков олефиновых ионов могут быть также достаточно высоки.
8.11. Карбоновые кислоты и их производные 261
?
R—«О* О
~н*
ОН
R-i-J ОН
-RCHCH2
ОН
онИ
-А
Схема 8.69
ОН
Чем длиннее алкильная цепь, тем выше интенсивность пика иона с m/z 61,
образующегося при миграции к карбоксильной группе двух атомов водорода
алкильной цепи. Наличие у ?-атома углерода алкильной группы не короче,
чем этильная, помимо сдвига перегруппировочных ионов с m/z 60 и 61 по
шкале масс (ионы с m/z 102, 103, 158, 159 на рис. 8.72,6 для 2-пропилнона-
новой кислоты), приводит к реализации двух перегруппировочных процессов по
обеим алкильным цепям в любой последовательности. Конечным результатом
этих перегруппировок вновь оказываются ионы с m/z 60. На схеме 8.70 эти
процессы представлены для случая 2-пропилнонановой кислоты, спектр которой
представлен на рис. 8.72, б.
С/о с5нп--|С/о ? 0>??-]? он
ОН ? ОН ^? ОН
<н7
<н7
-СзНб
ж\*
-С7Н14
ОН
—с7н14
он
m/zl5S
OHl"^
m/z 60
Схема 8.70
-CjHe
ОН Г
?
ОН
он
C3H7
m/zW2
В случае сложных эфиров распад М+' может быть инициирован спиртовым
атомом кислорода (аналогично простым эфирам, ионы с m/z 1Ъ на рис. 8.73, ? и
m/z 129 на рис. 8.73, в). Помимо указанных выше перегруппировочных процессов
по кислотной цепи при достаточной длине спиртовой цепи (не менее двух
атомов углерода) возможны еще две перегруппировки (схема 8.71). Первая из
них является типичным примером перегруппировки Мак-Лафферти, а вторая
заключается в переносе двух атомов водорода. Иногда ее называют «перегруп-
262 Глава 8. Основные направления фрагментации органических соединений
пировка Мак-Лафферти+1». Кстати, помимо сложных эфиров она характерна
для фрагментации тиоэфиров, амидов и фосфатов. Миграция гидрид-иона на
второй стадии перегруппировки Мак-Лафферти+1 осуществляется через
четырехчленное переходное состояние от того же /?-атома углерода, от которого
ушел атом водорода на первой стадии [35]. Следует, однако, отметить, что
признанного всеми механизма перегруппировки Мак-Лафферти+1 пока нет.
Некоторые исследователи считают, что второй атом водорода мигрирует из
любого доступного положения спиртовой цепи [82]. Вновь следует подчеркнуть,
что поскольку энергии ионизации олефинов, начиная с пропилена, могут быть
несколько ниже, чем энергии ионизации комплементарных карбоновых кислот,
интенсивности пиков олефиновых ионов могут быть выше, чем интенсивности
пиков альтернативных кислородсодержащих ионов. В частности, можно
отметить высокие интенсивности пиков алкеновых ионов с m/z 84 (перегруппировка
Мак-Лафферти по спиртовой цепи) и m/z 56 (перегруппировка Мак-Лафферти по
кислотной цепи) в спектре гексилгексаноата (рис. 8.73, в).
R О/ R Ш R ???
? ? ? ~н· ? -rchch2 ?
??—'-^ч ' ?
m/z 60 (Я=СНз)
?
V°H *гн- R 0H
о-сн2 йн
KHC-R
Схема 8.71
+
m/z 61 (R=CH3)
В предельном случае М+' сложных эфиров в результате перегруппировок
по кислотной и спиртовой цепям распадаются до М+* и МН+ уксусной кислоты.
Исключениями являются метиловые эфиры любых кислот, а также любые эфиры
?-метилзамещенных карбоновых кислот. На схеме 8.72 изображены
основные процессы фрагментации гексилгексаноата, спектр которого представлен на
рис. 8.73, е.
Масс-спектрометрическое поведение жиров (триглицеридов) и других ли-
пидов выходит за рамки данного пособия. Современные методы ионизации и
регистрации ионов позволили достичь значительных успехов в определении
молекулярных масс, состава, структуры и модификаций этого широчайшего
класса соединений. Для ознакомления с масс-спектрометрией липидов можно
обратиться к сборнику [369].
8.11. Карбоновые кислоты и их производные 263
СбНГ2"
[М-С5НП]+
m/z 129
+· \ \ +· / / / +
он \ \ о / / / он
I -С4Н8 II -СпНп II
CH2=C-0~CH2-C5Hii-« С4Н9-СН2-С-0-СН2-С5НЦ—^VQHg-O^-C-OH
m/z 144
СбН12
+·
О
СНз-С-ОН
m/z 60
-ОН"
сн3со
m/z 43
?"
-С4Н8
-СбН12
О
С4Н9-СН2-С-ОН
m/z 116
-С3Н7*
+
ОН
сн2=сн-с-он
m/z 73
Схема 8.72
-С4Н7
тДП7
-С4Н8
+
ОН
сн3-с-он
m/z 61
Молекулярные ионы дикарбоновых кислот менее устойчивы. Их пики в масс-
спектрах едва заметны. Основные первичные процессы связаны с
элиминированием одной или двух молекул воды, а в некоторых случаях —молекулы
С02 [370]. Ионы [М—Н20]+ далее могут элиминировать молекулы СО, С02
или радикал СООН', что свидетельствует о возможном образовании ионов со
структурой циклических ангидридов. Наличие алкильных заместителей в цепи
дикарбоновых кислот и их эфиров благоприятствует протеканию
перегруппировки Мак-Лафферти по обеим карбоксильным группам (m/z 74 и 102 на рис. 8.74).
100
90
80
ТО
во
SO
40 -I
ЗО -4
20 Ч
Ю 4
?9°/<>
JiLi
ILl
А
¦llilll.
A^U
•???
m/z
Рис. 8.74. Масс-спектр ЭУ диметилового эфира 2-этилоктандиовой кислоты
264 Глава 8. Основные направления фрагментации органических соединений
юо
90 -]
во А
?? -|
во 4
50 -\
АО Ч
ЗО А
20 4
ю н
?,^?
JL·
20 ЗО
ЮО ?
90 -
во -
7? -
во -
50 -
40 -
I I I
0 0 0
СО N г
? -?
19°Х>
J__^_
40
ЗЭ
1
—uL^
50
51
j ?_
ео
70
65
iLl—г_
во
m/z
??
¦ ¦ ' ¦¦
эо
Ц-
юо
Э1
I
LL г-
1Ю
120
130
119
J
4- 1
140
б
136
U
? ,
20 ЗО
Рис. 8.75. Масс-спектры электронного удара о-толуиловой (а) и л*-толуиловой кислоты (б)
Ароматические кислоты и сложные эфиры характеризуются интенсивными
пиками молекулярных ионов. Один из наиболее интенсивных пиков
принадлежит ацильному иону, который далее теряет молекулу СО с образованием
арильного катиона. Этот ион может образоваться и напрямую из М+" (рис. 8.75).
Замещенные ароматические кислоты являются отличными модельными
соединениями для изучения opmo-эффекта. Во многих случаях соответствующий пик
имеет максимальную интенсивность в спектре (см. пик иона с m/z 118 в спектре
о-толуиловой кислоты на рис. 8.75, а).
Для фенилалкановых кислот характерно отщепление молекулы воды. Пик
соответствующего иона может быть весьма интенсивен в спектре. Однако
максимальную интенсивность чаще имеют пики ионов, обусловленных бензильным
разрывом, перегруппировкой Мак-Лафферти, а также пик иона со структурой
стирола (рис. 8.76).
юо -,
??
80
70
60 -\
SO
40
ЗО
20 ?
??
о
20
1,%
lu
J—?-
90 ЮО 110 120 130 1-40 150 160 170
Рис. 8.76. Масс-спектр электронного удара 4-фенилбутановой кислоты
8.11. Карбоновые кислоты и их производные 265
ЮО ?
90 -
80 -
70 -
60 -
SO -
ло -
ЗО ->
20 -
ю -
о -
1,%
.1.. ,1,1,
A3
j tLA-
71
_L.
_l
94
i—, ¦
, Д_
Рис. 8.77. Масс-спектр электронного удара фенилбутирата
Для масс-спектров фениловых эфиров характерен пик иона с m/z 94,
имеющего структуру фенола и образующегося по механизму перегруппировки Мак-
Лафферти с выбросом молекулы кетена (рис. 8.77). Образование молекулярного
иона бензилового спирта в результате перегруппировочного процесса с
выбросом молекулы кетена характерно для фрагментации бензиновых эфиров жирных
кислот.
Алкильные эфиры о-фталевой кислоты являются весьма стабильными
соединениями. Их широчайшее использование в качестве пластификаторов привело к
значительному загрязнению окружающей среды. Фактически любая проба (вода,
почва, воздух, пищевые продукты и т. д.), контактировавшая с полимерными
материалами, содержит фталаты. Во многих странах они внесены в перечни
приоритетных загрязняющих веществ. Для соединений этого класса, за исключением
диметилового эфира, важнейшей характеристикой спектра электронного удара
является доминирующий пик иона с m/z 149. Именно по этому пику фталаты
можно легко детектировать в самых разнообразных смесях. Образование этого
иона идет по механизму, представленному на схеме 8.73.
? и-
C-OR '
C-OR
II
О
?
-OR'
yOR
С
О
Схема 8.73
?
[R-H]
!>
О
m/z 149
Интенсивность пиков молекулярных ионов ангидридов карбоновых кислот
низка, несколько увеличиваясь в случае ненасыщенных кислот. Кроме того, при
увеличении напуска образца в источник возрастает интенсивность пика иона
МН+. Основные пики в спектрах обусловлены ацильными ионами, которые
далее теряют молекулу СО (рис. 8.78). В случае смешанных ангидридов по
величинам m/z ацильных ионов легко определить, какими кислотами образован
ангидрид.
266 Глава 8. Основные направления фрагментации органических соединений
ЮО ?
ЭО -
80 -
70 -
во -
SO -
40 -
зо -
20 -
Ю -
О -
1,%
.1
1
н 1 " ¦ 1 ^J~
ST
U—, . ^ , ,
130
1 1 1 1
20 ЗО 40 SO ?? 70 вО ?? ЮО 11 О 120 130
m/z.
Рис. 8.78. Масс-спектр электронного удара ангидрида пропионовой кислоты
Для циклических ангидридов дикарбоновых кислот основной процесс
заключается в элиминировании молекулы СО2. Образующийся ион отщепляет далее
молекулу олефина или СО. Молекула СО может отщепиться и непосредственно
из М4*", причем интенсивность этого процесса может быть выше по сравнению
с отщеплением СО2 (m/z 86 и 70 на рис. 8.79). Для фталевого ангидрида
характерно последовательное элиминирование молекул СОг и СО.
Распад М+" амидов напоминает распад М+" сложных эфиров. Однако атом
азота создает свои особенности, поскольку в электронно-невозбужденном
состоянии заряд и неспаренный электрон с большей вероятностью локализованы
именно на нем. Интенсивности пиков молекулярных ионов невысоки, но эти
пики вполне различимы в спектрах.
Максимальной интенсивностью в масс-спектрах амидов низших кислот
обладает ион (OCNH^") с m/z 44. В случае более длинных алкильных цепей
С—С-разрывы приводят к гомологической серии ионов с m/z 58, 72, 86 и т. д.,
а аналогичные разрывы с миграцией атома водорода —к серии ионов с m/z 59,
73, 87 и т. д. В основной серии характеристических ионов с четными массами
наблюдаются повышенные интенсивности пиков ионов, отстоящих друг от
друга на 56 Да, начиная с m/z 72 для незамещенного амида (m/z 72, 128, 184
на рис. 8.80, а). Механизм их образования аналогичен описанному выше для
карбоновых кислот и их эфиров (схема 8.67а). Пики углеводородных ионов
значительно менее интенсивны, чем в спектрах кислот и сложных эфиров. На
юо -, 1,%
ЭО
80 ?
70
60
50
40
30
20
10
О
jU
Ik
70 8О
m/z
Рис. 8.79. Масс-спектр электронного удара ангидрида глутаровой кислоты
8.11. Карбоновые кислоты и их производные 267
100 ?
90 -|
80 -I
70 -|
во А
50 А
40 -I
30 А
20 4
40
100 ?
90 -|
60
70
60 Ч
50
40
30
20 ?
1 0
о
40
1,%
59
1 26
I
227
1 00
120 140
m /?
1 60
1 60
200
?,%
59
220
227
120 140
m /?
Рис. 8.80. Масс-спектры электронного удара тетрадеканамида (?) и ?,?-диэтилдеканами-
рисунке 8.80 представлены масс-спектры ЭУ тетрадеканамида и ?,?-диэтилде-
канамида.
Отщепление молекулярным ионом амидной группы с образованием ацильно-
го иона (процесс характерный для сложных эфиров) в случае незамещенных
амидов практически не идет. Несколько облегчается процесс разрыва этой
связи в М+* диалкиламидов, однако заряд предпочтительно остается на атоме
азота. Например, в спектре ?,?-диэтилдеканамида (рис. 8.80,6) ацильный пик
(m/z 155) едва заметен, тогда как интенсивность пика альтернативного аминного
иона (m/z 72) существенна. Следует отметить, что ацилий-катион все ясе
образуется при распаде амидов ароматических кислот.
Перегруппировка Мак-Лафферти по кислотной цепи наблюдается при
распаде амидов кислот, начиная с масляной, а по аминным цепям, начиная с
этильной. Для N-алкиламидов характерен обычный ?-разрыв с последующим
отщеплением молекулы кетена, а N-алкиламиды низших кислот могут
элиминировать молекулу кетена непосредственно из М+\ Достаточно благоприятен
для этих соединений также первоначальный ?-разрыв. На схеме 8.74 процессы
первоначального а- и /^-разрыва представлены для случая
?,?-диэтилдеканамида (рис. 8.80,6). Следует подчеркнуть, что это не единственные направления
образования ионов с m/z 58 и 72; другими могут быть также структуры и даже
состав ионов с этими величинами m/z, которые образуются при распаде М+'
амидов.
Молекулярные ионы амидов ароматических кислот достаточно устойчивы.
Основной процесс распада предполагает отщепление аминного радикала с
268 Глава 8. Основные направления фрагментации органических соединений
О
QH17-ra2-c-N-CH2--CH3 ~СНз
с2н5
9 4.
С8Н17~СН2-С-Ы-СН2-СНз
с2н5
-ьг
о
с8н17-сн-с
)г\ I
? +N-CH2-CH3
сн2
о
)г\ I
^SaS^ ш~сн2-снз
сн2
m/z 58
-С9Й18СО
? +N~CH2—CH3
СН2-СН2
HN-CH2-CH3
сн2—сн2
m/z 72
Схема 8.74
образованием ароильного иона, который далее теряет молекулу СО (m/z 105 и 77
на рис. 8.81). В спектрах N-фенилалкиламидов максимальный пик принадлежит
М+# аналина, который образуется при отщеплении молекулы кетена из
молекулярного иона.
Молекулярные ионы галогенангидридов карбоновых кислот не стабильны,
легко отщепляют атом галогена, в связи с чем их пики в спектрах не
наблюдаются. Максимальное значение m/z имеют ацильные ионы. Большая часть
ионного тока приходится на долю углеводородных фрагментов. Тем не менее
атом галогена сохраняется в составе ряда фрагментных ионов. На рисунке 8.82
в спектре октаноилхлорида можно выделить два дублета (m/z 78, 80 и m/z 91,
93) с характерным для одного атома хлора соотношением интенсивностей
юо
©о
80
ТО -j
во А
so -j
-*о
зо
20
1 О
20 ЗО -4-0 SO вО ?? вО 90 ??? ПО 120 130 140 ISO
rri/z
Рис. 8.81. Масс-спектр электронного удара ?,?-диметилбензамида
юо
90 ?
во
70 ?
во
50
40
ЗО
20
?.?,??... .?,?
u
40 50 ?? TO вО ЭО ЮО НО 120 130 140
m/z
Рис. 8.82. Масс-спектр электронного удара октаноилхлорида
8.11. Карбоновые кислоты и их производные 269
3:1. Первый из этих ионов образуется в результате перегруппировки Мак-
Лафферти, а второй эквивалентен иону с m/z 87, образующемуся при распаде
метиловых эфиров карбоновых кислот (схема 8.67а). Олефиновый ион с m/z 98
(рис. 8.82) образуется при одновременном отщеплении молекул НС1 и СО из
М4" (схема 8.75). Аналогичный процесс абсолютно не характерен для амидов и
сложных эфиров.
_ О
ГУ' 7И +
?
Схема 8,75
Химическая ионизация. Пики протонированных молекулярных ионов МН+
карбоновых кислот достаточно интенсивны в спектрах химической ионизации
метаном. Наиболее характерный процесс фрагментации связан с отщеплением
молекулы воды. Для ароматических кислот можно отметить также выброс из
МН+ молекулы СОг. Интенсивность пика [?? —?2?]+ резко возрастает при
наличии в молекуле второй карбоксильной группы [371] или (для ароматических
кислот) яртозаместителей с неподеленными парами электронов [372].
Например, отношение интенсивностей пиков ионов [?? —?2?]+/??+ для додека-
новой кислоты составляет 0,7, а для додекандиовой — 50. Геометрия молекулы
должна способствовать такому пространственному расположению этих
функциональных групп, при котором возможно их взаимодействие. В случае дикарбо-
новых кислот можно предположить образование ангидридов (схема 8.76), а в
случае ортозамещенных ароматических кислот—-взаимодействие заместителей
(схема 8.77).
Пики протонированных молекулярных ионов МН+ сложных эфиров имеют
значительную интенсивность в масс-спектрах химической ионизации водородом,
n~J(CH2)n ~Н2° . НО (СН2)„
о
Схема 8.76
А
ОН О
-С^ОН ^. /С— ОН ^v^CT
ОС
-Н20
? ^^ ^-хн ^ ^х
СС:
Схема 8.77
270 Глава 8. Основные направления фрагментации органических соединений
метаном, изобутаном. В двух последних случаях помимо пиков МН+
наблюдаются пики кластерных ионов, образующихся по механизму электрофильного
присоединения (разд. 5.2). Интенсивность процессов фрагментации
увеличивается с возрастанием экзотермичности процесса протонирования в ряду: водород >
метан > изобутан. Для метиловых эфиров основной процесс фрагментации МН+
обусловлен элиминированием молекулы спирта. Эта реакция аналогична
элиминированию воды из МН+ кислот. Присутствие второй карбоксильной группы
активирует этот процесс. С увеличением длины кислотной цепи метиловых
эфиров усиливаются процессы с образованием иона [М—Н]+ и последующим
отщеплением молекулы метилового спирта.
Для сложных эфиров с длинными цепями элиминирование молекулы спирта
подавлено, а основные процессы включают выброс молекулы алкена с
миграцией атома водорода от спиртового радикала, а также отщепление молекулы
кислоты с возникновением углеводородного карбокатиона (схема 8.78). Фактически
в этом процессе за протон конкурируют молекула кислоты и молекула олефина.
Результат этой конкуренции определяется величинами сродства к протону этих
соединений. Разветвления в спиртовой цепи, понижая СП соответствующего
олефина, приводят к увеличению интенсивности алкильного иона [373].
Образование иона протонированной кислоты можно объяснить также на основе
1,3-сигматропного сдвига.
ОН
+
??, ?
мнп
R-C— OCH2CH2R
J+
он он
1
1-1 '+ _. - +
R_C^-OH- R-C=0 ?'---CH2=CHR ** R-CH-CH3
Схема 8.78
В условиях химической ионизации дикарбоновых кислот метиловым спиртом
идет процесс этерификации [374]. Например, максимальный пик в спектре
себациновой кислоты принадлежит иону [МН—2Н2О+2СН3ОН]". Химическая
ионизация водой позволяет провести в газовой фазе обратный процесс —
гидролиз эфиров [375].
Для получения структурной информации об амидах можно использовать
триметилборат в качестве газа-реагента [376]. Ион-реагент (СНзОJВ+,
присоединяясь к атому азота, вызывает разрыв амидной связи, а, присоединяясь к
карбонильной группе, образует стабильный аддукт.
Химическая ионизация отрицательных ионов. Гидроксид-анион легко
отщепляет протон у карбоновых кислот. Образующийся карбоксилат-анион
устойчив. Часто его пик оказывается единственным в спектре. Аналогично протекает
8.11. Карболовые кислоты и их производные 271
химическая ионизация хлорид-анионом. Следует отметить, что наряду с карбок-
силат-анионом образуется и енолят-анион [377]. Дело в том, что, например,
для протона карбоксильной группы [378] и протона метильной группы [379]
в случае уксусной кислоты АН^СЛ = 348 и 363 ккал/моль. Изомерные ионы
имеют определенные различия в направлениях фрагментации (в частности,
карбоксилат-анионы теряют молекулу С02, а енолят-анионы — молекулу воды),
но в условиях активации соударением изомеризуются друг в друга [379].
Депротонирование последующих гомологов карбоновых кислот протекает
по карбоксильной группе, однако в результате прототропной таутомерии до
начала фрагментации идет изомеризация части ионов [М—Н]~ в форму енолята.
На схеме 8.79 представлены направления фрагментации депротонированного
молекулярного иона изомасляной кислоты. Потери атома водорода и метильного
радикала характерны для карбоксилат-аниона, а потеря молекулы метана и
образование гидроксид-аниона — для енолят аниона. Декарбоксилирование в данном
случае не идет, поскольку величина сродства к электрону для изопропильного
радикала отрицательна [380].
СИЗч /О" _н· СН3ч _ -сщ СН3ч /СГ
С=с' + СН-СОО —¦* С=С
сн/ \5 сн3' |4 ?/ \з
In
ю- ,-(сн3Jссо CH3sdax)H _-сн<_ Ш2=с=с/°
сн3' хон
Схема 8.79
Для длинноцепочечных гомологов жирных кислот в условиях химической
ионизации отрицательных ионов наиболее характерным процессом распада
является фрагментация, удаленная от места локализации заряда (разд. 3.2.1).
Изучению этого процесса был посвящен представительный ряд исследований
[102-106].
Сложные эфиры R^COOR в условиях химической ионизации гидроксид-
анионом распадаются с образованием заряженных фрагментов [М—Н]~,
[M-H-ROH]- и ^СОСГ [381, 382].
Благодаря такому набору фрагментных ионов химическая ионизация
отрицательных ионов может с успехом использоваться для установления состава
жиров. В спектрах триглицеридов наблюдаются четыре пика характеристических
ионов, на основании которых можно сделать вывод о молекулярной массе (ион
[М—Н]~) и природе жирных кислот (карбоксилат-анионы). Если в молекуле
триглицерида две ацильные группы обусловлены одной и той же кислотой,
интенсивность пика соответствующего карбоксилат-аниона примерно в два раза
выше, чем интенсивность пика аналогичного иона другой кислоты [383].
272 Глава 8. Основные направления фрагментации органических соединений
Фрагментация енолят-анионов сложных эфиров, активированная
соударениями, протекает по целому ряду направлений [384-386]. На схеме 8.80
представлены направления распада метилового эфира 2-этилмасляной кислоты. За
исключением отщепления метильного радикала все реакции распада можно
изобразить, исходя из комплекса кетена с метокси-анионом.
(С2Н5JС-С02СНз
-СНз
(СгНзЪС-ООг
/С2Н5ч
ОД'
с=с=о сн3ог
-(С2Н5JССО
-сн2о
-C2H5OCH3
-сн3он
Схема 8.80
СН30
(QH5)C--CHO
** С2Н5-С=(ХГ
_ \?=с=о
сн3-сн/
Ряд перегруппировочных процессов, характерных для енолятов сложных
эфиров, в газовой фазе протекает по механизмам, хорошо известным в классической
органической химии. Примером служит перегруппировка Кляйзена [387, 388]
для депротонированного аллилфенилацетата (схема 8.81) и конденсация Дикмана
[389, 390] для енолята диметилового эфира адипиновой кислоты (схема 8.82).
Вопросам фрагментации молекулярных анион-радикалов амидов посвящена
работа [391]. Основные направления распада этих частиц представлены на
схеме 8.83.
В условиях химической ионизации отрицательных ионов амиды
отщепляют прежде всего амидный атом водорода, для которого в случае ацетамида
А#°исл = 342 ккал/моль, против АЯ^СЛ = 375 ккал/моль для метильного протона
О
CH3oJ^
О
Схема 8.81
сн3о
_\ о
сн3о
- сн3он
сн3о'
Схема 8.82
8.11. Карбоновые кислоты и их производные 273
R1
R1 С NHR2
I
о~
J-R'CO
R2NH~
-R2
-C—NH
&
Схема 8.83
[391]. Основные направления фрагментации депротонированных молекулярных
ионов незамещенных, моно- и диалкилзамещенных по атому азота амидов
подробно описаны в работе [392]. Схема 8.84 отражает направления распада
незамещенных амидов. Число возможных направлений фрагментации возрастает
при введении алкильного заместителя к атому азота [392]. В случае амидов
ароматических кислот в спектре наблюдается также пик иона Аг~.
Если амидного протона нет, то образуется енолят-анион. На схеме 8.85
представлены основные направления распада депротонированного
молекулярного иона ?,?-диэтилацетамида в условиях активации соударением [392].
Богатство процессов фрагментации обусловлено возможной изомеризацией исходного
енолят-аниона до начала фрагментации в результате переноса протона.
RCH2CON'w [RCH2(HNCO)]
RCH2CONH
'-н* ^ ОН
-RCH3
NCO
RCHCONH
RCH2-C=N~ ^ [HO-(RCH2CN)] Z^RCHCN
Схема 8.84
Г)
2Н5
?
-?
? ?
сн3 '-
-СгИ»
-CH2CON(QiH5J
(CH2C»)N(C2H5J
CH3CONC2H5
-(CzHjbNH
>HCp
~°^° » (C2h5Jn"
-CiHsNCHCHj
CH3OON-CH-CH3—[CH3CX> (C2H5NCHCH3)]
Схема 8.85
СН3СО
-CHjCHO
>CH3CHN=CHCH3
274 Глава 8. Основные направления фрагментации органических соединений
Задача 8.65. Для каких из перечисленных ниже соединений возможна
перегруппировка Мак-Лафферти?
а) Ацетон;
б) гексаналь;
в) пропилбензол;
г) диэтилбензол;
д) пропионовая кислота;
е) метилпропионат;
ж) этилформиат;
з) пентанон-3;
и) бутилбутаноат;
к) октанол;
л) бутилбензол;
м) декан.
Задача 8.66. Как можно различить по масс-спектрам электронного удара
перечисленные ниже изомерные органические соединения?
а) Масляная и изомасляная кислоты;
б) 2-метилвалериановая и 3-метилвалериановая кислоты;
в) ?-фенилмасляная и /?-фенилмасляная кислоты;
г) децен-5-овая и децен-2-овая кислоты;
д) гексановая кислота и пропилпропионат;
е) метилоктаноат и октилацетат;
ж) этилбензоат и фенилпропионат;
з) этилацетат и метилпропионат;
и) бутилацетат и метилбутаноат;
к) метилбутаноат и валериановая кислота;
л) винилпропионат и этилакрилат;
м) 2,3,4-триметилдекановая и 5,6,7-триметилдекановая кислоты.
Задача 8.67. Какие перегруппировочные процессы возможны при
фрагментации перечисленных ниже соединений?
а) Бутиловый эфир масляной кислоты;
б) бутиловый эфир изомасляной кислоты;
в) этилпропионат;
г) изопропилпропионат;
д) 1-этилгексиловый эфир 2-этилвалериановой кислоты;
е) 2-(лд/>а-метилфенил)гексиловый эфир 6-фенилдодекановой кислоты;
ж) бензиновый эфир валериановой кислоты;
з) 3-фенилпропиловый эфира 4-фенилмасляной кислоты.
8.11. Карбоновые кислоты и их производные 275
Задача 8·68. Составьте схему фрагментации декановой кислоты, спектр
электронного удара которой представлен на рис. 8.83.
юо
90
80
70
во
50
40
зо ?
20
Ю
1,%
LJ
? ..U,
jJLL
20 ЗО *0 SO 60 70 80 90 ЮО 110 120 130 1<40 1SO 160 170
m/z
Рис· 8.83· Масс-спектр электронного удара декановой кислоты
Задача 8.69. Составьте схему фрагментации метилового эфира 2,4-диме-
тилгептановой кислоты, спектр электронного удара которой представлен на
рис. 8.84.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
о -J
?,%
u-Д
41
—?
55
LA
69
ill. ...?
88
К
1 1ц
1
] ^
129
—, L г* 1 , г—
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170
m /?
Рис· 8·84· Масс-спектр ЭУ метилового эфира 2,4-диметилгептановой кислоты
Задача 8.70. Составьте схему фрагментации амилового эфира валериановой
кислоты, спектр электронного удара которой представлен на рис. 8.85.
юо ? !»%
90 -|
80 А
70 ?
60 -|
50
40
30
20
10
43
57
-J.
70
103
85
172
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170
m/z
Рис. 8.85· Масс-спектр электронного удара амилового эфира валериановой кислоты
276 Глава 8. Основные направления фрагментации органических соединений
Задача 8.71. Составьте предполагаемые схемы фрагментации для
перечисленных ниже соединений.
а) 2-Метилбутановая кислота;
б) 3-метилбутановая кислота;
в) изопропилацетат;
г) этилпропионат;
д) октилпропионат;
е) пропилбензоат;
ж) бутиловый эфир триметилуксусной кислоты;
з) втор-бутилбензоат;
и) бензилацетат;
к) этиловый эфир фенилуксусной кислоты;
л) винилпропионат;
м) 4-метилоктановая кислота.
Задача 8.72. Идентифицируйте алифатическое соединение (нет циклов) по
спектру электронного удара (рис. 8.86).
ОО ?
90 -
80 -
70 -
60 -
SO -
40 -
зо -
20 -
Ю -
О -
1/М>
I
м. III
45
1L . .им
?-г
57 I
_j
1 , . L
? ??
L
20 ЗО
Ю SO ©? ?? 80
m/z
Рис. 8.86
m/z
18
26
27
28
29
30
; 31
39
41
42
43
/,% |
13,1
21,1
61,7
100
83,6
14,1
4,20
3,60
5,10
5,50
8,41 |
m/z
44
45
46
47
55
56
57
73
74
75
1 76
/, %
7,11
55,7
6,01
4,50
16,8
16,4 !
30,1
48,4 !
78,5
2,54
0,31
Задача 8.73. Идентифицируйте алифатическое соединение (нет циклов) по
спектру электронного удара (рис. 8.87)
юо
90
80
ТО Ч
60 -|
50
АО
ЗО -I
20 -|
10
о
1,°/о
so
m/z
Рис. 8.87
m/z
25
26
27
28
29
30
31
32
33
41
42
?, % ?
1,40
11,4
36,7
15,8
100
4,00
7,21
0,30
1,10
?,??
3,10
m/z
43
45
55
56
57
58
59
60
87
88
1 89
/,%
1,90
5,00
3,40
2,40
75,2
2,70
24,6
0,64
1,60
21,3
0,89
8.11. Карбоновые кислоты и их производные 277
Задача 8.74. Идентифицируйте алифатическое соединение (нет циклов) по
спектру электронного удара (рис. 8.88).
юо
90
80
70
60
50
40
ЗО
20
Ю
О
1,%
43
73
20 ЗО 40 50 60 70 80 90
m/z
Рис. 8.88
m/z
27
29
37
38
39
40
41
42
43
/,% 1
39,1
5,74
2,10
2,98
15,9
2,20
46,4
9,76
100
m/z
44
45
55
60
71
73
88
89
90
Д%
2,87
14,1
3,12
1,29
1,86
27,8
9,12
0,40
0,04
Задача 8.75.
(рис. 8.89).
1,%
Идентифицируйте соединение по спектру электронного удара
юо
90
80
70
60
50
40
ЗО
20
ю
о
42
60
87
20 ЗО 40 50
вО 70
m/z
вО 90 ЮО НО
Рис. 8.89
m/z
26
27
28
29
31
40
41
42
43
44
/,% 1
2,12
1,24
37,5
15,7
8,44
1,11
11,6
100
55,0
20,1
m/z
45
46
59
60
61
69
87
88
104
I 105
/, %
85,0
2,23
1,84
79,4
1,75
5,18
12,0
0,40
5,00
0,17
Задача 8.76. Идентифицируйте соединение по спектру электронного удара
(рис. 8.90)
юо
90
80
70
60
50
40
30
20
10
О
1,%
50
НИ, ?.
76
104
148
40 60 80 100 120 140
??1 /?
Рис. 8.90
m/z
26
27
28
29
36
37
38
39
44
49
50
51
52
/,% ?
7,01
3,27
4,17
2,13
4,17
22,0
24,3
4,17
3,27
10,1
60,5
4,17
7,01
m/z
53
61
73
74
75
76
77
104
105
106
148
149
I 150
/,%
4,72
3,27
7,01
24,3
14,7
90,1
9,01
100
7,61
0,45
13,0
1,19
0,11
278 Глава 8. Основные направления фрагментации органических соединений
Задача 8.77. Идентифицируйте соединение по спектру электронного удара
(рис. 8.91).
ЭО А
80 А
ТО А
во -|
SO A
АО А
ЗО А
20 А
10 А
о -L
19°Л>
и
14-3
1,1
во
_Ь-
1_
?
-4
1
L
88
? ,. . . .
m/z
27
28
29
30
31
41
42
43
44
45
55
/,% |
71,9
17,3
85,3
3,80
3,70
32,8
23,7
100
4,60
19,1
4,40 |
m/z
56
60
61
70
71
73
88
89
101
116
I 117
/,%
1,00
20,2
8,31
6,01
89,4
16,3
41,9
11,7 J
5,71
5,12
0,34
ЗО 40 50 60 70 80 ЭО 100110120
m/z
Рис. 8.91
Задача 8.78. Какие перегруппированные процессы протекают при
фрагментации под электронным ударом перечисленных ниже соединений?
а) N-Этилгексанамид;
б) N-пропилбутанамид;
в) N-изопропилпентанамид;
г) ?,?-диэтилбутанамид;
д) >1,М-диметил-2510-диэтилгексадеканамид.
Задача 8.79. Какие различия наблюдаются в спектрах электронного удара
перечисленных ниже изомеров?
а) N-Этилацетамид и ?,?-диметилацетамид;
б) бутанамид и N-метилпропанамид;
в) 2-метилпропанамид и N-этилацетамид;
г) N-фенилацетамид и амид фенилуксусной кислоты;
д) деканамид и N-амилпентанамид.
Задача 8.80. Объясните образование основных ионов и составьте схему
фрагментации под ЭУ N-гексилбутанамида (рис. 8.92).
1 00
90
80
70
60
50
40
30
20
1 0
ь%
43
AM
tlil.
1 01
128
1 71
30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180
m /z
Рис. 8.92. Масс-спектр электронного удара N-гексилбутанамида
8.11. Карбоновые кислоты и их производные 279
Задача 8.81. Идентифицируйте соединение по спектру электронного удара
(рис. 8.93).
ЮО ?
©О -
80 -
ТО -
60 -
SO -
40 -
ЗО -
20 -
Ю -
О -
I
,^
29
У
J ^.
лл
-" г-
57
¦¦¦I ,
73
-J
J 1
so во
m/z
Рис. 8.93
m/z
27
28
29
30
42
43
44
45
/,% ?
24,3
15,0
36,4
3,07
1,41
1,42
100
4,75 I
m/z
54
55
56
57
58
72
73
74
/,%
2,86
5,72
3,21
17,6 !
0,92
9,04
49,0
1,81
Задача 8.82. Идентифицируйте соединение по спектру электронного удара
(рис. 8.94). Пик молекулярного иона отсутствует. Обратите внимание на дублеты
с m/z 78-80 и 91-93.
100
90
80
70
60
50
40
30
20
10
0
1,%
,? »iiil|llh
43
57
71
1л_1
78
91
99
30
40
50
60
70
m /?
80
90
100
110
m/z
32
35
36
37
38
39
40
41
42
43
44
?,% ?
5,94
3,96
6,93
4,95
7,92
57,4
8,91
100
65,3
100
3,96
m/z
49
50
51
53
54
55
56
57
58
60
61
/,% |
2,97
2,97
3,96
5,94
2,97
57,4
30,6
46,5
1,98
0,99
0,99
m/z
63
65
69
70
71
72
73
74
77
78
79
I,% 1
10,8
3,96
8,91
27,7
28,7
1,98
0,99
0,99
2,97
39,6
5,94
m/z
80
81
91
93
98
99
100
105
106
107
1 108
/,%
14,8
2,97
15,1 !
5,02
8,12
57,8
3,86
4,95
5,94
1,98
1,97
Рис. 8.94
280 Глава 8. Основные направления фрагментации органических соединений
Задача 8-83.
(рис. 8.95).
Идентифицируйте соединение по спектру электронного удара
ЮО ?
90 А
80 А
70 А
во А
50 А
40 А
зо А
20 -|
10
о
.1,%
У
59
72
-UL
40
160
m/z
200
240
Рис. 8.95
m/z
41
43
44
55
56
57
59
60
71
72
73
74
85
86
87
100
/,% I
14,2
21,0
7,72
13,7
6,39
16,6
100
12,3
6,27
44,3
10,9
1,02
4,87
7,56
2,22
2,54
m/z
101
114
115
128
129
142
156
170
184
197
198
212
213
226
255
I 256
/,%
2,47
7,57
3,18
7,24
3,32
1,03
1,38
2,76
4,23
1,09
2,55!
4,78
1,11
2,34
3,15
0,55
Задача 8.84· Составьте предполагаемые схемы фрагментации
перечисленных ниже соединений под электронным ударом.
а) N-Этилбензамид;
б) N-этилпропанамид;
в) М-этил-М-пропилацетамид;
г) бромангидрид валериановой кислоты;
д) 2-метилпропаноилхлорид.
Задача 8.85.
(рис. 8.96).
Идентифицируйте соединение по спектру электронного удара
ЮО
90
80
70
во
50
40
ЗО
20
10
О
1,%
±JL
80 ЮО
m/z
m/z
27
29
38
39
40
41
50
51
52
57
63
?,% ?
6,58
29,1
1,95
7,13
1,02
1,05
1,40
5,00
1,38
13,8
2,10
m/z
64
65
66
77
91
92
93
94
149
150
I 151
I, %
2,36
8,28
8,19
6,30
1,49
3,41
100
7,13
18,0
1,68
0,10
Рис. 8.96
8.12. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоединения 281
8.12. Нитрилы, изонитрилы, нитросоединения,
гидразины, оксимы и диазосоединения
Электронный удар. Нитрилы характеризуются самыми высокими энергиями
ионизации среди функциональных соединений. В электронно-невозбужденном
состоянии заряд и неспаренный электрон локализованы на атоме азота. На
рисунке 8.97 представлен масс-спектр деканонитрила.
Молекулярные ионы очень неустойчивы, хотя их пики все-таки присутствуют
в спектрах. Более интенсивны (m/z 152 на рис. 8.97) пики ионов [М—Н]+. Атом
водорода в этом случае отщепляется от атома С2, для которого энергия связи
С—? близка к энергии аналогичной связи для аллильного положения [393].
При работе с нитрилами трудно избежать образования ионов МН+. ?-Разрыв не
характерен для нитрилов, поскольку должен приводить к четверной связи в
катионе CN+. Элиминирование молекулы HCN также не является доминирующим
направлением и протекает лишь при наличии в цепочке достаточно лабильных
атомов водорода.
Две основные гомологические серии ионов с m/z 26, 40, 54, 68 и 27, 41,
55, 69 оказываются изобарны углеводородным сериям (разд. 8.1 и 8.2). Ионы
первой серии образуются в результате разрывов С—С-связей по всей длине
углеводородной цепи, причем следует подчеркнуть, что обычные для других
классов соединений ?-разрывы не характерны для нитрилов. Наиболее
интенсивные пики в этой серии (??/? 96, ПО на рис. 8.97) обусловлены, по-видимому,
реакцией замещения с образованием циклических ионов (схема 8.86). Больший
размер цикла по сравнению с рассмотренными ранее аналогичными реакциями
аминов, тиолов и галогенидов объясняются геометрией тройной связи. Для того,
чтобы не было существенных искажений валентных углов, размер цикла должен
быть не менее 6, а наиболее вероятны циклы, в которых ? = 0-2.
Серия нечетноэлектронных ионов образуется в результате элиминирования
из М4" молекул олефинов. Для нитрилов эти реакции протекают достаточно
100 -?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
? -
и
2
I,0/.
} ,
I
0
I I
\ ?
30 40
41
I 5
I
? ? II
50
4 ?
I
II t l
60 7
39
||
?
?^
?
8
80
9
2 ?
?
??? щ I
90
б
II
100
1
1'
10
Jj
0
124
120
138
? ? ?
130 140
152
|.
150
m/z
Рис. 8.97. Масс-спектр электронного удара деканонитрила
282 Глава 8. Основные направления фрагментации органических соединений
R
· +
N
\сн2)/
R*
Схема 8.86
\—№)
№)„
активно, но оказываются не столь селективными, как это было описано для
других классов органических соединений [394]. Значительно влияние скелетных
перегруппировок и рандомизации атомов водорода. В частности, если ион с
m/z 41 образуется по механизму перегруппировки Мак-Лафферти (схема 8.87),
то потеря молекулы этилена из М+# может протекать с участием атомов
углерода из разных положений алкильной цепи [88]. Наиболее интенсивный пик
с m/z 97 в спектрах высших нитрилов обусловлен ионом циклической структуры
(схема 8.88).
?-·?#
?
+
??
Схема 8.87
_ RCH=CH2
Схема 8.88
- RCHCH2
CH2=C=NH
m/z 41
+·
??
m/z 97
Энергии ионизации изонитрилов несколько ниже, чем у изомерных нитрилов.
В электронно-невозбужденном состоянии заряд, по-видимому, расположен на
атоме азота, а неспаренный электрон —на атоме углерода. Молекулярный ион
имеет большую склонность к ?-разрыву с элиминированием радикала CN" и
образованием углеводородного иона алкановой серии (рис. 8.98). Достаточно
интенсивны также пики ионов, обусловленных выбросами из М+" частиц HCN
и НгОГ. Эти процессы позволяют различить изомерные нитрилы и изонитрилы
по их масс-спектрам ЭУ.
Следует упомянуть также, что достаточно высокую интенсивность имеет пик
иона с m/z 41, образующегося в результате перегруппировки Мак-Лафферти
(схема 8.89).
?
•с
R
СН
1+
RCHCH2
CH2=N=CH
Схема 8.89
8.12. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоединения 283
100 ?
90 -|
80 А
70 А
во А
50 А
40 -I
30
20 ?
10
1,%
41
57
20 25 30 35 40 45 50 55 60 65 70 75 80 85 90
m /?
Рис. 8.98. Масс-спектр электронного удара бутилизоцианида
100
90
80
70
60
50
40
30
20
ю Ч
1,%
94
+¦
70
121
30
40
50
60
80
110 120
130
90 100
m /z
Рис. 8.99. Масс-спектр электронного удара 4-фторбензонитрила
140
150
В отличие от алифатических аналогов ароматические нитрилы и изонитрилы
характеризуются очень близкими масс-спектрами. Пики М4" очень интенсивны.
На рисунке 8.99 представлен масс-спектр электронного удара
4-фторбензонитрила. Помимо пика молекулярного иона можно отметить лишь достаточно
интенсивный пик иона [М—HCN]+\ Ионы [М —Н]+ и [М —CN]+
характеризуются пиками с низкими интенсивностями. Для алкилзамещенных бензонитрилов
важную роль играет предварительный бензильный разрыв с образованием иона
тропилиевой структуры, который далее распадается с элиминированием
молекулы HCN.
Введение в молекулу нитрогруппы приводит к заметному увеличению
энергии ионизации. Благодаря высокому значению электроотрицательности нит-
рогруппа дестабилизирует катионы. В масс-спектрах алифатических нитросо-
единений пики М+'отсутствуют. Главный первичный процесс распада связан
с элиминированием нитрогруппы (рис. 8.100). В масс-спектрах доминируют
пики углеводородных ионов. Фактически отсутствует и серия гомологических
ионов. Минорный процесс может включать последовательное отщепление атома
кислорода и молекулы воды с образованием ионов со структурой нитрилов.
Аналогичным образом распадаются алициклические нитросоединения.
284 Глава 8. Основные направления фрагментации органических соединений
100
90
80
70
60
50
40
30
20 -I
?? ?
1,%
JJ
20 ЗО
50 ??
90 ЮО 110 120
m/z
Рис. 8.100. Масс-спектр электронного удара 1-нитрогексана
Наличие алкильного заместителя соответствующей длины инициирует
процесс по механизму перегруппировки Мак-Лафферти (схема 8.90). С увеличением
длины цепи возрастает интенсивность пика иона, возникающего в результате
миграции к нитрогруппе двух атомов водорода. Два иона с m/z 61 и 62
(для незамещенных по ?-положению нитросоединений) образуются таким же
образом (аналоги ионов с m/z 60 и 61 в спектрах кислот).
R4
СН
3>
но
ch2n
N
СН
л.
??)
о
но
А
-RCHCH2
ноГ
N.
О
R
СН'
J
Схема 8.90
СН"
R
,/
V
J
Пики молекулярных ионов ароматических нитросоединений достаточно
интенсивны (рис. 8.101). Помимо нитрогруппы ионы М+# элиминируют атом
кислорода и молекулу N0. Последний процесс становится возможным
благодаря установлению равновесия между двумя формами молекулярного иона
(схема 8.91). Выброс молекулы NO сопровождается последующим отщеплением
молекулы СО. Изомеризация в нитриты протекает и в случае алифатических
нитросоединений. ?-Разрыв в ионе такой формы обусловливает образование
иона с m/z 60 при распаде незамещенных в ?-положении нитроалканов [88].
FH^KNO^
m/z 125
^0_ F-H^-N^l*—; F-^-O-N^t
?
-NO
.-NO
-N02
F—<C )> +
m/z 95
Схема 8.91
m/zlll
-col
m/z 83
8.12. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоединения 285
1 00
90
80
70
60
50
40
30
20
1 О
О
100
90
60
70
60
50
40
30
20
1 О
О
1,%
ч1| ¦! ¦¦
J iL
90
m /?
?,%
51
iL
77
1 37
I
80
m /?
Рис. 8.101. Масс-спектры ЭУ 4-фторнитробензола (а) и 2-итротолуола (б)
Наличие двух и более нитрогрупп в молекуле не меняет основных
направлений распада. Высокую интенсивность в спектре могут иметь катионы NOj"
и NO+. Для алкилзамещенных нитробензолов наиболее характерен бензильный
разрыв. При наличии трех атомов углерода в цепи протекает перегруппировка
Мак-Лафферти (разд. 8.4).
Нитрогруппа активно взаимодействует с заместителями в оргао-положении,
содержащими атомы водорода [395]. В спектре о-нитротолуола максимальную
интенсивность имеет пик иона [М —ОН]+, который образуется по механизму,
аналогичному представленному на схеме 3.16. Этот факт позволяет легко
отличать opmo-нитросоединения от их мета- и иоря-изомеров. Ион [М — 0Н]+ в
случае онитротолуола элиминирует далее молекулу СО (m/z 92, рис. 8.101,6),
что свидетельствует об изомеризации исходной структуры иона [М—0Н]+;
процесс изомеризации предусматривает восстановление нитрогруппы. На схеме 8.92
подобный процесс восстановления, доказанный изотопными метками, показан на
примере фрагментации о-нитрофенилциклопропанов [396].
°ч —1 +
N-H
Схема 8.92
286 Глава 8. Основные направления фрагментации органических соединений
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
о J
I,%
I
U4—
4
I
aJLiiJ
5
Lj—r-
59
II
..I.I . ^.j^u.^ J
101
I
U . r-
144
, , u
a
—,
30 40 50 60 70 60 90 100 110 120 130 140 150
m/z
ioo -, I»%
90
80
70
60 ?
50
40 -|
30 A
20 ?
?? A
59
lliil . il.l
87
144
115
1
? 1 "? ? 1
30 40 50 60 70 80 90 100 110 120 130 140
100 -,
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
о -J
I,%
7
_Li ,1,1 llll , Ml
m/z
7
1
?1 il
k ... . Ill
07
J. Щ
121
J
в
136
? t
Ц
30 40 50 60 70 80 90 100 110 120 130 140
m/z
Рис. 8.102. Масс-спектры электронного удара 1,1-дибутилгидразина (?), 1,1-диэтил-2-
ewop-бутилгидразина (б) и 1-фенил-1-этилгидразина (в)
Молекулярные ионы замещенных гидразинов достаточно устойчивы
(рис. 8 Л 02). Заряд и неспаренный электрон могут быть локализованы на любом
из атомов азота (предпочтительнее на более замещенном). Как следствие,
первичная фрагментация идет по любому из заместителей. Может разорваться
как связь С—С (аминный распад), так и связь С—N. Этот процесс становится
особенно благоприятным для три- и тетраалкилтидразинов (ион с m/z 87 на
рис. 8.102, б). Для полиалкилированных гидразинов возможно также образование
иона R2N+ из первичного иона (фрагмент с m/z 72 на рис. 8.102,6).
Схема 8.93 отражает направления фрагментации 1,1-дибутилгидразина, масс-
спектр которого приведен на рис. 8.102, а.
8.12. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоединения 287
С3Н7-СН2
\+· -С3Н7
^N-NH2 —
СН2
С4Н9
m/z 144
-С4Н9
??9-?=??2
m/z 87
Qlty
^Йч
NH2
-С3Н6
СН<
m/z 101
-С4Н8
CH2=NH-NH2
m/z 45
Схема 8.93
CH3'
^N-NH2
m/z 59
-NH3
C5H10N
m/z 84
Основной первичный ион (m/z 101) распадается далее с элиминированием
молекул олефинов через четырехчленное или шестичленное переходное состояние
(схема 8.44 для аминов). Достаточно интенсивными могут быть пики алкильных
ионов (m/z 57 на рис. 8.102, а). Менее очевиден выброс молекулы аммиака (ион
m/z 84).
Для арилзамещенных гидразинов предпочтительно сохранение
ароматического ядра в заряженном фрагменте. В этом случае также возможен первоначальный
разрыв N—N-связи (рис. 8.102, в).
В работе [397] описан синтез индолов по Фишеру в условиях химической
ионизации (схема 8.94). Доказательством протекания реакции служит полная
идентичность спектров MIKES иона [?—??3]4" исходного фенилгидразона
ацетона и МН+ образующегося 2-метилиндола. Приведенная в работе схема
превращения является общей для реакции в растворе и в условиях химической
ионизации. Авторы [397] отмечают, что лишь часть молекулярных ионов
фенилгидразона ацетона трансформируется в индол, остальные же распадаются по
конкурентным направлениям. Это наблюдение совпадает с известным фактом о
незначительном выходе 2-метилиндола в реакции Фишера.
о©?
~н
?
Схема 8.94
288 Глава 8. Основные направления фрагментации органических соединений
1 00
90
80
70
60
50
40
30
20
10
100
90
80
70
60
50
40
30
20
10
1,%
1,%
30
иЦ
Ill
59
72
70
,,,,???,
40
50
60
80
m /z
73
90
1 01
90
100 110 120
_л
1 30
70 80
m /?
Рис. 8.103. Масс-спектры электронного удара оксим бутаналя (а) и оксим гептанона-4 (б)
Интенсивность пиков молекулярных ионов оксимов алифатических
альдегидов и кетонов мала (рис. 8.103). Наиболее важными процессами фрагментации
являются перегруппировка Мак-Лафферти и выброс алкильного радикала с
образованием циклического иона с m/z 72 (для альдоксимов). На схеме 8.95
представлены основные направления фрагментации оксима бутаналя, масс-
спектр которого приведен на рис. 8.103, я. В случае кетоксимов перегруппировка
Мак-Лафферти может идти последовательно по двум цепям (ионы с m/z 101 и
73 на рис. 8.103,6).
Пики М+' оксимов ароматических альдегидов и кетонов весьма интенсивны.
Основные первичные направления распада связаны с выбросом простейших
О*
ch3v-\+
? он
он
-СНз
-С
??^
ОН
? ?11?
-сн2СН2
+
ОН
m/z 72
¦+/
??
/6н
сн2^
m/z 59
ОН
—— CH2=C=NH
m/z 41
Схема 8.95
8Л 2. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоединения 289
100
90
80
70
60
50
40
30
20
10
1,%
146
90
105
77
U—L
40
51
50
63
60 70
80
1 18
90 100 110 120 130 140 150
m /z
Рис. 8.104. Масс-спектр электронного удара диазоацетофенона
частиц ОН', Н2О, CO, HCN, HCNO и т. д. (задача 8.90 для оксима ацетофенона).
Для протекания этих процессов зачастую необходима значительная перестройка
исходной структуры молекулы. Пики ионов [М —R]+ в спектрах альдоксимов
малоинтенсивны.
На схеме 8.96 представлены основные направления распада N-пропилимина
гептанона-4. Схема не претендует на полноту и включает прежде всего
перегруппировку Мак-Лафферти и вторичные перегруппировочные процессы через
четырехчленное переходное состояние, а также ?-разрывы. Все они очевидны,
исходя из информации, полученной при рассмотрении фрагментации других
классов органических соединений.
+ - СШ7 С3Н7\ +.
C3H7-C=NCH2-C2H5 « 3 7 C=NCH2-C2H5
с3н/
-с2н5
-СзН6
HCBNCH2-C2H5
-с3н6
?????
-СзН6
с3н7ч +
yO=N=CH2
с3н/
с3н6
+·
C3H7~HC=NCH2-C2H5
-с3н6
H2C=NCH2-C2H5
Схема 8.96
-с2н5
^C3H7-HC=N=CH2
-c2h5i
¦СзН6
?20=?=??2
Интенсивность пиков М+Ф диазокетонов существенно зависит от строения
молекулы и для многих ароматических представителей этого класса может
быть весьма высокой (рис. 8.104). Помимо очевидного отщепления радикала
CHN2 с образованием ацильного катиона, в спектрах диазокетонов наблюдается
выброс молекулы азота, сопровождающийся перегруппировкой Вольфа, хорошо
известной в органической химии с начала XX века (схема 8.97).
10—1113
290 Глава 8. Основные направления фрагментации органических соединений
R-C-CH-N=N ^ R^C-CH *> R-CH=C=0 ¦ —^->R-CH
о о
Схема 8.97
Зеллер с соавторами [398-401] установил, что перегруппировка протекает
для широкого круга алифатических диазокетонов и диазокетоэфиров при их
ионизации электронным ударом, а также в условиях термолиза и фотолиза.
Так трансформируются не только простейшие, но и более сложные
циклические диазокетоны [402^406]. Было доказано, что во всех изученных случаях
перегруппировка Вольфа является доминирующим процессом. С помощью
изотопных аналогов было установлено, что альтернативный механизм, связанный
с образованием эпоксидного цикла в качестве интермедиата, не реализуется
[401]. Перегруппировка Вольфа является основным направлением последующей
трансформации молекулярных ионов замещенных диазоацетофенонов [407, 408].
Если в алифатической цепи диазокетонов содержатся гетероатомы или ге-
тероатомные группировки, способные оказать анхимерное содействие
элиминированию молекулы азота диазогруппы, то в присутствии кислых агентов
такие диазокетоны могут подвергаться циклизации [409]. Это наблюдение было
положено в основу синтеза ряда ранее недоступных или труднодоступных
гетероциклических систем. Движущей силой подобной реакции служит прото-
нирование атома углерода диазокарбонильной группы (схема 8.98) с дальнейшей
нуклеофильной атакой по возникшему электронодефицитному центру.
СН2
* ¦· - -N2 4 +/ \
-X — №)„ -C-CHN2 + ~Х С= О
ft Ь ч 7
(СН2)„
Схема 8.98
Детальные масс-спектрометрические исследования на широком круге
диазокетонов, диазоэфиров и диазоамидов показали, что подобный процесс протекает
и в газовой фазе в условиях электронного удара и химической ионизации [9].
Химическая ионизация· Протонированные молекулярные ионы МН+ нитро-
и полинитроалканов достаточно устойчивы. Их пики имеют высокие
интенсивности в масс-спектрах химической ионизации метаном [410]. Это выгодно
отличает химическую ионизацию от электронного удара. Основные процессы
распада МН+ включают элиминирование молекул ЕЬО, UNO, HNO2. Еще более
надежную информацию о молекулярной массе алифатических нитросоединений
можно получить при использовании в качестве газа-реагента аммиака [411].
8.12. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоединения 291
В этом случае в спектрах доминируют пики ионов [M+NHU]"*". В спектрах
полинитроалканов высокую интенсивность может иметь пик иона NO^.
Химическая ионизация позволяет различать изомеры нитроароматических
соединений [412]. Поскольку для этой цели желательно помимо пика
молекулярного иона иметь представительный ряд фрагментов, лучшие результаты
получаются при использовании в качестве газа-реагента водорода, для которого
процесс ионизации характеризуется максимальной экзотермичностью (разд. 5.2).
Ключевыми направлениями распада, по которым можно идентифицировать
изомер, являются потери молекулы воды и азотсодержащих фрагментов (NO, HNO,
NO2, HNO2). В частности,, электронодонорные заместители с атомами водорода
в opmo-положении к нитрогруппе активизируют процесс элиминирования
молекулы воды из-за орто-эффекта.
Отщепление гидроксильного радикала очень выгодно при наличии электро-
нодонорного радикала в орто-шт лара-положениях к нитрогруппе, тогда как
потери азотсодержащих фрагментов благоприятны для л*еяш-изомеров.
Идентификация изомеров с электроноакцепторными группами проблематична [412].
Протонированные молекулярные ионы полинитроароматических соединений
характеризуются интенсивными пиками. Фрагментация не интенсивна и
протекает с выбросами ОН\ Н2О и N0 [413].
Особенностью нитроароматических соединений является их способность
восстанавливаться в амины в условиях химической ионизации протонирующими
газами-реагентами [412, 414, 415]. Благодаря этому процессу в спектрах
наблюдаются интенсивные пики протонированных молекулярных ионов
соответствующих аминов, которые ранее принимали за изобарные им ионы [МЫ—NO]".
Этот процесс активируется при повышении температуры и наличии паров воды
в источнике.
н+ гЯ^ -н2о
н ^ /?
с
Кн ?^??2
Ой
с-н
4??
н+ ГГЛ1 „ -н2о
?
Схема 8.99
жг ?2?^?
Первое сообщение о протекании в масс-спектрометре в условиях химической
ионизации перегруппировки Бекмана появилось в работе [416]. Результаты более
тщательного изучения этого процесса на целом ряде оксимов с ароматическими
и алифатическими радикалами [417] показали, что механизм масс-спектромет-
рических процессов аналогичен перегруппировке Бекмана, катализируемой кис-
292 Глава 8. Основные направления фрагментации органических соединений
лотами. Во всех случаях зарегистрирован высокий выход перегруппировочных
ионов. Как и в растворах, мигрирует заместитель, находящийся в исходной
молекуле в ан/тш-положении к гидроксильной группе (схема 8.99).
Химическая ионизация отрицательных ионов. Нитросоединения
характеризуются достаточно интенсивными пиками ионов М~~* в спектрах резонансного
захвата электрона. В работах [413, 418] детально изучены процессы
фрагментации молекулярных анион-радикалов.
Выше уже отмечалось, что перегруппировка Бекмана для оксимов,
протекающая в растворах под действием кислот, воспроизводится и в ионизационной
камере масс-спектрометра в условиях химической ионизации. Пока нет данных
об осуществлении подобного процесса в растворах под действием щелочей.
Возможно, причина здесь кроется в том, что гидроксид-анион оксимов является
чрезвычайно плохо уходящей группой. Между тем спектры активации
соударением депротонированных простейших оксимов обнаруживают максимальный по
интенсивности пик, обусловленный элиминированием молекулы воды. Авторы
[419] на основании исследования с помощью изотопных аналогов
образующегося фрагмента сделали вывод о протекании перегруппировки Бекмана
(схема 8.100). Усложнение структуры оксимов приводит к развитию конкурентных
направлений распада, а, например, для семикарбазонов перегруппировка вообще
не наблюдается.
Me2C=NO~ ^ ~CH2(Me)C=NOH > [(CH2=C=NMe) Он] ^^CH2=C=N-CH2
Схема 8.100
Перегруппировка Вольфа, подробно рассмотренная выше для положительных
ионов, протекает и в условиях химической ионизации отрицательных ионов
[420]. Например, депротонированный диазоацетофенон трансформируется в де-
протонированный арилкетен (схема 8.101), спектр активации соударением
которого оказывается полностью идентичным полученному при распаде этилового
эфира фенилуксусной кислоты. Вероятно, перегруппировка, как и для катион-
радикала, обусловлена миграцией карбаниона к электронодефицитному (хотя и
несущему отрицательный заряд) атому углерода.
- + - ~N2 /-^ _
PhC-C=N=N - Ph-G-C ** PhC=COT
II \?^
О О
Схема 8.101
Задача 8.86· Объясните образование иона m/z 60 из молекулярного иона
1-нитродекана.
8.12. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоединения 293
Задача 8.87. На рисунке 8.105 представлены масс-спектры о- и л/-нитро-
толуолов. Сделайте отнесение и объясните образование фрагментных ионов,
характеризующихся наиболее интенсивными пиками.
100
90
80
70
60
50
40
30
20
1 0
J 1,%
51
65
77
92
80 90 100 110
m /z
91
1 20
137
40
50
100 ?
90 A
80 -|
70
60
50
40 ?
30
20
10
I,%
51
J i .lit.
60 70
65
120 130 140
1L
107
_u
1 37
40
50
60
70
80
90
m /z
100 110 120 130 140
Рис. 8.105. Масс-спектры электронного удара ?-и л*-нитротолуолов
Задача 8.88. Объясните образование ионов с m/z 122, 109, 94, 93, 92, 81, 65
и составьте схему распада М+" онитрофенола (рис. 8.106).
1 0 0
9 0
8 0
7 0
6 0
5 0
4 0
3 0
2 0
1 0
1,%
3 9
6 5
5 3
8 1
9 3
1 0 9
J-
1 3 9
4 0
6 0
8 0 10 0 12 0
m /z
Рис. 8.106. Масс-спектр электронного удара о-нитрофенола
1 4 о
294 Глава 8. Основные направления фрагментации органических соединений
Задача 8.89· Составьте предполагаемые схемы фрагментации для
перечисленных ниже соединений.
а) и-Нитротолуол;
б) 3-хлорнитробензол;
в) 4-пропилнитробензол;
г) 3-нитрофенол;
д) 2-нитроантрацен;
е) тетраэтилгидразин;
ж) 1,2-диамилгидразин.
Задача 8.90. Объясните образование ионов, характеризующихся наиболее
интенсивными пиками, при распаде М+* оксима пентаналя (рис. 8.107).
Составьте схему фрагментации.
1 0 0
9 о
8 о
7 0
6 0
5 0
4 0
3 0
2 0
1 0
о
1,%
4 1
3 О
jJ ?!
L
5 9
7 2
4 О
5 О
6 0 7 0
m /?
8 О
9 О
1 О О
Рис. 8.107. Масс-спектр электронного удара оксима пентаналя
Задача 8.91
(рис. 8.108).
юо -, 1,%
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
1 0 -
0 -
'"'?
Идентифицируйте соединение по спектру электронного удара
30
А
40 50
5 О
jlh
76
6 О
70
m /z
80
"*-?—
90
1 03
100 110
m/z
27
37
38
39
49
50
51
52
61
62
63
/,% I
1,67
3,28
3,26
4,30
3,11
16,2
8,85
4,75
1,58
1,53
2,971
m/z
64
73
74
75
76
77
88
99
103
104
1 105
?%
1,53
1,75
4,95 !
8,60
35,3
5,55
1,01
1,42
100
8,18
0,24
Рис. 8.108
8.12. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоединения 295
Задача 8.92. Объясните образование ионов, характеризующихся наиболее
интенсивными пиками, при распаде М+* оксима ацетофенона (рис. 8.109).
Составьте схему фрагментации.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
с
..и 1
>1
L_
vjjL
J
77
L·
94
-ч!л
.
103
_J
Li,
119
..ili
135
_J
L
30
40
50
60
70
80 90
m /z
100 110 120 130 140
Рис. 8.109. Масс-спектр электронного удара оксима ацетофенона
Задача 8.93. Идентифицируйте соединение по спектру электронного удара
(рис. 8.110).
100
90 ?
80
70
60
50
40
30
20
10
1,%
50
40
60
75
80
143
155
100
120
m/z
140
160
180
201
200
m/z
46
48
49
50
51
52
61
62
63
64
73
? % ?
38,4
1,30
10,2
82,0
10,6
1,40
6,30
6,10
11,4
6,40
11,7
m/z
74
75
76
77
79
80
81
82
90
92
I 93
/,% |
47,5
81,5
69,0
4,70
10,5
1,60
10,4
1,50
1,20
3,30
1,03
m/z
105
106
107
117
119
128
129
130
131
143
1 144
/, % 1
1,20
0,90
1,10
2,40
2,20
1,10
2,70
1,20
2,60
18,7
1,20
m/z
145
155
156
157
158
171
173
201
202
203
1 204
/,%
18,3
53,4
4,30
52,2
3,50
6,10
5,901
41,6
3,30
40,6
3,10
Рис. 8.110
296 Глава 8. Основные направления фрагментации органических соединений
Задача 8.94· Идентифицируйте соединение по спектру электронного удара
(рис. 8.111).
