/












































































































































































































































































































































































































































































































































































































































Author: Лебедев А.Т.
Tags: другие физико-химические методы анализа (кроме оптических) аналитическая химия физика химия окружающая среда
ISBN: 978-5-94836-363-9
Year: 2013




Text
А.Т. Лебедев
Масс-спектрометрия
для анализа объектов
окружающей среды
Перевод с английского
под общей редакцией А.Т. Лебедева
ТЕХНОСФЕРА
Москва
2013
УДК 543.51
ББК 24.4
ЛЗЗ
ЛЗЗ Лебедев А.Т.
Масс-спектрометрия для анализа объектов окружающей среды
Москва: Техносфера, 2013. - 632с., ISBN 978-5-94836-363-9
Современная масс-спектрометрия является наиболее чувствительным,
информативным и надежным методом идентификации и количественного
определения экотоксикантов любого типа в образцах объектов окружающей
среды любой сложности. Хотя диапазон возможностей современной
масс-спектрометрии необычайно широк, многие из них остаются неизвестными
непрофессионалам. Основная цель книги - продемонстрировать, что самые
разные научные задачи, стоящие перед учеными разных специальностей, могут
быть решены масс-спектрометрически.
Книга предназначена, в первую очередь, для людей, работающих в смежных
дисциплинах (экология, геология, биология, гидрология, медицина и т.д.), а
также будет полезна студентам и аспирантам химических, физико-химических,
биологических и медицинских специальностей.
УДК 543.51
ББК 24.4
Переводчики:
К.А. Артеменко, Е.С. Бродский, С.А. Ильченко, А.И. Константинов, А.Т. Лебедев,
А.Ю. Лейкин, В.В. Лободин, П.В. Метальников, Е.В. Московец, А.С. Самохин,
Е.А. Чернецова
Обложка: автор идеи Маркос Эберлин (Университет Кампинос, Бразилия)
©2013, Лебедев А.Т.
©2013, ЗАО «РИЦ «Техносфера», оригинал-макет, оформление
ISBN 978-5-94836-363-9
ISBN 978-1-906799-12-0 (англ.)
Содержание
Список авторов 18
Список переводчиков 21
Предисловие к русскому изданию 22
Введение 23
Литература 28
Глава 1. Основные принципы масс-спектрометрии 29
1.1. Базовые аспекты 29
1.2. Ввод образца 32
1.3. Ионизация 33
1.3.1. Электронная ионизация 34
1.3.2. Химическая ионизация 35
1.3.3. Полевая ионизация 36
1.3.4. Ионизация электрораспылением 38
1.3.5. Химическая ионизация и фотоионизация при атмосферном давлении 39
1.3.6. Матрично-активированная лазерная десорбция/ионизация 40
1.4. Масс-анализаторы 40
1.4.1. Секторные приборы 40
1.4.2. Квадрупольные приборы 44
1.4.3. Ионные ловушки 46
1.4.4. Времяпролетные масс-спектрометры 46
1.4.5. Масс-спектрометрия ионного циклотронного резонанса
с преобразованием Фурье 47
1.4.6. Орбитальные ловушки 48
1.5. Детектирование ионов 49
Литература 49
Глава 2. Газовая хроматография/масс-спектрометрия — «рабочая лошадка»
для анализа объектов окружающей среды 52
2.1. Общие вопросы 52
2.2. Типы хроматограмм с регистрацией ионного тока .53
2.3. Скорость сбора данных 63
2.4. Какие соединения можно анализировать методом ГХ/МС 65
2.5. Количественный анализ 69
2.6. Выводы 73
Литература 73
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды 74
3.1. Введение 74
3.2. Методы и оборудование для мониторинга окружающей среды 75
3.2.1. Почему для мониторинга окружающей среды используется
жидкостная хроматография? 75
6 Содержание
3.2.2. Почему ЖХ/МС (жидкостная хроматография с масс-спектрометрическим
детектором) используется для мониторинга окружающей среды ? 76
3.3. Соединение ЖХс масс-спектрометром: проблемы и их решение 77
3.4. Ионизация при атмосферном давлении 79
3.5. Типы масс-спектрометров и их возможности для анализа образцов
из объектов окружающей среды 82
3.5.1. Приборы с низким разрешением 82
3.5.2. Приборы с высоким разрешением 85
3.6. Анализ образцов 86
3.7. Наиболее распространенные приложения 87
3.8. Подтверждающий анализ 87
3.9. Целевые подходы с использованием тандемной квадрупольной
масс-спектрометрии 89
3.10. Скрининговые и исследовательские методы с использованием
масс-спектрометров типа QTOF 95
3.11. Почему приборы типа QTOF используются для экологического контроля? 95
3.12. Исследовательская работа по изучению продуктов трансформации 99
3.13. Ближайшие и долговременные перспективы 100
Литература 101
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды 103
4.1. Введение 103
4.2. Тандемные масс-спектрометры 104
4.2.1. Масс-спектрометрия высокого разрешения 104
4.2.2. Тройные квадруполи 105
4.2.3. Ионные ловушки 106
4.2.4. Времяпролетная масс-спектрометрия (ВПМС) 108
4.2.5. Линейные ионные ловушки 108
4.2.6. Орбитрэп ПО
4.3. Приложения МС/МС к экологическому мониторингу 110
4.3.1. Новые загрязняющие вещества 113
4.3.2. Лекарственные препараты 115
4.3.3. Пестициды и гербициды 115
4.3.4. Аддукты ДНК и маркеры оксидативного стресса 118
4.3.5. Поверхностно-активные вещества и красители 120
4.3.6. Озон 120
4.4. Заключение 121
4.5. Замечание 122
Литература 122
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды 128
5.1. Введение 128
5.2. Базы данных масс-спектров и программы библиотечного поиска 132
5.3. Развитие баз данных масс-спектров 133
5.4. Базы данных масс-спектров 135
8 Содержание
5.5. Другие базы данных масс-спектров 137
5.6. Выбор диапазона сканирования в случае проведения поиска
по базам данных масс-спектров 139
5.7. Программное обеспечение, используемое для проведения поиска
по базам данных масс-спектров 140
5.8. Пример проведения правильной идентификации с помощью
программного обеспечения Agilent ChemStation 142
5.9. Пример проведения правильной идентификации с помощью
программного обеспечения NIST MS Search 148
5.10. Какой алгоритм библиотечного поиска является наилучшим? 152
5.11. Интерпретатор масс-спектров NIST 152
5.12. Использование базы данных масс-спектров ИЭ NIST при проведении
анализа методом высокоэффективной жидкостной хроматографии/масс-
спектрометрии 153
5.13. База данных индексов удерживания NIST 156
5.14. Программа для автоматического поиска компонентов и выделения
чистых масс-спектров AMDIS 158
5.15. Определение точных значений m/z 162
5.16. Дополнительное программное обеспечение, представляющее интерес
для масс-спектрометристов, работающих в области анализа объектов
окружающей среды 164
5.17. Заключение 168
Литература 168
Глава 6. Передовые методы на основе ГХ/МС 170
6.1. Режим быстрой ГХ/МС 170
6.1.1. Оптимизация газохроматографической составляющей
в условиях быстрой ГХ/МС 171
6.1.2. Вклад масс-спектрометрической составляющей при использовании
техники быстрой ГХ/МС 176
6.2. ГХ/ МС с интерфейсом ультразвуковых молекулярных пучков 179
6.3. Двумерная газовая хроматография — масс-спектрометрия ГХ х ГХ/МС 188
6.3.1. Принцип метода 188
6.3.2. Преимущества метода ГХ х ГХ/МС 191
6.4. Заключение 196
Литература 196
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки 198
7.1. Введение 198
7.2. Наиболее часто используемые методы
масс-спектрометрии в нормальных условиях 200
7.2.1. Десорбционная электрораспылительная ионизация (ДЭРИ, DESI)
и ионизация спреем с бумаги (paper spray ionization, PSI) 200
7.2.2. Прямой анализ в режиме реального времени (ПАРВ, DART) 202
7.2.3. Атмосферный зонд для анализа твердых образцов (ASAP) 203
7.2.4. Экстракционная электрораспылительная ионизация (ЭЭРИ, EESI) 204
Содержание
7.2.5. Низкотемпературная плазма (LTP) 205
7.2.6. Ионизация акустическим распылением в нормальных условиях —
easy ambient sonic-spray ionization (EASI) и ионизация акустическим
распылением в нормальных условиях с использованием эффекта
Вентури (Venturi easy ambient sonic-spray ionization, V-EASI) 206
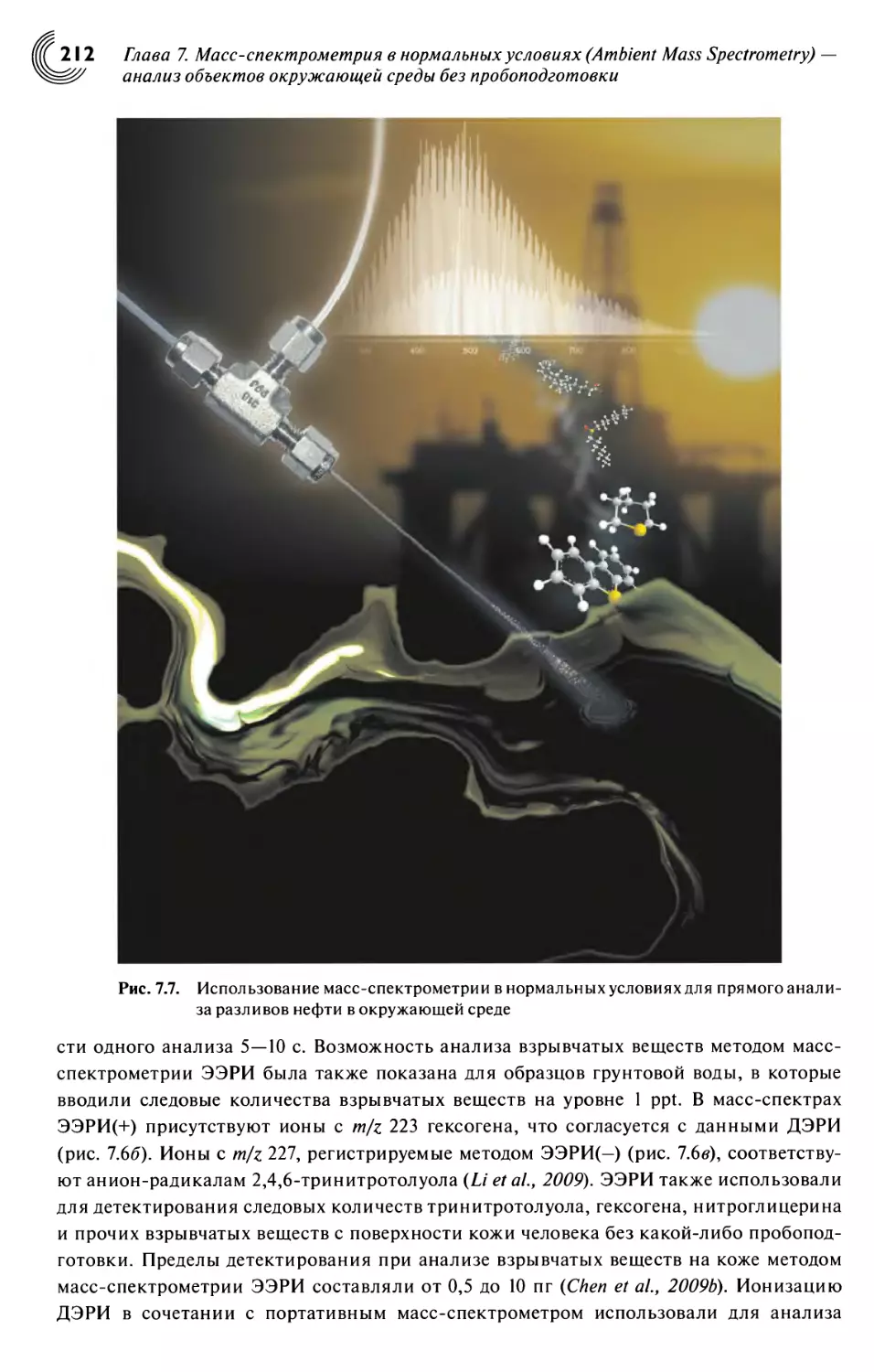
7.3. Применения в анализе объектов окружающей среды 208
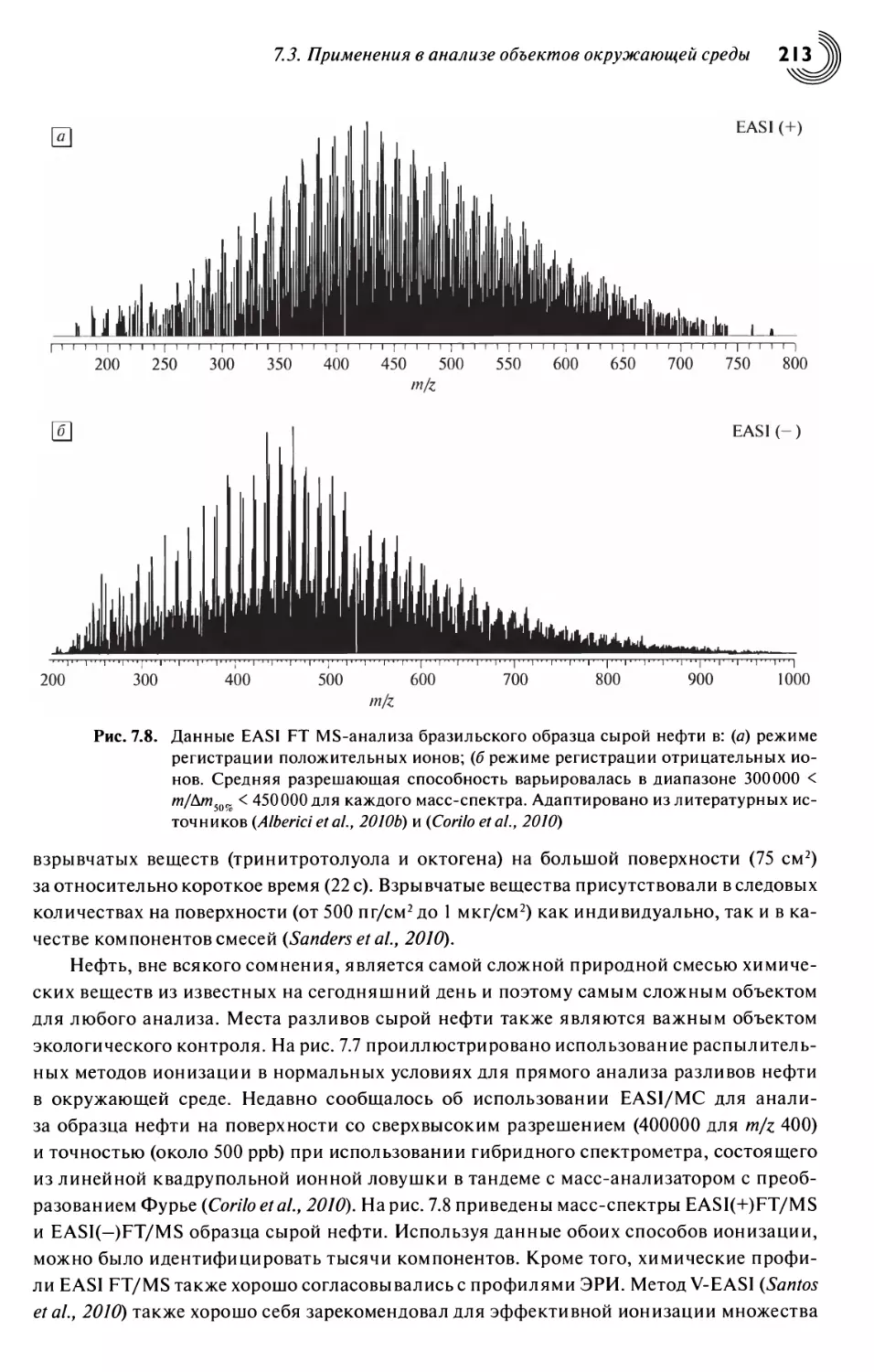
7.4. Заключение 215
Литература 216
Глава 8. Масс-спектрометрия десорбционной электрораспылительной ионизации 223
8.1. Введение 223
8.2. Экспериментальные установки и условия 224
8.3. Реакционная десорбция 227
8.4. Масс-спектрометрическое изображение поверхности с помощью ДЭРИ 229
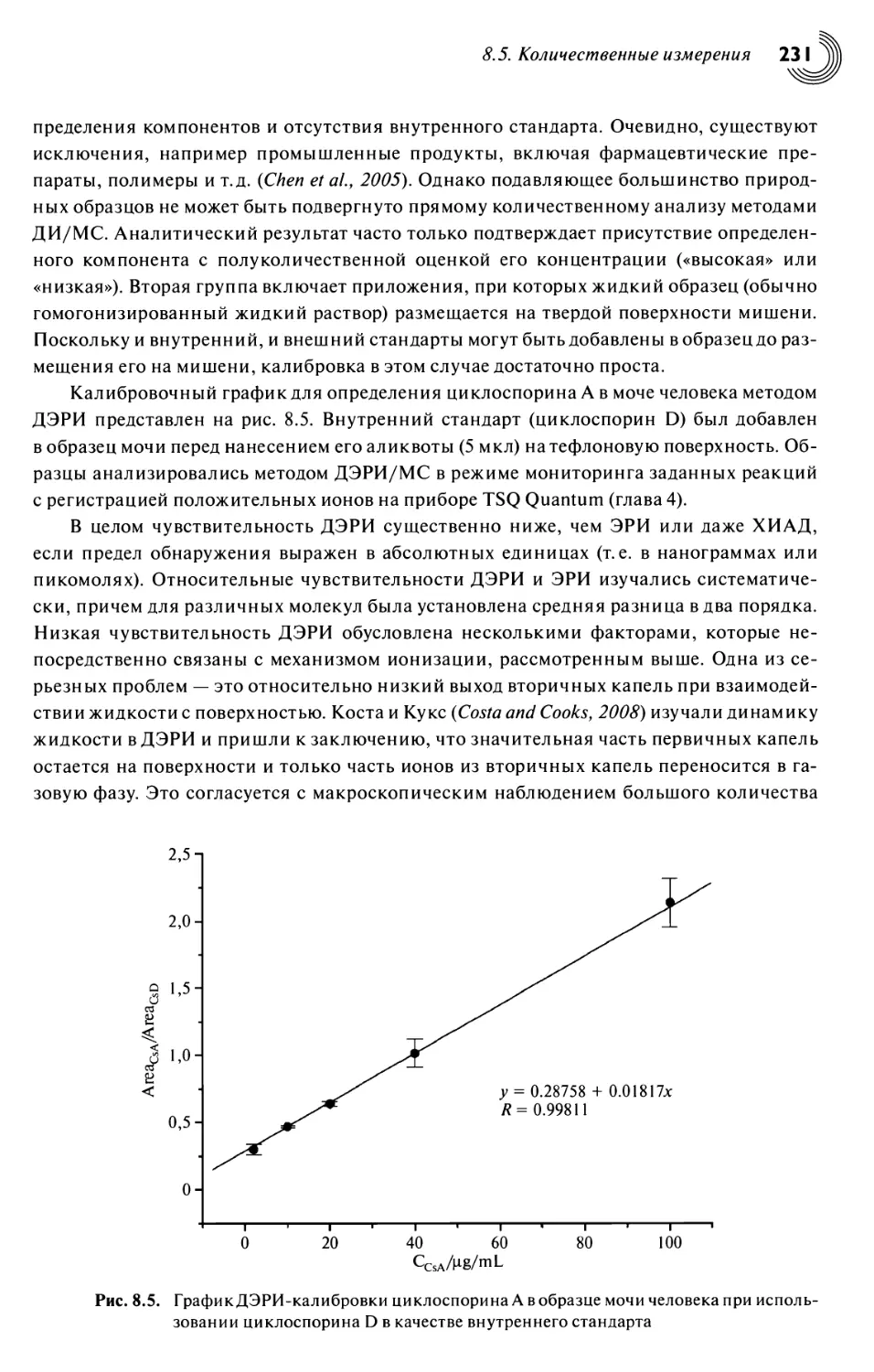
8.5. Количественные измерения 230
8.6. Анализ воды 232
8.7. Анализ аэрозолей 234
8.8. Прямой анализ 238
Литература 239
Глава 9. Миниатюрные масс-спектрометры для анализа объектов окружающей среды 242
9.1. Введение 242
9.2. Основы конструкции приборов 244
9.2.1. Методы ввода проб 244
9.2.2. Методы ионизации 249
9.2.3. Масс-анализаторы 254
9.2.4. Вакуумные системы 260
9.2.5. Детекторы 261
9.3. Экологические приложения для миниатюрных масс-спектрометров 262
9.3.1. Подводная масс-спектрометрия для регистрации летучих органических
соединений (ЛОС) и растворенных в воде газов 263
9.3.2. Мониторинг вулканических выбросов для контроля вулканической
активности 267
9.3.3. Детектирование перфторированных соединений 270
9.3.4. Анализ углеводородов 271
9.3.5. Детектирование пестицидов 273
9.3.6. Анализ полициклических ароматических углеводородов 275
9.3.7. Детектирование примесей в продуктах питания 277
9.4. Заключение 278
Литература 279
Глава 10. Масс-спектрометрия с индуктивно связанной плазмой
в экологическом анализе 288
10.1. Введение 288
10.2. Основы метода 289
10.3. Преимущества и недостатки 290
10.3.1. Спектральные интерференции 291
Содержание
10.3.2. Неспектральные интерференции (матричные эффекты) 292
10.4. Практическое применение 293
10.4.1. Анализ воздуха 293
10.4.2. Анализ воды 293
10.4.3. Анализ твердых образцов 295
10.4.4. Установление формы нахождения элементов 296
10.4.5. Определение изотопного состава 297
10.4.6. Лазерный пробоотбор в ИСП-МС 299
10.4.7. Стандартные образцы 299
Литература 300
Глава 11. Роль масс-спектрометрии в исследовании летучих органических соединений 306
11.1. Летучие органические загрязняющие вещества 306
11.1.1. Происхождение летучих органических веществ и их влияние
на окружающую среду 306
11.1.2. Воздействие ЛОС на здоровье человека 307
11.1.3. Экономический эффект биогенного загрязнения 308
11.2. Масс-спектрометрия летучих загрязнящих веществ 309
11.2.1. Протоколы пробоподготовки 310
11.2.2. Реакция переноса протона 312
11.2.3. Прямой анализ в режиме реального времени 314
11.3. Мониторинг летучих загрязняющих веществ в обычных условиях и при
пожарах 315
11.3.1. Определение антропогенных загрязняющих веществ 315
11.3.2. Определение биогенных загрязняющих веществ в окружающей среде 317
11.3.3. Влияние температуры на определение биогенных соединений 320
11.3.4. Анализ загрязняющих веществ, выделяемых при пиролизе различных
частей растений 323
11.4. Заключение и будущее направление работы 327
Благодарность 328
Литература 328
Глава 12. Идентификация и количественное определение токсикологически значимых
побочных продуктов дезинфекции воды методами масс-спектрометрии 333
12.1. Введение 333
12.2. Аналитические методы идентификации и количественного
определения ППД 339
12.2.1. Методы экстракции/концентрирования 339
12.2.2. Масс-спектрометрические методы детектирования. 343
12.2.3. Методы дериватизации 349
12.3. Что нас ждет в будущем? 352
Благодарность 353
Литература 353
Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде 360
13.1. Введение 360
13.2. Масс-спектрометрия и НПЗВ 363
Содержание
13.2.1. Газовая хроматография/масс-спектрометрия 364
13.2.2. Жидкостная хроматография/масс-спектрометрия 364
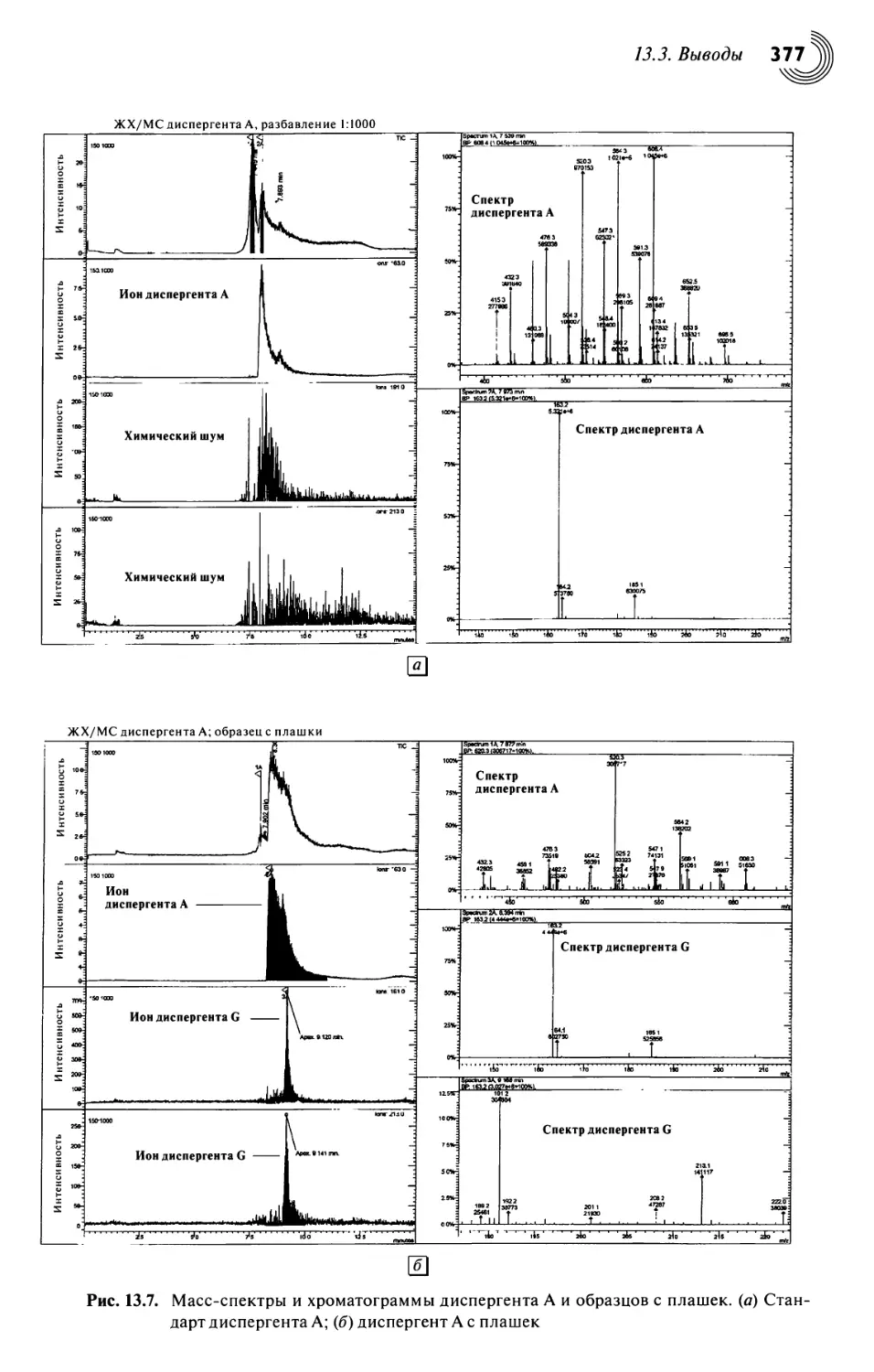
13.3. Выводы 376
Литература 378
Глава 14. Анализ пестицидов в образцах объектов окружающей среды современными
методами хроматомасс-спектрометрии 386
14.1. Введение 386
14.2. Анализ остаточных количеств пестицидов 388
14.3. ГХ/МС 391
14.4. ЖХ/МС 395
14.5. Тенденции 400
Благодарность 405
Литература 405
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны 409
15.1. Введение 409
15.1.1. Хлорированные диоксины в окружающей среде 409
15.1.2. Стадии ультраследового анализа объектов окружающей среды 411
15.1.2. Почему нужна масс-спектрометрия? 412
15.2. Подготовка проб для определения ХДД/ХДФ на следовом уровне 413
15.2.1. Методы экстракции 413
15.2.2. Очистка экстрактов 414
15.2.3. Замечания по контролю качества 414
15.3. Количественный анализ и метод изотопного разбавления 417
15.3.1. Внутренняя и внешняя градуировка 417
15.3.2. Особенности масс-спектрометрии с изотопным разбавлением 418
15.3.3. Стандартные образцы и другие средства оценки эффективности 419
15.3.4. Воспроизводимость, точность и неопределенность 420
15.4. Газохроматографические методы 420
15.4.1. Основные положения газовой хроматографии 420
15.4.2. Колонки, используемые для определения ХДД/ХДФ 421
15.5. Масс-спектрометрические методы 422
15.5.1. Масс-спектрометрия и интегрированные ГХ/МС-системы 422
15.5.2. Магнитные секторные приборы 424
15.5.3. Тандемный масс-спектрометр 426
15.5.4. Масс-спектрометр сверхвысокого разрешения 427
15.5.5. Методы ионизации для определения ХДД/ХДФ 427
15.5.6. Мониторинг заданных ионов (МЗИ) и мониторинг
заданных реакций (МЗР) 428
15.6. Полный набор аналитических методов для определения ХДД/ХДФ
и других стойких галогенированных соединений 428
15.7. Будущее многокомпонентных методов 430
15.7.1. Времяпролетный масс-спектрометр высокого разрешения 430
15.7.2. Автоматизация методов 431
15.7.3. Ограничения анализа 433
Содержание 15
Литература 434
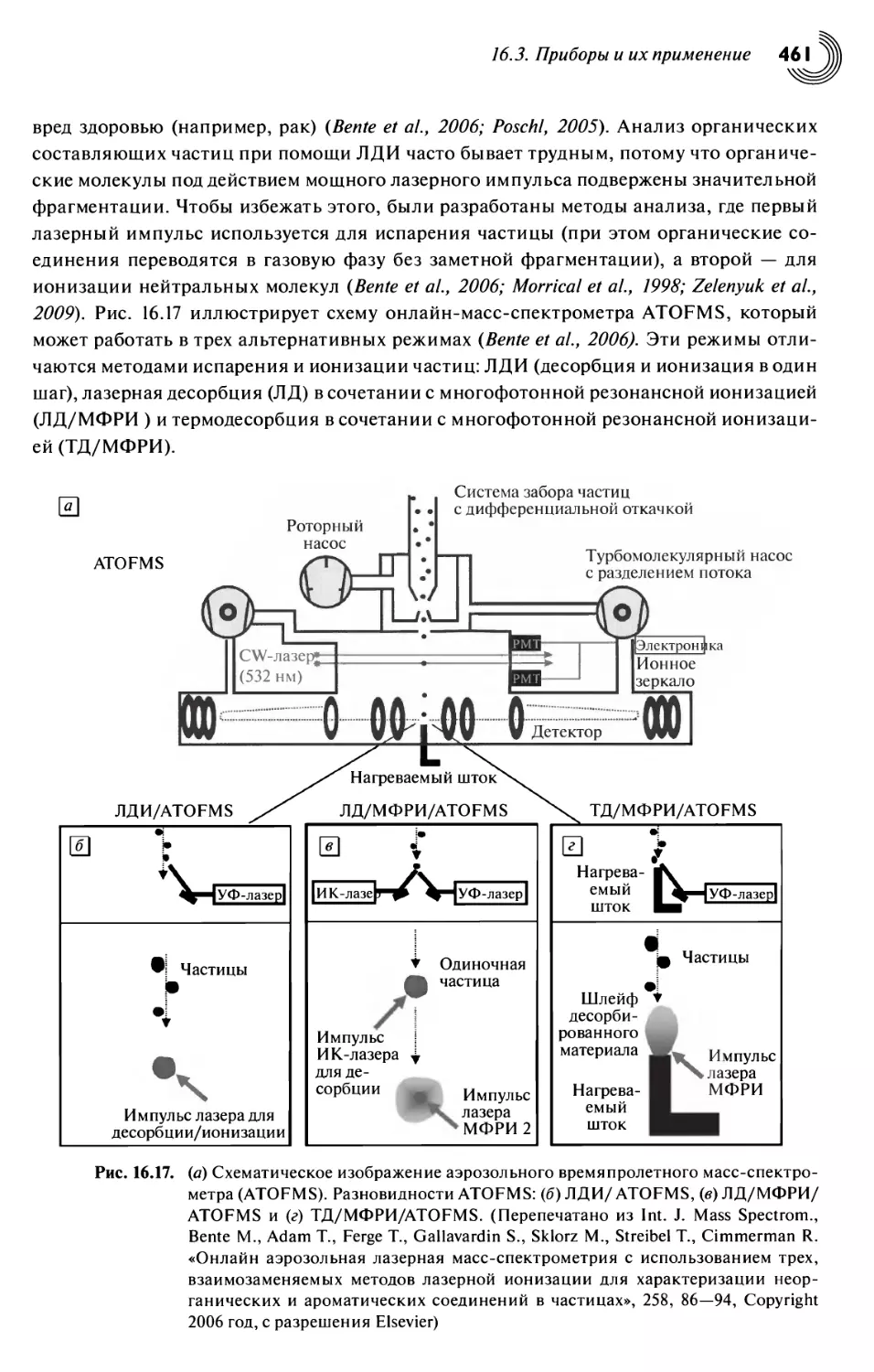
Глава 16. Масс-спектрометрия атмосферных аэрозолей 438
16.1. Введение 438
16.2. Свойства аэрозолей и их воздействие на окружающую среду 438
16.3. Приборы и их применение 443
16.3.1. Анализ атмосферных газов 444
16.3.2. Основные режимы работы приборов при анализе атмосферных частиц 447
16.3.3. Времяпролетные приборы для анализа частиц, работающие в режиме
онлайн 449
16.3.4. Применение масс-спектрометров дла анализа одиночных частиц
в исследованиях аэрозолей 452
16.3.5. Многоцелевые времяпролетные приборы для анализа частиц 460
16.3.6. Использование ионных ловушек для анализа одиночных частиц 463
16.3.7. Масс-спектрометры для анализа ансамблей частиц 466
16.4. Итоги и перспективы 470
Литература 471
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами 478
17.1. Введение 478
17.2. Источники ДНК для анализа ДНК-аддуктов 480
17.3. Подготовка образцов 481
17.4. Приборы для ВЭЖХ-ЭРИ/МС/МС-анализа ДНК-аддуктов 482
17.4.1. Валидация масс-спектрометрического метода 490
17.5. Примеры использования ВЭЖХ/МС/МС для анализа ДНК-аддуктов 491
17.5.1. Аддукты, образующиеся при воздействии ароматических аминов 491
17.5.2. Аддукты с нитрозаминами 492
17.5.3. Экзоциклические аддукты ДНК 494
17.5.4. Этанол/ацетальдегидные аддукты 494
17.5.5. Аддукты с полициклическими ароматическими
углеводородами (ПАУ) 495
17.5.6. ДНК-аддукты с эпоксидами 497
17.6. Новые подходы к повышению чувствительности аналитических методов
при анализе ДНК-аддуктов 498
17.6.1. Капиллярная ВЭЖХ/наноэлектроспрейная МС 499
17.6.2. МС-анализ с использованием ВЭЖХ-наноколонок и ионизацией
наноэлектроспреем 500
17.6.3. Очистка образцов в режиме онлайн 501
17.6.4. ЖХ/МС на микрочипах 504
17.7. Выводы 506
Благодарность 506
Литература 506
Глава 18. ИЦРФП-анализ сложных органических смесей. Петролеомика 515
18.1. Введение 515
18.2. МС-ИЦРФП 517
Содержание
18.3. Разрешение по массе, разрешающая способность
и точное измерение массы 521
18.4. Точная масса и дефект массы 524
18.5. Степень ненасыщенности (DBE) и Z-число 527
18.6. Масса в шкале Кендрика и дефект масс Кендрика 528
18.7. Визуализация данных: график масс Кендрика, диаграммы ван Кревелена,
распределение гетероатомных классов соединений, график зависимости
DBE от числа атомов углерода 530
18.7.1. График масс Кендрика 530
18.7.2. Диаграммы ван Кревелена 532
18.7.3. График распределения гетероатомных классов 533
18.7.4. График зависимости DBE от числа атомов углерода 534
18.8. Методы ионизации для МС-ИЦРФП-анализа сложных смесей 536
18.9. Применение МС-ИЦРФП для решения экологических задач 537
Литература 541
Глава 19. Применение масс-спектрометрии сверхвысокого разрешения с масс-
анализатором ионного циклотронного резонанса с преобразованием Фурье
для анализа природного органического вещества в объектах окружающей среды 547
19.1. Введение 547
19.1.1. Значение природного органического вещества и способы его анализа 547
19.1.2. Характеристики масс-спектров ПОВ и композиционное пространство 549
19.1.3. Визуализация данных сложных масс-спектров 551
19.1.4. Примеры анализа масс ПОВ 552
19.2. Материалы и методы 553
19.2.1. Масс-спектрометры ИЦР ПФ 553
19.2.2. Расчет масс для молекул, содержащих С, Н, N, О, S 553
19.3. Результаты и обсуждение 553
19.4. Заключение 563
Литература 563
Глава 20. Метод масс-спектрометрической визуализации (имиджинг) 568
20.1. Введение 568
20.2. Микрозондирование и молекулярная визуализация 568
20.3. Визуализация пространственного распределения молекулярного состава 570
20.4. Влияние матрицы 573
20.5. Применение масс-спектрометрии вторичных ионов (МСВИ) в биоанализе 575
20.6. Качество изображения и аналитический предел обнаружения 576
20.7. МС-визуализация как метод качественного анализа 577
20.8. МС-визуализация как точный аналитический метод 578
20.9. Идентификация и характеристика 580
20.10. МС-имиджинг в исследованиях объектов окружающей среды 581
20.11. Перспективы 584
Литература 585
Глава 21. Масс-спектрометрия изотопных отношений 587
21.1. Введение 587
Содержание 17
21.2. Величина 5 588
21.3. Причины вариаций распределения стабильных изотопов в природе 588
21.4. Масс-спектрометрия изотопных отношений в газах 590
21.4.1. Ионизация 591
21.4.2. Разделение по массам 592
21.4.3. Регистрация нескольких ионов 592
21.4.4. 17О-и HD-коррекция 593
21.5. Оборудование для подготовки проб и интерфейсы 593
21.5.1. Изотопный масс-спектрометр непрерывного потока 593
21.5.2. Элементный анализатор (сжигание и высокотемпературная
конверсия) — изотопный масс-спектрометр 595
21.5.3. ГХ/МСИО 595
21.5.4. ЖХ/МСИО 596
21.5.5. МСИО с многократными инжекциями через петлю 597
21.6. Некоторые приложения 597
21.6.1. Науки о Земле 597
21.6.2. Биология и экология 599
21.6.3. Археология и миграция людей 599
21.6.4. Контроль лекарств и наркотиков 601
21.6.5. Контроль допинга 601
21.6.6. Аутентичность продуктов питания и напитков 602
Литература 603
Дополнение 1. Основы и достижения масс-спектрометрии на основе орбитальной
ловушки ионов. Краткий обзор 605
Как устроена Орбитальная ловушка ионов 605
Семейство серийных масс-спектрометров Orbitrap 608
Современные тенденции практического использования Orbitrap 609
Пищевая безопасность и объекты окружающей среды 610
Заключение 611
Дополнение 2. Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды, пищевой
и сельскохозяйственной продукции 613
Список литературы 630
Список авторов
Rosana М. Alberici
ThoMSon Mass Spectrometry Laboratory
Institute of Chemistry
University of Campinas — UNICAMP
Campinas, Sao Paulo, Brazil
Aviv Amirav
School of Chemistry
Tel Aviv University
Tel Aviv, Israel
Konstantin A. Artemenko
Department of Physical and Analytical
Chemistry
Uppsala University
Uppsala, Sweden
Don Betowski
US Environmental Protection Agency
National Exposure Research Laboratory
Environmental Sciences Division
Las Vegas, Nevada, USA
Gerard Bondoux
Waters
Guyancourt, France
Ray Clement
Ontario Ministry of the Environment
Laboratory Services Branch
Toronto, Ontario, Canada
R. Graham Cooks
Department of Chemistry
Purdue University
West Lafayette, Indiana, USA
Marcos N. Eberlin
ThoMSon Mass Spectrometry Laboratory
Institute of Chemistry
University of Campinas — UNICAMP
Campinas, Sao Paulo, Brazil
Alexander B. Fialkov
School of Chemistry
Tel Aviv University
Tel Aviv, Israel
Z. Gabelica
Universite de Haute Alsace
Ecole Nationale Superieure de Chimie de
Mulhouse
Laboratoire Propre Integre — Groupe
Securite et Ecologie Chimiques
Mulhouse, France
Melissa Goggin
Department of Medicinal Chemistry and
the Masonic Cancer Center
University of Minnesota
Minneapolis, Minnesota, USA
Alexander Gordin
School of Chemistry
Tel Aviv University
Tel Aviv, Israel
M. Harir
Department of BioGeoChemistry and
Analytics
German Research Center for
Environmental Health
Helmholtz Zentrum Miinchen
Neuherberg, Germany
F. Hernandez
Research Institute for Pesticides and
Water
University Jaume I
Castellon, Spain
N. Hertkorn
Department of BioGeoChemistry and
Analytics
German Research Center for
Environmental Health
Список авторов
Helmholtz Zentrum Miinchen
Neuherberg, Germany
Andreas Hilkert
Thermo Fisher Scientific
Bremen, Germany
Klaus-Peter Hinz
Institute of Inorganic and Analytical
Chemistry
University of Giessen
Giessen, Germany
M. Ibanez
Research Institute for Pesticides and
Water
University Jaume I
Castellon, Spain
Tammy L. Jones-Lepp
US Environmental Protection Agency
Las Vegas, Nevada, USA
Jean-Marc Joumier
Waters
Guyancourt, France
Alexey Leykin
Intertech Corporation
Moscow Office
Moscow, Russia
Vladislav V. Lobodin
National High Magnetic Field Laboratory
Florida State University
Tallahassee, Florida, USA
Simin D. Maleknia
School of Biological, Earth and
Environmental Sciences
University of New South Wales
Sydney, New South Wales, Australia
Alan G. Marshall
National High Magnetic Field Laboratory
Florida State University
Tallahassee, Florida, USA
Simon Nelms
Thermo Fisher Scientific
Hemel Hempstead, UK
Robert J. Noll
Department of Chemistry
Purdue University
West Lafayette, Indiana, USA
Zheng Ouyang
Weldon School of Biomedical
Engineering
Purdue University
West Lafayette, Indiana, USA
T. Portoles
Research Institute for Pesticides and
Water
University Jaume I
Castellon, Spain
Eric J. Reiner
Ontario Ministry of the Environment
Laboratory Services Branch
Toronto, Ontario, Canada
Susan D. Richardson
US Environmental Protection Agency
National Exposure Research Laboratory
Athens, Georgia, USA
Ryan P. Rodgers
National High Magnetic Field Laboratory
Florida State University
Tallahassee, Florida, USA
Ph. Schmitt-Kopplin
Department of BioGeoChemistry and
Analytics
German Research Center for
Environmental Health
Helmholtz Zentrum Miinchen
20 Список авторов
Neuherberg, Germany
and
Department for Chemical—Technical
Analysis Research
Technische Universitat Miinchen
Freising-Weihenstephan, Germany
Gautam Sharma
Department of Chemistry and School of
Electrical and Computer Engineering
Purdue University
West Lafayette, Indiana, USA
Rosineide C. Simas
ThoMSon Mass Spectrometry Laboratory
Institute of Chemistry
University of Campinas — UNICAMP
Campinas, Sao Paulo, Brazil
O. David Sparkman
Department of Chemistry
Mass Spectrometry Facility
University of the Pacific
Stockton, California, USA
Bernhard Spengler
Institute of Inorganic and Analytical
Chemistry
Justus Liebig University of Giessen
Giessen, Germany
Zoltan Takats
Institute of Inorganic and Analytical
Chemistry
Justus Liebig University
Giessen, Germany
Anzhelika Talibova
Textronica AG/MS-ANALYTICA
Russia, Moscow
Michael Tokarev
Textronica AG/MS-ANALYTICA
Russia, Moscow
Natalia Tretyakova
Department of Medicinal Chemistry and
the Masonic Cancer Center
University of M innesota
Minneapolis, Minnesota, USA
D. Tziotis
Department of BioGeoChemistry and
Analytics
German Research Center for
Environmental Health
Helmholtz Zentrum Miinchen
Neuherberg, Germany
Список переводчиков
Глава 1 — авторский перевод А.Т. Лебедева
Глава 2 — авторский перевод А. Т. Лебедева
Глава 3 — перевод П. В. Метальникова
Глава 4 — перевод П. В. Метальникова
Глава 5 — перевод А. С. Самохина
Глава 6 — авторский перевод А.Т. Лебедева и К. А. Артеменко
Глава 7 — перевод Е. А.Чернецовой
Глава 8 — перевод С. А. Ильченко
Глава 9 — перевод П. В. Метальникова
Глава 10 — авторский перевод А. Ю. Лейкина
Глава 11 — перевод Е. В. Московца
Глава 12 — перевод А. Т. Лебедева
Глава 13 — перевод А. Т. Лебедева
Глава 14 — перевод А. Т. Лебедева
Глава 15 — перевод Е.С. Бродского
Глава 16 — перевод Е. В. Московца
Глава 17 — перевод Е. В. Московца
Глава 18 — авторский перевод В. В. Лободина
Глава 19 — перевод А. И. Константинова
Глава 20 — перевод Е. В. Московца
Глава 21 — перевод П. В. Метальникова
Редактирование — А. Т. Лебедев
Предисловие к русскому изданию
Современная масс-спектрометрия является наиболее чувствительным, информа-
тивным и надежным методом идентификации и количественного определения эко-
токсикантов любого типа в образцах объектов окружающей среды любой сложности.
Возможности работать с самыми разными аналитами (от химических элементов до
сложнейших биомолекул) в сложнейших матрицах без предварительного разделения,
действительно, ставят масс-спектрометрию в приоритетное положение по сравнению
с любыми другими методами анализа. Еще одним преимуществом масс-спектрометрии
является возможность получения информации о сотнях и тысячах аналитов в процес-
се единичного анализа одной пробы. При этом результаты масс-спектрометрических
исследований позволяют делать выводы, которые подготавливают административные
решения, влияющие на здоровье населения и экосистем в целом.
Бурное развитие метода в начале XXI века привело к созданию новых приборов,
новых методов ионизации образцов. Появилась возможность работать с минимальной
пробоподготовкой или вовсе без пробоподготовки. Популярным направлением стала
масс-спектрометрия в нормальных условиях, активные работы ведутся по миниатюри-
зации масс-спектрометров. Появление простых и чувствительных портативных масс-
спектрометров, работающих при нормальных условиях, станет прорывом в аналити-
ческой химии. При снижении стоимости таких масс-спектрометров они могут стать
бытовыми приборами.
Данная книга предназначена, в первую очередь, для людей, работающих в смежных
дисциплинах (экология, геология, биология, гидрология, медицина и т. д.). Она состоит
из 21 главы, написанной ведущими масс-спектрометристами из 12 стран мира, которые
постарались в простой форме рассказать о достижениях и потенциальных возможно-
стях метода для решения самых разнообразных экологических проблем. Хотя диапазон
возможностей современной масс-спектрометрии необычайно широк, многие из них
остаются неизвестными непрофессионалам. Поэтому основная цель — продемонстри-
ровать, что самые разные научные задачи, стоящие перед учеными разных специаль-
ностей, могут быть решены масс-спектрометрически.
Оригинальное издание вышло на английском языке. Русскоязычное издание, на
мой взгляд, еще более актуально, поскольку в России и в странах бывшего СССР про-
паганда возможностей масс-спектрометрии крайне необходима. Хотя Всероссийское
масс-спектрометрическое общество (www.vmso.ru) проводит регулярные конференции,
школы, издает журнал, выпустило несколько книг, дополнительная литература, осо-
бенно учебно-научная, очень нужна русскоязычному научному сообществу.
Я надеюсь, читатели (студенты, аспиранты, научные сотрудники) найдут в этом из-
дании много интересной и полезной информации, а масс-спектрометрия станет для них
неотъемлемым методом исследований.
А.Т. Лебедев
Если проблему нельзя решить с помощью масс-спектрометрии,
возможно, ее и не надо решать.
Фред МакЛафферти
Красота спасет мир.
Федор Достоевский
Введение
Делая в последнее время доклады по разным аспектам масс-спектрометрии на кон-
ференциях химиков, биологов, токсикологов, экологов, медиков в самых разных
странах, я обнаружил, что большинство исследователей не представляют уникаль-
ных возможностей этого метода даже в плане решения своих научных задач. Читая
лекции аспирантам МГУ имени М.В. Ломоносова, я также обратил внимание, что
молодые ученые относятся к масс-спектрометрии как к весьма сложному аналити-
ческому методу, предпочитая использовать для решения задач альтернативные под-
ходы: ЯМР, ИК-, УФ-спектроскопию. Так родилась идея написания этой книги, по-
скольку эффективность, надежность, простота и широта применимости современной
масс-спектрометрии поистине впечатляющи. Помимо классических наук (физика,
химия, биология) масс-спектрометрия чрезвычайно успешно используется в медици-
не, космических исследованиях, археологии, антропологии, антитеррористической
деятельности, экспертизе предметов искусства, экологии, допинг-контроле, метроло-
гии, ядерной физике и т.д. Благодаря масс-спектрометрии недавно родились новые
дисциплины: петролеомика, гуминомика, метабономика, протеомика. Фактиче-
ски трудно назвать раздел науки, в которой нельзя было бы эффективно применить
масс-спектрометрию.
Современная масс-спектрометрия — наиболее чувствительный, быстрый и ин-
формативный аналитический метод. Ей доступны любые аналиты: от химических эле-
ментов до сложнейших биологических молекул (белки, сахара, нуклеиновые кислоты).
Качественный и количественный анализ индивидуальных компонентов сверхсложных
смесей, состоящих из многих тысяч ингредиентов, может осуществляться быстро и без
какого-либо предварительного разделения. Вы можете отобрать пробу воды из реки,
лужи, бутылки и в течение пары часов проверить наличие в ней сотен индивидуаль-
ных веществ. Любое значимое с точки зрения экологии соединение может быть надежно
определено масс-спектрометрически. Если вы не нашли соответствующей готовой ме-
тодики в литературе, это означает лишь то, что это соединение до этих пор не интересо-
вало масс-спектрометристов.
Масс-спектрометрия обладает непревзойденной чувствительностью. Метод опе-
рирует фемто-, зепто- (10-15—10-21) молярными уровнями аналитов в образце. Если
принять во внимание число Авогадро (6,022-1023), становится понятно, что масс-
спектрометрия приближается к абсолютному теоретическому пределу анализа. Клас-
сические, не разгадываемые без волшебства задачи уровня нахождения ржаного зер-
нышка в мешке пшеницы или иголки в стоге сена могут быть легко решены с помощью
Введение
масс-спектрометрии. На самом деле, когда речь идет о возможности детектирования со-
единения на уровне 10-18, это значительно «круче», чем обнаружение одной-единствен-
ной иголки в миллионе стогов сена.
Разнообразие типов масс-спектрометров очень велико. Различные задачи ре-
ализуются с помощью разных детекторов, источников ионов и методов иониза-
ции. В частности, различие в размерах может быть прекрасно проиллюстрировано
рис. \а,б. Масс-спектрометр Mini 11.5 — хороший представитель переносных при-
боров (рис. Id), a KATRIN (рис. 1я) — самый большой на сегодняшний день прибор,
созданный для проведения сверхтонких измерений масс, например измерения массы
нейтрино (Kluge, 2010).
Рис. 1. а — Транспортирование масс-спектрометра через деревню Леопольдсхафен в ис-
следовательский центр Карлсруэ (с разрешения EJMS); б — Грэм Кукс выполня-
ет измерения с помощью портативного масс-спектрометра Mini 11.5. (Фото Jon
Dagleish)
Введение
Теория метода достаточно проста, также как и его основные применения. Фактиче-
ски уже выпускник школы может регистрировать спектры и работать с ними, особенно
когда речь идет о серийных образцах или поиске целевых аналитов.
Основная идея данной книги — рассказать о возможностях масс-спектрометрии
в решении экологических и родственных задач с демонстрацией того, как элегантно
могут быть разрешены весьма сложные вопросы. Книга дает ключ к возможному реше-
нию проблем, стоящих перед биологом, геологом, врачом, экологом, химиком, нефте-
химиком, криминалистом и т.д. Студенты, как, впрочем, и профессиональные масс-
спектрометристы, также найдут в ней достаточно много весьма полезных сведений,
которые расширят их горизонты в этой области, поскольку целый ряд современных ме-
тодов (петролеомика, имиджинг, изотопная и атмосферная масс-спектрометрия) описа-
ны простыми словами в форме лекции для начинающих. Широкий круг реальных при-
меров решенных задач красиво подкрепляет теорию различных методов исследования.
Зачастую результат эксперимента может быть удивительным и неочевидным для не-
посвященных. Например, измерив соотношение природных изотопов углерода, кисло-
рода и азота в образце, можно установить район производства наркотиков, виновника
разлива нефти, подлинность напитка или даже различить по остаткам костей древних
европейцев и американцев.
Помимо указанных превосходных технических характеристик масс-спектро-
метрия — это очень красивый метод. Я надеюсь, читатели смогут разделить со мной
это чувство, ознакомившись с богатым иллюстративным материалом книги, демон-
стрирующим результаты реальных исследований. Именно поэтому я использовал ци-
тату из «Идиота» Ф. М. Достоевского в качестве одного из эпиграфов к книге. Второй
эпиграф — высказывание «отца-основателя» органической масс-спектрометрии Фреда
МакЛафферти. Безусловно, будучи шутливым, оно, тем не менее, прекрасно отражает
возможности метода и диапазон его применимости.
Каждая из 21 главы повествует об отдельном направлении масс-спектрометрии
объектов окружающей среды. Главы 1—5 посвящены основам масс-спектрометрии.
Они раскрывают наиболее распространенные общие приемы. Материал этих глав по-
зволит легче схватывать идеи последующих разделов книги.
Основные принципы масс-спектрометрии коротко изложены в главе 1. Это форма
представления масс-спектров, системы ввода, анализаторы масс и методы ионизации,
играющие основную роль в экологической масс-спектрометрии.
Газовая хроматография/масс-спектрометрия (ГХ/МС) остается на сегодняшний
день наиболее эффективным методом качественного и количественного определения
наиболее распространенных загрязняющих веществ: фенолов, полициклических аро-
матических углеводородов, полихлорированных бифенилов, дибензодиоксинов и ди-
бензофуранов и т.д. Важнейшие аспекты этого комбинированного метода представлены
в главе 2.
С момента успешной стыковки масс-спектрометра с жидкостным хроматогра-
фом метод ЖХ/МС становится все более и более востребованным, поскольку огром-
ное число полярных и термолабильных соединений вошли в рабочий диапазон масс-
спектрометрии. Глава 3 повествует об основных деталях, особенностях и преимуществах
метода ЖХ/МС.
26 Введение
Курс на повышение чувствительности и надежности анализов привел ко все более
частому приложению тандемной масс-спектрометрии (МС/МС) к проблемам окружаю-
щей среды. Ранее этот метод использовался в основном в фундаментальных исследова-
ниях. Экологические приложения тандемной масс-спектрометрии рассмотрены в гла-
ве 4. Этот метод надежнее, чем обычная масс-спектрометрия. Кроме того, он позволяет
создавать различные автоматизированные программы для эффективного качественно-
го и количественного определения сотен экотоксикантов и(или) биомаркеров за один
ввод пробы. Метод МС/МС незаменим, когда речь идет об установлении структуры со-
единений в нецелевых анализах.
Поскольку за регистрацией всегда следует какая-либо процедура обработки масс-
спектра, эта часть эксперимента имеет особое значение. Сейчас в большинстве случаев
работы с экотоксикантами нет необходимости проводить установление структуры ана-
лита вручную. Представительные базы данных и постоянно совершенствующееся про-
граммное обеспечение — неотъемлемая часть современных масс-спектрометров. Имен-
но таким базам данных и компьютерным программам посвящена глава 5.
Следующий блок (главы 6—9) представляет инновации XXI века. Усовершенство-
ванные ГХ/МС-методы: ГХ/ГХ/МС и быстрая ГХ/МС (глава 6) расширили возможно-
сти метода. Первый из них делает возможным получение информации о тысячах ин-
дивидуальных компонентов сложнейших смесей за однократный ввод пробы, второй
позволяет сократить время анализа в 5—10 раз. Недавно внедренный интерфейс сверх-
звуковых молекулярных пучков продемонстрировал новые впечатляющие возможности
классического метода электронной ионизации и позволил расшить круг соединений,
которые можно эффективно анализировать ГХ/МС.
Долгие годы основным недостатком масс-спектрометрии считалась необходимость
пробоподготовки, зачастую весьма трудоемкой. Начало XXI века ознаменовалось рево-
люционными подвижками в преодолении этой трудности. Сегодня предложены более
40 масс-спектрометрических методов с ионизацией в нормальных (природных) услови-
ях с применением плазмы или распыления. Глава 7 посвящена наиболее эффективным
среди них: ДЭРИ, ПАРВ, EASY, ЭЭРИ и т. д. Во многих случаях процедура пробоподго-
товки может быть полностью устранена.
Исторически первый метод ионизации в нормальных условиях был назван десорб-
ционной электрораспылительной ионизацией (ДЭРИ, DESI). Он уникален по скорости,
кругу анализируемых молекул и области применения, включая, например, онлайн-де-
тектирование биомаркеров во время хирургических операций. Глава 8 посвящена воз-
можностям этой замечательной техники.
Другим недостатком масс-спектрометрии прошлых лет был размер приборов. Сей-
час производится много мощных и надежных настольных масс-спектрометров. Тем не
менее борьба за сокращение размеров не прекращается. Например, размер играет едва
ли не ключевую роль в создании приборов для размещения на космических кораблях.
В последние годы созданы портативные приборы (до нескольких килограммов), спо-
собные решать весьма сложные задачи. Один из авторов главы 9, посвященной мини-
атюризации, Грэм Кукс отметил в своей лекции на недавней международной масс-
спектрометрической конференции в Бремене (Cooks et al., 2010), что будет правильно,
если в недалеком будущем обычный человек сможет использовать масс-спектрометр
Введение
для замеров на «кухне», например для проверки качества пищевых продуктов, простых
экологических тестов или контроля здоровья.
С главы 10 начинается блок, посвященный наиболее значимым типам загрязнений
окружающей среды. Метод индуктивно связанной плазмы с масс-спектрометрическим
детектированием (ИСП МС) является наиболее быстрым и информативным для каче-
ственного и количественного определения химических элементов в пробах. Менее ми-
нуты достаточно для того, чтобы получить надежную информацию о наличии и уровнях
большинства химических элементов таблицы Д. И. Менделеева. Поскольку диапазон
линейности метода достигает 12 порядков, за один ввод пробы можно получить инфор-
мацию и о макроэлементах, и об элементах, присутствующих в ультраследовых количе-
ствах. Этому замечательному методу посвящена глава 10.
Достаточно большая группа химических соединений в объектах окружающей сре-
ды объединяется термином «летучие». Это связано исключительно с низкой величиной
давления их паров. В эту группу входят самые разнообразные органические вещества.
Опасность соединений этой группы для человека и экосистемы в целом лежит в широ-
ком диапазоне, а их присутствие должно контролироваться в атмосфере, воде, других
образцах и продуктах. Масс-спектрометрическому анализу таких соединений, которые
можно разделить на антропогенные и биогенные, посвящена глава 11.
Побочные продукты дезинфекции, возникающие при подготовке питьевой воды,
а также при эксплуатации плавательных бассейнов и утилизации отходов, представля-
ют собой серьезную экологическую проблему, нанося существенный вред здоровью че-
ловека. Не существует лучшего метода качественного и количественного определения
этих соединений, чем масс-спектрометрия. Об этом повествуется в главе 12.
Глава 13 посвящена опасным современным экотоксикантам: фармацевтическим
препаратам, наночастицам, металлоорганическим соединениям. Набор этих ксеноби-
отиков неуклонно возрастает год от года с появлением новых продуктов, часто не самых
экологичных. И вновь следует сказать, что для их детектирования не существует более
эффективного метода, чем масс-спектрометрия.
Пестициды всегда считались приоритетными загрязняющими веществами. Раз-
личные варианты ГХ/МС или ЖХ/МС показали свою эффективность в определении
этих экотоксикантов в объектах окружающей среды. Тем не менее в связи с широчай-
шим разнообразием структур и наличием очень жестких требований по чувствитель-
ности и надежности определение пестицидов продвинутыми хроматомасс-спектроме-
трическими методами представлено в главе 14.
Масс-спектрометрия является единственным аналитическим методом, способным
надежно определять суперэкотоксиканты: полихлорированные дибензодиоксины и ди-
бензофураны. Благодаря своей высочайшей токсичности и стабильности эти ксенобио-
тики должны контролироваться на постоянной основе. Более 30 лет им уделяется особое
внимание. Ежегодный представительный научный форум посвящен исключительно
этим соединениям. Детали масс-спектрометрического определения этих веществ пред-
ставлены в главе 15.
Загрязнение атмосферы является важным аспектом химии окружающей среды.
Глава 16 названа «Масс-спектрометрия атмосферных аэрозолей». Уникальные резуль-
таты, включая анализ единичных частиц, наглядно демонстрируют преимущество
масс-спектрометрии перед другими аналитическими методами в подобных анализах.
Введение
Что происходит, когда загрязняющее вещество попадает в организм человека? Воз-
можность масс-спектрометрического изучения взаимодействия экотоксикантов с ДНК
является предметом главы 17. Чрезвычайная сложность задачи требует высочайшей чув-
ствительности метода и возможности работать как с целевыми, так и нецелевыми ад-
дуктами ксенобиотиков с биомолекулами.
В четырех последних главах рассматриваются масс-спектрометрические подходы,
которые привели к созданию новых направлений научных исследований. В частности,
появилась возможность прямого анализа состава самых сложных смесей природных
соединений. Сверхвысокая разрешающая способность масс-спектрометрии с преоб-
разованием Фурье позволяет идентифицировать и оценить уровни тысяч индивиду-
альных родственных природных соединений без какого-либо предварительного разде-
ления. Главы 18 и 19 знакомят читателя с новейшими дисциплинами: петролеомикой
и гуминомикой.
Масс-спектрометрия изображений (имиджинг) — еще одно уникальное научное
направление, возникшее в самом конце XX века. Этот удивительный метод позволяет
детектировать и картировать распределение тысяч самых разнообразных соединений
в органах и тканях животных и растений, в минералах, овощах и фруктах, произведе-
ниях искусства. Стало возможным устанавливать распределение экотоксиканта, лекар-
ственного препарата или его метаболитов в целом организме лабораторного животного
или определять, какие белки и где конкретно генерируются организмом в качестве от-
клика на проникновение чужеродного вещества в организм. Эти аспекты рассматрива-
ются в главе 20.
Глава 21 посвящена изотопной масс-спектрометрии — красивому методу, базирую-
щемуся на существовании природных изотопов химических элементов. Соотношение
этих изотопов зависит от происхождения образца. Измерив отношения D/H или 13С/12С,
можно делать выводы о подлинности произведений искусства, определять, на каком за-
воде синтезировано данное вещество, устанавливать, природным или синтетическим
является полезный ингредиент (аскорбиновая или лимонная кислота) в соках и напит-
ках. Этот мощный метод полезен в криминалистике (анализ наркотиков) и в допинг-
контроле (различие между эндогенными и синтетическими стероидами).
Таким образом, я надеюсь, что читатель не только обнаружит в этой книге мно-
го полезной для своей будущей работы информации, но и просто получит удоволь-
ствие, знакомясь с фантастическими возможностями современной экологической
масс-спектрометрии.
Литература
[1]. Cooks R.G., Ifa D. R., Sharma G., Tadjimukhamedov F. Kh., Ouyang Zh. (2010). «Persrec-
tives and retrospectives in mass spectrometry: one view». European Journal of Mass Spec-
trometry, 16, 3: 283—300.
[2]. Kluge H.-Jurgen (2010). «High accuracy mass spectrometry for fundamental studies». Euro-
pean Journal of Mass Spectrometry, 16, 3: 269—282
ГЛАВА I
ОСНОВНЫЕ ПРИНЦИПЫ
МАСС-СПЕКТРОМЕТРИИ
Альберт Лебедев
1.1. Базовые аспекты
Для того чтобы получить масс-спектр, необходимо ввести образец в ионный ис-
точник масс-спектрометра, ионизировать молекулы (получить положительные или
отрицательные ионы), разделить эти ионы по величинам m/z (отношение их массы
к заряду) и установить число ионов с каждым m/z- Компьютер управляет всеми про-
цессами и проводит обработку данных. Принципиальная схема масс-спектрометра
представлена на рис. 1.1. Дополнительные узлы могут добавляться для решения более
сложных задач.
Таким образом, масс-спектрометрия является аналитическим методом, оперирую-
щим заряженными атомами и молекулами химических соединений. Принципиальным
моментом является ионизация молекул образца или перевод ионов, уже имеющихся
в растворе или в твердой матрице, в газовую фазу. Отношение массы к заряду (m/z) ио-
нов может быть точно измерено, что позволяет установить молекулярные массы ингре-
диентов образца. Можно также инициировать фрагментацию исходных молекулярных
ионов с получением набора фрагментных ионов. Эти ионы представляют собой наибо-
лее стабильные фрагменты исходной молекулы и позволяют делать выводы о ее струк-
туре. Интенсивности и величины m/z молекулярного и фрагментных ионов на графике
по осям у и х соответственно называются масс-спектром. Обычно график нормализу-
ется на 100% по наиболее интенсивному пику в спектре. Масс-спектр — уникальная
характеристика химического вещества. Для работы с масс-спектром необходимы толь-
ко атомные массы химических элементов, точнее природных изотопов этих элементов.
Поскольку атомные и молекулярные массы — простые и широко используемые характе-
ристики, масс-спектры значительно проще для интерпретации, чем ЯМР, И К, УФ, ЭПР
и другие типы спектров.
Рис. 1.1. Принципиальная схема масс-спектрометра
Глава 1. Основные принципы масс-спектрометрии
Рис. 1.2. Спектр электронной ионизации бензоилхлорида (NIST 27422)
Существуют определенные правила, определяющие направления фрагментации
химических соединений в масс-спектрометре. Поэтому на основе масс-спектра можно
установить молекулярную массу, элементный состав, присутствие определенных функ-
циональных групп, а зачастую и полную структуру аналита. Во многих случаях уже но-
вичок может успешно решать задачи даже без использования спектральных баз данных
и специального программного обеспечения.
Спектр электронной ионизации бензоилхлорида в классической графической фор-
ме представлен на рис. 1.2. Абсцисса соответствует массам ионов (точнее величинам
m/z), а ордината — относительным интенсивностям пиков этих ионов. Для новичка
проще принимать во внимание только наиболее интенсивные пики в спектре. Менее
интенсивные пики, следующие сразу за интенсивными, обусловлены наличием в со-
ставе молекул образца природных стабильных изотопов. Они играют важную роль при
установлении структуры и далее будут обсуждаться подробнее.
Пик иона с самой большой массой соответствует молекулярному иону анали-
та. В спектре на рис. 1.2 — это 140. Следует также обратить внимание на ион с m/z 142,
интенсивность пика которого составляет 1/3 от интенсивности пика иона с m/z 140.
Эти два пика обусловлены существованием двух природных изотопов хлора: 35С1 и 37С1.
Их природное соотношение ~ 3:1. Первичный фрагментный ион с m/z 105 образует-
ся при потере 35 единиц массы из молекулярного иона с m/z 140 или 37 единиц мас-
100-1
90-
80-
70-
60-
50-
40-
30-
20-
Ю-
0-М г-------h----1-
20 40 60 80
237
295
208
. 266
il 324 * 100
—।----1----1----1---1----i-J—г—А-------Д—।—-----------А
100 120 140 160 180 200 220 240 260 280 300 320
m/z
Рис. 1.3. Спектр электронной ионизации тетраэтилсвинца (NIST45993)
1.1. Базовые аспекты
сы из изотопного иона с m/z 142. Оба выброса обусловлены атомами хлора (35С1 и 37С1).
Первичный ион с m/z 105 теряет молекулу СО с образованием вторичного фрагментного
иона (m/z 77), который, однако, может образоваться и напрямую из молекулярного иона
при разрыве связи С-С. Даже человек с минимальным опытом работы по расшифровке
масс-спектров знает, что ион с m/z 77 может считаться визитной карточкой фенильной
группы (С6Н5+). Это стабильный в газовой фазе ион, который указывает на ароматиче-
скую природу аналита. Продукт следующей стадии фрагментации, ион с m/z 51, образу-
ется при отщеплении из фенил-катиона молекулы ацетилена (С2Н2). Измерение точной
массы (см. ниже) может надежно подтвердить сделанные выводы, поскольку дает воз-
можность установить элементный состав всех упомянутых выше ионов.
Существование природных изотопов химических элементов облегчает установле-
ние структуры и позволяет определить элементный состав анализируемого соединения
по его масс-спектру. На рис. 1.3 представлен хороший пример простоты и информатив-
ности масс-спектра, демонстрирующий важностьучета изотопных пиков. Три основных
природных изотопа свинца имеют атомные массы 206, 207 и 208 единиц с распростра-
ненностью 25,15%, 21,11% и 52,38% (грубо 1:1:2) соответственно. Тетраэтилсвинец —
топливная добавка. Будучи хорошо известным экотоксикантом, он запрещен в на-
9500
9000
8500
8000
7500
7000
6500
6000
5500
5000
4500
4000
3500
3000
2500
2000
1500
1000
500
0
m/z
Рис. 1.4. Масс-спектры электронной ионизации тетрахлорбифенила NIST 169679 (верх)
и трибромфосфина NIST 150824 (низ)
Глава 1. Основные принципы масс-спектрометрии
стоящее время в подавляющем большинстве стран. В области высоких масс, начиная
с m/z 324, в спектре отчетливо видны 5 триплетов (иногда искаженные) с соотношением
интенсивностей 1:1:2 и отстоящих друг от друга на 29 единиц массы. Эти группы ионов
обусловлены последовательными выбросами четырех этильных групп из молекулярно-
го иона РЬ(С2Н5)4. Детали работы с изотопными пиками представлены в (Lebedev, 2009;
Лебедев, 2003).
Хлор и бром часто присутствуют в молекулах приоритетных загрязняющих веществ
(пестициды, фармацевтические препараты, побочные продукты дезинфекции, анти-
пирены). Благодаря уникальному соотношению природных изотопов (35С1:37С1 ~ 3:1;
79Br:81Br ~ 1:1) присутствие этих элементов в анализируемых образцах может быть мо-
ментально установлено по масс-спектру. Для этого надо обращать внимание на мульти-
плеты, пики в которых отстоят друг от друга на две единицы массы. Число атомов этих
элементов в молекуле также однозначно определяется по изотопной картинке. Так, два
атома хлора в молекуле дадут изотопный кластер (триплет) с относительными интенсив-
ностями (3:1)2 = 9:6:1;три атома— (3:1)3 = 27:27:9:1 ит.д. Вид мультиплетовионоввза-
висимости от числа атомов хлора и брома в молекуле представлен на рис. 12.9 в главе 12.
Примеры масс-спектров экологически важных тетрахлорбифенила и трибромфосфина
представлены на рис. 1.4. Последовательные выбросы атомов хлора и брома с образо-
ванием характеристических изотопных кластеров ионов позволяют легко идентифи-
цировать эти токсиканты. Спектр трибромфосфина особенно прост, поскольку после-
довательное элиминирование трех атомов брома приводит к иону атомарного фосфора
с m/z 31. При этом дублете /иД 79 и 81 определяется двумя стабильными изотопами брома.
1.2. Ввод образца
Любой масс-спектрометрический эксперимент начинается с ввода образца в прибор.
На этой стадии наиболее серьезная проблема связана с тем, что большинство приборов
функционирует в условиях глубокого вакуума (10-5—10-6 торр). Существует несколько
подходов: прямой ввод, ввод через полупроницаемую мембрану, интерфейсы с много-
численными хроматографами (ГХ, ВЭЖХ, СКФХ, КЭ) и даже самостоятельное натека-
ние молекул или ионов из атмосферы в прибор, работающий в условиях пониженного
давления. Последний вариант будет детально рассматриваться в последующих главах,
посвященных миниатюризации и современным методам анализа без пробоподготовки
(главы 7—9).
В случае прямого ввода жидкий или твердый образец направляется в ион-
ный источник через вакуумный шлюз. Поскольку обычное давление в источнике
10-5—10"6 мм рт.ст., а образец может быть нагрет до 400°C и более, значительное число
органических соединений может быть переведено в газовую фазу и проанализировано.
Однако, если образец представляет собой смесь соединений с близкими летучестями,
зарегистрированный масс-спектр будет представлять собой суперпозицию спектров
компонентов. Этот факт может существенно усложнить процесс идентификации, при-
чем как компьютеризованный, так и производимый вручную.
Полупроницаемые мембраны могут с успехом применяться в экологическом мо-
ниторинге. Например, пусть вода протекает по трубке из полимерного материала.
1.3. Ионизация
Рис. 1.5. Вариант мембранного ввода с положением мембраны вне источника ионов
При этом липофильные соединения могут адсорбироваться на поверхности такого ма-
териала, просачиваясь через тонкие стенки. С наружной стороны они попадают в поток
инертного газа, доставляющего их непосредственно в ионный источник, где и проис-
ходит их детектирование. Подобные устройства успешно используются в онлайн-ре-
жиме для определения конкретных соединений в реках, промышленных сбросах и т.д.
(глава 9). Аналогичные системы используются для детектирования целевых соединений
в биологических жидкостях. К сожалению, метод не универсален и должен несколько
модифицироваться для каждого нового аналита. Вариант мембранного ввода представ-
лен на рис. 1.5.
Наиболее популярными являются системы ввода в виде различных интерфей-
сов между блоком разделения (хроматографы) и масс-спектрометром. Несколько глав
(2, 3, 6) посвящены подробному изложению вариантов такого комбинированного
метода — хроматомасс-спектрометрии.
1.3. Ионизация
Масс-спектрометрия регистрирует исключительно заряженные частицы. Ни молеку-
лы, ни радикалы не могут быть исследованы в своей исходной нейтральной форме. Сле-
довательно, молекулы образца должны быть ионизированы. Альтернативно ионы целе-
вого вещества, уже существующие в конденсированной фазе, могут быть каким-либо
образом переведены в газовую фазу. За столетнюю историю масс-спектрометрии описа-
но несколько десятков методов ионизации, причем следует отметить, что значительное
число новых атмосферных методов были предложены совсем недавно, уже в XXI веке.
Частота использования того или иного метода с годами изменяется. Одни из них про-
должают играть важную роль в современных исследованиях, другие уже прошли свой
«золотой период» и заменены более эффективными.
Все существующие методы могут быть грубо разделены на две группы: «мягкие»
и «жесткие». Это разделение основано на величине избытка внутренней энергии в об-
разующемся молекулярном ионе. Если эта величина мала, фрагментация незначи-
тельна и ион регистрируется как молекулярный. Такой процесс называется «мягкой
ионизацией». Определенный избыток внутренней энергии запускает процесс разрыва
конкретных химических связей в исходном молекулярном ионе, приводя к появлению
различных фрагментных ионов. Иногда молекулярный ион вообще не удается зареги-
Глава 1. Основные принципы масс-спектрометрии
стрировать. Подобный процесс называется «жесткой» ионизацией. Оба варианта по-
лезны. Первый дает информацию о молекулярной массе соединения и его элементном
составе (масс-спектрометрия высокого разрешения), второй позволяет получить струк-
турную информацию благодаря массам фрагментных ионов.
В этой главе будут рассмотрены только наиболее значимые для экологической масс-
спектрометрии методы ионизации. Несколько новых методов описаны в главах 7 и 8.
Для дополнительной информации может быть рекомендована книга (Westman-Brink-
malm and Brinkmalm, 2009) и литература, процитированная в ней.
1.3.1. Электронная ионизация
Электронная ионизация (традиционно называвшаяся «электронным ударом»), пред-
ложенная Демпстером еще в 1921 году (Dempster, 1921), исторически была первым и до
сих пор остается одним из наиболее востребованных методов ионизации. Она особен-
но важна для масс-спектрометрии образцов объектов окружающей среды. В процессе
электронной ионизации (ИЭ) газообразные молекулы образца должны оказаться в ион-
ном источнике, функционирующем в условиях высокого вакуума (10-5—10~6 мм рт.ст.).
Испускаемые катодом электроны, пролетая через ионный источник к противоэлектро-
ду, взаимодействуют с этими молекулами. В результате собственные электроны молекул
переходят на более высокие орбитали или совсем покидают молекулу с образованием
ионов. Процесс может быть представлен следующим уравнением:
М+ё = М+* + 2ё. (1.1)
Теряя электрон, молекула превращается в катион-радикал. Более 95% образую-
щихся ионов однозарядные, хотя образуются двух- и даже многозарядные ионы. Их ко-
личество зависит от природы молекул образца. Стандартная энергия ионизирующих
электронов — 70 эВ, что близко к максимуму сечения ионизации для большинства ор-
ганических молекул. Внутренняя энергия образующихся молекулярных ионов лежит
в диапазоне от 0 до 20 эВ, приводя к интенсивной фрагментации. В условиях высоко-
го вакуума отсутствуют ионно-молекулярные взаимодействия, т.е. перераспределение
энергии между частицами за счет их столкновений отсутствует. В результате образо-
вавшиеся молекулярные ионы ведут себя далее исключительно в зависимости от при-
обретенной в процессе ионизации внутренней энергии. Определенный избыток этой
энергии запускает соответствующий процесс фрагментации. Поскольку статистиче-
ски в результате ионизации образуются миллиарды и триллионы молекулярных ио-
нов с очень широким диапазоном внутренней энергии, активируются все возможные
пути фрагментации. Поэтому спектры электронной ионизации предоставляют богатую
структурную информацию и характеризуются высокой воспроизводимостью независи-
мо от типа масс-спектрометра. Последняя особенность позволила создать и эффектив-
но использовать библиотеки масс-спектров (глава 5).
Электронная ионизация — это универсальный метод, позволяющий исследовать
разные классы химических соединений. Однако у него есть существенный недостаток,
связанный с необходимостью перевода молекул образца в газовую фазу. Именно поэто-
му метод непригоден для полярных, термолабильных и тяжелых молекул. Тем не менее
электронная ионизация прекрасно справляется с анализом подавляющего большин-
ства экологически значимых соединений в самых разных матрицах.
1.3. Ионизация
Хотя в процессе ионизации электронами образуются и отрицательные ионы, их вы-
ходы существенно ниже, что лимитирует использование этого режима на практике.
1.3.2. Химическая ионизация
В 1966 г. Мансон и Филд описали «мягкий» метод ионизации (Munson and Field, 1966),
названный химической ионизацией (ХИ, chemical ionization, CI). Поскольку внутрен-
няя энергия образующихся в результате ХИ ионов обычно ниже 5 эВ, фрагментация
практически отсутствует. Иногда в спектре наблюдается исключительно пик молеку-
лярного иона. Наиболее полное описание процессов ХИ можно найти в книге А. Гарри-
сона (Harrison, 1992).
Основное различие между ИЭ и ХИ связано со значительно более высоким давле-
нием в источнике (~1 мм рт.ст.) во втором случае. Это давление достигается введением
так называемого газа-реагента, в качестве которого можно использовать практически
любое вещество (воду, аммиак, метан, бензол, гелий, этилендиамин и т.д.). Взаимодей-
ствие молекул газа-реагента с электронами высокой энергии (200—500 эВ) приводит
к ионизации. Благодаря высокому давлению образующиеся молекулярные ионы вза-
имодействуют с нейтральными молекулами. В результате самых разнообразных ион-
но-молекулярных реакций создается газовая плазма, существующая в динамическом
равновесии. Наиболее распространенные и реакционноспособные ионы в этой плазме
называются ионами-реагентами. Это могут быть молекулярные, фрагментные ионы
или ионы — продукты реакций исходных молекул газа-реагента. Уравнения (1.2) дают
представление о некоторых основных процессах в случае химической ионизации мета-
ном. Два иона (СН.+ — 44% и С2Н5+ — 30%) являются доминирующими ионами-реаген-
тами в образующейся плазме.
сн4 —> сн4+’, сн3+, сн2+-...
СН4++СН4^СН5+ + СН3-
СН3+ + СН4^С2Н5++н2 (1.2)
сн2+- + сн4 -> с2н3+ + н2 + н
с2н3++ сн4 —> с3н5++ н2
Когда молекула образца вводится в ионный источник, она взаимодействует с ио-
нами-реагентами. Возможны несколько типов реакций. Наиболее значимым является
протонирование (перенос протона). Если сродство к протону у аналита М выше, чем
у газа-реагента В (например метана в случае СН5+), протекает следующая реакция:
М + ВН+^МН+ + В.
(1.3)
Необходимо подчеркнуть два момента. Во-первых, протонирование приводит
к возникновению четноэлектронных катионов (протонированных молекул), кото-
рые значительно стабильнее катион-радикалов, образующихся в условиях ионизации
электронами. Во-вторых, избыток внутренней энергии МН+ может быть легко вычислен
по уравнению
Е = СП(М) — СП(В),
(1.4),
где СП — сродство к протону М и В соответственно.
Глава 1. Основные принципы масс-спектрометрии
Таким образом могут быть генерированы протонированные молекулы аналита с из-
вестной внутренней энергией.
Помимо протонирования возможны три других важных процесса ионизации.
Перенос заряда. Если молекула газа-реагента не имеет водородных атомов (напри-
мер Не), образуются катион-радикалы М+* аналита.
М+Х+‘—»М+‘+Х (1.5)
В отличие от ИЭ их внутренняя энергия может быть легко рассчитана на основе зна-
чений энергии ионизации (ЭИ) аналита и энергии рекомбинации (ЭР) газа-реагента.
Е = ЭР(Х+) — ЭИ(М) (1.6)
Электрофильное присоединение. Если сродство к протону молекулы аналита недоста-
точно велико, она может сформировать с ионом-реагентом стабильный комплекс.
М+Х+^МХ+ (1.7)
Например, пики ионов [М + NH4]+ и [М + NO]+ весьма интенсивны в условиях ХИ
аммиаком и окисью азота соответственно.
Отрыв аниона. Такой процесс протекает при взаимодействии ионов-реагентов пере-
носа протона с аналитами с низким СП.
АВ + Х+^АХ + В+ (1.8)
Наиболее распространенный пример — это образование ионов [М — Н]+ в условиях
химической ионизации алифатических углеводородов метаном.
В отличие от ИЭ в условиях ХИ образование отрицательных ионов может быть
весьма выраженным. Четыре основных процесса образования отрицательных ионов
аналита во многом напоминают процессы образования положительных ионов: перенос
протона (депротонирование), перенос заряда, нуклеофильное присоединение и нукле-
офильное замещение (Лебедев, 2003).
Химическая ионизация отрицательных ионов (ХИОИ, negative ion chemical ioniza-
tion, NICI) — это эффективный метод анализа соединений с высоким сродством к элек-
трону. В этом случае выигрыш в чувствительности по сравнению с режимом положи-
тельных ионов может быть весьма существенным. Хорошим примером использования
ХИОИ в экологических исследованиях является качественный и количественный ана-
лиз полихлорированных соединений (глава 15).
Недостатками химической ионизации являются необходимость перевода образца
в газовую фазу, слабо выраженная фрагментация и быстрое загрязнение источника ио-
нов. Тем не менее комбинация ХИ и ИЭ может быть очень успешной, поскольку первая
дает возможность зарегистрировать молекулярный ион, а вторая предоставляет исчер-
пывающую структурную информацию.
1.3.3. Полевая ионизация
Еще одним популярным «мягким» методом является полевая ионизация (ПИ, field ion-
ization, FI). Само явление было открыто Е. Мюллером в 1951 году. Он создал полевой
ионный микроскоп, наблюдая за появлением катионов вблизи металлической поверх-
ности в сильном электростатическом поле (Muller, 1951). Через несколько лет источ-
1.3. Ионизация
ник ПИ был использован в масс-спектрометре (Jnghram and Gomer, 1954). Тем не менее
успешное внедрение ПИ в систему ГХ/МС произошло только 20 лет спустя (Вескеу, 1969;
Damico and Barron, 1971). Метод основан на эффекте туннелирования электрона. Ио-
низация происходит, когда молекула образца в газовой фазе оказывается вблизи эмит-
тера, к которому приложен очень высокий потенциал (8—10 кВ). В качестве эмиттера
обычно используются острые металлические поверхности (лезвие бритвы, острие иглы)
или острые иголочки, образующиеся при пиролизе бензонитрила. Кривизна поверх-
ности приводит к очень высоким значениям плотности электрического поля (~1 В/А).
В результате электрон молекулы аналита туннелирует с образованием катион-радика-
ла практически без избытка внутренней энергии. Помимо того, этот катион не может
долго находиться рядом с положительно заряженным эмиттером (8—10 кВ) и выталки-
вается из источника за 10—12 с, т.е. в 106 раз быстрее, чем в случае ЭИ. Фрагментация ока-
зывается практически невозможной, а масс-спектр очень часто состоит исключительно
из пика молекулярного иона.
Если образец наносится непосредственно на поверхность эмиттера при атмосфер-
ном давлении, а затем эмиттер вводится в ионный источник и на него подается напря-
жение, протекает аналогичный процесс ионизации, а метод называется полевой де-
сорбцией (ПД, field desorption, FD). Этот метод был весьма популярен в 1970—1980-е гг.,
поскольку позволил впервые получить масс-спектры целого ряда нелетучих соедине-
ний. Механизм в этом случае связан с туннелированием электрона из твердофазного
образца с последующей десорбцией с эмиттера образовавшегося иона.
Некоторое время назад казалось, что ПИ утратила свое значение, но недавно по-
явившиеся приборы, способные попеременно снимать масс-спектры ЭИ и ПИ, вдохну-
ли новую жизнь в этот метод. Самые разнообразные экотоксиканты могут быть надеж-
Рис. 1.6. Масс-спектры электронной ионизации (верх) и полевой ионизации (низ) метил-
стеарата (с разрешения Waters Corp.)
38 Глава 1. Основные принципы масс-спектрометрии
но идентифицированы, когда их молекулярные ионы зарегистрированы в режиме ПИ,
а структурная информация получена в режиме ЭИ (рис. 1.6).
Еще три метода ионизации могут быть отмечены как наиболее приемлемые для
экологических исследований, связанных с определением полярных, термолабильных
и тяжелых молекул, т.е. таких, для которых предпочтительным методом разделения яв-
ляется ЖХ (глава 3).
1.3.4. Ионизация электрораспылением
Ионизация электрораспылением, или электроспрей (ЭРИ, electrospray ionization, ESI),
позволила совершить научный прорыв в область масс-спектрометрических исследо-
ваний нелетучих, нестабильных молекул, высокомолекулярных соединений, включая
природные полимеры (белки, сахара, нуклеиновые кислоты и т. д.). Основу для создания
метода заложили теоретические работы Доула {Dole et al., 1968). За пионерской работой
группы Л.Н. Галль {Александров и др., 1984) последовала публикация Дж. Фенна {Ya-
mashita and Fenn, 1984), который в 2002 году получил за создание электрораспыления
Нобелевскую премию.
Часто ионы аналита присутствуют уже в растворе, а если исходное полярное сое-
динение нейтрально, оно может быть легко переведено в ионную форму добавкой к его
раствору кислоты или основания. Для того чтобы использовать масс-спектрометрию
для регистрации этих ионов, надо всего лишь избавиться от молекул растворителя.
Для этой цели раствор аналита распыляется при атмосферном давлении в ионный ис-
точник прибора через тонкий капилляр (рис. 3.4 в главе 3), на который подается высо-
кое напряжение (3—7 кВ). Ионы одинаковой полярности в образующихся капельках
спрея стараются отойти друг от друга как можно дальше, распределяясь по поверхно-
сти сферической капли. Нагрев и поток инертного газа вызывают испарение раство-
рителя непосредственно из капелек спрея. В какой-то момент силы поверхностного
натяжения больше не могут компенсировать силы кулоновского расталкивания, вы-
зывая микровзрыв с образованием таких же капелек меньшего размера. Этот процесс
может повторяться многократно, приводя каждый раз к образованию капель мень-
шего размера. Предложено два основных механизма высвобождения ионов аналита.
Первый {Mora et al., 2000) базируется на том, что свободные ионы аналита образуются
исключительно благодаря последовательным микровзрывам, в результате которых ис-
ходные заряженные частицы мало-помалу теряют свою сольватную оболочку. Второй
ставит во главу угла ионное испарение за счет кулоновских взаимодействий {Iribarne
and Thomson, 1976). Серьезным аргументом в пользу ионного испарения служит не-
давнее исследование, проведенное группой Р. Зеноби методом ионной спектроскопии
{Chingin et al., 2010).
Существует несколько вариантов стыковки электрораспыления с жидкостным
хроматографом. Для улучшения процесса испарения могут использоваться несколько
дополнительных газовых потоков. Скорость потока элюента через колонку может со-
ставлять от нескольких миллилитров до нескольких нанолитров. В частности, режим
наноспрея с потоком 20—200 нЛ/мин. завоевал широкую популярность для анализа
биополимеров. В таком варианте достигаются сразу две цели: чувствительность значи-
тельно улучшается, а расход образца значительно уменьшается.
1.3. Ионизация
Важной особенностью ионизации электрораспылением является преимуществен-
ное образование многозарядных ионов. Поскольку анализаторы масс разделяют ионы
по их значениям m/z, спектры очень сложных соединений могут быть получены на до-
статочно простых приборах. Например, ион с массой 100000 дальтон и зарядом +100 бу-
дет зарегистрирован прибором в том же месте спектра, что и однозарядный ион с массой
1000. По этой причине электрораспыление успешно используется для исследования со-
единений с молекулярными массами от сотен до миллионов дальтон. Рекордная зареги-
стрированная молекулярная масса составляет ПО миллионов дальтон (Chen et al., 1995).
Авторам удалось детектировать молекулярные ионы ДНК бактериофага Т4 с зарядами
от 28000 до 35000.
Масс-спектрометрия с ионизацией электрораспылением обладает превосходной
чувствительностью, достигая аттомолярных (10-18) и даже зептомолярных (10-21) уров-
ней. С источником электрораспыления, помимо жидкостной хроматографии, успешно
сочетается и капиллярный электрофорез. Возможен и прямой ввод раствора образца.
Перечисленные преимущества сделали ионизацию электрораспылением одним из наи-
более эффективных и востребованных методов сегодняшнего дня.
1.3.5. Химическая ионизация и фотоионизация при
атмосферном давлении
Впервые о химической ионизации при атмосферном давлении (ХИАД, atmospheric pres-
sure chemical ionization, APCI) заявил Хорниг в 1973 году (Horning et al., 1973), а в 1974 она
уже была успешно использована в сочетании с ЖХ (Horning et al., 1974). Метод основан
на подаче растворенного образца в источник ионов, работающий при атмосферном дав-
лении. Высокое напряжение, приложенное к электроду в виде острой иглы, создает ко-
ронный разряд, который и генерирует ионную плазму (рис. 3.5 в главе 3). Большинство
этих ионов обусловлено молекулами растворителя и атмосферных газов: азота и кисло-
рода. В таких условиях самые разнообразные ионно-молекулярные реакции неизбеж-
ны. Так, реакция первичных ионов с молекулой воды может привести к образованию
катиона Н3О+ и аниона ОН-. Реакции этих ионов-реагентов с молекулами аналита ведут
к появлению протонированных МН+- и депротонированных [М — Н]--молекул, которые
вытягиваются в анализатор. ХИАД демонстрирует лучшие результаты по сравнению
с электрораспылением, когда речь идет об умеренно полярных или легких органиче-
ских соединениях. ХИАД превосходит электроспрей и в том, что касается толерант-
ности по отношению к солям и компонетам буферных растворов, часто используемых
в жидкостной хроматографии.
Если необходимо проанализировать сложную смесь неполярных соединений (на-
пример нефть, см. главу 18 — «Петролеомика»), эффективным может быть метод фотоио-
низации при атмосферном давлении (ФИАД, atmospheric pressure photoionization, APPI).
Фотоионизация традиционно использовалась в основном для установления физико-хи-
мических характеристик молекул. Первый коммерческий прибор был создан в 1956 году
(Lossing and Tanaka, 1956). Однако вариант метода при атмосферном давлении был пред-
ложен только в 1986 году, когда И.А. Ревельский (Revelskii, 1985, 1986) продемонстриро-
вал его успешное использование для решения проблем анализа объектов окружающей
среды. Дополнительные преимущества ФИАД для ЖХ/МС были представлены Роббом
в 2000 году (Robb etal., 2000). ФИАД требует распыления раствора аналита в ионный ис-
Глава 1. Основные принципы масс-спектрометрии
точник (рис. 3.6 в главе 3), где УФ-излучение вызывает ионизацию молекул аналита в ре-
зультате прямого или вторичных процессов. Образуются одновременно как положитель-
ные, так и отрицательные ионы. ФИАД особенно полезна при работе с неполярными
соединениями, когда альтернативные электрораспыление и ХИАД не столь эффектив-
ны. Процесс ионизации можно модифицировать, введя в систему избыток специально-
го реагента (допанта). Толуол, ацетон, некоторые другие простые молекулы легко иони-
зуются в условиях ФИАД, а затем передают заряд молекуле аналита. В таком варианте
правильнее называть процесс фотохимической ионизацией при атмосферном давлении.
1.3.6. Матрично-активированная лазерная
десорбция/ионизация
Еще один эффективный метод ионизации, созданный в 1980-х, был назван матрично-
активированной лазерной десорбцией/ионизацией (МАЛДИ, matrix assisted laser de-
sorption/ionization, MALDI). Его важность для науки была подтверждена присуждени-
ем за его создание (в тандеме с электрораспылением) Нобелевской премии в 2002 году.
Пока он не столь популярен в масс-спектрометрии окружающей среды, но в будущем
может оказаться весьма полезным. Метод основан на предварительной сокристаллиза-
ции молекул образца и матрицы. Облучение такого образца короткими лазерными им-
пульсами приводит к поглощению энергии лазера молекулами матрицы с практически
взрывным переносом этих молекул вместе с молекулами аналита в газовую фазу. По-
следующие реакции в образовавшейся плазме приводят к образованию ионов аналита.
Метод позволяет работать с очень сложными молекулами, включая природные и синте-
тические полимеры. МАЛДИ также является одним из наиболее эффетивных методов
для масс-спектрометрического имиджинга (глава 20).
1.4. Масс-анализаторы
После того как ионы образца получены, необходимо их разделить. Для этой цели исполь-
зуются масс-анализаторы. Существует несколько типов анализаторов. Каждый из них
имеет свои преимущества и недостатки. Поэтому, покупая масс-спектрометр, очень
важно представлять диапазон задач, которые вы собираетесь решать с его помощью.
1.4.1. Секторные приборы
Исторически магнитные секторные приборы были первыми масс-спектрометрами.
Эти приборы могут иметь один магнитный анализатор или несколько магнитных и элек-
тростатических анализаторов. Чем больше анализаторов имеет масс-спектрометр, тем
более сложные исследования могут осуществляться с его помощью. Схема секторного
масс-спектрометра прямой геометрии с двойной фокусировкой (магнитный анализатор
следует за электростатическим) представлена на рис. 1.7. Покидая источник, ионы уско-
ряются по направлению к анализаторам.
Формула 1.9 — уравнение разделения ионов в магнитном секторе:
(1.9)
1.4. Масс-анализаторы
где т — масса иона, е — элементарный заряд, z — число зарядов, В — напряженность
магнитного поля, R — радиус магнита, V — ускоряющее напряжение (обычно несколь-
ко кВ). Таким образом, магнитный анализатор может разделять ионы по их значениям
m/z при изменении напряженности поля, радиуса или ускоряющего напряжения, при-
чем предпочтительно изменять именно напряженность поля. Электростатический сек-
тор называется анализатором «энергий», поскольку пропускает только ионы с опреде-
ленной кинетической энергией (в обычном режиме напряжение электростатического
поля равно ускоряющему напряжению).
Объединение в приборе двух и более анализаторов позволяет проводить тандемные
масс-спектрометрические эксперименты (глава 4). Помимо этого, электростатический
анализатор (рис. 1.7) привносит дополнительную особенность — высокую разрешаю-
щую способность (разрешение по массе).
Разрешение по массе (Л) означает способность масс-спектрометра разделять два
иона с массами т и (т + А/эт). Формально для двух соседних пиков однозарядных ио-
нов равной интенсивности (рис. 1.8) разрешающая способность определяется как
R = т/кт. Другой часто используемый термин «разрешение по массе» определяется
как \т. Если \т = 1, то R — теоретический предел измеряемой массы на приборе (когда
еще возможно увидеть два разделенных сигнала от соседних ионов целочисленной мас-
сы). Единичное разрешение означает, что масс-спектрометр позволяет разделять ионы
с точностью до целочисленных масс. Например, разрешающей способности 1000 доста-
точно для разделения ионов с m/z 999 и m/z 1000. Рис. 1.8 иллюстрирует определение
разрешающей способности (10%-я ложбина). Следует отметить, что для разрешающей
способности других анализаторов обычно пользуются 50%-й ложбиной.
Возникает вопрос, зачем необходима высокая разрешающая способность (сот-
ни тысяч и даже миллионы), когда речь идет о молекулах с молекулярными массами
в пределах нескольких сотен единиц? Ответ заключается в том, что любой изотоп любо-
го химического элемента имеет свой уникальный дефект массы. Поскольку в качестве
стандарта был выбран основной изотоп углерода 12С (12,000000...), массы всех осталь-
2 БП
Источник ионов
Рис. 1.7. Принципиальная схема двухфокусного масс-спектрометра (прямая геометрия).
БП — бесполевое пространство
Глава 1. Основные принципы масс-спектрометрии
Рис. 1.8. Определение разрешающей способности масс-спектрометра
них изотопов нецелочисленны. Например, *Н — 1,00782506...; 14N — 14,00307407...; 16О—
15,99491475.... Измерив точную массу иона, можно установить его элементный состав,
поскольку не существует ионов с одинаковой массой и разным составом. Эти аспекты
будут детально рассмотрены в главе 18 при обсуждении петролеомики.
Важность высокого разрешения можно продемонстрировать на простейшем приме-
ре разделения мультиплета с целочисленной массой 28 дальтон. Три соединения с такой
молекулярной массой всегда присутствуют в фоне масс-спектрометра. Это азот, окись
углерода и этилен. Если разрешающая способность прибора ниже 500, молекулярные
ионы всех трех соединений регистрируются как один пик (рис. 1.9я). Постепенное уве-
личение разрешающей способности существенно меняет картину, приводя сначала
к дублету, а в конечном счете — к триплету (рис. 1.96, <?, г).
Чтобы разделить пики ионов составов СО и С2Н4 с точными масса-
ми 27,994915 и 28,03300 соответственно, необходима разрешающая способность
R = 28/(28,03300 - 27,994915) = 770, а для разделения пиков СО и N2 (масса молекулы
азота 28,006148 единиц) — 2500.
Установление элементного состава соединения, содержащего любые химические
элементы, за пару минут — важное преимущество масс-спектрометрии над трудоем-
ким определением состава с помощью классического элементного анализа. Тем не ме-
Рис. 1.9. Форма пика фоновых ионов с m/z 28 при разной разрешающей способности
прибора
1.4. Масс-анализаторы
нее следует отметить, что элементный анализ дает информацию о составе всего образца
(включая примеси), тогда как масс-спектрометрия устанавливает элементный состав
индивидуальных соединений (отдельно всех компонентов и примесей образца).
Принимая во внимание, что число возможных элементных составов резко возрас-
тает с увеличением массы иона, масс-спектрометрия высокого разрешения становится
крайне важной при использовании во время работы с биомолекулами методов электро-
распыления и МАЛДИ. В этих случаях для надежного определения точных масс необ-
ходимы очень высокая разрешающая способность и точность измерения.
Кстати, разрешение по массе не является синонимом точности измерения массы.
Первый из этих параметров определяет способность масс-спектрометра получить раз-
дельные изображения (пики) ионов с близкими массами, а второй — установить точное
и правильное значение m/z иона, обычно для последующего определения его элементно-
го состава. Не стоит думать, что увеличение разрешения всегда ведет к автоматическому
улучшению точности измерения масс. Как только разрешение оказывается достаточ-
ным для разделения пиков измеряемых ионов, бессмысленно увеличивать его далее, по-
скольку интенсивность сигналов будет уменьшаться с неизбежным уменьшением точ-
ности измерений масс.
Рис. 1.10 иллюстрирует важность применения масс-спектрометрии высокого раз-
решения для экологических исследований. Наблюдение широкого пика со временем
Рис. 1.10. Форма хроматографического пика в режиме SIM (см. главу 2) при использова-
нии разрешающей способности 3000 (широкий пик) и 10000 (два узких пика)
(с разрешения Thermo)
Глава 1. Основные принципы масс-спектрометрии
выхода 2,3,7,8-тетрахлордибензодиоксина на масс-хроматограмме (глава 2) по току
ионов с m/z 321,9 может привести к выводу о присутствии в образце значительных ко-
личеств этого суперэкотоксиканта. Тем не менее более точные измерения массы (4 де-
сятичных знака) наглядно демонстрируют, что этот широкий пик обусловлен супер-
позицией двух пиков ионов с m/z 321,8936 и 321,8678. Первый из них действительно
обусловлен 2,3,7,8-тетрахлордибензодиоксином, однако второй принадлежит значи-
тельно менее токсичному гептахлорбифенилу; т. е., возможно, экологическая ситуация
не настолько плоха, как могло показаться исходя из результатов анализа с недостаточ-
ным разрешением.
Измеряя массы с очень высоким разрешением, фактически можно измерять энер-
гии. Работает знаменитое уравнение Эйнштейна Е= тс2. В частности, энергия связи,
складывающаяся из сильных, слабых и электромагнитных взаимодействий, может быть
определена при измерении масс всей молекулы и ее составляющих.
Возвращаясь к секторным масс-спектрометрам, следует отметить следующие
аспекты. Современные приборы этого типа обладают великолепным разрешением
по массе, точностью измерения масс, чувствительностью, а также надежностью ко-
личественного определения. Их динамический диапазон остается непревзойденным,
а диапазон измеряемых масс уступает только времяпролетным приборам. К недостат-
кам этих анализаторов можно отнести небольшую скорость сканирования и достаточ-
но большие размеры.
1.4.2. Квадрупольные приборы
Прорыв масс-спектрометрии в область экологии произошел в 1970-х с внедрением ква-
друпольных приборов в широкую практику. Это устройство часто называют фильтром
масс. Оно состоит из четырех параллельных стержней (рис. 1.11), к которым приложено
переменное и постоянное напряжения, создающие квадрупольное поле, способное фо-
кусировать, удерживать и анализировать ионы. Низкая стоимость, небольшие размеры,
высокая скорость сбора данных, превосходный динамический диапазон, хорошая вос-
Ось поля
-(U + Kcoscor)
+(U + Kcoscor)
Рис. 1.11. Принципиальная схема квадрупольного анализатора
1.4. Масс-анализаторы
производимость и пониженные требования к вакуумной системе являются основными
достоинствами квадрупольных масс-спектрометров.
Благодаря этим характеристикам даже невысокое разрешение по массе не оста-
новило активного продвижения квадруполей на приборный рынок. Поскольку моле-
кулярные массы подавляющего большинства приоритетных загрязняющих веществ
редко превосходят 500 Дальтон, квадрупольные масс-спектрометры с разрешающей
способностью 1000 оказались идеальным инструментом для качественного и количе-
ственного определения этих соединений в самых разнообразных образцах. Неплохая
чувствительность квадруполей в режиме сканирования может быть увеличена при-
мерно в 100 раз при работе в режиме масс-фрагментографии (мониторинга заданных
ионов, см. главу 2).
Комбинация трех квадруполей (QqQ) позволяет использовать несколько режимов
тандемной масс-спектрометрии (МС/МС, глава 4). Этот подход активно применяется
в настоящее время для надежной идентификации и количественного определения за-
грязняющих веществ и природных соединений, включая пептиды и белки.
Предположим, у нас имеется смесь нескольких соединений. Можно ли установить
масс-спектрометрически их структуры без предварительного разделения (хроматогра-
фии), а введя непосредственно в ионный источник всю смесь? Действительно, магнит-
ные секторные и тройные квадрупольные приборы, ионные ловушки, приборы ионного
циклотронного резонанса, орбитальные ловушки (см. ниже) могут справиться с этой
задачей.
Использование «мягкого» метода ионизации (ХИ, ПИ, МАЛДИ, электрораспы-
ление) дает возможность получить молекулярные ионы компонентов смеси. Молеку-
лярные ионы каждого типа (рис. 1.12, белые, розовые или зеленые шарики) по очереди
проходят через первый квадруполь и направляются к следующему анализатору (Q3).
На пути следования они попадают в специальную ячейку (Q2), в которой их внутрен-
няя энергия повышается каким-либо образом для того, чтобы инициировать их фраг-
ментацию. Существует достаточно много реакций активации, хотя соударение с ато-
мами инертного газа остается наиболее популярным процессом. Фрагментные ионы
(рис. 1.12, синие, красные и черные шарики) исходного молекулярного иона (рис. 1.12,
зеленые шарики) достигают второго анализатора, в котором они разделяются по m/z,
т. е. происходит регистрация масс-спектра первого компонента образца. Далее про-
МС/МС
в пространстве
Рис. 1.12. Схема эксперимента МС/МС в пространстве. В качестве примера использован
тройной квадруполь (с разрешения Applied Biosystems)
46 Глава 1. Основные принципы масс-спектрометрии
цедура повторяется со вторым молекулярным ионом (например, розовые шарики
на рис. 1.12) и так далее. Когда будет осуществлена фрагментация всех молекулярных
ионов, анализ образца без его предварительного разделения будет завершен. Обыч-
но такой режим занимает значительно меньше времени, чем, например, ГХ/МС или
ЖХ/МС. Очевидным недостатком является невозможность анализировать изомеры.
Кстати, при наличии соответствующих конструкционных характеристик каждый
из фрагментных ионов может быть выделен аналогично молекулярному и после про-
цедуры активации фрагментации мы получим его масс-спектр. Этот метод называет-
ся МС3. Ионные ловушки способны зарегистрировать спектры до 10 поколений фраг-
ментных ионов. Этот метод называется МС". Детали тандемной масс-спектрометрии
будут рассмотрены в главе 4.
1.4.3. Ионные ловушки
Впервые ионная ловушка в качестве устройства для удерживания ионов была предложе-
на Паулем (Paul, Steinwedel, 1953) в 1953 году. Существует много разных ионных ловушек:
квадрупольные, цилиндрические, линейные и т.д. (подробнее см. главу 9). Принцип ра-
боты наиболее популярной цилиндрической ловушки мало отличается от такового для
квадруполя. Разница заключается в геометрии. Обычно ловушки состоят из двух торце-
вых и одного кольцевого электрода, которые создают рабочий объем для экспериментов
с ионами. Ловушки оказались еще дешевле и меньше по размеру, чем квадруполи. Более
того, сложные тандемные эксперименты (МС") могут осуществляться в любой ионной
ловушке, а при использовании низкой скорости сканирования разрешающая способ-
ность этих приборов может достигать 107 (March, 1998). Кстати, МС/МС-эксперименты
в ионных ловушках называются МС/МС во времени в противоположность МС/МС
в пространстве в тройных квадрупольных и магнитных секторных приборах. Эти осо-
бенности сделали ионные ловушки вполне конкурентоспособными квадруполям,
несмотря на меньший динамический диапазон и ухудшенные характеристики коли-
чественного анализа. В любом случае ионные ловушки позволяют работать с разноо-
бразными экотоксикантами в требуемых диапазонах концентраций. В экологических
исследованиях для повышения чувствительности и скорости сбора данных они обычно
стыкуются с газовым или жидкостным хроматографом и функционируют с разрешени-
ем в 1 единицу массы. Линейная квадрупольная ионная ловушка — еще один вариант
анализаторов этого типа. Она отлично зарекомендовала себя в качестве устройства для
инициирования фрагментации в режиме МС/МС.
1.4.4. Времяпролетные масс-спектрометры
Пусть у нас есть полая труба, а ионы с разными значениями m/z находятся на стартовой
линии около входа в эту трубу. Если, приложив потенциал (V), мы ускорим одновремен-
но все эти ионы, они полетят по трубе, причем их кинетическая энергия будет одинако-
вой (см. также главу 9).
eV=mv2/2 или m=2eV/v2 (1-Ю)
Это означает, что более тяжелые ионы достигнут конца трубы позже, чем более лег-
кие. Это основной принцип времяпролетной масс-спектрометрии (time-of-flight, TOF).
1.4. Масс-анализаторы
Хотя теоретические основы этого метода были заложены в середине XX века (Wolffand
Stephens, 1953), использование этих приборов для изучения самых разнообразных ор-
ганических соединений началось только через 40 лет. Проблемы были связаны с раз-
бросом по времени старта, энергии и в пространстве, а также с необходимостью иметь
очень быстрые детекторы, поскольку ионы с разной величиной m/z достигают детек-
тора с очень небольшой разницей по времени (если, конечно, труба имеет реальную
длину). Однако теперь при наличии современной электроники и после изобретения
рефлектрона (Mamyrin, 1973), применения фокусировки с временной задержкой (Wiley
and McLaren, 1955) и техники ортогонального ускорения (Dodonov et al., 1994, Dawson and
Guilhaus, 1989) времяпролетные приборы стали наиболее популярными, а их перспек-
тивы — самыми радужными. Разрешающая способность современных времяпролетных
масс-спектрометров достигает сотен тысяч, они обладают высокой точностью измере-
ния масс, теоретически неограниченным диапазоном масс. Их чувствительность до-
статочно высока, а скорость сбора данных непревзойденная. Например, прибор LECO
Pegasus IVD может регистрировать до 500 спектров в секунду. Эта особенность привела
к созданию быстрой хроматографии (глава 6). Кроме того, благодаря единовременному
старту ионов со всеми значениями m/z к детектору масс-спектры оказываются идентич-
ными вне зависимости оттого, в какой части хроматографического пика они были за-
регистрированы (рис. 2.6, глава 2). Это важное преимущество времяпролетных анали-
заторов по сравнению с другими масс-спектрометрами.
Времяпролетные анализаторы лучше других подходят для работы с импульсными
методами ионизации типа МАЛДИ, однако техника ортогонального ускорения позво-
ляет эффективно использовать их и в комбинации с непрерывными источниками ионов.
Времяпролетные приборы могут применяться и для МС/МС-экспериментов. При этом
они являются идеальным анализатором ионов-продуктов в различных гибридных при-
борах типа QTOF, IT-TOF.
1.4.5. Масс-спектрометрия ионного циклотронного резонанса
с преобразованием Фурье
История наиболее мощных анализаторов (масс-спектрометрия ионного циклотронного
резонанса с преобразованием Фурье, ИЦР ПФ, Fourier transform ion cyclotron resonance,
FTICR) связана с тремя датами. Принцип ионного циклотронного резонанса был пред-
ложен Лоуренсом в 1930-м году (Lawrence and Edelfsen, 1930), первый прибор был создан
Соммером в 1950 (Hippie et al., 1949), а в 1974 Маршалл и Комисаров впервые применили
преобразования Фурье (Comisarow and Marshall, 1974). В противоположность ранее опи-
санным анализаторам в масс-спектрометрах ионного циклотронного резонанса ионы
удерживаются в ячейке прибора благодаря приложенным перпендикулярным магнит-
ным и электростатическим полям. При этом ионы движутся по круговым траекториям,
перпендикулярным магнитному полю. Частоты движущихся в ячейке ионов зависят
от их значений m/z. Можно провести возбуждение ионов радиочастотным импульсом
и перевести их на резонансные орбиты. Движение ионов в ячейке генерирует во внеш-
них пластинах детектора наведенный ток, причем его частота точно соответствует зна-
чению m/z ионов в ячейке. Затухание этих наведенных токов регистрируется как времен-
ной сигнал, который может быть конвертирован в классическую форму масс-спектра
Глава 1. Основные принципы масс-спектрометрии
приложением преобразований Фурье. Поскольку ионы не покидают ячейку, процесс
возбуждения и детектирования может повторяться многократно. Подробнее этот метод
изложен в главе 18.
Метод ИЦР ПФ дает возможность инициировать каким-либо образом фрагмен-
тацию интересующих исследователя ионов и осуществить эксперимент тандемной
масс-спектрометрии. Кроме того, можно изучать реакции целевых ионов с разными
нейтральными или заряженными частицами. В этом случае речь идет об ионно-моле-
кулярных и ионно-ионных реакциях в газовой фазе.
Основным достоинством метода ионного циклотронного резонанса является его
сверхвысокая разрешающая способность. Именно благодаря ей элементный состав ио-
нов может быть установлен с высокой надежностью. Метод эффективен для решения
фундаментальных научных проблем, а на его базе были созданы новые прикладные
дисциплины, занимающиеся сверхсложными смесями природных соединений типа
нефтей и нефтепродуктов (глава 18) или гуминовых веществ (глава 19).
Основным недостатком масс-спектрометрии ионного циклотронного резонанса яв-
ляется очень высокая стоимость оборудования и его эксплуатации. Этот момент суще-
ственно сдерживает распространение этого фантастического метода.
1.4.6. Орбитальные ловушки
К концу XX века казалось, что будущее масс-спектрометрии будет связано исключи-
тельно с усовершенствованием известных типов анализаторов. Это мнение базирова-
лось на факте, что за несколько прошедших десятилетий не было предложено никаких
новых систем. Поэтому, когда Александр Макаров объявил о создании орбитальной ло-
вушки (рис. 1.13), это было воспринято в качестве революционного события (Makarov,
1999, 2000). Избегая технических деталей, не являющихся необходимыми для целей
данной книги, следует лишь отметить, что в орбитальных ловушках, как и в приборах
ИЦР, для измерения масс используются преобразования Фурье. Однако дорогостоящие
супермагниты в этом случае не нужны. Обладая высокой разрешающей способностью
Ионный
источник ХИАД
Линейная
ионная ловушка
С-ловушка
Рис. 1.13. Принципиальная схема орбитальной ловушки (с разрешения А. Макарова)
Литература
и высокой точностью измерения масс, орбитальные ловушки за 5 лет стали самыми вос-
требованными приборами на масс-спектрометрическом рынке.
1.5. Детектирование ионов
Конечный блок масс-спектрометра отвечает за детектирование ионов. В первых клас-
сических приборах использовалась фотопластинка. Ионы с одинаковым значением m/z
попадали на конкретный участок фотопластинки, а уровень засвечивания позволял
проводить количественные измерения.
Токи ионов с одной величиной m/z в конкретном эксперименте очень малы (10-9—
10-17 А) для прямого измерения. Поэтому детектированию предшествует процедура уси-
ления сигнала. Умножители генерируют вторичные токи, более мощные в 104—108 раз.
Когда ион проходит анализатор и попадает в детектор, он инициирует эмиссию вторич-
ных частиц, например фотонов или электронов. Детекторы конвертируют энергию вхо-
дящих частиц в ток, который измеряется соответствующими устройствами.
Электронные умножители могут быть разделены на две основные группы: с дискрет-
ным динодом (несколько раздельных динодов) и непрерывным динодом (непрерывная
эмиссионная поверхность с высокой способностью ко вторичной эмиссии, например
мультиканальная пластина). Несколько эффективных конструкций детекторов описаны
в (Westman-Brinkmalm and Brinkmalm, 2009). Для регистрации отрицательных ионов на пер-
вой стадии регистрирующей системы находится конверсионный динод. Отрицательный
ион, попадая на этот динод, генерирует разные частицы, включая положительные ионы,
которые регистрируются далее обычным образом. В случае приборов ионного цикло-
тронного резонанса и орбитальных ловушек процесс регистрации ионов недеструктивен
и основан на измерении наведенных токов. Важнейшими характеристиками детекто-
ров являются линейность, коэффициент умножения, время восстановления и уровень
шума. Дополнительная полезная информация о детекторах представлена в главе 9.
Литература
[1] . Aleksandrov M.L., Gall L.N., Krasnov V.N., Nikolaev V. I., Pavlenko V. A., Shkurov V.A.
(1984). «Ion extraction from solutions at atmospheric pressures: a mass spectrometric meth-
od of analysis of bioorganic compounds». Doklady Akademii Nauk SSSR. 277, 2: 379—383.
[2] . Beckey H. D., Hilt E., Maas A., Migahed M. D., and Ochterbeck E. (1969). «А method for
strong activation of field ion emitters». International Journal of Mass Spectrometry and Ion
Physics. 3, 1-2: 161-165.
[3] . Chingin K., Frankevich V., Balabin R. M., Barylyuk K., Chen H., Wang R., and Zenobi R.
(2010). «Direct access to isolated biomolecules under ambient conditions». Angewandte Che-
mie. International Edition., 49, 13: 2358—2361.
[4] . Chen R., Cheng X., Mitchell D.W., Hofstadler S.A., Wu Q., Rockwood A.L., Sher-
man M.G., and Smith R. D. (1995). «Trapping, Detection, and Mass Determination of
Coliphage T4 DNA Ions by Electrospray Ionization Fourier Transform Ion Cyclotron Reso-
nance Mass Spectrometry». Analytical Chemistry. 67, 7: 1159—1164.
Глава 1. Основные принципы масс-спектрометрии
[5] . Comisarow М.В., and Marshall A.G. (1974). «Fourier transform ion cyclotron resonance
spectroscopy». Chemical Physics Letters. 25, 2: 282—283.
[6] . Damico J. H., and Barron R. P. (1971). «Application of field ionization to gas-liquid chroma-
tography-mass spectrometry (GLC-MS) studies». Analytical Chemistry. 43, 1: 17—21.
[7] . Dawson J. H.J., and Guilhaus M. (1989). «Orthogonal-acceleration time-of-flight mass
spectrometer». Rapid Communications in Mass Spectrometry. 3, 5: 155—159
[8] . Dempster A. J. (1921). «Positive ray analysis of lithium and magnesium». Physical Reviews.
18,6:415-422.
[9] . Dodonov A. F., Chernushevich I. V., and Laiko V. V. (1994). Electrospray Ionization on a Re-
flecting Time-of-Flight Mass Spectrometer. In: Cotter R. J. Time-of-Flight Mass Spectrom-
etry. ACS Symposium Series 549, Washington, DC, 108—123.
[10] . Dole M., Mack L. L., Hines R. L., Mobley R.C., Ferguson L.D., and Alica M.B. (1968).
«Molecular beams of macroions». Journal of Chemical Physics. 49, 5: 2240—2249
[11] . Harrison A.G. (1992). Chemical Ionization Mass Spectrometry, CRC Press, Boca Raton,
USA.
[12] . Hippie J. A., Sommer H., and Thomas H. A. (1949). «А precise method of determining the
Faraday by magnetic resonance». Physical Reviews. 76, 12: 1877—1878
[13] . Horning E.C., Horning M.G., Carroll D. I., Dzidic I., and Stillwell R.N. (1973). «New
picogram detection system based on a mass spectrometer with an external ionization source
at atmospheric pressure». Analytical Chemistry. 45, 6: 936—943
[14] . Horning E.C., Carroll D. I., Dzidic I., Haegele K. D., Horning M.G., and Stillwell R.N.
(1974). «Liquid chromatograph-mass spectrometer-computer analytical systems: a contin-
uous-flow system based on atmospheric pressure ionization mass spectrometry». Journal
Chromatography A. 99, 13—21.
[15] . Inghram M.G., and Gomer R. (1954). «Mass spectrometric analysis of ions from the field
microscope». Journal of Chemical Physics. 22, 7: 1279—1280.
[16] . Iribarne J. V., and Thomson B.A. (1976). «On the evaporation of small ions from charged
droplets». Journal of Chemical Physics. 64, 6: 2287—2294.
[17] . Lawrence E. O., and Edelfsen N. E. (1930). «On the production of high speed protons». Sci-
ence. 72, 376-377
[18] . Lebedev A.T. (2009). Introduction to mass spectra interpretation: Organic chemistry. In: Ek-
man R., Silberring J., Westman-Brinkmalm A. M., and Kraj A. (Ed.), Mass spectrometry.
Instrumentation, interpretation and applications. John Wiley @ Sons Inc., Hoboken, New
Jersey.
[19] . Lossing F. P., Tanaka I. J. (1956). «Photoionization as a source of ions for mass spectrom-
etry». Journal of Chemical Physics. 25, 5: 1031 — 1034.
[20] . Makarov A. A., Mass Spectrometer. US Pat. 5,886,346, 1999.
[21] . Makarov A. (2000). «Electrostatic axially harmonic orbital trapping: a high-performance
technique of mass analysis». Analytical Chemistry. 72, 6: 1156—1162.
[22] . Mamyrin B. A., Karataev V. L, Shmikk D. V., and Zagulin V. A. (1973). «The mass reflec-
tron, a new non-magnetic time-of-flight mass spectrometer with high resolution». Zurnal
Experimentalnoi у Teoreticheskoi Physiki. 64, 1: 82—90
[23] . March R. E. (1998). «Quadrupole ion trap mass spectrometry: Theory, Simulation, Re-
cent Developments and Applications». Rapid Communications in Mass Spectrometry. 12, 21:
1543-1554
Литература
[24] . Mora J. F., Van Berkel G. J., Enke C. G., Cole R. B., Martinez-Sanchez M., and Fenn J. B.
(2000). «Electrochemical processes in electrospray ionization mass spectrometry». Journal
of Mass Spectrometry. 35, 8: 939—952.
[25] . Muller E.W. (1951). «Das Feldionenmikroskop». Zeitschriftfur Physic. Al31, 1: 136—142.
[26] . Munson M.S. B., Field F. H. (1966). «Chemical ionization mass spectrometry. I. General
introduction». Journal American Chemical Society. 88, 12: 2621—2630.
[27] . Paul W., and Steinwedel H. (1953). Ein neues Massenspektromrterohne Magnetfeld. Z. Natur-
forsch, A8: 448—450
[28] . Revelskii LA. (1985). The method of mass spectrometric analysis of the gas mixtures. USSR
Patent 1159412, Bulletin 47.
[29] . Revelskii I. A., Yashin Yu.S., Voznesenskii V. N., Kurochkin V. K., Kostyanovskii R.G.
(1986). Mass spectrometry with phjtjionization of n-Alkanes, Alkohols, Ketones, Esters and
Amines at atmospheric pressure. Izvestiya Akademii Nauk SSSR, Seriya Khimiya, 9:
1887-1892.
[30] . Robb D. R., Covey T. R., and Bruins A. P. (2000). «Atmospheric pressure photoionization:
an ionization method for liquid chromatography-mass spectrometry». Analytical Chemistry.
72, 15: 3653-3659
[31] . Westman-Brinkmalm A.M., and Brinkmalm G. (2009). A mass spectrometer's building
blocks. In: Ekman R., Silberring J., Westman-Brinkmalm A.M., and Kraj A. (Ed.), Mass
spectrometry. Instrumentation, interpretation and applications. John Wiley @ Sons Inc.,
Hoboken, New Jersey.
[32] . Wiley W. C., and McLaren I. H. (1955). «Time-of-flight mass spectrometer with improved
resolution». The review of Scientific Instruments. 26, 12: 1150—1157
[33] . Wolff M. M., and Stephens W. E. (1953). «А pulsed mass spectrometer with time disper-
sion». The review of Scientific Instruments. 24. 8: 616—617.
[34] . Yamashita M., and Fenn J. B. (1984). «Electrospray ion source: another variation on the
free-jet theme». The Journal of Physical Chemistry. 88, 20: 4451—4459.
[35] . A. T. Лебедев. Масс-спектрометрия в органической химии. Бином, 2003
ГЛАВА 2
ГАЗОВАЯ ХРОМАТОГРАФИЯ/
МАСС-СПЕКТРОМЕТРИЯ —
«РАБОЧАЯ ЛОШАДКА»
ДЛЯ АНАЛИЗА ОБЪЕКТОВ
ОКРУЖАЮЩЕЙ СРЕДЫ
А. Т Лебедев
2.1. Общие вопросы
Соединение газового хроматографа и масс-спектрометра было абсолютно логично, по-
скольку оба метода обладали примерно равной чувствительностью и успешно исполь-
зовались для анализа органических соединений. Газовый хроматограф разделяет ком-
поненты образца, а масс-спектрометр отвечает за их детектирование и идентификацию.
Однако благодаря синергетическому эффекту эта комбинация стала одним из наиболее
мощных аналитических методов современной науки. Основные принципы стыковки
были выдвинуты и реализованы в 1957 году. Единственной технической сложностью
объединения этих методов было рабочее давление. Если газовая хроматография работа-
ет в условиях атмосферного давления, то масс-спектрометрия требует условий высокого
вакуума. Разница достигает 8 порядков. В первых приборах вслед за набивной хрома-
тографической колонкой устанавливались специальные сепараторы, которые снижали
поток газа-носителя (30 мл/мин). Появление капиллярных колонок и более мощных
насосов устранило эту проблему. Теперь конец гибкой колонки из плавленого кварца
вводится непосредственно в ионный источник масс-спектрометра.
Более 20 лет потребовалось для того, чтобы состыковать масс-спектрометр с жид-
костным хроматографом. Подробнее о приборах и применении метода «жидкостная
хроматография/масс-спектрометрия» повествует Глава 3.
Безусловно, круг соединений, которые можно исследовать методом газовой хро-
матографии/масс-спектрометрии (ГХ/МС) значительно уже, чем тот, который можно
исследовать методом масс-спектрометрии индивидуально. По счастью, для подавляю-
щего большинства антропогенных загрязняющих веществ газовая хроматография при-
менима. Поэтому ГХ/МС является чрезвычайно эффективным аналитическим мето-
дом в экологических исследованиях.
Говоря о методах ионизации в ГХ/МС, следует отметить, что более 90% проводи-
мых анализов основано на электронной ионизации (ИЭ, глава 1), которая обеспечивает
интенсивную фрагментацию и позволяет эффективно применять компьютерные би-
2.2. Типы хроматограмм с регистрацией ионного тока
блиотеки масс-спектров (глава 5). Существенно реже, но весьма успешно применяются
химическая ионизация (ХИ) и полевая ионизация (ПИ). Основная цель этих методов —
регистрация пика молекулярного иона, поскольку он зачастую отсутствует в спектрах
электронной ионизации. В этом случае эти методы являются отличным дополнением
к ИЭ. Поочередная регистрация ИЭ- и ХИ- (ПИ-) спектров во время ГХ/МС-анализа
дает информацию и о молекулярном ионе, и о наиболее значимых фрагментных ионах
(рис. 1.6 в главе 1). Кроме того, поскольку в спектрах ХИ и ПИ часто присутствует лишь
один пик молекулярного иона, они могут характеризоваться лучшими пределами обна-
ружения при количественном определении аналитов.
Хроматография привносит в комбинированный метод ГХ/МС (ЖХ/МС) очень важ-
ный параметр — время выхода (удерживания). Благодаря этому параметру появляется
возможность различать изомеры, даже в тех случаях, когда их масс-спектры очень близ-
ки друг другу. Это особо значимо для экологических исследований, поскольку токсич-
ности изомеров могут отличаться в миллионы и миллиарды раз (например, бензапи-
рен и бенз[е]пирен; 2,3,7,8-тетрахлордибензодиоксин и его многочисленные изомеры).
Таким образом, время удерживания может играть ключевую роль в установлении (под-
тверждении) структуры аналита.
2.2. Типы хроматограмм с регистрацией ионного тока
Покидая колонку, элюат попадает в источник ионов масс-спектрометра, где осущест-
вляется ионизация. Образующиеся молекулярный и фрагментные ионы аналитов разде-
ляются в масс-анализаторе, причем осуществляется постоянная запись масс-спектров
в заданном диапазоне масс. Результирующая хроматограмма (рис. 2.1я) демонстрирует
последовательность пиков, обусловленных индивидуальными соединениями — ком-
понентами анализируемого образца. Каждый пик, как правило, охарактеризован не-
сколькими масс-спектрами. По абсциссе откладывается время выхода ингредиентов,
а по ординате — величина полного ионного тока (ПИТ), зарегистрированная в данный
момент времени. Другими словами, ПИТ — это абсолютное количество всех ионов,
образующихся в данный момент времени и регистрирующихся за одно сканирование
спектра во всем заданном диапазоне масс. Чем больше ионный ток, тем выше положе-
ние сигнала данного скана на оси ординат. Количественное определение достаточно
очевидно, так как площадь хроматографического пика пропорциональна количеству
данного компонента в образце.
Хроматограмма такого типа называется хроматограммой по полному ионному
току или ПИТ (рис. 2.1л). Компьютер обычно автоматически перестраивает ПИТ-
хроматограмму, чтобы устранить фоновые пики, дрифт нулевой линии, остаточные
сигналы растворителя, артефакты и т.д., создавая реконструированную хроматограм-
му по полному ионному току (РПИТ). Рис. 2.1 демонстрирует различия между ПИТ-
и РПИТ-хроматограммами. Почти все пики на РПИТ-хроматограмме разрешены до ну-
левой линии, что улучшает возможности как идентификации, так и количественного
определения. Таким образом, ГХ/МС позволяет проводить качественный и количе-
ственный анализ, оперируя с очень сложными смесями соединений без их предвари-
тельного разделения.
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
2е+007-
1,75е+007-
1,5е+007-
1,25е+007-
1е+007-
7,5е+006-
5е+006-
2,5е+006-
1100
Рис. 2.1. ГХ/МС-анализ. Органические загрязняющие вещества в образце природной
воды, (а) Хроматограмма по полному ионному току (ПИТ); (б) реконструирован-
ная хроматограмма по полному ионному току (РП ИТ)
Компьютерная обработка может существенно улучшить качество масс-спектров.
Наиболее распространенными вариантами являются усреднение спектров и вычитание
фона. В первом случае интенсивность пиков ионов с конкретной величиной m/z в масс-
спектрах по всему профилю хроматографического пика суммируется и делится на чис-
ло спектров. В частности, усреднение минимизирует искажение спектров (см. ниже).
На рис. 2.2 представлена процедура вычитания фона. Она позволяет детектировать, на-
пример, следовые количества аналита на фоне значительного матричного фона в слож-
ных образцах. Хотя два основных кластера пиков ДДЕ (m/z 246 и 318) едва различимы
в масс-спектре, зарегистрированном на вершине хроматографического пика (рис. 2.2а),
после процедуры вычитания сходимость очищенного спектра со стандартным (библио-
течным) составила 88 % (рис. 2.2в).
Фоновый спектр необходимо выбрать на расстоянии нескольких сканирований
справа или слева от пика аналита, в точке, в которой интенсивность пиков ионов ана-
лита близка к нулю. Сигналы ионов каждого значения m/z в фоновом спектре должны
быть вычтены из аналогичных сигналов (с тем же значением m/z) в спектре, зарегистри-
рованном на вершине хроматографического пика. Результирующий пик практически
не содержит фоновых сигналов. В этом случае идентификация по спектральным базам
данных становится значительно надежнее и эффективнее. Иногда таким образом мож-
но расчистить сигналы аналитов, присутствующих в пробе в следовых количествах.
Чтобы улучшить качество спектра, полезно также перед вычитанием фона провести
усреднение фонового спектра. В этом варианте интенсивности ионов с каждым значе-
2.2. Типы хроматограмм с регистрацией ионного тока
Caiper - sample "Zr2:l", 1436.61 s to 1436.61 s - ( 300.103 s to 300.103 s
300.103 s to 300.103 s )
85
1000т
800 -
600 -
400 -
200 -
If 9 207
к 1-75 igi I
|lHJJk hll. I
180 ----
246
200 220 240 260 280 300 320 340 360 380 400 420
278
302
318
361 379
405 421
160
Peak True - sample "Zr2:l", peak 351, at 1434.09 s
40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420 440
Library Hit - siniarity 882, "o,p'-DDE"
40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420 440
0
Рис.2.2. (а) Масс-спектр, зарегистрированный на вершине хроматографического пика;
(б) масс-спектр фона; (в) масс-спектр аналита после вычитания фона (степень
идентичности 88 %); (г) библиотечный (N 1ST) масс-спектр ДДЕ
нием m/z суммируются по двум спектрам, зарегистрированным справа и слева у подно-
жия хроматографического пика, а полученные величины делятся на два. Усредненный
спектр вычитается из спектра аналита, как уже описано выше.
Существует две основные стратегии использования ГХ/МС. Первая связана с де-
тектированием целевых соединений. В этом случае задача обычно заключается в коли-
чественном определении заранее выбранных аналитов. При этом все остальные компо-
ненты образца, как правило, игнорируются. Вторая стратегия называется скринингом,
нецелевым анализом или анализом полного спектра. Эта существенно более сложная
задача включает идентификацию всех компонентов образца с последующим выбором
среди них наиболее значимых исходя из целей анализа. Точное количественное опре-
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
деление в режиме скрининга, как правило, невозможно. Однако по результатам можно
сделать более или менее точные количественные оценки. Наиболее полезные варианты
ГХ/МС-анализа в рамках этих двух стратегий описаны далее.
Существует еще один крайне полезный момент, невозможный в обычной газовой
хроматографии. Он особенно хорош для целевого анализа и заключается в перестрое-
нии хроматограмм с использованием тока только заданных ионов, например, m/z 141
и 156 на рис. 2.3d. Будучи очень эффективен для детектирования в образце целевых со-
единений, такой вариант представления называется масс-хроматограммой, реконстру-
ированной ионной хроматограммой (reconstructed ion chromatogram, RIC), селективной
ионной хроматограммой (selected ion chromatogram, SIC) или хроматограммой по току
выбранных ионов (extracted ion current, EIC).
Например, ионысаиД 141 и 156 характерны для алкилнафталинов. Поскольку уров-
ни этих соединений в проанализированном образце природной воды были низки, их
Время, с 600 700 800 900 1000 1100 1200 1300 1400 1500
Спектр № 9597 11597 13597 15597 17597____ 19597 21597 23597 25597 27597
L______141 ______ 156~~I
Рис. 2.3. ГХ/МС-анализ. Хроматограммы образца природной воды с пиками органи-
ческих загрязняющих веществ, (а) Хроматограмма по полному ионному току;
(б) масс-хроматограмма по току ионов с m/z 141 (рыжий) и m/z 156 (зеленый), ха-
рактерных для метилнафталинов и диметилнафталинов соответственно
2.2. Типы хроматограмм с регистрацией ионного тока
1,8е+007
1,6е+007
1,4е+007
1,2е+007
1е+007
8е+00б
бе+006
4е+006
2е+006
550 600
650 700
Время, с
750 800 850
Рис. 2.4. Фрагмент ПИТ-хроматограммы (оранжевый) образца жира серого кита и масс-
хроматограммы потоку ионов с m/z 326 (зеленый), характерных для тетрахлорби-
фенилов, и m/z 360 (голубой), характерных для пентахлорбифенилов
пики отсутствуют на ПИТ-хроматограмме (рис. 2.Зя). Напротив, пики изомерных ме-
тил- и диметилнафталинов доминируют на масс-хроматограмме (рис. 2.3d). Становит-
ся возможным не просто детектирование, но и надежное количественное определение
этих соединений. Принимая во внимание, что построение масс-хроматограммы осно-
вано на перестроении исходной хроматограммы ПИТ и занимает лишь несколько се-
кунд компьютерного времени, эффективность такого подхода очевидна.
Хотя формально масс-хроматография не повышает чувствительности метода,
она позволяет наглядно выпятить инфомацию о целевых аналитах, скрытую на ПИТ-
хроматограмме пиками фона и мажорных компонентов. Масс-хроматограммы всегда ме-
нее сложны и более селективны, что делает этот подход очень полезным, когда речь идет
о многокомпонентных образцах. Очень часто целевой аналит выходит из колонки одно-
временно с мажорным компонентом. Результирующий масс-спектр оказывается мало-
пригодным для надежной идентификации, а пик на ПИТ-хроматограмме — неприемле-
мым для количественного определения. Пример такого случая представлен на рис. 2.4.
Образец жира серого кита был подвергнут экстракции дихлорметаном с последующей
простенькой очисткой. Жирные кислоты, продукты окисления жиров и холестериновые
производные составляют около 90% компонентов экстракта, проходящих через хрома-
тографическую колонку. Их пики доминируют на хроматограмме по полному ионному
току, закрывая значительно меньшие пики целевых ксенобиотиков. Обработка ПИТ-
хроматограммы с построением масс-хроматограмм по току характеристических ионов
позволяет выявить пики целевых соединений, например экологически значимых поли-
хлорированных бифенилов. На рис. 2.4 видно, что никаких проблем с измерением пло-
щадей соответствующих пиков и установлением уровней этих соединений нет.
Рис. 2.5 позволяет понять, насколько элегантно ГХ/МС преодолевает еще один не-
достаток ГХ, связанный с количественным определением соединений с близкими вре-
менами удерживания. Эта проблема возникает и при целевом, и при скриниговом ана-
лизе. В качестве примера можно привести результат анализа образца нефтепродуктов.
На рис. 2.5я представлен небольшой фрагмент ПИТ-хроматограммы с основным пиком
с временем выхода 555,262 секунд, а на рис. 2.5d — масс-спектр, зарегистрированный
на его вершине. Пики с m/z 43, 57, 71... обусловлены характеристическими ионами ал-
кильной серии. Однако ион с m/z 154 определенно принадлежит другому соединению.
Следовательно, данный спектр — суперпозиция масс-спектров двух соединений.
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
Рис. 2.5. ГХ/МС-анализ образца нефтепродуктов, (а) 14-секундный фрагмент ПИТ-
хроматограммы; (б) масс-спектр на вершине хроматографического пика (RT
555,262 с); (в) масс-хроматограммы по току ионов с m/z 154 и m/z 71; (г) масс-
спектр первого компонента (тетрадекан, молекулярный ион с m/z 198 едва заме-
тен) с временем выхода 554,719 с; (д) масс-спектр второго компонента (бифенил)
с временем выхода 555,562 с
2.2. Типы хроматограмм с регистрацией ионного тока
Масс-хроматограммы, построенные по току ионов с m/z 154 и 71 (рис. 2.5в), позволя-
ют увидеть, что времена выхода этих двух соединений очень близки и отличаются менее
чем на секунду (554,719 с и 555,562 с соответственно). Масс-спектры, зарегистрирован-
ные на вершине каждого из этих пиков после вычета фона с обеих сторон, позволяют
идентифицировать эти ингредиенты смеси как тетрадекан и бифенил, причем с высо-
кой степенью сходимости с библиотечными спектрами этих соединений. Площади пи-
ков на масс-хроматограммах дают возможность установить количество каждого из этих
компонентов в образце.
Масс-хроматограммы эффективны для подобных целей, если вершины двух пиков
отстоят не менее чем на 2 сканирования полного спектра. Таким образом, чем выше ско-
рость сканирования (сбора данных) масс-спектрометра, тем выше эффективность раз-
решения накладывающихся хроматографических пиков.
Существует вариант ГХ/МС-анализа, когда записи полного масс-спектра не тре-
буется. В этом случае масс-спектрометр настраивается на регистрацию только одного
или нескольких ионов с заранее выбранными значениями m/z. Этот метод называется
«мониторинг заданных (выбранных) ионов» (МЗИ, SIM) или «масс-фрагментография»
и используется для детектирования и количественного определения целевых аналитов
в сложных образцах (наиболее простой вариант целевого анализа). В этом случае ин-
формация о всех других компонентах образца теряется. Тем не менее, поскольку детек-
тор прибора в этом режиме постоянно измеряет лишь ток заданных ионов, а не скани-
рует несколько сотен различных величин m/z, интегральная чувствительность анализа
может быть повышена примерно на 2 порядка. Масс-фрагментография часто исполь-
зуется в экологических, криминалистических исследованиях, допинг-контроле, т.е.
в тех случаях, когда необходимо детектировать целевые соединения в ультра-следовых
количествах. Наличие пика заданного характеристического иона с известным временем
выхода на масс-фрагментограмме свидетельствует о присутствии данного соединения
Время,с
Рис. 2.6. SIM-анализ ПАУ в образце почвы (нафталин — m/z 128; аценафтен — m/z 154;
фенантрен и антрацен — m/z 178; флуорантен и пирен — m/z 202; бенз[а]антра-
цен и хризен — m/z 228; бенз[Ь]флуорантен, бенз[к]флуорантен и бенз[а]пирен —
m/z 252; бенз^Ы]перилен и индено[1,2,3-сб]пирен — m/z 276; дибенз[а,^антра-
цен — m/z 278; пердейтеронафталин — m/z 136 и пердейтерофенантрен — m/z 188
использованы в качестве внутренних стандартов)
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
в образце, а площадь пика позволяет провести его количественное определение. Однако,
поскольку вероятность присутствия в сложном образце веществ с близкими временами
выхода и имеющими в спектрах ионы с одинаковыми значениями m/z достаточно высо-
ка, результаты SIM не столь надежны, как результаты, полученные в режиме полного
сканирования спектра. Для повышения надежности используется одновременная реги-
страция нескольких характеристических ионов. Например, стандартные методы Агент-
ства по охране окружающей среды США требуют регистрации трех характеристических
ионов каждого целевого аналита. Аналогичный европейский документ (2002/657/ЕС)
еще жестче. Он требует четырех идентификационных точек (глава 3). Аналит считается
детектированным, если зарегистрированы все характеристические ионы, причем со-
отношения их пиков соответствуют этим соотношениям в стандартном масс-спектре
данного соединения.
Поскольку время выхода целевых компонентов всегда известно заранее, детектор
масс может переключаться для регистрации ионов с другими величинами m/z, как толь-
ко аналит был зарегистрирован (вышел из колонки). Такой подход позволяет создавать
автоматизированные программы для детектирования методом МЗИ серии аналитов
Рис. 2.7. SIM по току характеристических для станозолола ионов с m/z 560, 545 и 143 (об-
разец мочи) с низким разрешением R = 500 на приборе Agilent 5973 (с разрешения
Э. Вирюса)
2.2. Типы хроматограмм с регистрацией ионного тока
за один ввод пробы в прибор. На рис. 2.6 представлены результаты количественного
масс-фрагментографического определения 14 полициклических ароматических угле-
водородов (ПАУ) в образце почвы. Хотя на рис. 2.6 видны пики, обусловленные неко-
торыми другими компонентами образца, проблемы с идентификацией и количествен-
ным определением ПАУ нет, поскольку их времена выхода точно известны. Количество
целевых соединений в аналогичных автоматизированных программах анализа может
достигать сотен. Если одни и те же целевые аналиты должны быть определены во мно-
гих аналогичных образцах, очень полезным становится применение автосамплера.
В этом случае система ГХ/МС будет проводить последовательные анализы без участия
оператора.
Для улучшения пределов обнаружения и надежности определения исполь-
зуется масс-фрагментография высокого разрешения. На рис. 2.7 видно, что масс-
Рис. 2.8. SIM по току характеристических для станозолола ионов с m/z 560,3650; 545,3415
и 520,3642 (образец мочи) с высоким разрешением R = 10000 на приборе DSF ком-
пании Thermo Scientific (с разрешения Э. Вирюса)
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
хроматограммы по току ионов с m/z 560,545 и 143, характерных для станозолол а (спортив-
ный допинг, запрещенный WADA), едва ли предоставляют какое-либо доказательство
наличия данного соединения в пробе мочи (время выхода 26,95 мин.).
Улучшение качества сигнала в режиме высокого разрешения как для детектирова-
ния, так и для количественного определения, очевидно (рис. 2.8) Это обусловлено тем,
что запись масс-фрагментограммы по току ионов с величиной m/z, измеренной с не-
сколькими десятичными знаками, фактически позволяет детектировать только ионы
с определенным элементным составом. Эта особенность дает возможность существенно
сократить число мешающих ионов. Чтобы избежать проблем, связанных с небольшими
флуктуациями в работе прибора, ионы регистрируются в очень узком массовом диапа-
зоне, например m/z 560,3649—560,3651 (рис. 2.8).
Еще один красивый пример эффективности данного подхода представлен
на рис. 1.10 (глава 1). Он демонстрирует корректное установление уровня 2,3,7,8-тетра-
хлордибензодиоксина в анализируемом образце. МЗИ с высоким разрешением особен-
но полезен при анализе очень сложных матриц, когда требуется обнаружить ультрасле-
довые уровни экотоксикантов.
Еще одним усовершенствованием метода масс-фрагментографии для детектирова-
ния целевых аналитов является использование тандемной масс-спектрометрии с реги-
RT: 15.85-16.32
16.08
Время, мин.
Рис. 2.9. Мониторинг заданных реакций (МЗР) на примере характеристических процес-
сов фрагментации станозолола (560 —> 455, 560 —> 545 и 560 —> 387) (образец мочи)
на приборе Polaris Q (Thermo Scientific) (с разрешения Э. Вирюса)
2.3. Скорость сбора данных
страцией выбранных процессов фрагментации. Этот метод называется мониторингом
заданных реакций (МЗР, selected reaction monitoring, SRM или multiple reactions monitor-
ing, MRM). Он заключается в регистрации распада выбранного иона-предшественника
(зачастую молекулярного иона) с образованием известного иона-продукта. Подобные
процессы очень селективны и с учетом времени выхода аналита позволяют получить на-
дежные результаты качественного и количественного анализа даже при работе с самыми
сложными матрицами. В данном случае используется тандемная масс-спектрометрия
или МС/МС, которая будет подробно рассмотрена в главе 4. Рис. 2.9 демонстрирует
эффективность метода МС/МС на примере определения станозолола в том же образце
(сравн. рис. 2.7 и 2.8). В этом случае регистрируются три реакции фрагментации молеку-
лярного иона (m/z 560) с образованием фрагментных ионов с m/z 455, 545 и 387.
Как и МЗИ, МЗР позволяет детектировать большое число соединений за один ввод
образца в прибор. Это достигается последовательным изменением условий регистрации
для определения все новых и новых аналитов и дает возможность создавать автомати-
зированные программы. Так, на рис. 2.10 представлена обобщенная картина результата
определения 700 пестицидов в одном вводе образца в прибор. Результат получен с помо-
щью прибора ЖХ/МС, но сам принцип в этом случае остается тем же.
2.3. Скорость сбора данных
Скорость сканирования спектров (сбора данных) — еще один очень важный параметр
масс-спектрометра, уже упоминавшийся ранее. В идеале для получения надежных ка-
чественных и количественных результатов каждый хроматографический пик должен
быть охарактеризован 10—15 полными масс-спектрами. На рис. 2.11 представлен любо-
пытный пример катастрофического различия полученной формы хроматографического
пика аналита в зависимости от скорости записи масс-спектра. Поскольку количествен-
ное определение аналита осуществляется при измерении площади хроматографическо-
го пика, понятно, что измереннное с низкой скоростью сбора данных количество до-
декана в анализируемом образце будет абсолютно неправильным (рис. 2.11, красный).
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
В случае очень узких хроматографических пиков есть возможность совсем пропустить
важный для анализа ингредиентобразца. Напротив, при регистрации 250 спектров в се-
кунду (рис. 2.11) форма пика практически идеальна и приближается к таковой для ана-
логового сигнала.
Рис. 2.11 позволяет обсудить еще один аспект, связанный с качеством масс-спектров.
Низкие скорости сбора данных могут привести не только к неправильному количе-
ственному определению, но и создать проблемы для правильной идентификации со-
единения. В частности, при низкой скорости аналит (додекан) характеризуется только
двумя масс-спектрами (рис. 2.11). Если анализ проводится на сканирующем приборе,
например популярных в экологических исследованиях квадруполях, детектор реги-
стрирует ионы один за одним по возрастанию или убыванию их величин m/z. Если в это
время происходит изменение количества аналита, что справедливо для процесса хро-
матографического элюирования, число образовавшихся ионов будет больше либо для
ионов с низким, либо высоким m/z, в зависимости от работы с нисходящей или восхо-
дящей частью хроматографического пика. Самые качественные спектры регистрируют-
ся на вершине пика. Этот эффект может привести к проблемам поиска в спектральных
базах даннах и, как следствие, ошибкам в идентификации аналита. Для случая, изобра-
женного на рис. 2.11, для скана, стартовавшего в 7,4 с, будет наблюдаться определенная
дискриминация фрагментных ионов с низкой и высокой массой в пользу ионов средней
массы. Скан, стартовавший в 7,5 с, позволит зарегистрировать лишь несколько ионов
с низкой массой при полном отсутствии каких-либо ионов средней и большой массы,
так как вещества уже не будет в источнике ионов. Это — искажение масс-спектра (spec-
tral skewing).
Следует подчеркнуть, что проблема искажения масс-спектра подобного типа харак-
терна только для сканирующих приборов. Напротив, времяпролетные анализаторы,
в которых происходит одновременный отбор всех ионов в заданном диапазоне m/z, обе-
Рис. 2.11. Форма хроматографического пика додекана шириной 150 мс при скорости сбора
данных 10 и 250 спектров в секунду (с разрешения LECO Corp.)
2.4. Какие соединения можно анализировать методом ГХ/МС
Сканирующий МС
ГХ-пик
8000
6000
4000
2000
0
42,5 43,0 43,5
Время, с
8000
6000
4000
2000
О
Время, с
m/z
m/z
8000
6000
4000
2000
О
42,5 43,0 43,5
Время, с
Врем я пролетный МС
§ £ 1000 ]
£ х 750
о ш
О 500-
£ Ё 250 ’ I
О s п .1 L-uu.-hJ L.._lb. bo4il.. ill
60 80 100 120 140
m/z
m/z
m/z
Рис. 2.12. Масс-спектр дихлорбензола, зарегистрированный в разных местах хроматогра-
фического пика времяпролетным и сканирующим масс-спектрометрами (с раз-
решения LECO Corp.)
спечивают практически идентичные масс-спектры вдоль всего хроматографического
пика. Рис. 2.12 иллюстрирует это утверждение на примере дихлорбензола.
Очень высокая скорость сбора данных во времяпролетных приборах позволяет ре-
гистрировать несколько сот спектров в секунду, обусловливая высокую точность коли-
чественных определений. Кроме того, все спектры оказываются идентичными и неис-
каженными. Это очень важное преимущество таких анализаторов, благодаря которому
появился метод быстрой хроматографии. Его подробности рассмотрены в главе 6.
2.4. Какие соединения можно анализировать методом
ГХ/МС
Можно назвать три группы соединений, подвластных ГХ/МС: летучие, полулетучие,
а также нелетучие и термолабильные. Для прохождения хроматографической колонки
последним требуется предварительная дериватизация. Последующие примеры демон-
стрируют эффективность ГХ/МС для решения аналитических задач, связанных с эти-
ми тремя группами. Во всех случаях анализ был нецелевым, т.е. заранее было неиз-
вестно, какие соединения присутствуют в образцах, а в задачу входила идентификация
максимального их числа.
Коренные жители Чукотки ежегодно потребляют около 100 килограммов китово-
го жира на душу населения. Однако в последнее время участились жалобы на сильный
и неприятный запах, который не позволяет использовать в пищу некоторых добытых
животных. Масс-спектрометрический анализ образцов тканей таких «вонючих китов»
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
260000 -
240000
220000 :
200000
180000
160000
140000
120000
100000 7
80000 :
60000
40000 :
20000 - JUCjL
о ..........
TIC: KIT1B.D
5,00
6,00 7,00 8,00 9,00 ю'оо п'о'о
—А —. А
1з'оо 14*00 15'00 16,00
Время
Рис. 2.13. ПИТ-хроматограмма летучих соединений из жира «вонючего» серого кита. Сле-
ва направо: изобутаналь, бутаналь, 2-метилбутаналь, 3-метилбутаналь, 2-этил-
фуран, пентаналь, пентан-2-он, разветвленные гексанали (4 изомера), гексаналь
(максимальный пик), разветвленные октанали (3 изомера), октаналь, алкилке-
тоны (3 соединения), нонаналь
методом хедспейс (глава 11) выявил присутствие разнообразных карбонильных соеди-
нений, прежде всего альдегидов, в весьма значительных количествах (рис. 2.13). Не-
приятный запах альдегидов хорошо известен. Принимая во внимание, что уровни не-
фтяных углеводородов в «вонючих китах» были примерно в 10 раз выше, чем в «чистых»
особях, был сделан вывод о том, что наиболее вероятная причина запаха обусловлена
алифатическими альдегидами, образующимися при окислении алкильных цепей угле-
водородов в организме китов.
На рис. 2.14 представлена ПИТ-хроматограмма полулетучих соединений, экстра-
гированных дихлорметаном из листьев лекарственного растения Parilis. Все основ-
ные компоненты были надежно идентифицированы, поскольку их спектры уже при-
сутствовали в базе данных (табл. 2.1). Это пример успешной работы с полулетучими
соединениями.
Наиболее сложны для ГХ/МС нелетучие и полярные соединения. Хроматографи-
ческая часть объединенной ГХ/МС-системы лимитирует ее работу, поскольку соот-
ветствующие аналиты плохо проходят через хроматографическую колонку. Для них
4е+007 -
3,5е+007-
Зе+007 -
2,5е+007-
2е+007 -
1,5е+007-
1е+007 -
5е+006-
550 600
|--ПИТ|
Рис. 2.14. ПИТ-хроматограмма дихорметанового экстракта листьев Parilis
2.4. Какие соединения можно анализировать методом ГХ/МС
Таблица 2.1. Полулетучие соединения в дихлорметановом экстракте листьев Parilis
Пик № Название Время выхода, с
1 Этиловый эфир морфолин-4-карбоновой кислоты 554,993
2 Внутренний стандарт 566,293
3 2-Метил-5-(фуран-3-ил)-пент-1-ен-3-он 577,793
4 2-Метил-5-(фур-3-ил)-пент-1-ен-3-он (изомер) 587,793
5 Ледол 681,693
6 Аромадандрен 723,593
7 Изомерледола 729,493
8 Алло-аромадандрен 736,493
9 Лонжипинокарвон 790,793
4,20 4,25 4,30 4,35 4,40 4,45 4,50 4,55
Время, с
Рис. 2.15. Масс-хроматограммы образца мочи, содержащего метаболит нондралона: (а) без
дериватизации; (б) в виде монотриметилсилильного производного; (в) в виде
бис-триметилсилильного производного)
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
30000 -
27500 -
25000 -
22500 -
20000-
17500 -
15000-
Г\
---1----1-----1----1------1
440 460 480 500 520
Время, с
Рис. 2.16. Масс-хроматограммы по току наиболее интенсивных ионов в спектрах триэ-
тиленгликоля (верх, m/z 45) и его бис-триметилсилильного производного (низ,
m/z\\T)
предпочтительнее метод ЖХ/МС (глава 3). Однако простые процедуры дериватизации
зачастую позволяют преодолеть аналитические проблемы ГХ.
На рис. 2.15 дан пример значительного улучшения результатов при использовании
дериватизации аналита. Метаболит нондралона (очень важен для допинг-контроля)
плохо поддается прямому ГХ/МС-анализу (рис. 2.15я). Однако его монотриметилси-
лильное производное может быть детектировано сравнительно легко, причем с интен-
сивностью сигнала, большей в 20 раз (рис. 2.156, обратите внимание на интенсивность).
Введение второй триметилсилильной группы в молекулу позволяет повысить предел
обнаружения еще почти в 2 раза (рис. 2.15в).
Другой впечатляющий пример представлен на рис. 2.16. Он связан с детектировани-
ем триэтиленгликоля. Используемые с самыми разными целями полиолы все чаще ока-
зываются в объектах окружающей среды. Рис. 2.16 наглядно демонстрирует 100-кратное
увеличение высоты хроматографического пика с одновременным уменьшением его ши-
рины со 100 до 3 секунд в результате дериватизации. Кроме того, для количественно-
го определения используется характеристический ион с более высокой величиной m/z
(m/z 117 vs m/z 45). Это всегда предпочтительнее, поскольку вероятность наложения ио-
нов фона или других компонентов уменьшается. Таким образом, общий выигрыш в се-
лективности и чувствительности весьма впечатляющ. Многие аналиты (дикарбоновые
кислоты, полиамины) должны подвергаться дериватизации. Полезную информацию
о дериватизующих агентах для масс-спектрометрии можно найти в книге В. Заикина
и Дж. Хал кета (Zaikin and Halket, 2009).
2.5. Количественный анализ
Рис. 2.17. Анализ сигаретного дыма методом твердофазной микроэкстракции с последу-
ющим ГХ/ГХ/МС-определением (с разрешения LECO Corp.)
Если смесь оказывается слишком сложной, можно использовать разделение на двух
последовательных ГХ-колонках. Метод называется ГХ/ГХ/МС и будет подробно рассмо-
трен в главе 6. Пример эффективности такого анализа представлен на рис. 2.17. В этом
случае удалось надежно разделить, идентифицировать и количественно определить бо-
лее 100 индивидуальных компонентов сигаретного дыма. За разделением на длинной
неполярной колонке (осьх) следовало разделение на короткой полярной колонке (осьу).
Каждый пик (холм) обусловлен индивидуальным компонентом, количество которого
пропорционально размеру (объему) этого пика.
2.5. Количественный анализ
ГХ/МС (как и ЖХ/МС, глава 3) предоставляет отличные возможности для надежного
количественного определения индивидуальныхсоединений всложныхсмесях. Подроб-
ное описание соответствующих процедур можно найти в книге Вильяма Бадди (Budde,
2001). Для этих целей можно использовать ПИТ-хроматограммы, масс-хроматограммы
и масс-фрагментограммы. В качестве превосходного количественного метода зареко-
мендовал себя также мониторинг заданных реакций (режим МС/МС), подробно рас-
смотренный в главе 4.
В простейшем варианте площадь пика на ПИТ-хроматограмме или масс-
хроматограмме может служить мерой для определения уровня аналита в пробе. Однако
в этом случае целый ряд моментов может существенно исказить реальную картину. По-
этому, проводя количественный анализ, их необходимо принимать во внимание.
1. Наиболее частой проблемой является коэлюирование компонентов (напри-
мер, рис. 2.5). В сложном образце из объектов окружающей среды на ПИТ-
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
хроматограмме вообще редко встречаются полностью разрешенные пики. Часто
не два, а несколько компонентов, имея близкие времена выхода, приведут к по-
явлению широкого неразрешенного пика. Понятно, что, измерив его площадь,
ничего нельзя сказать об уровне какого-либо конкретного аналита в образце.
Эта проблема значительно менее серьезна в режиме масс-хроматографии, осо-
бенно в варианте высокого разрешения.
2. Эффективность ионизации существенно зависит от структуры соединения. Гру-
бо только 1 из 10000 молекул теряет свой электрон в процессе электронной ио-
низации. Это усредненная величина. На самом деле это может быть, например,
5 из 100000 или 7 из 10000 молекул. Таким образом, вводя в прибор одинаковые
количества двух разных соединений, можно получить на хроматограмме очень
разные по величине пики. Ошибка может достигать порядков.
3. Разные соединения характеризуются различной эффективностью фрагмента-
ции. Молекулярные ионы полициклических ароматических углеводородов со-
ставляют более 50 % в полном ионном токе, а молекулярные ионы хлоралканов —
менее 1 %. Это означает, что прямое использование масс-хроматограмм или
масс-фрагментограмм для количественного определения может в зависимости
от выбранного иона привести к абсолютно различным результатам. Для ПИТ-
(РПИТ-) хроматограмм этот аспект неважен.
4. Все приборы в какой-то степени демонстрируют дискриминацию по массе. На-
пример, сигнал от определенного числа ионов с небольшим значением m/z может
быть интенсивнее сигнала такого же количества ионов с высоким значением m/z-
Этот эффект важен для масс-хроматографии и масс-фрагментографии и менее
значим для режима ПИТ.
Для преодоления этих сложностей в целях надежного количественного определе-
ния могут применяться классические подходы аналитической химии: методы добавок,
внутреннего и внешнего стандартов и изотопного разбавления. Первый из них требует
повторного ввода образца после добавления к его аликвоте известного количества опре-
деляемого аналита. Концентрация образца в исходной пробе в этом случае может быть
рассчитана по формуле
Х = -^-, (2.1)
S, -S
где X — количество аналита в образце, А — количество аналита, добавленное ко второй
аликвоте, S и 51 — площадь пиков аналита до и после добавки.
Метод внешнего стандарта основан на предварительных экспериментах со стан-
дартными растворами целевого аналита. Последовательный ввод стандартных раство-
ров аналита в известной концентрации позволяет построить калибровочный график,
который должен быть линейным в требуемом диапазоне концентраций. По этому гра-
фику уровень аналита в реальном образце рассчитывается арифметически или гра-
фически. Если необходимо измерить уровни нескольких соединений, калибровочные
графики должны быть построены для каждого из них. Очевидный недостаток метода
связан с построением графика и анализом образца в независимых экспериментах. Не-
значительные изменения в работе прибора и различия в объеме вводимого образца
приводят к увеличению погрешности измерений. Кроме того, поскольку стандарты
2.5. Количественный анализ
Рис. 2.18. Метод изотопного разбавления для определения нафталина и фенантрена.
Масс-хроматограммы по току ионов с m/z 128 (нафталин — рыжий), m/z 136
(пердейтеронафталин — зеленый), m/z 188 (пердейтерофенантрен — красный),
m/z 178 (фенантрен — синий). Второй синий пик обусловлен присутствием в об-
разце изомерного фенантрену антрацена
не добавляются непосредственно в образец, измеряется лишь конечная концентрация
аналита без учета потерь при пробоподготовке. Чтобы учесть эти потери, калибровоч-
ные кривые должны строиться при введении стандарта в искусственно созданную ма-
трицу с последующей полной процедурой пробоподготовки.
С точки зрения надежности анализа наиболее точным является метод изотопного
разбавления. В этом случае в исходный образец добавляются изотопные аналоги целе-
вых аналитов, содержащие несколько изотопов D, |3С, l5N или других элементов. Уровни
добавок должны быть близки ожидаемым для целевых аналитов в данной пробе. Раз-
ница в величинах m/z характеристических ионов немеченого и меченого соединений
должна достигать нескольких единиц массы, чтобы избежать наложения на изотопные
пики природного аналита. Поскольку химические и физические свойства аналита и его
изотопного аналога очень близки, все процедуры пробоподготовки приводят к одина-
ковым потерям, а соотношение площадей пиков характеристических ионов на масс-
хроматограммах точно отражает соотношение уровней аналита и его изотопного ана-
лога в образце.
На рис. 2.18 представлены масс-хроматограммы, полученные при количественном
определении нафталина и фенантрена методом изотопного разбавления. Уровни целе-
вых соединений можно установить при сравнении площадей их характеристических пи-
ков и пиков соответствущих изотопных стандартов. Единственным недостатком метода
является высокая стоимость синтетических изотопных стандартов. По этой причине
изотопное разбавление применяется в наиболее важных экспериментах, как, например,
определение полихлорированных дибензодиоксинов и дибензофуранов (глава 15).
Наиболее часто используемый подход базируется на внутренних стандартах. В этом
случае площади характеристических пиков нескольких (или даже всех) целевых анали-
тов измеряются относительно площади характеристического пика специально добав-
ленного соединения (в известном количестве), называемого внутренним стандартом.
Добавлять стандарт можно как в исходную пробу, так и непосредственно перед вводом
образца в прибор. В первом случае можно учесть потери аналитов при пробоподготов-
ке, во втором случае измеряется только конечная концентрация целевых аналитов (по-
сле всех процедур с пробой). Оба варианта имеют определенные преимущества и не-
достатки. Хотя первый подход представляется более правильным, следует понимать,
Глава 2. Газовая хроматография-масс-спектрометрия — рабочая лошадка
для анализа объектов окружающей среды
что потери аналитов и внутреннего стандарта во время пробоподготовки могут быть
одинаковыми только в случае их близких физических и химических свойств. Поэто-
му усовершенствованный метод требует добавления к исходной пробе специального
стандарта, называемого суррогатом, а внутренний стандарт добавляется к готовому
образцу непосредственно перед его вводом в хроматограф. В этом случае структурная
близость стандарта и аналита не принципиальна. Однако структура суррогата должна
быть как можно ближе структуре целевых аналитов. Суррогат служит для целей кон-
троля качества и позволяет проверять факторы извлечения, надежность работы прибора
и оператора.
Теоретически в качестве внутреннего стандарта можно использовать любое веще-
ство. Тем не менее необходимо учесть следующие требования:
• инертность по отношению к компонентам образца,
• стандарт должен a priori отсутствовать в образце,
• значение m/z характеристического иона стандарта ложно быть близко соответ-
ствующим значениям целевых аналитов,
• его время выхода ложно быть близко соответствующим значениям целевых
аналитов,
• его фактор отклика к целевым аналитам должен лежать в диапазоне 0,1 — 10,
• концентрация стандарта должна находиться в диапазоне 1 — 10000% от уровней
целевых аналитов в пробе.
Наиболее приемлемы внутренние стандарты с изотопными метками (D, 13С, l5N, |8О
и т.д.), причем самыми популярными являются пердейтерированные полициклические
ароматические углеводороды. Они применяются в различных стандартных методи-
ках, включая методики определения экотоксикантов, созданные Агентством по охране
окружающей среды США.
Процедура начинается с приготовления стандартных растворов, содержащих из-
вестные количества всех целевых аналитов и внутренних стандартов. ГХ/МС-анализ
с последующим построением масс-хроматограмм по всем характеристическим ионам
дает возможность рассчитать факторы отклика (response factors, RF):
RF =
s.-Mx
SXM.K'
(2.2)
где RF — фактор отклика, M— количество аналита в стандартном растворе, 5 — интен-
сивность сигнала (площадь пика); индекс is относится к внутреннему стандарту, а ин-
декс х — к аналиту.
Фактор отклика может варьироваться в широком диапазоне в зависимости от при-
роды аналита и внутреннего стандарта. Чем ближе эта величина к 1, тем точнее будут
результаты анализа. Когда величины факторов отклика установлены, можно присту-
пать к анализу проб. Известное количество внутреннего стандарта добавляется в пробу
до или после всех процедур пробоподготовки (см. выше), и аликвота образца вводится
в ГХ/МС-систему. Количество аналита можно рассчитать по формуле
М = RF ^ ^is , (2.3),
^is
Литература 73
где М — количество аналита в образце, 5 — интенсивность сигнала аналита, RF — фак-
тор отклика, A/s — количество введенного внутреннего стандарта, 5is — интенсивность
сигнала внутреннего стандарта.
Для повышения надежности фактор отклика можно рассчитать для 2—3 характе-
ристических ионов. Результаты измерений усредняются с учетом того, что индивиду-
альные измерения по выбранным ионам должны давать близкие результаты, а соотно-
шение пиков этих ионов должно соответствовать таковому в стандартном масс-спектре
целевого аналита.
2.6. Выводы
Несмотря на то, что ЖХ/МС (глава 3) становится все более востребованным, ГХ/МС
пока остается наиболее активно использующимся методом для целей экологического
анализа. Он надежен, чувствителен, во многих случаях автоматизирован, успешно под-
креплен компьютерными библиотеками масс-спектров (глава 5) и программным обе-
спечением. В главе 6 речь пойдет о некоторых современных вариантах ГХ/МС.
Литература
[1] . Zaikin V., and Halket J. (2009). A handbook of derivatives for mass spectrometry, IM Publica-
tions, Charlton Mill, UK.
[2] . Budde W. L. (2001). Analytical mass spectrometry. Strategies for environmental and related ap-
plications. Oxford university press. 386 p.
ГЛАВА 3
ЖИДКОСТНАЯ
ХРОМАТОГРАФИЯ / МАСС-
СПЕКТРОМЕТРИЯ —
ОПТИМАЛЬНЫЙ МЕТОД
КАЧЕСТВЕННОГО
И КОЛИЧЕСТВЕННОГО
АНАЛИЗА ЗАГРЯЗНЕНИЙ
ОКРУЖАЮЩЕЙ СРЕДЫ
Жерар Бондю и Жан-Марк Жумье
3.1. Введение
Понимание влияния человеческой деятельности на окружающую среду, а также улуч-
шение экологической ситуации признаны ключевыми факторами, влияющими на здо-
ровье и благополучие нынешнего и будущих поколений. Большинство стран создало
организации и приняло научные программы по изучению кратко- и долговременных
последствий, вызываемых этими загрязнениями, а также по уменьшению вредных
выбросов и постепенному ограничению использования экологически опасных хими-
ческих веществ. В Европе во всех странах ЕС принята Рамочная водная директива.
Эта директива также соблюдается рядом других стран в сотрудничестве с Евросоюзом.
Программа REACH (регистрация, оценка, авторизация и ограничение химических
веществ) выполняется всеми европейскими компаниями, имеющими дело с химиче-
скими соединениями. В США Агентство по охране окружающей среды (АООС, ЕРА)
и Геологическая инспекция США (USGS) также разработали программы контроля кон-
центрации различных химических веществ в окружающей среде.
Все эти программы создают большой объем работы для химиков-аналитиков. В этой
главе обсуждается использование жидкостной хроматографии в сочетании с масс-
спектрометрией для качественного и количественного анализа загрязнений окружаю-
щей среды (вода, флора, фауна). Соединения, анализируемые этим методом, как пра-
вило, являются органическими молекулами. Они используются в сельском хозяйстве
(например пестициды, ветеринарные препараты), здравоохранении, бытовой химии,
а также в химической индустрии для производства различных товаров. Все эти соеди-
3.2. Методы и оборудование для мониторинга окружающей среды 75
нения заканчивают свой жизненный цикл в окружающей среде, для которой могут быть
опасны как сами соединения, так и продукты их трансформации.
Большинство таких соединений вредны и с точки зрения пищевой безопасности.
Регулируемые уровни и методы пробоподготовки в лабораториях, работающих в об-
ласти безопасности пищевых продуктов, несколько отличаются от принятых для ла-
бораторий, занимающихся анализом объектов окружающей среды. Поэтому, несмотря
на схожесть инструментальных аналитических методов, проблемы пищевой безопасно-
сти не будут затрагиваться в этой главе.
В первой части главы будут кратко представлены методы и приборы, использую-
щиеся для мониторинга состояния окружающей среды. Во второй части будут описаны
анализы образцов объектов окружающей среды наряду с обсуждением подтверждаю-
щих методов и научных подходов.
3.2. Методы и оборудование для мониторинга
окружающей среды
3.2.1. Почему для мониторинга окружающей среды
используется жидкостная хроматография?
Исторически наиболее интересной для экологического мониторинга группой веществ
были пестициды, большинство из которых могло анализироваться методом газовой хро-
матографии (ГХ) с различными видами детектирования, включая масс-спектрометрию.
ГХ есть почти во всех аналитических лабораториях, и они относительно дешевы. Одна-
ко ГХ — оптимальный метод для летучих и неполярных молекул. Полярные и нелету-
чие компоненты требуют предварительной дериватизации, что усложняет пробопод-
готовку. Кроме того, ГХ не подходит для термически неустойчивых молекул, так как
при разделении используются высокие температуры. Жидкостная хроматография (ЖХ)
по своей природе удобна для анализа полярных (так как они легко растворяются в вод-
ных растворах) и термически нестабильных молекул (так как разделение проводится
при температурах, близких к комнатной). Поэтому при использовании ЖХ не нужна де-
риватизация. Интересно, что, следуя тенденции синтеза пестицидов, разрушающихся
в биосфере, агрохимические компании сдвинули производство в сторону более поляр-
ных молекул, которые могут быть идентифицированы при помощи ЖХ (Barcelo и Неп-
nion, 1999). Принимая во внимание, что большинство образцов экологического мони-
торинга — это вода или пробы с большим содержанием воды (флора и фауна), можно
понять, почему ЖХ широко используется в экологических исследованиях.
В ЖХ для анализа образцов из окружающей среды практически всегда использу-
ются градиентные методы. Элюирующая сила растворителя увеличивается со временем
для обеспечения прохождения через колонку молекул, взаимодействие которых с не-
подвижной фазой сильнее.
ЖХ все еще находится в стадии совершенствования. Появление в 2004 году УЭЖХ
(ультраэффективной жидкостной хроматографии) позволило улучшить разрешение
и чувствительность при уменьшении времени анализа (рис. 3.1). УЭЖХ особенно полез-
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
100-1
вэжх
81 соединение
о л и-? Wнл A A-J?-г,,. Л' W'U 'ДО, А . д . кС./Ц\тт,.,.,
0,00 2,50 5,00 7,50 10,00 12,50 15,00 17,50 20,00 22,50 25,00
Рис. 3.1. Сравнение ВЭЖХ и УЭЖХ для скрининга пестицидов. Время анализа методом
УЭЖХ в три раза меньше, чем традиционного ВЭЖХ. При этом улучшены и чув-
ствительность, и разрешение
на для ЖХ/МС-систем, так как она обеспечивает большую производительность, луч-
шую чувствительность и меньшую интерференцию между ионами.
Однако ЖХ не может полностью заменить ГХ, все еще являющуюся наиболее под-
ходящим методом для многих молекул. А для некоторых соединений (таких как, напри-
мер, полихлорированные дибензодиоксины) — это единственный официально приня-
тый метод (глава 15).
3.2.2. Почему ЖХ/МС (жидкостная хроматография с масс-
спектрометрическим детектором) используется для
мониторинга окружающей среды ?
Как было обсуждено ранее, ЖХ — подходящий метод для разделения молекул, представ-
ляющих интерес с точки зрения охраны окружающей среды. Исторически основными
детекторами для ЖХ являлись рефрактометрический, ультрафиолетовый и флуорес-
центный. Рефрактометрический детектор несовместим с градиентной ЖХ и не может
обеспечить достаточные для экологических приложений чувствительность и специ-
фичность. УФ-детектор совместим с ЖХ, причем многие молекулы могут быть иден-
тифицированы с его помощью. Однако и он не обладает достаточной специфичностью.
К тому же его чувствительность может изменяться в широких пределах в зависимости
от коэффициента поглощения излучения. ЖХ с УФ-детектором широко использовалась
в прошлом и все еще используется сейчас в случаях, когда или не требуется достиже-
ния низких пределов обнаружения, или применяются соответствующие средства кон-
3.3. Соединение ЖХ с масс-спектрометром: проблемы и их решение
центрирования (для достижения нужного предела обнаружения) и очистки (для устра-
нения интерферирующих компонентов) пробы. Флуоресценция хороша для анализа
специфических молекул, таких, как полициклические ароматические углеводороды
(методы ЕРА 550 и 550.1) или карбаматы (метод ЕРА 531.2).
Масс-спектрометрия всегда обладала потенциалом для достижения необходимых
пределов чувствительности и специфичности, но соединение ЖХс МС — значительно
более сложная задача, чем соединение ГХ с МС. Разработка в конце 80-х годов источни-
ков ввода ионов при атмосферном давлении сделала соединение ЖХ с МС более про-
стым. Таким образом, был сделан решающий шаг для популяризации ЖХ/МС-метода
и разработки коммерческих приборов. Начиная с этого момента технические новшества
сделали ЖХ/МС универсальным методом экологического мониторинга.
3.3. Соединение ЖХ с масс-спектрометром:
проблемы и их решение
Фундаментальным свойством масс-спектрометрии является разделение ионов согласно
отношению их массы к заряду m/z. Это обычно происходит при высоком вакууме, обыч-
но ~10-5 торр (1 торр = 1 ммрт.ст.). На практике это означает, что молекула может быть
идентифицирована, только если она находится в газовой фазе и ионизована.
Процесс анализа достаточно прост
при соединении ГХ с МС, так как
анализируемое вещество находится
в газообразном состоянии еще до вво-
да в масс-спектрометр. Остающиеся
в этом случае этапы — это удаление
газа-носителя ГХ и ионизация моле-
кул, обычно достигаемая при помощи
электронов или методом химической
ионизации (см. главы 1 и 2).
При соединении ЖХ с МС-
анализируемые молекулы растворены
в мобильной фазе для хроматографи-
рования. Это делает «гибридизацию»
ЖХ и МС более сложной, что было
описано во многих публикациях (Nies-
sen, 2006) и хорошо изображено Патри-
ком Арпино (Arpino, 1982) (рис. 3.2). Су-
ществует несколько проблем, которые
будут описаны далее.
1. Газофазные ионы должны гене-
рироваться из соединений, ко-
торые либо представляют собой
сольватированные молекулы,
либо присутствуют в мобильной
Рис. 3.2. Соединение ВЭЖХ с МС: «сложная свадь-
ба», как это представил Патрик Арпино в 1982 году.
(Перепечатано с разрешения издательства Elsevier
из Trends in Analytical Chemistry, 1, 7, Arpino P.J.,
On-line liquid chromatography/mass spectrometry? An odd
couple! 154-158, 1982)
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
фазе уже в ионной форме. Распыление, десольватация и ионизация должны про-
исходить при атмосферном давлении. Этот процесс называется ионизацией при
атмосферном давлении (ИАД).
2. Ионы, образовавшиеся в источнике, должны быть эффективно введены в масс-
спектрометр, тогда как огромный объем газа, образующегося в процессе испаре-
ния растворителя, должен быть удален.
3. Нейтральные молекулы, которые могут загрязнять прибор, должны быть удале-
ны (насколько это возможно).
Все описанные процессы происходят в источнике масс-спектрометра и в ионной
оптике интерфейса, которая накапливает и фокусирует ионы в масс-спектрометр. За-
грязнение оптики при анализе сложных матриц являлось большой проблемой. В совре-
менных приборах спрей смещен от оси масс-спектрометра и источник оптимизирован
таким образом, что его загрязнение минимально. Это достигается, например, исполь-
зованием встречного газового потока или применением ортогональной, или двойной
ортогональной, геометрии (рис. 3.3). Конструкция интерфейсной оптики также важна
для чувствительности, надежности и воспроизводимости.
В идеале ЖХ-элюент должен быть летучим. Следует избегать нелетучих буферов,
таких, как фосфатные. Тем не менее современные источники ионов могут работать с не-
летучими буферами в течение нескольких часов до их последующей чистки.
Рис. 3.3. Конструкция источника ионов. Ионы, образовавшиеся в источнике, попадают
в экстрагирующий конус под прямым углом. Затем их разворачивают на 90° еще
раз, чтобы они попали в оптику интерфейса. Ионы движутся в направлении ин-
терфейса под действием электрических полей, тогда как нейтральные частицы
движутся в сторону более высокого вакуума и полностью удаляются до того, как
ионы дважды повернут на 90°. Поток азота предохраняет экстрагирующий конус
от загрязнения нейтральными частицами и солями. (Z-спрей, источник фирмы
Waters)
3.4. Ионизация при атмосферном давлении
3.4. Ионизация при атмосферном давлении
Термин «ионизация при атмосферном давлении» (ИАД) относится ко множеству
процессов ионизации, которые протекают в источнике при атмосферном давлении.
Эти процессы — так называемые «мягкие» методы ионизации. Они либо сопровождают-
ся небольшой фрагментацией, либо она совсем отсутствует. Это отличает ее от ГХ/МС,
так как ионизация электронами, использующаяся в ГХ/МС, инициирует интенсивную
фрагментацию.
При ИАД поток, поступающий из ЖХ, распыляется при помощи нагретого азота
с образованием спрея, состоящего из очень мелких капель, которые затем испаряются
в источнике ионов. Далее мы опишем различные виды ионизации.
ОтЖХ
Мультипротонированная
молекула аналита
Рис. 3.4. Схема электроспрея. Колонка ВЭЖХ соединена с зондом электроспрея, пред-
ставляющего собой металлический капилляр, обдуваемый азотом. Напряже-
ние прикладывается между острием зонда и конусом для отбора пробы. Первый
этап заключается в распылении пробы. При очень низких потоках (несколько
мкл/мин) разности потенциалов достаточно, чтобы создать спрей. При более
высоких потоках для поддержания стабильного распыления необходим поток
азота. ИАД-источники оборудованы подогревом для более быстрого испарения
мобильной фазы. В электрическом поле поверхность капли, содержащей ионы,
становится заряженной положительно или отрицательно (в зависимости от по-
лярности напряжения). Размер капли уменьшается вследствие испарения рас-
творителя, что ведет к возрастанию плотности заряда на ее поверхности. Силы
отталкивания между зарядами увеличиваются до тех пор, пока капля не лопнет.
Процесс повторяется до полного испарения мобильной фазы. Если позволяет
структура молекулы, то могут образовываться многозарядные ионы. Небольшие
молекулы становятся протонированными или депротонированными
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
Вспомога-
тельный газ
Нагревательный
блок (450—550°С)
Игла коронного разряда
К масс-
анали-
затору
Вакуум
Рис. 3.5. Схема ХИ АД. Капилляр ЖХ соединен с зондом ХИ АД, который представляет
собой стеклянную трубку, обтекаемую для распыления потоком азота. Источ-
ник оборудован подогревом, причем используется еще один поток азота для ис-
парения аналита и растворителя. Напряжение в несколько кВ приложено к игле,
находящейся рядом с зондом. Это так называемый электрод коронного разряда.
Под термином «коронный разряд» подразумевают разряд, возникающий около
проводника, находящегося под высоким напряжением. Это ведет к ионизации
и пробою газа, окружающего проводник. Огни святого Эльма являются приме-
ром коронного разряда в природе. В случае ХИАД газ, окружающий разрядный
электрод, состоит главным образом из паров мобильной фазы ЖХ, азота и ана-
лизируемых молекул. Молекулы мобильной фазы ионизируются разрядом и ре-
агируют с молекулами аналита в газовой фазе. На рисунке показаны молекулы
растворителя S, превращающиеся в ионы SH+, которые затем реагируют с моле-
кулами аналита М с образованием ионов МН+. Депротонированные молекулы
образуются при отрицательном напряжении на зонде
Электрораспыление (Cole и Cole, 1997): электрический потенциал (несколько кВ)
прикладывается к зонду (электрораспылительной игле). Капли аккумулируют элек-
трические заряды на своей поверхности, уменьшаясь затем в размере до полной десоль-
ватации вследствие испарения молекул растворителя и кулоновского взаимодействия
(рис. 3.4). Такая ионизация характерна для ионных и полярных соединений, которые
несут электрические заряды при типичных хроматографических условиях. Напряже-
ние, приложенное к игле, положительное при анализе положительно-заряженных ио-
нов (положительная ЭРИ) и отрицательное в случае анализа отрицательно-заряженных
ионов (отрицательная ЭРИ). Заряд, безусловно, зависит от pH хроматографической мо-
бильной фазы.
Химическая ионизация при атмосферном давлении (ХИАД): небольшие капли спрея
полностью испаряются в источнике. Напряжение в несколько кВ приложено к зонду
(зонду коронного разряда). Под действием этого напряжения молекулы газа, присут-
ствующего в источнике, ионизируются. Затем заряд от них передается молекулам ана-
лизируемого вещества. Опять же существуют режимы как положительной, так и от-
рицательной ионизации. ХИАД эффективнее для менее полярных молекул. Многие
компоненты могут быть ионизированы как ЭРИ, так и ХИАД.
3.4. Ионизация при атмосферном давлении
УФ-лампа
Блок нагрева
Газ-распылитель
hv
hv hv
К масс-
анали-
затору
Вакуум
Рис. 3.6. Схема ФИ АД. Источник содержит УФ-лампу. Ионизация совершается или сами-
ми фотонами, или посредством допантов (ацетон, толуол), добавляемых в малой
концентрации в мобильную фазу. Ионы могут представлять собой или катион-
радикалы М‘+, или протонированные молекулы МН+
Фотоионизация при атмосферном давлении (ФИАД) имеет сходство с ХИАД. Разни-
ца заключается в замене разрядного электрода лампой ультрафиолетового излучения.
Ионизация осуществляется с помощью УФ-фотонов: либо непосредственно анализиру-
емых молекул, либо путем переноса заряда от молекул мобильной фазы, первоначально
ионизованных этими фотонами. Для повышения эффективности процесса добавляют-
100-1
15,59
Отрицательный
электроспрей
х
и
X
о
х
Бентазон
0+-гг
Etalon 3
100-1
Положительный
электроспрей
х
и
X
о
х
8,00 9,00
Атразин
15
Изопротурон
2: Scan ES+
216+207+241
1,93е5
13,00 14,00 15,00 16,00 17,00 18,00 19,00
Время, мин.
&
х
S
X
х
х
&
х
X
X
х
х
Рис. 3.7. Переключение полярности в течение хроматографического анализа. Кислотные
пестициды (бетазон) регистрируются лучше в режиме регистрации отрицатель-
ных ионов, тогда как оснбвные требуют режима регистрации положительных ио-
нов. В современных приборах переключение быстрое: около 10 мс. Для относи-
тельно простых смесей это переключение можно запрограммировать, в то время
как для сложных оно должно осуществляться непрерывно
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
ся допанты. С практической точки зрения процесс ионизации сложен и может вести
к образованию различных ионов (протонированных молекул [М + Н]+, катион-ради-
калов М+) (рис. 3.6). ФИАД подходит для слабополярных соединений, таких, как ПАУ
(полициклические ароматические углеводороды). Сложность процесса ионизации де-
лает использование ФИАД более затруднительным, чем ХИАД или ЭРИ.
Смешанный режим. Чтобы анализировать образцы, содержащие молекулы, ионизи-
рующиеся в различных режимах (положительном или отрицательном, ЭРИ или ХИАД),
большинство приборов имеют опцию переключения полярности (рис. 3.7). Некоторые
фирмы предлагают комбинированные источники с ЭРИ, ХИАД и иногда с ФИАД. Ско-
рость переключения из режима в режим и влияние этих процессов на чувствительность
различны для разных моделей приборов.
3.5. Типы масс-спектрометров и их возможности
для анализа образцов из объектов окружающей
среды
Термин «масс-спектрометрия» относится к большому разнообразию методов и прибо-
ров. В этой главе будут кратко рассмотрены масс-спектрометры, применяющиеся для
экологического мониторинга. «Сердце» масс-спектрометра — это анализатор. Анализа-
тор разделяет ионы, находящиеся в газовой фазе, по соотношению массы к заряду.
Важными различиями между анализаторами являются разрешающая способ-
ность и точность измерения масс. Приборы с низким разрешением могут рассматри-
ваться как хроматографические детекторы, обладающие высокой чувствительностью
и высокой селективностью. Их основное предназначение — количественный анализ.
В ходе ГХ/МС-анализа ионизация электронами сопровождается четко установленной
и воспроизводимой фрагментацией, что обеспечивает возможность создания библи-
отек спектров и дает информацию о структуре соединений. В ходе ЖХ/МС-анализа
фрагментация, протекающая в источнике или ячейке столкновений, сильно зависит
от конструкции прибора. Следовательно, единственная возможность состоит в созда-
нии библиотек самими пользователями или компаниями —производителями прибо-
ров. В итоге ЖХ/МС-приборы низкого разрешения дают незначительную информацию
о структуре анализируемых соединений. Приборы высокого разрешения также могут
использоваться как чувствительные и селективные детекторы (что связано с высокой
точностью определения масс). Однако они также дают полезную информацию для
идентификации неизвестных компонентов (Ballogh, 2009). Далее мы рассматриваем
приборы, использующиеся в экологических приложениях.
3.5.1. Приборы с низким разрешением
Одиночные квадруполи иногда используются для мониторинга состояния окружа-
ющей среды, главным образом благодаря их низкой стоимости. По сравнению с УФ-
детекторами они обладают лучшей чувствительностью и лучшей селективностью, но их
характеристики достаточно заурядны. Поэтому аналитики вынуждены прикладывать
3.5. Типы масс-спектрометров и их возможности для анализа образцов
из объектов окружающей среды
Ячейка столкновений,
заполненная аргоном
5-40 эВ
Q2
Q1
Статичный режим
(настроен на один ион)
Статичный режим
(настроен на один ион)
Рис. 3.8. МЗР-эксперимент. Л/ЗР-режим обычно используется в тройных квадруполях
для количественного и подтверждающего анализа. Первый квадруполь настроен
на выбранный ион-предшественник, который диссоциирует в ячейке столкно-
вений. Второй квадруполь регистрирует только выбранный ион-продукт. Полу-
ченный сигнал используется для количественного анализа. Высокая скорость
сканирования позволяет регистрировать несколько Л/РЛ/-переходов в течение
нескольких миллисекунд
большие усилия и вкладывать больше денег, чтобы добиться приемлемой пробоподго-
товки и отличного хроматографического разделения.
Тандемные квадрупольные приборы, известные ранее как тройные квадруполи,
являются «рабочими лошадками» в любой лаборатории, занимающейся экологическим
мониторингом. Для рутинного количественного анализа они используются главным
образом в режиме мониторинга заданных реакций (МЗР, MRM, рис. 3.8), который обе-
спечивает достаточную чувствительность, селективность и линейность калибровоч-
ной кривой в широком диапазоне. С его помощью можно провести количественный
подтверждающий анализ многогокомпонентной смеси в течение нескольких минут.
Скрининговые методы позволяют обнаружить большее число соединений, а подтверж-
дающий анализ найденных скринингом соединений может быть осуществлен со следу-
ющей инжекцией. Важно помнить, что в режиме МЗР тандемный квадрупольный масс-
спектрометр детектирует только соединения, заданные заранее.
Важные приборные аспекты тандемных квадруполей
Скорость сканирования', для получения пика хорошей формы и воспроизводимого коли-
чественного анализа необходимо иметь от 10 до 15 точек измерения сигнала для каждо-
го хроматографического пика, чтобы точно определить его начало, конец и максимум.
Масс-спектрометр должен обладать достаточной скоростью сканирования, для того
чтобы удовлетворять этим требованиям. Скорость сканирования непосредственно свя-
зана со временем детектирования (dwell time) (время, затраченное на детектирование од-
ного MRM-перехода), а также с паузой между циклами сканирования (пауза предназна-
чена для очистки ячейки между двумя Л/ЯМ-экспериментами). Современные приборы
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
Рис. 3.9. Иллюстрация важности регистрации достаточного для количественного опре-
деления числа точек на хроматографическом пике. На хроматограмме А пока-
заны пики с восемью точками регистрации. Определение фронтов, вершины
и, следовательно, площади пика — не очень точное. На хроматограмме В показан
пик с четырнадцатью точками для обсчета. Количественное определение в этом
случае точнее
обеспечивают как минимум одну точку измерения в каждые 10 мс. Тем не менее умень-
шение времени детектирования может вести к ухудшению отношения сигнала к шуму.
С увеличением числа загрязняющих веществ использование многокомпонентных
методов становится необходимостью. Из-за большого числа анализируемых проб мето-
ды должны быть быстрыми, что предполагает использование УЭЖХ. Высокая произ-
водительность лаборатории диктуется необходимостью быстрого возврата инвестиций
и поддержания конкурентоспособности. Следовательно, скорость сканирования явля-
ется решающим параметром для тандемных квадруполей. Подтверждающий метод для
200 соединений требует как минимум двух MRM-переходов для каждого анализируемого
компонента, что означает наличие 400 MRM-переходов в течение 10-минутного анализа
с помощью УЭЖХ. Наилучший способ использовать преимущества УЭЖХ — это про-
граммировать переходы в зависимости от времени удерживания аналитов (рис. 3.10).
Тандемные квадрупольные масс-спектрометры имеют широкий диапазон линей-
ности калибровочной кривой — по крайней мере 5—6 порядков. При работе с ЖХ/МС
этот диапазон может быть сокращен вследствие влияния процессов ионизации. Однако
линейность все равно будет более чем достаточной для количественного анализа.
Последнее поколение тандемных квадрупольных приборов (например компании
Waters) отличается более эффективной ячейкой столкновений: такой, что накопленные
ионы и их фрагменты выталкиваются синхронизованно. Таким образом, эти приборы
обеспечивают хорошее качество тандемных спектров и дают информацию о матрице об-
разца без влияния последней на количественное определение.
Ионные ловушки широко используются как недорогая замена тройным квадрупо-
лям. Несмотря на прогресс в их разработке и появление линейных ловушек, чувстви-
3.5. Типы масс-спектрометров и их возможности для анализа образцов
из объектов окружающей среды
"4 Experiment Setup - c:\masslynx_data\quanpedia_pesticide.pro\acqudb\screening402 pesticides tqd.exp
File Edit View Options Toolbars Functions Help
□ В в Я1 X
g’ SIR MRM g MS Scan Parents g Daughters Neutral Loss Survey
Points Per Peak:
Total Run Time: |js.5O , -Г|
No. Type Information Time
1 MRM of 14 mass jjairs,Time 0.00 to 2.20, ES+
2 g MRM of 17 mass pairs, Time 1.70 to 2.95, ES+
3 g MRM of 7 mass pairs, Time 2.70 to 3.15, ES+
4 g MRM of 18 mass pairs, Time 3.05 to 3.50, ES+
5 g MRM of 15 mass pairs, Time 3.40 to 4.10, ES+
6 g MRM of 19 mass pairs, Time 4.00 to 4.45, ES+
7 g MRM of 11 mass pairs, Time 4.20 to 4.55, ES+
8 g MRM of 18 mass pairs, Time 4.40 to 4.75, ES+
9 g MRM of 18 mass pairs, Time 4.55 to 5.05, ES+
10 g MRM of 22 mass pairs, Time 4.75 to 5.20, ES+
11 g MRM of 8 mass pairs, Time 4.95 to 5.30, ES+
12 g MRM of 18 mass pairs, Time 5.15 to 5.45, ES+
13 g MRM of 14 mass pairs, Time 5.20 to 5.65, ES+
14 g MRM of 24 mass pairs, Time 5.35 to 6.00, ES+
15 0 MRM of 12 mass pairs, Time 5.70 to 6.05, ES+
16 g MRM of 9 mass pairs, Time 5.80 to 6.05, ES+
17 g MRM of 20 mass pairs, Time 5.95 to 6.25, ES+
18 И MRM of 16 mass pairs’Time 6 0510 6-30, ES+
19 g MRM of 7 mass pairs, Time 6.15 to 6.45, ES+
20 g MRM of 26 mass pairs, Time 6.25 to 6.65, ES+
21 g MRM of 30 mass pairs, Time 6.40 to 6.80, ES+
22 g MRM of 16 mass pairs, Time 6.70 to 7.05, ES+
23 g MRM of 7 mass pairs, Time 6.75 to 6.90, ES+
24 g MRM of 18 mass pairs, Time 6.90 to 7.30, ES+
25 g MRM of 14 mass pairs, Time 7.1 Oto 7.50, ES+
26 g MRM of 9 mass pairs, Time 7.35 to 8.50, ES+
Рис. 3.10. МС-метод для многокомпонентного скрининга. Аналитический метод должен
обеспечивать 10—15 точек регистрации для каждого хроматографического пика.
Для УЭЖХ ширина пика ~6 с. То есть для подтверждающего метода необходи-
мо регистрировать два MRM-перехода каждые 600 мс. При детектировании 200
компонентов время анализа вместе с паузой между переходами не должно пре-
вышать 1,5 мс, что почти невыполнимо технически для любого тройного ква-
друполя. Наилучшим решением в этом случае является программирование
переходов по времени с их перекрытием на случай сдвига времен удерживания
вследствие влияния матрицы
тельность и линейность калибровочной кривой все еще отстают от характеристик, до-
стижимых при помощи тройных квадруполей, работающих в Л/ЗР-режиме.
QTRAP является комбинацией квадрупольной и линейной ионной ловушек. Та-
кой прибор может использоваться как ловушка или как тройной квадруполь. В режиме
ловушки QTRAP может генерировать тандемные спектры высокого качества, которые
могут использоваться для подтверждения идентификации по созданной пользователем
библиотеке. Однако режим ионной ловушки не позволяет зарегистрировать достаточ-
ное для надежного количественного определения число спектров.
3.5.2. Приборы с высоким разрешением
Приборы с высоким разрешением, использующиеся для экологического мониторин-
га, — это или гибридные квадрупольно-времяпролетные (QTOF), или орбитальные
ловушки (компании Thermo Scientific). Разрешающая способность времяпролетных
приборов достигает 60000, а орбитрэпа — до 100000 по состоянию на 2010 год. В этих
приборах используются разные технические решения, но оба типа масс-спектрометров
дают полную информацию об образце при высоком разрешении и точности измерения
86 Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
массы не хуже 5 ррт или даже 1 ррт для приборов последнего поколения. Они исполь-
зуются или для детектирования специфических компонентов и их идентификации
при помощи библиотечного поиска по базам данных, или для определения продуктов
трансформации (продуктов трансформации экотоксикантов в окружающей среде) с по-
мощью точного измерения масс и интерпретации МС/МС-данных.
Каждый из приборов имеет свои преимущества и недостатки, связанные главным
образом со скоростью (совместимость с УЭЖХ), динамическим диапазоном (возмож-
ность получения масс-спектрометрической информации для следовых концентраций
в сложных матрицах) и экспериментами с фрагментацией для установления структу-
ры соединений. Программное обеспечение также является важным моментом, при-
нимая во внимание объем генерируемой информации. Несмотря на то, что эти масс-
спектрометры были разработаны для исследовательских целей, они стали широко
использоваться в целях рутинного скрининга. Однако тройные квадруполи все еще
остаются «рабочими лошадками» для рутинного количественного анализа.
Важно напомнить, что данный обзор касается только экологических приложений,
и приборы, редко используемые для таких приложений, могут иметь широкое примене-
ние в других областях, и наоборот.
3.6. Анализ образцов
Химики, работающие в области мониторинга окружающей среды, не имели бы про-
блем, если бы пестициды и другие компоненты измерялись в чистой воде. Однако реч-
ная вода даже после фильтрации и центрифугирования содержит большое количество
органических компонентов. Их концентрация выражается как «суммарный органиче-
ский углерод» и находится в пределах несколькихррт (миллионные доли = миллиграм-
мы на литр). Измерение пестицидов и эстрогенов на уровне ppt (триллионные доли) кон-
центраций проблематично по многим причинам.
Первая проблема — это пределы обнаружения. При прямой инжекции только самые
новые и наиболее чувствительные тандемные квадрупольные масс-спектрометры соот-
ветствуют требуемым пределам обнаружения. Менее чувствительные приборы требуют
предварительной экстракции и концентрирования анализируемых веществ. Эти этапы
пробоподготовки могут быть выполнены автоматически или вручную.
Другая сложность — это матричные эффекты (интерферирующие компоненты).
При использовании неспецифичных детекторов (как, например, УФ-детекторы) та-
кие компоненты могут регистрироваться как пики, имеющие одинаковые с аналитом
времена удерживания. При этом аналит может быть не определен на фоне этих пиков.
При использовании селективных детекторов таких, как масс-спектрометры, интерфе-
рирующие компоненты могут создавать два вида проблем:
• изменения в хроматографии: пики могут изменить форму или время удержива-
ния может измениться;
• конкурирующая ионизация — процесс, в результате которого чувствительность
снижается или увеличивается вследствие влияния интерферирующих компо-
нентов. Такое явление называется подавлением ионизации и компенсируется
разбавлением образца, улучшением разделения пиков, оптимизацией пробопод-
готовки или использованием внутренних стандартов.
3.8. Подтверждающий анализ
3.7. Наиболее распространенные приложения
Следующие виды анализов обычно выполняются с помощью ЖХ/МС:
1. Определение концентраций различных пестицидов в сточных и поверхностных
водах, питьевой воде, флоре и фауне. Требуемый предел обнаружения пестицидов
в воде обычно 10 ppt, что в 10 раз ниже максимально допустимых концентраций
в питьевой воде (директива Евросоюза 98/83/ЕС) как для исходных компонентов,
так и для их продуктов. Большинство аналитических лабораторий применяет
многокомпонентные методы анализа, используя тандемные квадруполи. Более
оснащенные лаборатории применяют для скрининга также и времяпролетные
масс-спектрометры (типа TOFw QTOF).
2. Определение остаточных количеств лекарственных средств и эстрогенов, вне-
сенных в окружающую среду человеком. Сейчас такие вещества привлекают осо-
бое внимание, так как изначально процессы очистки сточной и питьевой воды
не были оптимизированы для их удаления. Присутствие лекарств даже в низ-
ких концентрациях может быть опасным для экосистемы и здоровья человека.
Для экологического мониторинга предпочтительно использовать тандемные
масс-спектрометры вследствие их высокой чувствительности.
3. Детектирование наркотиков в водоемах или сточных водах является свидетель-
ством их использования.
4. Определение остаточных количеств средств персонального ухода и бытовой
химии: бактерициды (триклозан), консерванты и поверхностно-активные
вещества.
5. Идентификация перфторированных соединений, имеющих широкое примене-
ние в быту и промышленности и заканчивающих свой жизненный цикл в окру-
жающей среде. Они являются проблемными из-за влияния на эндокринную
систему и аккумулирования в организме. Группа, работающая над анализом но-
вых экотоксикантов, уделяет таким веществам особое внимание (сеть Норманна
http://www/norman-network.net). Изучение путей разложения и продуктов распада
различных загрязняющих веществ является областью активных исследований.
Проблемой является тот факт, что продукты трансформации могут быть более
токсичными, чем исходные вещества. Например, было показано, что разложение
триклозана (Buth и др., 2010) ведет к образованию диоксинов, включая наиболее
токсичные из них. Подобные исследования требуют использования приборов
высокого разрешения.
3.8. Подтверждающий анализ
Для улучшения надежности результатов идентификации веществ обычной практикой
является использование двух специфических МЗР-переходов. В 2002 году Еврокомиссия
опубликовала резолюцию (2002/657/ЕС) об аналитических методах и интерпретации
результатов определения остаточных количеств ветеринарных препаратов. Запрещен-
ный препарат считается идентифицированным, если его время удерживания находится
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
Таблица 3.1. Баллы идентификации для масс-спектрометрических методов анализа
МС-метод Баллы идентификации для одного иона
МС низкого разрешения 1,0
Ион-предшественник в МС" низкого разрешения 1,0
MRM-переход в МС" низкого разрешения 1,5
МС высокого разрешения 2,0
Ион-предшественник в МС" высокого разрешения 2,0
MRM-переход в МС" высокого разрешения 2,5
в приемлемых пределах и метод его детектирования оценен в 4 балла. В табл. 3.1 пере-
числены баллы идентификации для масс-спектрометрических методов анализа.
ВЭЖХ/МС/МС-метод с двумя МЗР-переходами считается подтверждающим, если
ионы количественного и качественного определения находятся в соответствующем со-
отношении и время удерживания находится в приемлемых пределах (рис. 3.11). Данный
документ был отредактирован и применен для других случаев. Например, резолюция
SANCO/10684/2009A, вступившая в действие 1 января 2010 года, описывает методы
валидации и контроля качества анализа остаточных количеств пестицидов в пище-
вых продуктах и кормах, определяя требования к идентификации для различных ти-
пов масс-спектрометров (табл. 3.2) и стандартные допустимые отклонения для отно-
сительных интенсивностей ионов (табл. 3.3). Критерии, разработанные для анализа
100
169
Количественный
переход
Подтверждающий
переход
▼
121
143
Рис. 3.11. Принцип подтверждающего анализа показан на примере деметон-5-метил суль-
фона. Ион-предшественник с m/z 263 выбирается первым квадруполем. Затем
ион попадает в ячейку столкновений, заполненную аргоном, где фрагменти-
рует с образованием ионов-продуктов. Для количественного анализа был вы-
бран переход с образованием наиболее интенсивного иона-продукта с m/z 169.
Для подтверждающего перехода был выбран следующий по интенсивности
ион-продукт с m/z 121. Соотношение между интенсивностями количественного
и подтверждающего переходов очень важно, так как оно также используется для
подтверждения
263
231
225 250 275 300 325
350
3.9. Целевые подходы с использованием тандемной квадрупольной
масс-спектрометрии
Таблица 3.2. Идентификационные требования для различных типов масс-спектрометров
с точки зрения валидации метода и контроля анализа пестицидов в продук-
тах питания и кормах (из SANCO/10684/2009A)
Режим МС Типичные систе- мы (примеры) Регистрация Требования к идентификации Соотноше- ние ионов
Одиночный Квадруполь, ион- Полный спектр, огра- Не менее 3 диагности- См.
МС (стан- дартное разрешение) ная ловушка, ВПМС ниченный диапазон масс, мониторинг вы- бранных ионов ческих ионов(пред- почтительно иметь молекулярные ионы) табл. 3.3
Одиночный ВПМС, орбитрэп, Полный спектр, огра- Не менее 2 диагности- См.
МС (высокое разрешение, высокая точ- ность измере- ния массы) МСИЦРПФ, маг- нитный секторный ниченный диапазон масс, мониторинг вы- бранных ионов ческих ионов(пред- почтительно иметь молекулярные ионы). Точность измерения массы не менее 5 ppm. По крайней мере один ион-фрагмент табл. 3.3
МС/МС Тройной квадру- поль, ионная ло- вушка, гибридный прибор (квад.- ВПМС, квад.- ионная ловушка) Мониторинг выбран- ной/выбранных реак- ций, полный спектр ионов-продуктов Не менее 2 ионов-продуктов См. табл. 3.3
Таблица 3.3. Допустимые по умолчанию отклонения точности относительных ионных
интенсивностей с точки зрения валидации метода и контроля анализа пе-
стицидов в продуктах питания и кормах (из SANCO/10684/2009A)
Относительная интенсивность (% от максимального пика в спектре) ГХ/МС с ИЭ (%) ГХ/МС с ХИ; ГХ/МС"/ЖХ/МС; ЖХ/МС" (%)
>50% ± 10% ±20%
> 20-50% ± 15% ±25%
> 10-20% ±20% ±30%
< 10% ±50% ±50%
остаточных количеств соединений в пищевых продуктах, традиционно используются
для подтверждающего анализа в других областях, например экологии и токсикологии.
Для количественного анализа важны и другие параметры, например качество бланка
(отсутствие привнесенного загрязнения) и линейность калибровки.
3.9. Целевые подходы с использованием тандемной
квадрупольной масс-спектрометрии
Целевые методы разрабатываются для проведения количественного анализа для зара-
нее определенного списка компонентов. Прибор функционирует в режиме МЗР, при
этом детектируются только молекулы, включенные в метод. Другие молекулы не будут
регистрироваться, даже если они присутствуют в пробе в высокой концентрации.
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
Таблица 3.4. МС/МС-методы определения фармацевтических препаратов в поверхност-
ной воде. Представлен типичный МС/МС-метод для подтверждающего ана-
лиза. В таблице указаны МЗР-переходы совместно с напряжением на конусе
(этот параметр источника необходим для оптимизации чувствительности)
и энергией столкновений. Подтверждающие переходы могут иметь другую
энергию столкновений
Компонент CV/CE MRM 1 (количеств.) CV/CE MRM 2 (подтвержд.)