100
90
80
70
60
50
40
30
20
10
О
1,%
90
-ill
63
¦ill,
JL.
117
30 40 50 60 70 80 90 100 110 120
m/z
Рис. 8.111
m/z
27
37
38
39
40
50
51
52
57
58
61
62
63
1 64
Л% ?
2,65
3,77
5,89
13,6
1,74
6,71
7,88
2,92
1,43
1,58
4,30
7,40
15,4
6,50!
m/z
65
74
75
76
87
88
89
90
91
114
116
117
118
| 119
/,% 1
3,44
2,25
5,23
3,84
2,95
4,69
32,5
49,9
6,71
2,01
67,3 !
100
8,52
0,32
Задача 8.95. Идентифицируйте соединение по спектру электронного удара
(рис. 8.112).
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 J
I,%
^-? ^
30
1 ill, .
50
Л-L
il 1
1 ? 1
Г6
Il_
9
2
J ¦
122
J r
j .
168
,
J ,
20
40
60
80
100
m/z
120
140
160
180
m/z
26
27
28
29
30
31
37
38
39
/,% 1
2,89
3,81
2,96
2,92
100
7,63
7,07
10,4
8,01
m/z
45
46
49
50
51
52
53
61
62
/,% 1
3,10
3,59
2,67
64,1
8,65
3,22
1,77
2,68
4,46
m/z
63
64
65
73
74
75
76
77
91
/, % 1
16,6
18,1
1,40
3,58
22,9
78,6
79,9
6,69
1,21
m/z
92
93
107
122
123
152
168
169
1 170
L%
44,0
2,92
1,03
36,3
2,57
2,55
94,4
7,02!
1,00
Рис. 8.112
8.13. Сульфоксиды, сульфоны, сульфокислоты 297
Задача 8·96. Идентифицируйте соединение по спектру электронного удара
(рис. 8.113).
100 ? 1»%
90 Ч
80 -|
70
60
50
40
30
20
10
О
57
л.
101
80 100
m/z
Рис. 8.113
m/z
27
28
29
30
31
32
39
41
42
43
/,% 1
8,71
16,7
17,9
37,5
4,60
5,83
3,80
15,4
2,96
12,4
m/z
44
45
55
57
58
59
70
71
72
87
/,% ?
4,62
41,6
2,43
11,6
1,54
2,93
5,01
1,97
3,13
3,361
m/z
99
101
102
103
142
144
145
146
/,% 1
8,30
100
6,34
0,12
1,31
33,6
3,23 !
0,12
8.13. Сульфоксиды, сульфоны, сульфокислоты
Электронный удар. Поскольку сера является (А+2)-элементом, ее
присутствие в молекулах соединений легко заметить по пику изотопного иона
(разд. 4.2). Сульфоксиды характеризуются достаточно интенсивными пиками
молекулярных ионов (рис. 8.114).
Рис. 8.114. Масс-спектры электронного удара дипропилсульфоксида (а), дифенилсульфок-
сида (б)
298 Глава 8. Основные направления фрагментации органических соединений
В отличие от кетонов первичные распады связаны не с ?-разрывами, а с
элиминированием гидроксильного радикала (m/z 117) и молекулы алкена (m/z 92
на рис. 8 Л14, а). Первый процесс подразумевает предварительную изомеризацию
М+Ф в енольную форму (схема 8.102). Второй протекает по механизму
перегруппировки Мак-Лафферти. Важнейшим вторичным процессом является ?-разрыв в
первичном перегруппировочном ионе с образованием четноэлектронного
фрагмента с m/z 63, единого для всех диалкилсульфоксидов (схема 8.102). Благодаря
достаточно необычной массе пик этого иона позволяет сразу предположить, что
анализируемым неизвестным образцом является сульфоксид. Простой разрыв
С—S-связи может идти с сохранением заряда на алкильном фрагменте (m/z 43
рис. 8.114, а). Этот ион может образоваться практически из любого
предшественника, представленного на схеме 8.102. Ион с m/z 41 имеет состав С3Н5. Для
сульфоксидов с более длинными алкильными цепями повышается доля пиков
углеводородных ионов, отчетливо просматривается алкильная серия.
+· +· -он* +
C3H7-S-C3H7 « C3H7-S=CH-C2H5 * C3H7-S=CH-C2H5
О m/zl34 ОН m/zm
-CsHeJ I-Сзн,
+· -с2н5* + +
C3H7-S-OH * CH2=S-OH HS=CH-C2H5
m/z 92 m/z 63 m/z 75
Схема 8.102
Разнообразие процессов фрагментации ароматических сульфоксидов
(рис. 8.114,6) указывает на существенную долю изомеризованных молекулярных
ионов. Составить схему фрагментации, как правило, не составляет труда,
учитывая, что наряду с изомеризацией М+# в сульфенатную форму идут
более сложные перестройки, в результате которых возможно элиминирование
частиц ОН', Н2О, CO, НСО', CS и т. д. На схеме 8.103 представлены четыре
простейшие изомерные структуры М+# дифенилсульфоксида. Все фрагмент-
ные ионы (рис. 8.114,6) могут быть образованы исходя из этих структур.
Масс-спектрометрии этих соединений посвящен обзор Р. А. Хмельницкого и
Ю. А. Ефремова [421].
О Схема 8.103
По сравнению с сульфоксидами фрагментация диалкилсульфонов протекает
по большему числу направлений. Однако эти направления достаточно
очевидны, и интерпретация спектра не представляет большой сложности. На
рисунке 8.115, ? представлен масс-спектр электронного удара бутилпропилсульфона.
8.13. Сульфоксиды, сульфоны, сульфокислоты 299
1 00
90
80
70
60
50
40
30
20
1 0
о
1,% . 43
57
1 09
1 64
f г^- . ' 1 —? 1 ' 1
90 100 110 120 130 140 150 160 170
m /z
7 б
94
30 40
50
60 70
1 00
90
60
70
60
50
40
30
20
1 О
О
1,%
_ш±
Лг
JJL
Ал.
JL
1 70
J_
*|— ?" ? '—? · ' "I 1 " 1 г*·"" г-
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170
m /?
Рис. 8.115. Масс-спектры ЭУ бутилпропилсульфона (а) и фенилэтилсульфона (б)
О
c3h7-s+
а
m/z 107
.-С4Н9
ОН
СзН7— St
О
m/z108
[м-он]+
m/z 147
|-ОН-
+·
О
ОН
m/z 109
С3Н7—S-C4H9
й
m/z164
-С3Н7
О
C4H9-S+
i
m/z 121
m/z 57
ОН
^-* C4H9-St
о
m/z 122
OH
C4H9-S +
OH
m/zl23
Схема 8.104
Схема 8.104 отражает основные направления фрагментации этого соединения
и объясняет образование ионов, характеризующихся наиболее интенсивными
пиками в спектре. Наряду с отщеплением гидроксильного радикала, М+* диал-
килсульфонов претерпевают разрыв С—S-связи с элиминированием алкильных
групп.
300 Глава 8. Основные направления фрагментации органических соединений
Наряду с перегруппировочным процессом с выбросом молекулы олефина
(см. сульфоксиды) эффективно протекает перегруппировка с миграцией двух
атомов водорода на сульфогруппу или простое отщепление алкильного
радикала (схема 8Л04). Прогнозировать, какой процесс окажется предпочтительнее,
достаточно сложно. Например, при распаде бутилпропилсульфона (рис. 8.115, а)
бутильный заместитель отщепляется в основном с миграцией двух атомов
водорода (m/z 109), а пропильный примерно с равной вероятностью выбрасывает
частицы С3Н5, СзНб и С3Н7 (m/z 121-123). Правило выброса максимального
алкила, как всегда, выполняется. Очень высокую интенсивность имеют пики
углеводородных ионов, доля тока которых в полном ионном токе еще более
возрастает с увеличением длины алифатических цепей.
Как и в случае арилсульфоксидов, для арилсульфонов протекают процессы
изомеризации молекулярного иона до его распада (схема 8.105). Наиболее
характерны миграции радикалов от атома серы к атому кислорода. В
спектрах могут наблюдаться интенсивные пики ионов АЮ+, АгОН+" и ArSO+.
Миграция алкильной группы в алкиларилсульфонах менее благоприятна, чем
арильной, однако и это направление распада реализуется (m/z 125 в спектре
фенилэтилсульфона на рис. 8.115,6). Кстати, последующий распад иона PhSO+
может протекать с выбросом молекулы СО (m/z 97 на рис. 8.115,6). Этот
процесс подразумевает существенную перестройку иона до распада. Пик иона
[ArSO—СО]+ может быть весьма интенсивен в спектрах диарилсульфонов,
когда пик родительского иона ArSO* оказывается максимальным в спектре.
Ион ArSO+ может элиминировать и молекулу CS. Однако этот процесс менее
благоприятен по сравнению с выбросом СО.
m/z 66
m/z 143
Ph-S-OCzHs ^ ph?0 -CQ > C5H5S+
О m/z 125 m/z 97
Схема 8.105
8.13. Сульфоксиды, сульфоны, сульфо1сислоты 301
юо
ЭО
80
70
во
SO
АО
зо
20
ю
1,°Л>
I I. lili
АО SO ?? 70 80 90 ЮО НО 120 130 ??? 150 1вО 170 1вО
m/z
Рис. 8.116. Масс-спектр электронного удара л-толилсульфоновой кислоты
Алкильные радикалы в алкиларилсульфонах могут отщепляться без миграции
или с миграцией одного или двух атомов водорода (m/z 141-143 на рис. 8.1 15,6).
Потеря молекулярным ионом диарилсульфонов молекулы SO2 является
заметным процессом и может сопровождаться элиминированием одного или двух
атомов водорода.
На рисунке 8.116 представлен масс-спектр электронного удара и-толилсуль-
фоновой кислоты. Все процессы фрагментации очевидны. Наличие
интенсивного пика иона с m/z 108 свидетельствует об изомеризации молекулярного
иона с миграцией толильной группы к атому кислорода (см. схему 8.105 для
арилсульфонов).
Задача 8.97. Определите состав фрагментных ионов и составьте схему
фрагментации дибутилсульфоксида, спектр которого представлен на рис. 8.117.
00 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
1 0 -
0 -I
I
I
I
ill
,,%
?
+
41
57
llilil,.|i.l.l
63
W
?
II—,—
?
J*-, 4.
9
¦?—
1 06
,—Li
"-•-? 1
145
?
—? ¦ ?- ,
162
? *' '
30 40 50 60 70 80 90 100 110 120 130 140 150 160
m /?
Рис. 8.117. Масс-спектр электронного удара дибутилсульфоксида
Задача 8.98. Предложите состав фрагментных ионов и составьте
схему фрагментации дифенилсульфоксида, спектр которого представлен на
рис. 8.114,6.
302 Глава 8. Основные направления фрагментации органических соединений
Задача 8.99. Идентифицируйте соединение, спектр ЭУ которого
представлен на рис. 8.118.
1 00
90
80
70
60
50
40
30
20
1 0
0
Ь%
78
4 1
57
63
1 17
1 34
I.
30
40
50
80
m /z
m/z
26
27
28
29
30
32
34
35
?,% ?
9,01
43,7
36,9
100
2,12
3,13
2,31
2,27
m/z
38
39
40
41
42
43
44
1 45
/,% 1
1,89
13,3
3,04
67,7
3,56
4,71
4,59
5,19
m/z
46
47
48
49
50
51
53
1 55
/,% |
2,94
4,89
2,37
2,44
1U
2,04
2,71
15,9
m/z
56
57
58
58
61
62
63
1 64
/,% 1
15,4
42,1
4,13
4,29
3,93
1,89
40,2
5,15
m/z
65
75
76
77
78
79
80
89
/,% 1
2,12
5,34
3,38
6,01
85,0
3,77
3,71
5,28
m/z
90
91
92
105
106
117
118
1 119
/,% |
2,01
1,11
2,41
1,25
5,79
10,0
1,94
1,00
m/z
134
135
136
/,%
8,34
0,62
0,40
Рис. 8.118
Задача 8.100. Предложите состав фрагментных ионов и составьте схему
фрагментации л-толилсульфоновой кислоты, спектр ЭУ которой представлен на
рис. 8.116.
Задача 8.101. Составьте предполагаемую схему фрагментации фенилнаф-
тилсульфона.
Задача 8.102. Идентифицируйте соединение, спектр ЭУ которого
представлен на рис. 8.119.
100
90
80
70
60
50
40
30
20
10
О
~|
ч
J
1,%
151
L
L h ,J
78
91
142
120
—4-i—\
, r
184
I
100 120
m/z
140 160 180
Рис. 8.119
m/z
! 39
40
41
42
43
44
50
51
52
65
66
i,% 1
19,4
2,13
54,8
4,21
98,1
5,10
11,6
45,7
4,33
4,33
3,18
m/z
75
77
78
79
91
92
94
105
117
118
|119
/, % 1
2,13
74,5
100
10,4
25,0
6,29
18,0
2,79
3,18
8,44
13,9
m/z
120
121
125
141
185
142
143
144
184
185
[_186
/,%
20,7
2,13
6,49
6,49
2,79
49,9
12,4
3,18
24,9
2,79
1,29
8.14. Элементоорганические соединения 303
Задача 8.103· Идентифицируйте соединение, спектр ЭУ которого
представлен на рис. 8.120.
ОО ?
90 -
во -
70 -
во -
SO -
4?'-
зо -
20 -
Ю -
?/*"?
I
i_J
лз
40 вО
108
I 15
I
, L
вО ЮО 120 140
m/z
Рис. 8.120
m/z
27
28
39
41
42
43
44
107
/,% 1
11,7
3,53
3,92
19,6
7,06
100
3,33
2,35 1
m/z
108
109
ПО
134
135
150
151
1 152
/,%
46,6
29,8
2,35
1,18
3,53
20,0
1,55
1,01
8.14. Элементоорганические соединения
Описание масс-спектрометрического поведения соединений этого класса
значительно реже встречается в литературе. В настоящем пособии будет дана
только общая характеристика фрагментации нескольких представителей этого
огромного класса соединений. До настоящего времени отсутствуют монографии,
посвященные этой теме, хотя, безусловно, пришло время написания такой
работы. Обобщение многочисленных результатов по масс-спектрометрии самых
разнообразных элементоорганических соединений было бы полезно по
нескольким причинам. Во-первых, поскольку элементоорганические соединения все
шире используются в современном обществе, знание закономерностей их
трансформации в газовой фазе в условиях масс-спектрометрических экспериментов
полезно с точек зрения и синтеза, и анализа, и прогнозирования химических
свойств, и применения, и метаболизма. Во-вторых, такая работа позволила бы
значительно расширить набор известных типов масс-спектрометрических
реакций и выявить новые законы фрагментации. В-третьих, необходимо отметить,
что многие соединения этого класса являются опасными экотоксикантами и
требуют постоянного контроля в объектах окружающей среды. Именно масс-
спектрометрия позволяет проводить надежное качественное и количественное
определение этих соединений в существующих природных формах на уровне
ультрамикроколичеств.
Качественное определение присутствия нестандартного для органических
соединений элемента, как правило, не вызывает особых сложностей в связи
с уникальной изотопной картиной и специфической массой такого элемента.
Например, кадмий имеет восемь изотопов, свинец —четыре, ртуть— семь (см.
приложение 1). Очень часто в спектре присутствует атомный пик элемента, что
еще более облегчает идентификацию. В частности, на рис. 8.121 представлен
304 Глава 8. Основные направления фрагментации органических соединений
100 ?
90 -|
80 -|
70 А
60 А
50 А
40 А
зо А
20 А
ю А
о -I
1,%
15
217
JJlL
202
232
20 40 60 80 100 120 140 160 180 200 220 240
m /?
Рис. 8.121. Масс-спектр электронного удара диметилртути
масс-спектр электронного удара диметилртути. Наряду с характерным кластером
изотопных пиков молекулярного иона и иона [М — СНз]+ высокую
интенсивность имеют пики всех изотопов ртути с массами 196-204. Отсутствие пиков
в области от 30 до 200 а. е. м. также свидетельствует о присутствии в составе
соединения тяжелого элемента.
Несколько более сложная картина наблюдается в спектре тетраэтилсвинца
(рис. 8.122), еще одного важного и опасного экотоксиканта, до сих пор иногда
применяющегося в качестве добавки к топливу (этилированный бензин). В
данном случае кластер молекулярного иона едва заметен (пики ионов с m/z 322-324),
а основной процесс фрагментации связан с последовательным отщеплением
этильных радикалов. Кстати, такая последовательность распада противоречит
правилу четноэлектронных ионов, т. е. приведенный пример является
исключением из этого правила (разд. 3.1.4).
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
* г ¦, , , ,
208
1
? 1 , 1 rJ 1 "
237
266
ц ,-Jll ,
295
J
|ц
324
„1
20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320
m/z
Рис. 8.122. Масс-спектр электронного удара тетраэтилсвинца
8.14. Элементоорганические соединения 305
100 ? ?,0/?
90 А
80 А
70 -J
60
50
40
30
20
10
0
27
40
80
121
Л
155
J
.183
??..
213
120
m /z
160
200
242
l.jlt.l ,
240
Рис. 8.123. Масс-спектр электронного удара триэтилоловохлорида
Правило четноэлектронных ионов плохо выполняется для элементоорганиче-
ских соединений, по-видимому, из-за участия d- и/-орбиталей элемента. Наряду
с элиминированием этильных радикалов протекает отщепление молекулы
этилена. Это особенно отчетливо видно по интенсивным пикам ионов с m/z 267 (ион
[(С2Н5J08РЬН]+) и m/z 209 (ион [208РЪН+]). Эти реакции хорошо согласуются
с правилами, изложенными в гл. 3, и аналогичны соответствующим реакциям
распада аминов, сульфидов, фосфинов и т. д. Эти перегруппировочные процессы
искажают характерную для свинца изотопную картину (см. приложение 1).
Следует обратить также внимание на пики ионов с m/z 57, 43, 41. Эти
фрагменты образуются в результате первоначального перегруппировочного процесса
с образованием новой связи С—С между двумя этильными радикалами, исходно
связанными через атом свинца. Аналогичные процессы протекают при
фрагментации полиалкил(арил)элементоорганических соединений кремния, германия,
олова и т. д. [15, 422, 423]. Если элемент (Е1) входит в состав цикла, может
протекать его элиминирование, сопровождающееся уменьшением размера цикла
(схема 8.106). Особенно характерен этот процесс для дигалогенпроизводных [422].
I ?
?' Ч
¦рйЛ1"
-?1??
Схема 8.106
(CH2nt
Еще более многолинейчатыми становятся спектры при наличии в молекуле
нескольких атомов полиизотопных элементов. На рисунке 8.123 представлен
масс-спектр электронного удара триэтилоловохлорида.
Два изотопа хлора и десять изотопов олова приводят к очень сложным
кластерам, хотя по сути весь процесс фрагментации связан лишь с отщеплениями
306 Глава 8. Основные направления фрагментации органических соединений
100 -?
90 Ч
80 Ч
70 А
во А
50 Ч
40 Ч
зо ч
20 Ч
ю ч
1,%
52
130
158
214
186
40
60
80
100
120 140
m /?
160
180
200
220
Рис. 8.124. Масс-спектр электронного удара бензолтрикарбонила хрома
этильных радикалов и атома хлора в любой последовательности. Группа пиков
в области m/z 120 обусловлена кластерами двухзарядного молекулярного иона и
иона Sn+.
Наличие атома галогена, гидрокси- или аминогруппы в органическом
заместителе, связанном с элементом, инициирует перегруппировочный процесс
элиминирования молекулы олефина из М+# или первичного иона [М—R]+ с
миграцией гетероатома или гетероатомной группировки к элементу (схема 8.107).
Эта перегруппировка аналогична приведенной для вторичного распада спиртов
(схема 8.18) или аминов (схема 8.44). Вероятность протекания процесса
определяется природой элемента, мигрирующей группы и остальных заместителей,
а также пространственным расположением элемента и мигрирующей группы
[19]. Как правило, лучше всего миграция протекает из ^-положения заместителя
(четырехчленное переходное состояние), однако атом фтора легко мигрирует из
любого положения [424].
«V,
-CH2CHR
X
E1=Z
Схема 8.107
Распад под ЭУ ?-комплексов с простыми лигандами протекает с
последовательным элиминированием лигандов. На рисунке 8.124 представлен масс-
спектр электронного удара бензолтрикарбонила хрома. Отчетливо
прослеживается серия пиков, отстоящих друг от друга на 28 а. е. м., что обусловлено
последовательным отщеплением молекул СО. Ион 130 теряет молекулу бензола,
образуя ион Cr+ (m/z 52).
Аналогично распадаются ?-комплексы с более сложными органическими
лигандами. Например, в спектре ферроцена (рис. 8.125) основные пики
обусловлены молекулярным ионом (m/z 186), а также ионами [М — CsHs]* (m/z 121) и
100
90
во
70
60
50
40
30
20
10
О
8.14. Элементоорганические соединения 307
1,%
121
56
_Ljl
186
60
80
100
120
m /z
140
160
180
200
Рис. 8.125. Масс-спектр электронного удара ферроцена
[М —2CsH5]+ (m/z 56);. последний является не чем иным, как атомарным ионом
железа.
Задача 8.104. Идентифицируйте соединение по масс-спектру ЭУ,
представленному на рис. 8.126.
1 0 0
9 О
80
7 О
6 О
50
4 О
30
2 О
1 О
О
? 1,%
4 5
л.
4 О
7 3
1 3 1
60
8 0 100
Ш /?
120 140
Рис. 8.126
m/z
29
! 31
43
44
45
46
58
59
69
72
/, % ?
1,83
1,28
6,82
1,52
14,6
0,95
1,54
3,45
1,68
2,311
m/z
73
74
75
115
131
132
133
146
147
1 148
/,%
100
8,37
3,36
1,40
24,9
3,91
1,71
8,59
1,44
0,60
Задача 8.105. Идентифицируйте неорганическое соединение по
масс-спектру ЭУ, представленному на рис. 8.127.
100
90
80
70
60
50
40
30
20
10
О
I
,%
127
208
J ,
335
I
.,—Д-.—
462
1
, , .1 ,
120
160
200
240
280 320
m /?
Рис. 8.127
360
400
440
480
308 Глава 8. Основные направления фрагментации органических соединений
Задача 8.106* Идентифицируйте элементоорганическое соединение по масс-
спектру ЭУ, представленному на рис. 8 Л 28.
100
90
80
70
60
50
40
30
20
10
о
1,%
39
58
97
123
Ш
Л
188
40
60
80
100
120
m /z
140
160
180
200
m/z
38
39
58
: 60
I 61
62
63
/,%
1,18
10,4
34,5
13,1
0,63
1,86
2,06
m/z
64
65
84
86
94
95
1 96
/,% 1
0,61
2,07
3,28
1,24
6,74
3,81
6,96
m/z
97
98
99
100
101
102
| 108
/>% 1
20,3
3,62
7,67
1,06
1,15
1,22
1,37
m/z
109
115
122
123
124
125
1 126
/,% 1
1,25
1,35
1,97
86,5
4,76
33,7
3,43]
m/z
127
128
129
130
135
136
1 160
/,% 1
5,38
7,63
10,3
1,54
1,50
2,14
1,19
m/z
162
173
175
184
185
186
1 187
/,% 1
1,76
4,82
1,93
1,06
0,83
8,92
2,31
m/z
188
189
190
191
192
194
/,%
100
11,1
39,1
6,14
5,74
2,34
Рис. 8.128
Задача 8.107. На рисунке 8.129 представлен масс-спектр электронного удара
запрещенного в настоящее время металлоорганического пестицида. Установите
его структуру.
100
90
80
70 -|
60 -|
50 Ч
40 -|
30
20 ?
10
о
1,%
29
202
237
? t 'ilibillti
266
20 40 60 80 100 120
140 160
m/z
180 200 220 240 260
Рис. 8.129
8.14. Элементоорганические соединения 309
Задача 8.108. Идентифицируйте элементоорганическое соединение по масс-
спектру ЭУ, представленному на рис. 8.130.
1 00 ?
90
80
70
60
50
40
30
20
1 О
О
1,%
52
7.8
i_ui
1 30
208
40 80 120 1 60 200
m /z
Рис. 8.130
Задача 8.109. Идентифицируйте элементоорганическое соединение по масс-
спектру ЭУ, представленному на рис. 8.131.
m/z
28
39
40
50
51
52
53
54
74
75
76
77
/,% 1
9,04
7,05
1,66
8,15
14,5
100
10,6
2,49
1,28
1,05
3,17
13,4 |
m/z
78
79
104
128
129
130
131
132
206
208
209
I 210
/,%
65,8
4,44
2,20
1,81
1,96!
33,6
6,05
1,01
1,20
25,3
6,22
0,91
100 ?
90 -
80 -
TO -
во -
SO -
40 -
зо -
20 -
1 О -
О -
1,%
--J,
55
4—Ч—
120
J-i
148
, I , ¦
204
п Д.
40 вО 80 100 120 140 160 180 200
m/z
Рис. 8.131
m/z
28
39
55
65
120
121
148
149
176
204
205
/, %
2,18
5,79
100
1,13
56,0
з,п
12,5
0,84
2,24
10,1
0,96
Задача 8.110. Идентифицируйте металлоорганическое соединение по масс-
спектру ЭУ, представленному на рис. 8.132.
1 00
90
80
70
60
50
40
30
20
1 О
О
1,%
223
208
238
il
253
268
200
21 О
220
230
240
m /?
250
260
270
280
Рис. 8.132
310 Глава 8. Основные направления фрагментации органических соединений
8.15· Аминокислоты, пептиды, белки
С тех пор как в 1958 г. Клаус Биман [425] впервые получил спектры
аминокислот, масс-спектрометрия шагнула далеко вперед.
Основной задачей Бимана было ввести полярные термолабильные молекулы
в ионный источник и провести их ионизацию. В последующие двадцать лет с
появлением химической ионизации, полевой десорбции и полевой ионизации
удалось добиться определенных успехов в анализе простых пептидов, однако
эксперименты были трудоемки, характеризовались плохой воспроизводимостью
и требовали проведения химической дериватизации исходных соединений для
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
30
А
1
I
-+ш
3
.
h
1_Ш *.
7
—1
0
1
-i_J . , ?
88
к 1
а
1 ' 1
30
100 ? 1,%
90 А
80 А
70 А
60 А
50 А
40 А
зо А
20 А
ю А
40
50
60
74
70 80
Ш /?
91
90
100
110
120
120
20
40
60
80
100
120
140
160
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
?,%
28
LJ
L
¦ ¦Jlli l II....I
56
и
61
75
?
J uJ
JU—
m /z
83
, LiilL...
104
ibl
116
¦ I., ¦
149
L· ,
II
в
-—n
20
40
60
80
100
120
140
160
m /z
Рис. 8.133. Масс-спектры ЭУ аминокислот аспарагина (?), фенилаланина (б) и метионина в)
8.15. Аминокислоты, пептиды, белки 311
ЮО ?
90 -
80 -
70 -
во -
50 -
40 -
ЗО -
20 -
ю -
О -
1,°Л>
,1
? ??
ЗО
|
? 1 It
. ? ? .??,???
44
, I ?—' ,
86
Ч , , г
а
1 1 1
Ю 20 ЗО 40 50 60 70 80 90 ЮО 110 120 130 140 150
m/z
ЮО -, l,%
90
80
70
60
50 -?
40 ?
ЗО -|
20
10 _L
74
—?—'—"?—
30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180
m/z
Рис. 8.134. Масс-спектры электронного удара метилового эфира лейцина (а) и N-ацетилизо-
лейцина (б)
повышения их летучести. Достижения масс-спектрометрии в этой области с 1958
до 1978 г. изложены в исчерпывающей монографии Б. В. Розынова [20].
Хотя ?-аминокислоты, являющиеся «кирпичиками» белков, можно
изобразить общей формулой RNH—CHR—СООН, разнообразие структур боковых
групп (R) приводит к высокой индивидуальности их спектров (рис. 8.133).
Среди общих направлений можно выделить аминный распад (элиминирование
боковой группы R и карбоксильной группы). Сохранение заряда на R (ионы R+)
характерно для ароматических аминокислот (тирозин, триптофан, фенилаланин).
В частности, в спектре последнего (рис. 8.133,6 —пик иона PhCH^ с m/z 91) —
второй по интенсивности в спектре. При достаточной длине цепи R возможна
перегруппировка Мак-Лафферти (треонин). Для увеличения летучести
соединений использовали их дериватизацию. Наиболее распространенными вариантами
являются алкилирование и ацилирование. На рисунке 8.134 представлены масс-
спектры электронного удара N-ацетилизолейцина и метилового эфира лейцина.
Спектры химической ионизации аминокислот столь же индивидуальны. Пики
протонированных молекулярных ионов аминокислот имеют достаточно высокую
интенсивность [426], особенно при использовании в качестве газа-реагента
изобутана [427]. Фрагментация, как обычно в условиях химической ионизации,
незначительна и существенно зависит от структуры аминокислоты, причем
отмечается, что фрагментация резко возрастает при увеличении температуры
источника [427].
312 Глава 8. Основные направления фрагментации органических соединений
В спектрах химической ионизации отрицательных ионов при использовании
в качестве иона-реагента ОН~ [428] доминируют пики депротонированных
молекулярных ионов [М —Н]~. При использовании С1~ помимо этих ионов
образуются также ионы [М+С1]~~, однако их пики исчезают из спектров
аминокислот, содержащих дополнительные полярные группы [429].
Возможности электронного удара и химической ионизации резко
уменьшаются при переходе к петидам в связи с еще меньшей летучестью и повышенной
термолабильностью этих соединений. Для анализа простейших пептидов этими
методами используются различные виды дериватизации, включая алкилирова-
ние, ацилирование, триметилсилилирование и т. д. [20]. Тем не менее по этим
спектрам уже можно было устанавливать последовательность аминокислотных
звеньев [20, 430, 431].
Однако реальный прорыв в масс-спектрометрии пептидов связан с
появлением метода бомбардировки быстрыми атомами (разд. 5.10). Благодаря ему
стало возможно достаточно легко устанавливать молекулярные массы и
последовательность аминокислотных звеньев олигопептидов с массами в несколько
тысяч дальтон. Еще больших успехов удалось добиться с помощью методов
электрораспыления (разд. 5.12) и матричной лазерной десорбционной ионизации
(разд. 5.14), которые позволили анализировать сложнейшие белковые молекулы
и сделали масс-спектрометрию ключевым методом новой науки — протеомики.
Именно эти три метода (ББА, МЛДИ и электроспрей) используются в настоящее
время для анализа пептидов и белков.
Рассмотрим плюсы и минусы этих трех методов применительно к анализу
пептидов и белков. Бомбардировка быстрыми атомами исторически была первым
эффективным методом для установления последовательности аминокислотных
звеньев в недериватизованных пептидах [432, 433]. ББА позволяет устанавливать
массы молекулярного и фрагментных ионов с большой точностью. Кроме того,
время анализа невелико. Однако поскольку диапазон регистрируемых масс
обычно не превышает 8000 Да, а для анализа необходимо иметь не менее
100 пикомолей чистого образца, ББА может применяться только для анализа
олигопептидов. л4-Нитробензиловый спирт оказался наилучшей матрицей для этой
цели. В качестве растворителя образца используют смесь воды и ацетонитрила с
возможными добавками метанола, уксусной и трифторуксусной кислот. ББА не
является мягким методом. Поэтому обычный спектр характеризуется значимой
фрагментацией молекулярного иона. Тем не менее для целей определения
последовательности аминокислотных звеньев в пептиде лучше использовать
тандемную масс-спектрометрию (гл. 7).
МЛДИ обладает высокой чувствительностью (уровень аттомолей, 10~18 М),
не требует сложной пробоподготовки и позволяет работать с гетерогенными
образцами. Метод позволяет установить молекулярную массу пептида или
белка в сотни тысяч дальтон с точностью 0,5-0,01%. Для него характерны
протонированные молекулярные ионы МН+ и аддукты с катионами щелочных
8.15. Аминокислоты, пептиды, белки 313
металлов [M+Na]+ и [М+К]+. В качестве матриц наиболее часто
используют 2,5-дигидроксибензойную и а-циано-4-гидроксикоричную кислоты. Первая
особенно успешно применяется для легких соединений с молекулярной массой
до 1000 Да, поскольку характеризуется незначительными фоновыми пиками в
области низких масс. Отмывание пробы, уже нанесенной на матрицу, от
примесей солей и ПАВ приводит к улучшению качества спектра, но приемлемого
результата можно достигнуть и с неочищенной пробой. К недостаткам метода
можно отнести тот факт, что некоторые компоненты неочищенных смесей могут
давать очень интенсивные пики в спектрах. Эти пики могут маскировать наличие
действительно важных для исследователя компонентов, т. е. МЛДИ не является
методом количественного анализа.
Электрораспыление в режиме ЖХ-МС (разд. 1.5 и 5.12) благодаря высокой
точности, возможности стыковки с квадрупольными анализаторами и
образованию многозарядных ионов позволяет устанавливать молекулярные массы
тяжелых пептидов и белков (в миллионы дальтон [7]), работать со смесями
соединений, надежно устанавливать последовательность аминокислотных звеньев
в условиях МС/МС. Практические наблюдения показывают, что для
электроспрея характерно присоединение одного протона на участок цепи аминокислот
массой в 1000 Да. Таким образом, белок с молекулярной массой 100 кДа можно
зарегистрировать на шкале масс в области m/z = 1000. К недостаткам метода
можно отнести требование высокой гомогенности пробы и отсутствия значимых
уровней солей (менее 1 ммоль).
Одним из наиболее популярных применений масс-спектрометрии для
анализа пептидов является определение последовательности аминокислотных
звеньев (первичная структура белка). Этот подход стал рутинным для пептидов
с молекулярной массой до 2500 Да. Нельзя сказать, что масс-спектрометрия
полностью вытеснила классические биохимические методы типа эдмановского
разложения. Однако возможности масс-спектрометрического подхода шире. В
частности, чувствительность эдмановского разложения невысока, пептиды перед
секвенированием должны быть хорошо очищены от примесей, а реагент Эдмана
не взаимодействует с модифицированными по терминальной аминогруппе
пептидами. Кроме того, поскольку эдмановское расщепление основано на
последовательном разрыве амидных связей с N-конца пептида с хроматографическим
определением образующихся аминокислот, любые посттрансляционные
модификации исходного пептида приведут к образованию нестандартных молекул.
Для их идентификации необходимо иметь богатейшую библиотеку стандартов
всех возможных модификаций аминокислот. Вопрос о месте присоединения и
структуре модифицирующей группы, напротив, легко может быть решен масс-
спектрометрически. Наилучших результатов можно добиться при использовании
комплекса методов. Например, эдмановское или энзиматическое расщепление
белка или тяжелого пептида на более мелкие фрагменты с последующим их
314 Глава 8. Основные направления фрагментации органических соединений
масс-спектрометрическим анализом, несомненно, будет наиболее эффективным
подходом [434].
Молекулярная масса белка является его ключевой физико-химической
характеристикой. Она отражает его аминокислотный состав и наличие
модифицирующих заместителей. Измерение массы позволяет выявить также
чистоту и гомогенность образца. Определение молекулярной массы пептидов не
представляет сложности. Для «легких» представителей этого класса
(молекулярная масса 1-8 кДа) можно использовать все три указанные выше метода
(ББА, МЛДИ, электрораспыление). Для полипептидов и белков с большими
молекулярными массами бомбардировка быстрыми атомами неприменима, но
два другие метода отлично справляются с задачей. МЛДИ с времяпролетным
анализатором позволяет получить непосредственно протонированный
молекулярный ион соединения с массой в сотни килодальтон. Максимальная точность
метода 0,01% может быть достигнута при правильном выборе внутреннего
стандарта. Спектр, полученный методом электрораспыления, характеризуется
набором многозарядных протонированных молекулярных ионов. В разд. 5.12
подробно описан метод расчета молекулярной массы соединения по такому
спектру. Благодаря возможности вычисления молекулярной массы, пользуясь
любой парой соседних пиков, можно провести очень точное измерение, усреднив
полученные значения.
В настоящее время, когда определены последовательности аминокислотных
звеньев миллионов различных пептидов и белков, полностью или частично
расшифрованы геномы самых разнообразных организмов, во многих случаях
определение молекулярной массы полипептида или белка позволяет
установить его структуру. Этот процесс достаточно эффективно осуществляется при
использовании компьютерных баз данных (например, SwissProt). Если белок с
полученной массой отсутствует в базах данных и существует необходимость
установления его структуры, оптимальным вариантом является энзиматическое
расщепление молекулы с последующим масс-спектрометрическим анализом его
фрагментов. Часть этих фрагментов могла быть изучена ранее, а
соответствующая информация присутствовать в компьютерных базах данных. С остальными
фрагментами надо вновь проводить процедуру биохимического расщепления
с установлением молекулярных масс и строения фрагментов уже второго
поколения. Когда молекулярные массы продуктов расщепления будут составлять
1-3 кДа, можно будет использовать процедуру масс-спектрометрического
определения последовательности аминокислотных звеньев.
8.15.1. Установление последовательности аминокислотных звеньев
в пептидах
Пептиды являются сложными молекулами, состоящими из тысяч атомов.
Поэтому количество возможных путей фрагментации их молекулярных ионов практи-
8.15. Аминокислоты, пептиды, белки 315
,Л У\ ? ^3 УЪ s7* У*ЪУЪ ~'?2 х*К<У1 s*\
' ,<;TT ,--' г'' г^2н Г Г Г+2Н Г' Г* Г+2Н Г*
о| ! С 1 О I I R j ol |? О
II ? II : ? II : ? II
CtNHt-CHrCtNH4-(^rC-tNHH-CH--C~OH
? +2?? ? j +2н| I I +2н!
af bf' c\''' ai' Ы'' c<i a$ hi cf' a4 Ъ% с*
Рис. 8.135. Основные направления фрагментации молекулярного иона пептида GSCRY
чески не ограничено и определяется конкретной структурой каждой
индивидуальной молекулы. Однако основные направления распада оказываются общими
для всех соединений этого класса. Они связаны с разрывами пептидных связей
вдоль скелета молекулы с образованием характеристических серий ионов [433,
435]. Безусловно, качественные масс-спектры требуют применения тандемной
масс-спектрометрии (гл. 7). Большинство белков состоит из 20 аминокислот,
представленных в таблице в приложении 2. В этой таблице наряду с
химической формулой представлены их трехбуквенный и однобуквенный шифры и
массы их остатков (фрагментов пептида). Поскольку на одном конце пептида
находится аминогруппа, а на другом — карбоксильная группа, масса пептида
равна сумме масс остатков аминокислот плюс 18 Да (вода). Поскольку лейцин
и изолейцин имеют одинаковые массы, для их идентификации приходится
использовать дополнительные пики (не связанные с расщеплением пептидной
связи). Для распознавания изобарных лизина и глутамина необходимо проводить
ацетилирование исходного пептида (лизин ацилируется, а глутамин нет), а затем
повторять масс-спектрометрический анализ. Эти моменты являются недостатком
масс-спектрического подхода по сравнению с биохимическими анализами.
Для установления последовательности аминокислотных звеньев в пептидах
используют метод тандемной масс-спектрометрии. Анализу, как правило,
подвергается однозарядный протонированный молекулярный ион пептида.
Благодаря предпочтительным разрывам конкретных связей молекулярного иона в
спектре доминируют пики ионов, образующихся в результате элиминирования одной,
двух, трех и т. д. аминокислот. Величины m/z фрагментных ионов указывают на
исходную последовательность аминокислот в пептиде.
Рассмотрим протонированный молекулярный ион пентапептида в общем
виде (рис. 8.135). Первая классификация общих фрагментов, образующихся при
распаде пептидов, была предложена Ропшторфом в 1984 г. [435]. Несколько
позже К. Биманом была предложена несколько отличная номенклатура [436]. В
зависимости от того, какая связь рвется и на какой части молекулы сохраняется
заряд (на N- или С-конце), выделяют несколько серий ионов. Фрагменты, в
которых заряд сохраняется на С-концевой части исходной молекулы, изоб-
G
H2N-CH
? ??.?
??
II :
•C-TNH-
+2H!
s
?
-CH-t
316 Глава 8. Основные направления фрагментации органических соединений
+ G + GO GOS
H2N=CHG H2N-CH-C=0 H2N-GH-C~NH3 H2N-OT-C-NH-CH-C=0
a\ b\ cx b2
+ + + + R О
YCH-COOH H3N-YCH-OOOH 0=C-HN-YCH-COOH H3N—iH_C-HN-YCH-OOOH
*i У\ х\ Уг
Рис. 8.136. Структуры некоторых фрагментов пептида GSCRY
ражаются символами ?, ? и Z, а фрагменты, в которых заряд сохраняется
на N-концевой части исходной молекулы, — символами А, В и С (рис. 8.135).
Различия в номенклатурах Ропшторфа и Бимана заключаются лишь в том, что во
втором случае ионы обозначаются строчными, а не заглавными буквами, а ионы
серий у и с образуются в результате не простого разрыва С—О-связи, а разрыва с
миграцией двух атомов водорода к заряженному фрагменту, что, кстати,
соответствует действительности. На рисунке 8.136 представлены структуры нескольких
фрагментных ионов серий а, Ь, с, х, у, ?, образующихся при распаде в результате
активации соударением МН" пентапептида (рис. 8.135).
Наиболее характеристичными при использовании низкоэнергетических
столкновений оказываются ионы серий у и Ь. Они образуются в результате
разрыва амидной связи, а их пики, как правило, доминируют в спектре. Например,
в спектре пентапептида GSCRY должны наблюдаться пики ионов с m/z 528, 441,
404, 338, 248, 182, 145, 58.
Реальный масс-спектр оказывается куда более сложным. Первичной задачей
является поиск двух ионов, масса которых отличается от массы протонированно-
го молекулярного иона на массу остатка какой-либо кислоты и на сумму масс
аминокислотного остатка и молекулы воды. Первый ион открывает у-серию,
а второй — fe-серию. Последующие пики ионов обеих серий отстоят друг от
друга на массы аминокислотных остатков (табл. 2 приложения). Практически
никогда не удается дойти до конца серий у и Ъ. В области низких масс
интенсивности соответствующих пиков очень низкие. Иногда пики отсутствуют
вовсе. Поэтому важно приступать к расшифровке последовательности с двух
сторон. Если удалось дойти до места, где серий у и Ъ начинают перекрываться,
задачу можно считать решенной. Большое количество пиков других ионов делает
масс-спектр многолинейчатым. Зачастую установить пики ионов серий у и Ъ не
так просто, поскольку их интенсивности существенно ниже, чем
интенсивности пиков ионов других типов. На интенсивности пиков характеристических
ионов влияют несколько факторов: различия в легкости разрыва пептидной
связи между разными аминокислотами, дополнительные выгодные направления
фрагментации конкретных аминокислот и эффект присутствия пролина.
Легкость разрыва пептидной связи параллельна легкости протонирования
данного атома азота. Чем легче протонируется конкретная пептидная связь,
8.15. Аминокислоты, пептиды, белки 317
сн
( СН2 О R О
4.6l-C-(NH-CH-C)^-0H +H+
(!?2 О R О
+ *М ? II I II
h3n -?он-сн>и-сн-с)-он
-с3н7-
>>Л + 1
—С4Н10
R
I
о
II
R
I
сн
Н + H-(NH-CH-Q— NH-CH
ап+1
?)
-R
СН2 О R О
II II I II +
CH-C-(NH~CH-C)—ОН + Н+
О R О
+ II I II
H2N =СН—C-(NH-CH-C )-ОН
R О СН2
I II II
? +H-(NH-CH-Q?r NH-CH
dn
Схема 8.108
тем более интенсивными будут пики, обусловленные ее разрывом. Кислотные
группы боковых цепей аминокислот могут стабилизировать положительный
заряд, поэтому разрывы пептидной связи рядом с глутаматным или аспар-
татным остатком нередко приводят к интенсивным пикам в спектре. Очень
часто повышенной интенсивностью обладают пики ионов, образовавшихся в
результате разрыва амидных связей в центральной части молекулы. Подробнее с
механизмами диссоциации пептидной связи можно познакомиться в работе [437]
и цитированной в ней статьях.
Если на N-конце молекулы находятся аргинин или лизин, в масс-спектре
будут доминировать пики ионов серии Ъ. Если эти аминокислоты замыкают
С-конец пептида, наиболее интенсивными будут пики ионов серии у [438].
Некоторые боковые группы аминокислот отвечают за специфические
направления фрагментации, не связанные с разрывом пептидных связей. В частности,
серии и треонин, содержащие гидроксильные группы в боковых цепях, легко
отщепляют молекулу воды. Аналогично, молекула сероводорода легко
отщепляется цистеиновым фрагментом. Интенсивность таких побочных ионов может
значительно превосходить интенсивность пиков основных характеристических
ионов.
На схеме 8.108 представлен процесс отщепления боковой группы с
образованием иона w. Аналогичным образом, с участием боковых групп образуются
318 Глава 8. Основные направления фрагментации органических соединений
НБ599
2.6·]
2 4
2.2
2.0
1.8
1.6
1.4 -I
1.2 \
1.1
R,P
,701
а2
226 1
\
ihk
МШШАъ
297 0 at
< '4 +
4513 ?224а5
jdiij.UJ i.m,lLuiJu,l.,i 1 ILIIJJiu.Lrli
100 200 300 400 500 600
800 900 1000 П00 1200 1300
Рис. 8.137. Спектр высокоэнергетической активации соударением МН+ олигопептида
RRKPQQFFGLM с m/z 1348,7
ионы серий d и ? (схема 8.108). Кстати, высокоэнергетические столкновения
приводят к спектру, в котором доминируют пики ионов серий а и d (рис. 8Л 37).
Регистрация этих ионов дает возможность подтверждать установленную по
основным ионам последовательность аминокислот, а также различать изомерные
лейцин и изолейцин.
Пролин обладает специфическими свойствами. Например, он препятствует
расщеплению пептидной связи трипсином, если связан с С-концом лизина или
аргинина. Аналогичный эффект наблюдается и в случае масс-спектрометри-
ческого распада. Пептидные связи пролина оказываются очень устойчивыми,
и соответствующие ионы серий у и Ъ могут отсутствовать в спектре. Этот
эффект пролина можно объяснить его структурой (?-пирролидинкарбоновая
кислота). Пирролидиновый цикл, включающий и ?-атом углерода, и аминогруппу
в пептидной цепи, имеет очень низкую основность. Его протонирование, а
следовательно, и соответствующий распад крайне затруднены.
Иногда для проверки или для более надежного установления сиквенса
пептиды дериватизуют и проводят повторный анализ. Например, обработка
пептида раствором НС1 в метаноле позволяет прометилировать все свободные
карбоксильные группы.
8.15. Аминокислоты, пептиды, белки 319
(GL)L(GL)LGSWSHWPAIVGHF-NH2
V V ? А I V G ? F(NH2)
? 1—г
?
~ (?)
(??*)
2069
1499
1612
1428 1446
1359 | /?
1503
/
?>,);,.]^|1^>4|кшД|<411.|д..,^гл^,1| Аь>, r.4.J,l,J„K»/i.?? ts ,·?>1?[1
2052
1711
1616
?/
1905
1768 1899
Il786
1400 1600
1600 1900
аЛ
J L
m/z
(Y+2)
(LG)
(LG)
I,%
?
4,,i,>ulliiaJJ
(В)
909 ? ввг
/ 996
ilblthjilJjJijl^.iiii.
600 900
1
m/z
(Y+2)
(PAI)
Рис. 8.138. Масс-спектр активации соударением протонированного молекулярного иона
пептида (GL)L(GL)LGSVVSHVVPAIVGHF-NH2
На рисунке 8.138 представлен реальный масс-спектр (МС/МС) МН+
пептида состава (GL)L(GL)LGSVVSHVVPAIVGHF-NH2. Гидроксильная группа
С-конца превращена в амидную. Над спектром обозначена последовательность
аминокислотных звеньев со стороны С-конца (серия Ь), а под спектром —со
стороны N-конца (серия у). Ион соответствующей серии может отсутствовать
в спектре. В этих случаях нельзя точно установить порядок пары (LG или GL)
аминокислотных звеньев. Поскольку серии у и b дополняют друг друга, иногда
можно установить точную последовательность по одной из серии. Например, в
серии у последовательность аминокислот внутри участка PAI не определяется.
Однако это легко сделать по серии Ь.
Как уже отмечалось выше, в случае тяжелых пептидов и белков процесс
определения последовательности аминокислотных звеньев проводится не с
самой молекулой, а с ее более мелкими фрагментами, которые получают путем
химического или энзиматического расщепления исходной молекулы.
320 Глава 8. Основные направления фрагментации органических соединений
Леддерное секвенирование является еще одним способом установления
последовательности аминокислотных звеньев в пептидах и белках. В этом
случае проводят контролируемое статистическое расщепление пептидных связей
химическими методами (например, эдмановское расщепление), причем каждый
последующий фрагмент отличается от предыдущего на одну аминокислоту.
Последующий МЛДИ-анализ полученной смеси (леддер секвенированного белка)
позволяет идентифицировать аминокислоты по разности масс между соседними
пиками в масс-спектре и таким образом устанавливать их последовательность
в исходной молекуле белка. Это достаточно быстрый и эффективный метод,
все активнее применяющийся в биохимических и масс-спектрометрических
лабораториях. К недостаткам этого метода можно отнести требование высокой
чистоты анализируемого пептида, а также высокий химический фон, образуемый
непрореагировавшими реагентами и побочными продуктами.
Задача 8.111. По спектру активации соударением МН+ пептида (рис. 8.139)
установите последовательность аминокислот по пикам ионов серий у и Ъ.
Принимайте во внимании только маркированные пики. Концевая гидроксильная
группа заменена на аминную.
? Г
^ ?,%
J1434 1547
1530
???^???^???
?—?—?—?—г
?
1871
1729
1658
16441
? ?
4?
г*
1772
1828
???.-.4?.
i
2479
1970
+Д
2084
2067
2166
УрпГ+гЛ
12197
2279
2350
?2325? 2453
и
+??+
1 1 (В)
(??*)
2737
2607
2566
m/z
(Y+2)
??*, ??%
'Too
258
I
171
.284
Да,
«***
412
1 I Г
653
540
ш
400 500
600
781
670^
909
670 \ 965
700 600 900 1
1008
1079
(В)
1207 1335
1303
^¦^^^^.Ufbui^MfUJ^.*^ m/z
1000 1100 1200 1Э0О 1400
Рис. 8.139. Масс-спектр активации соударением МН+олигопептида
(Y+2)
8.15. Аминокислоты, пептиды, белки 321
Задача 8.112. Определите последовательность аминокислотных звеньев в
масс-спектре активации соударением МН+ олигопептида, представленном на
рис. 8.140. Принимайте во внимании только маркированные пики. Концевая
гидроксильная группа заменена на аминную.
?
?
? г
?,%
1415
(мн+;
2659
2642
(В)
1748
1847
2358
1457
1512
1649174?
1585 Ц672>|
U*.
?
1842
?
2128
2015 2117
1970 ?
1944|
т_иЛ
Ш+J
2248
2229 / 2319
2495
2489
2376 ?
m/z
1400 1500 1600 1700 1600 1900 2000 2100 2200 2300 2400 2600 26CX
I I L_l I I I I L_l I I (Y+2)
i,%
,,,,,,, L,.?? i|H J
?|·||·|| III I I f I
00 200
1 ? ? ?
645 ( 690
543 \
302. \\ 532 . \ I 818 9"
ill ? li ? /912 98810\5 120J
? ?
1245
(В)
J L
1000 1100 . 1200 , 1300
13021358
Л^М m/z
I II (Y+2)
Рис. 8.140. Масс-спектр активации соударением МН+олигопептида
8.15.2. Пептидная карта масс
Картирование пептидов — обычная процедура анализа смеси продуктов протео-
литического расщепления белка. Наряду с масс-спектрометрическим
картированием хорошо известны хроматографические и электрофоретические
методы. Часто последовательность аминокислотных звеньев анализируемого белка
известна заранее. Использование конкретных ферментов позволяет расщепить
исходную молекулу на соответствующие фрагменты, молекулярные массы
которых можно вновь рассчитать заранее. Различие в массах между расчетом
и экспериментом означает, что данный фрагмент исходной молекулы
модифицирован. Пептидная карта масс позволяет проверить правильность сиквенса,
установить конформационные изменения в результате связывания лигандов,
выявить посттрансляционные модификации (природу заместителей и места
связывания), а также закрытые участки в структуре белка. Сравнение карт белков,
11—1113
322 Глава 8. Основные направления фрагментации органических соединений
сиквенс которых незначительно различается, дает возможность изучать мутации
и посттрансляционные модификации. Использование метода ЖХ-МС превносит
в картирование пептидов дополнительное измерение —время удерживания. Итак,
пептидная карта является «отпечатками пальцев» конкретного белка.
Работа над составлением пептидных карт немыслима без
компьютерного обеспечения. Учитывая важность правильной интерпретации результатов,
получаемых ежедневно в лабораториях всего мира, первостепенное
значение имеет доступность программного обеспечения. В данном случае
можно говорить о наиболее тесном и эффективном сотрудничестве ученых в
этой области. Программы для проведения и обработки результатов
картирования пептидов [439-445], а также конкретные результаты доступны, например
на сайтах http: //www,mann.embl-heidelberg.de и www.expasy.ch/
tools/peptident.html. Программы по анализу сиквенса белков входят в
общий пакет программного обеспечения основных приборопроизводительных
фирм. Эти программы, используя данные сиквенса белка в качестве основы,
позволяют рассчитывать молекулярные массы конкретных пептидных фрагментов
(например, после обработки данного белка трипсином). Сравнение полученных
результатов с расчетами дает возможность делать надежные выводы о составе
и структуре белка.
Фактически один из основных путей идентификации белка включает его
расщепление трипсином, масс-спектрометрический анализ смеси продуктов,
поиск по базам данных сиквенса белков всех теоретически возможных пептидов
(продуктов расщепления) для всех известных белков и, наконец, выбор
наиболее вероятного соединения, пептидная карта масс которого наиболее близка
полученной в эксперименте. Метод проверки установленного сиквенса включает
компьютерную программу (SEQUEST), реконструирующую модельный тандем-
ный масс-спектр, который далее сравнивается с реальным масс-спектром с
помощью корреляционных функций [445]. Широкое использование баз данных
и программного обеспечения привело к созданию метода [446], названного
молекулярным дроблением (shotgun). Поскольку тандемная масс-спектрометрия
дает возможность проводить анализ смесей, протеолитическому разложению
подвергается смесь белков. Полученная сложная смесь пептидов анализируется
масс-спектрометрически, а результаты автоматически обрабатываются
компьютером. Устранение процесса выделения и очистки приводит не только к резкому
сокращению времени анализа, но и исключает возможные химические
изменения структур белков во время этих процедур. Метод молекулярного дробления
особенно эффективен, если геном организма полностью расшифрован.
Работа с базами данных становится более эффективной при наличии «меток
сиквенса» [447]. Этот термин означает, что известна начальная
последовательность аминокислотных звеньев (две-три аминокислоты) с любого конца пептида
и его масса. Если такая метка позволяет подобрать хорошего кандидата в
пептидной базе данных, двойная масс-спектрометрия может быть использована
только для подтверждения выбора. Этим методом можно анализировать и мо-
8.15. Аминокислоты, пептиды, белки 323
дифицированные пептиды. Такой подход значительно сокращает время анализа
белков, поскольку расшифровка спектров тандемной масс-спектрометрии, часто
требующая длительного времени, в большинстве случаев опускается.
Если необходимо проанализировать модифицированный белок,
последовательность действий отличается незначительно. Сначала определяют
молекулярную массу исходного белка. Далее составляют пептидную карту, чтобы
обнаружить модифицированные пептиды. Затем ферментативно или химически
удаляют модифицирующие группы. Завершается процесс сравнительным масс-
спектрометрическим анализом модифицированных и нормальных пептидов.
Метод пептидной карты масс используется также для межвидовой
идентификации. В этом случае базы данных по белкам одного организма используются
для идентификации менее изученного организма [448]. В основе этого подхода
лежит правило: если организмы обладают аналогичными белками, места протео-
литического расщепления белка на олигопептиды также будут аналогичны.
8.15.3. Протеомика
Обсуждая масс-спектрометрию белков, нельзя не остановиться на протеомике.
Этой области знаний посвящена добрая половина современных масс-спектромет-
рических исследований и монографий [160, 449-458]. Последним достижениям
в этой области посвящен также один из последних номеров журнала масс-
спектрометрического общества США [459]. Протеомика занимается изучением
протеома, связанного с геном конкретного организма. Термины протеомика и
протеом были введены в мировую практику Марком Уилкинсоном в начале
90-х годов XX века. В определенной степени эти термины навеяны аналогией
с геномикой и геномом. Целью протеомики является установление составов,
структур, модификации и назначения всех белков, производимых геномом
организма. В отличие от привычной белковой химии протеомика изучает мульти-
белковые системы как часть всего организма. Работа ведется со смесями белков,
идентификация которых осуществляется благодаря частичному секвенированию
и широкому использованию баз данных. Это скорее системная, а не структурная
биология. Поэтому задача протеомики — охарактеризовать поведение системы в
целом, а не ее отдельных компонентов.
Хотя, расшифровав геном, можно знать a priori, какие белки может
произвести данный организм, протеомика развивается столь же быстро, что и
геномика. Расшифровка генома и даже знание количеств конкретных РНК в
клетке не дают надежной информации о количестве конкретных белков в клетке.
Белки различаются по устойчивости, реакционной способности и функциям;
часть из них может быть вовлечена в разнообразные быстрые и медленные
клеточные циклы. Эндогенные посттрансляционные модификации, мутации и
другие изменения под действием веществ из окружающей среды дополнительно
изменяют структуру и свойства исходных белков.
324 Глава 8. Основные направления фрагментации органических соединений
Протеомика базируется на четырех методах. Масс-спектрометрия является
одним из них и используется, как уже было отмечено выше, для установления
молекулярных масс белков и пептидов (продуктов расщепления белков), для
установления последовательности аминокислотных звеньев в пептидах, а также
природы и мест связывания модифицирующих групп. Наряду с масс-спектро-
метрией важную роль в исследованиях протеома играют базы данных по белкам,
меткам сиквенса (expressed sequence tag) и сиквенсу генома в целом, комплекс
программного обеспечения для коррелирования известных белковых сиквенсов
с данными масс-спектрометрических экспериментов, а также методы выделения
и очистки белков.
Говоря о приложениях протеомики, можно, прежде всего, выделить
следующие:
1. Прямой поиск (mining) —работа по идентификации максимального числа
белков в каждой пробе. Цель — получить информацию о протеоме напрямую,
а не опосредованно через информацию о сиквенсе (последовательности)
генов.
2. Поиск изменений состава белков организма (protein-expression profiling)
идентификация белков в образце в зависимости от состояния системы (организма,
клетки). Имеется в виду стадии размножения, роста, болезни или реакция
на воздействие лекарств, химических или физических факторов. Например,
сравнение белков здоровой и больной клеток позволяет получать важнейшую
информацию, которую в дальнейшем можно использовать в диагностике
болезней, выявлении мишеней для последующего медикаментозного лечения.
3. Установление межбелковых взаимодействий (protein-network mapping),
которые определяют, каким образом белки взаимодействуют друг с другом в
живом организме. Поскольку большинство функций организма обусловлено
не индивидуальным белком, а их совокупностью, информация о
взаимодействиях белков имеет первостепенное значение [460]. Ряд биохимических
подходов для изучения взаимодействий белок—белок был адаптирован для
применения метода молекулярного дробления [446]. Чувствительный метод
для установления межбелковых взаимодействий включает связывание белка с
твердой подложкой, используемой в качестве «наживки». Клеточный экстракт
выливается на такую подготовленную поверхность, и белки с высоким
сродством к иммобилизованному белку задерживаются. Метод позволяет
детектировать ассоциаты с константой связывания до 10~5, что
объясняется высокой концентрацией неподвижного компонента [460]. Модификацией
этого метода, когда прямо за связыванием проводится масс-спектрометриче-
ский анализ [187, 188], является метод поверхностной матричной лазерной
десорбционной ионизации (разд. 5.14).
Другим методом идентификации межбелкового связывания является
использование антител для соосаждения взаимодействующих белков. Обычно
8.16. Нуклеиновые кислоты 325
короткая последовательность аминокислот встраивается в анализируемый
белок, образуя эпитоп, для связывания которого имеются подходящие антитела.
За стадией осаждения следует стадия масс-спектрометрического анализа.
Изучению взаимодействий белок —белок и белок —лиганд посвящен ряд
специализированных статей и обзоров [461-466]. Результаты определений от
первичной до четвертичной структуры белков и межбелковых
взаимодействиях опубликованы в работе [467].
4. Картирование белковых модификаций направлено на установление того, в
какой момент, каким образом, по какой причине, в каком месте клетки
белки приобретают модифицирующие группы. Многие распространенные
посттрансляционные изменения белков определяют их пространственную
структуру, назначение и функциональные особенности. Модификации
обусловлены не только эндогенными причинами, но и влиянием лекарств,
химических соединений, поступающих в организм из окружающей среды.
Кстати, основополагающей характеристикой белка является информация о
том, в каком месте клетки он находится. Задачи белков ядра, цитоплазмы или
клеточной мембраны различны. Поэтому место локализация белка —первый
шаг определения его функционального назначения.
8.16. Нуклеиновые кислоты
Будучи еще более сложными, чем полипептиды и белки, биополимерами,
нуклеиновые кислоты долгое время были вне рамок досягаемости масс-спектро-
метрии. До 90-х годов XX века более или менее значимых результатов
удавалось добиться с использованием ББА (разд. 5.10) или плазменной десорбции
(разд. 5.8). Эти методы позволили получать спектры молекул с несколькими (до
10) нуклеотидами в цепи [468, 469]. Прорыв произошел с внедрением в практику
исследований электрораспыления (разд. 5.12) и матричной лазерной десорбци-
онной ионизации, МЛДИ (разд. 5.14). Несколько хороших обзоров посвящено
результатам анализов нуклеиновых кислот этими методами [470-472]. С каждым
годом масс-спектрометрия все более активно используется для установления
молекулярных масс и состава нуклеиновых кислот, последовательности нуклео-
тидов, а также для структурного анализа их аддуктов с самыми разнообразными
соединениями, включая лекарственные препараты, металлы, белки и вещества,
загрязняющие окружающую среду [473, 474]. Рекордная масса,
зарегистрированная масс-спектрометрически [7], принадлежит ДНК колифага Т4. Тем не менее
следует отметить, что получить качественный масс-спектр нуклеиновой кислоты
значительно сложнее, чем масс-спектр белка с такой же молекулярной массой.
Звенья цепи дезоксирибонуклеиновой кислоты (дезоксирибонуклеотиды)
состоят из моносахарида /J-D-2-дезоксирибофуранозы (VIII-7), фосфатной группы
и одного из четырех азотистых оснований (аденин —VIII-2, тимин —VIII-4,
гуанин —VIII-3, цитозин —VIII-5), представленных на рис. 8.141. Звенья цепи
326 Глава 8. Основные направления фрагментации органических соединений
NH2 ОН ОН
*Г"Ц N >Г^| ? ??^?™3
^?^?? ???^?^?? ??^?? HO^
ОН
?
НО'
А
УШ-2
???-3
ОН
VDI-5
???-6
ОН ОН
???-8
Рис. 8.141
НО
*' х'Ф --*' --*'* -'*'' »-*' Г'' г''' г''' г ** -'** г'*'
I i I iyi I I l?l I I It!
>OtP-!-Oj
! \Ь
?\ ?
N1
Ь
ki io k oi
?
ft
»?>·*2.··>^- %-™УхМУ}·-??
?
К,
i A
? ? I
*4o|
о
k u
!?.
OS
OH
Рис. 8.142. Основные направления фрагментации молекулярного иона гексамера ДНК
рибонуклеиновой кислоты (рибонуклеотиды) состоят из моносахарида /3-D-
рибофуранозы (VIII-8), фосфатной группы и одного из четырех азотистых
оснований (аденин, урацил — VIII-6, гуанин, цитозин), представленных на рис. 8.141.
Пуриновые основания (аденин и гуанин) присоединяют рибозный фрагмент по
атому азота N9, а пиримидиновые основания (тимин, цитозин, урацил) —по
атому азота N1. Фураноза присоединяет все основания по атому углерода С1
сахара. Образующиеся нуклеозиды соединяются друг с другом через гидрок-
сильные группы в положениях 3 и 5 фуранозного цикла посредством фосфатных
групп. На рисунке 8.142 представлен фрагмент молекулы нуклеиновой кислоты.
Для упрощения записи фуранозный фрагмент обозначают вертикальной чертой,
фосфатную группу часто обозначают буквой ? в круге, а основание первой
буквой его названия. Молекулу принято располагать по горизонтали, причем
на левом краю находится фураноза с незанятой гидроксильной группой в
положении 5, а на правом —в положении 3.