5-аминосалициловая кислота 26/15 153,9 > 136,0 26/20 153,9 > 108,0
Метронидазол 26/15 171,9 > 127,9 26/23 171,9 > 81,9
Парацетамол 26/16 151,9 > 110,0 26/24 151,9 >92,9
Амоксициллин 26/28 365,9 > 113,9 26/15 365,9 > 159,9
Ранитидин 26/17 315,9 > 176,0 26/24 315,9 > 123,9
Сальбутамол 26/20 240,0 > 148,0 26/10 240,0 > 222,1
Атенолол 34/19 266,9 > 190,1 34/25 266,9 > 145,0
Сульфапиридин 26/16 249,9 > 156,0 26/16 249,9 > 184,0
Цимитидин 26/15 252,9 > 152,0 26/15 252,9 > 117,0
Кодеин 45/25 299,9 >214,9 45/4 299,9 > 224,9
Габапентин 26/10 172,2 > 154,1 26/10 172,2 > 137,0
Амфетамин 18/10 135,9 > 119,0 18/16 135,9 >90,9
Триметоприм 42/22 290,9 > 230,0 42/22 290,9 > 123,0
Бензоилэкгонин 30/25 289,9 > 168,1 30/18 289,9 > 104,9
Ципрофлоксацин 35/17 332,0 >313,9 35/17 332,0 > 288,0
Сул ьфаметоксазол 26/16 253,9 > 156,0 26/21 253,9 > 107,9
Трамадол 15/15 264,1 > 246,0 15/15 264,1 >57,8
Кокаин 34/22 303,9 > 182,1 34/22 303,9 >81,9
Метопролол 35/17 268,1 > 115,9 35/20 268,1 >97,9
Хлорамфеникол 20/10 323,0 > 274,8 20/10 323,0 > 304,8
Доксициклин 30/17 445,1 > 427,9 30/25 445,1 >409,9
Пропранолол 34/18 259,9 > 183,1 34/16 259,9 > 116,0
Карбамазепин 26/19 236,9 > 194,1 26/19 236,9 > 192,1
Эритромицин-Н2О 26/15 716,4 > 558,2 26/34 716,4 > 158,1
Дилтиазем 35/20 415,0 > 178,0 35/20 415,0 >310,0
Валсартан 20/15 436,6 > 234,9 20/15 436,6 > 290,9
Амитриптилин 30/20 278,0 > 233,0 30/20 278,0 > 191,0
Симвастатин 25/10 419,0 > 284,9 25/10 419,0 > 199,0
Кофеин-б9 (1,3,7-триметил-б9) 34/16 204,0 > 144,0 — —
Фенацетин-этокси-1—|3С 34/15 180,9 > 139,0 — —
CV — напряжение на конусе, В; СЕ — энергия столкновений, эВ.
3.9. Целевые подходы с использованием тандемной квадрупольной
масс-спектрометрии
Табл. 3.4 демонстрирует МС/МС-метод обнаружения фармацевтических препара-
тов в поверхностных слоях воды (Kasprzyk-Hordern и др., 2007), а на рис. 3.12 представ-
лены хроматографические пики компонентов. Данный метод был создан для масс-
спектрометра Quattro Micro (Waters). Действующие вещества лекарственных препаратов
были экстрагированы из 1 л воды при концентрации 100 нг/л. Экстракт был сконцен-
трирован в 2000 раз. Объем инжекции составил 10 мкл, что соответствует анализу 20 мл
исходной воды. Для большинства компонентов селективность была превосходна и де-
тектирование пиков не представляло проблемы.
Когда список веществ достаточно длинен, используется термин «целевой скри-
нинг». Подтверждающий метод можно разработать, если число компонентов не превы-
шает 200. Для подтверждения идентичности компонентов используется второй пере-
ход МЗР. При этом можно определять количественно концентрации анализируемых
веществ. Программное обеспечение для обработки больших массивов данных должно
быть разработано в соответствии с директивами SANCO.
Дальнейшее увеличение числа компонентов возможно только при использовании
одного МЗР-перехода для каждого вещества (количество экспериментальных точек
F1 :MR М of 4 channels ,ES+
153.87>135 98
8 509е-Ю04
I । । । । I min
10(
Ciprofloxacin 11/^>12 16
10.89
F16.MRM of 2 channels. ES
332.00>313
2 494e+O0
F1 :MRM of 4 channels, ES+
171.910>127.940
4.734e*OO5
F18 MRM of 4 channels ,ES
Sulfamethoxazole_ 253.88>155.9
10.35 | 9.966e*O0
-j— min
%•
Paracetamol
4.89
F2 :MRM of 2 channels ,ES+
151.91 >109.95
1 289e+OO5
F18.MRM of 4 channels,ES
Tramadol 264.10>246.0
1.994e+00
V 98 13 96
8.57J0 64'
Amoxicillin
F3:MRMof2 channels,ES+
365.92>113.91
1.387e*OO4
10C
F20MRMof 2 channels,ES
Cocaine_ 303.89>182.0
11 46 I 5035e+00
Ranitidine F4:MR M of 2 channels ,ES+
6 08 315.86>176.00
I 7 883e*004
7 J 9
i | I Hi I IA| I I I I I I I [ I I I I I... mm
2 5 5.0 7 5 10.0 12.5 15 0 17 5
F19 MRMof 2 channels,ES
Metoprolol 268.10>115
11.45 ~l 1.273e+00
%-
25
50
7 5 10.0 12.5 15 0 17.5
Salbutamol
5 67
F5:MRM of 2 channels, ES+
239.98>147.98
2.575e+006
10(
%-
mm
F21 MRMof 2 channels,ES
Phenacetin-ethoxy-1-13C 180.94>139.0
12 00 "j 3.000e+00
Рис. 3.12. Хроматограммы компонентов из табл. 3.4. (Перепечатано с разрешения изда-
тельства Elsevier из Journal of Chromatography А, 1161, Kasprzyk-Hordern В., Din-
sdale R. M. and Guwy A. J. Multi-residue method for the determination basic/neutral
pharmaceuticals and illicit drugs in surface water by solid-phase extraction and utra per-
formance liquid chromatography — positive electrospray ionization tandem mass spec-
trometry, 132—145)
92
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
Рис. 3.13. Целевой скрининг пестицидов на тандемном квадрупольном приборе. Показа-
но разделение 400 пестицидов за 10 минут. Для подтверждения результатов про-
водится повторный анализ с регистрацией подтверждающих переходов. (Пере-
печатано с разрешения издательства Elsevier из Journal of Chromatography А, 1161,
Kasprzyk-Hordern В., Dinsdale R.M. and Guwy A.J. Multi-residue method for the
determination basic/neutral pharmaceuticals and illicit drugs in surface water by solid-
phase extraction and utra performance liquid chromatography — positive electrospray
ionization tandem mass spectrometry, 132—145)
колонка В
Рис. 3.14. Схема онлайн-концентрирования, которое позволяет параллельно подготавли-
вать несколько образцов. Проба объемом до 15 мл загружается на экстракцион-
ную колонку. На этапе промывки удаляются наиболее полярные компоненты.
В ходе хроматографического разделения первой пробы вторая проба загружа-
ется на другую экстракционную колонку, первая же в это время промывается
сильным органическим растворителем и уравновешивается. Система также по-
зволяет проводить прямой ввод проб через инжектор ЖХ. Такая конфигурация
может также работать и для последовательного анализа проб
3.9. Целевые подходы с использованием тандемной квадрупольной
масс-спектрометрии
на каждый пик не должно быть менее 10). В таком случае подтверждение достигается
повторным анализом.
Требуемые пределы обнаружения для проб воды обычно составляют несколько ppt.
Достижение таких уровней достигается концентрированием проб (рис. 3.13) или при ис-
пользовании наиболее чувствительных масс-спектрометров. В последнем случае кон-
центрирование не является обязательным.
Концентрирование проб проводится на обращенно-фазовых картриджах при исполь-
зовании больших объемов проб (до 1 л) (Brouwer, 2010). Исследуемые компоненты элюи-
руются органическими растворителями с последующим выпариванием и растворением
в мобильной фазе. В прибор инжектируется часть полученной пробы. Фактор концен-
трирования обычно составляет 500—1000 раз. Проблемы такого подхода связаны с транс-
портировкой больших емкостей с водой, значительными трудозатратами и стоимостью
картриджей. Описанный метод имеет и определенные преимущества, например гибкость
(можно включить очистку проб в процедуру их концентрирования), возможность анали-
за концентрата разными методами (ГХ/МС, ЖХ/МС). С улучшением чувствительности
приборов необходимый для анализа объем воды может снизиться до 50 мл.
Альтернативным подходом является онлайн-преконцентрирование (рис. 3.14), ког-
да весь образец загружается на колонку. Метод был разработан несколько лет назад для
экологического контроля с применением ВЭЖХ-систем, оснащенных оптическими
и электрохимическими детекторами (Brouwer, 2010). Метод по-прежнему широко ис-
пользуется. Он характеризуется очень низкими пределами обнаружения при использо-
вании меньших объемов инжекции (1 — 15 мл, в зависимости от прибора). В некоторых
случаях используются сменные картриджи, в других — короткие предколонки. Смен-
ные картриджи снижают риск загрязнения системы анализируемыми веществами.
100
Карбендазим
S/N 10415
Метолахлор
S/N 3916
Атразин
S/N 744
6,00 7.00 8.00 9.00 10.00 11,00 12,00 13.00 14.00 15.00
Время
Рис. 3.15. Чувствительность, достигнутая с помощью твердофазной онлайн-очист-
ки. Масс-хроматограммы 10 ppt пестицидов (инжекция 5 мл пробы в систему
со средними характеристиками). Даны отношения «сигнал/шум» (пик-к-пику)
для каждого сигнала без предварительной обработки спектра. Чувствитель-
ность более чем достаточна для большинства приложений
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
При использовании предколонок в последовательность анализа необходимо включать
дополнительный этап промывки.
Преимуществами этого анализа, очевидно, являются автоматизация и чувстви-
тельность. Крайне низкие пределы обнаружения могут быть достигнуты даже на систе-
мах среднего уровня (рис. 3.15). Дополнительным преимуществом является отсутствие
необходимости транспортировки и хранения больших емкостей с пробами, что делает
доставку образцов более простой и дешевой. Недостатки связаны со сложностью разра-
ботки универсальных для различных групп соединений методик, а также с обеспечени-
ем хорошего извлечения для этих анализируемых веществ. По этим причинам твердо-
фазная онлайн-экстракция применяется для специфических групп веществ, например
для кислотных гербицидов. Другой проблемой является вредное воздействие водных
проб на колонку, связанное, например, с высокой концентрацией гумусовых кислот.
Некоторые группы исследователей показали, что инжекция больших объемов проб
обеспечивает низкие пределы обнаружения даже для достаточно простых систем (Chi-
aia и Field, 2008). В упомянутой работе сообщается, что пределы обнаружения на уровне
ppt могут быть достигнуты даже на достаточно простых приборах и при анализе про-
блематичных образцов, как, например, сточные воды. Объем инжекции составляет все-
го лишь 4,5 мл. Вся пробоподготовка заключается в центрифугировании со скоростью
6000 об./мин в течение 30 мин.
Simetryn
253>171
214>124
8 ................
3 40 3 50 3 60
4 00 4 10
Время Время
Рис. 3.16. Прямой ввод питьевой воды с добавленной в нее смесью 0,2 ppt пестицидов
в УЭЖХ/МС-систему высокого уровня. Регистрируются как количественные,
так и подтверждающие переходы. Объем инжекции — 100 мкл
3.11. Почему приборы типа QTOFиспользуются для экологического контроля?
При использовании наиболее чувствительных систем возможен анализ питьевой
воды методом прямой инжекции. Объем инжекции для УЭЖХ/МС-систем составляет
около 100 мкл. Пробоподготовка фактически сводится к фильтрации и центрифугиро-
ванию. Другими преимуществами является минимизация расходов на преконцентри-
рование и оплату труда, простота в транспортировке проб и быстрота анализа. Также
устраняются дискриминации, связанные с процессом пробоподготовки. На рис. 3.16
представлены хроматограммы анализа питьевой воды, в которую была добавлена смесь
из 80 пестицидов в концентрации на уровне 0,2 ppt. Один переход был использован для
количественного анализа, а другой — для подтверждения результатов. Хотя объем ин-
жекции составлял лишь 100 мкл, все пестициды были легко идентифицированы.
Анализ непитьевой воды может быть затруднен из-за подавления сигнала или дру-
гих матричных эффектов, которые делают прямой анализ проблематичным. В таких
случаях необходима дополнительная пробоподготовка. При использовании очень чув-
ствительных систем можно ограничиться фильтрацией и центрифугированием в целях
очистки пробы от твердых частиц. Также можно использовать разбавление образца, что
является самым простым способом устранения матричных эффектов.
3.10. Скрининговые и исследовательские методы
с использованием масс-спектрометров типа
QTOF
Времяпролетные и гибридные квадрупольно-времяпролетные (QTOF) приборы дают
информацию о полном спектре с частотой 20—30 спектров в секунду, что идеально со-
четается с УЭЖХ. Такие масс-спектрометры обеспечивают измерение масс с высокой
точностью, что делает возможным получение сведений об элементном составе исследу-
емых компонентов (Ballogh, 2009). При использовании геометрии QTOF (рис. 3.17) мож-
но также фрагментировать ионы в ячейке столкновений. Полученные ионы-фрагменты
могут быть использованы для получения структурной информации. Первое поколение
приборов имело ограниченный диапазон линейности. Поэтому они в основном исполь-
зовались лишь для качественного анализа. Новейшие приборы, имеющие широкий
динамический диапазон (более 4 порядков), могут применяться и для количественного
анализа.
3.11. Почему приборы типа QTOF используются для
экологического контроля?
QTOF особенно ценен в качестве масс-спектрометра, используемого для быстрых скри-
нинговых и исследовательских целей, причем новейшие приборы этого типа могут при-
меняться как для качественного, так и для количественного анализа.
Между тандемными квадрупольными и времяпролетными приборами существует
фундаментальная разница в способе регистрации данных: МЗР-хроматограмма явля-
ется результатом прямой регистрации ионов, прошедших фильтрацию в квадруполе.
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
Кран для ввода
калибровочного
раствора
Спрей
калибровочного
раствора
Спрей
анализи-
руемого
раствора,
Запорный
кран и
сменный
конус
отбора
пробы
Проводник ионов
на основе Т-волны Линза
Разрешающий
квадруполь
Турбомолекулярный насос
с воздушным охлаждением
Безмасляный
роторный
насос
Т ранспортирующая
Ячейка оптика
столкновений
с Т-волной
гГТпППП
птшшггптлпг
......IIIIIIIIII
Рефлектрон 11 ► □
Детектор
Выталкивающий
электрод
Рис. 3.17. Геометрия прибора QTOF (квадрупольно-времяпролетного). В такой геометрии
квадруполь может изолировать ион для его последующей фрагментации в ячей-
ке столкновений. Ионы-продукты анализируются времяпролетным анализа-
тором. ВП может работать в режиме V, как показано на рисунке, или в режиме
W, если используется второй рефлектрон. Режим W обеспечивает лучшее разре-
шение при худшей чувствительности. Для целей скрининга первый квадруполь
работает в режиме пропускания всех ионов
Тогда как экстрагированная в узком окне масс-хроматограмма, полученная с помощью
времяпролетного масс-спектрометра, представляет собой сигнал, обработанный после
записи спектра. Тандемная квадрупольная масс-спектрометрия предназначена для де-
тектирования заранее определенных компонентов, тогда как времяпролетные приборы
предоставляют информацию о полном масс-спектре, которая в последствии подлежит
дальнейшей обработке. Таким образом, нецелевой компонент не может быть обнару-
жен при помощи тандемного квадрупольного прибора, работающего в режиме МЗР,
тогда как времяпролетный масс-спектрометр зарегистрирует этот компонент. Следо-
вательно, эффективность времяпролетного прибора строго зависит от программно-
го обеспечения, которое ответственно за экстракцию, фильтрацию и интерпретацию
данных. Идентификация компонентов осуществляется при помощи библиотечного
поиска по базам данных с точными значениями масс с использованием изотопных со-
отношений и времен удерживания (когда эта информация доступна). Эксперименты,
зависящие от полученных данных, могут быть осуществлены переключением в режим
МС/МС при обнаружении известного иона-предшественника в случае проведения це-
левого анализа. Полученный тандемный спектр может быть использован для подтверж-
дения идентификации. Главным ограничением для применения времяпролетного
прибора в целях скрининга является пробоподготовка. Одним из главных достоинств
использования приборов типа TOF и QTOF считается возможность проведения повтор-
3.11. Почему приборы типа QTOFиспользуются для экологического контроля?
ной обработки данных в целях обнаружения компонентов, пропущенных при первом
заходе. Если анализ проводится при помощи тандемных квадрупольных приборов, то
единственным способом обнаружить новые компоненты является изменение метода
с последующим повторным анализом.
Быстрый скрининг. Высокая скорость регистрации данных во времяпролетной
масс-спектрометрии делает возможным использование преимуществ УЭЖХ. Полу-
ченные данные обрабатываются, после чего проводится идентификация компонентов
с поиском по библиотечным базам данных. Идентификация основывается на измере-
нии точной массы, изотопных соотношений, а также на МС/МС-данных и времени
удерживания (когда такая информация доступна). Селективные масс-хроматограммы
могут быть получены в узком диапазоне масс. Интерпретация результатов значительно
упрощается, если точность и стабильность измерения массы достаточно высоки, чтобы
проводить экстракцию ионов в очень узком окне. Такой подход позволяет уменьшить
число ложноположительных результатов (рис. 3.18). Идентифицированный компонент
100-]
#-
0-
2 15
1 00 2 00
0
Monuron
m/z 199.0638
100 n
0
Monuron
m/z 199.0638
3.51
3 00 4 00
0 ..... II11 FlII | Ml11*1 R | Rl*P|l Wl fFRFF
1 00 2 00 3 00 4 00
215 Fenazox
m/z 199.0871
1 64
1 26
1 00 2 00
2 59 3 51
‘Mfbl ill mfrfl 11 ll 111
‘ 3 00 4 00
Fenazox
1OO1 m/z 199.0871
0-Lo|IWI1WWfl*>|4IM| l>4 . IM | I . U| I II
100 2 00 3 00 4 00
215 Cymoxamil
m/z 199.D831
Cymoxamil
m/z 199.0831
164
126^ 2?9 3 51
1 00 2 00 3 00 4 00
Время
Рис. 3.18. Демонстрация снижения числа ложноположительных результатов. В экспери-
ментах быстрого скрининга на ВП- и Q-ВП-приборах часто встречается ситуа-
ция, когда компоненты имеют одинаковые массы и не разделены хроматографи-
чески. При получении экстрагированной ионной хроматограммы в узком окне
число ложноположительных результатов значительно уменьшается, (а) Окно
экстракции 50 мДа; (б) окно экстракции 3 мДа
100-]
Время
0
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
будет количественно обработан при помощи калибровочной кривой, если такая кривая
существует.
Исследовательский анализ. Так как времяпролетный масс-спектрометр предостав-
ляет полный масс-спектр, с его помощью можно обнаружить совершенно неожидан-
ные компоненты, не присутствующие в базе данных. Процесс, описанный в недавно
опубликованных технических заметках (Waters, 2010), может быть обобщен следующим
образом:
1. Шаг 1: получите полный комплект данных УЭЖХ/МС и МС/МС. Специфиче-
ский режим анализа MSE, поддерживаемый специальным программным обе-
спечением (запатентовано компанией Waters), позволяет получить все МС-
Рис. 3.19. Подтверждение компонента с использованием тандемного спектра и точной
массы. На основании низкоэнергетического тандемного спектра (не показан)
при использовании точной массы и высокоэнергетического тандемного спек-
тра можно сделать вывод, что наиболее вероятная причина отравления пти-
цы — карбосульфан. Программа MassFragment моделирует возможные каналы
фрагментации карбосульфана (измеренная масса 381,2216) и сравнивает массы
теоретических фрагментов с экспериментальными. Если эти массы совпадают
в пределах 2 ppm, результат идентификации можно считать достоверным
3.12. Исследовательская работа по изучению продуктов трансформации
и МС/МС-данные в ходе одного анализа. Преимущество подхода, очевидно,
состоит в использовании лишь одной инжекции. Однако следует также отметить
возможность повторной обработки полученных ранее данных.
2. Шаг 2: выполните целевой поиск по библиотекам с использованием точных зна-
чений масс, изотопных карт ионов-продуктов и времен удерживания и создайте
список обнаруженных компонентов. Этот метод подобен быстрому скринингу.
3. Шаг 3: найдите неидентифицированные пики среди комплексных сигналов.
Программное обеспечение выполнит деконволюцию пиков и выделит хрома-
тографические пики. Пики, не присутствующие в стандартных образцах, пред-
ставляют наибольший интерес.
4. Шаг 4: идентифицируйте пики. Идентификация заключается в определении эле-
ментного состава при помощи значений точных масс, изотопных соотношений
и МС/МС-данных. Элементный состав будет представлен коротким списком воз-
можных опций. Программное обеспечение может теоретически моделировать
фрагментацию. Сравнение теоретической фрагментации с экспериментальной
поможет однозначному определению компонентов (рис. 3.19).
5. Шаг 5: добавьте идентифицированные в образце или описанные в литературе
компоненты в библиотеку.
6. Шаг 6: обработайте ранее записанные данные в целях обнаружения новых иден-
тифицированных компонентов.
3.12. Исследовательская работа по изучению
продуктов трансформации
Кроме определения хорошо известных экотоксикантов, необходимо регистрировать
продукты их трансформации, так как они могут оказаться более токсичными, чем роди-
тельские молекулы. В качестве примера возьмем фармацевтические препараты, которые
создают повод для растущего беспокойства о чистоте поверхностных, подземных вод,
а также питьевой воды. Пути метаболизма препаратов в организме человека и организ-
мах животных хорошо известны, однако их трансформация в окружающей среде все еще
является предметом изучения. Обычно исследования проводятся с помощью сочетания
ЖХ/МС/МС-анализа на масс-спектрометрах высокого разрешения типа Orbitrap или
QTOF и дополнительного анализа другими методами, например ЯМР. Исследование
обычно проводят для модельной среды — воды с осадочными слоями, находящимися
в анаэробных условиях. В один из образцов добавляется ксенобиотик, тогда как другой
служит бланком. Образцы анализируются с помощью системы ЖХ/масс-спектрометр
высокого разрешения. Возможный подход заключается в нахождении пиков, отсут-
ствующих в бланке. Другой подход заключается в составлении списка возможных
продуктов трансформации (исходя из химических структур исходных ксенобиотиков
и возможных химических реакций) с указанием элементного состава и точных масс со-
единений. По этому списку осуществляется поиск в образце с экотоксикантом компо-
нентов, отсутствующих в бланке при установленном пороге интенсивности сигнала. Та-
кие компоненты могут рассматриваться как возможные продукты трансформации (Kern
и др., 2009; Kosjek и др. 2008; Perez и Barcelo, 2010; Perez и др., 2007). Эти два подхода могут
Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
быть объединены. Могут быть использованы дополнительные возможности идентифи-
кации, например предсказанные времена удерживания. Так как масс-спектрометрия
обычно не помогает в различении изомеров, необходимо дополнительное подтверж-
дение. Такой эксперимент может быть осуществлен с использованием ЯМР-анализа
продуктов трансформации, выделенных с помощью преперативной ЖХ, или анализа
чистого стандарта (когда тот доступен). ЯМР обеспечит окончательное подтверждение
структуры изомерных продуктов трансформации.
Новейшие разработки в области приборостроения дают возможность разделения
изомеров методом спектрометрии ионной подвижности (Synapt, Waters). Спектрометры
ионной подвижности могут дифференцировать ионы одной и той же массы (изомеры)
в зависимости от их размеров и формы. Например, с помощью этого метода была осу-
ществлена однозначная идентификация изомеров фармацевтических препаратов (Dear
и др., 2010). Приложение этой методологии к характеризации продуктов трансформации
также возможно.
В целом процесс идентификации продуктов трансформации чрезвычайно сложен
и длителен. Качество масс-спектрометрической информации является критическим
фактором, особенно это касается точности измерения масс и изотопных соотношений.
Программное обеспечение для деконволюции спектров и моделирования возможных
структур также играет важную роль.
3.13. Ближайшие и долговременные перспективы
За последние 20 лет (со времени изобретения методов ввода при атмосферном давлении)
ЖХ/МС-методы завоевали ведущее положение в области экологического мониторинга.
Это стало возможным благодаря достижениям в разработке приборов и программно-
го обеспечения. Современные приборы намного проще в эксплуатации, намного более
чувствительны и надежны. 20 лет назад был необходим большой экспериментальный
опыт оператора для проведения ЖХ/МС-анализа на очень дорогостоящих приборах.
Позже ЖХ/МС-метод был пересмотрен вместе с его преимуществами и недостатками,
что позволило существенно ускорить разработку методов. Сегодня можно с уверенно-
стью сказать, что квадрупольные системы являются хроматографическими детектора-
ми, которые могут использоваться всеми лабораторными химиками.
Очевидно, что метод все еще развивается. Современные тандемные квадрупольные
приборы дают возможность получения данных о матрице образца методом МЗР. Это ин-
тересно с точки зрения понимания аналитических проблем, связанных с матричными
эффектами. QTOF-приборы при использовании соответствующего программного обе-
спечения позволяют проводить количественный анализ сотен компонентов наряду
со скринингом неожидаемых или совсем неизвестных веществ. Сейчас приборы такого
типа приобретаются в основном как дополнение к тандемным квадрупольным масс-
спектрометрам. Однако ожидается, что в течение нескольких последующих лет они бу-
дут широко использоваться для целей скрининга.
Непрерывное улучшение чувствительности приборов облегчит процесс анализа.
На сегодняшний день требуемые пределы обнаружения могут быть сравнительно лег-
ко достигнуты при соответствующей пробоподготовке. Лучшая чувствительность све-
Литература
дет пробоподготовку к простому разбавлению образца в целях уменьшения влияния
матрицы.
Новые разработки также будут связаны с новыми методами ввода проб. Например,
прямой ввод проб (ASAP, Waters) обеспечивает быстрый скрининг загрязняющих ве-
ществ в продуктах питания. С улучшением чувствительности эти методы, несомненно,
будут использоваться и для экологических целей. Количественный анализ будет прово-
диться только для выявленных в результате скрининга компонентов.
Другой перспективной областью приложения является непосредственный мони-
торинг водных потоков. Такие приложения имели место для ЖХ/УФ-систем, но ни-
когда для МС-детектирования (насколько это известно авторам). Такие подходы будут
полезны для изучения эволюции экотоксикантов в специфических средах, а также для
экстренного выявления внезапного загрязнения вод или даже для мониторинга водных
резервуаров с точки зрения национальной безопасности (Rosal и др., 2009). И, наконец,
может быть достигнута полная автоматизация анализа. Автоматические анализаторы
уже используются для контроля многих экологических параметров. «Черный ящик»
на основе ЖХ/МС будет следующим шагом в анализе органических загрязняющих ве-
ществ воды при низких уровнях обнаружения.
Литература
[1] . Arpino Р. J. (1982). «On-line liquid Chromatography/mass spectrometry? An odd couple!»
Trends in Analytical Chemistry, vol. I, n° 7.
[2] . Ballogh M. (2009). The Mass Spectrometry Primer. Waters Corporation, Massachusetts,
USA.
[3] . Barcelo D., Hennion M.-C. (1999). Trace Determination of Pesticides and their Degradation
Products in Water. Elsevier, Amsterdam, The Netherlands.
[4] . Brouwer E. (2010). On-line Trace enrichment in column liquid chromatography for the de-
termination of organic micropollutants in surface water. Thesis work presented on Feb 2
Free University of Amsterdam, The Netherlands.
[5] . Buth J. M., Steen P., SueperC., Bluementritt, Viekesland P. D., Arnold W., McNeil K. (2010).
Dioxin Photoproducts of Triclosan and Its Chlorinated Derivatives in Sediment Cores. En-
vironmental Science & Technology. 44: 4545—4551.
[6] . Chiaia A., and Field J. A. (2008). Large Volume (1,800 uL) Injection for the Direct Anal-
ysis of Illicit Drugs in Municipal Wastewaters. Environmental Science and Technology. 42,
8841-8848.
[7] . Cole A., Cole R. B. (1997). Electrospray Ionization Mass Spectrometry: Fundamentals, Instru-
mentation, and Applications. Wiley-Interscience, New York, USA.
[8] . Council Directive 98/83/ECof3 November 1998 on the quality of water intended for human
consumption.
[9] . Dear G.J., Munoz-Muriedas J., Beaumont C., Roberts A., Kirk J., Williams J. P., Cam-
puzano I. (2010). Sites of metabolic substitution: investigating metabolite structures utilis-
ing ion mobility and molecular modeling. Rapid Communications in Mass Spectrometry. 24,
21:3157-3162.
102 Глава 3. Жидкостная хроматография/масс-спектрометрия — оптимальный метод
качественного и количественного анализа загрязнений окружающей среды
[10] . Kasprzyk-Hordern В., Dinsdale R.M., Guwy A. J. (2007). Multi-residue method for the
determination of basic/neutral pharmaceuticals and illicit drugs in surface water by solid-
phase extraction and ultra performance liquid chromatography—positive electrospray ioni-
sation tandem mass spectrometry». Journal of Chromatography A, 1161, 132—145.
[11] . Kern S., Fenner K., Singer H. P., Schwarzenbach R. R, Hollender J. (2009). Identification
of Transformation Products of Organic Contaminants in Natural Waters by Computer-
Aided Prediction and High-Resolution Mass Spectrometry. Environmental Science & Tech-
nology. 43, 18: 7039-7046.
[12] . KosjekT., Zigon D., Kralj B., Heath E. (2008). The use of quadrupole-time-of-flight mass
spectrometer for the elucidation of diclofenac biotransformation products in wastewater.
Journal of Chromatography A, 1215 57—63.
[13] . Niessen W. M.A. (2006). Liquid Chromatography-Mass Spectrometry, Third Edition Taylor
& Francis Ltd.
[14] . Perez S., Barcelo D. (2009). First Evidence for Occurrence of Hydroxylated Human Me-
tabolites of Diclofenac and Aceclofenac in Wastewater Using QqLIT-MS and QqTOF-MS.
Analytical Chemistry. 280, 8135—8145.
[15] . Perez S., Eichhorn R, Barcelo D. (2007). Structural Characterization of Photodegradation
Products of Enalapril and Its Metabolite Enalaprilat Obtained under Simulated Environ-
mental Conditions by Hybrid Quadrupole-Linear lonTrap-MS and Quadrupole-Time-of-
Flight-MS. Analytical Chemistry. 79, 8293—8300.
[16] . Rosal C., Betowski D., Romano J., Neukom J., Wesolowski D., Zintek L. (2009). The de-
velopment and inter-laboratory verification of LC—MS libraries for organic chemicals of
environmental concern. Taianta. 79, 3: 810—817.
[17] . Waters technical brief. (2010). Detection and Identification of a Non-Targeted Unknown
Contaminant During ToF Screening for Pesticides. Ref 720003424EN.
ГЛАВА 4
ПРИМЕНЕНИЕ ТАНДЕМНОЙ
МАСС-СПЕКТРОМЕТРИИ ДЛЯ
АНАЛИЗА ЗАГРЯЗНЕНИЙ
ОКРУЖАЮЩЕЙ СРЕДЫ
Дон Бетовски
4.1. Введение
Озабоченность общества состоянием окружающей среды и начало экологическо-
го мониторинга совпали с развитием методологии «газовая хроматография/масс-
спектрометрия» (ГХ/МС). В 1970 году в США было создано Агентство по охране окру-
жающей среды (АООС, ЕРА). Очевидно, что для контроля состояния окружающей среды
были необходимы соответствующие методы анализа. Поэтому компьютеризированная
масс-спектрометрия в сочетании с обеспечивающей предварительное разделение газо-
вой хроматографией оказалась почти идеальным инструментом для решения задач та-
кого рода. Последующие 10—15 лет ГХ/МС-метод был «рабочей лошадкой» мониторин-
га состояния окружающей среды. Он не имел себе равных при контроле специфических,
заранее выбранных веществ. Принимая во внимание эффективное разделение летучих
и полулетучих соединений и воспроизводимую ионизацию, ГХ/МС-методы могут быть
стандартизованы с учетом соответствующих документов, регулирующих содержание
токсичных веществ в окружающей среде. ГХ/МС-методы широко используются и в на-
стоящее время. Однако регулирующие инстанции были обеспокоены тем фактом, что
многие важные полярные и нелетучие соединения не могут эффективно контролиро-
ваться при помощи ГХ/МС. Ранее соединения включались в список контролируемых
веществ именно благодаря возможности их анализа при помощи газовой хроматогра-
фии/масс-спектрометрии. Сейчас такие ограничения устранены.
Для анализа полярных и нелетучих соединений наиболее подходящим методом
представляется жидкостная (в особенности высокоэффективная жидкостная) хрома-
тография. Однако соединение ВЭЖХ с МС было нетривиальной инженерной задачей,
что обусловливало отставание развития этого метода по сравнению с ГХ/МС. После не-
скольких попыток разработать интерфейс, способный удалять элюент ВЭЖХ и вводить
ионы в масс-спектрометр, работающий в условиях вакуума, в конце 80-х был сделан
решающий прорыв. Двумя основными подходами, обеспечившими такой успех, были
термоспрей и его преемник — электроспрей. Эти методы совершили переворот в масс-
спектрометрии, обеспечив возможность анализа полярных и нелетучих загрязнений
окружающей среды (а также биомолекул) (см. главу 3).
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
£l°4
Ключевым моментом является тот факт, что и термоспрей, и электроспрей — ме-
тоды ионизации, которые одновременно обеспечивают и сопряжение ВЭЖХ с масс-
спектрометром. Это важное свойство, так как ГХ/МС-метод рассчитан на ионизацию
молекул, поступающих из хроматографа, электронами (ИЭ). Эта ионизация, протека-
ющая обычно при энергии электронов в 70 эВ, обладает высокой воспроизводимостью
и обеспечивает идентификацию соединения по его характерному масс-спектру (главы 1
и 5). Было создано множество библиотек, с помощью которых при соблюдении стандарт-
ных условий анализа вещества могут быть легко идентифицированы. Подтверждающий
этап в этом случае заключается в анализе стандартного образца идентифицированного
вещества для проверки совпадения времени удерживания (по оси ГХ) и масс-спектра
ИЭ (по оси МС).
Термоспрей и электроспрей ионизируют молекулы в более мягких условиях. Таким
образом, для одного и того же соединения вместо образования множества различных
ионов с получением специфического для данного вещества масс-спектра, как в случае
ионизации электронами, термоспрей и электроспрей обеспечивают генерирование ио-
нов лишь нескольких типов, а зачастую ионов только одного типа с массой, ассоцииру-
ющейся с молекулярной массой исходного соединения. В этом случае идентификация
неизвестных веществ и контроль a priori известных специфических соединений невоз-
можен. Учитывая, что время удерживания при использовании ЖХ не столь воспроизво-
димо и специфично, как в случае ГХ, возникла необходимость дополнительной инфор-
мации об ионах, образованных термо- или электроспреем.
4.2. Тандемные масс-спектрометры
4.2.1. Масс-спектрометрия высокого разрешения
Решение проблемы получения полезной информации об ионах, образующихся
в ЖХ/МС-интерфейсах, было найдено с помощью технологии МС/МС, известной так-
же как тандемная масс-спектрометрия. Данная концепция первоначально была реа-
лизована в масс-спектрометрах высокого разрешения при изучении переноса энергии
столкновений и выделении энергии в процессе диссоциации активированных ионов
(Shukla и др., 1990). Процесс диссоциации, активированной столкновениями (ДАС)
(также называемой диссоциацией, индуцированной столкновениями, ДИС), сыграл
важную роль для создания этих тандемных масс-спектрометров. Упомянутые приборы
высокого разрешения, обычно состоявшие из двух секторных анализаторов (электро-
статического и магнитного), были модифицированы для использования в качестве тан-
демных масс-спектрометров. Один из их анализаторов использовался для фокусировки
выбранных ионов-предшественников, а другой — для разделения ионов, генерирован-
ных в процессе ДАС. Обратная геометрия конфигурации ЕЕбыла применена для первых
МС/МС-экспериментов (Busch and Cooks, 1983). Различные режимы МС/МС в приборах
высокого разрешения могли быть реализованы путем согласованного сканирования
ускоряющего напряжения, магнитного и электростатического полей. Тандемная масс-
спектрометрия первоначально была использована не для жидкостной хроматографии,
а в сочетании с газовой хроматографией, бомбардировкой быстрыми атомами, полевой
ионизацией и другими методами ввода и ионизации соединений.
4.2.2. Тройные квадруполи
Тандемная масс-спектрометрия для экологического мониторинга стала быстро разви-
ваться после создания тройных квадруполей, тогда как приборы высокого разрешения
различных конфигураций использовались в основном для фундаментальных исследо-
ваний. В то время как ГХ/МС в приложении к экологическим задачам преимуществен-
но ограничивалась одиночным квадрупольным фильтром масс, в ГХ/МС/МС- и позже
ЖХ/МС/МС-приборах стали использовать три квадруполя (Yost and Enke, 1978; 1979).
На рис. 4.1 схематично представлена схема тройного квадруполя. Первый квадруполь
настроен на пропускание только ионов с одной величиной m/z. Второй квадруполь
представляет собой ячейку столкновений, работающую исключительно в радиоча-
стотном (RF) режиме. Этот квадруполь помещен в изолированную оболочку так, что-
бы была возможность создания в ней области повышенного давления. Таким образом,
выбранные ионы могут взаимодействовать в ячейке с молекулами (атомами) инертно-
го (как правило) газа. Между первым и вторым квадруполями существует некоторая
разность потенциалов. В результате анализируемые ионы могут получить энергию,
необходимую для их фрагментации. /?Г-режим позволяет транспортировать ионы, об-
разующиеся при столкновениях. Он также обеспечивает фокусировку таких ионов-
продуктов, минимизируя потери вследствие столкновений со стенками ячейки. Ино-
гда для лучшей фокусировки в целях увеличения трансмиссии в ячейке используются
мультиполи высших порядков (гексаполи и октаполи). Третий квадруполь работает
в обычном режиме сканирования напряжения и служит для получения спектра ионов-
продуктов, образовавшихся в ячейке столкновений. Режим, описанный выше, называ-
ют режимом сканирования ионов-продуктов (ранее использовалось название «режим
сканирования дочерних ионов»). Это наиболее популярный режим работы тройных
квадруполей. Другими режимами сканирования являются сканирование нейтральных
потерь и сканирование ионов-предшественников. В режиме сканирования нейтраль-
Источник
ионов
Квадру- Квадрупольная Квадру-
польный камера польный Электронный
фильтр масс столкновений фильтр масс умножитель
массу
ионов
Отсутствует в обычном масс-спектрометре
Рис. 4.1. Рабочая схема тройного квадруполя. Ионы движутся слева направо. Бук-
вы в кружках обозначают молекулярные ионы и их фрагменты. (Перепеча-
тано из Brandt H.R. and Gregg H.R., 1989. Expert system for triple quadrupole mass
spectrometry. Energy and Technology Review, с разрешения Национальной лаборато-
рии имени Лоуренса в Ливерморе)
106 Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
ных потерь первый и третий квадруполи сканируются согласованно со сдвигом по мас-
се, соответствующим массе нейтральной частицы, которая может быть элиминирована
ионом-предшественником при столкновениях (например, т = 28 соответствует потере
ионом молекулы СО). Этот режим помогает обнаружить соединения, теряющие общую
нейтральную частицу. Сканирование ионов-предшественников (ранее этот режим
называли «сканирование родительских ионов») осуществляется путем сканирования
первого квадруполя, в то время как третий квадруполь настроен на пропускание толь-
ко специфических ионов определенной массы. Этот режим позволяет идентифициро-
вать соединения, дающие при столкновениях один и тот же фрагментный ион (напри-
мер m/z 149 для фталатов).
4.2.3. Ионные ловушки
Масс-спектрометры, описанные выше, классифицируются как тандемные (или
МС/МС) в пространстве. Их работа основана на ряде последовательных процессов,
происходящих с ионами при их движении вдоль оси прибора. В противоположность им
существуют масс-спектрометры, характеризуемые как тандемные во времени. Наибо-
лее распространенным видом таких приборов является ионная ловушка. Все процессы
в ней происходят в одном физическом объеме, но они разделены во времени. Как сле-
дует из подписей к рис. 4.2, ионы удерживаются запирающими напряжениями. Гелий,
присутствующий в ловушке, охлаждает ионы так, что они накапливаются в основном
в центре ловушки. Так как и полное сканирование по массам, и МС/МС-эксперименты
осуществляются с помощью одного и того же устройства, функционирование прибора
в режиме МС/МС определяется лишь разработкой соответствующей электроники и ис-
пользованием соответствующих функций сканирования. Присутствующий вдовушке
гелий может быть использован как газ для индуцирования фрагментации.
Говоря коротко, выбранный ион-предшественник изолируется в ловушке, тогда
как все остальные захваченные ионы с другими величинами m/z удаляются либо при
помощи приложения к электродам широкополосных волновых форм (суперпозиции
синусоидальных напряжений всех частот, кроме резонансной частоты изолируемого
иона), либо путем сканирования радиочастотного напряжения. Так как столкновения
между атомами гелия и ионами без разрыва химических связей происходят в ловушке
постоянно, поступательная кинетическая энергия иона-предшественника должна быть
увеличена до значения, при котором химические связи разорвутся в результате возрас-
тания колебательной энергии. Эта кинетическая энергия не должна быть слишком вы-
сокой, иначе ион-предшественник будет выброшен из ловушки. Существуют два пути
увеличения поступательной энергии: нерезонансное и резонансное возбуждение. Нере-
зонансное возбуждение создать проще, но оно не будет обеспечивать селекцию по мас-
сам. Резонансное возбуждение реализовать сложнее, так как оно должно соотноситься
с частотой колебаний иона-предшественника. Тем не менее оно селективно возбуждает
резонансные колебания выбранного иона (Vartan, 2007). Этот процесс можно повторить
для иона, образовавшегося в ходе ДАС и изолированного одним из описанных выше
способов. Образовавшийся ион также может быть резонансно возбужден. Эта процеду-
ра может быть последовательно повторена несколько раз с получением МС"-спектров
(то есть фрагментные ионы из одного МС/МС-эксперимента являются ионами-предше-
4.2. Тандемные масс-спектрометры
Рис. 4.2. Схематическая диаграмма квадрупольной ионной ловушки. (Перепечатано
с разрешения издательства Elsevier из Trends in Analytical Chemistry, 17, 10 Creaser C.
and Stygall J. W. Recent developments in analytical ion trap mass spectrometry. 583—593)
ственниками для последующего МС/МС-эксперимента и так далее (Creaser and Stygall,
1998; de Hoffman, 1996). Ионы, представляющие интерес, выводятся из ловушки и детек-
тируются электронным умножителем.
Если режим сканирования ионов-продуктов может быть повторен несколько раз
для получения МС"-спектров, режимы сканирования нейтральных потерь или ска-
нирования ионов-предшественников в ионной ловушке отсутствуют. Следует под-
черкнуть, что, кроме возможности получения МС"-спектров, процесс ДАС в ловушке
в целом обладает высокой эффективностью (80—90%) (Creaser and Stygall, 1998; Johnson
et al., 1990).
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
4.2.4. Времяпролетная масс-спектрометрия (ВПМС)
В тандемной масс-спектрометрии времяпролетный масс-спектрометр — не отдельный
прибор, а один из анализаторов, служащий для изолирования ионов-предшествен-
ников и анализа фрагментов. ВП-масс-спектрометр в качестве второго анализатора
в тандемном приборе обладает высоким разрешением, хорошей точностью измерения
масс, высокой чувствительностью и быстротой сканирования (Hager, 2004). Когда ВП-
масс-спектрометр используется совместно с квадрупольным анализатором, установ-
ка имеет ортогональную конфигурацию, что позволяет устранить влияние скорости
ионов при их инжекции во времяпролетный анализатор (Guilhaus et al., 2000; Hager,
2004). Такая конструкция дает возможность использовать низкие начальные скорости
ионов, что ведет к высокому разрешению и линейности калибровки шкалы масс. Ко-
нечным результатом является очень хорошая точность измерения масс (не хуже 5 ppm)
(Leeetal., 1999).
ВП-приборы, как правило, используются вместе с квадрупольными фильтрами масс
(то есть как ^-ТОГили qQTOF), хотя могут быть соединены и с трехмерными ионными
ловушками в качестве первого анализатора (Michael et al., 1992). То есть МС"-спектры
могут быть получены до инжекции во времяпролетный анализатор. Такая конструкция
особенно полезна при определении структуры соединений, так как МС" дает спектры
нескольких поколений фрагментных ионов. Обеспечивающий высокое разрешение ВП-
анализатор предоставляет точную информацию о массах фрагментных ионов, что по-
могает детализировать структуру.
4.2.5. Линейные ионные ловушки
Использование масс-спектрометра совместно с жидкостным хроматографом имеет
некоторые недостатки, особенно если в качестве анализатора используются ионные
ловушки. Трехмерная ловушка и ЖХ-интерфейс (например источник электрораспы-
ления) не могут эффективно работать вместе, так как электрораспыление (ЭРИ) гене-
рирует непрерывный поток ионов, а ионная ловушка функционирует в импульсном ре-
жиме и к тому же имеет ограничения по максимальному числу анализируемых ионов.
Последняя проблема является возможной причиной потери чувствительности, а им-
пульсный режим ведет к низкой эффективности использования пробы (Сох et al., 1995;
Wilcox et al., 2002). Для исправления указанных недостатков ионных ловушек была пред-
ложена концепция линейной ионной ловушки (Cha et al., 2000). Во-первых, линейная
квадрупольная система имеет более эффективную систему транспортировки ионов, чем
система электростатических линз. Во-вторых, она может функционировать при более
высоких давлениях (что хорошо для соединения ловушки с интерфейсом ввода ионов
из атмосферы и с точки зрения снижения требований к давлению в масс-спектрометре).
Кроме того, при приложении на вход и выход линейной ловушки запирающих потенци-
алов и подаче на стержни квадруполя переменного напряжения радиочастотного диа-
пазона ионы могут накапливаться вдовушке (Cha et al., 2000). Таким образом, получаем
ловушку большей, чем трехмерная, емкости и без ограничений, вызываемых простран-
ственным зарядом. Амплитуды ионов выбранного отношения массы к заряду могут
быть резонансно возбуждены для эжекции или фрагментации. То есть можно изолиро-
вать ионы аналита, игнорируя ионы матрицы. В результате получаем улучшенное отно-
4.2. Тандемные масс-спектрометры
1,0 торр
7,0 мторр
0,75 мторр
Экрани- Отверстие Скиммер Qo
рующий для отбора
электрод пробы
Межквадру-
польная
линза (IQ)
Ц L2 Трехмерная
ионная ловушка
Рис. 4.3. Схема линейного квадрупольного интефейса. /0-отверстие (межквадрупольное)
является входом, a Lx — выходным отверстием квадруполя Qv Линза £2 фокуси-
рует ионы в трехмерную ловушку. (Перепечатано с разрешения Американского
химического общества из Cha В., Blades М. and Douglas D.J. (2000). «Ап interface
with a linear quadrupole ion guide for an electrospray-ion trap mass spectrometry system».
Analytical Chemistry. 72, 22: 5647—5654)
Линза 1
У|а |-Короткие стержни
Вторично-
электронный
умножитель
(ВЭУ)
- Апертура
Скиммер
V3
Конус
Проводник
Компенсатор
Длинные стержни
с приложенным
к ним радиочастот-
ным напряжением
Ограничитель
индуктивности
с приложенным
к ним радиочастот-
ным напряжением
и
Vlb
Блок
питания
ВЭУ
Линза 2
V2
Отклоняющая
линза
УЗ _|_0 Линза 3
Спрей
бар
Охлаж-
дение
ионов
Линзы для транс-
портировки ионов
ю-'
мбар
10
мбар
Ю“4
мбар
Аккумулирующий
квадруполь
2
мбар
10“2
мбар
10“5
мбар
Источ- Ц
ник
ионов
0
Зонд
Рис. 4.4. Схема Орбитрэпа с электрораспылением: а — вид сбоку; б — вид спереди. (С раз-
решения Американского химического общества из Hardman М. and Makarov А. А.
(2003). «Interfacing the orbitrap mass analyser to an electrospray ion source». Analytical
Chemistry. 75, 7: 1699—1705)
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
шение сигнала к шуму и разрешение при практически 100%-й эффективности исполь-
зования ионного сигнала (Senko et al., 1997). Линейная ионная ловушка может служить
первым анализатором в тандемном приборе, особенно с электроспрейным источником.
Например, линейная ловушка использовалась в тандеме с трехмерной (см. рис. 4.3).
И наоборот, линейная ловушка использовалась как последний анализатор в приборе
с квадруполем для изолирования ионов-предшественников и фрагментацией в ячейке
столкновений (Hager and Le Blanc, 2003). Для такой конфигурации сообщалось об уве-
личении чувствительности более чем в 500 раз по сравнению со стандартным тройным
квадруполем (Hager and Le Blanc, 2003).
4.2.6. Орбитрэп
Относительно новым прибором, использующим удерживание ионов в анализаторе для
получения спектров, является орбитальная ловушка (орбитрэп). Недостаточная разре-
шающая способность трехмерной ловушки и слишком высокая цена на другие приборы,
использующие удерживание ионов (например МС ИЦР ПФ), мотивировали разработку
масс-спектрометра типа орбитальной ловушки, основанного на динамическом удержи-
вании ионов в электростатических полях (Makarov, 2000). Так как для работы орбитрэпа
необходимо накопление ионов во внешних устройствах с последующим импульсным
вводом, он (так же как и трехмерная ловушка) использует преимущества включения
в ионно-оптическую схему линейных ловушек, расположенных между источником ЭРИ
и самим анализатором орбитрэпа (Hardman and Makarov, 2003) (см. рис. 4.4). В комби-
нации с линейной ловушкой орбитрэп может обеспечить разрешающую способность
150000 на половине высоты пика, точность измерения массы 1—3 мд для массового диа-
пазона до 2000 Да (Hardman and Makarov, 2003).
4.3. Приложения МС/МС к экологическому
мониторингу
Одним из первых приложений МС/МС к анализу загрязнений окружающей среды был
экспресс-скрининг с использованием различных режимов тройного квадруполя (Hunt
et al., 1985). Вместо стандартной экстракции, концентрирования и разделения при по-
мощи хроматографии Хант с коллегами помещал твердые матрицы и замороженные
водные образцы на подложке из стекловолокна в стеклянные трубки с последующим
нагреванием через твердотельный зонд. В результате был создан МС/МС-метод, состо-
ящий из одного эксперимента в режиме сканирования ионов-предшественников и семи
экспериментов в режиме сканирования нейтральных потерь в целях обнаружения при-
оритетных загрязняющих веществ. Им удалось сократить время полного анализа с не-
скольких часов до 25 минут. Такой подход оказался удобным для скрининга соедине-
ний, содержащихся в относительно чистых (простых) образцах. Однако необходимость
в дериватизации и трудности с анализом грязных, комплексных образцов свели этот
метод к использованию лишь в научных целях.
Применение тандемной масс-спектрометрии в ионных ловушках с ионизацией
электронами было продемонстрировано в двух работах (Nourse et al., 1992; Pyle et al.,
4.3. Приложения МС/МС к экологическому мониторингу
1997). Оба исследования заключались в мониторинге полициклических ароматических
углеводородов — группы соединений, тщательно контролируемых Агентством по охра-
не окружающей среды США. Нуре с коллегами использовали возможности МС" для из-
учения различных реакций пирена и антрацена в режиме ДАС. Таким способом авторы
хотели установить направления фрагментации обоих соединений и продемонстриро-
вать возможность установления структур с перспективой использования этого метода
для расшифровки структур неизвестных компонентов. Так как эти вещества вводились
в твердом виде, материала было достаточно для выполнения последовательных МС/МС-
экспериментов множество раз. Авторы смогли показать возможность осуществления
МС"-реакций вплоть до 10 раз (п = 10). Пайл с соавторами вводили образцы методом ГХ.
Они продемонстрировали применение резонансных и нерезонансных режимов ионной
ловушки. В резонансном режиме частота приложенного напряжения совпадала с ча-
стотой собственных колебаний изолированного иона-предшественника и полученные
фрагментные ионы образовывались только из него. В нерезонансном режиме частота
напряжения не совпадала с частотой собственных колебаний иона. Поэтому возбужде-
ние не было селективным и ионы-продукты также могли в свою очередь диссоцииро-
вать, учитывая наличие многократных столкновений. В спектрах, полученных в этих
экспериментах, наблюдалось большее число различных фрагментных ионов (Pyle et al.,
1997). Отличный пример для обоих режимов в сравнении с ДАС в тройном квадруполе
для пирена представлен на рис. 4.5.
Появление эффективных и надежных интерфейсов соединения масс-
спектрометров с жидкостными хроматографами сделало использование тандемной
масс-спектрометрии неизбежным. Так как ЖХ-интерфейсы с термоспреем, а сейчас
и с электроспреем обеспечивают неразрушающие для ионов условия ввода, необходимо
использовать ДАС для получения структурной информации. Эти режимы ДАС — не-
отъемлемая часть функций тандемных масс-спектрометров, изобретенных незадолго
до появления ЖХ-интерфейсов.
Первое применение тандемной масс-спектрометрии для контроля состояния
окружающей среды было осуществлено на тройном квадруполе с термоспреем в целях
идентификации красителей (Ballard and Betowski, 1986; Betowski and Ballard, 1984). Кра-
сители, выпускаемые промышленностью в большом объеме, представляют интерес,
так как в процессе их производства используются вредные химические соединения,
а также могут образовываться опасные побочные продукты. Красители не анализиро-
вались рутинным образом с помощью ГХ/МС, так как они нелетучи и имеют ионную
структуру. Напротив, термоспрей успешно использовался для анализа нескольких
классов красителей, молекулы которых вводились в масс-спектрометр в виде ионов.
Позже обнаружилось, что электрораспыление является более подходящим методом
ионизации для большинства красителей (Bruins et al., 1987а; Bruins et al., 1987b). В своей
ранней работе 1984 года Бетовски и Баллард обнаружили методом термоспрея неиз-
вестный краситель и несколько его потенциальных предшественников (Betowski and
Ballard, 1984). С помощью тройного квадруполя авторы получили спектры ДАС ионов
МН+ и смогли установить структуру исходного красителя и его предшественников,
которые присутствовали в составе анализируемого продукта. Можно считать эту ра-
боту одним из первых «криминалистических» исследований в области охраны окру-
жающей среды.
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
112
100%
Пирен, молекулярная масса 202;
нерезонансная ДИС
122
174
98
Д5 Щ111!58, И L2
146
150
170
90 100 НО 120 130 140 150 160 170 180 190 200 210
202
100%-|
Спектр ДИС
для TSQ 700
а
□
о
X
200-\
х
X
S
126
150^1
175
198
ПО 120 130 140 150 160 170 180 190 200 210
m/z
Рис. 4.5. Ионная ловушка: (а) нерезонансный режим при анализе пирена; (б) резонанс-
ный режим при анализе пирена; (в) TSQ 700: МС/МС-анализ пирена. (С разре-
шения издательства Elsevier из Journal of the American Society for Mass Spectrometry,
8, Pyle S.M., Betkowski L.D., Marcus A. B., Winnik W. and Brittain R. D. «Analysis of
polycyclic aromatic hydrocarbons by ion trap tandem mass spectrometry», 183—190)
4.3. Приложения МС/МС к экологическому мониторингу I 13
4.3.1. Новые загрязняющие вещества
Термин «новые загрязняющие вещества» с недавних пор используется химиками-эко-
логами. Этот термин необязательно означает, что такие вещества недавно поступили
в продажу и были представлены публике (Doughton, 2001). Это возможно, но главная
причина, почему соединения получили такое название, заключается в том, что появи-
лись аналитические приборы и методы для их идентификации. В начале списка этих
новых методов находятся ЖХ-интерфейсы, которые позволяют использовать масс-
спектрометрию для анализа таких неподатливых веществ. С их появлением возникла
необходимость в ДАС- и МС/МС-методах для извлечения полной информации об ис-
следуемых молекулах. Были сделаны попытки разработать в дополнение к тандемным
приборам недорогие масс-спектрометры, содержащие один анализатор (например ква-
друполь или времяпролетный анализатор), но обладающие возможностями осущест-
вления ДАС (Katta et al., 1991). В таком варианте ДАС обеспечивается увеличением элек-
трических потенциалов в области источника и повышенным давлением в этой части
прибора. В результате реализуется диссоциация всех анализируемых ионов одновре-
менно. В случае отсутствия сильного влияния матрицы такой подход видится неплохой
альтернативой тандемным приборам. Однако, так как селективность по ионам-пред-
шественникам отсутствует, результаты фрагментации сложно интерпретировать вслед-
ствие невозможности осуществления однозначного соответствия между ионами-про-
дуктами и ионами-предшественниками.
Существует много классов новых загрязняющих веществ. Одним из первых, отне-
сенных к «новым», стал класс соединений, влияющих на эндокринную систему. Такие
вещества нарушают баланс и выработку гормонов в организме (Colborn et al., 1993). Са-
мой распространенной группой являются эстрогены. Они могут иметь как природное,
так и искусственное происхождение. Например, p-эстрадиол — природный эстроген,
присутствующий в организме женщин в значительно более высокой концентрации, чем
у мужчин. Синтетические эстрогены, такие, как, например, этинилэстрадиол, широко
используются в медицине в конрацептических и терапевтических целях (Diaz-Cruz etal.,
1993; Sparrow, 1987). Диэтил эстрад иол (ДЭС) также широко использовался в гормональ-
ной терапии для предотвращения выкидышей и как промоутер роста для домашнего
скота (Martindale, 1982). Водоочистные сооружения не задерживают эти вещества. Та-
ким образом, эстрогены проникают в водную среду и могут достигать нанограммовых
и субнанограммовых концентраций (Rodriguez-Mozaz et al., 2004). Эти гормоны влияют
на рыбу, обитающую ниже по течению от водоочистных сооружений, вызывая ее феми-
низацию (Sole et al., 2003; Sumpter, 1995).
Анализ эстрогенов проводился методами ИФА (иммуноферментного анализа)
(Ниап and Sedlak, 2001; Mitani et al., 2005), ГХ/МС или ГХ/МС/МС (Kelly, 2000; Nakamura
et al., 2001; Xiao et al., 2001). Однако ИФА завышает концентрации (из-за недостаточной
селективности), а ГХ/МС требует дериватизации при подготовке образца, что усложня-
ет процедуру анализа. Поэтому чаще для анализа эстрогенов используется ЖХ/МС или
ЖХ/МС/МС. Родригес-Мозас (2004) и Митани (2005) соединили жидкостной хромато-
граф с установками для пробоподготовки и концентирования эстрогенов. При помощи
твердофазной эктракции (Rodriguez-Mozaz et al., 2004) и микротвердофазной эктракции
(Mitani etal., 2005) эти две группы смогли осуществить высокопроизводительный анализ
эстрогенов. Установка группы Катаоки и Митани представлена на рис. 4.6. Обе лабора-
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
у;; 7^
|П|1П11П11ГИ1Г11^1Г1^111.П11П1^|1Г|Д|1.11
Петля
птттттН
Игла для
инжекции
Адаптер для присо-
единения колонки •
Компьютер
Хроматографи-
ческая колонка
Мобиль-
ная фаза
от насоса
Тандем-
ный масс-
спектро-
метр
Инжектор
Автосамплер с 6 портами
"I Отходы
для управления
Капиллярная
ч колонка
для управления
Рис. 4.6. Схема системы твердофазной микроэкстракции с последующим ЖХ/МС/МС-
анализом в режиме реального времени: а — положение загрузки (микроэк-
стракция); б — положение инжекции (десорбция). (С разрешения издательства
Elsevier из Journal of Chromatography А, 1981, 2, Mitani К.; Fujioka Н.; Kataoka Н.
«Fully automated analysis of estrogens in environmental waters by in-tube solid phase
microextraction coupled with liquid chromatography — tandem mass spectrometry»,
218-224, 2005)
тории работали в режиме мониторинга заданной реакции (selected reaction monitoring,
SRM) (Rodriguez— Mozaz et al., 2004) или мониторинга нескольких выбранных реакций
(multiple reaction monitoring, MRM) (Mitanietal., 2005), регистрируя отрицательные ионы.
Режим SRM предполагает изолирование выбранного иона-предшественника в первом
квадруполе, его реакцию с нейтральным газом в ячейке столкновений и изолирование
одного выбранного иона-продукта в третьем квадруполе с последующим его детек-
тированием. MRM позволяет детектировать несколько ионов-продуктов в одном экс-
перименте. Предел количественного определения для этих систем составлял от 0,02
до 1,02 нг/л (Rodriguez-Mozaz et al., 2004).
Следует упомянуть о некоторых мерах предосторожности при работе с ЖХ/МС/МС.
При анализе сложных образцов экологического характера, если работа была выполнена
недостаточно тщательно, можно получить как ложноположительные, так и ложноотри-
цательные результаты. Ложноотрицательные результаты являются следствием эффек-
тов ионной суппрессии, тогда как ложноположительные данные могут быть вызваны
недостаточной селективностью метода (Reemtsma, 2001). Критерии для отсечения лож-
ноположительных и ложноотрицательных результатов следующие.
Время удерживания должно воспроизводиться с точность 1—2% от времени удер-
живания стандартного образца.
Относительные интенсивности для как минимум двух ионов, выбранных для ре-
гистрации SRM, не должны отличаться более чем на 20% от относительных интенсив-
ностей тех же ионов стандартного образца (Baronti et al., 2000; Lagana et al., 2000; Lopez de
Alda etal., 2003).
4.3.2. Лекарственные препараты
Так как многие из новых загрязняющих веществ — медицинские препараты (или, точ-
нее, активные медицинские ингредиенты), они заслуживают особого внимания. По-
скольку медикаменты предназначены для лечения людей и животных, их перечисление
среди экотоксикантов несколько противоречиво. Главная их опасность заключается
в экологическом ущербе, выраженном в основном в побочных эффектах, оказываемых
множеством препаратов на водные экосистемы. Активные фармацевтические ингре-
диенты непрерывно поступают из очистных сооружений и предприятий в природную
воду, так что рыба и другие водные животные постоянно плавают в таком «бульоне»
(Daughton and Ruhoy, 2009).
Для мониторинга медицинских препаратов необходимо знать их концентрации
вводе (например в стоках очистных сооружений или предприятий), нотакже важноопре-
делять концентрации в иле и грунте, абсорбирующих эти соединения из воды. Остано-
вимся на двух примерах такого мониторинга: анализе ила Тернсом сего коллегами (Ternes
etal., 2005) и анализе сточных вод, выполненном Гербхардтом и Шрёдером (Gerbhardtand
Schroder, 2007). Терне с соавторами был заинтересован в определении в иле активных
фармацевтических ингредиентов, включая антиэпилептический препарат карбамазе-
пин, цитостатические агенты, психиатрическое лекарство диазепам, а также йодирован-
ную контрастную среду. Препараты извлекались ультразвуковой экстракцией раствори-
телем или жидкостной экстракцией под высоким давлением. Очистка проводилась при
помощи сорбента С18 с обращенной фазой и силикагеля. Электрораспыление (для ней-
тральных фармацевтически активных соединений и йодированной контрастной среды
в режиме анализа положительных ионов) или химическая ионизация при атмосферном
давлении (для кислотных фармацевтически активных соединений в режиме анализа от-
рицательных ионов) были использованы на тройном квадруполе. Данные были получе-
ны в режиме MRM, в котором ион-предшественник изолировался в первом квадруполе,
а продукты ДАС, полученные во втором квадруполе, сканировались в квадруполе №3.
Для каждого соединения контролировались один или два иона-продукта. Пределы ко-
личественного определения для этих веществ находились в диапазоне 20—50 нг/л (Ternes
et al., 2005). Гербхардт и Шрёдер анализировали карбамазепин, диазепам, диклофенак
и клофибриновую кислоту в воде, прошедшей через различные стадии очистки. Они ис-
пользовали LTQ Orbitrap с электрораспылением, регистрируя ионы обеих полярностей.
Система использовалась как в режиме получения полного спектра с разрешением 300
мДа на полувысоте пика, так и в режиме ДАС с разрешением 60000 для m/z 400 и высокой
точностью определения массы. Рис. 4.7 иллюстрирует возможности орбитрэпа в режиме
ДАС высокого разрешения для идентификации выбранных ионов при анализе сложной
пробы сточной воды. По спектру отрицательных ионов оказалось возможным опреде-
лить клофибриновую кислоту и диклофенак, используя характеристические для SRM
массы ионов (Gerbhardt and Schroder, 2007). Эти компоненты постоянно присутствовали
в стоках станций по очистке воды в низких, но регистрируемых концентрациях (нг/л).
4.3.3. Пестициды и гербициды
Хлорацетанилиды, триазины, фенилмочевина и некоторые их метаболиты анализи-
ровались с помощью ГХ/МС/МС, ЖХ/МС и ЖХ/МС/МС (Dagnac et al., 2005). Ионные
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
Относительная интенсивность,
Рис. 4.7. Элюирование фармацевтических препаратов: (а) полный ионный токэкстракта сточ-
ных вод водоочистной станции в Аахен-Зоерс, полученный методом ЖХ/ЭРИ/МС
с концентрированием на С18-ТФЭ-картридже; (б) экстрагированная хроматограм-
ма по характеристическому иону клофибриновой кислоты; (в) экстрагированная
хроматограмма по характеристическому иону диклофенака; (г) экстрагированная
хроматограмма для иона-продукта клофибриновой кислоты, полученная методом
ЖХ/ЭРИ/МС/МС в режиме регистрации отрицательных ионов (время удержи-
вания 8,83 мин.; концентрация клофибриновой кислоты 0,02 нг/мл); (д) экстраги-
рованная хроматограмма для иона-продукта диклофенака, полученная методом
ЖХ/ЭРИ/МС/МС в режиме регистрации отрицательных ионов (время удержива-
ния 10,29 мин.; концентрация диклофенака 1,5 нг/мл). (С разрешения издательства
Elsevier из Journal of Chromatography А, 1160, 1—2, Gebhardt W. and Schroder H. Fr. «Liquid
Chromatography — (tandem) mass spectrometry for the follow-up of the persistanr pharma-
ceuticals during wastewater treatment and advanced oxidation», 34, 2007)
ловушки применялись совместно с ГХ и в одном случае — с ЖХ (в режиме МС/МС).
В нескольких случаях ЖХ использовалась вместе с одиночным квадруполем, источник
химической ионизации при атмосферном давлении (ХИАД) с системой ЖХ/одиночный
квадруполь, а источники ЭРИ и ХИАД с ЖХ/ионной ловушкой в тандемном режиме.
Поэтому поучительно сравнить пределы количественного определения для этих раз-
личных систем. Линурон детектировался лучше с ХИАД, которая более эффективна
для анализа неполярных соединений. Напротив, ЭРИ превосходно ионизирует кислот-
ные и основные компоненты.
Некоторые образцы настолько загрязнены материалами матрицы (гумусовыми кис-
лотами, фрагментами тканей), что преимущества ЖХ и МС/МС нивелируются. Сле-
довательно, в таких случаях требуется дополнительная очистка. Влияние матричных
компонентов на процессы ионизации — существенная проблема для количественного
анализа. Для ее решения Гервес и его коллеги (Gervais etal., 2008) применили твердофаз-
ную очистку в сочетании с ультраэффективной жидкостной хроматографией (УЭЖХ)
для анализа 31 пестицида в речной воде. Использование УЭЖХ позволяет существен-
но улучшить хроматографическое разделение. В этом случае конкуренция в процессе
ионизации между матрицей и аналитами минимизирована. Тройные квадруполи ис-
пользовались как тандемные масс-спектрометры. В процессе разработки метода особое
внимание следует уделять оптимизации напряжения на скиммере и энергии столкно-
вений в целях увеличения интенсивности ионов-предшественников и ионов-продуктов
соответственно. Пример такой оптимизации для дифлуфеникана продемонстрирован
на рис. 4.8. При использовании описанного подхода пределы количественного опреде-
Таблица4.1. Пределы количественного определения гербицидов для ГХ/МС/МС-
и ЖХ/МС/(МС)-методов (мкг/кг). (С разрешения издательства Elsevier из Jour-
nal of Chromatography А, 1067, 1—2, Dagnac Т., Bresteau S., Jeannot R., Mouvet C.,
Baran N. «Determination of chloroacetanilides, triazines and phenylureas and some
of their metabolites in soils by pressurized liquid extraction, GC-MS, LC-MS and
LC-MS/MS», 225-233, 2005)
Гербицид ГХ/МС/МС ЖХ/ХИАД/МС ЖХ/ЭРИ/МС/МС ЖХ/ХИАД/МС/С
Атразин 1,9 5,4 0,15 0,3
ДИА (диизопропилатразин) 1,6 22 4,5 11
ДЭА (диэтилатразин) 2,5 13 0,7 1,2
Метолахлор 0,3 6,3 0,3 0,3
Ацетохлор 0,9 22 0,7 1,5
Алахлор 2,2 — 1,5 1,5
дипм (ди изопропил мочевина) — 22 6 1,5
МИПМ (моноизопропил мочевина) — 11 0,7 0,7
Изопротурон — 4,8 0,7 3
Хлортолурон — 10 7,5 3
Линурон — — 7,5 0,7
Диурон — — 4,5 4,5
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
Рис. 4.8. Интенсивность сигнала для различных ионов-продуктов пестицида дифлуфе-
никана в зависимости от энергии столкновений. Данные получены в режиме
MRM для положительных ионов. (С разрешения издательства Elsevier из Journal
of Chromatography А, 1202, 2, Gervais G., Brosillon S., Laplanche A. and Hele C. «Ultra-
pressure liquid chromatography electrospray tandem mass spectrometry for multiresidue
determination of pesticides in water», 163—172, 2008)
ления для 31 пестицида оказались в диапазоне от 10 до 51 нг/л и для положительных,
и для отрицательных ионов (как показано в табл. 4.4 и статье Gervais etal., 2008).
Твердофазные сорбенты могут адсорбировать посторонние примеси в процессе экс-
тракции. Эти вещества могут усиливать или подавлять ионный сигнал. Гала-Гарсия с со-
трудниками (Gala-Garcia, 2010) для лучшей селективности при очистке триазинов и их
метаболитов с последующим их анализом посредством тандемной масс-спектрометрии
использовали молекулярно-отпечатанные полимеры (МОП), инкорпорированные
в сорбент. Авторы обнаружили, что применение МОП оказалось эффективным для
очистки и концентрирования аналитов, извлеченных из сложных образцов почв. Ква-
друпольная линейная ионная ловушка обеспечила высокую чувствительность анализа,
подтвержденную низкими пределами количественного определения в диапазоне нг/л
и нг/г (Gala-Garcia, 2010).
4.3.4. Аддукты ДНК и маркеры оксидативного стресса
Оценка риска для экотоксикантов может быть упрощена путем регистрации ДНК-
аддуктов этих веществ в тканях человека (подробнее это будет описано в главе 17). Де-
тектируя такие аддукты, можно получить физические свидетельства повреждений ДН К,
что невозможно с помощью мониторинга исходного вредного вещества. Используя
ВЭЖХ/ЭРИ/МС/МС и методы изотопного разбавления, Минз (Means et al., 2003) и его
сотрудники смогли определять ДНК-аддукты ароматических аминов на уровне 100 фем-
томолей, что означает присутствие одного аддукта на 6-107 неповрежденных нуклеоти-
дов. Использование изотопного разбавления минимизировало ошибки количественного
анализа, так как одна и та же реакция превращения иона-предшественника в ион-про-
дукт использовалась как для самого аддукта, так и для его изотопно-меченного аналога.
Фенге коллегами (Feng etal., 2009) использовали метод изотопного разбавления для
анализа стереоизомеров я//А?ш-бенз[а]пирендиол-эпоксидных (БПДЭ) аддуктов с помо-
щью системы УЭЖХ — тройной квадруполь в режиме MRM. Анти-ЬП1\Э — сильный
4.3. Приложения МС/МС к экологическому мониторингу
канцероген, образующийся из бенз[а]пирена. Он может реагировать с дезокси гуанози-
ном (dG) или с дезоксиаденозином (dA), приводя к возникновению стереоизомерных
ДНК-аддуктов (Willems et al., 2002). Режим MRM тройного квадруполя обеспечивает
достаточную специфичность, чтобы отделить четыре стереоизомера я//а?ш-БПДЭ-7У2<У(7
аддуктов от тетролов БПДЭ. Использование УЭЖХ с MRМ позволило сократить время
анализа с 60—100 мин. до 2—4 мин. Кроме уменьшения времени анализа, УЭЖХ дает
возможность сделать пики уже и интенсивнее. Таким образом, описанная система вкупе
с изотопно-меченными аналогами для количественного анализа — очень чувствитель-
ный и экспрессный метод определения четырех стереоизомеров ДНК-аддуктов (Feng
etal., 2009).
Вследствие высокой чувствительности и селективности, а также возможности ана-
лиза многих веществ одновременно ЖХ/МС/МС-методы предпочтительны при иден-
тификации маркеров окислительного стресса. Окислительный стресс в организме
выявляется в состоянии болезни. Однако измерить концентрации самих окислителей
(свободных радикалов и т.д.) in situ довольно сложно. Поэтому обычно соединения,
связанные с окислительным стрессом, определяются как маркеры заболевания в био-
логических жидкостях, таких как кровь, сыворотка и моча (Winnik and Kitchin, 2008).
Одним из наиболее распространенных классов маркеров окислительного стресса яв-
ляются изопростаны, связанные с перокислением липидов. Г2-изопростаны — проста-
гландины типа F2, образующиеся при окислении арахидоновой кислоты свободными
радикалами, были признаны индикаторами окислительного стресса (Winnik and Kitchin,
2008). Иммунохимические методы их определения страдают от низкой селективности
благодаря реакциям с другими метаболитами простагландинов, вследствие чего не яв-
ляются надежными. Другие изопростаны, такие как Г4-нейропростаны, также могут
быть маркерами окислительного стресса, например, нейродегенеративного заболева-
ния. Ян и его коллеги (Yan et al., 2007) описали ЖХ/МС/МС-метод анализа несколь-
ких изопростанов в режиме регистрации отрицательных ионов с изотопно-меченными
стандартами изопростанов с применением твердофазной экстракции. Полуавтоматиче-
ский ЖХ/МС/МС-метод для количественного определения Г2-изопростанов в плазме
и моче был описан Хашке (Haschke et al., 2007). Авторы использовали для экстракции
картридж, соединенный напрямую с ЖХ. Полезный ЖХ/МС/МС-метод с применени-
ем режима 57?Л/был разработан для идентификации в мышиной печени биологически
важных тиолов, участвующих в патологических процессах (Bouligand et al., 2006). Еще
один ЖХ/МС/МС-метод использовался для регистрации повышенных уровней гомо-
цистеина — маркера окислительного стресса, связанного с инсультом и другими забо-
леваниями (Tomaiuolo et al., 2006). Ароматические аминокислоты при окислении могут
участвовать в образовании маркеров окислительного стресса. Определение этих марке-
ров может обеспечить информацию о природе и происхождении повреждений, вызван-
ных окислительным стрессом (Orhan et al., 2005). З-Нитротирозин является конечным
продуктом окисления тирозина различными окислителями в живом организме. Риберг
и Кайдал (Ryberg and Caidahl, 2007) показали, что 3-нитротирозин — биомаркер более
чем 40 различных болезней. Авторы смогли разработать высокоселективный и высоко-
чувствительный ЖХ/МС/МС-метод его мониторинга, который с учетом отсутствия де-
риватизации при пробоподготовке можно считать идеальным при определении низких
концентраций 3-нитротирозина в плазме крови человека.
Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
4.3.5. Поверхностно-активные вещества и красители
Как было сказано ранее, красители были одними из первых веществ, анализируемых
при помощи ЖХ/МС/МС. Многие из них имеют ионные структуры, что требует ис-
пользование ЖХ. Поверхностно-активные вещества (ПАВ) принадлежат к другому
классу соединений. Они характеризуются высокой полярностью, что также обеспечи-
вает возможность их идентификации при помощи ЖХ. ПАВ всегда присутствуют в воде,
даже после ее очистки. Наличие ПАВ в воде было признано проблемой после того, как
ЖХ/МС/МС-методы стали использоваться для анализа сточных вод. Шрёдер (Schroder,
1993) был одним из первых, кто применил ЖХ/МС/МС-методы с ионизацией термо-
спреем к анализу ПАВ. Эти соединения классифицируются на две подгруппы: ионные
и неионные. Наиболее распространенные неионные ПАВ характеризуются ионами,
разница в массах которых равна 44 или 58, что соответствует мономерным звеньям це-
почек полигликолевых эфиров (Schroder, 1993). Распространенные ионные ПАВ — про-
изводные серной кислоты. Они были идентифицированы при помощи термоспрея в ре-
жиме отрицательных ионов.
Большой интерес вызывал мониторинг текстильных красителей в сточных водах
(Betowski et al., 1987) в связи как с эстетическими требованиями (отсутствие окрашен-
ной воды в водопроводе), так и проблемами, связанными с экологией и здравоохране-
нием (Clayson, 1962; Ниерег, 1969). Поэтому разработка метода непрерывного отбора проб
для мониторинга эффективности очистки воды от коммерческих красителей была не-
обходимой. Плам и Ре хорек (Plum and Rehorek, 2005) использовали ВЭЖХ с детектором
на основе диодной матрицы и ЭРИ-МС/МС для непрерывного мониторинга с целью
получения информации о биологически управляемом восстановлении азокрасителей.
Так как ион-парные реагенты применялись для хроматографического разделения ани-
онных красителей, метод катионной нейтрализации был использован перед вводом
пробы в ЭРИ-МС-интерфейс (так как нелетучие ионы могут блокировать интерфейс
и подавлять сигнал от ионов интересующих нас веществ). Использование ЖХ/МС/МС-
системы дает возможность контролировать исходный материал, а также неизвестные
промежуточные соединения, выделяемые аэробными и анаэробными бактериями
в процессе очистки воды (Plum and Rehorek, 2005).
4.3.6. Озон
Мониторингозона необходим по двум причинам: во-первых, для измерения концентра-
ции собственно озона, а во-вторых, для определения продуктов реакций, протекающих
с участием озона. Обе причины связаны с решением проблемы измерения в стратосфе-
ре уровня озона, предохраняющего Землю от вредного УФ-излучения. Так как озон —
сильный окислитель, он также потенциально вреден для здоровья людей вследствие его
воздействия на биомолекулы. Например, аккумулирование в организме окисленных
белков ассоциируется с различными заболеваниями, такими как катаракта и ревмато-
идный артрит (Kotiaho et al., 2000; Pirie, 1971). В ходе исследования водного озониро-
вания 22 аминокислот и небольших пептидов Котяхо с коллегами (Kotiaho et al., 2000)
идентифицировали эти окисленные соединения, используя ЭРИ-МС/МС. Успех этой
работы был обеспечен высокой чувствительностью ЭРИ-МС/МС. Из 22 аминокислот
только 4 образовывали окисленные формы: гистидин (His), триптофан (Тгр), тирозин
(Туг) и метионин (Met). Позже исследователи обнаружили, что реакционная способность
аминокислот при взаимодействии с озоном изменяется в порядке Met > Тгр > Туг > His.
В использованных в исследовании небольших пептидах в процесс окисления были во-
влечены те же аминокислоты, но порядок уменьшения реакционной способности был
несколько другим: Met > His > Тгр > Туг. Продукты окисления озоном исследованных
пептидов были настолько уникальны, что окисление озоном в сочетании с анализом
ЭРИ-МС/МС было предложено как метод определения структуры неизвестных пепти-
дов (Kotiaho et al., 2000).
Еще одно исследование было связано с изучением процесса очистки сточных вод
озоном и влиянием этого процесса на фармацевтические препараты, например на те-
трациклин (Dalmazjo et al., 2007). Вследствие широкого применения тетрациклина для
лечения инфекционных заболеваний людей и животных он может быть обнаружен
во входящих и исходящих потоках водоочистных сооружений (Karthikeyan and Meyer,
2006). Предполагалось, что очистка озоном сточных вод приведет к выпадению в осадок
тетрациклина и подобных ему соединений. Однако предположение не подтвердилось
и не было найдено продуктов реакции, образующихся при разрыве ароматических ко-
лец. Тем не менее применение ЖХ/МС" (в ионной ловушке) дало возможность иденти-
фицировать два важных полярных промежуточных продукта (Dalmazio et al., 2007).
4.4. Заключение
Использование тандемной масс-спектрометрии революционизировало химический
и биологический анализ. За изобретение электрораспыления профессор Джон Фенн
получил Нобелевскую премию (Fenn etal., 1989), однако применение этого метода не по-
лучило бы такого распространения, если бы не МС/МС. Одной из областей, сильно
изменившихся после этих изобретений, является экологический мониторинг. Успех
экологического мониторинга был обеспечен применением тщательно разработанно-
го метода ГХ/МС. В дальнейшем стало понятно, что ЖХ/МС/МС-методы также могут
быть полезны для целей мониторинга. Следует также отметить, что тандемная масс-
спектрометрия была реализована в различных вариантах, каждый из которых проде-
монстрировал полезность МС/МС-подхода. Такими вариантами являются приборы
высокого разрешения с обратной геометрией, тройные квадруполи, различные ионные
ловушки, разнообразные гибридные приборы, такие как квадруполь-ВПМС и недавно
появившийся масс-спектрометр типа орбитрэпа. Каждый из этих приборов обеспечи-
вает возможность получения структурной информации для идентификации соедине-
ния или количественного анализа путем детектирования специфической ионной пары
в процессе ДИС. Экологические приложения включают в себя анализ всей гаммы со-
единений: от токсичных загрязняющих веществ — пестицидов и гербицидов, до новых
компонентов, таких как лекарственные препараты и вещества для личного ухода, или
биологических материалов, содержащих ДНК-аддукты токсичных соединений и мар-
керы окислительного стресса, возникающие в результате загрязнения окружающей сре-
ды. Упомянутые аналитические методы обладают исключительной чувствительностью
к экотоксикантам и специфичностью анализа, недоступной для других методов. Факти-
чески чувствительность оборудования достигла того уровня, на котором ксенобиотики
122 Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
уже не несут опасности для окружающей среды. Однако все еще остаются нерешенные
проблемы, связанные с субтоксичными концентрациями загрязняющих веществ. Воз-
можно, экотоксиканты даже в таких концентрациях становятся токсичными при их
смешивании? Ответы на эти вопросы будут найдены по мере дальнейшего совершен-
ствования аналитического оборудования.
4.5. Замечание
Отдел исследований и разработок Агентства по охране окружающей среды (АООС)
США финансировал и направлял исследования, описанные в этой главе. Текст был про-
смотрен Агентством и одобрен для публикации. Упоминание торговых марок и коммер-
ческих продуктов не противоречит правилам АООС США.
Литература
[1] . Ballard J. М., and Betowski L. D. (1986). «Thermospray ionization and tandem mass spec-
trometry of dyes». Organic Mass Spectrometry. 21,9: 575.
[2] . Baronti C., Curini R., D'Ascenzo G., Di Corcia A., Centili A., and Samperi R. (2000).
«Monitoring natural and synthetic estrogens at activated sludge sewage treatment plants and
in a receiving river water». Environmental Science and Technology. 34, 24: 5059.
[3] . Betowski L. D., and Ballard J. M. (1984). «Identification of dyes by thermospray ionization
and mass spectrometry/mass spectrometry». Analytical Chemistry. 56, 13: 2604—2607.
[4] . Betowski L. D., Pyle S., Ballard J. M., and Shaul G.M. (1987). «Thermospray LC/MS/MS
analysis of wastewater for disperse azo dyes». Biomedical and Environmental Mass Spectrom-
etry. 14, 7: 343-354.
[5] . Bouligand J., Deroussent A., Paci A., Morizet J., and Vassal G. (2006). «Liquid chroma-
tography-tandem mass spectrometry assay of reduced and oxidized glutathione and main
precursors in mice liver». Journal of Chromatography B. 832, 1: 67—74.
[6] . Brand H. R., and Gregg H. R. (1989). «An expert system for triple quadrupole mass spec-
trometry». Energy and Technology Review. January: 1—7. Lawrence Livermore National
Laboratory, Livermore, CA. Available from National Technical Information Service, US
Department of Commerce, UCRL-52000-89-1.
[7] . Bruins A. P., Covey T. R., and Henion J. D. (1987a). «Ion spray interface for combined liquid
chromatography/atmospheric pressure ionization mass spectrometry». Analytical Chemistry.
59, 22: 2642-2646.
[8] . Bruins A. P., Weidolf L. O. G., Henion J. D.and Budde W. I. (1987b). «Determination of sul-
fonated azo dyes by liquid chromatography/atmospheric pressure ionization mass spectrom-
etry». Analytical Chemistry. 59, 22: 2647—2652.
[9] . Busch K. L., and Cooks R. G. (1983). Analytical applications of tandem mass spectrometry. In:
McLafferty F. W. (Ed.), Tandem Mass Spectrometry. John Wiley and Sons, New York.
[10] . Cha B., Blades M., and Douglas D. J. (2000). «An interface with a linear quadrupole ion
guide for an electrospray ion trap mass spectrometry system». Analytical Chemistry. 72, 22:
5647-5654.
Литература
[11] . Clayson D. В. (1962). Chemical Carcinogenesis. Churchill, London, UK.
[12] . Colborn T., vom Saal F. S., and Soto A. M. (1993). «Developmental effects of endocrine-
disrupting chemicals in wildlife and humans». Environmental Health Perspectives. 101, 5:
378.
[13] . Сох K.A., Cleven C. D., and Cooks R.G. (1995). «Mass shifts and local space charge ef-
fects observed in the quadrupole ion trap at higher resolution». International Journal of Mass
Spectrometry and Ion Processes. 144, 1—2: 47—65.
[14] . Creaser C.S., and Stygall J.W. (1998). «Recent developments in analytical ion trap mass
spectrometry». Trends in Analytical Chemistry. 17, 10: 583—593.
[15] . Dagnac T., Bristeau S., Jeannot R., Mouvet C., and Baran N. (2005). «Determination of
chloroacetanilides, triazines and phenylureas and some of their metabolites in soils by pres-
surised liquid extraction, GC—MS/MS, LC—MS and LC—MS/MS». Journal of Chroma-
tography A. 1067, 1-2: 225-233.
[16] . Dalmazio I., Almeida M. O., and Augusti R. (2007). «Monitoring the degradation of tetra-
cycline by ozone in aqueous medium via atmospheric pressure ionization mass spectrom-
etry». Journal of the American Society for Mass Spectrometry. 18,4: 679—687.
[17] . Daughton C. G. (2001). «Emerging pollutants, and communicating the science of environ-
mental chemistry and mass spectrometry: pharmaceuticals in the environment». Journal of
the American Society for Mass Spectrometry. 12, 10: 1067—1076.
[18] . Daughton C.G., and Ruhoy LS. (2009). «Environmental footprint of pharmaceuticals:
The significance of factors beyond direct excretion to sewers». Environmental Toxicology
and Chemistry. 28, 12: 2495—2521.
[19] . de Hoffmann E. (1996). «Tandem mass spectrometry: a primer». Journal of Mass Spectrom-
etry. 31,2: 129-137.
[20] . Diaz-Cruz M.S., Lopez de Aldea M.J., Lopez R., and Barcelo D. (2003). «Determination
of estrogens and progestogens by mass spectrometric techniques (GC/MS, LC/MS and
LC/MS/MS). Journal of Mass Spectrometry. 38, 9: 91.
[21] . Feng F., Wang X., Yuan H., and Wang H. (2009). «Ultra-performance liquid chromatog-
raphy-tandem mass spectrometry for rapid and highly sensitive analysis of stereoisomers
of benzo[a]pyrene diol epoxide—DNA adducts». Journal of Chromatography B. 877, 22:
2104-2112.
[22] . Fenn J. B., Mann M., Meng С. K., Wong S. F., and Whitehouse С. M. (1989). «Electrospray
ionization for mass spectrometry of large biomoleculesa. Science. 246, 4926: 64—71.
[23] . Garcia-Galan M.J., Diaz-Cruz M.S., Barcelo D. (2010). «Determination of triazines and
their metabolites in environmental samples using molecularly imprinted polymer extrac-
tion, pressurized liquid extraction and LC—tandem mass spectrometry». Journal of Hydrol-
ogy. 383, 1-2: 30-38.
[24] . Gebhardt W., and Schroder H. Fr. (2007). «Liquid chromatography—(tandem) mass spec-
trometry for the follow-up of the elimination of persistent pharmaceuticals during wastewa-
ter treatment applying biological wastewater treatment and advanced oxidation». Journal of
Chromatography A, 1160, 1—2: 34—43.
[25] . Gervais G., Brosillon S., Laplanche A., and Helen C. (2008). «Ultra-pressure liquid chro-
matography—electrospray tandem mass spectrometry for multiresidue determination of
pesticides in water». Journal of Chromatography A, 1202, 2: 163—172.
124 Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
[26] . Guilhaus М., Selby D., and Mlynski V. (2000). «Orthogonal acceleration time-of-flight
mass spectrometry». Mass Spectrometry Reviews. 19, 2: 65—107.
[27] . Hager J. W. (2004). «Review: Recent trends in mass spectrometer development». Analytical
and Bioanalytical Chemistry. 378, 4: 845—850.
[28] . Hager J. W., and Le Blanc J.C.Y. (2003). «Product ion scanning using a Q-q-Qlinearion-
trap (Q TRAPTM) mass spectrometer». Rapid Communication in Mass Spectrometry. 17, 10:
1056-1064.
[29] . Hardman M., and Makarov A.A. (2003). «Interfacing the Orbitrap mass analyzer to an
electrospray ion source». Analytical Chemistry. 75, 7: 1699—1705.
[30] . Haschke M., Zhang Y. L., Kahle C., Klawitter J., Korecka M., Shaw L.M., and Chris-
tians U. (2007). «HPLCatmospheric pressure chemical ionization MS/MS for quantifica-
tion of 15-F2t-isoprostane in human urine and plasma». Clinical Chemistry. 53, 3:489—497.
[31] . Huang С. H., and Sedlak D. L. (2001). «Analysis of estrogenic hormones in municipal
wastewater effluent and surface water using enzyme-linked immunosorbent assay and gas
chromatography/tandem mass spectrometry». Environmental Toxicology and Chemistry. 20,
1: 133.
[32] . Hueper W.C. (1969). Occupational and Environmental Cancers of the Urinary System. Yale
University Press, New Haven, Connecticut.
[33] . Hunt D. E, Shabanowitz J., Harvey T. M., and Coates M. (1985). «Scheme for the direct
analysis of organics in the environment by tandem mass spectrometry». Analytical Chemis-
try. 57, 2: 525-537.
[34] . Johnson J.V., Yost R.A., Kelley P. E., and Bradford D.C. (1990). «Tandem in-space and
tandem-in-time mass spectrometry: triple quadrupoles and qaudrupole ion traps». Analyti-
cal Chemistry. 62, 20: 2162—2172.
[35] . Karthikeyan K.G.,and Meyer M.T. (2006). «Occurrence of antibiotics in wastewater treat-
ment facilities in Wisconsin U. S. A. Science of the Total Environment. 361, 1—3: 196—207.
[36] . Katta V., Chowdhury S. K., and Chait В. T. (1991). «Use of a single-quadrupole mass spec-
trometer for collision induced dissociation studies of multiply charged peptide ions pro-
duced by electrospray ionization». Analytical Chemistry. 63, 2: 174—178.
[37] . Kelly C. (2000). «Analysis of steroids in environmental water samples using solid-phase ex-
traction and ion-trap gas chromatography—mass spectrometry and gas chromatography-
tandem mass spectrometry». Journal of Chromatography A. 872, 1—2: 309—314.
[38] . Kotiaho T., Eberlin M.N., Vainiotalo P., and Kostiainen R. (2000). «Electrospray mass
and tandem mass spectrometry identification of ozone oxidation products of amino acids
and small peptides». Journal of the American Society for Mass Spectrometry. 11,6: 526—535.
[39] . Lagana A., Bacaloni A., Fago G., and Marino A. (2000). «Trace analysis of estrogenic
chemicals in sewage effluent using liquid chromatography combined with tandem mass
spectrometry». Rapid Communication in Mass Spectrometry. 14, 6: 401.
[40] . Lee M.J., Monte S., Sanderson J., and Haskins N.J. (1999). «А preliminary study using fast
gradient liquid chromatography coupled to a quadrupole orthogonal time-of-flight mass
spectrometer». Rapid Communication in Mass Spectrometry. 13, 4: 216—221.
[41] . Lopez de Alda M. J., Diaz-Cruz S., Petrovic M., and Barcelo D. (2003). «Liquid chroma-
tography—(tandem) mass spectrometry of selected emerging pollutants (steroid sex hor-
mones, drugs and alkylphenolic surfactants) in the aquatic environment». Journal of Chro-
matography A. 1000, 1—2: 503—526.
Литература
[42] . Makarov А. А. (2000). «Electrostatic axially harmonic orbital a high-performance tech-
nique of mass analysis». Analytical Chemistry. 72, 6: 1156—1162.
[43] . Reynolds J. E. F., and Prasa A. B. (Eds). (1982). Martindale: The Extra Pharmacopoeia, 28th
edition (1982). «The Pharmaceutical Press, London.
[44] . McLafferty F. W., Bente P. F., Ill, Kornfeld R., Tsai S.-С., and Howe I. (1973). «Metasta-
ble ion characteristics. XXII. Collisional activation spectra of organic ions». Journal of the
American Chemical Society. 95, 7: 2120.
[45] . Means J.C., Olsen P. D., and Schoffers E. (2003). «Development of an isotope dilution
liquid chromatography/tandem mass spectrometry detection method for DNA adducts of
selected aromatic amines». Journal of the American Society for Mass Spectrometry. 14, 9:
1057-1066.
[46] . Michael S. M., Chien M., and Lubman D. M. (1992). «An ion trap storage/time-of-flight
mass spectrometer». Review of Scientific Instruments. 63, 10: 4277—4284.
[47] . Mitani K., Fujioka M., and Kataoka H. (2005). «Fully automated analysis of estrogens in
environmental waters by in-tube solid-phase microextraction coupled with liquid chroma-
tography-tandem mass spectrometry». Journal of Chromatography A. 1081, 2: 218—224.
[48] . Nakamura S., Sian T. H., and Daishima S. (2001). «Determination of estrogens in river wa-
ter by gas chromatography—negative-ion chemical-ionization mass spectrometry». Journal
of Chromatography A. 919, 2: 275—282.
[49] . Nourse B. D., Cox K. A., Morand K. L., and Cooks R. G. (1992). «Collisional activation of
pyrene and anthracene in an ion-trap mass spectrometer». Journal of the American Chemical
Society. 114, 6: 2010-2016.
[50] . Orhan H., Coolen S., and Meerman J. H. (2005). «Quantification of urinary o,o9-dity-
rosine, a biomarker for oxidative damage to proteins, by high performance liquid chroma-
tography with triple quadrupole tandem mass spectrometry: A comparison with ion-trap
tandem mass spectrometry». Journal of Chromatography B. 827, 1: 104—108.
[51] . Pirie A. (1971). «Formation of N'-formylkynurenine in proteins from lens and other sourc-
es by exposure to sunlight». Biochemical Journal. 125, 1: 203.
[52] . Plum A., and Rehorek A. (2005). «Strategies for continuous on-line high performance
liquid chromatography coupled with diode array detection and electrospray tandem mass
spectrometry for process monitoring of sulphonated azo dyes and their intermediates in
anaerobic—aerobic bioreactors». Journal of Chromatography A. 1084, 1—2: 119—133.
[53] . Pyle S. M., Betowski L. D., Marcus A. B., Winnik W., and Brittain R. D. (1997). «Analysis
of polycyclic aromatic hydrocarbons by ion trap tandem mass spectrometry». Journal of
American Society of Mass Spectrometry. 8, 2: 183—190.
[54] . Reemtsma T. (2001). «The use of liquid chromatography-atmospheric pressure ionization-
mass spectrometry in water analysis — Part II: Obstacles». Trends in Analytical Chemistry.
20, 10: 533.
[55] . Rodriguez-Mozaz S., Lopez de Alda M.J., and Barcelo D. (2004). «Picogram per liter level
determination of estrogens in natural waters and waterworks by a fully automated on-line
solid-phase extraction-liquid chromatography-electrospray tandem mass spectrometry
method». Analytical Chemistry. 76, 23: 6998—7006.
[56] . Ryberg H., and Caidahl K. (2007). «Chromatographic and mass spectrometric methods for
quantitative determination of 3-nitrotyrosine in biological samples and their application to
human samples». Journal of Chromatography B. 851, 1—2: 160—171.
126 Глава 4. Применение тандемной масс-спектрометрии для анализа загрязнений
окружающей среды
[57] . Schroder H.Fr. (1993). «Surfactants: non-biodegradable, significant pollutants in sewage
treatment plant effluents: Separation, identification and quantification by liquid chroma-
tography, flow-injection analysis—mass spectrometry and tandem mass spectrometry».
Journal of Chromatography A. 647, 2: 219—234.
[58] . Senko M. W., Hendrickson C. L., Emmett M. R., Shi S. D.-H., and Marshall A.G. (1997).
«External accumulation of ions for enhanced electrospray ionization Fourier transform ion
cyclotron resonance mass spectrometry». Journal of the American Society of Mass Spectrom-
etry. 8, 9: 970-976.
[59] . Shukla K., Qian K., Anderson S., and Futrell J. H. (1990). «Dynamics of tandem mass
spectrometry: a dynamics study of simple C-C bond cleavage in collision-activated dis-
sociation of polyatomic ions at low energy». Journal of the American Society of Mass Spec-
trometry. 1,1: 6—15.
[60] . Sole M., Raldua D., Piferrer E, Barcelo D., and Porte C. (2003). «Feminization of wild
carp, Cyprinus carpio, in a polluted environment: plasma steroid hormones, gonadal mor-
phology and xenobiotic metabolizing system». Comparative Biochemistry and Physiology
Part C: Toxicology and Pharmacology. 136, 2: 145.
[61] . Sparrow M.J. (1987). «Pill method failures». New Zealand Medical Journal. 100, 818: 102.
[62] . Sumpter J. P. (1995). «Feminized responses in fish to environmental estrogens». Toxicology
Letters. 82-83: 737-742.
[63] . Ternes T. A., Bonerz M., Herrmann N., Loffler D., Keller E., Lacida В. B., and Alder A. C.
(2005). «Determination of pharmaceuticals, iodinated contrast media and musk fragranc-
es in sludge by LC tandem MS and GC/MS». Journal of Chromatography A, 1067, 1—2:
213-223.
[64] . Tomaiuolo M., Vecchione G., Grandone E., Cocomazzi N., Casetta B., Di Minno G., and
Margaglione M. (2006). «А new method for determination of plasma homocystine by iso-
tope dilution and electrospray tandem mass spectrometry». Journal of Chromatography B.
842, 1:64-69.
[65] . Varian (2007). Varian: 500-MS IT Mass Spectrometer MS Workstation Version 6, Software
Operation Manual, 03-954077-00: Rev. 3. Varian, Inc., Walnut Creek, California, USA.
[66] . Wilcox В. E., Hendrickson C. L., and Marshall A. G. (2002). «Improved ion extraction from
a linear octopole ion trap: SIM ION analysis and experimental demonstration». Journal of
the American Society of Mass Spectrometry. 13, 11: 1304—1312.
[67] . Willems A. V., Deforce D. L., Van Den Eeckhout E.G., Van Peteghem W. E.C. H., De Leen-
heer A.P., and Van Bocxlaer J. F. (2002). «Analysis of benzo[a]pyrene diol epoxide-DNA
adducts by capillary zone electrophoresis—electrospray ionization—mass spectrometry in
conjunction with sample stacking». Electrophoresis. 23, 24: 4092.
[68] . Winnik W. M., and Kitchin K.T. (2008). «Measurement of oxidative stress parameters us-
ing liquid chromatography—tandem mass spectroscopy (LC—MS/MS)». Toxicology and
Applied Pharmacology. 233, 1: 100—106.
[69] . XiaoX.Y., McCalley D.V., and McEvoy J. (2001). «Analysis of estrogens in river water and
effluents using solid phase extraction and gas chromatography—negative chemical ionisa-
tion mass spectrometry of the pentafluorobenzoyl derivatives». Journal of Chromatography
A. 923, 1-2: 195-204.
Литература
[70] . Yan W., Byrd G. D., and Ogden M. W. (2007). «Quantitation of isoprostane isomers in hu-
man urine from smokers and nonsmokers by LC—MS/MS». Journal of Lipid Research. 48,
7: 1607-1617.
[71] . Yost R.A., and Enke C.G. (1978). «Selected ion fragmentation with a tandem quadrupole
mass spectrometer». Journal of the American Chemical Society. 100, 7: 2274—2275.
[72] . Yost R.A., and Enke C.G. (1979). «Triple quadrupole mass spectrometry for direct mixture
analysis and structure elucidation». Analytical Chemistry. 51, 12: 1251 A— 1264A.
ГЛАВА 5
ИСПОЛЬЗОВАНИЕ
СПЕЦИАЛИЗИРОВАННОГО
ПРОГРАММНОГО ОБЕСПЕЧЕНИЯ
И БИБЛИОТЕК МАСС-СПЕКТРОВ
В АНАЛИЗЕ ОБЪЕКТОВ
ОКРУЖАЮЩЕЙ СРЕДЫ
О.Д. Спаркман
5.1. Введение
Масс-спектрометрия играет ключевую роль в анализе органических веществ, начи-
ная с момента создания метода газовой хроматографии/масс-спектрометрии (ГХ/МС).
Производство коммерчески доступных хроматомасс-спектрометров, оборудован-
ных квадрупольным масс-анализатором (который иногда называют квадрупольным
фильтром масс), фирмой Finnigan Corporation в конце 1960-х годов положило начало
широкому использованию метода масс-спектрометрии в области анализа объектов
окружающей среды. Прибор, выпускаемый компанией Finnigan Corporation, постав-
лялся вместе с портативным компьютером, который использовался для регистрации
и хранения данных. В 1971 году Агентство по охране окружающей среды (Environmental
Protection Agency, ЕРА) приобрело шесть приборов фирмы Finnigan и разместило их
в своих лабораториях, расположенных на территории США. Данное событие можно
считать началом использования метода масс-спектрометрии в области анализа объек-
тов окружающей среды.
Начало производства портативных компьютеров оказало довольно сильное вли-
яние на развитие метода масс-спектрометрии. За несколько лет до этого ряд исследо-
вателей (среди которых следует отметить, например, Клауса Бимана) опубликовали
описание компьютерных алгоритмов обработки хроматомасс-спектральных данных,
реализованных на больших «суперкомпьютерах» (Biemann, 1962). Практически сра-
зу после начала производства портативных компьютеров компанией Digital Equipment
Corporation сотрудники ряда научных лабораторий (основываясь на ранее предложен-
ных алгоритмах) приступили к разработке компьютерных программ, предназначенных
для управления прибором, регистрации и обработки данных. Среди исследователей,
работающих в этом направлении, стоит отметить группу ученых из Школы медицины
Стэнфордского университета (Reynolds et al., 1970), группу ученых, возглавляемую Дже-
ком Холландом и Чаком Свили и работающую на факультете биохимии Мичиганско-
го университета (Sweeley et al., 1974), а также группу, возглавляемую Троком Уотсоном,
в Университете Вандербильта (Watson etal., 1973).
Длительное время регистрация и обработка данных осуществлялись без использо-
вания компьютеров. В те времена оператор должен был внимательно следить за процес-
сом записи хроматограммы по полному ионному току на ленту самописца. В тот момент,
когда хроматографический пик достигал максимума, оператор должен был нажать
на кнопку и начать процесс развертки масс-спектра (которая осуществлялась при ис-
пользовании магнитного секторного или квадрупольного масс-анализатора). При на-
жатии кнопки раздавался звуковой сигнал, другой звуковой сигнал информировал
о том, что масс-спектр был зарегистрирован и прибор готов к регистрации следующего
спектра. После этого оператор мог попытаться успеть зарегистрировать еще один масс-
спектр, прежде чем величина полного ионного тока не достигнет базовой линии. Масс-
спектр записывался на светочувствительную бумагу с помощью самописца, имеющего
три «пера», обладающих различной чувствительностью (рис. 5.1).
Листы бумаги с зарегистрированными масс-спектрами (рис. 5.2) могли упасть
на пол, их нужно было быстро собрать и вручную записать время удерживания соот-
ветствующего хроматографического пика. В то же время оператор должен был принять
решение о регистрации следующего масс-спектра и начать его развертку. Зарегистри-
рованные масс-спектры могли исчезнуть в случае использования люминесцентных
ламп или под воздействием солнечного света. По этой причине обработку зарегистри-
рованных масс-спектров проводили при свечах или использовали лампы накаливания
малой мощности. Шкала значений m/z отсутствовала, и каждый масс-спектральный
пик должен был быть отмечен вручную. Для определения относительной интенсивно-
сти масс-спектральных пиков было необходимо ввести поправку на уровень усиления
Рис. 5.1. Схематическое изо-
бражение процесса записи
масс-спектра на светочувстви-
тельную бумагу с использо-
ванием самописца. Для рас-
ширения динамического
диапазона детектора исполь-
зовалось три «пера», обладаю-
щих различной чувствитель-
ностью (которая определялась
жесткостью пружины). Рису-
нок воспроизведен с разреше-
ния издательства Wiley (опу-
бликован в McFadden W. Н.,
Ed. Techniques of Combined Gas
Chromatography/Mass Spectrome-
try: Application in Organic Analy-
sis’, Wiley Interscience: New York,
1973 и Watson J.T. and Spark-
man O. D. Introduction to Mass
Spectrometry: Instrumentation, Ap-
plications, and Strategies for Data
Interpretation’, Wiley: Chichester,
UK, 2007)
130 Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
300 400
Рис. 5.2. Пример масс-спектра, зарегистрированного на светочувствительную бумагу при
использовании самописца. Численная шкала значений m/z (расположенная вни-
зу рисунка) не была представлена в первых вариантах масс-спектрометров. Рису-
нок воспроизведен с разрешения издательства Wiley (опубликован в Watson J.T.
and Sparkman О. D. Introduction to Mass Spectrometry: Instrumentation, Applications, and
Strategies for Data Interpretation’, Wiley: Chichester, UK, 2007)
сигнала. Масс-спектр должен был быть нарисован от руки, прежде чем можно было бы
приступить к его интерпретации. Из хроматографических данных исследователю была
доступна только хроматограмма по полному ионному току.
В настоящее время построение масс-хроматограмм1 по выбранным значениям m/z
является одной из наиболее простых компьютерных операций, однако первое описание
такого подхода было удостоено публикации в научном журнале (Hites and Biemann, 1970).
О возможности подобного представления данных нельзя было даже мечтать во времена
регистрации масс-спектров с использованием самописцев.
В те времена, когда масс-спектры регистрировали только в аналоговом виде, было
практически невозможно использовать масс-спектральные базы данных для иденти-
фикации неизвестных органических соединений. Библиотечные масс-спектры храни-
лись в бумажном виде и содержали информацию только о восьми наиболее интенсив-
ных пиках, расположенных в порядке уменьшения интенсивностей (что было удобным
при проведении ручного поиска). Кроме того, были доступны атласы масс-спектров
(Stenhagen et al., 1969), в которых данные были представлены в графическом виде и мог-
ли быть использованы для визуального сопоставления. В первых компьютерных базах
1 При сборе ГХ/МС-данных фактически регистрируются только масс-спектры. Масс-
спектры регистрируются последовательно один за другим, диапазон сканирования и время реги-
страции одного спектра обычно не меняются в течение анализа. Полученные масс-спектры мо-
гут быть использованы впоследствии для построения хроматограммы по полному ионному току
(которая представляет собой зависимость суммы интенсивностей всех масс-спектральных пиков
от порядкового номера соответствующего спектра) или масс-хроматограмм (в этом случае по оси
ординат откладывают интенсивность ионов с определенным значением m/z или сумму интенсив-
ностей, отвечающих нескольким значениям m/z (глава 2)).
5.1. Введение
данных масс-спектры также были представлены в сокращенном виде (что объяснялось
ограниченными возможностями используемых в то время компьютеров). Один из наи-
более известных подходов к сокращению масс-спектральной информации был пред-
ложен Хайтсом и Биманом и заключался в том, что на каждом участке масс-спектра
шириной 14 атомных единиц массы (начиная со значения m/z, равного 5) рассматрива-
ли только два наиболее интенсивных пика. Компьютерные базы данных, содержащие
«полные» масс-спектры (включающие в том числе и изотопные пики), начали появлять-
ся только в середине 1980-х годов.
Хроматомасс-спектральные данные представляют собой набор последовательно
зарегистрированных масс-спектров, по этой причине можно сказать, что построение
любой хроматограммы заключается в «переорганизации» масс-спектральных данных.
Одиночный симметричный пик на хроматограмме по полному ионному току может
представлять собой случай неполного разделения нескольких компонентов (глава 2),
однако при регистрации данных с использованием самописцев установить этот факт
было практически невозможно. В этом случае масс-спектр, отвечающий максимуму
пика, зарегистрированного на хроматограмме по полному ионному току, представлял
собой суперпозицию масс-спектров соответствующих соединений. В связи с этим ра-
боты ряда исследователей были направлены на разработку компьютерных алгоритмов
автоматического поиска компонентов и выделения «чистых» масс-спектров (Biller and
Biemann, 1974; Colby, 1992; Stein, 1999). На сегодняшний день наиболее широко исполь-
зуемым программным обеспечением, предназначенным для автоматической обработки
хроматомасс-спектральных данных, является программа AM DIS (Automated Mass Spec-
tral Deconvolution and Identification System), разработанная сотрудниками Националь-
ного института стандартов и технологий США (NIST). Более подробная информация
о программном обеспечении AMDIS представлена в разделе 5.14.
Компьютерные программы, предназначенные для регистрации и обработки хро-
матомасс-спектральных данных, постоянно совершенствуются и в настоящее время
позволяют: проводить выделение «чистых» масс-спектров неполностью разделенных
компонентов; осуществлять поиск по базам данных масс-спектров; отображать полные
масс-спектры и структурные формулы соединений, представленных в базах данных;
устанавливать молекулярные массы многозарядных ионов; проводить поиск по базам
данных индексов удерживания; выполнять ряд других действий, облегчающих интер-
претацию данных, проведение количественного анализа и идентификацию. Сегодня
для запуска программ, предназначенных для регистрации и обработки хроматомасс-
спектральных данных, используют персональные компьютеры, работающие под управ-
лением операционной системы Microsoft Windows.
Наиболее важную роль в задаче установления структуры органических соеди-
нений в настоящее время играет масс-спектрометрия с ионизацией электронами
(ИЭ), которая относится к жестким методам ионизации. При использовании масс-
спектрометрии ИЭ наблюдается интенсивная фрагментация молекулярного иона
и инициируется большое число путей распада. Это приводит к тому, что масс-спектры
ИЭ могут, с одной стороны, иметь довольно сложный состав, а с другой — быть весь-
ма характерными, что позволяет в ряде случаев проводить однозначную идентифика-
цию неизвестных соединений (глава 1). Поскольку масс-спектрометрия ИЭ является
жестким методом ионизации, пик молекулярного иона отсутствует в масс-спектрах
132 Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
ИЭ порядка 20% соединений (около 40000 спектров), представленных в базе данных
масс-спектров NIST11. Сложный состав масс-спектров ИЭ, а также возможность от-
сутствия пика молекулярного иона приводят к необходимости использования авто-
матического поиска по базам данных при проведении идентификации неизвестных
органических соединений.
Создание электронных баз данных масс-спектров и программ библиотечного по-
иска было только одним из первых шагов компьютеризации процесса обработки хро-
матомасс-спектральных данных. Как будет показано далее в этой главе, использование
компьютерных программ позволяет увеличить надежность информации, полученной
при проведении ГХ/МС-анализа объектов окружающей среды. В настоящее время ком-
пьютерные программы позволяют выполнять различные операции, начиная с создания
автоматических отчетов при осуществлении контроля качества продукции и заканчи-
вая автоматическим поиском компонентов и выделением «чистых» масс-спектров коэ-
люируемых соединений.
5.2. Базы данных масс-спектров и программы
библиотечного поиска
При обсуждении вопросов библиотечного поиска необходимо разграничивать два по-
нятия: база данных масс-спектров и программа, предназначенная для поиска по базе
данных. База данных представляет собой набор масс-спектров различных соединений.
Для каждого соединения в базе данных, помимо масс-спектра, представлена также до-
полнительная информация, например название, молекулярная масса, структурная
формула и так далее. Программы библиотечного поиска предназначены для проведе-
ния автоматического сопоставления экспериментального масс-спектра со спектрами,
представленными в базе данных (при использовании специальных математических ал-
горитмов). Вне зависимости от используемого алгоритма возможности библиотечного
поиска лимитированы качеством масс-спектров, представленных в используемой базе
данных. Когда исследователь сообщает о том, что соединение было идентифицировано
с использованием базы данных масс-спектров, он должен указать не только саму базу
данных, но также и алгоритм и/или программу, используемые при проведении поис-
ка. Например, база данных — NIST11 (mainlib и replib), программа библиотечного поис-
ка — ChemStation Е.00.00.176 (алгоритм библиотечного поиска — РВМ). В приведенном
примере при выполнении поиска по базе данных NIST11 были использованы две под-
библиотеки mainlib и replib2 (однако можно использовать только одну из указанных под-
библиотек, например mainlib).
2 В базе данных NIST масс-спектры ИЭ представлены в двух подбиблиотеках: mainlib и replib.
Подбиблиотека mainlib содержит только один масс-спектр каждого соединения. Этот масс-
спектр обладает наилучшим качеством по сравнению с другими спектрами данного соединения,
представленными в базе данных NIST. В подбиблиотеке replib содержатся дополнительные
масс-спектры соединений, уже представленных в mainlib. Во многих случаях для соединений,
представляющих особое значение (например для загрязняющих веществ или запрещенных
препаратов), в базе данных представлено несколько масс-спектров ИЭ (при этом подразумевается,
что данные спектры не являются копией одного и того же масс-спектра, а были зарегистрированы
несколькими исследователями при различных экспериментальных условиях).
5.3. Развитие баз данных масс-спектров I
5.3. Развитие баз данных масс-спектров
В 1971 году вскоре после приобретения первых шести ГХ/МС-приборов Агентство
по охране окружающей среды США заключило контракт с Мемориальным институ-
том Баттеля на разработку системы, предназначенной для передачи данных по теле-
фонным линиям между портативным компьютером, подсоединенным непосредствен-
но к ГХ/МС-прибору, и удаленным большим «суперкомпьютером», используемым для
проведения поиска по базам данных. Используемый алгоритм библиотечного поиска
был разработан в Массачусетском технологическом университете Биманом и упоми-
нался ранее в этой главе. Названия соединений, масс-спектры которых были похожи
на экспериментальный масс-спектр неизвестного соединения, автоматически переда-
вались на портативный компьютер и были доступны пользователю. Соединения рас-
полагались в порядке уменьшения величины степени совпадения, рассчитанной между
библиотечным и экспериментальным масс-спектрами. Величина степени совпадения
(значение которой находилось в диапазоне от 0 до 1) также была доступна пользовате-
лю. Цель разработки такой системы заключалась в создании возможности проведения
предположительной идентификации неизвестных соединений без необходимости при-
влечения высококвалифицированных масс-спектрометристов. База данных Агентства
по охране окружающей среды состояла из масс-спектров, зарегистрированных в Центре
масс-спектральных данных в Олдермастоне. Данная библиотека масс-спектров объе-
диняла несколько небольших коллекций, принадлежащих ряду организаций (Амери-
канскому институту нефти, компании Dow Chemicals, Американскому обществу по ис-
пытанию материалов), а также некоторое количество других малочисленных наборов
масс-спектров. Изначально база данных содержала около 10600 спектров (при этом
для некоторых соединений было представлено более одного масс-спектра). Агентство
по охране окружающей среды добавило в эту базу данных порядка 600 масс-спектров
загрязняющих веществ окружающей среды (Heller and Milne, 1979). Существенным
ограничением системы, созданной Агентством по охране окружающей среды, а также
первых программ поиска по базам данных с использованием миникомпьютера была не-
обходимость возвращения исследователю спектра соединения, присланного для прове-
дения идентификации, т. е. база данных не пополнялась новыми спектрами.
Примерно в то же время сотрудники Национального института сердца и легких,
являющегося подразделением Национального института здравоохранения США, раз-
рабатывали аналогичную систему идентификации неизвестных соединений, исполь-
зующую несколько обновленную библиотеку масс-спектров, зарегистрированных
в Центре масс-спектральных данных в Олдермастоне. Портативный компьютер ис-
пользовался для последовательного ввода информации о каждом масс-спектральном
пике, введенное значение m/z и интенсивность пика передавались по телефонным
линиям на большой «суперкомпьютер». При этом портативный компьютер мог быть
не подключен к ГХ/МС-прибору, поскольку использовался только как устройство вво-
да-вывода и передачи информации. После ввода информации о каждом следующем
масс-спектральном пике на портативный компьютер передавалась информация о чис-
ле библиотечных масс-спектров, удовлетворяющих критериям поиска (разница между
интенсивностями пиков в экспериментальном и библиотечном масс-спектре не должна
была превышать определенное значение). Число рассматриваемых масс-спектральных
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
пиков увеличивалось до тех пор, пока количество соединений в списке возможных кан-
дидатов не уменьшалось до приемлемого значения. После этого пользователь мог по-
слать запрос и получить названия соединений из списка. Система масс-спектральной
идентификации, разработанная в Национальном институте здравоохранения, имела
несколько преимуществ по сравнению с системой, разработанной Агентством по охра-
не окружающей среды. Во-первых, при проведении поиска по базе данных можно было
задать только несколько масс-спектральных пиков, а также указать ряд дополнитель-
ных параметров поиска: значение номинальной молекулярной массы соединения, брут-
то-формулу или наличие определенных элементов, массу фрагментов, элиминируемых
из молекулярного иона в процессе распада, классификационный код паспорта безопас-
ности. Во-вторых, «полный» масс-спектр соединения, представленный в базе данных,
мог быть передан на портативный компьютер и доступен пользователю в числовом или
графическом виде. Поскольку окончательное решение относительно идентификации
принималось пользователем, считалось, что эта система была рассчитана на опытных
масс-спектрометристов. Еще одним преимуществом системы, разработанной в Наци-
ональном институте здравоохранения, было то, что библиотечные масс-спектры, по-
хожие на спектр неизвестного соединения, могли быть извлечены из базы данных и ис-
пользованы для облегчения процесса идентификации неизвестного соединения в том
случае, когда его масс-спектр отсутствовал в базе данных.
Приблизительно в 1975 году эти две системы были объединены. В проекте был ис-
пользован коммерческий большой «суперкомпьютер», а также современная по тем вре-
менам сеть передачи данных, позволяющая устанавливать соединение при помощи теле-
фонных звонков из многих городов США, а также других стран. В этот проект помимо
организаций, упомянутыхранее, оказалось также вовлечено Управление по контролю ка-
чества пищевых продуктов и лекарственных средств. Объединенная база данных состоя-
ла из расширенной версии оригинальной коллекции Центра масс-спектральных данных
в Олдермастоне, масс-спектров базы данных Wiley и масс-спектров, зарегистрированных
Агентством по охране окружающей среды. Общее число масс-спектров, содержащихся
в базе данных, составляло порядка 30000. Эта поисковая система, получившая название
Mass Spectral Search System (MSSS), позволяла передавать «полные» масс-спектры, а так-
же производить поиск по базе данных, учитывая дополнительные параметры. Переда-
ча данных осуществлялась по телефонным линиям. В то же время Национальное бюро
стандартов (которое позже было переименовано в Национальный институт стандартов
и технологий) предоставляло возможность годового использования базы данных, соз-
данной совместно Национальным институтом здравоохранения и Агентством по ох-
ране окружающей среды. В базе данных было представлено около 12500 масс-спектров
(для каждого соединения был представлен только один масс-спектр). В этой версии базы
данных масс-спектры были представлены в сокращенном виде (на каждом участке масс-
спектра шириной 14 атомных единиц массы присутствовали только два наиболее интен-
сивных пика). Для проведения поиска по базе данных необходимо было использовать
ранее зарегистрированный масс-спектр (поиск по другим параметрам был недоступен).
К 1978 году база данных масс-спектров, создаваемая совместно Национальным инсти-
тутом стандартов и технологий, Агентством по охране окружающей среды и Националь-
ным институтом здравоохранения (NIST/EPA/NIH), насчитывала 25556 масс-спектров.
Издание 1980 года содержало уже 34363 масс-спектра, а в 1994 году база данных была
5.4. Базы данных масс-спектров
расширена до 74828 спектров (12593 из которых были дополнительными масс-спектрами
соединений, уже включенных в базу данных). База данных NIST113 (выпущенная в мае
2011 года) содержит 243893 масс-спектра и 212961 соединений.
Параллельно с базой данных NIST/EPA/NIH развивалась база данных Wiley, ко-
торая берет свое начало с серии книг (под названием Atlas of Mass Spectra), изданных
в 1969 году под редакцией Стэнхагена, Абрахамсона и МакЛафферти. База данных была
издана тремя томами и содержала около 6800 масс-спектров, представленных в таблич-
ном виде (Sparkman, 2009).
Одновременно с выходом бумажной версии базы данных издательство Wiley также
выпустило электронную версию, содержащую масс-спектры в цифровом формате. Сле-
дующее четырехтомное издание (получившее название «Wiley Registry of Mass Spectral
Data») было опубликовано в 1974 году и содержало масс-спектры 18806 соединений,
представленные в графическом виде. Электронная версия базы данных также была до-
ступна и включала 5073 дополнительных масс-спектра соединений, уже представлен-
ных в бумажной версии. Эта версия электронной базы данных Wiley была установлена
на портативные компьютеры, поставляемые компаниями Hewlett Packard и Shimadzu
с коммерческими ГХ/МС-приборами. Это стало возможным благодаря созданию по-
исковой системы MSSS EPA/NIH/MSDC и покупки базы данных Wiley правительством
США. В 1989 году был издан семитомный сборник масс-спектров (под редакцией Мак-
Лафферти и Штауфера), получивший название The Wiley/NBS Registry of Mass Spectral
Data и содержащий масс-спектры 112272 различных соединений (73978 из которых были
представлены в базе данных Wiley, 6117 — в базе данных Национального бюро стандар-
тов и 32180 — в обеих базах данных). В последней версии базы данных Wiley (девятое
издание) содержится 669092 масс-спектра более чем 592000 соединений (база данных
доступна только в электронном формате).
5.4. Базы данных масс-спектров
На сегодняшний день существуют две неспециализированные, универсальные базы
данных масс-спектров ИЭ: база данных NIST/EPA/NIH (N1ST11, 2017), история соз-
дания которой была описана ранее в этой главе, и база данных Wiley (Мс Lafferty, 2010),
которая в настоящее время поддерживается и редактируется в Германии компанией
VCH Data Group (John Wiley and Sons)4. Для большинства соединений, находящихся
3 База данных NIST/EPA/NIH распространялась под различными названиями. В 1998 году
Национальный институт стандартов и технологий принял следующий формат названия: NISTYY
(где YY — две последние цифры года выпуска базы данных). Начиная с 1998 года было выпуще-
но несколько редакций базы данных NIST: NIST98, NIST02, NIST05 и NIST08. Последняя версия
базы данных получила название NIST11. Каждый раз, когда Национальный институт стандартов
и технологий выпускает новую версию базы данных, также издается новая версия программного
обеспечения MS Search, которая содержит ряд усовершенствований. Компания Agilent использу-
ет в названии базы данных NIST аббревиатуру Национального бюро стандартов (которое было
переименовано в Национальный институт стандартов и технологий) — NBS, а также число масс-
спектров, представленных в базе данных, например NBS128K.L.
4 Издательство John Wiley также предоставляет базу данных Wiley, объединенную с базой
данных NIST. Базы данных Wiley и NIST содержат ряд одинаковых спектров, однакоони не дубли-
руются в объединенной базе данных.
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
в этих двух базах данных, помимо названия представлена также структурная форму-
ла. В случае базы данных NIST11 дополнительные масс-спектры находятся в отдель-
ном файле. Однако некоторые производители масс-спектрального оборудования (на-
пример Agilent Technologies, Shimadzu или Waters Corporation) при использовании их
собственного программного обеспечения объединяют файлы, содержащие основные
и дополнительные масс-спектры, в один. В случае базы данных Wiley все масс-спектры
находятся в одном файле. Для каждого масс-спектра, представленного в базе данных,
помимо набора пар чисел (интенсивность и значение /и/z), присутствует структурная
формула, информация об основном и альтернативных названиях соединения, эле-
ментном составе, номинальной5 молекулярной массе, регистрационном номере CAS
и порядковом номере масс-спектра в базе данных. В зависимости от используемого
программного обеспечения информация, занесенная в поле Comments («Коммента-
рии»), также может быть использована при проведении библиотечного поиска. База
данных NIST/EPA/NIH поддерживается Центром масс-спектральных данных (рас-
положенным в городе Гейтесберг в США), являющимся подразделением Националь-
ного института стандартов и технологий. База данных NIST/EPA/NIH-индексируется
по структурным фрагментам6 молекулы. Каждый масс-спектр, представленный в базе
данных NIST/EPA/NIH, характеризуется уникальным номером NIST (связывающим
конкретный масс-спектр с данными, хранящимися в архиве института), который также
может быть использован при проведении поиска. В библиотеке NIST/EPA/NIH, кроме
всего прочего, присутствует информация о других базах данных, в которых представ-
лена информация о соответствующем соединении. Для большого числа масс-спектров
из базы данных NIST/EPA/NIH в поле «Contributor» представлена информация об ис-
следователе (и/или организации), который предоставил данный спектр. В базе данных
Wiley информация о происхождении масс-спектра также представлена, однако она со-
держится в поле Comments («Комментарии») в зашифрованном виде и не может быть
расшифрована пользователем.
Обычно внимание пользователей акцентируется на том, что база данных Wi-
ley содержит более чем в два раза больше соединений по сравнению с базой данных
NIST/EPA/NIH (в базах данных Wiley 9th и NIST11 представлено 663000 и 244000 масс-
спектров соответственно). Однако базы данных отличаются не только числом масс-
спектров. Другим важным аспектом является качество библиотечных масс-спектров.
Масс-спектры, представленные в базе данных NIST/EPA/NIH, постоянно проверяют-
ся, что позволяет находить и исключать «неправильные» спектры (масс-спектры, ко-
торые не соответствуют структуре; масс-спектры, для которых название и структура
не совпадают; масс-спектры, отвечающие не индивидуальному веществу, а смеси со-
единений и так далее), а также заменять их на более качественные (Ausloos et al., 1999).
Создатели базы данных Wiley 9th не сообщают о проведении подобной проверки масс-
5 Номинальная атомная масса элемента представляет собой массу наиболее распространен-
ного в природе изотопа, округленную до ближайшего целочисленного значения. Номинальная
молекулярная масса соединения рассчитывается как сумма номинальных масс элементов, входя-
щих в его состав.
6 В случае базы данных NIST термин, структурные фрагменты, (подструктуры) включает
в себя: гетероатомы, функциональные группы (фенильная, сложноэфирная, карбонильная груп-
пы и др.), число эквивалентов колец и двойных связей, а также другие фрагменты, использующи-
еся для описания молекулы.
спектров. Фрэд МакЛафферти, являющийся автором и редактором базы данных Wi-
ley 9th, отмечал, что если неправильный масс-спектр был опубликован для какого-либо
соединения, то он должен присутствовать в базе данных.
Было бы необоснованно говорить, что масс-спектры хорошего качества отсутству-
ют в базе данных Wiley 9th или что масс-спектры низкого качества отсутствуют в базе
данных NIST11. В базе данных Wiley присутствует большое число масс-спектров хоро-
шего качества, также как в базе данных NIST/EPA/NIH присутствует ряд масс-спектров
низкого качества, однако это число постоянно уменьшается благодаря работам, направ-
ленным на проверку качества библиотечных масс-спектров. Поскольку размер базы
данных Wiley 9th более чем в два раза превосходит размер базы данных NIST11, доста-
точно большое число соединений, присутствующих в базе данных Wiley, отсутствует
в базе данных N 1ST. Однако следует отметить, что некоторые соединения, представлен-
ные в базе данных NIST11, отсутствуют в последней версии базы данных Wiley. В связи
с этим наличие двух баз данных является оптимальным, если деятельность лаборато-
рии в основном связана с идентификацией неизвестных соединений. Несмотря на от-
носительно большой объем дискового пространства, необходимого для установки двух
баз данных (Wiley и NIST), такой подход является достаточно удобным. Вначале можно
проводить поиск с использованием одной только базы данных NIST11. Если коррект-
ные результаты не будут получены, поиск может быть выполнен с использованием объ-
единенной базы данных Wiley/NIST.
5.5. Другие базы данных масс-спектров
В дополнение к двум большим и универсальным базам данных существует ряд специ-
ализированных баз данных меньшего размера, представляющих интерес для ученых,
работающих в области анализа объектов окружающей среды. Одними из наиболее вос-
требованных являются библиотеки масс-спектров ИЭ пестицидов. Помимо баз данных
пестицидов, поставляемых производителями масс-спектрального оборудования, су-
ществует база данных, распространяемая издательством John Wiley and Sons и содержа-
щая 1238 масс-спектров ИЭ: Mass Spectra of Pesticides 2009 (автор — Рольф Кюнле; ISBN:
978-3-527-32488-0). Издательство Wiley также распространяет (в печатном и электрон-
ном виде) базу данных масс-спектров ИЭ, которая содержит помимо пестицидов другие
классы органических соединений и насчитывает 7840 масс-спектров и времен удержи-
вания: Mass Spectral Library of Drugs, Poisons, Pesticides, Pollutants and Their Metabolites
2007 (авторы — Ханс Маурер, Карл Пфлегер и Армин Вебер; ISBN: 978-3-527-32146-9),
эта база данных также известна под названием MPW. Для каждого соединения, входя-
щего в состав одной из двух вышеупомянутых баз данных, представлена информация
о структурной формуле вещества и времени удерживания. Все базы данных издатель-
ства Wiley распространяются в форматах, совместимых с программным обеспечением
производителей масс-спектрального оборудования, а также в формате программного
обеспечения NIST MS Search.
Компания Agilent Technologies предоставляет базу данных Stan Pesticides MS Library
(Agilent P/N G1028), содержащую 340 масс-спектров ИЭ пестицидов, а также две базы
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
данных фиксированных времен удерживания (retention time locking, RTL)7: Pesticides
RTL Library (G1672AA), содержащую 926 масс-спектров, и Japanese Positive List Pesticide
RTL Library (G1675AA), содержащую 431 масс-спектр. Компания Agilent также издает
базу данных масс-спектров среднелетучих органических соединений, насчитываю-
щую 273 спектра (Environmental Semi-VOAS RTL Library (GA1677AA)), и базу данных
масс-спектров токсичных веществ (которые могут присутствовать в воздухе рабочей
зоны), насчитывающую 171 масс-спектр (Indoor Air Toxics RTL Library (G1673AA)). По-
мимо указанных баз данных компания Agilent предоставляет базу данных вредных
органических веществ, содержащую 731 масс-спектр: Hazardous Chemical RTL Library
(G1671AA). Ни в одной из вышеупомянутых баз данных компании Agilent не представ-
лены структурные формулы соединений, однако для каждого соединения содержится
информация о названии, брутто-формуле, номинальной молекулярной массе и реги-
страционном номере CAS.
Все масс-спектральные базы данных компании Agilent предоставляются только
в формате, совместимом с программным обеспечением Agilent ChemStation. Упомяну-
тые базы данных могут быть сконвертированы в формат, используемый в программ-
ном обеспечении NIST MS Search, с помощью программы Lib2NIST (распространяемой
вместе с базой данных NIST/EPA/NIH и программным обеспечением NIST MS Search).
При открытии баз данных компании Agilent в программном обеспечении NIST MS
Search каждому масс-спектру автоматически будет поставлена в соответствие струк-
турная формула, однако это произойдет только в том случае, если база данных NIST
или Wiley также установлена и содержит масс-спектр рассматриваемого соединения
(это объясняется тем, что структура добавляется к масс-спектру в случае совпадения
номеров CAS). Базы данных пестицидов компании Agilent, а также база данных Mass
Spectra of Pesticides 2009, распространяемая издательством Wiley, не содержат ни одно-
го дополнительного соединения по сравнению с универсальными библиотеками NIST
или Wiley8.
Во многих компаниях, государственных и научно-исследовательских учреждени-
ях были созданы собственные внутрилабораторные базы данных, содержащие масс-
спектры не только наиболее распространенных органических соединений, но также
весьма редких соединений, встречающихся при проведении анализов объектов окру-
жающей среды, объектов криминалистики, добавок к полимерам и других объектов.
Агентство по охране окружающей среды США опубликовало методики, применяемые
при проведении анализов объектов окружающей среды, в основе которых лежит метод
ГХ/МС. В этих методиках четко указано, что при анализе целевых соединений база дан-
ных должна быть получена на том же приборе, на котором впоследствии будет прово-
диться анализ (что приводит к необходимости создания внутрилабораторных баз дан-
ных перед проведением рутинных анализов).
7 Базы данных фиксированных времен удерживания (retention time locking, RTL) упомина-
ются далее в этой главе совместно с программным обеспечением Agilent Deconvolution Reporting
Software.
8 Следует отметить, что в случае программного обеспечения Agilent ChemStation (в отличие
от любой другой программы библиотечного поиска) для того, чтобы получить доступ к структур-
ным формулам соединений, представленных в базе данных масс-спектров, необходимо вручную
указать файл, содержащий структурные формулы (используя режим ViewParametric Retrieval).
Структурные формулы и масс-спектры связаны между собой при помощи номеров CAS.
5.6. Выбор диапазона сканирования в случае проведения поиска
по базам данных масс-спектров
5.6. Выбор диапазона сканирования в случае
проведения поиска по базам данных масс-
спектров
Ситуация, при которой молекулярная масса соединения превышает верхнюю грани-
цу диапазона регистрируемых значений m/z, встречается весьма редко. Однако можно
найти довольно много библиотечных масс-спектров, для которых нижняя граница диа-
пазона сканирования была выбрана таким образом, что один или несколько довольно
интенсивных масс-спектральных пиков, обладающих низкими значениями m/z, про-
сто не были зарегистрированы. Хороший пример, иллюстрирующий данную проблему,
представлен на рис. 5.3.
При проведении анализа объектов окружающей среды нижняя граница диапазо-
на сканирования масс-спектрометра обычно составляет 35 единиц т/z- Использование
данного значения объясняется необходимостью исключить из масс-спектра пики, обу-
словленные присутствием воздуха (m/z28, 32) и воды (m/z 18). При использовании метода
выдувания и улавливания (purge-and-trap) диоксид углерода {m/z 44) также может ока-
зать мешающее влияние, в этихслучаях нижняя граница диапазона сканирования уста-
навливается равной 45. Однако увеличение нижней границы диапазона сканирования
Рис. 5.3. Масс-спектры ИЭ 2-хлор-1,3,5-тринитробензола. Нижняя граница диапазона
сканирования была равна 10 в случае масс-спектра, представленного вверху, и 40
в случае масс-спектра, представленного внизу. Отсутствие масс-спектрального
пика со значением m/z 30 в экспериментальном спектре гипотетически может
привести к неправильной идентификации, основанной на использовании баз
данных масс-спектров ИЭ
140 Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
всего на 15 единиц m/z может оказаться весьма существенным при проведении анализа
некоторых классов органических соединений, например алифатических аминов или
спиртов. В масс-спектрах алифатических аминов часто присутствует достаточно ин-
тенсивный пик со значением m/z 30, в масс-спектрах алифатических спиртов и эфиров
может присутствовать пик со значением m/z 31. Отсутствие интенсивных пиков в масс-
спектре со значениями m/z меньше 35 или 45 может привести к неудовлетворительным
результатам библиотечного поиска, а также усложнить ручную интерпретацию масс-
спектров. При проведении поиска по базам данных необходимо внимательно изучать
библиотечный масс-спектр возможного кандидата, чтобы оценить значение нижней
границы диапазона сканирования.
Следует также отметить, что попадание воздуха в хроматомасс-спектрометр (кото-
рое можно определить по интенсивным пикам воды {m/z 18), азота {m/z 28) и кислоро-
да (m/z 32)) оказывает негативное воздействие на неподвижную фазу и сокращает срок
службы хроматографической колонки.
5.7. Программное обеспечение, используемое для
проведения поиска по базам данных масс-
спектров
Как было отмечено ранее, использование компьютерных программ значительно упро-
щает процесс обработки масс-спектральных данных. В настоящее время одной из наи-
более востребованных функций программного обеспечения является поиск по базам
данных масс-спектров ИЭ (или, другими словами, библиотечный поиск). Это объяс-
няется тем, что ГХ/МС(ИЭ) является одним из наиболее популярных методов анализа
объектов окружающей среды.
Поиск по базам данных масс-спектров может значительно упростить задачу иден-
тификации неизвестного органического соединения. Однако корректность результата
идентификации, предложенного программой библиотечного поиска, должна быть про-
верена опытным масс-спектрометристом. В противном случае (несмотря даже на вы-
сокую степень совпадения экспериментального и библиотечного масс-спектров) ре-
зультатом идентификации по базе данных может оказаться соединение, которое ни при
каких условиях не могло быть найдено в исследуемом образце. Достаточно часто можно
встретить ситуацию, когда исследователи делают вывод о том, что неизвестное соеди-
нение, обнаруженное при анализе какого-либо образца, является соединением X, осно-
вываясь только на том, что соединение X возглавляло список возможных кандидатов,
полученный при проведении поиска по базе данных масс-спектров ИЭ. В таких ситуа-
циях важно помнить, что программы библиотечного поиска не проводят идентифика-
цию неизвестного соединения, а только помогают исследователю принять верное реше-
ние. Достоверность идентификации неизвестного соединения можно оценить, ответив
на следующие вопросы.
• Действительно ли масс-спектр неизвестного соединения присутствует в базе
данных? Или в базе данных присутствуют только соединения, обладающие по-
хожими масс-спектрами?
5.7. Программное обеспечение, используемое для проведения поиска
по базам данных масс-спектров
• Присутствуют ли в базе данных соединения, обладающие практически идентич-
ными масс-спектрами ИЭ, как, например, о-, м-, л-изомеры диметилбензола?
• Доступны ли данные, полученные с помощью других физико-химических ме-
тодов анализа? Например, известно ли время удерживания, точная молекуляр-
ная масса или точные массы фрагментных ионов (позволяющие подтвердить
элементный состав), зарегистрированы ли ИК-, УФ- или ЯМР-спектры, при-
сутствует ли соединение (выбранное в качестве наиболее вероятного кандидата)
в других базах данных, подтверждена ли величина молекулярной массы (номи-
нальной и/или моноизотопной) с помощью масс-спектрометрии с химической
ионизацией?
• Являются ли результаты библиотечного поиска разумными с точки зрения про-
исхождения образца?
• Проведен ли анализ неизвестного соединения и предполагаемого кандидата в од-
них и тех же условиях?
Как было отмечено ранее в этой главе, существуют два разных понятия — база
данных масс-спектров и программа библиотечного поиска. На сегодняшний день су-
ществуют два основных алгоритма поиска: алгоритм «вероятностного» сопоставления
спектров (probability-based matching, РВМ), предложенный МакЛафферти и использу-
емый в программном обеспечении компании Agilent Technologies (ранее носившей на-
звание Hewlett Packard) и Shimadzu Instruments, а также алгоритм (в основе которого ле-
жит модифицированный метод скалярного произведения), разработанный компанией
INCOS9 и модифицированный впоследствии сотрудниками Национального института
стандартов и технологий (в настоящее время этот алгоритм используется в программ-
ном обеспечении NIST MS Search). Кроме того, данная модификация метода скалярно-
го произведения используется в программном обеспечении, поставляемом с приборами
ряда производителей масс-спектрального оборудования. Третий алгоритм библиотеч-
ного поиска (являющийся еще одной модификацией метода скалярного произведения)
используется исключительно компанией Waters Corporation и реализован в программ-
ном обеспечении MassLynx. Однако компания Waters (также как и компания Agilent)
распространяет программу NIST MS Search наряду с собственным программным обе-
спечением (если приборы поставляются с базой данных NIST/EPA/NIH). Программное
обеспечение компаний Agilent и Waters позволяет передавать экспериментальный масс-
спектр непосредственно в программу NIST MS Search и использовать ее для проведения
библиотечного поиска.
Следует отметить, что подробное обсуждение всех особенностей и возможностей,
доступных при использовании программ библиотечного поиска (таких, например, как
Agilent ChemStation или NIST MS Search), выходит за рамки этой главы. В данной главе
дается краткий обзор доступных в настоящее время компьютерных программ и описы-
ваются их основные возможности.
Один из наиболее популярных вопросов, возникающих у исследователей, работа-
ющих в области идентификации органических соединений, — какой алгоритм библи-
9 В середине 1970-х годов компания INCOS разрабатывала и продавала конечным пользо-
вателям программное обеспечение, предназначенное для обработки масс-спектральных данных,
полученных на приборах различных производителей. Компания INCOS была приобретена фир-
мой Finnigan Corporation, сохранившей название программного обеспечения, которое, однако,
стало использоваться впоследствии только с оборудованием компании Finnigan.
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
отечного поиска является наилучшим? Наилучший алгоритм не должен вносить пута-
ницу в процесс идентификации. Однако идеального алгоритма библиотечного поиска
не существует. В представленных ниже примерах экспериментальные масс-спектры не-
скольких неизвестных органических соединений были использованы для проведения
поиска по базе данных NIST0810 (подбиблиотеки mainlib и replib) с помощью программ-
ного обеспечения Agilent ChemStation и NIST MS Search. В одном случае использование
программного обеспечения Agilent ChemStation позволило получить корректный ре-
зультат, в свою очередь интерпретация результатов библиотечного поиска, полученных
с помощью NIST MS Search, требовала вмешательства опытного масс-спектрометриста
(раздел 5.8). В другом примере алгоритм, используемый в программе NIST MS Search,
позволил провести правильную идентификацию, в то время как результаты, получен-
ные с использованием программного обеспечения Agilent ChemStation, были неодно-
значны (раздел 5.9).
5.8. Пример проведения правильной идентификации
с помощью программного обеспечения Agilent
ChemStation
Масс-спектр, отвечающий вершине пика, зарегистрированного на хроматограм-
ме по полному ионному току, представлен на рис. 5.4. При внимательном рассмо-
трении данного спектра можно установить, что ни один из зарегистрированных
масс-спектральных пиков не может отвечать пику молекулярного иона. Пики ионов
с массами 117 и 119 характеризуются наибольшими значениями m/z- Исходя из соот-
ношения интенсивностей этих пиков, можно предположить, что они отвечают одному
и тому же иону, имеющему в своем составе три атома хлора (то есть пик со значением
m/z, равным 119, является изотопным). Наличие пика со значением m/z, равным 97, мо-
жет быть объяснено отщеплением HF (номинальная молекулярная масса равна 20 Да).
Однако анализ изотопного распределения иона со значением m/z, равным 97, показы-
вает, что он содержит только два атома хлора. Изменение изотопного распределения
не могло быть вызвано элиминированием молекулы HF. Следует отметить, что отсут-
ствие пика молекулярного иона в экспериментальном масс-спектре снижает достовер-
ность библиотечного поиска.
Масс-спектр, представленный на рис. 5.4, был использован для проведения библио-
течного поиска по базе данных NIST08 (подбиблиотеки mainlib и replib) при использова-
нии РВМ-алгоритма, реализованного в программном обеспечении Agilent ChemStation
(Е.00.00.173). На рис. 5.5 представлены результаты библиотечного поиска. Первую и вто-
рую строчку в списке возможных кандидатов занимает хлороформ. Величина степени
совпадения (которая в данной программе имеет название Probability) может быть ис-
пользована для определения надежности идентификации. Если величина степени со-
впадения превышает 90, то сопоставляемые масс-спектры довольно похожи. Если вели-
чина степени совпадения не превышает 50, то существуют весьма серьезные различия
10 В данном случае использовали базу данных NIST08, поскольку работа была выполнена
до выхода базы данных NIST11.
5.8. Пример проведения правильной идентификации с помощью программного
обеспечения Agilent ChemStation
Рис. 5.4. Хроматограмма по полному ионному току и масс-спектр ИЭ, зарегистрирован-
ный в максимуме хроматографического пика, отмеченного вертикальной линией
между экспериментальным и библиотечным масс-спектрами, а к соответствующему
кандидату (из базы данных) следует относиться с настороженностью.
Степень совпадения, рассчитанная с помощью РВМ-алгоритма, для соединения,
возглавляющего список возможных кандидатов, составляет всего 38. Это не удивитель-
но, поскольку библиотечный масс-спектр хлороформа содержит только ряд пиков, при-
сутствующих в экспериментальном масс-спектре. Низкое значение степени совпадения
может также указывать на то, что экспериментальный масс-спектр представляет собой
суперпозицию нескольких спектров. Просматривая библиотечные масс-спектры, пред-
ставленные в списке возможных кандидатов, можно заметить, что экспериментальный
масс-спектр содержит пики, присутствующие в библиотечных спектрах двух соедине-
ний — хлороформа и 1,1,1-трихлорэтана. Также как и в случае хлороформа, только часть
пиков из масс-спектра 1,1,1-трихлорэтана (находящегося на 7 и 8 позициях в списке
возможных кандидатов) присутствует в экспериментальном спектре. Совместное рас-
смотрение библиотечных масс-спектров хлороформа и 1,1,1-трихлорэтана (рис. 5.5) по-
зволяет сделать вывод о том, что рассматриваемый хроматографический пик (рис. 5.4)
представляет собой случай неполного разделения этих двух соединений. Следует от-
метить, что 14 и 16 позицию в списке возможных кандидатов также занимают масс-
спектры хлороформа и 1,1,1-трихлорэтана (рис. 5.6).
Масс-спектр, отвечающий вершине хроматографического пика, зарегистриро-
ванного на хроматограмме по полному ионному току (рис. 5.4), был также использо-
ван для проведения библиотечного поиска по базе данных NIST08 при использовании
программного обеспечения NIST MS Search версии 2.0f. На первых четырех позициях
в списке возможных кандидатов находились масс-спектры 1,1,2-трихлорэтана (рис. 5.7).
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
Для всех библиотечных масс-спектров 1,1,2-трихлорэтана степень совпадения Match11
превышала 700 единиц.
Величина Probability (рассчитанная в программном обеспечении NIST MS Search)
для соединения, возглавляющего список возможных кандидатов, составила 84,1 %.
Обычно если значение величины Match превышает 700 одновременно для несколь-
ких библиотечных масс-спектров одного и того же соединения (представленных
в подбиблиотеках mainlib и replib), то вероятность правильной идентификации доста-
точно велика. Однако прежде чем сделать вывод, что неизвестное соединение явля-
ется 1,1,2-трихлорэтаном, нужно вручную сопоставить экспериментальный и библи-
отечный масс-спектр (рис. 5.7). Как видно из рис. 5.7, существует довольно большое
число различий между этими двумя масс-спектрами. В библиотечном масс-спектре
1,1,2-трихлорэтана присутствует группа пиков, отвечающих кластеру молекулярного
иона (ММ 132), изучение которой позволяет сделать вывод о присутствии трех атомов
хлора в молекуле. Эти пики отсутствуют в экспериментальном масс-спектре. В экс-
периментальном масс-спектре присутствует группа пиков (со значениями m/z, рав-
ными 117 и 119), которые отсутствуют в библиотечном масс-спектре. Основной (наи-
более интенсивный) пик имеет значение m/z, равное 83 в случае экспериментального
масс-спектра и 97 в случае библиотечного масс-спектра. Масс-спектр, полученный
при вычитании12 библиотечного масс-спектра (возглавляющего список возможных
кандидатов) из экспериментального, также был использован для проведения поиска
по базе данных; 1,1,1,3,3-пентахлор-2-пропанон возглавил список возможных канди-
датов, степень совпадения Match составила 580, из чего можно было сделать вывод о на-
личии существенных различий между спектрами. Принимая во внимание различия
между экспериментальным и библиотечным масс-спектром, исходные данные должны
быть рассмотрены повторно. Сопоставление спектров, зарегистрированных на левом
11 В программном обеспечении NIST MS Search используются три величины для определе-
ния степени совпадения масс-спектров: Match, Reverse Match и Probability. Величина Match может
изменяться в диапазоне от 0 до 999 и показывает, насколько экспериментальный масс-спектр по-
хож на библиотечный. При расчете величины Match учитывается совпадение как значений m/z, так
и интенсивностей масс-спектральных пиков. При этом во внимание принимаются пики, присут-
ствующие в обоих масс-спектрах. Пики, характеризующиеся большими значениями m/z, имеют
больший вес. Величина Reverse Match отличается тем, что при ее расчете во внимание принима-
ются только пики, присутствующие в библиотечном масс-спектре. Значения m/z, присутствующие
в экспериментальном и отсутствующие в библиотечном масс-спектре, не рассматриваются при
расчете величины Reverse Match. Это может оказаться полезным, если экспериментальный масс-
спектр представляет собой смесь спектров нескольких соединений. Величина Probability (которая
может изменяться в диапазоне от 0 до 99,9%) рассчитывается исходя из «уникальности» библи-
отечного масс-спектра по отношению к другим спектрам, представленным в списке возможных
кандидатов. Например, при проведении библиотечного поиска масс-спектра бензола величина
Probability составляет около 85 %, что объясняется тем, что масс-спектр бензола является достаточ-
но уникальным. Однако при проведении библиотечного поиска масс-спектра jw-диметилбензола
величина Probability составляет порядка 30%, что объясняется присутствием в базе данных масс-
спектров о-, м- и и-изомеров диметилбензола.
12 Одной из особенностей программного обеспечения NIST MS Search является возможность
проведения вычитания интенсивностей пиков, присутствующих в библиотечном масс-спектре,
из интенсивностей соответствующих пиков экспериментального спектра. Результат вычитания
не может быть отрицательным числом. Данная опция, как будет показано далее, может оказаться
полезной в тех случаях, когда экспериментальный масс-спектр представляет собой суперпозицию
масс-спектров нескольких соединений.
5.8. Пример проведения правильной идентификации с помощью программного
обеспечения Agilent ChemStation
Рис. 5.5. Результаты библиотечного поиска, полученные при использовании РВМ-
алгоритма, реализованного в программном обеспечении Agilent ChemStation.
Масс-спектр, расположенный вверху, отвечает максимуму хроматографического
пика, представленного на рис. 5.4. Масс-спектр, представленный внизу, является
библиотечным спектром соединения, занимающего первую — хлороформ (о), или
седьмую — 1,1,1-трихлорэтан (6), позицию в списке возможных кандидатов
и правом склонах рассматриваемого хроматографического пика (рис. 5.8), позволяет
сделать вывод о том, что на этом участке хроматограммы происходит наложение двух
органических соединений, наиболее интенсивные пики в масс-спектрах которых име-
ют значения m/z 83 и 97 соответственно. Построение масс-хроматограмм по указанным
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
Рис. 5.6. Список возможных кандидатов, полученный при использовании РВМ-
алгоритма. В число первых 16 кандидатов входит 3 масс-спектра хлороформа (за-
нимающего 1, 2 и 14 позиции) и 3 масс-спектра 1,1,1-трихлорэтана (занимающего
7, 8 и 16 позиции)
значениям m/z подтверждает факт наложения (рис. 5.8). Вычитание друг из друга масс-
спектров, отвечающих максимумам хроматографических пиков, зарегистрирован-
ных на масс-хроматограммах по ионам 83 и 97, позволило получить очищенные масс-
спектры (рис. 5.8).
Использование масс-спектра, представленного в нижней левой части рис. 5.8, для
проведения поиска по базе данных NIST08 (с использованием программного обеспече-
ния NIST MS Search) позволило получить список возможных кандидатов, верхние три
позиции в котором были заняты хлороформом (только три масс-спектра хлороформа
представлено в базе данных NIST08). Степень совпадения Match и Reverse Match была
равна 904—897 и 906—898 соответственно. Величина Probability составила 76,3%, что
объяснялось похожестью масс-спектров хлороформа и нескольких других соединений,
попавших в список возможных кандидатов. Аналогичное рассмотрение масс-спектра,
представленного в нижней правой части рис. 5.8, позволило получитьсписок возможных
кандидатов, верхние три позиции в котором были заняты 1,1,1-трихлорэтаном (только
три спектра 1,1,1-трихлорэтана представлено в базе данных NIST08). Степень совпаде-
ния Match и Reverse Match была равна 773—805 и 782—814 соответственно. Следует от-
метить, что видимые различия между экспериментальными масс-спектрами (очищен-
5.8. Пример проведения правильной идентификации с помощью программного
обеспечения Agilent ChemStation
|l-Scan 373(821 Б mint ЯТПШР IAd J У| Т1 |>-| QjOj
1 A Scar. <73 (6 216 mini 6JF0830D Dtdata
2 L Ус ar 135 (U 3 mini. IN I / JJ Will *Z5_
3 L Component at scon 1101 (13 361 tit ЦК
4 I sear 5(1(7 072 mn) IK 7i)_RAMP2 5_1
S L Усаг. tU (7.0/2 min) IN /Q.HAMI’Z 5_1
6 L Scan 136(6 396 mini IN 17J_HAMP2 5_‘
г I Яг.яп 23Я (H mmi 1/я|р?0
rromlit, repib. 2TJ/43 total 3p?cVa
100-
5)3 Ы 717 723 841
34 R 709 736 8С.1
5 Ы 667 C?« 8 5=
tj6 M €25 62/1 2 29
7 M €22 6% 2 32
±8 R 621 6E8 2 20
38 R €21 686 2 29
3 10 R 617 651 I 63
II Ы 597 915 0 74
1)12 Ы 572 614 163
13 M 558 574 0 17
14 Ы 551 551 013
15 Ы 525 539 0 04
E10 Ы 521 7ES 0 33
17 Ы 520 «33 0 0’
a19 R 515 521 0 32
тп19 R 514 535 0 32
s23 R 512 774 0 32
121 R 507 522 0 31
22 fl 507 520 0 31
s23 R 506 775 0 33
124R 505 677 0 3?
Ethane, 1,1,2-trichiora-
Ethane, 1.1.2-tnch wo-
Piup«n«.yl uIivikJb, 2^-di<-_
Ethane, 1,1,1 trichiora
Ethane, 2-brcrro-1, -di:hlc
Ethane, 1,1,1-liidi'oro-
Ethane, 1,1 1 tnchioro
Ethane, i.l-icnbro- -rr.ro
Mtlliriie, uxyjik[Ji_riJiD-
Ethane, 1,1 Jcnbto-i inn
Proparci: acd 2,2-dchlen
Ethane. 1,1-Jcnloro-2 2-cf
Propare, 1.2,2 tnebtoro-
Tricnkjromethan»
Propare l.12.2-tPtrErhk!n
Ethane. 1.1 2.2.tei'e: blow.
Ethane. 1,1,2,2-tefashlo’O
Methare, brotnodicl- arc-
MelhylphiMpI onyl dK'iluriJ
2,3-СкГ1хохэроту1 chtari
Trichloromethane
Tnchlu-oineiiia-ie
hiL* - 3CJ M.l LTl
Рис. 5.7. Результаты библиотечного поиска по базе данных NIST08 (подбиблиотеки main-
lib и replib), полученные при использовании программного обеспечения NIST
MS Search. Масс-спектр, расположенный вверху, отвечает максимуму хромато-
графического пика, представленного на рис. 5.4
G E*S й Я
Рис. 5.8. Масс-хроматограммы, построенные по току ионов с m/z (максимум соот-
ветствует 6,24 мин.) и 83 (максимум соответствует 6,19 мин.). Масс-спектры,
представленные на рисунке, были получены путем вычитания друг из друга
масс-спектров, отвечающих максимумам хроматографических пиков (зареги-
стрированных на масс-хроматограммах по ионам с m/z 83 и 97)
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
ными при использовании метода вычитания фона) и библиотечными масс-спектрами
(соединений, возглавляющих список возможных кандидатов) отсутствовали.
Приведенный пример показывает, чтотщательное изучение данных необходимо для
проведения правильной идентификации с использованием баз данных масс-спектров
ИЭ. Проведение правильной идентификации рассмотренной выше пары коэлюируемых
соединений (дающих только один пик на хроматограмме по полному ионному току)
требовало вмешательства специалиста, имеющего опыт обработки хроматомасс-спек-
тральных данных. Использование РВМ-алгоритма позволило сразу получить коррект-
ные результаты, в то время как использование программы NIST MS Search требовало
повторного и более тщательного рассмотрения полученных данных.
5.9. Пример проведения правильной идентификации
с помощью программного обеспечения NIST MS
Search
Принимая во внимание тот факт, что в предыдущем примере использование РВМ-
алгоритма было более предпочтительным, данный алгоритм был использован для
проведения библиотечного поиска еще одного масс-спектра, в котором нельзя было
однозначно установить, какой пик является молекулярным (рис. 5.9). Масс-спектр, от-
Рис. 5.9. Результаты библиотечного поиска, полученные при использовании РВМ-
алгоритма, реализованного в программном обеспечении Agilent ChemStation.
Масс-спектр, расположенный вверху, отвечает максимуму пика, зарегистриро-
ванного на хроматограмме по полному ионному току. Масс-спектр, представ-
ленный внизу, является библиотечным спектром этилбензола (представленного
в базе данных NIST08)
5.9. Пример проведение правильной идентификации с помощью программного
обеспечения NIST MS Search
вечающий вершине пика на хроматограмме по полному ионному току, был использо-
ван для проведения библиотечного поиска по базе данных NIST08 с РВМ-алгоритмом.
Так же как и в предыдущем примере, величины степени совпадения для первых 20
кандидатов были достаточно низкими, что могло указывать на то, что рассматрива-
емый экспериментальный масс-спектр отвечает смеси индивидуальных соединений.
На 3 позиции в списке возможных кандидатов располагался этилбензол. Кроме того,
5, 14 и 17 позиции также были заняты этилбензолом (четыре масс-спектра этилбензола
представлены в базе данных NIST08). На основании визуального сопоставления экспе-
риментального спектра с масс-спектром этилбензола, представленным в базе данных,
можно было предположить, что экспериментальный спектр отвечает смеси веществ,
одним из которых являлся этилбензол (рис. 5.9). К сожалению, масс-спектр коэлюи-
руемого соединения отсутствовал в списке первых 20 возможных кандидатов. Нали-
чие некоторых групп пиков в экспериментальном масс-спектре указывало на присут-
ствие атомов хлора и/или брома в составе соединения. В списке первых 20 возможных
кандидатов, полученных при использовании РВМ-алгоритма, присутствовало только
одно соединение, содержащее один-единственный атом хлора. В состав оставшихся 19
соединений не входили элементы, присутствие которых приводило бы к появлению
изотопного пика, отстоящего от основного масс-спектрального пика на 2 единицы m/z
|l£ ] i' |1 Scan 779 (12.983 mint 50P0830D DV J '£1| |Q |
s Src Name
1 E Scan 779 (12.983 min): 50r0830DD\dati
2 E Subtraction [Scan 779 <12963 mm) 50P
3 A Scan 779 (12 983 min). 50P0830D DWat;
4 E Subtraction. [Scan 777 (12.950 mm). эОР
5 A Scan 777 (12 950 min) . 50P0830D DWat.
6 A Scan 779 (12 983 min) 50РПЯЯ0П Dklat.
7 A Scan 7ОД (13 390 mm) 50Р(1ЯЗПГ) OWal Д
mair'ic rspub 219743 total spectra
106n
1000 90C 800 700 600 500
91
Масс-спектр отвечает
максимуму пика, зарегистри-
рованного на хроматограмме
по полному ионному току
„ М 66 ,, 95 106 117
__________З.ь ,4, 43 , ||. , , , | , ,L , , . П1,н?. I 1 4° , . .1,1. , . . 1.1
10 20 30 40 50 60 70 80 90 10С 110 120 130 140 150
Масс-спектр, полученный при вы-
читании спектра возглавляющего
список возможных кандидатов
из экспериментального спектра
61 97
. Ту«!,1____,|I,iT ,_,Ti ^.iIhi,
10 20 30 40 50 60 70 80 90 100
9 |LlM |R | Pr | Name -
al M 683 904 38 6 Ethylbenzene
e2 R 679 885 38 C Ethylbenzene
(яЗ R 660 863 38 6 Ethylbenzene
a4 R 658 863 38 6 Ethylbenzene
05 R 654 846 110 o-Xytene
06 R 646 834 8 17 Benzenepropanenitnle
Ы7 M 645 752 8 17 Benzenepropanenitnle
ПН R 633 774 8 17 Benzenepropanenitnle
9 M 630 668 1 70 Formamidine. N W dfcenzy
10 M 614 668 2 71 Guanidine, N nitro N’(phen
11 M 613 633 2 60 7-Cxabjcyclo[4 21]ncna-2,
12 M 605 652 1 94 \3-Cyclopentac*ene, 5 (1t
13 M 605 646 1 94 ci-Denzyi malanohydroxam
014 R 602 789 1 71 t ,3-Cyclopentscfene, 5-(1-i
015 R 600 670 1 58 2.8-Decadiyne
0I6 M 596 808 1 33 2,4-OctaUiyi e
V M 596 692 133 i.2.5-Oxadiazol-3-amne. 4
018 R 594 725 1 23 Benzene (2 chlornprcpyl)
tn1S R 590 606 1 33 2.4 Ocladiyne
20 M 586 608 0.92 2 Bromophenyl p pnenylpn
021 M 585 789 1 71 1,3-CydopentacSene, 5-(1-i
22 M 585 626 0 88 Benzene (1-propy heptyl)-
23 M 583 588 0.81 Benzenebutanoic acid, a-o
24 M 582 766 0 78 <,6-Hcotadien-3-yre. 5-mcj.
\ H»m.i7~Uigw / InUb- MI Hit Lilt
I-a-St.- i 77.1 (If- aS1 гип, Dulnl.l Subtraction xfi 2 MF=633 RmF=904 F? Fthylberzene
50
39 51 еь П
ol 27 39 I, , , , , ... . _
10 20 30 40 50 60 70 80
(mainfcj Ethylbenzene
Name Ethylbenzene
Formula СяНш
I
16 J ^110 120 130 ”140 150
Рис. 5.10. Результаты библиотечного поиска по базе данных NIST08, полученные при ис-
пользовании программного обеспечения NIST MS Search. В левом нижнем углу
представлен список возможных кандидатов. Масс-спектр, представленный
внизу, является библиотечным спектром этилбензола (возглавляющего список
возможных кандидатов). Масс-спектр, расположенный вверху, отвечает макси-
муму пика, зарегистрированного на хроматограмме по полному ионному току.
Масс-спектр, расположенный в середине, представляет собой результат вычи-
тания библиотечного масс-спектра этилбензола из экспериментального
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
namlib, replib. 219743 total spectra
1tK>l
# Src Name
1 Е Subtraction [Scan 779 (12.983 min). 50P
2 E Scan 779(12 983 min) 50P0830D Dkiatr
3 E Subtraction [Scan 779 (12 983 min] 50P
4 A Scan 779 (12 983 mm) 50P9E30D D'dati
5 E Subtraction [Scan 777 (12 950 min) 50P
6 A Scan 777 (12 950 mm) 50P0E30D D'dat
7 A Scan 779 Г12 983 mm) 50Г0РЗрП P^atjZl
# |l|m. |r .|Pr |Name -
El M 858 881 94 2 Ethane. 1,1,1.2-tetrachlor»
S2 R 857 870 94 2 Ethane, 1 1.1,2-tctrachloro-
b3 R 849 872 94 2 Ethane, 1,1,1,2-tetrachk>ri>
04 R 845 869 94 2 Ethane. 1.11,2-tetrachloro-
5 M 737 76? 4 RO Clnrethate
B6 R 665 682 0 66 Carbonochlondic acid, 2,2,;
7 M 632 650 017 Carbonic acid, 2-chlcroeth)
8 M 617 636 010 Caibonic acid, propaigyl2
нУ M 608 620 0 66 Carbonochlondic acid. 2.2.;
13 M 584 605 0 02 Carbonic acid, propyl 2 2.2
11 M 574 584 0.01 Carborrc acid, ethyl 2,2,2 ti
12 M 366 575 0.01 4-Cyanocartx>nytphenyl-(2
13 M 542 562 0.00 Carbonic acid, 3-pentyl 2,2
14 M Э35 552 0 00 Carbonic acid, butyl 2.2.2-ti
E15 M 518 567 0.00 Propanoic acid, 2.2,3-tiidil
16 M 504 516 0.00 2-Chloro-3-cyanocartonyl-t
17 M 487 491 0.00 Silane, dichbrofiuoromethy
18 M 477 496 0 00 3-Ctiloro-2-methy1cydopen
19 M 474 489 0 00 Carbonic acid, isobutvl 2,2.
23 M 470 494 0 DO Propane 1 2,2,3-tetrachlor.
21 M 467 580 ODO 2-Dichlormethytthiophene
s22 R 465 497 O DO Oxirane, (Irichlocometh/f).
23 M 464 469 0.00 2,2,2-Tnchloroethyl phosph
24 N 463 475 ODO 1-Butene 1 2-di5romo-
1001 Библиотечный масс-спектр соединения,
возглавляющего список возможных кан-
дидатов для идентификации соедине-
ния по спектру после вычитания
(marftb) Ethane, 1 1 1 2-tetrachloro-
Члте Etnane. l.i.i.2-tevathbro-
Fqrmula C2H2CI4
|ЛКЛ» 1РЛ PVI.V,r. 90/
Рис. 5.11. Результаты библиотечного поиска по базе данных NIST08, полученные при ис-
пользовании программного обеспечения NIST MS Search. При проведении по-
иска использовали масс-спектр, полученный путем вычитания библиотечно-
го спектра этилбензола из экспериментального, представленного на рис. 5.10.
В левом нижнем углу представлен список возможных кандидатов. Масс-спектр,
представленный внизу, является библиотечным спектром соединения, возглав-
ляющего список возможных кандидатов. Масс-спектр, расположенный вверху,
представляет собой результат вычитания библиотечного спектра этилбензола
из экспериментального спектра, отвечающего максимуму пика, зарегистриро-
ванного на хроматограмме по полному ионному току. В средней части представ-
лены эти же спектры в обращенном друг относительно друга варианте
(так называемые А + 2 элементы)13. На основании результатов библиотечного поиска,
полученных при использовании РВМ-алгоритма, можно заключить с высокой вероят-
ностью, что экспериментальный масс-спектр представляет собой суперпозицию масс-
спектров нескольких соединений, одно из которых — этилбензол, а другое или другие
соединения не могут быть установлены.
Перед тем как начать процесс интерпретации (в надежде получить какую-либо до-
полнительную информацию), было решено провести поиск по базе данных с исполь-
зованием программного обеспечения NIST MS Search. Первые четыре масс-спектра
в списке возможных кандидатов отвечали этилбензолу (рис. 5.10). Поскольку экспе-
риментальный масс-спектр отвечал смеси соединений, величина степени совпадения
Match была достаточно низкой (658—683), однако величина степени совпадения Reverse
Match составляла 863—904.
Следует отметить, что масс-спектр о-диметилбензола входил в список первых пяти
кандидатов. Спектры о-диметилбензола и этилбензола очень похожи, небольшое раз-
личие проявляется в том, что в случае о-диметилбензола интенсивность пика [М— 1]+
13 См. А.Т. Лебедев «Масс-спектрометрия в органической химии». Бином, 2003,
497 с. — Прим. ред.
5.9. Пример проведение правильной идентификации с помощью программного
обеспечения NIST MS Search
Рис. 5.12. Масс-хроматограммы, построенные по току ионов с m/z 131 и 106, однозначно
указывают на наложение пиков двух соединений
(нормированная на интенсивность пика молекулярного иона) значительно выше. Дан-
ное обстоятельство можно рассматривать как подтверждение того факта, что экспери-
ментальный масс-спектр отвечает смеси нескольких соединений, одним из которых яв-
ляется этилбензол. Спектр, полученный при вычитании библиотечного масс-спектра
этилбензола (находящегося на первой строчке таблицы возможных кандидатов) из экс-
периментального спектра, был использован для проведения поиска по базе данных с
помощью программы NIST MS Search (рис. 5.11). Первые четыре позиции в списке воз-
можных кандидатов занимали масс-спектры ИЭ 1,1,1,2-тетрахлорэтана (четыре спектра
1,1,1,2-тетрахлорэтана представлено в базе данных NIST08). При этом величина степени
совпадения Match составляла 839—852.
Величина Probability, рассчитанная для данного кандидата, составила 95,3 %. Такое
высокое значение величины Probability объясняется уникальностью соответствующе-
го спектра по сравнению с другими масс-спектрами, представленными в базе данных.
Более низкое значение величины Probability, полученное в случае этилбензола, объяс-
няется наличием в базе данных масс-спектров о-, м- и л-изомеров диметилбензола, об-
ладающих похожими спектрами.
Аналогично предыдущему примеру использование метода вычитания фона по-
зволило выделить очищенные масс-спектры коэлюируемых соединений (рис. 5.12),
использование которых увеличивает надежность результатов библиотечного поиска.
В данном случае использование алгоритма библиотечного поиска, реализованного
в программном обеспечении NIST MS Search, позволило установить, что эксперимен-
тальный масс-спектр представляет собой суперпозицию двух спектров индивидуаль-
ных соединений (этилбензола и 1,1,1,2-тетрахлорэтана). Результаты, полученные при
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
использовании РВМ-алгоритма, были менее определенными и требовали более тща-
тельного рассмотрения.
5.10. Какой алгоритм библиотечного поиска является
наилучшим?
Как показали два примера, рассмотренные выше, нельзя сказать, какой из алгоритмов
библиотечного поиска лучше (алгоритм, реализованный в программном обеспечении
NIST MS Search, или РВМ-алгоритм, используемый в программном обеспечении Agi-
lent ChemStation). Если результат библиотечного поиска, полученный, например, при
помощи программного обеспечения NIST MS Search, не является однозначным, можно
посмотреть, какие результаты будут получены при использовании других алгоритмов
(реализованных, например, в программном обеспечении, поставляемом вместе с при-
бором). Однако важно помнить, что при идентификации неизвестных соединений
окончательное решение принимает именно химик, а не база данных или программа би-
блиотечного поиска.
5.11. Интерпретатор масс-спектров NIST
NIST MS Search является свободно распространяемым программным обеспечением,
которое доступно для бесплатной загрузки на сайте http://chemdata.nist.gov (совмест-
но с демонстрационной версией базы данных масс-спектров NIST). Помимо про-
граммного обеспечения, предназначенного для проведения поиска по базам данных
масс-спектров, в распоряжении у пользователя окажутся различные вспомогательные
программы, например интерпретатор масс-спектров (MS Interpreter). Программа MS
Interpreter (рис. 5.13) требует для своей работы масс-спектр и ассоциированную с ним
структурную формулу (которые могут быть загружены непосредственно из программ-
ного обеспечения NIST MS Search). Масс-спектр отображается в нижней части окна
программы; в верхней части отображается структурная формула. При выборе опреде-
ленного масс-спектрального пика в верхней части окна отобразится возможная брут-
то-формула и структурная формула соответствующего фрагментного иона (при усло-
вии, что структурная формула была ассоциирована с исследуемым масс-спектром),
а также брутто-формула и молекулярная масса нейтральной частицы, элиминируемой
при фрагментации. Некоторые масс-спектральные пики отображаются серым цветом.
Это означает, что возможная структурная формула соответствующего фрагментного
иона интерпретатором не предложена (брутто-формула нейтральной частицы, элими-
нируемой при фрагментации также не установлена).
Изотопный калькулятор, реализованный в программе MS Interpreter, может быть
использован для получения информации о теоретически рассчитанном изотопном
распределении, характерном для конкретного фрагментного иона (исходя из соответ-
ствующей брутто-формулы). Информация об изотопном распределении представля-
ется как в цифровом, так и в графическом виде. При этом теоретически рассчитанное
и экспериментально полученное изотопные распределения могут быть сопоставлены.
5.12. Использование базы данных масс-спектров ИЭ NIST при проведении анализа
методом высокоэффективной жидкостной хроматографии/масс-спектрометрии
Рис. 5.13. Окно программы интерпретатора масс-спектров NIST
Кроме того, интерпретатор масс-спектров может быть использован для генерации все-
возможных брутто-формул, отвечающих фрагментному иону, имеющему определенное
значение m/z (при условии, что брутто-формула молекулярного иона известна). Чтобы
овладеть всеми возможностями программы MS Interpreter, может потребоваться доста-
точно много времени, однако использование данной программы значительно упростит
интерпретацию масс-спектров. Изначально эта программа была создана с целью облег-
чить оценку качества новых спектров, добавляемых в базу данных масс-спектров NIST.
Программа постоянно обновляется, следить за выходом новых версий можно на веб-
сайте NIST.
5.12. Использование базы данных масс-
спектров ИЭ NIST при проведении анализа
методом высокоэффективной жидкостной
хроматографии / масс-спектрометрии
Для целей анализов объектов окружающей среды возрастает роль метода высоко-
эффективной жидкостной хроматографии/масс-спектрометрии (ВЭЖХ/МС) (гла-
ва 3). Благодаря развитию метода времяпролетной масс-спектрометрии возможность
определения точной молекулярной массы доступна в настоящее время многим масс-
спектрометристам, работающим в этой области. Несмотря на то, что большинство
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
анализов объектов окружающей среды направлено на определение целевых аналитов,
существуют задачи по обнаружению и идентификации неизвестных (нецелевых) сое-
динений. Довольно часто при использовании метода ВЭЖХ/МС в экспериментальном
масс-спектре присутствует только один интенсивный пик, который отвечает прото-
нированной молекуле, иону [М — Н]+, депротонированной молекуле [М — Н]_ или иону,
образовавшемуся в результате присоединения к молекуле какой-либо заряженной ча-
стицы. Измерение точного значения m/z, доступное при использовании времяпролет-
ных масс-спектрометров (или же при совместном использовании квадрупольных масс-
спектрометров и программного обеспечения Cerno Biosciences MassWork, описание
которого будет представлено ниже), а также определение изотопного распределения
позволяют установить брутто-формулу неизвестного аналита. Полученная брутто-
формула может быть использована для поиска возможных кандидатов, содержащихся
в коммерческих базах данных масс-спектров ИЭ (таких, например, как Wiley Registry
или NIST/EPA/NIH). Обычно результатом такого поиска является список, состоя-
щий из большого числа соединений. При использовании базы данных масс-спектров
NIST/EPA/NIH для каждого кандидата также будут представлены альтернативные на-
звания, информация о дополнительных базах данных, в которых присутствует упоми-
нание данного соединения, и так далее. Если в результате поиска по брутто-формуле
с использованием программного обеспечения N 1ST MS Search был получен список из 10
Рис. 5.14. Структурная формула соединения и его масс-спектр, зарегистрированный с по-
мощью метода диссоциации, активируемой соударениями. В окне Substructure
Information представлена информация о вероятности присутствия (или отсут-
ствия) ряда структурных фрагментов в молекуле исследуемого соединения.
В правой части окна Substructure Information указаны рассматриваемые струк-
турные фрагменты (этот список может быть изменен пользователем)
5.12. Использование базы данных масс-спектров ИЭ NIST при проведении анализа
методом высокоэффективной жидкостной хроматографии/масс-спектрометрии
возможных кандидатов, среди которых только один присутствует в нескольких других
базах данных (в то время как остальные девять кандидатов не содержатся ни в одной
другой базе данных и/или для этих кандидатов представлено небольшое число альтер-
нативных названий), то соответствующее соединение является хорошим кандидатом
для предварительной идентификации. Для увеличения надежности идентификации
стандартный образец предполагаемого соединения может быть приобретен и проана-
лизирован в тех же условиях, а времена удерживания и масс-спектры (неизвестного
и предполагаемого соединения) сопоставлены друг с другом. При таком использовании
базы данных NIST/EPA/NIH идентификация основана только на сопоставлении брут-
то-формул аналита и вещества из базы данных (сопоставление масс-спектров не будет
производиться).
Соединения, которые могут быть обнаружены при проведении анализов объектов
окружающей среды, обычно известны и описаны в литературе. При проведении по-
иска соединений по заданной брутто-формуле помимо баз данных масс-спектров ИЭ
могут быть использованы и другие базы данных, например Chemical Abstracts, Merck
Index и др. В случае идентификации аналитов, основанной на поиске соединений, об-
ладающих заданной брутто-формулой, необходимо максимально использовать допол-
нительно доступную информацию об образце и оценивать корректность результатов
идентификации с точки зрения возможности присутствия рассматриваемого кандидата
в исследуемом объекте.
С другой стороны, база данных NIST/EPA/NIH и программное обеспечение NIST
MS Search могут быть использованы для поиска соединений по базе данных масс-
спектров, полученных методом диссоциации, активируемой соударениями (ДАС)14.
В случае масс-спектров ДАС могут быть использованы два алгоритма поиска Neutral
Loss Search или MS/MS Search. При использовании алгоритма Neutral Loss Search по-
иск по базе данных будет осуществляться при использовании информации о мас-
сах нейтральных фрагментов, элиминируемых из иона-предшественника в процессе
фрагментации.
Помимо всего прочего, программное обеспечение NIST MS Search позволяет про-
гнозировать присутствие или отсутствие ряда структурных фрагментов в составе мо-
лекулы неизвестного соединения. Для решения этой задачи используются корреля-
ционные взаимосвязи между структурой соединения и масс-спектром (Stein, 1995),
устанавливаемые при анализе списка возможных кандидатов (полученного в результате
библиотечного поиска). Такой подход (в случае отсутствия какой-либо другой доступ-
14 Активация ионов, обусловленная соударениями, обычно приводит к их распаду (диссо-
циации). Процесс диссоциации, активируемой соударениями, заключается в том, что ион, обла-
дающий определенной скоростью, сталкивается со свободно движущимися атомами (или молеку-
лами) инертного газа при давлении ~10_| Па. В качестве инертного газа могут быть использованы
одноатомные газы, такие как аргон, неон, гелий, или двухатомные газы, например азот. Если со-
ударения происходят в пространстве между источником ионов (находящимся при атмосферном
давлении) и областью масс-анализатора (находящейся при давлении 10-3 Па или ниже), данный
метод имеет название диссоциации, активируемой соударениями в ионном источнике (in-source
CAD). Если соударения происходят в камере соударений, расположенной в пространстве между
двумя масс-анализаторами, используют термин «тандемная масс-спектрометрия или масс-
спектрометрия/масс-спектрометрия (МС/МС)». Следует отметить, что термины «диссоциация,
активируемая соударениями», и «диссоциация, индуцируемая соударениями», являются взаимо-
заменяемыми (глава 4).
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
ной информации) может облегчить интерпретацию масс-спектров ДАС неизвестных
соединений.
Масс-спектр ДАС соединения (рис. 5.14) представлен в правой части главного окна
программы NIST MS Search. Изотопные пики имеют низкую интенсивность, из чего
можно сделать вывод о том, что в качестве иона-предшественника был выбран моно-
изотопный ион со значением m/z, равным 248. Структурная формула соединения также
представлена на рис. 5.14. Как видно из рисунка, число эквивалентов колец и двойных
связей равно шести, а в структуре молекулы можно выделить следующие фрагменты:
атомы кислорода и азота; фрагмент Et-O-СО; бензольное кольцо; насыщенный азотсо-
держащий гетероцикл; карбонильную группу; третичный атом азота. На рис. 5.14 в окне
Substructure Information представлена информация о фрагментах молекулы, присутствие
или отсутствие которых было установлено с помощью программного обеспечения NIST
MS Search с некоторой вероятностью. Присутствие всех указанных ранее фрагментов
молекулы было автоматически установлено программным обеспечением с высокой ве-
роятностью. Наличие информации об отсутствующих фрагментах (например, таких,
как незамещенный бензильный фрагмент, фенольный фрагмент, атомы кремния, азот-
содержащие ароматические циклы и так далее) облегчает задачу установления структу-
ры неизвестного соединения.
5.13. База данных индексов удерживания NIST
Довольно часто перед химиком-аналитиком, работающим в области анализа объектов
окружающей среды, встает задача создания новой методики ГХ/МС-анализа. В случае
отсутствия литературных данных эта задача может оказаться довольно трудоемкой.
База данных газохроматографических индексов удерживания NIST содержит 349103
значения индексов удерживания, полученных для 71169 индивидуальных соединений
(для 38639 из которых в базе данных NIST11 также присутствует масс-спектр ИЭ). Ин-
дексы удерживания, представленные в базе данных NIST, были получены из научных
публикаций и содержат, помимо самого значения индекса удерживания, информацию
о параметрах работы газового хроматографа, а также ссылку на соответствующую ста-
тью и ее название (рис. 5.15).
База данных масс-спектров NIST11 также включает в себя базу данных индек-
сов удерживания. Методы поиска информации, используемые в случае базы данных
масс-спектров NIST11, применимы и для базы данных индексов удерживания, одна-
ко возможности поиска ограничены. В случае приобретения базы данных индексов
удерживания как самостоятельного продукта используется специальное программное
обеспечение (NIST RI Search), облегчающее поиск информации, необходимой для раз-
работки условий газохроматографического разделения или идентификации неизвест-
ных соединений.
Программное обеспечение NIST RI Search открывает широкие возможности для
проведения поиска по базе данных газохроматографических индексов удерживания.
Можно, например, проводить поиск по значению индекса удерживания, названию со-
единений (так же как и в случае базы данных масс-спектров NIST11 для соединений
представлен ряд альтернативных названий), брутто-формуле, номинальной молекуляр-
5.13. База данных индексов удерживания NIST
2. Value: 1626.1 iu
Column Type: Capillary
Column Class: Standard nonpolar
Active Phase: DB-1
Column Length: 30 m
Carrier Gas: He
Column Diameter: 0.26 mm
Phase Thickness: 0.25 um
Data Type: Normal alkane RI
Program Type: Ramp
Start T: 120 °C
End T: 280 °C
Heat Rate: 8K/min
Start Time: 1 min
End Time: 22 min
Source: Manca, D; Ferron, L; Weber, J-P A System
for Toxicological Screening by Capillary
Gas Chromatography with Use of Drug
Retention Index Based on Nitrogen-
Containing Reference Compounds Clin.
Chem., 35(4), 1989, 601-607.
Рис. 5.15. Информация об индексе удерживания соединения, представленная в базе дан-
ных NIST
ной массе, регистрационному номеру CAS. Однако на результаты поиска не могут быть
наложены дополнительные ограничения. База данных индексов удерживания и соот-
ветствующая программа поиска оказывают существенную помощь при проведении
газохроматографического анализа новых (для исследователя) аналитов (поскольку для
каждого соединения, представленного в базе данных, доступна информация относи-
тельно используемых условий разделения). Например, при создании методики анализа
оловоорганических соединений можно провести поиск по базе данных с использова-
нием ограничения на число атомов олова, входящих в молекулу (a?Si > 0), что позволит
получить список из 32 соединений. Помимо всего прочего, в программе NIST RI Search
присутствует возможность проводить поиск соединений, имеющих схожую структур-
ную формулу с интересующим аналитом (Structure Similarity Search).
Если хроматографический пик, отвечающий неизвестному соединению, элюирует-
ся между пиками двух известных аналитов, то соответствующие индексы удерживания,
найденные в базе данных, могут быть использованы в качестве дополнительной инфор-
мации при идентификации. При анализе изомеров база данных индексов удерживания
позволяет определить порядок элюирования соединений. База данных индексов удер-
живания NIST, а также программа поиска NIST RI Search могут быть использованы как
в случае метода ГХ, так и ГХ/МС.
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
База данных масс-спектров NIST, а также база данных индексов удерживания NIST
может быть приобретена только у официальных дистрибьюторов, информацию о кото-
рых можно найти на сайте http://chemdata.nist.gov.
5.14. Программа для автоматического поиска
компонентов и выделения чистых масс-
спектров AMDIS
При использовании метода ГХ/МС существует вероятность того, что пик на хромато-
грамме по полному ионному току в действительности представляет собой случай на-
ложения двух или даже большего числа соединений. Кроме того, экспериментальные
масс-спектры могут быть искажены за счет высокого уровня фона. Каждая точка на хро-
матограмме по полному ионному току представляет собой масс-спектр, для регистра-
ции которого требуется время. Это приводит к тому, что в случае метода ГХ/МС коли-
чество точек на пик обычно в несколько раз ниже по сравнению с хроматограммами,
зарегистрированными при использовании одноканальных детекторов (таких как пла-
менно-ионизационный или электронозахватный). При использовании квадрупольного
Синонимы
Оценка
величины
индекса
удерживания
Ограничения
по величине
приведенных
индексов
удерживания
Рис. 5.16. Окно программного обеспечения NIST RI Search, в котором представлены ре-
зультаты поиска по названию соединения
5.14. Программа для автоматического поиска компонентов и выделения чистых
масс-спектров A MDIS
или магнитного секторного масс-спектрометра возникает еще одна проблема, заклю-
чающаяся в искажении масс-спектров, зарегистрированных на склонах хроматографи-
ческих пиков (глава 2). Искажение масс-спектров объясняется тем, что в случае метода
ГХ/МС при элюировании аналита из хроматографической колонки его концентрация
в источнике ионов быстро изменяется. Поскольку ионы, обладающие различными зна-
чениями m/z, регистрируются последовательно, относительная интенсивность пиков
в масс-спектрах, зарегистрированных на склонах хроматографических пиков, будет ис-
кажена (по сравнению с тем случаем, когда концентрация вещества в источнике ионов
остается постоянной в процессе развертки спектра). Искажение относительных интен-
сивностей масс-спектральных пиков может оказывать влияние на результаты библио-
течного поиска и затруднять интерпретацию данных. Среди других трудностей, часто
встречающихся при использовании метода ГХ/МС, следует отметить:
1) возможность неполного разделения пиков соединений, представляющих
интерес;
2) возможность наложения хроматографического пика соединения, представляю-
щего интерес, на пики мешающих компонентов матрицы;
3) возможность загрязнения масс-спектра аналита, обусловленного высоким фо-
ном хроматографической колонки.
Программное обеспечение AMDIS было разработано сотрудниками Национального
института стандартов и технологий в рамках работ, связанных с международной конвен-
цией о запрещении химического оружия. Программное обеспечение AMDIS использу-
ется для обработки ГХ/МС-данных (также могут быть обработаны ВЭЖХ/МС-данные)
и позволяет исключить посторонние пики из экспериментальных масс-спектров анали-
тов, внести поправку на искажение интенсивностей масс-спектральных пиков (возника-
ющее в случае сканирующих масс-анализаторов), а также выделить масс-спектры ана-
литов высокого качества, пригодные для проведения библиотечного поиска или ручной
интерпретации. Кроме того, программное обеспечение AMDIS позволяет осуществлять
поиск по базам данных масс-спектров и проводить количественный анализ. Ряд произ-
водителей масс-спектрального оборудования использует AMDIS напрямую или прово-
дит интеграцию в их собственное программное обеспечение. Например, AMDIS являет-
ся «сердцем» программного обеспечения Deconvolution Reporting System, разработанного
компанией Agilent и использующего базы данных RTL (Retention Time Locking).
Программное обеспечение AMDIS облегчает процесс нахождения следовых коли-
честв компонентов в сложных смесях органических соединений. На рис. 5.17 представ-
лено окно программы AMDIS, в верхней части которого отображена хроматограмма
по полному ионному току, зарегистрированная при анализе смеси модельных соеди-
нений (используемой в методике ЕРА-525 — «Анализ питьевой воды»), содержащей
добавки небольшого количества гербицидов. В случае ручной обработки данных для
обнаружения этих соединений необходимо последовательно просматривать зареги-
стрированные масс-спектры. При использовании программы AMDIS производится
автоматическая обработка данных, одним из первых этапов которой является поиск
пиков, на всех зарегистрированных масс-хроматограммах. Профили хроматографиче-
ских пиков, найденных на различных масс-хроматограммах (и обладающих близкими
временами удерживания) сопоставляются друг с другом. Если интенсивность сигнала
возрастает и уменьшается одновременно на нескольких масс-хроматограммах, то про-
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
граммное обеспечение делает вывод, что соответствующие значения m/z относятся
к одному и тому же соединению. Если хроматографические пики, отвечающие двум
различным соединениям, разрешены не полностью, а в масс-спектрах соединений при-
сутствует ряд общих значений m/z, то программное обеспечение AMDIS (в ряде случа-
ев) может корректно извлечь информацию об интенсивности соответствующих масс-
спектральных пиков.
В верхней части хроматограммы по полному ионному току, представленной
на рис. 5.17, находится ряд меток, имеющих форму треугольников. Каждый из треу-
гольников указывает на то, что программное обеспечение AMDIS обнаружило хрома-
тографический пик при данном времени удерживания и выделило чистый масс-спектр.
Значения m/z, присутствие которых обусловлено фоном матрицы или фоном прибора,
исключаются из очищенного масс-спектра. Как видно из рис. 5.17, компонент, имеющий
время удерживания 9,33 мин., не мог быть найден при анализе хроматограммы по пол-
ному ионному току, однако он был обнаружен программным обеспечением AMDIS.
При поиске очищенного масс-спектра (нижняя часть рис. 5.17) по базе данных NIST08
первую строчку в списке возможных кандидатов занимало соединение, относящееся
к классу гербицидов — А,А-диметил-А'-[3-(трифторметил)фенил]мочевина.
Рис. 5.17. Окно программного обеспечения AMDIS. В верхней части окна представлена
хроматограмма по полному ионному току. В левой части окна программы рас-
положено окно, отображающее фрагменты масс-хроматограмм, построенных
по характеристичным значениям m/z одного из обнаруженных компонентов.
В правой части окна представлены времена удерживания обнаруженных компо-
нентов (а также некоторая дополнительная информация). В нижней части окна
представлены неочищенный (нарисованный черным цветом) и очищенный (на-
рисованный белым цветом) масс-спектры. Присутствие масс-спектральных пи-
ков, отображаемых в виде пунктирных линий, не является однозначным (в дей-
ствительности данные пики могут отсутствовать в масс-спектре соединения)
5.14. Программа для автоматического поиска компонентов и выделения чистых
масс-спектров AMDIS
Рис. 5.18. Результаты автоматической обработки ГХ/МС-данных, отвечающих случаю
неполного разделения хроматографических пиков л-хлорфенола и нафталина,
полученные при использовании программного обеспечения AMDIS
Важной особенностью программного обеспечения AMDIS является возможность
проведения идентификации коэлюируемых соединений. При проведении целевого ана-
лиза могут быть использованы базы данных, предварительно созданные пользователем.
На рис. 5.18 представлены масс-спектры двух соединений (л-хлорфенола и нафталина),
хроматографические пики которых были разделены не полностью. Номинальная моле-
Рис. 5.19. Сравнение масс-спектров нафталина и л-хлорфенола
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
кулярная масса обоих соединений составляет 128 Да, причем пик молекулярного иона
является наиболее интенсивным пиком в их масс-спектрах ИЭ. Основное различие
между масс-спектрами соединений заключается в наличии изотопного пика (m/z 130)
в случае л-хлорфенола (рис. 5.19). Из данных, представленных на рис. 5.19, видно, что
времена удерживания аналитов совпадают (10,160 мин.). «Уточненные» времена удер-
живания, определенные программным обеспечением AMDIS путем интерполяции зна-
чений трех точек, находящихся вблизи максимума, составили 10,1587 и 10,1610 мин. для
л-хлорфенола и нафталина соответственно (разница составила 0,0024 мин. или 0,14 с).
Следует также отметить, что программное обеспечение AM DIS может быть использова-
но для проведения количественного анализа этих двух соединений даже в том случае,
если они оба присутствуют в образце.
Программа AMDIS поставляется вместе с программным обеспечением NIST MS
Search и базой данных масс-спектров NIST. Программа AMDIS входит в состав демон-
страционной версии базы данных NIST, а также может быть независимо загружена
с сайта http://chemdata.nist.gov.
5.15. Определение точных значений m/z
Большинство исследователей, работающих в области анализа объектов окружающей
среды, проводят измерение точной массы15 только при проведении научных исследо-
ваний. Основной причиной использования масс-спектрометрии высокого разрешения
в области анализа объектов окружающей среды является необходимость определения
полихлорированных дибензодиоксинов (главы 1, 15 и 18). Эти анализы проводятся
с использованием магнитных секторных масс-спектрометров с двойной фокусиров-
кой (как правило, данное оборудование используется в экологическом анализе только
при работе с диоксинами). Довольно часто при идентификации неизвестного соедине-
ния (обнаруженного, например, в токсичных отходах или загрязненных сточных во-
дах) оказывается, что подходящий кандидат отсутствует в базе данных масс-спектров
(даже использование алгоритма библиотечного поиска, реализованного в программе
NIST MS Search и получившего название Similarity Search16, порой не позволяет найти
15 В англоязычной литературе существует два термина (accurate mass и exact mass), которые
имеют различное значение, но одинаковый дословный перевод на русский язык (а именно «точная
масса»). Термин «accurate mass» следует переводить на русский язык как «точная измеренная
масса». Данный термин используется, когда значение m/z какого-либо иона определено
экспериментально с высокой точностью. Термин «exact mass» следует переводить на русский
язык как «точная рассчитанная масса». Данный термин используется, когда точное значение
m/z какого-либо иона было рассчитано исходя из его брутто-формулы, заряда и точных атомных
масс соответствующих элементов. Следует отметить, что оба термина обычно употребляются при
рассмотрении моноизотомной массы.
16 При проведении сопоставления масс-спектров ИЭ с использованием программного обе-
спечения NIST MS Search обычно используются два алгоритма Identity Search и Similarity Search.
Алгоритм Identity Search позволяет сказать, насколько экспериментальный и библиотечный масс-
спектры похожи друг на друга, поэтому его обычно используют при проведении идентификации
неизвестных соединений. Алгоритм Similarity Search используют, когда предполагается, что масс-
спектр неизвестного соединения отсутствует в базе данных. Результат поиска представляет собой
список соединений (которые могут быть гомологами или изомерами искомого соединения), имею-
щих похожий масс-спектр, что облегчает установление структуры неизвестного вещества.
5.15. Определение точных значений
соединения, обладающие «похожими» масс-спектрами). В ряде случаев информация
о брутто-формуле может быть получена при анализе изотопного распределения со-
ответствующего масс-спектрального пика, однако определение точного значения m/z
молекулярного иона увеличивает достоверность определения брутто-формулы. Воз-
можность измерения точного значения m/z доступна только при использовании масс-
спектрометров высокого разрешения. Обычно для этой цели используют магнитные
секторные приборы с двойной фокусировкой, устройство которых намного сложнее
по сравнению с квадрупольными масс-спектрометрами или ионными ловушками, ис-
пользуемыми наиболее часто при проведении ГХ/МС-анализов объектов окружающей
среды. Точное значение m/z может быть определено также с помощью времяпролетных
масс-спектрометров высокого разрешения, однако несмотря на то, что они проще в ис-
пользовании, их цена остается достаточно высокой для большого числа лабораторий.
В настоящее время благодаря программному обеспечению MassWorks, разработанному
компанией Cerno Bioscience, измерение точного значения m/z доступно и для квадру-
польных масс-спектрометров, работающих в режиме, обеспечивающем регистрацию
10—20 точек на масс-спектральный пик (Wang and Wu, 2010). Несколько эксперимен-
тальных масс-спектров перфтортрибутиламина (ПФТБА) усредняются программным
обеспечением MassWorks и используются для калибровки шкалы масс. Калибровка осу-
ществляется путем определения формы масс-спектральных пиков, имеющих различ-
ные значения m/z- Масс-спектры ПФТБА могут быть зарегистрированы до или после
анализа пробы (и храниться в отдельном файле), либо они могут быть зарегистрированы
в процессе анализа пробы (и храниться в том же файле). Последний вариант предпочти-
телен в случае использования масс-спектрометра Agilent 5975 MSD.
Измерение точных значений m/z значительно расширяет возможности исследова-
теля. Например, пик со значением m/z, равным 99 в масс-спектре 4-деканона, может
отвечать иону, имеющему состав С7Н15 или С6НИО (номинальная масса обоих фрагмен-
тов равна 99 Да). Рассчитанные точные массы фрагментных ионов составляют 99,1174
и 99,0810 Да соответственно, а разница между ними — 3,6-10-2 Да. Информация о том,
какой из двух ионов соответствует масс-спектральному пику со значением m/z, равным
99, может оказаться полезной при проведении идентификации неизвестных соедине-
ний. Особенностью последней версии программного обеспечения NIST MS Search
(поставляемого вместе с базой данных масс-спектров NIST11) является возможность
использовать информацию о точном значении молекулярной массы аналита при про-
ведении библиотечного поиска.
При использовании программного обеспечения MassWorks данные должны быть
получены в режиме регистрации профилей масс-спектральных пиков (profile mode).
Такой режим регистрации данных (рис. 5.20) поддерживается большинством квадру-
польных ГХ/МС-приборов, коммерчески доступных в настоящее время (за исключени-
ем оборудования компании Shimadzu). Один из существенных недостатков, связанных
с использованием данного режима, заключается в значительном увеличении размера
файлов. Два файла были зарегистрированы при анализе одного и того же образца, при
одних и тех же условиях. Один файл был получен при работе прибора в центроидном
режиме регистрации данных (centroid mode), другой — в режиме регистрации профилей
масс-спектральных пиков (profile mode). Размер файлов с данными составлял 962 кБ
и 43068 кБ (что приблизительно в 40 раз больше) соответственно.
Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
Рис. 5.20. Масс-спектры, полученные в режиме регистрации профилей масс-
спектральных пиков (profile mode) с использованием масс-спектрометра Agilent
5975. На вставке в увеличенном масштабе представлен участок масс-спектра
в диапазоне значений m/z 60—63
5.16. Дополнительное программное обеспечение,
представляющее интерес для масс-
спектрометристов, работающих в области
анализа объектов окружающей среды
Помимо компьютерных программ, описанных ранее в этой главе, можно выделить ряд
дополнительных программ, которые могут оказаться полезными исследователям, рабо-
тающим в лабораториях, занимающихся анализом объектов окружающей среды.
Канадская компания ACD/Labs, занимающаяся разработкой программного обе-
спечения, может предложить целый ряд продуктов, предназначенных для обработки
хроматографических и масс-спектральных данных, а также для облегчения процедуры
идентификации неизвестных органических соединений. Среди этих программ можно
выделить ACD/GC Simulator, ACD/ChromManager, ACD/MS Fragmenter, ACD/SpecMan-
ager и ACD/ChemSketch. Подробное описание всех продуктов ACD/Labs можно найти
на сайте компании: www.acdlabs.com.
1. Программное обеспечение ACD/GC Simulator моделирует гипотетическую хро-
матограмму, используя заданные условия газохроматографического разделения,
а также основываясь на рассчитанных температурах кипения аналитов. Кроме
того, программа ACD/GC Simulator предсказывает времена удерживания со-
единений (для новых условий анализа) и облегчает поиск оптимальных условий
разделения, основываясь на ранее полученных экспериментальных данных.
Хроматограмма, обрабатываемая в ACD/GC Simulator, может быть направлена
|69
5.16. Дополнительное программное обеспечение, представляющее интерес для масс-
спектрометристов, работающих в области анализа объектов окружающей среды
в программу ACD/ChromManager для последующей обработки и занесения ин-
формации в (пользовательскую) базу данных. С другой стороны, хроматограммы
могут быть переданы из программы ACD/ChromManager в программу ACD/GC
Simulator для проведения оптимизации или предсказания времен удерживания
(рис. 5.21).
2. Программное обеспечение ACD/ChromManager предназначено для обработ-
ки ГХ- и ГХ/МС-данных, создания баз данных и поиска по ним. Данная про-
грамма также может быть использована для работы с электрофореграммами
и хроматограммами, зарегистрированными с помощью метода ВЭЖХ с диодной
матрицей. Программа предоставляет широкие возможности по обработке хро-
матографических данных. Например, структурные формулы могут быть припи-
саны хроматографическим пикам, условия анализа могут быть ассоциированы
с хроматограммой, а хроматограммы и соответствующие условия анализа могут
храниться в пользовательской базе данных. Условия анализа впоследствии мо-
гут быть найдены в этой базе данных (в качестве параметров поиска можно ис-
пользовать условия работы оборудования, а также структурные формулы или их
фрагменты).
3. Программное обеспечение ACD/MS Fragmenter позволяет предсказывать фраг-
ментные ионы, принимая во внимание установленные и описанные в литературе
правила фрагментации. Использование программы ACD/MS Fragmenter облег-
чает установление направлений фрагментации молекулярного иона. Основные
направления распада и структурные формулы соответствующих фрагментных
ионов для удобства могут быть представлены в виде «древа фрагментации».
Г*- [> £*<"» Г—* г-а
Рис. 5.21. Окна программного обеспечения ACD/GC Simulator
166 Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
4. Программное обеспечение ACD/SpecManager предназначено для управления
данными и является надстройкой над программой ACD/ChromManager. В про-
грамме ACD/SpecManager можно создавать базы данных, содержащие масс-
спектры, а также хроматографические данные. Кроме того, можно работать
со спектральными данными, зарегистрированными с помощью методов ЯМР,
ИК, УФ, КР, а также данными, полученными с помощью термогравиметриче-
ского анализа, спектрометрии ионной подвижности, метода рентгеновской диф-
ракции и так далее. Программа ACD/SpecManager является «сердцем» продуктов
компании ACD/Labs. Эта программа предназначена для работы именно с хими-
ческими базами данных.
5. Программное обеспечение ACD/ChemSketch предназначено для рисования
структурных формул. Программа позволяет также давать названия химиче-
ским соединениям в соответствии с систематической номенклатурой, реко-
мендованной ИЮПАК (при условии, что молекула содержит не более 50 атомов
и не более трех колец, а в состав молекулы входят только следующие элемен-
ты: Н, С, N, Р, О, S, F, Cl, Br, I, Li, Na и К). Программа ACD/ChemSketch также
взаимодействует с программой ACD/Name, которая позволяет давать названия
соединениям не только в соответствии с рекомендациями ИЮПАК, но также
придерживаясь правил, установленных Международным союзом по биохимии
и молекулярной биологии, а также номенклатуры CAS. Кроме того, программа
ACD/Name позволяет автоматически создавать структурные формулы соеди-
нений, исходя из систематических или полусистематических (semi-systematic)
названий (в последнем случае поддерживаются основные органические со-
единения и большое число производных природного происхождения). Другим
расширением программного обеспечения ACD/ChemSketch является програм-
ма ACD/Name Chemist Version, которая позволяет давать соединениям систе-
матические названия в соответствии с номенклатурой ИЮПАК, создавать
структурные формулы при использовании систематических, торговых и триви-
альных названий, а также регистрационных номеров. Существует бесплатная
версия программного обеспечения ACD/ChemSketch, доступная на сайте ком-
пании ACD/Labs (однако эта версия программного обеспечения, как и большое
число бесплатных программ, может быть использована только физическим
лицом или для образовательных целей). В коммерческих или государственных
лабораториях должна использоваться полная платная версия ACD/ChemSketch
(рис. 5.22 и 5.23).
Компания ChemSW (США) предоставляет несколько программ, которые могут
оказаться полезными для масс-спектрометристов (www.chemsw.com). Среди них стоит
отметить MS Tools (набор программ, полезных при работе с данными, зарегистриро-
ванными на масс-спектрометрах высокого разрешения), ChemSite Pro (программа для
молекулярного моделирования), GC and GC/MS File Translator Professional (программа,
предназначенная для конвертирования данных, зарегистрированных при использо-
вании оборудования от разных производителей), MassSpec Calculator Professional (про-
грамма, облегчающая интерпретацию масс-спектров ИЭ) и ChromView (программа, ис-
пользуемая для добавления примечаний к хроматограммам и спектрам, помещаемым
в отчеты).
5.16. Дополнительное программное обеспечение, представляющее интерес для масс-
спектрометристов, работающих в области анализа объектов окружающей среды
I I III III II
File Edit Papes Tools Templates Options Qopuments AdrFpns I-tab ACDlabs Help
_ hJ_______ !_________====^___________________________
i — I I LI I I I —Ш_________________________ _
PLabLogn | SAMPLE SK2 | Modified <□ Page 272 £>| Fragments 7 | [C?H4]„ ' FW ’ 1C99> в1€59&+[28 05316)„
More.
Formula Weight
Рис. 5.22. Окно программного обеспечения ACD/ChemSketch, предназначенного для ри-
сования структурных формул
Рис. 5.23. Вариант представления структурной формулы в программном обеспечении
ACD/ChemSketch
168 Глава 5. Использование специализированного программного обеспечения
и библиотек масс-спектров в анализе объектов окружающей среды
5.17. Заключение
В дополнение к программному обеспечению, поставляемому с приборами, существу-
ет большое количество сторонних программ, использование которых может упростить
задачу обработки и интерпретации масс-спектральных данных. Вне зависимости
от сложности используемых компьютерных алгоритмов, программы не могут заменить
специалистов, которые в конечном итоге принимают решение о корректности результа-
тов идентификации неизвестных соединений, обнаруженных в объектах окружающей
среды. Несмотря на то, что число соединений, представленных в базах данных, будет
продолжать увеличиваться, оно не сможет сравниться с числом органических соеди-
нений, которые теоретически могут существовать (поскольку они никогда не будут до-
ступны для проведения анализа). Важно, чтобы химик-аналитик смог ответить на во-
прос: «Имеет ли полученный результат смысл?».
Литература
[1] . Ausloos В, Clifton C.L., Lias S.G., Mikaya A. I., Stein S.E., Tchekhovskoi D.V., Spark-
man O. D., Zaikin V., and Damo Z. (1999). «The critical evaluation of a comprehensive mass
spectral library». Journal of the American Society for Mass Spectrometry. 10, 2: 287—299.
[2] . Biemann K. (1962). Mass Spectrometry: Organic Chemical Applications. McGraw-Hill, New
York, USA (reprinted by the American Society for Mass Spectrometry, 1998).
[3] . Biller J. E., and Biemann K. (1974). «Reconstructed mass spectra, a novel approach for
the utilization of gas chromatography—mass spectrometer data». Analytical Letters. 7, 7:
515-528.
[4] . Colby B. N. (1992). «Spectral deconvolution for overlapping GC/MS components». Journal
of the American Society for Mass Spectrometry. 3, 5: 558—562.
[5] . Heller S.R., and Milne G.W.A. (1979). «The EPA—NIH chemical information system».
Environmental Science and Technology. 12,7: 798—803.
[6] . Hites R.A., and Biemann K. (1970). «Computer evaluation of continuously scanned mass
spectra of gas chromatographic effluents». Analytical Chemistry. 42, 8: 855—860.
[7] . McLafferty F. W. (2010). Wiley Registry of Mass Spectral Data. Ninth Edition. John Wiley and
Sons, Hoboken, New Jersey, USA.
[8] . NIST (2011). NIST/EPA/NIH Mass Spectral Database (NIST11). National Institute of
Standards and Technology, part of the United States Department of Commerce, Gaith-
ersburg, Maryland, USA. See http://chemdata.nist.gov (new version published every three
years).
[9] . Reynolds W. E., Bacon V. A., Bridges J. C., Coburn T. C., Halpern B., Lederberg J., Levin-
thak E. C., Steed E., and Tucker R. B. (1970). «А computer operated mass spectrometer sys-
tem». Analytical Chemistry. 42, 11: 1122—1129.
[10] . Sparkman O. D. (2009). «А review of electronic mass spectral databases from John Wiley
and Sons». Journal of the American Society for Mass Spectrometry. 20, 7: R18—R23.
[11] . Stein S. E. (1995). «Chemical structure identification by mass spectral library searching».
Journal of the American Society for Mass Spectrometry. 6, 6: 644—655.
Литература
[12] . Stein S. E. (1999). «An integrated method for spectrum extraction and compound identifi-
cation from gas chromatography/mass spectrometry data». Journal of the American Society
for Mass Spectrometry. 10, 7: 770—781.
[13] . Stenhagen E., Abrahamsson S., and McLafferty F. W. (1969). Atlas of Mass Spectral Data.
Wiley, New York, USA.
[14] . Sweeley C.C., Young N.D., Holland J.F., and Gates S.C. (1974). «Rapid computerized
identification in complex biological mixtures by gas chromatography—mass spectrometry».
Journal of Chromatography. 99: 507—517.
[15] . Wang Y, and Wu M. (2010). «The concept of spectral accuracy for MS». Analytical Chem-
istry. 82, 17: 7055-7062.
[16] . Watson J.T., Pelster D. R., Sweetman B.J., Frolich J.C., and Oates J. (1973). «Display
orientated data system for multiple ion detection with gas chromatography—mass spec-
trometry in quantifying biomedically important compounds». Analytical Chemistry. 45, 12:
2071-2078.
ГЛАВА 6
ПЕРЕДОВЫЕ
МЕТОДЫ
НА ОСНОВЕ
ГХ/МС
Константин А. Артеменко,
Александр Б. Фиалков, Александр Гордин, Авив Амирав
Альберт Т Лебедев
6.1. Режим быстрой ГХ/МС
Газовая хроматография — масс-спектрометрия стала незаменимым методом для ре-
шения многих экологических задач, в том числе при определении антропогенных за-
грязняющих веществ (глава 2). Большинство приоритетных загрязняющих веществ
являются полулетучими соединениями, что делает их идельными объектами для газо-
хроматографического разделения с последующим масс-спектрометрическим детекти-
рованием в газовой фазе.
Любой исследователь, использующий ГХ/МС, разумеется, стремится сократить
время, требуемое на проведение анализа. Меньшее время анализа автоматически оз-
начает уменьшение его стоимости, поскольку для получения того же аналитического
результата потребуется меньше рабочих ресурсов (человеко-часов). Помимо этого, для
выполнения таких же объемов работы требуется меньшее количество приборных часов,
а значит, и самих приборов, что особенно ценно в случае работы лаборатории в режиме
высокой пропускной способности и потокового анализа. Коммерчески это чрезвычайно
выгодно, поскольку за то же самое время можно проанализировать большее число за-
казных образцов.
В традиционных ГХ/МС-методах обычно используются капиллярные колонки1 дли-
ной около 30 м и внутренним диаметром (i.d.) 0,25 мм. Эти ГХ/МС-методы хорошо за-
рекомендовали себя, но скорость анализа на таких колонках невысока и составляет по-
рядка 60 минут, необходимых для удовлетворительного разделения сложных образцов.
Несмотря на преимущества сокращенного времени анализа, ни в коем случае нельзя за-
бывать и о результате анализа: основной целью быстрой ГХ/МС является ускорение ана-
лиза без потери качества получаемых аналитических данных. Такая задача вполне вы-
полнима, и на сегодняшний день 3—10-кратное сокращение времени анализа достижимо
1 Здесь и далее термин «ГХ-колонка» всегда подразумевает капиллярную колонку,
а не набивную.
6.1. Режим быстрой ГХ/МС I 71
для классических серийных анализов (например приоритетных загрязняющих веществ)
без потери качества данных. В зависимости от имеющегося оборудования и лабораторных
возможностей режим быстрой ГХ/МС может быть реализован за счет иструментальных
преобразований как хроматографической, так и масс-спектрометрической составляю-
щей (или же их обеих). Теоретически в процессе оптимизации ГХ/МС-метода, в том числе
с использованием специальных приемов разделения, возможно сокращение времени ана-
лиза без потерь хроматографического разрешения до определенного предела, после чего
дальнейшее уменьшение времени хроматографирования приведет к появлению накла-
дывающихся пиков. Однако в этом случае за счет обработки масс-спектрометрических
данных можно осуществить процесс деконволюции хроматографических пиков. Такие
возможности присущи именно масс-спектрометрическому методу, что позволяет значи-
тельно сократить время анализа при использовании комбинированного ГХ/МС-метода,
тогда как ПИД и другие детекторы для ГХ такой возможности не дают.
6.1.1. Оптимизация газохроматографической составляющей
в условиях быстрой ГХ/МС
Очевидно, что сократить время ГХ-анализа можно, используя короткие колонки: ана-
литы попросту находятся в колонке меньше времени, и анализ ускоряется. Однако эф-
фективность разделения, а именно разрешение и форма пиков, резко ухудшаются, если
не предприняты дополнительные меры оптимизации параметров хроматографирова-
ния. Эффективность хроматографического разделения обычно измеряется числом те-
оретических тарелок (N). Одинаковое число теоретических тарелок для колонок разной
длины (£) указывает на одинаковое разрешение этих двух колонок. Высота эквивалент-
ной теоретической тарелки (ВЭТТ, Н) рассчитывается как Н = L/N. Таким образом, при
необходимости использования более коротких колонок с сохранением той же эффек-
тивности разделения требуется уменьшить ВЭТТ за счет изменения других хроматогра-
фических параметров. Величина Я зависит от скорости газа-носителя и, внутреннего
диаметра (/.J.) d капиллярной колонки, коэффициента диффузии растворенного ком-
понента в газе-носителе D и термодинамических свойств системы фаза/растворенный
компонент, выражаемых константой К. ВЭТТ рассчитывается в соответствии с упро-
щенным уравнением Голэя (Gonnordetal., 1983):
„ 2D d2K2
H ——+-----и
и 96D
Для расчета минимального значения ВЭТТ данное уравнение следует продиф-
ференцировать. Минимальное значение ВЭТТ Ят|п = (J/2) • К оказывется прямо про-
порционально внутреннему диаметру колонки. Это означает, что для максимального
уменьшения ВЭТТ следует использовать колонки малого диаметра (рис. 6.1).
Другой очевидный способ сокращения времени хроматографирования — умень-
шение толщины пленки неподвижной фазы капиллярной ГХ-колонки, используемой
в анализе. Очевидно, хроматографируемому веществу требуется более короткое время
для равновесного распределения между газовой и конденсированной фазами в случае
использования более тонких пленок. Экспериментальное подтверждение изменения
времен удерживания в зависимости от внутреннего диаметра колонки и толщины плен-
ки неподвижной фазы продемонстрировано на рис. 6.1. Помимо сокращения времени
Глава 6. Передовые методы на основе ГХ-МС
Рис. 6.1. Влияние внутреннего диаметра колонки и толщины пленки на хроматографиче-
ские времена удерживания
удерживания в режиме быстрой хроматографии, стоит отдельно упомянуть улучшение
формы газохроматографических пиков, что является преимуществом также и для коли-
чественного анализа.
Уменьшения времени элюирования компонентов можно также достичь за счет уве-
личения потока газа-носителя. В газовой хроматографии линейный поток, измеренный
в см/с, отражает скорость, с которой газ-носитель проходит через колонку. Поскольку
хроматографируемые компоненты растворены в газе-носителе, то более высокие скоро-
сти потока ускоряют процесс элюирования аналитов из конденсированной фазы и со-
кращают общее время анализа. Как было показано выше, ВЭТТ и скорость потока га-
за-носителя связаны уравнением Голэя. Эта зависимость нелинейная (рис. 6.2), в связи
с чем так называемая оптимальная скорость потока может быть рассчитана в той точке,
где ВЭТТ — параметр Н — принимает минимальное значение, т.е. при максимальной
эффективности газохроматографической колонки. Как только линейная скорость по-
тока отклоняется от оптимального значения иор{ = %D/Kd, форма пиков и хроматографи-
ческое разрешение ухудшаются. При скоростях потока меньше оптимальной величины
аналиты слишком долго задерживаются в неподвижной фазе, что обеспечивает лучшее
разрешение, но ухудшает форму пиков и удлиняет анализ. При потоках газа-носителя,
Е
Е
0,10 мм
0,25 мм
0,32 мм
0,50 мм
0,75 мм
и/с m/s
Рис. 6.2. Кривые Голэя, рассчитанные для различных внутренних диаметров капилляр-
ных колонок
превышающих оптимум, аналиты слишком быстро элюируются из неподвижной фазы,
что сокращает время анализа, но резко ухудшает разделение компонентов. Важной осо-
бенностью узких колонок является возможность использования более высоких скоро-
стей потока газа-носителя без потерь разрешения. Последнее отражено на рис. 6.2, где
представлена зависимость ВЭТТ от скорости потока подвижной фазы, иными словами,
графическое изображение уравнения Голэя в виде так называемой кривой Голэя.
Как уже упоминалось, оптимальная линейная скорость потока достигается в точках
минимума кривых Голэя (точки А). В соответствии с рис. 6.2 оптимальная скорость потока
//^увеличивается при уменьшении внутреннего диаметра колонки i.d., например иор = 12
(точка А0 75) и 40 (точка Ао 10) см/с для колонок с внутренним диаметром 0,75 и 0,10 мм соот-
ветственно. Таким образом, более узкие колонки следует использовать с более высокими
потоками газа-носителя, что полностью соответствует концепции быстрой газовой хро-
матографии. Еще более важным является тот факт, что кривые Голэя для узких колонок
значительно более пологие. Это позволяет сильно увеличивать потоки газа-носителя без
существенной потери газохроматографического разрешения (сравните, например, ВЭТТ-
колонки с диаметром 0,10 мм при потоках газа-носителя 40 и 80 см/с).
Глава 6. Передовые методы на основе ГХ-МС
Таблица 6.1. Газохроматографические параметры и техника быстрой ГХ/МС
Изменяемый параметр Способ изменения для достижения миниального времени анализа
Длина колонки Использование более коротких колонок
Скорость нагрева термостата Использование ускоренных темпов нагрева
Внутренний диаметр колонки Использование более узких колонок увеличивает разрешение
Толщина неподвижной фазы (пленки) Более тонкие пленки улучшают хроматографическое разрешение
Скорость потока газа-носителя Увеличение скорости способствует ускоренному элюированию
Тип газа-носителя Использование водорода, если это возможно
Более пологие кривые Голэя с минимальными значениями величины ВЭТТ могут
быть также получены при замене стандартного газа-носителя — гелия — на водород
(Hair and Yost, 1989). Однако высокая горючесть и взрывоопасность водорода являются
серьезным препятствием для использования его в обычных лабораториях, практикую-
щих ГХ/МС-анализ.
Очевидно также, что использование более высоких скоростей нагревания в темпе-
ратурно-градиентной газовой хроматографии способствует ускорению элюирования
аналитов из колонки. Этот принцип в действительности работает, но ограничен в при-
менении характеристиками имеющегося оборудования, в частности термостата газово-
го хроматографа. Изящный способ осуществления сверхбыстрого нагрева газохрома-
тографической системы был разработан компанией Thermo Corporation. Основной узел
этой системы включает проволочный нагреватель, температурный сенсор и ГХ-колонку
малого диаметра, обмотанные вместе керамическим волокном, что позволяет достичь
скоростей нагрева колонки вплоть до 1200°С/мин. Такая технология получила название
«сверхбыстрая газовая хроматография». Она позволяет проводить полное хроматогра-
фическое разделение менее чем за три минуты.
В практике быстрой ГХ/МС используются колонки с внутренним диаметром
от 0,10 до 0,18 мм, длиной 20 м и меньше (см. подпись к рис. 6.3). Малая длина колонок
вкупе с небольшим внутренним диаметром и тонким покрытием неподвижной фазы
приводит к малой емкости таких колонок, то есть в практике быстрой ГХ/МС прихо-
дится использовать значительно меньшие количества образца в каждом вводе по срав-
нению с традиционной ГХ/МС. В связи с этим зачастую могут требоваться значительно
более высокие кратности деления потока во избежание перегрузки колонки, что яв-
ляется определенным требованием к используемому оборудованию. Стоит отметить,
что использование меньших количеств разделяемых компонентов никоим образом
не означает более низкую чувствительность быстрой ГХ/МС: она остается неизменной
благодаря значительно более острым и высоким пикам аналитов в сравнении с тради-
ционной ГХ/МС.
Для того чтобы использовать колонки большой емкости, в том числе и для анализа
методом быстрой ГХ/МС, была предложена техника быстрой ГХ при пониженном дав-
лении. В этом методе вместо коротких и узких колонок, напротив, используются колон-
ки большого диаметра, как правило, 0,53 мм, при длине около 10 м и толщине пленки
неподвижной фазы 0,25—1 мм. Выход из колонки присоединен к вакуумному насосу,
Abundance
10000000
9000000
8000000
7000000
6000000
5000000
4000000
3000000
2000000
1000000
5
6.00
Рис. 6.3. Сравнение традиционного (а) и быстрого (б) методов ГХ/МС для анализа состава
масла перечной мяты. Параметры: (а) длина колонки 25 м, внутренний диаметр
0,25 мм, толщина пленки неподвижной фазы 0,25 мкм; кратность деления потока
1/100, поток газа-носителя (гелия) 1 мл/мин, скорость нагрева 3°С/мин; (б) дли-
на колонки 10 м, внутренний диаметр 0,10 мм, толщина пленки неподвижной
фазы 0,10 мкм; кратность деления потока 1/200, поток газа-носителя (гелия)
0,6 мл/мин, скорость нагрева 20°С/мин. Прибор: хроматограф Trace GC Ultra,
присоединенный к масс-спектрометру Trace DSQ (квадруполь)
поэтому разделение компонентов происходит при пониженном давлении (low pressure
GC, LP/GC). Давление влияет на оптимальное значение скорости потока газа-носителя:
преобразование уравнения Голэя с учетом соотношения между скоростью потока газа
носителя и давления внутри колонки показывает, что более низкое давление приводит
к увеличению значений uopt (Hail’ and Yost, 1989). Поскольку масс-спектрометр работа-
ет в условиях высокого вакуума, он является идеальным детектором для осуществле-
ния режима ГХ при пониженном давлении (Mastovska et al., 2001). Данная технология,
ГХ/МС при пониженном давлении, помимо видимого сокращения времени анализа,
позволяет также проводить разделение компонентов при относительно низких темпе-
ратурах (благодаря пониженному давлению). Это особенно важно при анализе термо-
лабильных соединений, которые становятся доступными для анализа и не разлагаются,
что может иметь место в режиме ГХ/МС при нормальном давлении.
Глава 6. Передовые методы на основе ГХ-МС
6.1.2. Вклад масс-спектрометрической составляющей при
использовании техники быстрой ГХ/МС
Применение методов быстрой ГХ/МС даже при хорошем хроматографическом раз-
решении приводит к образованию очень узких пиков, что предъявляет определенные
требования к масс-спектрометрическому детектору. Для того чтобы форма пиков была
правильной и их можно было бы использовать в количественном анализе, необходима
более высокая скорость сбора данных (регистрации масс-спектров) по сравнению с тра-
диционным ГХ/МС-анализом. Данное утверждение хорошо иллюстрирует рис. 2.11
(глава 2) и комментарии к нему.
Общепринятая классификация техник ГХ/МС в зависимости от времени хрома-
тографирования (Hajslova and Cajka, 2008) представлена в табл. 6.2. Считается, что для
реконструирования хроматографического пика хорошего качества достаточно десяти
точек (масс-спектров) на его верхнюю половину, поэтому для различных типов ГХ/МС
возможен расчет минимально необходимой скорости записи данных (см. табл. 6.2). Ско-
рость сбора данных также является характеристикой масс-спектрометрического анали-
затора (Mastovskd and Lehotay, 2003). Этот параметр и приведен в табл. 6.3.
Если сравнить данные табл. 6.2 и 6.3, становится очевидной практическая непригод-
ность секторных приборов для задач быстрой ГХ/МС. Использование ионных ловушек
Таблица 6.2. Классификация ГХ/МС-методов в зависимости от продолжительности
анализа
Тип ГХ/МС-анализа Стандартное время хроматографирова- ния, мин. Полуши- рина пика (FWHM)’, мс Требуемая минимальная скорость записи данных, спектров/с
Традиционная >10 >1000 <10
Быстрая 1-10 200-1000 10-50
Очень быстрая 0,1-1 30-200 50-350
Сверхбыстрая <0,1 5-30 >350
FWHM — от англ., Full Width Half Maximum, — полная ширина на полувысоте пика
Таблица 6.3. Характеристики наиболее распространенных анализаторов, используемых
в ГХ/МС
Анализатор Верхний предел регистрируемых m/z Скорость записи данных, спектров/с (Гц) Разрешение
Квадрупольный 80-1050 Да 15—33 (для диапазона масс 300 Да) ~0,5Да
Ионная ловушка 650-1000 Да ~20 (для диапазона масс 300 Да) ~1 Да
Высокоскоростной время пролетный 1000 Да 100-500 1400 FWHM
Врем я пролетный высокого разрешения 1500 Да 200 50000 FWHM
Секторный 4000 Да 7 на декаду* До 80000
Декада — диапазон m/z, отличающихся на порядок (например 100—1000 или 50—500 m/z)
тоже нецелесообразно, тогда как квадрупольные анализаторы в принципе могут обеспе-
чить достаточную скорость записи данных. В качестве примера на рис. 6.3 представлены
результаты анализа эфирного масла методами традиционной и быстрой ГХ/МС с ква-
друпольным анализатором. Время анализа сократилось в шесть раз с 36 до 6 минут при
использовании техники быстрой ГХ/МС (Rubiolo et al., 2008). При использовании режи-
ма мониторинга заданных ионов (глава 2) скорость записи данных резко возрастает, что
позволяет использовать квадрупольный анализатор и в очень быстрой ГХ/МС. Однако
ГХ/МС-анализ с записью полных масс-спектров в режимах очень быстрой и сверхбы-
строй ГХ/МС возможен исключительно при использовании высокоскоростного время-
пролетного анализатора.
Однако не только высокая скорость записи данных делает времяпролетный ана-
лизатор наиболее подходящим для быстрой ГХ/МС. При использовании высоких
скоростей нагревания и потоков газа-носителя, превышающих оптимальные, хрома-
тографическое разрешение ухудшается. Это значит, что зачастую компоненты смеси
элюируются в виде перекрывающихся пиков, а записанные масс-спектры представля-
ют собой суперпозицию масс-спектров коэлюирующихся веществ. Это существенно за-
трудняет идентификацию по спектрам. Во избежание неверной идентификации компо-
нентов такие масс-спектры должны быть очищены от пиков коэлюирующихся веществ.
Один из способов очистки масс-спектров был уже описан в главе 2 и представляет собой
процедуру вычитания фона. Этот метод также является простейшим способом суще-
ственно улучшить масс-спектр, поскольку фоновые сигналы не изменяются в течение
всего времени выхода пика (или перекрывающихся пиков), которое является весьма
коротким в быстром ГХ/МС-анализе. Более сложной является очистка масс-спектра
конкретного аналита от пиков, принадлежащих коэлюирующемуся веществу, — так на-
зываемая деконволюция. Один из реализованных на сегодняшний день методов декон-
волюции представлен на рис. 6.4.
Рассмотрим хроматографический пик, полученный методом быстрой ГХ/МС
и представляющий собой накладывающиеся друг на друга пики двух веществ, X и Y.
Хроматограмма, выстроенная по полному ионному току (ПИТ), представляет собой пик
Рис. 6.4. Алгоритм деконволюции пиков
Глава 6. Передовые методы на основе ГХ-МС
неправильной формы, при этом не очевидно, что этот пик представляет собой супер-
позицию двух коэлюирующихся компонентов. Масс-спектры, записанные в течение
элюирования пика (спектры А на рис. 6.4), представляют собой суперпозицию масс-
спектров обоих компонентов X и Y. Для деконволюции пиков программное обеспече-
ние выстраивает масс-хроматограммы (глава 2) для всех значений m/z в регистрируемом
диапазоне. После этого находятся те конкретные m/z, масс-хроматограммы которых до-
стигают своих максимумов при одном и том же времени удерживания (например желтая
и синяя масс-хроматограммы на рис. 6.4). Только такие значения m/z (например m/z 57
и 41 на рис. 6.4, максимум при 171,9 с) и интенсивности соответствующих пиков исполь-
зуются для построения «восстановленного» масс-спектра (спектры В на рис. 6.4). Спектр
В вещества X, элюирующегося при 171,9 с, полностью очищен от пиков ионов с m/z 107
и 122, принадлежащих веществу Y с временем удерживания 172,5 с. Аналогичным об-
разом пики масс-хроматограмм, построеннных по m/z 107 и 122, достигают своего мак-
симума при 172,5 с и приписываются коэлюирующемуся веществу Y, восстановленный
масс-спектр которого (В) оказывается полностью очищенным от «чужих» пиков с m/z 57
и 41. Очищенные таким образом масс-спектры В используются далее программным обе-
спечением для сравнения с библиотечными спектрами (спектры С на рис. 6.4), а масс-
хроматограммы — для количественного определения. В случае если фрагментные ионы
присутствуют в спектрах обоих веществ с близкими временами удерживания, програм-
ма «делит» интенсивности таких ионов между ними в пропорции, соответствующей от-
ношению интенсивности этих фрагментых ионов в библиотечных спектрах идентифи-
цированных компонентов.
Как правило, деконволюция обеспечивает очень высокую степень совпадения очи-
щенных и библиотечных спектров (более 90% в примере на рис. 6.4). Вместе с тем для
успешной работы программ деконволюции в режиме быстрой ГХ/МС необходимо вы-
полнение нескольких требований. Во-первых, все компоненты должны элюироваться
в виде симметричного пика с одним максимумом, и этот максимум должен быть оди-
наковым для всех масс-хроматограмм каждого конкретного вещества. К сожалению,
данное требование зачастую невыполнимо из-за эффекта искажения спектра, харак-
терного для всех сканирующих анализаторов и описанного в главе 2. Таким образом,
несканирующие времяпролетные масс-анализаторы являются предпочтительными для
деконволюции, предоставляя одинаковые масс-спектры на протяжении всего времени
элюирования вещества1. Графическое объяснение эффекта искажения спектра пред-
ставлено на рис. 2.12. Во-вторых, сами пики должны быть как можно более гладкой
формы, иными словами, дифференцируемыми, что необходимо для автоматического
нахождения максимума пика. Более гладкая форма пика достижима при максималь-
ном количестве точек, формирующих пик, то есть максимальном числе масс-спектров,
записанных во время элюирования вещества. Таким образом, высокая скорость сбо-
ра данных является также необходимым условием работы программ деконволюции
(рис. 6.5). В-третьих, расстояние между максимумами (ART на рис. 6.4) двух пиков
должно быть достаточным для самой возможности деконволюции. Очевидно, что пики
наиболее правильной формы допускают наименьшую величину ART. Соответственно
чем выше скорость записи масс-спектров, тем более эффективной будет деконволюция.
1 Сам по себе алгоритм деконволюции работает и для сканирующих анализаторов, но только
при относительно длинных временах анализа (т.е. в режиме традиционной ГХ/МС).
6.2. ГХ/МС с интерфейсом ультразвуковых молекулярных пучков
Рис. 6.5. Влияние скорости записи масс-спектров на возможность автоматического на-
хождения пиков и деконволюции при анализе пестицидов, (а) I Гц, пики не най-
дены; (б) 20 Гц, найдено в результате деконволюции 8 веществ; (в) 40 Гц, найдены
все компоненты
В качестве примера на рис. 6.5 представлена работа алгоритма деконволюции смеси де-
вяти пестицидов, выходящих из колонки в течение 4 с, при скорости записи 1, 20 и 40
спектров/с. Скорость в 40 Гц позволяет разделить пики хлорпирифоса и паратиона,
в то время как оба этих вещества сливаются в один пик при 20 Гц. Помимо этого, масс-
спектр хлорпирифоса при 20 Гц содержит так много пиков коэлюирующихся компонен-
тов, что не может быть идентифицирован при библиотечном поиске.
Суммируя все вышесказанное, можно заключить, что наиболее подходящим для
поиска пиков, деконволюции и метода быстрой ГХ/МС в целом является высокоско-
ростной времяпролетный анализатор. Сверхбыстрая ГХ/МС возможна исключительно
с этим типом анализатора. Если анализируемая смесь содержит незначительное число
компонентов и они могут быть успешно деконволюированы, можно использовать очень
короткие (до 5 м) колонки и проводить анализ быстрее, чем за минуту. Такой потенциал
времяпролетного анализатора и возможностей сверхбыстрой ГХ/МС сделал возмож-
ным внедрение и разработку принципиально нового метода в хроматографии — дву-
мерной газовой хроматографии — масс-спектрометрии ГХ х ГХ/МС (см. ниже).
6.2. ГХ/МС с интерфейсом ультразвуковых
молекулярных пучков
Некоторые инновации позволяют не только увеличить скорость анализа, но и полу-
чить более качественные спектры с одновременным расширением числа соединений,
которые можно анализировать методом ГХ/МС. Как уже было сказано выше, наряду
со значительными аналитическими достоинствами ГХ/МС существуют определенные
компромиссы в функционировании обеих составляющих частей этого комбинирован-
ного метода. Так, вакуумные требования к масс-спектрометру обычно ограничивают
поток газа через колонку ГХ 1—2 мл/мин, что несколько сокращает диапазон анали-
зируемых соединений по сравнению, например, с ГХ с пламенно-ионизационным де-
тектором. С другой стороны, последовательный ввод в масс-спектрометр соединений
с увеличивающимися температурами кипения во время ГХ-анализа требует привязки
температуры источника ионов к этому параметру для наименее летучего компонента.
Это может ухудшить качество спектров более летучих соединений. Эти и некоторые
Глава 6. Передовые методы на основе ГХ-МС
другие проблемы совместимости частей в ГХ/МС могут быть устранены в ГХ/МС с ин-
терфейсом ультразвуковых молекулярных пучков (Supersonic Molecular Beams interface,
SMB GC/MS). При этом появляется и ряд дополнительных преимуществ (рис. 6.6). Тех-
ника ультразвуковых молекулярных пучков используется в системах ГХ/МС с 1990-х го-
дов (Amirav, 1991; Dagan and Amirav, 1994). Применение этого подхода описано в работах
(Fialkov et al., 2003; Fialkov et al., 2006; Fialkov et al., 2007; Amirav, et al., 2008; Fialkov et al.,
2008), а модель интерфейса «5975-SMB» (Aviv Analytical, Tel Aviv, Israel) производится
и может быть состыкована с приборами Agilent 5975 GC/MS и/или Agilent 7000.
Как показано на рис. 6.6, интерфейс SMB состоит из нескольких частей: трансфер-
ная линия с программируемой температурой и небольшой SMB-форсункой специаль-
ной формы (~0,1 мм в диаметре) на конце, вакуумная камера, в которой располагаются
форсунка и скиммер, SMB ионный источник пролетного типа и ионное зеркало (Fialkov
etal., 2006;Amirav, etal., 2008).
Перед форсункой элюат из хроматографической колонки смешивается с потоком
компенсирующего газа (Не), подающегося с атмосферным давлением. Поток газа с мо-
лекулами образца проходит через форсунку со скоростью около 100 мл/мин и расши-
ряется в вакуум, образуя ультразвуковой молекулярный пучок. Следующие характери-
стики этого пучка имеют первостепенную важность: все молекулы в пучке (атомы Не
и молекулы аналитов) имеют примерно одинаковую скорость, равную ультразвуковой
скорости атомов гелия; кинетическая энергия молекул образца, таким образом, увели-
чивается; колебательная энергия молекул образца существенно понижена в соответ-
ствии с понижением общей энергии в результате холодных (медленных) столкновений
внутри пучка (эквивалентно температуре от нескольких градусов до нескольких де-
сятков градусов Кельвина); молекулы образца распространяются более узким пучком
по сравнению с атомами Не (эффект струйного сепаратора). Внутренняя часть пучка
коллимируется скиммером и аксиально вводится в цилиндрический SMB-источник
Рис. 6.6. Схема прибора ГХ/МС с SM В
6.2. ГХ/МС с интерфейсом ультразвуковых молекулярных пучков
ионов пролетного типа, работающий в режиме электронной ионизации. Молекулы об-
разца летят вдоль центральной оси источника в бесполевом пространстве. При этом
электроны с энергией 70 эВ, эмитируемые длинным (~30 мм) катодом, пересекают пу-
чок в радиальном направлении, ионизируя его молекулы. Образовавшиеся ионы про-
должают двигаться вдоль центральной оси источника в том же направлении и с той же
скоростью, что и исходные молекулы. Они покидают источник и отклоняются ионным
зеркалом по направлению к квадрупольному анализатору. Фоновые ионы обычно пере-
мещаются не строго вдоль центральной оси. Поэтому они большей частью нейтрализу-
ются на стенках источника.
Можно перечислить несколько характеристик SMB, которые улучшают аналитиче-
ские возможности ГХ/МС:
• скорость газа в колонке может быть практически нелимитированной
(до 100 мл/мин);
• молекулы образца «колебательно охлаждены» перед процессом ионизации;
• неотъемлемой характеристикой источника является его «инертность»;
• ионный источник не имеет «хвостов» пиков;
• источник имеет очень малое время отклика (<100 микросекунд);
• большинство ионов фона не попадает в анализатор, что уменьшает шум и устра-
няет эффект «памяти».
Рис. 6.7 (левая часть) демонстрирует ухудшение хроматографических сигналов ме-
нее летучих веществ с уменьшением температуры источника ионов, что связано с уве-
личением времени нахождения молекул на внутренней поверхности источника. Понят-
но, что для ГХ/МС-анализа данной смеси температура источника не должна быть менее
250 °C.
Однако чем выше температура источника, тем ниже интенсивность пика молеку-
лярного иона в спектре (рис. 6.7, правая часть). Это связано с повышением в условиях
электронной ионизации (70 эВ) вероятности фрагментации молекулярного иона лю-
бого соединения при увеличении внутренней термической энергии соответствующей
молекулы, которая прямо пропорциональна температуре источника.
Следовательно, стандартный вариант ГХ/МС-анализа функционирует в условиях
компромисса с ухудшением качества и хроматограммы, и масс-спектра, которое в обо-
их случаях зависит от температуры источника ионов (Amirav et al., 2008). Это зачастую
является основной причиной получения в условиях ГХ/МС-анализа масс-спектров
с уменьшенной интенсивностью пика молекулярного иона по сравнению со спектром
из библиотеки NIST. Дело в том, что большинство спектров NIST получено в оптими-
зированных условиях, при меньшей температуре или в условиях прямого ввода образ-
ца. Следует отметить, что величина этого эффекта определяется структурой молекулы.
В частности, он невелик для небольших молекул с низкой теплоемкостью, включая,
например, октафторнафталин. Именно это является одной из причин использова-
ния октафторнафталина в качестве стандарта проверки чувствительности различных
ГХ/МС-систем.
Прибор ГХ/МС с SMB и источником ионов пролетного типа лишен описанного
выше недостатка, поскольку (i) молекулы образца пролетают область ионизации, не ка-
саясь стенок источника; (ii) молекулы образца, охлажденные в ультразвуковом пучке,
обладают очень низкой внутренней энергией (несколько градусов или десятков граду-
Глава 6. Передовые методы на основе ГХ-МС
Рис. 6.7. Влияние температуры источника на вид хроматограмм по полному ионному току
смеси алканов (левая часть) и вид масс-спектра электронной ионизации тетрако-
зана (правая часть). Хроматограммы иллюстрируют резкое уменьшение высоты
пиков с уменьшением температуры, а масс-спектр и соответствующая вставка
с правой стороны — эффект уменьшения интенсивности пика молекулярного
иона с ростом температуры. В данном случае был использован масс-спектрометр
Varian 1200 со стандартным источником ионов
m/z
сов Кельвина), что приводит к их минимальной фрагментации после ионизации элек-
тронами с энергией 70 эВ.
На рис. 6.8 проиллюстрирован эффект ИЭ (70эВ) холодных молекул («холодная»
ИЭ). Высокоинтенсивные пики молекулярных ионов наблюдаются в масс-спектрах
больших алканов: С40Н82(/иД 562) и даже С72Н|46(/иД 1011,2 (!)).
Хроматограммы ПИТ, представленные на рис. 6.8, также необычны. Левая часть ил-
люстрирует хроматограмму сверхбыстрого анализа, а правая — хроматограмму образца
соединений, которые значительно тяжелее аналитов, доступных стандартной ГХ/МС
(Fialkov et al., 2003; Amirav et al., 2008; Fialkov et al., 2008). Подобные хроматограммы сме-
си алканов Polywax 850 могут быть получены методом ГХ с плазменно-ионизационным
детектором (короткая колонка и большой поток газа-носителя). Стандартная ГХ/МС
6.2. ГХ/МС с интерфейсом ультразвуковых молекулярных пучков
8
8
6
Конец
анализа,
охлаж-
дение,
50 сек.
2
ok
о
Конец
цикла,
можно
вводить
новый
образец,
60 сек.
6
Polywax 850
60
Молеку-
лярные
ионы НС
видны в
условиях
66 ц
v72n 146
Конец
анализа в
условиях эд
стандарт-
ной
2,5
4
2
0
60
10
20
30
40
50
Рис. 6.8. Анализ алканов методом ГХ/МС с SM В. Левая часть иллюстрирует сверхбыстрый
анализ, а правая — анализ очень тяжелых соединений. Хроматограммы по пол-
ному ионному току (ПИТ) с маркировкой нескольких соединений представле-
ны в верхней части рисунка. Масс-спектры «холодной» электронной ионизации
алканов С40Н82 и С72Н146 представлены в нижней части. Образец Polywax 850
(Restek, США) был разбавлен ксилолом до концентрации 0,2 %
не позволяет получать информацию об алканах тяжелее, чем примерно С40. Фактиче-
ски надежное определение н-алканов в стандартных условиях ГХ/МС возможно толь-
ко до С28, поскольку спектры более тяжелых алканов практически не отличаются друг
от друга. При этом интенсивность пиков их молекулярных ионов ничтожна. В спектрах
разветвленных алканов эти пики и вовсе отсутствуют. Напротив, SMB ГХ/МС дает воз-
можность проанализировать всю смесь Polywax 850 (рис. 6.8). Газохроматографические
температуры элюирования уменьшаются на 20°С всякий раз при двукратном увеличе-
нии потока газа-носителя через колонку или двукратном уменьшении длины колонки
(Fialkov et al., 2003). Температуры элюирования могут быть уменьшены при использова-
нии более тонких пленок неподвижной фазы и более медленном повышении темпера-
туры колонки (см. выше). Так, для анализа образца Polywax 850 методом ГХ/МС с SMB
была использована колонка длиной 4 метра при скорости потока 32 мл/мин. Очевидно,
что сокращение времени выхода дает возможность проводить сверхбыстрый анализ.
На рис. 6.8 (левая часть) представлен результат сверхбыстрого анализа с использовани-
ем модуля быстрой ГХ, короткой колонки (1,5 метра) при скорости потока 35 мл/мин
и температурного градиента 440°С/мин, вместо стандартных ГХ-условий.
В режиме «высокая скорость потока с короткой колонкой» диапазон соединений,
которые можно анализировать методом ГХ/МС с SMB, значительно расширяется, при-
чем не только в сторону менее летучих, но и в сторону термолабильных, которые мо-
гут разлагаться в стандартных условиях ГХ/МС в инжекторе, колонке или источнике
184 Глава 6. Передовые методы на основе ГХ-МС
ионов. При работе с температурно программируемым инжектором температуры выхода
термолабильных соединений, попадающих в колонку, также уменьшаются, поскольку
эти термолабильные соединения выживают во время пребывания в инжекторе и ока-
зываются в колонке. После элюирования при пониженной температуре они попадают
в источник ионов пролетного типа без какой-либо деградации. Эффект скорости потока
на уровень деградации термолабильных соединений представлен на рис. 6.9. Оба анали-
за проведены на приборе ГХ/МС с SM В в идентичных условиях, но со скоростью потока
газа-носителя 16 мл/мин (верхняя часть) и 1 мл/мин (нижняя часть).
На нижней хроматограмме (1 мл/мин) хроматографические пики трипероксида три-
ацетона (ТАТР), нитроглицерина (NG) и гексогена (RDX) заметно менее интенсивны,
а пик тетранитрата пентаэритрита (PETN) отсутствует вовсе. Пик, который может при-
надлежать тетрилу (Tetryl), присутствует, но в соответствующем масс-спектре отсутству-
ет пик молекулярного иона тетрила (m/z 287). Следовательно, этот важный ингредиент
может быть идентифицирован как продукт деградации тетрила. Верхняя хроматограмма
(16 мл/мин) демонстрирует наличие в образце наряду с другими взрывчатыми вещества-
ми PETN и Tetryl. Масс-спектры «холодной» ИЭ (показаны только для тетрила) с интен-
сивными пиками молекулярных ионов подтверждают правильность идентификации.
Следует подчеркнуть, что в отсутствие в масс-спектре молекулярного иона сложно опре-
делить, присутствует ли в образце тетрил или продукт его деградации N-метилпикрамид
(Fialkov etal., 2003), да и надежно идентифицировать любой другой ингредиент.
Во многих случаях целью анализа является детектирование в сложном образце
следовых количеств целевых компонентов. Наиболее надежным методом для решения
подобной задачи является метод ГХ/МС/МС (глава 4). Вариант ГХ/МС с SMB расши-
ряет диапазон соединений, которые могут быть проанализированы, и дополнительно
повышает селективность и чувствительность ГХ/МС/МС-метода (Fialkov et al., 2007).
Это связано с несколькими аспектами. Можно указать на подавление шума и меньшие
потери вещества. Однако наиболее важным моментом является увеличение интенсив-
ности молекулярного иона (сравните «холодную» ИЭ, рис. 6.10д, и стандартную ИЭ,
рис. 6.106, при получении масс-спектра пестицида диазинона М+* 304). Есть несколько
преимуществ при использовании именно молекулярного иона в качестве иона-пред-
шественника в тандемной масс-спектрометрии. Прежде всего, это надежнее, поскольку
любой другой (фрагментный) ион может принадлежать гомологу, структурно близкому
соединению или любому другому компоненту с близким временем выхода. Во-вторых,
чем больше массы иона-продукта и иона-предшественника, тем меньше матричные
эффекты (Kochman, 2002). Более того, требуемая для активации соударением энергия
обычно также меньше. В результате образуется лишь один (или несколько) ионов-про-
дуктов (см. «холодную» ИЭ в МС/МС, рис. 6.10<?, с доминирующим в спектре пиком
фрагмента с m/z 179 при энергии соударений 5 эВ). Если молекулярный ион малоин-
тенсивен и в качестве иона-предшественника выбирается фрагментный ион с большой
массой (например фрагментный ион с m/z 179 в случае стандартной ИЭ диазинона), для
инициирования фрагментации требуется большая энергия соударений, поскольку, как
правило, четноэлектронные ионы стабильнее. В результате идут разрывы самых разных
связей, а интенсивность перераспределяется между большим числом фрагментных ио-
нов (рис. 6.10г). Сигнал в варианте «холодной» ИЭ в МС/МС может оказаться в несколь-
ко сот раз интенсивнее, чем в стандартном варианте МС/МС с ИЭ.
6.2. ГХ/МС с интерфейсом ультразвуковых молекулярных пучков 185
Интенсивность
50
100
200 250 Температура, °C
Рис. 6.9. Анализ смеси взрывчатых веществ методом ГХ/МС с SMB с увеличенным по-
током газа-носителя через колонку для снижения температур выхода компонен-
тов в целях предотвращения их деградации. Масс-спектры тетрила представле-
ны на вкладках. Колонка 6 метров, 0,25 мм (внутренний диаметр), 0,5 мкм фазы
DB5ms, температурная программа 35°С/мин. Температура трансферной линии
и форсунки 200°С, температура инжектора 180°С. Потоки через колонку состав-
ляли 16 мл/мин (верхний рисунок, А) и 1 мл/мин (нижний рисунок, В)
186 Глава 6. Передовые методы на основе ГХ-МС
10-
8-
6-
4-
2-
179
Diazinon, cold EI-MS/MS
Product ions scan
Precursor ion 304 (Molecular)
Product scan 50-350
Collision energy 5 eV
304, M
Diazinon, El
Ion source temp = 200°C
179
199 304,M
„vj, lД ., > ... ,1, .
200 250
□
178
X 16
100 150
Diazinon, cold EI-MS/MS
Product ions scan
Precursor ion 179 (fragment)
Product scan 50-350
Collision energy 50 eV
179
m/z
m/z
200 250
Рис. 6.10. Спектр «холодной» ИЭ (а) и стандартный масс-спектр ИЭ при температуре ис-
точника 200°C (б) диазинона. Вклад тока молекулярного иона в полный ион-
ный ток составляет 22,4% (а) и 1,6% (б). Правая часть иллюстрирует спектр
ионов продуктов (МС/МС) для диазинона, полученный при фрагментации
иона с m/z 304 с энергией соударений 5 эВ (в) и при фрагментации максималь-
ного фрагментного иона (m/z 179) с энергией соударений 50 эВ (г) в качестве
ионов-предшественников
Применение ГХ/МС/МС с SMB для анализа тестостерона в крови аллигатора
(рис. 6.11) — типичный пример целевого анализа следовых количеств аналита в сложной
матрице. Следует обратить внимание на тот факт, что в связи с расширенным диапазо-
ном веществ, которые можно анализировать методом ГХ/МС(-МС) с SMB, в отличие
от стандартного ГХ/МС-анализа, проводить дериватизацию тестостерона не требуется.
Пик тестостерона не виден на хроматограмме ПИТ, на которой доминирует пик холе-
стерина (рис. 6.11, средняя вставка). Масс-спектр в области выхода из колонки тестосте-
рона представляет собой лес пиков практически с каждым значением m/z вплоть до 400
(рис. 6.11, верхняя вставка). Небольшой пик, пробивающийся сквозь шум, наблюда-
ется в области выхода из колонки тестостерона на масс-хроматограмме, построенной
по току молекулярного иона тестостерона с m/z 288 (рис. 6.11, нижняя вставка). Нако-
нец, МС/МС-анализ с небольшой энергией соударений (6 эВ) с молекулярным ионом
тестостерона (m/z 288) в качестве иона-предшественника и фрагментного иона с m/z 124
в качестве иона-продукта позволяет зарегистрировать пик тестостерона, присутствую-
щего в образце на уровне 4 ppb, с хорошим отношением сигнал/шум (Fialkov et al., 2007).
В заключение следует отметить, что чем сложнее задача для ГХ/МС, тем очевиднее
возможности и преимущества ГХ/МС с SMB. Это повышенная чувствительность, улуч-
шенное качество масс-спектров, более широкий круг соединений, которые можно ана-
лизировать. В эту группу попадают вещества, с которыми, как считается, невозможно
работать методом ГХ/МС, что заставляет даже приверженцев этого метода переходить
на ЖХ/МС. Некоторые характеристики и приложения ГХ/МС с SMB остались за рам-
ками изложения. Сюда можно отнести кластерную химическую ионизацию (Fialkov
and Amirav, 2003), классическую электронную ионизацию (Gordin et al., 2008), ГХ/МС
6.2. ГХ/МС с интерфейсом ультразвуковых молекулярных пучков
4-
MS/MS:
288-124 (6 eV)
RSIM
288
।11111111111111111 । । ।
0 12 3 Время, мин.
0
1,0 2,0 3,0 4,0 5,0 Время, мин.
120 150 180 210 300 Температура, °C
Рис. 6.11. Анализ тестостерона в образце крови аллигатора из Флориды с устройством
1200-SMВ; колонка Varian VF5ms длиной 4 м с внутренним диаметром 0,25 мм
и толщиной фазы 0,1 цм. Скорость потока Не 12 мл/мин. МС/МС-эксперимент
проведен с молекулярным ионом недериватизованного тестостерона (m/z 288)
при энергии соударений 6 В. В качестве иона-продукта регистрировался ион
с m/z 124. На вставке с правой стороны приведены результаты параллельного
анализа того же образца. Они представляют собой масс-спектр в области выхо-
да из колонки тестостерона, хроматограмму ПИТ и масс-хроматограмму потоку
молекулярного иона тестостерона с m/z 288. Образцы и детальные инструкции
по проведению эксперимента были переданы Джоном Боуденом и Ричардом
Постом из университета Флориды, США. (Из Fialkov et al., 2007, с разрешения
Elsevier)
с SMB-метод анализа смесей углеводородов (видоа топлива) с новым вариантом опре-
деления изомеров (Fialkov and Amirav, 2008), применение (метод и программное обеспе-
чение) изотопного анализа для повышения эффективности идентификации соедине-
ний (Alon andAmirav, 2006), дополнительные инструментальные опции для упомянутой
быстрой ГХ, ГХ* ГХ/МС с пульсирующей модуляцией потока (Poliak et al., 2008; Poliak
etal., 2008a) и зонд для быстрого и простого пробоотбора с поверхности и прямого ввода
образца в систему быстрой ГХ/МС с SMB или МС с SMB (Poliak etal., 2010).
188 Глава 6. Передовые методы на основе ГХ-МС
6.3. Двумерная газовая хроматография —
масс-спектрометрия ГХ х ГХ/МС
6.3.1. Принцип метода
Принцип метода ГХх ГХ/МС основан на разделении компонентов смеси последова-
тельно на двух газохроматографических колонках. Компоненты вначале разделяют-
ся на первичной колонке в режиме традиционной ГХ и характеризуются первым вре-
менем удерживания на этой колонке (RT^. В процессе хроматографирования элюат
из первичной колонки делится на малые порции каждые несколько секунд. Порции
элюата отделяются друг от друга за счет быстрой заморозки компонентов в конденси-
рованную «каплю», которая сразу после формирования резко нагревается и поступает
во вторичную колонку. Аналиты, содержащиеся в каждой конденсированной порции,
очень быстро разделяются на вторичной колонке и характеризуются уже вторым вре-
менем удерживания (RT2). Полное газохроматографическое разделение на вторичной
колонке занимает всего несколько секунд, в течение которых формируется новая пор-
ция конденсированного элюата из первичной колонки. Эта новая порция вновь резко
нагревается и направляется на разделение во вторичную колонку, что замыкает цикл.
Выход вторичной колонки соединен с масс-спектрометром. Единственный анализатор,
совместимый со сверхбыстрой ГХ во вторичной колонке, — времяпролетный, поэтому
все коммерческие приборы двумерной газовой хроматографии — масс-спектрометрии
являются системами ГХ х ГХ/ВПМС. Пример двумерной хроматограммы ГХ х ГХ пред-
ставлен на рис. 6.12.
Две колонки систем ГХх ГХ/МС предназначены для наиболее полного разделения
компонентов смесей, поэтому в первичной и вторичной колонках используются совер-
шенно разные неподвижные фазы. Классическая система состоит из малоролярной или
неполярной первичной колонки, широко использумой в обычной ГХ/МС, и вторичной
колонки с полярной фазой, обеспечивающей совершенно иной, ортогональный прин-
цип разделения. Такая комбинация «неполярная/полярная» является наиболее рас-
Первичная колонка, RTj
Рис. 6.12. Схема записи хроматограммы в режиме ГХ х ГХ
6.3. Двумерная газовая хроматография — масс-<
пространенной для ортогонального разделения компонентов. В то же время опублико-
ваны варианты использования и других систем, в частности «неполярная/хиральная»
(Eljarrat Е. et al., 2008), «хиральная/полярная» (Bondajandi L.R. et al., 2005), «хираль-
ная/неполярная» (Bondajandi L. R. et al., 2005a) и другие.
Наиболее важным этапом в работе систем ГХ х ГХ/МС является разделение первич-
ного элюата на порции с последующим перенаправлением этих порций во вторичную
колонку. Данная операция осуществляется в специальном узле, называемом темпера-
турным модулятором, который является интерфейсом между обеими колонками. На се-
годняшний день существует два типа коммерчески доступных систем ГХх ГХ/ВПМС
с различными разновидностями модуляторов, работающих в режимах так называемой
четырехступенчатой или двухступенчатой модуляции. В обоих случаях при модуля-
ции используют газовые форсунки с горячим или холодным газом («горячая» и «холод-
ная» форсунки соответственно). Газ в холодной форсунке охлаждается жидким азотом.
Он необходим для эффективной криофокусировки элюата из первичной колонки. На-
гретый газ из горячей форсунки используется для испарения и перенаправления кон-
денсированной порции элюата во вторичную колонку. Четырехступенчатая модуляция
предполагает использование двух горячих и двух холодных форсунок, принцип ее рабо-
ты представлен на рис. 6.13. На этапе I обе холодные форсунки включены, обе горячие
выключены, а компоненты элюата конденсируются за счет охлаждения газом холодных
форсунок в первой зоне криофокусировки. На этапе II холодная форсунка №1 выклю-
Рис. 6.13. Схема четырехступенчатой модуляции. Римские цифры соответствуют этапам
модуляции
Глава 6. Передовые методы на основе ГХ-МС
чается и включается горячая форсунка №1, благодаря чему компоненты испаряются
из первой зоны криофокусировки и тут же конденсируются вновь во второй зоне кри-
офокусировки, поскольку холодная форсунка №2 продолжает работать. В это же вре-
мя дополнительные порции элюата могут пройти через первую зону криофокусировки
и также сконденсироваться во второй зоне (знак * на рис. 6.13). На этапе III холодная
форсунка №1 вновь включается, а горячая форсунка №1 выключается: благодаря этому
новые порции элюата, поступающие в модулятор, не могут пройти сквозь первую пару
форсунок и смешаться с порцией элюата, уже находящейся во второй зоне криофокуси-
ровки. Этот этап является ключевым для возможности анализа отделенных друг от дру-
га порций первичного элюата. Последний этап необходим для переноса первой порции
элюата для разделения во вторичную колонку. Это достигается за счет включения го-
рячей форсунки №2. После этого модулятор возвращается на этап I. Во время этапов I,
II и III второго цикла компоненты первой порции элюата должны успеть разделиться
на вторичной колонке и попасть в детектор. Это происходит в среднем за 5 секунд в режи-
ме свербыстрой ГХ/МС, а компоненты регистрируются времяпролетным анализатором.
Принцип двухступенчатой модуляции весьма схож с вышеописанным и детально
рассмотрен в работе (Ledford, 2002). В двухступенчатом варианте используется только
одна холодная и одна горячая форсунки, при этом холодная работает постоянно, а горя-
чая пульсирует каждые несколько секунд. Конец первичной колонки закручен внутри
модулятора в виде петли таким образом, что обе форсунки могут воздействовать на две
зоны первичной колонки одновременно (рис. 6.14).
При включении горячей форсунки порция элюата, сконденсированная во второй
зоне криофокусировки, поступает во вторичную колонку. В то же время сконденсиро-
ванная в первой зоне криофокусировки порция элюата продвигается по петле в сторону
второй зоны криофокусировки и конденсируется там при выключении горячей форсун-
ки. Время пульсации горячей форсунки подобрано таким образом, чтобы оно в точно-
сти равнялось времени, которое необходимо порции элюата для перемещения по задер-
живающей петле от первой до второй зоны криофокусировки.
Рис. 6.14. Двухступенчатый модулятор. Горячая форсунка перпендикулярна плоскости
рисунка
6.3. Двумерная газовая хроматография — масс-спектрометрия ГХ???ГХ/МС 191
Первичная колонка, RT]
Рис. 6.15. Двумерное и трехмерное представление ГХ х ГХ-хроматограмм
Нормальное время цикла двухступенчатой модуляции, как и в случае четырехсту-
пенчатого процесса, составляет около 5 секунд, и вторичная колонка также работает
в режиме сверхбыстрой ГХ/МС. Соответственно все ключевые рассуждения, приве-
денные нами выше касательно использования техники быстрой ГХ/МС, в полной мере
применимы и для вторичной колонки систем ГХ х ГХ/МС. Типичная вторичная колон-
ка очень короткая (<1 м), узкая (i.d. ~0,1 мм) и покрыта очень тонкой пленкой непод-
вижной фазы (~0,1 мкм). Вторичная колонка обычно помещается в отдельный термостат
с температурным режимом, настроенным таким образом, что в любой момент времени
температура внутри этого термостата была на -20—30 °C выше температуры основного.
Такой температурный режим дополнительно ускоряет разделение.
Графическое представление данных, полученных методом ГХ х ГХ/МС, схематично
представлено на рис. 6.15. Частота модуляции подбирается таким образом, чтобы за вре-
мя выхода каждого пика из первичной колонки формировалось от трех до шести порций
конденсированного элюата. Поскольку при традиционной ГХ типичная ширина пика
из первичной колонки составляет 10—30 с, частота модуляции составляет 0,1—0,5 Гц.
6.3.2. Преимущества метода ГХ х ГХ/МС
У метода ГХ х ГХ/МС имеются существенные преимущества перед традиционным ана-
лизом ГХ/МС. Во-первых, двумерная хроматография позволяет существенно лучше
разделять компоненты смеси, причем на основании не зависящих друг от друга физи-
ко-химических свойств (ортогональное разделение). Именно благодаря этому вещества,
которые элюируются одновременно с первичной колонки, зачастую могут быть разделе-
ны на вторичной колонке (рис. 6.15). Такая уникальная особенность ГХ х ГХ/МС упро-
щает деконволюцию пиков, очистку спектров и, как следствие, упрощает и повышает
надежность идентификации компонентов по их масс-спектрам при библиотечном по-
иске. Частным случаем очистки спектра является полное удаление пиков ионов непод-
вижной фазы ГХ-колонок, которые узкой полосой провляются в каждой ГХх ГХ/МС-
хроматограмме и не перекрываются с масс-спектрами аналитов (рис. 6.16).
Второй важной особенностью систем ГХх ГХ/МС является большая точность ре-
зультатов количественного анализа по сравнению с классической ГХ/МС. Такая же
особенность характерна и для одномерной быстрой ГХ/МС, при которой аналиты вы-
ходят в виде более острых и узких пиков. В двумерной хроматографии даже широкий
Глава 6. Передовые методы на основе ГХ-МС
Время удерживания на первичной колонке, с
Рис. 6.16. ГХ х ГХ/МС-анализ дизельного топлива (двумерный вид). Разделение проводи-
лось на первичной неполярной и вторичной полярной колонках
пик компонента при его элюировании с первичной колонки «разрезается» при модуля-
ции на несколько острых пиков во вторичной колонке, как показано на рис. 6.15. Сумма
интенсивностей таких «нарезанных» пиков обеспечивает значительно более высокую
точность и воспроизводимость количественного измерения компонента в сравнении
с интегрированием исходного широкого пика в одномерной ГХ/МС.
Третье важное преимущество ГХх ГХ/МС вновь связано с ортогональностью раз-
деления компонентов на двух колонках. Такой тип разделения позволяет группиро-
вать компоненты очень сложных смесей по классам, используя вид двумерной хрома-
тограммы. Отличным примером такой классификации служит рис. 6.16, на котором
прекрасно видны «кластеры» пиков, принадлежащих органическим веществам схо-
жей природы, которые элюируются в конкретных зонах хроматограммы. Например,
неполярные алканы элюируются при tR< 1 с во втором измерении, формируя кластер
2Б-хроматограммы. Низкокипящие малополярные производные нафталина форми-
руют кластер выше алканов при tR~ 1 с (вторичная колонка). Полярные дибензотио-
фены элюируются вверху 2Б-хроматограммы при /R>3 с (рис. 6.16). Такая особенность
ГХ х ГХ/МС позволяет легко различать компоненты со схожими масс-спектрами. Даже
если вещества имеют очень близкие спектры (например предельные спирты и изомер-
ные им алкены), они легко различаются по их положению на 2О-хроматограмме, по-
скольку классы предельных спиртов и алкены занимают на ней разные положения.
6.3. Двумерная газовая хроматография — масс-спектрометрия ГХ???ГХ/МС
Рис. 6.17. Двумерная хроматограмма по ПИТ легкого дизеля до (вверху) и после (внизу)
хлорирования в водном растворе гипохлорита натрия (Шайдулина, Лебедев,
2004)
Глава 6. Передовые методы на основе ГХ-МС
На практике при появлении в области элюирования, например, алкенов пика с масс-
спектром, который библиотечный поиск приписывает либо алкену, либо спирту, выбор
делают именно в пользу алкена.
Описанные преимущества ГХ х ГХ/МС позволяют проводить анализ исключитель-
но сложных смесей органических веществ, таких как нефти и нефтепродукты, смеси
экотоксикантов в объектах окружающей среды, смеси изомеров, а также веществ, при-
сутствующих в образце в следовых количествах. Рис. 6.16 и 2.17 (глава 2) демонстрируют
потенциал метода при анализе дизельного топлива и сигаретного дыма соответственно.
Нередко сама задача анализа, по сути, не требует четкой идентификации структур
всех компонентов смеси, достаточно идентифицировать изменения в структуре общих
кластеров. Такой тип анализа называют картированием, оно может с успехом приме-
няться во многих экологических задачах, а также в криминалистике.
На рис. 6.17 изображена ГХ х ГХ/МС-хроматограмма образца легкого дизеля в воде
до и после обработки гипохлоритом натрия (Шайдулина, Лебедев, 2004). Задача анализа
состояла в том, чтобы определить, какие классы углеводородов могут давать хлорпро-
изводные при обеззараживании питьевой воды хлором. Несмотря на то, что результа-
ты анализа позволили установить структуры более 2000 отдельных компонентов, ответ
на первоначальный вопрос практически очевиден при визуальном рассмотрении хро-
матограмм. Результаты понятны даже непрофессионалу, поскольку рис. 6.17 напрямую
указывает на реакционную способность различных классов углеводородов. В частно-
сти, картирование инертных алканов и нафтенов не указывает на какие-либо измене-
ния в их качественном и количественном составе при водном хлорировании. Около по-
ловины более реакционноспособных алкилбензолов превратились в соответствующие
хлорпроизводные. Наконец, наиболее активные алкилнафталины полностью исчезли,
превратившись в хлорпроизводные.
Другим изящным примером использования огромного потенциала ГХ х ГХ/МС яв-
ляется изобличение виновника нефтяного разлива (Лебедев, 2010). Например, в 2005 году
неподалеку от острова Сахалин нефтяной танкер потерпел аварию, в результате кото-
рой вытекшая нефть загрязнила несколько пляжей. Компания-судовладелец была го-
това заплатить штраф, тем не менее, согласно законодательным актам РФ, была прове-
дена химическая экспертиза. Однако результаты ГХ-анализа образцов нефти с пляжей
и с танкера оказались совершенно разными. Соответственно возник вопрос: а не были
ли загрязнены пляжи нефтью из другого источника? На рис. 6.18 представлены резуль-
таты анализа образцов нефти с пляжа и танкера методом ГХ х ГХ/МС. Видно, что хрома-
тограммы, построенные по ПИТ, выглядят совершенно по-разному для двух образцов.
Однако перестройка хроматограмм по ионам биомаркеров нефти (m/z 191 для тритерпа-
нов и m/z 217 для стеранов) не оставляет сомнений: хроматограммы одинаковы. Похожие
картинки были получены и для образцов с других пляжей. Результат анализа весьма на-
гляден и понятен любому. Причина, по которой хроматограммы, построенные по ПИТ,
выглядят по-разному, кроется в том, что образцы с пляжей были отобраны спустя 7 дней
после разлива. Процессы простого испарения и растворения некоторых компонентов,
а также фотохимической и биологической деградации нефти привели к тому, что состав
нефти сильно изменился по сравнению с изначальным. Тем не менее анализ методом
картирования с использованием масс-хроматограмм наиболее стабильных нефтяных
биомаркеров оказался весьма эффективным, а его результаты — убедительными.
6.3. Двумерная газовая хроматография — масс-спектрометрия ГХ???ГХ/МС
3D-хроматограмма, ПИТ
3D-масс-хроматограмма, m/z 217 тритерпаны
3D-масс-хроматограмма, m/z 191 тритерпаны
Рис. 6.18. ЗО-хроматограммы образцов нефти с танкера и с загрязненного пляжа по ПИТ,
масс-хроматограммы по току ионов с m/z 217 и тД 191
Глава 6. Передовые методы на основе ГХ-МС
6.4. Заключение
Новейшие разработки и усовершенствования классического ГХ/МС-анализа, описан-
ные в данной главе, с очевидностью свидетельствуют о том, что возможности метода
значительно шире и многограннее, чем предполагалось ранее. Внедрение недавно раз-
работанных технологий ультразвуковых молекулярных пучков и ГХх ГХ/МС в проце-
дуры экологического анализа, безусловно, позволит в скором будущем достигнуть впе-
чатляющих и важных результатов.
Литература
[1] . Alon Т., Amirav А. (2006). «Isotope Abundance Analysis Method and Software for Im-
proved Sample Identification with the Supersonic GC-MS». Rapid Communications in Mass
Spectrometry. 20, 17:2579—2588.
[2] . Amirav A. (1991). «Electron Impact and Surface Ionization Mass Spectrometry in Super-
sonic Molecular Beams». Organic Mass Spectrometry. 26, 1:1—17.
[3] . Amirav A., Gordin A., Poliak M., Alon T., Fialkov A. B. (2008). «Gas Chromatography
Mass Spectrometry with Supersonic Molecular Beams». Feature Article in Journal of Mass
Spectrometry. 43, 2:141 —163.
[4] . Bordajandi L.R., Korytar P., de Boer J., Gonzalez M.J. (2005). «Enantiomeric separation
of chiral polychlorinated biphenyls on beta-cyclodextrin capillary columns by means of
heart-cut multidimensional gas chromatography and comprehensive two-dimensional gas
chromatography. Application to food samples». Journal of Separation Sciences. 28, 2:163—71.
[5] . Bordajandi L. R., Ramos L., Gonzalez M.J. (2005). «Chiral comprehensive two-dimension-
al gas chromatography with electron-capture detection applied to the analysis of chiral poly-
chlorinated biphenyls in food samples». Journal of Chromatography A. 1078, (1—2): 128—35.
[6] . Dagan S., Amirav A. (1994). «Fast, High Temperature and Thermolabile GC-MS in Super-
sonic Molecular Beams». International Journal of Mass Spectrometry and Ion Processes. 133,
2:187-210.
[7] . Eljarrat E., Guerra P., Barcelo D. (2008). «Enantiomeric determination of chiral persistent
organic pollutants and their metabolites». TrAC Trends in Analytical Chemistry. 27, 847—861.
[8] . Fialkov A. B., Gordin A., Amirav A. (2003). «Extending the Range of Compounds Ame-
nable for Gas Chromatography Mass Spectrometry Analysis». Journal of Chromatography.
>4 991, 217-240.
[9] . Fialkov A. B., Steiner U., Jones L., Amirav A. (2006). «А New Type of GC-MS with Ad-
vanced Capabilities». International Journal of Mass Spectrometry. 251, 1:47—58.
[10] . Fialkov A. B., Steiner U., Lehotay S. J., Amirav A. (2007). «Sensitivity and Noise in GC-
MS: Achieving Low Limits of Detection for Difficult Analytes». International Journal of
Mass Spectrometry. 260, 1:31—48.
[11] . Fialkov A. B., Amirav A. (2008). «Hydrocarbons and Fuel Analysis with the Supersonic
GC-MS — The Novel Concept of Isomer Abundance Analysis». Journal of Chromatography.
A. 1195, 127-135.
Литература
[12] . Fialkov А. В., Amirav А. (2003). «Cluster Chemical Ionization for Improved Confi-
dence Level in Sample Identification». Rapid Communications in Mass Spectrometry. 17,
12:1326-1338.
[13] . Gonnord M.F., Guiochon G., Onuska F. I. (1983). «Narrow bore open tubular columns
for improvement of gas chromatographic analysis time». Analytical Chemistry. 55, 13:
2115-2120.
[14] . Gordin A., Amirav A., Fialkov A. B. (2008). «Classical Electron Ionization Mass Spectra
with GC-MS with Supersonic Molecular Beams». Rapid Communications in Mass Spec-
trometry. 22, 17:2660—2666.
[15] . Hail M. E., and Yost R. A. (1989). «Theoretical and Practical Aspects of Short Open Tubular
Columns at Subambient Pressures in Gas Chromatography/Mass Spectrometry». Analyti-
cal chemistry. 61, 21: 2402—2410.
[16] . Kochman M., Gordin A., Goldshlag P., Lehotay S. J., Amirav A. (2002). «Fast, High Sen-
sitivity, Multi-Pesticide Analysis of Complex Mixtures with the Supersonic GC-MS». Jour-
nal of Chromatography. A. 974, 185—212.
[17] . Лебедев А.Т. (2010). «Масс-спектрометрия в экотоксикологических исследовани-
ях». Токсикологический Вестник. 4:2—12.
[18] . Ledford Е. В. Jr. Method and apparatus for measuring velocity of chromatographic pulse.
PCT Patent Application No. PCT/US02/08488.
[19] . Mastovska K., Lehotay S.J., and Hajslova J. (2001). «Optimization and evaluation of low-
pressure gas chromatography—mass spectrometry for the fast analysis of multiple pesticide
residues in a food commodity». Journal of Chromatography. A 926, 291—295.
[20] . Mastovska K., Lehotay S.J. (2003). «Practical approaches to fast gas chromatography—
mass spectrometry». Journal of Chromatography. A1000, 153—180.
[21] . Poliak M., Kochman M., Amirav A. (2008). «Pulsed Flow Modulation Comprehensive
Two Dimensional Gas Chromatography». Journal of Chromatography. A. 1186, 189—195.
[22] . Poliak M., Fialkov A. B., Amirav A. (2008). «Pulsed Flow Modulation Two-Dimensional
Comprehensive Gas Chromatography Tandem Mass Spectrometry with Supersonic Mo-
lecular Beams». Journal of Chromatography. A1210, 108—114.
[23] . Poliak M., Gordin A., Amirav A. (2010). «Open Probe — A Novel Method and Device
for Ultra Fast Electron Ionization Mass Spectrometry Analysis». Analytical Chemistry. 82,
13:5777-5782.
[24] . Hajslova J., and Cajka T. (2008). «Gas chromatography in food analysis». In: Otle§ S. (ed.),
Handbook of Food Analysis Instruments. ISBN-13: 9781420045666, CRC Press, Taylor &
Francis Group, Boca Raton, FL, USA
[25] . Rubiolo P., Liberto E., Sgorbini B., Russo R., Veuthey J.-L., and Bicchi C. (2008). «Fast-
GC—conventional quadrupole mass spectrometry in essential oil analysis». Journal of Sep-
aration Science. 31, 1074—1084.
[26] . Шайдулина Г. M., Лебедев А.Т. (2004). «ГХ-ГХ/МС-изучения продуктов транс-
формации нефтяных углеводородов в условиях водного хлорирования». Масс-
спектрометрия, 1, 1:67—76.
ГЛАВА 7
МАСС-СПЕКТРОМЕТРИЯ
В НОРМАЛЬНЫХ
УСЛОВИЯХ (AMBIENT
MASS SPECTROMETRY) —
АНАЛИЗ ОБЪЕКТОВ
ОКРУЖАЮЩЕЙ СРЕДЫ
БЕЗ ПРОБОПОДГОТОВКИ
Розана М. Алберичи, Розинайде С. Симас и Маркос Н. Эберлин
7.1. Введение
Масс-спектрометрия (МС) стала мощным методом исследований, широко используе-
мым в любой отрасли химического анализа. Главной причиной такого ошеломляюще-
го успеха является уникальная возможность детектировать, измерять и характеризо-
вать атомы и молекулы различных типов, составов и размеров методом МС. Сочетание
высокой чувствительности, селективности и скорости анализа (sensitivity, selectivity,
speed — «3 S trademark of MS») — знак отличия масс-спектрометрии и общая характе-
ристика ее главных достоинств. Вместе с тем основными недостатками МС являются
зависимость регистрируемых спектров от состава матрицы образца и проявления эф-
фектов подавления ионов. Для того чтобы в области МС-ионизации и последующего
масс-спектрометрического анализа поступали аналиты в чистом виде, часто требуются
экстракция, дериватизация и хроматографическое разделение, а также другие способы
очистки анализируемой пробы. Для этих длительных манипуляций необходимо обе-
спечение доступа к разным аналитическим приборам и наличие опытного персонала.
Тем не менее, в зависимости от сформулированной цели анализа, во многих случаях
можно значительно минимизировать или даже полностью исключить предварительную
пробоподготовку и разделение образцов перед масс-спектрометрическим анализом.
Недавнее внедрение различных методов десорбции и ионизации, реализуемых в боль-
шинстве случаев в нормальных условиях путем прямой регистрации сигналов аналитов
в их исходных матрицах или на подложках, привело к существенному сокращению доли
пробоподготовки и предварительного разделения образцов в масс-спектрометрическом
анализе. Теперь эти методы стали частью области масс-спектрометрии, известной под
собирательным термином «масс-спектрометрия в нормальных условиях» (ambient mass
7.1. Введение
spectrometry; Alberici et al., 2010a). Развитие этой области началось с изобретения двух ме-
тодов — десорбционной электрораспылительной ионизации (ДЭРИ; desorption electro-
spray ionization, DESI; Takats etal., 2004a) и прямого анализа в режиме реального времени
(ПАРВ, direct analysis in real time, DART; Cody et al., 2005). После первых публикаций
о ДЭРИ и ПАРВ были изобретены десятки других методов ионизации в нормальных
условиях. Стали доступными коммерческие версии соответствующих ионных источ-
ников. Методы МС в нормальных условиях использовали для широкого спектра задач,
что привело клавинообразному росту числа публикаций об их эффективности в разных
областях (см. недавние обзоры: Cooks et al., 2006; Takats et al., 2005a; Venter et al., 2008; Van
Berkel et al., 2008; Harris et al., 2008; Manincke et al., 2008; Chen et al., 2009a; Ifa et al., 2009;
Weston, 2010; Ifa et al., 2010; Chen et al., 2010).
Главной привлекательной чертой масс-спектрометрии в нормальных условиях ста-
ло беспрецедентное упрощение и ускорение масс-спектрометрического анализа, вы-
полняемого in situ, в режиме реального времени, on line, с высокой производительностью
и малым потреблением образца и при отсутствующей или сведенной к минимуму про-
боподготовке. Соответственно, методы масс-спектрометрии в нормальных условиях
особенно привлекательны для анализа объектов окружающей среды и контроля без-
Рис. 7.1. Анализ объектов окружающей среды методами масс-спектрометрии в нормаль-
ных условиях
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
опасности в общественных местах. На рис. 7.1 проиллюстрированы общие возможности
использования МС в нормальных условиях как потенциально универсального метода
для решения различных задач анализа объектов окружающей среды. В данной главе об-
суждаются фундаментальные принципы, режимы работы, необходимое оборудование
и основные области применения типичных методов ионизации в нормальных условиях,
применяемых в анализе объектов окружающей среды.
7.2. Наиболее часто используемые методы
масс-спектрометрии в нормальных условиях
7.2.1. Десорбционная электрораспылительная ионизация
(ДЭРИ, DESI) и ионизация спреем с бумаги (paper spray
ionization, PSI)
Десорбционная электрораспылительная ионизация впервые описана в 2004 Куксом
и соавторами {Takats et al., 2004а). Ионизация ДЭРИ отличается от ионизации электро-
распылением {Takats et al., 2004b)', ее широко применяют, а ее дальнейшие возможности
исследуют, анализируя многие типы образцов и смесей (см. главу 8 с подробным опи-
санием ДЭРИ). Чаще всего молекулы аналитов ионизируют методом ДЭРИ напрямую
из их естественных матриц или со вспомогательных подложек — таких, как стекло,
бумага, металл, пластик и пластины для тонкослойной хроматографии (ТСХ) на ос-
нове силикагеля (рис. 7.1я) {Venter et al., 2008). Для анализа с использованием иониза-
ции ДЭРИ наиболее важными оптимизируемыми параметрами являются напряжение
на капилляре, состав растворителя и его pH, угол направления спрея, а также расстоя-
ния от капилляра до точки попадания спрея на поверхность образца и до входного от-
верстия в масс-спектрометр {Takats et al., 2005а; Costa and Cooks, 2008; Volnyet al., 2008).
В ДЭРИ ионы аналитов образуются в результате бомбардировки анализируемой
поверхности потоком положительно или отрицательно заряженных капель электро-
спрея с высоким зарядом, которые образуются из кислого или щелочного раствора.
В большинстве случаев в качестве растворителя используют смесь 1:1 метанол:вода или
чистый метанол. Очень тонкий слой растворителя распыляется по всей поверхности
образца. Окончательный процесс десорбции в ДЭРИ включает «отскакивание» обра-
зующихся микрокапель от поверхности. Образование ионов в ДЭРИ (после десорбции)
схоже с обычной ионизацией электрораспылением, то есть происходит за счет переноса
протона или катиона от таких катионов растворителя (S), как [S + Н]+ или [S + Na]+, или
путем отрыва протона такими анионами, присутствующими в растворе, как ОН- или
ОАс- (рис. 7.1л), или, например, при образовании анионных комплексов типа [М + С1]-.
Этот механизм десорбции/ионизации в ДЭРИ {Costa and Cooks, 2008; Harris et al., 2008)
обычно интерпретируют как процесс «сбора капель» (на поверхности мишени — прим,
переводчика) (drop pickup) или «сплэшинга» (splashing) (разбрызгивания капель при их
отскакивании от поверхности — прим, переводчика). Эффективность десорбции аналита
с поверхности и его перехода в слой растворителя и ионизации на основе электрора-
спыления являются главными параметрами, определяющими общую эффективность
ионизации ДЭРИ {Venteretal., 2008; Chipuk and Brodbelt, 2008).
Можно предположить, что ДЭРИ является одним из наиболее универсальных ме-
тодов десорбции/ионизации в нормальных условиях, с помощью которого можно ана-
лизировать широкий спектр соединений — от малых молекул до больших биомолекул
(Harris et al., 2008). При осуществлении дериватизации in-situ методом ДЭРИ можно ио-
низировать даже неполярные углеводороды (Wu et al., 2010). Как и для многих других
методов ионизации в нормальных условиях, ионные источники ДЭРИ адаптированы
для сочетания с большинством масс-спектрометров с ионизацией при атмосферном
давлении (ИАД) (Venter et al., 2008). В частности, известны работы о сочетании ДЭРИ
с линейными тройными квадруполями (Shin et al., 2007), ионными ловушками (Myung
et al., 2006), орбитальными ловушками (Ни et al., 2006), квадрупольно-времяпролетны-
ми (QTOF) масс-анализаторами (Weston etal., 2005), тандемными анализаторами ионной
мобильности в сочетании с времяпролетной или квадрупольно-времяпролетной ста-
дией (ion mobility-TOF, ion mobility-QTOF) (Kauppila et al., 2006) и масс-анализаторами
ионного циклотронного резонанса с преобразованием Фурье (Fourier transform ion
cyclotron resonance; FT-ICR) (Bereman et al., 2006). Возможность осуществления диссо-
циации, индуцированной столкновениями (collision-induced dissociation, CID), за счет
тандемной МС/МС-стадии, весьма привлекательна и важна для анализа методом масс-
спектрометрии ДЭРИ (Williams and Scrivens, 2005). При анализе сложных смесей для
ДЭРИ также важны высокое разрешение и высокая точность измерения масс (Ifa et al.,
2008). Пределы детектирования обычно составляют порядка 1—10 фмоль для малых мо-
лекул, тогда как относительная погрешность воспроизводимости количественного ана-
лиза составляет около 5—10% (Takats et al., 2005b). Уже сложившимися областями при-
менения ДЭРИ являются проведение анализов для судебно-медицинской экспертизы
(Ifa etal., 2009), имиджинг (глава 20, получение изображений молекулярных распределе-
ний на поверхностях — прим, переводчика) (Ifa etal., 2007; Eberlin etal., 2010a; Eberlin etal.,
2010b), метаболомика (Takats et al., 2005a), анализ лекарств (Williams and Scrivens, 2005),
анализ белков (Pasilis et al., 2008), исследование редокс-трансформаций (Benassi et al.,
2009), липидомика (Manincke et al., 2008) и анализ углеводородов (Wu et al., 2010). Реак-
ционная ДЭРИ с дериватизацией in-situ — это перспективный подход к анализу мало-
полярных или неполярных молекул, которые плохо ионизируются с помощью обычной
ДЭРИ (WuetaL, 2009).
Кукс и его сотрудники сообщили о варианте ЭРИ, называемом ионизирующим
спреем с бумаги (paperspray ionization, PSI) (Wangetal., 2010; Liu etal., 2010). В PSI пере-
нос аналитов осуществляется за счет напуска раствора в пористый материал бумаги,
вырезанной в форме макроскопического острия, и приложения поля высокого напря-
жения к этой бумаге для осуществления ионизации. Для переноса аналитов не требу-
ется пневматического усилия (рис. 7.20. При перемещении аналитов внутри бумаги
может происходить их химическое разделение, поэтому PSI включает сразу три разных
аналитических процесса: нанесение образца, его разделение и ионизацию. Благодаря
этим особенностям, PSI удобно использовать для клинических исследований, включая
неонатальный скрининг, терапевтический мониторинг лекарств и, возможно, в буду-
щем — персонализированную медицину. Анализ важных объектов окружающей сре-
ды на месте (например анализ воды) также представляется привлекательной областью
применения PSI.
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
Рис. 7.2. Схемы некоторых способов ионизации в нормальных условиях: (а) ДЭРИ,
(б) спрей с бумаги, (в) ПАРВ, (г) ASAP
7.2.2. Прямой анализ в режиме реального времени (ПАРВ,
DART)
3-5 kV
Коди и Ларамее (2005) изобрели новый метод масс-спектрометрии в нормальных усло-
виях — прямой анализ в режиме реального времени (ПАРВ, direct analysis in real time,
DART). На рис. 7.2в показана общая схема источника ПАРВ. Метод ПАРВ приобрел
большую популярность за счет возможности анализа газов, жидкостей и твердых образ-
цов и применимости к широкому кругу молекул различной полярности, находящихся
на различных поверхностях, например в бетоне, асфальте, человеческой коже, визитных
карточках, а также кожуре фруктов и овощей. Коммерчески доступные источники ПАРВ
были протестированы для решения многих задач, например в судебно-медицинской экс-
пертизе (Laramee et al., 2007; Jones et al., 2006), фармацевтике (Fernandez et al., 2006), пище-
вой химии (Haefligerand Jeckelman, 2007; Vailetal., 2007), анализе биологических образцов
(Pierce et al., 2007', Yew et al., 2008; Yu et al., 2009) и химическом анализе (Morlock and Ueda,
2007; Cody, 2009; Song et al., 2009; Wells et al., 2008). Описаны также автоматизированные
источники ПАРВ, позволяющие проводить количественный анализ (Yu etal., 2009).
При ионизации ПАРВ в режиме регистрации положительных ионов для более по-
лярных аналитов преимущественно образуются протонированные молекулы [М + Н]+,
но благодаря уникальному механизму ионизации, начальной стадией которого является
ионизация Пеннинга (см. ниже), за счет образования молекулярных ионов (М+’) можно
также ионизировать менее полярные или даже неполярные молекулы. Источник ПАРВ
7.2. Наиболее часто используемые методы масс-спектрометрии 203
в нормальных условиях
(рис. 7.2<?) состоит из нескольких отделений, через которые пропускают газ (Не или
N2), обычно со скоростью 1 л мин-1 (в DART SVP, последней модели источника ПАРВ,
скорость потока гелия составляет около 3 л мин-1 — прим, переводчика). Прилагаемый
потенциал в нескольких киловольт (1—5 кВ) обеспечивает тлеющий разряд, благодаря
которому образуются ионы, электроны и нейтральные частицы в возбужденном состо-
янии. Таким образом, плазма ПАРВ образуется преимущественно путем ионизации
в тлеющем разряде при атмосферном давлении (atmospheric pressure GD ionization) (Cody,
2009). После тлеющего разряда газ проходит через перфорированный промежуточный
электрод (рис. 7.2в (а); в последней версии источника DART этот электрод удален —
прим, переводчика), опциональный газонагреватель и решетчатый электрод (рис. 7.2в (Ь))
с изолирующим колпачком (рис. 7.2в (с)). Перфорированный промежуточный электрод
удаляет ионы из газового потока, а газонагреватель изменяет температуру газа (для тер-
модесорбции аналитов) от комнатной температуры до 250°C (Cody et al., 2005) или даже
до 500 °C (Cody, 2009). Решетчатый электрод (рис. 7.2в(Ь)), выступая в качестве отражате-
ля ионов, служит для удаления ионов противоположной полярности и предотвращения
снижения сигнала за счет ион-ионных рекомбинаций. На выходе из ионного источника
используется изолирующий колпачок, защищающий образец и оператора от соприкос-
новения с решетчатым электродом. Ионизация происходит, когда газ для ПАРВ контак-
тирует с образцом под углом 0° или отражается от поверхности образца под углом около
45° (Cody etal., 2005).
Первичным механизмом ПАРВ является ионизация Пеннинга (Cody et al., 2005;
Cody, 2009). Исходный поток газа подвергается электрическому разряду, образующийся
и затем нагретый поток газа содержит нейтральные атомы или молекулы газа (N). За счет
ионизации в тлеющем разряде электронное состояние частиц N изменяется с образова-
нием «метастабильных» частиц (N*), которые передают избыточную энергию молеку-
лам аналитов (М), обладающим меньшей энергией ионизации (ЭИ), генерируя, таким
образом, ион-радикалы М+’ и электроны е_ (уравнение 7.1).
N* + M^M+‘ +N +е- (7.1)
Гелий обладает долгоживущим 235-состоянием с внутренней энергией 19,8 эВ, пре-
вышающей энергии ионизации обычных атмосферных газов, поэтому частицы He*(23S)
могут эффективно ионизировать молекулы воды в окружающей атмосфере (уравне-
ние 7.2). Ионизированные молекулы, в свою очередь, могут реагировать с нейтральны-
ми молекулами воды (с типичным каскадом реакций, подобным химической иониза-
ции) с образованием протонированных кластеров воды [(H2O)z/ +Н]+. Эти кластерные
ионы переносят протон к молекулам М более полярных аналитов (уравнение 7.3).
He(23S) + лН2О -> [(Н2О)л_, + Н]+ + ОН" + He(lS') (7.2)
[(Н2О)„ + Н]+ + М [М + Н]+ + лН2О (7.3)
7.2.3. Атмосферный зонд для анализа твердых образцов (ASAP)
Атмосферный зонд для анализа твердых образцов (atmospheric solids analysis probe, ASAP;
McEwen et al., 2005; McEwen and Gutteridge, 2007) — это еще один метод ионизации для
масс-спектрометрии в нормальных условиях. Он создан путем модификации ионного
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
источника химической ионизации при атмосферном давлении (ХИАД, глава 3) таким
образом, чтобы подачу твердых или вязких жидких образцов можно было осуществлять
напрямую в горячий газ и поток капель ХИАД. В качестве зонда ASAP использовали
боросиликатный капилляр для измерения температур плавления веществ. Таким об-
разом, при ионизации ASAP зонд ХИАД функционирует в обычном режиме с распы-
лением растворителя либо может использоваться в качестве источника горячего потока
азота (обычно при температурах до 400—500°С), чтобы десорбировать аналиты с твердо-
го зонда с последующей ионизацией коронным разрядом (6 кВ). Следовательно, ASAP
может быть легко установлен на большинство коммерческих источников ионизации
при атмосферном давлении, а механизм ионизации ASAP является механизмом ХИАД,
который подходит наилучшим образом для не слишком больших молекул (с молекуляр-
ной массой <1000 Да) и аналитов средней полярности (McEwen et al., 2005). На рис. 7.2г
изображена основная схема источника ASAP с молекулами аналитов (М), вводимых
в область спрея ХИАД с помощью твердого зонда.
7.2.4. Экстракционная электрораспылительная ионизация
(ЭЭРИ, EESI)
Грэм Кукс и его коллеги представили метод ионизации в атмосферных условиях, назван-
ный экстракционной электрораспылительной ионизацией (ЭЭРИ, extractive electrospray
ionization, EESI). Этим методом можно ионизировать преимущественно более летучие
молекулы (Chen et al., 2006). Как предполагалось, ионизация ЭЭРИ решала проблему
ограничений ионизации ДЭРИ по отношению к анализу летучих аналитов (рис. 7.3л).
ЭЭРИ — это неинвазивный способ ионизации, основанный на ЭРИ, при реализации
которого пары, содержащие молекулы аналитов из раствора, распыляются в пото-
ке заряженных капель, образуемых ЭРИ. Молекулы аналитов попадают в капли ЭРИ
и ионизируются по механизму, подобному ЭРИ. Преимуществом ЭЭРИ, по сравнению
с исходным методом ЭРИ, является дизайн. Образцы вводят через отдельный канал, ко-
торый заземлен и расположен ортогонально по отношению к высоковольтному каналу
ЭРИ (Chen et al., 2006). Для менее полярных аналитов можно использовать ионизацию,
стимулируемую катионами Ag+ (Chen et al., 2007a). Шиа и др. (Changet al., 2002’, Fan et al.,
2005; Pan et al., 2007) ввели схожий метод, называемый ЭРИ со смешанными каплями
(fused droplet ESI, FD/ESI). Этот простой метод использовали для прямого получения
высококачественных масс-спектров биомолекул (таких, как пептиды и белки), раство-
ренных в воде. Метод ЭЭРИ использовали для анализа различных образцов, включая
определение среднелетучих компонентов таких сложных образцов, как молоко (Chen
et al., 2006), выдыхаемый воздух (Chen et al., 2007a), ароматы фруктов (Chen et al., 2007b),
взрывчатые вещества (Chen et al., 2009b), диэтилфталат в парфюмерных изделиях (Chingi
et al., 2009), диэтиленгликоль в зубной пасте (Ding et al., 2009) и натуральное оливковое
масло (Law et al., 2009; Law et al., 2010).
Механизм микроввода (Luque de Castro and Priego-Capote, 2007) может быть полезен
для анализа жидкостей с высокой вязкостью или сложных жидких смесей. При реа-
лизации такого способа ввода пробы образуется аэрозоль, состоящий из микрокапель
жидкости или раствора сложного состава, которые транспортируются в источник ЭРИ
в потоке воздуха или азота. Этот вариант ЭЭРИ (рис. 7.3л) предложен Зеноби с коллега-
ми (Chen et al., 2007) и называется экстракционной электрораспылительной ионизаци-
7.2. Наиболее часто используемые методы масс-спектрометрии
в нормальных условиях
2-5 кВ
2-5 кГц
Рис. 7.3. Схемы некоторых способов ионизации в нормальных условиях: а — ЭЭРИ, б —
НТП, в — EASI и г — VL-EASI. См. также рис. 7.2
ей с нейтральной десорбцией (ЭЭРИ/НД, neutral desorption EESI, ND/EESI). ЭЭРИ/НД
осуществляется за счет изначальной десорбции молекул образца в нейтральный поток
газа, а не в заряженный поток растворителя, как в ЭЭРИ, который смешивается со спре-
ем ЭРИ, что приводит к образованию масс-спектров образцов, похожих на спектры ЭРИ
{Weston, 2010). Главным достоинством метода является возможность анализа образцов
в очень сложных матрицах благодаря разделению процессов пробоотбора и ионизации
в пространстве и во времени. ЭЭРИ/НД использовали, например, для быстрого анализа
тканей живых организмов {Chen et al., 2007с), быстрого скрининга компонентов лекар-
ственных форм {Williams and Scrivens, 2008), определения аналитов в различных типах
образцов {Chen et al., 2007с), а также для детектирования следовых количеств нелетучих
взрывчатых веществ (таких, как тринитротолуол), которые легко накапливаются на по-
верхности кожи. Их пробоотбор осуществляли с помощью новейшего метода нейтраль-
ной десорбции при содействии газа (air-tight ND enclosure) для быстрого анализа мето-
дом тандемной масс-спектрометрии с ионизацией ЭЭРИ {Chen etal., 2009b). Ионизацию
ЭЭРИ/НД также использовали для определения взрывчатых веществ, десорбируемых
с поверхности кожи {Gu etal., 2010).
7.2.5. Низкотемпературная плазма (LTP)
Оуянг и сотрудники {Harper et al., 2008) впервые сообщили о методе ионизации в ат-
мосферных условиях, осуществляемой с использованием низкотемпературной плазмы
(НТП, low temperature plasma, LTP). На рис. 7.36 показана схема источника ионизации
НТП. Стеклянная трубка служит в качестве диэлектрического барьера. Внутри этой
трубки находится медная обмотка, служащая в качестве внешнего электрода, на кото-
рый подается переменный ток с высоким напряжением в диапазоне 3—5 кВ, с частотой
206 Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
осцилляции 2—5 кГц. Внутри стеклянной трубки коаксиально расположен заземлен-
ный внутренний электрод. Генерация плазмы внутри трубки осуществляется с помо-
щью газового разряда со скоростью потока газа, обычно, 0,4 л/мин. В качестве газа для
плазменного разряда использовали Не, Ar, N2 или даже воздух. Зонд НТП электрически
изолирован, что исключает возможность поражения оператора электрическим шоком.
Максимальная температура плазмы НТП не превышает 30°С, поэтому не вызывает тер-
мического повреждения большинства поверхностей или человеческой кожи. Благодаря
этому метод НТП/МС привлекателен для решения задач, требующих использования
низких температур для десорбции, например в точках контроля безопасности аэропор-
тов или для мобильных масс-спектрометров.
При ионизации НТП обычно образуются положительные ионы [М + Н]+ и/или М+‘,
или отрицательной ионы М-" и/или [М —Н]_, в зависимости от типа молекул. Метод
НТП/МС использовали для анализа взрывчатых веществ {Harper et al., 2008), пестици-
дов {Wiley et al., 2010), лекарств, наркотиков {Jackson etal., 2010) и даже вязких жидкостей,
например оливкового масла {Garcia-Reyes et al., 2009).
7.2.6. Ионизация акустическим распылением в нормальных
условиях — easy ambient sonic-spray ionization (EASI)
и ионизация акустическим распылением в нормальных
условиях с использованием эффекта Вентури (Venturi
easy ambient sonic-spray ionization, V-EASI)
В 1994 г. Хирабайяши и коллеги {1994; 1995; 1996) изобрели ионизацию акустическим
распылением (sonic spray ionization, SSI). Уникальность SSI состояла в новой концепции
ионизации для масс-спектрометрии: для образования ионов не требовалось напряже-
ний, излучения или нагрева. Заряженные капли образовывались просто путем распы-
ления подкисленного метанольного раствора аналита со скоростью звука. Заряд (как
отрицательный, так и положительный) этих очень мелких капель с ограниченной емко-
стью заряда образовывался за счет статистически несбалансированного распределения
катионов и анионов. Для десольватации капель не требовалось нагревание, поскольку
газообразные ионы аналитов [М + Н]+ и [М-Н]- образовывались из заряженных ка-
пель (в зависимости от природы аналита М) при комнатной температуре. Таким обра-
зом, ионы в SSI формировались только с помощью сжатого азота (или даже воздуха).
Эберлин с коллегами {Haddadetal., 2006; Haddadetal., 2008a) позже показали, что поток
очень маленьких биполярно (±) заряженных капель, образующийся за счет акустиче-
ского распыления метанольных растворов, также может эффективно десорбцировать
и ионизировать аналиты с поверхностей в нормальных условиях. Тем самым они ввели
в практику упрощенную ионизацию акустическим распылением в нормальных услови-
ях (easy ambient sonic-spray ionization, EASI, рис. 7.3e).
Таким образом, главными достоинствами метода EASI являются:
1) его простота благодаря тому, что для реализации метода нужен только сжатый
азот (или даже атмосферный воздух);
2) его способность к одновременному формированию и отрицательно, и поло-
жительно заряженных капель, благодаря которой не требуется переключать вы-
сокое напряжения от EASI(+) к EASI(-) или наоборот;
3) низкая концентрация заряда на поверхности капель позволяет снизить вклад
шума от растворителя (Haddad et al., 2006), тем самым обеспечивая высокие от-
ношения сигнал—шум для ионов аналитов (Van Berkeletal., 2008);
4) очень мягкая ионизация (Takats et al., 2003);
5) отсутствие термической деградации;
6) отсутствие вклада в сигнал электрических шумов и влияния процессов окисле-
ния, типичных для ЭРИ (Pasilis et al., 2008; Benassi et al., 2009) и других способов
ионизации, основанных на ЭРИ (Chen and Cooks, 2007; Boyset al., 2009).
Капли высоколетучего акустического спрея EASI глубоко проникают в матрицу
твердых образцов, тем самым обеспечивая высокую гомогенность пробоотбора и дли-
тельность регистрации устойчивых сигналов (Haddad et al., 2006). Таким образом, EASI
представляется идеальной для мобильных масс-спектрометров и в таком сочетании мо-
жет найти применение для решения большого числа экологических задач. Ионизацию
EASI, в основу которой легли технологии самых мягких способов ионизации (softest ion-
ization techniques, SSI), отличает также возможность детектирования нефрагментирован-
ных ионов аналитов. Такая исключительная мягкость процесса ионизации способствует
анализу лабильных молекул и сложных смесей по принципу «один компонент для коли-
чественного анализа — режим детектирования одного иона». Ионизация EASI включает
процесс растворения, и при регистрации положительных ионов EASI(+) обычно обра-
зуются ионы [М + Н]+ и/или [М + Na(K)]+, а в режиме регистрации отрицательных ионов
(-) — главным образом образуются ионы [М — Н]_. Однако, в отличие от ЭРИ, при EASI
анализе ионы М+* или М-’ никогда не наблюдали. EASI сочетали с масс-спектрометрией
с мембранным вводом (membrane introduction mass spectrometry; EASI/MIMS) (Haddad
et al., 2008a), тонкослойной хроматографией (thin-layer chromatography, EASI/TLC/MS)
(Haddadet al., 2008b), а также высокоэффективной тонкослойной хроматографией (high-
performance TLC, HPTLC) (Eberlin etal., 2009). Для наиболее селективного анализа мето-
дом EASI/МС в качестве селективных поверхностей использовали полимеры с молеку-
лярными отпечатками (molecularly imprinted polymers, MIP) (Figueiredo etal., 2010).
Ограничением EASI является сверхвысокая скорость потока спрея, из-за которой
может происходить сдувание образцов. Для образцов, закристаллизованных на поверх-
ностях, компактных твердых проб, а также вязких масел такое сдувание не является про-
блемой. Для летучих видов топлива, например, эту проблему можно решить путем уда-
ления растворителя в результате естественного испарения и сбора данных для полярных
маркеров в сухом остатке (Alberici et al., 2010b). Метод EASI/MC успешно применяли для
исследований различныханалитов и матриц, таких кактаблетированные лекарственные
препараты (Haddad et al., 2006), парфюмерные изделия (Haddad et al., 2008c), поверхност-
но-активные вещества (Saraiva et al., 2009), растительные масла (Simas et al., 2010), био-
дизели (Abdelnuretal., 2008; Eberlin etal., 2009, Albericietal., 2010c), прополис (Sawaya etal.,
2010), чернила (Lalli et al., 2010), сырая нефть (Corilo et al., 2010), а также поддельные де-
нежные купюры (Eberlin et al., 2010c). Недавно вышел обзор по применению этого метода
для анализа видов топлива (Alberici et al., 2010b).
В последнее время были описаны простейшие, в том числе и с точки зрения их ин-
струментального исполнения, методы ионизации в нормальных условиях, в основу ко-
торых легла ионизация EASI (Santos et al., 2010). Так, метод, включающий эффект Вен-
тури (Venturi self-pumping), получил название Venturi easy ambient sonic-spray ionization
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовка
Рис. 7.4. Ионизация V-EAS1 для анализа in situ органических экотоксикантов природных
вод
(V-EASI). Такая ионизация, как показано, может происходить в двойном режиме — как
для жидких (VL), так и для твердых (Vs) образцов (рис. 7.3г). В V-EASI используются
только комбинированные и превосходно сочетающиеся эффекты акустического по-
тока азота (или атмосферного воздуха), влияющие на всасывание растворов аналитов
и их ионизацию посредством SSI. Подобно EASI, V-EASI также является очень мяг-
кой ионизацией в нормальных условиях, при которой образуются сравнительно чи-
стые спектры, в которых доминируют сигналы одиночных молекул различных жидких
и твердых образцов. Как и в случае EASI, достоинством V-EASI является отсутствие на-
пряжений, нагревания и излучения, функционирование при комнатной температуре
при содействии только сжатого азота (или воздуха), не вызывающего возникновения
термических, электрических или разрядных шумов. Еще большая простота ионизации
V-EASI (по сравнению с EASI), для которой отсутствуют требования к электрической
мощности, обусловливает высокую применимость V-EASI в миниатюризованных масс-
спектрометрах. Устройство источника ионизации V-EASI представляется также иде-
альным для экологического анализа — анализа воды или, например, сухой или влажной
почвы, а также для продолжительного on-line экологического мониторинга сточных вод
в режиме реального времени. На рис. 7.4 проиллюстрировано использование V-EASI для
анализа in situ органических ксенобиотиков в природных водах.
7.3. Применения в анализе объектов окружающей
среды
Экологический анализ и меры по обеспечению безопасности обычно необходимо осу-
ществлять с высокой повторяемостью, чувствительностью, точностью и иногда непосред-
ственно на месте нахождения проб. Поэтому методы масс-спектрометрии с ионизацией
в атмосферных условиях могут существенно облегчить решение задач в этих областях хи-
мического анализа за счет их высокой простоты, воспроизводимости и производительно-
7.3. Применения в анализе объектов окружающей среды
сти. Несмотря на то, что общая методология МС в нормальных условиях только зарожда-
ется, ее методы уже высоко оценены как весьма перспективные для решения для решения
важных задач экологического анализа. Главной исходной целью являлся скрининг взрыв-
чатых веществ или вредных химикатов в воздухе, воде и на поверхностях {Mulligan et al.,
2006). Портативные масс-спектрометры уже являются предметом пристального изучения
для целей мониторинга проб воздуха, твердых и конденсированных сред в экологическом
анализе, и поэтому сочетание таких приборов с методами ионизации в нормальных усло-
виях является очень привлекательным для экологического анализа (глава 9).
В идеале анализ воздуха необходимо осуществлять путем анализа in situ, особенно
для определения высокотоксичных соединений. Показано, что с помощью ДЭРИ мож-
но детектировать пары веществ в воздухе. Так, образцы атмосферного воздуха, загряз-
ненного диметилметилфосфонатом (DMMP, dimethyl methylphosphonate), пропускали
через сорбирующий материал в целях предварительного концентрирования аналитов
для последующей десорбции и ионизации методом масс-спектрометрии ДЭРИ {Mulligan
etal., 2006). Возможность определения таких токсичных газов, как фосген, этиленоксид,
диоксид серы, акрилонитрил, синильная кислота, хлорциан, акролеин, формальдегид
и этилпаратион была продемонстрирована при использовании ДЭРИ и миниатюризо-
ванного масс-спектрометра. Полученные пределы детектирования различных анали-
тов составляли от 800 ppt до 3 ppm {Ouyang et al., 2009). ДЭРИ применяли для анали-
за полициклических ароматических углеводородов (ПАУ) в атмосферных аэрозолях.
Пределы детектирования ПАУ составляли около 10 пг при длительности пробоотбора
5 с. Селективное и быстрое определение ПАУ в атмосферных или горючих аэрозолях,
полученных, полученных из синтезированной в лаборатории биомассы, осуществляли
без проведения экстракции или предварительного концентрирования {Chen etal., 2008).
Детектирование следовых количеств неорганических веществ, особенно с полу-
чением информации об элементах, также является важной задачей экологических ис-
следований. Метод ЭЭРИ использовали для быстрого детектирования радиоактивных
неорганических веществ в образцах природных вод без какой-либо пробоподготовки
{Luo et al., 2010). Пределы детектирования соединений урана составляли около 1 пг/л.
При использовании ЭЭРИ можно было быстро определять изотопные отношения в со-
единениях урана, тем самым обеспечивая альтернативный способ скрининга образцов,
используемых для разделения и обогащения изотопов урана.
Использование гербицидов и пестицидов в сельском хозяйстве может приводить
к серьезному загрязнению грунтовых вод. При анализе смеси пестицидов, содержащей
10 нг А,А-диэтил-л/-толуамида, атразина и алахлора, нанесенных напрямую на поверх-
ность бумаги площадью 1 см2, соответствующие ионы аналитов были зарегистрированы
с m/z 192, 216 и 270 (рис. 7.5а) {Mulligan et al., 2006). Использование ДЭРИ в сочетании
с портативным масс-спектрометром также позволило проводить in situ детектирование
с различных поверхностей химических веществ, используемых в сельском хозяйстве.
Также можно детектировать следовые количества пестицидов in situ при использовании
ЭЭРИ {Lietal., 2009). В масс-спектре воды с территории сельскохозяйственных угодий,
в которую были добавлены паракват (200 нг/мл) и Р-циперметрин (45 нг/мл), зареги-
стрировали ионы с m/z 186 и 416, соответствующие ион-радикалу параквата и протони-
рованному Р-циперметрину (рис. 7.50. Предел детектирования составил 10 нг/мл для
параквата и 6 нг/мл для Р-циперметрина.
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
Анализ взрывчатых веществ in situ имеет особое значение для общественной без-
опасности и экологического контроля. Детектирование взрывчатых веществ на различ-
ных появерхностя проводили различными методами масс-спектрометрии в атмосфер-
ных условиях, включая ДЭРИ (Wells et al., 2008; Cotte-Rodmgues et al., 2005; Justes et al.,
2007), НТП (Harper etal., 2008), ПАРВ (Cody etal., 2005) и ЭЭРИ (Chen etal., 2007b). ДЭРИ
использовали для детектирования (Mulligan etal., 2006) гексогена (тринитрогексагидро-
1,3,5-триазин), адсорбированного на различных поверхностях, с превосходной чув-
ствительностью и селективностью. На рис. 7.6а показано МС-детектирование иона
с m/z 223, соответствующего протонированной молекуле [М + Н]+ гексогена. Показана
возможность детектирования гексогена (10 нг, нанесенных на бумажную поверхность
площадью 1 см2) при использовании портативного масс-спектрометра при длительно-
N, N - д и эт и л - м - то л у а м и д
Рис. 7.5. (а) Масс-спектр ДЭРИ(+) смеси пестицидов (диэтилтолуамид, атразин и ала-
хлор); (б) масс-спектр ЭЭРИ(+) водного параквата и Р-циперметрина из образца
воды с территории фермы. Адаптировано из литературных источников (Mulligan
etal., 2006) и (Lietal., 2009)
7.3. Применения в анализе объектов окружающей среды
RDX-DES1
Рис. 7.6. (а) Масс-спектр ДЭРИ(+) тринитрогексагидро-1,3,5-триазина (RDX); (б) масс-
спектр ЭЭРИ(+) тринитрогексагидро-1,3,5-триазина (RDX); (в) масс-спектр
ЭЭРИ(—) тринитротолуола (TNT). Адаптировано из литературных источников
(Mulligan et al., 2006) и (Liet al., 2009)
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовка
Рис. 7.7. Использование масс-спектрометрии в нормальных условиях для прямого анали-
за разливов нефти в окружающей среде
сти одного анализа 5—10 с. Возможность анализа взрывчатых веществ методом масс-
спектрометрии ЭЭРИ была также показана для образцов грунтовой воды, в которые
вводили следовые количества взрывчатых веществ на уровне 1 ppt. В масс-спектрах
ЭЭРИ(+) присутствуют ионы с m/z 223 гексогена, что согласуется с данными ДЭРИ
(рис. 7.60. Ионы с m/z 227, регистрируемые методом ЭЭРИ(—) (рис. 7.6в), соответству-
ют анион-радикалам 2,4,6-тринитротолуола {Li et al., 2009). ЭЭРИ также использовали
для детектирования следовых количеств тринитротолуола, гексогена, нитроглицерина
и прочих взрывчатых веществ с поверхности кожи человека без какой-либо пробопод-
готовки. Пределы детектирования при анализе взрывчатых веществ на коже методом
масс-спектрометрии ЭЭРИ составляли от 0,5 до 10 пг {Chen et al., 2009b). Ионизацию
ДЭРИ в сочетании с портативным масс-спектрометром использовали для анализа
200 250 300 350 400 450 500 550 600 650 700 750 800
m/z
m/z
Рис. 7.8. Данные EASI FT MS-анализа бразильского образца сырой нефти в: (а) режиме
регистрации положительных ионов; (б режиме регистрации отрицательных ио-
нов. Средняя разрешающая способность варьировалась в диапазоне 300000 <
/и/Л/и50% < 450000 для каждого масс-спектра. Адаптировано из литературных ис-
точников (Albericietal., 2010b) и (Corilo etal., 2010)
взрывчатых веществ (тринитротолуола и октогена) на большой поверхности (75 см2)
за относительно короткое время (22 с). Взрывчатые вещества присутствовали в следовых
количествах на поверхности (от 500 пг/см2 до 1 мкг/см2) как индивидуально, так и в ка-
честве компонентов смесей (Sanders et al., 2010).
Нефть, вне всякого сомнения, является самой сложной природной смесью химиче-
ских веществ из известных на сегодняшний день и поэтому самым сложным объектом
для любого анализа. Места разливов сырой нефти также являются важным объектом
экологического контроля. На рис. 7.7 проиллюстрировано использование распылитель-
ных методов ионизации в нормальных условиях для прямого анализа разливов нефти
в окружающей среде. Недавно сообщалось об использовании EASI/МС для анали-
за образца нефти на поверхности со сверхвысоким разрешением (400000 для m/z 400)
и точностью (около 500 ppb) при использовании гибридного спектрометра, состоящего
из линейной квадрупольной ионной ловушки в тандеме с масс-анализатором с преоб-
разованием Фурье (Corilo etal., 2010). На рис. 7.8 приведены масс-спектры EASI(+)FT/MS
и EASI(—)FT/MS образца сырой нефти. Используя данные обоих способов ионизации,
можно было идентифицировать тысячи компонентов. Кроме того, химические профи-
ли EASI FT/MS также хорошо согласовывались с профилями ЭРИ. Метод V-EASI (Santos
et al., 2010) также хорошо себя зарекомендовал для эффективной ионизации множества
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
m/z
Рис. 7.9. (а) Масс-спектр VL-EASI (+) метанольного раствора бензина, (б) керосина и сме-
сей бензина/керосина в пропорциях: (в) 50/50% (об.) и (г) 90/10% (об.). Адаптиро-
вано из литературного источника (Romao et al., 2011)
7.4. Заключение
полярных компонентов сырой нефти из раствора толуол:метанол, при этом регистриру-
емые данные были сходны с данными методов EASI и ЭРИ (см. также главу 18 для при-
меров из области петролеомики).
Метод Vl-EASI(+)/MC также использовали для определения полярных компонен-
тов образцов бензина и керосина (Wanderson et al., 2011). Ионизация образца бензи-
на происходила с образованием серии характеристичных ионов с m/z 94, 108, 122, 136,
150 и 164 (рис. 7.9л). Эти ионы соответствуют гомологической серии азотсодержащих
соединений (класс N), протонированных гомологов (от С6 до С(1) алкилпиридинов
([C5H5N(CH2),CH3+H]+). В спектрах VL-EASI(+)/MS керосина (рис. 7.90 детектировали
две гомологичные серии, относящиеся к соединениям азота. Первая серия была иден-
тична сигналам, наблюдаемым в бензине (ионы с m/z 122, 136 и 150), тогда как вторая
серия ионов (более интенсивных) соответствовала ионам алкилпиридинов С12—С17 с на-
фтеновым кольцом, присоединенным к пиридиновому циклу (протонированные моле-
кулы em/z 176, 190, 204, 218, 232 и 246). На рис. 7.9<? и 7.9г показаны VL-EASI масс-спектры
смесей бензина/керосина в пропорции 90/10 % (об.) и 50/50 % (об.), соответственно. Ана-
лиз этих спектров для типичных смесей бензин/керосин указывает на применимость
Vl-EASI для детектирования разбавления бензина керосином вплоть до содержания
10% об.
7.4. Заключение
За десятилетия развития классических методов ионизации в масс-спектрометрии на-
коплен большой объем знаний, который способствовал бурному возникновению мето-
дов масс-спектрометрии с ионизацией в нормальных условиях и их быстрому развитию.
Сейчас существует современный набор методов, основанных на различных механизмах
десорбции и ионизации. Этими методами можно работатьс различными типами матриц
и молекул, обладающих широким диапазоном масс и полярностей. Поэтому примене-
ние масс-спектрометрии в нормальных условиях с ее беспрецедентной скоростью, се-
лективностью, чувствительностью, а также простотой представляется перспективным
для анализа разнообразных образцов окружающей среды. Методы масс-спектрометрии
в нормальных условиях, позволяющие работать с жидкими образцами, кажутся особо
привлекательными для анализа вод, тогда как анализ сухой почвы, очевидно, наиболее
выигрышно проводить методами прямого анализа твердых образцов. В частности, пары
и более летучие компоненты жидких или твердых образцов удобнее всего анализиро-
вать при использовании ионизации ЭЭРИ.
Окончательная цель масс-спектрометрии — обеспечить анализ в нормальных ус-
ловиях «реального мира», причем таким образом, чтобы, с одной стороны, проводить
простой (без предварительной пробоподготовки или обработки образцов), быстрый,
селективный и высокочувствительный качественный и количественный химический
анализ, а с другой — анализировать объекты в их естественной среде и исходном ме-
стоположении — в тех случаях, когда использование масс-спектрометрии необходимо.
Эта цель стала теперь достижима благодаря методам ионизации в нормальных услови-
ях. Она привлекает многих исследователей перспективой появления новых областей
применения масс-спектрометрии в аналитической химии окружающей среды.
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
Литература
[1] . Abdelnur P.V., Eberlin L.S., de SaG.F., Souza V., and Eberlin M.N. (2008). «Single-shot
biodiesel analysis: nearly instantaneous typification and quality control solely by ambient
mass spectrometry». Analytical Chemistry. 80, 20: 7882—7886.
[2] . Alberici R. M., Simas R.C., Sanvido G.B., Romro W., Lalli P. M., Benassi M., Cun-
ha I.B.S., and Eberlin M.N. (2010a). «Ambient mass spectrometry: bringing MS into the
real world». Analytical and Bionalytical Chemistry. 398, 1: 265—294.
[3] . Alberici R. M., Simas R.C., Souza V., de Sa G. F., Daroda R.J., and Eberlin M.N. (2010b).
«Analysis of fuels via easy ambient sonic-spray ionization mass spectrometry». Analytica
Chimica Acta. 659, 1—2: 15—22.
[4] . Alberici R. M., Simas R.C., Abdelnur P.V., Eberlin M.N., Souza V., de Sa G.F., Daro-
da R.J. (2010c). «А highly effective antioxidant and artificial marker for biodiesel». Energy
and Fuels. 24, 12: 6522-6526.
[5] . Benassi M., Wu C., Nefliu M., Ifa D. R., Volny M., and Cooks R. G. (2009). «Redox trans-
formations in desorption electrospray ionization». International Journal of Mass Spectrom-
etry. 280, 1-3: 235-240.
[6] . Bereman M.S., Nyadong L., Fernandez F. M., and Muddiman D.C. (2006). «Direct high-
resolution peptide and protein analysis by desorption electrospray ionization Fourier trans-
form ion cyclotron resonance mass spectrometry». Rapid Communications in Mass Spectrom-
etry. 20, 22: 3409-3411.
[7] . Boys B.L., Kuprowski M.C., Noel J.J., and Konermann L. (2009). «Protein oxidative
modifications during electrospray ionization: solution phase electrochemistry or corona
discharge-induced radical attack?» Analytical Chemistry. 81, 10: 4027—4034.
[8] . Chang, D-Y., Lee, C-С., and Shiea J. (2002). «Detecting large biomolecules from high-salt
solutions by fused-droplet electrospray ionization mass spectrometry». Analytical Chemistry.
74, 11: 2465-2469.
[9] . Chen H., Venter A., and Cooks R. G. (2006). «Extractive electrospray ionization for direct
analysis of undiluted urine, milk and other complex mixtures without sample preparation».
Chemical Communications. 19: 2042—2044.
[10] . Chen H., Wortmann A., Zhang W., and Zenobi R. (2007a). «Rapid in vivo fingerprinting of
nonvolatile compounds in breath by extractive electrospray ionization quadrupole time-of-
flight mass spectrometry». Angewandte Chemie International Edition. 46, 4: 580—583.
[11] . Chen H., Sun Y, Wortmann A., Gu H., and Zenobi R. (2007b). «Differentiation of ma-
turity and quality of fruit using non invasive extractive electrospray ionization quadrupole
time-of-flight mass spectrometry». Analytical Chemistry. 79, 4: 1447—1455.
[12] . Chen H., Wortmann A., and Zenobi R. (2007c). «Neutral desorption sampling coupled to
extractive electrospray ionization mass spectrometry for rapid differentiation of biosamples
by metabolomic fingerprinting». Journal of Mass Spectrometry. 42, 9: 1123—1135.
[13] . Chen M., and Cook K. D. (2007). «Oxidation artifacts in the electrospray mass spectrom-
etry of a P peptide». Analytical Chemistry. 79, 5: 2031—2036.
[14] . Chen H., Li M., Zhang, Y-Р., Yang X., Lian, J-J., and Chen, J-M. (2008). «Rapid analysis
of SVOC in aerosols by desorption electrospray ionization mass spectrometry». Journal of
the American Society for Mass Spectrometry. 19, 3: 450—454.
[15] . Chen H., Gamez G., and Zenobi R. (2009a). «What can we learn from ambient ionization
techniques?» Journal of the American Society for Mass Spectrometry. 20, 11: 1947—1963.
[16] . Chen H., Hu B., Hu Y., Huan Y., Zhou Z., and Qiao X. (2009b). «Neutral desorption using
a sealed enclosure to sample explosives on human skin for rapid detection by EESI-MS».
Journal of the American Society for Mass Spectrometry. 20, 4: 719—722.
[17] . Chen, H-W., Hu B., and Zhang X. (2010). «Principles and application of ambient mass
spectrometry for direct analysis of complex samples». Chinese Journal of Analytical Chemis-
try. 38,8: 1069-1088.
[18] . Chingin K., Chen H., Gamez G., Liang Z., and Zenobi R. (2009). «Detection of diethyl
phthalate in perfumes by extractive electrospray ionization mass spectrometry». Analytical
Chemistry. 81, 1: 123—129.
[19] . Chipuk J. E., and BrodbeltJ.S. (2008). «Transmission mode desorption electrospray ioniza-
tion». Journal of the American Society for Mass Spectrometry. 19, 11: 1612—1620.
[20] . Cody R. B., Laramee J. A., and Durst H. D. (2005). «Versatile new ion source for the analysis
of materials in open air under ambient conditions». Analytical Chemistry. 77, 8: 2297—2302.
[21] . Cody R. B. (2009). «Observation of molecular ions and analysis of nonpolar compounds
with the direct analysis in real time ion source». Analytical Chemistry 81, 3: 1101 — 1107.
[22] . Cooks R. G., Ouyang Z., Takats Z., and Wiseman J. M. (2006). «Ambient mass spectrom-
etry». Science. 311, 5767: 1566—1570.
[23] . Corilo Y. E., Vaz B.G., Simas R.C., Lopes Nascimento H.D. L., Klitzke C.F., Perei-
ra R.C.L., Bastos W. L., Rodgers R. P., and Eberlin M.N. (2010). «Petroleomics by
EASI(+/—) FT-ICR MS». Analytical Chemistry. 82, 10: 3990—3996.
[24] . Costa A. B., and Cooks R.G. (2008). «Simulated splashes: Elucidating the mechanism of
desorption electrospray ionization mass spectrometry». Chemical Physics Letters. 464, 1—3:
1-8.
[25] . Cotte-Rodngues L, Takats Z., Talaty N., Chen H., and Cooks R.G. (2005). «Desorption
electrospray ionization of explosives on surfaces: sensitivity and selectivity enhancement by
reactive desorption electrospray ionization». Analytical Chemistry. 77, 21: 6755—6764.
[26] . Ding J., Haiwei G., Yang S., Li M., Li J., and Chen H. (2009). «Selective detection of dieth-
ylene glycol in toothpaste products using neutral desorption reactive extractive electrospray
ionization tandem mass spectrometry». Analytical Chemistry. 81, 20: 8632—8638.
[27] . Eberlin L.S., Abdelnur P.V., Passero A., de Sa G.F., Daroda R.J., Souza V, and Eber-
lin M.N. (2009). «Analysis of biodiesel and biodiesel-petrodiesel blends by high perfor-
mance thin layer chromatography combined with easy ambient sonic-spray ionization mass
spectrometry». Analyst. 134, 8: 1652—1657.
[28] . Eberlin L. S., Ifa D. R., Wu C., and Cooks R. G. (2010a). «Three-dimensional visualization
of mouse brain by lipid analysis using ambient ionization mass spectrometry». Angewandte
Chemie International Edition. 49, 34:873—876.
[29] . Eberlin L.S., Dill A. L., Costa A. B., Ifa D. R., Cheng L., Masterson T., Koch M.,
Ratliff T. L., and Cooks R. G. (2010b). «Cholesterol sulfate imaging in human prostate can-
cer tissue by desorption electrospray ionization mass spectrometry». Analytical Chemistry.
82,9:3430-3434.
[30] . Eberlin L.S., Haddad R., Sarabia Neto R.C., Cosso R.G., Maia D.R.J., Maldaner A.O.,
Zacca J. J., Sanvido G.B., Romro W., Vaz B.G., Ifa D. R., Manicke N.E., Dill A.,
218 Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
Cooks R. G., and Eberlin М. N. (2010с). «Instantaneous chemical profiles of banknotes by
ambient mass spectrometry». Analyst. 135, 10: 2533—2539.
[31] . Fernandez F. M., Cody R. B., Green M.D., Hampton C.Y., McGready R., Sengaloun-
deth S., White N.J., and Newton P. N. (2006). «Characterization of solid counterfeit drug
samples by desorption electrospray ionization and direct-analysis-in-real-time coupled to
time-of-flight mass spectrometry». ChemMedChem 1, 7: 702—705.
[32] . Figueiredo E.C., Sanvido G.B., Zezzi M.A., and Eberlin M.N. (2010). «Molecularly im-
printed polymers as analyte sequesters and selective surfaces for easy ambient sonic-spray
ionization». Analyst. 135, 4: 726—730.
[33] . Garcia-Reyes J. F., Mazzotti F., Harper J. D., Charipar N. A., Oradu S., Ouyang Z., Sindo-
na G., and Cooks R. G. (2009). «Direct olive oil analysis by low-temperature plasma (LTP)
ambient ionization mass spectrometry». Rapid Communications in Mass Spectrometry. 23,
21:3057-3062.
[34] . Gu H., Yang S., Li J., Hu B., Chen H., Zhang L., and Fei Q. (2010). «Geometry-independ-
ent neutral desorption device for the sensitive EESI-MS detection of explosives on various
surfaces». Analyst. 135, 4: 779—788.
[35] . Haddad R., Sparrapan R., and Eberlin M.N. (2006). «Desorption sonic spray ionization for
(high) voltage-free ambient mass spectrometry». Rapid Communications in Mass Spectrom-
etry. 20, 19: 2901-2905.
[36] . Haddad R., Sparrapan R., Kotiaho T., and Eberlin M.N. (2008a). «Easy ambient sonic-
spray ionization-membrane interface mass spectrometry for direct analysis of solution con-
stituents. Analytical Chemistry. 80, 3: 898—903.
[37] . Haddad R., Milagres H.M.S., Catharino R. R., and Eberlin M.N. (2008b). «Easy ambi-
ent sonic-spray ionization mass spectrometry combined with thin-layer chromatography».
Analytical Chemistry. 80, 8: 2744—2750.
[38] . Haddad R., Catharino R. R., Marques L.A., and Eberlin M.N. (2008c). «Perfume finger-
printing by easy ambient sonic-spray ionization mass spectrometry: nearly instantaneous
typification and counterfeit detection». Rapid Communications in Mass Spectrometry. 22, 22:
3662-3666.
[39] . Haefliger О. P., and Jeckelmann N. (2007). «Direct mass spectrometric analysis of flavors
and fragrances in real applications using DART». Rapid Communications in Mass Spectrom-
etry. 21,8: 1361 — 1366.
[40] . Harper J. D., Charipar N.A., Mulligan C.C., Zhang X., Cooks R.G., and Ouyang Z.
(2008). «Low-temperature plasma probe for ambient desorption ionization». Analytical
Chemistry. 80, 23: 9097—9104.
[41] . Harris G. A., Nyadong L., and Fernandez F. M. (2008). «Recent developments in ambient
ionization techniques for analytical mass spectrometry». Analyst. 133, 10: 1297—1301.
[42] . Hirabayashi A., Sakairi M., and Koizumi H. (1994). «Sonic spray ionization method for at-
mospheric pressure ionization mass spectrometry». Analytical Chemistry. 66,24:4557—4559.
[43] . Hirabayashi A., Sakairi M., and Koizumi H. (1995). «Sonic spray mass spectrometry».
Analytical Chemistry. 67,17: 2878—2882.
[44] . Hirabayashi A., Hirabayashi Y, Sakairi M., and Koizumi H. (1996). «Multiply-charged ion
formation by sonic spray». Rapid Communications in Mass Spectrometry. 10, 13:1703—1705.
[45] . Hu Q., Talaty N., Noll R.J., and Cooks R.G. (2006). «Desorption electrospray ionization
using an Orbitrap mass spectrometer: exact mass measurements on drugs and peptides».
Rapid Communications in Mass Spectrometry. 20, 22: 3403—3408.
[46] . Ifa D. R., Wiseman J. M., Song Q., and Cooks R. G. (2007). «Development of capabilities
for imaging mass spectrometry under ambient conditions with desorption electrospray ioni-
zation (DESI)». International Journal of Mass Spectrometry. 259, 1—3: 8—15.
[47] . Ifa D. R., Manicke N. E., Rusine A. L., and Cooks R. G. (2008). «Quantitative analysis of
small molecules by desorption electrospray ionization mass spectrometry from polytetra-
fluoroethylene surfaces». Rapid Communications in Mass Spectrometry. 22, 4: 503—510.
[48] . Ifa D. R., Jackson A. U., Paglia G., and Cooks R.G. (2009). «Forensic applications of
ambient ionization mass spectrometry». Analytical and Bioanalytical Chemistry. 394, 8:
1995-2008.
[49] . Ifa D.R., Wu C., Ouyang Z., and Cooks R.G. (2010). «Desorption electrospray ioniza-
tion and other ambient ionization methods: current progress and preview». Analyst. 135, 4:
669-681.
[50] . Jackson A. U., Garcia-Reyes J. F., Harper J. D., Wiley J. S., Molina-Dnaz A., Ouyang Z.,
and Cooks R. G. (2010). «Analysis of drugs of abuse in biofluids by low temperature plasma
(LTP) ionization mass spectrometry». Analyst. 135, 5: 927—933.
[51] . Jones R. W., Cody R. B., and McClelland J. F. (2006). «Differentiating writing inks using
direct analysis in real time mass spectrometry». Journal of Forensic Sciences. 51,4: 915—918.
[52] . Justes D. R., Talaty N., Cotte-Rodriguez I., and Cooks R. G. (2007). «Detection of explo-
sives on skin using ambient ionization mass spectrometry». Chemical Communications., 21:
2142-2144.
[53] . Kauppila T.J., Talaty N., Salo P.K., Kotiaho T., Kostiainen R., and Cooks R.G. (2006).
«New surfaces for desorption electrospray ionization mass spectrometry: porous silicon and
ultra-thin layer chromatography plates». Rapid Communications in Mass Spectrometry. 20,
14:2143-2150.
[54] . Lalli P. M., Sanvido G. B., Garcia J. S., Haddad R., Cosso R.G., Maia D. R. J., Zacca J. J.,
Maldaner A. O., and Eberlin M. N. (2010). «Fingerprinting and aging of ink by easy ambient
sonic-spray ionization mass spectrometry». Analyst. 135, 4: 745—750.
[55] . Laramee J. A., Cody R. B., Nilles J. M., and Durst H. D. (2007). «Forensic Analysis on the
Cutting Edge: new methods fortrace evidence analysis». In Blackleage R. D. (Ed.), Forensic
Applications of DART (Direct Analysis in Real Time) Mass Spectrometry. John Wiley & Sons,
NJ.
[56] . Law W. S., Chen H., Ding J., Yang S., Zhu L., Gomez G., Chingin K., Ren Y., and Ze-
nobi R. (2009). «Rapid characterization of complex viscous liquids at the molecular level».
Angewandte Chemie International Edition. 48, 44: 8277—8280.
[57] . Law W. S., Chen H.W., Balabin R., Berchtold C., Meier L., and Zenobi R. (2010). «Rapid
fingerprinting and classification of extra virgin olive oil by microjet sampling and extractive
electrospray ionization mass spectrometry». Analyst. 135, 4: 773—778.
[58] . Li M., Hu B., Li J., Chen R., Zhang X., and Chen H. (2009). «Extractive electrospray ion-
ization mass spectrometry toward in situ analysis without sample pretreatment». Analytical
Chemistry. 81, 18: 7724—7731.
220 Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
[59] . Liu J., Wang Н., Manicke N. Е., Lin J. M., Cooks R. G., and Ouyang Z. (2010). «Develop-
ment, characterization, and application of paper spray ionization». Analytical Chemistry.
82,6:2463-2471.
[60] . Luo M., Hu B., Zhang X., Peng D., Chen H., Zhang L., and Huan Y. (2010). «Extractive
electrospray ionization mass spectrometry for sensivite detection of uranyl species in natu-
ral water samples». Analytical Chemistry. 82, 1: 282—289.
[61] . Luque de Castro M.D., and Priego-Capote F. (2007). «Lesser known ultrasound-assist-
ed heterogeneous sample-preparation procedures». Trends in Analytical Chemistry. 26, 2:
154-162.
[62] . Manincke N. E., Wiseman J. M., Ifa D. R., and Cooks R. G. (2008). «Desorption electro-
spray ionization (DESI) mass spectrometry and tandem mass spectrometry (MS/MS) of
phospholipids and sphingolipids: ionization, adduct formation, and fragmentation». Jour-
nal of the American Society for Mass Spectrometry. 19, 4: 531—543.
[63] . McEwen C.N., McKay R.G., and Larsen B.S. (2005). «Analysis of solids, liquids, and
biological tissues using solids probe introduction at atmospheric pressure on commercial
LC/MS instruments». Analytical Chemistry. 77, 23: 7826—7831.
[64] . McEwen C., and Gutteridge S. (2007). «Analysis of the inhibition of the ergosterol pathway
in fungi using the atmospheric solids analysis probe (ASAP) method». Journal of the Ameri-
can Society for Mass Spectrometry. 18,7: 1274—1278.
[65] . Morlock G., and Ueda Y. (2007). «New coupling of planar chromatography with direct
analysis in real time mass spectrometry». Journal of Chromatography A 1143, 1—2: 243—251.
[66] . Mulligan C.C., Talaty N., and Cooks R.G. (2006). «Desorption electrospray ionization
with a portable mass spectrometer: in situ analysis of ambient surfaces». Chemical Com-
munications. 16: 1709—1711.
[67] . Myung S., Wiseman J.M., Valentine S.J., Takats Z., Cooks R.G., and Clemmer D. E.
(2006). «Coupling desorption electrospray ionization with ion mobility/mass spectrometry
for analysis of protein structure: Evidence for desorption of folded and denatured States».
The Journal of Physical Chemistry B. 110, 10: 5045—5051.
[68] . Ouyang Z., Noll R.J., and Cooks R.G. (2009). «Handheld miniature ion trap mass spec-
trometers». Analytical Chemistry. 81, 7: 2421—2425.
[69] . Pan С. T., Shiea J., and Shen S. C. (2007). «Fabrication of an integrated piezo-electric mi-
cro-nebulizer for biochemical sample analysis». Journal of Micromechanics and microengi-
neering. 17, 3: 659—669.
[70] . Pasilis S. P., Kertesz V, and Van Berkel G. J. (2008). «Unexpected analyte oxidation dur-
ing desorption electrospray ionization-mass spectrometry». Analytical Chemistry. 80, 4:
1208-1214.
[71] . Pierce C.Y, Barr J.R., Cody R. B., Massung R. F., Woolfitt A. R., Moura H., Thomp-
son H.A., and Fernandez F. M. (2007). «Ambient generation of fatty acid methyl ester
ions from bacterial whole cells by direct analysis in real time (DART) mass spectrometry».
Chemical Communications., 8: 807—809.
[72] . Romro W., Regiani T., Klitzke C. F., Vaz B.G., Sanvido G.B., Haddad R., Corilo Y. E.,
Augusti D. V., Pasa V. M. D., Pereira R.C.C., Castro E. V. R., Augusti R., and Eberlin M.N.
(Undated). Venturi easy ambient sonic-spray ionization mass spectrometry (VEASI—MS)
for crude oil destillates: fast gasoline, kerosene and diesel fingerprinting via polar natural
markers». Submitted for publication.
Литература
[73] . Sanders N. L., Kothari S., Huang G., Salazar G., and Cooks R.G. (2010). «Detection of
explosives as negative ions directly from surfaces using a miniature mass spectrometry».
Analytical Chemistry. 82, 12: 5313—5316.
[74] . Santos V.G., Regiani T., Dias F. F.G., Romro W., Klitzke C.F., Coelho E, and Eber-
lin M.N. (2010). «Development and applications of venturi easy ambient sonic-spray ioni-
zation (V-EASI) for easier than ever ambient mass spectrometry». Analytical Chemistry.
In press.
[75] . Saraiva S.A., Abdelnur P. V., Catharino R. R., NunesG., and Eberlin M.N. (2009). «Fabric
softeners: nearly instantaneous characterization and quality control of cationic surfactants
by easy ambient sonic-spray ionization mass spectrometry». Rapid Communications in Mass
Spectrometry. 23, 3: 357—362.
[76] . Sawaya A.C.H.F., Abdelnur P.V., Eberlin M.N., Kumazawa S., Ahn M.R., Bang K.S.,
Nagaraja N., Bankova V. S., and Afrouzan H. (2010). «Fingerprinting of propolis by easy
ambient sonic-spray ionization mass spectrometry». Taianta. 81, 1—2: 100—108.
[77] . Simas R.C., Catharino R. R., Cunha I.B.S., Cabral E.C., Barrera-Arellano D., Eber-
lin M.N., and Alberici R. M. (2010). «Instantaneous characterization of vegetable oils via
TAG and FFA profiles by easy ambient sonic-spray ionization mass spectrometry». Analyst.
135,4: 738-744.
[78] . Shin, Y-S., Drolet B., Mayer R., Dolence K., and Basile F. (2007). «Desorption electro-
spray ionization-mass spectrometry of proteins. Analytical Chemistry. 79, 9: 3514—3518.
[79] . Shieh, I-F., Lee, C-Y, L., and Shiea J. (2005). «Eliminating the interferences from TRIS
buffer and SDS in protein analysis by fused-droplet electrospray ionization mass spectrom-
etry». Journal of Proteome Research. 4, 2\ 606—612.
[80] . Song L., Dykstra A. B., Yao H., and Bartmess J. E. (2009). «Ionization mechanism of nega-
tive ion-direct analysis in real time: A comparative study with negative ion-atmospheric
pressure photoionization». Journal of the American Society for Mass Spectrometry. 20, 1:
42-50.
[81] . Takats Z., Nanita S. C., Cooks R. G., Schlosser G., Vekey K. (2003). «Amino acid clusters
formed by sonic spray ionization». Analytical Chemistry. 75, 6: 1514—1523.
[82] . Takats Z., Wiseman J.M., Gologan B., and Cooks R.G. (2004a). «Mass spectrometry
sampling under ambient conditions with desorption electrospray ionization». Science. 306,
5695:471-473.
[83] . Takats Z., Wiseman J. M., Gologan B., and Cooks R. G. (2004b). «Electrosonic spray ioni-
zation. A gentle technique for generating folded proteins and protein complexes in the gas
phase and for studying ion-molecule reactions at atmospheric pressure». Analytical Chem-
istry. 16, 14: 4050-4058.
[84] . Takats Z., Wiseman J. M., and Cooks R.G. (2005a). «Ambient mass spectrometry using
desorption electrospray ionization (DESI): instrumentation, mechanismsand applications
in forensics, chemistry, and biology». Journal of Mass Spectrometry. 40, 10: 1261 — 1275.
[85] . Takats Z., Cotte-Rodriguez I., Talaty N., Chen H., and Cooks R. G. (2005b). «Direct trace
level detection of explosives on ambient surfaces by desorption electrospray ionization mass
spectrometry». Chemical Communications. 15: 1950—1952.
[86] . Vail T., Jones P. R., and Sparkman O. D. (2007). «Rapid and unambiguous identification
of melamine in contaminated pet food based on mass spectrometry with four degrees of
confirmation». Journal of Analytical Toxicology. 31, 6: 304—312.
Глава 7. Масс-спектрометрия в нормальных условиях (Ambient Mass Spectrometry) —
анализ объектов окружающей среды без пробоподготовки
[87] . Van Berkel G. J., Pasilis S. P., and Ovchinnikova O. (2008). «Established and emerging at-
mospheric pressure surface sampling/ionization techniques for mass spectrometry». Jour-
nal of Mass Spectrometry. 43, 9: 1161 — 1180.
[88] . Venter A., Neflieu M., and Cooks R.G. (2008). «Ambient desorption mass spectrometry».
Trends in Analytical Chemistry. 27, 4: 284—290.
[89] . Volny M., Venter A., Smith S.A., Pazzi M., and Cooks R.G. (2008). «Surface effects and
electrochemical cell capacitance in desorption electrospray ionization». Analyst. 133, 4:
525-531.
[90] . Wang H., Liu J., Cooks R.G., and Ouyang Z. (2010). «Paper spray for direct analysis of
complex mixtures using mass spectrometry». Angewandte Chemie International Edition. 122,
5: 889-892.
[91] . Wells J. M., Roth M.J., Keil A. D., Grossenbacher J. W., Justes D. R., Patterson G. E., and
Barket J. Jr. (2008). «Implementation of DART and DESI ionization on a fieldable mass
spectrometer». Journal ofthe American Society for Mass Spectrometry. 19, 19: 1419—1424.
[92] . Weston D. J., Bateman R., Wilson I. D., Wood T. R., and Creaser C. (2005). «Direct analysis
of pharmaceutical drug formulations using ion mobility spectrometry/quadrupole-time-
of-flight mass spectrometry combined with desorption electrospray ionization». Analytical
Chemistry. 77, 23: 7572—7580.
[93] . Weston D.J. (2010). «Ambient ionization mass spectrometry: current understanding
of mechanistic theory; analytical performance and applications areas». Analyst. 135, 4:
661-668.
[94] . Wiley J.S., Garcia-Reyes J. F., Harper J. D., Charipar N.A., Ouyang Z., and Cooks R.G.
(2010). «Screening of agrochemicals in foodstuffs using low-temperature plasma (LTP) am-
bient ionization mass spectrometry». Analyst. 135, 5: 971—979.
[95] . Williams J. P., and Scrivens J.H. (2005). «Rapid accurate mass desorption electrospray
ionisation tandem mass spectrometry of pharmaceutical samples». Rapid Communications
in Mass Spectrometry. 19, 24: 3643—3650.
[96] . Williams J. P., and Scrivens J. H. (2008). «Coupling desorption electrospray ionisation and
neutral desorption/extractive electrospray ionisation with a travelling-wave based ion mo-
bility mass spectrometer for the analysis of drugs». Rapid Communications in Mass Spectrom-
etry.22,2: 187-196.
[97] . Wu C., Ifa D. R., Manicke N. E., and Cooks R. G. (2009). «Rapid, direct analysis of choles-
terol by charge labeling in reactive desorption electrospray ionization». Analytical Chemis-
try. 18: 7618-7624.
[98] . Wu C., Qian K., Nefliu M., and Cooks R. G. (2010). «Ambient analysis of saturated hydro-
carbons using discharge-induced oxidation in desorption electrospray ionization. Journal of
the American Society for Mass Spectrometry. 21, 2: 261—267.
[99] . Yew J.Y., Cody R. B., and Kravitz E. A. (2008). «Cuticular hydrocarbon analysis of an awake
behaving fly using direct analysis in real-time time-of-flight mass spectrometry. Proceed-
ings of the National Academy of Science of the United States of America. 105, 20: 7135—7140.
[100] . Yu S., Crawford E., Tice J., Musselman B., and Wu J.T. (2009). «Bioanalysis without sam-
ple cleanup or chromatography: the evaluation and initial Implementation of direct analy-
sis in real time ionization mass spectrometry for the quantification of drugs in biological
matrixes. Analytical Chemistry. 81, 1: 193—202.
ГЛАВА 8
МАСС-СПЕКТРОМЕТРИЯ
ДЕСОРБЦИОННОЙ
ЭЛЕКТРОРАСПЫЛ ИТЕЛЬНОЙ
ИОНИЗАЦИИ
Золтан Такач
8.1. Введение
Масс-спектрометрия (МС) традиционно является одним из фундаментально важных
методов химии окружающей среды благодаря высокой чувствительности, селективно-
сти и сравнительно простой обработке масс-спектрометрических данных. Метод по-
стоянно развивается, отвечая новым требованиям. Для анализа нелетучих и лабильных
веществ используются технологии распылительной ионизации и жидкостной хрома-
тографии в комбинации с тандемной масс-спектрометрией (ЖХ/МС/МС). Главными
недостатками МС уже в течение нескольких десятилетий остаются высокая стоимость
анализа, обусловленная чрезвычайно высокой стоимостью приборов, и сложность ана-
литических процедур подготовки образцов. В настоящее время требования к анализу
окружающей среды включают в себя не только расширение номенклатуры определяе-
мых веществ, но также существенное увеличение числа проб (по месту и времени от-
бора), причем в случае загрязнения суммарное число потенциально анализируемых со-
единений возрастает на несколько порядков. Увеличение частоты проведения анализов
приводит к удорожанию и усложнению проблемы, что не позволяет МС быть в первых
рядах методов мониторинга окружающей среды.
Возрастающие потребности в быстром и простом МС-анализе привели в середине
90-х годов к развитию так называемых МС-методов в нормальных (природных) условиях.
Общими чертами этих методов являются отсутствие необходимости модификации об-
разца, проведение измерений за несколько секунд и немедленная обратная связь с поль-
зователем (Chen et al., 2009', Van Berkel et al., 2008', Venter et al., 2008). Хотя развитие этого
подхода не снижает цену МС-прибора, существенно сокращается время анализа. Первым
методом с атмосферной (или прямой) ионизацией был прибор десобционной электро-
распылительной ионизации (DESI/MS) (Takats et al., 2004а). Феноменологически ДЭРИ
является методом ионизации при атмосферном давлении, в котором ионы десорбируются
с анализируемой поверхности под действием заряженных капель или ионов, образован-
ных электрораспылением. Схема эксперимента чрезвычайно проста: она состоит из пнев-
матического источника электрораспыления со спреем, направленным непосредственно
на анализируемую поверхность. Многозарядные капли растворителя, ударяясь о поверх-
Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
ность, генерируют вторичные заряженные капли, обогащенные молекулами вещества,
предварительно нанесенного на эту поверхность. Важно отметить, что распыляемый (об-
дувающий) газ играет ключевую роль в процессе ДЭРИ (Takats et al., 2005а).
Газовая струя не только увеличивает эффективность распыления, но также уско-
ряет капли. Поскольку ДЭРИ оптимально работает на поверхности изоляторов, на них
быстро накапливаются статические заряды (Takats et al., 2005b), следовательно, в отсут-
ствие высокоскоростного обдувающего газа кулоновское отталкивание существенно за-
трудняет столкновение первичных капель с поверхностью и формирование вторичных
капель (Volnyetal., 2008). Ионообразование осуществляется именно в этих вторичных за-
ряженных каплях по механизму, реализующемуся в обычной ионизации электрораспы-
лением. В результате ионы исследуемых молекул оказываются в газовой фазе. Этот про-
цесс был назван каплеподхватывающим механизмом, поскольку электрораспыленные
капли растворяют молекулы на поверхности и переносят их в воздушную фазу в процес-
се столкновения (Costa and Cooks, 2008; Venteretal., 2006). Согласно этому сценарию сле-
довало ожидать, что данные ДЭРИ должны быть весьма похожими на данные ЭРИ, что
действительно наблюдается во многих случаях. Пептиды, белки и их производные часто
регистрируются как многозарядные ионы или аддукты с ионами щелочных металлов
или растворителя. Однако, кроме этого механизма, существуют и альтернативные пути
ионообразования, приводящие к ионизации таких соединений, которые не ионизиру-
ются в электрораспылении даже в случае их высокой концентрации или в специальных
экспериментальных условиях. В качестве примеров можно привести полициклические
ароматические углеводороды (ПАУ), терпены, пестициды, хлорированные углеводо-
роды, стероидные гормоны, нитроароматические соединения и взрывчатые вещества
на основе нитросоединений ит.д. (Takats et al., 2005а). Их ионизация происходит по ме-
ханизму свободной капли с участием протонированных молекул растворителя. Ис-
точник электрораспыления генерирует значительное количество ионов растворителя
(буфера), которые, попадая на поверхность, могут вступать в реакцию обмена зарядом
с молекулами, находящимися на поверхности мишени. Десорбции исследуемых ионов
способствует общая заряженность поверхности, а также столкновения с электрораспы-
ленными каплями, кластерами и ионами. Кроме того, обмен зарядами в газовой фазе
является дополнительным механизмом образования ионов. В этом случае исследуемые
молекулы либо непосредственно испаряются с поверхности, либо подхватываются в аэ-
розоль вторичных капель (Cooks etal., 2004).
8.2. Экспериментальные установки и условия
Установка ДЭРИ и схема электрораспылителя представлены на рис. 8.1. Электрозвуко-
вой распылитель (Takats et al., 2004b) смонтирован в Т-образном соединителе (1/16’’ вну-
тренний диаметр, длина 50 мм, фирма Swagelok); капилляр распыляющего газа имеет
наружный диаметр 400 мкм, внутренний 250 мкм, длину 50 мм (плавленый кварц); на-
ружный диаметр капилляра для потока растворителя составляет 150 мкм, а внутрен-
ний — 100 мкм, сужаясь на выходе до 5 мкм. Капилляры зажимаются коаксиально с по-
мощью графитовых или полимерных (веспел) феррул соответствующего размера, как
показано на рис. 8.2. Капилляр растворителя выступает из газового на 0,1—0,2 мм. Рас-
8.2. Экспериментальные установки и условия
а
Графитовая феррула
с внутренним диаметром 0,2 мм
Т-образное соединение
из нержавеющей стали
Высокое
напряжение
(0-4 кВ)
Внутренний диаметр 0,25 мм
Внешний диаметр 0,4 мм
Капилляр из плавленого кварца
Внутренний диаметр 0,4 мм Длина 0,5 см
Феррула из супелтекса
0,1—0,2 мм
Спрей
Поток
образца
0,05 - 50 цл/мин
Соединение из
нержавеющей
стали
I/16 феррула
из нержавею-
щей стали
Внутренний диаметр 5—100 цм
Врешний диаметр 0,15 мм
Конический наконечник
Капилляр из плавленого кварца
£ = 50 см
б
N2давлением
8—25 атмосфер
1/16 трубка из
нержавеющей стали
Рис. 8.1. (а) Ионный источник ДЭРИ (прототип омниспрэя (Prosolia)), смонтированный
на линейной квадрупольной ловушке (LTQ) Thermo Finnigan. А — блок держате-
ля поверхности; В — электрозвуковой распылитель; С — 3-мерное юстировочное
устройство положения поверхности; D — 3-мерное юстировочное устройство
положения распылителя; Е — вращающее устройство распылителя; F — вход
в масс-спектрометр (Takats Z., Wiseman J. М. and Cooks R.G. (2005)). (б) Схема
электрозвукового распылителя. Источник схемы (a): Takats Z., Wiseman J. М. and
Cooks R. G. (2005). «Ambient mass spectrometry using desorption electrospray ionization
(DESI): instrumentation, mechanisms and applications in forensics, chemistry, and
biology». Journal of Mass Spectrometry. 40, 10: 1261 — 1275. С разрешения John Wiley
and Sons. Источник схемы (b): с разрешения от Takats Z., Wiseman J. M., Gologan B.
and Cooks R. G. Electrosonic spray ionization. «А gentle technique for generating folded
proteins and protein complexes in the gas phase and for studying ion molecule reactions
at atmospheric pressure». Analytical Chemistry. 76, 14: 4050—4058. Copyright (2004)
American Chemical Society)
Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
пылитель установлен на 3-мерном юстировочном вращающемся устройстве. Образцы
располагались либо непосредственно под струей вблизи атмосферного входа в МС, либо
на специальном держателе, который также мог юстироваться в трех измерениях, причем
движение в плоскости X—Y обычно управлялось компьютером, как в условиях получе-
ния масс-спектрометрических изображений (глава 20).
Атмосферный вход масс-спектрометра также следует причислить к составным эле-
ментам ДЭРИ. Многочисленные исследования показали, что атмосферный интерфейс,
выполненный из гибкого капилляра из нержавеющей стали для первичного сокраще-
ния потока, оптимален для приложений ДЭРИ. Поскольку зона воздействия струи для
оптимального взаимодействия должна быть менее 20 мм, практически все коммерче-
ские приборы требуют определенной модификации атмосферного входа для проведе-
ния экспериментов ДЭРИ.
В случае использования систем с подогреваемым входным капилляром требуется
удлиненный капилляр, а при использовании так называемых скиммерных входов (на-
пример, в устройствах Sciex) необходима более серьезная модификация, т.е. замена
оригинального входного устройства на устройство с подогреваемым капилляром (Denes
et al., 2009). Наиболее важные инструментальные параметры источника ДЭРИ сумми-
рованы в табл. 8.1, где указаны также оптимальные значения каждого параметра.
Оптимальные условия для ДЭРИ обычно определяются свойствами анализируемой
молекулы и, следовательно, механизмом ее ионизации. Основываясь на физико-хими-
ческих свойствах молекул, их можно разделить на две категории. Первая, так называ-
емая «электрораспылительная», объединяет молекулы, которые легко подвергаются
ионизации электрораспылением в режиме работы и с положительными, и с отрицатель-
ными ионами. Типичные представители этой категории — пептиды, белки, олигоса-
хариды, аминокислоты и сложные липиды. Другая категория, называемая «ХИАД-мо-
лекулы», объединяет вещества, для которых предпочтительна химическая ионизация
при атмосферном давлении (ВЭЖХ/ ХИАД/ МС). Эта группа включает алифатические
и ароматические углеводороды, нитроароматические соединения, пестициды, хлори-
Таблица 8.1. Оптимальные параметры ДЭРИ МС (источник: Takats Z., Wiseman J. М. and
Cooks R.G. (2005). «Ambient mass spectrometry using desorption electrospray
ionization (DES1): instrumentation, mechanisms and applications in forensics,
chemistry, and biology». Journal of Mass Spectrometry. 40, 10: 1261 — 1275. С разре-
шения John Wiley and Sons)
Параметр Тип аналита Оптимальные значения
Пептиды, белки, углеводы, нуклеиновые кислоты Взрывчатые вещества, липиды, ароматические углеводороды
Напряжение электрораспыления 1—4 кВ 3-8 кВ
Скорость потока 0,1—3 л/мин —
Скорость газа электрораспыления 350 м/с —
Температура подогрева капилляра 200-300 °C 200°С
Напряжение линзы 200-250 В 30—150 В для маленьких молекул
Расстояние до входного капилляра 1—2 мм 2—8 мм
Расстояние от образца до распылителя 1—2 мм 5—8 мм
Угол падения (а) 60-90° 20-50°
Угол сбора (р) <10° 10-15°
8.3. Реакционная десорбция
0 2 4 6 8
Напряжение спрея, кВ
Рис. 8.2. Зависимость интенсивности трехзарядных ионов мелиттина от (а) угла падения;
(б) напряжения электрораспыления (данные близки аналогичной зависимости
при ионизации электрораспылением); (в) давления распыляемого газа (изме-
ряемого в регуляторе); (г) расхода распыляемого раствора. Во всех случаях 10 нг
мелиттина помещались на поверхность полиметилметакрилата; 1 мкл/мин вод-
но-метанольного раствора распылялся в случаях (а)—(в); давление распыляюще-
го газа было 10 бар в случаях (а), (б) и (г); напряжение распыления составляло
3 кВ в случае (а), (в) и (г); угол падения 80° для (б)—(г); расстояние от распыли-
теля до поверхности всегда поддерживалось на уровне 1 мм (источник: Takats Z.,
Wiseman J.M. and Cooks R.G. (2005). «Ambient mass spectrometry using desorption
electrospray ionization (DESI):instrumentation, mechanisms and applications in
forensics, chemistry, and biology». Journal of Mass Spectrometry. 40, 10: 1261 — 1275. With
permission from John Wiley and Sons)
+ 16000 -
14000-
12000 -
= с 10000 -
5 g 8000-
X (D
£ £ 6000 -
х g 4000 •
§ 2000
0
0 50 100 150
Давление газа на входе
(унции на квадратный дюйм)
рованные углеводороды, стероидные липиды, жирные кислоты и их эфиры. В случае
«электрораспылительных молекул» ионизация происходит по «капле-подхватываю-
щуму» сценарию, при котором распределение капель по размерам и скоростям играет
ключевую роль. Зависимость интенсивности сигнала ДЭРИ от основных параметров
прибора представлена на рис. 8.2.
8.3. Реакционная десорбция
ДЭРИ предполагает уникальную возможность реакционной десорбционной иониза-
ции. Термин «реакционная десорбция» подразумевает эксперименты, при которых
в распыляемый раствор введены химически активные добавки, вступающие в быструю
реакцию с исследуемыми молекулами. Это химическое взаимодействие используется
для увеличения эффективности ионизации определенных молекул или для их марки-
Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
Рис. 8.3. Масс-спектр ионных компонентов, полученных реакционной ДЭРИ из анио-
на PhB(OH)3 при взаимодействии с (а) чистой поверхностью, (б) поверхностью
с нанесенной D-фруктозой, {в) с D-фруктозой на хлопковом тампоне, (ис-
точник: Chen Н., Cotte-Rodriguez I. and Cooks R.G. (2006). «cis-Diol functional
group recognition by reactive desorption electrospray ionization (DESI)». Chemical
Communications. 6: 597— 599. С разрешения The Royal Society of Chemistry)
ровки для последующей тандемной масс-спектрометрии. Одним из первых примеров
реакционной десорбции была дериватизация цис-1,2-диолов (например углеводы) при
использовании фенилборной кислоты {Chen et al., 2006). Реагент вводился в распыляе-
мый раствор ДЭРИ в концентрации 1—3 мМ при кислотности 9—10, что достигалось до-
бавкой гидроксида аммония. Фенилборная кислота реагирует со всеми компонентами,
содержащими фрагменты цис-1,2-диолов, с образованием соответствующих цикличе-
ских фенилборатных анионов (рис. 8.3).
Реакционная десорбция позволяет увеличить чувствительность путем превраще-
ния нейтральных диолов в анионы циклических фенилборатов, а также структурную
селективность, поскольку жесткие транс-1,2-диолы не подвергаются этой реакции.
Эта селективность была продемонстирована для изомеров углеводов и эстриолов. Даль-
нейшее развитие этого подхода связано с применением иодида М-метил-4 пиридин-
борной кислоты, добавляемого в растворитель ДЭРИ, для введения фиксированного
положительного заряда в аналит по аналогичной схеме {Zhang and Chen, 2010). Другие
примеры включают маркировку зарядом холестерина с использованием альдегида бета-
ина или образование нековалентных комплексов {Wu etal., 2009).
Чувствительность метода ДЭРИ в режиме регистрации отрицательных ионов для
взрывчатых веществ тринитрогексагидро-1,3,5-тразина (RDX) и октагидро-1,3,5,7-
тетранитро-1,3,5,7-тетразоцина была увеличена на 1—2 порядка при добавлении раз-
8.4. Масс-спектрометрическое изображение поверхности с помощью ДЭРИ
личных анионных веществ (трифтороуксусная кислота или летучие хлориды) в распы-
ляемый раствор (Cotte-Rodriguez et al., 2005).
Аналогичные результаты получены в случае ДЭРИ с регистрацией положительных
ионов при добавлении сильнощелочных первичных аминов для определения антима-
лярийного препарата (Nyadong et al., 2007). Образование нековалентных комплексов
в реакционной ДЭРИ было также использовано для анализа энантиомеров хиральных
соединений путем образования диастериомерных комплексов с разными картинами
фрагментации (Takats et al., 2005а).
8.4. Масс-спектрометрическое изображение
поверхности с помощью ДЭРИ
Подобно другим десорбционным ионизационным методам ДЭРИ также способна вос-
создать химическое изображение (глава 20) плоских поверхностей (Takats et al., 2004а;
Wiseman etal., 2006). Хотя это приложение имеет небольшой вклад в исследования окру-
жающей среды, его, безусловно, следует упомянуть ввиду потенципиальных возможно-
стей применения для целей здравоохранения и токсикологии. Для ДЭРИ-изображений
можно использовать срезы тканей или естественные плоские образцы (например ли-
стья) (Kertesz et al., 2008; Talaty et al., 2005).
Целью имиджинга является получение пространственно разрешенной масс-
спектрометрической информации с поверхности образца. В зависимости от простран-
ственного разрешения поверхность разбивается на пиксели, внутри каждого из которых
производится запись полного масс-спектра. При этом образец устанавливается гори-
зонтально и перемещается в плоскости под струей из распылителя. В отличие от изобра-
жения при помощи матрично-активированной лазерной десорбции/ионизации (МАЛ-
ДИ) движение образца в направлении Y (перпендикулярно направлению струи и сбору
ионов) оказывается непрерывным, поскольку струя не может быть остановлена после
каждого шага. Следовательно, такие изображения содержат индивидуальные линейные
срезы с соответствующим разрешением.
Распределение интенсивности каждого иона в спектрах визуализируется при обра-
ботке данных всего массива изображений. Интенсивности ионов хорошо коррелируют
с реальным распределением соответствующих молекул в сечении образца (рис. 8.4). ДЭ-
РИ-изображения широко используются для сканирования биологических образцов, осо-
бенно криогенно замороженных тканей (Dill et al., 2009). ДЭРИ-спектры криогенных се-
чений характеризуются в основном ионами соответствующих комплексов липидов, хотя
детектируются также и ксенобиотики (наркотики, пестициды и их метаболиты) (Wiseman
et al., 2008). Потенциальные приложения ДЭРИ-изображения срезов включают в себя
дифференцирование срезов по их гистологическому типу или представление распределе-
ния ксенобиотиков для фармакологических или токсикологических исследований.
Пространственное разрешение ДЭРИ уступает альтернативным технологиям по-
лучения изображений. Обычно разрешение составляет 100—200 мкм, однако недавно
было достигнуто значение 50 мкм (Kerteszand Van Berkel, 2008). Главная причина низкого
разрешения в уширении струи. Эта проблема преодолевается путем миниатюризации
распылителя. Эффективно также уменьшение расстояния между распылителем и по-
Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
□ СЬс^сс Сри . _ ' \ — LV аса 1 mm □ • h i * \ ’ LV m/z 885.6 Pl (38:4) □ ___ nvz 834.4 PS (40:6)
□ /СС Vcpupr * аса m/z888.8 ST (24:1) □ Cpu ’ ^aca m/z 906,8 ST (124:0) Q /cc ^Cpulr^y \aca m/z 788.5 PS (36:1)
z Cbc Xv m/z 281.4 (18:1) □ Cbc LV m/z 303.4 (20:4) □ Cbc ^-cc m/z 327.4 (22:6)
100%
О
Рис. 8.4. Изображения выбранных депротонированных молекул липидов [М — Н]_ из сре-
за мозга крысы, (а) Оптическое изображение коронарной секции мозга крысы:
сс — мозолистое тело; Сри — стриатум; СЬс — кора головного мозга; LV — бо-
ковой желудочек; аса — передняя часть передней комиссуры, (б)—(и) Ионные
изображения выбранных липидов. (Источник: Wiseman J. М., Ifa D. R., Song Q.
and Cooks R.G. (2006). «Tissue imaging at atmospheric pressure using desorption
electrospray ionization (DESI) mass spectrometry». Angewandte Chemie. 118, 43: 7346—
7350. С разрешения John Wiley and Sons)
верхностью и между поверхностью и вводом в масс-спектрометр. Приготовление об-
разца для ДЭРИ значительно проще по сравнению с альтернативными методологиями
получения масс-спектрометрических изображений. Образцы непосредственно распо-
лагают на держателе и анализируют без каких-либо дополнительных приготовлений.
8.5. Количественные измерения
Анализ объектов окружающей среды обычно требует строгого количественного опреде-
ления, поскольку любой результат, превышающий предельно допустимую, по местным
законам, концентрацию, может привести к правовым последствиям. С точки зрения ко-
личественного определения варианты десорбционной ионизации/масс-спектрометрии
(ДИ/МС) могут быть сгруппированы в две категории. Первая группа включает в себя
ДИ/МС-анализ необработанных образцов. В этом случае точное количественное
определение детектируемых аналитов часто не проводится из-за неравномерного рас-
пределения компонентов и отсутствия внутренного стандарта. Очевидно, существуют
исключения, например промышленные продукты, включая фармацевтические пре-
параты, полимеры и т.д. (Chen et al., 2005). Однако подавляющее большинство природ-
ных образцов не может быть подвергнуто прямому количественному анализу методами
ДИ/МС. Аналитический результат часто только подтверждает присутствие определен-
ного компонента с полуколичественной оценкой его концентрации («высокая» или
«низкая»). Вторая группа включает приложения, при которых жидкий образец (обычно
гомогенизированный жидкий раствор) размещается на твердой поверхности мишени.
Поскольку и внутренний, и внешний стандарты могут быть добавлены в образец до раз-
мещения его на мишени, калибровка в этом случае достаточно проста.
Калибровочный график для определения циклоспорина А в моче человека методом
ДЭРИ представлен на рис. 8.5. Внутренний стандарт (циклоспорин D) был добавлен
в образец мочи перед нанесением его аликвоты (5 мкл) на тефлоновую поверхность. Об-
разцы анализировались методом ДЭРИ/МС в режиме мониторинга заданных реакций
с регистрацией положительных ионов на приборе TSQ Quantum (глава 4).
В целом чувствительность ДЭРИ существенно ниже, чем ЭРИ или даже ХИАД,
если предел обнаружения выражен в абсолютных единицах (т.е. в нанограммах или
пикомолях). Относительные чувствительности ДЭРИ и ЭРИ изучались систематиче-
ски, причем для различных молекул была установлена средняя разница в два порядка.
Низкая чувствительность ДЭРИ обусловлена несколькими факторами, которые не-
посредственно связаны с механизмом ионизации, рассмотренным выше. Одна из се-
рьезных проблем — это относительно низкий выход вторичных капель при взаимодей-
ствии жидкости с поверхностью. Коста и Кукс (Costa and Cooks, 2008) изучали динамику
жидкости в ДЭРИ и пришли к заключению, что значительная часть первичных капель
остается на поверхности и только часть ионов из вторичных капель переносится в га-
зовую фазу. Это согласуется с макроскопическим наблюдением большого количества
Рис. 8.5. График ДЭРИ-калибровки циклоспорина А в образце мочи человека при исполь-
зовании циклоспорина D в качестве внутреннего стандарта
232 Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
капель, скользящих по поверхности. Следующая проблема лежит в отклонении вто-
ричных капель, вызываемом поверхностным зарядом и турбулентным профилем га-
зового потока (Venter et al., 2006). Эффективность сбора ионов при использовании то-
чечного атмосферного входа оказывается очень низкой. Средний характерный заряд
капли (заряд/масса или заряд/размер) также существенно ниже, чем в случае ЭРИ, что
проявляется в низкой ионизационной эффективности, необходимости высокой тем-
пературы для атмосферного интерфейса и сохранении капель даже вне атмосферного
входа. Эти внутренние проблемы ДЭРИ обусловили неудачу попыток существенного
увеличения чувствительности метода и привели к необходимости проведения пробо-
подготовки для целей следового анализа.
8.6. Анализ воды
Одним из многообещающих применений ДЭРИ/МС в области охраны окружающей
среды является анализ природных образцов воды. Технологически предлагаются раз-
личные подходы для анализа жидких проб, включая их непосредственное нанесение
и высушивание прямо на твердой подложке или размещение на пористом или волок-
нистом носителе (например на фильтровальной бумаге) и замораживание. Поскольку
для природоохранных целей требуется проведение качественного и количественного
определения экотоксикантов в ультраследовых количествах, зачастую чувствитель-
ности ДЭРИ/МС оказывается недостаточно. В связи с этим метод ДЭРИ/МС стал при-
меняться с различными способами подготовки образцов и, в частности, был изучен
процесс предварительного концентрирования. Маллиган с коллегами (Mulligan et al.,
2007) исследовали возможность детектирования следов пестицидов и остатков взрыв-
чатых веществ в грунтовой воде. Они смогли обнаружить гексоген, атразин и алахлор
на уровне ppm на поверхности фильтровальной бумаги, смоченной образцом анализи-
руемой воды, используя ДЭРИ/МС/МС с регистрацией положительных ионов. Во всех
случаях аналиты детектировались в виде протонированных молекул. Хотя измерения
с использованием смоченной фильтровальной бумаги притягательны своей простотой
и скоростью, детектируемые концентрации на несколько порядков превосходили ПДК,
установленные Агентством по охране окружающей среды США. Для увеличения чув-
ствительности метода образцы воды, содержащие следы взрывчатых веществ, подвер-
гались экстракции (1 мл водной пробы экстрагировался 100 мкл толуола в течение 90 с,
и 10 мкл аликвоты толуольного раствора помещались на тефлоновую подложку). Вы-
сохшее пятно анлизировалось ДЭРИ с регистрацией отрицательных ионов, используя
раствор метанола в воде (1:1) с 0,05 % НС1. Предел обнаружения для гексогена и тротила
составил 10 ppb и 1 ppb соответственно. Гексоген обнаруживался в виде иона [М + С1]-,
а тротил — в виде молекулярного анион-радикала М_. Твердофазная экстракция (ТФЭ)
для проб, содержащих по 300 ppb гексогена, алахлора и ацетохлора, была также опро-
бована в этих исследованиях. Стирол-дивинилбензольная сульфированная фаза была
использована для ТФЭ. Перед ДЭРИ-анализом с регистрацией положительных ионов
элюат высушивался на тефлоновой подложке. Все три модельные экотоксиканта были
успешно детектированы в указанных концентрациях, причем изобарные алахлор и аце-
тохлор были дифференцированы методом МС/МС.
8.6. Анализ воды
Возможность комбинации ТФЭ и ДЭРИ была подробно изучена (Denes et al., 2009).
Описанная аналитическая схема включает специализированный ТФЭ-метод, при кото-
ром элюирование с ТФЭ-картриджа было модифицировано в целях концентрирования
молекул аналита на твердой подложке. Фрит картриджа подвергался продувке горячим
азотом, который эффективно выпаривал элюат, концентрируя молекулы аналита на по-
верхности фрита. За этой стадией элюирования следовал ДЭРИ-анализ поверхности
фрита. Одно из преимуществ этой методики состоит в том, что практически любой объ-
ем водной пробы может быть обработан без каких-либо модификаций самого процесса
элюирования, что теоретически позволяет увеличить чувствительность на шесть —семь
порядков. Подготовка образца несколько усложняет метод, но полный цикл, тем не ме-
нее, позволяет проводить до 600 измерений в час, что на порядок превосходит произво-
дительность альтернативной ультраэффективной жидкостной хроматографии — масс-
спектрометрии (УЭЖХ/МС). ТФЭ/ДЭРИ-связка была усовершенствована добавлением
традиционного ЛЮЭР-коннектора с полипропиленовым шприцем, причем пакетиро-
вание осуществлялось не в барабане шприца, а в части Люэра. В этом случае фрит со-
прикасается только с концом шприца, что делает возможным ДЭРИ-анализ. Картрид-
жи и другое необходимое оборудование представлены на рис. 8.6.
Описано также 96-канальное элюирующее устройство в комплекте с источником
ДЭРИ. В варианте плоского ТФЭ отверстия диаметром 1 мм делаются в полипропи-
леновой плате и заполняются ТФЭ-волокном, а поверхность покрывается пористой
гидрофобной мембраной на передней стороне и гидрофильной мембраной на задней
стороне. Водные пробы, находящиеся в задней части, элюировались по направлению
к передней, где и проходило концентрирование аналита. Поскольку в ДЭРИ исполь-
зуются растворы, не смачивающие гидрофобные мембраны, обратное приникновение
аналита в пакет ТФЭ во время анализа минимально. Преимущества этого варианта
включают в себя простой сбор образца и портативность, что весьма критично для мо-
ниторинга окружающей среды.
Рис. 8.6. (а) Схема ТФЭ-картриджа; (б) схема платы образца: (I) полимерный фрит; (2)
пакет ТФЭ; (3) полипропиленовая трубка; (4) полипропиленовая пластина;
(5) полимерная мембрана; (6) ТФЭ-диски. (Источник: С разрешения Denes J.,
Katona М., Hosszu A., Czuczy N. and Takats Z. «Analysis of biological fluids by direct
combination of solid phase extraction and desorption electrospray ionization mass
spectrometry». Analytical Chemistry. 81, 4: 1669—1675. Copyright (2009) American
Chemical Society)
234 Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
Твердофазная микроэкстракция (ТФМЭ) была успешно применена с методом
ДЭРИ для анализа водных проб (Kennedy et al., 2010). Эта технология предлагает одно-
ступенчатую процедуру подготовки образца и увеличивает чувствительность на один-
два порядка.
Метод состоит в том, что подготовленное волокно погружается в водный образец
на несколько минут, ополаскивается и непосредственно анализируется ДЭРИ/МС.
При измерении первичный электрораспылитель поворачивался в сторону атмосфер-
ного входа в масс-спектрометр, а ТФМЭ-волокно помещалось в распыляемой струе.
Этот подход оказался эффективнее обычного, когда волокно располагалось на плоской
подложке. Волокно ТФМЭ передвигалось сквозь струю линейно с помощью юстиро-
вочного устойства под контролем компьютера. Поскольку молекулы аналита адсорби-
руются на стационарной фазе С18, органическая составляющая распыляющего раство-
ра ДЭРИ (обычно ацетонитрил), как правило, велика (75%). Ионная хроматограмма,
полученная для такого эксперимента, продемонстрировала линейное распределение
аналита вдоль волокна. Для целей количественного анализа необходимо обрабатывать
и интегрировать весь сигнал на ионной хроматограмме. Для различных органических
соединений, включая алкалоиды коки, предел обнаружения находится в диапазоне 10—
50 нг/мл. Метод был успешно прокалиброван (R2 = 0,997—0,999) для различных веществ
в диапазоне концентраций 10—500 нг/мл. Точность метода составила 38 % вблизи значе-
ний предела обнаружения и 7,6% для максимальных измеренных концентраций. Пол-
ное время анализа данным методом при мультиплексной подготовке образца составило
<1 мин., причем чувствительность и воспроизводимость данных сопоставимы с резуль-
татами ЖХ/МС или ГХ/МС. Более того, ТФМЭ-технология предлагает простой отбор
небольших проб в полевых условиях, что идеально подходит для мониторинга объектов
окружающей среды.
8.7. Анализ аэрозолей
Одним из интенсивно развивающихся приложений ДЭРИ/МС для изучения объектов
окружающей среды является анализ природных и техногенных аэрозолей. По существу,
на основе ДЭРИ было создано два подхода для анализа аэрозолей, аналогичных описан-
ным выше методикам анализа водных проб.
В первом случае аэрозоль, переносимый потоком воздуха, непосредственно анали-
зировался при его смешении с распыляемой струей (Donget al., 2007). В альтернативном
варианте аэрозольные частицы собирались на волокнистый фильтр, поверхность кото-
рого далее анализировалась (Chen etal., 2008; Lietal., 2009a). Количественные измерения
для аэрозольных проб (в отличие от водных) не могут быть выполнены корректно из-
за проблем с добавками правильных внутренних стандартов в аэрозоль или дополни-
тельной добавки внешнего стандарта в матрицу образца.
На рис. 8.7 представлена установка, предназначенная для анализа искусственных
аэрозолей порошкового рассеивателя в кипящем слое. Механизм ионизации включает
в себя подхватывание (поглощение) частиц аэрозоля электрораспыленными каплями.
Исследованные модели аэрозолей среди прочего включают порошки лекарствен-
ных препаратов, какао и искусственных подсластителей. Использование ДЭРИ оказа-
Aerosol particles
Рис. 8.7. Принципиальная схема установки ДЭРИ для анализа сухих частиц. Эмиттер
электрораспылителя (а) установлен в 4 мм от входного отверстия МС (в). Угол
между (а) и (в) составляет 60°. Газообразный азот (N2) способствует распылению
растворителя (S). Аэрозольные частицы генерировались в кипящем слое в труб-
ке (б). Угол между (а) и (б) составляет 90°. (Источник: Dong J., Rezenom Y. Н. and
Murray К. К. (2007). «Desorption electrospray ionization of aerosol particles». Rapid
Communications in Mass Spectrometry. 21, 24: 3995—4000. С разрешения John Wiley
and Sons)
лось успешным для каждого случая — как для детектирования, так и идентификации
основных компонентов этих аэрозолей. Хотя достоинства метода (пределы обнаружения
и количественного определения, точность, воспроизводимость) продемонстрированы
весьма наглядно, ясно, что прямое использование ДЭРИ/МС для анализа аэрозолей
возможно только в случае их высокой концентрации, а результаты дают информацию
только об их основных компонентах. Потенциальными приложениями этого метода
могут быть мониторинг промышленных аэрозолей для оценки безопасности и влияния
на здоровье работников или быстрый анализ дымов при пожаре (требуется быстродей-
ствующий МС или спектрометр ионной подвижности (СИП)).
Альтернативные подходы к анализу аэрозолей на основе ДЭРИ/МС осущест-
вляются при предварительном сборе частиц на фильтре и последующем их масс-
спектрометрическом анализе. Хотя такой подход не позволяет проводить непрерыв-
ный мониторинг, он предполагает практически любое концентрирование и, таким
образом, преодолевает проблему действительно низкой чувствительности ДЭРИ. Тех-
нология была успешно использована для полуколичественного определения уровня
ПАУ (Li et al., 2009а) и органических кислот (Li et al., 2009b) в аэрозолях, образующих-
ся при сжигании биомассы и в городских условиях. Аэрозоли собирались на мембра-
нах из кварцевого волокна. Приблизительно 0,2—0,5 м3 воздуха пропускались сквозь
Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
фильтр диаметром 90 мм для одного ДЭРИ-измерения. Далее из фильтра вырезался
участок 15 х 15 мм, который и анализировался ДЭРИ, используя подкисленный метанол
как распыляющий раствор. Пример спектра представлен на рис. 8.8.
Количественные измерения ПАУ проводились при внешней калибровке стандарт-
ным раствором, наносимым на кварцевый волокнистый фильтре последующей сушкой
и собственно ДЭРИ-анализом. Хотя калибровка была достаточна только для полуко-
личественного определения (R2 = 0,9714) в диапазоне 1 пг— 10 нг, данные представлен-
ных измерений абсолютного количества реальных городских аэрозолей имели точность
10—50%. Описанные оптимальные условия работы прибора (напряжение распыления,
геометрия ионного источника) однозначно предполагают включение в ионизацию ко-
ронного разряда, что может улучшить эффективность ионизации неполярных полуле-
тучих веществ. Аналогичная установка использовалась для определения содержания
органических кислот в аэрозольных продуктах при сжигании биомассы. Как чувстви-
тельность, так и точность определения аэрозольных частиц этим методом была лучше,
чем при определении ПАУ. Субпикограммовый предел обнаружения и 15%-я точность
были получены для щавелевой и олеиновой кислот. Органические кислоты оказывают-
ся идеальными объектами для ДЭРИ из-за наличия карбоксильной группы, при этом
оптимальные условия типичны именно для данного типа приборов.
m/z
Рис. 8.8. Типичный ДЭРИ/МС-спектр ПАУ из аэрозолей при сжигании рисовой соломы.
Асу: аценафтилен; Асе: аценафтен; Flo: флуорен; Ant: антрацен; Phe: фенантрен;
Flu: флуорантен; Руг: пирен; ВаА: бензо[а]антрацен; Chry: хризен; BbF: бензо[Ь]
флуорантен; BkF: бензо[к]флуорантен; ВаР: бензо[а]пирен;1пР: индено[1,2,3-
сфпирен; BgP: 6eH3o[g,h,i] перилен; DbA: дибенз [а,Ь]антрацен. (Источник:
Internationaljournal of Mass Spectrometry. 281, 1—2. Li M., Chen FL, Wang B. F., YangX.,
Lian J. J. and Chen J. M. «Direct quantification of PAHs in biomass burning aerosols by
desorption electrospray ionization mass spectrometry». 31—36. Copyright (2009), с раз-
решения Elsevier)
8.7. Анализ аэрозолей
Похожий подход представлен группой Laskin et al. (2010) для анализа органических
аэрозолей (ОА), получаемых при озонолизе паров D-лимонена. Вторичные ОА собира-
лись на ПТФЭ-пластину с использованием 10-ступенчатого микродроссельного одно-
родного разделителя аэрозолей (импактора). Осажденные образцы анализировались
немедленно или после отвердевания, используя коммерческий оригинальный ДЭРИ-
источник PROSOLIA. Сравнение параллельных измерений на приборах ДЭРИ и ЭРИ
показало, что ДЭРИ характеризуется более высокой интенсивностью и существенно бо-
лее широким диапазоном детектируемых вешеств. Этот эффект, практически противо-
положный обычному различию в чувствительности этих двух методов, был объяснен
нестабильностью продуктов озонолиза в кислотном растворе, использованном в ЭРИ-
экспериментах. Хотя такой же раствор использовался и в ДЭРИ-экпериментах, малое
время взаимодействия между аналитом и раствором (порядка миллисекунды) делает
влияние кислоты незначительным. Таким образом, был сделан вывод, что ДЭРИ обе-
спечивает улучшение результатов (по сравнению с автономным режимом) при анализе
химически неустойчивых аэрозолей, собираемых на твердых носителях.
Группа Ласкиной также представила альтернативный ДЭРИ-метод для анализа со-
бранных аэрозолей {Roach etal., 2010), названный нанодесорбционной электрораспыли-
тельной ионизацией (нано-ДЭРИ). Схема экспериментальной установки представлена
на рис. 8.9.
Метод не использует явление ДЭРИ (эффект столкновения многозарядных капель
о поверхность). Вместо этого проводится экстракция раствора с поверхности, совме-
щенная с наноэлектрораспылительной ионизационной МС. Нано-ДЭРИ решает про-
блему чувствительности ДЭРИ, заменяя стадию удара капель процессом экстракции.
Рис. 8.9. Принципиальная схема нано-ДЭРИ-источника для анализа органических
аэрозолей. (Источник: с разрешения Roach P.J., Laskin J. and Laskin A. (2010).
«Molecular characterization of organic aerosols using nanospray-desorption/electrospray
ionization-mass spectrometry». Analytical Chemistry. 82, 19: 7979—7986. Copyright
(2010) American Chemical Society)
Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
Как указано в начале данной главы, удары первичных капель о поверность вызывают
формирование вторичных капель с низкой эффективностью, только 1—3% всей рас-
пыляемой жидкости конвертируется во вторичные заряженные капли. Для сравнения
в случае нано-ДЭРИ образец практически не теряется на стадии взаимодействия жид-
кость-поверхность и весь экстракт конвертируется в полизаряженные капли, из ко-
торых и образуются ионы. Поскольку время взаимодействия аналита с раствором по-
прежнему ограничено (0,1—1 сек.), а эффективность ионизации увеличена на один— два
порядка, этот подход превосходит и ДЭРИ, и ЭРИ при анализе химически неустойчи-
вых компонентов, присутствующих на поверхности. Недостатком нано-ДЭРИ-метода
является тонкость в управлении. Для обеспечения контакта с жидкостью на поверхно-
сти зазор между капилляром и поверхностью должен поддерживаться с точностью в не-
сколько мкм.
8.8. Прямой анализ
Одним из уникальных свойств ДЭРИ является возможность неинвазивного пробоот-
бора произвольных образцов. Для целей экологического мониторинга и оценки риска
часто требуется сбор и анализ тысяч проб. Реальное число образцов обычно опреде-
ляется компромиссом между оптимальным числом, стоимостью и временем анализа.
В результате периодичность отбора проб оптимизируется во времени и пространстве.
Метод ДЭРИ в портативном варианте может обеспечить решение этих проблем путем
устранения стадий отбора, транспортировки и подготовки образцов, а также позволяя
осуществлять моментальные измерения в «полевых» условиях (1—5 сек.). Возможно соз-
дание полевого масс-спектрометра или спектрометра ионной подвижности для анали-
за растений, почвы, камней и т.д. непосредственно на месте. Поскольку ДЭРИ-анализ
окружающей среды является в основном полуколичественным, он никогда не заменит
лабораторные инструментальные исследования (ЖХ/МС, ГХ/МС). Однако потенци-
ально он играет роль предварительного скрининга, позволяя отобрать репрезентатив-
ные пробы для последующего лабораторного анализа. Этот скрининг может иметь два
варианта, которые направлены на выбор холостых образцов или образцов, в которых
регистрируется сигнал интересующего вещества. ДЭРИ-анализ может не иметь стро-
гого предела обнаружения при анализе природных образцов, однако чрезвычайно лег-
ко определить минимальную концентрацию, выше которой вероятность обнаружения
аналита близка к 100%. Если эта величина ниже предельно допустимой (безопасной)
концентрации для определенного вещества, бессмысленно отбирать пробы для лабора-
торного анализа, в которых не был зарегистрирован сигнал методом ДЭРИ. Тем не менее
возможности метода обычно позволяют оценивать реальные концентрации, т.е. на ос-
нове полученных результатов можно делать более значимые выводы.
Примеры реальных измерений загрязнений окружающей среды методом ДЭРИ уже
были представлены несколькими группами. Интересные результаты по ДЭРИ/ МС-
мониторингу мышьякорганических соединений на растениях и пище для животных
получены Лин и др. (Lin et al., 2010). Эти соединения долгое время добавлялись в пищу
домашним животным в качестве средства против простейших, что представляет собой
значительный риск как для пищи, так и для окружающей среды в целом. Установлен-
Литература
ные пределы обнаружения 10 различных соединений мышьяка находятся в диапазоне
2—40 пг/мм2 для случая обогащенных образцов. Разработанный метод может легко при-
меняться в полевых условиях или для предварительного скрининга образцов с после-
дующим анализом методами индуктивно связанной плазмы (ИСП)/МС или ЖХ/МС.
Хотя ДЭРИ/МС-технология не стала частью протокола серийных анализов объек-
тов окружающей среды, она уже была использована в ряде прикладных исследований.
Метод в его настоящем виде (т.е. как ионный источник для коммерческих аналитиче-
ских масс-спектрометров) может быть легко использован для высокопроизводитель-
ного анализа разнообразных образцов объектов окружающей среды, особенно при-
родной воды и собранных аэрозолей. Метод был успешно применен в комбинации
с ТФЭ-технологией для преодоления проблем чувствительности, связанных с влияни-
ем матрицы. Эта модификация схемы ДЭРИ существенно не сказывается на его высо-
кой производительности. Другим предсказуемым применением ДЭРИ/ МС является его
инкорпорирование в полевой миниатюрный масс-спектрометр или спектрометр ион-
ной подвижности. Судя по тенденциям миниатюризации, этот вариант может привести
к разработке коммерческого прибора уже в следующем десятилетии (см. гл. 9). Такие
приборы будут проводить анализ объектов окружающей среды «по мановению волшеб-
ной палочки», обнаруживая загрязнения на произвольных поверхностях в мгновение
ока.
Литература
[1] . Chen Н., Talaty N. N., Takats Z., and Cooks R. G. (2005). «Desorption electrospray ioniza-
tion mass spectrometry for high-throughput analysis of pharmaceutical samples in the ambi-
ent environment». Analytical Chemistry. 77, 21: 6915—6927.
[2] . Chen H., Cotte-Rodriguez I., and Cooks R. G. (2006). «cis-Diol functional group recogni-
tion by reactive desorption electrospray ionization (DESI)». Chemical Communications. 6:
597-599.
[3] . Chen H., Li M., Zhang Y. P., Yang X., Lian J. J., and Chen J. M. (2008). «Rapid analysis of
SVOC in aerosols by desorption electrospray ionization mass spectrometry». Journal of the
American Society for Mass Spectrometry. 19, 3: 450—454.
[4] . Chen H., Gamez G., and Zenobi R. (2009). «What can we learn from ambient ionization
techniques?» Journal of the American Society for Mass Spectrometry. 20, 11: 1947—1963.
[5] . Cooks R. G., Jo S.-С., and Green J. (2004). «Collisions of organic ions at surfaces». Applied
Surface Science. 231—232: 13—21.
[6] . Costa A.B., and Cooks R.G. (2008). «Simulated splashes: elucidating the mechanism of
desorption electrospray ionization mass spectrometry». Chemical Physics Letters. 464, 1—3:
1-8.
[7] . Cotte-Rodriguez I., Takats Z., Talaty N., Chen H., and Cooks R.G. (2005). «Desorption
electrospray ionization of explosives on surfaces: sensitivity and selectivity enhancement by
reactive desorption electrospray ionization». Analytical Chemistry. 77, 21: 6755—6764.
[8] . Denes J., Katona M., Hosszu A., Czuczy N., and Takats Z. (2009). «Analysis of biological
fluids by direct combination of solid phase extraction and desorption electrospray ionization
mass spectrometry». Analytical Chemistry. 81, 4: 1669—1675.
240 Глава 8. Масс спектрометрия десорбционной электрораспылительной ионизации
[9] . Dill A. L., Ifa D. R., Manicke N. Е., Ouyang Z., and Cooks R. G. (2009). «Mass spectromet-
ric imaging of lipids using desorption electrospray ionization». Journal of Chromatography B.
877, 26: 2883-2889.
[10] . Dong J., Rezenom Y. H., and Murray К. K. (2007). «Desorption electrospray ionization of
aerosol particles». Rapid Communications in Mass Spectrometry. 21, 24: 3995—4000.
[11] . Kennedy J. H., Aurand C., Shirey R., Laughlin B.C., and Wiseman J.M. (2010). «Cou-
pling desorption electrospray ionization with solid-phase microextraction for screening and
quantitative analysis of drugs in urine». Analytical Chemistry. 82, 17: 7502—7508.
[12] . Kertesz V., and Van Berkel G. J. (2008). «Improved imaging resolution in desorption elec-
trospray ionization mass spectrometry». Rapid Communications in Mass Spectrometry. 22,
17: 2639-2644.
[13] . Kertesz V., Van Berkel G. J., Vavrek M., Koeplinger K. A., Schneider В. B., and Covey T. R.
(2008). «Comparison of drug distribution images from whole-body thin tissue sections ob-
tained using desorption electrospray ionization tandem mass spectrometry and autoradiog-
raphy». Analytical Chemistry. 80, 13: 5168—5177.
[14] . Laskin J., Laskin A., Roach P.J., Slysz G.W., Anderson G.A., Nizkorodov S.A.,
Bones D. L., and Nguyen L.Q. (2010). «High-resolution desorption electrospray ionization
mass spectrometry for chemical characterization of organic aerosols». Analytical Chemistry.
82, 5: 2048-2058.
[15] . Li M., Chen H., Wang B. F., Yang X., Lian J. J., and Chen J. M. (2009a). «Direct quanti-
fication of PAHs in biomass burning aerosols by desorption electrospray ionization mass
spectrometry». International Journal of Mass Spectrometry. 281, 1—2: 31—36.
[16] . Li M., Chen H., YangX., Chen J.M., and LiC.L. (2009b). «Direct quantification of organ-
ic acids in aerosols by desorption electrospray ionization mass spectrometry». Atmospheric
Environment. 43, 17: 2717—2720.
[17] . Lin Z. Q., Zhao M.X., Zhang S. C., Yang C. D., and Zhang X. R. (2010). «In situ arsenic
speciation on solid surfaces by desorption electrospray ionization tandem mass spectrom-
etry». An alyst. 135,6: 1268-1275.
[18] . Mulligan C.C., MacMillan D.K., Noll R.J., and Cooks R.G. (2007). «Fast analysis of
high-energy compounds and agricultural chemicals in water with desorption electro-
spray ionization mass spectrometry». Rapid Communications in Mass Spectrometry. 21, 22:
3729-3736.
[19] . Nyadong L., Green M. D., De Jesus V. R., Newton P. N., and Fernandez F. M. (2007). «Re-
active desorption electrospray ionization linear ion trap mass spectrometry of latest-gener-
ation counterfeit antimalarials via noncovalent complex formation». Analytical Chemistry.
Tf 5:2150-2157.
[20] . Roach P. J., Laskin J., and Laskin A. (2010). «Molecular characterization of organic aer-
osols using nanospraydesorption/electrospray ionization-mass spectrometry. Analytical
Chemistry. 82, 19: 7979-7986.
[21] . Takats Z., Wiseman J.M., Gologan B., and Cooks R.G. (2004a). «Mass spectrometry
sampling under ambient conditions with desorption electrospray ionization». Science. 306,
5695:471-473.
[22] . Takats Z., Wiseman J. M., Gologan B., and Cooks R. G. (2004b). «Electrosonic spray ioni-
zation. a gentle technique for generating folded proteins and protein complexes in the gas
Литература
phase and for studying ion-molecule reactions at atmospheric pressure». Analytical Chem-
istry. 16, 14: 4050-4058.
[23] . Takats Z., Wiseman J.M., and Cooks R.G. (2005a). «Ambient mass spectrometry using
desorption electrospray ionization (DESI): instrumentation, mechanismsand applications
in forensics, chemistry, and biology». Journal of Mass Spectrometry. 40, 10: 1261 — 1275.
[24] . Takats Z., Cotte-Rodriguez I., Talaty N., Chen H., and Cooks R. G. (2005b). «Direct, trace
level detection of explosives on ambient surfaces by desorption electrospray ionization mass
spectrometry». Chemical Communications. 15: 1950—1952.
[25] . Talaty N., Takats Z., and Cooks R. G. (2005). «Rapid in situ detection of alkaloids in plant
tissue under ambient conditions using desorption electrospray ionization». Analyst. 130, 12:
1624-1633.
[26] . Van Berkel G. J., Pasilis S. P., and Ovchinnikova O. (2008). «Established and emerging at-
mospheric pressure surface sampling/ionization techniques for mass spectrometry». Jour-
nal of Mass Spectrometry. 43, 9: 1161 — 1180.
[27] . Venter A., Sojka P. E., and Cooks R.G. (2006). «Droplet dynamics and ionization mecha-
nisms in desorption electrospray ionization mass spectrometry». Analytical Chemistry. 78,
24: 8549-8555.
[28] . Venter A., Nefliu M., and Cooks R. G. (2008). «Ambient desorption ionization mass spec-
trometry». Trends in Analytical Chemistry. 27, 4: 284—290.
[29] . Volny M., Venter A., Smith S.A., Pazzi M., and Cooks R.G. (2008). «Surface effects and
electrochemical cell capacitance in desorption electrospray ionization». Analyst. 133, 4:
525-531.
[30] . Wiseman J. M., Ifa D. R., Song Q., and Cooks R. G. (2006). «Tissue imaging at atmospheric
pressure using desorption electrospray ionization (DESI) mass spectrometry». Angewandte
Chemie. 118,43: 7346-7350.
[31] . Wiseman J. M., Ifa D. R., Zhu Y, Kissinger C.B., Manicke N.E., Kissinger P.T., and
Cooks R. G. (2008). «Desorption electrospray ionization mass spectrometry: imaging drugs
and metabolites in tissues». Proceedings of the National Academy of Sciences of the United
States of America. 105, 47: 18 120—18 125.
[32] . Wu C., Ifa D. R., Manicke N. E., and Cooks R. G. (2009). «Rapid, direct analysis of choles-
terol by charge labeling in reactive desorption electrospray ionization». Analytical Chemis-
try. 81, 18: 7618-7624.
[33] . Zhang Y., and Chen H. (2010). «Detection of saccharides by reactive desorption electro-
spray ionization (DESI) using modified phenylboronic acids». International Journal of Mass
Spectrometry. 289, 2—3: 98—107.
ГЛАВА 9
МИНИАТЮРНЫЕ
МАСС-СПЕКТРОМЕТРЫ
ДЛЯ АНАЛИЗА ОБЪЕКТОВ
ОКРУЖАЮЩЕЙ СРЕДЫ
Гуатам Шарма, Роберт Дж. Нолл, Дженг Оуянг и Грэм Кукс
9.1. Введение
Загрязнение компанией Horizon Deepwater вод Мексиканского залива 636 миллионами
литров нефти — крупнейшая катастрофа такого рода в истории человечества (Camilli
et al., 2010). Извержение вулкана Эйяфьядлайёкюдль в Исландии выбросило в атмосфе-
ру облака вулканического пепла (Laursen, 2010). Два этих события являются примера-
ми эмиссии в окружающую среду огромных количеств потенциально вредных веществ
вследствие деятельности человека или природной активности. Незамедлительное
и долговременное влияние этих событий на окружающую среду подчеркивает необхо-
димость контроля загрязнений окружающей среды в целях принятия эффективных мер
в нужное время и в нужном месте. Традиционные аналитические процедуры в таких
случаях предполагают сбор образцов и их транспортировку в удаленную лабораторию.
Аналитические эксперименты, проводимые таким образом, подразумевают предвари-
тельное хранение, экстракцию, очистку и концентрирование. Эти этапы требуют се-
рьезных затрат времени и квалифицированного труда. Кроме того, пробы могут быть
загрязнены во время транспортировки или хранения, причем риск загрязнения воз-
растает с увеличением промежутка времени между отбором пробы и ее анализом. Ис-
пользование портативных аналитических приборов, с другой стороны, уменьшает эту
опасность и увеличивает эффективность анализа. Такие приборы должны обладать
способностью экспрессной генерации высококачественных данных, что должно быть
особенно полезным при исследовании динамических систем. Таким образом, суще-
ствует необходимость разработки портативных аналитических приборов, чувствитель-
ных к широкому спектру соединений и способных идентифицировать ксенобиотики
в окружающей среде в полевых условиях.
Разработка портативных приборов, мотивированная необходимостью быстрого
анализа в полевых условиях, является одним из наиболее активно развивающихся на-
правлений аналитической химии. Химические приборы, работающие в полевых ус-
ловиях, нужны для экспрессного детектирования и идентификации широкого круга
соединений, даже если они присутствуют в пробе в следовых количествах. Скорость
анализа — это один из наиболее важных параметров таких приборов. Желательно, что-
9.1. Введение
бы такое оборудование могло функционировать в широком диапазоне внешних усло-
вий. Такие приборы также должны детектировать исследуемые компоненты в сложных
матрицах с разрешением структурно и химически близких соединений. Существен-
ными критериями являются портативность прибора, простота в эксплуатации даже
для неспециалистов, минимальная пробоподготовка без потери чувствительности
и специфичности. Портативность имеется в виду здесь в широком смысле: подразуме-
вается возможность простой транспортировки прибора с применением механических
устройств, например автомобилей. Надежность и устойчивость — ключевые моменты
для них, принимая во внимание суровые климатические условия, в которых эти при-
боры должны работать.
Большое количество методов детектирования было проверено на предмет удов-
летворения упомянутым выше условиям. Например, были попытки применить метод
спектрометрии ионной подвижности (Bairn and Hill, 1982; Borsdorf and Eiseman, 2006; Hill
and Simpson, 1997). Этот метод активно используется и обладает высокой чувствитель-
ностью. Однако его химическая специфичность ограничена. Родственный метод масс-
спектрометрии дифференциальной ионной подвижности (Nazarov et al., 2006), в част-
ности в сочетании с простым масс-спектрометром, продемонстрировал определенный
потенциал для анализа сложных смесей (Tadjimukhamedov et al., 2010). Электрохимиче-
ские сенсоры (Knake et al., 2005) просты и оптимизированы для обнаружения специфи-
ческих токсичных газов (например угарного газа). Такие методы наиболее пригодны
для обнаружения одного или в крайнем случае нескольких компонентов. В противном
случае целевые аналиты могут перекрываться основными компонентами пробы. Ми-
ниатюрные комбинации газовых хроматографов с масс-спектрометрами уже были про-
тестированы (Contreras et al., 2008; Shortt et al., 2005). Однако они страдают от недоста-
точной надежности и низкой скорости анализа. Портативные КР-спектрометры были
разработаны Ван Дюйном и его коллегами (Young et al., 2004) и даже воплощены в ком-
мерческий продукт (например, DeltaNu, Thermo Fisher Scientific). Такой подход в сочета-
нии с многокомпонентными статистическими методами является многообещающим.
Тем не менее детектирование индивидуальных компонентов в смеси остается пробле-
матичным. Молекулярные имплантированные сенсоры (Alexanderetal., 2006) портатив-
ны и чувствительны. Однако они обеспечивают лишь подтверждение и количественное
определение ограниченного числа соединений.
Масс-спектрометры предлагают мощное сочетание универсальности, скорости
анализа и информативности, несмотря на то, что надежные миниатюрные прибо-
ры были разработаны лишь недавно. Режим МС/МС (McLuckey et al., 1988) помогает
установить идентичность данного компонента, обеспечивая необходимую специфич-
ность — качество, бесценное в полевых условиях ввиду снижения вероятности ошибки.
Лабораторные методы, основанные на масс-спектрометрии, служат для окончатель-
ного подтверждения результатов анализа, а масс-спектрометрия является «золотым
стандартом» для анализа микропримесей. Традиционно образцы доставлялись в лабо-
раторию. Затем проводилась достаточно сложная пробоподготовка, обеспечивающая,
тем не менее, хорошее качество результатов. Однако развитие методов ионизации в нор-
мальных условиях (Cooks et al., 2006; Huang et al., 2010c; Ifa et al., 2010; van Berkel et al.,
2008; Venter et al., 2008; Weston, 2010), которые требуют минимальной пробоподготовки,
или не требуют ее вовсе (будет обсуждаться далее в этой главе; смотрите также главы 7
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
и 8), а также сохранение характеристик масс-спектрометров при их миниатюризации
(Badman and Cooks, 2000; Diaz etal., 2001a; Diaz et al., 2001b; Gao et al., 2006; Gao et al., 2008b;
Geear et al., 2005; Mauclaire et al., 2004; Orient et al., 1997; White et al., 1998) делает масс-
спектрометрию многообещающим методом анализа в полевых условиях. Благодаря
этим тенденциям в последнее десятилетие значительного прогресса удалось достиг-
нуть в разработке портативных приборов (Ecelbergeretal., 2004; Gao et al., 2006; Gao etal.,
2008b; Laughlin et al., 2005; Patterson et al., 2002; Yang et al., 2008). Эта глава описывает ос-
новы миниатюризации масс-спектрометров и варианты использования таких приборов
для анализа объектов окружающей среды. Термин «миниатюрный» относится к прибо-
рам значительно меньшим по размеру по сравнению с традиционными лабораторными
масс-спектрометрами и весом порядка 10—30 фунтов (4,5—13,6 кг). Акцент будет сделан
на автономных приборах.
9.2. Основы конструкции приборов
Основные составные части миниатюрных масс-спектрометров — те же, что и у обычных
лабораторных приборов. А именно: устройство для ввода пробы, источник ионов, ана-
лизатор масс, детектор, вакуумная система, система контроля прибора и система реги-
страции данных (рис. 9.1) (Oyang and Cooks, 2009). В данной главе основные части мини-
атюрного масс-спектрометра будут обсуждаться в общем виде со ссылками на приборы
Mini 10 (Gao etal., 2006) и Mini 11 (Gao etal., 2008b) — миниатюрные масс-спектрометры,
которые были разработаны в лабораториях авторов данной главы в Университете Пер-
дью. В данном разделе будут описаны основные подсистемы приборов.
9.2.1. Методы ввода проб
Масс-спектрометрический анализ должен проводиться в вакууме. Следовательно, вы-
бор системы ввода проб играет важнейшую роль в функционировании прибора, так как
метод ввода образца в масс-спектрометр определяет газовую нагрузку для вакуумной
Процессы, связанные с ионами
Детектор
ионов
Масс-
анализатор
Источник
* ионов
откачка
Откачка
Высоковакуумная Форвакуум
------ откачка
Рис. 9.1. Блок-схема миниатюрного масс-спектрометра. (Источник: адаптировано
из Ouyang Z. and Cooks R.G. (2009). «Miniature mass spectrometers». Annual Review of
Analytical Chemistry. 2: 187—214. Copyright 2009 by Annual Reviews, Inc. Воспроизве-
дено с разрешения
Ввод
пробы
4---------1-
Ионизация в вакууме (Да/Нет) Рабочее давление
9.2. Основы конструкции приборов
системы. В случае миниатюрных масс-спектрометров этот выбор даже более критичен,
так как необходимость обеспечения малого веса установки и низкого энергопотребле-
ния налагает ограничения на выбор средств откачки. Тип образца и анализируемого
соединения диктует выбор метода ввода. Например, твердые сорбенты могут исполь-
зоваться для ввода паров аналита, а мембраны — для ввода проб, как в виде паров, так
и в виде водных растворов. Далее мы опишем различные методы ввода. Обратите вни-
мание, что мембранный ввод, ввод через твердый сорбент и через капилляр поставляют
в масс-спектрометр образцы в виде нейтральных частиц, тогда как интерфейсы дис-
кретного ввода при атмосферном давлении поставляют ионы, полученные вне прибора.
Мембранный ввод
Полупроницаемые мембраны селективно пропускают молекулы из воздуха или водных
смесей в вакуум. Это достигается адсорбцией аналитов на гидрофобной мембране с по-
следующей их диффузией (Lapack et al., 1990, 1991). Масс-спектрометрия с мембранным
вводом является удобным методом прямого непрерывного анализа воздушных и водных
проб при использовании портативных масс-спектрометров. Этот метод быстр, прост
и чувствителен. Он может использоваться для непрерывного мониторинга различных
экологически важных компонентов (Bierand Cooks, 1987; Bieretal., 1990;JanfeltetaL, 2006;
Patterson et al., 2002; Riter et al., 2002). На рис. 9.2 показана система мембранного ввода
для миниатюрного масс-спектрометра. В серии экспериментов по онлайн-анализу ле-
тучих органических компонентов в воздухе для ввода использовалась 15-сантиметровая
трубка, изготовленная из полидиметилсилоксана, с внешним диаметром 1,19 мм и вну-
тренним диаметром 0,64 мм (Gao et al., 2006). Миниатюрный насос (UNMP015D, KNF
Neuberger; скорость потока 1,3—1,6 л/мин.) использовался для прокачки пробы через
мембранную трубку. Летучие органические компоненты обладают высокой аффинно-
стью к гидрофобной полидиметилсилоксановой мембране. Они проходят через мембра-
ну в вакуумный интерфейс, где ионизируются и анализируются по массам.
Трубка-
мембрана
Рис. 9.2. Схематическая диаграмма миниатюрного масс-спектрометра Mini 10. По-
казаны: прямоугольная ловушка, источник ионов, электронный умножи-
тель и мембранный ввод. (Источник: перепечатано с разрешения из Gao L.,
Song Q. Y., Patterson G. E., Cooks R.G. and Ouyang Z. «Handheld rectilinear ion trap
mass spectrometer». Analytical Chemistry. 78: 5994—6002. Copyright 2006 American
Chemical Society)
Источник
ионов
Прямоугольная
ионная ловушка
Электронный
умножитель
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
Система ввода на основе твердого сорбента
Твердотельные сорбционные ловушки полезны для ввода газо- и парообразных проб (Keil
et al., 2008; Sokol et aL, 2008). На рис. 9.3 представлена схематическая диаграмма устрой-
ства ввода, использовавшегося при анализе газообразных токсичных промышленных
компонентов с помощью миниатюрного масс-спектрометра Mini 10 (Gaoet al., 2006). Та-
кая система состоит из трубки для концентрирования пробы, двух соленоидных клапа-
нов и миниатюрного насоса для подачи образца. Газовый поток, содержащий аналиты,
прокачивается через трубку при помощи миниатюрного насоса, а исследуемые молекулы
адсорбируются на твердофазном сорбенте. Выбор сорбента зависит от природы аналита.
Например, сорбент, содержащий тенакс, использовался для анализа ВТК (бензол, то-
луол, ксилол) (Sokol et al., 2008), тогда как при определении токсичных промышленных
выбросов трубка концентрирования содержала две фазы: тенакс и HayeSep С). Как по-
казано на временной диаграмме 9.3d, при быстром нагреве молекулы десорбируются
и проникают в вакуумную камеру масс-спектрометра. До сих пор во всех экспериментах
с сорбцией на твердом сорбенте применялась ионизация электронами.
0
0
Включено
Масс-анализ
Детектор
Катод
Нагреватель
Клапан 2
Клапан 1
0 15 30 45 60 75 90
Выключено
Выключено
Выключено
Выключено
Открыто для насоса
подачи пробы
Открыто для
подачи образца
.J L
Включено
Включено
Закрыто
Открыто для откачки
Рис. 9.3. а — Схематическая диаграмма миниатюрного масс-спектрометра, оборудован-
ного системой автоматической подачи проб на основе сорбционной трубки, б —
Временная диаграмма, демонстрирующая последовательность операций анализа
пробы. Соленоидные клапаны служат для ввода образца в сорбционную трубку
при содействии насоса. Выбирая соответствующий материал, можно адсорби-
ровать в сорбционной трубке исследуемый аналит с его последующим вводом
в масс-спектрометр при помощи быстрого нагрева трубки. Ловушка: прямо-
угольная ионная ловушка. Перепечатано с разрешения из Keil A., Hernandez-
Soto Н., Noll R.J., Fico М., Gao L., Ouyang Z. and Cooks R.G. «Monitoring of toxic
compounds in air using a handheld rectilinear ion trap mass spectrometer». Analytical
Chemistry. 80: 734—741. Copyright 2008 American Chemical Society)
9.2. Основы конструкции приборов
Система ввода на основе сорбционной ловушки представляет собой удобный ин-
терфейс между атмосферой и вакуумом и при соответствующем дизайне может обе-
спечивать высокие скорости ввода проб и может быть легко автоматизирована. Автома-
тизация ввода может успешно применяться для контроля определенных компонентов
в полевых условиях при минимальном вмешательстве человека. Смите коллегами {Smith
et aL, 2010) разработал масс-спектрометр с устройством ввода на основе твердого сор-
бента, который может быть использован на химическом предприятии для контроля
токсичных промышленных выбросов. Такая система может служить для раннего пред-
упреждения выбросов токсичных веществ в атмосферу.
Ввод при помощи капилляра
Ввод при помощи капилляра является разновидностью твердофазной микроэкстрак-
ции {Arthur and Pawliszyn, 1990) и масс-спектрометрии с мембранным вводом, впервые
представленной Эберлином и его коллегами {Meurer et al., 2002). Система состоит из ка-
пилляра, покрытого полидиметилсилоксаном. Капилляр подвергается контакту с ана-
лизируемой пробой, после чего аналиты вводятся непосредственно в масс-спектрометр.
Анализируемые молекулы десорбируются из капилляра при помощи нихромового на-
гревателя с последующей ионизацией электронами. Такой метод был использован для
миниатюрного масс-спектрометра с анализатором в виде цилиндрической ионной ло-
вушки (рис. 9.4). Он может быть применен для анализа летучих и полулетучих органи-
ческих компонентов с пределами обнаружения в несколько ppb для толуола и метилса-
лицилата {Riteret al., 2003).
Интерфейс дискретного ввода при атмосферном давлении
Традиционно масс-спектрометрический анализ проводился в высоком вакууме с ис-
пользованием ионизации электронами, которая ограничивает диапазон анализируе-
мых образцов. В последнее время были разработаны источники ионизации при атмос-
ГХ-септа толщиной
0,25 дюйма
Фланец QF 25
Пол иди метил -
силоксановый
капилляр
Держатель капилляра
для твердофазной
микроэкстракции
Рис. 9.4. Схематическая диаграмма системы ввода и концентрирования при помощи ка-
пилляра. Капилляр для твердофазной микроэкстракции вводится в вакуумную
систему через ГХ-прокладку посредством нескольких соединителей, сопряжен-
ных с вакуумным фланцем. Изолированная трубка из нержавеющей стали с внеш-
ним диаметром в 1/16 дюйма поддерживается под повышенным напряжением для
возможности резистивного нагрева нихромовой проволоки с нагреванием по-
лидиметилсилоксановой мембраны и десорбции молекул аналита. (Источник:
Riter L.S., Meurer Е.С., Cotte-Rodriguez I., Eberlin M.N. and Cooks R.G. (2003).
«Solid phase micro-extraction in a miniature ion trap mass spectrometer». Analyst. 128:
1119—1122. Воспроизведено с разрешения Королевского химического общества)
Нихромовый
нагреватель
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
ферном давлении, работающие на основе химической ионизации (ХИАД) (Carroll et al.,
1975; Horning et al., 1973), электрораспыления (ЭРИ) (Fenn et al., 1989; Ymashita and Fenn,
1984) и матрично-активированной лазерной десорбции/ионизации при атмосферном
давлении (АД-МАЛДИ) (Tanaka et al., 1988). При использовании недавно изобретенных
методов ионизации в нормальных условиях (например десорбционной электрораспы-
лительной ионизации, ДЭРИ (Takatsetal., 2004)) пробоподготовка отсутствует и образец
анализируется в обычных условиях. Новые методы существенно расширили диапазон
анализируемых соединений и сократили время анализа. Все методы с генерированием
ионов при атмосферном давлении требуют наличия интерфейса с высокой проводимо-
стью для эффективной транспортировки ионов в область вакуума. Для лабораторных
установок это достигается многостадийной дифференциальной откачкой при исполь-
зовании насосов с высокой скоростью откачки (обычно это сотни литров в секунду).
Это объясняется необходимостью поддержания соответствующего для масс-анализа
вакуума (Laughlin etal., 2005; Schwartzetal., 2002). Обеспечение таких высоких скоростей
откачки в миниатюрных приборах не представляется возможным из-за ограничений
на вес, размеры и потребляемую вакуумной системой мощность. Применение источни-
ков ионизации при нормальных условиях с интерфейсами низкой проводимости неиз-
бежно ведет к потерям в разрешении и чувствительности (Keiletal., 2007). Однако суще-
ствуют альтернативные механизмы соединения источников ионизации в нормальных
условиях с миниатюрными приборами.
Один из таких механизмов был предложен Гао (Gao et al., 2008а) в виде интерфейса
дискретной ионизации при атмосферном давлении (ДИАД, DAPI). Концепция ДИАД
состоит в кратковременном открытии канала большой проводимости с последующим
Ввод ионов
напряжения
У\П1ОЖИ 1СЛЬ
включен/выключен
Сигнал
Рис. 9.5. Иллюстрация обычного сканирования при масс-анализе с использованием
ДИАД в сочетании с миниатюрным масс-спектрометром. Интерфейс открыт
на короткое время (10—30 мс), в течение которого другие напряжения, за исклю-
чением радиочастотного, выключены. Масс-анализ начинается, только когда
давление, повышающееся во время ввода ионов, возвращается к нормальному
значению. (Источник: Gao L., 2009. Миниатюризация масс-спектрометра типа
ионной ловушки. Диссертация, Университет Пердью. Напечатано с разрешения)
9.2. Основы конструкции приборов
Рис. 9.6. Иллюстрация изменений
давления в вакуумной камере в тече-
ние масс-анализа. При открытии ин-
терфейса давление возрастает, делая
невозможным масс-анализ. После за-
крытия интерфейса и последующей
откачки давление достигает безопас-
ного уровня и масс-анализ начинает-
ся. (Перепечатано с разрешения Аме-
риканского химического общества
из: Gao L., Cooks R.G. and Ouyang Z.