Среди многочисленных процессов фрагментации молекулярных ионов
нуклеиновых кислот в масс-спектрометре особую ценность имеют те, которые
позволяют установить последовательность нуклеотидов. Аналогично
фрагментации пептидов была разработана классификация характеристических серий
фрагментных ионов, образующихся при распаде нуклеиновых кислот [475].
На рисунке 8.142 представлены основные серии ионов для гексамера. Се-
8.16. Нуклеиновые кислоты 327
рии а, Ь, с, d включают фрагменты со свободной гидроксильной группой в
положении 5 (левая часть молекулы). Серии w, ?, у, ? включают фрагменты
со свободной гидроксильной группой в положении 3 (правая часть молекулы).
Поскольку основная цепь нуклеиновых кислот характеризуется четырьмя типами
разрывов с каждой стороны (в отличие от пептидов с тремя типами разрывов),
обозначения d и w относятся не к побочным, как в случае пептидов, а к
основным ионам. Основание обозначается В„, где п — положение данного
основания относительно конца молекулы со свободной 5-гидроксильной группой.
Аббревиатура а4 —ВЗ(С) означает фрагментный ион серии а по четвертому
положению исходной молекулы, который дополнительно выбросил цитозин из
положения 3.
Достаточно сложно оказалось подобрать матрицу для МЛДИ-анализа
нуклеиновых кислот. В частности, хороших результатов удалось добиться в
замороженной водной матрице на медной подложке. При облучении образца лазером с
длиной волны 578 или 589 нм в газовую фазу переходят неразрушенные
молекулы, содержащие до 600 азотистых оснований в цепи [476-478]. Недостатком
этого подхода является плохая воспроизводимость. При использовании смеси
3-гидроксипиколиновой и пиколиновой кислот в качестве матрицы и лазера с
длиной волны 266 нм [479] удалось получить масс-спектры двойной спирали
нуклеиновой кислоты с 500 основаниями в цепи (m/z > 160000). Кстати, во
время пробоподготовки и анализа происходит денатурирование молекулы, поэтому в
спектрах проявляются только пики ионов одной цепи [480]. Еще в ранних
исследованиях методом МЛДИ было отмечено, что интенсивность масс-спектромет-
рического сигнала зависит от природы нуклеотидов. Самым высоким фактором
отклика характеризуется тимин, а РНК дают более интенсивные сигналы, чем
ДНК. Кроме того, МЛДИ позволяет получить спектры РНК более тяжелых по
сравнению с ДНК [481-483]. Эффект тимина объясняется меньшим сродством к
протону по сравнению с остальными нуклеотидами [484]. Поскольку первичные
реакции фрагментации инициируются протонированием азотистого основания,
за которым следует 1,2-транс-элиминирование (Е2) с разрывом С—N-гликозид-
ной связи [485, 486], тиминовые фрагменты оказываются более устойчивыми.
Поскольку процесс элиминирования включает атом водорода в положении 2
фуранозного цикла, наличие в этом положении гидроксильной группы в случае
РНК в определенной степени препятствует фрагментации, приводя к большей
стабильности молекулярных ионов РНК по сравнению с ДНК [486].
Еще одной проблемой анализа нуклеиновых кислот является необходимость
использования максимальной разрешающей способности прибора, поскольку
уже для олигомера с 15 основаниями существуют по одному изобарному иону
каждой целочисленной массы. Для олигомера с 20 основаниями необходимо
выбирать уже из трех изобарных ионов с каждой целочисленной массой [487].
Как и в случае белков, секвенирование нуклеиновых кислот позволяет не
только определить последовательность нуклеотидов, но и сделать вывод о
мутациях, других изменениях молекул [488]. Для установления последовательности
328 Глава 8. Основные направления фрагментации органических соединений
нуклеотидов в нуклеиновой кислоте с помощью активации соударением вновь
необходимо использовать максимальную разрешающую способность и точность
измерения масс, для того чтобы избежать неоднозначности в отнесении состава
фрагментов. Тем не менее спектры получаются очень сложными и требуют
значительного времени для интерпретации. В качестве примеров конкретных
исследований можно привести работы [475, 486, 489-491].
В связи со сложностью интерпретации спектров тандемной масс-спектромет-
рии для установления сиквенса активно используется леддерная техника,
аналогичная описанной для белков (разд. 8.15). Процесс включает последовательное
энзиматическое отщепление нуклеотидов от одного или другого конца цепи
с измерением разницы в массе. В данном случае требования к разрешающей
способности менее строги, хотя остаются достаточно жесткими [492, 493].
Наиболее простым методом секвенирования является процесс Зангера с масс-
спектрометрическим окончанием. Замена классической гель-хроматографии на
масс-спектрометрию привела к значительному сокращению времени анализа.
В этом случае требования к разрешающей способности прибора минимальны,
поскольку характеристические пики отстоят друг от друга примерно на 300 Да
[494, 495].
8.17. Масс-спектрометрический анализ
микроорганизмов
Проблема идентификации микроорганизмов возникла, как только стала понятна
взаимосвязь бактерий с заболеваниями. В классическом варианте
идентификацию гомогенных образцов микроорганизмов, полученных культивированием в
лабораторных условиях, проводили с помощью микроскопа. Современные
методы идентификации (включая автоматы для характеристики микроорганизмов по
набору энзиматических реакций со спектральным детектированием), хемотаксо-
номическая характеристика (основанная на анализе жирных кислот с помощью
газовой хроматографии), а также характеристика генов не лишены недостатков.
Такие анализы занимают много времени, а для их проведения необходимы
чистые виды (штаммы). Работа со смесями микроорганизмов значительно менее
эффективна и может привести к ложным результатам.
Масс-спектрометрия оказалась отличным методом для анализа
биологических образцов и позволяет преодолеть указанные затруднения. Современный
прибор дает возможность анализировать молекулы с молекулярной массой выше
107 Да, в том числе в неразделенных и неочищенных смесях (клеточные лизаты,
смеси пептидов и белков после различных расщеплений и т. д. [496, 497]). Для
проведения анализа требуются считанные минуты и фемто-аттомоли A0~15-
10~18) вещества. В частности, образец для детектирования маркеров широкого
8.17. Масс-спектрометрический анализ микроорганизмов 329
круга микроорганизмов в смешанных популяциях может быть проанализирован
в течение нескольких минут.
Можно выделить три типа масс-спектрометрического анализа
микроорганизмов. Наиболее примитивный подразумевает быстрый скрининг для
установления присутствия бактерий, вирусов, грибов, других микроорганизмов в
воде, воздухе, продуктах и т. д. Второй тип анализа включает идентификацию
микроорганизмов, иногда на уровне вида или штамма. Для этой цели масс-
спектрометрия, как правило, используется в комбинации с другими методами,
а для подготовки образца проводят лабораторное культивирование. Благодаря
обобщению накопленных в этой области данных и переходу к созданию
соответствующих библиотек масс-спектрометрия позволяет в кратчайшие сроки
эффективно идентифицировать смесевые препараты. Третий, наиболее
трудоемкий уровень исследований подразумевает установление состава и структуры
отдельных молекул в конкретных микроорганизмах.
Масс-спектрометрия была впервые применена для анализа микроорганизмов
по типу «отпечатков пальцев» в 70-х годах прошлого века. Использовали
пиролитическую масс-спектрометрию (разд. 5.15) с детектированием
низкомолекулярных (молекулярные массы ниже 150 Да) продуктов разложения или
контролируемый нагрев обезвоженных бактерий с регистрацией соединений
с молекулярными массами до 700 Да [498]. С внедрением в практику масс-
спектрометрии бомбардировки быстрыми атомами, методов лазерной десорбции,
электрораспыления, а также использование тандемной масс-спектрометрии
позволило проводить анализ смесей нелетучих органических соединений с
большими молекулярными массами. В результате появилась возможность
идентифицировать биомаркеры конкретных групп и индивидуальных микроорганизмов. В
качестве таких маркеров используются углеводы, гликолипиды, фосфолипиды,
пептиды, белки, нуклеиновые кислоты и т. д. [8, 499-501].
Самый ценный биомаркер, безусловно, представляют собой молекулы
нуклеиновых кислот, которые являются наиболее индивидуальным и полным
признаком объекта. Масс-спектрометрическое секвенирование генов пока не дает столь
эффективных результатов, как секвенирование пептидов и белков (разд. 8.15
и 8.16), однако измерение молекулярных масс продуктов расщепления генов
позволяет получать достоверную информацию о составе исходной молекулы.
Сравнение результатов с базами данных дает возможность распознавать
отдельные виды даже в смешанных популяциях. К сожалению, каждая клетка содержит
лишь одну молекулу ДНК, а количество молекул РНК не превышает 0,01% сухой
массы бактерий и 1% сухой массы спор.
Хорошими биомаркерами являются белки. Большинство белков
микроорганизмов имеют молекулярные массы в диапазоне 4000-15000 Да [502]. Они
составляют более 50% сухой массы микроорганизмов. С их помощью можно
различать очень близкие друг другу виды, а также подвиды, штаммы и их споры
[503]. Например, метод МЛДИ позволяет получить информацию о молекулярной
330 Глава 8. Основные направления фрагментации органических соединений
массе белка, а также молекулярных массах продуктов их энзиматического
расщепления. Уже по набору этих продуктов можно делать важные выводы о
принадлежности образца к тому или иному виду микроорганизмов. Для
экспрессного установления последовательности аминокислот в структуре пептидов,
полученных в результате расщепления белков, применяют тандемную масс-
спектрометрию.
Технически МЛДИ-анализ белков микроорганизмов мало отличается от
обычного анализа белков. Важнейшим различием является дополнительная процедура
по разрушению оболочки клеток. Однако эта стадия легко осуществима при
добавлении к образцу кислот [503], метанола [504] или этанола [505], причем
метанол стабилизирует образец лучше, чем, например, трифторуксусная кислота
[506]. Кроме того, следует подчеркнуть, что, поскольку подавляющее
большинство матриц МЛДИ является органическими кислотами, контакт
микроорганизмов с матрицей часто приводит к разрушению оболочек клеток без каких-либо
дополнительных стадий.
Расшифровка генома микроорганизма значительно облегчает и
увеличивает надежность белкового анализа. Например, многие пики в спектре МЛДИ
Sacsharomyces cerivasiae хорошо коррелировали с молекулярными массами
белков, предсказанных на основе генома данного микроорганизма [507]. Этот
подход активно используется и в других исследованиях [508, 509]. Повышенная
надежность такого варианта объясняется тем, что в зависимости от типа пробо-
подготовки и состояния анализируемого микроорганизма набор детектируемых
белков может быть различным. Однако все эти белки могут быть легко связаны
с ДНК организма. Тем самым надежность идентификации организма возрастает.
Очень широко используется в анализе белков компьютерный
поиск по базам данных, включая Интернет. Один из рабочих адресов —
www.expasy.ch/tools/peptident.html. В этих базах собраны данные о сотнях тысяч
белков, поэтому информация о молекулярной массе пептида или белка и о
начальной последовательности аминокислотных звеньев (метка сиквенса) дает
возможность вести эффективный компьютерный поиск.
При масс-спектрометрическом изучении целых (интактных)
микроорганизмов и их спор полезную информацию предоставляют полярные липиды. В этом
случае готовят суспензию клеток микроорганизма в растворителе или наносят
их на твердую подложку, например матрицу МЛДИ. За счет взаимодействия
со средой в этих условиях идет лизирование большинства бактериальных
клеток. Хотя состав жирных кислот может изменяться в жизненном цикле
организма, полярные головные группы фосфолипидов достаточно стабильны,
чтобы использовать их для таксономической характеристики. Полярные липиды
составляют около 5-8% сухой массы бактерий, их легко выделять и
анализировать, например, с помощью бомбардировки быстрыми атомами. В связи с этим
полярные липиды оказались наиболее удобным классом органических
соединений, анализ которых позволяет проводить идентификацию микроорганизмов.
8.17. Масс-спектрометрический анализ микроорганизмов 331
В частности, для грамотрицательных бактерий характерен фосфатидилэтано-
ламин, для грамположительных бактерий — фосфатидилглицерин, для грибов —
фосфатидилинозитол, для инкапсулированных вирусов — фосфатидилхолин, для
микроводорослей —сульфонолипиды. Для регистрации этих характеристических
головных групп полярных липидов используют тандемную масс-спектрометрию.
Выделяя молекулярный ион, проводя его активацию соударением и
регистрируя спектры дочерних ионов или спектры выбросов идентичных нейтральных
частиц, можно детектировать характеристическую группу и отнести образец к
тому или иному классу микроорганизмов.
Масс-спектрометрическое исследование вирусов включает не только
идентификацию вирусов, но и изучение связывания вирус—антитело, взаимодействия
белок—белок и динамических изменений белков [510]. Использование
электрораспыления дает возможность измерять молекулярные массы ДНК интактных
вирусов [7, 511]. Интереснейшим фактом является то, что вирус может не
погибнуть во время анализа. В частности, Сьюздаку [510] удалось инфицировать
листья табака вирусом табачной мозаики, подвергнутым предварительному масс-
спектрометрическому анализу.
Анализ белков капсид вирусов [512, 513] становится рутиной, поскольку
молекулярные массы этих соединений редко превышают 10000 Да. В условиях
МЛДИ при взаимодействии с кислой матрицей происходит денатурация белков,
так что для установления первичной структуры необходимо лишь смешать
солюбилизированный образец с материалом матрицы и провести анализ. Масс-
спектрометрическая идентификация вирусов по белкам их капсид достаточно
проста, поскольку последовательность аминокислот в вирусном пептиде и
последовательность нуклеотидов в ДНК уже известны для большинства
вирусов. Поэтому идентификации по величинам m/z продуктов энзиматического
расщепления является скорее компьютерной процедурой и легко осуществляется
в автоматическом режиме с использованием баз данных.
В последнее время возрастает важность экспрессной идентификации
патогенных микроорганизмов как части контртеррористических мероприятий. На
50-й конференции Масс-спектрометрического общества США в Орландо в июне
2002 г. этим вопросам была посвящена работа отдельной секции. Действительно,
масс-спектрометрия может быть крайне эффективной при достижении этих
целей. Получают спектр типа «отпечатков пальцев» и сравнивают его со
спектрами соответствующей библиотеки масс-спектров микроорганизмов, которые
потенциально могут быть использованы в качестве биологического оружия
[514]. Для анализа атмосферы можно использовать прямой забор проб воздуха
[515] или депонирование частиц из воздуха на полимерный материал [516].
Для определения микроорганизмов в воде [517, 518] хорошо зарекомендовали
себя специально подготовленные поверхности (аналог поверхностной матричной
лазерной десорбционной ионизации, разд. 5.14).
Глава 9
КОЛИЧЕСТВЕННЫЙ
МАСС-СПЕКТРОМЕТРИЧЕСКИЙ
АНАЛИЗ
С момента своего возникновения масс-спектрометрия была признана методом
количественного анализа. Для грубой количественной оценки можно
использовать площадь под кривой полного ионного тока, полученной при полном
испарении образца из штока прямого ввода. Важно только, чтобы кривая
испарения вещества не перекрывалась кривой испарения другого компонента
образца. Однако даже в последнем случае можно провести измерения, если
использовать для расчетов площади под кривой не всего ионного тока, а тока
характеристических для каждого из компонентов смеси ионов (разд. 9.1).
На рисунке 9.1 представлена кривая полного ионного тока, полученная при
полном испарении образца, состоящего из двух компонентов. В данном случае
можно говорить о термограмме образца. Компоненты образца существенно
отличаются по температуре испарения, поэтому плавный нагрев пробы позволяет
получить индивидуальные пики обоих ингредиентов. Масс-спектры компонентов
приведены на рис. 9.1,6 и в. Спектры не перекрываются и позволяют надежно
идентифицировать соединения, а площади под кривыми на термограмме дают
возможность провести количественную оценку состава смеси.
Если необходимо измерить абсолютное количество конкретного соединения
в препарате, оптимальным вариантом является предварительное построение
калибровочной кривой, связывающей количество анализируемого соединения
в граммах и площадь под кривой полного ионного тока или тока
характеристического иона. Однако прямой ввод может более или менее эффективно
использоваться только для анализа простых смесей.
Наилучшим инструментом для проведения количественного анализа смесей
органических соединений является хроматомасс-спектрометрия. В этом случае
на входе мы имеем неизвестную смесь органических соединений, а на выходе —
полную информацию о ее качественном и количественном составе. Масс-спектр
позволяет идентифицировать соединение, а площадь хроматографического
пика—оценить количество этого соединения в пробе. На рисунке 9.2 представлена
хроматограмма по полному ионному току смеси 46 приоритетных загрязняющих
веществ. Все пики разрешены у нулевой линии, их площади могут быть легко
и точно проинтегрированы.
Проблемы возникают при анализе сложных многокомпонентных систем,
например природных соединений. На рисунке 9.3 представлена хроматограмма
по полному ионному току дихлорметанового экстракта цветов тысячелистника.
Глава 9. Количественный масс-спектрометрический анализ 333
ТЮ; VTR30I4/I1 .D
I | ' * ? '* I I | I I I » |
104 (? .?-? О mln>: VF»30*/|-| .О
,ll, , J j , .1,1 ,. flfr , ,|1|?., ,.1I., , .?? -ЮЧ^Л-УхЧ
-IgfiOU' ¦-!/¦» Ч ¦ '_ I1"'"
¦^nfli., r ,-авгд'I'. ,1'.^я
кг* 1Эв С2.3*·* mln>: Vf»3DI4/|-l .ЕЭ
¦»^Г4'ая^Г^в'
тю»11Я|''ч'"|- . ча
Рис. 9.1. Кривая полного ионного тока двухкомпонентного образца (а), испаряемого из
штока прямого ввода, и масс-спектры компонентов (б и в)
?)
?!""!""!""!""!»"!
? ? ?
"?»4
?
^«
?) (?
3 ?
? ? l· ? ? ? ?
0 0 0 0 0 0 0
§ 8 §
0 0 0
**3
On
?
?
?*
9.1. Масс-хроматография 335
На этой хроматограмме более половины пиков недостаточно разрешены, а
часть пиков обусловлена двумя и более компонентами с близкими временами
выхода. В таком варианте прямое интегрирование площади пика приведет к
неправильным результатам.
Для проведения количественного хроматомасс-спектрометрического анализа
смесей органических соединений используют две родственные техники: масс-
хроматографию и масс-фрагментографию.
9.1. Масс-хроматография
Метод масс-хроматографии не связан ни с каким особым проведением
эксперимента. Ведется обычный хроматомасс-спектрометрический анализ с полным
сканированием масс-спектра в заданном диапазоне масс. Качественное определение
ингредиентов осуществляется по их масс-спектрам и временам выхода. Однако
для количественного определения компьютер строит хроматограммы по току
характеристических для каждого соединения ионов. Внешний вид
хроматограммы может полностью измениться по сравнению с исходной хроматограммой,
построенной по полному ионному току, поскольку все ионы, за исключением
заданных, игнорируются.
На рисунке 9.4 представлена хроматограмма по полному ионному току
дихлорметанового экстра^сга тюленьего жира и масс-хроматограммы по току
ионов с m/z 326, 360 и 394, характерных для пентахлор-, гексахлор- и гепта-
хлорбифенилов соответственно. Все пики на полученных масс-хроматограммах
хорошо разрешены, а их площади могут быть легко вычислены
интегрированием. Следует обратить внимание, что ни один из этих пиков не может быть
надежно зарегистрирован на хроматограмме по полному ионному току. Этот
факт тем более интересен, что речь не идет об увеличении чувствительности
метода. Фактически, меняется только форма представления результатов. Такая
форма характеризуется повышенной селективностью.
Масс-хроматография позволяет во многих случаях проводить качественный
и количественный анализ компонентов, которые не разделяются хроматогра-
фически. Если есть подозрение о наложении масс-спектров двух соединений
с близкими временами выхода, имеет смысл построить масс-хроматограммы
по ионам, характеризующимся наиболее интенсивными пиками. Как правило,
компоненты выходят не абсолютно одновременно. Пик на хроматограмме по
полному ионному току оказывается уширенным (рис. 9.5, я), но на
масс-хроматограммах отчетливо видно, что вершины пиков характеристических ионов,
принадлежащих двум компонентам, сдвинуты друг относительно друга (рис. 9.5, в).
Проведя вычитание масс-спектра, записанного на вершине второго пика, из
масс-спектра, записанного на вершине первого, можно получить
индивидуальный спектр первого компонента. Вычитание масс-спектра первого пика из масс-
336 Глава 9. Количественный масс-спектрометрический анализ
Abundance
TIC: NERPA1.D
1400000
1200000
???????-4
?????? ?
400000
IXj^Jol^uu^
Abundance
Ion 326.00 C25.70 to 326.70): NERPA1.D
Tlme->u
^Jl
?"? ? |
,л4
24.OO 24.SO 25.O0 25.SO 26.00 26.50 27.00 27.50 28.00 28.50 2?.??
T"l V p I I I Г-р1 It'll VI
I ^»
Abundance
Ion 360.00 C59.70 to 360.70): NERPA1.D
? ? ? i-i Сч \ ¦ T> ??-
? ? ? ? ? ? ? ? r ? ? ? ? ?- ? ? ? ? ?^? ? ?*?'?"? ? -? -^ ?*~?4 ? ' ' ' Г'*"' ¦ *' "I r\ ? ? '| ? ? ? ? | ?" ? ? ?") ?
24.?? 24.SO 25.?? 25.SO 26.00 26.SO 27.00 27.50 28.?? 28.SO 29.??
Abundance
Ion 394.00 C93.70 to 394.70): NERPA1.D
j~ -p· f*·
' ? V ? , , t , ,
24.00 24.50 25.00 25.50 26.00 26.50 27.?? 27.50 28.00 28.50 29.00
Рис. 9.4. Хроматограмма по полному ионному току дихлорметанового экстракта тюленьего
жира (а) и масс-хроматограммы по току ионов с m/z 326, 360 и 394, характерных для
пентахлор-, гексахлор- и гептахлорбифенилов соответственно F, в, г)
9.1. Масс-хроматография 337
спектра второго позволит получить чистый масс-спектр второго компонента. Для
количественного анализа необходимо просто проинтегрировать площадь пиков
на соответствующих масс-хроматограммах. Описанный пример иллюстрируется
рис. 9.5, а-д. Полученные после процедуры вычитания масс-спектры (рис. 9.5, г
и д) характеризуются хорошим совпадением с библиотечными и позволяют
надежно идентифицировать соединения.
Для увеличения надежности качественого и количественного анализа имеет
смысл строить масс-хроматограммы не по одному, а по 2-3 характеристическим
для данного соединения ионам. Расчет по любому из этих пиков с учетом
фактора отклика (см. ниже) дает правильный результат и позволяет провести
усреднение. Профиль всех трех пиков должен быть идентичен, а соотношение
их площадей должно соответствовать интенсивности пиков этих ионов в масс-
спектре (рис. 9.6, а). Если один из пиков оказывается уширенным (рис. 9.6,6),
или его интенсивность явно завышена (рис. 9.6 в), вероятно, другой компонент
смеси, имеющий близкое время удерживания, характеризуется пиком с такой же
величиной m/z. В этих случаях (9.6, бив) расчет надо вести по двум «чистым»
пикам.
В методике Агентства по охране окружающей среды США хроматомасс-
спектрометрического определения приоритетных органических экотоксикантов в
воде предложены по 10 наиболее интенсивных пиков ионов в спектрах каждого
определяемого соединения [519]. Такой набор позволяет выбрать несколько
характеристических ионов в каждом случае, избегая перекрываний с другими
компонентами смеси.
Стремление сделать качественный и количественный анализ более надежным
и точным, а также все более широкое применение быстрой хроматомасс-
спектрометрии [44] стимулируют создание все более мощных компьютерных
программ по деконволюции плохоразрешенных хроматографических пиков. В
программе Биллера—Бимана [520] компьютер строит масс-хроматограммы для
всех целочисленных значений масс сканируемого диапазона. Если несколько
масс-хроматограмм одновременно достигают максимума, независимо от полного
ионного тока в этом месте хроматограммы регистрируется хроматографический
пик. Все ионы, ток которых не достигает максимума в этот момент времени,
игнорируются. Таким образом строятся хроматограммы, разрешенные по
массе (mass-resolved chromatograms). Реконструированный масс-спектр также
содержит только пики ионов, проходящие через максимум в конкретный момент
времени. В этом спектре отсутствуют наложения со стороны колоночного
фона, который никогда не достигает максимума, и подавляющее большинство
наложений со стороны других компонентов смеси, характеризующихся близкими
временами удерживания. Для того чтобы хроматографические пики компонентов
были разрешенными, необходимо, чтобы их реальные максимумы отстояли друг
от друга хотя бы на три сканирования. Понятно, что увеличение скорости
сканирования приводит к более эффективному разрешению компонентов смесей.
vo
? .
??
«ft
W
t:
да
о
ц -4
ш!
о
§ о
? 10
О О
t
щи
1-е
«1*
•3!
1
^
л
V
?
fli
ь
4
ш ?
.· t
с г
?
0
Ifl
я
?
г
г
?
Ш
С
5
ю
•
«
fM
?
и J
?
¦I
г
*Н
г
*
*^
г
?
?
J
I " ' Ч " " I " " I" "
0
-?
;
\
f
>
«0
0
N
?
1
*2
S3
?
0
t
¦r
1
i
¦0
-N
?
0
?*
^r
1°
1
j8
r
§ °, ) 8 8 8 8 8 8 °»
О ! С 0 0 0 0 0 0 !
8
0 0 0 0 0
10 О Ю О (О
» « N г г
340 Глава 9. Количественный масс-спектрометрический анализ
mlik
m/z&
m/zC
Рис. 9.6. Возможные формы пиков
характеристических ионов: а — формы трех характеристических
пиков в эталонном образце; б — пик иона В на масс-хро-
матограмме уширен в связи с присутствием другого
компонента смеси с близким временем удерживания;
в — интенсивность пика иона А на масс-хроматограм-
ме завышена в связи с присутствием другого
компонента смеси с близким временем удерживания
Именно поэтому переход на времяпролетные анализаторы позволил значительно
повысить эффективность хроматомасс-спектрометрического анализа [44, 521,
522]. Метод Биллера—Бимана характеризуется надежными результатами и в
случае равных концентраций выходящих рядом компонентов, и в том случае,
когда один из них является минорной примесью. Несколько хуже оказываются
результаты, когда пики обусловлены структурно близкими соединениями
(например, изомерами). Более сложные в математическом отношении методы [523-525]
позволяют разделять хроматографические пики компонентов, различающиеся на
1,5 скана.
Современные времяпролетные приборы позволяют записывать до 500 полных
масс-спектров в секунду. Последующая математическая обработка результатов
(деконволюция) дает возможность получить хорошие масс-спектры
соединений, выходящих практически одновременно из хроматографа. На рисунке 9.7
представлена хроматограмма по полному ионному току смеси органических
соединений. Глаз человека может различить на этом 13-секундном отрезке
хроматограммы не более 7-8 пиков [526]. Однако современная процедура
?
11
13
секунды'
Рис. 9.7. Сколько пиков присутствует на этом 13-секундном участке хроматограммы?
9.2. Масс-фрагментография, мониторинг заданных ионов 341
Рис. 9.8. Компьютерная обработка позволяет выявить на представленном 13-секундном
участке хроматограммы и получить качественные масс-спектры 21 соединения
математической обработки с использованием масс-хроматографического подхода
однозначно показывает, что на этом участке хроматограммы имеются пики 21
соединения (рис. 9.8).
Таким образом, метод масс-хроматографии очень полезен и для
качественного, и для количественного анализа. Его достоинством является сохранение
всей информации, получаемой при использовании классического варианта хро-
матомасс-спектрометрии. Тем не менее существуют варианты проведения хро-
матомасс-спектрометрического анализа, когда чувствительность эксперимента
увеличивается на 1-2 порядка.
9.2. Масс-фрагментография,
мониторинг заданных ионов
(Selected Ion Monitoring, SIM)
Иногда задачей анализа является количественное определение конкретных уль-
трамикрокомпонентов смеси. Для повышения чувствительности метода можно
использовать технику масс-фрагментографии. В этом случае масс-спектрометр
не сканирует весь диапазон масс, а жестко настраивается на регистрацию ионов
с заданными значениями m/z. В чем причина улучшенных пределов обнаружения
в таком эксперименте? Представим себе, что в обычном сканирующем режиме
скорость развертки от m/z 50 до m/z 550 составляет 1 с. Даже без учета времени,
которое требуется для возвращения с верхней границы масс на нижнюю для
начала нового сканирования, прибор фиксирует конкретную целочисленную
массу лишь 1/500 долю секунды. Поскольку ионы генерируются непрерывно,
это означает, что детектора достигает 0,2% ионов каждого типа. Подавляющую
часть ионов сканирующий масс-спектрометр просто не может зарегистрировать,
поскольку «занят» определением других ионов. Понятно, что, если вместо
сканирования настроить прибор на регистрацию ионов только с определенным
342 Глава 9. Количественный масс-спектрометрический анализ
128
m/zm
32
Рис. 9.9. Масс-фрагментограммы дихлор-
метанового экстракта пробы природной
воды, m/z 178 —фенантрен, m/z 188 —пер-
дейтерированный фенантрен (внутренний
стандарт), m/z 181 — гексахлорциклогекса-
ны (а-, /?-, и 7-из°меры). Числа над m/z
означают ранги чувствительности
детектора. Площадь пика с учетом ранга
чувствительности прямо пропорциональна
количеству образца в пробе
значением m/z, все 100% ионов этого типа будут достигать детектора. Это
равносильно увеличению чувствительности в 500 раз. Попеременное пропускание
через анализатор ионов с двумя разными величинами m/z увеличит
чувствительность только в 250 раз, пяти ионов —в 100 раз. В любом случае метод
масс-фрагментографии дает по сравнению с классическим вариантом выигрыш в
чувствительности на два порядка. Метод хорошо зарекомендовал себя для
анализов суперэкотоксикантов (полихлорированных бифенилов, дибензодиоксинов и
дибензофуранов). На рисунке 9.9 представлены масс-фрагментограммы анализа
содержания в воде фенантрена, пердейтерированного фенантрена (внутренний
стандарт) и изомерных гексахлорциклогексанов, полученные при мониторинге
ионов с тремя значениями m/z.
Как правило, требуется определить в одной пробе несколько соединений.
Однако одновременный мониторинг большого числа характеристических ионов
приводит к закономерному уменьшению чувствительности. Поэтому
одновременно регистрируется не более 2-4 ионов, причем, поскольку время выхода
искомых соединений известно, можно вести мониторинг в течение узкого
интервала времени, а затем переключаться на регистрацию другого набора из
2-4 ионов, характерных для соединений, выходящих из колонки позже. Такой
последовательный процесс регистрации искомых соединений легко
программируется и может осуществляться в автоматическом режиме.
К сожалению, выигрыш в чувствительности сопровождается значительным
проигрышем в информативности и надежности метода. Во-первых, полностью
теряется информация о других компонентах смеси. Конечно, если исследователя
интересует исключительно одно конкретное соединение, этим можно прене-
m/Z 188 ?..?··?»<1«(«...··??·??.«??.???!»?????????!??!??!{?!
l!lt!tl..ilitili.H«..iiiHHlilitliii
32
m/zm Mllii!^
9.2. Масс-фрагментография, мониторинг заданных ионов 343
бречь, однако зачастую можно пропустить что-то важное. Во-вторых, существует
определенная вероятность неправильно измерить концентрацию или вообще
неверно определить присутствие в смеси какого-нибудь вещества. Поскольку
весь диапазон масс не сканируется, полный масс-спектр соединения не
регистрируется, а именно масс-спектр является уникальной характеристикой вещества. В
режиме масс-фрагментографии исследователь имеет время выхода соединения и
интенсивность пика одного или двух характеристических для этого соединения
ионов. Это, конечно, уже лучше, чем просто хроматография, когда время выхода
является единственным параметром. Однако в реальной смеси близкие времена
выхода могут иметь несколько соединений, причем с меньшей, но не равной
нулю, вероятностью эти соединения могут иметь характеристические пики с
одной и той же величиной m/z, В этом случае мы получим искаженный пик
искомого иона. В качестве примера, можно вновь обратиться к рис. 9.6,6 и е.
В худшем варианте определяемое соединение может отсутствовать в пробе, но
какое-либо вещество с близким временем выхода, образующее при
фрагментации ионы с такими же величинами m/z, приведет к появлению пиков на масс-
фрагментограмме. Поскольку метод мониторинга заданных ионов используется,
как правило, для определения действительно опасных соединений, цена такой
ошибки может быть велика.
Повысить надежность метода можно при использовании
масс-фрагментографии высокого разрешения. В этом случае магнитный анализатор пропускает
только ионы с заданным дефектом масс. Например, для детектирования диметил-
нитрозамина используют регистрацию иона с
массой 74,0480 Да. Безусловно, анализатор не
может пропускать исключительно ионы с
такой массой. Детектора достигнут все ионы с
массами в диапазоне 74,0480 ± ?, где ? может
достигать нескольких десятков миллионных
долей. Чем стабильнее работает прибор, тем
точнее будет проведен анализ. Хотя все равно
остается шанс регистрации ложного сигнала,
вероятность такого случая ничтожна.
Селективность такого метода очень высока. На
рисунке 9.10, а и б представлены масс-хроматограм-
мы низкого и высокого разрешения образца,
содержащего диметилнитрозамин. Комментарии
преимущества масс-фрагментографии высокого
разрешения излишни.
Вариантами масс-фрагментографии
являются анализы с использованием тандемной масс-
спектрометрии. Методы связанных
сканирований с регистрацией спектров дочерних ионов,
4
WL
Рис. 9.10. Масс-хроматограммы
низкого R = 500, m/z 74 (?) и
высокого R = 5000, m/z 74,0480
разрешения (б) образца,
содержащего диметилнитрозамин
344 Глава 9. Количественный масс-спектрометрический анализ
родительских ионов и выбросов идентичных нейтральных частиц (разд. 7.2)
фактически представляют собой методы мониторинга заданных ионов и
используются для поиска в смеси конкретных соединений. Регистрация пика
свидетельствует о присутствии искомого соединения в смеси, а измерение
площади соответствующего пика в спектре позволяет провести количественное
определение. Помимо экологических исследований [527] масс-фрагментография
и масс-спектрометрия в целом широко используется при разработке и
испытаниях лекарственных препаратов [528], а также в медицинских анализах, для
установления уровней исходных препаратов и их метаболитов в биологических
жидкостях и тканях [28].
9.3. Установление количества соединения в образце
по площади хроматографического пика
Простое интегрирование площади хроматографического пика (любого типа)
соединения не означает автоматического установления его концентрации в
анализируемом образце. Площадь пика зависит от нескольких параметров.
1. Сечение ионизации. Любое органическое вещество характеризуется своей
уникальной величиной сечения ионизации, т. е. количеством ионов,
образовавшихся из молекул в условиях эксперимента. В разделе 2.1 было сказано, что в
условиях электронного удара примерно одна из 10000 молекул ионизируется.
Эта величина зависит от структуры соединения и может изменяться в широком
диапазоне. Это означает, что одинаковые навески двух разных веществ после
ионизации приведут к возникновению разного количества ионов. Как следствие,
площади пиков что на хроматограмме в полном ионном токе, что на масс-
хроматограммах и масс-фрагментограммах будут существенно отличаться.
2. Дискриминация по массе. Анализатор масс любого типа пропускает ионы
с разными значениями m/z по-разному. В результате 100 ионов с m/z 56 дадут
пик с интенсивностью, отличной от пика 100 ионов с m/z 326. Этот процесс
мало сказывается на интенсивности пиков на хроматограммах по полному
ионному току, но важен для масс-хроматографии и масс-фрагментографии. Если
два соединения имеют существенно отличающиеся молекулярные массы, то
дискриминация по массе скажется и на интенсивностях хроматографических
пиков по полному ионному току.
3. Доля тока характеристического иона в полном ионном токе зависит от
природы соединения. Например, доля характеристического иона бенз[а]пирена
с m/z 252 в полном ионном токе этого соединения составляет 52%, тогда как
9.4. Метод внешнего стандарта 345
доля характеристического иона 2,3,7,8-тетрахлордибензодиоксина с m/z 324 —
менее 10%.
Эти сложности заставляют использовать для количественного масс-спектро-
метрического анализа специальные стандарты. Математическая зависимость,
связывающая площади пиков стандарта и искомого вещества, а также
известное количество используемого стандарта, позволяет проводить количественное
определение с высокой точностью.
9.4. Метод внешнего стандарта
Метод заключается в приготовлении стандартного раствора соединения,
которое требуется проанализировать. Известный объем этого раствора вводится в
прибор. Измеряется интенсивность сигнала, т. е. устанавливается зависимость
между количеством введенного соединения и интенсивностью его сигнала.
Затем, не изменяя никаких условий эксперимента, в прибор вводится такой
же объем анализируемой пробы, и вновь измеряется интенсивность сигнала
анализируемого соединения. В диапазоне линейности зависимости количество
образца —интенсивность регистрируемого сигнала рассчитать количество
анализируемого вещества в пробе можно, используя простейшую формулу 9.1:
M = kS, (9.1)
где М — количество определяемого соединения в объеме, введенном в прибор,
S — интенсивность сигнала определяемого соединения, к — фактор отклика,
равный отношению количества соединения, введенного в прибор из стандартного
раствора к интенсивности его сигнала (MCT/SCT).
Для электронного удара зависимость количество образца — интенсивность
регистрируемого сигнала сохраняет линейность в широком диапазоне (до 6
порядков). В случае других методов ионизации диапазон линейности уже в связи
с протеканием в источнике ионов побочных процессов. Поэтому для проведения
количественного анализа требуется построение калибровочного графика. Для
этого в прибор вводятся одинаковые объемы стандартных растворов,
содержащих разные известные количества анализируемого соединения. На основании
интенсивностей полученных сигналов строится график. В идеале, это должна
быть прямая, продолжение которой должно проходить через начало координат.
К сожалению, метод внешнего стандарта зачастую приводит к значительным
ошибкам. Это связано с невозможностью абсолютного совпадения объемов
пробы, вводимой в прибор, а также с изменениями работы масс-спектрометра
в течение проведения калибровок и анализа. Небольшие изменения, связанные с
величиной напряжения на различных линзах, с изменением потока газа-носителя
346 Глава 9. Количественный масс-спектрометрический анализ
или с делением потока в сплиттерном режиме, с давлением в источнике ионов
делают два последовательных анализа не абсолютно идентичными. Эти различия
возрастают при проведении анализа большой серии образцов в течение дня. В
связи с этим общепринятым является предпочтительное использование метода
внутреннего стандарта.
9.5. Метод внутреннего стандарта
В этом случае интенсивности сигналов анализируемых соединений
сравниваются с сигналом стандартного вещества, которое добавляется непосредственно в
образец, вводимый в прибор. Такой стандарт может использоваться для
одновременного определения многих соединений, присутствующих в анализируемом
образце.
Поскольку внутренний стандарт добавляется непосредственно в
анализируемую пробу, все перечисленные выше недостатки метода внешнего стандарта
устраняются. В качестве внутреннего стандарта может использоваться любое
вещество, важно только, чтобы оно было чистым, не реагировало с растворителем
и другими компонентами смеси и отсутствовало изначально в анализируемом
образце. В связи с этим широкое распространение, особенно для анализа
объектов окружающей среды, получили соединения, меченные изотопами D, 13С,
15N, 180 и т. д.
Анализ начинается с последовательного введения в прибор серии
стандартных растворов, с точно известным содержанием внутреннего стандарта
и соединений, которые планируется анализировать в дальнейшем. Количество
внутреннего стандарта в этих растворах, как правило, одно и то же, хотя это и
не обязательно. Его концентрация должна попадать в середину диапазона
потенциальных концентраций анализируемых ингредиентов в пробах. Концентрации
ингредиентов в стандартных растворах должны перекрывать весть диапазон их
предполагаемых концентраций в пробах. Обычно концентрационный диапазон
составляет 2-3 порядка. В этом диапазоне зависимость регистрируемого отклика
от концентрации линейна, т. е. фактор отклика, рассчитанный по формуле 9.2,
должен быть постоянным.
RF=WlT (9·2)
SxxMis
где RF — фактор отклика (response factor), M — количество соединения, в данном
стандартном растворе, S — интенсивность сигнала соединения; индекс is
относится к внутреннему стандарту, а индекс л: — к определяемому соединению.
Величина фактора отклика может изменяться в широком диапазоне в
зависимости от природы определяемого соединения и внутреннего стандарта. На
эту величину влияют прежде всего три фактора, рассмотренные в разд. 9.3. Для
9.5. Метод внутреннего стандарта 347
получения достоверных результатов фактор отклика должен быть как можно
ближе к единице. Если фактор отклика составляет менее ОД или более 10, для
анализа данного ингредиента желательно подобрать другой внутренний стандарт.
После завершения работ со стандартными растворами приступают к анализу
образцов. Для этого в образец добавляется точная навеска внутреннего
стандарта. Концентрация внутреннего стандарта должна примерно соответствовать
величине, использованной для определения факторов отклика. Такой вариант
количественного анализа можно использовать в любой из перечисленных выше
модификаций: хроматограмма по полному ионному току, масс-хроматография
или масс-фрагментография.
Расчет количества анализируемого вещества в пробе осуществляется по
формуле 9.3.
M = RFx^( (9.3)
где ? — количество анализируемого соединения в пробе, S — площадь пика
анализируемого соединения, RF — фактор отклика, Mis — количество введенного
внутреннего стандарта, Sis — площадь пика внутреннего стандарта. Для
измерения площадей пиков можно использовать масс-хроматограмму, масс-фрагменто-
грамму или хроматограмму по полному ионному току
Существуют разные варианты того, в какой момент лучше добавлять
внутренний стандарт к пробе. Эту процедуру можно осуществлять в любой момент
от отбора пробы до введения ее в прибор. Если внутренний стандарт по своей
природе очень близок определяемым соединениям, лучше добавлять его как
можно раньше. В этом случае на всех стадиях пробоподготовки (экстракция,
очистка, концентрирование) его потери будут адекватны потерям анализируемых
ингредиентов, и конечный результат будет точнее соответствовать реальной
концентрации вещества в пробе. Однако если свойства внутреннего
стандарта существенно отличны (например, он хуже экстрагируется используемым
растворителем или его летучесть значительно выше, чем у определяемых
компонентов), лучше добавлять его на поздних стадиях пробоподготовки. В
некоторых методиках количественного анализа помимо внутреннего стандарта,
который добавляется в пробу только перед вводом вещества в масс-спектрометр,
используют дополнительное вещество, называемое суррогатом. Его добавляют
перед началом любых операций пробоподготовки, а его химическая природа
должна быть близка природе определяемых компонентов. В этом случае
внутренний стандарт не теряется в процессе работы с пробой и служит точному
количественному определению соединений в готовой для ввода в прибор пробе.
Оценить же потери веществ при пробоподготовке можно исходя из разницы
между введенным и измеренным количеством суррогата.
Как уже упоминалось выше, в качестве внутренних стандартов удобно
использовать соединения, аналогичные по своей природе искомым, но содер-
348 Глава 9. Количественный масс-спектрометрический анализ
жащие стабильные изотопы. Во избежание перекрывания с изотопными пиками
немеченных соединений, предпочтительно работать с полидейтерированными
соединениями или с соединениями с большим количеством изотопов 13С.
Например, отличными стандартами являются пердейтерированные полициклические
ароматические углеводороды. Они инертны, достаточно стабильны, хорошо
экстрагируются, имеют высокую величину сечения ионизации и интенсивность
пика молекулярного иона около 50% в полном ионном токе. Это
позволяет использовать их для количественного определения самых разнообразных
соединений (например, приоритетных загрязняющих соединений в объектах
окружающей среды [519]).
9.5.1. Метод изотопного разбавления
Предельным вариантом метода внутренних стандартов является анализ, когда
каждому определяемому компоненту соответствует свой внутренний стандарт,
представляющий собой его изотопный аналог, например, пердейтерированное
производное. Такие соединения обладают практически идентичными физико-
химическими свойствами, тогда как величины m/z их характеристических ионов
различаются. Факторы отклика для таких стандартов будут равны единице.
Добавление такого стандарта до начала всех процедур пробоподготовки
позволит не только с минимальной погрешностью вычислить количество искомого
вещества в готовой пробе, но и, точно компенсировав все потери, неизбежные
в процессе пробоподготовки, надежно установить концентрацию вещества в
исходной пробе. Для конечного расчета используется формула 9.3, причем фактор
отклика принимается равным единице. Безусловно, такой подход является
предпочтительным для количественного хроматомасс-спектрометрического анализа.
Единственным серьезным недостатком является резкое удорожание анализа в
таком режиме. Стоимость меченных соединений очень высока и может достигать
сотен и тысяч долларов за миллиграмм (например, для полихлорированных
дибензодиоксинов, в которых все атомы углерода являются изотопами 13С).
9.6. Метод добавок
Количественный хроматомасс-спектрометрический анализ можно проводить,
используя классический метод аналитической химии. Для осуществления метода
добавок используют добавление к пробе известного количества анализируемого
вещества. Вновь, как и в случае метода внутреннего стандарта, эту процедуру
можно осуществлять на любой стадии пробоподготовки. Однако
предпочтительным вариантом является добавление этих веществ сразу после пробоотбора,
9.6. Метод добавок 349
поскольку потери вещества во время хранения пробы и пробоподготовки можно
будет оценить по результатам анализа.
Простейшим вариантом метода является деление пробы на две части и
добавление в одну из них известного количества анализируемых соединений.
Добавляется в 2-5 раз больше (по массе), чем их ожидаемое количество в пробе.
Поскольку часто нельзя заранее оценить уровни анализируемых соединений в
реальной пробе, во втором варианте проводят пробоподготовку и анализ первой
порции с добавлением внутреннего стандарта. Затем, грубо оценив
концентрации искомых компонентов, добавляют во вторую порцию нужные соединения в
нужных количествах (в 2-5 раза больше, чем было определено в первой порции
пробы). Кстати, внутренний стандарт можно не использовать, а приблизительно
оценить уровни компонентов в пробе по величине сигнала (фактически, метод
внешнего стандарта). Можно провести добавку непосредственно в готовую
пробу из первой порции и провести ее повторный анализ. Однако в этом случае
нельзя оценить потери веществ в результате пробоподготовки.
После анализа второй порции вещества оператор имеет два набора
результатов. Обозначим концентрацию в пробе искомого вещества за X. Пусть площадь
проинтегрированного пика, обусловленного этим компонентом, будет S. Тогда,
для второй порции пробы содержание искомого компонента будет X + А, а
площадь соответствующего пика S\. Решая пропорцию, получаем уравнение 9.4:
Глава 10
ОБОБЩЁННЫЕ ЗАДАЧИ
Задача ЮЛ На рисунке 10.1 представлен масс-спектр ЭУ органического
соединения. Установите, какому из перечисленных ниже соединений принадлежит
этот спектр.
а
б;
в'
г
д
е
ж
3
и
к
Бензойная кислота;
о-этилфенол;
о-метокситолуол;
и-этилфенол;
фенилэтиловый эфир;
л-толилметиловый эфир;
2-фенилэтиловый спирт;
1-фенилэтиловый спирт;
2,4-диметилфенол;
2,6-диметилфенол.
100
90 -I
80 А
70 -|
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1
1,%
39 51
_Ал
66
77
_?_!_
94
122
—,—
100
20 30 40 50 60 70 80
m/z
90
110 120 130
Рис. 10.1
Глава 10. Обобщенные задачи 351
Задача 10.2. Идентифицируйте изомерные соединения по спектрам
электронного удара (рис. 10.2).
I , %
3 9
лЬХ
6 4
9 2
2 0
4 О
6 0
— ,_
8 0
m /z
4-
1 0 О
1 2 8
1 2 О
100
90
80
70 -I
60 -|
50
40
30
20
10
О
4
1,%
50
65
73
60
70
80
93
м
-J-r
90
100
100
128
110
120
130
m/z
Рис. 10.2
352 Глава 10. Обобщенные задачи
Задача 10.3. Идентифицируйте соединение по спектру электронного удара
(рис. 10.3).
100
90
80
70
60
50
40
30
20
10
О
J,%
81
|l I|ll...i
141
I , J.uliilll
*
170
222
250
330
? '"" 1 ?*" 1—¦—? 1 ?" г
60 80 100 120 140 160 180 200 220 240 260 280 300 320 340
?? /?
m/z
60
61
62
63
64
79
80
81
82
85
86
89
90
91
92
1 93
/,% ?
19,8
75,7
100
44,9
2,73
51,0
16,1
50,8
15,8
2,13
1,53
2,37
15,4
18,9
2,37
6,90
m/z
104
106
110
111
112
115
116
117
118
119
140
141
142
143
144
1 164
/,% ?
5,47
5,29
3,91
6,90
3,63
3,42
7,69
7,69
7,22
4,19
6,45
46,7
11,6
44,9
5,90
1,121
m/z
165
166
167
169
170
171
172
173
220
221
222
223
224
225
248
1 249
/, % ?
3l28|
3,28
1,15
3,28
9,76
4,49
9,51
1,95
7,69
8,27
15,4
15,6
8,03
6,90
6,45
2,73
??/?
250
251
252
253
299
301
303
305
328
329
330
331
332
333
334
1 335
/, %
12,5
5,47
6,45
2,73
0,40
1,53
1,53
0,31
22,6
1,49
65,1
4,30
63,6
4,30
20,6
1,46
Рис. 10.3
Глава 10. Обобщенные задачи 353
Задача 10.4. Идентифицируйте соединение по спектру электронного удара
(рис. 10.4).
100
90
80
70
60
50
40
30
20
10
о
1,%
79
51
J _J
107
136
40 60 80 100 120
m /z
Рис. 10.4
140
m/z
39
43
50
51
57
58
63
65
77
78
79
I 80
/,% ?
4,33
2,41
2,68
9,10
2,68
1,47
1,69
1,69
21,1
4,97
85,9 !
5,66;
m/z
91
105
106
107
108
109
115
117
118
136
137
1 138
/,% 1
4,97
6,64
1,10
100
7,68
0,45
3,02
8,11
6,64
7,20
0,71
0,05
Задача 10.5. Идентифицируйте соединение по спектру электронного удара
(рис. 10.5).
100
90
80
70
60
50
40
30
20
10
0
1,%
172
65
39
93
0?
119
143
200
1 I I
20 40 60 80 100120140160180200
m/z
m/z
27
29
! 38
39
50
51
53
61
62
63
64
65
66
74
75
76
77
91
92
/,% ?
29,0
34,5
19,8
33,3
20,2
6,51
5,44
5,44
13,5
39,9
20,2
58,7
4,89
8,31
13,7
н,з
5,81
3,62
6,21
m/z
93
94
117
119
120
143
144
145
155
157
171
172
173
174
175
200
201
202
I 203
/, % 1
29,2
4,24
4,89
4,89
1,63
9,31
1,44
8,91
3,80
4,07
2,63
100 i
8,71 |
95,6
6,51
41,1
3,65
40,3
3,51
Рис. 10.5
12—1113
354 Глава 10. Обобщенные задачи
Задача 10.6. Идентифицируйте соединение по спектру электронного удара
(рис. 10.6).
100 ?
90
80 Ч
70 Ч
60 -|
50
40 -\
30
20
?? ?
о
20
1,%
91
117
5.1
65 77
¦ и .ty-i-li 1H...I. .U
105
L_J
136
4-
40 60 80 100 120 140
m/z
Рис. 10.6
m/z
31
39
41
1 45
46
50
51
52
57
63
65
66
/,% ?
16,3
12,0
5,24
6,18
3,00
5,24
14,0
4,03
3,00
6,51
18,0
2,13
m/z
11
78
79
80
89
91
92
93
103
104
105
I 106
/,% |
18,3
13,7
14,0
2,13
4,49
100
56,0
4,18
12,0
6,51
18,1
2,18
m/z
107
115
116
117
118
119
134
136
137
138
/,% |
2,13
6,00
6,58
78,0
70,0
6,34
2,13
26,0
2,59
0,16
Задача 10.7. Идентифицируйте изомерные 4-пропилфенол и фенилпропило-
вый эфир по спектрам электронного удара (рис. 10.7).
100 ?
90
80 -I
70 Ч
60 -I
50
40
30
20
10 Ч
1,%
77
-V hm-J Г^ 1 ШГ-
40 50 60 70 80
m /z
107
136
20
100 ?
90
80
70
60
50
40
30
20
10
0
30
?,%
90 100 110 120 130 140
94
-Ли—?-
,?? , L
136
20 30 40 50 60 70 80
m /z
90
100 110 120 130 140
Рис. 10.7
Глава 10. Обобщенные задачи 355
Задача 10.8.
(рис. 10.8).
Идентифицируйте соединение по спектру электронного удара
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
J
42
4_
с
Jl
>8
', '? ,'
86
100
I 115
I L
-L—,
20 40 60 80 100 120 140
m/z
Рис. 10.8
m/z
i 26
27
28
29
30
39
41
42
43
44
54
55
56
57
/,% 1
2,57
20,7
15,8
16,5
26,3
4,20
10,8
14,4
9,41
7,31
1,69
1,03
7,51
2,00
m/z
58
59
70
71
72
84
86
87
98
100
101
114
115
1 116
/,% 1
26,7
1,03
1,69
1,03
4,61
1,69
100
5,91
1,93 !
21,1
1,50
3,10
11,2
0,89
Задача 10.9. Идентифицируйте соединение по спектру электронного удара
(рис. 10.9).
100 ?
90-
80-
70-
60-
50-
40-
30-
20-
10-
0-
1,%
J
\к\
и
?
U
55
6
У
u,
7 8
73
3
116
»¦¦¦ ¦ ¦
"l
20 30 40 50 60 70 80 90 100110120
nVz
m/z
27
28
29
33
34
39
40
41
42
43
45
46
47
51
52
53
54
55
56
1 57
/, % 1
45,5
10,5
20,1
8,11
3,80
46,8
5,41
81,7
5,61
5,20
27,0
3,18
10,6
6,61
3,18
15,9
34,3
100
5,10
1,03
m/z
58
59
60
61
67
68
69
71
73
77
79
81
82
83
84
85
87
116
117
1 118
/,%
6,61
8,81
13,4
4,22
63,0
3,80
2,11
3,40
10,1
3,21
4,55
9,81
46,8
56,8
3,92
1,40
1,03
37,63
2,81
1,75
Рис. 10.9
356 Глава 10. Обобщенные задачи
Задача 10.10. Идентифицируйте соединение по спектру электронного удара
(рис. 10.10).
100 ?
90 -
80 -
70 -
60 -
50 -
40 -=
30 -
20 -
10 -
0 -
I
-Л
,%
55 |
39
ы
Ll|ll
J
63
Ll
90
?
»¦ , , , ¦¦¦
20 30 40 50 60 70 80 90 100110120130
m/z
Рис. 10.10
Задача 10.11. Идентифицируйте соединение по спектру электронного удара
(рис. 10.11).
m/z
25
26
27
28
29
35
37
38
39
40
41
42
43
45
48
49
50
L51
/,% ?
2,52
18,2
90,2
13,2
17,3
з,зз
5,38
6,21
28,3
2,32
11,7
1,60
26,5
2,30
1Д7
7,61
6,61
7,6l
m/z
53
54
55
56
61
62
63
64
65
75
77
90
91
92
93
126
128
| 130
Л% J
8,31
2,52
46,8
4,20
3,33
92,4
100
31,3
31,6
8,11
2,67
24,1
6,71
7,94
2,18
1,50
1,02
0,17
100
90
80
70
60
50
40
30
20
10
0
I,%
75
jLik L л ¦. L t
A 50
111
139
250
r
40 80 120 160
m/z
200 240
m/z
50
51
74
75
i 76
77
85
87
93
111
112
113
114
137
1 138
/,% 1
17,8
8,94
9,71
42,0
10,9
2,12
4,42
2,55
3,91
49,4
4,13
16,4
U2
2,23
2,45
m/z
139
140
141
142
150
151
152
215
217
250
251
252
253
254
1 255
/,%
100
7,84
32,5
2,54
1,24
1,31 I
3,45 !
4,74
1,54
10,8
1,54
7,20
1,03
1,20
0,17
Рис. 10.11
Глава 10. Обобщенные задачи 357
Задача 10.12. Идентифицируйте соединение по спектру электронного удара
(рис. 10.12). Малоинтенсивные пики ионов с m/z в области 199-203 обусловлены
примесями.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
Mi ?
109
jJUU
129
ш
1 IL
191
? I
К-
llL
40
80
120 160
m/z
200
m/z
27
28
i 37
38
39
49
51
61
63
73
75
82
83
84
85
| 87
/, % 1
9,31
3,70
4,20
4,80
9,61
9,01
2,70
4,80
1,70
5,50
6,41
2,50
12,7
1,50
8,31
1,40
m/z
93
95
107
108
109
110
111
112
113
117
119
121
127
129
131
1 153
/,% 1
10,9
11,0
38,3
1,20
100
3,30
41,6
1,50
6,81
12,0
11,8
3,90
11,9
15,9
4,00
2,40
m/z
155
188
189
190
191
192
193
194
195
224
226
227
228
229
230
1 232
/,% 1
2,90
2,70
50,1
6,21
82,9
4,90
38,6
1,60
5,20
2,70
5,40
0,18
3,61|
0,12j
1,02
0,10
Рис. 10.12
Задача 10.13. Идентифицируйте соединение по спектру электронного удара
(рис. 10.13). Объясните природу ионов с m/z 127 и 128.
100
90
80
70
60
50
40
30
20
10
0
? ?,%
1- . ¦- I- If ll_
175
tJ j
254
и ¦ ¦"¦ ¦
L
40 80 120 160 200 240 280
m/z
Рис. 10.13
m/z
47
49
59
61
j 79
81
103
105
127
128
158
160
162
173
174
175
176
/, % 1
4,2
1,4
6,0
2,0
6,1
6,0
5,4
5,3
6,1
6,4
1,0
1,9
0,9
48,0
1,06
80,0
1,76
m/z
111
178
179
217
219
221
223
252
253
254
255
256
257
258
259
260
1 261
/, % 1
37,3
0,82
5,32
1,62
3,78
2,70
0,54
37,5
0,83
100
2,20
91,7
2,02
33,3
0,73
4,17
0,09
358 Глава 10. Обобщенные задачи
Задача 10.14» Идентифицируйте соединение по спектру электронного удара
(рис. 10.14).
100
90
80
70
60
50
40
30
20
10
0
? ?,%
] 62
14
и
127
21
Ld
8
',' ,'
472
', , ,
50 100 150 200 250 300 350 400 450
m/z
Рис. 10.14
m/z
36
37
38
39
49
50
53
60
61
62
63
64
73
74
90
91
92
127
I 128
/,% ?
2,13
8,65
2,73
2,17
1,96
1,74
9,14
5,89
34,9
52,1
30,9
1,37
3,79
5,02
9,14
18,7
1,42
100
14,4
m/z
139
152
163
164
179
188
189
190
217
218
219
220
254
317
345
346
472
473
1 474
/, % 1
4,26
1,96
1,19
3,30
1,78
1,50
16,5
6,47
2,73
31,7
2,09
0,12
4,98
4,28
6,47
0,43
34,5
2,28
0,13
Задача 10.15. Идентифицируйте очень простое и распространенное
соединение по его спектру электронного удара (рис. 10.15).
00 СО О
о о о
? ? ?
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1>%,
4~
64
19
128
96
Ll^
L—r~
160
L-
L-
2
? 2?
224
щ
>б
L
40 80 120 160 200 240
m/z
m/z
32
64
65
66
96
98
128
129
130
132
160
161
162
164
I 192
/,% ?
1Д0
100
1,61
8,82
26,6
3,51
60,2
1,94
10,6
0,70
44,3
1,78
9,75
0,86
80,0
m/z
193
194
195
196
224
225
226
228
256
257
258
259
260
261
| 262
/,% 1
3,84
21,2
0,84
2,33
11,1
0,62
3,39
0,52
17,5
1,00
6,16
0,34
0,95
0,05
0,08
Рис. 10.15
Глава 10. Обобщенные задачи 359
Задача 10.16. Идентифицируйте соединение по спектру электронного удара
(рис. 10.16). Объясните образование иона с m/z 183.
100
90
80
70
60
50
40
30
20
10
о
1,%
262
1 83
108
??
80
120 160
m /?
200 240
Рис. 10.16
m/z
51
57
63
65
77
78
91
107
108
109
131
133
152
/, % ?
4Д2
2,18
3,54
1,03
2,89
1,32
2,12
11,6
32,0
2,90
3,17
2,46
9,961
m/z
153
157
170
181
182
183
184
185
186
261
262
263
1 264
?% 1
1,32
1,32
1,51
1,50
1,07
61,6
13,2
10,1
1,07
12,2
100
19,5
1,90
Задача 10.17.
(рис. 10.17).
Идентифицируйте соединение по спектру электронного удара
100 ?
80 А
70 А
604
504
404
30-4
20 А
10 А
0 -I
1Г%
29
JlL
40
66
94
122
, I.
60
80
100
120
m/z
m/z
27
28
29
30
41
43
48
65
, 66
/,% ?
35,6
10,0
100
2,12
1,48
2,54
2,80
1,94
56,0
m/z
68
76
93
94
95
96
122
123
I 124
/,%
2,66
1,27
2,56!
23,2 ?
0,70
1,13
5,16
0,27;
0,25
Рис. 10.17
360 Глава 10. Обобщенные задачи
Задача 10.18. Идентифицируйте соединение по спектру электронного удара
(рис. 10.18).
1 00
90
80
70
60
50
40
30
20
1 0
0
1,%
77 94
5 1
J_L
65
I ?
1 4 1
1 58
20 40 60 80 100120140160
m /z
Рис. 10.18
m/z
16
26
27
43
44
50
51
64
65
77
I 78
/,% 1
3,31
3,31
12,7
12,7
6,28
24,5
48,9
24,5
24,5
82,0
6,28
m/z
93
94
95
96
125
141
142
143
158
159
1 160
/,%
6,28
82,0
5,42
0,31
1,01
12,0
0,88
0,60
100
7,52
5,18
Задача 10.19.
(рис. 10.19).
Идентифицируйте соединение по спектру электронного удара
1 00
90
80
70
60
50
40
30
20
1 0
о
1,%
1 5
10 20
45
И*
79
6 1
30
4 0
? г*-
50 60
m /z
94
V 1
70 80 90 100
m/z
15
? 32
33
44
45
46
47
48
49
61
I 62
/, % 1
9,51
3,00
0,92
5,35
66,9
35,0
23,5
11,2
3,67
15,4
0,63
m/z
64
65
66
78
79
80
81
94
95
96
|__97
/,%
12,3
0,58
1,19
5,25
55,0
1,49
4,84
100
3,81
8,83
0,26
Рис. 10.19
Глава 10. Обобщенные задачи 361
Задача 10.20. Идентифицируйте соединение по спектру электронного удара
(рис. 10.20).
00
90
80
70
60
50
40
30
20
1 0
0
1,%
77
1 62
4
1 0 1
1 27
40 60 80 100 120 140 160
m /z Рис. 10.20
m/z
28
51
63
74
75
76
77
81
99
100
/,% ?
3,67
3,30
1,35
1,47
5,54
3,39
8,38
1,73
1,77
1,85
m/z
101
125
126
127
128
161
162
163
164
I 165
/,% |
5,82
2,88
19,9
36,4
5,19
1,03
100
10,8
32,9
3,60
Задача 10.21. Идентифицируйте соединение по спектру электронного удара
(рис. 10.21).
100
90
80 Ч
70 -|
60
50 ?
40
30
20 -\
10
1,%
63
50 |
л
и
1 77
Li
?_
92
и
1L
145
?—
120
m /z
_Щк
186
173
40
60
80
100
140
160
180
m/z
15
26
27
28
29
31
36
? 37
38
/,% ?
7,50
2,90
2,10
4,50
2,20
1,50
1,10
7,30
21,0
m/z
39
43
49
50
51
52
53
60
[61
/,% |
7,10
1,20
3,10
26,3
9,50
1,10
3,70
1,20
7,20
m/z
62
63
64
65
66
73
74
75
1 76
/,% 1
14,2
45,6
30,6
2,60
1,30
3,80
15,1
16,8
12,8
m/z
77
78
79
80
92
93
94
95
1 107
/,% 1
25,7
3,80
8,60
1,00
18,2
2,50
1,30
0,80
4,30
m/z
117
118
119
131
143
144
145
146
1 155
/,% 1
7,00
1,10
7,00
1,00
58,1
3,70
56,2
3,00
4,70
m/z
156
157
158
170
171
172
173
174
1 175
/,% 1
3,40
5,40
3,10
1,00
67,1
4,43
67,3
4,50
0,25
m/z
186
187
188
189
190
/,%
100
7,78
99,7
7,71
0,45
Рис. 10.21
362 Глава 10. Обобщенные задачи
Задача 10.22.
(рис. 10.22)
1 00
90
80
70
60
50
40
30
20
1 0
О
1,%
Идентифицируйте соединение по спектру электронного удара
30
40
50
50
70
96
60
m /z
70
80
90
1 00
Рис. 10.22
m/z
31
37
38
39
48
; 49
50
51
52
57
63
68
л% ?