Breaking the pumping speed barrier in mass
spectrometry: Discontinuous atmospheric
его закрытием в ходе цикла откачки, масс-анализа, удаления ионов и периодов восста-
новления для каждого режима сканирования. Ценой значительного уменьшения ра-
бочего цикла можно увеличить проводимость интерфейса дискретной ионизации при
атмосферном давлении по сравнению с традиционными интерфейсами непрерывной
ионизации. Ионы, сопровождаемые воздухом и парами пробы, увеличивают давление
в вакуумной камере. Однако ионы захватываются низковольтным радиочастотным на-
пряжением, тогда как молекулы воздуха и паров пробы откачиваются до достижения
необходимого давления (обычно это занимает 250—500 мс). После этого включается вы-
соковольтное радиочастотное напряжение для последующего масс-анализа. На рис. 9.5
показаны различные стадии цикла сканирования, включая ввод ионов и их анализ
(Gao, 2009). На рис. 9.6 изображен график изменения давления в ходе цикла анализа.
ДИАД был соединен с источниками ионизации в нормальных условиях и источником
с электрораспылением в миниатюрном масс-спектрометре (Gao etal., 2008а). Газообраз-
ные, жидкие и твердые пробы, ионизированные при помощи соответствующих внеш-
них источников ионов, были успешно введены в вакуумную камеру с последующим
масс-анализом.
9.2.2. Методы ионизации
Масс-анализаторы могут определять только отношение массы к заряду. Поэтому они
зависят от эффективности превращения молекул в газофазные ионы. Таким образом,
чувствительность к определенному аналиту определяется эффективностью ионизации
и эффективностью транспортировки ионов (Cech and Enke, 2001; Cole, 2000; Kebarle, 2000;
Kebarle and Tang, 1993; Lin and Sunner, 1994). В зависимости от того, где происходит иони-
зация — в вакууме или в атмосферных условиях, методы ионизации классифицируются
как внутренние или внешние. В миниатюрных масс-спектрометрах для большинства
аналитов требуются внешние методы, за исключением летучих компонентов, которые
могут быть введены посредством мембраны или сорбента.
О новом классе методов, называемых методами ионизации в нормальных условиях,
мы уже упоминали. Отсутствие пробоподготовки делает такие методы идеальными для
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
анализа в полевых условиях, когда условия для классической подготовки проб отсут-
ствуют. В данном разделе будут описаны различные методы ионизации, используемые
для миниатюрных масс-спектрометров. Мы начнем с внутренних методов.
Ионизация электронами
Это один из старейших и наиболее воспроизводимых методов ионизации, который, од-
нако, сейчас используется не столь широко {de Hoffman and Stroobant, 2007; Lamert et aL,
1976). Он состоит в ускорении электронов к анализируемым молекулам, передаче моле-
кулам достаточной для ионизации энергии и неизбежной последующей фрагментации.
Этот процесс происходит в вакууме и, как любой физический процесс, представляю-
щий собой мономолекулярную химическую реакцию, обладает хорошей воспроизводи-
мостью (глава 1).
Традиционно источниками электронов служат резистивно нагреваемые катоды.
Недостатками метода являются короткое время жизни катодов при высоком давле-
нии и высокое потребление энергии. Оба этих фактора несовместимы с эксплуатацией
в полевых условиях. Так как миниатюрные масс-спектрометры типа ионной ловушки
функционируют при достаточно высоких давлениях (в диапазоне мторр), поиск более
надежного метода ионизации оказался жизненно важным. Был предложен источник
с ионизацией электронами в тлеющем разряде (ИЭТР) {Gao et aL, 2007; Smith et al., 2010),
характеризующийся надежностью и низким потреблением энергии. На рис. 9.7 изобра-
жена схема такого источника.
Вакуумная камера (?) Датчик вакуума
m/z
Рис. 9.7. а — Конфигурация источника с тлеющим
разрядом; б — схематическая диаграмма прибора.
Разряд генерирует электроны, которые использу-
ются для ионизации; в — масс-спектр перфтортри-
бутиламина (ПФТБА), полученный на миниатюр-
ном масс-спектрометре с источником с тлеющим
разрядом. (Источник: для {а) и {б) Gao L., SongQ. Y.,
Noll R.J., Duncan J., Cooks R.G. and Zheng O.Y.
(2007). «Glow discharge electron impact ionization
source for miniature mass spectrometers». Journal of
Mass Spectrometry. 42: 675—680. Перепечатано с раз-
решения издательства John Wiley and Sons. Источник
для (e): Gao L., Sugiarto A., Harper J. D., Cooks R.G.
and Ouyang Z. «Design and characterization of a multi-
source hand-held tandem mass spectrometer». Analytical
Chemistry. 80: 7198—7205. Перепечатано с разреше-
ния Американского химического общества)
9.2. Основы конструкции приборов
ЭУ (электронный умножитель)
4000-
3000-
2000-
1000 -
0 =
Ацетон Никотин
59) (tn/z 163)
Бензол
{m/z 78)Толуол
7 {m/z 91)
' /Этилбензол
({m/z Ю5)
ж
40 60 80 100 120 140 160 180 200
m/z
Рис. 9.8. Схематическая диаграмма соединения источника ХИАД с миниатюрным масс-
спектрометром Mini 10.5. Mini 10.5 — вариант масс-спектрометра Mini 10 {Gao
etal., 2006) с усовершенствованным питанием для радиочастотного напряжения.
Прибор разработан для анализа небольших органических молекул. Игла нахо-
дится под напряжением 5 кВ и служит для инициирования коронного разряда.
Анализируемые молекулы ионизируются в потоке газа, вводимого в источник
ХИАД. При соответствующем выбора режима ДИАД ионы вводились в масс-
спектрометр для анализа, (б) Типичный масс-спектр, полученный на приборе
ХИАД/Mini 10.5 для бензола (125 ppb), толуола (150 ppb) и этилбензола (210 ppb)
в присутствии 0,1 % ацетона и 500ppb никотина в воздухе. (Источник: HuangG. М.,
Gao L., Duncan J., Harper J. D., Sanders N.L., Ouyang Z. and Cooks R.G. (2010a).
«Direct detection of benzene, toluene, and ethylbenzene at trace levels in ambient air by
atmospheric pressure chemical ionization using a handheld mass spectrometer». Journal
of the American Society for Mass Spectrometry. 21: 132—135. Рис. 1(a) и 1(d). Copyright
2010 American Society for Mass Spectrometry. Опубликовано Elsevier Inc. с разреше-
ния Springer Science + Business Media В. V.)
Химическая ионизация при атмосферном давлении (ХИАД)
Мы приступаем к обсуждению внешних источников ионизации. Первым бал разрабо-
тан метод химической ионизации при атмосферном давлении {Carrollet al., 1975; Horning
et al., 1973). Процесс ионизации инициируется коронным разрядом, который запускает
серию газофазных ионно-молекулярных реакций с участием молекул аналитов. Очень
острая игла, находящаяся под напряжением в 1—5 кВ, является источником коронного
разряда с силой тока 2—5 мкА. Этот разрядный ток ионизирует атомы и молекулы, нахо-
дящиеся вблизи иглы, которые затем реагируют с молекулами аналитов и ионизируют
их. При работе с положительными ионами ионизация, как правило, обусловлена прото-
нированием, в то время как при работе с отрицательными — депротонированием {Glish
and Vachet, 2003). Анализ методом ХИАД ограничивается полярными и относительно
неполярными молекулами с максимальной массой 1500 Да. Типичное подключение ис-
точника ионов ХИАД к миниатюрному масс-спектрометру показано на рис. 9.8л. До-
полнительную информацию о ХИАД вы можете найти в главах 1 и 3.
Ионизация электрораспылением (электроспрей, ЭРИ)
Электрораспыление как внешний метод ионизации был разработан Джоном Феном
в 1984 году {Fenn et al., 1989; Ymashita and Fenn, 1984)'.
1 Первая работа по использованию этого метода была опубликована коллективом под ру-
ководством Л.Н. Галль (Александров М.Л., Галль Л.Н., Краснов В. Н., Николаев В. И., Пав-
ленко В. А., Шкуров В. А. (1984). Ионная экстракция из растворов при атмосферном давлении:
масс-спектрометрический метод анализа бииорганических соединений. ДАН СССР. 277, 2: 379—
383). — Прим. ред.
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
0
Нано-ЭРИ
Рис. 9.9. (а) Схематическая диаграмма соединения источника наноэлектрораспыления
с миниатюрным масс-спектрометром. Нано-ЭРИ — вариант более популярного
источника ЭРИ. Дифференциация источников основана на диаметре капилляра
и скорости потока через капилляр, (б) Масс-спектр 200 ррт цитохрома С, полу-
ченного с помощью нано-ЭРИ. (Перепечатано с разрешения Американского хи-
мического общества из: Gao L., Sugiarto A., HarperJ.D., Cooks R.G. andOuyangZ.
«Design and characterization of a multisource hand-held tandem mass spectrometer».
Analytical Chemistry. 80: 7198—7205.)
m/z
Этот метод применяется для нелетучих соединений (смотрите также главы 1 и 3).
Небольшой капилляр с внутренним диаметром не более 250 мкм находится под напря-
жением 2—5 кВ. Раствор с пробой ионизируется при протекании через этот капилляр.
Этот метод «мягкой» ионизации практически не вызывает фрагментации.
Были также разработаны модификации электроспрея, а именно микроэлектро-
спрей и наноспрей. Критериями для такой классификации являются диаметр капил-
ляра и скорость потока раствора, однако различия не столь существенны. В типичных
источниках наноспрея используются кварцевые и стеклянные капилляры с диаметром
отверстия не более 10 мкм и потоками не более 100 нл/мин {Glish and Vachet, 2003; Wilm
and Mann, 1996). Типичная стыковка наноэлектроспрейного источника и миниатюрно-
го масс-спектрометра показана на рис. 9.9.
Другим вариантом электрораспыления является ионизация электроакустическим
распылением (ИЭАР). Ионизация акустическим распылением (ИАР) — метод, разра-
ботанный Хирабаяши с коллегами {Hirabayashi et al., 1994; 1995). В этом методе раствор
пробы в водно-метанольном растворе распыляется при атмосферном давлении через
кварцевый капилляр с коаксиальным капилляру потоком газа. При атмосферном дав-
лении в этом варианте образуются заряженные капли и ионы. ИЭАР — очень «мягкий»
метод. В частности, он может быть использован для идентификации нативных белков
по их зарядовым распределениям {Wiseman etal., 2007).
Десорбционная электрораспылителъная ионизация
ДЭРИ — ионизация, протекающая в нормальных условиях, была впервые проде-
монстрирована в 2004 году {Takats et al., 2004). Ключевым достоинством методов ио-
низации в нормальных условиях является отсутствие пробоподготовки. Посколь-
ку, работая в полевых условиях, желательно минимизировать пробоподготовку,
именно ионизация в нормальных условиях идеальна при работе с миниатюрными
масс-спектрометрометрами.
9.2. Основы конструкции приборов
а
Источник питания
высокого напряжения
Ввод в масс-
спектрометр при
атмосферном
Свободно перемещающаяся
на открытом воздухе
подложка с пробой
ДЭРИ, кларитин
Лоратидин
m/z
Рис. 9.10. Диаграмма типично-
го эксперимента с ДЭРИ, в кото-
ром водный раствор направляется
на поверхность изолятора (тефлон).
Десорбированные ионы попадают
в масс-спектрометр через интер-
фейс ввода при атмосферном давле-
нии. ДЭРИ был соединен с миниа-
тюрными масс-спектрометрами.
(б) Спектр таблетки кларитина, по-
лученный на миниатюрном масс-
спектрометре Mini И с источником
ДЭРИ без предварительной пробо-
подготовки. m/z 383 соответству-
ет пику действующего вещества —
лоратидина. (Источник для (а):
Takats Z., Wiseman J. М., Gologan В.
and Cooks R.G. (2004). «Mass
spectrometry sampling under ambient
conditions with desorption electrospray
ionization». Science. 306: 471—473.
Перепечатано с разрешения Аме-
риканской ассоциации продвиже-
ния науки (AAAS). Источник для
(б): Gao L., Sugiarto A., Harper J. D.,
Cooks R.G. and Ouyang Z. «Design
and characterization of a multisource
hand-held tandem mass spectrometer».
Analytical Chemistry. 80: 7198—
7205. Перепечатано с разреше-
ния Американского химического
общества.)
В ДЭРИ пневматический электроспрей направляется на поверхность с пробой.
Процесс включает в себя растворение аналита с последующим образованием вторич-
ных микрокапель. Вторичные капли, сформированные при взаимодействии первичных
заряженных микрокапель электроспрея с нейтральными молекулами аналита, анали-
зируются. Преимуществами ДЭРИ являются быстрота, широкий спектр анализируе-
мых веществ, чувствительность и отсутствие пробоподготовки. Типичное подключение
источника ДЭРИ к лабораторному масс-спектрометру продемонстрировано на рис. 9.10.
Интересная модификация этого метода связана с добавлением вепрей растворителя ре-
агентов для селективной ионизации. Этот метод известен как реакционная ДЭРИ (Chen
et aL, 2006; Wu etal., 2009).
Низкотемпературная плазма (НТП)
Зонд низкотемпературной плазмы — это прибор, работающий в обычных (атмосфер-
ных) условиях, в котором термически активированная десорбция и ионизация достига-
ются при помощи низкотемпературной плазмы (глава 7). Это один из источников ионов,
основанных на ионизации в плазме. Другими примерами являются плазменно-акти-
вированная десорбционная ионизация (Ratcliffe et aL, 2007) и послесвечение в потоке
при атмосферном давлении (Andrade et aL, 2008). Небольшой размер, малое потребле-
ние мощности и потенциал для прямого ввода без предварительной подготовки делают
плазменную ионизацию подходящим методом для портативных масс-спектрометров.
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
Рис. 9.11. (а) Схема источника ионизации в низкотемпературной плазме (НТП), со-
единенного с миниатюрным масс-спектрометром (Mini 10.5). Отсутствие не-
обходимости подготовки пробы, возможность использования для поддержания
плазмы воздуха, низкое энергопотребление и скорость анализа делают низ-
котемпературную плазму предпочтительным методом для работы в полевых
условиях, (б) Калибровочная кривая для детектирования меламина в цельном
молоке с источником НТП, соединенным с миниатюрным масс-спектрометром
Mini 10.5. (Источник: Huang G.M., Xu W., Visbal-Onufrak M.A., Ouyang Z. and
Cooks R. G. (2010). «Direct analysis of melamine in complex matrices using a handheld
mass spectrometer». Analyst. 135: 705—711. Перепечатано с разрешения Королев-
ского Химического Общества)
Стандартный ДИАД-интерфейс, используемый для присоединения источников иони-
зации при атмосферном давлении к масс-спектрометру {Gao et al., 2008а), может быть
модифицирован дополнительными средствами откачки в целях улучшения эффектив-
ности транспортировки ионов. Схема источника низкотемпературной плазмы, присо-
единенного к миниатюрному масс-спектрометру Mini 10.5, изображена на рис. 9.11.
9.2.3. Масс-анализаторы
Масс-анализатор — это блок масс-спектрометра, определяющий отношение массы
к заряду для ионов образца. Также как и остальные узлы масс-спектрометра, масс-
9.2. Основы конструкции приборов
анализатор определяет возможности прибора. Миниатюризация масс-спектрометра
непосредственно связана с миниатюризацией анализатора. Ранние исследования были
сфокусированы на миниатюрных анализаторах, сохраняющих адекватные харак-
теристики и потребляющих энергию на разумном уровне (Kaiser et al., 1991; Sinha and
Tomassian, 1991). Почти все типы анализаторов, за исключением орбитрэпа (Makarov,
2000), были миниатюризированы. Некоторые анализаторы более приспособлены для
миниатюризации. Масс-анализаторы могут быть разделены на два типа: ловушки
и анализаторы, работающие с пучком ионов (Glish and Vachet, 2003). Далее следует крат-
кое описание различных типов анализаторов.
Радиочастотные ионные ловушки
Масс-спектрометры, в которых радиочастотное напряжение используется для захва-
та и анализа ионов, называются радиочастотными ионными ловушками (March and
Todd, 2005). Способность ионных ловушек работать при давлениях, более высоких, чем
остальные анализаторы, делает их особенно ценными для анализа в полевых условиях,
так как более высокое давление предъявляет меньшие требования к вакуумной систе-
ме. Как уже было замечено, идентичность аналита может быть подтверждена с высокой
степенью достоверности с помощью МС/МС. Здесь будут обсуждаться классическая
квадрупольная ионная ловушка (КИЛ) Пауля, а также ее упрощенные модификации:
цилиндрическая и прямоугольная ионные ловушки (ЦИЛ и ПИЛ соответственно).
Квадрупольная ионная ловушка
Квадрупольная ионная ловушка (первоначально в гиперболической геометрии), была
изобретена Вольфгангом Паулем в 1953 году (Pauland Steinwedel, 1953). В КИЛ создается
трехмерное радиочастотное поле, удерживающее ионы. Вдовушке Пауля радиочастот-
ное напряжение для захвата и удерживания ионов прикладывается между торцевыми
и кольцевым электродами. Режим масс-селективной нестабильности, изобретенный
Стаффордом и его коллегами (Stafford et al., 1984), позволил использовать ловушку для
широкого спектра приложений. В зависимости от приложенного электрического поля,
ионы или удерживаются в ловушке, или удаляются из нее. Контроль электрического
поля, обычно с помощью увеличения амплитуды радиочастотного напряжения, позво-
ляет селективно удалять ионы из ловушки в зависимости от отношения их массы к заря-
ду. Ионы, удаленные из ловушки, попадают на детектор, генерируя первичный ионный
сигнал. Эксперименты МС/МС могут быть осуществлены при помощи удаления всех
ионов, за исключением выбранных, с последующей их фрагментацией методом диссо-
циации, индуцированной столкновениями, и детектированием фрагментов с помощью
еще одного сканирования в режиме масс-селективной нестабильности.
Шорт с коллегами (Short et al., 2001а) использовал модифицированную ионную
ловушку Saturn 2000 для подводного мониторинга летучих органических соединений
и растворенных газов. Этот масс-спектрометр содержал квадрупольную ионную ло-
вушку в качестве анализатора. Он весил 68 кг, потреблял мощность в 150 Вт, имел длину
1,35 м и диапазон масс до 650 а. е. м.
Цилиндрическая ионная ловушка
КИЛ имеет более простой аналог — цилиндрическую ионную ловушку, запатентован-
ную Лангмюром в 1962 году как прибор для удерживания ионов. Простота изготовле-
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
ния при достаточной точности и малых габаритах делает ЦИЛ идеальным кандидатом
в анализаторы {Badman et aL, 1998; Kornienko et aL, 1999; Quyang et aL, 1999). Она широко
использовалась в портативных масс-спектрометрах {Patterson etaL, 2002; RiteretaL, 2002;
Yang etal., 2008).
Прямоугольная ловушка
Прямоугольная ионная ловушка (ПИЛ) пришла на смену ЦИЛ {Ouyang et aL, 2004).
Она сочетает в себе большую емкость линейной ловушки (не следует путать двумерную
ловушку с двумерным квадрупольным масс-фильтром; см. далее) и простую геометрию
цилиндрической ионной ловушки. ПИЛ может оптимально функционировать при дав-
лениях выше 1 мторр {Keil et aL, 2008) и имеет в 40 раз большую емкость по сравнению
с цилиндрической ионной ловушкой того же сечения {Ouyanget aL, 2004).
ПИЛ используется как в Mini 10 {Gao et aL, 2006), так и в Mini 11 {Gao et aL, 2008b).
Масс-спектрометр Mini 11 весит примерно 5 кг вместе с батареями, потребляет 30 Вт
электроэнергии и имеет диапазон масс до 2000 Да. Его габариты — 22 х 12 х 18 см. При-
бор использовался для анализа углеводородов {Sokol et aL, 2008), фармацевтических
препаратов {Gao et aL, 2008b), перфторированных компонентов и полициклических
ароматических углеводородов {Sanders et aL, 2010b). Недавно были получены важные ре-
зультаты в режиме МС5 для А-аргинина на миниатюрном масс-спектрометре с прямоу-
гольной ионной ловушкой и источником акустического электрораспыления {Sokolet aL,
2008). Такие эксперименты демонстрируют необычайную специфичность, достигаемую
при использовании в миниатюрных масс-спектрометрах режима МС".
Квадруполъные масс-филътры
Квадрупольные масс-фильтры (КМФ) состоят из 4 стержней, на которые подается
переменное (радиочастотное) и постоянное напряжения. Принцип действия КМФ со-
стоит в пропускании специфичных значений m/z при заданном радиочастотном поле.
Увеличением постоянного и переменного напряжений можно последовательно про-
пускать на детектор ионы различных масс. КМФ — популярный для миниатюрных
масс-спектрометров анализатор. Он использовался как одиночный анализатор {Syms
et aL, 1996; Taylor et aL, 1998), так и в сборке из 9 идентичных анализаторов {Ferran and
Boumsellek, 1996; Orient et aL, 1997).
Шорт с коллегами использовали масс-спектрометр с квадрупольным анализатором
и мембранным вводом как основу для системы весом 33 кг, длиной 105 см, диапазоном
масс до 200 а. е. м. и энергопотреблением в 300 Вт. Этот прибор применялся для детекти-
рования различных растворенных газов, контроля потока сточных вод и изучения вод-
ных масс по глубине.
Линейная ионная ловушка
Линейная ионная ловушка {Douglas et aL, 2005) часто рассматривается как двумерная
версия трехмерной ловушки Пауля. В ней радиочастотный потенциал прикладывает-
ся к квадруполю, а постоянный потенциал — к торцевым электродам для запирания
ионов. Захваченные ионы могут эжектироваться из ловушки в аксиальном {Hager, 2002;
Schwartz et aL, 2002) или радиальном {Douglas et aL, 2005) направлении. Емкость линей-
ной ионной ловушки в 10 раз превышает емкость трехмерной ловушки Пауля. Другим
преимуществом ловушки данного типа является высокая эффективность захвата ионов
при инжекции их извне: более 50% по сравнению с 5 % у ловушки Пауля.
9.2. Основы конструкции приборов 257
Тороидальная ловушка
Миниатюрная версия тороидальной ионной ловушки, изобретенной Ламмертом с кол-
легами (Lammert et al., 2001), может быть описана как линейный квадруполь, согнутый
в окружность (Lammert et al., 2006). Захватывающее поле создается четырьмя электро-
дами: двумя торцевыми и двумя кольцевыми. Уменьшение размера анализатора в ра-
диальном направлении ведет к уменьшению амплитуды рабочего радиочастотного на-
пряжения (1 кВ вместо 15 кВ). Тороидальная ловушка при уменьшенном размере имеет
ту же емкость, что и коммерческая ионная ловушка с кольцевым электродом диаметром
1 см. Ионы в тороидальной ловушке находятся в однородном электрическом поле. Сооб-
щается о полученных на тороидальной ловушке спектрах единичного разрешения для
д-бутилбензола, ксенона и нафталина.
Коммерческий прибор Guardion-7 (Torion Technologies) представляет собой портатив-
ный газовый хроматограф с тороидальной ловушкой (Contreras etal., 2008). Его вес состав-
ляет 13 кг, габариты — 47 х 36 х 18 см, а максимальная потребляемая мощность — 80 Вт.
Ионная ловушка типа Halo
Ионная ловушка типа Halo, изобретенная Остином (Austin et al., 2007), представляет
собой крайне упрощенную версию тороидальной ионной ловушки. Эта ловушка еще
не была миниатюризирована. Она обладает большой емкостью при незначительном
влиянии пространственного заряда. Форма поля вдовушке может быть изменена варьи-
рованием потенциалов, приложенных к кольцевым электродам, что отличает ее от ло-
вушек других типов, для которых для изменения поля необходимо изменение формы
электродов.
Пути развития технологии ионных ловушек и взаимосвязь между ними представле-
ны на рис. 9.12.
Времяпролетные масс-анализаторы
Сейчас мы переходим от ловушек к анализаторам, работающим с пучками ионов, в ко-
торых ионы покидают источник, проходят через поле анализатора и достигают детек-
тора. В миниатюрных масс-спектрометрах используются времяпролетные анализаторы
(ВПА) (Cornish and Cotter, 1997). Из названия анализатора следует, что ионы разделяются
по их временам пролета. Ионы ускоряются разностью потенциалов и пролетают опреде-
ленное расстояние. Время, которое они затрачивают, и есть их время пролета, которое
позволяет различить ионы с разными массами (глава 1). Характеристики времяпролет-
ных приборов строго зависят от пути пролета и потенциалов на этой дистанции. Умень-
шение пути пролета и неоднородности электрического поля снижают разрешение при-
бора по массе. ВП-масс-анализаторы обычно обладают широким диапазоном масс.
На миниатюрном масс-спектрометре с МАЛДИ-источником был выполнен анализ бел-
ков массой до 50000 Да (рис. 9.13, Ecelbergeret al., 2004). Подробности об энергопотребле-
нии и весе данного прибора неизвестны.
Шимма и его коллеги (Shimma et al., 2010) недавно разработали миниатюрную вер-
сию многооборотного масс-спектрометра MULTUM-SII с неограниченным путем про-
лета ионов. Масса прибора составляет 35 кг, а габариты — 50 х 57 х 30 см. При разделении
парниковых газов диоксида углерода СО2 и закиси азота N2O, различающихся по массе
лишь на 0,0113 Да, разрешение составило 31600. Заявлен динамический диапазон при-
бора в 3 порядка.
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
3-мерная цилиндрическая
3-мерная гиперболическая
2-мерная прямоугольная
2-мерная торроидальная
2-мерная типа Halo
Рис. 9.12. Эволюция квадрупольных ионных ловушек в сторону упрощения конструк-
ции и увеличения емкости. (Ouyang Z. and Cooks R.G. (2009). «Miniature mass
spectrometers». Annual Review of Analytical Chemistry. 2: 187—214. Copyright 2009 by
Annual Reviews, Inc. Reproduced with permission of Annual Reviews, Inc. Image of
the toroidal ion trap: reprinted from International Journal of Mass Spectrometry, 212,
Lammert S. A., Plass W. R., Thompson С. V. and Wise M. B. Design, optimization and
initial performance of a toroidal rf ion trap mass spectrometer, 25—40, Copyright (2001),
with permission from Elsevier. Image of the halo ion trap: reprinted with permission
from Austin D. E., Wang M., Tolley S. E., Maas J. D., Hawkins A. R., Rockwood A. L.,
Tolley H.D., Lee E.D. and Lee M.L. «Halo ion trap mass spectrometer». Analytical
Chemistry. 79: 2927—2932. Перепечатано с разрешения Американского химиче-
ского общества)
Источник
+ 10 кВ
Пространство
лазер
Рис. 9.13. Миниатюрный времяпролетный анализатор с миниатюрным рефлектроном,
использующийся для регистрации спектров пептидов и белков при анализе
с помощью МАЛДИ. (Перепечатано из технического дайджеста лаборатории
прикладной физики Джона Хопкинса. Suitcase TOF: A man-portable time-of-flight
mass spectrometer, 25(1). # The Johns Hopkins University Applied Physics Laboratory)
9.2. Основы конструкции приборов
SIMION 6.0
CDFMS Geometry
Катод \
Рис. 9.14. Модельные вычисления CDFMS. Diaz J.A., Giese С. F. and Gentry W. R. «Sub-
miniature ExB sector-field mass spectrometer». Journal of the American Society for Mass
Spectrometry. 12: 619—632, Figure 5. Опубликовано Elsevier Science Inc. с разреше-
ния Springer Science + Business Media В. V.)
Секторные анализаторы
Традиционные секторные анализаторы отличаются высоким разрешением и высокой
точностью измерения массы. Эти характеристики достигаются на приборах с двойной
фокусировкой, в которых электрический и магнитный анализаторы расположены по-
следовательно, и позволяют компенсировать дисперсии по скорости. Таким образом,
достигается одновременная фокусировка по скорости и координатам (Badman and Cooks,
2000). Рис. 9.14 и 9.15 иллюстрируют два элемента двух различных масс-анализаторов
Рис. 9.15. Ионные траектории в циклоидальном фокусирующем анализаторе. (Пере-
печатано с разрешения издательства Elsevier из Trends in Analytical Chemistry,
21, Hemond H. and Camilli R., NEREUS: engineering concept for an underwater mass
spectrometer, 526—533)
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
с двойной фокусировкой: компактный масс-спектрометр с двойной фокусировкой,
разработанный Диасом и его коллегами (Diaz et al., 2001b) (рис. 9.14), и циклоидальный
масс-анализатор (Hemond and Camilli, 2002) (рис. 9.15).
9.2.4. Вакуумные системы
Масс-спектрометрический анализ должен проводиться в условиях высокого вакуума,
причем уровень вакуума определяет максимальное разрешение прибора. Вакуумная
система потребляет значительную мощность, а ее элементы — одни из самых тяжелых
узлов прибора. Так как потребляемая мощность и вес миниатюрного масс-спектрометра
ограничены, чрезвычайно важен тщательный выбор вакуумной системы с учетом ком-
промисса между характеристиками прибора и требованиями к его весу и мощности.
Различные типы насосов, применяемые в миниатюрных приборах, включают турбо-
молекулярные, форвакуумные и ионные. Одной из стратегий, применяемых для мини-
атюрных масс-спектрометров, является использование форвакуумного насоса только
для предварительной откачки системы перед отправкой прибора в «поле». При этом
в полевых условиях используются лишь легкие ионные насосы. Такая стратегия была
предложена Хембергером и воплощена в портативном масс-спектрометре типа «ионная
ловушка» (Yangetal., 2008). Хотя такая идея помогает снизить вес системы, вакуум может
поддерживаться лишь в течение короткого периода времени.
Далее мы рассмотрим вакуумные системы автономно работающих миниатюрных
масс-спектрометров. Первая версия миниатюрного масс-спектрометра Mini 5, пред-
ставляющего собой цилиндрическую ионную ловушку, была разработана в Универси-
тете Пердью (Patterson etal., 2002). В ней в качестве форвакуумного насоса использовался
четырехстадийный диафрагменный насос 813,4 ANDC (KNF Neuberger) со следующими
характеристиками: минимальное давление 400 мторр; скорость откачки 13 л/мин; габа-
риты 14 х 14x30 см; потребляемая мощность 15 Вт. Турбомолекулярным насосом слу-
жил ТРН/1КУ1\ (Pfeiffer) со скоростью откачки 60 л/с. Прибор Mini 8 (Laughlin etal., 2005)
также представлял собой цилиндрическую ионную ловушку и содержал трехстадийную
дифференциально откачиваемую вакуумную систему, обеспечивающую соединение
со многими источниками ионов, работающими при атмосферном давлении. Первая ва-
куумная стадия обеспечивалась небольшим роторным насосом (SD 2021, Alcatel Vacuum
Products, Inc., Hingham, MA, USA) соскорстью откачки 18 м3/ч при минимальном остаточ-
ном давлении 700 мторр. Вторая и третья стадии обеспечивались двумя турбомолеку-
лярными насосами AlcatelАТН-ЗЛ со скоростью откачки 30 л/с (для каждого из насосов).
Несколько стадий Холвека позволяли функционировать турбомолекулярным насосам
с высоким коэффициентом сжатия, что обеспечивало их работу при большем давлении
в выхлопе (до 30 торр), чем у большинства других турбомолекулярных насосов. Данное
обстоятельство позволило использовать в качестве форвакуумных насосов небольшие
двухстадийные диафрагменные насосы A^84.0 (KNFNeuberger, Trenton, NJ, USA). Базовое
давление на второй и третьей стадиях составляло 4 мторр и 2 • 10-5 торр, соответственно.
Новейшие миниатюрные приборы типа прямоугольных ионных ловушек могут функ-
ционировать при значительно более высоких давлениях. В Mini 10 (Gao et al., 2006) в ка-
честве форвакуумного насоса для поддержания давления ниже 2 торр использовался
двухстадийный диафрагменный насос (1091 -7V84.0-8.99, KNF Neuberger) со скоростью
откачки 5 л/мин. Этот насос весил 1,35 кг и непрерывно потреблял мощность в 10 Вт.
9.2. Основы конструкции приборов
Турбомолекулярный насос Pfeiffer TPDQ\ 1 (Pfeiffer Vacuum, Inc., Nashua, NH, USA) исполь-
зовался в качестве основного насоса и позволял поддерживать давление ниже 10-5 торр.
В новейшем миниатюрном приборе серии ионных ловушек Mini 11 (Gao et al., 2008b)
применялся двухстадийный диафрагменный насос 1220-TV84.0-9.00 (KNFNeuberger, Inc.,
Trenton, NJ, USA) весом в 0,8 кг, потребляемой мощностью в 6 Вт, давлением на выхлопе
3 торр и скоростью откачки 5 л/мин. Новый турбомолекулярный насос компании Сгеаге
Inc. (Hanover, NH, USA), поддерживаемый диафрагменным насосом, использовался для
откачки камеры прибора. Этот цилиндрический насос имел длину 10,5 см и диаметр
55 см, весил только 0,5 кг и работал с частотой вращения примерно 100000 об./мин, обе-
спечивая предельное давление в 10-6 торр и потребляя мощность в 10,5 Вт.
Шорт и его коллеги в приборе для подводного химического анализа (Short et al.,
2001а) применили насос V1QLP MacroTorr (Varian Vacuum Products, Lexington, MA, USA)
совместно с двумя соединенными последовательно диафрагменными насосами 7V84.0-
11.98 (KNF Neuberger, Trenton, NJ, USA). Оба форвакуумных насоса питались от посто-
янного напряжения в 24 В и потребляли мощность в 45 Вт. Эти насосы обеспечивали
выходное давление для насоса И70£Рв 1 торр. При использовании мембранного интер-
фейса ввода пробы при включенном потоке жидкости через мембрану давление в масс-
спектрометре составляло 10-5 торр. Диас и его коллеги в их масс-спектрометрической
системе для анализа вулканических газов использовали высоковакуумный турбомоле-
кулярный насос Alcatel АТН 30+ и форвакуумный насос AWT 84.4. Они смогли достичь
давления в диапазоне 10-5 торр (Diaz et al., 2010). В ГХ/МС-системе Guardian-1 от Torion
Technologies (Contreras et al., 2008) использовался двухстадийный диафрагменный насос
модели Pt/1781-84.0-8.05 (KNFNeuberger, Trenton, NJ, USA) и миниатюрный турбомолеку-
лярный насос (модель TPD Oil, Pfeiffer Vacuum, Inc., Nashua, NH, USA) co скоростью от-
качки 11 л/с при предельно достижимом давлении 5 • 10-4 торр.
9.2.5. Детекторы
Ионы после разделения покидают анализатор и достигают детектора, генерирующего
сигнал как отклик на ионный ток. Ток на детекторе обычно является мерой уровней
определяемых аналитов. В данном разделе будут рассмотрены детекторы различных
типов.
Цилиндр Фарадея
В простейшем виде цилиндр Фарадея представляет собой металлический цилиндр,
о который ударяются ионы. Ионы в процессе соударения нейтрализуются благодаря по-
тере или присоединению электрона. Ток протекает через резистор, образующий элек-
трическую цепь. Ток через резистор является мерой интенсивности ионного сигнала.
Очевидно, что цилиндр Фарадея является детектором с малым коэффициентом усиле-
ния. Камилли и Хеммонд (Camilli and Hammond, 2004), а также Шорт (Short et al., 2001а),
в своих масс-спектрометрах для подводного анализа использовали в качестве детекто-
ров цилиндры Фарадея.
Электронные умножители
Наиболее распространенными детекторами являются электронные умножители. В них
пучок ионов соударяется с поверхностью (динодом-конвертером) с генерацией вторич-
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
ных электронов (несколько электронов на один ион). Эти вторичные электроны соударя-
ются в свою очередь с поверхностью следующего динода с генерацией новых электронов
(опять же с небольшим коэффициентом усиления сигнала). Такой процесс повторяется
последовательно, и число электронов на каждой последующей стадии усиления воз-
растает. На конце детектора достигается коэффициент усиления обычно в пределах
106—108 электронов на один исходный ион (KoppenaaletaL, 2005). Полученный на выходе
из детектора сигнал может быть снова усилен дополнительными электронными устрой-
ствами и записан системой регистрации.
Диноды могут быть как дискретные, так и непрерывные. В первом случае прибор
известен как электронный умножитель с дискретными динодами, а во втором — как
электронный умножитель с непрерывным динодом (динод представляет собой поверх-
ность с высоким коэффициентом вторичной электронной эмиссии).
Разновидностью электронного умножителя непрерывного типа является многока-
нальная пластина. В таком детекторе в плоской пластине, изготовленной из материала
с достаточно высокой эмиссией вторичных электронов, с помощью фотолиграфической
технологии формируются микроканалы. Эти каналы имеют диаметр в несколько десят-
ков микронов и длину в несколько миллиметров (Koppenaal et aL, 2005). Принцип дей-
ствия такого детектора такой же, как и у умножителя с непрерывным динодом: ионы
соударяются с поверхностью с высокой эмиссией и генерируют вторичные электроны.
Коэффициент усиления составляет ~105 на один канал. Для одной пластины достигает-
ся усиление 102—104, а для сдвоенной сборки (шевронного типа) — 108.
Масс-спектрометр, способный детектировать отрицательные ионы, обеспечива-
ет лучшую специфичность, а следовательно, и более низкие пределы обнаружения для
определенных компонентов. Важные с точки зрения мониторинга окружающей среды
соединения часто детектируются в режиме отрицательных ионов более эффективно. Это,
например, нитросодержащие взрывчатые вещества, хлор- и фосфорорганические пести-
циды, галогеносодержащие соединения. Авторы этой главы работали с отрицательными
ионами на приборах Mini 10 (Sanders et aL, 2010a) и Mini 11. В этих случаях между масс-
анализатором и умножителем был помещен конвертирующий динод. Отрицательные
ионы соударяются с конверсионный динодом с генерацией вторичных положительных
и отрицательных ионов, а также электронов. Положительные ионы притягиваются к пе-
редней части электронного умножителя, который поддерживается под отрицательным
потенциалом (—1,4 кВ). Затем сигнал от этих положительных ионов усиливается через ка-
скад вторичных электронов в электронном умножителе. Преимущество такой конструк-
ции заключается в возможности регистрации и положительных и отрицательных ионов
с помощью одного и того же детектора путем правильного подбора напряжений на его
элементах. Более того, такая конструкция благодаря эффекту усиления на конвертирую-
щем диноде обеспечивает улучшенный сигнал и для положительных ионов.
9.3. Экологические приложения для миниатюрных
масс-спектрометров
В этом разделе будет описано широкое применение миниатюрных масс-спектрометров
для решения актульных проблем. Здесь мы опишем, в частности, аспекты, касающиеся
9.3. Экологические приложения для миниатюрных масс-спектрометров
мониторинга окружающей среды. При сравнении миниатюрных приборов с лаборатор-
ными необходимо учитывать разницу в их габаритах.
9.3.1. Подводная масс-спектрометрия для регистрации летучих
органических соединений (ЛОС) и растворенных в воде
газов
Изучение водных сред включает исследования протекающих в них биогеохимических
процессов, а также техногенного влияния человека. Характеризация этих химически
сложных экосистем является непростой задачей, требующей тщательных измерений,
так как они не были достаточным образом изучены ни пространственно, ни во времени.
Чувствительность масс-спектрометров к широкому ряду аналитов делает их удобными
для решения данной задачи и позволяет избежать необходимости использования мно-
гочисленных сенсоров, каждый из которых чувствителен только к одному или несколь-
ким соединениям. Хотя масс-спектрометры имеют много преимуществ вследствие их
универсальности, существует несколько проблем, связанных с их использованием: лю-
бая конструкция должна выдерживать огромные гидростатические давления под водой
и необходимость поддержания вакуума (для масс-спектрометрического анализа) при
таких условиях.
Интенсивные параллельные (но независимые) исследования были проведены Шор-
том и его коллегами (Belletal., 2007; Kibelka etal., 2004; Shortetal., 2000; 2001a; 2001b; 2006)
и Хеммондом с коллегами (Camilli and Hemond, 2004; Hemond and Camilli, 2002; Hemond
et al., 2008), которые сконструировали масс-спектрометры для подводных исследова-
ний. Группа Шорта использовала два различных масс-анализатора: квадрупольный
масс-фильтр с верхним пределом по массе в 200 Да (чувствительность для разных ком-
понентов от 1 до 5 ppb) и квадрупольную ионную ловушку с верхним пределом масс
650 Да и пределами обнаружения, меньшими 1 ppb (Kibelka et al., 2004; Short et al., 2001a).
Хеммонд и Камилли, в свою очередь, использовали циклоидальный масс-спектрометр
с диапазоном масс от 2 до 150 (Camilli and Hemond, 2004). МакМёртри и Смит (McMurtry
and Smith, 2001) изготовили другой масс-спектрометр для подводного анализа с вводом
образца посредством электрораспыления или мембраны и масс-анализатором с враща-
ющимся полем.
Масс-спектрометр с мембранным вводом и квадрупольным масс-анализатором
Шорта имел длину 1,4 м, вес 33 кг и потреблял мощность 100 Вт. Прибор типа квадру-
польной ионной ловушки имел длину 1,35 м, вес 69 кг и портеблял 150 Вт. Полученные
данные записывались в бортовой компьютер масс-спектрометра. Контроль в режиме
реального времени и визуализация собранных данных достигались посредством прово-
дного или беспроводного Интернет-соединения. Масс-спектрометры, оборудованные
этими анализаторами, были развернуты на нескольких платформах — автономных под-
водных платформах (АПП) (Kibelka et al., 2004), дистанционно управляемых платфор-
мах (Fries et al., 2001), надводных платформах (Wenner et al., 2004) и на причалах (Short
etal., 2001b).
Например, квадрупольный фильтр масс использовался для мониторинга распростра-
ненныхлетучихорганическихсоединений веточных водах водоочистной установки после
того, как вода подверглась предварительной фильтрации (Kibelka etal., 2004). Мониторинг
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
проводился в течение 30 ч., что свидетельствует об устойчивой работе масс-спектрометра.
На рис. 9.16 показаны спектры толуола и хлороформа (побочного продукта дезинфекции).
Масс-спектрометр с квадрупольным фильтром также использовался для изучения химии
растворенных газов вблизи гидротермальных источников (Kibelka etal., 2004). Контроли-
ровались следующие газы: азот, кислород, углекислый газ и аргон. Мониторинг позволил
обнаружить обогащение гидротермальных вод углекислым газом и обеднение кислоро-
дом. Прибор мог быть быстро установлен водолазами. На рис. 9.17 представлены результа-
ты определения растворенных газов, полученные с помощью этого прибора.
Загрязнение ЛОС из выхлопов моторных лодок контролировалось подводной ион-
ной ловушкой, расположенной на носу автономной подводной платформы в гавани с ин-
Время
Рис. 9.16. Анализ с помощью подводного линейного квадрупольного масс-спектрометра,
помещенного в резервуар со сточными водами в Санкт-Петербурге (США).
(а) Данные мониторинга по времени для иона с m/z 83, характеристичного
для хлороформа, побочного продукта дизенфекции. (б) Данные мониторинга
по времени для иона с m/z 91, характеристичного для толуола. Данные демон-
стрируют высокую надежность системы. (Перепечатано с разрешения издатель-
ства Elsevier из: Taianta, 64, Kibelka G. Р. G., Short R.T., Toler S. K., Edkins J. E. and
Byrne R. H. Field-deployed underwater mass spectrometers for investigations of transient
chemical systems, 961—969.)
9.3. Экологические приложения для миниатюрных масс-спектрометров
Рис. 9.17. Анализ растворенных газов с помощью квадрупольного масс-спектрометра
в воде геотермального источника в мелководной бухте Мексиканского залива.
Прибор может быть легко установлен водолазами. Контролировались следую-
щие соединения: N2 (m/z 28), О2 (m/z 32), Аг (m/z 40) и СО2 (m/z 44). Данные по-
зволяют делать выводы о химических процессах в воде, например об уменьше-
нии концентрации кислорода по сравнению с концентрацией углекислого газа.
(Перепечатано с разрешения издательства Elsevier из: Taianta, 64, Kibelka G. Р. G.,
Short R.T., Toler S. K., Edkins J. E. and Byrne R. H., Field-deployed underwater mass
spectrometers for investigations of transient chemical systems, 961—969)
тенсивным движением моторных судов. В проведенном исследовании ловушка работала
непрерывно на протяжении 72 часов, в результате чего были получены временные рас-
пределения ЛОС. В другом случае квадруполь был установлен на надводной платформе.
С помощью сочетания географической информации, полученной с помощью GPS (global
positioning system, системой глобального определения координат), с химической инфор-
мацией, полученной от квадрупольного масс-спектрометра, была составлена географи-
ческая карта распределения ряда химических веществ (Wenner et aL, 2004). Нанесение
этих данных на карту DOQQ (Digital Orthographic Quarter Quad, эти карты являются циф-
ровым изображением местности, сделанным с помощью аэрофотосъемки Геологиче-
ским топографическим агентством США с разрешением в 1 м) дает поверхностное рас-
пределение химических веществ в конкретном районе. На рис. 9.18 вы можете видеть
пример такой карты для распределений толуола и диметилсульфида.
Другое возможное приложение этого метода — мониторинг концентраций раз-
личных соединений в водоеме. Например, могут измеряться концентрации кислорода
и углекислого газа. Такая информация может быть использована для понимания про-
странственных изменений, касающихся фотосинтеза и дыхания организмов в водоемах.
Подводный масс-спектрометр нашел также применение для изучения распределения
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
Рис. 9.18. Иллюстрация пространственного распределения ЛОС от выхлопов мото-
ров судов в Bayboro Harbor, Санкт-Петербург, Флорида. Результаты нанесены
на DOQQ-карту. На карте GPS, совмещенной с результатами анализа, по ионам
с (a) m/z 91 (фрагмент толуола) и (б) с m/z 62 (вероятно, принадлежащему диме-
тилсульфиду) виден след от управляемого судна. Данные получены с высокой
плотностью, что было бы трудно сделать другими методами. То есть метод может
использоваться для мониторинга загрязнений. Перепечатано с разрешения из-
дательства Elsevier из: Trends in Analytical Chemistry, 23, Wenner P.G., Bell R.J.,
Van Amerom F. H. W., Toler S. K., Edkins J. E., Hall M. L., Koehn K., Short R.T. and
Byrne R.H., Environmental chemical mapping using an underwater mass spectrometer,
288-295)
растворенных газов. Концентрация кислорода была измерена в зависимости от глуби-
ны и хорошо согласовывалась с данными, полученными при помощи измерения про-
водимости, температуры, сенсоров глубины и уровня кислорода (Belief al., 2007). Такие
данные поддерживают идею широкого применения подводных масс-спектрометров.
Усилия по созданию подводного масс-спектрометра были предприняты и Массачу-
сетским технологическим институтом (МТИ). Разработанный Хеммондом и Камилли
NEREUS (novel, efficient, rapid evaluation of underwater spectra', новый, эффективный с бы-
строй обработкой подводных спектров) (Camilli R. and Hemond Н. F., 2004) — автономный
подводный масс-спектрометр, установленный на автономную подводную платформу
(АПП), был соединен с коммуникационной сетью для обеспечния химического анализа
в режиме реального времени. Его вес составляет 22 кг, а потребляемая мощность — 20 Вт
(в ждущем режиме менее — 2 Вт). Он используется для детектирования метаболических
газов и легких углеводородов. Прибор оборудован циклоидальным масс-анализатором,
источником ионов с ионизацией электронами и мембранным вводом. Рис. 9.19 иллю-
стрирует распределение кислорода по глубине, полученное при погружении платформы
на глубину до 18 м. Как видно из рисунка, данные, полученные в ходе экспедиции при-
9.3. Экологические приложения для миниатюрных масс-спектрометров
Время эксперимента, с
Рис. 9.19. Измерение уровня растворенного кислорода по глубине масс-спектрометром
NEREUS (внизу) и многозондовым методом (вверху) в ходе эксперимента на ав-
тономной подводной платформе. Хорошее согласие между двумя сигналами
очевидно. Небольшой локальный максимум на профиле, полученном много-
зондовым методом, — скорее всего артефакт вследствие дрейфа в температурно-
компенсационном контуре. (Из: Hemond Н. F., Mueller А. V. and Hemond М. «Field
testing of lake water chemistry with a portable and an AUV-based mass spectrometer»,
Journal of the American Society for Mass Spectrometry, 19, 1403—1410, Figure 6. Опубли-
ковано Elsevier Science Inc. с разрешения Springer Science + Business Media В. V.)
бором NEREUS и универсальным зондом, установленным на АПП, хорошо согласуют-
ся друг с другом (Hemond et al., 2008). Подобные эксперименты проводились также для
анализа распределения по глубине метана и азота. Такие исследования демонстрируют
способность масс-спектрометра анализировать растворенные в воде газы.
В будущем обе группы предусматривают оснащение многочисленных автономных
платформ портативными масс-спектрометрами, котрые будут служить как сеть для не-
прерывного мониторинга водной среды.
9.3.2. Мониторинг вулканических выбросов для контроля
вулканической активности
Вулканы несут потенциальные угрозы не только для окружающей их местности, но и
для атмосферы в целом и могут влиять на изменения глобального климата. Примером
этого было недавнее извержение вулкана в Исландии, когда следы выброшенного пепла
регистрировались на расстоянии до 3000 км. Вулканом были выброшены следующие
газы: водяной пар (Н2О), углекислый газ (СО2), диоксид серы (SO2), сероводород (H2S),
водород (Н2), пары соляной (НС1) и плавиковой (HF) кислот, метан (СН4) и угарный
газ (СО). Также был зарегистрирован выброс небольших количеств ртути (Hg), сероо-
киси углерода (COS), сероуглерода (CS2) и бромистоводородной кислоты (НВг). Диок-
сид серы, выброшенный в атмосферу, реагирует с парами воды с образованием серной
кислоты, разрушающей озоновый слой. Кроме того, диоксид серы блокирует проник-
новение на Землю ультрафиолетового излучения, что также влияет на климат, вызывая
глобальное похолодание. Выброшенный в больших количествах углекислый газ (парни-
ковый газ) способствует глобальному потеплению. Приведенные примеры показывают
значимость мониторинга вулканических газов.
Извержение может быть предсказано с помощью мониторинга эмиссии газов и ана-
лиза сейсмических данных. Мониторинг должен проводиться для достаточно большого
числа соединений непосредственно на месте, быть непрерывным и высокочувствитель-
ным. В настоящее время для таких задач используются электрохимические сенсоры, ко-
торые имеют ограничения по числу детектируемых веществ. Масс-спектрометрия пред-
ставляет собой более универсальный способ мониторинга вулканических газов ввиду
ее применимости к детектированию различных соединений. Кроме того, с помощью
масс-спектрометров можно детектировать гелий — важный предшественник вулкани-
ческой активности, который нельзя регистрировать другими методами, используемыми
вулканологами.
Внедрением масс-спектрометров в вулканологию интенсивно занимались Диас
и его коллеги (Arkin et aL, 2004; Diaz et al., 2001a; Diaz et aL, 2001b; Diaz et aL, 2002; Diaz et aL,
2010; Griffin et aL, 2008). Первые эксперименты были сделаны с коммерческим квадру-
польным масс-спектрометром весом 35 кг, потребляющим мощность в 250 Вт, диапазо-
ном масс от 1 до 300 Да при единичном разрешении (Diaz et aL, 2002), и оборудованным
источником ионов с ионизацией электронами. Для защиты прибора от опасных вулка-
нических газов использовался ввод через капилляр, разделенный на две секции. Пер-
вая была длиной 90 см и диаметром 125 мкм. Она понижала давление от атмосферного
до 1 мторр. Вторая имеладлину 3 м и диаметр 1,6 мм. Этот капилляр погружался в кратер
вулкана. Масс-спектрометр был установлен на вулкане Poas Vulcano и на вулкане Kilauea
на Гавайских островах для детектирования наиболее распространенных вулканических
газов. Первые эксперименты были успешны и хорошо коррелировали с данными назем-
ных обсерваторий (DiazetaL, 2002). Этот прибор использовался для регистрации ионов-
предшественников. Однако его применение было ограничено вследствие его массив-
ности. Научная группа перешла на конструирование более легких масс-спектрометров.
Исследователи Космического центра Кеннеди (КЦК) разработали портативный
масс-спектрометр для мониторинга атмосферных газов на высоте до 13 км над уровнем
моря. Такой прибор рассматривался как первый шаг в разработке приборов для монито-
ринга воздуха на Международной космической станции и, в частности, контроля опас-
ных газов вокруг космического шаттла перед его запуском (Arkin etal., 2004). Совместная
работа ученых из КЦК и группы Диаса (Griffin et aL, 2008) была посвящена мониторингу
газов непосредственно в кратере вулкана, а также газов вокруг него. Первоначально ис-
пользовался линейный квадруполь (вес 47 кг, диапазон масс 1—200 а.е. м.), названный
бортовым масс-спектрометром для контроля вулканической эмиссии. Он был установ-
лен на исследовательском самолете 1^5-57. Система ввода пробы такого прибора должна
9.3. Экологические приложения для миниатюрных масс-спектрометров
Рис. 9.20. Трехмерная карта концентрации воды (в относительных единицах, пА) вокруг
вулканов Irazu и Turrialba в Коста-Рике (географическое положение показано
на вставке), измеренной квадрупольным масс-спектрометром, установленным
на самолете Cessna Soloy 206. Обратите внимание, что более высокая концен-
трация в эмиссионном факеле находится ниже кальдеры и спускается в доли-
ну между двух вулканов. Эмиттируются также другие газы: диоксид углерода,
диоксид серы, сероводород и другие токсичные соединения. Польза от таких
трехмерных карт концентрации очевидна. (Из: Griffin Т. Р., Diaz J. A., Arkin С. R.,
Soto С., Curley С. Н. and Gomez О., «Three-dimensional concentration mapping of
gases using a portable mass spectrometer system», Journal of the American Society for
Mass Spectrometry, 19, 1411—1418, Figure 3. Опубликовано Elsevier Science Inc.
с разрешения Springer Science + Business Media В. V.)
функционировать в широком диапазоне температур и давлений, соответствующем на-
бору высоты от уровня моря до 44000 футов (13400 м). Конструкция такого интерфейса
имела решающее значение. Он состоял из двух диафрагм с дифференциальной откачкой
между ними. На этом масс-спектрометре был достигнут предел обнаружения в несколь-
ко ррт. Слегка модифицированная и более легкая модель такого прибора была исполь-
зована в Сан-Хосе (Коста-Рика), окруженном тремя вулканами: Pods, Irazu и Turrialba.
Масс-спектрометр, установленнный на самолете Cessna Soloy 206, обеспечивал про-
странственное разрешение, в 5 раз лучшее, чем ^^-57. Масс-спектрометрические дан-
ные были объединены с данными GPS и результатами измерений удаленными сен-
сорами, а затем отображены на картах в целях получения трехмерного изображения
вулканической эмиссии. В основном, мониторинг проводился для гелия, углекислого
газа, диоксида серы и водяного пара. На рис. 9.20 показан сигнал для водяного пара
около вулканов Irazu и Turrialba, полученный при облете самолета вокруг этих вулка-
нов. Для наземных измерений масс-спектрометр был установлен на 51/К (внедорожник)
в целях получения карты загрязнения окрестностей Сан-Хосе. Это еще одно приложе-
ние для портативного прибора (Griffin etal., 2008).
Последней разработкой группы Диаса был масс-спектрометр ULISSES (utilisation of
in situ instrumentation and remote sensing for the study of gaseous emissions at active volcanoes', ис-
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
пользование локального прибора и удаленных сенсоров для изучения газовой эмиссии
активных вулканов) весом в 10 кг и диапазоном масс от 1 до 100 а.е.м. (Diaz et al., 2010).
Этот прибор был использован до и после извержения вулкана Turrialba в начале января
2010 года. Ученые обнаружили изменение концентрации гелия в этот промежуток вре-
мени — от находящейся ниже предела обнаружения (несколькоррт) до 20ррт непосред-
ственно перед извержением, что подтверждает характеристичность этого газа как ин-
дикатора вулканической активности. Этот прибор также применялся для калибровки
и поверки спутниковых сенсорных систем удаленного мониторинга. Предполагается,
что прибор будет использоваться на недорогих беспилотных самолетах для мониторин-
га, калибровки и поверки. Это поможет избежать риска воздействия на пилота вулкани-
ческого извержения (Diazetal., 2010).
9.3.3. Детектирование перфторированных соединений
Галогенорганические соединения — класс веществ, которые загрязняют воздух, воду
и почву и которые желательно контролировать в этих средах. Эксперименты проводи-
лись с использованием масс-спектрометра Mini 10 с ПДМС-мембраной и источником
ионизации электронами. Пентафторанизол (C7H3F.O) служил модельным компонентом
как представитель класса поли- и перфторированных соединений. На рис. 9.21 пред-
ставлены МС- (9.21д) и МС/МС- (9.21 б) спектры. В тандемном спектре видны ион-пред-
Рис. 9.21. Анализ пентафторанизола на масс-спектрометре Mini 10. Пентафторанизол —
представитель многочисленного класса перфторированных и полифториро-
ванных компонентов, (а) Масс-спектр пентафторанизола, полученный с ис-
точником ионизации электронами, (б) Тандемный спектр пентафторанизола.
На вставке показаны основные фрагменты. (Перепечатано с разрешения IM
Publications LLP из European Journal of Mass Spectrometry. 16, 11—20, 2010)
9.3. Экологические приложения для миниатюрных масс-спектрометров
шественник (m/z 198) и ионы-продукты с m/z 183 и 167. Вставка на рис. 9.216 иллюстри-
рует молекулярную структуру иона-предшественника и разрывы связей, приводящие
к иону-продукту. Предел обнаружения составил 9 ppb, а линейный диапазон в интервале
от 9 до 100 ppb демонстрирует способность прибора Mini 10 детектировать фторирован-
ные соединения на уровне в несколькоppb. Спектры, полученные при помощи источни-
ка с ионизацией электронами, сравнимы со стандартными. Как было упомянуто ранее,
МС/МС улучшает специфичность метода. Этот вывод подтверждает детектирование
и идентификацию пентафторанизола на низких уровнях.
9.3.4. Анализ углеводородов
Экспрессная регистрация летучих органических соединений (ЛОС) является важной
задачей мониторинга окружающей среды с точки зрения охраны здоровья населения,
принимая во внимание их возможное негативное влияние. Примером может служить
ароматический углеводород бензол, который даже при воздействии в низких концен-
трациях ассоциируется с такими последствиями для здоровья человека, как лейкемия,
пластичная анемия и дефекты костных тканей (Hayes et al., 1997, Rin sky et al., 1987). Де-
тектирование углеводородов как в газообразном, так и в жидком виде возможно с по-
мощью масс-спектрометрии.
Анализ водной фазы
Портативные масс-спектрометры могут быть использованы для решения сложных за-
дач контроля загрязнения воды углеводородами при разливе нефти в море (Camilli and
Duryea, 2007; Camilli et al., 2010). Недавно такой подход использовался для решения та-
кой сложной задачи, как исследование разлива нефти из скважины Л/С252 Macondo
компании Deepwater Horizon в Мексиканском заливе (Camilli etal., 2010). Эта крупнейшая
в истории катастрофа была исследована с помощью масс-спектрометра с мембранным
вводом TETHYS (tethered yearlong spectrometer) (Camilli and Duryea, 2007) с диапазоном масс
от 1 до 200 а.е.м., потребляемой мощностью 25 Вт. Прибор весит 13 кг и обеспечивает
предел обнаружения ~1 ppb. Ранее он использовался для контроля естественных утечек
нефти вблизи берегов Калифорнии и в Мексиканском заливе (Camilli and Duryea, 2007).
При исследовании катастрофы в Мексиканском заливе прибор был установлен на ав-
тономной платформе Sentry для пространственного мониторинга следа разлива нефти
с использованием пробоотборника, соединенного с платформой (Camilli et aL, 2010).
След простирался на глубину до 1100 м, а его длина превышала 35 км.
Распределение углеводородных смесей по глубине представлено на рис. 9.22. Рас-
пределение демонстрирует вариации компонентов смесей, часто ассоциирующихся
с влиянием биологических эффектов в зависимости от глубины. Концентрация углево-
дородов на глубине 1100 м выше, чем на глубине 10 м.
Исследователи также измеряли с помощью масс-спектрометра концентрацию рас-
творенного в воде кислорода и пытались подтвердить данные методом титрования Вин-
клера и с помощью полярографии. Измерялась концентрация растворенного кислорода
как индикатора биодеградации углеводородов. В ходе анализа было установлено, что
на момент исследования уровень углеводородов практически не уменьшился. Это оз-
начает отсутствие существенной биодеградации углеводородов, то есть углеводороды
будут присутствовать в океане в течение некоторого времени.
Рис. 9.22. Масс-спектры, зарегистрированные на глубине 10 и 1160 м в ходе исследования
профиля по глубине масс-спектрометром с мембранным вводом TETHYS, уста-
новленным на платформе с кабелем для передачи данных. Исследования прово-
дились для оценки последствий разлива нефти в результате действий компании
Deepwater Horizon. Пики ионов ароматических углеводородов бензола и толуола
(m/z 78 и m/z 91 соответственно), вызывающих неблагоприятные биологические
эффекты, более выражены на глубине 1160 м, а не 10 м. (Перепечатано с разреше-
ния Американской ассоциации продвижения науки из: Camilli R., Reddy С. М.,
Yoerger D. R., Van Мооу В. A. S., Jakuba М. V., Kinsey J.C., Mcintyre С. Р., Sylva S. Р.
and Maloney J.V. «Tracking hydrocarbon plume transport and biodegradation at
Deepwater Horizon», Science, 2010, 330, 201—204)
Исследование было особенно актуально с точки зрения анализа влияния углево-
дородного загрязнения на морскую экосистему. Серийные измерения концентрации
многих соединений были необходимы для изучения нефтяного следа. Способность
масс-спектрометра проводить измерения для большого числа компонентов делает его
использование особенно ценным.
Анализ пара
Mini 10.5, разработанный в Университете Пердью, может успешно детектировать про-
стые углеводороды на следовом уровне порядка 0,2 ppb при использовании источника
химической ионизации при атмосферном давлении (ХИАД) (Huang et aL, 2010а). Ана-
лиз углеводородов проводился с помощью ЭРИ (Sokol et aL, 2008) и ХИАД (Huang et al.,
2010a). Концентрирование образца при использовании ЭРИ было достигнуто на трубке
с твердым сорбентом, а также при помощи мембранного ввода.
В работе Сокола и его коллег (Sokol et al., 2008) бензол и толуол регистрировались
ЭРИ на уровне 0,6 и 0,2 ppb соответственно, что ниже пределов допустимой эмиссии
(ПДЭ) (1 рр£для бензола и 2№ррт для толуола). Несколько типичных компонентов неф-
ти были обнаружены с помощью масс-спектрометра Mini 10.5, оснащенного источником
ЭРИ. Спектры представлены на рис. 9.23. Образцы вводились через ПДМС-мембрану.
Прибор Mini 10.5 регистрировал эти компоненты менее чем за 1 минуту. Способность
прибора детектировать соединения с неприятным запахом в воздухе без подготовки
пробы была подтверждена в эксперименте по регистрации паров из открытого сосуда
с бутилмеркаптаном.
9.3. Экологические приложения для миниатюрных масс-спектрометров
76 [M1 CS2
1000
800
600
400
200
0
Бланк - новая мембрана
50 60 70 80 90 100 НО 120 130
m/z
[HCNH] CH3CN
28 3
________
30 40 50 60 70 80 90 100 110
8
" 1400
? !200
>1000
800
600 ;
400 |
200 Ji
0 I-
30
32
178
Л.
m/z
40
80 90 100 110
800
600
400
200
О
20 30 40 50 60 70 80
m/z
30 40 50 60 70 80 90 100 110
m/z
800
x
§ 600
g 400
m
| 200
£
= o1----------------------------------
s 30 40 50 60 70 80 90 100 110
50 60 70
m/z
41 56 C4H9SH
m/z
£2000
>1500
g 1000
аз
о 500
S 60 70 80 90 100 110 120 130 140
45 55 65 75 85 95 105 115 125
m/z
m/z
Рис. 9.23. Анализ нескольких компонентов нефти на миниатюрном масс-спектрометре.
Представлены репрезентативные спектры бланка (новая мембрана), углеводо-
родов водной фазы и гетерозамещенных углеводородов: 50 ррт ацетона, 45 ррт
ацетонитрила, 28 ррт бензола, 24 ррт бутилмеркаптана, 45 ррт сероуглерода,
48 ррт дихлорметана, 9 ррт нафталина, 18 ррт толуола в воздухе. Спектры по-
лучены на масс-спектрометре Mini 10.5 с ионизацией электронами и вводом
через ПДМС-мембрану. Для каждого компонента могут быть зарегистрирова-
ны характеристические ионы, например молекулярный ион m/z 78 для бензола,
фрагментные ионы дихлорметана с характерными изотопными распределения-
ми и ионы с m/z 91 и 92 для толуола. (Источник: Sokol Е., Edwards К. Е., Qian К.
and Cooks R.G. (2008). «Rapid hydrocarbon analysis using a miniature rectilinear ion
trap mass spectrometer». Analyst. 133: 1064—1071. Перепечатано с разрешения Коро-
левского химического общества)
С помощью Mini 10.5 с источником ХИАД были проанализированы бензол, толуол
и этилбензол. Эти соединения могут быть детектированы одновременно в концентра-
циях, значительно меньших их ПДК. Предел обнаружения для бензола был 0,2 ppb, для
толуола — 0,5 ppb и для этилбензола — 0,7ppb. Анализ возможен даже в присутствии кон-
центрированных мешающих компонентов (0,1 % ацетона и 500ppb никотина), как пока-
зано на рис. 9.86. Такой эксперимент демонстрирует универсальность прибора и потен-
циал для анализа в полевых условиях, когда возможно присутствие мешающего фона.
9.3.5. Детектирование пестицидов
Гербициды и пестициды интенсивно используются в сельском хозяйстве, что может
вызвать загрязнение источников пресной воды через просачивание в грунтовые воды
(Budde, 2004). Таким образом, они формируют важный класс загрязняющих веществ,
которые должны контролироваться. Маллиган и его коллеги (Mulligan etal., 2006) в сво-
ей первой демонстрации источника ДЭРИ (десорбционной электрораспылительной
ионизации), присоединенного к миниатюрному масс-спектрометру Мини 8 с цилин-
дрической ионной ловушкой (Laughlin et aL, 2005), анализировали ДЭТА (А,А-диэтил-
л^-толуамид), алахлор (2-хлор-2',6'-диэтил-А-(метоксиметил)-ацетанилид) и атразин
(2-хлор-4-(этиламино)-6-(изопропиламино)-1,3,5-триазин), собранные с различных
поверхностей, в том числе с листьев и овощей. ДЭТА — ароматический амид, который
используется для отпугивания насекомых. Он присутствует во многих популярных
композициях и спреях-инсектицидах (Knepper, 2004). Алахлор — широко применяемый
анилидный пестицид, который трансформируется с образованием соединений, явля-
ющихся потенциальными загрязняющими веществами почвы и водных поверхностей
(Hogenboom et aL, 2000). Атразин — триазиновый гербицид, интенсивно использующий-
ся для контроля широколиственных сорняков (Mandelbaum etal., 1995).
Следовые количества всех этих компонентов могут быть проанализированы на раз-
личных поверхностях при помощи ДЭРИ на портативном масс-спектрометре весом
38 кг и габаритами 46x50x 38 (Laughlin et aL, 2005). Рис. 9.24 демонстрирует детекти-
рование 10 нг ДЭТА, нанесенных на бумагу (рис. 9.24г/), и со свежего стебля кукурузы
(рис. 9.240. Рис. 9.24<? иллюстрирует результат МС/МС-анализа иона с m/z 192, распа-
дающегося с образованием характеристичного фрагмента с m/z 119 вследствие потери
вторичного амина с массой 73 Да. Подобное исследование было проведено Маллиганом
и его коллегами (Mulligan et aL, 2007) для алахлора (рис. 9.25). На лабораторном прибо-
ре методом ДЭРИ/МС были получены данные вплоть до уровня фрагментации МС5.
Смесь, содержащая 10 нг ДЭТА, алахлора и атразина, была нанесена на фильтроваль-
m/z
Рис. 9.24. Масс-спектры ДЭРИ, полученные при регистрации положительных ионов,
для:(я) 10 нг ДЭТА, нанесенных на 1 см2 бумаги; (б) 10 нг ДЭТА, нанесенные
на лист кукурузы. Пикс m/z 192 соответствует (М + Н)+, а пик с m/z 214 обуслов-
лен компонентом из состава листа; (в) МС/МС-анализ иона с m/z 192, демон-
стрирующий характеристичный фрагменте m/z 119. (Источник: Mulligan С.С.,
Talaty N. and Cooks R.G. (2006). «Desorption electrospray ionization with a portable
mass spectrometer: in situ analysis of ambient surfaces». Chemical Communications.
1709—1711. Перепечатано с разрешения Королевского химического общества)
9.3. Экологические приложения для миниатюрных масс-спектрометров
100
[М + Н]
270
Алахлор
[М+ Н3О]+
288
100 150 200 250 300 350 400
m/z
Рис. 9.25. ДЭРИ/МС-анализ 10 нг алахлора, нанесенных на лист кукурузы. (Источник:
Mulligan С.С., Talaty N. and Cooks R.G. (2006). «Desorption electrospray ionization
with a portable mass spectrometer: in situ analysis of ambient surfaces». Chemical
Communications. 1709—1711. Перепечатано с разрешения Королевского химиче-
ского общества)
ную бумагу Ватмана 5 класса. В литературе указывается на трудности одновременного
анализа всех трех компонентов одним методом ионизации и в единственном режиме
детектирования (Thurman etal., 2001). Однако при использовании ДЭРИ все эти компо-
ненты регистрируются в одном сканировании в режиме решистрации положительных
ионов (m/z ДЭТА, алахлора и атразина 192, 216 и 270 соответственно, как и ожидалось).
Такой пример иллюстрирует пригодность ДЭРИ для анализа пестицидов с различных
поверхностей как поодиночке, так и в смеси.
9.3.6. Анализ полициклических ароматических углеводородов
ПАУ попадают в окружающую среду из выхлопов автомобилей, в результате процессов
сгорания и из промышленных отходов. ПАУ известны как вещества, загрязняющие по-
чвы. Анализ почвы обычно проводится в лаборатории методом ГХ/МС, включающим
несколько обременительных этапов. Джанфелт (Janfeltet al., 2006) разработал портатив-
ный масс-спектрометр с мембранным вводом и весом в 10 кг для анализа ПАУ, содержа-
щих до пяти ароматических колец (Frandsen etal., 2007). Масс-спектрометр представлял
собой миниатюрный мультиполь с диапазоном масс 4—300 а.е. м.
В этом методе анализируемый образец помещается в сосуд из нержавеющей стали
и вводится в нагреваемую ячейку масс-спектрометра. Анализируется газ из простран-
ства непосредственно над пробой без прямого контакта с мембраной. В лабораторных
экспериментах Франдсен и его коллеги (Frandsen et al., 2007) продемонстрировали ана-
лиз следующих ПАУ, содержащих от 3 до 5 ароматических колец: антрацен, бензантра-
цен, бензопирен, пирен, тетрацен и бензофлуорантен. Предел обнаружения для них
составлял несколько ррт при относительном стандартном отклонении 10% и времени
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
анализа от 5 до 10 минут. Рис. 9.26я демонстрирует профиль десорбции для смеси пи-
рена, бензантрацена и бенз[е]пирена в количестве 450ррт каждого. На рис. 9.266 пред-
ставлен масс-спектр, зарегистрированный примерно через 2 минуты после введения
флакона с пробой и непосредственно перед его извлечением. Эти результаты чрезвы-
чайно обнадеживающи с точки зрения возможности разработки метода скрининга ПАУ
в почве при использовании миниатюрного масс-спектрометра с мембранным вводом.
ПАУ могут быть количественно определены в воде при помощи масс-спектрометра
Mini 10, оснащенного мембраной для ввода воды, на уровне от нескольких ppb до не-
скольких ppm (Sanders et aL, 2010b). Пределы обнаружения обычно лежат в диапазоне
от 1 до 10 ppb, как показано в табл. 9.1. На рис. 9.27 представлена хронограмма полного
ионного тока и реконструированная ионная хронограмма для растворов, содержащих
следовые количества (ppb) нафталина.
m/z
Рис. 9.26. Миниатюрный масс-спектрометр с мембранным вводом может быть использован
для анализа ПАУ в почве. Проба помещается в сосуд и вводится в нагреваемую
ячейку. Анализируется газ над пробой, (а) Профиль для пирена (m/z 202), бензан-
трацена (m/z 228) и бенз[е]пирена (m/z 252). (б) Масс-спектр, зарегистрированный
через 2 минуты после ввода пробы в ячейку и непосредственно перед ее удалением.
(Источник: Frandsen Н., JanfeltC. and Lauritsen F. R. (2007). «Fast and direct screening
of polyaromatic hydrocarbon (PAH)-contaminated sand using a miniaturized membrane
inlet mass spectrometer (mini-MIMS)». Rapid Communications in Mass Spectrometry. 21:
1574—1578. Перепечатано с разрешения издательства John Wiley and Sons)
9.3. Экологические приложения для миниатюрных масс-спектрометров
Таблица 9.1. Количественный анализ для различных ПАУ в воде
Соедине- ние Фор- мула Молеку- лярный вес/г/моль Предел обнаруже- ния, ppb* Точка плавле- ния/°С Точка кипе- ния/°С Раство- римость в воде/мг/мл Коэффици- ент липо- фильности
Нафталин С,„н8 128,18 0,8 80 218 31 3,3
Аценафтен с,2н,„ 154,21 5,7 93 279 3,9 3,92
Антрацен с,4н10 178,24 1,9 215 340 0,0434 4,5
Фенантрен с,4н10 178,24 < 5,6 99 340 1,15 4,6
* Использовались различные виды калибровки. Показано наименьшее значение для предела
обнаружения.
80000
75000
70000
65000
60000
55000
50000
45000
40000
0:30 1:00 1:30 2:00 2:30
Рис. 9.27. Отклик прибора на водный раствор нафталина при анализе стандартов в 3, 9
и 30 ppb. Верхний рисунок: хронограмма полного ионного тока (сигнал/вре-
мя). Нижний рисунок: реконструированная ионная хронограмма по току ионов
с m/z 127—129. Молекулярный ион нафталина: m/z 128. Полное время хронограм-
мы — примерно 3 ч. (Источник: European Journal of Mass Spectrometry (16, 11—20,
2010). Перепечатано с разрешения IM Publications LLP)
9.3.7. Детектирование примесей в продуктах питания
Меламин — новый распространенный экотоксикант в продуктах питания. В настоящее
время неизвестно, оказывает ли он вредное воздействие на окружающую среду, одна-
ко его влияние на здоровье человека (например, при фальсификации продуктов) делает
проблему его анализа достаточно важной (Richardson, 2010). Два недавних происшествия
подчеркивают важность детектирования меламина в продуктах питания в целях заботы
о здоровье населения. Это подмешивание меламина в корм для домашних животных,
приведшее к токсикозу почек у кошек и собак в 2007 году, и загрязнение меламином ис-
кусственного детского молочного питания, вызвавшее образование почечных камней
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
Сз78
и отказу почек у новорожденных в 2008 году (Xin and Stone, 2008). Полное содержание
белка в продуктах питания определяется методом Кьельдаля (Wiles etal., 1998) с низкой
специфичностью, в котором определение белка основано на измерении полного содер-
жания азота. Меламин — промышленный химикат, богатый азотом (до 66,7 % от общего
веса), при добавлении в пищу «увеличивает» содержание белка (измеренное методом
Кьельдаля). Меламин реагирует с одним из его метаболитов — циануровой кислотой
с образованием плохо растворимого стабильного комплекса, который выпадает в осадок
в почечных канальцах и приводит к отказу почек.
Для борьбы с этой проблемой было разработано несколько инструментальных ме-
тодов: иммунохимические тест-системы (Kim et al., 2008), капиллярный зонный элек-
трофорез с диодной матрицей и с масс-спектрометрическим детектированием (Cook
et aL, 2005; Yan et aL, 2009), высокоэффективная жидкостная хроматография с ультра-
фиолетовым детектором (Pukkila et aL, 1987; Yokley et aL, 2000) и другие. Эти методы до-
статочно точны, но трудозатратны. Поэтому существовала необходимость в разработке
более простого и экспрессного альтернативного метода.
Методы ионизации в нормальных условиях, уже упомянутые ранее, не требуют
подготовки проб и значительно увеличивают производительность анализа. Так как
главной проблемой методов сейчас является их длительность, способы анализа в нор-
мальных условиях обладают значительным преимуществом. Хуанг с коллегами (Huang
et aL, 2010b) для детектирования меламина в пищевых продуктах соединили источ-
ник с низкотемпературной плазмой с портативным масс-спектрометром Mini 10.5.
Допустимые уровни меламина в США составляют 1 мкг/мл, тогда как в Европе это
2,5 мкг/мл. С помощью данной установки были достигнуты пределы обнаружения
в цельном молоке в 250 нг/мл, что значительно ниже требуемых законодательством.
Образцы рыбы, сухого молока, мочи и рыбной пасты были проанализированы на со-
держание меламина без дополнительной пробоподготовки. Полное время анализа од-
ной пробы составило 30 с, что в принципе позволяет анализировать на приборе 3000
проб за 24 часа. Такая производительность значительно выше, чем любого существу-
ющего ГХ/МС или ЖХ/МС-метода, даже при исключении времени на пробоподго-
товку. Сочетание источников ионизации в нормальных условиях с миниатюрными
масс-спектрометрами является привлекательным способом обнаружения случаев
фальсификации продуктов питания.
9.4. Заключение
В этой главе была продемонстрирована универсальность масс-спектрометрии как мето-
да анализа в полевых условиях. Разработка методов ионизации в нормальных условиях
позволяет ионизировать очень широкий круг соединений. Ионизация в нормальных ус-
ловиях и миниатюрные масс-спектрометры дополняют друг друга, а их соединение яв-
ляется важнейшим этапом дальнейшего развития масс-спектрометрии для работы в по-
левых условиях. Представляется, что дорогие крупногабаритные масс-спектрометры,
которые функционируют исключительно в лабораторных условиях, уступят дорогу не-
дорогим приборам, созданным для специальных приложений и доступным для массо-
вого производства.
Литература
[1] . Alexander С., Andersson H.S., Andersson L. 1., Ansell R.J., Kirsch N., Nicholls I.A.,
O’Mahony J., and Whitcombe M.J. (2006). «Molecular imprinting science and technology:
a survey of the literature for the years up to and including 2003». Journal of Molecular Recog-
nition. 19: 106—180.
[2] . Andrade F. J., Shelley J. T., Wetzel W. C., Webb M. R., Gamez G., Ray S. J., and Hieftje G. M.
(2008). «Atmospheric pressure chemical ionization source. 2. Desorption ionization for the
direct analysis of solid compounds». Analytical Chemistry. 80: 2654—2663.
[3] . Arkin C. R., Griffin T. P., Diaz J. A., Follistein D. W., Curley С. H., Floyd D. P., Naylor G. R.,
Haskell W. D., Blalock M., and Adams F. W. (2004). «А small mass spectrometer system for
in situ gas analysis». Trends in Analytical Chemistry. 23: 322—330.
[4] . Arthur C. L., and Pawliszyn J. (1990). «Solid-phase microextraction with thermal-desorp-
tion using fused-silica optical fibers». Analytical Chemistry. 62: 2145—2148.
[5] . Austin D. E., Wang M., Tolley S. E., Maas J. D., Hawkins A. R., Rockwood A. L., Tol-
ley H. D., Lee E. D., and Lee M. L. (2007). «Halo ion trap mass spectrometer». Analytical
Chemistry. 2927-2932.
[6] . Badman E. R., and Cooks R. G. (2000). «Special feature: perspective — miniature mass ana-
lyzers». Journal of Mass Spectrometry. 35: 659—671.
[7] . Badman E. R., Johnson R. C., Plass W. R., and Cooks R. G. (1998). «А miniature cylindri-
cal quadrupole ion trap: simulation and experiment». Analytical Chemistry. 70: 4896—4901.
[8] . Bairn M. A., and Hill H. H. (1982). «Tunable selective detection for capillary gas-chromatog-
raphy by ion mobility monitoring». Analytical Chemistry. 54: 38—43.
[9] . Bell R.J., Short R.T., Van Amerom F. H.W., and Byrne R.H. (2007). «Calibration of an
in situ membrane inlet mass spectrometer for measurements of dissolved gases and volatile
organics in seawater». Environmental Science and Technology. 41: 8123—8128.
[10] . Bier M. E., and Cooks R. G. (1987). «Membrane interface for selective introduction of vola-
tile compounds directly into the ionization-chamber of a mass spectrometer». Analytical
Chemistry. 59: 597—601.
[11] . Bier M.E., Kotiaho T., and Cooks R.G. (1990). «Direct insertion membrane probe for se-
lective introduction of organic compounds into a mass spectrometer». Analytica Chimica
Acta. 231: 175-190.
[12] . Borsdorf H., and Eiceman G. A. (2006). «Ion mobility spectrometry: principles and appli-
cations». Applied Spectroscopy Reviews. 41: 323—375.
[13] . Budde W. L. (2004). «Analytical mass spectrometry of herbicides». Mass Spectrometry Re-
views. 23: 1—24.
[14] . Camilli R., and Duryea A. (2007). «Characterizing marine hydrocarbons with in-situ mass
spectrometry». 2007Oceans. 1—7. IEEE.
[15] . Camilli R., and Hemond H.F. (2004). «NEREUS/Kemonaut, a mobile autonomous un-
derwater mass spectrometer». Trends in Analytical Chemistry. 23: 307—313.
[16] . Camilli R., Reddy С. M., Yoerger D. R., Van Mooy B.A. S., Jakuba M.V., Kinsey J.C.,
Mcintyre С. P., Sylva S. P., and Maloney J. V. (2010). «Tracking hydrocarbon plume trans-
port and biodegradation at Deepwater Horizon». Science. 330: 201—204.
[17] . Carroll D. L, Dzidic L, Stillwell R.N., Haegele K.D., and Horning E.C. (1975). «Atmos-
pheric pressure ionization mass spectrometry. Corona discharge ion source for use in liquid
280 Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
chromatograph-mass spectrometer-computer analytical system». Analytical Chemistry.
2369-2373.
[18] . Cech N. B., and Enke C. G. (2001). «Practical implications of some recent studies in elec-
trospray ionization fundamentals». Mass Spectrometry Reviews. 20: 362—387.
[19] . Chen H., Cotte-Rodriguez I., and Cooks R.G. (2006). «cis-diol functional group recog-
nition by reactive desorption electrospray ionization (DESI)». Chemical Communications.
597-599.
[20] . Cole R. B. (2000). «Some tenets pertaining to electrospray ionization mass spectrometry».
Journal of Mass Spectrometry. 35: 763—772.
[21] . Contreras J. A., Murray J. A., Tolley S.E., Oliphant J. L., Tolley H.D., Lammert S.A.,
Lee E. D., Later D.W., and Lee M. L. (2008). «Hand-portable gas chromatograph—toroidal
ion trap mass spectrometer (GC—TMS) for detection of hazardous compounds». Journal of
the American Society for Mass Spectrometry. 19: 1425—1434.
[22] . Cook H.A., Klampfl C.W., and Buchberger W. (2005). «Analysis of melamine resins by
capillary zone electrophoresis with electrospray ionization—mass spectrometric detection».
Electrophoresis, 26, 1576—1583.
[23] . Cooks R. G., Ouyang Z., Takats Z., and Wiseman J. M. (2006). «Ambient mass spectrom-
etry». Science. 311: 1566—1570.
[24] . Cornish T.J., and Cotter R. J. (1997). «High-order kinetic energy focusing in an end cap
reflectron time-of-flight mass spectrometer». Analytical Chemistry. 69: 4615—4618.
[25] . De Hoffmann E., and Stroobant V. (2007). «Mass Spectrometry: Principles and Applications».
John Wiley and Sons, Chichester, UK.
[26] . Diaz J. A., Giese C. F., and Gentry W. R. (2001a). «Portable double-focusing mass-spec-
trometer system for field gas monitoring». Field Analytical Chemistry and Technology. 5:
156-167.
[27] . Diaz J. A., Giese С. E, and Gentry W. R. (2001b). «Sub-miniature ExB sector-field mass
spectrometer». Journal of the American Society for Mass Spectrometry. 12: 619—632.
[28] . Diaz J. A., Giese С. E, and Gentry W. R. (2002). «Mass spectrometry for in-situ volcanic
gas monitoring». Trends in Analytical Chemistry. 21: 498—514.
[29] . Diaz J. A., Pieri D., Arkin C. R., Gore E., Griffin T. P., Fladeland M., Bland G., Soto C.,
Madrigal Y., Castillo D., Rojas E., and Achi S. (2010). «Utilization of in situ airborne MS-
based instrumentation for the study of gaseous emissions at active volcanoes». International
Journal of Mass Spectrometry. 295: 105—112.
[30] . Douglas D.J., Frank A. J., and Mao D. (2005). «Linear ion traps in mass spectrometry».
Mass Spectrometry Reviews. 24: 1—29.
[31] . Ecelberger S. A., Cornish T. J., Collins В. E, Lewis D. L., and Bryden W. A. (2004). «Suit-
case TOF: a man-portable time-of-flight mass spectrometer». Johns Hopkins APL Technical
Digest. 25: 14—19.
[32] . Fenn J. B., Mann M., Meng С. K., Wong S. E, and Whitehouse С. M. (1989). «Electrospray
ionization for mass spectrometry of large biomolecules». Science. 246: 64—71.
[33] . Ferran R. J., and Boumsellek S. (1996). «High-pressure effects in miniature arrays of quad-
rupole analyzers for residual gas analysis from 10-9 to 10-2 Torr». Journal of Vacuum Science
and Technology A — Vacuum Surfaces and Films. 14: 1258—1265.
[34] . Frandsen H., Janfelt C., and Lauritsen F. R. (2007). «Fast and direct screening of polyaro-
matic hydrocarbon (PAH)-contaminated sand using a miniaturized membrane inlet mass
spectrometer (mini-MIMS)». Rapid Communications in Mass Spectrometry. 21: 1574—1578.
[35] . Fries D. P., Short R.T., Langebrake L. L., Patten J.T., Kerr M.L., Kibelka G., Bur-
well D. C., and Jalbert J.C. (2001). «In-water field analytical technology: underwater mass
spectrometry, mobile robots, and remote intelligence for wide and local area chemical pro-
filing». Field Analytical Chemistry and Technology. 5: 121—130.
[36] . Gao L. (2009). Miniaturization of the Ion Trap Mass Spectrometer. PhD thesis, Purdue
University, Indiana, USA.
[37] . Gao L., Song Q. Y., Patterson G. E., Cooks R. G., and Ouyang Z. (2006). «Handheld recti-
linear ion trap mass spectrometer». Analytical Chemistry. 78: 5994—6002.
[38] . Gao L., Song Q.Y., Noll R.J., Duncan J., Cooks R.G., and Zheng O.Y. (2007). «Glow
discharge electron impact ionization source for miniature mass spectrometers». Journal of
Mass Spectrometry. 42: 675—680.
[39] . Gao L., Cooks R. G., and Ouyang Z. (2008a). «Breaking the pumping speed barrier in mass
spectrometry: discontinuous atmospheric pressure interface». Analytical Chemistry. 80:
4026-4032.
[40] . Gao L., Sugiarto A., Harper J. D., Cooks R. G., and Ouyang Z. (2008b). «Design and char-
acterization of a multisource hand-held tandem mass spectrometer». Analytical Chemistry.
80: 7198-7205.
[41] . Geear M., Syms R. R. A., Wright S., and Holmes A. S. (2005). «Monolithic MEMS quad-
rupole mass spectrometers by deep silicon etching». Journal of Microelectromechanical Sys-
tems. 14: 1156—1166.
[42] . Glish G. L., and Vachet R. W. (2003). «The basics of mass spectrometry in the twenty-first
century». Nature Reviews Drug Discovery. 2: 140—150.
[43] . Griffin T. P., Diaz J. A., Arkin C. R., Soto C., Curley С. H., and Gomez O. (2008). «Three-
dimensional concentration mapping of gases using a portable mass spectrometer system».
Journal of the American Society for Mass Spectrometry. 19: 1411 — 1418.
[44] . Hager J.W. (2002). «А new linear ion trap mass spectrometer». Rapid Communications in
Mass Spectrometry. 16: 512—526.
[45] . Harper J. D., Charipar N. A., Mulligan С. C., Zhang X. R., Cooks R. G., and Ouyang Z.
(2008). «Low-temperature plasma probe for ambient desorption ionization». Analytical
Chemistry. 80: 9097—9104.
[46] . Hayes R. B., Yin S. N., Dosemeci M., Li G. L., Wacholder S., Travis L. B., Li C. Y., Roth-
man N., Hoover R. N., and Linet M. S. (1997). «Benzene and the dose-related incidence of
hematologic neoplasms in China». Journal of the National Cancer Institute. 89: 1065—1071.
[47] . Hemond H., and Camilli R. (2002). «NEREUS: engineering concept for an underwater
mass spectrometer». Trends in Analytical Chemistry. 21: 526—533.
[48] . Hemond H. F., Mueller A. V., and Hemond M. (2008). «Field testing oflake water chemistry
with a portable and an AU V-based mass spectrometer». Journal of the American Society for
Mass Spectrometry. 19: 1403—1410.
[49] . Hill H. H., and Simpson G. (1997). «Capabilities and limitations of ion mobility spectrom-
etry for field screening applications». Field Analytical Chemistry and Technology. 1: 119—134.
[50] . Hirabayashi A., Sakairi M., and Koizumi H. (1994). «Sonic spray ionization method for
atmospheric-pressure ionization mass spectrometry». Analytical Chemistry. 66: 4557—4559.
282 Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
[51] . Hirabayashi A., Sakairi М., and Koizumi Н. (1995). «Sonic spray mass spectrometry».
Analytical Chemistry. 67: 2878—2882.
[52] . Hogenboom A.C., Niessen W. M.A., and Brinkman U.A.T. (2000). «Characterization of
photodegradation product of alachlor in water by on-line solid-phase extraction liquid chro-
matography combined with tandem mass spectrometry and orthogonal-acceleration time-
of-flight mass spectrometry». Rapid Communications in Mass Spectrometry. 14: 1914—1924.
[53] . Horning E.C., Horning M.G., Carroll D. I., Dzidic I., and Stillwel R.N. (1973). «New
picogram detection system based on a mass spectrometer with an external ionization source
at atmospheric pressure». Analytical Chemistry. 45: 936—943.
[54] . Huang G. M., Gao L., Duncan J., Harper J. D., Sanders N. L., Ouyang Z., and Cooks R. G.
(2010a). «Direct detection of benzene, toluene, and ethylbenzene at trace levels in ambi-
ent air by atmospheric pressure chemical ionization using a handheld mass spectrometer».
Journal of the American Society for Mass Spectrometry. 21:132—135.
[55] . Huang G.M., Xu W., Visbal-Onufrak M.A., Ouyang Z., and Cooks R.G. (2010b). «Direct
analysis of melamine in complex matrices using a handheld mass spectrometer». Analyst.
135: 705-711.
[56] . Huang M.Z., Yuan С. H., Cheng S. C., Cho Y.T., and Shiea J. (2010c). «Ambient ionization
mass spectrometry». Annual Review of Analytical Chemistry. 3: 43—65.
[57] . Ifa D. R., Wu С. P., Ouyang Z., and Cooks R.G. (2010). «Desorption electrospray ioniza-
tion and other ambient ionization methods: current progress and preview». Analyst. 135:
669-681.
[58] . Janfelt C., Frandsen H., and Lauritsen F. R. (2006). «Characterization of a mini membrane
inlet mass spectrometer for on-site detection of contaminants in both aqueous and liquid
organic samples». Rapid Communications in Mass Spectrometry. 20: 1441 — 1446.
[59] . Kaiser R. E., Cooks R. G., Stafford G. C., Syka J. E. P., and Hemberger P. H. (1991). «Op-
eration of a quadrupole ion trap mass spectrometer to achieve high mass charge ratios».
International Journal of Mass Spectrometry and Ion Processes. 106: 79—115.
[60] . Kebarle P. (2000). «А brief overview of the present status of the mechanisms involved in
electrospray mass spectrometry». Journal of Mass Spectrometry. 35: 804—817.
[61] . Kebarle P., and Tang L. (1993). «From ions in solution to ions in the gas-phase — the mech-
anism of electrospray mass spectrometry». Analytical Chemistry. 65: A972—A986.
[62] . Keil A., Talaty N., Janfelt C., Noll R. J., Gao L., Ouyang Z., and Cooks R.G. (2007). «Am-
bient mass spectrometry with a handheld mass spectrometer at high pressure». Analytical
Chemistry. 79: 7734-7739.
[63] . Keil A., Hernandez-Soto H., Noll R.J., Fico M., Gao L., Ouyang Z., and Cooks R.G.
(2008). «Monitoring of toxic compounds in air using a handheld rectilinear ion trap mass
spectrometer». Analytical Chemistry. 80: 734—741.
[64] . Kibelka G.P.G., Short R.T., Toler S.K., Edkins J.E., and Byrne R.H. (2004). «Field-de-
ployed underwater mass spectrometers for investigations of transient chemical systems».
Taianta. 64: 961—969.
[65] . Kim B., Perkins L. B., Bushway R. J., Nesbit S., Fan T., Sheridain R., and Greene V. (2008).
«Determination of melamine in pet food by enzyme immunoassay, high-performance liq-
uid chromatography with diode array detection, and ultra-performance liquid chromatog-
raphy with tandem mass spectrometry». Journal of AO AC International. 91: 408—413.
Литература
[66] . Knake R., Jacquinot P., Hodgson A.W. E., and Hauser P. C. (2005). «Amperometric sens-
ing in the gas-phase». Analytica Chimica Acta. 549: 1—9.
[67] . Knepper T. P. (2004). «Analysis and mass spectrometric characterization of the insect
repellent Bayrepel and its main metabolite Bayrepel acid». Journal of Chromatography A.
1046: 159-166.
[68] . Koppenaal D.W., Barinaga C.J., Denton M.B., Sperline R.P., Hieftje G.M., Schil-
ling G.D., Andrade F.J., and Barnes J. H. (2005). «MS detectors». Analytical Chemistry.
77: 418A—427A.
[69] . Kornienko O., Reilly P.T.A., Whitten W. B., and Ramsey J.M. (1999). «Micro ion trap
mass spectrometry. Rapid Communications in Mass Spectrometry. 13: 50—53.
[70] . Lamert J. B., Shurvell H. E, Verbit L., Cooks R. G., and Stout G. H. (1976). Organic Struc-
tural Analysis. Macmillan Publishing Co., New York, USA.
[71] . Lammert S. A., Plass W. R., Thompson С. V, and Wise M. B. (2001). «Design, optimization
and initial performance of a toroidal rf ion trap mass spectrometer». International Journal of
Mass Spectrometry. 212: 25—40.
[72] . Lammert S.A., Rockwood A. A., Wang M., Lee M.L., Lee E.D., Tolley S. E., Oli-
phant J.R., Jones J.L., and Waite R.W. (2006). «Miniature toroidal radio frequency ion
trap mass analyzer». Journal of the American Society for Mass Spectrometry. 17: 916—922.
[73] . Langmuir D. B., Langmuir R. V., Shelton H., and Wuerker R. F. (1962). Containment de-
vice. US Patent 3,065,640, USA.
[74] . Lapack M.A., Tou J.C., and Enke C.G. (1990). «Membrane mass spectrometry for the
direct trace analysis of volatile organic compounds in air and water». Analytical Chemistry.
62: 1265-1271.
[75] . Lapack M.A., Tou J.C., and Enke C.G. (1991). «Membrane extraction mass spectrom-
etry for the on-line analysis of gas and liquid process streams». Analytical Chemistry. 63:
1631-1637.
[76] . Laughlin В. C., Mulligan С. C., and Cooks R. G. (2005). «Atmospheric pressure ionization
in a miniature mass spectrometer». Analytical Chemistry. 77: 2928—2939.
[77] . Laursen L. (2010). «Iceland eruptions fuel interest in volcanic gas monitoring». Science.
328:410-411.
[78] . Lin B. W., and Sunner J. (1994). «Ion-transport by viscous-gas flow-through capillaries».
Journal of the American Society for Mass Spectrometry. 5: 873—885.
[79] . Makarov A. (2000). «Electrostatic axially harmonic orbital trapping: a high-performance
technique of mass analysis». Analytical Chemistry. 72: 1156—1162.
[80] . Mandelbaum R.T., Allan D. L., and Wackett L. P. (1995). «Isolation and characterization of
a Pseudomonas sp that mineralizes the s-triazine herbicide atrazine». Applied and Environ-
mental Microbiology. 61: 1451—1457.
[81] . March R. E., and Todd J. F. J. (2005). Quadrapole Ion Trap Mass Spectrometry. Wiley Inter-
Science, New Jersey, USA.
[82] . Mauclaire G., Lemaire J., Boissel P., Bellec G., and Heninger M. (2004). «M1CRA: a com-
pact permanent magnet Fourier transform ion cyclotron resonance mass spectrometer».
European Journal of Mass Spectrometry. 10: 155—162.
[83] . Mcluckey S.A., Busch K.L., and Glish G.L. (1988). Mass Spectrometry/Mass Spectrom-
etry: Techniques and Applications of Tandem Mass Spectrometry. VCH Publishers, New York,
USA.
G84
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
[84] . McMurtry G. М., and Smith S. J. (2001). «Mass SURFER: a low-power underwater mass
spectrometer for monitoring dissolved gas, solutes and large organic compounds. In: Pro-
ceedings of MTS/1EEE Conference Oceans 2001: MTS/IEEE: An Ocean Odyssey, Vol. 1,
pp. 259—263. Marine Technology Society, Columbia, MD, USA.
[85] . Meurer E.C., Tomazela D.M., Silva R.C., Augusto F., and Eberlin M.N. (2002). «Fiber
introduction mass spectrometry: fully direct coupling of solid-phase microextraction with
mass spectrometry». Analytical Chemistry. 74: 5688—5692.
[86] . Mulligan C.C., Talaty N., and Cooks R.G. (2006). «Desorption electrospray ionization
with a portable mass spectrometer: in situ analysis of ambient surfaces». Chemical Com-
munications. 1709—1711.
[87] . MulliganC.C., Macmillan D.K., Noll R. J., and Cooks R. G. (2007). «Fast analysis of high-
energy compounds and agricultural chemicals in water with desorption electrospray ioniza-
tion mass spectrometry». Rapid Communications in Mass Spectrometry. 21: 3729—3736.
[88] . Nazarov E.G., Miller R.A., Eiceman G. A., and Stone J. A. (2006). «Miniature differential
mobility spectrometry using atmospheric pressure photoionization». Analytical Chemistry.
78: 4553-4563.
[89] . Orient O.J., Chutjian A., and Garkanian V. (1997). «Miniature, high-resolution, quadru-
pole mass-spectrometer array». Review of Scientific Instruments. 68: 1393—1397.
[90] . Ouyang Z., and Cooks R.G. (2009). «Miniature mass spectrometers». Annual Review of
Analytical Chemistry. 2: 187—214.
[91] . Ouyang Z., Badman E. R., and Cooks R.G. (1999). «Characterization of a serial array of
miniature cylindrical ion trap mass analyzers». Rapid Communications in Mass Spectrom-
etry. 13: 2444-2449.
[92] . Ouyang Z., Wu G.X., Song Y.S., Li H.Y., Plass W.R., and Cooks R.G. (2004). «Rectilin-
ear ion trap: concepts, calculations, and analytical performance of a new mass analyzer».
Analytical Chemistry. 76: 4595—4605.
[93] . Patterson G.E., Guymon A. J., Riter L.S., Everly M., Griep-Raming J., Laughlin B.C.,
Zheng O. Y, and Cooks R.G. (2002). «Miniature cylindrical ion trap mass spectrometer».
Analytical Chemistry. 74: 6145—6153.
[94] . Paul W., and Steinwedel H. (1953). «*Ein Neues Massenspektrometer Ohne Magnetfeld».
Zeitschrift Fur Naturforschung Section A — a Journal of Physical Sciences. 8: 448—450.
[95] . Pukkila J., Peltonen K., and Savolainen T. (1987). «Determination of melamine in air by
high-performance liquidchromatography with ultraviolet detection». Journal of Chromatog-
raphy. 411: 409—414.
[96] . Ratcliffe L.V., Rutten F.J. M., Barrett D.A., Whitmore T., Seymour D., Greenwood C.,
Aranda-Gonzalvo Y, Robinson S.,and McCoustra M. (2007). «Surface analysis under am-
bient conditions using plasma-assisted desorption/ionization mass spectrometry». Analyti-
cal Chemistry. 79: 6094—6101.
[97] . Richardson S. D. (2010). «Environmental mass spectrometry: emerging contaminants and
current issues». Analytical Chemistry. 82: 4742—4774.
[98] . Rinsky R. A., Smith A. B., Hornung R., Filloon T. G., Young R. J., Okun A. H., and Land-
rigan P. J. (1987). «Benzene and leukemia — an epidemiologic risk assessment». New Eng-
land Journal of Medicine. 316: 1044—1050.
Литература
[99] . Riter L. S., Peng Y. A., Noll R. J., Patterson G. E., Aggerholm T., and Cooks R. G. (2002).
«Analytical performance of a miniature cylindrical ion trap mass spectrometer». Analytical
Chemistry. 74: 6154—6162.
[100] . Riter L.S., Meurer E.C., Cotte-Rodriguez I., Eberlin M.N., and Cooks R.G. (2003).
«Solid phase micro-extraction in a miniature ion trap mass spectrometer. Analyst. 128:
1119-1122.
[101] . Sanders N. L., Kothari S., Huang G. M., Salazar G., and Cooks R. G. (2010a). «Detection
of explosives as negative ions directly from surfaces using a miniature mass spectrometer».
Analytical Chemistry. 82: 5313—5316.
[102] . Sanders N.L., Sokol E., Perry R.H., Huang G.M., Noll R.J., Duncan J.S., and
Cooks R.G. (2010b). «Hand-held mass spectrometer for environmentally relevant ana-
lytes using a variety of sampling and ionization methods». European Journal of Mass Spec-
trometry. 16: 11—20.
[103] . Schwartz J.C., Senko M.W., and Syka J.E.P. (2002). «А two-dimensional quadrupole
ion trap mass spectrometer». Journal of the American Society for Mass Spectrometry. 13:
659-669.
[104] . Shimma S., Nagao H., Aoki J., Takahashi K., Miki S., and Toyoda M. (2010). «Minia-
turized high-resolution timeof-flight mass spectrometer MULTUM-S II with an infinite
flight path». Analytical Chemistry. 82: 8456—8463.
[105] . Short R. T., Fries D. P., Kerr M. L., and Byrne R. H. (2000). In-situ chemical analysis using
mass spectrometry on unmanned underwater vehicles. In: Proceedings of MTS/1EEE Con-
ference Oceans 2000 — Where Marine Science and Technology Meet, Vol. 1, pp. 605—
609. IEEE, USA.
[106] . Short R.T., Fries D. P., Kerr M.L., Lembke С. E., Toler S.K., Wenner P. G., and By-
rne R. H. (2001a). «Underwater mass spectrometers for in situ chemical analysis of the
hydrosphere». Journal of the American Society for Mass Spectrometry. 12: 676—682.
[107] . Short R.T., Fries D. P., Kibelka G.P. G., Kerr M.L., Toler S. K., Wenner P. G., and By-
rne R. H. (2001b). Field chemical analysis using real-time in-water mass spectrometry. In:
Proceedings of MTS/1EEE Conference Oceans 2001 — An Ocean Odyssey, Vol. 1,
pp. 256—258. Marine Technology Society, Columbia, MD, USA.
[ 108]. Short R. T., Toler S. K., Kibelka G. P. G., Roa D. T. R., Bell R. J., and Byrne R. H. (2006).
«Detection and quantification of chemical plumes using a portable underwater membrane
introduction mass spectrometer». Trends in Analytical Chemistry. 25: 637—646.
[109] . Shortt B. J., Darrach M. R., Holland P. M., and Chutjian A. (2005). «Miniaturized system
of a gas chromatograph coupled with a Paul ion trap mass spectrometer». Journal of Mass
Spectrometry. 40: 36—42.
[110] . Sinha M.P., and Tomassian A. D. (1991). «Development of a miniaturized, lightweight
magnetic-sector for a fieldportable mass spectrograph». Review of Scientific Instruments.
62: 2618-2620.
[111] . Smith J.N., Keil A., Likens J., Noll R.J., and Cooks R.G. (2010). «Facility monitoring
of toxic industrial compounds in air using an automated, fieldable, miniature mass spec-
trometer». Analyst. 135: 994—1003.
[112] . Sokol E., Edwards К. E., Qian K., and Cooks R.G. (2008). «Rapid hydrocarbon analysis
using a miniature rectilinear ion trap mass spectrometer». Analyst. 133: 1064—1071.
Глава 9. Миниатюрные масс-спектрометры для анализа объектов
окружающей среды
[113] . Sokol Е., Noll R. J., Cooks R. G., Beegle L. W., Kim H. I., and Kanik I. (2011). «Miniature
mass spectrometer equipped with electrospray and desorption electrospray ionization for
direct analysis of organics from solids and solutions». International Journal of Mass Spec-
trometry. 306: 187—195.
[114] . Stafford G. C., Kelley P. E., Syka J. E. P., Reynolds W. E., and Todd J. F. J. (1984). «Recent
improvements in and analytical applications of advanced ion trap technology». Interna-
tional Journal of Mass Spectrometry and Ion Processes. 60: 85—98.
[115] . Syms R. R. A., Tate T. J., Ahmad M. M., and Taylor S. (1996). «Fabrication of a microengi-
neered quadrupole electrostatic lens». Electronics Letters. 32: 2094—2095.
[116] . Tadjimukhamedov F. K., Jackson A. U., Nazarov E.G., Ouyang Z., and Cooks R.G.
(2010). «Evaluation of a differential mobility spectrometer/miniature mass spectrometer
system». Journal of the American Society for Mass Spectrometry. 21: 1477—1481.
[117] . Takats Z., Wiseman J. M., Gologan B., and Cooks R.G. (2004). «Massspectrometry sam-
pling under ambient conditions with desorption electrospray ionization». Science. 306:
471-473.
[118] . Tanaka K., Waki H., Ido Y., Akita S., Yoshida Y., Yoshida T., and Matsuo T. (1988). «Pro-
tein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spec-
trometry». Rapid Communications in Mass Spectrometry. 2: 151 — 153.
[119] . Taylor S., Tunstall J. J., Syms R. R. A., Tate T., and Ahmad M. M. (1998). «Initial results
for a quadrupole mass spectrometer with a silicon micromachined mass filter». Electronics
Letters. 34: 546-547.
[120] . Thurman E. M., Ferrer I., and Barcelo D. (2001). «Choosing between atmospheric pres-
sure chemical ionization and electrospray ionization interfaces for the HPLC/MS analysis
of pesticides». Analytical Chemistry. 73: 5441—5449.
[121] . Van Berkel G.J., Pasilis S. P., and Ovchinnikova O. (2008). «Established and emerging
atmospheric pressure surface sampling/ionization techniques for mass spectrometry».
Journal of Mass Spectrometry. 43: 1161 — 1180.
[122] . Venter A., Nefliu M., and Cooks R. G. (2008). «Ambient desorption ionization mass spec-
trometry». Trends in Analytical Chemistry. 27: 284—290.
[123] . Wenner P.G., Bell R.J., Van Amerom F. H.W., Toler S. K., Edkins J.E., Hall M.L.,
Koehn K., Short R.T., and Byrne R. H. (2004). «Environmental chemical mapping using
an underwater mass spectrometer». Trends in Analytical Chemistry. 23: 288—295.
[124] . Weston D.J. (2010). «Ambient ionization mass spectrometry: current understanding of
mechanistic theory; analytical performance and application areas». Analyst. 135: 661—668.
[125] . White A. J., Blamire M.G., Corlett C.A., Griffiths B.W., Martin D. M., Spencer S.B.,
and Mullock S. J. (1998). «Development of a portable time-of-flight membrane inlet mass
spectrometer for environmental analysis». Review of Scientific Instruments. 69: 565—571.
[126] . Wiles P.G., Gray 1. K., and Kissling R.C. (1998). «Routine analysis of proteins by Kjeldahl
and Dumas methods: review and interlaboratory study using dairy products». Journal of
AOAC International. 81: 620—632.
[127] . Wilm M.,and Mann M. (1996). «Analytical properties of the nanoelectrospray ion source».
Analytical Chemistry. 68: 1—8.
[128] . Wiseman J. M., Ifa D. R., and Cooks R.G. (Eds.). (2007). Electrosonic Spray Ionization.
Elsevier, Amsterdam, The Netherlands.
Литература
[129] . Wu С. Р., Ifa D. R., Manicke N.E., and Cooks R.G. (2009). «Rapid, direct analysis of
cholesterol by charge labeling in reactive desorption electrospray ionization». Analytical
Chemistry. 81: 7618—7624.
[130] . Xin H., and Stone R. (2008). «Tainted milk scandal. Chinese probe unmasks high-tech
adulteration with melamine». Science. 322: 1310—1311.
[131] . Yamashita M., and Fenn J. B. (1984). «Electrospray ion-source — another variation on the
free-jet theme». Journal of Physical Chemistry. 88, 20: 4451—4459.
[132] . Yan N., Zhou L., Zhu Z. F., and Chen X. G. (2009). «Determination of melamine in dairy
products, fish feed, and fish by capillary zone electrophoresis with diode array detection».
Journal of Agricultural and Food Chemistry. 57: 807—811.
[133] . Yang M., Kim T.Y, Hwang H.C., Yi S. K., and Kim D. H. (2008). «Development of a
palm portable mass spectrometer». Journal of the American Society for Mass Spectrometry.
19: 1442-1448.
[134] . Yokley R. A., Mayer L.C., Rezaaiyan R., Manuli M. E., and Cheung M.W. (2000). «Ana-
lytical method for the determination of cyromazine and melamine residues in soil using
LC—U V and GC—MSD». Journal of Agricultural and Food Chemistry. 48: 3352—3358.
[135] . Young M. A., Stuart D. A., Lyandres O., Glucksberg M. R., and Van Duyne R. P. (2004).
«Surface-enhanced Raman spectroscopy with a laser pointer light source and minia-
ture spectrometer». Canadian Journal of Chemistry—Revue Canadienne De Chimie. 82:
1435-1441.
ГЛАВА 10
МАСС-СПЕКТРОМЕТРИЯ
С ИНДУКТИВНО
СВЯЗАННОЙ ПЛАЗМОЙ
В ЭКОЛОГИЧЕСКОМ
АНАЛИЗЕ
А. Ю. Леи кин, С. Нэлмс
10.1. Введение
Активная человеческая деятельность привела к накоплению значительного количества
выбросов экзогенных веществ в окружающей среде (ОС). Проблема загрязнения ОС со-
стоит не только в накоплении органических загрязнителей, но также в выбросах ядо-
витых неорганических веществ. Некоторые простые неорганические молекулы, а также
некоторые химические элементы представляют большую угрозу для ОС даже в сравне-
нии с известными токсичными органическими соединениями. Например, предельно
допустимая концентрация (ПДК) кадмия в воздухе рабочей зоны (ПДКр ), составля-
ющая 0,01—0,02 мг/м3 (Huang, 2004), значительно ниже ПДК фосгена (0,08 мг/л) (Health
Protection Agency, 2007), являющегося боевым отравляющим веществом. В качестве дру-
гого примера можно привести бериллий, ПДКр з которого составляет 0,002 мг/м3 и будет
в дальнейшем снижена до 0,0002—0,00002 мг/м3 (Butler et aL, 2010).
В то же время все тяжелые металлы и многие другие токсичные элементы и их со-
единения широко используются не только в промышленности и машиностроении, но и
в производстве бытовых изделий, непосредственно контактирующих с человеком. На-
пример, соединения кадмия используются для производства цветных красок, наноси-
мых на стеклянную и керамическую посуду, контактирующую с продуктами питания.
Соли свинца, олова и бария используются в качестве модификаторов сушки лакокра-
сочных покрытий при производстве мебели. Неудивительно, что тщательный контроль
присутствия токсичных элементов на низком уровне стал широко востребованным.
Помимо низких пределов обнаружения (ПО), методы, используемые для анали-
за следовых концентраций элементов в ОС, должны обладать высокой производи-
тельностью. Низкая производительность явилась одним из факторов, который при-
вел к доминированию атомно-эмиссионной спектрометрии с индуктивно связанной
плазмой (ИСП-АЭС) над атомно-абсорбционной спектрометрией с электротермиче-
ской атомизацией (ААС-ЭТА). Другой современный многоэлементный метод — масс-
спектрометрия с индуктивно связанной плазмой (ИСП-МС) (Montaser, 1998; Thomas,
10.2. Основы метода
2004; Nelms, 2005; Hill, 2007) — обладает еще большей производительностью по сравне-
нию с ИСП-АЭС и демонстрирует аналогичные, а во многих случаях более низкие ПО
по отношению к ААС-ЭТА. В дополнение метод ИСП-МС значительно менее зависим
от матрицы по сравнению с ААС-ЭТА.
С момента первого применения ИСП-МС для экологического анализа в 1986 году
(Gray, 1986) метод нашел широкое применение как эффективный инструмент контроля
качества ОС. Опубликовано множество статей по использованию ИСП-МС для анали-
за объектов ОС. Обзор, посвященный конкретным случаям применения ИСП-МС для
анализа экологических образцов, ежегодно публикуется в журнале Journal of Analytical
Atomic Spectrometry в рубрике Atomic Spectrometry Update — Environmental analysis.
В настоящей главе приведены общие сведения по использованию ИСП-МС в эколо-
гическом анализе и указаны источники для дальнейшего поиска информации.
10.2. Основы метода
К настоящему моменту опубликовано несколько книг (Montaser, 1998; Thomas, 2004;
Nelms, 2005; Hill, 2007)\ в подробностях описывающих основные принципы работы
и конструкции различных типов приборов ИСП-МС. В этом разделе приведено упро-
щенное описание основ метода.
В большинстве случаев образцами для ИСП-МС являются водные растворы. Наи-
более подходящей средой для поддержания неорганических веществ в растворе явля-
ется 2%-я азотная кислота. Иногда для растворения некоторых элементов (Sn, Si, Cr,
Zr, W и др.) может потребоваться добавление соляной или фтористоводородной кислот.
Количество добавляемой НС1 и HF должно быть сведено к минимуму из-за спектраль-
ных наложений, вызываемых наличием хлора, и коррозионной активности НЕ Серную
и фосфорную кислоты обычно стараются полностью исключить из-за большого коли-
чества наложений, которые наблюдаются в присутствии этих кислот, и значительного
изменения вязкости раствора, влияющей на эффективность распыления.
Твердые образцы или жидкости, содержащие крупные частицы, должны быть мине-
рализованы перед анализом для полного растворения твердых частиц и перевода в рас-
твор содержащихся в них элементов. Минерализацию можно провести рядом методов,
включая озоление, автоклавное или микроволновое разложение с использованием кис-
лот и сплавление с последующим растворением плава (Dean, 2003). Благодаря активному
развитию лазерного пробоотбора стало возможным проведение прямого анализа твердых
образцов ИСП-МС (см. раздел 10.4.6). Сопряжение газовой хроматографии (ГХ) с ИСП-
МС позволяет проводить прямой анализ газообразных образцов (см. раздел 10.4.4).
1 Дополнительная литература на русском языке:
• Музгин В. Н., Емельянова Н. Н., Пупышев А. А. (1998). «Масс-спектрометрия с индуктивно свя-
занной плазмой — новый метод в аналитической химии». Аналитика и контроль. 2, 3—4: 3—25;
• Пупышев А.А., Суриков В.Т. (2006). «Масс-спектрометрия с индуктивно связанной плазмой».
Образование ионов. Екатеринбург: УРО РАН;
• Карандашев В. К., Туранов А. Н., Орлова Т.А., Лежнев А.Е., Носенко С. В., Золотарева Н.И.,
Москвина И.Р. (2007). «Использование метода масс-спектрометрии с индуктивно связанной
плазмой в элементном анализе объектов окружающей среды». Заводская лаборатория. Диагно-
стика материалов. 73, 1: 12—22.
Рис. 10.1. Общая схема конструкции ИСП-МС: 1 — распылительная камера с пневмати-
ческим распылителем; 2 — плазменная горелка; 3 — индуктор; 4 — пробоотбор-
ный конус (самплер); 5 — скиммер-конус; 6 — система ионной оптики и масс-
фильтр; 7 — детектор ионного тока
Традиционная ИСП-МС является масс-спектрометрией положительно заряжен-
ных ионов, образующихся в индуктивно связанной плазме. Плазма формируется вслед-
ствие поглощения аргоном высокочастотного электромагнитного излучения при про-
хождении через кварцевую горелку, помещенную в индуктор. В общих чертах принцип
работы ИСП-МС, представленный на рис. 10.1, состоит в следующем.
1. Анализируемый раствор переводится в аэрозоль, как правило, посредством пнев-
матического распылителя, установленного в распылительной камере.
2. Аэрозоль образца транспортируется в плазму по центральному каналу плазмен-
ной горелки.
3. В результате воздействия высокой температуры плазмы (-7000К) элементы, со-
держащиеся в образце, ионизируются с образованием положительно заряженных
ионов.
4. Ионы проходят через интерфейс в вакуумную часть спектрометра.
5. В вакуумной части ионы разделяются по величинам m/z.
6. Конечной стадией является детектирование величины ионного тока.
Разделение ионов по величинам m/z в ИСП-МС достигается посредством анали-
заторов, таких, как квадрупольный, магнитный секторный и времяпролетный (Batey
et al., 2005). Квадрупольные приборы ИСП-МС используются наиболее широко. Буду-
чи простыми в эксплуатации, быстрыми и относительно недорогими, они позволяют
успешно решать большинство аналитических задач. Секторные приборы демонстриру-
ют непревзойденную чувствительность и более низкие ПО в сравнении с квадруполь-
ными приборами. Для высокоточного определения изотопных отношений разработаны
мульти коллекторные приборы на основе магнитного анализатора.
10.3. Преимущества и недостатки
ИСП-МС стала очень популярным методом для экологического и других видов анализа
благодаря следующим характеристикам:
• чрезвычайно высокая чувствительность;
10.3. Преимущества и недостатки
• отличная стабильность сигнала в широком диапазоне концентраций;
• широкий динамический диапазон, составляющий несколько порядков величи-
ны (является большим преимуществом перед другими методами анализа, по-
скольку величины концентрации могут достаточно сильно варьироваться от об-
разца к образцу).
Тем не менее метод обладает рядом недостатков, особенно если речь идет об опреде-
лении примесей на уровне долей ppb.
10.3.1. Спектральные интерференции
Эта проблема обусловлена наложением сигналов различных ионов с одинаковой вели-
чиной m/z. Точное определение следовых концентраций некоторых элементов осложня-
ется как изобарными интерференциями, обусловленными наложением сигналов изотоп-
ных ионов с одинаковой номинальной массой (например 87Rb и 87Sr), так и различными
полиатомными наложениями (оксиды, аргиды, хлориды и др. ионы) и двухзарядными
ионами. Несколько сотен возможных ионов-интерферентов (May and Wiedmeyer, 1998\
могут присутствовать в масс-спектре в зависимости от состава образца.
Наиболее интенсивные интерференции наблюдаются в диапазоне ниже 80—
100 атомных единиц массы (а.е.м.). Даже в спектре особо чистой деионизованной воды
в этом диапазоне наблюдаются сигналы с интенсивностью от сотен до миллионов им-
пульсов в секунду (рис. 10.2). Наиболее проблемными изотопами являются 56Fe+ (на-
ложение 40Аг16О+ и 40Cal6O+), 80Se+ (40Ar2+), 75As (40Ar35Cl+), 5IV (35С116О+) и некоторые другие.
В квадрупольной ИСП-МС влияние спектральных наложений может быть учтено
с использованием уравнений математической коррекции или устранено с использова-
нием реакционно-столкновительных ячеек (Koyanagi et al., 2005)1 2. Пример подавления
Рис. 10.2. Масс-спектр деионизованной воды в диапазоне 35—65 а.е.м. без (серый)
и с (черный) использованием реакционно-столкновительной ячейки
1 Дополнительная литература на русском языке:
Пупышев А. А., Эпова Е. Н. (2001). «Спектральные помехи полиатомных ионов в методе масс-
спектрометрии с индуктивно связанной плазмой». Аналитика и контроль. 5, 4: 335—369.
2 Дополнительная литература на русском языке:
Лейкин А. К)., Акимович П.В. (2012). «Системы подавления спектральных интерференций в
масс-спектрометрии с индуктивно связанной плазмой». ЖАХ. 67, 8: 752—762.
Глава 10. Масс-спектрометрия с индуктивно связанной плазмой в экологическом
анализе
m/z
Рис. 10.3. Разрешение пиков 52Сг и 40Аг12С в секторном приборе ИСП-МС
спектральных интерференций с использованием реакционно-столкновительной ячей-
ки приведен на рис. 10.2. Избавиться от многих полиатомных интерференций можно,
применив высокое спектральное разрешение секторного прибора. В качестве приме-
ра можно привести разрешение пиков 52Сг и 40Аг12С (Адл = 0,021875 а.е. м.), достигнутое
на таком приборе (рис. 10.3).
10.3.2. Неспектральные интерференции (матричные эффекты)
Неспектральные интерференции представляют собой ряд явлений, обусловленных
присутствием матрицы и проявляющихся в подавлении сигнала (в некоторых случа-
ях — возрастании) и дрейфах, не зависящих от спектральных наложений (Evans and
Giglio, 1993; Horlick and Montaser, 1998). В экологическом анализе одну из основных
проблем ИСП-МС представляет возможное высокое содержание растворенного ве-
щества. При анализе концентрированных растворов имеет место существенное по-
давление сигнала по сравнению с растворами с более низкой концентрацией (Olivares
and Houk, 1986; Tan and Horlick, 1987; Allen et aL, 1997). В общем случае для анализа
методом ИСП-МС концентрация растворенного вещества должна быть ниже 0,2%
(2 г/л) (Houk, 1986; Douglas and Kerr, 1988). Матричные эффекты способствуют суже-
нию динамического диапазона метода, ухудшают ПО и иногда делают невозможным
прямой анализ. В таком случае может потребоваться отделение матрицы и/или пред-
варительное концентрирование аналитов. Если разбавление образца до приемлемой
концентрации матрицы невозможно, например как следствие изначально низкой
концентрации целевых аналитов, для компенсации матричных эффектов могут быть
использованы внутренние стандарты (Horlick and Montaser, 1998). Разбавление зача-
стую не является практичным, особенно при большом потоке образцов, поэтому ис-
пользование внутренних стандартов является обычной практикой при анализе мето-
дом ИСП-МС.
10.4. Практическое применение
10.4. Практическое применение
10.4.1. Анализ воздуха
Основная сложность при определении токсичных элементов в воздухе связана со сбо-
ром и подготовкой образца. Поскольку большинство неорганических веществ нелетучи
и находятся в воздухе в виде мелкодисперных частиц, необходимо собрать достаточное
количество образца для получения раствора с концентрацией на уровне или выше пре-
дела определения (Butler et al., 2008).
Вполне вероятно, что содержание экотоксикантов в частицах аэрозоля может суще-
ственно варьироваться в зависимости от размера частиц и их источника. Например, со-
держание Pt и Pd в аэрозольных частицах с размером 10 и 2,5 мкм и содержание каждого
металла в аэрозоле в целом оказалось равным 14/9/20 и 58/15/82 пг/м3 соответственно
(lavicoli et al., 2008). В этой связи крайне желательно определять не только общее содер-
жание загрязняющих веществ в аэрозоле, но также их распределение по частицам раз-
ного размера.
Наиболее подвижные и опасные частицы имеют размер менее 10 мкм. В связи с этим
последняя актуальная тенденция заключается в сборе частиц с размером менее 2,5 мкм.
Сбор таких микроскопических частиц является трудной задачей, поскольку частицы
проникают сквозь большинство фильтров. Для этих целей разрабатываются и произво-
дятся различные специальные устройства для извлечения аэрозольных частиц из воз-
духа (Butleret al., 2008; Gorbunov etal., 2009).
Процедура пробоотбора может длиться несколько часов, после чего фильтры извле-
каются из устройства и подвергаются отмывке или разложению с использованием под-
ходящих реагентов. Показано, что материал фильтра может существенно влиять на ПО
(Upadhyay et al., 2009). В общем случае считается, что в будущем при анализе воздуха
с использованием ИСП-МС ПО будут полностью лимитироваться контрольным опы-
том (Rasmussen etal., 2007).
Большие усилия прикладываются для разработки точных методов определения
элементов в аэрозольных частицах. На сегодняшний момент в США и Евросоюзе (ЕС)
действуют два стандартных метода: ЕРА 10-3.5 (US Environmental Protection Agency,
1999) и ISO 30011 (International Organization for Standardization, 2010), предназначенные
для определения металлов и некоторых других элементов в воздухе, с использованием
метода ИСП-МС.
10.4.2. Анализ воды
Анализ воды методом ИСП-МС является относительно простой задачей, поскольку
в большинстве случаев не требует предварительной пробоподготовки. Однако если вода
содержит большое количество взвешенных частиц и/или органического углерода, перед
анализом может потребоваться процедура разложения.
В США и ЕС для определения микроэлементов в воде приняты на вооружение мето-
ды ЕРА 200.8 (US Environmental Protection Agency) и ISO 17294-2 (International Organization
Глава 10. Масс-спектрометрия с индуктивно связанной плазмой в экологическом
анализе
for Standardization, 2003)1. Метод ЕРА 200.8 описывает процедуру определения Al, Sb, As,
Ba, Be, Cd, Cr, Co, Cu, Pb, Mn, Hg, Mo, Ni, Se, Ag, Tl, Th, U, V и Zn в питьевой и сточной
воде. В методе приводятся процедуры пробоподготовки, калибровки, проведения из-
мерений, коррекции спектральных наложений. В нем также установлены требования
к контролю качества и валидации прибора. Типичные значения ПО в соответствии с ме-
тодом ЕРА 200.8, достигаемые на квадрупольном ИСП-МС, представлены в табл. 10.1.
Линейный динамический диапазон простирается от долей мкг/л до нескольких тысяч
мг/л в зависимости от элемента (Thermo Scientific).
Таблица 10.1. Типичные ПО для квадрупольного ИСП-МС в соответствии с методом
ЕРА 200.8 (Thermo Scientific)
Элемент ПО (мкг/л) Элемент ПО (мкг/л) Элемент ПО (мкг/л)
Ag 0,01 Сг 0,02 Sb 0,02
Al 0,01 Си 0,01 Se 0,2
As 0,02 Hg 0,02 Th 0,004
Ba 0,02 Mn 0,007 Tl 0,002
Be 0,009 Mo 0,001 U 0,002
Cd 0,007 Ni 0,01 V 0,01
Co 0,005 Pb 0,002 Zn 0,03
Поскольку метод ЕРА 200.8 был внедрен в 1991 г., в нем не только не упоминается,
но и не разрешается использование таких современных возможностей ИСП-МС, как
подавление спектральных наложений с помощью реакционно-столкновительных яче-
ек. Такие «нестандартные» условия могут увеличить количество определяемых элемен-
тов, существенно понизить пределы обнаружения и расширить динамический диапа-
зон метода. Современные приборы ИСП-МС способны измерять содержание элементов
I и II групп на уровне мг/л несмотря на то, что определение этих элементов не одобрено
в методе ЕРА 200.8.
В отличие от ЕРА 200.8 европейские законы не оговаривают конкретных процедур
проведения измерений. Европейский стандарт ISO 17294 не предоставляет строгих ме-
тодических указаний и по большей части опирается на рабочие характеристики при-
бора. Метод в деталях описывает требуемые аналитические характеристики, такие как
правильность, точность, пределы обнаружения и т. д., но не дает четких указаний для
достижения упомянутых характеристик.
1 На территории РФ действуют следующие методы, использующие ИСП-МС для анализа
микроэлементного состава воды:
• НСАМ 480-Х (ИСП-МС-определение элементного состава природных и питьевых вод со степе-
нью минерализации до 2000 мг/л);
• НСАМ 481-Х (ИСП-МС-определение общей ртути в природных и питьевых водах со степенью
минерализации до 2000 мг/л);
• ФР.1.31.2000.00128 (МВИ элементного состава питьевой, природной и сточной воды методом
масс-спектрометрии с ионизацией в индуктивно-связанной плазме);
• методики НСАМ (научный совет по аналитическим методам) распространяются Всероссий-
ским институтом минерального сырья.
10.4.3. Анализ твердых образцов
Общеизвестно, что микроэлементный состав воды напрямую зависит от состава грун-
тов, донных отложений и ила. Накопленные донные отложения представляют собой
хороший источник информации о возможном времени сброса экотоксикантов. Высво-
бождение загрязняющих веществ из седиментов и ила в результате анаэробных про-
цессов может стать фактором существенного вторичного загрязнения. Поэтому анализ
состава донных отложений также важен, как и анализ плодородия почв (Worsfold, 2008).
Агентством по охране окружающей среды США (Environmental protection agency,
ЕРА) приняты на вооружение 2 метода: 6020А (US Environmental Protection Agency, 2007)
и ILM05.3 (US Environmental Protection Agency, 2006), использующие ИСП-МС для ана-
лиза почв, донных отложений, ила, твердых отходов и целого ряда других объектов ОС,
которые могут быть каким-либо образом минерализованы1. Аналогично тому, как это
сделано в методе ЕРА 200.8, методы 6020А и ILM05.3 строго регламентируют процедуры
проверки аналитических характеристик, таких как точность калибровки масс, спек-
тральное разрешение, стабильность и точность, пределы обнаружения, линейный ди-
намический диапазон, уровень интерференций и др.
Департамент окружающей среды Соединенного Королевства (UK Environment
Agency) внес поправки в схему сертификации средств мониторинга (Monitoring
Certification Scheme, MCERTS) для обеспечения качества результатов анализа объектов
окружающей среды. Схема MCERTS включает методики анализа почв. Процедура ана-
Таблица 10.2. Типичные ПО элементов в почве после экстракции царской водкой и ана-
лиза методом ИСП-МС (Spence, 2006)
Элемент ПО (мг/кг) Элемент ПО (мг/кг)
As* * 0,2 Мп* 0,1
В 0,4 Мо 0,1
Ва 0,05 Ni* 0,05
Be 0,1 Pb 0,05
Cd 0,05 Sb 0,02
Со* 0,1 Se* 0,3
Сг* 0,1 Sn 0,03
Си* 1 Т1 0,1
Fe* 1 V* 0,1
Hg 0,03 Zn* 1
* Для получения низких ПО требуется использование реакционно-столкновительных ячеек.
1 На территории РФ действуют следующие методы, использующие ИСП-МС для анализа
микроэлементного состава твердых объектов окружающей среды:
• НСАМ 499-С (определение элементного состава почв, грунтов и донных отложений атомно-
эмиссионным и масс-спектральным методами);
• НСАМ 500-С (определение элементного состава азотнокислых и ацетатно-аммонийных вытя-
жек из почв методом масс-спектрометрии с индуктивно-связанной плазмой);
• НСАМ 502-С (определение элементного состава образцов растительного происхождения (тра-
вы, листья) атомно-эмиссионным и масс-спектральным методами анализа);
• НСАМ 509-Х (ИСП-МС-анализ твердых полезных ископаемых Мирового океана).
Глава 10. Масс-спектрометрия с индуктивно связанной плазмой в экологическом
анализе
лиза состоит из стадий сушки, просеивания и разложения с использованием кислот,
например царской водки, с последующим анализом полученных растворов. Типичные
пределы обнаружения по описанной методике приведены в табл. 10.2. Следует иметь
в виду, что разные процедуры разложения с использованием различных реагентов при-
водят к получению различающихся результатов {Bettinelli et al., 2000). По этой причине
использование стандартных образцов для проверки правильности анализа является
нормальной и зачастую необходимой процедурой.
Элементный состав практически любого экологического объекта может дать по-
лезную информацию о качестве окружающей среды и потенциальных угрозах для нее.
Ниже приведены еще два примера задач, которые может решить ИСП-МС по результа-
там анализа проб объектов окружающей среды.
1. Содержание платиноидов в дорожной пыли служит индикатором интенсивно-
сти автомобильного движения и связанного с ним загрязнения. Элементы пла-
тиновой группы поступают в ОС с автомобильными выхлопами, прошедшими
через каталитический нейтрализатор {Zereini et aL, 2001). По литературным дан-
ным, концентрация Pt и Pd в дорожной пыли может находиться на уровне 101 —
1730 мг/кг и 28—174 мг/кг соответственно {Prohaska et aL, 2003; Wichmann et aL,
2007; Sutherland et aL, 2008).
2. Концентрация элементов в растениях может четко отражать содержание и биодо-
ступность этих элементов в почве. Способность некоторых растений концентри-
ровать определенные элементы из почвы широко используется для биорекульти-
вации {Beauchemin, 1989).
10.4.4. Установление формы нахождения элементов
Общеизвестно, что различные химические формы элементов существенно различаются
по биодоступности, подвижности и токсичности {Dopp et aL, 2004; Jenkins et aL, 2007).
Хорошим примером, иллюстрирующим это, является необходимый ряду организмов
Сг(Ш) и, напротив, канцерогенный и мутагенный Cr(VI) {TakahashietaL, 2004). He толь-
ко степень окисления, но и состав химического соединения, в виде которого существует
элемент, определяет его воздействие на окружающую среду. Например, известно, что
неорганические соединения мышьяка, в особенности As (III), являются более токсич-
Рис. 10.4. ИСП-МС XSeries 2, сопряженный с газовым хроматографом Trace GC Ultra
10.4. Практическое применение
250000
£ 200000
о
| 150000
| 100000
U 50000
Рис. 10.5. Хроматограмма образца, содержащего различные бутил- и фенилзамещенные
соединения олова и ртути, полученная с использованием ГХ-ИСП-МС
ными, чем органические (Ascaret al., 2008), и обладают наибольшей подвижностью в по-
чве (Bacon and Davidson, 2008). По этой причине для более полного понимания ущер-
ба окружающей среде важной задачей является определение конкретных химических
форм нахождения элемента.
Определение формы нахождения элемента достигается избирательной экстракцией
таких форм с последующим анализом экстрактов (Dean, 2003; Rao et al., 2008; Schmidt
et al., 2008; Popp et al., 2010). Более эффективным и удобным способом является сопря-
жение различных типов хроматографических систем (жидкостной, газовой, ионной,
эксклюзионной, капиллярного электрофореза) с ИСП-МС (рис. 10.4). Ряд недавних об-
зоров (Takahashi et al., 2004; Ray et al., 2004; Krupp et al., 2005) посвящен современному
состоянию методов определения форм нахождения элементов.
Наиболее востребовано определение форм нахождения таких элементов, как Hg, As,
Cr, Se, Sn и Sb. Среди других, менее «традиционных» элементов, в недавнем прошлом
добавленных в список, находятся I, Br, Р и S (Beauchemin, 1989; Ray etal., 2004; Krupp etal.,
2005). Определение форм нахождения последних используется для контроля содержа-
ния ряда пестицидов в окружающей среде (Profrock et al., 2004; Fidalgo-Usedet al., 2005).
Некоторые современные исследования демонстрируют возможность одновремен-
ного определения форм нахождения нескольких элементов. Например, ГХ-ИСП-МС ис-
пользовали для определения летучих металлоорганических соединений в газообразных
образцах различной природы. Рис. 10.5 иллюстрирует возможность одновременного
определения ряда олово- и ртутьорганических соединений с использованием ГХ-ИСП-
МС. Помимо вышеописанных веществ авторы определяли также другие элементоорга-
нические соединения, содержащие As, Bi, Sb и Pb (Maillefer et al., 2003). В работе (Duester
et al., 2005) описано использование pH-градиентной гидридной генерации в сочетании
с ГХ-ИСП-МС для одновременного определения форм нахождения Sn, As и Sb в почве.
10.4.5. Определение изотопного состава
Показано, что изотопный состав природных элементов может отличаться от состава эле-
ментов, поступающих в окружающую среду из антропогенных источников. Этот факт
делает изотопный анализ ценным инструментом определения источника загрязнения.
В этой связи очень востребовано определение изотопов свинца (Komarek et al., 2008).
В работе (Novak et al., 2003) приведены результаты исследования отношения 206РЬ/207РЬ
Глава 10. Масс-спектрометрия с индуктивно связанной плазмой в экологическом
анализе
в двух образцах болотного торфа — одного с возрастом 11 000 лет, не измененного за счет
антропогенного влияния, и другого, датированного 1880 годом. Оказалось, что с тече-
нием времени величина изотопного отношения изменилась с 1,193 до 1,168—1,178, что
является отчетливым индикатором антропогенного загрязнения. Другой пример пока-
зывает существенное отклонение изотопного состава свинца от природного в лишай-
нике в окрестностях медеплавильного завода (Spiro etal., 2004). Аналогичное исследова-
ние (Krachler et aL, 2004) показало, что несмотря на тот факт, что концентрация свинца
в пробах арктического льда оставалась практически неизменной с 1852 по 1974 гг. и со-
ставляла 6 и 9 пг/г соответственно, отношение 206РЬ/207РЬ изменилось с 1,147 до 1,169.
Этот факт является индикатором того, что в разные периоды времени свинец поступал
в окружающую среду из разных источников. В качестве других индикаторов антропо-
генного загрязнения можно привести цинк (Sivry et aL, 2008; Gioia et aL, 2008; Mattielli
etal., 2009) и, безусловно, радиоактивные изотопы.
Благодаря возрастающей роли атомной энергии и распространению радиоактив-
ных материалов определение радионуклидов стало широко востребовано. Изотопный
состав радиоактивных элементов может служить индикатором их источника — аварий
на АЭС, поизводства ядерного оружия и др. (Boulyga et aL, 2001). Определение радио-
нуклидов может быть осуществлено обычными радиометрическими методами, такими
как ос-спектрометрия или жидкостная сцинтилляционная радиометрия. Однако эти
методы часто не дают достаточной чувствительности по долгоживущим радиоизото-
пам в экологических образцах. Более того, с помощью этих методов не всегда возможно
определение отдельных изотопов элемента.
ИСП-МС является эффективной альтернативой традиционным методам опреде-
ления радионуклидов благодаря высокой чувствительности, хорошей точности и отно-
сительно несложной пробоподготовке (Morrow and Crain, 1995; Morrow and Crain, 1998).
Мульти коллекторные секторные приборы ИСП-МС в наилучшей степени подходят
для высокоточного изотопного анализа (Becker and Dietze, 2000). Однако эти приборы
весьма дороги, а сверхвысокая точность изотопного анализа не всегда востребова-
на. Квадрупольные масс-спектрометры обеспечивают удовлетворительную точность
определения изотопных отношений и достаточную чувствительность для детектиро-
вания трансурановых элементов. Таким образом, они становятся все более востребо-
ванными для решения такого рода задач (Liezers et aL, 1998; Muramatsu et aL, 1998; Boulyga
et aL, 2001; Ketterer et aL, 2008).
Лазер
Газ-носитель
Образец
Рис. 10.6. Схематическое представление конструкции и принципа работы ЛА
10.4. Практическое применение
10.4.6. Лазерный пробоотбор в ИСП-МС
Лазерная абляция (ЛА) позволяет осуществлять прямой микроотбор твердых образцов.
Упрощенная схема, иллюстрирующая принцип работы ЛА, представлена на рис. 10.6.
Луч лазера воздействует на поверхность образца, генерируя мелкодисперсный аэрозоль,
который транспортируется потоком газа в область индуктивно связанной плазмы. В со-
пряжении с ИСП-МС ЛА является мощным инструментом анализа различных объек-
тов. ЛА была впервые использована в геохимии и с тех пор нашла признание в ряде об-
ластей, включая анализ объектов окружающей среды (Gunther et al., 2000; Veinott, 2001;
Vogt and Latkoczy, 2005; Ghazi, 2007).
ЛА-ИСП-МС предоставляет следующие преимущества:
• минимальные трудозатраты на подготовку образца;
• минимальное количество образца, требующееся для анализа;
• возможность контроля изменения концентрации по площади/объему образца;
• возможность анализа различных типов образцов.
ЛА-ИСП-МС особенно полезна при анализе образцов, содержащих пространствен-
но зашифрованную информацию о состоянии окружающей среды на момент форми-
рования самого образца. Некоторые примеры таких носителей информации включают
кольца поперечного среза древесины (Monticelli et aL, 2009), раковины моллюсков, ко-
сти и т.д. (Ghazi, 2007). В качестве других примеров использования ЛА-ИСП-МС можно
привести анализ почв и отложений (Arroyo et al., 2009), льда (Reinhardt et al., 2001), изо-
топных отношений (Gasteletal., 1997; Hirata, 1997; Woodhead, 2008) и анализ частиц аэро-
золей (Rauch etal., 2002).
В последнее время особое внимание привлекли фемтосекундные лазеры (Fernandez
etal., 2007; Koch and Gunther, 2007; Sylvester, 2008). По сравнению с наносекундными лазе-
рами они предоставляют ряд преимуществ:
• более высокая эффективность испарения образца;
• более узкий диапазон размеров частиц;
• улучшенное пространственное разрешение;
• менее выраженное фракционирование элементов и индивидуальных изотопов
в процессе пробоотбора (Horn and Blanckenburg, 2007).
Основной трудностью при использовании ЛА для количественного анализа с по-
мощью ИСП-МС является необходимость применения калибровочных стандартов
в сходной с анализируемым образцом матрице. В литературе приводится множество
публикаций, посвященных разработке калибровочных стандартов и методик, дающих
правильные результаты для различных типов образцов (Sylvester, 2006; Butleretal., 2009).
10.4.7. Стандартные образцы
Как отмечалось ранее, даже стандартные методы не всегда предоставляют четкие ин-
струкции по проведению анализа. Различия в процедурах пробоподготовки и изме-
рений могут приводить к получению отличающихся результатов при анализе одного
и того же образца. Помимо этого, разные лаборатории могут разрабатывать свои соб-
ственные версии методик и нуждаются в критериях, подтверждающих правильность
разрабатываемых методик.
Глава 10. Масс-спектрометрия с индуктивно связанной плазмой в экологическом
анализе
Qoo
В таком случае правильность методики анализа определяется путем исследования
стандартного образца с сертифицированным содержанием определяемых компонен-
тов. Две наиболее известные организации, предоставляющие стандартные образцы для
анализа ряда материалов, — Национальный институт стандартов и технологии США
(National Institute of Standards and Technology, NIST) и Институт эталонных материалов
и измерений (Institute of Reference Materialsand Measurements, I RM M, www.irmm.jrc.be).
Образцы NIST для анализа микроэлементного состава объектов окружающей среды
можно найти на сайте www.nist.gov в разделах «Металлы в природных матрицах» (Metal
constituents in natural matrices), «Геологический материалы и руды» (Geological materials
and ores) и «Сельскохозяйственные материалы» (Agricultural materials). Эти материалы
представляют собой образцы почв, илов, отложений, воды, городской пыли, фильтров
с осажденными частицами и др. Образцы N1ST 2108, 2109 и 2701 представляют собой
стандарты для определения форм нахождения Cr(III)/Cr(VI) в воде и почве. Стандарты
синтетического стекла NIST 1872 и 1873 разработаны для проверки правильности опре-
деления методом ЛА-ИСП-МС. Стандарты BCR (Bureau Communautaire de Reference)
и ERM (European Reference Material), производимые IRMM, также представляют собой
различные типы экологических образцов для определения микроэлементного состава
и извлекаемых форм элементов.
Информацию практически по всем стандартным образцам, упоминавшимся в ли-
тературе, можно найти в базе данных GeoRem, доступной в Интернете по адресу www.
georem.mpch-mainz.gwdg.de. В дополнении к этому опубликованы два подробных обзора
(Jochum and Willbold, 2006; Jochum and Brueckner, 2008), дающих информацию по целому
ряду стандартных образцов, проанализированных за период 2004—2007.
Литература
[1] . Allen L. A., Leach J. J., Houk R. S. (1997). «Spatial location of the space charge effect in in-
dividual icon clouds using monodisperse dried microparticulate injection with a twin quad-
rupole inductively coupled plasma mass spectrometer». Anal. Chem. 69(13), 2384—2391.
[2] . Arroyo L., Trejos T., Gardinali P. R., Almirall J. R. (2009). «Optimization and validation of a
laser ablation inductively coupled plasma mass spectrometry method for the routine analysis
of soils and sediments». Spectrochim. Acta Part B: Atom. Spectr. 64(1), 16—25.
[3] . Ascar L., Ahumada I., Richter P. (2008). «Influence of redox potential on the availability of
arsenic species in soils and soil amended with biosolid». Chemosphere. 72(10), 1548—1552.
[4] . Bacon J. R., Davidson С. M. (2008). «Is there a future for sequential chemical extraction».
Analyst. 133(1), 25—46.
[5] . Batey J.H., Prohaska T., Horstwood M.S.A., Nowell G.M., Goenaga-lnfante H., Ei-
den G.C. (2005). «Mass spectrometers». In: Nelms S.M. (Ed.), Inductively coupled plasma
mass spectrometry handbook. CRC Press, Boca Raton.
[6] . Beauchemin D. (1989). «The ICP-MS approach to environmental studies». Mikrochim. Acta.
99(3-6), 273-281.
[7] . Becker J. S., Dietze H.-J. (2000). In: Meyers R.A. (Ed.), Encyclopedia of Analytical Chemis-
try. John Wiley, Chichester, UK. pp. 12947—12961.
[8] . Bettinelli M., Baffi C., Beone G.M., Spezia S. (2000). «Soils and sediments analysis by
spectroscopic techniques Part II: Determination of trace elements by ICP-MS». At. Spec-
trosc. 21(2), 60—70.
[9] . Boulyga S. F., Testa C., Desideric D., Becker J. S. (2001). «Optimisation and application of
ICP-MS and alpha-spectrometry for determination of isotopic ratios of depleted uranium
and plutonium in samples collected in Kosovo». J. Anal. At. Spectrom. 16(11), 1283—1289.
[10] . Butler O.T., Cooke J. M., Harrington C. F., Hill S.J., Rieuwerts J., Miles D. L. (2008).
«Atomic spectrometry update. Environmental analysis». J. Anal. At. Spectrom. 23(2),
249-286.
[11] . Butler O.T., Cooke J. M., Davidson C.M., Harrington C. F., Miles D. L. (2009). «Atomic
spectrometry update. Environmental analysis». J. Anal. At. Spectrom. 24(2), 131—177.
[12] . Butler O.T., Cairns W. R.L., Cooke J.M., Davidson C.M. (2010). «Atomic spectrometry
update. Environmental analysis». J. Anal. At. Spectrom. 25(2), 103—141.
[13] . Dean J. R. (2003). Methods for environmental trace analysis. John Wiley & Sons, Chichester,
UK.
[14] . Dopp E., Hartmann L.M., Florea A. M., Rettenmeier A. W., Hirner A.V. (2004). «Envi-
ronmental distribution, analysis and toxicity of organometalloid compounds». Crit. Rev.
Toxicol. 34(3), 301-333.
[15] . Douglas D. J., Kerr L. A. (1988). «Study of solids deposition on inductively coupled plasma
mass spectrometry samplers and skimmers». J. Anal. At. Spectrom. 3(6), 749—752.
[16] . Duester L.,. Diaz-Bone R.A, Kusters J., Hirner A.V. (2005). «Methylated arsenic, anti-
mony and tin species in soils». J. Environ. Monit. 7(12), 1186—1193.
[17] . Evans E.H., Giglio J. J. (1993). «Interferences in inductively coupled plasma mass spec-
trometry». J. Anal. At. Spectrom. 8(1), 1—18.
[18] . Fernandez B., Claverie F., Pecheyran C., Donard O. F.X. (2007). «Direct analysis of solid
samples by fs-LA-ICP-MS». TrAC-Trends Anal. Chem. 26(10), 951—966.
[19] . Fidalgo-Used N., Montes-Bayon M., Blanco-Gonzalez E., Sanz-Medel A. (2005). «De-
termination of organophosphorus pesticides in spiked river water samples using solid phase
microextraction coupled to gas chromatography with El-MS, ICP-MS detection». J. Anal.
At. Spectrom. 20(9), 876—882.
[20] . Gastel M., Becker J. S., Dietze H.-J. (1997). «Determination of long-lived radionuclides in
concrete matrix by laser ablation inductively coupled plasma mass spectrometry». Spectro-
chim. Acta Part В: Atom. Spectr. 52(14), 2051—2059.
[21] . Ghazi M. (2007). «Applications of laser ablation inductively coupled plasma mass spec-
trometry (LA-ICP-MS) in environmental forensic studies». In\ Murphy B.L., Morri-
son D. L. (Eds.), Introduction to environmental forensics. Second edition. Elsevier Academic
Press, Burlington, MA, USA.
[22] . Gioia S., Weiss D., Coles B., Arnold T., Babinski M. (2008). «Accurate and precise zinc
isotope ratio measurements in urban aerosols». Anal. Chem. 80(24), 9776—9780.
[23] . Gorbunov B., Priest N. D., Muir R.B., Jackson P. R., Gnewuch H. (2009). «А novel size-
selective airborne particle size fractionating instrument for health risk evaluation». Ann.
Occup. Hyg. 53(3), 225—237.
[24] . Gray A. (1986). «The evolution of the ICP as an ion source for mass spectrometry». J. Anal.
Atom. Spectrom. 1(6), 403—405.
Глава 10. Масс-спектрометрия с индуктивно связанной плазмой в экологическом
анализе
[25] . Gunther D., Horn I., Hattendorf В. (2000). «Recent trends and developments in laser abla-
tion-ICP-mass spectrometry». Fresenius’ J. Anal. Chem. 368(1), 4—14.
[26] . Health Protection Agency. (2007). HPA compendium of chemical hazards. Phosgene.
[27] . Hill S. J. (Ed.). (2007). Inductively coupled plasma spectrometry and its applications, second
ed. Blackwell Publishing Ltd., Oxford, UK.
[28] . Hirata T. (1997). «Ablation technique for laser ablation-inductively coupled plasma mass
spectrometry». J. Anal. At. Spectrom. 12(11), 1337—1342.
[29] . Horlick G. Montaser A. (1998). «Analytical characteristics of ICPMS». In’. Montaser A.
(Ed.), Inductively Coupled Plasma Mass Spectrometry. Wiley-VCH, N.-Y.
[30] . Horn I., Blanckenburg F. (2007). «Investigation on elemental and isotopic fractionation
during 196 nm femtosecond laser ablation multiple collector inductively coupled plasma
mass spectrometry». Spectrochim. Acta Part B: Atom. Spectr. 62(4), 410—422.
[31] . Houk R.S. (1986). «Mass spectrometry of inductively coupled plasmas». Anal. Chem. 58(1),
97A-105A.
[32] . Huang J. (2004). «Chinese National health standards for occupational exposure to cadmi-
um and diagnostic criteria of occupational chronic cadmium poisoning». Biometals. 17(5),
511.
[33] . lavicoli I., Восса В., Caroli S., Caimi S., Alimonti A., Carelli G., Fontana L. (2008). «Ex-
posure of rome city tram drivers to airborne platinum, rhodium, and palladium». J. Occup.
Env. Med. 50(10), 1158—1166.
[34] . International Organization for Standardization. (2003). ISO 17294-2:2003. Water quality —
Application of inductively coupled plasma mass spectrometry (ICP-MS) — Part 2: Determina-
tion of 62 elements.
[35] . International Organization for Standardization. (2010). ISO 30011:2010. Workplace air —
Determination of metals and metalloids in airborne particulate matter by inductively coupled
plasma mass spectrometry.
[36] . Jenkins R.O., Craig P.J., Francesconi K.A., Harrington C. F. (2007). «Environmental
and biological aspects of organometallic compounds». In’. Crabtree R. H., Mingos D. M. P.,
O’Hare D. (Eds.), Comprehensive Organometallic Chemistry III. 12, 603—661.
[37] . Jochum К. P., Brueckner S. M. (2008). «Reference materials in geoanalytical and environ-
mental research — Review for 2006 and 2007». Geostand. Geoanal. Res. 32(4), 405—452.
[38] . Jochum K.P., Willbold M. (2006). «Reference materials in geoanalytical research — Re-
view for 2004 and 2005». Geostand. Geoanal. Res. 30(3), 143—156.
[39] . Ketterer M. E., Szechenyi S. C. (2008). «Determination of plutonium and other transuranic
elements by inductively coupled plasma mass spectrometry: A historical perspective and
new frontiers in the environmental sciences». Spectrochim. Acta Part B: Atom. Spectr. 63(7),
719-737.
[40] . Koch J., Gunther D. (2007). «Femtosecond laser ablation inductively coupled plasma
mass spectrometry: achievements and remaining problems». Anal. Bioanal. Chem. 387(1),
149-153.
[41] . Komarek M., Ettler V., Chrastny V., Mihaljevic M. (2008). «Lead isotopes in environmen-
tal sciences: A review». Environ. Intern. 34(4), 562—577.
[42] . Koyanagi G. K., Bohme D. K., Bandura D. R. (2005). «Collision and reaction cells». In’.
Nelms S.M. (Ed.), Inductively coupled plasma mass spectrometry handbook. CRC Press,
Boca Raton.
Литература
[43] . Krachler М., Zheng J., Fisher D., Shotyk W. (2004). «Direct determination of lead isotopes
(206Pb, 207Pb, 208Pb) in arctic ice samples at picogram per gram levels using inductively
coupled plasma-sector field MS coupled with a high-efficiency sample introduction sys-
tem». Anal. Chem. 76(18), 5510—5517.
[44] . Krupp E., Seby E, Rodriguez R., Doimeadios M., Holliday A., Moldovan M., Kollensper-
ger G., Hann S., Donard O. F.X. (2005). «Trace metal speciation with ICP-MS detection».
In: Nelms S. M. (Ed.), Inductively coupled plasma mass spectrometry handbook. CRC Press,
Boca Raton.
[45] . Liezers M., Туе С. T., Mennie D., Koller D. (1998). «Ultra-low level (pg/L) Actinide deter-
minations and superior isotope ratio precisions by quadrupole ICP-MS». In: Morrow R. W.,
J. S. Crain (Eds.), Applications of inductively coupled plasma-mass spectrometry to radionu-
clide determinations, ASTM STP 1344. American Society for Testing and Materials, West
Conshohocken.
[46] . Maillefer S., Lehr C. R., Cullen W. R. (2003). «The analysis of volatile trace compounds in
landfill gases, compost heaps and forest air». Appl. Organmet. Chem. 17(3), 154—160.
[47] . Mattielli N., Petit J. C.J., Deboudt K., Flament P., Perdrix E., Taillez A., Rimetz-Plan-
chon J., Weis D. (2009). «Zn isotope study of atmospheric emissions and dry depositions
within a 5 km radius of a Pb-Zn refinery». Atmos. Environ. 43(6), 1265—1272.
[48] . May T.W., Wiedmeyer R. H. (1998). «А Table of Polyatomic Interferences in ICP-MS».
Atom. Spectr. 19(5), 150—155.
[49] . Montaser A. (Ed.). (1998). Inductively coupled plasma mass spectrometry. Wiley, N.-Y.
[50] . Monticelli D., Di Iorio A., Ciceri E., Castelletti A., Dossi C. (2009). «Tree ring microa-
nalysis by LA-1CP-MS for environmental monitoring: validation or refutation? Two case
histories». Microchim. Acta. 164(1—2), 139—148.
[51] . Morrow R.W., J.S. Crain (Eds.). (1995). Applications of inductively coupled plasma-mass
spectrometry to radionuclide determinations, ASTM STP 1291. American Society for Testing
and Materials, West Conshohocken.
[52] . Morrow R.W., J.S. Crain (Eds.). (1998). Applications of inductively coupled plasma-mass
spectrometry to radionuclide determinations, ASTM STP 1344. American Society for Testing
and Materials, West Conshohocken.
[53] . Muramatsu Y., Ruhm W., Yoshida S., Tagami K., Uchida S., Wirth E. (2000). «Concentra-
tions of 239Pu and 240Pu and their isotopic ratios determined by ICP-MS in soils collected
from the Chernobyl 30-km zone». Environ. Sci. Technol. 34(14), 2913—2917.
[54] . Nelms S.M. (Ed.). (2005). Inductively coupled plasma mass spectrometry handbook. CRC
Press, Boca Raton.
[55] . Novak M., Emmanuel S., Vile M. A., Erel Y, Veron A., Paces T., Wieder R. K., Vanicek M.,
Stepanova M., Bfizova E., Hovorka J. (2003). «Origin of lead in eight central European peat
bogs determined from isotope ratios, strengths, and operation times of regional pollution
sources». Environ. Sci. Technol. 37(3), 437—445.
[56] . Olivares J. A., Houk R. S. (1986). «Suppression of analyte signal by various concomitant
salts in inductively coupled plasma mass spectrometry». Anal. Chem. 58(1), 20—25.
[57] . Popp M., Hann S., Koellensperger G. (2010). «Environmental application of elemental spe-
ciation analysis based on liquid or gas chromatography hyphenated to inductively coupled
plasma mass spectrometry — A review». Anal. Chim. Acta. 668(2), 114—129.
304 Глава 10. Масс-спектрометрия с индуктивно связанной плазмой в экологическом
анализе
[58] . Profrock D., Leonhard Р., Wilbur S., Prange A. (2004). «Sensitive, simultaneous determi-
nation of P, S, Cl, Br and I containing pesticides in environmental samples by GC hyphen-
ated with collision-cell ICP-MS». J. Anal. At. Spectrom. 19(5), 623—631.
[59] . Prohaska T., Kollensperger G., Hann S., Stingeder G., Fitz W., Wenzel W. (2003). «Appli-
cation of ICP-MS in environmental science». In\ Holland G., Tanner S. D. (Eds.), Plasma
Source Mass Spectrometry: Applications and Emerging Technologies. The Royal Society of
Chemistry, Chambridge, UK.
[60] . Rao C. R. M., Sahuquillo A., Sanchez J. F. L. (2008). «А review of the different methods ap-
plied in environmental geochemistry for single and sequential extraction of trace elements
in soilsand related materials». Water air and soil pollution. 189(1—4), 291—333.
[61] . Rasmussen P. E., Wheeler A. J., Hassan N. M., Filiatreault A., Lanouette M. (2007). «Mon-
itoring personal, indoor, and outdoor exposures to metals in airborne particulate matter:
Risk of contamination during sampling, handling and analysis». Atmos. Environ. 41(28),
5897-5907.
[62] . Rauch S., Morrison G. M., Moldowan M. (2002). «Scanning laser ablation-ICP-MS track-
ing of platinum group elements in urban particles». Sci. Totl. Environ. 286(1—3), 243—251.
[63] . Ray S.J., Andrade F., Gamez G., McClenathan D., Rogers D., Schilling G., Wetzel W.,
Hieftje G. M. (2004). «Plasma-source mass spectrometry for speciation analysis: state-of-
the-art». J. Chromat. A. 1050(1), 3—34.
[64] . Reinhardt H., Kriews M., Miller H., Schrems O., Ludke C., Hoffmann E., Skole J. (2001).
«Laser ablation inductively coupled plasma mass spectrometry: a new tool for trace element
analysis in ice cores». Fresenius’ J. Anal. Chem. 370(5), 629—636.
[65] . Schmidt A. C., Haufe N., Otto M. (2008). «А systematic study on extraction of total arsenic
from down-scaled sample sizes of plant tissues and implications for arsenic species analy-
sis». Taianta. 76(5), 1233—1240.
[66] . Sivry Y., Riotte J., Sonke J. E., Audry S., Schafer J., Viers J., Blanc G., Freydier R., Du-
pre B. (2008). «Zn isotopes as tracers of anthropogenic pollution from Zn-ore smelters The
Riou Mort-Lot River system». Chem. Geol. 255(3—4), 295—304.
[67] . Spence B. (2006). «Choosing a multi-element technique for MCERTS compliant soil anal-
ysis». Int. Environ. Tech. 16(5), 41—44.
[68] . Spiro B., Weiss D.J., Purvis O. W., Mikhailova L, Williamson B.J., Coles B.J., Udachi V.
(2004). «Lead isotopes in lichen transplants around a Cu smelter in russia determined by
MC-ICP-MS reveal transient records of multiple sources». Environ. Sci. Technol. 38(24),
6522-6528.
[69] . Sutherland R.A., Pearson D.G., Ottley C. J. (2008). «Grain size partitioning of platinum-
group elements in road-deposited sediments: Implications for anthropogenic flux estimates
from autocatalysts». Environ. Poll. 151(3), 503—515.
[70] . Sylvester P.J. (2006). «Trends in analytical developments and earth science applications
in LA-ICP-MS and LA-MC-ICP-MS for 2004 and 2005». Geostand. Geoanal. Res. 30(3),
197-207.
[71] . Sylvester P.J. (2008). «LA-(MC)-ICP-MS trends in 2006 and 2007 with particular empha-
sis on measurement uncertainties». Geostand. Geoanal. Res. 32(4), 469—488.
[72] . Takahashi Y., Minamikawa R., Hattori K.H., Kurishima K., Kihou N., Yuita K. (2004).
«Arsenic behaviour in paddy fields during the cycle of flooded and non-flooded periods».
Env. Sci. Technol. 38(4), 1038—1044.
Литература
[73] . Tan S.H., Horlick G. (1987). «Matrix-effect observations in inductively coupled plasma
mass spectrometry». J. Anal. At. Spectrom. 2(8), 745—63.
[74] . Thermo Scientific (n.d.). US EPA method 200.8 using the XSeries 2 ICP-MS. Application
note 40722.
[75] . Thomas R. (2004). Practical guide to ICP-MS. Marcel Dekker Inc., N.-Y.
[76] . U. S. Environmental Protection Agency (n.d.). Method 200.8. Determination of trace ele-
ments in waters and wastes by inductively coupled plasma — mass spectrometry. (http://www.
epa.gov/sam/pdfs/E PA-200.8.pdf)
[77] . U. S. Environmental Protection Agency (1999). Compendium Method 10-3.5. Determina-
tion of metals in ambient particulate matter using inductively coupled plasma/mass spectrometry
(ICP/MS). (http://www.epa.gOv/sam/pdfs/EPA-IO-3.5.pdf)
[78] . U. S. Environmental Protection Agency (2006). Multi-media, multi-concentration inorganic
analysis, ILM05.4. (http://www.epa.gov/superfund/programs/clp/ilm5.htm)
[79] . U. S. Environmental Protection Agency (2007). Test methods for evaluating solid waste,
physical/chemical methods (SW-846). Method 6020A. Test methods for evaluating solid waste,
physical/chemical methods: Inductively coupled plasma — mass-spectrometry. (http://www.
epa.gov/sam/pdfs/EPA-6020a.pdf)
[80] . Upadhyay N., Majestic B. J., Prapaipong P., Herckes P. (2009). «Evaluation of polyurethane
foam, polypropylene, quartz fiber, and cellulose substrates for multi-element analysis of
atmospheric particulate matter by ICP-MS». Anal. Bioanal. Chem. 394(1), 255—266.
[81] . VeinottG.L (2001). «The use of laser ablation in environmental sciences». In: Sylvester P. J.
(Ed.), Laser-ablation-ICPMS in earth sciences Principle and applications. Mineralogical As-
sociation of Canada.
[82] . Vogt C., Latkoczy C. (2005). «Laser ablation ICP-MS». In: Nelms S. M. (Ed.), Inductively
coupled plasma mass spectrometry handbook. CRC Press, Boca Raton.
[83] . Wichmann H., Anquandah G.A. K., Schmidt C., Zachmann D., Bahadir M.A. (2007).
«Increase of platinum group element concentrations in soils and airborne dust in an urban
area in Germany». Sci. Totl. Env. 388(1—3), 121 — 127.
[84] . Woodhead J. D. (2008). «Isotope ratio determination in the earth and environmental sci-
ences: Developments and applications in 2006—2007». Geostand. Geoanal. Res. 32(4),
495-507.
[85] . Worsfold P. J., McKelvie I. D., Hanrahan G. (2008). «Environmental applications: waters,
sedimentsand soils». Comp. Anal. Chem. 54, 685—760.
[86] . Zereini F., Skerstupp B., Rankenburg K., Dirksen F., Beyer J.-M., Claus T., Urban H.
(2001). «Anthropogenic emission of platinum-group elements into the environment. Con-
centration, distribution and geochemical behavior in soils». J. Soils Sedim. 1(1), 44—49.
ГЛАВА 11
РОЛЬ МАСС-
СПЕКТРОМЕТРИИ
В ИССЛЕДОВАНИИ
ЛЕТУЧИХ
ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
Симин Ф. Малекниа
11.1. Летучие органические загрязняющие вещества
11.1.1. Происхождение летучих органических веществ и их
влияние на окружающую среду
Источники летучих органических загрязняющих веществ делятся на две основные
группы: антропогенную группу (от греческого «искусственный») и биогенную группу
(т.е. вырабатываемые живыми организмами или образующиеся в результате биологи-
ческих процессов). Термин «антропогенный» был впервые использован в техническом
смысле русским геологом Алексеем Петровичем Павловым; автор его первого исполь-
зования в английском языке — британский эколог сэр Артур Тэнсли (Bampton, 1999).
Данный обзор посвящен анализу воздействия биогенных летучих органических соеди-
нений (ЛОС) на окружающую среду, при этом иногда будет приводиться сравнение дан-
ного воздействия с воздействием антропогенных летучих загрязняющих веществ.
Биогенные ЛОС постоянно появляются в атмосфере в результате жизнедеятельно-
сти всех типов растительности. На присутствие ЛОС в воздухе указывают различные за-
пахи растений. Горы Голубого хребта, расположенного на восточном побережье США,
и Голубые горы в Сиднее, Австралия, славятся своей голубой дымкой. Эта дымка возни-
кает из-за наличия большого количества изопрена и разнообразных терпенов в лесном
воздухе. Эти вещества образуют аэрозоли, которые и вызывают рассеяние солнечного
света. Рассеяние света на коротких длинах волн более эффективно и приводит к появ-
лению синего оттенка у воздуха над лесом (Went, 1960). Биогенные летучие органиче-
ские соединения добавляют порядка одного тераграмма (1 Тгр = 1012 гр) к ежегодному
глобальному балансу углерода в биосфере. Данная оценка исключает вклад биогенного
метана и, как полагают, включает изопрен в качестве основного компонента. В миро-
вом масштабе доля биогенных летучих органических соединений значительно превы-
11.1. Летучие органические загрязняющие вещества 307
шает долю антропогенных выбросов, последние составляют около 10%. С другой сто-
роны, антропогенные выбросы Л ОС часто доминируют в городских районах (Goldstein
и Galbally, 2007). Лес площадью в 62000 км2 производит 3400 тонн терпенов в типичный
августовский день (Arno и Leif, 2009).
Выводы по поводу глобального потепления и изменению климата были основа-
ны на учете воздействия биогенных веществ (Denman et al., 2007; IPCC, 2007; Panuelas
и Straudt, 2010). С введением спутникового мониторинга началась новая эра непре-
рывного наблюдения за выбросами парниковых газов. Спутник наблюдения за парни-
ковыми газами, GOSAT, был запущен в начале 2009 года японским аэрокосмическим
агенством. Он вращается вокруг Земли на высоте 670 км и обеспечивает наблюдение
за парниковыми газами (диоксидом углерода и метаном) по всей планете, что значи-
тельно улучшает качество данных по сравнению с ситуацией, когда данные собираются
ограниченным числом наземных станций. Большие усилия прикладываются для улуч-
шения качества измерений на поверхности планеты, которые позволяют установить как
количество, так и тип биогенных летучих органических соединений, выделяемых при
температуре окружающей среды и при высоких температурах, характерных для лесных
пожаров (Maleknia et al., 2007; 2009а; 2009b).
Учет биогенных летучих органических соединений является критичным при рас-
смотрении жизнедеятельности растений: ЛОС влияют на рост растений, воспроизвод-
ство, защиту растений от стресса, от травоядных животных и патогенов. Биогенные ЛОС
включают в себя широкий спектр соединений от специфических изопреноидов расте-
ний (изопрен (С.Н8)), монотерпенов (С10Н16), сесквитерпенов (С15Н24) до разнообразных
химически активных соединений (органические кислоты, альдегиды, кетоны и спир-
ты). Диапазон биогенных летучих органических соединений и их концентрации зави-
сят в большой степени от суточного цикла (солнечного света и температуры) и других
факторов окружающей среды (например от влажности почвы и наличия питательных
веществ) (Fall, 1999; Guentheretal., 2000; Kesselmeier, Staudt, 1999). Другим основным источ-
ником биогенных ЛОС являются пожары и контролируемые отжиги леса, которые про-
водятся для контроля растительности в лесах (Crutzen и Andreae, 1990). Биогенные летучие
органические соединения могут существовать в атмосфере от минут до нескольких ча-
сов. Большинство из них быстро реагируют с озоном, с кислородсодержащими радика-
лами (например с гидроксильными радикалами), с нитрат-радикалами и превращаются
во вторичные продукты (Atkinson и Агеу, 2003). Наличие ЛОС изменяет химический и фи-
зический характер атмосферы, поэтому химический анализ ЛОС и определение степени
их воздействия на окружающую среду остается ключевой темой многих исследований.
11.1.2. Воздействие ЛОС на здоровье человека
Как биогенные, так и большинство антропогенных ЛОС, несмотря на их низкие кон-
центрации, могут вызвать хронические заболевания при длительном воздействии; при
этом симптомы заболевания могут развиваться медленно. Основными источниками
антропогенных ЛОС являются растворители, используемые в промышленных техно-
логиях и в быту; ЛОС образуются при сжигании ископаемого топлива на транспорте,
в промышленности и при генерации электроэнергии. Внутри помещений ЛОС могут
содержаться в краске, новой мебели, офисном оборудовании (например копировальных
аппаратах). Длительное воздействие этих типов ЛОС может способствовать развитию
308 Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
синдрома «больного здания» и целого ряда заболеваний (Bernstein et aL, 2008; Irigaray
et aL, 2007; Wang et aL, 2007; Yu и Crump, 1998). ЛОС, выбрасываемые в атмосферу при
сжигании биомассы (пиролитические продукты от пожаров и отжигов), а также пиро-
литические продукты городских объектов (печки, камины и пр.) вносят большой вклад
в атмосферное загрязнение. В последнее время значительное внимание уделяется иссле-
дованию связи хронических болезней легких (астмы et al.) и атмосферного загрязнения,
возникающего при лесных и городских пожарах. Ряд исследований показал увеличе-
ние числа пациентов, попавших в больницу с астмой, после крупных лесных пожаров,
а также корелляцию астмы с городским образом жизни (т.е. наличием загрязняющих
веществ от автотранспорта или бытовых/промышленных источников). Существует
прямая корреляция роста случаев заболевания астмой с увеличением концентрации ча-
стиц во вдыхаемом воздухе (Clinton et aL, 2006; Lipsett et aL, 1988; Swiston etaL, 2008).
Значительное количество различных естественных биогенных соединений широко
используется в фармацевтической и косметической промышленности. Разнообразные
терпены, в том числе эвкалиптол (1,8-цинеол), широко используются в парфюмерии
и в качестве ароматических добавок к косметическим или фармацевтическим продук-
там, таким как назальный спрей или средства для лечения простуды, а также для повы-
шения проницаемости кожи в составе жиро-растворимых соединений.
Другие терпены биогенного происхождения присутствуют в масле чайного дерева
и широко применяются в инсектицидных, антимикробных и противовоспалительных
препаратах (Brandet aL, 2001; Hawkes et aL, 2002).
Несмотря на широкое применение летучих органических соединений имеется весь-
ма ограниченная информация о взаимодействиях биогенных ЛОС с белками и влиянии
продуктов этого взаимодействия на организм (глава 17). Биологические исследования
выявили ряд побочных продуктов метаболизма, образующихся в результате взаимодей-
ствия 1,8-цинеола с цитохромом Р450 (этот гемопротеин участвует в жизненно важных
процессах, связанных с окислительным метаболизмом различных физиологически важ-
ных соединений и ксенобиотиков) (Duisken et aL, 2005). В недавнем исследовании in vitro
было обнаружено окисление метионина при взаимодействии кислородсодержащих тер-
пенов (например эвкалиптол и а-терпинеол) с серией пептидов и белков, содержащих
разнообразные аминокислотные последовательности (Maleknia и Адамс, 2007). Окисли-
тельное повреждение тканей может привести к различным воспалительным заболевани-
ям и астме. Связь между окислительными реакциями, вызванными ЛОС, и повреждени-
ем тканей до сих пор не была детально изучена. Исследования указали на прямую связь
раздражения верхних дыхательных путей с ЛОС и их фотохимическими продуктами ре-
акции с озоном. Например, мыши, подвергавшиеся воздействию ЛОС в течение 30 мин.
(время, достаточное для того, чтобы лимонен, изопрен или а-пинен прореагировали
с озоном), получали раздражение верхних дыхательных путей (Clausen et aL, 2001; Ror
etal., 2002). Для того чтобы понять связь между качеством воздуха и развитием астмы, не-
обходимы дополнительные исследования, которые позволят изучить механизм воздей-
ствия биогенных ЛОС и продуктов их трансформации в атмосфере на здоровье человека.
11.1.3. Экономический эффект биогенного загрязнения
Помимо биогенных загрязняющих веществ другая группа соединений, а именно воз-
никающих в результате пожаров, способствует появлению в атмосфере ряда токсичных
11.2. Масс-спектрометрия летучих загрязнящих веществ
соединений. Эти вещества появляются в атмосфере в результате пиролиза раститель-
ного масла и волокон (Maleknia et aL, 2009а). Дым от пожаров часто ухудшает качество
воздуха в областях, далеких от источника, и влияет на общее здоровье населения. Дру-
гим важным аспектом является то, что продукты пиролиза могут осаждаться на поверх-
ности различных сельскохозяйственных культур, что отрицательно влияет на качество
последних. Например, снижение качества вина во всем мире часто бывает связано с воз-
действием выбросов лесных пожаров (Maleknia и Adams, 2008; Pollnitz etal., 2004).
В последнее время особенно часто обсуждается экономическое воздействие за-
грязняющих веществ, возникающих в результате лесных пожаров, на винодельческую
промышленность. Многие ЛОС, связанные с лесными пожарами, например гваяколы,
образующиеся в результате пиролиза лигнина, являются общими при сгорании раз-
ных видов древесины. Вино, которое выдерживается в дубовых бочках, содержит от 10
до 100 мкг/л гваяколов — в данном случае они возникают при термической деструкции
лигнина и целлюлозы во время обгорания досок, из которых сделаны дубовые бочки
(Moreno и Azpilicueta, 2007). Следует отметить, что вина, получаемые из винограда, кото-
рый не подвергался воздействию дыма от пожаров, также содержат большое количество
летучих органических соединений, в том числе терпенов (п-цимол, линалоол, лимонен,
терпи ненов et al.), альдегидов, кетонов и спиртов от С5 до С9 (Girard etal., 2002). Однако
в винах, которые были сделаны из винограда, подвергшегося влиянию дыма, газовая
хроматография/масс-спектрометрия (ГХ/МС) выявила наличие гваяколов (метоксифе-
нолов), замещенных фенолов и некоторых других ЛОС, связанных с лесными пожарами.
В целом вина, содержащие высокие концентрации гваяколов, обладают неприятными
запахами и имеют вкусовые недостатки. Существует несколько доказательств того, что
определенные сложноэфирные соединения в вине маскируют его порчу, связанную
с воздействием дыма, даже несмотря на высокую концентрацию гваяколов (Maleknia
и Adams, 2008). Научное сообщество активно работает над разгадкой механизма влия-
ния ЛОС, образующихся при лесных пожарах, на качество вина.
11.2. Масс-спектрометрия летучих загрязнящих
веществ
Масс-спектрометрия (МС) играет важную важную роль в анализе ЛОС. Анализ атмос-
ферных выбросов ЛОС сложен в связи с их низкой концентрацией. Особенностью МС-
анализа ЛОС является работа с низкими пределами обнаружения: от нескольких частей
на миллион до нескольких частей на триллион по объему (ppmv до pptv). Летучие за-
грязняющие вещества образуют широкий спектр соединений, их точная идентифика-
ция и количественный анализ требуют комбинированного применения хроматографии
и МС-методов. ГХ/МС является наиболее широко используемым методом обнаружения
и количественной оценки ЛОС. ГХ/МС обеспечивает чувствительность в диапазоне pptv,
при этом необходимо концентрирование путем адсорбции ЛОС на подходящем сорбенте.
Наиболее широко используемым адсорбентом является тенакс (Buchem В. V., Нидерлан-
ды), состоящий из пористых полимерных смол на основе 2,6-дифенилфениленоксида,
который был специально разработан для улавливания летучих и полулетучих веществ
из воздуха. Агентство по охране окружающей среды США и Национальный институт
3 10 Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
охраны труда США рекомендуют использование адсорбента тенакс в стандартных ме-
тодиках анализа. Дополнительная процедура концентрирования может влиять на уро-
вень обнаружения некоторых соединений в связи с изменением эффективности их улав-
ливания, поскольку они могут значительно отличаться друг от друга давлением паров
и молекулярной массой. Это часто приводит к систематическим ошибкам (Helmig, 1997).
Процедура концентрирования становится существенной проблемой при проведении ко-
личественного определения ЛОС, включая широкий круг изомеров (например терпенов
с молекулярной формулой С10Н16). Еще одно преимущество ГХ/МС заключается в воз-
можности идентификации соединений с помощью поиска в базе данных масс-спектров
электронной ионизации и наличии электроных библиотек масс-спектров (глава 5). Ос-
новным недостатком ГХ/МС является сравнительно большое время для хроматографи-
ческого разделения соединений. Химический состав ЛОС был хорошо охарактеризован
с помощью ГХ/МС для целого ряда растительных материалов в целях выяснения их по-
тенциального применения в парфюмерии, в качестве лекарственных средств (Bignell etal.,
1997), а также в экологических исследованиях, в том числе там, где для выявления важ-
ных продуктов термического разложения использовался пиролиз (Faix et al.,1990; 1991).
Успехи в развитии атмосферной ионизации привели к широкому использовнию
двух ценных аналитических методов, в которых сочетается протонирование молекул
летучих соединений и масс-спектрометрия: реакция переноса протона (РПП, proton
transfer reaction, PTR) (Blake et aL, 2009; De Gouw и Warneke, 2007) и прямой анализ в ре-
жиме реального времени (ПАРВ, direct analysis in real time, DART) (Cody et al., 2005) для
непосредственного контроля ЛОС в воздухе без предварительного концентрирования
(глава 7). Метод PTR/MS был использован в атмосферных исследованиях для прямо-
го анализа биогенной эмиссии ЛОС в Средиземноморье (Hollinger et al., 2000), в север-
ных лесах (Ruuskanen, 2005), в Сьерра-Неваде (Lee et al., 2005) и саваннах (Hollinger et al.,
2000), в Австралии этот метод использовался для измерений ЛОС (Galbally et al., 2007;
Maleknia et al., 2009a), а также в целях определения загрязняющих веществ, возникаю-
щих при горении биомассы и при лесных пожарах (Karl et al., 2007). ПАРВ эффективен
для идентификации температурно-зависимой эмиссии ЛОС при горении различных
типов растительности (листья и древесина) (Maleknia et al., 2009b).
11.2.1. Протоколы пробоподготовки
Анализ летучих органических загрязнителей в низких концентрациях стал возможен
в результате крупных достижений в методах отбора и подготовки образцов. Специально
спроектированные сосуды используются для того, чтобы обесечить оптимальные ус-
ловия отбора и свести к минимуму деградацию образца. Разработаны также протоко-
лы отбора и концентрирования летучих веществ в низких концентрациях для ГХ- или
ГХ/МС-анализа (Hubschmann, 2009).
Склянки и контейнеры
Целый ряд контейнеров используется для отбора проб окружающей среды, включая
пластиковые контейнеры и контейнеры/склянки, изготовленные из боросиликатного
стекла (с силиконовыми септами, покрытыми тефлоном), а также металлические ка-
нистры. Пробы отбираются в виде газа (например, при мониторинге воздуха в закры-
тых помещениях или на открытых площадках), жидкости (например, при мониторинге
водопроводной воды, сточных вод, вод из природных или промышленных водоемов)
и твердой фазы (например, при анализе загрязненной почвы и мусора). Требования
к контейнерам зависят от процедур, разработанных различными правительственными
учреждениями для анализа конкретных видов летучих веществ. Для анализа жидких
или твердых образцов рекомендуется заполнение склянок полностью, чтобы свести
к минимуму влияние оставшегося в них воздуха.
Металлические канистры для отбора проб воздуха сделаны из высококачественной
нержавеющей стали (оксидированной хромом и никелем), что обеспечивает инертность
стенок контейнера и увеличивает стабильность летучих веществ. Другими популярными
контейнерами являются мешки для сбора газовых проб (например мешки Tedlar фир-
мы «Дюпон»), поверхность которых покрыта пленкой поливинилфторида, что делает их
устойчивыми к воздействию химических веществ. Как металлические канистры, так
и газовые мешки оборудованы соответствующими клапанами для откачки контейне-
ров перед отбором пробы. Контейнеры могут также заполняться газом высокой чистоты
(в основом азотом), чтобы избавиться от любых газовых примесей. После сбора образцов
склянки необходимо хранить при оптимальной температуре и влажности (4°C или ниже
в случае жидких или твердых образцов, чтобы сохранить летучие вещества). При этом
анализ следует проводить как можно быстрее (например не позже, чем через две недели
для металлических канистр), чтобы предотвратить потерю образца или его деградацию.
Протоколы отбора проб для ГХ/МС
Как описано выше, анализ летучих соединений при помощи ГХ/МС часто осуществля-
ется методом предварительного концентрирования. Один из самых распространенных
для выделения ЛОС из жидких или твердых образцов метод — это перенос в газовую
фазу в замкнутом пространстве над образцом. Выделение ЛОС из матрицы (жидкой или
твердой) происходит на основе эффекта распределения, который связан с парциальным
давлением веществ и проводит к установлению равновесия между уровнями соедине-
ний в матрице и в газовой фазе над образцом. Этот метод называют техникой сбора
в свободном пространстве (хедспейс, headspace, HS или HSGC) (рис. 11.1).
Методики отбора в свободном пространстве подразделяются на статические и ди-
намические. Статическая методика включает установление равновесия и перенос об-
разца, при этом ампулы нагревают, для того чтобы увеличить испарение летучих
веществ в газовую фазу. Динамический метод отбора, также называемый методом вы-
дувания-улавливания (purge & trap), включает поток нейтрального газа для траспорти-
ровки летучих веществ, испарившихся из матрицы. Эти летучие вещества улавливаются
соответствующими сорбентами (рис. 11.1). Ловушку затем нагревают, чтобы перенести
Таблица 11.1. Наиболее распространенные адсорбенты для отбора ЛОС
Сорбент Материал Применение
Активированный уголь углерод Летучие органические соединения
Карботрап Графитированный уголь Углеводороды от С4 до бифенилов
Тенакс пол и-2,6-дифен ил-п- фениленоксид Алифатические углеводороды, ароматические и полициклические углеводороды, альдегиды, ароматические амины и нитросоединения
Силикагель Диоксид кремния Высококипящие соединения
Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
Статический хедспейс
(ХС)
Динамический хедспейс
или выдувание-улавливание
Инжектор ГХ
ЛОС ИЗ
образцов воздуха
Адсорбент
ТФЭ
Ср = площадь ГХ-пика
fL — ь . rG
Н коэффициент распределения i
ЛОС, удерживаемые
в ловушке
Рис. 11.1. Варианты отбора проб для концентрирования летучих загрязняющих веществ
летучие вещества на хроматографическую колонку. Табл. 11.1 содержит перечень неко-
торых адсорбентов, пригодных для различных классов ЛОС.
Удобной альтернативой являются методы твердофазной экстракции (Mitra et aL,
1996) и твердофазной микроэкстракции (Pawliszyn, 2009). Эти варианты включают ис-
пользование адсорбента в виде коротких колонок или фильтров. Уникальным преиму-
ществом фильтров является возможность их прямого использования в ГХ-инжекторах.
Одноразовые картриджи для проведения экстракции, как и различные адсорбенты,
коммерчески доступны. Процесс извлечения ЛОС из картриджей прост и осуществля-
ется как в полевых условиях, так и в лаборатории.
11.2.2. Реакция переноса протона
Метод масс-спектрометрии с реакцией переноса протона (РПП/МС) был разработан
в середине 1990-х годов в лаборатории ВернераЛиндингера в Инсбруке, Австрия, и вско-
ре был доведен до стандартов общепринятого рабочего метода мониторинга атмосферы
(Hansel et aL, 1995). Детектирование атмосферных выбросов ЛОС на следовом уровне
стало возможым в результате использования техники селективного переноса протона
с применением Н3О+ (энергия сродства к протону равна 166,5 ккал/моль). Этот уни-
кальный процесс позволяет осуществлять прямой отбор проб воздуха без концентри-
рования, так как основные компоненты воздуха (N2, О2, СО2, СО, СН4 и О3) не вступают
в реакцию с ионами Н3О+ из-за их более низкого сродства к протону. РПП/МС идеально
подходит для мониторинга ЛОС в низких концентрациях в реальном времени: от не-
скольких частей на миллиард до нескольких частей на триллион по объему.
Первый прибор, в котором использовался метод переноса протона, был одиночным
квадруполем производства компании lonicon (Инсбрук, Австрия). Низкая разрешающая
способность квадрупольных масс-анализаторов на уровне одной единицы массы часто
11.2. Масс-спектрометрия летучих загрязнящих веществ
приводила к неоднозначной идентификации некоторых соединений; последующее при-
менение времяпролетных анализаторов (TOF/MS) с их высокой разрешающей способ-
ностью существенно улучшило идентификацию изобарных соединений (Graus et aL,
2010, Jordan etal., 2009, Tanimoto etal., 2007). Прибор, в котором сочеталась ионизация ме-
тодом переноса протона с квадрупольной ионной ловушкой, впервые был использован
в 2003 (Prazelleret al., 2003), а затем в 2005 (Warneke et al., 2005), хотя его чувствительность
была ниже, чем в случае использования линейных квадрупольных анализаторов. Тем не
менее возможность тандемных масс-спектрометрических экспериментов в ионных ло-
вушках способствует различению изобарныхсоединений, например метилвинил кетона
и метакролеина. Эта особенность ионных квадрупольных ловушек была использована
в 2010 году, когда линейная ионная ловушка (LIT) была применена для количественного
измерения изобарныхсоединений в полевых условиях (Levi etal., 2010).
Образование ионов-реагентов (Н3О+) в источнике ионов в РПП/МС основано
на применении газового разряда в полом катоде, причем труба дрейфа используется в ка-
честве реактора, где происходит ионизация ЛОС посредством переноса протона, а обра-
зовавшися ионы затем направляются в масс-анализатор (рис. 11.2я). Прибор, контроли-
рующий поток газа (контроллер потока), обеспечивает поток паров воды (8—10 мл/мин)
из пространства над водяным резервуаром для поддержания постоянного потока ио-
нов-реагентов. Напряжение (порядка 600 В) прилагается к трубе дрейфа длиной 9,2 см,
состоящей из ряда колец из нержавеющей стали, что обеспечивает однородное электри-
ческое поле для транспортировки ионов в анализатор. Пониженное напряжение трубы
дрейфа (~500 В) используется в некоторых экспериментах, чтобы ограничить фрагмен-
тацию ионов. Реакции переноса протона от Н3О+ к ЛОС (типичные скорости реакций
-106 имп/с) позволяют ионизировать ЛОС в следовых уровнях в воздухе в соответствии
с уравнением 11.1:
Нзо++ VOC = VOCH++ Н2о. (11.1)
Концентрация ЛОС рассчитывается путем применения кинетики псевдопервого
порядка (т.е. высокое соотношение концентрации Н3О+ в обьеме реактора к концентра-
ции ЛОС, чтобы проследить уровни [ЛОСН]+) в соответствии с уравнением 11.2.
[VOCH+] = [Н3О+]0{1 - exp(-k[VOC]Dt} - [H3O+]0£[VOC] Dt, (11.2)
где [Н3О+]0 представляет собой концентрацию первичных ионов-реагентов в отсутствие
ЛОС, к соответствует константе скорости реакции передачи протона от Н3О+ к ЛОС,
a Dt — время, которое требуется для ионов Н3О+, чтобы пройти трубку дрейфа (-105 мкс
со стандартными приборными параметрами). Оно определяется длиной трубки дрейфа,
температурным режимом дрейфа и напряжением на трубке). Константы скорости реак-
ций к известны для многих процессов переноса протона, например для монотерпенов к
варьируется от 2,2 до 2,5-10-9 см3/с (Tani et al., 2003).
Способы преобразования приборного сигнала в концентрацию ЛОС были хорошо
описаны еще в 1995 году, включая методологию и процедуру калибровки для измере-
ний биогенных ЛОС в лесах (Attapp etal., 2004). Для точного определения концентрации
прибор обычно калибруют с помощью специально приготовленной газовой смеси (Apel
Riemer Envoronmental Inc., Денвер, Колорадо, США), содержащей подходящий диапазон
ЛОС (каждый компонент в калибровочной смеси присутствует в концентрации в 1 ррт).
Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
Насос
Пары воды
Забор воздуха
Масс-анализатор
Н3О+/ЛОС->ЛОСН +
0
НС: разряд в полом катоде
IC: промежуточная камера
Электрод 1
Потенциал / ~
земли / Сеточныи электрод 2
Образец
0
ИТР
He/N2
П Нагреватель з
Метастабильный
। He/N2
Головка изолятора
Масс-анализатор
ИТР: источник тлеющего разряда
Рис. 11.2. Упрошенное представление методов (а) масс-спектрометрии с ионизацией при
переносе протона и (б) масс-спектрометрии с прямым анализом в режиме ре-
ального времени (DART/MS)
Калибровочный график получается путем последовательного разбавления калибровоч-
ного газа, при получении линейного отклика, при изменении концентрации. Чтобы из-
мерить фоновый сигнал (сигнал при отсутствии ЛОС), обычно используют сверхчистый
азот. Это позволяет точно рассчитать нижнюю границу обнаружения для каждого типа
ЛОС.
11.2.3. Прямой анализ в режиме реального времени
Ионные источники на основе метода DART (прямой анализ в режиме реального време-
ни) были использованы в 2005 году для атмосферного анализа веществ, находящихся не-
посредственно на поверхности (Cody etal., 2005). Сначала они применялись с времяпро-
летным анализатором с высокой разрешающей способностью AccuTOF-DART (JEOL,
Пибоди, штат Массачусетс, США), что позволяло устанавливать элементный состав ис-
следуемых веществ. Ионные источники, работающие на принципе DART, в настоящее
время используются как удобный интерфейс для ряда масс-анализаторов и могут быть
приобретены у компании lonSense (Saugus, МА, USA).
Процесс DART (рис. 11.20 осуществляется в потоке горячего газа (обычно гелия),
протекающего мимо электрода, на который подан высокий потенциал (для того чтобы
создать тлеющий разряд в атомосфере гелия), что приводит к появлению метастабиль-
ных атомов гелия или возбуженных молекул, которые реагируют с атмосферной водой
с образованием ионов Н3О+ и кластеров воды в соответствии с уравнениями 11.3—11.5.
He(23S) + Н2О -> Н2О+ + Не( 11S) + е~ (11.3)
11.3. Мониторинг летучих загрязняющих веществ в обычных условиях и при пожарах
Н20+ + Н20 —» Н30++ ОН
Н3О++ «Н2О^ [(Н,О)иН]+
(Н.4)
(Н.5)
Пространство в несколько сантиметров между источником ионизации DART и вхо-
дом в масс-спектрометр используется для размещения образцов. Одним из преиму-
ществ является то, что источник ионизации DART оснащен нагревателями для регули-
ровки температуры гелия (до 500°C), что в свою очередь позволяет нагревать образец.
Механизм ионизации молекул образца в DART включает перенос протона от ионов
Н3О+ к молекулам образца (т.е. механизм DART похож на РПП/МС) или процессы тер-
мической десорбции. DART применяется для анализа летучих и полулетучих веществ.
11.3. Мониторинг летучих загрязняющих веществ
в обычных условиях и при пожарах
Измерения уровней летучих загрязняющих веществ антропогенных и биогенных источ-
ников проводятся при анализе проб воздуха, собранного в городских районах, над по-
логом леса и т.д. Рис. 11.3 дает упрощенный вид экспериментальной установки для ана-
лиза летучих веществ, собранных в городских и лесных районах. Отбор окружающего
воздуха осуществляется через тонкостенную тефлоновую трубку на нескольких высотах
со скоростью потока примерно 10—50 л/мин, а собранный воздух затем делится на части
для проведения различных исследований. Общее время удерживания в линии отбора
проб (время прохождения пробы через трубку) измеряется путем введения в определен-
ное время стандартной смеси ЛОС в заборники, расположенные на определенной вы-
соте. В городских районах высокие здания или башни связи могут быть использованы
в качестве точек отбора проб. В лесных районах строятся специальные эксперименталь-
ные башни, которые затем используются различными исследовательскими группами.
Как правило, изменения типа и концентрации летучих органических соединений мо-
ниторируются в указанном интервале времени (от нескольких дней до нескольких не-
дель или месяцев). Окружающие условия (температура, скорость ветра, осадки) посто-
янно записываются и коррелируются с масс-спектральными данными для выявления
диапазона распространения ЛОС.
11.3.1. Определение антропогенных загрязняющих веществ
ЛОС являются важным компонентом загрязнения воздуха в городах в связи с их ролью
в формировании тропосферного озона, а также вследствие того, что такие соединения,
как бензол, имеют негативное влияние на здоровье человека. Ниже приведены результа-
ты нескольких исследований с использованием ГХ/МС и РПП/МС, которые позволяют
установить природу различных ЛОС и изменение их концентраций в городах по всему
земному шару. Большое исследование, проведенное в США, дает возможность сравнить
ЛОС в пробах воздуха, взятого в 28 городах (Baker et al., 2008). Пробы отбирались в ка-
нистры на протяжении пяти недель в течение каждого лета между 1999 и 2005 годами
и были проанализированы с помощью ГХ и ГХ/МС на предмет содержания ЛОС, окиси
углерода и метана. Уровни метана составили от 1,76±0,01 до 2,25±0,32 ppmv, а уровень
Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
Влажность
Температура
Ветер
Уровень
УФ-облучения
Рис. 11.3. Экспериментальная установка для анализа антропогенных и биогенных орга-
нических соединений в воздухе. Башня используется для того, чтобы устано-
вить линии забора проб и осуществить анализ воздуха на нескольких высотах.
При заборе проб воздуха на различных высотах измеряются температура, на-
правление и сила ветра, солнечное излучение. Каждая проба делится на не-
сколько частей для проведения различных анализов
Аналитическая станция:
> РПП/МС-анализ в режиме реального времени
-> Канистры для лабораторного анализа
СО варьировался от 150±22 до 640±270 ppbv. Диапазон изменений ЛОС в нескольких
крупных городах представлен на рис. 11.4. Эти соединения были разделены на четыре
группы: легкие алканы, высшие алканы С5—С8, алкены с ацетиленом и ароматические
углеводороды (бензол, толуол, этилбензол и о-ксилол). Было установлено, что этан —
это наиболее распространенный летучий экотоксикант. Его концентрация изменялась
в диапазоне от 560 ± 71 pptv до 8740 ± 2960 pptv.
Другие распространенные летучие ксенобиотики — это толуол, этан и ацетилен,
связанные с выбросами транспорта. Анализ воздуха в крупных городах (Балтиморе,
Чикаго и Лос-Анджелесе) показал самые высокие уровни толуола. Вещества с высокой
реакционной способностью, такие как бензол, толуол, этилбензол и ксилол, составили
значительную долю обнаруженных ненасыщенных углеводородов; их концентрация ва-
рьировалась в диапазоне между 1 и 4 ppbv.
Многолетние исследования 2004—2008 годов в штате Нью-Гемпшир, США, пока-
зали снижение уровня галогенированных растворителей, что было связано со сниже-
нием их использования в промышленности (Russo etal., 2010). Уровни тетрахлорэтиле-
на и трихлорэтилена снижались на 0,7±0,2 pptv и 0,3 ±0,05 pptv в год соответственно.
В этом исследовании пробы воздуха были собраны в канистры на территории обсерва-
тории AIRMAP университета штата Нью-Гемпшир и проанализированы с помощью ГХ
и ГХ/МС. Самые высокие концентрации летучих органических соединений были об-
условлены С2—С8-соединениями и оценивались в 109—1010 молекул/см2-с; эти уровни
наблюдались в середине и конце зимы с пиком в 2006 году.
Определения антропогенных выбросов ЛОС были проведены в нескольких местах
в Милане (Италия) в 1998, 2002 и 2003 годах (Steinbacher et aL, 2005). Забор проб осу-
ществлялся в сельской местности в 35 км к северу от центра города и в городском лан-
шафте в 5 км к северу от центра города. Пробы анализировались с использованием ГХ
и РПП/МС. Данное исследование показало низкую концентрацию толуола и бензола
по выходным дням, когда антропогенные выбросы были ниже из-за меньшего использо-
вания транспорта, а также вследствие снижения уровня промышленной деятельности.
11.3. Мониторинг летучих загрязняющих веществ в обычных условиях и при пожарах
Легкие алканы @ С5—С8-алканы Алкены + ацетилен □ Ароматические вещества
Рис. 11.4. Результаты двухлетнего исследования процентного составаЛОС в атмосфере Нью-
Йорка, Солт-Лейк-Сити и Филадельфии. Легкие алканы: этан, пропан, и-бутан
и изобутан; С5—С8-алканы: н-пентан, изопентан, гексан и октан; алкены + ацети-
лен: этилен, пропилен, 1-бутен и ацетилен; ароматические вещества: бензол, то-
луол, этилбензол и о-ксилол. Вклад алканов в ЛОС выше за второй год, когда все
образцы воздуха были собраны далеко от дорог, в то время как вклад ненасыщен-
ных углеводородов больше в первый год, когда образцы были собраны как на обо-
чинах дорог, так и на открытых участках. В течение 1999 года образцы собирались
на обочинах дорог, источником наиболее распространненых видов загрязняющих
веществ (ацетилен, изопентан и толуол) является транспорт. В 2004 году наиболее
распространенными экотоксикантами были этан, пропан и н-бутан — вещества,
не выделяемые траспортными средствами. (Перепечатано из Atmosph.Environ., 42,
Baker А. К., Beyersdorf A.J., Doesema L.A., Katzenstein A., Meinardi C., Simpson, IJ,
Blake, DR и Rowland F.S. «Измерения неметановых углеводородов в 28 городах Со-
единенных Штатов», 170—182. Copyright 2008, с разрешения Elsevier)
В другом исследовании антропогенные выбросы ЛОС определялись на высоте 200 м
в центре Лондона с конца сентября до начала ноября 2006 года (Nemitz et al., 2007). Забор
воздуха проводили через тефлоновую трубку длиной 45 м (наружный диаметр 9 мм) при
скорости потока 60 л/мин. Забранный воздух был проанализирован методом РПП/МС.
Были обнаружены следующие летучие экотоксиканты: метанол, ацетонитрил, ацеталь-
дегид, ацетон, изопрен, бензол, этилбензол и толуол. Выбросы бензола и ацетонитрила
достигали максимума в вечерние часы, что можно было соотнести с всплеском транс-
портного движения и включением отопления в домах (камины).
11.3.2. Определение биогенных загрязняющих веществ
в окружающей среде
Использование РПП/МС упростило определение уровней биогенных летучих органи-
ческих соединений в полевых условия. Метод РПП/МС работает в режиме сканирова-
3 18 Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
ния или режиме регистрации нескольких ионов. Экологические условия (температура,
скорость ветра, осадки) также непрерывно регистрируются для корреляции выбросов
ЛОС с масс-спектрометрическими данными. Пробы окружающего воздуха всасывают-
ся по пробоотборной трубке (тонкостенные тефлоновые трубки, 6 мм внешний диаметр)
на нескольких высотах (рис. 11.3) со скоростью забора примерно 10—50 л/мин, а затем
делятся на части для проведения различных исследований. Как правило, поток воз-
духа в источник ионов, работающий на принципе переноса протона, составляет от 50
до 250 мл/мин.
РПП/МС-анализ ЛОС в государственном заповеднике Баго, Новый Южный Уэльс,
Австралия, в ноябре 2006 г. (конец весеннего сезона в Южном полушарии) представлен
на рис. 11.5 (Maleknia и др ., 2009а). Доминирующие типы деревьев в эвкалиптовых лесах
со средней высотой деревьев 40 м — Е. delagatensis и Е. dalrympleana. Данные РПП/МС,
полученные для проб воздуха, взятых на высотах 15 м и 45 м, выявили две различные
модели, которые поддерживают гипотезу о разном распределении выбросов ЛОС выше
и ниже купола леса. Доминирующие виды ЛОС выше купола леса — это изопрен (m/z 69),
бутанон (m/z 73), гексеналь или монотерпеновый фрагмент (m/z 81), п-цимен (m/z 135)
и монотерпены (m/z 137), а также камфора (m/z 153). Ниже купола леса доминируют аце-
тальдегид (m/z45), муравьиная кислота (/иД47), ацетон (/этД59), уксусная кислота (/этД61),
гидроксиацетон (m/z75) и гексаналь или гексанон (m/z 101). Идентификация вышеупомя-
утых веществ основана на эталонном списке соединений, наиболее часто наблюдаемых
в РПП/МС-исследован иях (табл. 11.2). Ионы cm/z ИЗ и 127 могут быть связаны с продук-
тами окисления, а их идентификация требует дальнейших лабораторных экспериментов.
Использование ионных источников на основе РПП с новым поколением линей-
ных ионных ловушек (LIT) представлено в работе (Levi et aL, 2010). Методами тандем-
ной масс-спектрометрии было подтверждено наличие изобарных соединений и ионов
мешающих анализу, — химического шума. Исследование, в котором использовалась
специальная башня (PROPHET), построенная на территории биологической станции
в Университете штата Мичиган (UMBS), было проведено летом 2008 года. Лес в окрест-
ности станции представлен красным кленом, красным дубом, бумажной березой, белой
сосной и большой зубчатой осиной. Рис. 11.6 демонстрирует изменение концентрации
Рис. 11.5. Концентрация биогенных летучих веществ на высоте 15 м (внизу) и 45 м — вы-
соте купола леса — (вверху). Масс-спектрометрический анализ окружающего
воздуха проведен методом РПП/МС; пробы взяты в государственном заказнике
Баго, Новый Южный Уэльс, Австралия, в ноябре 2006 года
11.3. Мониторинг летучих загрязняющих веществ в обычных условиях и при пожарах
Таблица 11.2. Выделение биогенных летучих загрязняющих веществ в зависимости
от температуры (от температуры окружающей среды до высокой темпера-
туры горения) коррелирует с массами их протонированных молекул детек-
тированных методом РПП/МС. Некоторые сигналы могут быть связаны
с фрагментными ионами. Осколочные ионы (фрагменты) терпенов име-
ли величины m/z 81, 93, 95, 107, 121; фрагменты сесквитерпенов — m/z 149,
а ионы с m/z 57 связаны с трет-бутилом С4Н9+
Протонирован- Соединение
ная молекула
или фрагмент-
ный ион
28 Синильная кислота
31 Формальдегид
33 Метанол
42 Ацетонитрил
43 Пропен
45 Ацетальдегид
47 Муравьиная кислота
54 Акрилонитрил
57 Акролеин, бутены, метилтретбутиловый эфир, бутанол
59 Ацетон, глиоксаль
61 Уксусная кислота, гидроксиацетальдегид, метилфрмиат
69 Изопрен, фуран
71 Метилвинилкетон, метакролеин
73 Бутанон
75 Гидроксиацетон, 2-гидроксипропаналь
79 Бензол
81 Фрагмент гексеналя или терпенов
83 Гексенол, гексаналь, гексенилацетат, метилфуран, гидроксикетоны, образу-
ющиеся при окислении изопрена
85 Этилвинилкетон, циклопентанон, 2-метилбутен-2-аль, 2(5Н)-фуранон
87 2-Метилбутен-3-ол-2, пентаноны, метакриловая кислота, бутандион-2,3,
кротоновая кислота, бутиролактон
91 Пероксипропионилнитрат
93 Толуол
95 2-Винилфуран, фенол, диметилциклопентен
97 Фурфураль, 2-метилциклопентен-2-он-1
99 Гексеналь, фурфуриловый спирт, 3-метил-2(5Н)-фуранон
101 Гексаналь, гексанон, гидроксиацетон, гидропероксид изопрена
103 Нитрат перокиметакриловой кислоты
105 Стирол, нитрат пероксиизобутановой кислоты
107 С2-бензолы
111 5-метилфурфураль, дигидроксибензолы (пирокатехин, резорцин,
гидрохинон)
320 Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
Таблица 11.2. (окончание)
Протонирован- ная молекула или фрагмент- ный ион Соединение
113 Ди метил циклопентанон, 2-гидрокси-3-метилциклопентен-2-он-1
115 Гептаналь, 3-гидрокси-2-пентено-1,5-лактон
117 Гексановая кислота, 1-ацетилоксипропанон, инден
121 СЗ-бензолы, винилфенол
123 Диметилфенолы, этилфенол, бензойная кислота
125 2-Метоксифенол (гваякол), триметилциклопентенон, дигидрокситолуол
127 З-Метилинданон-1, 5-гидроксиметил-2-фуральдегид, 2-метил-З-гидрокси пирон-2
129 Октаналь, нафталин
135 С4-бензолы, п-цимол
137 Монотерпены (например пинены, лимонен, эвкалиптол)
139 Нопинон
143 Нонаналь
149 С5-бензол, 1-метокси-4-(2-пропенил)-бензол
151 Пиноальдегид
153 Камфора
изопрена за 10 дней. Эти изменения соответствовали суточному циклу (т.е. макси-
мальные концентрации изопрена наблюдались в максимуме температуры и ультрафи-
олетового излучения). Выбросы биомассы в РПП/МС-исследованиях часто измеряют
по интенсивности ионов с m/z 69, характерных для изопрена и 2-метил-3-бутен-2-ола
(МВО). Наличие МВО в выбросах нескольких видов североамериканских сосен вли-
яет на правильность анализа изопрена. Потеря воды протонированными молекулами
(С.НИО+) приводит к появлению сигнала с m/z 69. Белые сосны на территории биоло-
гической станции не выделяют МВО (Harley et al., 1998), т.е. надежная, идентификация
изопрена (рис. 11.6) основана на обнаружении двух ионов с m/z 69 (протонированные
молекулы С5Н9+) в МС-спектре и с m/z 41 (фрагмент, соответсвующий потере молекулы
С2Н4) в тандемном спектре (МС2). Метод МС2 с регистрацией иона с m/z 41 может быть
использован для установления вклада МВО в биомассу выбросов в смешанных лесах.
11.3.3. Влияние температуры на определение биогенных
соединений
Исследования выбросов ЛОС в зависимости оттемпературы предоставляют важную ин-
формацию о природе загрязняющих веществ, выбрасываемых в атмосферу на разных
этапах лесных пожаров. Растительное сырье из эвкалиптов использовалось для систе-
матического изучения ЛОС разными методами: ГХ/МС, PTR/MC и DART/MS (Maleknia
etal., 2007; 2009а; 2009b). Исследования показали, что температурный профиль ионных
сигналов в масс-спектрометре (т.е. выбросы ЛОС) могут быть соотнесены с точками
кипения и давления пара исследуемых соединений. Температурно-зависимые выбросы
11.3. Мониторинг летучих загрязняющих веществ в обычных условиях и при пожарах
Рис. 11.6. Временные профили изопрена, полученные при помощи РПП/линейной ион-
ной ловушки в режиме МС (m/z 69) и в режиме МС/МС (m/z 41), представлены
вместе с изменениями температуры окружающей среды и изменениями по-
тока широкополосного УФ-излучения. (Печатается с разрешения Levi Н.,
Mielke L. Н., Kerri A., Pratt К. A., Shepson Р. В., Scott A., McLuckey S. A, Wisthaler А.,
Henzel А. «Количественное определение биогенных летучих органических со-
единений в атмосфере с использованием масс-спектрометрии с ионизацией при
переносе протона в ионной ловушке». Anal. Chem., 82: 7952—7957. Copyright 2010
American Chemical Society)
летучих веществ могут быть подразделены на три основных класса по химическому со-
ставу соединений, выделяемых растениями в зависимости от температуры (т.е. от тем-
пературы окружающей среды до температуры, соответствующей пожарам).
Первый класс соединений включает в себя кислоты, альдегиды, кетоны, изопрен
и низкокипящие терпены, которые были обнаружены от температуры окружающей сре-
ды до 100°C. Терпены с относительно большой температурой кипения были отнесены
ко второму классу и обнаруживались до начала пиролиза. Третий класс включает ЛОС,
образующиеся при пиролизе растительных волокон при температуре выше 300°C (на-
пример ЛОС при пиролизе целлюлозы и лигнина).
Были проведены исследования температурной зависимости состава ЛОС для раз-
личных видов растительного материала (например листьев и древесины) с учетом как
свежих, так и состаренных материалов. ГХ/МС-анализ воздуха при нагревании до 40 °C
свежих и состаренных листьев растения Е. Grandis позволил установить ряд ЛОС (этил-
винилкетон, диэтилкетон, 2-этилфуран, гексаналь и гексенали), характерных для све-
жих листьев, в то время как несколько терпенов (а- и Р-пинены, а-фелландрен, эв-
калиптол, у-терпинен) были обнаружены при нагревании обоих типов листьев. Кроме
того, при DART/MC-анализе свежих листьев и стеблей Е. sideroxylon были обнаружены
тканеспецифические ЛОС (например, метанол и этанол были более распространены
в стеблях).
РПП/МС-анализ ЛОС, образующихся в результате нагревания свежих листьев
(Е. Grandis), показал, что концентрации метанола (m/z 33) и ацетальдегида (m/z 45) были
Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
наибольшими при температуре около 60°C. В то же время сигналы ионов с m/z 137
и m/z 153 (серия терпенов) монотонно увеличивались в широком диапазоне температур
от комнатной до 200 °C. Метод исследования температурной зависимости ЛОС с исполь-
зованием DART/МС был разработан для точного определения ряда специфических рас-
тительных терпенов до температуры 300°C (т.е. до начала пиролиза растительных во-
локон целлюлозы и лигнина). Температура гелия составляла 50, 100, 200 и 300 °C, кроме
того, растительные материалы были помещены непосредственно в источник ионизации
DART, что позволило вести прямое испарение соединений. Точные измерения массы
с использованием времяпролетного анализатора позволили установить элементный со-
став для ~30 соединений. В этих исследованиях были обнаружены как простые органи-
ческие соединения (например метанол и ацетон), так и серии монотерпенов (т.е. пинен,
m/z
Рис. 11.7. РПП/МС-анализ летучих загрязняющих веществ, образующихся при нагрева-
нии стареющих листьев эвкалипта Grandis. Использовался калориметр, который
измерял потерю массы. Температура нагревателя конуса (Тсн) была запрограм-
мирована на увеличение от комнатной до 900°C. Затененная область обозна-
чает диапазон масс, в котором преобладают продукты пиролиза. (Источник:
Int. J. Mass Spectrom., 279, Maleknia, SD, Bell, TL, Adams M. «Состав дыма при
пожарах эвкалиптовых лесов: зависимость химического состава выбросов био-
генных летучих органических соединений от температуры», 126—133. Copyright
2009, с разрешения Elsevier)
11.3. Мониторинг летучих загрязняющих веществ в обычных условиях и при пожарах
камфен, цимен, эвкалиптол), общие для многих видов растений, а также менее распро-
страненные сесквитерпены и флавоноиды.
Для анализа загрязняющих веществ, образующихся в результате пиролиза, был ис-
пользован калориметр, измеряющий потерю массы, сопряженный с РПП/МС (Maleknia
et al., 2009а). При нагревании растений от комнатной температуры до 600 °C непрерыв-
но записывались масс-спектры. Температурно-зависимый масс-спектрометрический
анализ стареющих листьев (£. Grandis) выявил два класса ЛОС (рис. 11.7). До наступле-
ния пиролиза (ниже 300°C) химический состав ЛОС остается примерно одинаковым
и хорошо коррелирует с естественным содержанием различных изопреноидов. Ионы
с m/z 137 и m/z 205 соответствуют протонированным молекулам монотерпенов и сескви-
терпенов. Кислородсодержащие монотерпены могут также давать вклад в сигнал ионов
с m/z 137 в результате потери молекулы воды. Другой фрагментный ион (m/z 81) является
общим для сесквитерпенов и монотерпенов; аналогично ионы с m/z 137 и m/z 149 могут
быть отнесены к фрагментам сесквитерпенов. Выше 300 °C выделение продуктов пиро-
лиза резко возрастает по сравнению с исходными терпенами (m/z 137, 149, 205). В част-
ности, при нагреве до 300°C интенсивность пиков ионов с m/z 61 (уксусная кислота,
гидроксиацетальдегид, метилформиат), с m/z 97 (фурфурол, 2-метилциклопентен-2-
он-1, диметилциклопентен) и с m/z 111 (5-метилфурфураль, дигидроксибензолы) уве-
личивается в десять раз. Табл. 11.2 содержит список соединений, массы которых со-
ответствуют протонированным молекулам окисленных альдегидов, кетонов, фуранов
и замещенным бензолам.
П.3.4. Анализ загрязняющих веществ, выделяемых при
пиролизе различных частей растений
РПП/МС-исследования показали, что среди соединений, образующихся при пироли-
зе целлюлозы и лигнина, преобладают высокотемпературные вещества. Основными
компонентами целлюлозы в хвойных деревьях и деревьях твердых пород являются га-
лактоглюкоманнан и глюкуронксилан. Пиролиз этих полисахаридов в начальной ста-
дии включает реакции дегидрирования, декарбоксилирования и декарбонилирования,
которые приводят к потере воды, СО и СО2. При температуре выше 300°C реакции де-
полимеризации приводят к разрыву гликозидных связей, в результате чего образуется
моносахарид левоглюкозан, который далее разлагается на более низкомолекулярные
соединения, включая фурфурали, 2,3-бутандион, дибензофуранолы и бензонафтофу-
ран. Для сравнения лигнин — природный полимер с поперечными сшивками — состо-
ит из различных фенилпропановых фрагментов, а его пиролиз происходит в широком
диапазоне температур. При нагревании до 300°С образуется формальдегид, а с повыше-
нием температуры — замещенные фенолы (гваяколы) (Butt, 2006; Evans and Miln, 1987;
Font etal., 2003; Jin etal., 2003; Saka, 2001; Tame et al.). Л и гни н в древесине обычно состоит
из сирингил (S) пропанового блока и гваяцил (G) пропанового блока. Отношение S/G
влияет на характеристику древесины (мягкая или жесткая).
ГХ/МС с набором масс-спектральных библиотек — идеальный метод точной
идентификации широкого спектра продуктов пиролиза. Целлюлоза и лигнин при-
сутствуют в растительных материалах в различных соотношениях. Таким образом,
пиролитическая хроматомасс-спектрометрия может быть использована для субтипи-
рования загрязняющих веществ в зависимости от растительного материала (например
Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
листьев, древесины или коры). Результаты пиролитической ГХ/МС листьев, древеси-
ны и коры Е. maculata представлены на рис. 11.8. Пирограммы выявляют различные
типы пиролитических продуктов для указанных выше растительных материалов.
Целый ряд соединений, которым соответствуют ГХ/МС-пики, является общим для
всех типов тканей растений, другие являются уникальными для конкретной ткани.
ЛОС, обнаруженные как в листьях, так и в стволовой древесине или коре, приведены
в табл. 11.3 и разделены на две основные группы. Пиролитические соединения из по-
лисахаридов отмечены как PS, пиролитические соединения, связанные с лигнином,
соответственно обозначены как сирингил (S) и гваяцил (G). Многие исследования
показывают, что загрязняющие вещества, образующиеся при пожарах, могут быть
Таблица 11.3. Список типичных соединений, которые получаются при пиролизе полиса-
харидов (PS) и при пиролизе двух подтипов лигнина (сирингил (S) и гвая-
цил (G)). ВУ — время удерживания
Тип ВУ Соединение Тип ВУ Соединение
PS 5,33 2-Пропеналь PS 34,95 2-Г идрокси-у-бутиролактон
PS 5,56 2,3-Бутандион Ph 35,89 п-Крезол
PS 7,56 3-Пентанон PS 36,47 5-Г идроксиметилдигидро фуранон-2
PS 8,44 Г идроксиацетальдегид PS 37,27 Не идентифицировано
PS 9,79 Уксусная кислота G 37,49 4-Метилгваякол
PS 11,17 Не идентифицировано Манноза 37,84 Не идентифицировано
PS 11,33 1-Г идроксипропанон-2 G 41,31 4-Этилгваякол
PS 12,5 Этиленгликоль PS 41,4 Не идентифицировано
PS 14,38 Пропионовая кислота PS 41,8 4-Г идрокси-3-метил- (5Н)-фуранон или 3-метил-2,4-фурандион
PS 15,03 Метилакрилат PS 42,95 1,4:3,6-Дигидро-ос-с1- глюкопираноза
PS 16,03 1-Г идроксибутанон-2 Ара- биноза 43,37 1,5-Ангидроарабинофураноза
PS 16,24 З-Гидроксипропаналь G 43,84 Винилгваякол
PS 16,54 Бутеналь-(3)-2-он G 44,88 Эвгенол
PS 16,87 Фуран-(ЗН)-2-он Ман- ноза 45,64 5-Г идроксиметилфуральде- гид-2
PS 17,69 Фуран-(2Н)-3-он S 46,17 Сирингол
PS 17,74 З-Фуральдегид PS 46,27 5-Г идроксиметилдигидро- фуран-2-он
PS 18,47 Бутандиаль G 47,21 Цис-изоэвгенол
PS 18,58 2-Г идроксибутанон-3-ал ь G 49,36 Транс-изоэвгенол
PS 19,29 Фуральдегид-2 Кси- лоза 49,52 1,5-Ангидро-[3-с1- ксилофураноза
PS 21,48 2,3-Дигидро-5- метилфуранон-2 S 49,91 4-Метилсирингол
PS 21,67 2-Фурил карбинол G 50,37 Ванилин
11.3. Мониторинг летучих загрязняющих веществ в обычных условиях и при пожарах
Таблица 11.3. (окончание)
Тип ВУ Соединение Тип ВУ Соединение
PS 22,07 1-Ацетокси пропанон-2 PS 50,62 неидентифицировано
PS 22,44 Тетрагидро-4- метилфуранон-3 или 2-этилбутаналь G 50,81 6-Г идрокси-7-метоксиинден (цис или транс)
PS 22,72 Дигидрометил- фуранон или диметилдигидрофуран G 51,23 6-Гидрокси-7-метоксиинден (цис или транс)
PS 23,06 2-Ацетилфуран S 52,85 4-Этилсирингол
PS 23,52 неидентифицировано G 53,59 Ацетогваякон
Еис 23,54 Лимонен S 55,11 4-Минилсирингол
PS 23,73 неидентифицировано G 55,58 Гваяцилацетон
PS 23,82 2-Циклопентендион-1,4 S 55,79 4-Аллил-[4-(2-пропенил)]- 2,6-диметоксифенол или пропилсирингол
Еис 24,62 Эвкалиптол S 57,7 4-Пропенилсирингол (цис)
PS 25,2 2-Г идроксициклопентенон S 59,04 6-Гидрокси-5,7-димето- ксиинден (цис или транс)
Кси- лоза 26,2 Дигидрометилфуранон Глю- коза 59,2 Левоглюкозан
PS 26,54 5-Метил-2-фуральдегид S 59,34 6-Гидрокси-5,7-димето- ксиинден (цис или транс)
Кси- лоза 26,64 Изомер 4-гидрокси-5,6- дигидро-(2Н)-пиранона-2 S 59,85 4-(1-Пропенил)-2,6- диметоксифенол (транс)
PS 27,37 З-Метилциклопентен-2- он-1 G 60,11 Дигидрокониферол
Кси- лоза 27,65 у-Бутиролактон, дигидро-2(ЗН)-фуранон S 61,03 Сирингиновый альдегид
Кси- лоза 28,11 2(5Н)-Фуранон G 61,12 Кониферол (цис)
PS 28,92 З-метил-5-метилиден- 2(5Н)-фуранон S 63,42 Ацетосирингол
PS 29,13 4-Гидрокси-5,6- дигидропиран G 63,99 Кониферол (транс)
PS 30,22 Метилдигидро-(2Н)- пиранон-2 G 64,67 Конифериловый альдегид
PS 30,89 неидентифицировано S 64,9 Сирингилацетон
Ph 31,88 Фенол S 65,92 Пропиосирингон
G 32,62 Гваякол, 2-метоксифенол S 68,98 Дигидросинапиновый спирт
PS 32,88 З-Метилтетрагидрофуран- дион-2,4 S 69,8 Синапиновый спирт (цис)
PS 34,06 Фурил гидроксиметил кетон S 72,66 Синапиновый спирт (транс)
Ph 34,18 о-Крезол S 73 Синапиновый альдегид
Ман- ноза 34,72 1,3-Диэтил-2-гидро- ксициклопентен-2-он-1
Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
Vkuh. jJaJjAI jlikL
10,0 20,0 30,0 40,0 50,0 60,0 70,0
Время, мин.
Рис. 11.8. Пирограммы Е. Maculata, демонстрирующие наличие различных продуктов пи-
ролиза в листьях, стволовой древесине и коре. Установка для пиролиза, состоя-
щая из пиролитического реактора с двумя камерами и микропечью, PY-20201D
(Frontier Lab, Япония), была оснащена пробоотборником и сопряжена с газо-
вым хроматографом НР6890 (Hewlett-Packard, США) и квадрупольным масс-
спектрометром (Agilent Technologies). Растительные образцы весом 100 мкг были
измельчены в криогенных условиях и затем подвергнуты пиролизу при 500°C
в потоке гелия в качестве газа-носителя
более точно охарактеризованы исходя из отношения целлюлозы к лигнину в данном
типе биомассы. В простейшем приближении разные типы растительности и деревьев
по всему миру могут быть охарактерированы и классифицированы по соотношению
целлюлозы к лигнину, что обеспечивает понимание того, какие ЛОС будут выброше-
ны в атмосферу в случае лесных пожаров.
11.4. Заключение и будущее направление работы
11.4. Заключение и будущее направление работы
В данном обзоре описаны разнообразные методики обнаружения летучих загрязняю-
щих веществ (как при нормальных условиях, так и при высоких температурах), кото-
рые указывают на исключительную эффективность применения масс-спектрометрии
для точного анализа ЛОС. Долгосрочный мониторинг антропогенных загрязняющих
веществ в городских районах предоставляет важные данные для сравнения антропоген-
ных и биогенных вкладов в ЛОС. Данные, представленные в этой главе, подтвержда-
ют предположение о том, что типы биогенных экотоксикантов коррелируют с типами
растительных материалов. Результаты этих исследований могут быть использованы для
разработки новых глобальных подходов к моделированию выбросов ЛОС от пожаров
путем сопоставления результатов масс-спектрального анализа с содержанием целлю-
лозы и лигнина в биомассе. При этом необходимо сравнение результатов с картами рас-
тительного покрова, которые получены при наблюдениях с воздуха.
Изменение климата и глобальное потепление, как ожидается, повлияет на строение
деревьев и их рост, которые в свою очередь влияют на выбросы ЛОС как при комнатной
температуре, так и при высоких температурах, типичных для лесных пожаров. Влияние
повышенной температуры и различной концентрации СО2 было исследованы для де-
ревьев, выращенных в контролируемых условиях для имитации влияния последствий
изменения климата на строение деревьев (Ghannoum et al., 2010). В этих исследованиях
эвкалипты показали более позитивный отклик на повышение уровня СО2: среди эвка-
липтов, выращенных при одинаковых температурах более высокие соответствовали
самым высоким концентрациям СО2 (рис. 11.9). Техника пиролитического ГХ/МС-
анализа в сочетании со статистическим анализом основных компонентов применяется
Кень/Тночь 26/18’С
[СО2] 280:400:640
30/22°C
280:400:640 цА/А
Рис. 11.9. Фотография растений Е. saligna (быстрорастущих видов). Различные условия
роста приводят к разнице в размере растения
Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
для изучения влияния изменения климата на строение деревьев и позволяет следить
за отношением целлюлозы к лигнину в зависимости от высоты дерева.
Предварительные результаты показали, что содержание целлюлозы и лигнина
у растений, выращенных в одинаковых условиях (например при близкой температуре
и содержании СО2), хорошо коррелируют друг с другом, что в дальнейшем позволяет
предположить, что небольшие вариации в содержании целлюлозы и лигнина мож-
но отслеживать, используя масс-спектрометрический анализ. Достижения в масс-
спектрометрии и области методов статистической оценки больших массивов данных
являются критически важными и будут продолжать оказывать большое влияние на точ-
ную идентификацию и количественное определение летучих загрязняющих веществ.
Благодарность
Исследования с использованеим PTR/MS в государственном заказнике Баго были
проведены во время полевых работ, организованных Т. Суни (в настоящее время ра-
ботающей в отделе астмосферных наук в Хельсинском университете) в сотрудничестве
с Т. Карлом, А. Гюнтером и А. Турнипсидом из Национального центра атмосферных
исследований (Boulder, СО). DART-эксперименты проводились в Университете Тихого
Океана. Особую благодарность автор выражает сотрудникам университета Д. Спаркма-
ну, Т. Вали, Т. Хокинсу и Р. Коди. Пиролитические ГХ/МС-исследования были про-
ведены в университете Гамбурга в сотрудничестве с Дж. Одерматом и А. Клинбергом.
Литература
[1] . Ammann С. Spirig A., Neftel A., Steinbacher М., Komenda М., Schaub А. (2004). «Cali-
bration and Application of PTR-MS for Biogenic VOC Measurements in a Deciduous For-
est», International Journal of Mass Spectrometry 239: 87—101.
[2] . Arno B., and Leif J. (2009). «Myrcene as a Natural Base Chemical in Sustainable Chemistry:
A Critical Review». Chem Sus Chem, 2: 1072—1095.
[3] . Atkinson R., Arey J. (2003). «Atmospheric degradation of volatile organic compounds».
Chemical Reviews, 103: 4605—4638.
[4] . Baker A. K., Beyersdorf A. J., Doesema L.A., Katzenstein A., Meinardi S., Simpson I. J.,
Blake D. R., Rowland F. S. (2008). «Measurements of nonmethane hydrocarbon in 28 Unit-
ed State cities». Atmospheric Environment, 42: 170—182.
[5] . Bampton M. (1999). Anthropogenic Transformation. In: Alexander D. E., and Fair-
bridge R.W. (Eds). Encyclopedia of Environmental Science. Kluwer Academic Publishers:
Dordrecht, The Netherlands.
[6] . Bernstein J. A., Alexis N., Bacchus H., Bernstein I.L., Fritz P., Horner E., et al. (2008).
«The health effects of nonindustrial indoor air pollution». Journal of Allergy and Clinical Im-
munology, 121(3), 585—591.
[7] . Bignell C.M., Dunlop P.J., Brophy J. J., Fookes C.J.R. (1997). «Volatile leaf oils of some
southwestern and southern Australian species of the genus Eucalyptus (Series I). Part XIV.
Subgenus Monocalyptus». Flavour and Fragrance Journal. 12 (3), 177—183.
Литература
[8] . Blake R. S., Monks P. S., Ellis A. M. (2009). «Proton-transfer reaction mass spectrometry».
Chemical Reviews, 109: 861—896.
[9] . Brand C., Ferrante A., Prager R.H., Riley T.V., Carson C.F., Finlay-Jones J. J., HartP. H.
(2001). «The water-soluble components of the essential oil of Melaleuca alternifolia (tea tree
oil) suppress the production of superoxide by human monocytes, but not neutrophils, acti-
vated in vitro». Inflammation Research, 50:213—219.
[10] . Butt D. E. A. (2006). «Formation of phenols from the low-temperature fast pyrolysis of ra-
diata pine (pinus radiata) Part 1. Influence of molecular oxygen». Journal of Analytical and
Applied Pyrolysis, 76: 38—47.
[11] . Clausen P.A., Wilkins С. K., Wolkoff P., Nielsen G.D. (2001). «Chemical and biological
evaluation of a reaction mixture of R-(+)-limonene/ozone, Formation of strong airway ir-
ritants». Environmental International, 26: 511—522.
[12] . Clinton N. E., Gong P., Scott K. (2006). «Quantification of pollutants emitted from very
large wildland fires in Southern California, USA. Atmospheric Environment, 40: 3686—3695.
[13] . Cody R. B., Laramee J. A., H. Dupont Durst H.D. (2005). «Versatile new ion source for
the analysis of materials in open air under ambient conditions». Analytical Chemistry, 77:
2297-2302.
[14] . Crutzen P.J., Andreae M.O. (1990). «Biomass Burning in the Tropics: Impact on Atmos-
pheric Chemistry and Biogeochemical Cycles». Science 250(4988): 1669—1678.
[15] . De Gouw J., Warneke C. (2007). «Measurements of volatile organic compounds in the
earth's atmosphere using proton-transfer reaction mass spectrometry». Mass Spectrometry
Reviews, 26: 223-257.
[16] . Denman K. L., Brasseur G., Chidthaisong A., Ciais P., Cox P. M., Dickinson R. E., Hau-
glustaine D., Heinze C., Holland E., Jacob D., Lohmann U., Ramachandran S., da Sil-
va Dias P. L., Wofsy S. C., Zhang X. (2007). Couplings between changes in the climate system
and biogeochemistry. In Solomon S., Qin D., Manning M., Chen Z., Marquis M., Av-
eryt К. B., Tignor M., Miller H. L. (Eds.), Climate Change 2007: The Physical Science Basis.
Cambridge University Press: New York.
[17] . Evans R.J., Milne T. A. (1987). «Molecular characterization of the pyrolysis of biomass. 2.
Applications». Energy and Fuels, 1 (1987). 123—137.
[18] . FaixO., Meier D., Fortmann I. (1990). «Thermal degradation products of wood. Gas chro-
matographic separation and mass spectrometric characterization of monomeric lignin de-
rived products». Hols als Roh- und Wekstoff, 48: 281—285.
[19] . FaixO., Fortmann L, Bremer J., Meier D. (1991). «Thermal degradation products of wood.
Gas chromatographic separation and mass spectrometric characterization of polysaccha-
ride derived products». Hols als Roh- und Wekstoff, 49: 213—219.
[20] . Fall R. (1999). Biogenic emissions of volatile organic compounds from higher plants. In:
Hewitt C. N. (Ed). Reactive Hydrocarbons in the Atmosphere. Academic Press: San Diego,
CA.
[21] . Font F., Esperanza M., Garcia A.N. (2003). «Toxic by-products from the combustion of
kraft lignin». Chemosphere, 52 (6): 1047—1058.
[22] . Galbally LE., Lawson S.J., Weeks LA., Bentley S.T., Gillett R.W., Meyer M., Gold-
stein A. H. (2007). «Volatile organic compounds in marine air at ICape Grim, Australia».
Environmental Chemistry, 4: 178—182.
330 Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
[23] . Ghannoum О., Phillips, N, Conroy J., Smith R.A., Attard R. D., Woodfield R., Lo-
gan B.A., Lewis J. D., Tissue D.T. (2010). Exposure to preindustrial, current and future
atmospheric CO2 and temperature differentially affects growth and photosynthesis in Eu-
caliptus. Global Change Biology. 16, 1:303—319.
[24] . Girard B., Fukumoto L., Mazza G., Delaquis P., Ewert B. (2002). «Volatile terpene con-
stituents in maturing gewiirztraminer grapes». British Columbia. Am. J. Enol. Viti. 53(2):
99-109.
[25] . Goldstein A. H., Galbally I. E. (2007). «Known and Unexplored Organic Constituents in
the Earth's Atmosphere». Environmental Science & Technology, 1515—1521.
[26] . Graus M., Markus Mtiller M., Hansel A. (2010). «High resolution PTR-TOF: Quantifica-
tion and formula confirmation of VOC in Real Time». Journal of the American Society for
Mass Spectrometry, 2\\ 1037—1044.
[27] . Guenther A., Geron C., Pierce T., Lamb B., Harley P., Fall R. (2000). «Natural emission
of non methane volatile organic compounds, carbon dioxide, and oxides of nitrogen from
north America». Atmospheric Environment, 34: 2205—2230.
[28] . Hansel A., Jordan A., Holzinger R., Prazeller P., Vogel W., Lindinger W. (1995). «Proton-
transfer reaction mass-spectrometry: online trace gas analysis at the ppb level». Interna-
tional Journal of Mass Spectrometry Ion Processes, 149/150: 609—619.
[29] . Harley P., Fridd-Stroud V, Greenberg J., Guenther A., Vasconcellos P. (1998). «Emissionof
2-methyl-3-buten-2-ol by pines: A potentially large natural source of reactive carbon to the
atmosphere». Journal of Geophysical Research 103: 25479—25486.
[30] . Hawkes D. B., Adams G.W., Burlingame A.L., Ortiz de Montellano P. R., De Voss J. J.
(2002). «Cytochrome P450cin (CYP176A), Isolation, Expression, and Characterization».
Journal of Biological Chemistry 277(31): 27725—27732.
[31] . Helmig D. (1997). «Ozone Removal Techniques in the Sampling of Atmospheric Volatile
Organic Trace Gases». Atmospheric Environment, 31: 3635—3651.
[32] . Holzinger R., Sandoval-Soto L., Rottenberger S., Crutzen P.J., Kesselmeier J. (2000).
«Emissions of volatile organic compounds from Quercus ilex L. measured by Proton Trans-
fer Reaction Mass Spectrometry under different environmental conditions». Journal of Geo-
physical Research , 105(D16): 20573—20579.
[33] . Hubschmann H.-J. (2008). Handbook of GC/MS: Fundamentals and applications. Second
Edition, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany.
[34] . IPCC. 2007. Climate change 2007: the physical science basis. Cambridge, UK: Cambridge
University Press.
[35] . Irigaray P., Newby J. A., Clapp R., Hardell L., Howard V., Montagnier L., et al. (2007).
«Lifestyle-related factors and environmental agents causing cancer: An overview». Biomed-
icine & Pharmacotherapy, 61(10), 640—658.
[36] . Jin Z., Akiyama T., Chung B. Y., Matsumoto Y, Liyama K., Watanabe S. (2003). «Changes
in lignin content of leaf litters during mulching». Phytochemistry, 64 (5): 1023—1031.
[37] . Jordan A., Haidacher S., Hanel, G., von Hartungen E., Mark L., Seehauser H., Schott-
kowsky R., Sulzer P., Mark T. D. (2009). «А High Resolution and High Sensitivity Proton-
Transfer-Reaction Time-of-Flight Mass Spectrometer (PTR-TOF-MS)». International
Journal of Mass Spectrometry, 286: 122— 128.
[38] . Karl T., Alex Guenther A., Yokelson R. J., Jim Greenberg J., Potosnak, M. Blake D. R., Ar-
taxo P. (2007). «The tropical forest and fire emissions experiment: Emission, chemistry, and
Литература 331
transport of biogenic volatile organic compounds in the lower atmosphere over Amazonia».
Journal of Geophysical Research, 112: DI 8302.
[39] . Kesselmeier J., Staudt M. (1999). «Biogenic volatile organic compounds (VOC): An over-
view on emission, physiology and ecology». Journal of Atmospheric Chemistry. 33: 23—88.
[40] . Lee A., Schade G. W., Holzinger R., Goldstein A. H. (2005). «А comparison of new meas-
urements of total monoterpene flux with improved measurements of speciated monoter-
pene flux». Atmospheric Chemistry and Physics 5 (2): 505—513.
[41] . Levi H. Mielke L.H., Kerri A. Pratt K.A., Shepson P. B., Scott A. McLuckey S.A.,
Wisthaler A., Hansel A. (2010). «Quantitative determination of biogenic volatile organic
compounds in the atmosphere using proton-transfer reaction linear ion trap mass spec-
trometry». Analytical Chemistry. 82: 7952—7957.
[42] . Lipsett M., Waller, K. Shusterman D., Thollaug S., Brunner W. (1988). «The respiratory
health impact of a large urban fire». American Journal of Public Health. 84(3): 434—438.
[43] . Maleknia S.D., Bell T. L., Adams M.A. (2007a). «Proton Transfer Reaction Mass Spec-
trometry (PTR-MS) of Reference and Plant-Emitted Volatile Organic Compounds». Inter-
national Journal of Mass Spectrometry. 206: 203—210.
[44] . Maleknia S.D., Adams M.A. (2007b). «Reactions of Oxygen-Containing Terpenes with
Peptides and Proteins», Proceedings of the 4th International Peptide Symposium. 334—335.
[45] . Maleknia S. D., Adams M.A. (2008). «Impact of Volatile Organic Compounds from Wild-
fires on Crop Production and Quality». Aspects of Applied Biology, 88: 93—97.
[46] . Maleknia S. D., BellT. L., Adams M.A. (2009a). «Eucalypt Smoke and Wildfires: Tempera-
ture Dependent Emissionsof Biogenic Volatile Organic Compounds», International Journal
of Mass Spectrometry 279: 126—133.
[47] . Maleknia S. D., Vail T. M., Cody R. B., Sparkman D.O., Bell T. L., Adams M.A. (2009b).
«Temperature Dependent Release of Volatile Organic Compounds of Eucalypts by Direct
Analysis in Real Time (DART) Mass Spectrometry», Rapid Communications in Mass Spec-
trometry 23:2241—2246.
[48] . Mitra S., Zhu N., Zhang X., Kebbekus B. (1996). «Continuous monitoring of volatile or-
ganic compounds in air emissions using an on-line membrane extraction-microtrap-gas
chromatographic system», Journal of Chromatography A, 736: 165—173.
[49] . Moreno N. J., and Azpilicueta C. A. (2007). «Binding of oak volatile compounds by wine
lees during simulation of wine ageing». LWT Food Sci. Technol. 40: 619—624.
[50] . Nemitz E., Langford B., House E., Phillips G., Famulari, Neil Cape J.N., Brian Davi-
son B., and Nick Hewitt N. (2007). «Micrometeorological Measurements of Anthropogenic
VOC Emissions from Urban Areas». In Volatile compounds in the polluted atmosphere, Pro-
ceedings of the 3rd ACCENT Barnsdale expert meeting.
[51] . Pawliszyn J. (2009). Handbook of Solid Phase Microextraction, Chemical Industry Press.
[52] . Penuelas J., Straudt M. (2010). «BVOCs and global change. Trends in Plant Science. 15(3):
133-143.
[53] . Pollnitz A P, Pardon К H, Sykes M, Sefton M A. (2004). «The effects of sample prepara-
tion and gas chromatograph injection technique on the accuracy of measuring guaiacol,
4-methylguaiacol and other volatile oak compounds in oak extracts by stable isotope dilu-
tion analyses». Journal of Agriculture & Food Chemistry. 52: 3244—3252.
Глава 11. Роль масс спектрометрии в исследовании летучих органических
соединений
[54] . Prazeller Р., Palmer Р.Т., Boscaini Е., Jobson Т., Alexander М. (2003). «Proton trans-
fer reaction ion trap mass spectrometer». Rapid Communications in Mass Spectrometry 17:
1593-1599.
[55] . Rohr A. C., Wilkins С. K., Clausen P.A., Hammer M., Nielsen G.D., WolkoffP., Speng-
ler J.D. (2002). «Upper airway and pulmonary effects of oxidation products of (+)-«-
pinene, d-limonene, and isoprene in balb/c mice». Inhalation Toxicology. 14(7): 663—684.
[56] . Russo R. S., Zhou Y., White M.L., Mao, H. Talbot R., Sive B.C. (2010). «Multi-year
(2004—2008) record of nonmethane hydrocarbons and halocarbons in New England: sea-
sonal variations and regional sources». Atmospheric Chemistry and Physics Discussions, 10:
1083-1134.
[57] . Ruuskanen T. M., Kolari P., Back J., Kulmala M., Rinne J., Hakola H., Taipale R.,
Raivonen M., Altimir N., Hari P. (2005). «On-line field measurements of monoterpene
emissions from Scots pine by proton-transfer-reaction mass spectrometry». Boreal Environ-
ment Research, 10(6): 553—567.
[58] . Saka S. (2001). «In: Hon D. N.-S., and Shiraishi N. (eds.). Wood and Cellulose Chemistry.
Marcel Dekker AG, Basal, Switzerland.
[59] . Steinbacher, M, Dommen J., Ordonez C., Reimann S., Gruebler F. C., Staehelin J., Prev-
ot A.S. H. (2005). «Volatile organic compounds in the Po Basin. Part A: Anthropogenic
VOCs». Journal of Atmospheric Chemistry, 51: 271—291.
[60] . Swiston J. R., Davidson W., Attridge S., Li G.T., Brauer, M., van Eeden S. F. (2008). «Wood
smoke exposure induces a pulmonary and systemic inflammatory response in firefighters.
European Respiratory Journal, 32: 129—138.
[61] . Tame N.M., Dlugogorski B.Z., Kennedy E. M. (2007). «Formation of dioxins and furans
during combustion of treated wood». Progress in Energy and Combustion Science, 33:
384-408.
[62] . Tani A., Hayward S., and Hewitt C. N. (2003). «Measurement of monoterpenes and related
compounds by proton transfer reaction-mass spectrometry». International Journal of Mass
Spectrometry, 223—224: 561—578.
[63] . Tanimoto H., Aoki N., Inomata S., Hirokawa J., Sadanaga Y. (2007). «Development of
a PTRTOFMS instrument for real-time measurements of volatile organic compounds in
air». International Journal of Mass Spectrometry, 263, 1—11.
[64] . Yu C., & Crump D. (1998). «А review of the emission of VOCs from polymeric materials
used in buildings». Building and Environment, 33(6), 357—374.
[65] . Wang S., Ang H. M., & Tade M.O. (2007). «Volatile organic compounds in indoor envi-
ronment and photocatalytic oxidation: State of the art». Environment International, 33(5),
694-705.
[66] . Warneke, C.; de Gouw J. A.; Lovejoy E.R.; Murphy P.C.; Kuster W.C.; Fall R. (2005).
«Development of proton-transfer ion trap-mass spectrometry: On-line detection and iden-
tification of volatile organic compounds in air». Journal American Society for Mass Spec-
trometry 16: 1316—1324.
[67] . Went F. W. (1960). «Blue hazes in the atmosphere». Nature, 187(4738): 642—643.
ГЛАВА 12
ИДЕНТИФИКАЦИЯ
И КОЛИЧЕСТВЕННОЕ
ОПРЕДЕЛЕНИЕ
ТОКСИКОЛОГИЧЕСКИ
ЗНАЧИМЫХ ПОБОЧНЫХ
ПРОДУКТОВ ДЕЗИНФЕКЦИИ
ВОДЫ МЕТОДАМИ
МАСС-СПЕКТРОМЕТРИИ
Сюзан Д. Ричардсон
12.1. Введение
Дезинфекция питьевой воды справедливо считается триумфом здравоохранения
XX века. До широкого внедрения этой процедуры миллионы людей умирали от болез-
ней, связанных с плохим качеством воды. Теперь население развитых стран ежеднев-
но получает качественную воду через национальные системы водоснабжения. Тем не
менее химическая дезинфекция стала источником непреднамеренного вреда для здо-
ровья. Сюда можно отнести риск новообразований, нарушение репродуктивной функ-
ции и проблемы развития (включая ранние выкидыши и дефекты у новорожденных).
Причиной являются химические соединения — побочные продукты дезинфекции
(ППД) — в питьевой воде (Richardson et al., 2007; Villanueva et al., 2004; Cantor et al., 2010;
Nieuwenhuijsen et aL, 2000; Waller et al., 1998; Savitzet aL, 2005). Миллионы людей подвер-
гаются воздействию ППД, не только потребляя химически подготовленную воду. Кон-
такт неизбежен во время приема душа, купания, посещения плавательного бассейна
(рис. 12.1). Для решения этих важных проблем здоровья во всем мире ведутся активные
исследования.
Химические дезинфектанты (хлор, озон, хлорамины, диоксид хлора) используются
для уничтожения патогенных микроорганизмов в питьевой воде. Однако эти реагенты
одновременно являются мощными окислителями и активно реагируют с природными
органическими соединениями (глава 19), которые всегда присутствуют в природной
воде благодаря разложению растительных материалов, а также с бромидами и иодидами,
которые также присутствуют в водоемах (реках, озерах, подземных горизонтах). При-
Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
Рис. 12.1. Варианты путей воздействия ППД
родные органические соединения, представленные в основном гуминовыми и фуль-
вокислотами, являются главными предшественниками ППД (схема 1), но с дезинфи-
цирующими агентами могут взаимодействовать и многочисленные антропогенные
загрязняющие вещества. Эти экотоксиканты оказываются в источниках питьевой воды
со сточными водами, со смывами с сельскохозяйственных угодий и т.д. Были описаны
ППД, связанные с фармацевтическими и антибактериальными препаратами, пестици-
дами, красителями тканей, эстрогенами, бисфенолом А, поверхностно-активными ве-
ществами на основе алкилфенолов, протекторами УФ-излучения и дизельным топли-
вом (Richardson etal., 2007). Обычно сами загрязняющие вещества напрямую реагируют
с дезинфицирующими агентами, но иногда в реакцию вступают уже продукты транс-
формации исходных ксенобиотиков в окружающей среде. Например, недавно показа-
Humic acid
Рис. 12.2. Образование ППД из природного органического вещества (гуминовых кислот);
теоретическая структрура гуминовой кислоты по Шультену и Шнитцеру. (Пе-
репечатано из Schulten H.-R. and Schnitzer М. (1993). A state of the art structural
concept for human substances. Naturwissenschaften, 80:29-30. С разрешения Springer
Science + Business Media B.V.)
12.1. Введение
но, что именно продукт микробного разложения толилфлуанида (популярный в Европе
фунгицид) отвечает за образование высококанцерогенного А-нитрозоди метилами на
(НДМА) при озонировании питьевой воды {Schmidt and Brauch, 2008).
Хлор, озон, хлорамины, диоксид хлора — наиболее популярные химические де-
зинфектанты, причем каждый из них характеризуется своим набором ППД питьевой
воды. Увеличивается популярность двух нехимических средств дезинфекции питьевой
воды: УФ-обработки и мембран обратного осмоса. Эти технологии определенно пер-
спективны с точки зрения снижения уровней ППД в питьевой воде. Однако недостатки
есть и у этих нехимических средств. Например, имеются предварительные данные, что
УФ-излучение может взаимодействовать с природными органическими соединениями,
приводя позднее к повышенным уровням ППД после проведения необходимого вто-
ричного дохлорирования (Liu et al., 2006). Обратный осмос приводит к проблеме ути-
лизации рассолов, образующихся после обработки воды. Этот метод все чаще исполь-
зуется на заводах опреснения, перерабатывающих морскую воду в питьевую. Основные
дезинфектанты и их структуры представлены в табл. 12.1.
Хлороформ и другие тригалометаны (ТГМ) были первыми ППД, идентифициро-
ванными в хлорированной питьевой воде в 1974 году (Rook, 1974; Bellaretal., 1974). Вскоре
после этого было установлено, что ТГМ могут вызывать рак у лабораторных животных
(NC1, 1976). В результате эти соединения были нормированы в США в 1979 году (U.S.
ЕРА, 1979), а позже и во многих других странах. Сейчас в США нормированы несколько
других ППД, включая 5 галогенуксусных кислот (ГУК), хлорит и бромат. Нормирован-
ные ТГМ иногда упоминаются как ТГМ4, ГУК — как ГУК5, а 9 наиболее распростра-
ненных хлорбромуксусных кислот — как ГУК9. ТГМ и ГУК образуются в основном при
использовании хлора и хлораминов; хлорит — диоксида хлора, а бромат — озона. Нор-
мируемые в США и Европейском Союзе ППД представлены в табл. 12.2.
Из четырех наиболее популярных современных дезинфектантов хлор обычно при-
водит к наиболее высоким уровням ТГМ и ГУК. Поскольку на станциях подготовки
питьевой воды хлором некоторые из указанных нормативов могли превышаться, мно-
гие были вынуждены использовать другие дезинфицирующие агенты. В частности,
хлор в качестве первичного дезинфектанта заменялся на озон, диоксид хлора, хлора-
мины или же использовалась УФ-обработка. В некоторых случаях хлор используется
для процедуры дохлорирования вслед за первичной обработкой воды диоксидом хлора,
озоном или ультрафиолетом. Это делается для поддержания требуемой концентрации
дезинфектанта в водораспределительных системах. Однако замена дезинфицирующе-
го агента может привести к возникновению новых проблем. Например, использова-
ние озона может заметно сократить или даже полностью устранить образование ТГМ
Таблица 12.1. Наиболее распространенные дезинфицирующие средства питьевой воды
Дезинфектант Химическая природа
Хлор С12 газообразный или НОС1 — хлорная вода
Монохлорамин NH2C1 (генерируется при взаимодействии аммиака с хлором; в меньших концентрациях образуются также дихлорамин и трихлорамин)
Озон Оз
Диоксид хлора сю2
УФ-обработка —
Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
и ГУК. Однако оно может резко повыситься уровень броматов, особенно когда при-
родные источники воды характеризуются повышенным содержанием бромидов. Вы-
сокие уровни бромидов и иодидов могут наблюдаться в природной воде (например
в реках) около морского побережья в связи с интрузией морской воды в природные ис-
точники, а иногда и в материковых водоемах благодаря окаменелой морской воде, когда
соли древних морей влияют на солевой состав поверхностных и грунтовых вод. Бромат
Таблица 12.2. Нормируемые побочные продукты дезинфекции’
Список Агентства по окружающей среде США ПДКа(мг/л)
Суммарное содержание тригалометанов (хлороформ, бромдихлорметан, дибромхлорметан, бромоформ) 0,08
Суммарное содержание 5 галогенуксусных кислот (хлор-, бром-, дихлор-, дибром и трихлоруксусные кислоты) 0,06
Бромат 0,01
Хлорит 1,0
Список Всемирной организации здравоохранения (ВОЗ) Целевое значение (мг/л)
Хлороформ 0,3
Бромдихлорметан 0,06
Дибромхлорметан 0,1
Бромоформ 0,1
Хлоруксусная кислота 0,02
Дихлоруксусная кислота 0,05б
Трихлоруксусная кислота 0,2
Бромат 0,01б
Хлорит 0,7б
Хлорат 0,7б
Дихлорацетонитрил 0,02б
Дибромацетонитрил 0,07
Хлорциан 0,07
2,4,6-Трихлорфенол 0,2
N-Нитрозодиметиламин (НДМА) 0,1
Стандарты Европейского Сообщества Стандартные значения (мг/л)
Общее содержание тригалометанов 0,1
Бромат 0,01»
Прочие нормативные документы ПДК (мг/л)
N-Нитрозодиметиламин (НДМА) 9', 10д
Список нормируемых соединений этого типа в России существенно отличается.
а Документы Всемирной организации здравоохранения (ВОЗ) поТГМ постулируют, что и уров-
ни отдельных ТГМ, и их сумма не должны превосходить указанных величин. Требования ВОЗ
можно найти на сайте: http://www.who.int/water_sanitation_health/dwq/gdwq3rev/en/.
6 Временные целевые значения.
“ Там, где это возможно без ущерба для дезинфекции, члены ЕС должны стремиться к меньшим
величинам.
[ Онтарио (Канада).
д Калифорния (США).
12.1. Введение
очень опасен, поскольку доказана его канцерогенность в экспериментах на лаборатор-
ных животных (Kurokawa et aL, 1986). N-нитрозодиметиламин — еще один канцеро-
ген, который может образовываться в значимых количествах при хлораминировании
воды (Mitch et aL, 2003; Richardson et aL, 2007). Иодсодержащие ТГМ и ГУК генотоксич-
ны и цитотоксичны для млекопитающих и человека (Richardson et aL, 2008а; Plewa et aL,
2004; Attene-Ramos etaL, 2010). Они также могут образовываться в повышенных концен-
трациях при хлораминировании (Richardson et aL, 2008а). Уровни экотоксикантов еще
одной группы ППД — бромнитрометанов — значительно увеличиваются при проведе-
нии предозонирования (Richardson et aL, 1999а; Krasner et al., 2006). Различия в составе
природной воды, включая концентрацию бромидов и иодидов, уровни природного ор-
ганического вещества, pH могут существенно влиять на образование различных хлор-,
бром- или иодсодержащих ППД.
За последние 30 лет была проведена большая работа по исследованию образования
ППД и их влиянию на здоровье. Сейчас установлено более 600 ППД (Richardson, 2011;
Richardson, 1998). Классы ППД представлены в табл. 12.3. Однако менее 100 из них из-
учены с точки зрения уровней образования и встречаемости в питьевой воде, а также
в токсикологических исследованиях. Уровни ППД в питьевой воде варьируются в диа-
пазоне от нанограммов до микрограммов в литре. Однако более 50% галогенсодержа-
щих (хлор, бром и иод) ППД, образующихся в результате хлорирования, и более 50%
ППД, образующихся в результате озонирования, остаются неидентифицированными
(Krasner et aL, 2006; Richardson et aL, 2008b), причем ничего не известно о токсичности
большинства ППД питьевой воды. Изучение влияния ППД на здоровье было связано
в основном с канцерогенностью, мутагенностью, генотоксичностью и цитотоксично-
стью. Есть данные, что типы рака, вызываемые нормируемыми ППД в экспериментах
с животными (в основном рак печени), не совпадают с наблюдаемыми в эпидемиоло-
гических исследованиях населения (в основном рак мочевого пузыря). Возможно, что
именно пока не нормированные соединения ответственны за этот эффект. Также не ис-
ключено, что проглатывание (основной путь в экспериментах с животными) не является
единственным важным путем воздействия ППД на организм.
Таблица 12.3. Примеры химических классов побочных продуктов дезинфекции (ППД)
Галогенированные ППД Негалогенированные ППД
Галометаны Нитрозамины
Галогенированные кислоты Ал ьдегиды
Галогенированные альдегиды Кетоны
Галогенированные кетоны Карбоновые кислоты
Галогенированные нитрилы —
Галогенированные амиды —
Галогенированные нитрометраны
Галогенированные фураноны (например MX) —
Галогенированные хиноны —
Галогенированные пирролы —
Галогенированные феназины —
Галогеноксиды (например хлораты, броматы, йодаты)
Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
Последние работы указывают, что другие виды жизнедеятельности, включая купа-
ние, принятие душа, посещение бассейнов с хлорированной водой, могут увеличивать
негативный эффект некоторых ППД {Ashley et al., 2005; Haddad et al., 2006; Leavens et al.,
2007; Xu and Weisel, 2005a and 2005b; Zwiener et aL, 2007; LaKindet aL, 2010; Richardson, 2005;
Richardson et al., 2010; Xu and Weisel, 2005a;2005b;Zwiener et al., 2007; Cardador and Gallego,
2011). Дело в том, что ППД оказываются в организме не только при питье воды. Некото-
рые из них могут проникать при дыхании или через кожу {Ashley etal., 2005;Хи and Weisel,
2005a, 2005b; Haddad et al., 2006; Leavens et al., 2007). Так, летучие ППД, легко перехо-
дящие из жидкой среды в газообразную (включая ТГМ), могут поступать в организм
при дыхании во время принятия душа или посещения крытых бассейнов. В последнем
случае неважно, плавает ли человек или сидит около бассейна. ТГМ и галокетоны де-
тектировались в выдыхаемом воздухе после купания, плавания или принятия душа
{Erdingeret aL, 2004; Хи and Weisel, 2004, 2005а, 2005b; Ashley et aL, 2005; Leavens etaL, 2007).
Эти пути воздействия официально признаны в настоящее время благодаря результатам
эпидемиологических исследований населения. Недавние результаты указывают также,
что именно эти альтернативные пути воздействия могут отвечать за увеличение риска
возникновения рака мочевого пузыря {Villanueva et aL, 2007), а посещение бассейнов
с хлорированной водой связано с возникновением респираторных заболеваний, вклю-
чая астму {LaKind et aL, 2010).
Появились также новые свидетельства в пользу того, что генетическая предраспо-
ложенность может играть роль в возникновении рака мочевого пузыря. Недавние эпи-
демиологические исследования, проведенные в Испании, показали, что люди с поли-
морфизмом, связанным с глутатион 5-трансферазой зета-1 {GSTZ1), или те, у которых
отсутствует одна или обе копии глутатион 5-трансферазы тета-1 {GSTT1), особенно вос-
приимчивы к раку мочевого пузыря, если уровень ТГМ в их питьевой воде превышает
49 мкг/л {Cantor et al., 2010). Около 29% населения Испании, вовлеченного в это иссле-
дование, имеет такую генетическую особенность. Она будет наблюдаться и у примерно
25 % жителей США.
Масс-спектрометрия — это ключевой метод идентификации ППД в питьевой воде
и в воде бассейнов. Она же часто используется и для количественного определения этих
соединений. Главной причиной популярности масс-спектрометрии является ее высокая
чувствительность, а также способность работать со сложными смесями (в стакане пи-
тьевой воды может присутствовать более 300 ППД). В этом случае масс-спектрометрия
обычно используется в тандеме с газовой, жидкостной или ионной хроматографией,
которые разделяют компоненты этих сложных смесей и дают возможность достигать
пределов обнаружения до нг/л.
Экстракция и концентрирование ППД обычно осуществляется перед масс-
спектрометрическим определением. Это позволяет увеличить исходные концентрации
ППД в питьевой воде. Дериватизация путем проведения химических реакций часто
используется для детектирования соединений с определенными функциональными
группами. Помимо питьевой воды уровни ППД измеряются в воде бассейнов, трубах
горячего водоснабжения, продуктах питания, крови и моче человека. В последующих
разделах будет приведено обсуждение этих аналитических методов. Будут также пред-
ставлены примеры того, как они используются для установления структур новых ППД
и для количественного определения токсикологически значимых соединений.
12.2. Аналитические методы идентификации и количественного определения ППД
12.2. Аналитические методы идентификации
и количественного определения ППД
12.2.1. Методы экстракции/концентрирования
Наиболее широко используемыми методами концентрирования являются твердофаз-
ная экстракция (solid phase extraction, SPE), твердофазная микроэкстракция (solid phase
microextraction, SPME), жидкость-жид костная экстракция (LLE), метод выдувания-
улавливания (purge-and-trap, Р&Т) и экстракция на смолах XAD (для больших коли-
честв воды). С недавних пор для концентрирования ППД используются также мембра-
ны обратного осмоса (Richardson etal., 2008b; Speth etal., 2008; Pressman etal., 2010).
Картриджи твердофазной экстракции в настоящее время являются наиболее по-
пулярными для концентрирования и количественного определения целевых ППД
(рис. 12.3). Эта популярность объясняется разнообразием адсорбционных фаз, которые
позволяют проводить экстракцию широкого круга химических соединений. Кроме
того, такая экстракция значительно быстрее, чем альтернативные (жидкость-жидкост-
ная и с использованием XAD-смол). Она также более автоматизирована, поскольку од-
новременно могут обрабатываться несколько образцов. Обычно экстракционные кар-
триджи SPE предварительно кондиционируются в органическом растворителе и/или
очищенной воде. Затем образец питьевой воды пропускается в условиях вакуумиро-
вания через картридж, после чего адсорбированные аналиты элюируются небольшим
количеством органического растворителя. В качестве примеров использования SPE
Рис. 12.3. Твердофазная экстракция (ТФЭ) (фотография Бенжамина Блаунта, Центр
по контролю и предотвращению заболеваний)
Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
можно привести процедуры экстракции/концентрирования галометанов, галоацето-
нитрилов, галопропанонов, а также галонитрометанов во время национального иссле-
дования распространения токсикологически значимых ненормируемых ППД (Krasner
etal., 2006; Weinbergetal., 2002). Твердофазная экстракция с использованием картриджей
Varian Bond Elut PPL позволяет детектировать эти ППД на уровнях ниже ppb (нг/л). SPE
также используется для экстрагирования нитрозами нов в рамках метода 521 (www.epa.
gov/microbes/m_52l.pdf), а в стыковке с ЖХ/МС/МС — для идентификации представи-
телей нового класса ППД — галохинонов (Zhao et al., 2010). В этом случае применялись
картриджи Oasis HLB, которые эффективны для извлечения из воды соединений высо-
кой полярности.
Для твердофазной микроэкстракции (SPME) используются волокна со специаль-
ным покрытием. Они не требуют применения органического растворителя (рис. 12.4).
Волокно твердофазной микроэкстракции обычно помещается в воду или биологиче-
скую жидкость, а иногда для улавливания летучих компонентов — в парофазный объем
над водой или биообразцом (headspace). Образец при этом перемешивается, а иногда на-
гревается. Органические соединения сорбируются волокном, которое затем помещается
непосредственно в инжектор газового хроматографа без использования элюирующего
растворителя (Ouyrang and Pawliszyn, 2006; Niri et al., 2008). Центры контроля и предот-
вращения заболеваний регулярно применяют SPME для количественного определе-
ния ППД и других ксенобиотиков в воде, крови и моче человека, поскольку этот под-
ход позволяет проводить в автоматическом режиме анализ тысяч образцов ежегодно.
Шприц
ТФМЭ
Газовый
шприц
Герметически
Вакуумиро-
ванная трубка
для забора
крови
Волокно
ТФМЭ
Парофазная ТФМЭ/
криофокусировка/
ГХ/МС МЗИ
Змл крови
закрытый образец + внутренний стандарт
Рис. 12.4. Слева: схема проведения твердофазной микроэкстракции (ТФМЭ) на приме-
ре образца человеческой крови. Справа: Combi Ра! автосамплер ТФМЭ (СТС
Analytics AG, Zwingen, Switzerland). (Схема и фотография Бенжамина Блаунта,
Центр по контролю и предотвращению заболеваний)
12.2. Аналитические методы идентификации и количественного определения ППД
В качестве свежего примера можно привести
определение методом ГХ/МС с твердофазной
микроэкстракцией ТГМ и иодсодержащих
ТГМ в питьевой воде и крови {Richardson et al.,
2008а; Silva et al., 2006).
Метод жидкость-жидкостной экстрак-
ции можно назвать наиболее старомодным.
Уже много десятилетий он используется для
извлечения из воды органических соедине-
ний (рис. 12.5). Метод основан на экстра-
гировании аналитов из воды органическим
растворителем в делительной воронке или
другом сосуде. Эффективность его зависит
от разницы в растворимости аналита в воде
и органическом растворителе. Наиболее по-
пулярными растворителями являются дих-
лорметан, гексан и метил-АиреАи-бутиловый
эфир (МТБЭ). Этот процесс в англоязычной
литературе часто называют «вытрясывани-
ем», поскольку он требует интенсивного пере-
мешивания водного образца и органического
Рис. 12.5. Процесс жидкость-жидкостной
экстракции больших объемов (объем образ-
цов воды I л)
растворителя для обеспечения максимального перехода аналитов в органическую фазу.
Иногда в водный образец до начала экстракции добавляется соль (например сульфат
натрия). Это увеличивает полноту извлечения, а сама процедура называется «высали-
ванием» {Richardson et aL, 2008а). Жидкость-жидкостная экстракция может быть более
трудоемкой, чем альтернативные варианты, но при этом для некоторых соединений она
может быть более эффективной. Именно поэтому она до сих пор широко использует-
ся. В частности, уровни извлечения из питьевой воды галофуранов, включая MX, были
максимальными при использовании этого метода в рамках исследований по нацио-
нальной программе выявления ППД (Weinburgetal., 2002; Krasner etal., 2006). Жидкость-
жидкостная экстракция оказалась оптимальным методом и при извлечении из питье-
вой воды кислот, содержащих иод {Richardson et al., 2008а).
Выдувание-улавливание (purge&trap, Р&Т) — весьма эффективная процедура для
извлечения из воды летучих соединений. Поток азота выдувает из воды аналиты, ко-
торые улавливаются на колонке, заполненной сорбентом (рис. 12.6). Затем ловушка на-
гревается и летучие соединения направляются в хроматографическую колонку для раз-
деления. Выдувание-улавливание — важнейший метод определения ТГМ (метод 524.2
Агентства по охране окружающей среды) (www.caslab.com/EPA-Methods/PDF/524_2.pdf).
Экстракция на смолах XAD — наиболее трудоемкий метод. Он требует серьезной
предварительной очистки используемой смолы всокслете {Richardson etal., 2008b; Richard-
son etal., 1994), причем может понадобиться несколько часов для проведения экстракции
из большого объема питьевой воды (рис. 12.7). Тем не менее это один из основных мето-
дов определения ППД в питьевой воде, поскольку достижимы факторы концентрирова-
ния более 10000, причем далее возможно получение полного масс-спектра, применение
масс-спектрометрии высокого разрешения, а также других масс-спектрометрических
Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
Рис. 12.6. Аппарат для проведения выдувания-улавливания (Purge-and-trap)
методов для идентификации ППД в следовых количествах. Этот метод также наиболее
эффективен для исследований, связанных с определением мутагенности, включая тест
Эймса и тест Комета на клетках млекопитающих, которые требуют больших факторов
концентрирования (Richardson etal., 2007). Использование XAD-смол позволило значи-
тельно расширить круг известных ППД.
Обратный осмос применялся в недавнем химико-токсикологическом исследова-
нии для предварительного концентрирования ППД, когда сохранение водной матри-
цы было необходимо для экспериментов на животных (Speth etal., 2008; Richardson etal.,
2008b; Pressman etal., 2010). Для идентификации ППД водные концентраты после прове-
дения обратного осмоса подвергались жидкость-жидкостной и твердофазной экстрак-
ции, а также экстракции на смолах XAD (в зависимости от класса). В результате удалось
12.2. Аналитические методы идентификации и количественного
Рис. 12.7. (а) Чистка смол XAD в аппарате Сокслета; (б) XAD-8 расположен над XAD-2;
(в) экстранирование органических соединений из питьевой воды
детектировать следовые количества ППД, причем некоторые из них не смогли бы быть
зарегистрированы без использования предварительной процедуры обратного осмоса
{Pressman et al., 2010).
12.2.2. Масс-спектрометрические методы детектирования
Масс-спектрометрия — ключевой метод идентификации неизвестных ППД и количе-
ственного определения целевых ППД в питьевой воде. Электронная ионизация и хими-
ческая ионизация традиционно используются в режиме ГХ/МС, а электрораспыление
и химическая ионизация при атмосферном давлении — в режимах ЖХ/МС, ультра-
эффективной жидкостной хроматографии/масс-спектрометрии и ионной хромато-
графии/масс-спектрометрии (главы 1—3). Масс-спектрометрия с мембранным вводом
применялась для предварительного концентрирования и введения образца в масс-
спектрометр в режиме онлайн. В некоторых случаях использовалась также спектро-
метрия приращения ионной подвижности. Для идентификации новых соединений
и подтверждения детектирования целевых аналитов желательно использовать полный
масс-спектр. В этом случае можно использовать библиотеки масс-спектров или ручную
расшифровку. Следует отметить, что запись полного масс-спектра, как правило, тре-
бует более высоких концентраций аналитов. С другой стороны, мониторинг заданных
ионов (глава 2) с регистрацией исключительно ионов с конкретной величиной m/z часто
Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
позволяет детектировать и проводить количественное определение целевых аналитов,
присутствующих в следовых количествах. В случае ЖХ/МС/МС мониторинг выбран-
ной реакции (selected reaction monitoring, SRM) или мониторинг нескольких реакций
(multiple reaction monitoring, MRM) используется для детектирования конкретного про-
цесса (или нескольких процессов) фрагментации молекулярного иона с образованием
известных фрагментных ионов. SRM- и MRM-режимы характеризуются улучшенными
пределами обнаружения аналитов благодаря уменьшению уровня фона, который в ус-
ловиях ЖХ/МС обычно выше (глава 3).
Типы и размеры масс-спектрометров варьируются в широких пределах (введение
и глава 1). Классические магнитные секторные масс-спектрометры могут работать в ре-
жимах низкого и высокого разрешения (глава 1), что очень полезно для установления
структуры новых соединений. Однако для большинства экологических исследований,
включая количественное определение ППД, наиболее популярны небольшие по раз-
меру квадрупольные приборы. Тройные квадрупольные масс-спектрометры часто
используются в режимах ЖХ/МС/МС или ГХ/МС/МС. Времяпролетные и квадру-
польно-времяпролетные приборы постепенно завоевывают популярность, однако их
использование в экологических исследованиях пока не реализовано должным образом.
Очень большие по размеру масс-спектрометры с преобразованием Фурье, обладающие
сверхвысоким разрешением, используются в основном для получения информации
о структуре предшественников ППД, природных органических соединениях (глава 19)
или о высокомолекулярных ППД.
ГХ/МС
ГХ/МС остается важнейшим методом идентификации и количественного определения
ППД (глава 2). Этому способствуют библиотеки масс-спектров (NIST и Wiley), опи-
санные в главе 5. Если спектр обнаруженного ППД отсутствует в библиотеке спектров,
химическая ионизация и масс-спектрометрия высокого разрешения используются для
получения структурной информации, а ручная интерпретация полного масс-спектра
дает возможность идентифицировать новые ППД. ГХ/МС — важнейший метод иденти-
фикации большинства известных на сегодняшний день более 600 ППД.
На рис. 12.8 представлена хроматограмма (ГХ/МС) типичного образца питьевой
воды. Как правило, наблюдается более 3000 пиков, обусловленных индивидуальными
ингредиентами. При использовании ГХ/ГХ/МС, позволяющего добиться разделения
компонентов с близкими временами выхода, число пиков становится еще большим.
Этот метод только-только начал применяться для анализа питьевой воды (глава 6).
Многие из этих пиков обусловлены ППД. Другие связаны с непрореагировавшим ор-
ганическим веществом, всегда присутствующим в природной воде. При проведении
скрининговых исследований по выявлению всего спектра ППД необходимо сравнивать
4.00 6.00 8.00 1(100 1200 14.00 16.00 18.00 2000 2200 24.00 26.00 28.00 3Q00 3200
Время, мни
Рис. 12.8. Хроматограмма экстракта питьевой воды. Метод ГХ/МС
12.2. Аналитические методы идентификации и количественного определения ППД 345
результаты анализа образцов обработанной дезинфицирующим агентом и необрабо-
танной (исходной) воды. В этом случае можно вычленить вклад природного органиче-
ского вещества и антропогенных экотоксикантов, например пестицидов.
Процесс идентификации новых ППД можно проиллюстрировать на примере двух
изомерных продуктов, недавно обнаруженных в питьевой воде и в воде плавательного
бассейна (Richardson etal., 2010; Pressman etal., 2010). Эти два соединения были обнаруже-
ны только в экстрактах, подвергшихся метилированию. В контрольном образце и в не-
дериватизованной пробе пики этих соединений отсутствовали. Времена выхода соеди-
нений различались (рис. 12.9, верх), хотя их масс-спектры были очень близки (типично
для изомеров). В обоих случаях наблюдались характерные триплеты с m/z 256/258/260
и 225/227/229, а также фрагментный ион с m/z 59 (рис. 12.9, низ). Спектры не были похо-
жи ни на один из присутствующих в спектральных библиотеках NIST и Wiley. Они так-
же не были ранее описаны в литературе. Потеря 31 единицы массы (как правило, ОСН3),
приводящая к иону с m/z 225, и присутствие пика иона с m/z 59 (обычно С(О)ОСН3) при-
сущи наличию в структуре фрагмента метилового эфира карбоновой кислоты, предпо-
ложительно с молекулярным ионом с m/z 256/258/260. Соотношение изотопных пиков
в кластере m/z 256/258/260 свидетельствовало о наличии одного атома брома и одного
атома хлора в молекуле. Такое соотношение обусловлено наложением благодаря двум
природным изотопам брома (79Вг и 8|Вг) и хлора (35С1 и 37С1). Кстати, число атомов хло-
ра и брома легко определяется по соотношению изотопных пиков, причем оно не за-
висит от наличия в молекуле большинства других элементов: углерода, водорода, азо-
та, кислорода и т.д. (рис. 12.10). Это весьма удобно, поскольку в случае присутствия
нескольких атомов этих галогенов в молекуле значительная доля молекулярной массы
может быть очень быстро установлена. Кстати, я нахожу эти кластеры весьма прият-
Диметиловый эфир z-бромхлорбутендиовой кислоты
Молекулярная масса = 256
m/z 225
m/z 197
m/z
Рис. 12.9. (а) Пики двух изомерных соединений на хроматограмме (ГХ/МС); (б) масс-
спектр одного из этих изомеров
346 Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
ными на вид, почти художественными. Они прекрасно представляют «красоту и мощь»
масс-спектрометрии1.
Таким образом, имеющаяся спектральная информация позволяла предположить,
что два неизвестных соединения могли быть диметиловыми эфирами бромхлорбутенди-
овой кислоты с моноизотопной молекулярной массой 256 Да. Измерение точной массы
с привлечением масс-спектрометрии высокого разрешения (10000) подтвердило брутто-
формулу такого соединения (С6Н6О4С1Вг). Измеренная точная масса максимального пика
изотопного кластера (ион с m/z 257,9116) всего на 0,0002 Да отличалась от теоретического
значения (m/z 257,9118). Это было дополнительным свидетельством в пользу диметиловых
эфиров бромхлорбутендиовой кислоты. Однако точное установление структуры сделать
исключительно на основании масс-спектрометрических данных не представлялось воз-
можным, как это часто случается со спектрами изомеров. Наиболее вероятными были
(Z-) и (Е-) изомеры, представляющие цис- и ягрянс-кислоты. По счастью, в образце при-
сутствовали оба изомера, т.е. задачей было только отнесение их пиков на хроматограм-
ме. С этой целью оба изомера были синтезированы, будучи коммерчески недоступными,
и проанализированы методом ГХ/МС в тех же условиях, что и экстракты образцов пи-
тьевой воды. Сравнение времени удерживания показало, что первым выходит Z-, а вто-
рым — Е-изомер. Их времена выхода были 16,8 и 16,9 мин. соответственно (рис. 12.9, верх).
ГХ/МС и ГХ/МС/МС популярны также для количественного определения ППД.
Мониторинг заданных ионов (SIM) или мониторинг заданных реакций (MRM) ис-
пользуются в режимах ГХ/МС и ГХ/МС/МС соответственно. Это увеличивает чувстви-
тельность, позволяя улучшить пределы обнаружения (глава 2). Метод 524.2 Агентства
по окружающей среде США использует ГХ/ИЭ-МС для определения ТГМ, а метод 521 —
ГХ/ХИ-МС/МС для определения нитрозаминов. Следует также отметить, что количе-
ственное определение многих ненормированных ППД в рамках Национального иссле-
дования распространенности ППД было проведено с помощью ГХ/МС (Krasner et al.,
2006; Weinberg et aL, 2002).
ЖХ/МС/МС
ЖХ/МС/МС и ультраэффективная жидкостная хроматография в сочетании с тандем-
ной масс-спектрометрией (УЭЖХ/МС/МС) становятся все более популярными для
идентификации и количественного определения высокополярных и высокомолекуляр-
ных ППД (Zwiener and Richardson, 2005). Так, именно ЖХ/МС/МС была использована
для идентификации первого галохинона (2,6-дихлор-1,4-бензохинон) в качестве ППД
питьевой воды (Qin et aL, 2010). Новый метод определения нитрозаминов был создан
на базе ЖХ/МС/МС. Он позволил выявить среди ППД два новых нитрозамина: нитро-
зопиперидин и нитрозодифениламин (Zhao etal., 2006). ЖХ/МС/МС крайне важна для
детектирования нитрозодифениламина, поскольку он термически нестабилен и не мо-
жет быть определен методом ГХ/МС. Дериватизующие агенты типа 2,4-динитрофенил-
гидразина (ДНФГ) применялись в сочетании с ЖХ/МС для детектирования высокопо-
лярных ППД (см. ниже).
Присутствие среди ППД высокомолекулярных соединений было установлено бла-
годаря ультрафильтрации через мембраны и определению величины общего органиче-
1 «Красота и мощь экологической масс-спектрометрии» — именно таким было первоначаль-
ное рабочее название всей книги. — Прим. ред.
12.2. Аналитические методы идентификации и количественного определения ППД
Без учета изотопных вкладов других элементов. — Прим. ред.
ского хлора (галогенов). Эти исследования показали, что >50% всего органического ма-
териала, содержащего хлор, имеет молекулярную массу более 1000 дальтон (Khiari et al.,
1997). Метод ЖХ/МС/МС с электрораспылением был использован для установления со-
ставаэтихсоединений. Для регистрации хлорсодержащих ППД с большими молекуляр-
ными массами природное органическое вещество вводилось в реакцию с 36С1-меченой
НОС1 (хлорная вода) (Zhang and Minear, 2002 and 2006). Эти эксперименты позволили
установить широкий разброс молекулярных масс со средней массой 2000 дальтон.
Спектры ионов-предшественников (глава 4) ионов хлора (m/z 35), брома (m/z 79 и 81)
и иода (m/z 127) были использованы для детектирования хлорированных, бромирован-
Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
ных и иодированных соединений соответственно (Zhang and Minear, 2005; Zhang et aL,
2008; Ding and Zhang, 2009). Например, регистрация ионов — предшественников иона
с m/z 127 методом УЭЖХ/ЭРИ-МС/МС позволила уточнить информацию о полярных
иодированных ППД в питьевой воде (Ding and Zhang, 2009). В частности, были идентифи-
цированы 17 соединений этой группы, причем некоторые из них ранее не были описаны.
Ионная хроматография/масс-спектрометрия с индуктивно-связанной
плазмой (ИХ/ИСП/МС) и ионная хроматография с электрораспылительной
масс-спектрометрией (ИХ/ЭРИ/МС).
Помимо органических ППД в питьевой воде присутствуют броматы, хлораты, йодаты
и хлориты. Они нелетучи и не могут быть определены ГХ/МС. Они также плохо разде-
ляются ЖХ, однако ионная хроматография справляется с этой задачей вполне успешно.
При нейтрал ьныхрН ГУКтакжеприсутствуютввидеанионовимогутбытьразделены ИХ.
Для этих анионов был создан целый ряд методик определения на базе ионной хромато-
графии/масс-спектрометрии с индуктивно-связанной плазмой (ИХ/ИСП/МС) и ион-
ной хроматографии с электрораспылительной масс-спектрометрией (ИХ/ЭРИ/МС).
Для определения следовых количеств этих аналитов следует проводить предваритель-
ную обработку пробы для удаления мешающих ионов (например сульфатов и хлоридов),
а также использовать предколонку перед вводом пробы в интерфейс масс-спектрометра.
Несколько методик были созданы Агентством по охране окружающей среды США
для регистрации бромата, канцерогенного аниона с предельно допустимой концентра-
цией в питьевой воде 10 мкг/л. Две из них базируются на масс-спектрометрии: метод 321.8
и метод 557. Первый из них использует ИХ/ИСП/МС с пределом обнаружения 0,3 мкг/л
(Creed et al., 1997); второй — ИХ/ЭРИ/МС/МС с пределом обнаружения 0,02 мкг/л (Zaf-
firo et al., 2009). Метод 557 может также использоваться для определения часто встреча-
ющихся хлорбромуксусных кислот (ГУК9) и дихлорпропановой кислоты с пределами
обнаружения в диапазоне от 0,015 до 0,20 мкг/л. Рель с коллегами опубликовали хоро-
ший обзор по использованию ИХ/ЭРИ/МС для определения ГУК и броматов (Roehletal.,
2002). Применение ионной хроматографии, включая ИХ/ЭРИ/МС и ИХ/ИСП/МС, для
определения ГУК изложено в обзоре Пауля и Баррона (Pauli and Barron, 2004).
Ши и Адамс недавно предложили быстрый ИХ/ИСП/МС-метод одновременного
количественного определения иодуксусных и бромуксусных кислот, йодатов и броматов
в питьевой воде, поверхностных и подземных водах, а также в плавательных бассейнах
(Shi and Adams, 2009). Его пределы обнаружения находятся на субмикрограммовых/литр
уровнях для иодированных ППД и единиц мкг/л для бромированных ППД. К сожале-
нию, метод не годится для хлорированных соединений из-за низкой чувствительности
ИСП/МС к хлору.
Масс-спектрометрия с мембранным вводом
Масс-спектрометрия с мембранным вводом (Membrane Introduction Mass Spectrometry,
MIMS) используется для прямого ввода аналитов в масс-спектрометр через полупрони-
цаемые мембраны (рис. 12.11). Это позволяет проводить измерения в режиме реального
времени с минимальной или даже совсем без пробоподготовки (глава 1). MIMS ранее
использовалась для измерения стабильности хлорциана в хлорированной и хлорами-
нированной воде (Na and Olson, 2004), для количественного определения хлорциана
и бромциана в питьевой воде (YangandShang, 2005), для измерения уровней хлораминов
12.2. Аналитические методы идентификации и количественного
Рис. 12.11. Интерфейс масс-спектрометра с мембранным вводом (схема и фотография
Е. Р. Блатчли III, Университет Пурду)
Выход в Ввод
масс-спектрометр инертного газа
и хлорбензолов в образцах воды {Riteret aL, 2001), для установления механизма и кине-
тики образования хлороформа в питьевой воде {Rios et al., 2000). Позже она использова-
лась для количественного определения летучих ППД в крытых плавательных бассейнах
{Weaver et al., 2009; Kristensen et al., 2010). В первом исследовании этой серии 11 летучих
ППД были идентифицированы в образцах из хлорированных бассейнов: монохлора-
мин, дихлорамин, трихлорамин, хлороформ, бромоформ, бромдихлорметан, дибром-
хлорметан, хлорциан, бромциан, дихлорацетонитрил и дихлорметиламин (Weaver et al.,
2009). Во втором исследовании в реальном времени осуществлялся мониторинг дина-
мики присутствия ТГМ в обогреваемых общественных плавательных бассейнах {Kris-
tensen et al., 2010). В этом случае прибор работал в автоматическом режиме более года
с короткими перерывами для замены катода каждые 6—8 недель. Мониторинг выявил
суточные циклы концентраций хлороформа и бромдихлорметана. Их уровни возрас-
тали после закрытия бассейна и уменьшались в рабочие часы.
Спектрометрия приращения ионной подвижности
Спектрометрия приращения ионной подвижности СПИМ (High-field asymmetric wave-
form ion mobility spectrometry (FAIMS)-MS) предоставляет еще один вариант разделения
аналитов. Он основан на различиях ионной подвижности в электростатическом поле
высокой и низкой напряженности {Guevremont et al., 2001). При использовании FAIMS
в тандеме с электрораспылительной масс-спектрометрией удается существенно сокра-
тить химический шум и улучшить процесс регистрации ППД. Пределы обнаружения
на уровне нескольких нанограммов в литре часто достижимы без предварительного
концентрирования, экстракции или дериватизации. ЭРИ/СПИМ/МС может приме-
няться для измерения уровней ГУК в питьевой воде и моче человека {Ells et aL, 2000;
Gabryelski et al., 2003); броматов, хлоратов и йодатов в питьевой воде {Barnett et al., 1999);
а также нитрозаминов в питьевой воде {Planas etal., 2008; Zhao etal., 2009) и в сбросах за-
водов по обработке сточных вод {Zhao etal., 2009).
12.2.3. Методы дериватизации
Для детектирования представителей некоторых классов ППД часто бывает необходима
процедура дериватизации. Некоторые реактивы, используемые для этой цели, перечис-
лены в табл. 12.4.
Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
Таблица 12.4. Реагенты для дериватизации ППД
Реагент Целевая функциональ- ная группа Метод анализа
Диазометан Карбоновые кислоты ГХ/МС
BF3 в метаноле Карбоновые кислоты ГХ/МС
1-(Пентафтрофенил)диазоэтан Карбоновые кислоты ГХ/МС
Пентафторбензилгидроксиламин (ПФБГА) Карбонильные группы (альдегиды, кетоны) ГХ/МС
Бис(триметилсилил)трифторацетамид (БСТФА) Спирты, фенолы ГХ/МС
N-Метилтриметилсилилтрифторацетамид (МСТФА) Спирты, фенолы ГХ/МС
5-Хлор-2,2,3,3,4,4,5,5-октафторпентил-хлорформиат (ХОФПХФ) Спирты, амины, карбоновые кислоты ГХ/ХИОИ/МС
2,4-Динитрофенилгидразин (ДНФГ) Карбонильные группы (альдегиды, кетоны) ЖХ/МС
4-Диметиламино-6-(4-метокси-1-нафтил)-1,3,5- триазин-2-гидразин (ДМНТГ) Карбонильные группы (альдегиды, кетоны) ЖХ/МС
О-Карбоксиметилгидроксиламин (КМГА) Карбонильные группы (альдегиды, кетоны) ЖХ/МС
ГХ/МС
Метилирование (часто диазометаном) позволяет детектировать карбоновые кислоты,
включая галогенсодержащие, методом ГХ/МС. Диазометан обычно генерируется с ис-
пользованием диазальда (Sigma-Aldrich) или другого реагента (рис. 12.12). Он реагиру-
ет с карбоновыми кислотами с образованием метиловых эфиров, которые более лету-
чи. В результате улучшается разделение и ГХ/МС-регистрация {Pressman et al., 2010).
1-Пентафторфенилдиазоэтан также показал себя эффективным реагентом деривати-
зации галогенуксусных кислот в воде {Berg et al., 2000). Для экстрагирования из воды
высокополярных карбонильных соединений с последующим ГХ/МС-определением ис-
пользуется о-(2,3,4,5,6-пентафторбензил)-гидроксиламин (ПФБГА, PFBHA) {Glaze and
Weinberg, 1993). После дериватизации альдегиды и кетоны хорошо экстрагируются гек-
саном. Следует отметить, что пики ПФБГА-производных легко детектируются в слож-
ных хроматограммах, поскольку при их фрагментации всегда образуются интенсивные
пики ионов с m/z 181, обусловленные частью молекулы, связанной с дериватизующим
агентом. Благодаря ПФБГА в озонированной питьевой воде удалось идентифицировать
широкий круг ранее неописанных альдегидов и кетонов {Richardson etal., 1999b; Glaze and
Weinberg, 1993). Успешное количественное определение нескольких альдегидов и кето-
нов удалось провести в рамках Национального исследования распространенности П ПД
{Krasner et al., 2006; Weinberg et al., 2002).
Силилирующие агенты типа бис(триметилсилил)трифторацетамида (БСТФА,
BSTFA) и А-метилтриметилсилилтрифторацетамида (МСТФА, MSTFA), иногда
используются для дериватизации фенолов и спиртов с последующим их ГХ/МС-
детектированием. Так, дериватизация BSTFA недавно позволила идентифицировать
методом ГХ/МС несколько ППД, образующихся при хлорировании воды, содержащей
парабены (эфиры п-гидроксибензойной кислоты). Последние стали распространен-
ными загрязняющими соединениями, будучи консервантами во многих косметиче-
12.2. Аналитические методы идентификации и количественного определения ППД
Рис. 12.12. Получение диазометана в реакции диазальда с КОН и карбитолом во внутрен-
ней трубке аппарата. Далее газообразный диазометан проходит через неболь-
шое отверстие во внутренней трубке во внешнюю, в которой находится метил-
третбутиловый эфир (МТБЭ). Раствор диазометана в МБТЭ вводится далее
в экстракт питьевой воды для метилирования карбоновых кислот. В реальных
условиях внешняя трубка погружена в лед
ских продуктах (кремы от солнца, гели для ванн, шампуни, зубные пасты) (Terasaki and
Makino, 2009).
В последние годы были разработаны реагенты на основе хлорформиатов. Они хоро-
шо зарекомендовали себя в качестве дериватизующих агентов для определения высоко-
полярных ППД в питьевой воде (Vincenti et al., 2005, 2010). Они позволяют проводить
прямую дериватизацию в воде ППД с несколькими гидроксильными, карбоксильными
и аминогруппами с образованием полизамещенных неполярных производных, легко
экстрагируемых из воды и доступных количественному определению методом ГХ/МС
с химической ионизацией отрицательных ионов. Вся процедура от исходного образца
до готовой к вводу в прибор пробы в гексане занимает менее 10 минут. Один из таких
352 Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
агентов, 5-хлор-2,2,3,3,4,4,5,5-октафторпентилхлорформиат, был успешно использо-
ван для установления структуры нескольких ранее неизвестных высокополярных ППД
в озонированной питьевой воде и в озонированных растворах фульво- и гуминовых
кислот (Vincenti et aL, 2010). Однако, в отличие от ранее упомянутых дериватизующих
агентов, такие хлорформиаты были лишь недавно предложены для решения конкрет-
ных задач и, будучи коммерчески недоступными, должны быть синтезированы непо-
средственно перед применением.
Спектры основных ППД после дериватизации (метиловые эфиры ГУК, ПФБГА-
производные альдегидов и кетонов, некоторые силильные производные) включены
в доступные библиотеки масс-спектров. К сожалению, часть спектров производных
пока требует ручной расшифровки.
ЖХ/МС
2,4-Динитрофенилгидразин (ДНФГ, DNPH) (Richardson et al., 2000; Zwiener et al.,
2002), О-(карбоксиметилгидроксиламин) (КМГА, CMHA) (Zwiener et al., 2003),
и 4-д иметиламино-6-(4-метокси-1 -нафтил)- 1,3,5-триазин-2-гидразин (ДМНТГ,
DMNTH) (Richardson and Karst, 2001) использовались для идентификации высокополяр-
ных ППД в питьевой воде методом ЖХ/МС(МС). Аналогично ПФБГА в случае ГХ/МС
эти реагенты позволяют перевести высокополярные альдегиды и кетоны из воды в под-
ходящий растворитель. Полученные производные способствуют ионизации аналитов
электрораспылением или ХИАД (глава 1), а также приводят к образованию характе-
ристических фрагментных ионов, имеющих диагностическое значение для МС/МС.
Дериватизация дифенилгидразином впервые была использована для детектирования
карбонильных соединений в воздухе. Позднее была разработана соответствующая
ЖХ/МС-методика для идентификации полярных карбонильных ППД в питьевой воде
(Richardson etal., 2000). Дериватизация ДМНТГ была специально разработана для опти-
мизации детектирования в режимах ЖХ/МС(ХИАД), ЖХ/УФ и ЖХ с флуоресцентным
детектором без экстракции и предварительного концентрирования пробы. Этот реагент
должен синтезироваться в лаборатории, поскольку его пока нет в продаже. Для галоге-
нированных карбонильных соединений предпочтительным реагентом является КМГА.
В отличие от ДНФГ и ДМ НТГ он не инициирует дегалогенирования в качестве побоч-
ной реакции (Zwieneretal., 2002).
12.3. Что нас ждет в будущем?
Скорее всего, масс-спектрометрия останется ключевым аналитическим методом для
идентификации и количественного определения ППД в питьевой воде. Новые приборы
появляются практически ежегодно, причем их чувствительность возрастает. Рутиной
становится детектирование фемтограммовых (10-15) количеств аналитов. Улучшение
пределов обнаружения, вероятно, будет продолжаться, а вот процедуры экстракции
и концентрирования, скорее всего, окажутся ненужными. Тем не менее помимо масс-
спектрометрии существуют альтернативные подходы, разработанные для количе-
ственного определения целевых ППД. Они могут оказаться в некоторых случаях более
предпочтительными, чем масс-спектрометрия. Так, группа Эммерта разрабатывает
недорогие приборы для измерения уровней ТГМ и ГУК практически в режиме реаль-
Литература
ного времени (Emmert et al., 2009). Они, в конечном счете, могут использоваться для ре-
гистрации нормируемых ППД непосредственно в распределительных системах питье-
вого водоснабжения. Вместо того, чтобы отбирать пробы, доставлять их в лабораторию,
проводить экстракцию и анализировать масс-спектрометрически, эти приборы могут
проводить измерения в реальном времени, позволяя компьютеру контролировать дозы
дезинфицирующих агентов для оптимизации процесса обеззараживания и минимиза-
ции образования ППД. Возможно также, что новые методы масс-спектрометрии прак-
тически не требующие пробоподготовки типа десорбционной электрораспылительной
ионизации (ДЭСИ, DESI) или прямого анализа в реальном времени (ПАРВ, DART)
(Harris etal., 2008), могут быть применены для этих же целей (глава?). Мы живем в очень
интересное время, когда замечательные масс-спектрометрические новинки появляют-
ся ежегодно.
Благодарность
Я хотела бы посвятить эту главу замечательному человеку, много лет назад научившему
меня масс-спектрометрии, — Мэри Джейн Маттина. Она работала вуниверситете Эмори,
в котором я была студенткой, а позже на экспериментальной сельскохозяйственной
станции в Коннектикуте, где она продолжала свои исследования с использованием
масс-спектрометрии. Я навсегда обязана ей за то, что она продемонстрировала мне
«красоту и мощь» масс-спектрометрии. Я хотела бы также поблагодарить Бена Блаунта
и Эрнста (Чипа) Блачли за великолепные фотографии, которые я использовала при под-
готовке этой главы, а также Юандалин Коффен, Хану Финк, Жанессу Хартман и Мата
Бладгуда за их тяжелую работу в моей лаборатории и разрешение использовать их рабо-
чие фотографии.
Этот материал был рецензирован в соответствии с существующими требованиями
Агентства по охране окружающей среды США и рекомендован к публикации. Исполь-
зование торговых названий или товаров не означает их поддержку или рекомендацию
к использованию со стороны Агентства по охране окружающей среды США.
Литература
[1] . Ashley D. L., Blount В. С., Singer Р. С., Depaz Е., Wilkes С., Gordon S., Lyu С., and Mas-
ters J. (2005). «Changes in blood trihalomethane concentrations resulting from differences
in water quality and water use activities». Archives of Environmental and Occupational Health.
60, 1:7-15.
[2] . Attene-Ramos M.S., Wagner E. D., and Plewa M.J. (2010). «Comparative human cell toxi-
cogenomic analysis of monohaloacetic acid drinking water disinfection byproducts». Envi-
ronmental Science & Technology 44, 19:7206—7212.
[3] . Barnett D.A., Guevremont R., and Purves R.W. (1999). «Determination of parts-per-tril-
lion levels of chlorate, bromate, and iodate by electrospray ionization/high-field asymmet-
ric waveform ion mobility spectrometry/mass spectrometry». Applied Spectroscopy. 53, 11:
1367-1374.
354 Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
[4] . BellarT.A., Lichtenberg J.J., and Kroner R. С. (1974). «The occurrence of organohalides in
chlorinated drinking waters». Journal of the American Water Works Association. 66: 703—706.
[5] . Berg M., Muller S. R., Muhlemann J., Wiedmer A., and Schwarzenbach R. P. (2000). «Con-
centrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural
waters in Switzerland». Environmental Science & Technology. 34, 13: 2675—2683.
[6] . Cantor К. P., Villanueva С. M., Silverman D.T., Figueroa J. D., Real F.X., Garcia-Clo-
sas M., Malats N., Chanock S., Yeager M., Tardon A., Garcia-Closas R., Serra C., Car-
rato A., Castano-Vinyals G., Samanic C., Rothman N., and Kogevinas M. (2010). «Poly-
morphisms in GSTT1, GSTZ1, and CYP2E1, disinfection by-products, and risk of bladder
cancer in Spain». Environmental Health Perspectives. 118, 11: 1545—1550.
[7] . Cardador M. J., and Gallego M. (2011). «Haloacetic acids in swimming pools: swimmer and
worker exposure». Environmental Science and Technology. 45: 5783—5790.
[8] . Creed J.T., Brockhoff C. A., and Martin T. D. (1997). (U.S. EPA Method 321.8. Determina-
tion of Bromate in Drinking Waters by Ion Chromatography Inductively Coupled Plasma Mass
Spectrometry) see www.epa.gov/microbes/m_321_8.pdf
[9] . Ding G.Y., and Zhang X. R. (2009). «А Picture of Polar Iodinated Disinfection Byprod-
ucts in Drinking Water by (UPLC/)ESI-tqMS». Environmental Science & Technology. 43:
9287-9293.
[10] . Ells B., Barnett D.A., Purves R. W., and Guevremont R. (2000). «Detection of nine chlo-
rinated and brominated haloacetic acids at part-per-trillion levels using ESI-FAIMS-MS».
Analytical Chemistry. 72, 19: 4555—4559.
[11] . Erdinger L., Kuhn, K.P, Kirsch, F, Feldhues R., Frobel T., Nohynek B., and Gabrio T.
(2004). «Pathways of trihalomethane uptake in swimming pools». International Journal of
Hygiene and Environmental Health. 207, 6: 571—575.
[12] . Emmert G. L., Geme G., Brown M. A., and Simone P. S. (2009). «А single automated in-
strument for monitoring total trihalomethane and total haloacetic acid concentrations in
near real-time». Analytica Chimica Acta. 656, 1—2: 1—7.
[13] . Gabryelski W., Wu F.W., Froese K. L. (2003). «Comparison of high-field asymmetric wave-
form ion mobility spectrometry with GC methods in analysis of haloacetic acids in drinking
water». Analytical Chemistry. 2003, 75: 2478—2486.
[14] . Glaze W., and Weinberg H. (1993). Identification and Occurrence of Ozonation By-Products
in Drinking Water. Awwa Research Foundation, Denver, CO.
[15] . Guevremont R., Ding L., Ells B., Barnett D.A., and Purves R.W. (2001). «Atmospheric
pressure ion trapping in a tandem FAIMS-FAIMS coupled to a TOFMS: Studies with elec-
trospray generated gramicidin S ions». Journal of the American Society for Mass Spectrom-
etry. 12: 1320-1330.
[16] . Haddad S., Tardif G. C., and Tardif R. (2006). «Development of physiologically based toxi-
cokinetic models for improving the human indoor exposure assessment to water contami-
nants: trichloroethylene and trihalomethanes». Journal of Toxicology and Environmental
Health, Part A. 69: 2095-2136.
[17] . Harris G. A., Nyadong L., and Fernandez F. (2008). «Recent developments in ambient ion-
ization techniques for analytical mass spectrometry». Analyst. 133: 1297—1301.
[18] . Khiari D., Krasner S. W., Hwang C. J., Chinn R., and Barrett S. E. (1997). «Effects of chlo-
rination and chloramination on the molecular weight distribution of natural organic mat-
ter and the production of high-molecular-weight disinfection by-products». Proceedings of
Литература
the 1996American Water Works Association Water Quality Technology Conference, American
Water Works Association: Denver, CO.
[19] . Krasner S.W., Weinberg H.S., Richardson S.D., Pastor S.J., Chinn R., Sclimenti M.J.,
Onstad G. D., and Thruston Jr. A. D. (2006). «The occurrence of a new generation of disin-
fection by-products». Environmental Science & Technology. 40: 7175—7185.
[20] . Kristensen G. H., Klausen M. M., Hansen V. A., and Lauritsen F. R. (2010). «On-line mon-
itoring of the dynamics of trihalomethane concentrations in a warm public swimming pool
using an unsupervised membrane inlet mass spectrometry system with off-site real-time
surveillance». Rapid Communications in Mass Spectrometry. 24: 30—34.
[21] . Kurokawa Y, Aoki S., Matsushima Y, Takamura N., Imazawa T., and Hayashi Y. (1986).
«Dose-response studies on the carcinogenicity of potassium bromate in F344 rats after
long-term oral administration». Journal of the National Cancer Institute. 77: 977—982.
[22] . LaKind J. S., Richardson S. D., and Blount В. C. (2010). «The good, the bad, and the vola-
tile: can we have both healthy pools and healthy people?» Environmental Science & Technol-
ogy. 44,9: 3205-3210.
[23] . Leavens T.L., Blount B.C., DeMarini D. M., Madden M. C., Valentine J. L., Case M.W.,
Silva L. K., Warren S. H., Hanley N. M., and Pegram P. A. (2007). «Disposition of bromodi-
chloromethane in humans following oral and dermal exposure». Toxicological Sciences. 99:
432-445.
[24] . Liu W., Cheung L. M., Yang X., and Shang C. (2006). «ТНМ, HAA, and CNC1 formation
from UV irradiation and chlor(am)ination of selected organic waters. Water Research 40,
10: 2033-2043.
[25] . Mitch W.A., Sharp J.O., Trussell R. R., Valentine R. L., Alvarez-Cohen L., and Sed-
lak D. L. (2003). «N-nitrosodimethylamine (NDMA) as a drinking water contaminant:
A review». Environmental Engineering Science. 20: 389—404.
[26] . Na C.Z., and Olson T. M. (2004). «Stability of cyanogen chloride in the presence of free
chlorine and monochloramine». Environmental Science & Technology. 38, 22: 6037—6043.
[27] . National Cancer Institute. (1976). Report on the carcinogenesis bioassay of chloroform
(CAS no. 67-66-3), MD: National Cancer Institute. TR-000 NTIS Rpt No PB264018
Bethesda, MD.
[28] . Nieuwenhuijsen M. J., Toledano M. B., Eaton N. E., Fawell J., and Elliott P. (2000). «Chlo-
rination disinfection byproducts in water and their association with adverse reproductive
outcomes: A review». Occupational and Environmental Medicine. 57: 73—85.
[29] . Niri V. H., Bragg L., and Pawliszyn J. (2008). «Fast analysis of volatile organic compounds
and disinfection by-products in drinking water using solid-phase microextraction-gas
chromatography/time-of-flight mass spectrometry». Journal of Chromatography A. 1201:
222-227.
[30] . Ouyrang G., and Pawliszyn J. (2006). «SPME in environmental analysis». Analytical and
Bioanalytical Chemistry. 386, 4: 1059—1073.
[31] . Pauli B., and Barron L. (2004). «Using ion chromatography to monitor haloacetic acids in
drinking water: a review of current technologies». Journal of Chromatography A. 1046, 1—2:
1-9.
[32] . Planas C., Palacios O., Ventura F., Rivera J., Caixach J. (2008). «Analysis of nitrosamines
in water by automated SPE and isotope dilution GC/HRMS — Occurrence in the different
356 Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
steps of a drinking water treatment plant, and in chlorinated samples from a reservoir and a
sewage treatment plant effluent». Taianta. 76, 4: 906—913.
[33] . Plewa M.J., Muellner M.G., Richardson S.D., Fasano F., Buettner K.M., Woo, Y.-T.,
McKague A. B., and Wagner E. D. (2008). «Occurrence, synthesis, and mammalian cell
cytotoxicity and genotoxicity of haloacetamides: an emerging class of nitrogenous drinking
water disinfection byproducts». Environmental Science & Technology. 42: 955—961.
[34] . Plewa M.J., Wagner E.D., Richardson S.D., Thruston A. D. Jr., Woo Y.-T., and McK-
ague A. B. (2004). «Chemical and biological characterization of newly discovered iodo-
acid drinking water disinfection byproducts». Environmental Science & Technology. 38, 18:
4713-4722.
[35] . Pressman J.G., Richardson S.D., Speth T. F., Miltner R.J., Narotsky M.G., Hunt-
er E.S. Ill, Rice G.E., Teuschler L. K., McDonald A., Parvez S., Krasner S.W., Wein-
berg H.S., McKague A.B., Parrett C.J., Bodin N., Chinn R., Lee C.-F.T., and Sim-
mons J. E. (2010). «Concentration, chlorination, and chemical analysis of drinking water
for disinfection byproduct mixtures health effects research: U.S. EPAs Four Lab Study».
Environmental Science & Technology. 44, 19: 7184—7192.
[36] . Qin F., Zhao Y.-Y, Zhao Y, Boyd J. M., Zhou W., and LiX.-F. (2010). «Atoxic disinfection
by-product, 2,6-dichloro-l,4-benzoquinone, identified in drinking water». Angewandte
Chemie- International Edition. 49, 790—792.
[37] . Richardson S. D. (1998). «Drinking water disinfection by-products», In: The Encyclopedia
of Environmental Analysis and Remediation, Vol. 3, pp. 1398—1421. John Wiley: New York.
[38] . Richardson S.D. (2002). «The role of GC-MS and LC-MS in the discovery of drinking
water disinfection by-products». Journal of Environmental Monitoring. 4: 1—9.
[39] . Richardson S. D. (2005). «New disinfection by-product issues: emerging DBPs and alterna-
tive routes of exposure». Global Nest. 7, 1: 43—60.
[40] . Richardson S. D. (2011). «Disinfection by-products: Formation and Occurrence of Drink-
ing Water Disinfection By-Products». In: Nriagu J.O. (Ed.), The Encyclopedia of Environ-
mental Health. Elsevier, Burlington, Vol. 2, pp. 110—136.
[41] . Richardson S. D., and Karst U. (2001). «А new tailor-made derivatizing agent for identi-
fying polar carbonyl DBPs in drinking water». Preprints of Extended Abstracts, American
Chemical Soceity National Meeting, Division of Environmental Chemistry, San Diego, CA.
[42] . Richardson S. D., Thruston A. D., Jr., Collette T. W., Patterson K. S., and Lykins B. W., Jr.
(1994). «Multispectral identification of chlorine dioxide disinfection byproducts in drink-
ing water». Environmental Science & Technology. 28: 592—599.
[43] . Richardson S. D., Caughran T. V, Poiger T., Guo Y, and Crumley F. G. (2000). «Applica-
tion of DNPH derivatization with LC/MS to the identification of polar carbonyl disinfec-
tion by-products in drinking water». Ozone Science & Engineering. 22, 6: 653—675.
[44] . Richardson S.D., Plewa M.J., Wagner E.D., Schoeny R., and DeMarini D. M. (2007).
«Occurrence, genotoxicity, and carcinogenicity of emerging disinfection by-products in
drinking water: a review and roadmap for research». Mutation Research. 636, 1—3: 178—242.
[45] . Richardson S.D., Fasano F., Ellington J. J., Crumley F.G., Buettner K.M., Evans J. J.,
Blount B.C., Silva L.K., Waite T.J., Luther G.W., McKague A. B., Miltner R.J., Wag-
ner E. D., and Plewa M. J. (2008a). «Occurrence and mammalian cell toxicity of iodinated
disinfection byproducts in drinking water». Environmental Science & Technology. 42, 22:
8330-8338.
Литература
[46] . Richardson S.D., Thruston A.D., Jr., Krasner S.W., Weinberg H.S., Miltner R.J., Nar-
otsky M.G., and Simmons J.E. (2008b). «Integrated disinfection byproducts mixtures
research: comprehensive characterization of water concentrates prepared from postchlo-
rinated and preozonated/postchlorinated drinking water». Journal of Toxicology and Envi-
ronmental Health, PartA.1V. 1165—1186.
[47] . Richardson S. D., Thruston A. D. Jr., CaughranT.V., Chen P. H., Collette T.W., FloydT. L.,
Schenck К. M., Lykins B.W., Jr., Sun, G.-R., and Majetich G. (1999a). «Identification of
new drinking water disinfection byproducts formed in the presence of bromide». Environ-
mental Science & Technology. 33: 3378—3383.
[48] . Richardson S. D., Thruston A. D. Jr., Caughran T.V., Chen P. H., Collette T.W., Floyd T. L.,
Schenck К. M., Lykins B.W., Jr., Sun, G.-R., and Majetich G. (1999b). «Identification of
new ozone disinfection by-products in drinking water». Environmental Science & Technol-
ogy. 33: 3368-3377.
[49] . Richardson S.D., DeMarini D. M., Kogevinas M., Fernandez P., Marco E., Lourencet-
ti C., Ballester C., Heederik D., Meliefste K., McKague A. B., Marcos R., Font-Ribera L.,
Grimalt J.O., and Villaneuva С. M. (2010). «What's in the pool? A comprehensive iden-
tification of disinfection by-products and assessment of mutagenicity of chlorinated and
brominated swimming pool water». Environmental Health Perspectives. 118, 11: 1523—1530.
[50] . Rios R. V. R. A., da Rocha L. L., Vieira T. G., Lago R. M., and Augusti R. (2000). «On-line
monitoring by membrane introduction mass spectrometry of chlorination of organics in
water, mechanistic and kinetic aspects of chloroform formation. Journal of Mass Spectrom-
etry. 35, 5: 618—624. Journal of Mass Spectrometry. 35: 618—624. Riter L.S., Charles L.,
Turowski M., and Cooks R.G. (2001). «External interface for trap-and-release membrane
introduction mass spectrometry applied to the detection of inorganic chloramines and
chlorobenzenes in water». Rapid Communications in Mass Spectrometry. 15, 23: 2290—2295.
[51] . Roehl R., Slingsby R., Avdalovic N., and Jackson P. E. (2002). «Applications of ion chro-
matography with electrospray mass spectrometric detection to the determination of envi-
ronmental contaminants in water». Journal of Chromatography A. 956: 245—254.
[52] . Rook J. J. (1974). «Formation of haloforms during chlorination of natural waters». Water
Treatment and Examination. 23: 234—243.
[53] . Savitz D. A., Singer P.C., Hartmann К. E., Herring A. H., Weinberg H. S., Makarushka C.,
Hoffman C., Chan R., and Maclehose R. (2005). Drinking Water Disinfection By-Products
and Pregnancy Outcome, Awwa Research Foundation, Denver, CO.
[54] . Schmidt С. K., and Brauch, H.-J. (2008). «/V,/V-dimethosulfamide as precursor for /V-ni-
trosodimethylamine (N DM A) formation upon ozonation and its fate during drinking water
treatment». Environmental Science & Technology. 42, 17: 6340—6346.
[55] . Schulten, H.-R., and Schnitzer M. (1993). «А State of the Art Structural Concept for Hu-
mic Substances». Naturwissenschaften. 80: 29—30.
[56] . Shi H. L., and Adams C. (2009). «Rapid IC-ICP/MS method for simultaneous analysis of
iodoacetic acids, bromoacetic acids, bromate, and other related halogenated compounds in
water». Talanta.79\ 523—527.
[57] . Silva L. K., Bonin M. A., McKague B., and Blount B.C. (2006). «Quantification of dichlo-
roiodomethane and bromochloroiodomethane in human blood by solid-phase microex-
traction coupled with gas chromatography-high resolution mass spectrometry». Journal of
Analytical Toxicology. 30, 9: 670—678.
358 Глава 12. Идентификация и количественное определение токсикологически
значимых побочных продуктов дезинфекции воды методами масс-спектрометрии
[58] . Speth Т. F., Miltner R.J., Richardson S.D., and Simmons J. E. (2008). «Integrated dis-
infection by-products mixtures research: Concentration by reverse osmosis membrane
techniques of disinfection by-products from water disinfected by chlorination and ozo-
nation/postchlorination». Journal of Toxicology and Environmental Health, Part A. 71, 17:
1149-1164.
[59] . Terasaki M., and Makino M. (2009). «Disinfection by-products of para-hydroxybenzoate
esters (parabens): synthesis and mass spectrometric study». Journal of Health Sciences. 55:
631-635.
[60] . U.S. Environmental Protection Agency. (1979). «National interim primary drinking water
regulations: Control of trihalomethanes in drinking water: Final rules». Federal Register.
44: 68624-68705.
[61] . U.S. Environmental Protection Agency. (2010). Unregulated Contamiinants Monitoring
Rule-2 (UCMR2) Occurrence Data (2008—2010); http://water.epa.gov/lawsregs/rules-
regs/sdwa/ucmr/data.cfm#ucmr2010.
[62] . Villanueva С. M., Cantor К. P., Grimalt J. O., Malats N., Silverman D., Tardon A., Garcia-
Closas R., Serra C., Carrato A., Castano-Vinyals G., Marcos R., Rothman N., Real F.X.,
Dosemeci M., and Kogevinas M. (2007). «Bladder cancer and exposure to water disinfec-
tion by-products through ingestion, bathing, showering and swimming in pools». American
Journal of Epidemiology. 165: 148—156.
[63] . Villanueva С. M., Cantor К. P., Cordier S., Jaakkola J. J. K., King W. D., Lynch C. F., Por-
ru S., and Kogevinas M. (2004). «Disinfection byproducts and bladder cancer—a pooled
analysis». Epidemiology. 15, 3: 357—367.
[64] . Vincenti M., Fasano F., Valsania M.C., Guarda P., and Richardson S.D. (2010). «Appli-
cation of the novel 5-chloro-2,2,3,3,4,4,5,5-octafluoro-l-pentylchloroformate derivatizing
agent for the direct determination of highly polar water disinfection byproducts». Analytical
and Bioanalytical Chemistry. 397 (1): 43—54.
[65] . Vincenti M., Biazzi, Ghiglione N., Valsania M. C., and Richardson S. D. (2005). «Compar-
ison of highly-fluorinated chloroformates as direct aqueous sample derivatizing agents for
hydrophilic analytes and drinking water disinfection by-products». Journal of the American
Society for Mass Spectrometry. 16, 6: 803—813.
[66] . Waller K., Swan S. H., DeLorenze G., and Hopkins B. (1998). «Trihalomethanes in drink-
ing water and spontaneous abortion». Epidemiology 9: 134—140.
[67] . Weaver W. A., Li J., Wen Y. L., Johnston J., Blatchley M.R. Ill, and Blatchley E. R. (2009).
«Volatile disinfection by-product analysis from chlorinated indoor swimming pools». Water
Research. 43: 3308—3318.
[68] . Weinberg H. S., Krasner S. W., Richardson S. D., and Thruston A. D., Jr. (2002). The oc-
currence of disinfection by-products (DBPs) of health concern in drinking water: results
of a nationwide DBP occurrence study. ЕРА/600/R02/068. www.epa.gov/athens/publica-
tions/reports/EPA_600_R02_068.pdf.
[69] . Xu X., and Weisel С. P. (2005a). «Dermal uptake of chloroform and haloketones during
bathing». Journal of Exposure Analysis and Environmental Epidemiology. 15: 289—296.
[70] . Xu X., and Weisel С. P. (2005b). «Human respiratory uptake of chloroform and haloketones
during showering». Journal of Exposure Analysis and Environmental Epidemiology. 15: 6—16.
[71] . YangX., and Shang C. (2005). «Quantification of aqueous cyanogen chloride and cyanogen
bromide in environmental samples by MIMS». Water Research. 39, 9: 1709—1718.
Литература
[72] . Zaffiro A.D., Zimmerman M., Pepich B.V., Slingsby R.W., Jack R. F., Pohl C.A., and
Munch D.J. (2009). U.S. EPA Method 557. Determination of haloacetic acids, bromate,
and dalapon in drinking water by ion chromatography electrospray ionization tandem mass
spectrometry (IC-ESI-MS/MS); http://water.epa.gov/scitech/drinkingwater/labcert/up-
load/met557.pdf.
[73] . Zhang X., and Minear R.A. (2006). «Removal of low-molecular weight DBPs and inor-
ganic ions for characterization of high-molecular weight DBPs in drinking water». Water
Research. 40, 2: 221—230.
[74] . Zhang X., Minear R.A., and Barrett S.E. (2005). «Characterization of high molecular
weight disinfection byproducts from chlorination of humic substances with/without co-
agulation pretreatment using UF-SEC-ESI-MS/MS». Environmental Science & Technol-
ogy. 39, 4: 963-972.
[75] . Zhang X., and Minear R.A. (2002). «Characterization of high molecular weight disinfec-
tion byproducts resulting from chlorination of aquatic humic substances». Environmental
Science & Technology. 36, 19: 4033—4038.
[76] . Zhang X., Talley J.W., Boggess B., Ding G., and Birdsell D. (2008). «Fast selective de-
tection of polar brominated disinfection byproducts in drinking water using precursor ion
scans». Environmental Science & Technology. 42, 17: 6598—6603.
[77] . Zhao Y, Qin F., Boyd J. M. Anichina J., and Li X.-F. (2010). «Characterization and deter-
mination of chloro- and bromo-benzoquinones as new chlorination disinfection byprod-
ucts in drinking water». Anal. Chem. 82, 4599—4605.
[78] . Zhao Y. Y, Liu X., Boyd J. M., Qin F., Li J., and Li X.-F. (2009). «Identification of A-nitro-
samines in treated drinking water using nanoelectrospray ionization high-field asymmetric
waveform ion mobility spectrometry with quadrupole time-of-flight mass spectrometry».
Journal of Chromatographic Sciences. 47: 92—96.
[79] . Zhao YY, Boyd J., Hrudey S.E., and Li X.-F. (2006). «Characterization of new nitrosa-
mines in drinking water using liquid chromatography tandem mass spectrometry». Envi-
ronmental Science & Technology. 40, 24: 7636—7641.
[80] . Zwiener C., Glauner T., and Frimmel F. H. (2002). «Method optimization for the determi-
nation of carbonyl compounds in disinfected water by DNPH derivatization and LC-ESI-
MS-MS». Analytical and Bioanalytical Chemistry. 372, 5—6: 615—621.
[81] . Zwiener C., Glauner T., and Frimmel F. H. (2003). «Liquid chromatography/electrospray
ionization tandem mass spectrometry and derivatization for the identification of polar car-
bonyl disinfection by-products». In: Ferrer L, Thurman E. M. (Eds.), Liquid Chromatogra-
phy/Mass Spectrometry, MS/MS and Time-of-Flight MS, ACS Symposium Series, Vol. 850,
American Chemical Society: Washington D. C., pp 356—375.
[82] . Zwiener C., and Richardson S. D. (2005). «Drinking water disinfection by-product analysis
by LC/MS and LC/MS/MS». Trends in Analytical Chemistry, 24, 7: 613—621. 23
[83] . Zwiener C., Richardson S.D., DeMarini D. M., Grummt T., Glauner T., and Frim-
mel F. H. (2007). «Drowning in disinfection byproducts? Assessing swimming pool water».
Environmental Science & Technology. 41: 363—372.
ГЛАВА 13
НОВЫЕ ТИПЫ
ПРИОРИТЕТНЫХ
ЗАГРЯЗНЯЮЩИХ
ВЕЩЕСТВ
В ОКРУЖАЮЩЕЙ
СРЕДЕ
Тамми Л. Джонс-Лепп
13.1. Введение
Эта глава посвящена использованию масс-спектрометрии для анализа новых типов
приоритетных загрязняющих веществ в окружающей среде. Будут рассмотрены некото-
рые металлоорганические соединения, фармацевтические препараты, наноматериалы
и поверхностно-активные вещества (ПАВ). Табл. 13.1 демонстрирует разнообразие но-
вых приоритетных экотоксикантов, значимых с экологической точки зрения. Однако
не стоит считать, что это исчерпывающий список. Скорее, это полезный для данной
главы перечень.
Что означает термин «новые приоритетные загрязняющие вещества (НПЗВ)»? На-
пример, в 3000 году до н.э. свинец всегда сопутствовал серебру при его добыче в сере-
бряных копях в Анатолии, а далее его выбрасывали за ненадобностью. Для населения,
жившего вблизи этих копей, свинец можно было считать НПЗВ, поскольку и вода и зе-
мельные угодья были заражены свинцом. Через 5 тысяч лет, в начале индустриальной
эпохи в США (1918—1919) общее производство синтетических органических продуктов
достигло 800 миллионов фунтов (USTC 1919). Этими конечными продуктами были кра-
сители, пигменты, фотореагенты, медикаменты, ароматизаторы, компоненты духов,
синтетические фенольные смолы, синтетические дубильные вещества, взрывчатые ве-
щества (USTC 1919). Однако в 2007 году объем синтетических органических продуктов
в США составлял уже около 27 триллионов фунтов (USEPA 2008). Должны ли все эти 27
триллионов фунтов считаться новыми приоритетными типами экотоксикантов? Ответ
дает современная концепция НПЗВ.
Идея НПЗВ возникла только после того, как стало понятно, что не любое произ-
водимое промышленностью вещество, является «хорошим» для окружающей среды.
Прецедент возник в 1962 году с выходом книги Рашель Карсон «Молчаливая весна»
(Carson, 1962). Она привлекла внимание публики к скрытым опасностям химических
13.1. Введение
Таблица 13.1. Категории новых приоритетных загрязняющих веществ, классификация
и доступные методы анализа1
Категория новых при- Соединения, включенные в категорию новых приоритетных загряз-
оритетных загрязняю- няющих веществ
щих веществ
Лекарственные • Повседневные лекарственные препараты
препараты/наркотики • Антидепрессанты • Заменители гормонов и ингибиторы овуляции • Некоторые бактерицидные и антимикробные препараты • Лекарства, отпускаемые по рецепту и безрецептурные препараты для человека и животных
Продукты ухода за телом • Мускусные соединения, некоторые бактерицидные и антимикробные препараты • Химические соединения продуктов гигиены • Продукты повседневной бытовой химии
Стероиды и гормоны • Анаболики • Заменители гормонов • Половые гормоны • Ингибиторы овуляции • Стероиды и фитостероиды • Использование с медицинскими целями и некоторые противозаконные применения
Поверхностно- • Нонилфенолы
активные вещества • Алкилфенолы • Линейные алкилэтоксилаты • Моноолеаты этоксилированного сорбита • Триолеаты этоксилированного сорбита
Металлоорганические • Медикаменты
соединения • Фунгициды • Моллюскоциды • Красители
Наноматериалы Противомикробные/противовирусные, косметические, солнцезащитные, радиофармацевтические средства
1 Таблица не претендует на полноту охвата. Исходная форма таблицы создана доктором Браяном
Энглертом из Интитута окружающей среды в Мариетте (Джорджия, США).
соединений, считавшихся хорошими и полезными. Прежде всего это касалосьдихлор-
дифенилтрихлорметилметана (ДДТ), который считался крайне полезным для сохра-
нения здоровья населения. Он убивал насекомых, переносящих малярию, уничтожал
постельных клопов, имел некоторые дополнительные достоинства. Однако сейчас об-
щеизвестно, что именно ДДТ ответственен за практически полное исчезновение бело-
голового орлана и других хищных птиц. Использование ДДТ в США было запрещено
14.06.1972 года (http://www.epa.gov/history/topics/ddt/01.htm). Хотя ДДТ не используется
в США уже почти 40 лет, его следы, а также следы известных продуктов его трансфор-
мации (дихлордифенилдихлорэтилена, ДДЕ и дихлордифенилдихлорэтана, ДДД) де-
тектируются масс-спектрометрически в объектах окружающей среды {McMahon et aL,
2006; Lubick, 2007).
Многие синтезируемые органические соединения, сослужившие человечеству хо-
рошую службу, тем не менее могут проявлять негативное воздействие на здоровье че-
Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
ловека и экосистемы в целом. Например, в современном мире целый ряд важных задач
решается с использованием оловоорганических соединений. Они применяются в каче-
стве средства против обрастания моллюсками в красках для покрытия днищ кораблей,
в качестве фунгицидов в сельском хозяйстве, в качестве строительных красок, в произ-
водстве обоев и половых покрытий, а также при промышленном получении пластмасс
(Hoch, 2001; Appel, 2004). Однако эти соединения разрушают эндокринную систему, яв-
ляются нейротоксинами, могут приводить к диабету и многим другим побочным нега-
тивным последствиям (Huggett et al., 1992; Eskeset al., 1999; Appel, 2004; Grun and Blumberg,
2006; Grote et aL, 2007; Grote et al., 2009; Moser et aL, 2009; Hobler et aL, 2010). Токсичность
различных оловоорганических соединений лежит в очень широком диапазоне. Именно
поэтому важно точно знать, какое именно соединение этого класса присутствует. Толь-
ко масс-спектрометрия в сочетании с хроматографией дает возможность идентифици-
ровать индивидуальные оловоорганические соединения. Напротив, измерение общего
содержания олова в образце не позволяет получить нужной информации. (Jones-Lepp
et aL, 1999; Morabito et aL, 2000; Moreno et aL, 2006).
Еще одним примером является использование фармацевтических препаратов для
лечения людей и животных. В результате они непреднамеренно попадают в окружаю-
щую среду (Ankley et aL, 2007; Kemper, 2008). В частности, появление антибиотиков в XX
веке привело к резкому сокращению смертности от бактериальных инфекций. Однако
в настоящее время признано, что все возрастающее применение этих препаратов ведет
к выработке резистентности к ним с непрогнозируемыми последствиями от дальнейше-
го использования этих полезных веществ (Guardabass et aL, 2002; Schwartz et aL, 2003; Da
Silva etal., 2006; Auerbach etaL, 2007; Kim and Aga, 2007; Schluter et al., 2007; Rosenblatt-Farrell,
2009; Szczepanowski et aL, 2009; Zhang et aL, 2009). В своем семилетием исследовании Кид
с соавторами продемонстрировали, как использование полезных лекарственных пре-
паратов может привести к негативным последствиям для окружающей среды (Kiddetal.,
2007). Они вносили в воду небольшого изолированного озерка незначительные коли-
чества синтетического эстрогена [17р-этинилэстрадиола (ЕЕ2)], используемого в про-
тивозачаточных таблетках, и продемонстрировали, что за семь лет даже очень незначи-
тельные количества этого синтетического препарата вызывают экологический коллапс
водной экосистемы, включая практически полное исчезновение популяций некоторых
видов рыб (Kidd et aL, 2007).
Все новые и новые химические материалы ежедневно создаются и используются для
разных целей человечеством. Одним из последних новых классов соединений являются
синтетические наночастицы. Природные наночастицы всегда существовали в окружа-
ющей среде. Они попадают на поверхность планеты благодаря природным явлениям,
например извержениям вулканов, или в результате непреднамеренной человеческой де-
ятельности (в частности выбросы, сопровождающие процессы сжигания). Тем не менее
недавним открытием стали специально синтезируемые наноматериалы (Owen and Handy,
2007; Lubick, 2008; Farr6etaL, 2009). Эти материалы могут считаться НПЗВ и обычно име-
ют нанометаллическую (серебряные, золотые, железные) или наноуглеродную (фуллере-
ны) природу. Размер их частиц составляет от 1 до 100 нанометров. С 2006 года Институт
Вудро Вильсона ведет учет коммерчески доступных продуктов на основе наноматериа-
лов (Woodrow, 2010). Количество таких продуктов возросло с 212 в 2006 году до 1015 в ав-
густе 2009 года (Woodrow, 2010). Большинство этих материалов содержит наночастицы
13.2. Масс-спектрометрия и НПЗВ
серебра. Следующими по распространенности являются наноуглеродные материалы.
Все эти продукты, безусловно, обладают важными достоинствами но, одновременно по-
бочные эффекты (последствия) их применения могут быть как положительными, так
и отрицательными {Colvin, 2003; Owen and Handy, 2007; Klaine et al., 2008).
В этой главе будут также рассмотрены диспергенты и роль масс-спектрометрии
в преодолении кризисных ситуаций в окружающей среде. В 2010 году произошла ава-
рия на глубоководной нефтяной скважине (Deepwater Horizon) в Мексиканском заливе.
Миллионы тонн нефти поступали в воды залива с апреля 2010 по 15 июля 2010 года, ког-
да течь наконец была устранена. Для смягчения негативных последствий разлива такого
масштаба к первому декабря 2010 года около 10 тысяч тонн диспергентов было распыле-
но или закачано под воду в районе нефтяного пятна (http://www.restorethegulf.gov/release
/2010/12/01/operations-and-ongoing-response-december-l-2010). На сегодняшний момент
неизвестно, было л и внесение такого количества диспергента в морскую экосистему по-
зитивным или негативным моментом. Это покажут только будущие наблюдения.
13.2. Масс-спектрометрия и НПЗВ
С самого начала масс-спектрометрия играла важную роль в детектировании фарма-
цевтических препаратов в объектах окружающей среды, что привело в итоге к внесе-
нию этой группы соединений в класс НПЗВ {Watts et al., 1983; Ternes, 1998; Daughton
and Ternes, 1999; Daughton, 2001; Daughton and Jones-Lepp, 2001). Большинство же методов
детектирования НПЗВ основано именно на масс-спектрометрии. Это связано прежде
всего с тем фактом, что химический состав большинства природных матриц очень сло-
жен. Поэтому исключительно масс-спектрометрия с ее высочайшей точностью изме-
рения масс и непревзойденной специфичностью может преодолеть сложности, связан-
ные с мешающим влиянием самых разнообразных компонентов реальных природных
образцов. В частности, в одном из первых наблюдений эстрогенов в окружающей среде
был использован метод ВЭЖХ с флуоресцентным детектором. Однако сами авторы от-
метили сложность идентификации целевых аналитов в связи с присутствием большого
числа разнообразных полярных соединений во фракции эстрогенов {Snyder et al., 1999).
Последующие исследования лидера этой научной группы (Шнайдер) с применени-
ем масс-спектрометрии позволили получить значительно более надежные результа-
ты и идентифицировать в водах того же озера ряд других лекарственных препаратов
(Vanderford et aL, 2003).
В настоящее время несколько типов масс-спектрометров используется в сочетании
с газовыми и жидкостными хроматографами: квадрупольные, времяпролетные, маг-
нитные секторные, ионные ловушки, тройные квадруполи, орбитальные ловушки (гла-
ва 1). Какой из этих приборов следует использовать для определения НПЗВ в природных
матрицах, зависит, от того какой метод разделения выбран (ГХ ил и ЖХ), какова должна
быть специфичность анализа, какой диапазон масс имеют аналиты и с какой точностью
должны быть измерены их молекулярные массы. Для дополнительного изучения необ-
ходимых основ масс-спектрометрии могут быть рекомендованы следующие источники:
(Me Lafferty, 1980; Busch etal., 1988; Barceld, 1996; Grayson, 2002; Herbert and Johnstone, 2003,
A. T. Лебедев, 2003 (прим, редактора)).
Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
13.2.1. Газовая хроматография/масс-спектрометрия
Как правило, для определения химических соединений с температурой кипения ниже
300°C и стабильных при этой температуре можно использовать комбинированный ме-
тод газовой хроматографии/масс-спектрометрии (ГХ/МС). В противоположность несе-
лективным вариантам детектирования (пламенно-ионизационный детектор, УФ- или
флуоресцентный детекторы) метод ГХ/МС дает возможность получить многочислен-
ные фрагментные ионы (электронная ионизация) аналита, что позволяет однозначно
его идентифицировать.
Для того чтобы через колонку ГХ прошли полярные и/или термолабильные экоток-
сиканты (металлоорганические соединения, фармацевтические препараты и т.д.), они
должны быть предварительно дериватизованы {Kelly, 2000; Moeder et al., 2000; Morabito
et al., 2000; Ternes et aL, 2002). В частности, метилирование диазометаном и триметил-
силилирование являются двумя основными методами трансформации гидроксильных
и карбоксильных функциональных групп фармацевтических препаратов {Ternes, 1998;
Moeder et al., 2000; Jones-Lepp et al., 2009). Для металлоорганических соединений наи-
более популярными процедурами являются образование гидридов, алкилирование
реактивами Гриньяра и реакции с тетраэтилборатом натрия (NaBEt4) {Morabito et al.,
2000). Методы дериватизации не лишены недостатков. Например, неполная дерива-
тизация приводит к неполному извлечению аналита из матрицы и последующему за-
нижению его уровня в образце. Часто стараются избегать использования диазометана,
обладающего токсичностью и взрывоопасностью. Триметилсилилирование, будучи
«безопасным», может привести к образованию моно- и ди-ТМС-производных, что вы-
зывает неоднозначность при идентификации и количественном определении. Именно
из-за подобных проблем дериватизация становится менее популярной. Предпочтитель-
ным для работы с полярными и/или термолабильными соединениями в объектах окру-
жающей среды становится метод ЖХ/МС.
13.2.2. Жидкостная хроматография/масс-спектрометрия
В предыдущем разделе было отмечено, что недостатком обычных ГХ/МС-методов яв-
ляются ограничения по свойствам соединений, доступных анализу. Многие НПЗВ —
полярные, термически нестабильные, гидрофобные, малолетучие соединения.
Эти особенности делают их идеальными кандидатами для метода ЖХ/МС. Стыковка
масс-спектрометра с жидкостным хроматографом была осуществлена более 30 лет на-
зад {Niessen, 2006). Если говорить вкратце, жидкая мобильная фаза из ЖХ распыляется,
заряжается и направляется в ионный источник, в котором поддерживается атмосфер-
ное давление. В большинстве случаев используется электрораспылительная ионизация
(ЭРИ). Заряженные молекулы аналита направляются к детектору, в вакуумированную
часть прибора, через систему горячих капилляров, ионных воронок, фильтров, ква-
друполей или гексаполей. Специфической особенностью ЖХ/МС обычно является
образование в ионном источнике исключительно молекулярного иона, что позволяет
установить молекулярную массу аналита (глава 3). Как правило, в режиме регистрации
положительных ионов генерируется протонированная молекула [М + Н]+, а в режиме
регистрации отрицательных ионов — депротонированная молекула [М-Н]-. Одна-
ко этот позитивный момент может одновременно являться ограничением, поскольку
13.2. Масс-спектрометрия и НПЗВ
при идентификации аналита в сложной матрице по одному иону вероятность ошибки
весьма высока. Например, метамфетамин (C|0H|6N, m/z 149,23, CAS 537-46-2) и N,N’-
диметилфенилэтиламин (C|0H|6N, m/z 149,23, CAS 1126-71-2; DMPEA; промышленное
вещество, используемое в качестве ароматизатора) являются изомерами с идентичной
молекулярной массой (149,23 Да), но несколько отличной структурой. В этом случае не-
обходимо использовать более специфичную масс-спектрометрическую технику иден-
тификации. Это тандемная масс-спектрометрия или МС/МС (глава 4). В таком варианте
ион-предшественник генерируется в источнике ЖХ/МС-прибора (обычно [М + Н]+ или
[М - Н]-), его энергия повышается в камере соударений (диссоциация, активированная
соударением (ДАС), collision induced dissociation — CID) и он фрагментирует с образо-
ванием ионов-продуктов. Для этой цели применяются тройные квадруполи, ионные
ловушки и магнитные секторные приборы. Ионы-продукты образуются при выбросе
различных фрагментов аналита, например [М + Н-Н2О]+ или [М + Н-СН3]+. Однако
ошибочная идентификация не исключена даже в режиме МС/МС. Например, и DMPEA
и метамфетамин в условиях ДАС распадаются с образованием мажорных характеристи-
ческих ионов-продуктов — m/z 105 [МН -NH(CH3)J+ и m/z 119 [МН -CH3NH2]+ соответ-
ственно. Однако при распаде обоих этих соединений образуется также первичный ион
с m/z 91. Если выбрать для регистрации метамфетамина ион с m/z 91, а не m/z 119 (в лите-
ратуре такие примеры приводятся), можно получить ложноположительный результат.
Еще один пример составляет пара MDMA (C(|H|5NO2, м.м. 193,25 Да, CAS 69610-10-2)
и кофеин (C8H|0N4O2, м.м. 194,19 Да, CAS 58-08-02). Хотя молекулярные массы этих сое-
динений различны, m/z 163 — наиболее важный ион-продукт в обоих случаях. Протони-
рованные молекулы MDMAcw/г 194 в условиях электрораспыления и активации соуда-
рением теряют молекулу метиламина с образованием иона с m/z 163,0, [МН -CH3NH2]+.
Протонированная молекула кофеина с m/z 195 в тех же условиях распадается с образо-
ванием основного иона-продукта с m/z 138, [МН-CH3NCO]+, однако m/z 163 — второй
по интенсивности фрагментный ион. Таким образом, если выбрать для мониторинга
ион с m/z 163, можно ошибочно принять MDMA за кофеин, поскольку времена выхода
этих соединений близки.
При использовании ЖХ/МС для одновременного определения целевых и неизвест-
ных соединений необходимо применить метод ЖХ/МС/МС для идентификации неиз-
вестного соединения, а затем правильно выбрать ионы-предшественники и ионы-про-
дукты для максимальной специфичности и точности определения целевых.
Металлоорганика
Металлоорганические соединения широко используются при производстве промыш-
ленных, сельскохозяйственных и потребительских товаров. Многие соединения этого
класса применяются в медицине (железоорганические и платинаорганические соеди-
нения — противоопухолевые препараты; борорганические соединения применяются
в нейтронозахватной терапии), средствах бытовой химии (дибутилолово, диметилолово,
октилолово — ингредиенты пластмасс), в сельском хозяйстве (трифенилолово — фун-
гицид, какодиловая кислота — контактный гербицид, фенилмышьяковые кислоты —
регуляторы роста животных), при обработке судов для предотвращения обрастания
моллюсками (трибутилолово и трифенилбор) (Huggett et al., 1992; Craig, 2003; Jones-Lepp
and Momplaisir, 2005; Allard et al., 2008; Oudijk, 2010). Биологические процессы трансфор-
мации, наряду с антропогенной активностью (например энергетика), также могут при-
366 Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
вести к появлению в окружающей среде металлоорганических соединений (например,
метилртуть и алкилсвинец) (Tessier and Turner, 1995). Очень важно определять именно
индивидуальные химические соединения, а не интегральное содержание элементов
в пробе. Это связано с крайне широким диапазоном токсикологических и биохимиче-
ских свойств металлоорганических соединений (Tessier and Turner, 1995; Newcombe et aL,
2010). В нескольких последующих подразделах будут рассмотрены некоторые типы ме-
таллоорганических соединений и масс-спектрометрические подходы для их идентифи-
кации и детектирования.
Оловоорганика
Как уже сказано выше, оловоорганические соединения в зависимости от природы
и числа алкильных групп, связанных с атомом олова, ответственны за целый ряд рас-
стройств эндокринной и нервной систем. Считается, что, как правило, оловооргани-
ческие соединения токсичнее, чем неорганические формы этого элемента (Eskes et aL,
1999; Grun and Blumberg, 2006; Moser et aL, 2009). Общая токсичность, при этом, увели-
чивается в следующем ряду: галогениды олова(П) и олова(1У) < монозамещенные < ди-
замещенные < тризамещенные оловоорганические соединения (Winship, 1988; Pelletier,
1995; GajdaandJancsd, 2010). Однако, различные оловоорганические соединения влияют
на различные органы. Например, триметилолово (ТМТ) и триэтилолово (ТЕТ) атакуют
различные части центральной нервной системы. ТМТ токсично для гиппокампуса и го-
могенетической коры головного мозга, а ТЕТ влияет на регионы спинного мозга (Gajda
andJancsd, 2010). Следовательно, для оценки риска и изучения превращений оловоорга-
ники в окружающей среде необходимо определять не только общее содержание олова,
но устанавливать точную структуру оловоорганических соединений. У олова 10 ста-
бильных природных изотопов. Это приводит к характерной масс-спектрометрической
картинке, помогая правильно детектировать и идентифицировать оловоорганические
соединения в образцах. Большинство методов определения оловоорганических соеди-
нений связано с ГХ/МС. Это означает, что определению должна предшествовать дери-
ватизация. Например, Томаидис и др. доводили образцы морской воды до pH 5 и про-
водили экстракцию оловоорганики с последующей дериватизацией тетраэтилборатом
натрия (STEB) в гексановом растворе (Thomaidis et aL, 2007). Далее дериватизованные
экстракты вводили в систему ГХ/МС. Пределы обнаружения для бутилолова составля-
ли около 2 пг в уколе (2000 нг/л при вводе 1 мкл пробы в прибор) в пересчете на олово.
Для соединений фенилолова пределы обнаружения были еще лучше, в особенности для
трифенилолова (1 пг) (Thomaidis et al., 2007). Сеговиа— Мартинес и др. добились еще бо-
лее низких пределов обнаружения: от 0,025 нг/л для трибутил и дифенилолова до 1 нг/л
для триэтилолова (Segovia-Martinez et aL, 2010). Их ГХ/МС-метод основан на in situ эти-
лировании с одновременной твердофазной микроэкстракцией из воздушной фазы над
жидкой пробой (хедспейс). Масс-спектры триэтил-, трибутил-, дифенил- и трифени-
лолова, полученные таким методом, представлены на рис. 13.1. В каждом спектре видна
характерная изотопная картина, обусловленная присутствием атома олова. Поскольку
метод базировался на дериватизации тетраэтилборатом натрия в спектре наблюдаются
пики характеристических ионов, включая дериватизованные: m/z 179 (SnEtH2), m/z 291
(SnBu3), m/z 151 (SnEtH2), m/zTSTl (SnEtBuH or SnEt3), m/z 235 (SnBu2H), m/z 263 (SnEtBu2),
m/z 303 (SnEtPh2), m/z 275 (SnHPh2), m/z 197 (SnPh), and m/z 351 (SnPh3) (Segovia-Martinez
et aL, 2010). Недавно разработан метод детектирования оловоорганики в других матри-
13.2. Масс-спектрометрия и НПЗВ
Интенсивность
5000000
4500000 -
4000000
3500000
3000000
2500000
207
179
149
101 120 139 158 177 195 214 231 251 270 288 305 321 338 356 371 387
2000000
1500000
1000000
500000
0
100 122 144 166 188 209 229 251 271 291 311 330 348 368 387
m/z
m/z
m/z m/z
Рис. 13.1. Масс-спектры (ГХ/МС) четырех оловоорганических соединений: триэтило-
лово, трибутилолово, дифенилолово и трифенилолово после проведения дери-
ватизации (+2 на формуле дифениолова означает степень окисления металла
(не заряд иона)). С разрешения Elsevier; L. Segovia-Martinez, A. Bouzas-Blanco,
Р. Campins-Falco, A. Seco-Torrecillas, Improving detection limits for organotin compounds
in several matrix water samples by derivatization-headspace-solid-phase microextraction
and GC/MS. Taianta, 80, figure 4, pgs. 1888—1893, 2010
цах (не только в воде). Оловоорганические соединения в образцах сетчатых моллюсков
(Nassarius reticulates) определялись количественно методом ГХ/МС путем твердофаз-
ной экстракции с последующей дериватизацией тетраэтилборатом натрия (Sousa et al.,
2009). Каннан и др. (Каппап et al., 2010) сообщили об обнаружении оловоорганики в до-
машней пыли. Они использовали модифицированный метод Соуза (Sousa et al., 2009).
Помимо ГХ/МС оловоорганические соединения анализировали и методом
ЖХ/МС. Поскольку упомянутые выше процедуры дериватизации всегда усложняют
анализ, были разработаны методики на базе ЖХ/МС, которые позволяют опустить эту
стадию (дериватизация и/или гидролиз) при определении оловоорганики, а также дру-
гих металлоорганических соединений. Сью и др. первыми использовали тандемную
масс-спектрометрию с химической ионизацией при атмосферном давлении (ХИАД)
в режиме мониторинга выбранной реакции (SRM) для определения оловоорганики
в стандартных образцах седи ментов (Siu et al., 1989). В другой ранней работе Куллен
и др. использовали масс-спектрометр Kratos MS 80 RFA с термораспылительным ин-
терфейсом для определения соединений бутилолова в морских образцах (Cullen et al.,
1990). Джонс-Лепп и др. (1999) первыми опубликовали данные, полученные в резуль-
тате быстрой экстракции и детектирования оловоорганики из образцов природных ис-
точников воды методом ЖХ/МС в ионной ловушке с ионизацией электрораспылени-
ем (Jones-Lepp et al., 1999). Существенной в этом случае была проблема нестабильности
процесса ионизации. Она была успешно преодолена при добавлении в мобильную фазу
Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
трополона в качестве стабилизатора. Благодаря этой добавке ионная ловушка реги-
стрировала трополоновые ионы-аддукты (Jones-Lepp et aL, 1999). Например, дихлорид
дифенилолова (м.м. 344 Да) легко идентифицировался по масс-спектру благодаря иону
с m/z 395, образующемуся при потере двух атомов хлора и присоединению трополоново-
го иона [М —2С1 + С7Н5О2]+, (рис. 13.2). Недавно описан новый метод твердофазной экс-
тракции с прямой стыковкой с ЖХ/МС в режиме ионизации электрораспылением или
химической ионизации при атмосферном давлении для идентификации оловооргани-
ческих соединений в пробах воды (Sun et al., 2009). Такая стыковка позволила одновре-
менно удалить большинство мешающих определению соединений матрицы и провести
предварительное концентрирование трибутилолово- и трифенилоловохлоридов. Спек-
тры оказались идентичными и в условиях ЭРИ, и в условиях ХИАД. Например, в слу-
чае трибутилоловохлорида максимальный пик в спектре принадлежал иону [М — С1]+
с m/z 291,1. Наряду с ним наблюдались пики фрагментных ионов, возникающих бла-
годаря элиминированию одной [М —C1,-C4HJ+, m/z 235,0, или двух [М -С1,-(С4Н8)2]+,
m/z 179,0, молекул бутена (Sun et aL, 2009). Сано и др. (Sano et aL, 2010) тестировали ме-
тод ЭРИ в сочетании с жидкостной хроматографией гидрофильных взаимодействий.
Их задачей было создание усовершенствованного быстрого хроматографического ме-
тода с масс-спектрометрическим детектированием в режиме электрораспылительной
ионизации. Полное время хроматографического цикла оказалось менее 15 минут, при-
чем удалось достигнуть хорошего разделения хлоридов трибутил- и трифенилолова. Од-
нако ионы, регистрируемые в этих условиях, отличались от ожидаемых a priori. Вместо
молекулярных ионов [М]+ или протонированных молекул [М + Н]+ образовывались
ионы аддуктов хлоридов трибутил- и трифенилолова с ацетонитрилом, [М + CH3CN]+
m/z
Рис. 13.2. Масс-спектр дифенилолова, полученный на ионной ловушке методом
ЖХ/ЭРИ/МС (с разрешения Агентства по охране окружающей среды США)
13.2. Масс-спектрометрия и НПЗВ
с m/z 332 и m/z 392 соответственно (Sano et al., 2010). Эти ионы возникали из-за исполь-
зования ацетонитрила на стадии экстракции и в качестве компонента мобильной фазы.
Пределы обнаружения хлоридов трибутил- и трифенилолова этим методом составили 3
и 6 нг/л соответственно.
Мышьякорганические соединения
Как уже отмечено выше, оловоорганические соединения считаются более токсичными,
чем элементарное олово. Однако в случае мышьяка это не так. Биометилирование мы-
шьяка считается механизмом детоксикации, который многие организмы используют
для снижения негативных эффектов от более токсичных неорганических соединений
этого элемента. Метилмышьяковая и диметилмышьяковистая кислоты, обнаружен-
ные в многочисленных объектах окружающей среды, оказались менее токсичными,
чем неорганические соединения мышьяка (Kaise et aL, 1989). Острая токсичность три-
метиларсиноксида оказалась аналогична таковой для арсенобетаина — наиболее рас-
пространенного соединения мышьяка в морских животных (Kaise et al., 1989; Newcombe
et al., 2010). Недавнее исследование по установлению биораспространенности арсено-
бетаина в организме человека комбинацией методов ЖХ/ИСП/МС и ЖХ/ЭРИ/МС по-
казало, что практически весь мышьяк, зарегистрированный методом ИСП, обусловлен
исключительно арсенобетаином. Мониторинг иона [М + Н]+, m/z 179, арсенобетаина
(м.м. 178,06 Да), образующегося в условиях ЭРИ, осуществлялся одновременно с мони-
торингом иона элементного мышьяка, m/z 75, в условиях ИСП. Рис. 13.3, демонстри-
рующий наложенные друг на друга хроматограммы ЖХ/ИСП/МС и ЖХ/ЭРИ/МС,
Рис. 13.3. Сравнение результатов, полученных методами ЖХ/ИСП/МС и ЖХ/ЭРИ/МС
для m/z 75 (общий мышьяк) и m/z 179 (арсенобетаин). Из J. Environ. Monitoring;
Fig. 2 from Newcombe et al. «Accumulation or production of arsenobetaine in humans,»
2010, 12, 832-837. С разрешения Royal Society of Chemistry
370 Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
подтверждает, что весь обнаруженный мышьяк находился в форме арсенобетаина
(Newcombe et aL, 2010).
Ртутъорганические соединения
Еще одним классом металлоорганических соединений, считающихся важными эко-
токсикантами, являются ртутьорганические соединения. В природе этот металл при-
сутствует в трех основных формах: элементарная ртуть (Hg°), неорганическая ртуть
(Hg2+) и органическая ртуть — (MeHg+). Токсичность всех трех форм варьируется в за-
висимости от их биодоступности для живых организмов, причем MeHg+ считается наи-
более токсичной биодоступной формой (Mahaffey etal., 1997). Биоцикл ртути достаточно
сложен. Она поступает в окружающую среду из природных или антропогенных источ-
ников, присутствует в атмосфере, затем выпадает на землю с дождем и твердыми ча-
стицами, оказывается в седиментах, почве, растительности и воде, где она превраща-
ется благодаря биотрансформации в MeHg+, накапливается в рыбе и в конечном счете
в организме человека (Aschner et al., 2010). В связи с существованием различных форм
ртути и их разными токсичностями очень важно уметь идентифицировать эти инди-
видуальные формы в сложных природных матрицах. Бесейро-Гонсалес и др. сообщи-
ли о результатах идентификации индивидуальных металлоорганических соединений
ртути, свинца и олова в образцах воды методом твердофазной микроэкстракции в ре-
жиме хедспейс с последующим ГХ/МС- и ГХ/МС/МС-определением (Beceiro-Gonzalez
et al., 2009). Ртуть, как и олово, имеет очень характерную изотопную картинку с семью
изотопами. Это делает масс-спектрометрическую идентификацию метилртути весьма
прямолинейной. Крупп и др. описали аналитический метод, позволяющий проводить
прямое детектирование ртути и комплексов метилртути с биологическими тиолами
в биоте (Krupp et aL, 2008). Используется одновременно ВЭЖХ/ЭРИ/МС на ионной ло-
вушке для идентификации и структурных исследований комплексов метилртути на мо-
лекулярном уровне и ИСП/МС для детектирования элементарной ртути (Krupp et aL,
2008). Эксперименты проводились с помощью двух высокоэффективных жидкостных
хроматографов в сочетании с ЭРИ/МС или ИСП/МС, а также с одним хроматографом
с разделением потока. 80% элюата направлялось в ЭРИ/МС, а 20% — в ИСП/МС. Ав-
торы сообщили об образовании комплекса цистеина с метилртутью. Методом ЭРИ/МС
была зарегистрирована его МН+ с m/z 338,01 (по изотопу максимальной интенсивности).
Другие металлоорганические соединения
Токсикологические характеристики, а также оценка влияния на окружающую среду
многих синтетических металлоорганических соединений, применяющихся в медицине
в качестве противоопухолевых препаратов или для других целей (ферроцены, платина-
органические и борорганические соединения) до сих пор детально не изучены (Cui etaL,
2003; Hann et aL, 2005; Lenz et aL, 2005; Allard et aL, 2008; Johnson et aL, 2008). Кюи с соавто-
рами (Cui et aL, 2003) использовал метод приращения ионной подвижности (asymmetric
waveform ion mobility spectrometry (FAIMS)) в сочетании с ЭРИ/МС для детектирования
цисплатина (м.м. 300,05 Да, Cl2H6N2Pt) в растворах. Они использовали способность ме-
тода фильтровать (разделять) ионы и добились значительного улучшения детектиро-
вания цисплатина благодаря 30-кратному уменьшению приборного шума. Этот метод
явно имеет потенциал для решения природоохранных задач.
Лекарственные препараты
В последние годы было надежно доказано, что фармацевтические препараты, как пред-
назначенные для человека, так и ветеринарные, оказываются в природной среде с экс-
крементами или вместе с бытовыми отходами (Daughton and Ternes, 1999; Daughton and
Jones-Lepp, 2001; Ankley et aL, 2007; Kemper, 2008; Kummerer, 2010). Для детектирования
этих соединений в объектах окружающей среды в целях оценки риска необходимы при-
емлемые, воспроизводимые и чувствительные аналитические методы определения.
Большинство подходов, позволяющих определять очень низкие уровни (мкг/л и ниже)
фармацевтических препаратов в сложных матрицах, основаны на масс-спектрометрии
(Daughton, 2001; Petrovic etaL, 2005; Hao etal., 2007; Wang, 2009; Richardson, 2010).
Изначально ГХ/МС использовалась для детектирования в природных образцах
неполярных, полярных (после дериватизации) фармацевтических препаратов, а так-
же стероидов и гормонов. В 1976 году Гаррисон и др. первыми сообщили об обнару-
жении лекарственных препаратов в окружающей среде (Garrison et al., 1976). НПЗВ
и другие «традиционные» экотоксиканты (ПАУ, фталаты, хлорированные алканы)
извлекались методом жидкость-жидкостной экстракции с последующим метилирова-
нием и ГХ/МС-определением (Garrison et al., 1976). Мёдер с сотрудниками разработали
ГХ/МС/МС-метод с твердофазной микроэкстракцией и соответствующей дериватиза-
цией для детектирования ибупрофена, кофеина, клофибриновой кислоты, парацетамо-
ла, феназона,карбамазепина гемфиброзила, напроксена, индометрацина, норэтистеро-
на, пропранолола и метапролола в воде (Moeder et al., 2000). Авторы добавляли 100 мкл
1Ч,О-бис(триметилсилил)-трифторацетамида ( BSTFA) к 500 мкл экстракта, нагревали
смесь в течение 1 часа при 40°С, упаривали до 250 мкл и инжектировали 1 мкл в прибор
для ГХ/МС-определения.
Однако многие препараты и гормоны являются полярными, термически неста-
бильными, гидрофобными и малолетучими соединениями. Эти особенности делают
их идеальными кандидатами для использования метода ЖХ/МС. Поэтому в настоящее
время практически все методы детектирования полярных фармацевтических препара-
тов в окружающей среде базируются на вариантах ЖХ/МС и ЖХ/МС/МС (Petrovic et aL,
2005; Kosjek et aL, 2007). Фаре и др. (Раггё et aL, 2001) разработали метод твердофазной
экстракции для тех же самых аналитов, что и Мёдер (Moeder et aL, 2000), но уже без де-
риватизации. Для анализа экстрактов они применили ЖХ/ЭРИ/МС в режиме регистра-
ции отрицательных ионов и показали, что такой подход имеет явные преимущества над
ГХ/МС, поскольку удается избежать стадии дериватизации.
Конли с сотрудниками описал быстрый метод детектирования широкого круга
распространенных лекарственных НПЗВ с помощью ультраэффективной жидкостной
хроматографии (УЭЖХ) в стыковке с тройным квадруполем в режиме ЭРИ (Conley et aL,
2008). Применение УЭЖХ позволило детектировать 13 препаратов и один метаболит ме-
нее чем за 5 минут, причем тандемная масс-спектрометрия дала возможность надежно
идентифицировать и количественно определять аналиты на уровне нг/л (Conley et aL,
2008). В работе продемонстрирована важность учета эффектов матрицы при исполь-
зовании методов ЭРИ/МС и представлено уравнение для выявления благоприятного
влияния или противодействия матрицы корректной регистрации аналита. Подобные
эффекты матриц отмечались и другими исследователями. Например, Янг с коллегами
показал, что в режиме ЖХ/ЭРИ/МС/МС на ионной ловушке наблюдается значительное
372 Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
подавление сигналов тетрациклинов во время анализа образцов сточной воды. При этом
на регистрацию сульфамидов эта матрица не влияла. Эффект подавления можно смяг-
чить при использовании внутреннего стандарта (Yang et al., 2004). Негативное влияние
матрицы при анализе сточных вод одной из больниц Швеции практически отсутство-
вало при детектировании сульфаметоксазола, но явно наблюдалось при определении
ципрофлоксацина {Lindberg et al., 2004).
В недавней публикации Лооса продемонстрирована эффективность и гибкость
ЖХ/МС/МС для целевого анализа {Loos et al., 2010). Авторы регистрировали 34 НПЗВ
(фармацевтические препараты, пестициды, хлорорганические соединения, перфтори-
рованные кислоты и т.д.) методами ЭРИ и ХИАД. Им пришлось работать и с положи-
тельными, и с отрицательными ионами, чтобы детектировать все 34 целевых аналита,
принадлежащих к самым разным классам органических соединений. Еще одно новше-
ство — применение жидкостной хроматографии в комплексе с орбитальной ловушкой
для проведения скрининговых природоохранных исследований {Ни et al., 2005). Орби-
тальные ловушки сочетают высокую разрешающую способность и точность измерения
масс с возможностью работы в режиме МС" {Ни et al., 2005). Сложность экологических
образцов обычно очень велика. Помимо целевых соединений они содержат значитель-
m/z 206.1551
C13H20NO
100 я
90 |
80 т
70-Е
Масс-спектр стандартного об-
разца метолахлороксалиновой
кислоты (орбитальная ловушка)
60 т
50 |
40 1
30 !
20 |
10 i
о Д................
100
....ц.....I......
150 200
m/z 278.1397
C15H20NO4
..........Ц..г
250
Рис. 13.4. Полный масс спектр высокого разрешения (режим регистрации отрица-
тельных ионов) метолахлороксалиновой кислоты в образце грунтовой воды
(верх) и в стандартном растворе (низ). Из J. Chromatogr. А, 1216 (3), figure 3 from
Hogenboom et al. «Accurate mass screening and identification of emerging contaminants
in environmental samples by liquid chromatography-hybrid linear ion trap orbitrap mass
spectrometry». 510—519, (2009). С разрешения Elsevier
ное число самых разнообразных НПЗВ. Хогенбум продемонстрировал успешное опре-
деление этим методом целевых аналитов с одновременной идентификацией несколь-
ких неизвестных НПЗВ, присутствовавших в образце грунтовой воды {Hogenboom et al.,
2009). Сначала были измерены точные массы всех неизвестных НПЗВ, а затем они были
сравнены со списком теоретических масс существующих НПЗВ. Если идеального соот-
ветствия не наблюдалось, авторы моделировали элементный состав таких неизвестных
соединений. MC''-эксперименты позволяли получать фрагментные ионы в линейной
ловушке с последующим измерением их точных масс в орбитальной ловушке. Составив
«схему фрагментации», они смогли связать все фрагментные ионы с конкретными ио-
нами-предшественниками, регистрируемыми в режиме обычного сканирования спек-
тра {Hogenboom et aL, 2009). Возможность точной идентификации нецелевых соедине-
ний в сложных экологических матрицах этим методом проиллюстрирована на рис. 13.4.
В верхней части рисунка представлен полный масс-спектр неизвестного соединения
в экстракте образца грунтовой воды с указанием точных масс ионов. После составления
«схемы фрагментации» авторы смогли идентифицировать это соединение как метолах-
лороксалиновую кислоту. В нижней части рисунка представлен масс-спектр, получен-
ный при введении в прибор стандартного раствора этой кислоты. Идентичность спек-
тров подтверждает правильность идентификации {Hogenboom et al., 2009).
Наноматериалы
Для правильной оценки риска для человека и окружающей среды наноматериалов не-
обходимо иметь надежные методы их детектирования в самых разных образцах. Соз-
данные наноматериалы, в особенности частицы наносеребра, уже часто используются
в продуктах косметики, в носках и нижнем белье, в стиральных машинах и т.д. Соот-
ветственно увеличивается вероятность попадания этих частиц в водную среду со сбро-
сами с заводов отходов обработки сточных вод или в результате случайных сбросов не-
очищенных вод {Geranio et aL, 2009; Howard, 2010; Weinberg et aL, 2011). Поскольку очень
мало известно о токсикологических характеристиках наноматериалов, первыми ша-
гами должны быть следующие: (1) определить, присутствуют ли в окружающей среде
созданные наноматериалы, (2) установить их концентрации, что, безусловно, связано
с аналитическими процедурами. Аналитические методы должны позволять различать
природные и искусственные наночастицы, дифференцировать их по размеру и отделять
искусственные частицы от всех остальных соединений, присутствующих в сложном
природном образце. Например, в некоторых работах ЖХ/МС использована для установ-
ления трансформации фуллеренов.
Изаксон с сотрудниками продемонстрировал надежное разделение и детектирова-
ние ряда фуллеренов (С60, 13С60, С70, С82, С88, С98) методом ЖХ/ЭРИ/МС (рис. 13.5). Пики
их молекулярных анион-радикалов М" были самыми интенсивными при ЭРИ в режиме
регистрации отрицательных ионов. Авторы успешно разделили и количественно опре-
делили каждый из указанных фуллеренов, причем их концентрации были достаточно
низкими для требуемых в экологических анализах. Так, предел обнаружения фуллере-
на С60 составил 0,0004 мкг/л при отношении сигнал/шум 3:1 {Isaacson et aL, 2007). Чен
с соавторами разработал метод твердофазной экстракции для извлечения и концентри-
рования фуллеренов из образцов природной воды. Метод позволил устранить мешаю-
щее влияние компонентов фона настолько, что экологически требуемые уровни опре-
деления фуллеренов легко достигались в варианте ЖХ/МС с источником ХИАД {Chen
Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
Рис. 13.5. ЖХ/ЭРИ/МС-хроматограммы для нескольких образцов фуллеренов в растворе
метанол/толуол (80:20). С60 (2 мкг/л в гомогенизированной матрице рыбы (да-
нио); 13С60 (10 мкг/л в гомогенизированной матрице рыбы (данио); С70 (10 мкг/л),
С82 (3,4 мкг/л), С88 (2,5 мкг/л), С98 (0,4 мкг/л). С разрешения Isaacson C.W.,
C.Y. Usenko, et al. (2007). «Quantification of Fullerenes by LC/ESI-MS and Its
Application to in Vivo Toxicity Assays». Analytical Chemistry 79(23): 9091—9097.
Copyright 2007 American Chemical Society
et al., 2008). Например, фуллерен C60 детектировался в виде молекулярного анион-ра-
дикала с m/z 720. Использованные экспериментальные условия позволяли детектиро-
вать единичные наночастицы {Chen et al., 2008). Изаксон с Бушаром состыковали метод
приращения ионной подвижности с фракционированием в потоке (AF4) с детектором
динамического рассеяния света в проточном режиме. Фракционированные таким об-
разом образцы попадали далее в источник фотоионизации при атмосферном давлении
{Isaacson and Bouchard, 2010). Этот подход позволил однозначно определять фуллерены
С60 во фракции каждой размерности, собранной в AF4.
В современной литературе нет данных о детектировании нанометаллических ча-
стиц (наносеребро, нанозолото, наножелезо), за исключением использования неспеци-
фических детекторов, например ИСП/МС, УФ/видимого света, флуоресцентного
{Howard, 2010; Weinberg et al., 2011). Хотя такие детекторы способны определить общее
содержание металла, в большинстве случаев различить природные и искусственные
наночастицы, а также подсчитать число частиц разных размеров невозможно. Явно не-
обходима стыковка неспецифических детекторов с методом AF4 или другой эксклюзи-
онной техникой. В таком варианте можно достичь улучшения в подсчете частиц разных
размеров и уменьшения мешающего влияния матричного фона {Howard, 2010).
Диспергенты нефтяных разливов
Диспергенты — это детергенты, состоящие из поверхностно-активных веществ (ПАВ),
растворенных в одном или нескольких растворителях. Они распыляются в месте не-
фтяного разлива для удаления нефти с поверхности и диспергирования по толще воды.
Диспергенты ускоряют деградацию компонентов нефти (алканов, ПАУ и т.д.) путем
разбавления и активации бактериального разложения, тем самым устраняя негативное
влияние нефти на окружающую среду (Lessard and DeMarco, 2000).
Как уже отмечено выше в этой главе, для борьбы с нефтяным разливом на глубоко-
водной нефтяной скважине (Deepwater Horizon) в Мексиканском заливе около 10 тысяч
тонн диспергентов (корексит 9500 и корексит 9527) были распылены с воздуха или внесе-
ны непосредственно под воду (http://www.restorethegulf.gov/release/2010/12/01/operations-
and-ongoing-response-december-1-2010). Именно благодаря масс-спектрометрии сотруд-
ники национальной исследовательской лаборатории воздействий Агентства по охране
окружающей среды США в Лас-Вегасе (Невада) смогли расшифровать химический со-
став примененных диспергентов. Используя ряд масс-спектрометрических методов, им
удалось установить структуры ингредиентов различных фракций диспергентов. В част-
ности, растворы корексита 9500 и корексита 9527 были приготовлены в воде (для опре-
деления летучих составляющих), гексане (для определения полулетучих составляющих)
и метаноле (для определения нелетучих составляющих). Для определения летучих ин-
гредиентов был использован ГХ/МС-анализ с вакуумной дистилляцией. Этот метод
особенно эффективен для соединений с диапазоном температур кипения от 180 до 240 °C
(Hiatt, 1995), http://www.epa.gov/epawaste/hazard/testmethods/sw846/new_meth.htm —
8261А)). Как показали анализы, летучие соединения обоих диспергентов были пред-
ставлены в основном низкокипящими растворителями (для упрощения воздушного
диспергирования), состоящими из легких алкилбензолов, простых алканов, нафталина
и 1,4-диоксана. Фракция полулетучих компонентов корексита 9500 была исследована
методом ГХ/МС. Одним из основных ингредиентов оказался бис(2-этилгексил)фумарат
(CAS 141-02-6). Это реагент для промышленного производства диоктилсульфосукци-
ната натрия (DOSS), который ранее был заявлен в качестве компонента корексита 9500
Национальной академией наук (NAS) (Committee on Understanding Oil Spill Dispersants:
Efficacy and Effects 2005)]. Меньший пик на хроматограмме был обусловлен изомером
бис(2-этилгексил)фумарата. Фракция нелетучих компонентов корексита 9500 была ис-
следована методом ЖХ/ЭРИ/МС на ионной ловушке и прямым анализом в реальном
времени (DART, глава 7) на времяпролетном масс-спектрометре. Метод DART дает точ-
ные величины молекулярных масс ионизируемых компонентов диспергента (Grange and
Sovocool, 2008), а ЖХ/ЭРИ/МС позволяет разделить и установить структуры неизвест-
ных компонентов методом тандемной масс-спектрометрии. Таким образом удалось вы-
явить основные ингредиенты этого типа. В режиме регистрации положительных ионов
идентифицированы неионогенные ПАВ (моно- и триолеаты этоксилированного сорби-
та) в соответствии с информацией NAS (Committee on Understanding Oil Spill Dispersants:
Efficacy and Effects 2005)], н-бутиловый эфир дипропиленгликоля и незначительные ко-
личества этоксилированного нонилфенола. В режиме регистрации отрицательных ио-
нов на тройном квадрупольном приборе было подтверждено присутствие DOSS [инфор-
мация NAS (Committee on Understanding Oil Spill Dispersants: Efficacy and Effects 2005)].
Масс-спектрометрия была также использована для обеспечения контроля каче-
ства при проведении токсикологического тестирования диспергентов (Judson et aL,
2010). Пробы с добавками (для токсикологического тестирования) и смеси морской
воды, нефти и диспергентов (рис. 13.6) анализировались как часть процедур контроля
Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
Рис. 13.6. Плашки для тестирования токсичности
качества теми же самыми методами, которые использовались для установления струк-
тур ингредиентов. Важность масс-спектрометрии была продемонстрирована во время
перекрестного контроля проб. В качестве первого примера представлен масс-спектр
и хроматограммастандартногообразца диспергента А (рис. 13.1а). Отчетливо прослежи-
вается серия ионов, обусловленных присутствием многочисленных ПАВ, в частности
олеатами оксиэтилированного сорбита. Одна из этих серий включает ионы с m/z 476,3,
m/z 520,3, m/z 564,3, m/z 608,4. Ионы в серии отличаются на 44 единицы массы, что об-
условлено фрагментом (-СН2СН2О-), указывающим на разное количество этих групп
в конкретных олеатах сорбита. В этом же спектре присутствует еще одна менее выра-
женная серия подобных ионов — m/z 503,3, 547,3, 591,3. В этой фракции неполярных
компонентов диспергента А присутствует соединение, обусловливающее интенсивный
пик с m/z 163,2, [М + Н]+ и меньший пик с m/z 185,2 [М + Na]+. Используя высокое разре-
шение времяпролетного прибора в режиме DART и специальное программное обеспе-
чение, ион m/z 163,2 был интерпретирован как [М + Н]+ 1-(этоксипропокси)пропанола-2
или 2-(2-бутоксиэтокси)этанола. Однако для однозначной идентификации необходимо
было иметь стандарты обоих веществ. Второй пример представлен на рис. 13.76. Пред-
ставлен масс-спектр и хроматограмма образца, который должен был содержать исклю-
чительно диспергент А. Однако и в масс-спектре и на хроматограмме видны пики, при-
надлежащие ионам, характерным для диспергента G: m/z 191 и m/z 213. Эти два примера
установления неизвестных соединений и перекрестного контроля замечательно демон-
стрируют эффективность масс-спектрометрии.
13.3. Выводы
В этой главе сделана попытка описать широкий круг масс-спектрометрических мето-
дов, применяющихся для экстракции и детектирования НПЗВ в сложных природных
13.3. Выводы
а
Рис. 13.7. Масс-спектры и хроматограммы диспергента А и образцов с плашек, (а) Стан-
дарт диспергента А; (б) диспергент А с плашек
Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
образцах. Многие из этих методов — это новые подходы, базирующиеся на более ста-
рых техниках эксперимента. В главу не вошли многочисленные варианты пробоотбора
и извлечения НПЗВ из образцов объектов окружающей среды. Читателю следует об-
ратиться к литературным источникам для получения информации по этим вопросам
(Jones-Lepp et aL, 2009; Richardson, 2010), поскольку при описании существующих масс-
спектрометрических методов детектирования НПЗВ рассматриваются вопросы пробо-
отбора и извлечения конкретных классов НПЗВ.
Благодарность. Автор благодарен своим коллегам из Агентства по охране окружаю-
щей среды США за их вклад в раздел о диспергентах: Майку Хиатту (летучие соедине-
ния методом ГХ/МС с вакуумной дистилляцией); Кардите Розаль (полулетучие и не-
летучие соединения); Джону Циммерману (полулетучие соединения методом ГХ/МС);
доктору Анди Гранжу (нелетучие соединения методом DART); а также докторам Дону
Бетовски и Уэйну Совоколу за их вклад в расшифровку масс-спектров в разделе о дис-
пергентах и других разделах этой главы.
Уведомление. Финансирование и проведение исследований, о которых шла речь
в этой главе, осуществлено Агентством по охране окружающей среды США через его
научно-исследовательский отдел. Материал был подвергнут соответствующей админи-
стративной проверке и одобрен для опубликования.
Литература
[1] . Allard Е., Passirani С., Garcion Е., Pigeon Р., Vessie'res A., Jaouen G., and Benoit, J.-P.
(2008). «Lipid nanocapsules loaded with an organometallic tamoxifen derivative as a novel
drug-carrier system for experimental malignant gliomas». Journal of Controlled Release. 130,
2: 146-153.
[2] . Ankley G.T., Brooks B.W., Huggett D. B., and Sumpter J. P. (2007). «Repeating his-
tory: pharmaceuticals in the environment». Environmental Science and Technology. 41, 24:
8211-8217.
[3] . Appel К. E. (2004). «Organotin compounds: toxicokinetic aspects». Drug Metabolism Re-
views. 36, 3—4: 763—786.
[4] . Aschner M., Onishchenko N., and Ceccatellie S. (2010). Toxicology of alkylmercury com-
pounds. In: Sigel A., Sigel H., and Sigel R. (Eds), Metal Ions in Life Sciences, Vol. 7,
pp. 1—575. Royal Society of Chemistry, Cambridge, UK.
[5] . Auerbach E.A., Seyfried E.E., and McMahon K. D. (2007). «Tetracycline resistance genes
in activated sludge wastewater treatment plants». Water Research. 41, 5: 1143—1151.
[6] . Barcelo D. (Ed.) (1996). Applications of LC-MS in Environmental Chemistry. Elsevier, Am-
sterdam, Netherlands.
[7] . Beceiro-Gonzalez E., Guimaraes A., and Alpendurada M.F. (2009). «Optimisation of
a headspace-solid-phase micro-extraction method for simultaneous determination of orga-
nometallic compounds of mercury, lead and tin in water by gas chromatography—tandem
mass spectrometry». Journal of Chromatography A. 1216, 29: 5563—5569.
[8] . Busch K., Glish G., and McLuckey S. (1988). Mass Spectrometry/Mass Spectrometry: Tech-
niques and Applications of Tandem Mass Spectrometry. VCH Publishers, New York, USA.
[9] . Carson R. (1962). Silent Spring. Houghton Mifflin, Boston, Massachusetts, USA.
Литература
[10] . Chen Z., WesterhofTP., and Herckes P. (2008). «Quantification of C60 fullerene concentra-
tions in water». Environmental Toxicology and Chemistry. 27, 9: 1852—1859.
[11] . Colvin V. (2003). «The potential environmental impact of engineered nanomaterials». Na-
ture Biotechnology. 21, 10: 1166—1170.
[12] . Committee on Understanding Oil Spill Dispersants: Efficacy and Effects N. R.C. (2005).
Oil Spill Dispersants: Efficacy and Effects». The National Academies Press, Washington,
DC, USA.
[13] . Conley J.M., Symes S.J., Kindelberger S.A., and Richards S.M. (2008). «Rapid liquid
chromatography—tandem mass spectrometry method for the determination of a broad mix-
ture of pharmaceuticals in surface water». Journal of Chromatography A. 1185, 2: 206—215.
[ 14]. Craig P. (2003). Organometallic Compounds in the Environment. John Wiley, Chichester, U K.
[15] . Cui M., Ding L., and Mester Z. N. (2003). «Separation of cisplatin and its hydrolysis prod-
ucts using electrospray ionization high-field asymmetric waveform ion mobility spectrom-
etry coupled with ion trap mass spectrometry». Analytical Chemistry. 75, 21: 5847—5853.
[16] . Cullen W. R., Eigendorf G. K., Nwata B. U., and Takatsu A. (1990). «The quantitation of
butyltin and cyclohexyltin compounds in the marine environment of British Columbia».
Applied Organometallic Chemistry. 4, 6: 581—590.
[17] . Da Silva M. F., Tiago I., Ven'ssimo A., Boaventura R. A. R., Nunes О. C., and Manaia С. M.
(2006). «Antibiotic resistance of enterococci and related bacteria in an urban wastewater
treatment plant». FEMS Microbiology Ecology. 55, 2: 322—329.
[18] . Daughton C. (2001). «Emerging pollutants, and communicating the science of environ-
mental chemistry and mass spectrometry: pharmaceuticals in the environment». Journal of
the American Society of Mass Spectrometry. 12, 10: 1067—1076.
[19] . Daughton C., and Jones-Lepp T. (Eds) (2001). Pharmaceuticals and Personal Care Products
in the Environment: Scientific and Regulatory Issues, ACS Symposium Series 791. American
Chemical Society, Washington, DC, USA.
[20] . Daughton C., and Ternes T. (1999). «Pharmaceuticals and personal care products in the
environment: agents of subtle change?» Environmental Health Perspectives. 107, 6: 907—938.
[21] . Eskes C., Honegger P., Jones-Lepp T., Varner K., Matthieu J., and Monnet-Tschudi F.
(1999). «Neurotoxicity of dibutyltin in aggregating brain cell cultures». Toxicology in Vitro.
13: 555-560.
[22] . Farre, М.1., Ferrer I., Ginebreda A., Figueras M., Olivella L., Tirapu L., Vilanova M., and
Barcelo, D. (2001). «Determination of drugs in surface water and wastewater samples by
liquid chromatography—mass spectrometry: methods and preliminary results including
toxicity studies with Vibrio fischeri». Journal of Chromatography A. 938, 1—2: 187—197.
[23] . Farre M., Gajda-Schrantz K., Kantiani L., and Barcelo, D. (2009). «Ecotoxicity and analy-
sis of nanomaterials in the aquatic environment». Analytical and Bioanalytical Chemistry.
393, 1:81-95.
[24] . Gajda T., and Jancso , A. (2010). «Organotins, formation, use, speciation, and toxicology».
In: Sigel A., Sigel H., and Sigel R. (Eds). Metal Ions in Life Sciences. Vol. 7, pp. 111—151.
Royal Society of Chemistry, Cambridge, UK.
[25] . Garrison A., Pope J., and Allen F. (1976). GC/MS analysis of organic compounds in domestic
wastewaters. In: Keith С. H. (Ed.), Identification and Analysis of Organic Pollutants in Wa-
ter, pp. 517—556. Arbor Science Publishers, Ann Arbor, Michigan, USA.
380 Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
[26] . Geranio L., Heuberger М., and Nowack В. (2009). «The behavior of silver nanotextiles dur-
ing washing». Environmental Science and Technology. 43, 21: 8113—8118.
[27] . Grange A.H., and Sovocool G.W. (2008). «Automated determination of precursor ion,
product ion, and neutral loss compositions and deconvolution of composite mass spectra
using ion correlation based on exact masses and relative isotopic abundances». Rapid Com-
munications in Mass Spectrometry. 22, 15: 2375—2390.
[28] . Grayson M. (Ed.) (2002). Measuring Mass: From Positive Rays to Proteins. Chemical Herit-
age Press, Philadelphia, USA.
[29] . Grote K., Hobler C., Andrade A.J.M., Grande S.W., Gericke C., Talsness C.E., Ap-
pel К. E., and Chahoud I. (2007). «Effects of in utero and lactational exposure to triphenyl-
tin chloride on pregnancy outcome and postnatal development in rat offspring». Toxicology.
238, 2-3: 177-185.
[30] . Grote K., Hobler C., Andrade A.J.M., Grande S.W., Gericke C., Talsness С. E., Ap-
pel К. E., and Chahoud I. (2009). «Sex differences in effects on sexual development in rat
offspring after pre- and postnatal exposure to triphenyltin chloride». Toxicology. 260, 1—3:
53-59.
[31] . Grun F., and Blumberg B. (2006). «Environmental obesogens: organotins and endocrine
disruption via nuclear receptor signaling». Endocrinology. 147, 6: 50—55.
[32] . Guardabassi L., Wong D., and Dalsgaard A. (2002). «The effects of tertiary wastewa-
ter treatment on the prevalence of antimicrobial resistant bacteria». Water Research. 36:
1955-1964.
[33] . Hann S., Stefanka Z., Lenz K., and Stingeder G. (2005). «Novel separation method for
highly sensitive speciation of cancerostatic platinum compounds by HPLC—ICP—MS».
Analytical and Bioanalytical Chemistry. 381, 2: 405—412.
[34] . Hao C., Clement R., and Yang P. (2007). «Liquid chromatography—tandem mass spec-
trometry of bioactive pharmaceutical compounds in the aquatic environment—a decade’s
activities». Analytical and Bioanalytical Chemistry. 387, 4: 1247—1257.
[35] . Herbert C., and Johnstone R. (2003). Mass Spectrometry Basics. CRC Press, Boca Raton,
Florida, USA.
[36] . Hiatt M. H. (1995). «Vacuum distillation coupled with gas chromatography/mass spectrom-
etry for the analysis of environmental samples». Analytical Chemistry. 67, 22: 4044—4052.
[37] . Hobler C., Andrade A.J.M., Grande S.W., Gericke C., Talsness C.E., Appel K.E.,
Chahoud L, and Grote K. (2010). «Sex-dependent aromatase activity in rat offspring after
pre- and postnatal exposure to triphenyltin chloride». Toxicology. 276, 3: 198—205.
[38] . Hoch M. (2001). «Organotin compounds in the environment — an overview». Applied Geo-
chemistry. 16, 7—8: 719—743.
[39] . Hogenboom A.C., van Leerdam J. A., and de Voogt P. (2009). «Accurate mass screening
and identification of emerging contaminants in environmental samples by liquid chroma-
tography-hybrid linear ion trap Orbitrap mass spectrometry». Journal of Chromatography
A. 1216,3:510-519.
[40] . Howard A.G. (2010). «On the challenge of quantifying man-made nanoparticles in the
aquatic environment». Journal of Environmental Monitoring. 12, 1: 135—142.
[41] . Hu Q., Noll R., Li H., Makarov A., Hardman M., and Cooks R. (2005). «The Orbitrap:
a new mass spectrometer». Journal of Mass Spectrometry. 40: 430—443.
Литература
[42] . Huggett R., Unger M., Seligman P., and Valkirs A. (1992). «The marine biocide tributyltin:
assessing and managing the environmental risks». Environmental Science and Toxicology.
26, 2: 232-237.
[43] . Isaacson C.W., and Bouchard D. (2010). «Asymmetric flow field flow fractionation of aque-
ous C60 nanoparticles with size determination by dynamic light scattering and quantifica-
tion by liquid chromatography atmospheric pressure photo-ionization mass spectrometry».
Journal of Chromatography A. 1217, 9: 1506—1512.
[44] . Isaacson C.W., Usenko C.Y., Tanguay R. L., and Field J. A. (2007). «Quantification of
fullerenes by LC/ESI-MS and its application to in vivo toxicity assays». Analytical Chemis-
try. 79, 23: 9091-9097.
[45] . Johnson A. C., Jorgens M. D., Williams R. J., Kiimmerer K., Kortenkamp A., and Sumpt-
er J. P. (2008). «Do cytotoxic chemotherapy drugs discharged into rivers pose a risk to the
environment and human health? An overview and UK case study». Journal of Hydrology.
348, 1-2: 167-175.
[46] . Jones-Lepp T. L., and Momplaisir, G.-M. (2005). «New applications of LC-MS and LC-
MS2 toward understanding the environmental fate of organometallics». Trends in Analytical
Chemistry. 24, 7: 590—595.
[47] . Jones-Lepp T., Varner K., McDaniel M., and Riddick L. (1999). «Determination of or-
ganotins in water by micro liquid chromatography-electrospray/ion trap mass spectrom-
etry». Applied Organometallic Chemistry. 13: 881—889.
[48] . Jones-Lepp T. L., Alvarez D. A., Englert B., and Batt A. L. (2009). «Pharmaceuticals and
hormones in the environment». In: Meyers R. A. (Ed.). Encyclopedia of Analytical Chemis-
try. John Wiley, Chichester, UK.
[49] . Judson R. S., Martin M.T., Reif D. M., Houck K. A., Knudsen T. B., RotrofTD. M., Xia M.,
Sakamuru S., Huang R., Shinn P., Austin С. P., Kavlock R. J., and Dix D. J. (2010). «Analy-
sis of eight oil spill dispersants using rapid, in vitro tests for endocrine and other biological
activity». Environmental Science and Technology. 44, 15: 5979—5985.
[50] . Kaise T., Yamauchi H., Horiguchi Y, Tani T., Watanabe S., Hirayama T., and Fukui S.
(1989). «А comparative study on acute toxicity of methylarsonic acid, dimethylarsinic acid
and trimethylarsine oxide in mice». Applied Organometallic Chemistry. 3, 3: 273—277.
[51] . Kannan K., Takahashi S., Fujiwara N., Mizukawa H., and Tanabe S. (2010). Organotin
compounds, including butyltins and octyltins, in house dust from Albany, New York, USA.
Archives of Environmental Contamination and Toxicology. 58, 4: 901—907.
[52] . Kelly C. (2000). «Analysis of steroids in environmental water samples using solid-phase
extraction and ion-trap gas chromatography—tandem mass spectrometry». Journal of Chro-
matography A. 872: 309—314.
[53] . Kemper N. (2008). «Veterinary antibiotics in the aquatic and terrestrial environment». Eco-
logical Indicators. 8: 1—13.
[54] . Kidd K.A., Blanchfield P.J., Mills K.H., Palace V. P., Evans R. E., Lazorchak J. M., and
Flick R. W. (2007). «Collapse of a fish population after exposure to a synthetic estrogen».
Proceedings of the National Academy of Sciences of the United States of America. 104, 21:
8897-8901.
[55] . Kim S., and Aga D. S. (2007). «Potential ecological and human health impacts of antibiotics
and antibiotic-resistant bacteria from wastewater treatment plants». Journal of Toxicology
and Environmental Health, Part B: Critical Reviews. 10, 8: 559—573.
382 Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
[56] . Klaine S.J., Alvarez Batley G.E., Fernandes T. F., Handy R. D., Lyon D.Y., Ma-
hendra S., McLaughlin M. J., and Lead J. R. (2008). «Nanomaterials in the environment:
behavior, fate, bioavailability, and effects». Environmental Toxicology and Chemistry. 27, 9:
1825-1851.
[57] . Kosjek T., Heath E., Petrovic M., and Barcelo, D. (2007). «Mass spectrometry for identify-
ing pharmaceutical biotransformation products in the environment». Trends in Analytical
Chemistry. 26, 11: 1076—1085.
[58] . Krupp E., Milne B., Mestrot A., Meharg A., and Feldmann J. (2008). «Investigation into
mercury bound to biothiols: structural identification using ESI-ion-trap MS and introduc-
tion of a method for their HPLC separation with simultaneous detection by ICP-MS and
ESI-MS». Analytical and Bioanalytical Chemistry. 390, 7: 1753—1764.
[59] . Kiimmerer K. (2010). «Pharmaceuticals in the environment». Annual Review of Environ-
ment and Resources. 35, 1: 57—75.
[60] . Lenz K., Hann S., Koellensperger G., Stefanka Z., Stingeder G., Weissenbacher N., Mah-
nik S.N., and Fuerhacker M. (2005). «Presence of cancerostatic platinum compounds in
hospital wastewater and possible elimination by adsorption to activated sludge». Science of
the Total Environment. 345, 1—3: 141 — 152.
[61] . Lessard R. R., and DeMarco G. (2000). «The significance of oil spill dispersants». Spill Sci-
ence and Technology Bulletin. 6, 1: 59—68.
[62] . Lindberg R., Jarnheimer, P.-А., Olsen B., Johansson M., and Tysklind M. (2004). «Deter-
mination of antibiotic substances in hospital sewage water using solid phase extraction and
liquid chromatography/mass spectrometry and group analogue internal standards». Che-
mosphere. 57, 10: 1479—1488.
[63] . Loos R., Locoro G., and Contini S. (2010). «Occurrence of polar organic contaminants in
the dissolved water phase of the Danube River and its major tributaries using SPE-LC-MS2
analysis». Water Research. 44, 7: 2325—2335.
[64] . Lubick N. (2007). «DDT’s resurrection». Environmental Science and Technology. 41, 18:
6323-6325.
[65] . Lubick N. (2008). «Risks of nanotechnology remain uncertain». Environmental Science and
Technology. 42, 6: 1821-1824.
[66] . Mahaffey K., Rice G., Schoeny R., Swartout J., and Keating M. (1997). Mercury Study
Report to Congress, EPA-452/R-97-009: 1 — 152. US Environmental Protection Agency,
Washington, DC, USA.
[67] . McLafferty F. (1980). Interpretation of Mass Spectra, 3rd edition. University Science Books,
Mill Valley, California, USA.
[68] . McMahon P. B., Dennehy K. F., Bruce B. W., Bohlke J. K., Michel R. L., Gurdak J. J., and
Hurlbut D. B. (2006). «Storage and transit time of chemicals in thick unsaturated zones
under rangeland and irrigated cropland, High Plains, United States». Water Resources Re-
search. 42, 3: W03413.
[69] . Moeder M., Schrader S., Winkler M., and Popp P. (2000). «Solid phase microextraction-
gas chromatography mass spectrometry of biologically active substances in water samples».
Journal of Chromatography A. 873: 95—106.
[70] . Morabito R., Massanisso P., and Quevauviller P. (2000). «Derivatization methods for the
determination of organotin compounds in environmental samples». Trends in Analytical
Chemistry. 19, 2-3: 113-119.
Литература
[71] . Moreno M.J., Pacheco-Arjona J., Rodrfguez-Gonzalez P., Preud’Homme H., Amour-
oux D., and Donard O. F.X. (2006). «Simultaneous determination of monomethylmercury,
monobutyltin, dibutyltin and tributyltin in environmental samples by multi-elemental-spe-
cies-specific isotope dilution analysis using electron ionisation GC-MS». Journal of Mass
Spectrometry. 41, 11: 1491 — 1497.
[72] . Moser V., McGee J., and Ehman K. (2009). «Concentration and persistence of tin in rat
brain and blood following dibutyltin exposure during development». Journal of Toxicology
and Environmental Health, Part A. IT 47—52.
[73] . Newcombe C., Raab A., Williams P. N., Deacon C., Haris P. I., Meharg A. A., and Feld-
mann J. (2010). «Accumulation or production of arsenobetaine in humans?» Journal of En-
vironmental Monitoring. 12, 4: 832—837.
[74] . Niessen W. (2006). Liquid Chromatography—Mass Spectrometry. Marcel Dekker, New York,
USA.
[75] . Oudijk G. (2010). «The rise and fall of organometallic additives in automotive gasoline.»
Environmental Forensics. 11: 17—49.
[76] . Owen R., and Handy R. (2007). «Formulating the problems for environmental risk assess-
ment of nanomaterials». Environmental Science and Technology. 41, 16: 5582—5588.
[77] . Pelletier E. (1995). «Environmental organometallic chemistry of mercury, tin, and lead:
present status and perspectives». In: Tessier A., and Turner D. R. (Eds), Metal Speciation
and Bioavailability in Aquatic Systems, Vol. 3, pp. 103—148. John Wiley and Sons, Chich-
ester, UK.
[78] . Petrovic M., Hernando M. D., Di'az-Cruz M.S., and Barcelo, D. (2005). «Liquid chroma-
tography-tandem mass spectrometry for the analysis of pharmaceutical residues in envi-
ronmental samples: a review». Journal of Chromatography A. 1067, 1—2: 1 — 14.
[79] . Richardson S. D. (2010). «Environmental mass spectrometry: emerging contaminants and
current issues». Analytical Chemistry. 82, 12: 4742—4774.
[80] . Rosenblatt-Farrell N. (2009). «The landscape of antibiotic resistance». Environmental
Health Perspectives. 117, 6: A244—A250.
[81] . Sano T., Takagi H., Nagano K., and Nishikawa M. (2010). «Analysis of triorganotin com-
pounds in water samples by hydrophilic interaction liquid chromatography—electrospray
ionization—mass spectrometry». Journal of Chromatography A. 1217, 26: 4344—4346.
[82] . Schliiter A., Szczepanowski R., Ptihler A., and Top E. (2007). «Genomics of IncP-1 anti-
biotic resistance plasmids isolated from wastewater treatment plants provides evidence for a
widely accessible drug resistance gene pool». FEMS Microbial Review. 31: 449—477.
[83] . Schwartz T., Kohnen W., Jansen B., and Obst U. (2003). «Detection of antibiotic-resistant
bacteria and their resistance genes in wastewater, surface water, and drinking water bio-
films». FEMS Microbiology Ecology. 43, 3: 325—335.
[84] . Segovia-Martinez L., Bouzas-Blanco A., Campins-Falco, P., and Seco-Torrecillas A.
(2010). «Improving detection limits for organotin compounds in several matrix water sam-
ples by derivatization-headspace-solid-phase microextraction and GC-MS». Taianta. 80,
5: 1888-1893.
[85] . Siu K. W. M., Gardner G. J., and Berman S. S. (1989). «Ion spray mass spectrometry/mass
spectrometry: quantitation of tributyltin in a sediment reference material for trace metals».
Analytical Chemistry. 61, 20: 2320—2322.
Глава 13. Новые типы приоритетных загрязняющих веществ в окружающей среде
[86] . Snyder S., Keith Т., Verbrugge D., Snyder E., Gross T., Kannan K., and Giesy J. (1999).
«Analytical methods for detection of selected estrogenic compounds in aqueous mixtures».
Environmental Science and Technology. 33: 2814—2820.
[87] . Sousa A., Laranjeiro F., Takahashi S., Tanabe S., and Barroso С. M. (2009). «Imposex and
organotin prevalence in a European post-legislative scenario: temporal trends from 2003 to
2008». Chemosphere. 77, 4: 566—573.
[88] . Sun Q., Chen Z., Yuan D., Megharaj M., and Naidu R. (2009). «On-line solid-phase ex-
traction coupled with liquid chromatography/electrospray ionization mass spectrometry for
the determination of trace tributyltin and triphenyltin in water samples». Rapid Communi-
cations in Mass Spectrometry. 23, 23: 3795—3802.
[89] . Szczepanowski R., Linke B., Krahn I., Gartemann, К.-H., Gotzkow T., Eichler W., Puh-
ler A., and Schluter A. (2009). «Detection of 140 clinically relevant antibiotic resistance
genes in the plasmid metagenome of wastewater treatment plant bacteria showing reduced
susceptibility to selected antibiotics». Microbiology, mic.0.028233-028230.
[90] . Ternes T. (1998). «Occurrence of drugs in German sewage treatment plants and rivers».
Water Research. 32: 3245—3260.
[91] . Ternes T., Andersen H., Gilberg D., and Bonerz M. (2002). «Determination of estrogens
in sludge and sediments by liquid extraction and GC/MS/MS». Analytical Chemistry. 74:
3498-3504.
[92] . Tessier A., and Turner D. R. (1995). Metal Speciation and Bioavailability in Aquatic Systems.
John Wiley, Chichester, UK.
[93] . Thomaidis N., Stasinakis A., Gatidou G., Morabito R., Massanisso P., and Lekkas T.
(2007). «Occurrence of organotin compounds in the aquatic environment of Greece». Wa-
ter, Air, Soil Pollution. 181, 1: 201—210.
[94] . USEPA (2008). 2006 Inventory Update Reporting: Data Summary, pp. 1—41. Office of
Pollution Prevention and Toxics, US Enviromental Protection Agency, Washington, DC,
USA.
[95] . USTC (1919). Census of Dyesand Coal—Tar Chemicals, Tariff Information Series, Vol. 22,
p. 110. United States Tariff Commission, Washington, DC, USA.
[96] . Vanderford B., Pearson R., Rexing D., and Snyder S. (2003). «Analysis of endocrine disrup-
tors, pharmaceuticals, and personal care products in water using liquid chromatography/
tandem mass spectrometry». Analytical Chemistry. 75: 6265—6274.
[97] . Wang J. (2009). «Analysis of macrolide antibiotics, using liquid chromatography—mass
spectrometry, in food, biological and environmental matrices». Mass Spectrometry Reviews.
28, 1:50-92.
[98] . Watts C. D., Crathorn B., Fielding M., and Steel С. P. (1983). Identification of non-volatile
organics in water using field desorption mass spectrometry and high performance liquid chroma-
tography. Proceedings of the Third European Symposium on Analysis of Organic Micropo-
llutants in Water. D. Reidel Publishing Company, Oslo, Norway.
[99] . Weinberg H., Galyean A., and Leopold M. (2011). «Evaluating engineered nanoparticles in
natural waters». Trends in Analytical Chemistry. 30, 1: 72—83.
[100] . Winship K. (1988). «Toxicity of tin and its compounds». Adverse Drug Reactions and Acute
Poisoning Reviews. 7, 1: 19—38.
[101] . Woodrow W. I. (2010). The Project on Emerging Nanotechnologies. Woodrow Wilson Insti-
tute, Washington, DC, USA.
Литература
[102] . Yang S., Cha J., and Carlson K. (2004). «Quantitative determination of trace concentra-
tions of tetracycline and sulfonamde antibiotics in surface water using solid-phase extrac-
tion and liquid chromatography/ion trap tandem mass spectrometry». Rapid Communica-
tions in Mass Spectrometry. 18: 2131—2145.
[103] . Zhang Y., Marrs C., Simon C., and Xi C. (2009). «Wastewater treatment contributes to
selective increase of antibiotic resistance among Acinetobacter spp». Science of the Total
Environment. 407: 3702—3706.
ГЛАВА 14
АНАЛИЗ ПЕСТИЦИДОВ
В ОБРАЗЦАХ ОБЪЕКТОВ
ОКРУЖАЮЩЕЙ СРЕДЫ
СОВРЕМЕННЫМИ
МЕТОДАМИ
ХРОМАТО МАСС-
СПЕКТРОМЕТРИИ
Ф. Эрнандес, М. Ибаньес, Т Портолес
14.1. Введение
Пестицид — это вещество или смесь веществ, предназначенных для уничтожения или
отпугивания вредителей (паразитов) в сельком хозяйтве, в домах и на приусадебных
участках. Класс пестицидов помимо инсектицидов включает также гербициды, фун-
гициды и некоторые другие вещества, используемые в борьбе с паразитами. Благода-
ря интенсивному использованию химических соединений в современном сельском
хозяйстве в последние десятилетия их уровни и ассортимент в объектах окружающей
среды значительно увеличились. Несмотря на очевидную пользу от использования пе-
стицидов, их применение может представлять определенную проблему для здоровья
человека и экосистемы в целом. Поскольку были выявлены негативные эффекты ранее
использовавшихся высокоустойчивых пестицидов, их регулирование во всех странах
стало строже, а производители были вынуждены разрабатывать менее персистентные
соединения, обладающие, кроме того, меньшей токсичностью для нецелевых организ-
мов флоры и фауны. Хотя современные типы пестицидов достаточно лабильны (хорошо
разлагаются в окружающей среде), их широчайшее использование не позволяет полно-
стью избежать их присутствия в окружающей среде.
В последние годы в связи с ростом озабоченности потенциальными негативными
эффектами, связанными с присутствием пестицидов в окружающей среде, создано до-
статочно много разнообразных аналитических методов для мониторинга и проведения
надежных измерений их уровней в воде, воздухе и почве. Поскольку ассортимент пе-
стицидов сегодня очень велик, наиболее эффективный подход связан с применением
мультикомпонентных методов анализа, которые могут определять как можно больше
соединений одновременно. Большинство разработанных методик оперируют десятка-
14,1. Введение
ми целевых соединений, хотя с появлением более быстрых и чувствительных методов
их число возросло до 100—200 и более. Обычно методы направлены на детектирование
целевых пестицидов. В результате теряется информация о многих других соединениях,
которые потенциально могут присутствовать в анализируемом образце.
Несмотря на превосходную работу современных аналитических приборов и по-
лезные новшества в подготовке проб, иногда в связи со специфическими физико-хи-
мическими характеристиками некоторых пестицидов для них должны использовать-
ся индивидуальные методы надежного количественного определения. Это замечание
справедливо, например, для гербицидов типа глифосата, глуфосината, параквата или
диквата, фунгицидов типа фосэтил-А1, или дитиокарбаматов, или регулятора роста
этефона. Эти соединения не могут быть включены в список аналитов для одновремен-
ного мультикомпонентного анализа. Их требуется определять индивидуально.
Разработка мультикомпонентных методик для анализа пестицидов представляет
собой сложную по нескольким причинам задачу. Основная проблема связана с разли-
чиями физико-химических характеристик индивидуальных пестицидов. Например,
они могут присутствовать в образцах в форме катионов, анионов или нейтральных ча-
стиц в широком диапазоне полярностей. В результате становится очень сложно создать
интегральный метод, включающий и пробоподготовку, и аналитические измерения
для всех них одновременно. Проблема усугубляется необходимостью вести мониторинг
продуктов трансформации (ПТ) исходных соединений, поскольку зачастую их уровни
в объектах окружающей среды значительно выше, чем самих пестицидов (Hernandez
et aL, 2008, Kolpin et aL, 2000, Kolpin et aL, 2001, Rebich et aL, 2004). Этот факт значитель-
но увеличивает число целевых аналитов. Большая полярность большинства ПТ влияет
на эффективность пробоподготовки (например концентрирования), а хроматографиче-
ское поведение является дополнительной трудностью при создании мультикомпонент-
ных методик. Среди ПТ пестицидов, все чаще встречающихся в воде, можно выделить
производные триазиновых гербицидов. В частности, нашей группой при анализе под-
земных вод часто регистирируются дезизопропилатразин, 2-ОН-тербутилазин, дезэ-
тилтербутилазин, дезэтил-2-ОН-тербутилазин и дезэтилтербуметон, причем уровни
этих ПТ существенно выше, чем исходных гербицидов (Marin etal., 2006, Hernandez etaL,
2008).
Еще одна проблема анализа остаточных количеств пестицидов связана с их очень
низкими уровнями в образцах, оченьжесткими нормативами (например ПДК0,1 мкг/л)
и сложностью матрицы природного образца. Это заставляет аналитиков разрабатывать
высокоселективные и высокочувствительные методики на базе усложненных инстру-
ментальных методов, например хроматографии (и газовой и жидкостной) с различны-
ми видами детекторов. Обычные детекторы (УФ- или флуоресцентный для ЖХ и ЭЗД,
ПИД или азотно-фосфорный для ГХ) обладают недостаточной чувствительностью или
не дают возможности надежно идентифицировать детектируемый сигнал. В настоящее
время общепризнанно, что однозначная идентификация вещества может быть прове-
дена исключительно масс-спектрометрически, поскольку именно этот метод предо-
ставляет надежную структурную информацию. Поэтому стыковка масс-спектрометра
с ГХ или ЖХ является оптимальным комбайном для определения следов пестицидов
в объектах окружающей среды. Два последних десятилетия предпочтение отдавалось
ГХ/МС, однако ЖХ/МС становится все популярнее. Причиной является то, что совре-
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
менные пестициды более полярны и менее летучи, а следовательно, определение их ПТ
и самих пестицидов эффективнее вести с использованием жидкостной хроматографии.
В настоящее время и ГХ/МС и ЖХ/МС, будучи комплементарны друг другу, востребо-
ваны для целей анализа следов пестицидов в равной мере.
14.2. Анализ остаточных количеств пестицидов
Анализ остаточных количеств пестицидов (АОКП) — это целое направление в анали-
тической химии, крайне востребованное в последние годы. Как и в смежных областях,
связанных с определением микроколичеств органических соединений, в этом случае
существует ряд требований, направленных на повышение надежности результатов.
Среди этих требований первостепенное значение имеет квалификация химика-анали-
тика, который должен досконально знать не только пробоподготовку и варианты ана-
лиза следовых количеств соединений, но также иметь опыт работы с современными
ГХ/МС- и ЖХ/МС-системами и набором масс-анализаторов. Для проведения АОКП
необходимо принимать во внимание несколько этапов: пробоотбор, обработка образца
(обычно экстракция и/или концентрирование), очистка полученных экстрактов и про-
ведение измерений хроматографическими методами (иногда на этой стадии требуется
дериватизация). Описание всех этих стадий выходит за рамки данной главы. Поэтому
для большинства из них приводится лишь короткое резюме, а основное внимание уде-
лено аналитическому применению ГХ/МС и ЖХ/МС.
Одним из важнейших аспектов является пробоотбор. Образцы почвы, воды и воз-
духа должны быть репрезентативными, в частности много внимания уделяется гомо-
генизированию. Отобранные образцы желательно хранить до анализа при температуре
< — 18°С.
Пробоподготовка зависит от природы матрицы и самих аналитов. В настоящее вре-
мя явно прослеживается тренд на упрощение соответствующих процедур, что оправ-
дано улучшениями селективности и чувствительности современных аналитических
приборов. Более не рекомендовано использование больших объемов растворителей
по причине их высокой стоимости, опасности для персонала и проблем с утилизацией
отходов. Мульти компонентные методы требуют применения простого и «универсаль-
ного» метода подготовки пробы, который бы позволял определять широкий круг соеди-
нений с различными физико-химическими свойствами.
Существует определенный набор процедур пробоподготовки для экстракции и кон-
центрирования пестицидов из образцов объектов окружающей среды. Твердофазная
экстракция (ТФЭ), пожалуй, наиболее востребована для работы с жидкими пробами
(например вода). Другие подходы базируются на экстракции растворителями (образцы
воды, почвы, биоты). Эффективность экстракционных методов может быть существен-
но улучшена при использовании жидкостной экстракции при повышенном давлении.
Твердофазная микроэкстракция (ТФМЭ) — одна из последних разработок, которая на-
шла широкое применение в последнее десятилетие для обработки водных проб. Обычно
она используется в комбинации с ГХ, хотя есть публикации и о вариантах ТФМЭ-ЖХ.
При работе со сложными матрицами перед ГХ/МС- или ЖХ/МС-анализом обыч-
но требуется очистка экстракта. Хотя эта стадия может стать причиной аналитических
14.2. Анализ остаточных количеств пестицидов 389
ошибок (большее число манипуляций с образцом) и снизить мультикомпонентность
метода, ее нельзя обойти во многих случаях, когда требуется удалить мешающие опре-
делению ингредиенты или улучшить чувствительность и пределы обнаружения. Обыч-
но для этих целей используется жидкость-жидкостная экстракция (ЖЖЭ) или препара-
тивная жидкостная хроматография.
В некоторых случаях для того чтобы сделать аналит более удобным для анализа,
необходима специальная обработка (например дериватизация). В частности, перед
ЖХ/МС-анализом глифосата проводят его дериватизацию флуоренилметилоксикарбо-
нилхлоридом (FMOC).
Как уже отмечалось во введении, уровни ПТ пестицидов в объектах окружающей
среды могут быть выше, чем самих пестицидов (Kolpin et al., 2000, Hernandez et al., 2008).
Иногда ПТ оказываются токсичнее родительских соединений. ПТ включены в списки
нормируемых для питьевой воды соединений в качестве «веществ, связанных с пести-
цидами» (Council Directive 98/83/ЕС). Кроме того, благодаря повышенной мобильности
в системе почва-вода, ПТ могут легче проникать в подземные воды. Поэтому наиболее
важные продукты трансформации должны включаться в методики аналитического
контроля, чтобы иметь более реалистичную картину загрязнения воды пестицидами.
Включение ПТ в мультикомпонентные методики представляет определенные слож-
ности по нескольким причинам: зачастую информация об этих соединениях весьма
скудна; существенно возрастает число аналитов, которые надо определить одновре-
менно; обычно ПТ полярнее исходных соединений, а их экстрагирование/концен-
трирование из воды сложнее; коммерческая доступность стандартных образцов этих
соединений оставляет желать лучшего. Кроме того, добавление новых аналитов в ме-
тодику на базе масс-спектрометрии приводит к увеличению числа мониторируемых
ионов (реакций). В результате наблюдается либо потеря чувствительности, либо ухуд-
шение качества регистрируемых пиков. Более того, больший разброс физико-химиче-
ских характеристик аналитов усложнит оптимизацию экспериментальных условий их
одновременного определения. Все же основной проблемой количественных методов
определения ПТ является их полярность, усложняющая стадии извлечения и концен-
трирования с использованием ТФЭ или ЖЖЭ (Hernandez et al., 2005а).
Число масс-спектрометрических методик (особенно МС/МС, глава 4) для целей
АОКП резко растет. При разработке новой методики следует оптимизировать и масс-
спектрометрические, и хроматографические условия, достигая удовлетворительного
разделения компонентов и адекватного сигнала детектора. На сегодняшний день бла-
годаря своей чувствительности и селективности ГХ/МС/МС и ЖХ/МС/МС наиболее
удобны для определения пестицидов и их ПТ в образцах окружающей среды. Тандемная
масс-спектрометрия может быть реализована в варианте МС/МС в пространстве (на-
пример тройные квадруполи в режиме мониторинга заданных реакций) или в варианте
МС/МС во времени (например ионные ловушки в режиме регистрации ионов-продук-
тов). В обоих случаях предварительно выбранный и выделенный ион-предшественник
подвергается фрагментации с регистрацией одного или нескольких ионов-продуктов
(глава 4).
Необходимость поверки аналитических методик общеизвестна. Для ме-
тодов АОКП для целей поверки полезными являются документы SANCO
(http://ec.europa.eu/food/plant/protection/resources/qualcontrol_en.pdf). Процедура обы ч но
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
включает оценку нескольких аналитических параметров, включая воспроизводимость,
правильность, селективность/специфичность, линейность, пределы определения и де-
тектирования. Правильность (обычно оцениваемая в экспериментах по извлечению
аналитов) и воспроизводимость (выраженная величиной стандартного отклонения)
обычно оцениваются при анализе бланка и образцов с добавками аналитов в разных
концентрациях (обычно ppb- или суб-ррЬ-уровни).
Для того чтобы провести точное количественное определение, необходимо прини-
мать во внимание матричные эффекты, которые могут существенно искажать резуль-
таты ЖХ/МС/МС-анализа. Искажения обусловлены влиянием коэкстрагированных
компонентов матрицы на эффективность ионизации аналита в условиях атмосферного
давления. Матричные эффекты обычно проявляются как резкое снижение чувстви-
тельности из-за подавления ионизации аналита соединениями матрицы. Иногда может
наблюдаться и рост аналитического сигнала. Матричный эффект различен для любой
комбинации матрицы аналит/вещество. Отдельные аспекты этого явления будут рас-
смотрены в разделе ЖХ/МС. Усиление или ослабление регистрируемого сигнала ана-
лита в результате эффекта матрицы может наблюдаться и при использовании ГХ/МС.
Усиление может быть результатом конкуренции компонентов матрицы и целевого пе-
стицида за доступные активные точки ГХ-системы (инжектор и колонка). Кроме того,
компоненты образца могут защитить аналит от деструкции в горячем инжекторе. Ос-
лабление сигнала связано, например, с аккумулированием нелетучих соединений
в хроматографической системе, что может привести к созданию новых активных точек.
В любом случае необходимо учитывать матричные эффекты, для того чтобы получать
правильные количественные результаты (Garrido Frenich etal., 2009).
Хотя большинство МС-методов обладают хорошей селективностью, некоторые ком-
поненты матрицы могут обладать характеристиками, подобными аналиту. Поэтому сле-
дует учитывать возможность появления и ложноположительных, и ложноотрицатель-
ных результатов. Методики количественного определения должны быть надежными.
Однако еще важнее, чтобы соединение было правильно идентифицировано, особенно
когда полученный результат превышает ПДК данного вещества.
В идеале идентификация и подтверждение должны быть надежными, объектив-
ными и не зависеть от субъективной интерпретации результатов аналитиком. Следова-
тельно, необходимы эффективные заранее разработанные правила. Несмотря на важ-
ность этого положения, насколько нам известно, на сегодня не существует каких-либо
специальных правил для подтверждения идентификации экотоксиканта в образцах
объектов окружающей среды. Вероятно, по этой причине обычно применяются ин-
струкции, разработанные для смежных анализов: WADA 2004, FDA 2001, SANCO 2010,
или постановление Европейской комиссии 2002/657/ЕС.
Один из самых детализированных критериев был предложен в Европейском Союзе
для подтверждения присутствия ксенобиотиков в живых животных и продуктах живот-
новодства (Decision, 2002/657/ЕС), которое основано на получении определенного числа
идентификационных очков (ИО). Этот критерий очень полезен для быстрого и надеж-
ного сравнения правильности масс-спектрометрических результатов. Число ИО зависит
от использованного подхода: МС или МС/МС, масс-спектрометрия высокого или низ-
кого разрешения. К сожалению, существующие инструкции базируются только на ко-
личественной (число ионов или ИО), а не на качественной стороне информации. А ведь
выбор конкретных ионов/реакций может быть неочевидным и привести к низкому
качеству результатов, недостаточному для надежного подтверждения идентификации
(Pozo et aL, 2006). Принято регистрировать по крайней мере 3 иона в режиме МЗИ или
2 реакции фрагментации в режиме МЗР. При этом должно приниматься во внимание
время удерживания, а отношение интенсивностей зарегистрированных ионов должно
соответствовать стандартному спектру аналита. Выполнение этих условий позволяет
получить информацию, достаточную для надежного подтверждения положительных
проб (детали в главе 3).
14.3. ГХ/МС
ГХ/МС — популярный метод определения многих приоритетных загрязняющих орга-
нических веществ (обычно летучих или полулетучих, термически стабильных и низко-
полярных) в объектах окружающей среды (Mekebri et al., 2008, van der Hoof et al., 1999,
Geerdinket aL, 2002, van Leewen etal., 2008, Santos etal., 2003). Он объединяет возможности
ГХ высокого разрешения в качестве метода разделения компонентов с выдающимися
характеристиками МС в качестве метода детектирования и установления структуры
(глава 2). Эти характеристики позволяют разрабатывать аналитические высокочувстви-
тельные и надежные методики. ГХ/МС стал одним из наиболее мощных методов АОКП
в воде, почве, седиментах, биоте и пищевых продуктах. Типичными целевыми аналита-
ми в этом случае являются хлорорганические инсектициды и акарициды, пиретроиды,
многочисленные фосфорорганические инсектициды, фунгициды и гербициды. Время-
затратная процедура дериватизации в некоторых случаях призвана улучшить хромато-
графические характеристики или увеличить стабильность и регистрируемость целевых
высокополярных и нелетучих соединений.
Что касается методов ионизации, то наиболее популярны в ГХ/МС электронная
(ИЭ) и химическая (ХИ), с явным преобладанием первой (глава 1). ИЭ обычно характе-
ризуется интенсивной фрагментацией, причем спектры воспроизводимы. Это упрощает
эффективное использование доступных библиотек масс-спектров для целей идентифи-
кации. Однако интенсивная фрагментация может быть и недостатком, поскольку чув-
ствительности ИЭ может не хватать в связи с низкой интенсивностью молекулярного
иона. Напротив, ХИ — мягкий метод ионизации, характеризующийся незначительной
фрагментацией, которая к тому же может контролироваться варьированием газа-реа-
гента (глава 1). ХИ облегчает установление молекулярной массы соединения, но предо-
ставляет существенно меньше структурной информации. Хотя ХИ обеспечивает для
ряда соединений лучшую, по сравнению с ИЭ, чувствительность и селективность, она
редко используется в мультикомпонентных методиках, будучи менее универсальным
методом. Вдобавок воспроизводимость результатов с применением ХИ ниже, что делает
проблематичным создание и использование соответствующих спектральных библио-
тек (глава 5).
Квадрупольные анализаторы наиболее часто применяются в ГХ/МС для целей
определения органических соединений благодаря своей низкой стоимости, компакт-
ности и простоте (Filho et aL, 2010, Gomez et aL, 2009). Они работают в режиме полного
сканирования спектра с получением классических воспроизводимых масс-спектров
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
с единичным разрешением. Однако этот режим редко применим для АОКП из-за недо-
статочной чувствительности. В этом случае необходимо работать в режиме мониторин-
га заданных ионов (МЗИ), когда регистрируются только ионы с заранее установленны-
ми величинами m/z. Обычно для каждого аналита выбираются 2—3 величины m/z для
проведения количественного определения (по наиболее интенсивному иону) и иденти-
фикации (по соотношению интенсивностей всех зарегистрированных). В этих условиях
чувствительность и селективность анализа улучшаются, но при этом теряется вся ин-
формация, которая была бы доступна в режиме полного сканирования спектра (глава 2).
ГХ/МС в режиме МЗИ обычно используется для количественного определения, тогда
как сообщения о надежной идентификации соединений этим методом крайне редки.
Несмотря на широкое применение этого метода для определения пестицидов в объектах
окружающей среды работа со следовыми уровнями в сложных матрицах оказывается
проблематичной в связи с мешающим влиянием ингредиентов.
Тандемная масс-спектрометрия более эффективна для целей АОКП благодаря по-
вышенной селективности и существенному сокращению фонового химического шума.
Условия МС/МС позволяют минимизировать или даже полностью устранить многочис-
ленные интерференции, влияющие на анализ на одиночном квадруполе, в связи с воз-
можностью выбора оптимальных иона-предшественника и иона-продукта (глава 4).
Ионные ловушки (ИЛ) и тройные квадруполи (QqQ) позволяют вести работу в режи-
ме МС/МС с великолепной чувствительностью и селективностью. Возможность реги-
стрировать спектры ионов-продуктов в сочетании с простотой использования и низкой
стоимостью сделали ИЛ хорошим методом для АОКП в сложных матрицах. Однако
возможность проведения двух стадий анализа методом МС/МС с использованием QqQ
дает возможность работы в режиме МЗР, который является наиболее селективным
и чувствительным для идентификации и количественного определения органических
экотоксикантов в объектах окружающей среды.
Можно отметить несколько различий между ИЛ и QqQ, когда речь идет о качествен-
ном и количественом определении органических соединений. QqQ чувствительнее
в режиме МС/МС, что позволяет добиваться низких пределов обнаружения для целе-
вых соединений. Подтверждение правильности идентификации достигается записью
полного спектра ионов-продуктов (ИЛ) или регистрацией нескольких реакций ион-
предшественник — ион-продукт в режиме МЗР (QqQ). С точки зрения качественного
анализа ИЛ может выглядеть предпочтительнее благодаря режиму МС" для выявления
всех направлений фрагментации, что очень важно при идентификации нецелевых со-
единений. ИЛ также обладают большей чувствительностью при полном сканировании
спектра в режиме одномерной МС. Недостатком ИЛ является недоступность информа-
ции об ионах-продуктах с величинами m/z ниже 30 % от массы иона-предшественника.