1,07
1,48
2,23
3,27
3,01
1,07
7,31
3,73
0,90
2,94
2,23
2,23
m/z
69
70
74
75
76
77
81
94
95
96
97
I 98
/,% 1
2,94
17,7
2,60
5,21
1,85
1,48
0,90
1,85
7,11
100
6,51 I
0,18
Задача 10.23. Идентифицируйте соединение по спектру электронного удара
(рис. 10.23).
100 ?
90 -
80 -
70 -
60-
50 -
40-
30 -
20-
10-
о -J
I,%
Lj
с
J
2 63
.. «ilii.ii
79
,????, | ?.,,
160
128
115
Oxjll ll
u
? ¦ , -l
L
40
60
80
100
m/z
120
140
160
m/z
33
37
38
39
45
50
51
52
/,% 1
1,10
1,28
2,57
9,17
6,60
10,3
12,7
17,1
m/z
58
61
62
63
64
65
66
67
/,% 1
1,16
2,93
6,71
14,3
5,14
2,68
1,50
6,29
m/z
69
73
74
75
76
77
78
1 79
/,% 1
5,37
0,99
8,25
8,07
4,11
5,50
1,89
13,8
m/z
80
81
82
85
86
87
88
89
/,% 1
8,62
1,28
1,65
1,10
2,38
3,48
1,65
5,83
m/z
93
98
99
101
102
113
114
1 115
/,%
2,20
2,20
1,47
3,48
5,25
2,38
4,40
46,4
m/z
116
117
126
127
128
129
134
1 158
/,% 1
18,3
1,83
8,07
13,8
77,0
7,60
4,59
4,03
m/z
159
160
161
162
/,%
11,2
100
12,0
4,96
Рис. 10.23
Глава 10. Обобщенные задачи 363
Задача 10.24. Идентифицируйте соединение по спектру электронного удара
(рис. 10.24).
1 00
90
80
70
60
50
40
30
20
1 0
0
1,%
21 8
91
65
Jf IL i
40 80 120 1 60 200
?? /?
Рис. 10.24
m/z
38
39
41
50
51
61
62
63
64
65
66
/,%
1,44
5,51
1,44
3,42
2,78
1,03
2,47
5,51
1,89
17,4
1,03
m/z
74
89
90
91
92
109
127
217
218
219
I 220
/,%
1,03
5,51
3,90
42,7
3,42
1,44
2,20
3,08
100
7,84
0,24
Задача 10.25. Идентифицируйте соединение по спектру электронного удара
(рис. 10.25).
100
90
80
70
60
50
40
30
20
10
0
1,%
39
63
89
115
144
172
20
40
60
80
100
m/z
120
140
160
180
m/z
15
16
26
27
28
29
/,%
1,22
1,35
1,82
7,86
8,88
6,28
m/z
39
50
51
62
63
I 64
/,% ?
4,36
2,21
3,03
2,88
6,93
1,49
m/z
65
74
75
76
77
1 87
/,%
3,11
2,01
2,79
1,30
2,52
1,78
m/z
88
89
90
101
113
1 114
/,%
3,46
9,97
1,47
2,13
3,73
7,31
m/z
115
116
117
126
127
1 128
/, %
82,69
31,40
2,75
3,88
7,56
4,36
m/z
129
141
143
144
145
| 157
Л% 1
1,89
0,98
8,20
100
11,3
3,66
m/z
171
172
173
174
/,%
2,37
58,1
7,72
0,61
Рис. 10.25
364 Глава 10. Обобщенные задачи
Задача 10.26. Идентифицируйте соединение по спектру электронного удара
(рис. 10.26).
100 л
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
о J
1,%
" ¦ "
83 96
1 "' Г ¦¦¦ '"' ? ^?
192
165 ?
60 80 100 120 140 160 180 200
m/z
Рис. 10.26
m/z
50
51
63
70
74
75
82
83
87
88
94
95
96
97
115
I 139
i,% 1
1,00
1,40
2,10
2,80
1,30
1,50
3,20
9,91
1,20
1,10
2,10
5,91
9,51
1,60
0,90
1,30
m/z
150
151
152
163
164
165
166
176
187
188
189
190
191
192
193
1 194
/, % 1
1,10
0,90
1,40
2,40
1,40
8,21
1,30
2,40
2,10
2,10
16,6
9,81
30,9
100
16,4
1,28
Задача 10.27. Идентифицируйте соединение по спектру электронного удара
(рис. 10.27).
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
31
ц—?
-4-
9
¦ и ? A J l| к J I
9
I-
\J
168
uJ
20 40 60
80 100 120 140 160
m/z
Рис. 10.27
m/z
31
37
51
55
56
61
68
69
74
75
79
80
84
1 86
/, % 1
23,8
2,49
1,22
2,90
2,44
3,82
4,76
6,58
4,53
9,99
7,23
6,54
4,80
2,58
m/z
93
98
99
100
106
117
118
124
137
148
149
168
169
1 170
/,%
11,8
7,80
73,0
4,46
1Д2
11,0
10,2 !
2,91
16,5
4,62
10,2
100
6,04
0,20
Глава 10. Обобщенные задачи 365
Задача 10.28. Идентифицируйте соединение по спектру электронного удара
(рис. 10.28).
1 оо
90
80
70
60
50
40
30
20
10
0
1,%
1 36
1 1 7
?. , l и ¦ !¦
JU i
1 84
40
80
1 20
m /?
160 200
Рис. 10.28
m/z
31
69
86
92
93
98
105
106
117
118
/,% I
6,01
5,00
7,01
6,01
5,00
3,00
7,01
3,00
31,0
3,00
m/z
136
137
155
156
165
166
183
184
185
1 186
/,% 1
63,0
8,01
8,01
0,45
4,00
0,27
5,00
100
7,01
0,41
Задача 10.29. Идентифицируйте сложный эфир по спектру электронного
удара (рис. 10.29).
1 00
90
80
70
60
50
40
30
20
1 0
о
20
1,%
9 1
1 04
1 64
40
60
80 1 00
m /z
, ,
120 140
1 60
m/z
28
32
39
43
44
50
51
52
63
65
73
/,% ?
20,4
2,88
5,14
90,0
2,19
1,72
4,98
1,15
2,07
4,78
1,20
m/z
77
78
79
89
91
92
103
104
105
164
1 165
/,%
5,17
5,06
1,74
1,01
17,3
3,04
4,96
100
8,8
2,20
0,24
Рис. 10.29
366 Глава 10. Обобщенные задачи
Задача 10.30. Идентифицируйте сложный эфир по спектру электронного
удара (рис. 10.30).
00 СО О
о о о
I I 1
70 -
60 -
50 -
40 -
30 -
20 -
10 -
о J
1,%
J
L л1|1 ili..,
65 79 I
J
U
u
91
К
u
)8
I
164
""? ?
L_
20 40 60
80 100 120 140 160
m/z
Рис. 10.30
m/z
26
27
28
29
30
39
41
50
51
52
57
58
63
64
65
66
L 75
/,% 1
5,30
22,0
6,41
42,8
1,20
15,2
3,60
5,81
15,2
3,90
46,2
1,90
6,71
2,30
17,8
1,10
1,10
m/z
76
77
78
79
80
89
90
91
92
105
106
107
108
109
164
165
1 166
/,%
1,20
14,7
3,70
15,9
1,40
8,21
27,4
100
7,61
3,12
1,24
9,13
84,1
6,35
25,0
2,75
0,22
Задача 10.31. Идентифицируйте сложный эфир по спектру электронного
удара (рис. 10.31).
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
j
? 65
91
U-ц-
164
20 40 60 80 100 120 140 160
m/z
Рис. 10.31
m/z
26
27
28
29
30
31
38
39
40
41
42
43
45
46
50
1 51
/, % 1
2,50
11,4
2,20
39,0
2,10
14,6
1,50
9,11
0,90
2,70
1,00
5,20
6,51
2,50
1,70
3,90
m/z
52
62
63
64
65
77
89
90
91
92
105
119
149
164
165
1 166
/,%
1,10
1,20
3,90
1,30
11,5
1,30
2,90
2,10
100
8,03
2,70
6,28 1
1,34
15,0
1,65
0,13
Глава 10. Обобщенные задачи 367
Задача 10.32. Идентифицируйте сложный эфир по спектру электронного
удара (рис. 10.32).
1 00
90
80
70
60
50
40
30
20
1 0
о
1,%
77
59
, .,1,1, il.
1 05
1 23
1 64
Ш . , , , ?. I
20 40 60 80 100 120 140 160
m /z
Рис. 10.32
m/z
27
39
41
I 42
43
50
51
52
59
76
77
78
/,% ?
3,90
3,10
5,71
2,10
11,3
4,06
11,8
1,00
18,9
2,50
31,6
3,70
m/z
79
104
105
106
122
123
124
146
149
164
165
J 166
/, % 1
2,60
1,10
100
8,27
20,4
30,8
2,40
1,10
3,78
15,3
1,65 !
0,13
Задача 10.33.
(рис. 10.33).
Идентифицируйте соединение по спектру электронного удара
1 00
90
80
70
60
50
40
30
2 0
1 0
о
1,%
20
JAr-
30
J.
60
40
50 60
m /z
7 3
70
—?—
80
90
m/z
18
J 26
27
28
29
38
39
40
41
42
43
/, % ?
?,??
2,00
16,4
4,40
9,31
1,60
8,01
1,30
16,8
16,0
14,8
m/z
45
55
60
61
62
71
73
74
87
88
I 89
/,%
12,9
5,50
100
2,20
0,40
2,30
30,0
1,50
1,80
3,12
0,14
Рис. 10.33
368 Глава 10. Обобщенные задачи
Задача 10.34. Идентифицируйте сложный эфир с молекулярной массой 116
по спектру электронного удара (рис. 10.34).
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
-J
L
43
J
?? . il
57
75
ll, г-Л
87
j
20 30 40 50 60 70 80 90 100110120
m /z Рис. 10.34
m/z
15
26
27
28
29
30
31
39
41
42
43
/,% |
1,56
3,48
27,3
8,63
60,9
2,75
8,45
5,47
14,8
9,99
20,4
m/z
55
56
57
58
59
74
75
76
86
87
| 116
/,%
0,96
1,65
100
3,38
4,99
3,58
52,1
1,77
1,36
14,2
1,62
Задача 10.35. На рисунке 10.35, а-в представлены масс-спектры
электронного удара изомерных метилциклогексанонов. Установите, какой спектр
соответствует какому изомеру.
110 12 0
Рис. 10.35
Глава 10. Обобщенные задачи 369
Задача 10.36. Идентифицируйте соединение с молекулярной
140,1201 Да, масс-спектр ЭУ которого представлен на рис. 10.36.
массой
100 ?
90 А
80 А
70 А
во А
50 А
40 А
зо А
20 А
ю -I
0 -
1,%
| 55
L
и
7
0 {
$3
98
111
II. J- ¦
40 50 60 70 80 90 100 110 120
m/z
Рис. 10.36
m/z
41
42
43
! 53
? 54
55
56
57
67
68
71
69
[ 70
/,% ?
42,3
22,7
14,5
6,32
9,87
37,7
14,9
3,24
6,54
3,29
2,22
13,1
22,4
m/z
79
81
82
83
84
95
96
97
98
99
111
112
I 140
/,% ?
2,89
5,44
1,37
18,8
5,62
1,02
3,59
13,9
100
6,52
9,14
0,70
2,96
Задача 10.37. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.37.
100
90
80
70
60
50
40
30
20
10
о
1,%
40
,13 5
108
лЛ.
ill.., h.
AL·
1 1 5
60
80 100
m /?
120 140
m/z
31
39
50
51
57
61
62
63
68
75
81
83
88
/,% ?
2,20
4,18
5,28
3,66
8,57
2,16
3,46
3,88
3,20
6,65
4,80
5,21
2,36
m/z
89
95
107
108
109
115
116
132
134
135
136
137
/,%
2,14
4,14
40,7
53,8
9,21
20,1
2,28
1,40
43,5
100
8,83
0,33
Рис. 10.37
370 Глава 10. Обобщенные задачи
Задача 10.38. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.38.
100
90
80
70
60
50
40
30
20
10
0
1,%
41
54
I. ...I
69
h U
82
96
1 10
20 40 60 80 100 120
m /z
Рис. 10.38
m/z
39
40
41
42
43
52
53
54
55
56
57
67
68
/,% ?
24,3
5,80
62,9
10,7
23,5
3,60
6,40
40,6
34,9
7,50
9,60
4,60
8,60
m/z
69
70
79
82
83
84
93
96
97
98
ПО
124
I 125
/,%
35,5
4,20
4,30
100
38,2
3,20 I
2,10 !
45,3
19,8
1,50
10,2
2,20
2,14
Задача 10.39. Идентифицируйте соединение по масс-спектру электронного
удара, представленному на рис. 10.39.
100
90
80
70
60
50
40
30
20
10
0
1,%
52
1 17
-«г
JL
41
182
20 40 60 80
100 120 140 160 180
m /z
m/z
39
50
52
53
54
65
66
90
91
115
/,% ?
2,33
3,16
62,0
7,06
1,76
U3
1,19
3,59
4,11
3,07
m/z
117
118
119
155
156
180
181
182
183
| 184
/,%
59,4
9,36
2,34 !
1,12
2,94
5,17
1,59
100
22,6
4,64
Рис. 10.39
Глава 10. Обобщенные задачи 371
Задача 10.40· Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.40.
100
90
80
70
60
50
40
30
20
10
0
1,%
75
51
L1
и
102
181
40 60 80 100 120 140 160 180
m/z
Рис. 10.40
m/z
37
38
39
48
49
50
51
52
61
62
63
64
73
74
75
1 76
/,% 1
15,2
16,6
и,з
2,32
12,0
47,0
54,4
8,99
5,21
6,19
5,95
2,47
6,48
20,4
73,9
35,3
m/z
11
79
81
91
92
99
100
101
102
103
104
181
182
183
184
1 185
/,% 1
3,10
3,17
2,98
ИЛ
11,1
8,13
8,26
5,61
100
8,42
1,75
87,4
7,08
87,1
7,12
0,21
Задача 10.41. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.41.
100
90
80
70
60
50
40
30
20
10
о
1,%
Lil, .ill.
40
60
74
¦llli ii . ?.? nil
T*
80
109
133
100
I^-UU
120
m/z
145
161
191
ild_
140
160
180
20Q
m/z
46
47
49
50
61
62
63
/,% 1
12,0
4,37
9,82
24,3
8,83
10,2
13,2
m/z
64
72
73
74
75
76
1 85
/,%
1,87
3,43
7,84
84,8
66,7
5,41
7,27
m/z
86
97
99
107
108
109
[ 110
/,% 1
5,97
4,31
6,34
3,90
4,72
100
6,78
m/z
111
112
119
121
133
134
1 135
/,%
33,1
2,21
2,23
1,77
80,3
4,93
51,1
m/z
136
137
145
146
147
148
J 149
/, % 1
3,01
8,78
52,9
3,69
35,3
2,42
5,97
m/z
161
162
163
164
165
175
| 177
/,% |
63,7
4,63
42,4
2,91
7,17
4,78
3,12
m/z
191
192
193
194
195
196
/,%
37,4
2,65
25,0
1,75
4,12
0,31
Рис. 10.41
372 Глава 10. Обобщенные задачи
Задача 10.42. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.42.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
¦?? ¦¦ 111
51
Jli, til
Hi il illlli
1Qi
124
—? 1 1
20 40 60 80 100 120 140 160 180
m /z
Рис. 10.42
m/z
37
38
39
50
51
52
69
70
73
74
75
76
/,% 1
3,20
4,40
3,30
18,5
21,7
1,80
2,40
9,71
2,00
7,91
8,91
8,51
m/z
77
78
95
96
97
104
105
106
123
124
125
1 126
/, %
33,7
2,20 !
7,81 !
75,4
5,00
1,30
31,2
2,40
17,7
100
7,61
0,45
Задача 10.43. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.43.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
I,
%
А
]
L
J
3
55 |
74
8
7
Ц 1 1 1
20 30 40 50 60 70 80 90 100110120
m /z
m/z
27
28
29
30
39
40
41
? 42
43
44
45
53
55
/,% ?
29,3
12,9
23,8
3,20
21,7
3,40
3,10
9,91
51,0
2,20
16,9
2,60
13,5
m/z
56
57
59
60
69
70
71
73
74
75
87
88
? 116
/,%
8,01
2,60
2,00
7,11
3,30
1,90
10,3
17,3
100
3,50
17,1
1,00
0,11
Рис. 10.43
Глава 10. Обобщенные задачи 373
Задача 10.44. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.44.
1 00
90
80
70
60
50
40
30
20
1 0
о
- 1,%
J
60
87
"Г " 1—¦—" ? 'г 1 1
40 50 60 70 80 90 100 110 120
m /?
Рис. 10.44
т/г
38
39
40
41
42
43
44
45
55
56
57
/,% 1
0,72
7,20
1,21
21,1
6,04
15,3
0,95
14,8
9,54
15,7
21,4 |
m/z
58
59
60
61
69
70
73
83
87
88
1 116
J,%
1,74
4,11
100
14,0
2,67
0,94
4,17
1,67
15,5
0,96
0,13
Задача 10.45. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.45.
100
90
80
70
60
50
40
30
20
10
о
1,%
57
123
+
178
Vlllh ' . ' '—l·
-?—
40 60 80 100 120 140 160 180
m /z
m/z
39
40
41
42
55
56
57
58
71
81
85
105
109
/,% I
10,0
50,1
3,50
5,54
12,5
18,0
100
4,58
2,37
5,39
2,54
2,87
2,03
m/z
119
121
122
123
124
125
136
145
149
161
178
179
1 180
/,%
2,87
17,8
2,50 ^
68,9
3,58
3,44
2,51
1,30
4,80
3,33
10,0
0,96
0,50
Рис. 10.45
374 Глава 10. Обобщенные задачи
Задача 10.46. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.46.
100 ? 1>%
90 -I
80 -|
70
60 ?
50
40 ?
30
20
10
1
30
41
liil
40
4^
57
63
LuL
92
106
? ?
131
148
50
60
70
80 90
m/z
100 110 120 130 140 150
m/z
26
27
28
29
30
32
34
37
д% I
5,93
41,7
13,9
63,6
2,71
2,41
2,27
2,74
m/z
38
39
40
41
42
43
44
1 45
/, % 1
3,19
25,7
5,74
95,9
9,44
93,8
5,14
6,21
m/z
46
47
48
50
53
55
56
1 57
/, % 1
3,59
7,72
2,18
4,22
3,82
20,5
21,8
67,6
m/z
58
59
61
63
64
65
66
1 73
/, % 1
4,13
2,54
5,32
42,4
11,2
2,76
1,29
2,57
m/z
75
76
77
89
90
91
92
1 93
/, % 1
3,18
5,41
5,59
37,7
5,29
7,44
100
4,46
m/z
94
103
105
106
107
108
131
1 132
/,% |
4,62
2,44
3,12
14,5
0,75
0,72
21,0
4,13
m/z
133
148
149
150
/, %
1,01
27,7
2,35
1,33
Рис. 10.46
Задача 10.47. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.47.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
1
¦??. ¦!,»¦
,???. .,.?
77
1,
Jl lll| .¦¦¦ , .
106
I
I ¦ ill.
135
?
L—j
20 40 60 80 100 120 140
Рис. 10.47
m/z
27
29
! 39
41
51
52
53
54
63
65
66
11
78
/,% 1
7,87
4,18
5,74
2,32
3,62
3,78
2,72
1,61
2,29
4,17
2,54
11,3
4,67
m/z
79
80
91
92
93
104
106
107
118
119
135
136
1 137
/, %
6,54
1,93
1,93
1,64
2,17
2,42
100
8,12
1,20
1,20
15,6
1,61
0,07
Глава 10. Обобщенные задачи 375
Задача 10.48. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.48.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
51
?.¦ I J
La
63
.¦'!""¦, ????
77
IL—
89
,,
1?±-
107
J
и-±т г
136
¦ , ?..
153
?-
J
40
50
60
70
80
90
100
m/z
110 120 130 140 150
160
m/z
26
27
28
29
30
31
32
37
/, %
1,92
6,84
15,3
14,0
29,5
12,8
6,84
3,02
m/z
38
39
40
41
46
49
50
? 51
/, % ?
6,37
17,0
3,39
8,28
12,7
2,24
25,1
36,5
??/?
52
53
55
57
61
62
63
? 64
?% ?
10,2
10,1
3,78
6,37
3,39
7,21
17,0
4,56
??/?
65
66
67
73
74
75
76
[ 77
/,%
4,87
4,56
4,10
2,64
10,3
7,21
9,18
100
??/?
78
79
80
86
87
89
90
? 91
/, % ?
33,5
25,4
1,92
1,92
1,44
36,2
5,16
2,64
??/?
92
94
95
105
106
107
108
? 109
/,%
1,92
5,20
1,92
11,4
16,2
52,2
15,1
1,20
??/?
124
125
136
137
152
153
154
? 155
/,%
2,90
1,02
13,6
2,61
3,90
30,0
2,43
0,26
Рис. 10.48
Задача 10.49. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.49.
1,%
100
90
80
70
60
50
40
30
20
10
0
Jll
40
77 92
64
ч-
123
X
153
60
80 100 120 140 160
m/z
Рис. 10.49
m/z
31
38
39
41
50
51
52
53
61
62
63
64
65
74
75
76
/,% 1
2,09
7,59
3,79
5,79
12,6
7,29
4,19
3,50
2,09
5,50
21,0
26,7
3,19
5,50
5,39
5,89]
m/z
11
78
79
80
92
93
95
96
107
108
123
124
137
153
154
155
/,%
46,5
4,39
2,39
2,79
46,5
3,39
10,3
0,66
10,0
0,78
28,6
2,32
4,39
100
8,24
0,91
376 Глава 10. Обобщенные задачи
Задача 10.50. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.50.
100
90
80
70
60
50
40
30
20
10
0
1,%
77
51
и- ?-?-? I"' Mil- J
107
40
60 80
m/z
122
100 120
m/z
27
29
30
38
39
43
45
50
51
52
53
61
63
64
65
74
75
/,% 1
4,23
13,2
2,12
3,39
9,80
5,64
2,87
7,92
21,0
6,51
3,39
5,64
5,64
3,39
6,13
2,12
2,12
m/z
77
78
79
80
91
92
93
94
104
105
106
107
108
121
122
123
| 124
/,% 1
67,5
13,0
16,5
1,90
3,39
5,64
5,64
2,12
9,80
9,80
7,92
46,5
3,44
8,87
100
8,51
0,26
Рис. 10.50
Задача 10.51. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.51.
100
90
80
70
60
50
40
30
20
10
0
1,%
44
-?
40
59
j tilL , uil
87
130
U Lb.
? ?
60 80 100 120
m/z
m/z
27
28
29
30
31
32
41
42
43
44
45
56
57
58
59
/,% 1
20,6
14,2
22,2
23,8
15,8
2,42
11,9
20,6
17,4
27,0
5,67
10,3
5,87
28,5
57,1
m/z
60
70
71
72
73
87
88
101
102
115
116
117
130
131
1 132
/,%
3,56
2,42
3,38
8,46
14,3
100
5,26
20,6
1,30
22,2
1,64
0,18
55,6
4,73
0,25
Рис. 10.51
Глава 10. Обобщенные задачи 377
Задача 10.52· Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.52.
100
90
80
70
60
50
40
30
20
10
0
1,%
119
103
78 91
51
63
JH Ы
I I
132
160
40 60 80 100 120 140 160
m/z
Рис. 10.52
m/z
39
41
50
51
52
53
61
62
63
64
65
74
75
76
77
78
/,% 1
22,0
5,24
13,7
28,4
13,4
4,17
3,76
8,28
21,5
5,24
16,9
6,07
4,17
5,24
27,0
43,8
m/z
79
89
90
91
92
102
103
104
105
119
120
132
133
160
161
I 162
/,% I
4,17
10,1
3,23
40,4
4,17
10,1
56,6
31,7
3,72
75,1
6,61!
12,4
1,24
100
10,7
0,65
Задача 10.53. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.53. Точная молекулярная масса соединения
составляет 115,0997 Да.
100
90
80
70
60
50
40
30
20
10
0
1,%
57
А
?1'1 -?»'- г'11111-! "?'» ?
73
100
jiL
30 40 50 60 70 80 90 100 110 120
m/z
Рис. 10.53
m/z
27
28
29
30
31
33
38
39
40
41
42
43
44
L54
/,% ?
7,83
7,83
6,21
1,84
2,33
3,27
1,24
14,1
3,88
33,6
44,4
19,5
2,10
8,53
m/z
55
56
57
58
69
73
74
82
83
84
98
100
101
1 115
/,%
7,41
11,1
29,6
7,41
2,93
100
3,72
1,84
4,03
4,68
2,93
10,2
0,62
1,00
378 Глава 10. Обобщенные задачи
Задача 10.54. Идентифицируйте соединение, масс-спектр ЭУ которого
представлен на рис. 10.54.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
(
.. .?.. 1, .1.1
35
^_^J
78
L—~_
91
106
lii. ..тк I
120
li
J
130
111,,, , u
148
{ ^
40 50 60 70 80 90 100 110 120 130 140 150 160 170
m/z
m/z
42
50
51
52
57
62
63
/,% 1
2,13
2,67
7,24
2,13
10,5
2,54
10,5
m/z
64
65
66
74
75
76
1 77
/,% 1
3,28
20,5
2,84
1,88
2,13
5,03
21,0
m/z
78
79
89
90
91
92
1 93
/,% 1
28,4
2,84
18,8
10,7
53,0
50,5
10,5
m/z
94
102
103
104
105
106
1 108
/,% 1
2,84
3,77
10,2
11,0
9,45
15,8
5,03
m/z
115
116
117
118
119
120
| 121
/,%
19,8
6,46
12,4
13,5
4,50
26,5
3,77
m/z
130
131
132
133
134
135
136
/,% 1
24,5
7,24
5,59
8,28
1Л2
1,29
2,841
m/z
146
148
149
150
165
166
/,%
2,13
100
10,3
0,64
5,02
0,52
Рис. 10.54
Задача 10.55. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.55.
100 ?
90 -
80 -
70 -
60 -
50 -
40 -
30 -
20 -
10 -
0 -
1,%
jU.
77
i
ч-*-
115
101
¦4-i
Ч-*
127
?—н—h—
1
L
40 60 80
100 120
m/z
140 160 180
Рис. 10.55
m/z
46
50
51
52
61
62
63
64
65
74
75
76
77
86
87
89
/,%
3,00
9,81
13,9
1,44
1,44
4,69
10,6
1,09
0,95
10,2
13,6
6,53
18,8 !
2,14
3,61
1,93
m/z
98
99
100
101
102
114
115
116
126
127
128
143
157
173
174
1 175
/, % 1
2,71
1,94
1,97
10,8
0,95
1,56
35,6
3,53
25,5
100
10,7
2,70
1,28
74,3
8,47
0,94
Глава 10. Обобщенные задачи 379
Задача 10.56. Идентифицируйте соединение, масс-спектр электронного
удара которого представлен на рис. 10.56.
100
90
80
70
60
50
40
30
20
10
о
1,%
40
51
77
_ulli
97
125
_JL_
60
80
100
120 140
m/z
160
180
218
200
220
m/z
27
38
39
45
50
51
52
l 53
/,% ?
3,48
0,96
5,88
1,50
9,51
36,1
2,31
2,44
m/z
63
65
69
74
75
76
77
]_78
/, % 1
1,63
5,34
1,12
2,48
2,39
3,25
42,5
3,98
m/z
81
97
98
99
109
125
126
1 127
/,% 1
2,19
18,2
1,15
0,84
1,74
100
7,49
4,78
m/z
151
152
153
154
217
218
219
| 220
/, %
1,28
4,56
3,97
1,44
1,19
25,2
3,63
1,44
Рис. 10.56
Задача 10.57. Идентифицируйте соединение по масс-спектру электронного
удара, представленному на рис. 10.57. Учтите, что кластер ионов с максимумом
m/z 439 обусловлен ионом [М—Ph]+.
100
90
80
70
60
50
40
30
20
10
0
1,%
-I
J
J
" ^ 1 * 4"—
г-Х }
208
2(
J
55
* . f
4
, J
39
\ 1 1
40
80
120 160
200
240 280
m /z
320 360 400 440 480 520
Рис. 10.57
Глава 11
РЕШЕНИЯ ЗАДАЧ
В данном разделе приводятся решения не всех задач. В некоторых случаях
решения очень просты. Для задач со многими вариантами разбирается ход
решения для 1-2 примеров.
Задача 1. Можно рассмотреть только наиболее интенсивные пики в спектре.
Это пики ионов с массами 60, 45, 43, 28 и 15. Самый тяжелый ион отражает
массу всей молекулы, а остальные — массы наиболее устойчивых фрагментов.
Таким образом, ион 45 образуется при потере молекулярным ионом 15 а. е. м., а
ион 43 — 17 а. е. м. Учитывая, что отщепляться могут реальные радикалы и
молекулы, вероятно, в первом случае отщепляется метильная группа, а во втором —
гидроксильная. Следовательно, органическая молекула с массой 60 Да имеет в
составе с одной стороны группу СНз, а с другой — группу ОН. Общая масса этих
фрагментов 32 Да. Вычитая 32 из 60, получаем массу неизвестного фрагмента,
который, вероятно, находится между этими группами. Его масса равна 28 Да.
Пик с такой массой тоже есть в спектре. Перебирая с использованием таблицы
Менделеева возможные варианты, получаем СО, С2Н4, NCH2 и N2. Зная брутто-
формулу соединения, получаем только один вариант: СО. Теперь соединяем три
фрагмента в единое целое.
СНз~СО—ОН, т. е. уксусная кислота
Задача 3.1. в) Основной первичный процесс распада аминов связан с
расщеплением С—С-связи между а- и ^-атомами углерода любой из алкильных
групп и сохранением заряда на атоме азота.
Как следует из представленной схемы, М+" emo/7-пентилизопропилпропил-
амина может отщеплять этильный радикал пропильной группы, один из
метальных радикалов изопропильной группы, метальный или пропильный радикалы
emop-пентильной группы (схема 11.1). По правилу выброса максимального ал-
кила, наиболее интенсивный пик должен принадлежать иону [? — ????]", менее
интенсивный — иону [М—С2Н5]4", а минимальный —иону [М—СНз]+. Следует,
однако, учесть, что вероятность выброса метильного радикала увеличивается,
поскольку могут отщепиться три разные метальные группы. Это приводит к
увеличению интенсивности пика соответствующего иона.
Глава И. Решения задач 381
СН3 +
С3Н7-СТ-Й-СН2-С2Н5
СН
СН3'
Т-СНз
снз+ и. сн3;
CH=N~CH2-C2H5 « 3 С3Н7- СН-Й-СН2-С2Н5 -^^ C3H7-CH=N-CH2-C2H5
сн сн гн
снз" ^сн3 шз'-снз сн3>н-сн3
?
|-С2Н5
снз
C3H7-CH-N=CH2
сн
сн3- -сн3
Схема 11.1
Задача 3.1. д) Основной первичный процесс распада спиртов обусловлен
расщеплением С—С-связи между атомом углерода, связанным с гидроксиль-
ной группой, и атомом углерода любой из алкильных групп. Как следует из
представленной схемы 11.2, М+* З-метилгептанола-3 может элиминировать
метальную, этильную или бутильную группы. Максимальную интенсивность будет
иметь пик [М — С4Н9]4", меньшую —пик [М—С2Н5]+, а минимальную — пик иона
[М—СН3]+. Поскольку все радикалы тривиальны, можно попытаться рассчитать
интенсивности первичных ионов, учитывая, однако, что интенсивность пика
иона [М —СН3]+ завышена. Принимая интенсивность пика иона [М —С4Н9]4"
(m/z 73) за 100%, получаем, что интенсивность пика иона [М —C2Hs]+ (m/z 101)
составит примерно 72% G3-^-101 ? 100%), а интенсивность пика иона [М —СН3]+
(m/z 115)-примерно 63% G3 -г 115 ? 100%).
ОН ОН
I
он
с-с2н5 ^?1с4н9-<|:-с2Н5-^^ C4H9-9
сн3 сн3 сн3
?
1-СНз
он
II
C4H9-C-QH5
Схема 11.2
Задача 3.2. а) Катионный и радикальный центры на атоме серы диэтил-
сульфида приведут к двум типам реакций. Благодаря тенденции к спариванию
электрона важную роль будет играть ?-распад с отщеплением атома водорода
382 Глава 11. Решения задач
или метильного радикала (схема 11.3). По правилу выброса максимального ал-
кила интенсивность пика иона [М —Н]+ будет значительно ниже интенсивности
пика [М —СН3]+. Катионный центр инициирует гетеролитический распад по
связи С—S, причем заряд может сохраняться и на углеводородном, и на
серосодержащем фрагменте. Перегруппировочный процесс через четырехчленное
переходное состояние с выбросом молекулы этилена может быть инициирован и
радикальным, и катионным центром.
CH3-CH2-S=:CH2 ^^-СНз-СНг-З-СНг-СНз-^^ CH3-CH2-S==CH-CH3
-C2H4
^-0>?5·
-ад
-OftS*
-С2Н4
HS=CH2 CH3-CH2-? CH3-CH2-SH СН3-СН2 н?=СН-СН3
Схема 11.3
Задача 3.2. д) Радикальный центр, расположенный на атоме хлора
(схема 11.4), вызовет ?-распад с образованием катиона СН2С1+. Однако хлор мало
активен в таких реакциях. Как следствие, интенсивность пика этого иона будет
низка. Катионный центр на атоме хлора инициирует притяжение электронной
пары связи С—С1, гетероциклический разрыв этой связи и образование алкиль-
ного иона. Высокая активность атомов галогенов в подобных реакциях приведет
к высокой интенсивности пика этого иона. Перегруппировочный процесс с
переносом атома водорода и отщеплением молекулы хлористого водорода приведет к
образованию нечетноэлектронного алкенового иона.
С4Н9
СН2=С1 СН_ СН2_С1 —
СНз^ ,
-на
С4Н8
Схема 11.4
Задача 3.3. а) Все альдегиды с незамещенным ?-углеродным атомом в
результате перегруппировки Мак-Лафферти распадаются с образованием иона
с m/z 44 (схема 11.5).
СН
\*
сн
-- о
сщ
с
сн2^ \н
сн,
V
сн)
сн2
+
но
+
но
-сн3снсн2
W \н
сщ"
\
но
п*
?
ch^Si
Схема 11.5
Глава 11. Решения задач 383
Задача 3.3. д) При распаде З-метилпентанона-2 в результате
перегруппировки Мак-Лафферти (схема 11.6) образуется катион-радикал енольной формы
метилэтилкетона и молекула этилена. Обратный вариант с образованием катион-
радикала пропилена и отщеплением енольной формы ацетона практически не
реализуется, поскольку ЭИ енола ниже чем ЭИ этилена на ~1 эВ.
™2 о ~н' > (СН2 но
[2 но -сн2сн2
НО
но
п*
II I >ч II с с
сн2 с сщ> с ^ чаь гн^ Vh
СН^ \СН3 СН^ \СН3 I СНз Г1 СТэ
/ J /
СН3 СН3 СТз Шз
СН2 Н? ч Г СЛО
СН2' С
"сн-^ \сн
^^-—-* сн2—сн2
3
сн3
Схема 11.6
Задача 3.4. в) Для протекания перегруппировки по механизму
Мак-Лафферти необходима миграция атома водорода от 7-атома углерода. В молекуле
гептен-5-она-2 7-атом углерода имеет sp2-гибридизацию. Связь С—? в этом
случае значительно прочнее, чем для ^р3-гибридизованного углерода. В результате
конкурентоспособность перегруппировки резко падает (схема 11.7).
СНз-СН /~^ СН3-СН —? +
9 О >Н' , \J НО -СН3СН=С=СН2 НО I
И ·" I^N II ¦* II *« »· I
СН2 с аУ С С /С
*caf 4?3 ^снГчсн3 сн2^ \сн3 сн2^ чсн3
Схема 11.7
Задача 4.1. Во всех случаях, поскольку рассматриваемый ион заведомо
имеет максимальную массу, а его элементного состава не приводится, можно
рассмотреть только выбросы нейтральных частиц.
а) Представленные фрагменты могут быть обусловлены выбросами
следующих частиц: 129 - [М-1] - [М-Н]+, 115 - [М-15] - [М-СН3]+,
113-[М-17]-[М-ОН]+, 102-[М-28]-[М-СО]+\ или [M-N2]+\
или [М-С2Н4]+·, 101-[М-29]--[М-НСО]+, или [М-С2Н5]+, или
[M-H,-N2]+, 97-[M-33]-[M-SH]+ или [М-СН3,-Н20]+. Следовательно,
ион 130 может быть молекулярным.
384 Глава 11. Решения задач
в) Ионы с массами 78 и 77 образоваться из иона 100 не могут, так как
отщепление 22 и 23 а. е. м. невозможно. Следовательно, ион с массой 100 не
молекулярный.
Задача 4.2. В отличие от предыдущей задачи, знание брутто-формулы
позволяет наряду с составом нейтральных частиц установить также степень
ненасыщенности иона и проверить, является ли он нечетноэлектронным.
а) Рассчитываем прежде всего степень ненасыщенности. R = 12—7,5+1 = 5,5.
Следовательно, ион четноэлектронный и молекулярным быть не может. Можно
проверить выбросы нейтральных частиц. Однако ответ на вопрос уже получен.
в) Рассчитываем степень ненасыщенности. i? = 8 — 6 + 1=3. Ион нечетно-
электронный, т. е. этот критерий позволяет считать его молекулярным. Теперь
необходимо проверить выбросы нейтральных частиц. CgHn — [? —?]+, С7Н9 —
[? —СНз]+, СбНп — [М—24] -? Такой процесс невозможен, следовательно, ион
CgHi2 молекулярным быть не может.
л) Рассчитываем степень ненасыщенности. R = 8 — 6,5 + 1,5 + 1 = 4.
Следовательно, по этому критерию наиболее тяжелый ион в последовательности
(C8H13N3O2) может быть молекулярным. Массы всех остальных ионов ниже,
чем у вероятного М+', и не попадают в запрещенные интервалы. Однако
ион C7H10NO3 содержит три атома кислорода, т. е. по четвертому критерию
приведенный в последовательности ион с максимальной массой не является
молекулярным.
Задача 4.3. а) Для расчета пятого пика (М+8) изотопной серии в случае
соединения с пятью атомами хлора необходимо вычислить пятый член формулы
биноминального разложения для а = 1; Ъ = 0,325; ? = 5.
п(п - 1)(и - 2)(л - 3)an~4b4 5x4x3x2x1^ 0,3254 . л„
4! = 24 = 0'°56
Таким образом, интенсивность пика М+8 будет составлять 5,6% от
интенсивности М.
Задача 4.4. г) Простейший расчет интенсивностей для мультиплета
молекулярного иона соединения с двумя атомами хлора и тремя атомами брома можно
провести следующим образом.
Пять атомов (А+2)-элементов дадут 6 сигналов в мультиплете.
Два атома хлора C + IJ дадут триплет с соотношением 9:6:1.
Три атома брома A + IK дадут квартет с соотношением 1:3:3:1.
Глава И. Решения задач 385
Перемножая матрично одно на другое, получаем:
(9:б:1)хA:3:3:1)= 9 :6:1
27 : 18 :3
27: 18 : 3
9: 6: 1
9 : 33 : 46 : 30 : 9 : 1
Таким образом, соотношение интенсивностей пиков ионов ?, ?+2, ?+4,
М+6, М+8 и М+10 будет 9 : 33 : 46 : 30 : 9 : 1.
Задача 4.5. Для решения задач удобно использовать данные табл. 4.2, либо
механически делить интенсивность пика М+1 на 1,1 и проверять полученное
число атомов углерода по интенсивности пика М+2 (табл. 4.2). При этом
необходимо помнить об азотном правиле и о возможном присутствии (А+2)-элемен-
тов. Если интенсивность М+* не 100%, необходимо осуществить нормирование
на 100%. Не стоит ожидать также абсолютного совпадения рассчитанных и
реальных интенсивностей пиков в спектре.
а) Для определения числа атомов углерода делим интенсивность пика иона
М+1 на 1,1 G,9 : 1,1 « 7). Для соединения с семью атомами углерода
интенсивность пика М+2 должна составить 0,25% (табл. 4.2), что примерно
соответствует значению, приведенному в задаче. Следовательно, в молекуле
соединения 7 атомов углерода.
в) Присутствие в спектре пика иона М+2 (m/z 136) с интенсивностью 33%
от ? (m/z 134) означает, что соединение содержит один атом хлора (табл. 4.1).
Интенсивность пика М+1 делим на 1,1. Получаем ? = 6. Наличие атома хлора
маскирует долю интенсивности пика М+2, обусловленную одновременным
присутствием двух атомов 13С в молекуле. В связи с этим для проверки нельзя
воспользоваться интенсивностью пика М+2 (табл. 4.2). Тем не менее проверить
результат можно по интенсивности пика М+3, обусловленного ионом,
содержащим по одному изотопу 13С и 37С1. Для соединения с 1 атомом хлора и 6 атомами
углерода его интенсивность должна составлять 1/3 от интенсивности пика М+1
или 6,6% от интенсивности пика М+2.
2,1/6,6 « 0,32; 2,1/33 та 0,064, т. е. совпадение вполне удовлетворительное.
Таким образом, соединение содержит 6 атомов углерода.
м) Пик молекулярного иона имеет интенсивность 33%. Принимаем эту
величину за 100% и нормируем интенсивности остальных пиков по М+\ Получаем
новую последовательность: М+' (m/z 107)- 100%, М+1 (m/z 108)-8,1%, М+2
(m/z 109) —0,24%. Поскольку молекулярная масса нечетная, вещество содержит
нечетное число атомов азота. Делим интенсивность пика А+1 на 1,1. Получаем
~7,4. Скорее всего соединение имеет 7 атомов углерода в молекуле, но на всякий
случай проверим варианты 8 и 6. Если атомов угаеродов 8, их масса 96. На
другие элементы остается 11 Да. Азот уже не подходит, а без азота масса не
13—1113
386 Глава 11. Решения задач
может быть нечетной. Если атомов углеродов 6, их масса 72. На другие элементы
остается 35. В этом случае должен быть 1 атом азота. Оставшиеся 21 а. е. м.
могут быть обусловлены составами ОН5 или FH^. Тогда полный состав будет
СбН5Ж) или C6H2NF. Проверим их по интенсивности пика ?+2. Шесть атомов
углерода дадут интенсивность 0,18%. Азот и фтор не имеют изотопов А+2.
Присутствие изотопа 180 приведет к увеличению интенсивности этого пика на
0,2%. Таким образом, для состава C6H5NO интенсивность пика М+2 должна
быть 0,38%, что заметно выше 0,24%, а рассчитанная интенсивность пика М+1
должна быть 6,6 + 0,4 = 7,0, что ниже значения, приведенного в условии задачи
(8,1). Для состава C6H2NF интенсивность пика М+1 должна быть тоже 7,0%, а
пика М+2 —0,18%. Это несколько ближе к условиям задачи.
Если в молекуле 7 атомов углерода, их масса 84. Оставшиеся 23 а. е. м. могут
быть обусловлены только азотом и водородом. Состав C7H9N. Интенсивность
пика М+1 должна быть 8,1, что точно соответствует условию, а интенсивность
пика М+2 —0,25%, что вновь практически совпадает с условием.
Следовательно, в качестве наиболее вероятного решения выбираем последнее. Вещество
содержит 7 атомов углерода.
Задача 4.6. Для решения удобно использовать данные табл. 4.2. При этом
необходимо помнить об азотном правиле и о возможном присутствии (А+2)-эле-
ментов. Если интенсивность М+* не 100%, необходимо осуществить
нормирование на 100%. Не стоит ожидать также абсолютного совпадения рассчитанных и
реальных интенсивностей пиков в спектре.
а) Поскольку интенсивность пика М+2 составляет только 0,4% от М+',
вещество не содержит атомов серы, кремния, хлора и брома, но может содержать
1 или 2 атома кислорода. По интенсивности пика М+1 определяем число атомов
углерода в молекуле: 7,7/1,1 = 7. Семь атомов углерода дадут интенсивность
пика М+2 0,2% (табл. 4.2). Еще 0,2% интенсивности пика М+2 должны быть
обусловлены 1 атомом кислорода. Сумма масс 7 атомов углерода и одного
атома кислорода дает 100. Оставшиеся 8 а. е. м. могут быть обусловлены только
атомами водорода. Следовательно, брутто-формула—C7HgO.
б) Интенсивность пика М+2 составляет только 0,1% от интенсивности
М+\ Это означает, что вещество не содержит атомов серы, кремния, хлора
и брома и маловероятно, что содержит 1 атом кислорода. Масса соединения
нечетная, следовательно, в составе нечетное число атомов азота. С учетом массы
соединения число атомов азота может быть 1 или 3. Азот —(А+1)-элемент. Это
надо помнить при подсчете числа атомов углерода по пику М+1. Делим 5,9
на 1,1. Получаем 5,37. Шесть атомов углерода даже с одним атомом азота по
массе не подходят. Пять атомов углерода и один атом азота дадут интенсивность
пика М+1, равную 1,1 ? 5 + 0,4 = 5,9, что точно соответствует условию
задачи. Интенсивность пика М+2 в этом случае будет 0,1% (табл. 4.2), что
вновь совпадает с величиной, приведенной в условии. Пять атомов углерода и
Глава И. Решения задач 387
один атом азота имеют суммарную массу 74. Оставшиеся 5 а. е. м. — водород.
Следовательно, брутто-формула соединения — C5H5N.
Задача 4.7. Хотя в условии задачи требуется идентифицировать соединение
по изотопным пикам молекулярного иона, на основании предоставленных
данных можно установить лишь брутто-формулу соединения, т. е. решить задачу
с точностью до изомера. Правда, в некоторых случаях решения оказываются
однозначными. Обычно же для установления структурной формулы соединения
необходима информация о фрагментных ионах. Для решения удобно
использовать данные табл. 4.2. При этом необходимо помнить об азотном правиле
и о возможном присутствии (А+2)-элементов. Если интенсивность М+* не
100%, необходимо осуществить нормирование на 100%. Не стоит ожидать также
абсолютного совпадения рассчитанных и реальных интенсивностей пиков в
спектре.
а) Наличие пика ?+2 с интенсивностью 98% от ? позволяет сделать вывод
о присутствии в молекуле образца атома брома (табл. 4.1). Учитывая, что по
условию задачи ион с массой 94 молекулярный, можно легко установить массу
остальной части молекулы: 94 — 79 = 15. По пику М+ 1 A,1%) можно сделать
вывод о наличии одного атома углерода. Следовательно, состав соединения —
СНзВг. В этом случае соединение определяется однозначно, так как не имеет
изомеров.
в) Интенсивность 4,8% пика ?+2 указывает на наличие в молекуле атома
серы или кремния (табл. 4.1). Кремний, однако, можно отвергнуть, так как в
противном случае интенсивность пика М+1 была бы больше, чем пика ?+2.
Следовательно, вещество содержит один атом серы. По пику М+1 можно
сделать вывод об отсутствии в молекуле атомов углерода, так как его интенсивность
@,8%) обусловлена исключительно изотопом 33S. Изотоп 34S ответственен за
4,4% интенсивности пика ?+2, следовательно, в составе соединения должен
присутствовать еще (А+2)-элемент, которым может быть только кислород. На
его долю приходится 32 а. е. м. F4 — 32 = 32). Такая масса соответствует двум
атомам кислорода. Эти атомы дадут прибавку 0,4% к интенсивности пика ?+2,
что полностью соответствует условию задачи D,4 + 0,4 = 4,8). Следовательно,
искомое вещество —диоксид серы.
е) В данном случае прежде всего необходимо нормировать интенсивности
изотопных ионов на 100%. Получаем новый ряд: M+*(m/z 46)—100%, М+1
(m/z 47) —2,2%, ?+2 (m/z 48) —0,2%. По интенсивности пика М+2 можно
сделать вывод о возможном присутствии одного атома кислорода. По пику
М+1 находим, что в составе 2 атома углерода B,2 : 1,1 = 2). Их вклад в
интенсивность пика М+2 нулевой. По-видимому, пик М+2 обусловлен именно
изотопом кислорода. В таком случае состав соединения — СгНбО. Это может
быть этиловый спирт или диметиловый эфир.
388 Глава И. Решения задач
Задача 4.8. Для решения удобно использовать формулу биноминального
разложения.
а) Два атома брома приводят к появлению пиков М+2 и ?+4.
Интенсивности этих пиков можно рассчитать по формуле квадрата суммы. Для
простоты можно считать, что изотопы 79Вг и 81Вг имеют равную природную
распространенность. Тогда A + 1 J = 1+2+1, т. е. соотношение пиков ?, ?+2
и ?+4 будет 1 : 2 : 1. Молекулярная масса дибромметана равна 172 Да (по
79Вг), следовательно, пики с m/z 172, 174 и 176 будут иметь интенсивности
50, 100 и 50%. Кроме того, в молекуле присутствует один атом углерода.
Каждому из трех указанных пиков будет соответствовать пик иона с массой
на единицу больше, что обусловлено изотопом 13С, с интенсивностью 1,1% от
интенсивности предшествующего пика, содержащего изотоп 12С. Общая картина
будет следующей: 172 E0%), 173 @,5%), 174 A00%), 175 A,1%), 176 E0%),
177 @,5%).
г) В случае сероуглерода расчет несколько сложнее из-за изотопа 33S. Начать
расчет следует с пиков М+2 и ?+4. Будем считать, что природное соотношение
изотопов 32S к 34S составляет 25 : 1 или 1 : 0,044. Для упрощения не
будем принимать в расчет изотоп 36S, природная распространенность которого
мала и составляет 0,11% (табл. 4Л). По формуле квадрата суммы получаем
A +0,044J = 1+0,088 + 0,0019. Таким образом, интенсивности пиков М, М+2
и М+4.будут 100%, 8,8% и 0,2% соответственно. Теоретически в интенсивность
пика М+2 будет вносить вклад пик иона 13C33S32S, однако его вклад составит
0,008 ? 0,011 ? 2 = 0,000176, т. е. менее 0,02%. Еще меньшим окажется вклад
пика иона 13C33S34S в интенсивность пика М+4. Поэтому, этими вкладами
можно пренебречь. Для расчета интенсивностей пиков М+1, М+3 и М+5
необходимо учесть вклад изотопов 13С и 33S. Интенсивность пика М+1 за счет
двух атомов 33S составит 1,6% от М, а за счет одного атома 13С — 1,1% от
М, т. е. суммарная интенсивность пика М+1 будет 2,7%. Интенсивность пика
М+3 будет 2,7% от интенсивности пика М+2, т. е. 8,8% ? 0,027 = 0,16%.
Аналогично, интенсивность пика М+5 будет 2,7% от интенсивности пика М+4,
т.е. 0,2% ? 0,027 = 0,0054%. Эта величина слишком мала для надежного
детектирования. В результате кластер пиков молекулярного иона будет выглядеть
следующим образом: 76 A00%), 77 B,7%), 78 (8,8%), 79 @,2%), 80 @,2%).
Задача 4.9. В отличие от предыдущих задач молекулярный ион не
обязательно оказывается первым в ряду представленных ионов. Пики более легких
ионов образуются за счет выброса атомов или молекул водорода из М+\ Если
интенсивность М+" не 100%, необходимо осуществить нормирование на 100%.
Не стоит ожидать также абсолютного совпадения рассчитанных и реальных
интенсивностей пиков в спектре.
а) Два первых иона серии (m/z 94 и 95) не могут быть молекулярными,
поскольку интенсивность пика иона с m/z 96 слишком велика, чтобы ее можно
Глава 11. Решения задач 389
было объяснить тяжелым изотопом какого-нибудь элемента. Не может быть
молекулярным и ион с m/z 97. В этом случае аномально интенсивен оказался
бы пик иона [М—Н]+. Такое соотношение интенсивностей пиков ионов М+#
и [М—Н]+ в масс-спектрометрии органических соединений не встречается.
Следовательно, единственным кандидатом в молекулярные ионы остается ион с
m/z 96. Интенсивность пика ?+2 составляет 0,2%, что указывает на отсутствие
в составе молекулы атомов хлора, брома, серы и кремния. Интенсивность пика
М+1 составляет 6,5%, что приблизительно соответствует 6 атомам углерода.
Можно уточнить интенсивность пика М+1, если учесть поправку,
обусловленную ионом [М—Н]+. Интенсивность пика этого фрагмента 7,1%. Учитывая, что
в молекуле 6 атомов углерода, изотопный пик этого фрагмента с целочисленной
массой, равной массе М+Ф, будет иметь интенсивность 0,47%. Это означает,
что интенсивность пика самого М+* составляет 99,53%. Для точного учета
интенсивностей пиков изотопных ионов необходимо нормировать приведенные в
задаче интенсивности пиков на 100%, однако, поскольку различия в этом случае
будут крайне малы (на уровне ошибки измерений), эту процедуру можно не
проводить.
Итак, по интенсивности пика М+1 сделан вывод о присутствии в
молекуле 6 атомов углерода. Пользуясь данными табл. 4.2, устанавливаем, что
интенсивность пика ?+2 в этом случае должна составлять 0,18%, что точно
соответствует условию задачи. Одновременно это означает, что в молекуле
отсутствуют атомы кислорода, а также другие (А+1)- и (А+2)-элементы. Масса
шести атомов углерода 72 Да. Оставшиеся 24 а. е. м. приходятся на А-элемен-
ты. На основании данных табл. 4.1 можно сделать вывод, что единственным
приемлемым вариантом является 1 атом фтора и 5 атомов водорода.
Следовательно, состав молекулы СбН5К Это ответ задачи. В данном случае можно
попытаться идентифицировать соединение. Учитывая, что максимальный пик в
спектре обусловлен молекулярным ионом, можно предположить, что речь идет
об ароматическом соединении фторбензоле.
ж) Таким же образом, что и в задаче 4.9, а) устанавливаем, что молекулярным
ионом может быть только ион с m/z 129. Остальные варианты нереальны,
поскольку этот ион имеет доминирующий пик в спектре и не может быть ни
изотопным к более легким, ни фрагментным к более тяжелым. Интенсивность
пика М+2 составляет 0,5% и указывает на отсутствие в составе молекулы
атомов хлора, брома, серы и кремния. Интенсивность пика М+1 составляет
10%, что приблизительно соответствует 9 атомам углерода. Можно уточнить
интенсивность пика М+1, если учесть поправку, обусловленную ионом [М—Н]+.
Интенсивность пика этого фрагмента 16%. Учитывая, что в молекуле 9 атомов
углерода, изотопный пик этого фрагмента с целочисленной массой, равной массе
М+', будет иметь интенсивность 1,6%. Это означает, что интенсивность пика
самого М+* составляет 98,4%. Для точного учета интенсивностей пиков
изотопных ионов необходимо нормировать приведенные в задаче интенсивности пиков
390 Глава 11. Решения задач
на 100%. Интенсивность пика М+1 будет 10,2%, а пика М+2 не изменится.
Нечетная масса М+* указывает на присутствие нечетного числа атомов азота.
Рассмотрим несколько вариантов.
Пусть в молекуле 9 атомов углерода и 1 атом азота. Они составят 122 Да.
Оставшиеся 9 а. е. м. могут быть обусловлены только водородом. Состав
соединения C9H7N. Интенсивность пика М+2 в этом случае 0,44% (табл. 4.2), что
примерно соответствует условию задачи.
Вариант с девятью атомами углерода и тремя атомами азота не проходит по
массе.
Возможно, что в молекуле 8 атомов углерода, а интенсивность пика М+1
выше теоретической из-за протекания ионно-молекулярных реакций. Вариант с
тремя атомами азота вновь не проходит по массе. Если в молекуле 8 атомов
углерода и 1 атом азота, их общая масса ПО Да. Оставшиеся 19 а. е. м. могут
быть обусловлены либо атомом фтора, либо тремя атомами водорода и одним
атомом кислорода. Оба эти варианта маловероятны с химической точки зрения.
Можно рассматривать более экзотические варианты с 5 или 7 атомами азота.
Тем не менее состав соединения C9H7N представляется наиболее вероятным и
соответствует хинолину.
Задача 4.10. а) Для расчета используем подход, изложенный в разд. 4.2.3
(табл. 11.1). Ион с m/z 120 может быть обусловлен только немеченным аце-
тофеноном. Поскольку интенсивность пика этого иона 15%, будем считать,
что в смеси 15 частей немеченного соединения. Исходя из этого, рассчитаем
интенсивности пиков ионов с m/z 121 и 122, обусловленных изотопными ионами
немеченного соединения. Поскольку чистое A00 частей) немеченное
соединение (табл. 7.2) характеризуется следующим спектром 120 A00%), 121 (8,8%),
122 @,54%), то 15 частей дадут-120 A5,0%), 121 A,32%), 122 @,08%).
Именно эти цифры представлены во второй строке табл. 11.1. В третьей строке
Таблица 11.1
Расчет доли молекул ацетофенона
с различным содержанием атомов дейтерия
Изотопомер
спектр
, *
Остаток 1
??
Остаток 2
d2
Остаток 3
*
Остаток 4
m/z 120
15,0
15,0
-
-
-
-
-
-
-
m/z 121
32,0
1,32
30,68
30,68
-
-
-
-
-
m/z 122
50,0
0,08
49,92
2,70
47,22
47,22
-
-
-
m/z 123
???
-
100
0,17
99,83
4,16
95,67
95,67
-
m/z 124
~K7
-
8,7
-
8,7
0,25
8,45
8,42
0,03
m/z 125
0,5
-
0,5
-
0,5
-
0,5
0,52
-0,02
Глава И. Решения задач 391
(остаток 1) представлен спектр образца после вычитания вклада немеченного
соединения. Теперь необходимо оценить долю монодейтерированного ацетофе-
нона. Интенсивность 30,68% пика с m/z 121 обусловлена М+" этого соединения.
Рассчитаем интенсивности изотопных пиков для такой доли этого продукта.
Получим следующий ряд: 121 C0,68%), 122 B,70%), 123 @,17%). Вновь проводим
вычитание этих интенсивностей из спектра (остаток 1) и получаем остаток 2, т. е.
спектр, обусловленный только соединениями с двумя и тремя атомами дейтерия
в молекуле. Повторяем эту процедуру еще дважды и получаем остаток 4, где
интенсивность 0,03% для иона 124 и отрицательный пик —0,02% для иона 125,
что соответствует уровню ошибки измерения. Таким образом, мы получили
следующий ряд: do —15 частей, d\ — 30,68 части, аг — 47,22 части, di — 95fil
части. Принимая сумму изотопомеров за 100%, получаем доли каждого: do —
8,0%, di-16,3%, rf2-25,0%, </3-50,7%.
Задача 4.11. а) Для расчета используем подход, изложенный в разд. 4.2.3
(табл. 11.2). Интенсивность пика иона [? —?]" составляет менее 1% от
интенсивности пика М4", т. е. им можно пренебречь. Ион с m/z 124 может быть
обусловлен только немеченным л-метоксифенолом. Поскольку интенсивность
пика этого иона 30%, будем считать, что в смеси 30 частей немеченного
соединения.
Исходя из этого, рассчитаем интенсивности пиков ионов с m/z 125 и 126,
обусловленных изотопными ионами немеченного соединения. Поскольку чистое
A00 частей) немеченное соединение (табл. 4.2) характеризуется следующим
спектром: 124 A00%), 125 G,7%), 126 @,65%), то 30 частей дадут- 124 C0,0%),
121 B,31%), 122 @,2%). Именно эти цифры представлены во второй строке
табл. 11.2. В третьей строке (остаток 1) представлен спектр образца после
вычитания вклада немеченного соединения. Теперь необходимо оценить долю
монодейтерированного w-метоксифенола. Интенсивность 57,69% пика с m/z 121
Таблица 11.2
Расчет доли молекул лара-метоксифенола
с различным содержанием атомов дейтерия
Изотопомер
Спектр
do
Остаток 1
dx
Остаток 2
1 d2
Остаток 3
di
Остаток 4
m/z 124
30,0
30,0
-
-
-
-
-
-
-
m/z 125
60,0
2,31
57,69
57,69
-
-
-
-
-
m/z 126
80,0
0,2
79,8
4,44
75,36
75,36
-
-
-
m/z 127
~L00
-
100
0,37
99,63
5,8
93,83
93,83
-
m/z 128
~1J
-
7,7
-
7,7
0,49
7,21
7,22
-0,01
m/z 129
0,6
-
0,6
-
0,6
-
0,6
0,61
-0,01
392 Глава И. Решения задач
обусловлена М+* этого соединения. Рассчитаем интенсивности изотопных
пиков для такой доли этого продукта. Получим следующий ряд 125 E7,69%),
126 D,44%), 127 @,37%). Вновь проводим вычитание этих интенсивностей из
спектра (остаток 1) и получаем остаток 2, т. е. спектр, обусловленный только
соединениями с двумя и тремя атомами дейтерия в молекуле. Повторяем эту
процедуру еще дважды и получаем остаток 4. Здесь получаем отрицательный
пик (—0,01%) для иона 128 и отрицательный пик (—0,01%) для иона 129. Эти
цифры соответствуют уровню ошибки измерения. Таким образом, мы получили
следующий ряд: do — 30 частей, d\— 57,69 части, аг — 75,36 части, d^ — 93,83
части. Принимая сумму изотопомеров за 100%, получаем доли каждого: do —
11,7%, di-22,5%, ??2-29,3%, d3-3695%.
Задача 4.12. а) Для расчета используем подход, изложенный в разд. 4.2,3
(табл. 11.3). Интенсивность пика иона [М—Н]+ составляет менее 1% от
интенсивности пика М+\ т. е. им можно пренебречь. Ион с m/z 156 может быть
обусловлен только немеченным 4-фтор-2-метоксианизолом. Поскольку
интенсивность пика этого иона 20%, будем считать, что в смеси 20 частей немеченного
соединения.
Таблица 11.3
Расчет доли молекул 4-фтор-2-метоксианизола
с различным содержанием атомов дейтерия
Изотопомер
Спектр
do
Остаток 1
dx
Остаток 2
d2
Остаток 3
m/z 156
20,0
20,0
-
-
-
-
-
m/z 157
100
1,76
98,24
98,24
-
-
-
m/z 158
12,0
0,15
11,85
8,65
3,20
3,20
-
m/z 159
?,?
-
?,?
0,73
0,27
0,28
-0,01
m/z 160
0,02
-
0,02
-
0,02 !
0,02
-
Исходя из этого, рассчитаем интенсивности пиков ионов с m/z 157 и 158,
обусловленных изотопными ионами немеченного соединения. Поскольку чистое
A00 частей) немеченное соединение (табл. 4.2) характеризуется следующим
спектром: 156 A00%), 157 (8,8%), 158 @,74%), то 20 частей дадут-156 C0,0%),
157 B,31%), 158 @,2%). Именно эти цифры представлены во второй строке
табл. 11.3. В третьей строке (остаток 1) представлен спектр образца после
вычитания вклада немеченного соединения. Теперь необходимо оценить долю
монодейтерированного 4-фтор-2-метоксианизола. Интенсивность 98,24% пика
с m/z 157 обусловлена М+" этого соединения. Рассчитаем интенсивности
изотопных пиков для такой доли этого продукта. Получим следующий ряд:
157 (98,24%), 158 (8,65%), 159 @,73%). Вновь проводим вычитание этих
интенсивностей из спектра (остаток 1) и получаем остаток 2, т. е. спектр,
Глава 11. Решения задач 393
обусловленный только молекулами с двумя атомами дейтерия. Повторяем эту
процедуру еще раз и получаем остаток 3. Здесь имеем лишь отрицательный пик
(—0,01%) для иона 159, что соответствует уровню ошибки измерения. Таким
образом, мы получили следующий ряд: do — 20 частей, d\— 98,24 части, аг —
3,20 части. Принимая сумму изотопомеров за 100%, получаем доли каждого:
d0 - 16,5%, d\ - 80,9%, d2 -2,6%.
Задача 4.13. Решать задачу удобно с помощью таблицы (разд. 4.2.3),
учитывая, что кластер молекулярного иона чистого немеченного (табл. 4.2) дифтор-
нафтилметилкетона имеет следующий вид: 206 A00%), 207 A3,2%), 208 A,0%),
209 @,03%).
Таблица 11.4
Расчет интенсивностей изотопных пиков в масс-спектре дейтерированного
дифторнафтилметилкетона
Изотопомер
do
*\
d2
dz
10% db
20% dx
30% d2
40% d3
??/? — d^
Нормализ. Yido — d^(%)
m/z 120
100
-
-
—
10,0
-
-
-
10,0
22,6
m/z 121
13,2
100
-
-
1,32
20,0
-
-
21,32
48,3
m/z 122
1,0
13,2
100
-
0,1
2,64
30,0
-
32,74
74,1
m/z 123
0,03
1,0
13,2
100
<0,01
0,2
3,96
40,0
44,16
100
m/z 124
—
0,03
1,0
13,2
-
<0,01
0,3
5,28
5,58
12,5
m/z 125
—
-
0,03
1,0
-
-
<0,01
0,4
0,4
0,7
m/z 126
—
—
-
0,03
-
-
0,01
0,01
0,02
Задача 4.14. Фторид, нитрил, амин.