Это связано с невозможностью приложения стабильного удерживающего потенциала
и к иону-предшественнику, и к ионам-продуктам. В заключение еще раз подчеркнем,
что ИЛ предпочтительнее для целей качественного анализа, a QqQ — для целей коли-
чественного анализа, хотя выбор достаточного числа реакций фрагментации для мони-
торирования в режиме МС/МС (по крайней мере два специфических процесса) делает
QqQ мощным методом и для подтверждения правильности идентификации. Опреде-
ленный тренд в АОКП, который можно выявить на основании публикаций последнего
десятилетия, связан с определенным сдвигом в сторону ГХ/МС/МС с QqQ.
14.3. ГХ/МС
Хорошей иллюстацией потенциала этого метода для анализа образцов из объектов
окружающей среды служит результат, полученный недавно в нашей лаборатории (Pi-
tarch et aL, 2007). Была разработана мульти компонентная методика скрининга, иден-
тификации и количественного определения микроколичеств органических загрязня-
ющих веществ в воде с помощью TX-(QqQ)MC/MC. В методике фигурировало около 50
соединений разных классов: 19 хлорорганических и фосфорорганических пестицидов,
6 гербицидов, 7 полихлорированных бифенилов, 16 ПАУ, 2 бромированных дифенило-
вых эфира, 3 октил/нонил-фенола и пентахлорбензол. Большинство этих соединений
включено в список приоритетных экотоксикантов в рамках Водного акта Европейско-
го Союза. ТФЭ на картриджах С18 использована для выделения аналитов, причем пять
изотопномеченых стандартов вносились в образец перед экстракцией в качестве сурро-
гатов. Анализ проводился методом ГХ/МС/МС в режиме ИЭ. Высокая селективность
и чувствительность в режиме МЗР позволила достичь удовлетворительных пределов
количественного определения на уровне 25 нг/л, а в некоторых случаях еще ниже. Пре-
делы обраружения методики находились в диапазоне 5—15 нг/л. Две МС/МС-реакции
MUE199
100-1
о-к-
7 50
MUE199
100-1
Симазин
2,2 мкг/л
8: MRM of 2 Channels EI+
201.01 > 173
3.02e3
Area
8.00
8.50 ' 9.00 ' 9.50
Кондиционирование:
6 мл МеОН
6 мл AcOEt:DCM (1:1)
6 мл МеОН
6 мл деионизованной воды
Промывание:
6 мл деионизованной воды
Элюирование:
5 мл AcOEt:DCM (1:1)
0-k-
8.00
8.50
Тербутилазин
1,2 мкг/л
MRM of 5 Channels EI+
214 >132
1 86еЗ
Area
I Упаривание при 40°С
1 в атмофере азота
Растворение, 0,5 мл гексана
Экстракт
MUE199
100-1
9.50
-1-п Time
10.00
16: MRM of 6 Channels EI+
11 82 198.9 >170.8
0-I...........I ' ' ' ' I.......I ' f ' '
11 00 11.50 12.00 12.50
MUE199
100-1
19 MRM of 2 Channels EI+
13 51 267 >158 9
134 “| 3 07e3
I Area
ГХ/QqQ/MC/MC
5?-
Хлорфенвинфос
1,0 мкг/л
* Образец разбавляется в 50 раз в случае
сточных вод с высоким содержанием органики
o-k-
12.50
। ..ргтт; । Time
13.00 13 50 14.00 14.50
a
a
Рис. 14.1. Схема последовательности операций определения следовых количеств ор-
ганических экотоксикантов в воде методом TX-(QqQ)MC/MC. ГХ/МС/МС-
хроматограммы, демонстрирующие детектирование симазина (2,2 мкг/л), тер-
бутилазина (1,2 мкг/л), хлорпирифоса (0,4 мкг/л) и хлорфенвинфоса (1,0 мкг/л)
в сточных водах завода по переработке бытовых отходов. DCM — дихлорметан
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
фрагментации регистрировались для каждого аналита, а отношение Q/q использова-
лось в качестве параметра подтвеждения идентификации. Параметр Q/q опредляет-
ся как отношение интенсивностей пиков мониторируемых реакций (или ионов), ис-
пользуемых для количественного определения (Q) и для подтверждения (q). В качестве
примера на рис. 14.1 представлена схема последовательности операций и ГХ/МС/МС-
хроматограммы для некоторых зарегистрированных аналитов: симазин (2,2 мкг/л),
тербутилазин (1,2 мкг/л), хлорпирифос (0,4 мкг/л) и хлорфенвинфос (1,0 мкг/л), кото-
рые были обнаружены в матрицах высокой сложности, например в необработанных
сточных водах завода по переработке бытовых твердых отходов (образец был разбавлен
в 50 раз перед процедурой ТФЭ в связи с очень высокой концентрацией органических
соединений). Рис. 14.2 иллюстрирует подтверждение присутствия двух из указанных
пестицидов (тербутилазина и хлорфенвинфоса) в данном образце. Наличие хроматогра-
фического пика с ожидаемым временем удерживания и выполнение условия правиль-
MUE188
MUE199
Стандарт 10 мкг/л
11 MRM of 5 Channels EI+
9 00;35.35
11 MRM of 5 Channels EI+
229 > 172
649
Area
Экстракт образца
11 MRM of 5 Channels El-
8 96;29 33
11 MRM of 5 Channels EI+
229> 172
415
Area
MUE188
MUE199
Отношение Q/q
для стандарта = 3,85
Отношение Q/q
для образца = 3,99
3.6% Z
9.00
5
Time
7.00
8.00
19 MRM of 2 Channels EI+
MUE188
О
S
О
...............
9.00 10.00 11.00
13.59.105.86
323 > 266 9
1 24еЗ
Area
1.2
5
8.00
7.00
10.00 11.00
13.51:97.75
Отношение Q/q
для образца = 1,48
323 > 266 9
1 ЗОеЗ
Area
12.00
13.00
Отношение Q/q
для стандарта = 1,60
14.00
15.00
7.5% Z
1.0pg/L
12.00
13.00
14.00
-i-i Time
15.00
q
q
5
q
5
q
Рис. 14.2. Подтверждение детектирования тербутилазина и хлорфенвинфоса в сточных
водах завода по переработке бытовых отходов. Q: мониторируемая реакция для
количественного определения; q: реакция для подтверждения идентификации.
< отношение Q/q внутри разрешенного диапазона; St: стандарт; W: образец
воды
ного отношения Q/q при сравнении со стандартом позволяет безошибочно определять
указанные экотоксиканты в образцах.
14.4. ЖХ/МС
До 1990-х именно неполярные вредные вещества составляли основу списков приори-
тетных экотоксикантов, причем в качестве предпочтительного метода анализа доми-
нировала ГХ/МС. На сегодняшний день большинство пестицидов представляет собой
полярные соединения, а их ПТ еще полярнее, менее летучи и обладают большей раство-
римостью в воде. Естественно, для работы с ними правильнее использовать ЖХ/МС.
После нескольких лет упорных работ трудности, связанные с переводом аналитов
из жидкой фазы в высоковакуумную газовую, были преодолены. Были созданы мето-
ды ионизации при атмосферном давлении (ИАД). Электрораспылительная ионизация
(ЭРИ) и химическая ионизация при атмосферном давлении (ХИАД), безусловно, наи-
более часто используются среди них для определения органических микропримесей
в образцах объектов окружающей среды (главы 1 и 3). Эти методы крайне полезны для
работы с широким кругом соединений без дериватизации, включая полярные (даже
ионные), нелетучие и термолабильные. Эти методы могут считаться в определенной
мере комплементарными, поскольку ХИАД обычно оперирует веществами с низкой
и средней полярностью, а ЭРИ — веществами от средней до высокой полярности. Бо-
лее того, оба метода обладают высокой чувствительностью, что обязательно в изучении
объектов окружающей среды, когда загрязняющие вещества присутствуют на уровнях
нг/л — мкг/л.
Важным достоинством ЖХ/МС является возможность прямого введения водной
пробы в хроматограф, которая обеспечивается высочайшей чувствительностью совре-
менных приборов (Greulich et aL, 2008). Однако стадия концентрирования перед инжек-
тированием пробы в систему ЖХ-ИАД/МС все-таки обычно практикуется для допол-
нительного повышения чувствительности. Чаще всего для этой цели используется ТФЭ
(онлайн или оффлайн). Онлайн-ТФЭ в отличие от оффлайн-варианта позволяет сокра-
тить объем образца без потери чувствительности. Она также минимизирует количество
манипуляций с образцом и объемы органических растворителей, легко автоматизиру-
ется, что облегчает ее использование в рамках программ мониторинга, когда требуется
анализировать большое число образцов.
Хотя обычные варианты ЖХ с УФ- или флуоресцентным детектором активно ис-
пользуются для определения органических ксенобиотиков в воде, в последнее десяти-
летие наблюдается очевидная тенденция к увеличению популярности ЖХ/МС. Изна-
чально использовали приборы с одним квадруполем. Однако низкая чувствительность
и высокий уровень фона (мешающий достижению требуемых пределов обнаружения),
а также ограниченная структурная информация мешали применению этих приборов
для целей АОКП. Безусловно, предпочтительнее использовать более чувствительный
и селективный метод МС/МС. Поэтому в настоящее время наиболее эффективным
методом для определения остаточных количеств пестицидов и их продуктов трансфор-
мации является ЖХ/МС/МС. Хотя ионные ловушки могут работать в режиме МС/МС
и играют значительную роль в анализе образцов объектов окружающей среды, высокая
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
чувствительность и устойчивость работы QqQ-систем в режиме МЗИ делает последние
идеальным анализатором для скрининга и количественного определения средне- и вы-
сокополярных пестицидов. Этим объясняется растущее в последние годы число пу-
бликаций по проблемам определения пестицидов и их ПТ (в основном в воде) методом
ЖХ/МС/МС с QqQ. Несмотря на превосходную работу этих приборов, не стоит забывать
о матричных эффектах, которые могут существенно повлиять на правильность опреде-
ления аналитов, т.е. матричные эффекты должны тщательно выявляться, а результаты
количественного определения соответствующим образом корректироваться (Dijkman
etal., 2001, Hernandezetal., 2005b, Marin etal., 2009, Matuszewskietal., 2003, Reemtsma, 2001).
Хорошим примером, раскрывающим потенциал ЖХ/МС/МС для целей АОКП
в водных образцах, является недавно разработанная методика определения 37 пестици-
дов (гербициды, фунгициды и инсектициды) в образцах природной и сточной воды с по-
мощью УЭЖХ/МС/МС (Marin etal., 2009). Для эффективной стыковки УЭЖХ и МС/МС
был использован QqQ-прибор с возможностью быстрого сбора данных для регистрации
трех переходов в режиме МЗР для каждого аналита. При этом для минимизирования
риска получения ложноположительных результатов из методики были исключены все
неспецифические переходы. Стадия концентрирования (фактор 100) включала офф-
лайн-ТФЭ с помощью картриджей OASIS HLB (рис. 14.За). Матричные эффекты были
изучены для различных типов образцов воды. Несколько изотопномеченых внутренних
стандартов были протестированы на предмет эффективности компенсации эффекта по-
давления сигнала большинства пестицидов, особенно в случае сточных вод. Методика
была поверена в рамках руководства SANCO Евросоюза по поверхностным, грунтовым,
питьевым и сточным (с завода переработки твердых бытовых отходов) водам для уровней
0,025 и 0,1 мкг/л. И точность, и воспроизводимость методики были удовлетворительны-
ми при работе с двумя уровнями добавок. Факторы извлечения составили 70—120% при
относительном стандартном отклонении менее 15% и превосходной чувствительности
(пределы обнаружения в диапазоне от 0,1 до 7 пг). Разработанная методика использо-
валась для анализа около 40 водных проб, включая поверхностные и подземные воды,
а также сточные воды с завода переработки твердых бытовых отходов. Несколько целе-
вых пестицидов были детектированы в широком диапазоне концентраций. Рис. 14.3d
иллюстрирует УЭЖХ/МС/МС-хроматограмму образца поверхностных вод. Высокая
чувствительность и селективность методики позволили определить несколько пести-
цидов (имидаклоприд, тиобенкарб, симазин и диурон) в очень низких концентрациях.
Надежное подтвеждение результатов основано на мониторинге трех реакций фрагмен-
тации (МЗР), оценке отношений Q/q и временах удерживания относительно стандартов.
На рис. 14.Зе представлен материал, доказывающий присутствие данных пестицидов.
Для наглядности представления приведены только 2 реакции фрагментации на каждый
аналит. Обратите внимание на отличное соответствие пераметра Q/q и времени удержи-
вания стандартов и целевых аналитов.
В работе (Marin et al., 2006) была представлена онлайн ТФЭ-ЖХ/МС/МС-методика
определения 18 полярных пестицидов и 9 их ПТ в воде. Учитывая существенные раз-
личия физико-химических характеристик аналитов, одновременное определение всех
аналитов по единой методике было невозможно. Было необходимо провести деривати-
зацию гептафторбутановой или муравьиной кислотой для достижения требуемой эф-
фективности ТФЭ и ЖХ. Благодаря высокой чувствительности стал возможен прямой
Скрининг
100 мл воды Цент (45( 5 mi + 1 м AGUAf 100-1 ри фугирование )0 об/мин в течение *- 4нут при необходимости) л НСООН (с) 3031 581 17440 3 MRM of 13 Channels ES+ 202 1 > 132 1 2 64е5 Area Симазин 0,02 мкг/л
+ 1 мл смеси суррогатов 100 1^4, ' 6” /MRM8°°I8CJ°°°ES. .. 6 43 232 9 > 72 1
Картриджи ТФЭ OASIS HLB, 200 мг по весу 100- 3337 ‘ 4 67е4 Area Диурон 0,003 мкг/л
Кондиционирование:
5 мл МеОН с AGUA! 5 мл ацетона ]00 5 мл НСООН 1% Высушивание в вакууме, 5 мин. ’ Элюирование 5 мл ацетона Выпаривание 0. ’ при 40°C в атмосфере азота Растворение )00 , в 1 мл 10%-го водного ацетони' 4 6 3031 Ти< 0,0 4 0 3031 4 12_ 1296 рила 0 6 00 8 00 10 00 7 MRM of 28 Channels ES+ 7 83 258 2 >125 1 66221 1 Юеб Area эбенкарб 15 мкг/л 0 6 00 8 00 10 00 1 MRM of 20 Channels ES+ 256 3 > 175 2 2 32е4 Area Имидаклоприд 0,015 мкг/л
Extract
0-
УЭЖХ/QqQ-MC/MC 4 00 6 00 8 00 10 00 Образец поверхностных вод
Подтверждение
Симазин
(Q/q\ = 2,0
(C/9Uple= 1.9
Dev = 5%J
3 MRM of 13 Channels ES+
5 73 _ 202 1 >124 1
11226 | 1 81e5
I Area
6 00 8 00 10 00
3 MRM of 13 Channels ES+
5 80 _ 202 1 >132 1
22022 1 3 54e5
I Area
AGUAS031 5 MRM of 19 Channels ES+ AGUAS039
Диурон
«Ж = 2,0
(0/?Uple= 2,1
Dev = 5%Z
6 00 8 00 10 00
5 MRM of 19 Channels ES+
6 43 _ 232 9 > 46 1
18174 | 2 72e5
1 Area
6 00 8 00 10 00
5 MRM of 19 Channels ES+
6 43 _ 232 9 > 72 1
35954 | 5 09e5
I Area
6 00 8 00 10 00
Тиобенкарб
(Q/q\ = 14,9
(G/?Uple = H.9
Dev = 20%^
Имидаклоприд
(Q/q\ = 1,6
(<2^Uple= 1’5
Dev= 6%^
Образец поверхностных вод 2,5 мкг/л стандарта
Рис. 14.3. (а) Схема последовательности операций определения методом УЭЖХ/МС/МС 37 пестицидов в образцах природных и сточных вод.
(б) УЭЖХ/МС/МС-хроматограммы, демонстрирующие селективное детектирование имидаклоприда, тиобенкарба, диурона и сима-
зина в образце поверхностных вод. (в) Подтверждение идентификации детектированных пестицидов (для простоты представлены
только две реакции фрагментации для каждого соединения)
□
14.4. ЖХ/МС
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
ввод 2 мл воды в ЖХ/МС/МС-систему. Метод был поверен с пределом количественно-
го определения 25 нг/л. Пределы обнаружения, как правило, были ниже 5 нг/л. Удов-
летворительные факторы извлекаемости (70—110%) были получены для большинства
аналитов в образцах поверхностных и подземных вод. Регистрация двух реакций для
каждого соединения методом МС/МС позволила надежно подтверждать правильность
идентификации на уровне предела количественного определения. Было проанализиро-
вано более 150 образцов воды (поверхностной и грунтовой), причем наиболее часто де-
тектируемыми оказались симазин, бромацил и ПТ триазиновых соединений: 2-гидрок-
си-тербутилазин, дезэтил-2-гидрокситербутилазин и дезэтилтербуметон. Эта работа
подтвердила необходимость включения ПТ в протоколы методов анализа, поскольку
несколько таких продуктов присутствовали в концентрациях выше, чем их исходные
соединения. Например, около 75 % триазинов в концентрации выше 0,1 мкг/л были про-
дуктами трансформации и только 25 % были исходными пестицидами. Важно отметить
также, что при регистрации только одной реакции фрагментации (обычно наиболее
значимой) было бы получено 5 % ложноположительных результатов.
Несмотря на огромный потенциал ЖХ/МС/МС и недавние успехи в области пробо-
подготовки специфические физико-химические характеристики некоторых пестици-
дов делают сложным или даже практически невозможным их включение в мультиком-
понентные методики. Это справедливо, например, для фосэтил-алюминия и этефона
(анионные соединения) или меламина (катионное соединение). Еще один интерес-
ный пример — используемый во всем мире гербицид глифосат. Его определение в воде
на уровне ниже 1 мкг/л осложнено его ионной природой, низкой летучестью, низкой
массой и отсутствием функциональных групп, которые могли бы облегчить его детек-
тирование. В связи со сложностью матрицы еще проблематичнее определение его остат-
ков в низких концентрациях (ниже 0,1 мг/кг) в почве. Недавно разработана методика
определения глифосата, продукта его трансформации аминометилфосфоновой кисло-
ты и глуфосината в почве и воде (Ibanez et al., 2005, Ibanez et aL, 2006). Метод включает
стадию дериватизации 9-флуоренметилхлорформиатом в боратном буфере (pH ~9), для
того чтобы сделать аналиты доступными для концентрирования ТФЭ и ЖХ-разделения.
В случае проб воды экстракт подкисляется до pH ~1 для остановки дериватизации и вво-
дится в онлайн-систему ТФЭ-ЖХ/МС/МС. Для проб почвы прямой ввод 50 мкл дери-
ватизованного подкисленного экстракта характеризовался 25% фактором извлекаемо-
сти и неприемлемыми величинами относительного стандартного отклонения. Кроме
того, наблюдался существенный матричный эффект и помехи на стадии дериватизации
(рис. 14.4 и табл. 14.1). Усложнение пробоподготовки для решения этой проблемы пред-
ставлялось нерациональным, поскольку оно сопровождается дополнительными ма-
нипуляциями с образцом, увеличением времени анализа и увеличением вероятности
получения аналитических ошибок. Калибровка со стандартами с учетом матрицы —
не очень надежный подход для образцов объектов окружающей среды, поскольку состав
матриц очень изменчив, а выбор репрезентативного матричного бланка проблематичен.
В связи с этим было решено протестировать метод ЖХ/МС/МС с изотопным разбавле-
нием с изотопномеченым глифосатом в качестве внутреннего стандарта. Как и ожида-
лось, извлекаемость и воспроизводимость определения глифосата резко улучшились,
а меченый аналог полностью компенсировал матричные эффекты (глава 2). Тем не ме-
нее наблюдалось значительное подавление ионизации, снижающее чувствительность
14.4. ЖХ/МС
Таблица 14.1. Матричные эффекты при определении глифосата, АМРА и глуфосината
в почве методом ЖХ/МС/МС. Роль разбавления перед проведением дери-
ватизации и корректировка результатов при использовании изотопномече-
ного глифосата в качестве внутреннего стандарта. Средние уровни извле-
каемости и относительное стандартное отклонение (ОСО) соответствуют
анализу 5 образцов почвы с добавками аналитов*
Соединение Без разбавления
Без стандарта Со стандартом
% Извлекаемость % ОСО % Извлекаемость % ОСО
Глифосат 25 79 97 6
АМРА 28 46 127 27
Глуфосинат 27 56 116 18
Соединение 10-кратное разбавление
Без стандарта Со стандартом
% Извлекаемость % ОСО % Извлекаемость % ОСО
Глифосат 83 24 98 3
АМРА 87 9 98 11
Глуфосинат 94 8 118 19
Соединение 20-кратное разбавление
Без стандарта Со стандартом
% Извлекаемость % ОСО % Извлекаемость % ОСО
Глифосат 83 23 91 11
АМРА 89 8 98 10
Глуфосинат 92 8 107 9
* Уровни добавок: 0,5 мг/кг (без разбавления), 5 мг/кг (10-кратное разбавление), 10 мг/кг (20-крат-
ное разбавление).
(площадь пика на рис. 14.40. Поэтому было исследовано разведение образцов почв
водой качества ВЭЖХ. Глифосат был добавлен в пять образцов различных почв, про-
экстрагирован раствором КОН и дериватизован с или без предварительного разбавле-
ния экстракта. По нашим данным (рис. 14.4, табл. 14.1), 10- и 20-кратное разбавление
экстракта почвы оказалось надежным способом проведения точного количественного
определения даже без использования изотопного аналога, поскольку в обоих случаях
извлекаемость аналита составила 90%. Изотопный аналог, тем не менее, улучшал от-
носительное стандартное отклонение, особенно для глифосата. Кроме того, исполь-
зование изотопного разбавления позволяет предотвратить неожиданные матричные
эффекты. Аналогичный эффект разбавления был получен в случае метаболита АМРА
и для гербицида глуфосината. Десятикратное разбавление экстракта с добавкой изотоп-
ных аналогов было выбрано в качестве метода определения глифосата и его метаболита
АМРА, поскольку приводило к улучшению пределов обнаружения. Для глуфосината,
напротив, изотопный аналог не улучшал ситуацию. Поэтому для его определения был
использован метод внешнего стандарта.
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
Рис. 14.4. ВЭЖХ/МС/МС-хроматограммы для селективного определения глифосата:
(а) стандарт 250 мкг/л; (б) эктракт образца почвы с добавкой 0,5 мг/кг; (в) деся-
тикратно разведенный экстракт образца почвы с добавкой 5 мг/кг; (г) двадцати-
кратно разведенный экстракт образца почвы с добавкой 10 мг/кг
Правильность метода была подтверждена для поверхностных и подземных вод с до-
бавками аналитов 50 и 500 нг/л, а также для почв с добавками 0,05 и 0,5 мг/кг. Пределы
обнаружения аналитов в воде были на уровне 0,005 мкг/л, а в почве — 0,005 мг/кг. Раз-
работанный метод был апробирован на образцах воды и почвы из разных регионов. Ко-
личественное определение сопровождалось регистрацией двух реакций фрагментации
для каждого соединения и оценкой отношения Q/q. Рис. 14.5 иллюстрирует (а) проце-
дуру анализа образца воды и ЖХ/МС/МС-хроматограммы для (б) образца грунтовых
вод, содержащих 304 нг/л глифосата и 87 нг/л АМРА, и (в) образца почвы, содержащей
0,28 мг/кг глифосата и 0,38 мг/кг АМРА.
14.5. Тенденции
Соединение хроматографических методов с масс-спектрометрическими на сегодняш-
ний день является наиболее эффективным и надежным аналитическим методом опре-
деления остаточных количеств пестицидов. Учитывая широкий диапазон свойств пе-
стицидов, и ГХ/МС, и ЖХ/МС должны использоваться, чтобы определять их, начиная
с неполярных/летучих и заканчивая полярными/нелетучими.
Образец воды 10 мл
Подкисление
+ 200 pL НС1 6М (pH ~ 1)
+ Потрясти и подождать 1 мин.
100 мкл внутреннего стандарта (НО мкг/л)
О
нкснДон
с'н2 о'н
сдон
Глифосат
Нейтрализация
+ 200 pL КОН 6М (pH - 6-7)
о
СНг-O-t-Cl
+ 0,6 мл боратного буфера 5%
+ 0,6 мл FMOC-C1 12000 ррт
FMOC
Оставить на ночь при комнатной температуре
Профильтровать через 0,45 мкм
нейлоновый фильтр
20 pL НС1 37% (pH - 1)
2 мл
ТФЭ-ЖХ/МС/МС
О О
CtVO-t-N сцКон
Л/\ сН Зн
Т ]сЗон
MRM of 8 Channels ES+
MU221
MU221
100-
%-
IS(Q)
395.1 >91.1
9.77еЗ
Area
SUELO242
MRM of 8 Channels ES+
MRM of 8 Channels ES+
8.33 395.1 >216.9
1174 IS (q)
1 Area
1По 741
%1 20676l IS (Q)
0^ . I . Дг...............I.....
SUELO242 MRM of 8 Channels ES+
IS(q)
395.1 >216.9
9 50e4
Area
MU221
10CH
M
MU221
MRM of 8 Channels ES+
392.1 > 88.1
Glyphosate (Q)
MRM of 8 Channels ES+
100q 7.41
%| 9037
SUELO242
SUELO242
MRM of 8 Channels ES+
392 1 >88.1
Glyphosate (Q)
/Л
392 1 >214
1“] 31333T Glyphosate (q)
«1__________ £ _____/______________________ Area
0^ । 1 1 1 1 м ' 1 i—1~ । |—। । । । । । । i-i-|
MU221 MRM of 8 Channels ES+
MRM of 8 Channels ES+
SUELO242
392.1 > 214
Glyphosate (q)
Area
334.1 >1119
AMPA (Q)
Ml
MRM of 8 Channels ES+
of 8 Channels ES+
AMPA (Q)
334 1 > 111.9
4 93e3
Area
334 1 > 179
AMPA (q)
। । i । । । । i । । । । i Time
15.00 20.00
MRM of 8 Channels ES+
SUELO242
334.1 > 179
5 34e3
Area
1 1 1 i Time
20.00
Образец грунтовых вод
0
Образец почвы
0
Рис. 14.5. (а) Схема последовательности операций для определения глифосата и АМРА
в воде методом ВЭЖХ/МС/МС. ЖХ/МС/МС-хроматограммы соответствую-
щие (б) образцу подземных вод с содержанием глифосата и АМРА на уровне 304
и 87 нг/л соответственно; (в) образцу почвы с содержанием глифосата и АМРА
на уровне 0,28 и 0,38 мг/кг соответственно. Q: реакция для количественного
определения; q; реакция для подтверждения идентификации
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
Одиночные квадрупольные анализаторы и ионные ловушки очень популярны для
АОКП методом ГХ/МС, причем они позволяют достигать весьма приемлемых резуль-
татов. Однако эти «типичные» ГХ/МС-анализаторы не столь успешны при работе в ре-
жиме ЖХ/МС (особенно квадруполи) из-за низкой чувствительности, высокого фона,
и недостатка структурной информации при работе с мягкими методами ионизации,
характерными для ЖХ/МС. В последнее десятилетие разработано много ЖХ/МС/МС-
методов для системы трех квадруполей QqQ. Превосходная чувствительность и селек-
тивность тандемной масс-спектрометрии делает этот метод чрезвычайно привлекатель-
ным для надежной идентификации и количественного определения. Обычно методики
оперируют ограниченным числом аналитов, выбранных предварительно для целевого
анализа до начала регистрации спектров. До настоящего времени такие методики вклю-
чали до нескольких десятков соединений. Этого количества, как правило, не хватает
для того, чтобы оценить качество окружающей среды. С появлением нового поколе-
ния QqQ-приборов (более быстрых и чувствительных), число аналитов, определяемых
в рамках одной методики, существенно возросло и достигло 100—200.
Комплексное использование ГХ/МС/МС и ЖХ/МС/МС с системой QqQ является
сегодня одним из самых мощных методов АОКП. Используя эти комплементарные ме-
тоды, можно проводить чувствительный и селективный анализ пестицидов в любых ма-
трицах. Регистрация по крайней мере двух реакций фрагментации для каждого аналита
необходима для их надежной идентификации и количественного определения в одной
инжекции. Однако следует помнить, что другие экотоксиканты, не включенные в мето-
дику, не будут детектированы, даже присутствуя в высоких концентрациях.
В более современных методиках ведут запись полного масс-спектра, а присутствие
любых соединений (без предварительной селекции) проверяется после завершения
программы регистрации спектров. Одиночные и тройные квадруполи, а также ион-
ные ловушки не обладают достаточной чувствительностью в режиме сканирования
полного масс-спектра. Кроме того, эти анализаторы дают информацию только о цело-
численной массе аналитов, что делает затруднительным идентификацию соединений,
присутствующих в следовых количествах. Напротив, времяпролетные (TOF) анализа-
торы обеспечивают требуемую селективность и чувствительность для эффективного
скрининга в широком диапазоне, сочетая высокую чувствительность и высокую раз-
решающую способность и позволяя измерять точные массы всех ионизировавшихся со-
единений. Поиск экотоксикантов в этом случае производится после завершения сбо-
ра масс-спектрометрических данных (Hernandez et al., 2005а). Преимуществом такого
«постцелевого» анализа является возможность детектирования большого числа ксено-
биотиков даже без использования стандартов и дополнительных анализов. Кроме того,
времяпролетные анализаторы обладают определенными характеристиками, которые
облегчают надежную идентификацию соединений. Масс-хроматограммы (глава 2), по-
строенные на базе полных спектров по точной массе в узком окне (~ ±0,01 Da), дают
возможность проводить высокочувствительный и селективный анализ. Идентифика-
ция детектированных соединений базируется на точной массе их (де)протонирован-
ных молекул, возможных фрагментных ионов, изотопном распределении и времени
удерживания (Hoogenboom et al., 1999, Petrovic et al., 2006, Sancho et al., 2006, Ibanez et al.,
2008). На рис. 14.6 представлены УЭЖХ-ТОГ-хроматограммы, позволившие совместно
с измерением точных масс детектировать и идентифицировать несколько пестицидов
14.5. Тенденции
Рис. 14.6. ВЭЖХ-ТОГ-хроматограммы (окно 20 мДа) и масс-спектры высокого разреше-
ния нескольких пестицидов в образце сточных вод, поступающих на завод по их
переработке, отобранных в июле 2005 года. Указана погрешность в мДа
в городских сточных водах, отобранных в июле 2005 года. Ошибки измерения массы
(в мДа) для молекулярных ионов и для фрагментных/изотопных ионов также представ-
лены на рисунке.
Наиболее эффективный и простой подход для целевого анализа методом ГХ/МС
с времяпролетным анализатором включает обработку результатов, которая должна со-
держать наиболее значимую информацию (время выхода, точные массы 2—5 ионов, от-
ношения Q/q), полученную при работе со стандартными образцами. Регистрация хотя
бы двух ионов с точными массами и отношением интенсивностей в заданном диапазоне
считается достаточной для надежной идентификации целевого аналита (Portoles et al.,
2007, European Commision Decision, 2002/657/ЕС). Возможность проведения скринин-
га для выявления присутствия большого числа органических экотоксикантов без ис-
пользования стандартных образцов является ключевым достоинством времяпролетных
анализаторов. Однако такой подход не очень эффективен для ГХ/МС, поскольку пред-
варительная информация о веществе будет лимитирована его химической формулой
и точной молекулярной массой. На базе этой информации могут быть зарегистрирова-
/4“4
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
ны только соединения с интенсивными пиками М+ в их спектрах ИЭ. Это наблюдает-
ся далеко не всегда, поскольку электронная ионизация характеризуется интенсивной
фрагментацией (глава 1). Следовательно, для надежного детектирования необходимо
знать массы основных фрагментных ионов. Чтобы облегчить идентификацию и детек-
тирование, привлекательным вариантом может быть использование мягких методов
ионизации типа ХИ или ХИАД (см. главу 1 о попеременном получении спектров ИЭ
и ПИ). В этом случае в зависимости от метода ионизации будут наблюдаться интенсив-
ные пики молекулярных ионов или протонированных молекул. Более детализирован-
ную информацию можно найти в (Portoles et al., 2010, Hernandez et al., 2010a).
Напротив, ЖХ/МС с широко используемой ионизацией электрораспылением ха-
рактеризуется интенсивными пиками (де)протонированных молекул. Регистрация
этого иона обычно является первым шагом процесса идентификации, который допол-
нительно выигрывает при наличии интенсивных фрагментных ионов. Недавно был соз-
дан эффективный подход, основанный на одновременном использовании двух режимов
сбора данных, различающихся энергией соударений (Hernandez etal., 2010b). Фрагмента-
ция осуществляется в камере соударений гибридного прибора QTOF. При низкой энер-
гии соударений фрагментация оказывается минимальной, а оператор получает инфор-
мацию о точной массе (де)протонированной молекулы. Напротив, при высокой энергии
соударений фрагментация усиливается, а в результате образуются интенсивные пики
фрагментных ионов. Этот подход, известный как MSE, позволяет за один ввод пробы
получать оптимальную структурную информацию о соединениях, присутствующих
в образце, и идентифицировать детектированные соединения с очень высокой степенью
надежности. На рис. 14.7 представлены масс-хроматограммы (в узком окне) и два вида
спектров (с низкой и высокой энергией соударений) антибиотика ципрофлоксацина, за-
регистрированного в образце сточных вод. Пик протонированной молекулы ([М + Н]+,
точная масса, m/z 332, 1423) присутствует в обоих спектрах, которые связаны с хромато-
графическим пиком с одним временем удерживания на обеих хроматограммах. Поми-
мо регистрации пика [М + Н]+ с минимальной погрешностью измерения точной массы
в спектре, зарегистрированном при высокой энергии соударений, присутствуют три
фрагментных иона, точные массы которых (с погрешностью менее 2 мДа) соответствуют
структуре ципрофлоксацина. Эти результаты дают высокую степень уверенности в пра-
вильности идентификации этого соединения.
Запись полного масс-спектра с регистрацией точных масс при удовлетворительной
чувствительности времяпролетных масс-анализаторов открывает возможность прове-
дения нецелевого анализа, т. е. идентификации любого загрязняющего вещества без ка-
кой-либо предварительной информации или селекции. Это трудоемкая и времязатрат-
ная задача, особенно когда неизвестное вещество присутствует в следовых количествах
в сложной матрице. В последние годы появились интересные разработки программ-
ного обеспечения для детектирования и последующей идентификации соединений.
Эти программы, безусловно, облегчат эту сложную задачу.
Использование ГХ-TOF и >KX-(Q)TOF для целевого и нецелевого скрининга — одно
из наиболее многообещающих направлений аналитической химии (Ibanez et al., 2008,
Hernandez et al., 2007). Комплементарное использование обоих методов открывает новые
перспективы для широкомасштабного скрининга, детектирования, идентификации
и структурного анализа многочисленных экотоксикантов. Комбинация этих методов
Литература
231,0570 (+1.4.mDa)
C12H8N2°2F
231 0584
288 1523
288,1512 (+ 1.1.mDa)
C16H19N3OF(-CO2)
100n
0-
ЮОп
CI-
2 00
338
292
2 TOF MS ES+
332 142 0 02D3
393100-
MSE с высокой
энергией
соударений
205 0789
245 1100
3 00 4 00 5 00
►04г
338
деЗЮОл
MSE с низкой
энергией
соударений
~i 111111111111111111 i'i~> Time
209 1380
2 00 3 00 4 00 5 00
ОХ
200
220
240
260
314,1305 (+0.8.mDa)
C17H17N3O2F(-H2O)
/ 2 TOF MS ES+
3 37e3
314 1313
332 1423
3151350
333 1450
I 305 1581 /15
О1
то 1349 332,1410 (+0.8 mDa) ,,
cI7h19n3o3f
Ципрофлоксацин
279 0936
332 1418
280
300
320
340
гтт m/z
360
РИС. 14.7. Детектирование и идентификация антибиотика ципрофлоксацина в образце
сточных вод методом МСЕ на приборе УЭЖХ-QTOF. Хроматограммы и спектры
при использования энергии соударений 4 эВ (низ) и повышения энергии в диа-
пазоне 15—40 эВ (верх)
будет весьма полезной и в областях анализа пищевых продуктов, допинг-контроля, ток-
сикологии и метаболомики (Portales et aL, 2009, Peters etal., 2010, Krauss et al., 2010).
Благодарность
Авторы благодарят за финансовую поддержку Министерство образования и науки Ис-
пании (проект CTQ2009-12347) и мэрию Валенсии (ведущая исследовательская группа
PROM ЕТЕО/2009/054), позволившую вести наши исследования в области анализа оста-
точных количеств пестицидов.
Литература
[1] . Council Directive 98/83/ЕС on the quality of water intended for human consumption, Of-
ficial Journal of European Communities, November 3, 1998.
[2] . Dijkman E., Mooibroek D., Hoogerbrugge R., Hogendoorn E., Sancho J.V., Pozo O.,
Hernandez F. (2001). «Study of matrix effects on the direct trace analysis of acidic pesticides
in water using various liquid chromatographic modes coupled to tandem mass spectrometric
detection». Journal of Chromatography A. 926, 1: 113—125.
[3] . European Commission Decision 2002/657/EC, implementing Council Directive 96/23/EC
concerning the performance of analytical methods and the interpretation of results. Official
Journal of European Communities 2002, L221, 8.
[4] . FDA, Guidance for industry: Mass spectrometry for confirmation of the identity of animal
drugs residues. Draft Guidance 118, Food And Drug Administration June 6, 2001.
[5] . Filho A.M., dos Santos F.N., Pereira P.A.d.P. (2010). «Development, validation and ap-
plication of a method based on Di-SPME and GC/MS for determination of pesticides of
Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
different chemical groups in surface and groundwater samples». Microchemical Journal. 96:
139-145.
[6] . Garrido Frenich A., Martinez Vidal J. L., Fernandez Moreno J. L., Romero Gonzalez R.
(2009). «Compensation for matrix effects in gas chromatography-tandem mass spectrometry
using a single point standard addition». Journal of Chromatography A. 1216: 4798—4808.
[7] . Geerdink R. B., Niessen W. M. A., Brinkman U. A. Th. (2002). «Trace-level determination
of pesticides in water by means of liquid and gas chromatography». Journal of Chromatogra-
phy A. 970: 65-93.
[8] . Gomez M.J., Gomez-Ramos M.M., Agiiera A., Mezcua M., Herrera S., Fernandez-Al-
ba A. R. (2009). «А new gas chromatography/mass spectrometry method for the simultane-
ous analysis of target and non-target organic contaminants in water». Journal of Chromatog-
raphy A. 1216:4071—4082.
[9] . Greulich K., Alder L. (2008). «Fast multiresidue screening of 300 pesticides in water for hu-
man consumption by LC/MS/MS». Analytical and Bioanalytical Chemistry. 391, 1: 183—97.
[10] . Hernandez F., Pozo O. J., Sancho J. V., Lopez F.J., Marin J.M., Ibanez M. (2005a). «Strat-
egies for quantification and confirmation of multi-class polar pesticides and transformation
products in water by LC/MS2 using triple quadrupole and hybrid quadrupole time-of-fiight
analyzers». TRAC-Trends in Analytical Chemistry. 24: 596—612.
[11] . Hernandez F., Sancho J. V., Pozo O. J. (2005b). «Critical review of the application of liquid
chromatography/mass spectrometry to the determination of pesticide residues in biological
samples». Analytical and Bioanalytical Chemistry. 382: 934—946.
[12] . Hernandez F., Portoles T., Pitarch E., Lopez F. J. (2007). «Target and non-target screen-
ing of organic micropollutants in water by solid-phase microextraction combined with gas
chromatography/high-resolution time-of-flight mass spectrometry». Analytical Chemistry.
19: 9494-9504
[13] . Hernandez F., Marin J. M., Pozo O. J., Sancho J. V., Lopez F. J., Morell I. (2008). «Pesticide
residues and transformation products in groundwater from a Spanish agricultural region on
the Mediterranean Coast». International Journal of Environmental Analytical Chemistry. 88,
6: 409-424.
[14] . Hernandez F., Portoles T., Pitarch E., Lopez F.J. (2010a). «Gas chromatography/high
resolution time-of-flight mass spectrometry: an advanced analytical tool to investigate the
presence of organic compounds at trace levels in environmental, food safety and toxicology
fields». Submitted for publication.
[15] . Hernandez F., Sancho J.V., Bijlsma L. (2010b). «Wide-scope screening of illicit drugs in
urban wastewater by UPLC-QTOF MS». In Castiglioni S., Zuccato E., Fanelli, R (Eds),
Illicit drugs in the environment: Occurrence, Analysis, and Fate using Mass Spectrometry. John
Wiley & Sons, Inc. in press.
[16] . Hogenboom A.C., Niessen W. M.A., Little D., Brinkman U.A.Th. (1999). «Accurate mass
determinations for the confirmation and identification of organic microcontaminants in
surface water using on-line solid-phase extraction liquid chromatography electrospray
orthogonal-acceleration time-of-flight mass spectrometry». Rapid Communication in Mass
Spectrometry. 13: 125—133.
[17] . Ibanez M., Pozo O. J., Sancho J. V., Lopez F. J., Hernandez F. (2005). «Residue determina-
tion of glyphosate, glufosinate and AM PA in water and soil samples by liquid chromatog-
Литература
raphy coupled to electrospray tandem mass spectrometry». Journal of Chromatography A.
1081: 145-155.
[18] . Ibanez M., Pozo O.J., Sancho J.V., Lopez F.J., Hernandez F. (2006). «Re-evaluation of
glyphosate determination in water by liquid chromatography coupled to electrospray tan-
dem mass spectrometry». Journal of Chromatography A. 1134: 51—55
[19] . Ibanez M., Sancho J.V., McMillan D., Rao R., Hernandez F. (2008). «Rapid non-tar-
get screening of organic pollutants in water by ultraperformance liquid chromatography
coupled to time-of-flight mass spectrometry». TRAC-Trends in Analytical Chemistry. 27:
481-489.
[20] . Kolpin D. W., Thurman E. M., Linhart S. M. (2000). «Finding minimal herbicide concen-
trations in ground water? Try looking for their degradates». Science of the Total Environment.
248: 115-122.
[21] . Kolpin D. W., Thurman E. M., Linhart S. M. (2001). «Occurrence ofcyanazine compounds
in groundwater: Degradates more prevalent than the parent compound». Environmental
Science and Technology. 35: 1217—1222.
[22] . Krauss M., Singer H., Hollender J. (2010). «LC-high resolution MS in environmental anal-
ysis: from target screening to the identification of unknowns». Analytical and Bioanalytical
Chemistry. 397: 943—951.
[23] . Marin J. M., Sancho J. V., Pozo O. J., Lopez F.J., Hernandez F. (2006). «Quantification and
confirmation of anionic, cationic and neutral pesticides and transformation products in wa-
ter by on-line solid phase extraction liquid chromatography-tandem mass spectrometry».
Journal of Chromatography A. 1133: 204—214.
[24] . Marin J. M., Gracia-Lor E., Sancho J. V., Lopez F. J., Hernandez F. (2009). «Application of
ultra-high-pressure liquid chromatography-tandem mass spectrometry to the determina-
tion of multi-class pesticides in environmental and wastewater samples». Study of matrix
effects Journal of Chromatography A. 1216: 1410—1420.
[25] . Matuszewski B.K., Constanzer M.L., Chavez-Eng C.M. (2003). «Strategies for the as-
sessment of matrix effect in quantitative bioanalytical methods based on HPLC/MS/MS».
Analytical Chemistry. 75, 3019—3030.
[26] . Mekebri A., Crane D. B., Blondina G.J., Oros D.R., Rocca J. L. (2008). «Extraction and
analysis methods for the determination of pyrethroid insecticides in surface water, sedi-
ments and biological tissues at environmentally relevant concentrations». Bulletin of Envi-
ronmental Contamination and Toxicology. 80: 455—460.
[27] . Peters R.J. B., Stolker A.A. M., Mol J. G. J., Lommen A., Lyris E., Angelis Y., Vonapar-
ti A., Stamou M., Georgakopoulos C., Nielen M.W. F. (2010). «Screening in veterinary
drug analysis and sports doping control based on full-scan, accurate-mass spectrometry».
TRAC-Trends in Analytical Chemistry. D01:10.1016/j.trac.2010.07.012.
[28] . Petrovic M., Gros M., Barcelo D. J. (2006). «Multi-residue analysis of pharmaceuticals in
wastewater by ultra-performance liquid chromatography-quadrupole-time-of-flight mass
spectrometry». Journal of Chromatography A. 1124: 68—81.
[29] . Pitarch E., Medina C., Portoles T., Lopez F. J., Hernandez F. (2007). «Determination of
priority organic micro-pollutants in water by gas chromatography coupled to triple quadru-
pole mass spectrometry». Analytica Chimica Acta. 583: 246—258.
408 Глава 14. Анализ пестицидов в образцах объектов окружающей среды
современными методами хроматомасс-спектрометрии
[30] . Portoles, Т., Pitarch Е., Lopez F.J., Sancho J.V., Hernandez F. (2007). «Methodical ap-
proach for the use of GC-TOF MS for screening and confirmation of organic pollutants in
environmental water». Journal of Mass Spectrometry. 42: 1175—1185.
[31] . Portoles T., Ibanez M., Sancho J. V., Lopez F.J., Hernandez F. (2009). «Combined use of
GC-TOF MS and UPLC-QTOF MS to investigate the presence of non-target pollutants in
a case of honeybee poisoning». Journal of Agricultural and Food Chemistry. 57: 4079—4090
[32] . Portoles, T., Sancho J. V., Hernandez F., Newton A., Hancock P. (2010). «Potential of at-
mospheric pressure chemical ionization source in GC-QTOF MS for pesticide residue
analysis». Journal of Mass Spectrometry. 45: 926—936.
[33] . Pozo O.J., Sancho J. V., Ibanez M., Hernandez F., Niessen W. M.A. (2006). «Confirmation
of organic micropollutants detected in environmental samples by liquid chromatography
tandem mass spectrometry: Achievements and pitfalls». TRAC-Trends in Analytical Chem-
istry. 25: 1030—1042
[34] . Rebich R. A., Coupe R. H., Thurman E. M. (2004). «Herbicide concentrations in the Mis-
sissippi River Basin — The importance of chloroacetanilide herbicide degradates». Science
of the Total Environment. 321: 189—199.
[35] . Reemtsma T. (2001). «The use of liquid chromatography-atmospheric pressure ionization-
mass spectrometry in water analysis — Part II: Obstacles». TRAC-Trends in Analytical
Chemistry. 20, 10: 533—542.
[36] . SANCO, Method validation and quality control procedures for pesticide residues analy-
sis in food and feed document no. Sanco/10684/2009 Supersedes document no. San-
co/3131/2007 Implemented by 01/01/2010).
[37] . Sancho J. V., Pozo O. J., Ibanez M., Hernandez F. (2006). «Potential of liquid chromatogra-
phy/time-of-flight mass spectrometry for the determination of pesticides and transforma-
tion products in water». Analytical and Bioanalytical Chemistry. 386: 987—997.
[38] . Santos F.J., Galceran M.T. (2003). «Modern development in gas chromatography-mass
spectrometry-based environmental analysis». Journal of Chromatography A. 1000: 125—151.
[39] . van der Hoof G. R., van Zoonen P. (1999). «Trace analysis of pesticides by gas chromatog-
raphy». Journal of Chromatography A. 843: 301—322.
[40] . van Leewen S. P. J., de Boer J. (2008). «Advances in the gas chromatographic determination
of persistent organic pollutants in the aquatic environment». Journal of Chromatography A.
1186: 161-182.
[41] . WADA, Identification criteria for qualitative analysis, WADA technical document
TD2003IDCR 1.2, January 1, 2004.
ГЛАВА 15
ОПРЕДЕЛЕНИЕ СТОЙКИХ
ГАЛОГЕНСОДЕРЖАЩИХ
СОЕДИНЕНИЙ:
ХЛОРИРОВАННЫЕ
ДИБЕНЗО-П-ДИОКСИНЫ
И ДИБЕНЗОФУРАНЫ
Рей Клемент и Эрик Райнер
15.1. Введение
15.1.1. Хлорированные диоксины в окружающей среде
Достаточно полное представление о загрязнении окружающей среды токсичными
химическими веществами сложилось более полувека назад после появления книги
«Безмолвная весна» (Carson, 1962), однако появление на фоне множества других про-
мышленных химических веществ такого токсиканта, как 2,3,7,8-тетрахлордибензо-л-
диоксин (ТХДД), привлекло всеобщее внимание и даже вызвало шок. После того как
изучение токсических свойств ТХДД показало, что доза менее 1 мкг/кг веса тела (ми-
крограмм — это 1 • 10-6 г) для крыс, или концентрация — 1 часть на млрд (ppb — 1 • 10-9)
вообще для животных, смертельна, огромные усилия исследователей были направлены
на разработку аналитических методов определения ТХДД в окружающей среде. Уже по-
сле первой декады исследований стало ясно, что ТХДД — это лишь один член группы
соединений, называемых «диоксинами», — семейства из 75 хлорированных дибензо-л-
диоксинов (ХДД), молекулы которых состоят из двух бензольных колец, соединенных
двумя мостиками из атомов кислорода, и содержат в качестве заместителей от одного
до восьми атомов хлора (рис. 15.1). Помимо ХДД к «диоксинам» относят семейство из 135
родственных им соединений — хлорированных дибензофуранов (ХДФ) (рис. 15.1), об-
ладающих такими же свойствами, в том числе и токсическими. Ситуация осложнилась
еще больше, когда в результате широкомасштабных исследований обнаружилось, что
высокотоксичными являются только те ХДД и ХДФ, которые замещены атомами хлора
в положениях 2, 3, 7 и 8, так что только их и следует определять. Из 210 ХДД и ХДФ толь-
ко 17 отвечают за токсичность образца (если не учитывать токсичность других типов
соединений, которые тоже могут присутствовать в образце). Стало ясно, что был необ-
ходим аналитический метод, который не только мог бы обеспечить предел обнаружения
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
2,3,7,8-Тетрахлордибензо-п-диоксин
3,3’,4,4’,5-Пентахлорбифенил (РСВ126)
Рис. 15.1. Структура ХДД, ХДФ и ПХБ. Существует 75 ХДД, 135 ХДФ, замещенных 1—8
атомами хлора, и 209 ПХБ с 1 — 10 атомами хлора
на уровне ppt (1 • 10-12) и даже ниже, но и обладал бы уникальной способностью выделить
17 токсичных ХДД и ХДФ из смеси в присутствии других относительно нетоксичных
конгенеров.
В результате многочисленных исследований было найдено, что ХДД и ХДФ могут
возникать не только в результате промышленных процессов, но и при неполном сгора-
нии материалов, содержащих углерод и хотя бы следы хлора. Иными словами, эти сое-
динения в следовых концентрациях могут обнаруживаться во всех природных объектах.
Кроме того, было найдено, что их токсический эффект для человека может проявлять-
ся не только в случае больших экспозиций, но и в результате длительного воздействия
ультраследовых концентраций в воде, пище, воздухе и предметах рабочей обстановки.
Стало необходимо разработать аналитический метод, который позволял бы надежно
определять концентрации всех 17 токсичных ХДД и ХДФ в любых объектах окружаю-
щей среды на уровне ppt и ниже (Clement, 1991; Clement and Tosine, 1988; Reiner et al., 2006;
Reiner, 2010; van den Bergetal., 2006).
Механизм действия всех диоксиноподобных соединений одинаков: их мишень —
арилуглеводородный рецептор (AHR) (Океу, 2007). Благодаря этому оказалось воз-
можным ввести специальные коэффициенты — «коэффициенты эквивалентной ток-
сичности», или просто «коэффициенты токсичности» (toxic equivalent factor — TEF),
с помощью которых можно охарактеризовать токсичность смесей ХДД/ХДФ/полихло-
15.1. Введение
рированных бифенилов (ПХБ) (van den Berg et al., 2006). Этот подход предполагает, что
наиболее токсичному конгенеру — 2,3,7,8-ТХДД, присваивается значение TEF, равное
единице, а всем другим конгенерам — значение TEF, которое соответствует их отно-
сительной токсичности по отношению к 2,3,7,8-ТХДД. В самой последней версии та-
блицы коэффициентов 2,3,7,8-тетрахлордибензофуран и 3,3’,4,4’,5-пентахлорбифенил
(РСВ126) имеют TEF, равные 0,1, а октахлордибензо-и-диоксин — 0,0001, т.е. их ток-
сичность ниже, чем у 2,3,7,8-ТХДД. Общая специфическая диоксиновая токсичность,
обусловленная ХДД/ХДФ, характеризуется эквивалентом токсичности (toxic equivalent
quantity (TEQ), который рассчитывается как сумма произведений концентраций всех 17
индивидуальных конгенеров ХДД/ХДФ на соответствующие величины ТЕЕ
15.1.2. Стадии ультраследового анализа объектов окружающей
среды
При определении ХДД/ХДФ в объектах окружающей среды на уровне концентраций
ppt и ниже специфика анализа большинства проб обусловлена главным образом слож-
ностью объектов. В них присутствуют сотни, а в некоторых случаях тысячи соединений
с концентрациями, большими, чем концентрации ХДД/ХДФ. Некоторые из этих соеди-
нений могут мешать определению ХДД/ХДФ, и их либо следует удалить в процессе под-
готовки проб к анализу, либо их сигнал должен подавляться детектирующей системой.
Даже при наличии селективного детектора для определения ХДД/ХДФ в сложных сме-
сях на уровне концентраций ppt и ниже обычно требуется предварительная химическая
обработка образцов окружающей среды. Самая чувствительная система детектирова-
ния может обеспечить столь низкие пределы обнаружения только после многоступен-
чатого разделения, процедура которого включена в общую схему анализа (см. ниже).
Методика определения ХДД/ХДФ должна предусматривать, кроме обычных стадий от-
бора проб, экстракции и очистки экстрактов, ряд операций по оценке качества, с тем
чтобы получаемые количественные результаты удовлетворяли заданным требовани-
ям по воспроизводимости и точности (Poole et al., 1990). Хотя все эти, как и описанные
ниже более детально (см. также гл. 2), элементы анализа очень важны, один из них име-
ет определяющее значение — это метод изотопного разбавления (isotope dilution — ID)
(Boyd etal., 2008; Hubschmann, 2001). После завершения всех операций по подготовке про-
бы семнадцать 2,3,7,8-замещенных ХДД/ХДФ должны быть разделены внутри общей
группы 210 конгенеров ХДД/ХДФ хроматографическим методом (de Boer, 1999). Нако-
нец, после всего этого осуществляется количественное определение с помощью высо-
кочувствительного и селективного детектора. Весь набор операций можно представить
в виде следующей последовательности:
• отбор представител ьных образцов;
• добавка к анализируемой пробе изотопных аналогов ХДД/ХДФ;
• выделение ХДД/ХДФ вместе с коэкстрагируемыми веществами из матрицы про-
бы с помощью одного или нескольких методов экстракции;
• удаление большей части мешающих коэкстрагируемых веществ из экстракта
пробы посредством химической обработки (см. ниже);
• доведение конечного экстракта (в соответствующем высококипящем раство-
рителе) до определенного небольшого объема для инструментального анализа
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
(для ультраследового определения конечный объем экстракта составляет обычно
от 10 до 50 мкл);
• ввод 1—5 мкл экстракта в ГХ/МС-систему;
• корректировка полученных данных на основе результатов по эффективности
экстракции внутренних стандартов, добавленных к пробе перед экстракцией;
• определение концентраций семнадцати 2,3,7,8-замещенных ХДД/ХДФ с учетом
всей имеющейся информации по контролю качества и градуировочных характе-
ристик масс-спектрометра.
15.1.2. Почему нужна масс-спектрометрия?
Из всех детекторов, доступных аналитику, только масс-спектрометр обладает такими
качествами, как перестраиваемая селективность и высокий детектирующий потенци-
ал, которые необходимы для определения ультраследовых концентраций ХДД/ХДФ
в сложных природных объектах. К слову, если бы можно было удалить все 100% ме-
шающих коэкстрагируемых соединений до окончательной стадии инструментального
анализа, то для определения ХДД/ХДФ почти во всех типах образцов даже на ультра-
следовом уровне было бы достаточно газового хроматографа (ГХ) с детектором захвата
электронов. К сожалению, это вряд ли достижимо при анализе объектов окружающей
среды. Главные трудности при использовании масс-спектрометрии для определения
ХДД/ХДФ — это большие начальные затраты, относительно дорогое обслуживание
и требование высокой квалификации персонала.
Для ультраследового определения ХДД/ХДФ почти всегда используются масс-
спектрометры высокого разрешения с двойной фокусировкой (МСВР) в сочетании с ГХ.
МСВР может не только селективно отделить ионы ХДД/ХДФ с характеристическими
массами от обычных фоновых ионов, но также и детектировать сигналы этих ионов,
Рис. 15.2. Современный электронный умножитель. Ионы из масс-спектрометра через
«щель» с правой стороны электронного умножителя ударяются о внутренние
стенки электронного умножителя. Специальное покрытие способствует вы-
биванию нескольких электронов при каждом соударении с поверхностью —
каскад электронов производит сигнал, который можно отличить от фонового
шума, даже если всего несколько ионов ударяется о поверхность
15.2. Подготовка проб для определения ХДД/ХДФ на следовом уровне
разделенных по точным массам, соответствующие фемтограммам (10-15 г) вещества,
с помощью высокочувствительного электронного умножителя (рис. 15.2) Кроме того,
МСВР позволяет разделять экстрагируемые из пробы нативные ХДД/ХДФ и их изотоп-
номеченые аналоги (см. ниже). Благодаря возможностям контроля качества и внутрен-
ней градуировки масс-спектрометрия имеет гигантское преимущество в проведении
количественного анализа почти перед всеми другими инструментальными методами.
15.2. Подготовка проб для определения ХДД/ХДФ
на следовом уровне
15.2.1. Методы экстракции
Методы экстракции проб для определения ХДД/ХДФ и других галогенированных со-
единений подробно описаны многими авторами (Antunes et al., 2008; Geens et al., 2010;
Focant and DePauw, 2002; Reiner, 2010; Richter et al., 1996). Конкретная методика зависит
от типа анализируемого образца. Для относительно «чистых» образцов воды, таких
как питьевые или поверхностные воды, содержащих сравнительно мало взвешенных
частиц, может использоваться жидкостная экстракция различными растворителями,
не смешивающимися с водой, хотя в современных методиках обычно такие образцы
просто фильтруют через диски для твердофазной экстракции (ТФЭ), которые количе-
ственно сорбируют все ХДД/ХДФ, тогда как вода и водорастворимые компоненты про-
сто проходят через фильтр. ХДД/ХДФ (и другие подобные им соединения) могут быть
сняты с экстракционного диска элюированием всего несколькими миллилитрами рас-
творителя. Помимо того, что это ускоряет и облегчает работу, требуется меньшее коли-
чество токсичного растворителя по сравнению с жидкостной экстракцией, так что ТФЭ
в этих случаях предпочтительнее. Образцы, содержащие большое количество взвешен-
ных частиц, вначале фильтруют, из фильтрата выделяют ХДД/ХДФ либо с помощью
ТФЭ, либо жидкостной экстракцией, а из отфильтрованных частиц — методами, соз-
данными для экстракции твердых материалов, описанными ниже. Следует отметить,
что, поскольку молекулы ХДД/ХДФ гидрофобны, они экстрагируются из водных ма-
триц количественно. Однако вследствие своей гидрофобности ХДД/ХДФ в водных об-
разцах преимущественно адсорбируются на твердых частицах, а не растворяются в воде.
Для твердых образцов, в том числе почв и донных отложений, воздушных фильтров,
золы мусоросжигателей, полиуретановых фильтров из пробоотборных устройств и др.,
в течение многих десятилетий предпочтение отдавалось экстракции в аппарате Сок-
слета. Этот метод очень эффективен, он широко используется и сейчас. Важно, чтобы
образцы были достаточно сухими, так как «мокрые» образцы могут заблокировать нор-
мальный проход растворителя через экстракционный пакет, что приводит к ухудше-
нию эффективности экстракции ХДД/ХДФ. В то же время сушить образцы перед экс-
тракцией следует осторожно, так как при перегревании возможны потери ХДД/ХДФ.
Экстракцию в сокслете обычно проводят сериями по 5—10 образцов (включая холостые
опыты) в течение ~12 ч., хотя эффективная экстракция может быть осуществлена за не-
сколько часов циркуляции растворителя через экстрактор с образцом. К недостаткам
этого метода можно отнести большой расход растворителя (100—200 мл) и большое ко-
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
личество стеклянной посуды, которую необходимо тщательно отмывать перед экстрак-
цией следующей партии образцов.
В более новом методе, назы ваемом жидкостной экстракцией под давлением (pressurised
liquid extraction (PLE) ил и флюидной экстракцией под давлением (pressurised fluid extraction
(PFE), эффективная экстракция достигается вследствие того, что растворитель действует
на образец при высокой температуре и высоком давлении, так что растворитель находится
в состоянии, близком к сверхкритическому (Antunes et al., 2008). Высокое давление пре-
пятствует кипению растворителя. Этот метод экстракции требует меньше растворителя
и меньше стеклянных деталей, чем сокслет. Эффективная экстракция может быть достиг-
нута за 30—60 мин., по сравнению с временем от нескольких часов до ~12 ч. для сокслета.
В методе 3545 Агентства по охране окружающей среды США описана процедура PLE при
температуре 150—170°С и давлениях 1500—2000 psi (10000—14000 кПа). Было показано,
что эффективность PLE экстракции ХДД/ХДФ из различных типов твердых образцов
эквивалентна экстракции в сокслете, однако в случае PLE требуется в 10 раз меньше рас-
творителя и менее 15 мин. для количественной экстракции ХДД/ХДФ.
15.2.2. Очистка экстрактов
Методики определения ХДД/ХДФ включают следующий набор операций по очистке
экстракта:
• удаление из экстракта липидов (если это необходимо) обработкой серной
кислотой,
• удаление мешающих соединений на кислотном/щелочном силикагеле,
• отделение от ХДД/ХДФ ортозамещенных полихлорированных бифенилов и дру-
гих мешающих соединений с аналогичными свойствами на активированном ок-
сиде алюминия,
• отделение ХДД/ХДФ от мешающих соединений с непланарной структурой
на активном угле.
Описаны также способы очистки, основанные на высокоэффективной жидкост-
ной хроматографии (ВЭЖХ), гель-хроматографии (GPC) и других методах. Эти методы
подробно описаны в ряде обзоров (Reiner, 2010; Reiner et al., 2006). Чтобы было понятно,
что дает процедура очистки, на рис. 15.3 показано, как выглядят колонки после очистки
экстракта из тканей рыбы. Колонки окрашиваются задерживаемыми ими коэкстраги-
руемыми органическими веществами, которые в противном случае могли бы помешать
определению ХДД/ХДФ (Lamparski et aL, 1979).
15.2.3. Замечания по контролю качества
Хотя МСВР позволяет обеспечить очень низкий предел обнаружения ХДД/ХДФ, этот
метод, как и любой аналитический метод, предназначенный для определения токсич-
ных органических соединений на уровне концентраций ppt или даже ppq (1 • 10-15), тре-
бует проведения большого количества анализов для контроля качества, чтобы иметь
уверенность, что воспроизводимость и точность аналитических результатов находятся
в заданных пределах (Covaci et aL, 2007; Van Bavel and Abad, 2008). Кроме того, важно,
чтобы был минимизирован риск получения ложноположительных и ложноотрица-
тельных результатов. Хотя основное внимание в этой главе уделяется собственно масс-
15.2. Подготовка проб для определения ХДД/ХДФ на следовом уровне
Рис. 15.3. Колонки с кислым и основным силикагелем от двух разных образцов после того,
как через них были пропущены экстракты. Разное окрашивание ясно показыва-
ет наличие значительного количества удаляемых компонентов образца, кото-
рые могут влиять на качество анализа
спектрометрическим методам, рассмотрим основные процедуры контроля качества, все
или часть из которых включены в аналитические методики определения ХДД/ХДФ:
• Обязательное проведение холостых опытов. При анализе ультраследовых кон-
центраций одна частица пыли, попавшая в образец перед экстракцией, может
привести к ложноположительному результату, особенно для тех типов образцов,
которые обычно считаются «чистыми», например питьевая вода. Специальные
холостые опыты нужны для проверки чистоты реагентов, стеклянной посуды,
контейнеров образцов, аналитической системы, области отбора образцов и даже
средств транспортировки и др.
• Специальное оборудование и приспособления. Многие из лучших лабораторий имеют
специальные рабочие места, лабораторные столы и вытяжные шкафы для подго-
товки проб для определения ХДД/ХДФ. Крайне важно иметь отдельную посуду
и оборудование для серий образцов, в которых ожидаются близкие концентрации
ХДД/ХДФ; так, стеклянная посуда, предназначенная для экстракции летучей
золы мусоросжигателей, не должна использоваться для анализа питьевой воды.
• Метод изотопного разбавления (ID). Всегда, на любой стадии анализа, когда это
возможно, должен использоваться метод ID (см. ниже).
• Анализ образцов-дубликатов. Периодически следует анализировать дубликаты
образцов.
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
• Межлабораторные сравнительные испытания (ILS). Лаборатории должны уча-
ствовать в международных сравнительных испытаниях или, по крайней мере,
обмениваться образцами с другими аккредитованными лабораториями.
• Аттестованные стандартные образцы (certified reference materials — CRMs). CRMs,
содержащие ХДД/ХДФ, выпускаются для широкого круга матриц. Анализ раз-
рабатываемых стандартных образцов должен быть постоянным элементом вну-
тренней программы контроля качества аналитических лабораторий.
• Оценка хроматографического разделения. Со временем происходит постепенное
падение эффективности газохроматографических (ГХ) колонок; фактически
один «грязный» образец может привести к значительному сдвигу времен удержи-
вания. ГХ-разрешение необходимо оценивать в каждом опыте, особенно прове-
ряя, что 2,3,7,8-замещенные ХДД/ХДФ отделяются от «нетоксичных» ХДД/ХДФ.
• Многоточечная градуировка ХДД/ХДФ. Градуировка МСВР аналитической систе-
мы должна производиться достаточно часто — обычно дважды в день. При этом
градуировка должна быть многоточечной, чтобы концентрации ХДД/ХДФ в ана-
лизируемых образцах попадали в интервал градуировки. Не следует экстраполи-
ровать градуировочную зависимость выше или ниже интервала градуировки.
• Оценка времени выхода газохроматографических пиков. Известны ГХ-времена
удерживания для всех 136 тетра-окта-ХДД/ХДФ. Любые пики, элюирующиеся
вне интервала этих времен удерживания, не могут быть отнесены к ХДД/ХДФ.
• Оценка масс-спектрометрических данных. Различными способами необходимо
удостовериться, что пики, наблюдаемые в ГХ/МСВР-анализе, действительно
относятся к ХДД/ХДФ. Конкретные способы определяются спецификой при-
меняемого метода анализа. Однако все эти способы должны обеспечивать уль-
траследовое детектирование ХДД/ХДФ, так как необходимы низкие значения
предела обнаружения. Поэтому МСВР осуществляется не в режиме регистрации
полного масс-спектра, а в режиме селективного детектирования нескольких вы-
бранных характеристических ионов, специфичных для различных ХДД/ХДФ
(см. ниже).
• Опытный персонал. Определение ХДД/ХДФ на уровне ультраследовых концен-
траций в образцах окружающей среды — эта задача по силам только аналитику
с большим опытом работы. Необходима постоянная проверка квалификации
новых сотрудников — их профессионализма и способности получать результаты
с требуемой точностью и воспроизводимостью.
Можно отметить пять основных критериев положительной идентификации пиков
ХДД/ХДФ (это общепринятые международные критерии при определении ХДД/ХДФ
методом ГХ/МСВР в режиме селективного детектирования выбранных ионов):
1 ) элюирование в правильном окне,
2 ) пик должен иметь гауссову форму,
3 ) отношение сигнала к шуму должно быть выше 3:1,
4 ) время выхода конгенеров, замещенных в положениях 2, 3, 7, 8, должно отли-
чаться от времени выхода соответствующих изотопномеченых аналогов не бо-
лее, чем на 2 с,
5 ) Изотопные отношения должны быть в пределах 15% от теоретической величины
(см. табл. 15.1).
15.3. Количественный анализ и метод изотопного
15.3. Количественный анализ и метод изотопного
разбавления
15.3.1. Внутренняя и внешняя градуировка
Градуировка — это одна из наиболее важных процедур для обеспечения точного и вос-
производимого количественного анализа. Неудачная градуировка может привести к по-
явлению выбросов и ухудшению воспроизводимости. Каждому аналиту соответствует
свой отклик детектора, и для получения хороших результатов отклик на молекулу ана-
лита должен быть постоянным во всем широком интервале концентраций. Постоянный
отклик соответствует линейной градуировочной зависимости (Y = тХ + Ь; где Y — сиг-
нал детектора, X — количество вещества, величина т — градуировочный коэффици-
ент, или отклик на единицу количества вещества). Масс-спектрометры по сравнению
с другими приборами имеют великолепный диапазон, в котором сохраняется линейная
градуировочная зависимость (до пяти порядков величины). Большой линейный диапа-
зон позволяет свести к минимуму число разбавлений образца. Если линейный диапа-
зон мал, то может использоваться и квадратичная градуировочная зависимость. Если не
используются изотопные стандарты, то применяется градуировка по внутреннему или
внешнему стандарту.
Градуировка по внешнему стандарту
Градуировка по внешнему стандарту — это самый простой вид градуировки, когда
строится градуировочная зависимость для ряда стандартных растворов с разными
концентрациями.
Определяется градуировочная зависимость так: А = rf С + Ь, где А — сигнал детек-
тора, С — количество определяемого вещества, rf — градуировочный коэффициент,
b — постоянная. При внешней градуировке по площади пика (сигнал детектора) с по-
мощью градуировочной зависимости находится количество определяемого аналита.
Внешняя градуировка используется, когда матричный эффект отсутствует или он на-
столько мал, что им можно пренебречь.
Градуировка по внутреннему стандарту
Градуировка по внутреннему стандарту производится так же, как и по внешнему, с той
разницей, что добавляется дополнительный внутренний стандарт, который служит для
введения поправки на потери при очистке или на дрейф сигнала детектора (глава 2).
В качестве внутреннего стандарта выбирают соединение, которое, как предполагает-
ся, отсутствует в анализируемом образце, обычно это изомер или аналог определяемого
соединения, не встречающийся в природе. Его можно добавлять к образцу на разных
стадиях: перед экстракцией, концентрированием, очисткой или вводом в прибор. Мо-
жет использоваться набор внутренних стандартов для контроля каждой стадии анализа.
Степень извлечения внутренних стандартов, оцениваемая сравнением их полученной
концентрации с исходной (обычно как процент от исходной), может служить критерием
для оценки качества. При градуировке по методу внутреннего стандарта площадь пика
аналита корректируют на степень извлечения внутреннего стандарта. Этот способ ис-
пользуется для уменьшения отклонений вследствие дрейфа сигнала, изменений сигнала
вследствие матричного эффекта или потерь при экстракции. Градуировка по внутрен-
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
нему стандарту дает наилучшие результаты, когда определяемое вещество и внутренний
стандарт имеют одни и те же свойства и ведут себя одинаково. Это значительно улучша-
ет воспроизводимость и точность определения. Если поведение внутреннего стандарта
и определяемого вещества различно, то воспроизводимость и точность могут быть даже
хуже, чем при использовании внешнего стандарта. Наилучшие результаты дает добавле-
ние изотопных аналогов определяемых соединений. Этот вариант называется методом
изотопного разбавления.
15.3.2. Особенности масс-спектрометрии с изотопным
разбавлением
Некоторые элементы встречаются в природе в виде двух и более изотопов в определен-
ных соотношениях. Например, стабильные изотопы углерода имеют атомную массу 12
и 13, причем содержание изотопа углерода-13 (13С) составляет 1,1 % от содержания изо-
топа 12С. Поскольку относительное содержание изотопа 13С столь мало, вероятность
того, что данный атом углерода представляет собой изотоп 13С, составляет около 1:100.
Молекулы ХДД/ХДФ содержат 12 атомов углерода. Специальными приемами синтеза
можно получить молекулу диоксина, в которой все атомы углерода будут изотопами 13С.
Если такой изотопномеченый диоксин, содержащий только 13С, добавить к образцу, со-
держащему нативные диоксины, этот синтетический диоксин будет иметь массу на 12
атомных единиц массы (Дальтон) больше, чем масса соответствующих нативных диок-
синов, изначально присутствующих в образце. В масс-спектрометре сигналы нативных
и изотопномеченых диоксинов четко различаются. Поскольку точно известно, какое
количество изотопномеченых диоксинов добавлено к образцу перед анализом, можно
определить процент их извлечения. Если принять, что химические и физические свой-
ства нативных и соответствующих изотопномеченых диоксинов однаковы на всех ста-
диях аналитического процесса, то факторы, влияющие на обнаружение аналита (низ-
кая эффективность извлечения, потери в процессе очистки и инжектирования в ГХ/
МСВР систему), могут считаться такими же, как и для соответствующих изотопноме-
ченых соединений. К счастью, изотопномеченые 13С-стандарты доступны для многих
ХДД/ХДФ, включая все семнадцать 2,3,7,8-замещенных конгенеров. Для ХДД/ХДФ
имеется также несколько стандартов, меченных изотопом 37С1. В процессе градуировки
устанавливают соотношение между нативными и мечеными соединениями и, таким об-
разом, любые потери в процессе подготовки образца к анализу, при концентрировании
или вследствие дрейфа сигнала автоматически учитываются, поскольку меченые и на-
тивные соединения испытывают эти влияния в одинаковой степени (глава 2).
Добавление различных изотопномеченых ХДД/ХДФ на разных стадиях анализа
дает ряд преимуществ:
• возможность прослеживать эффективность экстракции и каждой стадии подго-
товки пробы и анализа и корректировать результаты с учетом потерь и дрейфа
прибора;
• возможность определять эффективность экстракции изотопных внутренних
стандартов по методу внутреннего стандарта, а также осуществлять контроль ка-
чества на всех стадиях экстракции образца и очистки экстракта;
• изотопные внутренние стандарты могут быть использованы как маркеры хрома-
тографических времен удерживания для подтверждения правильной идентифи-
15.3. Количественный анализ и метод изотопного разбавления 419
кации аналитов. Во многих случаях это может быть не так важно для конгенеров,
не замещенных в положениях 2, 3, 7, 8, которые не подвержены биоаккумуля-
ции и не считаются токсичными, но все 2,3,7,8-замещенные конгенеры должны
быть правильно идентифицированы. Это может стать реальной проблемой при
анализе «грязных» образцов, когда времена удерживания могут сдвинуться назад
на 10 с и более;
• внутренние стандарты можно использовать для увеличения чувствительности
при низких концентрациях, поскольку они могут действовать как носитель,
транспортирующий ХДД и ХДФ с более низкими концентрациями через барье-
ры трехстадийной очистки и анализа. Они помогают также расширитьлинейный
участок градуировочной зависимости в области низких концентраций.
15.3.3. Стандартные образцы и другие средства оценки
эффективности
Стандартные образцы, межлабораторные испытания и аккредитация — важные пред-
посылки для получения точных и воспроизводимых результатов, поэтому они должны
быть включены в систему контроля качества каждой лаборатории.
Стандартные образцы
Имеется ряд стандартных образцов (certified reference materials, CRMs) донных отложе-
ний и биологических материалов для определения ХДД/ХДФ и ПХБ. К сожалению,
вследствие сильной гидрофобности этих соединений стандартные образцы для воды
отсутствуют. Нет также стандартных образцов для воздуха. ХДД/ХДФ и ПХБ легко
адсорбируются на частицах, и приготовить однородные стандартные образцы для воз-
духа и воды невозможно. Национальный институт стандартов и технологий (National
Institute of Standards and Technology, NIST), Правительственная химическая лаборато-
рия (Laboratory of the Government Chemist, LGC), Кембриджская изотопная лаборато-
рия (Cambridge Isotope Labs, CIL) и Веллингтонская лаборатория (Wellington laboratories)
выпускают стандартные образцы донных отложений и биологических материалов.
Эти стандартные образцы природных объектов прекрасно подходят для контроля ка-
чества, так как обычно их матрица содержит коэкстрагирующиеся вещества и аналиты,
которые присутствуют в концентрациях, характерных для природных образцов. Анализ
стандартных образцов на основе реальных матриц является наилучшим тестом на то,
что метод является стабильным и контролируемым.
Межлабораторные испытания
Межлабораторные испытания также позволяют определить, является ли метод ста-
бильным и статистически контролируемым, а также случаются ли выбросы и какова его
воспроизводимость. Анализ слепых проб помогает определить точность и качество ана-
литических стандартов и процедур, используемых для исключения наложений. Ежегод-
но проводится ряд межлабораторных испытаний и проверок для оценки технической
компетентности аналитических лабораторий, организуемых Комиссией по междуна-
родным интеркалибрационным испытаниям (Dioxin International Intercalibration Study,
Intercal) и Комиссией по обеспечению качества информации для морского мониторинга
окружающей среды в Европе (Quality Assurance of Information for Marine Environmental
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
Monitoring in Europe, QUASIMEME). Результаты, получаемые лабораториями, участву-
ющими в регулярных межлабораторных испытаниях или в проверках эффективности,
обычно отличаются более высокой точностью и воспроизводимостью, чем у лаборато-
рий, не участвующих в испытаниях.
Аккредитация
Большинство лабораторий, выполняющих рутинные анализы, аккредитуется по стан-
дарту ISO 17025 (ISO/IEC, 2005). Стандарт выдвигает ключевые требования к лабора-
ториям по обеспечению качества результатов анализа для возможности их дальней-
шего использования (Bethem etal., 2003). Стандарт ISO 17025 требует наличия системы
контроля качества, включая «руководство по качеству», детального документирова-
ния методов и результатов, обученного и опытного персонала, соблюдения условий
работы в лаборатории и системы оценки результатов (контрольные карты). Лабора-
тория также инспектируется опытными аудиторами для проверки соблюдения тре-
бований стандарта ISO 17025 и процедур статистического контроля. В большинстве
стран требуется аккредитация по ISO 17025, особенно при анализе образцов для целей
госконтроля.
15.3.4. Воспроизводимость, точность и неопределенность
Содержание хлорированных дибензо-л-диоксинов и дибензофуранов нормируется для
многих типов образцов: питьевой воды, промышленных выбросов, донных отложений,
почв, атмосферного воздуха и воздуха рабочих помещений, газовых выбросов, биоты
и пищевых продуктов. Вследствие высокой токсичности ХДД, ХДФ и диоксиноподоб-
ных ПХБ, которые имеют специфическую диоксиновую токсичность (ПХБ 77, 81, 105,
114, 118, 123, 126, 156, 157, 167, 169 и 189), необходимо, чтобы метод был чувствителен, то-
чен и воспроизводим. Этим требованиям удовлетворяет метод изотопного разбавления.
Поскольку он основан на определении соотношения нативных и меченых соединений,
для обеспечения точности чрезвычайно важны такие операции, как измерение исход-
ной массы образца и количества добавленных стандартов. Если нативные стандарты
достаточно хорошо охарактеризованы по каким-либо вторичным стандартам и коэф-
фициенты чувствительности определены правильно, то точность определения находит-
ся обычно в пределах ±10% от истинной величины. Неопределенность для отдельных
конгенеров — от 15 до 20%. Так как величины суммарного токсического эквивалента
TEQ представляют собой суммы токсических эквивалентов всех конгенеров, неопре-
деленность TEQ обычно ниже — менее 10% (корень квадратный из суммы квадратов
неопределенностей отдельных составляющих).
15.4. Газохроматографические методы
15.4.1. Основные положения газовой хроматографии
Газовая хроматография (ГХ) — важнейшая часть большинства аналитических систем
для определения ХДД/ХДФ, в которых масс-спектрометр служит хроматографическим
детектором. Функция газохроматографической колонки двоякая: 1) отделить ХДД/ХДФ
15.4. Газохроматографические методы 421
от любых возможных наложений коэктрагирующихся органических веществ, которые
еще остаются в экстракте даже после исчерпывающей очистки, и 2) разделить сем-
надцать 2,3,7,8-замещенных конгенеров и отделить их от остальных, не относящихся
к токсичным. Хроматографические времена удерживания специфических конгенеров
ХДД/ХДФ — ключевая характеристика для идентификации пиков. Чтобы обеспечить
наилучшее разделение в каждом отдельном анализе, времена удерживания ХДД/ХДФ
варьируют путем выбора соответствующей ГХ-колонки и подбора условий разделения.
Перечень ГХ-колонок, используемых в настоящее время, и их основные характеристики
приведены в разделе 15.4.2.
Чтобы ГХ часть аналитической системы для определения ХДД/ХДФ функциониро-
вала нормально, нужна эффективная очистка экстракта образца. Например, коэкстра-
гирующиеся вещества, удаляемые из экстракта очисткой на колонке с кислотно/основ-
ным силикагелем (рис. 15.3), могут вызвать серьезные нарушения в работе ГХ-колонки,
соединенной с масс-спектрометром, и даже привести ее в негодность. Чрезмерные
количества отдельных компонентов смеси, поступающей в колонку, также могут вли-
ять на времена удерживания ХДД/ХДФ. К счастью, наличие изотопных стандартов
позволяет оценить качество разделения на используемой колонке. В настоящее время
усилия ученых направлены на ускорение анализа без потери разделяющей способно-
сти ГХ-колонки (см. упомянутую ниже быструю ГХ) и на разработку хроматографиче-
ских методов, способных разделять сотни компонентов в образце помимо ХДД/ХДФ
(см. ГХ х ГХ, описанную ниже в разделе 15.7.1).
15.4.2. Колонки, используемые для определения ХДД/ХДФ
К сожалению, сейчас не существует ни колонки, способной разделить все ХДД/ХДФ
в одном опыте, ни колонки, которая могла хотя бы разделить все токсичные 2,3,7,8-за-
мещенные ХДД/ХДФ (de Boer, 1999; de Boer and Cofino, 2002; deBoer and Wells, 2006;
Ryan et aL, 1991). Обычно в качестве основной используется колонка с неполярной
неподвижной фазой (НФ) из полиметилсилоксана с 5% фенильных групп типа DB-
5, SPB-5, а если ее разделение недостаточно, то дополнительная колонка с полярной
НФ типа DB-225. В некоторых лабораториях используется НФ с приставкой MS (на-
пример DB-5MS, которая несколько отличается от обычной НФ DB-5). НФ DB-5MS
хорошо отделяет 2,3,7,8-ТХДФ от его ближайших соседей, чего не позволяет НФ
DB-5. К сожалению, некоторые другие коэлюирующиеся 2,3,7,8-замещенные изоме-
ры не разделяются на НФ DB-5MS, но разделяются на DB-5. Требуется по крайней
мере 186000 теоретических тарелок, чтобы отделить так называемую «критическую
пару» 1237/1238-изомеры от 2378-изомера (рис. 15.4), для этого нужна колонка 60 м х
0,25 мм х 0,25 мкм или 40 м х 0,18 мм х 0,18 мкм. ПХБ обычно анализируют на той
же колонке, что и ХДД/ХДФ. НФ с 5% фенильных групп может разделить группы
конгенеров от тетра- до октахлор без перекрывания, т. е. все тетра-ХДД/ХДФ элюиру-
ются до пентахлор-ХДД/ХДФ, а те — до гекса-ХДД/ХДФ, после которых элюируются
гепта-ХДД/ХДФ и окта-ХДД/ХДФ. Это показано в правой части рис. 15.4. Имеется
стандартная смесь для фиксации окон элюирования, которая содержит первый и по-
следний конгенер, элюирующийся в каждом окне (рис. 15.4). Фураны имеют более
широкие окна времен удерживания, чем диоксины, поэтому обычно именно фура-
ны используются в качестве граничных реперов, все соответствующие конгенеры
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
а
0
Oct26-10-Cs3wt
Oct26- 10-Cs3wt
2:Voltage SIR 20 Channels EI+
305.8987
8.5le5
100
%
17.36
2.3.7.8-TCDF
1.2.8.9-TCDF 18-91
Oct25-I0-Cs3wt
Oct25-10-Cs3wt
1,3.6.8-TCDF
100 ]f 2.3.7.8-TCDF
2:Voltage SIR 20 Channels EI+
305.8987
1.23e6
Oct26-10-Cs3wt 2:Voltage SI R 20 Channels E1 +
317.9389
8.0le6
Oct26-10-Cs3wt 2:Voltage SIR 20 Channels EI+
323.8834
2.3.7,8-TCDD 2‘66e5
15.93 1.2.3.7-TCDD/ I .,2,3.9-TCDD
100 1.2.3.8-TCDD |7 97 1,3.6.8-TCDD
Oct26- 10-Cs3wt
2:Voltage SIR 20 Channels EI+
335.9236
Oct25-10-Cs3wt
100
%
0
A = 1.3.4,6.8-Pcnta-CDF
В = 1.2.3.7,8- Penta-CDF
C = 2.3.4.7.8- Penta-CDF
D = 1,2.3.8.9- Penta-CDF
Oct25-1()-Cs3wt
3:Voltage SIR 20 Channels EI +
341.8568
2.76e6
4:VoltageSlR 16 Channels EI +
375.8178
100
%
0
E - 1.2,3,4.6.8 Hexa-CDF
F- 1,2,3.4.7.8 Hexa-CDF
G - 2.3.4.6,7,8 Hexa-CDF
H- 1,2.3.7.8.9-Hexa-CDF
Oct25-IO-Cs3wt
100
%
0
5:Voltage SIR 11 Channels EI +
29.01
1.2.3.4.6,7,8-Hepta-CDF
409.7788
1.98e6
100
%
0
17.80 |7.96
1.95e6
-2.3.7.8-TCDD
Oct25-10-Cs3wt
100
%
6:Voltage SIR 9 Channels EI +
443.7398
33.47 159e6
Oeta-CDF
16.00 16.50 17.00 17.50 18.00 18.50 19.00
16.00 18.00 20.00 22.00 24.00 26.00 28.00 30.00 32.00 34.00 36.00
Время
Время
0
Рис. 15.4. (а) Масс-хроматограмма в режиме МЗИ-стандартов тетрахлор-ХДД/ХДФ и со-
ответствующих |3С-изотопномеченых ХДД/ХДФ-аналогов, используемых для
определения эффективности извлечения после всей аналитической процедуры.
(б) Масс-хроматограммы стандартов ХДФ от тетра- до окта-, показывающие
границы окон времен удерживания ХДД/ХДФ в порядке их увеличения, на ко-
лонке DB-5. В табл. 15.1 показаны массы ионов, регистрируемых в каждом окне
для разных групп соединений
ХДД/ХДФ элюируются в пределах этих границ. Любые соединения, элюирующиеся
вне этих границ, не могут быть отнесены к диоксинам или фуранам с соответствую-
щей степенью хлорирования.
15.5. Масс-спектрометрические методы
15.5.1. Масс-спектрометрия и интегрированные
ГХ/МС-системы
Со времени открытия масс-спектрометрии более 100 лет тому назад она прошла путь
от очень сложного и специализированного исследовательского метода до обычного
15.5. Масс-спектрометрические методы
детектора для газового или жидкостного хроматографа. Сочетание газовой хромато-
графии (ГХ) и масс-спектрометрии (МС) стало прекрасным методом анализа много-
компонентных групп соединений, в частности ХДД/ХДФ, ПХБ и полибромированных
дифениловых эфиров (ПХБЭ), содержащих большое число конгенеров (210, 209 и 209
соответственно). Все они представляют собой стабильные ароматические соединения.
Масс-спектры ароматических соединений обычно очень просты, они содержат макси-
мальный пик молекулярного иона и несколько пиков осколочных ионов. Для достиже-
ния наибольшей чувствительности энергию ионизации понижают, насколько возмож-
но, чтобы минимизировать фрагментацию. В результате пик молекулярного иона всегда
имеет максимальную интенсивность при нескольких малоинтенсивных пиках осколоч-
ных ионов, а масс-спектры изомеров в каждой группе конгенеров практически иден-
тичны1. Таким образом, 17 токсичных ХДД/ХДФ должны быть отделены от остальных
193 нетоксичных конгенеров; капиллярная ГХ — идеальное средство для достижения
этого на ультраследовом уровне.
Все ГХ/МС-системы содержат ряд основных узлов: это система обработки данных
и контроля всей ГХ/МС-системы, система разделения (ГХ), устройство для соединения
ГХ (атмосферное давление) и МС (вакуум), источник ионов, масс-анализатор, детек-
тор/усилитель сигнала и вакуумная система. Все это обязательные элементы, и в боль-
шинстве случаев они аналогичны у разных приборов. Различия в основном состоят
в типе масс-анализатора. Как уже говорилось выше, система разделения требуется,
чтобы разделить изобарные компоненты, а также отделить определяемые соединения
от компонентов матрицы и мешающих соединений. Молекулы должны быть ионизо-
ваны, чтобы ими можно было управлять и анализировать по массам с помощью при-
ложенных напряжений, электрических и магнитных полей. Разделенные по массам,
а вернее по отношениям массы к заряду, ионы детектируются электронным умножите-
лем (рис. 15.2) или фотоумножителем. Ионы притягиваются высоким потенциалом кон-
версионного динода (10—20 кВ), испускающим электроны или фотоны, сигнал которых
регистрируется и усиливается. Вся масс-спектрометрическая система (источник ионов,
масс-анализатор и детектор) находится под вакуумом, чтобы минимизировать ионно-
молекулярные реакции и столкновения в процессе анализа ионов по массам, а также
исключить образование электрического разряда при образовании ионов и детектирова-
нии/усилении сигнала. Система обработки данных управляет всеми узлами, отслежи-
вает их состояние и регистрирует сигнал усилителя; она включает математическое обе-
спечение, которое позволяет аналитику обрабатывать данные и выдавать результаты.
В большинстве масс-спектрометрических систем основные узлы аналогичны (на-
пример система обработки данных, система разделения смесей, соединительная линия
между ГХ и МС, источник ионов, вакуумная система). Главное отличие — это масс-
анализатор. Есть ряд важных соображений при выборе подходящего масс-анализатора
для конкретных приложений. Он должен удовлетворять требованиям к чувствительно-
сти, селективности (определяющей точность и воспроизводимость) и скорости.
Выбор типа и режима работы масс-анализатора зависит от характера анализируе-
мых образцов и лимитируется необходимостью получения достаточно большого объ-
ема данных за время элюирования ГХ-пика. В некоторых случаях анализаторы не могут
1 Иногда при низких энергиях ионизации процессы фрагментации, характерные для неко-
торых изомеров, проявляются сильнее, например орто-эффект. — Прим, перев.
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
быть использованы в режиме сканирования полного масс-спектра, их можно только
переключать между выбранными ионами (мониторинг заданных ионов, МЗИ) для до-
стижения необходимой чувствительности и скорости детектирования целевых соедине-
ний (глава 2). Для определения ХДД/ХДФ важно, чтобы на протяжении элюирования
одного хроматографического пика получалось от семи до десяти измерений. Если полу-
чается меньше семи измерений, гауссовой формы пика может не наблюдаться, и пло-
щади пиков и соответствующие количественные результаты могут быть ошибочными.
Другой ключевой параметр — это разрешение R, т.е. способность прибора разделять
ионы с близкой массой. Разрешение определяется как АЛ//Л/ = (М1 - М^/М, где М}
и М — массы разделяемых ионов, М — целочисленная величина массы (номинальная
масса). Разрешения 10000 достаточно, чтобы исключить большую часть наложений для
2,3,7,8-ТХДД. Обычно стоимость масс-анализатора пропорциональна чувствительно-
сти, селективности (разрешению) и скорости работы анализатора.
15.5.2. Магнитные секторные приборы
Сегодня для определения ХДД/ХДФ чаще всего используются масс-спектрометры вы-
сокого разрешения (МСВР) с двойной фокусировкой, в которых масс-анализатор состо-
ит из сильного электромагнита и электростатического сектора. Магнитный секторный
масс-спектрометр — это наиболее чувствительный и селективный прибор с разреше-
нием от 1000 до 100000. ХДД/ХДФ обычно определяют на магнитных секторных при-
борах в режиме МЗИ при разрешении 10000 (рис. 15.5). Это наилучший компромисс
между разрешением и чувствительностью, и при использовании стандартной очистки
на колонке силикагель/А12О3/активированный уголь этот режим позволяет устранить
большинство наложений. Современные магнитные секторные приборы при разреше-
нии 10000 позволяют достичь уровня чувствительности ниже фемтограммов. Скорость
сканирования у них меньше, чем у квадруполей или у времяпролетных приборов, а сто-
Рис. 15.5. Магнитный секторный масс-спектрометр для определения ХДД/ХДФ. Слева —
газовый хроматограф, высокий куб в центре прибора — электромагнит. Слева
и справа от магнита — электростатические устройства для тонкой фокусировки
ионного пучка для получения более высокого разрешения
15.5. Масс-<
имость выше в 3—5 раз, для них требуется более обученный и квалифицированный
персонал. У магнитного секторного прибора ширина ионного пучка обратно пропор-
циональна разрешению. Магнитное поле изменяется или сканируется так, что пучки
разделенных по массам ионов последовательно фокусируются на детекторе, образуя
масс-спектр. Для сканирования также можно использовать ускоряющее напряжение.
Этот метод предпочтительнее в МСВР в режиме МЗИ, так как скорость сканирования
в этом случае намного выше, чем в случае сканирования магнитного поля. Однако этот
режим имеет тот недостаток, что при уменьшении ускоряющего напряжения уменьша-
ется и чувствительность. Поэтому вся хроматограмма разбивается на окна, в которых
регистрируются группы ионов, массы которых различаются не более чем на 30 %.
В табл. 15.1 приведены специфические ионы, регистрируемые для каждой характе-
ристической группы конгенеров оттетра- до окта-ХДД или ХДФ, атакже продолжитель-
ность регистрации, время задержки и теоретические значения изотопных отношений
Таблица 15.1. Программа регистрации ионов в МСВР для определения ХДД/ХДФ
(ХДФЭ — хлордифениловые эфиры)
Группа ионов m/z (ионы для количественного определения) Соединение Продолжи- тельность измерения, мс Время задерж- ки, мс Теоре- тическое изотопное отношение Допу- стимый интервал
1 303,9016; 305,8987; ТХДФ, 50 10 0,77 0,65-0,89
315,9419; 317,9389; 13С12-ТХДф 25 10 0,77 0,65-0,89
316,9824; ПФК 30 10 — —
319,8965; 321,8936; ТХДД, 50 10 0,77 0,65-0,89
327,8847; 37С1-ТХДД 25 10 — —
331,9368; 333,9339; 13с12-тхдд 25 10 0,78 0,65-0,89
375,8364 Гекса -ХДФЭ 25 10 — —
2 339,8597; 341,8567; ПеХДФ, 50 10 1,61 1,32-1,78
351,9000; 353,8970; 13С12-ПеХДФ, 25 10 1,56 1,32-1,79
354,9792; ПФК 30 10 — —
533,8576; 355,8546; 357,8517; ПеХДД, 50 10 1,55 1,32-1,78
367,8949; 369,8919; 13С12-ПеХДД, 25 10 1,56 1,32-1,79
409,7974 Гепта-ХДФЭ 25 10 — —
3 373,8208; 375,8178; ГкХДФ, 50 10 1,24 1,06-1,43
383,8369; 385,8610; 13С12-ГкХДФ, 25 10 0,52 0,44-0,60
389,8157; 391,8127; ГкХДД, 50 10 1,24 1,05-1,43
392,9760; ПФК, 30 10 — —
401,8559; 403,8529; 13С12-ГкХДД 25 10 1,25 1,06-1,23
445,7555 Окта-ХДФЭ 25 10 — —
4 407,7818; 409,7789; ГпХДФ, 50 10 1,04 0,88-1,19
417,8250; 419,8220; 13С12-ГпХДФ, 25 10 0,45 0,38-0,51
423,7766; 425,7737; ГпХДД, 50 10 1,03 0,88-1,19
435,8169; 437,8140; 13С12-ГпХДД, 25 10 1,04 0,88-1,20
442,9729; ПФК, 30 10 — —
479,7165 Нона-ХДФЭ 25 10 — —
5 441,7428; 443,7400; ОХДФ, 50 10 0,89 0,76-1,02
457,7377; 459,7348; ОХДД, 50 10 0,89 0,76-1,02
469,7779; 471,7750; 13С12-ОХДД, 25 10 0,89 0,76-1,03
480,9697 ПФК 30 10 — —
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
и соответствующие допустимые интервалы их изменения. В режиме МЗИ идет постоян-
ное переключение между величинами m/z регистрируемых ионов по очереди от низких
масс к высоким в данной группе ионов (окне). Каждый ион регистрируется в течение
определенного времени (dwell time). Ускоряющее напряжение увеличивается для пере-
хода к следующему регистрируемому иону с более высокой массой и выдерживается при
этом значении в течение некоторого времени (delay time) для стабилизации прибора и де-
тектора, пока все предыдущие ионы не пройдут через масс-спектрометр. Затем этот ион
регистрируется в течение заданного времени, и ускоряющее напряжение переключается
для регистрации следующего иона. После окончания цикла прибор захватывает ключе-
вую массу — ион из спектра внутреннего стандарта для градуировки шкалы масс (обыч-
но перфторкеросин — ПФК) и регулирует напряжение для минимизации дрейфа и изме-
нений чувствительности; затем стартует новый цикл. Каждое измерение откладывается
как масс-хроматограмма. Когда заканчивается время регистрации группы ионов в дан-
ном окне времен удерживания, магнитное поле переключается на следующую группу
ионов и начинается регистрация специфических ионов из этой группы. Этот процесс
продолжается, пока не будут зарегистрированы все заданные группы ионов.
15.5.3. Тандемный масс-спектрометр
Суть метода тандемной масс-спектрометрии (МС/МС) заключается в следующем. Вы-
бирается какой-либо ион в масс-спектре, которому затем сообщается дополнительная
энергия через стол кновения с молекулами газа — азота или аргона (Reineretal., 1991). Ион,
получивший дополнительную энергию, распадается, и осколочные ионы разделяются
вторым масс-анализатором (глава 4). Этот метод получил название «мониторинг мно-
жественных (заданных) реакций», МЗР (multiple reaction monitoring, MRM), поскольку
регистрируются специфические реакции фрагментации. Типичный тандемный масс-
спектрометр состоит из трех последовательных квадрупольных масс-фильтров. Первый
квадруполь служит для выбора иона-предшественника, который проходит во второй
квадруполь, служащий ячейкой соударений с атомами инертного газа. Этот квадру-
поль работает только в радиочастотном режиме. Все входящие или образующиеся в нем
ионы поступают в третий квадруполь, который настраивается так, чтобы пропускать
только определенные ионы. Диоксины и фураны образуют характерные фрагменты при
отщеплении от молекулярного иона СО и С1 (63 Da), что делает МС/МС очень селек-
тивным методом для этих соединений. Отношение сигнал/шум в МС/МС повышается
за счет уменьшения химического шума. Такие соединения, как ПХБ (которые отщепля-
ют С12) и полибромдифениловые эфиры, ПБДЭ (которые отщепляют Вг2), также могут
определяться тандемной масс-спектрометрией, однако, поскольку отщепление С12 и Вг2
характерно и для многих других галогенсодержащих соединений, МС/МС не являет-
ся для ПХБ и ПБДЭ столь же селективным методом, как для ХДД/ХДФ. Тандемная
масс-спектрометрия в режиме МЗР обычно позволяет осуществить несколько сканов
в секунду (Герц) и достичь предела обнаружения в несколько фемтограммов. Скани-
рование в тандемной масс-спектрометрии может производиться в различных режимах,
в том числе в режиме регистрации осколочных ионов (для выбранного в Q1 иона-пред-
шественника и сканировании Q3), режиме регистрации ионов-предшественников (ска-
нирование Q1 и пропускание определенного иона в Q3) или в режиме сканирования
нейтральных потерь (сканирование Q1 и Q3 с постоянным сдвигом по массе, например
15.5. Масс-спектрометрические методы 427
70 для идентификации полихлорированных соединений). Эти режимы сканирования
идеальны для обнаружения присутствия дополнительных соединений в экстрактах об-
разцов, но они на 1—2 порядка менее чувствительны, чем режим МЗР. Тандемные масс-
спектрометрические системы примерно в три раза дороже, чем обычные квадрупольные
приборы. Они сложнее в настройке и эксплуатации, поскольку приходится оптимизи-
ровать дополнительные параметры, такие как энергия столкновений и давление газа
для столкновений. В некоторых новых приборах это делается автоматически с помощью
предварительно запрограммированных процедур анализа, а также стандартов. Ионные
ловушки тоже могут работать в режиме МС/МС (Plomley et al., 2000), причем их стои-
мость меньше, чем тандемных квадрупольных приборов (глава 4).
15.5.4. Масс-спектрометр сверхвысокого разрешения
Масс-спектрометры ионного циклотронного резонанса с преобразованием Фурье
(FTICRMS) и орбитальные ловушки могут давать разрешение более 105—106 (глава 18).
FTICRMS осуществляется в ячейке, помещенной в сильный магнит (обычно >5 Тесла),
при сверхнизком давлении (<10-10торр). Ионы совершают циклическое движение в маг-
нитном поле с частотой со, где со = zeB/m. При приложении широкополосного радио-
частотного сигнала ионы возбуждаются и переходят на более высокие орбиты. Сфор-
мированный таким образом пакет ионов при движении генерирует переменный ток
в приемном контуре. Сигнал может быть разложен путем преобразования Фурье для
определения величин m/z ионов, находящихся в ячейке. Чем дольше ионы циркулируют
в ячейке, тем выше разрешение и точность измерения масс. В масштабе времени ГХ-
пика (1—2 с на скан) может быть получено разрешение 50000—100000 для ХДД/ХДФ
при точности измерения масс лучше 1 ррт и пределе обнаружения на уровне нескольких
пикограммов (Taguchietal., 2010).
15.5.5. Методы ионизации для определения ХДД/ХДФ
Ионизация молекул, поступающих в источник ионов масс-спектрометра, — одна из важ-
нейших стадий масс-спектрометрического анализа. При определении ХДД/ХДФ могут
использоваться режимы регистрации как положительных, так и отрицательных ионов.
Чаще всего в масс-спектрометрическом анализе используется электронная ионизация
при энергии электронов 70 эВ. Это наилучший компромисс между чувствительностью
и степенью фрагментации (важной при структурных исследованиях). При ультрасле-
довом определении ХДД/ХДФ обычно используют энергию электронов 30—40 эВ для
уменьшения фрагментации и увеличения сигнала молекулярного иона, в результате
чего улучшается чувствительность и понижается предел обнаружения.
Для получения ионов-реагентов, селективных по отношению к ХДД/ХДФ при хи-
мической ионизации (ХИ), использовался ряд различных газов-реагентов (глава 1). Ос-
новные варианты ХИ, применявшиеся для ионизации ХДД/ХДФ, — протонирование
и перезарядка. Многие из стойких галогенированных органических соединений, таких
как ХДД/ХДФ, легко ионизуются, причем чувствительность к ним в режиме регистра-
ции отрицательных ионов выше. К сожалению, 2,3,7,8-ТХДД — наиболее токсичный
из диоксинов — имеет в этом режиме чувствительность, на два порядка меньшую, чем
при ЭИ (Chernestova et al., 2002). Другие методы, обычно используемые в ЖХ/МС/МС
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
(Zhou etal., 2010а; 2010b), — химическая ионизация при атмосферном давлении (ХИАД)
и фотоионизация при атмосферном давлении (ФИАД) — также применялись для опре-
деления полигалогенированных соединений. При анализе методом ЖХ с обращенной
фазой С18 в условиях ФИАД чувствительность была такой же или чуть лучше, чем в ус-
ловиях ХИАД. Предел обнаружения был на уровне пг/мкл, но была возможность инжек-
тировать большие пробы.
15.5.6. Мониторинг заданных ионов (МЗИ) и мониторинг
заданных реакций (МЗР)
Для увеличения селективности при определении ХДД/ХДФ используются методы
МЗИ и МЗР. Из всех ионов, содержащихся в масс-спектре, регистрируется только не-
большая их часть, характеризующая целевые соединения, при этом масс-анализатор
по очереди выводит эти ионы для регистрации. Время регистрации каждого иона в этом
режиме намного больше, чем когда он регистрируется в режиме сканирования полного
масс-спектра. В результате чувствительность повышается в 10—100 раз (глава 2). В ме-
тоде МЗР регистрируются специфические реакции фрагментации, что снижает хими-
ческий шум (глава 4). Данные представляются в виде масс-хроматограмм ионов, об-
разующихся в этих реакциях. К сожалению, при этом теряется информация о других
соединениях в образце. Обычно при определении ХДД/ХДФ в режимах МЗИ и МЗР
регистрируют по два или несколько ионов из изотопного кластера, характерного для
галогенированных соединений. Как правило, это молекулярные ионы, а когда это воз-
можно, и осколочные ионы, образующиеся при распаде молекулярных. Критерии для
положительной идентификации включают: а) гауссову форму пика, б) одновременный
максимум хроматографических пиков для всех регистрируемых ионов данного соеди-
нения, в) отклонение хроматографического времени удерживания не более чем на ±2 с,
относительно соответствующего изотопного аналога в пределах данного интервала
времен удерживания. Отношение сигнала к шуму должно быть не менее 3:1, а отноше-
ние изотопных пиков должно отличаться от теоретического не более чем на ±15 %. ГХ/
МСВР в режиме МЗИ — это «золотой стандарт» метода определения ХДД/ХДФ, ди-
оксиноподобных ПХБ и полибромированных дифениловых эфиров (Kolic et al., 2009;
Reiner, 2010).
15.6. Полный набор аналитических методов для
определения ХДД/ХДФ и других стойких
галогенированных соединений
ХДД/ХДФ — первая и наиболее широко изученная группа загрязняющих веществ
окружающей среды, относящихся к стойким органическим загрязнителям (СОЗ). Ряд
организаций и юрисдикций, в том числе Международная организация по стандарти-
зации (ISO), Агентство по охране окружающей среды США (US ЕРА), Министерство
по окружающей среде Онтарио (МОЕ), Европейский Союз (EU) и Япония, использу-
ют аттестованные методики для определения ряда СОЗ; многие из этих методик могут
быть получены непосредственно у этих организаций или имеются в Интернете. Ряд
15.6. Полный набор аналитических методов для определения ХДД/ХДФ
и других стойких галогенированных соединений
методик определения галогенированных СОЗ, используемых в настоящее время, пере-
числен в табл. 15.2.
Таблица 15.2. Современные методы определения ХДД/ХДФ и других СОЗ, используемые
органами государственного регулирования
Аналиты/комментарии Метод Ссылка
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов с об- щим содержанием групп конгенеров в воде и сточных водах. Используется метод изотопного разбавления и ГХ/МСВР USEPA 1613 USEPA (1994b)
Бромированные дифениловые эфиры в воде, почве, донных отложениях и биологических тканях методом ГХ/МСВР USEPA 1614 US EPA(2007)
209 конгенеров ПХБ. Двенадцать диоксиноподбных ПХБ по версии ВОЗ методом ГХ/МСВР, остальные 197 — методом ГХ/МСНР US ЕРА 1668а US EPA(1999)
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов и общее содержание групп конгенеров в газовых выбросах мусоро- сжигателей. Используется метод изотопного разбавления и ГХ/МСВР US ЕРА 23 US EPA(1995)
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов и общее US ЕРА 8290 USEPA
содержание групп конгенеров в материалах и отходах. Ис- пользуется метод изотопного разбавления и ГХ/МСВР (SW-846) (1994a)
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов и общее содержание групп конгенеров в воде и сточных водах. Исполь- зуется метод изотопного разбавления и ГХ/МСВР. Можно использовать ГХ/МС как альтернативный метод. Двенадцать диоксиноподобных ПХБ по версии ВОЗ в объектах окружа- ющей среды методом ГХ/МСВР, можно использовать ГХ/МС как альтернативный метод. ISO 18073 ISO 2004
Двенадцать диоксиноподобных ПХБ по версии ВОЗ в объек- тах окружающей среды методом ГХ/МСВР. Можно использо- вать ГХ/МС как альтернативный метод. ISO 17858 ISO 2006a
Качество воды — определение некоторых полибромиро- ванных дифениловых эфиров в донных отложениях и осад- ках сточных вод. Метод использует экстракцию и газовую хроматографию/масс-спектрометрию. ISO 22032 ISO 2006b
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов, а также общее содержание групп конгенеров в газовых выбросах стационарных источников методом изотопного разбавления и ГХ/МСВР. EN 1948 European Standard (CEN, 1997)
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов, а также общее содержание групп конгенеров и 12 диоксиноподоб- ных ПХБ по версии ВОЗ методом изотопного разбавления и ГХ/МСВР. MOE 3418 Ontario Ministry of the Environment (2010)
Полибромированные дифениловые эфиры в образцах окружа- ющей среды методом ГХ/МСВР MOE 3430 Ontario Ministry of the Environment (2010)
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов, а также ENVCAN Environment
общее содержание групп конгенеров в сточных водах пред- приятий целлюлозно-бумажной промышленности методом изотопного разбавления и ГХ/МСВР l/RM/19 Canada (1992)
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
Таблица 15.2. (окончание)
Аналиты/комментарии Метод Ссылка
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов, а также общее содержание групп конгенеров в газовых выбросах мусо- росжигателей методом изотопного разбавления и ГХ/МСВР JIS КОЗИ JIS (1999а)
Семнадцать 2,3,7,8-замещенных диоксинов и фуранов, а также общее содержание групп конгенеров в сточных водах методом изотопного разбавления и ГХ/МСВР JIS К0312 JIS (1999b)
15.7. Будущее многокомпонентных методов
Большинство полигалогенированных соединений имеют близкие физические и хими-
ческие свойства. Обычно эти соединения неполярны, инертны, летучи или среднелету-
чи (могут быть разделены ГХ), способны к дальнему переносу и биоаккумуляции, они
токсичны или содержат токсичные компоненты. Они растворимы в неполярныхоргани-
ческих растворителях и могут экстрагироваться толуолом или гексаном. Вместе с ними
экстрагируются и другие компоненты матрицы, так что экстракт должен подвергаться
очистке для удаления коэкстрагирующихся соединений, которые могут вызвать иска-
жения хроматографического разделения и подавление сигнала в масс-спектрометре.
Очищенный экстракт может быть подвергнут фракционированию, чтобы разные
определяемые соединения распределились по отдельным фракциям. Этот подход сей-
час часто применяется при анализе объектов окружающей среды, поскольку помимо
ХДД/ХДФ существует много других экотоксикантов. Это, в первую очередь, полих-
лорированные бифенилы (ПХБ), полихлорированные дифениловые эфиры (ПХДЭ),
полибромированные дифениловые эфиры (ПБДЭ), полихлорированные нафталины
(ПХН), полициклические ароматические углеводороды (ПАУ), а также широкий круг
пестицидов и гербицидов. Благодаря фракционированию очищенного экстракта образ-
ца в каждой из фракций определяют только одну или несколько групп соединений, что
сильно упрощает последующую интерпретацию данных ГХ/МСВР. Однако продолжи-
тельность такого полного многокомпонентного анализа и его стоимость даже для одно-
го образца становятся слишком большими, так как для определения всех групп целевых
соединений требуется от трех до пяти отдельных ГХ/МСВР-анализов. Поэтому суще-
ствующая тенденция направлена на то, чтобы в обозримом будущем методики и про-
цедуры на основе ГХ/МСВР обеспечивали более быстрый и менее дорогой анализ, при
котором по возможности большее число соединений определялось бы в одном анали-
тическом опыте. Недавно разработанные методы позволяют достичь этих целей путем
использования быстрой ГХ (MacPherson et al., 1999; Reiner et al., 2000), времяпролетной
масс-спектрометрии (ВПМС), полной двумерной газовой хроматографии (ГХхГХ)
и автоматизации подготовки проб к анализу (глава 6).
15.7.1. Времяпролетный масс-спектрометр высокого
разрешения
Во времяпролетном масс-спектрометре ионы, которые ускоряются разностью по-
тенциалов, приобретают скорости, обратно пропорциональные корню квадратному
15.7. Будущее многокомпонентных методов
из их масс; таким образом, эти ионы могут анализироваться по массам путем изме-
рения времени, требуемого для пролета определенного расстояния в дрейфовом про-
странстве в вакууме. При импульсном прерывании напряжения в источнике ионов
образуются последовательные пакеты ионов, ускоренных до разных скоростей; ионы
разделяются в дрейфовом пространстве по массам, а их токи измеряются детекто-
ром. Масс-спектрометр ВП может сканировать очень быстро (до 500 Гц) даже полные
масс-спектры. В новых конструкциях используется система отражательных линз, так
что траектории ионов имеют вид V или W. Это значительно уменьшает дисперсию
скоростей (аналогично электростатическому сектору в МСВР с двойной фокуси-
ровкой) и позволяет достигать высокого разрешения. Прибор с одним отражателем
(V-траектории) обычно работает при разрешении 1000—2000. Прибор с большим чис-
лом отражателей может обеспечить значительно более высокое разрешение 7000—
100001. Времяпролетные приборы могут детектировать ХДД/ХДФ на уровне несколь-
ких пикограммов. В настоящее время скорость ограничивается системой обработки
данных, которая должна накапливать, регистрировать и обрабатывать данные в ГХ
масштабе времени. Приборы с высоким разрешением дают большие файлы гигабайт-
ных размеров.
Сейчас нет такой аналитической колонки, которая могла бы разделить все 210
ХДД/ХДФ или все 209 ПХБ (Adahchour et al., 2008; Focant et al., 2004b; Reiner et al., 2010).
Эти группы соединений приходится либо разделять на отдельные фракции, либо ана-
лизировать на двух колонках с разными неподвижными фазами. ГХх ГХ может разде-
лять сотни и тысячи соединений в сложных смесях при анализе объектов окружающей
среды (глава 6). Сочетание ГХхГХ с времяпролетным масс-спектрометром высокого
разрешения может обеспечить высокую чувствительность. Более того, высокая разде-
ляющая способность ГХх ГХ позволяет использовать менее дорогой ГХ-детектор, та-
кой как электронозахватный детектор (ЭЗД) или пламеннно-ионизационный детектор
(ПИД). Наибольшие надежды на ГХх ГХ, однако, связаны с ее возможностями значи-
тельно увеличивать число компонентов, которые можно определять в одном анализе, —
по крайней мере десятикратно по сравнению с обычной ГХ.
15.7.2. Автоматизация методов
Большинство стадий определения галогенсодержащих СОЗ может быть автоматизиро-
вано (Focant and DePauw, 2002; Focant et al., 2004d). Существует автоматическая система
PLE, которая позволяет экстрагировать пробы последовательно или параллельно. Раз-
работаны также автоматические системы очистки экстрактов. В этих устройствах ис-
пользуются заранее заполненные и затем запаянные колонки, что способствует мини-
мизации фонового загрязнения. Это особенно важно для соединений типа ПБДЭ, для
которых значительное фоновое загрязнение может существенно затруднить анализ. Ко-
лонки одноразовые, но могут использоваться и повторно (перезаполняться). Эти авто-
матические системы очистки состоят из ряда насосов и вентилей, которые распределяют
экстракт образца на разные колонки для очистки, подводят разные растворители или их
смеси для элюирования целевых аналитов на следующие колонки или в приемники для
1 Разрешение коммерческого ВП-прибора фирмы ЛЕКО серии HRT составляет
100000. —Прим. ред.
Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
Рис. 15.6. Автоматическая система очистки для определения ХДД/ХДФ и других типов
органических соединений. Колонки для очистки, показанные на рис. 15.3,
остались от очистки экстракта образца тканей рыбы на этой системе (FMS Inc.,
Waltham, Massachusetts, USA)
15.7. Будущее многокомпонентных методов
концентрирования перед ГХ/МС-анализом. Автоматические системы концентрирова-
ния имеют датчики для остановки процесса концентрирования при достижении нуж-
ного объема. На рис. 15.6 показана коммерческая система для автоматической очистки
экстрактов образцов рыбы для определения ХДД/ХДФ и ПХБ .
15.7.3. Ограничения анализа
Хотя образцы можно экстрагировать для одновременного определения в экстракте сра-
зу многих групп соединений методом ГХ/МСВР, при последующей очистке экстрактов
необходимо удалить основной массив компонентов матрицы, поскольку концентрации
галогенсодержащих СОЗ в большей части образцов обычно намного меньше, чем кон-
центрации других соединений, таких как гумусовые кислоты и липиды. Чтобы обеспе-
чить требуемый предел обнаружения, необходимо сконцентрировать экстракт образца
до нужной степени, причем требуется высокая степень концентрирования (105—106).
Что касается конечной детектирующей системы, то ГХ/МСВР в режиме МЗИ — это «зо-
лотой стандарт» для определения ХДД/ХДФ, диоксиноподобных ПХБ, ПБДЭ и ПХН.
Химическая ионизация отрицательных ионов также используется для определения
ПБДЭ и ПХБ. ГХх ГХ пока не применяется в рутинных анализах, так как обычно ис-
пользуемые ГХ/МСВР-системы не могут обеспечить достаточно быстрого сканиро-
вания без потери чувствительности, которая необходима для достижения требуемого
предела обнаружения. На современном уровне анализа предел обнаружения, требуе-
мый для полного определения ХДД/ХДФ и других галогенсодержащих СОЗ, находится
на уровне фемтограммов.
Что касается пределов обнаружения ХДД/ХДФ, которые существенно понизились
за последние 50 лет, может возникнуть вопрос, достигнута ли теперь точка, когда они
уже не могут понижаться дальше? Поскольку число Авогадро очень велико, такое коли-
чество вещества, как пикограмм, содержит очень много молекул. Для наиболее токсич-
ного из ХДД/ХДФ — 2,3,7,8-ТХДД, пикограмм — это 2 миллиарда молекул! Аттограмм
ТХДД (10—18 г) — это все еще 2000 молекул. При специальной технике и полной оптими-
зации всех стадий анализа определение таких количеств, как аттограмм еще возмож-
но (для стандарта!). В настоящее время определение столь малых количеств в образцах
окружающей среды при том, что они неизбежно теряются при экстракции, очистке
и ГХ/МСВР-анализе, кажется научной фантастикой. Казалось бы, можно достичь про-
извольно низких пределов обнаружения, экстрагируя все большее и большее количе-
ство образца. Однако проблема здесь (помимо времени и затрат) в том, что по мере того
как предел обнаружения опускается все ниже и ниже, все труднее и труднее становится
получить чистый холостой опыт.
Благодаря масс-спектрометрии и различным методам сочетания хроматографии
и масс-спектрометрии, ХДД/ХДФ в настоящее время можно реально определять почти
во всех типах образцов окружающей среды на уровне ppt (10-12) или ppq (10-15). Преде-
лы обнаружения будут понижаться и дальше, но реальные тенденции в ближайшем
будущем — это полностью автоматизированные быстрые аналитические системы, для
которых стоимость одного анализа на ХДД/ХДФ будет снижена от нынешних тысячи
долларов и выше до ста.
434 Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
Литература
[1] . AdahchourM., BeensJ., and Brinkman U.A.T. (2008). «Recent developments in the appli-
cation of comprehensive two-dimensional gas chromatography». Journal of Chromatography
A. 1186: 67-108.
[2] . Antunes P., Viana R, Vinhas T., Capelo J. L., Rivera J., and Gaspar E. M. S. M. (2008). «Op-
timization of pressurized liquid extraction (PLE) of dioxins-furans and dioxin-like PCBs
from environmental samples». Taianta. 75: 916—925.
[3] . Bethem R., Boison J., Gale J., Heller D., Lehotay S., Loo J., Musser S., Price P., and Stein S.
(2003). «Establishing the fitness for purpose of mass spectrometric methods». Journal of the
American Society for Mass Spectrometry. 14, 5: 528—541.
[4] . Boyd R. K., Basic C., and Bethem R.A. (2008). Trace Quantitative Analysis by Mass Spec-
trometry. Wiley, Chichester, England
[5] . Carson R. (1962). Silent Spring. Houghton Miffin, Boston, USA.
[6] . CEN. (1997). European Standard EN 1948 1997: Stationary Source Emissions, Determina-
tion of the Mass Concentration of PCDDs/PCDFs. CEN, Brussels, Belgium.
[7] . Chernestsova E.S., Revelsky A. I., Revelsky I. A., Mikhasenko I. A., and Sobolevsky T. G.
(2002). «Determination of polychlorinated dibenzo-p-diozins, dibenzofurans, and biphe-
nyls by gas chromatography/mass spectrometry in the negative chemical ionization mode
with different reagent gases». Mass Spectrometry Reviews. 21, 3: 373—387
[8] . Clement R. E. (1991). «Ultratrace dioxin and dibenzofuran analysis: 30 years of advances.
Analytical Chemistry. 63: 1130A—1139A.
[9] . Clement R. E., and Tosine H. M. (1988). «The gas chromatography/mass spectrometry de-
termination of chlorinated dibenzo-p-dioxins and dibenzofurans». Mass Spectrometry Re-
views. 7, 5: 593—636.
[10] . Covaci A., Voorspels S., Ramos L., Neels H., and Blust R. (2007). «Recent developments in
the analysis of brominated flame retardants and brominated natural compounds». Journal
of Chromatography A. 1153: 145—171.
[11] . de Boer J. (1999). «Capillary gas chromatography for the determination of halogenated
micro-pollutants». Journal of Chromatography A. 843: 179—198.
[12] . de Boer J., and Cofino W. P. (2002). «First world-wide interlaboratory study on polybromi-
nated diphenylethers (PBDEs)». Chemosphere. 46: 625—633.
[13] . de Boer J., and Wells D. E. (2006). «Pitfalls in the analysis of brominated flame retardants
in environmental, human and food samples — including results of three interlaboratory
studies». Trends in Analytical Chemistry. 24: 364—372.
[14] . Environment Canada. (1992). Reference Method for the Determination of PCDDs and
PCDFs in Pulp and Paper Mill Effluents, Report EPS l/RM/19. Environment Canada,
Gatineau, Canada.
[15] . Focant J. F., and DePauw E. (2002). «Fast automated extraction and clean-up of biological
fluids for polychlorinated dibenzo-p-dioxins, dibenzofurans and coplanar polychlorinated
biphenyls analysis». Journal of Chromatography B. 776: 199—212.
[16] . Focant J. F., Pirard C., and DePauw E. (2004a). «Automated sample preparation-fraction-
ation for the measurement of dioxins and related compounds in biological matrices: a re-
view». Taianta. 63: 1101 — 1113.
Литература
[17] . Focant J. F., Reiner E.J., MacPherson K., Kolic T., Sjodin A., Patterson D. G. Jr.,
Reese S.L., Dorman F. L., and Cochran J. (2004b). «Measurement of PCDDs, PCDFs,
and non-ortho-PCBs by comprehensive twodimensional gas chromatography—isotope di-
lution time-of-flight mass spectrometry (GC xGC—IDTOFMS)». Taianta. 63: 1231 — 1240.
[18] . GeensT., Ali N., Roosens L., Neels H., and Covaci A. (2010). «Analytical characteristics of
several new brominated flame retardants». Taianta. 81,4—5: 1865—1869.
[19] . Harrison A.G. (1992). Chemical Ionization Mass Spectrometry. CRC Press, Boca Raton,
Florida, USA.
[20] . Hubschmann H.J. (2001). Handbook of GC/MS, Fundamentals and Applications. Wiley-
VCH, Weinheim, Germany.
[21] . ISO. (2004). ISO 18073: Water Quality — Determination of Tetra- to Octa-chlorinated Dioxins
and Furans — Method Using Isotope Dilution HRGC/HRMS. International Organization for
Standardization, Geneva, Switzerland.
[22] . ISO. (2006a). ISO 17585: Water Quality — Determination of Dioxin-like Polychlorinatedbi-
phenyls — Method Using Gas Chromatography and Mass Spectrometry. International Organi-
zation for Standardization, Geneva, Switzerland.
[23] . ISO. (2006b). ISO 22032: Water Quality — Determination of Selected Polybrominated Di-
phenyl Ethers in Sediment and Sewage Sludge — Method Using Extraction and Gas Chro-
matography/Mass Spectrometry. International Organization for Standardization, Geneva,
Switzerland.
[24] . ISO/IEC. (2005). ISO 17025: 2005: General Requirements for the Competence of Testing
and Calibration Laboratories. International Organization for Standardization, Geneva,
Switzerland.
[25] . JIS. (1999a). K0311 Japanese Industrial Standard: Method for determination of tetra- through
octa- chlorodibenzop-doxins, tetra- through octa- chlorinated furans and coplanar polychlorin-
ated biphenyls in stationary source emissions. Japanese Standards Association, Tokyo, Japan.
[26] . JIS. (1999b). K0312 Japanese Industrial Standard: Method for determination of tetra- through
octa- chlorodibenzop-doxins, tetra- through octa- chlorinatedfurans and coplanar polychlorin-
ated biphenyls in industrial water and wastewater. Japanese Standards Association, Tokyo,
Japan.
[27] . Kolic T. M., Shen L., MacPherson K., Fayez L., Gobran T., Helm P., Marvin C.H., Ar-
senault G., Reiner E. J. (2009). «The analysis of halogenated flame retardants by gas chro-
matography — high resolution mass spectrometry in environmental samples». Journal of
Chromatographic Science. 47: 83—91.
[28] . Lamparski L. L., Nesterick T. J., and Stehl R. H. (1979). «Determination of part-per-tril-
lion concentrations of 2,3,7,8-tetrachlorodibenzo-p-dioxin in fish». Analytical Chemistry.
51: 1453-1458.
[29] . MacPherson K. A., Reiner E. J., ChenT., Brunato R., and Bogard M. A. (1999). «Optimiza-
tion of gas chromatographic parameters for reduced analysis times of chlorinated organic
compounds». Organohalogen Compounds. 40: 19—23.
[30] . Okey A. B. (2007). «An aryl hydrocarbon receptor odyssey to the shores of toxicology: The
Deichmann Lecture, International Congress of Toxicology-XI». Toxicological Sciences. 98,
1:5-38.
[31] . Ontario Ministry of the Environment. (2010a). The Determination of Polychlorinated Diben-
Zo-P-Dioxins, Polychlorinated Furans and Dioxin-like PCBs in Environmental Matrices by
436 Глава 15. Определение стойких галогенсодержащих соединений:
хлорированные дибензо-п-диоксины и дибензофураны
GC—HRMS. Method DFPCBE3418. Environment Ontario Laboratory Services Branch,
Toronto, Ontario, Canada.
[32] . Ontario Ministry of the Environment. (2010b). The Determination of Poly Brominated Di-
phenyl Ethers in Environmental Matrices by GC—HRMS. Method BDE-3430. Environment
Ontario Laboratory Services Branch, Toronto, Ontario, Canada.
[33] . Plomley J. B., Lausevic M., and March R. E. (2000). «Determination of dioxins/furans and
PCBs by quadrupole iontrap gas chromatography—mass spectrometry». Mass Spectrometry
Reviews. 19, 3: 305—365,
[34] . Poole S.K., Dean T.A., Oudsema J.W., and Poole C. F. (1990). «Sample preparation for
chromatographic separations: an overview». Analytical Chimica Acta. 236: 3—42.
[35] . Reiner E.J. (2010). «Analysis of dioxin and related compounds». Mass Spectrometry Re-
views. 29, 4: 526-559.
[36] . Reiner E. J., Schellenberg D. FL, and Taguchi V. Y. (1991). «Environmental applications for
the analysis of chlorinated dibenzo-p-dioxins and dibenzofurans using mass spectrome-
try/mass spectrometry». Environmental and Science Technology. 25: 110—117.
[37] . Reiner E. J., MacPherson K. A., Brunato R., Chen T., Bogard M. A., Boden A. R., and Lad-
wig G. (2000). «Analysis of persistent organic pollutants (POPs) using microbore columns».
Organohalogen Compounds. 45: 17—20.
[38] . Reiner E. J., Clement R. E., Okey A. B., and Marvin С. H. (2006). «Advances in the analyti-
cal techniques for polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans and
dioxin-like PCBs». Analytical and Bioanalytical Chemistry. 386: 791—806.
[39] . Reiner E.J., Boden A. R., Chen, T, MacPherson K.A., and Muscalu A. M. (2010). «Ad-
vances in the analysis of persistent halogenated organic compounds». LCGC Europe. 23, 2:
60-70.
[40] . Richter В. E., Jones B. A., Ezzell J. L., Porter N. L., Avdalovic N., and Pohl C. (1996). «Ac-
celerated solvent extraction: a technique for sample preparation». Analytical Chemistry. 68:
1033-1039.
[41] . Ryan J. J., ConacherH. B.S., Panapio L.G., Lau B. P.Y., Hardy J. A., and MasudaY. (1991).
«Gas chromatographic separations of all 136 tetra- to octa-polychlorinated dibenzo-p-di-
oxins and polychlorinated dibenzofurans on nine different stationary phases». Journal of
Chromatography. 541: 131 — 183.
[42] . Taguchi V.Y, Nieckarz R.J., Clement R.E., Krolik S., and Williams R. (2010). «Dioxin
analysis by gas chromatography—Fourier transform ion cyclotron resonance mass spec-
trometry (GC—FTICRM S)». Journal of the American Society for Mass Spectrometry. 21, 11:
1918-1921.
[43] . US EPA. (1994a). US EPA SW-846 Method 8290: Polychlorinated Dibenzodioxins and Poly-
chlorinated Dibenzofurans by High-Resolution Gas Chromatography/High-Resolution Mass
Spectrometry, Revision 0. United States Environmental Protection Agency, Washington,
DC, USA.
[44] . US EPA. (1994b). US EPA Method 1613: Revision B: Tetra-Through Octa-Chlorinated Di-
oxins and Furans by Isotope Dilution HRGC/HRMS, EPA 821-B94-0059. Office of Water,
United States Environmental Protection Agency, Washington, DC, USA.
[45 ]. U S E PA. (1995). US EPA Method 23: Determination of Polychlorinated Dibenzo-p-dioxins and
Polychlorinated Dibenzofurans from Municipal Waste Combustors. United States Environ-
mental Protection Agency, Washington, DC, USA.
Литература
[46] . US ЕРА. (1999). US ЕРА Method 1668: Revision A: Chlorinated Biphenyl Congeners in Water,
Soil, Sediment, and Tissue by HRGC/HRMS, EPA-821-R-00-002. Office of Water, United
States Environmental Protection Agency, Washington, DC, USA.
[47] . US EPA. (2007). US EPA Method 1614: Brominated Diphenyl Ethers in Water Soil, Sediment
and Tissue by HRGC/HRMS, EPA-821-R-07-005. Office of Water, United States Environ-
mental Protection Agency, Washington, DC, USA.
[48] . Van Bavel B., and Abad E. (2008). «Long-term worldwide QA/QC of dioxins and dioxin-
like PCBS in environmental samples». Analytical Chemistry. 80: 3956—3964.
[49] . Van den Berg M., Birnbaum L.S., Denison M., De Vito M., Farland W., Feeley M., Fie-
dler H., Hakansson H., Hanberg A., Haws L., Rose M., Safe S., Schrenk D., Toyama C.,
Tritscher A., Tuomisto J., Tysklind M., Walker N., and Peterson R. E. (2006). «The 2005
World Health Organization reevaluation of human and mammalian toxic equivalency fac-
tors for dioxins and dioxin-like compounds». Toxicology Science. 93: 223—241
[50] . Zhou S.N., Reiner E.J., Marvin C., Kolic T., Riddell N., Helm P., Dorman F., Mis-
selwitz M., and Brindle I. D. (2010a). «Liquid chromatography atmospheric pressure pho-
toionization tandem mass spectrometry for analysis of 36 halogenated flame retardants in
fish». Journal of Chromatography A. 1217, 633—664.
[51] . Zhou S.N., Reiner E.J., Marvin C., Helm P., Riddell N., Dorman F., Misselwitz M.,
Shen L., Crozier P., MacPherson K., and Brindle I.D. (2010b). «Development of liquid
chromatography atmospheric pressure chemical ionization tandem mass spectrometry for
analysis of halogenated flame retardants in wastewater». Analytical and Bioanalytical Chem-
istry. 396, 1311-1320.
ГЛАВА 16
МАСС-
СПЕКТРОМЕТРИЯ
АТМОСФЕРНЫХ
АЭРОЗОЛЕЙ
Клаус-Петер Хинц
16.1. Введение
Атмосферные газы, а также субмикронные и микронные твердые и жидкие частицы,
присутствующие в атмосфере, существенно влияют на атмосферные физические и хи-
мические процессы. Широкое обсуждение проблем, связанных с загрязнением воз-
духа, увеличением концентрации мелкодисперсной пыли в атмософере и изменением
климата, проблем аллергических реакций, связаных с наличием пыли в воздухе, и, на-
конец, проблем заражения инфекционными и другими заболеваниями, источником
которых являются помещения больниц, указывает на растущий интерес общества
к проблематике воздействия аэрозолей на окружающую среду и на здоровье человека.
Наличие субмикронных частиц в газовых смесях, используемых в нанотехнологии или
при проведении технологических процессов в специализированных чистых комнатах,
влияет на качество выпускаемой продукции. Свойства аэрозолей сильно варьируются
в пространстве и во времени из-за постоянного взаимодействия между аэрозольными
частицами и окружающей газовой фазой. Детальная характеризация аэрозольных ча-
стиц, определение твердых компонент аэрозольных частиц, а также мониторинг вза-
имодействия аэрозолей с компонентами газовой среды являются сложной аналитиче-
ской задачей. В данной главе описаны основные методы характеризации атмосферных
аэрозолей, а также последние методологичекие достижения. В частности, представлены
параметры основных измерительных приборов для непосредственного измерения аэро-
золей и даны примеры их применения.
16.2. Свойства аэрозолей и их воздействие
на окружающую среду
Смесь, состоящая из газов и взвеси жидких или твердых частиц, называется аэрозолем.
Диаметр аэрозольных частиц составляет от 1 нм до 100 мкм. Размер частицы является
важным параметром, поскольку он существенно влияет на время пребывания частицы
16.2. Свойства аэрозолей и их воздействие на окружающую среду
в окружающем газе (Jaenicke, 1988). Природные и антропогенные источники влияют
на состав атмосферных аэрозолей. Вулканические выбросы, горение биомассы, испаре-
ние с поверхности растений и океанов, пустынные бури — вот неполный список некото-
рых основных природных источников аэрозолей. Например, вулкан Эйяфьятлайокудль
(Исландия) весной 2010 года изверг гигантское облако пепла на высоту до 6 км над уров-
нем моря. Наличие этого облака существенно влияло на воздушное сообщение в Европе
в течение нескольких недель (рис. 16.1). Сжигание биомассы или ископаемых видов то-
плива в промышленности, выхлопы транспортных средств, дым от отопительных си-
стем в частных домах и сжигание растительности при проведении сельхозработ являют-
ся основными антропогенными процессами, приводящими к появлению газов и частиц
в атмосфере. В процессе горения образуется широкий спектр химических соединений.
При горении образуются разнообразные газовые компоненты: диоксид углерода (СО2,
парниковый газ), оксид азота, диоксид серы (SO2), озон, аммиак и летучие органические
соединения (ЛОС). Под влиянием солнечного света ЛОС могут окисляться и образовы-
вать твердые частицы и продукты конденсации. Результатом этих процессов является
формирование вторичных аэрозольных частиц, отличающихся от первичных, которые
выбрасываются в атмосферу непосредственно из указанных выше источников. Примеры
первичных аэрозольных частиц — это морская соль, минеральная пыль, пыль от транс-
портных средств, зола, сажа и разнообразная пыль биологического происхождения.
Пыль биологического происхождения может образовываться из измельченных обломков
растений, из микроорганизмов (бактерии, вирусы), а также из спор и пыльцы. Частицы,
Рис. 16.1. Облако пепла (слабо различимый коричневый шлейф внутри треугольника)
распространяется из вулкана Эйяфьятлайокудль (Исландия) на области Цен-
тральной Европы (картинка получена из NASA/GSFC, спутник наблюдения
MODIS, 15 апреля 2010, 13:30 UTC)
440 Глава 16. Масс-спектрометрия атмосферных аэрозолей
образовавшиеся в атмосфере, осаждаются на поверхность земли с атмосферными осад-
ками. Круговорот аэрозолей в атмосфере представлен на рис. 16.2 (Poschl, 2005).
Природа и концентрация газов и частиц, присутствующих в атмосфере, оказывают
влияние на возникновение и развитие таких атмосферных явлений, как туман, облака,
дым, смог. При этом местные, региональные или глобальные источники атмосферных
частиц оказывают влияние на масштабы и характер соответствующих атмосферных
явлений. Важными характеристиками взвешенных в воздухе частиц являются их раз-
мер, форма, оптические и электрические свойства, морфология, химический состав
и концентрация.
В некоторых случаях атмосферные частицы состоят из столь разнообразных хими-
ческих компонентов, что почти каждая частица является уникальной. Однако частицы
могут быть отсортированы по группам, где они имеют примерно одинаковые размеры
или схожий химический состав. Размер и химический состав отдельной частицы зави-
сит от ее происхождения и характера процессов ее превращения (процессов трансфор-
мации), произошедших с этой частицей в атмосфере. Размер частицы и ее химический
состав непосредственно влияют на характер реакций частицы с атмосферными газами,
поверхностью других частиц и определяют степень биологической активности. Другой
важной характеристикой совокупности частиц в атмофере является их концентрация,
которая варьируется от нескольких сотен частиц в см3 на полюсах Земли до 10000 ча-
стиц в см3 в зданиях. Значительно большие концентрации частиц регистрируются вбли-
зи транспортных развязок (50000—100000 в см3), концентрация в 1000000 частиц/см3
может быть обнаружена в дымовых трубах.
В следующих параграфах будут рассмотрены примеры влияния аэрозольных частиц
на окружающую среду и здоровье человека.
Размер, форма и химический состав частицы оказывают влияние на ее оптические
свойства, такие как рассеяние света и поглощение солнечной радиации. Таким обра-
зом, частицы непосредственно влияют на климат Земли. С другой стороны, частицы
могут выступать в качестве ядер конденсации при образовании облаков и таким обра-
зом косвенно влиять на климат. В течение последних 100 лет наблюдается существенное
Рис. 16.2. Атмосферный круговорот аэрозолей. (Из Poschl U. (2005). «Атмосферные аэро-
золи. Состав, превращение, влияние на климат и здоровье». Angewandte Chemie
International Edition. 44, 46: 7520—7540. Copyright Wiley-VCH Verlag GmbH & Co.
Воспроизводится с разрешения издательства)
16.2. Свойства аэрозолей и их воздействие на окружающую среду
увеличение выбросов парниковых газов (например СО2) и одновременное увеличение
усредненной глобальной температуры (Forster et al., 2007; Randall et al., 2007). Влияние
различных газов и частиц на изменение радиационного баланса системы Земля—атмос-
фера показано на рис. 16.3.
Аэрозольные частицы, попавшие в атмосферу в результате деятельности человека,
препятствуют глобальному потеплению, т.е. Земля охлаждается из-за присутствия ча-
стиц. Величины погрешностей при оценке влияния аэрозолей на климат Земли ука-
зывают на высокую степень неопределенности самих этих оценок, что отражает огра-
ниченное понимание того, как деятельность человека изменяет климат Земли. Одной
из причин этой неопределенности является неполная информация о содержании раз-
Радиационное воздействие на климат в 1750—2005 годах
Факторы, влияющие на радиационное воздействие
Радиационное воздействие (Вт/м2)
Рис. 16.3. Радиационное воздействие некоторых газов и твердых частиц на климатиче-
ские изменения (источник: «Изменение климата 2007»: Физические основы на-
уки. Вклад рабочей группы I в четвертый отчет об оценке межправительствен-
ной группы экспертов по изменению климата, FAQ 2.1, рисунок 2. Кембридж,
University Press. Воспроизводится с разрешения издательства)
442 Глава 16. Масс-спектрометрия атмосферных аэрозолей
нообразных химических веществ в отдельных частицах, так как химический состав су-
щественно влияет на радиационные свойства аэрозольных частиц.
Аэрозольные частицы также оказывают влияние на здоровье человека. Частицы
могут проникать в организм человека вместе с пищей, они могут попасть в легкие при
вдохе или повлиять на организм путем воздействия через кожу. Важные последствия для
здоровья, например, возникают в связи с аллергической реакцией на споры или пыльцу,
при попадании внутрь организма различных микроорганизмов, таких как бактерии или
вирусы, или микроволокнистых частиц, которые могут быть токсичными, вызвать кан-
церогенез и т.п. В регионах с высоким загрязнением воздуха наблюдается повышенная
смертность (Dockery et al., 1993; Peters et al., 1997; Pope et al., 1995).
Ухудшение здоровья в связи с загрязнением воздуха выхлопами транспортных
средств коррелирует с концентрацией аэрозольных частиц как снаружи траспортного
средства, так и внутри него. Измерения концентрации частиц как внутри, так и за преде-
лами транспортных средств показали, что концентрация частиц внутри транспортного
средства значительно зависит от внешней концентрации частиц, от типа транспортного
средства и от наличия кондиционера (Chan et al., 2002; Geiss et al., 2010). Всемирная ор-
ганизация здравоохранения (ВОЗ) определяет величину 50 мкг/м3 как предел допусти-
мой среднесуточной концентрации аэрозольных частиц с диаметром менее 10 микрон
(РМ10) (WHO, 2006). Экспериментальные исследования показали, что этот предел часто
может быть превышен при поездке в автомобиле. Это указывает на необходимость ис-
пользования более эффективных систем фильтрации воздуха в транспортных средствах
и мониторинга содержания аэрозолей в кабине, что, например, полезно для професси-
ональных водителей.
Высокая концентрация частиц в окружающем воздухе опасна не только при работе
на отрытом воздухе, но и при пребывании в жилом доме или в рабочем офисе. В зави-
симости от региона проживания люди проводят 65 % и более времени в зданиях (Leech
Время
Рис. 16.4. Концентрация частиц в воздухе жилой квартиры (число частиц в см3) в периоды
приготовления пищи и горения свечей в городе Мальме, зимой 2007 года. Дан-
ные получены A. Wierzbicka (Lund University, Швеция)
16.3. Приборы и их применение 443
etal., 2002). На рис. 16.4 представлены величины концентрации частиц в воздухе в одной
из жилых квартир в городе Мальме зимой 2007 во время разных этапов приготовления
пищи и при горении свечей. Значительное увеличение концентрации частиц наблюда-
лось во время этих событий. Кроме того, были определены другие важные источники
частиц внутри помещения. Похожие значения концентраций аэрозолей можно найти,
например, на открытом воздухе вблизи дорожно-транспортных развязок. Данные о воз-
можных высоких концентрациях частиц в закрытых помещениях важны для выбора
подходящей комбинации вентиляционной системы и фильтров.
Эти примеры указывают на необходимость детальной характеристики газообраз-
ных и твердых компонентов аэрозолей для лучшего понимания атмосферных процессов
и создания более совершенных методов снижения антропогенного загрязнения воздуха
(Anderson etal., 2003; Charlson etal., 1992; Kiehl, 1999).
Применение масс-спектрометрических методов для определения источников аэро-
зольных частиц и иследования химических превращений в атмосфере приводит к луч-
шему пониманию механизмов формирования частиц и улучшению оценки природного
и антропогенного факторов воздействия аэрозолей как на атмосферные процессы, так
и на здоровье человека. Однако до сих пор существует значительная потребность как
в надежных аналитических приборах, так и в разработанных методах их применения
в лабораторных и полевых условиях. Это необходимо для улучшения характеризации
самих аэрозолей и для правильной количественной оценки воздействия аэрозолей
на людей, животных, растения и климат. Совершенствование приборов для всесто-
роннего анализа аэрозолей также поможет улучшить методы контроля за содержанием
аэрозолей в технических и медицинских аспектах.
За последние 30 лет было разработано множество различных масс-спектро-
метрических приборов и методов для работы как в лабораторных, так и полевых усло-
виях. С их помощью стало возможным изучение одиночных частиц, ансамблей частиц
и атмосферных газов. Масс-спектрометрические методы широко используются в ис-
следованиях аэрозолей, причем многие из них могут быть применены для непосред-
ственного (in situ) анализа широкого спектра химических компонентов, содержащихся
в аэрозолях.
16.3. Приборы и их применение
В этом разделе будут описаны приборы и методы, используемые для установления ха-
рактеристик аэрозолей, с акцентом на методы анализа частиц в реальном времени. Бу-
дет описан один из примеров применения масс-спектрометрического анализа в реаль-
ном времени для непосредственного (онлайн) обнаружения газообразных компонентов
аэрозоля, потому что только одновременное изучение состава как атмосферных газов,
так и частиц может дать полное описание аэрозолей. Во время миграции в атмосфере
частицы, взвешенные в воздухе, испытывают различные физические и химические вза-
имодействия с окружающими газами и другими частицами. В результате коагуляции,
поглощения газа или химических реакций изменяются размер частиц, их структура
и состав. Эти процессы могут привести как к формированию новых частиц, так и к из-
менению свойств уже существующих частиц (например атмосферное старение частиц).
Глава 16. Масс-спектрометрия атмосферных аэрозолей
Образование вторичных органических аэрозолей (ВОА) происходит в результате ат-
мосферного окисления соединений, находящихся в газовой фазе (см. раздел 16.3.7, где
описаны массы-спектрометры, анализирующие группы частиц). Анализ атмосферных
газов необходим для понимания химических процессов в атмосфере, которые приводят
к превращению одних химических соединений в другие, что является важным момен-
том при образовании аэрозолей.
16.3.1. Анализ атмосферных газов
Аналитические приборы широко используются для контроля и изучения газовой со-
ставляющей аэрозолей. Например, следы газов (ЛОС, токсичных соединений) можно
обнаружить, используя масс-спектрометр с ионизацией при переносе протона (Blake
et aL, 2004) или ионную ловушку (Keil и др., 2008). Газовый анализ основных компонен-
тов воздуха, таких как азот, кислород и СО2, может быть выполнен также с помощью
квадрупольного фильтра масс в режиме электронной ионизации. Пример недавно раз-
работанной системы, которая может поместиться в рюкзаке, схематически показан
на рис. 16.5.
Система ULISS состоит из нагреваемого канала напуска, который служит для
поступления атмосферных газов, двухступенчатой системы откачки и анализатора
остаточного газа (АОГ). После прохода через систему напуска молекулы газов попа-
дают в ионизирующий промежуток в вакуумной камере (давление 10-5 мбар), где они
ионизируются электронами с энергией 70 эВ. Этот метод ионизации обеспечивает ка-
чественную и количественную информацию о присутствующих в атмосфере газах.
Образующиеся ионы анализируются с помощью квадрупольного фильтра масс и реги-
стрируются с помощью цилиндра Фарадея. Прибор потребляет энергию двух батарей
и может работать до 4 часов. Этот анализатор пригоден для соединений с молекулярной
массой до 100 а. е. м. (атомная единица массы, одна а. е. м. равна одной двенадцатой мас-
Рис. 16.5. Прототип компактного газового анализатора, который состоит из турбона-
соса (АТН30), двух миниатюрных диафрагменных насосов (KNF 84.4 и 84.3),
масс-анализатора (RGA 100 фирмы SRS), управляющей электроники и двух
12-вольтовых батарей. (Перепечатано из International J. Mass Spectrom. 2010,
Dias J. A., Pieri D., Arkin C. R., Gore E., Griffin T. P., Fladeland M., Bland G., Soto C.,
Madrigal Y., Castillo D., Rojas E. и Achi S. «Использование бортового масс-
спектрометра для изучения газообразных выбросов действующих вулканов».
295, 105—112, Copyright 2010, с разрешения Elsevier)
16.3. Приборы и их применение
сы изотопа углерода ,2С или 1,6606-10-27 кг. Например, изотоп ,2С имеет массу 12 а. е. м.).
Вес системы ULISS (от английской аббревиатуры «использование маловесящих локаль-
ных датчиков и дистанционного зондирования для изучения активных вулканических
выбросов») составляет около 10 кг. Прибор может переноситься одним человеком в рюк-
заке (рис. 16.6). Данная система была применена для мониторинга изменений уровня
газовых выбросов и для детектиривания характерных газов, появляющихся перед на-
чалом опасного периода вулканической активности. Такой прибор может быть очень
полезным для обеспечения раннего предупреждения о вулканических извержениях.
Примеры масс-спектров атмосферных газов до (вверху) и после (внизу) извержения вул-
кана Турриальба в Коста-Рике 5 января 2010 года представлены на рис. 16.7. Прибор был
установлен на борту самолета, а измерения проводились во время полетов над вулканом
Рис. 16.6. Полевые испытания прибора ULISSES на полигоне в районе вулкана Турриаль-
ба; система транспортируется в рюкзаке оператором вместе с блоком GPS отсле-
живания маршрута (красная линия) и блоком отбора проб. Оранжевые стрелки
отмечают вентиляционное отверстие (жерло), образовавшееся в время извер-
женя вулкана 5 января 2010 года. (Перепечатано из International J. Mass Spectrom.
2010, Dias J. A., Pieri, D, Arkin C. R., Gore E., Griffin T. P., Fladeland M., Bland G.,
Soto C., Madrigal Y., Castillo D., Rojas E. и Achi S. «Использование бортового масс-
спектрометра для изучения газообразных выбросов действующих вулканов».
295, 105—112, Copyright 2010, с разрешения Elsevier)
Глава 16. Масс-спектрометрия атмосферных аэрозолей
Рис. 16.7. Спектры, полученные с помощью масс-спекрометрической системы U LISSES.
Полный спектр до (вверху) и после (внизу) извержения вулкана Турриальба
5 января 2010 года. Присутствуют пики воздуха и водяных паров, а также не-
которых основных газообразных компонентов, которые указывают на прояв-
ление вулканичекой активности: SO2, H2S, СО2. После извержения наблюда-
ется интенсивный сигнал SO2. (Перепечатано из International J. Mass Spectrom.
2010, Dias J. A., Pieri, D, Arkin C.R., Gore E., Griffin T. P., Fladeland M., Bland G.,
Soto C., Madrigal Y., Castillo D., Rojas E. и Achi S. «Использование бортового масс-
спектрометра для изучения газообразных выбросов действующих вулканов».
295, 105—112, Copyright 2010, с разрешения Elsevier)
Турриальба. Спектры демонстрируют наличие пиков компонентов воздуха и водяного
пара, а также некоторых из основных маркеров вулканической активности (MBA): СО2,
SO2 и сероводород (H2S). Появление пика SO2 четко прослеживается после извержения,
в то же время интенсивность пика СО2 уменьшается.
16.3. Приборы и их применение
Данный пример показывает преимущество масс-спектрометрии для непосредсвен-
ного (in situ) мониторинга газовых компонентов в атмосфере в жестких окружающих
условиях. Масс-спектрометрические данные могут быть использованы в сочетании
с данными, полученными со спутников, и при автономном химическом анализе ча-
стиц, образующихся в атмосфере над местом извержения. Такое сочетание позволяет
лучше понять весь спектр геофизических явлений, характерных для вулканической де-
ятельности (см. также рис. 16.1). Портативные приборы идеально подходят для работы
в других областях, таких как обеспечение безопасности, или при мониторинге качества
воздуха.
16.3.2. Основные режимы работы приборов при анализе
атмосферных частиц
Методы анализа частиц можно разделить на две основные группы: интегральный ана-
лиз и анализ индивидуальных частиц. Интегральный анализтвердых частиц, как и ана-
лиз индивидуальных частиц, проводимый методом предварительного накопления, осу-
ществляются в два этапа — этап сбора частиц и этап исследования частиц. Сбор частиц
часто осуществляется с помощью фильтра и требует определенного времени. После от-
бора пробы фильтр транспортируется в лабораторию, где собранный материал может
быть проанализирован с помощью различных методов. Фильтрующие устройства, как
правило, малы и могут быть расположены непосредственно у анализируемого источ-
ника, в отличие от масс-спектрометра, который часто является слишком громоздким
и тяжелым для работы, особенно в полевых условиях. Рабочими методами, которые ис-
пользуются для определения конкретных химических составляющих (например неор-
ганических) собранных частиц, являются атомно-абсорбционная спектрометрия (AAS)
(Appel, 1993), рентгеновская эмиссия, индуцированная протонными пучками (PIXE)
(Eltayeb et al., 1993; Potocek, 1999), или ионная хроматография (IC) (Appel, 1993, Henning
etal., 2003).
Исследования одиночных частиц офлайн-методами (автономными) могут быть вы-
полнены с помощью сканирующего электронного микроскопа (СЭМ), просвечиваю-
щего электронного микроскопа (ПЭМ) (Ebert et al., 2002; 2004), электронно-зондового
рентгеновского микроанализа (ЕРХМA) (Xhofferetal., 1992) и лазерного микрозондового
масс-анализа (LAMMA, LMMS) (Kaufmann, 1986; Kolaitisetal., 1989; Тоигтапп и Kaufmann,
1993; Van Vaecku Gijbels, 1990a; Ван Vaecku Gijbels, 1990b; Wieser и Wurster, 1986). Отдельные
частицы обычно анализируются с помощью автоматизированных систем. Например,
автоматизированная система ЕРХМА может измерить в оптимальных условиях содер-
жание 12 химических элементов в 500 частицах за время, равное 2,5 часам (Van Grieken
и Xhoffer, 1992).
Отбор проб частиц с помощью фильтров и транспортировка фильтров в лабора-
торию занимают много времени и могут вызвать химические изменения в составе
соединений, из которых образованы частицы, либо привести к потере летучих ком-
понентов. Валовый анализ частиц дает количественную оценку массы различных
компнонентов, из которых состоят частицы. Степень воздействия частиц и влияние
их конкретных характеристик на здоровье человека — по-прежнему важный вопрос
исследований аэрозолей. Изучение одиночных частиц дает более детальную картину
Глава 16. Масс-спектрометрия атмосферных аэрозолей
Аэрозоль
Рис. 16.8. Схематическое изображение устройства онлайн-системы для анализа частиц,
включая систему ввода частиц, масс-спектрометр и систему обработки данных.
(Источник: Hinz К. Р. и Spengler В. (2007). «Приборы, обработка данных и коли-
чественная оценка в онлайн-масс-спектрометрии аэрозоля». J. Mass Spectrom.,
42: 843—860. С разрешения John Wiley & Sons, Ltd)
их состава и позволяет точнее определять эффект воздействия данного типа частиц
на здоровье человека или на окружающую среду. Тем не менее автономные методы
анализа могут дать ценную информацию о составляющих компонентах (например
AAS, IC и ЕРХМА) и морфологии частиц (например SEM). Следовательно, эти методы
(в сочетании с онлайн-измерениями) по-прежнему необходимы для всеобъемлющей
характеристики атмосферных аэрозолей (Broka etal., 2004; Dall'osto etal., 2006; Drewnick
et aL, 2003; Henning et aL, 2003; Hinz et aL, 2005; Kirshner et aL, 2003; Trimborn et aL, 2002;
Weber et aL, 2003).
Ограничения, которые присущи автономным (офлайн-) методам, определяют
спрос на устройства, которые могут анализировать атмосферные частицы в любом вы-
бранном месте (in situ) с высоким временным разрешением практически мгновенно (он-
лайн), не внося при этом существенных изменений в состав и строение частиц. Такие
приборы, способные работать в режиме реального времени, применяются во многих
лабораторных исследованиях и полевых экспериментах для лучшего понимания про-
цессов, происходящих в атмосфере, и для того, чтобы точнее предсказывать изменение
климата на Земле. Атмосферные частицы состоят из различных химических соедине-
ний. То, в каких пропорциях эти соединения смешаны, определяет реакционную спо-
собность частиц и их поведение в окружающей газовой среде. С помощью приборов,
способных работать в режиме реального времени, можно получить детальную инфор-
мацию об источниках разных частиц, о внутреннем составе различных типов частиц,
о химических реакциях, происходящих в частицах в атмосфере.
В следующих параграфах будут подробнее обсуждаться различные измеритель-
ные приборы для определения физических и химических свойств частиц, работаю-
щие в режиме реального времени. В принципе такой прибор состоит из заборного
устройства, которое обеспечивает поступление частиц из атмосферы внутрь прибора
и далее в устройство для измерения размера частицы, устройства для быстрого ис-
парения частицы, устройства для ионизации газовых компонент, образующихся при
испарении. Ионы, образовавшиеся при лазерной ионизации летучих соединений,
разделяются по массам в масс-анализаторе и регистрируются при помощи ионного
детектора, а полученные сигналы оцифровываются и хранятся в запоминающей си-
стеме (рис. 16.8).
16.3. Приборы и их применение
16.3.3. Времяпролетные приборы для анализа частиц,
работающие в режиме онлайн
Принципиальная схема онлайн-системы для анализа одиночных частиц представлена
на рис. 16.9. Аэрозольные частицы вводятся в основную вакуумную камеру прибора че-
рез систему забора частиц, где используется несколько камер дифференциальной от-
качки, которые разделены небольшими отверстиями диаметром от нескольких сотен
микрон до нескольких миллиметров. В системе забора частиц расширяющийся газ-
носитель откачивается насосами, в то время как частицы благодаря своей инерции бес-
препятственно пролетают через отверстия. При использовании такой системы забора
частиц возможен эффективный перенос частиц из окружающего воздуха в камеру с вы-
соким вакуумом (давление 10-6 мбар). Для оптимальной траспортировки частиц разных
размеров были разработаны различные контрукции систем забора частиц (рис. 16.10).
Существуют два основных типа конструкции: система, содержащая сопло и скиммер,
и система аэродинамических линз. В первом варианте частицы с размерами от 200 нм
до 2 мкм ускоряются до 200—400 м/с в результате градиента давления в системе диф-
ференциальной откачки. Система из аэродинамических линз состоит из небольшого
отверстия (диаметром 100—200 мкм), за которым следует ряд последовательных отвер-
стий на определенном расстоянии друг от друга. В этом случае может быть реализова-
на почти 100%-я эффективность транспортировки в оптимальном диапазоне размеров
Рис. 16.9. Схема прибора для анализа одиночных частиц в режиме онлайн с использова-
нием времяпролетной масс-спектрометрии с одновременной регистрацией по-
ложительных и отрицательных ионов
Рис. 16.10. Основные системы устройств ввода: (а) система напуска типа сопло-скиммер
и (б) система напуска, использующая аэродинамические линзы. (Источник:
Hinz К. Р. и Spengler В. (2007). «Приборы, обработка данных и количественная
оценка в онлайн-масс-спектрометрии аэрозоля». J. Mass Spectrom. 42: 843—860.
С разрешения John Wiley & Sons, Ltd)
частиц, который обычно охватывает десятикратное изменение диаметра частицы (Jayne
et aL, 2000; Zhang et aL, 2004). Система аэродинамических линз работает при уменьшен-
ном по сравнению с системой сопло—скиммер потоке газа на входе, и, следовательно,
скорость частиц в такой системе ниже (50—200 м/с). Таким образом, частицы пребывают
в вакууме до момента ионизации дольше, т.е. в этой системе возможна модификация
частицы, например испарение летучих компонентов и воды (Murphy, 2007; Tobias et aL,
2000).
Регистрация частиц в приборах для анализа одиночных частиц осуществляется
в вакуумной камере до момента лазерной ионизации частицы. В большинстве при-
боров используется два лазера непрерывного действия (типичная длина волны такого
лазера 532 нм), которые служат для обнаружения одиночных частиц и определения их
скорости. Свет, который рассеивают одиночные частицы при последовательном пере-
сечении двух лазерных лучей, детектируется фотоэлектронными умножителями (ФЭУ).
Измеряется время между двумя моментами рассеяния света, которое используется для
расчета скорости частиц, так как расстояние между двумя лазерными лучами извест-
но. Аэродинамический размер частиц может быть определен при использовании значе-
ний скоростей, измеренных для частиц с известными размерами (Hinds, 1999). Значение
скорости, полученное таким образом для каждой конкретной частицы, используется
для определения времени задержки для импульса ультрафиолетового (УФ) лазера (ти-
пичная длина волны УФ-лазера 193 или 337 нм). УФ лазерный импульс облучает ча-
стицу после того, как она пролетела область детектирования. Минимальное расстояние
между этой областью и областью ионизации зависит от времени внутренней задержки
между управляющим триггером и УФ лазерным импульсом и колеблется от нескольких
миллиметров до нескольких сантиметров. Интенсивное УФ лазерное излучение нагре-
вает частицу и ионизирует газобразные вещества, образовавшиеся при испарении. Воз-
никающие ионы регистрируются с помощью масс-спектрометра. Импульс УФ-лазера
дает также стартовый сигнал для цифрового осциллографа, который записывает и со-
храняет масс-спектры. Для онлайн-анализа частиц используются различные типы
масс-спектрометров.
Времяпролетные (ВП) масс-анализаторы чаще всего используются для исследова-
ний одиночных частиц в режиме реального времени. Эти анализаторы основаны на су-
ществовании прямого соотношения между массой иона и временем его пролета через
дрейфовую трубу. В таких ВП-масс-спектрометрах все ионы, образовавшиеся в середи-
не биполярной ускоряющей системы, изображенной на рис. 16.9, ускоряются в постоян-
ном электрическом поле до одинаковой потенциальной энергии Ej.
Ev = zeU. (11.1)
где z — это число некомпенсированных зарядов на частице, е — элементарный заряд,
U — ускоряющее напряжение. Время пролета t ионом расстояния 5 зависит от его массы
т, поскольку он обладает кинетической энергией Ек\
Ek = \/2m(s/t)2. (11.2)
Комбинация уравнений 1 и 2 дает возможность установить массу иона, измерив его
время пролета:
t2=Am + В, (11.3)
где А и В — параметры прибора.
Таким образом, ВП-масс-спектрометр состоит из области генерации ионов (ион-
ного источника или ионного промежутка), ускоряющей области, дрейфовой трубы
и детектора ионов. При анализе одиночных частиц ионы генерируются импульсом
УФ-лазера в центре ионного промежутка, который ограничен с обеих сторон сеточны-
ми электродами, используемыми для ускорения ионов. Образование ионов (например,
с использованием УФ-лазера) требует полного уничтожения частицы. Таким образом,
используя одну частицу, можно провести только одно измерение, и все ионы, образовав-
шиеся в результате воздействия лазерного импульса, должны быть обнаружены масс-
анализатором за время одного измерения. Одновременное определение положительных
и отрицательных ионов возможно при использовании двух противоположно направ-
ленных времяпролетных труб (рис. 16.9) (Erdmann et aL, 2005; Gard et aL, 1997; Hinz et aL,
1996; Lake etal., 2003). В качестве детектора ионов часто используются микроканальные
пластины (МКП), расположенные в конце времяпролетной трубы. В таком варианте
можно зарегистрировать все ионы, образующиеся из единственной частицы при одно-
кратном измерении. Масс-спектры как положительных, так и отрицательных ионов ре-
гистрируются и хранятся в цифровом осциллографе, а затем передаются в компьютер,
где они обрабатываются системой сбора данных при помощи специального программ-
ного обеспечения.
Для прямого анализа частиц были созданы различные ВП-масс-спектрометры.
Они были успешно применены во многих исследованиях как в лаборатории, так и в по-
левых условиях. Примерами портативных ВП-масс-спектрометров являются прибор
для аэрозольной времяпролетной масс-спектрометрии (АВПМС) (GardetaL, 1997), при-
бор для анализа частиц методом лазерной масс-спектрометрии (PALMS) (Thomson et aL,
2000), лазерный масс-анализатор взвешенных частиц в воздухе (LAMPAS) (Trimborn
Глава 16. Масс-спектрометрия атмосферных аэрозолей
et aL, 2000), быстрый масс-ан ал и затор единичных частиц (RSMS) (Rhoads et aL, 2003)
и времяпролетный масс-спектрометр для анализа одиночных частиц методом лазерной
абляции (SPLAT) (Zelenyuk etal., 2009). Для дополнительного изучения можно рекомен-
довать несколько обзоров и учебных материалов с подробным описанием различных
ВП-масс-спектрометров и сравнительным анализом их характеристик (Canagaratna
et aL, 2007; Hinz и Spengler, 2007; Johnston, 2000; Johnston и Wexler, 1995; McMurry, 2000;
Murphy, 2007; Nash et al. 2006; Zuess и Prater, 1999; Sallivan и Prater, 2005).
16.3.4. Применение масс-спектрометров дла анализа
одиночных частиц в исследованиях аэрозолей
В следующих параграфах будут описаны некоторые примеры использования масс-
спектрометров, работающих в режиме реального времени, дла анализа одиночных ча-
стиц при изучении атмосферных аэрозолей.
Целью измерений в полевых условиях являются определение источников частиц
и характеристика влияния деятельности человека на атмосферу. Чтобы собрать мак-
симально возможный обьем аэрозольных данных, в надежде получить наиболее пол-
ную информацию о распределении различных аэрозолей в условиях городской, сель-
ской, прибрежной и горной местности полевые измерения по всему миру проводятся
с использованием самых разных приборов. Например, долгосрочные наблюдения вре-
менных вариаций свойств аэрозолей осуществлялись в рамках программы Глобальной
службы наблюдения за атмосферой. Научно-исследовательская станция, расположен-
ная возле местечка Юнгфрауйох (JFJ, 3580 м над уровнем моря, Швейцария), выпол-
няет часть этой программы и вместе с сотнями глобальных и региональных станций
изучает влияние аэрозолей на климат и на качество воздуха в течение длительного пе-
Рис. 16.11. Лазерный масс-спектрометр LAMPAS 2 на террасе научно-исследовательской
станции JFJ (3580 м над уровнем моря, Швейцария); прибор сфотографирован
после выгрузки из вертолета 11 марта 2000 года
16.3. Приборы и их применение
риода времени. Эти долгосрочные измерения включают в себя короткие периоды, когда
для более детального исследования аэрозолей используются дополнительные прибо-
ры. Для проведения подобных измерений на территорию JFJ был доставлен лазерный
масс-спектрометр LAMPAS 2. Задачей данного прибора было определение химического
состава отдельных частиц аэрозоля, идентификация происхождения этих частиц и мо-
ниторинг фонового уровня аэрозолей в разных метеорологических условиях (рис. 16.11).
Во время проведения измерений на территории высотной альпийской станции JFJ
в Швейцарских Альпах с помощью LAMPAS 2 наблюдались разные направления эле-
ментарных воздушных потоков (иногда используется термин «парцелы»). Источники
аэрозольных частиц могут быть рассчитанны, используя обратный отсчет по отноше-
нию к погодным условиям и направлению атмосферных потоков. Зависимость состава
частиц от направления была подтверждена для всех обнаруженных типов аэрозольных
частиц. Рис. 16.12 демонстрирует пример одновременной регистрации положительных
и отрицательных ионов, образовавшихся при ионизации одиночной частицы, с четким
установлением минеральных компонентов.
При помощи этого прибора были идентифицированы минеральные компоненты,
характерные для аэрозолей: натрий (Na+, m/z 23), магний (Mg+, m/z 24), алюминий (Al+,
m/zll), кальций (Са+, тиД40), железо (Fe+, m/z 56), титан (Ti+/TiO+, m/z48/64), а также не-
которые другие оксиды. Спектр анионов был представлен фрагментами нитритов и ни-
тратов (NO2_, m/z 46 и NO3_, m/z 62), фосфатов (РО3_, m/z 79), хлоридов (Cl-, m/z 35/37)
и силикатов (SiO3"/SiO2_, m/z 76/60). Когда воздушные потоки достигают JFJ с юга, часто
регистрируются выбросы в Италии или Северной Африке (Сахара). Расчет обратных
траекторий воздушных потоков 23 марта 2000 года, когда JFJ находилась под влиянием
южных воздушных потоков, показал сильное влияние пыли из Сахары, что подвержда-
ется наблюдением большого числа минеральных частиц в воздухе станции в этот день.
100
75
50
25
Т----1--1--1--1--1-1—|--1—I—I—I—|—Г
20 50 m/z 100 150
Рис. 16.12. Одновременная регистрация положительных и отрицательных ионов при ис-
следовании одиночных минеральных аэрозольных частиц в окружающем воз-
духе (аэродинамический диаметр 1,3 мкм). Данные были получены в режиме
непрерывного наблюдения на станции JFJ 23 марта 2000 года. (Источник:
перепечатано из журнала J. Aerosol Sci., Hinz К. Р., Trimborn A., Weingartner Е.,
Henning S., Baltensperger U., Spengler В. «Композиция аэрозольных частиц
в Юнгфрауйох». 36, 123—145, Copyright 2005, с разрешения Elsevier)
Глава 16. Масс-спектрометрия атмосферных аэрозолей
В дополнение к минеральным на станции JFJ были зарегистрированы другие типы
частиц, состоящих из смеси углерода и солей, с добавлением вторичных соединений,
таких как аммоний (m/z 18 для NH4+), нитраты (m/z 46 для NO2“ и m/z 62 для NO3)
и сульфаты (m/z 80 для SO3“, m/z 97 для HSO4), аналогичные обычным для сельской
или городской местности (Held et al., 2002; Trimborn et al., 2000; Trimborn et al., 2002; Vogt
et al., 2003). Это указывает на то, что на состав атмосферы на станции JFJ иногда влия-
ют результаты деятельности человека в долинах Альп. Поступление загрязненного воз-
духа в свободную тропосферу может быть обусловлено тепловой конвекцией во время
северного ветра.
Масс-спектрометры для непрерывного аэрозольного анализа обычно приспособле-
ны для непрерывной работы в течение длительного периода времени. Использование
таких масс-спектрометров при анализе одиночных частиц позволяет записать от не-
скольких сотен до тысячи спектров в час в зависимости от концентрации частиц и эф-
фективности детектирования ионов прибором. Во время многолетних исследований
аэрозолей были получены от нескольких тысяч до миллионов спектров различных ча-
стиц. Оценка этих спектров в ручном режиме занимает очень много времени и поэтому
не является эффективной. Поэтому для сортировки частиц по конкретным параметрам
(например размер частиц, их химический состав или время записи спектров), а также
для извлечения конкретной полезной информации из полного набора данных исполь-
зуются различные компьютерные программы.
В одном из подходов к обработке спектров определяется изменение интенсивности
сигналов характеристических ионов, просуммированных с заданным временным раз-
решением по всем спектрам. Примером такого метода обработки результатов являет-
ся временная гистограмма химического состава, где данные суммировались в течение
1 часа (рис 16.13).
Относительное содержание ионов, характерных для вторичных компонентов (NH4+,
m/z 18; NO2_, m/z 46; NO3“, m/z 62; HSO4_, m/z 97), углеродистого материала (C2_, m/z 24;
C3_/+, m/z 36; C4_, m/z 48) и органических соединений (C7H7+, m/z 91; m/z +284 (org2); m/z
+299 (org 3)), определяется путем подсчета масс-спектров, которые содержат сигналы
этих ионов. Сигналы ионов, просуммированные внутри каждого временного интер-
вала, представляются в виде зависящей от времени ломаной кривой, которая отражает
временную зависимость содержания частиц с данным составом в данном месте. Полу-
ченные зависимости состава частиц от времени могут быть использованы для выбора
конкретных периодов наблюдений, представляющих интерес для дальнейшей стати-
стической оценки спектров (как, например, указано стрелками на рис 16.136) или для
корреляции измеренных характеристик аэрозолей с метеорологическими условиями
(такими как температура окружающей среды, давление, концентрация числа частиц)
или с различными атмосферными процессами (Erdmann etal., 2005; Hinz etal., 2006).
Другой подход к оценке масс-спектров одиночных частиц использует классифика-
ционные алгоритмы для определения основных химических классов, к которым при-
надлежат исследуемые частицы. При проведении классификации сходные спектры
обьединяются в один и тот же класс, поскольку частицы одного класса являются хи-
мически похожими. Классы отображаются в виде характерных (или модельных) масс-
спектров, что с точки зрения химического состава может быть интерпретировано как
разные виды аэрозольных частиц (Hinz и Spengler, 2007). Этот подход обеспечивает про-
16.3. Приборы и их применение
Дата и время
Рис. 16.13. Временные изменения уровней характеристических ионов, зарегистрирован-
ных прибором LAMPAS 2: (а) вторичные компоненты, (б) углеродистые матери-
алы, (в) органические вещества. Измерения сделаны на октрытом воздухе в Ис-
пре в декабре 2002 года. (Перепечатано из Int. J. Mass Spectrom., 2006, Hinz К. P.,
Erdmann N. Gruning C., Spengler В. «Параллельное сравнение характеристик
аэрозольных частиц двумя лазерными масс-спектрометрическими системами
LAMPAS 2 и SPASS». 258,151—166, Copyright 2006 год, с разрешения Elsevier)
456
Глава 16. Масс-спектрометрия атмосферных аэрозолей
2 1 L J 1 6 3 d 47 42 6I L 48 L J “ 0 ]280 97 Ц5 131 147 23 36 ... 1 (+) Соль О (i) 39 40 63 .Ki. .. '?8 . '?5
24 17 -Ik ,^.lL 1 L. > z 16 46 J 18 6 50 L $ .2 ]280 9] L .UL 1 >7 и—.—,——«—, . ,—— 1 2 22 18 1 36 24 1 4 t •8 ( Сажа co вторичными компонентами (2) .0 1 I2 84 l.i L. i,. .. и
Рис. 16.14. Модельные (характерные) спектры пяти классов частиц, определенные с по-
мощью нечеткого алгоритма для группы из 431 частиц. Данные были получе-
ны во время полевого эксперимента LACE 98 в городе Линденберг, Германия.
Аэродинамический диаметр частиц — примерно 1,0 мкм. (Источник: Aerosol
Science & Technol:. «Онлайн-анализ атмосферных частиц с помощью перенос-
ного лазерного масс-спектрометра». 33: 191—201. Copyright 2000. Mount Laurel,
NJ. Печатается с разрешения)
16.3. Приборы и их применение
стую характеризацию групп частиц. В качестве примера на рис. 16.14 представлено пять
характерных масс-спектров для 431 частицы, которые были охарактеризованы в про-
цессе полевых измерений 1 августа 1998 г. (Trimborn et al., 2000). Характерные (модель-
ные) спектры были рассчитаны с помощью метода нечеткой кластеризации С-средних
(Hinz etal., 1999).
Модельные спектры изображаются по аналогии с обычными масс-спектрами как
в случае положительных, так и в случае отрицательных ионов. Они могут быть соот-
несены с конкретным химическим составом по признаку присутствия тех или иных ио-
нов, следовательно, эти спектры представляют различные типы (классы) аэрозольных
частиц. Например, первый класс частиц на рис 16.14 можно интерпретировать как ча-
стицы соли, поскольку в их характерных масс-спектрах наблюдаются интенсивные сиг-
налы ионов натрия (m/z 23, Na+) и нитратов (m/z46, NO2_ и m/z^l, NO3). Третий класс —
это углеродные частицы, для спектров которых характерно распределение, типичное
для кластеров углерода как для положительной, так и для отрицательной полярностей
(С/;+ и С~, где п = 1—18). Пятый класс обьединяет частицы минеральной пыли. Их ха-
рактерные спектры похожи на спектр одиночной частицы, изображенный на рис. 16.12.
Этот спектр характеризуется теми же интенсивными сигналами как в спектрах по-
ложительных ионов, например натрий (m/z 23, Na+), магний (m/z 24, Mg+), алюминий
(m/z 27, А1+), кальций (m/z 40, Са+), железо (m/z 56, Fe+), так и в спектрах отрицательных
ионов: хлориды (m/z 35/37, С1_), нитраты (m/z 46 и 62, NO) и фосфаты (m/z 63, и 79 POv).
Два других класса частиц (классы 2 и 4) представляют собой смеси углеродистых и вто-
ричных компонентов.
В дополнение к информации о химическом составе в процессе разбиения всех ча-
стиц на классы классификационные алгоритмы выделяют частоту регистрации частиц,
принадлежащих тому или иному классу. Для наглядного представления различной
природы аэрозолей распространенность того или иного класса может быть отображена
в виде сектора с различной шириной на круговой диаграмме. Рис. 16.15 демонстрирует
распространенность разных аэрозольных классов. Данные были получены в результа-
те исследований на открытом водухе, в которых была проанализирована 431 частица
(рис. 16.14). Было установлено, что в исследованной совокупности аэрозолей с большим
избытком преобладают частицы соли при незначительном вкладе минеральных частиц
и частиц, состоящих из чистого углерода.
В процессе длительных атмосферных наблюдений аэрозольные частицы обнару-
живаются в широком диапазоне размеров. Исследования совокупности атмосферных
частиц с различными размерами являются важным дополнительным источником ин-
формации, который позволяет установить корреляцию между размером и химически-
ми характеристиками частицы. Как правило, классификация спектров выполняется
для каждого диапазона размеров отдельно, с последующим сравнением типичных для
каждого размера спектров. При использовании этой процедуры могут быть установле-
ны различия в модельных спектрах и, соответственно, составе аэрозолей в зависимости
от размеров частиц. Рис. 16.16 представляет пример такой оценки спектров одиночных
частиц в двух диапазонах размеров, демонстрируя корреляцию между составом частиц
и метеорологическими данными.
В рамках совместного проекта Lindenberg по характеризации аэрозолей в июле и ав-
густе 1998 года (LACE 98) многие исследовательские группы одновременно измеряли
Глава 16. Масс-спектрометрия атмосферных аэрозолей
Минеральная пыль
вторичными компонентами
18,0%
Соль
52,0%
Рис. 16.15. Относительное количество частиц, принадлежащих к разным классам; дан-
ные для группы из 431 отдельных частиц (аэродинамический диаметр частиц
1,0 мкм. Измерения проведены во время полевого эксперимента LACE 1 августа
1998 года
различные параметры аэрозолей. Прибор LAMPAS 2 был использован для определения
химического состава атмосферных частиц в различных аэрозолях в зависимости от их
размеров, а полученные результаты использованы для оценки влияния антропогенных
частиц на климат.
Обработка данных полевых измерений позволила провести оценку характерных
спектров частиц как функцию их размера. Эта оценка была сделана для пяти диапазо-
нов со средним аэродинамическим диаметром частицы от 0,2 до 1,5 мкм. Классифика-
ция спектров, зарегистрированных в определенные дни, показала наличие ежедневно
от 4 до 6 классов частиц, причем в каждом диапазоне размеров. Сравнение всех найден-
ных классов частиц привело исследователей к выводу, что подавляющее большинство
(около 90 %) частиц во все дни может быть описано всего лишь 10 основными классами.
Различие в составе аэрозолей состоит в основном в числе частиц, которые принадлежат
тому или иному основному классу (рис. 16.16). Кроме того, разделение на два основных
диапазона по размеру (на частицы с аэродинамичеким размером <0,8 мкм и >0,8 мкм)
позволило сравнить химический состав частиц в каждом «большом» диапазоне. Набор
из 10 основных классов был сокращен до четырех фундаментальных типов частиц на ос-
новании наличия общих основных компонент в масс-спектрах образцов (табл. 16.1).
Эти фундаментальные типы частиц могут быть интерпретированы как «минеральные
частицы», «частицы морской соли», «частицы, состоящие из смеси соли и углеродных
компонентов» и « чистые углеродные частицы».
Рис. 16.16 демонстрирует химический состав аэрозольных популяций в двух диа-
пазонах размеров частиц. Данные собирались на протяжении семи дней во время экспе-
римента LACE 98; при этом рассчитывались траектории воздушных потоков (парцелов),
которые в указанные дни проходили через точку измерений. Как сходство, так и значи-
тельное различие отмечены для соотношений между распределением частиц по классам
в зависимости от размера частиц и траектории парцелов. Ожидаемое появление частиц
морской соли в популяции частиц с размером более 0,8 мкм обьясняется перемещени-
ем соответствующих обьемов воздуха через море. Аэрозоли потоков воздуха, которые
перемещались над сельскими районами в течение длительного времени, характеризу-
ются высоким содержанием минеральных частиц (10 августа и 11 августа). Когда потоки
проходили через густонаселенные районы Западной Европы, наблюдалось повышение
16.3. Приборы и их применение
<0.8 цт >0.8 jim
Рис. 16.16. Относительное количество частиц, принадлежащих ктому или иному из клас-
сов, и метеорологические данные. Данные были получены во время полево-
го эксперимента LACE 98 в городе Линденберг, Германия. Высота столбцов
на диаграмме с цветными датами показывает процент (%), который составля-
ют частицы данного класса среди частиц всех классов. Частицы рассортирова-
ны по двум группам аэродинамичеких диаметров (D < 0,8 мкм и D > 0,8 мкм).
Соответствующие цветные линии показывают траектории элементарных
потоков воздуха (парцелов), которые были рассчитаны для различных дат.
(Источник: Trimborn A., Hinz К..Р. и Spengler В. «Онлайн-анализ атмосфер-
ных частиц с помощью переносного лазерного масс-спектрометра во время
LACE 98». Журнал геофизических исследований. 107, D21: 8132. Copyright 2002.
Американский Геофизический союз. Воспроизводится с разрешения Амери-
канского геофизического союза)
количества углеродсодержащих частиц и частиц, появление которых связано с выхлоп-
ными газами (например, 31 июля). Время, в течение которого обьем воздуха, содержа-
щий аэрозоль, совершает путешествие над конкретными областями (например над
городскими, сельскими и/или промышленными районами), оказывает существенное
влияние на количество некоторых классов аэрозолей. Корреляция между составом ча-
стиц и траекториями потоков воздуха или направлением ветра является ценным наблю-
дением, которое позволяет связать происхождение частиц, их траектории и химический
состав (Bein etal., 2007; Rhoads et al., 2003; Trimborn etal., 2002). Исследования такого типа
Глава 16. Масс-спектрометрия атмосферных аэрозолей
Табл. 16.1. Основные типы частиц, определенные во время эксперимента LACE 98, и их
предполагаемые оптические свойства. (Источник: Trimborn A., Hinz К.Р.
и Spengler В. Онлайн-анализ атмосферных частиц с помощью переносного
лазерного масс-спектрометра во время эксперимента LACE 98. J. Geophys.
Res. 107, D21: 8132. Copyright 2002 год. Американский геофизический союз.
Воспроизводится с разрешения Американского Геофизического Союза)
Тип частиц Тип спектра Оптические свойства
Тип 1 — неорганика Выхлоп Неорганическая пыль Рассеивание
Тип 2 — морская соль Старая морская соль Старая морская соль + органика Рассеивание
Тип 3 — соль + углерод Соль + С (К) Соль + С (Na) Рассеивание и абсорбция
Тип 4 — углерод С + соль Биогенный углерод С + вторичные компоненты Чистый углерод Значительная абсорбция
могут быть использованы для различения аэрозолей, образовавшихся при продолжи-
тельной транспортировке потоков воздуха и в результате влияния местных источников.
Важным дополнительным методом оценки характеристик, полученных при ис-
следовании одиночных частиц, является корреляция между химическим составом ча-
стиц и их оптическими свойствами (например показателем преломления), поскольку
эти свойства имеют важное значение для оценки их влияния на радиационный баланс.
Такая корреляция была найдена многими исследовательскими группами, которые уча-
ствовали в полевом эксперименте LACE 98 (итоговые данные представлены в табл. 16.1).
Обнаруженная корреляция между химическими и физическими (например оптически-
ми) свойствами отдельных частиц с одной стороны и метеорологическими данными
с другой может быть использована для создания более точных атмосферных моделей
и для улучшения надежности моделирования климата.
16.3.5. Многоцелевые времяпролетные приборы для анализа
частиц
Установка для онлайн-анализа частиц, описанная выше, использует для получения ха-
рактеристик одной аэрозольной частицы прямую (т. е. совершаемую за один шаг) лазер-
ную десорбцию/ионизацию (ЛДИ) в сочетании с ВП-масс-спектрометрией. Этот метод
особенно хорош для обнаружения неорганических соединений, сажи и пыли. С дру-
гой стороны, общий анализ органических компонентов аэрозоля представляет боль-
шой интерес, поскольку огромное количество органических веществ выбрасывается
в атмосферу в процессе сгорания топлива в автомобильных моторах, эксплуатации те-
пловых электростанций, а также при сжигании древесины. Органические соединения
могут вступать в реакции с различными газами в атмосфере или с веществами на по-
верхности частиц, превращаться в более химически активные соединения и тем самым
воздейстововать на здоровье человека.
Например, полициклические ароматические углеводороды (ПАУ), будучи рас-
пространенным классом органических соединений, могут оказать существенный
16.3. Приборы и их применение
вред здоровью (например, рак) (Bente et aL, 2006; Poschl, 2005). Анализ органических
составляющих частиц при помощи ЛДИ часто бывает трудным, потому что органиче-
ские молекулы под действием мощного лазерного импульса подвержены значительной
фрагментации. Чтобы избежать этого, были разработаны методы анализа, где первый
лазерный импульс используется для испарения частицы (при этом органические со-
единения переводятся в газовую фазу без заметной фрагментации), а второй — для
ионизации нейтральных молекул (Bente et aL, 2006; Morrical et aL, 1998; Zelenyuk et aL,
2009). Рис. 16.17 иллюстрирует схему онлайн-масс-спектрометра ATOFMS, который
может работать в трех альтернативных режимах (Bente et aL, 2006). Эти режимы отли-
чаются методами испарения и ионизации частиц: ЛДИ (десорбция и ионизация в один
шаг), лазерная десорбция (ЛД) в сочетании с многофотонной резонансной ионизацией
(ЛД/МФРИ ) и термодесорбция в сочетании с многофотонной резонансной ионизаци-
ей (ТД/МФРИ).
Рис. 16.17. (а) Схематическое изображение аэрозольного времяпролетного масс-спектро-
метра (ATOFMS). Разновидности ATOFMS: (б) ЛДИ/ ATOFMS, (в) ЛД/МФРИ/
ATOFMS и (г) ТД/МФРИ/ATOFMS. (Перепечатано из Int. J. Mass Spectrom.,
Bente M., Adam T., Ferge T., Gallavardin S., Sklorz M., Streibel T., Cimmerman R.
«Онлайн аэрозольная лазерная масс-спектрометрия с использованием трех,
взаимозаменяемых методов лазерной ионизации для характеризации неор-
ганических и ароматических соединений в частицах», 258, 86—94, Copyright
2006 год, с разрешения Elsevier)
462 Глава 16. Масс-спектрометрия атмосферных аэрозолей
Принципиальная схема такого онлайн-прибора похожа на ту, что изображена
на рис. 16.9. Частицы вводятся в масс-спектрометр через систему напуска и достигают
области регистрации, пересекая два луча от непрерывного лазера. Зарегистрированная
одиночная частица ионизируется одним из трех методов десорбции/ионизации. При ис-
пользовании метода одновременной десорбции/ионизации (ЛДИ) частицы облучаются
импульсами эксимерного УФ-лазера (X = 248 нм), а образующиеся ионы детектируются.
В методе ЛД/МФРИ на первом шаге импульс инфракрасного лазера (X = 10,6 мкм) ис-
пользуется для десорбции органических веществ, содержащихся в частице. На втором
шаге — ионизации — излучение мощного УФ-лазера (X = 248 нм) воздействует на де-
сорбированные молекулы, генерируя ионы. Процесс многофотонной резонансной
ионизации наиболее эффективен, когда существует сильное резонансное поглощение
УФ-излучения молекулами-мишенями, поэтому МФРИ хорошо подходит для анализа
ароматических соединений. При анализе частиц методом ТД/МФРИ происходит на-
Рис. 16.18. ЛДИ/АТОРМ8-спектры (а) положительных ионов и (б) отрицательных ионов
одиночной частицы древесной золы, аэродинамический диаметр частицы око-
ло 1,1 мкм; (в) органические соединения, обнаруженные при анализе прибором
ЛД/МФРИ/ATOFMS одной частицы древесной золы; (г) масс-спектр, полу-
ченный при ТД/МФРИ/АТОРМ8-анализе частиц, содержащихся в выхлопных
газах автомобилей. (Перепечатано из Int. J. Mass Spectrom., Bente M., Adam T.,
Ferge T., Gallavardin S., Sklorz M., Streibel T., Cimmerman R. «Онлайн-аэрозоль-
ная лазерная масс-спектрометрия с использованием трех взаимозаменяемых
методов лазерной ионизации для характеризации неорганических и аромати-
ческих соединений в частицах», 258, 86—94, Copyright 2006 год, с разрешения
Elsevier)
ЛД/МФРИ/ATOFMS одиночной частицы
0 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320
m/z
0 20 40 60 80 100 120 140 160 180 200 220 240
m/z
16.3. Приборы и их применение 463
грев стержня, который служит коллектором частиц и находится рядом с областью ио-
низации в источнике ионов. Частицы собираются на холодной поверхности стержня
в течение определенного времени (например 10 мин.), а затем испаряются при быстром
нагреве стерженя до температуры примерно 350°C. Десорбированные молекулы иони-
зируются импульсами УФ-излучения эксимерного лазера (X = 248 нм) в ходе МФРИ.
После ионизации положительные и отрицательные ионы ускоряются и направляются
из ионного источника во времяпролетную трубу в двух противоположных направлени-
ях. После отражения от ионного зеркала (зеркало применяется для улучшения разре-
шения) ионы детектрируются с помощью мульти коллекторной пластины. На рис. 16.18
приведены примеры обнаружения ионов во всех трех режимах работы прибора. Для из-
мерения частиц древесной золы были использованы ЛДИ и ЛД/МФРИ, в то время как
режим ТД/МФРИ был применен для исследования частиц в выхлопе бензинового дви-
гателя легкового автомобиля.
В спектрах частиц древесной золы в режиме ЛДИ (рис. 16.18а и б) были обнаруже-
ны сигналы катионов натрия, калия и железа, а также нитрат- и сульфат-анионов. До-
полнительные пики отрицательных ионов указывают на наличие углеродных класте-
ров в диапазоне малых масс. При этом только очень слабые пики были обнаружены для
органических соединений с большими массами. Разделение процесса анализа на эта-
пы десорбции (СО2-лазер, 10,6 мкм) и ионизации (эксимерный лазер, 248 нм (МФРИ))
позволило обнаружить органические молекулы, такие как ПАУ (рис. 16.18<?). Спектр
ТД/МФРИ-частиц в выхлопных газах автомобилей также указывает на наличие органи-
ческих соединений. Метод ТД/МФРИ может быть полезен для улучшения количествен-
ной оценки соединений со специфическими свойствами и для анализа мельчайших
частиц (вплоть до частиц диаметром около 10 нм), которые не могут быть обнаружены
с помощью метода лазерного рассеяния света, поскольку интенсивность рассеяния све-
та этими частицами слишком мала.
16.3.6. Использование ионных ловушек для анализа одиночных
частиц
Анализ одиночных частиц может также быть осуществлен с помощью масс-анализатора
типа ионной ловушки (Reilly et al., 1997). Ионная ловушка состоит из двух гиперболиче-
ских металл ическихторцевых электродов и гиперболического электрода в форме кольца.
Ионы захватываются в пространстве между этими тремя электродами и анализируются
с помощью комбинации переменного и постоянного электрических полей. Аналити-
ческие приборы, основанные на масс-анализаторах типа ионной ловушки, имеют не-
которые преимущества по сравнению с другими типами анализаторов. Они, как прави-
ло, очень компактны, потребляют меньше энергии и могут работать при более высоком
давлении (около 10-5 мбар), что снижает требования к мощности насосов, применяемых
для откачки (Keil и др., 2008). Кроме того, при исследовании одиночных частиц ионные
ловушки могут быть использованы для реализации тандемной масс-спектрометрии (то
есть для МС/МС- и МС/МС/МС-исследований), которая предоставляет структурную
информацию об отдельных типах органических соединений (Reilly et al., 1998). Рис. 16.19
демонстрирует недавно разработанный переносной масс-спектрометр на основе ионной
ловушки, который может работать в режиме реального времени и характеризовать со-
став отдельных частиц в воздухе.
Глава 16. Масс-спектрометрия атмосферных аэрозолей
Входное отверстие
Скиммер
Фотоумножитель
Компьютер
Ионная
ловушка
Электроника
контроля времени
Аэродинамическая
система линз
„ Насос
Диодные лазеры
Фотоумножитель
Конверсионный----
динод
Одноканальный
электронный
умножитель
Рис. 16.19. (а) Схематическое изображение прибора, использующего масс-спектрометр
на основе ионной ловушки. Аэрозольные частицы влетают через входное отвер-
стие и образуют хорошо коллимированный пучок на выходе из аэродинамиче-
ской системы линз. Пучок частиц проходит через скиммер в основную камеру
масс-спектрометра. Частицы пересекают два сфокусированных лазерных пуч-
ка, расположенных на расстоянии 2,7 см друг от друга; рассеянный частицами
свет регистрируется двумя отдельными ФЭУ. Время пролета между двумя луча-
ми регистрируется для установления скорости частиц; известная корреляция
скорости с аэродинамическим размером калиброванных частиц используется
для нахождения размеров исследуемых частиц. Импульс УФ-лазера синхро-
низован с моментом достижения частицей центра ионной ловушки с помощью
электроники, которая вычисляет время запуска УФ-лазера исходя из найденной
скорости. Ионы, полученные при лазерном облучении частицы, анализируются
в стандартной ионной ловушке, (б) Изображение прибора для изучения аэрозо-
лей, в котором в качестве масс-анализатора используется ионная ловушка. (Пе-
чатается с разрешения Harris W. A., Rejli Р.Т. A., Whitten W. В., Ramsey J.M. (2005).
«Переносной масс-спектрометр на основе ионной ловушки для измерений одиночных
частиц в режиме реального времени. Обзор приборов для научных исследований». 76:
064102-1-064102-8. Copyright 2005, Американский Институт физики)
16.3. Приборы и их применение
Частицы влетают в прибор через входное отверстие, проходят через аэродинамиче-
скую систему линз и входят в основную вакуумную камеру через скиммер. Далее инди-
видуальный размер и скорость движения каждой частицы детектируются с помощью
двух разделенных и сфокусированных лазерных лучей от диодных лазеров (X = 532 нм).
Свет, рассеянный частицей при прохождении через каждый лазерный луч, использует-
ся для определения размера частицы, который, в свою очередь, определяет ее скорость.
Когда частицы достигают центра ионной ловушки, они испаряются и ионизируются
в режиме ЛДИ импульсом сфокусированного УФ-лазера (используется лазер на нео-
дим-иттрий-алюминиевом гранате (Nd:YAG), X = 266 нм). Для того чтобы использовать
ионную ловушку при исследовании одиночных частиц, в кольцевом электроде имеются
четыре дополнительных отверстия: для входа и выхода частиц, а также луча УФ-лазера.
Выбранный диапазон масс в ионной ловушке (от m/z 30 до m/z 250) ограничивал мак-
симальную скорость сбора данных примерно до уровня 4 Гц. Система была успешно
применена для измерения как субмикронных частиц в воздухе лаборатории, так и для
Рис. 16.20. (а) Аэрозольный масс-спектр частиц, содержащих ацетаминофен; частицы по-
лучены путем распыления 50% (по объему) водно-метанольного (1:1) раствора;
(б) выделение ионов ацетаминофена с m/z 151; (в) спектр ДАС ионов с m/z 151,
энергия столкновения 1,0 и параметр радиочастотного поля в ловушке q = 0,3;
(г) МС/МС/МС-спектр ионов с m/z = 109; (d) МС/МС/МС/МС-спектр ионов
с m/z = 80. (Печатается с разрешения Harris W.A., Rejli Р.Т.А., Whitten W. В.,
Ramsey J. M. (2005). «Переносной масс-спектрометр на основе ионной ловушки для
измерений одиночных частиц в режиме реального времени. Обзор приборов для на-
учных исследований». 76: 064102-1-064102-8. Copyright 2005, Американский Ин-
ститут физики)
466 Глава 16. Масс-спектрометрия атмосферных аэрозолей
измерения распыленных синтетических частиц, содержащих ацетаминофен (C8H9NO2).
Ацетаминофен был выбран, чтобы продемонстрировать способность прибора осущест-
влять тандемный масс-спектрометрический анализ органических веществ в отдельных
частицах аэрозоля. Рис. 16.20 иллюстрирует пример такого тандемного анализа от-
дельных ацетаминофенсодержащих частиц. Масс-спектр на рис. 16.20я демонстрирует
наличие пика молекулярного иона ацетаминофена (m/z 151) после десорбции/иони-
зации одиночной частицы. После охлаждения соударениями с атомами гелия внутри
ловушки молекулярные ионы ацетаминофена были изолированы путем приложения
электрического сигнала специальной формы к торцевым электродам, в результате чего
был получен масс-спектр, представленный на рис. 16.206 Далее для инициирования
фрагментации молекулярного иона была использована диссоциация, активированная
соударениями. В результате образовалось несколько осколочных ионов (фрагментов),
которые позволили сделать выводы о структуре исследуемого вещества (рис. 16.20в, г, д).
Этот пример демонстрирует уникальную способность ионной ловушки определить со-
став и структуру органических соединений, содержащихся в одиночных частицах. Та-
ким образом, этот прибор очень полезен для характеризации органических частиц.
16.3.7. Масс-спектрометры для анализа ансамблей частиц
В качестве альтернативы детектированию одиночных аэрозольных частиц использу-
ется интегральный подход, в котором получаемые данные усредняются по нескольким
частицам (ансамблю частиц), формируя единый набор данных. Аэрозольные масс-
спектрометры (АМС) интегрального типа используют принцип тепловой возгонки
для выделения летучих веществ из частиц с последующей электронной ионизацией
(Canagaratna et al., 2007). Квадрупольные фильтры масс (КФ-АМС), времяпролетные
рефлектроны (ВП АМС) и ионные ловушки (ИЛ-АМС) были использованы для ана-
лиза ионов, полученных при ионизации электронами (Dekarlo et al., 2006; Drewnick et al.,
2005; Jayne et al., 2000; Kurten et al., 2007). Пример установки такого типа представлен
на рис. 16.21.
В АМС воздух, содержащий исследуемые частицы, вводится в вакуумную камеру
через систему аэродинамических линз, которая фокусирует поток субмикронных ча-
стиц в виде узкого потока. Сверхзвуковое расширение на выходе из системы аэродина-
мических линз ускоряет частицы в область измерения их размера, который определя-
ется при измерении времени пролета частиц через фиксированное расстояние между
вращающимся прерывателем и испарителем. Частицы испаряются при столкновении
с резистивно-нагретой поверхностью (600°C) испарителя, а испаренные вещества за-
тем подвергаются действию потока ионизирующих электронов (кинетическая энергия
электронов 70 эВ). Этот метод ионизации дает количественную информацию о возгоня-
мых компонентах частицы, то есть о тех, которые могут быть испарены в процессе очень
быстрого нагрева. Такими компонентами являются органические соединения, ионы
аммония, нитраты, хлориды и сульфаты. Трудновозгоняемые материалы, такие как
почвенная пыль, морская соль и элементарный углерод, в таком эксперименте не ре-
гистрируются. После образования ионы направляются в ортогональную ускоряющую
систему, где после приложения высоковольтного импульса они ускоряются в направ-
лении ВП-трубы масс-анализатора рефлектронного типа. АМС, как правило, работают
в режиме регистрации положительных ионов. Суммарные интенсивности сигналов, со-
16.3. Приборы и их применение
ответствующих ионам аммония (m/z 15, 16, 17), нитратов (m/z 30, 46), сульфатов (m/z 48,
64, 80, 81) и органики (большинство других ионов с m/z > И), конвертируются в массо-
вые концентрации с помощью калибровочных данных, полученных на основании ранее
измеренной эффективности ионизации различных компонентов аэрозолей (Allan et aL,
2004; Drewnick et aL, 2005). Пример зависимости массовой концентрации различных ти-
пов компонентов аэрозолей от времени представлен на рис. 16.22. Концентрации были
измерены одновременно на квадрупольном и времяпролетном АМС во время полевых
экспериментов в Нью-Йорке в январе 2004 года.
Зависимости массовой концентрации от времени, полученные при помощи этих
двух приборов, довольно хорошо коррелируют (рис. 16.22). Некоторые отличия, веро-
ятно, вызваны различной эффективностью прохождения ионов через масс-анализатор
кдетектору в этих типах приборов. Недавно были проведены исследования на ВП-масс-
спектрометре высокого разрешения. Этот масс-спектрометр способен напрямую диф-
ференцировать несколько разных типов фрагментов органических ионов (DeCarlo et aL,
2006). Высокое разрешение имеет важное значение для более точной характеристики
химического состава аэрозолей и, соответственно, для лучшего понимания атмосфер-
ных процессов. Различные типы АМС широко используются во многих полевых иссле-
дованиях для определения усредненных массовых концентраций летучих компонентов
аэрозолей и корреляции этих концентраций с метеорологическими характеристиками,
такими как температура и источник воздушных масс. Суммарные результаты исследо-
Рис. 16.21. Принципиальная схема ВП-АМС. (Взято из журнала «Аэрозольная наука и тех-
нология»: новый аэрозольный ВП-масс-спектрометр (ВП-АМС) — описание
инструмента и его первое развертывание в полевых условиях. 39: 637—658.
Copyright 2005. Mount Laurel, NJ. Печатается с разрешения издания)
Глава 16. Масс-спектрометрия атмосферных аэрозолей
ваний по определению массовых концентраций, полученные с помощью АМС в про-
цессе многолетних полевых экспериментов, представлены на рис. 16.23. Данные были
получены в разные сезоны в городских районах, в районах, расположенных с подве-
тренной стороны по отношению к городам, и отдаленных районах в Северном полуша-
рии (Jimenes et al., 2009; Zhang et aL, 2007).
Интегральные массовые концентрации и массовые доли летучих неорганических
и органических компонентов варьируются в зависимости от местоположения и време-
ни года. Высокая массовая доля органических соединений в аэрозолях представляет
особый интерес, потому что существует высокая неопределенность в описании источ-
ников и атмосферной эволюции этого типа аэрозолей. Первичные органические аэро-
золи выбрасываются в атмосферу непосредственно при сжигании ископаемого топлива
(органические аэрозоли с высоким углеводородным содержанием) и биомассы (орга-
нические аэрозоли, образующиеся при сжигании биомассы), в то время как вторичные
органические аэрозоли образуются при атмосферном окислении соединений, находя-
щихся в газовой фазе. Другим типом органических аэрозолей являются окисленные ор-
ганические аэрозоли (ООА) с высоким содержанием кислорода. Результаты различных
исследований показывают, что большая часть ООА имеет вторичное происхождение
1/17/2004
1/21/2004 1/25/2004
Дата и время
1/29/2004
Рис. 16.22. Временные зависимости, полученные с помощью ВП-МС, для легко возгоняе-
мых соединений, содержащих нитраты, сульфаты, аммоний, и для общего ко-
личества летучих органических соединений (соответственно, синий, красный,
оранжевый и зеленый цвета линий). Те же вещества, проанализированные
в квадрупольном масс-спектрометре (черный цвет линий). Оба типа измере-
ний сделаны за один и тот же промежуток времени во время полевых измерений
в январе 2004 года в Нью-Йорке. (Источник: «Наука аэрозолей и технологии:
новые времяпролетной масс-спектрометры аэрозолей (TOF-AMS) — описание
инструмента и его первое развертывание в полевых условиях». 39: 637—658.
Copyright 2005. Mount Laurel, NJ. Печатается с разрешения издателя)
16.3. Приборы и их применение
(Herndon et al., 2008; Zhang et al., 2005). Этот тип аэрозолей можно разделить на два под-
типа: легколетучие ООА и среднелетучие ООА.
Конкретный тип органических аэрозолей может быть установлен масс-
спектрометрически при использовании алгоритмов математической факторизации.
Факторный анализ (ФА) на основе различной фрагментации и с учетом соотношения
между кислородом и углеродом выявлет несколько типов ООА (см. вставку на рис. 16.23
для различных типов органических аэрозолей). Результаты этих исследований могут
быть интерпретированы с точки зрения значимости различных источников и эмисси-
онных процессов, особенно тех, которые приводят к появлению органической фракции
аэрозоля в различных регионах. Они могут быть использованы для уточнения клима-
тических моделей, уменьшая неопределенности в оценке влияния аэрозолей на атмос-
ферные процессы (рис. 16.3).
Неорганика: Сульфат Нитрат Аммоний Хлорид
Is
2
• Город
• Город по ветру
• Удаленные точки
Органика: И НОА Other ОА
Total ООА LV-OOA
Pinnacle Park, New York City, NY Manchester, UK Edinburgh,
(Winter) (Summer) (Winter) (Summer) UK
12.3 11.6 12.2 5.2 14.3 3.0
Chelmsford, Hyytiala,
UK
5.3 2.0
Taunus, Mainz,
Germany Germany
16.3 4.2
Houston, Mexico Duke Forest, Thompson Off Coast Chebogue, Cheju Okinawa, Fukue,
TX City NC Farm, NH New England, Canada Island, Japan Japan
US Korea
Рис. 16.23. Общая массовая концентрация (мкг/м3) и массовые доли летучих неорганиче-
ских и органических соединений, содержащихся в субмикронных аэрозолях.
Измерения в Северном полушарии были выполнены с помощью АМС в не-
скольких точках на поверхности, при этом были определены различные типы
органических компонентов аэрозолей, такие как органические аэрозоли угле-
водородного типа (НОА), другие органические аэрозоли (другие ОА), кисло-
родсодержащие органические аэрозоли (ООА), аэрозоли, содержащие органи-
ческие компоненты с низкой летучестью (LV-OOA), и аэрозоли, содержащие
органические компоненты со средней летучестью (SV-ООА). Врезка показы-
вает диапазон отношения кислорода к углероду в органических соединениях,
идентифицированных в разных точках. (Источник: Jimenes J. L. et al. (2009).
«Эволюция органических аэрозолей в атмосфере». Наука. 326: 1525—1529. Пе-
чатается с разрешения AAAS)
(470
Глава 16. Масс-спектрометрия атмосферных аэрозолей
16.4. Итоги и перспективы
Масс-спектрометрия в режиме реального времени превратилась в важный аналити-
ческий метод исследований атмосферных аэрозолей. Масс-спектрометрические си-
стемы обеспечивают селективность и чувствительность, необходимую для детальных
исследований аэрозолей. Применение масс-спектрометров, работающих в режиме ре-
ального времени, позволяет проводить анализ с высоким временным разрешением без
предварительного накопления образца. Потеря аналитической информации, таким
образом, сводится к минимуму. Самым главным преимуществом масс-спектрометров,
работающих в режиме реального времени, является возможность их непосредственно-
го использования в местах, представляющих интерес (например на море с помощью
судов, в горах, на больших высотах с помощью самолетов, в сельской или городской
местностях, а также в закрытых помещениях), позволяя делать мгновенный и деталь-
ный анализ химических веществ в содержащихся аэрозолях. Примеры, приведенные
в этой главе, отражают широкий спектр приложений таких приборов в исследованиях
аэрозолей. АМС, работающие в режиме реального времени, использовались во многих
полевых и лабораторных экспериментах, направленных на изучение источников аэро-
золей, для идентификации выбросов транспортных средств и промышленных пред-
приятий, для изучения характеристик биогенных аэрозолей (Russell, 2009). АМС так-
же успешно применяются в приложениях, связанных с изучением химии атмосферы
(Middlebrook et al., 2003), и для определения конкретных компонент аэрозолей, напри-
мер таких как органические соединения (Bente et aL, 2006; Jitnenes et al., 2009). Кроме
приложений, описанных в этой главе, есть еще две важные области применения масс-
спектрометрии для изучения объектов окружающей среды: охрана здоровья и обеспе-
чение безопасности.
Успешные приложения быстрого анализа атмосферных газов, одиночных частиц
и ансамблей частиц показывают высокий потенциал масс-спектрометрии для деталь-
ной характеристики аэрозолей. Будущие лабораторные и полевые исследования будут
включать в себя одновременную работу нескольких аэрозольных масс-спектрометров.