Задача 5.1. Прежде всего необходимо определить заряды ионов. Для этой
цели используем формулу 5.5. Ее можно применить к любой паре соседних
ионов. Например, проводя расчет по паре ионов 951,8 и 883,8, получаем,
? = (883,8 - 1)/(951,8 - 883,8) = 12,98. Это означает, что ион 951,8 имеет заряд
13. Аналогично, делая расчет для пары ионов 825,0 и 883,8, получаем ? = 14,01,
т. е. ион 883,8 имеет заряд 14. Ион 825,0 имеет заряд 15 и т. д.
Теперь можно рассчитать молекулярную массу пептида по любому из этих
ионов. Для этого используем формулу 5.4. Для иона 951,8 с зарядом 13 получаем
? = 13(951,8 - 1) = 12360,4. Для иона 883, 8 с зарядом 14 получаем 12359,2,
для иона 825,0 с зарядом 15 — 12360,0. Желательно провести этот расчет для
всех ионов в спектре и усреднить полученное значение.
Молекулярная масса цитохрома С (рис. 5.7)— 12 360,2 Да.
394 Глава 11. Решения задач
Задача 7.1. Прежде всего можно отметить высокое разрешение прибора
по родительским ионам при использовании в режиме МС/МС третьей или
четвертой (Q1) реакционной области. В таком режиме можно изучать и
низкоэнергетические, и высокоэнергетические процессы. Использование для
получения спектра дочерних ионов второй реакционной области позволяет улучшить
разрешение по дочерним ионам режима MIKES. Этот эффект наблюдается
благодаря квадруполю, который суживает широкие пики ионов, прошедших
через электростатический анализатор. Для записи спектров родительских ионов
настраивают квадруполь на пропускание выбранных дочерних ионов и
сканируют напряженность магнитного поля, используя для активации соударением
третью или четвертую реакционную область. Для записи спектров ионов,
выбрасывающих идентичные нейтральные частицы, одновременно сканируют
В и Q, при условии 2?2/g = const. Можно работать в режиме обычного
секторного двухфокусного прибора BE, настроив квадруполи на пропускание всех
ионов.
Помимо МС/МС-экспериментов прибор BEQQ позволяет работать в режиме
тройной масс-спектрометрии (МС/МС/МС). Например, ионы М* пропускают
через магнит. В камере соударений в реакционной области 2 они активируются
ц распадаются с образованием дочерних ионов. Электростатический анализатор
настраивается на пропускание только дочерних ионов М^. Эти ионы
распадаются после активации в реакционной области 3 или 4, после чего сканирование
квадруполя позволяет зарегистрировать спектр внучатых ионов для М^1- (он же
спектр дочерних ионов для М^).
Задача 8.1. а) Предпочтительные разрывы в М+* 4,10-диметилгексадекана
будут происходить в местах разветвлений (схема 11.8). В алкановой серии ионов
повышенными интенсивностями по сравнению с изомерным октадеканом
нормального строения будут характеризоваться пики ионов [М —С3Н7]" (m/z 211),
[M-C6Hi3]+ (m/z 169), С8Н+ (m/z 113) и С5Н+ (m/z 71). Три первых пика
будут отчетливо выделяться среди пиков алкановой серии, характеризующихся
экспоненциальным падением интенсивностей. Пик иона CsH^ (m/z 71) —один
из наиболее интенсивных в спектрах алканов любого строения.
с5нп
~С3Н7
- с6н13
Схема 11.8
Глава 11. Решения задач 395
Задача 8.2. б) В спектре 4-метилдекана аномально высокую интенсивность
среди пиков алкановой серии будут иметь ионы [М—СзН7]+ и CsH^. Второй
ион имеет массу 71 и не является характеристическим, поскольку его пик
один из наиболее интенсивных в спектрах алканов любого строения. Тем не
менее иногда интенсивность этого пика оказывается значительно выше пика
иона 57. Это может служить указанием на наличие разветвления в
соответствующем положении молекулы. В спектре 5-метилдекана аномально высокую
интенсивность среди пиков алкановой серии будут иметь ионы [М —С4Н9]+
и СбН^з. Помимо ионов алкановой серии для различения изомеров можно
использовать перегруппировочные нечетноэлектронные ионы. В первом случае
в спектре будет отчетливо проявляться пик иона [М — СзНв]", а во втором —
[M-C4H10]+\
Задача 8.3. а) Пики нечетноэлектронных ионов имеют повышенную
интенсивность в местах разветвлений углеродного скелета алканов. В случае 3,3-ди-
метилоктана в спектре будут отчетливо проявляться два пика, обусловленные
разрывами С—С-связи у атома углерода СЗ. По правилу выброса максимального
алкила интенсивность пика иона [М—СгНб]4" будет значительно ниже, чем
пика иона [М—С5Н12]4". Поскольку атом углерода СЗ имеет еще метальные
радикалы, теоретически возможно отщепление метильной группы и молекулы
метана. Однако конкурентоспособность этих процессов мала. Соответствующий
пик нечетноэлектронного иона [М—СКЦ]"*" будет либо едва заметен, либо
вообще отсутствовать в спектре.
Задача 8.4. В спектре отчетливо видны повышенные интенсивности пиков
ионов 84, 85, 154 и 155. Незначительный по интенсивности пик молекулярного
иона все же позволяет установить молекулярную массу алкана, равную 212 Да.
Таким образом, в спектре алкана с брутто-формулой С15Н32 наблюдаются
характеристические ионы, связанные с расщеплением С—С-связи у атома углерода С5.
Следовательно, на рис. 8.2 представлен масс-спектр 5-метилтетрадекана.
Задача 8.5. Молекулярная масса алкана равна 226, т. е. речь идет о димети-
лтетрадекане с брутто-формулой С16Н34. В ряду пиков ионов алкановой серии
аномально интенсивны пики ионов [М — СзНу]"*" с m/z 183 и CsH^ с m/z 71.
Поскольку таких (аномальных) ионов всего два, речь идет о симметричном
соединении или о соединении с двумя метальными группами у одного атома
углерода. В первом случае единственным вариантом является 4,11-диметил-
тетрадекан. Второй случай невозможен, поскольку отщепление пропильного
радикала означает разветвление у атома углерода С4. Однако наличие двух
метальных групп у этого атома привело бы к образованию максимального по
интенсивности пика фрагментного иона СбН^, а не СзН^. Таким образом, на
рисунке представлен спектр 4,11-диметилтетрадекана.
396 Глава 11. Решения задач
Задача 8.6. Для решения используем интенсивности пиков ионов
характеристической для алкинов серии. Повышенную интенсивность будет иметь
пик иона, образующийся по механизму, представленному на схеме 8.7. На
рисунке 8.7, ? повышенную интенсивность имеет ион с m/z 137, а на рис. 8.7,6 —
ион с m/z 165. Следовательно, в первом случае приведен спектр тетрадецина-5,
а во втором —тетрадецина-7.
Задача 8.7. Основные направления фрагментации молекулярного иона ме-
тилциклопентана связаны с отщеплением метильного радикала (как экзоцикли-
ческого, так и с участием атома углерода кольца), молекулы этилена и этильного
радикала. Следует также отметить образование ионов С3Н+в, С3Н+ и С2Н+#
(схема 11.9).
m/z2S
С5Н9
m/z 69
m/z 55
Схема 11.9
Задача 8.8. В спектре 8.11, а интенсивность пика иона 68 примерно в два
раза выше, чем пика иона 54. В спектре на рис. 8.11, б соотношение интенсивно-
стей прямо противоположно. Следовательно, на рис. 8.11,6 представлен спектр
4-метилциклогексена (ретродиеновый распад, схема 8.9). На рисунке 8.11, ?
представлен спектр 1-метил- или 3-метилциклогексена. Выбор между двумя
этими изомерами неочевиден. Поскольку в случае 3-метилциклогексена метильная
группа находится в аллильном положении, ее отщепление более благоприятно,
чем в случае 1-метилциклогексена, когда она находится в винильном положении.
Однако для такого выбора необходимо иметь спектры обоих изомеров.
Задача 8.9. а) Для пропилбензола основным первичным направлением
распада будет бензильный разрыв с отщеплением этильного радикала и
образованием иона тропилия (С7Н7*, m/z 91). Для изомерного кумола подобный
Глава 11. Решения задач 397
процесс приводит к отщеплению метильного радикала и образованию иона
CgH^". Кроме того, для первого соединения возможна перегруппировка Мак-Лаф-
ферти, протекающая через шестичленное переходное состояние и приводящая
к нечетноэлектронному фрагменту с m/z 92. Для изопропилбензола подобный
процесс не реализуется.
Задача 8.10. д) Распад алкилзамещенных полициклических ароматических
углеводородов протекает по тем же направлениям, что и углеводородов ряда
бензола. Основные направления фрагментации 2-пропилнафталина представлены на
схеме 11.10.
m/z ПО
С3Н7
?*
-с2н5·
СН2
(if-
m/z 141
ИС-0
~н1
-С2Н4
^
сн2
?
-н*
m/z 142
СН3
?*
с2н2
m/z 115
Схема 11.10
Задача 8.11. Уже по общему виду спектра можно сделать определенные
предположения о природе соединения. Доминирующие в спектре пики ионов 91
и 92 с большой вероятностью свидетельствуют о том, что образец является моно-
замещенным (бензольным) ароматическим соединением. Ион 91 имеет строение
тропилий-катиона, а ион 92 —строение молекулярного иона толуола. Высокая
интенсивность пика иона 92, как правило, свидетельствует о неразветвленной
цепи заместителя.
По кластеру молекулярного иона рассчитаем брутто-формулу соединения.
Прежде всего, необходимо нормировать интенсивности изотопных пиков на
100%. Получаем следующий ряд: 148 A00%), 149 A2,1%), 150 @,68%).
Невысокая интенсивность пика ?+2 означает, что соединение не содержит (А+2)-эле-
ментов. Расчет числа атомов углерода по пику М+1 дает 11 A2,1/1,1). Проверяя
по табл. 4.2, какую интенсивность будет иметь пик М+2 в спектре соединения
с 11 атомами углерода, получаем 0,67%, что практически совпадает с
условием задачи. Следовательно, соединение содержит 11 атомов углерода, масса
которых составляет 132 Да. Оставшиеся 16 а. е. м., могут быть обусловлены
только атомами водорода. Остальные варианты не имеют смысла. Брутто-фор-
мула соединения —CnHi6. Его степень ненасыщенности 4. Это соответствует
алкилбензолу. Учитывая первоначальное наблюдение (ионы 91 и 92), можно
398 Глава 11. Решения задач
сделать вывод, что соединение является пентилбензолом. Разветвление скелета
амильного заместителя маловероятно. В случае 2-фенилпентана бензильный
разрыв приводил бы к иону 105 (а не 91) и перегруппировка Мак-Лафферти
была сильно подавлена. В случае 3-фенилпентана бензильный разрыв приводил
бы к иону 119. Для 2-метил-1-фенилбутана перегруппировка Мак-Лафферти
протекала бы значительно менее интенсивно, а в спектре присутствовал
достаточно интенсивный пик иона 57. Однако его интенсивность в спектре составляет
всего 2%. Несколько сложнее выбрать между нормальным пентилбензолом и
изоамилбензолом. Тем не менее во втором случае в спектре должен был бы
наблюдаться достаточно интенсивный пик иона 43 (разрыв углеродной цепи в
месте разветвления). Поскольку пик этого иона в спектре отсутствует, можно
сделать однозначное отнесение — пентилбензол.
Задача 8.12. Этот процесс аналогичен, описанному для случая дифенилме-
тана (схема 8.13). Отщепление молекулярным ионом трифенилметана фенильной
группы приводит к образованию устойчивого дифенилметильного иона, который
может циклизоваться во флуоренил-катион с выбросом двух атомов водорода из
opmo-положений бензольных колец.
Задача 8.13. Столь высокая интенсивность пика молекулярного иона,
доминирующего в спектре, характерна для полициклических ароматических
соединений. Попробуем рассчитать брутто-формулу соединения по кластеру
молекулярного иона. Интенсивность пика ?+2 невысока, т. е. из (А+2)-элементов
в составе молекулы может быть только кислород. Далее рассчитываем число
атомов углерода. 15,8/1,1 = 14,4. Скорее всего, соединение содержит 14 атомов
водорода, но на всякий случай имеет смысл проверить варианты с 13 и 15
атомами. Если атомов углерода 15, их общая масса 180 Да, что больше
молекулярной массы соединения. Если атомов углерода 14, единственным вариантом
является соединение СнНю. Если атомов углерода 13, на долю других элементов
приходится 22 а. е. м. Поскольку присутствие одного атома азота невозможно
(азотное правило), остаются варианты Q3H22, С^Н^О и C13H3F. Последний
вариант явно маловероятен. Проверим три приемлемых варианта по пику ?+2.
Для состава СиНю его интенсивность должна составлять 1,10%, для состава
С13Н22—0,94%, а для состава С^НбО —1,14%. Наибольшие погрешности в
интенсивностях изотопных пиков М+1 и М+2 наблюдаются для состава С13Н22.
Кроме того, учитывая, что степень ненасыщенности такого соединения 3, оно
никак не может обладать столь стабильным молекулярным ионом. Оставшиеся
два варианта характеризуются очень высокими величинами степени
ненасыщенности A0 для СиНюи 11 для СиНбО). Выбор между этими вариантами
достаточно сложен. В спектре помимо кластера молекулярного иона, и пиков
ионов [М — Н]+ и [М — 2Н]+" присутствуют лишь три группы более или менее
интенсивных пиков ионов. Первая группа (m/z 150-152), а также ионы [М —Н]+
Глава 11. Решения задач 399
и [М—2Н]+# характерны для полициклических ароматических соединений
(см. рис. 8.12). Группа пиков (m/z 88 и 89) обусловлена двухзарядными ионами
М2+ и [М—2Н]2+, а группа пиков (m/z 75 и 76) — заряженными фрагментами
бензольного кольца. Обе эти группы вновь характерны для полициклических
ароматических соединений. Выбор легко можно было бы сделать, имея данные
масс-спектрометрии высокого разрешения. Поскольку этих данных нет, остается
попробовать нарисовать возможные структуры соединений состава С^НбО и
оценить основные направления их фрагментации. По типу фрагментации и
по более точному совпадению интенсивностей пиков изотопных ионов можно
сделать выбор в пользу состава СиНю. Такой состав среди ароматических
полициклических углеводородов имеют антрацен и фенантрен. Выбор между этими
изомерами только по спектру ЭУ невозможен. На рисунке 8.16 представлен масс-
спектр антрацена.
Задача 8.14· а) Основной первичный процесс распада спиртов идет с
расщеплением С—С-связи между атомом углерода, связанным с гидроксиль-
ной группой, и атомом углерода любой из алкильных групп.
Следовательно, М+" метилэтилпропилкарбинола может элиминировать метильную, этиль-
ную или пропильную группы. Максимальную интенсивность будет иметь пик
иона [М—СзНу]4", меньшую —пик иона [М—Сг^]-1-, а минимальную —пик
иона [М—СНз1+. Поскольку все радикалы тривиальны, можно попытаться
рассчитать интенсивности первичных ионов, учитывая, однако, что
интенсивность пика иона [М —СНз]+ будет завышена. Принимая интенсивность пика
иона [М — СзН7]+ (m/z 73) за 100%, получаем, что интенсивность пика иона
[М-С2Н5]+ (m/z 87) будет примерно 84% G3 ~ 87 ? 100%), а интенсивность
пика иона [M-CH3]+(m/z 101) - примерно 72% G3 -г 101 ? 100%).
Задача 8.15· а) Основной первичный процесс распада спиртов связан с
расщеплением С—С-связи между атомом углерода, связанным с гидроксильной
группой, и атомом углерода алкильных групп. В случае бутанола-1 первичными
фрагментными ионами будут [М—Н]+ и [М—С3Н7]4", причем по правилу
выброса максимального алкила интенсивность пика [М — СзНу]" будет значительно
выше. Интенсивность пиков спиртовой серии [М —С2Н5]+ и [М —СНз]+будет
очень низка. В случае бутанола-2 первичными фрагментами будут [М —Н]+,
[М — СНз]+и [М —С2Н5]+, причем их интенсивность возрастает в указанной
последовательности.
Задача 8.16. В этом случае определить элементный состав соединения
по изотопным пикам не удается, поскольку интенсивность предполагаемого
молекулярного иона (m/z 60) низка. К тому же пик иона [М —Н]+ имеет равную
интенсивность. Тем не менее простое деление интенсивности пика иона с m/z 61
на интенсивность пика иона с m/z 60 позволяет ориентировочно оценить, что в
составе вещества три атома углерода. Поскольку молекулярная масса соединения
невысока, интерпретация спектра не представляет большого труда.
400 Глава 11. Решения задач
Максимальный по интенсивности пик в спектре принадлежит иону с m/z 31.
Этот ион — представитель характеристической спиртовой серии. Два других иона
этой серии (m/z 45 и 59) также представлены в спектре заметными пиками,
причем важным фактом является значительно более высокая интенсивность
более тяжелого. Ион с m/z 42 обусловлен отщеплением от М+' 18 а. е. м., т. е.
молекулы воды. Можно предположить, что вещество является спиртом или
простым эфиром с молекулярной массой 60 Да. Этому условию удовлетворяют
только пропиловый и изопропиловый спирты, а также метилэтиловый эфир. В
спектре эфира и изопропанола максимальный по интенсивности пик по правилу
распада должен принадлежать иону [М—СНз]+. Однако ион с m/z 43 обладает
пиком с интенсивностью ниже 2%. Кроме того, интенсивность пика с m/z 31 в
спектрах этих соединений должна быть невелика. Пропанол, напротив, должен
характеризоваться максимальным по интенсивности пиком иона с m/z 31, низкой
интенсивностью пика иона с m/z 45 и заметным пиком иона с m/z 59.
Представленный спектр полностью соответствует этим условиям. Следовательно, на
рис. 8.27 представлен спектр пропанола.
Задача 8.17. б) 2-Метилциклогексанол и 4-метилциклогексанол можно
легко различить по спектрам электронного удара. Перегруппировочный процесс с
образованием наиболее устойчивого иона в первом случае приведет к двум
фрагментам с m/z 57 и 71, а во втором —только к фрагменту с m/z 57 (схема 11.11).
он он он
m/z 57 \^ m/z 71
+ +· +
он он он
m/z 51 ^f m/z 51
СН3
Схема 11.11
Задача 8.18· Попробуем установить молекулярный ион соединения.
Наиболее тяжелый ион в спектре (m/z 140) не может быть молекулярным, поскольку не
может обусловить образование фрагмента с m/z 130 (третье необходимое условие
для молекулярного иона). Следовательно, М+" данного соединения не стабилен
и в спектре ЭУ не проявляется. Молекулярные массы ундекана и деканаля
равны 156 Да, децена-1 —140 Да. Эти соединения не могут характеризоваться
Глава И. Решения задач 401
представленным на рис. 8.28 спектром. Остальные соединения, перечисленные
в условии, являются спиртами. Среди них только диэтилциклогексанол имеет
молекулярную массу 156 Да, а остальные спирты—158 Да. Отщепление воды
молекулярными ионами спиртов — хорошо известный процесс. Вероятно, ион с
m/z 140 обусловлен именно этим процессом. Поэтому можно исключить из
рассмотрения диэтилциклогексанол. Следовательно, соединение является жирным
спиртом. Рассмотрим гомологическую спиртовую серию ионов. Максимальный
пик в спектре принадлежит иону этой серии с m/z 59. Достаточно интенсивны
также пики ионов с m/z 31 и 129. Остальные ионы этой серии
характеризуются пиками очень низкой интенсивности. Как известно, основной первичный
процесс распада спиртов связан с расщеплением С—С-связи между атомом
углерода, связанным с гидроксильной группой, и атомом углерода алкильных
групп. Максимальной интенсивностью в случае деканола-2 и 3,4-диметилок-
танола-2 должен обладать пик иона с m/z 45, в случае деканола-3 — пики
ионов с m/z 59 и 129, в случае деканола-1 и 7-метилнонанола-1 — пик иона с
m/z 31, в случае З-метилнонанола-3 — пики ионов с m/z 73, 129 и 143, в случае
2,7-диметилоктанола-4 — пики ионов с m/z 87 и 101. Исходя из этих данных,
можно сделать однозначный вывод, что спектр на рис. 8.28 принадлежит дека-
нолу-3. Ион с m/z 31 образуется в результате вторичного перегруппировочного
процесса (схема 8.18) любого из двух указанных первичных фрагментов.
Задача 8.19. В качестве подсказки в условии указаны точные интенсивности
пиков изотопных ионов. В реальном спектре пики с интенсивностью 0,1%
от максимального и ниже не могут быть точно измерены. Молекулярный
ион, по-видимому, имеет массу 76 Да. По интенсивности пика М+1 можно
установить, что в составе молекулы, вероятно, три атома углерода B/58 =
0,034). Наиболее важные пики в спектре принадлежат ионам с массами 31,
45, 61. Два первых иона характерны для распада спиртов. Ион 61 обусловлен
отщеплением метильного радикала из М+\ По его изотопным пикам можно
установить, что в его составе два атома углерода и два атома кислорода.
Брутто-формула соединения С3Н8О2. Рассчитываем степень ненасыщенности
этого соединения: R = 3—4+1 = 0. Следовательно, в молекуле отсутствуют циклы
и кратные связи. Речь может идти о бифункциональном спирте или простом
эфире. Кроме того, понятно, что в молекуле присутствует метильная группа.
Это позволяет отвергнуть структуру пропандиола-1,3. Кроме того, маловероятны
изомерные структуры диметоксиметана и 2-метоксиэтанола-1. Таким образом,
необходимо выбрать между структурами пропандиола-1,2 и 1-метоксиэтанола-1.
Максимальный пик в спектре обусловлен ионом С2Н50+, который образуется
при отщеплении радикала состава СНзО из М+\ Этот катион действительно
должен быть наиболее устойчивым для обеих оставшихся структур. Ион 31
также не позволяет сделать однозначный выбор в пользу одной из этих структур.
Пожалуй, единственной возможностью для выбора является процесс отщепления
402 Глава 11. Решения задач
гидроксильного радикала из М+*. Для пропандиола-1,2 этот процесс
маловероятен, поскольку приводит к нестабильному иону, тогда как для полуацеталя,
которым является 1-метоксиэтанол-1, отщепление гидроксильной группы, хотя
и менее выгодно, чем альтернативный процесс с выбросом метоксигруппы,
однако должно протекать, поскольку образующийся фрагментный ион должен
быть весьма устойчивым (схема 11.12). Поскольку пик этого иона (m/z 59)
отсутствует в спектре, следует признать, что масс-спектр на рис. 8.29 принадлежит
пропандиолу-1,2.
СН3-СН--ОСН3
он*
Схема 11.12
СН3-СН=ОСН3
m/z 59
Задача 8.20. а) Пик М+' З-этилгексанола-3 будет малоинтенсивен. Он
может вовсе отсутствовать в спектре. Будет наблюдаться пик иона [М —НгО]",
причем он может иметь максимальную массу в спектре. Первичные процессы
распада (схема 11.13) связаны с элиминированием углеводородных радикалов
от ?-атома углерода. По правилу выброса максимального алкила пик иона
+
ОН
0>?5—с
- с2н5·
СзН7
m/z 101
-С2Н4
m/z 73
Н-уСН-СНз
сн±-сн2
он
+¦
он
с2н5—i— C2H5
С3Н7
m/z 130
-С3Н7
алкеновая серия
/я/г41,55,69,83,97
~СзНб . СН2=ОН
+
ОН
С2Н5—С—С2Н5
m/z 87
-С2Н4
+
ОН
СН—С2Н5
m/z 59
-С2Н4
m/z 31
Схема 11.13
Н-уСН2
CHL-CH2
ан
Глава 11. Решения задач 403
[М —СзН7]+ будет иметь большую интенсивность в спектре, а пик иона
[М — СгИб]" меньшую. Однако надо учесть, что в молекуле два этильных
радикала. Этот факт приведет к увеличению интенсивности пика [М — Сг^]"**.
Возможен ряд вторичных и даже третичных процессов с образованием перегруп-
пировочных ионов. Ион [М —СгНб]" может последовательно элиминировать в
любом порядке молекулы этилена и пропилена, а ион [М—СзЕ^]-1- две молекулы
этилена. В результате этих процессов будут возникать ионы спиртовой серии.
Помимо кислородсодержащих ионов будет регистрироваться ряд
углеводородных фрагментов алкеновой и алкановой серий. Интенсивности пиков этих
фрагментов могут быть достаточно высоки.
Задача 8.20. и) Спектр 3-этилфенола будет характеризоваться интенсивным
пиком молекулярного иона и максимальным по интенсивности пиком иона
[М —СНз]+. Этот ион далее может отщеплять молекулу СО или радикал НСО*.
Эти же частицы могут элиминироваться и непосредственно из молекулярного
иона («фенольный» распад). Однако предпочтительными будут процессы,
изображенные в правой части схемы фрагментации 3-этилфенола (схема 11.14).
[М-Н]+
m/z 121
[M-HCOf**
m/z 93
.+·
PhH
m/z П
-?"
-[M-CO]t
m/z 94
t ?1 ??
ЬГт9 ^7?
Схема 11.14
Задача 8.21. Для выбора между изомерными структурами необходимо
обратить внимание на пик иона с m/z 92, присутствующий только в спектре
на рис. 8.30,6. Он обусловлен отщеплением молекулы НВг из М+' благодаря
opmo-эффекту. Следовательно, этот спектр принадлежит 2-бромфенолу.
Задача 8.22. Очень много дает уже внешний вид спектра. Прежде всего,
пик иона 130 с интенсивностью в три раза меньше, чем пик иона 128, позволяет
сделать вывод о наличии одного атома хлора в молекуле. Этот вывод
подтверждается отщеплением 36 а. е. м. (ион 92) из М+'. Кроме того, поскольку пик
М+" максимальный в спектре, скорее всего соединение имеет ароматический
характер.
Рассчитаем элементный состав соединения. К сожалению, присутствие хлора
маскирует пик иона ?+2. По пику М+1 устанавливаем, что в молекуле
404 Глава И. Решения задач
6 атомов углерода. Этот же вывод можно сделать по пику иона М+3
относительно ?+2. Шесть атомов углерода и один атом хлора имеют массу
107 Да. Оставшиеся 21 а. е. м. могут быть обусловлены составами ОН5 и FH2. В
первом случае брутто-формула соединения будет C6H5OCI, во втором — C6H2FCI.
Степень ненасыщенности первой структуры R = 6 — 3 + 1=4, второй —
R = 6 — 2+1 = 5. Оба варианта могут иметь ароматическую природу и иметь
интенсивный пик молекулярного иона.
Первичный процесс распада М+Ф связан с элиминированием 28 и 29 Да
(ионы 99-102). Для состава C6H5OCI эти процессы могут быть обусловлены
выбросами частиц СО и НСО*. Однако для состава CoEbFCl частицы с такой
массой отщепиться не могут. Следовательно, брутто-формула соединения будет
C6H5OCI. Об ароматичности соединения помимо доминирующей интенсивности
пика М+" свидетельствует характерная серия фрагментных ионов с массами 92,
74, 75, 50, 51, 39. Учитывая, что все 6 атомов углерода должны участвовать
в образовании бензольного ядра, соединение может быть только хлорфенолом.
Этот вывод подтверждается «фенольным» типом распада (выбросы частиц СО и
НСО" из М+#). Для того чтобы установить взаимное расположение
заместителей, вновь надо обратить внимание на ион с m/z 92. Этот фрагмент обусловлен
выбросом молекулы НС1 из М+". Такой процесс активно протекает только в
случае opmo-изомера (см. предыдущую задачу). Следовательно, на рис. 8.31
представлен масс-спектр о-хлорфенола.
Задача 8.23. Поскольку энергия, переносимая на ионизируемую
молекулу спирта в результате ионно-молекулярной реакции, возрастает в ряду
изобутан < метан < водород, в этом же ряду возрастают интенсивности пиков
ионов, образующихся в результате вторичных, третичных и т. д. процессов
распада. Для изобутана характерны в основном лишь пики ионов, обусловленных
первичными процессами. Тем не менее пик молекулярного протонированного
иона оказывается нестабильным во всех трех случаях.
Задача 8.24. Для ответа на вопрос необходимо вспомнить основное
направление распада циклогексанолов (схема 8.22). Предварительно можно исключить
из рассмотрения 1-этилциклогексанол, так как пик иона [М —C2Hs]+ имеет очень
низкую интенсивность. При распаде 1-этилциклогексанола интенсивность этого
пика была бы значительно выше (80-100%). Основное направление распада
приводит к паре альтернативных фрагментов в зависимости от того, какая связь
рвется на первой стадии (CI—C2 или CI— C6). В случае 4-этилциклогексанола в
обоих случаях этильный заместитель не сохранится. Поэтому пик иона с m/z 85
должен иметь очень незначительную интенсивность. Напротив, для 3-этилцикло-
гексанола интенсивность этого пика должна быть выше, чем пика
альтернативного иона с m/z 57. Спектр на рис. 8.32 противоречит и тому, и другому варианту.
Для 2-этилциклогексанола первичные интенсивности этих пиков должны быть
примерно равны (схема 11.15). Однако в результате вторичных процессов часть
Глава 11. Решения задач 405
ионов с m/z 85 трансформируется далее. Таким образом, интенсивность пика
иона с m/z 57 в спектре будет в 3-4 раза выше интенсивности пика иона с m/z 85.
На рисунке 8.32 представлен масс-спектр 2-этилциклогексанола.
с2н5
+ +. +
ОН ОН ОН
J t -ggn JyWs _СзН; ^ Г
m/z57 ^^ w/z85
Схема 11.15
Задача 8.25. Соединение характеризуется достаточно устойчивым
молекулярным ионом, хотя интенсивные пики в области низких значений m/z
свидетельствуют об алифатической природе образца. Очень важно увидеть повышенную
интенсивность пика ?+2. Такая интенсивность свидетельствует о присутствии
атома серы или кремния. Ион с m/z 98 образуется в результате выброса 34 а. е. м.
из М+\ Такой процесс характерен для меркаптанов и связан с
отщеплением молекулы сероводорода. Дополнительным подтверждением тиольной или
сульфидной функции является гомологическая серия ионов 47, 61, 75, 89...
Интенсивности их пиков весьма высоки, причем, поскольку интенсивность пика
иона 89 выше, чем пика иона 75, можно сделать вывод о неразветвленном
углеродном скелете и наличии тиольной группы у первого атома углерода. На
рисунке 8.33 представлен спектр гептантиола-1.
Задача 8.26. Внешний вид спектра свидетельствует об ароматической
природе образца. По интенсивности изотопных пиков определяем состав молекулы.
На основании интенсивности пика М+1 находим, что число атомов углерода
10. Одновременное присутствие двух изотопов 13С в молекуле должно в этом
случае привести для пика ?+2 к интенсивности 0,54%. Оставшиеся 0,2%
интенсивности это пика, вероятно, обусловлены изотопом кислорода 180. В
этом случае состав соединения CioHgO. Степень ненасыщенности составляет
R = Ю-4+1 = 7.
Состава СнН^ хуже согласуется с интенсивностями изотопных пиков. Кроме
того, М+# такого соединения, например метилдигидронафталина, не сможет
элиминировать частицы с массами 28 и 29 Да. Если в составе молекулы 9 атомов
углерода, ее брутто-формула может быть C9H8N2, C9H20O, C9H4O2, C9H17F,
C9HOF. Варианты С9Н4О2 и C9HOF маловероятны с химической точки зрения.
Варианты с 20 и 17 атомами водорода представляют алифатические соединения,
пик молекулярного иона которых не может быть столь интенсивен. Вариант
C9H8N2 очень разумен с химической точки зрения (например, аминохинолин),
однако плохо согласуется с интенсивностью пика ?+2 (она должна быть почти
в 2 раза ниже). Маловероятны также выбросы частиц с массами 28 и 29 Да из
М4". Против присутствия атомов азота в молекуле свидетельствует отсутствие
более или менее интенсивных пиков ионов с четной массой в левой части
спектра.
406 Глава 11. Решения задач
Выбросы частиц с массами 28 и 29 Да из М+* обычно обусловлены «феноль-
ным» распадом (СО и НСО) или элиминированием этильного радикала и
молекулы этилена, причем последний процесс сопровождается перегруппировкой.
Однако спектр на рис. 8.34, а также брутто-формула соединения (CwHgO), не
позволяют разместить в составе молекуле этоксильный, пропилфенильный или
этиларилкетонный фрагменты. Вероятно, молекула содержит фенольную
функцию. В этом случае речь может идти о нафтоле. Масс-спектр электронного удара
не позволяет сделать вывод о точном положении гидроксильной группы. Более
экзотические варианты, например производные метилиндена, должны были бы
характеризоваться дополнительными фрагментными ионами. На рисунке 8.34
представлен масс-спектр нафтола-1.
Задача 8.27, б. В молекулярном ионе изопропилэтилового эфира
положительный заряд и неспаренный электрон локализованы на атоме кислорода.
Наиболее интенсивные пики обусловлены ионами, образующимися при отрыве
алкильных радикалов (?-распад, инициированный радикальным центром). И
изопропильный, и этильный радикалы будут элиминировать метальный
радикал (схема 11.16). Алкильные ионы образуются при инициировании распада
центром, несущим заряд. Вторичный изопропильный катион весьма устойчив.
Поэтому его пик будет иметь заметную интенсивность в спектре. Напротив,
первичный этильный катион нестабилен, а его пик будет едва заметен в спектре.
Интенсивность пиков алкоксильных ионов низка.
СН3ч+ СН3ч
СН3
СН3' ? ^ -снз/^сн3/
СН3
^СН~Ь'~СН2—СН3 х
/ ? \-сн<
L3
+
+
сн=о-сн2—сн3
СН2-СН3 СН3/
Схема 11.16
Задача 8.28. Основные направления фрагментации пропилфенилсульфида
представлены на схеме 11.17. Следует подчеркнуть, что ионы с m/z 11 могут
образоваться не только из М+', но и из фрагментов с m/z 123, ПО и 109. Ионы
с m/z 45 могут также образоваться другим путем.
^^ Ph-+Sk
+ -С2Н5* +¦ / m/zUO
Ph-S=CH2-. PI1-S-CH2-C2H5—(
ml\ 123 I -C3H7S* \=Э* Ph-S -^* m/z 65
\ + ? . m/z 109
HCS Ph+
m/z 45 m/z 77
Схема 11.17
I-Oflj·
Глава 11. Решения задач 407
Задача 8.29. а) В случае диэтилового эфира характеристическими фраг-
ментными ионами будут [М-СН3]+ (m/z 59), СН2=ОН+ (m/z 31), С2Н^ (m/z 29),
а для метилпропилового эфира —[М—С2Н5]+ (m/z 45), [?—?]" (m/z 73), C3H7"
(m/z 43), CH2=OH+ (m/z 31), CH+ (m/z 15).
Задача 8.30. Основные направления фрагментации emop-бутилпропилового
эфира представлены на схеме 11.18.
+ + +
С2Н5-СН=ОН С4Н9 С3Н7
/?/? 59 m/z 57 m/z 43
t-СзНб Х^^ У*
+ -сн3# +· -с2н5# +
С2Н5-СН=0-СН2-С2Н5 - С2Н5 -СН-О-СН2-С2Н5 -С2Н5 -Ш-0=СН2
m/z 101 СН3 СН3
m/z VI
1-С4Н8
+ -С3Н6 _ + + *
СН3-СН=ОН-« СН3~СН=0~СН2-С2Н5 НО=СН2
m/z 45 m/z 87 m/z 31
Схема 11.18
Задача 8.31. а) Наиболее важные направления распада диизопропилового
эфира, инициированные радикальным (ионы 87 и 45) и катионным центрами
(ионы 59 и 43), представлены на схеме 11.19.
сн— о—сн 3-+ ;сн— о=сн
сн/ чсн3 сн3х
m/z 102 m/z 87
-С3Нб
+ /Ш3
но=сн
m/z 59 m/z 43 m/z 45
Схема 11.19
Задача 8.32. Внешний вид спектра свидетельствует об ароматической
природе образца. Пик молекулярного иона доминирует в спектре, причем активно
идут процессы отщепления атома и молекулы водорода из М+#. Об
ароматичности соединения свидетельствуют также основные фрагментные ионы с m/z 51,
69, 77. 92. Высокая интенсивность пика иона [М —Н]+ несколько усложняет
расчет элементного состава образца по изотопным пикам.
408 Глава 11. Решения задач
Интенсивность пика М+1 составляет 15,8% от М. Следовательно, молекула
содержит 14-15 атомов углерода. Необходимо внести поправку в интенсивность
пика М+' за счет наложения со стороны изотопного пика иона [М —Н]+.
Интенсивность пика иона [М —Н]+ 65,1%. Следовательно, интенсивность изотопного
пика этого иона за счет одного изотопа 13С будет примерно 15% от этого
значения, т. е. почти 10%, а одновременное присутствие двух изотопов 13С в
молекуле приведет к пику А+2 с интенсивностью около 1% (табл. 4.2). Таким
образом, интенсивность пика молекулярного иона должна быть примерно 90%
A00— 10), а пика М+1—-14,8% A5,8 — 1). Теперь нормируем интенсивности
пиков кластера молекулярного иона на 100%. Получаем следующую серию:
186 A00%), 187 A6,3%), 188 E,2%), 189 @,7%). Такая интенсивность пика
?+2 практически однозначно означает присутствие в молекуле одного атома
серы. В этом случае надо еще раз провести корректировку интенсивностей
изотопных пиков за счет присутствия изотопов серы в ионе [М —Н]+. Если
интенсивность пика ? изменяется несущественно (на 0,8%), то интенсивность
пика М+1 должна была быть (до нормирования) не 14,8%, а 11,9% A4,8 —
65,1 ? 0,044). Интенсивность пика М+2 уменьшится примерно на 0,4% за
счет одновременного присутствия в молекуле одного атома 13С и одного атома
34S. Тогда имеем следующую серию: 186 (89,2%), 187 A1,91%), 188 D,8%),
189 @,61%), или после нормирования: 186 A00%), 187 A3,1%), 188 E,2%),
189 @,7%). Расчет элементного состава образца, исходя из этих интенсивностей,
дает CnHioS. Степень ненасыщенности R= 12 — 5 + 1 = 8. Остановимся на этом
варианте, хотя для полной корректности расчетов необходимо было бы исходя
из этого элементного состава пересчитать интенсивности пиков кластера М+*
с учетом наложений со стороны ионов [М —Н]+ и [М — 2Н]+\ Поскольку все
основные фрагментные ионы обусловлены распадом бензольных ядер, можно
предположить два варианта ответа: дифенилсульфид и меркаптобифенил.
Для выбора между двумя этими соединениями можно использовать
следующие особенности распада. Интенсивные пики ионов [М —Н]+ и [? — 2?]4"
не характерны для тиофенольных структур. В то же время подобные процессы
очень характерны для дифенилметана, дифениламина или дифенилфосфина.
Они связаны с циклизацией по opmo-положениям бензольных ядер. В данном
случае образуется молекулярный ион дибензотиофена (схема 11.20). Кроме того,
для тиофенолов характерны выбросы молекул CS и С2Н2. В данном случае
пики этих ионов малоинтенсивны. На рисунке 8.40 представлен масс-спектр
дифенилсульфида.
? ?
Схема 11.20
Глава 11. Решения задач 409
Задача 8.33. Основные первичные процессы фрагментации аминов связаны
с разрывами С—С-связей ?-углеродных атомов, причем действует правило
выброса максимального алкила (разд. 3.1.1).
а) При распаде молекулярного иона бутилпропилэтиламина максимальную
интенсивность среди первичных ионов будет иметь пик иона [? — 0$??]+,
меньшую — [М — СгН5]+, минимальную — [М — СНз]+.
Задача 8.34. Вторичные процессы распада аминов связаны с перегруппиро-
вочными процессами, которые идут через четырехчленное (реже шестичленное)
переходное состояние. В результате элиминируется молекула олефина. Заряд
сохраняется на атоме азота. Для протекания процесса необходима длина радикала
не менее двух атомов углерода.
а) Процессы распада бутилпропилэтиламина можно отобразить единой
схемой 11.21.
+ -СзН7*
CH2=N-CH2-QH5 СзН7-
сн2
СН3
m/z 100
С3Н7 -CH2-N-CH2-C2H5
СН-
-CH2-N-CH2-C2H5.
СН2
СН3
m/z ,143
_сн3*
-с2н5
СзН7-
-СН2—N=CH2
^Н2
СН3
m/z 114
C3H7-CH2-NH
СН2
-С4Н8
HN=CH2
-С2Н4
H2N=CH2
m/z 30
Схема 11.21
410 Глава 11. Решения задач
Ион [М — СзНу]* может претерпевать распад по пропильной группе с
выбросом молекулы пропилена или по этильной группе с выбросом молекулы этилена.
В результате образуются ионы с m/z 58 и 72. Первый из них далее отщепляет
молекулу этилена, а второй — пропилена. Оба процесса приводят к образованию
иона с m/z 30.
Ион [М—С2Н5]+ может претерпевать распад по бутильной группе с
выбросом молекулы бутилена или по этильной группе с выбросом молекулы этилена.
В результате образуются ионы с m/z 58 и 86. Первый из них далее отщепляет
молекулу этилена, а второй — бутилена. Оба процесса приводят к образованию
иона с m/z 30.
Ион [М—СН3]+ может претерпевать распад по бутильной группе с выбросом
молекулы бутилена или по пропильной группе с выбросом молекулы пропилена.
В результате образуются ионы с m/z 72 и 86. Первый из них далее отщепляет
молекулу пропилена, а второй — бутилена. Оба процесса приводят к образованию
иона с m/z 30.
Задача 8.35. Основные направления распада бутилизопропиламина
инициированы неспаренным электроном (схема 11.22). Все наиболее интенсивные
пики в спектре обусловлены ионами, образующимися в результате первичного
?-распада или вторичных перегруппировок. Эти вторичные перегруппировки
протекают через четырехчленное (ионы 44 и 30) или шестичленное (ион 58)
переходное состояние. Механизмы этих процессов представлены на схеме 8.44.
На схеме 11.22 не указаны направления фрагментации, связанные с
отщеплением от М+* атома водорода, поскольку интенсивность пика этого первичного
иона очень низка. Так как атом азота обладает очень высокой склонностью к
удержанию заряда, пики алкильных ионов имеют низкую интенсивность.
CH=NH-CH2-C3H7 ^L CH3-CH-NH-CH2-C3H7 "^ > CH3-CH-NH=CH2
СН3 СН3 СН3
m/z 100 m/z 115 m/z 12
-с4н8
-Сз^ -ОзНб
CH3-CH=NH2 CH3—CH2-NH=CH2 NH2=CH2
m/z 44 m/z 58 rn/z 30
Схема 11.22
Задача 8.36. Как и в случае бутилизопропиламина (задача 8.35) основные
направления распада метилдиизопропиламина инициированы неспаренным
электроном (схема 11.23). Все наиболее интенсивные пики в спектре обусловлены
Глава 11. Решения задач 411
ионами, образующимися в результате первичного ?-распада или вторичных
перегруппировок. Так как атом азота обладает очень высокой склонностью
к удержанию заряда, пики алкильных ионов низкую интенсивность. Ион 86,
малоинтенсивный пик которого присутствует в спектре, вероятно, обусловлен
незначительной примесью амина с пропильной группой нормального строения.
СН3-СН-N-СН-СНз *
I II I
СНз СН2СН3
m/z 114
-ОД
СНз-CH-NH
I II
СН3 СН2
m/z 72
-СзНб
+ ?
NH2=CH2
m/z 30
-^-CH3-CH-N-CH-CH3
III
CH3 CH3CH3
m/z 115
-H"
CH3-CH-N=C-CH3
III
CH3 CH3CH3
m/zlU
013 » CH3-CH-N=CH-CH3
-QHe
CH3
HN=C
I I
СНзСН3
m/z 72
Схема 11.23
I I
CH3 CH3
m/z100
-ОД
HN=CH-CH3
I
CH3
m/z 58
Задача 8.37. Для атома фосфора в отличие от атома азота помимо реакций
распада, инициированных радикальным центром, характерны процессы распада,
инициированные катионным центром. Этил-катион (m/z 29) может образоваться
практически из любого фрагмента, представленного на схеме 11.24.
-аь
HP—СН2—СН3 ¦* СН3—СН2— Р—СН2—СН3 ^СН3—СН2— Р=СН2
с2н5
m/z 90
—QHt
Н2Р—СН2—
m/z 62
-Ш
Н2Р=СН2
m/z 41
С2Н5
m/z 118
??—сн2—сн3
m/z 61
«"^^ СН2=НР-СН2—СН3
ти/г75
Схема 11.24
С2Н5
m/z 103
-С2Н4
СН3—СН2-НР=СН2
m/z 75
-СаН»
Н2Р=СН2
m/z 47
412 Глава 11. Решения задач
Задача 8.38. а) Основные процессы фрагментации М+* диэтиламина могут
быть отражены следующей схемой:
+ +· -СН*" +
С2Н5 + C2H5-NH-C2H5 - CH2=NH~C2H5
m/z 29 m/z 73 m/z 58
-н'х ~н
-С2Н4
+
CH3~CH=NH-C2H5 ^H4> CH3-CH=NH2 СН2=ЯН2
m/* 72 ~ m/z 44 тД 30
Схема 11.25
Задача 8.39. Интенсивный пик молекулярного иона и отчетливая серия
ионов с m/z 39, 51, 77 указывает на наличие ароматического фрагмента в
молекуле. Наряду с фенильной группой в молекуле имеется еще структурный фрагмент
с массой 44 Да. Поскольку в условии указано, что приведен спектр вторичного
амина, еще 15 Да принадлежит группе NH. Оставшиеся 29 а. е. м. должны быть
обусловлены этильной группой. Первичный процесс, приводящий к наиболее
стабильному иону в спектре, обусловлен отрывом метильной группы этильного
заместителя. Следовательно, на рис. 8.48 представлен спектр фенилэтиламина.
Задача 8.40. Молекулярный ион имеет массу 118 Да. Рассчитаем его
элементный состав, предварительно проведя нормирование на 100%. Получаем
следующий вид кластера М+#: 118 A00%), 119 F,6%), 120 @,2%). Отсюда
можно сделать вывод, что в молекуле 6 атомов водорода и отсутствуют другие
(А+1)- и (А+2)-элементы. На долю ?-элементов приходится 46 Да.
Возможными вариантами состава могут быть СбН^Р и C6HgF2. Степень ненасыщенности
в первом случае R = 6 — 7,5 + 0,5 + 1 = 0, а во втором — R = 6 — 5 + 1 =2.
Основные процессы распада связаны с выбросами 15 Да (метильная группа)
и 42 Да. По изотопному пику (m/z 77) можно рассчитать, что отщепляется
молекула пропилена. Пропильным фрагментом обусловлен, вероятно, и ион с
m/z 43. Первичный перегруппировочный процесс с потерей молекулы олефина,
не сопровождающийся интенсивным процессом отщепления
соответствующего алкильного радикала, характерен для фосфинов. В случае алкилфторидов
селективность этого процесса не может быть столь высокой. Следовательно,
речь идет о фосфине. Наличие атома фосфора можно подтвердить пиками с
m/z 33 и 34. Они обусловлены ионами РН^" и РН^~\ Малолинейчатость спектра
может свидетельствовать о симметричности структуры соединения. Учитывая,
что основным направлением распада является выброс молекулы пропилена,
можно сделать вывод о том, что образец является дипропилфосфином. Для
выбора между изомерными дипропилфосфином и диизопропилфосфином следует
обратить внимание на ион [М—СНз]+. Он образуется в результате ?-распада
Глава 11. Решения задач 413
изопропильного заместителя. Аналогичный процесс в случае пропильного
заместителя привел бы к элиминированию этильного радикала. Поскольку пик
иона [М—С2Н5]4" в спектре отсутствует, можно утверждать, что на рис. 8.49
представлен масс-спектр диизопропилфосфина.
Задача 8.41. Для решения необходимо составить схемы фрагментации
предложенных изомеров и найти различия в величинах m/z характеристических
фрагментов.
б) Дибутиламин и emop-бутилбутиламин. Для дибутиламина, молекула
которого симметрична, возможен только один первичный фрагментный ион
[М — С3Н7]"*". Молекулярный ион emop-бутилбутиламина помимо пропильного
радикала может отщеплять этильный и метальный (схема 11.26). Уже на этом
основании, не рассматривая весь спектр, можно различить два этих изомера.
CH=NH-CH2-C3H7 ч2 СНз-СН2-СН-ЫН-СН2-СзН7""СзН7> CH3-CH2-CH--NH=CH2
СН3 СН3 СН3
1-оъ
CH3-CH2-CH=NH~CH2-C3H7
Схема 11.26
в) Все первичные и вторичные фрагменты для дипентиламина и изопентил-
пентиламина будут идентичны. Однако для изопентилпентиламина возрастает
вероятность вторичного процесса, проходящего через шестичленное переходное
состояние (схема 11.27). В результате этого процесса интенсивность пика с
m/z 44 в спектре этого соединения будет выше.
СН3 /***
+ -с5н10 H-V+ \ -^?^+ -с* +
CH2=NH2- CH2^NH=CH2*-^ \?2-??-4?2—^ СН2=Ш-СНз
/иДЗО m/z U4 m/z 44
Схема 11.27
д) 1-Метилциклопентиламин и 2-метилциклопентиламин. Максимальный по
интенсивности пик фрагментного иона при распаде 1-метилциклопентиламина
будет соответствовать иону с m/z 70. Интенсивность пика [М—СНз]+
достаточно высока (схема 11.28). Процесс распада с разрывом цикла приводит к
иону с m/z 70. При распаде 2-метилциклопентиламина интенсивность пика
иона [М—СНз]+ будет низкой. Распад с раскрытием цикла в зависимости от
414 Глава 11. Решения задач
первоначального разрыва С]— С2 или Сi— C5 приведет к образованию ионов с
m/z 70 или 56 (схема 11.28), при этом путь 2 предпочтителен.
NH2 NH24 CH3 NH:
II 2.?\? U
СН3^
2
?2?? 3 и
m/z 70 m/z 84
NH, NH2 NH;
•2
CH3
m/z 56 N ' w/z70
Схема 11.28
и) В спектре дифениламина будет наблюдаться интенсивные пики ионов
[М-Н]+, [М—2Н]+\ Будет заметен и пик иона [М — ЗН]+. Эти ионы
обусловлены процессом внутримолекулярной циклизации (схема 8.47). Для 4-аминобифе-
нила такой процесс невозможен, зато большей интенсивностью будут обладать
пики ионов [M-HCN]+· И [M-H2CN]+.
Задача 8.42, а· Один из основных процессов фрагментации молекулярного
иона 1-бромгексана связан с образованием пятичленного бромониевого катиона
(схема 8.51). Интенсивность пиков этого иона ([М—CiHs]4", m/z 135, 137) будет
очень высока. Этот процесс полностью подавлен по стерическим причинам в
случае 3-бромгексана, а пик иона [М — С2Н5]4" едва заметен в спектре.
Задача 8.43. Внешний вид спектра позволяет сразу сделать два важных
вывода. Во-первых, в составе молекулы есть один атом хлора, во-вторых, высокая
интенсивность пика молекулярного иона и фрагментные ионы с m/z 77, 76, 51,
50, 39, 38 свидетельствуют об ароматической природе образца. В молекуле 6
атомов углерода (расчет по пику М+1 или по М+3). Оставшиеся 5 а. е. м. могут
быть обусловлены только атомами водорода. Состав CeHsCl. Хлорбензол.
Задача 8.44. Триплет молекулярного иона с соотношением пиков ионов
М/М + 2/М + 4 = 1/2/1 однозначно указывает на присутствие двух атомов брома
в молекуле. Соответственно пики 79 и 81 обусловлены двумя изотопами брома,
а пики 93 и 95 ионом СНгВг4", который образуется при отщеплении атома брома
из М+\ Дибромметан.
Задача 8.45. В спектре легко увидеть несколько групп пиков. В этих группах
наиболее интенсивные пики отстоят друг от друга на две единицы массы.
Это указывает на наличие в составе (А+2)-элементов. Легко определить, что
дублет пиков 61, 63 обусловлен ионом с одним атомом хлора (соотношение
интенсивностей пиков 3:1), триплет пиков 97, 99, 101 —ионом с двумя атомами
хлора (соотношение интенсивностей пиков 9 : 6 : 1), а квартет пиков 117, 119,
121, 123 —ионом с тремя атомами хлора (соотношение интенсивностей пиков
Глава 11. Решения задач 415
27 : 27 : 9 : 1). В последнем случае состав иона легко определить, поскольку
три атома хлора (изотопы 35С1) имеют массу 105. Оставшиеся 12 а. е. м. — атом
углерода, а состав иона ССЬ. Однако этот ион, хотя и обладает максимальной
массой в спектре, не может быть молекулярным, поскольку не может обусловить
образование следующего иона, содержащего только два атома хлора. Масса
атома хлора 35 Да, а разница в массах между этими ионами равна 20 Да
(четвертое необходимое условие для,определения молекулярного иона, разд. 4.1).
Следовательно, молекулярный ион данного соединения не стабилен, а его пик в
спектре не регистрируется. По изотопному пику (А+1)-иона с m/z 97
определяем, что в его составе два атома углерода. Тогда его состав CH3CCI2, а ион с m/z 61
образуется при отщеплении этим ионом молекулы НС1. Комбинируя
информацию по составу двух наиболее тяжелых ионов (ССЬи CH3CCI2), можно
предположить, что первый образовался из М+* при отщеплении метильного радикала,
а второй —атома хлора. Следовательно, брутто-формула соединения CH3CCI3.
Отщепление метильной группы возможно только из одной изомерной структуры
такого состава. На рисунке 8.54 представлен спектр 1,1,1-трихлорэтана.
Задача 8.46. В спектре легко увидеть несколько групп пиков. В этих
группах наиболее интенсивные пики отстоят друг от друга на две единицы
массы. Это указывает на наличие в составе (А+2)-элементов. Легко определить,
что триплет пиков 118, 120, 122 обусловлен ионом с двумя атомами хлора
(соотношение интенсивностей пиков 9 : 6 : 1), а квартеты пиков 141, 143,
145, 147 и 153, 155, 157, 159 —ионами с тремя атомами хлора (соотношение
интенсивностей пиков 27 : 27 : 9 : 1). Массы первых ионов более тяжелых
мультиплетов 188, 223 и 258 отстоят друг от друга на 35 Да. Следовательно,
эти ионы содержат 4, 5 и 6 атомов хлора соответственно. Проверить это можно
по формуле (разд. 4.2). Высокая интенсивность пиков кластера молекулярного
иона позволяет рассчитать число атомов углерода в молекуле. Расчет легко
провести по ионам с m/z 261 и 263. Кроме того, можно использовать для расчета
изотопные пики за счет атомов углерода 13С иона [М —С1]+. Во всех случаях
получается 4 атома углерода. Шесть атомов хлора (по 35С1) и четыре атома
углерода имеют суммарную массу 258 Да, что совпадает с молекулярной массой
образца. Брутто-формула соединения C4G6. Его степень ненасыщенности R =
4 — 3 + 1=2. Следовательно, вещество является диеном или алкином. Варианты
производных циклопропана, циклопропена и циклобутена можно исключить,
поскольку молекулярные ионы таких соединений не стабильны. Выбирая между
перхлорированными бутином-1, бутином-2, бутадиеном-1,2 и бутадиеном-1,3,
следует отдать предпочтение последнему. Его молекулярный ион очень устойчив,
а фрагментация заключается в последовательном отщеплении атомов и молекул
хлора. Разрывы винильных, а тем более кратных С —С-связей не выгодны.
Наличие трихлорметильной группировки в составе трех других соединений
неизбежно повлекло бы образование соответствующего иона, однако его пик
в спектре не наблюдается. Таким образом, на рис. 8.55 представлен масс-
416 Глава 11. Решения задач
спектр гексахлорбутадиена-1,3. Пики с m/z 129-132 обусловлены двухзарядными
молекулярными ионами.
Задача 8.47. Наиболее тяжелый ион в спектре имеет нечетную массу: либо в
молекуле нечетное число атомов азота, либо молекулярный ион нестабилен и его
пик в спектре отсутствует. Расчет числа атомов углерода в наиболее стабильных
ионах (m/z 119 и 69) по пикам А+1 показывает, что в первом ионе 2 атома
углерода, а во втором —один. Кроме того, в этих ионах нет других (А+1)-
и (А+2)-элементов. Для иона с m/z 69 состав достаточно очевиден. Это CF3.
Азотсодержащих ионов в спектре нет. Состав иона с m/z 119 в этом случае
должен быть C2F5. Этот ион четноэлектронный и молекулярным быть не может.
Поскольку все ионы в спектре обусловлены атомами углерода и фтора в
разных численных комбинациях, логично предположить, что и наиболее тяжелый
фрагментный ион образуется при элиминировании радикала, состоящего из этих
атомов. Наиболее простым и разумным вариантом является атом фтора. Если
бы речь шла о перфторированном производном пропана или более тяжелого
гомолога, в спектре наблюдались бы пики ионов с большим числом атомов
углерода. Следовательно, на рис. 8.56 представлен масс-спектр гексафторэтана.
Задача 8.48. В данном случае уже по условию пик молекулярного иона
в спектре отсутствует. Попытаемся установить элементный состав наиболее
тяжелых фрагментов. Очевидно, что во всех тяжелых фрагментах содержится
несколько атомов (А+2)-элементов (возможно хлора и брома). На эти элементы
указывают также пики ионов с m/z 35, 37 и 79, 81. Оценка числа атомов
углерода во всех фрагментах показывает, что они содержат лишь один атом
С и отличаются лишь набором галогенов. Три основные группы пиков, таким
образом, обусловлены ионами CClBr4"', СС^Вг" и CClBr^. Отсюда легко
сделать вывод, что на рис. 8.57 представлен масс-спектр дибромдихлорметана. Ион
с m/z 81,5, характеризующийся интенсивным пиком, обусловлен двухзарядным
ионом CCl2Br++. Пик однозарядного иона этого состава (m/z 163) является
максимальным в спектре. Другие ионы с нецелочисленными массами также
являются двухзарядными.
Задача 8.49. Основные направления фрагментации молекулярного иона
4-бромфенола связаны с отщеплением атома брома, молекулы НВг и «феноль-
ным» распадом (схема 11.29).
m/z 92 _CQy m/z 172 m/z 93 m/z 65
^ -c2H2
а41 _^^щ
Br Br
ml ? 144 m/z 143 /яД117
Схема 11.29
Глава 11. Решения задач 417
Задача 8.50. Отсутствие спектра в табличном виде не позволяет напрямую
установить элементный состав молекулярного иона. Однако наличие двух пиков
равной интенсивности ионов с массами, отличающимися на 2 Да, означает, что
в составе есть атом брома. Высокая интенсивность пика молекулярного иона,
а также присутствие в спектре пиков ионов 75, 51, 50, 39 свидетельствует
об ароматической природе соединения. Ион с m/z 95 — [? — Вг]+. Расчеты
с использованием интенсивностей изотопных пиков показывают, что в его
составе 6 атомов углерода, которые, видимо, входят в бензольное кольцо.
Оставшиеся 23 = 95 — 72 а. е. м. должны быть обусловлены моноизотопными
элементами. Единственным возможным составом соединения в этом случая
является CoHUBrF — бромфторбензол. К сожалению, по спектру нельзя установить
взаимное расположение атомов фтора и брома в молекуле. На рисунке 8.58
представлен масс-спектр 4-бромфторбензола.
Задача 8.51. Перегруппировка Мак-Лафферти протекает через шестичлен-
ное переходное состояние. Для ее реализация необходима длина цепочки,
связанной с ароматическим ядром или карбонильной группой, не менее трех атомов.
Разветвления в цепи, кратные связи могут препятствовать перегруппировке.
а) В молекулярном ионе З-метилоктанона-4 заряд и неспаренный электрон
локализованы на атоме кислорода. Оба алкильных заместителя имеют
достаточную длину для протекания перегруппировки. В одном случае возникает ион с
m/z 114, а во втором— с m/z 100 (схема 11.30).
+ t* +
ОН О ОН
СН--С---СН2— С3Н7 «~°2Н4 С2Н5—СН-С-СН2— С3Н7 "СзНб> С2Н5— СН-С-СН2
СНз СНз СНз
m/z 114 m/z 142 m/z 100
Схема 11.30
и) В молекулярном ионе децен-З-она-5 заряд и неспаренный электрон
локализованы на атоме кислорода. Оба алкильных заместителя имеют достаточную
длину для протекания перегруппировки. Однако перегруппировочный процесс
по заместителю с кратной связью невозможен, поскольку на второй стадии
должен проходить разрыв именно двойной связи С—С. Энергетически это
невыгодно. В масс-спектре будет наблюдаться только один пик перегруппировочного
иона [М-С4Н8]+ с m/z 98.
Задача 8.52, в. В молекулярном ионе бензилбутилкетона заряд и
неспаренный электрон могут быть локализованы как на атоме кислорода, так и на
бензольном кольце. В первом случае возможен процесс с отщеплением молекулы
пропилена и образованием енольной формы молекулярного иона бензилметил-
кетона (схема 11.31), тогда как перегруппировка по бензильному заместителю
14—1113
418 Глава 11. Решения задач
невозможна: она должна была бы сопровождаться миграцией фенильного атома
водорода, а этот процесс и образующийся ион энергетически не выгодны.
Однако во втором случае (заряд и неспаренный электрон локализованы на
ароматичесом ядре) миграция атома водорода с элиминированием молекулы
кетена приводит к образованию стабильного иона СуН^\
г>
? +· +
о он
и
I , ~ „С4Н8СО Ph— СН2-С-СН2— С3Н7 3 6> Ph— CH2-C-CH2
^^сн2
m/z 92 m/z 176 m/z 134
Схема 11.31
Задача 8.53. Необходимо представлять основные направления
фрагментации молекулярных ионов указанных изомеров. Во всех случаях существуют
фрагментные ионы, интенсивности пиков которых будут совершенно различны.
а) Изомерные пентаналь и 2-метилбутаналь можно очень легко различить по
спектру электронного удара на основании иона, образующегося в результате
перегруппировки Мак-Лафферти. Поскольку ?-атом углерода карбонильного
соединения сохраняется в составе фрагментного иона, метильная группа 2-ме-
тилбутаналя приведет к смещению пика перегруппировочного иона на 14 Да по
сравнению с пентаналем (с m/z 44 до m/z 58). Интенсивность обоих пиков будет
высока.
е) Гексен-2-аль и гексен-5-аль вновь легко различить по спектру
электронного удара на основании иона, образующегося в результате перегруппировки Мак-
Лафферти. Перегруппировка невозможна для гексен-2-аля, поскольку должна
протекать с разрывом кратной связи. Пик перегруппировочного иона (m/z 44)
в спектре отсутствует. Структура гексен-5-аля, напротив, благоприятствует
перегруппировке Мак-Лафферти. В данном случае первая стадия процесса
облегчается, так как мигрирует аллильный атом водорода (схема 11.32). Кроме того,
в результате последующего разрыва заряд может сохраняться как на енольном
(ЭИ 9,5), так и на бутадиеновом (ЭИ 9,1) фрагменте. Следствием этого будут два
интенсивных пика в спектре (m/z 44 и 54), причем пик бутадиенового фрагмента
будет выше благодаря меньшей энергии ионизации.
ОН
? м
сн2=сн-сн\_уо н. сн2=сн-а?) он ^сн^с-н
сн2 X * сщ) X v "А44
сн> н сн^ нХсн2==сн-сн-.сн2
m/z 54
Схема 11.32
Глава 11. Решения задач 419
Задача 8.54. Спектры изомерных 3,3-диметилгексаналя и 4,4-диметилгекса-
наля достаточно сильно различаются (рис. 8.65, а и б). Как правило, наиболее
полезными для установления структуры оказываются пики перегруппировочных
(нечетноэлектронных) ионов. На рисунке 8.65, ? пик перегруппировочного иона
(m/z 84) является максимальным в спектре, тогда как на рис. 8.65,6 он вообще
отсутствует. Ион 84 образуется в результате перегруппировки Мак-Лафферти,
которая протекает через шестичленное переходное состояние. Для реализации
процесса необходима длина углеродной цепочки не менее трех атомов и наличие
атома водорода у 7-атома углерода. В случае 4,4-диметилгексаналя такого атома
водорода нет, и перегруппировка не идет. В случае 3,3-диметилгексаналя пик
перегруппировочного иона также имеет нестандартное для альдегидов значение
(как правило, m/z 44), обусловленное ионом енольной формы ацетальдегида.
Дело в том, что на второй стадии перегруппировки за заряд и неспаренный
электрон конкурируют две молекулы. Наличие двух метальных групп у
атома углерода СЗ исходной молекулы приводит к образованию 3-замещенного
олефина, энергия ионизации которого оказывается значительно ниже, чем у
альтернативного енола (схема 11.33). В результате, пик олефинового иона
доминирует в спектре. Таким образом, на рис. 8.65, ? и б представлены спектры
3,3-диметилгексаналя и 4,4-диметилгексаналя соответственно.
СН3—СН2—ОЮб . СН3—СН2-СН ОН СН3—СН2-СН
I II -?' 2 ? ? ?
сн3—с ^с^ ¦* сн3—сГ^гс^ ** сн3—с +
сн/ СН2 н сн/ СН2 н сн/
m/z 84
Схема 11.33
Задача 8.55. В основе решения лежит тот же принцип, что и для
предыдущей задачи. Пик иона с m/z 104 обусловлен перегруппировкой Мак-Лафферти.
Интенсивности пиков альтернативных енольного и олефинового ионов
определяются энергиями ионизации этих соединений. При распаде 4-фенилбутаналя
конкурируют енольная форма ацетальдегида (ЭИ 9,5) и стирол (ЭИ 8,4). Поскольку
энергия ионизации последнего значительно ниже, в спектре наблюдается пик
стирольного иона, а пик кислородсодержащего фрагмента (m/z 44) отсутствует
вовсе (схема 11.34).
Ph-CH"V /0 „. Ph-CH ОН Ph-CH
I ч-^ || ~н » I ^ I ^ U
СН2 ^ СН2 ?^+С^ * СН2
сн2 н сн2 н m/z 104
Схема 11.34
420 Глава 11. Решения задач
Задача 8.56. г) Основное направление распада кетонов связано с
расщеплением С—С-связей с двух сторон от карбонильной группы. В случае бензил-
этилкетона по этому механизму образуются два ацильных и два углеводородных
иона. В паре ацильных ионов пик С2НзСО+ (m/z 57) будет более интенсивен по
правилу выброса максимального иона и в связи с последующей фрагментацией
альтернативного иона PhCtkCO" (m/z 119). В паре углеводородных ионов
безусловным лидером будет тропилий-катион (m/z 91). Он может обладать
максимальным пиком в спектре. Пик второго иона (m/z 29) значительно
менее интенсивен. Тропилий-катион далее теряет последовательно две молекулы
ацетилена с образованием ионов с m/z 65 и 39. Незначительную интенсивность
будет иметь фенил-катион (m/z 77) и продукт его вторичного распада —ион с
m/z 51. Единственный перегруппировочный процесс заключается в
элиминировании метилкетена с образованием нечетноэлектронного иона СуН^" (схема 11.35).
Схема 11.35
Задача 8.57. Основные направления фрагментации бутирофенона
представлены на схеме 11.36. В спектре доминируют пики ионов, обусловленных
ароматическим ядром. Перегруппировка Мак-Лафферти приводит к
образованию единственного нечетноэлектронного иона с m/z 120. Значительно большая
стабильность иона PhCO+ (m/z 105) по сравнению с ионом СзН7СО+ (m/z 71)
отчетливо проявляется при сравнении интенсивностей их пиков.
^PhCO+
m/z 105
?
+ Ph+ > m/z 51
m/z 77
Он
II .
Ph—C-CH2
m/z 120
Схема 11.36
Задача 8.58, е. Основное направление распада кетонов связано с
расщеплением С—С-связей с двух сторон от карбонильной группы. Поскольку дибутил-
кетон является симметричной молекулой, эти процессы приводят к образованию
не четырех, а двух ионов: С4Н9" и С4Н9СО+. Перегруппировка Мак-Лафферти
ъ
Ph—С-СН2—С2Н5
m/z 148
l-Cft
Глава И. Решения задач 421
может протекать последовательно по обоим заместителям с выбросами молекул
пропилена и приводить к иону со структурой енольной формы ацетона
(схема 11.37). Пики этих четырех ионов будут доминировать в спектре. Минорные
пики будут обусловлены углеводородными ионами, образующимися при распаде
бутил-катиона, и благодаря менее выгодным направлениям распада (например,
выброс этильного радикала из М4" при разрыве связи С2—СЗ).
с4н9со+ +о он он
m/zS5 \ II _аш II . _??? . II
? хСзН7-СН2-С-СН2->СзН7-Ь?^СНз-СН2-С-СН2 3^ * СН2-С-СНз
m/z 142 m/z 100 m/z5%
C4H9
m/z 51
Схема 11.37
Задача 8.59. Соединение обладает небольшой молекулярной массой. Пик
с m/z 58 удовлетворяет всем критериям для того, чтобы его можно было
рассматривать в качестве молекулярного. Интенсивность пика [М—Щ+ высока, что
несколько затрудняет расчет элементного состава молекулы по пикам изотопных
ионов. Тем не менее установить, что в молекуле три атома углерода, достаточно
легко, а два возможных состава — СзНбО и C3H3F. Интенсивные пики фрагмент-
ных ионов с m/z 28 и 29 позволяют отвергнуть фторсодержащее соединение.
Степень ненасыщенности кислородсодержащего соединения i? = 3 — 3 + 1 = 1.
Реальных изомеров с такой брутто-формулой всего 7. Для установлении
структуры необходимо обратиться к фрагментным ионам. Крайне низкая интенсивность
пиков с m/z 57, 31 и 43 позволяет отвергнуть структуры пропен-2-ола-1, ме-
тилоксирана и ацетона. Интенсивные пики фрагментных ионов с m/z 28 и 29 не
согласуются со структурой метилвинилового эфира. Молекулярный ион оксетана
рвется преимущественно по противолежащим связям цикла (аналогично цикло-
бутану). В спектре доминирует пик иона 28, но пик иона 29 малоинтенсивен.
Для циклопропанола характерно отщепление гидроксильной группы и молекулы
воды. Полностью соответствует картине фрагментации структура пропионового
альдегида. Наиболее важные ионы [М — Н]+ и НСО+ характерны для низших
альдегидов. Легко представить также образование ионов с m/z 26-29 в результате
самых разнообразных процессов. Следовательно, на рис. 8.67 представлен масс-
спектр пропаналя.
Задача 8.60. Соединение обладает небольшой молекулярной массой. Пик
с m/z 72 удовлетворяет всем критериям для того, чтобы его можно было
рассматривать в качестве молекулярного. По пику М+1 легко установить,
что в молекуле 4 атома углерода, а брутто-формула соединения С4ЩО. По
интенсивности пика иона 44 можно сделать вывод, что ион 43 (максимальный
422 Глава 11. Решения задач
пик в спектре) имеет состав С2Н3О и образуется в результате
элиминирования этильного радикала из М+". Среди возможных изомеров состава С4Н8О
только бутанон-2 распадается подобным образом. Исходя из структуры этого
соединения, легко объяснить также образование остальных ионов, пики которых
имеют значительную интенсивность в спектре. В частности, поскольку при
фрагментации кетонов образуется две пары ацильных и алкильных пиков, то ион
с m/z 43 образуется при элиминировании метильного радикала из М4", а ион с
m/z 29 представляет собой этил-катион. Следовательно, на рис. 8.68 представлен
масс-спектр бутанона-2.
Задача 8.61. г) Наиболее интенсивные пики в спектрах изомерных 2,4-ди-
метилциклогексанона и 3-этилциклогексанона будут обусловлены ионами с
разными величинами m/z. Основное направление распада алкилзамещенных
циклогексанонов связано с разрывом цикла и элиминированием алкильного
радикала, которому предшествует миграция атома водорода (схема 8.64). В
случае 2,4-диметилциклогексанона метильная группа из положения 4 будет
элиминироваться в составе нейтрального фрагмента, а два основных иона с
пиками примерно равной интенсивности будут иметь величины m/z 55 и 69.
В случае 3-этилциклогексанона подобный процесс приводит к образованию
основных ионов с величинами m/z 55 и 83, причем интенсивность последнего
пика должна быть выше (схема 11.38). Достаточно высокой интенсивностью
будет обладать также пик иона [М—С2Н5]4".
m/z 55
m/z 55
m/z S3
Схема 11.38
Задача 8.62. Общий вид спектра позволяет заключить, что соединение
принадлежит алифатическому ряду. Молекулярный ион имеет массу 100. Можно
предположить наличие атома брома (ионы 98 и 100), однако величины m/z
фрагментных ионов дают основание отвергнуть эту гипотезу. К сожалению,
Глава 11. Решения задач 423
крайне низкая интенсивность пика М+* не позволяет сделать вывод об
элементном составе соединения по изотопным пикам. Достаточно интенсивный
пик (m/z 82) обусловлен отщеплением молекулы воды из М+\ Этот процесс
характерен для спиртов и карбонильных соединений. Ион 82 предоставляет
также возможность рассчитать, что в его составе 6 атомов углерода (по иону
А+1, m/z 83). Следовательно, брутто-формула соединения СбН^О, а его
степень ненасыщенности 1. Этим параметрам удовлетворяют альдегиды, кетоны,
ненасыщенные или карбоциклические спирты. Невыраженная спиртовая серия
гомологических фрагментных ионов ставит под сомнение варианты спиртов.
Наиболее интенсивный пик в спектре (ион с m/z 44) характерен для альдегидов
с незамещенным ?-атомом углерода. Об этом же свидетельствует интенсивный
пик иона с m/z 56 (ион [М—44], разд. 8.10). Пик иона с m/z 44 не может иметь
столь высокую интенсивность в случае кетонов любого состава. Следовательно,
на рис. 8.69 представлен спектр одного из изомеров гексаналя. Весьма
затруднительно сделать вывод о месте разветвлений в углеродном скелете. Понятно, что
цепь не может иметь разветвлений в положении 2 (ион с m/z 44). Метальный
заместитель в положении 3 или 4 приводит к перераспределению интенсивностей
пиков ионов с m/z 44 и 56. Поскольку олефиновый фрагмент, конкурирующий
за заряд с енольной формой ацетальдегида в перегруппировке Мак-Лафферти,
оказывается в этих случаях разветвленным, его энергия ионизации ниже, чем
кислородсодержащего фрагмента. Как следствие, пик иона с m/z 56 становится
интенсивнее, чем пик иона с m/z 44. Таким образом, с большой степенью
вероятности можно считать углеродный скелет гексаналя неразветвленным. На
рисунке 8.69 представлен масс-спектр «-гексаналя.
Задача 8.63. Ион с m/z 100 хорошо проходит тест на то, чтобы быть
молекулярным. Его состав легко рассчитать по изотопным пикам. СбН^О, при
степени ненасыщенности 1. Наиболее интенсивные пики в спектре принадлежат
четноэлектронным ионам с массами 85, 57 и 43, а также перегруппировочному
иону с массой 58. Этот ион характерен для метилкетонов и а-метилальде-
гидов. Он образуется в результате перегруппировки Мак-Лафферти. Можно
допустить наличие в структуре молекулы одного из двух фрагментов СН2СОСН3
или СНзСНСНО. Эти фрагменты должны быть связаны с пропильным или
изопропильным заместителем. Следовательно, необходимо выбрать одно из
четырех возможных карбонильных соединений. Достаточно интенсивный пик
иона [М—СНз]+ позволяет исключить из рассмотрения оба альдегида. Напротив,
пик этого иона должен присутствовать в спектрах обоих метилкетонов. Сделать
выбор между гексаноном-2 и 4-метилпентаноном-2 действительно очень сложно.
Дойдя до этого места, можно считать, что задача решена. Полезным, но не
очень надежным критерием выбора может служить интенсивность пиков ионов
43 и 44. Расчет числа атомов углерода в ионе 43 по интенсивности пика 44
дает 2,7. Это означает, что пик 43 обусловлен ионами двух составов (СНзСО и
424 Глава 11. Решения задач
С3Н7), причем доля второго больше. Точный анализ интенсивностей пиков этих
ионов можно было бы легко сделать с помощью масс-спектрометрии высокого
разрешения. Учитывая, что пик иона СНзСО+ очень интенсивен в спектрах
метилкетонов, тот факт, что интенсивность пика иона С3Н7" еще выше означает,
что структура молекулы благоприятствуют образованию этого углеводородного
иона. На рисунке 8.70 представлен масс-спектр 4-метилпентанона-2.
Задача 8.64. Присутствие в спектре лишь нескольких интенсивных пиков
и отсутствие пиков в области низких масс свидетельствует об ароматической
природе соединения. Малолинейчатость спектра позволяет также вычислить
элементный состав для основных ионов (m/z 75, 95, 123, 218). Получаем, что
в составе ионов 75 и 95 по 6 атомов углерода (к сожалению, отсутствуют пики
А+2), состав иона 123 — C7H4OF, а иона 218, который является молекулярным —
Ci3HgOF2- Степень ненасыщенности двух последних ионов равна 5 и 9
соответственно. Разница в массах между ионами 123 и 218 составляет 95 Да, что,
кстати, соответствует массе второго по интенсивности иона в спектре. Разница в
массах между ионами 123 и 95 составляет 28 Да, что с учетом состава можно
интерпретировать как выброс молекулы СО. Тогда разница в массах между ионами
95 и 75 в 20 Да обусловлена выбросом молекулы HF. Вероятно, ион 95 имеет
структуру фторфенила, а ион 123 —фторбензоила. Тогда, соединение, скорее
всего, является ди(фторфенил)кетоном. Выявить расположение атомов фтора в
бензольных ядрах по масс-спектру невозможно. На рисунке 8.71 представлен
масс-спектр 4,4'-дифторбензофенона.
Задача 8.65. Перегруппировка Мак-Лафферти протекает через шестичлен-
ное переходное состояние в молекулярных ионах карбонилсодержащих
соединений и замещенных ароматических соединений. К месту локализации неспарен-
ного электрона мигрирует 7-атом водорода, после чего происходит разрыв между
а- и /3-атомами (С, N, О, S) цепи. Протеканию перегруппировки могут мешать
пространственные препятствия, наличие кратных связей между а- и /?-, /3- и 7~>
7- и 5-атомами цепи. Среди перечисленных соединений претерпевают
перегруппировку гексаналь, пропилбензол, этилформиат, бутилбутаноат, бутилбензол.
Задача 8.66. а) Масляная кислота имеет углеродную цепь достаточной
длины для протекания перегруппировки Мак-Лафферти. В молекуле изомасляной
кислоты цепь оказывается слишком короткой. В результате в спектре масляной
кислоты будет присутствовать интенсивный пик иона с m/z 60. В спектре
изомасляной кислоты его не будет.
д) Отличий будет много. Выводы можно сделать, рассматривая основные пе-
регруппировочные ионы. Перегруппировка Мак-Лафферти в гексановой кислоте
приведет к образованию иона с m/z 60. Достаточно высокой интенсивностью
будет обладать также пик иона (m/z 61), образующегося в результате миграции
Глава 11. Решения задач 425
двух атомов водорода. Для пропилпропионата перегруппировки Мак-Лафферти
и Мак-Лафферти+1 возможны только по спиртовой цепи. В его спектре будут
наблюдаться интенсивные пики перегруппировочных ионов с m/z 74 и 75.
л) Перегруппировка Мак-Лафферти позволяет различить изомерные винил-
пропионат и этилакрилат. В первом случае перегруппировка невозможна ни по
спиртовой, ни по кислотной цепи. Во втором — перегруппировочный процесс
по спиртовой группе приводит к потере молекулы этилена и образованию
нечетноэлектронного иона с m/z 72.
Задача 8.67. а) Бутиловый эфир масляной кислоты может претерпевать
перегруппировку Мак-Лафферти по кислотной (m/z 116) и спиртовой (m/z 88)
цепи (схема 11.39). Кроме того, возможна перегруппировка Мак-Лафферти+1
(m/z 89). Механизм этого процесса представлен на схеме 8.71. Конечным
продуктом является ион со структурой молекулярного иона уксусной кислоты
(m/z 60).
+ +· +·
ОН О О
с3н7—с-он« ~°4?7—с3н7—с-о-с4н9 "-°4?8—>с3н7—с-он
m/z 89 m/z 144 m/z 88
он
-QH8
сн2—с-о-с4н7
m/z116 m/z 60
Схема 11.39
г) Для изопропилпропионата перегруппировки Мак-Лафферти (m/z 74) и
Мак-Лафферти+ 1 (m/z 75) возможны лишь по спиртовой цепи (схема 11.40).
Кислотная цепь слишком коротка для реализации необходимого шестичленного
переходного состояния.
+ +. +.
ОН . ° СН3 °
с2н5—с-он « "СзН5* с2н5—с-о-сн" 3 "СзНб > с2н5— с-он
m/z 75 m/z П6 ЧСН3 m/z 74
Схема 11.40
Задача 8.68. Важнейшие характеристические направления распада
молекулярных ионов декановой кислоты включают образование перегруппировочных
ионов (m/z 129 и 73) по механизму, представленному на схеме 8.67.
Перегруппировка Мак-Лафферти приводит к иону с m/z 60. Двойная миграция
атомов водорода обусловливает образование иона с m/z 61. Эти четыре процесса
отражены на схеме 11.41. Кроме того, достаточно высокую интенсивность имеют
426 Глава 11. Решения задач
пики ионов кислотной (87, 101, 115...)? алкановой B9, 43, 57...) и алкеновой
D1, 55. 69...) гомологических серий. Эти ионы образуются по самым
разнообразным механизмам и могут быть первичными, вторичными, третичными
и т. д. Ион с m/z 129 образуется также по механизму, представленному на
схеме 8.68 с элиминированием пропильного радикала, включающего а-, /?- и
7-атомы углерода исходной молекулы. Этот ион имеет циклическую структуру
(схема 11.41). Аналогичный процесс с элиминированием этильного радикала,
включающего а- и /?-атомы углерода исходной молекулы, приводит к иону
кислотной гомологической серии с m/z 143.
+ +
он о
ch3--c=o« ~QHl6 с9н19-с-он—^
m/z 60 m/z 172
~QHiL
+
ОН
снз-с-он с н
m/z 61 2 5 m/z 129
Схема 11.41
Задача 8.69. Схема 11.42 отражает основные направления фрагментации
М+' метилового эфира 2,4-диметилгептановой кислоты. Перегруппировочные
ионы циклической структуры, образующиеся по механизму, представленному
на схеме 8.68, в данном случае имеют величины m/z 129 и 101. Первый
из них обусловлен отщеплением пропильного радикала с включением а- и
/3-атомов углерода и метильной группы в положении 2 исходной молекулы.
Второй обусловлен выбросом амильного радикала, включающего а-, /?-, 7-атомы
углерода и метальные группы в положениях 2 и 4 исходной молекулы. Ион с
m/z 101 образуется также по другому механизму (схема 8.67) и имеет линейную
структуру (схема 11.42). Перегруппировка Мак-Лафферти приводит к иону с
m/z 88, пик которого максимален в спектре. Ион с m/z 141 обусловлен ацильным
ионом [М—ОСНз]+. Достаточно высокую интенсивность имеют также пики
ионов кислотной (87, 101,115...), алкановой B9,43, 57...) и алкеновой D1, 55.
Глава 11. Решения задач 427
69...) гомологических серий. Эти ионы образуются по самым разнообразным
механизмам и могут быть первичными, вторичными, третичными и т. д.
+
ОН
+ ·
о
СН-С-ОСНз^5^С3Н7—СН-СН2—СН-С-ОСН3 "^"еСНзО--^ N
m/z 101
I
СН3
m/z 88
+·
ОН
сн2=с-с-осн3
СН3
m/z 101
I
СН3
m/z 172
-QHn
-оси
с3н7—сн-сн2—сн-с=о
I I
СН3 СН3
m/z 141
Схема 11.42
Задача 8.70. Ионы с m/z 85 и 115 образуются при фрагментации М+*
амилового эфира валериановой кислоты в результате ?-разрывов. Перегруппировка
Мак-Лафферти по спиртовой цепи обусловливает возникновение ионов с m/z 102
и 70 (схема 11.43).
Ь 5н ОН
С^-С^б^^1 С4Н9-^
шД85 m/??? m/z 130 m/z 61
-QH9
-ОД
10
C4H9-C-OH
m/z 103
C5H10
m/z 70
о ч
fell ± n
C4H9—C-0=CH2 C4H9— C-OH
m/z IIS m/z 102
Схема 11.43
-СзНб
fell
**СНз-С-ОН
m/z 60
Меньшая энергия ионизации иона (m/z 70) делает его пик значительно более
интенсивным по сравнению с альтернативным. Ион с m/z 103 появляется в
результате перегруппировки Мак-Лафферти+1. Перегруппировка Мак-Лафферти
по кислотной цепи приводит к иону с m/z 130, который далее распадается по
механизмам перегруппировок Мак-Лафферти и Мак-Лафферти+1 до ионов с
m/z 60 и 61. Кроме того, достаточно высокую интенсивность имеют пики ионов
кислотной (87, 101, 115...), алкановой B9, 43, 57...) и алкеновой D1, 55,
69...) гомологических серий. Эти ионы образуются по самым разнообразным
механизмам и могут быть первичными, вторичными, третичными и т. д.
428 Глава 11. Решения задач
Задача 8.71. а) Основные процессы фрагментации 2-метилбутановой
кислоты под электронным ударом представлены на схеме 11.44. Помимо указанных
ионов, характеризующихся максимальными по интенсивности пиками, в спектре
будут наблюдаться пики ионов кислотной, алкановой и алкеновой серий.
+
ОН
II
сн-с-он
I
сн3
«А 74
-С2Н4
t>
С2Н<—СН-С-ОН -°н\ СоН.—СН-С=5
СН3
m/z 102
I
СН3
m/z 85
-С4Н9
СООН"*"
m/z 45
Схема 11.44
-СО
С4Н9
m/z 57
и) Основные процессы фрагментации бензилацетата под электронным
ударом представлены на схеме 11.45.
+d
О
с7н?«—сн3 -с-осн2— рь -=^сн3 -с-б=сн2-^^но=сн2
m/z 91 m/z 150 m/z 73 m/z 31
""^2?2
C5H5 С6Н5
m/z 65 m/z 77
~~Q?h
C3H3
m/z 39
-C2H2
PhCH2OH+ *—??* Ph— CH-OH
m/z108
m/z 107
-CO
C4H3
m/z 51
CH,—C=0
m/z 43
C7H9
m/z 79
Схема 11.45
Ацетил-катион (m/z 43), тропилий-катион (w/z 91) и фрагмент с m/z 73
образуются в результате ?-разрывов. Последний из них далее теряет молекулу кетена
в результате вторичного перегруппировочного процесса через четырехчленное
переходное состояние (m/z 31). Такой процесс может протекать также
непосредственно в М+". В этом случае возникает фрагмент со структурой бензилового
спирта (m/z 108), который далее последовательно теряет атом водорода (m/z 107)
и молекулу СО (m/z 79). Фенильный (m/z 77) и тропилиевые ионы в результате
вторичных процессов образуют ароматическую серию ионов (m/z 39, 51, 65).
Глава 11. Решения задач 429
Задача 8.72. Молекулярный ион, вероятно, имеет массу 74 Да. По
изотопным пикам можно определить, что в составе молекулы три атома углерода и
два атома кислорода. Обратите внимание, что интенсивности изотопных пиков
несколько занижены по сравнению с теоретическими значениями в связи с
высокой интенсивностью пика иона [М — Н]+. Состав соединения СзНбОг, степень
ненасыщенности 1. Интенсивные пики ионов с m/z 45 и 73 характерны для
спиртов, кислот и эфиров. Поскольку циклические структуры исключаются по
условию задачи, можно рассмотреть лишь 1-гидроксипропанон-2, пропионовую
кислоту, этилацетат, этилформиат, 2-гидроксипропаналь, 3-гидроксипропаналь,
2-метоксиэтаналь. Для сложных эфиров и 2-метоксиэтаналя не характерно
отщепление гидроксильной группы и молекулы воды, тогда как пики ионов с
m/z 67 и 56 весьма интенсивны в спектре на рис. 8.86. Кроме того, ацетил-
катион (m/z 43) должен иметь максимальный пик в спектре метилацетата, а
этилформиат может претерпевать перегруппировку Мак-Лафферти с образованием
молекулярного иона муравьиной кислоты (m/z 46). Гидроксипропанон должен
иметь в спектре два доминирующих пика (ионы СН3СО" и СН2ОН4"). Для 2-гид-
роксипропаналя должен быть маловероятен выброс атома водорода, но весьма
благоприятен — выброс метильной группы. Поскольку пик иона [М —СНз]+ в
приведенном спектре отсутствует вовсе, альдегид такой структуры можно также
исключить из рассмотрения. Для фрагментации 3-гидроксипропаналя не
характерен ион с m/z 45, а максимальный пик в спектре должен быть обусловлен ионом
СН20Н+ (m/z 31). Единственным соединением, удовлетворяющим условию
задачи, является пропионовая кислота. Углеводородные ионы с m/z 26-29
обусловлены этильной группой. Карбоксильная группа обусловливает ион с m/z 45.
Отщепление гидроксильной группы и молекулы воды приводит к появлению
ионов с m/z 57 и 56. Ион [М —Н]+ образуется в результате отщепления атома
водорода метильной группы по механизму аллильного распада енольной формы
М+' (схема 11.46). Следовательно, на рис. 8.86 представлен спектр пропионовой
кислоты.
Ь ? ОН # ОН
СНз—СН2—С-ОН « ®J-OH-^C--OH ~Н' » СН2=СН-±С-ОН
Схема 11.46
Задача 8.73. Отсутствие в спектре пика ?+2 не позволяет установить
точный элементный состав соединения. По пику М+1 можно сделать вывод о
наличии в молекуле четырех атомов углерода. В спектре отсутствуют пики пере-
группировочных ионов. Высокая интенсивность пика иона [М — 31]+ при низкой
интенсивности пика иона с m/z 31 характерна для метиловых эфиров карбоновых
кислот. Пик иона с m/z 60 (изотопного для m/z 59) имеет интенсивность 2,2%,
т. е. ион содержит два атома углерода и обусловлен потерей этильной группы
430 Глава 11. Решения задач
из М+\ Объединение этих наблюдений приводит к однозначному варианту
решения. На рисунке 8.87 представлен спектр метилпропаноата.
Задача 8.74. Точные величины интенсивностей изотопных пиков позволяют
легко рассчитать элементный состав М"': С4Н8О2 при степени ненасыщенности
1. Интенсивный пик иона с m/z 73 связан с элиминированием метильного
радикала, а ион с m/z 71 —с элиминированием гидроксильного радикала из М+\
В спектре отсутствуют интенсивные пики перегруппировочных ионов, что
позволяет исключить из рассмотрения 4-гидроксибутанон-2, 1-метоксипропанон-2,
масляную кислоту, этилацетат, пропилформиат и изопропилформиат. Отсутствие
в спектре пика иона [М — ОСНз]+ дает возможность отвергнуть метилпропионат
и 1-гидроксибутанон-2. Пользуясь правилами распада альдегидов, спиртов и
простых эфиров, достаточно просто исключить любые альдегиды со спиртовой
или алкоксильной группой. Циклические соединения исключены по условию
задачи. Единственным вариантом, удовлетворяющим спектру, является изомас-
ляная кислота. Ион с m/z 45 обусловлен карбоксильной группой, с m/z 43 —
изопропильной группой. Ион [М —СНз]+ образуется в результате отщепления
метильной группы по механизму аллильного распада енольной формы М+*
(схема 11.47). Следовательно, на рис. 8.88 представлен спектр изомасляной
кислоты.
сн\ 1? СНз лг ?н · ?н
чсн-сн2—с-он « чС№1сн-^с--он ~СНз» сн=сн— с-он
СНз7 СНз7 СНз7
Схема 11.47
Задача 8.75. В составе наиболее тяжелого иона (m/z 104) всего три атома
углерода (по пику 105). Спектр позволяет также получить определенную
информацию о составе ионов с m/z 87 и 60. В первом вновь три атома углерода
(отщепление гидроксильного радикала), а во втором —два. Благоприятное
отщеплении 44 а. е. м. с одним атомом углерода возможно только в случае СОг.
Для протекания такого процесса необходима циклическая структура, например,
лактонов, ангидридов дикарбоновых кислот либо наличие в линейной молекуле
других гетероатомов, возможных центров локализации заряда и неспаренного
электрона (кетокислоты, дикарбоновые кислоты). Важную информацию можно
извлечь из иона с m/z 69. Он может возникнуть только в результате
последовательных выбросов гидроксильного радикала и молекулы воды. Следовательно,
в молекуле соединения скорее всего содержатся две гидроксильные группы,
причем, по крайней мере, одна из них карбоксильная. Это устраняет варианты
лактона и циклического ангидрида.
Глава 11. Решения задач 431
Три атома углерода, три атома кислорода и два атома водорода имеют
массу 86 Да, т. е. на остальные элементы приходится 18 а. е. м. Поскольку
присутствие атома азота невозможно (четная молекулярная масса), единственным
вариантом состава может быть С3Н4О4 при степени ненасыщенности 2. Так
как циклические структуры уже отвергнуты, остается три возможных варианта:
З-гидрокси-2-оксопропановая кислота, 2-гидрокси-З-оксопропановая кислота и
малоновая кислота.
Молекулярный ион З-гидрокси-2-оксопропановой кислоты не может
претерпевать перегруппировку Мак-Лафферти с выбросом СО2. Среди направлений
ее распада будут превалировать потери частиц СЩОН', НООСГ и
образование иона СНгОН*. Перегруппировка Мак-Лафферти с элиминированием СОг
возможна для М+# 2-гидрокси-З-оксопропановой кислоты, однако, значительной
интенсивностью должны обладать пики ионов [М — НООС]+ и [М — НСО]+.
Эти пики отсутствуют в приведенном спектре. Следовательно, на рис. 8.89
представлен масс-спектр малоновой кислоты. Все фрагментные ионы легко
объяснимы очевидными направлениями распада этого соединения.
Задача 8.76· В спектре ярко выражен выброс из М+* 44 Да. Однако в
данном случае благодаря высокой интенсивности пиков ключевых ионов легко
определить их элементные составы по пикам А+1 и А+2. Для иона с m/z 148,
по-видимому, молекулярного, состав СвЩОз; а для иона с m/z 104 —С7Н4О. В
спектре отчетливо прослеживается ароматическая серия (m/z 76, 50, 38), причем
такой ряд (не m/z 77, 51, 39) характерен для дизамещенного бензола. Если учесть
наличие в молекуле дизамещенного бензольного ядра, состав остальных
структурных элементов С2О3. Такой состав возможен только для фталевого ангидрида.
Следовательно, на рис. 8.90 представлен масс-спектр фталевого ангидрида.
Задача 8.77. Молекулярная масса соединения 116 Да, причем оно содержит
6 атомов углерода в молекуле (пик М+1). Интенсивные пики ионов с m/z 60 и
61 характерны для жирных кислот и их эфиров. В спектре задачи интенсивны
также пики перегруппировочных ионов с m/z 88 и 89. Отщепления 28 и
27 а. е. м. характеризуют перегруппировку Мак-Лафферти и Мак-Лафферти+1
для этиловых эфиров карбоновых кислот. Об этиловом эфире дополнительно
свидетельствует ацильный ион с m/z 71, образующийся при отщеплении этоксиг-
руппы из М+\ Поскольку в спектре наблюдается также достаточно интенсивный
пик нечетноэлектронного иона с m/z 60, вероятно, первичный ион с m/z 88
претерпевает еще одну перегруппировку с элиминированием молекулы этилена.
Такая перегруппировка возможна по кислотной цепи сложного эфира.
Отщепление этилена указывает на масляную кислоту. Следовательно, на рис. 8.91
представлен спектр этилбутирата.
432 Глава 11. Решения задач
Задача 8.78. а) Для N-этилгексанамида возможны последовательные
процессы по механизму перегруппировки Мак-Лафферти по кислотной цепи и по
алкильному заместителю у атома азота. Еще одна перегруппировка идет через
четырехчленное переходное состояние в ионе [М —СНз]+. Указанные процессы
представлены на схеме 11.48.
° '-с4н8 -су^
Он
II
СН2— C-NH-C2H5
гн\ Ь
С5Нц—C-NH-C2IV CH3—C-NH2
C5Hn—C-NH
r
О
с5ни-с-йн=сн2 -^^° , Йн2=сн2
Схема 11.48
Задача 8.79. д) Два этих изомера легко различить по фрагментным ионам,
образующимся в результате перегруппировки Мак-Лафферти. В спектре де-
канамида интенсивный пик будет обусловлен распадом по кислотной цепи
с выбросом молекулы октена (ион m/z 59). Для N-амилпентанамида
перегруппировка Мак-Лафферти протекает по обеим алкильным цепям. Распад по
кислотной цепи с выбросом молекулы пропена приводит к иону с m/z 129, а
по амильной группе —к иону с m/z 101 (выброс молекулы пентена). Пики этих
ионов будут достаточно интенсивны. Вторичные перегруппировочные процессы
в обоих указанных первичных ионах приведут к возникновению иона с m/z 59,
аналогичного образующемуся при распаде деканамида.
Задача 8.80. Разрыв связи С—N в М+" приводит к ацильному иону с
m/z 71. Этот же разрыв с миграцией атома водорода к азоту ведет к иону со
структурой гексиламина {m/z 101), дальнейший распад которого обусловливает
образование иона с m/z 30 (максимальный пик в спектре). Аминный тип распада
отвечает за возникновение двух фрагментных ионов (m/z 128 и 100). Дальнейший
распад первого из них приводит к иону с m/z 44, а второго — вновь к иону с
m/z 30. Результат перегруппировки Мак-Лафферти по кислотной цепи —ион с
m/z 143, а по аминному заместителю — ион с m/z 87. Ион с m/z 114 образуется в
результате /^-разрыва в аминном заместителе. Распадаясь далее, он превращается
в циклический фрагмент с m/z 44. Помимо приведенных на схеме 11.49 в
Глава 11. Решения задач 433
О
С3Н7+ « ~°° С3Н7с5 C3H7-C-NH -ОДбОО, ДН2
«Л 43 m/z 71^ ^ СН2—СН2 СН2—СН2
m/z 114 /иД44
-С4Н9
СН2—C-NH-СбНв ^ZSiL C3H7—C-NH-C6H13 ~СзН7'» C6H13NHCO ~СбН"» NH2CO
«/г 143 т/г 171 "^--^_ m/*128 «А44
C6H13NH2
QH12
?* 6н о
СН3—C-NH2 «"ОД* с3Н7—C-?? C3H7—C-NH=CH2 _?^?^_^ Йн2=СН2
тД59 тД87 т/^ 100 m/z 30
Схема 11.49
спектре отчетливо проявляются другие пики ионов аминной E8, 72, 86...),
перегрушгаровочной амидной G3, 87, 101...), алкановой и алкеновой серий.
Задача 8.81. Если считать, что М+* имеет массу 73 Да, соединение должно
содержать нечетное число атомов азота. Расчет изотопного кластера М+' (с
учетом наложения со стороны иона [М —Н]+) дает в качестве наиболее
вероятного варианта присутствие в составе трех атомов углерода и одного азота.
Вариантами элементного состава молекулы будут СзН7>Ю и C3H4NF. Степень
ненасыщенности указанных соединений 1 и 2 соответственно. Интенсивный пик
иона с m/z 44 в спектре характерен для фрагментации аминов и амидов. С
учетом возможных элементных составов ион с m/z 57 может образоваться только
при отщеплении аминогруппы из М+". Этот процесс наблюдается при распаде
под ЭУ первых членов ряда незамещенных по азоту амидов жирных кислот, но
абсолютно не характерен для аминов. На этом основании можно исключить из
рассмотрения состав C3H4NF. Единственным приемлемым вариантом остается
амид пропионовой кислоты. Ион с m/z 57 имеет состав С2Н5СО, а ион с m/z 44 —
NH2CO. На рисунке 8.93 представлен спектр пропанамида.
Задача 8.82. Дублеты, на которые обращается внимание в условии,
обусловлены ионом, содержащим один атом хлора (соотношение интенсивностей
пиков ~3 : 1). Следует также отметить, что первый из этих ионов, скорее всего,
перегруппировочный, поскольку имеет четную массу. В составе соединения
не должно быть атомов азота. В противном случае в области низких масс
наблюдались бы интенсивные пики с четной массой.
15—1113
434 Глава 11. Решения задач
Основные пики в спектре принадлежат ионам алкановой (возможно, ациль-
ной) и алкеновой серий. Поскольку интенсивные пики перегруппировочных
ионов не характерны для алкилгалогенидов, соединение должно принадлежать
к классам хлорангидридов, хлоркетонов или хлоральдегидов. Наиболее тяжелый
ион, не содержащий атома хлора, имеет массу 99 Да. По изотопному пику А+1
можно установить, что в его составе 6 атомов углерода. По всей видимости,
состав этого иона СбНцО, а образуется он при отщеплении атома хлора из
М+\ Тогда хлорсодержащие ионы (m/z 105-108) образуются при отщеплении
из М+" молекулы этилена и этильного радикала. Брутто-формула соединения
СбНцОС1. Столь интенсивный пик, обусловленный отщеплением атома хлора
из М4", значительно лучше согласуется со структурой хлорангидрида, чем со
структурами изомерных альдегидов или кетонов, для которых должны
доминировать распады, связанные с карбонильной группой. В спектре наблюдались бы
другие пики. Следовательно, речь идет о хлорангидриде гексановой кислоты.
Для того чтобы установить структуру углеводородного скелета, вновь
обратимся к хлорсодержащим фрагментам. Дублет с m/z 78, 80 обусловлен
перегруппировкой Мак-Лафферти и означает, что у ?-атома углерода разветвлений нет.
Дублет ионов с m/z 91, 93 образуется в результате перегруппировочного процесса
(схема 8.67) или аллильного разрыва енольной формы соединения (схема 11.47).
Регистрируемая величина m/z этого иона означает, что заместителей нет и
у /3-атома углерода. Выбор между изомерными гексановой и изогексановой
кислотами позволяет сделать еще один хлорсодержащий ион. Отщепление
этильной группы обусловлено процессом образования циклического пятичлен-
ного хлорониевого иона (схема 8.51). Любые разветвления в углеводородной
цепи подавляют этот процесс. Кроме того, отщепление этильной группы и
молекулы этилена вообще крайне маловероятно из М+в изогексаноилхлорида.
Следовательно, на рис. 8.94 представлен масс-спектр гексаноилхлорида.
Задача 8.83. Считая, что молекулярная масса соединения 255 Да, а
фрагменты основной гомологической серии имеют четные массы, можно сделать вывод,
что вещество является амином или амидом. Ионы с m/z 59 и 72, пики которых
максимальны в спектре, характерны для амидов жирного ряда.
Молекулярная масса 255 Да действительно соответствует гексадеканамиду. К сожалению,
низкая интенсивность пика М+* не позволяет рассчитать элементный состав
соединения по изотопным пикам с достаточной степенью надежности. Тем не
менее состав C16H33NO не противоречит интенсивности пика М+1. Несложно
сделать выводы и о структуре углеводородной цепи. Массы перегруппировочных
ионов ( m/z 59 и 72) характерны для неразветвленной углеводородной цепи у а-
и /3-атомов углерода. Повышенные интенсивности пиков ионов с m/z 128 и 184
в ряду пиков характеристической серии позволяют заключить, что углеродный
скелет молекулы неразветвлен (схема 8.67). Следующий ион с аномальной
интенсивностью должен иметь массу 240 Да. Однако процессы с элиминирова-
Глава 11. Решения задач 435
нием метильного радикала обычно подавлены более энергетически выгодными
процессами. Повышенная интенсивность пика иона с m/z 212 объяснима на
основе механизма элиминирования пропильного радикала, включающего ?-, ?-
и 7-атомы углерода исходной молекулы. Механизм этого процесса для случая
кислот представлен схемой 8.68. Следовательно, на рис. 8.95 представлен масс-
спектр нормального гексадеканамида.
Задача 8.84. а) Основные процессы распада М+' N-этилбензамида
представлены на схеме 11.50. Доминировать в спектре будут ионы с m/z 149, 105, 77.
О
Ph—C-NH-
m/z 149
-?*
-с2н5 -
PhCCT
m/z 105
~QH2.
> Ph
m/zTJ
+ -Oft,
C4H3
m/z 51
О
II +
Ph—С- NH=CH-CH3
m/z148
Ph—C-NH=CH2
m/z 134
Схема 11.50
Ph—C-NH2
m/z 121
г) Основные процессы распада М+* бромангидрида валериановой кислоты
представлены на схеме 11.51.
ь
II
С4Н9—С—Вг
m/z 164,166
-С3Н6
+
ОН
СН2—С-Вг
m/z 122, 124
> С4Н9СО
/иД85
-00
С4Н9
m/z 57
СН2=СН—С-Вг
m/z 135, 137
Схема 11.51
Задача 8.85. Считая ион с m/z 149 молекулярным, делаем вывод о нечетном
числе атомов азота в молекуле. Расчет элементного состава соединения по
изотопным пикам приводит к брутто-формуле C9H11NO. На ароматическую природу
соединения указывают пики характеристических фрагментов с m/z 91, 77, 65, 51,
39 и др. Доминирующий в спектре пик, вероятно, обусловлен ионом анилиновой
структуры, образующимся в результате перегруппировочного процесса. Состав
436 Глава 11. Решения задач
этого иона C6H7N можно вычислить по интенсивности пика А+1. Вторичные
ионЙ1 с m/z 66 и 65 образуются при элиминировании из М+" анилина частиц
HNC и H2NC\ Ион с m/z 120 образуется при элиминировании 29 Да из М+\
Учитывая, что в состав молекулы входит фрагмент PhNH, можно оценить два
варианта структур соединения: N-фенилпропанамид и 2-фениламинопропаналь.
Молекулярный ион альдегида должен помимо отщепления 29 Да (НССГ)
элиминировать также метильный радикал. Поскольку пика этого фрагментного иона
в спектре не наблюдается, можно сделать вывод, что на рис. 8.96 представлен
спектр N-фенилпропанамида.
Задача 8·86. Процесс возникновения иона с m/z 60 обусловлен
изомеризацией части молекулярных ионов нитродекана в нитритную форму. Механизм
трансформации представлен на схеме 11.52.
С9Н19—СН2—N02 * С9Н19— СН2—fr-NO -°9Hl9 » CH2=5-NO
m/z 60
Схема 11.52
Задача 8.87· На рисунке 8.105, ? представлен масс-спектр opmo-изомера, а
на рис. 8.105,6—л*ета-изомера. Помимо общих направлений распада в первом
случае активно идут процессы, приводящие к образованию ионов с m/z 120 и 92
(схемы 8.92 и 11.53), а во втором —ионов с m/z 107 и 93 (схема 11.53).
40-NO
Схема 11.53
Задача 8.88. Основные направления распада молекулярного иона онитро-
фенола представлены на схеме 11.54. Наличие нитрогруппы полностью
подавляет «фенольный» путь распада, который реализуется только после
предварительного элиминирования нитрогруппы, в ионе [?—N02]+ с m/z 93. Изомеризация
Глава 11. Решения задач 437
М+* в нитритную форму обусловливает возникновение ионов с m/z 109 и 81, а
орто-эффект — ионов с m/z 122 и 94.
Он
-NO
?
m/z109
-со
c5H5or
m/z 81
ОН I
ОН Г
-ОН*
отД 139
-??2
?02
О
tfo
/иД 120
ОН
¦^~ с5н5+
m/z 93
/яД 65
Схема 11.54
-со-* CsHtNO1"
/яД94
.^???2
С6Н40+
m/z 92
Задача 8.89. в) На схеме 11.55 представлены основные первичные и
вторичные направления распада л-пропилнитробензола. Они связаны с первоначальной
фрагментацией по обоим заместителям в бензольном ядре.
m/z 119
k
-no;
с7н7
тД91
с7нГ
m/z 90
4
-NO2*
C3H7
СбНГ
m/z 78
A
-CO
m/z 106
C3H7
/яД 149
Схема 11.55
/яД135
тяД 107
438 Глава 11. Решения задач
ж) Основные направления распада симметричного диамилгидразина связаны
с ?-распадом. Поскольку в М+* заряд и неспаренный электрон могут
равновероятно размещаться на любом из атомов азота, элиминироваться может или
бутильный, или амильный радикал (схема 11.56). Вторичный процесс приводит
к иону с m/z 30.
CH2=NH-NH-C5Hir
m/z 115
C5Hn-NH-NH-C5Hir
m/z 172
CH2=NH2
m/z 30
Схема 11.56
-^C5Hn— NH=NH
m/z101
Задача 8.90. Два наиболее важных процесса распада М+' оксима пентаналя
являются общими для всех оксимов алифатического ряда. Перегруппировка Мак-
Лафферти приводит к иону с m/z 59, который далее отщепляет молекулу воды и
превращается в ион с m/z 41 (схема 11.57). Отщепление молекулярным ионом
алкильного радикала (в данном случае этильного) сопровождается циклизацией
(ион с m/z 72). Интенсивный пик (m/z 43) обусловлен ионами двух составов:
С3Н7 и HCNO.
НИ'
СН3 /н^ он
НО N -Си" V N" -eft „
U1* U ~^cj
m/zl2 ? m/z 101 m/z 59
ОН
с3н7
m/z 43
t
HCNO
m/z 43
Схема 11.57
-н2о
CH2=C=NH
m/z 41
Задача 8.91. Нечетная масса наиболее тяжелого иона (вероятно, М+')
свидетельствует о нечетном числе атомов азота в молекуле. Высокая интенсивность
пика этого иона и две группы фрагментов в области 76 и 50 Да характерны
для ароматических соединений. Достаточно легко вычислить элементный
состав молекулы по изотопным пикам: C7H5N, при степени ненасыщенности 6.
Учитывая, что соединение ароматическое, можно уже на этой стадии оставить
лишь два варианта: бензонитрил и бензизонитрил. К сожалению, выбор между
двумя этими изомерами по представленному в задаче масс-спектру сделать
невозможно. На рисунке 8.108 спектр бензонитрил а.
Глава 11. Решения задач 439
Задача 8.92. Достаточно богатая схема фрагментации оксима ацетофенона
(схема 11.58) обусловлена возможностью отщепления из М+* самых
разнообразных частиц.
С4Н3* «-^Н2 Ph -
m/z 51
m/z 11
Ph—C-CH3
m/z 104
m/z 103
m/z 94
Схема 11.58
?2?4_^Ph— C-N=0
m/z 119
Ph-N=C-CH3
m/z№
Некоторые из этих процессов (ионы с m/z 103, 94) требуют значительной
перестройки исходной структуры молекулы. Механизмы этих структурных
изменений остаются мало изученными. Отщепление гидроксильной группы
сопровождается перегруппировкой Бекмана с миграцией заместителя, находившегося
в исходной молекуле в аниш-положении. Этот процесс подробно изучен в
условиях химической ионизации, когда отщепление молекулы воды является
доминирующей реакцией распада протонированного молекулярного иона.
Задача 8.93. Даже предварительный взгляд на спектр позволяет сделать
важные выводы. Соединение имеет ароматический характер (интенсивный пик
М+' и характерная серия ароматических фрагментов с m/z 76, 75, 50), содержит
один атом брома (дублет с соотношением 1:1 пиков М+* и ряда фрагментных
ионов) и нечетное число атомов азота (нечетная молекулярная масса).
Первичные фрагментные ионы обусловлены выбросами 16, 30 и 46 Да из М+\
что однозначно указывает на нитрогруппу. Дополнительным подтверждением
нитроароматической природы вещества является ион [?—??, —СО]4",
представленный в спектре дублетом с m/z 143, 145. Масса атома брома и нитрогруппы
79 + 46 = 125 Да. Оставшиеся 76 Да обусловлены бензольным ядром СбЩ.
Следовательно, мы имеем дело с бромнитробензолом. Установить расположение
заместителей по спектру, к сожалению, нельзя. На рисунке 8.110 представлен
спектр 3-бромнитробензола.
Задача 8.94. Нечетная масса и высокая интенсивность пика молекулярного
иона, а также набор фрагментных ионов (m/z 90, 76, 63, 51, 39) указывают
на ароматическое азотсодержащее соединение. По изотопным пикам (с учетом
наложения со стороны иона [М—Н]+) легко установить, что элементный состав
440 Глава 11. Решения задач
соединения CsHvN при степени ненасыщенности 6. Помимо фенильного ядра
все остальные фрагменты имеют состав C2H2N при степени ненасыщенности 2.
Выброс из М+* 27 Да согласуется со структурой нитрила, изонитрила, анилина
или этинилбензола. Интенсивный пик иона [М—Н]+ при отсутствии пиков
[М—Alk]+ характерен для мети л бензолов и позволяет отвергнуть структуры
изомерных этиниланилинов. В спектре N-этиниланилина должен присутствовать
пик HNCCH+ (m/z 40). В спектрах изомерных фенилацетонитрила и фенилизо-
ацетонитрила помимо пиков ионов, отмеченных в условии, достаточно
интенсивными были бы пики ионов с m/z 91 и 77. Поскольку пики ионов с m/z 40 и 77
отсутствуют в спектре, а интенсивность пика иона с m/z 91 крайне мала, можно
отвергнуть и N-этиниланилин, и фенилацетонитрил, и фенилизоацетонитрил.
Напротив, спектры изомерных метилбензонитрилов и метилбензоизонитри-
лов будут очень близки друг другу и спектру на рис. 8.111. Установить точное
расположение заместителей и нитрильную или изонитрильную природу
соединения по приведенному в условии спектру нельзя. На рисунке 8.111 представлен
масс-спектр 3-метилбензонитрила.
Задача 8.95. Интенсивные пики М+* (m/z 168) и фрагментных ионов
(m/z 92, 76, 75, 50) указывают на ароматическую природу вещества. Первичные
фрагментные ионы (m/z 152, 138, 122) обусловлены выбросами 16, 30 и 46 Да из
М+' и однозначно указывают на нитрогруппу. Поскольку молекулярная масса
вещества четная, в его состав должен входить еще один атом азота. Ион с
m/z 30 (максимальный пик в спектре) может иметь состав NO (характерно для
полинитроароматических соединений) или CH2NH2 (характерно для аминов).
Высокая интенсивность пика М+* позволяет использовать изотопные пики
для установления элементного состава соединения. Обращает на себя внимание
высокая интенсивность пика М+2 при сравнительно небольшой интенсивности
пика М+1. По пику М+1 можно еще раз убедиться, что соединение содержит
два атома азота и ароматическое ядро. Масса фрагмента Св^2 составляет
100 Да, а вклад изотопов 13С в интенсивность пика М+2 —0,2%. По пику
М+2 определяем, что молекула должна содержать 3-5 атомов кислорода. Тогда
элементный состав соединения может быть C6HN2O3F и C6H4N2O4. Вариант
с пятью атомами кислорода не проходит по массе. Вариант с тремя атомами
кислорода также маловероятен. Следовательно, здесь приведен спектр динитро-
бензола. Точное расположение заместителей установить нельзя. На рисунке 8.112
представлен масс-спектр 1,3-динитробензола,
Задача 8.96. Пики ионов с m/z 144 (вероятно, М+') и 101 достаточно
интенсивны, чтобы определить их элементный состав по изотопам. Для М+*
вариантами состава будут C8H20N2, C8HN2F, C9H5P и C9H17F, а для иона с
Глава 11. Решения задач 441
m/z 101 — CoHioF, C5HsNF и C5H13N2. Посколысу ион с m/z 101 обусловлен
выбросом из М+# частицы с массой 43 Да, можно исключить из рассмотрения
фосфорсодержащее соединение и вещество с элементным составом Cgffi^F.
Состав C9H17F может принадлежать либо алкену, либо нафтену. Спектры
этих классов органических соединений абсолютно отличны от представленного
на рис. 8.113. Следовательно, состав анализируемой молекулы CgH^o^ при
степени ненасыщенности 0. Речь может идти либо о диаминоалкане, либо об
алкилгидразине. Обращает на себя внимание тот факт, что в области низких
масс наибольшую интенсивность имеют пики ионов с нечетной массой (m/z 57,
45, 43, 41). Это позволяет отвергнуть вариант диамина. В спектрах этой группы
соединений доминирует аминная серия гомологических ионов (m/z 30, 44,
58...).
Не столь просто установить точную структуру алкилгидразина, поскольку в
составе молекулы может быть от одной до четырех алкильных групп разной
длины и разветвленности. Очень важно, что первичный распад идет фактически
по единственному направлению (выброс пропильного радикала). Учитывая, что
соединение содержит 8 атомов углерода, такая картина фрагментации возможна
только в случае, когда гидразин имеет два бутильных или изобутильных
заместителя.
Образование вторичного иона с m/z 30 можно легко объяснить исходя из
структуры симметричного дибутил- или диизобутилгидразина (схема 11.59). Для
несимметричного изомерного гидразина такой процесс невозможен.
Дополнительным свидетельством в пользу симметричного гидразина является отсутствие
пика иона [М—С3Н7, —NH3]"*" с m/z 84. Пик этого иона, хотя и имеет
незначительную интенсивность, является характеристичным для структуры 1,1-диал-
килгидразинов. Кроме того, заметную интенсивность должен иметь пик иона с
m/z 59 (рис. 8.102 и схема 8.91).
CH2=ife-NH -сзнтсннн _ m +
\Ч — + CH2=NH2
ЬР-СН-СНз
Схема 11.59
Сделать окончательный вывод в пользу 1,2-дибутил- или 1,2-диизобутилгид-
разина достаточно трудно. Можно ожидать большей интенсивности пика иона с
m/z 43 (С3Н7") в случае изобутильного изомера. Однако для использования этого
факта желательно иметь спектры обоих изомеров. На рисунке 8.113 представлен
спектр 1,2-дибутилгидразина.
Задача 8.97. Основные направления фрагментации дибутилсульфоксида
представлены на схеме 11.60. Ионы с m/z 145 и 89 обусловлены первоначальной
442 Глава 11. Решения задач
изомеризацией молекулы в енольную форму. Перегруппировка Мак-Лафферти
отвечает за появление иона с m/z 106, дальнейший распад которого приводит к
возникновению характеристичного для алкилсульфоксидов пика иона с m/z 63.
Достаточно интенсивны пики углеводородных ионов с m/z 57, 41, 29.
+ +# +· -он· +
QH9 C4H9-S-C4H9 ^=?i ОД—S=CH-CjH7 >ОД-8=еН~СзН7
ОН
m/z 51
СзН5+
m/z 41
О
m/z 162
-C4Hg
+· -С4Н7 +
QH9-S-OH ^-CH2=S-OH
m/z 106 m/z 63
Схема 11.60
m/z 145
-C4Hg
КЗ=СН-СзН7
m/z 89
Задача 8.98. Богатство направлений фрагментации подразумевает
многочисленные варианты изомеризации исходной структуры дифенилсульфоксида.
Изобразить эти структуры невозможно, тем не менее состав всех фрагментных
ионов (схема 11.61) достаточно очевиден.
[М-НСО]+-^ [М-СО]+*
m/z 173
[М-НО]+ =^*· [М-НзО]4"
m/z184
+ -CHS' +·
С11Н9-· Ph-S-Ph *
m/z 141 m/z 186
m/z202
m/z125
C0 - C5H5S+
m/z 97
ОД-
m/z 65
¦^°- Pho+
m/z 93
PhS
/иД 109
Схема 11.61
Ph
m/z 77
+ -C2H2
ОН3+
/и/г 51
Глава 11. Решения задач 443
Задача 8.99. Один из наиболее интенсивных пиков в спектре обусловлен
ионом с m/z 63. Этот ион достаточно редок для любых классов органических
соединений и характерен для сульфоксидов. Повышенные интенсивности пиков
А+2 подтверждают наличие в составе атома серы. Точные величины интен-
сивностей пиков изотопных ионов позволяют рассчитать элементный состав
соединения: CoH^SO. Предполагая, что образец является сульфоксидом, следует
обратить внимание на массы фрагментных ионов. Отщепление гидроксильной
группы (m/z 117) характерно для сульфоксидов. Два пика перегруппировочных
ионов (m/z 106 и 78) дают возможность предположить, что два алкильных
радикала представляют собой этильную и бутильную группы. Об этом же
свидетельствуют и пики алкильных ионов с m/z 57 и 29. Интенсивность второго
несколько завышена, поскольку этильный фрагмент может образовываться из
бутильного. Кроме того, часть ионного тока обусловлена изобарным ионом
НСО+. Ион с m/z 41 часто сопутствует бутильному фрагменту. Достаточно
сложно сделать выводы о структуре бутильного радикала, однако изобутильная
группа характеризовалась бы большей интенсивностью пика иона с m/z 43, а
emop-бутильная группа была бы не столь эффективна в перегруппировке Мак-
Лафферти (m/z 78). На рисунке 8.118 представлен масс-спектр бутилэтилсуль-
фоксида.
Задача 8.100. Спектр л-толуилсульфоновой кислоты малолинейчатый.
Среди первичных фрагментов можно выделить ион с m/z 108, обладающий
структурой л-крезола (схема 11.62). Максимальный пик в спектре обусловлен тропилий-
катионом (m/z 91). Ионы с m/z 155, 123, 107 образуются при отщеплении из
М+' частиц ОН", ОзН" и SO2H' соответственно. Два последних иона требуют
предварительной изомеризации сульфогруппы.
С5Н5 .
m/z 65
-с2н2
m/z 91
-SO3H
СН3
\J/—S0$i
m/z 172
>ЗН> ' —— CH3-<Q>-Sc?
m/z 155
-SQ2
-SQ2H·
СН3
m/z108
+·
ОН
CH3-^Q^O+
m/z 107
-со
m/z 79 m/z 123
Схема 11.62
444 Глава И. Решения задач
Задача 8.101. В спектре нафтилфенилсульфона будет достаточно много
пиков. На схеме 11.63 представлены лишь наиболее значимые первичные и
вторичные процессы распада.
Naft—5
m/z 159
C5H5S+
m/z 91
m/z 147
Nail—So
m/z 175
Схема 11.63
Задача 8.102. Общий вид спектра, представленного на рис. 8.119, позволяет
сделать вывод об ароматической природе образца (интенсивные пики ионов
М+* и m/z 91, 78, 77, 51). Учитывая высокую интенсивность пика М+2,
можно предположить наличие в молекуле атома серы или кремния. Точный
расчет элементного состава соединения дает единственный вариант: C9H12SO2.
Любые другие варианты значительно хуже согласуются с условиями. Серия
ароматических ионов с m/z 78, 77, 51 характерна для монозамещенной арома-
тики. Если в молекуле присутствует фенильная группа, то остальные фрагменты
молекулы можно изобразить брутто-формулой C3H7SO2. Отщепление 42 Да из
М+' (ион с m/z 142) может быть связано с элиминированием молекул кетена или
пропилена. Следующая группа фрагментных ионов (m/z 120, 119, 118) должна
быть обусловлена выбросами сульфогруппы и атомов водорода. Такие процессы
характерны для сульфонов. Два указанных процесса позволяют приписать
образцу структуру пропилфенилсульфона. Дополнительным подтверждением этого
служат фрагментные ионы PhSO+ (m/z 125), [PhSO-CO]+ (m/z 97), РЮН4"'
(m/z 94). Интенсивности пиков этих ионов не очень высоки, но они очень
полезны для идентификации. Тропилий-катион (m/z 91) образуется при отщеплении
этильного радикала ионом [М — S02]+". К сожалению, невозможно сделать
однозначный выбор между пропилфенилсульфоном и изопропилфенилсульфоном.
Очень высокая интенсивность пика иона с m/z 43 свидетельствует скорее в
Глава И. Решения задач 445
пользу изопропильного изомера. Тем не менее на рис. 8.119 представлен спектр
пропилфенилсульфона.
Задача 8.103. Поскольку в спектре присутствует лишь несколько пиков,
причем среди них нет характеристических для ароматических соединений,
можно предположить, что соединение содержит несколько одинаковых групп.
Высокая интенсивность пика М+2 свидетельствует о присутствии в молекуле
атома серы, а точный элементный состав соединения C6H14SO2. Отщепление
41 и 42 Да из М+* и образование фрагментных ионов с m/z 41-43 указывает
на пропильную группу, причем тип распада М+' с миграцией одного или
двух атомов водорода характерен для сульфонов. Структура дипропилсульфона
хорошо согласуется с приведенным спектром. Вновь по масс-спектру нельзя
точно установить пропильные или изопропильные группы входят в состав
молекулы. На рисунке 8.120 представлен спектр дипропилсульфона.
Задача 8.104. Малолинейчатость спектра и достаточно высокая
интенсивность пиков позволяет рассчитать элементный состав ионов с m/z 146, 131, 73.
Во всех трех случаях очевидно присутствие кремния. Состав основного иона в
спектре C3H9Si, а двух других C5H15S12 и C6Hi8Si2. Степень ненасыщенности
во всех случаях равна 0. Единственным вариантом структуры, удовлетворяющей
спектру, будет гексаметилдисилан. На рисунке 8.126 представлен масс-спектр
гексаметилдисилана.
Задача 8.105. Подсказка в условии задачи о неорганической природе
соединения несколько упрощает идентификацию. В спектре отчетливо видны три
группы пиков с идентичным изотопным распределением (m/z 462, 335, 208).
Разница в массах между этими группами 127 Да. Кроме того, пик иона с m/z 127
максимален в спектре. Иод является моноизотопным элементом с массой 127.
Поэтому можно сделать вывод, что в составе молекулы два атома иода. На
основании табл. 1 приложений можно легко определить, что группа пиков ионов
с m/z 206-208 обусловлена изотопами свинца. Следовательно, на рис. 8.127
представлен спектр иодида свинца.
Задача 8.106. На первый взгляд может показаться, что соединение содержит
атом хлора. Однако следует обратить внимание, что интенсивность пика М+2,
а также пиков А+2 для фрагментов с m/z 123, 97 и 58, несколько выше, чем
для изотопа 37С1. Кроме того, последняя пара ионов с m/z 58 и 60 не может
содержать атом хлора и по массе. Такое соотношение интенсивностей дублета во
всех указанных случаях определяется каким-то другим элементом. По таблице 1
приложений легко установить, что этим элементом является никель. Поскольку
никель не имеет изотопа А+1, можно предположить, что пик М+1 обусловлен
в основном изотопами 13С и, возможно, 15N. Наиболее подходящим является
446 Глава 11. Решения задач
вариант с десятью атомами углерода в молекуле. В этом случае элементный
состав соединения CioHwNi. Элементный состав иона с m/z 123 C5H5N1, а
основной первичный процесс распада М+" заключается в элиминировании
частицы С5Н5. Аналогичная частица отщепляется далее. В результате образуется
катион Ni+. Среди частиц такого состава наиболее вероятной является цикло-
пентадиенильный радикал. Следовательно, соединение является никелеценом.
Кстати, ферроцен, спектр которого представлен на рис. 8.125, характеризуется
аналогичными фрагментными ионами. Этот факт может служить косвенным
подтверждением предложенного решения. На рисунке 8.128 представлен масс-
спектр никелецена.
Задача 8.107. Основу соединения составляет полиизотопный элемент с
достаточно большой атомной массой. Четыре кластера пиков с максимумами
202, 230, 237 и 266 Да представляют собой М4" и три первичных фрагмента.
Разница в массах между М+' и фрагментами составляет 29, 35 и 64 Да. В
первом случае речь может идти об элиминировании этильного радикала или
частицы НСО', а во втором, отщепляется молекула НС1. Кстати, об этом
свидетельствует и заметное изменение соотношений интенсивностей пиков в
кластере. Максимальный пик обусловлен ионом с m/z 29, причем, поскольку ему
сопутствует фрагмент с m/z 27, этот ион может быть только этильным. Сумма
масс этильного радикала и атома хлора составляет 64 Да. Эта величина как раз
составляет разницу в массах между М"' и наиболее легким полиизотопным
фрагментным ионом. По таблице 1 приложения легко установить, что массы
и соотношения интенсивностей изотопов этого кластера соответствуют ртути
(см. также спектр на рис. 8.121). Следовательно, на рис. 8.129 представлен масс-
спектр этилмеркурхлорида, который использовался в качестве действующего
вещества пестицида под торговым названием гранозан.
Задача 8.108. Четыре основных пика в спектре обусловлены ионами с
m/z 208, 130, 52, 78. Предполагая, что наиболее тяжелый из этих ионов является
молекулярным, можно вычислить, что основные процессы распада связаны с
последовательным элиминированием двух частиц массой 78 Да (ионы с m/z 130
и 52). Ион с m/z 78 также представлен в спектре интенсивным пиком.
Воспользовавшись табл. 1 приложения, можно установить, что элемент с наиболее
распространенным изотопом с m/z 52 является хромом. Можно также подсчитать
соотношение интенсивностей пиков А+1 к А для ионов с m/z 208 и 130. В
первом случае получаем величину 24,5, а во втором—18,0. Вероятно,
первичный процесс включает элиминирование частицы с шестью атомами углерода.
Учитывая, что масса этой частицы 78 Да, можно сделать вывод, что речь идет о
составе СбНб, т. е. о молекуле бензола. Следовательно, молекула образца состоит
Глава 11. Решения задач 447
из атома хрома и двух молекул бензола. На рисунке 8.130 представлен масс-
спектр дибензолхрома.
Задача 8.109. Предполагая, что ион с m/z 204 является молекулярным,
можно рассчитать, что первые фрагментные ионы образуются при последовательном
элиминировании трех частиц с массой 28 Да (m/z 176, 148, 120). Такой характер
распада наблюдается в случае карбонилов металлов. Ион с m/z 120 отщепляет
65 Да с образованием иона с m/z 55, пик которого максимален в спектре. Этот
ион не содержит в составе углерода, так как интенсивность пика А+1 менее
1%. По-видимому, он обусловлен катионом элемента. По таблице 1 приложения
легко установить, что этим элементом является марганец. Этот элемент является
моноизотопным, поэтому пик иона с m/z 121 обусловлен исключительно
изотопом 13С в органическом фрагменте с m/z 65. Расчет показывает, что состав
этого фрагмента С5Н5. Резюмируя все сделанные выше наблюдения, можно
сделать вывод, что вещество, спектр которого представлен на рис. 8.131,—
циклопентадиенилтрикарбонил марганца.