Это позволит развить наши знания о взаимодействии газ —частица в атмосфере, улуч-
шить понимание процессов, приводящих к изменению климата, и путей воздействия
аэрозолей на здоровье человека. В настоящее время ведется ряд исследований по улуч-
шению работы масс-спектрометров и методов оценки данных. Недавно были разрабо-
таны миниатюрные АМС для проведения аэрозольных эскпериментов на самолетах.
Эти приборы были успешно протестированы для анализа одиночных частиц (Brands
et aL, 2011; Hinz et aL, 2010; Pratt et aL, 2009; Zelenyuk et aL, 2009). Они имеют малый раз-
мер, устойчивы к вибрации и просты в работе. Самолетные масс-спектрометры позво-
ляют проводить прямые измерения аэрозольных частиц на разных высотах и помогают
получать информацию о пространственном распределении атмосферных аэрозолей
в целях создания горизонтальных и вертикальных профилей соответствующих ансам-
блей аэрозолей.
Одновременная работа масс-спектрометров, измеряющих содержание компонентов
в газовой фазе, анализирующих одиночные частицы и характеризующих суммарные
свойства ансамблей частиц, необходима для исчерпывающей характеристики аэрозо-
лей, для понимания природы универсальных химических процессов в атмосфере и для
наиболее полной оценки влияния деятельности человека на климат Земли. Эти прибо-
ры предоставляют взаимнодополняющую информацию, а их совместная эксплуатация
имеет важное значение для получения полной картины об атмосферных аэрозолях.
Литература
[1] . Allan J.D., Delia А. Е., Сое Н., Bower K.N., Alfarra M.R., Jimenez J. L., Middle-
brook A. M., Drewnick E, Onasch T. B., Canagaratna M. R., Jayne J.T., and Worsnop D. R.
(2004). «А generalised method for the extraction of chemically resolved mass spectra from
Aerodyne aerosol mass spectrometer data». Journal of Aerosol Science. 35: 909—922.
[2] . Anderson T. L., Charlson R.J., Schwartz S.E., Knutti R., Boucher O., Rodhe H., and
Heintzenberg J. (2003). «Climate forcing by aerosols — a hazy picture». Science. 300:
1103-1104.
[3] . Appel B. R. (1993). Atmospheric sample analysis and sampling artefacts. In: Willeke K., and
Baron P. A. (Eds.), Aerosol Measurement, Ch 12. John Wiley and Sons, New York, USA.
[4] . Bein K. J., Zhao Y. J., Johnston M.V., and Wexler A. S. (2007). «Identification of sources of
atmospheric PM at the Pittsburgh supersite — Part III: Source characterization». Atmos-
pheric Environment. 41: 3974—3992.
[5] . Bente M., Adam T., Ferge T., Gallavardin S., Sklorz M., Streibel T., and Zimmermann R.
(2006). «An on-line aerosol laser mass spectrometer with three, easily interchangeable laser
based ionisation methods for characterisation of inorganic and aromatic compounds on par-
ticles». International Journal of Mass Spectrometry. 258: 86—94.
[6] . Blake R. S., Whyte C., Hughes С. O., Ellis A. M., and Monks P. S. (2004). «Demonstration
of proton-transfer reaction time-of-flight mass spectrometry for real-time analysis of trace
volatile organic compounds». Analytical Chemistry. 76: 3841—3845.
[7] . Brands M., KamphusM., BottgerT., Schneider J., Drewnick E, Roth A., Curtius J., Voigt C.,
Borbon A., Beekmann M., Bourdon A., Perrin T., and Borrmann S. (2011). «Characteri-
zation of a newly developed aircraft-based laser ablation aerosol mass spectrometer (ALA-
BAMA) and first field deployment in urban pollution plumes over Paris during MEGAPOLI
2009». Aerosol Science and Technology. 45, 1: 46—64.
[8] . Brock C.A., Hudson P. K., Lovejoy E.R., Sullivan A., Nowak J. B., Huey L.G., Coop-
er O. R., Cziczo D. J., de Gouw J., Fehsenfeld F. C., Holloway J. S., Hiibler G., Lafleur B. G.,
Murphy D. M., Neuman J. A., Nicks Jr. D. K., Orsini D.A., Parrish D. D., Ryerson T. B.,
Tanner D. J., Warneke C., Weber R. J., and Wilson J.C. (2004). «Particle characteristics fol-
lowing cloud-modified transport from Asia to North America». Journal of Geophysical Re-
search. 109: D23S26.
[9] . Canagaratna M.R., Jayne J.T., Jimenez J. L., Allan J. D., Alfarra M.R., Zhang Q.,
Onasch T. B., Drewnick E, Coe H., Middlebrook A., Delia A., Williams L. R., Trim-
born A. M., Northway M.J., DeCarlo P. E, Kolb С. E., Davidovits P., and Worsnop D. R.
(2007). «Chemical and microphysical characterization of ambient aerosols with the Aero-
dyne aerosol mass spectrometer». Mass Spectrometry Reviews. 26: 185—222.
[10] . Chan L.Y, Lau W. L., Lee S. C., and Chan C.Y. (2002). «Commuter exposure to particu-
late matter in public transportation modes in Hong Kong». Atmospheric Environment. 36:
3363-3373.
472 Глава 16. Масс-спектрометрия атмосферных аэрозолей
[11] . Charlson R. J., Schwartz S. Е., Hales J. M., Cess R. D., Coakley J. A. Jr., Hansen J. E., and
Hofmann D. J. (1992). «Climate forcing by anthropogenic aerosols». Science. 255:423—430.
[12] . Dall’Osto M., Harrison R. M., Beddows D. C.S., Freney E.J., Heal M.R., and Dono-
van R. J. (2006). «Single particle detection efficiencies of aerosol time-of-flight mass spec-
trometry during the North Atlantic marine boundary layer experiment». Environmental Sci-
ence and Technology. 40: 5029—5035.
[13] . DeCarlo P. F., Kimmel J. R., Trimborn A., Northway M.J., Jayne J.T., Aiken A.C.,
Gonin M., Fuhrer K., Horvath T., Docherty K. S., Worsnop D. R., and Jimenez J. L. (2006).
«Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer». Analytical
Chemistry. 78: 8281—8289.
[14] . Diaz J. A., Pieri D., Arkin C. R., Gore E., Griffin T. P., Fladeland M., Bland G., Soto C.,
Madrigal Y., Castillo D., Rojas E., and Achi, S. (2010). «Utilization of in situ airborne MS-
based instrumentation for the study of gaseous emissions at active volcanoes». International
Journal of Mass Spectrometry. 295: 105—112.
[15] . Dockery D.W., Pope C.A. Ill, Xu X., Spengler J. D., Ware J. H., Fay M.E., Ferris B.G.,
and Speizer F. E. (1993). «An association between air pollution and mortality in six U.S.
cities». The New England Journal of Medicine. 329: 1753—1759.
[16] . Drewnick E, Schwab J. J., Hogrefe O., Peters S., Husain L., Diamond D., Weber R., and
Demerjian K. L. (2003). «Intercomparison and evaluation of four semi-continuous PM2.5
sulfate instruments». Atmospheric Environment. 37: 3335—3350.
[17] . Drewnick F., Hings S.S., DeCarlo P. F., Jayne J.T., Gonin M., Fuhrer K., Weimer S.,
Jimenez J. L., Demerjian K. L., Borrmann S., and Worsnop D. R. (2005). «А new time-
of-flight aerosol mass spectrometer (TOF—AMS) — instrument description and first field
deployment». Aerosol Science and Technology. 39: 637—658.
[18] . Ebert M., Weinbruch S., Rausch A., Gorzawski G., Hoffmann P., Wex H., and Helas G.
(2002). «Complex refractive index of aerosols during LACE 98 as derived from the analysis
of individual particles». Journal of Geophysical Research. 107: 8121.
[19] . Ebert M., Weinbruch S., Hoffmann P., and Ortner H. M. (2004). «The chemical composi-
tion and complex refractive index of rural and urban influenced aerosols determined by
individual particle analysis». Atmospheric Environment. 38: 6531—6545.
[20] . Eltayeb M.A. H., Xhoffer С. E, Van Espen P.J., and Van Grieken R. E. (1993). «Sources
and composition of aerosol from Khartum, Sudan». Atmospheric Environment. T1W. 67—76.
[21] . Erdmann N., DellAcqua A., Cavalli P, Griining C., Omenetto N., Putaud J. P., Raes F.,
and Van Dingenen R. (2005). «Instrument characterization and first application of the sin-
gle particle analysis and sizing system (SPASS) for atmospheric aerosols». Aerosol Science
and Technology. 39: 377—393.
[22] . Forster P., Ramaswamy V., Artaxo P, Berntsen T., Betts R., Fahey D.W., Haywood J.,
Lean J., Lowe D. C., Myhre G., Nganga J., Prinn R., Raga G., Schulz M., and Van Dor-
land R. (2007). Changes in atmospheric constituents and in radiative forcing. In: Solomon S.,
Qin D., Manning M., Chen Z., Marquis M., Averyt К. B., Tignor M., and Miller H.L.
(Eds.), Climate Change 2007: The Physical Science Basis. Contribution of Working Group
I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change.
Cambridge University Press, Cambridge, UK and New York, USA.
Литература
[23] . Gard Е., Mayer J. E., Morrical B. D., Dienes T., Fergenson D. P., and Prather K. A. (1997).
«Real-time analysis of individual atmospheric aerosol particles: design and performance of
a portable ATOFMS». Analytical Chemistry. 69: 4083—4091.
[24] . Geiss O., Barrero-Moreno J., Tirendi S., and Kotzias D. (2010). «Exposure to particulate
matter in vehicle cabins of private cars. Aerosol and Air Quality Research. 10: 581—588.
[25] . Harris W. A., Reilly P.T. A., Whitten W. B., and Ramsey J. M. (2005). «Transportable re-
al-time single-particle ion trap mass spectrometer». Review of Scientific Instruments. 76:
064102-1-064102-8.
[26] . Held A., Hinz K. P., Trimborn A., Spengler B., and Klemm O. (2002). «Chemical classes
of atmospheric aerosol particles at a rural site in Central Europe during winter». Journal of
Aerosol Science. 33: 581—594.
[27] . Henning S., Weingartner E., Schikowski M., Gaggeler H. W., Gehrig R., Hinz K. P., Trim-
born A., Spengler B., and Baltensperger U. (2003). «Seasonal variation of water soluble ions
of the aerosol at the high-Alpine site Jungfraujoch (3580 m asl)». Journal of Geophysical
Research A. 108:4030.
[28] . Herndon S. C., Onasch T. B., Wood E. C., Kroll J. H., Canagaratna M. R., Jayne J. T., Za-
vala M. A., Knighton W. B., Mazzoleni C., Dubey M. K., Ulbrich I. M., Jimenez J. L., Sei-
la R., de Gouw J. A., de Foy B., Fast J., Molina L. T., Kolb С. E., and Worsnop D. R. (2008).
«Correlation of secondary organic aerosol with odd oxygen in Mexico City». Geophysical
Research Letters. 35: LI5804.
[29] . Hinds W. C. (1999). Aerosol Technology. John Wiley and Sons, New York, USA.
[30] . Hinz K. P., and Spengler B. (2007). «Instrumentation, data evaluation and quantification in
on-line aerosol mass spectrometry». Journal of Mass Spectrometry. 42: 843—860.
[31] . Hinz K. P., Kaufmann R., and Spengler B. (1996). «Simultaneous detection of positive and
negative ions from single airborne particles by real-time laser mass spectrometry». Aerosol
Science and Technology. 24: 233—242.
[32] . Hinz K. P., Greweling M., Drews F., and Spengler B. (1999). «Data processing in on-line
laser mass spectrometry of inorganic, organic or biological airborne particles». Journal of
the American Society for Mass Spectrometry. 10: 648—660.
[33] . Hinz, K..P, Trimborn A., Weingartner E., Henning S., Baltensperger U., and Spengler B.
(2005). «Aerosol single particle composition at the Jungfraujoch». Journal of Aerosol Sci-
ence. 36: 123—145.
[34] . Hinz K. R, Erdmann N., Griining C., and Spengler B. (2006). «Comparative parallel char-
acterization of particle populations with two laser mass spectrometric systems LAM PAS 2
and SPASS». International Journal of Mass Spectrometry. 258: 151—166.
[35] . Hinz K. P., Gelhausen E., and Spengler B. (2010). On-line characterization of aerosol parti-
cles with a compact mobile laser mass spectrometer LAMPAS 3. Proceedings of the Interna-
tional Aerosol Conference, Helsinki, 10H2.
[36] . Jaenicke R. (1988). Aerosol physics and chemistry. In: Fischer G. (Ed.), Landoldt—Bornstein
Numerical Data and Functional Relationships in Science and Technology, New Series
Group V: Geophysics and Space Research. Vol. 4. Meteorology Subvolume b. pp. 391—457.
Springer-Verlag, Berlin, Germany.
[37] . Jayne J. T., Leard D.C., Zhang X., Davidovits P., Smith K. A., KolbC. E., and Worsnop D. R.
(2000). «Development of an aerosol mass spectrometer for size and composition analysis of
submicron particles». Aerosol Science and Technology. 33: 49—70.
474 Глава 16. Масс-спектрометрия атмосферных аэрозолей
[38] . Jimenez J. L., Canagaratna М. R., Donahue N. М., Prevot A. S. Н., Zhang Q., Kroll J. H.,
DeCarlo P. E, Allan J. D., Coe H., Ng N. L., Aiken A. C., Docherty K. S., Ulbrich I. M.,
Grieshop A. P., Robinson A. L., Duplissy J., Smith J. D., Wilson K. R., Lanz V. A., Hue-
glin C., Sun Y. L., Tian J., Laaksonen A., Raatikainen T., Rautiainen J., Vaattovaara P.,
Ehn M., Kulmala M., Tomlinson J. M., Collins D. R., Cubison M. J., Dunlea E. J., Huff-
man J. A., Onasch T. B., Alfarra M. R., Williams P. L, Bower K., Kondo Y, Schneider J.,
Drewnick F., Borrmann S., Weimer S., Demerjian K., Salcedo D., Cottrell L., Griffin R.,
Takami A., Miyoshi T., Hatakeyama S., Shimono A., Sun J. Y, Zhang Y. M., Dzepina K.,
Kimmel J.R., Sueper D., Jayne J.T., Herndon S.C., Trimborn A. M., Williams L.R.,
Wood E. C., Middlebrook A. M., Kolb С. E., Baltensperger U., and Worsnop D. R. (2009).
«Evolution of organic aerosols in the atmosphere». Science. 326: 1525—1529.
[39] . Johnston M. V. (2000). «Sampling and analysis of individual particles by aerosol mass spec-
trometry». Journal of Mass Spectrometry. 35: 585—595.
[40] . Johnston M.V., and Wexler A. S. (1995). «MS of individual aerosol particles». Analytical
Chemistry. 67: 721A—726A.
[41] . Kaufmann R. L. (1986). Laser-microprobe mass spectrometry (LAMMA) of particulate mat-
ter. In: Spurny K. R. (Ed.), Physical and Chemical Characterization of Individual Airborne
Particles. Ch. 13. Ellis Horwood, Chichester, UK.
[42] . Keil A., Hernandez-Soto H., Noll R.J., Fico M., Gao L., Ouyang Z., and Cooks R.G.
(2008). «Monitoring of toxic compounds in air using a handheld rectilinear ion trap mass
spectrometer». Analytical Chemistry. 80: 734—741.
[43] . Kiehl J.T. (1999). «Solving the aerosol puzzle». Science. 283: 1273—1275.
[44] . Kirchner U., Vogt R., Natzeck C., and Goschnick J. (2003). «Single particle MS, SNMS,
SIMS, XPS, and FT IR spectroscopic analysis of soot particles during the AIDA campaign».
Journal of Aerosol Science. 34: 1323—1346.
[45] . Kolaitis L.N., Bruynseels F.J., Van Grieken R. E., and Andreae M.O. (1989). «Determi-
nation of methansulfonic acid and non-sea-salt sulfate in single marine aerosol particles».
Environmental Science and Technology. 23: 236—240.
[46] . Kiirten A., Curtius J., Helleis E, Lovejoy E. R., and Borrmann S. (2007). «Development
and characterization of an ion trap mass spectrometer for the on-line chemical analysis
of atmospheric aerosol particles». International Journal of Mass Spectrometry. 265: 30—39.
[47] . Lake D. B., Tolocka M. P., Johnston M. V., and Wexler A. S. (2003). «Mass spectrometry of
individual particles between 50 and 750 nm in diameter at the Baltimore supersite». Envi-
ronmental Science and Technology. 37: 3268—3274.
[48] . Leech J. A., Nelson W. C., Burnett R.T., Aaron S., and Raizenne M.E. (2002). «It’s about
time: a comparison of Canadian and American time-activity patterns». Journal of Exposure
Analysis and Environmental Epidemiology. 12: 427—432.
[49] . McMurry P. H. (2000). «А review of atmospheric aerosol measurements». Atmospheric En-
vironment. 34: 1959—1999.
[50] . Middlebrook A.M., Murphy D. M., Lee, S.-H., Thomson D. S., Prather K.A., Wen-
zel R. J., Liu, D.-Y, Phares D. J., Rhoads К. P., Wexler A. S., Johnston M. V., Jimenez J. L.,
Jayne J.T., Worsnop D. R., Yourshaw L, Seinfeld J. H., and Flagan R.C. (2003). «А com-
parison of particle mass spectrometers during the 1999 Atlanta Supersites Project». Journal
of Geophysical Research. 108, D7: 8424.
Литература
[51] . Morrical В. D., Fergenson D. P., and Prather K. A. (1998). «Coupling two-step laser desorp-
tion/ionization with aerosol time-of-flight mass spectrometry for the analysis of individual
organic particles». Journal of the American Society for Mass Spectrometry. 9: 1068—1073.
[52] . Murphy D. M. (2007). «The design of single particle laser mass spectrometers». Mass Spec-
trometry Reviews. 26: 150—165.
[53] . Nash D. G., Baer T., and Johnston M. V. (2006). «Aerosol mass spectrometry: an introduc-
tory review». International Journal of Mass Spectrometry. 258: 2—12.
[54] . Peters A., Wichmann H. E., Tuch T., Heinrich J., and Heyder J. (1997). «Respiratory effects
are associated with the number of ultrafine particles». American Journal of Respiratory and
Critical Care Medicine. 155: 1376—1383.
[55] . Pope C.A. HI, Thun M.J., Namboodiri M.M., Dockery D.W., Evans J.S., SpeizerF.E.,
and Heath C. W. Jr. (1995). «Particulate air pollution as a predictor of mortality in a pro-
spective study of U.S. adults». American Journal of Respiratory and Critical Care Medicine.
151:669-674.
[56] . Poschl U. (2005). «Atmospheric aerosols: composition, transformation, climate and health
effects». Angewandte Chemie International Edition. 44, 46: 7520—7540.
[57] . Potocek V. (1999). Aerosol analysis by a PIXE system. In: Spurny K. R. (Ed.), Analytical
Chemistry of Aerosols. Ch. 5. Lewis Publishers, Boca Raton, Florida, USA.
[58] . Pratt K.A., Mayer J. E., Holecek J.C., Moffet R.C., Sanchez R.O., Rebotier T. P., Furu-
tani H., Gonin M., Fuhrer K., Su Y., Guazzotti S., and Prather K. A. (2009). «Development
and characterization of an aircraft aerosol time-of-flight mass spectrometer». Analytical
Chemistry. 81: 1792-1800.
[59] . Randall D. A., Wood R. A., Bony S., Colman R., Fichefet T., Fyfe J., Kattsov V., Pitman A.,
Shukla J., Srinivasan J., Stouffer R.J., Sumi A., and Taylor К. E. (2007). «Climate mod-
els and their evaluation». In: Solomon S., Qin D., Manning M., Chen Z., Marquis M.,
Averyt К. B., Tignor M., and Miller H. L. (Eds.). Climate Change 2007: The Physical Science
Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmen-
tal Panel on Climate Change. Cambridge University Press, Cambridge, UK and New York,
USA.
[60] . Reilly P.T.A., Gieray R.A., Yang M., Whitten W. B., and Ramsey J. M. (1997). «Tandem
mass spectrometry of individual airborne microparticles». Analytical Chemistry. 69, 1:
36-39.
[61] . Reilly P.T. A., Gieray R. A., Whitten W. B., and Ramsey J. M. (1998). «Real-time charac-
terization of the organic composition and size of individual diesel engine smoke particles».
Environmental Science and Technology. 32: 2672—2679.
[62] . Rhoads K. P., Phares D. J., Wexler A. S., and Johnston M. V. (2003). «Size-resolved ultrafine
particle composition analysis». Journal of Geophysical Research. 108, D7: 8418.
[63] . Russell S.C. (2009). «Microorganism characterizazion by single particle mass spectrom-
etry». Mass Spectrometry Reviews. 28: 376—387.
[64] . Suess D.T., and Prather K. A. (1999). «Mass spectrometry of aerosols». Chemical Reviews.
99: 3007-3035.
[65] . Sullivan R. C., and Prather K. A. (2005). «Recent advances in our understanding of atmos-
pheric chemistry and climate made possible by on-line aerosol analysis instrumentation».
Analytical Chemistry. 7T. 3861—3886.
476 Глава 16. Масс-спектрометрия атмосферных аэрозолей
[66] . Thomson D. S., Schein М. Е., and Murphy D.M. (2000). «Particle analysis by laser mass
spectrometry WB-57F instrument overview». Aerosol Science and Technology. 33: 153—169.
[67] . Tobias H.J., Kooiman P. M., Docherty K. S., and Ziemann P.J. (2000). «Real-time chemi-
cal analysis of organic aerosols using a thermal desorption particle beam mass spectrom-
eter». Aerosol Science and Technology. 33: 170—190.
[68] . Tourmann J. L.,and Kaufmann R. (1993). «Laser microprobe mass spectrometry (LAM MS)
of coal mine dusts: single particle analysis and toxicity correlation». International Journal of
Environmental Analytical Chemistry. 52: 215.
[69] . Trimborn A., Hinz К. P., and Spengler B. (2000). «Online analysis of atmospheric particles
with a transportable laser mass spectrometer». Aerosol Science and Technology. 33: 191—201.
[70] . Trimborn A., Hinz К. P., and Spengler B. (2002). «On-line analysis of atmospheric parti-
cles with a transportable laser mass spectrometer during LACE 98». Journal of Geophysical
Research. 107, D21: 8132.
[71] . Van Grieken R., and Xhoffer C. (1992). «Microanalysis of individual environmental parti-
cles». Journal of Analytical Atomic Spectrometry. 7: 81—88.
[72] . Van Vaeck L., and Gijbels R. (1990a). «Laser microprobe mass spectrometry: potential and
limitations for inorganic and organic microanalysis, part 1: technique and inorganic ap-
plications». Fresenius’ Journal of Analytical Chemistry. 337: 743—754.
[73] . Van Vaeck L., and Gijbels R. (1990b). «Laser microprobe mass spectrometry: potential and
limitations for inorganic and organic microanalysis, part 2: organic applications». Frese-
nius' Journal of Analytical Chemistry. 337: 755—765.
[74] . Vogt R., Kirchner U., Scheer V., Hinz К. P., Trimborn A., and Spengler B. (2003). «Iden-
tification of diesel exhaust particles at an Autobahn, urban and rural location using single-
particle mass spectrometry». Journal of Aerosol Science. 34: 319—337.
[75] . Weber R., Orsini D., Duan Y., Baumann K., Kiang C. S., Chameides W., Lee Y. N., Brech-
tel F., Klotz P., Jongejan P., ten Brink H., Slanina J., Boring С. B., Genfa Z., Dasgupta P.,
Hering S., Stolzenburg M., Dutcher D. D., Edgerton E., Hartsell B., Solomon P., and Tan-
ner R. (2003). «Intercomparison of near real time monitors of PM2.5 nitrate and sulfate at
the U.S. Environmental Protection Agency Atlanta supersite». Journal of Geophysical Re-
search. 108: 8421.
[76] . WHO. (2006). WHO Air Quality Guidelines for Particulate Matter, Ozone, Nitrogen Dioxide
and Sulfur Dioxide. Global Update 2005. Summary of Risk Assessment. World Health Organi-
zation, Geneva, Switzerland.
[77] . Wieser P., and Wurster R. (1986). Application of laser-microprobe mass analysis to particle
collections. In: Spurny K. R. (Ed.), Physical and Chemical Characterization of Individual
Airborne Particles. Ch. 14. Ellis Horwood, Chichester, UK.
[78] . Xhoffer C., Wouters L., Artaxo P, Van Put A., and Van Grieken R. (1992). Characterization
of individual environmental particles by beam techniques. In: Buffies J., and Van Leeuwen H. P.
(Eds.), Environmental Particles. Ch. 3. Lewis Publishers, Chelsea, Michigan, USA.
[79] . Zelenyuk A., Yang J., Choi E., and Imre D. (2009). «SPLAT II: an aircraft compatible,
ultra-sensitive, high precision instrument for in situ characterization of the size and compo-
sition of fine and ultrafine particles». Aerosol Science and Technology. 43: 411—424.
[80] . Zhang X., Smith K.A., Worsnop D. R., Jimenez J. L., Jayne J.T., Kolb С. E., Morris J.,
and Davidovits P. (2004). «Numerical characterization of particle beam collimation: part II
integrated aerodynamic-lens-nozzle system». Aerosol Science and Technology. 38: 619—638.
Литература
[81] . Zhang Q., Worsnop D. R., Canagaratna M.R., and Jimenez J. L. (2005). «Hydrocarbon-
like and oxygenated organic aerosols in Pittsburgh: insights into sources and processes of
organic aerosols». Atmospheric Chemistry and Physics. 5: 3289—3311.
[82] . Zhang Q., Jimenez J. L., Canagaratna M. R., Allan J. D., Coe H., Ulbrich I., Alfarra M. R.,
Takami A., Middlebrook A. M., Sun Y. L., Dzepina K., Dunlea E., Docherty K., DeCar-
lo P. E, Salcedo D., Onasch T., Jayne J. T., M iyoshi T., Shimono A., Hatakeyama S., Takeg-
awa N., Kondo Y, Schneider J., Drewnick E, Borrmann S., Weimer S., Demerjian K.,
Williams P., Bower K., Bahreini R., Cottrell L., Griffin R.J., Rautiainen J., Sun J.Y.,
Zhang Y. M., and Worsnop D. R. (2007). «Ubiquity and dominance of oxygenated species
in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes».
Geophysical Research Letters. 34: LI3801.
ГЛАВА 17
ИСПОЛЬЗОВАНИЕ
МАСС-СПЕКТРОМЕТРИИ
ДЛЯ ИЗУЧЕНИЯ
ВЗАИМОДЕЙСТВИЯ ДНК
С ЭКОТОКСИКАНТАМИ
Наталья Третьякова и Мелисса Гоггин
17.1. Введение
Процесс образования ковалентных аддуктов дезоксирибонуклеиновой кислоты (ДНК)
при ее взаимодействии с канцерогенами или их химически активными метаболитами
является ранним критическим событием в развитии рака (Harris, 1991; Hecht, 1999; Rayski
и Williams, 1998). Многие канцерогены, находящиеся в окружающей среде, при попада-
нии в организм подвергаются метаболическому превращению (активации) посредством
ферментов, участвующих в биотрансформации ксенобиотиков, таких как, например,
цитохром Р450, монооксигеназа, сульфотрансфераза, N-ацетилтрансфераза и моно-
аминооксидаза. Электрофильные продукты этих ферментативных реакций, например
эпоксиды, соли диазония и нитрениевые ионы, способны вызвать химические изменил
нуклеофильных участков ДНК (Rayski и Williams, 1998). Другие химические вещества,
такие как а,р-ненасыщенные карбонильные соединения, химически активны по своей
природе и не требуют метаболической активации. Поскольку структурные модифика-
ции азотистых оснований изменяют структуру ДНК и, соответсвенно, характер водо-
родных связей, повреждения участков ДНК увеличивают вероятность нуклеотидных
нестыковок (т.е. отсутвие спаривания между нуклеотидами посредством водородных
связей) в процессе синтеза ДНК, что приводит к возникновению генетических повреж-
дений (рис. 17.2) (Harris, 1991; Rayski и Williams, 1998). В природе существует множество
механизмов репарации ДНК, результатом которых является удаление ДНК-аддуктов
и замена их на естественные азотистые основания (Delaney et al., 2005; Evans et al.,
2008a; Hang et al., 2003; Hang, 2004; Jones et al., 1999; Levin et al., 2000; Lindahl и Wood, 1999;
Niedernhofer et al., 2005; Singer и Grunberger, 1983), однако репарация ДНК не является эф-
фективной на 100%, что в некоторых случаях может привести к мутагенезу из-за оши-
бок синтеза при репарации.
Поскольку ДНК-аддукты играют центральную роль в химическом канцерогене-
зе, они могут быть использованы в качестве биомаркеров химического воздействия
на организм. Кроме того, количественный анализ ДНК-аддуктов позволяет найти ко-
17.1. Введение
Рис. 17.1. Структуры некоторых ДНК-аддуктов
личественный критерий, пригодный для оценки скорости образования активных про-
межуточных форм, которые связываются с биомолекулами. Появляются показатели,
которые позволяют сделать оценку биологически опасных доз для соответствующих
канцерогенов (Swenberg et aL, 2008). В настоящее время разработаны чувствительные,
точные и селективные методы для количественного анализа ДНК-аддуктов в биологи-
ческих образцах, которые могут быть использованы для выявления групп риска раз-
вития рака, а также для определения опасных для человека промышленных химиче-
ских веществ в окружающей среде (Farmer et al., 2005; Farmer и Singh, 2008; Кос и Swenberg,
2002; Swenberg et al., 2008). Типичные концентрации ДНК-аддуктов, присутствующие
в тканях в результате воздействия вредных антропогенных веществ, лежат в диапазоне
0,1—1 аддукт-нуклеотидов на каждые 108 ^модифицированных нуклеотидов (Otteneder
г Эндогенное^
или экзогенное
Ч соединение 2
Метаболи-
ческая
активация
Реакци-
онно-спо-
собный
метаболит
Метаболическая
детоксифи кация
ДНК ДНК-
аддукты
Апоптоз
Репликация м ута -
ДНК * иии
Восстановление ДНК
ДНК -аддукт,
выводящийся с мочой
ДНК
Рис. 17.2. Роль ДНК-аддуктов в химическом канцерогенезе
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
и Lutz, 1999). Таким образом, методы, используемые для анализа ДНК-аддуктов, долж-
ны быть сверхчувствительными и при этом способными дать точную количественную
оценку содержания измененных нуклеозидов в присутствии значительно большего
числа неизмененных нуклеозидов (степень избытка 106—109) (Кос и Swenberg, 2002).
17.2. Источники ДНК для анализа ДНК-аддуктов
Источниками ДНК для анализа ДНК-аддуктов могут служить ткани различных орга-
нов, кровь, клетки из ротовой полости и моча. Большее количество ДНК может быть
получено из, например, печени, однако ткани, как правило, не могут быть использова-
ны для биомониторинга ДНК-аддуктов в организме человека и в основном служат для
исследований животных. Поскольку кровь человека и щечные мазки получить легче,
они являются приоритетным источником ДНК для эпидемиологического анализа.
Типичные количества ДНК, необходимые для анализа ДНК-аддуктов методом высо-
Внутренний стандарт
Время
Рис. 17.3. Схема приготовления образца для масс-спектрометрического анализа ДНК-
аддуктов с использованием изотопномеченого внутреннего стандарта
17.3. Подготовка образцов
неэффективной жидкостной хроматографии/электрораспыления/тандемной масс-
спектрометрии (ВЭЖХ/ЭРИ/МС/МС), лежат в пределах между 5 и 200 мкг, в зависи-
мости от степени поражения ДНК и аналитической чувствительности метода. Кроме
того, концентрация ДНК-аддуктов может быть измерена в моче, содержащей депури-
нированные азотистые основания и нуклеозид-аддукты, которые генерируются при
репарации ДНК путем включения механизмов вырезания оснований или нуклеотидов
(Bransfield et aL, 2008; Egner et al., 2006; Hang et aL, 2003). Человеческая моча, однако, яв-
ляется сложной химической матрицей, содержащей большое количество солей и других
полярных соединений. Если эти соединения не удаляются в процессе подготовки об-
разца, они сильно мешают МС-анализу ДНК-аддуктов.
Для того чтобы высвободить ДНК-аддукты в виде отдельных модифицированных
азотистых оснований или нуклеозидов, ДНК подвергается термическому или энзи-
матическому гидролизу (рис. 17.3). В то время как многие ДНК-аддукты, включая
7-алкил-гуанин, 3-алкил-аденин и 8-нитро-гуанин, термически неустойчивы и могут
быть селективно высвобождены из структуры ДНК путем нагрева (Googin et aL, 2007,
Singer и Grunberg, 1983), другие повреждения ДНК стабильны и требуют ферментатив-
ного расщепления. Необходимо учитывать, что при ферментативном гидролизе хи-
мически модифицированных участков ДНК многие аддукты способны заблокировать
доступ фермента к этим участкам, что приводит к неполному расщеплению ДНК (Park
etal., 2003).
17.3. Подготовка образцов
Для проведения МС-анализа ДНК-аддуктов, присутствующих в ДНК-гидролизате,
как правило, требуется несколько этапов очистки образца. Должны быть удалены
немодифицированные нуклеозиды, белки, соли и другие компоненты образца, ко-
торые мешают МС-анализу (Farmer и Singh, 2008; Кос и Swenberg, 2002). Примеры ме-
тодов пробоподготовки, используемых при анализе ДНК-аддуктов, включают ультра-
фильтрацию, жидкость-жидкостную экстракцию (ЖЖЭ), твердофазную экстракцию
(ТФЭ), иммуноаффинную очистку и автономную (офлайн) ВЭЖХ. Ультрафильтрация
является простейшей формой очистки образца для удаления соединений с большим
молекулярным весом (белков, каркаса ДНК, который остается после депуринизации
аддуктов азотистых оснований, и т.д.). Имеющиеся в продаже одноразовые фильтры
(Centricon, Microcon) фирмы Millipore могут быть использованы для достижения бы-
строй и эффективной фильтрации. Фильтры Microcon предназначены для фильтра-
ции образцов с малым объемом (до 0,5 мл), а фильтры Centricon — для больших объ-
емов (20—50 мл).
В методологии ЖЖЭ ДНК-аддукты экстрагируются при помощи органических рас-
творителей, вто время как другие компоненты биологической матрицы остаются в вод-
ной фазе. Этот метод является эффективным и экономичным, но его использование
ограничено гидрофобными аддуктами, например такими, которые образуются при
взаимодействии ДНК с полициклическими ароматическими углеводородами (ПАУ)
и ароматическими аминами. В отличие от жидкость-жидкостной экстракции, метод
твердофазной экстракции применяется значительно шире благодаря большому разно-
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
образию имеющихся в продаже картриджей ТФЭ. В методе твердофазной экстракции
образцы загружаются на небольшую одноразовую колонку, содержащую стационар-
ную хроматографическую фазу, в условиях, которые содействуют полной адсорбции
анализируемого соединения. После промывки, при которой удаляются загрязнения,
анализируемые ДН К-аддукты элюируют более сильным растворителем. В зависимости
от структуры анализируемого вещества метод ТФЭ может использовать различные ме-
ханизмы взаимодействия анализируемого вещества и адсорбента: катионо-обменный,
анионо-обменный или смешанный. Иммуноаффинная очистка использует подобный
же принцип, с тем исключением, что обогащение анализируемого вещества достигает-
ся посредством его адсорбции на поверхности моноклональных или поликлональных
антител, прикрепленных к твердой подложке.
Хроматографическое разделение с использоваеним офлайн-ВЭЖХ может быть эф-
фективно использовано для обогащения анализируемого вещества и удаления большей
части примесей до этапа анализа этого вещества в масс-спектрометре. ВЭЖХ-фракцию,
содержащащую исследуемое соединение, испаряют в вакууме, снова растворяют
в небольшом количестве жидкости и наносят на колонку ВЭЖХ для последующего
ВЭЖХ/МС-анализа. Использование маркеров времени удерживания имеет решающее
значение для контроля за изменениями этого параметра в связи с изменениями темпе-
ратуры хроматографического разделения и состава матрицы. Хотя офлайн-ВЭЖХ обе-
спечивает оптимальную очистку и обогащение аналита, эта методология является тру-
доемкой и страдает от загрязнения предыдущими пробами анализируемого вещества
при использовании одной и той же колонки для анализа разных образцов.
17.4. Приборы для ВЭЖХ-ЭРИ/МС/МС-анализа
ДНК-аддуктов
Развитие метода электрораспылительной ионизации (ЭРИ) в 1980-е годы произвело ре-
волюцию в масс-спектрометрическом анализе ДНК-аддуктов, позволив осуществлять
высокочувствительные и прямые количественные определения различных поврежде-
ний ДНК без дериватизации (главы 1 и 3). Поскольку генерация ионов в ЭРИ является
конкурентным процессом с возможным насыщением сигналов, высокая концентрация
матричных ионов может подавлять ионизацию молекул аналита, коцентрация которых
ниже. К счастью, источник ЭРИ может быть непосредственно подсоединен к выходу
жидкостного хроматографа, который позволяет эффективно отделять молекулы анали-
зируемых веществ от многочисленных молекул матрицы. Источники ЭРИ генерируют
протонированные и депротонированные молекулы анализируемых веществ (в зависи-
мости от полярности проложенного напряжения). Следовательно, для анализа требует-
ся, чтобы анализируемые соединения содержали основные или кислотные функцио-
нальные группы. Поскольку азотистые основания в ДНК могут легко протонироваться
при pH < 5, ДНК-аддукты, как правило, анализируются в режиме регистрации положи-
тельных ионов. После того как ионы анализируемого вещества получены, они направ-
ляются в масс-анализатор для дальнейшего анализа.
Для анализа ДНК-аддуктов используются несколько типов масс-анализаторов,
в том числе одиночные и тройные квадруполи, ионные ловушки, орбитрэпы и время-
17.4. Приборы для ВЭЖХ-ЭРИ/МС-МС-анализа ДНК-аддуктов
пролетные (ВП) масс-спектрометры (Boyd et aL, 2008). Как будет описано ниже, раз-
личные типы масс-спектрометров различаются по своей разрешающей способности,
эффективности рабочего цикла, динамическому диапазону и по возможности осущест-
влять тандемный анализ (МС/МС). Масс-спектрометры различаются также по стоимо-
сти и простоте использования.
В квадрупольном фильтре масс (Q) используется переменное электромагнитное
поле, в котором только одна группа ионов с узким диапазоном значений отношения мас-
сы к заряду (m/z) обладает стабильными траекториями (Boyd et al., 2008; Farmer и Singh,
2008). Наиболее распространенным масс-спектрометром для анализа ДНК-аддуктов
является тройной квадруполь (QqQ). Этот прибор включает в себя два фильтра масс ((^
и Q3) и камеру столкновений, помещенную внутрь промежуточного квадруполя, на-
строенного на пропускание ионов в широком диапазоне масс (квадрупольный провод-
ник q2). QqQ-масс-анализаторы могут работать в режиме сканирования ионов-пред-
шественников или, по-другому, родительских ионов, в режиме мониторинга заданных
реакций (SRM), в режиме регистрации ионов-продуктов и нейтральных потерь. Все эти
режимы обеспечивают высокую специфичность обнаружения аддуктов (глава 4). В ре-
жиме SRM, который наиболее часто используется для количественного анализа аддук-
тов, параметры Qj устанавливаются таким образом, чтобы изолировать ионы-предше-
ственники с известным значением m/z, вычисленным для протонированных молекул
анализируемого вещества. Фрагментация этих ионов осуществляется посредством
диссоциации, активированной соударениями (ДАС, CID), с атомами инертного газа
во втором квадруполе (q2). Продукты диссоциации с заданным значением m/z проходят
через фильтр масс Q3 и направляются на ионный детектор. Тройные квадруполи явля-
ются основным типом масс-спектрометров, которые применяются для количественно-
го анализа следовых количеств веществ благодаря высокой эффективности их рабочего
цикла, хорошего разрешения (разрешающая способность достигает 1000), относительно
низкой стоимости и простоте в эксплуатации.
Ионные ловушки разделяют ионы не в пространстве, а во времени; при этом ионы
могут накапливаться для повышения чувствительности (Dass, 2001). Ионные ловушки
редко используются для следового количественного анализа в связи с низкой эффек-
тивностью их рабочего цикла в режимах мониторинга заданных ионов (SIM) или мо-
ниторинга заданных реакций (SRM), что сильно ограничивает их чувствительность
(Boyd et al., 2008). Тем не менее ионные ловушки могут накапливать ионы в широком
диапазоне значений m/z, обеспечивая превосходную чувствительность в режиме пол-
ного сканирования. Ловушки могут работать в режиме последовательной фрагмен-
тации МС" (п = 1 — 10), что позволяет получить детальную структурную информацию
об исследуемых соединениях. Недавно разработан гибридный 0^2/т03/т-прибор, кото-
рый может работать в режиме QqQ, сохраняя при этом преимущества ионной ловуш-
ки. В частности, этот прибор был использован для количественного анализа ДНК-
аддуктов (Baskerville-Abraham et aL, 2009; Goodenough et aL, 2007). Прибор Сд2/тОз/т
может работать в режиме улучшенного детектирования продуктов фрагментации
(EPI), когда ионы-предшественники проходят через фильтр масс Q1 и направляются
в ячейку соударений (квадруполь q2), где их фрагментация инициируется столкнове-
ниями с атомами инертного газа, как и в QqQ. Образовавшиеся фрагменты поступают
в ионную ловушку между q2 и Q3, хранятся там, а в определенное время извлекают-
484 Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
ся и анализируются при помощи фильтра масс Q3. Хотя некоторые авторы сообщают
о сравнимой чувствительности тройных квадруполей QqQ в режиме мониторинга не-
скольких заданных реакций (MRM) и гибрида QqQ с ловушкой, работающего в EPI
режиме (Boyd et al., 2008), тройные квадруполи QqQ чаще используются для анализа
ДНК-аддуктов.
Времяпролетные (ВП) масс-анализаторы имеют разрешающую способность 10000
или лучше, предоставляя более точную информацию о массах малых молекул, таких
как модифицированные нуклеозиды (Boyd et al., 2008). Однако такие приборы не могут
работать в режиме мониторинга заданных ионов для определения следовых количеств.
Как правило, значения пределов обнаружения (ПО) для qTOF-приборов в 10 раз ниже,
чем для тройного квадруполя QqQ, работающего в режиме мониторинга заданных ио-
нов или заданных реакций (SIM/SRM) (Lacorte и Fernandez-Alba, 2006).
Гибридные времяпролетные масс-спектрометры (QqTOF) состоят из квадру-
польного фильтра масс, ячейки соударений и времяпролетного анализатора (Lacorte
и Fernandez-Alba, 2006). QqTOF похож на QqQ и может работать в режиме, который похож
на SIM/SRM втройном квадруполе, нотолько с более высоким разрешением фрагментов
по массе (Lacorte и Fernandez-Alba, 2006). Гибридные времяпролетные масс-анализаторы
(QqTOF) использовались для анализа следовых количеств, но их основная область при-
менения — секвенирование пептидов/белков (Kristensen et al., 2000).
Магнитные секторные приборы, используемые в качестве масс-анализаторов име-
ют высокую разрешающую способность (10000 и выше) и могут быть легко соединены
с колонкой газового хроматографа (Boyd et aL, 2008). Эти анализаторы используются
в режиме мониторинга заданных ионов (SIM) для следового анализа органических со-
единений в окружающей среде, например таких, как хлорированные дибензодиоксины
(Buser et aL, 1985). Недостатком магнитных секторов является трудность их подсоеди-
нения к источникам ионизации, работающим при атмосферном давлении, например,
ЭРИ. Кроме того, они являются дорогими в установке и обслуживании.
В приборах ионного циклотронного резонанса анализ массы, фрагментация и де-
тектирование ионов происходят в одной ячейке (Dass, 2001). Использование масс-
спектрометеров ионного циклотронного резонанса с преобразованием Фурье (FTICR)
эффективно для анализа пептидов и белков в протеомике (Kang et aL, 2007). FTICR об-
ладает очень высокой разрешающей способностью и высокой чувствительностью, од-
нако имеет низкий рабочий цикл и высокую стоимость. Таким образом, FTICR обычно
не используется для количественного анализа следовых количеств органических ве-
ществ (Boyd et aL, 2008).
Орбитрэп является вариантом ионной ловушки, в которой ионы осциллируют во-
круг центрального электрода. Орбитрэп имеет очень высокую разрешающую способ-
ность (150000 и выше) и высокую точность определения массы (Makarov, 2000). Тем не
менее орбитрэпы относительно редко используются для выскочувствительного количе-
ствен ного анализа биологических образцов (Boydetal., 2008), вероятно, из-за их высокой
стоимости (Perry et aL, 2008). Гибрид орбитрэпа с линейной квадрупольной ловушкой
недавно был использован для количественного определения небольших молекул. Ана-
лиз, выполненный с помощью орбитрэпа, дал результаты, аналогичные тем, которые
были получены на API 4000 (QqQ), однако в этих исследованиях не были использованы
изотопномеченые внутренние стандарты (Zhang et aL, 2009).
Таблица 17.1. Применение методологии масс-спектрометрического анализа ДНК-аддуктов с добавлением изотопномеченого внутрен-
него стандарта in vivo для количественного определения ДНК-аддуктов. Сокращение «Нт»: нуклеотиды, МСИР — масс-
спектрометрия с изотопным разбавлением
Аддукт Метод Предел обнаружения Пробоподготовка Образцы Источник Ссылка
Аддукты природных соединений и лекарственных препаратов
4-OHE2-N7G в моче МСИР 70 пг Твердофазная экстракция МСХ или BondElut человек моча (Bransfield etal., 2008)
Mel-dG многоколоночная хрома- тография и МСИР 900 фг крысы печень, молоч- ные железы (Van den Driessche et aL, 2005)
dG-Tam многоколоночная хрома- тография и МСИР 1 аддукт/108 нт крысы печень, матка (Gamboa da Costa et aL, 2003b)
AFB1-N7G МСИР 0,02 пг Твердофазная экстракция OasisMCX или BondElut LRC, иммуноаффинная колонка человек моча (Egner et aL, 2006)
4-OH-E,-N3Ade кап ил-ЖХ—нано-ЭРИ 5 фмоль/г ткани Твердофазная экстракция, пред- варительная ВЭЖХ человек грудь (Zhang et aL, 2008)
Аддукты с ароматическими аминами
IQ-dG МСИР 6 фмоль С18 твердофазная экстракция крысы печень (Soglia et aL, 2001)
dG-C8-ABP МСИР 5 фмоль/ 300 мкг ДНК С18 твердофазная экстракция человек поджелудоч- ная железа (Ricicki et aL, 2005)
dG-C8-ABP многоколоночная хрома- тография и МСИР 0,72/107 нт ДНК-дайджест и онлайн много- колоночная хроматография мыши печень (Doerge et aL, 1999)
dG-C8-MeIQx МСИР 500 фг SepPak С18 твердофазная экстракция крысы печень (Paehler etal., 2002)
dG-N2-MeIQx МСИР 750 фг SepPak С18 твердофазная экстракция крысы печень (Paehler et aL, 2002)
17.4. Приборы для ВЭЖХ-ЭРИ/МС-МС-анализа ДНК-аддуктов 485
Таблица 17.1. (продолжение)
Аддукт Метод Предел обнаружения Пробоподготовка Образцы Источник Ссылка
NS-Ade-бензидин МСИР 22 пг SepPak С18 твердофазная экстракция (Means et aL, 2003)
N8-Ade-2- аминофлуорен МСИР 51 пг SepPak С18 твердофазная экстракция (Means et aL, 2003)
Аддукты с нитрозаминами
Гидроксиэтил-dG Капил-ЖХ-ЭРИ-МС/МС 100—150 фмоль 4 мм шприцевой фильтр крысы печень (Dennehy and Loeppky, 2005)
O6-POBdG МСИР 50 фмоль Strata-X С18 SPE мыши печень (Thomson etal., 2004)
РОВ аддукт МСИР 3 фмоль G, 1 фмоль dGuo, 100 амоль Thd, 2 фмоль Cyt StrataX твердофазная экстракция крысы печень/легкие (Lao et aL, 2006)
7-POB-Gua МСИР Strata-X твердофазная экстракция крысы печень/легкие (Stepanov and Hecht, 2009)
О6-метилбС многоколоночная хрома- тография и МСИР 24 фмоль O6-mdGuo крысы печень (Brink et aL, 2006)
О6-метил/О6-этил-бС многоколоночная хрома- тография и МСИР 0,03 O6-Me-dG/108 нт. 0,05 O6-Et-dG/108 нт. центрифугирование мыши печень (Churchwell et aL, 2006)
Экзоциклические аддукты
C-edC,C-edG, H-edA, H-edC, H-edG Капил-ЖХ-ЭРИ-МС/МС в режиме МЗР Нет данных 0,2 мкм фильтр, твердофазная экстракция Supelclean LC-C18 мыши кишечник (Williams et aL, 2006)
Таблица 17.1. (продолжение)
Аддукт Метод Предел обнаружения Пробоподготовка Образцы Источник Ссылка
gdG МСИР, капил-ЖХ-ЭРИ- МС/МС в режиме МЗР gdG: 1,5—2 пмоль 4 мм шприцевой фильтр крысы печень (Dennehy and Loeppky, 2005)
eGua МСИР, ЖХ-ЭРИ-МС/МС 1 пг Твердофазная экстракция SCX, С18-ОН человек моча (Chen and Chiu, 2005)
еА МСИР, ВЭЖХ-ЭРИ- МС/МС в режиме МЗР 2—3 пг Твердофазная экстракция С18-ОН человек моча (Chen and Chang, 2004)
EdA многоколоночная хроматография, МСИР, ХИАД-МС 8 пмоль Твердофазная экстракция Oasis HLB человек моча (Hille strom et aL, 2004)
l,N2-nponaHo-dG МСИР жх/мс/мс 0,015 фмоль/ мкг ДНК Центрифугирование (YM-10), твердофазная экстракция Oasis HLB крысы печень (Stout et aL, 2006)
Пропано-dG МСИР 4 аддукта/109 нт Твердофазная экстракция Strata-X человек легкие (Zhang et aL, 2007)
Сго-dG МСИР 0,2 фмоль Твердофазная экстракция Strata-X человек легкие/печень (Zhang et aL, 2006)
PdG, Et-dG МСИР 250 фг? Дайджест ДНК с жидкость-жид- костной экстракцией человек кровь (Matsuda etal., 2006)
l,N2-sdGuo многоколоночная хрома- тография, МСИР, 20 фмоль Экстракция хлороформом крысы печень (Loureiro etal., 2002)
EdC EdA EAde многоколоночная хрома- тография, МСИР, (ХИАД) 70 пмоль dAde, 100 пмоль dC, 17 пмоль dA Твердофазная экстракция OasisHLB человек моча (Hille strom et aL, 2006)
Аддукты ДНК-обусловленные этанолом/ацетальдегидом
1Ч7-Этил^С МСИР 0,25 фмоль Ym-30, Strata-X твердофазная человек печень (Chen et aL,
экстракция 2007а)
17.4. Приборы для ВЭЖХ-ЭРИ/МС-МС-анализа ДНК-аддуктов
Таблица 17.1. (продолжение)
Аддукт Метод Предел обнаружения Пробоподготовка Образцы Источник Ссылка
Ю-Этил-сЮ многоколоночная хрома- тография, МСИР 0,59 пг/мл мочи SepPak Cl8 твердофазная экстракция человек моча (Chao etal., 2006)
ЬР-Этил-бС МСИР 0,4 фмоль Твердофазная экстракция Stra- taX, OasisMAX человек печень (Wang et aL, 2006)
1Ч2-Этил-Сиа МСИР 0,05 фмоль в уколе, 3 аддук- та/109 нт Strata-X, OasisMAX твердофаз- ная экстракция человек лейкоциты (Chen et aL, 2007b)
C8-HEG МСИР 50 фмоль ВЭЖХ крысы печень (Nakaoet aL, 2002)
Аддукты ДНК с полициклическими ароматическими углеводородами
MP-dG, MP-dA МСИР 10 фмоль/2 фмоль в 100 мкг ДНК Oasis Н LB твердофазная экстракция крысы печень (Monien etal., 2008)
dG-N2-BPDE МСИР Не определено Не определено мыши печень, легкие, почки (Arlt et aL, 2008)
dG-N2-BPDE МСИР Oasis Н LB твердофазная экстракция мыши печень (Singh et aL, 2006)
dG-BPDE многоколоночная хрома- тография, МСИР 1/108 нт Дайджест ДНК мыши печень (Beland etal., 2005)
BP-6-N7Gua многоколоночная хрома- тография, МСИР 2,5 фмоль/1 мл мочи SepPak С8 и Strata-SCX твердо- фазная экстракция человек моча (Chen et aL, 2005)
1,3-бутадиен/эпоксид
THBG МСИР 3,5 фмоль YM-10 фильтрация мыши/ крысы печень/легкие (Powley et aL, 2005)
HMVK-dGuo МСИР 5 фмоль/200 мкг ДНК Centricon-10, ВЭЖХ крысы печень (Powley et aL, 2007)
Таблица 17.1. (окончание)
Аддукт Метод Предел обнаружения Пробоподготовка Образцы Источник Ссылка
N7-GA-Gua, N3-GA-Ade МСИР 1,5—2 аддукта/108 нт. Центрифугирование, фильтрация мыши печень/легкие/ почки (Gamboa da et aL, 2003a)
5/5-N7G-BD N7G-N6A-BD N7-HEG N7-HEG N7-HEG, Nl-HedA МСИР Нано-ЖХ—нано-ЭРИ- МС/МС многоколоночная хрома- тография, МСИР МСИР МСИР 6 аддуктов/109 нт 3 аддукта/109 нт 0,25 нг/ мл мочи 0,1 фмоль 0,5 фмоль YM-10 фильтрация, ВЭЖХ YM-10 фильтрация, SepPak С18 Твердофазная экстракция Центрифугирование YM-10 фильтрация YM-3 фильтрация, ВЭЖХ мыши/ крысы мыши/ крысы человек крысы крысы печень/легкие/ почки/мозг/ти- мус печень моча печень, сердце, селезенка, почки, толстая кишка, желу- док, легкие печень (Goggin et aL, 2009) (Goggin et aL, 2009) (Huang et al., 2008) (Marsden et al.,2007) (Tompkins etal., 2008)
Оксидативный стресс, вызванный аддуктами ДНК
8-oxo-dG 8-oxo-dG/A 8-OH-dG CEdG МСИР МСИР МСИР МСИР 2 фмоль 10 фмоль 8-oxodA 0,024 нг/мл 0,2 пг Очистка на иммуноаффинной колонке Oasis Н LB твердофазная экстракция SepPak Cl8 твердофазная экстракция Strata-X-C твердофазная экстракция мыши человек крысы простата моча моча (Pathak etal., 2008) (Evans et aL, 2008b) (Li et aL, 2005) (Synold et aL, 2008)
17.4. Приборы для ВЭЖХ-ЭРИ/МС-МС-анализа ДНК-аддуктов
490 Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
17.4.1. Валидация масс-спектрометрического метода
Как описано выше, ДНК-аддукты, присутствующие в биологических образцах, как
правило, подвергаются нескольким этапам обработки/обогащения до МС-анализа,
для того чтобы удалить из образца избыток веществ, ненужных для анализа (матрицу),
и свести к минимуму подавление сигнала в источниках ЭРИ. Даже в результате наибо-
лее эффективной очистки определенное количество анализируемого вещества теряется
в процессе переработки и восстановления. Эти потери могут значительно отличаться
от образца к образцу. Для повышения воспроизводимости и точности количественного
анализа, может быть использована методика изотопного разбавления (ИРМС) (главы 2
и 15). В тандемной масс-спектрометрии с изотопным разбавлением в качестве внутрен-
него стандарта для количественного определения исследуемого соединения использу-
ется полный химический и структурный аналог этого соединения, в котором часть ато-
мов, представленная натуральными изотопами, заменена на их стабильные изотопы,
которые очень редко встречаются в природе. Повышение воспроизводимости и точно-
сти достигается тогда, когда изотопномеченый стандарт добавляется в образец в начале
анализа. Изотопномеченый стандарт химически идентичен исследуемому соединению
(за исключением наличия тяжелых атомов в его структуре) и имеет близкий молеку-
лярный вес, что помогает проследить потери исследуемого соединения при переработке
и учесть влияние любых матричных эффектов и эффекта подавления сигнала в ионном
источнике при масс-спектрометрическом анализе. Количественное определение при
использовании стандарта в ВЭЖХ/МС/МС достигается путем сравнения площадей пи-
ков анализируемого соединения и внутреннего стандарта.
Для обеспечения точного количественного определения анализируемого вещества
в присутствии биологической матрицы количественные ВЭЖХ/МС/МС-методы долж-
ны быть проверены путем введения образца, который содержит матрицу и известные
количества анализируемого вещества и внутреннего стандарта. При проверке методики
на образцах, содержащих внутренний стандарт, используются одни и те же методы обо-
гащения и один и тот же метод ВЭЖХ/МС/МС-анализа. Результаты, как правило, выра-
жаются в виде графика, где измеренное количество анализируемого вещества сравнива-
ется с количеством, которое было добавлено в матрицу (рис. 17.4). Аналогичный подход
может быть использован для определения предела детектирования (когда отношение
сигнала к шуму > 3) и предела количественного обнаружения (ПКО) (когда отношение
сигнала к шуму > 10) анализируемых веществ в данной матрице. Точность метода и из-
менение этого параметра как в течение дня, так и в течение нескольких дней можно рас-
считать исходя из результатов повторных измерений, используя аналогичные образцы
с внутренним стандартом. В данном обзоре обсуждаются конкретные примеры приме-
нения МС с изотопным разбавлением для количественного определения ДНК-аддуктов
in vivo. Речь пойдет в основном о методе ВЭЖХ/ЭРИ-МС/МС. Будут обсуждены неко-
торые последние достижения в методах автономной (онлайн) обработки образцов, рас-
смотрено использование наноспрейных эмиттеров в ЕРИ, а также микрожидкостных
чипов. Метод ВЭЖХ/ЭРИ-МС/МС был использован для количественного определения
широкого спектра ДНК-аддуктов, в том числе образующихся при взаимодействии ДНК
с ароматическими аминами, нитрозаминами, полициклическими ароматическими
углеводородами и т.д. Метод изотопного разбавления был использован для количе-
ственного определения ДНК-аддуктов, которые образуются при воздействии канцеро-
Количество добавленного l,7V6-HMHP-dA в фемтомолях
Рис. 17.4. Валидация результатов многомерного хроматографического анализа
(ВЭЖХ/ЭРИ/МС/МС с регистрацией положительных ионов) 1,М6-(2-гидрокси-
3-гидроксиметилпропан-1,3-диил)-дезоксиаденозина в ДНК грызунов. (Пе-
чатается с разрешения Goggin М., Seneviratne U., Swenberg J. A., Walker В. Е.,
Tretyakova N. «Методы масс-спектрометрического анализа с применением
многомерной жидкостной хроматографии для количественного определения
экзоциклических dA ДНК-аддуктов в лабораторных животных, подвергнутых
действию 1,3-бутадиена». Химические исследования в токсикологии. 23, 808—812.
Copyright (2010) Американское химическое общество.)
генов, присутствующих в воздухе, еде, питье, и сигаретном дыме, а также химических
соединений, присутствующих в производственных помещениях. Ниже подробно опи-
саны конкретные методики, а типичные пределы обнаружения приведены в табл. 17.1.
17.5. Примеры использования ВЭЖХ/МС/МС
для анализа ДНК-аддуктов
17.5.1. Аддукты, образующиеся при воздействии
ароматических аминов
4-Аминобифенил (4-АВР) является канцерогеном и присутствует в сигаретном дыме.
В организме это соединение подвергается N-гидроксилированию и превращает-
ся в N-гидроксиаминобифенил, который способен связываться с ДНК. Воздействие
4-АВР на организм, как подозревают, вызывает рак поджелудочной железы (Ricickietal.,
2005). Основным ДНК-аддуктом, образующимся при воздействии 4-АВР, является
М-(дезоксигуанозин-8-ил)-4-АВР (dG-C8-4-ABP). В группе Пола Вороса из Северо-
Восточного университета (Бостон) был разработан метод количественного определения
4-АВР-аддуктов в ДНК, выделенной из поджелудочной железы человека (Ricicki et aL,
2005). В данном исследовании были использованы ткани поджелудочной железы 12 до-
норов (шесть мужчин и шесть женщин). К раствору выделенной ДНК был добавлен вну-
тренний стандарт (C8-dG-4-ABP-d9), образцы были очищены методом твердофазной
экстракции на картриджах Isolute С18 и проанализированы методом ВЭЖХ/МС/МС
(тройной квадруполь TSQ Quantum AM, Finnigan) в режиме микроэлектроспрея с ре-
гистрацией положительных ионов. Количественный анализ был выполнен методом
мониторинга заданных реакций с использованием переходов m/z 435 319 (для ионов
аналита) и m/z 444 —> 328 (для ионов внутреннего стандарта). При этом использовалась
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
калибровочная кривая, построенная по синтезированным стандартам. Уровни dG-
С8-АВР в шести образцах были ниже предела количественного определения, при этом
не было обнаружено никакой корреляции между наличием dG-C8-ABP и возрастом,
полом или курением/некурением. Самый высокий уровень аддуктов (60 dG-C8-ABP
на 108 нуклеотидов) был обнаружен у некурящего мужчины (Ricicki etal., 2005).
2-Амино-3-метилимидазо[4,5-е]хинолин (IQ) — гетероциклический ароматиче-
ский амин, обнаруженный в сигаретном дыме, а также в жареном мясе и рыбе. Это сое-
динение является канцерогеном для животных, т. е. чтобы определить степень его риска
для человека, необходимы чувствительные методы количественного анализа аддуктов
образующихся при взаимодействии ДНК и IQ. Основным аддуктом, который образу-
ется при воздействии IQ на ДНК, является C8-dG-IQ (обозначен цифрой 2 на рис. 17.1).
Соглиа и его коллеги разработали ВЭЖХ/МС/МС-метод для количественной оценки
IQ-аддуктов в ДНК, полученной из печени крыс, подвергавшихся воздействию доз IQ
от 0,05 до 10 мг/кг (Soglia et aL, 2001). В раствор, содержащий 300 микрограммов ДНК,
был добавлен внутренний стандарт C8-dG-IQ-d3, после чего ДНК была подвергнута
ферментативной обработке. После обогащения при помощи твердофазной экстракции
образцы были проанализириваны методом ВЭЖХ/МС/МС. Масс-спектрометр TSQ700,
оборудованный микроспрейным интерфейсом работал в режиме регистрации положи-
тельных ионов. Предел количественного обнаружения аддукта в этом методе был опре-
делен на уровне 6 фемтомоль/300 микрограмов ДНК. Частота возникновения аддукта
C8-dG-IQ была на уровне 3,5 аддуктов на 108 нуклеотидов при дозе IQ в 0,05 мг/кг веса,
и 39 аддуктов на 108 нуклеотидов при дозе IQ 10 мг/кг. Эти величины сопоставимы с по-
лученными при анализе данного аддукта методом постмаркировки с использованием
радиоактивного фосфора 32Р (Turesky et al., 1997), однако для обнаружения IQ-аддуктов
масс-спектрометрический метод является более точным и специфичным.
Эта же группа исследователей разработала ВЭЖХ/МС/МС-метод детектирования
2-амино-3,8-диметилимидазо-[4,5-/]хиноксалин (Ме^х)-индуцированных ДНК-
аддуктов (Paehleret al., 2002). MelQx образует аддукты с гуанином в положениях С8 и N2:
C8-dG-MeIQx и N2-dG-MeIQx. В организм самцов крыс IQ был инжектирован в количе-
стве 0,05; 0,5 или 10 мг IQ на кг массы тела. Крысы были умерщвлены через 24 часа после
приема препарата. ДНК была выделена из печени. Внутренние стандарты, C8-dG-IQ-d3
и N2-dG-IQ-d3 были добавлены к раствору, содержащему 250 микрограммов ДНК. По-
сле ферментативного расщепления MelQx-аддукты ДНК выделили с помощью твердо-
фазной экстракции и проанализировали методом ВЭЖХ/МС/МС (TSQ 7000, Finnigan).
Масс-спектрометр работал в режиме мониторинга заданных реакций. Предел обнару-
жения метода составил 0,5—1 аддукта на 108 нуклеотидов. Никаких аддуктов не было
обнаружено при дозе 0,05 мг IQ на кг веса; при дозах 0,5 и 10 мг IQ на кг было обнаружено
соответственно 0,45 и 3,06 аддукта на 107 нуклеотидов.
17.5.2. Аддукты с нитрозаминами
Молекулы N-нитрозодиэтаноламина (NDELA) метаболически превращаются в два
химических соединения, которые связываются с ДНК: 2-гидроксиэтилдиазониевые
ионы и глиоксаль (Dennehy и Loeppky, 2005). NDELA связывается с гуанином, образуя
О6-2-гидроксиэтил-2-дезоксигуанозин (OHEdG) и глиоксальдезокси гуанозин (gdG).
В исследовании (Dennehy и Loeppky, 2005) самцам крыс была введена однократная
17.5. Примеры использования ВЭЖХ/МС/МС для анализа ДНК-аддуктов
инъекция, содержащая 0,2; 0,4 или 0,8 миллимоль NDELA на один кг массы тела; жи-
вотных умерщвляли через 4 часа после воздействия, ДНК выделялась из печени. Об-
разцы (400 мкг ДНК) были подвергнуты гидролизу, далее в раствор был добавлен вну-
тренний стандарт (OHEdG-d4 и l3C,l5N2-gdG), раствор был отфильтрован и подвергнут
ЭРИ/МС/МС-анализу. Метод мониторинга заданных реакций был использован для
высокоселективного анализа ОН EG и gdG-аддуктов. Отсутствие аддуктов наблюда-
лось у животных, получавших 0 или 0,2 ммоль/кг NDELA, уровень аддуктов, не под-
дающийся количественной оценке, наблюдался при добавлении 0,4 ммоль NDELA на кг
веса. При добавлении 0,8 ммоль NDELA на кг веса уровни аддуктов были сопоставимы
с установленными методом 32Р-метки (Loeppky et al., 2002). Уровни gdG составляли 4,4
и 11 аддуктов на 106 нуклеотидов, в то время как OHEdG присутствовал на уровне 0,35
и 0,87 аддуктов на 106 нуклеотидов. В тех же образцах методом 32Р-метки были обнару-
жены уровни gdG 4—6 аддуктов на 106 нуклеотидов, а уровни ОН EG колебались от 2,3
до 24 аддуктов на 106 нуклеотидов (Loeppky et al., 2002).
Нитрозамином, специфичным для табака, является 4-(метилнитрозамино)-1-(3-
пиридил)-бутанон-1 (NNK). Это соединение является мощным канцерогеном, вызыва-
ющим рак легких у грызунов. Предполагается, что это соединение вносит свой вклад
в развитие рака легких у курильщиков. NNK может метаболизироваться в 4-гидрокси-
1-(3-пиридил)-бутанон-1, который взаимодействует с ДНК с образованием пири-
дилоксобутил (РОВ) аддуктов. Были разработаны МС-методы для количественного
определения четырех РОВ-аддуктов у животных, подвергшихся воздействию NNK
или 4-(метилнитрозамино)-1-(3-пиридил)-бутанола-1 (NNAL) (Lao et al., 2006). Этими
аддуктами являются О6-[4-(3-пиридил)-4-оксобут-1-ил]-2'-дезоксигуанозин (О6-РОВ-
dGuo), 7-[4-(3-пиридил)-4-оксобут-1-ил] гуанин (7-POB-Gua), О2-[4-(3-пиридил)-4-
оксобут-1-ил] цитозин (O2-POB-Cyt), О2-[4-(3-пиридил)-4-оксобут-1-ил]-тимидин (О2-
POB-dThd). ДНК выделялась из печени и легких крыс, которым втечение4дней вводился
либо физиологический раствор, либо NNK в количестве 5,2 мг на кг веса, либо NNK
в количестве 20,4 мг на кг веса. После проведения ферментативного гидролиза РОВ-
аддукты выделяли методом твердофазной экстракции с помощью картриджей Strata-X
и анализировали ВЭЖХ/МС/МС. 7-POB-Gua и O2-POB-dThd были самыми распростра-
ненными в обоих типах тканей. Следующим по значимости был аддукт O2-POB-Cyt, тог-
да как O6-POB-dGuo был наименее распространенным. Частота образования аддуктов
была выше в печени, чем в легких, за исключением аддукта O6-POB-dGuo, которого было
больше в легких. Пределы обнаружения были следующими: 3 фемтомоля для 7-POB-Gua,
1 фемтомоль для O6-POB-dGuo, 100 аттомолей для O2-POB-dThd и 2 фемтомоля для О2-
POB-Cyt в 1 миллиграмме ДНК. Метод количественной масс-спектрометрии с исполь-
зованием изотопых стандартов был использован для оценки образования РОВ-аддуктов
в ДНК из печени/легких крыс, подвергнутых хроническому воздействию NNK, (R)-
NNAL и (S)-NNAL (Lao etal., 2007). NNK может быть восстановлено до NNAL, который
может окисляться обратно в NNK (Lao etal., 2007). Данное исследование (Lao etal., 2007)
было первой попыткой провести количественный анализ РОВ-аддуктов в ДНК живот-
ных, подвергнутых воздействию NNAL (NNAL-экспозиция). Та же тенденция в уровнях
аддукта наблюдалась и для NNK: O2-POB-dThd являлся наиболее распространенным,
a O6-POB-dGuo — наименее распространеным аддуктом. В случае NNAL-экспозиции
уровни аддуктов были выше в легких по сравнению с печенью. Никаких существенных
494 Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
различий в уровнях аддуктов не было у крыс, подвергнутых воздействию NNK или (S)-
NNAL. У крыс, получавших (R)-NNAL, уровни аддукта были значительно ниже. NNAL
также может вступать в реакцию с ДНК с образованием пиридил гидроксибутил (PH В)
аддуктов. В лаборатории Лао были разработаны количественные ВЭЖХ/МС-методы
определения РНВ-аддуктов. Интересно, что более высокие уровни РНВ-аддуктов на-
блюдались у животных, получавших (R)-NNAL, но не (S)-NNAL или NNK (Upadhyaya
et al. 2008). Методы, разработанные для анализа РОВ и РНВ, недавно были использова-
ны для сравнения уровней аддуктов в митохондриальной и ядерной ДНК крыс, которые
подверглись воздействию NNK и NNAL (Stepanov и Hecht, 2009).
17.5.3. Экзоциклические аддукты ДНК
Экзоциклические этено-аддукты могут образовываться в ДН К в результате воздействия
экзогенных химических веществ, таких как винилхлорид. Альтернативным процессом
является взаимодействие с эндогенными продуктами перекисного окисления липидов.
Этено-ДНК-аддукты являются мутагенами и могут инициировать раковые заболева-
ния. Лоурейро и его коллеги разработали ВЭЖХ/МС-метод количественного анализа
1 ,N2-3TeHO-dG аддуктов in vivo (Loureiro etal., 2002). ДНК выделяли из печени самок крыс
линии Wistar, не подвергавшихся действию окислительных агентов. Масс-спектрометр
Quattro II работал в режиме регистрации положительных ионов. Количественный ана-
лиз осуществлялся в режиме мониторинга заданных реакций с использованием мечено-
го изотопами стандарта ([15N5] -l,N2-e-dGuo), который добавлялся к раствору ДНК. ДНК
ферментативно расщепляли, экстрагировали и анализировали методом ЖХ/МС/МС,
который позволил определить естественный уровень (1,7 аддуктов на 107 нуклеотидов)
l,N2-e-dGuo аддуктов в коммерческих образцах ДНК из тимуса теленка. В образцах
ДНК из печени крыс, не подвергшихся действию экзогенных химических веществ, этот
уровень составил 5,22 аддуктов на 107 нуклеотидов.
Метод ВЭЖХ/МС/МС с изотопным разбавлением был недавно применен для ко-
личественного определения 1,М6-этенодезоксиаденозин-аддуктов у мышей, которым
ввели уретан (Ham et al., 2004). В организме мышей уретан метаболически превращал-
ся в эпоксид винилкарбамата, который мог реагировать с ДНК с образованием 1,N6-
этено-2'-дезоксиаденозина (e-dA). Для улучшения чувствительности метода перед
ВЭЖХ/МС-анализом использовалась иммуноаффинная хроматография. Предел об-
наружения составил 2,5 фмоль, причем анализ показал пристуствие 2 аддуктов на 108
нормальных dA в ДНК тимуса теленка. Этот метод был также использован для срав-
нения уровней е-dA в ДН К диких животных и животных, у которых отсутствовал фер-
мент алкиладенин-ДН К-гликозилаза (AAG). Период полураспада е-dA в ДНК из легких
животных, у которых наблюдается дефицит фермента AAG, составил 185 ч, в то вре-
мя как у диких животных это время составило 28 часов. При этом предполагалось, что
ферментативная регенерация участков ДНК с е-dA происходит с участием AAG, а также
по другим механизмам (Ham et al., 2004).
17.5.4. Этанол/ацетальдегидные аддукты
Увеличение случев рака толстой кишки связано с употреблением алкоголя. Рак тол-
стой кишки может быть связан с влиянием ацетальдегида, активных форм кислорода
17.5. Примеры использования ВЭЖХ/МС/МС для анализа ДНК-аддуктов 495
(АФК) и 1-гидроксиэтильных радикалов. Все эти соединения возникают при метабо-
лизме этанола. В частности, было показано, что гидроксиэтильные радикалы образу-
ют С8-(1-гидроксиэтил)гуанин-аддукты (C8(l-HE)gua). Накаос коллегами разработали
ВЭЖХ/МС-метод количественного анализа C8(l-HE)gua у крыс, получавших этанол
(Nakao etal., 2002). Самцы крыс разновидности Sprauge-Dawley получали внутрикишеч-
ную инъекцию либо воды, либо этанола (5 г/кг). Через 3 или 12 часов после инъекции
этих животных умерщвляли, а их печень использовали для выделения РНК и ДНК.
Нуклеиновые кислоты гидролизировали в растворе IN НС1 с добавлением изотопно-
го внутреннего стандарта — 13C,15N2-C8-(l-HE)gua, а получившуюся смесь очищали
с помощью автономной (офлайн) ВЭЖХ. ВЭЖХ-фракции, содержащие аддукты, были
сконцентрированы и проанализированы с помощью ВЭЖХ/МС/МС на квадрупольном
масс-спектрометре Quattro II фирмы «Микромасс» в режиме мониторинга заданных
реакций с регистрацией положительных ионов. Количественный анализ проводили
по калибровочной кривой, построенной с использованием синтетических стандартов.
Хотя C8(l-HE)gua-aaayKT был детектирован в РНК и ДНК крыс, аналогичные уровни
этого аддукта наблюдались у крыс, не получавших этанол, а также в образцах коммер-
чески доступных ДНК из тимуса теленка (Nakao et al., 2002).
Ван и др. разработали разновидность количественного ВЭЖХ/МС/МС-анализа
ДНК-аддуктов, образовавшихся в результате воздействия ацетальдегида на печень че-
ловека (Wang et al., 2006). Ацетальдегид может образовать с ДНК аддукт 1Ч2-этилиден-
dGuo по положению N2 гуанина. Этот аддукт неустойчив на уровне нуклеозидов, но мо-
жет быть стабилизирован путем превращения его в стабильный 1Ч2-этил-сЮ-аддукт
восстановлением NaBH3CN. Для количественного определения данных аддуктов была
синтезирована пара 1Ч2-этил-сЮио и [151Ч5]-1Ч2-этил-сЮио. ДНК была гомогенизирова-
на в буфере, содержащем NaBH3CN, чтобы предотвратить окисление, ферментативно
расщеплена и проанализирована методом ВЭЖХ/МС/МС. В ДНК человеческой печени
средний уровень 1Ч2-этил-сЮ составил 534±245 фмоль аддукта/микромоль dGuo (от 257
до 1055). Аналогичные данные по воздействию алкоголя или табака на человека пока
отсутствуют. В будущем планируется использовать ВЭЖХ/МС/МС для изучения роли
ацетальдегида в процессе развития восприимчивости к раку у курильщиков и лиц, упо-
требляющих алкоголь.
17.5.5. Аддукты с полициклическими ароматическими
углеводородами (ПАУ)
Неполное сгорание органических материалов приводит к образованию ПАУ. Сот-
ни видов ПАУ были обнаружены в выхлопных газах автомобилей, в сигаретном дыме
и каменноугольной смоле. Многие из них являются канцерогенами для человека и жи-
вотных. Беланд и др. использовали ЖХ/ЭРИ/МС/МС для количественного анализа
БП-ДНК-аддуктов бенз[а]пирена in vivo (Beland et al., 2005). БП может быть метаболи-
чески превращен в транс-7,8-дигидрокси-анти-9,10-эпокси-7,8,9,10-тетрагидробенз[а]
пирен (BPDE), который может реагировать с ДНК с образованием аддуктов, напри-
мер 10-(дезоксигуанозин-1Ч2-ил)-7,8,9-тригидрокси-7,8,9,10-тетрагидро-бенз[а]пирена
(dG-BPDE) (помечен цифрой 3 на рис. 17.1). Ранее dG-BPDE-аддукты были обнаружены
с помощью 32Р-метки и иммунологическими методами. Для масс-спектрометрического
анализа этих аддуктов необходимо иметь 10—100 микрограммов ДНК. Метод
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
ВЭЖХ/МС/МС, разработанный Беландом и др. в 2005 г., был использован для анали-
за BPDE-аддуктов как in vitro, так и в образцах из мышиных или человеческих легких
(Beland et aL, 2005). Образцы ДНК (100 микрограммов) были ферментативно гидро-
лизированы, в раствор было добавлено 10 мкг внутреннего стандарта (dG-BPDE-d8),
анализ проведен с помощью капиллярной ВЭЖХ — тандемного квадрупольного масс-
анализатора. Количественный анализ dG-BPDE-аддуктов был проведен в режиме мо-
ниторирования заданных реакций, переход m/z 570 —> 257 использовался для анализа
dG-BPDE-аддукта, а переход m/z 577 —> 264 — для внутреннего стандарта. Количествен-
ный анализ ДНК-аддуктов проводился путем сравнения площади хроматографическо-
го пика соответствущего dG-BPDE с пиком внутреннего стандарта. С помощью этого
метода группе Беланда удалось обнаружить dG-BPDE-аддукты на уровне 7 dG-BPDE
аддуктов на 108 нуклеотидов у мышей, обрабатанных 0,5 мг [7,8-3Н] БП.
Эта же группа проанализировала ДНК из тканей курильщиков, бывших курильщи-
ков и некурящих. БП-аддукты были обнаружены лишь в одном образце, где они были
представлены на уровне 1,9 dG-BPDE аддуктов на 108 нуклеотидов. Таким образом,
уровень БП-аддуктов в печени человека был ниже предела обнаружения, доступного
для ВЭЖХ/МС/МС-метода (Beland et al., 2005). Хотя значительно большее количество
аддуктов (23,7 аддуктов на 108 нуклеотидов) было обнаружено в том же образце с помо-
щью метки 32Р, авторы предположили, что dG-BPDE-аддукты могут представлять собой
лишь небольшой процент от общего количества аддуктов, образованных при воздей-
ствии ПАУ, причем только часть из них детектируется другими методами.
Чен и его коллеги также изучали количественный состав БП-аддуктов при помощи
изотопного разбавления и ВЭЖХ/МС/МС (Chen et aL, 2005). Основное внимание было
уделено \7-(бенз[а]пирен-6-ил)-гуанину (BP-6-N7Gua), который образуется при одно-
электронном окислении БП и последующей реакции с гуанином. Эти аддукты могут
быть депуринированы и затем выведены с мочой. Поэтому группа Чена представила
количественный анализ этих аддуктов в моче человека в качестве метода для опреде-
ления экспозиции к БП и для оценки риска заболевания раком. Изотопномеченое со-
единение (15N5)BP-6-N7Gua было использовано в качестве внутреннего стандарта для
количественного определения аддуктов. Около 1,5 пикомолей внутреннего стандарта
было добавлено в образцы мочи (обьемом 15 мл), затем вещества были экстрагированы
ацетонитрилом, раствор очищен при помощи двухстадийной твердофазной экстракции
и проанализирован с помощью ВЭЖХ/МС/МС. Анализ осуществлялся в тройном ква-
друполе в режиме мониторинга заданных реакций, при этом были выбраны следующие
переходы: для аналита (m/z 402 —> 252), для внутреннего стандарта (m/z^Cl 252). Под-
тверждение результатов, полученных с помощью этого метода, проводилось с исполь-
зованием чистых проб мочи с добавлением 0,2; 1 и 2 пикомолей BP-6-N7Gua и 1,5 пи-
комолей внутреннего стандарта. Точность определения составила 94—102% с 2—4%
погрешностью. После проверки данный метод был использован для анализа мочи
работника коксового производства. В его моче было обнаружено 6 фемтомолей ВР-6-
N7Gua на один мг эквивалента креатинина, т. е. этот метод позволяет делать анализ ВР-
гуанин-аддуктов в моче у работающих с БП.
1-метилпирен (1-МР) является алкилированным ПАУ, который требует мета-
болического превращения в организме, для того чтобы стать химически активным
в форме 1-сульфоксиметилпирена (1-SMP). Для получения количественной оценки
17.5. Примеры использования ВЭЖХ/МС/МСдля анализа ДНК-аддуктов 497
1-МР-аддуктов аденина и гуанина у крыс, получавших 1-SMP, Мониен и сотрудники
сравнили метод масс-спектрометрии с использованием изотопного разбавления и метод
оснований на использовании радиоактивного изотопа фосфора 32Р (Monien et al., 2008).
Использование ультраэффективной наножидкостной хроматографии и мониторинга
заданных реакций в тройном квадруполе позволило достичь пределов обнаружения,
равных 3 молекулам MP-dGuo и 0,6 молекулам MP-dAdo на каждые 108 нуклеозидов.
Интересно, что в 3—4 раза большее количество аддуктов наблюдалось при использова-
нии МС с изотопным разбавлением, чем с радиоактивным 32Р (Monien etal. 2008). Кроме
того, пределы обнаружения метода с радиоактивным 32Р были в 2—9 раз выше.
17.5.6. ДНК-аддукты с эпоксидами
Соединение 1,3-бутадиен (БД) используется в промышленности и является распростра-
ненным токсичным соединением, присутствующим в городском воздухе и в сигаретном
дыме. В организме БД метаболизируется до соединений, которые могут реагировать
с нуклеофильными азотистыми основаниями или белками, например эпоксибутен
(ЕВ), диэпоксибутан (DEB) и эпоксибутандиол (EBD). Основным аддуктом in vivo явля-
ется \7-тригидроксибутилгуанин (THBG). ДНК-аддукты, возникающие при взаимо-
действии с эпоксидными соединениями, способны к спонтанной депуринизации с об-
разованием апуринизированных участков. Для определения THBG-аддуктов в ДНК
из печени и легких грызунов, которые подверглись воздействию БД, были разработаны
количественние МС-методы с использованием изотопного разбавления (Powley et aL,
2005). Образцы ДНК были проанализированы при помощи МС/МС в режиме монито-
ринга заданных реакций по переходам m/z 256 —> 152 для THBG и m/z 260 —> 156 для
13C4-THBG (внутренний стандарт). У мышей наблюдались чуть более высокие уровни
аддуктов по сравнению с крысами при том же уровне воздействия БД (Кос et al., 1999;
Tretyakova etal., 1998). Межвидовые различия в уровнях аддуктов могут быть обусловле-
ны увеличением метаболической активации БД у мышей или более низкой скоростью
детоксикации БД у мышей по сравнению с крысами.
Существует доказательство того, что THBG в основном образуется в реакции EBD,
но не DEB с ДНК. Тем не менее DEB является наиболее мутагенным метаболитом и счи-
тается главным канцерогенным производным БД. Таким образом, само по себе THBG
не может быть оптимальным биомаркером метаболического превращения БД в химиче-
ски активные эпоксидные соединения. Реакции DEB с ДНК вызывают сшивки ДНК-
ДНК, приводят к появлению ряда соединений: 1,4-бис-(гуан-7-ил)-2,3-бутандиола
(6nc-N7G-BD), 1-(гуан-7-ил)-4-(аден-1-ил)-2,3-бутандиола (N7G-N1A-BD), 1-(гуан-
7-ил)-4-(аден-3-ил)-2,3-бутандиола (N7G-N3A-BD), 1-(гуан-7-ил)-4-(аден-7-ил)-2,3-
бутандиола (N7GN7A-BD) и 1-(гуан-7-ил)-4-(аден-6-ил)-2,3-бутандиола (N7G-N6A-BD).
ДНК-Д НК-сшивки были охарактеризованы в лаборатории авторов настоящего обзора in
vitro (Park et al., 2004; 2005). Количественные методы, где использовались капиллярная
ВЭЖХ, электроспрей и тандемная масс-спектрометрия, были разработаны для анализа
ДНК-аддуктов in vivo (Goggin et al., 2007; 2008). Bnc-N7G-BD и N7G-N1A-BD были обна-
ружены при анализе ДНК из печени мышей при концентрациях БД до 625 частей на мил-
лион. Уровень 6nc-N7G-BD при этом был в 10 раз выше, чем N7G-N1A-BD (Goggin et al.,
2007; 2008). Дальнейшая оптимизация метода — в том числе использование автономной
(off-line) очистки образцов при помощи ВЭЖХ — позволила получить кривые доза—реак-
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
ция для вышеупомянутых ДНК-аддуктов у мышей/крыс, подвергавшихся воздействию
БД в концентрации 6,25—625 частей на млн в течение двух недель (Goggin etal., 2009).
Методы, где использовалась наножидкостная хроматография и наноэлектроспрей,
были необходимы для обнаружения N7G-N1A-BD в ДНК из печени крыс и мышей,
которые подвергались воздействию БД в концентрации ниже 625 частей на миллион
(Goggin et al., 2009). В лаборатории авторов также были недавно определены два экзоци-
клических DEB-DA-аддукта: 1,\6-(1-гидроксиметил-2-гидроксипропан-1, 3-диил)-2-
дезоксиаденозин (1, \6-альфаМНР-ЭА) и 1, \6-(2-гидрокси-3-гидроксиметилпропан-1,
3-диил)-2-дезоксиаденозин (1, N6-raMMaHMHP-DA). Для количественной оценки этих
аддуктов в ДНК мышей, подвергнутых воздействию БД, была разработана многомер-
ная хроматография. Предел обнаружения данного метода составил 1,1 l,N6-HMHP-dA
на 109 нуклеотидов, что достаточно для того, чтобы получить кривые доза—реакция для
мышей, подвергшихся воздействию БД в концентрации от 62,5 до 625 частей на млн.
Акриламид (АА) имеет множество применений: он сам используется для гель-
электрофореза, а его полимерная форма — для обработки воды и производства бетона.
Акриламид также является компонентом сигаретного дыма и считается канцерогеном
на основе результатов, полученных в исследованиях наживотных. Химически активным
метаболитом акриламида является эпоксидное производное гликозидамида (GA). Гам-
боа да Коста и его коллеги использовали однократную инъекцию АА или GA (20 мг/кг)
для взрослых и трехдневных мышей (Gamboa da Costa etal., 2003a). Через шесть часов по-
сле инъекции животных умерщвляли и экстрагировали ДНК из печени, легких и почек.
В полученный раствор ДН К добавлялся внутренний стандарт изотопномеченых соеди-
нений (l5N_), после чего ДНК подвергалась термическому гидролизу в нейтральной сре-
де, чтобы освободить N7-Gua, N7-Ade и N3-Ade аддукты ДНК. Эти вещества эксклю-
зионно фильтровались по размеру и анализировались ВЭЖХ/ЭРИ/МС/МС на тройном
квадруполе Quattro Ultima, который работал в режиме мониторинга заданных реакций.
Калибровочные кривые, полученные для изотопномеченых внутренних стандартов,
были использованы для точного количественного определения аддуктов. Во всех случа-
ях авторы наблюдали почти 100-кратное увеличение концентрации Ш-(2-карбамоил-2-
гидроксиэтил) гуанина (N7-GA-rya) по сравнению с №-(2-карбамоил-2-гидроксиэтил)
аденином (N3-GA-Ade). Организм взрослых мышей содержал в 1,2—1,5 раза больше GA,
чем АА аддуктов. В трехдневных мышах, интоксицированных GA, было обнаружено
в 5—7 раз больше аддуктов, поскольку в организме этих мышей отсутствовал в активной
форме фермент Р450 способный метаболизировать АА в GA.
17.6. Новые подходы к повышению
чувствительности аналитических методов
при анализе ДНК-аддуктов
Для количественного анализа ДНК-аддуктов in vivo в последнее десятилетие были раз-
работаны несколько новых подходов с улучшенной эффективностью ионизации. В этом
разделе мы рассмотрим три современных варианта повышения чувствительности ана-
литических методов: наноэлектроспрей, многомерная хроматография и хроматогра-
фическое разделение/ионизация в микрочипах. Использование наноэлектроспрея
49L)
17.6. Новые подходы к повышению чувствительности аналитических методов
при анализе ДНК-аддуктов
повышает чувствительность метода благодаря низким скоростям потоков, что связано
с пониженным химическим шумом, большей эффективностью ионизации и переноса
образца из ВЭЖХ-колонок в ионный источник.
Метод многомерной хроматографии соединяет непосредственную очистку образ-
ца с помощью ВЭЖХ-разделения и МС-анализ в режиме онлайн. Этот метод может по-
тенциально устранить этап автономной (офлайн) очистки образца, что экономит время
анализа и предотвращает потери образца. Наконец, последняя обсуждаемая методика
улучшения чувствительности включает в себя использование чипов. Чип представля-
ет собой кремниевую подложку, в которой проделан микроканал, заканчивающийся
наноэмиттером. Во многих случаях этот канал представляет себой короткую хромато-
графическую колонку, которая также заканчивается наноэмиттером. Использование
чипов позволяет увеличивать скорость анализа проб, так как чип устраняет хромато-
графический этап в ВЭЖХ-МС-анализе, а образцы могут поступать в масс-спектрометр
непосредственно через наноэлектроспрей (Wickremsinhe et al., 2005).
17.6.1. Капиллярная ВЭЖХ/наноэлектроспрейная МС
Метод капиллярной ВЭЖХ — наноэлектроспрей ной МС (capLC/nanoESI) связан с ис-
пользованием капиллярных колонок с внуренним диаметром 0,3—0,8 мм, которые рабо-
тают при скоростях потока 5—10 мкл/мин. Перед поступлением в источник ионизации,
которым в даном случае является наноэлектроспрей, поток растворителя из колонки
уменьшается при помощи разделителя потока. Капиллярная ЖХ/нано-ЭРИ/МС/МС-
метод с изотопным разбавлением был разработан для количественного определения
эстроген-модифицированных аддуктов аденина в тканях молочной железы человека
(Zhang et al., 2008). Ферментативное превращение эстрогена цитохромом Р450 приво-
дит к образованию 3,4-катехин-эстрогенхинонов, которые затем могут непосредственно
реагировать с ДНК с образованием аддуктов 4-гидроксиэстроген-2-\7-гуанина (4-ОН-
E2-N7Gua) и 4-гидроксиэстроген-1-№-аденина ^-OH-E^WAde). Авторы (Zhang et al.,
2008) использовали масс-спектрометр LCQ Deca фирмы ThermoFinnigan, в который
образец поступал из капиллярной ВЭЖХ-колонки (Waters). Для разделения использо-
валась специально упакованная колонка (тип хроматогафической фазы — Luna, размер
частиц 3 мкм, длина колонки — 120 мм, внутренний диаметр колонки — 75 мкм), которая
заканчивалась наконечником типа PicoFrit фирмы New Objective с внутренним диаме-
тром 15 мкм. Перед наноколонкой первоначальный поток (скорость потока 8 мкл/мин.)
был разделен на две части при помощи двух капилляров с разным внутренним диаме-
тром и длиной для достижения скорости потока 270 нл/мин на конце капилляра с вы-
соким гидродинамическим сопротивлением. Предел обнаружения составил 5 фмоль
4-OH-E)-N3-Ade на один грамм ткани. ДНК была извлечена из ткани, которая подвер-
глась ферментативной обработке. Полученную смесь обогащали методом твердофазной
экстракции и предварительно очищали на ВЭЖХ-колонке. После этого смесь анали-
зировалась методом капиллярной ЖХ/нано-ЭРИ/МС/МС. Были проанализированы
ткани молочной железы шести пациентов (три контроля и три больных раком молочной
железы). Во всех шести случаях был обнаружен 4-OH-E1-N3-Ade, однако широта иссле-
дования не была достаточной, чтобы определить повышения уровня 4-OH-E1-N3-Ade
у онкологических больных по сравнению с контрольной группой. Лью с соавт. (2005,
2006) разработали кап.ЖХ/нано-ЭРИ-метод для количественного определения двух
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
различных аддуктов ДНК. Первый метод был разработан для мониторинга акролеин-
dG (Acro-dG) аддуктов в ДНК тканей мозга человека. Acro-dG образуется в результате
реакции с гидроксил-2-ноненалем, который в свою очередь образуется при перекисном
окислении липидов (Liu et aL, 2005). Повышенное содержание продуктов перекисного
окисления липидов было обнаружено в тканях пациентов с болезнью Альцгеймера (AD)
(Liu etal., 2005).
Уровни Acro-dG сравнивались в образцах ДНК тканей головного мозга пациентов
с болезнью Альцгеймера и контрольной группы. ВЭЖХ-система UltiMate использова-
лась с ионной ловушкой фирмы Bruker Daltonics с микроэлектроспрейным эмиттером,
сделанным из заостренного капилляра из нержавеющей стали с внутренним диаме-
тром 50 микрон. При использовании этих методов предел обнаружения для Acro-DG
составил 31 аттомоль, что позволило исследователям наблюдать разницу в уровнях
аддукта в мозге пациентов с болезнью Альцгеймера по сравенению с соответствую-
щим контролем (5150±640, по сравнению с 2800±460 Acr-dG на 109 нуклеотидов) (Liu
et al., 2005). Другая методика была использована для измерения циклических 1,N2-
пропанодезоксигуанозиновых аддуктов (HNE-dG), образующихся в реакции с продук-
том перекисного окисления липидов транс-4-гидрокси-2-ноненаля (HNE). Была ис-
пользована описанная выше комбинация кап.ЖХ/нано-ЭРИ. Предел обнаружения для
чистого стандарта HNE-dG составил 24 аттомоль или 40 аддуктов на 109 нуклеотидов
в биологической матрице (Liu et aL, 2006). Ранее было показано, что уровни HNE были
повышены в тканях мозга пациентов с болезнью Альцгеймера. Поэтому авторы срав-
нивали уровень HNE-dG в контроле и в образцах, взятых из мозга пациетов с болезнью
Альцгеймера. Однако никаких статистически значимых различий зарегистрировать
не удалось. Таким образом, уровень HNE-dG может и не быть биомаркером оксидатив-
ного стресса при болезни Альцгеймера.
17.6.2. МС-анализ с использованием ВЭЖХ-наноколонок
и ионизацией наноэлектроспреем
Когда очистка и хроматографическое разделение элементов ДНК и ДНК-аддуктов
происходит в наноколонках, а ионизация — в наноэлектроспрее, используются хро-
матографические колонки, сделанные из полого кварцевого волокна (0,025—0,1 мм
внутренний диаметр), а поток растворителя проходит колонку со скоростью 100—
500 нанолитров/мин. В настоящее время этот метод разделения и ионизации ис-
пользуется в основном в протеомике (Lee et aL, 2009). Тем не менее Эмбрехтс и др.
использовали нано-ЖХ/нано-ЭРИ/МС/МС для анализа эстроген-ДНК-аддуктов:
17-ос-этинилэстрад иол-2'-дезоксиаденозина, эстрон-2'-дезокси гуанозина, эквиленин-
2'-дезоксигуанозина и эстрадиол-2'-дезокси гуанозина из тканей опухоли молочной
железы человека. Предел обнаружения составлял 200 фемтограммов, при этом, однако,
для количественного определения этих веществ авторы не использовали изотопномече-
ные внутренние стандарты (Embrechts et aL, 2003).
В лаборатории авторов данного обзора недавно был разработан метод количествен-
ного анализа продуктов химической связи 1-(гуан-7-ил)-4-(аден-1-ил)-2,3-бутандиола
(N7G-N1A-BD) с ДНК при воздействии БД на печень мышей (рис. 17.5). Хроматографи-
чекое разделение осуществлялась в ВЭЖХ системе nanoAquity фирмы Waters, при этом
использовалась колонка-ловушка Symmetry С18 (0,18 мм внутренний диаметр, 20 мм
17.6. Новые подходы к повышению чувствительности аналитических методов
при анализе ДНК-аддуктов
О 5 10 15 20
Рис. 17.5. Нано-ЖХ/нано-ЭРИ/МС/МС-анализ ДНК-аддукта N7G-N6A-BD из пече-
ни мышей, подвергнутых воздействию БД в концентрации 625 ppm в течение
2 недель. (Печатается с разрешения Goggin М., Seneviratne U., Swenberg J.А.,
Walker V. Е. and Tretyakova N. «Методы масс анализа с применением многомерной
жидкостной хроматографии для количественного анализа экзоциклических dA
ДНК-аддуктов в лабораторных животных, подвергнутых действию 1,3-бутади-
ена». Химические исследования в токсикологии. 23, 808—812. Copyright (2010) Аме-
риканское химическое общество)
(+4) N7G-N6A-BD
Внутренний стандарт
25 30 35 40 45
Время, мин.
длина колонки) и аналитическая колонка Atlantis dC18 (75 х Ю0 мкм). Анализ прово-
дился на масс-спектрометре «Ультра» TSQ фирмы ThermoFinnigan. При использовании
наноколонок наблюдалось почти 10-кратное увеличение чувствительности (а именно
3 аддукта на 109 нуклеотидов против 30 аддуктов на 109 нуклеотидов) по сравнению
с ЭРИ/МС/МС с обычными капиллярными колонками (колонки с внутренним диаме-
тром 0,3—0,8 мм). С помощью этой методики можно было обнаружить N7G-N1A-BD
в ДНК мышей, подверженных воздействию 62,5 ppm BD (Goggin et al., 2009).
17.6.3. Очистка образцов в режиме онлайн
Как уже отмечалось выше, количественный МС-анализ ДНК-аддуктов в биологиче-
ских образцах может быть затруднен из-за подавления сигнала образца ионами матрицы
(различные соли, белки, немодифицированные нуклеозиды), т.е. необходима глубокая
очистка образца до его введения в аналитическую ЖХ/МС-колонку. Эта очистка зани-
мает много времени и приводит к потере анализируемого вещества.
Использование многомерной хроматографии помогает уменьшить потери образца
и увеличить пропускную способность МС-анализа. Метод многомерной хроматогра-
фии предполагает загрузку образца в колонку-ловушку и последующий смыв солей
и загрязняющих веществ. После переключения вентиля образец смывается в анали-
тическую колонку для последующего хроматографического разделения и МС-анализа
(рис. 17.6). Многомерная хроматография успешно использовалась для количественного
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
Позиция А переключающего вентиля
Поступает из инжектора
с помощью насоса,
работающего в изокра-
в градиентном режиме
Позиция В переключающего вентиля
Поступает из инжектора
с помощью насоса,
работающего в изокра-
в градиентном режиме
Рис 17.6. Позиции переключающего вентиля в схеме многомерной жидкостной хрома-
тографии. В положении А образец загружается на колонку-ловушку (первое
хроматографическое измерение), затем колонка промывается. В положении В
второй насос смывает образец с колонки-ловушки на аналитическую колонку
(второе хроматографическое измерение), после разделения в аналитической ко-
лонке смесь поступает в источник ионов масс-спектрометра
17.6. Новые подходы к повышению чувствительности аналитических методов
при анализе ДНК-аддуктов
определения ДНК-аддуктов в биологических образцах несколькими группами (Beland
et al., 2004; Brink et al., 2006; Chao et al., 2007; Chao et al., 2008; Doerge et al., 2000; Hillestrom
et al., 2004; Hillestrom et al., 2006; Singh et al., 2009). Несколько примеров использования
этого метода будут рассмотрены ниже. Дорге с соавторами (Doerge et al., 2000) разрабо-
тали метод многомерного хроматографического анализа для определения содержания
ДНК-аддуктов, таких как e-dA и е-дезоксицитозин (e-dC) в печени и легких мышей,
подвергнутых воздействию уретана. В этом методе образец сначала вводился в колонку-
ловушку с обращенной фазой (тип фазы — Luna Cl8, 30 мм — длина колонки, 2 мм — ее
диаметр, 3 мкм — размер частиц), затем образец смывался по фракциям в аналитиче-
скую колонку (Luna С18, 150 мм — длина, 2 мм — диаметр, 3 мкм — размер частиц)
и анализировался в тандемном масс-спектрометре Quattro LC/QQQ, который работал
в режиме ЭРИ с регистрацией положительных ионов. Осуществлялся мониторинг за-
данных реакций потери дезоксирибозы протонированными молекулами нуклеозидов
e-dA и e-dC. Пределы обнаружения составляли 0,3—0,9 e-dA аддуктов на каждые 108 ну-
клеотидов и 0,7—1,8 e-dC аддуктов на каждые 108 нуклеотидов. ДНК из печени кон-
трольной группы мышей и мышей, которые подвергались воздействию уретана, содер-
жали соответственно 1,0 и 2,2 e-dA аддуктов на каждые 108 нуклеотидов и 1,0 и 2,7 e-dC
аддуктов на каждые 108 нуклеотидов. Чувствительность метода для e-dC была меньше,
чем для e-dA. В будущем авторы предполагают использовать иммуноаффинную он-
лайн-очистку для дополнительного повышения чувствительности метода.
Другой многомерный хроматографический метод был разработан Полсеном с кол-
легами для анализа e-dA-аддуктов в моче (Hillestrom et al., 2004). Двумерное разделение
осуществлялось на ловушке Luna (75 х 4,6 мм, 5 мкм) и аналитической колонке Synergi
Polar-RP (150x4,6 мм, 4 мкм). Анализ проводился на приборе API3000 QqQ с источ-
ником химической ионизации при атмосферном давлении. Пробы мочи (3 мл) пред-
варительно очищались твердофазной экстракцией (Oasis HLB). Предел определения
для этено-dA составил 600 аттомоль в уколе, или 0,7 пикомоль/л мочи. Однако авторы
не наблюдали увеличения уровня этено-dA в моче курильщиков по сравнению с мочой
некурящих (Hillestrom et al., 2004). Эта же группа модифицировала данный метод для из-
мерения аддуктов этено-dA в моче человека с пределом количественного определения
100 пМ e-dC (Hillestrom etal., 2006).
Бринк и соавт. (Brink etal., 2006) разработали методику с использованием многомер-
ной хроматографии для одновременного определения О6-метил-2'-дезоксигуанозина
(O6-Me-DG), 8-оксо-7,8-дигидро-2'-дезоксигуанозина (8-oxo-dG) и 1,М6-этено-2-
дезоксиаденозина в ДНК из печени крысы. Эти три аддукта могут образовываться
как эндогенно, так и при воздействии экзогенных химических веществ. Пределы ко-
личественного определения этих аддуктов колебались между 24 и 48 фемтомолями.
При этом для калибровки всех трех анализируемых веществ применялся только один
внутренний стандарт — меченный изотопами O6-Me-dG. Все три аддукта наблюдались
у крыс, которым вводился и диметилнитрозамин. Основное преимущество этого мето-
да состояло в том, что уровень наблюдаемого сигнала 8-oxo-dG (который являлся ар-
тефактом в данном исследовании) был намного ниже по сравнению с его уровнем при
анализе другим ЖХ/МС/МС-методом, в котором использовалась автономная (off-line)
очистка образца (Brink et al., 2006).
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
Об анализе 8-oxo-dG методом многомерной хроматографии сообщалось в работах
двух других лабораторий, где исследовалась ДНК печени крыс и мышей. В работе Чао
и др. (Chao et aL, 2008) продемонстрирована возможность удаления более 99 % dG из об-
разцов, если использовать в качестве ловушки колонку ODS-3 фирмы Intersil, а в каче-
стве аналитической колонки — колонку с покрытием из полиамина-П (длина колон-
ки 150 мм, диаметр 4,6 мм, фирма YMC America, Inc). Предел обнаружения составлял
0,13 аддукта на 106 dG в условиях, когда в колонку было введено 1,8 фмоля вещества, при
этом для анализа ДНК использовалось только 20 микрограмм печени мышей. Данный
предел обнаружения был ниже, чем фоновый уровень 8-oxo-dG в лимфоцитах человека
(0,3 аддукта на 106 dG). Низкий порог обнаружения в сочетании с небольшим временем
анализа делает этот метод идеальным для масс-спектрометрического анализа ДНК че-
ловека с высокой пропускной способностью (Chao et aL, 2008).
В недавнем исследовании, где использовался метод многомерной хроматографии,
было одновременно проанализировано содержание 8-oxo-dG и 8-оксо-7,8-дигидро-2-
дезоксиаденозина (8-oxo-dA) в ДНК из печени крыс (Singh etal., 2009). Предел обнаруже-
ния ДНК-аддуктов с помощью этого метода составил 5 фмоль. В то время как 8-oxo-dA
было обнаружено примерно в 30 раз меньше, чем 8-oxo-dG, артефактных 8-oxo-dA было
также меньше. Авторы предположили, что комбинация из 8-oxo-dG и 8-oxo-dA будет
давать более точную количественную оценку окисления ДНК (Singh etal., 2009).
Метод многомерной хроматографии был также использован для количественного
определения алкилгуаниновых аддуктов ДНК (Chao et aL, 2007). Авторы разработали
метод переключения колонок (многомерную хроматографию) для одновременного ана-
лиза Ю-метилгуанина (N7MeG) и \7-этилгуанина (N7EtG). Метод реализован с ко-
лонкой Nucleosil NH2 (35 мм — длина колонки, 4,6 мм — ее диаметр, 10 мкм — диаметр
частиц хроматографической фазы) в качестве ловушки, а в качестве аналитической ко-
лонки — ВЭЖХ-колонкой, в которой адсорбирующая поверхность частиц была моди-
фицирована полиамином-П (150 мм — длина колонки, 4,6 мм — ее диаметер, 5 мкм —
диаметр частиц, YMC America, Inc). Анализ при помощи этого метода занимал только
15 минут, а предел обнаружения составлял 0,42 фмоль в уколе для N7MeG и 0,17 фмоль
для N7EtG. Метод многомерной хроматографии был использован для построения кри-
вых доза—реакция для ДНК из печени гамбузии, которая подверглась воздействию
N-нитрозодиэтиламина и N-нитрозодиметиламина (Chao et aL, 2007). Авторы данного
исследования сообщают, что метод многомерной хроматографии показал более высо-
кую чувствительность, чем ранее разработанные ЖХ/МС/МС-методы. В лаборатории,
в которой работают авторы данного обзора, разработали метод многомерной хромато-
графии для обнаружения незначительных количеств экзоциклических ДНК-аддуктов
1,3-бутадиена: 1,М6-осНМНР-бАи l,N6-yHMHP-dA (GogginetaL, 2010\ рис. 17.7). При этом
был достигнут предел обнаружения 1,1 аддуктов l,N6-HMHP-dA на 109 нуклеотидов,
что позволило получить кривые доза—реакция для мышей, подвергнутых воздействию
биологических доз от 62,5 до 625 ррт.
17.6.4. ЖХ/МС на микрочипах
Масс-спектрометрия на чипах подразумевает использование кремниевого чипа с набо-
ром электрораспылительных форсунок, которые позволяют вести прямой ввод веще-
ства в прибор, т.е. без ВЭЖХ-колонки (Singh and Farmer, 2006). Преимуществом этого
17.6. Новые подходы к повышению чувствительности аналитических методов
при анализе ДНК-аддуктов
Рис 17.7. Многомерный хроматографический анализ (калил.ЖХ/ЭРИ/МС/МС с реги-
страцией положительных ионов) ДНК-аддукта l,N6-HMHP-dA из печени мы-
шей, подвергнутых действию БД в концентрации 200 ppm течение 2 недель
подхода является незначительный расход пробы, что снижает количество биологиче-
ской матрицы, оказывающейся в масс-спектрометре. Новая ЭРИ-игла используется для
каждого нового образца, что устраняет проблемы с переносом аналита. Недостатком яв-
ляется отсутствие хроматографического разделения. В результате аналит оказывается
в масс-спектрометре одновременно с веществами матрицы, т.е. возможны матричные
эффекты (подавление сигнала и многочисленные интерференции). В некоторых слу-
чаях простая жидкость-жидкостная экстракция образцов перед МС/МС на чипе может
существенно сократить эти нежелательные явления (Wickremsinhe et al., 2005).
В настоящее время МС на чипах используется в основном для анализа пептидов,
лекарственных препаратов или их метаболитов (Liu et al., 2009; Wickremsinhe et al., 2005).
По сведениям авторов обзора, только группа Вороса использовала МС-анализ на чи-
пах для количественного определения ДНК-аддуктов. Эта группа впервые сообщила
о нано-ЖХ/МС-методе на чипах для количественного определения ДНК-аддуктов,
образующихся при воздействии 2-амино-1-метил-6-фенилимидазо[4,5-Ь]пиридина
(Glick et al., 2007). В той же самой лаборатории недавно использован небольшой чип
для количественного определения 4-АВРсЮ-аддуктов в ДНК из печени крыс, которые
были подвергнуты воздействию 4-АВР (Randal et al., 2009). Авторы использовали ион-
ную ловушку Agilent LC/MSD ХСТ Ultra с чипом Zorbax 80SB-C18 с размером частиц
5 мкм. Этот чип состоял из колонки для обогащения (40 нл) и аналитической колонки
(75 мкм — внутренний диаметр, 43 мкм — длина). Два иона изолировались с последу-
ющим инициированием их фрагментации и регистрацией переходов, характерных для
dG-ABP и dG-ABP-d9 (m/z 435,4 —> 319 и m/z 444 —> 328 соответственно). Предел обнару-
жения метода — 2 dG-ABP на 108 нуклеотидов, причем требуется всего 1,25 мкг ДНК.
Метод применен для количественного определения dG-ABP в ДНК печени крыс, под-
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
вергнутых воздействию АВР интраперитональными инъекциями. dG-ABP был обнару-
жен на уровнях от 80 аддуктов на 108 нуклеотидов (Randall et aL, 2009).
17.7. Выводы
ВЭЖХ/ЭРИ/МС/МС с изотопным разбавлением является чувствительным и высоко-
селективным методом количественного определения ДНК-аддуктов в биологических
образцах. Эта техника превосходит другие методы, основанные на 32Р-метке или на флу-
оресценции, поскольку обеспечивает структурную информацию об анализируемом
веществе. Кроме того, использование внутренних стандартов с изотопным замещени-
ем делает метод высокоспецифичным, точным и воспроизводимым. Недавние усовер-
шенствования электроспрейных источников ионизации (использование наноколонок,
а также микрожидкостных чипов с наноэмиттерами для ЖХ-анализа) увеличили чув-
ствительность ЭРИ-МС-метода до уровня, близкого к чувствительности, которую демон-
стрируют методы, основанные на 32Р-метке. Использование многомерной хроматографии
привело к более качественной очистке образцов, в результате чего при уменьшении по-
давления ионного сигнала предел обнаружения стал ниже, а пропускная способность
выше. Повышенная чувствительность наноспрейных методов ионизации делает весьма
привлекательным их использование для количественного определения ДНК-аддуктов
с низким содержанием и прямогоанализаДНК-аддуктов вчеловеческихобразцах. Кроме
того, использование ЖХ-чипов, которые содержат несколько колонок, коммутирующее
устройство для переключения колонок и наноспрейный эмиттер, сокращает время ана-
лиза, затрачиваемое на образец, и уменьшает потери образца, т.е. увеличивает чувстви-
тельность метода. Хотя многие из методик, представленные в данном обзоре описывают
измерения ДНК-аддуктов у лабораторных животных, дальнейшие достижения в области
улучшения измерительных приборов (такие как наноэмиттеры, ЖХ-чипы и орбитрэп),
без сомнения, приведут к повышению чувствительности масс-спектрометрического ме-
тода количественного определения ДНК-аддуктов и более широкому применению этого
метода для оценки влияния различных экзогенных факторов на здоровье человека.
Благодарность
Авторы благодарят Боба Карлсона (Центр изучения рака, университет штата Минне-
сота) за помощь в подготовке рисунков для данной рукописи. Работа выполнена при
поддержке грантов от Национального института рака (С.А. 100670), Национального
института гигиены окружающей среды (ES 12689), Института воздействия на здоровье
(соглашение 05-12) и Американского химического совета (OLF-163.0).
Литература
[1] . Arlt V. М., Stiborova М., Henderson C.J., Thiemann М., Frei Е., Aimova D., Singh R.,
Gamboa da C. G., Schmitz O. J., Farmer P. B., WolfC. R., and Phillips D. H. (2008). «Meta-
17.7. Выводы
bolic activation of benzo[a]pyrene in vitro by hepatic cytochrome P450 contrasts with detoxi-
fication in vivo', experiments with hepatic cytochrome P450 reductase null mice». Carcino-
genesis. 29, 3: 656—665.
[2] . Baskerville-Abraham I. M., Boysen G., Troutman J. M., Mutlu E., Collins L., Dekrafft К. E.,
Lin W., King C., Chaney S.G., and Swenberg J. A. (2009). «Development of an ultraperfor-
mance liquid chromatography/mass spectrometry method to quantify cisplatin 1,2 intra-
strand guanine-guanine adducts». Chemical Research in Toxicology. 22, 5: 905—912.
[3] . Beland F. A., Churchwell M. I., Hewer A., Phillips D. H., da Costa G.G., and Marques M. M.
(2004). «Analysis of tamoxifen-DNA adducts in endometrial explants by MS and 32P-postla-
beling». Biochemical and Biophysical Research Communications. 320, 2: 297—302.
[4] . Beland E A., Churchwell M. I., Von Tungeln L. S., Chen S., Fu P. P., Culp S. J., Schoket B.,
Gyorffy E., Minarovits J., Poirier M.C., Bowman E.D., Weston A., and Doerge D. R.
(2005). «High-performance liquid chromatography electrospray ionization tandem mass
spectrometry for the detection and quantitation of benzo[a]pyrene-DNA adducts». Chemical
Research in Toxicology. 18,8: 1306—1315.
[5] . Boyd R. K., Basic C., and Bethem R. A. (2008). Tools of the trade V. Mass analyzers for quan-
titation: Separation of ions by m/z values. John Wiley & Sons, West Sussex, England, pp
266-301.
[6] . Bransfield L.A., Rennie A., Visvanathan K., Odwin S.A., KenslerT.W., Yager J. D., Fri-
esen M. D., and Groopman J. D. (2008). «Formation of two novel estrogen guanine adducts
and H PLC/MS detection of 4-hydroxyestradiol-N7-guanine in human urine». Chemical Re-
search in Toxicology. 21, 8: 1622—1630.
[7] . Brink A., Lutz U., Volkel W., and Lutz W. K. (2006). «Simultaneous determination of 06-
methyl-2'-deoxyguanosine, 8-oxo-7,8-dihydro-2'-deoxyguanosine, and l,7V6-etheno-2'-
deoxyadenosine in DNA using on-line sample preparation by HPLC column switching
coupled to ESI-MS/MS». Journal of Chromatography, B: Analytical Technologies in the Bio-
medical and Life Sciences. 830, 2: 255—261.
[8] . Buser H.R., Rappe C., and Bergqvist P.A. (1985). «Analysis of polychlorinated dibenzo-
furans, dioxins and related compounds in environmental samples». Environmental Health
Perspectives. 60, 293—302.
[9] . Chao M. R., Wang C. J., Chang L. W., and Hu C. W. (2006). «Quantitative determination of
urinary N7-ethylguanine in smokers and non-smokers using an isotope dilution liquid chro-
matography/tandem mass spectrometry with on-line analyte enrichment». Carcinogenesis.
27, 1: 146-151.
[ 10]. Chao M. R., Wang C. J., Yen С. C., Yang H. H., Lu Y. C., Chang L. W., and Hu C. W. (2007).
«Simultaneous determination of N7-alkylguanines in DNA by isotope-dilution LC-tan-
dem MS coupled with automated solid-phase extraction and its application to a small fish
model». Biochemical Journal. 402, 3: 483—490.
[11] . Chao M.R., Yen C.C., and Hu C.W. (2008). «Prevention of artifactual oxidation in de-
termination of cellular 8-oxo-7,8-dihydro-2'-deoxyguanosine by isotope-dilution LC-
MS/MS with automated solid-phase extraction». Free Radical Biology and Medicine. 44, 3:
464-473.
[12] . Chen H.J., and Chang С. M. (2004). «Quantification of urinary excretion of l,7V6-ethe-
noadenine, a potential biomarker of lipid peroxidation, in humans by stable isotope dilu-
tion liquid chromatography-electrospray ionization-tandem mass spectrometry: compari-
508 Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
son with gas chromatography-mass spectrometry». Chemical Research in Toxicology. 17, 7:
963-971.
[13] . Chen H.J., and Chiu W. L. (2005). «Association between cigarette smoking and urinary
excretion of LA^-ethenoguanine measured by isotope dilution liquid chromatography-
electrospray ionization/tandem mass spectrometry». Chemical Research in Toxicology. 18,
10: 1593-1599.
[14] . Chen L., Wang M., Villalta P.W., and Hecht S.S. (2007a). «Liquid chromatography-elec-
trospray ionization tandem mass spectrometry analysis of 7-ethylguanine in human liver
DNA». Chemical Research in Toxicology. 20, 10: 1498—1502.
[15] . Chen L., Wang M., Villalta P.W., Luo X., Feuer R., Jensen J., Hatsukami D. K., and
Hecht S.S. (2007b). «Quantitation of an acetaldehyde adduct in human leukocyte DNA
and the effect of smoking cessation». Chemical Research in Toxicology. 20, 1: 108—113.
[16] . Chen Y. L., Wang C.J., and Wu K. Y. (2005). «Analysis of N7-(benzo[a]pyrene-6-yl)gua-
nine in urine using two-step solid-phase extraction and isotope dilution with liquid chro-
matography/tandem mass spectrometry». Rapid Communications in Mass Spectrometry. 19,
7: 893-898.
[17] . Churchwell M. L, Beland F. A., and Doerge D. R. (2006). «Quantification of O6-methyl and
O6-ethyl deoxyguanosine adducts in C57BL/6N/Tk+/- mice using LC/MS/MS». Journal
of Chromatography, B: Analytical Technologies in the Biomedical and Life Sciences. 844, 1:
60-66.
[18] . Dass C. (2001). Mass Analysis and Ion Detection. Ed. Desiderio D. M., and Nibber-
ing N. M. M. New York: Wiley-Interscience, pp. 59—93.
[19] . Delaney J.C., Smeester L., Wong C., Frick L. E., Taghizadeh K., Wishnok J.S., Dren-
nan C. L., Samson L. D., and Essigmann J. M. (2005). «AlkB reverses etheno DNA lesions
caused by lipid oxidation in vitro and in vivo». Nature Structural & Molecular Biology. 12, 10:
855-860.
[20] . Dennehy M.K., and Loeppky R.N. (2005). «Mass spectrometric methodology for the
determination of glyoxaldeoxyguanosine and O6-hydroxyethyldeoxyguanosine DNA ad-
ducts produced by nitrosamine bident carcinogens». Chemical Research in Toxicology. 18,
3: 556-565.
[21] . Doerge D. R., Churchwell M.I., Fang J. L., and Beland F.A. (2000). «Quantification of
etheno-DNA adducts using liquid chromatography, on-line sample processing, and elec-
trospray tandem mass spectrometry». Chemical Research in Toxicology. 13, 12: 1259—1264.
[22] . Doerge D. R., Churchwell M.I., Marques M.M., and Beland F.A. (1999). «Quantitative
analysis of 4-aminobiphenyl-C8-deoxyguanosyl DNA adducts produced in vitro and in vivo
using HPLC-ES-MS». Carcinogenesis. 20, 6: 1055—1061.
[23] . Egner P. A., Groopman J. D., Wang J. S., Kensler T. W., and Friesen M. D. (2006). «Quanti-
fication of aflatoxin-Bl-N7-Guanine in human urine by high-performance liquid chroma-
tography and isotope dilution tandem mass spectrometry». Chemical Research in Toxicology.
19,9: 1191-1195.
[24] . Embrechts J., Lemiere F, Van D. W., Esmans E. L., Buytaert P., Van M. E., Kockx M., and
Makar A. (2003). «Detection of estrogen DNA-adducts in human breast tumor tissue and
healthy tissue by combined nano LC-nano ES tandem mass spectrometry». Journal of the
American Society for Mass Spectrometry. 14, 5: 482—491.
17.7. Выводы
[25] . Evans J. W., Chernikova S. B., Kachnic L. A., Banath J. P., Sordet O., Delahoussaye Y. M.,
Treszezamsky A., Chon В. H., Feng Z., Gu Y, Wilson W. R., Pommier Y, Olive P. L., Pow-
ell S.N., and Brown J.M. (2008a). «Homologous recombination is the principal pathway
for the repair of DNA damage induced by tirapazamine in mammalian cells». Cancer Re-
search. 68, 1: 257—265.
[26] . Evans M.D., Singh R., Mistry V., Sandhu K., Farmer P. B., and Cooke M.S. (2008b).
«Analysis of urinary 8-oxo-7,8-dihydro-purine-2'-deoxyribonucleosides by LC-MS/MS
and improved ELISA». Free Radical Research. 42, 10: 831—840.
[27] . Farmer P. B., Brown K., Tompkins E., Emms V. L., Jones D. J., Singh R., and Phillips D. H.
(2005). «DNA adducts: mass spectrometry methods and future prospects». Toxicology and
Applied Pharmacology. 207, 2 Suppl: 293—301.
[28] . Farmer P. B., and Singh R. (2008). «Use of DNA adducts to identify human health risk from
exposure to hazardous environmental pollutants: the increasing role of mass spectrometry
in assessing biologically effective doses of genotoxic carcinogens». Mutation Research. 659,
1-2: 68-76.
[29] . Gamboa da C.G., Churchwell M.I., Hamilton L. P., Von Tungeln L. S., Beland F.A.,
Marques M. M., and Doerge D. R. (2003a). «DNA adduct formation from acrylamide via
conversion to glycidamide in adult and neonatal mice». Chemical Research in Toxicology.
16, 10: 1328-1337.
[30] . Gamboa da C.G., Marques M. M., Beland F.A., Freeman J. P., Churchwell M. I., and Do-
erge D. R. (2003b). «Quantification of tamoxifen DNA adducts using on-line sample prepa-
ration and HPLC-electrospray ionization tandem mass spectrometry». Chemical Research
in Toxicology. 16, 3: 357—366.
[31] . Glick J., Zarbl H., and Vouros P. (2007). DNA adducts and you: When bad things happen to
good cells. Abstract presented at the 234th ACS National Meeting, Boston, MA, August
19-23, 2007.
[32] . Goggin M., Anderson C., Park S., Swenberg J., Walker V., and Tretyakova N. (2008).
«Quantitative high-performance liquid chromatography-electrospray ionization-tandem
mass spectrometry analysis of the adenine-guanine cross-links of 1,2,3,4-diepoxybutane
in tissues of butadiene-exposed B6C3F1 mice». Chemical Research in Toxicology. 21, 5:
1163-1170.
[33] . Goggin M., Loeber R., Park S., Walker V., Wickliffe J., and Tretyakova N. (2007). «HPLC-
ESF-MS/MS analysis of N7-guanine-N7-guanine DNA cross-links in tissues of mice ex-
posed to 1,3-butadiene». Chemical Research in Toxicology. 20, 5: 839—847.
[34] . Goggin M., Seneviratne U., Swenberg J. A., Walker V. E., and Tretyakova N. (2010). «Col-
umn switching H PLC-ESF-MS/MS methods for quantitative analysis of exocyclic dA ad-
ducts in the DNA of laboratory animals exposed to 1,3-butadiene». Chemical Research in
Toxicology. 23, 808—812.
[35] . Goggin M., Swenberg J. A., Walker V.E., and Tretyakova N. (2009). «Molecular dosimetry
of 1,2,3,4-diepoxybutane-induced DNA-DNA cross-links in B6C3F1 mice and F344 rats
exposed to 1,3-butadiene by inhalation». Cancer Research. 69, 6: 2479—2486.
[36] . Goodenough A. K., Schut H.A., and Turesky R.J. (2007). «Novel LC-ESI/MS/MS(n)
method for the characterization and quantification of 2'-deoxyguanosine adducts of the di-
etary carcinogen 2-amino-l-methyl-6-phenylimidazo[4,5-b]pyridine by 2-D linear quad-
rupole ion trap mass spectrometry». Chemical Research in Toxicology. 20, 2: 263—276.
Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
[37] . Ham A.J., Engelward В.Р., Кос Н., Sangaiah R., Meira L. В., Samson L. D., and Swen-
berg J. A. (2004). «New immunoaffinity-LC-MS/MS methodology reveals that Aag null
mice are deficient in their ability to clear l,7V6-etheno-deoxyadenosine DNA lesions from
lung and liver in vivo». DNA Repair (Amst). 3, 3: 257—265.
[38] . Hang B. (2004). «Repair of exocyclic DNA adducts: rings of complexity». Bioessays. 26, 11:
1195-1208.
[39] . Hang B., Chenna A., Guliaev A. B., and Singer B. (2003). «Miscoding properties of 1,7V6-
ethanoadenine, a DNA adduct derived from reaction with the antitumor agent 1,3-bis(2-
chloroethyl)-l-nitrosourea». Mutation Research. 531, 1—2: 191—203.
[40] . Harris С. C. (1991). «Chemical and physical carcinogenesis: advances and perspectives for
the 1990s». Cancer Research. 51, 18 Suppl: 5023s-5044s.
[41] . Hecht S. S. (1999). «Tobacco smoke carcinogens and lung cancer». Journal of the National
Cancer Institute. 91, 14: 1194—1210.
[42] . Hillestrom P. R., Hoberg A. M., Weimann A., and Poulsen H.E. (2004). «Quantifica-
tion of l,7V6-etheno-2'-deoxyadenosine in human urine by column-switching LC/APCI-
MS/MS». Free Radical Biology and Medicine. 36, 11: 1383—1392.
[43] . Hillestrom P. R., Weimann A., and Poulsen H. E. (2006). «Quantification of urinary ethe-
no-DNA adducts by column-switching LC/APCI-MS/MS». Journal of the American Soci-
ety for Mass Spectrometry. 17, 4: 605—610.
[44] . Huang С. C., Shih W. C., Wu C. F., Chen M. E, Chen Y. L., Lin Y. H., and Wu K. Y. (2008).
«Rapid and sensitive on-line liquid chromatographic/tandem mass spectrometric determi-
nation of an ethylene oxide-DNA adduct, N7-(2-hydroxyethyl)guanine, in urine of non-
smokers». Rapid Communications in Mass Spectrometry. 22, 5: 706—710.
[45] . Jones G.W., Hooley P, Farrington S.M., Shawcross S.G., Iwanejko L.A., and Strike P.
(1999). «Cloning and characterisation of the sagA gene of Aspergillus nidulans: a gene
which affects sensitivity to DNA-damaging agents». Molecular and General Genetics. 261,
2: 251-258.
[46] . Kang H., Pasa-Tolic L., and Smith R. D. (2007). «Targeted tandem mass spectrometry for
high-throughput comparative proteomics employing NanoLC-FTICR MS with external
ion dissociation». Journal of the American Society for Mass Spectrometry. 18,7: 1332—1343.
[47] . Кос H., and Swenberg J. A. (2002). «Applications of mass spectrometry for quantitation of
DNA adducts». Journal of Chromatography, B: Analytical Technologies in the Biomedical and
Life Sciences. 778, 1-2: 323-343.
[48] . Кос H., Tretyakova N.Y., Walker V. E., Henderson R. F., and Swenberg J. A. (1999). «Mo-
lecular dosimetry of N-7 guanine adduct formation in mice and rats exposed to 1,3-butadi-
ene». Chemical Research in Toxicology. 12, 7: 566—574.
[49] . Kristensen D. B., Imamura K., Miyamoto Y., and Yoshizato K. (2000). «Mass spectromet-
ric approaches for the characterization of proteins on a hybrid quadrupole time-of-flight
(Q-TOF) mass spectrometer». Electrophoresis. 21, 2: 430—439.
[50] . Lacorte S., and Fernandez-Alba A. R. (2006). «Time of flight mass spectrometry applied
to the liquid chromatographic analysis of pesticides in water and food». Mass Spectrometry
Reviews. 25, 6: 866—880.
[51] . Lao Y, Villalta P.W., Sturla S.J., Wang M., and Hecht S.S. (2006). «Quantitation of pyr-
idyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-
17.7. Выводы
performance liquid chromatography-electrospray ionization-tandem mass spectrometry».
Chemical Research in Toxicology. 19, 5: 674—682.
[52] . Lao Y, Yu N., Kassie E, Villalta P.W., and Hecht S.S. (2007). «Formation and ac-
cumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with
4-(methylnitrosamino)-l-(3-pyridyl)-l-butanone and enantiomers of its metabolite,
4-(methylnitrosamino)-l-(3-pyridyl)-l-butanol». Chemical Research in Toxicology. 20, 2:
235-245.
[53] . LeeJ.R., LeeS.J., KimT.W., Kim J.K., ParkH.S., Kim D.E., Kim К. P., and Yeo W. S.
(2009). «Chemical Approach for Specific Enrichment and Mass Analysis of Nitrated Pep-
tides. Analytical Chemistry.
[54] . Levine R. L., Yang I. Y, Hossain M., Pandya G. A., Grollman A. P., and Moriya M. (2000).
«Mutagenesis induced by a single l,A6-ethenodeoxyadenosine adduct in human cells».
Cancer Research. 60, 15: 4098—4104.
[55] . Li C. S., Wu K. Y, Chang-Chien G. P., and Chou С. C. (2005). «Analysis of oxidative DNA
damage 8-hydroxy-2'-deoxyguanosine as a biomarker of exposures to persistent pollutants
for marine mammals». Environmental Science and Technology. 39, 8: 2455—2460.
[56] . Lindahl T., and Wood R. D. (1999). «Quality control by DNA repair». Science. 286, 5446:
1897-1905.
[57] . Liu J., Chen C.F., Tsao C.W., Chang C.C., Chu C.C., and DeVoe D. L. (2009). «Poly-
mer microchips integrating solid-phase extraction and high-performance liquid chroma-
tography using reversed-phase polymethacrylate monoliths». Analytical Chemistry. 81, 7:
2545-2554.
[58] . Liu X., Lovell M. A., and Lynn В. C. (2005). «Development of a method for quantification
of acrolein-deoxyguanosine adducts in DNA using isotope dilution-capillary LC/MS/MS
and its application to human brain tissue». Analytical Chemistry. 77, 18: 5982—5989.
[59] . Liu X., Lovell M. A., and Lynn В. C. (2006). «Detection and quantification of endogenous
cyclic DNA adducts derived from trans-4-hydroxy-2-nonenal in human brain tissue by iso-
tope dilution capillary liquid chromatography nanoelectrospray tandem mass spectrom-
etry». Chemical Research in Toxicology. 19, 5: 710—718.
[60] . Loeppky R. N., Ye Q., Goelzer P., and Chen Y. (2002). «DNA adducts from N-nitrosodi-
ethanolamine and related beta-oxidized nitrosamines in vivo'. 32P-postlabeling methods for
glyoxal- and O6-hydroxyethyldeoxyguanosine adducts». Chemical Research in Toxicology.
15,4: 470-482.
[61] . Loureiro A. P., Marques S. A., Garcia С. C., Di M. P., and Medeiros M. H. (2002). «Devel-
opment of an on-line liquid chromatography-electrospray tandem mass spectrometry assay
to quantitatively determine l,A2-etheno-2'-deoxyguanosine in DNA». Chemical Research
in Toxicology. 15, 10: 1302—1308.
[62] . Makarov A. (2000). «Electrostatic axially harmonic orbital trapping: a high-performance
technique of mass analysis». Analytical Chemistry. 72, 6: 1156—1162.
[63] . Marsden D. A., Jones D. J., Lamb J. H., Tompkins E. M., Farmer P. B., and Brown K. (2007).
«Determination of endogenous and exogenously derived N7-(2-hydroxyethyl)guanine ad-
ducts in ethylene oxide-treated rats». Chemical Research in Toxicology. 20, 2: 290—299.
[64] . Matsuda T., Yabushita H., Kanaly R. A., Shibutani S., and Yokoyama A. (2006). «Increased
DNA damage in ALDH2-deficient alcoholics». Chemical Research in Toxicology. 19, 10:
1374-1378.
L Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
[65] . Means J.C., Olsen Р. D., and Schoffers Е. (2003). «Development of an isotope dilution
liquid chromatography/tandem mass spectrometry detection method for DNA adducts of
selected aromatic amines». Journal of the American Society for Mass Spectrometry. 14, 9:
1057-1066.
[66] . Monien В. H., Muller C., Engst W., Frank H., Seidel A., and Glatt H. (2008). «Time course
of hepatic 1-methylpyrene DNA adducts in rats determined by isotope dilution LC-MS/MS
and 32P-postlabeling». Chemical Research in Toxicology. 21, 10: 2017—2025.
[67] . Nakao L.S., Fonseca E., and Augusto O. (2002). «Detection of C8-(l-hydroxyethyl)gua-
nine in liver RNA and DNA from control and ethanol-treated rats». Chemical Research in
Toxicology. 15, 10: 1248-1253.
[68] . Niedernhofer L.J., Lalai A.S., and Hoeijmakers J. H. (2005). «Fanconi anemia (cross)
linked to DNA repair». Cell. 123,7: 1191-1198.
[69] . Otteneder M., and Lutz W. K. (1999). «Correlation of DNA adduct levels with tumor in-
cidence: carcinogenic potency of DNA adducts». Mutation Research. 424, 1—2: 237—247.
[70] . Paehler A., Richoz J., Soglia J., Vouros P., and Turesky R. J. (2002). «Analysis and quantifi-
cation of DNA adducts of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline in liver of rats
by liquid chromatography/electrospray tandem mass spectrometry». Chemical Research in
Toxicology. 15, 4: 551—561.
[71] . Park S., Anderson C., Loeber R., Seetharaman M., Jones R., and Tretyakova N. (2005).
«Interstrand and intrastrand DNA-DNA cross-linking by 1,2,3,4-diepoxybutane: role of
stereochemistry. Journal of the American Chemical Society. 127,41: 14355—14365.
[72] . Park S., Hodge J., Anderson C., and Tretyakova N. (2004). «Guanine-adenine DNA cross-
linking by 1,2,3,4-diepoxybutane: potential basis for biological activity». Chemical Research
in Toxicology. 17, 12: 1638-1651.
[73] . Park S., Seetharaman M., Ogdie A., Ferguson D., and Tretyakova N. (2003). 3'-Exonu-
clease resistance of DNA oligodeoxynucleotides containing O6-[4-oxo-4-(3-pyridyl)butyl]
guanine». Nucleic Acids Research. 31, 7: 1984—1994.
[74] . Pathak S., Singh R., Verschoyle R. D., Greaves P., Farmer P. B., Steward W. P., Mellon J. K.,
Gescher A. J., and Sharma R.A. (2008). «Androgen manipulation alters oxidative DNA ad-
duct levels in androgen-sensitive prostate cancer cells grown in vitro and in vivo». Cancer
Letters. 261, 1:74-83.
[75] . Perry R. H., Cooks R.G., and Noll R. J. (2008). «Orbitrap mass spectrometry: instrumenta-
tion, ion motion and applications». Mass Spectrometry Reviews. 27, 6: 661—699.
[76] . Powley M.W., Li Y., Upton P. B., Walker V. E., and Swenberg J. A. (2005). «Quantifica-
tion of DNA and hemoglobin adducts of 3,4-epoxy-l,2-butanediol in rodents exposed to
3-butene-l,2-diol». Carcinogenesis. 26, 9: 1573—1580.
[77] . Powley M.W., Walker V. E., LiY, Upton P. B., and Swenberg J. A. (2007). «The importance
of 3,4-epoxy-l,2-butanediol and hydroxymethylvinyl ketone in 3-butene-l,2-diol associ-
ated mutagenicity. Chemico-Biological Interactions. 166, 1—3: 182—190.
[78] . Rajski S. R., and Williams R. M. (1998). «DNA Cross-Linking Agents as Antitumor Drugs».
Chemical Reviews. 98, 8: 2723—2796.
[79] . Randall K., Argoti D., Paonessa J., Ding Y, Oaks Z., Zhang Y, and Vouros P. (2009).
LC-MS Detection of 4-ABP DNA Adduct Formation in Bladder Cells and Tissues. Ref Type:
Generic
17.7. Выводы
[80] . Ricicki Е.М., Soglia J. R., Teitel C., Kane R., Kadlubar E, and Vouros P. (2005). «De-
tection and quantification of N-(deoxyguanosin-8-yl)-4-aminobiphenyl adducts in human
pancreas tissue using capillary liquid chromatography-microelectrospray mass spectrom-
etry». Chemical Research in Toxicology. 18, 4: 692—699.
[81] . Singer B., and Grunberger D. (1983). Molecular Biology of Mutagens and Carcinogens. Ple-
num Press, New York and London.
[82] . Singh R., and Farmer P. B. (2006). «Liquid chromatography-electrospray ionization-mass
spectrometry: the future of DNA adduct detection». Carcinogenesis. 27, 2: 178—196.
[83] . Singh R., Gaskell M., Le Pla R.C., Kaur B., zim-Araghi A., Roach J., Koukouves G.,
Souliotis V. L., Kyrtopoulos S.A., and Farmer P. B. (2006). «Detection and quantitation
of benzo[a]pyrene-derived DNA adducts in mouse liver by liquid chromatography-tandem
mass spectrometry: comparison with 32P-postlabeling». Chemical Research in Toxicology.
19, 6:868-878.
[84] . Singh R., Teichert F., Verschoyle R. D., Kaur B., Vives M., Sharma R.A., Steward W. P.,
Gescher A.J., and Farmer P.B. (2009). «Simultaneous determination of 8-oxo-2'-
deoxyguanosine and 8-oxo-2'-deoxyadenosine in DNA using online column-switching
liquid chromatography/tandem mass spectrometry». Rapid Communications in Mass Spec-
trometry. 23, 1: 151 — 160.
[85] . Soglia J. R., Turesky R. J., Paehler A., and Vouros P. (2001). «Quantification of the heterocy-
clic aromatic amine DNAadduct N-(deoxyguanosin-8-yl)-2-amino-3-methylimidazo[4,5-
f]quinoline in livers of rats using capillary liquid chromatography/microelectrospray mass
spectrometry: a dose-response study». Analytical Chemistry. 73, 13: 2819—2827.
[86] . Stepanov L, and Hecht S.S. (2009). «Mitochondrial DNA adducts in the lung and liver
of F344 rats chronically treated with 4-(methylnitrosamino)-l-(3-pyridyl)-l-butanone and
(S)-4-(methylnitrosamino)-l-(3-pyridyl)-l-butanol». Chemical Research in Toxicology. 22,
2:406-414.
[87] . Stout M.D., Jeong Y.C., Boysen G., Li Y, Sangaiah R., Ball L.M., Gold A., and Swen-
berg J. A. (2006). «LC/MS/MS method for the quantitation of trans-2-hexenal-derived
exocyclic l,A2-propanodeoxyguanosine in DNA». Chemical Research in Toxicology. 19, 4:
563-570.
[88] . Swenberg J. A., Fryar-Tita E., Jeong Y. C., Boysen G., Starr T., Walker V. E., and Alber-
tini R.J. (2008). «Biomarkers in toxicology and risk assessment: informing critical dose-
response relationships». Chemical Research in Toxicology. 21,1: 253—265.
[89] . Synold T., Xi B., Wuenschell G.E., Tamae D., Figarola J. L., Rahbar S., and Ter-
mini J. (2008). «Advanced Glycation End Products of DNA: Quantification of A2-(l-
Carboxyethyl)-2'-deoxyguanosine in Biological Samples by Liquid Chromatography Elec-
trospray Ionization Tandem Mass Spectrometry». Chemical Research in Toxicology.
[90] . Thomson N.M., Mijal R.S., Ziegel R., Fleischer N.L., Pegg A. E., Tretyakova N.Y., and
Peterson L. A. (2004). «Development of a quantitative liquid chromatography/electrospray
mass spectrometric assay for a mutagenic tobacco specific nitrosamine-derived DNA ad-
duct, O6-[4-Oxo-4-(3-pyridyl)butyl]-2'-deoxyguanosine». Chemical Research in Toxicol-
ogy. 17, 12: 1600-1606.
[91] . Tompkins E.M., Jones D.J., Lamb J. H., Marsden D.A., Farmer P. B., and Brown K.
(2008). «Simultaneous detection of five different 2-hydroxyethyl-DNA adducts formed by
ethylene oxide exposure, using a high-performance liquid chromatography/electrospray
514 Глава 17. Использование масс-спектрометрии для изучения взаимодействия ДНК
с экотоксикантами
ionisation tandem mass spectrometry assay». Rapid Communications in Mass Spectrometry.
22, 1: 19-28.
[92] . Tretyakova N. Y., Chiang S. Y., Walker V. E., and Swenberg J. A. (1998). «Quantitative anal-
ysis of 1,3-butadiene-induced DNA adducts in vivo and in vitro using liquid chromatogra-
phy electrospray ionization tandem mass spectrometry». Journal of Mass Spectrometry. 33,
4: 363-376.
[93] . Turesky R.J., Box R. M., Markovic J., Gremaud E., and Snyderwine E.G. (1997). «Forma-
tion and persistence of DNA adducts of 2-amino-3-methylimidazo[4,5-f]quinoline in the
rat and nonhuman primates». Mutation Research. 376, 1—2: 235—241.
[94] . Upadhyaya P., Kalscheuer S., Hochalter J.B., Villalta P.W., and Hecht S.S. (2008).
«Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated
with 4-(methylnitrosamino)-l-(3-pyridyl)-l-butanone and enantiomers of its metabolite
4-(methylnitrosamino)-l-(3-pyridyl)-l-butanol». Chemical Research in Toxicology. 21, 7:
1468-1476.
[95] . Van den D. B., Esmans E. L., Van der L. A., Van D. W., Schaerlaken E., Lemiere F., Wit-
ters E., and Berneman Z. (2005). «First results of a quantitative study of DNA adducts
of melphalan in the rat by isotope dilution mass spectrometry using capillary liquid chro-
matography coupled to electrospray tandem mass spectrometry». Rapid Communications in
Mass Spectrometry. 19, 14: 1999—2004.
[96] . Wang M., Yu N., Chen L., Villalta P.W., Hochalter J.B., and Hecht S.S. (2006). «Identifi-
cation of an acetaldehyde adduct in human liver DNA and quantitation as V2-ethyldeoxy-
guanosine». Chemical Research in Toxicology. 19, 2: 319—324.
[97] . Wickremsinhe E. R., Ackermann B. L., and Chaudhary A. K. (2005). «Validating regulato-
ry-compliant wide dynamic range bioanalytical assays using chip-based nanoelectrospray
tandem mass spectrometry». Rapid Communications in Mass Spectrometry. 19, 1: 47—56.
[98] . Williams M. V, Lee S. H., Pollack M., and Blair LA. (2006). «Endogenous lipid hydroper-
oxide-mediated DNA-adduct formation in min mice». Journal of Biological Chemistry. 281,
15: 10127-10133.
[99] . Zhang N. R., Yu S., Tiller P., Yeh S., Mahan E., and Emary W. B. (2009). «Quantitation
of small molecules using high-resolution accurate mass spectrometers — a different ap-
proach for analysis of biological samples». Rapid Communications in Mass Spectrometry. 23,
7: 1085-1094.
[100] . Zhang Q., Aft R. L., and Gross M. L. (2008). «Estrogen carcinogenesis: specific identifica-
tion of estrogen-modified nucleobase in breast tissue from women». Chemical Research in
Toxicology. 21,8: 1509-1513.
[101] . Zhang S., Villalta P.W., Wang M., and Hecht S.S. (2006). «Analysis of crotonaldehyde-
and acetaldehyde-derived l,n(2)-propanodeoxyguanosine adducts in DNA from human
tissues using liquid chromatography electrospray ionization tandem mass spectrometry».
Chemical Research in Toxicology. 19, 10: 1386—1392.
[102] . Zhang S., Villalta P.W., Wang M., and Hecht S.S. (2007). «Detection and quantitation
of acrolein-derived l,V2-propanodeoxyguanosine adducts in human lung by liquid chro-
matography-electrospray ionization-tandem mass spectrometry». Chemical Research in
Toxicology. 20, 4: 565—571.
ГЛАВА 18
И ЦРФП-АНАЛИЗ
СЛОЖНЫХ
ОРГАНИЧЕСКИХ
СМЕСЕЙ.
ПЕТРОЛЕОМИКА
Владислав В. Лободин, Раян П. Роджерс, Алан Дж. Маршалл
18.1. Введение
Нефть является одной из наиболее сложных природных смесей органических соеди-
нений и, несмотря на более чем вековую историю активного промышленного исполь-
зования, ее состав еще не полностью известен. Будучи основным видом ископаемого
топлива, добываемого на планете, нефть и нефтепродукты вносят существенный вклад
в загрязнение окружающей среды. Кроме насыщенных и ароматических углеводородов,
нефть состоит из гетероатомных (N, О, S) и металлосодержащих (Ni, V) соединений,
которые могут составлять значительную часть в составе нефти и ее фракций.
Легкие летучие компоненты (в основном углеводороды) нефти тщательно изучены;
тогда как анализ нелетучих полярных гетероатомных соединений до недавнего времени
был ограничен из-за их сложности и отсутствия подходящих аналитических методов.
Тем не менее гетероатомные соединения, хотя они значительно менее распространены,
чем углеводороды, способны существенно влиять на свойства нефти и оказывать раз-
личное воздействие на окружающую среду (Headley et al., 2010; Headley et al., 2009a; Head-
ley et al., 2009b; Hughey et al., 2008; Kim et al., 2005).
Основной масс-спектрометрический подход для анализа проб окружающей среды,
как правило, включает предварительное хроматографическое разделение смеси соеди-
нений на индивидуальные компоненты. В идеальном случае каждое соединение элю-
ируется отдельным хроматографическим пиком и идентифицируется по масс-спектру.
Масс-спектр и хроматографическое время удерживания являются уникальными харак-
теристиками соединения. Успех такого подхода зависит от сложности смеси, эффектив-
ности хроматографического разделения, а также информационного содержания масс-
спектрометрических данных.
Современные гибридные методы, такие как двумерная газовая хроматогра-
фия/масс-спектрометрия (ГХ/ГХ/МС), позволяют успешно анализировать многоком-
понентные смеси, состоящие из нескольких тысяч соединений (глава 6). Однако что
если исследуемая смесь еще более сложная и состоит из миллионов компонентов? Раз-
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
деление такой смеси любым известным хроматографическим методом становится прак-
тически невозможным, а ее анализ, например, с помощью ГХ/МС или ЖХ/МС займет
много времени и все равно не будет полным. Кроме того, число возможных изомерных
структур резко возрастает с увеличением числа атомов в молекуле. Даже если все изо-
меры определенного химического состава разделены, концентрация некоторых из них
может быть ниже предела обнаружения.
В этой главе рассматривается альтернативный подход для комплексного анали-
за сложной смеси, который основан на использовании масс-спектрометрии ионного
циклотронного резонанса с преобразованием Фурье (МС-ИЦРФП). Благодаря МС-
ИЦРФП появилась возможность регистрировать массы ионов со сверхвысоким раз-
решением во всем диапазоне масс-спектра. Это, в свою очередь, позволяет разделять
и детектировать тысячи химически различных компонентов в одном масс-спектре. Та-
ким образом, МС-ИЦРФП в комбинации с соответствующими методами ионизации
представляет собой мощный аналитический метод для анализа таких сверхсложных
смесей, как сырая нефть, уголь, биотопливо, природные органические вещества и др.,
без предварительного хроматографического разделения. На самом деле, если рассма-
тривать МС-ИЦРФП просто как одномерный метод изократического разделения, то
он позволяет в теории различать более 500000 пиков. Хотя МС-ИЦРФП была исполь-
зована в сочетании с почти всеми известными методами ионизации, особенно хорошо
для анализа сложных смесей зарекомендовали себя неразрушающие, «мягкие» методы,
такие как электрораспыление (ЭРИ, ESI) и фотоионизация при атмосферном давлении
(ФИАД, APPI).
Нефть была одним из первых природных объектов, проанализированных с помо-
щью масс-спектрометрии, что существенно повлияло на эволюцию метода. Приборное
развитие масс-спектрометрии на ранних стадиях было в основном продиктовано спро-
сом со стороны нефтяной промышленности, и, например, первый коммерческий масс-
спектрометр (модель 21-101, Consolidated Engineering Corporation, США), построенный
в 1942 году, был продан Атлантической нефтеперерабатывающей компании (потомок
«Стандард Ойл»). Последующий интерес к масс-спектрометрии со стороны нефте-
химических и химических компаний явился залогом дальнейших приборных разра-
боток, установления стандартов, а также создания первой коллекции масс-спектров
(Meyerson, 1986).
Анализ нефти накладывает самые строгие требования на разрешающую спо-
собность масс-спектрометра, так как масса «дублетов» (разница между двумя близ-
ко расположенными пиками в масс-спектре) в нефти включает в себя большинство
вариантов, которые встречаются в протеомике, метаболомике и анализе других
сложных смесей. Таким образом, нефть представляет собой идеальный образец для
тестирования технических характеристик и оптимизации производительности МС-
ИЦРФП (Marshalletal., 2010). Возможность анализировать все химические компонен-
ты нефти, исследовать их взаимодействие и реакционную способность создала базу
новой дисциплины — петролеомики. Существует возможность различать нефть и не-
фтепродукты в соответствии с их геохимическим происхождением и степенью зрело-
сти, диапазоном дистилляции, методами добычи, каталитической обработки и т.д.
Если ГХ/МС обычно используется для анализа смесей летучих соединений, ИЦРФП
в основном применяется для детектирования менее летучих полярных компонентов.
Методом масс-спектрометрии ионного циклотронного резонанса с преобразованием
Фурье доказано, что нефть включает в себя более 100000 различных гетероатомных
органических соединений с элементным составом CcH/NnOoSs. Однако брутто-фор-
муле может соответствовать множество структурных изомеров, которые неразличи-
мы при измерении только их массы. Поэтому для более полного анализа сложных
органических объектов используются различные методы разделения сложной смеси
на несколько менее сложных, включая дистилляцию нефти на фракции и хромато-
графию. Последние достижения в области МС-ИЦРФП, а также ее роль в области
комплексного анализа смесей (петролеомика) были предметом нескольких обзорных
статей (Marshall and Hendrickson, 2008; Marshall and Rodgers, 2004; Rodgers et al., 2005)
и книги (Rodgers and Marshall, 2007). Подход, схожий с МС-ИЦРФП-анализом нефти,
применяется также в судебно-медицинской экспертизе, при анализе объектов окру-
жающей среды, пищевой промышленности, гуминовых и фульвокислот, угля и дру-
гих сложных природных смесей.
18.2. МС-ИЦРФП
Принципы ИЦРФП были впервые разработаны А.Дж. Маршаллом и М.Б. Комисароу
в 1974 году. Они заключались в применении преобразования Фурье (ФП) для регистра-
ции сигнала ионов в ответ на их одновременное резонансное возбуждение во всем диа-
пазоне масс-спектра (Comissarow and Marshall, 1974). Как и в случае ЯМР, применение
ФП для ИЦР привело к резкому сокращению времени регистрации спектра и сохране-
нию высокой разрешающей способности, присущей ИЦР МС. Здесь мы представляем
Ионно-циклотронное движение в ячейке ИЦР
Рис. 18.1. Схематическое изображение ионного циклотронного движения в ИЦР-ячейке
с квадратным поперечным сечением. Ион вращается в плоскости, перпендику-
лярной направлению пространственно однородного статического магнитного
поля, В
5 18 Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
краткий обзор принципов ИЦРФП. Более подробную информацию по теории метода
и приборным основам можно найти в работах {Marshall and Verdun, 1990; Marshall, 1996;
Marshall, 2000; Marshall and Hendrickson, 2002; Marshall etal., 1998).
Большинство ионизационных методов генерируют ионы непрерывно. Однако МС-
ИЦРФП включает в себя несколько последовательных стадий для каждой партии ионов
(например образование ионов, перенос ионов в ячейку ИЦР, возбуждение ионов, их де-
тектирование). В современных ИЦРФП-масс-спектрометрах ионизация образца проис-
ходит вне ячейки ИЦР. Ионы, как правило, накапливаются в ионной ловушке, а затем
переносятся в ячейку ИЦР в одну стадию.
Рис. 18.1 иллюстрирует движение ионов в ячейке ИЦР с квадратным сечением. Ион
(массой т, зарядом z и скоростью v) при движении в магнитном поле В, перпендикуляр-
ном его скорости г, совершает циклотронное движение за счет действия на него силы
Лоренца (FL = qVB), которая, в свою очередь, уравновешивается центробежной силой
(Fc= mv2/r)\
mv2 D /10 п
-----qvB. (18.1)
г
Т. к. угловая скорость, со (в рад/с), иона определяется как со = V/r, получаем
(0 = ^ (18.2)
т
Частота вращения иона, известная как циклотронная частота, равна v. = со/2л или
qB 1,53561 НО7 Я
V =-----=--------------, (1о.э)
2itm m/z
где vc в Гц, В в Теслах, т в Дальтонах, а нравно числу элементарных зарядов.
Таким образом, в отличие от других масс-анализаторов (за исключением орби-
трэпа), МС-ИЦРФП основана на измерении частоты (циклотронная частота), которая
не зависит от скорости иона. Поэтому все ионы с определенной величиной m/z имеют
одинаковую циклотронную частоту независимо от их кинетической энергии. В свою
очередь частота и время являются наиболее точно измеряемыми параметрами в физи-
ке {Diddams et al., 2004). Возможность измерения ИЦР частоты с точностью одна часть
на миллиард (ppb) потенциально передает такую же точность измерения и величине
m/z. Это свойство ИЦР оказывается особенно важным для точного определения массы
ионов неизвестных соединений. Тем не менее одной только точности измерения мас-
сы недостаточно — необходимо разделить все компоненты смеси по массе, чтобы быть
уверенным, что измеренная масса соответствует только одному химическому составу.
Как видно из уравнения 3, циклотронная частота ионов линейно пропорциональна век-
тору магнитной индукции (В). С ростом В возрастает абсолютная разница в циклотрон-
ных частотах (Av, в Гц) между ионами с различными величинами m/z и, следовательно,
увеличивается разрешение.
Сразу же после переноса ионов в ячейку их начальный радиус сравнительно мал,
а движение является некогерентным, т.е. не вызывает разности потенциалов на детек-
тирующих электродах и тока во внешней цепи. Для детектирования ионов, во-первых,
их радиус должен быть увеличен, чтобы они вращались в непосредственной близости
18.2. МС-ИЦРФП
Рис. 18.2. Вид временной (слева) и частотной области (справа) частотной развертки
(«чирп») для широкополосного возбуждения ионов. По материалам (Marshall
et aL, 2010)
от детектирующих пластин. Во-вторых, ионы с одним значением m/z первоначально
вращаются с одной частотой, но в разных фазах и, следовательно, не индуцируют ток
(сигнал) во внешней цепи, подключенной к детектирующим электродам. Приложение
высокочастотного напряжения на возбуждающие электроды-пластины с частотой, рав-
ной циклотронной частоте ионов, вызывает резонансное ускорение ионов с увеличени-
ем их скорости и радиуса. Этот процесс известен в ИЦРФП как возбуждение и приводит
к когерентному (в одной фазе) вращению ионов с определенным значением m/z. Воз-
буждение всех ионов за несколько миллисекунд может быть осуществлено путем приме-
нения короткого радиочастотного импульса, в диапазоне циклотронных частот ионов,
представляющих интерес. Широкополосное ионно-циклотронное возбуждение обыч-
но проводится через импульсную линейную частотную модуляцию («чирп») (рис. 18.2)
и происходит с относительно плоской магнитудой в широком диапазоне частот при ис-
пользовании относительно низких напряжений.
На рис. 18.3 представлено преобразование циклотронного движения в масс-спектр.
Детектирование ионов в ИЦРФП основано на регистрации ионного тока, вызванного
одновременным вращением всех ионов в ячейке (рис. 18.3а). Ток возрастает линейно
с увеличением числа ионов и циклотронного радиуса. Сигнал регистрируется посред-
ством измерения тока во внешней цепи, связанной с детектирующими пластинами
(рис. 18.3d). Общий сигнал (ионный ток во временной области) представляет собой су-
перпозицию сигналов от ионов с различными величинами m/z. Этот сигнал оцифро-
вывается и обрабатывается с помощью быстрого преобразования Фурье с получением
частотного спектра (рис. 18.Зе). Далее частотный спектр превращается в масс-спектр
(рис. 19.3г) с помощью приведенного уравнения калибровки.
Высокое разрешение и высокая точность определения массы являются наиболее
важными характеристиками ИЦРФП, необходимыми для анализа сложных смесей.
Как отмечалось выше, разрешающая способность ИЦРФП-масс-спектрометров воз-
растает с увеличением магнитного поля. Даже первые приборы ИЦРФП с постоянны-
ми магнитами и напряженностью поля не более 1 Тесла обеспечивали разрешающую
способность выше, чем магнитные секторные масс-спектрометры. Следующее поко-
ление приборов опиралось на низкие поля (~3 Тл), генерируемые сверхпроводящими
магнитами. Эти масс-спектрометры обладали значительно более высоким разреше-
нием, но еще недостаточным для получения высокого разрешения во всем диапазо-
не широкополосного масс-спектра нефти. В этом случае регистрировались отдельные
сегменты масс-спектра, которые впоследствии сшивались вместе, чтобы сформировать
широкополосный масс-спектр (Guan etaL, 1996).1ъкой подход являлся трудоемким, за-
520 Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
Рис. 18.3. Преобразование циклотронного движения ионов в масс-спектр, (а) Возбужде-
ние когерентного циклотронного вращения ионов, (б) регистрация сигнала, ин-
дуцируемого током ионов на детектирующих электродах, (в) частотный спектр,
полученный путем быстрого преобразования Фурье из оцифрованного во вре-
менной области сигнала, (г) масс-спектр, полученный из частотного спектра че-
рез уравнения калибровки. По материалам (Marshalland Hendrickson, 2008)
нимал много времени и был неудобен для комплексного анализа сложных смесей. По-
явление сверхпроводящих соленоидных ИЦР-магнитов с сильными полями (>7 Тл),
которые обеспечивают высокую временную стабильность (<50 ppb/час дрейфа поля)
и пространственную однородность (<10 ppm пиковое изменение в пределах измеряе-
мого объема), дало возможность регистрировать широкополосные масс-спектры вы-
сокого разрешения. Это достижение, в свою очередь, привело к быстрому развитию
приложений ИЦРФП для анализа сложных смесей (нефть, биотопливо, растворенное
органическое вещество).
Высокое разрешение и высокая точность измерения массы позволили разделять
сигналы изобарных соединений (соединения, имеющие одинаковую номинальную
массу, но разный элементный состав и, следовательно, различную точную массу)
и определять их брутто-формулу. Например, на рис. 18.4 приведены ИЦРФП-масс-
спектры отрицательных и положительных ионов, образующихся при электрораспыле-
нии североамериканской нефти без предварительного хроматографического разделе-
ния. Более 20000 компонентов определены в одном образце, причем для адекватной
оценки полученной информации важную роль занимают средства визуализации дан-
ных (обсуждаются ниже).
18.3. Разрешение по массе, разрешающая способность и точное измерение массы
Более 20000 различных компонентов зарегистрировано
с помощью ФП-ИЦР МС (9,4 Тесла)
Рис. 18.4. ИЦРФП (9,4 Тл) масс-спектры отрицательных (слева) и положительных (спра-
ва) ионов, образующихся при электрораспылении североамериканской неф-
ти. Сверхвысокое разрешение (т/кт^с. ~ 600000 на m/z 500, где Даи5()% — полу-
ширина пика) позволяет различать и идентифицировать тысячи химических
соединений
18.3. Разрешение по массе, разрешающая
способность и точное измерение массы
Спектральное разрешение чаще всего определяется как минимальная разница (в дан-
ном случае т2 — т) между двумя пиками одинаковой высоты, при которой они различи-
мы и едва не сливаются. В случае пиков правильной гауссовой формы это определение
эквивалентно ширине пика на половине его высоты (Д/и50%) (Marshall and Hendrickson,
2008).
Разрешающая способность в И ЦРФП, а также в других видах масс-спектрометров —
времяпролетных, ионной ловушки, орбитрэп и т.д. (за исключением магнитных сек-
торных приборов, для которых, как правило, используется метод 10%-й ложбины),
определяется как /и/Д/и50%. Для многозарядных ионов т может быть заменено на m/z
в приведенном выше определении. В реальном масс-спектре соседние пики часто имеют
разную высоту, поэтому для их надежного разделения требуется более высокое разреше-
ние, чем значение /и/Д/и50%. Стоит отметить, что для метода 10%-й ложбины, упомяну-
того выше, определяется разница между вершинами пиков равной высоты, перекрытых
на уровне 5 % от их высоты и образующих таким образом 10%-ю ложбину (отсюда назва-
ние) между пиками. Так как ширина пика гауссовой формы на 50% и 5% от его высоты
отличается приблизительно в два раза, разница такого же порядка (2 раза) наблюдает-
ся в разрешении, определяемом этими методами. Таким образом, важно указать метод
и величину m/z, для которой определено разрешение.
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
Масса
Рис. 18.5. Вершины двух пиков гауссовой формы равной высоты находятся на расстоянии
Даи50%. Разрешение по массе определяется как Даи50%, тогда как разрешающая
способность определяется как m/Sm5Q%
Недавно с помощью МС-ИЦРФП в нефти было определено более 100000 различных
химических соединений состава (CcH//N/OoSs) (McKenna etal., 2010а; McKenna etal., 2010b).
Это стало возможным именно благодаря сверхвысокому разрешению и точности изме-
рения масс, что характерно для этого метода. Стоит отметить, что разрешающая способ-
ность ИЦРФП в 10—100 раз выше, чем других масс-анализаторов (магнитный сектор-
ный, времяпролетный, орбитрэп) (MarshallandHendrickson, 2008). Рекорд в разрешающей
способности, достигнутый с помощью ФП-ИЦР, составляетт/Ат50% = 8000000 при ре-
гистрации 9+ зарядного иона бычьего убиквитина (MW= 8,6 кДа) (Shietal., 1998).
В случае анализа сложных органических смесей высокая разрешающая способ-
ность ИЦРФП необходима, чтобы различать сигналы ионов с очень близкими массами
(например 0,0034 Да — разница между изобарными ионами, различающимися по хими-
ческому составу как SH4 и С3). На рис. 18.6 показано разрешение двух часто встречаю-
щихся в нефтяных образцах дублетов (SH4 и С3). Разделение соответствующих изобар-
ных пиков позволяет определять различные химические классы в составе смеси, чего
невозможно достичь при использовании других масс-анализаторов. Обратите внима-
ние, что ИЦРФП обеспечивает высокое разрешение по всему спектру, а аналогичную
информацию по детальному элементному составу можно получить для каждой номи-
нальной массы в спектре (300 < m/z <900) (Rodgers etal., 2005).
Когда ионы переносятся в ячейку ИЦРФП, они движутся радиально (в плоско-
сти, перпендикулярной к магнитному полю) и вращаются с циклотронной частотой
(v). Чтобы ионы не покинули ячейку вдоль оси магнитного поля, они удерживаются
электрическим потенциалом, приложенным к концевым (удерживающим) электро-
дам. Это приводит к возникновению дополнительного колебательного движения ионов
между удерживающими электродами, сдвигу в резонансных частотах и появлению до-
полнительного члена (B/v2) в калибровочном уравнении, которое связывает m/z и v (Led-
ford etal., 1984).
18.3. Разрешение по массе, разрешающая способность и точное измерение массы 523
Рис. 18.6. Сегмент ИЦРФП-спектра ионизации электрораспылением с регистрацией по-
ложительных ионов образца тяжелой ближневосточной нефти. Решена важная
для петролеомики задача разделения изобарных ионов состава X + С3 и X + SH4
с разницей 0,0034 Да, где X — любой элементный состав)
А В
m/z = -+- (18.4)
v v
Калибровка масс-спектра заключается в присвоении точной массы пикам заведомо
известных ионов. Калибровка может быть внутренней (например использование ионов
известной массы в масс-спектре исследуемого образца) или внешней (т.е. использова-
ние предварительно полученной калибровочной кривой для калибровки масс-спектра
соединения, зарегистрированного в аналогичных условиях). Внутренняя калибровка,
как правило, в два раза точнее внешней калибровки и обеспечивает суб-ppm точность,
необходимую для анализа сложных смесей. Поскольку на всех стадиях регистрации
масс-спектра (возбуждение, регистрация и т.д.) ионы аналита и калибранта находят-
ся в идентичных электрическом и магнитном полях, использование внутренней кали-
бровки приводит к очень высокой точности определения масс во всем откалиброванном
диапазоне. Процедура калибровки в случае анализа нефти включает в себя внешнюю
калибровку, на основе которой с точностью ±5 ppm определяется наиболее интенсив-
ная гомологическая серия в спектре. Такая внешняя калибровка дает коэффициенты
А и В в калибровочном уравнении, которые впоследствии используются для внутренней
калибровки всех других неизвестных ионов аналита, обеспечивая высокую точность
(<1 ppm) в широком диапазоне масс {Rodgers etal., 2005).
Максимальное число ионов в ячейке ИЦР ограничено кулоновским взаимодействи-
ем, которое приводит к искажению пиков, их сдвигу и даже слиянию. Однако сложным
органическим смесям свойственен широкий динамический диапазон концентраций
(отношение интенсивного сигнала к слабому). Поэтому, чтобы достичь необходимого
соотношения сигнал/шум при детектировании слабоинтенсивных ионов, требуется
524 Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
55 молекулярных формул в одной номинальной массе
Образец асфальтенов
тяжелой нефти
9.4Т ESI ФТ-ИЦР МС
аи/Даи50% = 350000
Внутренний стандарт
629,00 629,10 629,20 629,30 629,40 629,50 629,60 629,70
m/z
Рис. 18.7. ИЦРФП-спектр отрицательных ионов, зарегистрированный при электро-
распылении образца асфальтенов, выделенных из южноамериканской тяжелой
нефти. 55 индивидуальных пиков зарегистрировано в сегменте шириной в одну
номинальную массу {Rodgers and Marshall, 2007)
продолжительная регистрация данных с накоплением и усреднением ста и более инди-
видуальных спектров.
На рис. 18.7 приведен ИЦРФП-спектр отрицательных ионов, зарегистрированный
при электрораспылении образца асфальтенов, выделенных из южноамериканской тя-
желой нефти. Отчетливо видны более 50 пиков в сегменте шириной в одну номиналь-
ную массу. Такая же высокая насыщенность сигналов ионов наблюдается для каждой
номинальной массы в диапазоне 200—1000 Да. Разница в значениях m/z между моноизо-
топным (12Сс) и изотопным (13С12Сс1) пиками в случае заданного иона позволяет опре-
делить его заряд z. Для однозарядных ионов U = 1, как показано на рис. 18.7) измеряе-
мое значение m/z соответствует массе иона в дальтонах. Точное измерение массы ионов
в диапазоне до ~500 Да позволяет однозначно определить их брутто-формулы {Kim etal.,
2006). Элементный состав для более тяжелых компонентов требует обработки данных
на основе регулярных повторений в масс-спектре и шкалы масс Кендрика (см. далее).
18.4. Точная масса и дефект массы
Атомные массы всех изотопов химических элементов определены с высокой точностью
(относительная погрешность 10-11). Это стало возможным благодаря ИЦР {Blaum, 2006).
В табл. 18.1 приведены точная массы и относительная распространенность элементов
и изотопов, часто встречающихся в органических соединениях {De Laeter et al., 2003).
Каждый изотоп имеет уникальную атомную массу, которая немного отличается («де-
18.4. Точная масса и дефект массы
фект» масс) от ближайшего целого значения (номинальная масса). Дефект масс являет-
ся уникальным для каждого изотопа.
Таблица 18.1. Точные массы и природная распространенность изотопов некоторых
элементов
Изотоп Точная масса (Да) Распростр., % Отн. распростр., %*
’Н 1.007825032 99.9885 100
2Н 2.014101778 0.0115 0.0115
|2С 12.00000000 98.93 100
|3С 13.00335484 1.07 1.08
l4N 14.00307401 99.632 100
l5N 15.0001089 0.368 0.369
|6О 15.99491463 99.757 100
|7О 16.9991312 0.038 0.038
|КО 17.9991603 0.205 0.205
19р 18.99840322 100 100
3|р 30.973762 100 100
32s 31.9720707 94.93 100
33s 32.97145843 0.76 0.80
34S 33.96786665 4.29 4.52
36S 35.96708062 0.02 0.02
35C1 34.96885272 75.78 100
37C1 36.96590262 24.22 31.96
79Br 78.9183361 50.69 100
K,Br 80.916289 49.31 97.28
l27I 126.904473 100 100
* Интенсивность нормализована к наиболее распространенному изотопу, принятому за 100%.
Рис. 18.8. Атомный дефект массы для отдельных изотопов распространенных химических
элементов. Поскольку каждый изотоп имеет уникальный дефект, достаточно
точные измерения массы позволяют определить элементный состав молекулы
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
Таблица 18.2. Распространенные дублеты масс в петролеомике
Дублет Дай, Да (аи/Даи50%) для и?Д500
Н16иО 0,1303 3,900
Н|6О2иС4 0,1150 4,400
H14hN 0,1065* 4,700
Н|2иС 0,0939 5,400
C2H8nS 0,0905 5,600
С2Н8 И 02 0,0728 6,700
H8N2 И С3 0,0687 7,300
Н80 И С2 0,0575 8,700
СН4О И S 0,0541 9,300
Н3О13С И S 0,0497* 11,000
С2Н6И N2 0,0490 11,000
H4N2 и о2 0,0476 11,000
Н8О2 и CN2 0,0463 11,000
CH3NO и 13CS 0,0460 11,000
H6NO и С3 0,0449* 12,000
CH513ChNO 0,0445 12,000
H2NO и S 0,0416* 13,000
С2Н5 и О13С 0,0409* 13,000
34S и О2Н2 0,0376 14,000
СН4 и О 0,0364 14,000
Н6О2 и C2N 0,0337* 15,000
13СН3 и О 0,0319* 16,000
N2O и CS 0,0290 18,000
С313С и HOS 0,0285* 18,000
Н5О2 и С213С 0,0256* 20,000
С2Н4 и N2 0,0252 20,000
H2N и О 0,0238* 22,000
НО и С 4 2 3 0,0211 24,000
О2и S 0,0178 29,000
С2Н3и ,3CN 0,0170 30,000
H6S и C2N 0,0159* 32,000
HN2h 13СО 0,0157* 32,000
С4иО3 0,0153 33,000
СН2 и N 0,0126* 40,000
13CH.S и C3N 0,0115 44,000
N2 и СО 0,0112 45,000
Н40ч и 4 3 2 2 0,0099 51,000
С2Н2и”С2 0,0089 57,000
|3СН и N 0,0081 62,000
Таблица 18.2. (окончание).
Дублет Дай, Да (аи/Даи5()%) для и?Д500
H3NSnC313C 0,0047 107,000
СН и 13С 0,0045* 112,000
C,N и 34SH4 2 4 0,0039* 129,000
|3С2и CN 0,0036* 139,000
H4s и С3 0,0034 148,000
C2HMS и NSI3C 0,0028 179,000
С4и l3CH3S 0,0011* 455,000
Разница масс дублетов при одновременном присутствии четно- и нечетноэлектронных ионов,
что часто наблюдается в условиях фотоионизации при атмосферном давлении.
Дефект масс для легких элементов (‘Н, 2Н, 13С, 14N и 15N) мал и слегка положителен,
но для большинства других элементов он отрицателен и растет с увеличением атомной
массы (рис. 18.8).
При образовании ядра из протонов и нейтронов часть массы нуклонов расходуется
на образование связи между ними (ядерная энергия связи). Согласно уравнению Эйн-
штейна ядерная энергия определяется уравнением Е= \тс\ в котором Д/и представля-
ет собой разницу между массой ядра и массой составляющих его протонов и нейтро-
нов. Дефект массы, используемый в масс-спектрометрии, связан с ядерной энергией,
но определяется как разность между точной массой атома и ближайшего целого числа
(на основе углеродной шкалы массы, 12С= 12,000000 Да) (Blaum, 2006).
Масса молекулы определенного изотопного состава может быть вычислена пу-
тем простого сложения массы входящих в нее атомов. Поскольку каждый атом имеет
уникальную массу с соответствующим дефектом массы, молекула также будет иметь
уникальную точную массу и уникальный дефект массы (Веупоп and Williams, 1963). Та-
ким образом, если измерить массу иона с высоким разрешением и высокой точностью,
можно надежно определить его элементный состав. В табл. 18.2 приведены некоторые
из наиболее часто встречаемых дублетов, определяющих разницу между изобарными
соединениями, а также соответствующая разница в массах (Д/и) и минимальная разре-
шающая способность анализатора, необходимая для их разделения.
18.5. Степень ненасыщенности (DBE) и Z-число
Определение элементного состава соединения является первым шагом в установле-
ние его структуры. Элементный состав (CHJWS^, полученный с помощью масс-
спектрометрии высокого разрешения, а также параметры регистрации спектра (метод
ионизации, полярность детектируемых ионов и т. д.) могут дать представление о хими-
ческой природе вещества. Разница между числом атомов водорода в исследуемой моле-
куле и ее полностью насыщенным ациклическим аналогом (CH2c+2+/NwOoS5) называется
«степенью ненасыщенности». Ее значение определяется количеством двойных и трой-
ных (нехарактерно для компонентов нефти) связей, циклов и ароматических колец.
Масс-спектрометристы и нефтехимики используют понятие «степень ненасы-
щенности», но рассчитывают и обозначают его по-разному. Нефтехимики обычно ис-
528 Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
пользуют Z-число, определяемое по элементному составу молекулы (СсН2с+_Х), в кото-
рой X — гетероатомы (N, О, S). Z-число можно легко вычислить, зная количество атомов
водорода и углерода в молекуле:
Z=h — 2c. (18.5)
Масс-спектрометристы предпочитают использовать степень ненасыщенности.
В англоязычной литературе этот термин также называется эквивалентом двойных свя-
зей (double bond equivalents) и обозначается DBE1. Мы будем использовать такое же обо-
значение. Значение DBE (целое или дробное) позволяет определить, является ли ион
четно- или нечетноэлектронным (McLafferty and Turecek, 1993). Для молекулы состава
CcH/NwOoSs DBE рассчитывается по уравнению 18.6.
h п
DBE(CcH„N„O„Ss) = C--+-+l (18.6)
Мы будем использовать DBE, потому что для насыщенных ациклических углево-
дородов (отсутствие колец) оно равно 0, тогда как Z-число для этих соединений будет
равно —2. В любом случае DBE и Z-число взаимосвязаны и легко могут быть вычислены
исходя из элементного состава, определенного с помощью ИЦРПФ. Уравнение 18.7 по-
зволяет вычислить Z-число, если DBE известно.
Z= —2(DBE) + л + 2
(18.7)
18.6. Масса в шкале Кендрика и дефект масс
Кендрика
В 1963 году Эдвард Кендрик, сотрудник компании «Эссо» (дочернее подразделение Экс-
он (Exxon)), предложил шкалу масс на основе 12СН2 = 14,00000 (вместо 14,01565 Да, как
в углеродной шкале, 12С = 12,00000) для упрощения определения элементного состава
изданных масс-спектрометрии высокого разрешения (Kendrick, 1963). В шкале Кендри-
ка все гомологи (соединения, отличающиеся на группу СН2) имеют одинаковый дефект
массы, что упрощает сортировку соединений в гомологические серии.
Ш кала Кендрика стала активно использоваться (Hsu et al., 1992; Hughey et al., 2001)
в петролеомике, когда стало возможным определять элементный состав компонентов
нефти с помощью масс-спектрометрии высокого разрешения. На рис. 18.9 представлены
два сегмента ИЦРФП спектра сырой нефти. Верхний сегмент отображает серию пиков,
отделенных друг от друга на 2,01565 Да (т.е. массу двух атомов водорода), для молекул,
принадлежащих одному гетероатомному классу и имеющих одинаковое число атомов
углерода, но разные величины DBE. Нижний сегмент демонстрирует гомологическую
серию ионов, принадлежащих к одному гетероатомному классу и имеющих одинаковое
значение DBE, но разное число атомов углерода. Ионы этой серии отличаются по соста-
ву на группы, кратные СН2, что соответствует массе 14,01565 Да. В этом случае преоб-
Подробнее о расчетах степени ненасыщенности см. (Лебедев, 2003).
18.6. Масса в шкале Кендрика и дефект масс Кендрика 529
Рис. 18.9. Часто встречаемые серии ионов в ИЦРФП-спектре нефти. Наличие цикла или
двойной связи снижает массу соединения на 2,01565 Да (вверху), а добавление
группы СН2 увеличивает массу на 14,011565 Да (внизу). Идентификация таких
интервалов в масс-спектре имеет важное значение для правильного установле-
ния элементного состава
разования масс-спектра из шкалы ИЮПАК (12С = 12,0000 Да) в шкалу Кендрика через
уравнение 18.8 особенно удобно:
Масса Кендрика = Масса ИЮПАК х (14,00000/14,01565). (18.8)
Шкала Кендрика конвертирует массу СН2-группы из 14,01565 точно в 14,00000. Та-
ким образом, последовательные члены гомологической серии (т.е. соединения, принад-
лежащие к одному гетероатомному классу с одинаковым DBE) будут отличаться в шкале
масс Кендрика на 14,00000 и могут быть легко опознаны по одинаковому дефекту масс
Кендрика (ДМК):
ДМК = НМК - масса Кендрика, (18.9)
где НМК — номинальная масса Кендрика, округленное значение массы Кендрика .
После того как химическая формула определена для нескольких гомологов с низкой
массой (<400 Да), элементный состав может быть определен для всех остальных соеди-
нений данной гомологической серии. Таким образом, возможно определение элемент-
ной формулы для более тяжелых соединений (>500 Да), для которых прямое измерение
исключительно на основе точного измерения массы имело бы большую неопределен-
ность (Hughey et al., 2001; Kim et al., 2006). Вышеупомянутый подход доказал свою реша-
ющую роль в идентификации компонентов сложных смесей.
530 Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
Масса и дефект масс Кендрика, кроме значительного упрощения определения эле-
ментного состава, также удобны для визуализации данных, потому что разные клас-
сы соединений (углеводороды, пептиды, углеводы и т.д.) имеют разную массу и дефект
масс Кендрика.
18.7. Визуализация данных: график масс Кендрика,
диаграммы ван Кревелена, распределение
гетероатомных классов соединений, график
зависимости DBE от числа атомов углерода
18.7.1. График масс Кендрика
С помощью МС-ИЦРФП в сложных образцах (нефть, биотопливо и т.д.) надежно уста-
навливаются тысячи химических формул. Это приводит к определенным трудностям
при представлении данных в компактной форме для их адекватной оценки. График
масс Кендрика (зависимость дефекта масс Кендрика от номинальной массы Кендрика)
дает возможность визуализации масс-спектрометрических данных посредством упоря-
дочения всех пиков в масс-спектре по дефекту масс и молекулярной массе.
На рис. 18.10 представлен график масс Кендрика для соединений северо-
американской нефти, принадлежащих к гетероатом ному О2 классу (объединяет все
соединения, имеющие в своем составе помимо углерода и водорода только два атома
кислорода). Таким образом, соединения, принадлежащие одному классу и имеющие
300
о 250
х
СЗ
200
X
о
8 150
л
S
е юо
о
50
Североамериканская нефть: Отсоединения
0
200 300 400 500 600 700 800
Номинальная масса Кендрика
Рис. 18.10. График зависимости дефекта масс Кендрика от номинальной массы Кендрика
для соединений О2-класса, детектируемых в виде отрицательных ионов мето-
дом МС-ИЦРФП при электрораспылении североамериканской нефти. Каждое
увеличение DBE сдвигает данные вверх по вертикали, в то время как введение
дополнительный СН2-группы приводит к сдвигу позиции по горизонтали
18.7. Визуализация данных: график масс Кендрика, диаграммы ван Кревелена,
распределение гетероатомных классов соединений, график зависимости DBE
от числа атомов углерода
одинаковое значение DBE, но разное количество СН2-групп, будут находиться на одной
горизонтальной линии. Однако соединения с разным DBE располагаются на разных
горизонтальных линиях, разделенных 0,013 единиц (разница дефекта массы Кендрика
для двух атомов водорода). Красные линии с обозначением «1О2» и «18 О2» соответствуют
соединениям с DBE = 1 и 18.
График масс Кендрика может быть более информативным, если каждая химиче-
ская формула будет отображаться в цвете, соответствующем относительному содержа-
нию соединений. Масс-спектр, тем самым, преобразуется в изображение. На рис. 18.11
представлен цветной график масс Кендрика для всех соединений, регистрируемых в се-
вероамериканской нефти с помощью МС-ИЦРФП. Такое отображение позволяет уви-
деть весь состав, включая относительную интенсивность компонентов, на одном рисун-
ке. Так как вертикальный сдвиг вверх соответствует повышению DBE (и, следовательно,
ароматичности), различия в степени ароматичности исследуемых образцов могут быть
быстро визуализированы.
График масс Кендрика предлагает ряд других преимуществ. Во-первых, «выброс»
данных (например шумы, помехи или нежелательные примеси) легко распознается,
если он выходит за рамки нормальных значений на графике. Во-вторых, если для тяже-
лых соединений невозможно определить химический состав посредством точного из-
мерения массы, то установление состава может быть проведено на основе выявленных
гомологических рядов. Другими словами, для тяжелых компонентов можно установить
массу посредством экстраполяции элементного состава от более легких членов гомоло-
Номинальная масса Кендрика
Отн. интенсивность (% общая)
Рис. 18.11. Цветной график масс Кендрика для всех компонентов североамериканской
нефти, зарегистрированных в виде отрицательных ионов методом МС-ИЦРФП
при электрораспылении
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
гических серий. Тем не менее графики масс Кендрика неспособны полностью обеспе-
чить полноту визуализации для всех классов соединений сразу. Для этого используются
диаграммы ван Кревелена.
18.7.2. Диаграммы ван Кревелена
Еще одним средством для графического отображения всех соединений в образце яв-
ляются диаграммы ван Кревелена. Диаграммы были впервые представлены в 1950 году
(Van Krevelen, 1950) в качестве средства для анализа общего состава угля, например мо-
лярное соотношение водорода и углерода (Н/С) в зависимости от соотношения кисло-
рода к углероду (О/С). Даже в 1950 году было очевидно, что образцы различного проис-
хождения можно отличить по расположению данных на таких графиках. Применение
МС-ИЦРФП для анализа сложных органических смесей привело к расширению идей
ван Кревелена для одновременного отображения всех брутто-формул регистрируемых
соединений. Изначально диаграммы ван Кревелена использовались для визуализации
данных анализа растворенного органического вещества в сточных водах, а затем нефти
(Kim etal., 2005; Wu etal., 2004c; Wu etal., 2005) и угля (Wu etal., 2004c; Wu etal., 2004b). Ди-
аграммы предоставляют простой графический способ демонстрации композиционных
отличий между образцами различной природы (уголь-нефть), географического проис-
хождения и переработки (Wu et aL, 2004с). Они также использовались для определения
биодеградации сырой нефти (Rodgers et al., 2005).
Отн. интенсивность (% общая)
Рис. 18.12. Диаграмма ван Кревелена: Н/С к О/С для Ол.-классов на основе данных МС-
ИЦРФП с регистрацией отрицательных ионов при электрораспылении образ-
ца североамериканской нефти
18.7. Визуализация данных: график масс Кендрика, диаграммы ван Кревелена,
распределение гетероатомных классов соединений, график зависимости DBE
от числа атомов углерода
SD-график ван Кревелена для нефти и угля
Рис. 18.13. Диаграммы ван Кревелена для N-, N0-, ЬЮ2-классов в образцах сырой нефти
и угольного экстракта (Wu etaL, 2004с)
Диаграммы ван Кревелена могут иметь разное обозначение осей (Н/С — О/С, или
N/С, или S/С) в зависимости от гетероатомов (О, N и S), представляющих интерес. Каж-
дая точка данных может быть окрашена для отражения относительной интенсивности
соответствующих соединений. Таким образом, с помощью диаграммы возможно гра-
фически разделять все гетероатомные соединения, а также отображать степень нена-
сыщенности и гомологию для каждого класса.
На рис. 18.12 представлена диаграмма ван Кревелена для органических соединений,
содержащих только атомы кислорода (Оу-классы) в североамериканской нефти. Все со-
единения с разным содержанием атомов О четко разделены по горизонтали (ось О/С),
а различия в DBE демонстрируются по вертикали (ось Н/С). При росте соотношения
Н/С степень ненасыщенности (т.е. DBE) уменьшается. Гомологи (соединения с тем же
DBE и числом гетероатомов, но разным количеством СН2-групп) отображаются диаго-
нальной зависимостью.
Еще один вариант диаграммы ван Кревелена демонстрирует графически различие
между образцами разного происхождения. Здесь оси диаграммы обозначаются как Н/С,
О/С и N/C (Wu et aL, 2004с). С ее помощью легко увидеть различия между составом сы-
рой нефти и угольного экстракта (рис. 18.13). Компоненты последнего характеризуются
меньшими значениями по оси Н/С, что и свидетельствует об их более ароматическом
характере (увеличение степени ненасыщенности и снижение доли гомологов), по срав-
нению с асфальтенами сырой нефти (Rodgers etaL, 2003).
18.7.3. График распределения гетероатомных классов
Определение элементного состава соединений обеспечивает три типа химической ин-
формации: принадлежность к гетероатомному классу (N/fOoSjt), степени ненасыщенно-
сти, а также определяет положение в гомологическом ряду (количество атомов углерода).
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
Гетероатомный класс объединяет все соединения с одинаковым набором гетероатомов
в их химической формуле. Например, углеводороды характеризуются отсутствием гете-
роатомов и формируют отдельный класс. Все соединения, имеющие в своем составе по-
мимо углерода и водорода один атом азота, образуют N-класс. Соединения, содержащие
два атома кислорода, — О2-класс и т.д.
Рис. 18.14 иллюстрирует относительное распределение гетероатомных классов, де-
тектируемых в североамериканской нефти. Вставка на рис. 18.14 демонстрирует относи-
тельное распределение соединений N-класса по степени ненасыщенности. На рис. 18.15
представлено относительное распределение соединений N-класса с DBE = 12 по числу
атомов углерода в их составе.
18.7.4. График зависимости DBE от числа атомов углерода
Среди различных возможных графических изображений состава сложных органических
смесей наиболее удобными являются графики для определенного гетероатомного клас-
са, отображающие зависимость DBE от количества атомов углерода. На рис. 18.16 показа-
но изображение двух наиболее распространенных классов (N, и О2), детектируемых в об-
разце вакуумного дистиллята канадского битума. Большинство компонентов ^-класса
лежат выше DBE = 9 (соответствует карбазолу), что свидетельствует об ароматическом
характере этих соединений. Напротив, соединения, которые составляют О2-класс (пред-
положительно карбоновые кислоты) в основном своем большинстве находятся ниже
DBE = 5 (бензойная кислота) и, скорее всего, соответствуют нафтеновым кислотам.
Аналогичные рисунки для N(- и О2-классов североамериканской нефти демонстри-
руют значительно более широкое распределение компонентов как по DBE (ароматич-
Рис. 18.14. Распределение гетероатомных классов, детектируемых методом МС-ИЦРФП
с регистрацией отрицательных ионов в образце североамериканской нефти
при электрораспылении. Вставка демонстрирует распределение соединений
N-класса по DBE (степени ненасыщенности)
18.7. Визуализация данных: график масс Кендрика, диаграммы ван Кревелена,
распределение гетероатомных классов соединений, график зависимости DBE
от числа атомов углерода
Рис.18.15. Относительное распределение соединений в образце североамериканской
нефти (N-класс, DBE = 12) по числу атомов углерода в их составе
ности), так и по числу атомов углерода (рис. 18.17). Это объясняется усеченным составом
дистиллята, который представлен компонентами, кипящими в узком диапазоне тем-
ператур. В целом графики зависимости DBE от количества атомов углерода оказались
чрезвычайно полезными для характеризации нефти, дистиллятов, серосодержащих по-
лициклических ароматических соединений {Mckenna et aL, 2010а; Mckenna et al., 2010b),
растворимых в воде компонентов {Stanford et al., 2007а), нефте-водяной эмульсии {Нет-
Североамериканская нефть
О2-класс
DBE= 5 (бензойная кислота)
20 30 40 50
Углеродное число
Отн. интенсивность (% общая)
Рис. 18.16. Графики зависимости DBE от количества атомов углерода для N(- и О2-классов
в вакуумном дистилляте канадского битума. Метод МС-ИЦРФП с регистра-
цией отрицательных ионов при электрораспылении
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
Рис. 18.17. Графики зависимости DBE от количества атомов углерода для Ц- и О2-классов
в североамериканской нефти. Метод МС-ИЦРФП с регистрацией отрицатель-
ных ионов при электрораспылении
mingsen et aL, 2006; Stanford et aL, 2007b; Stanford et aL, 2007c), нафтеновых кислот (Barrow
etaL, 2009; Headley etal., 2007), отложений в теплообменнике (Mapolelo et aL, 2009) и т.д.
18.8. Методы ионизации для МС-ИЦРФП-анализа
сложных смесей
Несмотря на существенный прогресс в МС-ИЦРФП и развитие многочисленных мето-
дов ионизации, количественный анализ остается одной из основных проблем в петро-
леомике. Ни один из известных методов ионизации не генерирует ионы с одинаковой
эффективностью для соединений различной природы.
Различные методы ионизации применялись в петролеомике одновременно с их
появлением. Электронная ионизация (ЭИ), химическая ионизация (ХИ) и полевая де-
сорбция (ПД) были первыми методами, использованными на ранних стадиях петро-
леомики. В настоящее время эти методы практически полностью заменены методами
ионизации при атмосферном давлении: электрораспылением (ЭРИ) и фотоионизаци-
ей при атмосферном давлении (ФИАД). ЭРИ обычно применяется для ионизации по-
лярных соединений и в зависимости от полярности ионного источника, как правило,
генерирует протонированные (регистрация положительных ионов) или депротониро-
ванные молекулы (регистрация отрицательных ионов). Недостатком электрораспы-
ления является неспособность ионизировать менее полярные соединения (например,
ароматические углеводороды, тиофены, фураны и т.д.). Для их анализа обычно исполь-
зуется ФИАД. Сверхвысокое разрешение особенно важно в случае ФИАД, потому что
18.9. Применение МС-ИЦРФП для решения экологических задач
m/z
Рис. 18.18. Масс-спектр ФИАД нафто[а]пирена с разделением двух близко расположен-
ных пиков, различающихся на 0,0045 Да (|3С и 12СН). По материалам (Rodgers
and Marshall, 2007)
процесс ионизации сопровождается одновременным образованием катион-радикалов
(М+’) и протонированных молекул ([М+Н]+). Так, для одного соединения в процессе ио-
низации могут образоваться два изобарных пика, отличающиеся по массе на 0,0045 Да
(13С и 12СН) (рис. 18.18). В действительности еще более узкий дублет масс в 0,0011 Да (С4
и 13CH3S) наблюдается в ФИ АД-спектрах. В настоящее время ни один другой тип масс-
анализатора, за исключением ИЦРФП, не способен разделить настолько близко рас-
положенные изобары.
Методы, в которых ионизация происходит в нормальных условиях (например ПАРВ
(Cody et al.,2005), ДЭРИ (Takats et al., 2004), EASI (Costa and Cooks, 2007) и др.) представ-
ляют еще одну группу «мягких» ионизационных методов, использование которых суще-
ственно упрощает пробоподготовку, а иногда и полностью ее исключает (глава 7). Такие
недеструктивные методы находят большой спрос для экспресс-анализа широкого спек-
тра соединений, а их сочетание с МС-ИЦРФП открывает возможность для ускоренного
анализа сложных органических смесей (Abdelnuret al., 2008; Albericietal., 2010; Coriloetal.,
2010; Eberlin et al., 2009; Rummel et al., 2010).
18.9. Применение МС-ИЦРФП для решения
экологических задач
Хроматографические методы (в первую очередь газовая хроматография) в сочетании
с масс-спектрометрией являются наиболее распространенным подходом для анализа
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
объектов окружающей среды. Однако ГХ/МС неспособна обеспечить детектирование
всех компонентов сложных органических смесей (например нефти), и ее использование
ограничивается анализом сравнительно легколетучих, низкомолекулярных соединений.
Тем не менее объединение газовой хроматографии со сверхвысоким разрешением МС-
ИЦРФП казалось логичным и впервые было осуществлено в 1980 году {Ledford et al., 1980).
Такая конфигурация открыла возможность анализа сложных смесей летучих органи-
ческих соединений с уникальной возможностью определения элементного состава для
всех образующихся ионов. ГХ/МС-ИЦРФП был использован для анализа видов топлива
{Szulejko and Solouki, 2002), диоксинов {Taguchi et al., 2010) и загрязняющих веществ в пи-
тьевой воде {Heffner et al., 2007). Однако, несмотря на указанные возможности ГХ/МС-
ИЦРФП, технические и эксплуатационные ограничения, стоимость, а также низкая
чувствительность стали препятствием для его широкого аналитического применения.
Напротив, ЖХ/МС-ИЦРФП позволяет работать с полярными нелетучими соединения-
ми, но страдает от плохого хроматографического разделения компонентов сложной смеси.
Благодаря сверхвысокому разрешению МС-ИЦРФП дает возможность анализи-
ровать различные соединения в сложной матрице без онлайн-хроматографического
разделения. В сочетании с различными методами ионизации (низковольтной ИЭ {Rod-
gers etal., 1999), полевой десорбцией {Stanford et al., 2007а), ЭРИ {Headley et al., 2010; Head-
ley et al., 2009a; Headley et al., 2009b; Hughey et al., 2008; Kim et al., 2005; Kujawinski, 2002)
и лазерной десорбцией/ионизацией (ЛДИ) {Aubriet and Carre, 2010)) он уже был успешно
использован для решения многих экологических задач, некоторые из которых обсужда-
ются в данном разделе.
Ископаемые виды топлива являются основным источником энергии на нашей пла-
нете, а годовое потребление сырой нефти в мире превышает 30 млрд баррелей (около
5 млрд м3 или 5 км3). Такой объем трудно себе представить, для сравнения средний годо-
вой расход воды в Москве-реке составляет только около 3,5 млрд м3.
В результате разливы углеводородов, а также продукты их неполного сгорания
являются основной причиной загрязнения почвы, вод (подземных и поверхностных)
и атмосферы. Так как нефть представляет собой очень сложную смесь органических
соединений, анализ загрязнения нефтяного происхождения является сложной зада-
чей для любого известного аналитического метода. В отличие от ГХ/МС (анализ низ-
комолекулярных летучих органических соединений: алканов, циклоалканов, моно-,
диароматических углеводородов и т.д.); МС-ИЦРФП (главным образом в сочетании
с ЭРИ и ФИАД) нацелен на анализ нелетучих гетероатомных (N, О, S) компонентов.
Из-за наличия полярных групп (-ОН, -СООН, >NH, -SO3H и др.) эти соединения об-
ладают поверхностно-активными свойствами, что приводит к их частичной раство-
римости в воде при контакте нефтяной и водной фаз. Такой контакт может произойти
случайно из-за утечки нефти и разливов или согласно технологии процесса (например
добыча нефти из нефтеносных песков путем экстракции). Все это приводит к загряз-
нению почвы, поверхностных и подземных вод. Природа загрязняющих веществ и их
судьба в окружающей среде были предметом детальных исследований с помощью раз-
личных аналитических методов, включая МС-ИЦРФП {Brandalet al., 2006; Headley et al.,
2010; Headley et al., 2009a; Headley et al., 2009b; Headley et al., 2007; Hemmingsen et al., 2006;
Hughey et al., 2007; Hughey et al., 2008; Kujawinski, 2002; Stanford et al., 2007a; Stanford et al.,
2007b; Stanford et al., 2007c).
18.9. Применение МС-ИЦРФП для решения экологических задач 539
Под воздействием окружающей среды сырая нефть подвергается абиотическим
и биотическим изменениям. Абиотические изменения включают потерю части не-
фтяных компонентов в результате испарения или окисления, тогда как биотические
связаны с микробиологической активностью. Относительная восприимчивость насы-
щенных и ароматических углеводородов к микробиологическим превращениям хорошо
известна, хотя определенные различия наблюдаются среди гомологов и между изомера-
ми. В целом нормальные алканы катаболизируются с максимальной скоростью. Затем
следуют разветвленные алканы, моноциклические насыщенные и моноароматические
углеводороды, полициклические нафтеновые и ароматические углеводороды и, нако-
нец, гетероатомные соединения (Kim et aL, 2005). Некоторые гетероатомные соедине-
ния, например ванадил порфирины, по-видимому, остаются неизменными даже в самой
биодеградированной нефти. Другие, такие как карбазолы, хинолины, дибензотиофены
и большие геомакромолекулы, подвергаются биоминерализации в различной степени.
Нафтеновые кислоты являются одним из наиболее изученных гетероатомных клас-
сов нефти. Для их анализа использовались различные методы ионизации: химическая
ионизация (Dzidic et al., 1988; Hsu et al., 2000), полевая десорбция (Stanford et al., 2007b),
бомбардировка быстрыми атомами (Fan, 1991; Hsu et al., 2000; Laredo et al., 2004) и хими-
ческая ионизация при атмосферном давлении (Hsu et al., 2000). При регистрации отри-
цательных ионов высокую эффективность для изучения этого класса соединений пока-
зало электрораспыление (Brandal et al., 2006; Headley et al., 2009a; Hemmingsen et aL, 2006;
Stanford et aL, 2007b; Headley et aL, 2007; Hughey et aL, 2008).
Нафтеновыми кислотами в органической химии называются карбоновые кислоты,
в которых одна или несколько карбоксильных групп соединены (прямо или через ал-
кильную цепь) с циклоалкановой частью (нафтены), состоящей из одного или несколь-
ких колец. Однако в нефтяной промышленности нафтеновые кислоты включают в себя
все карбоновые кислоты (циклические, ациклические и ароматические), найденные
в сырой нефти. Полярные группы (-СООН) и неполярные алкильные (циклоалкил,
арил) заместители определяют поверхностно-активные свойства этих соединений, что
приводит к особенно значимым загрязнениям контактирующих природных поверх-
ностей (почва, вода и т.д.). Карбоновые кислоты, помимо их природного содержания
в нефти, образуются также в результате биодеградации и окисления других нефтяных
компонентов, что, кстати, служит показателем степени деградации нефти в загрязнен-
ных образцах (Hughey et aL, 2008; Kim et aL, 2005).
Компоненты нефтяного загрязнения могут подниматься по пищевой цепи и нака-
пливаться в тканях живых организмов. Нафтеновые кислоты вызывают особую озабо-
ченность из-за их высокой растворимости в воде и высокой токсичности для различных
водных организмов, включая рыб.
Продукты микробной трансформации кислых и нейтральных гетероатомных со-
единений могут быть установлены с помощью МС-ИЦРФП (Kim etal., 2005). Некоторые
компоненты нефти и продукты их микробного разложения используются для оценки
степени биодеградации нефти в окружающей среде. Карбоновые кислоты являются
наиболее удобным объектом для этих целей. Оценка биодеградации проводится на ос-
нове сравнения относительной интенсивности О2-класса (кислоты) к О^классу (фено-
лы, спирты), а также соотношению ациклических карбоновых кислот (А) к нафтено-
вым (С) — А/С. Относительное содержание компонентов О^класса уменьшается при
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
биодеградации и из-за более предпочтительной деградации легких, более насыщенных
соединений, приводит к сдвигу DBE и числа атомов углерода в молекулах в сторону
больших значений. Ациклические карбоновые кислоты разлагаются быстрее, чем ци-
клические аналоги и, следовательно, А/С-соотношение уменьшается, когда степень
биодеградации увеличивается (Hughey et al., 2008).
Некоторые растения (водоросли Typha latifola) могут адаптироваться к загрязнению
и способны уменьшать концентрацию нафтеновых кислот в окружающей среде. Из-
за сложного характера матрицы этот процесс сложно распознать при низком разреше-
нии по массе. Некоторые компоненты с DBE = 1 (природныежирные кислоты растений)
при относительно высокой концентрации перекрываются в масс-спектре низкого раз-
решения с сигналом нафтеновых кислот.
Лабораторные исследования и результаты МС-ИЦРФП показали участие растений
в диссипации нафтеновых кислот (Headley et aL, 2009а). На рис. 18.19 показаны графики
(DBE к числу атомов углерода) для соединений О2-класса. Разница между абиотическим
контролем (биологическое разложение исключено) и растительной системой (биологи-
ческое разложение возможно), отражает изменения, связанные с деятельностью расте-
ний. Графики для пятого и двадцатого дня показывают, что растения эффективно по-
глощают нафтеновые кислоты.
Анализ воздуха и идентификация ксенобиотиков в атмосфере являются еще од-
ной областью применения МС-ИЦРФП. Органические соединения в атмосфере суще-
Абиотический
контроль — 60 мг/л
О2 класс
Отн. интенсивность (% общая)
Растительная система — Углеродное число
небольшая доза — 30 мг/л
Рис. 18.19. Графики зависимости DBE от числа атомов углерода для О2-класса: (а) абиоти-
ческий контроль, (б) растительная система (Headley etal., 2009а)
Литература
ствуют в виде аэрозолей (взвешенных мелких твердых частиц или капель жидкости).
При растворении в капельках воды они образуют туман или висящие облака. Исследо-
вание атмосферы, загрязненной в результате бытовой, транспортной и промышленной
деятельности (Mazzoleni et al., 2010; Wozniak et al., 2008), показало, что большинство об-
наруженных компонентов содержат гетероатомы (N, О, S) и имеют молекулярную мас-
су до 400 Да. Они характеризуются величинами DBE = 3—11 и широким диапазоном
числа углеродных атомов (3—26). Среди обнаруженных соединений были карбоновые
кислоты, ароматические соединения, фенолы, нитрофенолы, органические нитраты,
алкилбензолсульфонаты, органические сульфаты и окисленные монотерпены (напри-
мер сс-и Р-пинены) (Mazzoleni et al., 2010).
Помимо прикладных экологических исследований МС-ИЦРФП была использова-
на для решения задач судебно-медицинской экспертизы. С помощью ИЦРФП-спектров
электронной ионизации низкой энергии были проанализированы образцы с места
взрыва, в которых были идентифицированы взрывчатые вещества, а также их активные
и неактивные ингредиенты (Wu etal., 2002). Примером применения МС-ИЦРФП в кри-
миналистических целях является идентификация средств, используемых для поджога.
Масс-спектрометрия высокого разрешения обеспечивает уникальный спектр («отпеча-
ток пальца») для каждого нефтепродукта и делает возможным однозначную идентифи-
кацию использованной горючей жидкости, несмотря на сложность матрицы, присут-
ствие многочисленных компонентов и продуктов пиролиза (Rodgers et al., 2001).
МС-ИЦРФП использовалась для прямого анализа пищевых продуктов (вина и рас-
тительного масла) без предварительного разделения или очистки. При анализе вин
было идентифицировано более 30 природных флавоноидов. Существенные различия
в масс-спектрах ИЦРФП вин позволяют проводить их классификацию (Cooperand Mar-
shall, 2001). МС-ИЦРФП-анализ электрораспылением с регистрацией отрицательных
ионов образцов растительных масел (соевое, рапсовое, оливковое) обнаружил более
3000 различных брутто-формул. Еще 2000 элементных формул было идентифициро-
вано в спектре положительных ионов. Такой детальный анализ, основанный на детек-
тировании тысяч химических компонентов, позволяет сделать выводы о растительном
происхождении масла и определить фальсификацию (Wuetal., 2004а).
Литература
[1] . Abdelnur P.V., Eberlin L.S., De Sa G.F., De Souza V., and Eberlin M.N. (2008). «Single-
shot biodiesel analysis: Nearly instantaneous typification and quality control solely by ambi-
ent mass spectrometry». Analytical Chemistry. 80, 7882—7886.
[2] . Alberici R. M., Simas R.C., De Souza V., De Sa G.F., Daroda R.J., and Eberlin M.N.
(2010). «Analysis of fuels via easy ambient sonic-spray ionization mass spectrometry». Ana-
lytica Chimica Acta. 659, 15—22.
[3] . Aubriet F., and Carre V. (2010). «Potential of laser mass spectrometry for the analysis of en-
vironmental dust particles-A review». Analytica Chimica Acta. 659, 34—54.
[4] . Barrow M.P., Headley J.V., Peru К. M., and Derrick P.J. (2009). «Data Visualization for
the Characterization of Naphthenic Acids within Petroleum Samples». Energy & Fuels. 23,
2592-2599.
542 Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
[5] . Beynon J. Н., and Williams А. Е. (1963). Mass and Abundance Tables for Use in Mass Spec-
trometry, Amsterdam, Elsevier.
[6] . Blaum K. (2006). «High-accuracy mass spectrometry with stored ions». Physics Reports-
Review Section of Physics Letters. 425, 1—78.
[7] . Brandal O., Hanneseth A. M., Hemmingsen P.V., Sjoblom J., Kim S., Rodgers R. P., and
Marshall A.G. (2006). «Isolation and characterization of naphthenic acids from a metal
naphthenate deposit: Molecular properties at oil-water and air-water interfaces». Journal of
Dispersion Science and Technology. 27, 295—305.
[8] . Cody R. B., Laramee J. A., and Durst H. D. (2005). «Versatile new ion source for the analysis
of materials in open air under ambient conditions». Analytical Chemistry. 77, 2297—2302.
[9] . Comissarow M.B., and Marshall A.G. (1974). «Fourier Transform Ion Cyclotron Reso-
nance Spectroscopy». Chemical Physics Letters. 25, 282—283.
[10] . Cooper H.J., and Marshall A.G. (2001). «Electrospray ionization Fourier transform mass
spectrometric analysis of wine». Journal of Agricultural and Food Chemistry. 49, 5710—5718.
[11] . Corilo Y. E., Vaz B.G., Simas R.C., Nascimento H.D. L., Klitzke C. F., Pereira R.C.
L., Bastos W. L., Neto E. V. S., Rodgers R. P., and Eberlin M.N. (2010). «Petroleomics by
EASI(+/—) FT-ICR MS». Analytical Chemistry. 82, 3990—3996.
[12] . Costa A. B., and Cooks R.G. (2007). «Simulation of atmospheric transport and droplet-
thin film collisions in desorption electrospray ionization». Chemical Communications.
3915-3917.
[13] . De Laeter J. R., Bohlke J. K., De Bievre P., Hidaka H., Peiser H.S., Rosman K.J. R., and
Taylor P. D. P. (2003). «Atomic weights of the elements: Review 2000 — (IUPAC technical
report)». Pure and Applied Chemistry. 75, 683—800.
[14] . Diddams S.A., Bergquist J. C., Jefferts S. R., and Oates C.W. (2004). «Standards of time
and frequency at the outset of the 21st century». Science. 306, 1318—1324.