Задача 8.110. В данном случае решение не представляет сложности. В
спектре отчетливо проявляются 4 группы пиков с одинаковым изотопным
соотношением. Отстоят эти пики друг от друга на 15 а. е. м. Это могут быть
только метальные группы. Изотопная картина и массы B06-208 Да) изотопов
однозначно определяют свинец (см. также рис. 8.122 и 8.127). Следовательно, на
рис. 8.132 представлен масс-спектр тетраметилсвинца.
Задача 8.111. Пользуясь табл. 2 приложения и отметками в спектре на
рис. 8.139 можно получить следующую последовательность аминокислотных
звеньев: GLLKKLKKVAKKVLPKVVPVIAEKL-NH2.
Задача 8.112. Пользуясь табл. 2 приложения и отметками в спектре на
рис. 8.140 можно получить следующую последовательность аминокислотных
звеньев: GLLGAMFKVASKVLPHWPAITEHF-NH2.
Задача 10.1. Фенилэтиловый эфир.
Задача 10.2. а) о-Хлорфенол. б) л-Хлорфенол.
Задача 10.3. Трибромфенол.
Задача 10.4. 1-Фенилпропанол-1.
Задача 10.5. 4-Бромфенилэтиловый эфир.
Задача 10.6. З-Фенилпропанол-1.
Задача 10.7. а) 4-Пропилфенол. б) Фенилпропиловый эфир.
Задача 10.8. Диэтилпропиламин.
448 Глава И. Решения задач
Задача 10.9. Циклогексантиол.
Задача 10.10. 2,3-Дихлорбутан.
Задача 10.11. 4,4'-Дихлорбензофенон.
Задача 10.12. 3-Бром-1,1,1-трихлорпропан.
Задача 10.13. 1,2-Дибром-1,2-дихлорэтилен.
Задача 10.14. 2,4,6-Трииодфенол.
Задача 10.15. Сера (S8).
Задача 10.16. Трифенилфосфин.
Задача 10.17. Диэтилсульфон.
Задача 10.18. Бензолсульфоновая кислота.
Задача 10.19. Диметилдисульфид.
Задача 10.20. 2-Хлорнафталин.
Задача 10.21. и-Броманизол.
Задача 10.22. Фторбензол
Задача 10.23. Тионафтол.
Задача 10.24. 4-Иодтолуол.
Задача 10.25. Этоксинафталин.
Задача 10.26. Метилантрацен.
Задача 10.27. Пентафторбензол.
Задача 10.28. Пентафторфенол.
Задача 10.29. 2-Фенилэтиловый эфир уксусной кислоты.
Задача 10.30. Бензилпропионат.
Задача 10.31. Этиловый эфир фенилуксусной кислоты.
Задача 10.32. Изопропиловый эфир бензойной кислоты.
Задача 10.33. Масляная кислота.
Задача 10.34. Пропилпропионат.
Задача 10.35. На рисунке 10.35, а-в представлены 2-метил-, 3-метил-, 4-ме-
тилциклогексанон соответственно.
Задача 10.36. 2-Пропилциклогексанон.
Задача 10.37. З-Фторфенилацетонитрил.
Задача 10.38. Октанонитрил.
Задача 10.39. Хромоцен.
Задача 10.40. о-Бромбензонитрил.
Задача 10.41. 2,4-Дихлорнитробензол.
Задача 10.42. Бензоилфторид.
Задача 10.43. 2-Метилпентановая кислота.
Задача 10.44. З-Метилпентановая кислота.
Задача 10.45, Дибутилсульфон.
Задача 10.46. Бутилпропилсульфоксид.
Задача 10.47. 4-Пропиланилин.
Задача 10.48. 4-Нитробензиловый спирт.
Задача 10.49. 4-Нитроанизол.
Задача 10.50. 1 -Метил- 1-фенилгидразин.
Задача 10.51. 1,1-Диэтил-2-пропилгидразин.
Задача 10.52. а-Диазо-л*-метилацетофенон.
Задача 10.53. Оксим 4-метилпентан-2-она.
Задача 10.54. 2-Пропилнитробензол.
Задача 10.55. 2-Нитронафталин.
Задача 10.56. Дифенилсульфон.
Задача 10.57. Тетрафенилсвинец.
ПРИЛОЖЕНИЕ
Таблица 1
Природная распространенность и масса изотопов химических элементов
Элемент
Водоэод
Дейтерий
Гелий
Литий
Бериллий
Бор
Углерод
Азот
Кислород
1 Фтор
Неон
1 Натрий
Магний
| Алюминий
Кремний
1 Фосфор
Сера
Изотоп
?
2Н
3Не
4Не
6Li
7Li
9Ве
юв
"В
12С
13С
HN
15N
160
17о
180
19р
20Ne
21Ne
22Ne
23Na
24Mg
25Mg
26Mg
27 Al
28Si
2ySi
30Si
31p
32s
33S
34S
36s
Масса,
y.e.
1,007825
2,014102
3,016029
4,002603
6,015123
7,016005
9,012183
10,012938
11,009305
12,000000
13,003355
14,003074
15,000109
15,994915
16,999131
17,999159
18,998403
19,992439
20,993845
21,993845
22,989770
23,985045
24,985839
25,982595
26,981541
27,976928
28,976496
29,973772
30,973763
31,972072
32,971459
33,967868
35,967079
Интенсивность, %
(относительно
J3 изотопов)
99,985
0,015
0,0001
100,00
7,52
92,48
100,00
18,98
81,02
98,89
1,11
99,64
0,36
99,76
0,04
0,20
100,00
90,92
0,26
8,82
100,00
78,60
10,11
11,29
100,00
92,18
4,71
3,12
100,00
! 95,02
I 0,75
I 4,21
0,11
Интенсивность, %
(относительно
максимального
изотопа)
100,00
0,02
0,00
100,00
8,13
100,00
100,00
23,43
100,00
100,00
1,12
100,00
0,37
100,00
0,04
0,20
100,00
100,00
0,28
9,70
100,00
100,00
12,86
14,36
100,00 !
100,00
5,11
3,38
100,00
100,00
0,79
4,44
0,11
Приложение 451
Таблица 1. Продолжение
Элемент
Хлор
Аргон
Калий
Кальций
Скандий
Титан
Ванадий
Хром
Марганец
Железо
| Кобальт
Никель
Медь
Изотоп !
35С1
37С1
36 Аг
38 Аг
40 Аг
39К
40К
41К
^Са
42Са
43Са
44Са
46Са
48Са
45Sc
46Ti
47?
48Ti
49?
50Ti
50y
51v
50Cr
52Cr
53Cr
54Cr
55Mn
54Fe
56Fe
57Fe
58Fe
59Co
58Ni
60Ni
61Ni
62Ni
64Ni
63 Cu
65Cu
Масса,
y.e.
34,968853
36,965903
35,967546
37,962732
39,962383
38,963708
39,963999
40,961825
39,962591
41,958622
42,958770
43,955485
45,953689
47,952532
44,955914
45,952633
46,951765
47,947947
48,947870
49,944786
49,947161
50,943962
49,946046
51,940510
52,940651
53,938882
54,938046
53,939612
55,934939
56,935396
57,933278
58,933198
57,935347
59,930789
60,931059
61,928346
63,927968
62,929599
64,927792
Интенсивность, %
(относительно
J2 изотопов)
75,40
24,60
0,34
0,06
99,60
93,08
0,01
6,91
96,92
0,64
0,13
2,13
0,00
0,18
100,00
7,95
7,75
73,45
5,51
5,34
0,24
99,76
4,31
83,76
9,55
2,38
100,00
5,90
91,52
2,25
0,33
100,00
67,76
26,16
1,25
3,66
1,16
69,09
30,91
Интенсивность, %
(относительно
максимального
изотопа)
100,00
32,63
0,34
0,06
100,00
100,00
0,01
7,42
100,00
0,66
0,13
2,20
0,00
0,18
100,00
10,82
10,55
100,00
7,50
7,27
0,24
100,00
5,15
100,00
11,40
2,84
100,00
! 6,45
100,00
2,46
0,36
100,00
100,00
38,61
1,84 |
5,40 !
1,71
100,00
44,74
452 Приложение
Таблиц
\а 1. Продолжение
Элемент
Цинк
Галлий
Германий
Мышьяк
Селен
Бром
Криптон
Рубидий
Стронций
1 Иттрий
Цирконий
I
Изотоп
"Zn
66Zn
67Zn
68Zn
70Zn
69Ga
71 Ga
70Ge
72Ge
73Ge
74Ge
76Ge
75 As
74Se
76Se
77Se
78Se
80Se
82Se
79Br
81Br
78Kr
80Kr
82Ki
83Kr
84Kr
86Kr
85Rb
87Rb
84Sr
86Sr
87Sr
88Sr
89?
90Zr
91Zr
| 92Zr
^Zr
| 96Zr
Масса,
ye.
63,929145
65,926035
66,927129
67,924846
69,925325
68,925581
70,924701
69,924250
71,922080
72.923464
73,921179
75,921403
74,921595
73,922477
75,919207
76,919908
77,917304
79,916520
81,916709
78,918336
80,916290
77,920397
79,916375
81,913483
82,914134
83,911506
85,910614
84,911800
86,909184
83,913428
85,909273
86,908890
87,905625
88,905856
89,904708
90,905644
91,905039
93,906319
95,908272
Интенсивность, %
(относительно
?2 изотопов)
48,89
27,81
4,11
18.56
0,62
60,20
39,80
20,52
27,43
7,76
36,54
7,76
100,00
0,96
9,12
7,50
23,61
49,96
8,84
50,57
49,43
0,35
2,27
11,56
11,55
56,90
17,37
72,15
27,85
0,56
9,86
7,02
82,56
100,00
51,46
11,23
! 17,11
17,40
2,80
Интенсивность, %
(относительно
максимального
изотопа)
100,00
56,88
8,41
37,96
1,27
100,00
66,11
56,16
75,07
21,24
100,00
21,24
100,00
1,92
18,25
15,01
47,26
100,00
17,69
100,00
97,75
0,62
3,99
20,32
20,30
100,00
30,53
100,00
38,60
0,68
11,94
8,50
100,00
100,00
100,00
21,82
33,25
33,81
5,41
Приложение 453
Таблица 1. Продолжение
Элемент
Ниобий
Молибден
Рутений
| Родий
Палладий
Серебро
Кадмий
Индий
Олово
Изотоп
93Nb
^Мо
мМо
95Мо
96Мо
97Мо
98Мо
|00Мо
%Ru
98Ru
"Ru
iooRu
101 Ru
102Ru
104Ru
103Ro
,02Pd
104pd
,05Pd
106pd
108pd
nopd
107Ag
109Ag
106Cd
108Cd
110Cd
n,Cd
I,2Cd
113Cd
114Cd
,16Cd
113In
115In
| 1,2Sn
| 1,4Sn
| 115Sn
| 116Sn
| ,17Sn
Масса,
ye.
92,906378
91,906809
93,905086
94,905838
95,904675
96,906018
97,905405
99,907473
95,907596
97,905287
98,905937
99,042175
100,905581
101,904347
103,905422
102,905503
101,905609
103,904026
104,905075
105,903475
107,903894
109,905169
106,905095
108,904754
105,906461
107,904186
109,903007
110,904182
111,902761
112,904401
113,903361
115,904758
112,904056
114,903875
111,904823
113,902781
114,903344
115,901743
116,902954
Интенсивность, %
(относительно
J2 изотопов)
100,00
15,05
9,35
14,78
16,56
9,60
24,00
9,68
5,68
2,22
12,81
12,70
16,98
31,34
18,27
100,00
0,80
9,30
22,60
27,10
26,70
13,50
51,35
48,65
1,22
0,89
12,43
12,86
23,79
12,34
28,81
7,66
4,16
95,84
0,95
0,65
0,34
14,24
7,57
Интенсивность, %
(относительно
максимального
изотопа)
100,00
62,71
38,96
61,58
69,00
40,00
100,00
40,33
18,12
7,08
40,87
40,52
54,18
100,00
58,30
100,00
2,95
34,32
83,40
100,00
98,52
49,81
100,00
94,74
4,23
3,09
43,14
44,64
82,57
42,83
100,00
26,59
4,34
100,00
2,88
1,97
1,03
43,19
22,96
454 Приложение
Таблица L Продолжение
Элемент
Олово
Сурьма
Теллур
Иод
Ксенон
| Цезий
Барий
Лантан
Церий
| Празеодим
Изотоп
118Sn
Hi,Sn
,2°Sn
I22Sn
124Sn
121 Sb
,23Sb
120Te
122Te
,23Te
i24Te
125Te
,20Te
128Te
.зоТе
127j
124Xe
126Xe
128Xe
129Xe
130Xe
131Xe
132Xe
134Xe
136Xe
133Cs
130Ba
132Ba
134Ba
I35Ba
136Ba
137Ba
шВа
| ,38La
| mLa
| 136Ce
I 138Ce
| 140Ce
| 142Ce
| 141Pr
Масса,
y.e.
117,901607
118,903310
119,902199
121,903440
123,905271
120,903824
122,904222
119,904021
121,903055
122,904278
123,902825
124,904435
125,903310
127,904464
129,906229
126,904477
123,906120
125,904281
127,903531
128,904780
129,903509
130,905076
131,904148
133,905395
135,907219
132,905433
129,906277
131,905042
133,904490
134,905668
135,904556
136,905816
137,905236
137,907114
138,906355
135,907140
137,905996
139,905442
141,909249
140,907657
Интенсивность, %
(относительно
? изотопов)
24,01
8,58
32,97
4,71
5,98
57,25
42,75
0,09
2,46
0,87
4,61
6,99
18,71
31,79
34,49
100,00
0,10
0,09
1,92
26,44
4,08
21,18
26,89
10,44
8,87
100,00
0,10
0,10
2,42
6,59
7,81
11,32
71,66
0,09
99,91
0,19
0,25
88,48
11,07
100,00
Интенсивность, %
(относительно
максимального
изотопа)
72,82
26,02
100,00
14,29
18,14
100,00
74,67
0,26
7,13
2,52
13,37
20,27
54,25
92,17
100,00
100,00
0,37
0,33
7,14
98,33
15,17
78,76
100,00
38,82
32,99
100,00
0,14
0,14
3,38
9,20
10,90
? 15,80
100,00
0,09
100,00
1 0,21
| 0,28
100,00
12,51
100,00
Приложение 455
Таблица 1. Продолжение
Элемент
Неодим
Самарий
Европий
Гадолиний
1 Тербий
Диспрозий
1 Гольмий
Эрбий
| Тулий
Изотоп
142ш
143ш
144Nd
145Nd
146Nd
148Nd
150Nd
144Sm
,47Sm
148Sm
149Sm
150Sm
152Sm
154Sm
I51Eu
,53Eu
152Gd
154Gd
155Gd
156Gd
157Gd
158Gd
160Gd
159Tb
156Dy
158Dy
160Dy
1" Dy
i62Dy
163 Dy
I64Dy
,65Ho
162Er
164Er
i66Er
167Er
168Er
I70Er
169Tm
Масса,
y.e.
141,907731
142,909823
143,910096
144,912582
145,913126
147,916901
149,920900
143,912009
146,914907
147,914832
148,917193
149,917285
151,919741
153,922218
150,919860
152,921243
151,919803
153,920876
154,922629
155,922130
156,923967
157,924111
159,927061
158,925350
155,924287
157,924412
| 159,925203
160,926939
161,926805
162,928737
163,929183
164,930332
161,928787
163,929211
165,930305
166,932061
167,932383
169,935476
168,934225
Интенсивность, %
(относительно
? изотопов)
27,09
12,14
23,83
8,29
17,26
5,74
5,63
3,16
15,07
11,27
13,84
7,47
26,63
22,53
47,77
52,23
0,20
2,15
14,73
20,47
15,68
24,87
21,90
100,00
i 0,05
0,09
2,29
18,88
25,53
24,97
28,18
100,00
0,14
1,56
33,41
22,94
27,07
14,88
100,00
Интенсивность, %
(относительно
максимального
изотопа)
100,00
44,81
87,97
30,60
63,71
21,19
20,78
11,87
56,59
42,32
51,97
28,05
100,00
84,60
91,46
100,00
0,80
8,64
59,23
82,31
63,05
100,00
88,06
100,00
0,18
0,32 !
8,13
67,00
90,60
88,61
100,00
100,00
0,42
4,64
100,00
68,66
81,02
44,54
100,00
456 Приложение
Таблица 1. Продолжение
Элемент
Иттербий
Лютеций
Гафний
Тантал
Вольфрам
Рений
Осмий
Иридий
Платина
1 Золото
Изотоп
168 Yb
170Yb
"I yb
172 уь
173 уь
,74Yb
176Yb
175Lu
I76Lu
174ffi
"«Of
177Gf
178Gf
179Gf
180Gf
181Ta
180W
182W
183W
184W
186W
185Re
187Re
1840s
186Os
187Os
188Os
189Os
190Os
1920s
i9iIr
,93Ir
190pt
I92pt
194pt
| 195Pt
1 196Pt
198pt
1 l97Au
Масса,
y.e.
167,933908
169,934774
170,936338
171,936393
172,938222
173,938873
175,942576
174,940785
175,940065
173,940065
175,941420
176,943233
177,943710
178,945827
179,946561
180,948014
179,946727
181,948225
182,950245
183,950953
185,954377
184,952977
186,955765
183,952514
185,953852
186,955762
187,955850
188,958156
189,958455
191,961487
190,960603
192,962942
189,959937
191,961049
! 193,962679
194,964785
195,964947
197,967879
196,966560
Интенсивность, % ;
(относительно
? изотопов)
0,14
3,03
14,31
21,82
16,13
31,84
12,73
97,40
2,60
0,20
5,23
18,55
27,23
13,79
31,07
99,99
0,13
26,31
14,28
30,64
28,64
37,07
62,93
0,02
1,59
1,64
13,20
16,10
26,40
41,00
38,50
61,50
0,01
0,78
! 32,80
33,70
25,40
7,23
100,00
Интенсивность, %
(относительно
максимального
изотопа)
0,44
9,52
44,94
68,53
50,66
100,00
40,06
100,00
2,67
0,64
16,83
59,70
87,64
44,38
100,00
100,00
0,42
85,87
46,61
100,00
93,47
58,91
100,00
0,05
3,88
4,00
32,19
39,27
64,39
100,00
62,60
100,00
0,03
2,31
97,33
100,00
75,37
21,45
100,00
Приложение 457
Таблица 1. Продолжение
Элемент
Ртуть
Таллий
Свинец
Висмут
1 Торий
Уран
Изотоп
196Hg
,s*Hg
199Hg
200Hg
201Hg
202Hg
204Hg
203-pj
205 jj
204pb
206pb
207pb
208pb
209Bi
232Tr
234tj
235u
238U
Масса,
y.e.
195,965812
197,966760
198,968269
199,968316
200,970293
201,970632
203,973481
202,972336
204,974410
203,973037
205,974455
206,975885
207,976641
208,980388
232,038054
234,040947
235,043925
238,050786
Интенсивность, %
(относительно
? изотопов)
0,15
10,02
16,84
23,13
13,22
29,80
6,85
29,50
70,50
1,37
25,15
21,11
52,38
100,00
100,00
0,01
0,71
99,28
Интенсивность, %
(относительно
максимального
изотопа)
0,50
33,62
56,51
77,62
44,36
100,00
22,92
41,84
100,00
2,62
48,01
40,30
100,00
100,00
100,00
0,01
0,71
100,00
458 Приложение
Таблица 2
Масса и структура наиболее распространенных аминокислот
Аминокислота
Алании
Аргинин
Аспарагин
Аспарагиновая кислота
Цистеин
Глутаминовая кислота
Глутамин
Глицин
Гистидин
Изолейцин
Лейцин
Лизин
Метионин
Орнитин
Фенилаланин
Пролин
Серии
Треонин
Символ
Ala
А
Arg
R
Asn
?
Asp
D
Cys
С
Glu
?
Gin
Q
Gly
G
His
?
He
I
Leu
L
Lys
К
Met
?
Orn
О
Phe
F
Pro
?
Ser
s
Thr
?
Структура остатка
СНз
—NH— CH— CO—
CH^iCH^-NH-C-NHa
-NH—CH—CO NH
CH2-CONH2
—NH—CH—CO—
ЩгСООН
1
—NH—CH—CO—
CH2-SH
1
—NH—CH—CO—
CH2-CH2— COOH
—NH—CH—CO—
CH2-CH2— CONH2
—NH—CH—CO
—NH—CH2—CO—
—NH—CH—CO—
СН(СНз)СН2— СНз
—NH—CH—CO—
СН2-СН(СНзJ
—NH—CH—CO—
CH2-(CH2J-NH2
—NH—CH—CO—
CH2—CH2-S— CH3
—NH—CH—CO—
CH2-(CH2J-NH2
—NH—CH—CO—
CH2-Ph
1
—NH—CH—CO—
—N-CH— CO—
CH2-OH
1
—NH—CH—CO—
СН(ОН)СНз
—NH—CH—CO—
Масса
71,03711
156,10110
114,04292
115,02694
103,00918
129,04259
128,05857
57,02146
137,05891
113,08406
113,08406
128,09496
131,04048
114,07931
147,06841
97,05276
87,03202
101,04767
Приложение 459
Таблица 2. Продолжение
Аминокислота
Триптофан
Тирозин
Валин
Символ
Тгр
W
ТУг
?
Val
V
Структура остатка
—NH— СН— СО—
сн2-^ У-он
— NH— CH— СО—
СН(СНзJ
—NH—СН—СО—
Масса
186,07931
163,06332
99,06841
Таблица 2а
Масса и структура менее распространенных аминокислот
Аминокислота
а-аминомасляная
кислота
Аминоэтилцистеин
а-аминоизомасляная
кислота
Карбокси-
метилцистеин
Дегидроаланин
Дегидро-а-
аминомасляная
кислота
Гидроксилизин
Гидроксипролин
Изовалин
Норлейцин
2-Пиперидин-
| карбоновая кислота
Пироглутаминовая
кислота
1 Саркозин
Символ
Aba
AECys
Aib
CMCys
Dha
Dhb
Hyl
Hyp
Iva
nLeu
Pip
pGlu
Sar
Структура остатка
сн2-сн3
—NH—CH—CO—
CH2-S— (CH2J"-NH2
—NH—CH—CO—
CH3
—NH—C—CO—
1
CH3
CH2-S—CH2—COOH
— NH—CH—CO—
CH2
II
—NH—C—CO—
CH—CH3
II
—NH—C—CO—
CH2- CH2- CH(OH)CH2- NKfe
— NH—CH—CO
OH
—N CH—CO—
CH2-CH3
—NH—С(СНз)—CO—
CH2-(CH2J-CH3
—NH—CH—CO—
—N—CH—CO—
N—CH—CO—
—ВДСНэ)—СНг— СО—
Масса
85,05276
146,05138
85,05276
161,01466
69,02146
83,03711
144,08987
113,04767
99,06841
113,08406
111,06841
111,03203
71,03711
460 Приложение
Таблица 3
Величины m/z, состав и структуры фрагментных ионов, характерных для распада
органических соединений, рассмотренных в пособии
m/z
15 1
16
17
18
19
70
76
77
28
29
30
31
32
33
34
Состав (структура) ионов :
СН3
О
NH2 S
СН4
ОН
Н20
NH4
F
Н30
Аг
HF
с2н2
CN
С2Н3
HCN
СО
N2
CH2N
с2н4
с2н5
сно
SiH
СН20
CH2NH2
NO
СН2ОН
С?
S
о2
СН3ОН
NHOH
СН50
HS
РН2
CH2F
| H2S
1 РН3
Соединения, для которых характерны данные ионы
Алкилпроизводные
Кислородсодержащие соединения 1
Азотсодержащие соединения
Алкилпроизводные
1Сислородсодержащие соединения |
Кислородсодержащие соединения |
Амины 1
Фторсодержащие соединения
Полиолы 1
Аргон (двухзарядный ион) |
Алкилфториды 1
Ароматические соединения 1
Азотсодержащие ароматические соединения |
Ароматические соединения 1
Азотсодержащие ароматические соединения 1
Ангидриды, циклические кетоны 1
Азот (воздух) 1
Алкиламины, этиленимины ]
Алкилпроизводные |
Алкилпроизводные 1
Ароматические альдегиды, фенолы |
Кремнийорганические соединения 1
Метоксипроизводные |
Амины 1
Нитросоединения
Алифатические спирты, эфиры, ацетали
Фторсодержащие соединения
Тиолы, сульфиды, полисульфиды
Кислород (воздух)
Спирты
Оксимы
Спирты, полиолы, ацетали
Тиолы, сульфиды
Фосфины
Фторсодержащие соединения
Тиолы, сульфиды
Фосфины
Приложение 461
Таблица 5. Продолжение
m/z
35
! 36
37
38
39
40
41
42
43
44
45
46
Состав (структура) ионов
35С1
H3S
Н35С1
37С1
Н37С1
С3Н2
С3Нз
Аг
С3Н4
с3н5
CH2CNH
С3Н6
СН2СО
CH2NCH2
С3Н7
CH3CHNH
СН3СО
C2F
SiCH3
С3Н8
СН2СНОН
со2
CS
CH3SiH
CH3CHNH2,
CH2NHCH3
CONH2
CH3CHOH
СН3СН20
CH3OCH2
соон
(CH3JNH
CH3NNH2
CHS
CH3SiH2
CH3CH2OH
CH2S
N02
Соединения, для которых характерны данные ионы
Хлорсодержащие соединения 1
Тиолы, сульфиды 1
Хлорсодержащие соединения 1
Хлорсодержащие соединения |
Хлорсодержащие соединения 1
Ароматические соединения с электроноакцепторными заме- 1
стителями
Алкены, диены, ацетилены, ароматические соединения |
Аргон (воздух) 1
Ароматические соединения
Алкилпроизводные, алициклы, алкеновая серия
Нитрилы 1
Алкилпроизводные, алкены, алициклы |
Ацетилпроизводные, циклические кетоны 1
Этиленимины |
Алкилпроизводные |
Нитрилы 1
Ацетильные производные, кетоны, циклические спирты ]
Перфторуглеводороды 1
Кремнийпроизводные
Алкилпроизводные |
Альдегиды, циклические спирты, сложные эфиры |
Кислоты, ангидриды, воздух
Тиофенолы, диарилсульфиды, 1
Силаны, силациклоалканы
Амины
Амиды
Спирты, эфиры
Этоксипроизводные
Простые эфиры
Карбоновые кислоты, сложные эфиры
Диметиламинопроизводные
Метилалкилгидразины
Ароматические тиолы, сульфиды
Алкилсиланы
Этиловые эфиры кислот
Тиолы, сульфиды
Нитросоединения
462 Приложение
Таблица 3. Продолжение
m/z
47
48
49
50
?
52
53
54
55
56
57
58
Состав (структура) ионов
с2н7о
сн3о2
CH2SH
C2H4F
СН2РН2
CH3SH
so
CH3PH2
СН35С1
CH3SH2
СН^5С1
С4Н2
СН37С1
CF2
C4H3
CHF2
CHfCl
С4Н4
С4Н5
с4н6
CH2CH2CN
С4Н7
СН2СНСО
C2H5CN
C3F
С4Н8
CH2CHCHNH2
С2Н4СО
С4Н9
С2Н5СО
СН2СНСНОН
NH2CHCO
C3H2F
CH2C(OH)CH3
СНзСНСНОН
С2Н6СО
C3H8N
CH4NCO
1 C2H2S
Соединения, для которых характерны данные ионы
Диолы, алкоксиспирты 1
Ацетали
Тиолы, сульфиды 1
Алкилфториды
Фосфины |
Тиолы, метилсульфиды 1
Сульфоксиды, сульфоны 1
Фосфины 1
Хлорсодержащие соединения 1
Метилсульфиды 1
Хлоралкильные производные
Ароматические, полиненасыщенные соединения 1
Хлорсодержащие соединения 1
Перфторуглеводороды 1
Ароматические, полиненасыщенные соединения 1
Перфторуглеводороды 1
Хлоралкильные производные
Ароматические, полиненасыщенные соединения |
Ненасыщенные углеводороды 1
Ненасыщенные углеводороды, циклоалкены 1
Алифатические нитрилы |
Алкилпроизводные, алициклы, алкеновая серия 1
Циклические кетоны |
Нитрилы
Перфторуглеводороды
Алкилпроизводные, алициклы 1
Циклоалкиламины
Циклические кетоны
Алкилпроизводные
Этилкетоны, производные пропионовой кислоты
Циклоалканолы
| а-Аминокислоты
Фторуглеводороды
Кетоны
а-Метилальдегиды
Циклоалканолы
Алкиламины
Амиды
Сульфиды
Приложение 463
Таблица 3. Продолжение
m/z
Ь9
60
61
62
63
64
65
66
67
68
69
Состав (структура) ионов
С3Н6ОН
С2Н5ОСН2
СН3ОСО
CH2C{OH)NH2
CH2CHNHOH
C2H7N2 I
C2H3S
SiH(CH3J
CH2C[OHJ
HOCHCHOH
C3HgO
C2H6NO
CH2ONO
C2H4S
CH3C{OHJ
C3H90
C2H5S
C2H6P
CH2N02H
HO(CH2JOH
C2H5PH2
C2H5SH
CH2N(OHJ
с5н3
CH2SOH
C2HfCl
C5H4
s2
so2
C5H5
C2Hf CI
C5H6
S2H2
C5H7
C5H8
C4H6N
C5H9
C4H50
C4H7N
1 CF3
Соединения, для которых характерны данные ионы
Спирты 1
Простые эфиры 1
Метиловые эфиры кислот 1
Амиды карбоновых кислот 1
Альдоксимы 1
Алкилгидразины I
Тиацикланы |
Алкилсиланы |
Алифатические карбоновые кислоты, их эфиры |
Углеводы I
Спирты, полиолы, простые эфиры > С5 I
Амиды карбоновых кислот 1
Алифатические нитросоединения 1
Циклические сульфиды |
Алифатические карбоновые кислоты, их эфиры |
Спирты, полиолы, простые эфиры > С5 I
Тиолы, сульфиды 1
Фосфины 1
Нитроалканы 1
Полиолы 1
Фосфины 1
Сульфиды 1
Нитроалканы |
Ароматические, полиненасыщенные соединения |
Диалкилсульфоксиды 1
Алкилхлориды 1
Ароматические, полиненасыщенные соединения |
Сера, дисульфиды 1
Сульфоны, сульфокислоты 1
Ароматические соединения, металлоцены |
Алкилхлориды
Ароматические, полиненасыщенные соединения
Алкилдисульфиды
Ацетилены, диены, циклоалканы
Циклоалканы, циклоалканолы
Нитрилы
Алкилпроизводные, алициклы, алкеновая серия
Алициклические спирты, кетоны
Нитрилы
Фторуглеводороды
464 Приложение
Таблица 3. Продолжение
m/z
\ 70 !
71
72
73
74
75
76
77
78
79
Состав (структура) ионов
С5Н10
с4н6о
C4H8N
С5Н11
С3Н7СО
с4н8о
C4H10N
C2H6NCO
С4Н90
С3Н502
СН2СОСН2ОН
C2H7NCO
C3H9N2
(CH3KSi
СбН2
с3н6о2
C2H4N02
NH2CHCOOH
C3H6S
с6н3
с4нпо
с3н7о2
NH2CHC(OHJ
C3H7S
(CH3JSiOH
C3H8P
C6H4
C3H7PH2
C3H7SH
с6н5
c&fc\
СбН$
C5H4N
C2H5SOH
C6H7
C5H5N
C3Hf CI
1 79Br
Соединения, для которых характерны данные ионы
Алкилпроизводные, алициклы, алкены |
Алициклические кетоны |
Алициклические амины 1
Алкилпроизводные 1
Кетоны, алициклические спирты |
Кетоны, альдегиды, алициклические спирты |
Амины |
Амиды |
Алифатические спирты, эфиры |
Алифатические карбоновые кислоты, их эфиры
Углеводы 1
Амиды
Диалкилгидразины
Триметилсилильные производные
Ароматические, полиненасыщенные соединения
Алифатические карбоновые кислоты, их эфиры
Алифатические нитросоединения
?-Аминокислоты ]
Циклические сульфиды
Ароматические, полиненасыщенные соединения
Спирты, полиолы, простые эфиры > С$
Карбоновые кислоты, их эфиры, ацетали
?-Аминокислоты
Тиолы, сульфиды
Триметилсилильные эфиры
Алкилфосфины
Ароматические, полиненасыщенные соединения
Алкилфосфины
Диалкилсульфиды
Монозамещенные бензолы, ароматические,
полиненасыщенные соединения
Алкилхлориды
Монозамещенные бензолы
Ароматические амины
Этилалкилсульфоксиды
Ароматические, полиненасыщенные соединения
Ароматические амины
Алкилхлориды
Бромсодержащие соединения
Приложение 465
Таблица 3. Продолжение
m/z
80
81
82
8?
84
85
86
87
88
89
90
Состав (структура) ионов
СбН8
CH3SSH
Н79Вг
СбН9
с5н5о
81Вг
СбНю
C4H8CN
CHCF3
С35С12
?81 Br
СбНц
СН35С12
С4Н7СО
с6н12
с5н8о
C5H10N
с6н13
С4Н9СО
С3Н7С(ОН)СН2
C5H12N
C4H10S1
с5н„о
с3н7со2
C4HnN2
C3H9NCO
с4н8о2
C3H6N02
C4H8S
С5Н130
С4Н902
C4H9S
(CH3KSiO
С4НюР
с7н6
С4Н9РН2
Соединения, для которых характерны данные ионы
Замещенные циклогексены |
Метилдисульфиды |
Бромсодержащие соединения |
Циклоалкены, диены, ацетилены, |
Алициклические спирты, эфиры, альдегиды 1
Бромсодержащие соединения |
Монозамещенные циклогексаны, полизамещенные цикло-
пентаны, циклоалканолы |
Алифатические нитрилы |
Трифторметилалканы |
Полихлорсодержащие соединения |
Бромсодержащие соединения
Алкилпроизводные, алициклы, алкеновая серия |
Полихлорсодержащие соединения |
Алициклические спирты, эфиры, альдегиды, кетоны |
Алкилпроизводные, алициклы, алкены 1
Алициклические кетоны |
Алициклические амины |
Алкилпроизводные |
Кетоны, алициклические спирты
Пропилалкилкетоны
Алифатические амины
Силациклоалканы
Спирты, эфиры
Алифатические карбоновые кислоты, их эфиры
Диалкилгидразины
Амиды
Алифатические карбоновые кислоты, их эфиры
Алифатические нитросоединения
Циклические сульфиды
Спирты, полиолы, простые эфиры > С5
Карбоновые кислоты, их эфиры, ацетали
Сульфиды, тиолы
Триметилсилильные эфиры
Алкилфосфины
Ароматические соединения, замещенные бензонитрилы
Алкилфосфины
16—1113
466 Приложение
Таблица 3. Продолжение
m/z
91
92
93
94
95
96
97
98
99
100
101
Состав (структура) ионов
С7Н7(Тг+)
C6H5N
Q
с7н8
C6H6N
C3H7SOH
С7Н9
С6Н50
СН|9Вг
С6Н5ОН
с7Нц
СН^Вг
C5H10CN
С7Нв
С5Н9СО
\^Чн
С7Н14
с5н10со
C6H12N
С7Н15
С5Н11СО
С6НпО
О
CH3CONHCHCO
С5Н12СО
сбн12о
C6H14N
C4H10NCO
C2F4
с5н9о2
С6Н130
1 C5H9S
Соединения, для которых характерны данные ионы
Ароматические соединения, монозамещенные бензолы I
Производные анилина |
1-хлоралканы
Ароматические соединения, монозамещенные бензолы 1
Производные анилина |
Пропилалкилсульфоксиды |
Циклодиены, терпены 1
Производные фенола
Алкилбромиды
Фениловые эфиры, бензопирены |
Диены, циклоалкены, ацетилены 1
Алкилбромиды 1
Алифатические нитрилы 1
Алкилпроизводные, алициклы, алкеновая серия |
Алициклические кетоны 1
Алифатические нитрилы
Алкилпроизводные, алициклы, алкены |
Алициклические кетоны |
Алициклические амины |
Алкилпроизводные
Алифатические кетоны 1
Алициклические спирты
Диалкилмалеаты
N-ацетил-а-аминокислоты
[ Алифатические кетоны
Алициклические спирты
Алифатические амины
Алифатические амиды
Перфторалканы
Алифатические карбоновые кислоты, их эфиры
Алифатические спирты, эфиры. кетоны
| Алициклические тиолы, сульфиды 1
Приложение 467
Таблица 3. Продолжение
m/z
102
103
104
105
106
107
108
109
110
111
112
Состав (структура) ионов
С6Н5ССН
С5Н10О2
C5H12NO
C4H8N02
H2NCHCO2C2H5
СбН5С2Н2
с6н15о
С5Нп02
C5H11S
LI
^^сн=сн2
С5НпРН2
С6Н5С2Н4
С6Н5СО
С5Н^С1
QH5C2H5
C6H5NHCH2
^Чн2
C4H9SOH
С8Нц
с7н6он
С2Н19Вг
с8н12
с7н7он
с8н13
PhS
С6Н4(ОНJ
C6H12CN
QH15
С6НпСО
QHi6
с7н12о
C7H14N
Соединения, для которых характерны данные ионы
Ароматические производные 1
Алифатические карбоновые кислоты, их эфиры 1
Алифатические амиды 1
Алифатические нитросоединения 1
Этиловые эфиры ?-аминокислот ?
Алкенилбензолы |
Спирты, полиолы, простые эфиры 1
Алифатические карбоновые кислоты, их эфиры 1
Тиолы, сульфиды 1
о-замещенные бензолы
/?-фенилэтиловые эфиры, алкенилбензолы
Алифатические фосфины
Алкилбензолы, алкилароматические спирты |
Бензоилпроизводные |
Хлоралканы
Алкилбензолы 1
Алкиланилины
о-замещенные фениловые эфиры
Бутилалкилсульфоксиды
Циклодиены, терпены
Алкилфенолы
Бромсодержащие соединения
Циклодиены, терпены
Бензиловые и толиловые эфиры
Диены, циклоалкены, ацетилены
! Фенилсульфиды, фенилсульфоксиды
Алкоксифенолы (алкил ^ СгН5)
Алифатические нитрилы
Алкилпроизводные, циклоалканы, алкены
Алициклические кетоны
Алкилпроизводные, циклоалканы, алкены
Алициклические кетоны
Алициклические амины
468 Приложение
Таблица 3. Продолжение
m/z
113
114
! 115
116
117
118
119
120
121
122
123
124
125
126
Состав (структура) ионов
с8н17
C7H16N
С9Н7
СбНцС>2
C6Hi3NO
C6HUS
С6Н12О2
C6H,4NO
С6Н5С3Н4
с7н17о
СбН^Ог
(CH3KSiOOC
С35С13
С6Н5С3Н5
С6Н5С3Н6
СН3СбН4СО
CF3CF2
C35ClfCl
С9Н12
C6H5C(OH)CH2
C8H10N
СрНв
с8н9о
&r
^Anh
C3H?9Br
C6H5COOH
с8нпо
С6Н5СООН2
С3Н^Вг
C7H14CN
С9Н17
C6H5SO
С9Н18
C6H5SOH
Соединения, для которых характерны данные ионы
Алкилпроизводные 1
Алифатические амины 1
Нафталины, индены 1
Алифатические карбоновые кислоты, их эфиры
Алифатические амиды
Алициклические тиолы, сульфиды
Алифатические карбоновые кислоты, их эфиры 1
Алифатические амиды
Алкенилбензолы, замещенные стиролы, циклоалкилбензо- 1
лы
Спирты, полиолы, простые эфиры 1
Алифатические карбоновые кислоты, их эфиры
Триметилсилильные эфиры ?-аминокислот ?
Трихлорметильные производные
Алкенилбензолы
Алкилбензолы
Толуиловые кислоты и их эфиры
Перфторалканы 1
Трихлорметильные производные
алкилбензолы 1
фенилалкилкетоны
Алкиланилины
Терпены 1
Замещенные фенолы, метоксибензолы 1
Производные салициловой кислоты
о-Нитроанилины
Бромсодержащие соединения
Производные бензойной кислоты
Терпены
Производные бензойной кислоты
Бромсодержащие соединения
Алифатические нитрилы
Алкилпроизводные, циклоалканы, алкены
Алкилфенилсульфоксиды
Алкилпроизводные, циклоалканы, алкены
Алкилфенилсульфоксиды
Приложение 469
Таблица 3. Продолжение
m/z
127
128
129
131
132
133
135
137
138
140
141
147
149
155
207
221
1 281
Состав (структура) ионов
С9Н19
СюН7
I
CioHg
C8H18N
HI
С7Н1302
C3F5
С10Н12
Q0H13
ю
?
81В^
C8H16CN
С10Н20
С10Н21
С11Н9
CH2I
СцН,5
C4H11SI2O2
С8Н503
СиНц
C5H15S13O3
C6H,7Si303
C7H21SI3O4
Соединения, для которых характерны данные ионы
Алкилпроизводные
Нафталины 1
Иодсодержащие соединения
Нафталины 1
Алифатические амины
Иодсодержащие соединения
Алифатические карбоновые кислоты, их эфиры |
Перфторалканы
Алкенилбензолы
Алкилбензолы |
Монозамещенные адамантаны
Алкилбромиды
Алкилбромиды
Алифатические нитрилы
Алкилпроизводные, циклоалканы, алкены
Алкилпроизводные
Алкилнафталины
Алкилиодиды
Полиалкилбензолы
Полидиметилсилоксаны (фаза колонки)
Эфиры о-фталевой кислоты
Алкилнафталины
Полидиметилсилоксаны (фаза колонки)
Полидиметилсилоксаны (фаза колонки)
Полидиметилсилоксаны (фаза колонки)
470 Приложение
Таблица 4
Масса и состав наиболее распространенных частиц, элиминируемых молекулярными ионами
органических соединений
Масса
1
2
3 1
15
16
17
18
19
20
26
27
28
29
30
31
32
33
34
1 35
Состав
?
2Н
ЗН
СН3
??2
О
СН4
??3
ОН
Н20
F
HF
н2+н2о
С2Н2
CN
С2Н3
HCN
С2Н4
СО
?2
H+HCN
с2н5
нсо
сн2о
с2н6
N0
СН30
СН3ОН
S
СН3+Н20
HS
CH2F
H2S
135ci
Соединения, М+ * которых элиминируют данные частицы
Большинство органических соединении 1
Силаны, фосфины, полициклические ароматические углеводороды |
Фосфины, бензиловые спирты, соединения Аг X Аг 1
Алкилпроизводные, циклоалкилпроизводные 1
Ароматические амиды |
Сульфоксиды, ароматические нитросоединения |
Алкилпроизводные (редко), элементоорганические соединения 1
Амины, полиамины (редко) 1
Карбоновые кислоты, оксимы, некоторые спирты, сульфоны,
сульфоксиды, о-нитроалкилбензолы |
Спирты, альдегиды, кетоны, простые эфиры 1
Фторпроизводные 1
Фторпроизводные ]
Алифатические спирты (часто термически) 1
Ароматические соединения |
Ароматические нитрилы 1
Ароматические соединения, этиловые эфиры кислот 1
Нитрилы, ароматические амины |
Функциональные этилпроизводные, цикланы, циклоалкены 1
Фенолы, алициклические кетоны, диариловые эфиры 1
Диазосоединения, ароматические азосоединения
Ароматические амины |
Алкилпроизводные, алициклические соединения 1
Фенолы, ароматические альдегиды, диариловые эфиры
Метоксиарилы, ацетали
Алкилпроизводные
Ароматические нитросоединения
Метоксипроизводные, полиолы
Простые и сложные эфиры
Сульфиды, тиофенолы
Некоторые спирты
Сульфиды, тиофенолы
Фторпроизводные
Тиолы, сульфиды
Хлорпроизводные
Приложение 471
Таблица 4, Продолжение
Масса
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
Состав
Н35С1
2Н20
37С1
Н37С1
С3Нз
С3Н4
CH2CN
С3Н5
с3н6
СН2СО
С3Н7
СНзСО
HNCO
с3н8
СНзСНО
со2
CS
CONH2
С2Н5О
соон
HCS
(CH3JNH
С2Н5ОН
н2о+с2н4
н2о+со
N02
CH2S
??02
C2H4F
CH3S
CH3SH
SO
HF+C2H5
CH35C1
H20+CH3OH
CF2
Соединения, М+# которых элиминируют данные частицы
Хлорпроизводные 1
Полиолы, сахара |
Хлорпроизводные 1
Хлорпроизводные |
Некоторые алленовые и пропаргильные производные |
Ароматические соединения, алкенилариловые эфиры 1
Нитрилы 1
Пропиловые эфиры кислот, пропиламиды |
Функциональные пропилпроизводные, цикланы, циклоалкены 1
Ацетилпроизводные, алициклические кетоны |
Алкилпроизводные 1
Ацетилпроизводные 1
Циклические амиды 1
Алкилпроизводные 1
Альдегиды ( с образованием стабилизированного иона) 1
Ангидриды 1
Тиофенолы, диарилсульфиды
Ароматические амиды
Этоксипроизводные 1
Карбоновые кислоты, ?-аминокислоты
Тиофенолы
Диметиламинопроизводные
Простые и сложные эфиры (этиловые)
Первичные спирты с длинной цепью
Некоторые карбоновые кислоты
Нитропроизводные
Метиларилсульфиды
Алифатические нитросоединения
Фторпроизводные
Некоторые метилсульфиды, тиолы J
Некоторые метилсульфиды, тиолы
Ароматические сульфоксиды
Фторпроизводные
Хлорпроизводные
Полиолы, метиловые эфиры полиолов
Трифторметильные производные
472 Приложение
Таблица 4. Продолжение
Масса
51
53
54
55
56
57
58
59
60
61
62
63
64
65
67
68
69
70
71
72
Состав
CHF2 ?
CHfCl
С4Н5
С4Н6
с3н2о
С4Н7
С4Н8
СНзСНСО
С4Н9
С2Н5СО
CH3NCO
С4Н10
С2Н6СО
NO+CO
с3н7о
сн3со2
С3Н7ОН
СН3ОН+СО
СН3СООН
C2H5S
H2S-|-C2H4
C2HfCl
(СН3ОНJ
so2
C2HfCl
с5н7
с5н8
с5н9
CF3
С5Н10
С3Н6СО
1 с5нп
С3Н7СО
C2H5NCO
C5Hi2
С3Н8СО
Соединения, М+ * которых элиминируют данные частицы
Фторпроизводные 1
Хлорпроизводные |
Некоторые алленовые и пропаргильные соединения |
Ароматические соединения, алкенилариловые эфиры, циклоалкены |
Ненасыщенные алициклические кетоны |
Бутиловые эфиры кислот, бутиламиды |
Функциональные бутилпроизводные, цикланы, циклоалкены 1
Пропионильные производные, алициклические кетоны |
Алкилпроизводные |
Некоторые этилкетоны, алициклические кетоны
Циклические амиды |
Алкилпроизводные 1
Метилкетоны ( с образованием стабилизированного иона) |
Ароматические нитросоединения
Пропоксипроизводные
Метиловые эфиры карбоновых кислот, некоторые алкилацетаты |
Простые и сложные эфиры (пропиловые) |
Некоторые метиловые эфиры карбоновых кислот |
Некоторые ацетаты |
Этилсульфиды |
Тиолы |
Хлорпроизводные
Полиолы, сахара, их метиловые эфиры
Сульфоны, алкилсульфонаты
Хлорпроизводные
Некоторые алленовые и пропаргильные соединения
Ароматические соединения, алкенилариловые эфиры, циклоалкены,
терпены
Амиловые эфиры кислот, амиламиды
Трифторметилпроизводные
Функциональные амилпроизводные, цикланы, циклоалкены
? Бутирильные производные, алициклические кетоны
Алкилпроизводные
Некоторые пропилкетоны, алициклические кетоны
Циклические амиды
Алкилпроизводные
1 Некоторые кетоны и альдегиды
Приложение 473
Таблица 4. Продолжение
Масса
73
74
76
77
78
79
80
81
82
88
91
93
95
127
Состав
с4н9о
с2н5со2
(CH3KSi
С4Н90Н
(CH3KSiH
H2S + Q3H6
Ph
PhH
79Br
H79Br
81Br
H81Br
(CH3LSi
C7H7
с6н5о
CH^9Br
CH^Br
I
Соединения, М+ * которых элиминируют данные частицы
Бутоксипроизводные 1
Метиловые эфиры некоторых карбоновых кислот, некоторые пропио-
наты
Триметилсилильные производные
Простые и сложные бутиловые эфиры ]
Триметилсилильные производные
Тиолы
Некоторые фенилпроизводные
Некоторые фенилпроизводные ]
Бромпроизводные
Бромпроизводные
Бромпроизводные
Бромпроизводные
Политриметилсилильные производные
Бензильные и толильные производные
Феноксипроизводные
Бромпроизводные
Бромпроизводные
Иодпроизводные
474 Приложение
? cia ci3 ?4 сц а6
1
1
ilk 11L
CIBr CI2Br CI3Br CIBr2 CI2Br2 CI3Br2
11 Jiil
CIBfj CfeBrj Br BCj Br3 Br4
?ib
it
Рис. 1. Графический вид кластеров ионов с разным содержанием атомов хлора и брома
СПИСОК ЛИТЕРАТУРЫ
1. Measuring mass. From positive rays to proteins, Ed. by M. A. Grayson, Chemical Heritage
Press, Philadelphia, 2002, 149 p.
2. Дж. Бейнон, Масс-спектрометрия и ее применение в органической химии. — М.: Мир,
1964, 701 с.
3. Т. А. Леман, М. М. Берси, Спектрометрия ионно-циклотронного резонанса. —М.: Мир,
1980, 215 с.
4. S. Т. Graul, R. R. Squires, Mass Spectrom. Rev., 1988, 7, 263.
5. P. Snyder, Biochemical and biotechnological applications of electrospray ionization mass
spectrometry, ACS, Washington D. C. 1995, 601 p.
6. M. Karas, F. Hillenkamp, Advances in Mass Spectrom., Ed. by P. Longevialle, Heyden &
Son, London, 1989, p. 416.
7. R. Chen, X. Cheng, D. W. Mitchell, S. A. Hofstadler, Q. Wu, A. L. Rockwood, M. G.
Sherman, R. D. Smith, Anal. Chem., 1995, 67, 1159.
8. Mass Spectrometry for the characterization of microorganisms, Ed. by С Fenselau, ACS,
Washington D. С 1994, 240 p.
9. A. T. Lebedev, Mass Spectrom. Rev., 1991, 10, 91.
10. H. Schwarz, in Proc. 15th International Mass Spectrometry Conference, Barcelona, 2000,
p. 11.
11. F. W. McLafferty, D. M. Horn, Y. Ge, B. Cerda, E. Fridriksson, K. Breuker, in Proc. 15th
International Mass Spectrometry Conference, Barcelona, 2000, p. 12.
12. А. А. Полякова, Молекулярный масс-спектральный анализ органических соединений. —
М.: Химия, 1983, 248 с.
13. А. А. Полякова, Р. А. Хмельницкий, Введение в масс-спектрометрию органических
соединений. —Л.: Химия, 1966, 204 с.
14. Н. А. Шеховцов, Магнитные масс-спектрометры. —М.: Атомиздат, 1971, 232 с.
15. П. Б. Терентьев, Масс-спектрометрия в органической химии. —М.: Высшая школа,
1979, 223 с.
16. Н. С. Вульфсон, В. Г. Заикин, А. И. Микая, Масс-спектрометрия органических
соединений.—М.: Химия, 1986, 312 с.
17. В. Г. Заикин, А. И. Микая, В. М. Вдовин, Масс-спектрометрия малых циклов. —М.:
Наука, 1983, 159 с.
18. П. Б. Терентьев, А. П. Станкявичюс, Масс-спектрометрический анализ биологически
активных азотистых оснований. — Вильнюс: Мокслас, 1987, 280 с.
19. Химия несольватированных ионов в газовой фазе / Под ред. Г. А. Толстикова. — Уфа,
1987, 183 с.
20. Б. В. Розынов, Масс-спектрометрия в органической химии. Применение в анализе
аминокислот, пептидов, белков., Итоги науки и техники. Органическая химия. Том
2.-М.: ВИНИТИ, 1978, 179 с.
21. В. Г. Заикин, А. И. Микая, Химические методы в масс-спектрометрии органических
соединений. —М.: Наука, 1987, 200 с.
22. В. В. Тахистов, Органическая масс-спектрометрия. —Л.: Наука, 1990, 223 с.
476 Список литературы
23. В. В. Тахистов, Практическая масс-спектрометрия органических соединений. —Л.:
ЛГУ, 1977, 278 с.
24. И. Г. Зенкевич, Б. В. Иоффе, Интерпретация масс-спектров органических
соединений.-Л.: Химия, 1986, 175 с.
25. Р. А. Хмельницкий, Е. С. Бродский, Масс-спектрометрия загрязнений окружающей
среды.-М.: Химия, 1990, 182 с.
26. В. И. Хвостенко, Масс-спектрометрия отрицательных ионов в органической химии. —
М.: Наука, 1981, 159 с.
27. А. Т. Лебедев, Задачник по масс-спектроскопии органических соединений. —М.: МГУ,
1991, 103 с.
28. О. С. Анисимова, Л. Ф. Линдберг, Ю. Н. Шейнкер, Масс-спектрометрия в
исследовании метаболизма лекарственных препаратов. —М.: Медицина, 1978, 168 с.
29. Р.А.Хмельницкий, Е.С.Бродский, Хроматомасс-спектрометрия. — М.: Химия, 1984,
216 с.
30. В. А. Исидоров, И. Г. Зенкевич, Хроматомасс-спектрометрическое определение следов
органических веществ в атмосфере. —Л.: Химия, 1982, 136 с.
31. Физические основы масс-спектрометрии: Методы ионизации / Под ред. Г. А. Толстико-
ва. —Уфа: Башкирский филиал АН СССР, 1985, 120 с.
32. Г. Будзикевич, К. Джерасси, Д. Уильяме, Интерпретация масс-спектров органических
соединений. —М.: Мир 1966, 323 с.
33. Р. Джонстон, Руководство по масс-спектрометрии для химиков-органиков. —М.: Мир,
1975, 236 с.
34. Дж. Чепмен, Практическая органическая масс-спектрометрия. —М.: Мир, 1988,
216 с.
35. F. W. McLafferty, F. Turecek, Interpretation of mass spectra, Mill Valey: University Science
Books, 1993, 422 p.
36. В. Г. Заикин, А. В. Варламов, А. И. Микая, Н. С. Простаков, Основы
масс-спектрометрии органических соединений. —М.: Наука/Интерпериодика, 2001, 286 с.
37. G. Hoch, В. Kok, Arch. Biochem. Biophys., 1963, 101, 160.
38. V. Т. Virkki, R. A. Ketola, ?. Ojala, Т. Kotiaho, V. Komppa, A. Grove, S. Facchetti,
Anal. Chem. 1995, 67, 1421.
39. M. E. Bier, R. G. Cooks, Anal. Chem., 1987, 59, 597.
40. M. Soni, S. Bauer, J. W. Amy, P. Wong, R. G. Cooks, Anal. Chem. 1995, 67, 1409.
41. M. E. Cisper, C.G.Gill, L. E. Townsend, P. H. Hemberger, Anal. Chem., 1995, 67,
1413.
42. F. R. Lauritsen, S. Gylling, Anal. Chem., 1995, 67, 1418.
43. J. C. Holmes, F. A. Morrell, Appl. Spectrosc, 1957, 11, 86.
44. T. A. Ternes, J. Hany, W. Baumann, R. Nagel, Fresenius J. Anal. Chem., 1995,351, 790.
45. Current practice of gas chromatography-mass spectrometry, Ed. by W. M. A. Niessen, Marcel
Dekker Inc., NY, 2001, 528 p.
46. W. H. McFadden, H. L. Schwartz, S. Evans, J. Chromatogr., 1976, 122, 389.
47. Liquid Chromatography-Mass Spectrometry, Ed. By W. M. A. Niessen, Global View
Publishing, Pittsburg, PA, 1999.
48. R. Willoughby, E. Sheehan, S. Mitrovich, A Global View of LC/MS, Global View Publishing,
Pittsburg, PA, 2002.
49. J. D. Pinkston in Practical supercritical fluid chromatography and extraction, ed. by M. Caude,
D. Thiebaut, Harwood Acad. Publ., Amsterdam, 1999, p. 161.
50. F. Sadoun, H. Virilizier, P. J. Arpino, J. Chromatogr., 1993, 647, 351.
51. B. Hurugaverl, K. J. Voorhees, S. J. Luca, J. Chromatogr., 1993, 633, 17.
52. J. W. Jorgenson, K. D. Lukacs, Anal. Chem., 1981, 53, 1298.
53. J. A. Olivares, N. T. Nguyen, С R. Yonker, R. D. Smith, Anal. Chem., 1987, 59, 1230.
54. R. D. Smith, J. H. Wahl, D. R. Goodlett, S. A. Hofstadler, Anal. Chem. 1993, 65, 574A.
Список литературы 477
55. A. Hirabayashi, ?. Sakairi, M. Kpizumi, Anal. Chem., 1994, 66, 4557.
56. A. Hirabayashi, M. Sakairi, M. Kpizumi, Anal. Chem., 1995, 67, 2878.
57. T. Arinobu, H. Hattori, H. Seno, A. Ishii, O. Suzuki, J. Am. Soc. Mass Specrrom., 2002, 13,
204.
58. C. A. Monnig, J. W. Jorgenson, Anal. Chem., 1991, 63, 802.
59. J. F. Banks, T. Dresch, Anal. Chem., 1996, 68, 1480.
60. S. A. Hofstadler, F. D. Swanek, D. С Gale, A. G. Ewing, R. D. Smith, Anal.chem., 1995,
67, 1477.
61. А. Бейкер, Д. Беттеридж, Фотоэлектронные спектры. —?.: Мир, 1975, 200 с.
62. R. J. Waugh, J. H. Bowie, M. L. Gross, Austr. J. Chem., 1993, 46, 693.
63. А. Т. Лебедев, А. Г. Казарян, В. А. Бакулев, Ю. ?. Шафран, В. С. Фалько, В. Г. Лукин,
В. С. Петросян, Химия Гетероцикл. Соед., 1987, 7, 941.
64. F. W. McLafferty, ?. Ge, S. К. Sze, Han Bin Oh, H. Zhai, M. EINagger, V. Zabrouskov,
Proc.50th ASMS Conf. On Mass Spectrometry & Allied Topics', Orlando, FL, 2002,
A020721.
65. H. К. Ляпина, В. С. Симаков, И. И. Фурлей, А. Д. Улендеева, А. С. Воробьев, Г. А. Тол-
стиков, Изв. АН СССР, сер. Химия, 1987, 1688.
66. Н. М. Rosenshtock, ?. В. Wallenstein, A. Warhaftig, H. Eyring, Proc. Natl. Acad. Sci USA,
1952, 38, 667.
67. R. A. Marcus, J. Chem. Phys., 1952, 20, 359.
68. А. И. Бродский, Физическая химия. Т. 2. —?.: Госхимиздат, 1948, с. 942.
69. А. Т. Lebedev, Т. N. Alekseeva, V. A. Bakulev, ?. Yu. Kolobov, V. S. Petrosyan,
Org. Mass Specrrom., 1988, 23, 825.
70. F. Compernolle, M. Dekeirel, Org. Mass Specrrom., 1971, 5, 427.
71. J.-L. Aubagnac, P. Campion, P. Guenot, Org. Mass Spectrom., 1978, 13, 571.
72. K. Biemann, Mass Spectrometry, Organic Chemical Applications, New-York, McGraw Hill,
1962, 370 p.
73. W. Benz, Massenspectrometrie Organischer Verbindungen, Leipzig, Akademische Verlags-
geseschaft, 1969, 369s.
74. O. A. Ponomarev, V. V. Takhistov, in Recent Advances in Analytical Techniques, Ed. by
M. Atta-ur-Rahman, Harwood Acad. Publ., Reading, UK, 2000, p. 369.
75. G. S. Hammond, J. Am. Chem. Soc, 1955, 77, 334.
76. U. I. Zahorszky, Org. Mass Specrrom., 1979, 14, 66.
77. D. P. Stevenson, Disc. Faraday Soc, 1951, 10, 35.
78. ?. ?. Audier, Org. Mass Specrrom., 1969, 2, 280.
79. A. G. Harrison, С D. Finney, J. A. Sherk, Org. Mass Specrrom., 1971, 5, 1313.
80. J. A. Gilpin, F. W. McLafferty, Anal. Chem., 1957, 29, 990.
81. A. G. Harrison, Org. Mass Specrrom., 1970, 3, 549.
82. D. G. I. Kingston, J. T. Bursey, ?. ?. Bursey, Chem. Rev., 1974, 74, 215.
83. M. Kami, A. Mandelbaum, Org. Mass Specrrom., 1980, 15, 53.
84. F. H. Field, in Mass Spectrometry, Ed. A. Maccoll, MTP Int. Rev. Sci. Butterworths, 1972,
p. 133.
85. R. D. Bowen, B. J. Stapleton, D. H. Williams, Chem. Commun., 1978, 24.
86. P.F.Bente III, F. W. McLafferty, D. J. McAdoo, С Lifshitz, J. Phys. Chem., 1975, 79,
713.
87. M. K. Hoffman, J. С Wallace, J. Am. Chem. Soc, 1973, 95, 5064.
88. H. Budzikievicz, C. Djerassi, D. H. Williams, Mass Spectrometry of Organic Compounds
San Fransisco: Holden Day, 1967, 690 p.
89. M. H. Green, R. J. Cooks, J. M. Schwab, R. B. Roy, J. Am. Chem. Soc, 1970,92,3076.
90. J. M. Pechine, Org. Mass Specrrom., 1971, 5, 705.
91. H. Suming, C. Yaozu, J. Longfrei, X. Shuman, Org. Mass Specrrom., 1986, 21, 7.
478 Список литературы
92. ?. ?. Green, in Topics in stereochemistry, ed. N. L. Allinger, E. L. Eliel, Wiley Interscience,
1976, p. 35.
93. A. Mandelbaum, in Stereochemistry, ed. N. B. Kagan, Thieme, Stuttgart, 1977, 1, 137.
94. A. Mandelbaum, Mass Spectrom. Rev., 1983, 2, 223.
95. Application of mass spectrometry to organic stereochemistry, Ed. by J. S. Splitter, F. Turecek,
VCH Publishers Inc., NY, 1994, 705 p.
96. H. Schwarz, Top. Curr. Chem., 1978, 73, 232.
97. A. T. Lebedev, T. N. Alekseeva, T. G. Kutateladze, S. S. Mochalov, Yu. S. Shabarov,
V. S. Petrosyan, Org. Mass Spectrom., 1989, 24, 149.
98. A. T. Lebedev, I. V. Dianova, S. S. Mochalov, V. V. Lobodin, T. Yu. Samguina, R. A. Gazza-
eva , T. Blumental, J. Am. Soc. Mass Spectrom., 2001, 12, 956.
99. S. Hammerum, Mass Spectrom. Rev., 1988, 7, 123.
100. J. B. Lambert, H. F. Shurvell, D. A. Lightner, R. G. Cooks, Organic Structural Spectroscopy
Prentice-Hall Inc.; New Jersey 1998, 568 p.
101. T. Clark, J. Am. Chem. Soc, 1987, 109, 6838.
102. N. J. Jensen, K. B. Tomer, M. L. Gross, P. A. Lyon in Desorption Mass Spectrometry ed. by
P. A. Lyon ACS, Washington, D. C. 1985, p. 194.
103. R. L. Cerny, K. B. Tomer, M. L. Gross, Org. Mass Spectrom., 1986, 21, 655.
104. K. B. Tomer N. J. Jensen, M. L. Gross, Anal. Chem., 1986, 58, 2429.
105. J. Adams, M. L. Gross, J. Am. Chem. Soc, 1989, 111, 435.
106. J. Adams, Mass Spectrom. Rev., 1990, 9, 141.
107. J. B. Lambert, H. F. Shurvell, D. A. Lightner, R. G. Cooks, Organic Structural Spectroscopy,
Prentice-Hall Inc., New Jersey, 1998, 568 p.
108. А. Т. Лебедев, Л. Г. Сагинова, Т. ?. Алексеева, О. А. Малошицкая, Ю. С. Шабаров,
В. С. Петросян, Вестник МГУ, сер. 2, Химия, 1990, 31, 425.
109. А. Т. Лебедев, А. Г. Казарян, П. А. Шарбатян, А. М. Сипягин, В. Г. Карцев, В. С.
Петросян, ЖОрХ, 1988, XXIV, 1393.
ПО. F. P. Lossing, I. J. Tanaka, Chem. Phys., 1956, 25, 1031.
111. M. S. B. Munson, F. H. Field, J. Am. Chem. Soc, 1966, 88, 2621.
112. A.G.Harrison, Chemical Ionization Mass Spectrometry, CRC Press, Boca Raton, 1992,
208 p.
113. В. Л. Тальрозе, А. К. Любимова, ДАН СССР 1952, 86, 909.
114. A. J. Dempster, Philos. Mag., 1916, 31, 438.
115. J. S. Brodbelt, Mass Spectrom. Rev., 1997, 16, 91.
116. E. Burrows, Mass Spectrom. Rev., 1995, 14, 107.
117. T. Donovan, J. Brodbelt Org. Mass Spectrom., 1992, 27, 9.
118. E. S. Eichmann, J. S. Brodbelt, Org. Mass Spectrom., 1993, 28, 737.
119. H. Suming, С Yaozu, J. Longfrei, X. Shuman, Org. Mass Spectrom., 1985, 20, 719.
120. V. K. Nanayakkara, H. I. Kenttamaa, in Proc 42nd ASMS Conf. On Mass Spectrom & Allied
Topics, Chicago, 1994, 737.
121. E. J. Alvarez, J. S. Brodbelt, J. Mass Spectrom., 1995, 30, 625.
122. С S. Kreaser, B. L. Williamson, J. Chem. Soc, Perkin Trans. II, 1996, 3, 427.
123. G. J. Shaw, G. Eglinton, J. ?. ?. Quirke, Anal. Chem., 1981, 53, 2014.
124. D. V. Bowen, F. H. Field, Org. Mass Spectrom., 1974, 9, 195.
125. S. C. Subba Rao, C. Fenselau, Anal. Chem., 1978, 50, 511.
126. С. С Van de Sande, F.van Gaever, P.Sandra, J. Monstrey, Z. Naturforsch., 1977, 32B,
573.
127. B. L. Jelus, B. Munson, С Fenselau, Biomed. Mass Spectrom., 1974, 1, 96.
128. T. Keough, A. J. DeStefano, Org. Mass Spectrom., 1981, 16, 527.
129. B. Munson, Anal. Chem., 1977, 49, 722A.
130. D. F. Hunt, T. M. Harvey, W. C. Brumley, J. F. Ryan III, J. W. Russell, Anal. Chem., 1982,
54, 492.
Список литературы 479
131. D. F. Hunt, J. F. Ryan III, Anal. Chem., 1972, 44, 1306.
132. D. F. Hunt, С N. McEwen, R. A. Upham, Anal. Chem., 1972, 44, 1292.
133. R. V. Hodges, J. L. Beauchamp, Anal. Chem., 1976, 48, 825.
134. D. A. Peake, M. L. Gross, Anal. Chem., 1985, 57, 115.
135. R. С Burnier, G. D. Byrd, B. S. Freiser, Anal. Chem., 1980, 52, 1641.
136. J. H. Bowie, Mass Spectrom. Rev., 1990, 9, 349.
137. A. K. Bose, H. Fujiwara, B. N. Pramanik, Tetrahedron Lett., 1979, 4017.
138. R. С Dougherty, Anal. Chem., 1981, 53, 625A.
139. H. P. Tannenbaum, J. D. Roberts, R. С Doughrty, Anal. Chem., 1975, 47, 49.
140. J. H. Bowie, Mass Spectrometry Specialists Reports, Royal Society of Chemistry, London,
1989, 10, 145.
141. R. С Dougherty, Positive and negative chemical ionization mass spectrometry in Mass
Spectrometry in Environmental Sciences, ed. By F. W. Karasek, O. Hutzinger, S. Safe,
Plenum Press, New-York, 1985, p. 77.
142. D. F. Hunt, G. С Stafford, F. W. Crow, Anal. Chem., 1976, 48, 2098.
143. M. A. Baldwin, F. W. McLafferty, Org. Mass Spectrom., 1973, 7, 1353.
144. A. Dell, D. H. Williams, H. R. Morris, G. A. Smith, J. Feeney, G. С. К. Roberts, J. Am.
Chem. Soc, 1975, 97, 2497.
145. A. P. Bruins, Biomed. Mass Spectrom., 1981, 8, 31.
146. H. D. Beckey, Principles of Field Ionization and Field Desorption Mass Spectrometry,
Pergamon, London 1977.
147. A. Otsuki, H. Shiraishi, Anal. Chem., 1979, 51, 2329.
148. T. Matsuo, H. Matsuda, I. Katakuse, Anal. Chem., 1979, 51, 1329.
149. D. L. Smith, Anal. Chem., 1983, 55, 2391.
150. R. D. Macfarlane, D. F. Torgerson, Science, 1976, 191, 920.
151. B. Sundqvist, R. D. Macfarlane, Mass Spectrom. Rev., 1985, 4, 421.
152. D. M. Hercules, R. J. Day, K. Balasanmugam, T. A. Dang, С. Р. Li, Anal. Chem., 1982, 54,
280A.
153. M. Barber, R. S. Bordoli, G. J. Elliott, R. D. Sedgwick, A. N. Tyler, Anal. Chem., 1982, 54,
645A.
154. R. E. Honig, in Advances in Mass Spectrometry, Ed. By R. M. Elliott, Heyden & Son,
London, 1962, vol. 2, p. 25.
155. A. Benninghoven, D. Jaspers, W. Sichtermann, Appl. Phys., 1976, 11, 35.
156. A. Benninghoven, W. K. Sichtermann, Anal. Chem., 1978, 50, 1180.
157. Г. Д. Танцырев, ?. ?. Николаев, ЖЭТФ, 1971, 13, 473.
158. Г. Д. Танцырев, Н. А. Клейменов, ДАН СССР, 1973, 213, 649.
159. К. L. Busch, S. ?. Unger, A. Vincze, R. G. Cooks, T. Keough, J. Am. Chem. Soc, 1982,
104, 1507.
160. G. Siuzdak, Mass Spectrometry for Biotechnology, Academic Press Inc., San Diego, CA,
1996, 161 p.
161. Desorption Mass Spectrometry, Ed. P. A. Lyon, ACS, Washington DC, 1985, 326 p.
162. Y. Ito, I. Takeuchi, D. Ishii, M. Goto, J. Chromatogr., 1985, 346, 161.
163. S. Pleasance, M. Thibault, M. Mosely, L. Deterding, K.Tomer, J. Jorgenson, J. Am. Soc.
Mass Spectrom., 1990, 1, 312.
164. J. B. French, B. A. Thomson, W. R. Davidson, N. M. Reid, J. A. Buckley, in Mass
Spectrometry in Environmental Sciences, Ed. by F. W. Karasek, O. Hutzinger, S. Safe, Plenum Press,
New-York, 1985, p. 101.
165. С ?. Whitehouse, R. N. Dreyer, M. Yamashita, J, B. Fenn, Anal. Chem., 1985,57, 675.
166. M. Dole, R. L. Hines, R. С Mack, R. С Mobley, L. D. Ferguson, M. B. Alice, J. Chem.
Phys., 1968, 49, 2240.
167. S. J. Gaskell, J. Mass Spectrom., 1997, 32, 677.
168. M. Gamero-Castano, J. Fernandes de la Mora, J. Mass Spectrom., 2000, 35, 790.
480 Список литературы
169. P. Kebarle, J. Mass Spectrom., 2000, 35, 804.
170. J. Femandes de la Mora, G. J.van Berkel, С G. Enke, R. B. Cole, M. Martinez-Sanchez,
J. B. Fenn, J. Mass Spectrom., 2000, 35, 939.
171. Applied electrospray mass spectrometry, Ed. by B. N. Pramanik, A. K. Ganguly,
M. L. Gross, Marcel Dekker Inc., 2002, 464 p.
172. Electrospray Ionization Mass Spectrometry, Ed. by R. B. Cole, Global View Publishing,
USA, 1997.
173. Applied Electrospray Mass Spectrometry, Ed. by B. N. Pramanik, A. K. Ganguly,
M. L. Gross, Global View Publishing, 2002.
174. G. Hopfgartner, T. Wachs, K. Bean, J. Henion, Anal. Chem., 1993, 65, 439.
175. M. S. Wilm, M. Mann, Int. J. Mass Spectrom. Ion Processes, 1994, 136, 167.
176. M. A. Tito, K. Tars, K. Valegard, J. Hajdu, С V Robinson, J. Am. Chem. Soc, 2000, 122,
3550.
177. M. Karas, D. Bachmann, U. Bahr, F. Hillenkamp, Int. J. Mass Spectrom. Ion Proa, 1987,
78, 53.
178. К. Тапака, ?. Waki, ?. Ido, S. Akita, Y. Yoshida, T. Yoshida, Rapid Commun. Mass
Spectrom., 1988, 2, 151.
179. M. Karas, D. Bachmann, F. Hillenkamp, Anal. Chem., 1985, 57, 2935.
180. M. Karas, F. Hillenkamp, Anal. Chem., 1988, 60, 2299.
181. B. Spengler, R. S. Cotter, Anal. Chem., 1990, 62, 793.
182. F. Hillenkamp, S Berkenkamp, A. Leisner, Proc.50th ASMS Conf. On Mass Spectrometry &
Allied Topics, Orlando, FL, 2002, A020361.
183. R. Zenobi, R. Knochenmuss, Mass Spectrom. Rev., 1999, 17, 337.
184. M. Karas, M. Gluckmann, J. Schafer, J. Mass Spectrom., 2000, 35, 1.
185. R. Knochenmuss, A. Stortelder, K. Breuker, R. Zenobi, J. Mass Spectrom., 2000, 35,
1237.
186. D. Spengler, D. Kirsch, R. Kaufmann, J. Phys. Chem., 1992, 96, 9678.
187. T. W. Hutchens, Т. Т. Yip, Rapid Commun. Mass Spectrom., 1993, 7, 576.
188. B. A. Bruenner, T. T. Yip, T. W. Hutchens, Rapid Commun. Mass Spectrom., 1996, 10,
1797.
189. G. L. Jr. Wright, L. H. Cazares, S. M. Leung, S. Nasim, B. Adam, T. Yip, P. F. Schellham-
mer, L. Gong, A. Vlahou, Prostate Cancer and Prostatic Deseases, 1999, 2, 264.
190. M. Merchant, S. R. Weinberger, Electrophoresis, 2000, 21, 1164.
191. S. Petruk, ? Sedkov, S. Smith, S. Tillib, V. Kraevski, T. Nakamura, E. Canaani, C. Croce,
A. Mazo, Science, 2001, 249, 1331.
192. K. Sato, K. Sasaki, M. Tsao, K. Yamaguchi, Cancer Letters, 2002, 176, 199.
193. G. Ball, S. Mian, F. Holding, R. O. Allibone, J. Lowe, S. Ali, G. Li, S. McCardle, I. O. Ellis,
C. Creaser, R. С Rees, Bioinformatics, 2002, 18, 395.
194. P. А. Хмельницкий, И. М. Лукашенко, Е. С. Бродский, Пиролитическая масс-спектро-
метрия высокомолекулярных соединений. —М.: Химия, 1980, 279 с.
195. С. S. Gutteridge, L. Vallis, H. J. H. MacFie, in Computer-Assisted Bacterial Systematics, Ed.
by M. Goodfellow, D. Jones, F. Priest, London, Academic Press, 1985, 369.
196. R. Goodacre, D. B. Kell, Analytica Chimica Acta, 1993, 279, 17.
197. P. Boccini, P. Traldi, J. Mass Spectrom., 1998, 33, 1053.
198. J. Meili, F. С Walls, R. McPherron, A. L. Burlingame, J. Chromatogr. Sci., 1979,
17, 29.
199. E. O. Lawrence, N. E. Edelfsen, Science, 1930, 72, 376.
200. H. Sommer, H. A. Thomas, J. A. Hippie, Phys. Rev., 1949, 76, 1877.
201. A. G. Marshall, M. B. Comisarow, G. Parisod, J. Chem. Phys., 1979, 71, 4434.
202. L. Schweikhard, N. С Hill, G. M. Alber, A. G. Marshall, in Proc.12 Intern. Mass Spectrom.
Conf. Amsterdam, 1991, p. 76.
Список литературы 481
203. D. A. Laude, S. С. Beu, in Experimental mass spectrometry, Ed. by D. H. Russel, Plenum
Press, NY, 1994, p. 188.
204. A. G. Marshall, F. R. Verdun, Fourier Transforms in NMR, optical and mass spectrometry
Elsevier, Netherlands, 1990, 450 p.
205. T. Dienes, S. J. Pastor, S. Scherch, J. R. Scott, J. Yao, S. Cui, C. L. Wilkins,
Mass Spectrom. Rev., 1996, 15, 163.
206. С Weickhardt, F. Moritz, J. Grotemeyer, Mass Spectrom. Rev., 1996, 15, 139.
207. A. G. Marshall, C. L. Hendrickson, G. S. Jackson, Mass Spectrom. Rev., 1998, 17, 1.
208. P. H. Dawson, Mass Spectrom. Rev., 1986, 5, 1.
209. R. G. Cooks, G. L. Glish, S. A. McLuckey, R. E. Kaiser, Chemical & Engineering News,
1991, 69, 26.
210. J. Am. Soc. Mass Spectrom., 2002, 13, № 5.
211. J. Am. Soc. Mass Spectrom., 2002, 13, № 6.
212. M. С Wiley, I. H. McLaren, Rev. Sci. Instrum., 1955, 26, 1150.
213. Б. А. Мамырин, В. И. Каратаев, Д. В. Шмикк, В. А. Загулин, Журнал экспер, и теор.
физики, 1973, 64, 82.
214. A. F. Dodonov, I. V. Chernushevich, V. V. Laiko, in Time-of-Flight Mass Spectrometry
(Chapter 7), Ed. by R. J. Cotter, ACS Symposium Series 549, Washington, DC, 1994,
108.
215. J. H. J. Dawson, M. Guilhaus, Rapid Commun. Mass Spectrom., 1989, 3, 155.
216. K. L. Busch, G. L. Glish, S. A. McLuckey, Mass spectrometry/mass spectrometry.
Techniques and applications of tandem mass spectrometry, VCH Publishers, Inc., USA,
1988, 333 p.
217. F. W. Aston, Proc. Cambridge Philos. Soc, 1919, 19, 317.
218. F. W. McLafferty, P. F. Bente, R. Kornfeld, S. С Tsai, I. J. Howe, J. Am. Chem. Soc, 1973,
95, 2120.
219. R. S. Bordoli, R. H. Bateman, Int. J. Mass Spectrom., Ion Proc, 1992, 122, 243.
220. F. W. McLafferty, P.J.Todd, D. C. McGilvery, M.A.Baldwin, J. Am. Chem. Soc, 1980,
102, 3360.
221. G. L. Glish, P. J. Todd, Anal. Chem., 1982, 54, 842.
222. R. С Dunbar, in Gas Phase Ion Chemistry, Ed. by M. Bowers, Academic: New York, 1979,
vol. 2.
223. R. B. Cody, B. S. Freiser, Anal. Chem., 1979, 51, 547.
224. M. A. Mabud, M. J. Derkey, R. G. Cooks, Int. J. Mass Spectrom. Ion Proc, 1985, 67,
285.
225. A. T. Lebedev, V. A. Bakulev, R. N. Hayes, J. H. Bowie, Rapid Comm. Mass Spectrom.,
1991, 5, 234.
226. D.F.Hunt, W. M. Bone, J. Shabanowitz, J. Rhodes, J. MBallard, Anal. Chem., 1981, 53,
1704.
227. A. G. Marshall, Т. С L. Wang, T. L. Ricca, J. Am. Chem. Soc, 1985, 107, 7893.
228. J. W. Gauthier, R. R. Trautman, D. B. Jacobson, Anal. Chim. Acta, 1991, 246, 211.
229. J. H. Bowie, T. Blumenthal, J. Am. Chem. Soc, 1975, 97, 2959.
230. T. A. Lehman, M. M. Bursey, J. R. Hass, Org. Mass Spectrom., 1983, 18, 373.
231. S. Villeneuve, P. С Burgres, Org. Mass Spectrom., 1986, 21, 733.
232. D. V. Zagorevskii, Coord. Chem. Rev., 2002, 225, 5.
233. J. K. Terlouw, H. Schwarz, Angewandte Chemie (Intern. Edition), 1987, 26, 805.
234. J. L. Holmes, Mass Spectrom. Reviews, 1989, 8, 513.
235. С Wesdemiotis, F. W. McLafferty, Chem. Rev., 1987, 87, 485.
236. H. Schwarz, Pure Appl. Chem., 1989, 61, 685.
237. J. L. Holmes, Adv. Mass Spectrom., 1989, 11, 53.
238. F. W. McLafferty, Science, 1990, 247, 925.
239. С A. Shalley, G. Hornung, D. Schroder, H. Schwarz, Chem. Soc. Rev., 1998, 27, 91.
482 Список литературы
240. J. К. Terlow, Adv. Mass Spectrom., 1989, 11, 984.
241. ?. ?. Cordero, С. Wesdemiotis, in Biological Mass Spectrometry: Present and Future, Ed.
by T. Matsuo, R. M. Caprioli, M. L. Gross, Y. Seyama, New York, Wiley, 1994, 119.
242. P. С Burgers, J. L. Holmes, A. A. Mommers, J. K. Terlouw, Chem. Phys. Lett., 1983,
102, 1.
243. С Wesdemiotis, R. Feng, E. R. Williams, F. W. McLafFerty, Org. Mass Spectrom., 1986,
21, 689.
244. J. K. Terlouw, P. С Burgers, B. L. M.vanBaar, T. Weiske, H. Schwarz, Chimia 1986, 40,
357.
245. D. V. Zagorevskii, J. L. Holmes, Organometallics, 1992, 11, 3224.
246. M. J. Gaillard, A. G. de Pinho, J. С Poizat, J. Remillieux, R. Saoudi, Phys. Rev., 1983,
A28, 1267.
247. G. I. Gellene, R. F. Porter, J. Chem. Phys., 1984, 81, 5570.
248. G. I. Gellene, R. F. Porter, High Temp. Sci., 1984, 17, 171.
249. F. Turecek J. Mass Spectrom., 1998, 33, 779.
250. D. V. Zagorevskii, J. L. Holmes, Mass Spectrom. Rev., 1994, 13, 133.
251. D. V. Zagorevskii, J. L. Holmes, Mass Spectrom. Rev., 1999, 18, 87.
252. M. J. Cohen, F. W. Karasek J. Chromatogr. Sci., 1970, 8, 330.
253. Y. Raneko, M. R. Megill, J. B. Hasted, J. Chem. Phys., 1966, 45, 3741.
254. H. W. Revercomb, E. A. Mason, Anal. Chem., 1975, 47, 970.
255. G.von Helden, M.-T. Hsu, P. R. Kemper, M. T. Bowers, J. Chem. Phys., 1991, 93, 3835.
256. J. M. Hunter, J. L. Fue, E. J. Roscamp, M. F. Jarrold, J. Phys. Chem., 1994, 98, 1810.
257. G.von Helden, M.-T. Hsu, P. R. Kemper, M. T. Bowers, J. Chem. Phys., 1991, 93, 3835.
258. D. E. Clemmer, M. F. Jarrold, J. Mass Spectrom., 1997, 32, 577.
259. H. H. Hill, W. F. Siems, R. H. St. Louis, D. G. McMinn, Anal. Chem., 1990, 62, 1201A.
260. ? ?. Chen, ?. ?. Hill, D. P. Wittmer, Int. J. Mass Spectrom. Ion Proc, 1996, 154, 1.
261. D.F.Hagen, Anal. Chem., 1979, 51, 870.
262. Q. N. Porter, Mass Spectrometry of Heterocyclic Compounds John Wiley & Sons, USA,
1985, 966 p.
263. K. Hiraoka, P. Kebarle, J. Am. Chem. Soc, 1976, 98, 6119.
264. D. F. Hunt, Т. М. Harvey, Anal. Chem., 1975, 47, 1965.
265. V. Dommes, F. Wirtz-Peitz, W. H. Kunau, J. Chromatogr. Sci., 1976, 14, 360.
266. K. Levsen, Fundamental aspects of organic mass spectrometry Weinheim, New-York: Verlag
Chemie, 1978, 312 p.
267. F. H. Field, J. Am. Chem. Soc, 1968, 90, 5649.
268. H. Budzikiewicz, E. Busker Tetrahedron, 1980, 36, 255.
269. D. A. Peake, M. L. Gross, Anal. Chem., 1985, 57, 115.
270. С. Ранг, О. Эйзен, А. Мюрисепп Инфракрасные и масс-спектры ненасыщенных
углеводородов.—Таллин.: Варгус, 1977, 616 с.
271. Е. Busker, H. Budzikiewicz, Org. Mass Spectrom., 1979, 14, 222.
272. F. H. Field, M. S. B. Munson, J. Am. Chem. Soc, 1968, 89, 4272.
273. M. J. Buchanan, G. Olerich, Org. Mass Spectrom., 1984, 19, 486.
274. P. N. Rylander, S. Meyerson, J. Am. Chem. Soc, 1956, 78, 5799; S. Meyerson, Org. Mass
Spectrom., 1989, 24, 267.
275. W. J. Simonsick, R. A. Hites, Anal. Chem., 1984, 56, 2949.
276. D. Berthomieu, H. Audier, J.-P. Denhez, С Monteiro, P. Mourgues, Org. Mass Spectrom.,
1991, 26, 271.
277. S. B. Hawthorne, D. J. Miller, Anal. Chem., 1985, 57, 694.
278. R. Chai, A. G. Harrison, Anal. Chem., 1983, 55, 969.
279. M. Oehme, Anal. Chem., 1983, 55, 2290.
280. R. A. J. O'Hair, J. H. Bowie, Rapid Comm. Mass Spectrom., 1989, 3, 293.
484 Список литературы
316. W. Gerrard, M. F. Lappert, ?. ?. Silver, Proc. Chem. Soc, 1957, 19.
317. U. Svanholm, V. D. Parker, Chem. Commun., 1972, 645.
318. U. Svanholm, V. D. Parker, J. Chem. Soc, Perkin Trans., 1974, 2, 169.
319. R. J. Waugh, R. N. Hayes, P. С ?. Eichinger, ?. ?. Downard, J. ?. Bowie, J. Am. Chem.
Soc, 1990, 112, 2537.
320. P. С ?. Eichinger, J. H. Bowie, J. Chem. Soc, Perkin Trans. II, 1988, 497.
321. M. D. Rozeboom, J. P. Kiplinger, J. E. Bartmess, J. Am. Chem. Soc, 1984, 106, 1025.
322. P. С ?. Eichinger, J. H. Bowie, J. Chem. Soc, Perkin Trans. II, 1987, 1499.
323. E. J. Reiner, R. A. Poirier, M. R. Peterson, I. G. Csizmadia, A. G. Harrison, Can. J. Chem.,
1986, 64, 1652.
324. M. J. Raftery, J. H. Bowie, Austr. J. Chem., 1988, 41, 1477.
325. J. J. Eisch, С A. Kovacs, J. Organomet. Chem., 1971, 30, 97.
326. J. J. Eisch, S. K. Dua, С A. Kovacs, J. Org. Chem., 1987, 52, 4437.
327. L. B. Reeks, S. Dua, J. H. Bowie, Rapid Comm. Mass Spectrom., 1993, 7, 282.
328. F. W. McLafferty, Anal. Chem., 1962, 34, 2.
329. F. W. McLafferty, Anal Chem., 1962, 34, 16.
330. A. G. Harrison, P. H. Lin, Can. J. Chem., 1975, 53, 1314.
331. H. W. Leung, A. G. Harrison, Can. J. Chem., 1976, 54, 3439.
332. W. G. Liauw, A. G. Harrison, Org. Mass Spectrom., 1981, 16, 388.
333. A. G. Harrison, F. I. Onuska, C. W. Tsang, Anal. Chem., 1981, 53, 1183.
334. H. W. Leung, A. G. Harrison, J. Am. Chem. Soc, 1980, 102, 1623.
335. П. А. Шарбатян, А. Т. Лебедев, А. М. Сипягин, В. Г. Карцев, В. С. Петросян, Химия
Гетероцикл. Соед., 1982, 3, 334.
336. А. Т. Лебедев, П. А. Шарбатян, А. М. Сипягин, В. Г. Карцев, В. С. Петросян, Химия
Гетероцикл. Соед., 1982, 7, 919.
337. Е. О. Oswald, P. W. Albro, J. D. McKinney, J. Chromatogr., 1974, 98, 363.
338. F. J. Biros, R. С Dougherty, J. Dalton, Org. Mass Spectrom., 1972, 6, 1161.
339. Т. С Cairns, E. G. Siegmund, Anal. Chem., 1981, 53, 1599.
340. E. L. White, ?. ?. Bursey, Bio. Environ. Mass Spectrom., 1989, 18, 413.
341. G. R. B. Webster, K. Olie, O. Hutzinger, in Mass Spectrometry in Environmental Sciences,
Ed. by F. W. Karasek, O. Hutzinger, S. Safe Plenum Press, New-York, 1985, p. 257.
342. D. P. Ridge, J. L. Beauchamp, J. Am. Chem. Soc, 1974, 96, 3595.
343. F. W. Crow, A. Bjorseth, К. Т. Knapp, R. Bennett, Anal. Chem., 1981, 53, 619.
344. E. A. Stemmler, R. A. Hites, Electron Capture Negative Ion Mass Spectra of Environmental
Contaminants and Related Compounds, VCH Publishers, New-York, 1988.
345. H. R. Buser, Anal. Chem., 1986, 58, 2913.
346. H. R. Buser, С Rappe, P. A. Berqvist, Environ. Health Perspect., 1985, 60, 293.
347. D. F. Hunt, T. M. Harvey, J. W. Rassell, J. Chem. Soc, Chem. Commun., 1975, 151.
348. G. R. B. Webster, D. A. Birkholz, in Mass Spectrometry in Environmental Sciences, Ed. by
F. W. Karasek, O. Hutzinger, S. Safe Plenum Press, New-York, 1985, p. 209.
349. G. Sundstrom, B. Jansson, in Mass Spectrometry in Environmental Sciences, Ed. by
F. W. Karasek, O. Hutzinger, S. Safe Plenum Press, New-York, 1985, p. 297.
350. G. A. Eiceman, in Mass Spectrometry in Environmental Sciences, Ed. by F. W. Karasek,
O. Hutzinger, S. Safe Plenum Press, New-York, 1985, p. 311.
351. F. I. Onuska, in Mass Spectrometry in Environmental Sciences, Ed. by F. W. Karasek,
O. Hutzinger, S. Safe Plenum Press, New-York, 1985, p. 405.
352. J. H. Beynon, R. A. Saunders, A. E. Williams Appl. Spectrosc, 1960, 14, 95.
353. J. Seibl, Z. Gaumann, Helv. Chim. Acta, 1963, 46, 2857.
354. M. L. Gross, F. W. McLafferty, J. Am. Chem. Soc, 1971, 93, 1267.
355. A. T. Lebedev, R. N. Hayes, J. H. Bowie, Rapid Comm. Mass Spectrom., 1991, 5, 160.
356. H. M. Fales, C. Fenselau, J. H. Duncan, Org. Mass Spectrom., 1976, 11, 669.
357. J. V. Headley, A. G. Harrison, Can. J. Chem., 1985, 63, 609.
Список литературы 483
281. G. J. Currie, J.H.Bowie, R. A. Massy-Westropp, G.W.Adams, J. Chem. Soc, Perkin
Trans. II, 1988, 403.
282. E. L. Eliel, J. D. McCollum, S. Meyerson, P. N. Rylander, J. Am. Chem. Soc, 1961, 83,
2481.
283. О. С. Чижов, А. Я. Отт, Успехи биологической химии. —М.: Наука, 1978, с. 151.
284. В. И. Каденцев, И. А. Трушкина, О. С. Чижов, А. Г. Таймуразов, Биоорганическая
химия, 1988, 14, 1317.
285. Т. Radford, D. С. DeJongh, Carbohydrates in Biomedical Application of Mass Spectrometry,
First Supplementary Volume, ed. G. R. Waller, О. С Dermer, Wiley Interscience, New-York,
1980.
286. A. M. Hogg, T. L. Nagabhusan, Tetrahedron Lett., 1972, 4827.
287. R. C. Dougherty, J. D. Roberts, W. W. Binkley, O. S. Chizhov, V. I. Kadentsev,
A. A. Solovyanov, J. Org. Chem., 1974, 39, 451.
288. A. K. Ganguly, N. F. Cappuccino, H. Fujiwara, A. K. Bose, J. Chem. Soc, Chem. Commun.,
1979, 148.
289. O. S. Chizhov, V. I. Kadentsev, A. A. Solovyanov, P. F. Leonowich, R. С Dougherty,
J. Org. Chem., 1976, 41, 3425.
290. V. N. Reinhold, S. A. Carr, Anal. Chem., 1982, 54, 499.
291. K. K. Mock, M. Davey, J. S. Cottrell, Biochem. Biophys. Res. Commun., 1991, 177,
644.
292. K. L. Duffin, J. K. Welply, E. Huang, J. D. Henion, Anal. Chem., 1992, 64, 1440.
293. V. N. Reinhold, В. В. Reinhold, С. Е. Costello, Anal. Chem., 1995, 67, 1772.
294. A. S. Weiskopf, P. Vouros, D. J. Harvey, Rapid Comm. Mass Spectrom., 1997, 11,
1493.
295. W. Chai, V. Piskarev, A. M. Lawson, J. Am. Soc. Mass Spectrom., 2002, 13, 670.
296. B. Munson, T.-M. Feng, H. D. Ward, R. K. Murray, Jr., Org. Mass Spectrom., 1987, 22,
606.
297. E. S. Eichmann, E. Alvarez, J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 1992, 3, 535.
298. H. Suming, С Yaozu, J. Longfrei, X. Shuman, Org. Mass Spectrom., 1985, 20, 719.
299. S. Daishima, Y. Ilida, F. Kanda, Anal. Sci., 1991, 7, 203.
300. G. L. Glish, R. G. Cooks, J. Am. Chem. Soc, 1978, 100, 6720.
301. A. Maquestiau, R. Flammang, M. Flammang-Barbieux, H. Mispreuve, Tetrahedron, 1980,
36, 1993.
302. О.А.Реутов, А. Л. Курц, К. П. Бутин, Органическая химия. —М.: МГУ, 1999,
с. 285.
303. R. N. Hayes, J. С. Sheldon, J. H. Bowie, D. E. Lewis, J. Chem. Soc, Chem. Commun.,
1984, 1431.
304. R. Houriet, D. Stahl, F. J. Winkler, Environ. Health Perspect., 1980, 36, 63.
305. M. J. Raftery, J. H. Bowie, J. С Sheldon, J. Chem. Soc, Perkin Trans. II, 1988, 563.
306. R. W. Thies, E. P. Seitz, J. Chem. Soc, Chem. Commun., 1976, 846.
307. P. С ?. Eichinger, J. H. Bowie, Int. J. Mass Spectrom. Ion Processes, 1992, 117, 1.
308. J. M. Denis, J. M. Conia, Tetrahedron Lett., 1972, 4593.
309. P. D. Bartlett, R. H. Rosewald, J. Am. Chem. Soc, 1934, 56, 1990.
310. S.K. Dua, R. B. Whait, M.J.Alexander, R.N.Hayes, A. T. Lebedev, P. С. Н. Eichinger,
J. H. Bowie, J. Am. Chem. Soc, 1993, 115, 5709.
311. S. K. Dua, M. J. Alexander, J. H. Bowie, Org. Mass Spectrom., 1993, 28, 1155.
312. M. I. Gorfinkel, L. Yu. Ivanovskaia, V. A. Koptyug, Org. Mass Spectrom., 1969, 2, 273.
313. P. С ?. Eichinger, J. H. Bowie, R. N. Hayes, J. Am. Chem. Soc, 1989, 111, 4224.
314. В. И. Каденцев, В. Д. Соковых, О. С. Чижов, Изв. АН СССР, сер. Химия, 1978,
1949.
315. E.E.Kingston, J. ?. Beynon, J. G. Liehr, P. Meyrant, R. Flammang, A. Maquestiau, Org.
Mass Spectrom., 1985, 20, 351.
Список литературы 485
358. R. Chai, A. G. Harrison, Anal. Chem., 1983, 55, 969.
359. I. Jardine, С Fenselau, Org. Mass Spectrom., 1975, 10, 748.
360. G. A. Olah, D. H. O'Brien, M. J. Calin, J. Am. Chem. Soc., 1967, 89,3582.
361. R. F. Foster, W. Tumas, J. I. Brauman, J. Chem. Phys., 1983, 79, 4644.
362. M. J. Raftery, J. H. Bowie, Int. J. Mass Spectrom. Ion Proc, 1987, 79, 267.
363. M. B. Stringer, J. H. Bowie, J. L. Holmes, J. Am. Chem. Soc, 1986, 108, 3888.
364. M. B. Stringer, J. H. Bowie, G. J. Currie, J. Chem. Soc, Perkin Trans. II, 1986, 1821.
365. D. F. Hunt, A. B. Giordani, J. Shabanowicz, G. Rhodes, J. Org. Chem., 1982, 47, 738.
366. S. Chowdhury, A. G. Harrison, J. Am. Chem. Soc, 1988, 110, 7345.
367. A. Marshall, M. Tkaczyk, A. G. Harrison, J. Am. Soc Mass Spectrom., 1991, 2, 292.
368. R. Ryhage, E. Stenhagen, in Mass Spectrometry of Organic Ions, Ed. by F. W. McLafferty,
Academic Press, New York, 1963, p. 403.
369. Mass Spectrometry of Lipids, Ed. by R. C. Murphy, Global View Publishing, Pittsburg,
PA, 1993.
370. J. L. Holmes, Org. Mass Spectrom., 1970, 3, 1505.
371. A. G. Harrison, R. К. М. R. Kallury, Org. Mass Spectrom., 1980, 15, 277.
372. H. Ichikawa, A. G. Harrison, Org. Mass Spectrom., 1978, 13, 389.
373. J. A. Herman, A. G. Harrison, Can. J. Chem., 1981, 59, 2133.
374. W. Chai, G. Wang, Z. Xu, J. Pan, S. Duan, Org. Mass Spectrom., 1983, 18, 64.
375. S. B. Hawthorne, D. J. Miller, Appl. Spectrosc, 1986, 40, 1200.
376. V. K. Nanayakkara, H. I. Kenttamaa, in Proc 42nd ASMS Conf. On Mass Spectrom & Allied
Topics, Chicago, 1994, 737.
377. J. J. Grabowski, X. Cheng, J. Am. Chem. Soc, 1989, 111, 3106.
378. M. Fujio, R. T. Mclver, R. W. Taft, J. Am. Chem. Soc, 1981, 103, 4017.
379. R. A. J. O'Hair, S. Gronert, C. H. DePuy, J. H. Bowie, J. Am. Chem. Soc, 1989, 111,
3105.
380. M. B. Stinger, J. H. Bowie, P. С ?. Eichinger, G. J. Currie, J. Chem. Soc, Perkin Trans. II,
1987, 385.
381. A. L. C. Smit, F. H. Field, J. Am. Chem. Soc, 1977, 99, 6471.
382. A. P. Bruins, Anal. Chem., 1979, 51, 967.
383. W. С Brumley, J. A. Sphon, in Applications of Mass Spectrometry in Food Science, Ed. by
J. Gilbert, Elsevier, London, 1987.
384. D. F. Hunt, J. Shabanowicz, A. B. Giordani, Anal. Chem., 1980, 82, 386.
385. S. W. Froelicher, R. E. Lee, R. R. Squires, B. S. Fraser, Org. Mass Spectrom., 1985,
20, 4.
386. R. N. Hayes, J. H. Bowie, J. Chem. Soc, Perkin Trans. II, 1986, 1827.
387. A. Frater, Helv. Chim. Acta, 1975, 58, 442.
388. R. E. Ireland, R. H. Mueller, A. K. Willard, J. Am. Chem. Soc, 1976, 98, 2868.
389. D. J. Burinsky, R. G. Cooks, J. Org. Chem., 1982, 47, 4864.
390. M. J. Raftery, J. H. Bowie, Org. Mass Spectrom., 1988, 23, 719.
391. J.H.Bowie, in Experimental Mass Spectrometry, Ed. by D.H.Russell. Plenum Press,
New-York, 1994, p. 1.
392. M. J. Raftery, J. H. Bowie, Int. J. Mass Spectrom. Ion Proc, 1988, 85, 167.
393. R. F. Pottie, F. P. Lossing, J. Am. Chem. Soc, 1961, 83, 4737.
394. F. W. McLafferty, Anal. Chem., 1962, 34, 26.
395. H. Schwarz, Topics Current Chem., 1978, 73, 231.
396. G. Depke, W. Klose, H. Schwarz, Org. Mass Spectrom., 1983, 18, 495.
397. G. L. Glish, R. G. Cooks, J. Am. Chem. Soc, 1978, 100, 6720.
398. K. P. Zeller, H. Meier, E. Muller, Lieb. Ann. Chem., 1971, 749, 178.
399. K. P. Zeller, H. Meier, E. Muller, Org. Mass Spectrom., 1971, 5, 373.
400. K. P. Zeller, H. Meier, E. Muller, Tetrahedron, 1972, 28, 5831.
401. K. P. Zeller, Org. Mass Spectrom., 1975, 10, 317.
486 Список литературы
402. О. С. DeJongh, R. Y. Van Fossen, L. R. Dusold, M. P. Cava, Org. Mass Spectrom., 1970,
3, 31.
403. P. H. Kinson, B. V. Trost, Tetrahedron Lett., 1969, 1075.
404. K. Undheim, O. Thorstad, G. Hvistendahl, Org. Mass Spectrom., 1971, 5, 73.
405. O. Thorstad, K. Undheim, G. Hvistendahl, Org. Mass Spectrom., 1974, 9, 548.
406. G. Hvistendahl, K. Undheim, Org. Mass Spectrom., 1972, 6, 217.
407. C. W. Thomas, L. L. Leveson, 1978, 13, 39.
408. H. К. Караханова, Т. А. Сергеева, А. Т. Лебедев, ?. С. Яшина, В. Г. Ларионова,
В. Г. Карцев, В. С. Петросян, Вестн. МГУ, сер. Химия, 1988, 29, 290.
409. В. Г. Карцев, А. М. Сипягин, Л. И. Кирковский, Т. Г. Денисова, в Алифатические
соединения в органическом синтезе / Под ред. И. К. Коробицина. — Л., 1985, 152 с.
410. О. S. Chizhov, V. I. Kadentsev, G. G. Palmbach, К. I. Burstein, S. A. Shevelev, A. A. Fein-
silberg, Org. Mass Spectrom., 1978, 13, 611.
411. J. A. Ballantine, J. D. Barton, J. F. Carter, J. P. Davies, K. Smith, G. Stedman,
E. E. Kingston, Org. Mass Spectrom., 1988, 23, 1.
412. A. G. Harrison, R. K. M. R. Kallury, Org. Mass Spectrom., 1980, 15, 284.
413. J. Yinon, Forensic Mass Spectrometry, CRC Press, Boca Raton, FL, 1987.
414. J. J. Brophy, V. Diakiw, R. J. Goldsack, D. Nelson, J. S. Shannon, Org. Mass Spectrom.,
1979, 14, 117.
415. J. Yinon, M. Laschever, Org. Mass Spectrom., 1981, 16, 264.
416. R. K. M. R. Kallury, P. L. M. K. Rao, Org. Mass. Spectrom., 1977, 12, 411.
417. A. Maquestiau, Y. Van Haverbeke, R. Flammang, P. Meyrant, Org. Mass Spectrom., 1977,
15, 80.
418. E. A. Stemmler, R. A. Hites, Biomed. Environ. Mass Spectrom., 1987, 14, 417.
419. G. W. Adams, J. H. Bowie, J. Chem. Soc, Perkin Trans. II, 1989, 2159.
420. A. T. Lebedev, R. N. Hayes, J. H. Bowie, J. Chem. Soc, Perkin Trans. II, 1991, 1127.
421. P. А. Хмельницкий, Ю. А. Ефремов, Успехи химии, 1977, 46, 83.
422. В. Н. Бочкарев, Дисс. канд. хим. наук, М., 1975, 209 с.
423. А. Е. Чернышев, Дисс. канд. хим. наук, М., 1985, 209 с.
424. В. Н. Бочкарев, Т. Ф. Слюсаренко, А. Н. Поливанов, А. А. Вернадский, ЖОХ, 1980, 50,
1783.
425. К. Biemann, J. Seibl, F. Gapp, Biochem. Biophys. Res. Commun., 1959, 1, 307.
426. С W. Tsang, A. G. Harrison, J. Am. Chem. Soc, 1976, 98, 1301.
427. M. Meot-Ner, F. H. Field, J. Am. Chem. Soc, 1973, 95, 7207.
428. A. L. С Smit, F. H. Field, J. Am. Chem. Soc, 1977, 99, 6471.
429. M. Vairamani, R. Srinivas, G. K. Viswandha, Rao Biomed. Environ. Mass Spectrom., 1988,
17, 299.
430. A. A. Kiryushkin, H. M. Fales, T. Axenrod, E.J.Gilbert, G. W. A. Milne, Org. Mass
Spectrom., 1971, 5, 19.
431. С V. Bradley, I. Howe, J. H. Beynon, Biomed. Mass Spectrom., 1981, 8, 85.
432. C.V.Bradley, D.H.Williams, M. R. Hanley, Biochem. Biophys. Res. Commun., 1982,
104, 1223.
433. D. F. Hunt, A. M. Buko, J. M. Ballard, J. Shabanowitz, A. B. Giordani, Biomed. Mass
:Spectrom., 1981, 8, 397.
434. B. T. Chait, R. Wang, R. С Beavis, S. B. Kent, Science, 1993, 262, 89.
435. R Roepstorff, J. Fohlmann, Biomed. Mass Spectrom., 1984, 11, 601.
436. K. Biemann, in Methods in Enzimology ed.by J. A. McCloskey, Academic Press, San Diego,
1990, vol. 193, 866.
437. M. J. Poke, Da Ren, С Wesdemiotis, J. Mass Spectrom., 2000, 35, 1391.
438. A. L. McCormack, A. Somogyi, A. R. Dongr'e, V. H. Wysocki, Anal. Chem., 1993, 65,
2859.
439. J. R. Yates, S. Speicher, P. R. Griffin, T. Hunkapiller, Anal. Biochem., 1993, 214, 397.
Список литературы 487
440. ?. Mann, P. Hojrup, P. Roepstorff, Biol. Mass Spectrom., 1993, 22, 338.
441. P. James, M. Quadrioni, E. Carafoli, G. Gonnet, Biochem. Biophys. Res. Commun., 1993,
195, 58.
442. P. James, M. Quadrioni, E. Carafoli, G. Gonnet, Protein Sci., 1994, 3, 1347.
443. S. D. Patterson, D. Thomas, R. A. Bradshow, Electrophoresis, 1996,17, 877.
444. J. K. Eng, A. L. McCormack, J. R. Jates III, J. Am. Soc. Mass Spectrom., 1994, 5, 976.
445. J. R. Jates III, J. K. Eng, A. L. McCormack, D. Schieltz Anal. Chem., 1995, 67, 1426.
446. A. L. McCormack, D. M. Schieltz, B. Goode, S. Yang, G. Barnes, D. Drubin, J. R. Yates,
Anal. Chem., 1997, 69, 767.
447. M. Mann, M. S. Wilm, Anal. Chem., 1994, 66, 4390.
448. S. J. Cordwell, V. С Wasinger, A. Cerpa Poljak, M. W. Duncan, I. Humphrey-Smith, J. Mass
Spectrom., 1997, 32, 370.
449. New Methods in Peptide Mapping Character of Proteins, Ed. by W. S. Hancock, Global View
Publishing, Pittsburg, PA, 1996.
450. Mass Spectrometry of Biological material, Ed. by B. S. Larsen, С N. McEwen, Global View
Publishing, Pittsburg, PA, 2nd edition, 1998.
451. Peptide and Protein Drug Analysis, Ed. by R. E. Reid, Global View Publishing, Pittsburg,
PA, 2000.
452. A. P. Snyder, Interpreting Protein Mass Spectra: A Comprehensive Resource Global View
Publishing, Pittsburg, PA, 2000.
453. Mass Spectrometry of Proteins and Peptides, Ed. by J. R. Chapman, Global View Publishing,
Pittsburg, PA, 2000.
454. M. Kinter, N. E. Sherman, Protein Sequencing and Identification Using Tandem MS Global
View Publishing, Pittsburg, PA, 2000.
455. D. C. Liebler, Introduction to Proteomics Global View Publishing, Pittsburg, PA, 2002*
456. J. Roboz, Mass Spectrometry in Cancer Research Global View Publishing, Pittsburg,
PA, 2002.
457. The Protein Protocols Handbook, Ed. by J. M. Walker, Global View Publishing, Pittsburg,
PA, 2nd Edition, 2002.
458. Modern Protein Chemistry: Practical Aspects, Ed. by G. С Howard, W. E. Brown, Global
View Publishing, Pittsburg, PA, 2002.
459. J. Am. Soc. Mass Spectrom., 2002, 13, № 7.
460. ?. ?. Phizicky, S. Fields, Microbiol. Rev., 1995, 59, 94.
461. R. R. О Loo, D. R. Goodlett, R. D. Smith, J. A. Loo, J. Am. Chem. Soc, 1993, 115,
4391.
462. J. A. Loo, Mass Spectrom. Rev., 1997, 16, 1.
463. E. C. Huang, B. N. Pramanic, A. Tsarbopoulos, P. Reichert, A. K. Gauguly, P. P. Trotta,
T. Nagabhushan, J. Am. Soc. Mass Spectrom., 1993, 4, 624.
464. B. N. Pramanik, P. L. Bartner, U. A. Mirza, ? ?. Liu, A. K. Ganguly, J. Mass Spectrom.,
1998, 33, 911.
465. C. V. Robinson, E. E. Chung, B. B. Kragelund, J. Knudsen, R. T. Aplin, F. M. Poulsen,
С ?. Dobson, J. Am. Chem. Soc, 1996, 118, 6643.
466. J. R. Yates III, J. Mass Spectrom., 1998, 33, 1.
467. T. B. Farmer, R. M. Caprioli J. Mass Spectrom., 1998, 33, 697.
468. P. F. Crain, Mass Spectrom. Rev., 1990, 9, 505.
469. B. Spengler, Y. Pan, R. J. Cotter, L.-S. Kan, Rapid Comm. Mass Spectrom., 1990,4,99.
470. J. A. McCloskey, P. F. Crain, Int. J. Mass Spectrom. Ion Proc, 1992, 118/119, 593.
471. M. C. Fitzgerald, L. M. Smith, Ann. Rev. Biophys. Biomol. Struct., 1995, 24, 117.
472. K. K. Murray, J. Mass Spectrom., 1996, 31, 1203., A. L. Burlingame, R.K.Boyd,
S. J. Gaskell Anal. Chem., 1996, 68, 599.
473. J. L. Beck, M. L. Colgrave, S. F. Ralph, ?. ?. Sheil, Mass Spectrom. Rev., 2001,20,61.
488 Список литературы
474. С. G. Huber, ?. Oberaher, Mass Spectrom. Rev., 2001, 20, 311.
475. S. A. McLuckey, G. J. Van Berkel, G. L. Glish, J. Am. Soc. Mass Spectrom., 1992,
3, 60.
476. R. W. Nelson, M. J. Rainbow, D. E. Lohr, P. Williams, Science, 1989, 246, 1585.
477. R. W. Nelson, R. M. Thomas, P. Williams, Rapid Comm. Mass Spectrom., 1990, 4, 348.
478. D. M. Schieltz, С Chou, C. Luo, R. M. Thomas, P. Williams, Rapid Comm. Mass Spectrom.,
1992, 6, 631.
479. K. Tang, N. I. Taranenko, S. L. Allman, L. Y. Chang, С. Н. Chen, Rapid. Comm. Mass
Spectrom., 1994, 8, 727.
480. K. Tang, S. L. Allman, С ?. Chen, L. Y. Chang, M. Schell, Rapid Comm. Mass Spectrom.,
1994, 8, 183.
481. E. Nordhoff, R. Cramer, M. Karas, F. Hillenkamp, F. Kirpekar, K. Kristiansen, P. Roepstorff,
Nucleic Acids Res., 1993, 21, 3347.
482. E. Nordhoff, A. Ingendoh, R,Cramer, A. Overberg, B. Stahl, M. Karas, F. Hillenkamp,
Rapid Comm. Mass Spectrom., 1992, 6, 771.
483. F. Kirpekar, E. Nordhoff, K. Kristiansen, P. Roepstorff, A. Lezius, S. Hahner, M. Karas,
F. Hillenkamp, Nucleic Acida Res., 1994, 22, 3866.
484. F. Greco, A. Liguori, G. Sindona, N. Uccella, J. Am. Chem. Soc, 1990, 112, 9092.
485. L. Zhu, G. R. Parr, M. С Fitzgerald, С ?. Nelson, L. M. Smith, J. Am. Chem. Soc, 1995,
117, 6048.
486. E. Nordorff, M. Karas, R. Cramer, S. Hahner, F. Hillenkamp, F. Kirpekar, A. Lezius, J. Muth,
С Meier, J. W. Engels, J. Mass Spectrom., 1995, 30, 99.
487. S. C. Pomerantz, J. A. Kowalak, J. A. McCloskey, J. Am. Soc Mass Spectrom., 1993, 4,
204.
488. С G. Huber, H. Oberaher, Mass Spectrom. Rev., 2001, 20, 311.
489. S. A. McLuckey, S. Habibi-Goudarzi, J. Am. Soc. Mass Spectrom., 1994, 5, 740.
490. J.Ni, S. С Pomerantz, J. Rozenski, Y.Zhang, J. A. McCloskey, Anal. Chem., 1996, 68,
1989.
491. R. S. Brown, J. J. Lennon, Anal. Chem., 1995, 67, 3990.
492. С M. Bentzley, M.V.Johnston, B. S. Larsen, S. Gutteridge, Anal. Chem., 1996, 68,
2141.
493. R. P. Glover, G. M. A. Sweetman, P. B. Farmer, G. С. К. Roberts, Rapid Comm. Mass
Spectrom., 1995, 9, 897.
494. T. A. Shaler, Y. Tan, J. N. Wickham, K. J. Wu, С. Н. Becker, Rapid Comm. Mass Spectrom.,
1995, 9, 942.
495. L. Chang, K. Tang, M. Schell, С Ringelberg, K. J. Matteson, S. L. Allman, С. Н. Chen,
Rapid Comm. Mass Spectrom., 1995, 9, 772.
496. E. С Liu, S. A. Hofstadler, J. A. Bresson, H. R. Udseth, T. Tsukuda, R. D. Smith,
A. P. Snyder, Anal. Chem., 1998, 70, 1797.
497. F. Xiang, G. A. Anderson, T. D. Veenstra, M. S. Lipton, R. D. Smith, Anal. Chem., 2000,
72, 2475.
498. J. P. Anhalt, С Fenselau, Anal. Chem., 1975, 47, 219.
499. C. Fenselau, P. A. Demirev, Mass Spectrom. Rev., 2001, 20, 157.
500. J. O. Lay, Mass Spectrom. Rev., 2001, 20, 172.
501. T. Krishnamurthy, U,Rajamani, P. L. Ross, R. Jabhour, H. Nair, J. Eng, J. Yates, ?. ?. Davis,
D. C. Stahl, T. D. Lee, J. Toxicol-Toxin Rev., 2000, 19, 95.
502. P. A. Demirev, Y. P. Ho, V. Ryzhov, С Fenselau, Anal. Chem., 1999, 71, 2732.
503. V Ryzhov, ? Hathout, С Fenselau, Appl. Environ. Microbiol., 2000, 66, 3824.
504. Z. P. Wang, L. Russon, L. Li, D. С Roser, S. R. Long, Rapid Comm. Mass Spectrom., 1998,
12, 456.
505. A. J. Madonna, F. Basile, I. Ferrer, M. A. Meetani, J. C. Rees, K. J. Voorhees, Rapid Comm.
Mass Spectrom., 2000, 14, 2220.
Список литературы 489
506. ?. A. Winkler, J. Uher, S. Сера, Anal. Chem., 1999, 71, 3416.
507. B. Eliasi, С Fenselau, Anal. Chem., 2001, 73, 5228.
508. P. Demirev, V. Ryzhov, С Fenselau, Anal. Chem., 1999, 71, 2732.
509. P. A. Demirev, J. S. Lin, E. J. Pineda, С Fenselau, Anal. Chem., 2001, 73, 4566.
510. G. Siuzdak J. Mass Spectrom., 1998, 33, 203.
511. S. D. Fuerstenau, W. H. Benner, Rapid Comm. Mass Spectrom., 1995, 9, 1528.
512. J. J. Thomas, B. Falk, С Fenselau, J. Jackman, J. Ezzel, Anal. Chem., 1998, 70, 3863.
513. M. A. Tito, K. Tars, K. Valegard, J. Hajdu, С V. Robinson, J. Am. Chem. Soc, 2000, 122,
3550.
514. C. S. Hayek, F. J. Pineda, O. W. Doss, J. S. Lin, Johns Hopkins APL Technical Digest, 1999,
20, 363.
515. R. A. Gieray, P. T. A. Reilly, M. Yang, W. B. Whitten, J. M. Ramsey, J. Microbiol. Methods,
1997, 29, 191.
516. P. F. Scholl, M. A. Leonardo, A. M. Rule, M. A. Carlson, M. D. Antoine, T. J. Buckley, Johns
Hopkins APL Technical Digest, 1999, 20, 343.
517. A. H. Brockman, R. Orlando, Anal. Chem., 1995, 67, 4581.
518. R. W. Nelson, J. Krone, A. Bieber, P. Williams, Anal. Chem., 1995, 67, 1123.
519. B. S. Middleditch, S. R. Missler, ?. ?. Hines, Mass Spectrometry of priority pollutants,
Plenum Press, New-York, 1981, 308 p.
520. J. E. Biller, K. Biemann, Anal. Lett., 1974, 7, 515.
521. T. A. Ternes, W. Baumann, R. Nagel Fresenius, J. Anal. Chem., 1995, 354, 237.
522. P. A. Leclercq, С A. Cramers, Mass Spectrom. Rev., 1998, 17, 37.
523. R. G. Dromley, M. J. Stefik, Т. С Rindfleisch, A. M. Duffield, Anal. Chem., 1976, 48,
1368.
524. R. J. Ghosh, R. Anderegg, Anal. Chem., 1989, 61, 2118.
525. E. J. Karjalainen, U. P. Karjalainen, Proc. 14th IMSC, Tampere, Finland, 1997, 88.
526. F. Glisson, CD version of the Proc. GC-GC-TOF-MS Workshop, Prague, 13-15 November,
2002.
527. W. L. Budde, Analytical mass spectrometry. Strategies for environmental and related
applications, American Chemical Society, Washington, D. C, 2001, 386 p.
528. H. А. Клюев, ЖАХ, 2002, 57, 566.
ОГЛАВЛЕНИЕ
Благодарности 4
Предисловие 5
Введение 10
Глава 1. Система ввода образца 12
1.1. Баллон напуска 12
1.2. Прямой ввод 13
1.3. Мембранный ввод (Membrane Inlet Mass Spectrometry, MIMS) 14
1.4. Хроматомасс-спектрометрия, ГХ-МС (GC-MS) 15
1.5. Жидкостная хроматография—масс-спектрометрия, ЖХ-МС (LC-MS) 20
1.5.1. Ленточный транспортер (Moving Belt) 21
1.5.2. Прямой ввод жидкости (Direct Liquid Introduction, DLI) 22
1.5.3. Поток частиц (Particle Beam) 23
1.5.4. Термораспыление или термоспрей (Thermospray, TSP) и плазморас-
пыление или плазмаспрей (Plasmaspray) 24
1.6. Сверхкритическая флюидная хроматография—масс-спектрометрия, СФХ-
МС (Supercritical Fluid Chromatography/Mass Spectrometry, SFC/MS) 25
1.7. Капиллярный электрофорез—масс-спектрометрия (Capillary Electrophoresis/Mass
Spectrometry, CE/MS) 27
Глава 2. Физические основы процесса масс-спектрометрического распада 28
2.1. Электронный удар или электронная ионизация, ЭУ (Electron Impact, Electron
Ionization, EI) 28
2.2. Физические основы масс-спектрометрического распада 33
2.3. Метастабильные ионы 38
2.4. Полуколичественная теория масс-спектрометрического распада 40
Глава 3. Основные правила и подходы к интерпретации масс-спектров 42
3.1. Стабильность ионов и нейтральных частиц 42
3.1.1. Правило выброса максимального алкильного радикала 44
3.1.2. Правило Стивенсона (правило Стивенсона— Одье) 46
3.1.3. Правила распада четноэлектронных ионов 49
3.1.4. Правило степеней свободы 50
3.1.5. Прочность химических связей 51
3.1.6. Структурные и стереохимические факторы 51
3.1.7. орто-Эффекг 55
3.2. Концепция локализации заряда и неспаренного электрона 56
3.2.1. Фрагментация, удаленная от места локализации заряда 61
Глава 4. Практические основы интерпретации масс-спектров 62
4.1. Молекулярный ион 62
4.2. Определение элементного состава ионов на основании изотопных пиков .. 65
4.2.1. Азотное правило 73
Оглавление 491
4.2.2. Определение содержания изотопа 13С в природных образцах 76
4.2.3. Расчет изотопной чистоты соединений 76
4.3. Фрагментные ионы 79
4.3.1. Гомологические серии ионов 79
4.3.2. Выбросы простейших нейтральных частиц 84
4.3.3. Наиболее интенсивные пики в спектре 84
4.4. Библиотеки масс-спектров 85
4.5. Использование для интерпретации дополнительной масс-спектральной
информации 86
4.6. Схема фрагментации 88
Глава 5. Альтернативные методы ионизации образца 92
5.1. Ионизация фотонами (Photoionization) 92
5.2. Химическая ионизация, ХИ (Chemical Ionization, CI) 93
5.3. Химическая ионизация отрицательных ионов (Negative Ion Chemical
Ionization, NICI) 96
5.4. Пульсирующая химическая ионизация (Pulsed Positive, Negative Ion Chemical
Ionization Mass Spectrometry) 98
5.5. Десорбционная (прямая) химическая ионизация, ДХИ (Direct (Desorption)
Chemical Ionization, DO) , 98
5.6. Полевая ионизация (Field Ionization, FI) 100
5.7. Полевая десорбция (Field Desorption, FD) 100
5.8. Плазменная десорбционная масс-спектрометрия (Plasma Desorption Mass
Spectrometry, PDMS) 101
5.9. Лазерная десорбционная масс-спектрометрия (Laser Desorption Mass
Spectrometry, LDMS) 102
5.10. Бомбардировка быстрыми атомами, ББА (Fast Atom Bombardment, FAB); вто-
ричноионная масс-спектрометрия, ВИМС (Secondary Ion Mass Spectrometry,
SIMS) 102
5.10.1. Проточный (динамический) вариант бомбардировки быстрыми
атомами (Continuous Flow or Dynamic Fast Atom Bombardment) 105
5.11. Химическая ионизация при атмосферном давлении (Atmospheric Pressure
Chemical Ionization, APCI) 106
5.12. Электрораспыление, электроспрей (Electrospray Ionization, ESI) 107
5.13. Ультразвуковое распыление (Sonic Spray) 112
5.14. Матричная лазерная десорбционная ионизация, МЛДИ (Matrix-Assisted
Laser Desorption/Ionization, MALDI) 112
5.15. Пиролитическая масс-спектрометрия (Pyrolysis/Mass Spectrometry, Py/MS) 121
Глава 6. Разделение и регистрация ионов 124
6.1. Магнитный секторный масс-спектрометр 124
6.2. Электростатический анализатор. Двухфокусный секторный масс-
спектрометр 126
6.3. Масс-спектрометрия высокого разрешения, МСВР 127
6.4. Масс-спектрометрия с преобразованиями Фурье (Fourier Transform Mass
Spectrometry, FT-MS) .' 129
6.5. Квадрупольный анализатор 135
6.6. Ионная ловушка 136
6.7. Времяпролетный анализатор 138
6.8. Детектирование ионов 141
Глава 7. Тандемная масс-спектрометрия, МС/МС 144
7.1. Активация ионов 145
492 Оглавление
7.1.1. Активация соударением или диссоциация, индуцированная
столкновениями (Collisional Activation, CA; Collision-Induced Dissociation,
CID) 145
7.1.2. Фотодиссоциация (Photodissociation) 147
7.1.3. Поверхностно-индуцированная диссоциация (Surface-Induced
Dissociation, SID) 148
7.2. Анализаторы ионов в тандемной масс-спектрометрии 148
7.2.1. Система трех квадруполей 148
7.2.2. Магнитные секторные приборы 150
7.2.3. Ионная ловушка 162
7.2.4. Масс-спектрометрия с преобразованиями Фурье 163
7.2.5. Приборы продленной геометрии 164
7.2.6. Приборы гибридной геометрии 166
7.3. Инверсия заряда (Charge Inversion) 168
7.4. Нейтрализация—реионизация (Neutralization—Reionization Mass Spectrometry,
NRMS) 170
7.5. Спектрометрия ионной подвижности (Ion Mobility Spectrometry) 172
Глава 8. Основные направления фрагментации важнейших классов органических
соединений 176
8.1. Алканы 176
8.2. Алкены и диены 181
8.3. Алкины 185
8.4. Алициклические углеводороды 188
8.4.1. Циклоалкены 190
8.5. Ароматические углеводороды 192
8.6. Спирты, фенолы, тиолы 199
8.6.1. Спирты 199
8.6.2. Фенолы 210
8.6.3. Тиолы 211
8.7. Простые эфиры и сульфиды 217
8.8. Амины и фосфины 225
8.9. Алкилгалогениды, арилгалогениды 234
8.10. Карбонильные соединения 243
8.11. Карбоновые кислоты и их производные 258
8.12. Нитрилы, изонитрилы, нитросоединения, гидразины, оксимы и диазосоеди-
нения 281
8.13. Сульфоксиды, сульфоны, сульфокислоты 297
8.14. Элементоорганические соединения 303
8.15. Аминокислоты, пептиды, белки 310
8.15.1. Установление последовательности аминокислотных звеньев в
пептидах 314
8.15.2. Пептидная карта масс 321
8.15.3. Протеомика 323
8.16. Нуклеиновые кислоты 325
8.17. Масс-спектрометрический анализ микроорганизмов 328
Глава 9. Количественный масс-спектрометрический анализ 332
9.1. Масс-хроматография 335
9.2. Масс-фрагментография, мониторинг заданных ионов (Selected Ion
Monitoring, SIM) 341
9.3. Установление количества соединения в образце по площади хроматографи-
ческого пика 344
Оглавление 493
9.4. Метод внешнего стандарта 345
9.5. Метод внутреннего стандарта 346
9.5.1. Метод изотопного разбавления 348
9.6. Метод добавок 348
Глава 10. Обобщенные задачи 350
Глава 11. Решения задач 380
Приложение 450
Список литературы 475
Научное издание
Методы в химии
Лебедев Альберт Тарасович
МАСС-СПЕКТРОМЕТРИЯ В ОРГАНИЧЕСКОЙ ХИМИИ
Редактор Почкаева Т. И.
Художник Инфантэ Ф.
Технический редактор Бленцева Т. Н.
Оригинал-макет подготовлен в пакете ДОбХ 2?
Лапко О. Г.
Лицензия на издательскую деятельность №06331
от 26 ноября 2001 г.
Подписано в печать 25.02.03 г. Формат 70 ? 100/16
Гарнитура Тайме. Бумага офсетная. Печать офсетная.
Усл. печ. л. 39,99. Тираж 5000 экз. Заказ 1113
Издательство «БИНОМ. Лаборатория знаний»
Телефон @95)955-0398 E-mail:lbz@aha.ru
Отпечатано с готовых диапозитивов
в полиграфической фирме «Полиграфист»
160001, г. Вологда, ул. Челюскинцев, 3
нию импульсных вариантов вольтамперометрии, химически
модифицированных электродов, электрохимических сенсоров, методам
детектирования определяемых компонентов в потоке.
Для студентов, аспирантов и преподавателей химических и
химико-технологических специальностей вузов, а также для специалистов,
работающих в области аналитической химии и аналитического приборостроения.
119071, Москва, а/я 32,
БИНОМ. Лаборатория знаний
Тел./факс @95) 955-0536,
955-0398
955-0429
E-mail: lbz@aha.ru
&
ИЗДАТЕЛЬСТВО
«БИНОМ.
Лаборатория знаний»
Ш&;
fcjlj
SI11MI
Ш^ШИИШ!
СКОРО ПОЯВИТСЯ:
Карпов Ю. ?., Савостин А. П., Глинская И. В. Методы пробоотбора
и пробоподготовки — М.: БИНОМ Лаборатория знаний, 15 л. (Методы
в химии).
В курсе лекций в соответствии с учебной программой изложены
различные способы и схемы пробоотбора природных и технических
материалов, используемых в металлургическом производстве. Подробно
рассмотрены вопросы разложения материалов, а также методы разделения
компонентов и концентрирования микропримесей для их последующего
количественного определения. Проведены сопоставления методов и выбор
оптимального из них для данных условий с целью повышения точности и
чувствительности определения.
Для студентов и преподавателей химических факультетов вузов.
&
ИЗДАТЕЛЬСТВО
«БИНОМ.
Лаборатория знаний»
119071, Москва, а/я 32,
БИНОМ. Лаборатория знаний
Тел./факс @95) 955-0536,
955-0398
955-0429
E-mail: lbz@aha.ru