[15] . Dzidic I., Somerville A.C., Raia J. C., and Hart H.V. (1988). «Determination of Naph-
thenic Acids in California Crudes and Refinery Wastewaters by Fluoride-Ion Chemical
Ionization Mass-Spectrometry». Analytical Chemistry. 60, 1318—1323.
[16] . Eberlin L. S., Abdelnur P.V., Passero A., De SaG.F., Daroda R.J., De Souza V., and Eb-
erlin M.N. (2009). «Analysis of biodiesel and biodiesel-petrodiesel blends by high perfor-
mance thin layer chromatography combined with easy ambient sonic-spray ionization mass
spectrometry». Analyst. 134, 1652—1657.
[17] . Fan T. P. (1991). «Characterization of Naphthenic Acids in Petroleum by Fast-Atom-Bom-
bardment Mass-Spectrometry». Energy & Fuels. 5, 371—375.
[18] . Guan S. H., Marshall A. G., and Scheppele S. E. (1996). «Resolution and chemical formula
identification of aromatic hydrocarbons and aromatic compounds containing sulfur, ni-
trogen, or oxygen in petroleum distillates and refinery streams». Analytical Chemistry. 68,
46-71.
[19] . Headley J. V., Armstrong S. A., Peru К. M., Mikula R. J., Germida J. J., Mapolelo M. M.,
Rodgers R. P., and Marshall A. G. (2010). «Ultrahigh-resolution mass spectrometry of sim-
ulated runoff from treated oil sands mature fine tailings». Rapid Communications in Mass
Spectrometry. 24, 2400—2406.
[20] . Headley J.V., Peru К. M., Armstrong S.A., Han X. M., Martin J.W., Mapolelo M.M.,
Smith D. F., Rogers R. P., and Marshall A.G. (2009a). «Aquatic plant-derived changes in
Литература
oil sands naphthenic acid signatures determined by low-, high- and ultrahigh-resolution
mass spectrometry». Rapid Communications in Mass Spectrometry. 23, 515—522.
[21] . Headley J. V., Peru К. M., and Barrow M. P. (2009b). «Mass Spectrometric Characteriza-
tion of Naphthenic Acids in Environmental Samples: A Review». Mass Spectrometry Re-
views. 28, 121 — 134.
[22] . Headley J. V., Peru K.M., Barrow M.P., and Derrick P.J. (2007). «Characterization of
naphthenic acids from Athabasca oil sands using electrospray ionization: The significant
influence of solvents». Analytical Chemistry. 79, 6222—6229.
[23] . Heffner C., Silwal I., Peckenham J.M., and Solouki T. (2007). «Emerging technologies
for identification of disinfection byproducts: GC/FT-ICR MS characterization of solvent
artifacts». Environmental Science & Technology. 41, 5419—5425.
[24] . Hemmingsen P.V., Kim S., Pettersen H.E., Rodgers R. P., Sjoblom J., and Marshall A. G.
(2006). «Structural characterization and interfacial behavior of acidic compounds extract-
ed from a North Sea oil». Energy & Fuels. 20, 1980—1987.
[25] . Hsu C.S., Dechert G.J., Robbins W.K., and Fukuda E.K. (2000). «Naphthenic acids in
crude oils characterized by mass spectrometry». Energy & Fuels. 14, 217—223.
[26] . Hsu C. S., Qian K. N., and Chen Y. N. C. (1992). «An Innovative Approach to Data-Analy-
sis in Hydrocarbon Characterization by Online Liquid-Chromatography Mass-Spectrom-
etry». Analytica Chimica Acta. 264, 79—89.
[27] . Hughey C. A., Galasso S. A., and Zumberge J. E. (2007). «Detailed compositional compari-
son of acidic NSO compounds in biodegraded reservoir and surface crude oils by negative
ion electrospray Fourier transform ion cyclotron resonance mass spectrometry». Fuel. 86,
758-768.
[28] . Hughey C.A., Hendrickson C. L., Rodgers R. P., Marshall A. G., and Qian K.N. (2001).
«Kendrick mass defect spectrum: A compact visual analysis for ultrahigh-resolution broad-
band mass spectra». Analytical Chemistry. 73, 4676—4681.
[29] . Hughey C. A., Minardi C.S., Galasso-Roth S.A., Paspalof G. B., Mapolelo M.M., Rodg-
ers R. P., Marshall A.G., and Ruderman D. L. (2008). «Naphthenic acids as indicators of
crude oil biodegradation in soil, based on semi-quantitative electrospray ionization Fou-
rier transform ion cyclotron resonance mass spectrometry». Rapid Communications in Mass
Spectrometry. 22, 3968—3976.
[30] . Kendrick E. (1963). «А Mass Scale Based on Ch2 = 14.0000 for High Resolution Mass
Spectrometry of Organic Compounds». Analytical Chemistry. 35, 2146—2154.
[31] . Kim S., Kramer R.W., and Hatcher P. G. (2003). «Graphical method for analysis of ultra-
high-resolution broadband mass spectra of natural organic matter, the van Krevelen dia-
gram». Analytical Chemistry. 75, 5336—5344.
[32] . Kim S., Rodgers R. P., and Marshall A. G. (2006). «Truly «exact» mass: Elemental compo-
sition can be determined uniquely from molecular mass measurement at similar to 0.1 mDa
accuracy for molecules up to similar to 500 Da». International Journal of Mass Spectrometry.
251,260-265.
[33] . Kim S., Stanford L. A., Rodgers R. P., Marshall A. G., Walters С. C., Qian K., Wenger L. M.,
and Mankiewicz P. (2005). «Microbial alteration of the acidic and neutral polar NSO com-
pounds revealed by Fourier transform ion cyclotron resonance mass spectrometry». Organic
Geochemistry. 36, 1117—1134.
544 Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
[34] . Kujawinski Е.В. (2002). «Electrospray ionization Fourier transform ion cyclotron reso-
nance mass spectrometry (ESI FT-ICR MS): characterization of complex environmental
mixtures». Environmental Forensics. 3, 207—216.
[35] . Laredo G. C., Lopez C. R., Alvarez R. E., Castillo J. J., and Cano J. L. (2004). «Identifica-
tion of naphthenic acids and other corrosivity-related characteristics in crude oil and vacu-
um gas oils from a Mexican refinery. Energy & Fuels. 18, 1687—1694.
[36] . Ledford E.B., Rempel D. L., and Gross M.L. (1984). «Space-Charge Effects in Fourier-
Transform Mass-Spectrometry — Mass Calibration. Analytical Chemistry. 56, 2744—2748.
[37] . Ledford E. B., White R. L., Ghaderi S., Wilkins C. L., and Gross M. L. (1980). «Coupling
of Capillary Gas-Chromatograph and Fourier-Transform Mass-Spectrometer. Analytical
Chemistry. 52, 2450-2451.
[38] . Mapolelo M. M., Stanford L.A., Rodgers R. P., Yen A.T., Debord J. D., Asomaning S., and
Marshall A. G. (2009). «Chemical Speciation of Calcium and Sodium Naphthenate Depos-
its by Electrospray Ionization FT-ICR Mass Spectrometry». Energy & Fuels. 23, 349—355.
[39] . Marshall A. G. (1996). «Ion cyclotron resonance and nuclear magnetic resonance spectros-
copies: Magnetic partners for elucidation of molecular structure and reactivity». Accounts of
Chemical Research. 29, 307—316.
[40] . Marshall A.G. (2000). «Milestones in Fourier transform ion cyclotron resonance mass
spectrometry technique development». International Journal of Mass Spectrometry. 200,
331-356.
[41] . Marshall A. G., Blakney G. T., Beu S. C., Hendrickson C. L., Mckenna A. M., Purcell J. M.,
Rodgers R. P., and Xian F. (2010). «Petroleomics: a test bed for ultra-high-resolution Fou-
rier transform ion cyclotron resonance mass spectrometry». European Journal of Mass Spec-
trometry. 16, 3 67—3 71.
[42] . Marshall A. G., and Hendrickson C. L. (2002). «Fourier transform ion cyclotron resonance
detection: principles and experimental configurations». International Journal of Mass Spec-
trometry. 215, 59—75.
[43] . Marshall A.G., and Hendrickson C. L. (2008). «High-Resolution Mass Spectrometers».
Annual Review of Analytical Chemistry. 1, 579—599.
[44] . Marshall A. G., Hendrickson C. L., and Jackson G. S. (1998). «Fourier transform ion cyclo-
tron resonance mass spectrometry: A primer». Mass Spectrometry Reviews. 17, 1—35.
[45] . Marshall A.G., and Rodgers R. P. (2004). «Petroleomics: The next grand challenge for
chemical analysis». Accounts of Chemical Research. 37, 53—59.
[46] . Marshall A.G., and Verdun F. R. (1990). Fourier Transforms in NMR, Optical, and Mass
Spectrometry: A User's Handbook, New York, Elsevier Science.
[47] . Mazzoleni L. R., Ehrmann B.M., Shen X.H., Marshall A.G., and Collett J. L. (2010).
«Water-Soluble Atmospheric Organic Matter in Fog: Exact Masses and Chemical Formula
Identification by Ultrahigh-Resolution Fourier Transform Ion Cyclotron Resonance Mass
Spectrometry». Environmental Science & Technology. 44, 3690—3697.
[48] . Mckenna A. M., Blakney G.T., Xian F., Glaser P. B., Rodgers R.P., and Marshall A.G.
(2010a). «Heavy Petroleum Composition. 2. Progression of the Boduszynski Model to the
Limit of Distillation by Ultrahigh-Resolution FT-ICR Mass Spectrometry». Energy & Fu-
els. 24, 2939-2946.
[49] . Mckenna A. M., Purcell J. M., Rodgers R. P., and Marshall A.G. (2010b). «Heavy Petro-
leum Composition. 1. Exhaustive Compositional Analysis of Athabasca Bitumen HVGO
Литература
Distillates by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry: A Defini-
tive Test of the Boduszynski Model». Energy & Fuels. 24, 2929—2938.
[50] . Mclafferty F. W., and Turecek F. (1993). Interpretation of Mass Spectra, Sausalito, CA, Uni-
versity Science Books.
[51] . Meyerson S. (1986). «Reminiscences of the Early Days of Mass-Spectrometry in the Petro-
leum-Industry». Organic Mass Spectrometry. 21, 197—208.
[52] . Purcell J. M., Rodgers R. P., Hendrickson C. L., and Marshall A.G. (2007). «Speciation
of nitrogen containing aromatics by atmospheric pressure photoionization or electrospray
ionization Fourier transform ion cyclotron resonance mass spectrometry». Journal of the
American Society for Mass Spectrometry. 18,1265—1273.
[53] . Rodgers R. P., Blumer E.N., Freitas M.A., and Marshall A.G. (1999). «Jet fuel chemical
composition, weathering, and identification as a contaminant at a remediation site, deter-
mined by Fourier transform ion cyclotron resonance mass spectrometry». Analytical Chem-
istry. 71,5171-5176.
[54] . Rodgers R.P., Blumer E.N., Freitas M.A., and Marshall A.G. (2001). «Compositional
analysis for identification of arson accelerants by electron ionization Fourier transform ion
cyclotron resonance high-resolution mass spectrometry». Journal of Forensic Sciences. 46,
268-279.
[55] . Rodgers R. P., Klein G. C., Wu Z., and Marshall A. G. (2003). «Environmental applications
of ESI FT-ICR mass spectrometry: The identification of polar N, S and О containing PAH
species in crude oil and coal extracts». Preprints of Symposia — American Chemical Society,
Division of Fuel Chemistry. 48, 758—759.
[56] . Rodgers R.P., and Marshall A.G. (2007). «Petroleomics: Advanced Characterization of
Petroleum-Derived Materials by Fourier Transform ion Cyclotron Resonance Mass Spec-
trometry (FT-ICR MS)». IN Mullins О.C., Sheu E.Y., Hammani A., and Marshall A.G.
(Eds.) Asphaltenes, Heavy oils, and Petroleomics. New York, Springer.
[57] . Rodgers R. P, Schaub T. M., and Marshall A.G. (2005). «Petroleomics: MS returns to its
roots». Analytical Chemistry. 77, 20a—27a.
[58] . Rummel J. L., Mckenna A. M., Marshall A. G., Eyler J. R., and Powell D. H. (2010). «The
coupling of direct analysis in real time ionization to Fourier transform ion cyclotron reso-
nance mass spectrometry for ultrahigh-resolution mass analysis». Rapid Communications in
Mass Spectrometry. 24, 784—790.
[59] . Shi S.D. H., Hendrickson C. L., and Marshall A.G. (1998). «Counting individual sulfur
atoms in a protein by ultrahigh-resolution Fourier transform ion cyclotron resonance mass
spectrometry: Experimental resolution of isotopic fine structure in proteins». Proceedings of
the National Academy of Sciences of the United States of America. 95, 11532—11537.
[60] . Stanford L. A., Kim S., Klein G. C., Smith D. F., Rodgers R. P, and Marshall A. G. (2007a).
«Identification of water-soluble heavy crude oil organic-acids, bases, and neutrals by elec-
trospray ionization and field desorption ionization Fourier transform ion cyclotron reso-
nance mass spectrometry». Environmental Science & Technology. 41, 2696—2702.
[61] . Stanford L.A., Rodgers R. P., Marshall A.G., Czarnecki J., and WuX.A. (2007b). «Com-
positional characterization of bitumen/water emulsion films by negative- and positive-ion
electrospray ionization and field desorption/ionization Fourier transform ion cyclotron
resonance mass spectrometry». Energy & Fuels. 21, 963—972.
&
Глава 18. ИЦРФП анализ сложных органических смесей. Петролеомика
[62] . Stanford L. A., Rodgers R. Р., Marshall A. G., Czarnecki J., Wu X. A., and Taylor S. (2007c).
«Detailed elemental compositions of emulsion interfacial material versus parent oil for nine
geographically distinct light, medium, and heavy crude oils, detected by negative- and pos-
itive-ion electrospray ionization Fourier transform ion cyclotron resonance mass spectrom-
etry». Energy & Fuels. 21,973—981.
[63] . Szulejko J. E., and Solouki T. (2002). «Potential analytical applications of interfacing a GC
to an FT-ICR MS: Fingerprinting complex sample matrixes». Analytical Chemistry. 74,
3434-3442.
[64] . Taguchi V.Y., Nieckarz R.J., Clement R.E., Krolik S., and Williams R. (2010). «Diox-
in Analysis by Gas Chromatography-Fourier Transform Ion Cyclotron Resonance Mass
Spectrometry (GC-FT ICR MS)». Journal of the American Society for Mass Spectrometry. 21,
1918-1921.
[65] . Takats Z., Wiseman J. M., Gologan B., and Cooks R. G. (2004). «Mass spectrometry sam-
pling under ambient conditions with desorption electrospray ionization». Science. 306,
471-473.
[66] . Van Krevelen D.W. (1950). «Graphical-Statistical Method for the Study of Structure and
Reaction Processes of Coal». Fuel. 29, 269—284.
[67] . Wozniak A. S., Bauer J. E., Sleighter R. L., Dickhut R. M., and Hatcher P. G. (2008). «Tech-
nical Note: Molecular characterization of aerosol-derived water soluble organic carbon us-
ing ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance
mass spectrometry». Atmospheric Chemistry and Physics. 8, 5099—5111.
[68] . Wu Z.G., Hendrickson C.L., Rodgers R. P., and Marshall A.G. (2002). «Composition of
explosives by electrospray ionization Fourier transform ion cyclotron resonance mass spec-
trometry». Analytical Chemistry. 74, 1879—1883.
[69] . Wu Z.G., Rodgers R. P., and Marshall A.G. (2004a). «Characterization of vegetable oils:
Detailed compositional fingerprints derived from electrospray ionization Fourier transform
ion cyclotron resonance mass spectrometry». Journal of Agricultural and Food Chemistry. 52,
5322-5328.
[70] . Wu Z.G., Rodgers R.P., and Marshall A.G. (2004b). «Compositional determination of
acidic species in Illinois no. 6 coal extracts by electrospray ionization Fourier transform ion
cyclotron resonance mass spectrometry». Energy & Fuels. 18, 1424—1428.
[71] . Wu Z.G., Rodgers R. P., and Marshall A.G. (2004c). «Two- and three-dimensional van
Krevelen diagrams: A graphical analysis complementary to the Kendrick mass plot for sort-
ing elemental compositions of complex organic mixtures based on ultrahigh-resolution
broadband Fourier transform ion cyclotron resonance mass measurements». Analytical
Chemistry. 76, 2511-2516.
[72] . Wu Z.G., Rodgers R.P., Marshall A.G., Strohm J. J., and Song C. (2005). «Comparative
compositional analysis of untreated and hydrotreated oil by electrospray ionization Fourier
transform ion cyclotron resonance mass spectrometry». Energy & Fuels. 19, 1072—1077.
ГЛАВА 19
ПРИМЕНЕНИЕ МАСС-
СПЕКТРОМЕТРИИ
СВЕРХВЫСОКОГО РАЗРЕШЕНИЯ
С МАСС-АНАЛИЗАТОРОМ
ИОННОГО ЦИКЛОТРОННОГО
РЕЗОНАНСА С ПРЕОБРАЗОВАНИЕМ
ФУРЬЕ ДЛЯ АНАЛИЗА
ПРИРОДНОГО ОРГАНИЧЕСКОГО
ВЕЩЕСТВА В ОБЪЕКТАХ
ОКРУЖАЮЩЕЙ СРЕДЫ
Ф. Шмитт-Копплин, М. Харир,Д. Тциотис, Д. Габелика, Н. Херткорн
19.1. Введение
19.1.1. Значение природного органического вещества и способы
его анализа
Природное органическое вещество (ПОВ) образуется в результате общих термодинами-
ческих и кинетических процессов из молекул геохимического или (в конечном счете)
биогенного происхождения и относится к самым гетерогенным веществам на Земле
(Hertkorn et al., 2008; Koch et al., 2005). Оно неизменно участвует во всех биогеохимиче-
ских процессах и находится во всех природных средах (в почвах, водах, атмосфере). ПОВ
определяет химическую специфику и круговорот питательных веществ, следовых эле-
ментов и органических молекул (Tipping, 2002). В ходе синтеза и биогеохимического пре-
образования (диагенезиса) природного органического вещества характерные для со-
ответствующих экосистем оригинальные биомаркеры, обычно образующиеся из таких
природных соединений, как, например, углеводы, (поли)фенолы, пептиды/белки и ли-
пиды, постепенно сглаживаются под действием биотических (например фотосинтез,
гетеротрофный микробный метаболизм) и абиотических (таких как фотохимические
и катализируемые металлами окислительно-восстановительные реакции) процессов
( 548 Глава 19. Применение масс-спектрометрии сверхвысокого разрешения смасс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
(Kujawinski et al., 2004; 2009). ПОВ связывает мир живой и неживой природы от нано-
уровня до уровня континентов (Battin et al., 2009; Hedges and Oades, 1997; Hertkorn et al.,
2007) и играет ключевую роль в переносе энергии и вещества в водных средах. Оно со-
стоит из полифункциональных молекул и из-за своей огромной сложности остается не-
достаточно охарактеризованным, несмотря на то, что в ПОВ сосредоточен самый боль-
шой на Земле запас органического углерода.
Масс-спектрометрия ионного циклотронного резонанса с преобразованием Фу-
рье (ИЦР ПФ) (Marshall et al., 1998) с очевидностью показала огромную сложность
природного органического вещества и открыла беспрецедентную возможность непо-
средственно фиксировать тысячи молекулярных формул индивидуальных веществ
в составе ПОВ (Hertkorn et al., 2008; Kim et aL, 2006; Koch et al., 2007; Kujawinski, 2002;
Kujawinskiand Behn, 2006; Reemtsma, 2009; Stenson etal., 2003; Wittetal., 2009). В последнее
десятилетие масс-спектрометрия ИЦР ПФ с ионизацией электрораспылением (ЭРИ)
в режиме регистрации положительных [ЭРИ(+)] и отрицательных [ЭРИ(—)] ионов ак-
тивно использовалась для анализа молекулярного состава ПОВ и для объяснения хи-
мических и биологических изменений в широком ряду природных сред, в числе ко-
торых поровые воды глинистых минералов (Huclier-Markai et al., 2010), поровые воды
осадков континентального шельфа (Schmidt et al., 2009), воды поверхностных пресно-
водных водоемов и горных рек (Sleighter et al., 2009), морские воды (D’Andrilli et al., 2010;
Hertkorn etal., 2006; Koch etal., 2005; Koprivnjak etal., 2009; Kujawinski etal., 2009; Reemtsma
et al., 2008), термальные воды (Kovacs et al., 2010), пирогенный углерод (Hockaday et al.,
2006), а также органическое вещество хондритов (Schmitt-Kopplin et al., 2010b). Кроме
того, фотохимические и биохимические изменения в молекулярном составе ПОВ так-
же были отдельно описаны для органических серосодержащих соединений в богатых
железом поровых водах шахт (Herzprung et al., 2010), для органосульфатов как компо-
нентов фотохимического смога во вторичных органических аэрозолях (Schmitt-Kopplin
et al., 2010a), а также для растворенного органического вещества в пресных водах
и эстуариях (Gonsior et al., 2009). При этом неоднократно подтверждалось, что арома-
тические соединения в растворенном органическом веществе морских, пресноводных
и эстуариевых сред более подвержены фоторазложению, чем алифатические (Gonsior
et al., 2009; Kujawinski et aL, 2009).
Фракционирование ПОВ методами жидкостной хроматографии высокого разре-
шения, электрофореза в свободном потоке и спектрометрии ионной подвижности по-
казало большое различие в структурах индивидуальных фракций (Capley et aL, 2010;
Gasparetal., 2009; GasparetaL, 2010; Koch etal., 2008; Stenson, 2008). Молекулярный состав
и структура ПОВ, определенные методом масс-спектрометрии ИЦР ПФ, очевидно за-
висят от:
1) природной экосистемы — источника происхождения ПОВ;
2) используемого метода выделения ПОВ (Koprivnjak et aL, 2009);
3) метода ионизации для последующей регистрации масс-спектра.
Поскольку молекулы ПОВ полифункциональны и характеризуются значительны-
ми размерами, в Фурье-преобразованных масс-спектрах, полученных с использовани-
ем комплементарных методов ионизации, таких как фотоионизация при атмосферном
давлении (ФИАД), лазерная ионизация при атмосферном давлении (ЛИАД) или хи-
мическая ионизация при атмосферном давлении (ХИАД), наблюдаются значительные
19.1. Введение
расхождения (Chilom et al., 2008; Hertkorn et al., 2008; Huclier-Markai et al., 2010) (см. также
главы 1 и 3).
19.1.2. Характеристики масс-спектров ПОВ и композиционное
пространство
В масс-спектрах, получаемых без фрагментации или разделения ионов по их подвиж-
ности, всем изомерным структурам молекулярного иона соответствует один пик. Соот-
ветственно композиционное пространство молекул представляет собой часть всего про-
странства молекулярных структур, получаемую в результате объединения изомеров.
Преобразование полного пространства структур молекул в композиционное простран-
ство эквивалентно его проекции с высоким разрешением наось значений молекулярных
масс и означает существенное снижение количества данных при сохранении ключевой
информации о составе молекул. Композиционное пространство квантовано в соответ-
ствии с законами образования химических связей и расширяется с увеличением раз-
мера молекулы и количества составляющих ее элементов. Квантованность композици-
онного пространства подразумевает фундаментальные ограничения местонахождения
и взаимного расположения сигналов в масс-спектре. Специфические разности масс,
которые современная масс-спектрометрия ИЦР ПФ позволяет успешно устанавливать,
задают некоторый интервал между соседними пиками С,H,N,О,S-содержащих молекул
внутри кластеров, соответствующих номинальным массам, и между такими кластера-
ми во всем композиционном пространстве (Hertkorn et al., 2008; Kim et al., 2006).
Масс-спектры низкого разрешения ПОВ обычно представляют собой довольно про-
тяженные огибающие сигналов с максимальной интенсивностью в районе 350—700 Да и
содержат несколько регулярных серий, соответствующих разнице в числе таких струк-
турных единиц, как, например, метиленовые группы (Д/и = ±14 Да) и степень ненасы-
щенности1 или по-другому эквиваленты двойной связи (DBE: Д/и = ±2 Да). При ограни-
ченном разрешении линейные комбинации серий метиленовых групп и эквивалентов
двойной связи невозможно отличить от серий кислородсодержащих фрагментов, таких
как Н2О (Д/и = ±18 Да), СО (Д/и = ±28 Да) и СО2 (Д/и = ±44 Да). Высококачественные
масс-спектры ИЦР ПФ природного органического вещества, напротив, демонстрируют
от нескольких тысяч до десятков тысяч разрешенных сигналов, разница между которы-
ми часто составляет миллидальтоны.
Сигналы, соответствующие двум молекулам одного класса и близкой структуры,
например гомологам (соединения, отличающиеся на СН2-группу, Д/и = ±14,01565 Да),
далеко отстоят друг от друга на шкале масс (рис. 19.1). В целом любые два иона, разли-
чающиеся по своему положению менее чем на 2,1057 Да (±DBE), независимо от своего
положения в спектре и от своего состава (С Н О Z * где Z — любой элемент или ком-
г х п т q 2.2
бинация элементов) неизбежно будут относиться к двум различным классам химиче-
ских структур. Это ключевое правило относится к любой паре пиков в пределах любого
кластера ионов с одной целочисленной массой, независимо от элементного состава. Таким
образом, внешне неприметные различия в интенсивностях и положениях пиков в масс-
спектрах ИЦР ПФ сложных объектов, таких как ПОВ, в действительности отражают су-
1 А.Т. Лебедев. Масс-спектрометрия в органической химии. Бином, Москва,
2003. — Прим. ред.
Рис. 19.1. Фундаментальные характеристики композиционного пространства молекул, содержащих атомы С, Н, О. (а) Построение компози-
ционного пространства в результате объединения изомеров эквивалентно проекции всего структурного пространства на ось m/z
с высоким разрешением, (б) При массах ионов менее 1,5 кДа композиционное пространство молекул, состоящих из атомов С, Н, О,
упорядочивается в виде серий отдельных кластеров с одинаковой целочисленной массой, а добавление либо удаление типичных
структурных фрагментов, таких как Н2О или СН2, соответствует переходам между отдельными кластерами, (в) В кластере с одинако-
вой целочисленной массой пики с низким значением массы соответствуют структурам с дефицитом водорода, а пики с высоким зна-
чением массы — кислород-дефицитным структурам. Регулярные интервалы между соседними пиками отражают специфику С,Н,О-
содержащих структурных единиц, (г) Классические фрагменты молекул (красный: Дай > ±2,1057 Да; ±DBE) и основные разности масс
Дай между соседними пиками (голубой/черный: Дай < ±2,1057 Да) определяют композиционное пространство С,Н,О; меньшие значе-
ния разностей масс всегда отвечают большим различиям в составе. (Печатается с разрешения: Hertkorn N., Frommberger М., Schmitt-
Kopplin Ph., Witt M., Koch B. and Perdue E. M. «Natural organic matter and the event horizon of mass spectrometry» . Analytical Chemistry. 80,
8908—8919. Copyright 2008 American Chemical Society)
Глава 19. Применение масс-спектрометрии сверхвысокого разрешения смасс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
19.1. Введение
шественные (или даже кардинальные) различия в их составе и молекулярной структу-
ре. Это означает, что наиболее значимая, специфическая для данного ПОВ структурная
информация содержится в его масс-спектре в последовательностях пиков с интервалом
порядка миллидальтон и меньше, доступных только для масс-спектрометрии сильно-
польного ИЦР с Фурье-преобразованием.
Высококачественный масс-спектр ИЦР ПФ природного органического вещества
покрывает значительную часть математически возможного композиционного про-
странства, охватывая довольно большой диапазон молекулярных масс и соотношений
элементов {Hertkorn etal., 2008). Непосредственный анализ масс-спектров, а также мате-
матические методы анализа и схемы обработки данных, такие как анализ масс Кендрика
и построение диаграмм ван Кревелена, позволяют сопоставить все внутреннее строение
квантованного композиционного пространства и данные, получаемые в результате об-
работки масс-спектров ИЦР ПФ сложных образцов. Таким образом, концепция компо-
зиционного пространства предлагает практическое руководство и классификационные
критерии для обработки многолинейчатых масс-спектров сложных веществ {Gougeon
etal., 2009; Schmitt-Kopplin etal., 2010b).
19.1.3. Визуализация данных сложных масс-спектров
Наиболее общими методами представления значений m/z являются построение диа-
грамм ван Кревелена {van Krevelen, 1950) и диаграмм масс Кендрика {Hughey et al., 2001;
Kendrick, 1963). На диаграммах ван Кревелена (см. также главу 18) элементные соотно-
шения, рассчитанные из найденных брутто-формул, располагаются по двум или трем
осям; чаще всего изображаются двумерные диаграммы Н/С (по оси у) — О/С (по оси х)
(рис. 19.3; Schmitt-Kopplin et al., 2010a; 2010b). Возможно дополнение построенной таким
образом диаграммы другими элементными соотношениями (например N/С или S/C)
и увеличение ее размерности {Kim et al., 2003; Wu et al., 2004). Соотношение Н/С пока-
зывает относительную степень алифатичности и, соответственно, ароматичности ве-
щества и коррелирует, например, с количеством эквивалентов двойной связи (DBE),
приходящихся на один атом углерода, в то время как отношение О/С связано со степе-
нью окисления вещества. Индекс ароматичности был введен {Koch and Dittmar, 2006)для
дифференцирования водород-дефицитных молекул, типа моно- и полиароматических.
Диаграммы дефекта масс Кендрика (ДМК) (глава 18) иллюстрируют серии моле-
кул, различающихся только небольшим специфическим фрагментом. Чаще всего та-
ким фрагментом выступает метиленовая группа (СН2) — наиболее распространенный
структурный блок органических веществ. Так называемая масса Кендрика рассчиты-
вается исходя из того, что она должна иметь целочисленное значение для выбранного
за основу фрагмента. Так, масса Кендрика для СН2 равняется 14,00000 Да (согласно
ИЮПАК масса метиленовой группы составляет 14,01565 Да), и добавление произволь-
ного количества метиленовых групп к любой выбранной молекуле оставит без измене-
ния десятичную часть значения массы Кендрика для всей серии молекул. Для анализа
на основе метиленовых звеньев коэффициент преобразования молекулярных масс ИЮ-
ПАК в массы Кендрика (МК) составляет 14,00000/14,01565 = 0,998883.
МК(СН2) = т(ИЮПАК) х 14,00000/14,01565 = т(ИЮПАК) х 0,998883
[Г 552 Глава 19. Применение масс-спектрометрии сверхвысокого разрешения с масс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
Затем определяются номинальная масса Кендрика как целая часть и дефект масс
Кендрика (ДМК) как разность:
ДМК(СН2) = номинальная МК(СН2) - МК(СН2).
Таким образом, молекулы, относящиеся к одной гомологической серии и различа-
ющиеся только числом метиленовых звеньев, относятся к одному классу соединений
и обладают одним и тем же значением КМД, одинаковым числом гетероатомов и экви-
валентов двойной связи. Но сам класс химических соединений может оставаться не-
ясным, поскольку анализ масс Кендрика не требует определения элементного состава
анализируемого объекта и, соответственно, молекулярных формул.
Анализ масс Кендрика может осуществляться и на основании других, отличных
от метиленовых звеньев, структурных единиц, например таких, как СО2 {Hertkorn et al.,
2006).
Диаграммы Кендрика изображают в координатах ДМК (осьу) — МК (осьх). Веще-
ства одного класса на них располагаются на одной горизонтальной линии, а вещества,
не являющиеся гомологами, различаются между собой характерными для их классов
величинами ДМК (рис. 19.6; Hughey et al., 2001; Wu et al., 2004). Авторы этой главы стре-
мятся расширить классический метод масс Кендрика, используя не одну, а одновремен-
но несколько базовых структур, таких как СО2, О, Н2, NH(x = 0—2), SO. (z = 0—4) для
построения одной целостной расчетной модели, представленной в виде сети (рис. 19.10).
Альтернативные алгоритмы и подходы базируются на точных разностях масс, соответ-
ствующих определенным «строительным блокам» ПОВ. Авторы одного из них опреде-
ляли массы таких блоков на основании анализа периодически повторяющихся после-
довательностей сигналов, обнаруженных в масс-спектре {Kunenkov et al., 2009; Grinhut
et al., 2010). Согласно другому подходу для описания химического строения вторичных
органических аэрозолей использовали среднее значение степени окисления атомов
углерода {Krollet al., 2011). Эти способы анализа данных предлагают новые возможности
для качественного и количественного описания сложных органических молекулярных
систем в целом и ПОВ в частности {Meija, 2006).
19.1.4. Примеры анализа масс ПОВ
Три примера ПОВ, которые хотя и являются сложными органическими смесями, но су-
щественно различаются по составу и генезису, позволяют проиллюстрировать разные
подходы к оценке масс-спектрометрических данных. Растворенное органическое ве-
щество реки Суванни (США) представляет собой сильно преобразованный почвенный
фильтрат с протяженным композиционным пространством С,Н,О-содержащих моле-
кул. Во вторичных органических аэрозолях под воздействием явно выраженных абио-
тических реакций создается значительное молекулярное разнообразие с большой долей
гетероатомов (N, S) в высоких степенях окисления. ПОВ внеземного происхождения
на примере растворенного органического вещества хондритов отражает наиболее бога-
тую совокупность генезиса при крайне высоких и крайне низких температурах и про-
должительном облучении в пределах всего спектра электромагнитных волн. Органи-
ческое вещество хондритов консолидировано в виде гетерогенного материала, по своей
сложности сравнимого с биомолекулами и, возможно, превышающего сложность био-
молекул, найденных на Земле {Schmitt-Kopplin etal., 2010b).
19.3. Результаты и обсуждение
19.2. Материалы и методы
19.2.1. Масс-спектрометры ИЦР ПФ
Приведенные здесь масс-спектры сверхвысокого разрешения были получены
на спектрометрах ИЦР ПФ Apex 12 Qe и SolariX компании Bruker (г. Бремен, Герма-
ния), оснащенных сверхпроводящим магнитом с индукцией поля 12 Тл и источни-
ком электрораспыления Apollo II. Перед введением в источник все исследованные
экстракты были разбавлены метанолом до стандартной концентрации в несколь-
ко частей на миллион. Электрораспыление осуществляли при скорости потока
120 мкл/ч, давлении распыляющего газа 1,4 бар и давлении осушающего газа 1 бар
(250°С). Спектры записывали в интервале значений m/zот 100 до 2000 Да, количество
точек в спектре во временном диапазоне равнялось 4000000. При записи спектров
количество сканирований для каждого образца составило от 1000 до 5000. В диапа-
зоне значений m/z 150—1000 Да наблюдалось от нескольких тысяч до десятков тысяч
пиков, соответствующих соединениям с разным элементным составом. Оказалось
возможным точно определить брутто-формулы для соединений с массой до 800 Да,
поскольку в рутинных измерениях ошибка не превышает значения 100 млрд-1 в слу-
чае внутренней калибровки (по известным пикам, соответствующим ионам жирных
кислот в режиме регистрации отрицательных ионов и примесям в растворителе в ре-
жиме регистрации положительных ионов).
19.2.2. Расчет масс для молекул, содержащих С, Н, N, О, S
Спектры ИЦР ПФ были представлены втабличном виде при соотношении сигнал/шум,
равном 2. При помощи программного обеспечения собственной разработки для каждо-
го иона в пакетном режиме обработки данных рассчитывали брутто-формулу. На рас-
считанные брутто-формулы накладывались понятные ограничения химического ха-
рактера (такие как азотное правило; О/С < 1; Н/С < 2п + 2, где п — количество атомов
углерода; число атомов углерода, кислорода, азота и серы не превышало некоторых вы-
бранных максимальных значений). При этом происходило автоматическое сравнение
теоретически возможных изотопных распределений.
19.3. Результаты и обсуждение
Высококачественный масс-спектр ИЦР ПФ высокого разрешения ПОВ содержит де-
сятки тысяч пиков, примерно для третьей части которых могут быть подобраны брут-
то-формулы, отвечающие строгим условиям. При индукции поля 12 Тл масс-спектры
ИЦР ПФ были записаны в режиме широкополосного сканирования с разрешающей
способностью 400000 (для m/z 400). Эти параметры достаточны для того, чтобы одно-
значно представить все химическое пространство CHONS в виде разрешенных индиви-
дуальных пиков вплоть до значений масс порядка 500 Да {Hertkorn et al., 2008; Kim et al.,
2006). Сложные ионно-молекулярные реакции в распыляемой фазе способствуют по-
явлению однозарядных ионов в стандартных условиях ионизации, и только снижение
( 554 Глава 19. Применение масс-спектрометрии сверхвысокого разрешения смасс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
концентрации приводит к тому, что многозарядные ионы ПОВ также становятся замет-
ны (Gaspar et al., 2009).
Масс-спектры ИЦР ПФ типичного ПОВ, снятые в режиме электрораспыления с ре-
гистрацией отрицательных ионов, приведены на рис. 19.2 на примере фракции фуль-
вокислот растворенного органического вещества реки Суванни (SRFA), водораствори-
мой фракции (SWOC) вторичных органических аэрозолей и растворимого в метаноле
вещества углеродистого хондрита (COM) (Hertkorn etal., 2008; Schmitt-Kopplin etal., 2010a;
2010b). Молекулярно-массовое распределение SRFA имеет характер больцмановского,
оно типично для органического вещества суши и обусловлено, вероятно, двумя проти-
воположными тенденциями. Число возможных изомерных структур для конкретного
элементного состава стремительно возрастает с увеличением молекулярной массы, что
ведет к относительному росту интенсивности пиков при переходе от низких молеку-
лярных масс к высоким. С другой стороны, интенсивность пиков более тяжелых ионов
снижается вследствие меньшей летучести молекул с высокими молекулярными масса-
ми (соответственно, меньшего присутствия их ионов в газовой фазе). Более того, веро-
ятность обнаружить немногочисленные гетероатомы (к которым в данном случае не от-
носятся атомы Н, С, О) растет с увеличением молекулярной массы и ведет к большей
неоднородности состава в пределах кластера с одной целочисленной массой. Отсюда
следует, что для более высоких масс интенсивность сигналов в пределах кластера будет
распределена между большим количеством ионов, что ведет к снижению интенсивно-
сти, приходящейся на один ион.
Аналогично интенсивность сигналов в пределах любого спектрального интерва-
ла распределена в виде периодически повторяющихся серий кластеризованных пиков
с общей разностью масс (рис. 19.2), и так вплоть до кластеров, обусловленных ионами
с одинаковой целочисленной массой (Reemtsma et al., 2006). Интенсивности пиков ио-
нов часто обусловлены числом изомеров, которые могут соответствовать данной моле-
кулярной формуле. Число обладающих химическим смыслом изомерных структур, со-
ответствующее данному составу С,Н,О, максимально для средних значений Н/С и О/С
(Hertkorn et al., 2007). Любой ион включает все изомеры, поскольку они обладают иден-
тичными молекулярными массами и составом. Следовательно, наиболее интенсивные
пики в пределах серий кластеров, типичных для масс-спектров ПОВ, должны представ-
лять собой суперпозиции ионов с составом с наибольшим числом изомеров, т. е. со сред-
ними значениями соотношений Н/С и О/С.
Масс-спектрометрия ИЦР ПФ, для которой и разрешающая способность, и точ-
ность определения массы исключительно высоки, позволяет различать серии ионов
СНО и CHOS вплоть до значений масс 800 Да (рис. 19.26 и в) (Schmitt-Kopplin etal., 2010а).
Сера в составе ПОВ участвует в самых разнообразных абиотических (например фото-
окисление вторичных органических аэрозолей, абиогенный синтез сложных органиче-
ских молекул) и биологических окислительно-восстановительных процессах, которые,
к примеру, существенно влияют на ПОВ донных отложений и грунтовых вод (Herzsprung
et al., 2010; Schmitt-Kopplin et aL, 2010a). Диаграммы ван Кревелена (рис. 19.2в и 19.3) де-
монстрируют широкое разнообразие молекул в составе ПОВ. Множества точек, соот-
ветствующих определенным элементным составам, занимают протяженные интервалы
значений Н/С и О/С даже для отдельных кластеров, отвечающих целочисленным моле-
кулярным массам.
19.3. Результаты и обсуждение
Тысячи индивидуальных брутто-формул, полученных для SRFA, WSOC и СОМ,
также могут быть представлены в виде диаграмм Н/С — m/z (рис. 19.3), отражающих
степень молекулярного разнообразия по мере изменения значений молекулярных
масс. На таких разрешенных по массе диаграммах все установленные брутто-форму-
лы представлены отдельно, в то время как на диаграммах ван Кревелена одинаковые
значения элементных соотношений, отвечающие различным брутто-формулам, будут
приходиться на одну и ту же точку. Так, молекуле состава С10Н)8О8 (с номинальной мас-
сой 266 Да) соответствуют те же элементные соотношения (Н/С = 1,8 и О/С = 0,8), что
и молекулам состава С15Н27О12 (с номинальной массой 398 Да) и С20Н36О16 (с номинальной
Рис. 19.2. (а) Сверху вниз — масс-спектры SRFA, WSOC и СОМ в диапазонах масс 150—
600 Да, 402—416 Да и часть спектра в области целочисленной массы 411 Да; (б) се-
рии ионов состава СНО (синие и красные пики) и CHOS (светло- и темно-зеленые)
в спектре WSOC с целочисленной массой 411 Да (разность масс между сериями
СНО и CHOS кт = 2,4 мДа) и (в) эти же четыре молекулярные серии на диаграмме
ван Кревелена, разность между пиками внутри каждой серии номинально соот-
ветствует замене СН4 на атом кислорода (Aw = 36,4 мДа). (Печатается с разреше-
ния: Schmitt-Kopplin Ph., KissG., Dabek-Zlotorzynska E., GelencserA., Hertkorn N.,
Harir M., Hong Y. and Gebefiigi I. «Analysis of the unresolved organic fraction in at-
mospheric aerosols with ultrahigh-resolution mass spectrometry and nuclear magnetic
resonance spectroscopy: organosulfates as photochemical smog constituents». Analytical
Chemistry. 82, 8017—8026. Copyright 2010 American Chemical Society)
Глава 19. Применение масс-спектрометрии сверхвысокого разрешения с масс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
массой 530 Да). С другой стороны, визуальное представление данных в виде диаграмм
ван Кревелена позволяет видеть дискретное распределение молекул, что редко для до-
статочно хорошо преобразованного ПОВ с существенно нивелированным содержанием
характерных биомаркеров.
Данные по SRFA демонстрируют преимущественно серии ионов состава СНО и по-
следовательности, типичные для поверхностных вод с растворенным органическим
веществом суши, таким как окисленные лигнины и таннины. Для WSOC диаграмма
Н/С — m/z имеет такой же плавный вид, но при этом в ней значительно больше точек
серий CHOS, которые легко распознать по расщеплению Д/и = 3,4 мДа по сравнению
с сериями СНО (замена С3 на H4S; рис. 19.20. Кроме этих сульфированных вторичных
аэрозолей, в WSOC обнаружены молекулы состава CHNO, являющиеся продуктами го-
рения биомассы {Schmitt-Kopplin etal., 2010а).
150 250 350 m/z «50 560 660
H/C
05
Рис. 19.3. SRFA (вверху), SWOC (посередине) и COM (внизу): разрешенные по массе диа-
граммы (Н/С vs m/z) (слева) и диаграммы ван Кревелена (Н/С vs О/С) (справа).
В крайнем правом ряду показано число пиков молекулярных ионов с уста-
новленными брутто-формулами для составов СНО (синим), CHOS (зеленым),
CHNO (оранжевым) и CHNOS (красным); точки на диаграммах отмечены тем
же цветом; данные получены из масс-спектров ИЦР ПФ, снятых в режиме реги-
страции отрицательных ионов. (Печатается с разрешения: Schmitt-Kopplin Ph.,
Kiss G., Dabek-Zlotorzynska E., Gelencser A., Hertkorn N., Harir M., Hong Y. and
Gebefiigi I. «Analysis of the unresolved organic fraction in atmospheric aerosols with ul-
trahigh-resolution mass spectrometry and nuclear magnetic resonance spectroscopy: or-
ganosulfates as photochemical smog constituents». Analytical Chemistry. 82, 8017—8026.
Copyright 2010 American Chemical Society)
CHO
CHOS
CHNO
CHNOS
CHO CHOS CHON CHONS
19.3. Результаты и обсуждение
Разделение спектральных данных в соответствии с установленными молекуляр-
ными сериями представляет собой мощный инструмент визуального представления
состава различных сложных материалов (рис. 19.4). Представление всех спектральных
данных на одном рисунке часто страдает из-за существенного наложения пиков ионов
на широких областях композиционного пространства (рис. 19.3). Разделение общей
спектральной картины для ПОВ на отдельные серии пиков молекулярных ионов, за-
нимающих характерную область на диаграмме ван Кревелена, позволяет определить
класс и структуру вещества. Например, сравнение молекулярных серий состава CHOS
и CHNO для SWOC показывает наличие довольно насыщенных соединений соста-
ва CHOS (преимущественно среднеокисленных), в то время как соединения состава
CHNO отличаются большей ароматичностью и менее окислены. В составе WSOC со-
единения состава CHOS чаще всего представляют собой эфиры серной кислоты и али-
фатических спиртов терпеноидного происхождения, образующиеся в результате фото-
окисления, в то время как молекулы состава CHNO представляют собой обогащенные
азотом конденсированные полиароматические структуры, образующиеся в результате
горения биомассы {Schmitt-Kopplin et al., 2010а). Из трех исследованных ПОВ наиболь-
шее разнообразие химических структур, вплоть до почти полного отсутствия отдельных
кластеров на диаграмме ван Кревелена, демонстрирует СОМ, что отражает его космиче-
ское происхождение с абсолютным доминированием (в отличие от SRFA и SWOC) про-
цессов абиотического синтеза.
Распределение интенсивностей пиков, которое не отражают на стандартной диа-
грамме ван Кревелена, может быть задано, например, цветом или радиусом пятна,
Рис. 19.4. Раздельное представление масс-спектральных данных ИЦР ПФ на диаграммах
ван Кревелена SRFA (вверху), SWOC (посередине) и СОМ (внизу) для серий мо-
лекулярных ионов состава СНО, CHOS, CHNO и CHNOS, показывающее специ-
фическое распределение точек на диаграмме, характерное для каждого ПОВ.
(Печатается с разрешения: Schmitt-Kopplin Ph., Kiss G., Dabek-Zlotorzynska E.,
Gelencser A., Hertkorn N., Harir M., Hong Y. and Gebefiigi I. «Analysis of the unresolved
organic fraction in atmospheric aerosols with ultrahigh-resolution mass spectrometry and
nuclear magnetic resonance spectroscopy: organosulfates as photochemical smog constitu-
ents». Analytical Chemistry. 82, 8017—8026. Copyright 2010 American Chemical Society)
Глава 19. Применение масс-спектрометрии сверхвысокого разрешения смасс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
когда его площадь коррелирует с интенсивностью соответствующего пика (рис. 19.4).
Для обычных ПОВ непропорционально большие пики могут относиться к примесям
или экзогенным веществам. В частности, органическое вещество СОМ различается
по характеристическим картинам в масс-спектре: (1) довольно непрерывное и протя-
женное распределение пиков молекулярных ионов, вероятно, отражающее процессы
абиотического «энтропийного» синтеза разнообразных химических структур; (2) инди-
видуальные резко выраженные пики, скорее всего связанные с примесями земного про-
исхождения (такими как продукты микробного разложения субстрата СОМ).
Для СОМ близкое подобие диаграмм СНО и CHNO, с одной стороны, и CHOS
и CHNOS, с другой, ведет к предположению об эволюции во времени индивидуальных
молекулярных серий СОМ: от изначально цельного «ядра» соединений состава CHNO
под воздействием изменений, вызванных на поздних стадиях контактом с водой, отде-
ляется ряд растворимых, по большей части алифатических соединений состава СНО.
Затем взаимодействие растворенной восстановленной серы минерального происхожде-
ния с соединениями состава СНО и CHNO привело к образованию молекул с составом
CHOS и CHNOS (Schmitt-Kopplin etal., 2010b).
Ионизация электрораспылением (ЭРИ) позволяет определить широкий диапазон
преимущественно полярных соединений. При использовании химической ионизации
при атмосферном давлении (ХИАД) ионизуются менее полярные молекулы меньших
молекулярных масс, в то время как при использовании фотоионизации при атмосфер-
ном давлении (ФИАД) преимущественно ионизуются ароматические и другие молеку-
лы, содержащие л-связь. Следовательно, селективность анализа зависит от выбранного
метода ионизации и полярности ионов. В случае анализа ПОВ степень комплементар-
ное™ между режимами регистрации положительных и отрицательных ионов, а также
между методами ионизации особенно велика. Всесторонняя характеристика SRFA
Рис. 19.5. Диаграммы ван Кревелена, отражающие селективность методов ионизации
в режимах регистрации положительных (слева) и отрицательных (справа) ионов.
(Печатается с разрешения: Hertkorn N., Frommberger М., Schmitt-Kopplin Ph.,
Witt M., Koch В. and Perdue E.M. «Natural organic matterand the event horizon of
mass spectrometry». Analytical Chemistry. 80, 8908—8919. Copyright 2008 American
Chemical Society)
19.3. Результаты и обсуждение
КМД (Да) на базе СН2
0.4
0.38
0.36
0.34
0.32
0.3
• ”
о о с
ч: 1
□
б
tnntumi
•***mtm
ШИНИН,
it
200-700
ESlpos
□ APCIpos
| APPI pos
L APCI_APPI
L APCIESI
4 APPI ESI
• APCI APPI
its888 88/8 ♦'
DO ^♦♦♦♦♦♦♦♦♦'•’♦’Dl?'!?
° ♦,88888888s *'•>
а*г**3333**Г:11Й-
*3333333333333*•
□ d * ♦ ♦ •+* ^4*4.* *ЛЛЛЛ»°и*
° • * it ci et 88888* • • • •"
88ili+tiiJtti?88aa
□ □□□□□□□□□
a ♦ ♦ Ш88888шу
Л "//ЛА* ’Л’Л’А "Л"
>*DODDn(?DOODD' * "
V* *• *-♦•♦****<*<**"
ESI
Г->00—700
ESIneg
□ APCIneg
И APPI neg
•j<APCI_APPl
| APCIESI
APP1_ES1
♦ APCI APPI
300 400 500 600 400 500 600 700
Масса Кендрика (Да) на базе СН2
Рис. 19.6. Применение к SRFA метода масс Кендрика на базе метиленовых звеньев демон-
стрирует резко выраженную селективность в зависимости от метода ионизации.
Маленькими серыми точками отмечены положения в композиционном про-
странстве С,Н,О, не занятые пиками ионов. (Печатается с разрешения: Hert-
korn N., Frommberger М., Schmitt-Kopplin Ph., Witt M., Koch В. and Perdue E.M.
«Natural organic matterand the event horizon of mass spectrometry». Analytical Chemis-
try. 80, 8908—8919. Copyright 2008 American Chemical Society)
Число атомов углерода
Число атомов углерода
Число атомов углерода
Рис. 19.7. Средние значения OSC в зависимости от числа атомов углерода в молекуле ПОВ,
поданным масс-спектров ИЦР ПФ отрицательных ионов SRFA, WSOC и СОМ
Глава 19. Применение масс-спектрометрии сверхвысокого разрешения с масс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
показывает, что в режиме регистрации положительных ионов множества точек на диа-
грамме ван Кревелена, определенных методами ХИАД и ФИАД, пересекаются более
чем на две трети, в то время как для методов ХИАД и ЭРИ — менее чем на одну пятую;
с другой стороны, в режиме регистрации отрицательных ионов все три метода иониза-
ции дают около половины общих точек (рис. 19.5, Hertkorn et al., 2008).
В результате анализа спектральных данных для SRFA методом масс Кендрика
на базе метиленовых звеньев (СН2) семь групп номинально идентичных молекулярных
соединений состава С,Н,О, различающихся предпочтительным методом ионизации,
выстраиваются в нерегулярные ряды на диаграмме ДМК (рис. 19.6). Избирательность
ионизации SRFA в режимах регистрации положительных и отрицательных ионов суще-
ственно различается. Предпочтительный метод ионизации соединений в составе SRFA
связан со значением дефекта масс Кендрика: соединения одного класса преимуще-
ственно ионизуются одним и тем же или немногими одинаковыми методами. В преде-
лах одного класса решающее значение для селективности имеет величина молекуляр-
ной массы (Hertkorn et al., 2008).
Недавно было предложено использовать среднюю степень окисления атомов угле-
рода (OSC) для оценки меры окисленности атмосферного ПОВ (Krolletal., 2011). Для мо-
лекул состава С,Н,0 величина OSC может быть рассчитана непосредственно из элемент-
ного состава согласно уравнению OSC = 2О/С - Н/С. В ПОВ, содержащем гетероатомы
2.5
X
g 1.5
х
о
о 1
X
Е—
о
0.5
0
0
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Отношение О/С
Атмосферные Сульфинированные
РОА Атмосферные SOA
SOA
Обогащенные
карбоксильны
группами
алициклические
молекулы в море
ПОВ
суши
и воды
Сажа ВС
0.00 0.10 0.20 0.30 0.40 0.50 0.60 0.70 0.80 0.90 1.00
Отношение О/С
Рис. 19.8. Типичные диаграммы ван Кревелена для различных ПОВ (слева) и различных
классов биомолекул (справа). Расположение на диаграмме связывает состав ПОВ
с составом известных биомолекул. Диапазоны элементных отношений, характер-
ные для различных ПОВ, включая первичные органические аэрозоли (РОА), вто-
ричные органические аэрозоли (SOA) и продукты их сульфирования (Sulfonated
SOA), органическое вещество вод суши и моря ПОВ (NOM) и CRAM (обогащен-
ные карбоксильными группами алициклические молекулы), а также конденси-
рованный и окисленный пирогенный углерод (ВС), представлены на диаграммах
наряду с диапазонами, типичными для разных классов природных соединений
и других молекулярных предшественников ПОВ. (Печатается с разрешения:
Gougeon R. D., Lucio М., Frommberger М., Реугоп D., Chassagne D., Alexandre Н.,
Feuillat Е, Voilley A., Cayot Р., Gebefugi L, Hertkorn N., and Schmitt-Kopplin P. (2009).
«The chemodiversity of wines can reveal a metabologeography expression of cooperage oak
wood». Proceedings of the National Academy of Sciences of the United States of America. 106,
9174—9179; Schmitt-Kopplin Ph., KissG., Dabek-Zlotorzynska E., Gelencser A., Hert-
korn N., HarirM., Hong Y. and Gebefiigi I. «Analysis of the unresolved organic fraction in
atmospheric aerosols with ultrahigh-resolution mass spectrometry and nuclear magnetic
resonance spectroscopy: organosulfates as photochemical smog constituents». Analytical
Chemistry. 82, 8017—8026. Copyright 2010 American Chemical Society)
19.3. Результаты и обсуждение
Эффект метода ионизации
Эффект полярности ионов
m/z Содержание
4 кислорода
DBE
Ароматичность
Рис. 19.9. Предложенное здесь представление масс-спектральных данных базируется
на методе главных компонент с соответствующими зависящими от массы па-
раметрами. В настоящее время такие скорее эстетические, чем всеобъемлющие
параметры, как m/z, степень ароматичности и содержание кислорода, лучше
всего дискриминируют объединенные данные масс-спектров SRFA, получен-
ных в режимах регистрации положительных и отрицательных ионов с исполь-
зованием методов ионизации ЭРИ, ХИАД и ФИАД
(таком как WSOC и СОМ), сера и азот могут находиться в широком диапазоне степеней
окисления (от —2 до +6 для серы и от —3 до +5 для азота), и дополнительный анализ
функциональных групп методами спектроскопии ИК и ЯМР или альтернативными
методами масс-спектрометрии мог бы быть полезен для более точного определения со-
ответствующих степеней окисления углерода. Диаграммы, на которые нанесены оце-
ненные величины О8сдля молекулярных серий состава СНО, CHOS, CHNO и CHNOS
(рис. 19.7), крайне полезны для понимания структуры и состава ПОВ.
Наземные ПОВ занимают значительный диапазон в композиционном пространстве
и часто содержат очень важную дополнительную информацию о биомаркерах, позволя-
ющую оценить относительный вклад различных биогеохимических реакций в форми-
рование данного ПОВ (рис. 19.8).
Биомаркеры в спектрах наземного и пресноводного ПОВ часто проявляются в виде
интенсивных пиков молекулярных ионов, которым могут быть найдены соответствия
в базе данных Киотской энциклопедии генов и геномов при помощи транслятора Mass-
Глава 19. Применение масс-спектрометрии сверхвысокого разрешения с масс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
Рис 19.10. Трехмерный граф, построенный на основе анализа масс-спектральных дан-
ных по молекулам состава С,Н,0 для ПОВ р. Суванни. Каждому узлу отвеча-
ет экспериментально найденная молекулярная масса; соединяющие их ребра
разных цветов представляют собой визуализированные и оптимизированные
по расстояниям разности масс, связанные с изменением числа тех или иных
структурных единиц
TRIX (www.masstrix.org; Suhre and Schmitt-Kopplin, 2008). Расшифровка изменяющихся
со временем индивидуальных маркеров ПОВ (Zhang et al., 2011) является первым шагом
в направлении сближения биогеохимического подхода и подхода с точки зрения био-
разнообразия, учитывающего специфические для организмов метаболиты и метаболи-
ческие цепочки. Будущие всесторонние исследования структуры ПОВ и его функций
в окружающей среде будут опираться на полный набор спектральных методов анализа
структур органического вещества, метаболомику и системную биологию. Детализиро-
ванный сетевой анализ масс-спектральных данных ПОВ р. Суванни представляет со-
бой род искусства наглядного представления сложных массивов данных (рис. 19.9) и де-
монстрирует богатство информационных связей и новые возможности качественного
и количественного представления состава сложных органических молекул, как это по-
казано на рис. 19.10.
Теория графов широко используется в биоинформатике и хемометрике благодаря
способности эффективно анализировать и визуально представлять подробные модели.
С ее помощью множество процессов реального мира может быть удобно представлено
в виде диаграмм с набором точек (узлов, вершин) и линий (ребер), соединяющих свя-
занные точки; при этом математическое обобщение зависит от концепции данного гра-
фа (Bondy and Murty, 2007).
При помощи описанной модели была сделана попытка обнаружить зависимость
функционального и структурного пространства ПОВ от различных условий, таких как
сезонные изменения, стадия диагенеза или биогеохимической трансформации.
19.4. Заключение
Разнообразные современные методы графического представления сложных масс-
спектров создают новые возможности для классификации ПОВ на молекулярном уров-
не. В будущем глубокие исследования роли биоразнообразия при ясном понимании
структуры и роли ПОВ могут привести к новому, цельному пониманию биоразнообра-
зия и биогеохимии. В настоящее время в метаболомике и системной биологии начинают
применяться современные биоинформационные методы визуализации данных, кото-
рые представляют количественную и функциональную информацию о специфических
для организмов метаболитах в виде интегральной системы метаболических связей. Но-
вые методики построения моделей на основе комплексных данных, такие как модели-
рование системы структурных связей, только начинают развиваться и обещают даль-
нейший прогресс в анализе и понимании структур сложных органических молекул.
Литература
[1] . Battin T.J., Luyssaert S., Kaplan L.A., Aufdenkampe A. К., Richter A., and Tranvik L.J.
(2009). «The boundless carbon cycle». Nature Geoscience. 2: 598—600.
[2] . Bondy J. A., and Murty U.S.R. (2007). Graduate Texts in Mathematics: Graph Theory.
Springer, New York, USA.
[3] . Capley E. N., Jeremiah D., Tipton J. D., Alan G. Marshall A. G., and Stenson A. C. (2010).
«Chromatographic reduction of isobaric and isomeric complexity of fulvic acids to enable
multistage tandem mass spectral characterization». Analytical Chemistry. 82: 8194—8202.
[4] . Chilom G., Chilom O., and Rice J. A. (2008). «Exploring the high-mass components of hu-
mic acid by laser desorption ionization mass spectrometry». Rapid Communications in Mass
Spectrometry. 22: 1528—1532.
[5] . D’Andrilli J., DittmarT., Koch В. P., Purcell J. M., Marshall A. G., and Cooper W.T. (2010).
«Comprehensive characterization of marine dissolved organic matter by Fourier transform
ion cyclotron resonance mass spectrometry with electrospray and atmospheric pressure pho-
toionization». Rapid Communications in Mass Spectrometry. 24: 643—650.
[6] . Gaspar A., Kunenkov E.V., Lock R., Desor M., Perminova I., and Schmitt-Kopplin P.
(2009). «Combined utilization of ion mobility and ultra-high-resolution mass spectrometry
to identify multiply charged constituents in natural organic matter». Rapid Communications
in Mass Spectrometry. 23: 683—688.
[7] . Gaspar A., Harir M., Hertkorn N., and Schmitt-Kopplin, Ph. (2010). «Preparative free-flow
electrophoretic offline ESI-Fourier transform ion cyclotron resonance/MS analysis of Su-
wannee River fulvic acid». Electrophoresis. 31: 2070—2079.
[8] . Gonsior M., Peake В. M., Cooper W.T., Podgorski D., D’Andrilli J., and CooperW. J. (2009).
«Photochemically induced changes in dissolved organic matter identified by ultrahigh reso-
lution Fourier transform ion cyclotron resonance mass spectrometry». Environmental Science
and Technology. 43: 698—703.
[9] . Gougeon R.D., Lucio M., Frommberger M., Peyron D., Chassagne D., Alexandre H.,
Feuillat F., Voilley A., Cayot P., Gebefiigi L, Hertkorn N., and Schmitt-Kopplin P. (2009).
«The chemodiversity of wines can reveal a metabologeography expression of cooperage oak
Глава 19. Применение масс-спектрометрии сверхвысокого разрешения смасс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
wood». Proceedings of the National Academy of Sciences of the United States of America. 106:
9174-9179.
[10] . Grinhut T., Lansky D., Gaspar A., Hertkorn N., Schmitt-Kopplin P., Hadar Y., and
Chen Y. N. (2010). «Novel software for data analysis of Fourier transform ion cyclotron
resonance mass spectra applied to natural organic matter». Rapid Communications in Mass
Spectrometry. 24: 2831—2837.
[11] . Hedges J. I., and Oades J. M. (1997). «Comparative organic geochemistries of soils and ma-
rine sediments». Organic Geochemistry. 27: 319—361.
[12] . Hertkorn N., Benner R., Frommberger M., Schmitt-Kopplin P., Witt M., Kaiser K.,
Kettrup A., and Hedges J. I. (2006). «Characterization of a major refractory component of
marine dissolved organic matter». Geochimica et Cosmochimica Acta. 70: 2990—3010.
[13] . Hertkorn N., Ruecker C., Meringer M., Gugisch R., Frommberger M., Perdue E.M.,
Witt M., and Schmitt-Kopplin P. (2007). «High-precision frequency measurements: in-
dispensable tools at the core of the molecular-level analysis of complex systems». Analytical
and Bioanalytical Chemistry. 389: 1311 — 1327.
[14] . Hertkorn N., Frommberger M., Schmitt-Kopplin, Ph., Witt M., Koch B., and Perdue E. M.
(2008). «Natural organic matter and the event horizon of mass spectrometry». Analytical
Chemistry. 80: 8908—8919.
[15] . Herzsprung P., Hertkorn N., Friese K., and Schmitt-Kopplin P. (2010). «Photochemical
degradation of natural organic sulfur compounds (CHOS) from iron-rich mine pit lake
pore waters — an initial understanding from evaluation of single-elemental formulae using
ultra-high-resolution mass spectrometry». Rapid Communications in Mass Spectrometry. 24:
2909-2924.
[16] . Hockaday W. C., Grannas A. M., Kim S., and Hatcher P. G. (2006). «Direct molecular evi-
dence for the degradation and mobility of black carbon in soils from ultrahigh-resolution
mass spectral analysis of dissolved organic matter from a fire-impacted forest soil». Organic
Geochemistry. 37: 501—510.
[17] . Huclier-Markai S., Landesman C., Rogniaux C.H., Monteau F., Vinsot A., and Gram-
bow B. (2010). «Nondisturbing characterization of natural organic matter (NOM) con-
tained in clay rock pore water by mass spectrometry using electrospray and atmospheric
pressure chemical ionization modes». Rapid Communications in Mass Spectrometry. 24:
191-202.
[18] . Hughey C.A., Hendrickson C.L., Rodgers R.P., and Marshall A.G. (2001). «Kendrick
mass defect spectrum: a compact visual analysis for ultrahigh-resolution broadband mass
spectra». Analytical Chemistry. 73: 4676—4681.
[19] . Kendrick E. (1963). «А mass scale based on CH2 = 14.0000 for high resolution mass spec-
trometry of organic compounds». Analytical Chemistry. 35: 2146—2154.
[20] . Kim S., Kramer R.W., and Hatcher P. G. (2003). «Graphical method for analysis of ultra-
high-resolution broadband mass spectra of natural organic matter, the van Krevelen dia-
gram». Analytical Chemistry. 75: 5336—5344.
[21] . Kim S., Rodgers P. R., and Marshall A. G. (2006). «Truly «exact» mass: elemental composi-
tion can be determined uniquely from molecular mass measurement at ~0.1 mDa accuracy
for molecules up to -500 Da». International Journal of Mass Spectrometry. 251: 260—265.
Литература
[22] . Koch В. Р., and Dittmar Т. (2006). «From mass to structure: an aromaticity index for high-
resolution mass data of natural organic matter». Rapid Communications in Mass Spectrom-
etry. 20: 926-932.
[23] . Koch В. P., Witt M. R., Engbrodt R., Dittmar T., and KattnerG. (2005). «Molecular formu-
lae of marine and terrigenous dissolved organic matter detected by electrospray ionization
Fourier transform ion cyclotron resonance mass spectrometry». Geochimica et Cosmochi-
micaActa. 69: 3299—3308.
[24] . Koch В. P., Dittmar T., Witt M., and Kattner G. (2007). «Fundamentals of molecular for-
mula assignment to ultrahigh resolution mass data of natural organic matter». Analytical
Chemistry. 79: 1758—1763.
[25] . Koch B.P., Ludwichowski K. U., Kattner G., Dittmar T., and Witt M. (2008). «Advanced
characterization of marine dissolved organic matter by combining reversed-phase liquid
chromatography and FT-ICR-MS». Marine Chemistry. 111: 233—241.
[26] . Koprivnjak J.-F., Pfromm P. H., Ingall E., Vetter T. A., Schmitt-Kopplin P., Hertkorn N.,
Frommberger M., Knicker H., and Perdue E.M. (2009). «Chemical and spectroscopic
characterization of marine dissolved organic matter isolated using coupled reverse osmo-
sis-electrodialysis». Geochimica et Cosmochimica Acta. 73: 4215—4231.
[27] . Kovacs K., Gaspar A., Sajgo C., Schmitt-Kopplin P., and Tombacz E. (2010). «Compari-
sons of humic substances isolated from thermal water and surface water by electrospray ion-
ization Fourier transform ion cyclotron resonance mass spectrometry». European Journal of
Mass Spectrometry. 16:625—630.
[28] . Kroll J. H., Donahue N. M., Jimenez J. L., Kessler S. H., Canagaratna M. R., Wilson K. R.,
Altieri K.R., Mazzoleni L. R., Wozniak A.S., Bluhm H., Mysak E.R., Smith J. R.,
Kolb С. E., and Worsnop R.W. (2011). «Carbon oxidation state as a metric for describing
the chemistry of atmospheric organic aerosol». Nature Chemistry. 3: 133—139.
[29] . Kujawinski E.B. (2002). «Electrospray ionization Fourier transform ion cyclotron reso-
nance mass spectrometry (ESI FT-ICR MS): characterization of complex environmental
mixtures». Environmental Forensics. 3: 207—216.
[30] . Kujawinski E. B., and Behn M. D. (2006). «Automated analysis of electrospray ionization
Fourier transform ion cyclotron resonance mass spectra of natural organic matter». Analyti-
cal Chemistry. 78: 4363—4373.
[31] . Kujawinski E.B., Del Vecchio R., Blough N.V., Klein G.C., and Marshall A.G. (2004).
«Probing molecular-level transformations of dissolved organic matter: insights on photo-
chemical degradation and protozoan modification of DOM from electrospray ionization
Fourier transform ion cyclotron resonance mass spectrometry». Marine Chemistry. 92:
23-37.
[32] . Kujawinski E.B., Longnecker K., Blough N.V., Del Vecchio R., Finlay L., Kitner J. B.,
and Giovannoni S. J. (2009). «Identification of possible source markers in marine dissolved
organic matter using ultrahigh resolution mass spectrometry». Geochimica et Cosmochimica
Acta. 73: 4384-4399.
[33] . Kunenkov E., Kononikhin A., Perminova I., Hertkorn N., Gaspar A., Schmitt-Kopplin,
Ph., Popov I., Garmash A., and Nikolaev E. (2009). «Total mass difference statistics algo-
rithm: a new approach to identification of high mass building blocks in electrospray ioniza-
tion Fourier transform ion cyclotron mass spectrometry data of natural organic matter».
Analytical Chemistry. 81: 10106—10115.
566 Глава 19. Применение масс-спектрометрии сверхвысокого разрешения смасс-
анализатором ионного циклотронного резонанса с преобразованием Фурье для
анализа природного органического вещества в объектах окружающей среды
[34] . Marshall A. G., Hendrickson С. L., and Jackson G. S. (1998). «Fourier transform ion cyclo-
tron resonance mass spectrometry: a primer». Mass Spectrometry Review. 17: 1—35.
[35] . Meija J. (2006). «Mathematical tools in analytical mass spectrometry». Analytical and Bio-
analytical Chemistry. 385: 486—499.
[36] . Reemtsma T. (2009). «Determination of molecular formulas of natural organic matter mol-
ecules by (ultra-) high resolution mass spectrometry — status and needs». Journal of Chro-
matography A. 1216:3687—3701.
[37] . Reemtsma T., These A., Springer A., and Linscheid M. (2006). «Fulvic acids as transition
state of organic matter: indications from high resolution mass spectrometry». Environmen-
tal Science and Technology. 40: 5839—5845.
[38] . Reemtsma T., These A., Linscheid M., Leenheer J., and Spitzy A. (2008). «Molecular and
structural characterization of dissolved organic matter from the deep ocean by FT-ICR-
MS, including hydrophilic nitrogenous organic molecules». Environmental Science and
Technology. 42: 1430—1437.
[39] . Schmidt F., Elvert M., Koch В. P., Witt M., and Hinrichs, K-U. (2009). «Molecular char-
acterization of dissolved organic matter in pore water of continental shelf sediments». Geo-
chimica et Cosmochimica Acta. 73: 3337—3358.
[40] . Schmitt-Kopplin, Ph., Kiss G., Dabek-Zlotorzynska E., Gelencser A., Hertkorn N.,
Harir M., Hong Y, and Gebefiigi I. (2010a). «Analysis of the unresolved organic fraction
in atmospheric aerosols with ultrahighresolution mass spectrometry and nuclear magnetic
resonance spectroscopy: organosulfates as photochemical smog constituents». Analytical
Chemistry. 82: 8017—8026.
[41] . Schmitt-Kopplin, Ph., Gabelica Z., Gougeon R.D., Fekete A., Kanawati B., Harir M.,
Gebefuegi I., Eckel G., and Hertkorn N. (2010b). «Murchison meteorite, 40 years after
unprecedented molecular diversity of extraterrestrial organic matter». Proceedings of the
National Academy of Sciences of the United States of America. 107: 2763—2768.
[42] . Sleighter R.L., McKee G.A., and Hatcher P. G. (2009). «Direct Fourier transform mass
spectral analysis of natural waters with low dissolved organic matter». Organic Geochemis-
try. 119-125.
[43] . Stenson A. (2008). «Reversed-phase chromatography fractionation tailored to mass spec-
tral characterization of humic substances». Environmental Science and Technology. 42:
2060-2065.
[44] . Stenson A.C., Marshall A.G., and Cooper W.T. (2003). «Exact masses and chemical for-
mulas of individual Suwannee River fulvic acids from ultrahigh resolution electrospray ion-
ization Fourier transform ion cyclotron resonance mass spectra». Analytical Chemistry. 75:
1275-1284.
[45] . Suhre K., and Schmitt-Kopplin P. (2008). «MassTRIX: Mass TRanslator Into Pathways».
Nucleic Acids Research. 36, Web Server issue: W481—W484.
[46] . Tipping E. (2002). «Cation Binding by Humic Substances». Cambridge University Press.
Cambridge, UK.
[47] . Van Krevelen D.W. (1950). «Graphical statistical method for the study of structure and
reaction processes of coal». Fuel. 29: 269—284.
[48] . Witt M., Fuchser J., and Koch В. P. (2009). «Fragmentation studies of fulvic acids using col-
lision induced dissociation Fourier transform ion cyclotron resonance mass spectrometry».
Analytical Chemistry. 81: 2688—2694.
Литература
[49] . Wu Z.G., Rodgers R.P., and Marshall A.G. (2004). «Two- and three-dimensional van
Krevelen diagrams: a graphical analysis complementary to the Kendrick mass plot for sort-
ing elemental compositions of complex organic mixtures based on ultrahigh-resolution
broadband Fourier transform ion cyclotron resonance mass measurements». Analytical
Chemistry. 76: 2511—2516.
[50] . Zhang F., Zhang J., Schmitt-Kopplin P., Fekete A., Wu Y., Gaspar A., and Hertkorn N.
(Undated). «Characterization of dissolved organic matter during and after cyanobacterium
(Microcystis aeruginosa) bloom in Taihu Lake, China». Organic Geochemistry. Submitted
for publication.
ГЛАВА 20
МЕТОД МАСС-
СПЕКТРОМЕТРИЧЕСКОЙ
ВИЗУАЛИЗАЦИИ
(ИМИДЖИНГ)
Бернхард Шпенглер
20.1. Введение
Химические взаимодействия веществ в окружающей среде не только зависят от их хи-
мических и термодинамических свойств, но также определяются степенью их колока-
лизации и динамикой (пространственной и временной) соответствующих молекуляр-
ных процессов, причем на весьма сложном уровне. Поэтому крайне важно разработать
аналитические методы, которые в состоянии описать состав исследуемых образцов
не только в качественном и количественном аспектах, но также и в пространственном
контексте, что обеспечит пространственную информацию о взаимодействиях различ-
ных веществ и о локальном накоплении важнейших соединений. Применение про-
странственного химического анализа важно при исследованиях биологических тканей,
органов или клеток, образцов почвы, поверхности раздела фаз объектов окружающей
среды или мембран. В этой главе описываются достижения в молекулярной визуализа-
ции с помощью масс-спектрометрии (МС), методологические принципы и стандартные
приложения этого метода в области биологических и экологических наук.
20.2. Микрозондирование и молекулярная
визуализация
Общий состав гомогенизированной биологической ткани или образца, полученного при
анализе окружающей среды, зачастую неинформативен для выяснения основных прин-
ципов, которые управляют биохимическими, токсикологическими или транспортны-
ми процессами. Для изучения многих явлений необходимо применять методы, которые
позволяют получить не усредненную, а локальную аналитическую информацию, как
по поверхности исследуемого образца, так и по его глубине. Одним из таких методов
является микрозондирование, которое позволяет получать данные о молекулярном или
элементном составе замкнутых или локализированных областей. Другой метод основан
на визуализации состава поверхности методом сканирования с получением оптических
20.2. Микрозондирование и молекулярная визуализация 569
или ионно-оптических изображений. Методы могут быть разрушающими или нераз-
рушающими, что влияет на возможность изучения живых организмов без изменения
их состояния. И, самое главное, они могут быть узкоспециализированными (целевыми)
или общими (неспецифическими). Анализ с использованием целевых методов иногда
требует прикрепления конкретной молекулярной метки к выбранному для анализа со-
единению, тогда как неспецифические методы способны обнаруживать большое коли-
чество ранее неизвестных соединений без предварительного мечения.
Методы, основанные на микрозондировании или на получении изображений,
предоставляют информацию о локализации отдельных билогических маркеров в тка-
нях или в почве. Распределение маркеров в инфицированных тканях или загрязненных
образцах почвы предоставляет информацию, которая важна для понимания состояний
и процессов в естественной среде. Типичная цель таких исследований — это количе-
ственный анализ неорганических веществ, таких как фосфор, железо, селен или ко-
бальт; анализ пестицидов, гормонов (при передаче их из организма в другой организм
или окружающую среду), токсичных загрязняющих веществ, наркотиков, соединений
и их метаболитов, липидов, пептидов, белков или углеводов. Информация о распределе-
нии соединений в сложной среде, например такой, как почва или биологический орган,
может быть получена путем измерения локальной концентрации данных соединений.
Очевидно, что локальный анализ, основанный на получении изображений, должен да-
вать приемлемый уровень аналитической точности. Помимо получения информации
о пространственном распределении индивидуальных соединений, так называемые то-
понимические исследования изучают колокализацию ансамблей молекул соединений.
Локализация молекул илихимическихэлементов методом построения изображений
является общим принципом нескольких классических методов, таких как радиоактив-
ный иммуноанализ или флюоресцентная микроскопия. Существенным недостатком
этих методов является то, что изображаемые молекулы должны быть заранее известны
и промаркированы. Поэтому эти методы не могут использоваться для изучения про-
странственного распределения неизвестных соединений. Ограничение, связанное с не-
обходимостью маркировки, можно преодолеть, если химический состав в определенном
месте образца определяется напрямую, без какой-либо специальной обработки иссле-
дуемых соединений. Такой пространственный анализ в принципе можно осуществить
с помощью масс-спектрометрии (МС). Масс-спектрометры для анализа микроскопи-
ческих количеств вещества (микропробы) использовались на протяжении многих лет.
В 1970-х был разработан один из первых приборов для лазерного МС-анализа микро-
проб. Этот прибор позволил определить элементный и молекулярный состав биологи-
ческих образцов {Hillenkamp et al., 1975) с приемлемым пространственным разрешением.
Для получения ионов в этом случае применялся метод лазерной десорбции/ионизации
(ЛДИ). В ЛДИ хорошо сфокусированное излучение импульсного УФ-лазера использу-
ется для испарения и ионизации биологических или синтетических материалов. Обра-
зовавшиеся при облучении микроскопической области образца ионы анализировались
во времяпролетном (ВП) масс-спектрометре {Spengler et al., 1989). Подобные приборы
до сих пор используются в нескольких лабораториях по всему миру, хотя ЛДИ как метод
поверхностной ионизации значительно уступает по чувствительности и диапазону ре-
гистрируемых молекулярных масс другому методу — матрично-активированной лазер-
ной десорбции/ионизации (МАЛДИ). Масс-спектрометрия вторичных ионов (МСВИ),
Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
в частности применение ВП-МСВИ для построения ионных изображений, является
еще одним методом элементного и молекулярного анализа с пространственным раз-
решением (Benninghoven, 1994). МСВИ позволяет проанализировать атомы и небольшие
молекулы поверхностного слоя синтетических или биологических образцов; при этом
анализируется очень тонкий поверхностный слой с высоким поперечным разрешени-
ем в несколько микрометров. С появлением новых источников первичных ионов, где
используются ускоренные кластеры, МСВИ-анализу стали доступны и высокомолеку-
лярные соединения. Это позволило анализировать и получать изображения липидов
и фрагментов больших биомолекул (Toubouletal., 2005).
С появлением новых эффективных методов — МАЛДИ {Karas и НШепкатр, 1988)
и ионизации электрораспылением (ЭРИ) (Фенн et al., 1989), основной интерес анали-
тической биохимии как науки переместился в направлении изучения больших молекул
(например белки) в биологических образцах. Пространственный МС-анализ стал попу-
лярен в 90-е годы (главы 1 и 3). Метод построения МАЛДИ-изображений впервые был
описан в 1994 году (Spengler et al., 1994) и нашел широкое применение в исследованиях
биологических образцов в последующие годы (Stoeckli et al., 2001).
20.3. Визуализация пространственного
распределения молекулярного состава
Процедура перевода масс-спектрометрических данных в микроаналитические изобра-
жения требует значительных усилий по обработке и хранению данных. Упрощенный
подход показан на рис. 20.1. Хорошо сфокусированный луч импульсного лазера или сфо-
кусированный поток первичных ионов направляется в определенную точку образца,
прикрепленного к подвижной платформе. Полученные масс-спектры хранятся вместе
с информацией о позиции лазерного луча (потоке первичных ионов) на исследуемом об-
разце. Диаметр фокусного пятна лазера или ионного пучка на поверхности определяет
достижимое пространственное разрешение. Диаметр фокусного пятна обычно опреде-
ляется выбранным шагом сканирования. После анализа одной точки образца платфор-
ма, на которую помещен образец, перемещается на один шаг; соответственно в другом
подходе перемещение точки сканирования происходит за счет перепозиционирования
лазерного луча или ионного пучка. В большинстве лазерных приборов лазерный луч
фиксируется, а перемещается платформа, в то время как МСВИ-анализ с высоким раз-
решением осуществляется перемещением первичного ионного луча, а анализ с низким
разрешением — при движении платформы. Масс-спектр регистрируется для каждой но-
вой позиции луча или потока первичных ионов.
После окончания сканирования интересующей области образца в двух измерениях
и завершения записи масс-спектров, каждый из которых соответствует определенному
местоположению луча, весь набор данных используется для формирования изображе-
ний. Для поиска интересующих масс (т.е. химических соединений) используется спе-
циальное программное обеспечение быстрого просмотра сохраненных масс-спектров.
Это принципиально иной подход по сравнению с классическим, когда создается изо-
бражение на основе пространственного распределения заранее известных веществ
20.3. Визуализация пространственного распределения молекулярного состава
Рис. 20.1. Схема получения МС-изображений при лазерной десорбции/ионизации. Опре-
деленное число импульсов наносекундной длительности хорошо сфокусиро-
ванного лазера используется для анализа (десорбции и ионизации) материала
образца на поверхности. Ионы, образованные при облучении одной микроско-
пической области образца, анализируются масс-спектрометром; полученные
спектры записываются и хранятся вместе с информацией о позиции лазерного
пятна. После завершения анализа в одной точке образец перемещается и лазер
облучает следующую точку образца, пока не проанализируется вся его площадь.
Используя набор сохраненных данных, МС-изображения создаются путем пре-
образования интенсивности выбранных ионных сигналов для каждой позиции
лазерного луча в значения яркости соответствующих пикселей, используя от-
тенки серого цвета
(как например в флюоресцентной микроскопии) и где аналитическая информация
об образце ограничена заранее выбранным соединением.
Интесивность пиков, соответствующих определенной массе, может быть использо-
вана для построения изображений с помощью оттенков серого цвета. В таких изобра-
жениях яркость пикселя определяется величиной относительной интенсивности пика.
Значения, соответствующие белому (пики с самой высокой интенсивностью сигнала)
и черному (пики с самой низкой интенсивностью сигнала) могут быть определены пу-
тем нормализации интенсивности пиков по всему множеству пикселей на одном изо-
бражении либо по всему набору изображений.
Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
Синтезированные изображения (псевдоцветные изображения) могут быть постро-
ены на основе изображений с оттенками серого цвета. Такие изображения могут быть
использованы для увеличения динамического диапазона интенсивностей, который
может быть различим человеческим глазом. Более важным преимуществом исполь-
зования цвета является возможность объединить три изображения, где используются
оттенки серого и которые соответственно получены для трех разных соединений, и пе-
реопределить их в три цвета: в первом изображении в качестве шкалы интенсивности
использовать интенсивность красного цвета, во втором — зеленого и в третьем — сине-
го, построив таким образом синтезированное КЗС-изображение. На полученной таким
способом картинке можно легко увидеть колокализацию или сегрегацию соединений.
Наличие двух веществ в одном и том же пикселе, закодированных соответственно зеле-
ным и красным цветами, приведет к появлению желтого пикселя (рис. 20.2).
Помимо изображений, демонстрирующих распределение интенсивностей для од-
ного соединения, изображения могут быть созданы на основе сложных сочетаний
и корреляций сигналов. В будущем этот принцип может привести к новому способу ви-
зуализации масс-спектрометрических данных: например, изображение распределения
сигналов отдельных веществ может быть заменено синтезированным изображением ха-
рактерных особенностей распределения множества ионных сигналов.
При программной обработке изображений первостепенное значение имеет сохране-
ние прямой связи обработанного изображения и первоначальных аналитических дан-
ных. Используемое программное обеспечение, таким образом, должно давать пользова-
телю возможность вернуться из обработанного цветного изображения к лежащему в его
основе необработанному изображению в шкале серого цвета и далее к индивидуальным
масс-спектрам, которые формируют данное изображение. Этот принцип управления
качеством является обязательным условием использования МС-имиджинга в качестве
аналитического метода.
Рис. 20.2. Модельное цветное (КЗС) изображение, составленное из трех изображений се-
рого цвета. Колокализация двух веществ, одно из которых представлено зеленым
сигналом определенной яркости, а другое — красным сигналом определенной
яркости, приводит к появлению желтых пикселей. КЗС-изображения позволяют
в принципе описать локальное распределение трех различных соединений, вы-
бранных из МС-набора данных, полученных при сканировании образца
20.4. Влияние матрицы
В отличие от МСВИ-изображений, МАЛДИ МС-визуализация требует применения
стандартной матрицы для получения масс-спектров образца. Наличие матрицы оказы-
вает негативное влияние на линейность сигнала и часто приводит кего подавлению, что,
однако, считается приемлемым ограничением в обычном МАЛДИ-МС-анализе. Нали-
чие матрицы создает еще одну проблему, которая связана с ограничением простран-
ственного разрешения. Так как матрица и исследуемое вещество должны быть смешаны
в жидкой фазе для достижения оптимальной аналитической чувствительности, матрица
должна быть нанесена на поверхность образца в жидком состоянии. Такой способ нане-
сения обязательно приводит к потере пространственного разрешения, так как молеку-
лы аналита мигрируют из их первоначального положения в образце в жидкую микро-
каплю с матрицей, где они и обнаруживаются при анализе. Поэтому необходимо найти
компромисс между аналитической чувствительностью и потерей пространственного
разрешения. Как и в обычном МАЛДИ-МС-методе, целевые соединения, доступные
МАЛДИ-МС, должны быть включены в виде примесей в растущие кристаллы матрицы
после высыхания раствора. Это означает, что для получения изображений с высоким
разрешением раствор матрицы следует наносить в виде микрокапель, которые, с одной
стороны, содержат достаточно растворителя, чтобы не влиять на процесс инкорпориро-
вания аналита в кристаллическую структуру высохшей матрицы, а с другой стороны,
достаточно малы, чтобы избежать перемешивания жидкости и исследуемого вещества
по площади образца. Приемлемый компромисс был достигнут при использовании спе-
циального метода пневматического распыления, когда в качестве ускоряющего газа
использовался азот, а также при использовании вакуумного напыления. В последнем
случае микроскопический слой сухой матрицы выращивается на поверхности образ-
ца в вакууме путем возгонки молекул из кристаллов матрицы, на втором этапе обра-
зец помещается во влажную атмосферу и микроскопический слой матрицы постепенно
рекристаллизуется прямо на поверхности во влажной атмосфере (Bouschen et al., 2010).
Вследствие ограничения миграции исследуемых молекул по поверхности образца оба
метода позволяют получить эффективное пространственное разрешение порядка 2—3
микрон. Хотя в обычной масс-спектрометрии МАЛДИ-оптимизация взаимодействия
матрица — аналит представляет интерес главным образом для достижения высокой
чувствительности, многие другие микроскопические явления оказывают значительное
воздействие на качество результатов при получении хорошо разрешенных изображений
с помощью МАЛДИ-МС. Эти микроскопические явления включают в себя сегрегацию
соединений (эффект местного обогащения) в сложных образцах и разделение анали-
тов и ионных соединений на поверхности образца. Наблюдать эти явления можно при
анализе изображений с высоким разрешением, которые получаются при использовании
стандартных методов подготовки поверхности образца (рис. 20.3). Распределение пеп-
тида (вещество Р) в микрокристаллах высохшей капли, содержащей раствор 2,5-диги-
дроксибензойной кислоты (DHB) — полулярной МАЛДИ-матрицы, — демонстрирует
расположение молекул аналита, главным образом внутри микрокристаллов матрицы.
Ионы щелочных металлов (например К+), которые, как известно, уменьшают эффектив-
ность ионизации в МАЛДИ и тем самым снижают достижимую чувствительность, как
видно из представленной картинки, исключаются из кристаллов матрицы в процессе
Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
Матрица DHB
Пептид:
(вещество Р)
Примеси соли:
калий
Рис. 20.3. Первое полученное МАЛДИ/МС-изображение (Spengler et aL, 1994), демон-
стрирующее распределение пептида (вещество Р) в высушенной капле DHB-
матрицы. Изображение имеет пространственное разрешение порядка 1 мкм
и получено при помощи ВП-масс-спектрометра, в котором сканирование об-
разца осуществлялось с высокой точностью. На этом изображении видно, что
ионы солей отделены от молекул аналита. Такое разделение указывает на то,
что именно молекулы аналита влючались в виде примесей в кристаллическую
структуру микрокристаллов матрицы. Это означает, что сокристаллизация ис-
следуемого вещества и материала матрицы является уникальным и методологи-
чески важным шагом очистки
кристаллизации и мигрируют во внешние области, где структура твердого вещества,
образованного при высыхании матрицы, более аморфна. В результате этого перерас-
пределения интенсивности вещества Р и К+ на рис. 20.3 оказываются почти идеально
инвертированными по отношению друг к другу.
С микроскопической точки зрения нанесение матрицы, очевидно, приводит
к очистке образца от ионов щелочных металлов и определяет высокий выход десорбции
и ионизации биомолекул, расположенных в областях, свободных от солей щелочных
металлов. В обычной технике МАЛДИ-МС, когда используется лазерное пятно диаме-
тром в несколько сотен микрометров, этот эффект в основном остается незамеченным,
так как лазерный луч покрывает и кристаллические, и аморфные области. В результате
в масс-спектрах наблюдаются сигналы как от аналита, так и от ионов щелочных метал-
лов. Таким образом, взаимное разделение двух этих типов аналитов не обнаруживается.
При использовании хорошо сфокусированного лазера с пятном порядка одного микро-
на для достижения высокого разрешения, как, например, в методе микроанализа при
использовании сканирующего МАЛДИ-МС-прибора (сМАЛДИ), эти микроскопиче-
ские явления могут быть хорошо видны.
Включение аналита в кристаллическую матрицу является предварительным усло-
вием для высокочувствительного МС-анализа веществ, которые принадлежат к тому
или иному классу. При МАЛДИ-МС или СМАЛДИ-МС-визуализации сложных об-
разцов, таких как биологические ткани или образцы грунта, матрица должна быть до-
бавлена к образцу таким образом, чтобы произошло смешение матрицы с исследуемым
веществом (пусть даже на короткий срок); далее должны быть обеспечены условия,
которые позволили бы расти кристаллам матрицы, каждый из которых «легирован»
молекулами аналита. Очевидно, что при этом обязательно происходит размывание
концентрации первоначально локализованного аналита, что приводит к появлению
новых пиков (артефактов), которые обьясняются пространственным разделением (се-
20.5. Применение масс-спектрометрии вторичных ионов (МСВИ) в биоанализе
грегацией) веществ при кристализации. Таким образом, чтобы получить высокое про-
странственное разрешение и одновременно высокую аналитическую чувствительность,
необходимо придумать достаточно хорошее компромиссное решение. Некоторые опти-
мальные решения были найдены для техники пострения МС-изображений с помощью
СМАЛДИ, где было продемонстрировано пространственное разрешение в 3 микрона
и низкий предел обнаружения (Bouschen etal., 2010). Альтернативные методы получения
МС-изображений, не требующие применения матрицы, находятся в стадии разработ-
ки. Примером может служить получение МС-изображений методом лазерной десорб-
ции/ионизации с использованием инфракрасного лазера.
Достижимое пространственное разрешение, очевидно, является ключевым крите-
рием полезности и применимости метода МС-изображений. Можно довольно легко най-
ти техническое решение для достижения пространственного разрешения в диапазоне 50
микрон для метода МС-изображений с использованием МАЛДИ. Значительно больше
усилий требуется для того, чтобы создать лазерный ионно-оптический инструмент,
который позволяет анализиривать с высоким разрешением клеточные структуры, что
соответствует достижению эффективного разрешения в диапазоне микрометров. В ли-
тературе были описаны лишь очень немногие примеры, демонстрирующие эффектив-
ное пространственное разрешение лучше 10 микрон (Chaurand et al., 2007; Guenther et al.,
2011; Rompp et al., 2010). Этот режим высокого пространственного разрешения в методе
получения МС-имиджинга был назван сканирующим лазерным МАЛДИ-микрозонди-
рованием (СМАЛДИ-МС) (Spengler и Hubert, 2002). Он отличается от обычного метода
МС-имиджинга с низким разрешением не только по своим аналитическим целям, но и
в связи с тем, что при таком малом лазерном пятне у материала, который подвергается
испарению и ионизации, проявляются совершенно иные механические свойства. Пер-
вая демонстрация, указывающая на то, что методика получения избражений с помощью
МАЛДИ-МС, тем не менее, может использоваться для анализа биомолекул, даже когда
лазерное пятно на поверхности имеет поперечный размер всего 1 микрон, появилась
в 1994 году (Spengler et al., 1994) (рис. 20.3).
20.5. Применение масс-спектрометрии вторичных
ионов (МСВИ) в биоанализе
Бомбардировка поверхности в высоком вакууме с помощью быстрых атомов или ио-
нов приводит к образованию вторичных ионов и их десорбции с поверхности. Техника
МСВИ использовалась на протяжении многих лет для получения информации об эле-
ментном составе поверхностных атомных слоев твердых веществ. В отличие от МАЛ-
ДИ, предел обнаружения в МСВИ в высокой степени зависит от молекулярного веса
анализируемого вещества, поскольку механизмы закачки энергии в поверхностный
слой при ионной бомбардировке и при поглощении фотонов достаточно сильно раз-
личаются. Использование классических источников первичных ионов (цезиевые или
жидкометалличесткие ионные пушки) для МСВИ-анализа приводит к сильному уве-
личению пределов обнаружения при увеличении молекулярного веса анализируемого
вещества. Это ограничивает рабочий диапазон метода соединениями с молекулярным
весом от нескольких сотен до одной тысячи дальтон. На практике органические молеку-
Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
лы регистрируются (и, соответственно, идентифицируются) в спектрах МСВИ, главным
образом в виде набора мелких фрагментных, но не молекулярных ионов. Новые раз-
работки в области ионных источников значительно изменили эту ситуацию. Исполь-
зование добавленной матрицы (по аналогии с МАЛДИ) привело к значительному рас-
ширению доступного диапазона масс. Этот метод называют матрично-активированной
МСВИ (Wuu Odom, 1996). Он был использован для анализа небольших биоорганических
молекул и получения МСВИ-изображений. При этом лимитирующие этот метод про-
странственные эффекты были аналогичны тем, которые наблюдаются в случае МАЛ-
ДИ. Применение многозарядных кластеров в качестве первичных ионов позволило
значительно улучшить воспроизводимость и расширить диапазон масс анализируемых
молекул. Использование многозарядных кластеров висмута (Brunelle и Laprevote, 2009)
или ионов С60 (Carado et al., 2008) в качестве первичных ионов привело в существенному
улучшению предела обнаружения для крупных биомолекул. Многие биомолекулы все
еще определяются в МСВИ посредством анализа ионов-продуктов, однако при исполь-
зовании многозарядных кластеров в качестве первичных ионов такие классы веществ,
как фосфолипиды, углеводы и пептиды, были впервые определены в образцах, где их
концентрация соответствует естественному уровню. Однако даже с учетом этих новых
технологий белки по-прежнему не регистрируются в МСВИ как при общем анализе, так
и в режиме сканирования поверхности при получении изображений. МСВИ, однако,
является эфективным методом МС-визуализации, когда необходимо проанализировать
элементный состав поверхности или, например, когда необходимо описать морфоло-
гию проб окружающей среды по отношению к микроэлементам, пестицидам или дру-
гим небольшим молекулам. Самым большим преимуществом МСВИ по отношению
к лазерным методам получения МС-изображений, безусловно, является более высокое
пространственное разрешение, которое может достигать нескольких нанометров, в за-
висимости от аналитического предела обнаружения для данного вещества. Недостатком
МСВИ является отсутствие опции МС/МС в коммерческих приборах. Это не позволяет
охарактеризовать и идентифицировать молекулярные структуры в сложных смесях.
20.6. Качество изображения и аналитический предел
обнаружения
Процесс кристаллизации матрицы является не единственным ограничением достижи-
мого пространственного разрешения. Для всех МС-методов визуализации существует
внутреннее ограничение, которое состоит в том, что при анализе меньшего по размеру
пятна в выбранной области задействовано квадратично меньшее число молекул анали-
та. Например, уменьшение размера исследуемой области от 10 до 1 мкм может привести
к 100-кратному ухудшению аналитического предела обнаружения; в реальности умень-
шение чувствительности зависит от степени уменьшения так называемого эффекта
химического шума (падение отношения сигнал—шум может быть меньше, чем 100 раз
при 10-кратном уменьшении поперечного размера пятна, если уровень шума также
уменьшается). Несмотря на то, что масс-спектрометрические анализаторы становятся
все более и более чувствительными, не существует никаких сомнений, что существует
абсолютный предел полезного пространственного разрешения. В случае классических
20.7. МС-визуализация, как метод качественного анализа
поверхностно-чувствительных методов визуализации (например МСВИ) можно под-
считать, что образец размером 1 микрон в диаметре содержит не более 200000 молекул
среднего молекулярного веса. Принимая во внимание, что пробы окружающей среды,
как правило, — достаточно сложные системы, состоящие из сотен и тысяч различных
веществ, ситуация может достигнуть такого уровня, что только несколько молекул со-
единения, которое необходимо охарактеризовать, будет присутствовать в данной малой
области. В дополнение статистическое ограничение на то, что считается регистриру-
емым сигналом, требует наличия минимального количества ионов данного вещества
в одном спектре. МАЛДИ-МС находится в несколько лучшем положении в этом отно-
шении, так как этот метод не является реальным поверхностным аналитическим ме-
тодом. МАЛДИ-МС-анализирует не только первый монослой, но некий слой глубиной
до 300 нм при использовании лазерного пятна в 1 мкм.
В целом может быть показано, что независимо от выбранного масс-спектро-
метрического метода МС-изображения с пространственным разрешением значительно
меньше, чем 1 мкм, не будут давать какую-то исключительно полезную информацию
при изучении распределения веществ в типичных биоаналитических или экологиче-
ских образцах.
20.7. МС-визуализация как метод качественного
анализа
Метод получения МС-изображений с точки зрения других аналитических мето-
дов все еще находится в зачаточном состоянии. Результаты экспериментов по МС-
визуализации, опубликованные в первые годы, главным образом были хорошо вос-
приняты потому, что цветные изображения, которые содержат узнаваемые структуры,
немедленно создавали некую аналитическую ценность для человеческого глаза, в от-
личие от представления данных в табличном виде, что часто подвергается справедливой
критике со стороны большинства ученых. Несмотря на то, что метод МС-имиджинга
оказался перспективным, он все еще не позволяет справедливо судить о воспроизво-
димости и аналитической важности полученных результатов. Изображения на основе
масс-спектрометрических данных указывают на распределение веществ с неким моле-
кулярным весом в образцах, но природа этих веществ и однозначность регистрируемых
сигналов часто остаются неизвестными. Метод МС-изображений, вследствиеуказанных
технических и методологических проблем, остается по сути качественным и описатель-
ным (можно даже сказать: «импрессионистским») а не проверенным (валидированным)
методом аналитической химии. При использовании МАЛДИ-МС для визуализации
распределения молекул в биологических срезах были получены исключительно каче-
ственные результаты, причем с низким пространственным разрешением, низким раз-
решением по массе и низкой точностью измерения масс. В МСВИ-изображениях каче-
ственные результаты были получены для диапазона довольно низких масс, когда эти
ионы играют роль маркеров больших биомолекул.
При исследовании реальных биоаналитических образцов или проб окружающей
среды было продемонстрировано пространственное разрешение на уровне микро-
метров или даже субмикрометров. Высокое пространственное разрешение в МСВИ
Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
Рис. 20.4. МСВИ-изображения двух простейших во время спаривания (сопряжения).
В зоне сопряжения, которая формируется двумя мембранами, концентрация
фосфатидилхолина ниже (справа), в то время как общая концентрация фосфо-
липидов (в центре) остается постоянной. Рисунок (слева) показывает предпо-
лагаемое распределение фосфатидилхолина (белые кружки) и фосфатидилэта-
ноламина (черные кружки). (Источник: Ostrowski S.G., van Bell С.Т., Winograd N.
Ewing A.G. (2004). «Масс-спектрометрическое изображение сильно изогнутой
мембраны во время спаривания Tetrahytnena». Science. 305: 71—73. Печатается
с разрешения AAAS)
позволяет проводить измерения и визуализировать биохимические процессы в кле-
точной мембране при условии, что сигналы целевых молекулярных ионов превышают
предел обнаружения. Впечатляющим примером является визуализация спаривания
Tetrahymena thermophila во время слияния мембран клеток двух микроорганизмов, отно-
сящихся к простейшим, в результате которого присходит обмен генетическим матери-
алом (Ostrowski et aL, 2004). На рисунке изображена зона слияния двух мембран, внутри
которой наблюдается значительное снижение концентрации фосфатидилхолина по от-
ношению к общей концентрации фосфолипидов (рис. 20.4). Также наблюдается значи-
тельное перераспределение липидов в зоне слияния мембран. В этом исследовании изо-
бражаемый ионный сигнал был идентифицирован как фрагмент фосфатидилхолина,
причем эта идентификация была произведена не из непосредственно МСВИ-спектров,
а из данных, которые были получены независимо с помощью ЭРИ-МС/МС-анализа го-
могенизированных образцов. При этом предполагаемая идентичность двух сигналов,
полученных двумя разными методами, выводилась из идентичности значений масс
(хотя данные были получены с низким разрешением). Детальная характеризация типов
фосфатидилхолинов непосредственно в мембране была невозможна, так как наблюдае-
мый фрагментный ион соответствовал только головной части группы, а не всей молеку-
ле, состоящей в том числе из изомерных жирных кислот.
20.8. МС-визуализация как точный аналитический
метод
Кроме МСВИ, только метод получения МАЛДИ-изображений при сканировании
с высоким разрешением (СМАЛДИ), позволяет достичь разрешения на клеточном
и субклеточном уровне. В отличие от МСВИ массы веществ, анализируемых мето-
20.8. МС-визуализация как точный аналитический метод
дом СМАЛДИ/МС-изображений не ограничены по верхнему пределу. Так, например,
СМАЛДИ-исследование распределения веществ при анализе клеток, полученных
из тканей человеческой почечно-клеточной карциномы при диаметре лазерного фо-
куса 1 микрометр, выявило большое число сигналов вплоть до молекулярной массы
в 15000дальтон(/?ои$сЛелetal., 2010). Прорывваналитическойточности МС-изображений
был достигнут путем сочетания высокого пространственного разрешения, высоко-
го разрешения по массе и точности измерения масс. Это позволило добиться высокой
селективности благодаря построению изображений в узком диапазоне масс. При этом
имелась возможность выяснения природы неизвестных пиков по МС/МС-спектрам
и сохранялось нативное состояние ткани, поскольку анализ проводился с помощью
ионного источника, работающего при атмосферном давлении (Guenther et al., 2011; Rompp
et aL, 2010). В этих условиях МС-имиджинг показал себя в качестве надежного и весь-
ма специфичного метода гистологического анализа фосфолипидов и нейропептидов.
Типы и подтипы тканей могут быть дифференцированы и охарактеризованы с про-
странственным разрешением до 3 мкм. Метод МАЛДИ-МС-изображений в качестве
общего метода анализа поверхности способен выявить сотни сигналов, характерных
для данного типа ткани, что значительно расширяет возможности классических узко-
специализированных (то есть основанных на маркировке) гистохимических методов.
Распределение нейропептидов в гипофизе мыши было получено с высоким разре-
шением по массе, высокой точностью измерения масс и высоким пространственным
разрешением (Guenther et aL, 2011). Проведенное исследование позволило дифферен-
цировать отдельные клетки, идентифицировать пептиды непосредственно в клетках
Oxytocin
РОМС 76-85
ADH
AVP-NP11 154-168 AVP-NPII 151-168
РОМС 103-120
Ас-РОМС 124-136 DiAc-POMC 124-136
Рис. 20.5. Распределение восьми нейропептидов в гипофизе мыши. Данные получены при
обработке МС-спектров с высоким разрешением (как по массе, так и по про-
странству). МАЛДИ-МС-изображения тонких срезов гипофизных тканей мозга
мыши. (Для получения дополнительной информации о подобном исследовании
см. Guenther S., Rompp A., Kummer W. и Spengler В. «Использование МАЛДИ-
МС-источника при атмосферных условиях для визуализации нейропептидов
в мышином гипофизе с пятимикроным пространственным разрешением и вы-
сокой массовой точностью». Int. J. Mass Spectrom., 2011, 305: 228—237)
Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
путем проведения МС/МС-анализов выбранных родительских ионов и достичь про-
странственного разрешения в 5 мкм. Рис. 20.5 иллюстрирует распределение восьми
нейропептидов, взятых из полного набора данных, и демонстрирует полезность общего
анализа, выполненного методом МАЛДИ-МС-изображений. Обнаруженные пептиды
были специфическим образом модифицированы или расщеплены в гипофизе после
их биосинтеза. Метод МС-изображений позволяет провести быстрый просмотр масс-
спектрометрических данных, при этом неожиданные изменения известных соедине-
ний или неизвестные соединения могут быть легко распознаны и отображены.
20.9. Идентификация и характеристика
Почти все масс-спектрометрические методы получения изображений, опубликованные
до сих пор, были ограничены полуколичественным или только качественным пред-
ставлением и давали двумерное распределение интенсивности сигналов в выбранном
интервале масс. При этом МС-изображения не позволяли идентифицировать и оха-
рактеризовывать вещества, которым соответствовали те или иные пики. Кроме того,
аналитическая однородность выбранного интервала масс не гарантировалась при ус-
У5 Уб У7 У8 У9 У10
Уп У12 У13 У14 У15 У16
600 800 100 1200 1400 1600 1800 2000
m/z
Рис. 20.6. Пример изображений, полученых при использовании СМАЛДИ МС/МС, визу-
ализации тонких срезов тканей гипофиза мыши. Изображения получены при
обработке фрагментных ионов нейропептида AEEEAVWGDGSPEPSPRE-NH2
(РОМС 103—120, m/z 1940,8626). Размер шага сканирования (ширина пикселей
и высота) составлял 10 мкм. В качестве масс-спектрометра использовался орби-
трэп (Thermo Fisher Scientific GmbH, Бремен), оснащенный МАЛДИ-источни-
ком с высоким пространственным разрешением. (Более подробную информа-
цию можно найти в работе GuentherS., RomppA., Kummer W. и Spengler В. (2011).
«Использование МАЛДИ-МС-источника при атмосферных условиях для ви-
зуализации нейропептидов в мышином гипофизе с пятимикроным простран-
ственным разрешением и высокой массовой точностью». /nt. J. Mass Spectrom.,
2011,305: 228-237)
20.10. МС-имиджинг в исследованиях объектов окружающей среды
ловии, когда использовались МС-приборы с низким разрешением по массе. Таким об-
разом, в подобных исследованиях аналитическая проверка была невозможна. В силу
этого необходимо было использовать классические масс-спектрометрические методы
идентификации веществ в качестве параллельного метода, используя гомогенизиро-
ванный материал тканей. Идентичность между сигналом, полученным при построении
МС-изображения, с одной стороны, и веществом, которое было идентифицировано при
параллельном анализе, с другой стороны, подразумевалось, но не могло быть строго до-
казано. Это ограничение было устранено в текущей методологии, поскольку компонен-
ты сигнала с определенной массой теперь можно структурно идентифицировать в про-
цессе получения изображения поверхности образца благодаря высокому разрешению
масс-анализатора, высокой точности измерения массы и возможности регистрации
МС/МС-спектров. Таким образом, стала вполне достижима непосредственная харак-
теризация соединений в процессе МС-имиджинга (рис. 20.6). Применение источников
ионов, которые позволяют получать изображения с высоким пространственным раз-
решением, и высокоточных масс-спектрометров, использующих преобразование Фурье
(МСПФ), позволяет идентифицировать отдельные компоненты образца напрямую (то
есть без МС/МС-анализа), с помощью комбинаторного анализа данных. Этот анализ
позволяет делать надежные выводы о типе и структуре соединения по его точной экс-
периментальной массе и элементному составу (Spengler, 2004).
20.10. МС-имиджинг в исследованиях объектов
окружающей среды
В настоящее время техника МАЛДИ-МС-изображений доказала свою полезность и ана-
литическую пригодность в основном в области исследования тканей млекопитающих.
Этот метод, однако, имеет большой потенциал во многих других областях, в том чис-
ле в исследованиях объектов окружающей среды. В настоящее время техника МСВИ,
в частности нано-МСВИ, уже доказала свою полезность: нано-МСВИ была успеш-
но применена к исследованию сорбционных явлений в почве, для изучения возраста
и происхождения образцов или загрязняющих веществ. Нано-МСВИ-исследования
были проведены на основе анализа элементов, низкомолекулярных соединений или
маркеров больших молекул и типов бактерий. В технике нано-МСВИ имеется возмож-
ность установления изотопного сдвига, что позволяет использовать изотопные метки
для анализа переходных явлений, временных изменений или миграции веществ. Одним
из примеров применения нано-МСВИ является анализ азотфиксирующих морских ци-
анобактерий на уровне одной клетки. Этот анализ указал на механизм поглощения как
углерода (в виде НСО3), так и азота (в виде N2) из окружающей среды (рис. 20.7). Масс-
спектрометрические изображения были получены при сканировании с шагом 150 нм.
По сравнению с методом получения МС-изображений, основанным на МСВИ,
МАЛДИ-визуализация позволяет анализиривать (био)органические молекулы, что мо-
жет найти применение в исследованиях объектов окружающей среды, давая более вы-
сокую аналитическую точность по сравнению с методом, основанным на регистрации
низкомолекулярных фрагментов, как в случае МСВИ-изображений. При исследовании
сорбционных процессов в почвах нецелевой масс-спектрометрический анализ с полу-
Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
Рис. 20.7. Изображения области, содержащей примерно две клетки хвоста бактерии
Trichodestniutn, получение методом трансмиссионной электронной микроскопии
(ТЭМ). Изображения после (а) 8 часовой и (г) 24-часовой инкубации с Н13СО3
и l5Nr Соответствующие нано-МСВИ-изображения демонстрируют процент
фиксированного l5N после (б) 8-часовой и (д) 24-часовой инкубации, а также
процент |3С после (в) 8 часовой и (е) 24-часовой инкубации. Стрелки указыва-
ют на корреляцию между цианофициновыми гранулами, которые были обна-
ружены с помощью ТЭМ, и «горячими» точками в нано-МСВИ-изображениях,
которые содержат '^-обогащенные вещества. (Источник: Finzi-Hart J.A, Pett-
Ridge J., Weber P. K, Popa R., Fallon S. J., Gunderson T., Hutcheon I. D., Nealson К. H.,
Kapone D.G. (2009). «Фиксация и распространение азота и углерода в цианобак-
териях Trichodestniutn при использовании вторично-ионной масс-спектрометрии
с нанометровым расрешением». Труды Национальной академии наук. 106: 6345—
6350, печатается с разрешения)
чением молекулярных изображений представляет большой интерес. Почва является
сложнейшей матрицей из частиц, микроорганизмов и жидкостей, взаимодействующих
друг с другом через многочисленные типы агрегатов и посредством гигантского числа
путей обмена веществ. Поглощение и деградация загрязняющих веществ в почве яв-
ляются одним из самых сложных аналитических вопросов экологии. Поглощение ги-
дрофобных органических веществ недоступными участками органического вещества,
как известно, является основной причиной повышенной сорбции этих веществ в почве
(Bohm и During, 2010). Исследование этих явлений должно включать в себя стандартный
масс-спектрометрический анализ представляющих интерес соединений с помощью
концентрирования образцов методом твердофазной микроэкстракции. Последующий
20.10. МС-имиджинг в исследованиях объектов окружающей среды
масс-спектрометрический анализ образцов с получением МС-изображения полезен для
локализации критических соединений на уровне агрегатов (рис. 20.8). Методы МАЛДИ
или ЛДИ МС-изображений имеют явные преимущества в этом отношении по сравне-
нию с МСВИ-изображениями, так как они позволяют получать данные не только из по-
верхностных слоев агрегатов, но и из более глубоких слоев частиц и скрытых полостей.
Другие методы, применяемые для исследования проб окружающей среды, исполь-
зуют источники ионов с десорбционной электрораспылительной ионизацией и масс-
спектрометрическую визуализацию с пространственным разрешением (ДЭРИ-МСВ).
Этот метод использует пучок заряженных капель растворителя, которые образуются
при дроблении капель раствора при электрораспылении. После электрораспыления
микрокапли увлекаются струей газообразного азота и направляются на поверхность
исследуемого образца. Если первичный ионный пучок имеет достаточно малый диа-
метр, образец может быть просканирован. Этот метод поверхностного МС-анализа спо-
собен предоставить большое количество ценных молекулярных изображений (глава 8
по ДЭРИ).
Другой перспективной техникой МС-микроанализа с пространственным разреше-
нием является низкотемпературная плазма (НТП). В этом методе газ-реагент ионизи-
руется в газовом атмосферном разряде и направляется к поверхности образца. Экспе-
рименты показали наличие вторичных ионов органических молекул в масс-спектрах,
что указывает на возможность сбора полезной информации при микросканировании
образца. НТП-МС является очень мягкой техникой ионизации, которая работает при
Рис. 20.8. Схема сорбционных процессов в почве при старении образца и в процессах пере-
носа гидрофобных органических веществ внутри агрегатов. Исследования про-
ведены путем сочетания техники твердофазной микроэкстракции МС и МС-
томографии с использованием сканирующего микрозонда на основе лазерной
десорбции/ионизации (СЛДИ-МСВ). К — коэффициент распределения; х — ги-
дрофобный органический экотоксикант; DOC — растворенный органический
углерод; г — твердая фаза; F — волокна. (Источник: предоставлено During R.A.)
Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
Рис. 20.9. Поток низкотемпературной плазмы (НТП), направленный к поверхности паль-
ца. Масс-спектрометрия с использованием источника ионов, в котором приме-
няется НТП, позволяет определить молекулярный состав на поверхности жи-
вых существ без причинения боли или видимого разрушения
атмосферном давлении и может быть применена для анализа поверхности живых орга-
низмов (поверхностей растений, кожи животных и т.д.) без какого-либо вреда для них
(рис. 20.9).
20.11. Перспективы
МС-имиджингявляется очень перспективной техн и кой, которая все еще находится в ста-
дии развития. До настоящего времени МС-визуализация использовалась в биомедицин-
ских приложениях для анализа распределения биомолекул в срезах биологических тка-
ней с низким пространственным разрешением (например МАЛДИ-МС-визуализация)
и при исследованиях поверхностей материалов, основанных на элементном и изотоп-
ном масс-спектрометрическом анализе (метод МСВИ-изображений). В экологической
микробиологии новая технология, основанная на методе нано-МСВИ-изображений,
уже показала большой потенциал для изучения элементарных химических процессов,
происходящих при поглощении веществ почвой. Метод МАЛДИ-МС-визуализации
при атмосферном давлении также весьма перспективен для расширения области при-
менения МС-методов, например в область анализа больших молекул в окружающей
среде и для изучения межфазных процессов в биосфере. МС-визуализация имеет боль-
шие перспективы в ботанике и других областях науки, связанных с сельским хозяй-
ством. Современная МС-визуализация сочетает высокое пространственное разрешение
до уровня нескольких микрометров, высокое разрешение по массе и высокую точность
измерения масс. Все это необходимо для надежной идентификации веществ при анали-
зе сложных природных образцов. Методы МС-имиджинга, где используются некоторые
Литература 585
новые методы ионизации при атмосферном давлении (такие как ДЕРИ или НТП), также
являются очень перспективными для быстрого и непосредственного (in situ) анализа об-
разцов в естественных условиях.
Литература
[1] . Benninghoven А. (1994). «Chemical analysis of inorganic and organic surfaces and thin
films by static time-offlight secondary ion mass spectrometry (ToF-SIMS)». Angewandte
Chemie International Edition. 33: 1023—1043.
[2] . Bohm L., and Diiring R. A. (2010). «Partitioning of polycyclic musk compounds in soil and
aquatic environment-experimental determination of К-DOC». Journal of Soils and Sedi-
ments. 10: 708—713.
[3] . Bouschen W., Schulz O., Eikel O., and Spengler B. (2010). «Matrix vapour deposition/re-
crystallization and dedicated spray preparation for high-resolution scanning microprobe
MALDI imaging mass spectrometry (SM ALDI-MS) of tissue and single cells». Rapid Com-
munications in Mass Spectrometry. 24: 355—364.
[4] . Brunelle A., and Laprevote O. (2009). «Lipid imaging with cluster time-of-flight secondary
ion mass spectrometry». Analytical and Bioanalytical Chemistry. 393: 31—35.
[5] . CaradoA., Passarelli M.K., Kozole J., Wingate J. E., Winograd N., and Loboda A. V. (2008).
«С60 secondary ion mass spectrometry with a hybrid-quadrupole orthogonal time-of-flight
mass spectrometer». Analytical Chemistry. 80: 7921—7929.
[6] . Chaurand P., Schriver К. E., and Caprioli R. M. (2007). «Instrument design and characteri-
zation for high resolution MALDI-MS imaging of tissue sections». Journal of Mass Spec-
trometry. 42: 476—489.
[7] . Fenn J. B., Mann M., Meng С. K., Wong S. F, and Whitehouse S. M. (1989). «Electrospray
ionization for mass spectrometry of large biomolecules». Science. 246, 64—71.
[8] . Guenther S., Rompp A., Kummer W., and Spengler B. (2011). «АР-MALDI imaging of neu-
ropeptides in mouse pituitary gland with 5 m spatial resolution and high mass accuracy».
International Journal of Mass Spectrometry. 305: 228—237.
[9] . Hillenkamp F., Unsold E., Kaufmann R., and Nitsche R. (1975). «Laser microprobe mass
analysis of organic materials». Nature. 256: 119—120.
[10] . Karas M., and Hillenkamp F. (1988). «Laser desorption ionization of proteins with molecu-
lar masses exceeding 10 000 Daltons». Analytical Chemistry. 60: 2299—2301.
[11] . Ostrowski S.G., Van Bell C.T., Winograd N., and Ewing A. G. (2004). «Massspectrometric
imaging of highly curved membranes during tetrahymena mating». Science. 305: 71—73.
[12] . Rompp A., Guenther S., Schober Y, Schulz O., Takats Z., Kummer W., and Spengler B.
(2010). «Histology by mass spectrometry: label-free tissue characterization obtained
from high-accuracy bioanalytical imaging». Angewandte Chemie International Edition. 49:
3834-3838.
[13] . Spengler B. (2004). «De novo sequencing, peptide composition analysis, and composition-
based sequencing: a new strategy employing accurate mass determination by Fourier trans-
form ion cyclotron resonance mass spectrometry». Journal of the American Society of Mass
Spectrometry. 15: 703—714.
586 Глава 20. Метод масс-спектрометрической визуализации (имиджинг)
[14] . Spengler В., and Hubert М. (2002). «Scanning microprobe matrix-assisted laser desorption
ionization (SMALDI) mass spectrometry: instrumentation for sub-micrometer resolved
LDI and MALDI surface analysis». Journal of the American Society of Mass Spectrometry.
13: 735-748.
[15] . Spengler B., Karas M., Bahr U., and Hillenkamp F. (1989). «Laser mass analysis in biol-
ogy». Berichte der Bunsengesellschaftfur Physikalische Chemie. 93: 396—402.
[16] . Spengler B., Hubert M., and Kaufmann R. (1994). MALDI ion imagingand biological ion im-
aging with a new scanning UV-laser microprobe. Proceedings of the 42nd ASMS Conference
on Mass Spectrometry and Allied Topics, Chicago, Illinois, 29 May—3 June 1994, S. 1041.
[17] . Stoeckli M., Chaurand E., Hallahan D. E., and Caprioli R. M. (2001). «Imaging mass spec-
trometry: a new technology for the analysis of protein expression in mammalian tissue».
Nature Medicine. 7: 493—496.
[18] . Touboul D., Kollmer E, Niehuis E., Brunelle A., and Laprevote O. (2005). «Improvement
of biological time-offlight-secondary ion mass spectrometry imaging with a bismuth cluster
ion source». Journal of the American Society of Mass Spectrometry. 16: 1608—1618.
[19] . Wu K. J., and Odom R. W. (1996). «Matrix-enhanced secondary ion mass spectrometry: a
method for molecular analysis of solid surfaces». Analytical Chemistry. 68: 873—882.
ГЛАВА 21
МАСС-
СПЕКТРОМЕТРИЯ
ИЗОТОПНЫХ
ОТНОШЕНИЙ
Анжелика Талибова, Михаил Токарев и Андреас Хилькерт
21.1. Введение
Изотопы — это атомы, содержащие в своих ядрах одинаковое число протонов (атом-
ный номер), но различное число нейтронов. Некоторые изотопы радиоактивны. По-
этому они называются радиоизотопами или радионуклидами. Другие не подвержены
радиоактивному распаду. Их называют стабильными изотопами. Например, водород
существует в виде двух стабильных изотопов, 'Н и 2Н (дейтерий), и одного радиоактив-
ного 3Н (тритий). Углерод встречается в виде двух стабильных изотопов, 12С и 13С, и пяти
радиоактивных. Наиболее известным радиоактивным изотопом углерода является изо-
топ с массой 14 (14С).
Отношение стабильных изотопов каждого элемента в различных природных объ-
ектах неодинаково. Было проведено множество исследований относительного содержа-
Таблица21.1. Стабильные изотопы и их распространенность для основных элементов
Элемент Изотоп Распространенность, %
Водород ‘Н 99,985
2Н 0,015
Углерод |2С 98,89
|3С Ml
Азот l4N 99,63
l5N 0,37
Кислород |6О 99,759
|7О 0,037
|8О 0,204
Сера 32s 95
33s 0,76
34S 4,22
36S 0,014
Глава 21. Масс-спектрометрия изотопных отношений
ния изотопов. Главным приложением метода измерения изотопных отношений явля-
ется определение происхождения объекта (вещества). Исторически первые измерения
изотопных отношений были использованы в геохимии и геологических науках. В на-
стоящее время анализ изотопов является эффективным аналитическим методом, осо-
бенно при определении аутентичности продуктов питания. Табл. 21.1 содержит пере-
чень изотопов и их распространенности для наиболее часто используемых элементов.
21.2. Величина 8
Изотопное обогащение 8 измеряется в промилле (%о) и выражается через единицы СИ
по формуле
8'3С [%0] = (l3C/l2CMm/),/('3C/'2CJ - 1)-1000%.
Основные международные стандарты перечислены в табл. 21.2. Параметр R обо-
значает отношение тяжелого изотопа к более легкому соответствующего элемента.
8-величины этих стандартов равны нулю по определению. Так как некоторые их этих
первичных стандартов коммерчески уже недоступны, были предложены вторичные
стандарты с точно известными обогащениями 8 (Faure, 1986).
Таблица 21.2. Международные принятые стандарты легких изотопов
н Стандарт усредненной морской воды (SMOW) R (D/H) = 0,001558
с Пи-ди белемнит (PDB) R (|3С/|2С) = 0,0112372
N Атмосферный воздух (AIR) R (l5N/l4N) = 0,0036765
О Стандарт усредненной морской воды (SMOW) R (|8О/16О) = 0,0020052
S Триолит каньона Диабло (CTD) R (34S/32S) = 0,0450045
21.3. Причины вариаций распределения стабильных
изотопов в природе
Распределение изотопов в природе не гомогенно. Глобальный геохимический цикл
и природные процессы регулируют реакции изотопного обогащения органических
и неорганических соединений в природе (рис. 21.1).
Кинетические изотопные эффекты обычно ассоциируются с такими физическими
процессами, как испарение, диффузия, реакции диссоциации и почти все биологиче-
ские реакции. Кроме того, физические свойства молекул, содержащих более тяжелые
изотопы, отличаются от более легких аналогов (равновесное и термодинамическое
фракционирование). Эффекты равновесного фракционирования наблюдаются в хими-
ческих реакциях и в процессе фазовых переходов.
Температура — главный физический фактор, влияющий на распределение изотопов
кислорода и водорода. Любая водная система в природе имеет уникальный изотопный
21.3. Причины вариаций распределения стабильных изотопов в природе
Рис. 21.1. Схема фракционирования в глобальном атмосферном цикле воды
состав. С этой точки зрения изотопный состав кислорода и водорода в растениях и ор-
ганизмах животных отличен от изотопного состава осадков (Faure, 1986; Sharp, 2007).
Отношение ,3С/|2С в биологических объектах зависит практически исключительно
от механизма фотосинтеза, который используется растениями для усвоения углекис-
лого газа СО2 — цикла Кальвина, или С3, со средними 5,3С —26/—28%о и цикла Хэтча
и Слэка, или С4, со средними 513С —12/—14%о (Smith and Epstein, 1971). Около 85% всех
сосудистых растений используют тип фотосинтеза С3. Тропические растения обычно
Океанический планктон—12 to—22%о XDIC— 30to0%o
Рис. 21.2. Изотопные отношения для углерода в основных компонентах земных экоси-
стем. DIQ. растворенный неорганический углерод
Глава 21. Масс-спектрометрия изотопных отношений
Таблица 21.3. Величины 8 l5N азота для различных образцов
Образец 8,5NAir/%o
Атмосферный азот 0
Растения с метаболизмом С3 от -8,0 до +4,0
Океанический планктон от -3,0 до +18,0
nh4 от +4,0 до +20,0
Неорганические удобрения от -4,0 до +4,0
Органические удобрения от +6,0 до +30,0
Коровье молоко от +1,0 до +3,0
Таблица 21.4. Величины 8 13С углерода для различных образцов
Образец 8i3CVpdD/%o
Атмосферный СО2 в воздухе в настоящее время (среднее значение) -7,8
Морской СаСО3 +1,0
Графит органического происхождения -24,5
Алмаз магматического происхождения -4,5
Метан (продукт микроорганизмов) -50,0
Древесина (ель) -26,3
Кукуруза(початок) -П,9
имеют метаболизм Хэтча и Слэка. Некоторые растения засушливых регионов (напри-
мер ананас, кактусы или ваниль) используют кислотный метаболизм Crassulacean, ко-
торый близок к метаболизму С4. Они называются С4Л/-растениями и имеют углеродное
обогащение, промежуточное между 813С С3 и С4 (рис. 21.2).
Почвенные микроорганизмы фракционируютизотопы азота во время ассимиляции
и в процессе метаболизма (табл. 21.3). Биохимические реакции азотного цикла, такие,
как нитрификация или аммонизация, могут сопровождаться сильной вариацией 815N
(от —10 до —40 815N%o по сравнению с воздухом). Например, при ассимиляции аммония
водными водорослями 8 изменяется от —27 до 0%о. С другой стороны, фракционирова-
ние при минерализации близко к нулю (+1 до —1%о). Соответственно, использование
синтетических удобрений с обогащением, близким к 0%о, ведет к изменению изотоп-
ного состава азота в сельскохозяйственных культурах. 8I5N и 8|3С в растениях зависят
от их вида, типа почвы и разновидности удобрения (с минеральным или органическим
азотом) (Francois etal., 2002; Galimov, 1985). Табл. 21.4 содержит 8-величины наиболее рас-
пространенных объектов окружающей среды.
21.4. Масс-спектрометрия изотопных отношений
в газах
Изотопы легких элементов, как правило, измеряются на газовых изотопных масс-
спектрометрах. Прибор состоит из источника ионов, в котором происходит ионизация
21.4. Масс-спектрометрия изотопных отношений в газах
ТС фильтром
по энергии
источник ионов Универсальный тройной коллектор
для отбора N2, СО, О2, СО2, SO2
Рис. 21.3. Масс-спектрометр для измерения отношений стабильных изотопов, состоящий
из источника ионов, магнита и набора цилиндров Фарадея для одновременной
регистрации изотопов. (Перепечатано с разрешения Thermo Fisher Scientific)
молекул газа и ускорение образовавшихся ионов, вакуумной камеры с магнитом для
разделения ионов и системы детекторов типа цилиндра Фарадея для измерения количе-
ства индивидуальных изотопов (рис. 21.3).
При анализе на таком масс-спектрометре исследуемые молекулы должны быть
переведены в газовую фазу. В настоящее время используется множество приставок для
превращения пробы в газ при помощи сжигания, химического окисления, пироли-
за или уравновешивания. Анализируемые газы (СО2, N2, СО, Н2 и SO2) ионизируются
электронами (глава 1). Ионы разделяются в секторном магнитном анализаторе согласно
их отношениям m/z и детектируются с точностью превосходящей единицы ррт. Источ-
ники ионов современных масс-спектрометров изотопных отношений имеют оптималь-
ную для ионизации легких газов конструкцию.
21.4.1. Ионизация
Источники ионов, использующиеся в масс-спектрометрах для измерения изотопных
отношений, имеют конструкцию А. Нира (Nier, 1947). Источник состоит из ионизаци-
онной камеры, катода, ловушки, постоянного магнита и набора электродов ионно-элек-
тронной оптики. Большая часть образовавшихся в источнике ионов попадает в сектор-
ный магнитный анализатор через его входную щель.
Молекулярные ионы образуются при взаимодействии молекул с электронами,
эмиттируемыми горячим катодом, изготовленным из вольфрама, рения или торирован-
ного иридия. Для оптимальной ионизации молекул электроны ускоряются до энергии
в 70-150 эВ.
Положительнозаряженные ионы, такие как СО2+’, N2+’ и СО+’, экстрагируются
из ионизационной камеры с помощью электростатических линз. Затем ионы ускоряют-
ся высокой разностью потенциалов (ускоряющим напряжением) в 2—10 кВ, фокусиру-
ются и вводятся в масс-анализатор.
Глава 21. Масс-спектрометрия изотопных отношений
21.4.2. Разделение по массам
Магнитное поле, обеспечивающее отклонение ионов, генерируется электромагнитом.
Когда ион массы т и с зарядом е ускоряется разностью потенциалов V и отклоняется
магнитным полем с индукцией В, он имеет радиус траектории г:
г = \4\5(2тУ/еУ'5/В.
Если В и ^постоянны, а ион — однозарядный, то
г ~ /И0’5.
Следовательно, более легкий ион имеет радиус отклонения меньше, чем более тяже-
лый. Новейшие приборы имеют диапазон измерения до 130 m/z с разделением соседних
изотопов.
Электромагниты коммерческих приборов обеспечивают магнитную индукцию
до 0,75 Т с возможностью быстрого переключения магнитного поля с массы Н2 на массу
СО и от N2 к СО2 (см. табл. 21.5).
Таблица 21.5. Наиболее часто используемые для определения изотопных отношений
массы
8 для изотопного отношения Газ Массы измеряемых изотопологов, m/z
82Н Н2 2,3
8I5N N2 28, 29, 30
8|8О со 28, 29, 30
8|3С,818О со2 44,45,46
834S so2 64,66
21.4.3. Регистрация нескольких ионов
Отдельные коллекторы (цилиндры Фарадея) размещаются в фокальной плоскости для
одновременного детектирования всех нужных ионных пучков в целях определения
изотопных отношений. В большинстве масс-спектрометров изотопных отношений ис-
пользуется базисная конструкция с двумя цилиндрами Фарадея большого размера и од-
ним небольшим цилиндром Фарадея между ними (универсальный тройной коллектор).
Такая конструкция позволяет измерять изотопные отношения для СО2, N , СО и SO2.
Для измерения отношения HD/H2 добавляются еще два цилиндра Фарадея. В не-
которых системах для Н2 используется та же индукция магнитного поля, что и для СО
и N2. В этом случае цилиндр Фарадея для регистрации Н2 располагается в отдельной
фокальной плоскости на меньшем радиусе. При использовании магнитного поля с су-
щественно более слабой индукцией HD коллекторы располагаются в той же фокальной
плоскости, что и универсальный тройной коллектор.
В современных масс-спектрометрах изотопное отношение и величины 8 вычисля-
ются автоматически и сохраняются программным обеспечением (Sharp, 2007).
21.5. Оборудование для подготовки проб и интерфейсы 593
21.4.4. ,7О- и HD-коррекция
В некоторых приложениях ионы одной массы, но с разным набором изотопов реги-
стрируются одним и тем же коллектором. Ионы с массой 45 для определения 13С в СО2
являются смесью ионов 13С16О2+ и |2С17О16О+. Значит, информация о |3С должна быть
скорректирована с учетом вклада 17О. Первая корректировка для вклада |7О в СО2 была
предложена Хэрмоном Крэгом в 1957 голу (Craig, 1957):
813С [%о] = 1,0676 • 845(СО2) - 0,0338 • 818О.
Новейшей коррекцией по 17О является линеаризованная коррекция (Brand et al.,
2010).
При исследовании водорода в источнике ионов в результате ионно-молекулярных
реакций образуются ионы Н3+, которые имеют ту же массу, что и ионы HD+. Поэтому
в измерение изотопных отношений может быть внесена ошибка. Вклад Н3+ может быть
оценен в дополнительных экспериментах, если известно количество введенного газа Н2.
Эта поправка (Н3+-фактор) используется для коррекции.
Программное обеспечение для масс-спектрометра использует специальные алго-
ритмы для коррекции 17О и Н3+.
21.5. Оборудование для подготовки проб
и интерфейсы
Для ввода газов в масс-спектрометр при измерении изотопных отношений используют-
ся два основных способа. Первый — это классическая система двойного ввода, постав-
ляющая газы, полученные и очищенные в вакуумном генераторе, в условиях вязкого
течения. Второй способ — это интерфейс с непрерывным ламинарным потоком газа-
носителя. В этом разделе мы уделим внимание интерфейсу с непрерывным потоком
и системе подготовки.
21.5.1. Изотопный масс-спектрометр непрерывного потока
Масс-спектрометр непрерывного потока для измерений изотопных отношений требует
в качестве газа-носителя Не. Гелий служит для ввода анализируемого газа и референс-
ных газов через открытый интерфейс с делением потока. Этот метод был впервые пред-
ложен Мэтьюсом и Хэйесом (Matthews D. Е. and Hayes J. М., 1978). Он открыл широкие
возможности для разработки приборов on-line пробоподготовки и интерфейсов к ним.
В настоящее время в МСИО сочетают газовую и жидкостную хроматографию (ГХ и ЖХ)
с системами подготовки пробы и методом непрерывного потока.
Открытый интерфейс с делением потока обычно состоит из небольшой трубки
с капилляром внутри нее для защиты постоянного потока гелия от контакта с возду-
хом. Второй капилляр служит для ввода образца в масс-спектрометр, а третий — для
ввода образца из системы пробоподготовки в открытый интерфейс (рис. 21.4). Следует
упомянуть о двух основных методах подготовки проб для режима с непрерывным по-
током. Первый — это метод объемного анализа стабильных изотопов (ОАСИ). В нем
Глава 21. Масс-спектрометрия изотопных отношений
Открытый интерфейс
с делением потока
Рис. 21.4. Открытый интерфейс с делением потока для измерения изотопных отношений
в непрерывном потоке. (Перепечатано с разрешения Thermo Fisher Scientific)
для преобразования аналитов в простые газы с последующим их хроматографиче-
ским разделением используются элементные анализаторы со сжиганием пробы или
пиролизом (восстановление углерода при высокой температуре). Второй метод — изо-
топный анализ, оптимизированный для соединения (ИАОС). В этом методе сложные
смеси сначала разделяются при помощи ГХ или ЖХ с последующим онлайн-преобра-
зованием микропробы для анализа каждого компонента после хроматографического
разделения (рис. 21.5).
ОАСИ
Реактор
Конверсия в элементном
анализаторе
Открытый интерфейс
с делением потока
Разделение газов
с помощью ГХ
для измерения
изотопных отношений
ОАСИ: б2Н и б|8О, а также б|3С, 6I6N и 634S в одном эксперименте
ИАОС
ГХ разделение
сложных смесей
Открытый интерфейс
с делением потока
Капиллярный реактор
Конверсия в режиме
он-лайн
V11VI41 pviivivi р
для измерения
изотопных отношений
ИАОС: 5|3С, 82Н, SI5N или SI8O величины индивидуальных соединений в смеси
Рис. 21.5. Сравнение ОАСИ и ИАОС. (Перепечатано с разрешения Thermo Fisher Scientific).
(ОАСИ — объемный анализ стабильных изотопов)
21.5. Оборудование для подготовки проб и интерфейсы
21.5.2. Элементный анализатор (сжигание
и высокотемпературная конверсия) —
изотопный масс-спектрометр
Элементные анализаторы — наиболее распространенные приборы для приложений
с непрерывным потоком. Как правило, используются два основных метода: сжигание
при помощи обычного элементного анализатора ЭА для определения l3C, ,5N и 34S и вы-
сокотемпературное преобразование посредством использования специфического ЭА
(ВТП/ЭА) для объемного анализа ,8О и D. Оба метода также реализуемы на гибридных
коммерческих системах (Flash НТ) с полностью автоматизированным переключением
между различными модулями сжигания и разделения.
Типичное количество аналитов в пробе при объемном анализе изотопных отно-
шений углерода или кислорода составляет 10—100 мкг. В зависимости от содержания
аналита и матрицы размер пробы для объемного анализа может варьироваться, осо-
бенно при ВТП, которая дает информацию о |8О и D для всех органических и многих
неорганических проб, таких как нитраты, фосфаты, сульфаты и другие компоненты,
включая воду. Для анализа воды и других жидкостей необходимо наличие инжектора
с автосамплером. Необходимое для анализа О и Н количество воды может составлять
всего лишь 0,1 мкл.
21.5.3. ГХ/МСИО
ГХ используется совместно с МСИО для анализа изотопного состава индивидуаль-
ных соединений в пробе. Изотопная информация о каждом соединении в смеси может
быть использована для определения происхождения образца, а также для изучения
распределения и деградации органического вещества. Все соединения, выходящие
из ГХ-колонки, окисляются в капиллярном керамическом реакторе сжигания до СО2,
Рис. 21.6. Концепция современной ГХ/МСИО-системы. ПИД — пламенно-ионизацион-
ный детектор. (Перепечатано с разрешения Thermo Fisher Scientific)
Глава 21. Масс-спектрометрия изотопных отношений
N2 и Н20 при температуре 1000°C или трансформируются в СО и Н2 в капиллярном пи-
ролизном реакторе при температуре 1450°C. Молекулы вида NOx, образующиеся в про-
цессе окисления, могут быть восстанавлены до N2 во втором капиллярном восстанавли-
вающем реакторе при температуре 650°C. Недавние усовершенствования в технологии
ГХ-сжигания показали, что наличие восстанавливающего реактора больше не является
обязательным. Новые окислительно-восстановительные реакторы гарантируют как от-
сутствие NO*, так и полное сгорание при температуре 1000 °C. Образующаяся в ходе сжи-
гания вода удаляется системой автоматической откачки.
При анализе изотопов азота перед МСИО размещается ловушка с жидким азотом
для удаления молекул СО2, которые мешают определению (рис. 21.6).
21.5.4. ЖХ/МСИО
ЖХ/МСИО-приборы были разработаны для изотопного анализа органических соедине-
ний, которые невозможно исследовать при помощи ГХ без дополнительной пробоподго-
товки и дериватизации. Метод дает возможность получить информацию о 513С для всех
растворимых в воде молекул (сахаров, аминокислот, органических кислот и спиртов).
Смесь органических соединений разделяется с помощью высокоэффективной
жидкостной хроматографии, сопровождающейся химическим микроокислением в мо-
бильной фазе. Все органические вещества трансформируются в СО2 по отдельности
в окисляющием реакторе, содержащем смесь пероксодисульфата и фосфорной кислоты,
при температуре 100°C. В качестве мобильной фазы можно использовать только воду
и натрий-фосфатный буфер (PBS). После окисления газ СО2 каждого соединения вы-
деляется через мембрану и используется для анализа, тогда как жидкая фаза удаляется
(Krummen et aL, 2004).
Порции СО2 каждого вещества осушаются в потоке гелия при пропускании через
полимерную мембрану (Nafiori) и вводятся через открытый интерфейс с делением потока
в МСИО, как показано на рис. 21.7.
Открытый интерфейс
с делением потока
Рис. 21.7. ВЭЖХ с химическим реактором и интерфейсом. (Перепечатано с разрешения
Thermo Fisher Scientific)
21.6. Некоторые приложения
21.5.5. МСИО с многократными инжекциями через петлю
Система для подготовки пробы GasBench и интерфейс (рис. 21.8) предназначены для ана-
лиза любых газов (в частности атмосферных СО2 и N2) и паров соединений (карбонатов,
растворенного органического углерода, органических веществ, воды), а также паров, об-
разованных при трансформации соединений. После пробоподготовки in vitro анализи-
руемый газ переносится из флакона в петлю фиксированного объема (10, 100 и 250 мкл)
в слабом потоке гелия. Из петли пробы поступают в ГХ-колонку для отделения анализи-
руемого газа от других газов. Вода удаляется с помощью двух газовых систем продувки.
Открытый интерфейс с делением потока переносит аналит в МСИО. Наиболее часто ис-
пользуемые приложения — это Н2/Н2О- и СО2/Н2О-уравновешивание для определения
8D и 5,8О в воде и кислотная деградация карбонатов для измерения 5|3С и 8,8О.
Анализ 5D для воды может служить примером цикла операций системы GasBench.
Измерение величин 5D и 5,8О для природной воды, воды из фруктового сока и вина —
одно из самых популярных приложений системы GasBench. Пробы воды объема 0,2 мл
переносят во флаконы с закручивающимися крышками. Во флакон вставляется плати-
новый катализатор, необходимый для переноса молекулярной информации в Н2 или СО2
в газовой фазе над водой. Флаконы продуваются смесью газов Н2/Не и СО2/Не, подава-
емыми из автосамплера, в течение 5 минут при газовом потоке в 100 мл/мин. Реакция
уравновешивания для отношения D/Н длится 40 минут, а |8О/,6О — 18 часов (рис. 21.9).
Этот метод может быть напрямую использован для измерения отношений ,8О/,6О в во-
дной фракции фруктового сока, вина и других напитков без предварительного выделе-
ния воды.
Рис. 21.8. GasBench с многократной инжекцией через петлю и интерфейс. (Перепечатано
с разрешения Thermo Fisher Scientific)
21.6. Некоторые приложения
21.6.1. Науки о Земле
Данные о составе стабильных изотопов могут быть успешно использованы при реше-
нии различных задач в области наук о Земле. Соответствующие приложения могут быть
классифицированы по нескольким типам.
Глава 21. Масс-спектрометрия изотопных отношений
200 мкл
пробы
воды
2% Н2---
в гелии
Закрутите
крышку
флакона
» Перенесите 200 мкл
пробы во флакон
» Вставьте платиновый катализатор » Поместите все флаконы
» Закройте флакон в автосам пл ер
» Все флаконы автоматически
продуваются
Не
Перенос D из Н2О
в Н2 в объеме
над пробой
2
» Уравновешивать
в течение 40 минут
Анализ
» Старт серии
Рис. 21.9. Уравновешивание Н2/Н2О и СО2/Н2О во флаконах. (Перепечатано с разрешения
Thermo Fisher Scientific)
• Гидрология. Изотопное обогащение для водорода и кислорода специфично для
каждого водного резервуара. Измерения стабильных изотопов могут служить
основой для изучения эволюции водных ресурсов Земли, исследования осадков
и определения циклов различных гидрологических систем (рис. 21.10). Более
того, данные об изотопах помогают сделать выводы о пригодности природных
водных источников для потребления человеком.
• Палеоклиматология. Более 60 лет измерения 8|8О и 8,3С в карбонатах, глине
и торфе, а также определение 8|8О и 8D в ледяных кернах используются для вос-
становления условий окружающей палеосреды.
• Термометрия. Термометрия стабильных изотопов сыграла важную роль при
исследовании температур формирования горных пород, минералов и других
геосистем.
• Происхождение вещества, массоперенос и метаморфизм. Изотопные отношения
используются как маркеры для изучения происхождения горных пород и жидких
сред.
• Механизмы реакций. Различия в изотопном составе могут быть обнаружены
в разных системах и в результате физических процессов, таких как диффузия
и рекристаллизация (Sharp, 2007).
21.6. Некоторые приложения
Рис. 21.10. Вариации изотопов кислорода в водах Северной Америки
21.6.2. Биология и экология
В биологиии и экологии измерение стабильных изотопов используется главным обра-
зом при исследовании миграции птиц и других животных, а также при создании мо-
делей питания в экосистемах. Изотоп 13С часто используется для определения главных
источников питания животных, например в экосистемах, в которых присутствуют рас-
тения с метаболизмом по типу С3 и С4. Интенсивное фракционирование изотопов азота
(8I5N) в пищевых цепочках позволяет определить длину и структуру трофических цепей
для животных. Рис. 21.11 иллюстрирует метод дифференциации материковых и при-
брежных популяций птиц на основе измерения различий в изотопном составе серы
и водорода в их перьях.
21.6.3. Археология и миграция людей
Исследования распределения стабильных изотопов в человеческих останках предостав-
ляют информацию о миграции людей и могут служить свидетельством перемещений
популяций и освоения ими территорий. Например, рис. 21.12 иллюстрирует различия
в изотопных отношениях для углерода, кислорода и водорода в останках континенталь-
ных викингов и викингов побережья.
Глава 21. Масс-спектрометрия изотопных отношений
+ Материковые
♦ Прибрежные
14
12 *
10
i 8
сю 6 ♦
4 А
♦ * А
2 ♦
♦
О1--------------------------------------------------------------------
-165 -145 -125 -105 -85 -65 -45
5D/%o
Рис. 21.11. Вариации изотопов серы и водорода в перьях птиц иллюстрируют миграцию
животных (откуда и куда мигрируют птицы). (Средние величины взяты из
Kelly, 2005)
Рис. 21.12. Изотопный состав ногтей викингов (зеленые квадраты — данные по углероду;
черные ромбы — данные по азоту)
Таблица 21.6. Вариации изотопов углерода и азота в героине, произведенном в различных
странах
Образец 8'’СГРМ/%0 б|8О /%О air'
Героин из Колумбии -33,8 -о,з
Героин из Мексики -32,4 -0,6
Героин из Кореи -35,7 —2,5
Героин из Афганистана -29,5 +1,2
Героин из Лаоса -31,8 +2,9
21.6. Некоторые приложения
21.6.4. Контроль лекарств и наркотиков
Идентификация географического происхождения препаратов, используемых в крими-
нальных целях, в особенности полученных из природных источников (кокаин, опиа-
ты, марихуана и т.д.), представляет большой интерес в глобальном масштабе. В насто-
ящее время специальные полицейские подразделения, расследующие наркоторговлю,
успешно используют изотопный состав азота и углерода для определения происхожде-
ния наркотиков (табл. 21.6).
21.6.5. Контроль допинга
Измерение отношений изотопов углерода используется для определения природы экс-
трагированнных из мочи стероидов (то есть являются ли они эндогенными или фарма-
цевтическими). Точно такой же подход используется и при контроле допинга лошадей.
Основная идея подхода основана на различии в изотопном составе между фармацев-
тическими стероидами, использующимися для стимуляции атлетов, и человеческими
метаболическими стероидами (рис. 21.13).
Рис. 21.13. Метаболизм стероидов. DHEA — дегидроэпиандростерон. DHT — дегидро-
тестостерон
Глава 21. Масс-спектрометрия изотопных отношений
21.6.6. Аутентичность продуктов питания и напитков
На уровне современных знаний измерение стабильных изотопов является наиболее эф-
фективным способом установления географического происхождения, а также фактов
фальсификации продуктов питания, напитков, вина и алкоголя. Хорошими примерами
использования методов определения отношений стабильных изотопов могут служить
различия в 813С для этанола различного происхождения (табл. 21.7).
МСИО — многообещающий инструмент изучения происхождения продуктов
питания и сырья (рис. 21.14), а также установления фальсификации этих продуктов
(рис. 21.15). В частности, этот метод анализа позволяет вскрыть добавление искусствен-
но синтезированной лимонной кислоты или кислоты, полученной из других фруктов,
во фруктовые соки.
Определение 8|8О в водных фракциях соков позволяет различить соки, полученные
напрямую из фруктов, и соки из натуральных концентратов (см. табл. 21.8).
Таблица 21.7. Величины 8|3С для этанола
Образец и его происхождение §,3СГРМ/%о
Синтетический этанол -33,8
Этанол из кукурузы -13,2
Этанол из зерен пшеницы -23,9
Этанол из сахарного тростника -10,8
Гидролизный этанол -20,7
160 п
ф ф Апельсин
_ Грейпфрут
156 -
154 -
152 -
150 -
ф Лимон
,, Л Малина
Клубник^
• Черная смородина
148 -
Ф Коммерческий С3
ф Ананас
Коммерческий С4,
С4 + Ц
146 -|----------------1--------1--------1---------1—
-27 -25 -23 -21 -19 -17
813CVPDB/%o
--15 Дз
Рис. 21.14. Средние значения SI3C для лимонной кислоты из различных фруктов и синте-
зированной разными производителями. (Данные из Jamin et al., 2005)
23,5
23
22,5
♦ Тунис
20,5
♦ Италия
♦ Греция
$ Cvpdb/%0
Рис. 21.15. 8|3С и SI8O для глицерина из оливкого масла различного происхождения (Ита-
лия, Греция и Тунис). (Данные взяты из Fronza etal., 2001)
Этот метод наиболее эффективен в сочетании с ГХ/МС и анализом микропримесей
химических элементов. Главным ограничением для использования методов стабильных
изотопов для установления аутентичности продуктов питания является отсутствие баз
данных по распределению изотопов в пищевых ингредиентах.
Таблица 21.8. Величина б|8О для водных фракций натуральных фруктов
Образец и его происхождение §™OVSMOW/%o
Яблочный сок из России —3,8
Апельсиновый сок из Турции 2,5
Гранатовый сок из Азербайджана —0,1
Литература
[1] . Brand W.A., Assonov S.S., and Coplen T. В. (2010). «Correction for the 17O interference in
8(I3C) determinations when analyzing CO2 with stable isotope mass spectrometry». 1UPAC
Technical Report, Pure Applied Chemistry. 82: 1719—1733.
[2] . Craig H. (1957). «Isotopic standards for carbon and oxygen and correction factors for mass-
spectrometric analysis of carbon dioxide». Geochimica et Cosmochimica Acta. 12: 133—149.
[3] . Faure G. (1986). Principles of Isotope Geology. John Wiley and Sons, Chichester, UK.
[4] . Francois L., Faure H., and Probst, J.-L. (2002). «The global carbon cycle and its changes
over glacial-interglacial cycles». Global and Planetary Change. 33, 1—2: vii—viii.
[5] . Fronza G., Fuganti C., Grasselli P., Serra S., Reniero F., and Guillou C. (2001). 813C-
and 8l8O-values of glycerol of food fats». Rapid Communications in Mass Spectrometry. 15:
763-766.
[6] . Galimov E. M. (1985). The Biological Fractionation of Isotopes. Academic Press, New York,
USA.
604 Глава 21. Масс-спектрометрия изотопных отношений
[7] . Jamin Е., Martin F., Santamaria-Fernandez R., and Lees M. (2005). «Detection of exog-
enous citric acid in fruit juices by stable isotope ratio analysis». Journal of Agricultural and
Food Chemistry. 53: 5130—5133.
[8] . Kelly J. F. (2005). «Combining isotopic and genetic markers to identify breeding origins of
migrant birds». Ecological Applications. 15: 1487—1494.
[9] . Krummen M., Hilkert A.W., Juchelka D., Duhr A., Schliiter, H.-J., and Pesch R. (2004).
«А new concept for isotope ratio monitoring liquid chromatography/mass spectrometry».
Rapid Communications in Mass Spectrometry. 18: 2260—2266.
[10] . Matthews D. E., and Hayes J. M. (1978). «Isotope-ratio-monitoring gas chromatography-
mass spectrometry». Analytical Chemistry. 50: 1465—1473.
[11] . Nier A. O. (1947). «А mass spectrometer for isotope and gas analysis». Reviews in Scientific
Instrumentation. 18: 398—411.
[12] . Sharp Z. (2007). Principles of Stable Isotope Geochemistry. Pearson Education Inc., USA.
[13] . Smith B.N., and Epstein S. (1971). «Two categories of 13C/I2C ratios for higher plants». Plant
Physiology. 380—384.
ДОПОЛНЕНИЕ I
ОСНОВЫ И ДОСТИЖЕНИЯ
МАСС-СПЕКТРОМЕТРИИ
НА ОСНОВЕ ОРБИТАЛЬНОЙ
ЛОВУШКИ ИОНОВ.
КРАТКИЙ ОБЗОР
Alexander Makarov
Thermo Fisher Scientific, Hanna-Kunath-Str. 11, 28199 Bremen, Germany
Tel: +49 421 54 93 410, Fax: +49 4215493 396,
Alexander.makarov@thermofisher.com
Аналитическая химия получила свое развитие в значительной степени благодаря от-
крытиям в сфере масс-спектрометрии. Высокое разрешение, чувствительность, кото-
рыми обладают современные масс-спектрометры, стали неотъемлемой частью при ре-
шении аналитических задач как в разных исследованиях, таки в рутин ной лабораторной
практике. Последней новинкой в семье масс-спектрометров стал анализатор Orbitrap,
который дал возможность лабораториям получать результаты с ультра-высоким разре-
шением. Оборудование, созданное на базе технологии Orbitrap, закрепило свои позиции
в таких областях как протеомика, метаболомика и анализ метаболитов, а также пользу-
ется популярностью в области биоанализа, липидомике, антидопинговом анализе и в
анализах на остаточное количество пестицидов и лекарственных средств в пищевых
продуктах, сырье и объектах окружающей среды.
Развитие первых опытов движения заряженных частиц в электростатических полях
привело к созданию Александром Макаровым в 2005 году серийного масс-спектрометра
с анализатором орбитальная ловушка ионов (ORBITRAP) с Фурье преобразованием
сигнала. С тех пор несколько тысяч приборов Orbitrap были произведены, установлены
и с успехом используются в множестве аналитических лабораторий во всем мире.
Как устроена Орбитальная ловушка ионов
Orbitrap состоит из трех электродов, как показано на рис. Д1.1. Рисунок демонстрирует
как стандартную ловушку, представленную в 2005 году, и, так называемую, высоко-по-
левую ловушку, представленную в 2011 году. Внешние электроды имеют форму чаши
и разделены между собой промежутком толщиной в волос, в виде центрального кольца
Дополнение 1. Основы и достижения масс-спектрометрии на основе орбитальной
ловушки ионов. Краткий обзор
Рис. Д1.1. Схема орбитальной ионной ловушки
из диэлектрика. Когда ионы попадают вдовушку, они начинают движение по устойчи-
вым траекториям, которые объединяют три циклических движения:
• вращательное движение вокруг центрального электрода с частотой вращения а)ф;
• радиальное движение с частотой сог (между максимальными и минимальными
радиусами внутри пространства между электродами);
• осевые колебания вдоль центрального электрода с частотой со.
Даже при том, что траектория иона принимает форму сложной спирали, важно
отметить, что осевое движение остается абсолютно независимым от вращательного
движения.
Чтобы обеспечить круговую траекторию движения ионов, тангенциальная скорость
ионов должна быть приведена к такой величине, чтобы центробежная сила компенсиро-
вала силу, созданную радиальным электрическим полем.
Для радиуса R этой спирали, частота углового вращения о)ф описывается формулой:
Частота радиальных колебаний (отписывается формулой
(1)
(2),
где частота осевых колебаний (о:
со =
(3),
где е заряд электрона (1,602-10-19 С).
Вращательная и радиальная частоты показывают зависимость от начального ради-
уса R. С другой стороны, осевая частота абсолютно независима от всех начальных ско-
Как устроена Орбитальная ловушка ионов
ростей и координат ионов. Поэтому, только осевая частота может использоваться для
определения отношений массы к заряду, m/z-
Поскольку невозможно поместить источник ионов внутрь ловушки, ионы долж-
ны генерироваться снаружи анализатора Orbitrap. Чтобы ионы могли быть захвачены,
нужно их «электродинамически сжать». Ионы, входящие в захватывающее их элек-
трическое поле, подвергаются воздействию возрастающего поля. Потенциал внешних
электродов остается постоянным, в то время как потенциал центрального электрода по-
нижается (для положительно заряженных ионов). Попавшие в поле ионы не могут вер-
нуться назад к точке входа, поскольку за время, прошедшее с момента их входа в ловуш-
ку, между ними и местом входа образовался потенциальный барьер. Поскольку, сначала
захватываются ионы с меньшими значениями m/z, а более тяжелые ионы запаздывают,
время возрастания поля выбирается таким, чтобы обеспечить наиболее широкий диа-
пазон значений m/z захваченных ионов (обычно 30—50 мкс). Сжатие останавливается,
когда траектория движения ионов достигает желаемого радиуса (обычно на одинаковом
расстоянии между внешним и центральным электродами) и потенциал центрального
электрода стабилизируется. Ввиду того, что вращательные и колебательные частоты
очень сильно зависят от энергий ионов, углов и начального положения (уравнения 1—2),
каждый пакет ионов распределяется по угловым и радиальным координатам и форми-
рует тонкое вращающееся кольцо.
Частоты аксиальной осциляции ионов могут быть измерены по наведенному току
на внешних электродах ловушки. Потому как, ионные пакеты гармонично осцилли-
руют вдоль оси, разница в наведенных токах между электродами детектируется диф-
ференциальным усилителем. Широкополосный частотный сигнал оцифровывается
и конвертируется путем преобразования Фурье в спектр отношений массы к заряду.
Процесс детектирования ничем не отличается от используемого в масс-
спектрометрии ионно-циклотронного резонанса. Существенная разница заключается
в том, что благодаря природе электрического поля падение разрешающей способности
с ростом m/z происходит гораздо медленнее по сравнению с ИЦР.
Однако, для того чтобы можно было детектировать наведенные ионные токи, не-
обходимо сконцентрировать все ионы с одинаковым m/z в компактные пакеты, размеры
которых значительно меньше в аксиальном направлении, чем амплитуда колебаний.
Кроме того, ионы генерируются непрерывно источником ионов, а вводится вдовушку
должны в виде коротких импульсов. Для этого необходимо использовать устройство,
внешнее по отношению к орбитальной ловушке, где ионы захватываются газонапол-
ненной искривленной линейной ионной ловушкой (С-образной ловушкой). К конце-
вым и центральным электродам устройства прикладываются импульсные потенциалы
так, что ионы оказываются в сильном поле. Вероятность столкновительной диссоци-
ации ионов минимизируется за счет того, что ионы находятся у выходного отверстия.
Ионы с индивидуальными величинами отношений массы к заряду инжектируются
в орбитальную ловушку в виде плотных пакетов в неосевое входное отверстие и начина-
ют когерентные аксиальные осцилляции.
В идеальных условиях ионы могут оставаться в ловушке сколько угодно. К сожа-
лению, столкновения с остаточными газами приводят к рассеиванию ионов и лими-
тируют это время до нескольких секунд. При давлении в ловушке 10-10 мБар за вре-
мя < 1 с можно разрешать изотопные пакеты малых и среднеразмерных белков, а при
Дополнение 1. Основы и достижения масс-спектрометрии на основе орбитальной
ловушки ионов. Краткий обзор
дальнейшем улучшении вакуума доступны измерения белков с массами в десятки
килоДальтон.
Другим эффектом, воздействующим на уменьшение разрешения и чувствительно-
сти орбитальной ловушки ионов, является, безусловно, образование пространственного
заряда. Однако, этот эффект также минимизируется конструкцией ловушки.
Если проводить фрагментацию ионов внутри ловушки за счет столкновения с мо-
лекулами газа или, например, освещения импульсами лазерного излучения, фрагменты
до 50 % по массе будут падать на центральный электрод, в то время как с массами больше
50% — на внешний электрод, вследствие того, что фрагменты будут обладать такой же
скоростью как ионы-прекурсоры и, соответственно, их энергии будут пропорциональ-
ны их m/z и их орбиты будут вытягиваться в длинные эллипсы. Поэтому во всех практи-
ческих применениях Orbitrap используется только как детектор точных масс.
Семейство серийных масс-спектрометров Orbitrap
Поскольку МС/МС в орбитальной ловушке проводить невозможно, первые приборы
на их основе были гибридными, где первым анализатором является линейная ква-
друпольная ионная ловушка, а между ней и Orbitrap используется С-образная ловуш-
ка. Такая геометрия позволяет выбирать ионы-прекурсоры и разнообразные методы
фрагментации.
Три года спустя появилась отдельно стоящая орбитальная ловушка, в которой пер-
вый масс-анализатор был заменен на простое мультипольное устройство трансфера
ионов из ионного источника. Еще через три года появилась версия гибрида, в котором
на первой стадии используется квадрупольный масс-анализатор, позволяющий, благо-
даря высокой скорости, делать выборку ионов. Выбранные ионы затем могут фрагмен-
тироваться в высокоэнергетичной камере соударений (HCD). Заполнение ионами HCD
или С-образной ловушки, в то время как Orbitrap детектирует ионы из предыдущего
:.......Finnigan LTQ™ Linear Ion Trap.....:
API Ion source
Linear Ion Trap
Differential pumping
Рис. Д1.2. Схема линейной квадрупольной ионной ловушки/орбитальной ловушки ионов
Современные тенденции практического использования
Рис. ДЕЗ. Схема трибридного масс-спектрометра
цикла, позволяет сосредоточить большую часть времени на накоплении ионов (70—95 %
цикла), благодаря чему возрастает чувствительность без принесения в жертву скорости
анализа и качества спектров.
В 2013 году новое поколение орбитальных ловушек впервые ввело в масс-спектро-
метрическую терминологию слово «трибридный». Новый прибор соединяет в себе ор-
битальную ловушку ионов, на которой можно достигнуть разрешения 450000, с квадру-
польным анализатором и двухкамерной квадрупольной линейной ионной ловушкой
(рис. ДЕЗ).
Естественно, в процессе совершенствования технологии основные аналитические
параметры — разрешение, точность измерения массы, диапазон масс, чувствитель-
ность — постоянно улучшаются.
Это позволяет постоянно расширять области практического использования прибо-
ров Orbitrap для аналитических измерений.
Современные тенденции практического
использования Orbitrap
Технология Orbitrap явилась платформой для множества практических применений
в различных сферах анализа. Высокое разрешение по массам, точность измерения масс
и широкий динамический диапазон позволяют использовать масс-спектрометры на его
основе для характеризации сложных матриц.
Гибридная и трибридные версии приборов дают возможность проведения много-
уровневых МСП экспериментов с целью выяснения детальной структуры сложных
молекул, например, для идентификации белков без их предварительного гидролиза
на пептиды.
В тоже время, относительно простые одностадийные приборы с успехом использу-
ются для множества применений, требующих точного измерения масс органическихсо-
единений — анализа метаболитов и фармакокинетики, скрининга неизвестных соеди-
Дополнение 1. Основы и достижения масс-спектрометрии на основе орбитальной
ловушки ионов. Краткий обзор
нений и остаточных количеств экзогенных примесей — пестицидов, микротоксинов,
ветеринарных препаратов и множество других задач.
Пищевая безопасность и объекты окружающей
среды
В контексте пищевой безопасности и анализа объектов окружающей среды, чем более
сложной является матрица образца, в котором нужно определять экзогенные компо-
ненты, и чем ниже требуемый предел обнаружения, тем более высокая требуется селек-
тивность. Эти требования определяют возможность правильного анализа, позволяю-
щего избегать ложно-положительные или ложно-отрицательные результаты.
Все большее количество исследований токсинов, пестицидов и остаточных коли-
честв ветеринарных препаратов основывается на масс-спектрометрии высокого разре-
шения (A. Kaufmann, Р. Butcher, К. Maden, S. Walker and М. Widmer, Semi-targeted residue
screening in complex matrices with liquid chromatography coupled to high resolution mass
spectrometry: current possibilities and limitations, Analyst, 136, 1898—1909 (2011)).
Было показано сколь важным является точность измерения массы при исследова-
нии сложных матриц образца (L. Alder, A. Steinborn and S. Bergelt, Suitability of an Orbitrap
mass spectrometer for the screening of pesticide residues in extract of fruits and vegetables, J.
AOAC Inti. 94, 1 — 13 (2011). M. Kellmann, H. Muenster, P. Zomer and H. Mol, Full scan MS in
comprehensive qualitative and quantitative residue analysis in food and feed matrices: how much
resolving power is required, J. Am. Soc. Mass Spectrom., 20, 1464—1476 (2009). H.G.J. Mol,
R.C.J. Van Dam, P. Zomer and P.P.J. Mulder, Screening of plant toxins in food, feed and
botanicals using full-scan high-resolution (Orbitrap) mass spectrometry, Food Addit. Contain.,
28, 1405-1423 (2011)).
Результаты, полученные при различных величинах разрешения (от 10,000 до 100,000
на половине высоты на m/z 200), показали, что для надежного определения точной мас-
сы (отклонение менее 2 ррт) анализируемых соединений в сверхмалых количествах
в сложной матрице требуется высокое разрешение. При использовании разрешения
меньше чем 50000, увеличивается ошибка определения массы, вследствие интерферен-
ции соэлюируемых соединений с такой же номинальной массой, как у анализируемого
соединения.
Для увеличения производительности анализа с масс-спектрометром Orbitrap ис-
пользовалась система хроматографии в турбулентном потоке. В этом хроматографи-
ческом методе анализируемые соединения отделяются от матрицы на специфических
колонках с большим размером частиц наполнения. При этом относительно небольшие
молекулы анализируемых соединений удерживаются фазой, в то время как большие мо-
лекулы — такие как белки или липиды — вымываются из колонки. Такая полностью
автоматизированная система анализа использовалась для быстрого скрининга и под-
тверждения по точным массам пестицидов на уровнях концентраций много ниже, чем
самые жесткие требования Европейского Союза (Т. Bernsmann, Р. Fuerst and М. Godula,
Quick screening of priority b-agonists in urine using automated TurboFlowTM-LC/Exactive
mass spectrometry, Food Addit. Contain., 28, 1352—1363 (2011).
Заключение
Возможность записывать масс-спектры высокого разрешения как положительно,
так и отрицательно заряженных ионов в одной хроматографической разгонке, позво-
ляет детектировать на самом низком уровне такие загрязнители как биогенные амины,
морские биотоксины и микотоксины.
Источником загрязнений могут быть и упаковочные материалы. Одним из при-
меров может служить определение бензофенона, методика быстрого, высокочувстви-
тельного и высокоселективного определения которого по высокому разрешению была
разработана.
Orbitrap может быть соединен с источником DART, позволяющим проводить пря-
мой анализ с поверхности образца. Такая система была использована для анализа оста-
точных количеств пестицидов и требует гораздо меньше трудозатрат и времени по срав-
нению с традиционными методами.
Помимо анализа «плохих» компонентов, на котором концентрируются большин-
ство аналитиков, интерес представляет и анализ «хороших» компонентов. Например,
антационины, ответственные за красный, пурпурный или темно-синий цвета многих
фруктов, обладают положительным воздействием на функцию обучаемости человека.
Благодаря высокой селективности при высоком разрешении, анализ на Orbitrap
в 200 раз более чувствителен по сравнению с тройными квадрупольными масс-
спектрометрами и позволяет дифференцировать изобарные компоненты, массы кото-
рых различаются всего на 21 мДа с линейным динамическим диапазоном в 4 порядка.
(W. Mullen, S. Larcombe, К. Arnold, Н. Welchman and A. Crozier, Use of accurate mass full
scan mass spectrometry for the analysis of anthocyanins in berries and berry-fed tissues, J. Agric.
Food Chem., 58, 3910-3915 (2010)).
Заключение
Масс-анализаторы Orbitrap стали мощным прибавлением в арсенале масс-спектро-
метрии, увеличив достоверность и селективность для исследовательского и рутинного
анализа в широком диапазоне применений от идентификации компонентов до следово-
го анализа в сложных матрицах в протеомике, исследовании метаболизма, допинговом
контроле или детектировании загрязнителей в пищевых продуктах и объектах окружа-
ющей среды.
Технология орбитальной ловушки ионов, без сомнения, будет развиваться за счет
увеличения скорости сканирования, разрешения, точности определения массы
и чувствительности.
ДОПОЛНЕНИЕ 2
ВЫСОКОПРОИЗВОДИТЕЛЬНЫЕ
ХРОМАТОМАСС-СПЕКТРОМЕТРЫ
SHIMADZU UFMS: ПРИМЕНЕНИЕ
ДЛЯ АНАЛИЗА ОБЪЕКТОВ
ОКРУЖАЮЩЕЙ СРЕДЫ,
ПИТЬЕВОЙ ВОДЫ, ПИЩЕВОЙ
И СЕЛЬСКОХОЗЯЙСТВЕННОЙ
ПРОДУКЦИИ
Ведущий мировой производитель аналитического оборудования Shimadzu Corporation
является лидером в разработке технологий и оборудования для реализации методов
быстрой и сверхбыстрой хроматографии и хроматомасс-спектрометрии, которые по-
зволяют кардинальным образом сократить время анализа и тем самым существенно
увеличить производительность работы аналитических лабораторий. С момента своего
основания в 1875 году SHIMADZU уделяет особое внимание защите окружающей сре-
ды и повышению качества жизни населения. Большое значение специалисты Shimadzu
Corporation придают методической поддержке своих пользователей. Ежегодно публику-
ется большое количество примеров практического применения приборов и сборников
методических рекомендаций по использованию аналитических инструментов в раз-
личных областях науки и промышленности.
За последние нескольколет компания выпустила на рынок ряд уникальных высоко-
производительных хроматомасс-спектрометров, объединенных в линейку под общим
названием UFMS (Ultra Fast Mass Spectrometry): газовые квадрупольные хроматомато-
масс-спектрометры GCMS-QP2010Ultra и GCMS-TQ8030, жидкостный квадрупольный
масс-спектрометр LCMS-2020 и две модели тандемных квадрупольных жидкостных
масс-спектрометров LCMS-8030 и LCMS-8040.
Благодаря своим техническим характеристикам GCMS-QP2010Ultra (рис. Д2.1)
занимает особое место на рынке газовых хроматомасс-спектрометров, что в первую
очередь связано с быстродействием квадрупольного масс-селективного детектора. За-
патентованная технология Advanced Scanning Speed Protocol™ (ASSP™) обеспечивает
непревзойденную на сегодняшний день максимальную скорость сканирования, со-
ставляющую 20000 а. е. м./с. При этом, в отличие оттрадиционных квадрупольных масс-
анализаторов, с ростом скорости не происходит снижения чувствительности, которая
Дополнение 2. Высокопроизводительные хроматомасс-спектрометры
Shimadzu UFMS: применение для анализа объектов окружающей среды,
питьевой воды, пищевой и сельскохозяйственной продукции
Рис. Д2.1.
является одной из самых высоких среди приборов аналогичного класса (S/N > 850, EI,
Scan 1 пг OFN m/z Til).
В дополнение к сверхбыстрому масс-селективному детектору GCMS-QP2010Ultra
оснащен сверхмощной системой вакуумирования, включающей дифференциаль-
ный турбомолекулярный насос суммарной производительностью 364 л/с. Вследствие
этого величина потока газа-носителя через аналитическую колонку может достигать
15 мл/мин, что делает возможным использование для анализа любых типов капилляр-
ных колонок, включая макрокапиллярные колонки (wide-bore capillary columns). Более
того, мощная система вакуумирования позволяет одновременно подключать две капил-
лярные колонки к интерфейсу масс-спектрометра.
Данный хроматомасс-спектрометр может оснащаться как традиционной системой
ионизации электронным ударом (EI), так и системами положительной и отрицательной
химической ионизации (CI и NCI). Основные технические характеристики прибора
приведены втабл. Д2.1.
Определение пестицидов в питьевой воде, объектах окружающей среды и пище-
вой продукции остается одним из приоритетных направлений прикладной аналити-
ческой химии. Так, например, в Японии количество пестицидов, содержание кото-
рых в пищевых продуктах и объектах окружающей среды должно контролироваться
в обязательном порядке, составляет более 240. В настоящее время наиболее широко
используемым методом определения остаточного содержания пестицидов являет-
Таблица Д2.1. Основные технические характеристики GCMS-QP2010Ultra
Диапазон определяемых масс: 1,5-1090 m/z
Разрешение: 2 М (FWHM)
Методы ионизации: Электронный удар (EI) Положительная химическая ионизация (CI) Отрицательная химическая ионизация (NCI)
Максимальная скорость сканирования: 20000 а. е. м./сек.
Чувствительность: EI: S/N > 850, Scan, 1 пг OFN, m/z 272 CI: S/N > 500, Scan, 100 пг бензофенона, m/z 183 NCI: S/N > 500, Scan, 100 фг OFN, m/z 272
Система вакуумирования: Дифференциальный турбомолекулярный насос суммарной производительностью 364 л/сек
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
ся именно газовая хроматомасс-спектрометрия. Поэтому наряду с традиционными
коммерческими библиотеками масс-спектров, такими как NIST и WILEY, газовые
масс-спектрометры Shimadzu могут комплектоваться специализированными библио-
теками масс-спектров пестицидов собственной разработки. Уникальная особенность
этих библиотек заключается в том, что они содержат масс-спектры, полученные как
в режиме ионизации электронным ударом, так и в режиме отрицательной химической
ионизации. Использование отрицательной химической ионизации при определе-
нии ряда пестицидов в образцах со сложной матрицей (экстрактах из почвы, расте-
ний и т. п.) позволяет существенно увеличить чувствительность анализа [1]. В случае
одинаковой чувствительности анализа в режимах EI и NCI последний является более
предпочтительным при анализе образцов со сложной матрицей, поскольку дает воз-
можность значительно снизить интерферирующее влияние компонентов матрицы,
тем самым увеличивая соотношение сигнал/шум и, соответственно, чувствитель-
ность и селективность анализа.
Как правило, при определении пестицидов необходимо детектировать значительное
число (до 30—40) индивидуальных соединений в одном образце, что обуславливает про-
должительное время анализа при использовании стандартных капиллярных колонок
и традиционных хроматографических режимов. Так, например, при анализе смеси из 30
пестицидов с использованием стандартной 30-метровой капиллярной колонки время
определения составляет порядка 45—60 минут. Быстрая хроматография и хроматомасс-
спектрометрия с использованием коротких и узких капиллярных колонок позволяют
кардинально сократить время анализа при сохранении требуемой эффективности раз-
деления [2]. Поскольку в случае быстрой хроматографии и хроматомасс-спектрометрии
полная ширина на половине высоты хроматографического пика (FWHM) может со-
ставлять порядка 0,5 с [3]. Это обуславливает использование детекторов с максимально
Таблица Д2.2. Определяемые пестициды
№№ пика Определяемое соединение №№ пика Определяемое соединение
1 DDVP 15 Benthiocarb
2 Etridiazole 16 Chloropyrifos
3 Chloroneb 17 Captan
4 МСРР methyl ester 18 Pendimethalin
5 ВРМС 19 Isofenphos
6 Pencycuron 20-1 Isoprothiolane
7 Bethrodine 20-2 Napropamide
8 CAT 21 Butamifos
9-1 TPN 22 Flutolanil
9-2 Propyzamide 23 Isoxathion
10 Diazinon 24 Mepronil
11 IBP 25 CNP
12 Tolclofbs-methyl 26 Pyridaphenthion
13 Terbucarb 27 Iprodion
14 МЕР 28 EPN
Q 616 Дополнение 2. Высокопроизводительные хроматомасс-спектрометры
Shimadzu UFMS: применение для анализа объектов окружающей среды,
питьевой воды, пищевой и сельскохозяйственной продукции
Таблица Д2.3. Условия анализа
Прибор: Shimadzu GCMS-QP2010Ultra
Хроматографическая колонка: DB-1, 10 м (дл.) х 0,10 мм (вн. д.) х 0,10 мкм (п. н. ф.) (Agilent Technologies, США)
Температурная программа: 60°С (1,5 мин.) - 170°С @ 90°С/мин - 250°С @ 20°С/мин. (3 мин.)
Температура инжектора: 280 °C
Температура интерфейса: 280 °C
Газ-носитель: гелий, режим программирования давления на входе в колонку
Давление газа-носителя: 680 кПа
Режим ионизации: электронный удар (EI)
Режим ввода пробы: без деления потока
Режим измерения: SCAN
возможным быстродействием [4]. На сегодняшний день Shimadzu GCMS-QP2010Ultra
является самым быстродействующим газовым квадрупольным хроматомасс-спек-
трометром, что делает его лучшим в своем классе для реализации быстрой газовой
хроматомасс-спектрометрии.
Ниже приведен пример определения содержания остаточного количества 28 пе-
стицидов (табл. Д2.2) в воде методом быстрой газовой хроматомасс-спектрометрии [5].
Общее время анализа при этом составило немногим более 5 минут.
Таблица Д2.4. Чувствительность в режиме SCAN (предел обнаружения (пг) был установ-
лен при соотношении сигнал/шум, равном 3
№№ пика Определяемое соединение S/N = 3 (nr) №№ пика Определяемое соединение S/N = 3 (nr)
1 DDVP 4,4 15 Benthiocarb 1,9
2 Etridiazole 6,3 16 Chloropyrifos 6,1
3 Chloroneb 3,5 17 Captan 39,7
4 МСРР methyl ester 3,3 18 Pendimethalin 32,6
5 ВРМС 2,9 19 Isofenphos 9,8
6 Pencycuron 2,1 20-1 Isoprothiolane 9,2
7 Bethrodine 3,8 20-2 Napropamide 9,8
8 CAT 4,7 21 Butamifos 36,0
9-1 TPN 5,1 22 Flutolanil 1,4
9-2 Propyzamide 12,0 23 Isoxathion 47,9
10 Diazinon 13,8 24 Mepronil 4,9
11 IBP 6,0 25 CNP 39,3
12 Tolclofbs-methyl 1,5 26 Pyridaphenthion 33,6
13 Terbucarb 1,2 27 Iprodion 20,6
14 МЕР 21,9 28 EPN 41,3
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
Таблица Д2.5. Воспроизводимость результатов в режиме быстрой хроматомасс-спектро-
метрии (результаты получены при анализе стандартных образцов пестици-
дов (200 мкг/л) в пятикратной повторности)
№№ пика Определяемое соединение CV, % №№ пика Определяемое соединение CV, %
1 DDVP 1,04 15 Benthiocarb 0,50
2 Etridiazole 5,22 16 Chloropyrifos 5,46
3 Chloroneb 3,85 17 Captan 4,63
4 МСРР methyl ester 7,44 18 Pendimethalin 7,42
5 ВРМС 3,28 19 Isofenphos 4,70
6 Pencycuron 5,28 20-1 Isoprothiolane 2,83
7 Bethrodine 5,47 20-2 Napropamide 4,31
8 CAT 3,47 21 Butamifos 5,24
9-1 TPN 7,20 22 Flutolanil 0,99
9-2 Propyzamide 2,12 23 Isoxathion 5,54
10 Diazinon 3,90 24 Mepronil 3,03
11 IBP 4,64 25 CNP 4,68
12 Tolclofos-methyl 1,91 26 Pyridaphenthion 4,60
13 Terbucarb 3,77 27 Iprodion 5,29
14 МЕР 5,50 28 EPN 5,16
На рис. Д2.2 и Д2.3 показаны хроматограмма по полному ионному току и приме-
ры калибровочных кривых для некоторых пестицидов, а в табл. Д2.4 и Д2.5 приведе-
ны результаты, демонстрирующие чувствительность анализа и воспроизводимость
результатов.
Таким образом, по сравнению с традиционной газовой хроматомасс-спектроме-
трией быстрая хроматомасс-спектрометрия обеспечивает сокращение времени анали-
за в 5—15 раз без потери эффективности разделения, чувствительности, селективности
и воспроизводимости.
В тех случаях, когда исследуемый образец содержит большое количество опреде-
ляемых соединений и/или характеризуется очень сложной матрицей, разрешающая
способность хроматографической системы может быть недостаточна даже при ис-
пользовании капиллярных колонок большой длины (> 50 метров). Для анализа таких
сложных многокомпонентных смесей SHIMADZU предлагает системы двумерной га-
Рис. Д2.2
Дополнение 2. Высокопроизводительные хроматомасс-спектрометры
Shimadzu UFMS: применение для анализа объектов окружающей среды,
питьевой воды, пищевой и сельскохозяйственной продукции
Рис. Д2.3
зовой хроматографыи/хроматомасс-спектрометрии. Система MDGC-2010 (рис. Д2.4)
построена на базе газового хроматографа GC-2010Plus и хроматомасс-спектрометра
GCMS-QP2010Ultra. В системе используются две капиллярные колонки с различными
характеристиками (например с различной полярностью неподвижной фазы). Те компо-
ненты анализируемой смеси, которые не могут быть эффективно разделены на первой
колонке, при помощи пневматического переключателя газовых потоков направляются
во вторую колонку, где и происходит окончательное разделение. Таким образом, полу-
чают разрешение пиков, которое не может быть достигнуто при традиционном анализе
на одной капиллярной колонке. Система MDGC-2010 использует инновационную тех-
нологию многократного переключения газовых потоков («вырезания» хроматографиче-
ских пиков, multiple-heart-cutting) при помощи усовершенствованного пневматического
переключателя Динса (Multi-Deans Switch). В отличие от традиционного переключателя
Динса при использовании усовершенствованной модели не происходит сдвига времен
удерживания элюируемых компонентов во время разделения на первой колонке даже
в случае «вырезания» большого числа (до 10) пиков. При этом разделение на второй ко-
лонке также характеризуется превосходной воспроизводимостью времен удерживания.
Конструкция мультипереключателя Динса отличается чрезвычайно малым мертвым
объемом и предусматривает использование деактивированных капилляров. Следстви-
ем этого является анализ с высокой чувствительностью и воспроизводимостью резуль-
Рис. Д2.4
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
The Loop Modulator assembly, shown in accumulation
mode, hot jet off
Key: (I) Cold jet assembly
(2) hot jet assembly
(3) modulator tube
(4) column holder
(5) Kapton film tensioner
(6) cold gas jet
Bottom view of the Loop Modulator assembly
Рис. Д2.5
татов. Существует и более экономичное решение, когда обе колонки и пневматический
переключатель установлены в термостате одного хроматомасс-спектрометра, оснащен-
ного дополнительным атмосферным детектором.
Другой подход к реализации техники двумерной хроматомасс-спектрометрии —
комплексная двумерная хроматомасс-спектрометрия (Comprehensive GCxGCMS), впер-
вые предложенная Дж. Филлипсом в 1989 году [6, 7], основан на использовании специ-
ального криогенного модулятора производства Zoex Corporation, США (рис. Д2.5л,0.
При этом используются две хроматографические колонки разной длины и с разными
неподвижными фазами. В отличие от традиционной техники с «вырезанием» пиков
используется только один масс-селективный детектор. Компоненты анализируемой
смеси разделяются на первой длинной колонке и поступают в криогенный модулятор,
который отделяет небольшие фракции элюата, фокусирует их в узкую зону и направ-
ляет во вторую короткую колонку. Каждый такт модулятора генерирует отдельную
«быструю» хроматограмму с чрезвычайно узкими пиками, для регистрации которых
необходимо использовать быстродействующий масс-селективный детектор. Именно
поэтому на сегодняшний день техника Comprehensive GCxGCMS наиболее полно реа-
9.050 9.075 9.100 9.125 9.150 9.175 9.200 9.225 9.250 9.275 9.300 9.325 9.350 9.375
Рис. Д2.6
Дополнение 2. Высокопроизводительные хроматомасс-спектрометры
Shimadzu UFMS: применение для анализа объектов окружающей среды,
питьевой воды, пищевой и сельскохозяйственной продукции
лизована на базе газовых хроматомасс-спектрометров производства SHIMADZU в со-
четании со специализированным программным обеспечением ChromSquare™, которое
наглядно представляет результаты двумерного разделения.
Ниже приведен пример использования комплексной двумерной хромато-
масс-спектрометрии для определения содержания полихлорированных бифени-
лов (ПХБ) [8] на базе хроматомасс-спектрометра GCMS-QP2010Ultra с установлен-
ным криогенным двухстадийным (холод/тепло) модулятором Zoex Corporation.
Периодичность работы модулятора составляла 1 такт за 8 секунд. Для разделения
образца на первой стадии применяли 30-метровую капиллярную колонку ВРХ-
1 (0,25 мм (вн. д.) х 0,25 мкм (п.н.ф.), SGE Europe Ltd, Великобритания), для фи-
нального разделения — колонку для быстрой хроматографии ВРХ-50 (1,0 м (дл.) х
х 0,15 мм (вн. д.) х 0,15 мкм (п.н.ф.), SGE Europe Ltd). Измерения проводили при
ионизации электронным ударом (EI) и отрицательной химической ионизации (NCI)
в режимах сканирования (SCAN) и мониторинга отдельного иона (SIM). Для оценки
количественных характеристик анализа использовали эталонный образец, содержа-
щий ПХБ и диоксины, растворенные в концентрации 10—250 пг/г в масле. На рис. Д2.6
показаны SIM-хроматограммы, полученные при анализе стандартного образца тетра-
Рис. Д2.7
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
ПХБ (140 ррт) и эталонного образца. Ширина пиков на «быстрой» хроматограмме
составляла ~350 мс, при этом для получения достоверных количественных результа-
тов на каждый пик приходилось не менее 20—30 измерений. На рис. Д2.7 приведены
калибровочные кривые для стандартных образцов ПХБ-28 и ПХБ-114 (4—140 ррт,
суммировались площади пиков целевого компонента всех «быстрых» хроматограмм).
При измерении в режиме SCAN был выбран диапазон масс 34—500 а.е. м., при этом
на каждый пик приходилось не менее 20 измерений.
Как видно из итогового двумерного изображения, полученного при анализе эта-
лонного образца в режиме SCAN с использованием ионизации электронным ударом,
компоненты матрицы образца оказывают существенное интерферирующее воздей-
ствие на результаты анализа (рис. Д2.8). Идентификация ПХБ осуществляется пу-
тем выделения целевого иона при помощи программного обеспечения ChromSquare
(рис. Д2.9). Альтернативой этому является использование отрицательной химической
ионизации для кардинального снижения интерферирующего влияния компонентов
матрицы. На рис. Д2.10 приведены результаты анализа эталонного образца и стандарт-
ного образца смеси ПХБ в режиме SCAN (34—500 а. е. м.) с использованием отрицатель-
ной химической ионизации. Очевидно, что в данном случае селективность анализа
существенно выше и идентификация целевых соединений не требует дополнительной
программной обработки. На базе Shimadzu Comprehensive GCxGCMS в настоящее вре-
мя разработаны аналитические методики для определения содержания полихлориро-
ванных бифенилов и полибромдифениловых эфиров в питьевой и природной воде [9],
остаточного содержания пестицидов в воде, почве, сельскохозяйственной продукции,
продуктах питания и др.
Рис. Д2.8
Дополнение 2. Высокопроизводительные хроматомасс-спектрометры
Shimadzu UFMS: применение для анализа объектов окружающей среды,
питьевой воды, пищевой и сельскохозяйственной продукции
Рис. Д2.9
Линейку UFMS высокопроизводительных масс-спектрометров SHIMADZU про-
должают жидкостные тандемные квадрупольные масс-спектрометры LCMS-8O3O
и LCMS-8040.
Квадрупольные масс-анализаторы отлично зарекомендовали себя как наиболее
универсальные и чувствительные хроматографические детекторы в системах тра-
диционной ВЭЖХ, однако до недавнего времени их применение в системах быстрой
и сверхбыстрой хроматографии было ограничено вследствие недостаточного бы-
стродействия. Попытки увеличить скорость работы квадрупольных масс-детекторов
приводили к закономерному падению их чувствительности. Решением проблемы
стал разработанный и запатентованный SHIMADZU комплекс технологий под об-
щим названием UF (UltraFast): UFscanning™, UFswitching™ и UFsensitivity™. Выпу-
щенный первым одинарный квадрупольный масс-спектрометр LCMS-2020, в кон-
струкции которого впервые были реализованы эти технологии, и сегодня остается
абсолютным рекордсменом среди одинарных квадрупольных масс-спектрометров
по таким показателям, как скорость сканирования, время переключения полярности
и чувствительность.
За счет технологии UFsensitivity™ и системы ионной оптики Qarray достигаются
высокая чувствительность, великолепная воспроизводимость результатов и широкий
линейный диапазон. Технология UFscanning™ обеспечивает корреляцию между на-
пряжением, приложенным к стержням квадруполя, скоростью сканирования и вели-
чиной m/z определяемых ионов, что гарантирует высочайшую скорость сканирования
(до 15000 а.е. м./с) во всем диапазоне масс без потери чувствительности. Технология
UFswitching™ обеспечивает сверхбыстрое (15 мс) переключение режимов положитель-
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
| Position. (13.767,1.560) | Value: 1624860.0000
State Mass Spetlrum View | Substate: »xel Mass Epectrum
Рис. Д2.10
ной и отрицательной ионизации, что позволяет практически одновременно регистри-
ровать положительные и отрицательные ионы.
Развитие технологии сверхбыстрой квадрупольной масс-спектрометрии стало
основой создания тройного квадрупольного масс-детектора LCMS-8O3O. В дополне-
ние к уже апробированным UF-технологиям специалисты SHIMADZU использо-
вали в конструкции ячейки столкновения этого прибора еще одну инновационную
технологию — UFsweeper™. Ячейки столкновения традиционной конструкции всегда
являлись «узким местом» в тандемной масс-спектрометрии вследствие потери иона-
ми скорости при столкновении с молекулами газа-реагента и наличия взаимных по-
мех при регистрации нескольких MRM-переходов (cross-talk). Ячейка столкновения
LCMS-8O3O практически лишена этих недостатков. Технология UFsweeper™ обеспе-
чивает формирование в ячейке столкновения псевдопотенциальных поверхностей,
что эффективно ускоряет ионы. Тем самым достигается высочайшая эффективность
Дополнение 2. Высокопроизводительные хроматомасс-спектрометры
Shimadzu UFMS: применение для анализа объектов окружающей среды,
питьевой воды, пищевой и сельскохозяйственной продукции
столкновительной диссоциации (CID) и сверхбыстрый транспорт ионов без потерь
во второй квадруполь, что существенно снижает потери чувствительности и взаим-
ные помехи.
Сочетание сверхбыстрых квадрупольных масс-анализаторов и технологии
UFsweeper™ позволило добиться высокой производительности тандемного масс-
спектрометрического анализа: максимальная скорость работы LCMS-8030 в режиме
регистрации MRM-переходов составляет 500 MRM-переходов в секунду. При этом па-
уза между регистрациями — всего 1 мс. В сочетании со сверхбыстрым переключением
полярности это гарантирует надежное и точное детектирование хроматографических
пиков шириной менее 1 с. Для хорошей воспроизводимости результатов в программ-
ном обеспечении масс-спектрометра предусмотрен режим синхронизации регистрации
MRM-переходов со временем выхода хроматографического пика. Более того, техноло-
гия синхронизированного сканирования со скоростью 15000 а.е.м./с позволяет одно-
временно с регистрацией MRM-переходов получать полные масс-спектры продуктов
Таблица Д2.6. Основные технические характеристики LCMS-8030/8040
LCMS-8030 LCMS-8040
Диапазон определяе- мых масс: 10-2000 m/z 10-2000 m/z
Разрешение: < 0,7 М (FWHM) < 0,7 M (FWHM)
Методы ионизации: Электроспрей (ESI) (стандартно) Химическая ионизация при атмосферном давлении (APCI) (опционально) Сдвоенная система ионизации (DUIS) (опционально) Электроспрей (ESI) (стандартно) Химическая ионизация при атмосферном давлении (APCI) (опционально) Сдвоенная система ионизации (DUIS) (опционально)
Максимальная ско- рость сканирования: 15000 а.е. м./с 15000 а.е.м./с
Время переклю- чения полярности ионизации: 15 мс 15 мс
Скорость работы в режиме MRM: 500MRM/C 555 MR М/с
Чувствительность в режиме MRM: S/N > 200:1 (RMS, 1 пг резерпина, w/z609,3 > 195; ESI) S/N > 10000:1 (RMS, 1 пг резерпи- на, m/z 609,3 > 195; ESI+)
Режимы работы MS: QI Scan/SIM Q2 Scan/SIM QI Scan/SIM Q2 Scan/SIM
Режимы работы MS MS: MRM Product ion scan Precursor ion scan Neutral loss scan Synchronized Survey Scanning™: MRM Product ion scan MRM Product ion scan Precursor ion scan Neutral loss scan Synchronized Survey Scanning™: MRM Product ion scan
Уровень перекрест- ных помех (cross-talk): < 0,003%
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
-CMS-8D4D
Рис. Д2.11
фрагментации (режим работы MRM/SCAN), что существенно увеличивает точность
и информативность анализа.
Тройной квадрупольный масс-спектрометр LCMS-8040 (рис. Д2.11) является даль-
нейшим развитием модели LCMS-8O3O, он также оснащен фирменными технология-
ми UFscanning™ и UFswitching™. Существенное преобразование в новой модели пре-
терпели технология ускорения ионов в ячейке столкновений, получившая название
UFsweeper™ II, система вакуумирования и конструкция ионной оптики UF-Lens™.
Засчетэтогодостигнуто пятидесятикратное увеличение чувствительности анализа в ре-
жиме регистрации MRM-переходов по сравнению с моделью LCMS-8O3O (S/N > 10000:1,
Таблица Д2.7. Условия анализа
Прибор: LCMS-8030 с ВЭЖХ LC-20 Prominence XR
Колонка: Shim-pack FC-ODS (2,0 мм вн. д. х 150 мм дл., 3 мкм)
Подвижная фаза А: Ацетат аммония, 5 мМ/вода
Подвижная фаза В: Ацетат аммония, 5 мМ/метанол
Градиент: 30% В (0 мин) - 70%В (5 мин.) - 90% В (15-20 мин.) -30% В (20,01-25 мин.)
Скорость потока: 0,2 мл/мин.
Объем пробы 2,0 мкл
Температура колонки: 40 °C
Напряжение на капилляре источника ионизации: +4,5 кВ (ESI, режим положительной ионизации) -3,5 кВ (ESI, режим отрицательной ионизации)
Поток газа-распылителя: 1,5 л/мин.
Поток газа-осушителя: 10,0 л/мин.
Температура линии десольватации: 250 °C
Температура источника ионизации: 400 °C
Напряжение на линии десольватации/ системе ионной оптики: По умолчанию
Дополнение 2. Высокопроизводительные хроматомасс-спектрометры
Shimadzu UFMS: применение для анализа объектов окружающей среды,
питьевой воды, пищевой и сельскохозяйственной продукции
ТаблицаД2.8. Условия измерения в режиме регистрации MRM-переходов и результаты
анализа стандартных образцов
Определяемое соединение Поляр- ность MRM-переход Энергия соударе- ния, эВ CV, % (*) S/N (*) Минимально определяемая концентрация (мкг/л)п(*)
Thiamethoxam + 292,00211,10 -10 4,3 18 0,28
Acetamiprid + 223,05> 126,05 -20 4,5 36 0,14
Halosulfuron-methyl + 435,05> 182,10 -20 6,6 29 0,17
Ethoxysulfron + 399,10>261,00 -16 6,1 22 0,23
Simazine + 202,10> 124,15 -20 6,3 11 0,45
Cyclosulfamron + 422,05>261,05 -20 2,9 83 0,06
Metalaxyl + 280,00>220,15 -15 2,0 40 0,13
Azoxystrobin + 404,10>372,05 -15 2,4 26 0,19
Mepronil + 270,05>l 19,10 -25 5,9 13 0,38
Propyzamide - 254,10>228,05 +15 5,5 13 0,38
Cumyluron + 303,05> 185,15 -15 4,3 12 0,42
Bensulide - 396,10>213,10 + 10 6,7 13 0,38
Tebuconazole + 308,10>70,05 -25 4,2 18 0,28
Propiconazole + 342,00>69,10 -25 4,8 10 0,50
Diazinon + 305,05> 169,15 -20 6,9 12 0,42
Butamifbs + 333,05> 180,15 -10 4,1 27 0,19
Isoxathion + 314,00> 105,05 -15 3,9 18 0,28
Pencycuron + 329,05> 125,10 -25 5,9 25 0,20
Triflumizole + 346,05>278,15 -10 4,6 15 0,33
Oxaziclomefone + 376,05>190,20 -15 з,з 25 0,20
Pyributicarb + 331,05>181,15 -15 1,9 16 0,31
(*) — на основании измерений в 6-кратной повторности
1 пг резерпина, ESI+). Основные технические характеристики LCMS-8030 и LCMS-8040
приведены в табл. Д2.6.
Для интенсификации и облегчения работы аналитических лабораторий
SHIMADZU предлагает готовые пакеты аналитических методик (Method Packages)
к жидкостным хроматомасс-спектрометрам. Пакеты методик представляют собой
комплексное решение аналитической задачи, включая метод-файлы с условиями хро-
матографического разделения и работы масс-спектрометра в режимах регистрации
MRM-переходов и MRM/Product ion Scan, калибровочные данные и библиотеки масс-
спектров продуктов фрагментации. Также пакеты методик содержат подробное опи-
сание процедуры пробоподготовки, состава подвижной фазы и хроматографической
колонки. По желанию пользователя вместе с пакетами методик могут поставляться
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
0.0 2.5 5.0
7.5 10.0 12.5 15.0 min
292.00>211.10 (+)
223.05>126.05 (+)
435.05>182.00 (+)
399.10>261.00 (+)
202.10>124.15 (+)
422.05>261.05 (+)
280.00>220.15 (+)
404 10>372 05(»)
270.05>119.10 (+)
303.05>185.15 (+)
342.00>69.10 (+)
305.05>169.15 (+)
333.05>180.15 (+)
314.00>105.05 (+)
329.05>125.10 (+)
346.05>278.15 (+)
376.05>190.20 (+)
331.05>181.15 (+)
254 10>228 05 (-)
396.10>213.10 (-)
Thiamethoxam
Acelamiprid
Halosulfuron-methyl
Ethoxysulfuron
Simazine
Cyclosulfamuron
Metalaxyl
Azoxyskob n
Meproml
Cumyluron
Propiconazole
Diazinon
Butamifos
Isoxalhion
Pencycuron
Tnflumizole
Oxaziclomefone
Pynbuticarb
Propyzarrde
Bensulide
Рис. Д2.12
Таблица Д2.9. Основные технические характеристики GCMS-TQ8030
Диапазон определяемых масс: 1,5-1090 m/z
Разрешение: 1,5 М (FWHM)
Методы ионизации: Электронный удар (EI) Положительная химическая ионизация (CI) Отрицательная химическая ионизация (NCI)
Максимальная скорость сканирования: 20000 а.е. м./сек.
Скорость работы в режиме MRM: > 600 MRM/сек.
Чувствительность: EI, Scan: S/N > 600, 1 пг OFN, m/z 272 EI, MRM: S/N > 3000, 100 фг OFN, m/z 272>241 CI, MRM: S/N > 1500, 1 пгбензофенона, m/z 193 > 110 NCI, SIM: S/N > 400, 10 фг OFN, m/z 212
Режимы работы MS: QI Scan/SIM Q2 Scan/SIM
Режимы работы MS/MS: MRM Product ion scan Precursor ion scan Neutral loss scan Scan MRM
Система вакуумирования: Дифференциальный турбомолекулярный насос суммарной производительностью 364 л/сек
628 Дополнение 2. Высокопроизводительные хроматомасс-спектрометры
Shimadzu UFMS: применение для анализа объектов окружающей среды,
питьевой воды, пищевой и сельскохозяйственной продукции
Рис. Д2.13
соответствующие стандартные образцы и колонка. В настоящее время пользователям
предлагается около десяти вариантов пакетов методик для использования в различных
областях, в том числе и пакет для анализа питьевой и природной воды (определение 80
индивидуальных соединений).
Ниже показан пример анализа природной воды на содержание остаточного коли-
чества пестицидов с использованием пакета методик. Условия и результаты приведены
в табл. Д2.7 и Д2.8.
На рис. Д2.12 приведена хроматограмма, полученная при анализе стандартных об-
разцов пестицидов в режиме регистрации MRM-переходов, а на рис. Д2.13 — примеры
калибровочных кривых. Образцы сточных вод были отобраны, профильтрованы че-
рез фильтр 0,45 мкм и далее проанализированы в соответствии с условиями, описан-
ными выше. На рис. Д2.14 приведены примеры анализа нативных образцов и образцов
с добавками стандартов пестицидов (конечная концентрация 10 и 50 мкг/л). В реаль-
ных образцах содержание пестицидов обнаружено не было, в образцах же с извест-
15000- 10000- 202.10>124.15(+) 'l S/N=151 /, § i 1. 1.... 1.... 305.05>169.15(+) S/N=136 n !\ ! I 400007 300007 314.00>105.05(4-) S/N =203 i I I 2000007 1500007 346.05>278.15(4-) S/N =593 [ ( I I | I
5000- ! । Spiked cond: 10 pg/L Guideline target: 30 pg/L 5000 7 I ' Spiked comf.: 1p pg/L Guideline target 50 pg/L I 20000- I ' Spiked cone.: W pg/L Guideline target 180 pg/L 1000007 Spiked cone! 50'pg/L Guideline talget 500 pg/L I '
2500 7 100007 500007
о- °7 0- —J °7 J V.
65 ' ' 70 ' 1 1 1 I 1 1 1 1 I 1 11.5 12.0 'n's' ' ' '12'0' 13.0 13.5
Рис. Д2.14
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
Рис. Д2.15
Таблица Д2.10. Определения остаточного содержания пестицидов в экстракте из образ-
цов сельскохозяйственной продукции
Определяемое соединение Концентрация,ppb
Дифениламин 227,50
Кадусафос 79,77
Диазенон 29,75
Фтал ид 27,89
Ципродинил 314.96
Фольпет 428,62
Флудиоксонил 194,94
Гексазинон 1,06
Фосмет 93,52
Боскалид 226,29
питьевой воды, пищевой и сельскохозяйственной продукции
ними добавками пестициды идентифицировались с превосходным соотношением
сигнал/шум.
Технологии, разработанные при создании масс-спектрометров GCMS-QP2010Ultra
и LCMS-8030/8040, получили свое дальнейшее развитие в еще одной модели линейки
UFMS — тандемном квадрупольном газовом хроматомасс-спектрометре GCMS-TQ8030
(рис. Д2.15). Комбинация технологий ASSP™ и UFsweeper™ позволила добиться фан-
тастической производительности тандемного масс-спектрометрического анализа:
максимальная скорость работы GCMS-TQ8030 в режиме регистрации MRM-переходов
составляет более 600 MRM-переходов в секунду с паузой между регистрациями ме-
нее 1 мс. При этом чувствительность анализа в режиме регистрации MRM достигает
впечатляющей величины: отношение сигнал/шум составляет более 3000 (EI, 100 фг
OFN, m/z 272>241). Благодаря быстродействию GCMS-TQ8030 позволяет реализовать
практически любые комбинированные режимы измерения: SCAN/SIM, MRM/SCAN,
MRM/SIM, MRM/Product ion Scan, MRM/Precurson ion Scan. Основные технические ха-
рактеристики прибора приведены в табл. Д2.9.
Возможности GCMS-TQ8030 проиллюстрированы ниже на примере определения
содержания пестицидов в экстракте из образцов сельскохозяйственной продукции [10].
Анализ проводили на колонке Rxi-5Sil (20 м (дл.) х 0,15 мм (вн. д.) х 0,15 мкм (п.н.ф.),
Restek, США), которая позволяет после оптимизации режима измерения разделить бо-
лее 50 индивидуальных пестицидов менее чем за 12 минут (рис. Д2.16). Использовали
комбинированный режим быстрой хроматомасс-спектрометрии MRM/Product ion Scan.
Результаты анализа представлены в табл. Д2.10 и демонстрируют, что быстрая тандем-
ная газовая хроматомасс-спектрометрия дает точные и исчерпывающие качественные
и количественные данные в ходе одного измерения за короткое время.
Режим регистрации MRM-переходов обеспечивает высокую чувствительность и се-
лективность анализа и используется для количественной оценки величины остаточного
содержания пестицидов. Информация, полученная в режиме Product ion Scan, исполь-
зуется для гарантированной идентификации определяемого соединения.
Таким образом, на сегодняшний день SHIMADZU предлагает для химиков-анали-
тиков линейку хроматомасс-спектрометров, которые представляют собой квинтэссен-
цию передовых технологий в области масс-спектрометрии и позволяют решать любые
сложные задачи с высочайшей эффективностью и производительностью.
Список литературы
[1] . J.-F. Focant et al., Shimadzu LCGC. The Application book, March 2009.
[2] . A. van Es, High Speed Narrow Bore Capillary Gas Chromatography, Hiithig, Heidelberg
1992.
[3] . H.-U Baier, L. Mondello. Die schnelle Gaschromatographie in der Lebensmittelanalytik in
Schnellmethoden zur Beurteilung von Lebensmitteln und deren Rohstoffen Кар. 3.2, Behrs Ver-
lag 2004.
[4] . J. Hinshaw, LCGC (2002), vol. 15, p. 152.
[5] . Pesticide Analysis using Fast-GC/MS, Shimadzu Application Note No. M190, LA146-E014
Высокопроизводительные хроматомасс-спектрометры Shimadzu UFMS:
применение для анализа объектов окружающей среды, питьевой воды,
пищевой и сельскохозяйственной продукции
[6] . John В. Phillips, Zaiyou Liu. «Chromatographic Technique and Apparatus», U.S. Patent
No. 5.135.549; August 4, 1992.
[7] . John B. Phillips, Zaiyou Liu. «Apparatus and Method for Multi-dimensional Chemical Sep-
aration» U.S. Patent No. 5,196,039.
[8] . H.-U. Baier, J.-F. Focant. Comprehensive GCxGC (qMS): Qualitative and Quantitative
Analysis of PCBs using EI and NCI Mode with a Rapid Scanning Quadrupole Mass Spec-
trometer, Shimadzu LCGC The Application book, March 09.
[9] . H.-U. Baier. Comprehensive GCxGC(q)MS-NCI analyis of environmental samples: Quali-
tative and Quantitative Analysis of PCBs and PBDEs, Shimadzu LCGC The Application book,
2010.
[10] . С. Креер, Х.-У. Байер. Анализ пестицидов, Labor&More, 02, 2012.
Производство книг на заказ
Издательство «Техносфера»
тел.(495)234-01-10
e-mail: knigi@technosphera.ru
Реклама в книгах:
• модульная
• статьи
Подробная информация о книгах на сайте
www.technosphera.ru
Лебедев Альберт Тарасович
Масс-спектрометрия
для анализа объектов окружающей среды
Компьютерная верстка - С.С. Бегунов
Дизайн - М.А. Костарева
Корректор - Н.А. Шипиль
Выпускающий редактор - О.Н. Кулешова
Ответственный за выпуск - С.А. Орлов
Подписано в печать 12.09.2013.
Формат 70x100/16. Печать офсетная.
Гарнитура Ньютон
Печ.л. 39,5. Тираж 1000 экз., Зак. №
Бумага офсет №1, плотность 65 г/м2.
Издательство «Техносфера»
Москва, ул. Краснопролетарская, д. 16, стр.2
Отпечатано в соответствии с предоставленными материалами
в ООО «ИПК Парето-Принт»,
г. Тверь, www.pareto-print.ru